

Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 11/14/33. The contractual start date was in December 2012. The final report began editorial review in August 2016 and was accepted for publication in April 2017. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Andreas Goebel reports grants from the National Institute for Health Research (NIHR) Medical Research Council (MRC) Efficacy and Mechanism Evaluation (EME) programme, other funds from Biotest AG, Germany, and grants from Biotest AG during the conduct of the study. Furthermore, he received personal fees from Biotest AG and Axsome Therapeutics outside the submitted work. Mick Serpell reports grants from NIHR EME programme (a MRC and NIHR partnership), grants for additional funding obtained by the Pain Relief Foundation Liverpool, and non-financial support was received from Biotest UK Ltd, which provided the active study medication at no cost during the study. He has also received research support, consulting fees or honoraria in the past 3 years from Astellas Pharma, Grünenthal Ltd, NAPP and Pfizer. Nick Shenker reports NIHR funding for researchers' time during the conduct of the study.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2017. This work was produced by Goebel et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2017 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
The text in the following chapter is reproduced from Goebel et al. 1 and includes minor additions and formatting changes to the original text. © Goebel et al. ; licensee BioMed Central Ltd. 2014. This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
Background
Complex regional pain syndrome (CRPS) is post-traumatic pain in a limb and is associated with sensory, motor, autonomic, skin and bone changes. 2,3 CRPS can resolve spontaneously, but if spontaneous resolution does not occur early, it is less likely to occur later. Many patients with CRPS have no effective method to relieve their ongoing pain. 4 Those patients with CRPS of moderate to severe pain intensity were the target group for this study and report, on average, a very poor quality of life (QoL) and they usually cannot work. 5
Immunoglobulin treatment for chronic pain is a novel technology. 6 In a first, open trial we found that low-dose intravenous immunoglobulin (IVIg) may be effective for relieving pain in some patients with CRPS (11 participants: 3 had > 70% pain relief, 2 had > 25%< 70%, and 6 had 0–25% relief, following a variable number of low-dose infusion repeats). 7 We later showed that, in one patient, repeat treatments provided reproducible effects. 8 In a UK single-centre crossover randomised placebo-controlled trial,9 a single, low-dose (0.5 g/kg) infusion of IVIg significantly reduced pain in patients with CRPS (n = 13, pain intensity on an validated 11-point Numeric Rating Scale (NRS) higher than 4 (NRS 0 = no pain, 10 = pain as bad as you can imagine10); these patients had the disease for between 0.5 and 2.5 years. The treatment difference was 1.55 NRS points [95% confidence interval (CI) 1.29 to 1.82 points; p < 0.001]. In a responder analysis (12 patients had received treatment), three patients had ≥ 50% less pain (4.5, 5 and 5 NRS points) after IVIg when compared with after saline treatment, and two patients had 29% and 31% less pain (2 and 2.5 NRS points less pain). One patient had 25% less pain (2 NRS points less pain) after saline than those patients having IVIg treatment. The average effect duration was 5 weeks. There was also a significant overall reduction of CRPS-related, non-painful symptoms and, in responders, improved sleep and global improvement, with few adverse events (AEs) (headaches and pain increases for < 3 days). Post-infusion questionnaires showed successful blinding of patients and study doctors.
We conducted a trial to explore whether or not subcutaneous immunoglobulin, in weekly self-administration at home, over 1 year would provide sustained pain relief in those who initially responded to 0.5 g/kg IVIg. 8 Five patients with at least 2 NRS points less pain after IVIg in the earlier randomised controlled trial (RCT) were invited to take part. Of these patients, one declined participation and a second patient developed metastasising colon cancer; therefore, three patients participated. By August 2011, two patients who had the disease for 6 years and 5 years at study entry and baseline pain intensities of NRS 7 and 6 points, had experienced sustained pain reduction of > 70% for 12 and 3.5 months, respectively. The third patient, who had had 31% relief in the RCT, showed no benefit. The two responding patients reported major improvement in their QoL; EuroQol-5 Dimensions (EQ-5D) scores11 improved from 0.26 and 0.30 at baseline to 0.66 and 0.65 at 12/3 months, and both patients experienced reduced interference of daily functioning by their pain [Brief Pain Inventory (BPI)12 interference scores (pain interference = the impact of pain on activities of daily life) improved from 7.7 and 6.1 at baseline to 1.4 and 0 at 12/3 months].
The aforementioned evidence provides proof of concept for the efficacy of low-dose immunoglobulin treatment for moderate to severe CRPS in reducing pain, with an advantageous side effect profile. The data also suggest that this treatment may improve QoL and pain interference. Because the numbers of treated patients have been small and most research was conducted in a single centre, it was important to confirm these findings in a larger group of patients and across several centres to gain confidence about both efficacy and effect size of this novel technology and to demonstrate its generalisability.
Chapter 2 Objectives
Primary objective
To gain, within 44 months, definite proof of the clinical efficacy and a more confident estimate of the effect size of low-dose IVIg treatment to reduce pain in patients with moderate or severe CRPS.
Secondary objectives
To achieve better understanding of this technology, including:
-
stability of effect with repeat administration
-
factors predicting a beneficial response
-
effects on additional outcome parameters including stimulus evoked pain, pain interference, QoL and short-term risk profile
-
health economics evaluation
-
creation of a bank of biological samples at the University of Liverpool for future CRPS serum autoantibody and serum substances research.
Chapter 3 Methods
Study design and participants
The LIPS is a Phase II multicentre, randomised, double-blind, placebo-controlled, parallel group trial with an open extension. The open extension was an optional trial element, in which participants who had completed the parallel, blinded phase could receive one or two doses of IVIg. This open-label extension was included to account for the participant’s preferences and optimise recruitment and retention rates.
Patients were given the patient information sheet (see Appendix 1) to read at least 24 hours before the screening visit, at which time they provided informed consent (see Appendix 2).
Eligible patients had moderate or severe CRPS of between 1 and 5 years’ duration. A mean pain intensity of ≥ 5 on an 11-point (0–10) NRS over the first seven daily entries after screening and a recorded pain intensity during this period that did not drop below 4 were required for eligibility.
The protocol has been published previously. 1
Eligibility criteria
Inclusion criteria
-
Diagnosis of CRPS I or II according to Budapest research criteria13 (see Appendix 3).
-
Duration of the disease of between 1 and 5 years and a mean pain intensity of ≥ 5 on an 11-point (0–10) NRS over the first seven daily entries after screening (the first entry is the day after the screening visit), and a recorded pain intensity that never drops below four during this period, is required for eligibility. At least six out of seven entries of an average of 24 hours of pain intensity are required in the pain diaries.
-
Failure to respond (poor efficacy or unacceptable side effects) to drugs recommended for the treatment of neuropathic pain,14 including pregabalin or gabapentin, a tricyclic antidepressant, and mild and strong opioids (when not contraindicated or refused by the patient).
-
Previous specialised pain physiotherapy15 (when not contraindicated or refused by the patient).
-
Willingness to confirm the use of adequate birth control while on the trial in the case of premenopausal women without evidence for an inability to become pregnant.
-
Willingness to not start any other treatment for CRPS during the parallel part of the trial.
-
Aged ≥ 18 years.
Exclusion criteria
Any individuals meeting any of the following were excluded from the study:
-
Other significant chronic pains that, in the view of the study doctor, may make assessment of the pain arising from CRPS difficult.
-
Recent initiation of a new therapy for CRPS that, in the view of the study doctor, may change the patient’s pain level during the period of participation in the trial.
-
Unstable medical conditions.
-
Litigation. Patients in litigation will be excluded only if conclusion of that litigation is imminent during the course of the study.
-
Pregnant or breastfeeding patients.
-
Complete immunoglobulin A deficiency.
-
Rare contraindications to IVIg therapy as per summary of product characteristics.
-
Receiving IVIg for other reasons.
-
Previous enrolment in CRPS IVIg/subcutaneous immunoglobulin trials.
-
Ongoing drug or alcohol abuse.
-
Psychiatric or mental health disorder that could, in the judgement of the site investigator, interfere with successful study participation.
-
Unwillingness or inability to complete daily diaries, or inability to understand the questionnaires being used.
-
Cancer other than basal cell carcinoma within the last 5 years. However, those patients who have received definitive treatment, such as curative surgery, > 6 months ago with no known recurrence can be included.
-
A history of hypercoagulable or thrombophilic clotting abnormalities.
-
A history of thrombembolic events: ischaemic stroke, confirmed myocardial infarction, pulmonary embolism, deep-vein thrombosis except when immobility related (e.g. after injury or operation).
-
Unstable angina.
-
Renal failure or serum creatinine > 1.5 times the upper limit of normal at screening.
-
Any medical condition that, in the opinion of the investigator, would make it unsafe for the patient to participate or that would interfere with assessment of the outcome measures.
-
Participation in another interventional trial within 3 months of randomisation. Participation in non-interventional studies is not a reason for exclusion.
The study time frame was scheduled with the date of randomisation defined as day 0 and a screening visit was conducted maximally 3 weeks before this (day –21). Patients received blinded infusions on days 1 and 22. Participants who decided to receive open-label infusions received infusions on days 43 and 64. Paper diaries documenting the patients’ self-reported daily pain score were completed from day 2 to day 43 and a weekly pain score was documented for a further 9 weeks to explore the duration of drug and unspecific treatment effects. Participants who received open-label infusions documented a daily pain score to the point of the final infusion they received (1 or 2 open-label infusions) and a weekly pain score 9 weeks thereafter.
Participating centres and recruitment dates
The study was conducted across seven UK-based specialist secondary care pain clinics, and participants were recruited from these clinics or were referred to the clinics from a UK network of pain clinics or patient identification centres. Recruitment commenced in August 2013 at the lead centre and a delay in contract negotiations at a number of recruiting centres resulted in an initial slower-than-anticipated rate of recruitment. However, recruitment targets were met within the 24th month of the project and recruitment was ahead of target by the 27th month of the project. This resulted in the trial over-recruiting and the ending of recruitment 1 month ahead of the study schedule.
The seven centres involved in the multicentre study and dates of active recruitment were:
-
The Walton Centre NHS Foundation Trust, Liverpool, UK (August 2013 to October 2015)
-
Guy’s and St Thomas’s NHS Foundation Trust, London, UK (January 2014 to September 2015)
-
Royal National Hospital for Rheumatic Diseases, Bath, UK, and University West of England, Bristol, UK (May 2014 to October 2015)
-
Queen Elizabeth University Hospital, Glasgow, UK (January 2014 to September 2015)
-
Norwich and Norfolk University NHS Trust, Norwich, UK (March 2014 to September 2015)
-
Cambridge University Hospitals NHS Foundation Trust, Cambridge, UK (March 2014 to July 2015)
-
University Hospital of Leicester NHS Trust, Leicester, UK (April 2014 to June 2015).
The following patient identification centres were set up to refer potential participants to the seven study centres.
-
King’s College London NHS Foundation Trust, London, UK.
-
Central Manchester University Hospitals NHS Foundation Trust, Manchester, UK.
-
Royal National Orthopaedic Hospital NHS Trust, Stanmore, UK.
-
Royal Devon and Exeter NHS Foundation Trust, Exeter, UK.
-
Plymouth Hospitals NHS Trust, Plymouth, UK.
-
University Hospitals Southampton NHS Foundation Trust, Southampton, UK.
-
City Hospitals Sunderland NHS Foundation Trust, Sunderland, UK.
-
The Dudley Group NHS Foundation Trust, Dudley, UK.
-
Tameside Hospital NHS Foundation Trust, Tameside, UK.
Interventions
A full description of the assessments utilised in the trial is documented in the published study protocol. 1 The visit schedule and assessments conducted are outlined in Table 1 and summarised below.
| Study week/month | Visit 1: screen (day –21 to day –10) | Telephone to confirm eligibility (day –11 to day –1) | Day 0 | Day 1 | Day 2 (+ up to 3 day) | Day 5 (+/– 2 days) | Day 22 (+/– 1 day) | Day 23 (+ up to 3 days) | Day 26 (+/– 2 days) | Day 43 (+/– 1 day) | Day 64 (+/– 1 day) | Day 85 (+ up to 3 days) | Day 148 (+/– 1 day) | Withdrawal |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Randomisation | Visit 2 (first blinded infusion) | Telephone | Telephone | Visit 3 (second blinded infusion) | Telephone | Telephone | Visit 4 (obligatory, with optional first open infusion) | Visit 5 (this visit is only for second open infusion) | Telephone | Telephone: end of trial | ||||
| Registration/demographics | ✗ | |||||||||||||
| Informed consent | ✗ | |||||||||||||
| Eligibility form | ✗ | |||||||||||||
| Randomisation form | ✗ | |||||||||||||
| Medical history | ✗ | |||||||||||||
| CRPS history | ✗ | |||||||||||||
| Limb examination | ✗ | ✗ | ✗ | ✗ | ||||||||||
| Limb temperature, limb volume | ✗ | ✗ | ||||||||||||
| Safety bloods (U&E, FBC, serum-Ig, LFT) | ✗ | |||||||||||||
| Pregnancy test (beta HCG) | ✗ | |||||||||||||
| Pregnancy test (urine) | ✗ | |||||||||||||
| Screening pain diaries (average 24-hour pain intensity only) | a | ✗ (over telephone) | b | |||||||||||
| Detailed (blind) diary, weeks 1–3 (average pain intensity, pain unpleasantness, sleep quality). Patients who consent will receive daily prompting text reminders during days 2–42 | a | b | ||||||||||||
| Detailed (blind) diary, weeks 4–6 (average pain intensity, pain unpleasantness, sleep quality). Patients who consent will receive daily prompting text reminders during days 2–42 | a | b | ||||||||||||
| Detailed (open) diary, weeks 7–9 (average pain intensity, pain unpleasantness, sleep quality). For patients who receive open-label infusion only | a | b | ||||||||||||
| LIPS detailed (open) dairy, weeks 10–12 (average pain intensity, pain unpleasantness, sleep quality). For patients who receive open-label infusion only | a | c | ||||||||||||
| Simplified pain diaries (weekly pain intensity scores only) | a (patients who do not receive open-label infusion) | c (patients who do not receive open infusion) | a (patients who do receive open infusions) | c (patients who do receive open infusions) | a | |||||||||
| Questionnaires | ||||||||||||||
| Expectation from treatment | ✗ | |||||||||||||
| EQ-5D-5L | ✗ | ✗ | ✗ | |||||||||||
| McGill Pain Questionnaire (Short Form) pain descriptors | ✗ | ✗ | ✗ | |||||||||||
| BPI | ✗ | ✗ | ✗ | |||||||||||
| HADS | ✗ | ✗ | ✗ | |||||||||||
| Pain catastrophising (Sullivan’s catastrophising scale) | ✗ | ✗ | ✗ | |||||||||||
| Global Impression of Change Scale | ✗ | ✗ | ✗ | ✗ | ||||||||||
| Health/social care utilisation | ✗ | |||||||||||||
| Patient-developed measures | ✗ | ✗ | ✗ | |||||||||||
| Stanford Presenteeism Scale | ✗ | ✗ | ||||||||||||
| Neglect-like symptoms | ✗ | ✗ | ||||||||||||
| Vital signs (pulse, blood pressure, before and after infusion) | ✗ | ✗ | ✗ | ✗ | ||||||||||
| Treatment infusion administration | ✗ | ✗ | ✗ | ✗ | ||||||||||
| Research bloods (30 ml) | ✗ | ✗ | ||||||||||||
| Quantitative sensory testing (subset of 40 patients only) | ✗ | ✗ (if not done on visit 1) | ✗ | |||||||||||
| Concomitant and CRPS pain treatments medications | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||||
| Concomitant therapies | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||||
| AEs form | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||||||
| Patient medication guess | ✗ | ✗ | ✗ | ✗ | ||||||||||
| Physician medication guess | ✗ | ✗ | ✗ | |||||||||||
| Research nurse medication guess | ✗ | ✗ | ✗ | |||||||||||
| Withdrawal form | ✗ |
The experimental intervention was 0.5 g/kg Intratect TM IVIg infusion, in combination with ongoing normal standard treatment for CRPS.
Interventions were available in 5 g/100 ml and 10 g/200 ml bottles of Intratect™ IVIg infusion (active) or matching placebo. The volume prescribed was within normal clinical doses per unit weight determined (see Appendix 4).
Participants were scheduled to receive infusion of the active or matching placebo on day 1 post randomisation and day 22 post randomisation. The protocol allowed for up to 5 working days from randomisation to the first infusion to account for exceptional circumstances, such as the participant presenting with symptoms that made it unsafe for them to receive the infusion.
Participants who consented to the open-label extension phase received the IVIg on days 43 and 64 post randomisation. The protocol allowed for infusions to occur –1/+1 day from the scheduled date. However, primary outcome timelines remained fixed from the date of randomisation regardless of when the infusion was received. These intervals of 3 weeks between infusions were scheduled in line with usual clinical practice, which accommodates the half-life of IVIg of about 3 weeks.
Safety bloods were collected at the screening visit as part of the eligibility criteria. When consent was provided, additional research bloods were taken at screening and on day 64. CRPS-specific assessments including Limb Assessments and Quantitative Sensory Assessments, vital signs and pregnancy tests were conducted and questionnaires were completed by the participant at set infusion time points, and the paper pain score diaries were retrieved and distributed. Participants who agreed to the open-label extension and received a second infusion were requested to return their daily pain diary and weekly diary to the study team via post. Study staff contacted participants twice following each infusion to confirm adherence to the pain questionnaires and to document any AEs experienced.
All data were collected on trial-specific source data worksheets and transcribed onto electronic case report forms (eCRFs) by authorised trial staff. The eCRF was designed within the Elsevier InferMed MACRO (version 4; Elsevier, London, UK) system which is regulatory compliant (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use Guideline for Good Clinical Practice,16 US Food and Drug Administration 21 CRF Part 11,17 European Commission Clinical Trial Directive18). The eCRF was created in collaboration with the trial statisticians and the investigators, and maintained by the King’s Clinical Trials Unit (KCT). It was hosted on a dedicated secure server within King’s College London.
A trial manager was employed to conduct onsite/central monitoring of the data and was responsible for raising and resolving queries to ensure quality assurance.
All causes for withdrawal from randomised treatment were reported at days 22 and 43 post randomisation. The prevalences of AEs and reactions were reported descriptively at 22 and 43 days post randomisation. If given open-label infusion, AEs reported from 43 to 85 days post randomisation were tabulated separately for reports rather than being reported with blinded AEs. Serious adverse events (SAEs) were monitored for 21 days after the final dose of IVIg/placebo or until resolution.
The study end was defined as the last participant’s final study contact at day 148 for those who consented to receive open-label infusions, or at day 64 for those who declined the open-label extension.
Outcomes
The primary outcome was the average 24-hour pain intensity over 37 days, recorded in paper pain diary entries for the previous 24 hours and collected on days 6 to 42 (day 1 = day of first infusion). The trial initially utilised a text messaging system to collect the NRS pain scores from patients who consented as a supplementary means of gathering the primary outcome data; however, the system was abandoned by agreement of the Trial Steering Committee (TSC) owing to a high compliance rate of the paper diaries being completed and returned. In addition, there was a low response rate of the text message scores sent by patients who had consented to the system and the site staff experienced technical difficulties in setting patients up to use the system.
Secondary outcomes were the pain interference measured using the interference subscale of the BPI12 and QoL measured using the EuroQol-5 Dimensions, five-level version (EQ-5D-5L). 19
All other outcomes were exploratory.
A list of the measures that were used is given below (for measurement times see Table 1).
-
Detailed daily pain diaries (three items: pain unpleasantness,10 average 24-hour NRS pain intensity, last 24-hour sleep quality20), and simplified weekly (weekly NRS pain intensity) pain diaries.
-
AEs.
-
BPI12 (diagram, worst pain intensity and interference scales only).
-
Concomitant medications.
-
Concomitant therapies.
-
Patient weight.
-
Skin temperature measured with a surface thermometer.
-
Limb volume measured with a water bath technique.
-
EQ-5D-5L. 19
-
Expectations from treatment. 21
-
Functional items and fatigue suggested by, and developed together with, patient group (five scales).
-
Patient Global Impression of Change. 22
-
Hospital Anxiety and Depression Scale. 23
-
Health and social care utilisation.
-
Limb examination recording Budapest CRPS signs and any additional abnormalities on inspection, and sensory (cotton wool, pinprick, cold fork) and motor (observation of active range) examination.
-
McGill Pain Questionnaire (Short Form). 24
-
Quantitative sensory testing in 40 patients with stimulus evoked pain, excepting thermosensitivities (only in three trial centres).
-
Sullivan’s Pain Catastrophising Scale. 25
-
Work interference (Stanford Presenteeism Scale). 26
-
Neglect-like symptoms in CRPS. 27
-
Patient recommended scale (see Appendix 5).
Randomisation and blinding
Participants were randomly assigned (1 : 1) to IVIg or placebo by site staff who were authorised to request randomisation via an independent online randomisation system based at the KCT within the Institute of Psychology, Psychiatry and Neuroscience. Allocation was at the level of the individual patient via block randomisation with randomly varying block sizes, stratified by centre.
The trial was double blinded. Supplies of the study medication dispensed on day 1 and day 2 post randomisation were blinded (and the IVIg prescribed on days 43 and 64 was open label). Blinding was achieved by preparing the investigational medicinal product/active and placebo solution (0.1% albumin in normal saline) into bottles of identical appearance, including the labelling and the batch numbers/expiry dates. By adding albumin to the placebo, the solutions were indistinguishable in colour and foaming of the solution. The study drug was delivered to the designated pharmacy contact with a removable section on the bottle and secondary packaging which informed the pharmacy staff of the true contents (active or placebo). This section was removed when dispensing in order to maintain blinding. Study medication was prescribed by an authorised study physician in accordance with the protocol, using a trial-specific prescription.
Blinding was maintained by utilising the services of an external pharmaceutical project management company, ModePharma, which centrally monitored study medication and instructed the distribution of the study medication from the Aseptic Manufacturing Pharmacy Unit at Royal Liverpool and Broodgreen Hospital, Liverpool, UK. With the exception of the pharmacy site staff, the staff at ModePharma and Royal Broadgreen Hospital and the Director of KCT, all other research team members involved in the study were blinded to the treatment allocation. The analysing statistician was subgroup unblinded only until analyses were complete, at which point the trial was fully unblinded.
In the event of an urgent need to unblind the treatment of participants, site staff were informed of the contact details of a code break service that was utilised through Guy’s Medical Toxicology Unit. However, the service was not utilised and no participants were unblinded throughout the duration of the trial.
Statistical analysis
The sample size was based on the following assumptions based from the pilot study:9 122 participants were required to detect a difference in pain score of 1.2 using a two-sample t-test, assuming 5% statistical significance, 85% power and a common standard deviation of 2.2 (as in our previous study9). Assuming 10% loss to follow-up and a 5% non-compliance increased this number to 152 participants. We intended to collect 37 measurements of pain intensity (the primary outcome) per participant and analyse the outcome using a mixed-effects regression model. Therefore, the sample size was reduced based on these extra measurements. From the pilot study,9 the correlation between a patient’s measures was assumed to be 0.7; hence the multiplying factor was [1 + (37 – 1) × 0.7)/37 = 0.71]. Therefore the total required sample size was calculated at 152 × 0.71 = 108 participants, 54 patients per study arm. 28
Primary analysis
The primary outcome was analysed using a mixed model to establish any difference between pain scores after IVIg and placebo. The stratification factor (study centres) was a fixed effect. The model efficiently modelled the repeated measurements data. Modelling assumptions were checked (e.g. residuals) and all analyses were performed on an intention-to-treat basis. Every effort was made to reduce loss to follow-up using fixed-point telephone calls. Participants who provided any outcome data were included and no primary outcome were omitted from the primary analysis; however, they were included within a sensitivity analysis along with participants who were incorrectly consented into the trial for not meeting the inclusion criteria.
Secondary analysis
As a secondary analysis, we calculated the proportion of patients in each arm who achieved 50% or 30% pain relief based on the average pain level entered on day 6–42, compared with their baseline level of pain (the average pain level recorded during the first 7 days of the screening period). Using these proportions, we calculated the number needed to treat with IVIg so that one additional patient will achieve 50% pain relief.
Possible changes in treatment effect over time and association between disease duration, psychological baseline measurements, allergy status/low baseline immunoglobulin G (IgG) plasma level, IgG increase, and treatment response, and any association between psychological measurements with the primary outcome was investigated using exploratory plots and regression models with interaction terms. Change in McGill Pain Questionnaire-Short Form,24 descriptor terms, limb temperature and quantitative sensory testing changes before and after IVIg or placebo treatments on affected versus/contralateral sides, pain interference and QoL outcomes were investigated using either standard regression models or mixed models. In those who decided to receive both open infusions and who had at least 30% or 2 NRS points average pain relief from 6 to 20 days after their last open infusion compared with baseline, the time between the last open infusion and the first period with average weekly pain equalling or exceeding baseline –1 NRS point was calculated as the IVIg effect duration. As the study ended on day 148 (12 weeks after the second open infusion), later effects were not recorded.
The statistical tests were conducted using Stata® version 14 (StataCorp LP, College Station, TX, USA).
A Data Monitoring Committee was formed, which had access to the unblinded data and monitored the progress of the trial in terms of safety and ethics issues. A blinded interim analysis was performed for futility and safety after half of the patients completed the trial and reported to the TSC that the trial should continue based on pre-agreed stopping rules.
Patient and public involvement
In preparation for the application to the National Institute for Health Research (NIHR), the early design had been sent to several patients with CRPS and comments were integrated. The final study protocol for the full proposal was then sent to 18 patients with CRPS who had previously agreed to be contacted for this purpose. These patients were a subgroup of patient participants in the ‘Liverpool CRPS pathway group’, a regional support group. Responses mostly related to convenience of attendance and additional outcomes, and have been implemented. Further suggestions from a patient participant on the NIHR review board were also taken on board. Patient information sheets were reviewed for acceptability by the same Liverpool patient group, and suggestions were implemented.
A patient representative with a history of CRPS volunteered to join the TSC and regularly attended these meetings. The patient representative offered invaluable advice and guidance throughout the trial.
The plain English summary was also forwarded to our patient representative on the TSC. Minor suggestions raised were addressed prior to submission.
Ethics
The study was approved by the National Research Ethics Service Committee East of England-Hatfield on 6 June 2012 and each site was granted site-specific approval from its NHS Research and Development department before trial commencement. This trial is registered with ISRCTN42179756.
Protocol changes
Original protocol (version 1.1) is dated 2 May 2012 and the final version of the protocol is available online (www.nets.nihr.ac.uk/projects/eme/111433) (Table 2).
| Substantial amendment number | Summary of changes |
|---|---|
| Substantial amendment 1, protocol version 2.0 (19 October 2012) | Sponsor contact |
| The new details reflect a change to the legal representative for one of the sponsors | |
| 7.1 Primary outcome measure | |
|
The new text describes an extended period for 24-hour pain diary from 15 to 37 days. An additional description of the text prompting system is described to improve compliance This change reverts back to the original design agreed by the funders as the TSC felt it more robust |
|
| 7.2 Secondary outcome measures | |
|
The EQ-5D will now be used as a measure of QoL The rewording clarifies the two parts to the secondary outcomes. The secondary outcomes and the exploratory outcomes. There were additional missing references for the assessments to be used that have now been included. The standard gamble was removed on the advice of a health economist |
|
| 8.2 Exclusion criteria | |
|
Serum IgA levels previously defined as an exclusion criterion have now been redefined An additional exclusion criterion has been included to prevent the inclusion of individuals that have participated in an intervention trial within the past 3 months |
|
| 9 Screening recruitment and consent | |
| Additional text has been included for clarification | |
| 10.3 Selection and timing of dose for each participant | |
| Additional details included to clarify the inclusion of participants that are non-compliant for the infusion visit | |
| 10.6 Packaging and labelling of investigational medicinal product | |
| Study name and EudraCT number now included on label | |
| 10.11 Concomitant medications | |
| Additional information is provided on how to proceed if there is a change in a participant’s condition with regards to CRPS and trial intervention | |
| 10.12.2 Biochemistry | |
| Additional tests have been included for biochemistry | |
| 11.3 Implementation procedures | |
| Clarification of the implementation procedures, including a checklist for the site nurses to go through before a participant can be randomised | |
| 14.1.1 Efficacy safety | |
|
Treatment stopping rules agreed and included The reporting of AEs has also been clarified |
|
| 14.2 Sample size calculation | |
|
The sample size calculation was adjusted to account for the increased number of days included for the measurement of the primary outcome This change reverts back to the original design agreed by the funders as the TSC felt it more robust |
|
| 15.3 Withdrawal of participants | |
| The following points were included to clarify discontinuation of participants in the study | |
| 16.2 Monitoring Quality Control and Assurance Safety | |
|
Changes have been made to the representatives in the TSC and the DMEC Addition of PI video The video is intended to standardise the explanation of the trial across sites. As the outcome is subjective this is felt to be of importance |
|
| Substantial amendment 2, protocol version 3.0 (11 April 2013) | REC |
| Updated REC address | |
| 7.2 Secondary and exploratory outcome measures | |
| Removal of time-trade-off scale | |
| 8.1 Inclusion criteria | |
| Rewording of inclusion criteria 4: previous specialised pain physiotherapy (when not contraindicated or refused by the patient) | |
| 10 Study medication | |
| Clarification of drug/placebo availability, drug/placebo labelling and packaging. Changes to the blinding procedure | |
| 10.12.3 Pregnancy | |
|
Addition of urine pregnancy test at visit 4 for females wanting open-label drug Summary of study procedures Clarification to study procedures PI video Addition of slides |
|
| Substantial amendment 3, protocol version 4.0, (1 July 2013) | 7.2 Secondary outcome measures |
| Neglect-like symptoms in CRPS questionnaire have been added to the list of measures to be used within the secondary and exploratory outcome measures | |
| 14 Statistical considerations | |
| Allergy status and low-baseline IgG plasma level have been added to secondary analysis | |
| Substantial amendment 4, protocol version 5.0, (4 October 2013) |
Protocol (throughout) Additional site: Leicester Minor corrective changes Participant information sheet Amendment to common, occasional and rare side effect of IVIg |
| Non-substantial amendment 5, protocol version 5.1 (7 July 2014) | 10.3 Selection and timing of dose for each participant |
| Clarification of an allowance of 5 working days instead of 5 days for first infusion and inclusion for analysis |
Chapter 4 Results
In total, 121 participants were screened, of whom 111 participants were randomised to one of the two trial arms. A total of 56 participants were randomised to placebo and 55 to IVIg. Two of these participants did not receive their first infusion and supplied no outcome pain data. Three further participants did receive their first infusion but also did not supply any outcome pain data. Therefore, all five of these participants are excluded from the primary analysis.
In addition, three participants were randomised in error. Two of these participants had an average baseline pain score (over the first 7 days) of < 5 and one participant had the disease for < 12 months. These three participants (all randomised to IVIg) are excluded from the primary analysis.
The primary analysis was performed on 103 participants, with 53 in the placebo group and 50 in the IVIg group. This is summarised in Figure 1.
FIGURE 1.
Participant flow diagram.
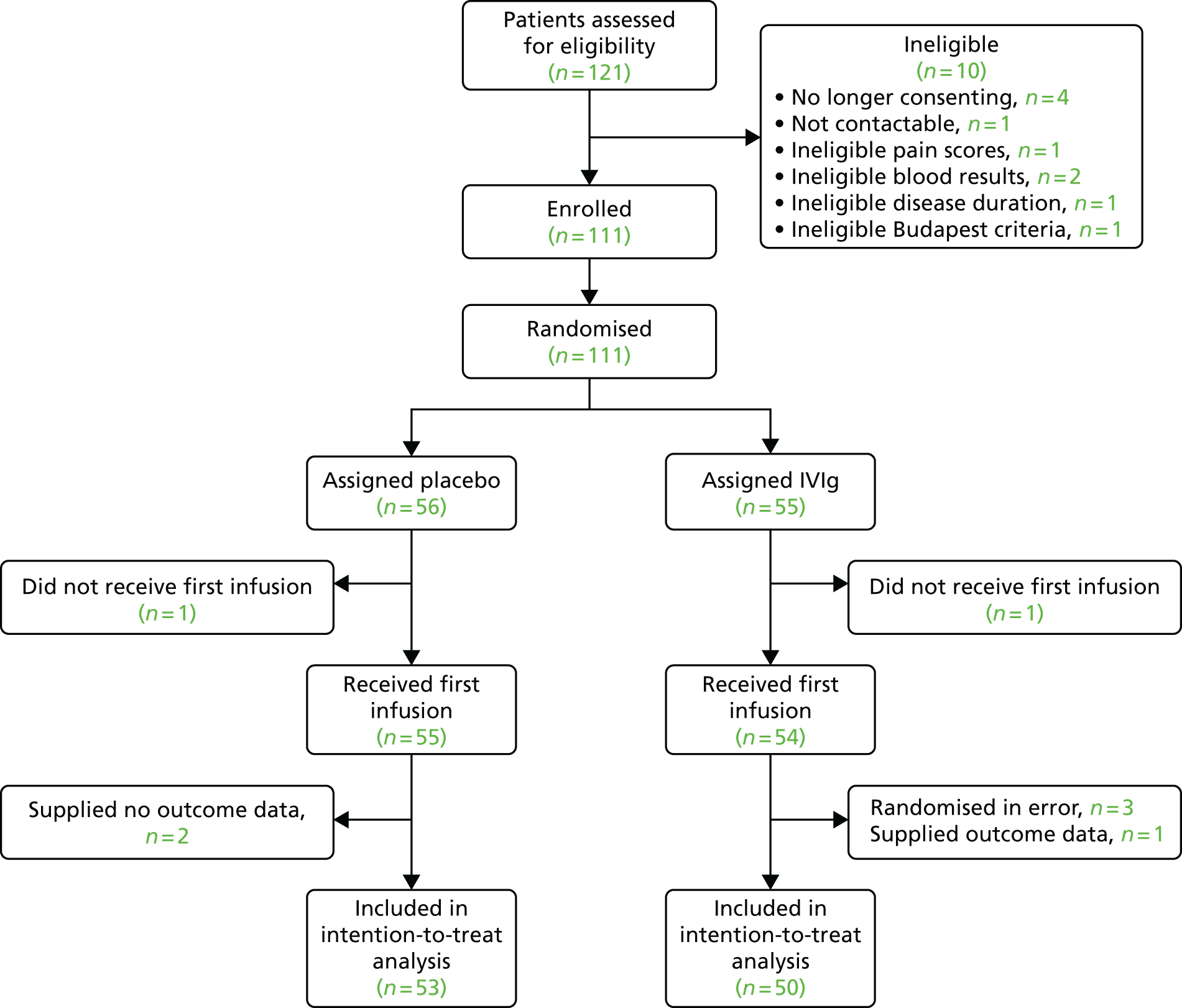
Withdrawals from study medication
Twelve participants withdrew their consent to use the study medication before the end of the blinded phase (day 42). This includes the two participants who did receive their first infusion and the three participants who received their first infusion but did not supply any outcome data. The remaining seven participants received their first infusion and completed their pain diaries for at least 2 weeks and are included in the primary analysis. Only one of these 12 participants received a second infusion. Further details regarding these participants can be found in Table 3.
| Day | Treatment | Reason for withdrawal | First infusion? | Second infusion? |
|---|---|---|---|---|
| 0 | Placebo | Refused further participation | No | No |
| 1 | IVIg | Participant unable to travel | No | No |
| 6 | Placebo | AE (participant withdrew) | Yes | No |
| 15 | Placebo | AE (participant withdrew) | Yes | No |
| 20 | IVIg | Other (alternative treatment sought: high-dose capsaicin patch) | Yes | No |
| 20 | IVIg | Participant unable to travel | Yes | No |
| 21 | IVIg | Refused further participation | Yes | No |
| 21 | IVIg | AE (participant withdrew) | Yes | No |
| 22 | Placebo | AE (team withdrew) | Yes | No |
| 22 | IVIg | AE (team withdrew) | Yes | No |
| 33 | IVIg | Refused further participation | Yes | No |
| 36 | IVIg | AE (participant withdrew) | Yes | Yes |
Baseline characteristics
Baseline characteristics for the 111 randomised participants are shown in Table 4. It is clear that balance has been achieved for most variables, although there is a slight sex imbalance.
| Trial arm | ||
|---|---|---|
| Placebo (n = 56) | IVIg (n = 55) | |
| Age (years) | 41.0 (12.5) | 43.7 (11.6) |
| Sex (female) | 42 (75.0%) | 35 (63.6%) |
| Ethnicity | ||
| Asian | 0 (0.0%) | 2 (3.6%) |
| White | 55 (98.2%) | 53 (96.4%) |
| Other | 1 (1.8%) | 0 (0.0%) |
| Disease duration (years) | 2.5 (1.2) | 2.3 (1.2) |
| CRPS type | ||
| I | 49 (87.5%) | 47 (85.5%) |
| II | 6 (10.7%) | 6 (10.9%) |
| Undecided | 1 (1.8%) | 2 (3.6%) |
| Average baseline pain | 7.40 (1.10) | 7.43 (1.13) |
| Site | ||
| Bath | 4 (7.1%) | 6 (10.9%) |
| Cambridge | 5 (8.9%) | 4 (7.3%) |
| Glasgow | 9 (16.1%) | 9 (16.4%) |
| Leicester | 3 (5.4%) | 3 (5.5%) |
| Liverpool | 16 (28.6%) | 15 (27.3%) |
| London | 14 (25.0%) | 14 (25.5%) |
| Norwich | 5 (8.9%) | 4 (7.3%) |
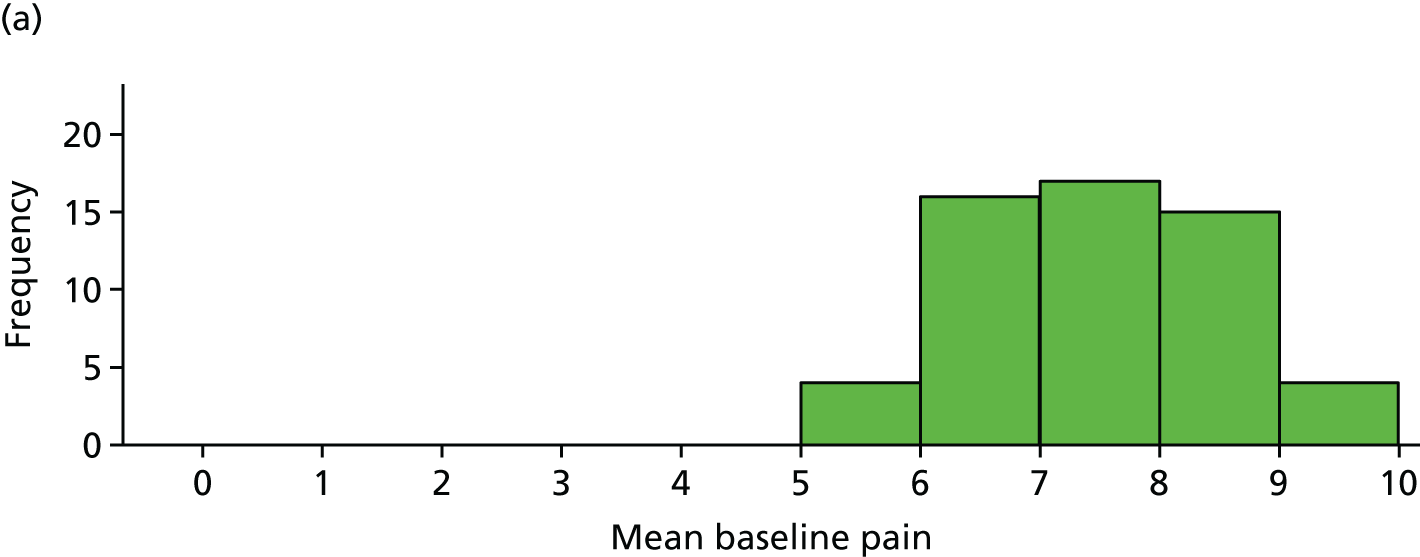
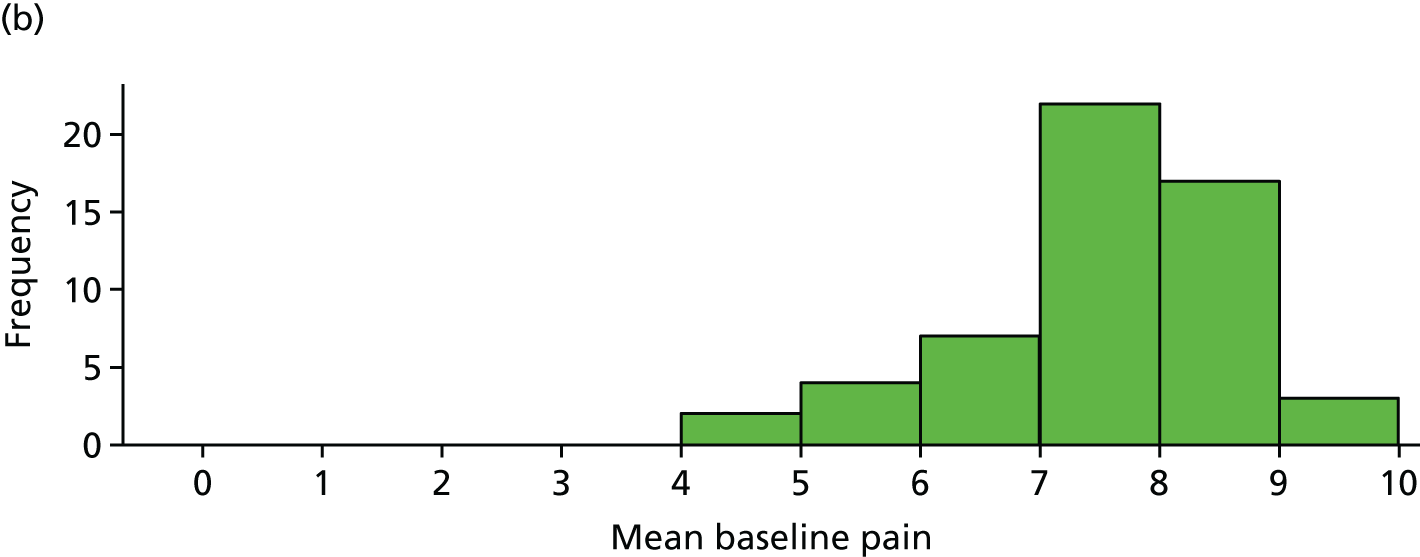
The distribution of age, disease duration and mean baseline pain by trial arm is shown in Figures 2–4.
FIGURE 2.
Distribution of age by trial arm. (a) Placebo and (b) IVIg.
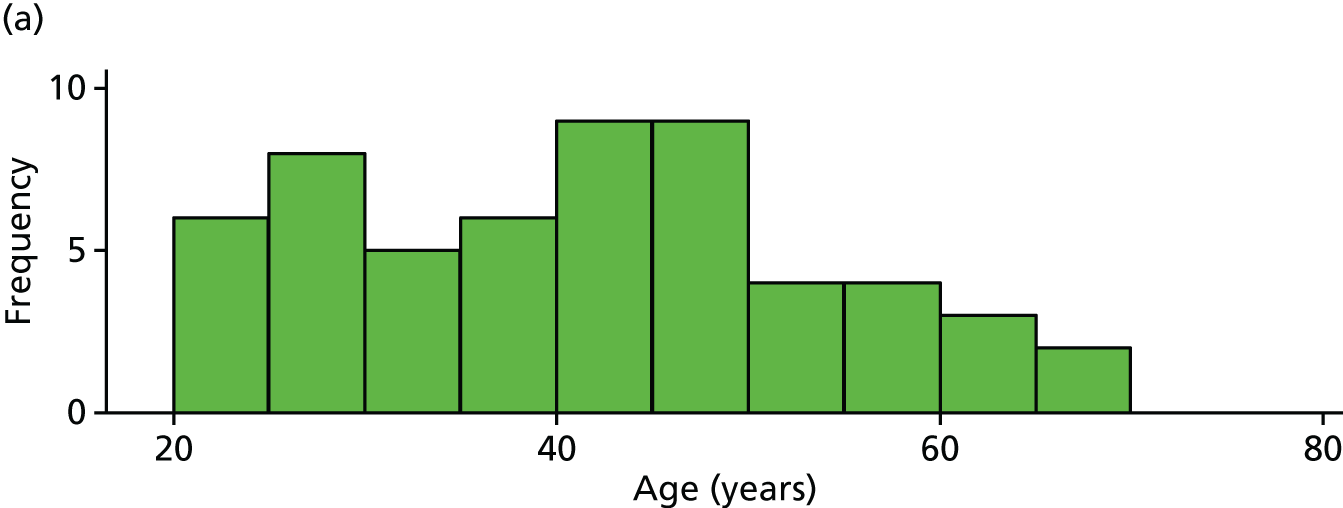
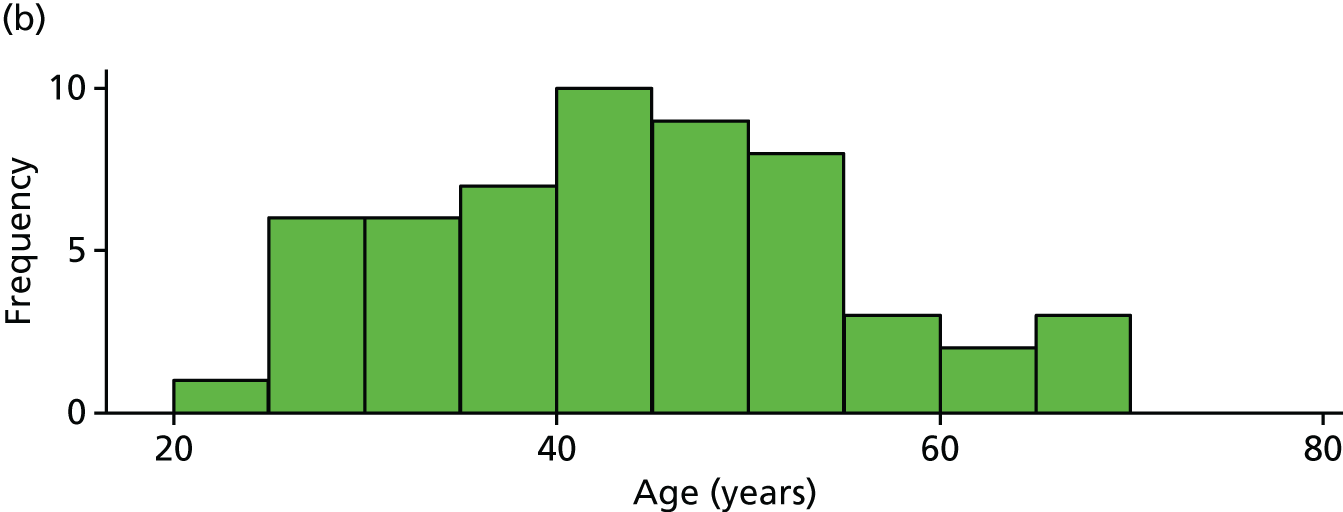
FIGURE 3.
Distribution of disease duration by trial arm. (a) Placebo and (b) IVIg.
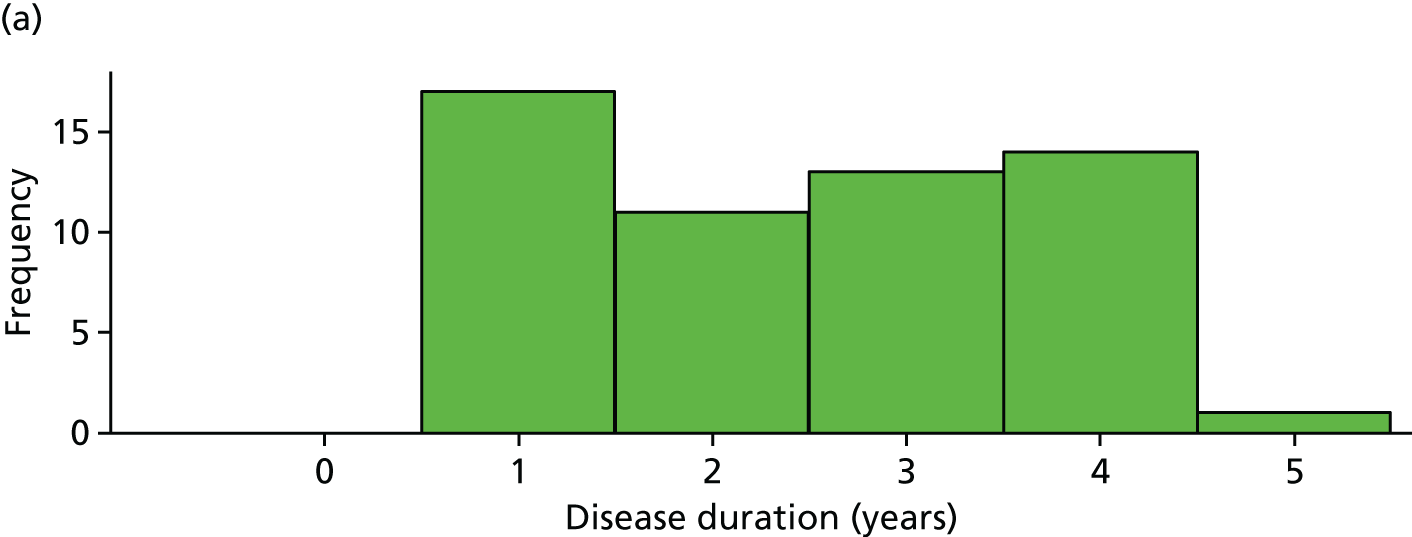
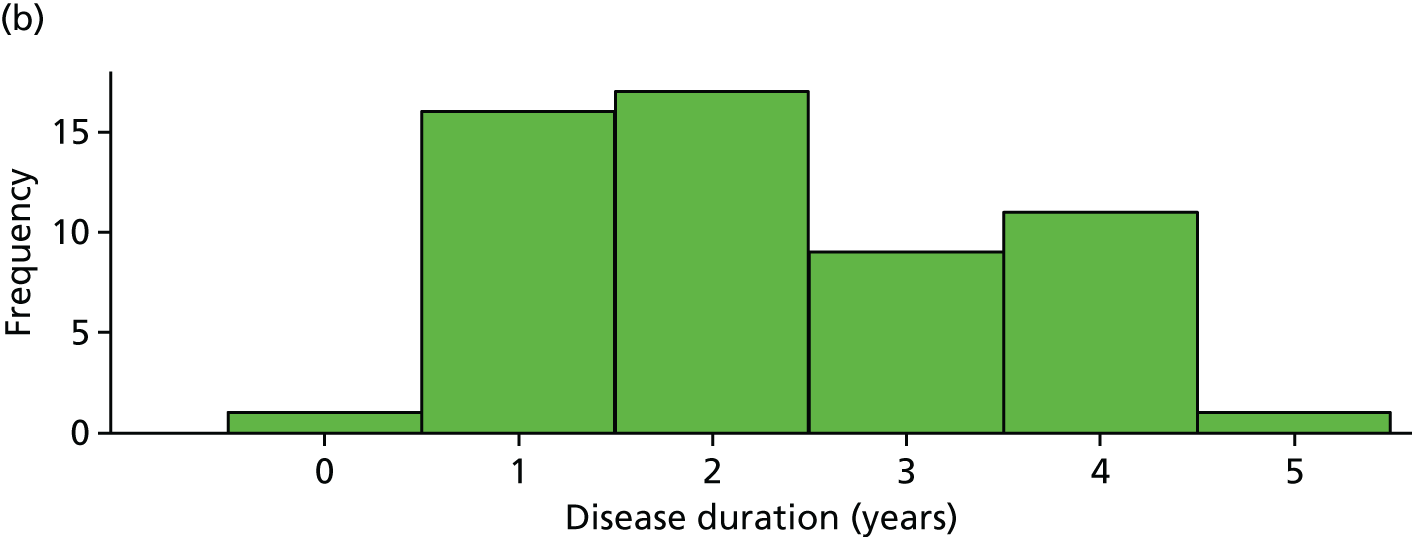
FIGURE 4.
Distribution of average baseline pain by trial arm. (a) Placebo and (b) IVIg.
Time to infusion
Time to first infusion is shown in Table 5 for all randomised participants (n = 111). There were seven early and three late infusions. Two participants did not receive their first infusion as detailed earlier.
| Trial arm | ||
|---|---|---|
| Placebo | IVIg | |
| Time to first infusion (days) | ||
| 0 | 6 | 1 |
| 1 | 48 | 51 |
| 4 | 1 | – |
| 7 | – | 1 |
| 8 | – | 1 |
| (No infusion) | 1 | 1 |
| Total | 56 | 55 |
The protocol states the following: in exceptional circumstances, when a randomised participant does not attend the first infusion on day 1, delay of the first infusion up to 5 working days is acceptable (section 10.3 of protocol). Specifically, one participant did attend their first infusion on day 1 but presented with a high fever and it was not safe to proceed. This participant recovered and had their infusion on day 8 (i.e. a delay of 5 working days).
One participant was randomised 12 days too early owing to a site administrative error. The TSC was consulted and it was agreed that their randomisation date could be ‘corrected’.
Table 6 shows the time to second infusion for all randomised participants (n = 111). Most infusions were administered between days 21 and 23, although there were four infusions later than this. Eleven participants withdrew from treatment medication and did not receive a second infusion.
| Trial arm | ||
|---|---|---|
| Placebo | IVIg | |
| Time to second infusion (days) | ||
| 21 | 6 | 5 |
| 22 | 40 | 33 |
| 23 | 4 | 8 |
| 24 | – | 1 |
| 26 | 1 | – |
| 29 | – | 1 |
| 36 | 1 | – |
| (No infusion) | 4 | 7 |
| Total | 56 | 55 |
Participant follow-up (blinded phase: days 6–42)
Table 7 shows the number of (non-missing) recorded pain scores for each participant. Only five participants produced no outcome pain score data as detailed earlier. All other participants recorded at least 14 values.
| Trial arm | ||
|---|---|---|
| Placebo | IVIg | |
| Number of recorded pain scores | ||
| 14 | – | 1 |
| 15 | – | 1 |
| 16 | 2 | 1 |
| 17 | – | 2 |
| 19 | – | 1 |
| 34 | – | 1 |
| 35 | 1 | 1 |
| 36 | 9 | 5 |
| 37 | 41 | 40 |
| (None) | 3 | 2 |
| Total | 56 | 55 |
Primary analysis (n = 103)
Average pain scores (over days 6–42) for each participant, by trial arm, are shown in Figure 5 for the 103 participants included in the primary analysis. It is clear that the pain scores are very similar for each group.
FIGURE 5.
Average pain score for each participant by trial arm. (a) Placebo and (b) IVIg.
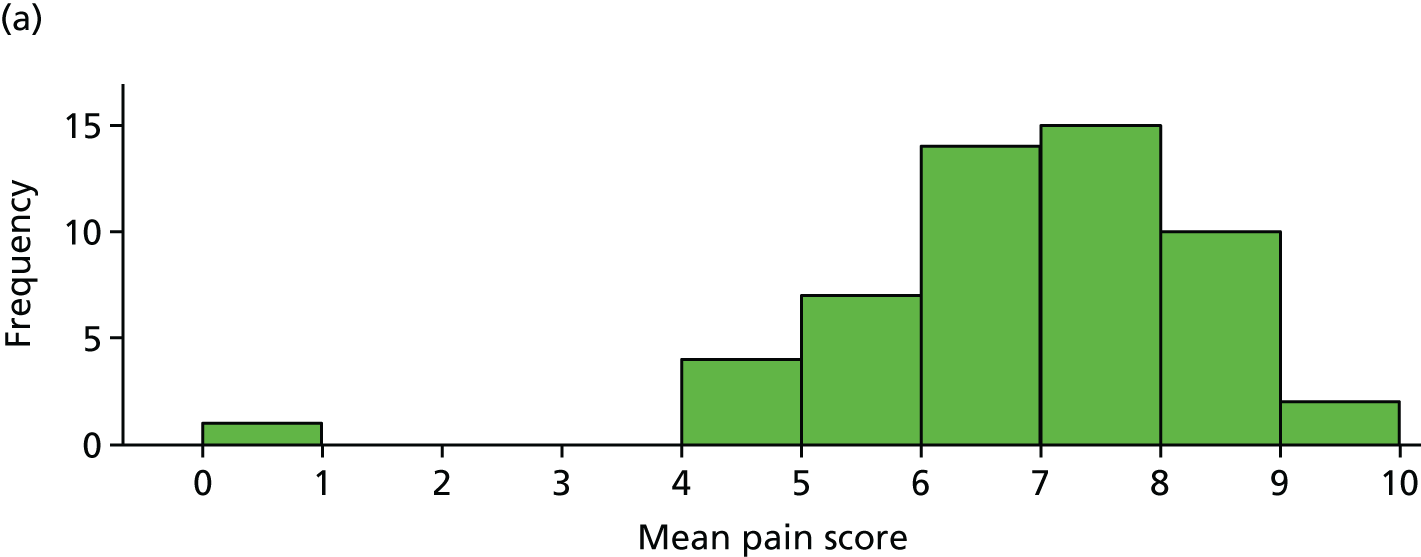
Fitting the primary analysis mixed model produced the treatment effect estimate in Table 8. The average pain score in the IVIg group was 0.27 units (95% CI –0.24 to 0.80 units) higher than in the placebo group. The 95% CI includes 0 and the corresponding p-value is relatively large (p = 0.30). Therefore, there is no significant evidence of a treatment effect at the 5% level. The full model results and residual plots are shown in Table 9 and Figure 6.
| Variable | Coefficient (95% CI) | p-value |
|---|---|---|
| Average pain (IVIg – placebo) | 0.27 (–0.25 to 0.80) | 0.30 |
| Variable | Coefficient (95% CI) | p-value |
|---|---|---|
| Average pain (IVIg – placebo) | 0.27 (–0.25 to 0.80) | 0.30 |
| Site (Cambridge) | 0.06 (–1.22 to 1.34) | 0.88 |
| Site (Glasgow) | 0.14 (–0.94 to 1.23) | |
| Site (Leicester) | 0.07 (–1.32 to 1.46) | |
| Site (Liverpool) | –0.38 (–1.38 to 0.63) | |
| Site (London) | 0.03 (–0.99 to 1.05) | |
| Site (Norwich) | –0.24 (–1.63 to 1.15) | |
| Intercept | 6.99 (6.07 to 7.92) | < 0.001 |
| σ u | 1.33 (1.16 to 1.53) | |
| σ 2 | 1.08 (1.05 to 1.10) |
FIGURE 6.
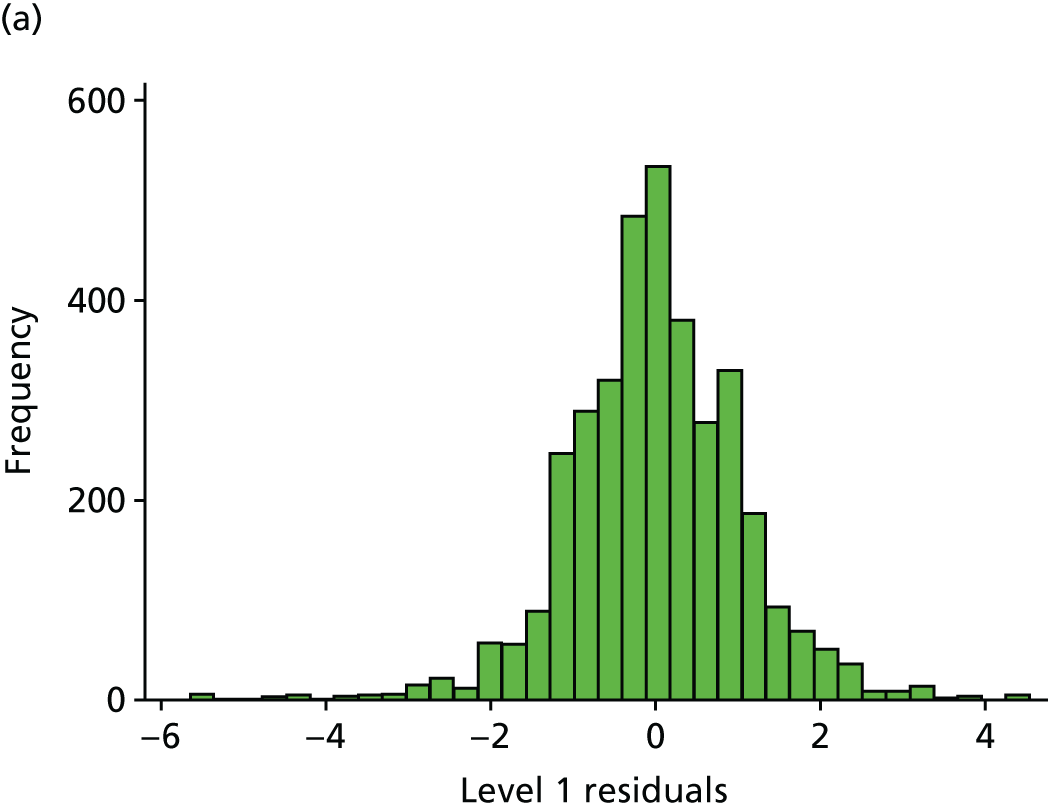
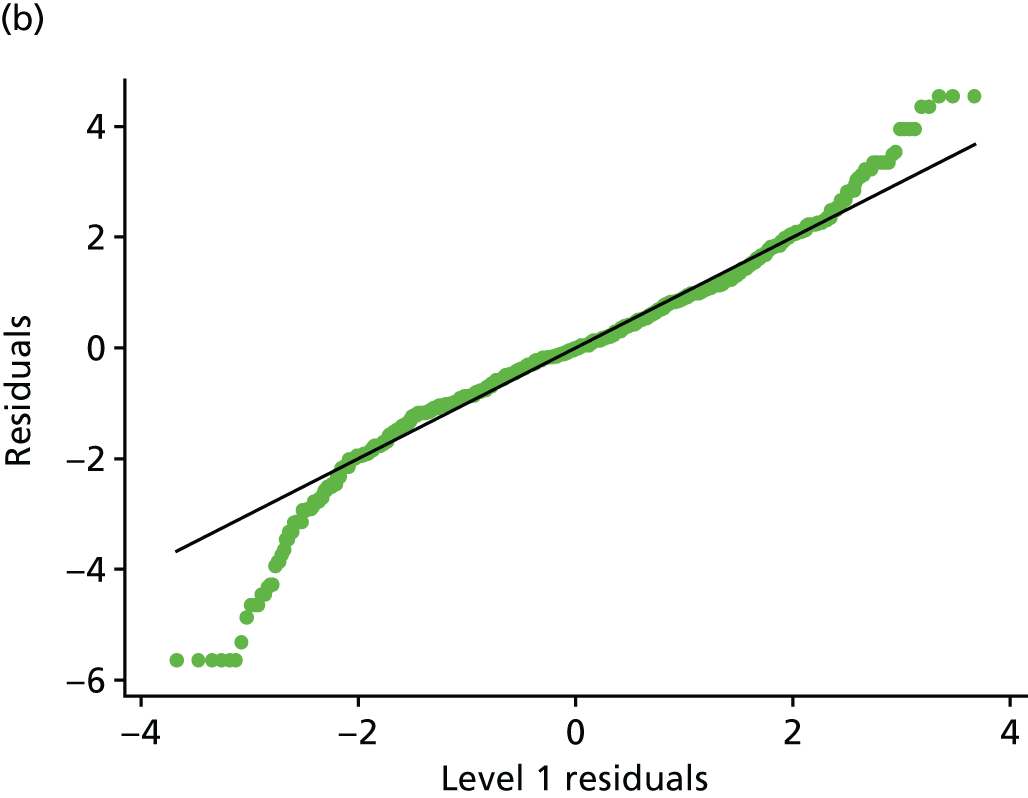
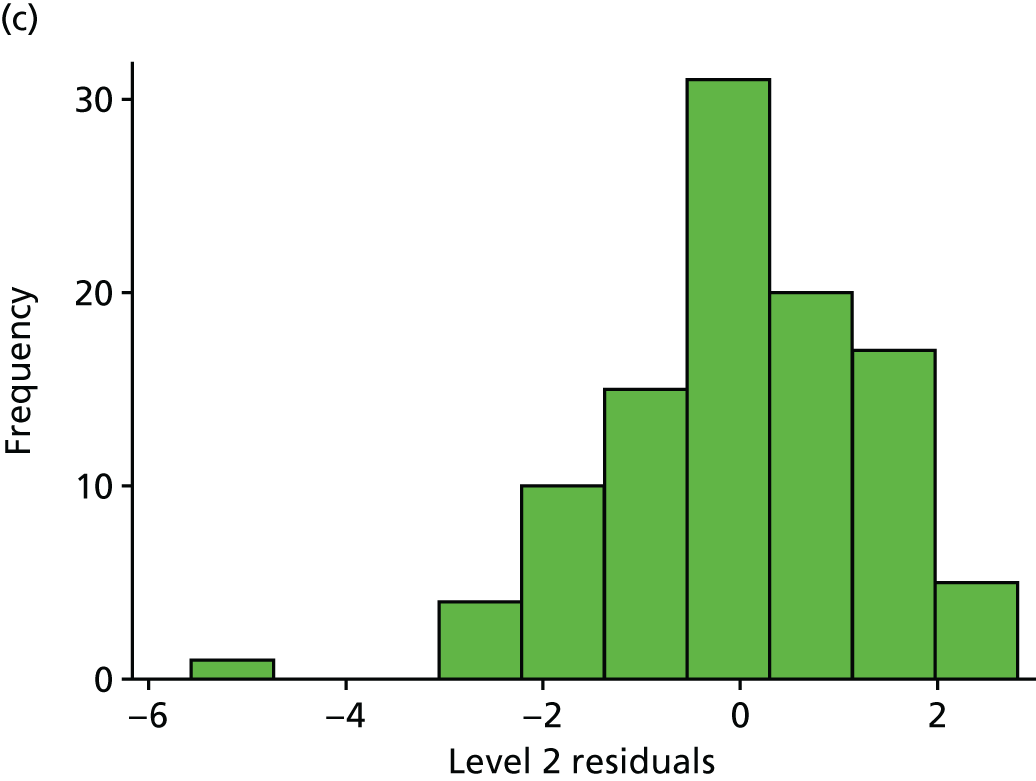
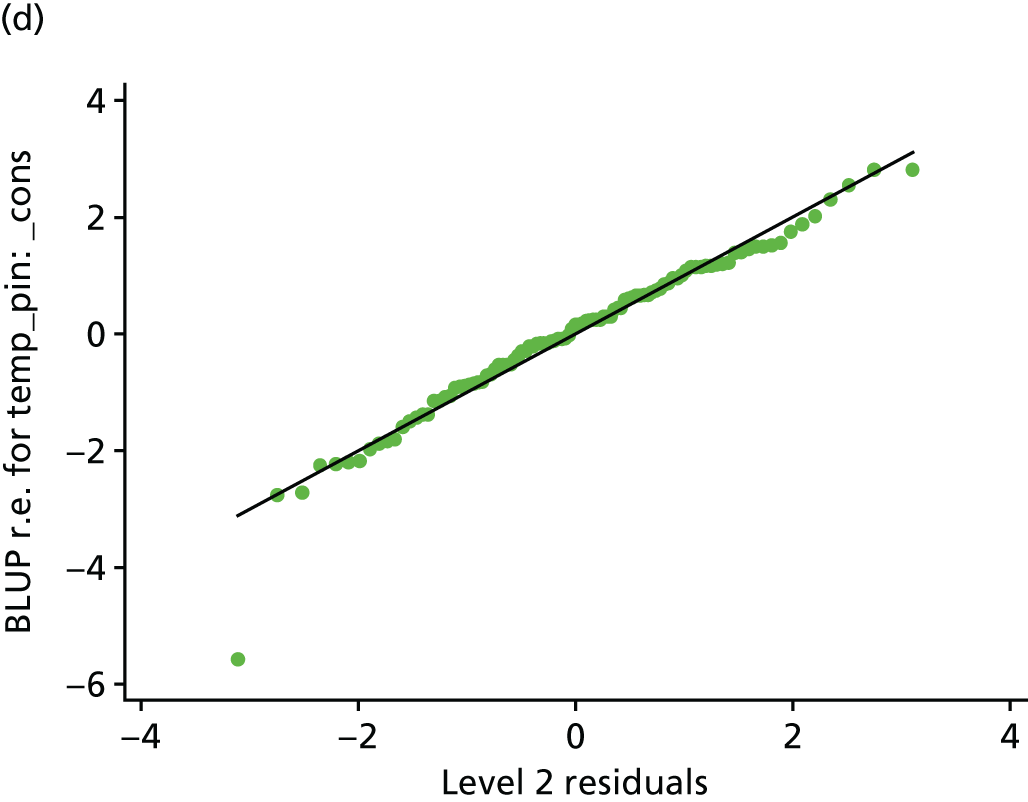
Histogram and normal plots for the level 1 (measurements) and 2 (participant) residuals. (a) Level 1 residual histogram; (b) level 1 residual normal plot; (c) level 2 residual histogram; and (d) level 2 residual normal plot.
As a check, a t-test was used to compare the mean pain scores across trial arms. This produced a treatment effect of 0.28 (p = 0.30), which, as expected, is very similar to that from the primary analysis. The mixed model was also refitted with an analysis of covariance adjustment for average baseline pain. This produced a treatment effect of 0.23 (p = 0.22), which, again, is similar to that of the primary analysis.
One participant recorded very low pain scores (mean pain = 0.9 from 37 measurements). Omitting this participant from the primary analysis reduces the placebo treatment effect by one-third (to 0.17).
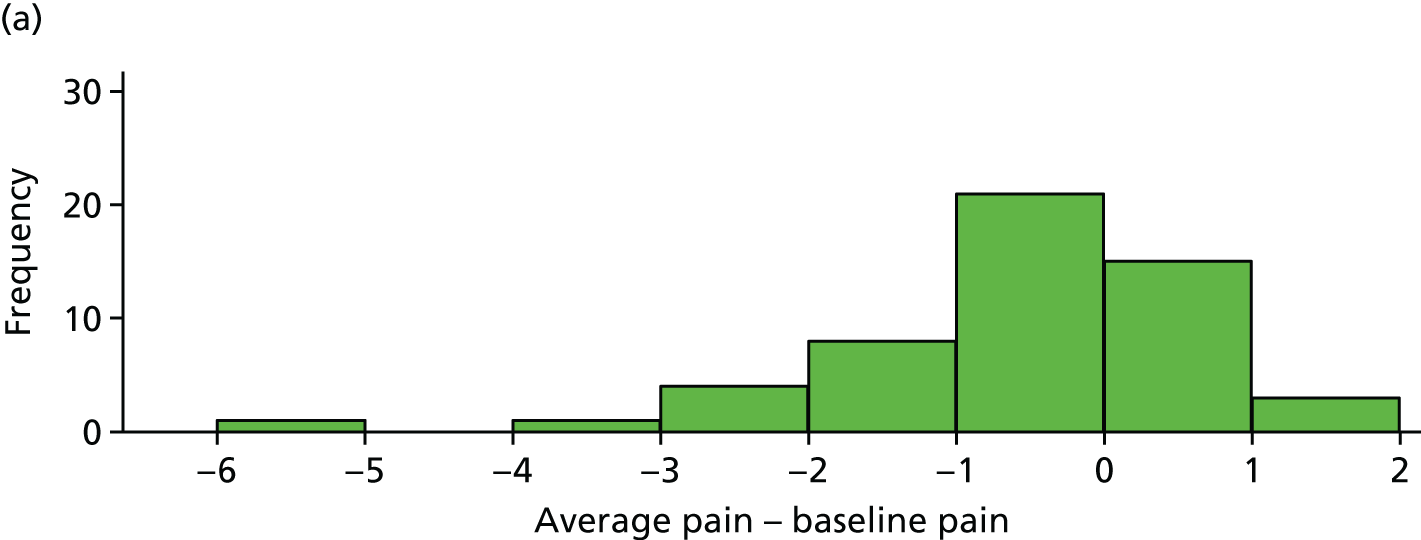
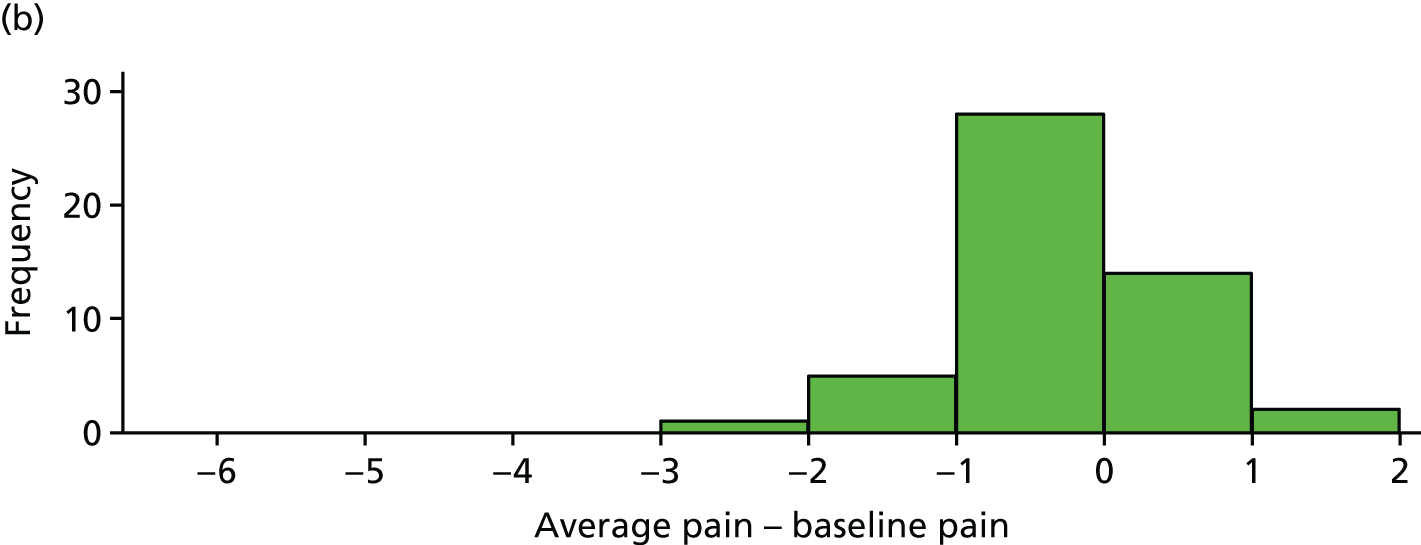
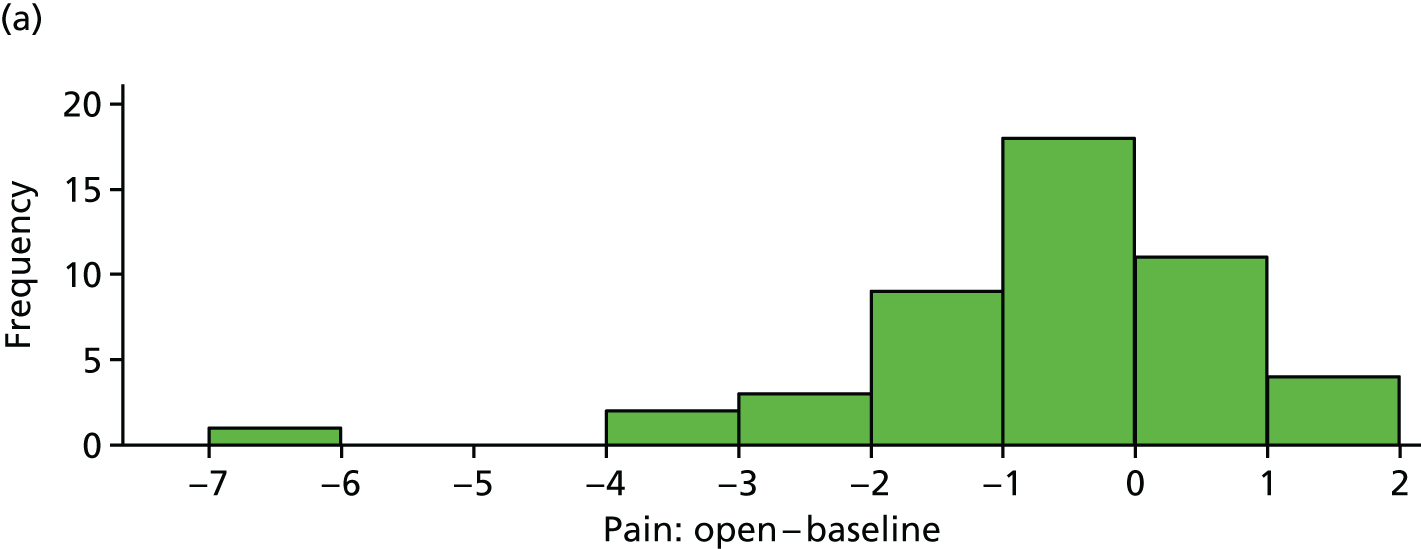
Absolute pain reduction
The difference between average pain score and average baseline pain score is plotted for each participant in Figure 7. These differences are also summarised in Table 10.
FIGURE 7.
Difference between average pain and average baseline pain for each participant. (a) Placebo and (b) IVIg.
| Variable | Trial arm | |
|---|---|---|
| Placebo | IVIg | |
| Mean (SD) difference | –0.55 (1.17) | –0.32 (0.74) |
Percentage pain reduction
Average pain scores were also plotted against average baseline pain scores. The outlying participant (average pain = 0.9) has been removed from Figure 8 for clarity.
FIGURE 8.
Average pain vs. average baseline pain for each participant. (a) Placebo and (b) IVIg.
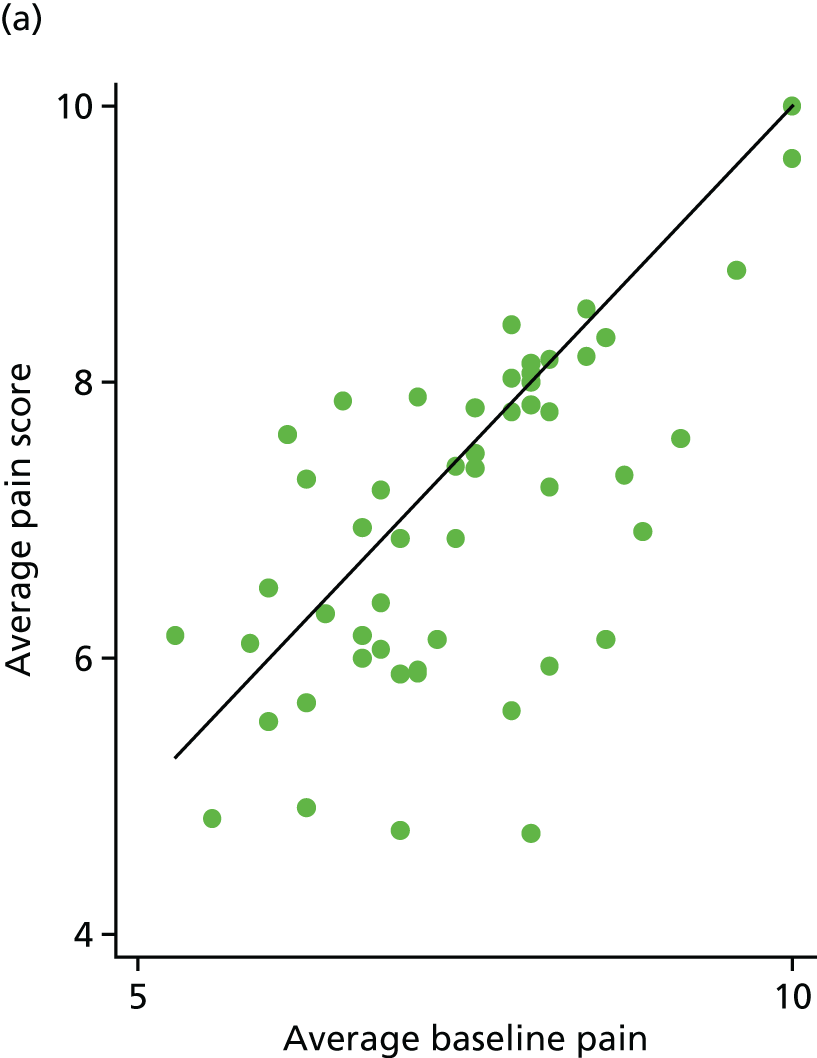
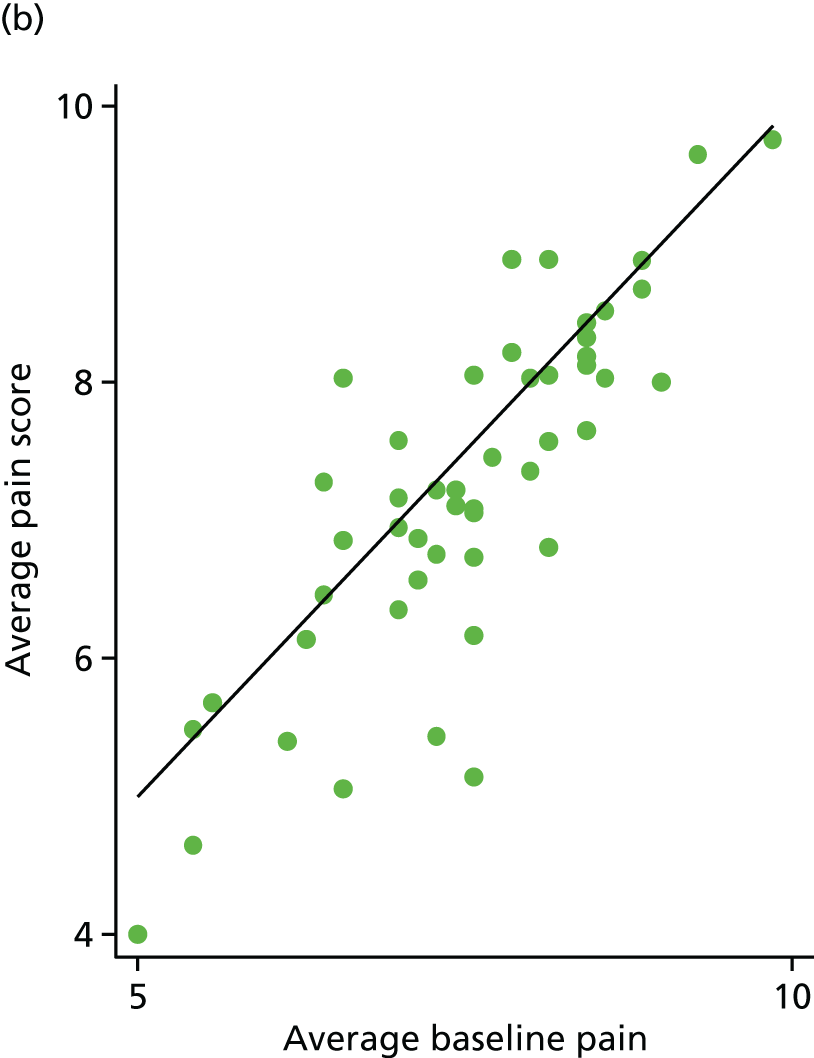
Sixty-nine (67%) participants had lower pain scores following treatment. This was very similar in both arms [35/53 (66%) for the placebo arm and 34/50 (68%) for the IVIg arm]. Four participants achieved 30% pain reduction: three in the placebo arm and one in the IVIg arm. Only one participant in the placebo arm achieved 50% pain reduction.
Sensitivity analyses
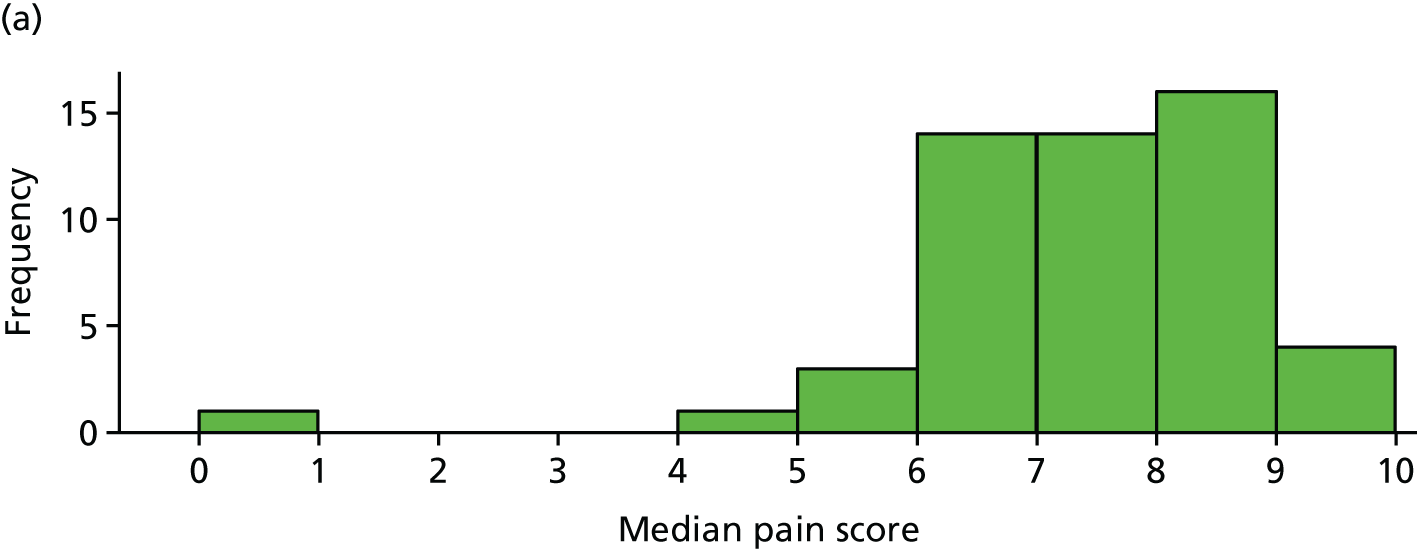
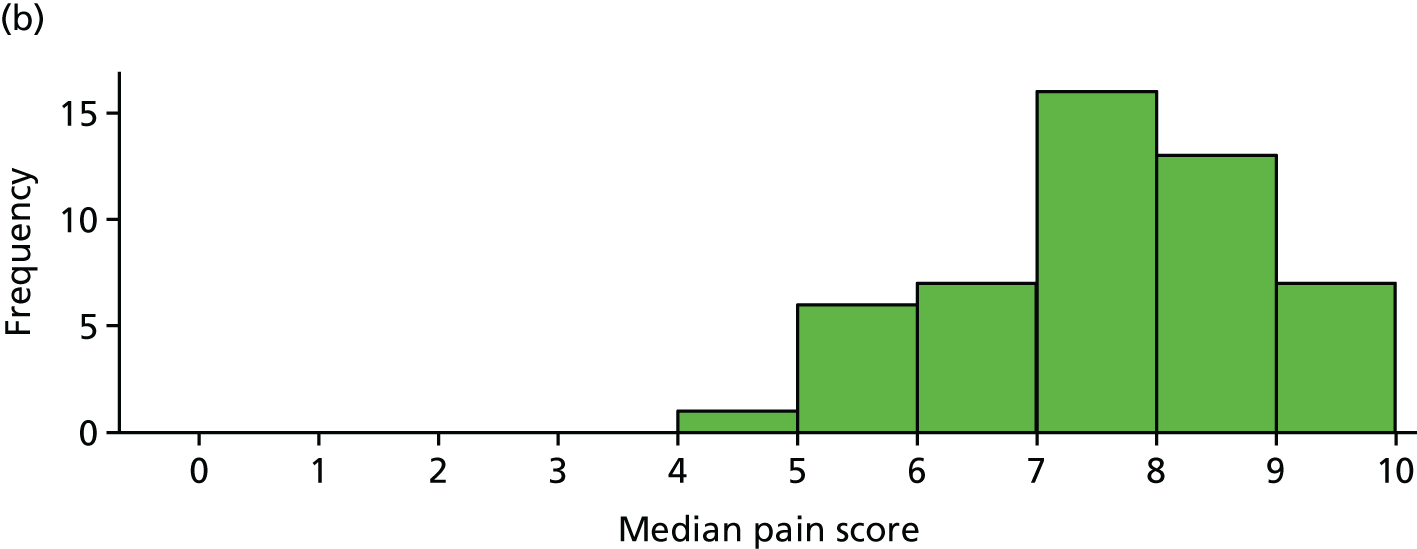
Primary analysis using median pain scores (n = 103)
Median pain scores (over days 6–42) for each participant, by trial arm, are shown in Figure 9 for the 103 participants included the primary analysis. This plot is similar to that for mean pain, which is unsurprising as the means and medians are highly correlated (p = 0.97).
FIGURE 9.
Median pain score for each participant by trial arm. (a) Placebo and (b) IVIg.
A t-test was used to compare these median pain scores across trial arms (with no adjustment for number of values). This produced a treatment effect shown in Table 11.
| Variable | Coefficient (95% CI) | p-value |
|---|---|---|
| Average pain (IVIg – placebo) | 0.23 (–0.35 to 0.80) | 0.44 |
Per-protocol analysis (n = 100)
The per-protocol analysis excludes three participants who received their first infusion between days 4 and 8, although it does include the seven participants who received this infusion on day 0. The IVIg effect is slightly larger but the conclusions are unchanged (Table 12).
| Variable | Coefficient (95% CI) | p-value |
|---|---|---|
| Average pain (IVIg – placebo) | 0.32 (–0.21 to 0.84) | 0.24 |
All participants (n = 111)
A further analysis was performed including all randomised participants. However, the five participants that did not produce any outcome pain data are essentially excluded from the model. The IVIg effect is very similar to that from the primary analysis and the conclusions are again unchanged (Table 13).
| Variable | Coefficient (95% CI) | p-value |
|---|---|---|
| Average pain (IVIg – placebo) | 0.23 (–0.28 to 0.75) | 0.37 |
Exploratory analyses
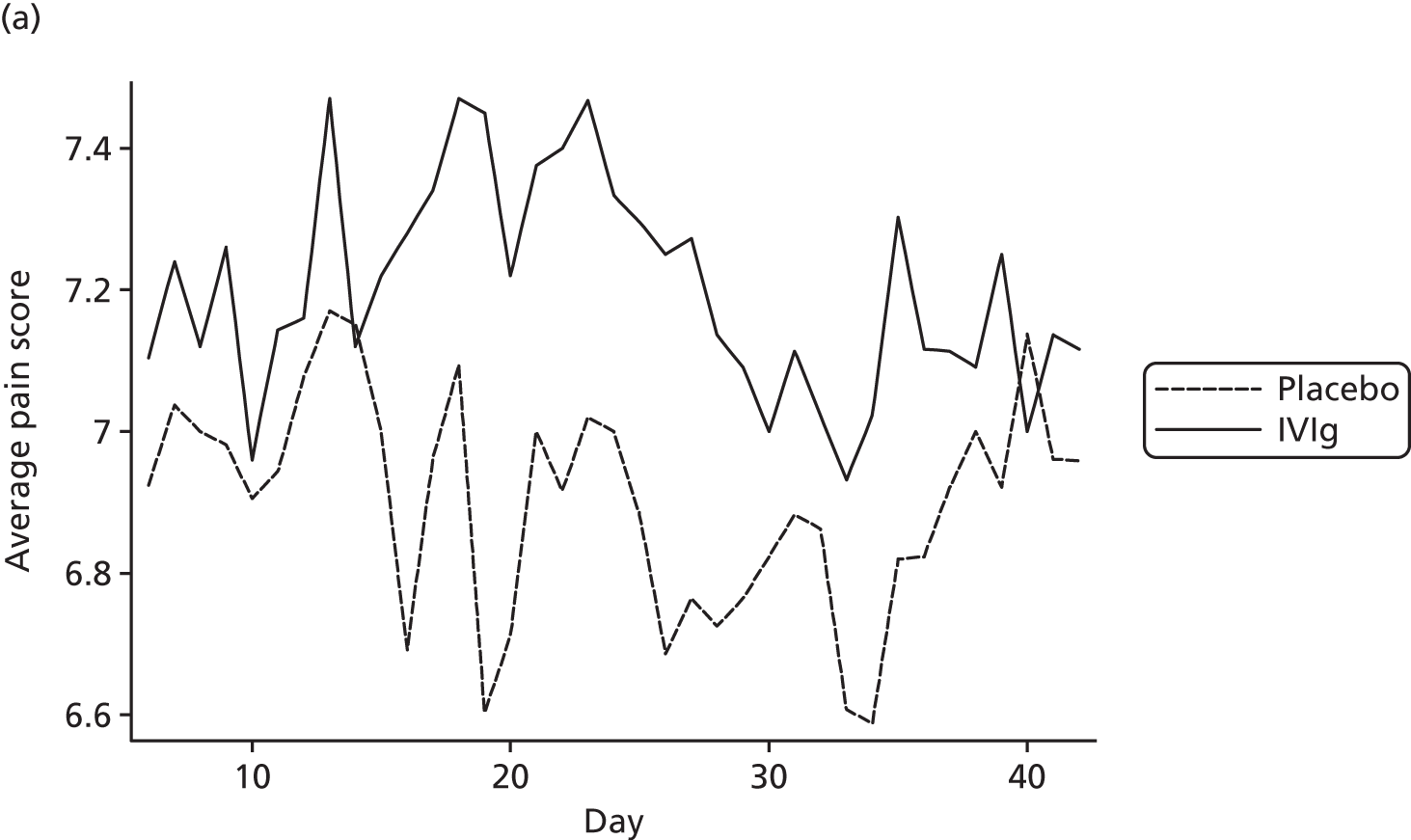
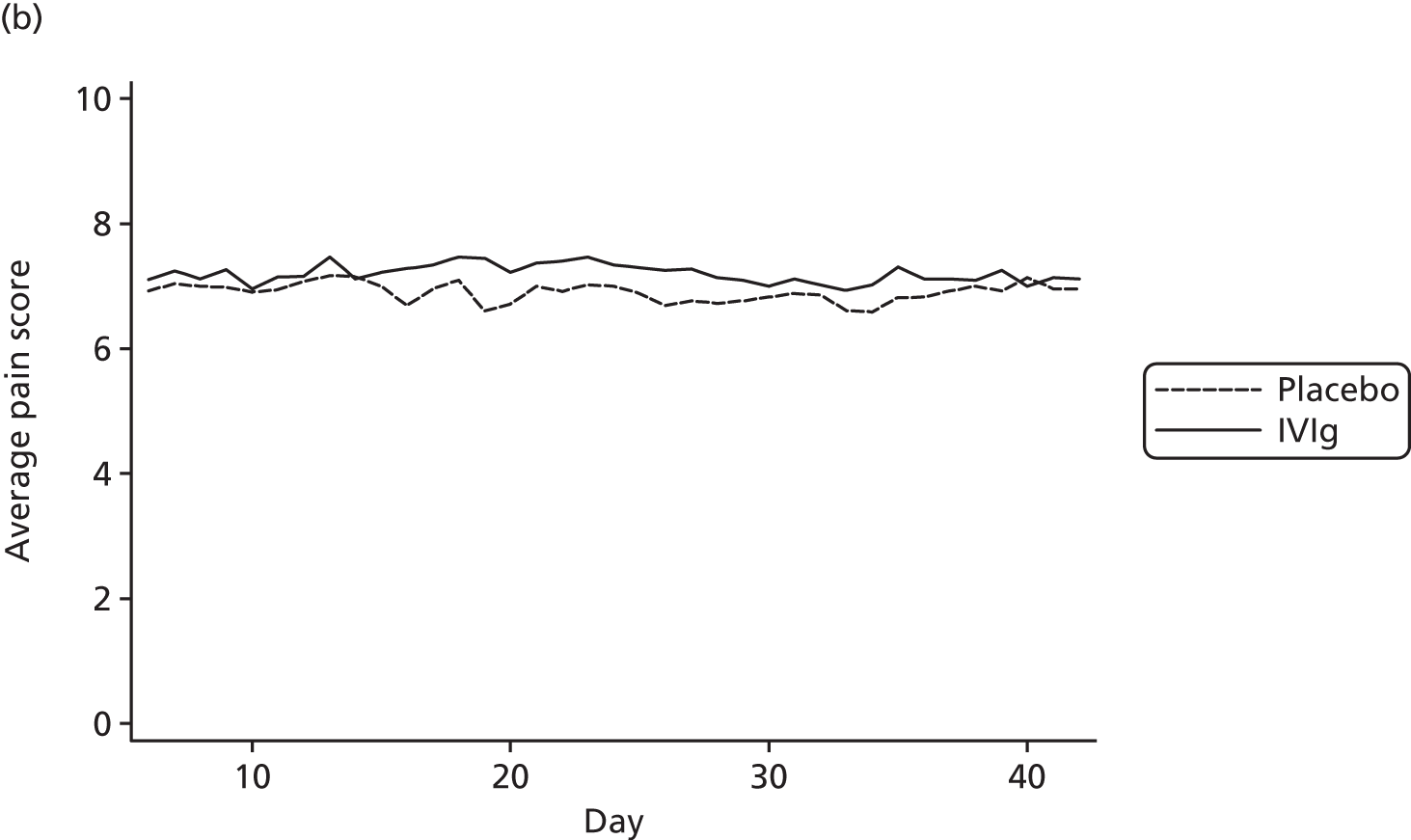
Pain scores over time
The average pain scores for each day, by trial arm, are shown in Figure 10. This indicates that there is clear separation between the pain scores in the trial arms on most days, although the difference is relatively small. In addition, it can be seen that average pain score in both groups is fairly constant over time.
FIGURE 10.
Average pain for each day by trial arm (different y-axes) with (a) showing truncated pain score at 6.6.
Treatment effect by site (n = 103)
The primary analysis was repeated with a separate treatment effect for each site. The treatment effects for each site are shown in Table 14. We note that pain scores are lower for the placebo arm in all centres except Glasgow; however, there is no statistical evidence for a difference in treatment effects (p = 0.68), but we note that this study was not powered for such a comparison.
| Average pain (IVIg – placebo) | Number of participants | Coefficient (95% CI) |
|---|---|---|
| Bath | 9 | 0.90 (–0.83 to 2.64) |
| Cambridge | 8 | 1.10 (–0.73 to 2.93) |
| Glasgow | 17 | –0.61 (–1.87 to 0.65) |
| Leicester | 6 | 0.86 (–1.25 to 2.97) |
| Liverpool | 30 | 0.03 (–0.92 to 0.98) |
| London | 27 | 0.41 (–0.58 to 1.41) |
| Norwich | 6 | 0.75 (–1.36 to 2.87) |
Treatment effect by complex regional pain syndrome type (n = 100)
The primary analysis was repeated with a separate treatment effect for CRPS type I and II. Three participants with ‘undecided’ CRPS type were omitted from this analysis. The treatment effects for each CRPS type are shown in Table 15. We note that pain scores are lower for IVIg for participants with CRPS type II. In addition, there is weak evidence for a difference in treatment effects (p = 0.02) between the two CRPS types.
| Variable | Number of participants | Coefficient (95% CI) |
|---|---|---|
| CRPS type I | 88 | 0.44 (–0.11 to 0.98) |
| CRPS type II | 12 | –1.51 (–3.00 to –0.03) |
Treatment effect by number of infusions (n = 103)
The primary analysis was repeated with a separate treatment effect for patients who either did or did not receive their second (blind) infusion. The treatment effects for both groups are shown in Table 16 but there is no statistical evidence for a difference in these effects (p = 0.29).
| Number of infusions | Number of participants | Coefficient (95% CI) |
|---|---|---|
| 1. First only | 6 | –1.38 (–4.29 to 1.53) |
| 2. Both blind | 97 | 0.32 (–0.21 to 0.86) |
This analysis was extended to include whether or not participants received open-phase infusions. Again, there is no statistical evidence for a difference in treatment effects (p = 0.74) (Table 17).
| Number of infusions | Number of participants | Coefficient (95% CI) |
|---|---|---|
| 1. First blind only | 6 | –1.38 (–4.25 to 1.49) |
| 2. Both blind | 8 | 0.34 (–1.60 to 2.28) |
| 3. Both blind and first opena | 15 | 0.30 (–1.14 to 1.74) |
| 4. All | 74 | 0.29 (–0.31 to 0.90) |
Treatment effect by disease duration (n = 103)
The primary analysis was repeated with a separate treatment effect for each disease duration (1–5 years). There is little evidence to suggest that treatment effect depends on disease duration (p = 0.33).
This analysis was repeated, treating disease duration as a continuous variable (Table 18). The interaction term (treatment × duration) was negative (–0.32, 95% CI –0.77 to 0.13), suggesting that treatment might be effective for those of longer disease duration. However, this term was not statistically significant (p = 0.16).
| Disease duration (years) | Number of patients | Coefficient (95% CI) |
|---|---|---|
| 1 | 30 | 0.90 (–0.04 to 1.85) |
| 2 | 26 | 0.47 (–0.58 to 1.52) |
| 3 | 22 | –0.56 (–1.66 to 0.54) |
| 4 | 23 | –0.03 (–1.12 to 1.06) |
| 5 | 2 | 1.12 (–2.45 to 4.70) |
Treatment effect by psychiatric medical history (n = 103)
The primary analysis was repeated with a separate treatment effect for psychiatric medical history (yes/no). There is no evidence for a difference in treatment effects (p = 0.25).
Treatment effect by allergy status (n = 103)
The primary analysis was repeated with a separate treatment effect for allergy status (yes/no). There is no evidence for a difference in treatment effects (p = 0.49).
Treatment effect by low baseline IgG plasma level (n = 103)
The primary analysis was repeated with a separate treatment effect for low IgG plasma level (< 10/≥ 10 mg/dl). There is no evidence for a difference in treatment effects (p = 0.19).
Treatment effect adjusted for disease duration (n = 103)
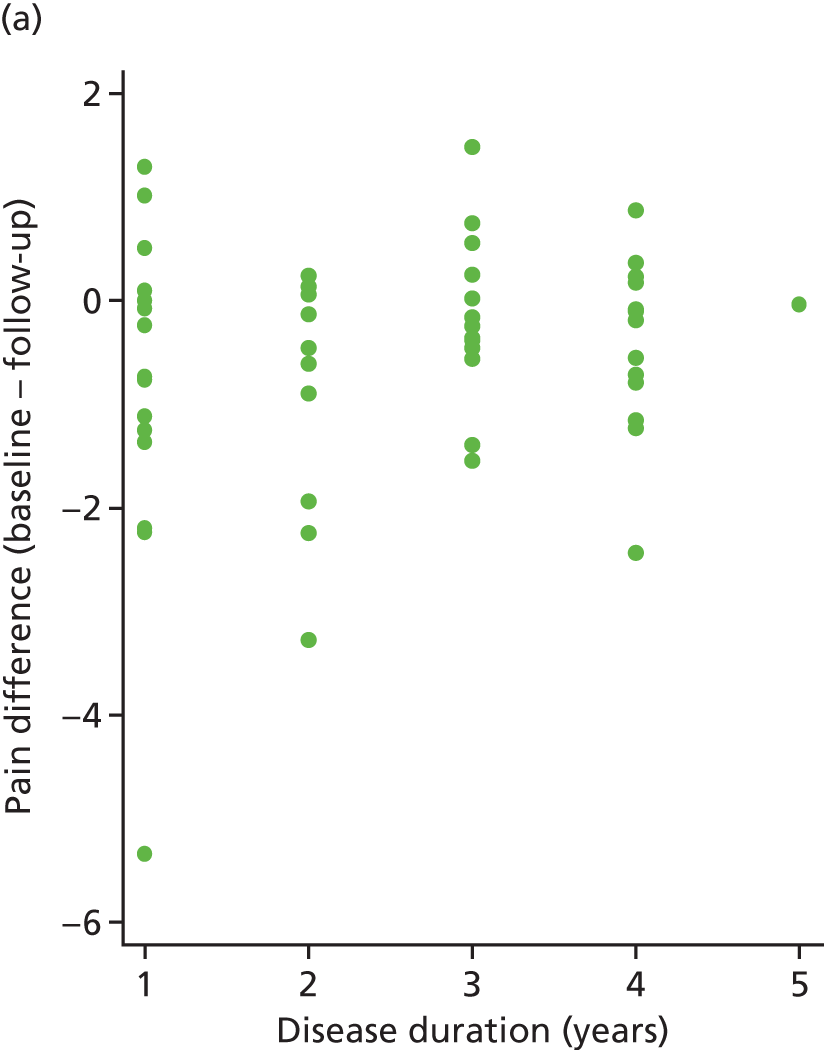
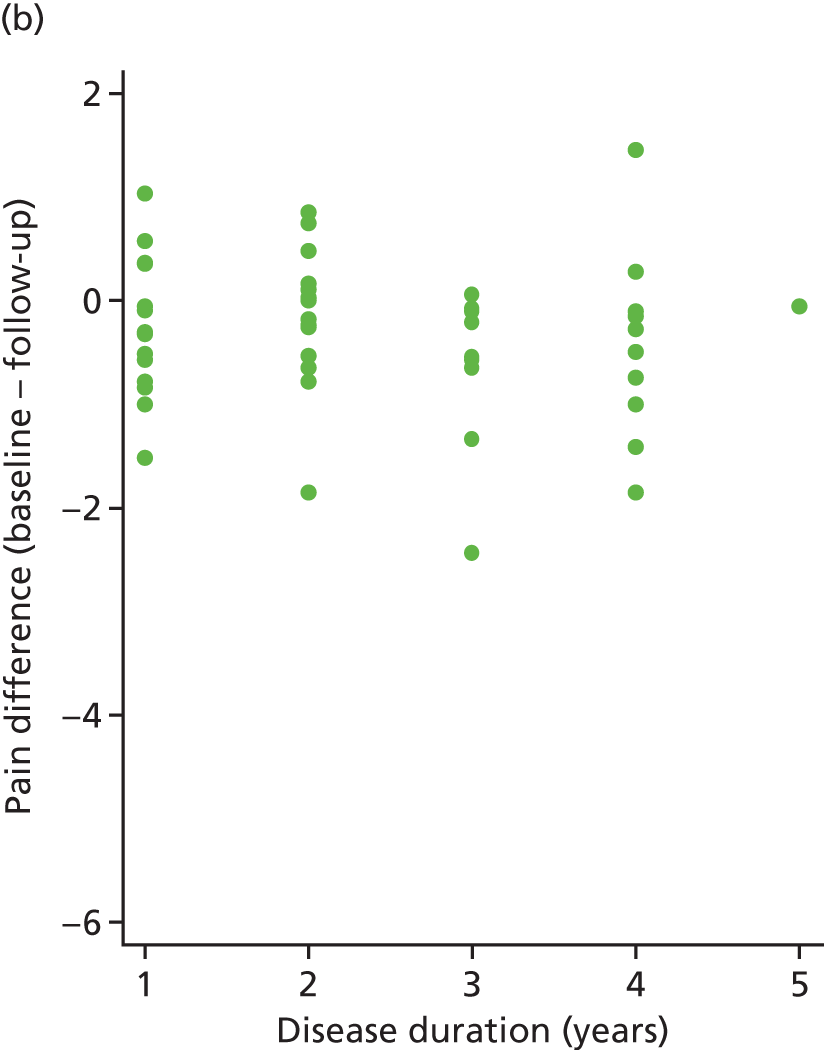
The primary analysis model was refitted after adjusting for disease duration. The treatment effect changed little (0.27, 95% CI –0.26 to 0.79).
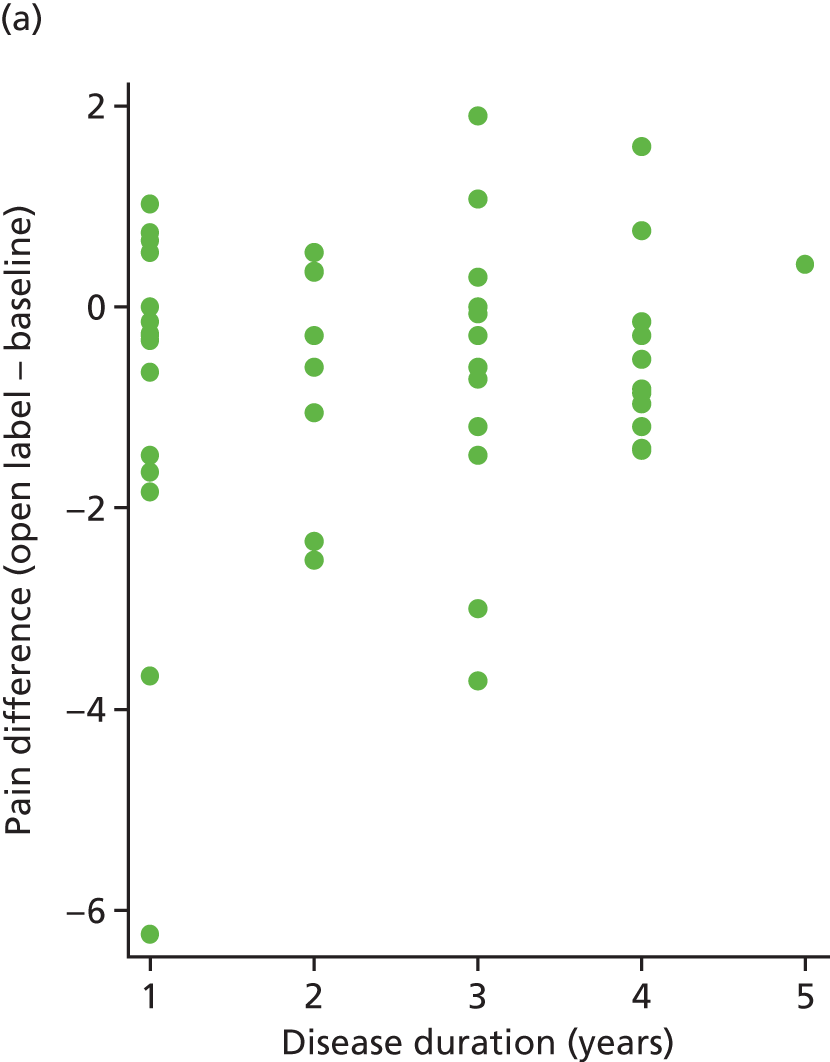
The correlation between pain difference (blinded phase – baseline) and disease duration was calculated for both arms. These values were 0.18 (placebo) and –0.12 (IVIg). These relationships can be seen in Figure 11.
FIGURE 11.
Pain difference (baseline – follow-up) vs. disease duration. (a) Placebo and (b) IVIg.
Open-label results
Withdrawals from study medication (open phase)
Out of the 111 randomised participants, 12 withdrew their consent to use the study medication during the blinded phase. A further 13 participants withdrew their consent to use the study medication during the open phase. All 13 participants received their first open infusion but none received their second. Further details regarding these participants can be found in Table 19.
| Day | Treatment | Reason for withdrawal | Open infusion 1? | Open infusion 2? |
|---|---|---|---|---|
| 44 | Placebo | AE (participant withdrew) | Yes | No |
| 50 | Placebo | AE (participant withdrew) | Yes | No |
| 51 | Placebo | AE (participant withdrew) | Yes | No |
| 59 | Placebo | AE (participant withdrew) | Yes | No |
| 62 | Placebo | AE (participant withdrew) | Yes | No |
| 62 | IVIg | AE (team withdrew) | Yes | No |
| 63 | Placebo | AE (participant withdrew) | Yes | No |
| 64 | IVIg | AE (participant withdrew) | Yes | No |
| 64 | IVIg | AE (participant withdrew) | Yes | No |
| 64 | Placebo | AE (participant withdrew) | Yes | No |
| 64 | Placebo | AE (team withdrew) | Yes | No |
| 69 | Placebo | Other (circumstance change) | Yes | No |
| 71 | IVIg | AE (team withdrew) | Yes | No |
Open-label infusions
Out of the 99 participants who did not withdraw from study medication during the blinded phase, 90 received their first open infusion (Table 20). A total of 77 participants received their second open infusion, including one participant who did not receive their first open infusion. This was due to the participant having to travel abroad owing to a family emergency. However, the participant was eager to have the open-label infusions. The chief investigator was consulted and it was agreed that, on compassionate grounds, the participant would be able to attend for the second open-label infusion at the set time point.
| Trial arm | |||
|---|---|---|---|
| Placebo (n = 56) | IVIg (n = 55) | ||
| Did participant receive first open infusion? | Yes | 49 (87.5%) | 41 (74.5%) |
| Did participant receive second open infusion? | Yes | 39 (69.6%) | 38 (69.1%) |
Two of the ‘ineligible’ participants received both infusions; these participants are excluded from the following analyses, which use the ‘primary analysis’ sample.
Average pain score by day
Average pain scores for each day, by trial arm, are shown in Figure 12 based on all available data at each time point for days 1 to 84. Note that the number of participants decreases for the open-label period (Table 21).
FIGURE 12.
Average pain for each day by trial arm (different y-axes) with (a) showing truncated at 6.
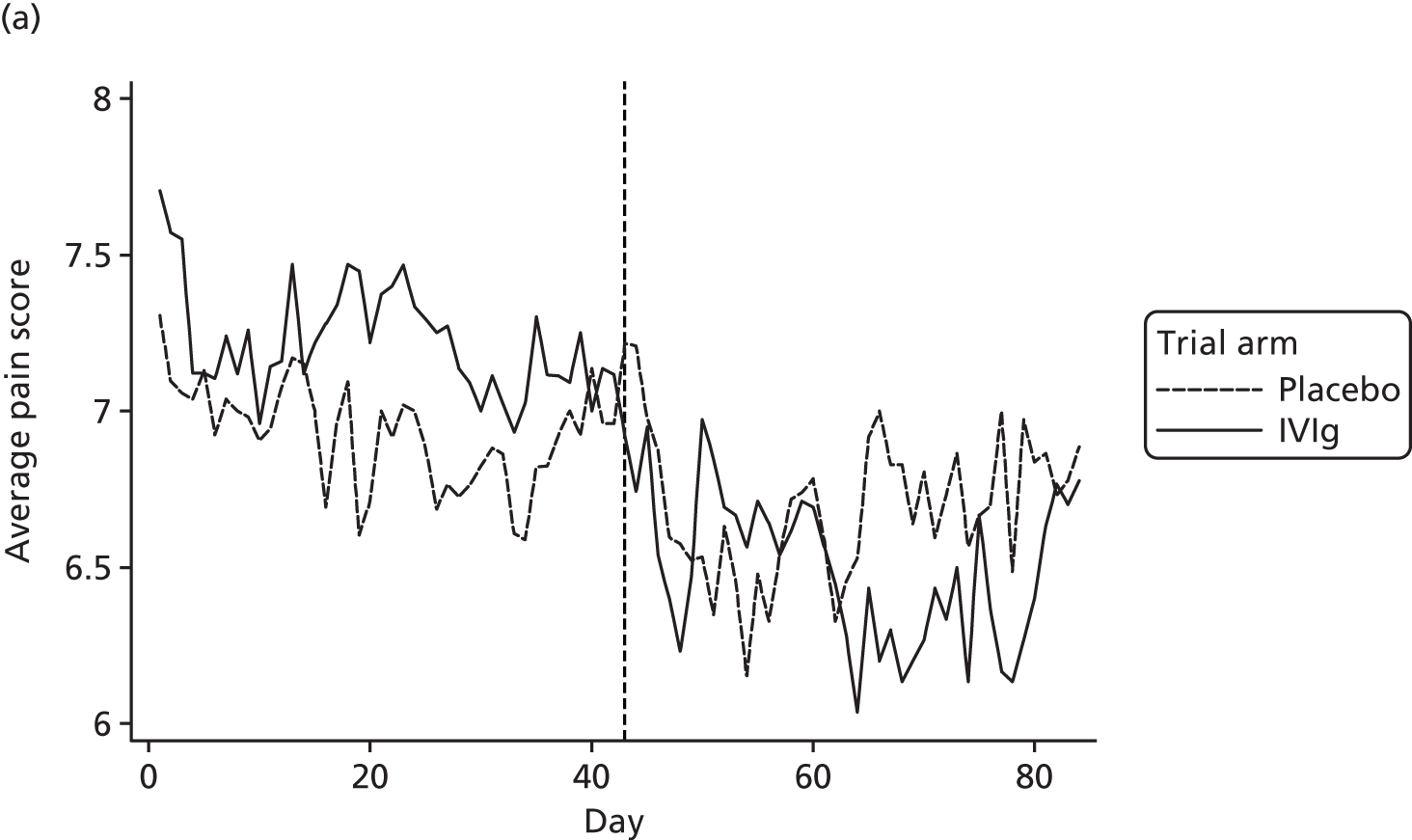
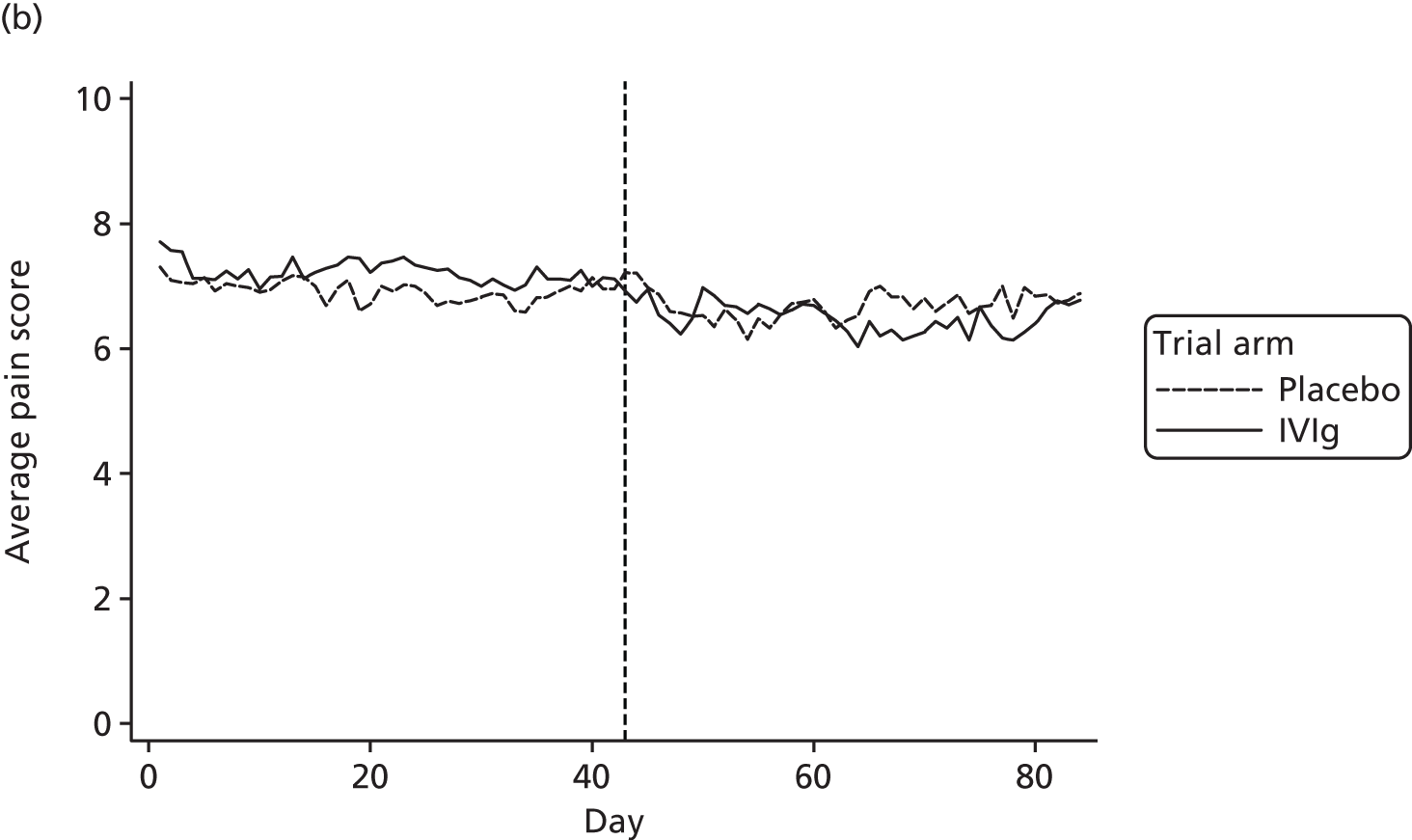
| Study group | Baseline | Day | |||
|---|---|---|---|---|---|
| 1 | 22 | 43 | 64 | ||
| Placebo | 7.5/8 (n = 52) | 7.3/7 (n = 49) | 6.9/7.5 (n = 48) | 7.2/8 (n = 46) | 6.5/7 (n = 34) |
| IVIg | 7.5/8 (n = 50) | 7.7/8 (n = 44) | 7.4/8 (n = 45) | 6.8/7 (n = 39) | 6.0/7 (n = 28) |
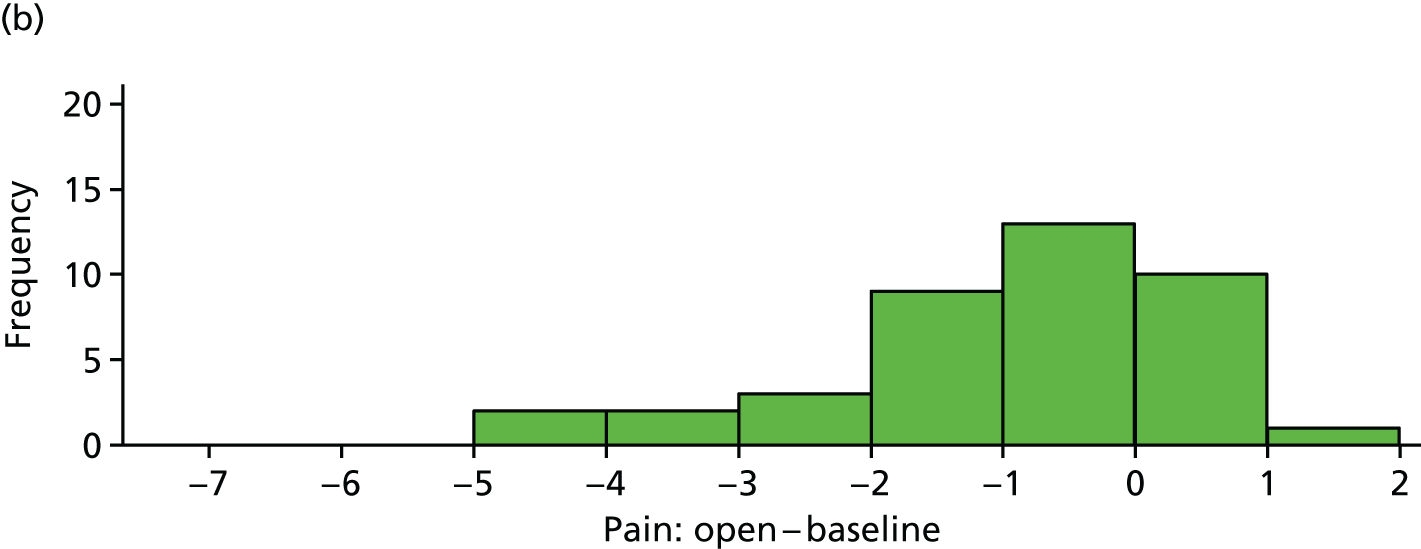
Reduction in pain score: open versus blinded
The difference between (average) open-label pain score and blinded pain score is plotted in Figure 13 for each participant. These differences are also summarised in Table 22.
FIGURE 13.
Difference between open-label pain and blinded pain for each participant. (a) Placebo and (b) IVIg.
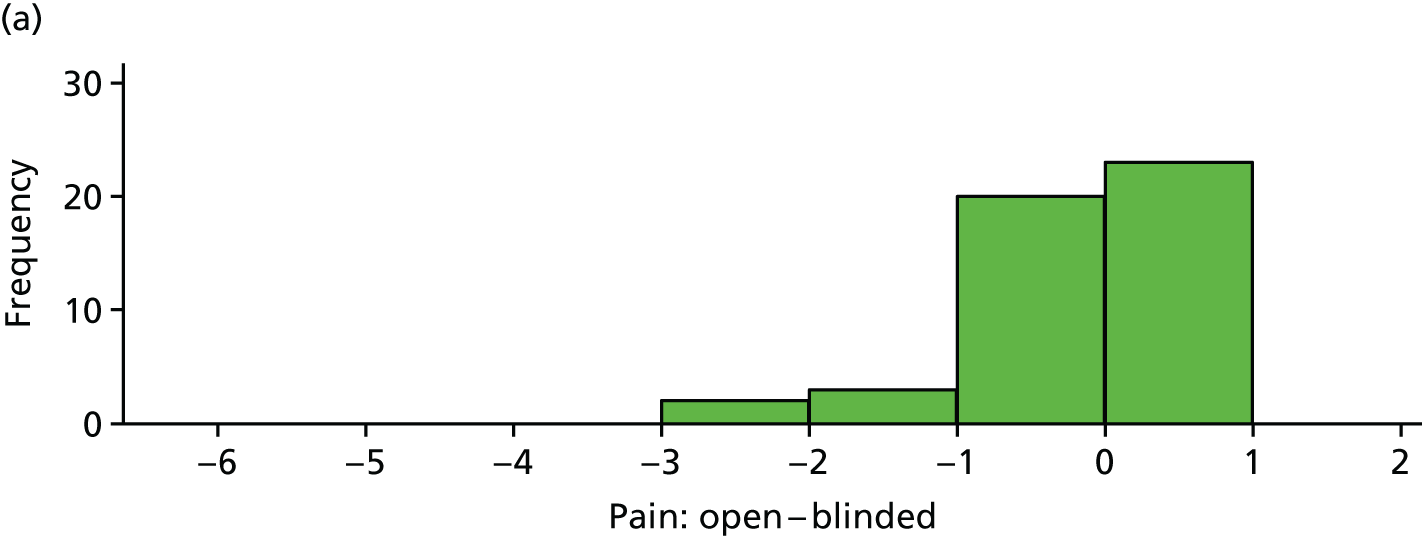
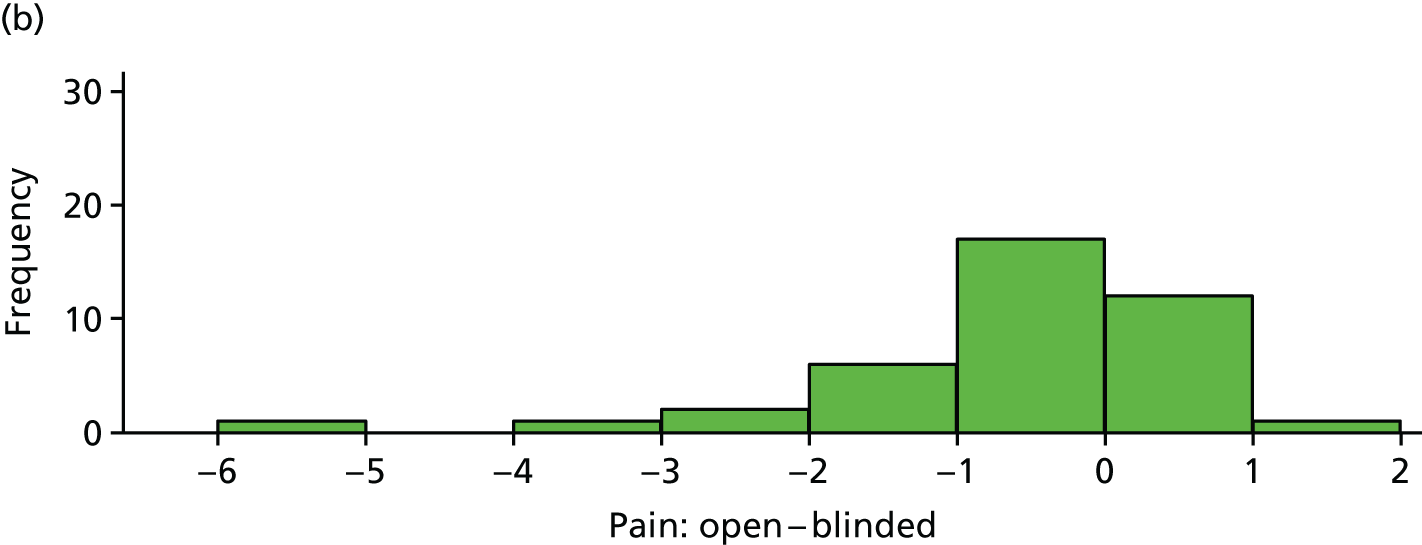
| Variable | Trial arm | |
|---|---|---|
| Placebo (n = 48) | IVIg (n = 40) | |
| Mean (SD) difference | –0.13 (0.76) | –0.58 (1.25) |
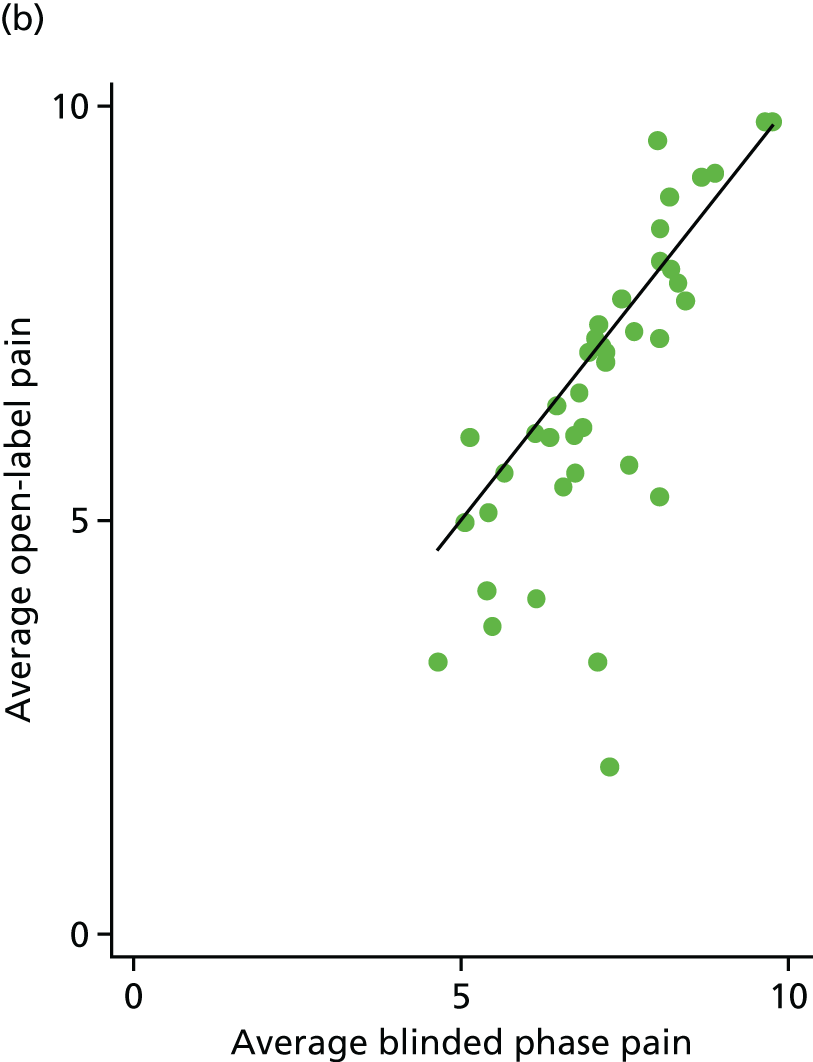
The average open-label pain scores were also plotted against the average blinded phase pain scores for 88 participants (Figure 14). Fifty-two (59%) participants had lower pain scores in the open phase [25/48 (52%) for the placebo arm and 27/40 (68%) for the IVIg arm].
FIGURE 14.
Average open-label pain vs. average blinded phase pain for each participant. (a) Placebo and (b) IVIg.
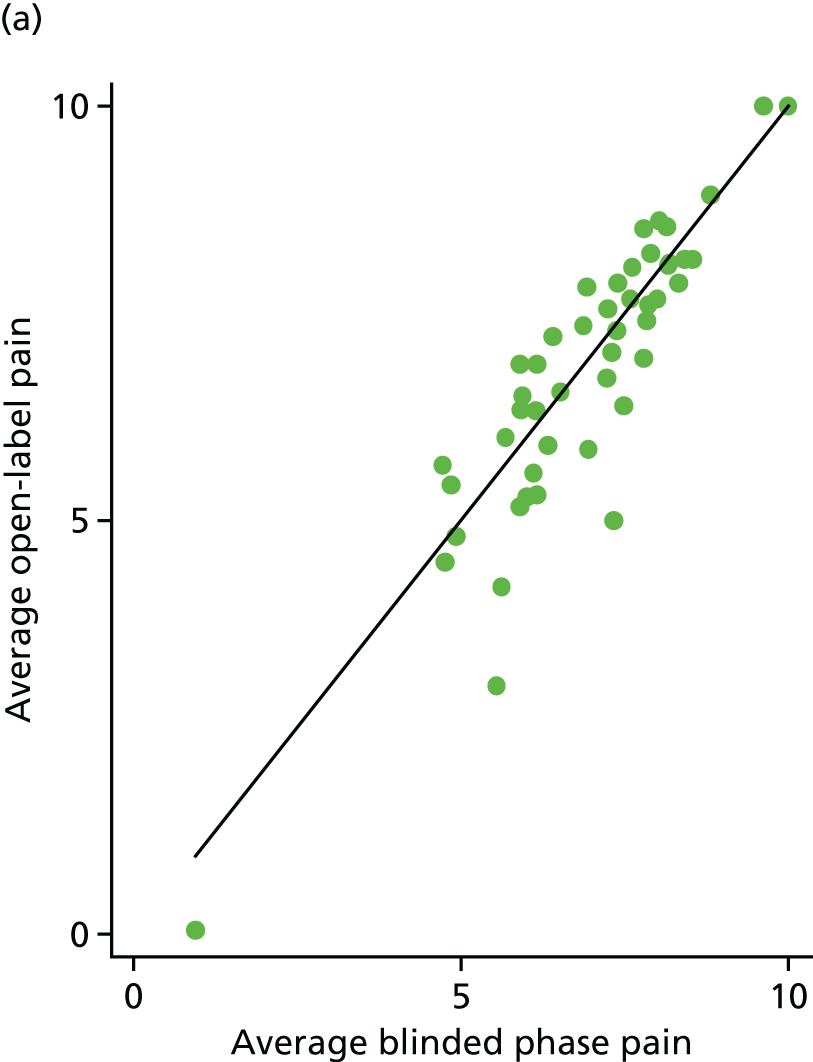
Reduction in baseline pain score: open versus baseline
The difference between average baseline pain score and average open-label pain score is plotted in Figure 15 for each participant. These differences are also summarised in Table 23.
FIGURE 15.
Difference between open-label pain and baseline pain for each participant. (a) Placebo and (b) IVIg.
| Variable | Trial arm | |
|---|---|---|
| Placebo (n = 48) | IVIg (n = 40) | |
| Mean (SD) difference | –0.67 (1.46) | –0.92 (1.37) |
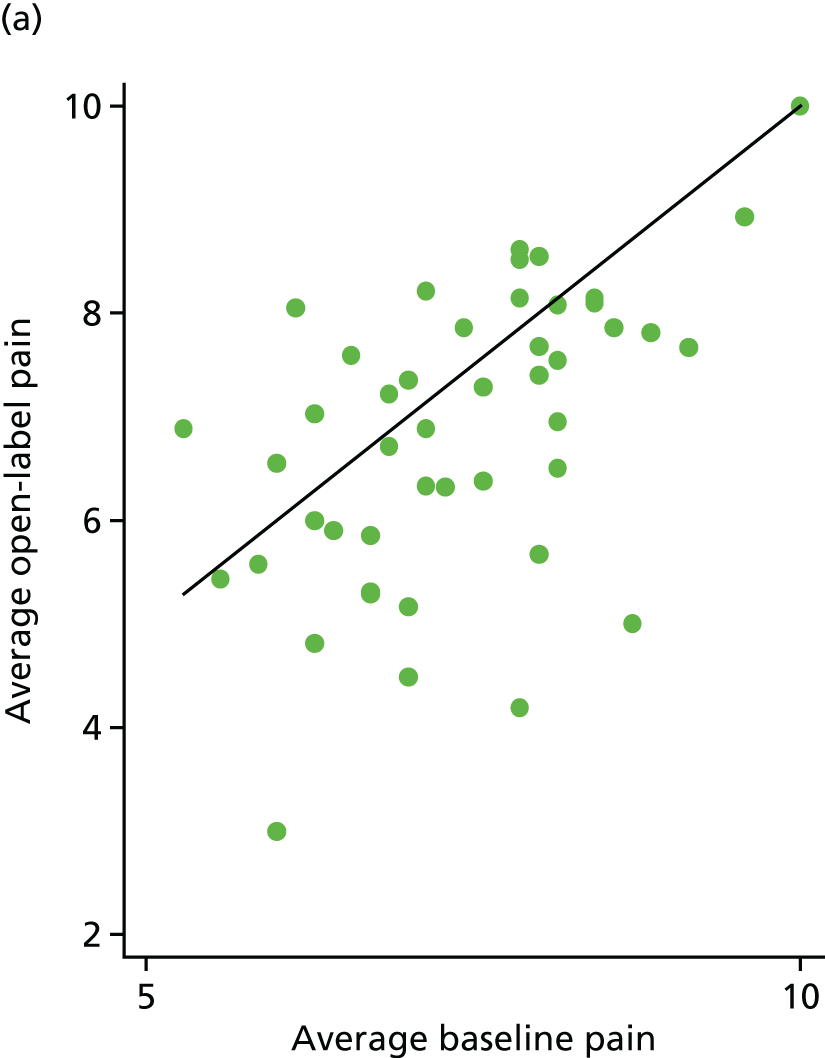
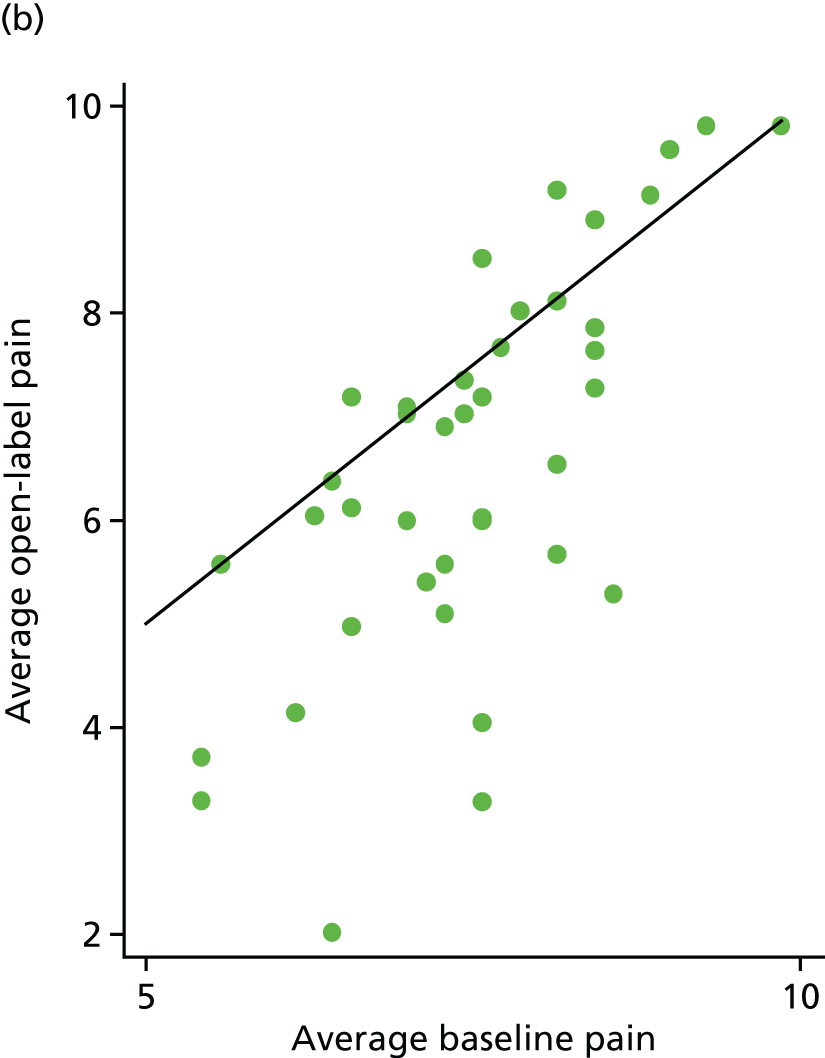
Average pain scores from the open phase were plotted against average baseline pain scores for 88 participants (Figure 16). Sixty-two (70%) participants had lower pain scores in the open phase [33/48 (69%) for the placebo arm and 29/40 (73%) for the IVIg arm].
FIGURE 16.
Average open-label pain vs. average baseline pain for each participant. (a) Placebo and (b) IVIg.
Comparison of pain relief: blind and open
Table 24 shows the number of participants achieving ‘considerable’ pain relief compared with that at baseline. This is based on the 88 participants who provided both blinded and open-phase pain scores.
| Pain relief | Trial arm | |||
|---|---|---|---|---|
| Placebo (n = 48) | IVIg (n = 40) | |||
| Blinded | Open | Blinded | Open | |
| ≥ 2 points | 5 | 6 | 1 | 8 |
| 30–39% | 1 | 1 | 1 | 6 |
| 40–49% | 1 | 2 | 0 | 1 |
| 50–69% | 0 | 1 | 0 | 2 |
| ≥ 70% | 1 | 1 | 0 | 0 |
Fourteen participants achieved ≥ 2 points’ pain relief in the open-label phase and they are described in Table 25.
| IVIg | Pain reduction | Sex | Duration (years) | IgG | Disorder of the immune system and allergies |
|---|---|---|---|---|---|
| Placebo | 2.33 | Male | 2 | 9.7 | Yes |
| Placebo | 2.51 | Female | 2 | 8.8 | No |
| Placebo | 3 | Female | 3 | 10.4 | No |
| Placebo | 3.67 | Female | 1 | 10.4 | Yes |
| Placebo | 3.71 | Male | 3 | 13 | No |
| Placebo | 6.24 | Female | 1 | 9.36 | Yes |
| IVIg | 2 | Female | 4 | 10.4 | No |
| IVIg | 2.14 | Male | 2 | 11.8 | No |
| IVIg | 2.19 | Female | 4 | 9.8 | No |
| IVIg | 2.48 | Male | 3 | 10.07 | No |
| IVIg | 3.29 | Male | 3 | 13.1 | No |
| IVIg | 3.52 | Female | 4 | 8.8 | Yes |
| IVIg | 4.29 | Male | 4 | 10.3 | No |
| IVIg | 4.40 | Male | 2 | 6.2 | No |
Open-label pain relief by disease duration (n = 88)
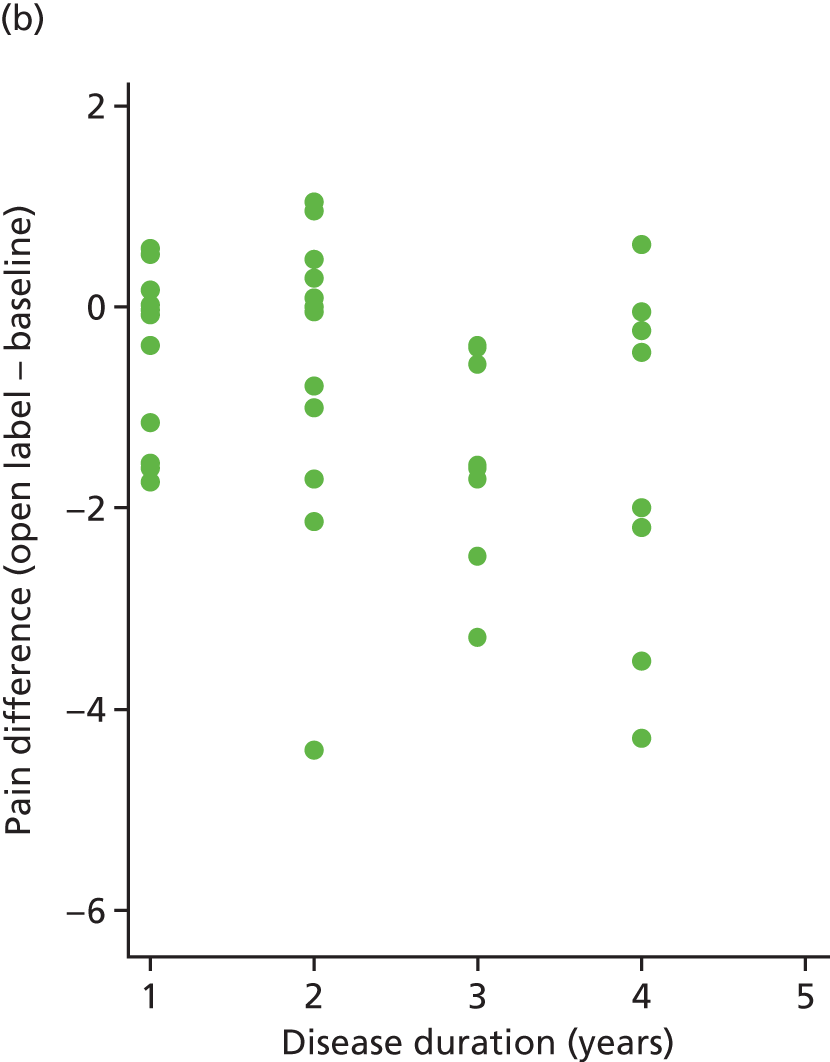
The correlation between the difference of (average) follow-up and baseline pain was calculated for both arms. These values were 0.13 (placebo) and –0.33 (IVIg). These relationships can be seen in Figure 17.
FIGURE 17.
Pain difference (open label – baseline) vs. disease duration. (a) Placebo and (b) IVIg.
Secondary outcomes
The analyses of EQ-5D-5L and BPI are adjusted for site. The differences (and CIs) and p-values for visit 4 (v4) values are obtained from these adjusted analyses. The differences and CIs for visit 1 (v1) and visit 3 (v3) values are unadjusted. All analyses are based on the primary analysis data set (n = 103), that is, they relate to the blinded phase.
EuroQol-5 Dimensions, five-level version
There is little difference in EQ-5D between trial arms (Table 26 and Figures 18–20).
| Variable | Trial arm | IVIg – placebo | p-value | |||
|---|---|---|---|---|---|---|
| Placebo | IVIg | |||||
| n | Mean (SD) | n | Mean (SD) | Difference (95% CI) | ||
| v1. EQ-5D-5L | 53 | 0.34 (0.28) | 50 | 0.33 (0.26) | –0.01 (–0.12 to 0.09) | |
| v3. EQ-5D-5L | 53 | 0.37 (0.29) | 45 | 0.35 (0.25) | –0.02 (–0.13 to 0.09) | |
| v4. EQ-5D-5La | 51 | 0.37 (0.29) | 43 | 0.41 (0.27) | 0.03 (–0.08 to 0.15) | 0.58 |
FIGURE 18.
Baseline EQ-5D. (a) Placebo and (b) IVIg.
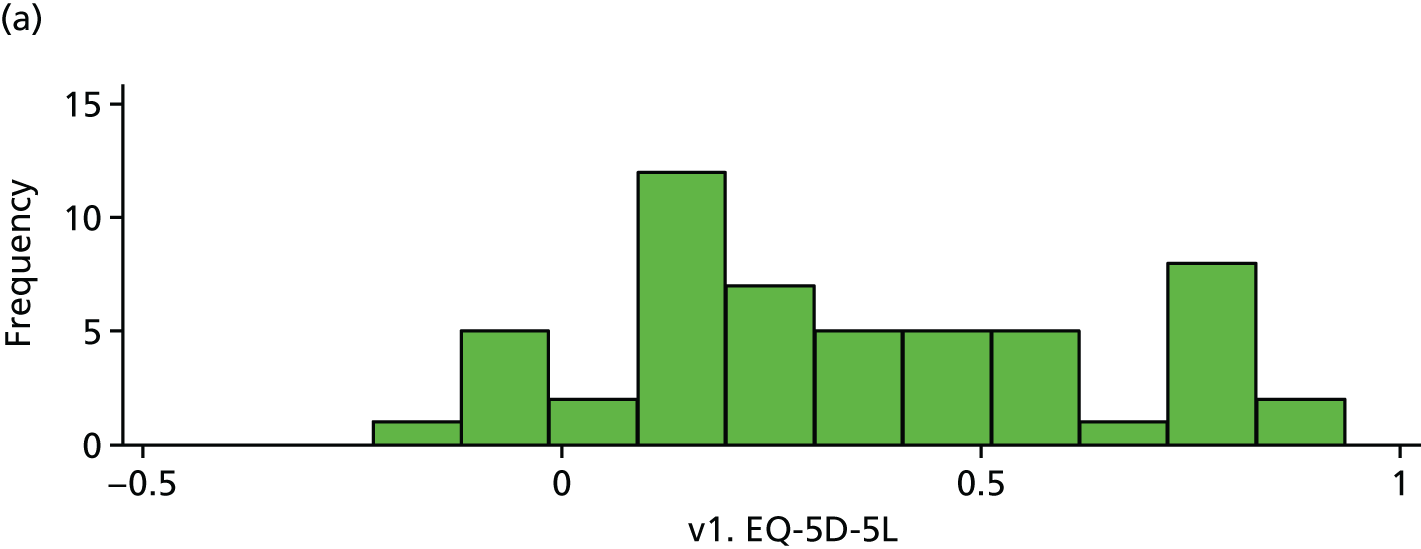
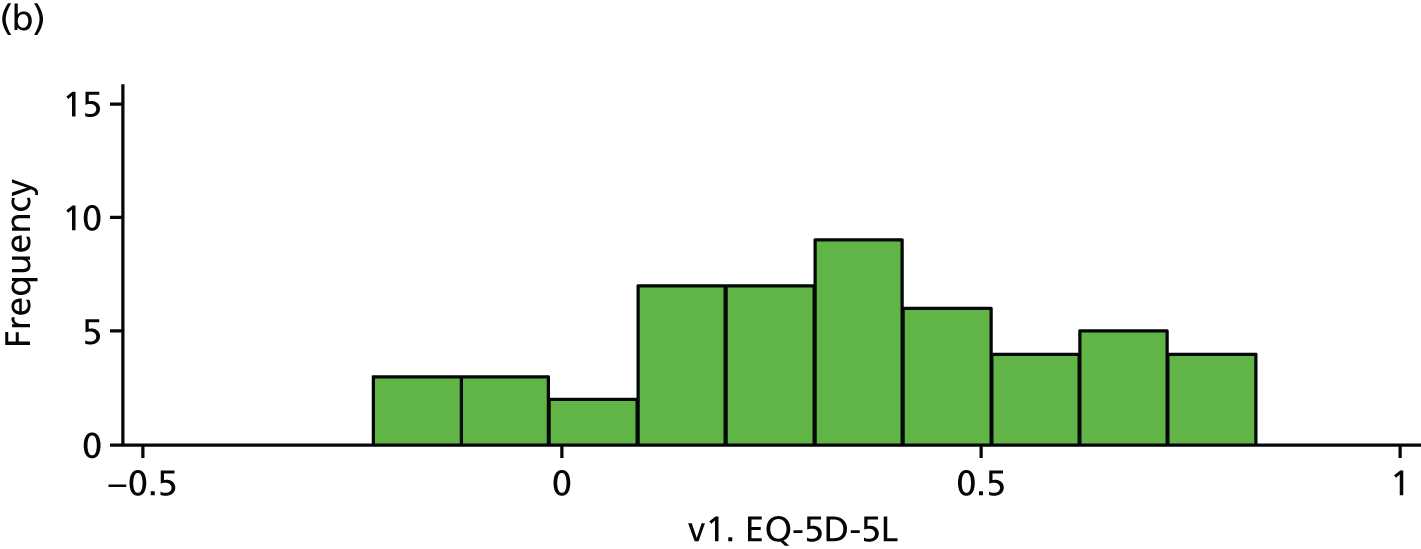
FIGURE 19.
EQ-5D (visit 4). (a) Placebo and (b) IVIg.
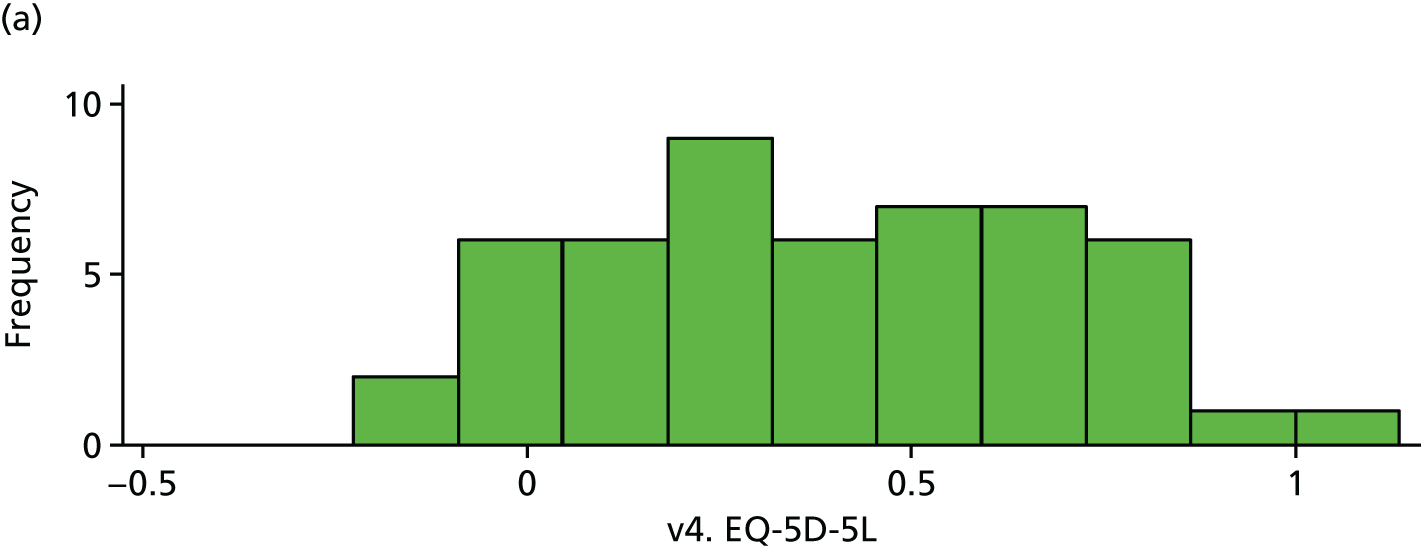
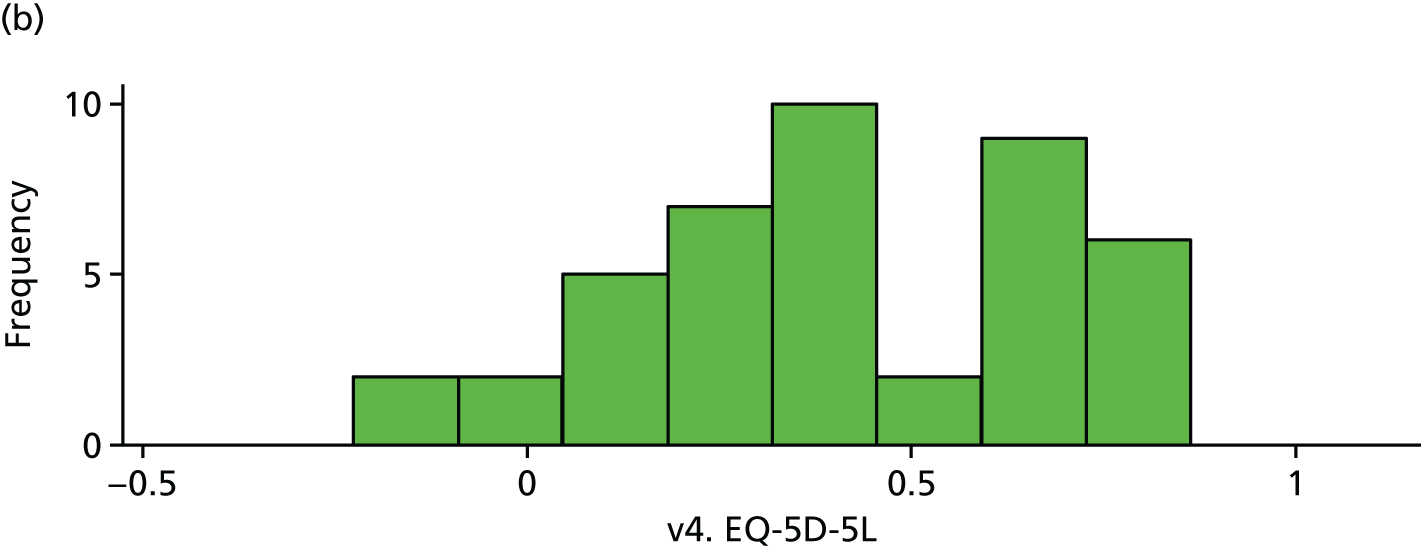
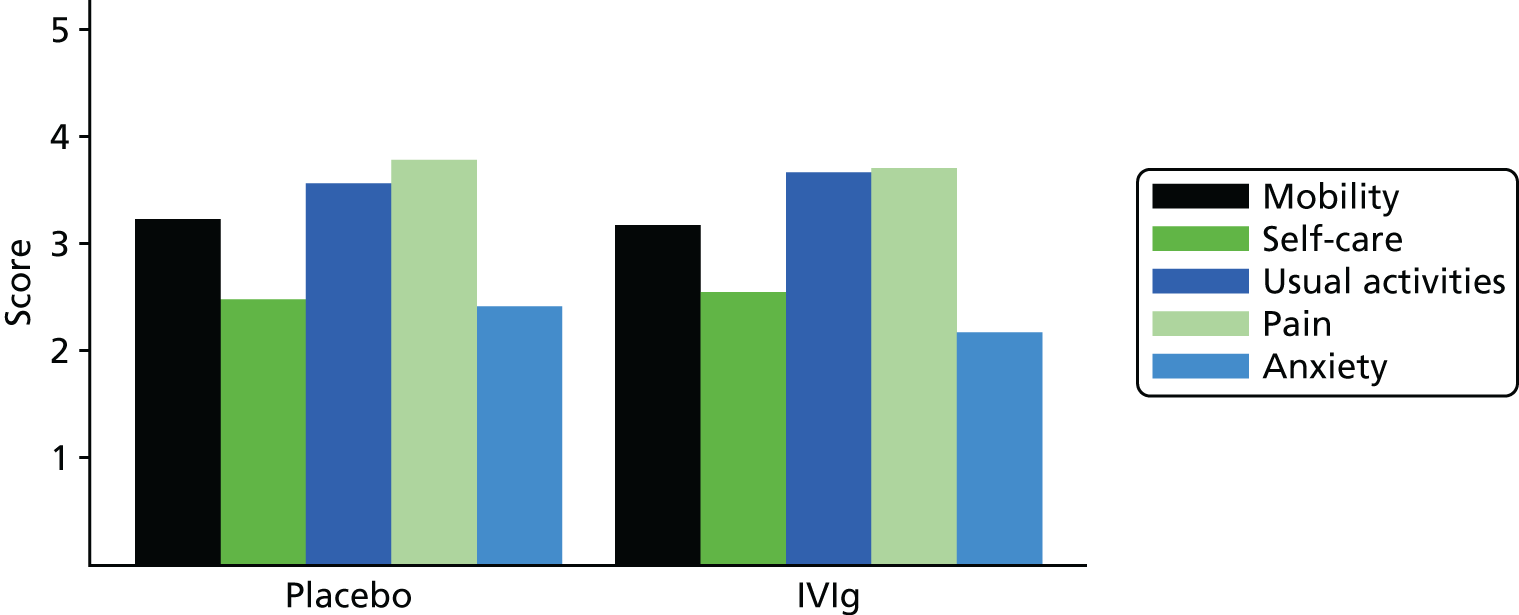
FIGURE 20.
EQ-5D domains (visit 4).
Brief Pain Inventory
There is no evidence of a difference in BPI between the trial arms (Table 27 and Figures 21 and 22).
| Variable | Trial arm | IVIg – placebo | p-value | |||
|---|---|---|---|---|---|---|
| Placebo | IVIg | |||||
| n | Mean (SD) | n | Mean (SD) | Difference (95% CI) | ||
| v1. BPI interference | 53 | 7.31 (1.63) | 50 | 7.43 (1.64) | 0.12 (–0.52 to 0.76) | |
| v3. BPI interference | 53 | 6.98 (1.94) | 45 | 7.34 (1.30) | 0.36 (–0.32 to 1.03) | |
| v4. BPI interferencea | 51 | 6.89 (2.08) | 44 | 7.24 (1.54) | 0.35 (–0.43 to 1.13) | 0.38 |
FIGURE 21.
Baseline BPI. (a) Placebo and (b) IVIg.
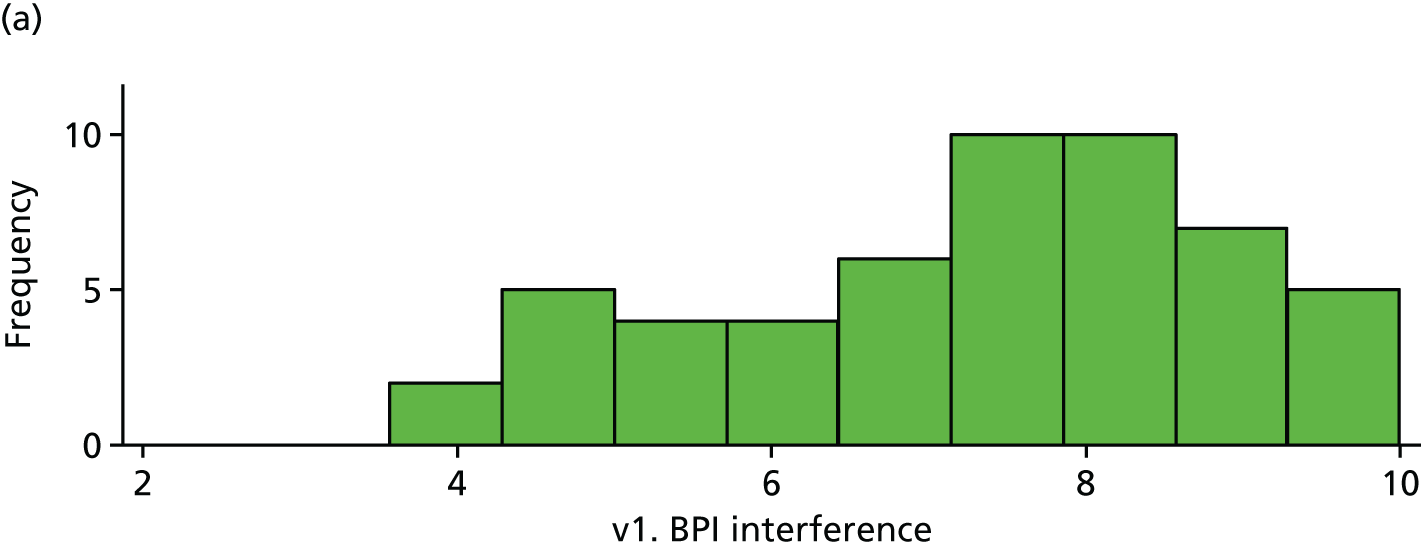
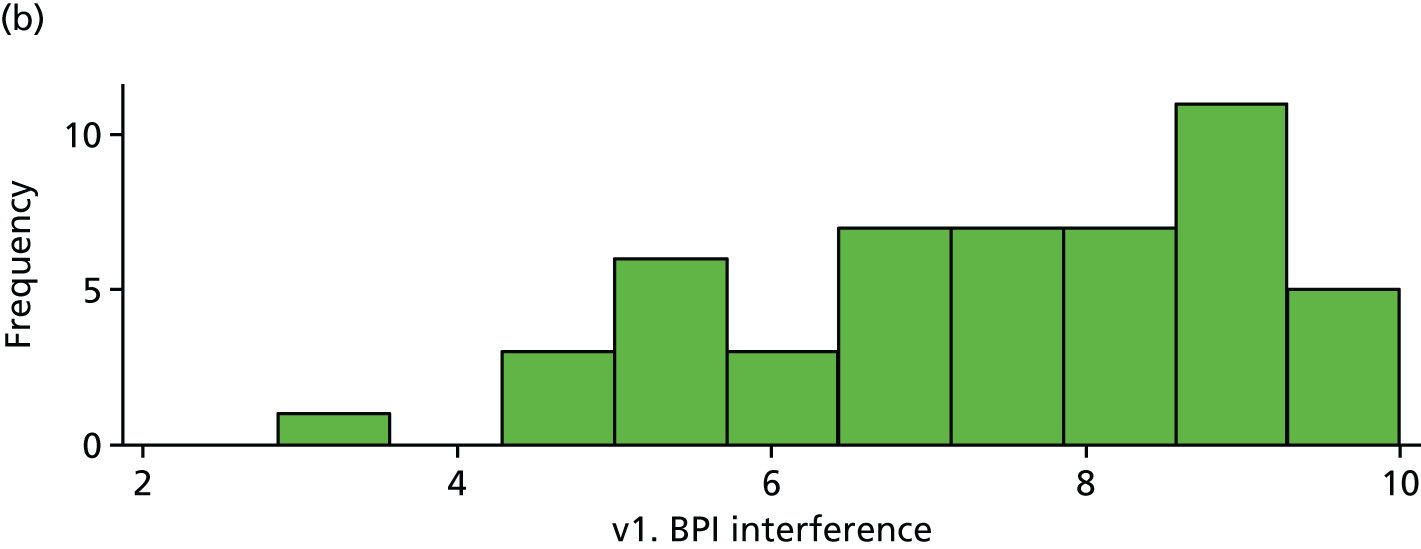
FIGURE 22.
The BPI (visit 4). (a) Placebo and (b) IVIg.
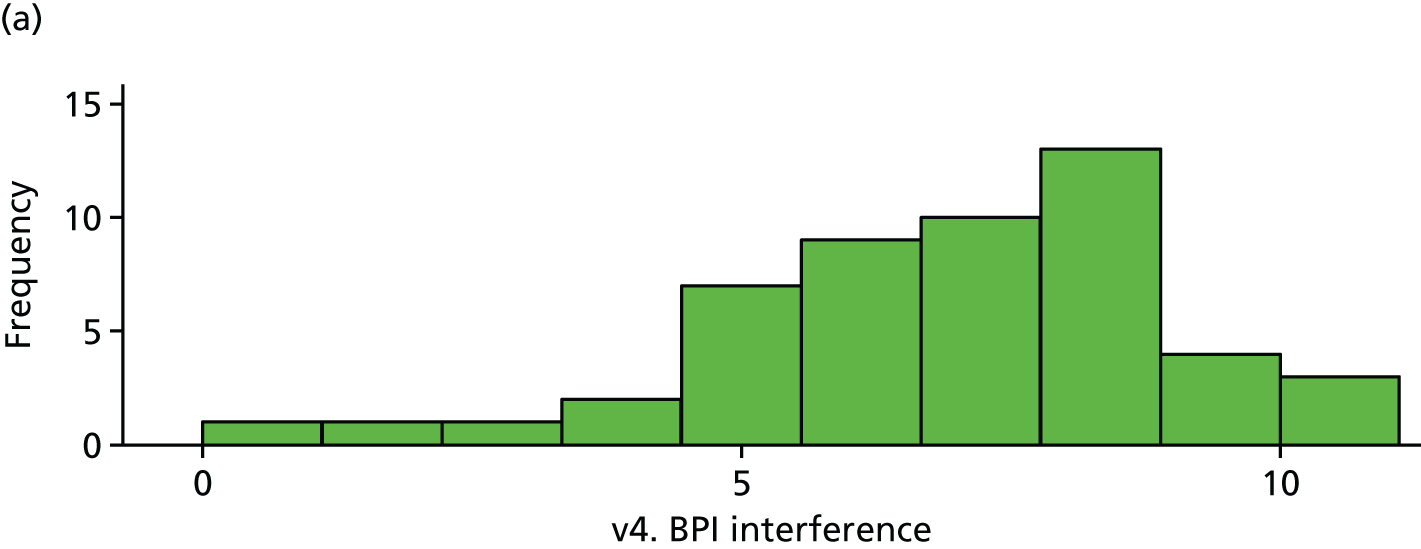
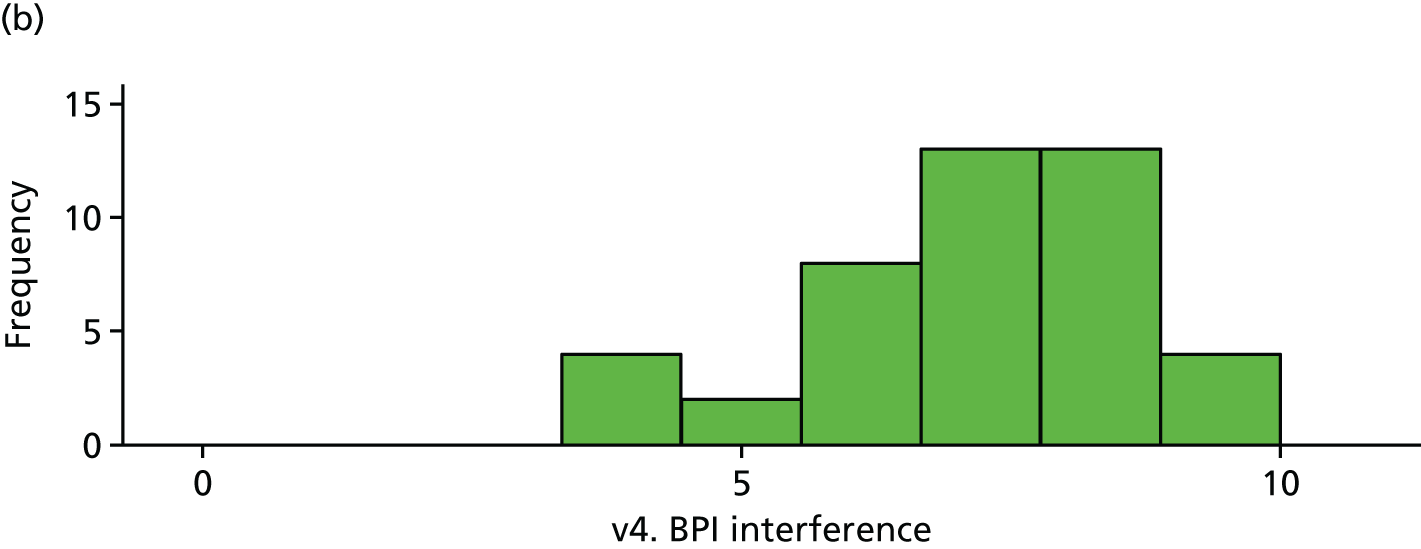
All differences (and CIs) and p-values for the exploratory outcomes are unadjusted. All analyses are based on the primary analysis data set (n = 103).
The odds ratio (from ordinal regression) is 1.22 (95% CI 0.57 to 2.59; p = 0.61).
Limb temperature and volume
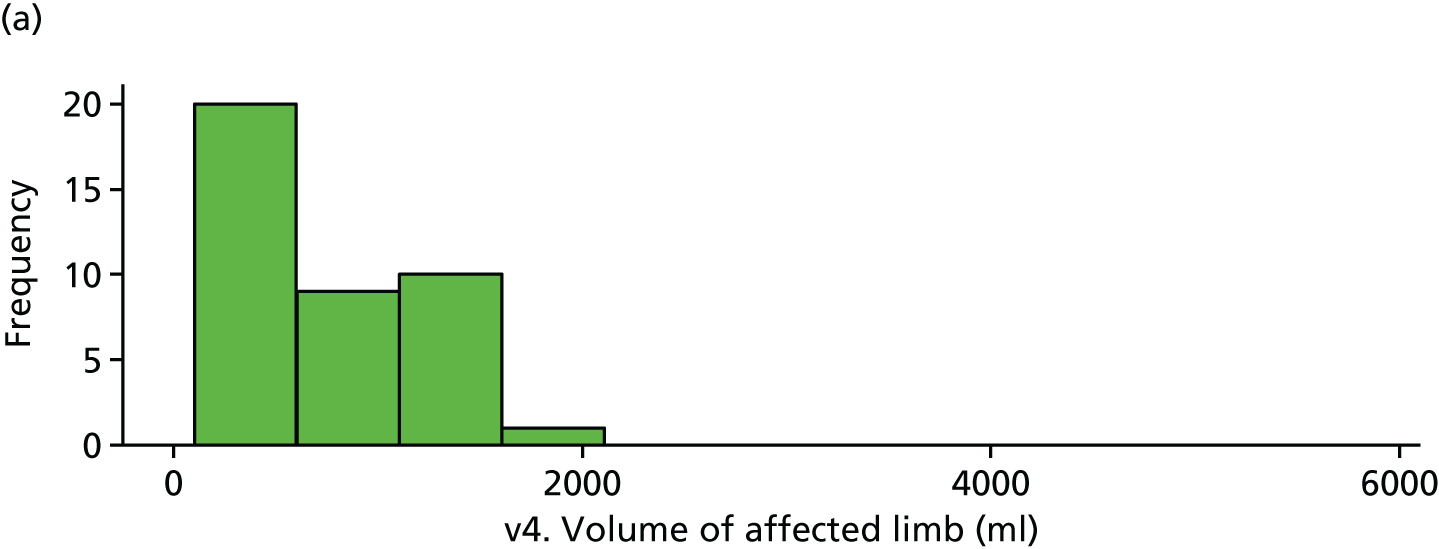
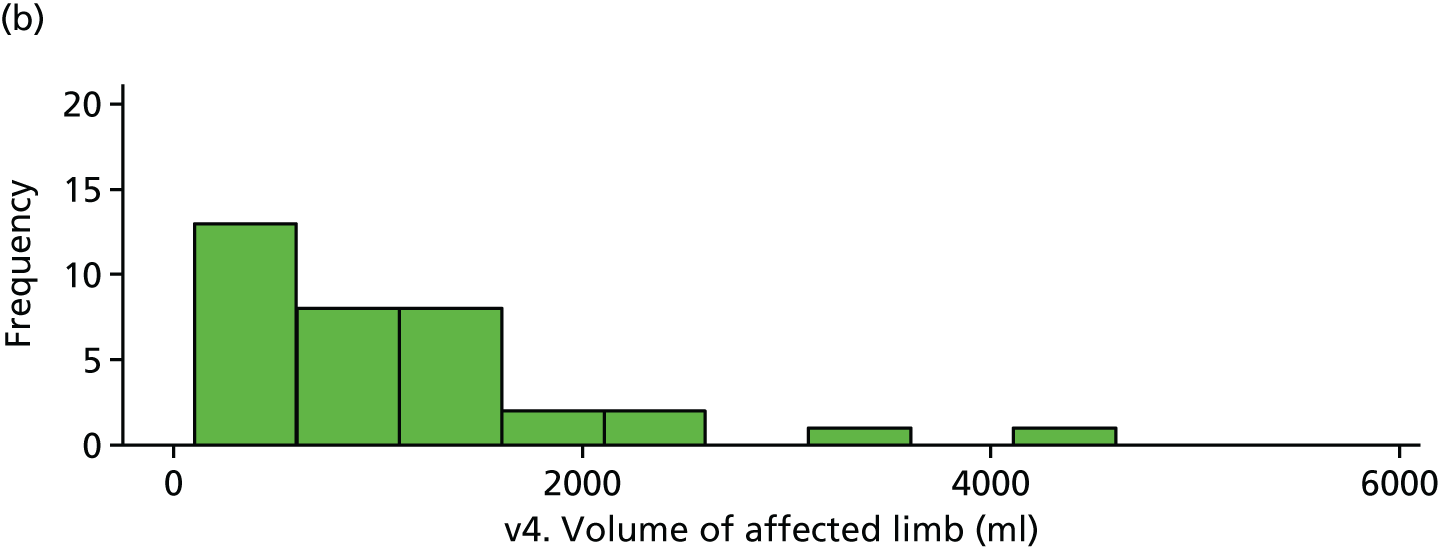
Limb volume was very skewed and, hence, a cube root transformation was applied before analysis. There is weak evidence of a difference in affected limb volumes at visit 4 (Tables 28 and 29 and Figure 23).
| Variable | Trial arm | Difference (95% CI) | p-value | |||
|---|---|---|---|---|---|---|
| Placebo | IVIg | |||||
| n | Mean (SD) | n | Mean (SD) | |||
| Cube root volume | ||||||
| v1. Non-affected limb | 43 | 9.32 (1.93) | 40 | 9.50 (2.44) | 0.18 (–0.78 to 1.14) | |
| v1. Affected limb | 43 | 9.34 (1.94) | 40 | 9.52 (2.49) | 0.18 (–0.79 to 1.15) | |
| v4. Non-affected limb | 40 | 8.96 (2.17) | 35 | 9.30 (2.48) | 0.34 (–0.73 to 1.41) | 0.525 |
| v4. Affected limb | 40 | 8.70 (2.05) | 35 | 9.72 (2.60) | 1.02 (–0.05 to 2.09) | 0.061 |
| Temperature | ||||||
| v1. Non-affected limb | 44 | 29.55 (2.61) | 44 | 29.87 (2.69) | 0.32 (–0.80 to 1.45) | |
| v1. Affected limb | 44 | 28.88 (3.44) | 44 | 28.96 (3.33) | 0.09 (–1.35 to 1.52) | |
| v4. Non-affected limb | 42 | 29.29 (2.33) | 36 | 29.06 (2.31) | –0.23 (–1.28 to 0.82) | 0.661 |
| v4. Affected limb | 42 | 28.68 (3.02) | 36 | 27.92 (2.63) | –0.75 (–2.04 to 0.53) | 0.247 |
| Variable | Trial arm | Difference (95% CI) | p-value | |||
|---|---|---|---|---|---|---|
| Placebo | IVIg | |||||
| n | Mean (SD) | n | Mean (SD) | |||
| v2. Participant weight (kg) | 53 | 81.77 (20.96) | 50 | 84.47 (19.64) | 2.71 (–5.25 to 10.66) | 0.501 |
| v3. Participant weight (kg) | 52 | 82.13 (20.96) | 45 | 85.32 (20.07) | 3.18 (–5.13 to 11.49) | 0.449 |
| v4. Participant weight (kg) | 49 | 81.67 (20.64) | 39 | 85.11 (20.51) | 3.44 (–5.34 to 12.22) | 0.438 |
FIGURE 23.
Volume of affected limb (visit 4). (a) Placebo and (b) IVIg.
Other questionnaires (Tables 30–35 and Figure 24)
| Variable | Trial arm | Difference (95% CI) | p-value | |||
|---|---|---|---|---|---|---|
| Placebo | IVIg | |||||
| n | Mean (SD) | n | Mean (SD) | |||
| McGill: continuous pain | ||||||
| v1. McGill: continuous pain | 52 | 6.20 (1.67) | 50 | 6.09 (1.80) | –0.11 (–0.79 to 0.57) | |
| v3. McGill: continuous pain | 51 | 5.57 (2.06) | 45 | 5.99 (1.93) | 0.42 (–0.40 to 1.23) | |
| v4. McGill: continuous pain | 50 | 5.36 (2.24) | 44 | 5.23 (2.46) | –0.12 (–1.09 to 0.84) | 0.802 |
| McGill: intermittent pain | ||||||
| v1. McGill: intermittent pain | 52 | 5.68 (2.34) | 50 | 5.94 (2.09) | 0.26 (–0.61 to 1.14) | |
| v3. McGill: intermittent pain | 52 | 5.00 (2.49) | 45 | 5.44 (2.42) | 0.45 (–0.54 to 1.44) | |
| v4. McGill: intermittent pain | 50 | 4.93 (2.65) | 44 | 4.86 (2.44) | –0.07 (–1.12 to 0.98) | 0.894 |
| McGill: neuropathic pain | ||||||
| v1. McGill: neuropathic pain | 53 | 5.80 (2.17) | 50 | 6.04 (1.69) | 0.24 (–0.53 to 1.00) | |
| v3. McGill: neuropathic pain | 53 | 5.18 (2.13) | 45 | 5.47 (1.77) | 0.29 (–0.50 to 1.08) | |
| v4. McGill: neuropathic pain | 51 | 4.92 (2.41) | 44 | 5.10 (2.06) | 0.18 (–0.74 to 1.10) | 0.697 |
| McGill: affective | ||||||
| v1. McGill: affective | 52 | 5.16 (2.17) | 50 | 5.21 (2.33) | 0.05 (–0.83 to 0.94) | |
| v3. McGill: affective | 51 | 4.72 (2.54) | 45 | 5.44 (2.28) | 0.73 (–0.25 to 1.71) | |
| v4. McGill: affective | 50 | 4.52 (2.65) | 44 | 4.89 (2.45) | 0.37 (–0.68 to 1.42) | 0.484 |
| McGill: total score | ||||||
| v1. McGill: total score | 52 | 5.74 (1.63) | 50 | 5.88 (1.54) | 0.14 (–0.49 to 0.76) | |
| v3. McGill: total score | 51 | 5.14 (1.93) | 45 | 5.60 (1.74) | 0.46 (–0.29 to 1.21) | |
| v4. McGill: total score | 50 | 4.96 (2.22) | 44 | 5.03 (2.03) | 0.07 (–0.80 to 0.95) | 0.872 |
| Variable | Trial arm | Difference (95% CI) | p-value | |||
|---|---|---|---|---|---|---|
| Placebo | IVIg | |||||
| n | Mean (SD) | n | Mean (SD) | |||
| HADS anxiety | ||||||
| v1. HADS anxiety | 53 | 10.68 (4.59) | 50 | 10.38 (4.77) | –0.30 (–2.13 to 1.53) | |
| v3. HADS anxiety | 53 | 9.64 (4.75) | 45 | 9.93 (4.71) | 0.29 (–1.61 to 2.20) | |
| v4. HADS anxiety | 51 | 9.78 (4.88) | 44 | 9.41 (4.57) | –0.37 (–2.30 to 1.57) | 0.706 |
| HADS depression | ||||||
| v1. HADS depression | 53 | 10.98 (4.19) | 50 | 9.40 (3.55) | –1.58 (–3.11 to –0.06) | |
| v3. HADS depression | 53 | 10.43 (4.39) | 45 | 10.36 (3.84) | –0.08 (–1.75 to 1.59) | |
| v4. HADS depression | 51 | 9.94 (4.47) | 44 | 9.91 (3.65) | –0.03 (–1.71 to 1.65) | 0.973 |
| Variable | Trial arm | Difference (95% CI) | p-value | |||
|---|---|---|---|---|---|---|
| Placebo | IVIg | |||||
| n | Mean (SD) | n | Mean (SD) | |||
| PCS rumination | ||||||
| v1. PCS rumination | 53 | 9.42 (4.38) | 50 | 9.24 (4.27) | –0.18 (–1.87 to 1.52) | |
| v3. PCS rumination | 53 | 8.77 (4.87) | 45 | 9.84 (4.24) | 1.07 (–0.78 to 2.92) | |
| v4. PCS rumination | 51 | 8.29 (5.25) | 44 | 8.18 (4.62) | –0.11 (–2.14 to 1.92) | 0.913 |
| PCS magnification | ||||||
| v1. PCS magnification | 53 | 4.32 (3.24) | 50 | 3.84 (3.11) | –0.48 (–1.72 to 0.76) | |
| v3. PCS magnification | 53 | 3.51 (2.94) | 45 | 3.67 (2.77) | 0.16 (–1.00 to 1.31) | |
| v4. PCS magnification | 51 | 3.84 (3.06) | 44 | 3.66 (2.87) | –0.18 (–1.40 to 1.03) | 0.764 |
| PCS helplessness | ||||||
| v1. PCS helplessness | 53 | 11.94 (5.61) | 50 | 12.42 (5.98) | 0.48 (–1.79 to 2.74) | |
| v3. PCS helplessness | 53 | 11.08 (6.25) | 45 | 11.58 (6.11) | 0.50 (–1.99 to 2.99) | |
| v4. PCS helplessness | 51 | 10.53 (6.22) | 44 | 10.18 (5.92) | –0.35 (–2.83 to 2.14) | 0.782 |
| PCS total score | ||||||
| v1. PCS total score | 53 | 25.68 (12.19) | 50 | 25.50 (12.41) | –0.18 (–4.99 to 4.63) | |
| v3. PCS total score | 53 | 23.36 (13.25) | 45 | 25.09 (11.76) | 1.73 (–3.33 to 6.80) | |
| v4. PCS total score | 51 | 22.67 (13.96) | 44 | 22.02 (12.49) | –0.64 (–6.08 to 4.79) | 0.814 |
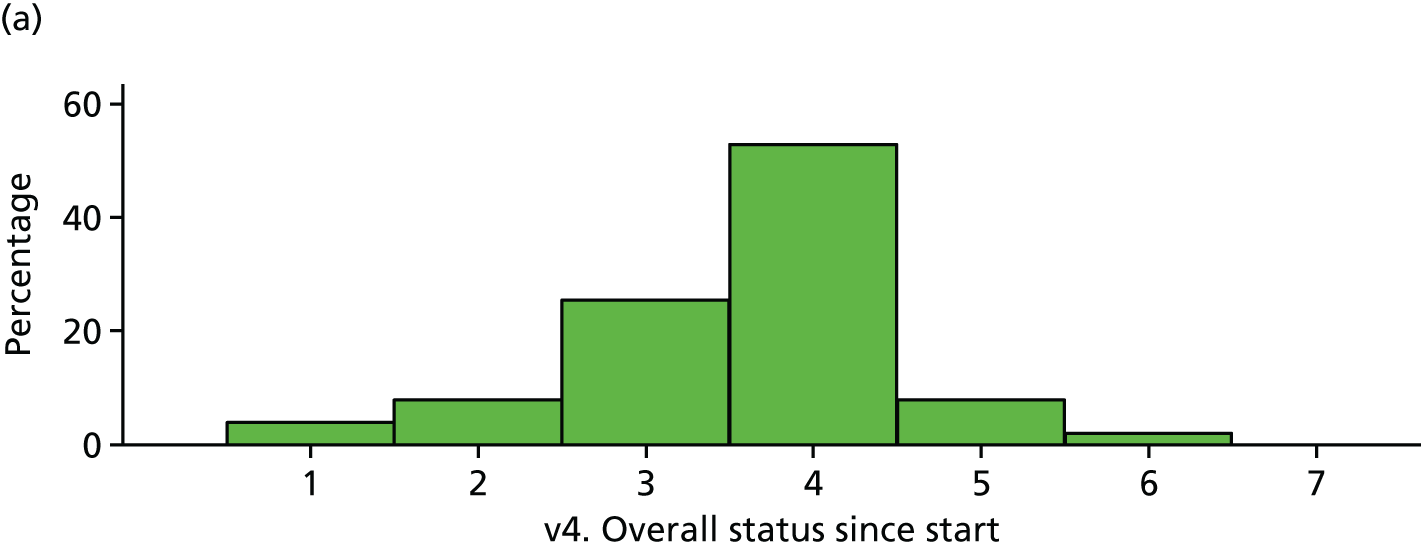
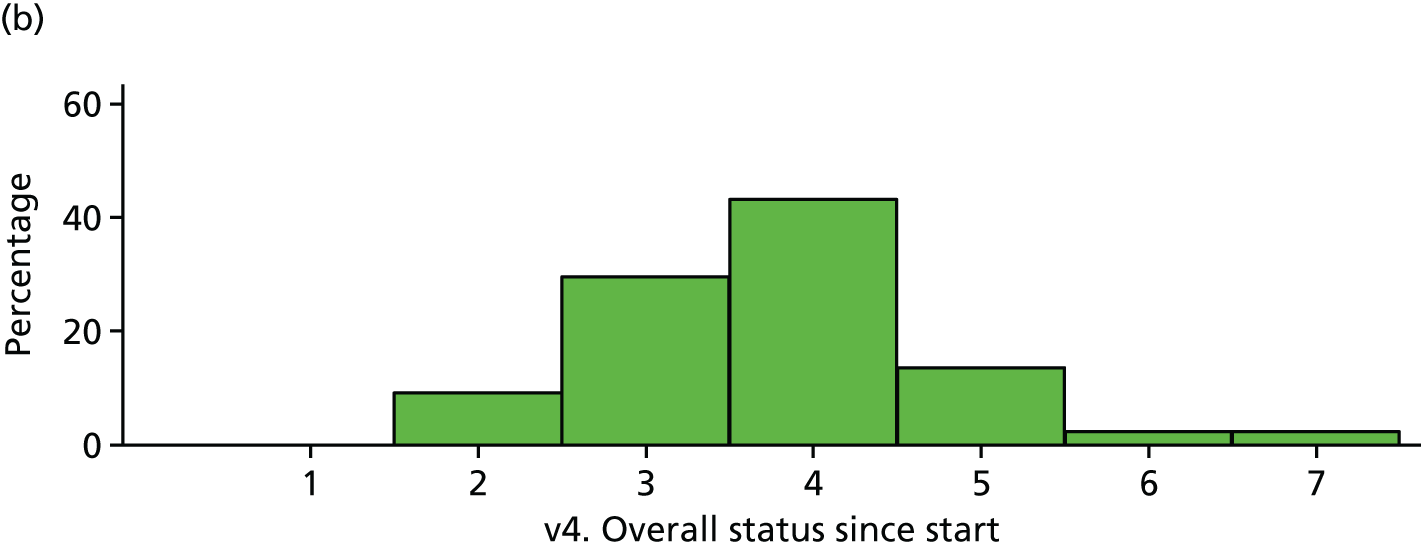
| Visit 4 | Trial arm | |
|---|---|---|
| Placebo | IVIg | |
| Very much improved | 2 | 0 |
| Much improved | 4 | 4 |
| Minimally improved | 13 | 13 |
| No change | 27 | 19 |
| Minimally worse | 4 | 6 |
| Much worse | 1 | 1 |
| Very much worse | 0 | 1 |
| Total | 51 | 44 |
| Variable | Trial arm | Difference (95% CI) | p-value | |||
|---|---|---|---|---|---|---|
| Placebo | IVIg | |||||
| n | Mean (SD) | n | Mean (SD) | |||
| v1. SPS total score | 19 | 19.63 (3.35) | 16 | 19.44 (3.14) | –0.19 (–2.44 to 2.06) | |
| v4. SPS total score | 19 | 20.00 (3.35) | 12 | 19.83 (2.86) | –0.17 (–2.56 to 2.22) | 0.888 |
| Variable | Trial arm | Difference (95% CI) | p-value | |||
|---|---|---|---|---|---|---|
| Placebo | IVIg | |||||
| n | Mean (SD) | n | Mean (SD) | |||
| v1. NLS mean score | 50 | 4.04 (1.37) | 49 | 3.92 (1.40) | –0.12 (–0.67 to 0.43) | |
| v4. NLS mean score | 47 | 3.57 (1.37) | 41 | 3.61 (1.10) | 0.05 (–0.48 to 0.58) | 0.856 |
FIGURE 24.
Participant global impression of change (visit 4). (a) Placebo and (b) IVIg.
A total of six SAEs were reported overall by six patients across the blinded and open-label phases of the trial. There were no suspected unexpected serious adverse reactions reported (Tables 36–39).
| Variable | Trial arm | Difference (95% CI) | |||
|---|---|---|---|---|---|
| Placebo | IVIg | ||||
| n | Mean (SD) | n | Mean (SD) | ||
| v2. Expected pain level | 53 | 5.25 (2.38) | 50 | 5.52 (1.68) | 0.27 (–0.53 to 1.08) |
| Variable | Trial arm | |||
|---|---|---|---|---|
| Placebo | IVIg | |||
| N | n (%) | N | n (%) | |
| v1. Contacted GP or health care professional | 53 | 41 (77.4) | 50 | 32 (64.0) |
| v1. Admitted as inpatient during last 6 weeks | 53 | 2 (3.8) | 49 | 2 (4.1) |
| v1. Referred as outpatient during last 6 weeks | 53 | 18 (34.0) | 50 | 11 (22.0) |
| v1. Made contact with other facility during last 6 weeks | 53 | 8 (15.1) | 48 | 7 (14.6) |
| v1. Prescribed medication during last 6 weeks | 53 | 13 (24.5) | 49 | 8 (16.3) |
| v1. Contact with social service during last 6 weeks | 53 | 2 (3.8) | 50 | 2 (4.0) |
| SAE number | TRT | SAE | Code | Intensity | Related | Resolved |
|---|---|---|---|---|---|---|
| 1 | Placebo | Headache | 10. Neurological | Severe | Probably | Yes |
| Vomiting | 4. Gastro Intestinal | Moderate | Probably | Yes | ||
| 2 | IVIg | Headache | 10. Neurological | Severe | Probably | Yes |
| SAE number | SAE | Code | Intensity | Related | Resolved |
|---|---|---|---|---|---|
| 3 | Aseptic meningitis | 10. Neurological | Moderate | Probably | Yes |
| Associated symptoms documented | |||||
| Headache | 10. Neurological | Moderate | Probably | Yes | |
| Photopia | 10. Neurological | Moderate | Probably | Yes | |
| 4 | Increased heart rate | 10. Neurological | Moderate | Probably | Yes |
| 5 | Aseptic meningitis | 10. Neurological | Severe | Probably | Yes |
| 6 | Deep-vein thrombosis | 7. Neurological | Severe | Probably | Yes |
Chapter 5 Discussion
In our Phase III RCT, treatment with two low doses of IVIg (0.5 g/kg/treatment) did not change patients’ pain. No patient treated with IVIg reported substantial pain reduction, which contrasted with results in earlier studies. 7,9 In addition, the overall placebo response was small.
Trial participants had characteristics typical of patients with long-standing CRPS cared for in a secondary or tertiary setting, including an average age of mid-40s, female-to-male ratio of 2–3 : 1, high pain intensity, high pain interference,29 low QoL5 and cold limbs. 30
The lack of effect on the primary outcome (pain intensity) was underpinned by the finding of no significant effects on mood, pain impact, QoL and limb temperature.
None of the predefined prognostic markers predicted a beneficial treatment response in this trial. The unexpected, post hoc finding of significant pain reduction in the rare group of patients with CRPS II may be due to random effects and would need prospective confirmation.
Potential reasons for the discrepancy between the results in this and prior trials include selection bias and random effects. Although this trial included patients with longer disease durations than the earlier RCT,9 we found no correlation between disease duration and treatment outcome.
The present standard of care for patients with long-standing CRPS includes physical rehabilitation, psychological support and treatment with drugs and devices, but many patients still experience a high level of pain while on treatment. 31 The findings of this study add to earlier negative trials of immune-modulating treatments, including lenalidomide, intrathecal steroids, infliximab and oral steroids. 32–35 These negative findings should be considered on the background of recent in vivo and in vitro results on functionally active, non-inflammatory autoantibodies in CRPs. 36–38 The target of effective immune-modulating therapy may be the production of such autoantibodies, rather than inflammatory processes. 39 Alternative immune-modulating technologies, such as plasma exchange therapy, disease modifying antirheumatic drugs, high-dose immunoglobulin or B-cell ablation therapies, should be explored; initial results have been encouraging, but RCTs are needed. 40–42 This remains an area of large unmet need, a fact that is also underpinned by the recently discussed, patient-driven, generally futile approaches to achieve pain relief by surgical removal of the painful limb. 43
The trial strengths include the multicentre set-up, the size, the swift recruitment to plan, the high patient retention (low withdrawal) and the high patient adherence. Patient demographics and disease characteristics are typical for those with long-standing CRPS, as seen in secondary and tertiary care. 29 As patients engaged well, they provided comprehensive data aiding the data quality and statistical analysis. A much higher attrition had been anticipated (see Chapter 3) from experience in academic trials of this size. A comprehensive patient recruitment and retention strategy was essential to this achievement; details will be published separately. Blinding was tested and was successful.
The study is limited by the upper disease duration of 5 years and the lower cut-off point of 1 year, the short dosing, and the lack of a high-dose treatment arm. Although the study was well balanced overall, there was some imbalance in the health resource use, with a higher use in the placebo arm. The trial was not powered for the individual subgroup evaluations.
Chapter 6 Conclusion
In conclusion, contrary to our expectation, low-dose immunoglobulin treatment did not produce pain relief in patients with long-standing moderate and severe CRPS of up to 5 years duration. Additional research might include the assessment of treatment effects in patients with severe concomitant autoimmunity or in patients with CRPS type II. Because recent laboratory findings have suggested that there might be an autoantibody contribution in many CRPS, alternative immune modulating treatment or higher-dose IVIg treatment should be further examined.
Using comprehensive recruitment strategies, the completion of this trial for a rare condition within 2 years as per the plan in a UK setting is encouraging, and should reassure investigators wishing to perform clinical trials in this and other rare chronic pain conditions that this is possible in a feasible time frame.
Acknowledgements
Trial Steering Committee
Professor Richard Hughes (chairperson), Mrs Wendy Hall (patient representative), Dr Mick Serpell (principal investigator representative), Dr Paul Farquhar-Smith (independent), Dr Phil Wiffen (independent) and Professor Turo Nurmikko (non-voting).
Data safety and monitoring board
Dr Siraj Misbah (chairperson), Zoe Hoare (independent statistician) and Professor Solomon Tesfaye (independent member).
Study centre research teams
-
The Walton Centre NHS Foundation Trust, Liverpool.
Dr Praveen Ganey (subinvestigator), Dr Kiran Sachane (subinvestigator), Dr Sumit Gulati (subinvestigator), Dr Helen Banks (subinvestigator), Dr Heike Ardnt (subinvestigator), Diane Wood (pharmacist), Clare Charters (pharmacist), Louise Pate (research nurse), Kate O’Hanlow (research nurse), Helen Leggett (research nurse), Imelda O’Brien (research nurse), Deborah Davies (research nurse) and Loretta Kerr (data input).
-
Guys and St Thomas’s Trust, London.
Dr David Pang (subinvestigator), Emma Hudson (research manager), Samuel Wesley (research administrator), Verena Eberhardt (research nurse), Lisa Hurley (clinical trials technician), Chi Kai Tam (pharmacist), Angela Cape (pharmacist) and Tanya Richards (data manager).
-
Royal National Hospital for Rheumatic Diseases and University of the West of England.
Poppy Hocken (research nurse), Dr Philip Hamann (subinvestigator), Dr Shyama Pilapitiya (subinvestigator), Dr Sarah Tansley (subinvestigator), Dr Chloe Lapraik (subinvestigator), Dr Shabina Habibi (subinvestigator), Rosemary Wood (clinical research nurse manager), Jan Ball (clinical research nurse), Sarah Cole (clinical research nurse), Nicola Fulstow (clinical research nurse), Kate Moloney (pharmacist), Sarah Murdoch (pharmacist), Jill MacDonald (pharmacist), Margaret MacMillan (pharmacy assistant), Samantha Williams (pharmacy technician) and Katarina Brop (pharmacy technician).
-
Queen Elizabeth University Hospital, Glasgow.
Deborah McGlynn (research nurse), Karen Duffy (research nurse), Lynn Melone (research nurse), Anissa Benchiheub (research nurse), Victoria Patterson (research nurse), Andrew Dougherty (research nurse), Naomi Hickey (research nurse), Linda Wilson (research nurse), Dr Jonathon McGhie (subinvestigator), Marie Callaghan (research nurse), Barbara McLaren (research nurse), Collin Roddin (pharmacist) and Helen Hamilton (pharmacy technician).
-
Norwich and Norfolk University NHS Trust, Norwich.
Gill Pout (research nurse), Jocelyn Price-Keshet (research nurse), Kerrie Self (lead nurse), Helen O’Driscoll (research nurse), Nikki Hunt (research nurse), David Tomlinson (research nurse), Mandy Burrows (research nurse), Ben Booth (research nurse), Praveen Dhillon (subinvestigator), Gail Healey (study pharmacist) and Janette Guymer (administrative support).
-
Cambridge University Hospitals NHS Foundation Trust, Cambridge.
Alison Mitchell (research nurse), Tracey Drayton (research nurse), Dr Andra Negoescu (subinvestigator), Dr Maeve McLaughlin (subinvestigator), Naval Vyse (clinical trials coordinator pharmacy) and Mark Bolton (senior clinical trials technician).
-
University Hospital of Leicester NHS Trust, Leicester.
Margaret Weston (administrator), Sue Cockburn (research nurse), Sandra Kazembe (research nurse), Dr Yehia Kamel (subinvestigator), Afroz Ghumra (clinical trials assistant – pharmacy), Rakhee Rawal (trial pharmacist), Farzana Khalifa (clinical trial co-ordinator – pharmacy) and Mahendra Raval (clinical trial assistant pharmacy).
King’s Clinical Trials Unit
Beverly Alao-White (trial management strategic lead), Amaka Ejibe (trial manager), Peter Lloyd Morgan (trial manager), Evangelos Georgiou (senior clinical software analyst), Janice Jimenez (administration manager), Olivier Pressey (data manager) and Dominic Stringer (data manager).
Patient identification centres
-
Dr Adam Woo, King’s College London NHS Foundation Trust, London, UK.
-
Dr Rachel Gorodkin, Central Manchester University Hospitals NHS Foundation Trust, Manchester, UK.
-
Dr Helen Cohen, Royal National Orthopaedic Hospital NHS Trust, Stanmore, UK.
-
Dr Richard Haigh, Royal Devon and Exeter NHS Foundation Trust, Exeter, UK.
-
Dr Anthony Davies and Plymouth Hospitals NHS Trust, Plymouth, UK.
-
Dr Cathy Price, University Hospitals Southampton NHS Foundation Trust, Southampton, UK.
-
Dr Kiran Koneti, City Hospitals Sunderland NHS Foundation Trust, Sunderland, UK.
-
Professor Jonathon Raphael and Dr Duarte, The Dudley Group NHS Foundation Trust, Dudley, UK.
-
Dr Basil Ousta, Tameside Hospital NHS Foundation Trust, Ashton-under-Lyne, UK.
The authors would also like to thank the following for their invaluable help:
Shakeel Herwitker (Assistant Director – manufacturing services), Aseptic Manufacturing Pharmacy Unit at Royal Liverpool and Broodgreen Hospital, Professor G Sprotte (Emeritus Professor in Pain Medicine, Wuerzburg/Germany), Olivier Gupta (Director, Modepharma), Julie Williams (Administrator, Pain Relief Foundation), Brenda Hall (Administrator, Pain Relief Foundation), Dave Watling (The Walton Centre NHS Trust), Dr Malcom Steiger (The Walton Centre NHS Trust), Mrs. Maria Thornton (The Walton Centre NHS Trust), Alex Astor (University of Liverpool), David McVey (University of Liverpool), James Fox (University of Liverpool), Steve Potter (University of Liverpool), John Roberts (University of Liverpool), Professor Munir Pirmohamed (University of Liverpool) Karen Wilding (University of Liverpool), Dr Andrea Wartenberg-Demand (Biotest, Germany), Professor Artur Bauhofer (Biotest, Germany) John Hodkinson (Biotest, UK) and Mr Chris Hyde (Biotest, UK) eSMS Emergency Code Break Service, NIHR Clinical Research Network: North West Coast.
The study was sponsored by The Walton Centre NHS Foundation Trust and the University of Liverpool.
Biotest UK Ltd provided the active study medication at no cost but had no other input into the design or implementation of the study and did not participate in the preparation of this publication. The funders/Biotest UK had no role in the study design, data collection, data analysis, data interpretation or writing of the report.
Contributions of authors
The authors would like to thank all of the collaborators who contributed to the LIPS trial.
Andreas Goebel (Chief Investigator, Pain Research Institute, Clinical Sciences Centre, Liverpool) wrote the first draft, conceived of the trial and contributed to the design and the acquisition, analysis and interpretation of trial data.
Jatinder Bisla (LIPS Trial Manager, King’s College London, KCT, London) wrote the first draft, contributed to data acquisition and interpretation.
Roy Carganillo (Research Nurse, Pain Management and Neuromodulation Centre, Guys and St Thomas’ Hospital, London) contributed to data acquisition and interpretation.
Claire Cole (Research Coordinator, Pain Research Institute, Clinical Sciences Centre, Liverpool) contributed to the data acquisition.
Bernhard Frank (Subinvestigator, The Walton Centre NHS Foundation Trust, Liverpool) contributed to the trial design, data acquisition, analysis and interpretation.
Rima Gupta (Drug Monitor, Modepharma, Beckenham) contributed to the trial design.
Mairi James (Research Coordinator, Queen Elizabeth University Hospital, Glasgow) contributed to data acquisition and interpretation.
Joanna Kelly (Strategic Data Management Lead, King’s College London, KCT, London) contributed to the trial design and data interpretation.
Candy McCabe (Principal Investigator, Royal National Hospital for Rheumatic Diseases, Bath, and University of the West of England, Bristol) contributed to the trial design, data acquisition, analysis and interpretation.
Holly Milligan (Research Coordinator, Pain Research Institute, Clinical Sciences Centre, Liverpool) contributed to the data acquisition.
Caroline Murphy (Operational Director, King’s College London, KCT, London) contributed to the trial design and data interpretation.
Nick Padfield (Principal Investigator, Pain Management and Neuromodulation Centre, Guys and St Thomas’ Hospital, London) contributed to the trial design, data acquisition, analysis and interpretation.
Ceri Phillips (Health Economics Advisor, Swansea University, Swansea) contributed to the trial design.
Helen Poole (Psychological Advisor, Liverpool John Moores University, Liverpool) contributed to the trial design, and data interpretation.
Mark Saunders (Principal Investigator, Norwich and Norfolk University NHS Trust, Norwich) contributed to the trial design, data acquisition, analysis and interpretation.
Mick Serpell (Principal Investigator, Queen Elizabeth University Hospital, Glasgow) contributed to the trial design, data acquisition, analysis and interpretation.
Nick Shenker (Principal Investigator, Cambridge University Hospitals, Cambridge) contributed to the trial design, data acquisition, analysis and interpretation.
Karim Shoukrey (Principal Investigator, University Hospital of Leicester NHS Trust, Leicester) contributed to the trial design, data acquisition, analysis and interpretation.
Lynne Wyatt (Research Nurse, The Walton Centre NHS Foundation Trust, Liverpool) contributed to data acquisition and interpretation.
Gareth Ambler (Trial Statistician, University College London, London) wrote the first draft, contributed to the trial design leading the statistical aspects, led the data analysis and contributed to the data interpretation.
All authors reviewed, revised and approved the final version of the manuscript.
Publication
Goebel A, Shenker N, Padfield N, Shoudrey K, McCabe C, Serpell M, et al. Low-dose intravenous immunoglobulin treatment for complex regional pain syndrome (LIPS): study protocol for a randomized controlled trial. Trials 2014;15:404.
Data sharing statement
All available data can be obtained by contacting the corresponding author.
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, NETSCC, the EME programme or the Department of Health. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the EME programme or the Department of Health.
References
- Goebel A, Shenker N, Padfield N, Shoudrey K, McCabe C, Serpell M, et al. Low-dose intravenous immunoglobulin treatment for complex regional pain syndrome (LIPS): study protocol for a randomized controlled trial. Trials 2014;15. https://doi.org/10.1186/1745-6215-15-404.
- Goebel A. Complex regional pain syndrome in adults. Rheumatology 2011;50:1739-50. https://doi.org/10.1093/rheumatology/ker202.
- Marinus J, Moseley GL, Birklein F, Baron R, Maihöfner C, Kingery WS, et al. Clinical features and pathophysiology of complex regional pain syndrome. Lancet Neurol 2011;10:637-48. https://doi.org/10.1016/S1474-4422(11)70106-5.
- Forouzanfar T, Köke AJ, van Kleef M, Weber WE. Treatment of complex regional pain syndrome type I. Eur J Pain 2002;6:105-22. https://doi.org/10.1053/eujp.2001.0304.
- Kemler MA, Furnée CA. Economic evaluation of spinal cord stimulation for chronic reflex sympathetic dystrophy. Neurology 2002;59:1203-9. https://doi.org/10.1212/01.WNL.0000028686.74056.E3.
- Goebel A. Immunoglobulin responsive chronic pain. J Clin Immunol 2010;30:103-8. https://doi.org/10.1007/s10875-010-9403-8.
- Goebel A, Netal S, Schedel R, Sprotte G. Human pooled immunoglobulin in the treatment of chronic pain syndromes. Pain Med 2002;3:119-27. https://doi.org/10.1046/j.1526-4637.2002.02018.x.
- Goebel A, Mishbah S, McKiver K, Haynes L, Burton J, Philips C, et al. Immunoglobulin maintenance therapy in longstanding complex regional pain syndrome, an open study. Rheumatology 2013;52:2091-3. https://doi.org/10.1093/rheumatology/ket282.
- Goebel A, Baranowski AP, Maurer K, Ghiai A, McCabe C, Ambler G. Intravenous immunoglobulin treatment of complex regional pain syndrome: a randomized trial. Ann Intern Med 2010;152:152-8. https://doi.org/10.7326/0003-4819-152-3-201002020-00006.
- Dworkin RH, Turk DC, Farrar JT, Haythornthwaite JA, Jensen MP, Katz NP, et al. Core outcome measures for chronic pain clinical trials: IMMPACT recommendations. Pain 2005;113:9-19. https://doi.org/10.1016/j.pain.2004.09.012.
- Brooks R. EuroQol: the current state of play. Health Policy 1996;37:53-72. https://doi.org/10.1016/0168-8510(96)00822-6.
- Tan G, Jensen MP, Thornby JI, Shanti BF. Validation of the Brief Pain Inventory for chronic non-malignant pain. J Pain 2004;5:133-7. https://doi.org/10.1016/j.jpain.2003.12.005.
- Harden RN, Bruehl S, Stanton-Hicks M, Wilson PR. Proposed new diagnostic criteria for complex regional pain syndrome. Pain Med 2007;8:326-31. https://doi.org/10.1111/j.1526-4637.2006.00169.x.
- Dworkin RH, O’Connor AB, Backonja M, Farrar JT, Finnerup NB, Jensen TS, et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain 2007;132:237-51. https://doi.org/10.1016/j.pain.2007.08.033.
- Oerlemans HM, Oostendorp RA, de BT, Goris RJ. Pain and reduced mobility in complex regional pain syndrome I: outcome of a prospective randomised controlled clinical trial of adjuvant physical therapy versus occupational therapy. Pain 1999;83:77-83. https://doi.org/10.1016/S0304-3959(99)00080-9.
- Guideline for Good Clinical Practice. Geneva: ICH; 1996.
- Title 21 – Food and Drugs. Chapter I – Food and Drug Administration. Department of Health and Human Services. Subchapter A – General Part 11 Electronic Records; Electronic Signatures. Silver Spring, MD: US Food and Drug Administration; 1997.
- European Commission Clinical Trial Directive . Medicines for Human Use (Clinical Trials) Regulations 2008 n.d. www.opsi.gov.uk/si/si2004/20041031.htm.
- Herdman M, Gudex C, Lloyd A, Janssen M, Kind P, Parkin D, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res 2011;20:1727-36. https://doi.org/10.1007/s11136-011-9903-x.
- Cappelleri JC, Bushmakin AG, McDermott AM, Sadosky AB, Petrie CD, Martin S. Psychometric properties of a single-item scale to assess sleep quality among individuals with fibromyalgia. Health Qual Life Outcomes 2009;7. https://doi.org/10.1186/1477-7525-7-54.
- Wasan AD, Kong J, Pham LD, Kaptchuk TJ, Edwards R, Gollub RL. The impact of placebo, psychopathology, and expectations on the response to acupuncture needling in patients with chronic low back pain. J Pain 2010;11:555-63. https://doi.org/10.1016/j.jpain.2009.09.013.
- Farrar JT, Young JP, LaMoreaux L, Werth JL, Poole RM. Clinical importance of changes in chronic pain intensity measured on an 11-point numerical pain rating scale. Pain 2001;94:149-58. https://doi.org/10.1016/S0304-3959(01)00349-9.
- Zigmond AS, Snaith RP. The hospital anxiety and depression scale. Acta Psychiatr Scand 1983;67:361-70. https://doi.org/10.1111/j.1600-0447.1983.tb09716.x.
- Dworkin RH, Turk DC, Revicki DA, Harding G, Coyne KS, Peirce-Sandner S, et al. Development and initial validation of an expanded and revised version of the Short-form McGill Pain Questionnaire (SF-MPQ-2). Pain 2009;144:35-42. https://doi.org/10.1016/j.pain.2009.02.007.
- Sullivan LJ, Bishop SR, Pivik J. The Pain catastrophizing scale: development and validation. Psychological Assess 1995;7:524-32. https://doi.org/10.1037/1040-3590.7.4.524.
- Koopman C, Pelletier KR, Murray JF, Sharda CE, Berger ML, Turpin RS, et al. Stanford presenteeism scale: health status and employee productivity. J Occup Environ Med 2002;44:14-20. https://doi.org/10.1097/00043764-200201000-00004.
- Frettlöh J, Hüppe M, Maier C. Severity and specificity of neglect-like symptoms in patients with complex regional pain syndrome (CRPS) compared to chronic limb pain of other origins. Pain 2006;124:184-9. https://doi.org/10.1016/j.pain.2006.04.010.
- Machin D, Campbell MJ, Tan S-B, Tan S-H. Sample Size Tables for Clinical Studies 2008. https://doi.org/10.1002/9781444300710.
- Veldman PH, Reynen HM, Arntz IE, Goris RJ. Signs and symptoms of reflex sympathetic dystrophy: prospective study of 829 patients. Lancet 1993;342:1012-16. https://doi.org/10.1016/0140-6736(93)92877-V.
- Bruehl S, Maihöfner C, Stanton-Hicks M, Perez RS, Vatine JJ, Brunner F, et al. Complex regional pain syndrome: evidence for warm and cold subtypes in a large prospective clinical sample. Pain 2016;157:1674-81. https://doi.org/10.1097/j.pain.0000000000000569.
- Goebel AB, Barker CH, Turner-Stokes L, . Complex Regional Pain Syndrome in Adults. UK Guidelines for Diagnosis, Referral and Management in Primary and Secondary Care. London: Royal College of Psychiatrists; 2012.
- Dirckx M, Groeneweg G, Wesseldijk F, Stronks DL, Huygen FJ. Report of a preliminary discontinued double-blind, randomized, placebo-controlled trial of the anti-TNF-alpha chimeric monoclonal antibody infliximab in complex regional pain syndrome. Pain Practice 2013;13:633-40. https://doi.org/10.1111/papr.12078.
- Munts AG, van der Plas AA, Ferrari MD, Teepe-Twiss IM, Marinus J, Van Hilten JJ. Efficacy and safety of a single intrathecal methylprednisolone bolus in chronic complex regional pain syndrome. Eur J Pain 2010;14:523-8. https://doi.org/10.1016/j.ejpain.2009.11.004.
- Barbalinardo S, Loer SA, Goebel A, Perez RS. The treatment of longstanding complex regional pain syndrome with oral steroids. Pain Med 2016;17:337-43.
- Manning DC, Alexander G, Arezzo JC, Cooper A, Harden RN, Oaklander AL, et al. Lenalidomide for complex regional pain syndrome type 1: lack of efficacy in a phase II randomized study. J Pain 2014;15:1366-76. https://doi.org/10.1016/j.jpain.2014.09.013.
- Tékus V, Hajna Z, Borbély É, Markovics A, Bagoly T, Szolcsányi J, et al. A CRPS-IgG-transfer-trauma model reproducing inflammatory and positive sensory signs associated with complex regional pain syndrome. Pain 2014;155:299-308. https://doi.org/10.1016/j.pain.2013.10.011.
- Kohr D, Singh P, Tschernatsch M, Kaps M, Pouokam E, Diener M, et al. Autoimmunity against the β2 adrenergic receptor and muscarinic-2 receptor in complex regional pain syndrome. Pain 2011;152:2690-700. https://doi.org/10.1016/j.pain.2011.06.012.
- Dubuis E, Thompson V, Leite MI, Blaes F, Maihöfner C, Greensmith D, et al. Longstanding complex regional pain syndrome is associated with activating autoantibodies against alpha-1a adrenoceptors. Pain 2014;155:2408-17. https://doi.org/10.1016/j.pain.2014.09.022.
- Wigerblad G, Bas DB, Fernades-Cerqueira C, Krishnamurthy A, Nandakumar KS, Rogoz K, et al. Autoantibodies to citrullinated proteins induce joint pain independent of inflammation via a chemokine-dependent mechanism. Ann Rheum Dis 2016;75:730-8. https://doi.org/10.1136/annrheumdis-2015-208094.
- Aradillas E, Schwartzman RJ, Grothusen JR, Goebel A, Alexander GM. Plasma exchange therapy in patients with complex regional pain syndrome. Pain Physician 2015;18:383-94.
- Blaes F, Dharmalingam B, Tschernatsch M, Feustel A, Fritz T, Kohr D, et al. Improvement of complex regional pain syndrome after plasmapheresis. Eur J Pain 2015;19:503-7. https://doi.org/10.1002/ejp.572.
- Goebel A, Jones S, Oomman S, Callaghan T, Sprotte G. Treatment of long-standing complex regional pain syndrome with therapeutic plasma exchange: a preliminary case series of patients treated in 2008-2014. Pain Med 2014;15:2163-4. https://doi.org/10.1111/pme.12601.
- Midbari A, Suzan E, Adler T, Melamed E, Norman D, Vulfsons S, et al. Amputation in patients with complex regional pain syndrome: a comparative study between amputees and non-amputees with intractable disease. Bone Joint J 2016;98–B:548-54. https://doi.org/10.1302/0301-620X.98B4.36422.
Appendix 1 Patient information sheet
Appendix 2 Consent form
Appendix 3 Research diagnostic criteria (the ‘Budapest Criteria’) for complex regional pain syndrome
General definition of the syndrome
Complex regional pain syndrome describes an array of painful conditions that are characterised by a continuing (spontaneous and/or evoked) regional pain that is seemingly disproportionate in time or degree to the usual course of any known trauma or other lesion. The pain is regional (not in a specific nerve territory or dermatome) and usually has a distal predominance of abnormal sensory, motor, sudomotor, vasomotor, and/or trophic findings. The syndrome shows variable progression over time.
To make the clinical diagnosis, the following criteria must be met:
-
Continuing pain, which is disproportionate to any inciting event.
-
Must report at least one symptom in all four of the following categories:
-
sensory – reports of hyperaesthesia and/or allodynia
-
vasomotor – reports of temperature asymmetry and/or skin colour changes and/or skin colour asymmetry
-
sudomotor/oedema – reports of oedema and/or sweating changes and/or sweating asymmetry
-
motor/trophic – reports of decreased range of motion and/or motor dysfunction (weakness, tremor, dystonia) and/or trophic changes (hair, nail, skin).
-
-
Must display at least one sign at time of evaluation in two or more of the following categories:
-
sensory – evidence of hyperalgesia (to pinprick) and/or allodynia (to light touch and/or temperature sensation and/or deep somatic pressure and/or joint movement)
-
vasomotor – evidence of temperature asymmetry (> 1 °C) and/or skin colour changes and/or asymmetry
-
sudomotor/oedema – evidence of oedema and/or sweating changes and/or sweating asymmetry
-
motor/trophic – evidence of decreased range of motion and/or motor dysfunction (weakness, tremor, dystonia) and/or trophic changes (hair, nail, skin)
-
-
There is no other diagnosis that better explains the signs and symptoms.
Appendix 4 Weight-determined dosing guide
| Weight (kg) range | Dose (g) to be administered | Kits to be dispensed (ml) | Volume to be administered (ml) | Maximum hourly infusion rate (ml/hour) | |
|---|---|---|---|---|---|
| 100 | 200 | ||||
| 35.5–45.4 | 20 | – | 2 | 400 | 88–113 |
| 45.5–55.4 | 25 | 1 | 2 | 500 | 113–138 |
| 55.5–65.4 | 30 | – | 3 | 600 | 138–163 |
| 65.5–75.4 | 35 | 1 | 3 | 700 | 163–188 |
| 75.5 | 40 | – | 4 | 800 | 188–213 |
| 85.5–95.4 | 45 | 1 | 4 | 900 | 213–238 |
| 95.5–105.4 | 50 | – | 5 | 1000 | 238–263 |
| 105.5–115.4 | 55 | 1 | 5 | 1100 | 263–288 |
| 115.5–125.4 | 60 | – | 6 | 1200 | 288–313 |
| 125.5–135.4 | 65 | 1 | 6 | 1300 | 313–338 |
| 135.5–145.4 | 70 | – | 7 | 1400 | 338–363 |
| 145.5–155.4 | 75 | 1 | 7 | 1500 | 363–388 |
| 155.5–165.4 | 80 | – | 8 | 1600 | 388–413 |
Appendix 5 Patient-recommended scale
| 1. | How much use do you have of your limb overall? | 0 | Full use |
| 1 | |||
| 2 | |||
| 3 | |||
| 4 | |||
| 5 | |||
| 6 | |||
| 7 | |||
| 8 | |||
| 9 | |||
| 10 | No use | ||
| 777 | Not available or not applicable | ||
| 888 | Not done | ||
| 999 | Unknown | ||
| 2. | Can you move your limb__________ than before your treatment? | 0 | Much better |
| 1 | Better | ||
| 2 | Same | ||
| 3 | Less | ||
| 777 | Not available or not applicable | ||
| 888 | Not done | ||
| 999 | Unknown | ||
| 3. | Has lack of energy affected you/interfered with your activities over the last week? | 0 | Not at all |
| 1 | |||
| 2 | |||
| 3 | |||
| 4 | |||
| 5 | |||
| 6 | |||
| 7 | |||
| 8 | |||
| 9 | |||
| 10 | Completely | ||
| 777 | Not available or not applicable | ||
| 888 | Not done | ||
| 999 | Unknown | ||
| 4. | What was your average pain intensity at rest over the last week? | 0 | No pain |
| 1 | |||
| 2 | |||
| 3 | |||
| 4 | |||
| 5 | |||
| 6 | |||
| 7 | |||
| 8 | |||
| 9 | |||
| 10 | Pain as bad as you can imagine | ||
| 777 | Not available or not applicable | ||
| 888 | Not done | ||
| 999 | Unknown | ||
| 5. | Over the last week, has movement of your limb caused pain? | 0 | None |
| 1 | Mild | ||
| 2 | Moderate | ||
| 3 | Severe | ||
| 777 | Not available or not applicable | ||
| 888 | Not done | ||
| 999 | Unknown |
List of abbreviations
- AE
- adverse event
- BPI
- Brief Pain Inventory
- CI
- confidence interval
- CRPS
- complex regional pain syndrome
- eCRF
- electronic case report form
- EME
- Efficacy and Mechanism Evaluation
- EQ-5D
- EuroQol-5 Dimensions
- EQ-5D-5L
- EuroQol-5 Dimensions, five-level version
- IgG
- immunoglobulin G
- IVIg
- intravenous immunoglobulin
- KCT
- King’s Clinical Trials Unit
- MRC
- Medical Research Council
- NIHR
- National Institute for Health Research
- NRS
- Numeric Rating Scale
- QoL
- quality of life
- RCT
- randomised controlled trial
- SAE
- serious adverse event
- TSC
- Trial Steering Committee