

Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 13/94/15. The contractual start date was in April 2016. The final report began editorial review in December 2019 and was accepted for publication in June 2020. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2021. This work was produced by Lotery et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2021 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
Background
Central serous chorioretinopathy (CSCR), predominantly affecting men of working age, is the fourth most common vision-threatening retinopathy. 1–4 Fluid spontaneously gathers under the retina causing a neurosensory retinal detachment, which leads to permanent vision loss in up to one-third of cases. 5 The disease course is variable; some cases spontaneously resolve, up to 50% develop chronic CSCR [subretinal fluid (SRF) for > 3 months] and others resolve then recur or affect the fellow eye. 2,4
Characterised by widespread retinal abnormalities, including localised retinal detachments and a thickened choroid with dilated vessels, chronic CSCR leads to a permanent reduction in vision. 1 Abnormalities of choroidal circulation are thought to play a pivotal role in disease pathogenesis. Distinct choroidal abnormalities, including excess dilatation and hyperpermeability, can be seen in patients, and primate studies demonstrate choroidal endothelial cell defects. 2,6 Corticosteroids, both endogenous and exogenous, are the most significant risk factor for CSCR, with mineralocorticoid receptor overactivation in rodents producing a CSCR phenotype. 2,7 The cause of CSCR is unknown, although occurrence within families suggests heritable factors and Schubert et al. 8 identified the first genetic determinants when they identified variation in the cadherin gene in male patients with CSCR. A recent genome-wide association study identified mutations in complement factor H to be significantly associated with chronic CSCR. 9,10
Treatments with some evidence of benefit include half-dose verteporfin photodynamic laser therapy (PDT) and subthreshold micropulse laser treatment. 11–13 However, these treatments are not available in most NHS hospitals and verteporfin for PDT is not licensed for this indication. Furthermore, PDT can have side effects that reduce vision, including retinal scarring, which is permanent, and choroidal ischaemia, which can be temporary. 14 Anti-vascular endothelial growth factor (VEGF) agents used to treat neovascular macular degeneration have not shown efficacy for treating CSCR. 15 Therefore, standard care for CSCR is often observation only; there is no gold standard treatment.
Rationale
Research into the aetiology of CSCR has opened new avenues for potential treatments. In a rat model of CSCR, Zhao et al. 16 showed that one of the features of the condition, choroidal vasodilation, was induced by aldosterone acting via an endothelial vasodilatory potassium channel KCa2.3. Aldosterone is a mineralocorticoid receptor (MR) activator. Blockade of this pathway prevented aldosterone-induced choroidal thickening. This work was translated to a clinical setting. We treated two patients with chronic CSCR with the oral MR antagonist eplerenone for 5 weeks and observed an improvement in visual acuity as well as resolution of retinal detachment and choroidal vasodilation. The observed benefit was maintained for 5 months after the patients stopped taking eplerenone. 16 These results identified MR signalling as a pathway controlling choroidal vascular bed relaxation and provide a pathogenic link with CSCR. This research supports the hypothesis that blockade of MRs could be used as a treatment for CSCR. A subsequent pilot study of 13 patients with chronic CSCR found that eplerenone was associated with a significant reduction in central macular thickness (CMT) and SRF and an improvement in visual acuity in some patients. 17
A small number of double-masked randomised controlled trials (RCTs) investigating eplerenone to treat CSCR have shown some benefit in reducing SRF and/or improving visual acuity. 18–20 However, the studies were limited by being conducted at single centres and with too few patients to draw meaningful conclusions.
In summary, there are several small studies suggesting efficacy of eplerenone in treating CSCR but there have been no adequately powered placebo-controlled RCTs to conclusively demonstrate whether or not eplerenone is a safe and effective treatment. The VICI trial provides evidence for whether or not eplerenone is an effective treatment and contributes to the knowledge gap on the natural history of the disease. This trial is especially important because ophthalmologists are prescribing eplerenone off licence for CSCR without robust evidence of its efficacy.
Aim and objectives
The aim of this study was to compare the efficacy and safety of eplerenone with usual care versus placebo with usual care for chronic CSCR for 12 months in a Phase III randomised placebo-controlled clinical trial.
Primary objective:
-
to evaluate whether best corrected visual acuity (BCVA) following eplerenone therapy with usual care is superior to placebo with usual care in eyes with chronic CSCR.
Secondary objectives:
-
to evaluate whether or not eplerenone treatment with usual care is better than placebo with usual care for resolution of SRF
-
to describe the safety profile of eplerenone treatment with usual care (compared with placebo with usual care)
-
to evaluate whether or not participant-reported visual function improves with eplerenone treatment with usual care compared with placebo with usual care
-
to describe how the choroid responds to treatment in CSCR
-
to describe how retinal pigment epithelium (RPE) function changes over 1 year in CSCR as measured by autofluorescence (AF)
-
to evaluate how low-luminance visual acuity (LLA) changes with eplerenone treatment.
Mechanistic substudy objectives:
-
to explore treatment response by conducting imaging studies of retina and choroid
-
to generate a biobank of DNA, serum and plasma for future mechanistic studies.
Chapter 2 Methods
Study design
The VICI trial was a Phase III, parallel, randomised (1 : 1 ratio), multicentre, double-masked trial, stratified by site (i.e. participating hospital) and BCVA score, comparing the superiority of oral eplerenone plus usual care with placebo plus usual care in patients with CSCR.
The trial was registered with the International Standard Randomised Controlled Trial Number (ISRCTN) registry on 6 May 2016 (identifier: ISRCTN92746680). Ethics approval was granted by the Wales Research Ethics Committee (REC) 1 on 30 April 2016 (identifier: 16/WA/0069). Health Research Authority (HRA) approval was granted on 21 July 2016. A Clinical Trials Authorisation was granted by the Medicines and Healthcare products Regulatory Agency (MHRA) on 31 March 2016 [European Union Drug Regulating Authorities Clinical Trials Database (EudraCT) 2016–000113–70]. Recruitment opened on 14 December 2016 and closed on 28 February 2018.
The trial was managed by a trial management group, coordinated and monitored by the Bristol Trials Centre Clinical Trials and Evaluation Unit (BTC-CTEU) and sponsored by the University Hospital Southampton NHS Foundation Trust. Trial conduct was overseen by an independent Trial Steering Committee (TSC) that received recommendations from an independent Data Monitoring and Steering Committee (DMSC) (see Appendix 2).
The trial protocol is available online and has been published. 21,22
Changes to study design after commencement of the study
The trial commenced recruitment on protocol version 4.0, dated 5 October 2016. During recruitment, version 5.0, dated 26 January 2017, was approved and follow-up version 6.0, dated 20 March 2018, was approved. Between the end of follow-up and database lock, version 7.0, dated 19 March 2019, was approved.
Changes made to the protocol after recruitment commenced are described below.
Between version 4.0 and version 5.0 the phase of the trial was corrected from a Phase II to a Phase III trial; fundus photography was added to the trial schema, having been omitted from previous versions in error; fasting blood glucose, which had been removed from the trial schema in the original protocol but had been left in the main body of text in error, was removed. Between version 5.0 and version 6.0 the optical coherence tomography angiography (OCT-A) image collection schedule was updated to allow collection of the first OCT-A images at any follow-up time point if it was missed at baseline; the reference safety information was updated to remove myocardial infarction from the list of expected events in line with the publication of updated summary of product characteristics (SmPC) for eplerenone in November 2017; a minor correction was made to the wording of the sample size justification to confirm that the sample size allowed for a drop of up to 15% rather than 10%. Between version 6.0 and version 7.0 secondary outcome 10 was split into secondary outcomes 10 and 15 to separate outcomes related to disease resolution from disease recurrence, and a minor clarification was made to secondary outcome 12 to confirm that fundus fluorescein angiography (FFA) phenotypes would be classified at both baseline and 12 months. A subgroup analysis looking at the interaction of the presence or absence of new vessels and treatment was removed.
Because of the bilateral nature of CSCR and the systemic nature of the intervention, we originally conceived of the trial potentially studying two eyes in one participant. As we set up the data collection tools and the database of the study it became clear that outcomes such as resolution of CSCR could not be accommodated in such a design and we concluded that only one study eye per participant, chosen at baseline, should be analysed. The plan of analysis in the protocol and the statistical analysis plan (SAP) are consistent with analysing only one eye per participant.
After the data were analysed the trial management team became aware of a recently published meta-analysis of MR antagonists in CSCR. 23 This prompted us to conduct an updated meta-analysis to include our data, which was not planned in the trial protocol or SAP.
Participants
Eligibility criteria
Participants were eligible to enter the study if all inclusion criteria and none of the exclusion criteria applied.
Inclusion criteria:
-
Age 18–60 years.
-
Visual impairment due to CSCR of at least 4 months’ duration, defined as comprising the following:
-
– subfoveal presence of SRF on optical coherence tomography (OCT)
-
– characteristic appearance of CSCR on FFA and indocyanine green angiography (ICGA)
-
– investigator believes that there is sufficient evidence from patient history, case note documentation or appearance of the macula that CSCR has been present for at least 4 months.
-
-
Women must have a negative pregnancy test and be willing to use effective contraception* for the duration of participation in the trial and for 3 months after or be surgically sterile or post menopausal for > 12 months.
-
Ability to provide written informed consent.
The following applied to the study eye only:
-
Early Treatment Diabetic Retinopathy Study (ETDRS) BCVA score of 54–85 letters
-
clear ocular media and adequate pupillary dilatation to permit photography.
*This includes progestogen-only oral hormonal contraception, where inhibition of ovulation is not the primary mode of action; male or female condom with or without spermicide cap; diaphragm or sponge with spermicide; combined (oestrogen- and progestogen-containing) hormonal contraception associated with inhibition of ovulation (oral, intravaginal or transdermal); progestogen-only hormonal contraception associated with inhibition of ovulation (oral, injectable, implantable); intrauterine device; intrauterine hormone-releasing system; bilateral tubal occlusion; vasectomised partner; and sexual abstinence. There were no special precautions/contraceptive requirements for male participants with female partners of child-bearing potential.
If both eyes presented with CSCR at baseline, the clinical trial site decided which was the study eye and this eye had retinal imaging performed first. The study eye was usually the eye with the most active disease/most SRF, which was identified by OCT imaging, and subsequent investigations such as fluorescein and ICGA were performed initially on this eye. If a patient presented with one affected eye at baseline and the fellow eye subsequently developed CSCR, the eye first affected was the study eye.
Exclusion criteria:
-
hyperkalaemia (serum potassium level of > 5.0 mmol/l)
-
hepatic or renal impairment: severe hepatic insufficiency (Child–Pugh Class C) or severe renal insufficiency [estimated glomerular filtration rate (eGFR) < 30 ml/minute/1.73 m2]
-
pregnancy or breastfeeding
-
known allergy to fluorescein or indocyanine green
-
receiving potassium-sparing diuretics, potassium supplements or inhibitors of cytochrome P450 3A4 (CYP3A4) (e.g. amiodarone, diltiazem, fluconazole, itraconazole, ketoconazole, ritonavir, nelfinavir, saquinavir, clarithromycin, telithromycin, erythromycin, verapamil, spironolactone and nefazodone) (patients taking furosemide were eligible)
-
receiving non-steroidal anti-inflammatory drugs (e.g. ibuprofen, naproxen)
-
receiving the combination of an angiotensin-converting enzyme inhibitor and an angiotensin receptor blocker
-
receiving lithium, ciclosporin or tacrolimus
-
hypersensitivity or known allergy to eplerenone or any excipients
-
known hereditary problems of galactose intolerance, Lapp lactase deficiency or glucose-galactose malabsorption
-
receiving high doses (> 75 mg) of aspirin.
The following additional exclusion criteria applied to the study eye only:
-
evidence of choroidal neovascularisation (CNV)
-
previous or current treatment with eplerenone for any reason or previous or current treatment with PDT/any anti-VEGF therapy/any intra-ocular steroid use/thermal laser therapy for CSCR
-
presence of any other disease that could cause retinal fluid or SRF to accumulate (e.g. diabetic retinopathy,* polypoidal choroidal vasculopathy, dome-shaped maculopathy or choroidal haemangioma) or affect visual acuity
-
myopia of > –6 dioptres.
*Diabetes alone was not an exclusion criterion.
Setting
Patients were identified and recruited from outpatient ophthalmology clinics at 22 secondary and tertiary care NHS trusts in the UK between December 2016 and February 2018. Sites were required to have SPECTRALIS® (Heidelberg Engineering Ltd, Hemel Hempstead, UK) imaging equipment to minimise heterogeneity in retinal imaging between sites. Broad coverage of England and Northern Ireland was achieved, making the trial accessible to a wide population of patients with CSCR. The following NHS trusts participated in the trial:
-
University Hospital Southampton NHS Foundation Trust
-
Belfast Health and Social Care Trust
-
Bradford Teaching Hospitals NHS Foundation Trust
-
Brighton and Sussex University Hospitals NHS Trust
-
City Hospitals Sunderland NHS Foundation Trust
-
East Lancashire Hospitals NHS Trust
-
Frimley Health NHS Foundation Trust
-
Guy’s and St Thomas’ NHS Foundation Trust
-
King’s College Hospital NHS Foundation Trust
-
Leeds Teaching Hospitals NHS Trust
-
Manchester University NHS Foundation Trust
-
Moorfields Eye Hospital NHS Foundation Trust
-
Newcastle upon Tyne Hospitals NHS Foundation Trust
-
Oxford University Hospitals NHS Foundation Trust
-
Royal Liverpool and Broadgreen University Hospitals NHS Trust
-
Royal Wolverhampton NHS Trust
-
Sheffield Teaching Hospitals NHS Foundation Trust
-
Southend University Hospital NHS Foundation Trust
-
Torbay and South Devon NHS Foundation Trust
-
University Hospitals Bristol NHS Foundation Trust
-
University Hospitals Coventry and Warwickshire NHS Trust
-
York Teaching Hospital NHS Foundation Trust.
Conduct
Screening was a two-stage process. Patients were initially screened against criteria that could be assessed from medical records. Eligible patients were provided with a patient information leaflet (PIL) and where possible were given at least 24 hours to consider participating in the trial. Written informed consent was obtained from interested patients to undergo trial-specific screening assessments required to assess full eligibility. If participants were eligible following assessments and did not withdraw consent, they were randomised into the trial. Eligibility was determined by experienced ophthalmologists at participating sites. Consent was taken by experienced research nurses or ophthalmologists at the sites.
Interventions
Randomised participants received a dose of 25 mg/day of oral eplerenone or matching placebo for 1 week. If tolerated (blood serum potassium level of ≤ 5.0 mmol/l), the dose was increased to 50 mg/day of oral eplerenone for up to 12 months or until complete resolution of SRF or a blood serum potassium level of > 5.0 mmol/l. Because the interventions were masked, participants in both treatment arms followed the same dose escalation schedule.
Eplerenone was administered as 25-mg or 50-mg tablets that were overencapsulated to match the placebo capsules. The placebo capsules were backfilled with lactose, which was chosen because lactose is a constituent of eplerenone tablets. No restrictions were placed on taking the eplerenone or placebo capsules; capsules could be taken at any time of day, with or without food.
Participants took the capsules home in the form of one bottle of 10 25-mg capsules (1 week’s supply) or one, two or three bottles of 36 50-mg capsules (1, 2 or 3 months’ supply, respectively) depending on the length of time between follow-up visits.
Treatment decisions
Safety criterion for dose escalation and continuation of study drug
Hyperkalaemia (high level of blood serum potassium) is a common side effect of eplerenone. In this trial, in which eplerenone was administered off licence, a conservative safety threshold of ≤ 5.0 mmol/l for serum blood potassium level was applied. Potassium levels were assessed prior to randomisation as part of the eligibility criteria. Dose escalation at 1 week and continuation of the 50-mg dose at 4 weeks and 3, 6 and 9 months was dependent on having a blood serum potassium level of ≤ 5.0 mmol/l. If serum potassium level was > 5.0 mmol/l, the participant was withdrawn from treatment for the remainder of the trial and invited to continue with follow-up to the 12-month time point.
Central serous chorioretinopathy resolution and recurrence treatment criteria
Resolution of CSCR (absence of SRF on OCT) was assessed by the treating clinician at 4 weeks and 3, 6, 9 and 12 months post randomisation. If CSCR resolved, participants stopped treatment and were invited to continue with follow-up. If at a later follow-up visit SRF recurred, participants restarted treatment (provided that the serum potassium threshold criterion was met; see Safety criterion for dose escalation and continuation of study drug) and repeated the week 1 dose escalation (restarting week 1) and week 4 (restarting week 4) follow-up visits for safety monitoring purposes before returning to their original follow-up schedule; for example, if a participant restarted treatment at the 6-month follow-up and repeated the dose escalation schedule, they would next attend their 9-month post-randomisation follow-up visit.
Usual care
Eplerenone and placebo were administered alongside usual care. Additional therapies for CSCR were permitted at the discretion of the treating clinician (e.g. PDT, thermal laser therapy, anti-VEGF agents). The recommended criterion for deciding to administer an alternative therapy was a decrease in BCVA score of ≥ 15 letters from baseline (i.e. a sight-threatening event); however, this was not mandated.
Outcomes
Primary outcome
The primary outcome is BCVA score at the 12-month visit, adjusted for baseline BCVA score, measured using validated ETDRS vision charts, with measurements made in accordance with a standardised protocol for trials in medical retina. Refracted visual acuity was measured at baseline, 4 weeks and 3, 6, 9 and 12 months.
Secondary outcomes
-
Low-luminance visual acuity. This is measured immediately after measuring BCVA by interposing a neutral density filter of 2 log units into the trial lens of the eye being tested and recording the number of letters read.
-
Central subfield retinal thickness (CSRT) as measured by OCT recorded at 12 months, including CSRT measured at interim visits and adjusted for baseline CSRT.
-
Change in subretinal fluid thickness (SRFT) as measured by OCT.
-
Systemic and ocular adverse events (AEs) at any time during the 12-month follow-up period.
-
Proportion of patients with macular atrophy of the RPE defined as hypoautofluorescence at 12 months.
-
Area change in macular RPE hypoautofluorescence at 12 months.
-
Choroidal thickness as measured by enhanced depth imaging (EDI) OCT at 12 months, adjusted for baseline choroidal thickness. Measurements made subfoveally.
-
Proportion of patients with reduced choroidal permeability on ICGA at 12 months.
-
Time to resolution of SRF.
-
Classification of all study eyes as complete, partial or no resolution of SRF at each time point of the study. Partial resolution of SRF is defined as a decrease of > 25% of CMT from baseline. A non-responder is defined as a study eye with an increase in SRF or decrease in SRF of ≤ 25% from baseline.
-
Patient-reported visual function using the National Eye Institute Visual Function Questionnaire-25 (NEI-VFQ-25)24 will be assessed at baseline and 12 months.
-
Classification of all study eyes by each FFA phenotype, such as ‘smoke stack’, ‘ink-blot’ and ‘chronic epitheliopathy’ at baseline and 12 months.
-
Classification of all study eyes as early, late or non-responder. An early responder is defined as a study eye with complete or partial resolution of subfoveal SRF by 3 months. A late responder is defined as a study eye with complete or partial resolution of subfoveal SRF after 6 months.
-
Incidence of CSCR in the fellow eye as measured by OCT, FFA, ICGA or AF.
-
Time to recurrence of SRF. Recurrence is defined as the appearance of new SRF in a study eye after complete resolution of SRF at any point.
See Table 1 for secondary outcomes grouped by corresponding objective.
| Objective | Corresponding secondary outcome(s) |
|---|---|
| (b) to evaluate whether or not eplerenone treatment with usual care is better than placebo with usual care for resolution of SRF | (3) Change in SRF thickness as measured by OCT |
| (9) Time to resolution of SRF | |
| (10) Classification of all study eyes as complete, partial or no resolution of SRF at each time point of the study | |
| (13) Classification of all study eyes as early, late or non-responder | |
| (c) to describe the safety profile of eplerenone treatment with usual care (compared with placebo with usual care) | (4) AEs at any time during the 12-month follow-up period |
| (d) to evaluate whether or not participant-reported visual function improves with eplerenone treatment with usual care compared with placebo with usual care | (11) Patient-reported visual function using the NEI-VFQ-25 at 12 months, adjusted for baseline |
| (e) to describe how the choroid responds to treatment in CSCR | (7) Choroidal thickness as measured by EDI OCT over a 12-month period |
| (8) Proportion of patients with reduced choroidal permeability on ICGA at 12 months | |
| (f) to describe how RPE function changes over 1 year in CSCR as measured by AF | (5) Proportion of patients with macular atrophy of the RPE defined as hypoautofluorescence at 12 months |
| (6) Area change in macular RPE hypoautofluorescence at 12 months | |
| (g) to evaluate how LLA changes with eplerenone treatment | (1) LLA score over a 12-month period |
Assessments
Best corrected visual acuity
Best corrected visual acuity was measured as follows. Participants were seated at 4 metres from the ETDRS chart. The right eye was refracted first, followed by the left eye, with the eye that is not tested covered with an occluder. Rooms were darkened such that, with the ETDRS chart light switched off, not more than 161.4 lux fell on the centre of the chart. Full aperture trial lenses were used with a trial frame. At baseline, initial acuity was measured with the participants’ own spectacles or unaided if spectacles were not worn, followed by refraction. At follow-up visits the refraction from the previous visit was used as a starting point.
If the initial acuity was ≥ 40 letters read correctly, participants were refracted at 4 metres, or at 1 metre if < 40 letters were read correctly. If a participant was unable to read ≥ 20 letters at 4 metres, the test was repeated at 1 metre with an additional + 0.75 dioptre lens added to the trial frame, to correct for the closer testing distance, and participants were asked to read from only the first six rows. Participants were instructed to read the letters one line at a time until the number of correctly identified letters in the smallest line of letters was lower than three. The same procedure was repeated with the fellow eye and a binocular measurement was recorded. All letters correctly identified by the participant were circled on the score sheet for each eye and then for the binocular record.
If measured at 4 metres, the visual acuity score is the number of letters read correctly plus 30 (i.e. the maximum number of letters that could be read correctly at 1 metre). If measured at 1 metre, the visual acuity score is the total number of letters read. If the subject cannot identify any of the letters at a testing distance of 1 metre, detection of hand movements or light perception in the eye of interest is recorded.
Low-luminance visual acuity
Low-luminance visual acuity is performed after the BCVA test using the same lenses and lighting conditions. LLA can be a more sensitive measure of deterioration in visual acuity than BCVA under normal light conditions. 25 A base 10 neutral density filter (to reduce luminance 100-fold) was held up close to the trial frame. The visual acuity measurement is repeated at 4 metres. If < 20 letters are read correctly, the participant is tested at 1 metre with + 0.75 dioptres added to the trial frame. Letters read are recorded and LLA scores calculated in the same way as BCVA scores.
Retinal image capture
Details of the retinal image capture specifications can be found in Appendix 3.
National Eye Institute Visual Functioning Questionnaire-25
The NEI-VFQ-25 was administered using a print version. Research teams were supplied with the NEI-VFQ-25 in two formats: self-administered and interviewer-administered formats. Participants were offered the option of completing the questionnaire themselves (self-administered) or having the questions read to them and their answers recorded by a member of the research team (interviewer-administered). Participant choice of format was not recorded. Local research teams entered participants’ responses from the NEI-VFQ-25 into a secure internet-based database. 26
Adverse events
Serious adverse events (SAEs) and non-serious AEs were recorded throughout the 12-month follow-up period. Participants’ general practitioners were notified of their participation, with a request to inform the local research team about any suspected AEs or reactions.
At each follow-up visit participants were asked to report any AEs experienced since the previous visit. All events were recorded on a case report form (CRF) that coded them by Medical Dictionary for Regulatory Activities (MedDRA) version 14.1 categories. SAEs were reviewed by the local treating clinician, who was masked to the allocation. The treating clinician made the decision on whether or not the SAE was related to the intervention.
Adverse events were reviewed at least monthly by the Trial Management Group and twice per year by the DMSC, which gave recommendations of continuation of the trial to an independent TSC. Unexpected SAEs that were causally related to the intervention were subject to expedited reporting to the REC, MHRA and DMSC.
Sample size
We calculated that 45 patients in both groups would be sufficient to detect a difference of five or more letters in BCVA score at 12 months between the eplerenone and placebo groups with 90% power and 5% significance. This assumed the following:
-
a standard deviation (SD) of change in BCVA score of nine letters11
-
the correlation between baseline and any follow-up BCVA score was 0.5 letters27
-
data were collected from a minimum of two follow-up assessments per participant28
-
the correlation between BCVA score on follow-up visits was 0.8 letters.
The target sample size was 104, which allowed for < 15% drop-out over the 12-month period. There were no changes to the sample size during the trial.
Randomisation
Participants were randomly allocated to either the eplerenone with usual care group or the placebo with usual care group in a 1 : 1 ratio. The randomisation was stratified by centre and visual acuity level (low, ETDRS BCVA score of 54–67 letters; high, ETDRS BCVA score of 68–85 letters) and used block randomisation with block sizes of two and four. Allocations were generated by computer, in advance of starting the trial, by a statistician at the co-ordinating centre and supplied to the Newcastle Specials Pharmacy Production Unit (Newcastle Upon Tyne, UK). Randomisation occurred only once information used to identify a participant uniquely and confirm eligibility had been entered into a secure internet-based randomisation system, accessible only to approved trial personnel. Randomisation was performed by local ophthalmologists or delegated members of the local trial team (e.g. research nurses or trial co-ordinators) within 4 weeks of screening.
Masking
Participants, clinicians, research nurses, outcome assessors, pharmacists, the trial management team and statisticians were masked to the allocations. Masking was implemented by overencapsulation of eplerenone/placebo to produce identical capsules. Capsules were supplied in bottles with labelling that was identical except for a unique bottle number. Bottle numbers were assigned against an allocation list. Only personnel at the manufacturing pharmacy and the trial database manager had access to the allocation list for the purposes of producing and managing the investigational medicinal product (IMP). The trial manager and site pharmacists were able to unmask the allocation if requested, for example in the event of a SAE where knowledge of the allocation would affect the patient’s care. The final decision to unmask lay with the chief investigator or co-lead investigator.
Data collection and follow-up schedule
Participants were followed up for 12 months (except where participants withdrew from follow-up). Data were collected at baseline and 1 week, 4 weeks and 3, 6, 9 and 12 months post randomisation (Table 2) and at 1 week and 4 weeks post restarting the IMP, if applicable. All prospective data were collected face to face during outpatient clinics. Images captured by OCT, AF, FFA, ICGA and colour fundus photography (CFP) taken during research visits were sent in pseudonymised format to the Network of Ophthalmic Reading Centres UK for independent grading by trained assessors masked to the allocations (see Multimodal image grading for central serous chorioretinopathy for details). Retrospective data (pre-screening eligibility and medical history) were collected by local research teams from medical notes and by asking participants for information. Data were recorded on paper CRFs and entered into a secure internet-based database. 26
| Variable | Study period | |||||||
|---|---|---|---|---|---|---|---|---|
| Enrolment | Allocation | Post allocation | Close-out | |||||
| –t1 | 0 | t 1 | t 2 | t 3 | t 4 | t 5 | t 6 | |
| Enrolment | ||||||||
| Eligibility pre screen | ✓ | |||||||
| Informed consent | ✓ | |||||||
| Post-consent eligibility screen | ✓ | |||||||
| Allocation | ✓ | |||||||
| Intervention | ||||||||
| Eplerenone | ||||||||
| Placebo | ||||||||
| Assessment | ||||||||
| Medical history | ✓ | |||||||
| Ophthalmic history | ✓ | |||||||
| Concomitant medicationsa | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |
| Pregnancy test (women only) | ✓ | |||||||
| FFA | ✓ | ✓ | ||||||
| ICGA | ✓ | ✓ | ||||||
| AF | ✓ | ✓ | ||||||
| Fundus photography | ✓ | ✓ | ||||||
| BCVA (and binocular BCVA) score | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| LLA score | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| EDI OCTb | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| OCT angiographyc | ✓ | ✓ | ||||||
| DNA, serum and plasmad | ✓ | |||||||
| HbA1ce | ✓ | ✓ | ||||||
| Thyroid function testse | ✓ | ✓ | ||||||
| Full blood counte | ✓ | ✓ | ||||||
| Liver function testse | ✓ | ✓ | ||||||
| Urea and electrolytes (including potassium)e,f | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |
| Blood pressure | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |
| Heart rate | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |
| Slit lamp examination | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| AEs | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |
| NEI-VFQ-25 | ✓ | ✓ | ||||||
Multimodal image grading for central serous chorioretinopathy
Central serous chorioretinopathy results in fluid accumulation in the subretinal space and is often accompanied by pigment epithelial detachments, which is caused by fluid in the subpigment epithelial space. An important feature of CSCR is the presence of a thicker than average choroid, which may be diffusely or focally thickened. CFP, FFA, ICGA, AF and OCT images were used to determine the presence, extent and severity of CSCR. Measurements of disease extent and severity were made using both enface (two-dimensional) and tomographic images. Both study eyes and fellow eyes were scrutinised for the presence of features of CSCR. In addition, if any other pathology was found to be present, this was recorded for both study eyes and fellow eyes. All imaging modalities were graded at baseline and 12 months. In addition, OCT images were graded at 4 weeks and 3, 6, and 9 months.
Colour fundus photography
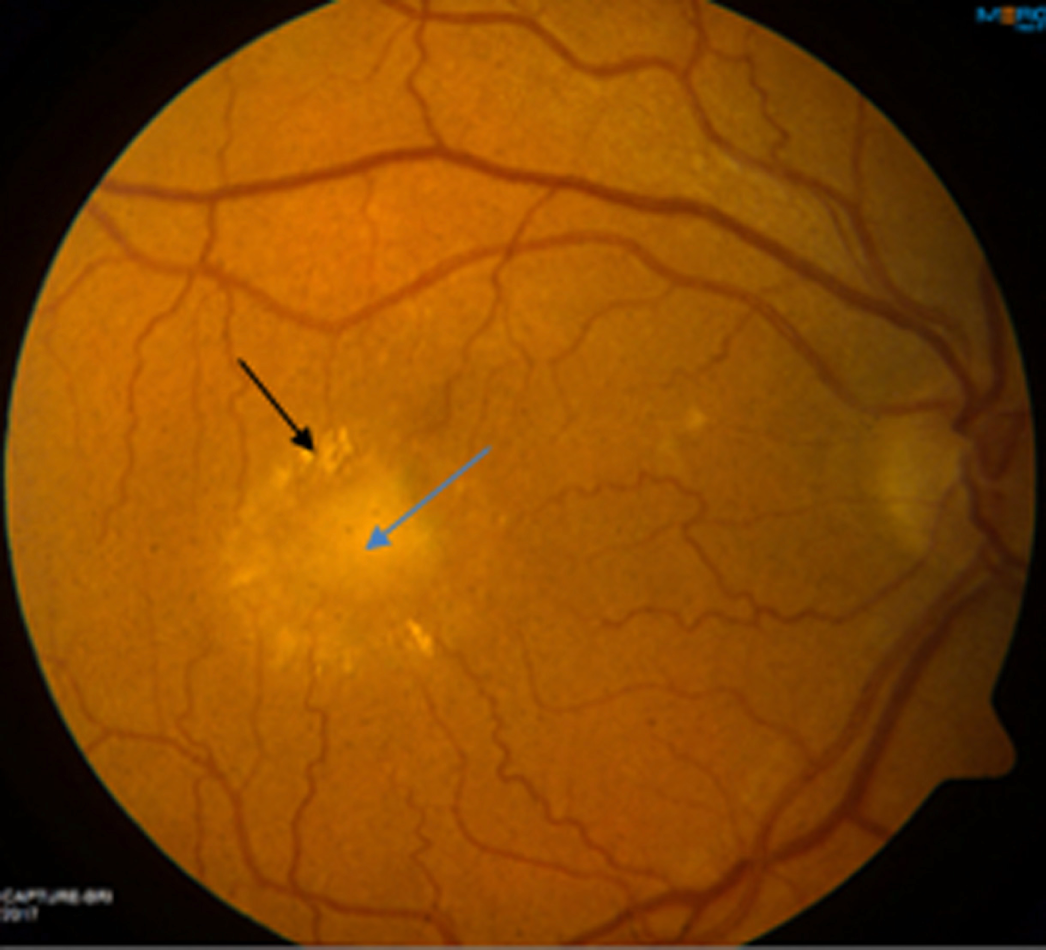
The presence of bullous collections of SRF, foci of hypo- and hyperpigmentation and exudate, which are common features of CSCR, was recorded. The colour image in Figure 1 shows an example of a region of SRF with a surrounding area of exudate seen as streaks. The colour images were used ad hoc to help adjudicate on retinal features seen with other imaging modalities if these were not clear from a single imaging modality.
FIGURE 1.
Colour fundus imaging. The colour image shows a region of SRF (blue arrow) with a surrounding area of exudate seen as streaks (black arrow). For scale, the optic disc is approximately 1.5 mm in diameter.
Fundus fluorescein angiography
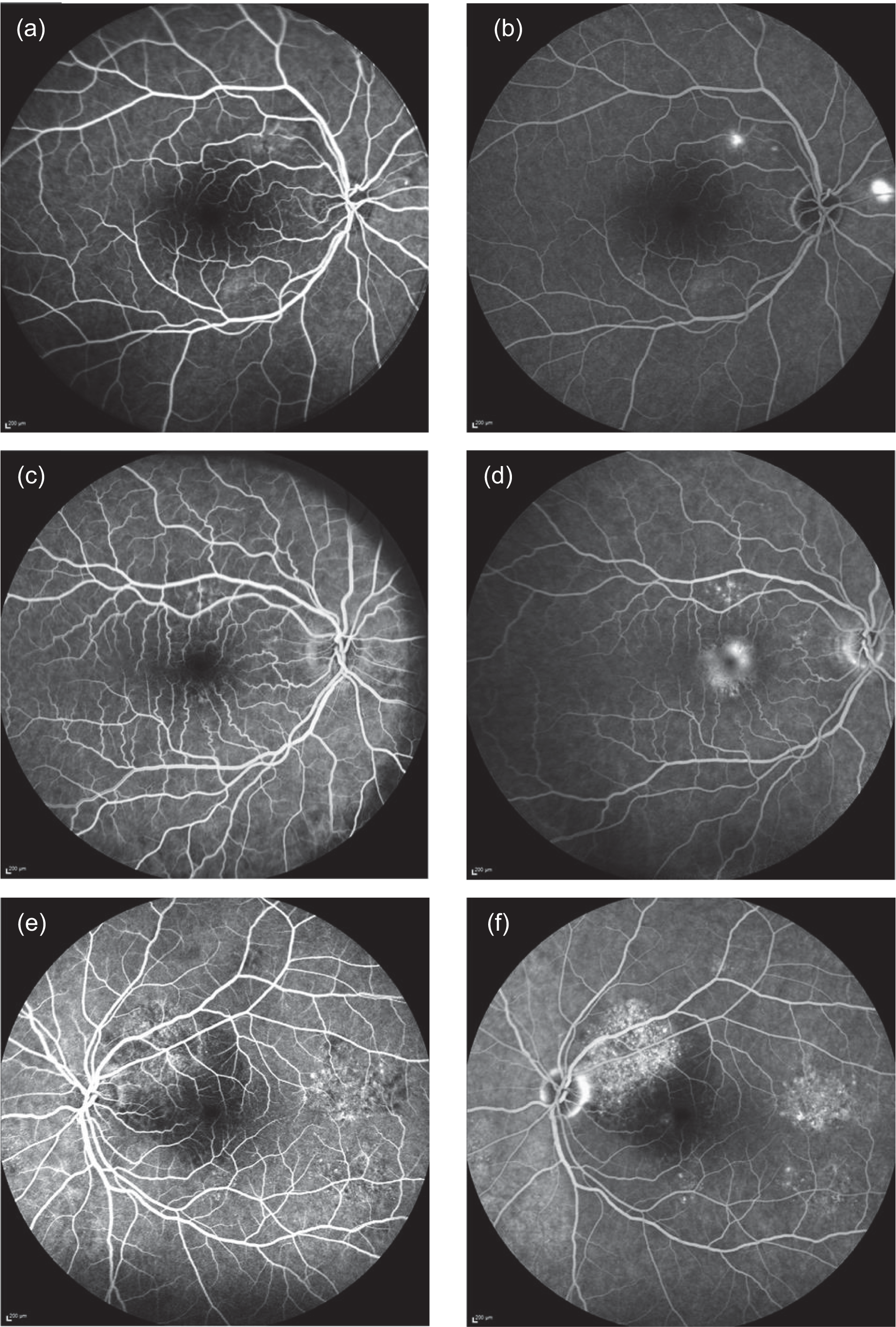
The transit of fluorescein through retinal vessels can be seen clearly, and under normal conditions the dye remains in the retinal circulation. The choroidal circulation is leaky and, as a result, fluorescein enters the extravascular space and is seen as a low-intensity diffuse background level of fluorescence similar to the appearance of ground glass. In CSCR, leakage of fluorescein occurs and results in the appearance of a fluorescein leak during the angiographic frame, which was recorded. The appearance (phenotype) of the leak can vary. A single circular region of leak is termed ‘ink-blot’. Leaks can sometimes track vertically; these have the appearance of a ‘smoke stack’. A third variety, termed ‘chronic epitheliopathy’, appears as speckled diffuse hyperfluorescence, representing a very chronic disease state with no definite morphological features (see Figure 2 for examples). FFA phenotypes (ink-blot, smoke stack, chronic epitheliopathy or no leakage visible) were recorded at baseline and 12 months to address secondary outcome 12. In Figure 2a, two areas of early hyperfluorescence superotemporal and nasal to the optic disc are seen. In the later frame of the angiogram (Figure 2b), these areas of hyperfluorescence increase in size and hyperfluorescence while the fluorescence in the normal retinal blood vessels fades. This is described as a ‘smoke stack’ form of leakage. In a different patient, early (Figure 2c) and late (Figure 2d) frames of a fluorescein angiogram show some pinpoint leakage superior to the fovea, which increases slightly in size in Figure 2d, and a central area of increased hyperfluorescence in the fovea appears in Figure 2d. This is described as an ‘ink blot’ form of leakage. Finally, in a different patient, diffuse areas of chronic retinal pigment epitheliopathy are seen in both early (Figure 2e) and late (Figure 2f) frames of the fluorescein angiogram. These areas of punctate hyperfluorescence are seen superotemporal to the optic disc and temporal and inferotemporal to the fovea.
FIGURE 2.
Fundus fluorescein angiogram phenotypes. (a) Early-frame smoke stack; (b) late-frame smoke stack; (c) early-frame ink-blot; (d) late-frame ink-blot; (e) early-frame chronic epitheliopathy; and (f) late-frame chronic epitheliopathy.
Other FFA variables were graded but were not specified as secondary outcomes, including the timing, morphology and spatial distribution of the leaks. Leaks can occur during any stage of the angiogram. Leaks that occurred within the first 30 to 60 seconds were classified as ‘early’ and those that occurred after 60 seconds were classified as ‘late’. The morphology or pattern of leakage was also determined. Leaks can be at multiple sites of the fundus and, consequently, grading required classification as unifocal or multifocal. The spatial distribution of the leak was also determined as central or peripheral (central within the macular arcades and peripheral beyond macular arcades).
Indocyanine green angiography
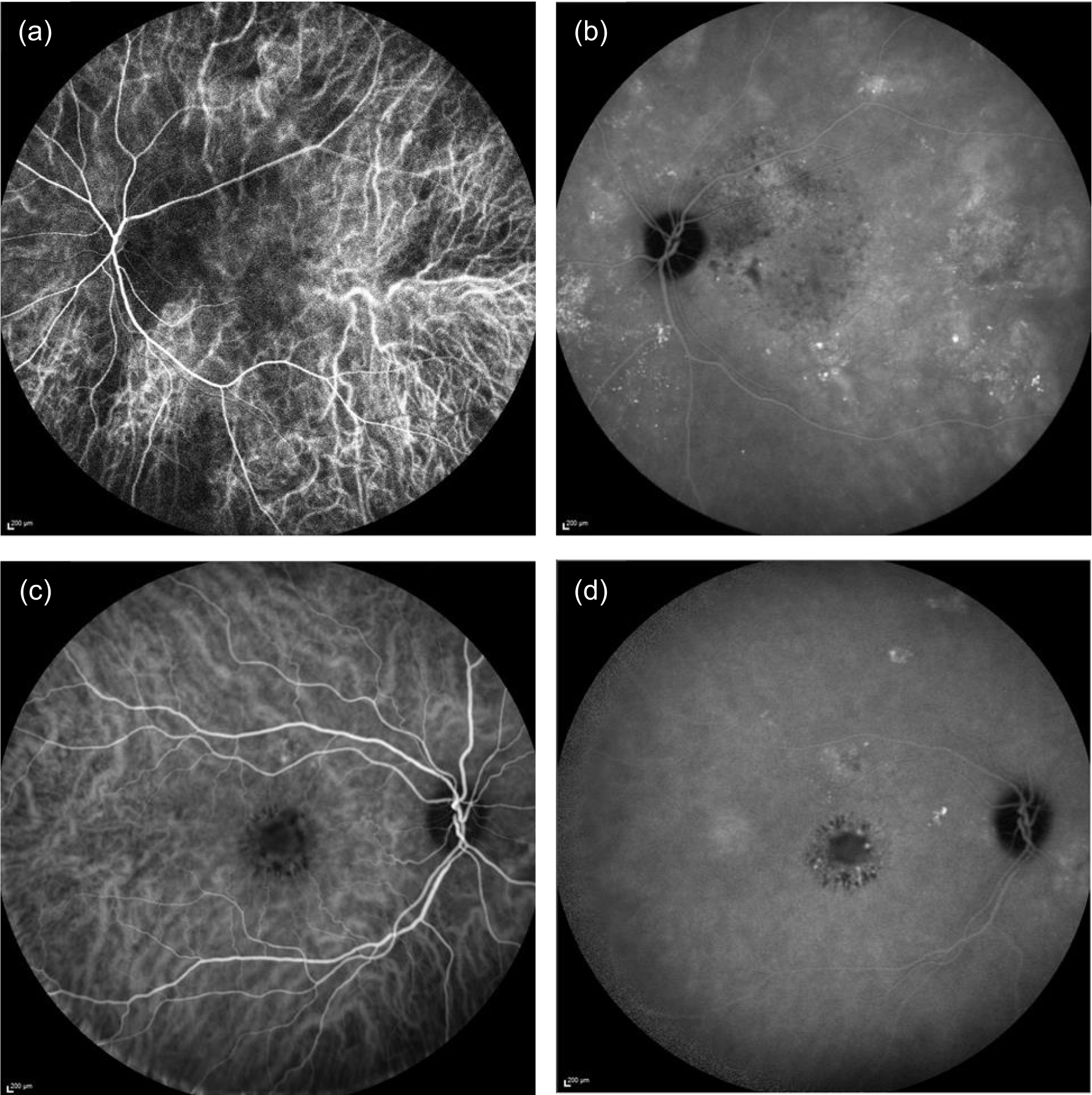
The dye indocyanine green binds tightly to large plasma proteins and remains within the choroidal vessels. Consequently, the morphology of the choroidal circulation is better observed with this dye. Because CSCR is primarily a choroidal disease, considerable attention was given to grading of ICGA images. The inclusion in the angiographic run of the arterial phase of the indocyanine green is important and graders were asked to state whether or not appropriate frames were captured. ICGA images were used to identify reduced choroidal permeability at 12 months (secondary outcome 8), which was recorded as ‘yes’ or ‘no’. Figure 3 shows examples of ICGA images taken early and late in the run. In the early frame of the ICGA the retinal vessels are seen extending from the optic nerve (oval dark area at the right of the frame) sweeping across superior and inferior to the macula forming arcades (Figure 3a and c). The choroidal vessels are seen throughout the frame as linear bands of hypercyanescence. These vessels are broader and the cyanescence is of lesser intensity than in the retinal vessels. There is a focal area of punctate cyanescence in the central macula. The late frame of the ICGA shows multiple foci of hypercyanescence at the fovea, in the macular arcades and in the superior part outside the macular arcade, representing leakage from choroidal vessels.
FIGURE 3.
Early- and late-frame indocyanine green angiograms. (a) Early frame showing abnormally dilated choroidal vasculature with flow voids; (b) late frame showing multiple foci of leakage with staining of the RPE (punctate hypercyanescence); (c) early frame showing normal-looking choroidal vasculature; and (d) late frame showing leakage from choroidal vessels with areas of hypercyanescence and punctate staining of the RPE.
Other key features of interest that were graded but did not contribute to the secondary outcomes were spots (circular regions of hypercyanescence) and plaques (irregular regions of hypercyanescence), the number of plaques and the area of the largest plaque. We also recorded whether or not the position of the largest plaque was subfoveal. Temporal characterisation was also important and abnormal hypercyanescence was classified as appearing ‘early’ or ‘late’ and with a spatial distribution ‘macular’ or ‘peripheral’. The visibility of the individual choroidal vessels in the early and late ICGA frames and the presence of dilated vessels anywhere in the angiographic frame was noted.
Autofluorescence
Autofluorescence imaging was used to determine the proportion of participants with macular atrophy (defined as the presence of hypoautofluorescence) of the RPE at 12 months and the area change in macular RPE hypoautofluorescence at 12 months, adjusted for baseline (secondary outcomes 5 and 6, respectively).
Other key features were graded but were not secondary outcomes. Abnormalities of blue AF were identified by macular grid location. Both hypo- and hyperautofluorescence were identified, and the total area was measured. Correspondence between AF abnormalities and near infrared was examined.
Optical coherence tomography
Optical coherence tomography allows the assessment of the relationships between the retina, RPE and choroid in unprecedented detail. The phenotypic characterisation of the vascular choroid, including details of the vascular profiles and retinochoroidal interface, has been markedly enhanced, permitting assessments of the presence of choroidal varices, engorged dilated choroidal vessels and diffuse thickening of the overall choroidal structure. In addition, changes in the outer retinal layers – namely, presence of SRF, disorganisation and/or shedding of the photoreceptors into the subretinal space, and intactness of the external limiting membrane and ellipsoid zone – could be ascertained. Changes at the level of the RPE/Bruch’s membrane, such as thinning or thickening of this structure or presence of small foci of pigment epithelial detachments, were also identified. Among other descriptive measures, the presence of outer retinal atrophy resulting in increased transmission of the OCT signal was captured.
Optical coherence tomography images can be used for measurements such as of the vertical height of pockets of fluid as well as of the thickness of tissue compartments. In addition, automated outputs of central macular volume are available with the current generation of OCT capture systems. For the VICI trial, measurements were made on the foveal image and included the height of SRF and choroidal thickness at the fovea and the thickest point.
Optical coherence tomography images were used to measure CSRT (µm) at baseline, 4 weeks and 3, 6, 9 and 12 months (secondary outcome 2); the change in SRFT (µm) at 12 months, adjusted for baseline (secondary outcome 3); and the thickness of the choroid (µm; measured subfoveally) at 12 months (secondary outcome 7). In addition, OCT was used to assess the resolution or reduction of SRF (secondary outcomes 9, 10) and the time taken for reduction or resolution of SRF to occur (secondary outcome 13).
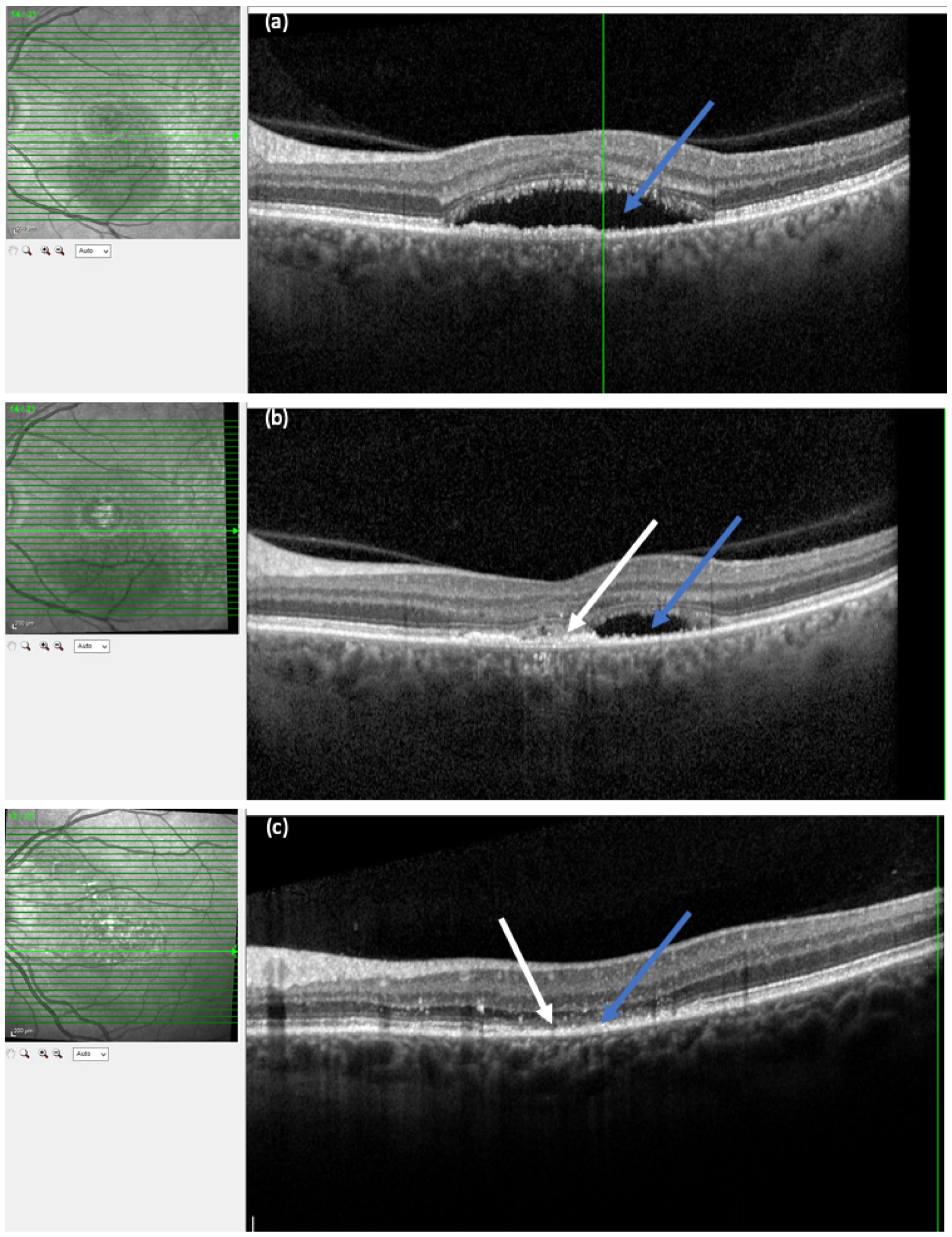
Example OCT images are shown in Figure 4. The OCT image in Figure 4b, a cross-section of the fovea (bold line of the raster shown in the reflectance image), displays the neurosensory retina with the multiple layers of neural cells. The innermost fine hyper-reflective band is the internal limiting membrane. The outer two bright bands represent the outer regions of the photoreceptors and the RPE. The choroid is the crescent-shaped area located on the outside of the RPE. Vascular profiles, surrounded by intervascular pillars, are visible within the choroid. In the central region of the OCT image in Figure 4b there is a region of dark hyporeflectivity separating the neurosensory retina from the pigment epithelium, which represents the collection of SRF.
FIGURE 4.
Optical coherence tomography images. (a) Baseline OCT image of an eye with CSCR. The dark area lying between the retina and the pigment epithelium (blue arrow) represents a region with SRF. (b) OCT image of the same eye at 12 months. The area of SRF has reduced (blue arrow). The white arrow shows an area of residual disturbance of the RPE and outer retinal layers, seen as an irregular region of hyper-reflectivity. (c) OCT image at 12 months from another participant showing complete resolution of SRF (blue arrow). The retina is thinned and the outer nuclear band (white arrow) has almost disappeared in the region where SRF was originally present.
Detecting complications of central serous chorioretinopathy
Optical coherence tomography was used to monitor and detect onset of CNV, which is a complication of CSCR. The onset of CNV can be detected by OCT and is seen as a splitting of the RPE from Bruch’s membrane and the appearance of hyper-reflective material where SRF is no longer clear but becomes turbid owing to leakage of whole blood components.
The parameters graded and their definitions are available as separate documents on the National Institute for Health Research (NIHR) project web page. 21
All imaging modalities could be used by the treating clinicians to diagnose CSCR in the fellow eye (secondary outcome 14).
Biobank samples
Pre randomisation, consenting participants donated approximately two 10-ml samples of blood in ethylenediaminetetraacetic acid vacutainers and one 10-ml sample of blood in clot-activating gel vacutainers. Samples were posted at room temperature to the University of Southampton and processed into DNA, plasma and serum components, which were stored at –80 °C for use in future ethics-approved studies. Donating blood samples to the biobank was an optional aspect of study participation.
Adherence monitoring
Participants’ adherence to the IMP regimen was monitored throughout the trial. Participants were asked to return used bottles of capsules from their previous follow-up visit at their next follow-up visit (if applicable; see Safety criterion for dose escalation and continuation of study drug and Central serous chorioretinopathy resolution and recurrence treatment criteria). Returned capsules were counted by clinical trials pharmacists and recorded in a secure web-based IMP management system. 22 The number of expected capsules consumed was calculated as the number of days from the date the participant was informed to continue on the IMP to the date of the follow-up visit at which the capsules were returned, or the date of the follow-up visit at which the capsules should have been returned if the follow-up visit was missed. (If a visit was missed, no IMP could be prescribed for the subsequent period and monitoring adherence did not apply.) If participants took > 70% of the expected number of capsules in a follow-up period, they were categorised as adherent. During protocol development, the adherence threshold was discussed by the trial management group and a consensus was reached, defining satisfactory adherence as > 70% of pills that should have been taken in accordance with the protocol. The threshold was subsequently discussed and agreed at an investigators’ meeting.
If a participant failed to return used bottles of capsules, the bottles were categorised as ‘lost’. It was assumed that ‘lost’ bottles contained no capsules because the participant had taken them all and discarded the bottle. A sensitivity analysis to test the effect of this assumption on the primary outcome was performed. This was performed by using multiple imputation to impute the number of pills remaining in bottles confirmed as ‘lost’.
Emergency unmasking
In the event of a medical emergency, research personnel followed working instructions to facilitate unmasking of a participants’ allocation by the trial manager at the BTC-CTEU (during working hours) and local site pharmacists (outside working hours).
-
The internet-based IMP management system that was used by sites and the BTC-CTEU to allocate, track and account for bottles of IMP throughout the trial had an unmasking function. On completion of a minimum data set (name of person requesting unmasking; reason for unmasking; person doing the unmasking) the system would unmask a participants’ allocation. The unmasking function was role-restricted to users who were delegated pharmacy or trial administrator permissions.
-
To enable emergency unmasking by out-of-hours on-call pharmacists who were not registered on the IMP management system, a generic username and password specific to each site were created and disclosed only to site pharmacists. This ensured that, in a medical emergency, local pharmacy personnel would be able to unmask a participant without needing to register and await approval from the trial manager.
-
To enable unmasking in the event of internet failure, local pharmacies were provided with sealed code-break envelopes for the 25-mg-dose bottles of IMP. Every participant, irrespective of overall time on IMP, was allocated an initial bottle of 25-mg capsules at randomisation but would not be guaranteed to receive subsequent bottles of IMP. Therefore, it was necessary to produce code-break envelopes for the 25-mg bottles only. Pharmacists were instructed to look up the first bottle number allocated to the participant in the event of an unmasking event. Envelopes were manufactured by Newcastle Specials Pharmacy Production Unit. Code-break envelopes were retrieved by the BTC-CTEU at the end of the trial and inspected for evidence of tampering.
Unmasking events were monitored by the trial manager. If a participant’s allocation was unmasked via the above options 1 or 2, an automated e-mail alert to the trial e-mail address was triggered. In the event of unmasking via option 3, local pharmacists were required to report having unmasked allocation to the trial manager at the earliest opportunity. Code-break envelopes were returned to the co-ordinating centre at the end of the trial.
Statistical methods
The analysis and safety populations comprised all participants randomised into the study, excluding those who withdrew and were unwilling for their data already collected to be used. All analyses were directed by a pre-specified SAP (see the NIHR project web page)21 and performed on an intention-to-treat basis. Continuous variables are summarised using the mean and SD or, if the distribution was skewed, the median and interquartile range (IQR). Categorical variables are summarised as number and percentage.
The intention was to adjust all models for the two stratification factors included in the randomisation: visual acuity level (low, ETDRS BCVA score of 54–67 letters; high, ETDRS BCVA score of 68–85 letters) as a fixed effect and centre as a random effect. Owing to small numbers of patients at some centres, it was not always possible to estimate a random effect for each centre. If this was the case, centres with a small number of patients were combined (e.g. centres with one or two patients were grouped and centres with three or four patients were grouped). For time-to-event outcomes it was not possible to fit centre as a random effect, so this was accounted for by using a clustered sandwich estimator to adjust the standard errors of the estimates for clustering within centre.
The primary outcome and other longitudinal outcomes (LLA and CSRT) were analysed using linear mixed-effects methods, with treatment effects presented as mean differences (MDs) with 95% confidence intervals (CIs). Both time and time by treatment interactions were added to each of the models as fixed effects to allow the treatment effect at 12 months to be estimated. Baseline values measured before treatment had started were also added to each model as a fixed effect. Difference variance/covariance structures were assessed and the structure that provided the best fit was used to model within-patient errors. Likelihood ratio tests determined that an unstructured covariance structure provided the best fit in all longitudinal models. Time-to-event outcomes were analysed using proportional hazards parametric survival models for interval-censored data, and treatment estimates presented as hazard ratios (HRs) and 95% CIs. Complete resolution of SRF and complete or partial resolution of SRF were censored at the time of last follow-up if SRF did not resolve completely or partially. Disease recurrence was censored at time of last follow-up if disease did not recur. Continuous outcomes measured at 12 months [SRFT, patient-reported visual function (NEI-VFQ-25) and choroidal thickness] were compared using linear regression, adjusted for baseline values, with treatment estimates presented as MDs. For all methods outlined, underlying assumptions were checked using standard methods (e.g. tests for normality, residual plots) and if assumptions were not valid then alternative methods of analysis were sought.
A pre-specified exploratory analysis was performed for the primary outcome, BCVA score, to assess the effect of adherence and treatment. Two indicators, one for treatment (on treatment vs. not on treatment) and one for adherence (adhered vs. did not adhere), were generated for each post-randomisation time point at which BCVA was assessed. If a patient was not on treatment owing to disease resolution, they were considered adherent; if a patient was not on treatment for any other reason, they were deemed non-adherent. Interactions between treatment group and these indicators were added to the model and indicator status-specific treatment effects were estimated. A sensitivity analysis reassessing the effect of adherence and treatment on BCVA score after imputing pill counts for lost bottles was also performed.
A sensitivity analysis pre-specified in the SAP but not in the protocol was performed for time-to-event outcomes – complete resolution of SRF and complete or partial resolution of SRF – because an initial review of baseline data found imbalances between treatment groups in SRF. This was performed by adding baseline SRF level to the models.
A post hoc sensitivity analysis of BCVA, CSRT, SRFT and choroidal thickness was performed, which involved adjusting for whether or not the participant had received PDT. These were performed as the proportion of patients receiving PDT throughout the trial differed between the treatment groups. For outcomes measured at multiple time points (BCVA and CSRT), an indicator was added to the model that was updated to ‘yes’ at the point at which the participant received PDT. For outcomes measured at 12 months only (SRFT and choroidal thickness), an indicator for whether or not the participant had received PDT in the study eye at any point throughout the trial was added to the model.
For hypothesis tests, two-tailed p-values of < 0.05 were considered statistically significant. Likelihood ratio tests were used in preference to Wald tests. No formal adjustment was made for multiple testing. However, formal statistical comparisons were not made for outcomes with low event rates and consideration has been given to the number of tests performed when interpreting the results.
All analyses were performed in Stata® version 15.1 (StataCorp LP, College Station, TX, USA).
In all tables, missing data are described in footnotes. Rules for imputing missing data were outlined in the SAP and depended on the level of missing data. Multiple imputation was required for NEI-VFQ-25 score, choroidal thickness and SRFT analyses because missing data at 12 months accounted for 10%, 9% and 8% of observations, respectively. Imputation by chained equations was used to generate multiple complete data sets (using the Stata -ice- command) and results were combined using Rubin’s rules.
To set our findings in context, we did a systematic review and meta-analysis, searching for other trials comparing eplerenone and placebo [identified using medical subject headings (MESH) by searching for the following: ‘central serous choroidal retinopathy’ AND (‘eplerenone’ OR ‘mineralocorticoid receptor antagonists’) AND ‘publication type = randomized controlled trial’]. We included trials that investigated eplerenone treatment in patients with chronic CSCR. Trials were excluded if they did not investigate eplerenone or included only patients with acute CSCR. We assessed the risk of bias29 and re-analysed data reported by the trials to obtain treatment effects for BCVA score and SRF thickness using the same metric (i.e. the difference between groups in the change in BCVA score and SRF thickness from baseline to the end of treatment). If it was not possible to calculate the SD of the mean change from baseline for a group, an average of the SDs from the other studies was used. The treatment effects were synthesised in a fixed-effects meta-analysis to estimate weighted MDs between eplerenone and placebo groups according to the inverse variance method.
Patient and public involvement
Before the trial commenced, a representative (Geraldine Hoad) from the Macular Society (URL: www.macularsociety.org; accessed 2 September 2020) provided input on the PIL to ensure that the information provided to patients was acceptable and clear.
A patient and public involvement (PPI) group comprising VICI trial participants was formed during the trial after all participants had been recruited. Participants from nine sites, selected by geographical proximity to London where the meetings would be held, were invited to join the PPI group by local research teams. Two PPI meetings were held: one during follow-up and one when the results were available. An agenda was prepared for the first meeting, with input from Caroline Barker and Francesca Lambert (PPI facilitators, University Hospital Southampton NHS Trust).
Topics covered at the first meeting were as follows:
-
How did you find out about the VICI trial?
-
Decision-making processes behind taking part in research.
-
Getting to your trial visits. (We aimed to find out what made attending regular follow-up visits possible and whether or not there were barriers to attending visits.)
-
Taking the study tablets regularly. (We aimed to find out what methods participants used to remind themselves to take the study drug daily.)
-
Introduction to the biobank.
-
Taking part in future events as a public representative.
A second PPI meeting was held after the analysis stage to develop a results leaflet that was understandable and acceptable to trial participants.
Chapter 3 Results: trial cohort
Screened patients
In total, 402 patients were screened for the trial (Figure 5). During the initial eligibility screening stage from patient medical records, 97 patients were found to be ineligible. Eighty-two initially eligible patients were not approached for consent. Among patients who were eligible and given a PIL, 44 declined to consent to trial-specific screening assessments and participation in the trial. A total of 164 initially eligible patients consented to trial-specific screening assessments, of whom 64 were ineligible. One patient who was eligible and initially gave consent withdrew shortly before randomisation. See Figure 5 for reasons why patients were excluded from the trial at each screening stage. There was variation in the ratio of screened to randomised participants per site because of the different methods of screening employed by sites. Therefore, the proportions of screened patients classified as eligible and consenting should be interpreted with caution.
FIGURE 5.
The VICI trial Consolidated Standards of Reporting Trials (CONSORT) flow diagram. CTIMP, clinical trial of investigational medicinal product. Reproduced with permission from Lotery et al. 30 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) license, which permits others to copy and redistribute the material in any medium or format, for non-commercial use, with no derivatives, provided the original work is properly cited. See: https://creativecommons.org/licenses/by-nc-nd/4.0/. The figure includes formatting changes to the original.
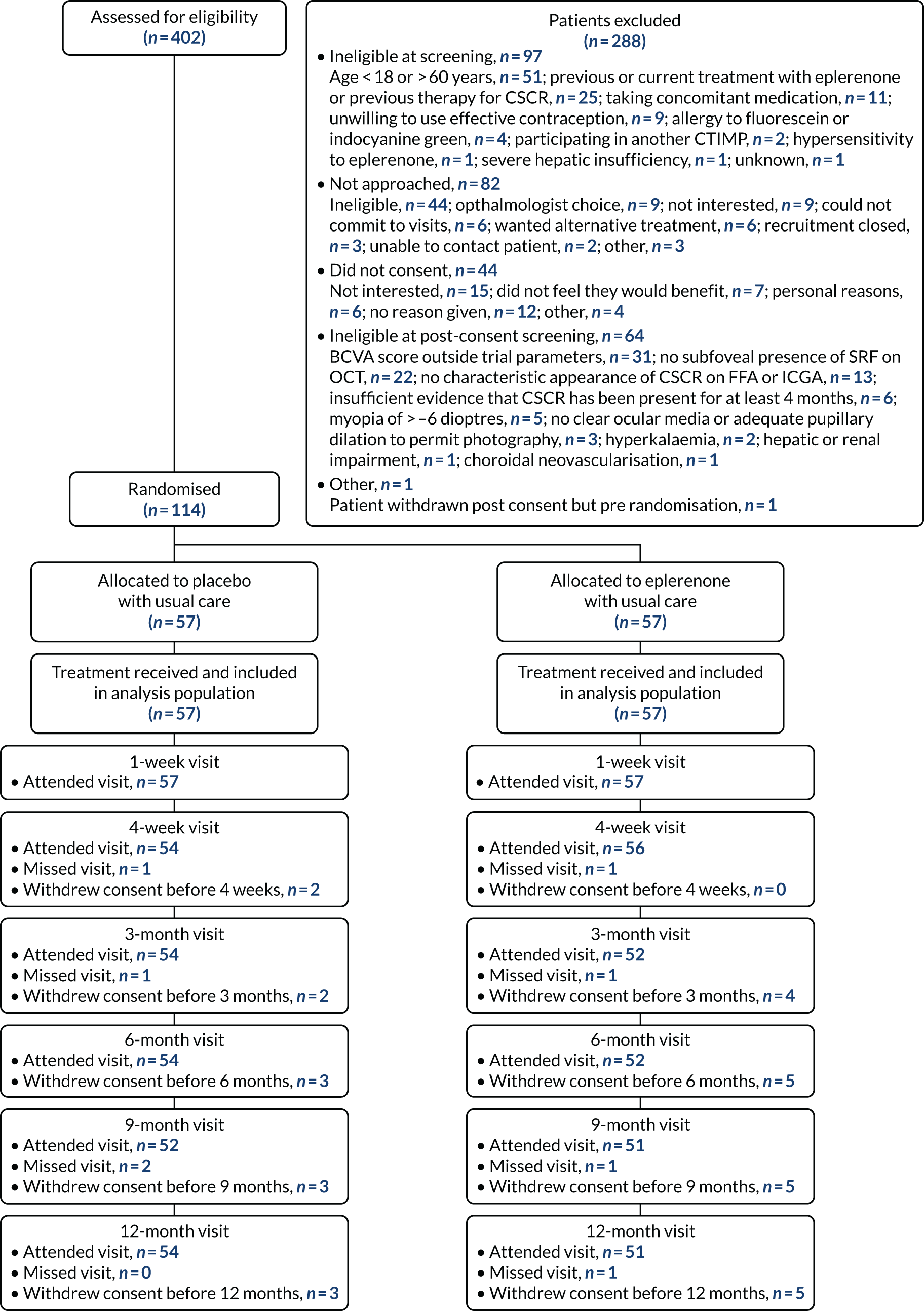
Randomised patients
The first site started to recruit (i.e. to screen patients) 14 December 2016. Recruitment closed 28 February 2018. A total of 114 participants were randomised between 11 January 2017 and 22 February 2018 (Figure 6). Follow-up (i.e. last patient, last visit) was completed on 28 February 2019.
FIGURE 6.
Predicted and actual recruitment.
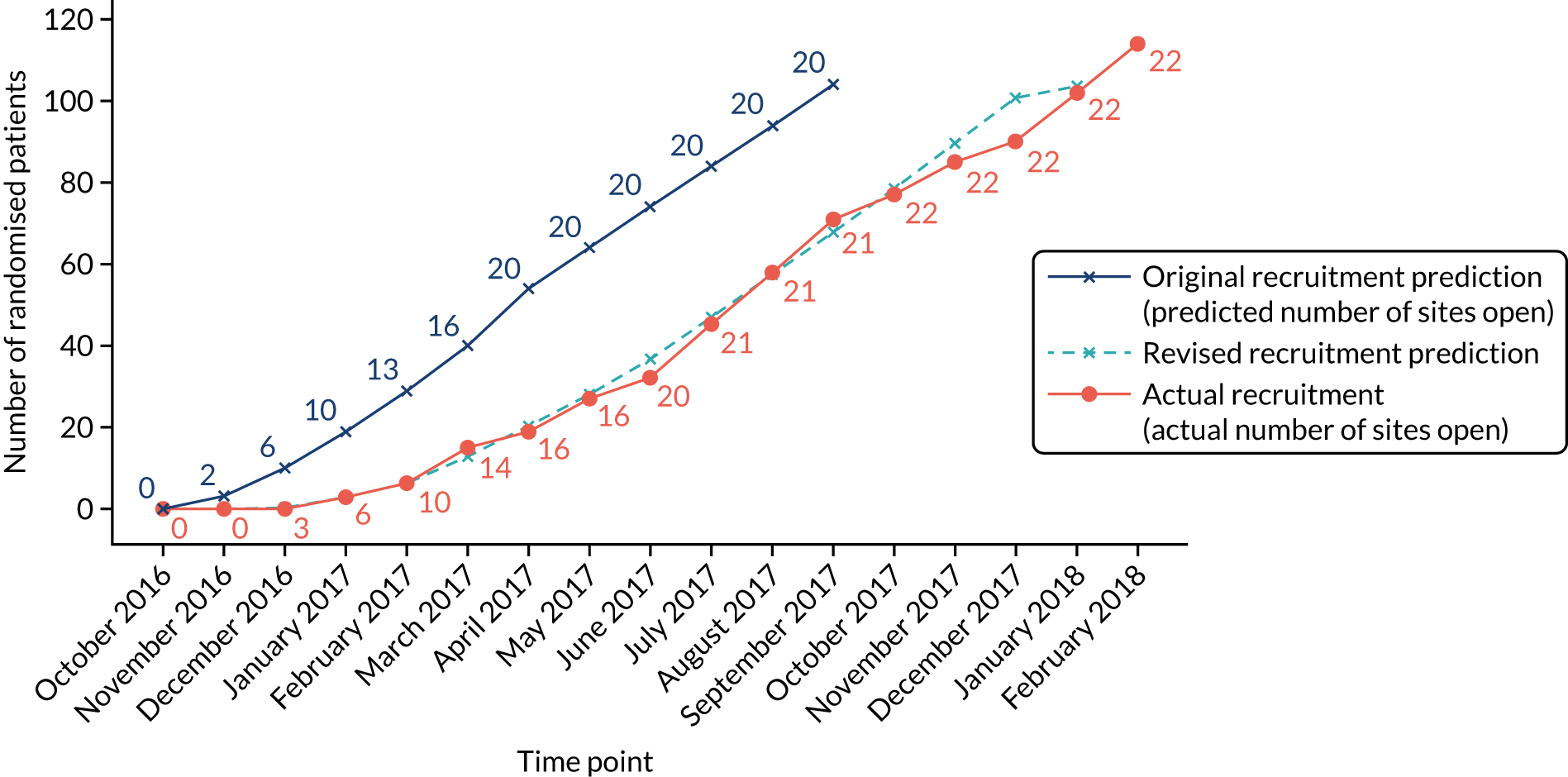
The trial over-recruited by 10 participants for ethical and logistical, but not methodological, reasons. At the point of reaching the recruitment target of 104 participants, participants who had already consented and were undergoing screening continued to enter the trial if eligible up to the point of recruitment closing.
Recruitment to the trial
It was estimated that sites would recruit one incident patient every 2 months (0.5 participants per month) plus one additional patient recruited from a pool of prevalent patients with CSCR already under hospital review. These estimates were based on investigators’ estimates of the average number of patients with CSCR who present at their clinics annually. All 22 sites contributed at least one randomised participant to the trial (see Appendix 4, Table 33).
There was a 3-month delay in trial set-up and recruitment started 3 months late. The initial projected recruitment rate was revised to take the delay into account. The actual recruitment rate was very close to the revised target recruitment rate (see Figure 6). One site contributed 17% of the total randomised participants (see Appendix 4, Table 33).
Recruitment was monitored frequently by the trial management group using monthly monitoring reports as well as weekly monitoring by the trial manager via the trial database. Sites that were identified as having difficulties with recruitment were discussed at trial management group meetings and contact would be made with the local research team to identify and resolve their barriers to recruitment where possible.
Representativeness of the participants
Baseline characteristics of patients are presented in Table 3 (separately for those who were ineligible at initial screening, eligible at initial screening but who did not consent, consented and underwent trial-specific screening assessments but who were not randomised because they were found to be ineligible, and randomised). On average, patients who were ineligible at initial screening were older than all other patients, who were of similar age. A lower proportion of men were ineligible at screening (65%) and a higher proportion were eligible but did not consent (85%) compared with those who were consented (75%).
| Characteristic | Ineligible at initial screening (n = 97) | Eligible, not consented (n = 126) | Consented, ineligible at full screen (n = 65) | Randomised (n = 114) |
|---|---|---|---|---|
| Age (years) at screening,a mean (SD) | 59.8 (13.0) | 46.8 (8.9) | 48.1 (7.7) | 48.6 (7.6) |
| Male, n/N (%) | 56/86 (65.1) | 99/116 (85.3) | 47/63 (74.6) | 85/114 (74.6) |
| Ethnicity, n/N (%) | ||||
| White | 2/2 (100.0) | - | 21/22 (95.5) | 99/114 (86.8) |
| Asian | 0/2 (0.0) | - | 0/22 (0.0) | 13/114 (11.4) |
| Mixed | 0/2 (0.0) | - | 0/22 (0.0) | 1/114 (0.9) |
| Other | 0/2 (0.0) | - | 1/22 (4.5) | 1/114 (0.9) |
Participant withdrawals
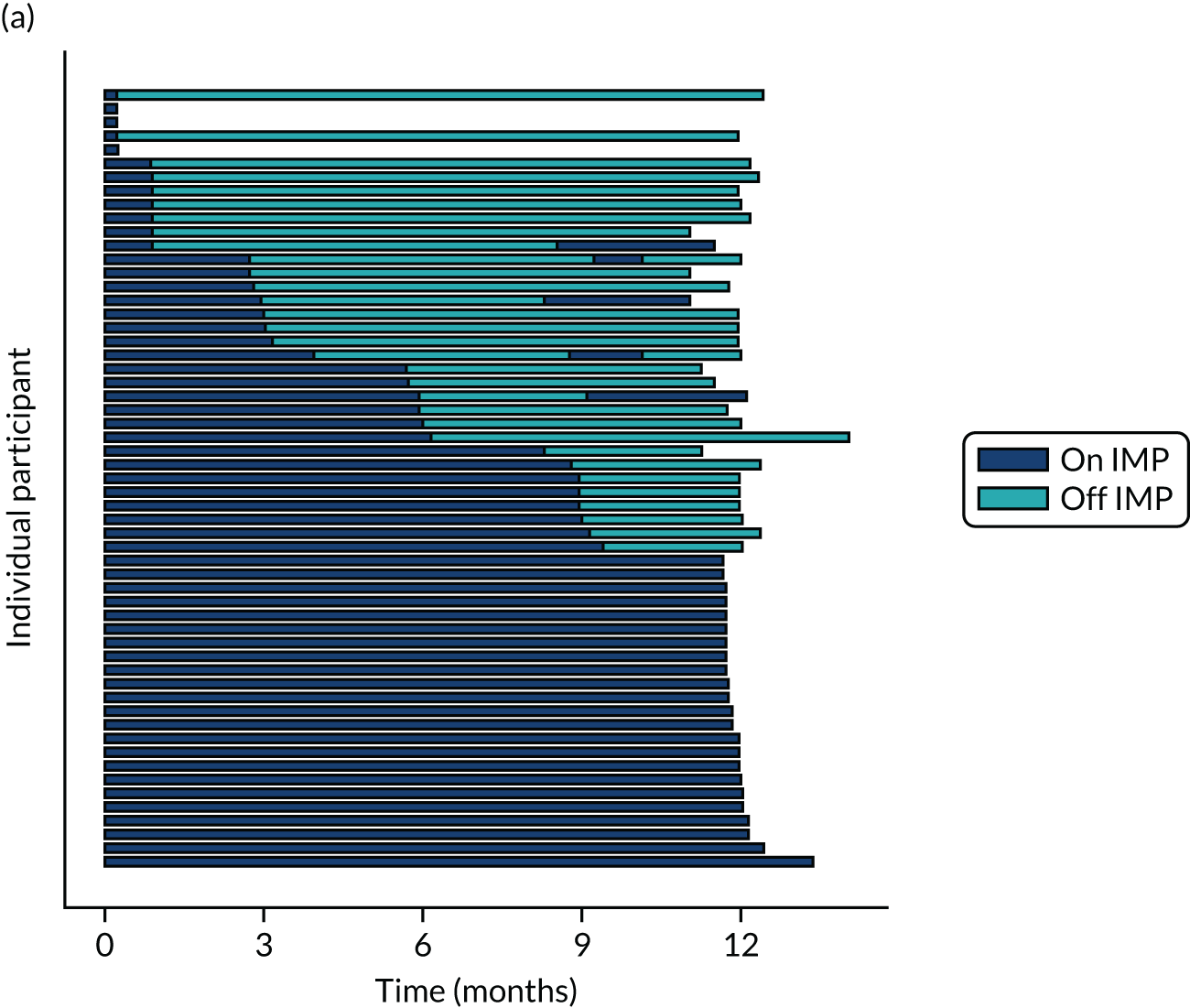
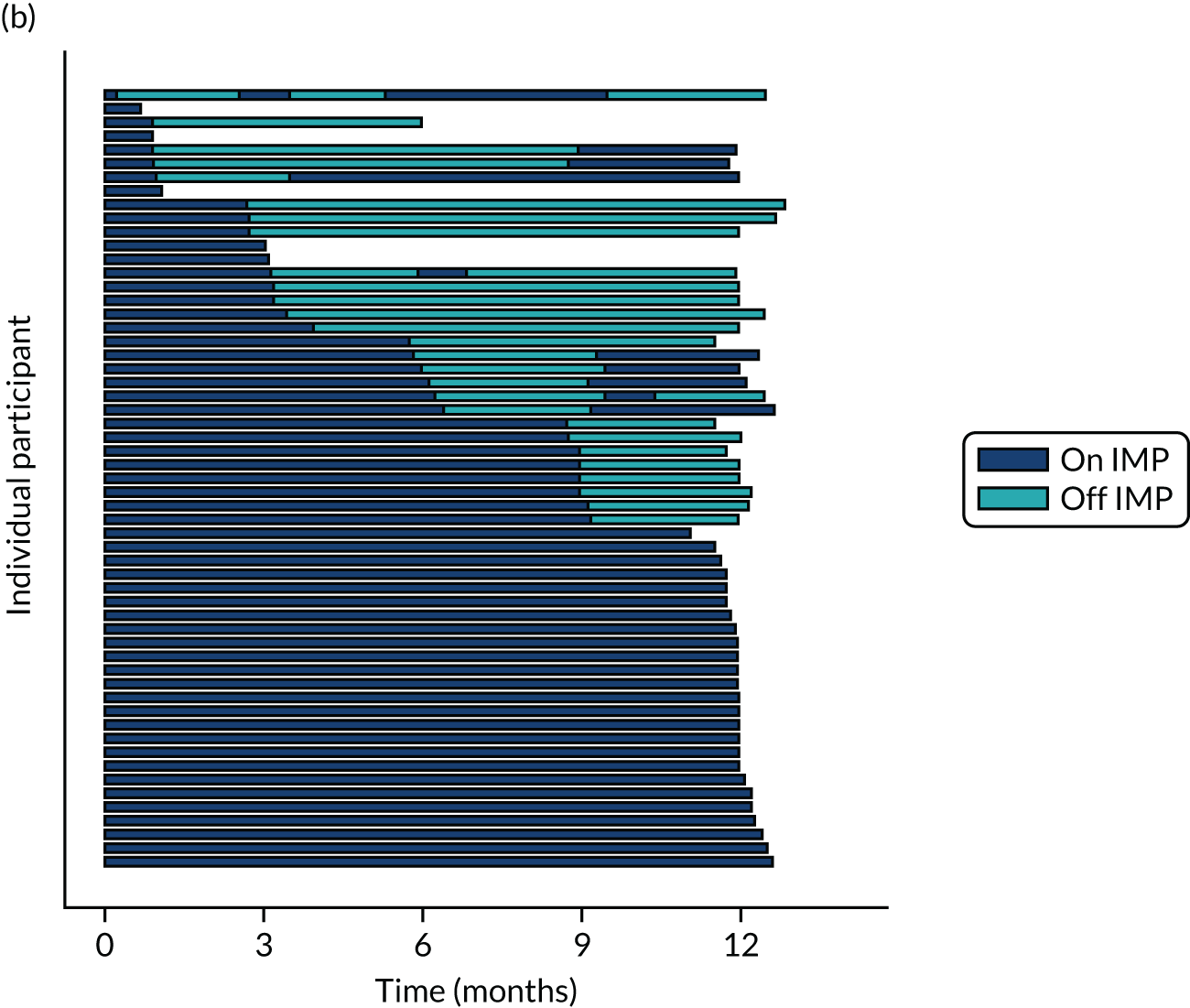
Twenty-six participants permanently ceased taking the study drug during the 12-month follow-up period; 18 out of 26 participants agreed to continue with follow-up to 12 months and 8 out of 26 participants withdrew from follow-up before 12 months. The reasons for stopping the study drug and subsequent withdrawal from follow-up, if applicable, are detailed in Table 4. Time to stopping the study drug is shown in Appendix 4, Figure 21. Follow-up time classified as ‘on’ or ‘off’ IMP for each participant is shown in Figure 7.
| Reason for withdrawal from treatment | Randomised to placebo (n = 57), n/N (%) | Randomised to eplerenone (n = 57), n/N (%) | Overall (n = 114), n/N (%) |
|---|---|---|---|
| Any | 12/57 (21) | 14/57 (25) | 26/114 (23) |
| Clinician withdrawal | 9/12 (75) | 8/14 (57) | 17/26 (65) |
| Hyperkalaemia | 8/9 (89) | 8/8 (100) | 16/17 (94) |
| SAE | 0/9 (0) | 0/8 (0) | 0/17 (0) |
| Othera | 1/9 (11) | 0/8 (0) | 1/17 (6) |
| Patient withdrawal | 2/12 (17) | 5/14 (36) | 7/26 (27) |
| Wants standard treatment | 1/2 (50) | 0/5 (0) | 1/7 (14) |
| Otherb | 1/2 (50) | 5/5 (100) | 6/7 (86) |
| Withdrawn from follow-up | 3/12 (25) | 5/14 (36) | 8/26 (31) |
FIGURE 7.
Time participants were on and off the IMP during the trial. (a) Placebo; and (b) eplerenone. Reproduced with permission from Lotery et al. 30 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) license, which permits others to copy and redistribute the material in any medium or format, for non-commercial use, with no derivatives, provided the original work is properly cited. See: https://creativecommons.org/licenses/by-nc-nd/4.0/. The figure includes formatting changes to the original.
Protocol deviations
Few protocol deviations occurred during the trial (Table 5). All participants were dispensed the correct drug and followed the correct dosage regimen at all time points. Two participants, one in the eplerenone group and one in the placebo group, were informed to continue taking the study drug despite having a serum potassium level > 5.0 mmol/l. In both cases the deviation was detected by the trial manager through remote central monitoring and the participants were informed to stop taking the study drug 3 weeks after the blood test result. The most common protocol deviation was visits attended outside of the specified visit window. This deviation occurred equally between groups. Distributions of follow-up visits around the visit windows are shown in Appendix 4, Figure 22.
| Protocol deviation | Randomised to placebo (n = 57) | Randomised to eplerenone (n = 57) | Overall (n = 114) |
|---|---|---|---|
| Any, n/N (%) | 34/57 (60) | 33/57 (58) | 67/114 (59) |
| Patient did not receive allocated drug, n/N (%) | 0/57 (0) | 0/57 (0) | 0/114 (0) |
| Patient ineligible but randomised, n/N (%) | 0/57 (0) | 0/57 (0) | 0/114 (0) |
| Missed visit, events/patients (%) | 4/3 (5) | 5/3 (5) | 9/6 (5) |
| Visit attended but outside visit window,a events/patients (%) | 53/30 (53) | 62/31 (54) | 115/61 (54) |
| Serum potassium level > 5.0 mmol/l but patient informed to continue treatment,b n/N (%) | 1/8 (13) | 1/8 (13) | 2/16 (13) |
| Incorrect dosage regimen followed, n/N (%) | 0/57 (0) | 0/57 (0) | 0/114 (0) |
| Patient prescribed more medication than required at any study visit, n/N (%) | 2/57 (4) | 2/57 (4) | 4/114 (4) |
| Disease resolved but patient informed to continue treatment,c n/N (%) | 0/57 (0) | 1/57 (2) | 1/114 (1) |
Emergency unmasking
No participant was unmasked before the end of the trial. Code-breaking envelopes (see Chapter 2, Emergency unmasking) were inspected by the trial manager on receipt at the BTC-CTEU. No evidence of tampering was detected. Out of 196 envelopes expected to be returned, three were reported missing from two sites.
One participant’s allocation was unmasked after the final database lock (which defined the end of the trial)21 at the request of the local principal investigator. The participant still had chronic SRF and wanted to request eplerenone treatment if he had not already been taking it in the trial.
Participants and optometrists’ beliefs about allocations
At the end of the 12-month follow-up visit, each participant and the optometrist who measured BCVA were asked whether or not they knew which group the participant had been allocated to (Table 6). Ten participants claimed to know their allocation: five in the placebo group and five in the eplerenone group. Two and four participants guessed correctly in the placebo and eplerenone groups, respectively. The reason for the belief about their allocation was not collected. No optometrists claimed to have guessed the participants’ allocation.
| Variable | Randomised to eplerenone (n = 57), n/N (%) | Randomised to placebo (n = 57), n/N (%) |
|---|---|---|
| Patient guessed allocation | 5/54 (9.3) | 5/51 (9.8) |
| Patient guessed correctly | 2/5 (40.0) | 4/5 (80.0) |
| Optometrist guessed allocation | 0/54 (0.0) | 0/51 (0.0) |
Numbers analysed
The analysis population consisted of all 114 randomised participants; no participant who withdrew did not consent for their data to be used. The number of participants included in the analysis of each outcome is presented in Table 7.
| Outcome | Number included in analysis, n (%) |
|---|---|
| BCVA score at 12 months (primary) | 111 (97) |
| AEs | 114 (100) |
| Complete resolution of SRF | 114 (100) |
| Complete or partial resolution of SRF | 114 (100) |
| Any CSCR | 114 (100) |
| New CSCR | 114 (100) |
| Change in SRFT at 12 months | 114 (100)a |
| Choroidal thickness at 12 months | 114 (100)b |
| NEI-VFQ-25 at 12 months | 114 (100)c |
| CSRT at 12 months | 111 (97) |
| LLA at 12 months | 109 (96) |
| Reduced choroidal permeability at 12 months | 103 (90) |
| Macular atrophy of RPE at 12 months | 102 (89) |
| Area change in macular RPE hypoautofluorescence at 12 months | 8 (7) |
| Recurrence of SRF | 42 (37) |
Adherence to the investigational medicinal product regimen
All participants in the eplerenone and placebo groups adhered to the IMP regimen between baseline and week 1 according to the definition of taking > 70% of the expected number of capsules in that time period, as described in Chapter 2, Adherence monitoring. This is important as decisions to increase the dose from 25 mg to 50 mg were made on the assumption that the participant had been adherent and potassium levels remained at ≤ 5.0 mmol/l. At subsequent time periods, at least 80% of participants were adherent (according to the same definition) and adherence was well balanced between the groups (Table 8). Out of 497 bottles dispensed to participants in the placebo group and 559 bottles dispensed to participants in the eplerenone group, 46 and 57 were not returned, respectively (see Appendix 4, Table 34). It was assumed that these bottles were discarded empty (i.e. the participant had taken all the pills and discarded the bottle), so the capsule counts were recorded as zero.
| Time point | Randomised to placebo (n = 57), n/N (%) | Randomised to eplerenone (n = 57), n/N (%) |
|---|---|---|
| Baseline | 57/57 (100) | 57/57 (100) |
| 1 week | 52/52 (100) | 53/56 (95) |
| 4 weeks | 42/45 (93) | 43/47 (91) |
| 3 months | 36/37 (97) | 37/39 (95) |
| 6 months | 31/31 (100) | 29/34 (85) |
| 9 months | 22/24 (92) | 22/26 (85) |
| Restarting | 5/5 (100) | 14/14 (100) |
| Restarting week 1 | 5/5 (100) | 11/13 (85) |
| Restarting week 4 | 3/3 (100) | 11/11 (100) |
Additional treatments administered as usual care
Clinicians were permitted to administer usual care for CSCR during the 12-month follow-up period at their discretion (Table 9). No participants received thermal laser therapy. One participant in the placebo group received subthreshold laser therapy in their study eye while prescribed the study drug. In the placebo group six patients received nine PDT treatments (i.e. some patients received PDT more than once) in their study eye; two of these six patients were prescribed the study drug at the time of administration. In the eplerenone group three participants each received one PDT treatment; one of these participants received PDT in their study eye and was prescribed the study drug at the time of administration.
| Additional therapy | Randomised to placebo (n = 57), treatments/patients (%) | Randomised to eplerenone (n = 57), treatments/patients (%) | ||||
|---|---|---|---|---|---|---|
| Therapy received | Received in study eye | Received while on study IMP | Therapy received | Received in study eye | Received while on study IMP | |
| PDT | 9/6 (10.5) | 9/6 (10.5) | 3/2 (3.5) | 3/3 (5.3) | 1/1 (1.8) | 1/1 (1.8) |
| Thermal laser therapy | 0/0 (0.0) | 0/0 (0.0) | 0/0 (0.0) | 0/0 (0.0) | 0/0 (0.0) | 0/0 (0.0) |
| Other therapya | 1/1 (1.8) | 1/1 (1.8) | 1/1 (1.8) | 0/0 (0.0) | 0/0 (0.0) | 0/0 (0.0) |
Participant characteristics and data at baseline
The mean participant age at baseline was 48.7 ± 7.6 years (placebo, 49.9 ± 7.9 years; eplerenone, 47.4 ± 7.1 years) and approximately three-quarters were male. The median duration of CSCR before randomisation was 9 months overall and was well balanced between groups [placebo, 9 months (IQR 6–18 months); eplerenone, 8 months (IQR 6–22 months)]. See Table 10 for baseline demography, blood test results, medical history and ocular examination data by group and overall, and Table 11 for baseline outcome data by group and overall.
| Characteristic | Randomised to placebo (n = 57) | Randomised to eplerenone (n = 57) | Overall (n = 114) |
|---|---|---|---|
| Age at randomisation (years), mean (SD) | 49.9 (7.9) | 47.4 (7.1) | 48.7 (7.6) |
| Male, n/N (%) | 43/57 (75) | 42/57 (74) | 85/114 (75) |
| Ethnicity, n/N (%) | |||
| White | 53/57 (93) | 46/57 (81) | 99/114 (87) |
| Asian | 4/57 (7) | 9/57 (16) | 13/114 (11) |
| Mixed | 0/57 (0) | 1/57 (2) | 1/114 (1) |
| Other | 0/57 (0) | 1/57 (2) | 1/114 (1) |
| Systolic blood pressure (mmHg),a median (IQR) | 132 (125.0–146.0) | 129 (121.0–141.0) | 130 (122.0–144.0) |
| Diastolic blood pressure (mmHg),a median (IQR) | 80 (75.0–88.0) | 80 (72.5–88.5) | 80 (73.0–88.0) |
| Heart rate (b.p.m.),a median (IQR) | 68 (60.0–76.0) | 73 (66.0–80.0) | 72 (63.0–78.0) |
| TSH/thyrotropin (mlU/l),a median (IQR) | 2 (1.1–2.2) | 2 (1.2–2.2) | 2 (1.1–2.2) |
| Thyroxine (pmol/l),b median (IQR) | 14 (13.0–17.0) | 14 (13.0–16.0) | 14 (13.0–16.0) |
| Triiodothyronine (nmol/l),c median (IQR) | 5 (4.4–4.8) | 5 (4.3–4.8) | 5 (4.3–4.8) |
| HbA1c (mmol/mol),d median (IQR) | 35 (33.5–37.0) | 36 (33.0–39.0) | 36 (33.0–39.0) |
| Hct (l/l),e mean (SD) | 0.4 (0.0) | 0.4 (0.0) | 0.4 (0.0) |
| Platelets (× 109/l),f median (IQR) | 250 (222.5–287.5) | 259 (221.0–290.0) | 254 (222.0–290.0) |
| WBC (× 109/l), median (IQR) | 7 (5.0–8.0) | 7 (5.6–7.7) | 7 (5.0–7.9) |
| Serum creatinine (µmol/l), mean (SD) | 78 (14.9) | 79 (13.2) | 78 (14.0) |
| Urea (mmol/l), mean (SD) | 5 (1.2) | 5 (1.2) | 5 (1.2) |
| Potassium (mmol/l), mean (SD) | 4 (0.3) | 4 (0.4) | 4 (0.3) |
| Sodium (mmol/l), median (IQR) | 140 (139.0–141.0) | 141 (139.0–142.0) | 141 (139.0–142.0) |
| Chloride (mmol/l),g mean (SD) | 102 (2.7) | 102 (2.6) | 102 (2.6) |
| Bicarbonate (mmol/l),h mean (SD) | 27 (3.3) | 25 (2.2) | 26 (2.9) |
| eGFR (ml/minute/1.73 m2), median (IQR) | 82 (60.0–90.0) | 84 (71.0–90.0) | 84 (66.0–90.0) |
| Bilirubin (µmol/l),f median (IQR) | 8 (7.0–12.0) | 8 (6.0–11.0) | 8 (6.0–11.0) |
| ALT (units/l),i median (IQR) | 22 (19.0–29.0) | 28 (21.0–39.0) | 24 (20.0–35.0) |
| Albumin (g/l),f median (IQR) | 45 (41.0–47.0) | 45 (41.0–47.0) | 45 (41.0–47.0) |
| Protein (g/l),j median (IQR) | 73 (70.0–76.0) | 72 (69.0–76.0) | 72 (70.0–76.0) |
| Smoking, n/N (%) | |||
| Current | 10/57 (18) | 12/57 (21) | 22/114 (19) |
| Ex | 16/57 (28) | 25/57 (44) | 41/114 (36) |
| Never | 31/57 (54) | 20/57 (35) | 51/114 (45) |
| Heart failure, n/N (%) | 0/57 (0) | 0/57 (0) | 0/114 (0) |
| Myocardial infarction, n/N (%) | 1/57 (2) | 0/57 (0) | 1/114 (1) |
| History of angina, n/N (%) | 0/57 (0) | 0/57 (0) | 0/114 (0) |
| CCS class: no angina, n/N (%) | 57/57 (100) | 57/57 (100) | 114/114 (100) |
| NYHA class, n/N (%) | |||
| 0 | 56/57 (98) | 57/57 (100) | 113/114 (99) |
| I | 1/57 (2) | 0/57 (0) | 1/114 (1) |
| Transient ischaemic attack, n/N (%) | 0/57 (0) | 0/57 (0) | 0/114 (0) |
| Stroke, n/N (%) | 1/57 (2) | 0/57 (0) | 1/114 (1) |
| DVT, n/N (%) | 0/57 (0) | 1/57 (2) | 1/114 (1) |
| PE, n/N (%) | 0/57 (0) | 1/57 (2) | 1/114 (1) |
| Claudication, n/N (%) | 0/57 (0) | 0/57 (0) | 0/114 (0) |
| Diabetes, n/N (%) | |||
| None | 55/57 (96) | 54/57 (95) | 109/114 (96) |
| Oral | 1/57 (2) | 2/57 (4) | 3/114 (3) |
| Non-insulin injections | 1/57 (2) | 1/57 (2) | 2/114 (2) |
| BCVA score, n/N (%) | |||
| Low (54–67 letters) | 7/57 (12) | 7/57 (12) | 14/114 (12) |
| High (68–85 letters) | 50/57 (88) | 50/57 (88) | 100/114 (88) |
| CSCR duration (months), median (IQR) | 9 (6.0–18.0) | 8 (6.0–22.0) | 9 (6.0–19.0) |
| Family history of CSCR, n/N (%) | 1/57 (2) | 0/57 (0) | 1/114 (1) |
| Pupils abnormal, n/N (%) | 0/57 (0) | 0/57 (0) | 0/114 (0) |
| Cornea abnormal,k n/N (%) | 3/57 (5) | 0/57 (0) | 3/114 (3) |
| Anterior chamber cells present, n/N (%) | 0/57 (0) | 0/57 (0) | 0/114 (0) |
| Anterior chamber flare present, n/N (%) | 0/57 (0) | 1/57 (2) | 1/114 (1) |
| IOP measurement (mmHg), median (IQR) | 15 (14.0–18.0) | 15 (13.0–17.0) | 15 (13.0–17.0) |
| Lens status, n/N (%) | |||
| Phakic | 57/57 (100) | 55/57 (96) | 112/114 (98) |
| Pseudophakic | 0/57 (0) | 2/57 (4) | 2/114 (2) |
| NUC grade,l n/N (%) | |||
| NUC-0 | 47/57 (82) | 51/55 (93) | 98/112 (88) |
| NUC-1 | 9/57 (16) | 4/55 (7) | 13/112 (12) |
| NUC-2 | 1/57 (2) | 0/55 (0) | 1/112 (1) |
| COR grade,l n/N (%) | |||
| COR-0 | 56/57 (98) | 55/55 (100) | 111/112 (99) |
| COR-1 | 1/57 (2) | 0/55 (0) | 1/112 (1) |
| CEN,e n/N (%) | 2/57 (4) | 2/55 (4) | 4/112 (4) |
| PSC grade: PSC-0,l n/N (%) | 57/57 (100) | 55/55 (100) | 112/112 (100) |
| Macula abnormal, n/N (%) | 55/57 (96) | 55/57 (96) | 110/114 (96) |
| Peripheries abnormal,m n/N (%) | 1/57 (2) | 5/57 (9) | 6/114 (5) |
| Disc abnormal, n/N (%) | 0/57 (0) | 0/57 (0) | 0/114 (0) |
| Cup disc ratio,a median (IQR) | 0.3 (0.2–0.3) | 0.3 (0.2–0.3) | 0.3 (0.2–0.3) |
| Cataract surgery, n/N (%) | 0/57 (0) | 2/57 (4) | 2/114 (2) |
| Outcome | Randomised to placebo (n = 57) | Randomised to eplerenone (n = 57) | Overall (n = 114) |
|---|---|---|---|
| BCVA score, median (IQR) | 78 (73.0–82.0) | 77 (73.0–80.0) | 78 (73.0–81.0) |
| LLA score,a median (IQR) | 64 (57.0–67.0) | 57 (50.0–64.0) | 60 (52.5–65.0) |
| Choroidal thickness (µm),b median (IQR) | 461 (381.5–534.5) | 447 (398.0–509.0) | 447 (389.0–521.0) |
| SRFT (µm), median (IQR) | 119 (88.0–178.0) | 147 (93.0–196.0) | 134 (90.0–194.0) |
| CSRT (µm), median (IQR) | 322 (280.0–394.0) | 360 (290.0–406.0) | 349 (280.0–401.0) |
| Macular atrophy of RPE, n/N (%) | 3/55 (5) | 2/56 (4) | 5/111 (5) |
Biobank sample collection
A total of 109 participants donated DNA, plasma and serum samples to the biobank at the University of Southampton for use in future ethically approved studies.
Chapter 4 Results: visual function outcomes
Best corrected visual acuity
Median BCVA score increased by about four letters in both groups during follow-up (Figure 8). Modelled mean BCVA score at 12 months was 80.4 letters (SD 4.6 letters) in the eplerenone group and 79.5 letters (SD 4.5 letters) in the placebo group. Mean BCVA score at 12 months did not differ between the groups (eplerenone minus placebo: 1.73 letters, 95% CI –1.12 to 4.57 letters; p = 0.24; Table 12 and Figure 8). In the exploratory analysis, adherence to treatment did not change the effect of eplerenone on BCVA score, irrespective of whether or not pill counts were imputed for lost IMP bottles (Tables 13 and 14). In the post hoc analysis, which adjusted for photodynamic therapy administered during follow-up, the effect of eplerenone on BCVA score was unchanged (see Appendix 4, Table 35).
FIGURE 8.
Primary outcome: BCVA score over time. Reproduced with permission from Lotery et al. 30 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) license, which permits others to copy and redistribute the material in any medium or format, for non-commercial use, with no derivatives, provided the original work is properly cited. See: https://creativecommons.org/licenses/by-nc-nd/4.0/. The figure includes formatting changes to the original.
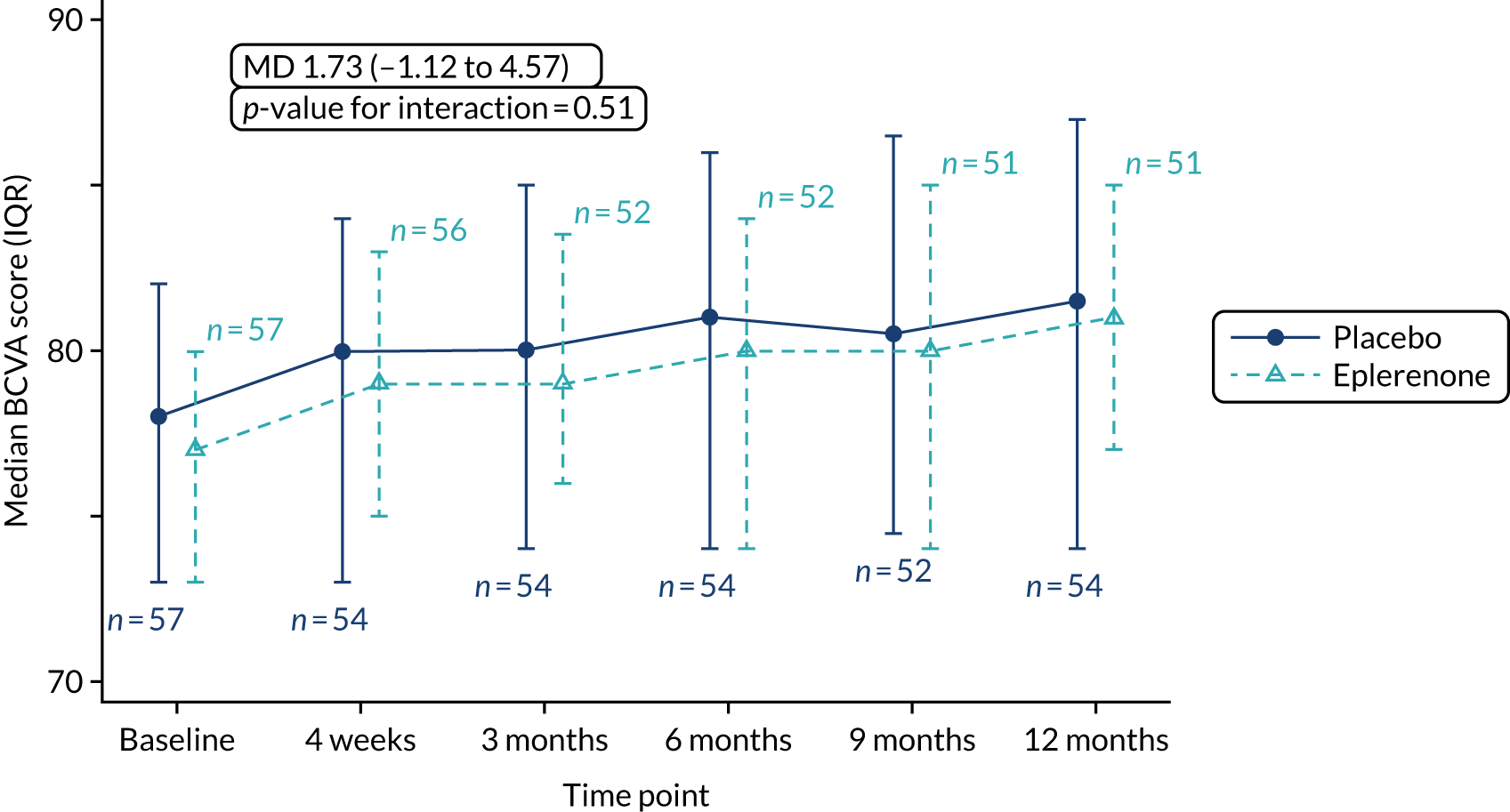
| Time point | Randomised to placebo (n = 57), median (IQR) | Randomised to eplerenone (n = 57), median (IQR) | Effect, MD (95% CI) | p-value |
|---|---|---|---|---|
| Baseline | 78 (73.0–82.0) | 77 (73.0–80.0) | ||
| 4 weeksa | 80 (73.0–84.0) | 79 (75.0–83.0) | ||
| 3 monthsb | 80 (74.0–85.0) | 79 (76.0–83.5) | ||
| 6 monthsb | 81 (74.0–86.0) | 80 (74.0–84.0) | ||
| 9 monthsc | 81 (74.5–86.5) | 80 (74.0–85.0) | ||
| 12 monthsd | 82 (74.0–87.0) | 81 (77.0–85.0) | 1.73 (–1.12 to 4.57) | 0.236 |
| Test for treatment × time interaction | 0.511 |
| Treatment status | Randomised to placebo (n = 57), median (IQR) | Randomised to eplerenone (n = 57), median (IQR) | MD (95% CI) | p-value for interaction |
|---|---|---|---|---|
| On treatment and adhered | 79 (69.0–87.0) | 80 (74.0–83.0) | 1.28 (–2.24 to 4.80) | 0.816 |
| On treatment and did not adhere | 84 (70.0–90.0) | 81 (76.5–86.0) | –0.30 (–10.10 to 9.50) | |
| Not on treatment (disease resolution) | 85 (78.0–87.0) | 85 (81.0–88.5) | 2.20 (–2.12 to 6.53) | |
| Not on treatment (other reason) | 77 (77.0–82.0) | 81 (75.0–84.0) | 4.81 (–2.51 to 12.13) |
| Treatment status | Primary analysis | Sensitivity analysis | ||
|---|---|---|---|---|
| MD (95% CI) | p-value for interaction | MD (95% CI) | p-value for interaction | |
| On treatment and adhered | 1.28 (–2.24 to 4.80) | 0.816 | 1.12 (–2.51 to 4.75) | 0.795 |
| On treatment and did not adhere | –0.30 (–10.10 to 9.50) | –0.67 (–9.60 to 8.26) | ||
| Not on treatment (disease resolution) | 2.20 (–2.12 to 6.53) | 2.42 (–1.92 to 6.75) | ||
| Not on treatment (other reason) | 4.81 (–2.51 to 12.13) | 4.96 (–2.36 to 12.28) | ||
Exploratory analyses of the effects of age and confluent/granular hyperautofluorescence on BCVA score in the trial cohort are shown in Appendix 4, Figure 23. Neither factor influenced BCVA score (age: –0.16 letters per year of age, 95% CI –0.35 to 0.04 letters per year of age; presence of confluent/granular hyperautofluorescence: –1.75 letters, 95% CI –4.78 to 1.28 letters).
Low-luminance visual acuity
Median LLA score appeared to be lower in the eplerenone group at baseline but the IQRs for the groups clearly overlapped (see Appendix 4, Table 36). Therefore, median LLA score appeared to improve more in the eplerenone group (9 letters) than in the placebo group (1 letter) (Figure 9). The modelled LLA score mean at 12 months in the eplerenone group was 63.9 letters (SD 4.8 letters) and in the placebo group was 65.3 letters (SD 3.7 letters). Mean LLA score at 12 months did not differ between the groups (eplerenone minus placebo: 0.69 letters, 95% CI –3.79 to 5.02 letters, p = 0.79).
FIGURE 9.
Low-luminance visual acuity over time. Reproduced with permission from Lotery et al. 30 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) license, which permits others to copy and redistribute the material in any medium or format, for non-commercial use, with no derivatives, provided the original work is properly cited. See: https://creativecommons.org/licenses/by-nc-nd/4.0/. The figure includes formatting changes to the original.
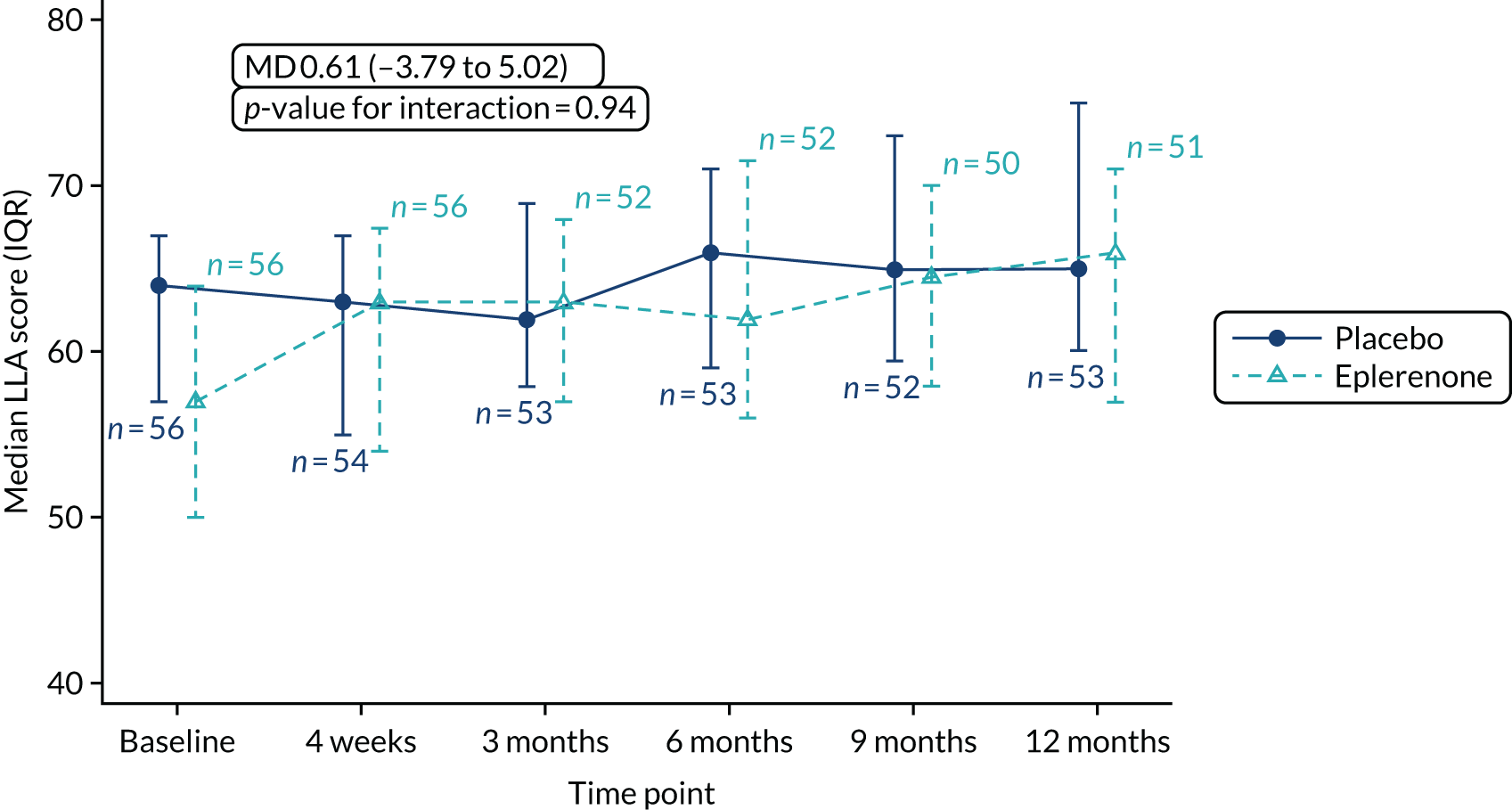
Patient-reported visual function using the NEI-VFQ-25 questionnaire
Higher NEI-VFQ-25 scores represent better visual function. Median NEI-VFQ-25 scores and IQRs at baseline and 12 months for eplerenone and placebo groups, overall and for each questionnaire domain, are shown in Appendix 4, Table 37. There were no differences between groups [modelled means were 86.5 letters (SD 5.3 letters) for the eplerenone group and 89.1 letters (SD 4.4 letters) for the placebo group]. Mean NEI-VFQ-25 score at 12 months, adjusted for baseline score, did not differ between the groups (eplerenone minus placebo:–2.39 letters, 95% CI –5.45 to 0.68 letters; p = 0.13) (Figure 10).
FIGURE 10.
Effect size for patient-reported visual function.
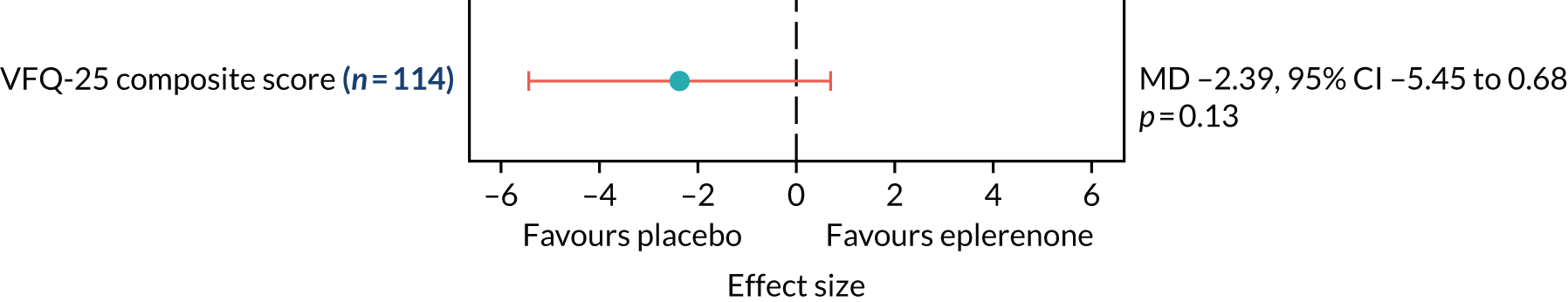
Resolution and recurrence of central serous chorioretinopathy
Classification of CSCR in study eyes as having resolved completely, resolved partially or not having responded (based on resolution of SRF) to IMP at scheduled time points is shown in Table 15, by eplerenone or placebo group. CSCR could resolve completely at one time point but then recur. Figure 11 shows follow-up time by group for each participant, classified as not completely resolved or completely resolved. Table 16 shows the number of participants’ fellow eyes with any, and new, CSCR.
| Visit | Classification | Randomised to placebo (N = 57), n/N (%) | Randomised to eplerenone (N = 57), n/N (%) |
|---|---|---|---|
| 4 weeks | Complete resolution | 2/54 (3.7) | 4/56 (7.1) |
| Partial resolution | 4/54 (7.4) | 8/56 (14.3) | |
| Non-responder | 48/54 (88.9) | 44/56 (78.6) | |
| 3 months | Complete resolution | 6/54 (11.1) | 5/52 (9.6) |
| Partial resolution | 12/54 (22.2) | 15/52 (28.8) | |
| Non-responder | 36/54 (66.7) | 32/52 (61.5) | |
| 6 months | Complete resolution | 11/54 (20.4) | 8/52 (15.4) |
| Partial resolution | 10/54 (18.5) | 16/52 (30.8) | |
| Non-responder | 33/54 (61.1) | 28/52 (53.8) | |
| 9 months | Complete resolution | 13/52 (25.0) | 9/51 (17.6) |
| Partial resolution | 10/52 (19.2) | 14/51 (27.5) | |
| Non-responder | 29/52 (55.8) | 28/51 (54.9) | |
| 12 months | Complete resolution | 16/54 (29.6) | 8/51 (15.7) |
| Partial resolution | 13/54 (24.1) | 14/51 (27.5) | |
| Non-responder | 25/54 (46.3) | 29/51 (56.9) |
FIGURE 11.
Time participants did or did not have complete resolution of SRF during the trial. Reproduced with permission from Lotery et al. 30 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) license, which permits others to copy and redistribute the material in any medium or format, for non-commercial use, with no derivatives, provided the original work is properly cited. See: https://creativecommons.org/licenses/by-nc-nd/4.0/. The figure includes formatting changes to the original.
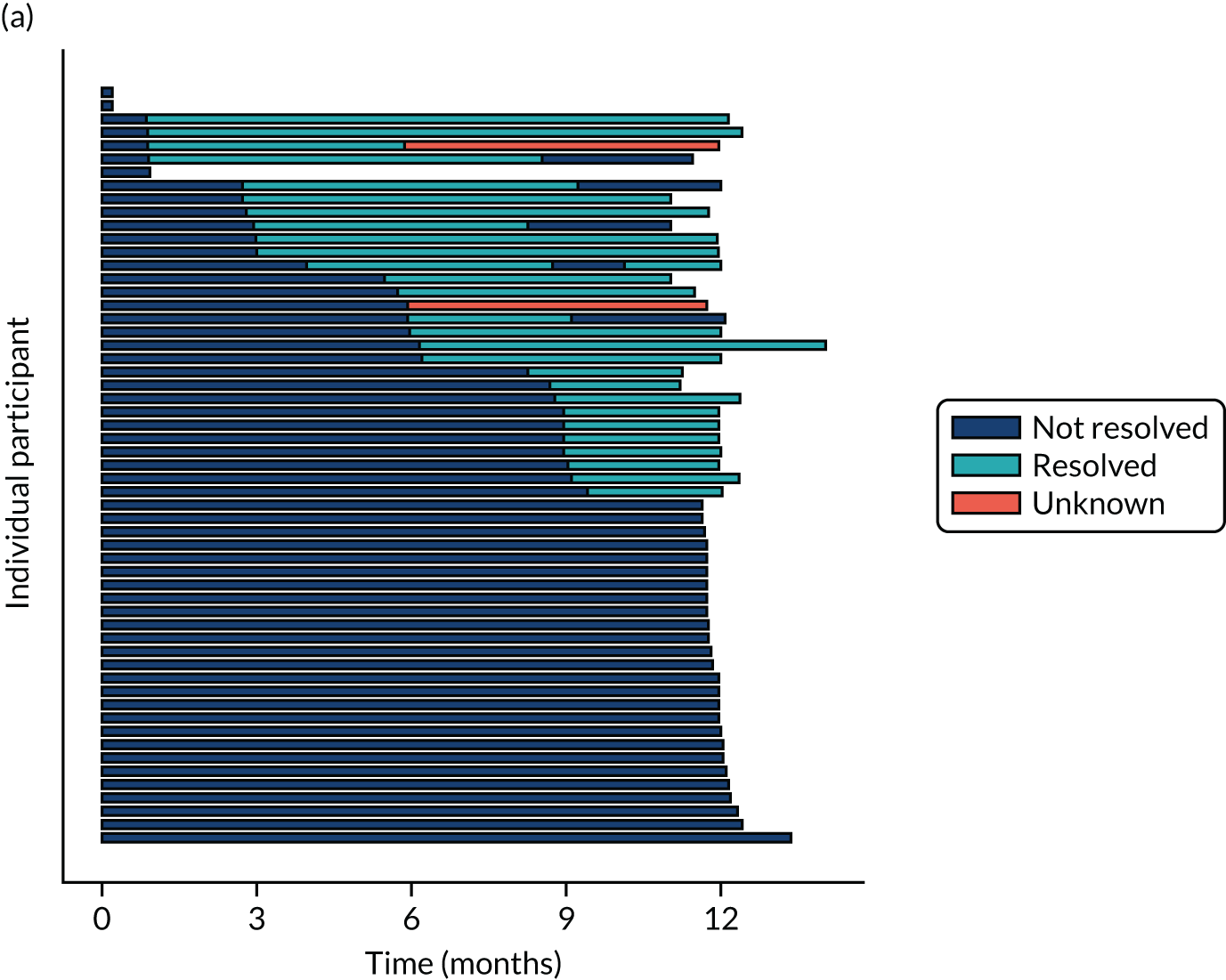
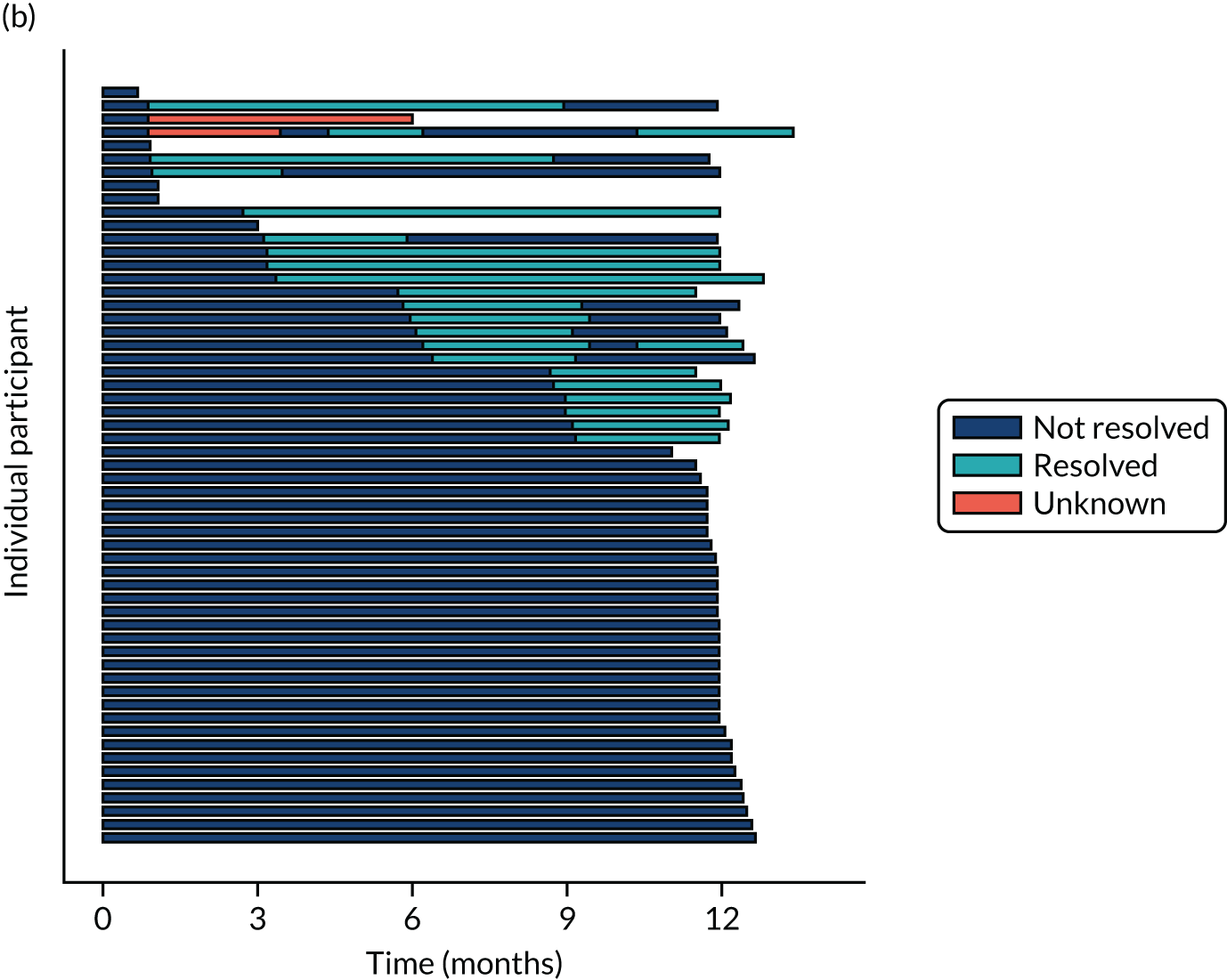
| Outcome | Randomised to placebo (N = 57) | Randomised to eplerenone (N = 57) | HR (95% CI) | p-value |
|---|---|---|---|---|
| Complete resolution of SRF at any time during follow-up, n/N (%) | 24/54 (44.4) | 18/56 (32.1) | ||
| Estimated median time (days) to complete resolution of SRF, median (IQR) | 458.2 (214.1–702.2) | 603.3 (313.1–893.5) | 0.78 (0.41 to 1.51) | 0.463 |
| Recurrence of SRF, n/N (%) | 11/24 (45.8) | 10/18 (55.6) | ||
| Estimated median time (days) to recurrence of SRF,a median (IQR) | 192.1 (136.6–247.6) | 182.5 (117.7–247.3) | 1.10 (0.45 to 2.66) | 0.836 |
| Complete or partial resolution of subfoveal SRF, n/N (%) | 38/54 (70.4) | 38/57 (66.7) | ||
| Estimated median time (days) to complete or partial resolution of subfoveal SRF, median (IQR) | 184.2 (122.3–246.0) | 141.1 (57.9–224.4) | 1.23 (0.75 to 2.01) | 0.418 |
| Any CSCR in fellow eye, n/N (%) | 8/57 (14.0) | 8/57 (14.0) | ||
| New CSCR in fellow eye at any time during follow-up, n/N (%) | 4/57 (7.0) | 5/57 (8.8) |
Table 16 shows the number of participants in each group with complete resolution of SRF at any time during follow-up, with recurrence of SRF after complete resolution and with complete or partial resolution of SRF at any time during follow-up. The table also describes estimates by group of median times to complete resolution of SRF, recurrence of SRF and complete or partial resolution of SRF, and hazard ratios (HRs) for the effects of eplerenone versus placebo on time to complete resolution (HR 0.78, 95% CI 0.41 to 1.51; p = 0.463), time to recurrence of SRF (after complete resolution) (HR 1.10, 95% CI 0.45 to 2.66; p = 0.836) and time to complete or partial resolution (HR 1.23, 95% CI 0.75 to 2.01; p = 0.418). Note that estimated median times to complete resolution of SRF are outside the period of follow-up (i.e. > 1 year) because < 50% of participants experienced complete resolution of SRF during the study. See Appendix 4, Table 38, for data contrasting the results of the primary and sensitivity analyses, adjusting for baseline SRF, for time to complete, and complete or partial, resolution of SRF.
Chapter 5 Results: lesion morphology outcomes
Findings from optical coherence tomograms
Median SRFT and choroidal thickness at baseline and 12 months in each group are shown in Table 17. In addition, these tables (see also Figure 13) show the treatment effects of eplerenone versus placebo, both of which favoured the placebo group (eplerenone minus placebo: SRFT –48.08 µm, 95% CI –82.73 to –13.43 µm, p = 0.01; choroidal thickness –38.53 µm, 95% CI –64.74 to –12.31 µm, p = 0.004).
| Outcome | Randomised to placebo (N = 57), median (IQR) | Randomised to eplerenone (N = 57), median (IQR) | MDa (95% CI) |
|---|---|---|---|
| SRFT (µm) | |||
| Change | –70 (–122.0 to –4.0) | –45 (–140.0 to 56.0) | |
| Baseline | 119 (88.0 to 178.0) | 147 (93.0 to 196.0) | |
| 12 monthsb | 61 (0.0 to 111.0) | 89 (23.0 to 196.0) | 48.1 (13.5 to 82.8) |
| Choroidal thickness (µm) | |||
| Baselinec | 460.5 (381.5 to 534.5) | 447.0 (398.0 to 509.0) | |
| 12 monthsd | 444.0 (375.0 to 524.0) | 495.5 (423.0 to 534.0) | 38.5 (12.3 to 64.7) |
Median CSRT in each group at each time point is shown in Appendix 4, Table 39, and Figure 12. There were no differences between the groups at 12 months, as evidenced by no difference in the estimated treatment effect of eplerenone versus placebo on CSRT, adjusted for baseline CSRT (eplerenone minus placebo: 24.4 µm, 95% CI –7.9 to 56.6 µm; p = 0.142) (Figure 13).
FIGURE 12.
Central subfield retinal thickness over time. Reproduced with permission from Lotery et al. 30 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) license, which permits others to copy and redistribute the material in any medium or format, for non-commercial use, with no derivatives, provided the original work is properly cited. See: https://creativecommons.org/licenses/by-nc-nd/4.0/. The figure includes formatting changes to the original.
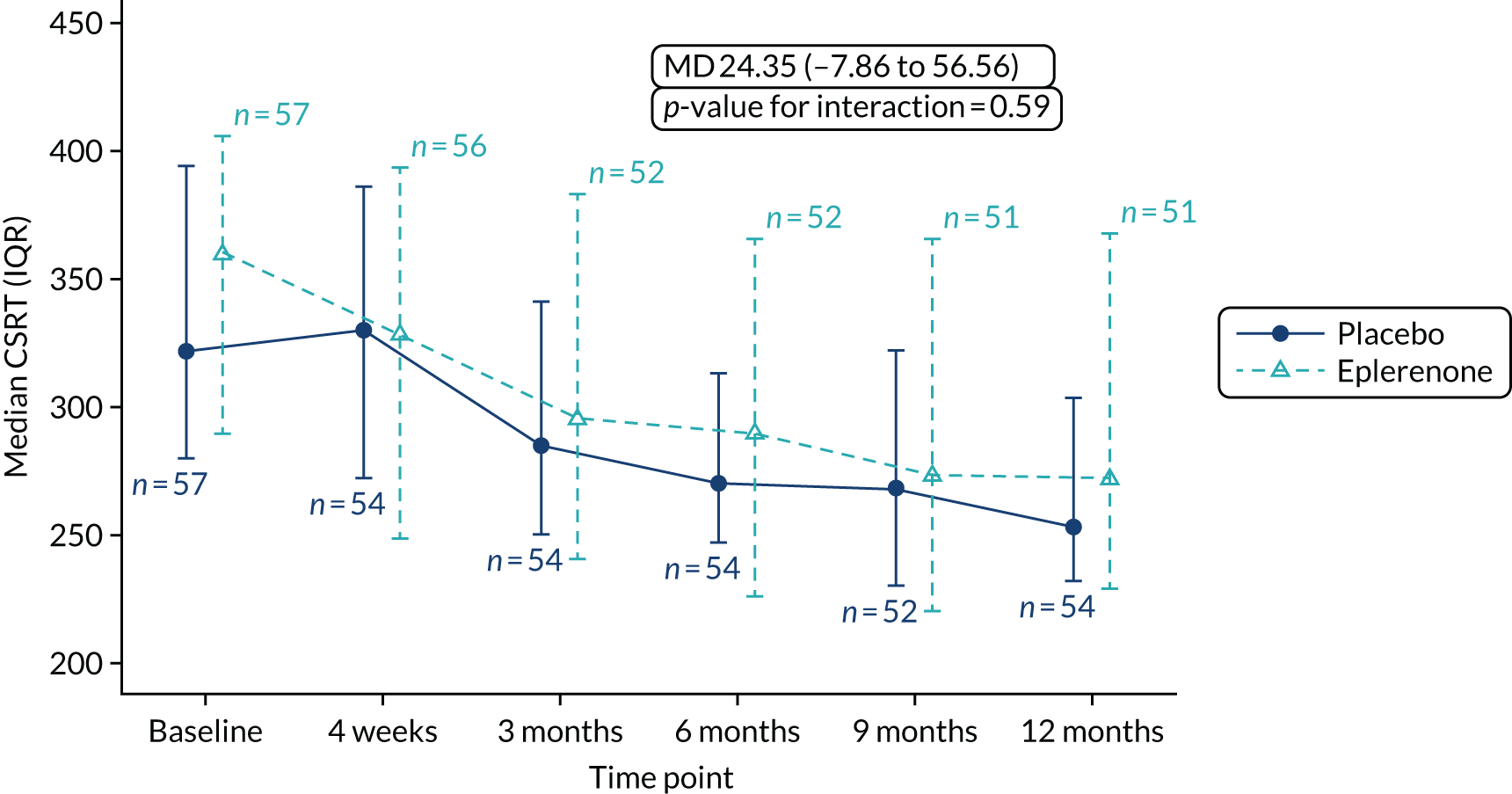
FIGURE 13.
Effect sizes of CSRT, SRFT and choroidal thickness.
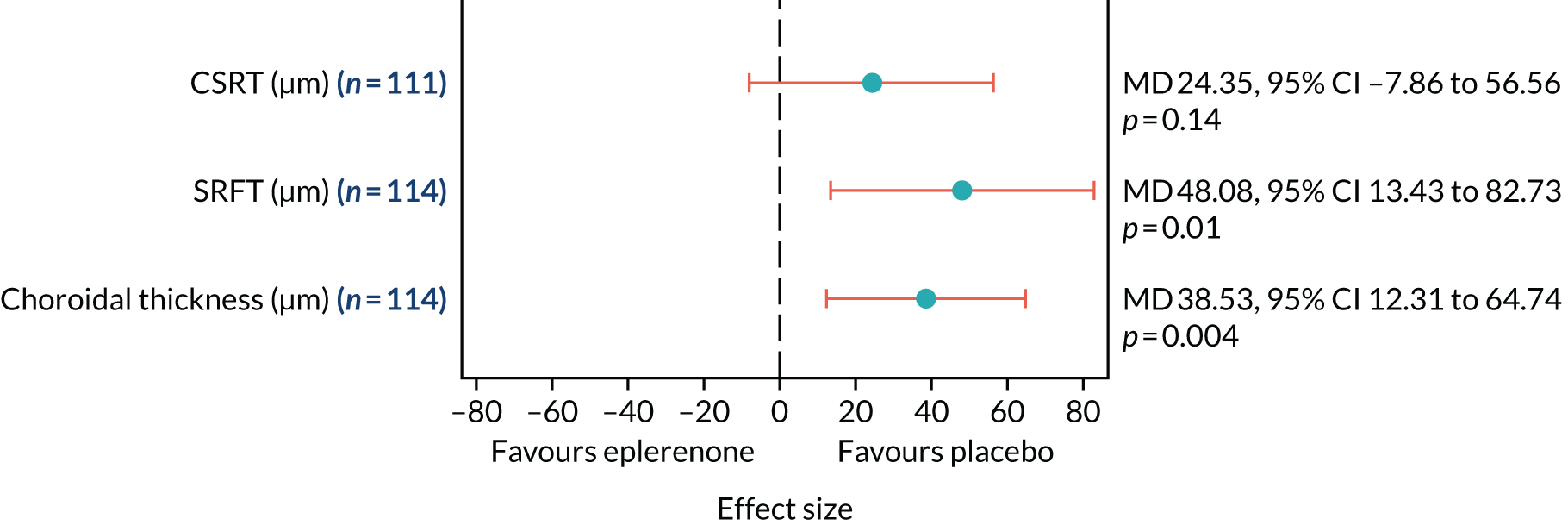
The choroid of fellow eyes was usually graded as thickened, despite clinical judgements that CSCR was mainly absent in fellow eyes (see Table 16). A post hoc analysis of choroidal thickness in fellow eyes, carried out to investigate the consistency of the effect favouring placebo in study eyes (because eplerenone is a systemic treatment), is shown in Table 18. The effects of eplerenone versus placebo on choroidal thickness for study and fellow eyes are contrasted in Figure 14. The post hoc analysis shows that the fellow eye effect was consistent with the study eye effect.
| Time point | Choroidal thickness (µm), median (IQR) | p-value | ||
|---|---|---|---|---|
| Randomised to placebo (N = 57) | Randomised to eplerenone (N = 57) | MD (95% CI) | ||
| Baselinea | 429 (365.5–486.0) | 386 (328.0–477.0) | ||
| 12 monthsb | 466 (416.0–554.0) | 475 (390.0–525.0) | 30.05 (–0.48 to 60.58) | 0.056 |
FIGURE 14.
Choroidal thickness in study eye and fellow eye. Reproduced with permission from Lotery et al. 30 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) license, which permits others to copy and redistribute the material in any medium or format, for non-commercial use, with no derivatives, provided the original work is properly cited. See: https://creativecommons.org/licenses/by-nc-nd/4.0/. The figure includes formatting changes to the original.
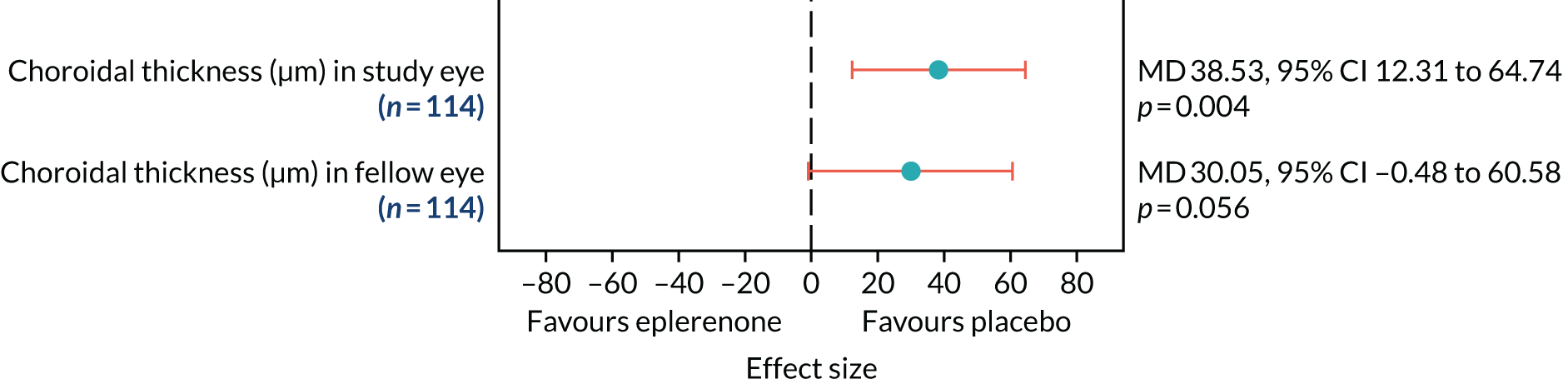
Post hoc analyses of CSRT and choroidal thickness were also carried out to take account of having had PDT administered to the study eye during follow-up. See Appendix 4, Table 40, for data contrasting the results of the primary analyses for these outcomes and the post hoc analyses, which were very similar.
Fluorescein and indocyanine green angiographic and retinal pigment epithelium autofluorescence outcomes
Study eyes with FFA images available at both baseline and 12 months (n = 104) were included in the FFA analysis. All study eyes showed leakage at baseline. The most common baseline phenotype in both groups was ink-blot, followed by chronic epitheliopathy and then smoke stack. Table 19 shows the FFA phenotype of study eyes at baseline cross-tabulated by phenotype at 12 months in the eplerenone and placebo groups. In both groups at 12 months, approximately half of phenotypes remained the same as at baseline, approximately one-third converted to a different leakage phenotype and just under one-fifth showed no visible leakage.
| Phenotype at baseline | Phenotype at 12 months, n/N (%) | |||
|---|---|---|---|---|
| Smoke stack | Ink-blot | Chronic epitheliopathy | No early or late leakage visible | |
| Randomised to placebo | ||||
| Smoke stack | 0/54 (0.0) | 0/54 (0.0) | 1/54 (1.9) | 0/54 (0.0) |
| Ink-blot | 2/54 (3.7) | 15/54 (27.8) | 11/54 (20.4) | 6/54 (11.1) |
| Chronic epitheliopathy | 0/54 (0.0) | 2/54 (3.7) | 15/54 (27.8) | 2/54 (3.7) |
| Randomised to eplerenone | ||||
| Smoke stack | 0/50 (0.0) | 1/50 (2.0) | 1/50 (2.0) | 2/50 (4.0) |
| Ink-blot | 0/50 (0.0) | 19/50 (38.0) | 6/50 (12.0) | 5/50 (10.0) |
| Chronic epitheliopathy | 0/50 (0.0) | 6/50 (12.0) | 8/50 (16.0) | 2/50 (4.0) |
In both the eplerenone and placebo groups the proportion of participants with reduced choroidal permeability at 12 months was low (Table 20). Statistical comparisons of treatment effects were not performed as fewer than 10 events occurred in total.
| Randomised to placebo (n = 57), n/N (%) | Randomised to eplerenone (n = 57), n/N (%) | |
|---|---|---|
| Proportion of patients with reduced choroidal permeability at 12 months | 3/54 (5.6) | 1/49 (2.0) |
Table 21 shows the area of macular RPE hypoautofluorescence (mm2) at baseline and 12 months and the area change at 12 months compared with baseline in the eplerenone and placebo groups. Only eight patients in total had macular RPE hypoautofluorescence at baseline and/or 12 months, so no statistical comparisons between groups were made.
| Outcome | Randomised to placebo (n = 57) | Randomised to eplerenone (n = 57) |
|---|---|---|
| Macular atrophy of RPE, n/N (%) | ||
| Baseline | 3/55 (5.5) | 2/56 (3.6) |
| 12 months | 3/53 (5.7) | 4/49 (8.2) |
| Baseline and/or 12 months | 3/51 (5.9) | 5/49 (10.2) |
| Area (mm2) of macular RPE hypoautofluorescence, median (IQR) | ||
| Baseline | 0.03 (0.03–0.04) | 0.72 (–0.73–2.10) |
| 12 months | 0.12 (0.08–0.21) | 0.00 (0.00–0.73) |
| Change | 0.15 (0.11–0.25) | 0.72 (0.00–2.10) |
Chapter 6 Safety
Mean serum potassium levels at all time points in both groups during follow-up are shown in Figure 15. A potassium level of > 5.0 mmol/l was observed in eight participants in each group (14%), resulting in permanent discontinuation of the IMP; the time points when these observations were recorded are shown in Table 22.
FIGURE 15.
Serum potassium levels at all time points in the placebo and eplerenone groups. Reproduced with permission from Lotery et al. 30 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) license, which permits others to copy and redistribute the material in any medium or format, for non-commercial use, with no derivatives, provided the original work is properly cited. See: https://creativecommons.org/licenses/by-nc-nd/4.0/. The figure includes formatting changes to the original.
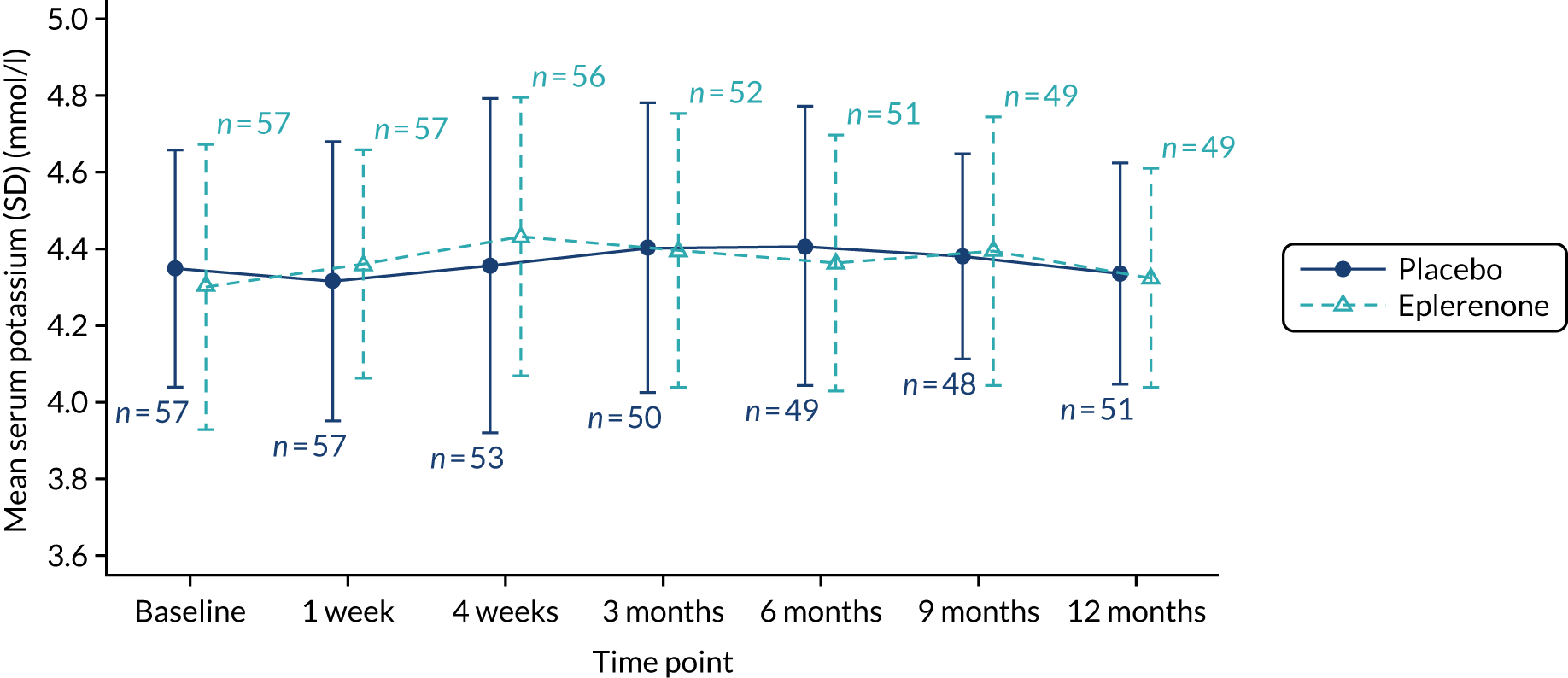
| Time point | Randomised to placebo (N = 8), n/N (%) | Randomised to eplerenone (N = 8), n/N (%) |
|---|---|---|
| 1 week | 4/8 (50.0) | 0/8 (0.0) |
| 4 weeks | 4/8 (50.0) | 2/8 (25.0) |
| 3 months | 0/8 (0.0) | 3/8 (37.5) |
| 9 months | 0/8 (0.0) | 2/8 (25.0) |
| Restarting week 1 | 0/8 (0.0) | 1/8 (12.5) |
There was one unexpected SAE (gastric sleeve surgery), which occurred in the placebo group and was not considered to be related to the IMP. There were two expected SAEs, neither of which was considered to be related to the IMP and both of which occurred in participants in the placebo group. Tables 23 and 24 tabulate expected SAEs and all AEs, respectively, by group. AEs are categorised by the MedDRA system organ class.
| SAE | Maximum intensity | Relatedness | Allocation |
|---|---|---|---|
| Acute diverticulitis | Moderate | Not related | Placebo |
| Myocardial infarction | Severe | Not related | Placebo |
| AE | Randomised to placebo (N = 57) | Randomised to eplerenone (N = 57) | ||||||
|---|---|---|---|---|---|---|---|---|
| All AEs | SAEs | All AEs | SAEs | |||||
| Patients (AEs) | % | Patients (SAEs) | % | Patients (AEs) | % | Patients (SAEs) | % | |
| Any AE (anticipated or otherwise) | 31 (72) | 54 | 3 (3) | 5 | 30 (95) | 53 | 0 (0) | 0 |
| Anticipated AEs listed in study protocol | ||||||||
| Study eye AE | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Decrease in visual acuity ≥ 15 letters | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Incident CNV | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Non-study eye AE | 0 (0) | 0 | 0 (0) | 0 | 1 (2) | 2 | 0 (0) | 0 |
| Decrease in visual acuity ≥ 15 letters | 0 (0) | 0 | 0 (0) | 0 | 1 (1) | 2 | 0 (0) | 0 |
| Incident CNV | 0 (0) | 0 | 0 (0) | 0 | 1 (1) | 2 | 0 (0) | 0 |
| Infections and infestations | 3 (4) | 5 | 0 (0) | 0 | 8 (10) | 14 | 0 (0) | 0 |
| Infection | 3 (4) | 5 | 0 (0) | 0 | 8 (10) | 14 | 0 (0) | 0 |
| Pharyngitis | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Pyelonephritis | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Eosinophilia | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Hypothyroidism | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Metabolism and nutrition disorders | 9 (9) | 16 | 0 (0) | 0 | 8 (8) | 14 | 0 (0) | 0 |
| Dehydration | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Hypercholesterolaemia | 1 (1) | 2 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Hyperkalaemia | 8 (8) | 14 | 0 (0) | 0 | 8 (8) | 14 | 0 (0) | 0 |
| Hypertriglyceridaemia | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Hyponatraemia | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Insomnia | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Nervous system disorders | 8 (10) | 14 | 0 (0) | 0 | 7 (9) | 12 | 0 (0) | 0 |
| Dizziness | 3 (3) | 5 | 0 (0) | 0 | 4 (4) | 7 | 0 (0) | 0 |
| Headache | 6 (7) | 11 | 0 (0) | 0 | 3 (5) | 5 | 0 (0) | 0 |
| Hypoaesthesia | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Syncope | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Cardiac disorders | 2 (2) | 4 | 1 (1) | 2 | 3 (5) | 5 | 0 (0) | 0 |
| Atrial fibrillation | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Left ventricular failure | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Myocardial infarction | 1 (1) | 2 | 1 (1) | 2 | 0 (0) | 0 | 0 (0) | 0 |
| Tachycardia | 1 (1) | 2 | 0 (0) | 0 | 3 (5) | 5 | 0 (0) | 0 |
| Vascular disorders | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Arterial thrombosis limb | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Hypotension | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Orthostatic hypotension | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Cough | 2 (2) | 4 | 0 (0) | 0 | 3 (3) | 5 | 0 (0) | 0 |
| Gastrointestinal disorders | 6 (10) | 11 | 0 (0) | 0 | 9 (19) | 16 | 0 (0) | 0 |
| Constipation | 0 (0) | 0 | 0 (0) | 0 | 1 (1) | 2 | 0 (0) | 0 |
| Diarrhoea | 2 (3) | 4 | 0 (0) | 0 | 2 (2) | 4 | 0 (0) | 0 |
| Flatulence | 0 (0) | 0 | 0 (0) | 0 | 1 (1) | 2 | 0 (0) | 0 |
| Nausea | 5 (7) | 9 | 0 (0) | 0 | 4 (7) | 7 | 0 (0) | 0 |
| Vomiting | 0 (0) | 0 | 0 (0) | 0 | 4 (8) | 7 | 0 (0) | 0 |
| Skin and subcutaneous tissue disorders | 1 (1) | 2 | 0 (0) | 0 | 5 (5) | 9 | 0 (0) | 0 |
| Angioedema | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Hyperhidrosis | 0 (0) | 0 | 0 (0) | 0 | 1 (1) | 2 | 0 (0) | 0 |
| Pruritus | 0 (0) | 0 | 0 (0) | 0 | 2 (2) | 4 | 0 (0) | 0 |
| Rash | 1 (1) | 2 | 0 (0) | 0 | 2 (2) | 4 | 0 (0) | 0 |
| Musculoskeletal and connective tissue disorders | 5 (8) | 9 | 0 (0) | 0 | 11 (11) | 19 | 0 (0) | 0 |
| Back pain | 2 (3) | 4 | 0 (0) | 0 | 3 (3) | 5 | 0 (0) | 0 |
| Muscle spasms | 0 (0) | 0 | 0 (0) | 0 | 1 (1) | 2 | 0 (0) | 0 |
| Musculoskeletal pain | 5 (5) | 9 | 0 (0) | 0 | 7 (7) | 12 | 0 (0) | 0 |
| Renal impairment | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Cholecystitis | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Gynaecomastia | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| General disorders and administration site conditions | 0 (0) | 0 | 0 (0) | 0 | 2 (2) | 4 | 0 (0) | 0 |
| Asthenia | 0 (0) | 0 | 0 (0) | 0 | 1 (1) | 2 | 0 (0) | 0 |
| Malaise | 0 (0) | 0 | 0 (0) | 0 | 1 (1) | 2 | 0 (0) | 0 |
| Investigations | 0 (0) | 0 | 0 (0) | 0 | 1 (1) | 2 | 0 (0) | 0 |
| Blood creatinine levels increased | 0 (0) | 0 | 0 (0) | 0 | 1 (1) | 2 | 0 (0) | 0 |
| Blood glucose levels increased | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Blood urea levels increased | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Epidermal growth factor receptor levels decreased | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Any anticipated AE | 25 (46) | 44 | 1 (1) | 2 | 28 (75) | 49 | 0 (0) | 0 |
| Any unanticipated AE | 16 (26) | 28 | 2 (2) | 4 | 13 (20) | 23 | 0 (0) | 0 |
Chapter 7 Systematic review of eplerenone versus placebo for central serous chorioretinopathy
The results of the literature search are shown in the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) flow diagram (Figure 16). We assessed the full text of five trials for eligibility. Two trials were excluded for reasons described in Figure 16. Data were extracted for the remaining three trials; their characteristics are shown in Table 25, together with the characteristics of the VICI trial. Two trials were identified as primary publications that were published after the VICI trial had been set up. 19,20 A third trial was identified from a previous meta-analysis of MR antagonists in CSCR that compared eplerenone with placebo. 18,23 These three trials had been reviewed by the TSC when they were published; the TSC concluded that the new information reported did not make the VICI trial redundant.
FIGURE 16.
The PRISMA flow diagram of literature screened for meta-analysis. Reproduced with permission from Lotery et al. 30 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) license, which permits others to copy and redistribute the material in any medium or format, for non-commercial use, with no derivatives, provided the original work is properly cited. See: https://creativecommons.org/licenses/by-nc-nd/4.0/. The figure includes formatting changes to the original.
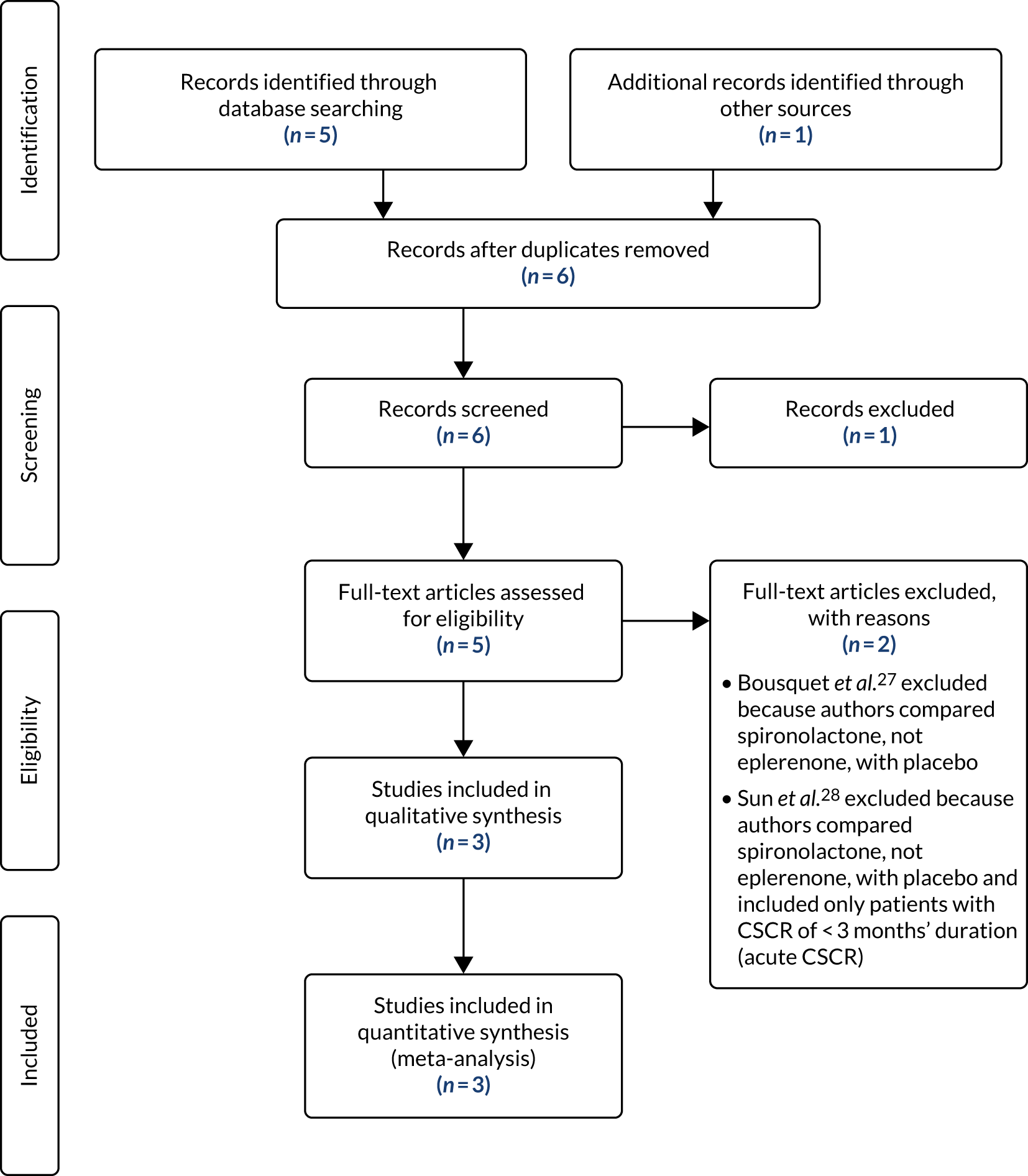
| Study | Registered in advance of starting recruitment | Primary outcome prespecified | Prespecified primary outcome matches reported primary outcome | SAP | Unit of analysis error |
|---|---|---|---|---|---|
| Rahimy et al.20 | Yesa | Yes | Nob | None identified | Yesc |
| Pichi et al.19 | No | Cannot tell | Cannot tell | None identified | No |
| Schwartz et al.18 | Yesd | Yes | Yes | None identified | Yese |
| VICI | Yes | Yes | Yes | Yes | No |
The four included trials compared eplerenone and placebo in 40, 21, 19 and 111 eyes over 1, 2, 3 and 12 months of treatment, respectively. Risk-of-bias assessments for the included trials are shown in Figures 17 and 18. We pooled the treatment effects that we re-estimated from data reported by the trials for BCVA score and SRFT. For BCVA score, the pooled difference between the eplerenone and placebo groups was 0.06 logMAR (equivalent to 3.0 letters, 95% CI –0.09 to –0.02 letters; equivalent to –4.5 to –1.0 ETDRS letters; I2 = 31%) (Table 26 and Figure 19). For SRFT, the pooled difference between eplerenone and placebo groups was 26.7 µm (95% CI –63.1 to 9.8 µm; I2 = 31%) (Table 27 and Figure 20).
FIGURE 17.
Risk-of-bias assessment of trials included in the meta-analysis of eplerenone for CSCR: BCVA outcome. 1, Rahimy et al. ;20 2, Pichi et al. ;19 3, Schwartz et al. ;18 4, VICI trial. +, low risk of bias; ?, some concerns about bias; !, some concerns about bias; I, high risk of bias. Reproduced with permission from Lotery et al. 30 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) license, which permits others to copy and redistribute the material in any medium or format, for non-commercial use, with no derivatives, provided the original work is properly cited. See: https://creativecommons.org/licenses/by-nc-nd/4.0/. The figure includes formatting changes to the original.

FIGURE 18.
Risk-of-bias assessment of trials included in the meta-analysis of eplerenone for CSCR: SRF outcome. 1, Rahimy et al. ;20 2, Pichi et al. ;19 3, Schwartz et al. ;18 4, VICI trial. +, low risk of bias; ?, some concerns about bias; !, some concerns about bias; I, high risk of bias. Reproduced with permission from Lotery et al. 30 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) license, which permits others to copy and redistribute the material in any medium or format, for non-commercial use, with no derivatives, provided the original work is properly cited. See: https://creativecommons.org/licenses/by-nc-nd/4.0/. The figure includes formatting changes to the original.

| Study | Time period | Randomised to placebo | Randomised to eplerenone | Weight (%) | MD (95% CI) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Number of eyes (number of patients) | MD | SD | Number of eyes (number of patients) | MD | SD | ||||
| Pichi et al.19 | Baseline to 1 month | 20 (20) | –0.05 | 0.18 | 20 (20) | –0.05 | 0.13 | 12.3 | 0.00 (–0.10 to 0.10) |
| Rahimy et al.20 | Baseline to 2 months | 6 (5) | 0.03 | 0.04 | 15 (10) | –0.06 | 0.08 | 43.7 | –0.09 (–0.14 to –0.04) |
| aSchwartz et al.18 | Baseline to 3 months | 6 (5) | –0.19 | 0.11 | 13 (12) | –0.22 | 0.11 | 10.6 | –0.03 (–0.13 to 0.07) |
| VICI | Baseline to 12 months | 54 (54) | –0.06 | 0.16 | 52 (52) | –0.10 | 0.15 | 33.4 | –0.05 (–0.10 to 0.01) |
| All | 86 (84) | 100 (100) | 100.0 | –0.06 (–0.09 to –0.02) | |||||
FIGURE 19.
Forest plot for meta-analysis of trials evaluating eplerenone for CSCR: BCVA. WMD, weighted mean difference.
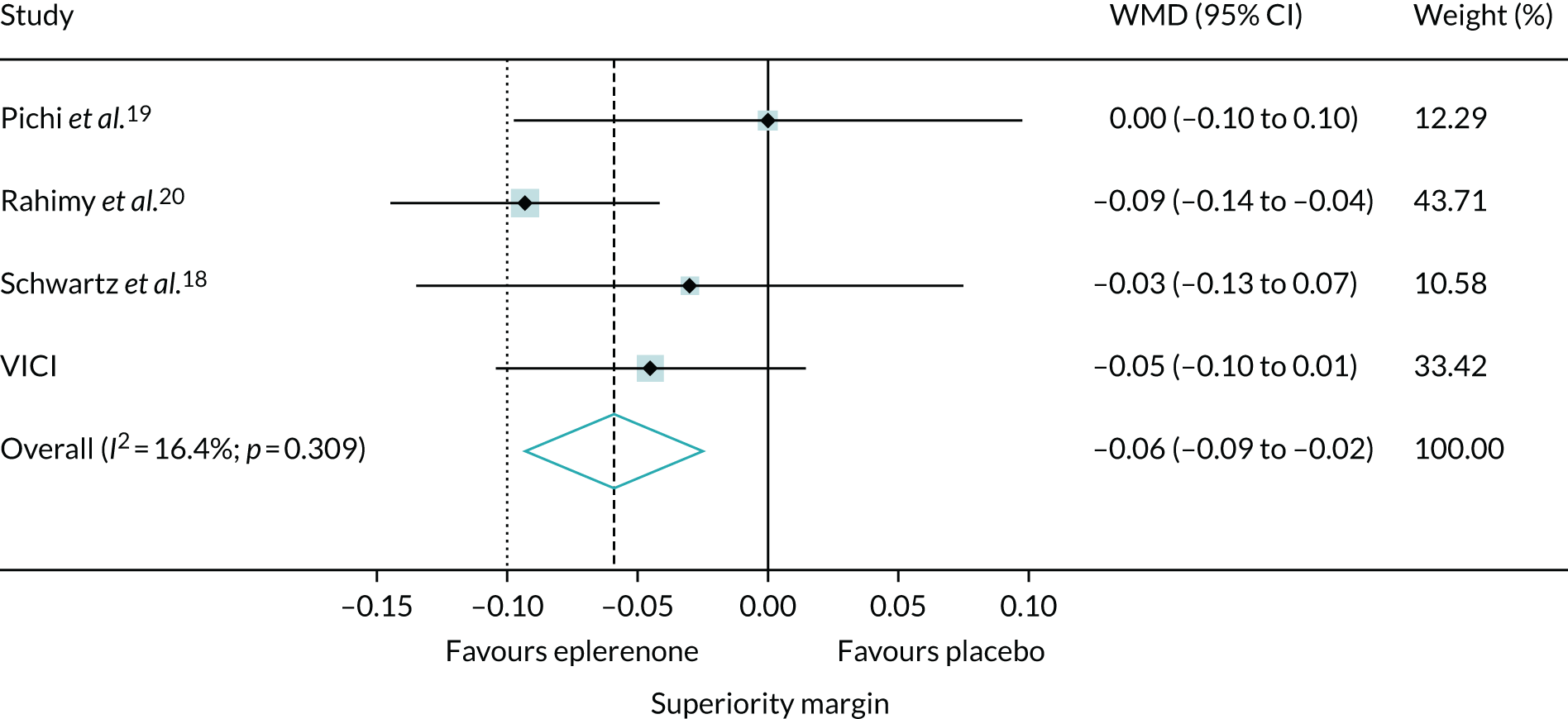
| Study | Time period | Randomised to placebo | Randomised to eplerenone | Weight (%) | MD (95% CI) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Number of eyes (number of patients) | MD | SD | Number of eyes (number of patients) | MD | SD | ||||
| Pichi et al.19 | Baseline to 1 month | 20 (20) | 24.0 | 297.5 | 20 (20) | –183.5 | 189.4 | 5.6 | –207.5 (–362.1 to -53.0) |
| Rahimy et al.20 | Baseline to 2 months | 6 (5) | 36.4 | 60.3 | 15 (10) | –87.5 | 97.1 | 28.1 | –123.9 (–192.8 to -55.1) |
| aSchwartz et al.18 | Baseline to 3 months | 6 (5) | –99.6 | 178.9 | 13 (12) | –74.8 | 143.0 | 5.0 | 24.8 (–138.1 to 76.6) |
| VICI | Baseline to 12 months | 54 (54) | –68.0 | 106.6 | 51 (51) | –38.0 | 134.3 | 61.4 | 30.0 (–16.6 to 76.6) |
| All | 86 (84) | 99 (93) | 100.0 | –26.7 (–63.1 to 9.8) | |||||
FIGURE 20.
Forest plot for meta-analysis of trials evaluating eplerenone for CSCR: SRFT (µm). WMD, weighted mean difference. Reproduced with permission from Lotery et al. 30 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) license, which permits others to copy and redistribute the material in any medium or format, for non-commercial use, with no derivatives, provided the original work is properly cited. See: https://creativecommons.org/licenses/by-nc-nd/4.0/. The figure includes formatting changes to the original.
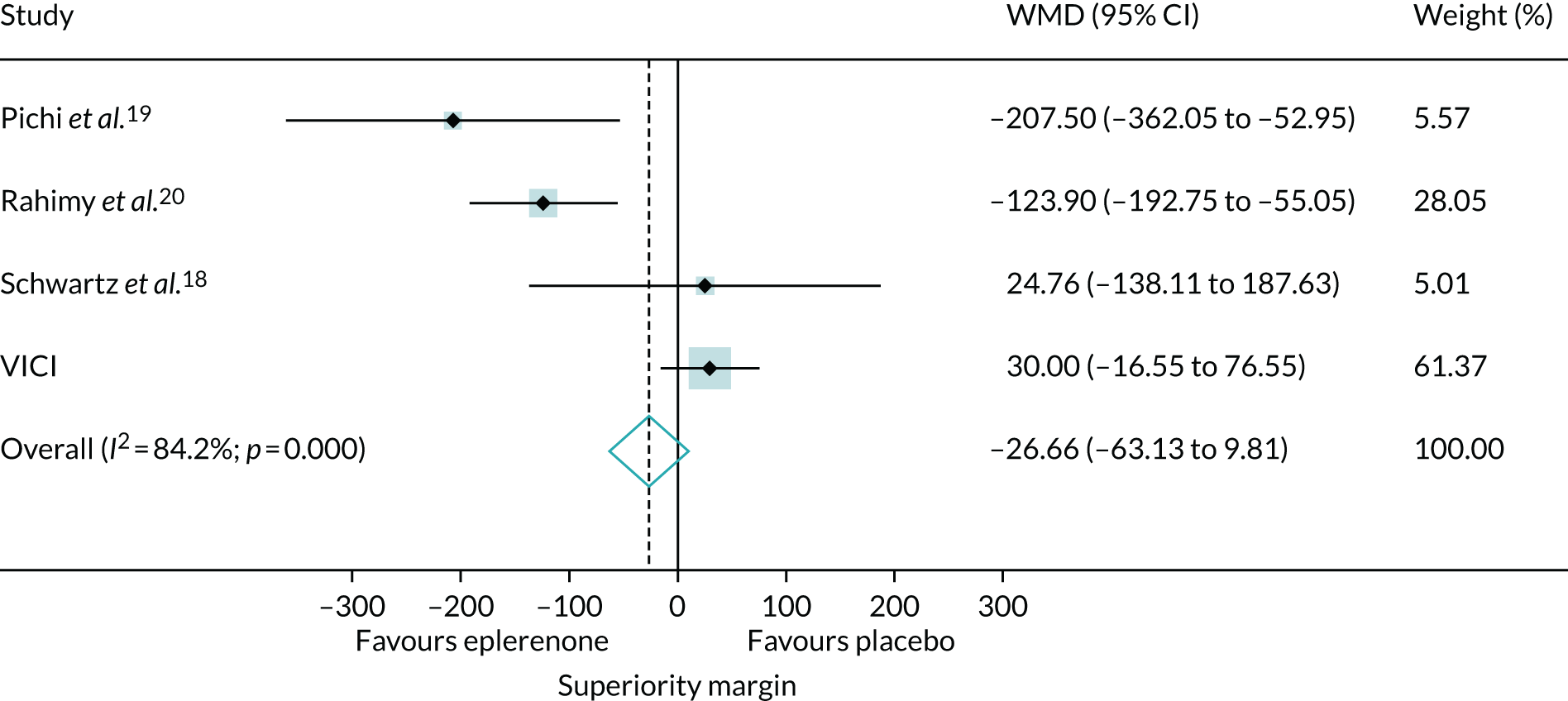
Chapter 8 Patient and public involvement
Amendments to the patient information leaflet
The PIL was amended following review by a representative from the Macular Society. Minor clarifications were made to the wording to make it more concise and understandable to patients. The full list of possible side effects was moved from the main body of the text to an appendix at the end as it was suggested that the list could be overwhelming for patients. The main potential side effect, hyperkalaemia, was retained in the main body of text.
Patient and public involvement group
Eight participants (five men and three women) from six sites opted to join the PPI group and attended the first meeting on 21 June 2018. Five of the original eight PPI group members (two men and three women) from three sites attended the second meeting on 22 June 2019.
Feedback from the first patient and public involvement meeting
How did you find out about the VICI trial?
Participants were people who had been diagnosed with CSCR either for some time or recently. Four participants had been referred to hospital other than their local hospital to take part in the trial. All participants reported being informed about the trial face-to-face, not via postal invitation. All participants agreed that they were not keen on the idea of receiving information about participating in a clinical trial by post in the first instance and, had that have been their first mode of contact, they may not have agreed to take part.
Decision-making processes behind taking part in research
Reasons why participants decided to take part in the trial included a fear of having to give up driving (paired with a belief that the trial intervention might speed up recovery), a lack of other treatments or clinical trials to participate in, that they were already taking other medications and did not see a problem with taking one more, not being keen on the idea of laser treatment (with the perceived possibility of permanent side effects), and wanting to help with research and further knowledge for others. The participants reported they were aware that they could stop taking part in the trial at any time: if it was not working for them, they could withdraw. The time commitment, list of potential side effects detailed in the PIL and additional imaging required were negative factors that participants reported could have put them off taking part.
Getting to your trial visits
Some of the participants worked part-time and were able to schedule hospital visits in their non-working days. Participants found communicating directly with research nurses to schedule appointments much easier than going through the normal clinic appointments system. They reported that research nurses showed a lot of flexibility with appointments and tried to accommodate last-minute changes if required.
One participant had been signed off work for several months and found attending appointments easy but admitted that it may become more complicated once they returned to work. Another participant reported having a very accommodating employer and would not have found it easy to take part in the trial if that was not the case.
All participants found being reimbursed for travel and hospital parking very helpful. One participant had a taxi booked and paid for directly by the research team, which they reported allowed them to avoid stress associated with getting to the hospital and claiming expenses. Some participants (depending on which hospital they attended) were given separate waiting areas and/or food and refreshments, which made their hospital visits more enjoyable. One participant reported enjoying the social aspect of the hospital visits as they came to know the staff quite well. Approximately half the participants lived close to their hospital, so travel was not much of an issue. Others lived a considerable distance away, with one participant requiring their partner to drive them to appointments, which affected both their lives.
One of the main issues with hospital appointments that almost all the participants reported as inconvenient and frustrating was the length of time taken to receive the prescription from the hospital pharmacy. Waiting times were reported to be up to a few hours. One participant would leave the hospital and return later in the day to collect their prescription. If considering participation in a future trial, they reported that this factor would put them off.
Taking the study tablets regularly
Participants reported working out what method worked best for them for remembering to take the study medication each day. Ensuring that the method fitted well around their lifestyle was important for taking the medication. The lack of restrictions on taking the tablets in terms of time of day and with or without food, and having to take only one tablet per day, reportedly made adhering to the regimen easy.
Introduction to the biobank
Participants were reminded of the biobank described in Chapter 2, Biobank samples. Most participants were unable to clearly recall any discussion about the biobank with the research teams when they joined the study and had only vague memories about donating blood samples. However, one participant remembered it clearly because they opted not to donate samples owing to concerns about confidentiality and not knowing what would happen to their samples. All participants agreed that information provision from the PIL or the research teams about the biobank samples had not been clear enough; they recommended producing a separate PIL about biobank samples for future research to make it very clear that extra samples would be taken, where they would be stored, for how long, who would be responsible for the samples and what would happen to them.
Taking part in future events as a public representative
The participants were invited to attend the next VICI trial investigators (see Appendix 1, Table 28) meeting to feed back on their experiences of taking part in a clinical trial. No participants took up this offer. Participants were keen to take part in another PPI group meeting because they reported enjoying the experience of the first meeting. They were also keen to help with dissemination activities such as helping to design participant results leaflets.
Participant input from the second patient and public involvement meeting
The PPI group were presented with the trial results and asked which secondary outcomes they thought were important to include in the results leaflet for trial participants.
The PPI group advised including in the leaflet the results on (1) SRFT and (2) estimated time to resolution and recurrence. The group members suggested omitting the result for the NEI-VFQ-25 patient-reported outcome from the leaflet because they reported feeling that the questionnaire was aimed at people with much worse vision problems than theirs. They did not think that the NEI-VFQ-25 was a good tool for assessing the impact of CSCR on their quality of life. They also suggested leaving the potassium results out of the leaflet because there was no difference between the groups and the results were not likely to be of interest to participants.
The PPI group members were asked for their preference on receiving a short (one sheet of bifold A4 paper) or long (two sheets of bifold A4 paper) version of the results leaflet. All preferred the short version, suggesting that people would prefer brief, simple information.
Chapter 9 Discussion
Main findings
Study conduct
Recruitment to the trial was challenging, as illustrated in Figure 6. Chronic CSCR is an uncommon condition and sites had to be vigilant to identify potentially eligible patients. Screening for eligibility had to be done in two stages because some eligibility criteria required tests that were not carried out as part of usual care. Therefore, eligibility was first assessed on other criteria, consent for the additional tests of eligibility was obtained, the additional tests were carried out and final eligibility was confirmed.
Most patients initially approached about the trial were willing in principle to take part and consented to the additional tests (see Figure 5). About one-third of the patients having further tests for eligibility were found to ineligible. Almost half of those found to be ineligible after further tests were ineligible because their BCVA score was too good; despite having appeared to be eligible at the initial screen on the basis of their presenting visual acuity, visual acuity improved with refraction. This phenomenon arose because fluid in the retina (thickening the retina) causes a hyperopic shift, and refraction with a stronger positive lens improved visual acuity. We could have used a lower level of visual acuity as an initial eligibility criterion (and we considered amending the visual acuity criterion), but this would have restricted the pool of patients at the initial screen.
When designing the trial, we were uncertain about whether or not participants would accept the use of a placebo (although this was a condition of the award), not knowing their allocation and continuing on masked IMP for 12 months. Given the finding that eplerenone is not beneficial in treating CSCR, we believe that it was the correct decision to use a placebo control because this prevents ‘bias in measurement of the outcome’. 29 We were also able to show that the placebo was effective in masking the allocation from both participants and research personnel.
We specified the manufacture and delivery of the amounts of different doses of the IMP to match the original trial schedule, timetabling a second manufacturing batch to achieve the expiry time required for the duration of the trial. The second batch was also adjusted to take account of estimates of the proportion of participants with complete resolution and recurrent CSCR, which affected the total volume of low-dose tablets required; these parameters were unknown at the outset. Because recruitment was delayed by 3 months, the timing of the second manufacturing batch became critical because of low stock of 25-mg tablets and the risk that the stock would pass the expiry date. In addition, we had to shift low-dose IMP already despatched to one site to another site to ensure the availability of lose-dose IMP for all participants in whom CSCR had resolved and might have recurred. The lessons we learned (see Lessons for the future) include that it is advisable to manufacture substantially more IMP than required and to try to foresee risks such as a delay in recruitment. In this instance, the cost of the active drug ingredient was low and the total cost of the IMP would not have been greatly increased by ordering a larger volume of tablets.
Study results
The VICI trial has shown that eplerenone did not result in an improvement in BCVA score of ≥ 5 letters compared with placebo over a 12-month period. LLA, which can be a more sensitive test for deterioration in visual function in patients with age-related macular degeneration,25 was similar in both groups after 12 months, supporting the finding that eplerenone did not improve visual acuity compared with placebo. Time to complete resolution of SRF, and subsequent recurrence of CSCR in participants in whom CSCR resolved, did not differ between groups. Point estimates for time to complete resolution of SRF and recurrence of CSCR favoured placebo, but not significantly so.
Measurements of retinal morphology from the various imaging modalities captured showed no benefit of eplerenone. Unexpectedly, two morphological outcomes, SRFT and choroidal thickness, significantly favoured placebo. One possible explanation is the effect of eplerenone on renal function, because eplerenone is known to induce hyponatraemia. Loss of sodium from the urine and secondary reductions in blood sodium content has the potential to cause movement of fluid into extravascular compartments. However, we found no evidence of any difference in sodium level between the groups at any time point during follow-up. Because grading of baseline OCTs showed choroidal thickening in fellow eyes even when examining ophthalmologists considered the eyes not to have CSCR, and because eplerenone is a systemic treatment affecting both eyes, we also carried out a post hoc analysis of choroidal thickness to investigate the consistency of the treatment effect in study and fellow eyes. The treatment effect for the fellow eye was very similar to the treatment effect for the study eye but did not reach statistical significance, probably because there was less choroidal thickening in fellow eyes, which may have caused a floor effect.
The treatment effects for BCVA, time-to-event outcomes and key morphological outcomes were unaltered in additional sensitivity, exploratory and post hoc analyses. The failure to find a difference cannot be attributed to limitations of the trial: the primary analysis included data for 97% of randomised participants, participants attended 94% of all scheduled visits and there were no protocol deviations affecting the treatment comparison.
Potential adverse effects were captured at all follow-up visits. Eplerenone is known to increase blood potassium levels but we found no difference between the groups in the number of patients exceeding the prespecified threshold defining hyperkalaemia (an expected AE). Qualitatively, the profiles of sodium levels during follow-up were very similar in the two groups. The low frequencies of other complications precluded formal comparisons between the groups.
In summary, we found no evidence of benefit or harm of eplerenone, apart from the differences between the groups in SRFT and choroidal thickness that favoured placebo.
We sought to place the findings of the VICI trial in the context of similar trials by searching systematically for trials and pooling data for trials that were sufficiently similar. Unsurprisingly, given the relative size of the included studies, the treatment effects for BCVA for the VICI trial and the pooled estimates from the meta-analysis are consistent: VICI point estimates lie within the 95% CIs for the pooled estimates. The upper 95% confidence limit for the pooled BCVA score estimate (4 letters) also excludes the target difference that the VICI trial was powered to detect (5 letters). The SRF thickness results, however, show substantial heterogeneity and the VICI treatment effect is in the opposite direction to the results of the other three trials. We have no explanation for this inconsistency. One possibility is that eplerenone has a short-lived effect on SRF, which may have been missed in VICI because SRF was measured only at baseline and 12 months (the follow-up durations in the other three trials were 1, 2 and 3 months). Measuring SRF more frequently during follow-up would have allowed a more detailed comparison between the included trials in terms of response of SRFT to treatment with eplerenone and placebo.
We have concerns about the quality of the three small trials (see Table 25), even though the risk-of-bias assessments do not indicate obvious sources of bias. Despite publication in 2016, one trial was not registered; we were unable to find a published protocol or prespecified analysis plan for any of the trials; and ways in which the data were analysed and the treatment effect estimates calculated were unusual in the trials, potentially indicating bias from selection of the reported result. The SD estimates for BCVA score for one trial appeared too small in relation to the other trials, potentially giving the trial undue weight in the meta-analysis. 20 The trials were not assessed as being at risk of this bias because we reanalysed the published data to provide a consistent treatment effect for the meta-analysis.
Patient and public involvement
Input from representatives of the Macular Society was invaluable for producing a PIL that was understandable and acceptable to participants. Moving the list of potential drug side effects from the main body of the text to an appendix made the PIL less overwhelming and improved the flow of text while ensuring that this important information was still available to potential participants to aid their decision-making. This PPI-guided PIL format is being applied in new studies coordinated by the BTC-CTEU. However, despite the relocation of the side effects, the PPI group still reported the listed side effects as off-putting when considering whether or not to participate in the trial.
An unusual aspect of the PPI group formed for the VICI trial was the recruitment of active trial participants during the trial follow-up period. We assembled the group in this way because attendance at follow-up visits and adherence to the intervention was high and loss to follow-up and withdrawal rates were low. This presented an opportunity to explore why a RCT was going well, something that is rarely investigated or documented. There was mild concern that (1) we risked unmasking if participants compared their symptoms or disease resolution status during the meeting and (2) participants would withdraw from the trial if they became upset by discussions at the meeting. To address point (1), participants were reminded at the beginning of the meeting of the masked design of the trial and why it is important, and they were asked not to discuss and compare their CSCR symptoms. The PPI group were happy with this request and respected it. The risk of point (2) occurring was considered low. However, careful planning ahead of the meeting ensured that agenda items were appropriate and non-inflammatory. Two experienced PPI facilitators were consulted to ensure that the language used in the meeting was appropriate and constructive.
An important point raised by the PPI group was to ensure that any substudies, such as the substudy in this trial in which participants donated samples to a biobank for future research, are clearly explained to patients prior to obtaining consent. Although information about the biobank samples was included in the PIL and was separated from the main trial information in the consent form, it was advised that more could be done to make this type of information apparent. The suggestions made by the group have been taken onboard and will be implemented in future studies. The outcome of the PPI meeting was shared with teams at participating sites.
The feedback received from some members of the PPI group that the NEI-VFQ-25 was, in their view, not suitable for assessing visual-function-related quality of life in patients with CSCR was taken on board. At present, a quality of life assessment tool specifically for patients with CSCR is an unmet need. Further research is required to develop and validate an appropriate assessment tool.
The PPI group partly attributed the ease of taking the study medication to the lack of restrictions on taking the capsules with regard to time of day and food intake. This is an important consideration for future studies as flexibility could minimise burden on participants and lead to improved adherence. However, such flexibility is not always possible depending on the guidelines for the study drug. Where restrictions are mandated by guidelines it could be useful to identify alternative strategies that make adherence easier, such as sending regular text message reminders to participants or implementing online or paper diaries.
Gaining PPI input on the content of the results leaflets from a subset of trial participants was highly beneficial because the perceived priority of different results differed between them and the professional stakeholders. We highly recommend engaging with trial participants or members of the public when preparing dissemination documents for a lay audience. We also learned from the participants that the NEI-VFQ-25 was, in their opinion, not an appropriate patient-reported outcome measure (PROM) for assessing their quality of life. It would be beneficial in future studies of CSCR to use a PROM that is sensitive enough to detect improvements in quality of life in patients with CSCR.
Strengths and limitations
The VICI trial had several strengths. All but three participants contributed to the primary analysis and participants attended almost all scheduled visits. There were no protocol deviations compromising the treatment comparisons. The trial was powered to detect a clinically important difference in BCVA targeted by several large multicentre trials of treatments for retinal conditions and,31–33 in fact, had more power than anticipated owing to participants attending almost all scheduled visits.
Limitations included the need to discontinue treatment if CSCR completely resolved during follow-up or an elevated serum potassium level was detected. These issues may have reduced the observed treatment effect but were required to safeguard participants. Hyperkalaemia was detected in 14% of participants using the potassium threshold adopted for the study of 5.0 mmol/l but, in all but one of these patients, the absolute potassium level that triggered discontinuation of IMP was below the threshold for reducing the dose or withdrawing the treatment as specified in the SmPC for eplerenone for use in its licensed indication (5.5–5.9 mmol/l: reduce dose or withdraw treatment if on lowest dose; ≥ 6.0 mmol/l: withdraw treatment). We adopted the lower threshold for the trial because we wanted to ensure the safety of participants who did not have heart failure and therefore had no opportunity to benefit from eplerenone for this indication. However, participants in the trial were younger and fitter than patients who are usually prescribed eplerenone for heart failure and, with hindsight, we think that the established potassium threshold would have been more appropriate. We discontinued IMP when CSCR completely resolved because, when this happened, participants had no opportunity to benefit from the IMP but would have been at risk from possible side effects. Discontinuing IMP when CSCR completely resolved allowed us to compare recurrence of CSCR after complete resolution. We observed no difference in time to recurrence between groups.
The main protocol deviation was study visits taking place outside the visit window, which occurred equally between the two groups. The main reasons for this were participants taking holidays and having work commitments. Occasionally, study visits were delayed owing to staffing issues at sites. Owing to the controlled supply of the study drug (i.e. dispensing only enough to last between follow-up visits) and the safety monitoring required, we were not able to expand the visit windows. Such issues are likely to occur in future studies in this population. We will investigate strategies to make follow-up more efficient to reduce the amount of time participants spend in clinics and the resources research teams need to attribute to follow-ups.
Our inability to control the use of co-treatments could have introduced bias if these were used differentially by group. We observed that more participants in the placebo group were treated with PDT than in the eplerenone group. However, co-interventions (including PDT) were used infrequently. A post hoc analysis that adjusted for the imbalance between groups in the proportion of study eyes treated with PDT produced results that did not differ from the results of the primary analysis. We believe that our results are generalisable to a wider population of patients with CSCR. The visual acuity eligibility criterion for the trial meant that some patients, who would otherwise have been eligible, were excluded because their BCVA score was too good (see Main findings, Study conduct). However, we have no reason to believe that the effects of eplerenone in these patients would have been any different. This view is supported by the consistent effect of eplerenone on choroidal thickness in the study and fellow eyes of participants, as fellow eyes could be considered to represent similarly less severe CSCR (i.e. those patients whose BCVA score was too good). In this study, fellow eyes were judged by ophthalmologists not to have CSCR, but grading of OCTs showed considerable choroidal thickening. The same logic would apply to patients who did not meet the trial criterion for chronicity. Requiring eligible participants to have SRF tended to shift the threshold towards chronicity. We required the duration of the current CSCR episode to be ≥ 4 months but did not consider whether or not a participant had had a previous episode of CSCR, and if so how many, or how quickly a previous episode or episodes had resolved. In other respects, the characteristics of the trial participants were similar to the wider CSCR population.
Neovascular age-related macular degeneration (nAMD) is a common phenocopy of CSCR. To avoid nAMD cases being inadvertently recruited into the trial, we did not recruit any patients aged > 60 years because nAMD usually presents at a later age than this. In addition, we checked for nAMD with fluorescein angiography and ICGA at baseline. Presence of CNV at baseline was an exclusion criterion, as was a history of previous anti-VEGF therapy. These criteria, we believe, stopped any patients with nAMD phenocopies entering the study.
We reduced the risk of patients with dome-shaped maculopathy being recruited with our exclusion criterion of myopia of > –6 dioptres. Patients on MEK inhibitors can develop SRF similar to CSCR. However, no patients on such drugs were recruited.
Lessons for the future
There are two lessons for the future in relation to manufacture of an IMP for trials.
The first lesson is that triallists should obtain quotes for IMP manufacture from several pharmacies. We obtained three quotes, which varied by ≈ £100,000 for the same specification. Triallists are rarely experts with respect to preparation/formulation of the drug of interest as an IMP or issues relating to manufacture of an effective placebo and may have to choose a supplier with little information about the track record of specific pharmacies. At the outset, it would have been helpful to have a checklist of issues to consider and a list of ‘accredited’ pharmacies. From its oversight of a wide range of studies, the NIHR may be well placed to help triallists in this respect. In this trial, budgeting for the IMP was made unnecessarily complicated because the manufacturing pharmacy did not distinguish the cost of the active drug from the cost of manufacturing the placebo and over-encapsulation of both drug and placebo. This prevented the sponsor from attributing the cost of the IMP to treatment versus research cost categories at the time of preparing the grant application. Triallists require manufacturing pharmacies to itemise research quotes accordingly.
Given the problems we experienced with low stock and expiry times, the second lesson relates to the amount of IMP (drug and placebo) ordered from the manufacturing pharmacy. We recommend that triallists order substantially more IMP than required simply on the basis of the number of scheduled doses. This recommendation is tempered by the need to weigh up the cost of wastage against the risks to the trial of running out or the expense of moving stock around. Triallists also need to consider potential threats to the supply of IMP owing to issues such as a delayed start to recruitment or slower than projected recruitment.
There is also a lesson for future trials on treatments for CSCR that relates to visual acuity. The fact that visual acuity in eyes with CSCR can be improved by prescribing additional positive optical power raises the question of how a visual acuity eligibility criterion should be defined, or if eligibility needs to defined in terms of visual acuity at all. One possibility would be to specify the visual acuity criterion with the most up-to-date refraction prescription preceding the onset of CSCR, but this would be difficult to implement. An alternative would be to not define eligibility in terms of visual acuity. This is attractive because visual acuity does not capture well the visual problems that patients experience.
If visual acuity does not reflect the visual problems that patients experience, there is also a question about whether or not it should be the primary outcome in trials of CSCR. This question is particularly relevant when considering a trial of PDT because PDT might be expected to reduce BCVA scores by a few letters but still be effective in treating the condition (i.e. avoiding the distortion symptoms about which patients complain and avoiding future costs to the NHS of managing the condition). There needs to be a debate involving all stakeholders about the pros and cons of choosing different outcomes (e.g. BCVA, retinal fluid, health-related quality of life, psychometric test of visual distortion) as the primary outcome. Other functional measures of vision such as microperimetry also need to be considered.
The disadvantage of not using visual acuity as an eligibility criterion or as the primary outcome is the difficulty in putting CSCR in context alongside other visually disabling conditions. The challenge in CSCR trials is that the range of visual acuity loss varies widely. Some patients have severe visual loss, but these patients may be excluded from entering trials owing to irreversible structural damage. Patients more representative of the majority of CSCR patients may have refracted visual acuities in the normal visual range. Other functional measures such as microperimetry may be a better end point for future clinical trials. However, microperimetry is not used routinely in clinical practice and so many eye departments do not have access to such equipment. A future study correlating structural changes (e.g. SRF) with microperimetry would be useful. If they were strongly correlated, then presence of SRF could be considered as a trial end point because SRF is easily measured with OCT equipment, which is readily available in eye departments.
Future research
We believe that there would be considerable value in following the VICI trial cohort for longer. As described in Strengths and limitations, participants could have had previous episodes of chronic CSCR before the one that qualified them for the trial; some had complete resolution during the trial and some of those had a recurrence of CSCR during the trial. The natural history of resolution and recurrence of CSCR has not been well characterised. Notwithstanding the fact that we did not characterise episodes of CSCR before recruitment, the trial cohort represents the most comprehensively documented CSCR cohort, and information about the subsequent CSCR experience of participants (e.g. via an annual follow-up) would be valuable to both ophthalmologists and future patients.
With respect to other potential treatments, the PLACE trial raised the possibility that half-fluence PDT could be effective. 12 We do not consider this single trial to be sufficient evidence to conclude that half-fluence PDT is effective because of flaws in the trial, notably the lack of masking. Nevertheless, we believe that the trial should be replicated. It is difficult to know what capital investment would be required to do such a trial in the UK. Most UK centres have PDT equipment (it was previously used to treat nAMD), but it may have been decommissioned or be obsolete. 34 Verteporfin is expensive but the patent expires in October 2020. Moreover, if multiple patients attend one clinic, one vial of verteporfin can be used to treat three or four patients with half-fluence PDT. If such a trial were to be designed, we recommend that microperimetry be considered as the primary outcome. However, investment in microperimetry equipment would probably be needed.
Chapter 10 Conclusion
The VICI trial found no evidence of a clinically important benefit of eplerenone for the treatment of CSCR, either in the primary outcome or in a wide range of secondary outcomes including retinal morphology. It will be important to review the use of eplerenone for treating CSCR. Future trials of other potential interventions should be investigated. CSCR can be a devastating condition for people who are affected, who are often of working age. It is also a challenging condition for ophthalmologists to manage and uses considerable NHS resources through repeat clinic attendances and investigations.
Acknowledgements
Contribution of authors
Professor Andrew Lotery (https://orcid.org/0000-0001-5541-4305) (Professor of Ophthalmology and Chief Investigator) conceived the trial, obtained funding, managed the trial with the Trial Management Group, developed the retinal image grading protocols and managed the grading process, interpreted the data and co-authored the first draft of the report.
Professor Sobha Sivaprasad (https://orcid.org/0000-0001-8952-0659) (Consultant Ophthalmologist) obtained funding, designed the trial, was a member of the Trial Management Group, developed the retinal image grading protocols and managed the grading process, interpreted the data and co-authored the first draft of the report.
Dr Abby O’Connell (https://orcid.org/0000-0001-7598-927X) (Trial Manager) managed all aspects of the trial including monitoring and co-authored the first draft of the report.
Miss Rosie A Harris (https://orcid.org/0000-0002-2405-667X) (Medical Statistician) was responsible for data management, analysed the data and co-authored the first draft of the report.
Dr Lucy Culliford (https://orcid.org/0000-0002-9255-6617) (Research Fellow) obtained funding, designed the trial, oversaw trial management and co-authored the first draft of the report.
Ms Angela Cree (https://orcid.org/0000-0002-1987-8900) (Senior Research Manager) obtained funding, oversaw the biobank, was a member of the Trial Management Group and co-authored the first draft of the report.
Ms Savita Madhusudhan (https://orcid.org/0000-0002-8203-0929) (Consultant Ophthalmologist) developed the retinal image grading protocols and managed the grading process.
Ms Helen Griffiths (https://orcid.org/0000-0003-3908-884X) (Research Technician) processed the biobank samples.
Miss Lucy Ellis (https://orcid.org/0000-0001-8179-5172) (Trial Manager) was responsible for trial management during trial set-up.
Professor Usha Chakravarthy (https://orcid.org/0000-0002-2606-3734) (Consultant Ophthalmologist) provided expert input on the trial, developed the retinal image grading protocols and managed the grading process, interpreted the data and co-authored the first draft of the manuscript.
Professor Tunde Peto (https://orcid.org/0000-0001-6265-0381) (Consultant Ophthalmologist) provided expert input on the trial, developed the retinal image grading protocols and managed the grading process and interpreted the data.
Professor Chris A Rogers (https://orcid.org/0000-0002-9624-2615) (Professor of Medical Statistics) obtained funding, designed the trial and interpreted and analysed the data.
Professor Barnaby C Reeves (https://orcid.org/0000-0002-5101-9487) (Professor of Health Services Research) obtained funding, designed the trial, managed the trial, interpreted the data and co-authored the first draft of the report.
Publications
Willcox A, Culliford L, Ellis L, Rogers CA, Cree A, Chakravarthy U, et al. Clinical efficacy of eplerenone versus placebo for central serous chorioretinopathy: study protocol for the VICI randomised controlled trial. Eye 2019;33:295–303.
Lotery A, Sivaprasad S, O’Connell A, Harris RA, Culliford L, Ellis L, et al. Eplerenone for chronic central serous chorioretinopathy in patients with active, previously untreated disease for more than 4 months (VICI): a randomised, double-blind, placebo-controlled trial. Lancet 2020;395:294–303.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to anonymised data may be granted following review.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, NETSCC, the EME programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the EME programme or the Department of Health and Social Care.
References
- Daruich A, Matet A, Behar-Cohen F. Central serous chorioretinopathy. Dev Ophthalmol 2017;58:27-38. https://doi.org/10.1159/000455267.
- Daruich A, Matet A, Dirani A, Bousquet E, Zhao M, Farman N, et al. Central serous chorioretinopathy: recent findings and new physiopathology hypothesis. Prog Retin Eye Res 2015;48:82-118. https://doi.org/10.1016/j.preteyeres.2015.05.003.
- Wang M, Munch IC, Hasler PW, Prunte C, Larsen M. Central serous chorioretinopathy. Acta Ophthalmol 2008;86:126-45. https://doi.org/10.1111/j.1600-0420.2007.00889.x.
- Gemenetzi M, De Salvo G, Lotery AJ. Central serous chorioretinopathy: an update on pathogenesis and treatment. Eye 2010;24:1743-56. https://doi.org/10.1038/eye.2010.130.
- Loo RH, Scott IU, Flynn HW, Gass JD, Murray TG, Lewis ML, et al. Factors associated with reduced visual acuity during long-term follow-up of patients with idiopathic central serous chorioretinopathy. Retina 2002;22:19-24. https://doi.org/10.1097/00006982-200202000-00004.
- Yoshioka H, Katsume Y. Experimental central serous chorioretinopathy. III: ultrastructural findings. Jpn J Ophthalmol 1982;26:397-409.
- Nicholson BP, Atchison E, Idris AA, Bakri SJ. Central serous chorioretinopathy and glucocorticoids: an update on evidence for association. Surv Ophthalmol 2018;63:1-8. https://doi.org/10.1016/j.survophthal.2017.06.008.
- Schubert C, Pryds A, Zeng S, Xie Y, Freund KB, Spaide RF, et al. Cadherin 5 is regulated by corticosteroids and associated with central serous chorioretinopathy. Hum Mutat 2014;35:859-67. https://doi.org/10.1002/humu.22551.
- Schellevis RL, van Dijk EHC, Breukink MB, Altay L, Bakker B, Koeleman BPC, et al. Role of the complement system in chronic central serous chorioretinopathy: a genome-wide association study. JAMA Ophthalmol 2018;136:1128-36. https://doi.org/10.1001/jamaophthalmol.2018.3190.
- Kaye RA, Cree AJ, Lotery AJ. A genome-wide complement for central serous chorioretinopathy. JAMA Ophthalmol 2018;136:1136-7. https://doi.org/10.1001/jamaophthalmol.2018.3199.
- Chan WM, Lai TY, Lai RY, Liu DT, Lam DS. Half-dose verteporfin photodynamic therapy for acute central serous chorioretinopathy: one-year results of a randomized controlled trial. Ophthalmology 2008;115:1756-65. https://doi.org/10.1016/j.ophtha.2008.04.014.
- van Dijk EHC, Fauser S, Breukink MB, Blanco-Garavito R, Groenewoud JMM, Keunen JEE, et al. Half-dose photodynamic therapy versus high-density subthreshold micropulse laser treatment in patients with chronic central serous chorioretinopathy: the PLACE trial. Ophthalmology 2018;125:1547-55. https://doi.org/10.1016/j.ophtha.2018.04.021.
- Gawęcki M. Micropulse laser treatment of retinal diseases. J Clin Med 2019;8. https://doi.org/10.3390/jcm8020242.
- Salehi M, Wenick AS, Law HA, Evans JR, Gehlbach P. Interventions for central serous chorioretinopathy: a network meta-analysis. Cochrane Database Syst Rev 2015;12. https://doi.org/10.1002/14651858.CD011841.pub2.
- Chung YR, Seo EJ, Lew HM, Lee KH. Lack of positive effect of intravitreal bevacizumab in central serous chorioretinopathy: meta-analysis and review. Eye 2013;27:1339-46. https://doi.org/10.1038/eye.2013.236.
- Zhao M, Célérier I, Bousquet E, Jeanny JC, Jonet L, Savoldelli M, et al. Mineralocorticoid receptor is involved in rat and human ocular chorioretinopathy. J Clin Invest 2012;122:2672-9. https://doi.org/10.1172/JCI61427.
- Bousquet E, Beydoun T, Zhao M, Hassan L, Offret O, Behar-Cohen F. Mineralocorticoid receptor antagonism in the treatment of chronic central serous chorioretinopathy: a pilot study. Retina 2013;33:2096-102. https://doi.org/10.1097/IAE.0b013e318297a07a.
- Schwartz R, Habot-Wilner Z, Martinez MR, Nutman A, Goldenberg D, Cohen S, et al. Eplerenone for chronic central serous chorioretinopathy-a randomized controlled prospective study. Acta Ophthalmol 2017;95:e610-e618. https://doi.org/10.1111/aos.13491.
- Pichi F, Carrai P, Ciardella A, Behar-Cohen F, Nucci P. Central Serous Chorioretinopathy Study Group . Comparison of two mineral corticosteroids receptor antagonists for the treatment of central serous chorioretinopathy. Int Ophthalmol 2017;37:1115-25. https://doi.org/10.1007/s10792-016-0377-2.
- Rahimy E, Pitcher JD, Hsu J, Adam MK, Shahlaee A, Samara WA, et al. A randomized double-blind placebo-control pilot study of eplerenone for the treatment of central serous chorioretinopathy (ecselsior). Retina 2017;38:962-9. https://doi.org/10.1097/IAE.0000000000001649.
- National Institute for Health Research (NIHR) . Clinical Efficacy and Mechanistic Evaluation of Eplerenone for Central Serous Chorioretinopathy the VICI Study 2020. www.journalslibrary.nihr.ac.uk/programmes/eme/139415/#/ (accessed 29 April 2020).
- Willcox A, Culliford L, Ellis L, Rogers CA, Cree A, Chakravarthy U, et al. Clinical efficacy of eplerenone versus placebo for central serous chorioretinopathy: study protocol for the VICI randomised controlled trial. Eye 2019;33:295-303. https://doi.org/10.1038/s41433-018-0212-2.
- Wang SK, Sun P, Tandias RM, Seto BK, Arroyo JG. Mineralocorticoid receptor antagonists in central serous chorioretinopathy: a meta-analysis of randomized controlled trials. Ophthalmol Retina 2019;3:154-60. https://doi.org/10.1016/j.oret.2018.09.003.
- RAND Health Care . Visual Function Questionnaire (VFQ-25) n.d. www.rand.org/health/surveys_tools/vfq.html (accessed 29 April 2020).
- Hutton D, Smith N, Cappel-Porter H, Saw C, Rogers CA. Development of the ‘GeneSYS’ database system to support trial data capture and conduct. Trials 2013;14. https://doi.org/10.1186/1745-6215-14-S1-P66.
- Sterne JAC, Savović J, Page MJ, Elbers RG, Blencowe NS, Boutron I, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ 2019;366. https://doi.org/10.1136/bmj.l4898.
- Bousquet E, Beydoun T, Rothschild PR, Bergin C, Zhao M, Batista R, et al. Spironolactone for nonresolving central serous chorioretinopathy: a randomized controlled crossover study. Retina 2015;35:2505-15. https://doi.org/10.1097/IAE.0000000000000614.
- Sun X, Shuai Y, Fang W, Li J, Ge W, Yuan S, et al. Spironolactone versus observation in the treatment of acute central serous chorioretinopathy. Br J Ophthalmol 2018;102:1060-5. https://doi.org/10.1136/bjophthalmol-2017-311096.
- Group CR, Martin DF, Maguire MG, Ying GS, Grunwald JE, Fine SL, et al. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N Engl J Med 2011;364:1897-908. https://doi.org/10.1056/NEJMoa1102673.
- Lotery A, Sivaprasad S, O’Connell A, Harris RA, Culliford L, Ellis L, et al. Eplerenone for chronic central serous chorioretinopathy in patients with active, previously untreated disease for more than 4 months (VICI): a randomised, double-blind, placebo-controlled trial. Lancet 2020;395:294-303. https://doi.org/10.1016/S0140-6736(19)32981-2.
- Chakravarthy U, Harding SP, Rogers CA, Downes SM, Lotery AJ, Culliford LA, et al. IVAN study investigators . Alternative treatments to inhibit VEGF in age-related choroidal neovascularisation: 2-year findings of the IVAN randomised controlled trial. Lancet 2013;382:1258-67. https://doi.org/10.1016/S0140-6736(13)61501-9.
- Frison L, Pocock SJ. Repeated measures in clinical trials: analysis using mean summary statistics and its implications for design. Stat Med 1992;11:1685-704. https://doi.org/10.1002/sim.4780111304.
- Sivaprasad S, Prevost AT, Vasconcelos JC, Riddell A, Murphy C, Kelly J, et al. Clinical efficacy of intravitreal aflibercept versus panretinal photocoagulation for best corrected visual acuity in patients with proliferative diabetic retinopathy at 52 weeks (CLARITY): a multicentre, single-blinded, randomised, controlled, phase 2b, non-inferiority trial. Lancet 2017;389:2193-203. https://doi.org/10.1016/S0140-6736(17)31193-5.
- Reeves BC, Harding SP, Langham J, Grieve R, Tomlin K, Walker J, et al. Verteporfin photodynamic therapy for neovascular age-related macular degeneration: cohort study for the UK. Health Technol Assess 2012;16. https://doi.org/10.3310/hta16060.
Appendix 1 The VICI trial investigators
| Location | Investigator(s) |
|---|---|
| Centre | |
| University of Southampton, Southampton, UK | Andrew Lotery, Angela Cree, Helen Griffiths |
| BTC-CTEU, University of Bristol, Bristol, UK | Barnaby C Reeves, Chris A Rogers, Abby O’Connell, Rosie A Harris, Lucy Ellis, Lucy Culliford, Samir Bellani |
| Network of Ophthalmic Reading Centres, Liverpool, UK | Savita Madhusudhan |
| University of Lausanne, Lausanne, Switzerland | Francine Behar-Cohen |
| Site | |
| University Hospital Southampton NHS Foundation Trust, Southampton, UK | Andrew Lotery, Suresh Thulasidharan, Catrin Watkins, Thea Sass, Rebecca Kaye |
| NIHR Moorfields Biomedical Research Centre, Moorfields Eye Hospital NHS Foundation Trust, London, UK | Sobha Sivaprasad, Deepthy Menon, Qin Neville |
| Centre for Vision Sciences, Belfast Health and Social Care Trust, Queen’s University of Belfast, UK | Tunde Peto, Usha Chakravarthy, Rebecca Denham, Karen Gillvray |
| Ophthalmology Department, East Lancashire Hospitals NHS Trust, Blackburn, UK | Salwa Abugreen, Natalie Nixon, Mohammed Alarbi |
| Ophthalmology, Bradford Teaching Hospitals NHS Foundation Trust, Bradford, UK | Faruque Ghanchi, Zeid Madanat, Nicola Hawes |
| Sussex Eye Hospital, Brighton and Sussex University Hospitals NHS Trust, Brighton, UK | Edward Hughes, Campbell Keir, Krystian Kisza |
| Bristol Eye Hospital, University Hospital Bristol NHS Foundation Trust, Bristol, UK | Clare Bailey, Phillippa Hazlewood, Julie Cloake |
| Eye Department, Frimley Health NHS Foundation Trust, Frimley, UK | Geeta Menon, Manju Chandran, Abigail Raguro |
| Department of Ophthalmology, Guy’s and St Thomas’ NHS Foundation Trust, London, UK | Moin Mohamed, Wei Sing Lim |
| Laser and Retina Research Unit, King’s College Hospital, Kings’ College Hospital NHS Foundation Trust, London, UK | Haralabos Eleftheriadis, Stefanos Efraimidis |
| Leeds Teaching Hospitals NHS Trust, Leeds, UK | Martin McKibbin, Raj Mukherjee, Joanne Wilson |
| St Paul’s Eye Unit, Royal Liverpool and Broadgreen NHS Trust, Liverpool, UK | Pauline Lenfestey, Simon Harding, Kelly Haigh |
| Manchester Royal Eye Hospital, Manchester University NHS Foundation Trust, Manchester, UK | Ramandeep Chhabra, Mania Horani, Raisa-Marie Platt |
| Newcastle upon Tyne Hospitals NHS Foundation Trust, Newcastle upon Tyne, UK | James Talks, Devanga Bhatia, Violet Andrews |
| Oxford Eye Hospital, Oxford University Hospitals NHS Foundation Trust, Oxford, UK | Susan Downes, Ivy Samuel, Daniel Buttress |
| Department of Ophthalmology, University Hospitals Coventry and Warwickshire, Coventry, UK | Sergio Pagliarini, Linzi Randle, Jeanette Allison |
| Sheffield Teaching Hospitals NHS Foundation Trust, Sheffield, UK | Christopher Brand, Maria Edwards |
| Eye Unit, Southend University Hospital NHS Foundation Trust, Southend, UK | Niral Karia, Maria Shipman, Elridge Thompson |
| Sunderland Eye Infirmary, City Hospitals Sunderland NHS Foundation Trust, Sunderland, UK | Ajay Kotagiri, David Steel, Steven Dodds |
| Ophthalmology Department, Torbay and South Devon NHS Foundation Trust, Torbay, UK | Stephen Turner, Yinka Osoba, Sharon Criddle |
| New Cross Hospital, Royal Wolverhampton NHS Trust, Wolverhampton, UK | Yit Yang, Niro Narendran, Meena Karpoor |
| Department of Ophthalmology, York Teaching Hospital NHS Foundation Trust, York, UK | Richard Gale, Archana Airody, Alison Grice-Holt |
Appendix 2 Trial Oversight Committee members
Trial Steering Committee
(Chairperson: September 2016 to April 2017) Professor Susan Downes, Consultant Ophthalmologist, Oxford Eye Hospital, Oxford, UK.
(Chairperson: April 2017 to trial end) Mr Ben Burton, Clinical Director of Research, James Paget University Hospital, Great Yarmouth, UK.
Dr Gabriella Czanner, Lecturer in Ophthalmic Statistics, University of Liverpool, Liverpool, UK.
Dr Dolores Conroy, Director of Research, Fight for Sight, London, UK.
Professor Theresa McDonagh, Independent Professor of Heart Failure and Consultant Cardiologist, King’s College Hospital, London, UK.
Mrs Blanche Morrissey, Patient Representative, Buckinghamshire, UK.
Miss Veronica Rossi, Patient Representative, London, UK.
Mr Mandeep Bindra, Consultant Ophthalmologist and Vitreo-retinal Surgeon, Stoke Mandeville Hospital, Aylesbury, UK.
Mr Quresh Mohammed, Consultant Ophthalmologist, Cheltenham General Hospital, Cheltenham, UK.
Data Monitoring and Safety Committee
(Chairperson: September 2016 to September 2018) Professor Toby Prevost, Professor of Medical Statistics, Imperial College London, London, UK.
(Chairperson: September 2018 to end of trial) Mr Mark Dayer, Consultant Cardiologist, Musgrove Park Hospital, Taunton, UK.
Professor Simon Taylor, Professor of Ophthalmology, Royal Surrey County Hospital, Guildford, UK.
Professor Baljean Dhillon, Professor of Clinical Ophthalmology, University of Edinburgh, Edinburgh, UK.
Appendix 3 Retinal image capture specifications
| OCT | Image specifications per eye |
|---|---|
| 1 horizontal line scan | High resolution; ART 100 frames |
| 1 vertical line scan | High resolution; ART 100 frames |
| 1 volume scan | 31 sections; 30° × 25°; ART 15 frames; HS |
| 1 EDI volume scan | 19 sections; 30° × 15°; ART 25 frames; HS |
| 1 pre-set 7 lines |
| AF imaging | Specifications per eye |
|---|---|
| 1 AF image field 1 (disc centred) | High resolution; 55° lens; ART 30 frames |
| 1 AF image field 2 (macula centred) | High resolution; 55° lens; ART 30 frames |
| Time point | FFA and ICGA images captured |
|---|---|
| Prior to injection | Red-free, fellow eye, then study eye |
| Injection | Timer started when fluorescein is injected rapidly (< 5 seconds). One field 2 image of the study eye was taken at the beginning of the injection and one at the end |
| Arterial filling phase | A minimum of eight field 2 images of the study eye as soon as the retinal arteries start to fill with the dye and throughout the first 45 seconds |
| 45 seconds to 2 minutes | Field 1, field 2, field 3, field 4 and field 5 images of the study eye |
| 2–3 minutes | Field 1, field 2, field 3, field 4 and field 5 images of the fellow eye |
| 5–7 minutes | Field 1 and field 2 images of the study eye; field 1 and field 2 images of the fellow eye |
| 10–11 minutes | Field 1 and field 2 images of the study eye; field 1 and field 2 images of the fellow eye |
| CFP | Specifications |
|---|---|
| Calibration image | Mono; whole disc and macula in view |
| Fundus reflex | Mono; the rest should be captured in stereo |
| Optic disc | Stereo; centre the temporal edge of the optic disc at the intersection of the ocular crosshairs |
| Macula | Stereo; shift the camera towards the temporal area from field 1M without vertical adjustment. The ocular crosshairs should be positioned approximately 1/8–1/4 disc diameter above the centre of the fovea |
| Fundus reflex (anterior segment) | Taken to document media opacities. To obtain the largest possible fundus reflex image the photographer should turn the focusing knob all the way forward and then adjust focus by manually moving the camera closer or further away from the patient |
Appendix 4 Additional results tables and figures
| Site | Months open (n) | Screened (n) | Consented (n) | Randomised (n) | Withdrawn from treatment (n) | Withdrawn from follow-up (n) |
|---|---|---|---|---|---|---|
| Moorfields Eye Hospital NHS Foundation Trust | 14 | 30 | 22 | 19 | 0 | 0 |
| University Hospital Southampton NHS Foundation Trust | 14 | 33 | 19 | 8 | 3 | 1 |
| Manchester University NHS Foundation Trust | 14 | 19 | 8 | 7 | 3 | 1 |
| East Lancashire Hospitals NHS Trust | 13 | 13 | 5 | 1 | 0 | 0 |
| Torbay and South Devon NHS Foundation Trust | 13 | 26 | 9 | 5 | 1 | 0 |
| Leeds Teaching Hospitals NHS Trust | 13 | 15 | 10 | 8 | 5 | 0 |
| York Teaching Hospital NHS Foundation Trust | 12 | 12 | 4 | 1 | 0 | 0 |
| Bradford Teaching Hospitals NHS Foundation Trust | 12 | 14 | 4 | 4 | 0 | 0 |
| Sheffield Teaching Hospitals NHS Foundation Trust | 12 | 19 | 4 | 2 | 1 | 0 |
| City Hospitals Sunderland NHS Foundation Trust | 12 | 18 | 5 | 3 | 0 | 0 |
| Brighton and Sussex University Hospitals NHS Trust | 11 | 7 | 7 | 5 | 1 | 0 |
| Royal Liverpool and Broadgreen University Hospitals NHS Trust | 11 | 14 | 5 | 2 | 1 | 0 |
| University Hospitals Bristol NHS Foundation Trust | 11 | 12 | 8 | 6 | 2 | 2 |
| Newcastle upon Tyne Hospitals NHS Foundation Trust | 11 | 26 | 7 | 7 | 1 | 1 |
| Belfast Health and Social Care Trust | 10 | 53 | 8 | 6 | 3 | 2 |
| Frimley Health NHS Foundation Trust | 10 | 14 | 12 | 5 | 1 | 1 |
| Southend University Hospital NHS Foundation Trust | 8 | 20 | 6 | 6 | 0 | 0 |
| University Hospitals Coventry and Warwickshire NHS Trust | 8 | 20 | 7 | 6 | 3 | 0 |
| Guy’s and St Thomas’ NHS Foundation Trust | 7 | 4 | 3 | 1 | 0 | 0 |
| King’s College Hospital NHS Foundation Trust | 7 | 2 | 2 | 1 | 0 | 0 |
| Oxford University Hospitals NHS Foundation Trust | 7 | 25 | 8 | 6 | 0 | 0 |
| Royal Wolverhampton NHS Trust | 4 | 6 | 6 | 5 | 1 | 0 |
| Total | 234 | 402 | 169 | 114 | 26 | 8 |
FIGURE 21.
Time to stopping IMP for the first time.
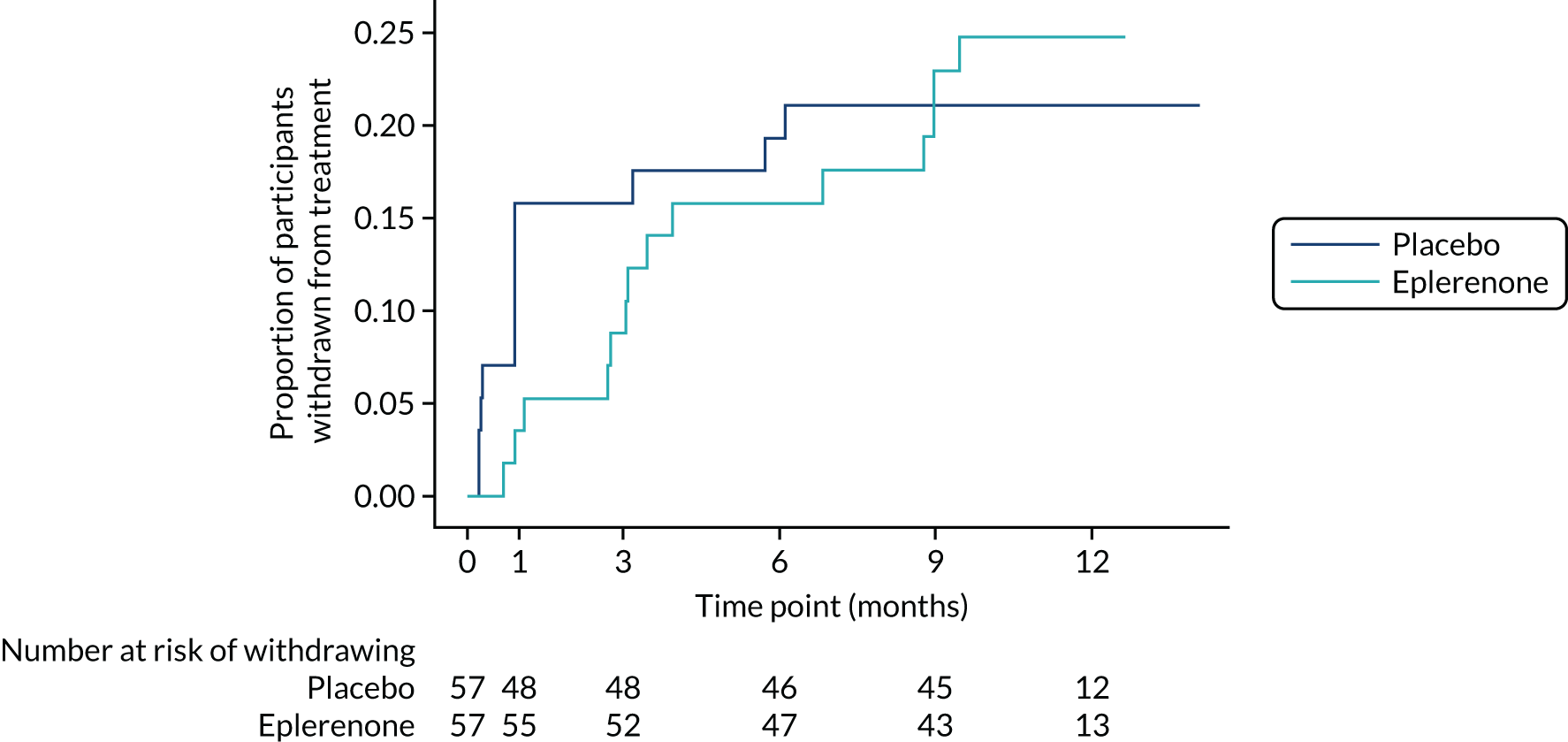
FIGURE 22.
Distributions of follow-up visit attendances around specified visit windows. (a) Week 1 visit; (b) week 4 visit; (c) month 3 visit; (d) month 6 visit; (e) month 9 visit; and (f) month 12 visit. Vertical dashed lines represent visit windows (week 1 ± 1 day; week 4 ± 5 days; all other follow-up visits ± 10 days). Green bars represent visits that occurred inside the window; dark blue bars represent visits that occurred outside the window. Mean time to visit by group: 1 week – placebo, 8 days (SD 2.4 days); eplerenone, 8 days (SD 2.9 days); 4 weeks – placebo, 29 days (SD 4.0 days); eplerenone, 30 days (SD 4.0 days); 3 months – placebo, 91 days (SD 6.8 days); eplerenone, 92 days (SD 9.5 days); 6 months – placebo, 180 days (SD 7.5 days); eplerenone, 182 days (SD 6.7 days); 9 months – placebo, 271 days (SD 7.9 days); eplerenone, 275 days (SD 9.5 days); 12 months – placebo, 363 days (SD 14.5 days); eplerenone, 366 days (SD 9.9 days). Reproduced with permission from Lotery et al. 30 This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) license, which permits others to copy and redistribute the material in any medium or format, for non-commercial use, with no derivatives, provided the original work is properly cited. See: https://creativecommons.org/licenses/by-nc-nd/4.0/. The figure includes formatting changes to the original.
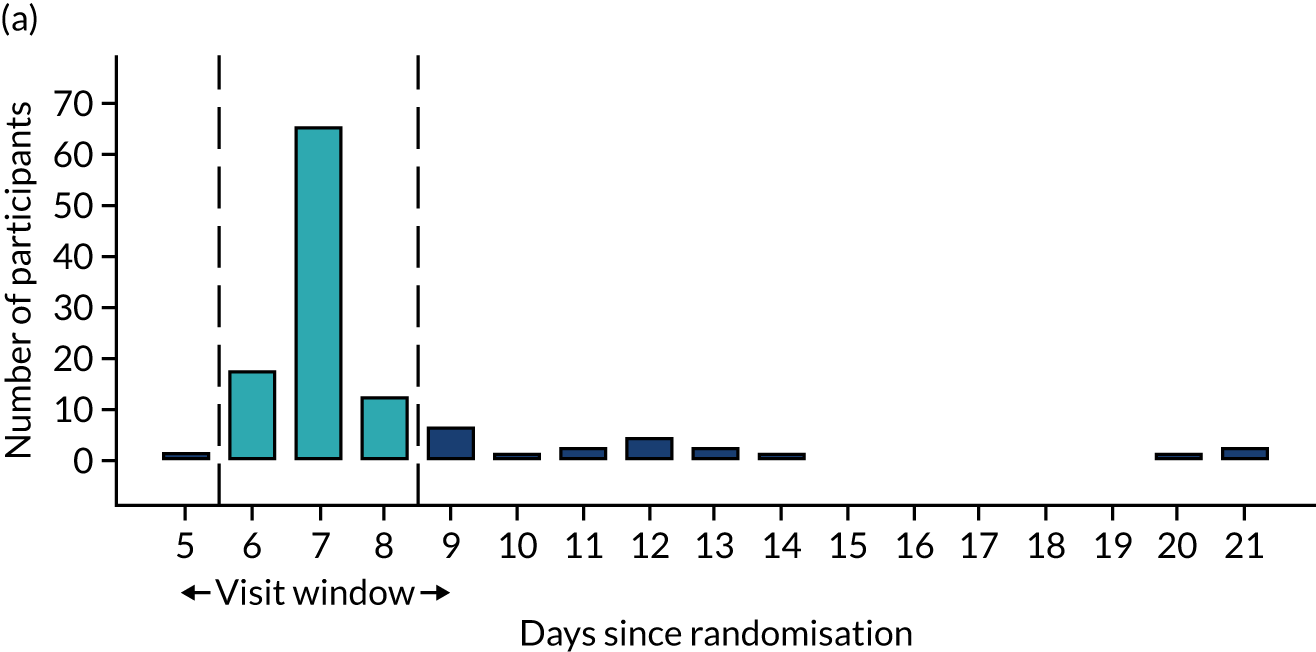
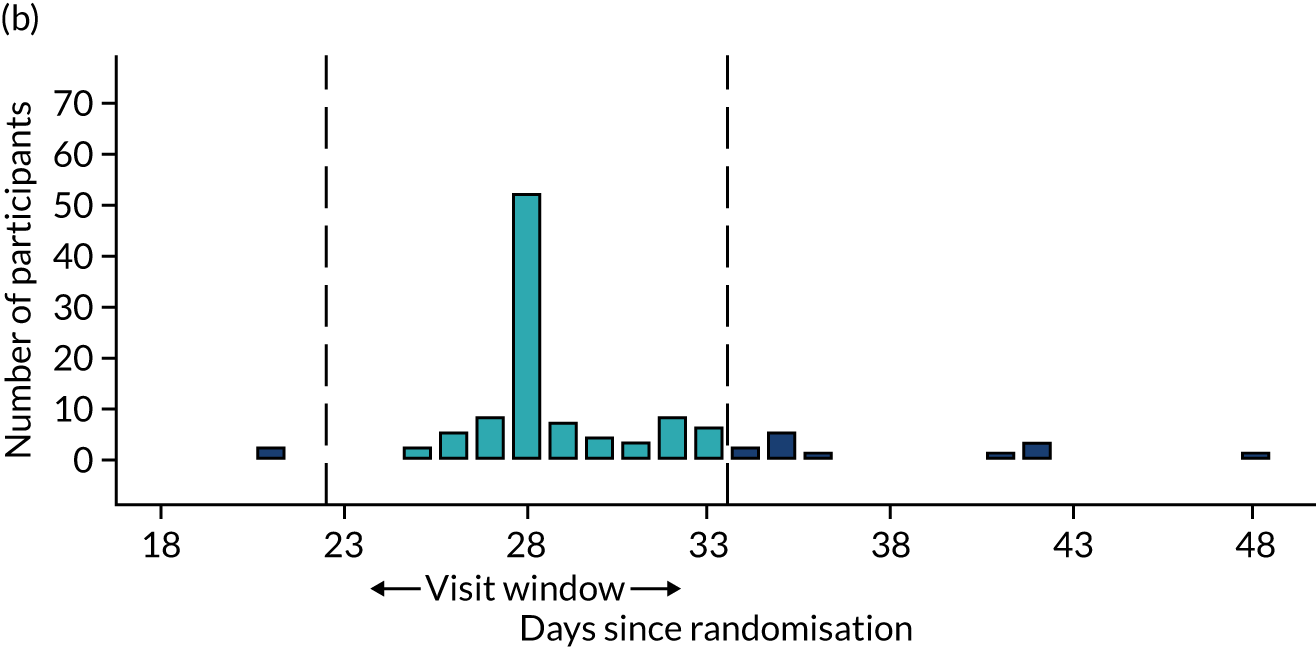
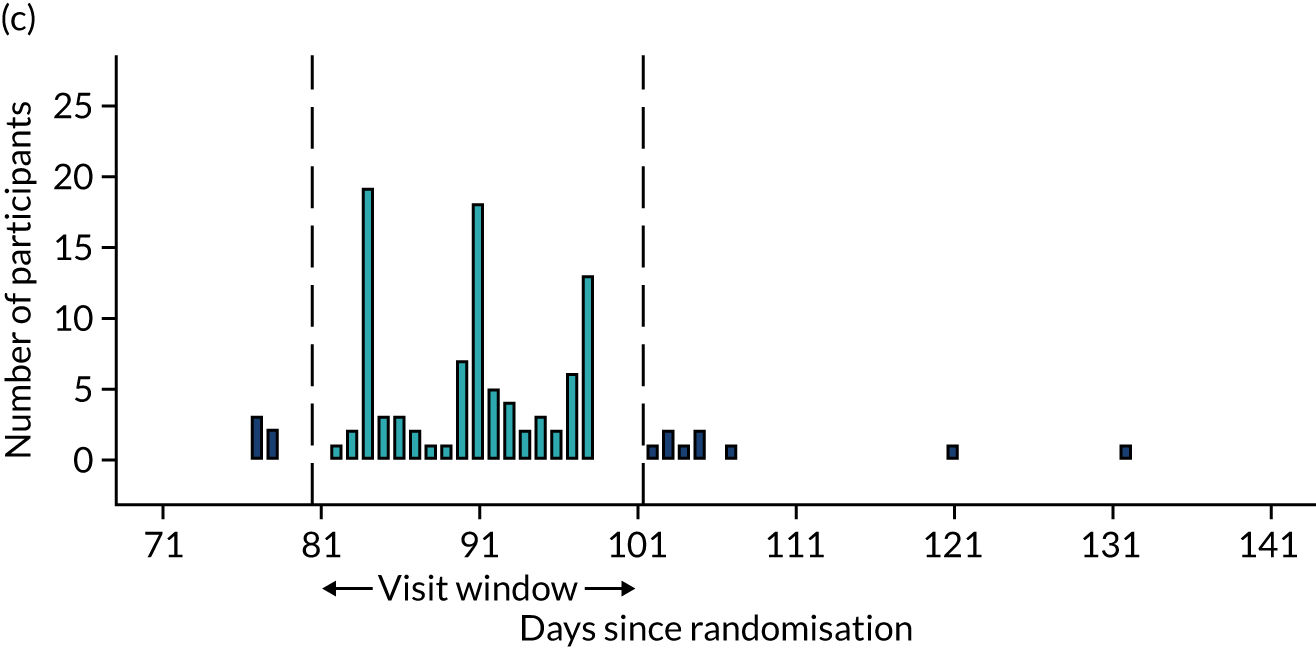
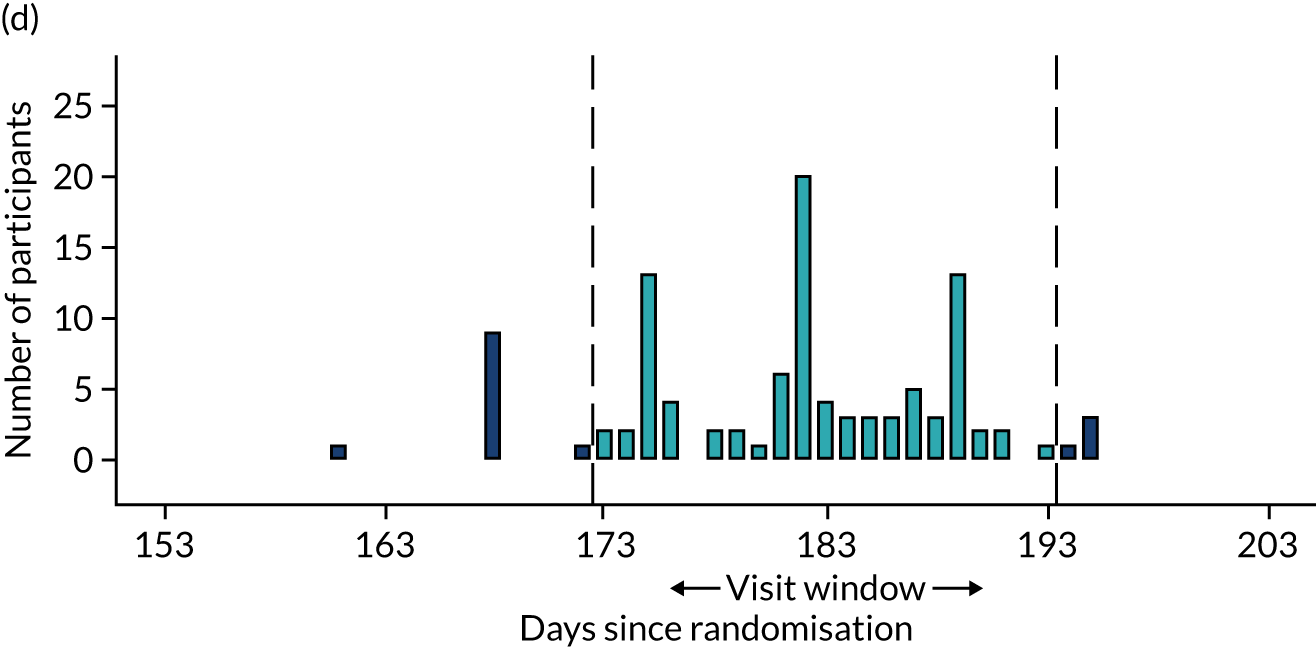
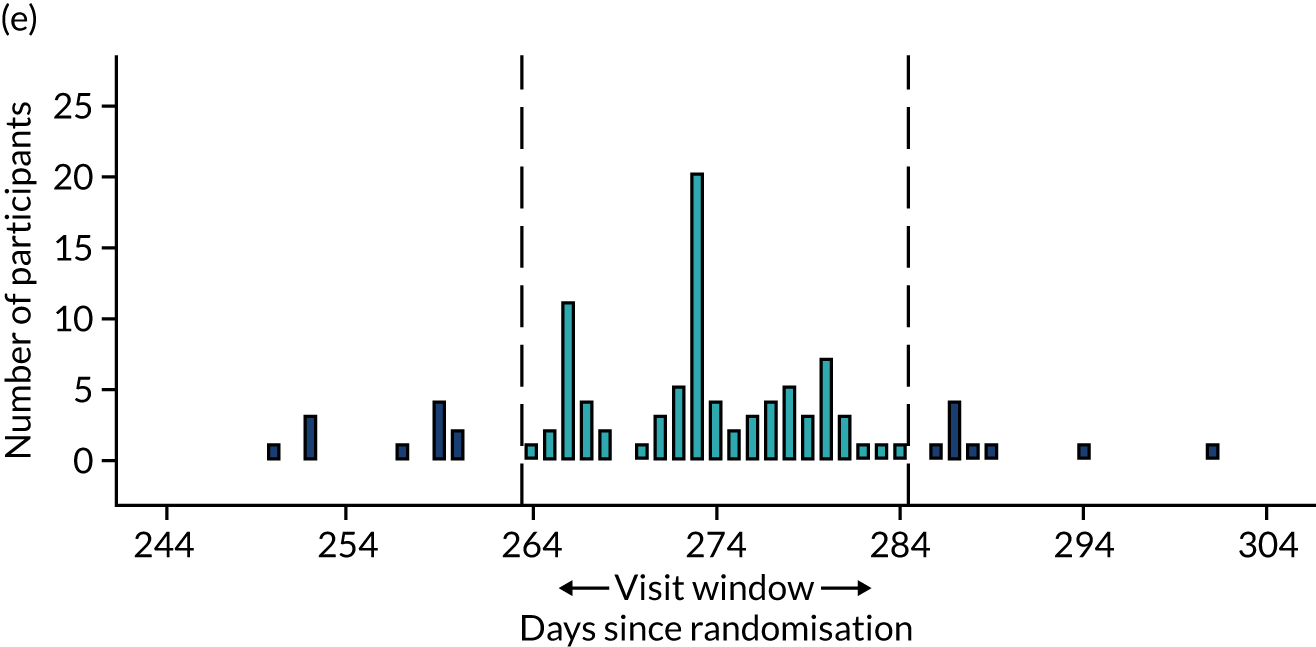
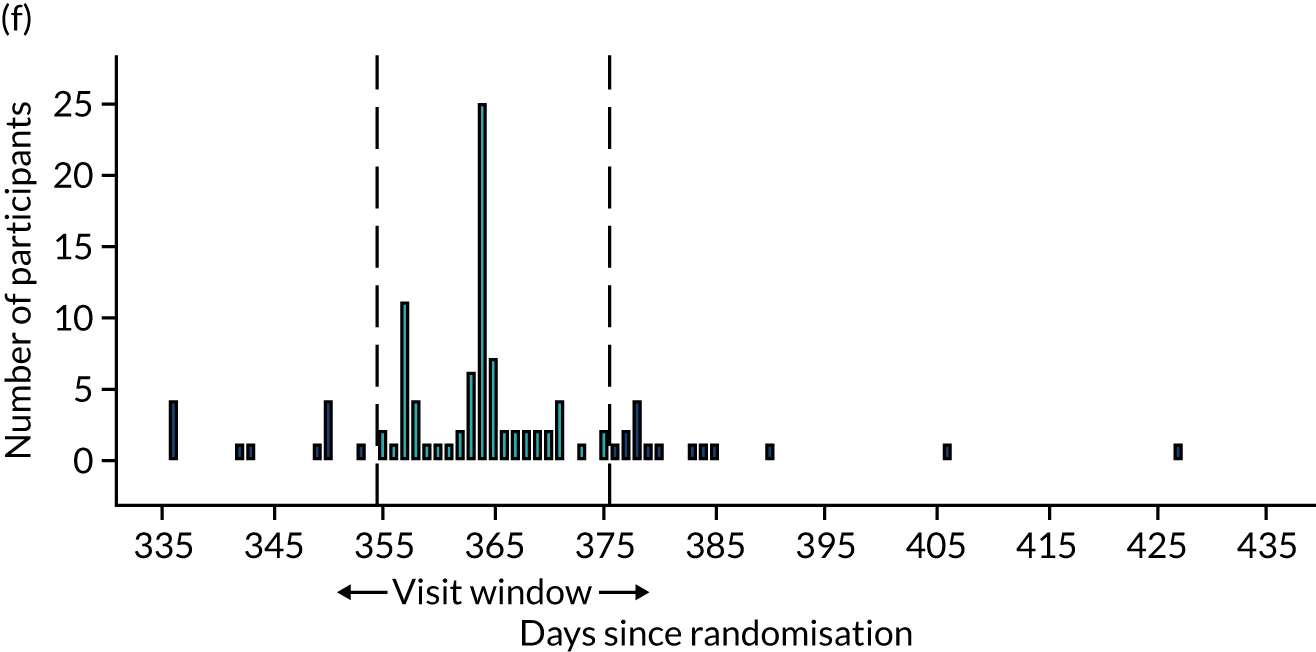
| Time point | Randomised to placebo, n/N (%) | Randomised to eplerenone, n/N (%) |
|---|---|---|
| Baseline | 2/57 (3.5) | 3/57 (5.3) |
| 1 week | 2/52 (3.8) | 3/56 (5.4) |
| 4 weeks | 20/92 (21.7) | 18/94 (19.1) |
| 3 months | 13/111 (11.7) | 7/117 (6) |
| 6 months | 5/94 (5.3) | 12/106 (11.3) |
| 9 months | 2/72 (2.8) | 8/78 (10.3) |
| Restarting | 0/5 (0) | 0/14 (0) |
| Restarting week 1 | 0/7 (0) | 0/14 (0) |
| Restarting week 4 | 2/7 (28.6) | 6/23 (26.1) |
| Outcome | Primary analysis | Analysis adjusted for PDT | ||
|---|---|---|---|---|
| MD (95% CI) | p-value | MD (95% CI) | p-value | |
| BCVA score | 1.73 (–1.12 to 4.57) | 0.236 | 1.63 (–1.23 to 4.48) | 0.266 |
FIGURE 23.
Exploratory analyses of the effects of age and confluent/granular hyperautofluorescence in the overall trial cohort.
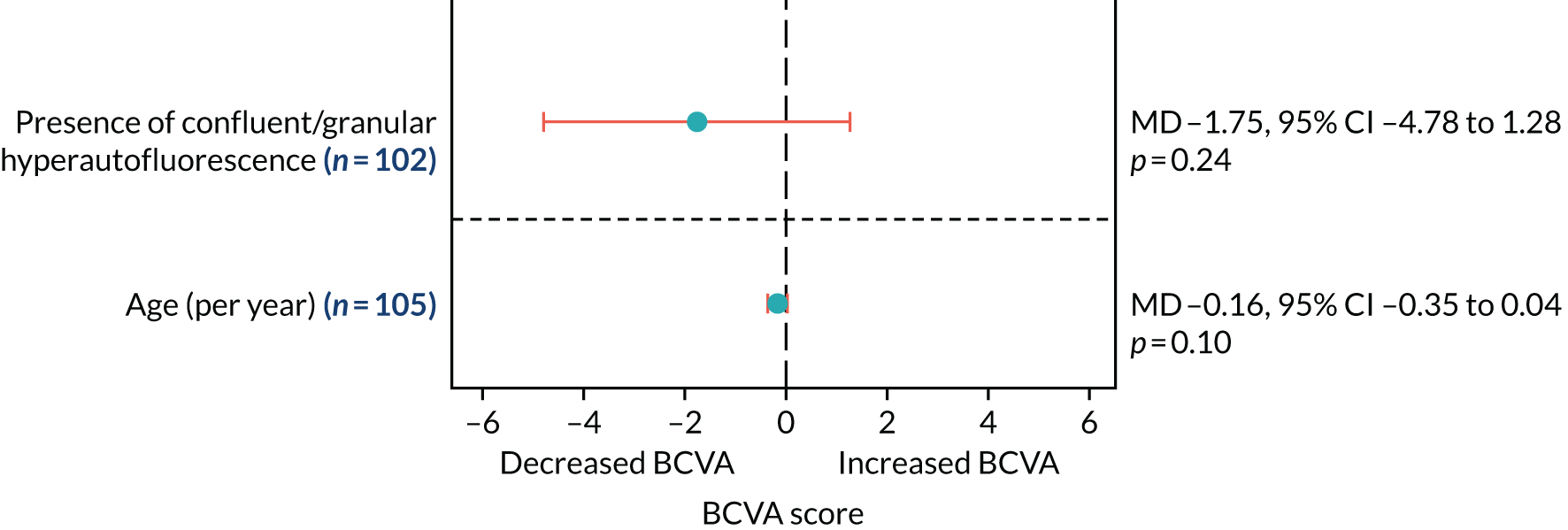
| Time point | Randomised to placebo (n = 57), median (IQR) | Randomised to eplerenone (n = 57), median (IQR) | Effect, MD (95% CI) | p-value |
|---|---|---|---|---|
| Baselinea | 64 (57.0–67.0) | 57 (50.0–64.0) | ||
| 4 weeksb | 63 (55.0–67.0) | 63 (54.0–67.5) | ||
| 3 monthsc | 62 (58.0–69.0) | 63 (57.0–68.0) | ||
| 6 monthsc | 66 (59.0–71.0) | 62 (56.0–71.5) | ||
| 9 monthsd | 65 (59.5–73.0) | 65 (58.0–70.0) | ||
| 12 monthse | 65 (60.0–75.0) | 66 (57.0–71.0) | 0.61 (–3.79 to 5.02) | 0.785 |
| Test for treatment × time interaction | 0.944 |
| Variable | Visit | Randomised to placebo (n = 57), median (IQR) | Randomised to eplerenone (n = 57), median (IQR) | Effect, MD (95% CI)a | p-value |
|---|---|---|---|---|---|
| Total score | Baselineb | 87 (80.3–91.3) | 89 (81.2–92.0) | ||
| 12 monthsc | 92 (86.1–94.6) | 89 (83.7–93.3) | –2.39 (–5.45 to 0.68) | 0.127 | |
| Subscale | |||||
| Near vision | Baselineb | 75 (66.7–83.3) | 83 (66.7–91.7) | ||
| 12 monthsc | 83 (75.0–100.0) | 83 (66.7–91.7) | |||
| Distance vision | Baselineb | 92 (83.3–100.0) | 100 (83.3–100.0) | ||
| 12 monthsc | 92 (83.3–100.0) | 100 (83.3–100.0) | |||
| General health | Baselineb | 75 (50.0–75.0) | 75 (50.0–75.0) | ||
| 12 monthsc | 75 (50.0–75.0) | 75 (50.0–75.0) | |||
| General vision | Baselineb | 70 (60.0–80.0) | 60 (60.0–80.0) | ||
| 12 monthsc | 80 (60.0–80.0) | 80 (60.0–80.0) | |||
| Driving | Baselined | 92 (83.3–100.0) | 92 (83.3–100.0) | ||
| 12 monthse | 92 (83.3–100.0) | 92 (83.3–100.0) | |||
| Peripheral vision | Baselineb | 100 (75.0–100.0) | 100 (75.0–100.0) | ||
| 12 monthsc | 100 (100.0–100.0) | 100 (75.0–100.0) | |||
| Colour vision | Baselineb | 100 (100.0–100.0) | 100 (100.0–100.0) | ||
| 12 monthsc | 100 (100.0–100.0) | 100 (100.0–100.0) | |||
| Ocular pain | Baselineb | 88 (75.0–100.0) | 88 (75.0–100.0) | ||
| 12 monthsc | 100 (75.0–100.0) | 88 (75.0–100.0) | |||
| Vision specific | |||||
| Role difficulties | Baselineb | 88 (75.0–100.0) | 88 (62.5–100.0) | ||
| 12 monthsc | 100 (75.0–100.0) | 88 (62.5–100.0) | |||
| Dependency | Baselineb | 100 (91.7–100.0) | 100 (91.7–100.0) | ||
| 12 monthsc | 100 (100.0–100.0) | 100 (100.0–100.0) | |||
| Social functioning | Baselineb | 100 (100.0–100.0) | 100 (100.0–100.0) | ||
| 12 monthsc | 100 (100.0–100.0) | 100 (100.0–100.0) | |||
| Mental health | Baselineb | 75 (62.5–87.5) | 75 (56.3–87.5) | ||
| 12 monthsc | 88 (75.0–93.8) | 81 (75.0–87.5) | |||
| Outcome | Primary analysis | Analysis adjusted for baseline SRF | ||
|---|---|---|---|---|
| HR (95% CI) | p-value | HR (95% CI) | p-value | |
| Time to complete resolution of SRF | 0.78 (0.41 to 1.51) | 0.46 | 0.79 (0.38 to 1.64) | 0.53 |
| Time to complete or partial resolution of SRF | 1.23 (0.75 to 2.00) | 0.42 | 1.08 (0.72 to 1.62) | 0.70 |
| Time point | Randomised to placebo (n = 57), median (IQR) | Randomised to eplerenone (n = 57), median (IQR) | Effect, MD (95% CI) | p-value |
|---|---|---|---|---|
| Baseline | 322 (280.0–394.0) | 360 (290.0–406.0) | ||
| 4 weeks | 330 (272.0–386.0) | 328 (248.5–393.5) | ||
| 3 months | 285 (250.0–341.0) | 295 (240.5–383.0) | ||
| 6 months | 270 (247.0–313.0) | 290 (226.0–366.0) | ||
| 9 months | 268 (230.0–322.0) | 273 (220.0–366.0) | ||
| 12 months | 253 (232.0–303.0) | 272 (229.0–368.0) | 24.35 (–7.86 to 56.56) | 0.142 |
| Test for treatment × time interaction | 0.585 |
| Outcome | Primary analysis | Analysis adjusted for PDT | ||
|---|---|---|---|---|
| MD (95% CI) | p-value | MD (95% CI) | p-value | |
| CSRT (µm) at 12 months | 24.35 (–7.86 to 56.56) | 0.142 | 23.23 (–9.11 to 55.58) | 0.163 |
| Choroidal thickness (µm) | 38.53 (12.31 to 64.74) | 0.004 | 36.69 (9.91 to 63.47) | 0.007 |
| SRFT (µm) | 48.14 (13.51 to 82.78) | 0.007 | 45.88 (10.67 to 81.09) | 0.011 |
List of abbreviations
- AE
- adverse event
- AF
- autofluorescence
- BCVA
- best corrected visual acuity
- BTC-CTEU
- Bristol Trials Centre Clinical Trials and Evaluation Unit
- CFP
- colour fundus photography
- CI
- confidence interval
- CMT
- central macular thickness
- CNV
- choroidal neovascularisation
- CRF
- case report form
- CSCR
- central serous chorioretinopathy
- CSRT
- central subfield retinal thickness
- DMSC
- Data Monitoring and Steering Committee
- EDI
- enhanced depth imaging
- eGFR
- estimated glomerular filtration rate
- ETDRS
- Early Treatment Diabetic Retinopathy Study
- FFA
- fundus fluorescein angiography
- HR
- hazard ratio
- ICGA
- indocyanine green angiography
- IMP
- investigational medicinal product
- IQR
- interquartile range
- LLA
- low-luminance visual acuity
- MD
- mean difference
- MedDRA
- Medical Dictionary for Regulatory Activities
- MHRA
- Medicines and Healthcare products Regulatory Agency
- MR
- mineralocorticoid receptor
- nAMD
- neovascular age-related macular degeneration
- NEI-VFQ-25
- National Eye Institute Visual Function Questionnaire-25
- NIHR
- National Institute for Health Research
- OCT
- optical coherence tomography
- OCT-A
- optical coherence tomography angiography
- PDT
- photodynamic laser therapy
- PIL
- patient information leaflet
- PPI
- patient and public involvement
- PRISMA
- Preferred Reporting Items for Systematic Reviews and Meta-Analyses
- PROM
- patient-reported outcome measure
- RCT
- randomised controlled trial
- REC
- Research Ethics Committee
- RPE
- retinal pigment epithelium
- SAE
- serious adverse event
- SAP
- statistical analysis plan
- SD
- standard deviation
- SmPC
- summary of product characteristics
- SRF
- subretinal fluid
- SRFT
- subretinal fluid thickness
- TSC
- Trial Steering Committee
- VEGF
- vascular endothelial growth factor