

Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 12/66/15. The contractual start date was in July 2014. The final report began editorial review in July 2017 and was accepted for publication in March 2018. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Sobha Sivaprasad reports grants and non-financial support from Bayer HealthCare, Bayer Plc (Reading, UK) during the conduct of the study and has received research grants, travel fees and advisory board honoraria from Bayer HealthCare, Novartis Pharmaceuticals UK Ltd (Frimley, UK), Allergan Ltd (Marlow, UK) and Roche Holding AG (Basel, Switzerland); grants and advisory board honoraria from Boehringer Ingelheim (Ingelheim am Rhein, Germany); and advisory board honoraria from Heidelberg Engineering GmbH (Heidelberg, Germany), outside the submitted work. Philip Hykin reports grants, personal fees and non-financial support from Bayer HealthCare during the conduct of the study.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2018. This work was produced by Sivaprasad et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2018 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
Background
Diabetes mellitus is a major public health problem that affects 415 million people worldwide. 1 Diabetic retinopathy is the most common complication of diabetes mellitus. With the increasing prevalence of diabetes mellitus globally, diabetic retinopathy is emerging as the leading cause of avoidable blindness worldwide. The progression and severity of diabetic retinopathy can be delayed by optimal control of medical risk factors such as hyperglycaemia, hypertension and hyperlipidaemia. 2 However, despite better control of these well-established risk factors, good uptake of established national diabetic retinopathy screening programmes in many countries and improved patient awareness, diabetic retinopathy remains a significant morbidity, indicating the need for alternative management options for this condition. 3–5 The two vision-threatening complications of diabetic retinopathy are diabetic macular oedema (DMO) and proliferative diabetic retinopathy (PDR). DMO is caused by accumulation of excess extracellular fluid in the macula. PDR is characterised by growth of new blood vessels on the retina and, if left untreated, these blood vessels can bleed and scar the retina, causing severe visual loss because of vitreous haemorrhage, retinal detachment and neovascular glaucoma (NVG). Approximately 110,000 people in the UK have PDR and, of these, 14,000 have severe visual loss in both eyes, highlighting the need to address this prevalent public health problem. 5,6
Mechanisms of management of proliferative diabetic retinopathy
Multiple molecular mechanisms are involved in the pathogenesis of diabetic retinopathy. However, all lead to a final common pathway of retinal hypoxia and consequent increased levels of angiogenic growth factors, principally vascular endothelial growth factor (VEGF). 7 So the aim of treatment options for PDR is either to increase the oxygen availability to the retina or to decrease the VEGF levels.
Panretinal laser photocoagulation (PRP) has been the mainstay of treatment for DMO and PDR for > 40 years based on a robust evidence base. 8,9 In PRP, laser burns are applied to the peripheral retinal tissue to destroy the peripheral retina to reduce retinal oxygen consumption. This reduction in hypoxic drive results in decreased growth factor production, principally VEGF, which in turn causes retinal new vessel regression. 10,11 About 60% of patients respond to laser with total regression of new vessels within 3 months. 12 However, many need supplemental PRP and 4.5% require vitrectomy following PRP. 13 Approximately 13% develop visual loss because of the development or worsening of pre-existing macular oedema. 14 In addition, PDR may lead to transient or permanent loss of visual function, including peripheral visual field defects, night vision loss, loss of contrast sensitivity, and progression of visual loss in nearly 5% of individuals despite appropriate treatment. 15 Peripheral visual field loss may have an effect on the overall visual field required for safe driving. A proportion of patients treated with repeated PRP fail the driving standards in the long term. 16,17 There is therefore an unmet need for an alternative treatment option that could either replace or delay the need for laser treatment for PDR.
Recently, intravitreal treatments targeting VEGF, such as aflibercept (Eylea®, Regeneron, Tarrytown, NY, USA/Bayer Pharma AG, Berlin, Germany), ranibizumab (Lucentis®, Genentech, S. San Francisco, CA, USA/Roche, Basel, Switzerland), bevacizumab (Avastin®, Genentech, S. San Francisco, CA, USA/Roche, Basel, Switzerland) and pegaptanib (Macugen®, Eyetech, New York, NY, USA/Pfizer, New York, NY, USA), have introduced a paradigm shift in the treatment of a wide array of ocular diseases, including neovascular age-related macular degeneration, DMO and retinal vein occlusions. Anti-VEGF treatment has superseded laser treatment and is now the standard of care in patients with centre-involving DMO. However, our therapeutic options for PDR remain limited to laser despite several clinical and preclinical studies indicating that VEGF is the key causative factor of retinal neovascularisation (NV). Evidence that VEGF is a key stimulus for ocular NV was demonstrated by the injection of VEGF into the eye of a non-human primate, which stimulated growth and permeability of new vessels on the retina simulating PDR and also induced NVG. 18 There is also clear evidence that hypoxia from decreased retinal perfusion produces VEGF. 19,20 Levels of VEGF messenger ribonucleic acid (mRNA) and protein were shown to be elevated in a manner that is spatially and temporally consistent with the role for VEGF in the growth of new vessels. 21 Moreover, it has been hypothesised that high VEGF from ischaemic retina may be a positive feedback for capillary closure from leucostasis induced by VEGF. 22 The VEGF levels are highest in ocular fluid in patients with PDR than in those with other retinal diseases. 23 Vitreous VEGF levels were also observed with active PDR and decreased in eyes with quiescent PDR or eyes previously treated with PRP. There is therefore a lot of evidence that VEGF plays a critical role in the pathogenesis of PDR. Evidence in support of a direct role of anti-VEGF agents in blocking retinal new vessel growth has also been reported using soluble VEGF receptors, anti-VEGF aptamers and VEGF receptor 1-neutralising antisera. 24 Recent evidence also indicates that monthly anti-VEGF treatment can reduce the severity and delay the progression of diabetic retinopathy over 24 months. 25 Several case series using different anti-VEGF agents have shown that anti-VEGF therapy is effective in causing transient regression of retinal NV in PDR. We conducted a review of literature on this topic before the start of this study and concluded that, although the current evidence points towards the potential for anti-VEGF treatment for PDR to obviate or delay the need for laser treatment, the efficacy, safety and cost-effectiveness of this treatment relative to PRP were unclear. 26
As anti-VEGF has superseded macular laser treatment as the treatment of choice for DMO, it is advantageous for both PDR and DMO to be treated with anti-VEGF agents as doing so will reduce health-care burden, patient burden and potentially improve patient outcomes. At the inception of this study, there were two multicentre trials evaluating the efficacy of ranibizumab in PDR. However, these studies included only treatment-naive eyes with high-risk PDR. This group is less prevalent in the NHS because of prompt referrals and treatment of early PDR thanks to our established screening programmes. The majority of our patients being reviewed in the NHS have eyes that have been previously treated with PRP to evaluate the need for further PRP for persistent or new NV. It was therefore necessary to do a similar study in the UK to assess the benefit of anti-VEGF therapy in our patient cohort with PDR. There are enough pre-clinical and short-term clinical data to support an adequately powered trial to compare the efficacy, safety and cost-effectiveness of anti-VEGF therapy with PRP (standard of care) in PDR.
Choice of anti-vascular endothelial growth factor agent in proliferative diabetic retinopathy
The anti-VEGF agents that are currently available include pegaptanib, ranibizumab, bevacizumab and aflibercept. Although pegaptanib is a selective VEGF-A 165 inhibitor, both ranibizumab and bevacizumab are humanised monoclonal antibodies that inhibit all known isomers of VEGF-A.
Aflibercept (previously VEGF Trap-Eye) is a 115-kD decoy receptor fusion protein. 27 Aflibercept is capable of binding both VEGF and placental growth factor. The receptor sequences of the aflibercept provide powerful VEGF binding (140 times that of ranibizumab) and the molecule’s intermediate size of 110 kD (compared with 48 kD for ranibizumab and 148 kD for bevacizumab) creates a 1-month intravitreal binding activity that exceeds both ranibizumab and bevacizumab. 28 The pivotal Phase III studies that investigated the efficacy and safety of aflibercept in wet age-related macular degeneration (VIEW 1 and 2 trials) showed that monthly and bimonthly aflibercept were non-inferior to monthly ranibizumab at preventing vision loss (< 15-letter loss) with comparable vision gains and safety. 29 Year 2 treatment involved both as-needed treatment and mandatory injections every 3 months and this regimen maintained the vision gains from the first year, with an average of 4.2 injections of aflibercept and 4.7 injections of ranibizumab, suggesting a longer durability of aflibercept over ranibizumab. Aflibercept also has higher binding affinity to VEGF than ranibizumab and bevacizumab. A recently published comparative study of the three intravitreal agents in DMO also showed a more rapid and sustained effect of aflibercept over 2 years. 30 In addition, aflibercept is already licensed for use in DMO based on the VIVID (Intravitreal aflibercept injection in vision impairment due to DME) and VISTA (Study of intravitreal aflibercept injection in patients with diabetic macular edema) studies. These studies also proved the beneficial effect of aflibercept in improving the diabetic retinopathy severity scale. 31 So there is sufficient evidence that aflibercept is as effective and has a longer duration of action than other anti-VEGF agents. Given that PDR is a progressive disease, an agent with a longer duration of action is preferable so we chose aflibercept as the agent of choice for this study. The NHS-discounted cost of aflibercept is similar to the discounted NHS cost of ranibizumab that is available in the public domain.
In summary, based on the existing research at the time of the grant application, we planned to conduct a robust trial with adequate sample size to evaluate the efficacy, safety and cost-effectiveness of aflibercept in a UK patient cohort with PDR over 12 months. In addition, we compared the ocular and systemic effects of this drug with PRP. In the mechanistic substudy, we evaluated the changes induced by aflibercept on retinal new vessels, capillary non-perfusion and retinal intravascular oxygen saturation.
Objectives
To compare the efficacy, safety and cost-effectiveness of intravitreal aflibercept with standard of care, PRP for PDR for 52 weeks, in a Phase IIb randomised active-controlled clinical trial.
Primary objective
To evaluate whether or not mean change in best corrected visual acuity (BCVA) following intravitreal aflibercept therapy is non-inferior to PRP in eyes with PDR at 52 weeks as measured by Early Treatment Diabetic Retinopathy Study (ETDRS) letters.
Secondary objectives
-
To measure the effect of intravitreal aflibercept therapy, relative to PRP, on additional visual function and quality-of-life outcomes, including:
-
uniocular and binocular Esterman visual fields defects
-
binocular visual acuity and low-luminance visual acuity
-
visual acuity outcomes in terms of visual gain or loss
-
contrast sensitivity using Pelli–Robson charts
-
vision-related quality of life measured by the National Eye Institute Visual Function Questionnaire (NEI-VFQ-25) and the Retinopathy-Dependent Quality-of-Life Questionnaire (RetDQoL)
-
diabetic retinopathy treatment satisfaction outcomes using the retinopathy treatment satisfaction questionnaire (RetTSQ)
-
generic health-related quality of life (HRQoL) using the EuroQol-5 Dimensions, three-level version (EQ-5D-3L), and capability using the Investigating Choice Experiments Capability Measure for Adults (ICECAP-A)
-
health and social care service use using a Client Service Receipt Inventory (CSRI).
-
-
To estimate incremental cost-effectiveness of intravitreal aflibercept versus standard PRP treatment at 52 weeks, where effectiveness is measured in terms of BCVA and quality-adjusted life-year (QALY) gain.
-
To determine the proportions of treatment-naive and post-PRP-treated eyes that do not require PRP through 52 weeks after basic treatment of three loading doses of aflibercept in the aflibercept arm and after initial completion of PRP in the PRP arm.
-
To compare the regression pattern at 12 weeks and the regression and reactivation patterns at 52 weeks between arms.
-
To compare the proportion of patients with one-step and three-step improvement or worsening of diabetic retinopathy between treatment arms at 12 and 52 weeks as per schedule of assessment.
-
To explore the difference in safety profile between intravitreal aflibercept and PRP at 52 weeks, in terms of proportion of patients developing macular oedema [defined as a central subfield thickness (CST) of > 300 µm on spectral-domain optical coherence tomography (SD-OCT) because of clinical evidence of macular oedema], any de novo or increase in existing vitreous haemorrhage, de novo or increasing tractional retinal detachment, NVG, and the requirement for vitrectomy. The indication for vitrectomy will be reported.
Objective for substudy on mechanistic evaluation
-
To explore whether or not intravitreal aflibercept compared with PRP causes measurable regression of retinal NV at 12 and 52 weeks in terms of decimal disc area units in four-field colour photographs and fundus fluorescein angiography (FFA).
-
To explore differences in the mean change in retinal vessel calibre and oxygen saturation in eyes treated with intravitreal aflibercept compared with PRP at 12 and 52 weeks.
-
To explore whether or not intravitreal aflibercept reduces angiographically quantifiable areas of retinal non-perfusion compared with PRP through 52 weeks.
Chapter 2 Methods
Study design
This is a multicentre, prospective, individually randomised, single-masked, active-controlled non-inferiority trial with concurrent economic evaluation that compared the clinical efficacy and cost-effectiveness of intravitreal aflibercept versus PRP in patients with treatment-naive PDR or post-PRP active retinal NV at 52 weeks. A subset of the participants also took part in a mechanistic evaluation substudy.
The study was registered on 10 July 2014 (as ISRCTN 32207582). Clinical trial authorisation was given by the Medicines and Healthcare products Regulatory Agency (MHRA): European Clinical Trials Database (EudraCT) 2013-003272-12. Ethics approval was granted by the National Research Ethics Service Committee London – South East; 14/LO/0203. Recruitment commenced in August 2014 and was completed to time and target in November 2015.
The trial protocol is available at www.journalslibrary.nihr.ac.uk/programmes/eme/126615. The trial steering and data monitoring committees provided independent oversight.
Setting and locations
The study was conducted at the 22 NHS clinical sites listed below. The sites were chosen based on previous clinical trial experience or by the estimated volume of potentially eligible patients. Interested sites completed a site feasibility questionnaire.
-
Birmingham and Midland Eye Centre, Sandwell and West Birmingham NHS Foundation Trust, Birmingham.
-
Brighton and Sussex University Hospitals NHS Trust, Brighton.
-
Bristol Eye Hospital, Bristol.
-
Essex County Hospital, Colchester.
-
Frimley Park Hospital NHS Foundation Trust, Surrey.
-
Hillingdon Hospitals NHS Foundation Trust, London.
-
James Paget University Hospital, Great Yarmouth.
-
King’s College Hospital, London.
-
Leicester Royal Infirmary, Leicester.
-
Maidstone & Tunbridge Wells NHS Trust, Kent.
-
Moorfields Eye Hospital NHS Foundation Trust, London.
-
Princess Alexandra Hospital, Harlow.
-
Royal Bolton Hospital NHS Trust, Greater Manchester.
-
Royal Liverpool & Broadgreen University Hospitals NHS Trust, Liverpool.
-
Royal Victoria Hospital and Queen’s University, Belfast.
-
Royal Victoria Infirmary, Newcastle upon Tyne.
-
St James’s University Hospital, Leeds.
-
Sunderland Eye Infirmary, Sunderland.
-
Torbay Hospital, South Devon.
-
University Hospital Southampton NHS Foundation Trust, Southampton.
-
Wolverhampton & Midland Counties Eye Infirmary, Wolverhampton.
-
York Hospital NHS Trust, York.
Participants
Inclusion criteria
-
Subjects of either sex aged ≥ 18 years.
-
Diagnosis of diabetes mellitus (type 1 or type 2).
-
BCVA in the study eye better than or equal to 54 ETDRS letters (Snellen visual acuity 6/18).
-
PDR with no evidence of previous PRP, or the presence of new or persistent retinal NV despite prior PRP that (1) requires treatment in the opinion of the investigator and (2) has sufficient space in the peripheral retina to perform more PRP treatment. In patients with both eye involvement, the eye with no PRP or the least number of PRP burns will be randomised as the study eye. If both eyes have had no previous PRP, the eye with the better visual acuity will be randomised as the study eye.
-
Media clarity, pupillary dilatation and subject co-operation sufficient for adequate fundus photographs. Eyes with mild preretinal haemorrhage or mild vitreous haemorrhage that does not interfere with clear visualisation of the macula and optic disc are considered eligible for this study.
-
Ability to give informed consent.
-
Female subjects should use effective contraception, be post-menopausal for at least 12 months prior to trial entry, or be surgically sterile.
Exclusion criteria
The following exclusions apply to the study eye only (i.e. they may be present for the non-study eye):
-
coexistent ocular disease that may interfere with visual outcome
-
moderate or dense vitreous haemorrhage that prevents clear visualisation of the macula and/or optic disc or prevents PRP treatment
-
significant fibrovascular proliferation or tractional retinal detachment in the posterior pole
-
prior vitrectomy
-
presence of macular oedema at baseline confirmed by SD-OCT as a CST of > 320 µm [SPECTRALIS optical coherence tomography (OCT) (Heidelberg Engineering, Germany)] because of the presence of morphological evidence of diffuse or cystoid oedema
-
other causes of retinal NV
-
iris or angle NV and NVG
-
anticipated need for cataract extraction or vitrectomy within the next 12 months
-
known allergy to fluorescein or any components of aflibercept formulation
-
previous intravitreal anti-VEGF or steroid treatment for DMO in the last 4 months
-
PRP in the last 8 weeks
-
aphakia
-
uncontrolled glaucoma as per investigator’s judgement
-
severe external ocular infection.
Exclusion criteria also apply to systemic conditions as follows:
-
The participant should not have a glycated haemoglobin (HbA1C) level of > 12%. As a precautionary measure, normal health-care providers will be informed if any patient with a HbA1C level of more than 8% is identified using a standard letter, directing the provider to the current National Institute for Health and Care Excellence (NICE) guidelines32 on the management of diabetes mellitus to ensure optimal follow-up.
-
The subject should not have a blood pressure (BP) of > 170/110 mmHg. As a precautionary measure, normal health-care providers will be informed if any patient with a BP of > 150/90 mmHg is identified. A standard letter will be provided directing the provider to the current NICE guidelines on the management of hypertension in patients with diabetes mellitus to ensure optimal follow-up of these patients.
-
A medical condition that, in the opinion of the investigator, would preclude participation in the study.
-
Myocardial infarction, stroke, transient ischaemic attack, acute congestive cardiac failure or any acute coronary event within 6 months of randomisation.
-
Dialysis or renal transplant.
-
Pregnant women.
-
Women of child-bearing potential who do not agree to use effective contraception during the study and for at least 3 months after the study has finished. Effective contraception is defined as one of the following:
-
barrier method – condoms or occlusive cap with spermicides
-
true abstinence – when it is in line with the preferred and usual lifestyle of the subject. Periodic abstinence (e.g. calendar, ovulation, symptothermal, post-ovulation methods) and withdrawal are not acceptable methods of contraception
-
female sterilisation – have had tubal ligation or bilateral oophorectomy (with or without hysterectomy)
-
male partner sterilisation – the vasectomised male partner should be the only partner for the female participant
-
use of established oral, injected or implanted hormonal methods of contraception and intrauterine device
-
Breastfeeding women.
-
-
Males who do not agree to use an effective form of contraception for the duration of the study and for 3 months after the study has finished. Effective contraception is defined as one of the following:
-
Barrier method – condoms or occlusive cap with spermicides.
-
True abstinence – when it is in line with the preferred and usual lifestyle of the subject. Periodic abstinence and withdrawal are not acceptable methods of contraception.
-
Male sterilisation (vasectomy).
-
-
Participation in an investigational trial involving an investigational medicinal product (IMP) within 30 days of randomisation.
Randomisation
Randomisation was completed via a bespoke web-based randomisation system hosted at the King’s Clinical Trials Unit (KCTU) on a secure server (www.ctu.co.uk). Patients were randomised 1 : 1 at the level of the individual using the method of minimisation incorporating a random element. The minimisation factors were PDR status (naive PDR and non-naive PDR), HbA1c level (< 8%, 8–10%, > 10%), diastolic BP (> 90 vs. ≤ 90 mmHg), BCVA (54–69 vs. ≥ 70 letters) and trial site. After informed consent was signed, a patient identification number (PIN) was generated by registering the patient on the MACRO electronic case report form (eCRF) system (InferMed Elsevier Macro v4.0, London, UK) and this PIN was used in the randomisation system. This unique PIN was also recorded on all source data worksheets and used to identify the patient throughout the study. Only authorised staff were allowed to log in to the randomisation system. Once a patient was randomised, the system automatically generated e-mails to key staff within the study. E-mails were also sent to site pharmacies to alert them to a patient’s treatment group – aflibercept or PRP therapy. The pharmacy department used this alert to cross-check the trial prescription to ensure that aflibercept was being dispensed for the correct patient. Additional e-mails were also generated from the randomisation system and sent to key trials staff, with or without treatment allocation information, depending on their role in the study.
Masking
The research optometrists were the primary outcome assessors and they conducted the visual acuity tests at screening, 12 and 52 weeks. They were masked to treatment allocation throughout the study. The optometrists received the participants into the visual acuity lanes with a visual acuity-specific source data worksheet that included the PIN and details of the study eye and non-study eye to be refracted, but with no previous records or case report forms by which the patient’s treatment arm could be identified. The optometrists also assessed the secondary outcome measure of contrast sensitivity and low-luminance visual acuity using the same technique of masking as above. At all other visits, visual acuity examiners were provided with a copy of the refraction log to conduct an open aperture visual acuity test in both eyes with the previous refraction. At these time points, visual acuity tests were also conducted by unmasked professionals and these data were not used for the primary outcome analysis. The other tests of secondary outcome measures, including visual fields and OCT scans, were performed by masked technicians. The technicians received the patients into the visual field and OCT room using the specific source data worksheet that provided details of the patient’s PIN and eye to be examined. After every visit, the completed source data worksheets were kept with the principal investigator’s team. The participants were also advised at enrolment that they must not discuss the study arm they were in with these assessors. The retinal photographs at screening, and at 12 and 52 weeks and FFA at screening and 52 weeks were graded by masked graders in the independent reading centres within the Network of Ophthalmic Reading Centres UK (NetwORC UK). The photographers were trained to take the photographs as per the standard operating procedures (SOPs) for this study. The graders in the Reading Centre were trained and quality assured to grade diabetic retinopathy based on the ETDRS grading system as required for this study. These masking procedures avoided both performance and detection bias.
Intervention
Intravitreal aflibercept (Bayer plc, Reading, UK) dosed at 2 mg/0.05 ml is approved by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) for wet age-related macular degeneration, DMO, retinal vein occlusion related macular oedema and myopic choroidal NV. Study eyes assigned to the aflibercept arm received an intravitreal injection of aflibercept 2 mg/0.05 ml at baseline, 4- and 8-week visits.
Patterns of regression
The treatment response was defined by degree of regression of retinal NV as assessed by colour photographs as per predefined SOP. The categories of regression patterns are summarised in Figure 1.
FIGURE 1.
Classification of patterns of regression. NVD, neovascularisation of the disc; NVE, neovascularisation elsewhere.

Week-12 retreatment
At 12 weeks, the patients were reviewed and categorised into three groups depending on treatment response to the first three injections. Figure 2 shows the retreatment plan at week 12 following first review of regression pattern.
FIGURE 2.
Retreatment plan at week 12 following the first review of regression pattern. NR, no regression; PR, partial regression; TR, total regression. Reproduced from Sivaprasad et al. 33 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) licence, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/.
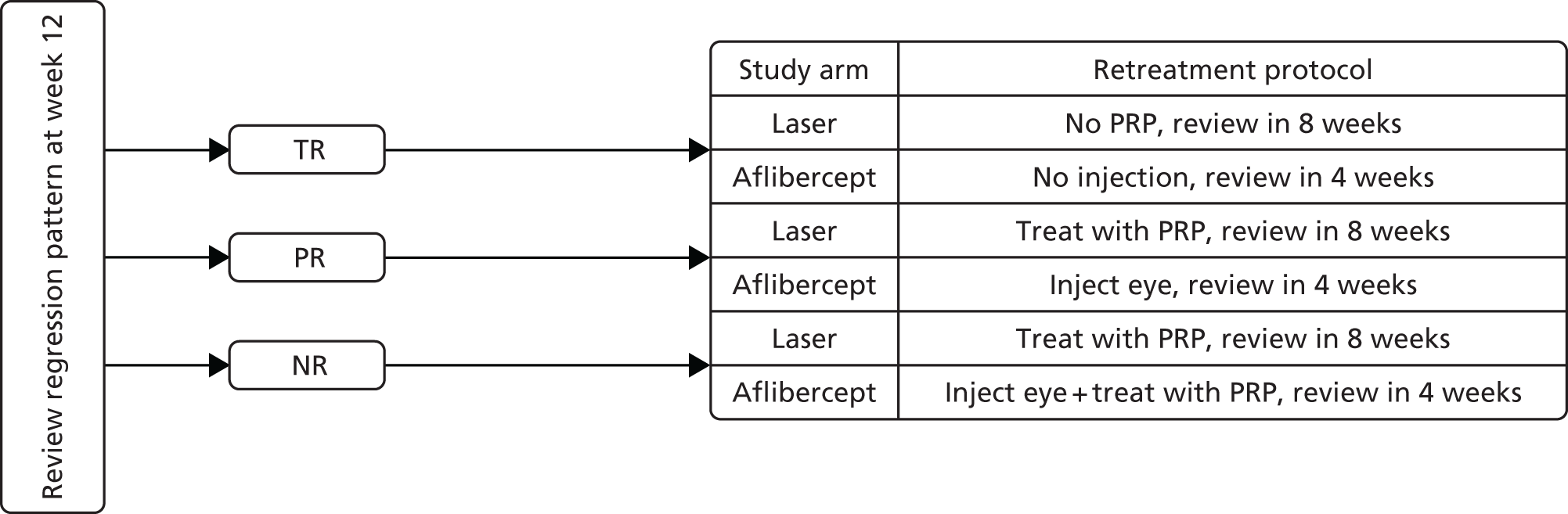
If an eye had experienced adverse effects from prior intravitreal injection, retreatment with intravitreal aflibercept was at the discretion of the investigator. In addition, if any future treatment with aflibercept was contraindicated based on a previous adverse reaction, treatment with PRP for PDR was at the investigator’s discretion after discussion with the chief investigator or his/her designee.
Week-16 to week-48 retreatment
All patients in the aflibercept arm were reviewed on a 4-weekly basis. From week 16, further treatment was determined by both regression and reactivation of NV on clinical examination with adequate visualisation of the entire retina and by comparing the four-field colour photographs or wide-field imaging carried out in the previous visit. The treatment responses were categorised into four groups (no regression, partial regression, reactivation and total regression) and the retreatment was based on the protocols shown in Figure 3.
FIGURE 3.
Summary of retreatment plan from week 16 to 48. NR, no regression; PR, partial regression; RAc, reactivation; RP, regression pattern; TR, total regression. Reproduced from Sivaprasad et al. 33 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) licence, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/.
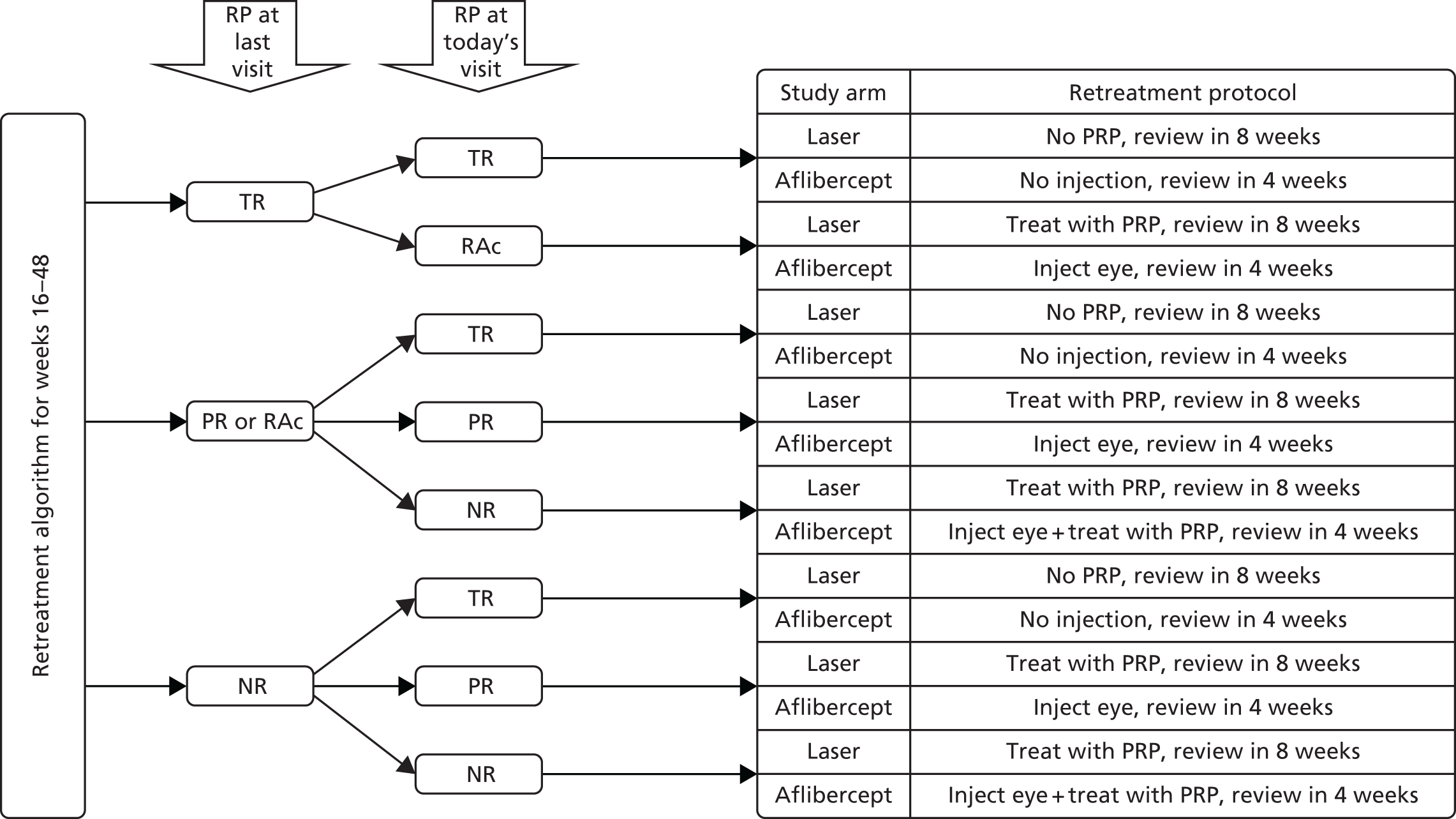
Further fields of colour retinal photographs or FFA could be performed at any visit if there was any doubt that a clinical feature represented retinal NV.
Aflibercept injections were deferred if an eye had experienced adverse effects from prior intravitreal injection. In this circumstance retreatment with intravitreal aflibercept was at the discretion of the investigator. In addition, if any future treatment with aflibercept was contraindicated based on a previous adverse reaction, treatment with PRP for PDR was also at the investigator’s discretion. Treatment with aflibercept or PRP could also be deferred in cases of total vitreous haemorrhage with no clear view of the fundus until the fundus was sufficiently well visualised to permit subsequent intraocular injection, in an eye that developed a rhegmatogenous retinal detachment or required surgical intervention for tractional retinal detachment threatening the fovea. Aflibercept injections could be resumed following surgical repair. Aflibercept injections could also be deferred if the interval between visits was < 4 weeks or if the intraocular pressure remained > 30 mmHg despite apraclonidine (Iopidine 1%) eye drops. In such scenarios, the participant could be prescribed Iopidine eye drops for a week and rescheduled for aflibercept injection within 1 week if the intraocular pressure was < 30 mmHg. PRP was deferred in the aflibercept arm in the ‘no regression’ category if the medium was too hazy to allow PRP or if, in the opinion of the investigator, PRP was not deemed necessary at that visit.
Comparator
The comparator was PRP, the current standard of care. Patients in the PRP group were initiated on PRP, which was completed in fractionated 2-weekly sessions. From week 12, all patients in the PRP arm were assessed for treatment response every 8 weeks and categorised using exactly the same categories as the intervention arm (as shown in Figure 1). Figures 2 and 3 give the summary of further treatment in the PRP arm. PRP treatment could be carried out using any PRP delivery system including indirect PRP. If PRP had to be performed as a day-case it was not recorded as a serious adverse event (SAE) despite hospitalisation of the patient. PRP was deferred in the PRP arm if the medium was too hazy to perform the procedure or if, in the opinion of the investigator, the eye had received adequate PRP and there was insufficient space for further fill-in PRP.
Assessments
The study assessments for the aflibercept arm of the study are shown in Table 1 and for the PRP arm in Table 2.
| Time point | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Screening | Baseline | Week 4 | Week 8 | Week 12 | Week 16 | Week 20 | Week 24 | Week 28 | Week 32 | Week 36 | Week 40 | Week 44 | Week 48 | Week 52 | Withdrawal | |
| Visit window | Day –15 to day 0 | Day 0 | ± 10 days | ± 10 days | ± 10 days | ± 10 days | ± 10 days | ± 10 days | ± 10 days | ± 10 days | ± 10 days | ± 10 days | ± 10 days | ± 10 days | ± 10 days | |
| Visit number | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | |
| Informed consent | ✗ | |||||||||||||||
| Inclusion/exclusion review | ✗ | |||||||||||||||
| Medical and ocular history | ✗ | |||||||||||||||
| Blood test – HbA1Ca level | ✗ | ✗ | ✗ | |||||||||||||
| Pregnancy testb | ✗ | |||||||||||||||
| Standard ophthalmic examination and tonometry | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| Blood pressurec | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||
| ETDRS visual acuity tests in both eyesd | ✗ (+R) | ✗ | ✗ | ✗ | ✗ (+R) | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ (+R) | ✗ (+R) |
| Binocular vision acuity | ✗ | ✗ | ✗ | |||||||||||||
| Low-luminance acuity in both eyes | ✗ | ✗ | ✗ | |||||||||||||
| Pelli Robson contrast sensitivity tests in both eyes | ✗ | ✗ | ✗ | |||||||||||||
| Esterman driving visual fields tests – uniocular (in study eye) and binocular | ✗ | ✗ | ✗ | |||||||||||||
| Questionnaires: NEI-VFQ-25, CSRI, RetTSQ, RetDQoL, EQ-5D-3L, ICECAP-A | ✗ | ✗ | ✗ | |||||||||||||
| SD-OCT in both eyes | ✗ | ✗ | ✗ | ✗ | ||||||||||||
| CFP 7-field or wide-field in study eye onlye | ✗ | ✗ | ✗ | ✗ | ||||||||||||
| FFA in study eye onlye | ✗ | ✗ | ✗ | |||||||||||||
| CFP 4-field or wide-fielde | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||||||
| Concomitant medication review | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| Adverse event review | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| Randomisationf | ✗ | |||||||||||||||
| Review of regression in the study eye | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||||
| Aflibercept injection in study eye only | ✗ | ✗ | ✗ | ✗ ± L (+/–) | ✗ ± L (+/–) | ✗ ± L (+/–) | ✗ ± L (+/–) | ✗ ± L (+/–) | ✗ ± L (+/–) | ✗ ± L (+/–) | ✗ ± L (+/–) | ✗ ± L (+/–) | ✗ ± L (+/–) | |||
| Post-injection checkg | ✗ | ✗ | ✗ | ✗ (+/–) | ✗ (+/–) | ✗ (+/–) | ✗ (+/–) | ✗ (+/–) | ✗ (+/–) | ✗ (+/–) | ✗ (+/–) | ✗ (+/–) | ✗ (+/–) | |||
| Treatment allocation guess formh | ✗ | ✗ | ||||||||||||||
| Study completion formi | ✗ | ✗ | ||||||||||||||
| Mechanistic evaluation substudy (Moorfields Eye Hospital only) | ||||||||||||||||
| Retinal oximetryj both eyes | ✗ | ✗ | ✗ | ✗ | ||||||||||||
| Assessments | Time point | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Screening | Baseline | Week 4 | Week 12 | Week 20 | Week 28 | Week 36 | Week 44 | Week 52 (final visit) | Withdrawal | |
| Visit window | (Day –15 to day 0) | Day 0 | ± 10 days | ± 10 days | ± 10 days | ± 10 days | ± 10 days | ± 10 days | ± 10 days | |
| Visit number | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
| Informed consent | ✗ | |||||||||
| Inclusion/exclusion review | ✗ | |||||||||
| Medical and ocular history | ✗ | |||||||||
| Blood test – HbA1Ca level | ✗ | ✗ | ✗ | |||||||
| Pregnancy testb | ✗ | |||||||||
| Standard ophthalmic examination + tonometry in both eyes | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| Blood pressurec | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||
| ETDRS visual acuity tests in both eyesd | ✗ (+R) | ✗ | ✗ | ✗ (+R) | ✗ | ✗ | ✗ | ✗ | ✗ (+R) | ✗ (+R) |
| Low-luminance visual acuity in both eyes | ✗ | ✗ | ✗ | |||||||
| Binocular vision acuity | ✗ | ✗ | ✗ | |||||||
| Pelli–Robson contrast sensitivity tests in both eyes | ✗ | ✗ | ✗ | |||||||
| Esterman driving visual fields tests – uniocular (study eye) and binocular | ✗ | ✗ | ✗ | |||||||
|
Questionnaires: NEI-VFQ-25, Ret TSQ, RetDQoL, EQ-5D-3L ICECAP-A, and CSRI |
✗ | ✗ | ✗ | |||||||
| SD-OCT macular thickness protocol in both eyes | ✗ | ✗ | ✗ | ✗ | ||||||
| CFP 7-field or wide-field in study eye onlye | ✗ | ✗ | ✗ | ✗ | ||||||
| FFA in study eye onlye | ✗ | ✗ | ✗ | |||||||
| CFP 4-field or wide-fielde in study eye only | ✗ | ✗ | ✗ | ✗ | ||||||
| Concomitant medication review | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| Adverse event review | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| Randomisationf | ✗ | |||||||||
| Review of regression in study eye only | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||
| PRP treatment in study eye only | ✗ | ✗ (+/–) | ✗ (+/–) | ✗ (+/–) | ✗ (+/–) | ✗ (+/–) | ✗ (+/–) | |||
| Treatment allocation guess formg | ✗ | ✗ | ||||||||
| Study completion formh | ✗ | ✗ | ||||||||
| Mechanistic evaluation substudy (Moorfields Eye Hospital only) | ||||||||||
| Retinal oximetry in both eyesi | ✗ | ✗ | ✗ | ✗ | ||||||
The flow of patients in both arms of the study at the initiation of the trial are shown in Figure 4.
FIGURE 4.
Summary of flow of patients at screening/baseline. a, The number of sites was increased to 22 from 17 during the conduct of the trial as shown in the list of protocol amendments (see Table 4). CLARITY, clinical efficacy and mechanistic evaluation of aflibercept for proliferative diabetic retinopathy.
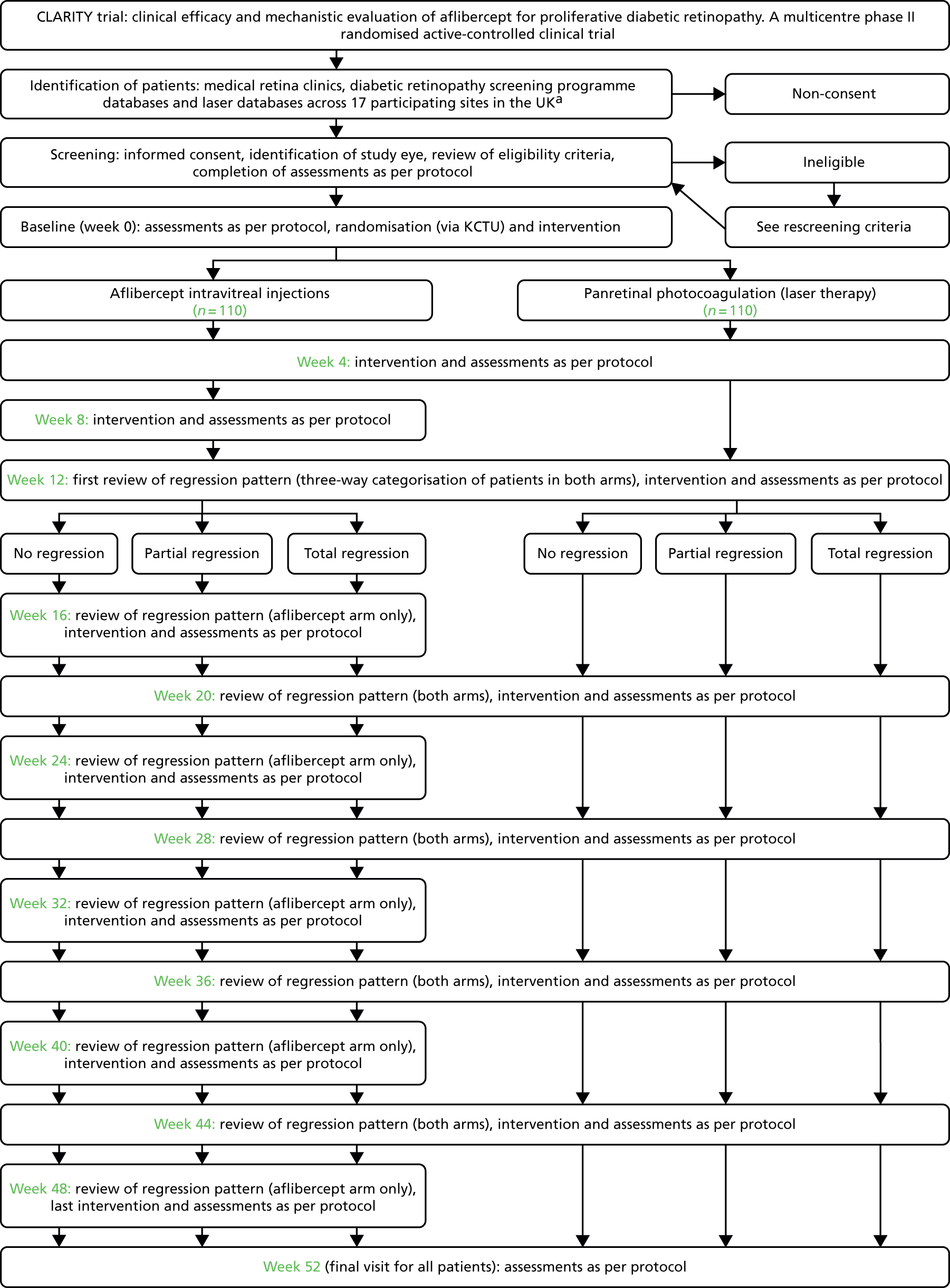
Participant demographics and other baseline characteristics
This information was retrieved from the participant, hospital medical records or the general practitioner. Data included age, sex and ethnic background. Data were also collected on diabetic history and management, ocular history and treatment, other clinically relevant medical history and their management in the last 12 months, and concomitant medication.
Visual acuity tests
The visual acuity tests were performed using the validated ETDRS vision charts in accordance with SOPs. 34,35 Refracted visual acuity was carried out in both eyes at screening, weeks 12 and 52 and at the point of withdrawal. For all other visits, visual acuity was tested with the previous protocol refraction and not included in the primary outcome analysis. Binocular visual acuity and low-luminance acuity were tested at screening and 52 weeks and at the point of withdrawal. The visual acuity scores were recorded in full in the eCRF.
Contrast sensitivity tests
The Pelli–Robson chart was used to test contrast sensitivity in both eyes at screening, week 52 and at the point of withdrawal as per SOP. 36 The total contrast sensitivity scores in both eyes were recorded in the eCRF.
Driving fields tests
Uniocular (study eye) and binocular Esterman fields were carried out at screening, week 52 and at the point of withdrawal. 37
Standard ophthalmic examination
A standard ophthalmic examination using slit-lamp biomicroscopy, tonometry and dilated fundus examination was carried out in both eyes at all visits. Gonioscopy was performed if neovascularisation of the angle (NVA), neovascularisation of the iris (NVI) or NVG was suspected. The grade of diabetic retinopathy and the presence or absence of macular oedema as assessed by the investigator were recorded in the eCRF.
Spectral-domain optical coherence tomography
The CST in both eyes was recorded from SD-OCT thickness map at screening, 12 and 52 weeks and at the point of withdrawal. This test was repeated at any visit at the investigator’s discretion. If treatment of DMO was planned, OCT was carried out for confirmation of DMO and monitoring treatment. Any SD-OCT machine was used for the study but the same model of SD-OCT was used for each individual throughout the period of the study.
Colour fundus photography and fundus fluorescein angiography
Seven-field colour fundus photography (CFP) was performed to assess the severity level of diabetic retinopathy and area of retinal NV at screening, weeks 12 and 52 and at the point of withdrawal. FFA was carried out at screening, week 52 and at the point of withdrawal. Four-field photography or Optos wide-field imaging was performed at all other visits to evaluate regression and reactivation patterns. In the PRP arm, these included visits at weeks 20, 28, 36 and 44 and in the aflibercept arm at weeks 4, 8,16, 20, 24, 28, 32, 36, 40, 44 and 48. Additional fields for colour photographs and FFA may have been performed to determine the presence or absence of NV in either eye at any of these visits as per local practice or investigator discretion. The 7-field photographs performed at screening, weeks 12 and 52 and withdrawal were read by masked graders at the Independent Reading Centres in NetwORC UK.
Blood pressure
Blood pressure was measured at each study visit except baseline and week 4 for both arms and week 8 for the aflibercept arm. Patients were not considered eligible for the study if the BP was recorded as > 170/110 mmHg at screening.
If the BP was > 170/110 mmHg, the patient was allowed to be rescreened, at least 1 month after the last screening visit, if the parameter was brought under control and the other inclusion/exclusion criteria were still met.
If the BP was > 150/90 mmHg but ≤ 170/110, the participant was eligible but the normal health-care provider was informed via a standard letter directing the provider to the NICE guidance of management of BP in patients with diabetes mellitus. All randomised patients continued to be followed up by their normal health-care provider and remained in the study and underwent all study assessments and treatment as per protocol.
Glycated haemoglobin
The blood samples for HbA1c level assessment were processed at local laboratories or in accordance with local practice. The HbA1c assessment was performed at screening and at the final visit. If the test was performed within 3 months of the visit, it was not repeated. At screening, participants who had a HbA1c level of > 12% at the start of the study were not eligible and could be rescreened after 3 months and randomised if the parameter was brought under control and the other inclusion/exclusion criteria were met. Any patients identified with a HbA1c level of > 8% at either screening or final visit were referred for follow-up with their normal health-care provider. A standard letter directing the health-care provider to the NICE guidelines for management of diabetes mellitus was provided. If in the opinion of the principal investigator the result was classed as clinically and significantly abnormal this was recorded as an adverse event and was followed up accordingly.
Questionnaires
The following participant satisfaction, generic health-related and vision-specific quality-of-life questionnaires were administered at screening, week 52 and where necessary at the point of withdrawal: RetTSQ,38 NEI-VFQ-25,39 RetDQoL,40 EQ-5D-3L,41 ICECAP-A42 and CSRI. Please see Chapter 4 for details.
Mechanistic tests (Moorfield’s Eye Hospital only)
Forty participants (20 in each arm) who consented for the mechanistic evaluation underwent oximetry tests in both eyes at baseline, weeks 12 and 52 and at the point of withdrawal. A within-visit flexibility of +10 days was allowed for patients to complete these tests. Please see Chapter 5 for details.
Independent Reading Centres in NetwORC UK
The NetwORC provided each site with a manual giving instructions and guidance on how to acquire and transfer the colour retinal photographs completed at screening, weeks 12 and 52 and at the point of withdrawal. The images were anonymised to study PIN and included the time point at which the image was collected. The images were transferred to the reading centre via compact disc, secure file transfer protocol or another suitably secure medium agreed by the reading centre and the chief investigator. The images were accompanied by a transmittal log that required the patient’s date of birth as an identifier. Sites ensured that all PINs and dates on images complement the information recorded on the transmittal log and that all images were captured, exported and submitted in accordance with the requirements of the study imaging protocol. The reading centre sent reports regularly to the KCTU throughout the study with an overview of what had been received and what was outstanding from each of the sites. The reading centres evaluated the images and the results were transferred to the chief investigator and trial manager in the KCTU. The trial manager transferred the data to the study statistician.
Data collection and management
It was the responsibility of the principal investigator at each site to identify the personnel responsible for data collection and handling, including those who had access to the trial database, and to ensure the completeness of the delegation log. All source documents were prepared in advance by the trial team and these forms were signed by the researcher who completed the assessments. The principal investigator at each site was also responsible for the accuracy of all data entered in the worksheets in accordance with good clinical practice and the Data Protection Act 199843, ensuring that the source data worksheets were filed in a suitably secure location to ensure that source data verification could be undertaken throughout the study. Source data worksheets were reconciled at the end of the trial with the patients’ NHS medical notes in the recruiting centres. During the trial, critical clinical information was written into patients’ medical notes to ensure that informed medical decisions could be made in the absence of the study team. Trial-related clinical letters were copied to the medical notes during the trial.
Randomisation and collection of data on completion at each visit were performed by the unmasked research staff. The randomisation details were e-mailed to the pharmacy, the delegated unmasked researcher, the principal investigator at that site and the UK trial manager. The study data were entered into a study-specific eCRF created in collaboration with the trial statistician and the chief investigator, and maintained by the KCTU using the InferMed MACRO database system hosted on a dedicated secure server within King’s College London [MACRO electronic data capture (EDC) V4 at www.ctu.co.uk]. This system was regulatory compliant, supported real-time data cleaning and reporting, and had data discrepancy functionality and database lock functionality. A series of logic and range checks were built into the system to reduce the possibility of erroneous data being entered. The system also contained an audit trail of all notable events associated with the trial (including inserts, updates and deletions) and this provided a clear and complete audit trail throughout the trial. The trial manager was responsible for providing usernames and passwords to permitted local study personnel. Only those authorised by the trial manager were able to use the system.
Monitoring and site visits
The first site visit was a prerecruitment visit for training of the protocol, adverse event reporting, MACRO and randomisation and data entry training and outline of good clinical practice. The optometrists were certified at Moorfields Eye Hospital. The trial photographers were certified by the Central Angiographic Resource Facility (CARF) or by Optos. The trial manager ensured that all relevant documentation and certifications were complete before a greenlight letter was sent to each site to initiate the study. All procedures were detailed in an operation manual. On-site monitoring visits were conducted routinely; more monitoring visits were arranged if problems were identified (e.g. poor data collection or under-reporting of primary end point data) or when a site requested a visit to discuss specific issues (including data collection, screening patients, recruitment and staff training). The main areas of focus included consent, SAEs, essential documents in study site files, and source data verification on selected outcomes including the primary outcome measure. A monitoring report was prepared after each monitoring visit and the points raised in the monitoring report were then addressed with sites remotely or at the next onsite monitoring visit. A data management plan and a monitoring plan were designed to ensure that the relevant data were monitored methodologically. In addition, the unblinded statisticians performed central statistical monitoring by reviewing event rates, unusual trends and data anomalies.
In addition, all adverse events and SAEs were monitored at each data monitoring committee meeting. All SAEs were also sent to the drug manufacturer (Bayer Plc, Reading, UK) and to the sponsor. The SAE data were collected on paper SAE report forms and faxed to the KCTU. Summary details of SAEs were also transcribed to the adverse event section of the eCRF.
The principal investigator provided an electronic signature for each patient case report form once all queries were resolved and immediately prior to database lock.
Multiple systematic approaches were therefore instituted to ensure that all outcome data were as accurate and complete as possible.
Outcomes
Primary outcomes
The primary outcome was change in BCVA from screening to 52 weeks in the study eye measured in ETDRS letter score at 4 metres: difference in means.
Secondary outcomes
To measure the effect of intravitreal aflibercept therapy, relative to PRP on additional visual function and quality-of-life outcomes including:
-
percentage of uniocular and binocular Esterman efficiency scores at 52 weeks – difference in proportions
-
binocular visual acuity and low-luminance visual acuity at 52 weeks – difference in means
-
visual acuity outcomes in terms of visual gain or loss – difference in proportions
-
contrast sensitivity measured using the Pelli–Robson chart at 52 weeks – difference in means
-
change from baseline in vision-related quality of life measured using NEI-VFQ-25 and RetDQol at 52 weeks – difference in means
-
change from baseline in diabetic RetTSQ scores at 52 weeks – difference in means
-
gain in HRQoL from baseline over the 52 weeks study period – difference in means.
Cost-effectiveness
From a public sector multiagency perspective, covering health and social care services, we assessed the cost-effectiveness of intravitreal aflibercept compared with PRP. The incremental costs and effects of alternative arms were compared in the primary cost-effectiveness analysis, with effectiveness measured in terms of BCVA. We also undertook a secondary exploratory cost–utility analysis (i.e. cost per QALY analysis). Evidence is mixed but points to the EQ-5D-3L not being sufficiently sensitive to be useful in studies of visual impairment. 44,45 Published sources of national unit costs at 2016 price year were used to calculate the total cost of health and social care service use and ophthalmic-related drug use over 52 weeks. 46 The costs of aflibercept injection treatment in the intervention arm and PRP laser treatment in the comparator arm were calculated using published national unit costs from the Department of Health46 and the British National Formulary (BNF)47 at 2016 price year. We employed ‘bootstrapping’ to overcome the skewed data and produced cost-effectiveness acceptability curves (CEACs) to quantify uncertainty. 48
We used sensitivity analysis to explore whether or not the estimated effect (‘change in BCVA score’) and cost of aflibercept injection treatment relative to PRP laser treatment were sensitive to the key variable factor of our analysis – the pricing of aflibercept. In our opinion, the pricing of aflibercept is the most important variable because the NHS Patient Access Scheme (PAS) could provide the drug at a range of prices.
We undertook a subgroup analysis to explore the potential effect of heterogeneity defined in terms of DMO in the fellow eye. The subgroup analysis was performed by the presence of DMO at baseline (no DMO in both eyes vs. DMO presence in at least one eye). In this study, DMO was an exclusion criterion for the study eye so DMO in at least one eye is defined as presence of DMO in the fellow eye only. The incremental costs and effects of alternative arms were reported within each of the categories of the subgroup variable, and the incremental cost-effectiveness ratio (ICER) (i.e. cost per change in BCVA) was calculated for both of these categories of the subgroup variable.
Measurement of other outcomes
From a methodological perspective, we compared the performance of the EQ-5D-3L (a generic, preference-based HRQoL measure) with the vision-specific HRQoL measures (NEI-VFQ-25 and RetDQoL). We included the ICECAP-A measure as an alternative to EQ-5D-3L focusing on capability to see how this measure compared in terms of sensitivity in this patient group.
Anatomical outcomes
-
To compare the regression patterns of new vessels at 12 weeks and the regression and reactivation patterns between treatment arms at 52 weeks: means and proportions.
-
To compare the proportion of patients with one-step and three-step improvement or worsening of diabetic retinopathy between treatment arms at 12 and 52 weeks: difference in proportions. 49
Treatment-related outcomes
To determine the proportions of treatment-naive and post-PRP-treated eyes that do not require PRP through 52 weeks after basic treatment of three loading doses of aflibercept in the aflibercept arm and after initial completion of PRP in the PRP arm: difference in proportions.
Safety profile
To explore the difference in safety profiles between intravitreal aflibercept and PRP at 52 weeks, in terms of proportion of patients developing macular oedema (defined as a CST of > 300 µm on SD-OCT because of clinical evidence of macular oedema), any de novo or increase in existing vitreous haemorrhage, de novo or increasing tractional retinal detachment, NVG, and the requirement for vitrectomy for various indications: difference in proportions.
To determine the systemic safety events including the Anti-Platelet Triallists’ Collaboration (APTC) events between arms. 50
Mechanistic evaluation
-
To explore whether intravitreal aflibercept, compared with PRP, causes measurable regression of retinal NV at 12 and 52 weeks in terms of decimal disc area units in 4-field colour photographs and FFA: difference in means.
-
To explore differences in the mean change in retinal vessel calibre and oxygen saturation in eyes treated with intravitreal aflibercept compared with PRP at 12 and 52 weeks: difference in means.
-
To explore whether intravitreal aflibercept reduces angiographically quantifiable areas of retinal non-perfusion compared with PRP through 52 weeks: means and proportions.
Sample size calculation
The sample size calculation was performed using nQuery Advisor v4.0 software (Statistical Solutions, Saugus, MA, USA). The primary outcome is the change in BCVA ETDRS letter score from baseline to 52 weeks. Based on the objectives of this study and the potential deleterious effects on visual function of PRP, a non-inferiority margin of 5 letters was judged to be clinically acceptable. 17,33,34,51–53 In addition, this margin is less than the lower limit of the 95% confidence interval (CI) for the comparison of PRP with observation. This helped to ensure that aflibercept is superior to observation alone in the event that it was found to be non-inferior to PRP. In the wider patient population, if aflibercept was no more than 5 letters worse than PRP, it would therefore be defined as being non-inferior. The sample size was based on providing a 95% CI for the between-arm difference in mean change in visual acuity that would be sufficiently narrow to detect non-inferiority (by the CI lying entirely above the margin) with high power, while keeping a false declaration of non-inferiority to 5% through use of a statistical test applied at the two-sided 5% level of significance.
The standard deviation (SD) of the change in visual acuity, after adjustment for baseline, was estimated to be 10.3, based on a relevant trial. 54 There was no issue of clustering of outcomes from eyes within subject effects because only one eye per subject was selected for the study.
With 110 patients (one eye per subject) randomised per group (total 220 patients), 182 had to be followed up to 52-week outcome [allowing for 17% dropout or per protocol (PP) exclusion]. This provided 90% power to detect non-inferiority using a two-sided 95% CI from an analysis of covariance (ANCOVA) test with adjustment for baseline visual acuity.
For a continuous secondary outcome, with 182 subjects followed up, we can detect effects of size 0.42 SDs difference between means with 80% power using a two-sided t-test at the 5% significance level. For binary outcomes, we had at least 77% power to detect a difference in proportions of 0.2 using a chi-squared test at the 5% significance level.
Collection and analysis of outcome data
Baseline comparability of randomised groups
Baseline descriptions of participants by treatment and overall were summarised and included age, sex, diabetes mellitus history and management, ocular history and treatment, other clinically relevant medical history and their management in the last 12 months, and concomitant medication. No significance testing was carried out, as any differences found may have been generated by chance and not for hypothesised reasons.
Continuous variables such as contrast sensitivity and macular thickness were summarised using means and SD and/or medians and IQR for variables presenting a skewed distribution. Categorical variables such as proportion of patients with one- and three-step improvement or worsening of diabetic retinopathy were described using numbers and percentages.
Comparison of rates of adherence and follow-up
Compliance rates and attrition rates were compared and reported by arm. High compliance and low attrition rates were anticipated for this study on the basis of previous clinical trial experience.
Analysis covariates
Stratifiers
Covariates were predefined according to the International Conference on Harmonisation (ICH) E9 guideline, which recommends considering factors on which randomisation has been stratified as these factors tend to be predictive of outcome, and therefore the need for adjustment of the minimisation stratifiers. 55 Randomisation stratifiers as shown in Figure 5, as covariates were planned to improve the precision of the estimated treatment effects.
FIGURE 5.
Randomisation stratifiers.
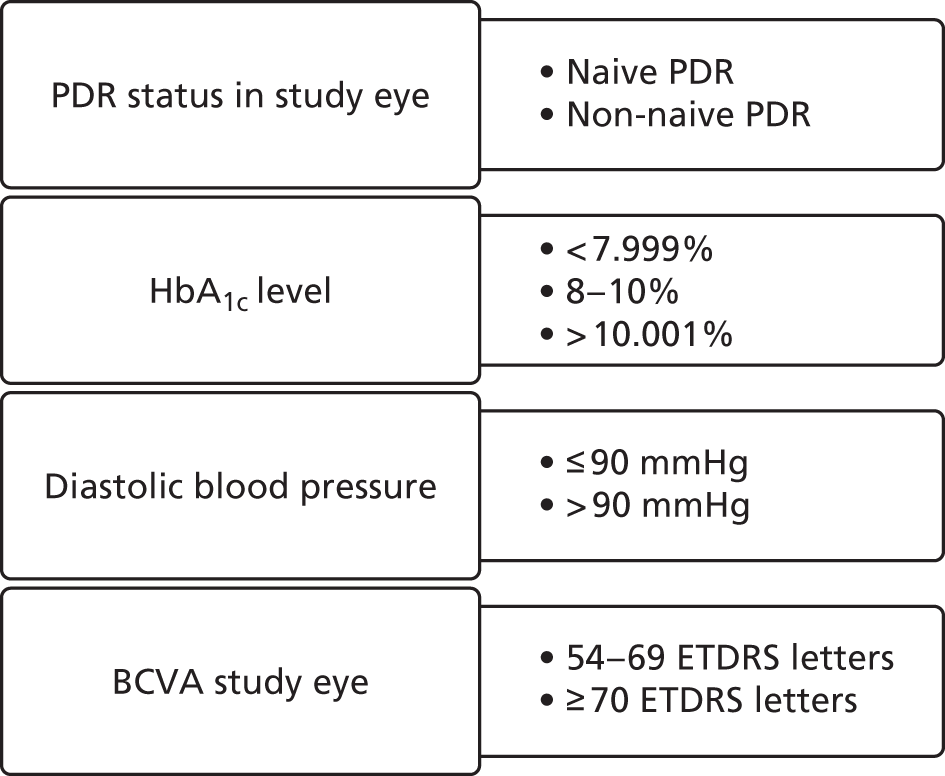
Baseline
The corresponding baseline measure for a continuous outcome is often predictive of the outcome at follow-up and so retinal thickness and BCVA at baseline were included as additional covariates when modelling these continuous outcomes.
Primary outcome analysis
Statistical model
The primary efficacy measure was the change from baseline in refracted BCVA in the study eye, using the ETDRS letter score at 52 weeks. As the analysis approach for continuous outcomes takes advantage of covariate adjustment for the baseline of the outcome, the primary end point can equivalently be regarded to be each participant’s 52-week measurement. This is convenient because then those with a 52-week outcome, but whose baseline measurement is missing, are not regarded to be missing the end point. The primary outcome may therefore be referred to below as the 52-week visual acuity, rather than the change in visual acuity from baseline to 52 weeks.
The primary outcome was analysed using a linear mixed-effects (LME) model, with an unstructured variance–covariance matrix which incorporated the 12-week and 52-week post-baseline measurements of the refracted BCVA outcome. This mixed model had a mix of random- and fixed-effect terms. The random effects in the model were participant, represented as a random intercept at each follow-up time point, allowing for within-participant correlation in the two adjusted post-baseline outcomes. The fixed effects in the model were the main effect terms for arm, baseline PDR status (naive vs. non-naive PDR), HbA1c (< 8%, ≥ 8% to ≤ 10% and > 10%), and diastolic BP (≤ 90 mmHg vs. > 90 mmHg), ‘time’ (12 weeks vs. 52 weeks), the baseline of the outcome and its missing indicator required for the missing indicator method. 56,57 The other fixed effects included in the model were the interactions between ‘time’ and each of the other fixed effects in the model. This model allowed the treatment effect to be estimated at 12 weeks and at the primary time point of 52 weeks, adjusting for time-specific effects of baseline BCVA and of other baseline clinical covariates that were chosen for minimisation stratifiers.
Intention-to-treat strategy
Outcome data were valid and included if the BCVA measure was refracted. All randomised subjects who provide at least one post-baseline valid measurement were included.
The achieved trial sample comprised those study participants who consented to participate and were actually randomised into this trial. This randomised trial sample was also the trial intention-to-treat (ITT) population. The ITT principle states that every subject will be analysed according to the treatment group to which they were randomised. In this trial, subjects’ data were analysed in accordance with Intention to Treat Strategy,58 under which at least one analysis is recommended to be based on the ITT population.
The trial ITT population comprised all randomised participants, regardless of eligibility (inclusion/exclusion) error, post-randomisation withdrawal, and whether the correct study treatments or other interventions were received. On 20 November 2015, The Data Monitoring and Ethics Committee (DMEC) discussed the circumstances under which the BCVA score at either 12 or 52 weeks would not reflect the underlying visual status of a participant. In particular, recent vitreous haemorrhages that might cause low BCVA scores which would then return to normal for the patient, either spontaneously or through appropriate clinical management (vitrectomy), were discussed. The challenge was that any such measurements occurring at week 12 or 52 could artificially induce very large negative changes in BCVA which would have enormous influence in statistical analysis – specifically by leading to very large inflations in the standard deviation for the change from baseline. This could have profound implications for the ability of this non-inferiority trial to achieve its objectives, which rely on the 95% CI for the difference between randomised groups in the change from baseline falling within prespecified bounds (the non-inferiority margin). As such values do not intrinsically reflect the underlying visual status of the patient, the DMEC proposed that the trial steering committee (TSC) consider amending the primary analysis population to exclude from analysis any BCVA measurement at 12 or 52 weeks, that is both > 3SD below the mean at that timepoint (including all measurements) and taken within 3 months of occurrence of a vitreous haemorrhage. Endophthalmitis was also considered as a comparable cause (to vitreous haemorrhage), but, as this is rare and unlikely to occur in a trial of this size, the definition of primary outcome analysis measurements had not encompassed this. The absolute number of measurements excluded was expected to be small.
Per-protocol analysis
For the analysis of the primary outcome, the mixed-effects models were refitted in a reduced PP population, excluding patients found to be ineligible at entry, those patients who received the alternative treatment to that allocated up to the end point, and those not receiving at least the minimal randomised treatment up to and including the 8-week visit (whether because of discontinuation, exclusion or another reason for missing a randomised treatment in this period). This was prior to the point of treatment stratification from the 12-week visit, which included a stratum for patients who required no further treatment.
Rationale
The main reason for having a PP set came from the fact that this was a non-inferiority trial and so the use of the full analysis set is generally not conservative. As Lesaffre59 states, ‘dropouts and a poor conduct of the study might direct the results of the two arms towards each other’. Although this can be interpreted as an indication that the PP analysis is the conservative choice for non-inferiority studies, Garrett60 states that ‘The perceived conservative nature of the PP population appears to be much more a reflection of reduced patient numbers than the presence of bias, while bias can be in either direction depending on the pattern of violations’. Moreover, with two active treatments it may be more likely that any bias affecting both treatments would be reduced in comparison with a placebo-controlled trial.
Prominence
Non-inferiority was planned to be declared only if both ITT and the PP analysis were supportive of a non-inferiority conclusion at 52 weeks. This is supported by the Committee on Proprietary Medical Products61 and in several other papers. 59,62
The requirement to declare non-inferiority in both the ITT and the PP analyses promotes the adherence to treatment protocol and the minimisation of exclusions, maintaining power. Non-inferiority was also assessed secondarily in ITT and PP populations at 12 weeks from the same models. Non-inferiority was declared if the estimated 95% CI for the difference in means was wholly above the margin of –5 letters in both ITT and PP analysis models primarily at 52 weeks and secondarily at 12 weeks.
Superiority
If non-inferiority was concluded, superiority was planned to be assessed from the ITT LME model by reporting the p-value from the two-sided test of the hypothesis of a zero difference in population means using a 5% significance level without need for correction for multiple testing.
Subgroup analysis
Subgroup analysis was performed by baseline retinopathy status (naive and non-naive PDR), HbA1c (< 8%, ≥ 8% to ≤ 10% and > 10%), diastolic BP (≤ 90 mmHg vs. > 90 mmHg) and BCVA (54–69 vs. ≥ 70 letters). The p-value for the between-subgroup comparison of effectiveness was obtained from the ITT LME model from the interaction between arm and the subgroup variable at 52 weeks. The treatment effect and associated 95% CI were reported within each of the categories of the subgroup variable.
Sensitivity to missing data
An expert missing-data group concluded that, rather than statisticians reacting to missing data at the end of a trial, there should be comprehensive, proactive planning for handling missing data at the stage of designing trials. 63 The group recommended that there should be consideration of missing data mechanisms (e.g. missing at random) and, if the missing data may be informative, that appropriate sensitivity analyses should be undertaken to investigate the robustness of the inferences to the different assumptions made by the main analysis. It has also been recommended that analyses allowing for non-response and low intervention uptake (or compliance) are best specified in advance and included in the analysis plan. 64 As it was expected that compliance would be high from the fear of loss of sight, and as non-inferiority is concluded only when declared in both a compliant PP population and a less compliant ITT population, the focus was on the handling of missing data.
The main reasons for dropout in this study were thought to be related to patients’ comorbidities, such as hypertension and hyperlipidaemia, and less connected with underlying visual acuity. Such reasons mean that dropout may not depend on visual acuity and, as data on BP and HbA1c were collected, it was possible to explore the association between these data and dropout to improve the interpretation of the sensitivity analysis results. The primary outcome of refracted BCVA was collected only at two post-baseline measurement points, and there were limited serial data. Nevertheless, as described in Sensitivity analysis to use of concomitant treatments, a sensitivity analysis was undertaken to assess the possibility of alternative plausible values of treatment effect arising from potential mishandling of missing data in the primary analysis model.
The LME model for the primary outcome analysis described above was the first of a two-part approach called the intention-to-treat strategy,64 in which a second analysis examined the sensitivity of the results to missing data in the full randomised ITT population. This met the ideal objectives of ITT. The approach to missing data taken for this study followed the recently published implementation paper of the ITT strategy. 62 The few cases with values that were affected substantially to become unrepresentative temporarily because of vitreous haemorrhage were included among the measurements with missing visual acuity data. This approach was then also applied again to the PP population so that the non-inferiority conclusion could be reassessed under the sensitivity analysis.
For the sensitivity analysis, we prespecified a range for BCVA from –20 to +20 letters over which the mean of the ‘unobserved outcome data’ might depart (or be different) from the mean of the ‘observed outcome data’. 62 In other words, this range can be thought of as how much a typical subject with missing data may on average have had a different estimated treatment effect from the corresponding subject with the outcome data observed (given the same baseline covariates and follow-up data in the LME model). The range (–20 to +20) was chosen to represent both negative and positive departures that could potentially arise as the ‘net effect’ of alternative reasons that may be unknown, such as dropout because of no anticipated further improvement or dropout because of no improvement so far together with no anticipated achievable improvement.
This range of 40 letters (from –20 to +20) was generously wide for exploring sensitivity of the main results to departures from the missing at random (MAR) assumption because 20 letters (as the maximum departure in either direction) is larger than the detectable between-arm treatment effect of 3 lines (15 letters) seen in superiority trials (difference in means), a sizeable shift in the mean of the distribution for dropouts than completers.
At the end of the trial, the fractions of individuals with missing data for visual acuity at 52 weeks were available in each arm: fi (for intervention) and fc (for control). The parameter representing excess visual acuity in those missing compared with those observed, δ, took values by passing across the range –20 to +20. Three scenarios were undertaken within the sensitivity analysis. 61,62 These reflected whether departures from the MAR assumption applied within the intervention arm only (aflibercept), within the comparator arm only (PRP) or within both arms equally and in the same direction (thereby potentially cancelling out across the sensitivity range, if the dropout rate were to be the same in both arms).
Scenario 1: the treatment effect from the LME model will be increased by fiδ.
Scenario 2: the treatment effect from the LME model will be increased by –fcδ.
Scenario 3: the treatment effect from the LME model will be increased by (fi – fc)δ.
Sensitivity analysis to use of concomitant treatments
The use of concomitant treatments was monitored by the DMEC. A sensitivity analysis was not undertaken to examine the robustness of the 52-week PP analysis to the use of concomitant treatments as there were none.
Sensitivity analysis to adjust for site in the primary outcome model
Sites were added as a covariate in the primary outcome analysis model.
Secondary outcome analysis
Analysis of continuous outcomes
The analyses of continuous secondary outcomes were compared between arms at 52 weeks using ANCOVA or the LME model, as the primary outcome, adjusting for baseline PDR status (naive PDR and non-naive PDR), HbA1c (< 8%, ≥ 8% to ≤ 10% and > 10%), diastolic BP (≤ 90 mmHg vs. > 90 mmHg), and BCVA (54–69 vs. ≥ 70 letters), and where collected, the baseline of BCVA with the associated missing indicator. ‘Time’ (12 weeks vs. 52 weeks) was represented as a categorical contrast in main effect form and in interaction with all other fixed effects. For those outcomes where there were no post-baseline measures taken on the outcome before 52 weeks, ANCOVA was used instead.
Analysis of binary outcomes
For the binary outcomes, such as the proportion of patients with a three-step improvement of diabetic retinopathy, chi-squared tests were used. Safety outcomes were also reported as unadjusted patient proportions and rates within and between arms with 95% CIs using exact methods where appropriate (Wilson’s method with no continuity correction). Furthermore, CST and macular volume were corrected in the analysis plan to the use of the LME model instead of ANCOVA as there were two time points (12 weeks and 52 weeks) to take into account. In a DMEC meeting on 14 October 2016, it was decided that the number of patients with, and number of events of, vitreous haemorrhage in the study eye and in the non-study eye would be analysed and reported as a separate table from the adverse events tables.
There was a list of outcomes that required derivations:
-
The NEI-VFQ-25 questionnaire is a validated tool for vision-related quality of life. It consists of a base set of 25 vision-targeted questions representing 11 vision-related subscales, plus an additional single-item general health rating question. The overall composite score is computed as the simple average of the vision-targeted subscale scores, excluding the general health rating question. The overall score can range from 0 (worst possible score) to 100 (best).
-
The RetTSQ is a diabetic retinopathy treatment satisfaction questionnaire consisting of 13 items asking participants to rate different aspects of treatment. It can be scored as total score or as two subscales (one covering negative experiences and the other one positive aspects of treatment). The total score for treatment satisfaction was calculated by summing the scores for items 1 to 13, with a possible range of 0 to 78, where higher scores represent more satisfaction. For the positive and negative subscales, the possible ranges are from 0 to 42 and from 0 to 36, respectively.
-
The RetDQoL is a questionnaire designed to measure individualised quality of life in people with diabetic retinopathy. It consists of two overview items using a 7-point scale and 26 domain-specific items. The first overview item asks participants to complete the statement ‘In general, my present quality of life is – ’. Answers to this question range from ‘excellent,’ scored as 3, through ‘neither good nor bad,’ scored as 0, to ‘extremely bad,’ scored as –3. The second item asks how quality of life is affected by diabetic eye problems; answers include ‘very much better’ (scored –3), ‘much better’ (–2), ‘better’ (–1), ‘the same’ (0) and ‘worse’ (1). For the domain-specific items, the participant indicates where items are not applicable to them and, for the items that are applicable, they first rate the impact of diabetic eye problems on each aspect of life and then rate the importance of each aspect of life to their quality of life. The impact and importance ratings for each applicable item are then multiplied to obtain a weighted impact score with a range from –9 to 3. A more negative average weighted impact (AWI) score indicates a more negative impact of diabetic retinopathy on quality of life; a positive score would indicate a positive impact of diabetic retinopathy on quality of life. A total score, the AWI, can be obtained by summing the weighted impact scores of all applicable domain-specific items and dividing the result by the number of applicable domains.
-
The EQ-5D-3L is a generic HRQoL measure that consists of two pages. The EQ-5D-3L descriptive system comprises five dimensions (mobility, self-care, usual activities, pain/discomfort, anxiety/depression) and the EuroQol Visual Analogue Scale (EQ VAS), which records the respondent’s self-rated health on a vertical visual analogue scale. Each of the five dimensions has three levels (no problems, some problems and extreme problems). The health state preference values (utilities) for EQ-5D-3L profiles were based on time trade-off valuations by members of the UK general public.
-
The ICECAP-A42 is a brief questionnaire that measures the ability of an individual to carry out activities and is a measure of adult capability. The original website has Stata® (StataCorp LP, College Station, TX, USA) code that was used for the scoring of the scale. The questionnaire consists of only five items classified from 1 to 4.
If there was existing syntax code to derive a variable within the KCTU then this was used. Otherwise new code was developed by the trial statistician and verified by the senior statistician.
Missing items in scale and subscales
The number (%) of patients with complete data for each scale was reported. If scales provide, missing value guidance was used.
Use of data transformation
It was not anticipated that any continuous outcomes would need to be considered for transformation, because the sample size was reasonably large for group comparisons in the main trial analyses. Assumptions of normality and constant variance required by the models were examined using residual and other diagnostic plots. If relevant, and necessary (e.g. in the mechanistic evaluation where sample size is reduced) a log-transformation could be considered, because this retains a sensible interpretation for inferences in relative terms between arms. If an absolute interpretation was needed then data transformation would not be undertaken; instead, a non-parametric bootstrap method for obtaining CIs was considered. 65 For the mechanistic evaluation, non-parametric methods were also considered.
Defining outliers
Outliers are observations that have extreme values relative to other observations detected under the same conditions. An outlier was defined here as a data point at least 4 SDs from the mean of its distribution of values observed across other patients. This definition was applied to the transformed scale for those outcomes that were log-transformed.
A ‘bivariate outlier’ was defined here as the difference between successive serial data points of the same measure being at least 4 SDs from the mean of these differences. Simple plots of successive pairs of serial measures were used through the 24-month period to assist in identifying outliers.
Handling outliers
Outliers were identified for further investigation by looking at the distributions of the data through histograms, scatterplots or box plots. Univariate tests for the compatibility of the distribution with a normal distribution were not undertaken since they can be too sensitive to departures that are often not relevant for the comparison of means (the central limit theorem).
Once an outlier was found, a blinded member of the team with sufficient clinical experience was involved in the decision as to whether a data value was impossible versus implausible versus plausible. If the outlier was impossible it would be set to missing and a list of these occurrences then appended to the statistical analysis plan. If an outlier was clinically plausible, the outlier remained. If an outlier was clinically implausible (but possible), it was not ignored or deleted and was retained for ITT analysis.
If outliers remained in the distribution of a variable then data transformations or non-parametric methods of analysis were considered.
Sensitivity analyses were undertaken to check whether the outlier was influential by obtaining results with and then without the inclusion of the outlier. If the conclusions were changed, these were documented.
Analysis methods for secondary outcomes
All study analyses were based on tests that are two-sided, including the two-sided 95% CIs assessed at the 5% significance level. Table 3 shows the method of analysis chosen for each secondary measure.
| Types of variables | Outcomes | Methods |
|---|---|---|
| Continuous | ||
| BCVA measured in ETDRS letter score at 12 weeks | LME model | |
| Binocular visual acuity at 52 weeks | ANCOVA | |
| Low-luminance deficit score at 52 weeks | ANCOVA | |
| Contrast sensitivity measured using Pelli–Robson chart at 52 weeks | ANCOVA | |
| Percentage of unilateral and binocular Esterman efficiency score at 52 weeks | ANCOVA | |
| Diabetic retinopathy treatment satisfaction questionnaire (RetTSQ) at 52 weeks | ANCOVA | |
| Vision-related quality of life measured using NEI-VFQ-25 and RetDQoL at 52 weeks | ANCOVA | |
| HRQoL (QALYs derived using the EQ-5D-3L index scores, ICECAP-A index change scores and total costs derived using the CSRI) at 52 weeks | Bootstrapping | |
| Central subfield thickness measured at 12 and 52 weeks | LME model | |
| Macular volume measured at 12 and 52 weeks | LME model | |
| Categorical | ||
| Visual acuity outcomes in terms of visual gain or loss at 52 weeks | Chi-squared test | |
| One-step, two-step or three-step or more levels change of diabetic retinopathy at 12 and 52 weeks. (Those participants who are close to the floor or ceiling of the scale will not be physically able to improve or worsen by certain amount) | Chi-squared test | |
| Regression pattern of new vessels at 12 weeks and the regression and reactivation patterns of retinal NV on colour photographs and fluorescein angiography at 52 weeks | Chi-squared test | |
| Participants not requiring supplemental panretinal photocoagulation in naive PDR patients at 52 weeks | Chi-squared test | |
| Participants not requiring supplemental panretinal photocoagulation in post-treatment PRP patients at 52 weeks | Chi-squared test | |
| Participants developing macular oedema, any de novo or increase in existing vitreous haemorrhage, de novo or increasing tractional retinal detachment, NVG and requirement for vitrectomy at 12 weeks and 52 weeks | Chi-squared test | |
Handling multiple comparisons
Significance tests were used sparingly and were restricted, where possible, to addressing stated hypotheses. Secondary outcomes, as well as the primary outcome, were summarised using an effect size with a 95% CI. Interpretation for those secondary outcomes that did not directly address the stated study hypotheses was more cautious.
Ethics approval and monitoring
The study was granted approval by the National Research Ethics Committee Service London – South East (14/LO/0203). Clinical Trials Authorisation was given by the MHRA (11518/0013/001-0001) and the European Union Drug Regulating Authorities Clinical Trials (EudraCT) number was 2013-003272-12. The trial was run using the SOPs of the sponsor, Moorfields Eye Hospital NHS Foundation Trust. The sponsor provided the oversight of the study and the KCTU collaborated with the sponsor to ensure efficient delivery of the study. The trial had two committees overseeing its conduct: the TSC and the Trial Management Group (TMG). In addition, there was an independent DMEC to ensure the safety of patients in the trial and review operational issues such as recruitment. The membership of the DMEC is provided in the Acknowledgements.
The DMEC was the only group to review interim analyses broken down by treatment groups during recruitment and follow-up of patients in the trial. The DMEC performed interim safety analyses. The interim reports contained details of patient recruitment, demographic and baseline characteristics, the intervention, primary safety end points, the primary efficacy end point and other end points identified by the DMEC including adverse events and SAEs.
The TSC consisted of four independent members, two lay members and key members of the TMG, in addition to clinical and methodological experts. The membership of the TSC is provided in the Acknowledgements. The TSC had overall responsibility for the scientific integrity and quality of the trial. This involved conducting the trial to the standards set out in the guidelines for good clinical practice, adhering to the protocol as far as possible and holding responsibility for overall patient safety, as well as considering new relevant information as it arose throughout the duration of the trial. The TSC was also responsible for considering any recommendations made by the DMEC. The TSC met throughout the trial to monitor the progress and qualities of the trial (review the recruitment rate and consider protocol amendments). The TMG was responsible for the day-to-day running of the trial, meeting fortnightly during the setting up of the clinical efficacy and mechanistic evaluation of aflibercept for proliferative diabetic retinopathy (CLARITY) trial and the early stages of recruitment and then approximately monthly for the remainder of the trial. This group consisted of the chief investigator, co-lead, trial manager, research fellow, statisticians, sponsor representative, data manager and the KCTU operations manager. The role of the TMG was to monitor the conduct and ensure progress of the trial according to the study protocol and to take appropriate action to safeguard participants and the trial itself.
Patient and public involvement
The study design, patient information sheets and consent forms for the main trial and mechanistic substudy were discussed with the North East London Diabetes Research Network patient and public involvement group, who provided feedback and support for the study. A user representative was a grant co-applicant and two other user representatives were members of the TSC and as part of this role made suggestions for the use of laser instead of PRP to describe the comparator.
Reporting
The trial was reported in accordance with the Consolidated Standards of Reporting Trials (CONSORT) statement.
Summary of changes made to protocol
Table 4 summarises the changes made to the protocol. In addition, the statistical analysis plan was amended after publication of the protocol. Version 5.0 was amended to version 5.1 as a result of the DMEC meeting held on 3 November 2015, in open session, and the DMEC recommendation to the TSC, accepted at the TSC meeting on 20 November 2015.
| Amendment code/number | Classification (as confirmed by sponsor) | Brief description/purpose of amendment | Category A/B/C (for NHS R&D purposes) | Details of documents submitted, version number and date | ||
|---|---|---|---|---|---|---|
| Substantial | Non-substantial/minor clarification | New | Revised | |||
| SA1 | ⊠ | □ | Labelling of IMP | Royal Free Label Approval Form V3 (31 July 2014) | Notice of Substantial Amendment V1 | |
| SA2 | ⊠ | □ | Wording | Letters of invitation to participant V1 (10 October 2014) |
|
|
| SA3 | ⊠ | □ | Addition of following five sites, as main NHS sites:
|
Notice of Substantial Amendment V3 | ||
| SA4 | ⊠ | □ | The inclusion of qualified trained injectors, for example nurse to work on the study |
|
||
| MA1 | □ | ⊠ | Wording of CSRI Questionnaire p. 25 | C | CLARITY questionnaire – V2 | |
The TSC requested confirmation that vitreous haemorrhage will be reported as an outcome by arm, and this has been confirmed to be present in Table 3; the number of measurements excluded from the analysis, by arm, will be reported.
Version 5.1 of the protocol was amended to version 5.2 as a result of the DMEC meeting held on 4 October 2016.
It was decided that study site should be taken out of the primary and secondary outcome LME and ANCOVA models as a covariate since the models would be parameter-heavy with as many as 22 sites having recruited, and where some sites have only included one or a few patients who, at follow-up, could contribute to estimating site rather than to treatment effect. For transparency, sites will then be added as a covariate in a sensitivity analysis. In response to a query by the principal investigator, it was clarified, in the open session of the DMEC meeting, that the actual categorised baseline values of stratifying covariates, rather than those used in the randomisation, which included errors, will be used in the outcome models, so that any baseline confounding by these is more fully adjusted for, and analyses are consistent with subgroup analyses using the same categorisations of these covariates. There was agreement for this approach as the trial employs minimisation. The outcome ‘A small number of summary measures will be calculated and reported to represent the pattern and frequency of randomised treatment received over time’ was added as a process outcome.
Chapter 3 Clinical results
Between August 2014 and November 2015, 290 patients were assessed for eligibility and 232 randomly assigned to receive intravitreal aflibercept (n = 116) or PRP (n = 116).
The trial was conducted in 22 NHS trusts. Recruitment was completed ahead of schedule in 17 months. Table 5 shows the total number of participants recruited from each site and recruitment by calendar month.
| Site | Year | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2014 | 2015 | Site total | |||||||||||||||
| August | September | October | November | December | January | February | March | April | May | June | July | August | September | October | November | ||
| Wolverhampton | – | – | – | – | – | – | – | – | – | – | – | 1 | 0 | 1 | 0 | 2 | 4 |
| Hillingdon Hospitals, London | – | – | – | – | – | – | – | 2 | 1 | 2 | 4 | 4 | 3 | 3 | 0 | 0 | 19 |
| Liverpool | – | – | – | – | – | 3 | 0 | 0 | 0 | 2 | 2 | 1 | 2 | 2 | 0 | 0 | 12 |
| Belfast | – | – | – | – | – | – | – | – | – | 4 | 0 | 0 | 2 | 0 | 1 | 2 | 9 |
| Moorfields Eye Hospital, London | 1 | 0 | 3 | 0 | 2 | 3 | 4 | 3 | 3 | 1 | 1 | 8 | 1 | 2 | 7 | 1 | 40 |
| Kent | – | – | – | – | – | – | – | – | – | – | – | 3 | 0 | 3 | 1 | 2 | 9 |
| Newcastle upon Tyne | – | – | – | 2 | 1 | 0 | 1 | 2 | 0 | 1 | 1 | 0 | 1 | 1 | 3 | 0 | 13 |
| Great Yarmouth | – | – | – | – | 1 | 0 | 1 | 1 | 1 | 0 | 2 | 2 | 0 | 3 | 0 | 1 | 12 |
| Colchester | – | – | 2 | 2 | 2 | 0 | 0 | 2 | 1 | 0 | 1 | 1 | 1 | 1 | 0 | 0 | 13 |
| Sunderland | – | – | – | 4 | 2 | 1 | 2 | 1 | 2 | 1 | 2 | 2 | 0 | 0 | 0 | 1 | 18 |
| Bristol | – | – | 1 | 1 | 0 | 1 | 1 | 2 | 0 | 0 | 0 | 1 | 1 | 1 | 3 | 1 | 13 |
| Leeds | – | 1 | 1 | 0 | 1 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 2 | 0 | 2 | 12 |
| Surrey | – | – | – | – | 1 | 1 | 2 | 1 | 2 | 0 | 1 | 2 | 0 | 0 | 0 | 3 | 13 |
| Southampton | – | – | – | – | – | – | 1 | 0 | 1 | 3 | 2 | 1 | 0 | 0 | 1 | 0 | 9 |
| Harlow | – | – | – | – | – | – | – | – | – | – | – | – | – | 1 | 3 | 3 | 7 |
| Birmingham | – | – | – | – | – | – | – | – | – | – | – | – | – | 3 | 1 | 2 | 6 |
| Brighton | – | – | – | – | – | – | – | – | – | – | 2 | 1 | 1 | 0 | 0 | 0 | 4 |
| King’s College Hospital | – | – | – | – | – | – | – | – | – | – | – | – | 4 | 2 | 1 | 1 | 8 |
| Leicester | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | 1 | 1 |
| York | – | – | – | – | – | – | – | – | – | – | – | 2 | 2 | 1 | 1 | 0 | 6 |
| Bolton | – | – | – | – | – | – | – | – | – | – | – | – | – | – | 1 | 2 | 3 |
| South Devon | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | 1 | 1 |
| Total | 1 | 1 | 7 | 9 | 10 | 10 | 12 | 15 | 12 | 14 | 18 | 30 | 19 | 26 | 23 | 25 | 232 |
| Total cumulative | 1 | 2 | 9 | 18 | 28 | 38 | 50 | 65 | 77 | 91 | 109 | 139 | 158 | 184 | 207 | 232 | |
| No sites having randomised | 1 | 2 | 4 | 6 | 8 | 9 | 10 | 11 | 11 | 12 | 13 | 16 | 17 | 19 | 20 | 22 | |
The actual recruitment period was on target to the planned original target of 18 months. Figure 6 shows the actual monthly recruitment compared with the original target.
FIGURE 6.
Cumulative monthly accrual of patients into the CLARITY trial.
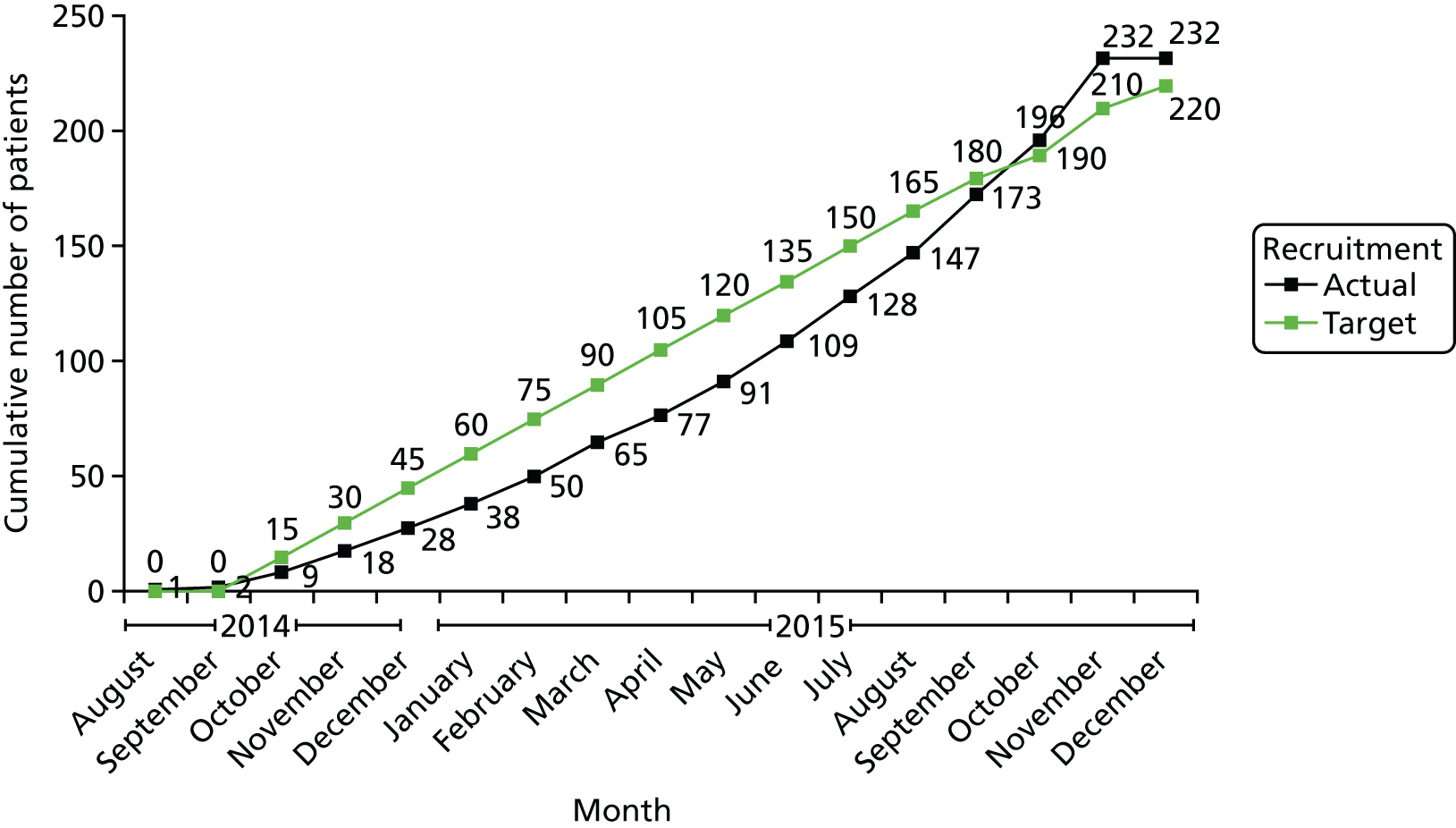
Trial milestones
Study start date was August 2014 and was planned to involve 17 recruiting sites around the UK. The sample size was 220 participants. The eligible patients were identified from the medical retina clinics. New patients were also referred in from the diabetic retinopathy screening programmes. As these patients had to be seen in retinal clinics within 2 weeks of referral and treated within 4 weeks, it was necessary for the trial team to communicate well with the referral team to ensure that these patients were identified before they were treated with PRP in the clinic.
Success in trial recruitment and management
The National Institute for Health Research (NIHR) recognises the need for good trial management and recommends that a clinical trials unit collaborate in the conduct of the study. A multidisciplinary team at the KCTU was funded for the study. As a collaborator on the CLARITY study, the unit ensured the appointment of a dedicated trial manager – a crucial step for the success of this study. The KCTU also has a trial methodologist, data manager, operations manager and statisticians who met with the chief investigator, co-lead and the trial manager for initial brainstorming sessions and then on a regular basis to ensure the smooth running of the study. This group approach and shared ownership were very useful in the successful delivery of the study. During the early part of the set-up phase of the study, the team met often to develop the protocol, obtain all the regulatory approvals and prepare the IMP delivery system. The study source documents and case report forms were user-friendly and we paid significant attention to detail in order to capture all of the required outcome measures. Following the approvals, meetings focused on targeting and solving barriers to site set-up, identifying and managing recruitment hurdles and monitoring recruitment. During these meetings, we evaluated the reasons for initial delay in recruitment.
The start-up delays at some sites were associated with the following:
-
delay in completion of training and certification of the staff for the study
-
IT system delay
-
lack of approved devices to do the study assessments
-
delay in site approval process
-
patients being treated with PRP at first attendance itself to maintain national time to treatment target
-
patients referred from diabetic retinopathy screening programmes not being targeted
-
other retinal consultants preferred standard of care to intervention
-
research team and clinical team were not at the same area and so patients were being missed.
Recruitment strategies used
The target recruitment rate for the study was one to two patients per month per centre, based on the original 17 centres recruiting and a target recruitment figure of 220. We decided to open five further recruiting centres to counteract initial slow recruitment. We made no changes to the inclusion criteria and the recruitment time was not extended.
The trial management meetings enabled us to pay attention to the trial pathway and ensured that all barriers were tackled in time as we progressed. The trial manager and the chief investigator were always available to provide guidance and support to the clinical sites. The sites were chosen based on previous clinical trial experience or by the estimated volume of potentially eligible patients. Interested sites completed a site feasibility questionnaire. Some sites required support from the UK Clinical Research Network to be able to recruit patients for the study.
Recruitment was monitored closely. We sent out a recruitment league table in a newsletter to all sites every month initially so that the sites were aware of the progress of the study. The chief investigator also e-mailed or telephoned the principal investigators of clinical sites that were recruiting below target. The trial manager had to travel to all sites to monitor the study at the sites.
Screening and eligibility
A total of 290 patients were assessed for eligibility. Of these, 58 were excluded either because they did not meet the inclusion/exclusion criteria (n = 51) or because they were not eligible or not keen (n = 7). The remaining eligible patients (n = 232) were randomised into the trial.
Randomisation
A summary of the recruitment and randomisation across the 22 sites has already been detailed in Table 5. Randomisation was balanced across the treatment groups and hospital sites, and within strata.
Withdrawals
Table 6 shows the numbers of participants who did not complete week 52 in both arms. In total, 21 out of 232 (9.1%) did not complete week 52: 9 from the aflibercept arm and 12 from the PRP arm. Table 7 shows the time to withdrawal and reason for withdrawal, for all patients. The withdrawals were requested by the participants themselves. The withdrawals were balanced between arms.
| Visit | Treatment arm | Total | |
|---|---|---|---|
| Aflibercept | PRP | ||
| Baseline | 2a | 6 | 8 |
| 4 weeks | 1 | 1 | 2 |
| 8 weeks | 1 | 0 | 1 |
| 12 weeks | 0 | 1 | 1 |
| 16 weeks | 1 | 0 | 1 |
| 20 weeks | 0 | 0 | 0 |
| 24 weeks | 2 | 0 | 2 |
| 28 weeks | 1 | 1 | 2 |
| 32 weeks | 1 | 0 | 1 |
| 36 weeks | 0 | 1 | 1 |
| 40 weeks | 0 | 0 | 0 |
| 44 weeks | 0 | 2 | 2 |
| Total | 9 | 12 | 21 |
| Date withdrawn | Date randomised | Weeks in trial | Reason |
|---|---|---|---|
| 4 March 2015 | 4 March 2015 | 0 | Screen failure |
| 1 April 2015 | 1 April 2015 | 0 | Participant no longer wishes to take part |
| 30 September 2015 | 30 September 2015 | 0 | Lost to follow-up/to be determined |
| 8 October 2015 | 8 October 2015 | 0 | Lost to follow-up/to be determined |
| 24 November 2015 | 24 November 2015 | 0 | When informed of being randomised to the laser arm no longer wanted to take part as she had already received laser treatment and ideally wanted the injection arm |
| 26 November 2015 | 26 November 2015 | 0 | Participant no longer wishes to take part |
| 2 July 2015 | 13 May 2015 | 7 | Participant no longer wishes to take part |
| 9 September 2015 | 11 June 2015 | 12 | Death of participant |
| 20 November 2015 | 24 June 2015 | 21 | Not eligible for anti-VEGF injections on clinical trial, moved to NHS |
| 27 November 2015 | 1 May 2015 | 30 | Lost to follow-up. Stopped attending all appointments |
| 10 November 2015 | 10 March 2015 | 36 | Death of participant |
| 22 December 2015 | 16 January 2015 | 48 | Unable to locate/contact participant |
Baseline characteristics
Baseline characteristics were well balanced between treatment groups, as shown in Table 8. A total of 123 (53%) treatment-naive and 109 (47%) non-naive patients were recruited. Mean baseline BCVA was 81.4 (SD 8.1) ETDRS letters. The proportion of patients with baseline BCVA 54–69 and ≥ 70 ETDRS letters was 9% and 91%, respectively. Table 9 shows the baseline BCVA.
| PRP (%; N = 116) | Aflibercept (%; N = 116) | |
|---|---|---|
| Age | 50.8 (13.2) | 51.5 (14.6) |
| Women | 44 (38%) | 33 (28%) |
| Men | 72 (62%) | 83 (72%) |
| Diabetes | ||
| Type 1 | 51 (44%) | 54 (47%) |
| Type 2 | 65 (56%) | 62 (53%) |
| Medication | ||
| Insulin only | 53 (46%) | 61 (53%) |
| Oral hypoglycaemic agents only | 24 (21%) | 26 (22%) |
| Insulin and oral hypoglycaemic agents | 39 (34%) | 29 (25%) |
| Diet controlled | 0 (0%) | 0 (0%) |
| Best corrected visual acuity (ETDRS letters) | ||
| 54–69 | 11 (9%) | 10 (9%) |
| ≥ 70 | 105 (91%) | 106 (91%) |
| Lens status (study eye) | ||
| Clear lens | 80 (69%) | 68 (59%) |
| Visually insignificant cataract | 26 (22%) | 37 (32%) |
| Visually significant cataract | 0 (0%) | 0 (0%) |
| Pseudophakia | 10 (9%) | 10 (9%) |
| Macular oedema (study eye) | ||
| No macular oedema | 87 (75%) | 87 (76%) |
| Non-central macular oedema | 28 (24%) | 27 (23%) |
| Central macular oedema | 1 (1%) | 1 (1%) |
| Central subfield thickness (µm) | 271.6 (28.1) | 275.3 (30.9)a |
| Total volume (mm3) | 8.94 (0.88) | 8.99 (1.09)a |
| Proliferative diabetic retinopathy | ||
| Treatment naive | 63 (54%) | 60 (52) |
| Previously treated active PDR | 53 (46) | 56 (48%) |
| Previous anti-VEGF therapy | 5 (4) | 6 (5) |
| Previous intravitreal steroid therapy | 0 (0) | 1 (1) |
| HbA1C | ||
| < 8% (< 63.90 mmol/mol) | 44 (38%) | 41 (35%) |
| 8–10% (63.9–85.8 mmol/mol) | 48 (41%) | 51 (44%) |
| > 10% (> 85.81 mmol/mol) | 24 (21) | 24 (21) |
| Blood pressure (diastolic), % (n) | ||
| ≤ 90mmHg | 102 (88%) | 101 (87%) |
| > 90mmHg | 14 (12%) | 15 (13%) |
| BCVA | PRP, % (n) | Aflibercept, % (n) |
|---|---|---|
| Baseline | N = 116 | N = 116 |
| ≥ 83 letters | 54% (63) | 48% (56) |
| 70–82 letters | 36% (42) | 43% (50) |
| 54–69 letters | 9% (11) | 9% (10) |
| 38–53 letters | 0% (0) | 0% (0) |
| ≤ 37 letters | 0% (0) | 0% (0) |
The trial flow
The CONSORT flow diagram (Figure 7) shows the participant flow through the study period.
FIGURE 7.
Participant flow through the study period. 66 a, ITT population included in the sensitivity analysis. Reprinted from The Lancet, Vol. 389, Sivaprasad S et al. ,66 Clinical efficacy of intravitreal aflibercept versus panretinal photocoagulation for best corrected visual acuity in patients with proliferative diabetic retinopathy at 52 weeks (CLARITY): a multicentre, single-blinded, randomised, controlled, phase 2b, non-inferiority trial, pp. 2193–203, © 2017, with permission from Elsevier.
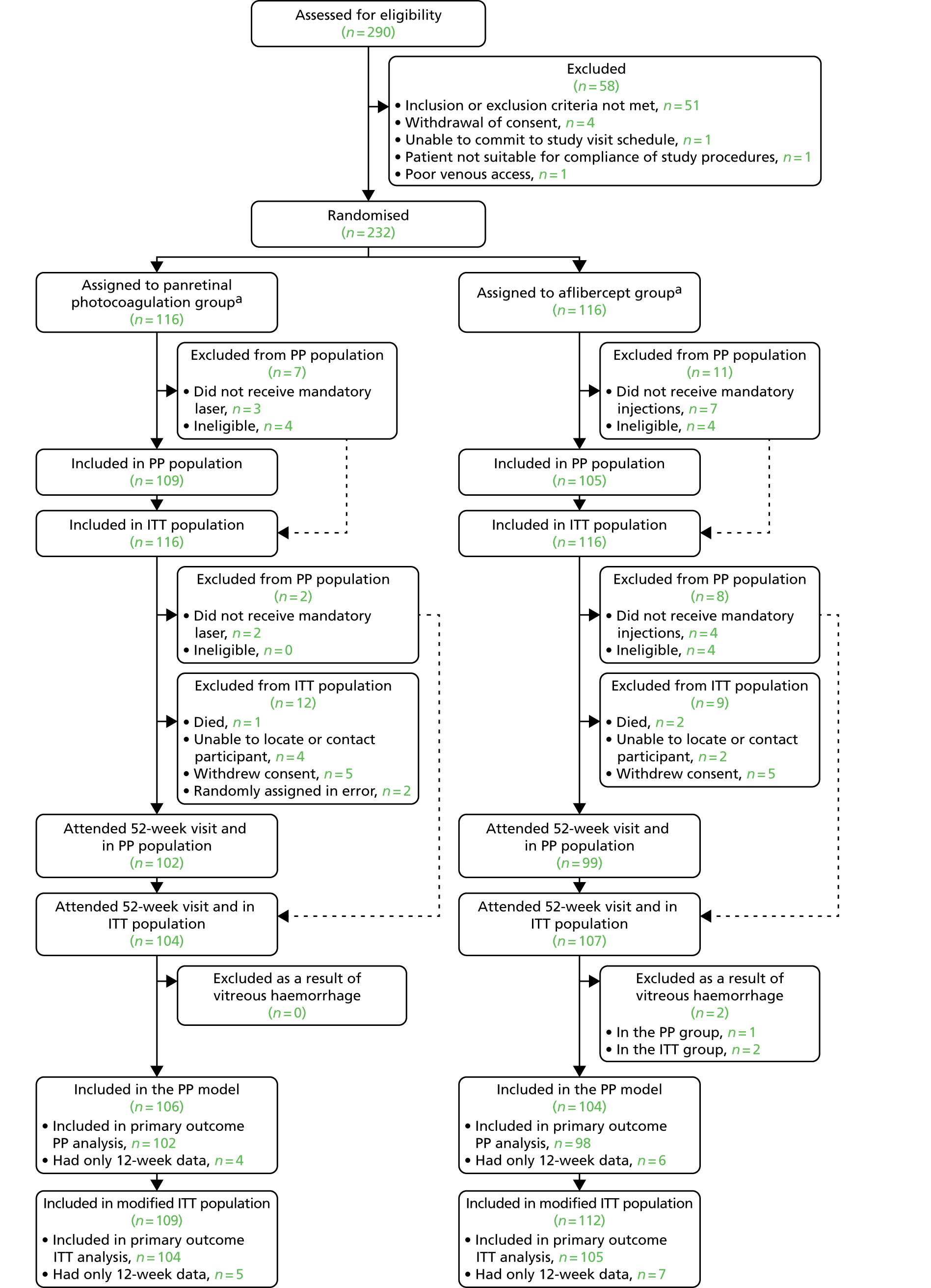
Derivation of the intention-to-treat model and per-protocol populations
Patients included in the prespecified ITT LME model were derived as follows:
-
The BCVA data were available for 211 patients of 232 randomly assigned patients (n = 107 in the aflibercept arm and n = 104 in the PRP arm) at 52 weeks and for 214 patients at 12 weeks (n = 109 in the aflibercept arm and n = 105 in the PRP arm).
-
A total of four patients in the PRP arm at 12 weeks and two patients in the aflibercept arm at 52 weeks were excluded because of the presence of vitreous haemorrhage within 3 months of BCVA recordings and BCVA was more than 3 SD below the mean at that time point (including all measurements).
-
There were 198 patients with BCVA available at both 12 and 52 weeks. A total of 11 patients had BCVA recorded at 52 weeks and not 12 weeks (n = 8 in the PRP arm and n = 3 in the aflibercept arm). In addition, there were 12 patients who had BCVA recorded at 12 weeks but not at 52 weeks (n = 5 in the PRP arm and n = 7 in the aflibercept arm).
-
There were therefore 221 patients who contributed to the analysis in the LME model for the ITT strategy (n = 109 in the PRP arm and n = 112 in the aflibercept arm).
-
A total of 18 patients did not meet the PP definition and were not included in the PP population (n = 214). This included 11 (9.5%) patients in the aflibercept arm and 7 (6.0%) in the PRP arm, with 4 patients in the aflibercept arm and 4 in the PRP arm not being compliant with the eligibility criteria and a further 7 patients in the aflibercept arm and 3 in the PRP arm not receiving initial mandatory treatment requirements. There were therefore 210 patients who contributed to the PP analysis in the LME model (n = 106 in the PRP arm and n = 104 in the aflibercept arm).
Primary outcome
The primary outcome at 52 weeks showed that aflibercept was both non-inferior and superior to PRP in terms of BCVA in both ITT and PP populations (Table 10 and Figure 8). The 95% CI for the adjusted difference between arms fell both above the prespecified acceptable non-inferiority margin of –5 letters and above the superiority margin of 0 letters, at both 12 and 52 weeks.
| n | Mean (SD) | Change from baseline, mean (SE) | Adjusted difference between groups (95% CI) | p-value | ||||
|---|---|---|---|---|---|---|---|---|
| PRP | Aflibercept | PRP | Aflibercept | PRP | Aflibercept | |||
| Baseline | 116 | 116 | 81.9 (8.0) | 80.9 (8.3) | ||||
| At 12 weeks | ||||||||
| Intention to treat | 101 | 109 | 81.3 (7.8) | 82.6 (9.6) | –0.8 (0.4) | 1.4 (0.5) | 2.1 (0.5 to 3.7)a | 0.0100 |
| Per protocol | 99 | 102 | 81.3 (7.9) | 82.7 (9.7) | –0.9 (0.4) | 1.5 (0.6) | 2.3 (0.6 to 3.9)b | 0.0074 |
| At 52 weeks | ||||||||
| Intention to treat | 104 | 105 | 79.1 (9.7) | 82.4 (10.1) | –3.0 (0.7) | 1.1 (0.6) | 3.9 (2.3 to 5.6)a | < 0.0001 |
| Per protocol | 102 | 98 | 79.3 (9.3) | 82.6 (10.1) | –2.9 (0.7) | 1.3 (0.6) | 4.0 (2.4 to 5.7)b | < 0.0001 |
FIGURE 8.
Primary outcome at 52 weeks (ITT and PP).

Three sensitivity analyses on the population with completed follow-up at 52 weeks were carried out, adjusting for sites, outliers and missing data. No patients were offered anti-VEGF treatment for macular oedema in the PRP arm so sensitivity analysis for concomitant treatments was not required. When sites were considered, the adjusted difference in BCVA between arms remained significant at 4.1 letters (95% CI 2.4 to 5.7 letters; p < 0.0001) and 4.1 letters (95% CI 2.4 to 5.7 letters; p < 0.0001) in the ITT and PP populations, respectively. A total of 207 and 198 patients remained after outliers in the ITT and PP populations, defined as less than or more than 4 SDs, were removed. This sensitivity analysis showed the adjusted difference in BCVA between arms as significant at 4.0 letters (95% CI 2.7 to 5.4 letters; p < 0.0001) in the ITT and 4.1 letters (95% CI 2.7 to 5.5 letters; p < 0.0001) in the PP population. The sensitivity analysis for missing data also confirmed a superiority effect in both ITT (n = 232) and PP populations (n = 214) for three prespecified alternative scenarios.
The primary outcome was also analysed at 12 weeks and the superiority of aflibercept was noted as early as 12 weeks in both the ITT and PP population (Figure 9).
FIGURE 9.
Visual outcome at 12 weeks in the (a) ITT population; and (b) PP population.
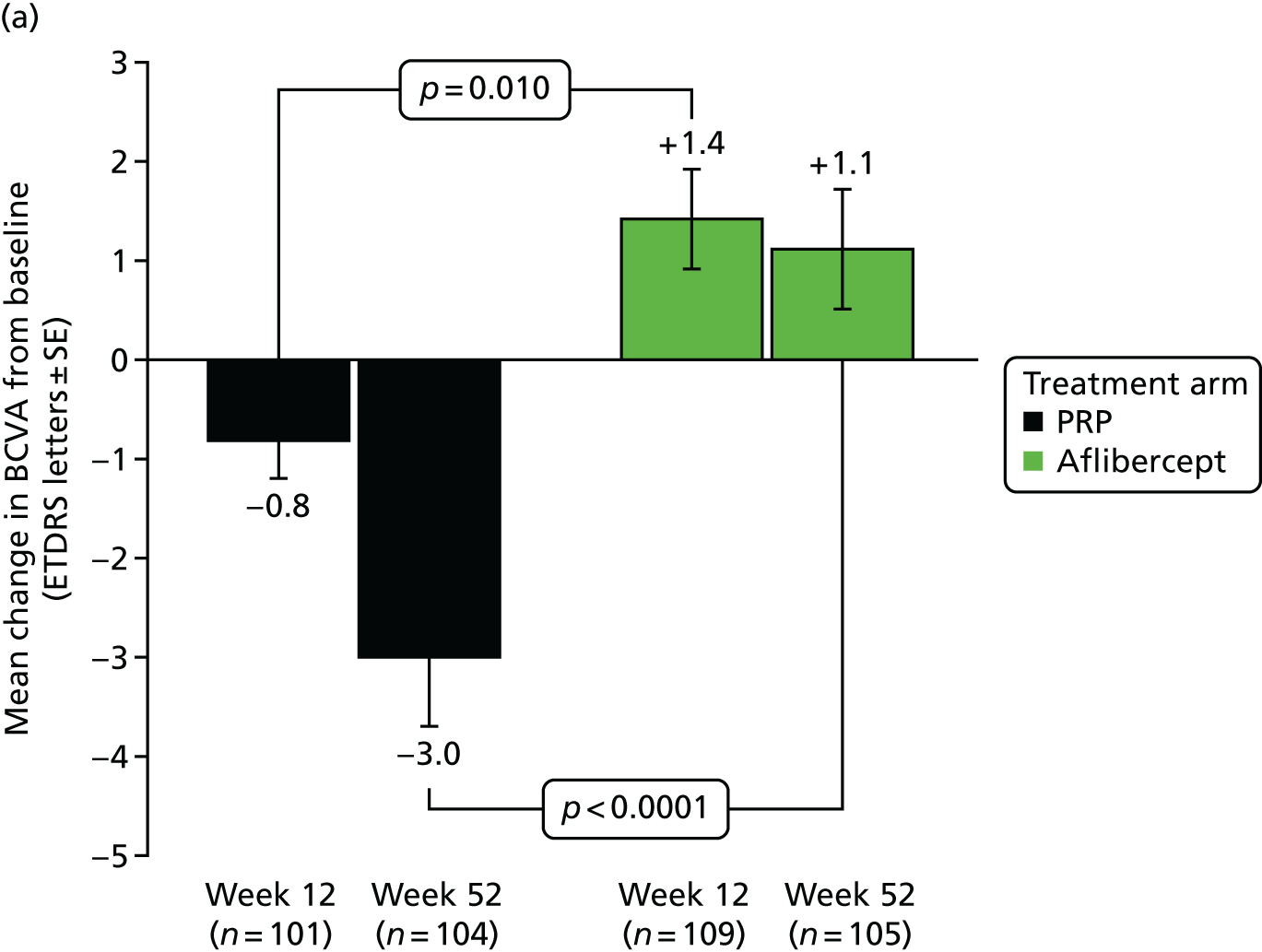
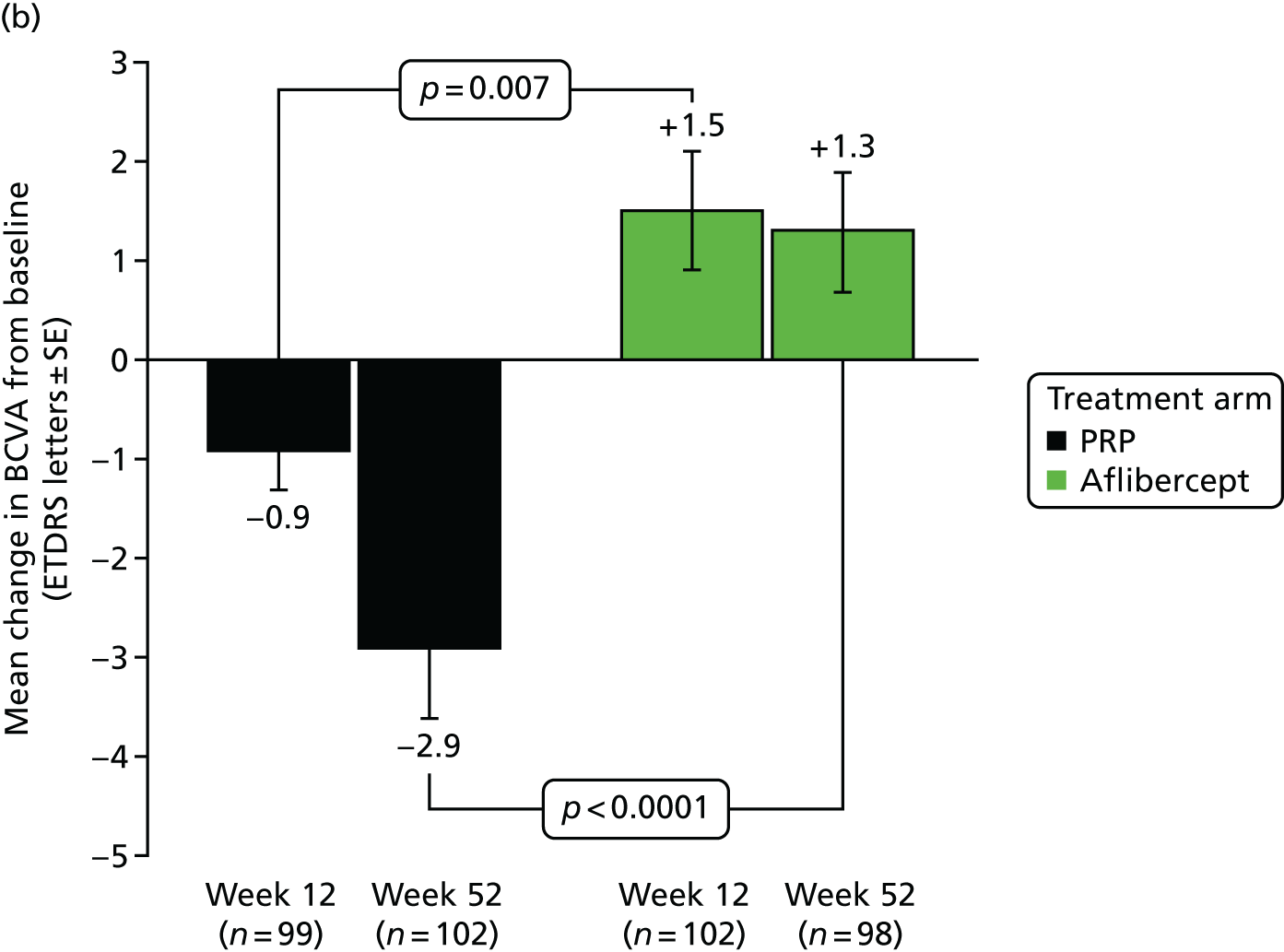
Subgroup analysis of BCVA in both the ITT and PP populations at 52 weeks was carried out based on the baseline PDR status and HbA1c categories (Table 11).
| Subgroups of BCVA | Mean (SD); n | Change from baseline, mean (SE) | Adjusted difference between arms (95% CI) | p-value for interaction between subgroups | ||
|---|---|---|---|---|---|---|
| PRP | Aflibercept | PRP | Aflibercept | |||
| PDR status | ||||||
| Baseline | ||||||
| Naive | 83.2 (7.4); 63 | 81.7 (9.1); 60 | – | – | – | – |
| Non-naive | 80.3 (8.5); 53 | 80.1 (7.3); 56 | – | – | – | – |
| ITT at 52 weeks | ||||||
| Naive | 80.7 (8.7); 54 | 84.6 (11.1); 54 | –2.6 (0.7) | 2.1 (0.8) | 4.5 (2.3 to 6.7) | 0.48 |
| Non-naive | 77.3 (10.5); 50 | 80.1 (8.3); 51 | –3.5 (1.2) | 0.1 (0.9) | 3.3 (1.0 to 5.7) | – |
| PP at 52 weeks | ||||||
| Naive | 80.6 (8.7); 53 | 84.9 (10.9); 51 | –2.7 (0.7) | 2.3 (0.9) | 4.8 (2.5 to 7.1) | 0.36 |
| Non-naive | 77.9 (9.7); 49 | 80.0 (8.6); 47 | –3.2 (1.2) | 0.2 (1.0) | 3.2 (0.8 to 5.6) | – |
| HbA1c levels | ||||||
| Baseline | ||||||
| < 8% | 82.9 (7.6); 44 | 80.3 (8.5); 41 | – | – | – | – |
| ≥ 8% to ≤ 10% | 81.7 (8.6); 48 | 82.0 (7.1); 51 | – | – | – | – |
| > 10% | 80.3 (7.6); 24 | 79.8 (10.2); 24 | – | – | – | – |
| ITT at 52 weeks | ||||||
| < 8% | 80.2 (7.8); 41 | 83.4 (8.1); 36 | –3.0 (1.0) | 2.4 (0.7) | 4.5 (1.9 to 7.2) | 0.35 |
| ≥ 8% to ≤ 10% | 78.6 (11.6); 42 | 82.9 (8.7); 47 | –3.4 (1.3) | 0.4 (0.9) | 3.9 (1.6 to 6.4) | – |
| > 10% | 77.8 (9.2); 21 | 79.8 (14.7); 22 | –2.3 (1.1) | 0.7 (1.8) | 2.9 (–0.6 to 6.5) | – |
| PP at 52 weeks | ||||||
| < 8% | 80.2 (7.8); 41 | 83.0 (8.2); 33 | –3.0 (1.0) | 2.6 (0.7) | 4.6 (1.9 to 7.3) | 0.49 |
| ≥ 8% to ≤ 10% | 79.3 (10.8); 40 | 83.6 (8.5); 43 | –3.2 (1.3) | 0.7 (1.0) | 4.1 (1.5 to 6.7) | – |
| > 10% | 77.8 (9.2); 21 | 79.8 (14.7); 22 | –2.3 (1.1) | 0.7 (1.8) | 2.9 (–0.7 to 6.5) | – |
Table 12 shows the change in BCVA in patients categorised into diastolic BP > 90 mmHg versus those ≤ 90 mmHg. There was no significant difference in BCVA outcome between patients who presented with BCVA 54–69 letters than those who presented with 70 letters or better.
| Subgroups of BCVA | Mean (SD); n | Change from baseline, mean (SE) | Adjusted difference between arms (95% CI) | p-value for interaction between subgroups | ||
|---|---|---|---|---|---|---|
| PRP | Aflibercept | PRP | Aflibercept | |||
| Diastolic blood pressure | ||||||
| Baseline | ||||||
| ≤ 90 mmHg | 81.9 (8.2); 102 | 80.9 (8.0); 101 | – | |||
| > 90 mmHg | 81.8 (6.5); 14 | 81.6 (10.4); 15 | – | |||
| ITT at 52 weeks | ||||||
| ≤ 90 mmHg | 78.8 (10.2); 92 | 83.0 (10.1); 91 | –3.1 (0.8) | 1.3 (0.6) | 4.2 (2.5 to 5.9) | 0.67 |
| > 90 mmHg | 81.1 (3.8); 12 | 83.1 (9.9); 14 | –2.4 (1.2) | –0.4 (2.2) | 2.4 (–2.3 to 7.0) | – |
| PP at 52 weeks | ||||||
| ≤ 90 mmHg | 79.1 (9.8); 90 | 82.4 (10.2); 85 | –3.0 (0.8) | 1.5 (0.7) | 4.2 (2.4 to 6.0) | 0.66 |
| > 90 mmHg | 81.1 (3.8); 12 | 83.5 (10.3); 13 | –2.4 (1.2) | 0.2 (2.3) | 2.7 (–2.0 to 7.5) | – |
| BCVA | ||||||
| Baseline | ||||||
| 54–69 letters | 64.4 (4.4); 11 | 61.7 (4.3); 10 | – | |||
| ≥ 70 letters | 83.7 (5.8); 105 | 82.8 (5.9); 106 | – | |||
| ITT at 52 weeks | ||||||
| 54–69 letters | 59.6 (9.1); 9 | 61.9 (15.6); 8 | –4.2 (3.4) | –0.9 (5.0) | 0.5 (–5.1 to 6.0) | 0.60 |
| ≥ 70 letters | 80.9 (7.5); 95 | 84.1 (7.4); 97 | –2.9 (0.7) | 1.3 (0.5) | 4.0 (2.4 to 5.7) | – |
| PP at 52 weeks | ||||||
| 54–69 letters | 61.0 (8.5); 8 | 61.3 (16.7); 7 | –2.5 (3.3) | –1.0 (5.7) | –1.0 (–6.9 to 5.0) | 0.91 |
| ≥ 70 letters | 80.9 (7.5); 94 | 84.2 (7.4); 91 | –2.9 (0.7) | 1.5 (0.6) | 4.2 (2.5 to 5.9) | – |
Secondary outcomes
The proportion of patients with a ≥ 10-letter improvement and able to do so with a baseline BCVA of ≤ 90 was 5% (5/101) in the aflibercept arm compared with 2% (2/95) in the PRP arm (difference between arms was 2.8%, 95% CI –3.1% to 9.1%; p = 0.45). The proportion of patients with a ≥ 10-letter worsening was 5% (5/107) in the aflibercept arm compared with 15% (16/104) in the PRP arm (difference between arms was 10.7%, 95% CI 2.6% to 19.3%; p = 0.009). There were 5% (5/107) of patients with a ≥ 15-letter worsening in the aflibercept arm and 6% (6/104) in the laser arm (difference between arms was 1.1%, 95% CI –5.5% to 7.9%; p = 0.72) (Figure 10).
FIGURE 10.
Best corrected visual acuity (≥ 10 letters) improvement and worsening at week 52.
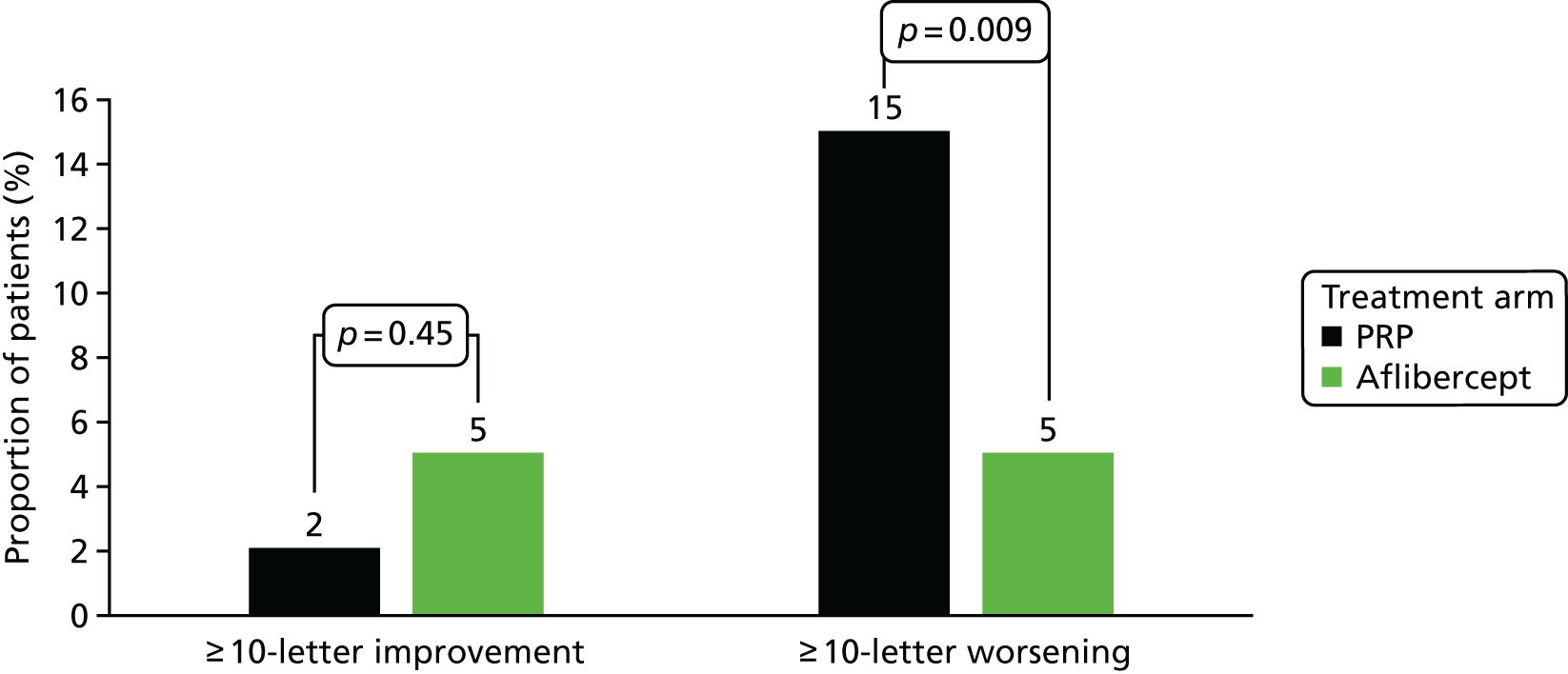
Binocular Esterman scores showed significant worsening in the PRP arm. This was also reflected in lower binocular visual acuity scores in the PRP arm. Other visual function tests did not vary between arms. Table 13 shows changes in visual function between baseline and week 52.
| Visual function outcomes | Mean (SD); n | Change from baseline, mean (SE) | Adjusted difference between arms (95% CI) | p-value | ||
|---|---|---|---|---|---|---|
| PRP | Aflibercept | PRP | Aflibercept | |||
| Low-luminance deficit score | ||||||
| Baseline | 68.8 (10.2); 116 | 69.2 (9.8); 116 | – | – | 2.0 (–0.9 to 4.8) | 0.18 |
| 52-week | 65.2 (14.3); 103 | 67.8 (15.5); 107 | –3.4 (1.0) | –1.5 (1.1) | ||
| Binocular visual acuity | ||||||
| Baseline | 84.3 (6.9); 116 | 84.2 (7.9); 116 | – | – | 2.3 (0.6 to 3.9) | 0.007 |
| 52-week | 82.8 (8.9); 104 | 84.9 (9.6); 106 | –1.8 (0.6) | 0.5 (0.5) | ||
| Contrast sensitivity (Pelli–Robson chart) | ||||||
| Baseline | 32.6 (5.1); 116 | 32.2 (6.1); 116 | – | – | 0.55 | |
| 52-week | 31.7 (5.6); 103 | 32.1 (6.8); 107 | –1.0 (0.5) | –0.5 (0.5) | ||
| Unilateral Esterman score % | ||||||
| Baseline | 11.2 (12.6); 112 | 13 (13.9); 112 | – | – | –1.9 (–4.3 to 0.5) | 0.12 |
| 52-week | 15.3 (15.5); 100 | 14.4 (13.9); 99 | 3.9 (0.9) | 1.9 (0.8) | ||
| Bilateral Esterman score % | ||||||
| Baseline | 5.6 (10.9); 115 | 7.4 (10.7); 114 | – | – | –3.0 (–5.1 to –0.8) | 0.007 |
| 52-week | 9.1 (14.4); 102 | 7.0 (9.3); 102 | 3.2 (0.8) | 0.2 (0.8) | ||
The NEI-VFQ-25 scores did not show significant differences between arms (Table 14).
| Vision related quality of life measured using VFQ-25 (0–100) | Mean score (SD); n | Change from baseline, mean (SE) | Adjusted difference between arms (95% CI) | p-value | ||
|---|---|---|---|---|---|---|
| PRP | Aflibercept | PRP | Aflibercept | |||
| Baseline | ||||||
| General health | 48.0 (23.2); 115 | 45.5 (23.0); 116 | – | – | – | – |
| General vision | 74.4 (14.9); 115 | 70.5 (16.4); 116 | – | – | – | – |
| Ocular pain | 89.0 (17.4); 115 | 87.1 (18.5); 116 | – | – | – | – |
| Near activities | 84.5 (18.1); 115 | 82.6 (20.4); 116 | – | – | – | – |
| Distance activities | 88.8 (14.4); 115 | 86.4 (18.3); 116 | – | – | – | – |
| Vision specific: social functioning | 96.5 (10.0); 115 | 92.5 (16.5); 115 | – | – | – | – |
| Vision-specific: mental health | 76.9 (22.5); 115 | 74.4 (24.7); 116 | – | – | – | – |
| Vision-specific: role difficulties | 85.7 (23.5); 115 | 78.3 (29.6); 116 | – | – | – | – |
| Vision-specific: dependency | 92.3 (18.3); 115 | 87.6 (24.5); 116 | – | – | – | – |
| Driving | 86.5 (25.6); 84 | 85.0 (27.0); 83 | – | – | – | – |
| Colour vision | 96.7 (10.7); 115 | 95.5 (13.8); 116 | – | – | – | – |
| Peripheral vision | 92.0 (15.4); 115 | 89.1 (21.2); 115 | – | – | – | – |
| VFQ-25 composite | 87.5 (12.7); 115 | 84.3 (15.1); 116 | – | – | – | – |
| 52 weeks | ||||||
| General health | 46.6 (24.0); 103 | 46.9 (21.9); 104 | –1.0 (2.0) | 2.2 (1.9) | 1.9 (–3.0 to 6.9) | 0.44 |
| General vision | 73.4 (15.2); 103 | 71.5 (14.7); 103 | –2.4 (1.6) | 0.2 (1.6) | –0.2 (–4.0 to 3.6) | 0.92 |
| Ocular pain | 87.1 (17.5); 103 | 88.2 (17.1); 103 | –2.5 (2.0) | 1.0 (1.9) | 1.8 (–2.7 to 6.2) | 0.43 |
| Near activities | 83.1 (21.5); 103 | 81.1 (21.2); 104 | –2.2 (2.1) | –1.6 (1.8) | –0.5 (–5.4 to 4.5) | 0.85 |
| Distance activities | 88.4 (16.3); 103 | 85.5 (20.0); 104 | –1.1 (1.6) | –1.1 (1.7) | –1.3 (–5.6 to 2.9) | 0.55 |
| Vision-specific: social functioning | 96.4 (11.0); 103 | 92.4 (17.9); 104 | –0.2 (1.4) | –0.2 (1.6) | –2.3 (–6.0 to 1.3) | 0.21 |
| Vision-specific: mental health | 78.8 (21.5); 103 | 75.2 (23.4); 104 | 0.2 (2.0) | 0.5 (2.1) | –1.4 (–6.4 to 3.6) | 0.58 |
| Vision-specific: role difficulties | 79.9 (27.5); 102 | 80.3 (28.0); 104 | –6.6 (3.0) | 1.9 (2.8) | 3.8 (–3.0 to 10.6) | 0.27 |
| Vision-specific: dependency | 91.4 (18.7); 103 | 88.2 (25.3); 103 | –1.6 (2.1) | 0.4 (2.1) | –0.8 (–6.1 to 4.5) | 0.77 |
| Driving | 85.3 (26.5); 76 | 85.8 (25.6); 72 | –2.4 (1.8) | –0.9 (1.5) | 1.5 (–3.2 to 6.3) | 0.53 |
| Colour vision | 96.8 (12.0); 103 | 94.7 (15.9); 103 | –0.2 (1.3) | –0.2 (1.5) | –1.1 (–4.6 to 2.4) | 0.52 |
| Peripheral vision | 90.9 (16.4); 102 | 88.0 (21.2); 104 | –1.0 (1.8) | –1.0 (2.0) | –1.5 (–5.9 to 3.0) | 0.52 |
| VFQ-25 composite | 86.5 (14.2); 103 | 84.3 (16.8); 104 | –1.8 (1.3) | –0.1 (1.2) | 0.3 (–2.9 to 3.6) | 0.84 |
The RetTSQ scores showed that patient satisfaction scores were significantly better in the aflibercept arm and the adjusted mean difference was 3.0 (95% CI 0.4 to 5.5; p = 0.022). There are no thresholds for clinically meaningful change for the RetTSQ. 67 However, in the validation of the RetTSQ a three-point difference was observed between those with very good visual acuity and those in the category below (five-category visual acuity variable). 38 The quality-of-life change assessed using RetDQoL did not show any difference between arms (Table 15).
| Patient-reported outcomes | Mean (SD); n | Change from baseline, mean (SE) | Adjusted difference between arms (95% CI) | p-value | ||
|---|---|---|---|---|---|---|
| PRP | Aflibercept | PRP | Aflibercept | |||
| Treatment satisfaction (RetTSQ) | ||||||
| Baseline | ||||||
| Positive subscale | 37.2 (5.8); 111 | 37.0 (6.2); 111 | – | – | – | – |
| Negative subscale | 26.7 (7.5); 113 | 26.3 (7.9); 110 | – | – | – | – |
| Total score | 64.0 (11.7); 111 | 63.3 (12.6); 109 | – | – | – | – |
| 52-week | ||||||
| Positive subscale | 38.7 (4.9); 101 | 39.3 (4.4); 103 | 1.1 (0.5) | 2.6 (0.7) | 0.8 (–0.4 to 2.0) | 0.18 |
| Negative subscale | 27.6 (7.5); 101 | 29.2 (6.7); 103 | 0.7 (0.7) | 3.1 (0.8) | 2.0 (0.3 to 3.7) | 0.023 |
| Total score | 66.2 (11.1); 100 | 68.4 (9.8); 103 | 1.8 (1.0) | 5.5 (1.3) | 3.0 (0.4 to 5.5) | 0.022 |
| Quality of life (RetDQoL) | ||||||
| Baseline | ||||||
| Overview item I | 1.4 (1.1); 114 | 1.3 (1.2); 116 | – | – | – | |
| Overview item II | –1.5 (1.2); 114 | –1.4 (1.0); 116 | – | – | – | |
| Impact score | –1.5 (1.8); 114 | –1.8 (2.0); 116 | – | – | – | |
| 52-week | ||||||
| Overview item I | 1.5 (1.1); 104 | 1.3 (1.2); 104 | 0.1 (0.1); 102 | 0.0 (0.1); 104 | –0.2 (–0.4 to 0.1) | 0.27 |
| Overview item II | –1.4 (1.2); 104 | –1.3 (1.1); 104 | 0.2 (0.1); 102 | 0.1 (0.1); 104 | 0.0 (–0.3 to 0.3) | 0.99 |
| Impact score | –1.4 (1.9); 100 | –1.7 (2.4); 103 | 0.2 (0.2); 99 | 0.0 (0.2); 103 | –0.3 (–0.7 to 0.2) | 0.28 |
Anatomical outcomes
Macular thickness and volume (Table 16) significantly increased in the PRP arm compared with the aflibercept arm. The proportion of patients with new-onset centre-involving macular oedema also increased significantly in the PRP arm.
| OCT outcomes | Mean (SD); n | Change from baseline, mean (SE) | Adjusted difference between arms (95% CI) | p-value | ||
|---|---|---|---|---|---|---|
| PRP | Aflibercept | PRP | Aflibercept | |||
| Central subfield thickness | ||||||
| Baseline | 275.3 (30.9); 116 | 271.6 (28.1); 115 | – | |||
| 12-week | 289.2 (41.2); 102 | 257.7 (28.3); 110 | 15.0 (2.9) | –14.0 (1.8) | –29.3 (–38.5 to –20.0) | < 0.0001a |
| 52-week | 298.2 (59.5); 103 | 263.4 (32.1); 106 | 24.0 (5.5) | –8.9 (2.3) | –32.7 (–42.0 to –23.4) | < 0.0001b |
| Previously untreated | ||||||
| Baseline | 274.6 (28.8); 63 | 270.7 (28.9); 59 | < 0.0001c | |||
| 52-week | 300.7 (58.4); 53 | 260.7 (29.6); 54 | 27.5 (7.6) | –12.2 (2.6) | –40.1 (–52.5 to –27.6) | – |
| Previously treated | ||||||
| Baseline | 276.2 (33.5); 53 | 272.6 (27.5); 56 | < 0.0002d | |||
| 52-week | 295.6 (61.0); 50 | 266.3 (34.6); 52 | 20.3 (8.1) | –5.5 (3.8) | –26.8 (–40.5 to –13.0) | – |
| Macular volume | ||||||
| Baseline | 8.99 (1.09); 116 | 8.94 (0.88); 115 | ||||
| 12-week | 9.16 (0.96); 101 | 8.39 (0.87); 110 | 0.18 (0.04) | –0.53 (0.04) | –0.71 (–0.86 to –0.56) | < 0.0001 |
| 52-week | 9.13 (1.14); 103 | 8.52 (0.95); 106 | 0.22 (0.07) | –0.40 (0.06) | –0.62 (–0.77 to –0.47) | < 0.0001 |
| Previously untreated | ||||||
| Baseline | 8.94 (1.14); 63 | 8.84 (0.67); 59 | < 0.0001 | |||
| 52-week | 9.17 (1.24); 53 | 8.36 (0.77); 54 | 0.35 (0.10) | –0.47 (0.07) | –0.82 (–1.02 to –0.63) | – |
| Previously treated | ||||||
| Baseline | 9.04 (1.04); 53 | 9.05 (1.05); 56 | 0.001 | |||
| 52-week | 9.09 (1.04); 50 | 8.70 (1.10); 52 | 0.08 (0.11) | –0.32 (0.09) | –0.40 (–0.63 to –0.18) | – |
The proportion of patients with macular oedema at 52 weeks assessed by the clinical investigator was significantly higher in the PRP arm than in the aflibercept arm (Table 17).
| Macular oedema | PRP, % (n) (N = 104) | Aflibercept, % (n) (N = 105a) | p-valueb |
|---|---|---|---|
| No macular oedema | 71% (74) | 89% (93) | 0.007 |
| Non-central macular oedema | 21% (22) | 9% (9) | |
| Central macular oedema | 8% (8) | 3% (3) |
Treating investigators determined regression and reactivation patterns of retinal new vessels to decide retreatment based on predefined criteria. Table 18 shows the proportion of patients with each regression pattern in each arm.
| PRP, % (n) | Aflibercept, % (n) | p-valuea | |
|---|---|---|---|
| At 12 weeks | N = 105 | N = 110 | |
| Total regression | 24% (25) | 74% (81) | < 0.0001 |
| Partial regression | 55% (58) | 25% (28) | |
| No regression | 21% (22) | 1% (1) | |
| At 52 weeks | N = 104 | N = 107 | |
| Total regression | 34% (35) | 64% (68) | < 0.0001 |
| Partial regression | 44% (46) | 17% (18) | |
| No regression | 7% (7) | 2% (2) | |
| Reactivation | 15% (16) | 18% (19) |
A significant proportion of eyes showed total regression of retinal new vessels in the aflibercept arm compared with the PRP arm. The difference in proportions of total regression favouring the aflibercept arm was 30% (95% CI 16% to 42%; p < 0.0001) at 52 weeks.
NetwORK UK, masked to treatment allocation, graded ETDRS diabetic retinopathy severity scores from colour fundus photographs obtained at baseline, 12 weeks and 52 weeks. 14 Of patients with gradable photographs (n = 227), 175 (77%) were graded low-risk PDR (levels 61 and 65) and 52 (23%) high-risk PDR (levels 71 and 75). Three eyes were graded below level 61. Improvement from diabetic retinopathy severity score is difficult to assess in lasered eyes and so the improvement of the level of remaining retinopathy was graded. A significantly higher proportion of patients in the PRP arm remained at PDR (level 61 or above) than in the aflibercept arm at both 12 and 52 weeks.
Treatment outcomes
The proportion of patients who received treatment in accordance with protocol was 94% (109/116) in the aflibercept arm and 97% (113/116) in the PRP arm. The treatment allocation guess form, which measures the success of masking of primary assessors to treatment allocation, was reported for 210 participants. Assessors guessed correctly for 15% (32/210), incorrectly for 10% (20/210), and were unable to tell for 75% (158/210) of participants.
By 52 weeks, aflibercept arm patients received a mean (SD) of 4.4 (1.7) injections (95% CI 4.1 to 4.7) [median (IQR) 4.0 (3.0 to 5.0)] including the three mandated loading doses. The mean number of aflibercept injections in treatment-naive patients was 4.6 (1.6) [median (IQR) 4 (3.0 to 6.0)] while non-naive patients received a mean number of injections of 4.1 (1.8), [median (IQR) 4.0 (3.0 to 4.8)]. A total of two (1.6%) patients required supplemental PRP in the aflibercept arm.
In the PRP arm, 78 (69%) received multispot laser and the remaining received single-spot laser. The type of laser delivery was not recorded for three patients. From week 12, 75 patients (65%) in the PRP arm required supplemental PRP.
Safety outcomes
A breakdown of the ocular adverse events reported is shown in Table 19. Trials such as CLARITY are not designed to detect reasonably small effects in secondary safety outcomes because adverse events are typically sparsely categorically distributed and sample size is set to provide power instead for the primary outcome. The ratio of false-positive significant findings to genuine significant findings is consequently larger than for primary outcomes. We therefore use p-values as a guide only, with caution, and we use 95% CIs whose width demonstrate the extent of the lack of information around the effects between arms in adverse event rates. Twice as many patients in the PRP arm (18%) developed vitreous haemorrhage as patients in the aflibercept arm (9%; p = 0.034). This 9% higher PRP rate had a wide 95% CI from 1% to 18%. The percentage of patients requiring vitrectomy was 6% in the PRP arm compared with 1% in the aflibercept arm (p = 0.066). This 5% higher PRP rate had a 95% CI from –0% to 11%. Later phase trials may be helpful to validate these results and to add to the body of safety evidence around these and other preliminary safety event results. There were no cases of NVI, NVA or NVG in either eye observed in a study of this size, where absence of evidence (low rates) means that there is low precision in estimated rate differences; this would be ameliorated for study treatments taken forwards in larger and longer later phase trials. There were no cases of endophthalmitis in the study. Table 20 shows the frequency of non-ocular adverse events in both groups. Twice as many patients in the aflibercept arm (n = 8) had APTC-defined events than in the PRP arm (n = 4), representing a 3% excess (Table 21). This was not statistically significant (p = 0.25) and the 95% CI for the between-arm difference was –3% to 10%. Three patients died during the trial (two in the aflibercept arm and one in the PRP arm).
| PRP (n = 116) | Aflibercept (n = 116) | p-valuea | |
|---|---|---|---|
| Endophthalmitis | 0 (0%) | 0 (0%) | – |
| Inflammationb | 3 (3%) | 9 (8%) | 0.075 |
| Visual disturbancesc | 11 (9%) | 10 (9%) | 0.82 |
| Ocular discomfortd | 4 (3%) | 6 (5%) | 0.52 |
| Cornea related problemse | 0 (0%) | 5 (4%) | 0.060 |
| Retinal tear | 0 (0%) | 1 (1%) | – |
| Progression of cataract | 1 (1%) | 0 (0%) | – |
| Elevation in IOP | 0 (0%) | 1 (1%) | – |
| Iris neovascularisation | 0 (0%) | 0 (0%) | – |
| Neovascular glaucoma | 0 (0%) | 0 (0%) | – |
| Vitreo–retinal interface abnormalitiesf | 1 (1%) | 2 (2%) | – |
| Subconjunctival haemorrhage | 0 (0%) | 1 (1%) | – |
| Increasing severity of diabetic retinopathy | 0 (0%) | 1 (1%) | – |
| Macular oedema | 2 (2%) | 0 (0%) | – |
| Retinal detachment | 0 (0%) | 0 (0%) | – |
| New or increasing vitreous haemorrhage | 21 (18%) | 10 (9%) | 0.034 |
| Vitreous haemorrhage requiring vitrectomy | 7 (6%) | 1 (1%) | 0.066 |
| System organ class of adverse events, % per participant | PRP, % (n) (N = 116) | Aflibercept, % (n) (N = 116) | p-valuea |
|---|---|---|---|
| Cardiac | 6% (7) | 8% (9) | 0.60 |
| Metabolism and endocrine | 8% (9) | 7% (8) | 0.80 |
| Infections/infestations | 33% (38) | 29% (34) | 0.57 |
| Respiratory | 3% (3) | 6% (7) | 0.20 |
| Ear and labyrinth | 0% (0) | 2% (2) | – |
| Surgical | 1% (1) | 2% (2) | – |
| Gastrointestinal | 7% (8) | 9% (11) | 0.47 |
| Genitourinary | 3% (3) | 5% (6) | 0.50 |
| Haematological | 3% (3) | 3% (4) | 1 |
| Hypersensitivity immune system | 3% (3) | 3% (4) | 1 |
| General disorders | 2% (2) | 5% (6) | 0.28 |
| Investigations | 0% (0) | 2% (2) | – |
| Musculoskeletal | 6% (7) | 9% (10) | 0.45 |
| Neoplasm | 0% (0) | 2% (2) | – |
| Neurological | 7% (8) | 11% (13) | 0.25 |
| Reproductive | 2% (2) | 2% (2) | – |
| Vascular | 4% (5) | 4% (5) | 1 |
| Injury | 9% (11) | 6% (7) | 0.33 |
| PRP (n = 116) | Aflibercept (n = 116) | p-valuea | |
|---|---|---|---|
| Non-fatal myocardial infarction | 3 (3%) | 3 (3%) | 1.00 |
| Non-fatal stroke | 0 (0%) | 3 (3%) | 0.25 |
| Vascular death | 1 (1%) | 2 (2%) | 1.00 |
| Unknown death | 0 (0%) | 0 (0%) | – |
| Any ATPC event | 4 (3%) | 8 (7%) | 0.24 |
Pregnancy
Two trial participants in the aflibercept arm and three in the PRP arm became pregnant despite being advised on double contraception. One patient’s partner became pregnant in the aflibercept arm (Table 22). Aflibercept was stopped in both the patients who were randomised to aflibercept, but none of them required supplemental PRP during the trial. The patient whose partner became pregnant did not need further treatment as per retreatment guidelines. All pregnancies were followed up after the trial and there were no adverse events reported among the mothers or babies.
| Arm | Site | SAE details | Randomisation date | Weeks from randomisation to report of pregnancy |
|---|---|---|---|---|
| PRP | Sunderland Eye Infirmary | Pregnancy | 21 November 2014 | 50.6 |
| Aflibercept | Moorfields Eye Hospital | Pregnancy | 29 July 2015 | 28.7 |
| PRP | King’s College Hospital | Pregnancy | 10 August 2015 | 22.3 |
| PRP | Royal Victoria Hospital | Pregnancy | 24 August 2015 | 28.1 |
| Aflibercept | James Paget Hospital | Partner of participant | 15 September 2015 | 54.3 |
| Aflibercept | Essex County Hospital | Pregnancy | 25 September 2015 | 6.6 |
Chapter 4 Health economics evaluation
Background
In this project, aflibercept was evaluated as an alternative treatment to PRP in PDR without macular oedema. The clinical efficacy results showed that aflibercept is superior to PRP in terms of visual acuity change at 52 weeks. In this chapter, we report the economic evaluation of aflibercept as a treatment option for PDR.
Objective
Drawing on the Consolidated Health Economic Evaluation Reporting Standards (CHEERS) checklist,68 we report cost-effectiveness of intravitreal aflibercept compared with PRP.
Methods
Economic evaluation
From a public sector multiagency perspective that covers health and social care services, follow-up at 52 weeks was based on data collected from the CLARITY study, a Phase IIb, single-blind, non-inferiority trial on adults (aged ≥ 18 years) with type 1 or 2 diabetes mellitus and previously untreated or post-laser treated active PDR. Study participants were recruited from 22 UK ophthalmic centres.
Cost of intervention (aflibercept) and comparator (panretinal photocoagulation)
We included both the purchase price of aflibercept and the injection procedure cost. PRP was costed using the published national mean unit cost. 46 Aflibercept injection treatment in the intervention arm and PRP in the comparator arm were costed using published national unit costs from the Department of Health46 and BNF47 at 2016 price year.
Costing service use
We explored patterns of hospital- and community-based service use over the 52-week follow-up study period, using a CSRI. 69 National unit costs from published sources at 2016 price year were used to calculate health and social care service use costs. 46,48,70,71 We bootstrapped differences in cost to produce a 95% CI around these differences. 48
Measurement of outcomes
In our primary cost-effectiveness analysis, we used BCVA as the measure of effectiveness. Change in BCVA score from baseline was calculated for intervention and comparator groups and the difference in change in BCVA score between groups was assessed.
For HRQoL, we collected EQ-5D-3L data at baseline and at 52 weeks post baseline. The EQ-5D-3L index scores were calculated at baseline and 52 weeks for both the intervention and comparator groups. Potential QALY gains were then calculated using the standard ‘area under the curve’ (AUC) method. 72,73 QALY is a generic preference-based measure weighing the quantity of life (i.e. survival or the number of additional life-years) by the quality of life of participants, experienced at baseline and 52-week follow-up.
From a methodological perspective, we compared the performance of the EQ-5D-3L (a generic, preference-based HRQoL measure) with the vision-specific (non-preference-based) HRQoL measures (NEI-VFQ-25 and RetDQoL). We included the ICECAP-A measure as an alternative to EQ-5D-3L, focusing on capability to see how this measure compared in terms of sensitivity in this patient group. 74 We did not collect information on activities of daily living (ADL), which would have told us something about how treatment of this condition with aflibercept versus PRP affects home and work life.
Cost-effectiveness analysis
From a public sector multiagency perspective, which covers health and social care services, we assessed the cost-effectiveness of intravitreal aflibercept compared with conventional PRP. The incremental costs and effects of alternative arms were compared in the primary cost-effectiveness analysis with effectiveness measured in terms of BCVA. Published sources of national unit costs at 2016 price year were used to calculate the total cost of health and social care service use and ophthalmic-related drugs use over 52 weeks by patients in each arm of the study. We employed ‘bootstrapping’ to overcome the skewed data and produced CEACs to quantify uncertainty. For the cost-effectiveness analysis and cost–utility analysis, difference in cost and effects (change in BCVA and QALY) between arms of the study were adjusted for differences in baseline cost and effects. 75 We undertook this adjustment by applying linear regression in IBM SPSS Statistics 22 (IBM Corporation, Armonk, NY, USA) with baseline cost and trial arm as the only covariates for cost adjustment, baseline BCVA and trial arm as the only covariates for change in BCVA adjustment, and baseline EQ-5D-3L index score and trial arm as the only covariates for QALY adjustment. 76
Secondary exploratory cost–utility analysis
We undertook a secondary exploratory cost–utility analysis, that is, a cost per QALY analysis. Evidence is mixed but points to the EQ-5D-3L not being sufficiently sensitive to be useful in studies of visual impairment. 44,45
Sensitivity analysis
We used sensitivity analysis to explore whether or not the estimated effect (change in BCVA score) and cost of aflibercept injection treatment relative to PRP laser treatment were sensitive to the key variable factor of our analysis – the pricing of aflibercept. This is most important variable factor because the NHS PAS can make the drug available at a range of prices.
Subgroup analysis
We undertook a subgroup analysis to explore the potential effect of heterogeneity defined in terms of DMO. The subgroup analysis was performed by the presence of DMO at baseline (no DMO in both eyes vs. DMO presence in at least one eye). The incremental costs and effects of alternative arms were reported within each of the categories of the subgroup variable, and the ICER (i.e. cost per change in BCVA) was calculated for both of these categories of the subgroup variable.
Results
Economic sample
Economic evaluation was undertaken on 202 participants (101 per arm) with complete cost and outcome data. This represents 96.7% of the clinical sample included in primary outcome ITT analysis. The economic sample was made up of male (n = 134) and female (n = 68) participants with an overall mean age of 51.3 years (SD 13.4 years). There was no statistically significant difference in the EQ-5D-3L index score at baseline (p > 0.05) or ICECAP-A score at baseline (p > 0.05).
Cost of intervention (aflibercept) and comparator (panretinal photocoagulation)
The list price for aflibercept was £81645 with an administration cost of £182 per injection procedure (Department of Health and Social Care, 2016). PRP laser treatment costs £131 per procedure. 44 All participants in both groups received FFA, which costs £117 per angiogram. 45 A full list of hospital- and community-based and ophthalmic related medication unit costs and their sources are shown in Appendix 3. All unit costs were at 2016 price year.
Frequency and cost of service use over 52 weeks
Table 23 shows the frequency of contacts with primary and secondary care health services and other services used by 202 participants in the intervention and comparator arms of the CLARITY trial over the 52-week study period.
| Care sector and services | Treatment arm, mean, median (minimum, maximum) | Mann–Whitney U-test p-valuea | |
|---|---|---|---|
| Aflibercept (n = 101) | PRP (n = 101) | ||
| NHS primary care sector and other community-based services | |||
| GP | 2.87, 2 (0, 41) | 2.33, 1 (0, 25) | 0.273 |
| Practice nurse | 0.98, 0 (0, 7) | 1.66, 0 (0, 30) | 1.000 |
| District nurse | 0.09, 0 (0, 8) | 0.03, 0 (0, 2) | 0.996 |
| Diabetic clinic (non-hospital based) | 0.78, 0 (0, 6) | 1.21, 0 (0, 12) | 0.574 |
| Social worker | 0.07, 0 (0, 7) | 0.00, 0 (0, 0) | 0.317 |
| Counsellor | 0.56, 0 (0, 40) | 0.08, 0 (0, 5) | 0.676 |
| Dietitian | 0.22, 0 (0, 3) | 0.42, 0 (0, 12) | 0.612 |
| Optician | 0.47, 0 (0, 6) | 0.36, 0 (0, 3) | 0.306 |
| Chiropodist | 0.90, 0 (0, 20) | 1.24, 0 (0, 12) | 0.339 |
| Physiotherapist | 0.71, 0 (0, 18) | 0.30, 0 (0, 13) | 0.412 |
| Occupational health therapist | 0.19, 0 (0, 16) | 0.03, 0 (0, 3) | 0.316 |
| Alternative therapist | 0.01, 0 (0, 1) | 0.10, 0 (0, 10) | 0.994 |
| Other services | 0.14, 0 (0, 5) | 0.16, 0 (0, 9) | 0.715 |
| NHS secondary care sector | |||
| Inpatient stays (bed-days) | |||
| Ophthalmology inpatient stays (bed-days) | 0.08, 0 (0, 2) | 0.03, 0 (0, 3) | 0.181 |
| Other inpatient stays (bed-days) | 5.26, 0 (0, 301) | 0.65, 0 (0, 22) | 0.039b |
| Total inpatient stays (bed-days) | 5.34, 0 (0, 301) | 0.68, 0 (0, 22) | 0.015b |
| Outpatient visits and procedures | |||
| Ophthalmology outpatient visits and procedures | 2.71, 1 (0, 15) | 2.42, 1 (0, 14) | 0.632 |
| Diabetic clinic (hospital-based) outpatient visits | 1.32, 0 (0, 40) | 0.90, 0 (0, 30) | 0.635 |
| Diabetic consultant outpatient visits | 0.83, 0 (0, 40) | 0.78, 0 (0, 30) | 0.695 |
| Renal consultant outpatient visits | 0.47, 0 (0, 16) | 0.15, 0 (0, 8) | 0.171 |
| Other outpatient visits and procedures | 0.58, 0 (0, 11) | 0.57, 0 (0, 12) | 0.821 |
| Total outpatient visits and procedures | 5.91, 3 (0, 81) | 4.82, 3 (0, 60) | 0.576 |
| Other hospital services | 0.18, 0 (0, 10) | 0.17, 0 (0, 12) | 0.319 |
| FFAb | 1.00, 1 (1, 1) | 1.00, 1 (1, 1) | 1.000 |
| Aflibercept injection | 4.49, 4 (3, 13) | 0.01, 0 (0, 1) | 0.000b |
| PRP laser treatment | 0.03, 0 (0, 2) | 2.77, 3 (1, 7) | 0.000b |
| Ophthalmology-related medications | 6.50, 6 (0, 33) | 3.61, 3 (0, 18) | 0.000b |
Calculation of total mean service use costs over 52 weeks
Table 24 shows the mean cost of health and social care service use and ophthalmic related drug use over 52 weeks of study follow-up. The price year of all costs was 2016. No discount rate was applied as the study ran for 12 months.
| Care sector and services | Treatment arm, mean (SD) | Mean difference in £ (95% CI bootstrapped) | |
|---|---|---|---|
| Aflibercept (n = 101) | PRP (n = 101) | ||
| NHS primary care sector and other community-based services | |||
| GP | 103.37 (180.26) | 83.76 (132.38) | 19.60 |
| Practice nurse | 11.29 (17.41) | 19.83 (53.20) | –8.53 |
| District nurse | 1.63 (14.69) | 0.33 (2.44) | 1.31 |
| Diabetic clinic (non-hospital based) | 8.60 (13.62) | 13.36 (25.25) | –4.76 |
| Social worker | 5.48 (55.03) | 0.00 (0.00) | 5.48 |
| Counsellor | 23.70 (172.60) | 3.33 (22.76) | 20.38 |
| Dietitian | 4.57 (13.15) | 8.73 (35.79) | –4.16 |
| Optician | 12.10 (20.98) | 9.27 (15.85) | 2.83 |
| Chiropodist | 18.92 (55.36) | 25.99 (55.81) | –7.07 |
| Physiotherapist | 11.41 (42.83) | 4.75 (24.11) | 6.65 |
| Occupational health therapist | 4.14 (35.30) | 0.65 (6.57) | 3.49 |
| Alternative therapist | 0.47 (4.73) | 4.70 (47.26) | –4.23 |
| Other services (e.g. podiatrist, psychologist) | 4.11 (26.02) | 2.94 (19.48) | 1.17 |
| Total primary care and other community-based services cost | 209.79 (300.13) | 177.64 (221.88) | 32.15 (–38.17 to 106.06) |
| NHS secondary care sector | |||
| Inpatient stays | |||
| Ophthalmology inpatient stays | 34.18 (169.15) | 11.50 (115.62) | 22.67 |
| Other inpatient stays | 1538.27 (7079.39) | 400.36 (1702.85) | 1137.91 |
| Total inpatient stays | 1572.45 (7073.91) | 411.86 (1704.04) | 1160.59 |
| Outpatient visits and procedures | |||
| Ophthalmology outpatient visits and procedures | 362.69 (479.54) | 353.50 (506.06) | 9.19 |
| Diabetic clinic (hospital-based) outpatient visits | 265.07 (843.84) | 191.75 (657.68) | 73.32 |
| Diabetic consultant outpatient visits | 146.81 (655.81) | 141.48 (543.63) | 5.34 |
| Renal consultant outpatient visits | 75.26 (328.32) | 24.75 (144.14) | 50.50 |
| Other outpatient visits and procedures | 260.92 (869.43) | 59.45 (170.23) | 201.48 |
| Total outpatient visits and procedures | 1110.75 (1860.34) | 770.93 (1284.18) | 339.82 |
| Other hospital services | 38.37 (202.90) | 71.67 (538.44) | –33.30 |
| Total secondary care cost | 2721.57 (7472.42) | 1254.47 (2630.01) | 1467.10 (146.07 to 3202.77) |
| Total primary care, other community-based services and secondary care cost | 2931.37 (7518.60) | 1432.11 (2699.44) | 1499.26 (111.61 to 3192.23) |
| Total intervention delivery cost | 4597.07 (1676.17) | 490.05 (194.40) | 4107.02 (3799.65 to 4455.21) |
| Total cost | 7528.44 (7766.09) | 1922.16 (2660.83) | 5606.28 (4212.53 to 7435.52) |
| Ophthalmology-related medication cost | 247.16 (660.38) | 378.92 (1188.59) | –131.76 (–408.71 to 116.13) |
| Grand total costa | 7775.59 (7787.54) | 2301.08 (3003.90) | 5474.52 (4028.26 to 7362.63) |
| Grand total costb | 7775.89 (985.94) | 2301.31 (948.65) | 5474.57 (5210.79 to 5749.82) |
Total mean costs of service use included primary care consultations, secondary care consultations and other community-based services, for example social work. Table 24 shows that the mean adjusted total cost (including ophthalmic-related drug cost) per participant was £7775.89 (SD £985.94) for the aflibercept group and £2301.31 (SD £948.65) for the PRP group. The difference in mean total cost between the two arms was £5474.57 (bootstrapped 95% CI £5210.79 to £5749.82). Primary care and other community-based costs accounted for a very small proportion of total mean costs (2.7% in the intervention group and 7.7% in the comparator group). Hospital-based costs, specifically the cost of aflibercept or PRP, accounted for the majority of total service use costs.
Measurement of outcome
Table 25 shows mean and SD scores for BCVA (our primary measure of effectiveness) in the intervention and comparator groups over 52 weeks. The far-right column shows the difference in mean adjusted change scores between arms with the bootstrapped 95% CI (5000 replications). We observed an adjusted 3.93-point difference on the BCVA (bootstrapped 95% CI 3.84 points to 4.02 points) between study arms in our economic sample. This represents a statistically significant difference between groups and is exactly the same as the difference observed in the ITT clinical sample analysis.
| Measure | Treatment arm, mean (SD) | Difference in mean change scores between groupsa,b (bootstrapped 95% CI) | |||||
|---|---|---|---|---|---|---|---|
| Aflibercept (n = 101) | PRP (n = 101) | ||||||
| Baseline | 52 weeksa | Change in mean score between baseline and 52 weeksa | Baseline | 52 weeksa | Change in mean score between baseline and 52 weeksa | ||
| BCVA score | 81.48 (7.51) | 82.42 (7.19) | 0.94 (0.32) | 82.13 (7.98) | 79.14 (7.64) | –2.99 (0.34) | 3.93 (3.84 to 4.02) |
Table 26 shows the mean and SDs for EQ-5D-3L index scores and QALYs over 52 weeks in the intervention and comparator arms. At baseline, there was no statistically significant difference in mean EQ-5D-3L index scores between intervention and comparator arms (p = 0.774 > 0.05). Compared with UK national population norms, our EQ-5D-3L index scores at baseline were just a little lower than the population mean of 0.85 with similar SD. 77 The far-right column shows a difference in mean incremental adjusted QALY of –0.022 between groups (using AUC methods) with the bootstrapped 95% CI (–0.080 to 0.034) and this difference is not statistically significant. We undertook a secondary exploratory cost–utility analysis, that is, a cost-per-QALY analysis. The results are shown in Table 26. The results speak for themselves; a mean adjusted cost difference of £5475 is divided by an extremely small and not statistically significant mean adjusted QALY difference of –0.022. This yields a cost per QALY of –£252,827, in which we do not have much confidence. Given that a positive significant difference was observed in the BCVA for the intervention group, we interpreted this as the EQ-5D-3L not being sufficiently sensitive in this context.
| Measure | Treatment arm, mean (SD) | Incremental mean QALYs between groupsa,b (bootstrapped 95% CI) | |||||
|---|---|---|---|---|---|---|---|
| Aflibercept (n = 101) | PRP (n = 101) | ||||||
| Baseline | 52 weeksa | QALY over 52 weeksa | Baseline | 52 weeksa | QALY over 52 weeksa | ||
| EQ-5D-3L index score | 0.788 (0.292) | 0.784 (0.178) | 0.785 (0.235) | 0.817 (0.218) | 0.798 (0.133) | 0.807 (0.175) | –0.022 (–0.080 to 0.034) |
Table 27 shows the mean and SDs for ICECAP-A capability index scores and change in mean ICECAP-A index score over 52 weeks in the intervention and comparator arms. We observed no statistically significant change from baseline scores between groups using the ICECAP-A measure of capability as an alternative to QALYs as shown in the far-right column of Table 27.
| Measure | Treatment arm, mean (SD) | Difference in mean change scores between groupsa,b (bootstrapped 95% CI) | |||||
|---|---|---|---|---|---|---|---|
| Aflibercept (n < 101) | PRP (n < 101) | ||||||
| Baseline | 52 weeks | Change in mean score between baseline and 52 weeks | Baseline | 52 weeks | Change in mean score between baseline and 52 weeksa | ||
| ICECAP-A capability index scores | 0.861 (0.167) | 0.855 (0.151) | –0.006 (0.141) | 0.878 (0.137) | 0.868 (0.148) | –0.010 (0.157) | 0.004 (–0.006 to 0.035) |
Vision specific quality-of-life measures
As shown in the far-right columns of Tables 28 and 29, we observed no statistically significant difference in mean scores for NEI-VFQ-25 and RetDQoL between arms over 52 weeks. Table 28 shows the mean NEI-VFQ-25 composite scores, change in mean NEI-VFQ-25 composite score between study time points and difference in mean change scores between groups over 52 weeks. Table 29 shows mean RetDQoL scores for overview items 1 and 2 and AWI score, change in mean score between study time points and difference in mean change scores between groups over 52 weeks.
| Measure | Treatment arm, mean (SD) | Difference in mean change scores between groupsa,b (bootstrapped 95% CI) | |||||
|---|---|---|---|---|---|---|---|
| Aflibercept (n = 101) | PRP (n = 101) | ||||||
| Baseline | 52 weeks | Change in mean score between baseline and 52 weeks | Baseline | 52 weeks | Change in mean score between baseline and 52 weeksa | ||
| NEI-VFQ-25 composite scores | 84.57 (15.26) | 84.86 (15.42) | 0.29 (11.44) | 88.04 (12.75) | 86.30 (14.28) | –1.74 (13.39) | 2.03 (–1.31 to 5.56) |
| Measure | Treatment arm, mean (SD) | Difference in mean change scores between groupsa,b (bootstrapped 95% CI) | |||||
|---|---|---|---|---|---|---|---|
| Aflibercept (n < 101) | PRP (n < 101) | ||||||
| Baseline | 52 weeks | Change in mean score between baseline and 52 weeks | Baseline | 52 weeks | Change in mean score between baseline and 52 weeksa | ||
| RetDQoL Overview items 1 score | 1.360 (1.194) | 1.340 (1.148) | –0.020 (1.054) | 1.449 (1.095) | 1.510 (1.142) | 0.061 (1.225) | –0.081 (–0.371 to 0.251) |
| RetDQoL Overview items 2 score | –1.380 (1.033) | –1.310 (1.107) | –0.070 (1.174) | –1.520 (1.186) | –1.367 (1.205) | –0.153 (1.327) | 0.083 (–0.289 to 0.402) |
| RetDQoL AWI score | –1.700 (1.981) | –1.667 (2.330) | –0.033 (1.939) | –1.581 (1.926) | –1.376 (1.921) | –0.205 (1.718) | 0.172 (–0.373 to 0.658) |
Cost-effectiveness analysis
In our primary cost-effectiveness analysis using BCVA as our measure of outcome, we divided the difference in mean adjusted total cost between arms by the difference in mean adjusted change in BCVA between arms to gain an ICER of £1392.99 for aflibercept as compared with PRP laser treatment. Figure 11 is a cost-effectiveness plane, a scatterplot of the joint distribution of incremental cost and effect (BCVA) between the two arms. Figure 11 shows the 5000 bootstrapped replications used to generate a 95% CI around our mean point estimate ICER. All bootstrapped points appear in the north-east quadrant (more effective but more costly) of our cost-effectiveness plane. Accompanying this plane is a CEAC (Figure 12) that shows that at the threshold of £1400, our hypothetical societal willingness to pay, there is a probability of 56.60% of aflibercept being cost-effective at its list price of £816.
FIGURE 11.
Cost-effectiveness plane: using BCVA as the measure of effectiveness.
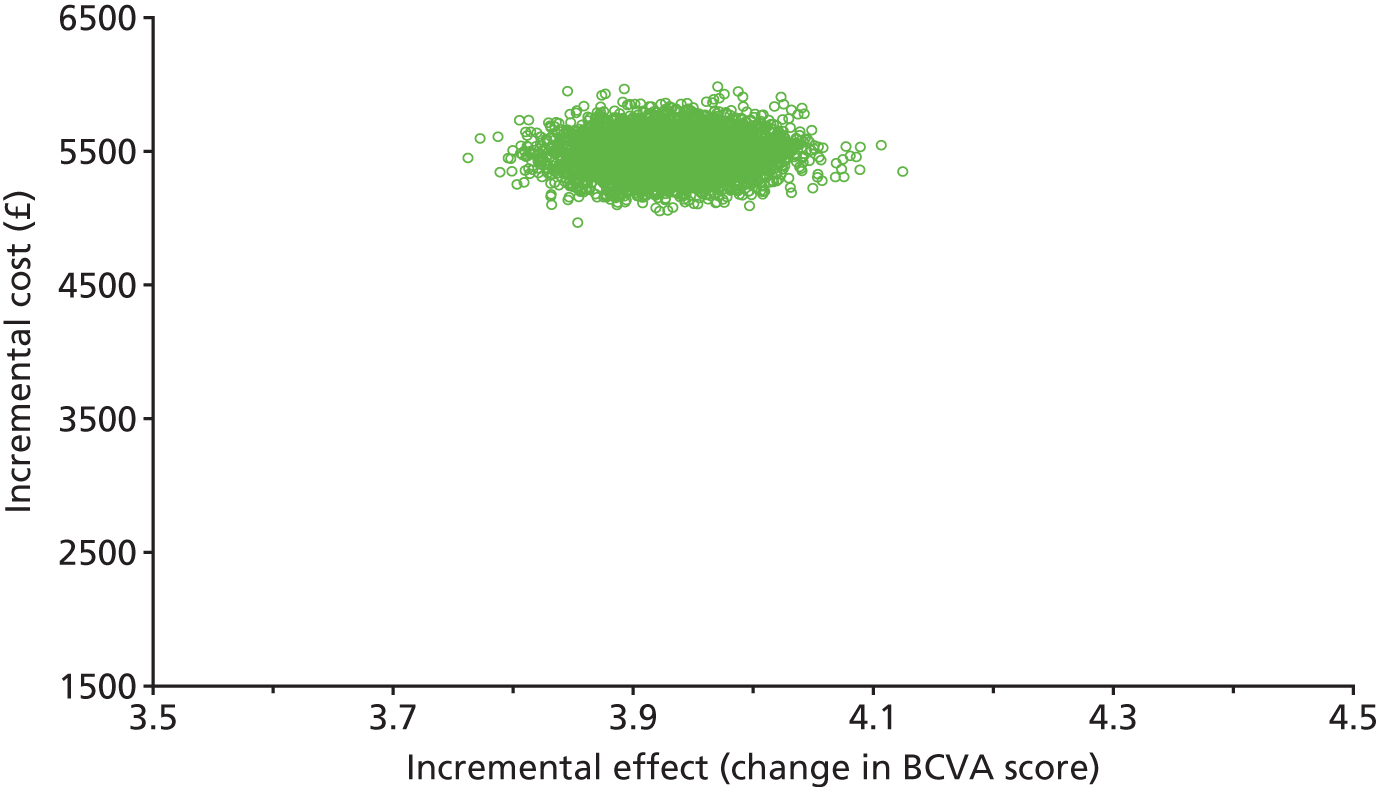
FIGURE 12.
Cost-effectiveness acceptability curve.
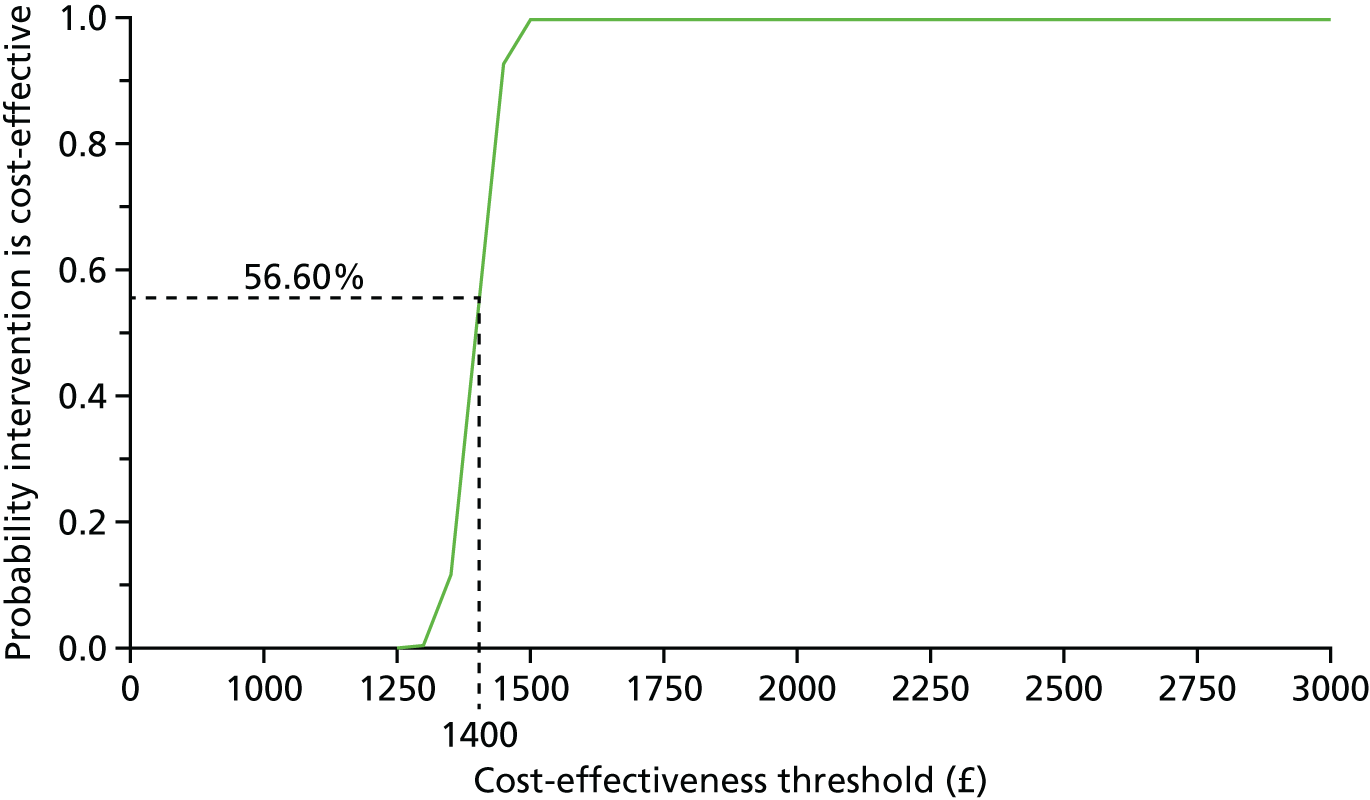
Sensitivity analysis
The most important variable factor is the pricing of aflibercept. The NHS PAS could make the drug available at a range of prices to the NHS. In Table 30 we show how the ICER and probability of cost-effectiveness vary as a discount is applied to the list price of aflibercept. We have used a hypothetical societal willingness-to-pay threshold of £1400 to mimic the NICE threshold used in cost-per-QALY calculations. If society is willing to pay £1400 for an additional 1-point improvement in BCVA, then aflibercept has a 56.60% probability of being cost-effective at the list price of £816. From 20% through to 100% PAS, results showed 100% probability of aflibercept being cost-effective at the hypothetical societal willingness-to-pay threshold of £1400.
| Discount applied to aflibercept (%) | Aflibercept cost (£), a | Aflibercept injection procedure cost (£), b | Total cost of aflibercept and aflibercept injection procedure (£), c (c = a + b) | PRP laser treatment cost (£) | Cost difference between groupsa (£) | Effect difference between groups, change in BCVA letter scorea | ICER [cost (£) per unit change in BCVA letter score] | Interpretation of ICER results |
|---|---|---|---|---|---|---|---|---|
| 0 | 816.00 | 182.00 | 998.00 | 131.00 | 5474.57 | 3.93 | 1392.99 |
Aflibercept intervention is more effective and more costly (Non-dominance – trade-off) |
| 20 | 652.80 | 182.00 | 834.80 | 131.00 | 4774.83 | 3.93 | 1214.94 |
Aflibercept intervention is more effective and more costly (Non-dominance – trade-off) |
| 40 | 489.60 | 182.00 | 671.60 | 131.00 | 4075.21 | 3.93 | 1036.92 |
Aflibercept intervention is more effective and more costly (Non-dominance – trade-off) |
| 60 | 326.40 | 182.00 | 508.40 | 131.00 | 3375.59 | 3.93 | 858.91 |
Aflibercept intervention is more effective and more costly (Non-dominance – trade-off) |
| 80 | 163.20 | 182.00 | 345.20 | 131.00 | 2675.86 | 3.93 | 680.86 |
Aflibercept intervention is more effective and more costly (Non-dominance – trade-off) |
| 100 | 0.00 | 182.00 | 182.00 | 131.00 | 1976.24 | 3.93 | 502.85 |
Aflibercept intervention is more effective and more costly (Non-dominance – trade-off) |
Subgroup analysis
We performed a subgroup analysis by the presence of DMO at baseline [no DMO in both eyes at baseline, n = 126, vs. DMO in at least one eye (in this study, this applies to fellow eye only as DMO was an exclusion in the study eye) at baseline, n = 76]. Table 31 shows that the incremental costs and effects of alternative arms were £5552.25 (bootstrapped 95% CI £5217.24 to £5873.87) and 3.98 (bootstrapped 95% CI 3.87 to 4.09) for the ‘no DMO in both eyes at baseline’ group, and £5351.48 (bootstrapped 95% CI £4894.25 to £5805.31) and 3.86 (bootstrapped 95% CI 3.70 to 4.02) for the ‘DMO in at least one eye at baseline’ group. This yields an ICER of £1394.27 per change in BCVA score (bootstrapped 95% CI £1305.87 to £1484.50) and £1387.45 per change in BCVA score (bootstrapped 95% CI £1395.86 to £1441.65) for the ‘no DMO in both eyes at baseline’ group and the ‘DMO in at least one eye at baseline’ group, respectively. The subgroup analysis showed there to be no meaningful difference in the cost per change in BCVA when subgroup analysis of this kind was undertaken.
| Number | Analysis method,a aflibercept unit cost of £816 (list price) | Incremental costb (£) | Incremental effectb (change in BCVA score) | ICER [cost per unit change in BCVA score (£)] | Interpretation of ICER results | Probability of cost-effectiveness (%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| At £900 | At £1050 | At £1400 | At £1500 | At £1600 | ||||||
| 1 | Primary cost-effectiveness analysis – cost and effect (change in BCVA score) | 5475 | 3.93 | 1393 |
Aflibercept intervention is more effective and more costly (Non-dominance – trade-off) |
0.00 | 0.00 | 56.60 | 99.70 | 100.0 |
| 2a |
Subgroup analysis – split by presence of macular oedema at baseline (Macular oedema baseline = no macular oedema in both eyes at baseline) |
5552 | 3.98 | 1394 |
Aflibercept intervention is more effective and more costly (Non-dominance – trade-off) |
0.00 | 0.00 | 56.08 | 98.78 | 100.0 |
| 2b |
Subgroup analysis – split by presence of macular oedema at baseline (Macular oedema baseline = macular oedema in at least one eye at baseline) |
5351 | 3.86 | 1387 |
Aflibercept intervention is more effective and more costly (Non-dominance – trade-off) |
0.00 | 0.00 | 57.28 | 95.88 | 99.96 |
| Number | Analysis method,a sensitivity analyses | Incremental costb (£) | Incremental effectb (change in BCVA score) | ICER [cost per unit change in BCVA score (£)] | Interpretation of ICER results | Probability of cost-effectiveness (%) | ||||
| At £900 | At £1050 | At £1400 | At £1500 | At £1600 | ||||||
| 3 | 0% discount applied to aflibercept | 5475 | 3.93 | 1393 |
Aflibercept intervention is more effective and more costly (Non-dominance – trade-off) |
0.00 | 0.00 | 56.60 | 99.70 | 100.0 |
| 4 | 20% discount applied to aflibercept | 4775 | 3.93 | 1215 |
Aflibercept intervention is more effective and more costly (Non-dominance – trade-off) |
0.00 | 0.00 | 100.0 | 100.0 | 100.0 |
| 5 | 40% discount applied to aflibercept | 4075 | 3.93 | 1037 |
Aflibercept intervention is more effective and more costly (Non-dominance – trade-off) |
0.00 | 63.54 | 100.0 | 100.0 | 100.0 |
| 6 | 60% discount applied to aflibercept | 3376 | 3.93 | 859 |
Aflibercept intervention is more effective and more costly (Non-dominance – trade-off) |
86.66 | 100.0 | 100.0 | 100.0 | 100.0 |
| Number | Analysis method,a aflibercept unit cost of £816 (list price) | Incremental costb (£) | Incremental effectb (QALY) | ICER [cost per QALY (£)] | Interpretation of ICER results | Probability of cost-effectiveness (%) | ||||
| At £40,000 | At £80,000 | At £160,000 | At £320,000 | At £600,000 | ||||||
| 7 | 80% discount applied to aflibercept | 2676 | 3.93 | 681 |
Aflibercept intervention is more effective and more costly (Non-dominance – trade-off) |
100.0 | 100.0 | 100.0 | 100.0 | 100.0 |
| 8 | 100% discount applied to aflibercept | 1976 | 3.93 | 503 |
Aflibercept intervention is more effective and more costly (Non-dominance – trade-off) |
100.0 | 100.0 | 100.0 | 100.0 | 100.0 |
| 9 | Secondary exploratory cost–utility analysis – cost and effect (QALYs) | 5475 | –0.022 | –252,827 |
Aflibercept intervention is less effective and more costly (Dominance – Intervention dominated) |
0.00 | 0.08 | 2.42 | 8.64 | 13.98 |
Table 31 summarises our health economics findings. All analyses used bootstrapping with 5000 replications. In the far-right column of Table 31, we show how the probability that aflibercept is cost-effective increases when society is willing to pay more to achieve a 1-point improvement in BCVA.
Chapter 5 Mechanistic evaluation
Objectives
-
To explore whether intravitreal aflibercept compared with PRP causes measurable regression of retinal NV at 12 and 52 weeks in terms of decimal disc area units in 4-field colour photographs and FFA: difference in means.
-
To explore differences in the mean change in retinal vessel calibre and oxygen saturation in eyes treated with intravitreal aflibercept compared with PRP at 12 and 52 weeks: difference in means.
-
To explore whether intravitreal aflibercept reduces angiographically quantifiable areas of retinal non-perfusion compared with panretinal photocoagulation through 52 weeks: means and proportions.
Participants for the mechanistic study
The mechanistic substudy was carried out on 40 patients recruited at Moorfields Eye Hospital. These patients also underwent retinal oximetry in addition to the other study assessments carried out at baseline, 12 and 52 weeks. A total of 20 patients were in the aflibercept arm and 20 were in the PRP arm. A total of 20 patients were treatment-naive and 20 patients had been previously treated with PRP but had persistent new vessels at baseline.
Retinal oximetry
Retinal oximetry was performed using the retinal oximeter (Oxymap T1 device connected to Topcon TRC50-DX fundus camera; Oxymap ehf., Reykjavik, Iceland). 78
The retinal oximeter consists of a fundus camera with an attached image splitter as well as a digital camera that captures images at two wavelengths, 605 nm and 586 nm. Optical density is sensitive to oxygen saturation at 605 nm but not at the reference wavelength of 586 nm. The oxyhaemoglobin saturation (SO2) of a vessel can therefore be calculated because the optical density ratio at these wavelengths has been shown to have an approximately inverse linear relationship with SO2. Although the saturation data are not absolute, the oximeter has been shown to give reproducible results and to be sensitive to changes in oxygen saturation. The comparison of data in the same eye before and after treatment is therefore valid. 79
Optic disc-centred images were captured through dilated pupils. The images obtained were 1200 × 1600 pixels and covered a 50° field of central retina. The images were captured for both eyes but only the study eye was used in this analysis. Images were analysed by two independent observers (Luke Nicholson and Roxanne Crosby-Nwaobi) using the Oxymap Analyser software (Oxymap ehf., Reykjavik, Iceland) version 2.5, which automatically detects vessels > 8 pixels in diameter. The area analysed was selected by centring quadrant lines on the optic disc. An initial central circle was used to delineate the optic disc. Two additional measurement circles, an inner and outer circle, two and four times the diameter of the central circle, respectively, were then demarcated. The area between the circles centred on the optic disc was analysed. The width of this ring was two disc diameters (DDs). Vessels beyond the area of analysis, certain areas where vessel detection would prove inaccurate (branching, overlapping or intersecting) and segments of vessels < 19 pixels in length were excluded. Vessels to be measured were selected manually. Measurements were first taken to yield arteriolar SO2 (SaO2) by quadrant. This was repeated for venules to yield venular SO2 (SvO2) by quadrant. Overall saturation was computed by averaging saturation values of each quadrant. Because Oxymap software measurements are calibrated to non-diabetic young individuals, results are relative to that calibration, occasionally resulting in SO2 measurements > 100%. These values were not truncated to 100%, per established oximetry protocol. The retinal oximetry data streams were extracted from the source data file. The data streams underwent multistep preprocessing to eliminate missing or invalid data. The data were inspected for (1) interrupted regions of the recording (as noted in the research record) and (2) regions where it was not able to properly measure saturations. The entire data epoch was rejected if the data stream failed one or more of these checks or if continuous measurements were not available. Data collected from the Oxymap software included the mean oxygen saturation in the identified segments of first and second branch retinal arterioles (SaO2) and venules (SvO2) within 0.5–1.0 optic DDs from the disc margin, and their difference, the Sa – vO2. The mean arterial and venous diameters were also recorded.
Ultrawide field colour fundus photography
All 40 patients underwent CFP and fluorescein angiography using the Optos 200TX (Optos Plc, Dumfermline, UK) ultrawide field system.
The 200° images were captured with steering, that is aligning images with three axes, eyes looking at centre, superiorly and inferiorly. The pattern of vessel regression was assessed by one investigator (JR) and confirmed by a second investigator (SS or LN) (Table 32).
| Ring | Original area per segment, DA | Enlargement factor | Modified area per segment, DA |
|---|---|---|---|
| M | 2.00 | 1.08 | 1.85 |
| 1 | 6.25 | 1.20 | 5.21 |
| 2 | 10.42 | 1.34 | 7.78 |
| 3 | 14.58 | 1.54 | 9.47 |
| 4 | 18.75 | 1.81 | 10.36 |
| 5 | 22.92 | 1.97 | 11.63 |
Ultrawide field fundus fluorescein angiography
The FFA images were acquired after intravenous bolus infusion of 5 ml of 20% fluorescein sodium. The images were acquired at transit phase (up to 45 seconds), arteriovenous phases (3–4 minutes) and late frames at 5–7 minutes. A single investigator (LN) identified the best macular-centred fluorescein angiography (FA) image in the arteriovenous phase from the FA series of each eligible eye. A correction factor was applied for the flattening of the three-dimensional image to a two-dimensional image using the Optos V2 Vantage Pro software (Dunfermline, Scotland). Capillary non-perfusion was defined as an area of the capillary network that failed to fill with fluorescein by the late arteriovenous phase, with minimum linear dimension of at least 63 µm (width of a retinal venule at the disc margin).
In order to assess spatial location and quantity of non-perfusion and area of new vessels, a concentric rings template was applied to each image in accordance with previously described methods. 80 In brief, this validated method incorporates a macular ring with a radius of 2.5 DDs and five additional concentric rings (rings 1–5), each with a 2.5-DD increment in radius. Each of the six rings (ring M and rings 1–5) was divided into 12 segments. Each segment is graded as ungradeable, not perfused or perfused (if ≥ 50% of the segment is involved). In addition to quantifying non-perfusion, the concentric rings method allows documentation of location of non-perfusion. The area of each cell in each concentric ring was modified based on the enlargement factor identified using 3D printed model eyes. 81 The enlargement factors for rings M, 1, 2, 3, 4 and 5 were 1.08, 1.20, 1.34, 1.54, 1.81 and 1.97, respectively. The modified area of each segment in each ring is represented in Table 32.
Statistical analysis
The oximetry measurements, new vessel area and area of retinal non-perfusion were described using means and SD at cross-sectional time points within each arm, and the change from baseline to follow-up time points in each arm was estimated by the mean change and its standard error (SE). The outcomes were compared between aflibercept and PRP arms at both 12 and 52 weeks for oximetry and area of new vessels and at 52 weeks for area of retinal non-perfusion using ANCOVA. If the data contained highly skewed outcomes, the Mann–Whitney U-test was used instead and data were also described using medians and interquartile ranges (IQRs). The statistical significance was set at 0.05.
Mechanistic results
The baseline characteristics of the 40 patients were evenly matched. Retinal oximetry measurements reveal fairly similar SaO2 and SvO2 saturations in both arms at every visit. The mean Sa – vO2 at baseline was 36.7% in the PRP group and 33.4% in the aflibercept group. At week 12, the Sa – vO2 was 36.1% and 35.5% for the PRP and aflibercept group, respectively. At week 52, the Sa – vO2 increased in the PRP arm to 39.7% and minimally decreased in the aflibercept arm to 32.5%. However, the –4.0% (95% CI –8.8% to 0.8%) adjusted difference in the change from baseline between the two groups was not found to be statistically significant (p = 0.10). Mean arterial and venous diameter decreased in both groups at week 52, again with no significant difference between groups. Table 33 shows the detailed retinal oximetry values in the whole cohort and Tables 34 and 35 show the oximetry values in the treatment-naive and non-naive groups, respectively.
| Sa – vO2 | Treatment arm, mean (SD); n | Change from baseline, mean (SE) | Adjusted difference between arms (95% CI) | p-value | ||
|---|---|---|---|---|---|---|
| PRP | Aflibercept | PRP | Aflibercept | |||
| Mean arterial SaO2 (%) | ||||||
| Baseline | 98.0 (9.7); 19 | 98.0 (9.8); 19 | ||||
| 12 weeks | 98.6 (10.0); 18 | 100.4 (10.2); 18 | 0.7 (2) | 2.3 (1.3) | 1.6 (–2.9 to 6.2) | 0.47 |
| 52 weeks | 102.5 (12.8); 18 | 98.9 (7.1); 18 | 3.7 (2) | 0.9 (1.3) | –3.0 (–7.8 to 1.9) | 0.22 |
| Mean venous SaO2 (%) | ||||||
| Baseline | 63.2 (7.0); 19 | 67.2 (8.4); 19 | ||||
| 12 weeks | 63.8 (7.8); 18 | 66.7 (6.6); 18 | 0.4 (1.8) | –0.3 (1.4) | 1.0 (–3.2 to 5.1) | 0.64 |
| 52 weeks | 63.4 (7.9); 18 | 67.7 (4.4); 18 | 0.3 (1.4) | 0.4 (1.4) | 2.0 (–1.4 to 5.5) | 0.23 |
| Mean arterio venous SaO2 (%) | ||||||
| Baseline | 36.7 (10.2); 19 | 33.4 (8.2); 19 | ||||
| 12 weeks | 36.1 (14.7); 18 | 35.5 (6.7); 18 | –0.3 (2.5) | 1.9 (1.4) | 1.8 (–4.1 to 7.6) | 0.54 |
| 52 weeks | 39.7 (11.9); 18 | 32.5 (6.9); 18 | 2.1 (2.1) | –0.9 (1.2) | –4.0 (–8.8 to 0.8) | 0.10 |
| Mean arterial diameter (µm) | ||||||
| Baseline | 113.5 (10.2); 19 | 111.8 (16.3); 19 | ||||
| 12 weeks | 111.0 (8.5); 18 | 108.8 (11.0); 18 | –3.3 (2.1) | –2.7 (3.6) | –1.3 (–7.3 to 4.8) | 0.67 |
| 52 weeks | 107.4 (9.3); 18 | 109.3 (14.4); 18 | –5.5 (2.1) | –3.4 (2.4) | 2.0 (–3.8 to 7.8) | 0.48 |
| Mean venous diameter | ||||||
| Baseline | 143.4 (13.1); 19 | 146.7 (16.6); 19 | ||||
| 12 weeks | 144.9 (14.8); 18 | 144.1 (12.3); 18 | 0.2 (2.6) | –3.3 (4.6) | –1.7 (–10.4 to 7.0) | 0.69 |
| 52 weeks | 140.5 (12.7); 18 | 143.9 (15.4); 18 | –3.8 (2.0) | –1.4 (3.8) | 2.8 (–5.0 to 10.7) | 0.47 |
| Sa – vO2 | Treatment arm, mean (SD); n | Change from baseline, mean (SE) | Adjusted difference between arms (95% CI) | ||
|---|---|---|---|---|---|
| PRP | Aflibercept | PRP | Aflibercept | ||
| Mean arterial SaO2 (%) | |||||
| Baseline | 96.4 (10.7); 11 | 99.3 (9.0); 13 | |||
| 12 weeks | 98.2 (8.0); 11 | 102.2 (10.9); 12 | 1.8 (2.1) | 2.8 (1.2) | 0.2 (–5.4 to 5.8) |
| 52 weeks | 101.5 (13.8); 10 | 100.7 (6.5); 13 | 4.0 (1.8) | 1.4 (1.4) | 2.7 (–2.0 to 7.3) |
| Mean venous SaO2 (%) | |||||
| Baseline | 61.9 (8.4); 11 | 69.1 (9.0); 13 | |||
| 12 weeks | 65.0 (8.4); 11 | 69.0 (6.5); 12 | 3.2 (2.2) | 0.0 (2.1) | 0.2 (–5.4 to 5.8) |
| 52 weeks | 62.0 (9.4); 10 | 69.1 (4.4); 13 | 0.5 (1.8) | –0.1 (1.7) | 2.7 (–2.0 to 7.3) |
| Mean arteriovenous SaO2 (%) | |||||
| Baseline | 35.3 (10.7); 11 | 33.7 (8.6); 13 | |||
| 12 week | 34.1 (10.4); 11 | 35.7 (7.9); 12 | –1.2 (2.9) | 1.5 (1.8) | 2.3 (–4.0 to 8.6) |
| 52 weeks | 39.3 (10.7); 10 | 33.3 (6.8); 13 | 2.7 (2.4) | –0.5 (1.5) | –4.0 (–9.2 to 1.2) |
| Mean arterial diameter (µm) | |||||
| Baseline | 114.3 (12.2); 11 | 111.3 (17.6); 13 | |||
| 12 weeks | 112.3 (9.2); 11 | 108.8 (11.5); 12 | –2.0 (3.0) | –2.0 (4.6) | –2.3 (–10.2 to 5.7) |
| 52 weeks | 108.4 (9.1); 10 | 109.7 (15.7); 13 | –5.0 (2.9) | –1.6 (2.1) | 2.8 (–3.6 to 9.3) |
| Mean venous diameter (µm) | |||||
| Baseline | 146.4 (11.8); 11 | 142.6 (12.9); 13 | |||
| 12 weeks | 145.4 (12.4); 11 | 145.9 (13.6); 12 | –1.0 (3.4) | 2.4 (4.3) | 1.9 (–8.5 to 12.4) |
| 52 weeks | 144.5 (8.4); 10 | 145.0 (15.7); 13 | –3.8 (1.6) | 2.3 (2.7) | 5.4 (–1.9 to 12.8) |
| Sa – vO2 | Treatment arm, mean (SD); n | Change from baseline, mean (SE) | Adjusted difference between arms (95% CI) | ||
|---|---|---|---|---|---|
| PRP | Aflibercept | PRP | Aflibercept | ||
| Mean arterial SaO2 (%) | |||||
| Baseline | 100.3 (8.3); 8 | 95.2 (11.6); 6 | |||
| 12 weeks | 99.3 (13.3); 7 | 96.7 (8.2); 6 | –0.9 (3.9) | 1.5 (3.2) | 1.9 (–5.2 to 9.0) |
| 52 weeks | 103.7 (12.3); 8 | 94.4 (7.0); 5 | 3.5 (11.8) | –0.2 (3.4) | –1.0 (–7.3 to 5.3) |
| Mean venous SaO2 (%) | |||||
| Baseline | 65.0 (4.0); 8 | 62.9 (4.9); 6 | |||
| 12 weeks | 61.7 (6.9); 7 | 61.9 (4.0); 6 | –4.0 (2.7) | –1.0 (0.9) | 1.9 (–5.2 to 9.0) |
| 52 weeks | 65.1 (5.5); 8 | 64.2 (1.9); 5 | 0.1 (6.6) | 1.6 (2.8) | –1.0 (–7.3 to 5.3) |
| Mean arteriovenous SaO2 (%) | |||||
| Baseline | 38.7 (9.9); 8 | 32.6 (8.1); 6 | |||
| 12 weeks | 39.1 (20.4); 7 | 35.3 (4.1); 6 | 0.9 (4.7) | 2.7 (2.5) | 3.0 (–10.5 to 16.5) |
| 52 weeks | 40.1 (14.0); 8 | 30.6 (7.5); 5 | 1.4 (10.6) | –1.9 (1.9) | –4.2 (–16.5 to 8.1) |
| Mean arterial diameter (µm) | |||||
| Baseline | 112.4 (7.1); 8 | 112.9 (14.5); 6 | |||
| 12 week | 109.0 (7.4); 7 | 108.9 (11.1); 6 | –5.2 (3.0) | –4.0 (6.3) | 0.2 (–11.4 to 11.8) |
| 52 weeks | 106.2 (10.0); 8 | 108.1 (11.7); 5 | –6.2 (9.6) | –8.2 (6.8) | 0.6 (–13.2 to 14.3) |
| Mean venous diameter (µm) | |||||
| Baseline | 139.2 (14.6); 8 | 155.3 (21.6); 6 | |||
| 12 weeks | 144.2 (19.0); 7 | 140.6 (8.9); 6 | 2.0 (4.0) | –14.8 (9.6) | –8.4 (–27.9 to 11.1) |
| 52 weeks | 135.4 (15.8); 8 | 141.0 (16.2); 5 | –3.8 (11.7) | –11.1 (11.5) | 0.7 (–19.9 to 21.2) |
The mean total area of retinal non-perfusion increased in both PRP and aflibercept groups with no statistically significant difference between groups. At baseline, the total area of retinal non-perfusion was 125.1 disc areas in the PRP group and 131.2 disc areas in the aflibercept group. At week 52, this increased to 156.1 disc areas and 158.4 disc areas in the PRP and aflibercept groups, respectively. The proportion of eyes that experienced an increase in retinal non-perfusion of more than 20 disc areas was 51.6%, while 22.6% had an increase in retinal non-perfusion of 5–20 disc areas and 25.8% did not experience a significant increase (< 5 disc area change). This is further detailed in Table 36.
| Sa – vO2 | Treatment arm, mean (SD); n | Change from baseline, mean (SE) | Adjusted difference between arms (95% CI) | p-value | ||
|---|---|---|---|---|---|---|
| PRP | Aflibercept | PRP | Aflibercept | |||
| M + 1 | ||||||
| Baseline | 15.4 (12.2); 18 | 16.9 (15.6); 18 | 0.24 | |||
| 52 weeks | 19.6 (14.7); 18 | 14.8 (11.7); 17 | 4.0 (3.6) | –1.2 (2.6) | –5.0 (–13.4 to 3.5) | |
| Treatment naive | ||||||
| Baseline | 16.0 (11.0); 11 | 12.3 (8.0); 12 | ||||
| 52 weeks | 18.2 (17.5); 9 | 16.5 (8.9); 11 | 4.3 (5.9) | 3.0 (2.0) | –2.2 (–14.6 to 10.2) | |
| Non-naive | ||||||
| Baseline | 14.5 (14.7); 7 | 26.1 (23.1); 6 | ||||
| 52 weeks | 21.1 (12.2); 9 | 11.7 (16.1); 6 | 3.7 (3.9) | –9.4 (5.0) | –11.3 (–24.3 to 1.6) | |
| 2 + 3 + 4 | ||||||
| Baseline | 109.6 (48.7); 18 | 114.3 (65.7); 18 | 0.23 | |||
| 52 weeks | 136.4 (62.1); 18 | 143.6 (53.7); 17 | 23.5 (11.5) | 39.2 (9.7) | 17.4 (–11.8 to 46.5) | |
| Treatment naive | ||||||
| Baseline | 116.5 (55.1); 11 | 117.3 (69.1); 12 | ||||
| 52 weeks | 146.6 (69.6); 9 | 149.1 (62.6); 11 | 37.7 (16.6) | 37.0 (12.4) | 2.9 (–38.6 to 44.4) | |
| Non-naive | ||||||
| Baseline | 98.9 (37.7); 7 | 108.3 (64.1); 6 | ||||
| 52 weeks | 126.3 (55.9); 9 | 133.5 (34.5); 6 | 5.2 (13.5) | 43.5 (17.1) | 30.8 (–11.6 to 73.3) | |
| Total | ||||||
| Baseline | 125.1 (55.1); 18 | 131.2 (75.4); 18 | 0.46 | |||
| 52 weeks | 156.1 (74.7); 18 | 158.4 (54.5); 17 | 27.5 (13.3) | 38.0 (9.6) | 12 (–20.7 to 44.7) | |
| Treatment naive | ||||||
| Baseline | 132.5 (60.7); 11 | 129.7 (74.3); 12 | ||||
| 52 weeks | 164.8 (85.4); 9 | 165.6 (63.6); 11 | 41.9 (19.7) | 40.0 (12.3) | 0.3 (–47.6 to 48.2) | |
| Non-naive | ||||||
| Baseline | 113.3 (46.9); 7 | 134.4 (84.8); 6 | ||||
| 52 weeks | 147.4 (66.2); 9 | 145.1 (33.1); 6 | 8.9 (15.3) | 34.0 (16.7) | 21.3 (–21.6 to 64.3) | |
The total area of new vessels was reduced from baseline with treatment in both arms with more rapid and significant regression in the aflibercept arm at 12 weeks (p = 0.019). By 52 weeks, the PRP arm also showed similar regression of new vessel area (p = 0.45). This is further detailed in Table 37.
| Sa – vO2 | Treatment arm, mean (SD); n | Change from baseline, mean (SE) | p-valuea | ||
|---|---|---|---|---|---|
| PRP | Aflibercept | PRP | Aflibercept | ||
| Total new vessel area (mm2) | |||||
| Baseline | 1.54 (1.75); 19 | 1.68 (1.99); 18 | |||
| 12 weeks | 1.13 (1.36); 19 | 0.00 (0.00); 18 | –0.42 (0.16) | –1.68 (0.47) | 0.019 |
| 52 weeks | 0.35 (0.42); 16 | 0.11 (0.23); 18 | –1.03 (0.41) | –1.57 (0.45) | 0.45 |
| Total new vessel area: naive subgroup (mm2) | |||||
| Baseline | 1.22 (1.28); 11 | 1.09 (1.34); 12 | |||
| 12 weeks | 0.98 (1.09); 11 | 0.00 (0.00); 12 | –0.25 (0.22) | –1.09 (0.39) | |
| 52 weeks | 0.45 (0.48); 9 | 0.04 (0.06); 12 | –0.93 (0.52) | –1.05 (0.39) | |
| Total new vessel are: non-naive subgroup (mm2) | |||||
| Baseline | 1.98 (2.28); 8 | 2.87 (2.63); 6 | |||
| 12 weeks | 1.33 (1.73); 8 | 0.00 (0.00); 6 | –0.66 (0.25) | –2.87 (1.07) | |
| 52 weeks | 0.23 (0.33); 7 | 0.25 (0.38); 6 | –1.17 (1.8) | –2.62 (1.03) | |
Chapter 6 Discussion
Summary of findings
The CLARITY study showed that intravitreal aflibercept monotherapy was superior to standard PRP treatment for PDR through 52 weeks; the effect was achieved with a median of one aflibercept injection only in the 40 week post-loading phase, indicating that aflibercept is a feasible new approach for compliant patients. The superior BCVA findings were supported by significantly better binocular visual acuity and binocular Esterman scores in the aflibercept arm. These observations have significant impact on eligibility to retain a driving licence. In the UK, the Driver and Vehicle Licensing Agency has designated both a minimum visual acuity and Esterman visual field standard to maintain a valid driving licence. 83 With advances in laser technology and techniques, there are reports with short follow-up suggesting that modern-day laser techniques and technology, such as multispot laser, have reduced the prevalence of visual field loss with PRP. 84,85 However, our study shows that, despite 69% of the study cohort being treated with multispot laser, aflibercept is associated with a lower risk of visual field loss than modern-day laser at 52 weeks, in keeping with findings noted in the recent ranibizumab trial in PDR at 2 years. 53 Other visual outcomes that measured adverse effects of PRP, such as contrast sensitivity and low-luminance visual acuity, were not significantly different between arms, although removing outliers suggested greater preservation of low-luminance visual acuity letter score by 52 weeks in the aflibercept arm.
Panretinal photocoagulation remains an effective treatment in preventing severe visual loss. 52 On average, the visual acuity remained very stable over 52 weeks but twice as many people lost 10 or more letters when treated with PRP. In contrast, the proportion of patients who lost visual acuity was minimal in the aflibercept arm. However, the study reiterates the Protocol S study in that PRP is not a one-off procedure: 65% of patients require supplemental therapy despite advances in laser technology.
Aflibercept also improved the level of diabetic retinopathy severity, which resulted in a higher proportion of eyes with total regression of new vessels than PRP. This anatomical effect should also be considered when choosing between anti-VEGF and PRP as a first-line option in PDR. As aflibercept is licensed for DMO, the findings of this study indicate that aflibercept is also effective in the management of PDR in the first year, allowing the use of a single agent to address both of these sight-threatening complications of diabetes mellitus.
These data are likely to promote a paradigm shift in the treatment of PDR.
Economic evaluation
Our objective was to evaluate the cost-effectiveness of intravitreal aflibercept therapy for PDR versus standard care, PRP.
Economic evaluation was undertaken on 202 participants (101 per arm) with complete cost and outcome data. This represents 96.7% of the clinical sample included in primary outcome ITT analysis. From a public sector multiagency perspective that covers health and social care services, treatment with aflibercept costs more in terms of total resource use (mean adjusted total additional cost per patient = £5475, bootstrapped 95% CI £5210.79 to £5749.82) than PRP laser treatment over the 52-week follow-up period. Sensitivity analysis in which we varied the costs of aflibercept from the list price to reflect possible NHS PAS showed this to be the case at any price because of the additional cost of administering aflibercept. Participants who received aflibercept gained some benefit in BCVA (mean = 3.93, bootstrapped 95% CI 3.84 to 4.02) but at an increased cost. No statistically significant difference was found in self-reported generic HRQoL (EQ-5D-3L) or in terms of capability (ICECAP-A). It may be that these measures were not sufficiently sensitive to pick up any changes over the 52-week follow-up period between arms. We have undertaken a secondary exploratory cost–utility analysis (i.e. cost-per-QALY analysis). Evidence is mixed, but points to the EQ-5D-3L not being sufficiently sensitive to be useful in studies of visual impairment. 44,45 The results of our study speak for themselves: a mean adjusted cost difference of £5475 is divided by an extremely small and not statistically significant mean adjusted QALY difference of –0.022. This yields a cost per QALY of –£252,827, in which we do not have much confidence. Given that a positive significant difference was observed in the BCVA for the intervention group, we interpreted this as the EQ-5D-3L not being sufficiently sensitive in this context. Interestingly, the vision-specific self-reported health-related QoL measures (RetDQoL and NEI-VFQ-25, non-preference-based) also showed no statistically significant difference over the study period between arms. If society is willing to pay £1400 for an additional 1-point improvement in BCVA, then aflibercept has a 56.60% probability of being cost-effective at the list price of £816.
Mechanistic evaluation
This section is adapted from Nicholson et al. 82 ©The Authors, 2018. This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/.
Our study on 40 patients (20 in each arm) did not reveal any significant change in retinal oximetry between arms. This may be because of the small sample size. The study was not powered to detect important clinically significant differences between arm. The study was exploratory and hypothesis driven to decide on sample size of future studies in this area if changes observed were worth pursuing. The SaO2 and SvO2 at baseline were both high compared with the normative data on retinal oximetry, in keeping with reports on retinal oximetry in PDR. In addition, the standard deviations of the values for both SaO2 and SvO2 are similar to reports on PDR, suggesting that the non-significant changes are unlikely to be errors in measurement. After accounting for test–retest variability, we would require a mean of 5% change in SO2 in the arteries and veins to confidently report a treatment effect. 78 This was not achieved in either the treatment-naive group or the post-PRP-treated non-naive group in either arm. Therefore, although there may be an overall change in the distribution of oxygen supply and consumption with either treatment, our study results show that these treatments do not affect the overall oxygen exchange in the retina. Guduru et al. 86 reported a correlation between retinal capillary non-perfusion and retinal arterial SaO2. Our study participants in the mechanistic study did show significant areas of capillary non-perfusion but both aflibercept and PRP failed to show any difference in change in capillary non-perfusion at 52 weeks, with patients in both arms showing a continuing increase in area of total non-perfusion. This increase in non-perfusion was not associated with a corresponding change in retinal oximetry values, emphasising that retinal oximetry represents only global change in oxygen and may not be accurate enough to measure local changes in redistribution of oxygen demand and supply. 87
Strengths and limitations
This study included patients presenting prospectively to the medical retina clinics where patients are managed in a real-life setting so the study allowed accurate testing of the hypothesis. The study also included both treatment-naive eyes and eyes with persistent NV post-initial PRP and was therefore representative of the UK population who would benefit from an alternative treatment to PRP. Patients who have been previously treated are often excluded from ophthalmology trials and, therefore, this is the first study that demonstrated the impact of anti-VEGF in this indication. We excluded eyes with macular oedema to avoid any confounding effect on the primary outcome analysis. Therefore, the study provided the true benefit of anti-VEGF agent in PDR. Participants underwent thorough phenotyping including clinical, qualitative and objective assessment and an independent Reading Centre also further graded the retinal images to ensure a robust validation of the study anatomical outcomes.
The study was slow to recruit initially but we successfully met the target by increasing recruitment of five clinical trial sites in time. The main reason for the slow recruitment in the initial phase was the additional burden on busy clinics, the provision of PRP being available on the same day as the retinal consultation, and the perception that anti-VEGF therapy is short-acting. The increase in performance across the sites in the later half of the study may be because of the availability of new data on the effectiveness of anti-VEGF on diabetic retinopathy and so the research team became more reassured that anti-VEGF agents may indeed be a useful treatment for PDR too.
The robust randomised controlled trial (RCT) design, high statistical power and excellent retention rates are particular strengths of this study. The study patients are representative of the PDR population and, therefore, these findings can be generalised to clinical practice for the first year of therapy. Retreatment criteria used in CLARITY were very similar to those followed in the ranibizumab trial17 and were determined by treating investigators at each study visit. Compliance with treatment (94% aflibercept arm and 97% PRP arm) was very good in CLARITY, indicating that these retreatment criteria can be easily applied to routine clinical practice.
The safety evaluation of aflibercept in CLARITY revealed no new concerns on this drug. There were no statistically significant differences in APTC events or other systemic adverse events between arms at 52 weeks, substantiating the reports of previous intravitreal aflibercept studies in diabetic patients. Long-term studies on anti-VEGF in PDR patients are required for meta-analysis of the safety of these agents in PDR.
The limitation of this study is that it was a Phase IIb study with follow-up for only 52 weeks. To date, the only other well-designed study on anti-VEGF for PDR included patients with DMO and so the treatment regimen was preplanned to be more intense than this study. 11 However, as a 5-year study, it will provide long-term outcomes of ranibizumab in PDR, information on the disease-modifying effect of anti-VEGF and the long-term compliance of patients. The study also excluded patients with significant fibrovascular proliferation or tractional retinal detachment in the posterior pole and eyes with prior vitrectomy. We have limited experience of treating severe retinopathy at the posterior pole with anti-VEGF therapy and so we decided that this group is best avoided as risks may outweigh benefits.
Approximately 75% of the PDR patients included in this study had low-risk PDR. This mirrors the study population in Protocol S suggesting that PRP is now initiated in patients with low-risk PDR.
We did not collect information on ADL that would have told us something about how treatment of this condition with aflibercept versus PRP affects home and work life.
This study used a 12-month recall period for contacts with health and social care services, which is considered reasonable but towards the boundary of recall for patients. 88
For the economic evaluation, we were primarily interested in our health economic analysis on how the cost per change in BCVA compared with the cost per QALY and hence decided to use complete-case analysis where full data were available for these outcome measures. We do not impute costs and, therefore, this left us with 202 participants (this represents 96.7% of the clinical sample included in primary outcome ITT analysis).
Comparison with existing literature
The DRCR.net performed a multicentre (55 sites), randomised clinical trial comparing panretinal photocoagulation with 0.5 mg of intravitreal ranibizumab in 305 patients with PDR. 52,53 PRP was performed at baseline and ranibizumab was given at baseline and week 4 pro re nata. Eyes with DMO in both groups were eligible to receive ranibizumab. The primary outcome was change in BCVA and the secondary outcomes included area under the visual acuity curve, peripheral visual field loss (as measured on Humphrey automated visual field testing), incidence of vitrectomy, development of DMO, and persistent or new neovascularisation. Improvements in BCVA for the ranibizumab and PRP groups were +2.2 and +0.2 letters, respectively (95% CI –0.5 to + 5.0). The group receiving ranibizumab experienced less peripheral visual field sensitivity loss (–23 dB vs. –422 dB, 95% CI 213 dB to 531 dB; p < 0.001), fewer vitrectomies (4% vs. 15%, 95% CI 4% to 15%; p < 0.001), and a lower incidence of DMO (9% vs. 28%). Ranibizumab-treated eyes required a median of seven injections through year 1 and 10 injections through year 2. Forty-five per cent of eyes in the PRP group required additional laser and 53% of eyes required ranibizumab for DMO. The authors concluded that ranibizumab may be a reasonable alternative to PRP through 2 years. The decreasing number of injections in year 2 suggests that some disease modulation occurs after 1 year of ranibizumab therapy.
Comparison with other economic evaluation studies
A US study exploring the cost-effectiveness of intravitreal ranibizumab compared with PRP for PDR found that over 2 years, compared with PRP, 0.5 mg of ranibizumab was within the US$50,000 to US$150,000 per QALY range frequently cited as cost-effective in the USA for eyes presenting with PDR and vision-impairing DMO, but not for those with PDR without vision-impairing DMO. 88 The US study measured costs associated with ocular treatments and potential complications only, and based quality-of-life adjustments in QALY calculations on utility weights by mapping from Brown et al. 89 for a 2-year period. Sensitivity analysis explored the effect on the ICER of time trade-off responses from trial participants. Brown et al. 89 did not collect preference-based utility data directly from patients.
Implications for practice and research recommendations
In addition to improving the visual acuity, intravitreal aflibercept also reduced the level of diabetic retinopathy severity and resulted in a higher proportion of eyes with total regression of new vessels than PRP, fewer visually disabling vitreous haemorrhages and fewer cases of incident macular oedema. These anatomical effects should be considered when choosing between anti-VEGF and PRP as a first-line option in PDR. As aflibercept is licensed for DMO, the findings of this study indicate that aflibercept is also effective in the management of PDR in the first year, allowing the use of a single agent to address both of these sight-threatening complications of diabetes mellitus in patients presenting with both PDR and DMO.
We recommend a longer study investigating the efficacy and effects of this treatment, including safety events over a duration of up to 5 years to inform future treatment guidelines for this indication.
Future health economics studies in this area should ensure the inclusion of a range of outcome measures (vision specific and generic, as we did in this study) and should model costs and benefits over a longer period. Although the CLARITY study was limited to 1 year, the rate of complications was significantly higher in the PRP arm in terms of new-onset macular oedema and vitreous haemorrhage by 52 weeks. A similar study, Protocol S,53 showed that, over 2 years, significantly more vitrectomies were performed in the PRP arm. Cataract surgery is also frequently needed after vitrectomy. Similarly, more patients in the PRP arm need to be treated with anti-VEGF therapy for macular oedema. The costs of these complications have to be taken into account when modelling costs over a 5-year period or longer.
Some physicians who prefer anti-VEGF therapy over laser will probably use bevacizumab although data for these drugs are not yet available. Confirmatory studies have to be done before translating the effect of ranibizumab and aflibercept studies to bevacizumab as the number of injections varied between Protocol S and CLARITY and the study designs of these two studies were not identical. Therefore, the CLARITY study results should be used cautiously if other anti-VEGF agents are used to treat PDR.
If there is an intention in a future economic evaluation study to use QALYs as an outcome measure to allow comparison with, for example, the NICE payer threshold of £20,000 per QALY, then feasibility work is necessary to identify ways of measuring utility relating to visual impairment that make the QALY a meaningful outcome measure in this context.
Chapter 7 Conclusion
In conclusion, this is the second study to show non-inferiority of anti-VEGF therapy to PRP and the first study to show superiority of an anti-VEGF agent to PRP in eyes with PDR. If society is willing to pay £1400 for an additional 1-point improvement in BCVA, then aflibercept has a 56.60% probability of being cost-effective at the list price of £816. The trial outcomes are further augmented by the higher satisfaction scores of patients treated with aflibercept than with PRP. However, this is a 52-week study while PDR is a lifelong condition and so longer-term studies are required to evaluate long-term patient compliance and the disease-modifying effect of anti-VEGF agents in PDR. Therefore, the clinical effectiveness and cost-effectiveness of the different anti-VEGF agents in PDR have to be evaluated in Phase III clinical trials for global adoption of anti-VEGF as a treatment adoption for PDR.
Acknowledgements
The authors would like to thank the following for their invaluable help:
Essential support and guidance was provided by the KCTU, the service user group, the TSC (chairperson Alistair Laidlaw) and the DMEC (chairperson Sarah Walker). We would also like to thank the Royal Free London NHS Foundation Trust Pharmacists and members of the independent UK Network of Ophthalmic Reading Centres (NetwORC UK) for their support of the trial. We would like to thank the lay panel in the North East Diabetes Research Network and Mr Richard Lane OBE (Officer of the Most Excellent Order of the British Empire), lay representative grant co-applicant.
We would also like to express thanks to the NIHR Local Clinical Research Networks.
Finally, and most importantly, we would like to thank all the patients who took part in the study and the follow-up survey.
Support
The study was sponsored by NIHR Moorfields Biomedical Research Centre and supported by the UK Clinical Research Network. The research was supported by the NIHR Biomedical Research Centre at Moorfields Eye Hospital NHS Foundation Trust and University College London Institute of Ophthalmology, the NIHR Moorfields Clinical Research Facility and the UK Clinical Research Collaboration-registered KCTU at King’s Health Partners, which is part funded by the NIHR Biomedical Research Centre for Mental Health at South London and Maudsley NHS Foundation Trust and King’s College London. We would also like to thank Maggie Shergill, the Research Manager (Monitoring) and the other team members of the NIHR Evaluation Trials and Studies Coordinating Centre (NETSCC).
Contributions of authors
All authors reviewed, revised and approved the final version of the manuscript.
Sobha Sivaprasad (Professor, Consultant Ophthalmologist) was the chief investigator and co-authored the first draft.
Philip Hykin (Consultant Ophthalmic Surgeon) was a co-applicant, was involved in the design and recruitment of the study and co-authored the first draft.
A Toby Prevost (Professor, Medical Statistics) was a co-applicant, was involved in the design and statistical analysis of the study and co-authored the first draft.
Joana Vasconcelos (Research Fellow, Medical Statistics) was involved in the design and statistical analysis of the study and co-authored the first draft.
Amy Riddell (Trial Manager) was involved in all aspects of the study’s management and monitoring and co-authored the first draft.
Jayashree Ramu (Research Fellow, Moorfields Eye Hospital) was involved in the design and recruitment of the study and co-authored the first draft.
Caroline Murphy (King’s Clinical Trial Unit) was a co-applicant and was involved in the design and trial management and co-authored the first draft.
Joanna Kelly (King’s Clinical Trial Unit) was a co-applicant, was involved in the design and data management and co-authored the first draft.
Rhiannon Tudor Edwards (Professor, Health Economics) was a co-applicant and was involved in the design, management, analysis and interpretation of the health economics part of the study, the co-authoring of the first draft and the writing up of the report.
Seow Tien Yeo (Research Fellow, Health Economics) was involved in the management, analysis and interpretation of the health economics part of the study, the co-authoring of the first draft and the writing up of the report.
James Bainbridge (Professor, Consultant Ophthalmic Surgeon) was a co-applicant and was involved in the design of the study.
David Hopkins (Diabetologist) was a co-applicant and was involved in the design of the study.
Beverley White-Alao (King’s Clinical Trial Unit) was involved in the protocol development and study management and co-authored the first draft.
Publications
Sivaprasad S, Prevost AT, Bainbridge J, Edwards RT, Hopkins D, Kelly J, et al. Clinical efficacy and mechanistic evaluation of aflibercept for proliferative diabetic retinopathy (acronym CLARITY): a multicentre phase IIb randomised active-controlled clinical trial. BMJ Open 2015;5:e008405.
Sivaprasad S, Prevost AT, Vasconcelos JC, Riddell A, Murphy C, Kelly J, et al. Clinical efficacy of intravitreal aflibercept versus panretinal photocoagulation for best corrected visual acuity in patients with proliferative diabetic retinopathy at 52 weeks (CLARITY): a multicentre, single-blinded, randomised, controlled, phase 2b, non-inferiority trial. Lancet 2017;389:2193–203.
Nicholson L, Crosby-Nwaobi R, Vasconcelos JC, Prevost AT, Ramu J, Riddell A, et al. Mechanistic evaluation of panretinal photocoagulation versus aflibercept in proliferative diabetic retinopathy: CLARITY substudy. Invest Ophthalmol Vis Sci 2018;59:4277–84.
Data-sharing statement
Data are archived at Moorfields Eye Hospital. All data requests should be submitted to the corresponding author for consideration. Access to anonymised data may be granted following review.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslifes You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, NETSCC, the EME programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the EME programme or the Department of Health and Social Care.
References
- Diabetes.co.uk . Diabetes Prevalence n.d. www.diabetes.co.uk/diabetes-prevalence.html (accessed July 2018).
- Antonetti DA, Klein R, Gardner TW. Diabetic retinopathy. N Engl J Med 2012;366:1227-39. https://doi.org/10.1056/NEJMra1005073.
- Abbate M, Cravedi P, Iliev I, Remuzzi G, Ruggenenti P. Prevention and treatment of diabetic retinopathy: evidence from clinical trials and perspectives. Curr Diabetes Rev 2011;7:190-20. https://doi.org/10.2174/157339911795843168.
- Scanlon PH. Why do patients still require surgery for the late complications of Proliferative Diabetic Retinopathy?. Eye 2010;24:435-40. https://doi.org/10.1038/eye.2009.320.
- Sivaprasad S, Gupta B, Gulliford MC, Dodhia H, Mohamed M, Nagi D, et al. Ethnic variations in the prevalence of diabetic retinopathy in people with diabetes attending screening in the United Kingdom (DRIVE UK). PLOS ONE 2012;7. https://doi.org/10.1371/journal.pone.0032182.
- Sivaprasad S, Gupta B, Gulliford MC, Dodhia H, Mann S, Nagi D, et al. Ethnic variation in the prevalence of visual impairment in people attending diabetic retinopathy screening in the United Kingdom (DRIVE UK). PLOS ONE 2012;7. https://doi.org/10.1371/journal.pone.0039608.
- Arden GB, Sivaprasad S. Hypoxia and oxidative stress in the causation of diabetic retinopathy. Curr Diabetes Rev 2011;7:291-304. https://doi.org/10.2174/157339911797415620.
- Early Treatment Diabetic Retinopathy Study (ETDRS) Research Group . Early photocoagulation for diabetic retinopathy. ETDRS report number 9. Ophthalmology 1991;98:766-85. https://doi.org/10.1016/S0161-6420(13)38011-7.
- The Diabetic Retinopathy Study Research Group . Preliminary report on effects of photocoagulation therapy. Am J Ophthalmol 1976;81:383-96. https://doi.org/10.1016/0002-9394(76)90292-0.
- Stefánsson E. Ocular oxygenation and the treatment of diabetic retinopathy. Surv Ophthalmol 2006;51:364-80. https://doi.org/10.1016/j.survophthal.2006.04.005.
- Aiello LM. Perspectives on diabetic retinopathy. Am J Ophthalmol 2003;136:122-35. https://doi.org/10.1016/S0002-9394(03)00219-8.
- Bailey CC, Sparrow JM, Grey RH, Cheng H. The National Diabetic Retinopathy Laser Treatment Audit. III. Clinical outcomes. Eye 1999;13:151-9. https://doi.org/10.1038/eye.1999.42.
- Flynn HW, Chew EY, Simons BD, Barton FB, Remaley NA, Ferris FL. Pars plana vitrectomy in the Early Treatment Diabetic Retinopathy Study. ETDRS report number 17. The Early Treatment Diabetic Retinopathy Study Research Group. Ophthalmology 1992;99:1351-7. https://doi.org/10.1016/S0161-6420(92)31779-8.
- Brucker AJ, Qin H, Antoszyk AN, Beck RW, Bressler NM, . Diabetic Retinopathy Clinical Research Network . Observational study of the development of diabetic macular edema following panretinal (scatter) photocoagulation given in 1 or 4 sittings. Arch Ophthalmol 2009;127:132-40. https://doi.org/10.1001/archophthalmol.2008.565.
- Mackie SW, Walsh G. Contrast and glare sensitivity in diabetic patients with and without pan-retinal photocoagulation. Ophthalmic Physiol Opt 1998;18:173-81. https://doi.org/10.1016/S0275-5408(97)00083-5.
- Subash M, Comyn O, Samy A, Qatarneh D, Antonakis S, Mehat M, et al. The effect of multispot laserpanretinal photocoagulation on retinal sensitivity and driving eligibility in patients with diabetic retinopathy. JAMA Ophthalmol 2016;134:666-72. https://doi.org/10.1001/jamaophthalmol.2016.0629.
- Chakravarthy U, Harding SP, Rogers CA, Downes SM, Lotery AJ, . IVAN Study Investigators . Ranibizumab versus bevacizumab to treat neovascular age-related macular degeneration: one-year findings from the IVAN randomized trial. Ophthalmology 2012;119:1399-411. https://doi.org/10.1016/j.ophtha.2012.04.015.
- Adamis AP, Shima DT, Tolentino MJ, Gragoudas ES, Ferrara N, Folkman J, et al. Inhibition of vascular endothelial growth factor prevents retinal ischemia-associated iris neovascularization in a nonhuman primate. Arch Ophthalmol 1996;114:66-71. https://doi.org/10.1001/archopht.1996.01100130062010.
- Lange CAK, Stavrakas P, Luhmann UFO, de Silva DJ, Ali RR, Gregor ZJ, et al. Intraocular oxygen distribution in advanced proliferative diabetic retinopathy. Am J Ophthalmol 2011;152:406-12. https://doi.org/10.1016/j.ajo.2011.02.014.
- Shima DT, Adamis AP, Ferrara N, Yeo KT, Yeo TK, Allende R, et al. Hypoxic induction of endothelial cell growth factors in retinal cells: identification and characterization of vascular endothelial growth factor (VEGF) as the mitogen. Mol Med 1995;1:182-93.
- Miller JW, Adamis AP, Shima DT, D’Amore PA, Moulton RS, O’Reilly MS, et al. Vascular endothelial growth factor/vascular permeability factor is temporally and spatially correlated with ocular angiogenesis in a primate model. Am J Pathol 1994;145:574-84.
- Campochiaro PA, Aiello LP, Rosenfeld PJ. Anti-vascular endothelial growth factor agents in the treatment of retinal disease: from bench to bedside. Ophthalmology 2016;123:S78-S88. https://doi.org/10.1016/j.ophtha.2016.04.056.
- Aiello LP, Avery RL, Arrigg PG, Keyt BA, Jampel HD, Shah ST, et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med 1994;331:1480-7. https://doi.org/10.1056/NEJM199412013312203.
- Aiello LP, Pierce EA, Foley ED, Takagi H, Chen H, Riddle L, et al. Suppression of retinal neovascularization in vivo by inhibition of vascular endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc Natl Acad Sci USA 1995;92:10457-61. https://doi.org/10.1073/pnas.92.23.10457.
- Ip MS, Domalpally A, Hopkins JJ, Wong P, Ehrlich JS. Long-term effects of ranibizumab on diabetic retinopathy severity and progression. Arch Ophthalmol 2012;130:1145-52. https://doi.org/10.1001/archophthalmol.2012.1043.
- Salam A, Mathew R, Sivaprasad S. Treatment of proliferative diabetic retinopathy with anti-VEGF agents. Acta Ophthalmol 2011;89:405-11. https://doi.org/10.1111/j.1755-3768.2010.02079.x.
- Weidle UH, Schneider B, Georges G, Brinkmann U. Genetically engineered fusion proteins for treatment of cancer. Cancer Genomics Proteomics 2012;9:357-72.
- Papadopoulos N, Martin J, Ruan Q, Rafique A, Rosconi MP, Shi E, et al. Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF trap, ranibizumab and bevacizumab. Angiogenesis 2012;15:171-85. https://doi.org/10.1007/s10456-011-9249-6.
- Schmidt-Erfurth U, Kaiser PK, Korobelnik JF, Brown DM, Chong V, Nguyen QD, et al. Intravitreal aflibercept injection for neovascular age-related macular degeneration: ninety-six-week results of the VIEW studies. Ophthalmology 2014;121:193-201. https://doi.org/10.1016/j.ophtha.2013.08.011.
- Wells JA, Glassman AR, Ayala AR, Jampol LM, Bressler NM, Bressler SB, et al. Aflibercept, bevacizumab, or ranibizumab for diabetic macular edema: two-year results from a comparative effectiveness randomized clinical trial. Ophthalmology 2016;123:1351-9. https://doi.org/10.1016/j.ophtha.2016.02.022.
- Brown DM, Schmidt-Erfurth U, Do DV, Holz FG, Boyer DS, Midena E, et al. Intravitreal aflibercept for diabetic macular edema: 100-week results from the VISTA and VIVID studies. Ophthalmology 2015;122:2044-52. https://doi.org/10.1016/j.ophtha.2015.06.017.
- Type 2 Diabetes in Adults: Management. London: National Institute for Health and Care Excellence.; n.d.
- Sivaprasad S, Prevost AT, Bainbridge J, Edwards RT, Hopkins D, Kelly J, et al. Clinical efficacy and mechanistic evaluation of aflibercept for proliferative diabetic retinopathy (acronym CLARITY): a multicentre phase IIb randomised active-controlled clinical trial. BMJ Open 2015;5. https://doi.org/10.1136/bmjopen-2015-008405.
- Rosser DA, Cousens SN, Murdoch IE, Fitzke FW, Laidlaw DA. How sensitive to clinical change are ETDRS logMAR visual acuity measurements?. Invest Ophthalmol Vis Sci 2003;44:3278-81. https://doi.org/10.1167/iovs.02-1100.
- Ferris FL, Kassoff A, Bresnick GH, Bailey I. New visual acuity charts for clinical research. Am J Ophthalmol 1982;94:91-6. https://doi.org/10.1016/0002-9394(82)90197-0.
- Elliott DB, Bullimore MA, Bailey IA. Improving the reliability of the Pelli–Robson contrast sensitivity test. Clin Vis Sci 1991;6:471-5.
- Esterman B. Functional scoring of the binocular field. Ophthalmology 1982;89:1226-34. https://doi.org/10.1016/S0161-6420(82)34647-3.
- Brose LS, Bradley C. Psychometric development of the retinopathy treatment satisfaction questionnaire (RetTSQ). Psychol Health Med 2009;14:740-54. https://doi.org/10.1080/13548500903431485.
- Mangione CM, Lee PP, Gutierrez PR, Spritzer K, Berry S, Hays RD. National Eye Institute Visual Function Questionnaire Field Test Investigators . Development of the 25-item National Eye Institute Visual Function Questionnaire. Arch Ophthalmol 2001;119:1050-8. https://doi.org/10.1001/archopht.119.7.1050.
- Brose LS, Bradley C. Psychometric development of the individualized Retinopathy-Dependent Quality of Life Questionnaire (RetDQoL). Value Health 2010;13:119-27. https://doi.org/10.1111/j.1524-4733.2009.00589.x.
- Janssen MF, Birnie E, Bonsel GJ. Quantification of the level descriptors for the standard EQ-5D three-level system and a five-level version according to two methods. Qual Life Res 2008;17:463-73. https://doi.org/10.1007/s11136-008-9318-5.
- Al-Janabi H, Peters TJ, Brazier J, Bryan S, Flynn TN, Clemens S, et al. An investigation of the construct validity of the ICECAP-A capability measure. Qual Life Res 2013;22:1831-40. https://doi.org/10.1007/s11136-012-0293-5.
- The Data Protection Act 1998. London: The Stationery Office; 1998.
- Macedo AF, Ramos PL, Hernandez-Moreno L, Cima J, Baptista AMG, Marques AP, et al. Visual and health outcomes, measured with the activity inventory and the EQ-5D, in visual impairment. Acta Ophthalmol 2017;95:e783-e791. https://doi.org/10.1111/aos.13430.
- Luo N, Wang X, Ang M, Finkelstein EA, Aung T, Wong TY, et al. A vision ‘bolt-on’ item could increase the discriminatory power of the EQ-5D Index score. Value Health 2015;18:1037-42. https://doi.org/10.1016/j.jval.2015.08.002.
- NHS Reference Costs 2015 to 2016. London: Department of Health and Social Care; 2016.
- British National Formulary. London: BMJ Group and Pharmaceutical Press; 2016.
- Briggs AH, Bousquet J, Wallace MV, Busse WW, Clark TJ, Pedersen SE, et al. Cost-effectiveness of asthma control: an economic appraisal of the GOAL study. Allergy 2006;61:531-6. https://doi.org/10.1111/j.1398-9995.2006.01038.x.
- Early Treatment Diabetic Retinopathy Study (ETDRS) Research Group . Grading diabetic retinopathy from stereoscopic color fundus photographs – an extension of the modified Airlie House classification. ETDRS report number 10. Ophthalmology 1991;98:786-80. https://doi.org/10.1016/S0161-6420(13)38012-9.
- Antiplatelet Trialists’ Collaboration . Collaborative overview of randomised trials of antiplatelet therapy – I: prevention of death, myocardial infarction, and stroke by prolonged antiplatelet therapy in various categories of patients. BMJ 1994;308:81-106. https://doi.org/10.1136/bmj.308.6921.81.
- Martin DF, Maguire MG, Ying GS, Grunwald JE, Fine SL, . CATT (Comparison of Age-related macular degeneration Treatments Trials) Research Group . Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N Engl J Med 2011;364:1897-908. https://doi.org/10.1056/NEJMoa1102673.
- Stewart MW. Treatment of diabetic retinopathy: Recent advances and unresolved challenges. World J Diabetes 2016;7:333-41. https://doi.org/10.4239/wjd.v7.i16.333.
- Gross JG, Glassman AR, Jampol LM, Inusah S, Aiello LP, . Writing Committee for the Diabetic Retinopathy Clinical Research Network . Panretinal photocoagulation vs intravitreous ranibizumab for proliferative diabetic retinopathy: a randomized clinical trial. JAMA 2015;314:2137-46. https://doi.org/10.1001/jama.2015.15217.
- González VH, Giuliari GP, Banda RM, Guel DA. Intravitreal injection of pegaptanib sodium for proliferative diabetic retinopathy. Br J Ophthalmol 2009;93:1474-8. https://doi.org/10.1136/bjo.2008.155663.
- ICH Harmonised Tripartite Guideline . Statistical principles for clinical trials. International Conference on Harmonisation E9 Expert Working Group. Stat Med 1999;18:1905-42.
- White IR, Thompson SG. Adjusting for partially missing baseline measurements in randomized trials. Stat Med 2005;24:993-1007. https://doi.org/10.1002/sim.1981.
- D’Agostino RB, Massaro JM, Sullivan LM. Non-inferiority trials: design concepts and issues – the encounters of academic consultants in statistics. Stat Med 2003;22:169-86. https://doi.org/10.1002/sim.1425.
- White IR, Carpenter J, Horton NJ. Including all individuals is not enough: lessons for intention-to-treat analysis. Clin Trials 2012;9:396-407. https://doi.org/10.1177/1740774512450098.
- Lesaffre E. Superiority, equivalence, and non-inferiority trials. Bull NYU Hosp Jt Dis 2008;66:150-4.
- Garrett AD. Therapeutic equivalence: fallacies and falsification. Stat Med 2003;22:741-62. https://doi.org/10.1002/sim.1360.
- Committee for Proprietary Medicinal Products . Points to consider on switching between superiority and non-inferiority. Br J Clin Pharmacol 2001;52:223-8. https://doi.org/10.1046/j.1365-2125.2001.01397-3.x.
- White IR, Horton NJ, Carpenter J, Pocock SJ. Strategy for intention to treat analysis in randomised trials with missing outcome data. BMJ 2011;342. https://doi.org/10.1136/bmj.d40.
- Burzykowski T, Carpenter J, Coens C, Evans D, France L, Kenward M, et al. Missing data: discussion points from the PSI missing data expert group. Pharm Stat 2010;9:288-97. https://doi.org/10.1002/pst.391.
- White IR, Kalaitzaki E, Thompson SG. Allowing for missing outcome data and incomplete uptake of randomised interventions, with application to an internet-based alcohol trial. Stat Med 2011;30:3192-207. https://doi.org/10.1002/sim.4360.
- Carpenter J, Bithell J. Bootstrap confidence intervals: when, which, what? A practical guide for medical statisticians. Stat Med 2000;19:1141-64. https://doi.org/10.1002/(SICI)1097-0258(20000515)19:9<1141::AID-SIM479>3.0.CO;2-F.
- Sivaprasad S, Prevost AT, Vasconcelos JC, Riddell A, Murphy C, Kelly J, et al. Clinical efficacy of intravitreal aflibercept versus panretinal photocoagulation for best corrected visual acuity in patients with proliferative diabetic retinopathy at 52 weeks (CLARITY): a multicentre, single-blinded, randomised, controlled, phase 2b, non-inferiority trial. Lancet 2017;389:2193-203. https://doi.org/10.1016/S0140-6736(17)31193-5.
- Ramu J, Chatziralli I, Yang Y, Menon G, Bailey C, Eckstein M, et al. Health-related quality of life, visual function and treatment satisfaction following intravitreal dexamethasone implant for diabetic macular edema. Patient Prefer Adherence 2017;11:579-86. https://doi.org/10.2147/PPA.S132859.
- Husereau D, Drummond M, Petrou S, Carswell C, Moher D, Greenberg D, et al. Consolidated Health Economic Evaluation Reporting Standards (CHEERS) statement. Cost Eff Resour Alloc 2013;11. https://doi.org/10.1186/1478-7547-11-6.
- Ridyard CH, Hughes DA. Methods for the collection of resource use data within clinical trials: a systematic review of studies funded by the UK Health Technology Assessment program. Value Health 2010;13:867-72. https://doi.org/10.1111/j.1524-4733.2010.00788.x.
- Curtis L, Burns A. Unit Costs of Health and Social Care 2016. Canterbury: Personal Social Services Research Unit (PSSRU), University of Kent; 2016.
- NHS Choices . Acupuncture on the NHS n.d. www.nhs.uk/conditions/Acupuncture/Pages/Introduction.aspx#NHS (accessed 18 June 2017).
- Glick HA, Doshi JA, Sonnad SS, Polsky D. Economic Evaluation in Clinical Trials. Oxford: Oxford University Press; 2007.
- Drummond MF, Sculpher MJ, Claxton K, Stoddart GL, Torrance GW. Methods for the Economic Evaluation of Health Care Programmes. Oxford: Oxford University Press; 2015.
- Grewal I, Lewis J, Flynn T, Brown J, Bond J, Coast J. Developing attributes for a generic quality of life measure for older people: preferences or capabilities?. Soc Sci Med 2006;62:1891-901. https://doi.org/10.1016/j.socscimed.2005.08.023.
- Manca A, Hawkins N, Sculpher MJ. Estimating mean QALYs in trial-based cost-effectiveness analysis: the importance of controlling for baseline utility. Health Econ 2005;14:487-96. https://doi.org/10.1002/hec.944.
- Laramée P, Wonderling D, Cahen DL, Dijkgraaf MG, Gouma DJ, Bruno MJ, et al. Trial-based cost-effectiveness analysis comparing surgical and endoscopic drainage in patients with obstructive chronic pancreatitis. BMJ Open 2013;3. https://doi.org/10.1136/bmjopen-2013-003676.
- Kind P, Dolan P, Gudex C, Williams A. Variations in population health status: results from a United Kingdom national questionnaire survey. BMJ 1998;316:736-41. https://doi.org/10.1136/bmj.316.7133.736.
- Hardarson SH, Harris A, Karlsson RA, Halldorsson GH, Kagemann L, Rechtman E, et al. Automatic retinal oximetry. Invest Ophthalmol Vis Sci 2006;47:5011-16. https://doi.org/10.1167/iovs.06-0039.
- Hardarson SH, Gottfredsdottir MS, Halldorsson GH, Karlsson RA, Benediktsson JA, Eysteinsson T, et al. Glaucoma filtration surgery and retinal oxygen saturation. Invest Ophthalmol Vis Sci 2009;50:5247-50. https://doi.org/10.1167/iovs.08-3117.
- Nicholson L, Vazquez-Alfageme C, Ramu J, Triantafyllopoulou I, Patrao NV, Muwas M, et al. Validation of concentric rings method as a topographic measure of retinal nonperfusion in ultra-widefield fluorescein angiography. Am J Ophthalmol 2015;160:1217-25.e2. https://doi.org/10.1016/j.ajo.2015.09.003.
- Nicholson L, Vazquez-Alfageme C, Clemo M, Luo Y, Hykin PG, Bainbridge JW, et al. Quantifying retinal area in ultra-widefield imaging using a 3-dimensional (3-D) printed eye model. Ophthalmol Retina 2017;2:65-71. https://doi.org/10.1016/j.oret.2017.03.011.
- Nicholson L, Crosby-Nwaobi R, Vasconcelos JC, Prevost AT, Ramu J, Riddell A, et al. Mechanistic evaluation of panretinal photocoagulation versus aflibercept in proliferative diabetic retinopathy: CLARITY substudy. Invest Ophthalmol Vis Sci 2018;59:4277-84. https://doi.org/10.116/iovs.17-23509.
- Visual Disorders: Assessing Fitness to Drive. Swansea: Driver and Vehicle Licensing Agency; n.d.
- Muqit MM, Wakely L, Stanga PE, Henson DB, Ghanchi FD. Effects of conventional argon panretinal laser photocoagulation on retinal nerve fibre layer and driving visual fields in diabetic retinopathy. Eye 2010;24:1136-42. https://doi.org/10.1038/eye.2009.308.
- Muqit MM, Young LB, McKenzie R, John B, Marcellino GR, Henson DB, et al. Pilot randomised clinical trial of Pascal TargETEd Retinal versus variable fluence PANretinal 20 ms laser in diabetic retinopathy: PETER PAN study. Br J Ophthalmol 2013;97:220-7. https://doi.org/10.1136/bjophthalmol-2012-302189.
- Guduru A, Martz TG, Waters A, Kshirsagar AV, Garg S. Oxygen saturation of retinal vessels in all stages of diabetic retinopathy and correlation to ultra-wide field fluorescein angiography. Invest Ophthalmol Vis Sci 2016;57:5278-84. https://doi.org/10.1167/iovs.16-20190.
- Jørgensen CM, Bek T. Lack of differences in the regional variation of oxygen saturation in larger retinal vessels in diabetic maculopathy and proliferative diabetic retinopathy. Br J Ophthalmol 2017;101:752-7. https://doi.org/10.1136/bjophthalmol-2016-308894.
- Hutton DW, Stein JD, Bressler NM, Jampol LM, Browning D, Glassman AR. Diabetic Retinopathy Clinical Research Network . Cost-effectiveness of intravitreous ranibizumab compared with panretinal photocoagulation for proliferative diabetic retinopathy: secondary analysis from a diabetic retinopathy clinical research network randomized clinical trial. JAMA Ophthalmol 2017;135:576-84. https://doi.org/10.1001/jamaophthalmol.2017.0837.
- Brown MM, Brown GC, Sharma S, Busbee B. Quality of life associated with visual loss: a time tradeoff utility analysis comparison with medical health states. Ophthalmology 2003;110:1076-81. https://doi.org/10.1016/S0161-6420(03)00254-9.
- Prescription Cost Analysis, England – 2016. Leads: NHS Digital; 2017.
Appendix 1 The CLARITY study group and resource centres
The CLARITY study group thanks all the patients who participated in the study, and all site investigators and research teams.
| Sites | Principal investigators |
|---|---|
| Moorfields Eye Hospital NHS Foundation Trust, London | Sobha Sivaprasad |
| Hillingdon Hospitals NHS Foundation Trust, London | Sheena George |
| Sunderland Eye Infirmary, Sunderland | Maged Habib |
| Royal Victoria Infirmary, Newcastle upon Tyne | James Talks |
| Essex County Hospital, Colchester | Jignesh Patel |
| Bristol Eye Hospital, Bristol | Adam Ross |
| Frimley Park Hospital NHS Foundation Trust, Surrey | Geeta Menon |
| Royal Liverpool & Broadgreen University Hospitals NHS Trust, Liverpool | Amira Stylianides |
| James Paget University Hospital, Great Yarmouth | Ben Burton |
| St James’s University Hospital, Leeds | Martin McKibbin |
| Royal Victoria Hospital and Queen’s University, Belfast | Usha Chakravarthy |
| Maidstone and Tunbridge Wells NHS Trust, Kent | Luke Membrey |
| University Hospital Southampton NHS Foundation Trust, Southampton | Andrew Lotery |
| King’s College Hospital, London | Haralabos Eleftheriadis |
| Princess Alexandra Hospital, Harlow | Priya Prakash |
| Birmingham and Midland Eye Centre, Sandwell and West Birmingham NHS Foundation Trust, Birmingham | Bushra Mushtaq |
| York Hospital NHS Trust, York | Richard Gale |
| Wolverhampton and Midland Counties Eye Infirmary, Wolverhampton | Ajay Bhatnagar |
| Brighton and Sussex University Hospitals NHS Trust, Brighton | Robert Purbrick |
| Royal Bolton Hospital NHS Trust, Greater Manchester | Simon Kelly |
| Leicester Royal Infirmary, Leicester | Theo Empeslidis |
| Torbay Hospital, South Devon | Olayinka Osoba |
Resource centres
We would also like to thank the following resource centres for their support of the trial:
King’s College Clinical Trial team: Gill Lambert, Beverley White-Alao, Oliver Pressey, Negin Sarafraz-Shekary, Evangelos Georgiou and Janice Jimenez.
The Royal Free London NHS Foundation Trust Pharmacists team: Nicky Heath and Sabina Melander.
The North East Diabetes Research Network’s lay panel.
Patient-reported outcomes consultant to the study: Professor Clare Bradley, Royal Holloway, University of London.
Mechanistic Evaluation team: Luke Nicholson, Roxanne Crosby-Nwaobi, Lauren Leitch-Devlin at the NIHR Moorfields Clinical Research Facility.
Certifications for visual acuity and contrast sensitivity: Catherine Grigg and Katherine Binsted at Moorfields Eye Hospital, London.
Members of the NetwORC UK: Professor Usha Chakravarthy, Professor Tunde Peto, Professor Simon Harding, Dr Pauline Lenfestey, Ms Savitha Madhusudhan, Clare Newell, Michelle McGaughey, Vittorio Silvestri, Karleigh Kelso, Barbra Hamill, Graham Young, Irene Leung, Peter Blows, Frank Picton, David Parry and Sophie Leach.
Appendix 2 The CLARITY study committees
Trial steering committee
Independent chairperson: Mr Alistair Laidlaw (Guy’s and St Thomas’ NHS Foundation Trust, London); independent members: Graham A Hitman (Blizard Institute, Barts and The London School of Medicine and Dentistry, London), Winfried Amoaku (University Hospital, Queen’s Medical Centre, University of Nottingham), Gillian Hood (NIHR Clinical Research Network, North West London); and lay representatives: Daniel Preece and Paul Burns.
Data monitoring and ethics committee
Sarah Walker (Oxford University, Oxford, UK; Chairperson), Niral Karia (Southend NHS Trust) and Evelyn Mensah (Central Middlesex NHS Trust, UK).
Trial management group members
Sobha Sivaprasad, Philip Hykin, A Toby Prevost, Joana Vasconselos, Amy Riddell, Beverley White-Alao, Caroline Murphy, Joanna Kelly and the sponsor representative (Moorfields Eye Hospital).
Appendix 3 Additional health economics data
| Health-care resource | Unit | Unit cost (£) | Details and source |
|---|---|---|---|
| Primary care and other community-based contacts (e.g. social worker, podiatrist, psychologist) | Consultation | 8.00–79.00 | Costed by professionc,e,f |
| Inpatient hospital stays for various reasons | Procedure | 48–7487 | Costed by type of inpatient activityd |
| Outpatient visits/procedures for various reasons | Visit/procedure | 29–3859 | Costed by type of outpatient visit/procedured |
| FFA | Procedure | 117 | Costed by type of outpatient procedured |
| PRP laser treatment outpatient procedure | Procedure | 131 | Costed by type of outpatient procedured |
| Aflibercept injection outpatient procedure | Procedure | 182 | Costed by type of outpatient procedured |
| Aflibercept injection, 0.1-ml vial, 40 mg/ml | Medication | 816 | Costed by type of medicationg |
| Ophthalmology-related medication | Medication | Various | Costed by type of medicationg,h |
Appendix 4 Summary of patient and public involvement
Patients and the public were involved throughout the CLARITY study as follows:
-
Design – the application had a lay co-applicant (Richard Lane, OBE) who helped design the study and contributed to the study grant application.
-
North East London Lay Member panel of the diabetes research network – three lay members of the panel contributed to the design of the study, design and content of the patient information sheets and the consent form. They also contributed to the contents of the letter to the participants informing them of the results.
-
The Trial Steering Committee had two lay members and both were service users and contributed at the TSC meetings.
-
Report – Mr Richard Lane contributed to the content of final report by contributing to the lay summary.
Appendix 5 Patient information sheet and consent form
List of abbreviations
- ADL
- activities of daily living
- ANCOVA
- analysis of covariance
- APTC
- Anti-Platelet Triallists’ Collaboration
- AUC
- area under the curve
- AWI
- average weighted impact
- BCVA
- best corrected visual acuity
- BNF
- British National Formulary
- BP
- blood pressure
- CARF
- Central Angiographic Resource Facility
- CEAC
- cost-effectiveness acceptability curve
- CFP
- colour fundus photography
- CI
- confidence interval
- CLARITY
- clinical efficacy and mechanistic evaluation of aflibercept for proliferative diabetic retinopathy
- CONSORT
- Consolidated Standards of Reporting Trials
- CSRI
- client service receipt inventory
- CST
- central subfield thickness
- DD
- disc diameter
- DMEC
- Data Monitoring and Ethics Committee
- DMO
- diabetic macular oedema
- eCRF
- electronic case report form
- EDC
- electronic data capture
- EMA
- European Medicines Agency
- EQ-5D-3L
- EuroQol-5 Dimensions, three-level version
- ETDRS
- Early Treatment Diabetic Retinopathy Study
- EudraCT
- European Union Drug Regulating Authorities Clinical Trials
- FA
- fluorescein angiography
- FDA
- US Food and Drug Administration
- FFA
- fundus fluorescein angiography
- HbA1c
- glycated haemoglobin
- HRQoL
- health-related quality of life
- ICECAP-A
- Investigating Choice Experiments Capability Measure for Adults
- ICER
- incremental cost-effectiveness ratio
- ICH
- International Conference on Harmonisation
- IMP
- investigational medicinal product
- IQR
- interquartile range
- ITT
- intention to treat
- KCTU
- King’s Clinical Trials Unit
- LME
- linear mixed-effects
- MAR
- missing at random
- MHRA
- Medicines and Healthcare products Regulatory Agency
- mRNA
- messenger ribonucleic acid
- NEI-VFQ-25
- National Eye Institute Visual Function Questionnaire
- NetwORC UK
- Network of Ophthalmic Reading Centres UK
- NICE
- National Institute for Health and Care Excellence
- NIHR
- National Institute for Health Research
- NV
- neovascularisation
- NVA
- neovascularisation of the angle
- NVG
- neovascular glaucoma
- NVI
- neovascularisation of iris
- OBE
- Officer of the Most Excellent Order of the British Empire
- OCT
- optical coherence tomography
- PAS
- Patient Access Scheme
- PDR
- proliferative diabetic retinopathy
- PIN
- patient identification number
- PP
- per protocol
- PRP
- panretinal photocoagulation
- QALY
- quality-adjusted life-year
- RCT
- randomised controlled trial
- RetDQoL
- Retinopathy-Dependent Quality-of-Life Questionnaire
- RetTSQ
- Retinopathy Treatment Satisfaction Questionnaire
- SAE
- serious adverse event
- SD
- standard deviation
- SD-OCT
- spectral-domain optical coherence tomography
- SE
- standard error
- SOP
- standard operating procedure
- TMG
- Trial Management Group
- TSC
- Trial Steering Committee
- VEGF
- vascular endothelial growth factor