
Notes
Article history
The research reported in this issue of the journal was commissioned and funded by the HTA programme on behalf of NICE as project number 08/96/01. The protocol was agreed in July 2009. The assessment report began editorial review in February 2010 and was accepted for publication in July 2010. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the referees for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
none
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2011. This work was produced by Picot et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This journal is a member of and subscribes to the principles of the Committee on Publication Ethics (COPE) (http://www.publicationethics.org/). This journal may be freely reproduced for the purposes of private research and study and may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NETSCC, Health Technology Assessment, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2011 Queen’s Printer and Controller of HMSO
Chapter 1 Background
Description of underlying health problem
Multiple myeloma (MM) is a type of cancer. The cancer (myeloma) tends to be located at more than one site where there is bone marrow, such as the pelvis, spine and ribs, which is why it is known as MM. 1 MM occurs when a plasma cell begins to proliferate in an unregulated way. Plasma cells are a specialised component of the bone marrow and immune system and they normally produce specific antibodies to fight infection. In MM the myeloma cells produce large quantities of one type of abnormal antibody – monoclonal immunoglobulin protein (M-protein). 2 As the abnormal myeloma cells build in number, the normal functions of bone marrow become impaired to varying degrees of severity because the abnormal myeloma cells may disrupt the function of normal cells, and because the space available for normal bone marrow may be reduced.
In the early stages of MM there may not be any symptoms or a range of symptoms may be present, which are not specific to MM, such as fatigue, weight loss and increased infections. A common presenting symptom of MM is bone pain, and/or bone fracture due to lytic bone lesions. Lytic bone lesions are a typical feature of MM and are caused because the malignant plasma cells impair normal bone repair functions. MM cells both produce and influence chemokines and cytokines, which causes bone resorption to become uncoupled from bone formation such that resorption predominates. 3
The most common finding on clinical investigation is anaemia. 4 This occurs because the presence of proliferating myeloma cells in the bone marrow negatively impacts on the ability of the bone marrow to produce red blood cells, leading to a reduction in red blood cells in the circulation, which contributes to the symptom of fatigue. Likewise, circulating numbers of other cells produced in the bone marrow are also reduced. The reduction in normal white blood cells and the antibodies these produce (hypogammaglobulinaemia) leads to an increased risk of infection, while the reduction in platelets contributes to easy bruising and other bleeding.
Other common findings on clinical investigation are M-protein, which is secreted by the myeloma cells, and an excess of calcium in the blood (hypercalcaemia), which occurs as a result of bone destruction. 5 The presence of M-protein in serum may increase blood viscosity, which is associated with an increased risk of thrombosis. A high level of serum protein (hyperproteinaemia), M-protein and light chains may also contribute to renal failure. The aetiology of this is generally multifactorial, and hypercalcaemia is another common contributing factor.
Multiple myeloma is one of a number of lymphoproliferative diseases classified by the World Health Organization (WHO) International Classification of Diseases 10th edition (ICD-10) as malignant neoplasms of lymphoid, haematopoietic and related tissue. 6 The exact aetiology of MM is unknown but it is clear that the malignant cells arise from a single plasma cell. Therefore, research has focused on gaining an understanding of the chain of events that occurs between haematopoietic stem cells giving rise to B lymphocytes in the bone marrow, and these B cells subsequently differentiating to form plasma cells. 7,8
Normally, plasma cells would contain a pair of each of the 22 autosomal (non-sex) chromosomes. Myeloma cells, however, display a variety of genetic abnormalities. Common abnormalities of MM cells include aneuploidy (an abnormal number of chromosomes) and translocations (exchange of material between two different chromosomes). When aneuploidy is present, monosomies (one copy of a chromosome) are more common than trisomies (three copies of a chromosome). One of the most common monosomies is the loss of one copy of chromosome 13, which is associated with a shorter survival and lower response rate to treatment. 9,10 Of the translocations t(11;14)(q13;q32) and t(4;14)(p16.3;q32) are the most common; the former is associated with improved survival, whereas the latter is an indication of an unfavourable prognosis. 9,10 The genetic abnormalities underlying cases of MM can be identified by cytogenetic techniques, such as conventional karyotype analysis and fluorescence in situ hybridisation.
Prognosis
Myeloma is not curable, but can be treated with a combination of supportive measures and chemotherapy to improve survival and quality of life (QoL). A range of factors affects prognosis. These include factors related to burden of disease [e.g. beta2-microglobulin (β2-microglobulin)], characteristics of the myeloma cells’ biology (e.g. the type of cytogenetic abnormality present), the microenvironment surrounding the myeloma cells (e.g. bone marrow microvessel density), patient-related factors (e.g. age and performance status) and treatment response factors [e.g. whether complete response (CR) is achieved with initial therapy]. 5 Because of the number of factors that affect prognosis, survival of patients from the point of diagnosis varies from months to over a decade. 4 In the UK and Ireland, median survival increased from around 2 years in the 1980s and early 1990s to around 4 years in the late 1990s. 11 There is evidence from some cohorts of patients that novel therapies can extend median survival time to 8 years. 12
Epidemiology
Multiple myeloma is the second most common haematological cancer after lymphoma in the UK. In 2007 there were 3357 new diagnoses of MM in England,13 with the highest incidence among those aged 75–79 years (Table 1). In Wales, in the 3 years from 2004 to 2006, an average of 252 new MM diagnoses were recorded. 14 MM is rare before the age of 40 years. The average incidence rates were higher in men than in women, and higher for both sexes in Wales compared with England (Table 2). There are ethnic differences in incidence rates that have been observed in data from the USA; in black people (African American and other black people, but not Hispanic people) the incidence of MM is about twice that of white people, whereas in Asian people the incidence is lower than that of white people. 15 The statistical information team at Cancer Research UK has used incidence and mortality data for 2001–5 to estimate the lifetime risk of developing MM, which is 1 in 148 for men and 1 in 186 for women in the UK. 16 There are currently approximately 10,000–15,000 people living with MM in the UK. 17
| Age group (years) | Nos.a | Ratesb | ||
|---|---|---|---|---|
| Males | Females | Males | Females | |
| 20–24 | 2 | 1 | 0.1 | 0.1 |
| 25–29 | 3 | 4 | 0.2 | 0.2 |
| 30–34 | 9 | 4 | 0.5 | 0.2 |
| 35–39 | 11 | 7 | 0.6 | 0.4 |
| 40–44 | 26 | 17 | 1.3 | 0.9 |
| 45–49 | 47 | 31 | 2.7 | 1.7 |
| 50–54 | 96 | 51 | 6.3 | 3.3 |
| 55–59 | 158 | 91 | 10.3 | 5.7 |
| 60–64 | 214 | 174 | 15.1 | 11.7 |
| 65–69 | 239 | 176 | 22.2 | 15.2 |
| 70–74 | 300 | 217 | 32.7 | 20.9 |
| 75–79 | 324 | 248 | 44.8 | 26.8 |
| 80–84 | 256 | 238 | 53.2 | 32.3 |
| 85+ | 173 | 240 | 50.2 | 31.7 |
| Men | Women | |
|---|---|---|
| England | 6.0 | 3.9 |
| Wales | 6.8 | 4.9 |
The risk factors for developing MM are not well defined but there is evidence for involvement of genetic factors because the first-degree relatives of people with MM are at greater risk of developing MM and related conditions than the first-degree relatives of people without MM. 8,18 Epidemiological studies have looked for evidence of a causal link between a range of potential environmental risk factors and MM but, in general, these have not produced consistent results. 4,8
Diagnosis and staging
Multiple myeloma is typically diagnosed in secondary care using a combination of tests such as urine tests, blood tests, bone marrow examination, imaging, plain radiograph and/or magnetic resonance imaging. If necessary, further tests can be conducted to find out the stage of disease. 1 There are two systems for staging MM. The Durie–Salmon19 (DS) staging system, which has been in use since 1975, is one of the systems but this is gradually being replaced by an updated system, the International Staging System (ISS). 20 This new system is based on measurement of two serum proteins, β2-microglobulin and albumin (Table 3). A patient with stage I disease will not necessarily proceed linearly through disease stages. Stage III disease can be reached without a requirement to pass through stage II first. It is also noteworthy that staging does not have a significant influence on treatment. If MM is symptomatic, treatment is required irrespective of disease stage.
| Stage | Criteria | |
|---|---|---|
| DS19 | ISS20 | |
| I | All of the following:
|
|
| II | Fitting neither stage I nor stage III | Not stage I or III:
|
| III | One or more of the following:
|
|
| Subgroup for each stage |
A – If relatively normal renal function (serum creatinine value < 2.0 mg/100 ml) B – If abnormal renal function (serum creatinine value ≥ 2.0 mg/100 ml) |
|
Current service provision
The aim of treatment for MM is to extend the duration and quality of survival by alleviating symptoms and achieving disease control while minimising the adverse effects of the treatment. 21 First-line treatment aims to achieve a period of stable disease (plateau phase) for as long as possible, prolonging survival and maximising QoL. In England and Wales the choice of first-line treatment depends on a combination of factors, including age, comorbidity, social factors and performance status of the patient. High-dose chemotherapy (HDT) with autologous stem-cell transplantation (SCT) will be offered if appropriate for the patient. However, the British Society for Haematology (BSH) guidelines on the diagnosis and management of MM (2005)22 state that (p. 428) ‘Although high-dose is recommended where possible, the majority of patients will not be able to receive such therapy because of age, specific problems or poor performance status’. For those patients who are not able to withstand such an intensive type of treatment, single-agent or combination chemotherapy (which is less intensive) may be offered as a first-line treatment. Patients eligible for HDT will get initial chemotherapy to reduce disease burden before transplant.
Typically, combination therapies include chemotherapy with an alkylating agent (such as melphalan or cyclophosphamide) and a corticosteroid (such as prednisolone or dexamethasone). The treatment recommended by the 2005 guidelines for patients who are unable to receive intensive treatment was either melphalan or cyclophosphamide, given either with or without prednisolone. 22 More recent treatment options may also include drugs such as thalidomide23–25 (Thalidomide Celgene®, Celgene, Uxbridge, UK) and bortezomib26 (Velcade®, Janssen–Cilag, High Wycombe, UK). Such drugs are being investigated in ongoing clinical trials, such as the Medical Research Council (MRC)-funded Myeloma IX study,27 which has compared thalidomide in combination with cyclophosphamide and dexamethasone (CTDa) against the standard drug combination of melphalan with prednisolone (MP).
The BSH guideline on the diagnosis and management of MM is being revised and updated. The draft of these revised guidelines28 contains a recommendation that, for older and/or less fit patients in whom high-dose therapy is not planned, the initial therapy should consist of either a thalidomide-containing regimen in combination with an alkylating agent and steroid [such as thalidomide in combination with MP (MPT) or CTDa] or bortezomib in combination with melphalan and prednisolone (VMP). The draft revised guideline indicates that the choice of first-line therapy should take into account patient preference, comorbidities and the toxicity profile of the treatments. 28
After first-line treatment most patients will show a response. Response is usually assessed based on changes in serum levels of M-protein and/or urinary light chain excretion, and ranges from partial to complete remission, but almost all patients will eventually relapse. A minority of patients will have disease that proves resistant to primary treatment.
In addition to chemotherapy, patients also require concomitant supportive therapy to control the symptoms of the disease, including bisphosphonates to treat bone disease, erythropoietin to treat anaemia, antibiotics to treat infections and various types of pain medication. Prophylaxis against thrombosis is recommended in the thalidomide summary of product characteristics (SPC) for the first 5 months that patients receive thalidomide. 29 In the UK this recommendation for prophylaxis against thrombosis is followed, but there is less agreement about whether to continue with prophylaxis for the entire duration of thalidomide therapy. Therefore, clinical practice is likely to vary. Side effects of treatment may result in discontinuation or change of chemotherapy treatment.
UK clinical experts have indicated that the most common combination therapy used as a first-line treatment for patients who are not able to withstand high-dose therapy is CTDa. The second most common therapy is MPT, with the ratio of patients on CTDa to those on MPT being approximately 2 : 1, although in some areas the ratio may be nearer 3 : 1. Intolerance to thalidomide limits its use in some patients, and occurrence of peripheral neuropathy limits the duration of treatment in some patients (clinical opinion expert advisor). VMP is not widely used as a first-line treatment, but may be used in the subgroup of patients who have renal impairment or failure at presentation. Use of MP is declining, but this is still used in patients who cannot tolerate thalidomide or where the use of thalidomide is contraindicated (clinical opinion expert advisor).
As noted above (see Description of underlying health problem) there is some evidence that myeloma that is characterised by a high-risk cytogenetic abnormality can demonstrate a poor response to conventional treatment. However, although there is interest in the use of cytogenetic data as a prognostic indicator, the incorporation of cytogenetic data into decisions about treatment choice is not currently supported in the UK. 22,28
When patients relapse after first-line treatment most will receive a second-line treatment. The choice of second-line treatment is individualised to the patient and, in theory, a patient could receive the same therapy that they received as a first-line treatment, particularly if this had been effective and the remission had lasted a long time. However, in current UK practice many patients will receive bortezomib monotherapy as a second-line treatment because, as noted below, this has been recommended by the National Institute for Health and Clinical Excellence (NICE). Similarly when patients relapse after second-line treatment the treatment recommended by NICE for this patient group is lenalidomide.
In addition to the BSH guidelines on the diagnosis and management of MM,22 two NICE technology appraisals have been completed for MM. NICE (TA12930) has previously recommended bortezomib monotherapy for relapsed MM as a possible treatment for progressive MM for people:
-
whose MM has relapsed for the first time after having one treatment, and
-
who have had a SCT, or who are unsuitable to receive one.
The National Institute for Health and Clinical Excellence has also recommended lenalidomide (a structural derivative of thalidomide) when used in combination with dexamethasone as a possible treatment for MM when people have already received at least two other treatments (TA17131). Neither of these NICE appraisals considered first-line therapy for MM.
One technology appraisal is in development – denosumab for the treatment of bone metastases from solid tumours and MM – but the scope of this appraisal was not available at the time of writing (January 2010). A draft scope for consultation was issued in March 2010.
The National Institute for Health and Clinical Excellence has also published Guidance on Cancer Services – Improving Outcomes in Haematological Cancers – The Manual. 32 This document covers all haematological cancers, including MM, and makes recommendations for service delivery and organisation. Some information about current service costs are included but these relate to the haematological cancer service as a whole.
Description of technology under assessment
Two interventions are being considered in this assessment:21 bortezomib in combination therapy with an alkylating agent and a corticosteroid, and thalidomide in combination therapy with an alkylating agent and a corticosteroid. The scope of this review allows for the inclusion of bortezomib or thalidomide when used in combination with any alkylating agent and any corticosteroid. This may therefore include drug combinations that are not covered by the licences for bortezomib and thalidomide, for example CTDa.
Place of the interventions in the treatment pathway
In this assessment bortezomib and thalidomide are being considered for use in combination therapy with an alkylating agent and a corticosteroid as a first-line treatment for MM in patients who are not eligible for HDT with autologous SCT.
Bortezomib
Bortezomib (Velcade®, manufacturer Janssen–Cilag, High Wycombe, UK) is a proteasome inhibitor that is specific for the 26S proteasome of mammalian cells and it has been designed to inhibit the chymotrypsin-like activity of this proteasome. Inhibition of the proteasome by bortezomib affects cancer cells in a number of ways, resulting in cell cycle arrest and apoptosis, which causes a reduction in tumour growth. 33
Bortezomib is administered by injection. It was initially granted a marketing authorisation in the European Union in 2004 as a therapy for patients with MM who had received at least two prior lines of treatment. Subsequently, in 2005, the indication was extended to enable treatment, earlier in the course of the disease, for relapsed MM in patients who have progressed after receiving at least one previous line of treatment. 34
In 2008 the marketing authorisation for bortezomib was extended further for the following indication: ‘Velcade in combination with melphalan and prednisone is indicated for the treatment of patients with previously untreated MM who are not eligible for high-dose chemotherapy with bone marrow transplant’ (p. 2). 34
The SPC for bortezomib33 recommends nine 6-week treatment cycles for combined therapy with VMP. During these treatment cycles bortezomib is administered as a 3- to 5-second bolus intravenous injection through a peripheral or central intravenous catheter at a dose of 1.3 mg/m2 of body surface area, followed by a flush with sodium chloride 9 mg/ml (0.9%) solution for injection. In the first four cycles of treatment, bortezomib is administered twice weekly. For cycles 5–9, bortezomib is administered once weekly. Melphalan (9 mg/m2) and prednisone (60 mg/m2) are both administered orally on days 1, 2, 3 and 4 of the first week of each cycle. The dose and total number of cycles may change depending on the patient’s response to treatment and on the occurrence of certain side effects. Because the licence for bortezomib does not cover its use in combination with agents other than melphalan and prednisone the SPC does not provide dosage information for any other alkylating agents or corticosteroids.
The net price for a 3.5-mg vial of bortezomib is £762.38. 35 Full details of the estimated drug costs associated with the use of bortezomib as a first-line treatment for MM are described within our independent economic evaluation (see Chapter 5, SHTAC data sources, Estimation of costs).
Thalidomide
Thalidomide is an immunosuppressive agent with antiangiogenic and other activities that are not fully characterised. It is also a non-barbiturate centrally active hypnotic sedative. Although the precise mechanism of action is unknown and under investigation, the effects of thalidomide are immunomodulatory, anti-inflammatory and antineoplastic. 29
Thalidomide (formerly known as Thalidomide Pharmion) is taken orally. It was granted a marketing authorisation in 2008 for use in combination with melphalan and prednisone as first-line treatment for patients with untreated MM, aged ≤ 65 years or who were ineligible for HDT. Because thalidomide is a known human teratogen it must be prescribed and dispensed according to the Thalidomide Pharmion Pregnancy Prevention Programme.
The SPC for thalidomide29 recommends an oral dose of 200 mg per day, taken as a single dose at bedtime to reduce the impact of somnolence. However, the advisory group for this review has indicated that treatment usually starts with a lower dose, which is gradually increased if the patient can tolerate this. In the UK, most patients who are ineligible for HDT and SCT are likely to receive a 100-mg dose. A maximum number of 12 cycles of 6 weeks is recommended. Thromboprophylaxis should also be administered for at least the first 5 months of treatment, especially in patients with additional thrombotic risk factors. The dose and total number of cycles may change depending on the patient’s response to treatment and on the occurrence of certain side effects.
The SPC does not recommend particular doses or dosing schedule for melphalan and prednisone when administered in combination with thalidomide (licensed indication). Because the licence for thalidomide does not cover its use in combination with agents other than melphalan and prednisone the SPC does not provide dosage information for any other alkylating agents or corticosteroids.
The net price of a 50-mg × 28-capsule pack of thalidomide is £298.48. 35 Full details of the estimated drug costs associated with the use of thalidomide as a first-line treatment for MM are described within our independent economic evaluation (see Chapter 5, SHTAC data sources, Estimation of costs).
Chapter 2 Definition of the decision problem
This section states the key factors that will be addressed by this assessment, and defines the scope of the assessment in terms of these key factors in line with the definitions provided in the NICE scope. 21
Decision problem
Two interventions are included within the scope of this assessment. These are (1) bortezomib in combination with an alkylating agent and a corticosteroid and (2) thalidomide in combination with an alkylating agent and a corticosteroid. In both cases the focus of this assessment is the use of these combination chemotherapies for the first-line treatment of MM.
The population that is being considered by this assessment is people with previously untreated MM, for whom HDT with SCT is not appropriate. If sufficient evidence is available consideration will be given to specific patient subgroups, for example patients with different prognostic factors such as β2-microglobulin, performance status and stage, patients whose MM has different cytogenetic features, and patients who have a comorbidity such as renal impairment. Additionally, if the evidence allows, consideration will be given to the number of treatment cycles and continuation rules for treatment.
The interventions will be assessed when compared with melphalan or cyclophosphamide in combination with prednisolone or dexamethasone. The NICE scope also allows for the interventions to be compared with one another. In this assessment we will include interventions using prednisone as well as prednisolone. Prednisone, which is not used in the UK, is converted into the biologically active steroid prednisolone by the liver. 36 Prednisone and prednisolone are equally effective, they are used in the same manner, and doses are largely equivalent.
The clinical outcomes of interest include overall survival (OS), progression-free survival (PFS), time to progression (TTP), response rates, health-related quality of life (HRQoL), and adverse effects (AEs) of treatment. Other outcomes of interest, such as duration of treatment, or second-line treatments received may also be reported. Outcomes for the cost-effectiveness assessment will include direct costs based on estimates of health-care resources associated with the interventions as well as consequences of the interventions, such as treatment of AEs.
Overall aims and objectives of assessment
The aim of this health technology assessment (HTA) is to systematically assess the evidence on the clinical effectiveness and cost-effectiveness of bortezomib or thalidomide in combination regimens with an alkylating agent and a corticosteroid for the first-line treatment of MM. 21
Chapter 3 Methods
The a priori methods for systematically reviewing the evidence of clinical effectiveness and cost-effectiveness are described in the research protocol (Appendix 1), which was sent to our expert advisory group for comment. None of the comments we received identified specific problems with the methods of the review. The methods outlined in the protocol are briefly summarised below.
Search strategy
The search strategies were developed and tested by an experienced information specialist. The strategies were designed to identify studies reporting clinical effectiveness, cost-effectiveness, HRQoL, resource use and costs, epidemiology and natural history.
The following databases were searched for published studies and ongoing research from 1999 (earliest use of thalidomide for MM37 and earliest description of bortezomib as a potential cancer therapy38) to December 2009: MEDLINE, MEDLINE In-Process, EMBASE, Web Of Science, BIOSIS, Centre for Reviews and Dissemination (CRD) Database of Abstracts of Reviews of Effects (DARE), HTA, NHS Economic Evaluation Database (NHS EED) and Cochrane Central Register of Controlled Trials (CENTRAL). Bibliographies of articles and grey literature sources were also searched. Reference lists within drug manufacturers’ submissions (MSs) to NICE were searched for any additional studies that met the inclusion criteria. Our expert advisory group was asked to identify additional published and unpublished references. Searches were restricted to English language. Further details, including an example search strategy, can be found in Appendix 2.
Inclusion and exclusion criteria
Study design
-
For the systematic review of clinical effectiveness, randomised controlled trials (RCTs) were eligible for inclusion. In addition, evidence from good-quality observational studies was also eligible for consideration if the data from available RCTs were incomplete (e.g. absence of data on outcomes of interest).
-
For the systematic review of cost-effectiveness economic evaluations (such as cost-effectiveness studies, cost–utility studies, cost–benefit studies) were eligible for inclusion.
-
Abstracts or conference presentations of studies were eligible for inclusion only if sufficient details were presented to allow an appraisal of the methodology and the assessment of results to be undertaken.
-
Case series, case studies, narrative reviews, editorials and opinions were excluded, as were non-English-language studies. Systematic reviews and clinical guidelines were used only as a source of references.
Intervention(s)
-
Bortezomib in combination with an alkylating agent and a corticosteroid for first-line treatment of MM.
-
Thalidomide in combination with an alkylating agent and a corticosteroid for first-line treatment of MM.
-
Studies of treatment with either bortezomib or thalidomide as a single agent were excluded.
Comparator(s)
-
Interventions described above compared with each other.
-
Melphalan or cyclophosphamide in combination with prednisolone/prednisone or dexamethasone.
-
Other chemotherapy regimens or SCT were excluded.
Population
-
People with previously untreated MM who are not candidates for HDT with SCT.
-
Studies of MM patients who had received previous treatment(s) were excluded.
Outcomes
Studies were included if they reported on one or more of the following outcomes:
-
overall survival
-
progression-free survival (deaths counted as events)
-
time to progression (deaths are excluded from the calculation of this outcome)
-
response rates
-
health-related quality of life
-
cost-effectiveness [such as incremental cost per quality-adjusted life-year (QALY) gained]
-
adverse events of treatment were reported when available within the trials that met the inclusion criteria.
Response definitions
-
Response to treatment is usually assessed based on changes in serum levels of M-protein and/or urinary light chain excretion. Two different systems for categorising response are included in this report, the European Group for Blood and Marrow Transplantation (EBMT) criteria39 and the Intergroupe Francophone du Myélome (IFM) criteria. 23 Where there are differences in the two systems, in general the EBMT criteria require a slightly greater improvement. For example, in the definition of partial response (PR) one of the IFM requirements is more than a 75% reduction in 24-hour urinary light chain excretion, whereas one of the EBMT criteria for PR is a 90% decrease in urinary light chain excretion. The EBMT and IFM criteria for judging response are provided in Appendix 3.
Adverse event definitions
-
Two slightly different National Cancer Institute (NCI) criteria have been used to grade AEs, the NCI Common Terminology Criteria for Adverse Events (CTCAE) version 4, and the NCI Common Toxicity Criteria (CTC) version 2. The NCI CTCAE version 4 grades AEs on a five-point scale (1–5) and the NCI CTC version 2 grades AEs on a six-point scale, as 0 is included (0 = no AE or within normal limits). Events of a higher grade are more serious than those at a lower grade, with a grade 1 event described as ‘mild’, grade 2 ‘moderate’, a grade 3 event would be considered ‘severe’, while a grade 4 event could be ‘life threatening’. Grade 5 is reserved for deaths related to an AE.
Inclusion and data extraction process
Studies were selected for inclusion in the systematic reviews of clinical effectiveness and cost-effectiveness through a two-stage process. Literature search results (titles and abstracts) were screened independently by two reviewers to identify all of the citations that might meet the inclusion criteria. Full manuscripts of selected citations were then retrieved and assessed by one reviewer against the inclusion/exclusion criteria and checked independently by a second reviewer. Discrepancies were resolved by discussion, with involvement of a third reviewer when necessary.
Data from included studies were extracted by one reviewer using a standardised data extraction form and each data extraction was checked for accuracy by a second reviewer. Again discrepancies in the extracted data were resolved by discussion, with involvement of a third reviewer when necessary.
Critical appraisal strategy
The quality of included clinical effectiveness studies was assessed using the CRD criteria. 40 Quality criteria were applied by one reviewer and checked by a second reviewer with any disagreements resolved by consensus and involvement of a third reviewer where necessary.
Methods of data synthesis
Clinical effectiveness and cost-effectiveness studies were synthesised through a narrative review with tabulation of results of included studies. Results of included RCTs were meta-analysed if appropriate (more than one trial with populations, interventions and outcomes believed to be sufficiently similar) and possible (adequate data reported). For time-to-event analyses (OS and PFS) the log-hazard ratio (HR) and its standard error (SE) for each outcome were used to calculate a summary HR and 95% confidence interval (CI) using the Cochrane Collaboration Review Manager 5.0.23 software. However, as the SEs of the log-HRs were not reported by the RCTs, these had to be estimated using the methods and Microsoft Excel spreadsheet of Tierney and colleagues. 41
This report contains reference to confidential information provided as part of the NICE appraisal process. This information has been removed from the report and the results, discussions and conclusions of the report do not include the confidential information. These sections are clearly marked in the report.
Chapter 4 Clinical effectiveness
Results of the systematic review of clinical effectiveness
Quantity and quality of research available
Titles and, where available, abstracts of a total of 1436 records were screened and full copies of 40 references were retrieved. Of these, six were excluded after inspection of the full article (see Appendix 4). Two of these articles were excluded because they were not clinical trial reports, two were abstracts that were excluded because they described maintenance therapy with thalidomide, another abstract was excluded because it did not report on any of the outcomes of interest, and a sixth abstract described a systematic review with meta-analysis. Five full papers described four RCTs that met the inclusion criteria of the review (Figure 1 and Table 4). Each RCT was described by at least one full paper, with linked abstracts also being available. As the full papers provided the most complete data these were the primary source of information for the review.
FIGURE 1.
Flow diagram of reference screening processes. a, Additional information was received from the trialists providing details for one RCT only described in conference abstracts. The additional details allowed us to appraise the study methodology and make judgements about study quality. Results from this RCT could therefore be considered for inclusion in the systematic review of clinical effectiveness. b, Outcomes from these studies could not be included because of insufficient details about study methodology and insufficient details about study quality. These studies, which are all ongoing, are briefly summarised below (see Ongoing studies).

| Study detailsa |
VISTA trial Multicentre RCT at 151 centres in 22 countries in Europe, North and South America, Asia 682 enrolled |
Facon et al. 23 IFM 99/06 trial Multicentre RCT at 73 centres in France, Belgium, and Switzerland 447 enrolled to all three groups |
Hulin et al. 59 IFM 01/01 trial Multicentre RCT at 44 centres in France and Belgium 232 enrolled |
Palumbo et al. 24 GIMEMA network Multicentre RCT at 54 centres in Italy 331 enrolled (255 followed up) |
MMIX Trial: non-intensive pathway49,52 Multicentre RCT (AiC/CiC information has been removed) in the UK (AiC/CiC information has been removed) |
| Median follow-up |
Abstract 25.9 months60 Abstract 36.7 months61 |
51.5 monthsb | 47.5 monthsb |
38.4 months MPTc 37.7 months MP |
(AiC/CiC information has been removed)d (AiC/CiC information has been removed) |
| Intervention |
VMP: n = 344 9 × 6-week cycles of bortezomib (1.3 mg/m2) on days 1, 4, 8, 11, 22, 25, 29 and 32 during cycles 1–4 and on days 1, 8, 22 and 29 during cycles 5–9 + MP melphalan (9 mg/m2) plus prednisone (60 mg/m2) on days 1–4 of each cycle |
MPT: n = 125 Thalidomide < 400 mg daily for 12 MP cycles (i.e. 72 weeks) + MP 12 × 6 week cycles of melphalan 0.25 mg/kg and prednisone 2 mg/kg on 4 days per cycle |
MPT: n = 115 Thalidomide 100 mg daily for 72 weeks + MP 12 × 6 week cycles of melphalan 0.2 mg/kg and prednisone 2 mg/kg on days 1–4 of each cycle |
MPT: n = 167 (129 followed up) Thalidomide 100 mg daily for six MPT cycles (i.e. for 24 weeks) + MP 6 × 4 week cycles of melphalan 4 mg/m2 and prednisone 40 mg/m2 on days 1–7 of each cycle |
CTDa: (AiC/CiC information has been removed) Thalidomide: 50 mg daily for 4 weeks, increasing every 4 weeks by 50-mg increments to 200 mg daily + Cyclophosphamide: 500 mg once a week, on days 1, 8, 15 and 22 of each cycle Dexamethasone: 20 mg daily on days 1–4 and 15–18 of each cycle Cycle length 4 weeks, to maximal response, but with a minimum–maximum number of cycles of 6–9 |
| Comparator |
MP: n = 338 MP: 9 × 6-week cycles of melphalan (9 mg/m2) plus prednisone (60 mg/m2) on days 1–4 of each cycle |
MP: n = 196 MP: 12 × 6 week cycles of melphalan 0.25 mg/kg and prednisone 2 mg/kg on 4 days per cycle Third arm did not meet inclusion criteria |
MP + placebo: n = 117 Placebo daily for 72 weeks + MP 12 × 6 week cycles of melphalan 0.2 mg/kg and prednisone 2 mg/kg on days 1–4 of each cycle |
MP: n = 164 enrolled (126 followed up) MP: 6 × 4 week cycles of melphalan 4 mg/m2 and prednisone 40 mg/m2 on days 1–7 of each cycle |
MP: (AiC/CiC information has been removed) MP: Six to nine cycles of melphalan 7 mg/m2 and prednisolone dose 40 mg on days 1–4 of a 4-week cycle |
| Key attributes of participants |
Not candidates for HDT with SCT because of age 65 years or over, or co-existing conditions Newly diagnosed and previously untreated Measurable disease |
Aged between 65 and 75 years Previously untreated MM at DS stage II or III Patients younger than 65 years were included if they were ineligible for high-dose treatment Patients with DS stage I MM who met criteria of high-risk stage I disease |
Aged at least 75 years Newly diagnosed MM at DS stage II or III Patients with DS stage I MM who met criteria of high-risk stage I disease Patients with non-secretory or oligosecretory MM allowed |
Older than 65 years of age Younger participants included if unable to undergo transplantation Previously untreated DS stage II or III MM Measurable disease |
Aged at least 18 years Newly diagnosed symptomatic MM or non-secretory MM |
| Selected baseline characteristics |
Median age (years, range): VMP 71 (57–90) MP 71 (48–91) |
Age ≥ 70 years: MPT 50/125 (40%) MP 84/196 (43%) |
Age ≥ 80 years: MPT 43/113 (38%) MP + placebo 40/116 (34%) |
Median age (years): MPT 72 MP 72 |
(AiC/CiC information has been removed) |
| Gender (M : F) |
VMP 175 : 169 (51% : 49%) MP 166 : 172 (49% : 51%) |
MPT 63 : 62 (50% : 50%) MP 109/87 (56% : 44%) |
MPT 43 : 70 (38% : 62%) MP + placebo 61 : 55 (53% : 47%) |
NR | (AiC/CiC information has been removed) |
| Disease stage (DS or ISS criteria) |
ISS stage: Stage I VMP 19%, MP 19% Stage II VMP 47%, MP 47% Stage III VMP 35%, MP 34% |
DS stage II or III MPT 112/125 (90%) MP 177/196 (91%) |
DS stage II or III MPT 100/113 (89%) MP + placebo 107/116 (93%) |
DS stage II or III MPT 129/129 (100%) MP 126/126 (100%) Calculated by reviewer |
(AiC/CiC information has been removed) |
| Performance statuse |
Karnofsky performance status ≤ 70: VMP 122 (35%) MP 111 (33%) |
WHO performance index 3–4: MPT 10/125 (8%) MP 13/196 (7%) |
WHO performance index 3–4: MPT 9/113 (8%) MP 7/116 (6%) |
WHO performance index 3–4: MPT 9/129 (7%) MP 6/126 (4%) |
WHO performance index 3: (AiC/CiC information has been removed) WHO performance WHO performance index 4: (AiC/CiC information has been removed) |
| Bone lesions present |
VMP 224/343 (65%) MP 222/336 (66%) |
MPT 90/125 (76%) MP 154/196 (79%) |
MPT 87/113 (78%) MP + placebo 93/116 (82%) |
NR | (AiC/CiC information has been removed) |
| Primary outcome(s) | Time to disease progression | OS | OS | Response rates, PFS | OS, PFS, response |
| Secondary outcomes | Rate of CR, duration of response, time to subsequent myeloma therapy, OS, PFS | Response, PFS, survival after progression, toxicity | Safety, response rates, PFS | OS, time to first evidence of response, prognostic factors, frequency of any grade 3 or higher AEs | QoL, skeletal-related events, height loss, toxicity, proportion receiving bortezomib–dexamethasone as ‘early rescue’ on induction chemotherapy, or at relapse |
One ongoing RCT, the Myeloma IX (MMIX) Trial, which is a UK-based MRC collaborative RCT with two treatment pathways, appeared to meet the inclusion criteria of the review and was described in conference abstracts. The search for studies of clinical effectiveness identified four abstracts for this RCT,42–45 with a further three abstracts identified in the MSs. 46–48 Four of the abstracts,42–44,47 were excluded because they described the intensive pathway or thalidomide maintenance treatment that did not meet the inclusion criteria for this review. Three abstracts45,46,48 described the non-intensive pathway that met the inclusion criteria. The final results from the 3-year median follow-up have not yet been published. However, because of this RCT’s potential relevance to our inclusion criteria, the Clinical Trials Research Unit (CTRU) at the University of Leeds, who are coordinating the RCT, provided the trial protocol,49 additional background information50,51 and trial baseline data,52 and have also made the results from the non-intensive treatment pathway53–58 available to NICE and the authors of this report in academic confidence. As the trial protocol and results provided directly by the CTRU provided the most complete and up-to-date data these were used as the primary source of information for the review.
Four additional ongoing RCTs were described in conference abstracts but it was unclear whether these met the inclusion criteria for this review. These ‘unclear’ studies are briefly described later in this chapter (see Ongoing studies). The total number of records assessed at each stage of the systematic review screening process is shown in the flow chart in Figure 1.
One of the included RCTs evaluated VMP (VISTA trial),26 while three RCTs evaluated MPT [IFM 01/01 trial,59 IFM 99/06 trial,23 and the Gruppo Italiano Malattie Ematologiche dell’Adulto (GIMEMA – Italian Group for Adult Hematologic Diseases) trial24]. The fifth RCT, the MMIX Trial (non-intensive pathway), evaluated CTDa. The comparator in all five of the included RCTs was MP.
Bortezomib in combination with melphalan and prednisone (VISTA trial)
The RCT investigating VMP was a randomised (1 : 1), open-label, Phase III trial conducted in 151 centres in 22 countries in Europe, North and South America, and Asia. The RCT enrolled 682 participants and was funded by two industry sponsors (see Table 4).
Patients received nine 6-week cycles of melphalan (at a dose of 9 mg/m2 of body surface area) and prednisone (at a dose of 60 mg/m2) on days 1–4, alone or in combination with bortezomib (at a dose of 1.3 mg/m2), by intravenous bolus on days 1, 4, 8, 11, 22, 25, 29 and 32 during cycles 1–4 and on days 1, 8, 22 and 29 during cycles 5–9. The dose of bortezomib or melphalan was reduced if there was any prespecified haematological toxic effect or grade 3 or 4 non-haematological effect. Patients with myeloma-associated bone disease received bisphosphonates unless such therapy was contraindicated.
Patients were eligible if they had newly diagnosed, untreated, symptomatic, measurable myeloma and were not candidates for HDT plus SCT because of age (≥ 65 years) or co-existing conditions. Measurable disease was defined as the presence of quantifiable M-protein in serum or urine, or measurable soft-tissue or organ plasmacytomas. Over 80% of patients had ISS stage II or III disease, about one-third had a Karnofsky performance score of ≤ 70%, and over 60% had lytic bone lesions. Most participants were white. No exclusion criteria for study entry were stated.
During the 54-week treatment period, blood and urine samples were collected every 3 weeks. After completion of treatment, samples were collected every 8 weeks until disease progression. Patients were followed after disease progression at least every 12 weeks for survival and subsequent myeloma therapy.
The primary outcome measure was time to disease progression. The study was powered at 80% for the primary outcome but no power calculations were reported for patient subgroups. Secondary outcomes were rate of CR, duration of response time, time to subsequent myeloma therapy, OS and PFS. Disease progression was defined by EBMT criteria and assessed by investigators. The sponsors also determined progression with the use of a computer algorithm that applied EBMT criteria. Data are presented in the published paper from the assessment by investigators and from the algorithmic analysis. TTP, time to subsequent myeloma therapy and OS were analysed from randomisation to the event of interest.
Thalidomide in combination with melphalan and prednisone (IFM and GIMEMA trials)
All three of the included RCTs investigating MPT were multicentre trials. The number of centres ranged from 44 to 73 and all were located in one or more European countries (France, Belgium, Switzerland, Italy). The IFM RCT by Facon and colleagues23 was the largest, recruiting 447 patients; however, only 321 participants are reported on here because this trial had a third arm (reduced-intensity SCT), which is not relevant to this review as the intervention does not meet the inclusion criteria. The GIMEMA group RCT by Palumbo and colleagues24 enrolled 331 participants, and the remaining IFM RCT, Hulin and colleagues,59 enrolled 232 participants (see Table 4). All of the RCTs received free thalidomide for the study from the drug manufacturers but other funding costs were met by grants from other sources (see Appendix 5).
The dosing schedules of the RCTs varied in terms of overall length and the drug doses used. Hulin and colleagues59 and Facon and colleagues,23 the two IFM RCTs, had 72-week treatment periods consisting of 12 six-week treatment cycles. The treatment period in the GIMEMA group RCT by Palumbo and colleagues24 was shorter, lasting for 24 weeks and consisting of six 4-week treatment cycles. The intervention in each RCT was MPT. Thalidomide was prescribed as a set 100-mg daily dose in the RCTs by Hulin and colleagues59 and Palumbo and colleagues,24 whereas a 400-mg daily dose was the goal of Facon and colleagues (if this could be tolerated). 23 In the two IFM RCTs,23,59 doses were described according to body weight. The dosing schedule of MP (on days 1–4 of each 6-week treatment cycle) and prednisone dose (2 mg/kg prednisone) were the same in both RCTs, while the melphalan doses differed slightly (Hulin and colleagues,59 0.2 mg/kg of melphalan; Facon and colleagues,23 0.25 mg/kg of melphalan). Palumbo and colleagues24 described drug doses according to body surface area. Melphalan (4 mg/m2) and prednisone (40 mg/m2) were taken on days 1–7 of each 4-week treatment cycle. All RCTs allowed thalidomide dose adjustments. In each RCT the comparator was MP alone (no thalidomide prescribed), provided in the same manner as in the MPT arms as described (also see Table 4). Only one RCT, Hulin and colleagues,59 included a placebo in place of thalidomide in the comparator arm.
As mentioned earlier, to be included in this systematic review, RCTs had to report on treatment of participants with MM who were not eligible for HDT with SCT and who had not been previously treated. All participants in each RCT met these criteria. The two IFM RCTs differed in the target age range of participants: Hulin and colleagues59 focused on people aged at least 75 years, whereas Facon and colleagues23 focused on people aged between 65 and 75 years, with younger patients being eligible for inclusion providing they were not eligible for HDT. Palumbo and colleagues24 focused on people who were older than 65 years of age without specifying any upper age limit, and, like Facon and colleagues,23 did include participants who were younger than 65 years, providing that they were unable to undergo SCT. All RCTs included people whose MM was at DS stage II or III, and the two IFM RCTs23,59 also included patients with DS stage I MM if they met the criteria for high-risk stage I disease. The percentage of participants in the IFM23,59 and GIMEMA24 RCTs with a WHO performance status score of 3 or 4 ranged from 4% to 8%. Over three-quarters of the participants in the IFM RCTs had bone lesions but this information was not reported by Palumbo and colleagues24 (see Table 4). None of the RCTs reported on the ethnicity of the participants.
All three RCTs specified their exclusion criteria. In the two IFM RCTs23,59 these were almost identical, the only difference being that Hulin and colleagues59 excluded anyone with a history of venous thrombosis during the previous 6 months in addition to the other exclusions [anyone with previous neoplasms (except basocellular cutaneous or cervical epithelioma); primary or associated amyloidosis; a WHO performance index of 3 or higher, if unrelated to MM; substantial renal insufficiency with creatinine serum concentration of 50 mg/l or more; cardiac or hepatic dysfunction; peripheral neuropathy; HIV infection, or hepatitis B or C infections]. Palumbo and colleagues24 listed fewer exclusion criteria. Two were similar to those of the IFM RCTs (exclusion of people with another cancer or any grade 2 peripheral neuropathy) and one was novel to this RCT (exclusion of people with psychiatric disease). Palumbo and colleagues24 also stated that abnormal cardiac function, chronic respiratory disease, and abnormal liver or renal functions were not criteria for exclusion.
The timing of clinic visits during the RCTs varied. Palumbo and colleagues24 monitored response to treatment every 4 weeks, whereas visits were scheduled every 6 weeks for the RCT by Hulin and colleagues59 until treatment completion or study withdrawal. Facon and colleagues23 saw participants after inclusion at 3 months, 6 months and then every 6 months thereafter until withdrawal from the RCT. When scheduled clinic visits ended (after withdrawal or end of treatment), Palumbo and colleagues24 continued to assess participants every 2 months, and the two IFM RCTs23,59 continued to assess participants every 6 months.
Overall survival was the primary outcome measure for the two IFM RCTs. 23,59 Both RCTs were powered at 80% for the primary outcome but recruitment was stopped early in both RCTs because interim analyses had demonstrated a clear survival advantage. The secondary outcomes of these RCTs were response rates,23,59 PFS,23,59 survival after progression,23 toxicity23 and safety. 59 Facon and colleagues23 report some of their outcomes for more than one follow-up period. OS, PFS and survival after progression analyses were reported for a data point of 8 January 2007; these outcomes were also reported along with all other outcomes for the earlier date point of 8 October 2005. In contrast, the primary outcomes of the RCT by Palumbo and colleagues24 were stated as response rates and PFS. A power calculation was reported for the response outcome. The secondary outcomes of this RCT were OS, time to first evidence of response, prognostic factors, and frequency of any grade 3 or higher AEs.
Thalidomide in combination with cyclophosphamide and attenuated dexamethasone (MMIX Trial)
The MMIX RCT non-intensive pathway evaluated CTDa in comparison with MP. Participants were randomised in a 1 : 1 ratio to receive either CTDa or MP. Within each treatment arm, participants were also randomised to bisphosphonate treatment with either sodium clondronate or zoledronic acid. This multicentre RCT was conducted [academic-in-confidence (AiC) and/or commercial-in-confidence (CiC) information has been removed] in the UK (AiC/CiC information has been removed) (see Table 4). The RCT was funded by a core grant from the MRC, with some other funding provided by five industry sponsors and one charitable sector sponsor (see Appendix 5).
The treatment period with CTDa in the intervention arm was designed to be between 24 and 36 weeks, equivalent to a minimum of six, or a maximum of nine, 4-week treatment cycles. Thalidomide was prescribed as a daily starting dose of 50 mg with the aim that this would be increased every 4 weeks by 50 mg to a maximum of 200 mg. During each 4-week treatment cycle 500 mg of cyclophosphamide was taken once a week on days 1, 8, 15 and 22, and dexamethasone 20 mg was taken daily on days 1–4 and days 15–18 of each cycle. Participants in the comparator arm received MP (melphalan 7 mg/m2 and prednisolone 40 mg) on days 1–4 of each 4-week cycle. Dose adjustments were permitted in both RCT arms.
In common with the other included RCTs, patients were eligible if they were newly diagnosed with symptomatic MM or non-secretory MM and had not received previous treatment for myeloma (other than local radiotherapy). The MMIX non-intensive pathway was designed for older (generally ≥ 70 years of age) or less fit patients (who could be younger than 70 years) but strict age restrictions were not in place to ensure that fit older patients were not excluded from the intensive therapy arm. (AiC and CiC information has been removed.) Exclusion criteria included asymptomatic MM, solitary plasmacytoma of bone and extramedullary plasmacytoma (without evidence of myeloma). People with acute renal failure were excluded but those with a history of ischaemic heart disease or psychiatric disorder could be considered for inclusion at the discretion of the clinician. Further details of exclusion criteria can be found in Appendix 5.
Overall survival, PFS, and response were the co-primary outcomes and power calculations were provided for both survival and response. Secondary outcomes were QoL, skeletal-related events, height loss, toxicity (thromboembolic events, renal toxicity, haematological toxicity, graft-versus-host disease) and proportion receiving bortezomib–dexamethasone as ‘early rescue’ on induction chemotherapy, or at relapse.
Quality assessment of included studies
The outcome of the quality assessment of included RCTs is summarised in Table 5.
| Study | Randomisation sequence | Allocation concealment | Balanced baseline characteristics | Blinding | Dropout imbalance | More outcomes than reported | ITT analysis | Missing data accounted for |
|---|---|---|---|---|---|---|---|---|
| San Miguel et al.26,60 | NR | NR | Yes | No | ? | No | Yes | ?a |
| Facon et al.23 | NR | NR | ? | NR | ? | No | Yes | ? |
| Hulin et al.59 | NR | Yes | Yes | ? | ? | No | Yes | ? |
| Palumbo et al.24 | Yes | Yes | Yes | No | ? | No | Yes | ? |
| MMIX49,50,52 | Yes | Yes | Yes | No | NR | No | Yes | ? |
Bortezomib in combination with melphalan and prednisone
The VISTA study of VMP versus MP was an RCT, with randomisation stratified according to baseline levels of β2-microglobulin, serum albumin and region. However, no details are given on the methods used to generate random numbers or conceal allocation to treatment group, and therefore it is not possible to know whether the RCT is at risk of selection bias due to unbalanced confounding factors and failure to adequately conceal allocation. Baseline demographics and disease characteristics are reported to be well balanced between the two groups. The RCT is described as open label, which suggests that researchers and/or participants were not blinded. As bortezomib is administered intravenously the researchers may have felt blinding was not possible. However, for objective outcomes, such as OS, risk of bias is low regardless of lack of blinding. There is no evidence that more outcomes were measured than reported by study authors. The authors did not report whether there were any unexpected imbalances in dropouts between the groups. TTP, time to subsequent myeloma therapy, and OS from randomisation were analysed in the intention-to-treat (ITT) population (all randomised patients). For TTP analyses, data from patients for whom there was no disease progression were censored at the last assessment, or at the start of subsequent therapy. Although not explicitly stated it is assumed that deaths without disease progression were not included in the outcome of TTP. Details of censoring in terms of number of patients with censored data and reasons for censoring in each group are not given. The response analysis was not ITT as seven patients in each group could not be evaluated for a response: five did not receive the study drug; three patients in the VMP arm and six patients in the MP arm had no measurable disease at baseline on the basis of assessment by a central laboratory (although the patients met the eligibility criteria of measurable disease according to evaluation by a local laboratory).
Thalidomide in combination with melphalan and prednisone
All of the included studies were RCTs of MPT versus MP. However, one of the three RCTs, Facon and colleagues,23 did not report on the methods used to generate random allocations or how the allocations were concealed. Without this information we cannot be certain that the randomisation method balanced out confounding factors or that allocation bias has been avoided in this RCT. Hulin and colleagues59 did not report on the method used to generate the randomisation sequence but the central allocation of patients should have provided adequate allocation concealment. Palumbo and colleagues24 were the only authors to report sufficient information about randomisation and allocation concealment, allowing this RCT to be judged at low risk from unbalanced confounding factors and low risk of allocation bias.
All three MPT RCTs reported on the baseline characteristics of participants according to treatment group. Hulin and colleagues59 provided an indication that statistical testing had been used to test the similarity of the groups at baseline, and reported that the only statistically significant difference was for sex (more female participants in MPT group, p = 0.03). However, Facon and colleagues23 did not report on whether the groups had been judged to be similar at baseline. Palumbo and colleagues24 stated that baseline demographics and other characteristics of the two groups were balanced.
One of the three MPT RCTs, Palumbo and colleagues,24 was not blinded, and this was clearly stated by the authors. One of the RCTs, Hulin and colleagues,59 involved the use of a placebo in the comparator arm, which suggests blinding may have been in place although this was not explicitly stated. The third MPT RCT did not report whether blinding was in place or not. In each RCT some of the outcomes were objective (e.g. survival) and therefore the risk of bias for these would be low, regardless of whether blinding was in place or not.
There was no evidence in any of the MPT RCTs that more outcomes were measured than were reported. But for each of the three MPT RCTs it was unclear whether there were any unexpected imbalances in dropouts between groups because none of the RCT authors commented on this. 23,24,59
All of the MPT RCTs stated that an ITT analysis had been conducted but the details of these analyses and methods used to account for missing data were unclear due to poor reporting. Hulin and colleagues59 stated that an ITT analysis was conducted, but in this case the ITT analysis appears to have excluded three randomised participants who discontinued before study treatment (two in the MPT group and one in the MP group). Facon and colleagues23 stated that an ITT analysis was conducted and, from the numbers provided in the results for OS and PFS, but not response, their ITT analysis appears to have included all patients randomised, including those who were not treated as assigned. Palumbo and colleagues24 stated an ITT analysis had been conducted at 6 months (the only outcome point eligible for inclusion in this review) but at the time of analysis not all randomised participants had been enrolled for 6 months. Therefore, 76 out of the 331 randomised participants (38 in each arm) were not included in the analysis of 6-month data. As these RCTs reported time-to-event data, such as OS and PFS, it was expected that some data would be censored. However, only one RCT, Hulin and colleagues,59 stated when data on patients who were alive were censored in the survival analysis and when data on patients without disease progression were censored for the analysis of PFS. One of the MPT RCTs, Facon and colleagues,23 marked the position of censored data on the survival plots but none of the RCTs reported details of how many participants’ data were censored, and for what reason (e.g. censored due to withdrawal, censored due to death from an unrelated cause such as a car accident or censored as event of interest not experienced). It is not possible to determine whether the amount and pattern of censoring was comparable between the groups and whether this had any effect on outcomes.
Thalidomide in combination with cyclophosphamide and attenuated dexamethasone
The MMIX study of CTDa versus MP was an RCT (with randomisation) that used a minimisation algorithm, stratified by centre, haemoglobin, corrected serum calcium, serum creatinine and platelets. 49,52 No details are reported on the methods used to generate random numbers; however, allocation to treatment groups was adequately concealed by the use of an automated 24-hour telephone system. The RCT is therefore at a low risk of selection bias. (AiC/CiC information has been removed.) The RCT was not a blinded RCT, but, as already noted for the other included RCTs, the risk of bias is low for the objective outcomes. There is no evidence that more outcomes have been measured during the RCT than are reported. The authors did not report whether there were any unexpected imbalances in dropouts between the groups. All summaries and analyses were by ITT unless stated otherwise and ITT was defined as all patients randomised, with the exception of those misdiagnosed. For the QoL data the analysis includes all patients who agreed to take part in the QoL study. Patients with missing follow-up data or who had not experienced progression were censored on the last date they were known to be alive and progression free. OS was calculated from initial randomisation to death. Patients with missing follow-up data, or not known to have died at time of analysis, were censored on the last date they were known to be alive. It was not reported whether the amount and pattern of censoring was comparable between the groups. PFS was calculated from random assignment to progression or death. There was no other censoring of data.
Assessment of effectiveness
Overall survival
Overall survival was a secondary outcome in the VISTA RCT of VMP versus MP (Table 6) and was calculated from randomisation. A statistically significant survival benefit for VMP compared with MP is reported in an abstract60 after a median follow-up of 25.9 months (HR = 0.64, p = 0.0032). Three-year survival rates in a more recent abstract61 after a median follow-up of 36.7 months were 68.5% versus 54%, respectively. At the earlier median follow-up of 16.3 months, reported in the published paper,26 median OS had not been reached. However, San Miguel and colleagues26,60 stated that a survival benefit was associated with bortezomib because 45 patients (13%) in the VMP group had died in comparison with 76 patients (22%) in the MP group (HR 0.61, p = 0.008) (despite 43%60 of MP patients receiving subsequent bortezomib therapy after disease progression – see Table 21). The most recent abstract reports that median OS is 43.1 months in the MP group but not estimable in the VMP group. 61
| Study | Median follow-up (months) | Treatment arms | HR and p-value | |
|---|---|---|---|---|
| San Miguel et al.26 VISTA | ||||
| VMP (n = 344) | MP (n = 338) | |||
| OS (abstract61) | 36.7 | Not estimable | 43.1 months | 0.653; 0.0008 |
| OS (abstract60) | 25.9 | NR | NR | 0.64; 0.0032 |
| OS26 | 16.3 | Median survival not reached | Median survival not reached | 0.61; 0.008 |
| Deaths26 | 45/344 (13%) | 76/338 (22%) | ||
| Three-year survival rate (abstract61): % | 68.5 | 54.0 | NR | |
| Three-year survival rate (abstract60): % | 72 | 59 | NR | |
| Facon et al.23 IFM 99/06 | ||||
| MPT (n = 125) | MP (n = 196) | |||
| OS:a median (SE, IQR) |
51.5 (IQR 34.4–63.2) |
51.6 (4.5, 26.6 to ‘not reached’) months | 33.2 (3.2, 13.8 to 54.8) months | 0.59 (95% CI 0.46 to 0.81); 0.0006 |
| Deaths | 62/125 (50%) | 128/196 (65%) | ||
| Hulin et al.59 IFM 01/01 | ||||
| MPT (n = 113) | MP + placebo (n = 116) | |||
| OS: median (95% CI) | 47.5 | 44.0 (33.4 to 58.7) months | 29.1 (26.4 to 34.9) months | 0.68 (95% CI not reported); 0.028 |
| Deathsb | 58/113 (51%) | 76/116 (65.5%) | 0.03 | |
Two23,59 of the three RCTs investigating MPT versus MP alone reported OS as their primary outcome. Both RCTs calculated OS from randomisation but only one of them, Hulin and colleagues,59 explained that data on patients who were alive at the time of analysis were censored in the survival analysis on the last date they were known to be alive. For the third RCT,24 OS was a secondary outcome and was not eligible for inclusion in this systematic review because participants received maintenance therapy with thalidomide after the six 4-week cycles of MPT were completed.
A statistically significant difference in OS in favour of the MPT group was found by both RCTs (see Table 6). Facon and colleagues23 reported their results after a median follow-up of 51.5 months. In the MPT group there were 62 events (deaths) and median survival was 51.6 months [interquartile range (IQR) 26.6 to ‘not reached’], whereas in the MP group, where there were 128 events, median survival was 33.2 months (IQR 13.8–54.8). The difference in OS was statistically significant, with an estimated HR for median OS in favour of MPT of 0.59 (95% CI 0.46 to 0.81, p = 0.0006). When adjusting for prognostic factors (e.g. WHO performance index; β2-microglobulin, albumin, etc.) the results showed that MPT remained the superior treatment in terms of the specified outcome OS (HR 0.49, 95% CI 0.33 to 0.73, p = 0.0002) (see Appendix 5). Similarly Hulin and colleagues,59 reporting after a slightly shorter median follow-up of 47.5 months, found that the median survival of 44 months (95% CI 33.4 to 58.7) in the MPT group was statistically significantly longer than in the MP + placebo group where median survival was 29.1 months (95% CI 26.4 to 34.9). In this RCT the reported HR for median OS in favour of MPT was 0.68 (95% CI for the HR not reported, p = 0.028).
As noted above, neither RCT reported on the amount of censored data, or the reasons for this. It is therefore not possible to determine whether censored data had any impact on the outcome of OS.
The MMIX RCT49 OS outcome was not eligible for inclusion in this systematic review because participants were entered into a second randomisation to receive either maintenance therapy with thalidomide or no maintenance therapy after they had completed first-line treatment with either CTDa or MP.
Two MPT versus MP RCTs23,59 reported OS outcome data that met the inclusion criteria of the review. A fixed-effects meta-analysis was conducted and, as can be seen in Figure 2, the I2-test suggests there is little or no heterogeneity between the two RCTs for this outcome. The summary OS HR was 0.62 (95% CI 0.50 to 0.77) in favour of MPT.
FIGURE 2.
Melphalan, prednisolone/prednisone plus thalidomide vs MP OS.

The Facon study23 CIs shown in Figure 2 obtained from Review Manager are slightly different to those reported by the published paper and shown in Table 6. The difference arises from the estimating method41 used to obtain the SEs for the log-HRs needed to undertake the meta-analysis.
Deaths during treatment
In the VISTA RCT26 of VMP versus MP, death rates during treatment were similar for the VMP group and the MP group (5% and 4%, respectively) (Table 7). San Miguel and colleagues26 also report that treatment-related deaths were similar in the two groups, but the time at which these deaths occurred is not reported (treatment-related deaths VMP 1% and MP group 2%).
| Study | Treatment arms | p-value | |
|---|---|---|---|
| San Miguel et al.26 VISTA | |||
| VMP (n = 344) | MP (n = 338) | ||
| Deaths during treatment (%) | 5 | 4 | NR |
| Treatment-related deaths (%) | 1 | 2 | NR |
| Facon et al.23 IFM 99/06 | |||
| MPT (n = 124) | MP (n = 193) | ||
| Toxic death | n = 0 | n = 4 (2%), all due to infection | NR |
| Early death – in first 3 months of treatment (n, %) | 3/124 (2) | 13/193 (7) | NR |
| aHulin et al.59 IFM 01/01 | |||
| MPT (n = 113) | MP + placebo (n = 116) | ||
| Toxic death (intestinal perforation) | n = 1 | n = 1 | NR |
| Early death – after 1 month of treatment | n = 3 | n = 3 | NR |
| Early death – after 3 months of treatment | n = 5 | n = 6 | NR |
| MMIX49,53,54 | |||
| CTDa (AiC/CiC information has been removed) | MP (AiC/CiC information has been removed) | ||
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
The two RCTs of MPT that report OS23,59 also provide some information about the deaths that occurred. Facon and colleagues23 provided very limited information, commenting on only toxic deaths (no definition is provided but the term toxic death usually refers to a treatment-related death) and deaths within the first 3 months of treatment (see Table 7). In the MPT group there were no toxic deaths and only three deaths in the first 3 months of treatment. In the MP group there were both more toxic deaths (four deaths all due to infection) and more early deaths (13 deaths) but, as no statistical comparison between the arms is reported, it is not known whether these differences were statistically significant.
Hulin and colleagues59 reported only one toxic death in the MPT group and one in the MP + placebo group. Both of these toxic deaths were caused by intestinal perforation. The number of early deaths was also very similar between the groups. In the MPT group three deaths were reported after 1 month of treatment, and five deaths after 3 months of treatment. In the MP + placebo group, three deaths were reported after 1 month of treatment and six after 3 months of treatment. For both study arms it is not clear whether the number of deaths reported after 3 months is a cumulative value, i.e. including the deaths reported after 1 month of treatment, or whether these are additional deaths that have occurred in months 2 and 3.
(AiC/CiC information has been removed.)
Response to treatment
Various response to treatment rates are reported as secondary outcomes in the VISTA RCT of VMP (Table 8),26 although the analysis is not ITT as previously explained. The time at which response was assessed is not reported. Rates of PR or better (according to EBMT criteria, Appendix 3) were 71% in the VMP group and 35% in the MP group (p < 0.001), and the CR rates were 30% and 4%, respectively (p < 0.001). The rate of PR was 40% in the VMP group and 31% in the MP group and minimal response (MR) rates were 9% and 22%, respectively. Stable disease rates were 18% in the VMP group and 40% in the MP group, and progressive disease rates were 1% and 2%, respectively.
| Study | Treatment arms | p-value | |
|---|---|---|---|
| San Miguel et al.26 VISTA | |||
| VMP (n = 344): n/N (%) | MP (n = 338): n/N (%) | ||
| Rate of PR or better | 238/337 (71) | 115/331 (35) | < 0.001 |
| Rate of CR | 102/337 (30) | 12/331 (4) | < 0.001 |
| Rate of PR | 136/337 (40) | 103/331 (31) | NR |
| MR | 32/337 (9) | 72/331 (22) | NR |
| Stable disease | 60/337 (18) | 113/331 (40) | NR |
| Progressive disease | 3/337 (1) | 7/331 (2) | NR |
| Facon et al.23 IFM 99/06 | |||
| MPT (n = 125): n/N (%) | MP (n = 196): n/N (%) | ||
| CR at 12 months | 10/75 (13) | 4/165 (2) | 0.0008 |
| At least PR at 12 months | 57/75 (76) | 57/165 (35) | < 0.0001 |
| At least very good PR at 12 months | 35/75 (47) | 11/165 (7) | < 0.0001 |
| Hulin et al.59 IFM 01/01 | |||
| MPT (n = 115): n/N (%) | MP + placebo (n = 117): n/N (%) | ||
| CR at 12 months | 7/107 (7) | 1/112 (1) | < 0.001 |
| At least PR at 12 months | 66/107 (62) | 35/112 (31) | < 0.001 |
| At least very good PR at 12 months | 23/107 (21) | 8/112 (7) | < 0.001 |
| Palumbo et al.24 GIMEMA | |||
| MPT (n = 167, 129 analysed): n/N (%) | MP (n = 164, 126 analysed): n/N (%) | Absolute difference MPT–MP: % (95% CI) | |
| CR at 6 months | 20/129 (15.5) | 3/126 (2.4) | 13.1 (6.3 to 20.5) |
| Complete or PR at 6 months | 98/129 (76.0) | 60/126 (47.6) | 28.3 (16.5 to 39.1) |
| PR | 78/129 (60.4) | 57/126 (45.2) | 15.2 (3.0 to 26.9) |
| Near CR | 16/129 (12.4) | 6/126 (4.8) | NR |
| 90–99% M-protein reduction | 11/129 (8.5) | 6/126 (4.8) | NR |
| 50–89% M-protein reduction | 51/129 (39.5) | 45/126 (35.7) | NR |
| MR at 6 months | 7/129 (5.4) | 21/126 (16.7) | –11.2 (–19.2 to –3.6) |
| No response at 6 months | 7/129 (5.4) | 19/126 (15.1) | –9.7 (–17.4 to –2.2) |
| Progressive disease at 6 months | 10/129 (7.8) | 21/126 (16.7) | –8.9 (–17.2 to –0.8) |
| Not available | 7/129 (5.4) | 5/126 (4.0) | NR |
| MMIX49,53,54 | |||
| CTDa (AiC/CiC information has been removed) | MP (AiC/CiC information has been removed) | p-value | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
All three RCTs investigating MPT reported on response to treatment (see Table 8). 23,24,59 The two IFM RCTs23,59 reported the response at 12 months as a secondary outcome, and response was judged according to their own criteria. These criteria are very similar, but not identical, to the EBMT criteria that were used in the RCT by Palumbo and colleagues24 to assess response at 6 months, which was the primary outcome of this RCT (see Appendix 5). Facon and colleagues23 stated that all analyses were undertaken on an ITT basis; it is therefore unclear why response to treatment outcomes are reported for only 60% of the MPT group (75 of the 125 participants enrolled to this group) and 84% of the MP group (165 of 196 enrolled). Hulin and colleagues59 did not indicate that all analyses were ITT (only survival analyses were clearly stated to be ITT) but response to treatment is reported for 93% of the MPT group and 96% of the MP + placebo group. Palumbo and colleagues24 reported on all of those who contributed to the 6-month follow-up results. However, as noted earlier, not all of the randomised participants contributed data to this outcome because some participants had not achieved 6 months of follow-up when these data were analysed.
At 12 months, statistically significant differences in CR in favour of the MPT group were observed in both the IFM RCTs. 23,59 Facon and colleagues23 reported 13% of 75 participants in the MPT group had achieved CR at 12 months in comparison with just 2% of 165 participants in the MP group (p = 0.008). Caution must be applied in interpreting these results, however, which appear to be based on a small proportion of the participants. The difference between the groups reported by Hulin and colleagues59 was less marked but still statistically significant (MPT 7% of 107 participants CR versus 1% of 112 participants in MP + placebo group, p < 0.001). Palumbo and colleagues24 reported an absolute difference in CR MPT–MP at 6 months of 13% (95% CI 6.3 to 20.5).
When response categories were combined, the percentage of participants in the IFM RCT MPT groups achieving at least a PR at 12 months was double the percentage achieving this level of response in the MP group (Facon and colleagues,23 MPT 76% vs MP 35%, p < 0.0001; Hulin and colleagues,59 MPT 62% vs MP + placebo 31%, p < 0.001). At 6 months in the RCT by Palumbo and colleagues24 there was a difference in favour of the MPT group of 28.3% (95% CI 16.5 to 39.1) for participants achieving either a CR or PR.
Each MPT RCT reported on a subcategory of participants with a PR. In the two IFM RCTs, only one subcategory of participants was reported on who were described as having a very good PR. 23,59 These participants had more than a 90% decrease in monoclonal protein in serum and urine. Palumbo and colleagues24 reported on three subcategories of participants with PR. 24 Those with a near CR had disappearance of M-protein from serum and urine but still detectable by immunofixation (immunofixation positive); the remaining participants with a PR were divided into those with a 90–99% M-protein reduction and those with a 50–89% M-protein reduction. Facon and colleagues23 reported at least a very good PR at 12 months in 35 out of 75 participants (47%), which was statistically significantly better than in the MP group where only 7% (11/165) of participants met the criteria (p < 0.001). Hulin and colleagues59 also reported a statistically significant difference in favour of the MPT group at 12 months when 21% (23/107) met the criteria for at least a very good PR, in comparison to 7% (8/112) in the MP group (p < 0.001). Palumbo and colleagues24 report greater proportions of participants in the MPT group than in the MP group at each subcategory of PR after 6 months of follow-up. Of the 78 participants (60.4%) in the MPT group with a PR most (n = 51) had experienced a 50–89% M-protein reduction, 11 participants had a 90–99% M-protein reduction, and 16 participants had a near CR. In contrast, only 57 (45.2%) of MP group participants achieved a PR with the majority (n = 45) having a 50–89% M-protein reduction, six participants having a 90–99% M-protein reduction, and six participants achieving a near CR.
Facon and colleagues23 and Hulin and colleagues59 gave no details about participants who achieved less than a PR at 12 months. Palumbo and colleagues,24 however, provided information on each of the remaining three EBMT categories – minimal response at 6 months, no response at 6 months, and progressive disease at 6 months – as well as indicating the proportion of data that was not available (see Table 8). There were greater proportions of participants from the MP group than the MPT group in the final three categories.
The MMIX RCT49,53,54 assessed maximal response after induction chemotherapy with either CTDa or MP. Response was categorised using EBMT definitions (see Appendix 3). Response was one of the three co-primary outcomes of this RCT. (AiC/CiC information has been removed) (Table 8).
Risk ratios for complete response
Three MPT versus MP RCTs23,24,59 reported CR outcome data that could be meta-analysed. A fixed-effects meta-analysis was conducted and, as can be seen from Figure 3, the I2-test suggests that there is little or no heterogeneity between the three RCTs for this outcome. The outcome is reported as a risk ratio (RR) because a summary relative risk was required for the cost-effectiveness model (see Chapter 5, SHTAC data sources, Complete response). The Facon results were entered using the original group sizes to generate a conservative estimate of overall treatment effect for use in the cost-effectiveness model. The overall effect for the outcome of CR favours MPT (RR 5.49, 95% CI 2.55 to 11.83).
FIGURE 3.
Melphalan, prednisolone/prednisone plus thalidomide vs MP CR.

Risk ratios for the outcome of CR were also obtained for the single RCTs for the VMP versus MP comparison and the CTDa versus MP comparison using the data reported in Table 8 and Review Manager software. These RRs were needed for the cost-effectiveness model [CR VMP vs MP RR 8.35, 95% CI 4.68 to 14.89, (AiC/CiC information has been removed)].
Other time-to-event data
The VISTA RCT26 of VMP was the only included RCT to report time to disease progression (TTP) and this was the primary outcome of this RCT. TTP was calculated from randomisation to disease progression. Data from patients in whom there was no disease progression were censored at the last assessment or at the start of subsequent therapy. Although not explicitly stated, it is assumed that this outcome does not include deaths where there was no disease progression (these events would be included in the outcome of the PFS section: see Progression-free survival). Median TTP was significantly longer in the VMP group than in the MP group (VMP group 20.7 months vs MP group 15.0 months, HR = 0.54, p < 0.001). The median time to first response (partial or better) was 1.4 months in the VMP group and 4.2 months in the MP group (p < 0.001), and 4.2 months and 5.3 months for CR (p < 0.001), respectively (Table 9). The median duration of response (according to EBMT criteria) was 19.9 months in the VMP group and 13.1 months in the MP group; the median duration of response among patients who had a CR was 24 months in the VMP group and 12.8 months in the MP group. Time to subsequent myeloma therapy and treatment-free interval were reported in the published paper26 as 20.8 months and 9.4 months, respectively, in the MP group; these times were not reached for the VMP group. In the abstracts reporting longer follow-up,60,61 time to next therapy was 28.1 months in the VMP group and 19.2 months in the MP group (p < 0.000001, HR 0.53); the treatment-free intervals were 16.6 months and 8.4 months (p < 0.00001, HR 0.54), respectively, after a median follow-up of 25.9 months. 60 After a median follow-up of 36.7 months,61 the treatment-free interval was 17.6 months in the VMP group and 8.4 months in the MP group (HR 0.54, p < 0.0001).
| Study | Treatment arms | HR and p-value | |
|---|---|---|---|
| aSan Miguel et al.26 VISTA | |||
| VMP (n = 344): months | MP (n = 338): months | ||
| TTP median (from computer algorithm analysis) | 20.7 | 15.0 | 0.54; < 0.001 |
| Median time to first response (PR or better) | 1.4 | 4.2 | < 0.001 |
| Median time to CR | 4.2 | 5.3 | <0.001 |
| Median duration of CR or PR | 19.9 | 13.1 | NR |
| Median duration of CR | 24 | 12.8 | NR |
| Median time to subsequent myeloma therapy | Not reached | 20.8 | 0.52; < 0.001 |
| Treatment-free interval | Not reached | 9.4 | NR |
| Time to next therapy from abstracts60,61 | 28.1 (n NR) | 19.2 (n NR) | 0.53; < 0.000001 |
| Treatment-free interval from abstract60 | 16.6 (n NR) | 8.4 (n NR) | 0.54; < 0.00001 |
| Treatment-free interval from abstract61 | 17.6 | 8.4 | 0.543; < 0.0001 |
| Palumbo et al.24 GIMEMA | |||
| MPT: months | MP: months | ||
| Time to PR, median (range, days) | 1.4 (22–200) | 3.1 (25–210) | NR |
Of the MPT RCTs, only Palumbo and colleagues24 reported on the length of time it took to observe a PR to treatment (see Table 9). In the MPT treatment arm the median time to PR was 1.4 months (range 22–200 days) but in the MP arm it took longer to reach the median time to PR of 3.1 months but responses occurred within a very similar range of 25–210 days.
The MMIX RCT49 TTP outcome was not eligible for inclusion in this systematic review because participants were entered into a second randomisation to either maintenance therapy with thalidomide or no maintenance therapy after they had completed first-line treatment with either CTDa or MP.
Progression-free survival
Progression-free survival in the VISTA RCT of VMP was defined by San Miguel and colleagues26 as the time between randomisation and either disease progression or relapse from CR by EBMT criteria, or death due to any cause, whichever occurred first. Median PFS by investigator assessment based on central laboratory data and applying EBMT criteria was 21.7 months in the VMP group and 15.2 months in the MP group (HR 0.56, p < 0.001) – see Table 10.
| Study | Median follow-up (months) | Treatment arms | HR and p-value | |
|---|---|---|---|---|
| San Miguel et al.26 VISTA | ||||
| VMP (n = 344) | MP (n = 338) | |||
| PFS (months): median | 16.3a | 21.7 | 15.2 | 0.56; < 0.001 |
| PFS (months): median (reported in Janssen–Cilag submission to NICE)b | 16.3 | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| Facon et al.23 IFM 99/06 | ||||
| MPT (n = 125) | MP (n = 196) | |||
| PFS (months): median (SE) | 51.5 | 27.5 (2.1) | 17.8 (1.4) | 0.51 (95% CI 0.39 to 0.66); 0.0001c |
| Hulin et al.59 IFM 01/01 | ||||
| MPT (n = 113) | MP + placebo (n = 116) | |||
| PFS (months): median (95% CI) | 47.5 | 24.1 (19.4 to 29.0) | 18.5 (14.6 to 21.3) | 0.62; 0.001 |
The two included IFM RCTs23,59 reported on PFS and both calculated PFS from randomisation to either progression, or death without progression (Table 10). Hulin and colleagues59 censored data on patients who had not experienced progression to the last day that they were known to be alive and progression free. Facon and colleagues23 did not comment on methods for censoring data.
After a median follow-up of 51.5 months, 92 of the 125 participants in the MPT group of the Facon and colleagues23 RCT had either experienced disease progression or they had died. The median PFS of the MPT group was 27.5 months (SE 2.1). In comparison, in the MP group 171 of 196 participants had disease progression or had died and the median PFS was 17.8 months (SE 1.4). The difference in PFS was statistically significant (p = 0.001) with a HR for median PFS in favour of MPT of 0.51 (95% CI 0.39 to 0.66).
Hulin and colleagues59 also found that the difference in PFS between MPT and MP + placebo groups after a median follow-up of 47.5 months was statistically significant with a HR of 0.62 (p = 0.001). In the MPT group median PFS was 24.1 months (95% CI 19.4 to 29.0) in comparison to 18.5 months (95% CI 14.6 to 21.3) in the MP + placebo group.
The event-free24 and progression-free49 survival outcomes reported by Palumbo and colleagues24 and the MMIX RCT49 were not eligible for inclusion in this systematic review because participants received maintenance therapy with thalidomide after first-line treatment had been completed.
Two MPT versus MP RCTs23,59 reported PFS outcome data that was included in a fixed-effects meta-analysis. As can be seen in Figure 4, the I2-test suggests there is little or no heterogeneity between the two RCTs for this outcome. The summary PFS HR was 0.56 (95% CI 0.46 to 0.67) in favour of MPT.
FIGURE 4.
Melphalan, prednisolone/prednisone plus thalidomide vs MP PFS.
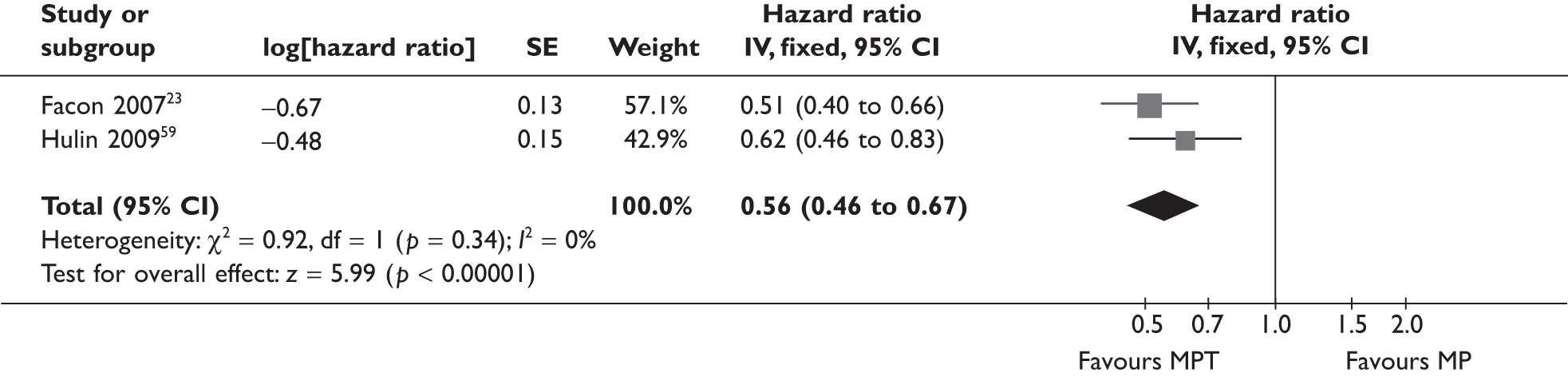
The Facon study CIs shown in Figure 4 obtained from Review Manager are slightly different to those reported by the published paper and shown in Table 10. The difference arises from the estimating method used to obtain the SEs for the log-HRs needed to undertake the meta-analysis.
Quality of life
The VISTA RCT included a QoL assessment (Table 11) that has been reported only in an abstract. 62 The abstract states that the aim of the study was to describe the rate of patients who experienced a sustained HRQoL improvement after best response and the overall HRQoL impact of best response. A sustained HRQoL improvement was defined as a change in score of at least five points for at least two consecutive cycles after best response (CR, PR or MR). After best-response onset, patients in the VMP arm had a higher sustained HRQoL improvement rate than those in the MP arm in 14 of the 15 European Organisation for Research and Treatment of Cancer quality-of-life questionnaire C30 (EORTC QLQ-C30) scores.
| Study | Treatment arms | p-value | |
|---|---|---|---|
| VMP (n = 344, no. analysed NR) | MP (n = 338, no. analysed NR) | ||
| Dhawan et al.62 VISTA | |||
| Sustained response in QLQ-C30 domainsa (%) | |||
| Cognitive functioning | 27 | 28 | NR |
| Nausea/vomiting | NR | NR | 0.0095b |
| Appetite loss | NR | NR | 0.0170 |
| Diarrhoea | NR | NR | 0.0082 |
| Global health | 49 | 40 | Not statistically significant |
| Pain | 40 | 32 | Not statistically significant |
| Insomnia | 32 | 24 | Not statistically significant |
The MMIX RCT49 assessed QoL but it was not possible to include data in this systematic review because some of the participants were entered into a second randomisation to either maintenance therapy with thalidomide or no maintenance therapy after they had completed first-line treatment with either CTDa or MP.
Adverse events
This section summarises AEs reported by RCTs, concentrating on the events that require active management and/or have the greatest impact on patient QoL. The AEs that have been omitted from each table are listed in the table footnotes and the complete AE data for each RCT can be found in the data extraction forms in Appendix 5.
Adverse events reported by San Miguel and colleagues in the VISTA RCT26 of VMP were graded with the use of the NCI CTCAE (version 3). Occurrence of any AE and grade 4 AE was similar in the two groups, although grade 3 events were more common in the VMP group (53% vs 44%, p = 0.02) (Table 12). Haematological toxic events were the most frequently reported AEs and were also similar in the two groups. Peripheral sensory neuropathy was reported more frequently in the VMP group but, by the data cut-off point, 74% of peripheral neuropathy events had either resolved (56%) or decreased by at least one toxicity grade (18%) within a median of 2 months. All grade 3 and grade 4 gastrointestinal events were more frequent in the VMP group than in the MP group (19% vs 5%, no p-value given). Incidence of deep-vein thrombosis (DVT) was low and similar in the two groups.
| Study | Treatment arms | p-value | |
|---|---|---|---|
| VMP (n = 340) | MP (n = 337) | ||
| San Miguel et al.26,60 VISTA | n (%) | n (%) | |
| Any eventa | 338 (99) | 326 (97) | NR |
| Grade 3 | 181 (53) | 148 (44) | 0.02 |
| Grade 4 | 96 (28) | 92 (27) | NR |
| Serious AEs | 46 | 36 | NR |
| Haematological eventsb | |||
| Thrombocytopenia | 178 (52) | 159 (47) | NR |
| Neutropenia | 165 (49) | 155 (46) | NR |
| Anaemia | 147 (43) | 187 (55) | NR |
| Leucopenia | 113 (33) | 100 (30) | NR |
| Lymphopenia | 83 (24) | 58 (17) | NR |
| Gastrointestinal events of grade 3 and grade 4 c | 19 | 5 | NR |
| Infections | |||
| Pneumonia | 56 (16) | 36 (11) | NR |
| Herpes zoster | 45 (13) | 14 (4) | NR |
| Nervous system disorders | |||
| Peripheral sensory neuropathy | 151 (44) | 16 (5) | NR |
| Neuralgia | 121 (36) | 5 (1) | NR |
| Dizziness | 56 (16) | 37 (11) | NR |
| Other conditionsc | |||
| Fatigue | 98 (29) | 86 (26) | NR |
| DVT | 4 (1) | 6 (2) | NR |
The two IFM RCTs23,59 did not report which system was used to grade toxic effects and AEs to treatment and therefore caution must be applied when comparing the results of these two RCTs with each other, and with the RCT reported by Palumbo and colleagues. 24 Neither IFM RCT describes whether all AEs that occurred have been reported, or whether only a subset of AEs is reported in the trial publication. Palumbo and colleagues24 used the NCI CTC (version 2) to grade AEs and all grade 3–4 events reported by patients or observed by investigators were reported. However, only AE reporting of infections from Palumbo and colleagues24 can be included here as the majority of AEs were reported for the whole trial period, which included administration of thalidomide maintenance therapy in the MPT group. AEs are summarised in Table 13. Facon and colleagues23 analysed safety at the October 2005 date point after 36.8 months of follow-up, a shorter follow-up than for the outcomes of OS, PFS and survival after progression.
| Study | Treatment arms | p-value | |
|---|---|---|---|
| a,bFacon et al.23 IFM 99/06 | n (%) | n (%) | |
| MPT (n = 124) | MP (n = 193) | ||
| Grade 3 and 4 AEs – after 36.8 months of follow-up | |||
| Haematological: | |||
| anaemia | 17 (14) | 27 (14) | 0.94 |
| neutropenia | 60 (48) | 51 (26) | < 0.0001 |
| thrombocytopenia | 17 (14) | 19 (10) | 0.29 |
| thrombosis or embolism | 15 (12) | 8 (4) | 0.008 |
| Peripheral neuropathy | 7 (6) | 0 | 0.001 |
| Somnolence/fatigue/dizziness | 10 (8) | 0 | < 0.0001 |
| Infection | 16 (13) | 18 (9) | 0.32 |
| Gastrointestinal: | |||
| nausea | 1 (1) | 2 (1) | |
| constipation | 13 (10) | 0 | < 0.0001 |
| Any grade ≥ 3 non-haematological toxic effect | 52 (42) | 30 (16) | < 0.0001 |
| b,cHulin et al.59 IFM 01/01 | n (%) | n (%) | |
| MPT (n = 113) | MP + placebo (n = 116) | ||
| Peripheral neuropathy: | 0.003 | ||
| grade 1 | 20 (18) | 19 (16) | |
| grade 2 | 21 (19) | 4 (3) | |
| grade 3 | 2 (2) | 2 (2) | |
| Neutropenia grade 3 or 4 | 26 (23) | 10 (9) | 0.003 |
| Thrombosis or embolism grade 3 or 4 | 7 (6) | 4 (3) | 0.33 |
| Somnolence grades 2–4 | 7 (6) | 3 (3) | 0.19 |
| Constipation grades 2–4 | 19 (17) | 12 (10) | 0.16 |
| Nausea/vomiting grades 2–4 | 3 (3) | 5 (4) | 0.5 |
| dPalumbo et al.24 GIMEMA | n/N (%) | n/N (%) | |
| MPT | MP | ||
| Grades 3–4 infections | 12/129 (10) within the first 4 months of treatment | 2/126 (2); timing of occurrence unknown | 0.01 |
Four types of haematological event (at grade 3 and 4) were reported by Facon and colleagues. 23 There were no statistically significant differences in the occurrence of anaemia (14% both groups, p = 0.94) or thrombocytopenia (MPT group 14%, MP group 10%, p = 0.29). A statistically significant difference was reported for neutropenia, which occurred in a greater proportion of MPT patients than MP patients (48% vs 26%, p < 0.0001). Hulin and colleagues59 also reported that a statistically significantly greater proportion of participants in the MPT group experienced neutropenia (grades 3 and 4) than in the MP group (23% vs 9%, p=0.003) but did not report on any other haematological events.
Both IFM RCTs23,59 reported the occurrence of grades 3 and 4 thrombosis or embolism. Facon and colleagues23 found that the greater proportion of patients with grades 3 and 4 thrombosis or embolism in the MPT group was a statistically significant difference in comparison with the MP group (MPT 12% vs MP 4%, p = 0.008). In contrast, there was no statistically significant difference in this AE in the Hulin and colleagues RCT59 (MPT 6% vs MP 3%, p = 0.33).
Peripheral neuropathy occurred statistically significantly more frequently in the MPT groups of both IFM RCTs but the reporting of this differed. Facon and colleagues23 reported on the occurrence of grades 3 and 4 peripheral neuropathy in both groups (MPT 6% vs zero events in the MP group, p = 0.01). Facon and colleagues23 also stated that peripheral neuropathy was observed in 69 (55%) patients in the MPT group, with the majority of cases (n = 62) being grade 1 or 2, and the remainder grade 3 (n = 7), with no grade 4 events. The equivalent data for the MP group were not provided. In contrast, Hulin and colleagues59 reported on each grade of peripheral neuropathy separately for each group. The proportion of patients with peripheral neuropathy was reported to be statistically significantly greater in the MPT group than the MP group (p = 0.003), although it was not clear whether the p-value related to peripheral neuropathy in general or grade 1 peripheral neuropathy in particular. Most cases of peripheral neuropathy were of grade 1 or 2 (grade 1 peripheral neuropathy MPT 18%, MP 16%, grade 2 peripheral neuropathy MPT 19%, MP 3%). Severe peripheral neuropathy was less common with 2% of both groups experiencing grade 3 peripheral neuropathy, and no grade 4 events reported.
Facon and colleagues23 report what appears to be a composite outcome described as somnolence/fatigue/dizziness (grades 3 and 4). This occurred in 8% of the MPT group, statistically significantly more than the MP group where no one had these symptoms at this grade (p < 0.0001). In contrast, Hulin and colleagues59 reported on the single outcome of somnolence but over a wider severity range (grades 2–4) and found no statistically significant difference between the groups (MPT 6% vs MP 3%, p = 0.19).
The incidence of grade 3 and 4 infections was reported by two RCTs23,24 and details of the infections contributing to this outcome were provided. Facon and colleagues23 reported no statistically significant difference in the number of patients with infections of grades 3 and 4 (MPT n = 16 patients, 13% vs MP n = 18 patients, 9%, p = 0.32). However, it is clear, although not explicitly stated, that some patients must have experienced more than one grade 3 or 4 infection, because the reported numbers of individual infections sum to 20 (see Appendix 5). In the 6-month period of treatment in the Palumbo and colleagues RCT24 eligible for inclusion in the review, there were statistically significantly more infections in the MPT group than the MP group (MPT 10% all within the first 4 months vs MP 2%, p = 0.01). Hulin and colleagues59 did not report this outcome, other than stating that the higher incidence of neutropenia in the MPT group did not translate into more frequent severe infections.
Gastrointestinal events of nausea and vomiting when reported were also infrequent events (see Table 13). Constipation was the most commonly reported gastrointestinal AE. Facon and colleagues23 reported that only participants in the MPT group experienced constipation at grades 3 and 4, which was a statistically significant difference (p < 0.0001) in comparison to the MP group where no grade 3 and 4 constipation AEs were reported. Hulin and colleagues59 reported on constipation AEs of grades 2–4, and the difference between the groups was not statistically significant (MPT 17% vs MP 10%, p = 0.16).
Overall, Facon and colleagues23 found that non-haematological toxic effects of grade 3 or higher were statistically significantly more likely in the MPT group than the MP group (MPT 42% vs MP 16%, p < 0.0001).
The MMIX IX protocol49 does not indicate which system would be used to grade AEs. It is also not clear whether all AEs that occurred related to induction chemotherapy are presented in the results that have been made available. 54 AEs are summarised in Table 14.
| Study | Treatment arms | p-value | |
|---|---|---|---|
| CTDa (safety population) | MP (safety population) | ||
| MMIX54 | |||
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | ||
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | ||
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | ||
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | ||
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | ||
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | ||
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | ||
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | ||
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
(AiC/CiC information has been removed.)
Discontinuation or withdrawal due to AEs
In addition to the reporting of AEs in general above (see Adverse events, Tables 12–14) some of the included RCTs also reported on the patients who discontinued study medication, or withdrew from the study as a consequence of AEs (Tables 15 and 16).
| Study | Treatment arms | p-value | |
|---|---|---|---|
| VMP (n = 340) | MP (n = 337) | ||
| San Miguel et al.26 VISTA: (n, %) | |||
| Discontinued treatment due to AEs | 50 (15) | 47 (14) | NR |
| Discontinued treatment due to treatment-related events | 37 (11) | 35 (10) | NR |
| Discontinuations of bortezomib alone | 63 (19) | – | |
| Study | Treatment arms | p-value | |
|---|---|---|---|
| aFacon et al.23 IFM 99/06 | |||
| MPT | MP | ||
| Discontinuation of thalidomide because of toxic effects: | 56/124 (45%) | NR | NR |
| peripheral neuropathy | n = 23 | NR | NR |
| thrombosis | n = 7 | NR | NR |
| somnolence, dizziness or fatigue | n = 8 | NR | NR |
| cutaneous toxic effects | n = 4 | NR | NR |
| psychiatric complications | n = 1 | NR | NR |
| Withdrawn because of other reasons: | n = 13 | NR | NR |
| haematological toxic effects | n = 5 | NR | NR |
| infection | n = 7 | NR | NR |
| stroke | n = 1 | NR | NR |
| bHulin et al.59 IFM 01/01 | |||
| MPT (n = 113) | MP + placebo (n = 116) | ||
| Withdrawals due to adverse events/toxicity: | n = 48 (42.5%)c | n = 15 (12.9%)c | NR |
| peripheral neuropathy | n = 12 | n = 3 | NR |
| neurological events (non-peripheral) | n = 10 | n = 1 | NR |
| thrombosis/embolism | n = 7 | n = 1 | NR |
| haematological events | n = 7 | n = 6 | NR |
| digestive events | n = 4 | n = 2 | NR |
| cardiac events | n = 3 | n = 1 | NR |
| rash | n = 2 | n = 0 | NR |
| other | n = 3 | n = 1 | NR |
| Dose reduction required because of AEs | n = 20 (17.7%)c | n = 3 (2.6%)c | NR |
| Palumbo et al.24 GIMEMA | |||
| MPT | MP | ||
| Unable to complete six cycles due to AEs | 17/129 (13.2%)c | 4/126 (3.2%)c | NR |
| Thalidomide discontinuation required | 43 (33.3%)c patients after a median of 2.1 months | N/A | N/A |
| Thalidomide dose reduction to 50 mg required | 37 (28.7%)c patients after a median of 4 months | N/A | N/A |
In the VISTA RCT,26 50 patients (15%) in the VMP group and 47 patients (14%) in the MP group discontinued treatment due to AEs (see Table 15), including 37 patients (11%) and 35 patients (10%), respectively, who had treatment-related events. San Miguel and colleagues26 provide no further details. Bortezomib alone was discontinued in an additional 63 patients (19%).
For the MPT RCTs it was not clear from the study reports how many of the AEs that led to discontinuation or withdrawal had already been included in the general reporting of AEs (see Table 13). It also seemed clear from data reported that some discontinuations and withdrawals were due to events not specified in the general reporting of AEs (see Table 13), for example discontinuation of thalidomide due to cutaneous effects,23 and withdrawals due to cardiac events59 and rash59 (see Table 16).
Two of the RCTs, by Hulin and colleagues59 and Palumbo and colleagues,24 reporting on withdrawals due to AEs/toxicity and inability to complete six cycles of treatment due to AEs, respectively, reported the outcome for both study groups. AEs led to more withdrawals from treatment in the MPT group than the MP group but the differences were not tested statistically (Hulin and colleagues,59 MPT 42.5% vs MP plus placebo 12.9%; Palumbo and colleagues24 MPT 13.2% vs MP 3.2%). Palumbo and colleagues24 also report that discontinuation of thalidomide was required by 43 patients (33.3%) after a median of 2.1 months. It is not clear, but presumably these 43 patients included the 17 in the MPT group who were unable to complete the six treatment cycles. Facon and colleagues23 report discontinuation of thalidomide in the MPT group among 45% of the participants who discontinued because of toxic effects, but do not report on discontinuations in the MP arm due to AEs.
In addition to discontinuation of thalidomide due to AEs, two RCTs reported that reductions in the dose of study drug were required by 17.7% of the MPT group vs 2.6% of the MP group in the Hulin and colleagues RCT59 and in 28.7% of the MPT group after a median of 4 months of treatment in the Palumbo and colleagues RCT,24 where dose reductions required in the MP arm were not reported.
Withdrawals from study due to any reason
A supplementary appendix to the VISTA RCT publication26 reports on the numbers of patients withdrawn from the study with reasons (Table 17). Numbers are similar in the two groups overall and for treatment-related events, death and other non-specified reasons. Withdrawal due to patient choice and maintenance of CR is higher in the VMP group, while withdrawal due to progressive disease is higher in the MP group (no p-values are given).
| Study | Treatment arms | p-value | |
|---|---|---|---|
| VMP (N = 340) | MP (N = 337) | ||
| San Miguel et al.26 VISTA: (n, %) | |||
| Patients still receiving assigned protocol at data cut-off point | 47 (14) | 33 (10) | NR |
| Total discontinued treatment | 139 (41) | 166 (49) | NR |
| Discontinued due to progressive disease | 24 (7) | 72 (21) | NR |
| Discontinued due to AEsa | 50 (15) | 47 (14) | NR |
| Discontinued due to patient choice | 32 (9) | 18 (5) | NR |
| Discontinued due to death | 14 (4) | 17 (5) | NR |
| Discontinued due to maintenance of CR | 9 (3) | 1 (< 1) | NR |
| Other reasons for discontinuation | 10 (3) | 11 (3) | NR |
The IFM RCTs report on the proportion of participants withdrawn from the study, and provide some information on the reasons for the withdrawals. It is not clear whether withdrawal data in Facon and colleagues23 are reported for the initial analysis date of October 2005 (median follow-up 36.8 months), or the later date of 2007 (median follow-up of 51.5 months). Hulin and colleagues59 report withdrawals for the median follow-up of 47.5 months. The majority of participants from both RCTs had been withdrawn from study treatment arms at the point of data analysis. In Facon and colleagues’ RCT23 93 participants (75%) were withdrawn from the MPT arm, and 151 participants (78%) from the MP arm. Facon and colleagues23 do not report the reasons for these withdrawals but do indicate what proportion of withdrawn participants went on to receive a second-line treatment, and of those who had not received another treatment, how many had died and how many were still alive (Table 18). Hulin and colleagues59 had 88.5% of participants withdraw from the MPT arm and 93.1% withdraw from the MP + placebo arm of their RCT. Most withdrawals in the MPT arm were due to toxicity (48 of 100 withdrawals) whereas in the MP + placebo arm most withdrawals were due to disease progression (69 of 108 withdrawals) (see Table 18). A similar pattern was reported by Palumbo and colleagues24 for the initial six treatment cycles (before the introduction of thalidomide maintenance therapy), where the most common reason for participants in the MPT group being unable to complete the six treatment cycles was AEs, but in the MP group progressive disease was the main reason. No statistical comparisons of the data are reported within any of the RCTs.
| Study | Treatment arms | p-value | |
|---|---|---|---|
| aFacon et al.23 IFM 99/06: (n, %) | |||
| MPT | MP | ||
| Not withdrawnb | 31/124 (25) | 42/193 (22) | NR |
| Withdrawn and not receiving second-line treatment | 38/124 (31) | 25/193 (13) | NR |
| up to death | 11/38 (29) | 24/25 (96) | NR |
| still alive | 27/38 (72) | 1/25 (4) | NR |
| Withdrawn and having received second-line treatment | 55/124 (44) | 126/193 (65) | NR |
| cHulin et al.59 IFM 01/01 | |||
| MPT | MP + placebo | ||
| Withdrawals overall: | n = 100/113 (88.5%) | n = 108/116 (93.1%) | NR |
| due to disease progression | n = 37 | n = 69 | NR |
| due to death | n = 6 | n = 16 | NR |
| due to consent withdrawal | n = 9 | n = 8 | NR |
| due to toxicity (details above) | n = 48 | n = 15 | NR |
| Palumbo et al.24 GIMEMA | |||
| MPT | MP | ||
| Unable to complete six cycles: | 32/129 (25%) | 31/126 (25%) | NR |
| due to AEs (as noted above) | 17/32 | 4/31 | NR |
| due to progressive disease | 9/32 | 16/31 | NR |
| because withdrew consent | 3/32 | 2/31 | NR |
| because lost to follow-up | 3/32 | 7/31 | NR |
| due to protocol violations | 0/32 | 2/31 | NR |
(AiC/CiC information has been removed.)
Withdrawals from the MMIX Trial of CTDa are shown in Table 19.
| Study | Treatment arms | Total | |
|---|---|---|---|
| CTDa | MP | ||
| MMIX52 | |||
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
Duration and intensity of first-line treatment
San Miguel and colleagues26 reported that in the VISTA RCT26 treatment lasted for a median of eight cycles in the VMP arm and seven cycles in the MP arm (Table 20). This is equivalent to approximately 11.5 months and 10 months, respectively.
| Study | Treatment arms | p-value | |
|---|---|---|---|
| San Miguel et al.26 VISTA | |||
| VMP (n = 340) | MP (n = 337) | ||
| Median number of treatment cycles | 8 (46 weeks) | 7 (39 weeks) | NR |
| Facon et al.23 IFM 99/06 | |||
| MPT | MP | ||
| Median duration of treatment (IQR) | 11 months (5–15) | NR | NR |
| Initial daily dosea of thalidomide ≤ 200 mg | n = 64/124 (52%) (includes nine participants receiving initial dose of 100 mg) | NR | NR |
| Initial daily dose of thalidomide > 200mg | n =60/124 (48%) (includes five participants receiving initial dose of 300 mg) | NR | NR |
| No change of dose throughout first-line treatment | n = 66/124 (36 at ≤ 200 mg/day; 30 at > 200 mg/day) | NR | NR |
| Dose increased during first-line treatment | n = 11/124 | NR | NR |
| Dose reduced during first-line treatment | n = 47/124 | NR | NR |
| Hulin et al.59 IFM 01/01 | |||
| MPT | MP + placebo | ||
| Median duration of treatment | 13.5 months | 18 months | NR |
| MMIX52,53 | |||
| CTDa | MP | ||
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
The median duration of treatment of 11 months in the MPT arm of one of the MPT RCTs, Facon and colleagues,23 was similar to that of the VISTA RCT. The duration of treatment was not reported for the MP trial arm. Facon and colleagues23 also reported on the intensity of treatment with thalidomide. The aim was for participants to achieve a 400-mg daily dose of thalidomide if it could be tolerated. Although not explicitly reported, it appears unlikely (see Table 20) that many participants received 400 mg for the majority of the treatment period. Approximately 29% (36/124 participants) received < 200mg/day for the duration of first-line treatment, and 47/124 of participants had their dose reduced during treatment. Only 11 participants were able to tolerate having their thalidomide dose increased during treatment.
Hulin and colleagues59 had a treatment period of 72 weeks (about 18 months), but while the median duration of treatment in the MP group was 18 months, the median duration of treatment in the MPT group was only 13.5 months (see Table 20). The trial authors do not comment on this.
(AiC/CiC information has been removed.)
Second-line treatments received by trial participants
San Miguel and colleagues26 reported that in the MP group 57% of participants started second-line therapy within 2 years, in comparison with 35%26 (updated to 38% in a more recent abstract60) in the VMP group. It is not clear what the denominator in these calculations is, the total number of randomised participants or the number of surviving participants. Over one-half of the participants in each group received either thalidomide or lenalidomide as a second-line therapy (Table 21).
| Study | Treatment arms | |||
|---|---|---|---|---|
| San Miguel et al.26 VISTA | ||||
| VMP | MP | |||
| Started second-line treatment within 2 years26 (%) | 35 | 57 | ||
| Outcomes from abstract61 at median follow-up of 36.7 months | ||||
| n = 178 | n = 233 | |||
| Received subsequent therapy containing: | ||||
| Bortezomib (n, %) | 43 (24) | 116 (50) | ||
| Thalidomide (n, %) | 81 (46) | 110 (47) | ||
| Lenalidomide (n, %) | 57 (32) | 30 (13) | ||
| Overall response rate to subsequent therapy: | ||||
| Bortezomib (%) | 47 | 59 | ||
| Thalidomide (%) | 41 | 53 | ||
| Lenalidomide (%) | 59 | 52 | ||
| Outcomes from abstract60 at median follow-up of 25.9 months | ||||
| n = 129 | n = 194 | |||
| Required subsequent therapy (%) | 38 | 57 | ||
| Received bortezomib (%) | 16 | 43 | ||
| Received thalidomide (%) | 49 | 44 | ||
| Received lenalidomide (%) | 19 | 6 | ||
| aSubsequent therapy and number of patients who received it | ||||
| CR (%) | PR (%) | CR (%) | PR (%) | |
| Bortezomib or bortezomib combination (n = 105) | 6 | 33 | 10 | 45 |
| Thalidomide combination (n = 149) | 4 | 44 | 3 | 52 |
| Lenalidomide combination (n = 37) | 4 | 52 | 0 | 55 |
| bFacon et al.23 IFM 99/06: (n, %) | ||||
| MPT | MP | |||
| Second-line treatment administered | 55/124 (44) | 126/193 (65) | ||
| Second-line treatment thalidomide alone or in combination | 10/55 (18) | 55/126 (44) | ||
| Second-line treatment VAD | 15/55 (27) | 42/126 (33) | ||
| Second-line treatment dexamethasone | 7/55 (13) | 12/126 (10) | ||
| Second-line treatment alkylating agent-based regimens | 14/55 (25) | 13/126 (10) | ||
| Bortezomib | 7/55 (13) | 3/126 (2) | ||
| Other | 2/55 (4) | 1/126 (1) | ||
| Hulin et al.59 IFM 01/01: (n, %) | ||||
| MPT | MP + placebo | |||
| Disease progression occurrence | 72/113 (64) | 84/116 (72) | ||
| Second-line treatment administeredc | 61/72 (85) | 70/84 (83) | ||
| Thalidomide | 16/72 (22) | 53/84 (63) | ||
| Bortezomib | 22/72 (31) | 28/84 (33) | ||
| Lenalidomide | 11/72 (15) | 9/84 (11) | ||
| Thalidomide and/or lenalidomide | 25/72 (35) | 59/70 (83) | ||
| Thalidomide and/or lenalidomide and/or bortezomib | 38/72 (53) | 68/81 (83) | ||
Two of the three RCTs of MPT versus MP provided data on second-line treatment that could be included in the review23,59 (as participants in the RCT by Palumbo and colleagues24,25 received maintenance therapy with thalidomide, second-line treatment data have not been included here). Second-line treatment was administered to 65% of the MP group in comparison with 44% of the MPT group in the RCT reported by Facon and colleagues. 23 Hulin and colleagues59 reported disease progression occurrence in 156 participants overall, with more participants with disease progression in the MP + placebo group than the MPT group (72% vs 64%). Second-line treatment was administered to a similar proportion of participants with disease progression in each arm. In both RCTs, thalidomide (alone or in combination) was the most commonly administered second-line treatment in the MP group, with about a fifth of participants in the MPT groups of these RCTs receiving thalidomide again as second-line therapy. The most commonly administered second-line treatment in the MPT group reported by Facon and colleagues23 was a combination of vincristine, doxorubicin and dexamethasone (VAD). Only 13% of MPT arm participants received bortezomib. In contrast, Hulin and colleagues59 reported that 31% of participants in the MPT arm received bortezomib as a second-line treatment (see Table 21).
Survival time after disease progression
Median survival time after disease progression was longer by approximately 2 months in participants in the MPT groups than for those in the MP groups in the two RCTs that reported this outcome. 23,59 However, in the one RCT that reported a statistical comparison this difference was not statistically significant (MPT 11.5 months vs MP 9.9 months, p = 0.89) (Table 22).
| Study | Treatment arms | p-value | |
|---|---|---|---|
| Facon et al.23 IFM 99/06 | |||
| MPT (n = 83) | MP (n = 154) | ||
| Survival time after progression: median (SE) | 13.4 (2.3) months | 11.4 (1.9) months | |
| After median follow-up of 51.5 months | 52 deaths/83 patients | 111 deaths/154 patients | |
| Hulin et al.59 IFM 01/01 | |||
| MPT | MP | ||
| Survival time after progression: median | 11.5 months | 9.9 months | 0.89 |
Subgroup analysis
Subgroup analysis in the VISTA RCT took place between VMP and MP participant subgroups defined by participant baseline characteristics. Results were presented only for the outcome of time to disease progression. These results showed that for each of the seven prespecified subgroups (age, sex, race, baseline β2-microglobulin level, baseline albumin level, region and disease stage) and single post hoc subgroup (baseline creatinine clearance) the risk of disease progression in the VMP arm was lower than for participants in the equivalent subgroups in the MP arm (i.e. time to disease progression was shorter in the MP subgroups than the VMP subgroups). It is not clear whether the RCT was powered for these subgroup analyses and therefore caution should be applied when interpreting the results.
No subgroup analysis data were eligible for inclusion in this review from the RCTs of MPT versus MP. Facon and colleagues23 state in the discussion section of their paper that post hoc analyses for three subgroups were conducted but the results of these are not presented. No subgroup analyses are reported by Hulin and colleagues59 and the subgroup analyses for PFS and OS reported by Palumbo and colleagues24,25 are not eligible for inclusion due to the use of thalidomide maintenance in the MPT group of this RCT.
The MMIX Trial protocol49 states that ‘Subgroup analysis may by chance generate false negative/positive results. Those carried out will be interpreted with caution and treated as hypothesis-generating’ (p. 57). (AiC/CiC information has been removed.)
SHTAC review of clinical effectiveness in manufacturers’ submissions
Celgene Ltd (thalidomide manufacturer) and Janssen–Cilag Ltd (bortezomib manufacturer) submitted reports to NICE. The clinical effectiveness evidence presented in these reports has been briefly appraised (see Appendix 7). A discussion of the economic models and cost-effectiveness results included in the MSs can be found in Chapter 5 (see Review of the Janssen–Cilag submission to NICE and Review of the Celgene submission to NICE).
The manufacturers both conducted systematic reviews of the clinical effectiveness evidence; however, only Janssen–Cilag presented this within the main body of the MS. Celgene reported a systematic review only as part of the appendix, which described the meta-analysis they had undertaken. Both manufacturers supplied search strategies and reported on the details of the searches undertaken. Neither manufacturer appeared to have searched for ongoing studies although conference proceedings were included in their searching.
The MSs differ in the clinical effectiveness evidence that has been included, and the evidence in each submission also differs to that included in the SHTAC systematic review (see Results of the systematic review of clinical effectiveness, above). These differences can be seen in Table 23, which shows which studies have been included. In addition to the available published evidence, Janssen–Cilag included data from the clinical study reports of the bortezomib RCT.
| Triala | Janssen–Cilag | Celgene | SHTAC |
|---|---|---|---|
| San Miguel et al.26,60,61 (VISTA) | ✓ | ✓ | ✓ |
| Facon et al.23 (IFM 99/06) | ✓ | ✓ | ✓ |
| Hulin et al.59 (IFM 01/01) | ✓ | ✓ | ✓ |
| Palumbo et al.24 (GIMEMA) | ✓ | In sensitivity analysis only | Only data prior to the start of thalidomide maintenance therapy |
| bNordic myeloma study group63,64 | ✓ | In sensitivity analysis only | ✗ (ongoing study, designated unclear) |
| bHOVON 4965–67 | ✓ | In sensitivity analysis only | ✗ (ongoing study, designated unclear) |
| b,cMMIX49,52 | ✓ | ✗ | ✓ |
The conclusions on the clinical effectiveness of MPT and VMP of the two MSs and the SHTAC systematic review (based on narrative summaries of trial outcomes) are broadly similar. Owing to the differences in the trials included and the different methodologies employed between the SHTAC meta-analyses and the manufacturers’ mixed-treatment comparisons (MTCs), it has not been possible to draw meaningful comparisons between them.
Ongoing studies
The clinical effectiveness search for studies identified seven abstracts and two ClinicalTrials.gov records that described four ongoing studies, each comparing MPT with MP. It is not clear whether these studies meet the inclusion criteria of this systematic review.
Two abstracts63,64 and a ClinicalTrials.gov record (identifier NCT00218855) describe an ongoing study that recruited participants in Norway, Sweden and Denmark between 2002 and 1 May 2007. This study has not been reported on in detail because insufficient details about the study were provided (e.g. drug doses for MP unknown, number of participants in each study arm unknown), and there were also insufficient details presented to allow judgements about study quality to be made. Some information presented differed between the two abstracts, and the ClinicalTrials.gov description of the study indicates that patients receive thalidomide maintenance treatment, so it is therefore unclear whether this study meets the inclusion criteria of this review.
The second ongoing study, HOVON 49, is described in three abstracts65–67 and, again, it is unclear whether this study meets the inclusion criteria for this review because patients could receive thalidomide maintenance treatment. The study recruited participants in the Netherlands starting in 2002 but participant accrual was stopped early (date not reported) due to the publication of other RCTs showing a positive outcome for thalidomide-treated participants.
The third ongoing study, described in a single abstract,68 compares MP with MPT but also includes a second randomisation at the end of induction therapy to maintenance therapy with either dexamethasone, or dexamethasone plus thalidomide.
If the full publications describing the three studies above report outcome data for participants at a time point prior to maintenance therapy then these data would be eligible for inclusion. Similar data have already been included in this review from the study by Palumbo and colleagues24 and the MMIX study,49,53,54 which both incorporated maintenance therapy.
The fourth ongoing RCT described in a conference abstract and a ClinicalTrials.gov record (identifier NCT00934154)69 is an RCT initiated by the Turkish Myeloma Study Group that allows participants from the MP arm to cross over to the MPT arm if insufficient response to MP is obtained, with response being evaluated at every other cycle. It is not clear whether this crossover RCT will meet the inclusion criteria of the review.
Summary of clinical effectiveness
-
Five RCTs23,24,26,49,59 met the inclusion criteria of the systematic review; four23,24,26,59 have been published in full papers, one has only been reported in abstracts but additional information has been provided by the trialists. 49 One RCT26 examined the effectiveness of bortezomib in combination with MP, three RCTs23,24,59 examined the effectiveness of MPT, and one RCT49 examined CTDa. The comparator in all five RCTs was MP, and the comparator of one RCT also included a placebo in place of thalidomide.
-
Four further trials, published only in abstract form,63–69 provided insufficient details to allow a judgement about whether they are likely to meet the inclusion criteria of this review (so these were excluded from the systematic review).
VMP versus MP alone
-
The quality of the RCT26 was difficult to determine. Risk of allocation bias and of unbalanced confounding factors could not be judged because details on these aspects were not reported. Most, but not all, analyses had followed the ITT principle but the methods used to account for any missing data were not described. It was not possible to determine whether the amount and pattern of censored data was similar between trial arms.
-
Time to disease progression was the primary outcome of the RCT and a statistically significant effect in favour of the VMP group was reported.
-
Overall survival was a secondary outcome. A survival advantage for the VMP arm in comparison with the comparator MP was reported.
-
Statistically significantly more participants in the VMP group achieved CR, or achieved a PR or better. This outcome was not analysed by ITT principles.
-
Median PFS was statistically significantly longer in the VMP group than in the MP group.
-
Limited data on HRQoL was available. This indicated that, after the onset of best response, participants treated with VMP had a higher sustained HRQoL improvement rate in 14 of the 15 EORTC QLQ-C30 scores than those participants receiving therapy with MP.
-
Adverse events occurred in both trial arms. Although the occurrence of any AE and any grade 4 AE was similar in the two groups, there was a statistically significant increase in grade 3 AEs in the VMP group.
-
Subgroup analyses were conducted. The RCT may not have been powered for these analyses so the results, which indicate that the reported benefits of bortezomib for TTP apply to each of the seven subgroups of participants assessed, should be interpreted with caution.
MPT versus MP alone
-
The quality of the three RCTs23,24,59 was variable. Risk of allocation bias could not be judged for one RCT, and the risk of allocation bias and of unbalanced confounding factors could not be judged for another RCT because the necessary details were not reported. Although all RCTs stated that ITT analyses had been conducted, the details of these analyses and the methods used to account for missing data were, in general, poorly described. It was not possible to determine whether the amount and pattern of censored data was similar between trial arms for any of the included RCTs.
-
Overall survival was a primary outcome in two of the RCTs. 23,59 Both reported a survival advantage for the MPT arm in comparison with the comparator MP alone. As expected, meta-analysis of the OS data from two RCTs of MPT versus MP provided a HR in favour of MPT for the OS outcome. The third RCT of MPT included maintenance therapy with thalidomide and therefore OS, which was a secondary outcome of this RCT, was not eligible for inclusion in the review.
-
Response to treatment was the primary outcome of one RCT24 (at a 6-month time point) and a secondary outcome in two RCTs23,59 (both at a 12-month time point). At 6 months more participants in the MPT group achieved CR or achieved a PR or better, but a p-value for the comparison is not reported. At 12 months, two RCTs reported that a statistically significant greater proportion of participants had achieved CR or had achieved at least a PR. However, it was noteworthy that in one of these RCTs the numbers of participants contributing data to this outcome was low. Outcomes for CR from three RCTs were combined by meta-analysis, which confirmed that MPT was superior in comparison with MP in terms of the proportion of patients achieving CR.
-
Two RCTs23,59 reported a statistically significant advantage in the MPT group in comparison with the MP group for the outcome of PFS. The PFS data were combined by meta-analysis, which confirmed that MPT was superior in comparison with MP for this outcome.
-
Adverse events were reported in different ways so it was difficult to summarise the results across the RCTs. Because one RCT had included maintenance therapy with thalidomide, few AE data could be included, so the majority of the data come from just two RCTs. AEs with a statistically significant greater occurrence in the MPT arm that was reported by two RCTs included neutropenia and peripheral neuropathy. One RCT found that overall non-haematological toxic effects were statistically significantly more likely in the MPT group. For the outcomes of thrombosis or embolism, somnolence, constipation and infections, the results were inconsistent between RCTs (no significant difference in incidence reported by one RCT, but statistically significantly more in the MPT arm reported by the other RCT). This inconsistency may be a consequence of the different methods of reporting AEs. Some outcomes were only reported by one RCT, such as anaemia and thrombocytopenia (no statistically significant differences).
CTDa versus MP
-
This RCT49 was judged to be at low risk from allocation bias and bias due to unbalanced confounding factors. Analyses had been conducted by ITT principles and some information was provided on the methods used to handle missing data. It was not reported whether the amount and pattern of censoring was comparable between the groups.
-
Response was one of three co-primary outcomes of this RCT. (AiC/CiC information has been removed.) The remaining two co-primary outcomes, OS and PFS, and also the HRQoL outcomes, were not eligible for inclusion because participants were randomised to maintenance therapy with thalidomide after induction chemotherapy and this treatment did not meet the inclusion criteria of the systematic review.
-
Adverse events occurred in both RCT arms. (AiC/CiC information has been removed.)
-
Subgroup analyses were conducted although numerical data were not presented. (AiC/CiC information has been removed.)
Chapter 5 Economic analysis
Introduction
The aim of this section is to assess the cost-effectiveness of first-line treatments for people with MM, who are ineligible for HDT with SCT, compared with existing treatments. The economic evaluation comprises:
-
a systematic review of the cost-effectiveness of either bortezomib or thalidomide in combination with an alkylating agent and a corticosteroid (see Systematic review of existing cost-effectiveness evidence)
-
a systematic review of studies of the HRQoL of people with MM (see Systematic review of HRQoL studies)
-
a critical appraisal of the submissions from manufacturers received as part of the NICE appraisal process (see Review of the Janssen–Cilag submission to NICE (Bortezomib) and Review of the Celgene submission to NICE (Thalidomide), and
-
a de novo economic model and cost-effectiveness evaluation developed by SHTAC (see SHTAC independent economic assessment).
Systematic review of existing cost-effectiveness evidence
Methods for the systematic review of cost-effectiveness
A systematic literature search was undertaken to identify economic evaluations for first-line treatment with either bortezomib or thalidomide in combination with an alkylating agent and a corticosteroid in people with MM, who are ineligible for HDT with SCT, compared with existing treatments. The details of the search strategy and the methods for the systematic review of cost-effectiveness studies are outlined in Chapter 3 and Appendix 1.
Results of the systematic review of cost-effectiveness
Searches for economic evaluations identified the titles and abstracts of 183 potentially relevant studies. The full text of seven papers was retrieved for further consideration, with none of the studies meeting the a priori inclusion criteria. A summary of the selection process and the reasons for exclusion are presented in Figure 5 and a list of excluded studies is presented in Appendix 8. Two studies were excluded as they assessed a different intervention and/or population group from that specified in the research protocol. 70,71 Although five studies reported as abstracts appeared to meet the a priori inclusion criteria,72–76 they did not contain sufficient information on the methods used and the results to justify formal data extraction or critical appraisal. Given the apparent relevance of these five studies, a brief summary of the abstracts is presented below.
FIGURE 5.
Flow chart of identification of studies for inclusion in the review of cost-effectiveness. a, The five abstracts provided insufficient details of methods and results to allow inclusion in a formal systematic review. However, as the abstracts met other inclusion criteria they are discussed for information.

Deniz and colleagues72 estimated the lifetime health and cost consequences of MPT compared with MP in people in Scotland with previously untreated MM. They developed a Markov model for a cohort of patients receiving a course of MPT or MP, conceptualising the disease by four health states: preprogression without AE, preprogression with AE, progressive disease and death. Progression between health states as well as treatment duration, dose and AE risks were derived from a long-term RCT (see Chapter 4, Results of the systematic review of clinical effectiveness). 23 Patient cohorts received a maximum of 12 six-week cycles of treatment, until progression or treatment-limiting toxicity. The abstract indicates that health-state utilities associated with disease states and AEs were obtained from the literature, but no sources are provided. Thalidomide costs were from UK list prices and routine disease management costs reflected current practice in Scotland. Costs and health outcomes were discounted at 3.5% per annum. The model estimated improvements in health outcomes with MPT with a median TTP of 25 months versus 12 months with MP. Estimated median OS was 4.03 years with MPT versus 2.88 years with MP. These translated to a gain of 0.91 QALYs for MPT (3.24 QALYs) compared with MP (2.32 QALYs). There were increased costs with MPT of £25,199 per patient compared with £8935 per patient for MP, leading to an incremental cost-effectiveness ratio (ICER) of £17,847 per QALY and £14,803 per life-year gained. The authors state that sensitivity analyses showed that these results were consistent through changes in model parameters, although no information is presented. The authors conclude that MPT improves PFS and OS compared with MP and the results are cost-effective. A similar study comparing lifetime health and cost consequences of MPT compared with MP was completed for untreated MM patients in Wales. 74 While this evaluation used the same clinical outcomes for OS and PFS, it used slightly different QALY gains (0.9 QALYs) and lifetime costs specific to managing the disease in Wales (£16,937 per patient for MPT vs £1524 per patient for MP). The study produced a slightly more favourable ICER of £17,002 per QALY and £13,346 per life-year gained. It was reported that sensitivity analyses showed that findings were robust, with 95% of outcomes between £12,750 and £26,500 per QALY gained. Both studies were funded by the manufacturer of thalidomide.
De Abreu Lourenco and colleagues75 assessed whether MPT was cost-effective compared with MP for people in Australia who had been newly diagnosed with MM as part of an application to the Pharmaceutical Benefits Advisory Committee (PBAC). They extrapolate Kaplan–Meier survival curves from an unspecified Phase III study to a lifetime horizon to estimate the mean survival time. Costs included drugs, medical services and treatment for thalidomide-related AEs. These data were incorporated into a cost-effectiveness analysis adopting an Australian health-care system perspective, with costs and benefits discounted at 5% (AUS$2008). The modelled analysis estimated an incremental gain in average survival of 1.47 years and 1.14 QALYs with an associated average incremental cost of AUS$23,953. This results in an ICER of AUS$20,998. The authors concluded that the analysis had resulted in a positive recommendation from PBAC to fund thalidomide for the treatment of patients newly diagnosed with myeloma.
Yoong and colleagues73 estimated the cost-effectiveness of bortezomib in combination with MP (VMP) compared with MP and MPT in previously untreated people with MM in Canada who are unsuitable for SCT. Clinical outcomes originated from the VISTA study26 for VMP compared with MP and from an unspecified indirect comparison of VMP and MPT. The economic model projected OS over a 10-year horizon for VMP, MP and MPT using data from relevant studies and survival HRs. Resource use data included costs of drugs, outpatient cancer clinic, management of AEs, supportive care and subsequent lines of treatment, although sources were not specified. The discounted QALYs were 3.51 for VMP, 2.84 for MP and 3.29 for MPT. The total cost of treatment per patient was CAN$59,117 for VMP, CAN$27,026 for MP and CAN$52,226 for MPT. The ICER for VMP versus MP was CAN$48,294 per QALY gained, and it was CAN$31,975 per QALY gained for VMP versus MPT. The study states that sensitivity analyses showed that survival difference was the most influential factor. The authors concluded that the VMP regimen indicates good value for money, and it is being adopted by public cancer agencies in Canada.
Wang and colleagues76 also compared the cost-effectiveness of VMP, MPT and MP as first-line therapy for people with MM in the USA who were ineligible for autologous SCT. A lifetime (20 years) Markov model from the US payer’s perspective was developed with seven health states respresenting periods of treatment response (stable disease/MR, PR or CR), treatment-free interval, progressive disease, second-line treatment and death. Monthly transition probabilities were estimated from the VISTA trial data for VMP and MP,26 and from the IFM 99/06 trial for MPT. 23 Costs included drug and medical costs, treatment-related AEs, second-line treatment and resource utilisation during treatment-free intervals and progressive disease. All costs were adjusted to 2009 and presented in US dollars. State-specific utility estimates were derived from patient-level European Quality of Life-5 Dimensions (EQ-5D) data from the VISTA RCT. 26 Cost and health outcomes were discounted at 3%. The discounted QALY was 2.99 for VMP, 2.09 for MP and 2.95 for MPT. The total costs were US$110,870 for VMP, US$57,864 for MP and US$129,902 for MPT. The ICER of VMP versus MP was US$56,109 per QALY gained. VMP was dominant compared with MPT (greater benefit and lower cost). One-way sensitivity analyses were reported to show that the ICERs were robust, with the key drivers being the HR for VMP versus MP for the transition between second-line treatment and the HRs for MPT versus MP for treatment discontinuation. The authors concluded that VMP is cost-effective compared with MP in the USA.
Summary
The systematic review of cost-effectiveness showed that there were no fully published economic evaluations assessing the use of either bortezomib or thalidomide in combination with an alkylating agent and a corticosteroid as first-line treatment for people with MM who are ineligible for HDT. Five economic evaluations published as abstracts only were identified. 72–76 Of these evaluations, three compared MPT with MP72,74,75 and two compared VMP with MPT and MP. 73,76 All three studies showed additional benefits from MPT compared with MP at additional cost, with cost per QALY gained ranging from £17,002 to £17,84772,74 in the UK and being AUS$20,998 in Australia. 75 The two economic evaluations assessing VMP, MPT and MP showed that additional benefits were provided by VMP compared with MPT and by VMP and MPT compared with MP. The studies showed ICERs ranging from CAN$48,29473 to US$56,10976 per QALY gained for VMP compared with MP and CAN$31,975 per QALY gained73 and dominance76 for VMP compared with MPT. All of the studies had the involvement of the manufacturer of the interventions.
Systematic review of health-related quality-of-life studies
A systematic review was undertaken to assess the HRQoL of people suffering from and/or treated for MM. The aim was to provide data to populate the lifetime economic model with utilities to calculate QALYs. Although the methods used, and the process for their application, were similar to those described in Chapter 3 and appendix 1, there were some variations. The selection criteria used to assess the titles and abstracts of studies and the full papers of those retrieved were modified. Although the primary focus of the review was on people with previously untreated MM who were not candidates for HDT with SCT, it was thought that there would be limited HRQoL data available. As a consequence, the selection criteria were broadened. Studies were included if they assessed the HRQoL of people with previously untreated MM who were not candidates for HDT with SCT using either a generic preference-based utility measure (e.g. the EQ-5D) or the EORTC QLQ-C30 disease-specific measure. Although the EORTC QLQ-C30 is a disease-specific measure rather than a generic preference-based measure, it is commonly used to assess HRQoL in cancer and mapping studies are available to convert this measure to other HRQoL utility values (i.e. EQ-5D). In addition, studies were included if they assessed the HRQoL of people with MM irrespective of treatments received as long as a generic preference-based measure was used.
Generic preference-based methods generate a HRQoL score using a choice-based method, such as time trade-off or standard gamble, which values patients’ HRQoL on a scale between 0 (death) and 1 (perfect health). 77 These measures use a generic questionnaire that can be used for most health conditions or diseases. The EQ-5D is the preferred measure of HRQoL in adults by NICE78 and has been used and validated in many different patient populations. The EQ-5D consists of five dimensions of health: mobility, self-care, ability to undertake usual activities, pain and discomfort, and anxiety and depression. HRQoL utility values are generated for patients’ responses using an algorithm derived from a large UK population study.
The search strategy identified 208 papers that were potentially relevant. The titles and abstracts were screened with the full text of 18 papers retrieved for further inspection. After checking the retrieved papers, six studies met the inclusion criteria: five full papers and one abstract. A summary of the selection process and the reasons for exclusion are presented in Figure 6 and a list of excluded studies in Appendix 9. The 12 studies were excluded owing to the use of an inappropriate measure of QoL,79–87 assessment of different population groups,80,82 or insufficient details due to publication as an abstract only. 88–90 The six studies included in the systematic review are summarised in Table 24. No generic preference-based QoL studies were found for newly diagnosed and untreated patients who were ineligible for HDT. Three studies focused on newly diagnosed and untreated patients; however, they were assessed either on the EORTC QLQ-C30 non-generic preference-based measure91,92 and/or received treatment not included in the current evaluation. 92,93
| Gulbrandsen et al.91 | Mujica-Mota et al.94 | Slovacek et al.95 | Strasser-Weippl and Ludwig92 | Uyl-de Groot et al.96 | Van Agthoven et al.93 | |
|---|---|---|---|---|---|---|
| Publication year | 2004 | 2004 | 2008 | 2008 | 2005 | 2004 |
| Country | Denmark, Sweden and Norway | USA | Czech Republic | Austria | Netherlands | Belgium and the Netherlands |
| Study type | Two prospective studies using QoL questionnaire with comparison to reference population through regression | Utility mapping study. | QoL observational cohort study | QoL substudy within an RCT | Prospective, longitudinal cohort study | Cost–utility study based on an RCT |
| Study population | 424 patients with newly diagnosed MM | 202 patients with relapsed and refractory MM | 32 patients with MM | 92 patients with recently diagnosed and previously untreated MM (ECOG performance status ≤ 3) | 51 patients newly diagnosed with MM either untreated or undergoing first-line treatment | 261 patients with undiagnosed and untreated MM |
| Study population age | < 60 years for people treated with HDM with ABSCS, > 60 years for those treated with MP and 18–93 years for reference population | NR | Mean age 60 years (range 53–67 years) | Median age 66 years (range 43–84 years) | Mean (SD) 53 years (7.2) | Median age: intensive chemotherapy group 55 years (range 38–65); myeloablative therapy group 56 years (range 32–65) |
| Comparator population | Randomly selected Norwegian adults as a reference population (n = 3000) | No comparator | No comparator | Age- and gender-adjusted reference population (no details provided) | No comparator | UK general public (no details provided) |
| Intervention(s) | HDM with ABSCS (n = 221) and MP (n = 203) | Bortezomib (Velcade) | HDT (melphalan) followed by autologous transplantation of blood stem cells (PBPCT) | Continuous or intermittent prednisolone plus vincristine, melphalan, cyclophosphamide, prednisolone, interferon-α-2b (VMCP-IFNα-2b) for induction therapy | Vincristine, adriamycin and dexamethasone/vincristine, adriamycin and methyl prednisone (VAD/VAMP) chemotherapy, followed by HDM and then PSCT | Intensive chemotherapy compared with intensive chemotherapy followed by myeloablative chemotherapy with autologous stem cell rescue |
| Included QoL instrument used | EORTC QLQ-C30 | Elements of the EORTC QLQ-C30 and MY24, FACT-Fatigue and FACT/GOG-Ntx were mapped to the EQ-5D | EQ-5D and EQ-5D VAS | EORTC QLQ-C30 | EORTC QLQ-C30 and EQ-5D | EQ-5D |
| Time period when HRQoL instruments administered | QoL was assessed at baseline, 1, 6, 12, 24 and 36 months | NR | NR | Baseline only | Baseline (2 weeks’ post induction therapy), day of hospital discharge after HDM (T2), 1 month post discharge after HDM (T3), day of hospital admission for PSCT (T4), day of discharge following PSCT (T5), 6 months (T6) and 12 months (T7) post discharge for PSCT | 6, 12, 18 and 24 months |
| Methodology of collecting QoL data | The 30-item questionnaire was administered within the two studies and by postal questionnaire to the reference population | NR | Postal questionnaire with voluntary and anonymous response | Patients in the RCT were invited to take part and provided with the questionnaires at their first study visit of the trial | Questionnaires were either handed to patients in hospital wards or mailed to their homes. Reminders were sent where not returned within a month | NR |
| Results | At diagnosis MM patients had significantly impaired QoL on all scores compared with the reference population, except for diarrhoea. Pain and fatigue, reduced physical functioning, limitations in role functioning and reduced overall QoL were the most distressing problems. After start of treatment, small to moderate improvements in mean QoL scores were observed in most domains | Utility scores appeared similar across patient groups as defined by serological response to bortezomib, with an overall utility score of 0.65 |
For people treated with HDT and PBPCT the global QoL was 0.689 on EQ-5D and 0.666 on EQ-5D VAS By age group the EQ-5D was 0.815 for people aged 40–49 years, 0.742 for those 50–59 years, 0.642 for those 60–69 years and 0.615 for those 70–79 years |
Study showed low levels of functional QoL scores and increased symptom scores in patients with active disease at start of first-line therapy. It was felt that measures such as pain, fatigue, physical functioning were important Patients have significant impairment of physical and psychosocial dimensions at baseline compared with the health reference population |
Mean absolute scores (SD) on EQ-5D at baseline after VAD/VAMP and mean change scores from baseline: baseline 0.52 (0.33); T2, 0.03; T3, 0.14; T4, 0.14; T5, –0.14; T6, 0.12; T7, 0.17 Mean absolute scores (SD) on EQ-5D at baseline and 12-month follow-up for patients who proceeded to PSCT: baseline – patients who proceeded to 12-month follow-up 0.60 (0.33); 12-month follow-up patients with baseline 0.77 (0.13); 12 month follow-up all patients 0.79 (0.18) |
Utility values on EQ-5D:
|
FIGURE 6.
Flow chart of identification of studies for inclusion in the review of QoL studies.
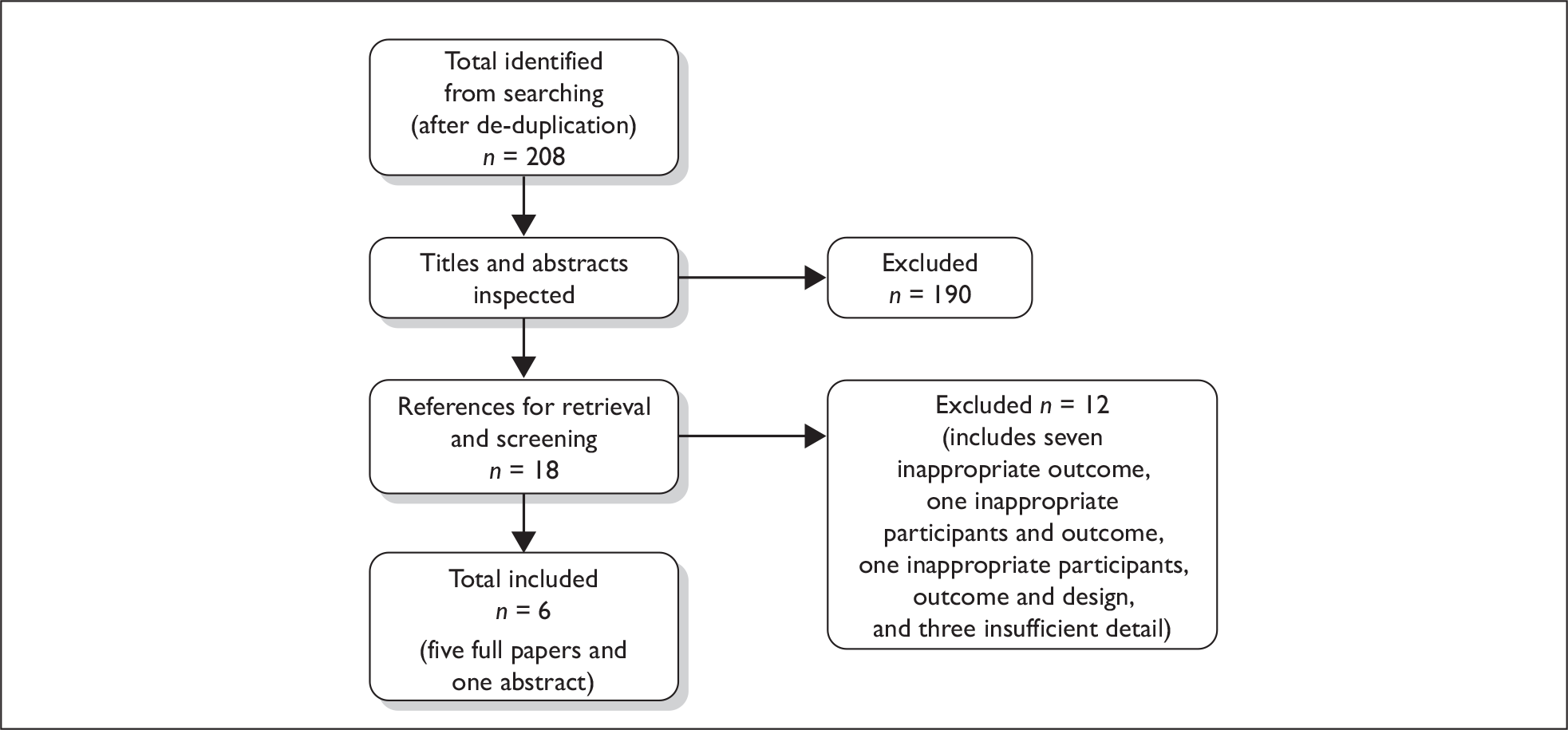
Generic preference-based measures of HRQoL (i.e. EQ-5D) were assessed in four studies. 93–96 These four studies evaluated the EQ-5D among people with MM who were receiving either second-line or subsequent treatment,94,96 where treatment status was unclear95 or who had received or were receiving treatment not included in this evaluation. 93,95,96 Two studies reported HRQoL for patients receiving interventions included in this evaluation,91,94 using the EORTC QLQ-C30 to assess patients newly diagnosed with MM receiving MP91 and patients with relapsed and refractory MM receiving bortezomib. 94 The remainder of this section examines the six studies in more detail, providing an indication of the HRQoL of people with MM at different stages during their treatment.
Uyl-de Groot and colleagues96 investigated the HRQoL of patients with newly diagnosed MM who were treated in a tandem transplantation programme. All patients were scheduled for intensive treatment with vincristine, adriamycin and dexamethasone/vincristine, adriamycin and methyl prednisone (VAD/VAMP) chemotherapy followed by high-dose melphalan (HDM) and transplantation of whole blood stem cells and, finally, re-infusion of the previously collected peripheral stem cells. The EQ-5D questionnaire was completed, at several time points, by 51 patients with a mean age of 53 years. Table 25 shows the EQ-5D utility estimates at different time points. The utility estimates vary between 0.38 and 0.69, with the lower utility estimates during treatment periods or immediately after discharge of treatment. The longer term QoL estimates after discharge of treatment range from 0.64 to 0.69.
| QoL measure | Baseline | Discharged HDT | 1 montha | Admitted PBSCT | Discharged PBSCT | 6 monthsb | 12 monthsb |
|---|---|---|---|---|---|---|---|
| EQ-5D value | 0.52 | 0.55 | 0.66 | 0.66 | 0.38 | 0.64 | 0.69 |
Slovacek and colleagues95 analysed the effect of selected demographics, and psychosocial and health aspects on HRQoL in MM survivors treated with HDT (melphalan) followed by autologous peripheral blood progenitor cell transplantation (PBPCT). Thirty-two patients of a mean age of 60 years completed the EQ-5D questionnaire. The EQ-5D estimate was 0.689.
Mujica-Mota and colleagues94 mapped HRQoL measurements from EORTC QLQ-C30 estimates to the EQ-5D utility measure for patients with relapsed and refractory MM from the SUMMIT-1 trial. Few details are given in this abstract. The authors stated that the utility scores appear similar across patient groups as defined by serological response to bortezomib, with an overall utility score of 0.65.
Van Agthoven and colleagues93 estimated the cost–utility of intensive chemotherapy versus intensive chemotherapy followed by myeloablative chemotherapy with autologous stem cell rescue in newly diagnosed and untreated patients with MM. There were 129 patients in the intensive chemotherapy arm and 132 in the myeloablative arm and all were less than 65 years old. Little detail was given on the methodology or results. The authors state that patients in an undefined state following intentionally curative primary therapy would have HRQoL 19.5% lower than those in the general population, i.e. 0.644.
Strasser-Weippl and colleagues92 evaluated baseline HRQoL in elderly patients recently diagnosed with MM who were previously untreated. Ninety-two patients (of median age 66 years) participated in the HRQoL substudy of an RCT of continuous or intermittent prednisolone plus vincristine, melphalan, cyclophosphamide, prednisolone, interferon-α-2b (VMCP-IFN-α-2b) for induction therapy. They used the EORTC QLQ-C30 questionnaire for these patients and compared them with a reference population for the general population of same age and gender (see Appendix 10 for observed scores). The study found a significant impairment of physical and psychosocial dimensions of QoL in patients with MM at baseline compared with a healthy reference population. Low psychosocial QoL at baseline was associated with poor prognosis.
Gullbrandsen and colleagues91 compared HRQoL scores of MM patients at diagnosis and over time with the scores of a reference population. Patients from two prospective Nordic Myeloma Study Group trials for HDM with autologous blood stem cell support (ABSCS) and MP completed the EORTC QLQ-C30 questionnaire. There were 221 patients for HDM who were < 60 years old and 203 patients for MP who were > 60 years old. The reference population consisted of 3000 randomly selected adults from the Norwegian population (see Appendix 10). At diagnosis, the most distressing problems were pain and fatigue, reduced physical functioning, limitations in role functioning and reduced overall HRQoL. These differences from the reference population were statistically significant, and large or moderate according to the rating systems. After the start of treatment, small to moderate improvements in mean QoL scores were observed for most domains.
Summary and conclusions of the health-related quality-of-life review
The systematic review did not find any generic preference-based HRQoL studies that were directly related to the population of interest. The utility estimates from HRQoL studies in patients with MM who had intensive therapy vary between 0.38 and 0.69, with the lower utility estimates during treatment periods or immediately after discharge from treatment. 96 The longer-term HRQoL estimates after discharge from treatment range from 0.64 to 0.69. This may indicate that HRQoL is lower during the treatment period and improves after treatment has finished. Furthermore, long term HRQoL may be stable over time. It is unclear whether patients with CR following treatment have a higher HRQoL than those with other responses.
Review of the Janssen–Cilag submission to NICE (bortezomib)
A structured data extraction form was used to guide the review of the Janssen–Cilag submission to NICE (see Appendix 11). The MS reports the total costs, the QALYs gained and the cost-effectiveness associated with the interventions under consideration in the appraisal. The model evaluates lifetime costs and benefits for bortezomib in combination with MP (VMP), for previously untreated MM patients not eligible for HDT–SCT, compared with MPT, CTDa and MP. The perspective of the analysis is clearly stated as being that of the NHS and PSS, capturing direct costs and benefits only.
Modelling approach
A decision-analytic cost–utility model, developed in Microsoft Excel, was used in this submission. The model uses a cohort of newly diagnosed myeloma patients treated with MP as the baseline treatment. Treatment effects for VMP, MPT and CTDa are then modelled over time by adjusting the baseline patient experience via HRs. A survival model appears to be used, which estimates OS and PFS curves for each of the comparators. The model also includes further lines of treatment (second- and third-line) to estimate the total treatment costs.
The analytic framework was based on a variant of Quality-Adjusted Analysis of Time Without Symptoms or Toxicity (Q-TWiST97) using partitioned survival analysis, and utilises the area under, and the difference between, time-to-event curves to estimate mean durations spent within the disease states of interest.
Survival is partitioned into three different states: (1) prior to response to treatment; (2) response but no progression; and (3) post progression. Death represents the final state. The time to response or death was estimated from life tables constructed directly from the VISTA trial patient-level data. 26 PFS for MP was estimated from a meta-analysis of the MP arms of included RCTs to compute MP PFS values at 6, 12, 18 and 24 months. PFS was extrapolated beyond 24 months, assuming an exponential survival distribution, using the hazard rate for all time periods beyond 24 months equal to the hazard rate calculated between months 18 and 24. OS for MP was estimated in a similar way to PFS, but using 48 months of summary survival data from the MP arms of the included RCTs.
For the comparator treatments, relative HRs were taken from the random effects results of the meta-analysis that used OS and PFS summary data. OS and PFS hazard rates were computed for each time period by multiplying the VMP–MP, MPT–MP and CTDa–MP HRs by the appropriate hazard rate for that time period. The computed hazard rates were then used to generate the VMP, MPT and CTDa OS and PFS life tables that extend out to the end of the 30-year lifetime horizon of the model.
The HRs were estimated using a piece-wise constant hazard model using derived survival data from the Kaplan–Meier curves for each of the included RCTs. HRs were estimated at 48 months for OS for each of the RCTs, except the VISTA Trial, which had only 36 months’ follow-up. For estimation of the OS hazard for thalidomide, data from five RCTs were used, which included RCTs that had included thalidomide maintenance. Data were synthesised using Bayesian meta-analysis with fixed and/or random effects models. Results from the random effects model were used in the cost-effectiveness model. (AiC/CiC information has been removed.)
Following the first-line therapy, and upon disease progression, it was assumed that the second-line treatment would consist of bortezomib plus high-dose dexamethasone (HDD), CTDa or HDD. Most patients received CTDa after first-line VMP and bortezomib and HDD for all other first-line therapies. All patients received lenalidomide plus dexamethasone as third-line treatment.
Adverse events were included in the analysis by estimating the incidence of AEs (grades 3 and 4) across the RCTs for each of the comparators and combining this with the unit costs of treating the AEs. Unit costs were mostly based upon those used in a previous NICE report for lenalidomide (TA171). 31 The most common AEs for MPT were non-haematological toxicity, neutropenia and deep venous thrombosis; for MP they were neutropenia, anaemia and thrombocytopenia; and for VMP they were neutropenia, oedema, leucopenia and thrombocytopenia.
Assumptions
The manufacturer’s model makes the following assumptions:
-
Dose of thalidomide of 150 mg per day for MPT and 167 mg per day for CTDa.
-
Adverse events are included in the model as the cost of treating them; the incidence of AEs does not influence the treatment duration, efficacy or patient utility.
-
Costs included for second- and third-line treatments. Most patients who received VMP as first-line treatment receive CTDa as second-line treatment and most who did not receive VMP as first-line treatment do receive it as second-line treatment.
-
Thalidomide RCTs that included maintenance therapy with thalidomide were included in the meta-analysis.
Appraisal of the manufacturer’s cost-effectiveness analysis
The Janssen–Cilag MS was appraised for methodological quality and generalisability to the UK NHS using a checklist adapted from the NICE reference case requirements78 and the Philips and colleagues checklist (see Appendix 12). 98 The submission meets all of the requirements for methodological quality and generalisability, except that it did not provide any evidence that the economic model had been validated.
The evaluation provided a clear statement of the decision problem to be addressed, including the population, which appeared to follow the scope for the appraisal issued by NICE. The comparators included (VMP, CTDa, MP and MPT) were appropriate as these are being routinely used or considered for use within the NHS in England and Wales. The perspective for the model was the NHS and PSS. A survival modelling methodology was used which seemed appropriate given the clinical nature of MM. The lifetime horizon used in the model reflects NICE guidance. The model structure was clearly presented with a description and justification of the key assumptions and data inputs used. Measures of clinical effectiveness are from a systematic review of RCTs with an MTC. Benefits for the model are measured in QALYs using the EQ-5D for measuring utility. All benefits and costs are discounted at 3.5% as outlined in NICE guidance. 78 Data on PPS were extrapolated from observed data using an exponential distribution. Uncertainty was assessed through a one-way deterministic sensitivity analysis and probabilistic sensitivity analysis (PSA). It was unclear if the model had been fully validated, as no details were provided.
Estimation of quality-adjusted life-years
Health-related quality of life utility values are assigned to each of the states: prior to response to treatment, response to treatment without progression, and post progression, based on a study evaluating chemotherapy followed by SCT in people with MM. 93 For the response state, a utility value of 0.81 was used, based on the utility of the general public at an age (median 54 years) corresponding to that of the patients in the study. A utility value of 0.64 was applied to the post-progression disease state. A utility value of 0.77 was applied to patients prior to the response to treatment. The submission considered this the most appropriate source of utility values because it is the only study that reports utility values according to response and progression status and, secondly, utility values were derived using the EQ-5D rather than the less methodologically robust indirect mapping approaches used in the other studies. However, as shown in the systematic review of HRQoL, there are several other more relevant HRQoL studies. In particular, it is unlikely that patients with MM would have the same HRQoL as the general population.
Estimation of costs
Treatment unit costs and doses were based on the British National Formulary (BNF) No. 5735 and MIMS 2009. 99 The duration of treatment was based upon the mean treatment duration in the trials and was assumed to incorporate discontinuation of treatment due to progression, death and AEs. The duration of treatment with MP was seven cycles as per the VISTA trial. 26 For bortezomib, (AiC/CiC information has been removed) vials were used per patient (VISTA trial26); the reason why the number of vials used is far fewer than the full treatment course of 52 vials is not given. The submission used an average dose of 150 mg per day for thalidomide obtained from the five MPT RCTs included in the meta-analysis of the MS. Within the CTDa combination, a daily dose of 167 mg was used for thalidomide. This is the weighted average as per protocol escalating dose from the MMIX RCT prior to the maintenance phase. A mean duration of treatment with thalidomide of 315 days was used, based on the duration reported in the MPT RCTs.
The resource use cost for the management of first-line MM was assumed to be the same for patients receiving VMP, MPT, CTDa and MP. There was an outpatient cost of £102 per visit and a total of nine outpatient visits. In addition, patients receiving VMP had this outpatient cost each time they were administered bortezomib.
Cost-effectiveness results
Table 26 shows the base-case results from the submission. The ICER for VMP versus MP is estimated to be £10,498. Furthermore the ICERs of VMP versus MPT and VMP versus CTDa are estimated to be £11,907 and £10,411, respectively. The submission states that the incremental analysis shows extended dominance of MTP over CTDa. However, the assessment group has found an error in the calculation of third-line costs for CTDa (correct cost £24,978 instead of £16,652). Correction of this error resulted in an ICER of £51,552 per QALY gained for CTDa versus MP.
| Treatment | Mean QALYs | Mean cost (£) | ICER vs MP (cost/QALY, £) | ICER (cost/QALY, £) vs next best option with lower cost |
|---|---|---|---|---|
| MP | 2.86 | 54,434 | – | – |
| CTDa | 3.07 | 56,668 | 10,905 | 10,905 |
| MPT | 3.41 | 59,322 | 8912 | 7724 |
| VMP | 4.03 | 66,676 | 10,498 | 11,907 |
One-way sensitivity analyses were undertaken for a limited number of parameters, including different survival distributions for OS and PFS, alternative HRs for OS, dose and duration of thalidomide, utilities, time horizon and discounting rate. The results are generally robust to changes in the sensitivity analyses. The model is most sensitive to the following parameters: underlying MP survival hazard, HRs for OS, dose of thalidomide, and duration of treatment with thalidomide in the MPT arm.
A PSA was undertaken using Monte Carlo simulation with 10,000 iterations. All parameters in the model were included except medication costs. For the PSA, at the £20,000 and £30,000 willingness to pay thresholds, VMP has the highest probability of being cost-effective: 64% and 75%, respectively.
Two scenario analyses were conducted. Scenario A did not include the costs of subsequent therapy after first-line treatment. In this scenario, the cost-effectiveness results were less favourable for each of the treatments and the ICERs increase to £48,437, £16,956 and £21,099 per QALY gained for CTDa, MPT and VMP compared with MP, respectively. Scenario B assumed the same second-line therapies as those treated with MP in the VISTA26 RCT. The results were similar for this scenario to the base-case analyses.
Summary of general concerns
-
Hazard ratio used for OS for thalidomide was derived from a meta-analysis that included RCTs with thalidomide maintenance.
-
The utility estimates were from a study with the wrong population, i.e. younger patients who received high-dose therapy. Furthermore, patients who had responded to treatment were assumed to have the same utility as the general population.
-
There was an error in the calculation of third-line costs for CTDa.
-
There was no evidence provided of model validation.
Review of the Celgene submission to NICE (thalidomide)
Overview
A structured data extraction form was used to guide the review of the Celgene submission to NICE (see Appendix 11). The submission states that its objective is to provide an evaluation comparing the costs and benefits of MPT with those of VMP and MP in patients with MM who are older than 65 years or who are ineligible to receive HDT. The evaluation has two stages. First, a short unsystematic review examines the literature for any relevant cost-effectiveness models in general and specifically in previously untreated MM patients who are not eligible for HDT. The review of cost-effectiveness studies indicates that a literature search was undertaken, although no details of the search strategy or methods for the review are provided. Searches identified five publications, with only one having relevance to the scope of the appraisal. The study by Deniz and colleagues72 compared MPT with MP as first-line treatment for MM in Scotland and provided the basis for the model developed for the submission by Celgene. Second, an economic model has been developed using data on the clinical effectiveness of MPT23,59 and VMP26 through a Bayesian MTC. The perspective of the economic evaluation is stated as being that of the NHS and PSS, including direct costs and benefits only. The analysis takes a lifetime horizon (30 years), presenting costs and outcomes (i.e. years of life gained and QALYs gained) for the three treatment arms of MPT, MP and VMP and an incremental analysis of costs and outcomes for MP and VMP when compared with MPT.
Modelling approach
A Markov model was developed to compare the difference in the progression of MM and in the costs of treatment when managed with the three different treatment options of MPT, VMP or MP through a series of different health states. It was developed from a model produced by Deniz and colleagues,72 which compared MPT and MP as first-line treatment for MM in Scotland.
The model has four different health states that are defined by the stage of disease progression or the occurrence of AEs. The four states are preprogression without AEs, preprogression with AEs, post progression and an absorbing state of death. All patients start in the preprogression without AEs state and move to other states if their condition worsens or they incur an AE. As MM is a progressive condition, people can only move to a worse state or remain in the same state. The submission provides limited discussion of the rationale for the approach or of the basis for the transition probabilities used to determine progression between states and other approaches assessing the phases of treatment may reflect variations in HRQoL more closely.
The model has a cycle length of 6 weeks (42 days) with a maximum of 12 cycles for MPT and MP and nine cycles for VMP. The cycle length and the number of cycles correspond to those used in clinical RCTs. 23,26,59 The time horizon used in the model equates to a lifetime horizon, although characteristics of the cohort used in the model are not clearly stated. The consequences of a shorter time horizon of 5 years were examined in the sensitivity analysis.
Treatment effects were calculated from a random effects Bayesian MTC of data originating from three RCTs. 23,26,59 The MTC was undertaken despite differences in the dosage used in the RCTs comparing MP with MPT. It used measures of survival time before and after progression as the primary outcomes. TTP and PFS were used and assumed to be equivalent. The outcome from the MTC was a measure of the risk of progression, provided through the percentage of patients experiencing PFS at 6-month intervals up to 30 months, with extrapolation beyond this point using an exponential distribution. It was assumed that post-progression survival (PPS) would be the same irrespective of preprogression treatment, with the different arms assumed to receive the same alternative treatment after progression (i.e. second- and third-line treatments). PPS was calculated by combining the MPT, MP and MEL100 (VAD, cyclophosphamide and melphalan 100 mg/m2) arms from the IFM 99/06 trial to create an average survival curve. 23 Average survival at different time points was then extrapolated with an exponential distribution. Treatment interruptions or discontinuations were encompassed in the trial efficacy data for MP and MPT, with no alteration to costs in the base case. Changes in cost were encompassed in sensitivity analyses through a reduction in dose as they are likely to reflect clinical practice. No data were available for VMP on discontinuation.
Adverse events were included for people on active treatment only if they were treatment related and considered to be clinically significant (i.e. grade 3 or above or occurred in 2% or more of patients in either arm). Those associated with disease progression were not incorporated into the model. The treatment-related AEs were included in the model through an estimate of the risk of AEs per cycle based on trial data. 23,26 The effects of AEs on HRQoL were also included in the model. A literature search revealed no HRQoL data specific to MM, and so HRQoL decrements were obtained for different patient populations. Costs of AEs were also included.
Assumptions
The manufacturer’s model makes the following additional assumptions:
-
Post-progression survival is modelled to be the same across different treatment strategies.
-
Patients assumed to discontinue first-line treatment upon disease progression.
-
Deaths can only occur at or after progression and are assumed to be due to disease-related deterioration.
-
Adverse events are included in the model as a utility decrement at the time of the event and the cost of treating them. They are assumed to not affect the disease progression rate or OS, or treatment duration, efficacy or dose.
-
Assumes venous thromboembolism (VTE) antithrombotic prophylaxis for 5 months for patients receiving MPT with no resultant risk in incidence of VTEs, and antiviral prophylaxis for VMP.
Appraisal of the manufacturer’s cost-effectiveness analysis
The Celgene MS was appraised for methodological quality and generalisability to the UK NHS using a checklist adapted from the NICE reference case requirements78 and the Philips and colleagues checklist (see Appendix 12). 98 Although the economic evaluation lacked detail on some criteria, it adhered to the scope of the appraisal and followed the many aspects of the NICE reference case.
The evaluation provided a clear statement of the decision problem to be addressed, which appeared to follow the scope for the appraisal issued by NICE. Despite stating that the model focused on first-line therapy for people with MM who are ineligible for HDT and/or are aged over 65 years, insufficient details were provided of the population cohort used in the model itself. The comparisons of MP, MPT and VMP were appropriate as these are being routinely used or considered for use within the NHS in England and Wales. The setting for the evaluation was England and Wales and the perspective for the model was that of the NHS and PSS. A Markov modelling methodology was used which was developed from a previous evaluation. 72 The methodology seemed appropriate given the progressive nature of MM through distinct stages. The lifetime horizon (30 years) used in the model reflects NICE guidance.
Although the model structure was presented, limited details are given linking the model structure to the baseline risk of the condition. The submission outlines and justifies the assumptions used in the model and the different benefit, resource and cost inputs and their sources. Measures of clinical effectiveness are from a systematic review of RCTs with an MTC. Benefits for the model are measured in QALYs using the EQ-5D for measuring utility. All benefits and costs are discounted at 3.5%, as outlined in NICE guidance. 78 Data on PPS were extrapolated from observed data using an exponential distribution. While uncertainty has been assessed through a one-way deterministic sensitivity analysis, no probabilistic analysis or model validation processes were undertaken. As a consequence, the analysis provides only a partial assessment of the uncertainty in the model with the possibility of correlation between parameters and difficulty in summarising the implications of uncertainty.
Estimation of quality-adjusted life-years
No systematic review was undertaken to identify HRQoL values associated with the benefits of the treatment, but a literature search was conducted to identify utility decrements for AEs. The HOVON 24 study,93 an RCT of intensive chemotherapy followed by myeloblative therapy with autologous stem cell rescue compared with intensive chemotherapy, provided HRQoL data using the EQ-5D to assess the benefits of treatment for people with MM. Although not directly relevant in terms of the population and treatments included in the scope for the technology appraisal, it does provide an indication of the possible utilities for managing people with MM when specific assumptions are applied. The utility values used were 0.64 for people not responding to treatment and 0.81 for people who did respond (using general public utility for same age group). A utility value of 0.77 at 24 months was used for those who continue to respond to treatment with intensive chemotherapy and had not progressed. An assumption was made that preprogression patients and post-progression patients matched responders and non-responders in the HOVON trial. 93 However, other more relevant HRQoL studies (see Systematic review of health-related quality-of-life studies, above) show that the utility values used in the MS are higher than would be experienced by people with MM, whether newly diagnosed (0.52), undergoing treatment (0.38–0.55) or after treatment at 6 months (0.64) and 12 months (0.69).
The literature search for utility decrements for AEs did not identify specific values for people with MM and so utility values from different population groups were used (e.g. breast, colon and rectal cancer). Average per cent reduction in utility by each AE was calculated from these values and applied to the cohort in the model.
Estimation of costs
Resources and costs were obtained from several sources. NHS resources were from an unpublished survey of UK haematologists by Celgene Ltd. Inpatient, outpatient and day-case hospitalisation costs were derived from NHS Reference Costs,100 including inpatient and day-care costs for disease-related complications and treatment-related AEs, outpatient consultations and disease monitoring tests and treatment care costs in primary care. Costs of medicines were from the BNF (No. 57)35 and costs of blood transfusions from Wilson and colleagues101 with costs inflated to 2008. 102 When on active treatment, patients receive the mean observed treatment dose from the trials. Other resource use and cost data were provided for outpatient consultations, disease monitoring and treatment of AEs/complications. No indirect costs were included in the model. The costs of AEs were calculated by combining resource use data from the survey of haematologists with unit costs to estimate total costs. These costs and trial data on the frequency of AEs23 were then used to calculate a weighted average cycle cost. The methods for deriving resources and costs used and the sources were clearly described.
Cost-effectiveness results
The submission reports the benefits [i.e. TTP, patients progressed, deaths, proportion of patients with AEs, median OS, mean survival in years of life (life-years) and total QALYs] and the total costs (i.e. medication, monitoring and management of AEs) separately for each treatment pathway in the model.
Comparison of the benefits used for the model showed considerable benefit for those receiving MPT or VMP over MP on median TTP, median OS, total life-years and total QALYs. In contrast, more people receiving MPT (43.2%) or VMP (40.9%) suffered AEs compared with those receiving MP (13.4%). The total costs of the different treatment strategies used within the model showed considerable variation between MP (£1365) and VMP (£42,616). The cost of the medications was the main reason for these differences.
The base-case analyses (Table 27) produced two comparisons, MPT versus MP and VMP versus MPT, with differing outcomes. When compared with MP, MPT had an ICER of £18,188 per life-year gained and £23,381 per QALY gained. In contrast, the comparison of VMP with MPT showed that VMP produced a small benefit in additional life-years and QALYs at a large additional cost (£21,483). The resultant ICERs were £200,237 per life-year gained and £303,845 per QALY gained.
| Treatment | Mean QALYs | Mean cost (£) | ICER vs MP (cost/QALY, £) | ICER (cost/QALY, £) vs next best option with lower cost |
|---|---|---|---|---|
| MP | 2.43 | 1365 | – | – |
| MPT | 3.28 | 21,133 | 23,381 | 23,381 |
| VMP | 3.35 | 42,616 | 45,024a | 303,845 |
The submission assessed uncertainty through one-way deterministic sensitivity analyses. No PSA was conducted as the manufacturer stated that the efficacy of MPT and VMP were essentially the same and that the cost differences would be the key driver for the model. The submission included a number of one-way sensitivity analyses for parameter values and model structure. The parameters with the greatest effect on the model results were for the changes in treatment efficacy with a range of £16,586–33,275 per QALY gained for MPT versus MP and a range of £148,873–1,000,435 per QALY gained for VMP versus MPT.
The submission concludes that MPT represents a cost-effective use of NHS resources compared with MP as a first-line therapy for people with MM who are not eligible for HDT and/or are aged over 65 years. In contrast, when comparing MPT and VMP the manufacturer stated there was negligible clinical benefit from VMP at an additional cost that resulted in the ICERs exceeding £300,000 per QALY. When these findings were assessed through sensitivity analysis, the ICERs were reasonably robust.
Summary of general concerns
-
The economic evaluation focuses on the effectiveness of first-line treatment for people with MM who were ineligible for HDT, reflecting the scope for the NICE technology appraisal. Exclusion of second- or third-line treatment options may oversimplify the evaluation with consequences for the incremental benefits and costs that would result from different possible options available. Given that first-line treatment may, in part, determine subsequent treatment options, it would be helpful to include these in the evaluation.
-
All deaths are assumed to be caused by disease-related deterioration and occur only at or after progression. In practice, deaths may and do occur prior to progression and, as such, the evaluation may overestimate the benefits that are accrued.
-
Post-progression survival was the same irrespective of preprogression treatment, which would affect the incremental benefits.
-
No HRQoL studies relevant to the evaluation were identified by the manufacturer and utility values from comparisons of different MM populations using alternative management strategies were used.
Comparison of manufacturers’ results
The manufacturers’ economic models had similar structures but used different methodology: one used a survival model and the other a Markov model. Both models compared first-line treatment with VMP, MPT and MP. Janssen–Cilag also included CTDa as a comparator. The ICERs produced by the Janssen–Cilag and Celgene submissions vary considerably from £11,907 to £303,845 per QALY gained for VMP versus MPT. These differences stem from the number of vials used for treatment with bortezomib, the HRs for thalidomide and the inclusion of second- and third-line treatments.
SHTAC independent economic assessment
Overview
We developed a new model to estimate the costs, benefits and cost-effectiveness of MPT, VMP and CTDa compared with MP, in newly diagnosed patients with MM ineligible for HDT–SCT. CTDa was included in order to compare all relevant comparators; however, there are limitations to the effectiveness data, as the effectiveness estimate for OS was not statistically significant and the MMIX RCT included a second randomisation to thalidomide maintenance for some patients. The model was populated with clinical effectiveness data from the included RCTs in our systematic review of effectiveness (Chapter 4), HRQoL data from a systematic review of HRQoL studies (see Systematic review of health-related quality-of-life studies) and cost data derived from published studies (where available) and from national and local NHS unit costs.
The economic evaluation was from the perspective of the NHS and PSS, as only these direct costs were included. The model estimates the lifelong costs and benefits from each of the treatments. The costs and benefits were discounted at 3.5%, as recommended by NICE. 78 The base-price year for the costs was 2009. The intervention effect in terms of improvement in OS and PFS was derived from the systematic review of effectiveness reported in Chapter 4 (see Overall survival and Progression-free survival). The outcome of the economic evaluation is reported as cost per QALY gained.
Description of the SHTAC model
A survival model was used to compare the cost-effectiveness estimates of VMP, CTDa and MPT versus MP. The model uses a survival analysis approach to estimate the mean OS and PFS for each of the interventions for a cohort of patients with newly diagnosed MM. The model consisted of cycles of 6 weeks in length to be consistent with the cycle lengths used for chemotherapy treatment. A lifetime horizon of 30 years was modelled to capture all clinical events using partitioned survival analysis for OS and PFS. Two survival curves were constructed for OS and PFS (Figure 7b), based on the derived probability of death and progression in each model cycle, respectively. The mean time spent in each state was calculated from the survival curves for OS and PFS (see Figure 7a).
FIGURE 7.
The survival model adopted for the cost-effectiveness model. Treatment and post-treatment health states refer to first-line treatment.

Survival was classified into three health states, and the mean time spent in each state is as follows:
-
Treatment (Ttreat) is the mean duration of first-line treatment.
-
Post treatment (TPost_treat) is the mean time from stopping first-line treatment until progression, i.e. TPFS – Ttreat
-
Post progression (Tprog) is the time from disease progression until death, i.e. TOS – TPFS, where TOS is mean OS and TPFS is mean PFS.
Each health state was associated with a HRQoL utility estimate that was multiplied by the length of time spent in that state. The total QALYs over the lifetime of a patient were calculated by aggregating the estimated QALYs from each health state.
Due to lack of data on subsequent therapies, it was unclear how the subsequent therapies affected HRQoL and survival and therefore second-line therapy is only included in the model as a cost.
The methodology used for deriving the parameters for the survival curves for the alternative treatments is as follows:
-
Construct the baseline survival curves for MP using the adjusted event probability for each time interval.
-
Construct the survival curves for other treatments by using the event probability for each time interval; i.e. event probability for MP multiplied by HR for treatment option.
For the baseline MP treatment, OS and PFS at regular time points were derived for each of the included studies from our meta-analysis of the clinical RCTs. The data from the RCTs were combined to form baseline MP OS and PFS curves. These curves provided the probability of an event (death or disease progression), i.e. hazard rate, for MP in each time interval (see Baseline MP curves).
The treatment effects for the other interventions compared with MP (HRs) were taken from our systematic review of clinical effectiveness (see Assessment of effectiveness). As the HR of the treatments versus MP varied over time, a constant HR was not appropriate. We estimated the HR for each 6-monthly period for each of the treatments versus MP.
The hazard rate for death was derived for each of the treatments by multiplying the baseline MP probability of death by the HRs for each time interval. The hazard rate for disease progression was derived in a similar manner. This method provided a closer fit to the trial data than approximations, such as fitting distributions. Parameters used in the model and the data sources used to derive them are described in more detail below (see SHTAC data sources). The methodology used for deriving the survival curves is described in more detail in Appendix 13.
The costs in the model comprise drug treatment, consultation, monitoring costs, and costs for treating AEs. Patients remained on drug treatment unless their disease progressed or they died. All patients who had not died received second-line therapy and this was assumed to start at the mean time of disease progression for the cohort. Third-line therapy was not included as it was assumed that most patients would receive lenalidomide, irrespective of the initial treatment. Costs used in the model are described in more detail below (see Estimation of costs).
A list of the model assumptions is given below. Assumptions are applied to all treatment options unless explicitly stated otherwise. All assumptions were tested in sensitivity analyses.
The model includes the following assumptions:
-
For bortezomib, each patient receives one vial per administration.
-
Costs included for second-line treatments. Most patients who received VMP as first-line treatment receive CTDa as second-line treatment and most who did not receive bortezomib as first-line treatment receive it as second-line treatment.
-
Costs and outcomes of third-line and subsequent treatments are assumed to be the same between arms.
-
Patients discontinue first-line treatment upon disease progression.
-
Health-related quality of life is better for those with CR than those with less than CR and is assumed to improve when patients stop treatment.
-
AEs are not modelled explicitly in the model for patient outcomes, i.e. OS and PFS, but are included as additional cost for treating the AEs in the model.
In each cycle the total costs and QALYs are calculated by multiplying the individual costs and HRQoL by the number of people in the cohort still alive for each of the treatments. The total lifetime costs and QALYs are calculated by aggregating the costs and QALYs for all cycles. The total discounted QALY gain and cost of treatments are calculated. Thus, the cost-effectiveness of each of the treatments is calculated,
Evaluation of uncertainty
The evaluation of the cost-effectiveness of treatment for MM is based on uncertain information about variables, such as the clinical effect, HRQoL and resource use. This uncertainty was evaluated using deterministic and probabilistic sensitivity analyses. One-way deterministic sensitivity analyses were conducted to evaluate the influence of individual parameters on the model results and test the robustness of the cost-effectiveness results to variations in the structural assumptions and parameter inputs (see Deterministic sensitivity analysis, below).
Multiparameter uncertainty in the model was addressed using PSA (see Probabilistic sensitivity analysis, below). 103 In the PSA, probability distributions are assigned to the point estimates used in the base-case analysis. The model is run for 1000 iterations, with a different set of parameter values for each iteration, by sampling parameter values at random from their probability distributions. The uncertainty surrounding the cost-effectiveness of the treatment is represented on a cost-effectiveness acceptability curve (CEAC) according to the probability that the intervention will be cost-effective at a particular willingness-to-pay threshold. Appendix 14 reports the parameters included in the PSA, the form of distribution used for sampling each parameter, and the upper and lower limits assumed for each variable.
Model validation
The SHTAC model was validated by checking the model structure, calculations and data inputs for technical correctness. The structure was reviewed by clinical experts for appropriateness for the disease and its treatment. The SHTAC model was checked for internal consistency against the MS economic models by running the SHTAC model with the inputs used in MS models to ensure similar results. The robustness of the model to changes in input values was tested using sensitivity analyses to ensure that any changes to the input values produced changes to the results of the expected direction and magnitude. Finally, the model results were compared with those from the MSs.
SHTAC data sources
Baseline MP curves
The baseline MP OS curve was generated using the MP OS curves from the RCTs included in our systematic review of clinical effectiveness (see Chapter 4, Overall survival). Survival probabilities (at 6-month intervals) were extracted from a scanned copy of the Kaplan–Meier plots for each MP group using the digitising software Engauge104 (Appendix 13). A weighted average of the survival probabilities for each time point was calculated to provide a summary MP OS curve (Table 28 and Figure 8) using the number of participants in the trials as weights.
FIGURE 8.
Summary curve for MP OS obtained from weighted average of individual MP curves.

| Trial MP arm | No. in MP arm | Time (months) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 6 | 12 | 18 | 24 | 30 | 36 | 42 | 48 | 54 | 60 | 66 | 72 | ||
| Facon et al.23 | 196 | 0.88 | 0.78 | 0.72 | 0.63 | 0.55 | 0.48 | 0.39 | 0.31 | 0.26 | 0.23 | 0.19 | 0.17 |
| Hulin et al.59 | 116 | 0.90 | 0.83 | 0.71 | 0.62 | 0.47 | 0.39 | 0.38 | 0.30 | 0.28 | |||
| Palumbo et al.24 | 164 | 0.96 | 0.84 | 0.75 | 0.71 | 0.65 | 0.59 | 0.53 | 0.49 | 0.49 | 0.36 | ||
| VISTA Trial105 | 338 | 0.92 | 0.82 | 0.76 | 0.69 | 0.64 | 0.54 | 0.51 | 0.41 | ||||
| MMIX53 | 426 | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| MP weighted averagea | 0.90 | 0.79 | 0.72 | 0.65 | 0.56 | 0.48 | 0.42 | 0.36 | 0.31 | 0.27 | 0.17 | 0.17 | |
A baseline MP PFS curve was generated using the PFS curves from the trial data included in our systematic review of clinical effectiveness (see Chapter 4, Progression-free survival) in a similar way to the baseline OS curves – see Appendix 13 and Table 29. A weighted average of the PFS probabilities for each 6-month time point was calculated to provide a summary MP PFS curve (Figure 9) using of participants in the trials as weights.
| Study MP arm | No. in MP arm | Time (months) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 6 | 12 | 18 | 24 | 30 | 36 | 42 | 48 | 54 | ||
| Facon et al.23 | 196 | 0.77 | 0.63 | 0.49 | 0.34 | 0.22 | 0.14 | 0.08 | 0.06 | 0.05 |
| Hulin et al.59 | 116 | 0.8 | 0.66 | 0.51 | 0.33 | 0.17 | 0.12 | 0.06 | 0.05 | 0.05 |
| Palumbo et al.24 | 164 | 0.88 | 0.60 | 0.41 | 0.28 | 0.22 | 0.19 | 0.18 | 0.18 | |
| VISTA trial26 | 338 | 0.77 | 0.63 | 0.43 | 0.28 | |||||
| MP weighted averagea | 0.78 | 0.63 | 0.46 | 0.30 | 0.21 | 0.15 | 0.11 | 0.10 | 0.05 | |
FIGURE 9.
Summary curve for PFS obtained from weighted average of individual PFS data in MP arms of trials.
The probability of an event at each time interval for the MP treatment arm (hazard OS and hazard PFS) is calculated from the baseline MP OS and MP PFS curves. These probabilities are shown in Table 30. The hazard rate for an event for MP per cycle is estimated for each time point, ti:
| Months | Cycles | Hazard rate | |
|---|---|---|---|
| OS | PFS | ||
| 6 | 4.4 | 0.024 | 0.056 |
| 12 | 8.7 | 0.030 | 0.048 |
| 18 | 13.0 | 0.021 | 0.070 |
| 24 | 17.4 | 0.023 | 0.094 |
| 30 | 21.7 | 0.034 | 0.067 |
| 36 | 26.1 | 0.035 | 0.067 |
| 36+ | 26+ | 0.028 | 0.067 |
where s(t) is the survival function over time t.
For OS, few individuals were followed up for more than 36 months, and so a constant hazard rate was assumed after 36 months using the hazard rate in the first 36 months. For PFS, few individuals were followed up for more than 24 months, and so a constant hazard rate was assumed after 24 months using the hazard rate in the first 24 months. The methodology used to derive the survival curves is described in more detail in Appendix 13.
Overall survival and progression-free survival hazard ratios for treatments versus MP
The relative effectiveness of the treatments versus MP for OS and PFS were represented as HRs. The HRs were obtained from the Kaplan–Meier plots in the trial publications (see Overall survival and Progression-free survival). As the HR of the treatments versus MP varied over time, a constant HR was not appropriate. We derived the HR for each 6-monthly period for each of the treatments versus MP.
The HR for each treatmentj versus MP at each time point ti is,
The HRs for the MPT trials summary were combined using simple weighted averages of the proportion of surviving patients in each trial arm at each time point, weighted by numbers of patients in the trial. The HRs were assumed to be constant after 36 months for OS and 24 months for PFS as there were few patients with more than this length of follow-up in the trials. HRs of OS and PFS are shown in Tables 31 and 32, respectively.
| Months | Facon et al.23 MPT | Hulin et al.59 MPT | SHTAC MPT trials summary | San Miguel et al.26 VMP | MMIX54 CTDa |
|---|---|---|---|---|---|
| 0–6 | 0.52 | 0.95 | 0.67 | 1.00 | (AiC/CiC information has been removed) |
| 6–12 | 0.57 | 0.50 | 0.55 | 0.30 | (AiC/CiC information has been removed) |
| 12–18 | 0.49 | 0.91 | 0.71 | 0.60 | (AiC/CiC information has been removed) |
| 18–24 | 0.56 | 0.64 | 0.59 | 0.85 | (AiC/CiC information has been removed) |
| 24–30 | 0.64 | 0.33 | 0.46 | 0.70 | (AiC/CiC information has been removed) |
| 30–36 | 0.74 | 0.79 | 0.76 | 0.46 | (AiC/CiC information has been removed) |
| 36+ | 0.59 | 0.64 | 0.62 | 0.62 | (AiC/CiC information has been removed) |
| Months | Facon et al.23 MPT | Hulin et al.59 MPT | SHTAC MPT trials summary | San Miguel et al.26 VMP | MMIX54 CTDa |
|---|---|---|---|---|---|
| 0–6 | 0.36 | 0.61 | 0.45 | 0.47 | (AiC/CiC information has been removed) |
| 6–12 | 0.61 | 0.76 | 0.67 | 0.62 | (AiC/CiC information has been removed) |
| 12–18 | 0.49 | 0.70 | 0.57 | 0.74 | (AiC/CiC information has been removed) |
| 18–24 | 0.70 | 0.51 | 0.62 | 0.48 | (AiC/CiC information has been removed) |
| 24+ | 0.55 | 0.62 | 0.58 | 0.58 | (AiC/CiC information has been removed) |
The event rate at each time interval for MPT, VMP and CTDa was estimated by multiplying the risk of death or progression by the HR for each cycle. The effects of using alternative HRs were evaluated in sensitivity analyses. It should be noted that the MMIX RCT included a second randomisation to maintenance therapy with thalidomide for some patients after first-line therapy and there were no data available for OS and PFS for patients who did not have maintenance therapy.
Complete response
Complete response outcome data for each treatment option is described in Chapter 4 (see Response to treatment). For each treatment option, the relative risk of CR compared with MP was derived using Review Manager 5. The CR rate for MP was estimated using the trial data by simple weighted average of the MP arm using the number of trial participants as the weight. CR for the other treatment options was derived by multiplying the MP CR rate by the relative risk. Table 33 shows the CR data used in the model for MP, VMP, MPT and CTDa.
| Treatment | CR (%) |
|---|---|
| MP | 2.6 |
| MPT | 14.2 |
| VMP | 21.7 |
| CTDa | (AiC/CiC information has been removed) |
Health-related quality of life
Although our systematic review of HRQoL studies (see Systematic review of health-related quality-of-life studies) did not find any generic preference-based HRQoL studies of people with untreated MM who were not eligible for HDT with SCT, it did identify two studies that assessed HRQoL in this group using the EORTC QLQ-C30. A targeted search was therefore conducted for studies that mapped data from the EORTC QLQ-C30 onto the EQ-5D to enable the estimation of health state values based on EORTC QLQ-C30 data. The EORTC QLQ-C30 is the most commonly used instrument to measure the HRQoL of cancer patients. Two studies were identified. 106,107
McKenzie and van der Pol107 used an ordinary least squares (OLS) regression analysis with data from an RCT of palliative therapies for 199 patients with inoperable oesophageal cancer, with an average age of 74.8 years. The regression results for the mapping are shown in Table 34.
| Dimension | Coefficient |
|---|---|
| Global QoL | 0.0016 |
| Physical functioning | 0.0004 |
| Role functioning | 0.0022 |
| Emotional functioning | 0.0028 |
| Cognitive functioning | 0.0009 |
| Social functioning | 0.0002 |
| Fatigue | –0.0021 |
| Nausea | –0.0005 |
| Pain | –0.0024 |
| Dyspnoea | 0.0004 |
| Insomnia | 0.00004 |
| Appetite loss | 0.0003 |
| Constipation | 0.0001 |
| Diarrhoea | –0.0003 |
| Financial difficulties | –0.0006 |
| Constant | 0.2376 |
Kontodimopoulos and colleagues106 used an OLS regression with data from 48 patients with gastric cancer, split into equal subgroups by age, sex and chemotherapy scheme. Three scales were significant predictors (p < 0.05 or better) of EQ-5D indices: physical functioning, emotional functioning and global health status. The regression results for the mapping are shown in Table 35.
| Dimension | Coefficient |
|---|---|
| Physical functioning | 0.00508 |
| Emotional functioning | 0.00313 |
| Global health status | 0.00546 |
| Constant | –0.18143 |
Our systematic review of HRQoL studies found two studies in the population of interest but these used the EORTC QLQ-C30. 91,92 For both studies, we mapped the EORTC QLQ-C30 HRQoL scores to the EQ-5D using each of the mapping algorithms described above (Tables 36 and 37).
| Mapping algorithm | MM | Reference population |
|---|---|---|
| McKenzie and van der Pol107 | 0.59 | 0.82 |
| Kontodimopoulos et al.106 | 0.58 | 0.88 |
| Mapping algorithm | Reference population | Time (months) | |||||
|---|---|---|---|---|---|---|---|
| 0 | 1 | 6 | 12 | 24 | 36 | ||
| McKenzie and van der Pol107 | 0.81 | 0.55 | 0.58 | 0.68 | 0.68 | 0.68 | 0.69 |
| Kontodimopoulos et al.106 | 0.86 | 0.52 | 0.58 | 0.69 | 0.69 | 0.69 | 0.71 |
Gulbrandsen and colleagues91 provide HRQoL at different time points. Based on this study, it appears that HRQoL is lower during the treatment period and improves after treatment has finished and this is consistent with HRQoL results from Uyl-de Groot and colleagues. 96 Long-term HRQoL appears to be stable over time. In addition, the utility estimates from the HRQoL studies in populations treated with HDT are similar to those from Gulbrandsen and colleagues. 91
The accuracy of the mapping studies was assessed for the study by Uyl-de Groot and colleagues,96 which reported EORTC QLQ-C30 and EQ-5D results. Figure 10 shows the comparison between the EQ-5D utility estimates using the two mapping methods compared with the EQ-5D data from Uyl-de Groot and colleagues. For these data, the mapping algorithm by McKenzie and van der Pol provides the better fit and for most time points is a good fit to the data.
FIGURE 10.
Comparison of results from mapping studies from EORTC QLQ-C30 to EQ-5D with EQ-5D data from Uyl-de Groot and colleagues. 96

We suggest that the most appropriate source of HRQoL data for the treatment period and post-treatment values is from Gulbrandsen and colleagues91 from the mapping by McKenzie and van der Pol. These utility estimates are shown in Table 37. The utility estimates for the treatment period are for the 1-month time point, i.e. 0.58, and for post treatment (and post progression) is an average of the 6- to 36-month time points, i.e. 0.68.
Complete response
Health-related quality-of-life data from the MMIX RCT57 were analysed to determine whether patients with CR had a better HRQoL after response than those with other levels of response. EORTC QLQ-C30 data were available for 0, 3, 6 and 12 months after initial treatment commenced. We mapped the EORTC QLQ-C30 data to EQ-5D health utilities using the algorithm from McKenzie and van der Pol. 107 For the first three periods, the EQ-5D utility scores were similar for both CTDa and MP groups, and similar to those from Gulbrandsen and colleagues. 91 (AiC/CiC information has been removed.)
In the model we estimate the utility for the post-treatment health state (until disease progression) as a weighted average of those who had a CR (AiC/CiC information has been removed) and those with a lesser response (AiC/CiC information has been removed).
Estimation of costs
Drug costs
Drug unit costs and doses were based on the BNF (No. 57). 35 Duration of treatment was based on recommendations from the SPC,29,33 expert clinical opinion and the published trials. A summary of the dose and duration of treatment for each of the comparators is given in Table 38.
| Parameter | Melphalan | Prednisolone | Bortezomib | Thalidomide | Cyclophosphamide | Dexamethasone | Source |
|---|---|---|---|---|---|---|---|
| Drug dose | 9 mg/m2 | 60 mg/m2 | 1.3 mg/m2 | 150 mg/day | 250 mg/m2/week | 20 mg/day for 4 days every 28 | BNF,35 SPC,29,33 VISTA,26 MMIX49 and clinical expert opinion |
| No. cycles | 8a | 8a | 9 | 8 | 7 | 7 | |
| Cycle length (weeks) | 6 | 6 | 6 | 6 | 6 | 6 | |
| Duration | 10–12 months | 10–12 months | 54 weeks | 10–12 months | 6–8 months | 6–8 months | |
| Days of cycle | Days 1–4 | Days 1–4 |
Cycles 1–4: 1, 4, 8, 11, 22, 25, 29, 32 Cycles 5–9: 1, 8, 22, 29 |
Daily | 4 doses/cycle | 30 doses/course | |
| Unit costs | Melphalan £11.46 for 25-tablet pack (2 mg); total cost £126.24 | Prednisolone £20 for 50-tablet pack (25 mg); total cost £25.71 | Bortezomib £762.68 per 3.5-mg vial; total cost £39,643.76 | £298.48 per 28-tablet (50-mg) pack; total cost £10,745.80 | £12.44 per 100-tablet pack (50 mg); total cost £44.41 | £13.92 per 100-tablet pack (2 mg); total cost £58.46 | BNF35 |
The duration of treatment varied between seven cycles for CTDa and nine cycles for VMP. We assumed that MP would be given for the same number of cycles as thalidomide and bortezomib when it was given in combination with them. The SPC of thalidomide states that a maximum number of 12 cycles of 6 weeks each should be used, as used in the trial by Facon and colleagues. 23 However, one of our clinical experts advised that a shorter duration of eight cycles was more representative of clinical practice.
The dose of thalidomide was assumed to be 150 mg, based upon the dosages used in the IFM RCT (100 mg)59 and the MMIX RCT (200 mg). 49 The dose recommended by the SPC is 200 mg per day, but one of our clinical experts advised that, in practice, few patients are able to tolerate such a high dose. Bortezomib is administered as a 3- to 5-second bolus intravenous injection. The cost of the 3.5-mg vial is £762.68. The cost of bortezomib administration was £153.40. 100
The total cost for bortezomib depends on the wastage from the vial. In the NICE appraisal of bortezomib for relapsed MM,30 the appraisal committee considered the issue of vial sharing. They expressed a number of concerns including issues related to maintenance of best aseptic practice and the practical constraints of patient numbers and geographical locations of myeloma centres. The Committee was not convinced that vial sharing could be considered either safe or routinely achievable in practice across the NHS.
One of our clinical experts advised that they attempted to administer bortezomib in groups of three persons to minimise wastage. However, this may not be possible in smaller units. In the base-case analysis we assumed that only one vial would be used per patient and then varied this assumption in a scenario analysis.
Patients on thalidomide also received thromboprophylaxis for 5 months in the form of low-molecular-weight heparin (dalteparin 5000 units once daily subcutaneously)28 at a total cost of £428.88. In addition to chemotherapy, patients also require treatment with other medication, such as bisphosphonates, but the cost for these was assumed to be similar across all interventions, and has therefore not been included in the model costs.
Second-line treatment
Following disease progression after first-line therapy, patients receive second-line treatment. Based on clinical advice, NICE guidance,30 trial data and assumptions used in the Janssen–Cilag submission, it was assumed that most individuals would receive bortezomib as second-line therapy unless they had already received it as first-line therapy. HDD and CTDa were also used as these are common second-line treatments in the UK. 30 Most patients who had VMP as first-line treatment had CTDa as second-line treatment. The dose for HDD was 40 mg per day and the cost of treatment was £189.31. The assumed distribution of second-line treatments following first-line treatment is shown in Table 39. For all treatments, 60% of patients received second-line treatment, based upon the number of patients still alive at the time corresponding to mean PFS.
| Second-line treatment (%) | First-line treatment | |||
|---|---|---|---|---|
| MP | MPT | VMP | CTDa | |
| Bortezomib + HDD | 70 | 70 | 15 | 70 |
| CTDa | 15 | 15 | 70 | 15 |
| HDD | 15 | 15 | 15 | 15 |
Consultations
Based on clinical advice, we assumed patients receive on average one consultation every month during their treatment period and one consultation every three months thereafter. The outpatient consultation cost was £121.11 (reference cost code 370: medical oncology follow-up consultation). 100
Monitoring tests
The monitoring tests used for the management of MM, based on those used for the MMIX RCT,49 are shown in Table 40 with their unit costs.
| Test | Unit cost (£) | Costs source |
|---|---|---|
| Full blood count | 3.02 | Southampton University Hospital Trust 2009108 |
| Biochemistry (calcium, creatinine, albumin and uric acid) | 5.15 | |
| Protein electrophoresis | 13.85 | |
| Immunoglobin (IgA, IgG, IgM) | 41.55 | |
| Urinary light chain excretion | 13.85 |
Adverse events
For each comparator, the incidence of AEs was estimated using evidence from the RCTs included in our systematic review of clinical effectiveness (see Chapter 4, Adverse events). AEs included in the model were treatment-related serious (grades 3 and 4) AEs and the incidence was taken from the VISTA trial26 for VMP, from the IFM 99/06 trial for MPT23 and from the MMIX trial for CTDa. The IFM 99/06 trial was used for MPT as this trial had more comprehensive reporting than the other MPT trials. For MP, a weighted average was calculated using data from the MP arm from each of these trials.
Although AE data is consistently reported across studies as percentage patients, the types of AEs reported differed between the studies. This summary extracts key AEs (haematological, gastrointestinal, infections, neuropathy and thrombosis) for use within the model (and is not a comprehensive analysis of all AEs). Gastrointestinal AE numbers for MMIX were calculated from constipation grades 3 and 4 as reported and other gastrointestinal AEs (grade not specified but proportion calculated for grades 3 and 4). Total infection for the VISTA study was calculated by totalling figures for pneumonia and herpes zoster (which assumes that there were no others). Infections were not specified for other studies. The definition of haematological AEs may not be exactly consistent across studies but gives an indication of possible rates for thrombocytopenia/cytopenia. AE data were not available for the MMIX RCT for the incidence of neutropenia and anaemia and for these AEs we have assumed the same incidence for CTDa as for MPT. Where events of grades 3 and 4 were not reported separately, we assumed there were twice as many grade 3 as grade 4 events, as this was the ratio for the total numbers of grade 3 and 4 AEs.
The unit costs of treating AEs were estimated, based on those used in a NICE technology appraisal for lenalidomide (TA171)31 and the Celgene MS [see Review of the Celgene submission to NICE (Thalidomide) and Appendix 12]. The NICE technology appraisal for lenalidomide31 collected information on the proportion of patients who would receive treatment, the location where treatment would be administered, and treatments administered for each specific disease-related complication. The unit cost of inpatient and day-case treatment for the AE was calculated from CHKS (Caspe Healthcare Knowledge Systems) data, which contains individual patient-level data from most UK hospital trusts, and NHS reference cost data. This report did not include all relevant AE costs. The Celgene MS used a similar methodology to calculate unit costs and these were used for AEs of infection, dizziness or fatigue (Table 41). There was no distinction made in that report between the costs of grade 3 and 4 AEs and so for these AEs we have assumed equal costs for grades 3 and 4. We used the cost of diarrhoea for the cost of gastrointestinal AEs as this cost was between the costs of nausea and constipation. The unit costs for treating the AEs are shown in Table 42.
| AE (%) | VISTA trial26 | Facon et al.23 99/06 | MMIX54 | MP weighted average | |||
|---|---|---|---|---|---|---|---|
| VMP | MP | MPT | MP | CTDa | MP | ||
| Haematological | |||||||
| Thrombocytopenia | 37 | 30 | 14 | 10 | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | 19 |
| Neutropenia | 40 | 38 | 48 | 26 | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | 34 |
| Anaemia | 19 | 28 | 14 | 14 | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | 23 |
| Gastrointestinal | 20 | 5 | 11 | 3 | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | 3 |
| Nervous system | |||||||
| Peripheral neuropathy | 14 | 0 | 6 | 0 | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | > 1 |
| Dizziness/fatigue | 9 | 1 | 8 | 0 | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | 1 |
| Infections | 10 | 7 | 13 | 9 | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | 7 |
| Thrombosis | 1 | 1 | 12 | 4 | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | 2 |
| AE | Unit cost (£) | Source | |
|---|---|---|---|
| Grade 3 | Grade 4 | ||
| Thrombocytopenia | 164.37 | 683.62 | TA17131 |
| Neutropenia | 190.86 | 354.30 | TA17131 |
| Anaemia | 384.75 | 551.63 | TA17131 |
| Gastrointestinal | 830.84 | 1302.90 | TA17131 |
| Peripheral neuropathy | 174.75 | 317.37 | TA17131 |
| Dizziness/fatigue | 172.24 | 172.24 | Celgene MS |
| Infection | 1018.01 | 1018.01 | |
| DVT | 347.17 | 1014.29 | TA17131 |
The total costs of treating AEs were estimated by multiplying each AE incidence by the appropriate unit cost for that AE.
Results of SHTAC independent economic evaluation
This section reports the cost-effectiveness results for a typical person with MM who received treatment with bortezomib in combination with MP or thalidomide in combination with MP compared with those receiving MP. Results for costs and QALYs are presented for each treatment, with costs and benefits discounted at 3.5%. 78 The survival curves for OS from the model are shown in Figure 11. The results show increased survival for MPT, VMP and CTDa versus MP. The cost-effectiveness is presented as incremental cost per QALY compared with existing treatment with MP. The summary results of the non-discounted treatment effects are shown in Table 43. In the base-case analysis, OS varied from 4.20 years for MP to 6.66 years for MPT. Survival for MPT is slightly longer than for VMP. The cost-effectiveness results for CTDa should be treated with caution, (AiC/CiC information has been removed). The summary results of the undiscounted costs are shown in Table 44 for each treatment. First-line treatment costs ranged from £112 for MP to £43,824 for VMP. Second-line treatment costs were the same for MP, MPT and CTDa, and about £10,000 lower for VMP. The total costs of the treatments ranged from £23,248 for MP to £59,644 for VMP.
FIGURE 11.
Overall survival curves for MP, MPT, VMP and CTDa.

| Health state | Duration (years) | |||
|---|---|---|---|---|
| MP | MPT | VMP | CTDa | |
| Treatment | 0.92 | 0.92 | 1.04 | 0.81 |
| Post treatment | 0.88 | 2.13 | 2.00 | 1.37 |
| Post progression | 2.39 | 3.61 | 3.60 | 2.52 |
| OS | 4.20 | 6.66 | 6.64 | (AiC/CiC information has been removed) |
| Cost | Costs (£) | |||
|---|---|---|---|---|
| MP | MPT | VMP | CTDa | |
| First-line treatment | 112 | 10,316 | 43,824 | 8691 |
| Second-line treatment | 17,695 | 17,695 | 7786 | 17,695 |
| Monitoring | 5075 | 7248 | 7375 | 5318 |
| AEs | 365 | 563 | 658 | 507 |
| Total | 23,248 | 35,822 | 59,644 | 32,211 |
The baseline discounted cost-effectiveness results are shown in Table 45. Each of the treatments is more expensive than MP, with the additional cost ranging from £8,600 (CTDa) to more than £35,000 (VMP) over a patient lifetime. The incremental cost-effectiveness versus MP for MPT, VMP and CTDa figures are £9135, £29,820 and £33,031 per QALY gained, respectively.
| Treatment | QALY | Cost (£) | ICER vs MP (£/QALY) | ICER (cost/QALY) vs next best option |
|---|---|---|---|---|
| MP | 2.42 | 21,439 | – | – |
| CTDa | 2.68 | 29,983 | 33,031 | 33,031 |
| VMP | 3.62 | 57,168 | 29,820 | 28,937 |
| MPT | 3.64 | 32,598 | 9135 | Dominates VMP |
Each comparator is presented in successive rows ordered by the number of QALYs generated. Each option is then compared to the next best option. In summary the incremental analysis suggests extended dominance of MPT over CTDa, and MPT dominates VMP as it is more effective and cheaper (Figure 12). The comparison of VMP versus MPT suggests that VMP and CTDa are unlikely to be cost-effective treatment options at the conventional willingness-to-pay threshold of £20,000–30,000 per QALY gained. However, there is much uncertainty around the results for CTDa because the OS effectiveness estimates were not statistically significant and the results from the MMIX RCT included those of participants who had received thalidomide maintenance therapy.
FIGURE 12.
Cost-effectiveness plane for treatments MP, CTDa, VMP and MPT.

Sensitivity analysis
Deterministic sensitivity analysis
One-way deterministic sensitivity analyses were performed, in which model parameters were systematically and independently varied, using a realistic minimum and maximum value. The sensitivity analysis investigated the effect of uncertainty around the model assumptions, structure and parameter values on the cost-effectiveness results, in order to highlight the most influential parameters. The effects of uncertainty in multiple parameters were addressed using PSA, which is reported later in this chapter (see Probabilistic sensitivity analysis). Where possible, the parameters were varied according to the ranges of the CIs of these parameters, based on the published estimates. Where these data were not available an alternative suitable range was chosen. The same ranges were used in the deterministic analyses and PSA and these are described in Appendix 14.
Tables 46–48 show the results of the deterministic sensitivity analyses for each of the treatments versus MP for the most influential parameters. Other parameters, such as AE cost, CR rate and utility values, were varied in the sensitivity analyses but were found to only have a negligible effect on the results. The cost-effectiveness results are fairly robust to changes in parameters in the deterministic sensitivity analysis. For each of the treatments, the model results are most sensitive to the HR for OS, cost and dosage of the treatment and the overall baseline survival curve used for MP. The deterministic sensitivity results for MPT versus MP are shown in Table 46 and varied between £6445 and £22,749 per QALY gained. MPT dominates VMP for all parameters, except the VMP treatment effectiveness for OS (HR). Using the higher CI for OS, the cost-effectiveness estimate of VMP versus MPT is £44,928 per QALY gained.
| Parameter | Baseline | Upper value | Lower value | Upper value ICER (£/QALY) | Lower value ICER (£/QALY) | Range |
|---|---|---|---|---|---|---|
| HR for OS | 0.62 | 0.82 | 0.5 | 22,749 | 6445 | 16,304 |
| Dosage thalidomide (mg/day) | 150 | 200 | 100 | 11,765 | 6504 | 5261 |
| MP OS baseline curvea | 0.028 | 0.039 | 0.02 | 11,230 | 7779 | 3451 |
| Unit cost thalidomide (£) | 298.48 | 358.18 | 238.78 | 10,713 | 7557 | 3156 |
| Second-line treatment, bortezomib MPb (%) | 70 | 80 | 60 | 7772 | 10,497 | 2725 |
| Second-line treatment, bortezomib MPTb (%) | 70 | 80 | 60 | 10,440 | 7830 | 2610 |
| No. of cycles, MPT | 8 | 9 | 7 | 10,282 | 7976 | 2306 |
| Parameter | Baseline | Upper value | Lower value | ICER (£/QALY) | ||
|---|---|---|---|---|---|---|
| Upper value | Lower value | Range | ||||
| HR for OS | 0.62 | 0.83 | 0.51 | 87,665 | 20,440 | 67,225 |
| MP OS baseline curvea | 0.028 | 0.039 | 0.02 | 37,791 | 24,778 | 13,013 |
| Unit cost bortezomib (£) | 762.38 | 914.86 | 609.90 | 33,779 | 25,862 | 7917 |
| Discount rate benefits (%) | 3.5 | 5 | 2 | 33,795 | 26,081 | 7714 |
| Utility progression | 0.68 | 0.75 | 0.61 | 27,788 | 32,173 | 4385 |
| No. of cycles VMP | 9 | 10 | 8 | 31,801 | 27,748 | 4052 |
| Cost of bortezomib administration (£) | 153.40 | 199.41 | 107.38 | 31,632 | 28,009 | 3623 |
| Parameter | Baseline | Upper value | Lower value | ICER (£/QALY) | ||
|---|---|---|---|---|---|---|
| Upper value | Lower value | Range | ||||
| HR for OS | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | –29,210 | 16,897 | 46,107 |
| MP OS baseline curve a | 0.028 | 0.039 | 0.02 | 49,240 | 24,621 | 24,619 |
| Thalidomide dose (mg/day) | 150 | 200 | 100 | 43,501 | 22,561 | 20,940 |
| Second-line treatment, bortezomibb MP (%) | 70 | 80 | 60 | 26,596 | 39,466 | 12,870 |
| Second-line treatment, bortezomibb CTDa (%) | 70 | 80 | 60 | 39,385 | 26,677 | 12,708 |
| Unit cost thalidomide (£) | 298.48 | 358.18 | 238.78 | 39,483 | 26,978 | 12,505 |
| No. of cycles CTDa | 7 | 8 | 6 | 39,313 | 26,749 | 12,564 |
The deterministic sensitivity results for VMP versus MP are shown in Table 47 and varied between £20,440 and £87,665 per QALY gained. VMP is dominated by MPT for all parameters, except the MPT treatment effectiveness for OS (HR). This is also the case if the model assumes that vials for bortezomib can be shared, rather than assuming one vial per patient. Using the lower CI for OS, the cost-effectiveness estimate of VMP versus MPT is £34,015 per QALY gained.
The deterministic sensitivity results for CTDa versus MP are shown in Table 48 and varied between –£29,210 and £16,897 per QALY gained. (AiC/CiC information has been removed.)
Scenario analysis
In addition to the sensitivity analyses four alternative scenarios were undertaken to investigate the uncertainty around structural assumptions (Table 49).
| Scenario | ICER (cost per QALY gained, £) vs MP | ||
|---|---|---|---|
| MPT | VMP | CTDa | |
| Base-case analysis | 9135 | 29,820 | 33,031 |
| Scenario A | 9699 | 37,711 | 33,828 |
| Scenario B | 9330 | 22,533 | 33,307 |
| Scenario C | 24,276 | 29,820 | 33,031 |
| Scenario D | 20,605 | 71,223 | 80,382 |
Scenario A – no subsequent therapies
The base-case scenario included the cost of second-line therapy. This scenario investigates the cost-effectiveness of first-line therapy only without including the subsequent treatment costs. In this case, MPT and CTDa are slightly less cost-effective versus MP, and VMP is considerably less cost-effective. The cost-effectiveness estimate for VMP versus MP increases to £37,711 per QALY gained. MPT continues to dominate VMP.
Scenario B – vial sharing
The base-case scenario assumes that it is not possible for patients to share vials of bortezomib. This scenario investigates the cost-effectiveness where patients do share vials of bortezomib. With vial sharing and no wastage, bortezomib becomes more cost-effective versus MP, with an ICER of £22,533 per QALY gained. MPT continues to dominate VMP.
Scenario C – inclusion of thalidomide maintenance trials
The base-case scenario uses the efficacy for MPT using only RCTs that did not include thalidomide maintenance. This scenario investigates the cost-effectiveness using the estimate for MPT efficacy from a meta-analysis that includes trials with thalidomide maintenance. Janssen–Cilag conducted a MTC for MPT efficacy with trials that included thalidomide maintenance and derived a HR (AiC/CiC information has been removed) for MPT versus MP. Using this HR makes MPT less cost-effective with an ICER of £24,276 per QALY gained versus MP. In addition, MPT no longer dominates VMP, with an ICER of £32,774 for VMP versus MPT.
Scenario D – treatment effectiveness beyond the end of trial
The base-case scenario extrapolates beyond the end of the trial by assuming a constant HR for the treatment effectiveness compared with MP. Although this is a standard methodological assumption, it is unclear how the treatment effectiveness changes beyond the end of the trial. This scenario investigates an alternative assumption whereby there is no treatment benefit for VMP, MPT and CTDa over MP, i.e. the event rates for these treatments are the same as for MP after the end of the trial. Using this assumption has a large effect on the model results, and all treatments are less cost-effective compared with MP. The ICERs for each of the treatment options more than double to £20,605 (MPT), £71,223 (VMP) and £80,382 (CTDa) per QALY gained versus MP. MPT continues to dominate VMP.
There are two additional scenario analyses for treatment duration and treatment discontinuations in Appendix 15.
Probabilistic sensitivity analysis
In the PSA, all parameters were sampled probabilistically from an appropriate distribution using similar ranges as used in the deterministic sensitivity analyses. The parameters sampled were discount rate, number of treatment cycles, utility values, CR rate, cost of AEs, parameters for the survival curves and the proportions of patients receiving bortezomib as second-line therapy. The distribution assigned to each variable included in the PSA and the parameters of the distributions are reported in Appendix 14.
One thousand simulations were run. The PSA results are presented in Table 50 and show similar results to the deterministic analyses (Tables 46–48). The scatter plots for cost and health outcomes for the treatment options for the PSA are shown in Figure 13. The CEAC is shown in Figure 14, and indicates that at the £20,000 and £30,000 willingness-to-pay thresholds MPT has the highest probability of being cost-effective of 0.95 and 0.95, respectively.
| Result | MP | MPT | VMP | CTDa |
|---|---|---|---|---|
| Total cost (£) | 21,620 | 33,050 | 57,545 | 30,371 |
| Total QALY | 2.44 | 3.68 | 3.66 | 2.70 |
| Incremental cost vs MP (£) | – | 11,495 | 35,991 | 8816 |
| Incremental QALY vs MP | – | 1.26 | 1.24 | 0.28 |
| ICER vs MP (£) | – | 9124 | 29,102 | 31,612 |
FIGURE 13.
Scatter plots of the costs and health benefits from PSA for MP, MPT, VMP and CTDa.

FIGURE 14.
Cost-effectiveness acceptability curve from the PSA.

Summary of cost-effectiveness
-
A systematic search of the literature found five abstracts of economic evaluations of treatment for patients with previously undiagnosed MM, who were ineligible for HDT-SCT. None of the studies contained sufficient information for critical appraisal. Three of the abstracts compared MPT with MP in patients in Scotland, Wales and Australia. Each abstract concluded that MPT was a cost-effective alternative to MP. Two abstracts compared VMP, MPT and MP in Canadian and US patients. Both studies concluded that the VMP regimen was cost-effective compared with MP and MPT. The latter study stated that VMP dominated MPT (i.e. more effective at a lower cost). All studies were industry funded.
-
A systematic review of studies of QoL for patients with MM identified six studies: only two of these studies were for the population of interest and both studies did not include generic preference-based utility measures; the other four QoL studies provided utility estimates for patients with MM who had intensive therapy.
-
Two manufacturers submitted evidence to be considered for the appraisal of bortezomib and thalidomide treatment. Janssen–Cilag, the manufacturer of bortezomib, constructed a survival model that estimated OS and PFS based on treatment effects from a MTC of the RCTs. They included second- and third-line treatment. The base-case results from the submission found all treatments (VMP, MPT and CTDa) to be cost-effective. The ICER for VMP versus MP is estimated to be £10,498. Furthermore, the ICERs of VMP versus MPT and VMP versus CTDa are estimated to be £11,907 and £10,411, respectively.
-
Celgene, the manufacturer of thalidomide, constructed a Markov model with health states for preprogression (with or without AEs), post progression and death. They assumed that survival after disease progression was the same irrespective of first-line treatment. Treatment effects for disease progression were calculated from a random effects MTC. The base-case results from the submission estimated an ICER of £23,381 per QALY gained for MPT versus MP and £303,845 per QALY for VMP versus MPT.
-
The authors of this report developed an independent survival model. The survival model consisted of two survival curves which estimated the mean time to death and disease progression. These survival durations were used to derive the time spent in three health states: treatment, post treatment and progression. Utility values were applied to these health states to estimate total QALYs for each treatment option. Costs were included for medications and outpatient costs and AEs. The model base-case results showed increased survival for each of the treatments compared with MP at an increased cost. The OS was marginally longer for MPT than for VMP at a considerably lower cost. The cost-effectiveness estimates for MPT, VMP and CTDa versus MP were £9135, £29,820 and £33,031 per QALY gained, respectively. However, MPT dominated VMP as it was cheaper and more effective.
-
The effects of a range of parameter values in the economic model were evaluated in sensitivity analyses. The model results were found to be robust to changes in the parameter values. The model results are most sensitive to changes in the parameter values of the HRs for OS.
-
The PSA estimated the probability of each of the treatments to be cost-effective at the £20,000 and £30,000 willingness-to-pay thresholds. MPT has the highest probability of being cost-effective, with probabilities of 0.95 and 0.95, respectively.
Chapter 6 Assessment of factors relevant to the NHS and other parties
Bortezomib is already used as a monotherapy within the NHS for patients with relapsed MM and therefore oncology departments will have experience of administering this treatment. However, increased use of bortezomib will result in an increase in staff time to cover its administration. Some clinicians will also have experience of treating patients with thalidomide because of the UK-based MMIX RCT of CTDa versus MP. It is not clear whether there will be additional resource implications with increased use of thalidomide because of the requirement that it is prescribed and dispensed according to the Thalidomide Pharmion Pregnancy Prevention Programme.
Chapter 7 Discussion
Statement of principal findings
Clinical effectiveness
-
Five RCTs were included in the systematic review of clinical effectiveness. One examined the effectiveness of VMP, three examined the effectiveness of MPT, and one examined the effectiveness of CTDa. The comparator in all RCTs was MP. Two RCTs had a maintenance phase with thalidomide which followed the initial treatment phase. The maintenance phase did not meet the inclusion criteria. Reporting on the results of these RCTs was therefore limited to outcomes that had been reported at a time point prior to the start of maintenance therapy with thalidomide.
-
Judgements about aspects of study quality could not be made for some studies because of a lack of detailed reporting in the published papers. Consequently, there is uncertainty about some aspects of study quality. In particular, it was not possible to determine whether the amount and pattern of censoring in the RCTs was comparable between study groups.
-
Overall survival was increased in the intervention group in comparison to the groups receiving MP in both of the MPT versus MP RCTs that provided data for this outcome. Meta-analysis of the OS data from two RCTs of MPT versus MP confirmed the superiority of MPT and was in agreement with a published meta-analysis of three MPT versus MP trials. 109 OS was also increased in the single VMP versus MP RCT. Because OS data for the single RCT of VMP are not as mature as those for the two RCTs of MPT it was not possible to determine whether OS was greater with MPT or VMP.
-
More participants in the intervention arms of the included RCTs achieved a CR to treatment than in the MP comparator arms. The difference was reported to be statistically significant in four of the included studies, with a fifth study not reporting a p-value. It should be noted, however, that the proportion of participants achieving a CR to treatment was not assessed according to ITT principles in one RCT, which reported data for only approximately three-quarters of the enrolled participants, and the proportions of data missing from each trial arm appeared to be unequal but no explanation for this was provided. The remaining four RCTs reported results for approximately 95% or more of the participants and the proportion of data missing from each arm seemed comparable. A meta-analysis of the CR outcome data from three MPT versus MP RCTs confirmed that MPT was superior in comparison with MP in terms of the proportion of patients achieving a CR. As there were only single trials for VMP versus MP and for CTDa versus MP no meta-analyses for these comparisons were undertaken.
-
Progression-free survival was reported to be statistically significantly longer in the intervention group in comparison with the groups receiving MP in both of the MPT versus MP RCTs that provided data on this outcome, and the single VMP versus MP RCT. Only the RCT of VMP versus MP reported on time to disease progression, which was the primary outcome of this trial. There was a statistically significant difference in median time to disease progression in favour of the VMP group.
-
Adverse events occurred with all treatments. Some AEs were statistically significantly increased in trial intervention arms. The combination of bortezomib and MP was associated with a statistically significant increase in grade 3 AEs in comparison with the MP group. AE outcomes for thalidomide in combination with MP varied between the RCTs, but the two trials which reported on peripheral neuropathy reported a statistically significant increase in this AE in the treatment groups receiving thalidomide.
Cost-effectiveness
-
A systematic search of the literature found five abstracts of economic evaluations of treatment for patients with previously undiagnosed MM, ineligible for HDT–SCT. None of the studies contained sufficient information for critical appraisal.
-
A systematic search for published studies of QoL for patients with MM identified six studies: only two of these studies were for the population of interest and neither study included generic preference-based utility measures; the other four QoL studies provided utility estimates for patients with MM who had intensive therapy.
-
Two manufacturers submitted evidence to be considered for this review for bortezomib and thalidomide treatment. Janssen–Cilag, the manufacturer of bortezomib, constructed a survival model that estimated OS and PFS based on treatment effects from a MTC of the trials. The base-case results from the submission found all interventions to be cost-effective, with ICERs of less than £11,000 per QALY gained versus MP for MPT, VMP and CTDa.
-
Celgene, the manufacturer of thalidomide, constructed a Markov model with health states for preprogression (with or without AEs), post progression and death. The base-case results from the submission estimated MPT to be cost-effective compared with MP, while the ICER for VMP versus MPT was more than £300,000 per QALY gained.
-
The authors of this report developed an independent survival model. From this independent model, the incremental cost-effectiveness versus MP for MPT, VMP and CTDa is estimated as £9135, £29,820 and £33,031 per QALY gained, respectively. However, MPT dominated VMP as it was cheaper and more effective. The model results are most sensitive to changes in the parameter values of the HRs for OS. The PSA showed that MPT has the higher probability to be cost-effective at the £20,000 and £30,000 willingness-to-pay thresholds.
Discussion of cost-effectiveness results
The results for the manufacturers’ and SHTAC’s economic analyses are shown in Table 51. The results of the analyses vary considerably. The costs vary substantially between the analyses; for example the cost of MP varies between £1365 for the Celgene submission to £54,434 for the Janssen–Cilag submission. The costs from the Celgene analysis were lower as they had not included any subsequent treatment costs, whereas the SHTAC analysis included costs for second-line treatment and the Janssen–Cilag analysis included costs for second- and third-line treatment.
| Result | Analysis | MP | MPT | VMP | CTDa |
|---|---|---|---|---|---|
| Total cost (£) | SHTAC | 21,439 | 32,598 | 57,168 | 29,983 |
| Janssen–Cilag | 54,434 | 59,322 | 66,676 | 56,668 | |
| Celgene | 1365 | 21,133 | 42,616 | – | |
| Total QALY | SHTAC | 2.42 | 3.64 | 3.62 | 2.68 |
| Janssen–Cilag | 2.86 | 3.41 | 4.03 | 3.07 | |
| Celgene | 2.43 | 3.28 | 3.35 | – | |
| Incremental cost vs MP (£) | SHTAC | – | 11,159 | 35,729 | 8544 |
| Janssen–Cilag | – | 4888 | 12,242 | 2234 | |
| Celgene | – | 19,768 | 41,251 | – | |
| Incremental QALY vs MP | SHTAC | – | 1.22 | 1.20 | 0.26 |
| Janssen–Cilag | – | 0.55 | 1.17 | 0.21 | |
| Celgene | – | 0.85 | 0.92 | – | |
| ICER vs MP (£ per QALY) | SHTAC | – | 9135 | 29,820 | 33,031 |
| Janssen–Cilag | – | 8912 | 10,498 | 10,905 | |
| Celgene | – | 23,381 | 45,024a | – |
The incremental costs for MPT versus MP vary between £4888 (Janssen–Cilag) and £19,768 (Celgene). The Celgene submission uses higher dosages of thalidomide (238 mg/day) for longer periods (11 cycles) than the other two analyses. The incremental costs for VMP versus MP vary between £12,242 (Janssen–Cilag) and £41,251 (Celgene). These differences are largely due to the assumptions around the number of vials of bortezomib used, with Janssen–Cilag assuming a mean of (AiC/CiC information has been removed) vials used per person, whereas the mean number of vials used is over 40 in the SHTAC and Celgene economic evaluations. The incremental costs for CTDa versus MP vary between £2234 (Janssen–Cilag) and £8544 (SHTAC), and these differences are due to an error in the cost calculation for third-line therapy for CTDa in the Janssen–Cilag analysis.
The total QALY estimates between the studies are reasonably similar with estimates for all treatment arms varying between 2.42 and 4.03 QALY. The incremental QALY estimates for MPT versus MP vary widely and these differences are due to the estimates chosen for the HR for OS compared with MP. The incremental QALY estimates for MPT versus MP range from 0.55 (Janssen–Cilag) to 1.22 (SHTAC).
The different assumptions and methodology described above results in a range of estimates for the cost-effectiveness of the treatment options. The ICER for MPT versus MP varies between £9135 (SHTAC) and £23,381 (Celgene) per QALY gained. The ICER for VMP versus MP varies between £10,498 (Janssen–Cilag) and £44,838 (Celgene) per QALY gained. The ICER for CTDa versus MP varies between £10,905 (Janssen–Cilag) and £33,031 (SHTAC) per QALY gained.
The results have also been estimated for VMP versus MPT. These results also vary widely for the reasons given above. The ICER for VMP versus MPT was estimated as £11,907 (Janssen–Cilag) and £303,845 (Celgene) per QALY gained. For the SHTAC economic analysis, MPT dominated VMP, i.e. cheaper and more effective, for the base case and most sensitivity analyses.
Strengths and limitations of the assessment
The review has the following strengths:
-
The systematic review and economic evaluation have both been carried out independent of any vested interest, and the results are presented in a consistent and transparent manner.
-
The project was undertaken following the established methodology and principles for conducting a systematic review. The methods used were set out in a research protocol (see Appendix 1), which drew on the NICE scope to define the research question, inclusion and quality assessment criteria, data extraction process and the other methods to be used during the evidence synthesis. The research protocol was circulated to clinical experts and agreed with NICE before the project started.
-
An advisory group reviewed and commented on drafts of the protocol and the final report.
-
A de novo economic model has been developed following recognised guidelines. The main results have been summarised and presented. The model structure and data inputs are clearly presented in this report. This should facilitate replication and testing of our model assumptions.
-
Clinical evidence to populate the model has been extracted from reasonable quality RCTs included in the systematic review. The effect of treatment on OS and PFS was assessed using appropriate measures to model cost and outcome differences over the model time horizons.
In contrast, the review also has certain limitations:
-
Only two RCTs contributed data on OS following treatment with thalidomide and MP. The doses of thalidomide used differed between the two RCTs, as did the ages of the included participants, and the 72-week treatment period is not reflective of typical UK practice. It is therefore difficult to assess what the impact of MPT on OS would be when prescribed according to UK clinical practice to a typical MM patient in England and Wales.
-
Only one RCT contributed data on OS following treatment with bortezomib and MP and the published peer-reviewed follow-up data are immature. At the data-analysis cut-off date in the published paper not all patients had completed their assigned treatment.
-
No evidence on OS or PFS following treatment with CTDa met the inclusion criteria for the systematic review of clinical effectiveness because the only included RCT that assessed CTDa had a second randomisation to maintenance therapy with thalidomide for some participants after the completion of first-line treatment.
-
No head-to-head trials were identified which compared bortezomib in combination with an alkylating agent and a corticosteroid with thalidomide in combination with an alkylating agent and a corticosteroid.
-
Assessment of the impact of treatment on QoL was very limited. Data on HRQoL could only be included from one RCT, the study of VMP versus MP. Although one of the RCTs that assessed MPT versus MP reported on HRQoL these outcomes could not be included in the systematic review of clinical effectiveness because this RCT has included the use of thalidomide maintenance therapy in the later part of the RCT.
-
There were limited data available for meta-analysis. Furthermore, most studies did not report all the data items that were necessary to enable meta-analysis to be conducted. These missing data items were therefore estimated using published methods. A MTC was not carried out because of doubts about the validity of doing so due to potential differences in participant characteristics, delivery of MP treatment in the comparator arms, and length of follow-up. Furthermore, CTDa could not have been included in such an analysis because the single RCT that assessed CTDa included randomisation to maintenance therapy for some participants.
-
For pragmatic purposes in the economic model, analyses were included for CTDa, although the OS and PFS data included some patients who had received thalidomide maintenance from the MMIX RCT as no other data were available for CTDa.
-
Where possible, the data included in the model are in the public domain. However, some data for OS and PFS were extracted from an MS where these were not reported in sufficient detail in published sources and these are reported as AIC and CIC, as appropriate.
-
There were few HRQoL studies for the population of interest and these were only disease-specific HRQoL studies, using the EORTC QLQ-C30 measure. It was necessary to derive EQ-5D utility estimates using a mapping algorithm.
Uncertainties
-
It is not clear whether participants in the European trials reflect the population of patients that would receive these treatments in the UK. The participants in these trials, in general, had a better performance status than the participants in the UK MMIX clinical trial who are likely to more accurately reflect the typical UK MM patient who is ineligible for HDT with SCT.
-
It is not clear for OS and PFS outcomes how much data has been censored and for what reason. Therefore, it was not possible to determine whether the amount and pattern of censored data was comparable between the trial groups. Whether censoring had any effect on the reported outcomes is unknown.
-
Alterations in the doses of study drug were permitted in all studies and the target doses of thalidomide varied between the included RCTs. Although some trials provided some details on the duration and intensity of treatment it is not clear whether these dose alterations had a significant effect on the outcomes.
-
Duration of MPT treatment in the two IFM RCTs23,59 was longer than would be generally considered necessary or desirable in the UK. It is not certain what impact a shorter treatment period would have had on trial outcomes.
-
Very limited data from subgroup analyses were available for the comparisons of VMP versus MP, and CTDa versus MP, and no subgroup data were available from the RCTs of MPT versus MP. The outcomes from the available subgroup analyses should be interpreted with caution.
-
Concern was expressed by a clinical advisor that the incidence of AEs may be underestimated by the clinical trials. In particular, the incidence of peripheral neuropathy occurring with thalidomide was believed to be lower in the trials than that observed in UK clinical practice. Peripheral neuropathy, if it develops, can worsen quickly, and can be irreversible. This may be managed by dose modification or may limit the duration of treatment with thalidomide or with bortezomib for some patients and there can be a need for long-term treatment of neuropathic pain with gabapentin. Peripheral neuropathy can also preclude later treatment with bortezomib. Similarly the incidence of somnolence/dizziness/fatigue that occurs with thalidomide treatment may have been underestimated.
-
The second-line and other subsequent treatments received by participants in the included RCTs were variable. They did not reflect current UK practice in which most patients in the UK will receive bortezomib as their second-line therapy. This is owing to NICE guidance30 that recommends bortezomib only as a second-line therapy. The impact of second-line and later therapies on trial outcomes is unknown.
-
There is some uncertainty around the appropriate dosage for thalidomide. The daily dosages in the RCTs varied between 100 and 200 mg per patient. The SPC for thalidomide in the electronic Medicines Compendium states a daily dose of 200 mg. However, our clinical expert advised that, in practice, most patients will not be able to tolerate such a high dose and a lower dose of 100 mg is more common. In the economic analysis, we took the conservative assumption that the dose would be 150 mg. Lower dosages will result in more favourable cost-effectiveness estimates.
-
It is unclear what effect second-line and subsequent treatment has on patient survival and HRQoL. In the RCTs, there was a large number of different treatments given for second-line treatment. In the absence of appropriate data, we included second-line treatment as a cost and did not model its effect on health outcomes.
-
There was considerable heterogeneity in the reporting of AEs in the RCTs. For this reason, the cost of treating AEs was included in the model but any short-term utility decrements due to the AEs were not included.
-
The cost-effectiveness results for CTDa should be treated with caution, as the effectiveness estimates from the MMIX RCT include patients who received thalidomide maintenance therapy. (AiC/CiC information has been removed.)
Chapter 8 Conclusions
Implications for service provision
Service provision is unlikely to change greatly, although there will be additional intravenous administration to cover if bortezomib use is extended.
Suggested research priorities
Head-to-head trials of combination chemotherapy regimens containing bortezomib versus regimens containing thalidomide are desirable. For the results of such a trial to be easily generalisable to UK clinical practice drug doses and treatment periods should reflect those in widespread use in the UK. All trials of first-line therapy for MM in patients who are ineligible for HDT and SCT should include assessments of patient HRQoL in response to treatment.
The patients in the RCTs included in the clinical effectiveness review received a variety of second-line and subsequent treatments. This does not reflect current UK practice, which is that most patients receive bortezomib as their second-line therapy. If research is conducted to assess the impact of second-line treatments on patient outcomes it would also be desirable to assess whether the sequence of treatment, for example first-line therapy with a thalidomide-containing regimen followed by second-line treatment with a bortezomib-containing regimen, or vice versa, has any impact on patient outcomes.
Acknowledgements
We would like to thank members of our advisory group who provided expert advice and comments on the protocol and a draft of this report: Professor Graham Jackson, Honorary Clinical Professor of Clinical Haematology, Northern Institute for Cancer Research, Newcastle University (Professor Jackson was a Chief Investigator on the MMIX Trial); Dr Alastair Smith, Consultant Haematologist, Southampton General Hospital (Dr Smith has received honoraria from Ortho-Biotech and Celgene during the past 2 years for invited lectures, was a local investigator in the MMIX Trial, undertakes unpaid patient and carer education, and reviews research grant applications for Myeloma UK); Mrs Joan Smith, Secretary, North Manchester & Bury Myeloma Support Group; and Paul Tappenden, Senior Research Fellow, Health Economics and Decision Science, School of Health and Related Research (ScHARR), University of Sheffield.
We would also like to thank the MMIX Trial team for providing unpublished data for this report, in particular: Professor Anthony Child, Professor Gareth Morgan, Dr Sue Bell, Dr Walter Gregory, Mrs Kim Cocks and Mr Alex Szubert.
We are also grateful to Susan Baliss, Finance Manager – Costing, Southampton University Hospitals NHS Trust; Karen Welch, Information Scientist, SHTAC, University of Southampton, for generating and running the literature searches; and Jonathan Shepherd, Principal Research Fellow, SHTAC, for reviewing a draft of this report.
Contributions of authors
J Picot (Research Fellow) developed the research protocol, drafted the background section, assisted in the development of the search strategy, assessed studies for inclusion, extracted data from and quality assessed included studies, synthesised evidence, drafted and edited the final report, and project managed the study.
K Cooper (Senior Research Fellow) developed the research protocol, assessed studies for inclusion, extracted data from, and quality assessed, included studies, synthesised evidence, developed the economic evaluation and drafted the report.
J Bryant (Principal Research Fellow) developed the research protocol, assisted in the development of the search strategy, assessed studies for inclusion, extracted data from and quality assessed included studies, synthesised evidence and drafted the report.
AJ Clegg (Professor) developed the research protocol, assessed studies for inclusion, extracted data from, and quality assessed, included studies, synthesised evidence and drafted the report.
Disclaimers
The views expressed in this publication are those of the authors and not necessarily those of the HTA programme or the Department of Health.
References
- Cancer Research UK . Myeloma 2009. www.cancerhelp.org.uk/type/myeloma/about/what-is-myeloma (accessed 28 May 2010).
- Munshi NC, Longo DL, Anderson KC, Fauci AS, Kasper DL, Longo DL, et al. Harrison’s principles of internal medicine. London: McGraw-Hill; 2008.
- Ashcroft AJ, Davies FE, Morgan GJ. Aetiology of bone disease and the role of bisphosphonates in multiple myeloma. Lancet Oncol 2003;4:284-92.
- Dispenzieri A, Lacy MQ, Greipp PR, Greer JP, Foerster J, Rodgers GM, et al. Wintrobe’s clinical hematology. Philadelphia, PA: Lippincott, Williams & Wilkins; 2009.
- Munshi N, Anderson KC, Govindam R. Devita, Hellman & Rosenberg’s cancer: principles & practice of oncology. Philadelphia, PA: Lippincott, Williams & Wilkins; 2009.
- World Health Organization . International Classification of Diseases 10 (ICD-10) 2004.
- Gonzalez D, van der Burg M, Garcia-Sanz R, Fenton JA, Langerak AW, Gonzalez M, et al. Immunoglobulin gene rearrangements and the pathogenesis of multiple myeloma. Blood 2007;110:3112-21.
- Morgan GJ, Davies FE, Linet M. Myeloma aetiology and epidemiology. Biomed Pharmacother 2002;56:223-34.
- Fonseca R, Barlogie B, Bataille R, Bastard C, Bergsagel PL, Chesi M, et al. Genetics and cytogenetics of multiple myeloma: a workshop report. Cancer Res 2004;64:1546-58.
- Dispenzieri A, Rajkumar SV, Gertz MA, Fonseca R, Lacy MQ, Bergsagel PL, et al. Treatment of newly diagnosed multiple myeloma based on Mayo stratification of myeloma and riskadapted therapy (mSMART): consensus statement. Mayo Clin Proc 2007;82:323-41.
- Quinn M, Wood H, Cooper N, Rowan S. Cancer atlas of the United Kingdom and Ireland 1991–2000. Gosport: Palgrave Macmillan; 2005.
- Zangari M, van Rhee F, Anaissie E, Pineda-Roman M, Haessler J, Crowley J, et al. Eightyear median survival in multiple myeloma after total therapy 2: roles of thalidomide and consolidation chemotherapy in the context of total therapy 1. Br J Haematol 2008;141:433-44.
- Office for National Statistics (ONS) . Cancer Statistics Registrations. Registrations of Cancer Diagnosed in 2007, England 2010.
- Office for National Statistics . Cancer Incidence and Mortality in the United Kingdom and Constituent Countries, 2004–06 2009.
- National Cancer Institute . SEER Cancer Statistics Review, 1975–2006 2009. http://seer.cancer.gov/csr/1975_2006/.
- Statistical Information Team. Cancer Research UK; 2009.
- NHS Choices . Multiple Myeloma n.d. www.nhs.uk/Conditions/Multiple-myeloma/Pages/ (accessed 23 November 2009).
- Kristinsson SY, Goldin LR, Björkholm M, Koshiol J, Turesson I, Landren O. Genetic and immune-related factors in the pathogenesis of lymphoproliferative and plasma cell malignancies. Haematologica 2009;94:1581-9.
- Durie BG, Salmon SE. A clinical staging system for multiple myeloma. Correlation of measured myeloma cell mass with presenting clinical features, response to treatment, and survival. Cancer 1975;36:842-54.
- Greipp PR, San MJ, Durie BG, Crowley JJ, Barlogie B, Blade J, et al. International staging system for multiple myeloma. J Clin Oncol 2005;23:3412-20.
- National Institute for Health and Clinical Excellence (NICE) . Bortezomib and Thalidomide for the First-Line Treatment of Multiple Myeloma n.d.
- Smith A, Wisloff F, Samson D, UK MF. Nordic Myeloma Study Group, British Committee for Standards in Haematology . Guidelines on the diagnosis and management of multiple myeloma 2005. Br J Haematol 2006;132:410-51.
- Facon T, Mary JY, Hulin C, Benboubker L, Attal M, Pegourie B, et al. Melphalan and prednisone plus thalidomide versus melphalan and prednisone alone or reduced-intensity autologous stem cell transplantation in elderly patients with multiple myeloma (IFM 99–06): a randomised trial. Lancet 2007;370:1209-18.
- Palumbo A, Bringhen S, Caravita T, Merla E, Capparella V, Callea V, et al. Oral melphalan and prednisone chemotherapy plus thalidomide compared with melphalan and prednisone alone in elderly patients with multiple myeloma: randomised controlled trial. Lancet 2006;367:825-31.
- Palumbo A, Bringhen S, Liberati AM, Caravita T, Falcone A, Callea V, et al. Oral melphalan, prednisone, and thalidomide in elderly patients with multiple myeloma: updated results of a randomized controlled trial. Blood 2008;112:3107-14.
- San Miguel JF, Schlag R, Khuageva NK, Dimopoulos MA, Shpilberg O, Kropff M, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med 2008;359:906-17.
- Low E. MRC Myeloma IX. Edinburgh: Myeloma UK; 2006.
- Bird J, Owen R, d’Sa S, Snowden J, Littlewood T, Pratt G, et al. Guidelines on the diagnosis and management of multiple myeloma. London: UKMF/BCSH; 2010.
- European Medicines Agency (EMEA) . Summary of Product Characteristics for Thalidomide Celgene 2009.
- National Institute for Health and Clinical Excellence (NICE) . Bortezomib Monotherapy for Relapsed Multiple Myeloma (TA 129) 2007.
- National Institute for Health and Clinical Excellence (NICE) . Lenalidomide for the Treatment of Multiple Myeloma in People Who Have Received at Least One Prior Therapy (TA 171) 2009.
- National Institute for Health and Clinical Excellence (NICE) . Guidance on Cancer Services – Improving Outcomes in Haematological Cancers – the Manual 2003.
- European Medicines Agency (EMEA) . Summary of Product Characteristics for Velcade 2009.
- European Medicines Agency (EMEA) . Assessment Report for Velcade 2008.
- British Medical Association and Royal Pharmaceutical Society of Great Britain . British National Formulary. 2009.
- Jenkins JS, Sampson PA. Conversion of cortisone to cortisol and prednisone to prednisolone. Br Med J 1967;2:205-7.
- Singhal S, Mehta J, Desikan R, Ayers D, Roberson P, Eddlemon P, et al. Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med 1999;341:1565-71.
- Teicher BA, Ara G, Herbst R, Palombella VJ, Adams J. The proteasome inhibitor PS-341 in cancer therapy. Clin Cancer Res 1999;5:2638-45.
- Blade J, Samson D, Reece D, Apperley J, Bjorkstrand B, Gahrton G, et al. Criteria for evaluating disease response and progression in patients with multiple myeloma treated by high-dose therapy and haemopoietic stem cell transplantation. Myeloma Subcommittee of the European Group for Blood and Marrow Transplant. Br J Haematol 1998;102:1115-23.
- Centre for Reviews and Dissemination (CRD) . Systematic Reviews. CRD’s Guidance for Undertaking Reviews in Health Care 2009.
- Tierney JF, Stewart LA, Ghersi D, Burdett S, Sydes MR. Practical methods for incorporating summary time-to-event data into meta-analysis. Trials 2007;8.
- Davies FE, Child J, Hawkins K, Bell S, Brown J, Drayson MT, et al. Newly diagnosed myeloma pateints are at risk of venous thrombotic events: high risk patients need to be identified and receive thromboprophylaxis: the MRC experience. Blood 2004;104.
- Morgan GJ, Jackson GH, Davies FE, Drayson MT, Owen RG, Gregory WM, et al. Maintenance thalidomide may improve progression free but not overall survival; results from the Myeloma IX Maintenance Randomisation. Blood 2008;112.
- Morgan GJ, Davies FE, Owen RG, Rawstron AC, Bell S, Cocks K, et al. Thalidomide combinations improve response rates: results from the MRC IX study. Blood 2007;110.
- Morgan GJ, Davies FE, Gregory WM, Bell SE, Szubert AJ, Cocks K, et al. The Addition of Thalidomide to the Induction Treatment of Newly Presenting Myeloma Patients Increases the CR Rate Which Is Likely to Translate into Improved PFS and OS n.d.
- Owen RG, Rawstron AC, Drayson MT, Davies FE, Jackson GH, Ross FM, et al. MRD studies in MM: data from the MRC Myeloma IX trial. Clin Lymphoma Myeloma 2009;9.
- Owen RG, Child JA, Jackson GH, Davies FE, Drayson MT, Ross FM, et al. MRC Myeloma IX: Preliminary Results from the Intensive Pathway 2009.
- Owen RG, Morgan GJ, Jackson H, Davies FE, Drayson MT, Ross FM, et al. MRC Myeloma IX: Preliminary Results from the Non-Intensive Study 2009.
- Medical Research Council (MRC) Myeloma IX . Myelomatosis Therapy Trial for Patients of All Age Groups 2009.
- MRC Myeloma IX Trial Background Information: Induction Chemotherapy for Non-Intensive Pathway Only 2009.
- MRC Myeloma IX Trial Background Information: Randomisation to Maintenance 2009.
- MRC Myeloma IX Trial Baseline Data: Information on Induction Chemotherapy for Nonintensive Pathway Only 2009.
- Interpretation of the MRC ‘Myeloma IX’ Results for the Non-Transplant (non-Intensive) Group 2009.
- MRC Myeloma IX Trial Outcomes: Results from the Non-Transplant (non-Intensive) Group 2009.
- Myeloma IX EQ-5D and EORTC QLQ-C30 Data Summary for NICE 2009.
- MRC Myeloma IX Trial Outcomes: EQ-5D Summaries from the Non-Transplant (nonintensive) Group 2009.
- MRC Myeloma IX Trial Outcomes: EORTC QLQ C30 Summaries from the Non-Transplant (non-Intensive) Group 2009.
- MRC Myeloma IX Trial Outcomes: EORTC QLQ C30 Graphs from the Non-Transplant (nonintensive) Group 2009.
- Hulin C, Facon T, Rodon P, Pegourie B, Benboubker L, Doyen C, et al. Efficacy of melphalan and prednisone plus thalidomide in patients older than 75 years with newly diagnosed multiple myeloma: IFM 01/01 trial. J Clin Oncol 2009;27:4848-57.
- San Miguel JF, Schlag R, Khuageva NK, Dimopoulos MA, Shpilberg O, Kropff MH, et al. Updated follow-up and results of subsequent therapy in the Phase III VISTA trial: bortezomib plus melphalan-prednisone versus melphalan-prednisone in newly diagnosed multiple myeloma. Blood 2008;112.
- Mateos M-V, Richardson PG, Schlag R, Khuageva NK, Dimopoulos MA, Shpilberg O, et al. Bortezomib Plus melphalan–prednisone Continues to Demonstrate a Survival Benefit Vs melphalan–prednisone in the Phase III VISTA Trial in Previously Untreated Multiple Myeloma After 3 years’ Follow-up and Extensive Subsequent Therapy Use n.d.
- Dhawan R, Robinson D, Meunier J, Regnault A, Rosa K, Cakana AZ, et al. Sustained Health-Related Quality of Life (HRQoL) Improvement in Newly Diagnosed Multiple Myeloma Patients Treated With Bortezomib Melphalan Prednisone Versus Melphalan Prednisone: Results from the VISTA Trial n.d.
- Gulbrandsen N, Waage A, Gimsing P, Turesson I, Juliusson G, Abildgaard N, et al. A randomised placebo controlled study with melphalan/prednisone vs melphalan/prednisone/ thalidomide: quality of life and toxicity. Haematologica 2008;93.
- Waage A, Gimsing P, Juliusson G, Turesson I, Fayers P. Melphalan-prednisone-thalidomide to newly diagnosed patients with multiple myeloma: a placebo controlled randomised phase 3 trial. Blood 2007;110.
- Wijermans P, Schaafsma M, van Norden Y, Ammerlaan R, Wittebol S, Sinnige H, et al. Melphalan plus prednisone versus melphalan plus prednisone plus thalidomide in induction therapy for multiple myeloma in elderly patients: final analysis of the Dutch Cooperative Group HOVON 49 Study. Blood 2008;112:241-2.
- Wijermans P, Schaafsma M, Van Norden Y, Ammerlaan R, Sonneveld P, Wittebol S, et al. Melphalan + prednisone vs melphalan + prednisone + thalidomide in induction therapy for multiple myeloma in elderly patients: first interim results of the Dutch cooperative group HOVON. Haematologica 2008;93.
- Wijermans PW, Zweegman S, van Marwijk KM, van der Griend R, Lokhorst H, Schaafsma M, et al. MP versus MPT in elderly myeloma patients: the final outcome of the HOVON-49 study. Clin Lymphoma Myeloma 2009;9.
- Sacchi S, Marcheselli R, Pozzi S, Bari A, Ciarcia O, Masini L, et al. A randomized phase II study (GISL-MM03 Trial) with oral melphalan plus prednisone (MP) versus melphalan, plus prednisone plus thalidomide (MPT) for newly diagnosed elderly patients with multiple myeloma. Haematologica 2009;94.
- Beksac M, Haznedar R, Firatli-Tuglular T, Ozdogu H, Aydogdu I, Konuk N, et al. Addition of Thalidomide (T) to Oral Melphalan Prednisone (MP) in Patients With Multiple Myeloma: Initial Results of a Randomized Trial from the Turkish Myeloma Study Group n.d.
- Sampson FC, Beard SM, Scott F, Vandenberghe E. Cost-effectiveness of high-dose chemotherapy in first-line treatment of advanced multiple myeloma. Br J Haematol 2001;113:1015-19.
- Cecchi M, Caccese E, Messori A, Orsi C, Tendi E. Cost-effectiveness of bortezomib in multiple myeloma. Pharm World Sci 2007;29:485-6.
- Deniz B, Facon T, Singer I, Micallef-Eynaud P, Joseph I, Shearer A, et al. Economic evaluation of thalidomide combined with melphalan and prednisone in previously untreated multiple myeloma in Scotland. Blood 2008;112.
- Yoong K, Attard C, Jivraj F, Shustik C, Reece D. Cost effectiveness analysis of bortezomib in previously untreated multiple myeloma patients in Canada. Value Health 2009;12.
- Joseph I, Facon T, Lewis P, Deniz HB, Caro JJ. Cost effectiveness of thalidomide combined with melphalan and prednisone in previously untreated myltiple myeloma in Wales. Value Health 2009;12.
- De Abreu Lourenco R, Colman S, Lee C. Thalidomide plus melphan and prednisone for Australian patients newly diagnosed with multiple myeloma is cost effective when compared with melphalan and prednisone alone. Value Health 2009;12.
- Wang S, Huang H, Shi H, Duh M, Chen K. The Cost Effectiveness of Bortezomib for the Initial Treatment of Multiple Myeloma in the United States n.d.
- Brazier J, Ratcliffe J, Salomon JA, Tsuchiya A. Measuring and valuing health benefits for economic evaluation. Oxford: Oxford University Press; 2007.
- National Institute for Health and Clinical Excellence (NICE) . Guide to the Methods of Technology Appraisal 2008.
- Sherman AC, Simonton S, Latif U, Plante TG, Anaissie EJ. Changes in quality-of-life and psychosocial adjustment among multiple myeloma patients treated with high-dose melphalan and autologous stem cell transplantation. Biol Blood Marrow Transplant 2009;15:12-20.
- Lee SJ, Richardson PG, Sonneveld P, Schuster MW, Irwin D, San Miguel JF, et al. Bortezomib is associated with better health-related quality of life than high-dose dexamethasone in patients with relapsed multiple myeloma: results from the APEX study. Br J Haematol 2008;143:511-19.
- Sherman AC, Simonton S, Latif U, Spohn R, Tricot G. Psychosocial adjustment and quality of life among multiple myeloma patients undergoing evaluation for autologous stem cell transplantation. Bone Marrow Transplant 2004;33:955-62.
- Gulbrandsen N, Wisloff F, Brinch L, Carlson K, Dahl IM, Gimsing P, et al. Health-related quality of life in multiple myeloma patients receiving high-dose chemotherapy with autologous blood stem-cell support. Med Oncol 2001;18:65-77.
- Multiple myeloma: QALY gains from optimal therapy. Drugs Ther Perspect 2000;16:12-6.
- Ellis K, Smith AG. An evaluation of quality of life (QOL) in patients after treatment for multiple myeloma (MM). Br J Haematol 2005;129.
- Thomas ML. Quality of life in persons with multiple myeloma: a descriptive study. Blood 2001;98.
- Deniz B, Morgan G, Schey S, Ishak J, Dale P, Shearer A, et al. Economic evaluation of lenalidomide combined with dexamethasone for the treatment of multiple myeloma in the UK. Blood 2008;112:836-7.
- Belch A, Reece DE, Bahlis NJ, White D, Teixeira B, Camacho F, et al. Bortezomib [VELCADE (TM)], pegylated liposomal doxorubicin [DOXIL/CAELYX (R)] and dexamethasone in the treatment of previously untreated multiple myeloma patients: impact on quality-of-life. Blood 2007;110:1058-9A.
- Dhawan R, Meunier J, Regnault A, Robinson D, Rosa K, Cakana A, et al. Impact of complete response on quality of life in newly diagnosed multiple myeloma patients. Clinical Lymphoma Myeloma 2009;9.
- Meunier J, Regnault A, Robinson D, Rosa K, San Miguel JF, van de Velde H, et al. Impact of tumor response on health-related quality of life (HRQoL) in newly diagnosed multiple myeloma patients treated with velcade/melphalan/prednisone (V-MP): results from the VISTA trial. Value Health 2009;12.
- Petrucci MT, Calabrese E, Levi A, Federico V, Ceccolini M, Rizzi R, et al. Costs and quality of life of multiple myeloma (MM) in Italy: the Co.Mim Study. Value Health 2009;12.
- Gulbrandsen N, Hjermstad MJ, Wisloff F. Nordic Myeloma Study Group . Interpretation of quality of life scores in multiple myeloma by comparison with a reference population and assessment of the clinical importance of score differences. Eur J Haematol 2004;72:172-80.
- Strasser-Weippl K, Ludwig H. Psychosocial QOL is an independent predictor of overall survival in newly diagnosed patients with multiple myeloma. Eur J Haematol 2008;81:374-9.
- van Agthoven M, Segeren CM, Buijt I, Uyl-de Groot CA, van der Holt B, Lokhorst HM, et al. A cost–utility analysis comparing intensive chemotherapy alone to intensive chemotherapy followed by myeloablative chemotherapy with autologous stem-cell rescue in newly diagnosed patients with stage II/III multiple myeloma: a prospective randomised phase III study. Eur J Cancer 2004;40:1159-69.
- Mujica-Mota R, Bagust A, Haycox A, Dhawan R, Dubois D. Mapping health-related quality of life (HRQOL) measurements into generic utility measures (EQ-5D): a case study with bortezomib (VELCADE). Value Health 2004;7.
- Slovacek L, Slovackova B, Pavlik V, Hrstka Z, Macingova Z, Jebavy L, et al. Health-related quality of life in multiple myeloma survivors treated with high dose chemotherapy followed by autologous peripheral blood progenitor cell transplantation: a retrospective analysis. Neoplasma 2008;55:350-5.
- Uyl-de Groot CA, Buijt I, Gloudemans IJ, Ossenkoppele GJ, Berg HP, Huijgens PC. Health related quality of life in patients with multiple myeloma undergoing a double transplantation. Eur J Haematol 2005;74:136-43.
- Gelber RD, Goldhirsch A, Cole BF. Evaluation of effectiveness: Q-TWiST. The International Breast Cancer Study Group. Cancer Treat Rev 1993;19:73-84.
- Philips Z, Ginnelly L, Sculpher M, Claxton K, Golder S, Riemsma R, et al. Review of guidelines for good practice in decision-analytic modelling in health technology assessment. Health Technol Assess 2004;8.
- Monthly Index of Medical Specialties (MIMS). London: Haymarket Medical Media Ltd; 2009.
- Department of Health . NHS Reference Costs 2008.
- Wilson J, Yao GL, Raftery J, Bohlius J, Brunskill S, Sandercock J, et al. A systematic review and economic evaluation of epoetin alpha, epoetin beta and darbepoetin alpha in anaemia associated with cancer, especially that attributable to cancer treatment. Health Technol Assess 2007;11.
- Curtis L. Unit costs of health and social care. University of Kent, Canterbury: Personal Social Services Research Unit; 2008.
- Briggs A, Sculpher M, Claxton K. Decision modelling for health economic evaluation. Oxford: Oxford University Press; 2006.
- Engauge Digitizer – digitizing software n.d.
- Johnson & Johnson Pharmaceutical Research & Development and Millennium: The Takeda Oncology Company . Clinical Study Report 3: Survival and Subsequent Therapy Update to Clinical Study Report MMY-3002 2009.
- Kontodimopoulos N, Aletras VH, Paliouras D, Niakas D. Mapping the cancer-specific EORTC QLQ-C30 to the preference-based EQ-5D, SF-6D, and 15D instruments. Value Health 2009;12:1151-7.
- McKenzie L, van der Pol M. Mapping the EORTC QLQ C-30 onto the EQ-5D instrument: the potential to estimate QALYs without generic preference data. Value Health 2009;12:167-71.
- Contracting Unit, Southampton University Hospital Trust 2009.
- Hicks LK, Haynes AE, Reece DE, Walker IR, Herst JA, Meyer RM, et al. A meta-analysis and systematic review of thalidomide for patients with previously untreated multiple myeloma. Cancer Treat Rev 2008;34:442-52.
- Mehta J, Duff SB, Gupta S. Cost effectiveness of bortezomib in the treatment of advanced multiple myeloma. Manag Care Interface 2004;17:52-61.
- Kumar SK, Therneau TM, Gertz MA, Lacy MQ, Dispenzieri A, Rajkumar SV, et al. Clinical course of patients with relapsed multiple myeloma. Mayo Clin Proc 2004;79:867-74.
- Dimopoulos MA, Richardson P, Schlag R, Khuageva NK, Shpilberg O, Kastritis E, et al. Bortezomib-melphalan-prednisone (VMP) in newly diagnosed multiple myeloma patients with impaired renal function: cohort analysis of the Phase III VISTA study. Blood 2008;112:608-9.
- Harousseau JL, Palumbo A, Richardson P, Schlag R, Dimopoulos MA, Shpilberg O, et al. Superior outcomes associated with complete response: analysis of the Phase III VISTA study of bortezomib plus melphalan-prednisone versus melphalan-prednisone. Blood 2008;112.
- San Miguel J, Schlag R, Khuageva N, Dimopoulos M, Shpilberg O, Kropff M, et al. Superior efficacy with bortezomib plus melphalan-prednisone (VMP) versus melphalan-prednisone (MP) alone in previously untreated multiple myeloma (MM): results of the Phase III MMY- 3002 VISTA study. Haematologica 2008;93.
- Janssen–Cilag . Bortezomib and Thalidomide for the First-Line Treatment of Multiple Myeloma: Bortezomib (Velcade) 2009.
- Celgene Ltd . Multiple Technology Appraisal of Bortezomib and Thalidomide for the First-Line Treatment of Multiple Myeloma 2009.
- Celgene Ltd . A Survey of Resource Utilisation Among Haematologists in Wales 2007.
- Drummond MF, Jefferson TO. Guidelines for authors and peer reviewers of economic submissions to the BMJ. The BMJ Economic Evaluation Working Party. BMJ 1996;313:275-83.
Appendix 1 Report methods for synthesis of evidence of clinical effectiveness and cost-effectiveness as described in the research protocol
A systematic review of the evidence for clinical effectiveness and cost-effectiveness will be undertaken following the general principles outlined in Systematic reviews: CRD’s guidance for undertaking reviews in health care. 40
Search strategy
A search strategy will be developed and tested by an experienced information specialist. The strategy will be designed to identify studies reporting clinical effectiveness, cost-effectiveness, HRQoL, resource use and costs, epidemiology and natural history.
A draft search strategy for MEDLINE will be adapted for other databases. Literature will be identified from several sources, including electronic databases, bibliographies of articles, and grey literature sources. Reference lists contained within manufacturers’ submissions to NICE will be searched for any additional studies that meet the inclusion criteria. Experts will be contacted to identify additional published and unpublished references. A comprehensive database of relevant published and unpublished articles will be constructed using Reference Manager software.
All databases will be searched from 1999 (earliest use of thalidomide for MM37 and earliest description of bortezomib as a potential cancer therapy38) to the current date. Searches will be restricted to English language and updated around December 2009.
Inclusion and exclusion criteria
| Interventions |
|
| Participants |
People with previously untreated MM who are not candidates for HDT with SCT (Studies of patients with MM who have received previous treatment(s) will not be included) |
| Comparators | Interventions described above will be compared with each other and the following comparators:
|
| Outcomes | Studies will be included if they report on one or more of the following outcomes:
|
| Design |
The following types of study will be eligible for inclusion:Studies published as abstracts or conference presentations will be included only if sufficient details are presented to allow an appraisal of the methodology and the assessment of results to be undertaken Systematic reviews and clinical guidelines will be used as a source of references Case series, case studies, narrative reviews, editorials and opinions will be excluded Non-English-language studies will be excluded |
Inclusion and data extraction process
Studies will be selected for inclusion through a two-stage process. Literature search results (titles and abstracts) will be screened independently by two reviewers to identify all citations that may meet the inclusion criteria. Full manuscripts of selected citations will be retrieved and assessed by one reviewer against the inclusion/exclusion criteria and checked independently by a second reviewer. Discrepancies will be resolved by discussion, with involvement of a third reviewer when necessary.
Data will be extracted by one reviewer using a standardised data extraction form and will be checked for accuracy by a second reviewer. Discrepancies will be resolved by discussion, with involvement of a third reviewer when necessary.
Quality assessment strategy
The quality of included clinical effectiveness studies will be assessed using NHS CRD (University of York) criteria. 40 Methodological quality of economic evaluations will be assessed based on recognised criteria for appraising economic evaluations. 80,110 Quality criteria will be applied by one reviewer and checked by a second reviewer with any disagreements resolved by consensus and involvement of a third reviewer where necessary.
Methods of analysis/synthesis
Clinical effectiveness and cost-effectiveness studies will be synthesised through a narrative review with tabulation of results of included studies. Where appropriate the results from individual clinical effectiveness studies will be synthesised through meta-analysis, with causes of heterogeneity of results examined. The systematic review may explore the possibility of conducting an indirect comparison of thalidomide and bortezomib used in combination with an alkylating agent and a corticosteroid versus a common comparator. The specific methods for meta-analysis and for the detection and investigation of heterogeneity will depend upon the particular outcome measure under consideration.
Report methods for economic analysis
The cost-effectiveness of bortezomib or thalidomide used in combination with an alkylating agent and a corticosteroid for first-line treatment of MM will be assessed through a review of previous cost-effectiveness studies and, if appropriate, through the development of a decision-analytic model. The purpose of the review is to identify recent relevant evaluations, in order to analyse the methodological approaches undertaken, and to discern whether, and how, existing models can be adapted for use in the current project.
Model structure
Where necessary, a de novo decision-analytic model will be developed to assess the cost-effectiveness of bortezomib and thalidomide. The exact structure of the model will be designed to reflect important clinical events over the course of the disease and will be validated through discussion with expert advisors. Modelling will be conducted according to accepted methodology for economic evaluations. 78,98 The perspective will be the NHS and PSS. Costs and benefits will be discounted using standard rates (3.5%). 78 The model will be developed using standard software such as Microsoft Excel and Tree-Age Pro.
The model will contain a hypothetical cohort of individuals and will estimate changes in disease progression, morbidity and mortality for the MM treatments under consideration. The time horizon for the model will be 15 years, which, for the majority of patients in the hypothetical cohort, is likely to be equivalent to a lifetime horizon.
Whilst de novo modelling is planned, the possibility of adapting an existing published model along the lines of the proposed model will be explored through contact with experts in the field.
Clinical effectiveness data
The parameters of the model will be informed primarily by the systematic review of effectiveness studies. Additional targeted searches will be undertaken to identify specific data to populate the model. These will include searches for data on the epidemiology and natural history of MM; the HRQoL impacts of disease stages and the adverse effects of treatment; the cost of treatment and health-care costs. Where these data cannot be identified through searches, estimates will be based on information supplied by our expert advisory group and others.
Baseline disease progression will be predicted using trial data where available or good-quality observational studies (such as the Mayo Clinic study, which has followed cohorts of patients with MM over a 13-year period111). Treatment effect will be modelled over time by adjusting the baseline prediction of treatment pathway and disease progression, based upon reported hazard ratios (HRs) in the systematic review for time to progression to more severe states, and OS.
Costs and resource estimation
The resources necessary for providing the treatments will be estimated from the systematic review of effectiveness, and from discussion with expert advisers. Unit costs for these resources will be developed based on data in published sources such as the Unit Costs of Health and Social Care, PSS Research Unit (PSSRU). 102 Data on the cost of assessing and treating MM will be sought from Southampton University Hospitals Trust (SUHT), which routinely supplies SHTAC with cost data and clinical expertise. Information on resource use and costs will also be derived from sponsor submissions to NICE, as appropriate.
Outcomes
The model will provide a cost-effectiveness analysis, reporting the costs of treatments under consideration in the appraisal and their long-term consequences in terms of life-years saved and QALYs gained and additional costs. Results will be expressed in terms of ICERs (e.g. incremental costs per QALY gained).
Uncertainty in model parameters and structure will be investigated through one-way deterministic and probabilistic sensitivity analyses where appropriate and feasible. The key variables to be explored will include treatment effect estimates (e.g. OS and disease progression), baseline disease progression estimate, treatment costs and HRQoL. Cost-effectiveness acceptability curves (CEACs) will be generated in any probabilistic sensitivity analysis (PSA) to illustrate the probability of the treatment being cost-effective over a range of willingness-to-pay values.
Handling the company submission(s)
All data submitted by the manufacturers/sponsors will be considered if received by the technology assessment report (TAR) team no later than 15 October 2009. Data arriving after this date will not be considered. If the data meet the inclusion criteria for the review they will be extracted and quality assessed in accordance with the procedures outlined in this protocol. Any economic evaluation included in the company submission, provided it complies with NICE’s advice on presentation, will be assessed for clinical validity, reasonableness of assumptions and appropriateness of the data used in the economic model.
Any ‘commercial-in-confidence’ data taken from a company submission will be underlined and highlighted in red in the assessment report (followed by an indication of the relevant company name in brackets unless it is obvious from the context). Any ‘academic-in-confidence’ data will be highlighted in yellow.
Appendix 2 Example MEDLINE search strategies for clinical effectiveness and cost-effectiveness
Clinical effectiveness
-
(bortezomib or velcade).mp.
-
thalidomid*.mp.
-
thalidomide/
-
or/1-3
-
exp multiple myeloma/
-
exp Plasmacytoma/
-
exp Paraproteinemias/
-
(myeloma* or (multiple adj myeloma*) or plasmacytom* or plasmocytom* or MGUS or (monoclonal adj gammopath*)).mp.
-
or/5-8
-
4 and 9
-
randomized controlled trial/
-
randomized controlled trial.pt.
-
controlled clinical trial/
-
controlled clinical trial.pt.
-
clinical trial.pt.
-
exp Clinical Trial/
-
random*.tw.
-
exp Research Design/
-
(systematic$adj2 review$).mp.
-
(systematic$adj2 overview$).mp.
-
(meta analy* or metaanaly*).ti,ab,pt.
-
exp meta analysis/
-
((hand or manual or computer or electronic or database) adj2 search*).ti,ab.
-
(open adj label*).tw.
-
double-blind method/
-
single-blind method/
-
((singl* or doubl* or tripl* or trebl*) adj5 (blind* or mask*)).tw.
-
exp cohort studies/
-
cohort*.ti,ab.
-
or/11-29
-
10 and 30
-
limit 31 to (english language and humans and yr=“1999 -Current”)
-
(editorial or comment or letter).pt.
-
32 not 33
-
from 34 keep 1-381
Cost-effectiveness
-
exp economics/
-
exp economics hospital/
-
exp economics pharmaceutical/
-
exp economics nursing/
-
exp economics medical/
-
exp “Costs and Cost Analysis”/
-
Cost Benefit Analysis/
-
value of life/
-
exp models economic/
-
exp fees/and charges/
-
exp budgets/
-
(value adj2 (money or monetary)).tw.
-
(economic adj2 burden).tw.
-
(expenditure* not energy).tw.
-
budget*.tw.
-
(economic* or price* or pricing or financ* or fee* or pharmacoeconomic* or pharma economic* or pharmaco-economic*).tw.
-
(decision adj1 (tree* or analys* or model*)).tw.
-
Resource Allocation/
-
(unit cost or unit-cost or unit-costs or unit costs or drug cost or drug costs or hospital costs or health-care costs or health care cost or medical cost or medical costs).tw.
-
((value or values or valuation) adj2 (money or monetary or life or lives or costs or cost)).tw.
-
(cost adj2 (util* or effective* or efficac* or benefit* or cosequence* or analys* or minimi* or saving* or breakdown* or lowering or estimate* or variable* or allocation* or control* or illness* or affordable* or instrument* or technolog* or fee* or charge* or charges)).tw.
-
Markov Chains/
-
Monte Carlo Method/
-
exp Decision Support Techniques/
-
(resource adj2 (use* or utili* or allocat*)).tw.
-
or/1-25
-
(bortezomib or velcade).mp.
-
thalidomid*.mp.
-
thalidomide/
-
or/27-29
-
exp multiple myeloma/
-
exp Plasmacytoma/
-
exp Paraproteinemias/
-
(myeloma* or (multiple adj myeloma*) or plasmacytom* or plasmocytom* or MGUS or (monoclonal adj gammopath*)).mp.
-
or/31-34
-
26 and 30 and 35
-
multiple myeloma/ec
-
*multiple myeloma/
-
26 and 38
-
“multiple myeloma”.ti.
-
26 and 40
-
36 or 37 or 39 or 41
-
limit 42 to (english language and humans and yr=“1999 -Current”)
-
(editorial or comment or letter).pt.
-
43 not 44
Appendix 3 Response criteria
| EBMT, IBMTR and ABMTR criteriaa | IFM criteriab | |
|---|---|---|
| CR requires all of the following: | Absence of the original monoclonal paraprotein in serum and urine by immunofixation, maintained for a minimum of 6 weeks. The presence of oligoclonal bands consistent with oligoclonal immune reconstitution does not exclude CR | Absence of the original monoclonal protein in serum and urine by immunofixation. No confirmation needed |
| Less than 5% plasma cells in a bone marrow aspirate and also on trephine bone biopsy, if biopsy is performed. If absence of monoclonal protein is sustained for 6 weeks it is not necessary to repeat the bone marrow, except in patients with non-secretory myeloma where the marrow examination must be repeated after an interval of at least 6 weeks to confirm CR | Less than 5% of plasma cells in a bone marrow aspirate. No confirmation needed | |
| No increase in size or number of lytic bone lesions (development of a compression fracture does not exclude response) | ||
| Disappearance of soft tissue plasmacytomas | Disappearance of soft tissue plasmacytomas | |
| VGPR | More than a 90% decrease in monoclonal protein in serum and urine. No confirmation needed | |
| PR requires all of the following: | More than 50% reduction in the level of the serum monoclonal paraprotein, maintained for a minimum of 6 weeks | More than a 50% reduction in the concentration of serum monoclonal protein. No confirmation needed |
| Reduction in 24-hour urinary light chain excretion either by > 90% or to < 200 mg, maintained for a minimum of 6 weeks | More than a 75% reduction in 24-hour urinary light chain excretion. No confirmation needed | |
| For patients with non-secretory myeloma only, > 50% reduction in plasma cells in a bone marrow aspirate and on trephine biopsy, if biopsy is performed, maintained for a minimum of 6 weeks | ||
| More than 50% reduction in the size of soft tissue plasmacytomas (by radiography or clinical examination) | Reduction in the size of soft tissue plasmacytomas | |
| No increase in size or number of lytic bone lesions (development of a compression fracture does not exclude response) | ||
| MR | 25–49% reduction in the level of the serum monoclonal paraprotein maintained for a minimum of 6 weeks | |
| 50–89% reduction in 24-hour urinary light chain excretion, which still exceeds 200 mg/24 hours, maintained for a minimum of 6 weeks | ||
| For patients with non-secretory myeloma only, 25–49% reduction in plasma cells in a bone marrow aspirate and on trephine biopsy, if biopsy is performed, maintained for a minimum of 6 weeks | ||
| 25–49% reduction in the size of soft tissue plasmacytomas (by radiography or clinical examination) | ||
| No increase in the size or number of lytic bone lesions (development of a compression fracture does not exclude response) | ||
| EBTM – no change, IFM – stable disease | Not meeting the criteria of either MR or progressive disease | Not meeting the criteria of CR, PR or progressive disease |
| Plateau | Stable values (within 25% above or below value at the time response is assessed) maintained for at least 3 months | |
| Relapse from CR; requires at least one of the following: | Reappearance of serum or urinary paraprotein on immunofixation or routine electrophoresis, confirmed by at least one further investigation and excluding oligoclonal immune reconstitution | |
| Greater than 5% plasma cells in a bone marrow aspirate or on trephine bone biopsy | ||
| Development of new lytic bone lesions or soft tissue plasmacytomas or definite increase in the size of residual bone lesions (development of a compression fracture does not exclude continued response and may not indicate progression) | ||
| Development of hypercalcaemia (corrected serum calcium > 11·5 mg/dl or 2·8 mmol/l) not attributable to any other cause | ||
| Progressive disease (for patients not in CR); requires at least one of the following: | A greater than 25% increase in the level of the serum monoclonal paraprotein, which must also be an absolute increase of at least 5 g/l and confirmed by at least one repeated investigation | A greater than 25% increase in the concentration of serum monoclonal protein, which must also be an absolute increase of more than 5 g/l and confirmed by at least one repeated assessment |
| A greater than 25% increase in the 24-hour urinary light chain excretion, which must also be an absolute increase of at least 200 mg/24 hours and confirmed by at least one repeated investigation | A greater than 50% increase in the 24-hour urinary light chain excretion, confirmed by at least one repeated assessment | |
| A greater than 25% increase in plasma cells in a bone marrow aspirate or on trephine biopsy, which must also be an absolute increase of at least 10% | ||
| Definite increase in the size of existing bone lesions or soft tissue plasmacytomas | A confirmed increase in the size of existing bone lesions or soft tissue plasmacytomas | |
| Development of new bone lesions or soft tissue plasmacytomas (development of a compression fracture does not exclude continued response and may not indicate progression) | Development of new bone lesions or soft tissue plasmacytomas | |
| Development of hypercalcaemia (corrected serum calcium > 11·5 mg/dl or 2·8 mmol/l) not attributable to any other cause | Development of hypercalcaemia, not attributable to any cause other than MM |
Appendix 4 Table of excluded studies
| Excluded reference | Reason for exclusion |
|---|---|
| Anon. Melphalan prednisone thalidomide versus melphalan prednisone in patients aged ≥ 75 years with untreated multiple myeloma: preliminary results of the randomized, double-blind, placebo-controlled IFM 01–01 trial. Clin Lymphoma Myeloma 2007;7:455–6 | Not a clinical trial report |
| Anon. Thalidomide added to standard therapy prolongs overall survival in newly diagnosed multiple myeloma patients over age 75. Oncology 2008;22:87 | Not a clinical trial report |
| Morgan GJ, Jackson GH, Davies FE, Drayson MT, Owen RG, Gregory WM, et al. Maintenance thalidomide may improve progression free but not overall survival: results from the Myeloma IX Maintenance Randomisation. Blood 2008;112:Abstract 656 | Thalidomide maintenance |
| Morgan GJ, Davies FE, Owen RG, Rawstron AC, Bell S, Cocks K, et al. Thalidomide combinations improve response rates: results from the MRC IX study. Blood 2007:110:Abstract 3593 | Thalidomide maintenance |
| Davies FE, Child JA, Hawkins K, Bell S, Brown J, Drayson MT, et al. Newly diagnosed myeloma patients are at risk of venous thrombotic events – high risk patients need to be identified and receive thromboprophylaxis: the MRC experience. Blood 2004;104:Abstract 2395 | Outcomes |
| Kapoor P, Rajkumar SV, Dispenzieri A, Lacy MQ, Dingli D, Kyle R, et al. Melphalan and prednisone (MP) versus melphalan, prednisone and thalidomide (MPT) as initial therapy for previously untreated elderly and/or transplant ineligible patients with multiple myeloma: a meta-analysis of randomized controlled trials. 51st ASH Annual Meeting and Exposition, New Orleans, LA, 5–8 December 2009, Abstract no. 615 | Meta-analysis (insufficient details in abstract) |
Appendix 5 Clinical effectiveness included studies – data extraction forms
Data extracted by JB, extraction checked by JP.
| Reference and design | Intervention | Participants | Outcome measures |
|---|---|---|---|
|
Author: San Miguel et al. 26 VISTA trial Abstracts for follow-up data60,61 Year: 2008 Countries: 22 countries in Europe, North and South America, and Asia Study design: Multicentre RCT Setting: Secondary care No. of centres: 151 centres Recruitment dates: December 2004 to September 2006 Funding: Supported by Johnson & Johnson Pharmaceutical Research & Development and Millennium Pharmaceuticals |
Intervention: Nine 6-week cycles of melphalan (9 mg/m2) plus prednisone (60 mg/m2) on days 1–4, plus bortezomib (1.3 mg/m2 by intravenous bolus) on days 1, 4, 8, 11, 22, 25, 29 and 32 during cycles 1–4 and on days 1, 8, 22 and 29 during cycles 5–9 Control: Nine 6-week cycles of melphalan (9 mg/m2) plus prednisone (60 mg/m2) on days 1–4 Treatment discontinued on withdrawal of patient’s consent, disease progression or the occurrence of unacceptable toxic effects Dose of melphalan or bortezomib reduced if any prespecified haematological toxic effect or grade 3/4 non-haematological toxic effect; bortezomib-associated neuropathic pain and peripheral sensory neuropathy managed with use of established dose- modification guidelines (referenced) Other interventions used: Patients with myeloma-associated bone disease received bisphosphonates unless such therapy was contraindicated (referenced) |
No. of participants: 682 (VMP: 344, MP: 338) Sample attrition/dropout: Not clearly or explicitly described – nos. provided for AE data but reasons for all withdrawals not given Timing of withdrawals: NR Sample crossovers: None Inclusion criteria for study entry: Newly diagnosed untreated symptomatic measurable myeloma patients not candidates for HDT plus SCT because of age (≥ 65 years) or co-existing conditions. Measurable disease defined as presence of quantifiable M-protein in serum or urine or measurable soft tissue or organ plasmacytomas Exclusion criteria for study entry: None stated Characteristics of participants: Age (years): Median (range): VMP 71 (57–90), MP 71 (48–91) Age < 65: VMP 14 (4%), MP 9 (3%) Age ≥ 75: VMP 107 (31%), MP 101 (30%) Gender (M : F): VMP 175 : 169 (51% : 49%), MP 166 : 172 (49% : 51%) Ethnicity: White VMP 304 (88%), MP 295 (87%) Asian VMP 33 (10%), MP 36 (11%) Black VMP 5 (1%), MP 7 (2%) Other VMP 2 (1%), MP 0 Region: Europe VMP 79%, MP 78% North America VMP 9%, MP 9% Other VMP 11%, MP 13% Karnofsky performance status ≤ 70: VMP 122 (35%), MP 111 (33%) Myeloma type: IgG VMP 64%, MP 62% IgA VMP 24%, MP 26% IgD VMP 1%, MP 1% IgM VMP 1%, MP1% Light chain VMP 8%, MP 8% Biclonal VMP 2%, MP 2% Lytic bone lesions, no./total no.(%): VMP 224/343 (65%), MP 222/336 (66%) Median plasma cells on bone marrow biopsy: VMP 40%, MP 41% ISS: Stage I VMP 19%, MP 19% Stage II VMP 47%, MP 47% Stage III VMP 35%, MP 34% Serum β2-microglobulin level (mg/l): Median (range): VMP 4.2 (1.7–21.6), MP 4.3 (0.6–60.9) < 2.5 VMP 12%, MP 12% 2.5–5.5 VMP 55%, MP 55% > 5.5 VMP 33%, MP 33% Albumin level (g/dl): Median (range): VMP 3.3 (1.3–4.7), MP 3.3 (1.4–5.0) < 3.5 VMP 58%, MP 62% ≥ 3.5 VMP 42%, MP 38% Haemoglobin (g/l): Median (range) VMP 104 (64–159), MP 106 (73–165) Platelet count/mm3 median (range): VMP 221,500 (68,000–515,000), MP 221,500 (33,000–587,000) Creatinine clearance (%): < 30 ml/minute VMP 6%, MP 5% 30–60 ml/minute VMP 48%, MP 50% > 60 ml/minute VMP 46%, MP 46% History of cardiac condition: VMP 121 (35%), MP 105 (31%) |
Primary outcomes: Time to disease progression Secondary outcomes: Rate of CR, duration of response, time to subsequent myeloma therapy, OS PFS (reported in supplemental appendix) Method of assessing outcomes: Response to treatment and disease progression assessed using EBMT criteria and previously validated computer algorithm, on basis of M-protein in serum and urine Definitions from appendix: PFS is time between randomisation and either disease progression or relapse from CR or death Blood and 24-hour urine samples collected every 3 weeks during 54-week treatment phase and then every 8 weeks until disease progression. Other efficacy assessment included bone marrow examination and skeletal survey as required by EBMT or on basis of clinical/biochemical measurements Relapse from CR defined as reappearance of M-protein on immunofixation Seven prespecified and one post hoc subgroups defined (not data extracted) AEs: Graded by National Cancer Institute’s Common Terminology Criteria for AEs (version 3.0). No further details given Safety evaluated throughout study and until 30 days after administration of a study drug Length of follow-up: Not specifically stated. Patients followed for survival and subsequent myeloma therapy at least every 12 weeks after disease progression Median follow-up at data cut-off point not reported |
| Results | |||
|---|---|---|---|
| Primary outcomes | VMP (n = 344) | MP (n = 338) | p-value |
| TTP median (from trial investigators) | 24 months | 16.6 months | < 0.001, HR = 0.48 |
| TTP median (from computer algorithm analysis) | 20.7 months | 15.0 months | < 0.001, HR = 0.54 |
| Comments | |||
|
HR in favour of VMP was 0.48 (independent of age, sex, race, baseline β2-microglobulin level, baseline albumin level, region, ISS or creatinine clearance) HR using algorithmic analysis was 0.54 HR for each subgroup of patients (seven prespecified and one post hoc) was lower for VMP than MP, indicating lower risk of progression in the VMP group as assessed by investigators. However, the study may not have been powered to show this for subgroups |
|||
| Secondary outcomes | VMP (n = 377) | MP (n = 331) | p-value |
| Response rates using EBMT criteria | |||
| Rate of PR or better | 238 (71%) | 115 (35%) | < 0.001 |
| Rate of CR | 102 (30%) | 12 (4%) | < 0.001 |
| Rate of PR | 136 (40%) | 103 (31%) | NR |
| MR | 32 (9%) | 72 (22%) | NR |
| Stable disease | 60 (18%) | 113 (40%) | NR |
| Progressive disease | 3 (1%) | 7 (2%) | NR |
| Response rates using IURC (post hoc analysis) | |||
| Rate of PR or better | 251 (74%) | 128 (39%) | < 0.001 |
| Rate of CR | 111 (33%) | 13 (4%) | < 0.001 |
| Rate of VGPR | 28 (8%) | 13 (4%) | NR |
| Rate of PR | 112 (33%) | 102 (31%) | NR |
| Stable disease | 79 (23%) | 192 (58%) | NR |
| Progressive disease | 3 (1%) | 7 (2%) | NR |
| Time to eventa | |||
| Median time to first response (PR or better) | 1.4 months | 4.2 months | < 0.001 |
| Median time to CR | 4.2 months | 5.3 months | < 0.001 |
| Median duration of CR or PR | 19.9 months | 13.1 months | NR |
| Median duration of CR | 24 months | 12.8 months | NR |
| Median time to subsequent myeloma therapy | Not reached (based on 344 patients) | 20.8 months (based on 338 patients) | < 0.001, HR = 0.52 |
| Started second-line treatment within 2 years | 35% | 57% | NR |
| Survivalb | |||
| VMP (n = 344) | MP (n = 338) | ||
| Treatment-free interval | Not reached | 9.4 months | NR |
| Deaths after median follow-up of 16.3 months | 45 (13%) | 76 (22%) | 0.008, HR = 0.61 |
| Median OS | Not reached | Not reached | |
| Median PFS | 21.7 months | 15.2 months | < 0.001, HR = 0.56 |
| At data cut-off point | |||
| Patients still receiving assigned protocol | 47 (14%) | 33 (10%) | |
| Results from abstract61 | |||
| Median OS after median follow-up of 36.7 months | Not estimable | 43.1 months | NR |
| Three-year OS rate | 68.5% | 54.0% | NR |
| Risk of death after median follow-up of 36.7 months | Risk reduced by 35% in VMP group compared to MP group | 0.0008, HR = 0.653 | |
| Received subsequent therapy | 178 (52%) | 233 (69%) | NR |
| Median time to subsequent therapy | 28.1 months | 19.2 months | < 0.0001, HR 0.527 |
| Median treatment-free interval | 17.6 months | 8.4 months | < 0.0001, HR 0.543 |
| Median survival from start of subsequent therapy | 30.2 months | 21.9 months | 0.21, HR = 0.815 |
| Results from abstract60 | |||
| Survival after median follow-up of 25.9 months | NR | NR | 0.0032, HR = 0.64 |
| Three-year survival rates | 72% | 59% | NR |
| Time to next therapy | 28.1 months | 19.2 months | 0.000001, HR = 0.53 |
| Treatment-free interval | 16.6 months | 8.4 months | 0.00001, HR = 0.54 |
| Required subsequent therapy | 38% | 57% | NR |
| IURC, International Uniform Response Criteria. | |||
| a Time-to-event data determined by computer algorithm using EBMT criteria. | |||
| b Data based on 344 patients in VMP group and 338 patients in MP group. | |||
| HR after median follow-up of 16.3 months was 0.61 in favour of VMP (p = 0.008). | |||
| From abstract60 | |||
|
HR for survival after median follow-up of 25.9 months was 0.64 in favour of VMP (p = 0.0032). HR for time to next therapy was 0.53 in favour of VMP (p < 0.000001). HR for treatment-free interval was 0.54 in favour of VMP (p < 0.00001). |
|||
| Results from abstract61 | VMP (n = 344, no. analysed NR) | MP (n = 338, no. analysed NR) | p-value |
|---|---|---|---|
| Sustained response in QLQ-C30 domains | |||
| Cognitive functioning | 27% | 28% | NR |
| Nausea/vomiting | NR | NR | 0.0095 |
| Appetite loss | NR | NR | 0.0170 |
| Diarrhoea | NR | NR | 0.0082 |
| Global health | 49% | 40% |
Not statistically significant |
| Pain | 40% | 32% |
Not statistically significant |
| Insomnia | 32% | 24% |
Not statistically significant |
| Comments | |||
| The aim of the study was to describe the rate of patients who experienced a sustained HRQoL improvement after best response and the overall HRQoL impact of best response. A sustained HRQoL improvement was defined as a change in score of at least five points for at least two consecutive cycles after best response (CR, PR or MR). The rate of sustained improvement and the time to sustained improvement were calculated in the population of patients who were followed for at least two cycles after best response (n = 363). All EORTC domain scores were similar at baseline across the study arms. Worse health was reported in all domains with VMP arm at best tumour response onset. However, after best response onset, patients in the VMP arm had a higher sustained HRQoL improvement rate than those in the MP arm in 14 of the 15 EORTC QLQ-C30 scores. The differences for nausea and diarrhoea remained significant in the Cox models when adjusted for baseline score, score at best response, and type of response (CR, PR or MR) | |||
| AEs | VMP (n = 340) | MP (n = 337) | p-value |
|---|---|---|---|
| Median no. of treatment cycles | 8 (46 weeks) | 7 (39 weeks) | NR |
| Death rates during treatment | 5% | 4% | NS |
| Treatment-related deaths | 1% | 2% | NS |
| Rate of serious AEs | 46% | 36% | NR |
| Discontinued treatment due to AEs | 50 (15%) | 47 (14%) | NR |
| Discontinued treatment due to treatment-related events | 37 (11%) | 35 (10%) | NR |
| Additional discontinuations (bortezomib) | 63 (19%) | – |
| AEs, no. (%)a | Total | Grade 3 | Grade 4 | Total | Grade 3 | Grade 4 | |
|---|---|---|---|---|---|---|---|
| Any event | |||||||
| 338 (99%) | 181 (53%) | 96 (28%) | 326 (97%) | 148 (44%) | 92 (27%) |
0.02 for grade 3 NR for grade 4 |
|
| Haematological events b | NR | ||||||
| Thrombocytopenia | 178 (52%) | 68 (20%) | 58 (17%) | 159 (47%) | 55 (16%) | 47 (14%) | |
| Neutropenia | 165 (49%) | 102 (30%) | 34 (10%) | 155 (46%) | 79 (23%) | 49 (15%) | |
| Anaemia | 147 (43) | 53 (16%) | 9 (3%) | 187 (55%) | 66 (20%) | 26 (8%) | |
| Leucopenia | 113 (33%) | 67 (20%) | 10 (3%) | 100 (30%) | 55 (16%) | 13 (4%) | |
| Lymphopenia | 83 (24%) | 49 (14%) | 18 (5%) | 58 (17%) | 30 (9%) | 7 (2%) | |
| Gastrointestinal events: all | 19% | 5% | NR | ||||
| Nausea | 164 (48%) | 14 (4%) | 0 | 94 (28%) | 1 (<1%) | 0 | |
| Diarrhoea | 157 (46%) | 23 (7%) | 2 (1%) | 58 (17%) | 2 (1%) | 0 | |
| Constipation | 125 (37%) | 2 (1%) | 0 | 54 (16%) | 0 | 0 | |
| Vomiting | 112 (33%) | 14 (4%) | 0 | 55 (16%) | 2 (1%) | 0 | |
| Infections | NR | ||||||
| Pneumonia | 56 (16%) | 16 (5%) | 6 (2%) | 36 (11%) | 13 (4%) | 4 (1%) | |
| Herpes zoster | 45 (13%) | 11 (3%) | 0 | 14 (4%) | 6 (2%) | 0 | |
| Nervous system disorders | NR | ||||||
| Peripheral sensory neuropathy | 151 (44%) | 43 (13%) | 1 (<1%) | 16 (5%) | 0 | 0 | |
| Neuralgia | 121 (36%) | 28 (8%) | 2 (1%) | 5 (1%) | 1 (< 1%) | 0 | |
| Dizziness | 56 (16%) | 7 (2%) | 0 | 37 (11%) | 1 (< 1%) | 0 | |
| Other conditions | NR | ||||||
| Pyrexia | 99 (29%) | 8 (2%) | 2(1%) | 64 (19%) | 6 (2%) | 2 (1%) | |
| Fatigue | 98 (29%) | 23 (7%) | 2 (1%) | 86 (26%) | 7 (2%) | 0 | |
| Anorexia | 77 (23%) | 9 (3%) | 1 (< 1%) | 34 (10%) | 4 (1%) | 0 | |
| Asthenia | 73 (21%) | 20 (6%) | 1 (< 1%) | 60 (18%) | 9 (3%) | 0 | |
| Cough | 71 (21%) | 0 | 0 | 45 (13%) | 2 (1%) | 0 | |
| Insomnia | 69 (20%) | 1 (< 1%) | 0 | 43 (13%) | 0 | 0 | |
| Peripheral oedema | 68 (20%) | 2 (1%) | 0 | 34 (10%) | 0 | 0 | |
| Rash | 66 (19%) | 2 (1%) | 0 | 24 (7%) | 1 (<1%) | 0 | |
| Back pain | 58 (17%) | 9 (3%) | 1 (<1%) | 62 (18%) | 11 (3%) | 1 (< 1%) | |
| Dyspnoea | 50 (15%) | 11 (3%) | 2 (1%) | 44 (13%) | 5 (1%) | 3 (1%) | |
| Hypocalaemia | 44 (13%) | 19 (6%) | 3 (1%) | 25 (7%) | 8 (2%) | 2 (1%) | |
| Arthalgia | 36 (11%) | 4 (1%) | 0 | 50 (15%) | 2 (1%) | 1 (< 1%) | |
| DVT | 4 (1%) | 3 (1%) | 0 | 6 (2%) | 2 (1%) | 0 | |
| Discontinuations | VMP (n = 340) | MP (n = 337) | p-value |
|---|---|---|---|
| Total discontinued treatment | 139 (41%) | 166 (49%) | NR |
| Discontinued due to progressive disease | 24 (7%) | 72 (21%) | NR |
| Discontinued due to AEs | 50 (15%) | 47 (14%) | NR |
| Discontinued due to patient choice | 32 (9%) | 18 (5%) | NR |
| Discontinued due to death | 14 (4%) | 17 (5%) | NR |
| Discontinued due to maintenance of CR | 9 (3%) | 1 (< 1%) | NR |
| Other reasons for discontinuation | 10 (3%) | 11 (3%) | NR |
| Methodological comments |
|---|
|
Allocation to treatment groups: Randomisation (1:1) was stratified according to baseline levels of β2-microglobulin (< 2.5, 2.5–5.5 or > 5.5 mg/l), serum albumin (< 3.5 or ≥ 3.5 g/dl) and region (North America, Europe or other region) Blinding: Not stated but study described as open-label Comparability of treatment groups: Baseline demographic and disease characteristics stated to be well-balanced between groups (no p-values given) Method of data analysis: TTP, time to subsequent myeloma therapy and OS analyses from randomisation to event of interest. Differences between groups compared using stratified log-rank tests with ITT analysis (all randomised patients). Distributions estimated using Kaplan–Meier method. For time-to-progression analyses, data from patients in whom there was no disease progression were censored at the last assessment or at the start of subsequent therapy. HRs estimated using stratified Cox proportional hazards model for ITT and subgroups defined according to baseline characteristics (seven prespecified analyses according to age, sex, race, baseline β2-microglobulin level, baseline albumin level, region, disease stage and post hoc creatinine clearance). Response rates were analysed in patients who could be evaluated for a response (not ITT) and compared between groups using stratified Cochran–Mantel–Haenszel chi-squared test. Treatment differences tested at a two-sided alpha level of 0.05. Safety population was all randomised patients who received at least one dose of study drug Sample size/power calculation: Sample size of 340 patients per group was determined to provide a power of 80% to detect a 33% improvement in time to progression in patients receiving VMP compared with MP. Three interim analyses planned using O’Brien–Fleming method. On basis of third analysis (data cut-off 15 June 2007), the data and safety monitoring committee recommended that the trial be stopped since the prespecified statistical boundary (an alpha level of 0.0108) for the primary end point of TTP has been crossed (HR in bortezomib group 0.54, p < 0.001). Data from the third analysis are presented. Not clear if study powered for subgroup analyses Attrition/dropout: Not explicitly reported but withdrawals given in AE data. Not all reasons for discontinuations in VMP group reported |
| General comments |
|
Generalisability: Patients ≥ 65 so probably generalisable in terms of population Outcome measures: Defined and graded Intercentre variability: Not stated Conflict of interests: Data collected by sponsors and analysed in collaboration with senior academic authors who vouch for the completeness and accuracy of the data and analyses. Eleven of 21 authors report conflicts of interest |
Quality criteria40
| Criteria for assessment of risk of bias in RCTs | Answera | Notes and comments |
|---|---|---|
| Was the method used to generate random allocations adequate? | NR | |
| Was the allocation adequately concealed? | NR | |
| Were the groups similar at the outset of the study in terms of prognostic factors, e.g. severity of disease? | Yes | No p-values given |
| Were the care providers, participants and outcome assessors blind to treatment allocation? If any of these people were not blinded, what might be the likely impact on the risk of bias (for each outcome)? | No | Open label |
| Were there any unexpected imbalances in dropouts between groups? If so, were they explained or adjusted for? | Unclear | |
| Is there any evidence to suggest that the authors measured more outcomes than they reported? | No | |
| Did the analysis include an ITT analysis? If so, was this appropriate and were appropriate methods used to account for missing data? | Yes | No details given in published paper but MS states that missing data were imputed using last-observation-carried-forward method |
Additional outcomes/comments/notes
| Outcomes from abstract,61 median follow-up of 36.7 months | VMP (n = 178) | MP (n = 233) | p-value |
|---|---|---|---|
| Received subsequent therapy containing | |||
| Bortezomib | 43 (24%) | 116 (50%) | |
| Thalidomide | 81 (46%) | 110 (47%) | |
| Lenalidomide | 57 (32%) | 30 (13%) | |
| Overall response rate to subsequent therapy | |||
| Bortezomib | 47% | 59% | |
| Thalidomide | 41% | 53% | |
| Lenalidomide | 59% | 52% | |
| Outcomes from abstract60 | VMP (n = 129) | MP (n = 194) | p-value | ||
|---|---|---|---|---|---|
| Received bortezomib | 16% | 43% | |||
| Received thalidomide | 49% | 44% | |||
| Received lenalidomide | 19% | 6% | |||
| Subsequent therapy and no. of patients who received ita | CR | PR | CR | PR | |
| Bortezomib or bortezomib combination (n = 105) | 6% | 33% | 10% | 45% | |
| Thalidomide combination (n = 149) | 4% | 44% | 3% | 52% | |
| Lenalidomide combination (n = 37) | 4% | 52% | 0 | 55% | |
Data extracted by JOP, extraction checked by JB.
| Reference and design | Intervention | Participants | Outcome measures |
|---|---|---|---|
|
Author: Facon et al. 23 IFM 99/06 Year: 2007 Countries: France, Belgium and Switzerland Study design: Multicentre RCT Setting: Not stated, appears to be secondary care No. of centres: 73 IFM centres. No. in each country not stated Recruitment dates: 22 May 2000 to 8 August 2005 Funding: Sponsored by the Centre Hospitalier et Universitaire de Lille; by a research grant from the French Ministry of Health; and by the Swiss Group for Clinical Cancer Research (SIAK). Laphal, and later Pharmion, supplied free thalidomide |
MPT: Oral thalidomide not exceeding 400 mg daily taken throughout the 12 MP cycles. Thalidomide stopped on day 4 of the last MP cycle. Advice was to initiate thalidomide at a dose of 200 mg per day, increasing to 400 mg per day after 2–4 weeks in the absence of severe adverse effects. Initial dose defined as the greatest dose used in the first four weeks of treatment plus MP for 12 × 6-week cycles comprising melphalan 0.25 mg/kg and prednisone 2 mg/kg on 4 days (days 1–4) per cycle. Both drugs taken orally Control, MP only: MP for 12 × 6-week cycles comprising melphalan 0.25 mg/kg and prednisone 2 mg/kg on 4 days (days 1–4) per cycle. Both drugs taken orally The trial had a third arm, reduced intensity stem-cell transplant using melphalan 100 mg/m2, which has not been data extracted Dose reductions: Thalidomide dose modification allowed at discretion of local investigators Thalidomide temporarily stopped if patients developed DVT or pulmonary embolism but treatment resumed once patients had undergone therapeutic anticoagulation No thromboprophylaxis prospectively planned Treatment stopped: Thalidomide stopped for any non-haematological grade 3 or 4 toxic effects Other interventions used: Clondronate orally, 1040 mg per day continuously to all patients |
No. of participants: 447 to all three groups (one group NR on here) MPT: 125 assigned (but one died before treatment initiation) MP + placebo: 196 assigned (but three died before treatment initiation) Sample attrition/dropout: Not clearly described – nos. withdrawn provided but reasons for withdrawals not provided for each type of event, e.g. death, progression, toxicity, etc. Timing of withdrawals: NR Sample crossovers: None Inclusion criteria for study entry: Generally patients aged between 65 and 75 years of age with previously untreated MM at stage II or III (DS criteria). Prior treatment with minimum-dose radiotherapy to localised lesions for symptom relief allowed. Additionally, patients younger than 65 years were included if they were ineligible for high-dose treatment. Patients with DS stage I MM who met criteria of high-risk stage I disease also eligible (criteria not listed, reference provided) Exclusion criteria for study entry: Previous neoplasms (except basocellular cutaneous or cervical epithelioma); primary or associated amyloidosis; a WHO performance index of 3 or higher, if unrelated to MM; substantial renal insufficiency with creatinine serum concentration of 50 mg/l or more; cardiac or hepatic dysfunction; peripheral neuropathy; HIV infection, or hepatitis B or C infections Characteristics of participants (as assigned, includes those who died before treatment): Age ≥ 70 years: MPT 50/125 (40%); MP 84/196 (43%) Gender (M : F): MPT 63 : 62 (50% : 50%); MP 109/87 (56% : 44%) Ethnicity: NR Immunoglobulin A isotype: MPT 25/125 (20%); MP 43/196 (22%) DS stage II or III: MPT 112/125 (90%); MP 177/196 (91%) DS substage B: MPT 12/125 (10%); MP 15/196 (8%) ISS stage 1: MPT 38/112 (34%); MP 61/182 (34%) ISS stage 2: MPT 42/112 (38%); MP 67/182 (37%) ISS stage 3: MPT 32/112 (29%); MP 54/182 (30%) WHO performance index 3–4: MPT 10/125 (8%); MP 13/196 (7%) Bone lesions: MPT 90/125 (76%); MP 154/196 (79%) β2-microglobulin ≥ 3.5 mg/l: MPT 69/112 (62%); MP 110/182 (60%) Albumin < 35 g/l: MPT 24/125 (19%); MP 45/194 (23%) Creatinine ≥ 20 mg/l: MPT 11/124 (9%); MP 13/196 (7%) Calcium ≥ 105 mg/l: MPT 17/125 (14%); MP 40/196 (20%) C-reactive protein ≥ 6 mg/l: MPT 50/114 (44%); MP 85/173 (49%) Lactate dehydrogenase ≥ 300 U/l: MPT 65/107 (61%); MP 116/175 (66%) Chromosome 13 deletion: MPT 49/101 (49%); MP 72/147 (52%) Translocation (11;14): MPT 11/58 (19%); MP 11/95 (12%) Translocation (4;14): MPT 10/57 (18%); MP 7/95 (7%) |
Primary outcome: OS Secondary outcomes: Response, PFS, survival after progression and toxicity Method of assessing outcomes: Visits after inclusion at 3 months, 6 months, and every 6 months thereafter until withdrawal from the trial. At every visit, response was assessed. After withdrawal from trial, patient treatment and status updated every 6 months. These data also requested at other specific points for patients still alive at last known status For achievement of response there had to be improvement in bone pain and performance status, correction of hypercalcaemia, and no increase in size or no. of lytic bone lesions Response definitions: CR – absence of the original monoclonal protein in serum and urine by immunofixation, fewer than 5% plasma cells in a bone marrow aspirate, and the disappearance of soft tissue plasmocytomas Very good PR – more than 90% decrease in monoclonal protein in serum and urine PR – reduction in the size of soft-tissue plasmocytomas, more than a 50% reduction in the concentration of serum monoclonal protein, and more than a 75% reduction in 24-hour urinary light chain excretion Progressive disease– at least one of a greater than 25% increase in serum monoclonal protein concentration, which must also be an absolute increase of more than 5 g/l, confirmed by at least one repeated assessment; a greater than 50% increase in the 24-hour urinary light chain excretion, confirmed by at least one repeated assessment; a confirmed increase in the size of existing bone lesions or soft tissue plasmocytomas; development of new bone lesions or soft tissue plasmocytomas or the development of hypercalcaemia, not attributable to any cause other than MM Stable disease – patient not meeting criteria of CR, PR or progressive disease The best response at 12 months was defined as the highest amount of disease improvement achieved by a patient at any follow-up visit while on treatment, from randomisation to month 15, except if progressive disease had occurred during that period without response assessment at 12 months (between 9 and 15 months) AEs: Method of monitoring or assessing NR. Reported for the safety population (all those randomised but excluding those who died before receiving treatment) Length of follow-up: Not clearly stated but a 2-year follow-up appears to have been planned. Outcomes reported for two date points with median follow-ups of 36.8 months (IQR 20.8–51.2 months) in October 2005, and 51.5 months (IQR 34.4–63.2 months) in January 2007 |
| Results Primary outcomes |
MPT | MP | p-value |
|---|---|---|---|
| OS, median (SE, IQR) | 51.6 months (4.5, 26.6 to ‘not reached’) | 33.2 months (3.2, 13.8–54.8) | 0.0006 |
| No. of deaths/no. of patients after median follow-up of 51.5 months (IQR 34.4–63.2) | 62/125 (50%)a | 128/196 (65%)a | |
| Toxic death | n = 0 | n = 4 (2%), all due to infection | |
| Early death – in first 3 months of treatment | 3/124 (2%) | 13/193 (7%) | |
| a Percentages calculated by reviewer. | |||
| Comments | |||
| HR for median OS in favour of MPT = 0.59 (95% CI 0.46 to 0.81). When adjusting for prognostic factors (e.g. WHO performance index; β2-microglobulin, albumin, etc.) the results showed that MPT remained the superior treatment (HR 0.49, 95% CI 0.33 to 0.73, p = 0.0002). At the initial analysis (median follow-up 36.8 months) no difference in OS was recorded as a function of initial thalidomide dose (≤ 200 mg per day vs > 200 mg per day, p = 0.93). | |||
| Secondary outcomes | MPT | MP | p-value |
| PFS, median (SE, no. of events/no. of patients) after median follow-up of 51.5 months | 27.5 months (2.1, 92/125) | 17.8 months (1.4, 171/196) | 0.0001 |
| Survival time after progression, median (SE, no. of events/no. of patients) after median follow-up of 51.5 months | 13.4 months (2.3, 52/83) | 11.4 months (1.9, 111/154) | |
| At least PR at 12 months | 57/75 (76%) | 57/165 (35%) | < 0.0001 |
| At least very good PR at 12 months | 35/75 (47%) | 11/165 (7%) | < 0.0001 |
| CR at 12 months | 10/75 (13%) | 4/165 (2%) | 0.0008 |
| Not withdrawn (still on first-line treatment; first-line treatment ceased as planned and no further treatment, alive without progression, not withdrawn for other reason) | 31/124 (25%) | 42/193 (22%) | |
| Withdrawn and not receiving second-line treatment | 38/124 (31%) | 25/193 (13%) | |
| - up to death | 11/38 (29%) | 24/25 (96%) | |
| - still alive | 27/38 (72%) | 1/25 (4%) | |
| Withdrawn and having received second-line treatment | 55/124 (44%) | 126/193 (65%) | |
| Comments | |||
|
HR for median PFS in favour of MPT = 0.51 (95% CI 0.39 to 0.66) after median follow-up of 51.5 months. At the initial analysis (median follow-up 36.8 months), no difference in PFS was recorded as a function of initial, maximum or average thalidomide doses (p = 0.22, p = 0.75 and p = 0.92, respectively) Details of second-line treatments given are presented in the table following the Quality criteria table below |
|||
| AEs and safety | MPT | MP | p-value |
| Discontinuation of thalidomide because of toxic effects | 56/124 (45%) | ||
| Peripheral neuropathy | n = 23 | ||
| Thrombosis | n = 7 | ||
| Somnolence, dizziness or fatigue | n = 8 | ||
| Cutaneous toxic effects | n = 4 | ||
| Psychiatric complications | n = 1 | ||
| Withdrawn because of other reasons | n = 13 | ||
| Haematological toxic effects | n = 5 | ||
| Infection | n = 7 | ||
| Stroke | n = 1 | ||
| Grade 3 and 4 AEs, nos. of patients (%) | MPT (n = 124) | MP (n = 193) | p-value |
| Haematological | |||
| Anaemia | 17 (14%) | 27 (14%) | 0.94 |
| Neutropenia | 60 (48%) | 51 (26%) | < 0.0001 |
| Thrombocytopenia | 17 (14%) | 19 (10%) | 0.29 |
| Severe haemorrhage | 0 | 3 (1.5%) | Too few events to be clinically meaningful |
| Thrombosis or embolism | 15 (12%) | 8 (4%) | 0.008 |
| Peripheral neuropathy | 7 (6%) | 0 | 0.001 |
| Somnolence/fatigue dizziness | 10 (8%) | 0 | < 0.0001 |
| Infection | 16 (13%) | 18 (9%) | 0.32 |
| Fever of unknown origin | 1 (1%) | 2 (1%) | |
| Pneumonia | 9 (7%) | 5 (2.5%) | |
| Septicaemia | 4 (3%) | 6 (3%) | |
| Meningitis | 2 (2%) | 0 | |
| Other | 1 (1%) | 6 (3%) | |
| Herpes zoster | 3 (2.5%) | 6 (3%) | |
| Cardiac | 2 (2%) | 1 (0.5%) | Too few events to be clinically meaningful |
| Arrhythmia | 2 (2%) | 0 | |
| Myocardial infarction/angina | 0 | 0 | |
| Cardiac failure | 0 | 1 (0.5%) | |
| Hypertension | 0 | 0 | |
| Gastrointestinal | |||
| Nausea | 1 (1%) | 2 (1%) | Too few events to be clinically meaningful |
| Constipation | 13 (10%) | 0 | < 0.0001 |
| Mucositis | 0 | 1 (0.5%) | Too few events to be clinically meaningful |
| Bleeding | 0 | 2 (1%) | |
| Any grade ≥ 3 non-haematological toxic effect | 52 (42%) | 30 (16%) | < 0.0001 |
| Comments | |||
| Note that Facon et al. analysed safety at the October 2005 date point after 36.8 months of follow-up, a shorter follow-up than for the outcomes of OS, PFS and survival after progression analyses. In the MPT group 15 patients experienced 17 episodes of thrombosis or pulmonary embolism. Thalidomide was resumed in 8 of the 15 patients with thrombosis after full anticoagulation, and without recurrence in seven patients (one patient had three episodes). In the MPT group 69 (55%) of patients experienced peripheral neuropathy. Of these the majority, 62 patients, had grade 1 or 2 peripheral neuropathy and seven patients had grade 3 peripheral neuropathy (these are the seven noted above), and none had grade 4 | |||
| Median duration of treatment (IQR) | 11 months (5–15) | ||
| Methodological comments | |||
|
Allocation to treatment groups: States randomly assigned in a 3 : 2 : 2 ratio (MP : MPT : Mel100; Mel100 arm not included here). No further details Blinding: No details provided Comparability of treatment groups: Not described and no evidence presented to indicate whether similarity of groups had been statistically tested (although methods describe how this would be done). Visual inspection of the data suggests groups similar for most baseline characteristics reported on Method of data analysis: Parameters generally described by number and percentage of patients. Distributions of parameters assessed at inclusion compared between treatment groups using chi-squared tests for categoric variables and Kruskal–Wallis rank test for continuous variables (although no evidence from such tests presented as noted above). Best response rates at 12 months compared using the chi-squared test or Fisher’s exact test when necessary. Curves for OS, PFS and survival after progression calculated from randomisation and from progression (for survival after progression) using the Kaplan–Meier method. Time-to-event data expressed as median (SE and IQR). Comparison between treatment groups and HRs for death, progression or death without progression, or death after progression were estimated through the unstratified proportional hazards model, with 95% CI. AE rates compared through chi-squared test or Fisher’s exact test when necessary. Comparisons of OS between groups were adjusted on prognostic factors using a stepwise multivariate proportional hazards model, by forward selection with likelihood ratio test. All analyses carried out on an ITT population (not defined but numbers presented in tables indicate true ITT). AEs were analysed on the safety population (all those who received treatment, i.e. not including those who died before start of treatment). Confirmatory analysis on the primary end point was done on the per-protocol population at the first follow-up but data NR. Authors of this study do not report if it was necessary to censor any data and if so how this was done Sample size/power calculation: Sample size was estimated to be 500 patients (for the three arms of the trial) to guarantee, in a two-sided test, a power of 80% to detect an increase in the median survival time of 18 months (with an accrual time of 3 years and additional follow-up of 2 years). Power calculation assumed a median survival time of 30 months in the control group and used the Bonferroni correction for a global type I error rate of 5%. Slightly fewer than 500 patients were recruited (447 overall) because recruitment was stopped earlier than planned (although this is not explicitly stated) in August 2005 when a clear survival advantage for MPT was found. The authors do not comment on any possible implications of the recruitment shortfall Attrition/dropout: Withdrawals reported (see outcomes above) |
|||
| General comments | |||
|
The ITT analysis included patients not treated per protocol: MP – six protocol violations at inclusion; three protocol violations during follow-up; MPT – 0 protocol violations at inclusion; two protocol violations during follow-up Generalisability: This trial focuses on patients 65–75 years and the results may therefore only be applicable to patients in this age bracket Outcome measures: Methods for grading of AEs not described Intercentre variability: No comments made regarding possible intercentre variability Conflict of interests: States that the study sponsor had no role in study design, data collection, data analysis, data interpretation or writing of the report. Three authors had received scientific adviser board and lecture fees from Pharmion, Celgene and Janssen–Cilag. The remaining authors had no conflict of interest to declare |
|||
Quality criteria40
| Criteria for assessment of risk of bias in RCTs | Answera | Notes and comments |
|---|---|---|
| Was the method used to generate random allocations adequate? | NR | |
| Was the allocation adequately concealed? | NR | |
| Were the groups similar at the outset of the study in terms of prognostic factors, e.g. severity of disease? | Unclear | Baseline characteristics provided but no p-values and no statement indicating whether groups were similar |
| Were the care providers, participants and outcome assessors blind to treatment allocation? If any of these people were not blinded, what might be the likely impact on the risk of bias (for each outcome)? | NR | |
| Were there any unexpected imbalances in dropouts between groups? If so, were they explained or adjusted for? | Unclear | No comments made by authors of paper on this |
| Is there any evidence to suggest that the authors measured more outcomes than they reported? | No | |
| Did the analysis include an ITT analysis? If so, was this appropriate and were appropriate methods used to account for missing data? | Unclear | ITT analysis was conducted but no indication of whether missing data had to be accounted for and, if so, how this was done |
Additional outcomes/comments/notes
| Outcomes | MPT | MP | p-value |
|---|---|---|---|
| Initial daily dose of T 200 mg or less | n = 64/124 (52%) | ||
| Initial daily dose of T 200 mg or more | n = 60/124 (48%) | ||
| Initial daily dose of T 100 mg | n = 9 | ||
| Initial daily dose of T 300 mg | n = 5 | ||
| No change of dose throughout first-line treatment | n = 66/124 (36 at ≤ 200 mg/day; 30 at > 200 mg/day) | ||
| Dose increased during first-line treatment | n = 11/124 | ||
| Dose reduced during first-line treatment | n = 47/124 | ||
| Second-line treatment administered | 55/124 (44%) | 126/193 (65%) | |
| Second-line treatment thalidomide alone or in combination | 10/55 (18%) | 55/126 (44%) | |
| Second-line treatment VAD | 15/55 (27%) | 42/126 (33%) | |
| Second-line treatment dexamethasone | 7/55 (13%) | 12/126 (10%) | |
| Second line treatment alkylating agent-based regimens | 14/55 (25%) | 13/126 (10%) | |
| Second-line treatment bortezomib | 7/55 (13%) | 3/126 (2%) | |
| Second-line treatment other | 2/55 (4%) | 1/126 (1%) | |
| T, thalidomide. | |||
| Comments | |||
| Less than one-half of the patients at first progression on MP received rescue with thalidomide alone or in combination. Only 12 patients given MP or MPT underwent a transplant. These outcomes reported for the shorter median follow-up of 36.8 months | |||
Data extracted by JOP, extraction checked by JB.
| Reference and design | Intervention | Participants | Outcome measures |
|---|---|---|---|
|
Author: Hulin et al. 59 IFM 01/01 Year: 2009 Countries: France and Belgium Study design: Multicentre RCT Setting: Not stated; appears to be secondary care No. of centres: 44 IFM centres (39 in France, 5 in Belgium) Recruitment dates: 10 April 2002 to 22 December 2006 Funding: Sponsored by the Centre Hospitalier Universitaire de Nancy; by a research grant from the French Ministry of Health; by Laphal; by Pharmion; and by Celgene, which supplied free experimental treatment (thalidomide or placebo) for the study |
MP + T: Oral thalidomide 100 mg daily dose at bedtime for 72 weeks plus MP for 12 × 6-week cycles comprising melphalan 0.2 mg/kg on days 1–4; prednisone 2 mg/kg on days 1–4. Control – MP+placebo: Oral placebo at bedtime for 72 weeks plus MP for 12 × 6-week cycles comprising melphalan 0.2 mg/kg on days 1–4; prednisone 2 mg/kg on days 1–4. Dose reductions: Dose reduction to 50 mg per day of thalidomide or placebo allowed at investigator discretion in event of patient intolerance to 100 mg/day dose, especially in case of mild or moderate peripheral neuropathy (grade 1 or 2) No other dose reductions allowed Treatment stopped: Thalidomide stopped for symptomatic peripheral neuropathy (grade 3 or 4) confirmed by electromyogram Experimental treatment stopped and unblinded in the event of any non-haematological grade 3 or 4 AEs or disease progression before 72 weeks Other interventions used: Clondronate orally, 1040 mg per day continuously to all patients. No thromboprophylaxis prospectively planned Transfusions of red blood cells and platelets, and the administration of neutrophil growth factors or erythropoiesis-stimulating agents permitted as required Plasmapheresis at initial treatment and radiotherapy to localised lesions to relieve symptoms during the treatment phase permitted |
No. of participants: 232 (229 received treatment) MP + T: 115 (113 received treatment) MP + placebo: 117 (116 received treatment) Sample attrition/dropout: Three discontinued before treatment (failed inclusion criteria); 208 withdrawn (MP + T n = 100; MP + placebo n = 108) from study for other reasons (details in results) Timing of withdrawals: NR Sample crossovers: None Inclusion criteria for study entry: At least 75 years of age with newly diagnosed MM at stage II or III (DS criteria). Patients with DS stage I MM who met criteria of high-risk stage I disease also eligible (criteria not listed; reference provided). Patients with non-secretory or oligosecretory MM allowed Exclusion criteria for study entry: Previous neoplasms (except basocellular cutaneous or cervical epithelioma); primary or associated amyloidosis; a WHO performance index of 3 or higher, if unrelated to MM; substantial renal insufficiency with creatinine serum concentration of 50 mg/l or more; clinically significant cardiac or hepatic dysfunction; clinically significant peripheral neuropathy; history of venous thrombosis during the previous 6 months; HIV infection, or hepatitis B or C infections Characteristics of participants (only for participants who received treatment): Age ≥ 80 years: MP + T 43/113 (38%) MP + placebo 40/116 (34%) Gender (M : F): MP + T 43 : 70 (38% : 62%); MP + placebo 61/55 (53% : 47%) Ethnicity: NR Immunoglobulin A subtype: MP + T 31/113 (28%) MP + placebo 34/116 (30%) DS stage II or III: MP + T 100/113 (89%) MP + placebo 107/116 (93%) DS substage B: MP + T 8/113 (7%) MP + placebo 14/116 (12%) ISS stage 1: MP + T 25/98 (25%) MP + placebo 26/104 (25%) ISS stage 2: MP + T 39/98 (40%) MP + placebo 47/104 (45%) ISS stage 3: MP + T 34/98 (35%); MP + placebo 31/104 (30%) WHO performance index 3–4: MP + T 9/113 (8%); MP + placebo 7/116 (6%) Bone lesions: MP + T 87/113 (78%); MP + placebo 93/116 (82%) β2-microglobulin ≥ 3.5 g/dl: MP + T 70/101 (69%); MP + placebo 73/107 (68%) Albumin < 3.5 g/dl: MP + T 27/110 (25%) MP + placebo 34/113 (30%) Clearance creatinine ≤ 30 ml/minute: MP + T 11/105 (11%) MP + placebo 16/105 (15%) Significant comorbidity: MP + T 70/113 (62%) MP + placebo 69/116 (60%) Electromyogram abnormal: MP + T 17/54 (31%); MP + placebo 22/58 (38%) |
Primary outcomes: OS Secondary outcomes: Safety, response rates, PFS. Method of assessing outcomes: Visits every 6 weeks until treatment completion or study withdrawal Response assessed at 3, 6, 12 and 18 months After end of treatment or withdrawal from trial, patient status assessed every 6 months All clinical responses required documentation of improvement from baseline in bone pain and performance status, correction of hypercalcaemia, and no increase in size or number of lytic bone lesions Response definitions: CR – absence of the original monoclonal protein in serum and urine by immunofixation, fewer than 5% plasma cells in a bone marrow aspirate, and the disappearance of soft tissue plasmocytomas Very good PR – more than 90% decrease in monoclonal protein in serum and urine PR – reduction in the size of soft-tissue plasmocytomas, a more than 50% reduction in the concentration of serum monoclonal protein, and a more than 75% reduction in 24-hour urinary light chain excretion Progressive disease – at least one of a higher than 25% increase in serum monoclonal protein concentration constituting an absolute increase of more than 5 g/l, confirmed by at least one repeated assessment; a higher than 50% increase in the 24-hour urinary light chain excretion, confirmed by at least one repeated assessment; a confirmed increase in the size of existing bone lesions or soft-tissue plasmocytomas; development of new bone lesions or soft-tissue plasmocytomas; or the development of hypercalcaemia, not attributable to any cause other than MM Stable disease – patient not meeting criteria of CR, PR or progressive disease The best response at 12 months was defined as the best improvement achieved by a patient at any time on treatment, from random assignment to month 15 AEs: Safety issues related to thalidomide closely monitored at every visit. Explanation of grading of AEs NR Length of follow-up: Not explicitly stated. Median follow-up 47.5 months at time of data analysis in October 2008 |
| Results Primary outcomes |
MP + T | MP + placebo | p-value |
|---|---|---|---|
| OS, median (95% CI) | 44.0 months (33.4 to 58.7) | 29.1 months (26.4 to 34.9) | 0.028 |
| Overall deaths | 58/113 (51%) | 76/116 (65.5%) | 0.03 |
| Death – myeloma progression considered major cause | n = 36 | n = 54 | |
| Toxic death (intestinal perforation) | n = 1 | n = 1 | |
| Early death – after 1 month of treatment | n = 3 | n=3 | |
| Early death – after 3 months of treatment | n = 5 | n = 6 | |
| Comments | |||
| HR for median OS in favour of MP+T = 0.68 | |||
| Secondary outcomes | MP + T | MP + placebo | p-value |
| PFS, median (95% CI) | 24.1 months (19.4 to 29.0) | 18.5 months (14.6 to 21.3) | 0.001 |
| At least PR | 66/107 (62%) | 35/112 (31%) | < 0.001 |
| At least very good PR | 23/107 (21%) | 8/112 (7%) | < 0.001 |
| CR | 7/107 (7%) | 1/112 (1%) | < 0.001 |
| Disease progression occurrence | 72/113 (64%) | 84/116 (72%) | |
| Comments | |||
| HR for median PFS in favour of MP+T = 0.62 | |||
| AEs and safety | MP + T | MP + placebo | p-value |
| Peripheral neuropathy grade 1 | 20/113 (18%) | 19/116 (16%) | 0.003 reported. Although aligned in table with grade 1, appears more likely that this relates to all peripheral neuropathy |
| Peripheral neuropathy grade 2 | 21/113 (19%) | 4/116 (3%) | |
| Peripheral neuropathy grade 3 | 2/113 (2%) | 2/116 (2%) | |
| Neutropenia grade 3 or 4 | 26/113 (23%) | 10/116 (9%) | 0.003 |
| Thrombosis or embolism grade 3 or 4 | 7/113 (6%) | 4/116 (3%) | 0.33 |
| Somnolence grades 2–4 | 7/113 (6%) | 3/116 (3%) | 0.19 |
| Depression grades 2–4 | 8/113 (7%) | 3/116 (3%) | 0.11 |
| Constipation grades 2–4 | 19/113 (17%) | 12/116 (10%) | 0.16 |
| Nausea/vomiting grades 2–4 | 3/113 (3%) | 5/116 (4%) | 0.5 |
| Oedema grades 2–4 | 15/113 (13%) | 8/116 (7%) | 0.11 |
| Comments | |||
| There is contradictory information in text and Table 3 of this paper. For peripheral neuropathy grades 1 and 2 text states 21 (19%) grade 1 and 20 (18%) grade 2 in MP + T group but table has these the other way around (as shown here). For the MP + placebo group table states 17% with peripheral neuropathy grade 1, whereas text states 16%. Text appears correct as 19/116 is 16.4%. For neutropenia (grades 3 or 4) text states 25 (22%) for MP + T group but table has 26 (23%). There were no peripheral neuropathy events reported at grade 4 | |||
| Withdrawals | MP + T(n = 113) | MP + placebo (n = 116) | p-value |
| Withdrawals due to AEs/toxicity | n = 48 (42.5%)a | n = 15 (12.9%)a | |
| Peripheral neuropathy | n = 12 | n = 3 | |
| Neurological events (non-peripheral) | n = 10 | n = 1 | |
| Thrombosis/embolism | n = 7 | n = 1 | |
| Haematological events | n = 7 | n = 6 | |
| Digestive events | n = 4 | n = 2 | |
| Cardiac events | n = 3 | n = 1 | |
| Rash | n = 2 | n = 0 | |
| Other | n = 3 | n = 1 | |
| Dose reduction required because of AEs | n = 20 (17.7%)a | n = 3 (2.6%)a | |
| Median duration of treatment | 13.5 months | 18 months | |
| a Percentages calculated by reviewer. | |||
| Comments | |||
| There is contradictory information in text and Figure 1. Text states that nine MP + T group participants withdrew due to neurological events (non-peripheral), whereas Figure 1 shows 10 participants. Data provided on timing of withdrawal due to toxicity but appear to be for study overall, not by group: within 3 months, nine patients; within 6 months, 23 patients; within 12 months, 38 patients. Also unclear which patients are included, as patient numbers given alongside timing of withdrawals sum to 70, but only 63 patients (48 MP + T and 15 MP + placebo) withdrew due to toxicity. | |||
| Withdrawals overall | n = 100 (88.5%)a | n = 108 (93.1%)a | |
| Due to disease progression | n = 37 | n = 69 | |
| Due to death | n = 6 | n = 16 | |
| Due to consent withdrawal | n = 9 | n = 8 | |
| Due to toxicity (details above) | n = 48 | n = 15 | |
| a Percentages calculated by reviewer. | |||
| Methodological comments | |||
|
Allocation to treatment groups: Described as random in a 1 : 1 ratio with assignments provided centrally. No further details Blinding: Not explicitly described but assume this is a blinded study due to use of a placebo and statement that, if experimental treatment stopped due to grade 3–4 AEs or disease progression, unblinding occurred. Issue of patients taking thalidomide needing to comply with a risk-management programme (which would mean patients not blind to study drug) is not discussed. All patients may have been subject to the same protocol Comparability of treatment groups: Groups described as well balanced except for sex as there were more female participants in the MP + T group (p = 0.03) Method of data analysis: Parameters described by number and percentage of patients. Distributions of parameters assessed at inclusion compared between treatment groups using chi-squared tests for categoric variables and Kruskal–Wallis rank test for continuous variables. Best response rates at 12 months compared using the chi-squared test. OS calculated from random assignment to death from any cause. Data on patients alive at the time of analysis were censored in the survival analysis on the last date they were known to be alive. PFS calculated from random assignment to progression or death. Patients who had not experienced progression were censored on the last date they were known to be alive and progression free. Survival estimated with Kaplan–Meier product limit method and curves were compared with the stratified log-rank test on an ITT basis. HRs estimated by stratified Cox proportional hazards model for the ITT population. AEs compared between groups using the chi-squared test. ITT not defined Sample size/power calculation: Sample size was estimated to be 280 patients to guarantee, in a two-sided test, a power of 80% to detect an increase in the median survival time of 6 months. Power calculation assumed a median survival time of 22 months in the control group and a global type I error rate of 5%. Fewer than 280 patients were recruited, presumably because recruitment was stopped earlier than planned (although this is not explicitly stated) in December 2006 when a clear survival advantage for MPT was found in the IFM 99/06 trial and because the French Autorisation Temporaire d’Utilisation had made MPT available for newly diagnosed myeloma patients ineligible for high-dose therapy. The authors do not comment on any possible implications of the recruitment shortfall Attrition/dropout: Reasons for withdrawals reported (see outcomes above). After loss from the trial of those with disease progression, due to deaths, and withdrawals due to toxicity very few participants remained (MPT n = 8; MP + placebo n = 13) |
|||
| General comments | |||
|
Substantial renal insufficiency with creatinine serum concentration of 50 mg/l or more was an exclusion criterion. At baseline 13% of patient had severe renal failure (creatinine clearance < 30 ml/minute) Generalisability: This trial focuses on patients 75 years and older and the results may therefore only be applicable to patients in this age range. Authors state doses of melphalan and thalidomide were lower than had been used in similar trials with patients 65–75 years of age Outcome measures: Methods for grading of AEs not described Intercentre variability: No comments made regarding possible intercentre variability Conflict of interests: Two authors had consultant or advisory roles with Pharmion, Celgene and Janssen–Cilag for which they had been compensated. Three authors had received honoraria from Pharmion, Celgene and Janssen–Cilag |
|||
Quality criteria40
| Criteria for assessment of risk of bias in RCTs | Answera | Notes and comments |
|---|---|---|
| Was the method used to generate random allocations adequate? | NR | |
| Was the allocation adequately concealed? | Yes | Participants assigned centrally |
| Were the groups similar at the outset of the study in terms of prognostic factors, e.g. severity of disease? | Yes | Only difference was in proportion of women |
| Were the care providers, participants and outcome assessors blind to treatment allocation? If any of these people were not blinded, what might be the likely impact on the risk of bias (for each outcome)? | Unclear | Not explicitly stated but use of placebo suggests blinding in place. Plus text states that treatment was unblinded on participant withdrawal. However, those on thalidomide may have had to comply with a risk management programme but this is not discussed. Most outcomes were objective therefore risk of bias low |
| Were there any unexpected imbalances in dropouts between groups? If so, were they explained or adjusted for? | Unclear | Overall withdrawals similar – but greater withdrawals due to toxicity of thalidomide in MP + T group. Authors describe toxicity as acceptable |
| Is there any evidence to suggest that the authors measured more outcomes than they reported? | No | |
| Did the analysis include an ITT analysis? If so, was this appropriate and were appropriate methods used to account for missing data? | Unclear | Analysis described as ITT, although ITT was not defined |
Additional outcomes/comments/notes
| Outcomes | MPT | MP + placebo | p-value |
|---|---|---|---|
| Rescue treatment administered | 131 (84%) of 156 patients presenting with disease progression. Rate similar in the two groups (as in row below) | ||
| Prescription of any type of novel agent as rescue treatment after progression | 61/72 (85%) | 70/84 (83%) | |
| Thalidomide | 16/72 (22%) | 53/84 (63%) | |
| Bortezomib | 22/72 (31%) | 28/84 (33%) | |
| Lenalidomide | 11/72 (15%) | 9/84 (11%) | |
| Thalidomide and/or lenalidomide | 25/72 (35%) | 59/70 (83%) | |
| Thalidomide and/or lenalidomide and/or bortezomib | 38/72 (53%) | 68/81 (83%) | |
| Survival time after progression, median | 11.5 months | 9.9 months | 0.89 |
| Comments | |||
| Inevitably most patients (84% as noted above) in the study who had disease progression went on to have further treatment. The possible effects of the different rescue treatment on the outcomes of OS and survival after progression are not commented on by the authors of this paper. Survival after progression was described as ‘similar in the two groups’ by the authors who state that this strongly suggests the first-line treatment is of major importance in this population of elderly patients. The impact of the initial treatment on treatment decisions at progression are not commented on | |||
Data extracted by JOP, extraction checked by JB.
| Reference and design | Intervention | Participants | Outcome measures |
|---|---|---|---|
|
Author: Palumbo et al. 24 Year: 2006 Linked to later publication: Palumbo et al. 25 2008 Country: Italy Study design: Multicentre RCT Setting: Not stated, appears to be secondary care No. of centres: 54 Recruitment dates: January 2002 to May 2005 Funding: Supported by Associazione Italiana Ricerca Cancro, Milan; Associazione Italiana Leucemie, Rome; Compagnia di S. Paolo, Turin; Fondazione Neoplasie Sangue Onlus, Turin; Ministero Università Ricerca Scientifica e Tecnologica, Rome; Consiglio Nazionale delle Ricerche, Rome. Pharmion supplied free thalidomide for the study |
MPT: Thalidomide 100 mg daily dose administered continually during the six MPT cycles plus MP six cycles, each cycle repeated every 4 weeks: oral melphalan 4 mg/m2 on days 1–7; oral prednisone 40 mg/m2 on days 1–7 Note: After 6 × 4-week cycles of MPT, thalidomide was continued at 100 mg per day as maintenance therapy. This does not meet the inclusion criteria of the review, therefore only outcomes to 24 weeks are data extracted here Control, MP: Six cycles, each cycle repeated every 4 weeks: oral melphalan 4 mg/m2 on days 1–7; oral prednisone 40 mg/m2 on days 1–7 Note: In the control group there was no planned maintenance therapy Dose reductions: Dose reduced by 50% on the occurrence of any non-haematological grade 2 toxic effect Treatment stopped: Thalidomide stopped for any non-haematological grade 3 toxic effects Other interventions used: No anticoagulation prophylaxis was given initially but the protocol was amended December 2003 and enoxaparin at 40 mg per day was delivered subcutaneously during the first four cycles of therapy |
No. of participants: 331 overall (MPT : 167; MP: 164). However, only 255 had been followed up for 6 months or more at time of the initial analysis included here (no thalidomide maintenance) MPT : 129 MP : 126 Sample attrition/dropout: Of the 331 overall, 76 follow-up less than 6 months (MPT n = 38; MP n = 38). Of the 255 followed up for 6 months 63 had not completed six cycles (MPT n = 32; MP n = 31) (details in results) Timing of withdrawals: NR Sample crossovers: In the MP (control) group patients with progressive disease or relapse were permitted to cross over to receive thalidomide as salvage treatment Inclusion criteria for study entry: Older than 65 years of age, or younger but unable to undergo transplantation, with previously untreated stage II or III (DS criteria) MM and measurable disease (not defined) Exclusion criteria for study entry: Another cancer; psychiatric disease; any grade 2 peripheral neuropathy Abnormal cardiac function, chronic respiratory disease, and abnormal liver or renal functions were not criteria for exclusion Characteristics of participants (for those included in initial analysis): Median age, years: MPT 72; MP 72 Age < 65 years: 4/129 (3%); MP 3/126 (2%) Age 65–70 years: 49/129 (38%); MP 51/126 (41%) Age 71–75 years: 44/129 (34%); MP 37/126 (29%) Age 76–80 years: 26/129 (20%); MP 28/126 (22%) Age > 80 years: MPT 6/129 (5%); MP 7/126 (6%) Gender (M : F): NR Ethnicity: NR M-protein IgG class: MPT 83/129 (64%); MP 73/126 (58%) M-protein IgA class: MPT 31/129 (24%); MP 37/126 (29%) Bence-Jones protein: MPT 15/129 (12%); MP 16/126 (13%) DS stage IIA: MPT 50/129 (39%); MP 49/126 (39%) DS stage IIB: MPT 4/129 (3%); MP 3/126 (2%) DS stage IIIA: MPT 64/129 (50%); MP 62/126 (49%) DS stage IIIB: MPT 11/129 (8%); MP 12/126 (10%) WHO performance index ≥ 3: MPT 9/129 (7%); MP 6/126 (4%) Bone marrow plasmacytosis %, median (range): MPT 45 (5–95); MP 46 (5–95) Serum β2-microglobulin mg/l, median (range): MPT 116 patients, 3.7 (0.36–40); MP 110 patients, 3.7 (0.2–37.5) β2-microglobulin ≤ 3.5 mg/l: MPT 53/129 (41%); MP 53/126 (42%) β2-microglobulin > 3.5 mg/l: MPT 63/129 (49%); MP 57/126 (45%) β2-microglobulin data missing: MPT 13/129 (10%); MP 16/126 (13%) Plasma C-reactive protein mg/l, median (range): MPT 105 patients, 2.53 (0.005–157); MP 100 patients, 2.0 (0.001–128) Haemoglobin g/l, median (range): MPT 125 patients, 106 (73–147); MP 122 patients, 102 (67–155) Serum creatinine mg/l, median (range): MPT 129 patients, 8 (5.6–102); MP 125 patients, 8 (6–68) Calcium mmol/l, median (range): MPT 115 patients, 2.25 (1.22–3.17); MP 118 patients, 2.27 (1.09–2.72) |
Primary outcomes: Response rates and PFS Secondary outcomes: OS, time to first evidence of response, prognostic factors, frequency of any grade 3 or higher AEs Method of assessing outcomes: Visits every 4 weeks during chemotherapy regimens to monitor response to treatment by measurement of protein in serum and urine. Assessments every 2 months thereafter. Response rate assessed at 6 months and confirmed after a further 6 weeks. Bone marrow plasmacytosis and skeletal disease were included in response evaluation Response definitions used criteria of the EGBMT/IBMTR: CR – disappearance of myeloma protein in serum and urine and negative immunofixation PR – at least 50% reduction of myeloma protein in serum and a 90% decrease in urine. Near CR (subcategory of PR) – disappearance of myeloma protein in serum and urine and positive immunofixation MR – serum myeloma protein reduction of 25–49% and in urine of 50–89%. No response – reduction in myeloma protein of 24% or less Progressive disease – an increase of 25% or greater in myeloma protein AEs: Assessed at each visit and graded according to the National Cancer Institute Common Toxicity Criteria (version 2). Causes of death were recorded as attributable to myeloma, study drugs, other causes or a combination of these. Thromboembolism was assessed by clinically objective evidence of thrombosis and use of ultrasound echography Length of follow-up: Data analysed after median follow-up of 38.4 months (range 0.23–69.45 months; SD 16.5 months) in the MPT group and 37.7 months (range 0–72.34 months; SD 17.1 months) in the MP group. Due to use of thalidomide as maintenance therapy only 6-month data eligible for inclusion in review |
| Results | |||
|---|---|---|---|
| Primary outcomes | MPT | MP | Absolute difference: MPT – MP (95% CI) |
| Complete or PR at 6 months | 98/129 (76.0%) | 60/126 (47.6%) | 28.3% (16.5 to 39.1) |
| Complete response | 20/129 (15.5%) | 3/126 (2.4%) | 13.1% (6.3 to 20.5) |
| PR | 78/129 (60.4%) | 57/126 (45.2%) | 15.2% (3.0 to 26.9) |
| Near CR | 16/129 (12.4%) | 6/126 (4.8%) | |
| 90–99% myeloma protein reduction | 11/129 (8.5%) | 6/126 (4.8%) | |
| 50–89% myeloma protein reduction | 51/129 (39.5%) | 45/126 (35.7%) | |
| MR | 7/129 (5.4%) | 21/126 (16.7%) | –11.2% (–19.2 to –3.6) |
| No response | 7/129 (5.4%) | 19/126 (15.1%) | –9.7% (–17.4 to –2.2) |
| Progressive disease | 10/129 (7.8%) | 21/126 (16.7%) | –8.9% (–17.2 to –0.8) |
| Not available | 7/129 (5.4%) | 5/126 (4.0%) | |
| Secondary outcomes | MPT | MP | p-value |
| Time to PR, median (range) | 1.4 months (22–200 days) | 3.1 months (25–210 days) | |
| AEs and safety | MPT | MP | p-value |
| Grade 3–4 infections | 12/129 (10%) within the first 4 months of treatment: | 2/126 (2%), timing of occurrence unknown: | 0.01 |
| Pneumonia | 6 (5%) patients | 2 (2%) patients | |
| Upper respiratory tract | 2 (2%) patients | 0 patients | |
| Herpes zoster | 1 (1%) patient | ||
| Fever of unknown origin | 3 (2%) patients | ||
| Comments | |||
| Full AE reporting not data extracted because period that this covered and timing of the occurrence of the events was NR (therefore unable to distinguish between events occurring during the first 6 months of treatment and those occurring later during thalidomide maintenance) | |||
| Withdrawals | MPT | MP | p-value |
| Unable to complete six cycles owing to: | 32/129 (25%) | 31/126 (25%) | |
| AEs | 17/32 (13.2%)a | 4/31 (3.2%)a | |
| progressive diseases | 9/32 | 16/31 | |
| withdrew consent | 3/32 | 2/31 | |
| Lost to follow-up | 3/32 | 7/31 | |
| Protocol violations | 0/32 | 2/31 | |
| Thalidomide discontinuation required | 43 (33.3%)a patients after a median of 2.1 months | ||
| Thalidomide dose reduction to 50 mg required | 37 (28.7%)a patients after a median of 4 months | ||
| Methodological comments |
|---|
|
Allocation to treatment groups: A simple randomisation sequence was generated by a centralised computer. Registration to the trial was via the internet to centralised database. An automated assignment procedure concealed from the investigators randomly allocated patients to treatments Blinding: Study described as unblinded Comparability of treatment groups: For patients included in the 6-month follow-up states baseline demographics and other characteristics of the two groups were balanced but results of any statistical tests to confirm this not presented. Comparability of patient groups in final analysis NR Method of data analysis: For the analysis of the 6-month follow-up data times of observation were censored on 15 June 2005. Analysis was undertaken on an ITT basis (this is not defined). The absolute difference (with 95% CI) of the proportion of patients in each response category between the two groups was calculated with ci analysis, version 2.1.1. Methods for analysis of data NR here also described. The incidence of any AE was compared by the chi-squared test or Fisher’s exact test when cell counts were lower than five. The analyses were performed with sas (version 8.2) Sample size/power calculation: Sample size was estimated to be 380 patients (190 per arm) to detect a 10% increase in CR in the MPT arm (from 5% to 15%), with an α error of 0.05 and a β error of 0.10. Fewer than 380 patients were recruited because at the second interim analysis (timing of this not stated) there were statistically significant improvements for the MPT group in response rate and prolongation of event-free survival compared with the MP group. In addition enrolment was falling. The steering committee therefore decided to stop the trial in May 2005 when 331 patients had been randomised (87% of planned sample size). The authors do not comment on any possible implications of the recruitment shortfall Attrition/dropout: Reasons for withdrawals, but not timing of withdrawal, reported (see outcomes above) |
| General comments |
|
Generalisability: This trial focuses on patients 65 years and older and the results may therefore be applicable only to patients in this age range Outcome measures: Due to the use of thalidomide as a maintenance therapy only the first 6 months of data are eligible for inclusion. It is not clear whether the observed results would have been maintained longer term. Only some of the AEs were reported with an indication of when they occurred. It was not possible to extract all AE data and thus AEs are likely to be underrepresented in the data extraction Intercentre variability: No comments made regarding possible intercentre variability Conflict of interests: Two authors had received scientific adviser board and lecture fees from Pharmion and Celgene. However, their association with Celgene involved lenalidomide only, and not thalidomide. The other authors declared they had no conflict of interest |
Quality criteria40
| Criteria for assessment of risk of bias in RCTs | Answera | Notes and comments |
|---|---|---|
| Was the method used to generate random allocations adequate? | Yes | Generated by computer |
| Was the allocation adequately concealed? | Yes | Participants assigned centrally |
| Were the groups similar at the outset of the study in terms of prognostic factors, e.g. severity of disease? | Yes | Although no statistical evidence of similarity presented authors state groups were comparable and this appears to be the case |
| Were the care providers, participants and outcome assessors blind to treatment allocation? If any of these people were not blinded, what might be the likely impact on the risk of bias (for each outcome)? | No | States study is unblinded |
| Were there any unexpected imbalances in dropouts between groups? If so, were they explained or adjusted for? | Unclear | Overall withdrawals similar – but greater withdrawals due to toxicity of thalidomide in MPT group and greater withdrawals due to progression in MP group |
| Is there any evidence to suggest that the authors measured more outcomes than they reported? | No | |
| Did the analysis include an ITT analysis? If so, was this appropriate and were appropriate methods used to account for missing data? | Unclear | Analysis described as ITT, although ITT was not defined. |
Additional outcomes/comments/notes
| Nothing to add. |
Data extracted by JOP, extraction checked by JB.
| Reference and design | Intervention | Participants | Outcome measures |
|---|---|---|---|
|
Author: MMIX Trial (from investigators),49,50,52–54 Davies et al. ,42 MRC myeloma info guide,27 Owen et al. 48 Year: 2009 Country: UK Study design: Multicentre RCT Setting: Hospitals (AiC/CiC information has been removed) Recruitment dates: June 2003 to November 2007 Funding: Core grant from the MRC. Unrestricted educational grants provided by Pharmion, Novartis, Bayer-Schering, Chugai and Ortho Biotech. Leukaemia Research Fund supported some of the biological studies |
CTDa (cyclophosphamide, thalidomide, attenuated dexamethasone): Cyclophosphamide: Once a week, 500 mg orally (on days 1, 8, 15 and 22) Dexamethasone: Days 1–4 and 15–18 of each cycle, 20 mg daily (orally) Thalidomide: Daily, 50 mg daily for 4 weeks, increasing every 4 weeks by 50-mg increments to 200 mg daily Cycle length 4 weeks, to maximal response, but with a minimum–maximum no. of cycles of 6–9 Control, MP: Daily once a day by mouth for days 1–4 of a 4-week cycle. No. of cycles 6–9 Melphalan: 7 mg/m2 Prednisolone: dose 40 mg Note: After completion of induction chemotherapy, eligible patients entered a second randomisation to thalidomide maintenance or no maintenance. The initial randomisation to chemotherapy was not maintained, although initial chemotherapy was a stratification factor. As maintenance therapy does not meet the inclusion criteria of the review only outcomes from the induction chemotherapy are data extracted here Dose modification: MP: Treatment delay indicated by neutrophil and platelet counts. Melphalan reduced to 5 mg/m2 if serum creatinine >200 µmol/l CTDa: Treatment-related cytopenias led to omission of cyclophosphamide for one course, then dose reduction, e.g. to 400 mg or 300 mg. Cyclophosphamide omitted if serum creatinine is >300 µmol/l despite vigorous hydration Thalidomide stopped if a thromoboembolic event occurred. Under good anticoagulant control thalidomide could be started again at 50 mg, with escalation to 100 mg Thalidomide stopped for a cycle then reintroduced at 50 mg if grade 3–4 toxicity occurred In rare instance of intolerance to low-dose dexamethasone dose reduction or omission of one of the 4-day pulses per cycle was permitted Treatment stopped: CTDa: Pregnancy or suspected pregnancy (including in male patient’s partner) also led to stopping of thalidomide Other interventions used: In addition to the randomisation to CTDa or MP, participants were also randomised to either sodium clodronate 1600 mg daily or zoledronic acid 4 mg by infusion every 3–4 weeks. Treatment continued indefinitely, or at least until disease progression Thromboprophylaxis: Physicians were advised to consider full anticoagulation with warfarin or low-molecular-weight heparin for all patients at high risk of VTE42 Provision of thalidomide had to meet the approved process for thalidomide risk management and pregnancy prevention |
(AiC/CiC information has been removed) Timing of withdrawals: NR Sample crossovers: None Inclusion criteria for study entry: At least 18 years of age with newly diagnosed symptomatic MM or non-secretory MM (criteria provided). Provided written informed consent. Prepared to use contraception. Negative pregnancy test Exclusion criteria for study entry: Asymptomatic MM. Solitary plasmacytoma of bone. Extramedullary plasmacytoma (without evidence of myeloma). Previous or concurrent active malignancies, except surgically removed basal cell carcinoma of the skin or other in situ carcinomas. Previous treatment for myeloma except local radiotherapy to relieve bone pain or spinal cord compression; prior bisphosphonate treatment; low-dose corticosteroids, up to four single doses of corticosteroids (total dose 1 g methylprednisolone, 200 mg dexamethasone or 1.25 g prednisolone). Past history of ischaemic heart disease or psychiatric disorders – exclusion at discretion of clinician. Acute renal failure (unresponsive to 72 hours rehydration, creatinine > 500 µmol/l or urine output < 400 ml/day or requirement for dialysis) (AiC/CiC information has been removed) |
Primary outcomes: OS PFS Response Secondary outcomes: QoL Skeletally related events Height loss Toxicity (thromboembolic events; renal toxicity; haematological toxicity; graft-versus-host disease) Proportion receiving bortezomib–dexamethasone as ‘early rescue’ on induction chemotherapy, or at relapse Method of assessing outcomes: Response was assessed at the end of randomised induction chemotherapy Patients were followed up locally 4-weekly during chemotherapy, then 3-monthly thereafter. Central follow-up was 3-monthly until disease progression then annually thereafter QoL was assessed with the EORTC QLQ-C30, QLQ-MY24 and the EQ-5D at: Pre-initial randomisation (when patient unaware of treatment allocation); at 3, 6 and 12 months post initial randomisation and annually thereafter until maintenance randomisation or 5 years post initial randomisation A diary card was also used daily from initial randomisation for 3 months Indicators of skeletally related events collected at 3-monthly intervals Response definitions: Response assessments were according to the modified EBMT/IBMTR definitions (i.e. EMBT criteria plus the categories of very good PR, and early death). Reference provided Survival time calculated from randomisation to date of death from any cause. If patient still alive or lost to follow up they will be censored at date last known to be alive PFS – from randomisation to date of disease progression or death. Disease progression – relapse from CR (if patient had achieved this) or progressive disease (EBMT criteria) if not in CR AEs: SAEs, Hickman line infection, renal toxicity, sensory neuropathy, motor neuropathy, constipation, somnolence, infection, rash, elevated alkaline phosphatase, hypothyroidism, postural hypotension, thromboembolic events, osteonecrosis of the jaw, haematological toxicity, and pregnancy/suspected pregnancy summarised by trial arm/treatment group Length of follow-up (AiC/CiC information has been removed): The cut-off date for final analysis was 5 October 2009 |
| Results | |||
|---|---|---|---|
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| Comments | |||
|
Outcomes of OS, PFS and survival after progression: TTP not data extracted because after first-line chemotherapy participants meeting eligibility criteria were entered into a second randomisation to thalidomide maintenance therapy, which does not meet the inclusion criteria for this review (AiC/CiC information has been removed) |
|||
|
Secondary outcomes QoL |
CTDa | MP | p-value |
| Comments | |||
| (AiC/CiC information has been removed) | |||
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | ||
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | ||
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | ||
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | ||
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | ||
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | ||
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | ||
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | ||
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| Comments | |||
|
Data supplied by MMIX trialists states that AEs relate to induction chemotherapy. Denominator for AEs calculated by reviewer (AiC/CiC information has been removed) |
|||
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| Comments | |||
| (AiC/CiC information has been removed). No other withdrawals appear to be reported | |||
| Methodological comments | |||
|
Allocation to treatment groups: Conducted by a central trials office using an automated 24-hour telephone system. Random assignment in a 1 : 1 ratio to CTDa or MP and bisphosphonate (sodium clodronate or zoledronic acid). Allocations concealed until interventions assigned. Randomisation used a minimisation algorithm and was stratified by centre, haemoglobin, corrected serum calcium, serum creatinine, platelets Blinding: Not blinded Comparability of treatment groups: (AiC/CiC information has been removed) Method of data analysis: All summaries and analyses by ITT unless otherwise stated. The per-protocol population may also be used if deemed appropriate. ITT defined as all patients randomised, with the exception of those misdiagnosed. Only patients who withdraw consent for the study, or for whom no written informed consent was received are not included in the ITT population. The QoL population includes all randomised patients agreeing to take part in the QoL study. All hypothesis tests are two-sided and at the 5% significance level; p-values < 0.05 considered statistically significant. Primary end points will be ranked according to clinical relevance, i.e. survival and PFS have equal ranking and response a lower ranking. OS calculated from initial randomisation to death. Patients with missing follow-up data, or not known to have died at time of analysis will be censored on the last date they were known to be alive. PFS calculated from random assignment to progression or death. Patients with missing follow-up data or who had not experienced progression were censored on the last date they were known to be alive and progression free. There was no other censoring of data. Cox’s proportional hazards models used to compare chemotherapy groups while adjusting for bisphosphonate treatment group and the minimisation factors. Models will be constructed for OS and PFS. The proportional hazards assumptions will be assessed by plotting hazards over time for each treatment arm. Kaplan–Meier and adjusted curves will be constructed for each chemotherapy group. Response outcomes centrally reviewed and only responses from the central review will be reported. Subgroup analyses will be conducted (six subgroups defined) Sample size/power calculation: Sample size was based on testing the hypothesis that CTDa is superior to MP in terms of OS and PFS. It was anticipated that 850 patients (425 per group) would be randomised to induction chemotherapy in the non-intensive pathway. 204 patients (102 per group) would provide 80% power at a 5% significance level to detect a 15% absolute difference in 5-year survival (two-tailed test). This was based on the assumption of 15% 5-year survival in the MP group. 152 events would be required for these analyses. Sample size reached. For response, if 182 patients (91 per group) were entered the trial would be powered to detect an increase in the number of patients achieving a CR from 20% with MP as induction chemotherapy to 40% with CTDa (with 80% power at a 5% level of significance). The anticipated number per group (425) would provide more than 80% power to detect this difference. No power calculation was made for subgroups Attrition/dropout: Not reported on (apart from some data on participants withdrawing consent during induction chemotherapy) |
|||
| General comments | |||
|
The results of this study have not yet been fully published. The assessment team has had early access to data but these have not been peer reviewed. It is possible that some data, particularly those on QoL may alter as analyses are finalised Generalisability: Likely to be generalisable as the study took place in the UK Outcome measures: Methods for grading of AEs not described Intercentre variability: Not discussed Conflict of interests: States none |
|||
Quality criteria40
| Criteria for assessment of risk of bias in RCTs | Answera | Notes and comments |
|---|---|---|
| Was the method used to generate random allocations adequate? | Yes | An automated 24-hour telephone system was used but no further information |
| Was the allocation adequately concealed? | Yes | Participants assigned centrally |
| Were the groups similar at the outset of the study in terms of prognostic factors, e.g. severity of disease? | Yes | Not specifically stated but appear to be from baseline characteristics provided |
| Were the care providers, participants and outcome assessors blind to treatment allocation? If any of these people were not blinded, what might be the likely impact on the risk of bias (for each outcome)? | No | |
| Were there any unexpected imbalances in dropouts between groups? If so, were they explained or adjusted for? | NR | |
| Is there any evidence to suggest that the authors measured more outcomes than they reported? | No | |
| Did the analysis include an ITT analysis? If so, was this appropriate and were appropriate methods used to account for missing data? | Unclear | Analysis was ITT, with ITT defined. Unclear how missing data was accounted for |
Additional outcomes/comments/notes
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | |
| (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) |
Appendix 6 Karnofsky performance status and WHO performance status scores
Karnofsky performance status
-
100% – normal, no complaints, no signs of disease
-
90% – capable of normal activity, few symptoms or signs of disease
-
80% – normal activity with some difficulty, some symptoms or signs
-
70% – caring for self, not capable of normal activity or work
-
60% – requiring some help, can take care of most personal requirements
-
50% – requires help often, requires frequent medical care
-
40% – disabled, requires special care and help
-
30% – severely disabled, hospital admission indicated but no risk of death
-
20% – very ill, urgently requiring admission, requires supportive measures or treatment
-
10% – moribund, rapidly progressive fatal disease processes
-
0% – death.
WHO performance status scores
-
0 –Asymptomatic (fully active, able to carry on all pre-disease activities without restriction).
-
1 – Symptomatic but completely ambulatory (restricted in physically strenuous activity but ambulatory and able to carry out work of a light or sedentary nature; for example, light housework, office work).
-
2 – Symptomatic, < 50% in bed during the day (ambulatory and capable of all self-care but unable to carry out any work activities; up and about more than 50% of waking hours).
-
3 – Symptomatic, > 50% in bed, but not bedbound (capable of only limited self-care, confined to bed or chair 50% or more of waking hours).
-
4 – Bedbound (completely disabled, cannot carry on any self-care, totally confined to bed or chair).
-
5 – Death.
Appendix 7 SHTAC data summary of manufacturers’ submissions of clinical effectiveness
SHTAC peer review of clinical effectiveness in Celgene’s submission for bortezomib and thalidomide for MM
Comprehensiveness of ascertainment of published studies
Clinical effectiveness
-
The MS contains a narrative summary of trials, with the methods and results of each trial presented separately. Tabulation of details on study design and methodology, baseline characteristic of participants, efficacy outcomes, subgroups, second-line therapy, and AEs is presented in Appendix 2 of the MS (pp. 134–9).
-
There is no formal systematic review of clinical effectiveness evidence in the main body of the MS. However, a systematic review was conducted to identify clinical effectiveness evidence for the Bayesian meta-analysis that is reported in Appendix 4 of the MS, which was presented as a separate document.
-
– Were databases and dates of searches specified?
-
– Appendix 4 of the MS clearly reports search dates, search strategies and databases searched.
-
-
– Were search strategies supplied?
-
– Yes.
-
-
– Was enough detail provided to be reproducible?
-
– Sufficient detail was provided in Appendix 4 of the MS for the searches to be reproducible.
-
-
– Did they search/report on ongoing studies?
-
– No searches for ongoing studies are reported.
-
-
Did they search for conference proceedings?
-
– Conference proceedings were included in the searching.
-
-
– How much of the data is CIC/AIC?
-
– The submission contains little CIC information and no AIC information. CIC information is located only on p. 54 and pp. 84–6 of the submission and all relates to the same clinical study report (CSR).
-
-
Cost-effectiveness
The MS economic evaluation section states that a literature search was conducted to identify cost-effectiveness models, but no search strategy is presented. There does not appear to have been a search for QoL data.
Searches identified
-
XXX clinical trials (details)
The systematic review conducted as part of the MTC and reported in Appendix 4 included the following RCTs (the references are as cited in Celgene’s submission116):
-
IFM 99/06 (Facon 2007 and Facon 2004 abstract); IFM 01/01 (Hulin 2009, three Hulin 2007 abstracts); GIMEMA (Palumbo 2008, Palumbo 2006, and Palumbo abstracts of 2004–8); Nordic study (Gulbransen 2008 abstract, Waage 2007 abstract, and Nordic Myeloma Study Group PowerPoint slide presentation 2009); HOVON 49 (two Wijermans 2008 abstracts, Wijermans 2008 ASH PowerPoint presentation ‘Final analysis. The HOVON 49 study’); VISTA (San Miguel 2008, San Miguel 2008 abstracts, San Miguel 2007 abstract; Palumbo 2008 abstract; Harousseau 2008 abstract). The studies identified in the systematic review are the same as those reported on in the main submission document. The submission document also recognises the Myeloma IX study is ongoing but the study was not included as complete data were not available.
-
What study types (X RCTs, X cohort studies, etc.)?
-
– The included studies were RCTs.
-
-
Did any meet our inclusion criteria that we have not already included?
-
– The identified studies published as full papers (IFM 99/06, IFM 01/01, GIMEMA and VISTA) are included in the SHTAC systematic review. As the GIMEMA study included maintenance therapy with thalidomide, the SHTAC review only includes outcomes reported for the period prior to the start of maintenance therapy. SHTAC also identified abstracts reporting on the Nordic myeloma group study, and the HOVON 49 study but the powerpoint presentations had not been identified. Owing to the limited reporting of methodological details and outcome data these studies were not included in the SHTAC systematic review but have been briefly mentioned as ongoing studies.
-
Clinical analysis
-
Any major differences in evidence reported?
-
– The MS includes a narrative summary for individual trials, with tabulation of the studies’ characteristics and results located in MS Appendix 2. There was no quality assessment of the trials. AEs are also presented separately for each trial.
-
-
Are their conclusions are similar to ours?
-
– Although the Celgene MS (but not the MTC) included the OS outcome from Palumbo and colleagues,24 which SHTAC excluded due to the use of thalidomide maintenance treatment in the MPT arm, the conclusions (based on narrative summary) on the clinical effectiveness of MPT and VMP are broadly similar. MPT and VMP treatments both show better OS and PFS than MP. The conclusions from the MS MTC were the same. A summary statement on response outcomes from the included trials is not provided. The MS presents an indirect comparison (as noted below), which suggests that MPT provides better PFS outcomes than VMP at 6, 12 and 18 months but the credibility intervals cross 1. The MS finds subgroup data variable and insufficient, so no conclusions have been drawn.
-
-
Any indirect comparisons?
-
– The MS included an indirect comparison to enable comparison of MPT and VMP as there are no head-to-head trials for this comparison. Not all of the studies identified by the systematic review were included in the meta-analysis and indirect comparison. The base case excluded the GIMEMA trial (on the basis of a different regimen of thalidomide not consistent with the label, and due to cross over to thalidomide in the MP arm after disease progression), and the Nordic and HOVON 49 trials (insufficient information in abstracts for meta-analysis). These three studies were included in a sensitivity analysis (using information from slide presentations for Nordic and HOVON 49 trials).
-
-
Any differences in outcome measures?
-
– The MS reports on the same outcome measures as the SHTAC review. Outcome data were not reported from the studies included in the systematic review presented in MS Appendix 4.
-
-
Any extra AE info?
-
– Adverse event information was restricted to that reported in trial publications.
-
Interpretation
-
Does their interpretation of the clinical data match their analyses?
-
– Limited analyses in main MS document (mainly just narrative summary) but where analyses are presented, for example MTC, the interpretation of the clinical data broadly matches these.
-
Questions
-
Any areas of uncertainty/discrepancy compared with the SHTAC review?
-
– The MS presents a narrative summary of the Palumbo and colleagues24 study as well as the Nordic and HOVON 49 trials (as cited in Celgene’s submission116), which have been reported only in abstract form. However, these three studies were not included in the base-case MTC, and therefore the data in the base-case MTC more closely match the data included in the SHTAC review.
-
– The SHTAC excluded most of the data from Palumbo and colleagues24 because participants in the MPT group received thalidomide maintenance therapy. In contrast, this study was excluded from the MS MTC because the thalidomide regimen was inconsistent with the label and because participants could cross over to thalidomide at disease progression. SHTAC do not believe that on this latter point the study differs substantially from the IFM trials,23,59 where participants received treatment after disease progression that could include thalidomide, and where a greater proportion of participants in the MP groups received thalidomide at this point than in the MPT group.
-
SHTAC peer review of clinical effectiveness in Janssen–Cilag’s submission for bortezomib and thalidomide for MM
Comprehensiveness of ascertainment of published studies
Clinical effectiveness
The MS contains a systematic review of clinical effectiveness evidence in the main body of the report. Summary details on trial size, interventions, inclusion criteria, efficacy end points, and duration of treatment are tabulated. Trials were critically appraised. Results from the included studies were tabulated.
-
Were databases and dates of searches specified?
-
– The MS briefly summarises the searches and clearly reports search dates, search strategies and databases searched in Appendix 1. Searches were conducted in two phases, before and after the finalisation of the scope of the appraisal.
-
-
Were search strategies supplied?
-
– Yes.
-
-
Was enough detail provided to be reproducible?
-
– Sufficient detail was provided for the searches to be reproduced.
-
-
Did they search/report on ongoing studies?
-
– No searches for ongoing studies are reported.
-
-
Did they search for conference proceedings?
-
– Conference proceedings were included in the searching.
-
-
How much of the data is CIC/AIC?
-
– The submission contains both CIC and AIC information. CIC data appears on the following pages: 32–4; 60; 61; 67; 70; Appendix 4 (from VISTA); References: all three clinical study reports (J&J, Velcade CSR 1, CSR 2, CSR 3). AIC data appears on pages 2; 3; 33–6; 40–41; 43–47; 59–60; 66; Appendices 7, 8 and 11.
Cost-effectiveness
A review of economic evaluations was conducted, reported in detail in Appendix 9. The review sought to identify any economic evaluations and resource use studies assessing the first-line therapy of patients with MM with regimens included in the NICE scope, as well as others, for example VAD that did not form part of the final scope. The review included studies assessing first-line chemotherapy regimens but also included induction/mobilisation regimens prior to transplantation. Appendix 9 reports on 30 studies, but the MS states that none of these cost-effectiveness studies included bortezomib-based regimens in the patient group of interest. There does not appear to have been a search for QoL data.
Searches identified
-
XXX clinical trials (details)
The systematic review included the following RCTs:
-
VMP versus MP VISTA (Dimopoulos et al. 2008;112 Harousseau et al. 2008;113 San Miguel et al. 2008;60 San Miguel et al. 2008;114 San Miguel et al. 200826).
-
MPT versus MP IFM 99/06 (Facon et al. 200723); GIMEMA [Palumbo et al. 2006 (not in MS reference list, presume24) and 200825]; IFM 01/01 (Hulin et al. 200959); HOVON 49 (Wijermans et al. 200864); Gulbrandsen et al. 2008 (Gulbrandsen et al. 200866)].
-
MP versus CTDa (maintenance treatment: thalidomide only) MRC Myeloma IX study (non-intensive arm) (Owen 2009; Morgan 2009, not in MS reference list).
-
What study types (X RCTs, X cohort studies, etc.)?
-
– The included studies were RCTs.
-
-
Did any meet our inclusion criteria which we have not already included?
-
– The identified studies published as full papers (IFM 99/06, IFM 01/01, GIMEMA, and VISTA) are included in the SHTAC systematic review. As the GIMEMA study included maintenance therapy with thalidomide the SHTAC review only includes outcomes reported for the period prior to the start of maintenance therapy. SHTAC also identified abstracts reporting on the Nordic myeloma group study (Gulbrandsen), and the HOVON 49 study. Due to the limited reporting of methodological details and outcome data these studies were not included in the SHTAC systematic review but have been briefly mentioned as ongoing studies. Abstracts for the MRC Myeloma IX study were identified, but not the two cited by the MS, the second of which is not referenced in the MS.
-
Clinical analysis
-
Any major differences in evidence reported?
-
– The MS includes a narrative summary and tabulation of the studies’ characteristics. The main efficacy results are very briefly summarised and tabulated. Trials were subject to critical appraisal using a modification of the CONSORT Assessment Framework. The VISTA study is additionally presented in more detail including some data that is not in the public domain. A small amount of non-RCT evidence from Phase I/II trials of bortezomib is presented.
-
-
Are their conclusions are similar to ours?
-
– Although the Janssen–Cilag MS systematic review included more studies than SHTAC, the conclusions (based on narrative summary) on the clinical effectiveness of MPT and VMP are broadly similar. The results from meta-analysis and indirect comparison are more difficult to compare with the SHTAC results because of additional data used in the MS and the different methodology (MS winbugs MTC, SHTAC pairwise meta-analysis). For the comparisons of MPT versus MP and MPV versus MP the direction of the overall effect is the same, although the magnitude differs. It appears that the MS MTC indicates a greater difference in effect in favour of MPV over MPT than the SHTAC pairwise estimates suggest.
-
-
Any indirect comparisons?
-
– The MS included an indirect comparison to enable comparison of MPT and VMP as there are no head-to-head trials for this comparison. The studies identified by the systematic review were included and, in addition, unpublished updated survival data from the VISTA trial were also included in the meta-analysis and indirect comparison.
-
-
Any differences in outcome measures?
-
– The MS reports on the same outcome measures as the SHTAC review.
-
-
Any extra AE info?
-
– Adverse event information was restricted to that reported in trial publications.
-
Interpretation
-
Does their interpretation of the clinical data match their analyses?
-
– The interpretation of clinical data appears to match the analyses that have been undertaken.
-
Questions
-
Any areas of uncertainty/discrepancy compared with the SHTAC review?
-
– The MS has included final data from the Palumbo and colleagues24 study, which SHTAC did not include, as well as the Nordic and HOVON 49 trials, which have only been reported in abstract form and were therefore not included by SHTAC (with the HOVON 49 trial designated ‘unclear’ because of the use of thalidomide maintenance therapy). The impact of including these studies within the MTC presented by the MS is uncertain and SHTAC cannot determine what the outcomes would have been had these data been excluded from the MTC.
-
Appendix 8 Table of excluded studies for systematic review of cost-effectiveness
| Excluded reference | Reason for exclusion |
|---|---|
| Sampson FC, Beard SM, Scott F, Vandenberghe E. Cost-effectiveness of high-dose chemotherapy in first-line treatment of advanced multiple myeloma. Br J Haematol 2001;113:1015–19 | Participants and intervention |
| Deniz B, Facon T, Singer I, Micallef-Eynaud P, Joseph I, Shearer A, et al. Economic evaluation of thalidomide combined with melphalan and prednisone in previously untreated multiple myeloma in Scotland. Blood 2008;112:835 | Abstract |
| Cecchi M, Caccese E, Messori A, Orsi C, Tendi E. Cost-effectiveness of bortezomib in multiple myeloma. Pharm World Sci 2007;29:485–6 | Participants |
| Yoong K, Attard C, Jivraj F, Shustik C, Reece D. Cost effectiveness analysis of bortezomib in previously untreated multiple myeloma patients in Canada. Value Health 2009;12:A272 | Abstract |
| Joseph I, Facon T, Lewis P, Deniz HB, Caro JJ. Cost effectiveness of thalidomide combined with melphalan and prednisone in previously untreated multiple myeloma in Wales. Value Health 2009;12:A271 | Abstract |
| De Abreu Lourenco R, Colman S, Lee C. Thalidomide plus melphalan and prednisone for Australian patients newly diagnosed with multiple myeloma is cost effective when compared with melphalan and prednisone alone. Value Health 2009;12:A381 | Abstract |
| Wang S, Huang H, Shi H, Duh M, Chen K. The cost effectiveness of bortezomib for the initial treatment of multiple myeloma in the United States. #1379. 51st ASH Annual Meeting and Exposition, New Orleans, LA, 5–8 December 2009 | Abstract |
Appendix 9 Table of excluded studies for systematic review of health-related quality of life
| Excluded reference | Reason for exclusion |
|---|---|
| Sherman AC, Simonton S, Latif U, Plante TG, Anaissie EJ. Changes in quality-of-life and psychosocial adjustment among multiple myeloma patients treated with high-dose melphalan and autologous stem cell transplantation. Biol Blood Marrow Transplant 2009;15:12–20 | Outcome |
| Lee SJ, Richardson PG, Sonneveld P, Schuster MW, Irwin D, San Miguel JF, et al. Bortezomib is associated with better health-related quality of life than high-dose dexamethasone in patients with relapsed multiple myeloma: results from the APEX study. Br J Haematol 2008;143:511–19 | Participants and outcome |
| Sherman AC, Simonton S, Latif U, Spohn R, Tricot G. Psychosocial adjustment and quality of life among multiple myeloma patients undergoing evaluation for autologous stem cell transplantation. Bone Marrow Transplant 2004;33:955–62 | Outcome |
| Gulbrandsen N, Wisloff F, Brinch L, Carlson K, Dahl IM, Gimsing P, et al. Health-related quality of life in multiple myeloma patients receiving high-dose chemotherapy with autologous blood stem-cell support. Med Oncol 2001;18:65–77 | Participants and outcome |
| Multiple myeloma: QALY gains from optimal therapy. Drugs Ther Perspect 2000;16:12–16 | Outcome |
| Ellis K, Smith AG. An evaluation of quality of life (QOL) in patients after treatment for multiple myeloma (MM). Br J Haematol 2005;129:192 | Outcome |
| Thomas ML. Quality of life in persons with multiple myeloma: a descriptive study. Blood 2001;98:4971 | Outcome |
| Deniz B, Morgan G, Schey S, Ishak J, Dale P, Shearer A et al. Economic evaluation of lenalidomide combined with dexamethasone for the treatment of multiple myeloma in the UK. Blood 2008;112:836–7 | Outcome |
| Belch A, Reece DE, Bahlis NJ, White D, Teixeira B, Camacho F, et al. Bortezomib [VELCADE (TM)], pegylated liposomal doxorubicin [DOXIL/CAELYX (R)] and dexamethasone in the treatment of previously untreated multiple myeloma patients: impact on quality-of-life. Blood 2007;110:A1058–9 | Outcome |
| Petrucci MT, Calabrese E, Levi A, Federico V, Ceccolini M, Rizzi D, et al. Costs and quality of life of multiple myeloma (MM) in Italy: the Co. Mim Study. Value Health 2009;12:A265 | Abstract |
| Meunier J, Regnault A, Robinson D, Rosa K, Miguel JFS, van de Velde H, et al. Impact of tumor response on health-related quality of life (HRQOL) in newly diagnosed multiple myeloma patients treated with velcade/melphalan/prednisone (V-MP): results from the Vista Trial. Value Health 2009;12:A284 | Abstract |
| Dhawan R, Meunier J, Regnault A, Robinson D, Rosa K, Cakana A, et al. Impact of complete response on quality of life in newly diagnosed multiple myeloma patients. Clin Lymphoma Myeloma 2009;9:S58 | Abstract |
Appendix 10 Health-related quality-of-life studies – data extraction forms
Reference
-
Gulbrandsen and colleagues (2004). 91
-
Data extracted by KC; extraction checked by AC.
Study characteristics
Research question
-
What are the stated objectives of the study?
To compare QoL scores of MM patients at diagnosis and over time with the scores of a reference population.
-
Describe the type of study and study design.
Two prospective studies using a QoL questionnaire with comparison to a reference population through regression.
-
Was the sample from (1) the general population, (2) patients with the disease of interest, (3) individuals with knowledge of the disease, (4) other?
Patients from two prospective Nordic Myeloma Study Group trials: high-dose melphalan (HDM), and melphalan and prednisone (MP)
-
What are the characteristics of the baseline cohort for the evaluation?
| Age | < 60 years old for HDM; > 60 years old treated with MP |
| Sex | NR |
| Race (if appropriate) | NR |
| Indication/disease | Newly diagnosed MM |
| Other characteristics (sample size) | 221 patients for HDM and 203 patients for MP. QoL was also estimated for reference Norwegian population, consisting of 3000 randomly selected adult individuals (18–93 years) |
| QoL instrument | EORTC QLQ-C30 at baseline and at 1, 6, 12, 24 and 36 months |
| Utility values (Y/N) | N |
| Treatment effect (if reported) | NR |
Country/setting
-
What is the country and setting for the evaluation?
Denmark, Sweden and Norway.
Data sources
Effectiveness
-
Were the QoL data derived from: a single (observational) study, a review/synthesis or combination of previous studies, expert opinion?
Two trials.
Results
Results reported as mean difference between scores of all newly diagnosed MM patients and age- and gender-adjusted reference population.
| Mean score difference | 95% CI for the difference | p-value | |
|---|---|---|---|
| Functioning scales | |||
| Global QoL | –24.3 | –21.7 to –26.9 | < 0.001 |
| Physical functioning | –34.3 | –31.8 to –36.7 | < 0.001 |
| Role functioning | –48.4 | –45.4 to –51.3 | < 0.001 |
| Social functioning | –21.0 | –18.1 to –23.9 | < 0.001 |
| Emotional functioning | –14.0 | –11.7 to –16.4 | < 0.001 |
| Cognitive functioning | –5.8 | –3.5 to –8.1 | < 0.001 |
| Symptom scales | |||
| Nausea/vomiting | 5.6 | 4.1 to 7.1 | < 0.001 |
| Pain | 26.7 | 23.4 to 29.9 | < 0.001 |
| Fatigue | 19.1 | 16.3 to 21.9 | < 0.001 |
| Single items | |||
| Sleep disturbance | 6.1 | 2.8 to 9.3 | < 0.001 |
| Appetite loss | 15.4 | 13.1 to 17.7 | < 0.001 |
| Diarrhoea | –1.1 | –3.5 to 1.2 | 0.349 |
| Constipation | 10.4 | 7.7 to 13.1 | < 0.001 |
| Dyspnoea | 6.9 | 4.2 to 9.6 | < 0.001 |
| Financial impact | 5.3 | 2.7 to 8.0 | < 0.001 |
Change in most important functioning and symptom scales during the first 3 years for patients who received MP (values estimated from graphs).
| Reference group | 0 months | 1 month | 6 months | 12 months | 24 months | 36 months | |
|---|---|---|---|---|---|---|---|
| Global QoL | 70 | 46 | 52 | 60 | 60 | 60 | 60 |
| Physical functioning | 78 | 46 | 51 | 60 | 60 | 60 | 63 |
| Role functioning | 85 | 43 | 45 | 58 | 59 | 61 | 66 |
| Social functioning | 81 | 70 | 70 | 76 | 76 | 75 | 72 |
| Fatigue | 30 | 51 | 48 | 38 | 41 | 40 | 42 |
| Pain | 27 | 52 | 38 | 30 | 33 | 33 | 33 |
-
Were the methods for deriving these data adequately described (give sources if using data from other published studies)?
Yes. The EORTC QoL questionnaire was used.
Mapping
-
If a model was used, describe the type of model (e.g. regression) or other conversion algorithm.
Not applicable.
Conclusions/implications
-
Give a brief summary of the author’s conclusions from their analysis.
At diagnosis, the most distressing problems were pain and fatigue, reduced physical functioning, limitations in role functioning and reduced overall QoL. These differences from the reference population were statistically significant, and large or moderate, according to the rating systems. After the start of treatment, small to moderate improvement in mean QoL scores was observed for most domains.
-
What are the implications of the study for the model?
This study indicates that QoL is worse initially at diagnosis but improves after end of treatment. Long-term QoL appears stable but is lower than for the reference population.
Reference
-
Mujica-Mota and colleagues (2004). 94
-
Data extracted by KC; extraction checked by AC.
Study characteristics
Research question
-
What are the stated objectives of the study?
To map HRQoL measurements into generic utility measures (EQ-5D).
-
Describe the type of study and study design.
Utility mapping study; limited details of statistical mapping process provided.
-
Was the sample from (1) the general population, (2) patients with the disease of interest, (3) individuals with knowledge of the disease, (4) other?
Patients with relapsed and refractory MM.
-
What are the characteristics of the baseline cohort for the evaluation?
| Age | NR |
| Sex | NR |
| Race (if appropriate) | NR |
| Indication/disease | Patients with relapsed and refractory MM |
| Other characteristics (sample size) | Sample size of SUMMIT-1 trial (n = 202) identified but not all of the sample used for mapping study |
| QoL instrument | EORTC QLQ-C30 and MY24, FACT Fatigue and GOG-Ntx mapped to EQ-5D |
| Utility values (Y/N) | Y |
| Treatment effect (if reported) | NR |
Country/setting
-
What is the country and setting for the evaluation?
Although the setting is not stated, the SUMMIT-1 trial was undertaken in the USA.
Data sources
Effectiveness
-
Were the QoL data derived from: a single (observational) study, a review/synthesis or combination of previous studies, expert opinion?
Phase II trial.
Results
Utility scores appear similar across patient groups as defined by serological response to Velcade, with an overall utility score of 0.65.
-
Were the methods for deriving these data adequately described (give sources if using data from other published studies)?
Limited details of the methods or results are reported in the abstract.
Questions relevant to the EQ-5D were identified from EORTC and FACT, and five summary measures of severity, corresponding to the five EQ-5D dimensions, were obtained. The summary measures were transformed into the corresponding EQ-5D scale for each dimension.
Mapping
-
If a model was used, describe the type of model (e.g. regression) or other conversion algorithm.
Not applicable.
Conclusions/implications
-
Give a brief summary of the author’s conclusions from their analysis
Method used to derive utility scores from reported HRQoL outcomes is a feasible and sensitive option for providing valid estimates of patient well-being for terminal conditions.
-
What are the implications of the study for the model?
Study provides a post-treatment utility measure for relapsed or refractory MM patients post treatment with Velcade. This is not the patient group or intervention for the evaluation.
Reference
-
Slovacek and colleagues (2008). 95
-
Data extracted by KC; extraction checked by AC.
Study characteristics
Research question
-
What are the stated objectives of the study?
To analyse an effect of selected demographics, psychosocial and health aspects on QoL in MM survivors treated with high-dose chemotherapy followed by autologous PBPCT.
-
Describe the type of study and study design.
Observational study. Mailed QoL questionnaire.
-
Was the sample from (1) the general population, (2) patients with the disease of interest, (3) individuals with knowledge of the disease, (4) other?
Patients with MM scheduled to be treated with high-dose chemotherapy (single dose of melphalan) followed by PBPCT.
-
What are the characteristics of the baseline cohort for the evaluation?
| Age | Mean 60 years (53–67 years) |
| Sex | 18 M, 14 F |
| Race (if appropriate) | NR |
| Indication/disease | MM treated with HDT followed by autologus PBPCT |
| Other characteristics (sample size) | Total n = 32 |
| QoL instrument | EQ-5D and EQ-5D VAS |
| Utility values (Y/N) | Y |
| Treatment effect (if reported) | Not applicable |
Country/setting
-
What is the country and setting for the evaluation?
University Hospital, Hradec Kralove, Czech Republic. All patients scheduled for intensive treatment of MM.
Data sources
Effectiveness
-
Were the QoL data derived from: a single (observational) study, a review/synthesis or combination of previous studies, expert opinion?
Single observational study.
Results
-
Summarise the results.
The global QoL in respondents with MM treated with HDT followed by autologous PBPCT was 0.689 for EQ-5D and 0.666 for EQ-5D VAS.
For individual dimensions, 59% had trouble with mobility, 19% had trouble with self-care, 81% had difficulty with their normal activity, 69% had medium to serous pain, and 59% had medium to serious anxiety/depression.
The study also presented QoL results by age.
| QoL measure | Age (years) | |||
|---|---|---|---|---|
| 40–49 | 50–59 | 60–69 | 70–79 | |
| EQ-5D score | 0.815 | 0.742 | 0.642 | 0.615 |
| EQ-5D VAS | 0.775 | 0.673 | 0.604 | 0.712 |
-
Were the methods for deriving these data adequately described (give sources if using data from other published studies)?
Yes, the Czech version of the EuroQol EQ-5D questionnaire was used in the study (Slovacek 2005 reference no. 2). The EQ-5D questionnaire was mailed to respondents with a covering letter. The QoL was analysed for the effect of age, sex, level of education, marital status, number of associated diseases, smoking, abuse, religion and time lapse from PBPCT.
Mapping
-
If a model was used, describe the type of model (e.g. regression) or other conversion algorithm.
Not applicable.
Conclusions/implications
-
Give a brief summary of the author’s conclusions from their analysis.
Global QoL was at a low level for all studied patients and reduces with increasing age. Smokers and former smokers have lower QoL than non-smokers.
-
What are the implications of the study for the model?
The study assesses a different population group and intervention than assessed in the NICE appraisal.
Reference
-
Strasser-Weippl and Ludwig (2008). 92
-
Data extracted by AC; extraction checked by KC.
Study characteristics
Research question
-
What are the stated objectives of the study?
To evaluate the prognostic importance of baseline QoL and whether QoL at onset of therapy is a truly independent prognostic factor. To identify which dimensions of QoL are important predictors for outcome in patients with MM.
-
Describe the type of study and study design.
Substudy within an RCT of continuous or intermittent prednisolone plus vincristine, melphalan, cyclophosphamide, prednisolone, interferon-α-2b (VMCP-IFN-α-2b) for induction therapy. Maintenance therapy of IFN-α-2b with or without prednisolone twice weekly.
-
Was the sample from (1) the general population, (2) patients with the disease of interest, (3) individuals with knowledge of the disease, (4) other?
Elderly patients recently diagnosed with MM who were previously untreated (ECOG performance status of ≤ 3, adequate organ function) (n = 92).
-
What are the characteristics of the baseline cohort for the evaluation?
| Age (years) | Median (range): 66 (43–84) |
| Sex | M/F: 51 : 41 |
| Race (if appropriate) | NR |
| Indication/disease | MM – DS stage: I – 5 (5.4%); II – 26 (28.3%); III – 61 (66.3%) |
| Other characteristics (sample size) | n = 92 |
| QoL instrument | EORTC QLQ-C30 |
| Utility values (Y/N) | N |
| Treatment effect (if reported) | RCT showed similarity between two treatment arms with respect to response rate, PFS and OS. No data are presented |
Country/setting
-
What is the country and setting for the evaluation?
Vienna, Austria.
Data sources
Effectiveness
-
Were the QoL data derived from: a single (observational) study, a review/synthesis or combination of previous studies, expert opinion?
A substudy within an RCT.
Results
Observed, age/gender equivalent expected mean scores and deviations for myeloma patients at baseline.
| Observed | Expected | Observed – expected | p-value | |
|---|---|---|---|---|
| Global QoLa | 47.28 | 70.63 | –22.3 | 1.60 × 1015 |
| Physicala | 58.74 | 80.75 | –22.01 | 2.26 × 1010 |
| Rolea | 58.4 | 87.04 | –28.64 | 3.82 × 1015 |
| Emotionala | 66.67 | 83.61 | –16.94 | 1.30 × 107 |
| Cognitivea | 78.44 | 82.89 | –4.45 | 0.04 |
| Sociala | 71.2 | 82.34 | –11.14 | 0.001 |
| Fatigueb | 49.14 | 30.17 | 18.97 | 6.0 × 109 |
| Painb | 47.64 | 25.92 | 21.72 | 8.5 × 108 |
| Nausea/vomitingb | 13.04 | 4.18 | 8.86 | 0.001 |
| Dyspnoeab | 32.25 | 19.41 | 12.84 | 4.1 × 105 |
| Insomniab | 32.61 | 25.79 | 6.82 | 0.035 |
| Appetite lossb | 28.99 | 7.30 | 21.69 | 1.9 × 108 |
| Constipationb | 22.71 | 15.30 | 7.41 | 0.024 |
| Diarrhoeab | 8.79 | 9.62 | –0.83 | 0.64 |
| Financial difficultiesb | 12.59 | 10.66 | 1.93 | 0.22 |
-
Were the methods for deriving these data adequately described (give sources if using data from other published studies)?
Yes.
Mapping
-
If a model was used, describe the type of model (e.g. regression) or other conversion algorithm.
Regression techniques were used to evaluate QoL as a prognostic indicator in relation to outcomes such as survival.
Conclusions/implications
-
Give a brief summary of the author’s conclusions from their analysis.
Study showed low levels of functional QoL scores and increased symptom scores in patients with active disease at start of first-line therapy, supporting previous reports of severe and significant impairment of QoL in MM patients. Although independent of age and gender, they did reflect parameters of disease activity that were thought to be linked to individual psychological factors. It was felt that physical measures of QoL, such as pain, fatigue, physical functioning and global QoL, were particularly important.
There is a significant impairment of physical and psychosocial dimensions of QoL in patients with MM at baseline compared with a healthy reference population. Low psychosocial QoL at baseline is associated with poor prognosis.
-
What are the implications of the study for the model?
The study provides baseline measures of QoL on the EORTC QLQ-C30 for patients recently diagnosed with MM who have not undergone treatment. If these can be mapped to utility measures it may provide a source for the model.
Reference
-
Uyl-de Groot and colleagues (2005). 96
-
Data extracted by AC; extraction checked by KC.
Study characteristics
Research question
-
What are the stated objectives of the study?
To investigate the subjective well-being of patients with newly diagnosed MM who were treated in a tandem transplantation programme.
All patients were scheduled for the following treatment protocol: two courses of VAD or VAMP chemotherapy, HDM and transplantation of whole blood stem cells, collection of r-met HuG-CSF mobilised peripheral blood progenitor cells by leucopheresis and, finally, HDT (busulfan/cyclophosphamide) followed by reinfusion of the previously collected peripheral stem cells (PSCT).
-
Describe the type of study and study design.
Prospective, longitudinal questionnaire study.
-
Was the sample from (1) the general population, (2) patients with the disease of interest, (3) individuals with knowledge of the disease, (4) other?
Patients with MM irrespective of previous treatment regimes who were scheduled for intensive treatment between March 1997 and December 1998, whether at the start of the treatment protocol or who were undergoing treatment and had not passed the last two measurement points for QoL (started between March 1995 and September 1996).
-
What are the characteristics of the baseline cohort for the evaluation?
| Age (years) | All patients (n = 51): mean (SD) 53 (7.2); median (minimum/maximum) 54 (31/65) |
| Patients in analysis (n = 25): mean (SD) 53 (8.2); median (minimum/maximum) 55 (31/65) | |
| Sex | All patients (n = 51): male 31 (61%); female 20 (39%) |
| Patients in analysis (n = 25): male 16 (64%); female 9 (36%) | |
| Race (if appropriate) | NR |
| Indication/disease | MM |
| DS stage [n (%)] | |
| All patients (n = 51): Ia 12 (24); IIa 4 (8); IIIa 32 (63); IIIb 3 (6) | |
| Patients in analysis (n = 25): Ia 8 (32); IIa 1 (4); IIIa 15 (60); IIIb 1 (4) | |
| Other characteristics (sample size) | n = 51; 35 from the start of the treatment protocol and 16 partially completed treatment |
| QoL instrument | EORTC QLQ-C30, EQ-5D |
| Data collected at baseline (2 weeks post induction therapy); day of hospital discharge after HDM (T2); 1 month after discharge after HDM (T3); day of hospital admission for PSCT (T4); day of discharge following PSCT (T5); 6 months post discharge following PSCT (T6); 12 months post discharge following PSCT (T7) | |
| Utility values (Y/N) | Y |
| Treatment effect (if reported) | NR |
Country/setting
-
What is the country and setting for the evaluation?
Local referring hospitals and the academic hospital at the VU University Medical Centre, Amsterdam, the Netherlands.
Data sources
Effectiveness
-
Were the QoL data derived from: a single (observational) study, a review/synthesis or combination of previous studies, expert opinion?
A single observational study.
Results
Mean absolute scores (SD) at baseline after VAD/VAMP (baseline) and mean change scores from baseline
| Baseline (n = 25) | T2 (n = 22) | T3 (n = 24) | T4 (n = 15) | T5 (n = 14) | T6 (n = 15) | T7 (n = 12) | |
|---|---|---|---|---|---|---|---|
| EORTC QLQ-C30 functioning scales | |||||||
| Physical | 50 (28) | –2 | 13b | 13a | –19a | 13a | 20a |
| Role | 41 (29) | 2 | 18b | 14 | –26b | 19a | 20 |
| Emotional | 72 (22) | 3 | 10a | 6 | 0 | 0 | 1 |
| Cognitive | 76 (25) | –11 | 8 | 1 | –6 | 3 | 3 |
| Social | 59 (30) | 5 | 12 | 6 | –23a | 10 | 13a |
| Global QoL | 58 (23) | –11a | 3 | 10a | –17b | 7 | 4 |
| EORTC QLQ-C30 symptoms | |||||||
| Fatigue | 55 (29) | 7 | –15a | –13 | 10 | –13 | –6 |
| Nausea/vomiting | 11 (25) | 26b | 2 | –1 | 27a | –1 | 4 |
| Pain | 37 (29) | –7 | –8 | –10 | 4 | –9 | –11 |
| Appetite loss | 22 (31) | 40b | 2 | –4 | 43b | –4 | –3 |
| Diarrhoea | 18 (31) | 25b | –1 | 0 | 36b | –2 | 3 |
| Disease/treatment-related symptoms | |||||||
| Pain in back | 43 (37) | –6 | –14a | –7 | –21a | –7 | –11 |
| Soreness of mouth | 9 (20) | 26b | 1 | –11 | 36b | –2 | –6 |
| Change in taste | 20 (32) | 23a | 6 | –9 | 21 | –4 | –8 |
| Diminished sexual interest | 52 (40) | 11 | –1 | –27a | –12 | –20 | –22 |
| Pain in bones | 35 (35) | –20a | –4 | –7 | –21a | –9 | –6 |
| EuroQol utility | 0.52 (0.33) | 0.03 | 0.14a | 0.14 | –0.14 | 0.12 | 0.17 |
Mean absolute scores (SD) at baseline and at 12 months’ follow-up for the patients who proceeded to PSCT 12 months’ follow-up
| Baseline: patients who proceeded to 12 months’ follow-up (n = 12) | 12 months’ follow-up | ||
|---|---|---|---|
| Patients with baseline (n = 12) | All patients (n = 26) | ||
| EORTC QLQ-C30 functioning scales | |||
| Physical | 65 (28) | 85 (15) | 78 (19) |
| Role | 53 (32) | 72 (18) | 71 (21) |
| Emotional | 74 (19) | 74 (20) | 78 (19) |
| Cognitive | 79 (21) | 82 (21) | 85 (17) |
| Social | 69 (27) | 82 (25) | 82 (25) |
| Global QoL | 66 (23) | 70 (16) | 69 (19) |
| EORTC QLQ-C30 symptoms | |||
| Fatigue | 42 (30) | 35 (27) | 30 (26) |
| Nausea/vomiting | 1 (5) | 6 (13) | 3 (9) |
| Pain | 28 (30) | 17 (17) | 15 (16) |
| Appetite loss | 6 (13) | 3 (10) | 1 (7) |
| Diarrhoea | 0 (0) | 3 (10) | 1 (7) |
| Disease/treatment-related symptoms | |||
| Pain in back | 31 (39) | 19 (17) | 22 (19) |
| Soreness of mouth | 11 (22) | 6 (13) | 4 (11) |
| Change in taste | 14 (22) | 6 (13) | 7 (22) |
| Diminished sexual interest | 56 (38) | 33 (40) | 40 (38) |
| Pain in bones | 19 (30) | 14 (17) | 17 (7) |
| EuroQol utility | 0.60 (0.33) | 0.77 (0.13) | 0.79 (0.18) |
-
Were the methods for deriving these data adequately described (give sources if using data from other published studies)?
Yes. The EORTC QLQ-C30 and the EQ-5D are outlined as are the methods for their application.
Mapping
-
If a model was used, describe the type of model (e.g. regression) or other conversion algorithm.
Not applicable.
Conclusions/implications
-
Give a brief summary of the author’s conclusions from their analysis.
The authors found an improvement in subjective well-being on the EORTC QLQ-C30 and the EQ-5D for patients who were able to complete the treatment programme. There was a trend towards improved functioning and reduced symptoms. There were declines associated with the provision of treatment; however, improvements did occur with time.
-
What are the implications of the study for the model
Although the study provides utility outcomes for MM patients, these are related to a different patient group and to different treatment regimens.
This study indicates that QoL is worse initially at diagnosis and treatment but improves after end of treatment. Long-term QoL appears stable but is lower than for the reference population.
Study characteristics
Research question
-
What are the stated objectives of the study?
Estimate the cost–utility of intensive chemotherapy versus intensive chemotherapy followed by myeloablative chemotherapy with autologous stem cell rescue in newly diagnosed patients with MM.
-
Describe the type of study and study design.
Cost–utility study based on a RCT in patients ≤65 years old with previously untreated MM. Trial of intensive chemotherapy versus intensive chemotherapy followed by myeloablative chemotherapy with autologous stem cell rescue.
-
Phase I VAD remission–induction therapy (three to four cycles at 28-day intervals), Phase II cyclophosphamide and autologous stem cell collection, Phase III intensive melphalan (two cycles at 8-week intervals), Phase IV peripheral blood SCT for patients in myeloablative group (cyclophosphamide/total body irradiation), Phase V maintenance with interferon-α-2a.
-
Was the sample from (1) the general population, (2) patients with the disease of interest, (3) individuals with knowledge of the disease, (4) other?
Patients with undiagnosed and untreated MM.
-
What are the characteristics of the baseline cohort for the evaluation?
| Age (years) | Mean (range): intensive chemotherapy 55 (38–65); myeloablative 55 (32–65) |
| Sex | Intensive chemotherapy 74 M, 55 F; myeloablative therapy 81 M, 51 F |
| Race (if appropriate) | NR |
| Indication/disease | Newly diagnosed MM and stage II or II A/B disease; in intensive arm 32/129 stage IIA; 89/129 stage IIIA; 8/129 stage IIIB; myeloblative arm 26/132 stage IIA; 92/132 stage IIIA; 11/132 stage IIIB |
| Other characteristics (sample size) | 129 in intensive chemotherapy arm and 132 in myeloablative treatment arm |
| QoL instrument | EQ-5D assessed up to 24 months and then assumed to be stable until 36 months |
| Utility values (Y/N) | Y |
| Treatment effect (if reported) | Median OS in myeloablative treatment group 47 months vs 50 months in intensive chemotherapy group (p = 0.41) |
Country/setting
-
What is the country and setting for the evaluation?
Holland and Belgium.
Data sources
Effectiveness
-
Were the QoL data derived from: a single (observational) study, a review/synthesis or combination of previous studies, expert opinion?
Single study.
Results
Authors state that patients in an undefined state following intentionally curative primary therapy would have a QoL 19.5% lower than those in the general population (0.8), i.e. QoL is 0.644 [0.8 – (0.195 × 0.8)].
Utility values for the different treatment groups
| Time from randomisation (months) | Intensive chemotherapy | Myeloablative treatment |
|---|---|---|
| 6 | 0.81 | 0.65 |
| 12 | 0.80 | 0.62 |
| 18 | 0.81 | 0.69 |
| 24 | 0.77 | 0.75 |
-
Were the methods for deriving these data adequately described (give sources if using data from other published studies)?
Limited detail given on methodology or results in present study. Reference given for more detail: Segeren CM. Intensive therapy in MM. Thesis. Rotterdam: Erasmus University; 2002.
Mapping
-
If a model was used, describe the type of model (e.g. regression) or other conversion algorithm.
Not applicable.
Conclusions/implications
-
Give a brief summary of the author’s conclusions from their analysis.
Cost-effectiveness of myeloma therapy after 3 years of follow-up seems not to be favoured by myeloablative treatment with autologous stem cell rescue. Cost per QALY at 3 years: intensive €37,328; myeloablative €51,357.
-
What are the implications of the study for the model?
Although the study assessed the QoL in newly diagnosed and untreated people with MM, it focused on interventions not included in the current evaluation. It provides an indication of the QoL following curative treatment and over 2-year period of treatment.
Appendix 11 Cost-effectiveness data extraction forms for manufacturers’ submissions
Reference
Janssen–Cilag (2009). 115
Research question
-
What are the stated objectives of the evaluation?
To provide a cost-effectiveness analysis, reporting the total costs associated with the interventions under consideration in the appraisal and the QALYs gained (p. 49).
Funding source
-
Janssen–Cilag.
Study population
-
What definition was used for [condition]?
The patient population is newly diagnosed patients ineligible for HDT–SCT in line with the scope of the appraisal.
-
What are the characteristics of the baseline cohort for the evaluation?
The characteristics of the baseline cohort are not specified but the authors report that they are reflective of the UK population and the trial evidence.
Interventions and comparators
-
What interventions/strategies were included?
Bortezomib in combination therapy with an alkylating agent and a corticosteroid. Thalidomide in combination therapy with an alkylating agent and a corticosteroid
-
Was a no treatment/supportive care strategy included?
Compared with melphalan + prednisone (MP)
-
Describe interventions/strategies.
Bortezomib + melphalan + prednisone (VMP). Thalidomide + melphalan + prednisone (MPT). Thalidomide + cyclophosphamide + dexamethasone (CTDa)
Analytical perspective
-
What is the perspective adopted for the evaluation [health service, health and PSS, third party payer, societal (i.e. including costs borne by individuals and lost productivity]?
UK NHS and PSS.
Study type
-
Cost-effectiveness/cost–utility/cost–benefit analysis?
Cost–utility.
Institutional setting
Where is/are the intervention(s) being evaluated usually provided?
NHS inpatient care.
Country/currency
-
Has a country setting been provided for the evaluation? What currency are costs expressed in and does the publication give the base year to which those costs relate?
UK, pounds sterling, 2008–9 costs.
Effectiveness
-
Were the effectiveness data derived from: a single study, a review/synthesis of previous studies or expert opinion?
A meta-analysis was conducted to estimate the treatment effects.
-
Give the definition of treatment effect used in the evaluation.
The treatment effects for VMP, MPT and CTDa were estimated using constant HRs for PFS and OS relative to MP.
-
Give the size of the treatment effect used in the evaluation.
| (AiC/CiC information has been removed) |
Intervention costs
-
Were the cost data derived from: a single (observational) study, a review/synthesis of previous studies or expert opinion?
Treatment unit costs are based on the BNF No. 5735 and MIMS 2009. 99
-
Were the methods for deriving these data adequately described (give sources if using data from other published studies)?
Yes.
-
List the direct intervention costs used in the evaluation – include resource estimates (and sources for these estimates, if appropriate) as well as sources for unit costs used.
Summary of costs
| Dose | Duration of treatment | Unit cost | |
|---|---|---|---|
| Bortezomib | 1 × 3.5-mg vial | Mean no. of vials used in the VISTA trial: (AiC/CiC information has been removed) (J&J, Velcade CSR 1) | £762 |
| Thalidomide |
CTDa arm: 167 mg per day MPT arm: 150 mg per day |
315 days | £298 per 28 tablets |
| Enoxaparin as thromboprophylaxis | 40 mg per day | 6 months (four cycles of thalidomide) | 40 mg/0.4 ml: 10 syringes = £40.36 |
| Melphalan |
9 mg/m2 Four doses/cycle; 28 doses/course |
Seven cyclesa | £11.46 for 25 tablets of 2 mg |
| Prednisone |
60 mg/m2 Four doses/cycle; 28 doses/course |
Seven cyclesb | £20 for 56 tablets of 25 mg; £0.98 for 28 tablets of 5mg |
| Cyclophosphamide |
500 mg Four doses/cycle; 30 doses/course |
Seven and a half cyclesb | US$12.44 for 100 tables of 50 mg |
Indicate the source for individual cost values (if appropriate).
Other direct costs (costs incurred directly in treating patients)
-
Were the cost data derived from: a single (observational) study, a review/synthesis of previous studies expert opinion?
Costs for subsequent treatment and AEs were from previous studies. Incidence of the included AEs was from the RCTs.
-
Were the methods for deriving these data adequately described (give sources if using data from other published studies)?
Yes.
-
List the costs used in the evaluation – if quantities of resource use are reported separately from cost values, show sources for the resource estimates as well as sources for unit costs used.
Upon disease progression, patients have second-line treatment. Costs for second- and third-line treatment are shown in Table 28 of the MS.
The unit costs of treating AEs (Table 25) were applied to the incidence of AEs (Table 24) to obtain the total cost of treating AEs (Table 26). Unit costs of AEs are mainly from NICE TA17131 for lenalidomide.
Unit costs of AEs
| AE | Cost (£) | Care setting |
|---|---|---|
| Anaemia | 430.53 | Day case |
| Deep venous thrombosis | 199.00 | Outpatient |
| Haematological | 455.00 | Day case |
| Infection | 685.00 | Day case |
| Leukopenia | 470.00 | Day case |
| Lymphopenia | 470.00 | Day case |
| Neurological | 580.00 | Day case |
| Neutropenia | 470.00 | Day case |
| Non-haematological toxicity (≥ grade 3) | 97.00 | Outpatient |
| Oedema (peripheral) | 0.85 | Outpatient |
| Peripheral neuropathy | 97.00 | Outpatient |
| Thrombocytopenia | 547.89 | Day case |
Indicate the source for individual cost values (if appropriate).
Indirect costs (costs due to lost productivity, unpaid inputs to patient care)
-
Were indirect costs included?
None.
-
Describe how indirect costs were estimated (e.g. how days of lost productivity were estimated and how those days were valued).
Not applicable (indicate the source for individual cost values if appropriate).
Health-state valuations/utilities (if study uses QoL adjustments to outcomes)
-
Were the utility data derived from: a single (observational) study, a review/synthesis of previous studies or expert opinion? Were the methods for deriving these data adequately described (give sources if using data from other published studies)?
Single study (Van Agthoven et al. 2004).
-
List the utility values used in the evaluation:
| Utilities | For prior to response to treatment state | For response state | For post-progression state |
|---|---|---|---|
| EQ-5D (UK weights) – (Van Agthoven et al. 2004) | 0.77 | 0.81 | 0.64 |
Modelling
-
If a model was used, describe the type of model used (e.g. Markov state transition model, discrete event simulation). Was this a newly developed model or was it adapted from a previously reported model? If an adaptation, give the source of the original.
A cost–utility decision-analytic model was used in this economic evaluation. The model, developed using Excel, considers a cohort of newly diagnosed myeloma patients and defines a baseline response, disease progression and survival based on treatment with MP. Treatment effects for VMP, MPT and CTDa are then modelled over time by adjusting this baseline patient experience via HRs. Further lines of treatment (second- and third-line) are taken into consideration to estimate the total treatment costs.
The analytic framework was based on a variant of Quality-Adjusted Analysis of Time Without Symptoms or Toxicity (Q-TWiST97) using partitioned survival analysis, and utilises the area under and the difference between time-to-event curves to estimate mean durations spent within the disease states of interest.
-
What was the purpose of the model (i.e. why was a model required in this evaluation)?
Not reported. However, model needed to extrapolate trial data over patient lifetime.
-
What are the main components of the model (e.g. health states within a Markov model)? Are sources for assumptions over model structure (e.g. allowable transitions) reported? List them if reported.
Survival is partitioned into three different states: (1) prior to response to treatment; (2) response but no progression; and (3) post-progression. Death represents the final state. The steps to estimate the mean periods in these states are described below and the approach is presented schematically in Figure 15:
-
Step 1 Estimate mean OS (µOS) from start of treatment until death.
-
Step 2 Estimate mean PFS (µPFS) from start of treatment until progression or death.
-
Step 3 Estimate mean survival after progression (µPROG) as µOS – µPFS.
-
Step 4 Estimate mean time until response (µPreRESP) from start of treatment until response, progression, or death (include all patients, such that non-responders will either have event time as that of progression or death, or will be censored if they drop out).
-
Step 5 Estimate mean time from response to progression or death (µDOR) as µPROG – µPreRESP.
FIGURE 15.
Partitioned survival framework. TTRPD, time to response or disease progression.

To determine QALYs over the life of a patient, utilities for the following health states were assigned:
-
from start of treatment until response (uPRE)
-
from response to progression (ur)
-
from progression to death (uPROG).
-
Extract transition probabilities for [natural history/disease progression] model and show sources (or refer to table in text).
None stated.
-
What is the model time horizon?
30-year time horizon.
-
What, if any, discount rates have been applied in the model? Same rate for costs and outcomes?
3.5%.
Results/analysis
-
What measure(s) of benefit were reported in the evaluation?
Cost per QALY gained.
-
Provide a summary of the clinical outcome/benefits estimated for each intervention/strategy assessed in the evaluation:
| CTDa | MPT | MP | VMP | |
|---|---|---|---|---|
| QALYs (discounted) | 3.07 | 3.41 | 2.86 | 4.03 |
Provide a summary of the costs estimated for each intervention/strategy assessed in the evaluation:
| CTDa | MPT | MP | VMP | |
|---|---|---|---|---|
| Costs (discounted) (£) | 56,668 | 59,322 | 54,434 | 66,676 |
-
Synthesis of costs and benefits – are the costs and outcomes reported together (e.g. as cost-effectiveness ratios)? If so, provide a summary of the results.
The ICER for VMP versus MP is estimated to be £10,498. Furthermore, the ICERs of VMP versus MPT and VMP versus CTDa are estimated to be £11,907 and £10,411, respectively.
Base-case results
| VMP vs MP | VMP vs MPT | VMP vs CTDa | MPT vs MP | MPT vs CTDa | CTDa vs MP | |
|---|---|---|---|---|---|---|
| Incremental QALYs | 1.17 | 0.62 | 0.96 | 0.55 | 0.34 | 0.20 |
| Incremental cost (£) | 12,241 | 7353 | 10,007 | 4888 | 2654 | 2234 |
| Incremental ICER (£) | 10,498 | 11,907 | 10,411 | 8912 | 7724 | 10,905 |
-
Give results of any statistical analysis of the results of the evaluation.
Survival curves are presented for PFS and OS.
-
Was any sensitivity analysis performed – if yes, what type(s) [i.e. deterministic (one-way, two-way, etc.) or probabilistic]?
One-way sensitivity analyses and PSAs have been undertaken. Two alternative scenario analyses have also been undertaken.
-
What scenarios were tested in the sensitivity analysis? How do these relate to structural uncertainty (testing assumptions over model structure such as relationships between health states), methodological uncertainty (such as choices of discount rate or inclusion of indirect costs) or parameter uncertainty (assumptions over values of parameters in the model, such as costs, QoL or disease progression rates)?
One-way sensitivity analyses have been undertaken for a limited number of analyses, including different survival distributions for OS and PFS, alternative HRs for OS, dose and duration of thalidomide, utilities, time horizon and discounting rate.
A PSA was undertaken using Monte Carlo simulation with 10,000 iterations. All parameters in the model were included except medication costs.
-
Give a summary of the results of the sensitivity analysis: did they differ substantially from the base-case analysis? If so, what were the suggested causes?
The results are generally robust to changes in the sensitivity analyses. The model is most sensitive to the following parameters: underlying MP survival hazard, HRs for OS, dose of thalidomide, and duration of treatment with thalidomide in the MPT arm.
For the PSA, at the £20,000 and £30,000 willingness-to-pay thresholds, VMP has the highest probability of being cost-effective: 64% and 75%, respectively.
Two scenarios were conducted:
Scenario A assumes there is no subsequent therapy after first-line treatment:
| Mean QALYs | Mean cost (£) | ICER vs MP (£) | |
|---|---|---|---|
| MP | 2.86 | 13,888 | – |
| CTDa | 3.07 | 23,810 | 48,437 |
| MPT | 3.41 | 23,188 | 16,956 |
| VMP | 4.03 | 38,574 | 21,099a |
Scenario B assumes that the same second-line therapies as those treated with MP in the VISTA trial. The results were similar for this scenario to the base-case analyses.
Conclusions/implications
-
Give a brief summary of the author’s conclusions from their analysis.
Base-case results from the model demonstrated that VMP is more costly, but more effective than comparator treatments.
-
What are the implications of the evaluation for practice?
None stated.
Reference
Celgene (2009). 116
Research question
-
What are the stated objectives of the evaluation?
To compare the costs and benefits of adding thalidomide (T) to the combination melphalan and prednisolone (MP) with those of MP alone and bortezomib in combination with melphalan and prednisolone (VMP) in patients with MM older than 65 years or who are ineligible for HDT.
Funding source
Celgene Ltd.
Study population
-
What definition was used for [condition]?
Patients with untreated MM aged 65 years and over or who are ineligible for HDT.
-
What are the characteristics of the baseline cohort for the evaluation?
The characteristics of the baseline cohort are not discussed.
Interventions and comparators
-
What interventions/strategies were included?
MPT compared with MP alone and with VMP.
-
Was a no treatment/supportive care strategy included?
No. Comparisons as defined above.
-
Describe interventions/strategies
Comparisons defined above.
Analytical perspective
-
What is the perspective adopted for the evaluation [health service, health and PSS, third-party payer, societal (i.e. including costs borne by individuals and lost productivity)]?
NHS and PSS.
Study type
-
Cost-effectiveness/cost–utility/cost–benefit analysis?
Cost–utility analysis.
Institutional setting
-
Where is/are the intervention(s) being evaluated usually provided?
Patients were treated in several settings. Although the majority of care was provided as day-case and outpatient care, there was some provision of care within inpatient and primary care. The effect of the setting is taken into account in resources and costs.
Country/currency
-
Has a country setting been provided for the evaluation? What currency are costs expressed in and does the publication give the base year to which those costs relate?
The evaluation is for England and Wales, with costs expressed as pounds sterling. The base year for costs appears to be 2008, although some costs are for 2007–9 and 2009.
Effectiveness
-
Were the effectiveness data derived from: a single study, a review/synthesis of previous studies or expert opinion? Give the definition of treatment effect used in the evaluation.
Treatment effects were calculated from a Bayesian MTC of data originating from three trials,23,26,59 using survival time before and after progression as the primary outcomes. PFS was assumed to be equivalent to TTP. The percentage of patients at 6-month intervals was used with extrapolation using an exponential distribution. Treatment interruptions/reductions were included in sensitivity analysis for MPT through reduction in average dose. AEs were included through data from two trials. 23,26 PPS was reported as if patients had changed treatments from their original treatment to a similar but different treatment. PPS was calculated by combining the MPT, MP and MEL100 arms from the IFM 99/06 trial to create an average survival curve. 23 Average survival at different time points was then extrapolated through an exponential distribution. Treatment interruptions/discontinuations were encompassed in the trial efficacy data, with no alteration to costs in the base case. Differences in costs were assessed.
-
Give the size of the treatment effect used in the evaluation.
Meta-analysed odds ratios of PFS for MPT compared with MP and VMP – random effects:
| Comparison | Point estimate | 95% CI |
|---|---|---|
| MPT vs MP – 6 months | 2.63 | 1.03 to 7.01 |
| MPT vs VMP – 6 months | 1.08 | 0.23 to 5.21 |
| MPT vs MP – 12 months | 2.15 | 0.92 to 5.1 |
| MPT vs VMP – 12 months | 1.07 | 0.25 to 4.47 |
| MPT vs MP – 18 months | 2.10 | 0.83 to 5.24 |
| MPT vs VMP – 18 months | 1.02 | 0.22 to 4.81 |
| MPT vs MP – 24 months | 2.19 | 0.9 to 5.07 |
| MPT vs VMP – 24 months | 0.85 | 0.2 to 3.52 |
| MPT vs MP – 30 months | 2.70 | 1.1 to 6.55 |
Meta-analysed odds ratios of PPS for MPT compared with MP – random effects:
| Comparison | Point estimate | 95% CI |
|---|---|---|
| MPT vs MP – 6 months | 1.04 | 0.37 to 2.81 |
| MPT vs MP – 12 months | 1.05 | 0.41 to 2.66 |
| MPT vs MP – 18 months | 1.15 | 0.47 to 3.03 |
| MPT vs MP – 24 months | 0.99 | 0.38 to 2.68 |
| MPT vs MP – 30 months | 1.15 | 0.43 to 3.06 |
Intervention costs
-
Were the cost data derived from: a single (observational) study, a review/synthesis of previous studies or expert opinion?
Resources and costs were obtained from several sources. NHS resources were obtained from an unpublished survey of UK Haematologists by Celegene Ltd. 117 Inpatient, outpatient and day-case hospitalisation costs were derived from NHS reference costs. 100 Costs of medicines were from the BNF No. 5735 and costs of blood transfusions from Wilson et al. 101 with costs inflated to 2008. 102
-
Were the methods for deriving these data adequately described (give sources if using data from other published studies)?
Yes.
-
List the direct intervention costs used in the evaluation – include resource estimates (and sources for these estimates, if appropriate) as well as sources for unit costs used.
Medication and preparation costs used in model base case:
| Medication | Drug acquisition cost (£) | Cost per mg (£) | Dosing (mg/day)a | Drug acquisition cost per cycle (£) | Preparation costs (£) |
|---|---|---|---|---|---|
| Bortezomib | 762.40 for 1 × 3.5-mg vial | 217.83 | 1 vial | 6099.04 (cycles 1–4), 3049.52 (cycles 5–9) | 159.93 per dose |
| Melphalan | 11.46 for 25 × 2 mg | 0.229 | 0.25 | 16.23 | 0 |
| Prednisone | 1.95 for 28 × 5 mg | 0.014 | 2 | 7.87 | 0 |
| Thalidomide | 298.48 for 28 × 50 mg | 0.213 | 238.1 | 2132.04 | 13.64 per cycle |
Indicate the source for individual cost values (if appropriate).
Other direct costs (costs incurred directly in treating patients)
-
Were the cost data derived from: a single (observational) study, a review/synthesis of previous studies or expert opinion?
Other resource use and cost data were provided for outpatient consultations, disease monitoring and treatment of AEs/complications. Resources and costs used and their sources are outlined below.
-
Were the methods for deriving these data adequately described (give sources if using data from other published studies)?
Yes. The costs of AEs were calculated by combining resource use data from the survey of haematologists with unit costs to estimate total costs. These costs and trial data on the frequency of AEs23 were then used to calculate a weighted average cycle cost. AE management costs were calculated for the entire time horizon for each AE through addition of the average medication and treatment cost (weighted for setting of care). This was then multiplied by the proportion of occurrence of AEs over the total number of AEs in the treatment arm. The costs per specific AE are then summed to provide an average cost per AE, which is applied at the time of the AE.
-
List the costs used in the evaluation – if quantities of resource use are reported separately from cost values, show sources for the resource estimates as well as sources for unit costs used.
Unit costs and mean number of regular outpatient consultations and disease monitoring tests:
| Cost (£) | Source | Frequency (mean no. of assessments per year) | |||
|---|---|---|---|---|---|
| Preprogression | Post progression | ||||
| Active treatment | Off active treatment | ||||
| Outpatient | 82 | OP | 12 | 6 | 12 |
| Tests to monitor therapy response and disease status | |||||
| Routine blood counts (FBC) | 2.99 | H | 10.7 | 7.1 | 20.1 |
| Clotting | 2.99 | H | 1.1 | 0.4 | 3.9 |
| INR monitoring | 2.99 | H | 2.9 | 0.4 | 2.6 |
| Biochemistry (U&Es) | 1.34 | P | 9.7 | 6.6 | 17.3 |
| Liver function tests | 1.34 | P | 7.6 | 5.1 | 14.6 |
| Erythrocyte sedimentation rate | 2.99 | H | 1.4 | 0.9 | 2.6 |
| Plasma viscosity | 1.34 | P | 0.3 | 0.3 | 1.6 |
| Uric acid (urate) | 1.34 | P | 1.4 | 0.9 | 2.7 |
| Immunoglobulin | 1.34 | P | 6.4 | 4.9 | 9.7 |
| Paraprotein measurements | 1.34 | P | 7.6 | 6.1 | 11.1 |
| Protein electrophoresis | 1.34 | P | 6.7 | 5.1 | 9.6 |
| Serum β2-microglobulin | 1.34 | P | 3.0 | 2.0 | 5.0 |
| Serum erythropoietin level | 1.34 | P | 0.1 | 0.1 | 0.5 |
| Immunofixation | 1.34 | P | 3.4 | 2.9 | 4.8 |
| Creatinine clearance | 1.34 | P | 0.7 | 0.4 | 2.3 |
| Glomerular filtration rate | 1.34 | P | 3.3 | 2.7 | 7.1 |
| Serum-free light chains | 1.34 | P | 2.9 | 1.7 | 4.1 |
| Routine urinalysis | 1.34 | P | 1.7 | 1.0 | 4.4 |
| 24-hour urine measurement | 1.34 | P | 1.3 | 1.0 | 3.0 |
| 24-hour urine for creatinine | 1.34 | P | 0.6 | 0.1 | 1.4 |
| Total urine protein (24 hour) | 1.34 | P | 1.4 | 0.4 | 3.2 |
| Urine protein electrophoresis/light chains | 1.34 | P | 2.7 | 2.1 | 4.9 |
| Urine immunofixation | 61.70 | Assumption | 1.0 | 1.0 | 2.1 |
| Skeletal survey by X-ray | 18.56 | Assumption | 0.1 | 0.0 | 1.6 |
| Skeletal survey by X-ray individual sites | 18.56 | Assumption | 0.1 | 0.1 | 1.6 |
| Bone marrow aspirate | 1.34 | P | 0.2 | 0.1 | 2.1 |
| Bone marrow trephine biopsy | 1.34 | P | 0.2 | 0.1 | 2.0 |
| Bacterial investigation | 7.52 | P | 0.4 | 0.3 | 1.6 |
| Calcium | 1.34 | P | 6.0 | 1.0 | 20 |
| Albumin | 1.34 | P | 6.0 | 1.0 | 20 |
Unit costs used in the model analyses:
| Average unit cost (£)a | |
|---|---|
| Anaemia – grade 3/4 | 358.07 |
| Thrombocytopenia – grade 3/4 | 379.71 |
| Neutropenia – grade 3/4 | 772.13 |
| Leucopenia – grade 3/4 | 573.62 |
| Lymphopenia – grade 3/4 | 1480.55 |
| Peripheral neuropathy – grade 3 | 856.99 |
| Thrombosis or embolism – grade 3/4 | 661.59 |
| Somnolence/fatigue/dizziness – grade 3/4 | 147.94 |
| Fever of unknown origin – grade 3/4 | 1,195.37 |
| Pneumonia – grade 3/4 | 12,734.34 |
| Septicaemia – grade 3/4 | 2740.69 |
| Meningitis – grade 3/4 | 857.98 |
| Herpes zoster – grade 3/4 | 383.82 |
| Constipation – grade 3/4 | 1277.13 |
| Lung disorder – grade 5 | 971.46 |
| Septic shock – grade 5 | 784.53 |
Indicate the source for individual cost values (if appropriate).
Indirect costs (costs due to lost productivity, unpaid inputs to patient care)
-
Were indirect costs included?
No.
-
Describe how indirect costs were estimated (e.g. how days of lost productivity were estimated and how those days were valued).
Not applicable.
Indicate the source for individual cost values (if appropriate).
Health-state valuations/utilities (if study uses QoL adjustments to outcomes)
-
Were the utility data derived from: a single (observational) study, a review/synthesis of previous studies expert opinion. Were the methods for deriving these data adequately described (give sources if using data from other published studies)?
The HOVON study,93 a RCT of intensive chemotherapy followed by myeloblative therapy with autologous stem cell rescue compared with intensive chemotherapy, provided QoL data using the EQ-5D. A literature search was conducted for utility decrements for AEs, with utility values from different population groups used (e.g. breast, colon and rectal cancer). Average per cent reduction in utility by each AE was calculated from these values.
-
List the utility values used in the evaluation.
Utility values were 0.64 for people not responding to treatment and 0.81 for people who did respond (using general public utility for same age group). A utility value of 0.77 at 24 months was presented for those who continue to respond to treatment with intensive chemotherapy. An assumption was made that preprogression patients and post-progression patients matched responders and non-responders in the HOVON trial. 93 A 0.77 utility score was used for those who had not progressed at the end of 2 years.
Indicate the source for individual cost values (if appropriate).
Modelling
-
If a model was used, describe the type of model used (e.g. Markov state transition model, discrete event simulation). Was this a newly developed model or was it adapted from a previously reported model? If an adaptation, give the source of the original.
A lifetime Markov model developed from the evaluation presented by Deniz and colleagues,72 which compared MPT to MP in first-line treatment for MM in Scotland. The model was updated to include comparison with VMP.
-
What was the purpose of the model (i.e. why was a model required in this evaluation)?
Not stated.
-
What are the main components of the model (e.g. health states within a Markov model)? Are sources for assumptions over model structure (e.g. allowable transitions) reported – list them if reported.
Model structure
The model tracks the progress of patients with MM as managed with MPT, VMP or MP through four Markov states, specifically (1) preprogression without AE, (2) preprogression with AE, (3) post progression and (4) death. Patients start in the preprogression without AE health state and may move to a worse state or remain in the same state. Patients receive first-line treatment with MPT or MP for up to 12 six-week cycles or VMP for up to nine cycles. If the patients experience a serious treatment-related AE, they enter preprogression with AE state with no risk of additional AE. History of AEs does not determine progression. Death can only occur at progression or after progression and is assumed to be disease-related deterioration. Cycle length 6 weeks (42 days), equivalent to dosing cycle in trials. 23,26,59
Resources and costs
-
Dose reductions, treatment interruptions and discontinuations are modelled as a reduction in costs.
-
When on active treatment, patients receive the mean observed treatment dose from the trials.
-
Routine management resources were estimated by UK haematologists by progression status and costed using publicly available data and applied to relevant cohorts until the end of the time horizon.
Adverse events
Only costs of treatment-related serious (grade 3 and above) AEs or AEs that occurred in ≥ 2% in treatment arms are included for those on active treatment. Rates are taken from the trials. 23,26 Risk of AE was estimated from the mean time alive per patient from Kaplan–Meier survival curves, with average patients alive during a 6-month period calculated and average duration on treatment calculated. Progression is the point of stopping treatment. (AiC/CiC information has been removed.) The magnitude and duration of the risk reductions were used to calculate the relative reduction in utility value over the complete time horizon of the AE in cycles. These values were weighted according to relative frequency and summed to produce the average total relative disutility per AE which was applied to the cohort experiencing AEs.
Assumptions
-
Only patients on active treatment at risk of AEs.
-
Costs of managing AE considered separately.
-
AE disutility applied at time of the event.
-
No discontinuation through AEs, implicitly included in dosing, duration and efficacy of treatment.
-
Deaths from AEs are through OS.
Average treatment duration applied in the base-case model was 12 months for MP and MPT, despite treatment interruptions/discontinuations meaning median treatment duration was less (AiC/CiC information has been removed). This assumption increases costs not efficacy. No data on discontinuation for VMP were available.
Progression
-
Post-progression survival is modelled to be the same across treatment strategies.
-
Patients assumed to discontinue active treatment upon disease progression.
-
Adverse events assumed not to affect progression rate.
Concurrent medication
Assumes VTE antithrombotic prophylaxis for patients receiving MPT with no resultant risk in incidence of VTEs and antiviral prophylaxis for VMP.
-
Extract transition probabilities for [natural history/disease progression] model and show sources (or refer to table in text).
None stated.
-
What is the model time horizon?
Lifetime horizon.
-
What, if any, discount rates have been applied in the model? Same rate for costs and outcomes?
Costs and benefits were discounted at 3.5%.
Results/analysis
-
What measure(s) of benefit were reported in the evaluation?
Cost per life-year gained and cost per QALY.
-
Provide a summary of the clinical outcome/benefits estimated for each intervention/strategy assessed in the evaluation:
-
Provide a summary of the costs estimated for each intervention/strategy assessed in the evaluation:
-
Synthesis of costs and benefits – are the costs and outcomes reported together (e.g. as cost-effectiveness ratios)? If so, provide a summary of the results.
Base-case results calculated by model (discounted):
| MPT vs MP | MPT vs VMP | |
|---|---|---|
| Incremental life-years | 1.09 | 0.11 |
| Incremental QALYs | 0.85 | 0.07 |
| Incremental costs (£) | 19,768 | 21,483 |
| Incremental cost per life-year gained (£) | 18,188 | 200,237 |
| Incremental cost per QALY gained (£) | 23,381 | 303,845 |
-
Give results of any statistical analysis of the results of the evaluation.
None.
-
Was any sensitivity analysis performed? If yes, what type(s) [i.e. deterministic (one-way, two-way, etc.) or probabilistic].
Yes.
-
What scenarios were tested in the sensitivity analysis? How do these relate to structural uncertainty (testing assumptions over model structure such as relationships between health states), methodological uncertainty (such as choices of discount rate or inclusion of indirect costs) or parameter uncertainty (assumptions over values of parameters in the model, such as costs, QoL or disease progression rates)?
One-way deterministic sensitivity analysis examined time horizon (5 years), risk progression (2.5% and 97.5% CIs from MTC), resource use and costs (monitoring, AE and all costs varied by ± 100%), AE rates and utility (scores varied by ± 10%), PFS and OS (expanded MTC).
-
Give a summary of the results of the sensitivity analysis. Did they differ substantially from the base-case analysis? If so, what were the suggested causes?
One-way deterministic sensitivity analysis (discounted):
| MPT vs MP | MPT vs VMP | |||
|---|---|---|---|---|
| Incremental cost/LYG (£) | Incremental cost/QALY (£) | Incremental cost/LYG (£) | Incremental cost/QALY (£) | |
| Base case | 18,188 | 23,381 | 200,201 | 303,790 |
| No discounting | 14,892 | 19,355 | 153,339 | 226,033 |
| Time horizon (5 years) | 41,703 | 49,134 | 613,900 | 1,241,139 |
| Efficacy | ||||
| 2.5% CI MTC | 25,836 | 33,275 | 482,097 | 1,000,435 |
| 97.5% CI MTC | 12,916 | 16,586 | 106,683 | 148,873 |
| Monitoring costs and AE costs | ||||
| +100% | 18,538 | 23,831 | 199,749 | 303,103 |
| –100% | 18,005 | 23,145 | 200,427 | 304,133 |
| Utility scores | ||||
| +10% increase | 18,188 | 22,961 | 200,201 | 305,666 |
| –10% decrease | 18,188 | 23,816 | 200,201 | 302,045 |
Single trial analyses (discounted):
| MPT vs MP | MPT vs VMP | |||
|---|---|---|---|---|
| Incremental cost/LYG (£) | Incremental cost/QALY (£) | Incremental cost/LYG (£) | Incremental cost/QALY (£) | |
| IFM 99/06 | 16,603 | 21,285 | MPT dominates | MPT dominates |
| IFM 01/01 | 9404 | 12,067 | 1,430,625 | 7,234,876 |
| GIMEMA | 23,648 | 30,882 | 314,357 | 462,088 |
Conclusions/implications
-
Give a brief summary of the author’s conclusions from their analysis.
The analyses indicate that MPT represents a cost-effective use of NHS resources when compared with MP in England and Wales for managing previously untreated MM patients aged ≥ 65 years or ineligible for HDT.
-
What are the implications of the evaluation for practice?
The authors estimate that the eligible incident and prevalent population will increase from 3196 in 2010 to 15,929 in 2014. With the assumption that MPT currently has no market share and that its market share will grow from 60% in 2010 to 70% by 2014, the authors estimate that the incremental budget impact of using thalidomide (based on the proposed total annual costs with MPT and MP minus the total annual costs of managing patients with MP alone) will rise from £32M in 2010 to £44.8M in 2014. As MPT is estimated to have a market share of 54% currently, the incremental budget impact of increasing the market share to 60% in 2010 and 70% by 2014 will result in an incremental total annual cost of £3.2M in 2010 rising to £10.2M in 2014.
Appendix 12 Critical appraisal checklist of economic evaluation
The quality of the cost-effectiveness studies was assessed using a critical appraisal checklist based on that by Drummond and Jefferson,118 Philips and colleagues,98 and the NICE reference case.
| Item | Celgene116 | Janssen–Cilag115 | |
|---|---|---|---|
| 1 | Is there a clear statement of the decision problem? | Y | Y |
| 2 | Is the comparator routinely used in UK NHS? | Y | Y |
| 3 | Is the patient group in the study similar to those of interest in UK NHS? | ? | Y |
| 4 | Is the health-care system or setting comparable to UK? | Y | Y |
| 5 | Is the perspective of the model clearly stated? | Y | Y |
| 6 | Is the study type and modelling methodology reasonable? | Y | Y |
| 7 | Is the model structure described and does it reflect the disease process? | ? | Y |
| 8 | Are assumptions about model structure listed and justified? | Y | Y |
| 9 | Are the data inputs for the model described and justified? | Y | Y |
| 10 | Is the effectiveness of the intervention established based on a systematic review? | Y | Y |
| 11 | Are health benefits measured in QALYs? | Y | Y |
| 12 | Are health benefits measured using a standardised and validated generic instrument? | Y | Y |
| 13 | Are the resource costs described and justified? | Y | Y |
| 14 | Have the costs and outcomes been discounted? | Y | Y |
| 15 | Has uncertainty been assessed? | Y | Y |
| 16 | Has the model been validated? | N | N |
Appendix 13 Methodology used for disease projection
The methodology used for estimating survival curves for the alternative treatments is as follows:
-
Derive a baseline survival curve for MP. This curve is derived by calculating the event probability for each time interval, by calculating a weighted average of the trial MP arms using number of participants in the trial as a weight.
-
Derive HRs for each of the treatments versus MP at different time points for each trial. Combine HRs for treatments with more than one trial.
-
Construct the baseline survival curves for MP using the event probability for each time interval.
-
Construct the survival curves for other treatments by using the event probability for each time interval, i.e. event probability for MP multiplied by HR.
For MP treatment, OS and PFS at regular time points were estimated for each of the included studies from our meta-analysis of the clinical trials. The data from the trials were combined to form baseline MP, OS and PFS curves through a weighted average, using number of patients in the trials as the weight. We estimated the hazard rate for MP for each 6-monthly period (Table 52). The hazard rate for death for MP per cycle is estimated for each time point ti:
| Months | Cycles | Survival OS | Hazard OS |
|---|---|---|---|
| 6 | 4.35 | 0.90 | 0.024 |
| 12 | 8.69 | 0.79 | 0.030 |
| 18 | 13.04 | 0.72 | 0.021 |
| 24 | 17.38 | 0.65 | 0.023 |
| 30 | 21.73 | 0.56 | 0.034 |
| 36 | 26.07 | 0.48 | 0.035 |
| 36+ | 26+ | 0.028 |
where s(t) is the survival function over time t.
The treatment effects for the other interventions compared with MP were taken from our clinical review (see Chapter 4, Assessment of effectiveness). As the HR of the treatments versus MP varied over time, a constant HR was not appropriate. A similar methodology was used for estimating OS and PFS; however, only OS is described in this appendix.
We derived the HR for each 6-monthly period for each of the treatments versus MP.
The hazard rate for death for each of the treatments per cycle is estimated for each time point ti:
where s(t) is the survival function over time t.
The HR (HR) for each intervention j versus MP at each time point ti is:
The HR was assumed to be constant after 36 months for OS as there were few patients with more than this length of follow-up in the trials. This HR was estimated for each of the treatments versus MP at 36 months’ follow-up for OS.
The hazard rate for death for each of the treatments per cycle was also assumed to be constant after 36 months and is given by:
where s(t) is the survival function and t is 36 months (26.1 cycles).
The methodology is illustrated for OS for VMP with data from the VISTA trial. Table 53 shows the hazards and the HRs derived from the VISTA trial for OS.
| Months | Cycles | Survival s(t) | Hazard h(t) | HR | ||
|---|---|---|---|---|---|---|
| MP | VMP | MP | VMP | |||
| 0 | 0 | 1.00 | 1.00 | |||
| 6 | 4.35 | 0.92 | 0.92 | 0.019 | 0.019 | 1.00 |
| 12 | 8.69 | 0.82 | 0.89 | 0.026 | 0.008 | 0.30 |
| 18 | 13.04 | 0.76 | 0.85 | 0.017 | 0.010 | 0.60 |
| 24 | 17.38 | 0.69 | 0.78 | 0.022 | 0.019 | 0.85 |
| 30 | 21.73 | 0.64 | 0.74 | 0.018 | 0.013 | 0.70 |
| 36 | 26.07 | 0.54 | 0.69 | 0.036 | 0.017 | 0.46 |
| 36+ | 26+ | 0.54 | 0.69 | 0.023 | 0.014 | 0.62 |
To generate the survival curves for each of the treatments the baseline death rate in each time period for MP was multiplied by the HR to give the new death rate for the alternative treatment. This method provided a closer fit to the trial data than approximations, such as fitting distributions.
The survival curves were constructed by multiplying the survival in the previous time point by the proportion who survived in the current time interval, using the estimated hazards for MP and the HR for the other interventions.
Thus the survival function s(t) is given by:
To demonstrate the fit from this method we derive the VMP survival curves using the trial MP curves and compare with the original trial curves. Figure 16 shows the MP and VMP survival curves derived for the model against the trial data from the VISTA trial. As can be seen in the figure, the derived survival curves in the model closely match both treatments during the trial period.
FIGURE 16.
MP and VMP survival curves derived for the model against trial data from the VISTA trial.

In the model, instead of using the MP trial data, the MP baseline data are used with the same method as described above. Figure 17 shows the MP and VMP survival curves derived for the model using the baseline combined MP curves.
FIGURE 17.
Melphalan + prednisolone/prednisone (MP) and VMP survival curves for the model using combined baseline MP curves.

Appendix 14 Parameters included in the sensitivity analyses
The ranges used for the deterministic analyses and PSAs are reported in this appendix. Where appropriate, parameters were assigned a distribution in the PSA. Distributions were chosen according to the methodology suggested by Briggs and colleagues. 103 They suggest that the normal distribution is a ‘candidate distribution for representing the uncertainty in any parameter in the model’. Further, they suggest the beta distribution for binomial outcomes, where parameters can vary between zero and one, for example probabilities, and the gamma distribution for costs where parameters are non-negative.
-
Discount rates were varied between 2% and 5% for costs and benefits in the deterministic sensitivity analyses.
-
The number of cycles for each of the treatments varied between seven and nine. For each of the interventions, we assumed a range between one fewer than the mean to one more than the mean. The number of cycles was assumed to follow a normal distribution.
-
Second-line treatment was varied according to the proportion who had bortezomib and HDD. For MP, MPT and CTDa, the proportion varied between 60% and 80% and for VMP, the proportion varied between 5% and 25%.
-
The range for the utility values was assumed to be ± 10% of the mean utility values, based on the uncertainty in the utility values from the MMIX Trial. We analysed the HRQoL data from the MMIX Trial. (AiC and/or CiC information has been removed.) Utility values were sampled from a beta distribution.
-
The CR data were obtained from the trials and SEs were derived. Values were sampled from a beta distribution.
-
The costs for AEs, bortezomib administration and consultation were assumed to vary within the range ± 30% of the mean and were sampled from a gamma distribution.
-
The ranges for the HRs and event rates for the MP survival curves were taken from the trial data. Values were sampled from a log-normal distribution.
-
The costs for AEs, bortezomib administration and consultation were varied within ± 30% of the mean for the deterministic sensitivity analysis.
Parameters and distributions for the deterministic and probabilistic sensitivity analyses
| Name | Mean | CI | SE | Distribution | |
|---|---|---|---|---|---|
| Higher | Lower | ||||
| Discount rate | |||||
| Discount rate costs (%) | 3.5 | 5.0 | 2.0 | NA | |
| Discount rate benefits (%) | 3.5 | 5.0 | 2.0 | NA | |
| Cycles of treatment | |||||
| cycle_MP | 8 | 9 | 7 | 0.5102 | Log normal |
| cycle_MPT | 8 | 9 | 7 | 0.5102 | Log normal |
| cycle_VMP | 9 | 10 | 8 | 0.5102 | Log normal |
| cycle_CTDa | 7 | 8 | 6 | 0.5102 | Log normal |
| Subsequent treatment, Bort. | |||||
| Sub_treat_Bort_MP | 70 | 80 | 60 | 5.1020 | Log normal |
| Sub_treat_Bort_MPT | 70 | 80 | 60 | 5.1020 | Log normal |
| Sub_treat_Bort_VMP | 15 | 25 | 5 | 5.1020 | Log normal |
| Sub_treat_Bort_CTDa | 70 | 80 | 60 | 5.1020 | Log normal |
| Utility values | |||||
| u_treatment | 0.58 | 0.639 | 0.522 | 0.030 | Beta |
| u_response | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | Beta |
| u_progression | 0.68 | 0.748 | 0.612 | 0.035 | Beta |
| CR | |||||
| CR_MP | 0.026 | 0.035 | 0.017 | 0.005 | Beta |
| CR_MPT | 0.142 | 0.307 | 0.066 | 0.084 | Beta |
| CR_VMP | 0.217 | 0.386 | 0.121 | 0.087 | Beta |
| CR_CTDa | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | Beta |
| AEs (£ per cycle) | |||||
| cAE_MP | 45.63 | 59.32 | 31.94 | 6.98 | Gamma |
| cAE_MPT | 70.40 | 91.52 | 49.28 | 10.78 | Gamma |
| cAE_VMP | 73.16 | 95.11 | 51.21 | 11.20 | Gamma |
| cAE_CTDa | 72.45 | 94.19 | 50.72 | 11.09 | Gamma |
| Other | |||||
| Cost of bortezomib administration (£) | 153.40 | 199.42 | 107.38 | 23.4796 | Gamma |
| Outpatient appointment medical oncology (£) | 121.11 | 157.44 | 84.78 | 18.5372 | Gamma |
| Survival curve parameters | |||||
| Multipliers | |||||
| MP OS baseline curve | 0.028 | 0.039 | 0.020 | 0.0041 | Log normal |
| MP PFS baseline curve | 0.067 | 0.070 | 0.060 | 0.0036 | Log normal |
| HR OS MPT | 0.62 | 0.82 | 0.50 | 0.0714 | Log normal |
| HR OS VMP | 0.62 | 0.83 | 0.51 | 0.0714 | Log normal |
| HR OS CTDa | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | Log normal |
| HR PFS MPT | 0.58 | 0.77 | 0.49 | 0.0612 | Log normal |
| HR PFS VMP | 0.58 | 0.76 | 0.48 | 0.0612 | Log normal |
| HR PFS CTDa | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | (AiC/CiC information has been removed) | Log normal |
| Cost of treatments | |||||
| Unit cost bortezomib (£) | 762.38 | 914.86 | 609.90 | 77.7939 | NA |
| Unit cost thalidomide (£) | 298.48 | 358.18 | 238.78 | 30.4571 | NA |
| Dosage thalidomide (mg/day) | 150 | 200 | 100 | 25.5102 | Log normal |
Appendix 15 Additional scenario analyses
Scenario 1
The base-case analysis uses the number of cycles of treatment with bortezomib as per the protocol for the VISTA study, i.e. nine cycles. After discontinuation of treatment for some patients due to death or disease progression, this equates to about 48 vials per individuals. This scenario investigates the cost-effectiveness through treatment for a shorter number of treatment cycles, i.e. four cycles, with no loss of efficacy. A reduced number of treatment cycles equates to about 31 vials. In this scenario, bortezomib becomes more cost-effective, with an ICER of £19,039 per QALY gained versus MP and £332,546 per QALY gained versus MPT.
| VMP vs MP | VMP vs MPT | |||
|---|---|---|---|---|
| Base case | Scenario | Base case | Scenario | |
| Incremental QALY | 1.20 | 1.26 | –0.02 | 0.04 |
| Incremental cost (£) | 35,749 | 24,002 | 24,542 | 12,592 |
| ICER (£/QALY) | 29,820 | 19,039 | –1 million | 322,546 |
Scenario 2
The base-case analysis uses the number of cycles of treatment with thalidomide as suggested by clinical experts, i.e. eight cycles. This scenario investigates the cost-effectiveness through treatment for a longer number of treatment cycles as used in the MPT IFM trials, i.e. 12 cycles. In this scenario, MPT becomes slightly less cost-effective compared with MP, with an ICER of £13,619 per QALY gained (base case £9135).
List of abbreviations
- ABSCS
- autologous blood stem cell support
- AE
- adverse event
- AiC
- academic-in-confidence
- BNF
- British National Formulary
- BSH
- British Society for Haematology
- CEAC
- cost-effectiveness acceptability curve
- CI
- confidence interval
- CiC
- commercial-in-confidence
- CR
- complete response
- CRD
- Centre for Reviews and Dissemination
- CSR
- clinical study report
- CTC
- Common Toxicity Criteria
- CTCAE
- Common Terminology Criteria for Adverse Effects
- CTDa
- cyclophosphamide, thalidomide and attenuated dexamethasone
- CTRU
- Clinical Trials Research Unit
- DS
- Durie–Salmon staging system
- DVT
- deep-vein thrombosis
- EBMT
- European Group for Blood and Marrow Transplantation
- EORTC
- European Organisation for Research and Treatment of Cancer quality-of-life
- QLQ-C30
- questionnaire C30
- EQ-5D
- European Quality of Life-5 Dimensions
- GIMEMA
- Gruppo Italiano Malattie Ematologiche dell’Adulto (Italian Group for Adult Hematologic Diseases)
- HDD
- high-dose dexamethasone
- HDM
- high-dose melphalan
- HDT
- high-dose chemotherapy
- HR
- hazard ratio
- HRQoL
- health-related quality of life
- HTA
- health technology assessment
- IBMTR
- International Bone Marrow Transplant Registry
- ICD-10
- International Classification of Diseases 10th edition
- ICER
- incremental cost-effectiveness ratio
- IFM
- Intergroupe Francophone du Myélome
- IQR
- interquartile range
- ISS
- International Staging System
- ITT
- intention to treat
- MIMS
- Monthly Index of Medical Specialties
- MM
- multiple myeloma
- MMIX
- Myeloma IX Trial
- MP
- melphalan plus prednisolone/prednisone
- MPT
- melphalan plus prednisolone/prednisone plus thalidomide
- MR
- minimal response
- MRC
- Medical Research Council
- MS
- manufacturer’s submission
- MTC
- mixed-treatment comparison
- NCI
- National Cancer Institute
- NICE
- National Institute for Health and Clinical Excellence
- NIHR
- National Institute for Health Research
- NR
- not reported
- NS
- not significant
- OLS
- ordinary least squares
- OS
- overall survival
- PBAC
- Pharmaceutical Benefits Advisory Committee
- PBPCT
- peripheral blood progenitor cell transplantation
- PFS
- progression-free survival
- PPS
- post-progression survival
- PR
- partial response
- PSA
- probabilistic sensitivity analysis
- PSCT
- peripheral stem cell transplantation
- PSS
- Personal Social Services
- QALY
- quality-adjusted life-year
- QoL
- quality of life
- RCT
- randomised controlled trial
- RR
- risk ratio
- SCT
- stem cell transplantation
- SD
- standard deviation
- SE
- standard error
- SHTAC
- Southampton Health Technology Assessments Centre
- SPC
- summary of product characteristics
- SUHT
- Southampton University Hospitals Trust
- TAR
- technology assessment report
- TTP
- time to progression
- VAD
- vincristine, doxorubicin and dexamethasone
- VAD/VAMP
- vincristine, adriamycin and dexamethasone/vincristine, adriamycin and methyl prednisone
- VAS
- visual analogue scale
- VMCP-IFNα2b
- vincristine, melphalan, cyclophosphamide, prednisolone, interferon-α2b
- VMP
- bortezomib (Velcade®) plus melphalan plus prednisolone/prednisone
- VTE
- venous thromboembolism
- WHO
- World Health Organization
All abbreviations that have been used in this report are listed here unless the abbreviation is well known (e.g. NHS), or it has been used only once, or it is a non-standard abbreviation used only in figures/tables/appendices, in which case the abbreviation is defined in the figure legend or in the notes at the end of the table.
Note
This monograph is based on the Technology Assessment Report produced for NICE. The full report contained a considerable number of data that was deemed commercial-in-confidence and academic-in-confidence. The full report was used by the Appraisal Committee at NICE in their deliberations. The full report with each piece of commercial-in-confidence and academic-in-confidence data removed and replaced by the statement ‘commercial-in-confidence and academic-in-confidence information (or data) removed’ is available on the NICE website: www.nice.org.uk.
The present monograph presents as full a version of the report as is possible while retaining readability, but some sections, sentences, tables and figures have been removed. Readers should bear in mind that the discussion, conclusions and implications for practice and research are based on all the data considered in the original full NICE report.
Notes
Health Technology Assessment programme
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
Prioritisation Group
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Imti Choonara, Professor in Child Health, Academic Division of Child Health, University of Nottingham
Chair – Pharmaceuticals Panel
-
Dr Bob Coates, Consultant Advisor – Disease Prevention Panel
-
Dr Andrew Cook, Consultant Advisor – Intervention Procedures Panel
-
Dr Peter Davidson, Director of NETSCC, Health Technology Assessment
-
Dr Nick Hicks, Consultant Adviser – Diagnostic Technologies and Screening Panel, Consultant Advisor–Psychological and Community Therapies Panel
-
Ms Susan Hird, Consultant Advisor, External Devices and Physical Therapies Panel
-
Professor Sallie Lamb, Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick
Chair – HTA Clinical Evaluation and Trials Board
-
Professor Jonathan Michaels, Professor of Vascular Surgery, Sheffield Vascular Institute, University of Sheffield
Chair – Interventional Procedures Panel
-
Professor Ruairidh Milne, Director – External Relations
-
Dr John Pounsford, Consultant Physician, Directorate of Medical Services, North Bristol NHS Trust
Chair – External Devices and Physical Therapies Panel
-
Dr Vaughan Thomas, Consultant Advisor – Pharmaceuticals Panel, Clinical
Lead – Clinical Evaluation Trials Prioritisation Group
-
Professor Margaret Thorogood, Professor of Epidemiology, Health Sciences Research Institute, University of Warwick
Chair – Disease Prevention Panel
-
Professor Lindsay Turnbull, Professor of Radiology, Centre for the MR Investigations, University of Hull
Chair – Diagnostic Technologies and Screening Panel
-
Professor Scott Weich, Professor of Psychiatry, Health Sciences Research Institute, University of Warwick
Chair – Psychological and Community Therapies Panel
-
Professor Hywel Williams, Director of Nottingham Clinical Trials Unit, Centre of Evidence-Based Dermatology, University of Nottingham
Chair – HTA Commissioning Board
Deputy HTA Programme Director
HTA Commissioning Board
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
-
Department of Public Health and Epidemiology, University of Birmingham
-
Professor of Clinical Pharmacology, Director, NIHR HTA programme, University of Liverpool
-
Professor Ann Ashburn, Professor of Rehabilitation and Head of Research, Southampton General Hospital
-
Professor Peter Brocklehurst, Professor of Women’s Health, Institute for Women’s Health, University College London
-
Professor Jenny Donovan, Professor of Social Medicine, University of Bristol
-
Professor Jonathan Green, Professor and Acting Head of Department, Child and Adolescent Psychiatry, University of Manchester Medical School
-
Professor John W Gregory, Professor in Paediatric Endocrinology, Department of Child Health, Wales School of Medicine, Cardiff University
-
Professor Steve Halligan, Professor of Gastrointestinal Radiology, University College Hospital, London
-
Professor Freddie Hamdy, Professor of Urology, Head of Nuffield Department of Surgery, University of Oxford
-
Professor Allan House, Professor of Liaison Psychiatry, University of Leeds
-
Dr Martin J Landray, Reader in Epidemiology, Honorary Consultant Physician, Clinical Trial Service Unit, University of Oxford
-
Professor Stephen Morris, Professor of Health Economics, University College London, Research Department of Epidemiology and Public Health, University College London
-
Professor Irwin Nazareth, Professor of Primary Care and Head of Department, Department of Primary Care and Population Sciences, University College London
-
Professor E Andrea Nelson, Professor of Wound Healing and Director of Research, School of Healthcare, University of Leeds
-
Professor John David Norrie, Chair in Clinical Trials and Biostatistics, Robertson Centre for Biostatistics, University of Glasgow
-
Dr Rafael Perera, Lecturer in Medical Statisitics, Department of Primary Health Care, University of Oxford
-
Professor Barney Reeves, Professorial Research Fellow in Health Services Research, Department of Clinical Science, University of Bristol
-
Professor Martin Underwood, Professor of Primary Care Research, Warwick Medical School, University of Warwick
-
Professor Marion Walker, Professor in Stroke Rehabilitation, Associate Director UK Stroke Research Network, University of Nottingham
-
Dr Duncan Young, Senior Clinical Lecturer and Consultant, Nuffield Department of Anaesthetics, University of Oxford
-
Dr Tom Foulks, Medical Research Council
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
HTA Clinical Evaluation and Trials Board
-
Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick and Professor of Rehabilitation, Nuffield Department of Orthopaedic, Rheumatology and Musculoskeletal Sciences, University of Oxford
-
Professor of the Psychology of Health Care, Leeds Institute of Health Sciences, University of Leeds
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Keith Abrams, Professor of Medical Statistics, Department of Health Sciences, University of Leicester
-
Professor Martin Bland, Professor of Health Statistics, Department of Health Sciences, University of York
-
Professor Jane Blazeby, Professor of Surgery and Consultant Upper GI Surgeon, Department of Social Medicine, University of Bristol
-
Professor Julia M Brown, Director, Clinical Trials Research Unit, University of Leeds
-
Professor Alistair Burns, Professor of Old Age Psychiatry, Psychiatry Research Group, School of Community-Based Medicine, The University of Manchester & National Clinical Director for Dementia, Department of Health
-
Dr Jennifer Burr, Director, Centre for Healthcare Randomised trials (CHART), University of Aberdeen
-
Professor Linda Davies, Professor of Health Economics, Health Sciences Research Group, University of Manchester
-
Professor Simon Gilbody, Prof of Psych Medicine and Health Services Research, Department of Health Sciences, University of York
-
Professor Steven Goodacre, Professor and Consultant in Emergency Medicine, School of Health and Related Research, University of Sheffield
-
Professor Dyfrig Hughes, Professor of Pharmacoeconomics, Centre for Economics and Policy in Health, Institute of Medical and Social Care Research, Bangor University
-
Professor Paul Jones, Professor of Respiratory Medicine, Department of Cardiac and Vascular Science, St George‘s Hospital Medical School, University of London
-
Professor Khalid Khan, Professor of Women’s Health and Clinical Epidemiology, Barts and the London School of Medicine, Queen Mary, University of London
-
Professor Richard J McManus, Professor of Primary Care Cardiovascular Research, Primary Care Clinical Sciences Building, University of Birmingham
-
Professor Helen Rodgers, Professor of Stroke Care, Institute for Ageing and Health, Newcastle University
-
Professor Ken Stein, Professor of Public Health, Peninsula Technology Assessment Group, Peninsula College of Medicine and Dentistry, Universities of Exeter and Plymouth
-
Professor Jonathan Sterne, Professor of Medical Statistics and Epidemiology, Department of Social Medicine, University of Bristol
-
Mr Andy Vail, Senior Lecturer, Health Sciences Research Group, University of Manchester
-
Professor Clare Wilkinson, Professor of General Practice and Director of Research North Wales Clinical School, Department of Primary Care and Public Health, Cardiff University
-
Dr Ian B Wilkinson, Senior Lecturer and Honorary Consultant, Clinical Pharmacology Unit, Department of Medicine, University of Cambridge
-
Ms Kate Law, Director of Clinical Trials, Cancer Research UK
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
Diagnostic Technologies and Screening Panel
-
Scientific Director of the Centre for Magnetic Resonance Investigations and YCR Professor of Radiology, Hull Royal Infirmary
-
Professor Judith E Adams, Consultant Radiologist, Manchester Royal Infirmary, Central Manchester & Manchester Children’s University Hospitals NHS Trust, and Professor of Diagnostic Radiology, University of Manchester
-
Mr Angus S Arunkalaivanan, Honorary Senior Lecturer, University of Birmingham and Consultant Urogynaecologist and Obstetrician, City Hospital, Birmingham
-
Dr Diana Baralle, Consultant and Senior Lecturer in Clinical Genetics, University of Southampton
-
Dr Stephanie Dancer, Consultant Microbiologist, Hairmyres Hospital, East Kilbride
-
Dr Diane Eccles, Professor of Cancer Genetics, Wessex Clinical Genetics Service, Princess Anne Hospital
-
Dr Trevor Friedman, Consultant Liason Psychiatrist, Brandon Unit, Leicester General Hospital
-
Dr Ron Gray, Consultant, National Perinatal Epidemiology Unit, Institute of Health Sciences, University of Oxford
-
Professor Paul D Griffiths, Professor of Radiology, Academic Unit of Radiology, University of Sheffield
-
Mr Martin Hooper, Public contributor
-
Professor Anthony Robert Kendrick, Associate Dean for Clinical Research and Professor of Primary Medical Care, University of Southampton
-
Dr Nicola Lennard, Senior Medical Officer, MHRA
-
Dr Anne Mackie, Director of Programmes, UK National Screening Committee, London
-
Mr David Mathew, Public contributor
-
Dr Michael Millar, Consultant Senior Lecturer in Microbiology, Department of Pathology & Microbiology, Barts and The London NHS Trust, Royal London Hospital
-
Mrs Una Rennard, Public contributor
-
Dr Stuart Smellie, Consultant in Clinical Pathology, Bishop Auckland General Hospital
-
Ms Jane Smith, Consultant Ultrasound Practitioner, Leeds Teaching Hospital NHS Trust, Leeds
-
Dr Allison Streetly, Programme Director, NHS Sickle Cell and Thalassaemia Screening Programme, King’s College School of Medicine
-
Dr Matthew Thompson, Senior Clinical Scientist and GP, Department of Primary Health Care, University of Oxford
-
Dr Alan J Williams, Consultant Physician, General and Respiratory Medicine, The Royal Bournemouth Hospital
-
Dr Tim Elliott, Team Leader, Cancer Screening, Department of Health
-
Dr Joanna Jenkinson, Board Secretary, Neurosciences and Mental Health Board (NMHB), Medical Research Council
-
Professor Julietta Patrick, Director, NHS Cancer Screening Programme, Sheffield
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Disease Prevention Panel
-
Professor of Epidemiology, University of Warwick Medical School, Coventry
-
Dr Robert Cook, Clinical Programmes Director, Bazian Ltd, London
-
Dr Colin Greaves, Senior Research Fellow, Peninsula Medical School (Primary Care)
-
Mr Michael Head, Public contributor
-
Professor Cathy Jackson, Professor of Primary Care Medicine, Bute Medical School, University of St Andrews
-
Dr Russell Jago, Senior Lecturer in Exercise, Nutrition and Health, Centre for Sport, Exercise and Health, University of Bristol
-
Dr Julie Mytton, Consultant in Child Public Health, NHS Bristol
-
Professor Irwin Nazareth, Professor of Primary Care and Director, Department of Primary Care and Population Sciences, University College London
-
Dr Richard Richards, Assistant Director of Public Health, Derbyshire County Primary Care Trust
-
Professor Ian Roberts, Professor of Epidemiology and Public Health, London School of Hygiene & Tropical Medicine
-
Dr Kenneth Robertson, Consultant Paediatrician, Royal Hospital for Sick Children, Glasgow
-
Dr Catherine Swann, Associate Director, Centre for Public Health Excellence, NICE
-
Mrs Jean Thurston, Public contributor
-
Professor David Weller, Head, School of Clinical Science and Community Health, University of Edinburgh
-
Ms Christine McGuire, Research & Development, Department of Health
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
External Devices and Physical Therapies Panel
-
Consultant Physician North Bristol NHS Trust
-
Reader in Wound Healing and Director of Research, University of Leeds
-
Professor Bipin Bhakta, Charterhouse Professor in Rehabilitation Medicine, University of Leeds
-
Mrs Penny Calder, Public contributor
-
Dr Dawn Carnes, Senior Research Fellow, Barts and the London School of Medicine and Dentistry
-
Dr Emma Clark, Clinician Scientist Fellow & Cons. Rheumatologist, University of Bristol
-
Mrs Anthea De Barton-Watson, Public contributor
-
Professor Nadine Foster, Professor of Musculoskeletal Health in Primary Care Arthritis Research, Keele University
-
Dr Shaheen Hamdy, Clinical Senior Lecturer and Consultant Physician, University of Manchester
-
Professor Christine Norton, Professor of Clinical Nursing Innovation, Bucks New University and Imperial College Healthcare NHS Trust
-
Dr Lorraine Pinnigton, Associate Professor in Rehabilitation, University of Nottingham
-
Dr Kate Radford, Senior Lecturer (Research), University of Central Lancashire
-
Mr Jim Reece, Public contributor
-
Professor Maria Stokes, Professor of Neuromusculoskeletal Rehabilitation, University of Southampton
-
Dr Pippa Tyrrell, Senior Lecturer/Consultant, Salford Royal Foundation Hospitals’ Trust and University of Manchester
-
Dr Nefyn Williams, Clinical Senior Lecturer, Cardiff University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Interventional Procedures Panel
-
Professor of Vascular Surgery, University of Sheffield
-
Consultant Colorectal Surgeon, Bristol Royal Infirmary
-
Mrs Isabel Boyer, Public contributor
-
Mr Sankaran Chandra Sekharan, Consultant Surgeon, Breast Surgery, Colchester Hospital University NHS Foundation Trust
-
Professor Nicholas Clarke, Consultant Orthopaedic Surgeon, Southampton University Hospitals NHS Trust
-
Ms Leonie Cooke, Public contributor
-
Mr Seumas Eckford, Consultant in Obstetrics & Gynaecology, North Devon District Hospital
-
Professor Sam Eljamel, Consultant Neurosurgeon, Ninewells Hospital and Medical School, Dundee
-
Dr Adele Fielding, Senior Lecturer and Honorary Consultant in Haematology, University College London Medical School
-
Dr Matthew Hatton, Consultant in Clinical Oncology, Sheffield Teaching Hospital Foundation Trust
-
Dr John Holden, General Practitioner, Garswood Surgery, Wigan
-
Dr Fiona Lecky, Senior Lecturer/Honorary Consultant in Emergency Medicine, University of Manchester/Salford Royal Hospitals NHS Foundation Trust
-
Dr Nadim Malik, Consultant Cardiologist/Honorary Lecturer, University of Manchester
-
Mr Hisham Mehanna, Consultant & Honorary Associate Professor, University Hospitals Coventry & Warwickshire NHS Trust
-
Dr Jane Montgomery, Consultant in Anaesthetics and Critical Care, South Devon Healthcare NHS Foundation Trust
-
Professor Jon Moss, Consultant Interventional Radiologist, North Glasgow Hospitals University NHS Trust
-
Dr Simon Padley, Consultant Radiologist, Chelsea & Westminster Hospital
-
Dr Ashish Paul, Medical Director, Bedfordshire PCT
-
Dr Sarah Purdy, Consultant Senior Lecturer, University of Bristol
-
Dr Matthew Wilson, Consultant Anaesthetist, Sheffield Teaching Hospitals NHS Foundation Trust
-
Professor Yit Chiun Yang, Consultant Ophthalmologist, Royal Wolverhampton Hospitals NHS Trust
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Pharmaceuticals Panel
-
Professor in Child Health, University of Nottingham
-
Senior Lecturer in Clinical Pharmacology, University of East Anglia
-
Dr Martin Ashton-Key, Medical Advisor, National Commissioning Group, NHS London
-
Dr Peter Elton, Director of Public Health, Bury Primary Care Trust
-
Dr Ben Goldacre, Research Fellow, Division of Psychological Medicine and Psychiatry, King’s College London
-
Dr James Gray, Consultant Microbiologist, Department of Microbiology, Birmingham Children’s Hospital NHS Foundation Trust
-
Dr Jurjees Hasan, Consultant in Medical Oncology, The Christie, Manchester
-
Dr Carl Heneghan, Deputy Director Centre for Evidence-Based Medicine and Clinical Lecturer, Department of Primary Health Care, University of Oxford
-
Dr Dyfrig Hughes, Reader in Pharmacoeconomics and Deputy Director, Centre for Economics and Policy in Health, IMSCaR, Bangor University
-
Dr Maria Kouimtzi, Pharmacy and Informatics Director, Global Clinical Solutions, Wiley-Blackwell
-
Professor Femi Oyebode, Consultant Psychiatrist and Head of Department, University of Birmingham
-
Dr Andrew Prentice, Senior Lecturer and Consultant Obstetrician and Gynaecologist, The Rosie Hospital, University of Cambridge
-
Ms Amanda Roberts, Public contributor
-
Dr Gillian Shepherd, Director, Health and Clinical Excellence, Merck Serono Ltd
-
Mrs Katrina Simister, Assistant Director New Medicines, National Prescribing Centre, Liverpool
-
Professor Donald Singer, Professor of Clinical Pharmacology and Therapeutics, Clinical Sciences Research Institute, CSB, University of Warwick Medical School
-
Mr David Symes, Public contributor
-
Dr Arnold Zermansky, General Practitioner, Senior Research Fellow, Pharmacy Practice and Medicines Management Group, Leeds University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Mr Simon Reeve, Head of Clinical and Cost-Effectiveness, Medicines, Pharmacy and Industry Group, Department of Health
-
Dr Heike Weber, Programme Manager, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Psychological and Community Therapies Panel
-
Professor of Psychiatry, University of Warwick, Coventry
-
Consultant & University Lecturer in Psychiatry, University of Cambridge
-
Professor Jane Barlow, Professor of Public Health in the Early Years, Health Sciences Research Institute, Warwick Medical School
-
Dr Sabyasachi Bhaumik, Consultant Psychiatrist, Leicestershire Partnership NHS Trust
-
Mrs Val Carlill, Public contributor
-
Dr Steve Cunningham, Consultant Respiratory Paediatrician, Lothian Health Board
-
Dr Anne Hesketh, Senior Clinical Lecturer in Speech and Language Therapy, University of Manchester
-
Dr Peter Langdon, Senior Clinical Lecturer, School of Medicine, Health Policy and Practice, University of East Anglia
-
Dr Yann Lefeuvre, GP Partner, Burrage Road Surgery, London
-
Dr Jeremy J Murphy, Consultant Physician and Cardiologist, County Durham and Darlington Foundation Trust
-
Dr Richard Neal, Clinical Senior Lecturer in General Practice, Cardiff University
-
Mr John Needham, Public contributor
-
Ms Mary Nettle, Mental Health User Consultant
-
Professor John Potter, Professor of Ageing and Stroke Medicine, University of East Anglia
-
Dr Greta Rait, Senior Clinical Lecturer and General Practitioner, University College London
-
Dr Paul Ramchandani, Senior Research Fellow/Cons. Child Psychiatrist, University of Oxford
-
Dr Karen Roberts, Nurse/Consultant, Dunston Hill Hospital, Tyne and Wear
-
Dr Karim Saad, Consultant in Old Age Psychiatry, Coventry and Warwickshire Partnership Trust
-
Dr Lesley Stockton, Lecturer, School of Health Sciences, University of Liverpool
-
Dr Simon Wright, GP Partner, Walkden Medical Centre, Manchester
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Expert Advisory Network
-
Professor Douglas Altman, Professor of Statistics in Medicine, Centre for Statistics in Medicine, University of Oxford
-
Professor John Bond, Professor of Social Gerontology & Health Services Research, University of Newcastle upon Tyne
-
Professor Andrew Bradbury, Professor of Vascular Surgery, Solihull Hospital, Birmingham
-
Mr Shaun Brogan, Chief Executive, Ridgeway Primary Care Group, Aylesbury
-
Mrs Stella Burnside OBE, Chief Executive, Regulation and Improvement Authority, Belfast
-
Ms Tracy Bury, Project Manager, World Confederation of Physical Therapy, London
-
Professor Iain T Cameron, Professor of Obstetrics and Gynaecology and Head of the School of Medicine, University of Southampton
-
Professor Bruce Campbell, Consultant Vascular & General Surgeon, Royal Devon & Exeter Hospital, Wonford
-
Dr Christine Clark, Medical Writer and Consultant Pharmacist, Rossendale
-
Professor Collette Clifford, Professor of Nursing and Head of Research, The Medical School, University of Birmingham
-
Professor Barry Cookson, Director, Laboratory of Hospital Infection, Public Health Laboratory Service, London
-
Dr Carl Counsell, Clinical Senior Lecturer in Neurology, University of Aberdeen
-
Professor Howard Cuckle, Professor of Reproductive Epidemiology, Department of Paediatrics, Obstetrics & Gynaecology, University of Leeds
-
Professor Carol Dezateux, Professor of Paediatric Epidemiology, Institute of Child Health, London
-
Mr John Dunning, Consultant Cardiothoracic Surgeon, Papworth Hospital NHS Trust, Cambridge
-
Mr Jonothan Earnshaw, Consultant Vascular Surgeon, Gloucestershire Royal Hospital, Gloucester
-
Professor Martin Eccles, Professor of Clinical Effectiveness, Centre for Health Services Research, University of Newcastle upon Tyne
-
Professor Pam Enderby, Dean of Faculty of Medicine, Institute of General Practice and Primary Care, University of Sheffield
-
Professor Gene Feder, Professor of Primary Care Research & Development, Centre for Health Sciences, Barts and The London School of Medicine and Dentistry
-
Mr Leonard R Fenwick, Chief Executive, Freeman Hospital, Newcastle upon Tyne
-
Mrs Gillian Fletcher, Antenatal Teacher and Tutor and President, National Childbirth Trust, Henfield
-
Professor Jayne Franklyn, Professor of Medicine, University of Birmingham
-
Mr Tam Fry, Honorary Chairman, Child Growth Foundation, London
-
Professor Fiona Gilbert, Consultant Radiologist and NCRN Member, University of Aberdeen
-
Professor Paul Gregg, Professor of Orthopaedic Surgical Science, South Tees Hospital NHS Trust
-
Bec Hanley, Co-director, TwoCan Associates, West Sussex
-
Dr Maryann L Hardy, Senior Lecturer, University of Bradford
-
Mrs Sharon Hart, Healthcare Management Consultant, Reading
-
Professor Robert E Hawkins, CRC Professor and Director of Medical Oncology, Christie CRC Research Centre, Christie Hospital NHS Trust, Manchester
-
Professor Richard Hobbs, Head of Department of Primary Care & General Practice, University of Birmingham
-
Professor Alan Horwich, Dean and Section Chairman, The Institute of Cancer Research, London
-
Professor Allen Hutchinson, Director of Public Health and Deputy Dean of ScHARR, University of Sheffield
-
Professor Peter Jones, Professor of Psychiatry, University of Cambridge, Cambridge
-
Professor Stan Kaye, Cancer Research UK Professor of Medical Oncology, Royal Marsden Hospital and Institute of Cancer Research, Surrey
-
Dr Duncan Keeley, General Practitioner (Dr Burch & Ptnrs), The Health Centre, Thame
-
Dr Donna Lamping, Research Degrees Programme Director and Reader in Psychology, Health Services Research Unit, London School of Hygiene and Tropical Medicine, London
-
Professor James Lindesay, Professor of Psychiatry for the Elderly, University of Leicester
-
Professor Julian Little, Professor of Human Genome Epidemiology, University of Ottawa
-
Professor Alistaire McGuire, Professor of Health Economics, London School of Economics
-
Professor Neill McIntosh, Edward Clark Professor of Child Life and Health, University of Edinburgh
-
Professor Rajan Madhok, Consultant in Public Health, South Manchester Primary Care Trust
-
Professor Sir Alexander Markham, Director, Molecular Medicine Unit, St James’s University Hospital, Leeds
-
Dr Peter Moore, Freelance Science Writer, Ashtead
-
Dr Andrew Mortimore, Public Health Director, Southampton City Primary Care Trust
-
Dr Sue Moss, Associate Director, Cancer Screening Evaluation Unit, Institute of Cancer Research, Sutton
-
Professor Miranda Mugford, Professor of Health Economics and Group Co-ordinator, University of East Anglia
-
Professor Jim Neilson, Head of School of Reproductive & Developmental Medicine and Professor of Obstetrics and Gynaecology, University of Liverpool
-
Mrs Julietta Patnick, Director, NHS Cancer Screening Programmes, Sheffield
-
Professor Robert Peveler, Professor of Liaison Psychiatry, Royal South Hants Hospital, Southampton
-
Professor Chris Price, Director of Clinical Research, Bayer Diagnostics Europe, Stoke Poges
-
Professor William Rosenberg, Professor of Hepatology and Consultant Physician, University of Southampton
-
Professor Peter Sandercock, Professor of Medical Neurology, Department of Clinical Neurosciences, University of Edinburgh
-
Dr Philip Shackley, Senior Lecturer in Health Economics, Sheffield Vascular Institute, University of Sheffield
-
Dr Eamonn Sheridan, Consultant in Clinical Genetics, St James’s University Hospital, Leeds
-
Dr Margaret Somerville, Director of Public Health Learning, Peninsula Medical School, University of Plymouth
-
Professor Sarah Stewart-Brown, Professor of Public Health, Division of Health in the Community, University of Warwick, Coventry
-
Dr Nick Summerton, GP Appraiser and Codirector, Research Network, Yorkshire Clinical Consultant, Primary Care and Public Health, University of Oxford
-
Professor Ala Szczepura, Professor of Health Service Research, Centre for Health Services Studies, University of Warwick, Coventry
-
Dr Ross Taylor, Senior Lecturer, University of Aberdeen
-
Dr Richard Tiner, Medical Director, Medical Department, Association of the British Pharmaceutical Industry
-
Mrs Joan Webster, Consumer Member, Southern Derbyshire Community Health Council
-
Professor Martin Whittle, Clinical Co-director, National Co-ordinating Centre for Women’s and Children’s Health, Lymington