

Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 12/206/09. The contractual start date was in December 2014. The final report began editorial review in January 2020 and was accepted for publication in October 2020. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2021. This work was produced by Wilson et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2021 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
Idiopathic pulmonary fibrosis: a condition with great unmet need
Idiopathic pulmonary fibrosis (IPF) is a chronic, progressive, fibrotic lung disease characterised by symptoms of breathlessness, cough and bibasilar fine late inspiratory crepitations. It is diagnosed at a multidisciplinary team (MDT) meeting following confirmation of a pattern of usual interstitial pneumonia (UIP), which is identified from high-resolution computerised tomography (HRCT) scanning or histopathological review of lung biopsy, as defined by international criteria,1 once all known causes of interstitial lung disease (ILD) are excluded.
Idiopathic pulmonary fibrosis is a progressive, and usually fatal, lung disease with a 5-year survival rate of 20–40%. 2 At 7.44 per 100,000 person-years,3 the incidence of IPF is similar to that of subarachnoid haemorrhage. 4 The mortality of IPF is increasing at a rate of approximately 5% per year [rate ratio 1.05, 95% confidence interval (CI) 1.05 to 1.06]3 and more people die from IPF each year than from ovarian cancer, leukaemia or mesothelioma. 3 IPF is responsible for nearly 10,000 admissions to hospital per year in the UK, with an annual 5% increase in hospitalisations over the last decade. 5 A review of a US claims database revealed that, between 2001 and 2008, the direct cost for patients with IPF was US$26,000/person-year, twice as high as for controls. 6 The increasing incidence and rising mortality and morbidity represent a considerable unmet public health need. 7
At the time of designing the research protocol, only oxygen and lung transplantation were recommended by guidelines. 8 Immunosuppressive therapy, the mainstay treatment for more than a decade, had recently been proven to be harmful and was no longer advised. 9 Warfarin, which had been previously shown to reduce mortality in an open-label study,10 was shown not to be beneficial in a placebo-controlled trial. 11 N-acetylcysteine, also part of standard care based on evidence of preserved lung function when prescribed with prednisolone and azathioprine,12 was being evaluated and was shown not to be beneficial by the time the first patient was recruited. 13 Pirfenidone14 and BIBF-1120,15 renamed nintedanib, have been shown to improve forced vital capacity (FVC), but not mortality.
Unfortunately, the current situation is not much better. Current international guidelines conditionally recommend pirfenidone (Esbriet, Roche Holding AG, Basel, Switzerland), nintedanib (Ofev®, Boehringer Ingelheim, Brackness, UK) and anti-acid therapy. 16 A pooled analysis of Phase III placebo-controlled trials showed pirfenidone to reduce all-cause mortality17 and, correspondingly, there is evidence that nintedanib reduces mortality; both treatments are recommended by the National Institute for Health and Care Excellence (NICE) for people with moderately severe disease only [i.e. FVC between 50% and 80% of the predicted normal value (FVC per cent predicted)]. 18 However, a recent systematic review and network meta-analysis of randomised and quasi-randomised controlled trials showed that neither pirfenidone nor nintedanib significantly reduced mortality or acute exacerbations. 19 Evaluation of other possible therapeutic interventions is required.
Potential beneficial effect of co-trimoxazole
A review20 of the medical literature revealed two clinical trials20,21 of co-trimoxazole (SEPTRIN®; Essential Generics Ltd, Egham, UK; Chemidex Generics Ltd, Egham, UK) used in people with IPF. In one study of 20 patients, 3 months’ treatment with 960 mg of co-trimoxazole twice daily improved the primary end point of shuttle walk test distance, as well as FVC and Medical Research Council (MRC) Dyspnoea Scale score. Active treatment showed significant improvements in FVC and shuttle walk test distance.
In the Treating Idiopathic Pulmonary Fibrosis with the Addition of Co-trimoxazole (TIPAC) trial,21 we evaluated the effect of taking 960 mg of co-trimoxazole twice daily for 12 months in 181 patients with idiopathic interstitial pneumonia (IIP), 166 of whom had IPF. There was no effect on FVC (the primary end point) or other lung function measurements; however, we found that co-trimoxazole improved quality of life [in terms of the St George’s Respiratory Questionnaire (SGRQ) score] and reduces the percentage of patients requiring an increase in oxygen therapy. Furthermore, a health economic cost–utility analysis found that co-trimoxazole may be cost-effective22 in the intention-to-treat (ITT) analysis from a societal perspective. The incremental cost-effectiveness ratio for quality-adjusted life-years (QALYs) gained was £22,012, with a 54.5% probability of being < £30,000, which is below the upper limit considered ‘acceptable’ by NICE. 18
In a per-protocol (PP) analysis, the co-trimoxazole-treated group demonstrated significant reductions in mortality compared with the placebo-treated group (3/53 vs. 14/65; odds ratio 0.21, 95% CI 0.06 to 0.78), as well as improvements in QALYs and a reduced need for oxygen therapy. The findings were similar when confined to IPF and were not influenced by baseline immunosuppressive therapy in a subgroup analysis. There were reductions in non-infection-related deaths (placebo, n = 7; active, n = 3) as well as infection-related deaths, suggesting that co-trimoxazole may have both disease-modifying and anti-infective roles. The results were even more striking when considering patients with impaired lung function. Patients with a FVC of ≤ 75% predicted were nearly twice as likely to be admitted to hospital or die as patients with a FVC of > 75% predicted, with a borderline significant (p = 0.053) treatment effect in this subgroup using these combined end points (post hoc sensitivity analysis on an ITT basis from the TIPAC trial). 21
In a retrospective review of people with IPF receiving high-dose corticosteroid and mechanical ventilation for respiratory failure, Oda et al. 23 reported that more survivors were receiving co-trimoxazole than non-survivors. Administration of co-trimoxazole was significantly associated with a good prognosis and the dose of co-trimoxazole was related to survival, with higher doses (960 mg taken three times per day) producing better outcomes than lower doses.
Aetiology of idiopathic pulmonary fibrosis and potential mechanisms of co-trimoxazole
As the pathogenesis of IPF is unknown, the potential mechanisms of action of co-trimoxazole are uncertain. Co-trimoxazole is a broad-spectrum antibiotic with bactericidal effects against respiratory pathogens, with the role of infection in IPF becoming more evident. However, it may have non-antimicrobial effects, targeting cellular processes that have been implicated in the pathogenesis of IPF.
Antimicrobial effects
The role of infection in the pathogenesis of IPF has not been fully evaluated. 8 Infection is common in patients with IPF – even in those not receiving immunosuppression. In the TIPAC trial,21 we found that, of the patients in the placebo group not receiving prednisolone, 62% had an infection during the study. 24 In a meta-analysis25 of patients allocated to the placebo group from clinical trials of patients with IPF, the mean reported rate of pneumonia among studies not permitting immunosuppression was 37.1 per 1000 patient-years, which is higher than in those with chronic obstructive pulmonary disease (COPD). 26 Mortality from IPF increases in the winter even when recognised infection is not considered to be the cause of death. 27
In a study of bacterial culture from bronchial washings,28 more than one-third of patients with IPF were colonised with pathogenic bacteria or Pneumocystis jirovecii,29 the majority of whom were sensitive to co-trimoxazole. Garzoni et al. 30 evaluated the lung microbiota by sequencing bacterial 16S ribosomal ribonucleic acid (rRNA) genes. The study showed that the lungs of patients with ILD are not sterile. Subsequently, two independent groups of researchers, also using sequencing of bacterial 16S rRNA genes, have shown that bacterial load31,32 and lung microbiota profiles enriched with Streptococcus and Staphylococcus spp. 33 predict poor outcomes in IPF. The stability of the lung microbiota and the response to antibiotic therapy are unknown in IPF.
There is also evidence that innate immune responses may be abnormal in IPF, potentially increasing susceptibility to infection. In particular, alveolar macrophages from patients with IPF express a functional deficiency in their ability to kill intracellular bacteria,34 predisposing them to bacterial infection or colonisation. There is evidence that the expression of toll-like receptor (TLR) 2, which has a key role in Gram-positive pathogen sensing, is altered in IPF,35 and TLR9, which stimulates pro-inflammatory cytokine release, is upregulated in biopsies of rapidly deteriorating patients with IPF. 36
Co-trimoxazole has a broad spectrum of activity and is effective against most of the non-anaerobic bacteria in the airways of patients with IPF, including P. jirovecii, Streptococcus spp. and Staphylococcus spp.
Non-antimicrobial effects
Co-trimoxazole has beneficial effects in patients with granulomatous polyarthritis37 that are greater if treatment is started early and is not related to infection. 38 These potentially immunomodulatory effects have been poorly studied. Sulfamethoxazole has a structure similar to other sulfonamides, such as dapsone and sulfapyridine, which are known to have effects on neutrophil chemotaxis39 and superoxide production. 40 In vitro studies have shown that co-trimoxazole and its individual components (trimethoprim and sulfamethoxazole) inhibit neutrophil post-phagocytic myeloperoxidase-mediated protein iodination. 41 In other studies42,43 assessing the effects of different antimicrobial agents on neutrophil respiratory burst, co-trimoxazole and trimethoprim inhibited the chemiluminescence response at therapeutic concentrations.
Oxidant stress has been implicated in alveolar epithelial injury44 and epithelial–mesenchymal transition,45 and IPF patients have increased concentrations of 8-isoprostane in exhaled breath condensate. 46 Neutrophils play an important role in causing oxidant stress in IPF47 and neutrophilic alveolitis features frequently. 48 Furthermore, higher than normal neutrophil counts in sputum are associated with worse lung function49 and the percentage of bronchoalveolar lavage fluid (BALF) neutrophils at diagnosis is an independent predictor of mortality. 50
Thus, co-trimoxazole may inhibit neutrophil activation and reduce neutrophil-derived oxidative stress. These potential non-antimicrobial effects of co-trimoxazole would be predicted to have beneficial effects in IPF independently of and/or in addition to its antimicrobial actions.
Potential risks of co-trimoxazole
Co-trimoxazole has been licensed and prescribed to patients with respiratory disease for decades; hence the risks of this drug are well established. Many patients infected with the human immunodeficiency virus (HIV) receive long-term co-trimoxazole prophylaxis against Pneumocystis spp. without serious adverse reactions (SARs).
The drug is contraindicated in patients with hypersensitivity to sulfonamides or trimethoprim, in those with severe liver or renal failure, and in infants. Serious risks include hypersensitivity reactions, bone marrow depression (reduced by co-administration of folic acid) and crystalluria (reduced by adequate fluid intake), all of which occur extremely rarely. In the TIPAC trial,21 co-trimoxazole, when compared with placebo, increased the number of gastrointestinal adverse reactions (ARs), the severity of a rash and the level of serum creatinine. There were no significant differences in other adverse effects except infection and hyperkalaemia. Co-trimoxazole increased serum potassium concentration, even in those patients not taking antikaliuretic drugs. The magnitude of this change was small; however, in 5.7% of individuals the potassium level reached > 5.5 mmol/l, with potential clinical significance. 51 There is a well-recognised risk of drug interactions, which we managed by increased monitoring or drug exclusion. There is a theoretical risk of the development of antimicrobial resistance; however, co-trimoxazole is already prescribed on a long-term basis for the prophylaxis of a P. jirovecii infection, and IPF is sufficiently rare that any selection in IPF patients will make only a tiny addition to the total resistance burden in the population.
Although co-trimoxazole has recognised ARs, like many other pharmacological interventions (pirfenidone causes nausea in one-third of patients and photosensitivity reaction six times more commonly than placebo14), these adverse events (AEs) are also common in patients receiving placebo, so the presence of an AE will not necessarily result in study unblinding: treatment allocation will not be obvious.
Rationale for current study
As the TIPAC trial21 was powered to detect differences in FVC, all other end points were exploratory, but are nonetheless intriguing because no other study in IPF has shown this magnitude of effect on survival. Importantly, international prescribing practices in IPF have changed since the TIPAC trial21 was completed, with the cessation of corticosteroid treatment and commencement of antifibrotic therapies. An evaluation of efficacy in a clinical trial that is adequately powered on a clinically relevant end point is required before this treatment can be considered in clinical practice. In addition, it is important to explore the mechanism of action of co-trimoxazole so that this medication can be suitably targeted, newer therapies can be considered and further studies can be designed.
Rational for primary outcome
All-cause hospitalisation and death have been recommended by the Pulmonary Fibrosis Foundation summit of end points for Phase III clinical trials in IPF. 52 Others have advocated FVC as an end point, claiming that mortality requires unfeasibly large studies in mild to moderate IPF. 53 The situation is, however, different when evaluating patients with more severe disease. In a meta-analysis of placebo data of all clinical trials of IPF,54 annual mortality in studies selecting only mild–moderate patients was 8%, but in those including moderate to severe patients, annual mortality was 19%, in keeping with the data from the TIPAC trial21 and the epidemiology of the disease. 3 The event rate of mortality and hospitalisations is even higher when selecting patients with severe disease (up to 16% in 3 months55). The outcome measure of all-cause death, non-elective hospitalisation or transplant also meets the European Medicines Agency’s criteria for composite end points, as hospitalisation is an important predictor of mortality. 56 Hospitalisation can be easily and reliably assessed without patient involvement and is the least likely outcome to be influenced by withdrawal from the study, or unintentional or unavoidable unblinding of patients.
Rational for biomarkers
Although the mechanism of IPF is not fully elucidated, it is considered that a trigger or triggers result in alveolar epithelial injury with chaotic epithelial repair, fibroblast proliferation and collagen deposition, which distorts the lung architecture. We assessed markers of infection/inflammation, monocyte activity, neutrophil activity, alveolar epithelial injury, fibro proliferation, pulmonary hypertension and bronchial epithelium (mucus-associated cancer antigens).
Aims and objectives
The primary aim of the study was to determine the clinical efficacy of co-trimoxazole in patients with moderate to severe IPF (defined as FVC of ≤ 75% predicted). A secondary mechanistic aim was to evaluate the effect of co-trimoxazole on biomarkers of disease progression. An exploratory aim was to assess whether or not the mechanistic properties relate to clinical efficacy.
The primary objective
To compare the time to death (all causes), lung transplant or first non-elective hospital admission between co-trimoxazole and the placebo in patients with moderate to severe (FVC of ≤ 75% predicted) IPF during a median treatment period of 27 months (range 12–42 months).
Secondary objectives
To compare the clinical efficacy between the co-trimoxazole and placebo in patients with moderate to severe (FVC of ≤ 75% predicted) IPF during a median treatment period of 27 months (range 12–42 months) in terms of:
-
time to respiratory-related death, lung transplant or first respiratory-related hospital admission
-
time to respiratory-related and all-cause hospital admission
-
time to respiratory-related and all-cause death
-
QALYs
-
health-related quality of life
-
cough-related quality of life
-
breathlessness and Cough Symptom Scores
-
lung function
-
oxygen saturations.
Mechanistic objectives
To compare blood biomarkers between the co-trimoxazole and placebo in patients with moderate to severe (FVC of ≤ 75% predicted) IVF at baseline and after 12 months of treatment in terms of markers of:
-
infection/inflammation
-
monocyte activity
-
neutrophil activity
-
alveolar epithelial injury
-
fibroproliferation
-
pulmonary hypertension
-
bronchial epithelium.
Chapter 2 Research methods/design
Parts of this chapter have been reproduced with permission from the study protocol, which has been published in an open-access peer-reviewed journal. 57 This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated. The text below contains minor additions and formatting changes to the original text.
This was a Phase II, double-blind, placebo-controlled, parallel-group, randomised multicentre study of oral co-trimoxazole when added to standard care, with outcomes assessed during a median treatment period of 27 months (range 12–42 months). The aim was to recruit 330 patients with moderate or severe IPF (initially defined as FVC of ≤ 70% predicted but changed to ≤ 75% following a protocol amendment). Initially, patients had to be diagnosed within 2 years of study entry, but this requirement was removed (see Table 2). Patients continued on treatment from randomisation until withdrawal, death, first non-elective admission for any cause or the end of the study follow-up period, with a minimum duration of 12 months. The end of the trial was defined as 12 months following the last trial visit of the last patient randomised. Patients, therefore, had a variable study duration ranging from 12 to 42 months. The visit schedule was designed to align with clinical care and patients were reviewed approximately every 3 months throughout the study. Table 1 shows the study schedule and Figure 1 is the flow diagram of the study design schedule.
FIGURE 1.
Study design and schedule for the Efficacy and Mechanism Evaluation of Treating Idiopathic Pulmonary Fibrosis with the addition of Co-trimoxazole (EME-TIPAC) trial. A subgroup of 50 patients (randomised on a 1 : 1 basis to receive active and placebo treatments) will undergo a bronchoscopy for bronchoalveolar lavage at baseline, 3 months and hospitalisation (if clinically indicated) for molecular analysis, differential cell count, quantitative microbiology culture, P. jirovecii identification through polymerase chain reaction, and alveolar epithelial cell injury marker, neutrophil function markers and collagen turnover marker analysis. FBC, full blood count; FEV1, forced expiratory volume in 1 second; G6PD, glucose-6-phosphate dehydrogenase; LFT, liver function test; U&E, urea and electrolytes.
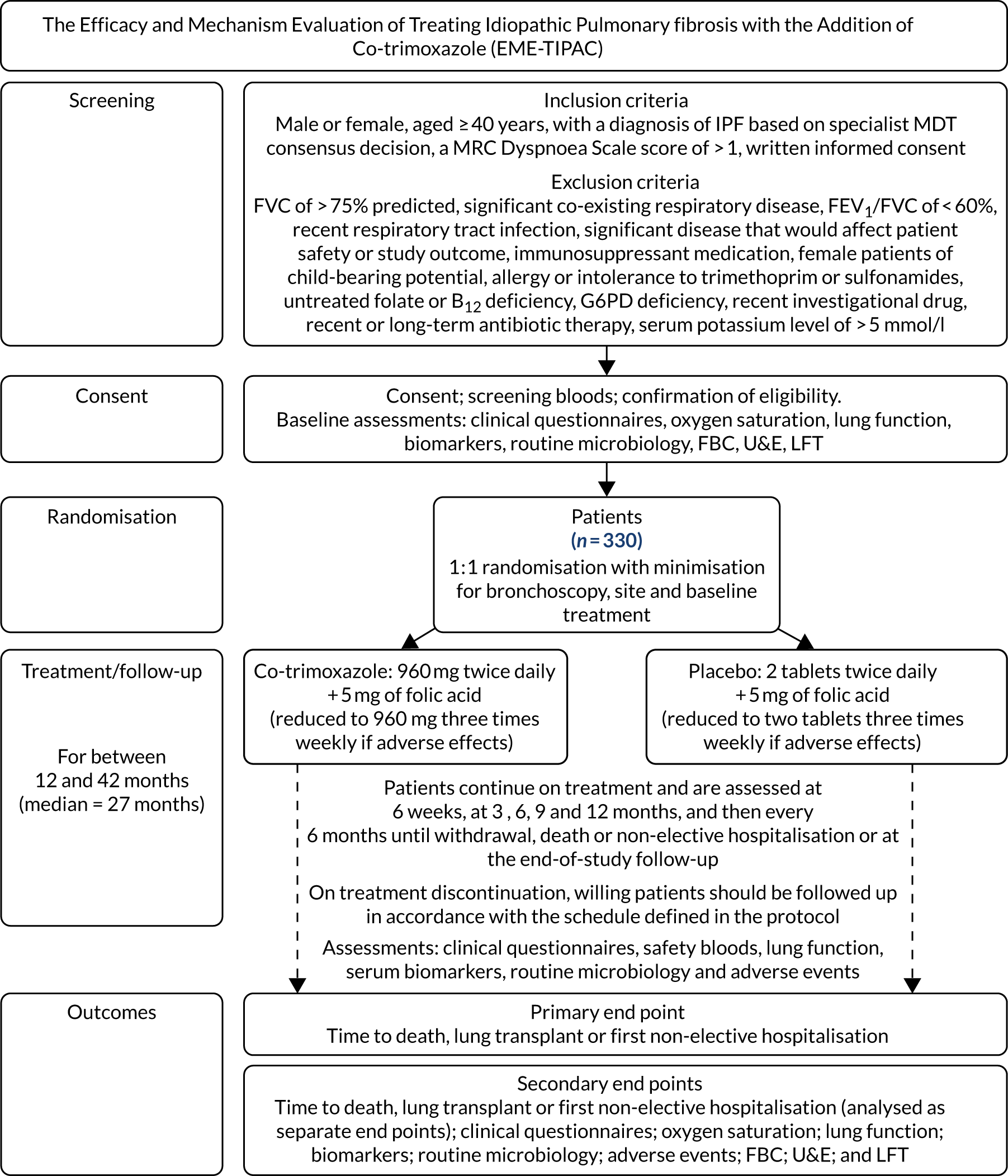
| Study event | Time point | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Enrolment | Randomisation | Post allocationa | Close-out | ||||||
| –28 to –1 day | 0 | 6 weeksb | 3 months | 6 months | 9 monthsb | 12 months | Every 6 months | End of study or first non-elective admission | |
| Informed consent | ✗ | ||||||||
| Demographics, etc. | ✗ | ||||||||
| Entry criteria | ✗ | ||||||||
| Allocation | ✗ | ||||||||
| IMP dispensed | ✗ | ✗ | ✗ | ✗ | ✗ | ||||
| Safety bloodsc (FBC, U&Es, LFTs) | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| B12, folate, G6PDd | ✗ | ||||||||
| DNA | ✗ | ||||||||
| Biomarkers | ✗ | ✗ | ✗ | ✗ | ✗ | ||||
| K-BILD score, MRC Breathlessness Score, EQ-5D score, Cough Symptom Score, Global Rating of Concept Scale | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||
| Leicester Cough Questionnaire | ✗ | ✗ | |||||||
| Full lung function | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||
| Microbiology (as clinically indicated) | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| AEs | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||
| BALF (subgroup) | ✗ | ✗ | ✗ | ||||||
The study was conducted in accordance with Good Clinical Practice (GCP). The study protocol received ethics approval from Surrey Borders Research Ethics Committee (reference 14/LO/1800) on 24 November 2014 and Medicines and Healthcare products Regulatory Agency (MHRA) clinical trial authorisation (13630/0008/001–0001) on 19 December 2014. All patients provided written informed consent, which included consent to inform the patient’s general practitioner (GP) of involvement in the study. Separate consent was obtained to provide blood for genetic analysis. The study was registered on the International Standard Randomised Controlled Trials Number (ISRCTN) registry on 29 January 2015 as ISRCTN17464641. The first patient was randomised in April 2015 and the last patient was randomised in April 2018.
Figure 1 provides a schematic representation of study design and schedule. The face-to-face study assessments were carried out on patients at recruitment/baseline, and at 6 and 12 months, as shown in Table 1.
Methods
Study setting
The study was conducted in UK secondary care centres that either met the specifications required for specialist ILD centre status or worked in association with specialist centres.
Sites had to have the facilities for research staff to undertake all of the measurements and store the samples required for the study unless, in exceptional circumstances, approval for the site to be excluded from some aspects of the study was granted by the chief investigator prior to site enrolment. The local principal investigator (PI) was responsible for the conduct of the study at their site in accordance with the protocol and for the safety and medical care of study patients. The investigators had to demonstrate the potential for recruiting the required number of suitable patients within the agreed recruitment period. They were responsible for ensuring that they had adequately trained staff to conduct the trial and for completing delegations of responsibility logs. Both the investigator and the local trust legal representative signed the site agreements. GCP training was required for all staff responsible for trial activities. The frequency of repeat training was dictated by the requirements of their employing institution or was conducted 2-yearly where the institution has no policy. The PI or delegate was required to document and explain any deviation from the approved protocol and to communicate this to the trial team at Norwich Clinical Trials Unit (NCTU).
Study population
Patient inclusion criteria
-
Male or female, aged ≥ 40 years.
-
A diagnosis of IPF based on a multidisciplinary consensus undertaken at a specialist centre (or a MDT otherwise meeting the criteria of a specialist centre) following a review of an appropriate clinical history, characteristic features of UIP on thoracic HRCT and/or UIP histology confirmed by surgical lung biopsy, according to the contemporaneous international guidelines. 8
-
Patients could receive oral prednisolone up to a dose of 10 mg per day, antioxidant therapy, pirfenidone, nintedanib or other licensed medication for IPF. Patients were receiving a stable treatment regimen for at least 4 weeks to ensure that baseline values were representative.
-
A MRC Dyspnoea Scale score of > 1 to exclude asymptomatic patients.
-
Able to provide informed consent.
Patient exclusion criteria
-
Forced vital capacity of > 75% predicted.
-
A recognised significant co-existing respiratory disease, defined as a respiratory condition that exhibits a clinically relevant effect on respiratory symptoms and disease progression, as determined by the PI following a multidisciplinary discussion. For example, patients with bronchiectasis were included only if this was deemed to be traction bronchiectasis as a result of IPF.
-
Patients with obstructive airways disease, defined as a forced expiratory volume in 1 second (FEV1)/FVC of < 60%.
-
Patients with a self-reported respiratory tract infection within 4 weeks of screening, defined as two or more of cough, sputum or breathlessness, and requiring antimicrobial therapy, were not eligible because of the difficulty of obtaining reliable baseline lung function.
-
Significant medical, surgical or psychiatric disease that, in the opinion of the patient’s attending physician, would affect patient safety or influence the study outcome, including liver [e.g. serum transaminase more than three times upper limit of normal (ULN), bilirubin more than two times ULN (unless the patient has Gilbert’s syndrome) and renal failure (e.g. creatinine clearance rate of < 30 ml/minute/1.73m2)].
-
Patients receiving immunosuppressant medication (except low-dose prednisolone) including azathioprine and mycophenolate mofetil. Immunosuppression is not advised for people with IPF. 9 Moreover, combining azathioprine with co-trimoxazole increases the potential for patients to develop neutropenia.
-
Female patients of child-bearing potential. Non-child-bearing potential was defined as follows: postmenopausal female patients with at least 12 months of spontaneous amenorrhoea or 6 months of spontaneous amenorrhoea with a serum follicle-stimulating hormone concentration of > 40 mIU/ml, or female patients who had had a hysterectomy, bilateral salpingectomy or bilateral oophorectomy at least 6 weeks prior to enrolment.
-
Known allergy or intolerance to trimethoprim or sulfonamides or their combination, for safety reasons.
-
Untreated folate or B12 deficiency. This was to ensure that no bone marrow or neurological adverse effects occured with folate therapy to B12-deficient individuals.
-
Known glucose-6-phosphate dehydrogenase (G6PD) deficiency or G6PD deficiency measured at screening in males of African, Asian or Mediterranean descent. Sulfonamides are recognised to increase the risk of haemolysis in individuals with G6PD deficiency. The prevalence of G6PD deficiency is higher in males of African, Asian or Mediterranean descent than in those of other ethnic backgrounds. However, the risk of haemolysis is low even in populations with high prevalence. 58
-
Receipt of an investigational drug or biological agent within the 4 weeks prior to study entry or five times the drug half-life, whichever is longer.
-
Receipt of short-course antibiotic therapy for respiratory and other infections within 4 weeks of screening.
-
Patients receiving long-term (defined as > 1 month of therapy) prophylactic antibiotics, as this may have had an impact on lung microbiota. Such patients could enrol in the Efficacy and Mechanism Evaluation of Treating Idiopathic Pulmonary Fibrosis with the addition of Co-trimoxazole (EME-TIPAC) trial, if this was supported by their clinician, after a wash-out period of 3 months.
-
Serum potassium level of > 5.0 mmol/l because of the potentially increased risk of hyperkalaemia in patients taking co-trimoxazole in combination with potassium-sparing diuretics (including angiotensin-converting enzyme inhibitors or angiotensin receptor blockers).
No exceptions to the stated eligibility criteria were permitted. Patients could be entered into other observational studies with the prior agreement of the Trial Management Group (TMG) of both studies.
Recruitment
Patients were mainly identified by review of ILD MDT meeting minutes or summaries. Patient identification took place through screening patient registries, hospital medical records and databases of research-interested patients or clinical details. Recruitment strategies included the following:
-
Patients were approached by the clinical care team directly when they attended the hospital outpatient clinic, at which point they were given an ethics-approved invitation letter on hospital headed paper that provided an overview of the study and a patient information leaflet. The clinic staff then arranged a subsequent recruitment visit.
-
The clinic team mailed the invitation letter, with or without a patient information sheet, with a reply form detailing a range of methods for the interested potential patients to contact the local trial team to arrange a screening appointment.
-
When patients were due to attend the clinic for a routine appointment in the near future, the clinical care team mailed an invitation letter on hospital headed paper that provided an overview of the study and a patient information sheet. This was timed so that the patient received these documents at least 24 hours before the forthcoming routine clinic assessment visit so that the potential patient would have sufficient time to read the information and decide whether or not they wished to consent at the subsequent clinic visit. If the patient provided written informed consent, screening for eligibility and baseline assessments were undertaken at the routine clinic visit.
-
For centres with access to a volunteer database, the researchers mailed the invitation letter and reply form directly to the volunteer.
Potential patients could be contacted by telephone between 3 and 7 days after the mailing of the letter to ensure that they had received it.
Interventions
Patients were randomised on a 1 : 1 basis to receive one of the following treatments for between 12 and 42 months (median 27 months):
-
Oral (non-proprietary) 960 mg of co-trimoxazole twice daily, taken as two tablets of 480 mg twice daily. Patients received the medication in containers containing 1 month’s supply every 3 months for the first 6 months and then every 6 months thereafter.
-
Oral placebo tablets (manufactured by the pharmacy at Guy’s and St Thomas’s Hospital to be identical to 480 mg of co-trimoxazole), taken as two tablets twice daily. Patients received the medication in containers (identical to those containing the active treatment) containing 1 month’s supply every 3 months for the first 6 months and then every 6 months thereafter.
Patients with a baseline serum potassium level between 4.7 and 5.0 mmol/l who were aged ≥ 66 years and taking potassium-sparing diuretics were required to have an extra blood test for safety 1 week after starting trial treatment owing to the increased risk of hyperkalaemia.
All patients received 5 mg of folic acid orally daily. Treatments were given in addition to standard care as defined by NICE guidelines. 18
In the TIPAC trial,21 withdrawal (29% of patients in the active treatment group and 8% of patients in the placebo group) was mostly due to ARs. For this reason, patients from both the co-trimoxazole and the placebo treatment groups had the option to reduce the dose to two tablets (i.e. 960 mg or two placebo tablets) plus 5 mg of folic acid three times weekly in the following circumstances:
-
if the patient developed gastrointestinal adverse effects or rash
-
if the patient had a potassium level > 5.0 mmol/l and < 5.5 mmol/l (i.e. grade 1 hyperkalaemia)
-
if the patient developed any other AE that, in the opinion of the local PI, required a dose reduction.
The dosing interval was to ensure that the dosing was spread throughout the week (e.g. Monday, Wednesday and Friday, or equivalent). Once a patient had a dose reduction, no re-escalation was permitted, even if the AE leading to the reduction resolved.
Drug preparation and supply
Europe-wide tendering for investigational medicinal product (IMP) manufacture for the trial was undertaken in June 2014. Following a successful bid, the tender to manufacture IMPs was awarded to Guy’s and St Thomas’ NHS Foundation Trust Pharmacy Manufacturing Unit (GSST PMU) (London, UK). The licence granted to GSST PMU by the MHRA under the requirements of EU Directive 2001/20EC was MA (IMP) 11387.
A dose of 480 mg of co-trimoxazole or matching placebo tablets was packaged in a white opaque high-density polyethylene plastic container that was sealed with a child-resistant/tamper-evident cap and labelled in compliance with applicable regulatory requirements, including EU GMP Annex 13 Investigational Medicinal Products, (Vol. 4, Annex 13; www.gmp-compliance.org/guidelines/gmp-guideline/eu-gmp-annex-13-investigational-medicinal-products; accessed October 2020). Each container contained a 31-day supply, which equated to 124 tablets. In addition, reduced-dose containers were produced containing only 26 tablets for those patients who were reducing to two tablets once a day, three times per week.
The active and placebo IMP containers appeared identical, except that the containers were coded with treatment group 1 or 2, to differentiate between active and placebo packs, on a tear-off strip. When the tear-off strip was removed, the packs appeared identical.
Randomisation was performed centrally with secure database-generated e-mail correspondence by NCTU to research pharmacists only. This enabled both the trial team at NCTU and the local research team (including the PI) at the site to remain fully blinded to the allocation. The ‘semi-blind’ pharmacist receiving the e-mail allocated the IMP according to treatment group 1 or 2 and was unblind to the group (1 or 2), but blind to the intervention.
The IMPs and folic acid were dispensed by the hospital pharmacy to the patient directly or, at baseline, in situations where it would prevent the patient from having to return to the hospital to collect the IMP, by a health-care courier to the patient’s home.
Pharmacies were required to maintain up-to-date accountability, dispensing and destruction logs for inspection by the NCTU and regulatory authorities during the trial. IMPs and folic acid sent by courier (or other signed-for delivery services) to patients required signature on receipt. Patients were advised to store their medication at < 25 °C, but there was no temperature monitoring after dispatch from the third party. Patients were dispensed an initial 3 months’ supply at baseline, with the same amount dispensed 3 months later. They were then given 6 months’ supply, that is six bottles, on each subsequent occasion.
Assignment of intervention
The allocated treatment for a patient was generated by a computer-written code using minimisation under the supervision of the study statistician. Minimisation was performed using Taves’ method with the factors measured at baseline: (1) study site, (2) whether or not the patient had consented to take part in the bronchoscopy substudy (yes/no) and (3) whether or not the patient was receiving licensed antifibrotic medication for IPF at the screening visit (yes/no). To decide on the treatment allocation, the code calculated the number of patients in each group who had the same characteristics as the patient awaiting allocation; patients were allocated to the intervention with the smaller number with a high probability. If the numbers were the same, then simple randomisation was used.
The patients were allocated to the intervention by a process embedded in the web-based data management system. The randomisation code was saved in the study database for later decoding and for emergency unblinding purposes. When a patient was randomised, an e-mail was sent to the appropriate local pharmacy, who prepared the medication pack.
Blinding
This was a double-blind study. The placebo and active treatments appeared identical and were dispensed in identical containers containing the same number of tablets. Other than in instances requiring emergency unblinding or unblinding for safety reporting, all trial patients, care providers, outcome assessors and data analysts remained blind throughout the study.
The decision to unblind a single case was made when knowledge of an individual’s allocated treatment was required to enable treatment of severe adverse event(s) [SAE(s)].
Where possible, requests for emergency or unplanned unblinding of individuals were made through the trial manager and the agreement of the chief investigator was sought. However, in circumstances in which there was insufficient time to make this request or for agreement to be sought, the treating clinician made the decision to unblind immediately. This was done using the study database (local PIs and the chief investigator had special logins that allowed unblinding and that were closely audited within the database management system) or by the chief investigator, who authorised unblinding by the Data Management Team. All instances of unblinding were recorded and reported to NCTU by the local PI, including the identity of all recipients of the unblinding information.
Compliance and adherence
Compliance with study treatment, in the form of returned tablet counts, was monitored as part of drug accountability at each visit.
Concomitant medication
Patients were permitted to receive N-acetylcysteine and antioxidants, prednisolone (up to a dose of 10 mg per day) and licensed treatments for IPF. All concomitant medications were recorded at baseline and any change in concomitant medication was recorded at each visit.
Patients were permitted to receive other medications (e.g. for other conditions), but non-permitted therapies included amiodarone, azathioprine, mycophenolate mofetil, cyclophosphamide, methotrexate, D-penicillamine, colchicine, clozapine, methenamine, dapsone, interferon gamma, ciclosporin, mercaptopurine, repaglinide, pyrimethamine, lamivudine, typhoid vaccination or unlicensed medication.
Therapy requiring caution or increased monitoring included digoxin, warfarin, phenytoin, sulfonylureas and procainamide hydrochloride. Increased monitoring of potassium levels was required if patients commenced medication that increases serum potassium concentration.
Treatment discontinuation
Individual patients stopped treatment early for the following reasons:
-
any non-elective hospitalisation or lung transplant (meeting the primary end point)
-
a serum potassium level of > 5.5 mmol/l
-
co-trimoxazole-related haematological disease (e.g. blood dyscrasia or thrombocytopenia)
-
unacceptable treatment toxicity or an AE
-
intercurrent illness that prevented further treatment
-
any change in the patient’s condition that, in the clinician’s opinion, justified the discontinuation of treatment
-
withdrawal of consent for treatment by the patient.
Patients who discontinued protocol treatment for any of the above reasons remained in the trial for the purpose of follow-up and data analysis, unless they requested otherwise. The patients were invited to continue follow-up in the trial, although they no longer took the study drug. However, whenever the patient no longer wished to be followed up either, this view was respected and the patient was withdrawn entirely from the trial. Data that were already collected were kept and included in analyses according to the ITT principle for all patients who stop follow-up early. Patients provided consent so that follow-up information on overall or hospital-free survival could be obtained from medical records using the NHS number, for example through their GP, if required. Research teams were asked to account for the vital status and details of admission to hospital for all patients, regardless of whether they had withdrawn from the intervention or study assessments. Patients who stopped taking the study drug or withdrew from the study were not replaced.
Patients who were admitted to hospital non-electively were deemed to have met the primary end point. From that point onwards, patients ceased to have follow-up measurements taken, but survival status was reported at the end of the study.
Outcomes
Primary outcomes
The primary outcome was the time to death (all causes), transplant or first non-elective hospital admission. This was defined as the time from randomisation to death, lung transplant or first non-elective hospital admission for any reason. Details of patients admitted to hospital or dying were captured by examining SAE reports, hospital patient databases and the tracing of patients missing appointments by contacting their primary care physician, as required. Non-elective admission was defined as a hospital stay lasting more than 24 hours that had not been arranged more than 24 hours prior to admission. Death could not be influenced by unintentional unblinding and we believe it is unlikely that admission to hospital was influenced by this either. Treatment or absence of treatment with co-trimoxazole rarely causes conditions that would require hospitalisation and, given the costs and other implications of admission to hospital, hospitalisation was likely to be because of clinical need.
Secondary outcomes
The individual components of the primary outcome – time to death (all causes), transplant and time to first non-elective hospital admission – were analysed separately as secondary outcomes. In addition, respiratory-related events were analysed separately from non-respiratory-related events.
Extracts from the case report form (CRF) were forwarded to an independent review committee. This committee, chaired by a consultant respiratory physician with experience of undertaking clinical trials, also included a consultant respiratory physician, a nurse consultant specialising in ILD and a primary care physician. Each independently reviewed whether or not the event was respiratory related based on the CRF listing. If no consensus could be achieved, then the chairperson had the casting vote.
The following measurements were undertaken at baseline, 3 and 6 months, and then 6-monthly throughout the study. Every effort was made to collect data at time points within a 2-week window (i.e. 2 weeks before or after the visit schedule) until 6 months, then within a 1-month window thereafter. However, as the study visits were aligned with routine clinical care assessments, this scheduling was not always possible and values obtained outside these windows were captured along with the assessment date. In addition, a final assessment was made at the end of the study if a patient had not had a primary outcome measurement undertaken within 2 months of the end of the study.
Questionnaires
The King’s Brief Interstitial Lung Disease questionnaire
The King’s Brief Interstitial Lung Disease (K-BILD) questionnaire59 is a validated tool describing health status during the past 2 weeks in people with ILD. This 15-question self-completed questionnaire has a mean [standard deviation (SD)] score of 53 units (26 units) in IPF and a minimum clinically significant difference of 3.9 units. 60 It evaluates three dimensions (psychological, breathless and activity, and chest symptoms) on a seven-point Likert scale. Total score ranges from 0 units (worst health status) to 100 units (best health status). 61
The Medical Research Council Dyspnoea Scale
The modified MRC Dyspnoea Scale62 is commonly used to assess breathlessness and response is classified on a five-point scale: grade 0 (dyspnoea with strenuous exercise); grade 1 (dyspnoea when hurrying or walking up a slight hills); grade 2 (walk slower than people or has to stop for breath); grade 3 (stops for breath after walking 100 yards); and grade 4 (too breathless to leave house or breathless when dressing).
EuroQol-5 Dimensions, five-level version
The EuroQol-5 Dimensions, five-level version (EQ-5D-5L),63 is a validated global health status instrument containing five dimensions: mobility, self-care, usual activities, pain/discomfort and anxiety/depression. Each dimension is answered on a five-level scale (1, no problems; 2, slight problems; 3, moderate problems; 4, severe problems; and 5, severe problems/unable to do) and the score is the sum of the dimensions.
The Leicester Cough Questionnaire
The Leicester Cough Questionnaire (LCQ) is a valid, repeatable, 19-item self-completed quality-of-life measure of chronic cough that is responsive to change. 64 The questionnaire captures cough according to the physical, psychological and social domains, and the total score ranges between 0 (worst health status) and 100 (best health status). It has been validated in IPF with a median total score of 15.4 (range 6.95–20.88). 65
Cough Symptom Score
We captured Cough Symptom Score (CSS) on a visual analogue scale (100 mm in length) to record patients’ score of cough to assess their overall symptoms of cough over the preceding 2 weeks, with 0 meaning that they were not bothered by cough at all and 100 referring to cough that is the worst it can be.
Lung function
Lung function was measured at recruitment and at 6 and 12 months using spirometry performed to American Thoracic Society/European Respiratory Society standards. 66 Spirometry is a routine part of the clinical assessment of people with ILD and is usually measured at all clinical assessments. The FVC and FEV1 measures were obtained as part of the spirometry assessment. The predicted equations, derived by Crapo et al. ,67 were used to calculate the predicted normal value and percentage predicted values for FVC and FEV1. Where spirometry was contraindicated or patients were not able to complete spirometry, this was omitted.
Gas transfer measurements were measured at recruitment and at 6 and 12 months using spirometry performed to American Thoracic Society/European Respiratory Society standards. 68 The diffusing capacity of the lung for carbon monoxide (DLCO) was obtained and the percentage predicted values were calculated using the predicted values obtained from the equations derived from the European Coal and Steel Community. 69
Forced vital capacity and DLCO are both components of prognostic modelling algorithms,56,70 are frequently utilised in clinical trials14 and are part of routine care. These were the main lung function outcomes.
Peripheral blood
Peripheral blood was taken at baseline, at 3, 6 and 12 months, and at the end of the study. The peripheral blood samples were stored throughout the study. Blood was collected in ethylenediaminetetraacetic acid (EDTA) and serum Vacutainers® (Fisher Scientific UK Ltd, Loughborough, UK), centrifuged, and the supernatant aliquoted and stored locally at –70/–80 °C. Periodically, samples were couriered (CitySprint, Loughborough, UK) in a BioTherm 45 (Intelsius UK, York, UK) with dry ice for next-day delivery (category B biological samples) to the Department of Laboratory Medicine at the Norfolk and Norwich University Hospital and stored at –70/–80 °C until the end of the study. Serum of matching baseline and 12-month (± 60 days) samples was analysed for the following biomarkers.
Measures of infection/inflammation
C-reactive protein (CRP) is an acute-phase protein that is present in serum in increased concentrations in patients with inflammation. It is routinely used along with clinical parameters to monitor patients with respiratory tract infection and is a significant prognostic indicator for survival in patients with IPF. 71 It was measured by an immunoturbidimetric assay on the Cobas® 6000 (Roche Diagnostics GmbH, Mannheim, Germany) using latex particles coated with monoclonal anti-CRP antibodies.
Monocyte activity marker
Chemokine ligand 18 (CCL18) has a role in immune cell trafficking and predicts outcome in pulmonary fibrosis. 72 CCL18 released from alveolar macrophages increases collagen production from fibroblasts. In a post hoc review73 of numerous biomarkers collected in trials evaluating pirfenidone (CAPACITY14 and ASCEND74 trials), blood CCL18 levels were the most consistent predictor of disease progression as assessed by absolute change in percentage predicted FVC over 12 months. CCL18 was analysed using enzyme-linked immunosorbent assays (ELISAs) purchased from Bio-Techne Ltd (Abingdon, UK). Monocyte chemotactic protein 1 (MCP-1; also known as CCL2) is another monocyte activity marker and its levels are increased in people with IIP. 75 MCP-1 is significantly correlated with interstitial lung lesions. In addition, a monoclonal antibody that neutralises the fibrotic activities of MCP-1 has been used in a clinical trial. 76 MCP-1 was measured by ELISA following the manufacturer’s instructions.
Neutrophil activity
Myeloperoxidase (MPO) is almost exclusively expressed in neutrophils and its release into serum is a marker of neutrophil activation and degranulation. Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) has an important role in regulating the survival of immune cells, including neutrophils, and has been shown to be a potential marker of IPF. 77 As osteoprotegerin (OPG) is a decoy receptor of TRAIL, we measured its concentration to interpret the findings of TRAIL. MPO and TRAIL were purchased from R&D Systems, Inc. (Minneapolis, MN) and OPG was purchased from Biomedica Medizinprodukte GmbH (Vienna, Austria). All three ELISAs, i.e. for MPO, TRAIL and OPG, were performed following the manufacturers’ instructions.
Alveolar epithelial injury
Markers of alveolar epithelial cell injury were measured, given the importance of injury in the pathogenesis and prognosis of IPF, and the fact that both infection and neutrophil activation may result in injury to the epithelium. Surfactant proteins (SPs) are among the most widely evaluated biomarkers in IPF. Surfactant protein D (SP-D) is a marker of epithelial injury78 and is produced only by alveolar epithelial cells. It is elevated in BALF and serum in patients with IPF79 and predicts mortality. 80 SP-D ELISAs (R&D Systems, Inc.) were measured in accordance with the manufacturer’s instructions.
Fibro proliferation
Matrix metalloproteinase 7 (MMP-7), a profibrotic metalloproteinase, has consistently been shown to be elevated in BALF and plasma of patients with IPF. 81 MMP-7 is related to disease severity82,83 and is an independent predictor of mortality. 84 MMP-7 was measured by ELISA (R&D Systems, Inc.) in accordance with the manufacturer’s instructions.
Heat shock protein 47 (HSP47) is a collagen-specific molecular chaperone involved in intracellular processing of procollagen. Its concentration is higher in animal models of fibrosis and a number of other fibrotic conditions. HSP47 is able to distinguish between acute interstitial pneumonia and stable IPF,85 and its concentration in lung fibroblasts predicts survival in fibrotic lung disease. 86 HSP47 (Novus Biologicals Littleton, CO, USA) failed to pass performance validation and, therefore, was not analysed. Briefly, no internal quality controls (IQCs) were provided with the kit and sample pools were used. A high pool was found to be above the top standard [upper limit of quantitation (ULOQ)], but a serial dilution failed to provide a result, as all results up to 64-fold dilution produced results above the ULOQ. The percentage of coefficient of variation (CV) of duplicates was up to 22.5%.
Pulmonary hypertension
The development of pulmonary hypertension is a frequent and significant event for people with IPF as it corresponds to a deterioration in symptoms and disease control. We measured pro-brain natriuretic protein (pro-BNP) as a measure of pulmonary arterial hypertension,87 given that pro-BNP predicts disease progression and mortality in IPF. 88 Pro-BNP was analysed on the Cobas® 6000 following the manufacturer’s instructions. Pro-BNP was measured using an electrochemiluminescence immunoassay (ECLIA; Roche Diagnostics GmbH] using microparticles coated with monoclonal anti-pro-BNP antibodies.
Bronchial epithelium carbohydrate antigen 19-9 (CA19-9) and cancer antigen 125 (CA-125) are tumour markers. Raised concentrations of CA19-9 were highly predictive of progressive fibrosis, and rising concentrations of CA-125 predicted both disease progression and overall survival. 89 Levels of CA19-9 and CA-125 were measured by ECLIA with kits manufactured by Roche Diagnostics GmbH.
The assay ranges, sensitivity, reference range and units are given in Appendix 1, Table 23.
For all assays, performance (intra-assay and interassay) had a IQC of < 10%, except for SP-D (< 20%). External quality controls (EQCs) for CA19-9 and CA-125 were also added (through a sample swap with Norwich and Norfolk University Hospitals NHS Foundation Trust) and the interassay CV was < 9 %. Overall accuracy of the EQCs compared with the provided target was 105% ± 7 % for CA-125 and 100% ± 5% for CA19-9.
Routine microbiology
Sputum was obtained, where possible, and sent for local microbiological culture and sensitivity testing. For all patients, a nasal swab was sent for viral culture, if clinically indicated. All microbiology laboratories followed a common protocol for sputum processing and susceptibility testing of the bacteria recovered. Assessment of urinary Legionella and pneumococcal antigens or serology for atypical respiratory pathogens were not undertaken routinely as these are not helpful in asymptomatic individuals.
Safety
The following were measured at baseline, 6 weeks, 3, 6, 9 and 12 months, and then 6-monthly for the duration of the study, as well as at the end of the study/at hospitalisation [ideally within a 1- or 2-month window (as above)]:
-
full blood count and differential white cell count
-
urea and electrolytes
-
liver function.
Unless the patient was otherwise due to attend a clinic visit at the 6-week and 9-month time points as part of their standard care, the safety bloods for these visits were performed at the patient’s primary care surgery and the patient followed up by telephone (to check for AEs and any change in concomitant medication). Abnormal routine laboratory values were considered to be AEs if they were outside the normal reference range for the local laboratory.
Measurements during the first non-elective admission
An entry was made on the patient’s medical records and patients were asked to carry a card detailing their involvement in the study, with research nurse and study co-ordinator contact details, to maximise ascertainment of questionnaires, blood samples and routine microbiology in the same manner as undertaken at the routine visits during their admission to hospital.
Bronchoscopy substudy
We had planned a substudy to investigate the effects of co-trimoxazole on BALF in a subset of 50 patients, who volunteered to undergo bronchoscopy at baseline and after 3 months of treatment. Bronchoalveolar lavage (BAL) is the only appropriate method of sampling the lower airways, which are the principal region of disease in IPF. The alternative methods (spontaneous or induced sputum analysis) would have confounded the results by upper airway contamination90 and, to our knowledge, none of the available biomarkers of lung injury and inflammation has been evaluated in sputum. Bronchoscopy with BAL is safe in patients with IPF with a risk of SAEs of < 1 : 1000 at experienced sites. In a study of 281 patients with ILD undergoing BAL, no events necessitated therapy. 91
Bronchoscopies were performed as per current British Thoracic Society guidelines. 92 All bronchoscopies were to be through the oral route to avoid nasal contamination. BAL was undertaken by instilling four 50-ml aliquots of sterile saline through the bronchoscope wedged in a segment of the middle lobe. The material was recovered by gentle suction and aliquots were taken. BALF was placed on ice and strained through sterile gauze prior to centrifugation (at 310 g for 5 minutes at 4 °C) to collect the cell pellet. The supernatant was aspirated and snap frozen at –80 °C in 1-ml aliquots. The cell pellet was washed and resuspended. In addition, bronchoscope washing was stored for sequencing of deoxyribonucleic acid (DNA) derived from bacterial 16S rRNA genes.
Unfortunately, bronchoscopy with BAL was undertaken in only two people for the purposes of the study. We initially planned that this procedure would be undertaken in only two sites (Royal Papworth Hospital NHS Foundation Trust and the Royal Brompton Hospital) to maximise internal consistency as these sites have significant experience with this procedure for the purposes of research. However, despite doubling the number of sites available to offer research bronchoscopy for people interested in participating in this substudy (by adding Aintree University Hospital and University Hospitals Coventry & Warwickshire), and with plans for expansion to another eight trusts, we could still not recruit into the substudy. The Data Monitoring Committee (DMC) recommended abandoning the substudy on 14 June 2017 on the grounds of futility.
The lack of interested patients was thought to be due to the invasive nature of the procedure. Following discussion on 1 August 2017, the Trial Steering Committee (TSC) acknowledged the efforts that had been undertaken to improve recruitment by the chief investigator and the study team, but agreed with the DMC’s recommendation and, as a result, the substudy was closed to recruitment. None of the samples was analysed, as the results would have been meaningless.
Pharmacovigilance
Adverse events
This trial complied with the UK NHS Health Research Authority’s guidelines for reporting AEs (URL: https://hra.nhs.uk/approvals-amendments/managing-your-approval/safety-reporting/; accessed 10 March 2021). AEs were defined as any untoward event that occurred following consent into the study. All patients were asked about AEs at each study visit or telephone call: at 6 weeks, at 3, 6, 9 and 12 months, and then 6-monthly for the duration of the study, in addition to at the end of the study/at hospitalisation. The details of AEs were recorded in the CRF. Patients were notified of recognised ARs to co-trimoxazole and encouraged to contact the local study centre if they experienced these. All AEs were followed up until resolution.
Definitions of harm for the trial were adapted from Directive 2001/20/EC (European Commission), International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) E2 A entitled ‘Clinical Safety Data Management: Definitions and Standards for Expedited Reporting’, ICH GCP E6 and the EU’s CT-3 (v 2011/C 172/01). 93
All AEs were assessed by the local PI or delegate as to whether or not they met the criteria of a SAE, as defined in the protocol. SAE definitions included AEs that:
-
resulted in death
-
were life-threatening
-
required hospitalisation or prolonged existing hospitalisation
-
resulted in persistent or significant disability or incapacity
-
were a congenital anomaly or birth defect
-
were another important medical condition.
All SAEs were assessed by the local PI for their severity (mild, moderate or severe) and relatedness (unrelated, unlikely to be related, possibly related, probably related or definitely related), as determined by the clinical context of the event, including the association with the timing of onset of the event. SAEs that were deemed to be possibly, probably or definitely related to the trial IMP were categorised as SARs. SARs that did not relate to trial end points, including any that resulted from a possible interaction between co-trimoxazole and folic acid, were notified to NCTU within 24 hours of the investigator becoming aware of the event. As death and non-elective hospital admission formed part of the primary end point for the trial, hospital admissions were not reported separately through SAE reporting unless the death or non-elective hospital admission was treatment related, in the opinion of the local investigator. Hospitalisations and deaths that were not related to trial treatment and, therefore, did not require reporting as SAEs for the trial were recorded on the CRF. The chief investigator reviewed all SAEs reported to the NCTU and confirmed the assessment of causality and relatedness.
All SARs were assessed for expectedness against the MHRA-approved reference safety information (RSI) by the chief investigator. Any SARs deemed unexpected were to be classified as a suspected unexpected serious adverse reaction (SUSAR). SUSARs were to be reported to the ethics committee sponsor by e-mail and to the regulatory authorities using the electronic SUSAR web portal within 7 days if fatal or life-threatening and within 15 days if otherwise.
Study monitoring
Prior to commencement of the trial, a quality management and monitoring plan (QMMP) was produced, which detailed all planned and systematic actions established to ensure that the EME-TIPAC trial was performed and that the data were generated, documented and reported in accordance with the principles of GCP and applicable regulatory requirements. The QMMP was reviewed annually during the trial by the NCTU Quality Management Group and updated when appropriate.
Any findings identified during monitoring that caused concern were to be discussed with the chief investigator and/or the TMG, with discussions recorded and stored in the trial master file.
Central quality control procedures included a formal, documented site assessment procedure; the signing of a PI statement agreeing to the responsibilities of the role; the review of delegation logs, PI curricula vitae and GCP certificates; regular trial team meetings to review data and recruitment; and a review of anonymised screening logs, trial drug accountability and dispensing logs at the NCTU at periodic intervals. The trial database was also programmed to prevent the randomisation of ineligible patients, with liver function, renal function, G6PD and lung function tests validated in real time.
Quality control procedures at clinical sites included formal site initiation training (either in person or by teleconference), electronic CRF review, and the periodic checking of essential documents, investigator site files (ISFs) and pharmacy site files (PSFs).
In addition, central monitoring was performed throughout the trial and documented by the trial team in an annual monitoring report that was provided to the trial sponsor and the TMG. Central monitoring including the following actions:
-
Collection of dispensing and accountability logs from all participating pharmacies and the subsequent reconciliation of these with data contained in the CRF.
-
Protocol compliance checks, for example, checking that no dose re-escalations occurred (through a review of accountability logs) or checking adherence to the protocol-defined non-IMP regimen while patients were on active treatment.
-
An additional centralised eligibility review of patients’ age, FVC per cent predicted, and folate, B12 and serum potassium levels to identify any ineligible patients.
-
Collection of delegation logs for review and cross-referencing against consent forms and the database to ensure that only appropriately delegated members of staff were performing trial-related activities.
-
Collection of ISF and PSF checklists to ensure that sites were working to current trial documentation.
-
Ongoing review of CRF data for errors, inconsistencies and missing key data points.
-
A review of overall data accuracy and completeness for each site to flag issues and escalate where applicable.
-
The cross-referencing of visit dates against expected visit dates to ensure that sites were carrying these out in accordance with the protocol visit schedule.
-
A review of all AE data to ensure that these were coded using Medical Dictionary for Regulatory Activities (MedDRA) terminology and to identify any under-reported SAEs.
-
Patients provided consented to enable the NCTU to hold a copy of the consent form to ensure that the correct version had been used, the correct staff members had undertaken consent and the consent form had been completed appropriately.
-
Out-of-hours contact details provided to patients were tested by the trial team outside working hours for the top three recruiting sites during the trial.
On-site monitoring was performed at the top 10 recruiting sites between 2017 and 2018. The on-site monitoring visits, in addition to activities performed during central monitoring, involved source data verification of a sample of patients at the site, checks to ensure that documentation was completed according to GCP, a review of the clinic notes to check for unreported notable or serious events, a pharmacy inspection, and ISF and PSF review.
Risk-based central statistical monitoring (CSM) was performed by the trial statistician, or a delegate, prior to each DMC meeting, with the aim of identifying potential recording and entry errors, procedural errors and possible fraud. Blinded CSM results were to be discussed with the trial manager prior to each DMC meeting to enable any issues to be resolved prior to the meeting, if possible, or escalated to the chief investigator.
Direct access to patient records
Participating investigators agreed to allow trial-related monitoring, including audits, Research Ethics Committee review and regulatory inspections, by providing access to source data and other trial-related documentation, as required. Patient consent for this was obtained as part of the informed consent process for the trial.
Sample size
The primary outcome measure was unplanned hospitalisation-free survival, which is a composite end point of the time to death (all causes) or first non-elective (all-cause) hospital admission. The study duration was estimated to be 30 months’ recruitment phase and an additional 12 months’ follow-up after the last patient was recruited (a total of 42 months after the first patient was enrolled), which approximated to a median patient study duration of 27 months. The trial was designed to have 80% power (two-sided test, significance level of 5%) to show a change in hospitalisation-free survival from a median value of 28.8 months in the placebo group to 51.1 months in the co-trimoxazole group [hazard ratio (HR) of 0.56] over this study period, assuming that 264 patients were randomised. This was based on a sensitivity analysis of patients from the TIPAC trial21 with reduced lung function (i.e. FVC< 70% predicted) using an ITT analysis.
With regard to the power of the mechanistic studies, we assumed that 264 patients would provide data for the mechanistic aspect. This would provide 80% power to detect a difference of 6.7 mg/l in CRP concentration based on a SD of 19.38 mg/dl,71 of 0.51 ng/ml in MMP-7 concentration based on a SD of 1.48 ng/ml82 and of 99 ng/ml in SP-D concentration based on a SD of 212 ng/ml. 94 It was not possible to undertake a power calculation for the change in the microbiota. However, co-trimoxazole is effective against many of the organisms detected in BAL from routine culture and genotyping techniques and, therefore, we expected, within a proposed group of 50 patients, to be able to detect a change in the flora.
Statistical analysis
A statistical analysis plan (SAP) was produced and agreed with the TSC and DMC prior to analysis (see Appendix 1):
-
primary outcome –
-
time from randomisation to death (all causes), lung transplant or first non-elective hospital admission for any reason
-
-
secondary efficacy outcomes –
-
time from randomisation to death (all causes)
-
time from randomisation to first non-elective hospital admission for any reason
-
time from randomisation to lung transplantation
-
the K-BILD health-related quality-of-life questionnaire score, the MRC Breathlessness Score, the EuroQol-5 Dimensions (EQ-5D) quality-adjusted life-years assessment, CSS and quality-of-life LCQ score
-
lung function, including assessment by spirometry (FVC) and total DLCO
-
-
secondary outcome measures for safety (measured at local hospital laboratories) –
-
full blood count
-
urea and electrolytes
-
liver function
-
AEs including SAEs.
-
Additional analyses
In addition to the efficacy analyses, analyses were planned to attempt to correlate the change in clinical outcomes with the change in mechanical parameters. However, this was not undertaken given the findings of the study.
Analysis population and missing data
The analyses populations were defined as:
-
ITT – all randomised individuals regardless of adherence
-
PP – all randomised individuals who adhered to the study medication to within 80% (based on pill counts)
-
modified per protocol (mPP) – all randomised individuals who adhered to the high-dose regime
-
safety population – all patients randomised who received at least one dose of the study treatment.
The primary outcome analysis should not be subject to missing data, although the data will be incomplete due to right censoring; this was explicitly allowed for in the Cox proportional hazard modelling.
Missing secondary and mechanistic outcomes data were multiply imputed to increase the precision of the treatment effect estimates. Sensitivity analyses were conducted to assess the impact of the multiple imputations and a complete-case analysis was also conducted. All imputations were examined to ensure that sensible values were being generated. Imputation models contained baseline measures, outcome measures and factors predictive of missing data. For the imputation, a chained equation approach was used with the values of the outcomes at 12 months and at baseline. In addition, gender, randomisation group, body mass index and baseline IPF medications were included in the imputation model. As there was a high percentage of missing data (mainly because of death) at 12 months, a total of 45 imputed data sets were created. The analysis was run on each data set and then the results were combined using Rubin’s equations.
Individuals who met the primary end point or withdrew consent for collection of any outcome were censored at the last observation point; for example, data on the time until first hospitalisation were censored at the time of death.
Efficacy analyses
The primary outcome was analysed using a Cox proportional hazards model adjusted for the variables included in the minimisation algorithm: baseline licensed IPF medication and site (adjustment for bronchoscopy was planned but not undertaken given the number of patients who underwent this procedure). The results are presented as the Kaplan–Meier estimate of the survival function for each treatment group separately and the median time to outcome was estimated. The treatment effect size was the hazard ratio and was estimated with 95% CIs and a p-value.
The time until death and time until non-elective hospital admission were also analysed using Cox proportional hazards models adjusted for the variables included in the minimisation algorithm. Furthermore, they were also presented as the Kaplan–Meier estimates with hazard ratios, 95% CIs and a p-value.
At each relevant time point after 6 weeks post randomisation, the K-BILD, EQ-5D, LCQ, spirometry (FVC absolute value, FVC per cent predicted, FEV1 absolute and FEV1 per cent predicted) and DLCO scores were analysed using a linear model to compare the average values between the treatment groups, adjusted for the variables baseline licensed IPF medication and site as a random effect. The effect size was the mean difference and is presented with 95% CIs and p-values.
In addition to the above, a repeated measures model was undertaken including all post-randomisation observation for all individuals. An additional random effect for patients was included in the model. An overall p-value was given for treatment versus control, as well as at each time point. To control for multiplicity, the comparison at each time point was corrected using a Bonferroni adjustment.
The MRC Breathlessness Score and CSS were analysed using a Mann–Whitney U-test to compare the distribution of the scores between the treatment groups. A generalised effect size was estimated and presented with 95% CIs and a p-value.
Safety analysis
The safety analysis was based on the predefined population (as above). Summary tables are presented for incidence rates (number of patients experiencing at least one event) of AEs and SAEs, coded according to MedDRA (Medical Dictionary for Regulatory Activities). Tables of change from baseline are presented for the blood and other clinical laboratory assessments.
Mechanistic analysis
The same linear mixed model for the analysis of K-BILD scores was used for the biomarkers.
There were three protocol amendments, which are summarised in Table 2.
| Protocol version | Date | Summary of changes |
|---|---|---|
| 1.3 | 12 December 2014 | N/A – first submitted version |
| 2.0 | 2 February 2015 | Major changes (those not relating to administrative, typographical and formatting corrections) included:
|
| 3.0 | 16 May 2016 | Major changes (those not relating to administrative, typographical and formatting corrections) included:
|
Trial oversight committees
A TSC with independent members oversaw the conduct and progress of the trial. The committee met by teleconference every 6 months for the duration of the study and comprised the following individuals:
-
Professor Ron du Bois (chairperson) – no affiliation – retired
-
Dr Kim Harrison – Swansea University
-
Dr Sanjay Agrawal – University of Leicester
-
Professor Ann Millar – University of Bristol.
On 3 August 2016, University Hospitals of Leicester NHS Trust was added as a recruiting site with Dr Felix Woodhead as PI. As Dr Agrawal held a substantive contract with the same trust, to meet the National Institute for Health Research (NIHR)’s definition of independence (i.e. that the TSC member is not part of an institution acting as a recruiting centre), Professor Ann Millar joined the TSC to maintain the presence of three independent members on the TSC.
An independent DMC oversaw the safety of patients within the trial. The committee met by teleconference at least annually for the duration of the study and comprised the following individuals:
-
Dr Nik Hirani – University of Edinburgh
-
Professor Sarah Pett – University College London
-
Dr Jack Bowden – University of Bristol.
All TSC and DMC members were required to complete a Terms of Reference form and declare any potential competing interests.
Breaches and protocol deviations
Breaches of trial protocol or GCP were recorded and reported to the trial sponsor. Protocol deviations were recorded on the NCTU non-conformance database and were included in TMG, DMC and TSC reports.
A summary of breaches and protocol deviations is given in Appendix 1, Table 24. Patients who were the subject of a protocol deviation remained in the ITT population, the safety population and the PP population (if compliance criteria were met).
Chapter 3 Results
Screening and recruitment
Screening for patients started in April 2015 and ended in April 2018. The last follow-up assessment was in April 2019. A total of 54 sites screened patients and those sites are given in Table 3. The largest recruiting sites were Norwich, Royal Brompton, South Manchester and Aintree. A graph of recruitment against projected recruitment is given in Figure 2. The graph demonstrates that the study had a slower than expected start to recruitment, but that after around 7 months’ delay, recruitment ran parallel to the projected rate of recruitment.
| Site number | Site | Number screened | Number signing consent form | Number eligible | Number randomised | Date site opened |
|---|---|---|---|---|---|---|
| 1 | Norwich | 244 | 31 | 29 | 29 | 1 April 2015 |
| 2 | Papworth | 71 | 23 | 16 | 15 | 19 January 2016 |
| 3 | Royal Brompton | 116 | 26 | 20 | 20 | 21 September 2015 |
| 4 | Sheffield | 37 | 12 | 9 | 8 | 23 July 2015 |
| 5 | Birmingham | 25 | 19 | 18 | 18 | 6 August 2015 |
| 6 | Heart of England | 7 | 7 | 6 | 6 | 20 December 2016 |
| 7 | North Midlands | 16 | 15 | 12 | 12 | 14 July 2015 |
| 8 | Bristol | 14 | 14 | 14 | 14 | 18 August 2015 |
| 9 | University Hospital Wales | 10 | 10 | 9 | 9 | 2 March 2017 |
| 11 | Newcastle | 155 | 13 | 12 | 12 | 30 August 2016 |
| 12 | Gateshead | 7 | 6 | 6 | 6 | 8 December 2015 |
| 13 | Salford | 10 | 3 | 3 | 3 | 4 August 2015 |
| 14 | South Manchester | 40 | 35 | 27 | 25 | 25 September 2015 |
| 15 | Aintree | 159 | 27 | 23 | 21 | 10 March 2016 |
| 17 | Lancashire | 13 | 9 | 7 | 7 | 12 October 2015 |
| 18 | Aberdeen | 8 | 8 | 7 | 7 | 22 February 2016 |
| 19 | Greater Glasgow & Clyde | 5 | 5 | 3 | 3 | 8 October 2015 |
| 20 | Peterborough & Stamford | 0 | 0 | 0 | 0 | 15 December 2015 |
| 22 | Oxford | 10 | 10 | 8 | 8 | 11 August 2016 |
| 24 | Imperial College | 32 | 4 | 4 | 4 | 5 November 2015 |
| 25 | NHS Tayside | 12 | 12 | 12 | 12 | 6 November 2015 |
| 27 | Royal Devon & Exeter | 16 | 4 | 4 | 4 | 2 May 2017 |
| 28 | Hull & East Yorkshire | 22 | 19 | 14 | 14 | 25 February 2016 |
| 29 | Nottingham | 14 | 6 | 6 | 6 | 27 November 2017 |
| 30 | Cambridge University Hospitals | 8 | 3 | 2 | 2 | 6 August 2015 |
| 31 | Leicester | 6 | 6 | 6 | 6 | 3 August 2016 |
| 32 | University College London | 14 | 5 | 4 | 3 | 9 December 2015 |
| 33 | Blackpool, Fylde and Wyre | 42 | 10 | 10 | 10 | 3 July 2015 |
| 34 | Shrewsbury & Telford | 17 | 5 | 5 | 5 | 23 September 2015 |
| 35 | Sherwood Forest Hospitals | 7 | 5 | 3 | 2 | 11 November 2015 |
| 36 | St George’s University | 12 | 9 | 7 | 7 | 25 January 2016 |
| 39 | Worcestershire | 1 | 1 | 1 | 1 | 14 July 2016 |
| 40 | Western Health & Social Care | 22 | 9 | 7 | 7 | 22 October 2015 |
| 41 | Royal Wolverhampton | 8 | 8 | 6 | 6 | 12 April 2016 |
| 42 | Southampton | 14 | 9 | 8 | 8 | 22 November 2016 |
| 44 | Morecambe Bay | 32 | 9 | 8 | 8 | 22 June 2016 |
| 47 | Mid Cheshire | 1 | 0 | 0 | 0 | 12 May 2016 |
| 48 | Calderdale & Huddersfield | 6 | 3 | 3 | 3 | 8 April 2016 |
| 50 | South Tyneside | 8 | 8 | 5 | 5 | 15 June 2016 |
| 53 | Forth Valley | 4 | 1 | 1 | 1 | 17 November 2017 |
| 54 | Coventry | 60 | 11 | 5 | 5 | 6 March 2017 |
| Total | 1305 | 420 | 350 | 342 | ||
FIGURE 2.
Graph of actual and projected recruitment.
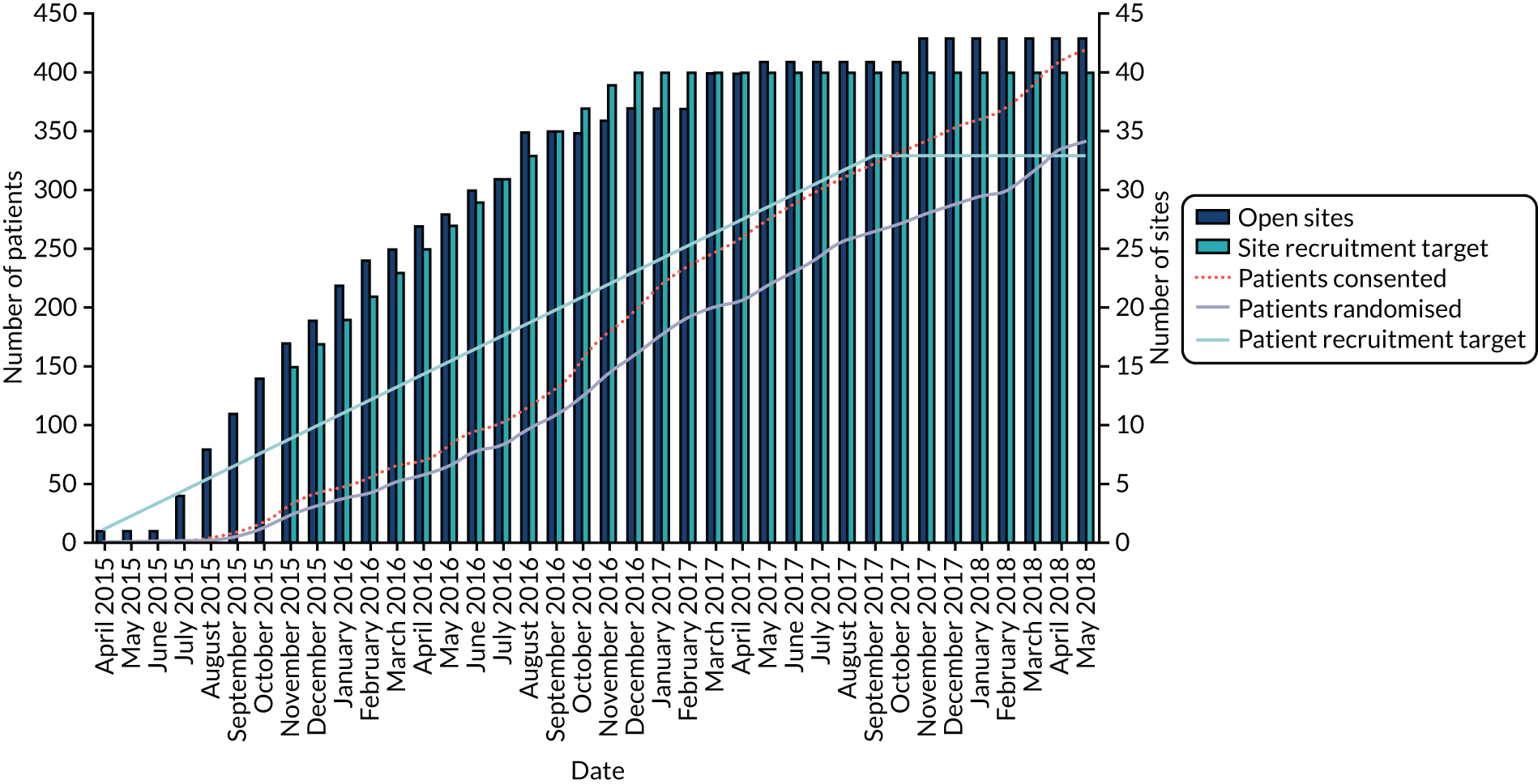
Eligibility violations
One patient was randomised in error. The patient had a lung function that was too high and violated the inclusion criteria. The decision was made to exclude this person from the analysis.
Patient flow
The Consolidated Standards of Reporting Trials (CONSORT) flow chart for this trial, in respect of the primary outcome, is provided in Figure 3. In summary, a total of 1305 patients were screened, of whom 420 were assessed for eligibility, 349 met the criteria and 342 were randomised. Of the 342 randomised patients, 172 were randomised to the placebo group and 170 were randomised to the active treatment group. One patient, randomised to the active treatment group, was randomised in error and their data were not analysed. A total of 58 randomised patients dropped out of the study (the reasons are given in Appendix 1, Table 25). A total follow-up of 394.6 person-years was observed with 164 events.
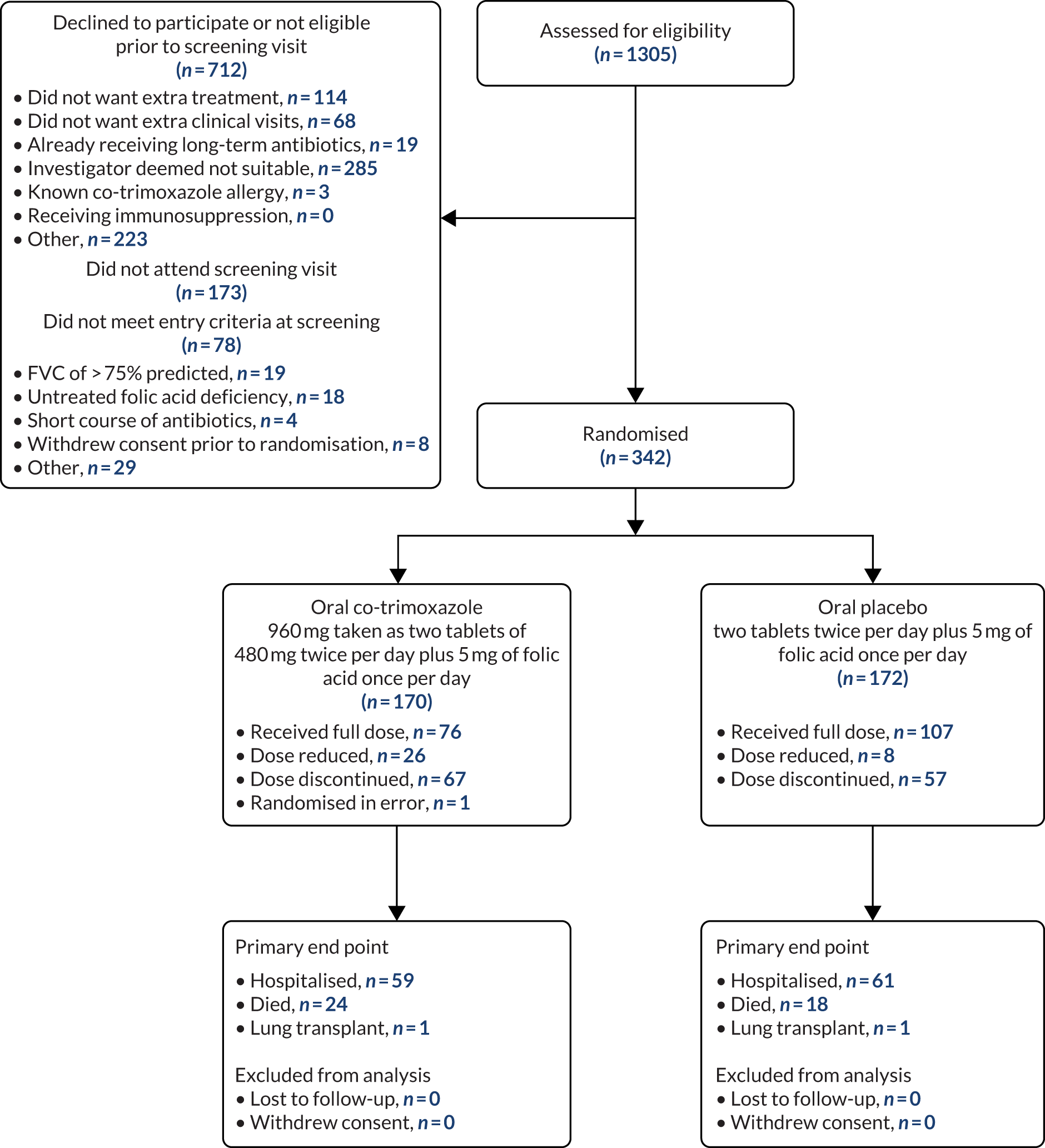
Patient withdrawal
A total of 58 (17%) patients withdrew from the study: 32 (19%) from the active treatment group and 26 (15%) from the placebo group. The reasons for withdrawal are given in Appendix 1, Table 25.
Baseline characteristics
Overall, the baseline characteristics and other factors at baseline were balanced between the two treatment groups, as shown in Table 4. The mean age was 71.9 years for active treatment patients and 70.7 years for placebo patients. Fewer male patients than female patients were randomised in the active treatment group (n = 138, 81.7%) compared with the placebo group (n = 157, 91.3%); this difference was due to chance. More patients with diabetes mellitus were randomised to the active treatment group (n = 40, 23.7%) than to the placebo group (n = 25, 14.5%); this difference was due to chance. The average FVC per cent predicted was 56.2 l in the active treatment group, compared with 55.2 l in the placebo group.
| Baseline characteristic | Treatment group | |
|---|---|---|
| Active treatment | Placebo | |
| Number of patients in group | 169 | 172 |
| Male patients, n (%) | 138 (81.7) | 157 (91.3) |
| Age in years, mean (SD) | 71.9 (7.8) | 70.7 (7.1) |
| Smoking status, n (%) | ||
| Never smoked | 59 (34.9) | 56 (32.6) |
| Ex-smoker | 109 (64.5) | 114 (66.3) |
| Current smoker | 1 (0.6) | 2 (1.2) |
| Comorbidities, n (%) | ||
| COPD | 6 (3.6) | 6 (3.5) |
| Bronchiectasis | 2 (1.2) | 7 (4.1) |
| Ischaemic heart or angina | 38 (22.5) | 44 (25.6) |
| GORD | 69 (40.8) | 62 (36.0) |
| Diabetes mellitus | 40 (23.7) | 25 (14.5) |
| Osteoporosis | 11 (6.5) | 11 (6.4) |
| Pulmonary hypertension | 13 (7.7) | 10 (5.8) |
| Anxiety or depression | 17 (10.1) | 23 (13.4) |
| Medications, n (%) | ||
| Pirfenidone | 71 (42.0) | 66 (38.4) |
| N-acetylcysteine | 8 (4.7) | 7 (4.1) |
| Other antioxidants | 3 (1.8) | 5 (2.9) |
| Prednisolone | 12 (7.1) | 10 (5.8) |
| Nintedanib | 56 (33.1) | 61 (35.5) |
| Proton pump inhibitor | 87 (51.5) | 78 (45.3) |
| Lung tests, mean (SD) | ||
| Absolute value | ||
| FVC (l) | 2.2 (0.6) | 2.3 (0.5) |
| FEV1 (l) | 1.9 (0.5) | 1.9 (0.4) |
| FEV1/FVC ratio | 0.8 (0.1) | 0.8 (0.1) |
| DLCO (mmol/minute/kPa) | 3.6 (1.8) | 3.7 (1.5) |
| Per cent predicted | ||
| FVC | 56.2 (8.9) | 55.2 (10.0) |
| FEV1 | 61.5 (9.3) | 60.0 (10.6) |
| DLCO | 43.3 (20.2) | 44.5 (18.0) |
| Minimisation factor, n (%) | ||
| Number on licensed IPF medication | 126 (74.6) | 127 (73.8) |
| Study site, n (%) | ||
| Norwich | 14 (8.3) | 15 (8.7) |
| Papworth | 7 (4.1) | 8 (4.7) |
| Royal Brompton | 10 (5.9) | 10 (5.8) |
| Sheffield | 4 (2.4) | 4 (2.3) |
| Birmingham | 8 (4.7) | 10 (5.8) |
| Heart of England | 3 (1.8) | 3 (1.7) |
| North Midlands | 6 (3.6) | 6 (3.5) |
| Bristol | 8 (4.7) | 6 (3.5) |
| University Hospital Wales | 4 (2.4) | 4 (2.3) |
| Newcastle | 6 (3.6) | 6 (3.5) |
| Gateshead | 3 (1.8) | 3 (1.7) |
| Salford | 2 (1.2) | 1 (0.6) |
| South Manchester | 13 (7.7) | 12 (7.0) |
| Aintree | 11 (6.5) | 10 (5.8) |
| Lancashire | 3 (1.8) | 4 (2.3) |
| Aberdeen | 3 (1.8) | 4 (2.3) |
| Greater Glasgow & Clyde | 2 (1.2) | 1 (0.6) |
| Oxford | 3 (1.8) | 5 (2.9) |
| Imperial College | 1 (0.6) | 3 (1.7) |
| NHS Tayside | 6 (3.6) | 6 (3.5) |
| Royal Devon & Exeter | 2 (1.2) | 2 (1.2) |
| Hull & East Yorkshire | 7 (4.1) | 7 (4.1) |
| Nottingham | 3 (1.8) | 3 (1.7) |
| Cambridge University Hospitals | 1 (0.6) | 1 (0.6) |
| Leicester | 2 (1.2) | 4 (2.3) |
| University College London | 1 (0.6) | 2 (1.2) |
| Blackpool, Fylde and Wyre | 6 (3.6) | 4 (2.3) |
| Shrewsbury & Telford | 3 (1.8) | 2 (1.2) |
| Sherwood Forest Hospitals | 0 (0.0) | 2 (1.2) |
| St George’s University | 3 (1.8) | 4 (2.3) |
| Worcestershire | 0 (0.0) | 1 (0.6) |
| Western Health & Social Care | 4 (2.4) | 3 (1.7) |
| Royal Wolverhampton | 3 (1.8) | 3 (1.7) |
| Southampton | 5 (3.0) | 3 (1.7) |
| Morecambe Bay | 4 (2.4) | 4 (2.3) |
| Calderdale & Huddersfield | 2 (1.2) | 1 (0.6) |
| South Tyneside | 3 (1.8) | 2 (1.2) |
| Forth Valley | 1 (0.6) | 0 (0.0) |
| Coventry | 2 (1.2) | 3 (1.7) |
| Outcome measures | ||
| MRC, n (%) | ||
| 1: not troubled by breathlessness apart from on strenuous exercise | 6 (3.6) | 7 (4.1) |
| 2: short of breath when hurrying on the level or walking up a slight hill | 72 (43.1) | 84 (49.1) |
| 3: walks slower than contemporaries on level ground because of breathlessness or has to stop for breath when walking at own pace | 50 (29.9) | 39 (22.8) |
| 4: stops for breath after walking about 100 yards or after a few minutes on level ground | 27 (16.2) | 31 (18.1) |
| 5: too breathless to leave the house or breathless when undressing | 12 (7.2) | 10 (5.8) |
| MRC score, median (IQR) | 3.00 (2.0–3.00) | 2.00 (2.00–3.00) |
| EQ-5D utility score, mean (SD) | 0.67 (0.20) | 0.69 (0.22) |
| CSS, mean (SD) | 39.37 (27.45) | 40.89 (26.58) |
| LCQ score, mean (SD) | ||
| Total | 16.08 (3.55) | 15.76 (3.73) |
| Physical | 5.22 (1.06) | 5.12 (1.04) |
| Psychological | 5.37 (1.37) | 5.32 (1.49) |
| Social | 5.39 (1.41) | 5.36 (1.39) |
| K-BILD score, mean (SD) | ||
| Psychological | 55.16 (14.88) | 54.92 (17.11) |
| Breathless | 37.68 (15.30) | 38.90 (14.30) |
| Chest | 62.95 (20.83) | 62.55 (20.67) |
| Total | 53.70 (9.71) | 53.55 (10.64) |
Most patients were on some form of medication, with almost 50% being on proton pump inhibitors at baseline (51.5% in the active treatment group and 45.3% in the placebo group). Approximately 40% of patients were taking pirfenidone: 42% in the active treatment group and 38.4% in the placebo group.
The baseline characteristics of patients included in the PP and mPP samples are given in Table 5 and Appendix 1, Table 26.
| Baseline characteristic | Treatment group | |
|---|---|---|
| Active treatment | Placebo | |
| Number of patients in group | 120 | 124 |
| Male patients, n (%) | 104 (86.7) | 116 (93.5) |
| Age in years, mean (SD) | 70.90 (7.89) | 70.62 (6.95) |
| Smoking status, n (%) | ||
| Never smoked | 37 (30.8) | 42 (33.9) |
| Ex-smoker | 83 (69.2) | 81 (65.3) |
| Current smoker | 0 (0.0) | 1 (0.8) |
| Comorbidities, n (%) | ||
| COPD | 4 (3.3) | 2 (1.6) |
| Bronchiectasis | 1 (0.8) | 4 (3.2) |
| Ischaemic heart disease or angina | 28 (23.3) | 31 (25.0) |
| GORD | 47 (39.2) | 42 (33.9) |
| Diabetes mellitus | 32 (26.7) | 21 (16.9) |
| Osteoporosis | 7 (5.8) | 9 (7.3) |
| Pulmonary hypertension | 10 (8.3) | 5 (4.0) |
| Anxiety or depression | 11 (9.2) | 17 (13.7) |
| Medications, n (%) | ||
| Pirfenidone | 51 (42.5) | 53 (42.7) |
| N-acetylcysteine | 7 (5.8) | 6 (4.8) |
| Other antioxidants | 2 (1.7) | 4 (3.2) |
| Prednisolone | 9 (7.5) | 9 (7.3) |
| Nintedanib | 42 (35.0) | 40 (32.3) |
| Proton pump inhibitor | 62 (51.7) | 52 (41.9) |
| Lung tests, mean (SD) | ||
| Absolute value | ||
| FVC (l) | 2.31 (0.56) | 2.24 (0.53) |
| FEV1 (l) | 1.93 (0.45) | 1.89 (0.43) |
| FEV1/FVC ratio | 0.84 (0.07) | 0.85 (0.11) |
| DLCO (mmol/minute/kPa) | 3.70 (1.91) | 3.73 (1.70) |
| Per cent predicted | ||
| FVC | 56.54 (8.98) | 54.37 (10.40) |
| FEV1 | 61.50 (9.50) | 59.70 (11.15) |
| DLCO | 43.67 (21.46) | 44.47 (19.48) |
| Minimisation factor, n (%) | ||
| Number on licensed IPF medication | 92 (76.7) | 93 (75.0) |
| Study site, n (%) | ||
| Norwich | 9 (7.5) | 11 (8.9) |
| Papworth | 6 (5.0) | 6 (4.8) |
| Royal Brompton | 9 (7.5) | 8 (6.5) |
| Sheffield | 3 (2.5) | 3 (2.4) |
| Birmingham | 6 (5.0) | 6 (4.8) |
| Heart of England | 2 (1.7) | 2 (1.6) |
| North Midlands | 3 (2.5) | 6 (4.8) |
| Bristol | 7 (5.8) | 3 (2.4) |
| University Hospital Wales | 3 (2.5) | 3 (2.4) |
| Newcastle | 4 (3.3) | 5 (4.0) |
| Gateshead | 3 (2.5) | 2 (1.6) |
| Salford | 2 (1.7) | 1 (0.8) |
| South Manchester | 10 (8.3) | 10 (8.1) |
| Aintree | 7 (5.8) | 7 (5.6) |
| Lancashire | 3 (2.5) | 4 (3.2) |
| Aberdeen | 3 (2.5) | 4 (3.2) |
| Greater Glasgow & Clyde | 1 (0.8) | 1 (0.8) |
| Oxford | 3 (2.5) | 5 (4.0) |
| Imperial College | 0 (0.0) | 3 (2.4) |
| NHS Tayside | 3 (2.5) | 2 (1.6) |
| Royal Devon & Exeter | 2 (1.7) | 1 (0.8) |
| Hull & East Yorkshire | 4 (3.3) | 7 (5.6) |
| Nottingham | 1 (0.8) | 2 (1.6) |
| Cambridge University Hospitals | 1 (0.8) | 1 (0.8) |
| Leicester | 2 (1.7) | 2 (1.6) |
| University College London | 0 (0.0) | 0 (0.0) |
| Blackpool, Fylde and Wyre | 2 (1.7) | 2 (1.6) |
| Shrewsbury & Telford | 2 (1.7) | 0 (0.0) |
| Sherwood Forest Hospitals | 0 (0.0) | 2 (1.6) |
| St George’s University | 2 (1.7) | 3 (2.4) |
| Worcestershire | 0 (0.0) | 1 (0.8) |
| Western Health & Social Care | 4 (3.3) | 3 (2.4) |
| Royal Wolverhampton | 2 (1.7) | 2 (1.6) |
| Southampton | 4 (3.3) | 1 (0.8) |
| Morecambe Bay | 2 (1.7) | 2 (1.6) |
| Calderdale & Huddersfield | 0 (0.0) | 0 (0.0) |
| South Tyneside | 2 (1.7) | 2 (1.6) |
| Forth Valley | 1 (0.8) | 0 (0.0) |
| Coventry | 2 (1.7) | 1 (0.8) |
| Outcome measures | ||
| MRC, n (%) | ||
| 1: not troubled by breathlessness apart from on strenuous exercise | 5 (4.2) | 4 (3.3) |
| 2: short of breath when hurrying on the level or walking up a slight hill | 53 (44.5) | 61 (49.6) |
| 3: walks slower than contemporaries on level ground because of breathlessness or has to stop for breath when walking at own pace | 36 (30.3) | 26 (21.1) |
| 4: stops for breath after walking about 100 yards or after a few minutes on level ground | 21 (17.6) | 23 (18.7) |
| 5: too breathless to leave the house or breathless when undressing | 4 (3.4) | 9 (7.3) |
| MRC score, median (IQR) | 3.00 (2.0–3.00) | 2.00 (2.00–4.00) |
| EQ-5D utility score, mean (SD) | 0.67 (0.20) | 0.69 (0.22) |
| CSS, mean (SD) | 37.92 (27.73) | 40.92 (27.03) |
| LCQ score, mean (SD) | 0.67 (0.19) | 0.69 (0.24) |
| Total | 16.33 (3.38) | 15.86 (3.67) |
| Physical | 5.33 (1.00) | 5.16 (1.04) |
| Psychological | 5.41 (1.37) | 5.37 (1.46) |
| Social | 5.44 (1.36) | 5.38 (1.39) |
| K-BILD score, mean (SD) | ||
| Psychological | 54.64 (14.62) | 55.07 (16.71) |
| Breathless | 38.26 (14.60) | 38.80 (14.69) |
| Chest | 62.57 (21.66) | 62.47 (21.05) |
| Total | 53.57 (9.71) | 53.61 (10.49) |
Adherence/treatment received
Compliance data were available for 339 (99%) patients. The average (SD) compliance in the active treatment group was 81.4% (22.8%), compared with 85.5% (21.7%) in the placebo group. The percentage of patients who met the 80% threshold was roughly equal in both treatment groups: 120 (71.9%) in the active treatment group compared with 125 (72.1%) in the placebo group.
Dose reduction occurred in 31 out of 167 (19%) patients in the co-trimoxazole group and 16 out of 172 (9%) patients in the placebo group. This generally occurred in the first visits at either 3 or 6 months. Data are presented in Appendix 1, Table 27. Only three patients stopped taking medication because of increased potassium levels: one in the active treatment group and two in the placebo group.
The percentage of individuals who were prescribed concomitant medication is given in Table 6. Overall, the percentage of individuals is similar in both treatment groups, with the only significant differences being in more antifungal and oral supplements in the placebo group and more endocrinological drugs in the active treatment group.
| Drug class | Treatment group, n (%) | p-value | |
|---|---|---|---|
| Active treatment (N = 169) | Placebo (N = 172) | ||
| Antibiotic | 52 (30.8) | 61 (35.5) | 0.36 |
| Antifungal | 1 (0.6) | 8 (4.7) | 0.019 |
| Antioxidant | 31 (18.3) | 40 (23.3) | 0.26 |
| Antiviral | 3 (1.8) | 3 (1.7) | 0.98 |
| Neurological | 47 (27.8) | 48 (27.9) | 0.98 |
| Cardiovascular | 96 (56.8) | 97 (56.4) | 0.94 |
| Dermatological | 8 (4.7) | 7 (4.1) | 0.76 |
| Diabetic | 27 (16.0) | 23 (13.4) | 0.50 |
| Endocrinological | 60 (35.5) | 43 (25.0) | 0.035 |
| Gastroenterological | 115 (68.0) | 127 (73.8) | 0.24 |
| Haematological | 16 (9.5) | 23 (13.4) | 0.26 |
| Mouth/oral | 1 (0.6) | 4 (2.3) | 0.18 |
| Musculoskeletal | 33 (19.5) | 33 (19.2) | 0.94 |
| Ocular | 10 (5.9) | 4 (2.3) | 0.095 |
| Oral supplements | 12 (7.1) | 24 (14.0) | 0.039 |
| Respiratory | 96 (56.8) | 94 (54.7) | 0.69 |
| Urogenital | 17 (10.1) | 24 (14.0) | 0.27 |
| Vaccination | 9 (5.3) | 4 (2.3) | 0.15 |
Primary outcome
A total of 164 events occurred for the primary outcome: 80 in the placebo group and 84 in the active treatment group. The total exposure time was 394.6 years, roughly an average of 1.2 person-years. The rate of events was 0.45 (84/185.6) per person-year in the active treatment group and 0.38 per person-year (80/209.1) in the placebo group. The estimated unadjusted hazard ratio was 1.2 (95% CI 0.9 to 1.6) and when adjusting for the factors used in the minimisation algorithm was virtually unchanged at 1.2 (95% CI 0.9 to 1.6), as shown in Table 7. The time to event is displayed graphically in Figure 4. This demonstrates some crossing of the survival curves, but, overall, the proportional hazards assumption was found not to be violated (p = 0.976). The median survival time was 531 days in the active treatment group and 709 days in the placebo group.
| Primary outcome | Treatment group | Hazard ratio (active treatment vs. placebo) | Total exposure time (years) | |||
|---|---|---|---|---|---|---|
| Active treatment (N = 169) | Placebo (N = 172) | |||||
| Total exposure time (years) | Number of events to date (incidence) | Total exposure time (years) | Number of events to date (incidence) | |||
| ITT | 185.6 | 84 | 209.1 | 80 | Unadjusted:a 1.2 (95% CI 0.9 to 1.6); p = 0.328 | 394.6 |
| Adjusted:b 1.2 (95% CI 0.9 to 1.6); p = 0.319 | ||||||
| PP | 132.6 | 59 | 132.0 | 64 | Unadjusted:a 0.9 (95% CI 0.7 to 1.3); p = 0.700 | 264.6 |
| Adjusted:b 0.9 (95% CI 0.7 to 1.3); p = 0.760 | ||||||
| mPP | 92.5 | 44 | 123.7 | 62 | Unadjusted:a 0.9 (95% CI 0.6 to 1.4) | 216.2 |
| Adjusted:b 0.9 (95% CI 0.6 to 1.4) | ||||||
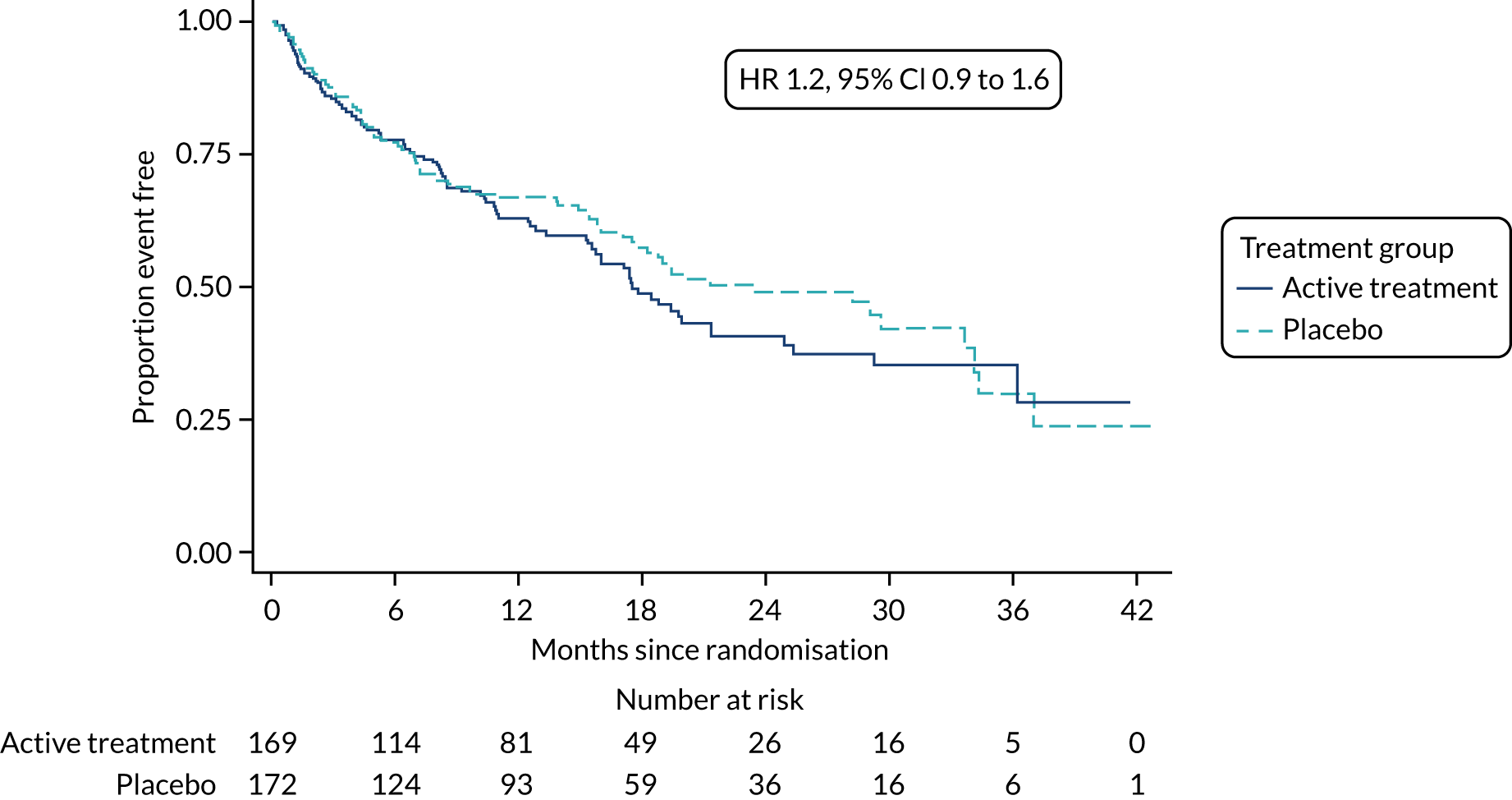
Per-protocol analysis
A total of 123 events occurred for the primary outcome: 64 in the placebo group and 59 in the active treatment group. The total exposure time was 264.6 years, roughly an average of 1.08 person-years. The rate of events was 0.44 (59/132.6) per person-year in the active treatment group and 0.48 per person-year (64/132) in the placebo group. The estimated unadjusted hazard ratio was 0.9 (95% CI 0.7 to 1.3) and when adjusting for the factors used in the minimisation algorithm it was virtually unchanged at 0.9 (95% CI 0.7 to 1.3), as shown in Table 7. The time to event is displayed graphically in Figure 5. This figure demonstrates some crossing of the survival curves, but overall the proportional hazards assumption was found not to be violated (p = 0.9908).
FIGURE 5.
Kaplan–Meier estimate of time to event: PP.
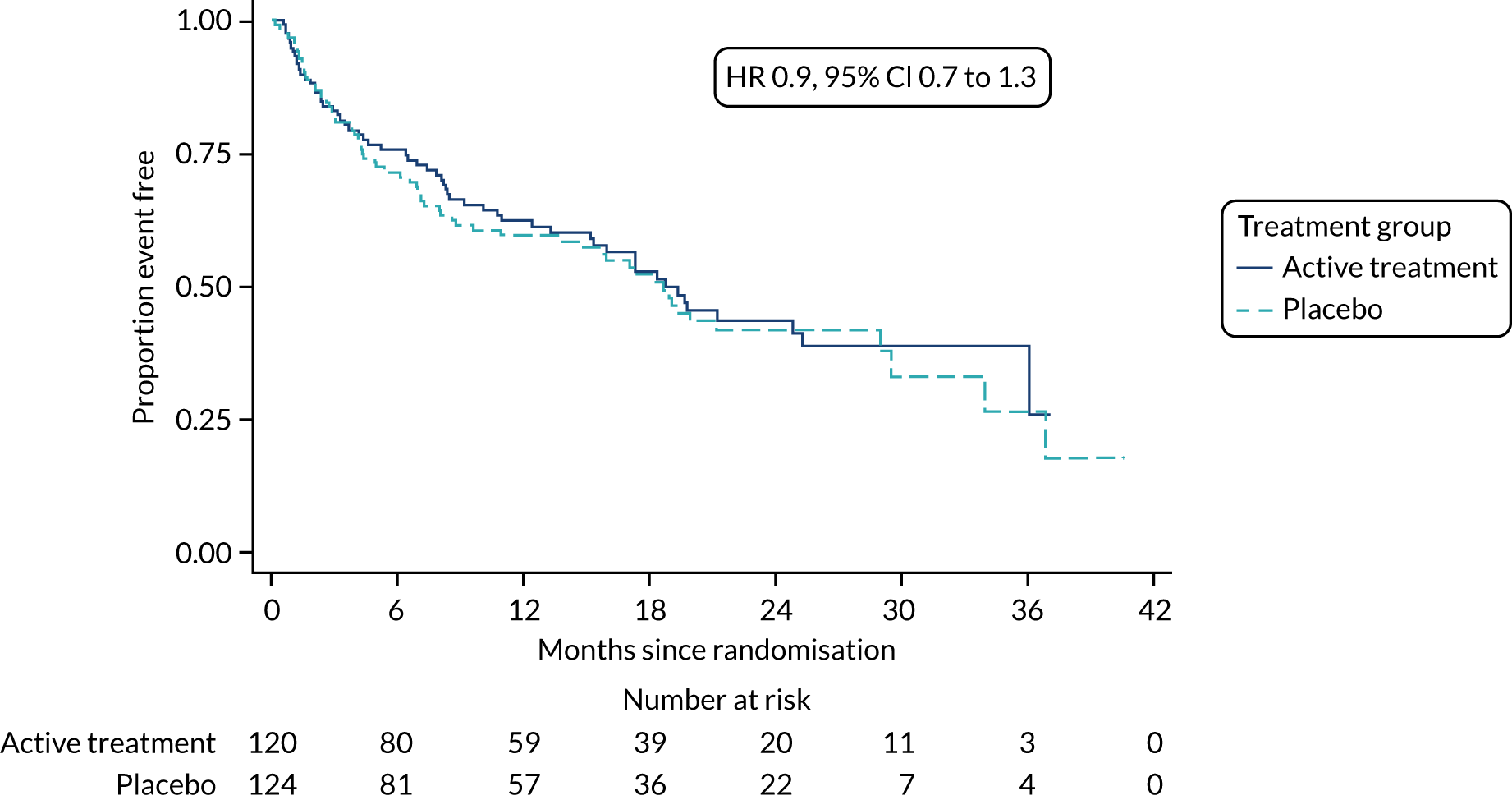
Modified per-protocol analysis
A total of 106 events occurred for the primary outcome: 44 in the placebo group and 62 in the active treatment group. The total exposure time was 216.2 years, roughly an average of 1.02 person-years. The rate of events was 0.48 (44/92.5) per person-year in the active treatment group and 0.50 per person-year (62/123.7) in the placebo group. The estimated unadjusted hazard ratio was 0.9 (95% CI 0.6 to 1.4) and when adjusting for the factors used in the minimisation algorithm it was virtually unchanged at 0.9 (95% CI 0.6 to 1.4), as shown in Table 7. The time to event is displayed graphically in Figure 6. This demonstrates some crossing of the survival curves, but overall the proportional hazards assumption was found not to be violated (p = 0.3193).
FIGURE 6.
Kaplan–Meier estimate of time to event: mPP.
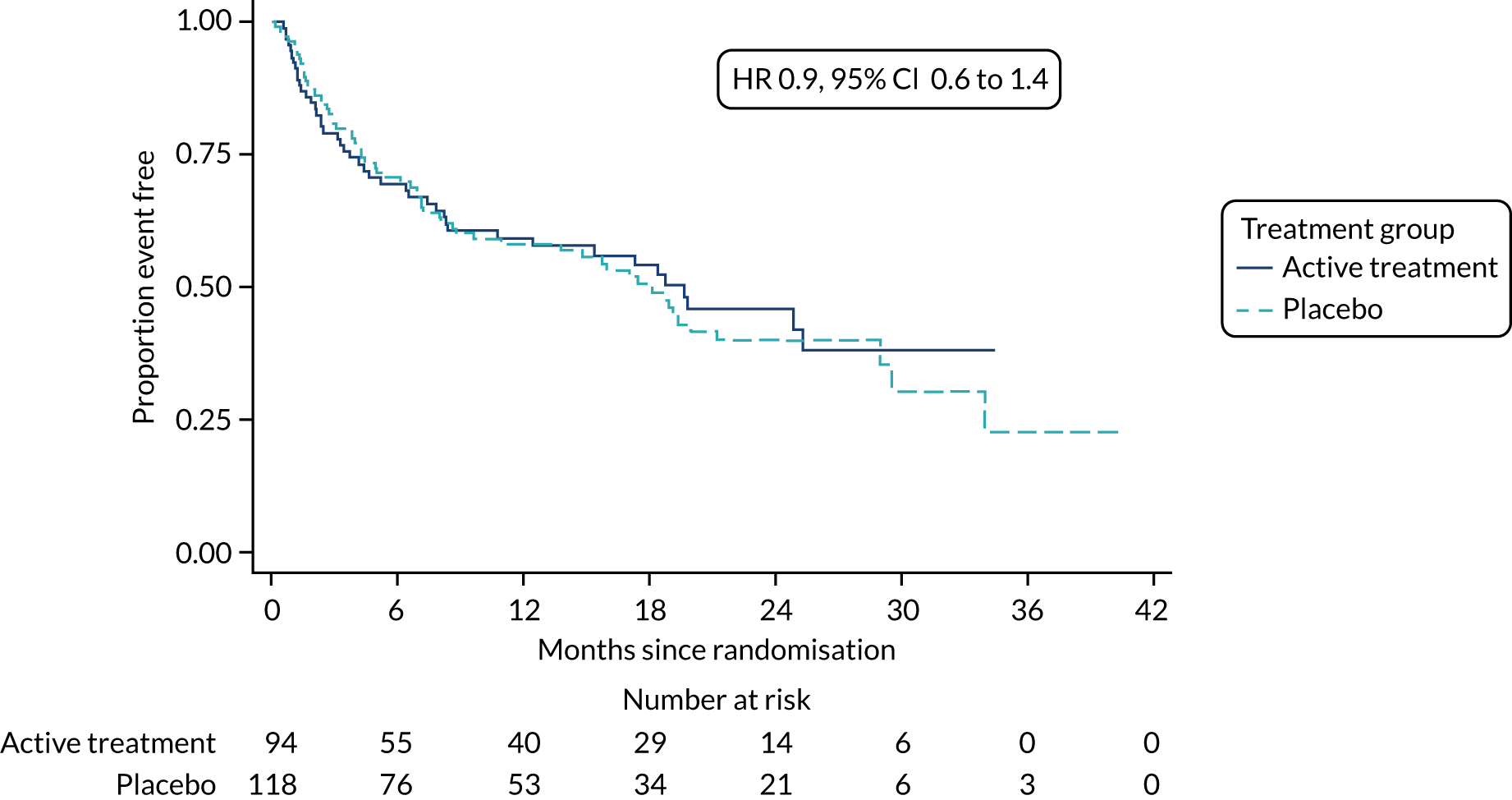
Secondary outcomes
Time-to-event outcomes
Intention to treat
The individual component events of the primary outcome are given in Table 8 in the same format as that of the primary outcome. The number of events is reduced compared with the primary outcome so that the CIs are correspondingly wider. The total exposure time is the same for all outcomes as once one event had occurred the follow-up was censored for all other events. The number of deaths was higher in the active treatment group than in the placebo group, with 24 and 18 deaths, respectively, but the difference was not significant (HR 1.5, 95% CI 0.8 to 2.8; p = 0.167). Respiratory-related deaths were, similarly, higher in the active treatment group than in the placebo group, with 20 and 17 deaths, respectively, but the difference was not significant (HR 1.4, 95% CI 0.7 to 2.6; p = 0.343). Hospital admissions were roughly equal in both the active treatment and placebo groups, 59 and 61, respectively; but the difference was not significant (HR 1.1, 95% CI 0.7 to 1.5; p = 0.754). Respiratory-related hospitalisations were roughly equal in both the active treatment and placebo groups, 40 and 42, respectively, but the difference was not significant (HR 1.0, 95% CI 0.7 to 1.6; p = 0.827). In all cases, the unadjusted and adjusted analyses were practically the same. As the Kaplan–Meier plot would provide a biased estimate due to informative censoring, a cumulative incidence plot is provided for each outcome in Figures 7 and 8. A separate analysis was not carried out for lung transplant as only one event occurred in each treatment group.
| Secondary outcome | Treatment group | Hazard ratio (active treatment vs. placebo) | |||
|---|---|---|---|---|---|
| Active treatment (N = 169) | Placebo (N = 172) | ||||
| Total exposure time (years) | Number of events to date (incidence) | Total exposure time (years) | Number of events to date (incidence) | ||
| ITT | |||||
| Deaths censored at date of primary event (if primary event not death) | 185.6 | 24 | 209.1 | 18 | Unadjusted:a 1.5 (95% CI 0.8 to 2.8); p = 0.167 |
| Adjusted:b 1.5 (95% CI 0.8 to 2.8); p = 0.169 | |||||
| Respiratory-related death (censored at time of primary event) | 185.6 | 20 | 209.1 | 17 | Unadjusted:a 1.4 (95% CI 0.7 to 2.6); p = 0.343 |
| Adjusted:b 1.4 (95% CI 0.7 to 2.6); p = 0.352 | |||||
| Non-elective hospital admissions (all cause) | 185.6 | 59 | 209.1 | 61 | Unadjusted:a 1.1 (95% CI 0.7 to 1.5); p = 0.754 |
| Adjusted:b 1.1 (95% CI 0.7 to 1.3); p = 0.731 | |||||
| Respiratory-related hospitalisation (censored at time of primary event) | 185.6 | 40 | 209.1 | 42 | Unadjusted:a 1.0 (95% CI 0.7 to 1.6); p = 0.857 |
| Adjusted:b 1.0 (95% 0.7 to 1.6); p = 0.827 | |||||
| PP | |||||
| Deaths censored at date of primary event (if primary event not death) | 132.6 | 13 | 132.0 | 12 | Unadjusted:a 1.1 (95% CI 0.5 to 2.4); p = 0.824 |
| Adjusted:b 1.1 (95% CI 0.5 to 2.4); p = 0.812 | |||||
| Respiratory-related death (censored at time of primary event) | 132.7 | 12 | 132 | 11 | Unadjusted:a 1.1 (95% CI 0.5 to 2.5); p = 0.821 |
| Adjusted:b 1.1 (95% CI 0.5 to 2.5); p = 0.821 | |||||
| Non-elective hospital admissions (all cause) | 132.6 | 45 | 132.0 | 51 | Unadjusted:a 1.1 (95% CI 0.7 to 1.7); p = 0.581 |
| Adjusted:b 0.9 (95% CI 0.6 to 1.4); p = 0.644 | |||||
| Respiratory-related hospitalisation (censored at time of primary event) | 132.6 | 29 | 132.0 | 35 | Unadjusted:a 0.8 (95% CI 0.5 to 1.3); p = 0.451 |
| Adjusted:b 0.8 (95% CI 0.5 to 1.4); p = 0.487 | |||||
| mPP | |||||
| Deaths censored at date of primary event (if primary event not death) | 92.5 | 11 | 123.7 | 12 | Unadjusted:a 1.2 (95% CI 0.5 to 2.8); p = 0.597 |
| Adjusted:b 1.3 (95% CI 0.6 to 2.9); p = 0.585 | |||||
| Respiratory-related death (censored at time of primary event) | 92.5 | 10 | 123.7 | 11 | Unadjusted:a 1.2 (95% CI 0.5 to 2.9); p = 0.630 |
| Adjusted:b 1.2 (95% CI 0.5 to 2.9); p = 0.624 | |||||
| Non-elective hospital admissions (all cause) | 92.5 | 32 | 123.7 | 49 | Unadjusted:a 0.9 (95% CI 0.5 to 1.3); p = 0.487 |
| Adjusted:b 0.9 (95% CI 0.6 to 1.3); p = 0.511 | |||||
| Respiratory-related hospitalisation (censored at time of primary event) | 92.5 | 23 | 123.7 | 34 | Unadjusted:a 0.9 (95% CI 0.5 to 1.5); p = 0.635 |
| Adjusted:b 0.9 (95% 0.5 to 1.5); p = 0.657 | |||||
FIGURE 7.
Cumulative incidence function: death only.
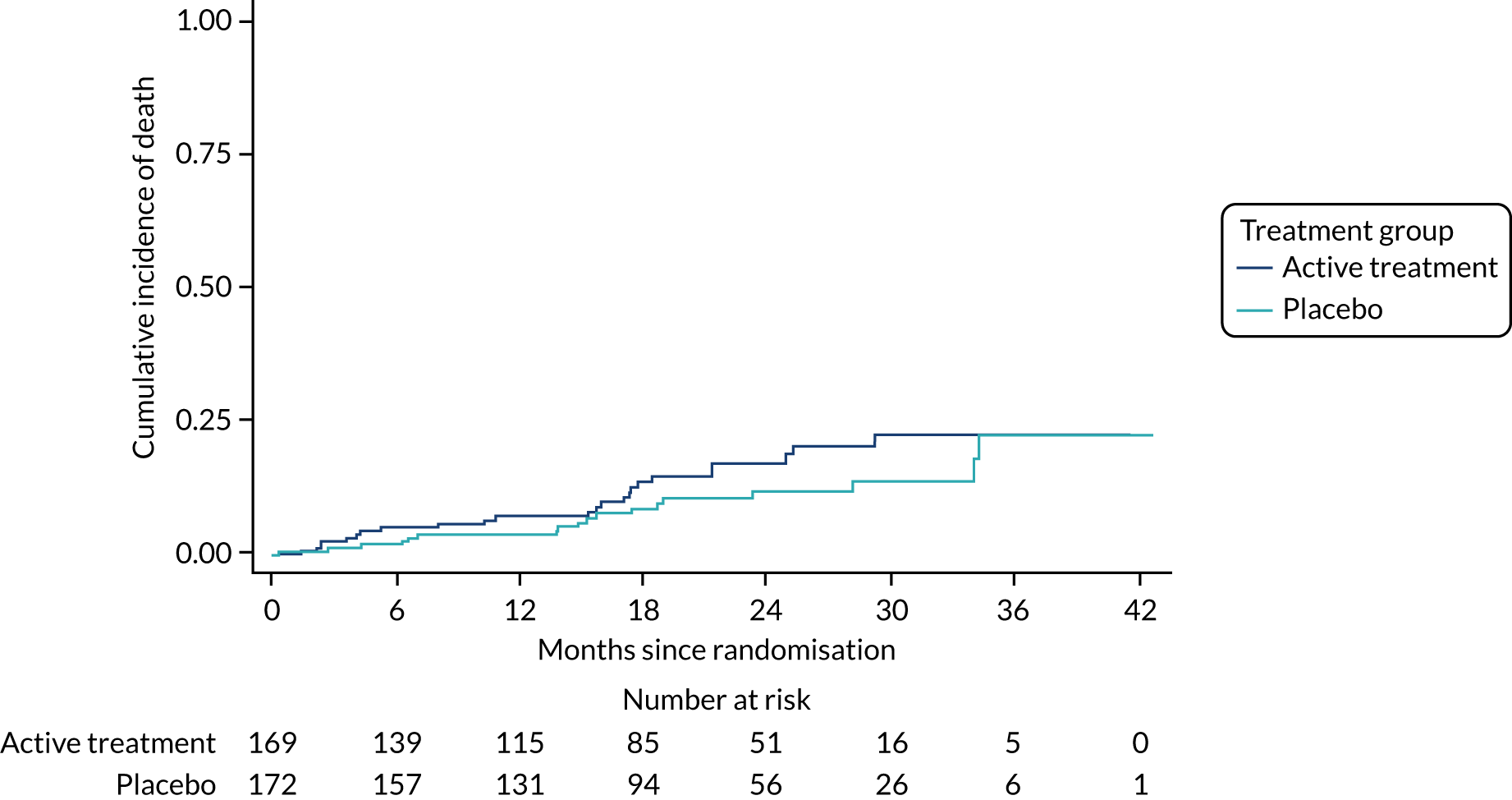
FIGURE 8.
Cumulative incidence function: hospitalisation only.
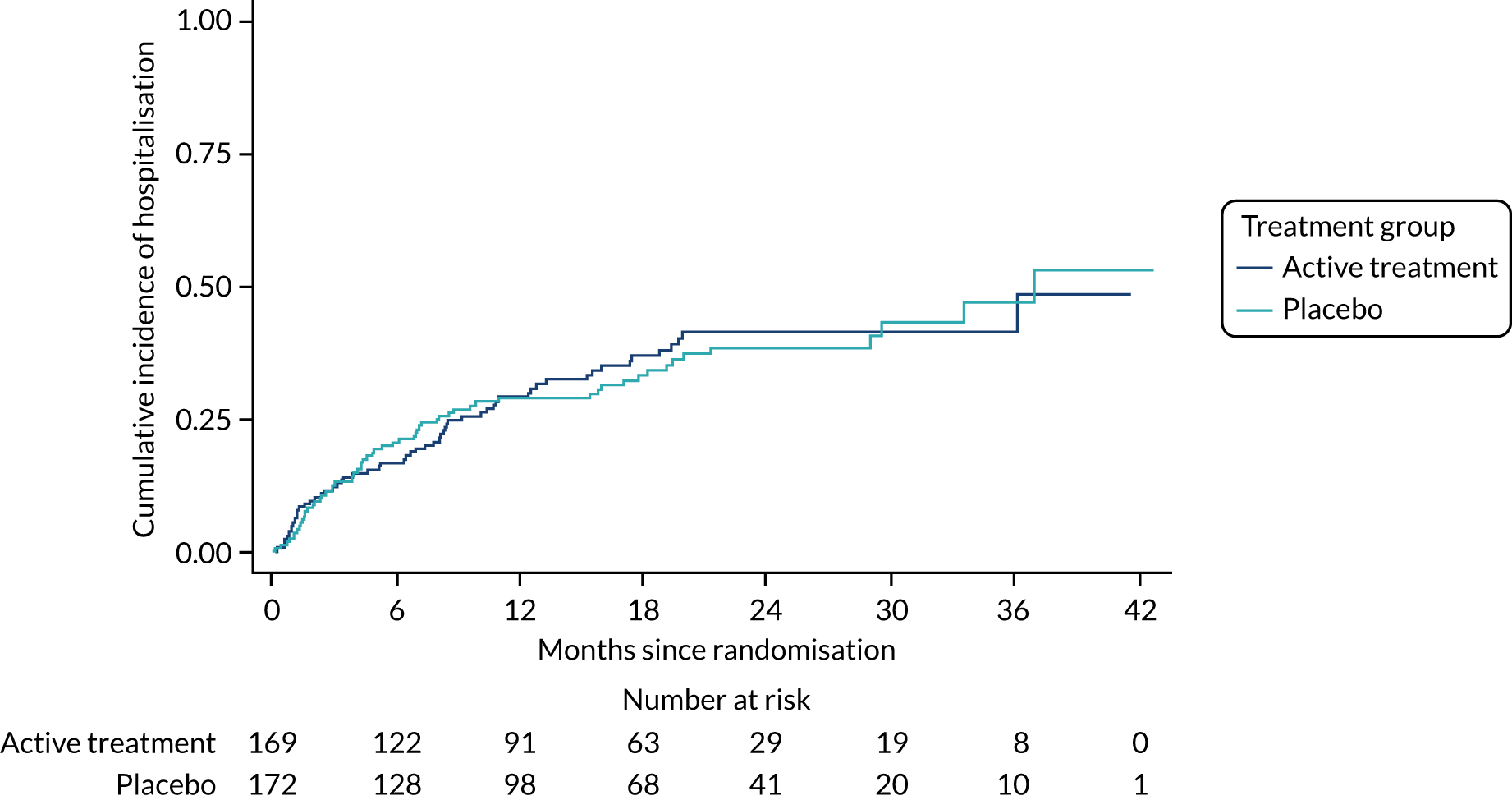
Per protocol
The individual component events of the primary outcome are given in Table 8 in the same format as that of the primary outcome. The number of events is reduced compared with the primary outcome so that the CIs are correspondingly wider. The total exposure time is the same for all outcomes as once one event had occurred the follow-up was censored for all other events. The number of deaths was higher in the active treatment group than in the placebo group, with 13 and 12 deaths, respectively, but the difference was not significant (HR 1.1, 95% CI 0.5 to 2.4; p = 0.824). Respiratory-related deaths were similarly higher in the active treatment group than in the placebo group, with 12 and 11 deaths, respectively; however, the difference was not significant (HR 1.1, 95% CI 0.5 to 2.5; p = 0.821). Hospital admissions were roughly equal in both the active treatment and placebo groups, with 45 and 51 hospitalisations, respectively, but the difference was not significant (HR 1.1, 95% CI 0.7 to 1.7; p = 0.581). Respiratory-related hospitalisations were roughly equal in both the active treatment and placebo groups, 29 and 35, respectively, but the difference was not significant (HR 0.8, 95% CI 0.5 to 1.3; p = 0.451). In all cases, the unadjusted and adjusted analyses were practically the same. As the Kaplan–Meier plot would provide a biased estimate as a result of informative censoring, a cumulative incidence plot is provided for each outcome in Figures 9 and 10. A separate analysis was not done for lung transplant as only one event occurred in each treatment group.
FIGURE 9.
Cumulative incidence function: death only – PP.
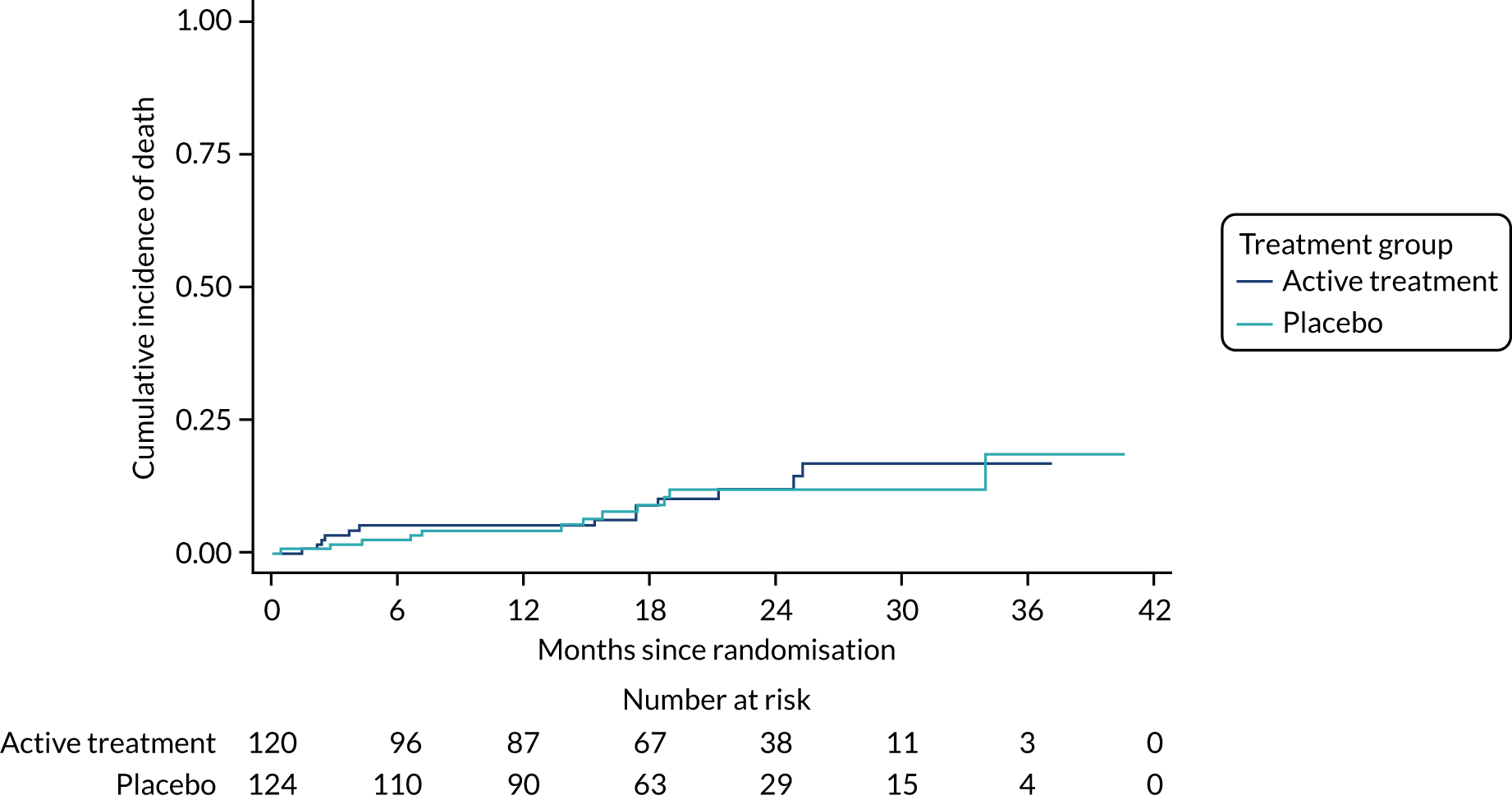
FIGURE 10.
Cumulative incidence function: hospitalisation only – PP.
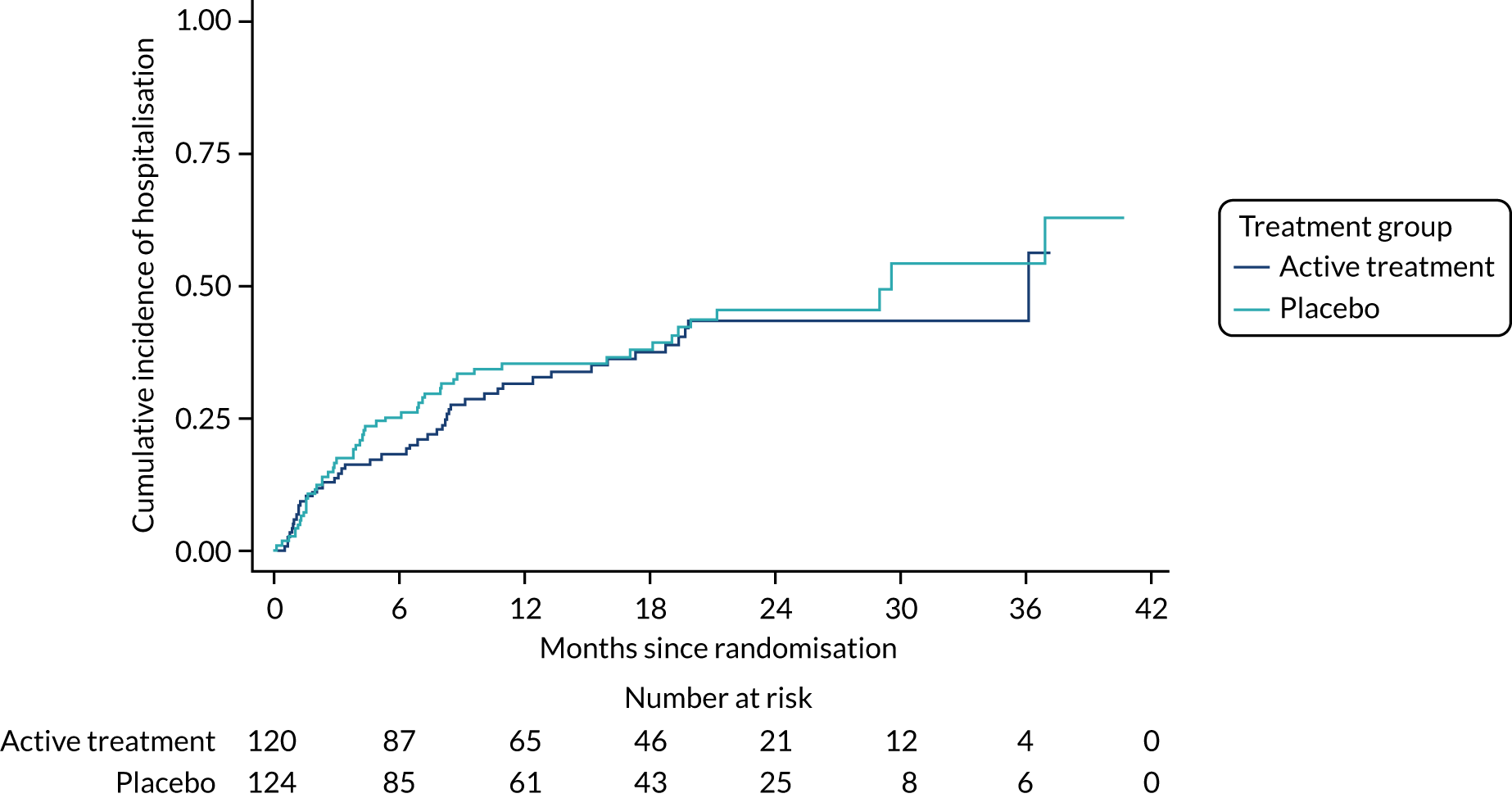
Modified per protocol
The individual component events of the primary outcome are given in Table 8 in the same format as that of the primary outcome. The number of events is reduced compared with the primary outcome so that the CIs are correspondingly wider. The total exposure time is the same for all outcomes as once one event had occurred the follow-up was censored for all other events. The number of deaths was about the same in the active treatment group and the placebo group, 11 and 12, respectively, but the difference was not significant (HR 1.2, 95% CI 0.5 to 2.8; p = 0.597). Respiratory-related deaths were similarly higher in the active treatment group than in the placebo group, with 10 and 11, respectively, but the difference was not significant (HR 1.2, 95% CI 0.5 to 2.9; p = 0.630). Hospital admissions were roughly equal in both the active treatment and placebo groups, 32 and 49, respectively, but the difference was not significant (HR 0.9, 95% CI 0.5 to 1.3; p = 0.487). Respiratory-related hospitalisations were roughly equal in both the active treatment and placebo groups, 23 and 34, respectively, but the difference was not significant (HR 0.9, 95% CI 0.5 to 1.5; p = 0.635). As the Kaplan–Meier plot would provide a biased estimate as a result of informative censoring, a cumulative incidence plot is provided for each outcome in Figures 11 and 12. A separate analysis was not done for lung transplant as only one event occurred in each treatment group.
FIGURE 11.
Cumulative incidence function: death only – mPP.
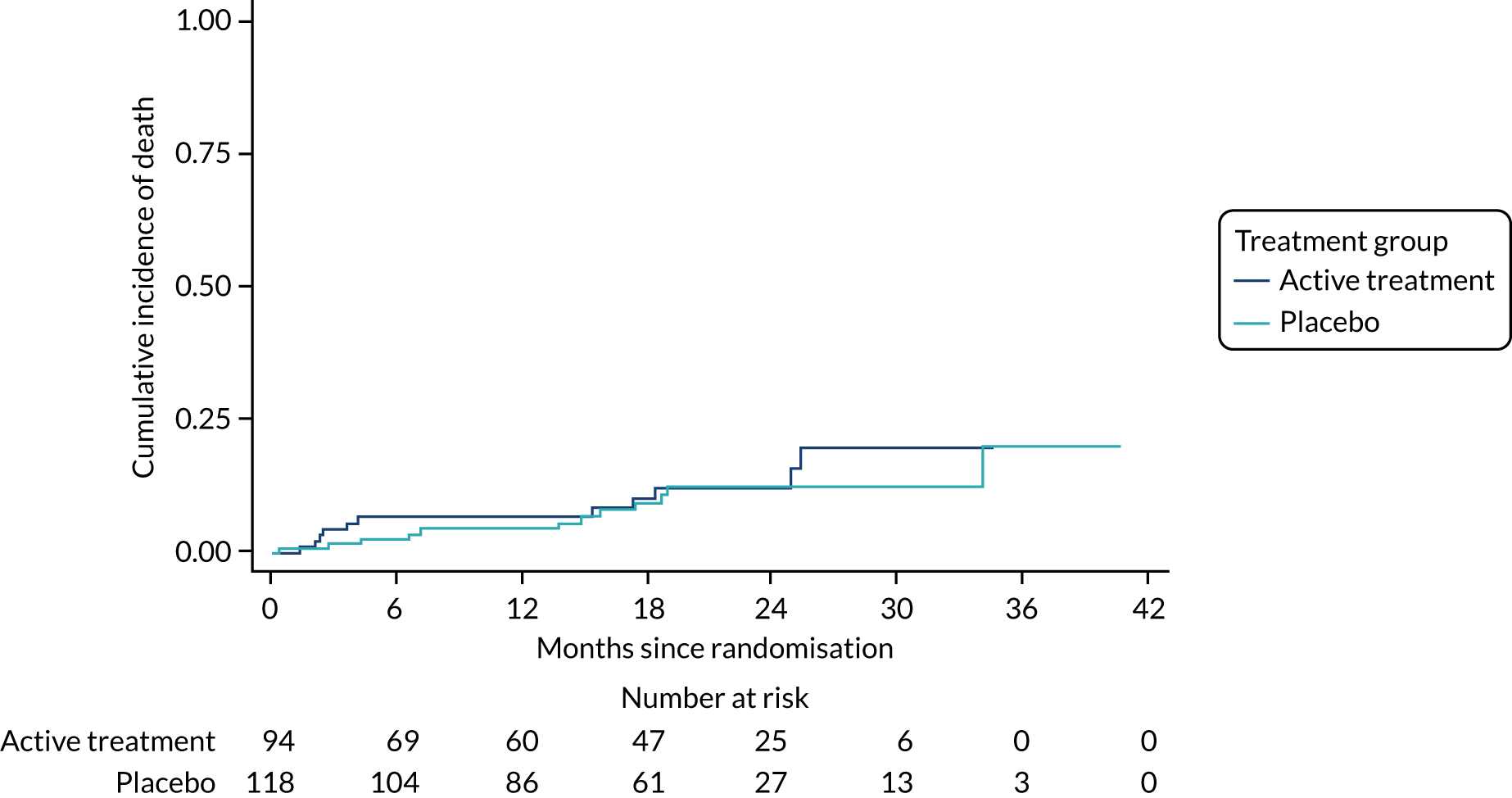
FIGURE 12.
Cumulative incidence function: hospitalisation only – mPP.
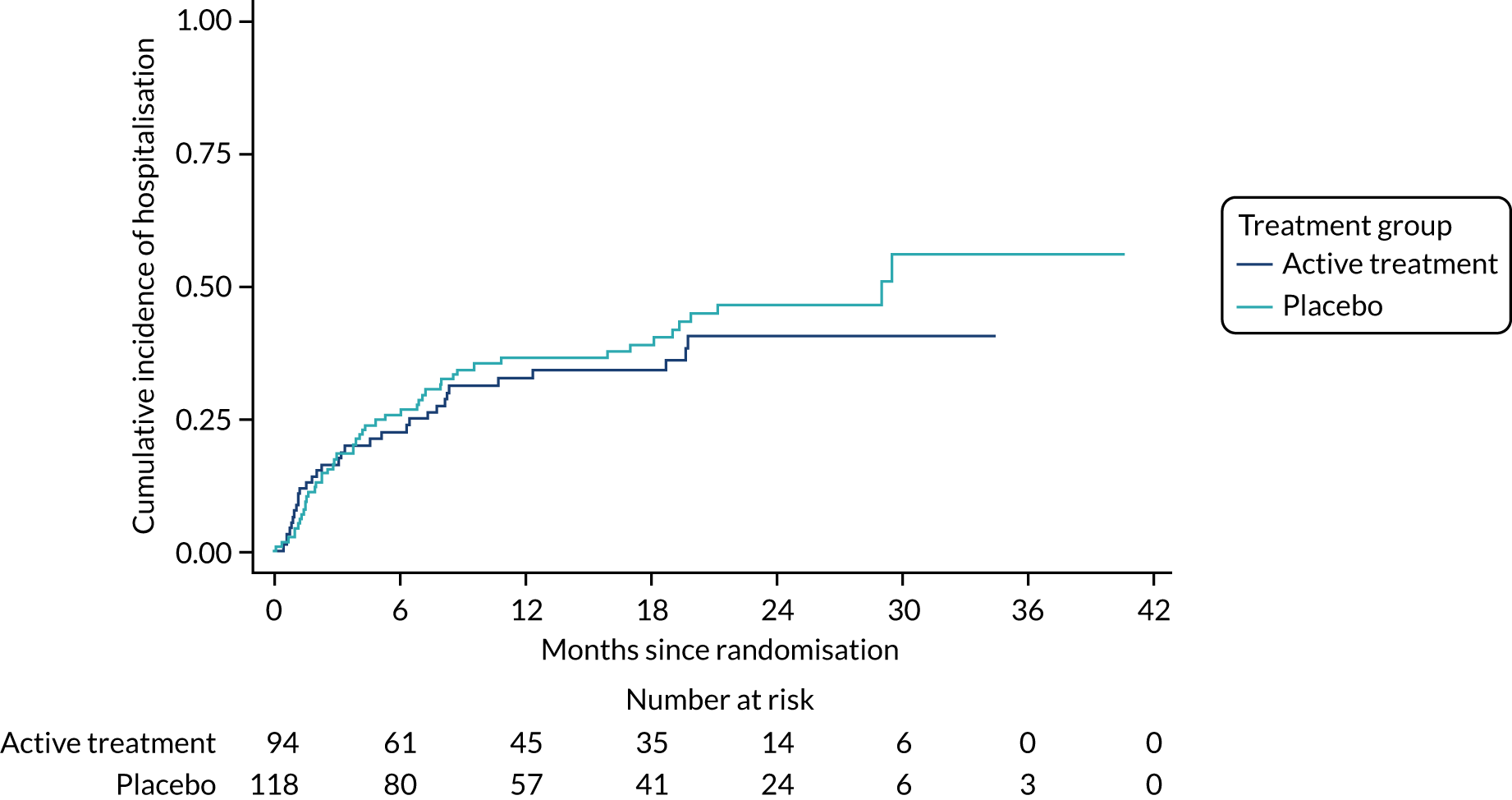
Questionnaire outcomes at 12 months
Intention-to-treat results
The questionnaire data were available for approximately 160 individuals at 12 months post randomisation, with more data available in the placebo group than in the active treatment group. For the LCQ, the mean score in the active treatment group was 15.37, compared with 14.59 in the placebo group. This mean difference of –0.75 was in favour of the active treatment group, but was not significant (p = 0.267). After accounting for baseline score, the difference decreased slightly (–0.60, 95% CI –1.56 to 0.36) and was still non-significant (p = 0.219). None of the scores for components of the LCQ was significantly different between the treatment groups, but all of the mean differences were in favour of the active treatment group.
The MRC score had the same median value in both the active treatment and placebo groups and neither the difference between groups (p = 0.9359) nor the change from baseline was significant (p = 0.2932). The average CSS was 44.74 in the active treatment group, compared with 49.69 in the placebo group; this difference was not significant (p = 0.243), nor did this change after adjusting for baseline (p = 0.570). The K-BILD questionnaire had average total scores of 50.32 in the active treatment group and 50.74 in the placebo group; the score difference was not significant (p = 0.834), nor did this change after adjusting for baseline (p = 0.932). None of the scores for components of the K-BILD questionnaire was significant. The results are shown in Table 9.
| Outcome | Treatment group | Analysis | ||||||
|---|---|---|---|---|---|---|---|---|
| Active treatment | Placebo | Unadjusteda | Adjustedb | |||||
| n | Mean (SD) | n | Mean (SD) | Mean difference (95% CI) | p-value | Mean difference (95% CI) | p-value | |
| LCQ score | ||||||||
| Total | 69 | 15.37 (3.99) | 71 | 14.59 (4.00) | –0.75 (–2.08 to 0.58) | 0.267 | –0.60 (–1.56 to 0.36) | 0.219 |
| Physical | 69 | 4.88 (1.22) | 72 | 4.70 (1.16) | –0.18 (–0.57 to 0.21) | 0.357 | –0.12 (–0.4 to 0.17) | 0.428 |
| Psychological | 69 | 5.16 (1.43) | 75 | 4.86 (1.51) | –0.28 (–0.77 to 0.2) | 0.254 | –0.26 (–0.63 to 0.11) | 0.171 |
| Social | 69 | 5.33 (1.49) | 75 | 5.05 (1.49) | –0.27 (–0.76 to 0.22) | 0.282 | –0.23 (–0.58 to 0.12) | 0.199 |
| MRC score, n (%) | 72 | 86 | ||||||
| 1 | 3 (4) | 3 (3) | ||||||
| 2 | 27 (38) | 27 (31) | ||||||
| 3 | 17 (24) | 31 (36) | ||||||
| 4 | 19 (26) | 20 (23) | ||||||
| 5 | 3 (4) | 3 (3) | ||||||
| MRC score, median (IQR) | 72 | 3.00 (2.00–4.00) | 86 | 3.00 (2.00–4.00) | 0.9359 | 0.2932 | ||
| CSS | 72 | 44.74 (27.01) | 84 | 49.69 (26.68) | 5.08 (–3.45 to 13.6) | 0.243 | 2.22 (–5.45 to 9.9) | 0.57 |
| EQ-5D-5L utility scorec | 103 | 0.41 (0.36) | 118 | 0.45 (0.35) | 0.04 (–0.05 to 0.13) | 0.37 | 0.03 (–0.06 to 0.11) | 0.55 |
| K-BILD score | ||||||||
| Psychological | 71 | 49.73 (17.92) | 85 | 51.86 (16.89) | 2.00 (–3.52 to 7.51) | 0.477 | 1.45 (–3.02 to 5.93) | 0.525 |
| Breathless | 72 | 34.37 (17.42) | 86 | 34.96 (14.55) | 0.88 (–4.12 to 5.89) | 0.729 | –0.53 (–4.41 to 3.34) | 0.787 |
| Chest | 72 | 59.86 (20.26) | 86 | 56.75 (22.82) | –3.42 (–10.25 to 3.42) | 0.327 | –2.00 (–7.76 to 3.76) | 0.497 |
| Total | 71 | 50.32 (12.26) | 85 | 50.74 (11.20) | 0.40 (–3.31 to 4.11) | 0.834 | 0.12 (–2.76 to 3.01) | 0.932 |
Per-protocol results
The questionnaire data were available for approximately 110 individuals at 12 months post randomisation, with more data available in the placebo group than in the active treatment group. The LCQ total score was borderline statistically significantly different between the active treatment and placebo groups. The mean score in the active treatment group was 15.91, compared with 14.30 in the placebo group; this mean difference of –1.53 (95% CI –3.11 to 0.04) was in favour of the active treatment group and was of borderline statistical significance (p = 0.057). After accounting for baseline score, the difference decreased slightly to –1.24 (95% CI –2.37 to –0.11), but was significant (p = 0.032). None of the components of the LCQ was significantly different between the active treatment and placebo groups in the unadjusted analyses, but all of the mean differences were in favour of the active treatment group. In the adjusted analyses, the physiological score was significantly different in favour of the active treatment group, with a mean difference of –0.44 (95% CI –0.85 to –0.03; p = 0.037).
The MRC score had the same median value in both treatment groups and was not significantly different (p = 0.8363); the change from baseline was also not significantly different (p = 0.4482). The average CSS was 44.80 in the active treatment group, compared with 49.56 in the placebo group; this difference was not significant (p = 0.402), nor did this alter after adjusting for baseline (p = 0.855). The K-BILD questionnaire had average total scores of 51.47 in the active treatment group and 50.82 in the placebo group; this difference was not significant (p = 0.759), nor did this alter after adjusting for baseline (p = 0.881). Of the components of the K-BILD questionnaire, only the chest component was significant, with a mean score of 62.46 in the active treatment group compared with 54.62 in the placebo group; this mean difference of –8.41 (95% CI –16.05 to –0.76; p = 0.031) was in favour of the active treatment group and, although it reduced to –6.85 (95% CI –13.29 to –0.41) after adjusting for baseline, it was still significant (p = 0.037). The results are shown in Table 10.
| Outcome | Treatment group | Analysis | ||||||
|---|---|---|---|---|---|---|---|---|
| Active treatment | Placebo | Unadjusted | Adjusted | |||||
| n | Mean (SD) | n | Mean (SD) | Mean difference (95% CI) | p-value | Mean difference (95% CI) | p-value | |
| LCQ score | ||||||||
| Total | 53 | 15.91 (3.86) | 46 | 14.30 (4.08) | –1.53 (–3.11 to 0.04) | 0.057 | –1.24 (–2.37 to –0.11) | 0.032 |
| Physical | 53 | 5.05 (1.18) | 47 | 4.68 (1.17) | –0.36 (–0.82 to 0.11) | 0.131 | –0.26 (–0.62 to 0.09) | 0.141 |
| Psychological | 53 | 5.34 (1.39) | 49 | 4.78 (1.56) | –0.53 (–1.1 to 0.05) | 0.074 | –0.5 (–0.93 to –0.06) | 0.024 |
| Social | 53 | 5.52 (1.46) | 49 | 4.95 (1.52) | –0.54 (–1.13 to 0.04) | 0.068 | –0.44 (–0.85 to –0.03) | 0.037 |
| MRC score | ||||||||
| 1 | 2 (4) | 2 (4) | ||||||
| 2 | 22 (42) | 20 (36) | ||||||
| 3 | 14 (25) | 18 (32) | ||||||
| 4 | 14 (25) | 12 (21) | ||||||
| 5 | 3 (5) | 4 (7) | ||||||
| Median (IQR) | 3.00 (2.00–4.00) | 3.00 (2.00–4.00) | 0.8363 | 0.4482 | ||||
| CSS | 44.80 (28.76) | 49.56 (26.77) | 4.42 (–5.92 to 14.75) | 0.402 | –0.9 (–10.51 to 8.72) | 0.855 | ||
| EQ-5D-5L utility scorea | 77 | 0.43 (0.37) | 84 | 0.40 (0.37) | –0.04 (–0.14 to 0.07) | 0.491 | –0.03 (–0.13 to 0.06) | 0.492 |
| K-BILD score | ||||||||
| Psychological | 54 | 51.38 (16.65) | 55 | 52.34 (16.57) | 0.99 (–5.33 to 7.31) | 0.759 | –0.41 (–5.76 to 4.94) | 0.881 |
| Breathless | 55 | 35.17 (17.60) | 56 | 35.08 (15.42) | 0.58 (–5.58 to 6.75) | 0.853 | –1.7 (–6.61 to 3.21) | 0.498 |
| Chest | 55 | 62.46 (18.81) | 56 | 54.62 (21.91) | –8.41 (–16.05 to –0.76) | 0.031 | –6.85 (–13.29 to –0.41) | 0.037 |
| Total | 54 | 51.47 (11.80) | 55 | 50.82 (11.04) | –0.66 (–5.01 to 3.68) | 0.765 | –1.61 (–5.12 to 1.9) | 0.369 |
Modified per-protocol results
The questionnaire data were available for approximately 90 individuals at 12 months post randomisation, with more data available in the placebo group than in the active treatment group (Appendix 1, Table 28). For the LCQ total score, the mean in the active treatment group was 15.67 compared with 14.13 in the placebo group; this mean difference of –1.47 (95% CI –3.20 to 0.26) was in favour of the active treatment group, but was not significant (p = 0.096). After accounting for baseline score, the difference decreased slightly to –1.43 (95% CI –2.72 to –0.14), but was significant (p = 0.029). None of the components of the LCQ was significantly different between the active treatment and placebo groups in the unadjusted analysis, but all of the mean differences were in favour of the active treatment group. In the adjusted analysis, all of the components were close to significance, but only the physiological score was significantly different, in favour of the active treatment group, with a mean difference of –0.54 (95% CI –1.04 to –0.05; p = 0.030).
The MRC score had the same median value in both the active treatment and placebo groups and was not significantly different (p = 0.6585). In addition, the change from baseline was not significantly different (p = 0.4859). The average CSS was 43.13 in the active treatment group, compared with 49.83 in the placebo group; this difference was not significant (p = 0.287), nor did this change after adjusting for baseline (p = 0.668).
The K-BILD questionnaire had average total scores of 51.49 in the active group and 50.77 in the placebo group; this difference was not significant (p = 0.805), nor did this change after adjusting for baseline (p = 0.485). None of the scores for components of the K-BILD questionnaire was significant.
Clinical measurement outcomes at 12 months
Intention-to-treat results
The lung function data were available for approximately 140 individuals at 12 months post randomisation, with more data available in the placebo group than in the active treatment group. The results are shown in Table 11.
| Outcome | Treatment group | Analysis | ||||||
|---|---|---|---|---|---|---|---|---|
| Active treatment | Placebo | Unadjusted | Adjusted | |||||
| n | Mean (SD) | n | Mean (SD) | Mean difference (95% CI) | p-value | Mean difference (95% CI) | p-value | |
| Absolute | ||||||||
| FVC (l) | 63 | 2.26 (0.53) | 77 | 2.23 (0.51) | –0.02 (–0.19 to 0.15) | 0.81 | –0.01 (–0.09 to 0.07) | 0.80 |
| FEV1 (l) | 63 | 1.86 (0.43) | 77 | 1.86 (0.42) | 0 (–0.14 to 0.14) | 1.0 | –0.02 (–0.08 to 0.05) | 0.62 |
| DLCO (mmol/minute/kPa) | 50 | 3.49 (1.75) | 60 | 3.71 (1.50) | 0.19 (–0.39 to 0.77) | 0.51 | 0.30 (–0.26 to 0.85) | 0.30 |
| Per cent predicted | ||||||||
| FVC | 63 | 54.02 (8.87) | 77 | 53.64 (9.12) | –0.54 (–3.56 to 2.47) | 0.72 | –0.55 (–2.56 to 1.45) | 0.59 |
| FEV1 | 63 | 57.83 (9.68) | 77 | 58.15 (10.42) | 0.16 (–3.23 to 3.55) | 0.93 | –0.65 (–2.77 to 1.46) | 0.55 |
| DLCO | 50 | 40.22 (17.68) | 60 | 43.17 (16.32) | 2.51 (–3.67 to 8.68) | 0.43 | 3.94 (–2.35 to 10.24) | 0.22 |
The absolute FVC had a mean of 2.26 l in the active treatment group, compared with 2.23 l in the placebo group. This difference was not significant in either the unadjusted (p = 0.81) or the adjusted (p = 0.80) analysis. The per cent predicted FVC was 54.02% in the active treatment group and 53.64% in the placebo group; this was not significant in either the unadjusted (p = 0.72) or the adjusted (p = 0.59) analysis.
The absolute FEV1 had a mean of 1.86 l in the active treatment group compared with 1.86 l in the placebo group. This difference was not significant in either the unadjusted (p = 1.00) or the adjusted (p = 0.62) analysis. The per cent predicted FEV1 was 57.83% in the active treatment group and 58.15% in the placebo group; this was not significant in either the unadjusted (p = 0.93) or the adjusted (p = 0.55) analysis.
The absolute DLCO had a mean of 3.49 mmol/minute/kPa in the active treatment group, compared with 3.71 mmol/minute/kPa in the placebo group. This difference was not significant in either the unadjusted (p = 0.51) or the adjusted (p = 0.30) analysis. The per cent predicted DLCO was 40.22% in the active treatment group and 43.17% in the placebo group; this was not significant in either the unadjusted (p = 0.43) or the adjusted (p = 0.22) analysis.
Per-protocol results
The lung function data were available for approximately 100 individuals at 12 months post randomisation, with more data available in the placebo group than in the active treatment group. The results are shown in Table 12.
| Outcome | Treatment group | Analysis | ||||||
|---|---|---|---|---|---|---|---|---|
| Active treatment | Placebo | Unadjusted | Adjusted | |||||
| n | Mean (SD) | n | Mean (SD) | Mean difference (95% CI) | p-value | Mean difference (95% CI) | p-value | |
| Absolute | ||||||||
| FVC (l) | 48 | 2.21 (0.49) | 50 | 2.27 (0.52) | 0.05 (–0.16 to 0.25) | 0.65 | 0.04 (0.06 to 0.14) | 0.42 |
| FEV1 (l) | 48 | 1.83 (0.39) | 50 | 1.90 (0.42) | 0.08 (–0.08 to 0.25) | 0.33 | 0.03 (–0.05 to 0.12) | 0.44 |
| DLCO (mmol/minute/kPa) | 39 | 3.37 (1.92) | 40 | 3.69 (1.47) | 0.28 (–0.47 to 1.03) | 0.46 | 0.45 (–0.24 to 1.15) | 0.20 |
| Per cent predicted | ||||||||
| FVC | 48 | 52.75 (8.65) | 50 | 54.02 (9.41) | 0.90 (–2.73 to 4.53) | 0.63 | 0.6 (–1.76 to 2.96) | 0.62 |
| FEV1 | 48 | 56.54 (9.17) | 50 | 59.17 (11.05) | 2.68 (–1.43 to 6.80) | 0.20 | 0.95 (–1.70 to 3.59) | 0.48 |
| DLCO | 39 | 38.47 (19.03) | 40 | 42.22 (14.83) | 3.46 (–4.09 to 11.02) | 0.37 | 5.81 (–1.72 to 13.35) | 0.131 |
The absolute FVC had a mean value of 2.21 l in the active treatment group, compared with 2.27 l in the placebo group. This difference was not significant in either the unadjusted (p = 0.65) or the adjusted (p = 0.42) analysis. The per cent predicted FVC was 52.57% in the active treatment group and 54.02% in the placebo group; this was not significant in either the unadjusted (p = 0.63) or the adjusted (p = 0.62) analysis.
The absolute FEV1 had a mean value of 1.83 l in the active treatment group, compared with 1.90 l in the placebo group. This difference was not significant in either the unadjusted (p = 0.33) or the adjusted (p = 0.44) analysis. The per cent predicted FEV1 was 56.54% in the active treatment group and 59.17% in the placebo group; this was not significant in either the unadjusted (p = 0.20) or the adjusted (p = 0.48) analysis.
The absolute DLCO had a mean value of 3.37 mmol/minute/kPa in the active treatment group, compared with 3.69 mmol/minute/kPa in the placebo group. This difference was not significant in either the unadjusted (p = 0.46) or the adjusted (p = 0.20) analysis. The per cent predicted DLCO was 38.47% in the active treatment group and 42.22% in the placebo group; this was not significant in either the unadjusted (p = 0.37) or the adjusted (p = 0.13) analysis.
Modified per-protocol results
The lung function data were available for approximately 80 individuals at 12 months post randomisation, with more data available in the placebo group than in the active treatment group. The results are shown in Appendix 1, Table 29.
The absolute FVC had a mean value of 2.23 l in the active treatment group, compared with 2.26 l in the placebo group. This difference was not significant in either the unadjusted (p = 0.83) or the adjusted (p = 0.30) analysis. The per cent predicted FVC was 52.49% in the active treatment group and 53.98% in the placebo group; this was not significant in either the unadjusted (p = 0.47) or the adjusted (p = 0.39) analysis.
The absolute FEV1 had a mean value of 1.84 l in the active treatment group, compared with 1.89 l in the placebo group. This difference was not significant in either the unadjusted (p = 0.58) or the adjusted (p = 0.70) analysis. The per cent predicted FEV1 was 56.16% in the active treatment group and 59.20% in the placebo group; this was not significant in either the unadjusted (p = 0.18) or the adjusted (p = 0.29) analysis.
The absolute DLCO had a mean value of 3.71 mmol/minute/kPa in the active treatment group, compared with 3.69 mmol/minute/kPa in the placebo group. This difference was not significant in either the unadjusted (p = 0.99) or the adjusted (p = 0.37) analysis. The per cent predicted DLCO was 41.44% in the active treatment group and 42.22% in the placebo group; this was not significant in either the unadjusted (p = 0.77) or the adjusted (p = 0.21) analysis.
Repeated measures analysis
Questionnaire outcomes
Medical Research Council score
As the MRC score is ordinal, the repeated measures analysis reduces to a Mann–Whitney U-test at each time point. The p-value has been inflated using the Bonferroni correction. The median MRC score is 3 in each group at 3 months and remains roughly constant over time, dropping only in the last measurement point, however, the number of individuals at that point is very small (Table 13). The difference between the intervention and placebo groups is not significant at any time point.
| Time point (months) | Treatment group | p-value (Bonferroni corrected) | |||
|---|---|---|---|---|---|
| Active treatment | Placebo | ||||
| Median (IQR) | n | Median (IQR) | n | ||
| 3 | 3.00 (2.00–4.00) | 122 | 3.00 (2.00–4.00) | 142 | 1.000 |
| 6 | 3.00 (2.00–4.00) | 111 | 3.00 (2.00–3.00) | 118 | 1.000 |
| 12 | 3.00 (2.00–4.00) | 72 | 3.00 (2.00–4.00) | 86 | 1.000 |
| 18 | 3.00 (2.00–4.00) | 43 | 3.00 (2.00–4.00) | 53 | 1.000 |
| 24 | 3.00 (2.00–4.00) | 23 | 2.00 (2.00–3.00) | 30 | 1.000 |
| 30 | 3.00 (2.00–4.00) | 13 | 3.00 (2.50–4.00) | 12 | 1.000 |
| 36 | 1.50 (1.00–2.00) | 2 | 2.50 (1.50–3.50) | 4 | 1.000 |
Cough Symptom Score
The mean CSS increased steadily in the placebo group over time until 24 months, when this trend started to change (Table 14). However, there were relatively few patients by this time point. In the active treatment group, the mean CSS remained roughly constant over time until 24 months. The interaction between treatment and time was not significant (p = 0.0829). The score difference was significant at 18 months (p = 0.023) in favour of the active treatment group, but this was not consistent over time. However, all of the time points, except the last, favoured the active treatment group and, overall, the difference was significant, with a mean difference of 5.65 (95% CI 0.25 to 11.06; p = 0.040).
| Time point (months) | Treatment group | Difference (95% CI) | p-value (Bonferroni corrected) | |||
|---|---|---|---|---|---|---|
| Active treatment | Placebo | |||||
| Mean (SD) | n | Mean (SD) | n | |||
| 3 | 43.69 (26.78) | 122 | 44.16 (28.29) | 139 | 1 (–8.07 to 10.07) | 1 |
| 6 | 40.11 (26.18) | 106 | 47.49 (28.69) | 116 | 6.77 (–2.89 to 16.44) | 0.415 |
| 12 | 44.74 (27.01) | 72 | 49.69 (26.68) | 84 | 4.47 (–6.63 to 15.58) | 1 |
| 18 | 46.02 (29.70) | 42 | 58.37 (24.99) | 51 | 14.97 (1.29 to 28.65) | 0.023 |
| 24 | 39.73 (27.25) | 22 | 48.57 (25.68) | 30 | 11.84 (–5.78 to 29.46) | 0.494 |
| 30 | 43.62 (27.16) | 13 | 54.58 (26.75) | 12 | 10.55 (–13.7 to 34.81) | 1 |
| 36 | 75.00 (7.07) | 2 | 41.25 (38.38) | 4 | –21.16 (–71.61 to 29.29) | 1 |
| Overall difference | 5.65 (0.25 to 11.06) | 0.04 | ||||
King’s Brief Interstitial Lung Disease Psychology
The average K-BILD Psychology score decreased slightly over time. The interaction between treatment and time was not significant (p = 0.5132). At no time point was the score difference between the active treatment and placebo groups significant. No overall score difference was observed (p = 0.9543) (see Appendix 1, Table 30).
King’s Brief Interstitial Lung Disease Breathless
The average K-BILD Breathless score remained similar over time. The interaction between treatment and time was not significant (p = 0.3135). At no time point was the score difference between the active treatment and placebo groups significant. No overall score difference was observed (p = 0.2084) (see Appendix 1, Table 31).
King’s Brief Interstitial Lung Disease Chest
The average K-BILD Chest score remained similar over time. The interaction between treatment and time was not significant (p = 0.9781). At no time point was the score difference between the active treatment and placebo groups significant. No overall score difference was observed (p = 0.9381) (see Appendix 1, Table 32).
King’s Brief Interstitial Lung Disease Total
The average K-BILD Total score remained similar over time (Table 15). The interaction between treatment and time was not significant (p = 0.6532). At no time point was the difference between the active treatment and placebo groups significant. No overall score difference was observed (p = 0.7828).
| Time point (months) | Treatment group | Difference (95% CI) | p-value (Bonferroni corrected) | |||
|---|---|---|---|---|---|---|
| Active treatment | Placebo | |||||
| Mean (SD) | n | Mean (SD) | n | |||
| 3 | 52.61 (11.19) | 122 | 53.31 (11.49) | 142 | 0.83 (–2.98 to 4.63) | 1 |
| 6 | 53.20 (11.18) | 111 | 53.50 (11.15) | 117 | 0.4 (–3.55 to 4.35) | 1 |
| 12 | 50.32 (12.26) | 71 | 50.74 (11.20) | 85 | 0.46 (–3.89 to 4.81) | 1 |
| 18 | 50.59 (11.61) | 43 | 50.21 (12.87) | 52 | 0.17 (–4.83 to 5.17) | 1 |
| 24 | 54.15 (16.79) | 22 | 51.59 (10.31) | 30 | –3.01 (–9.15 to 3.13) | 1 |
| 30 | 53.75 (14.45) | 13 | 49.57 (11.32) | 12 | 0.99 (–7.07 to 9.06) | 1 |
| 36 | 66.35 (9.69) | 2 | 62.30 (11.22) | 4 | 4.48 (–11.59 to 20.55) | 1 |
| Overall difference | 0.35 (–2.17 to 2.88) | 0.783 | ||||
EuroQol-5 Dimensions
The average EQ-5D score remained similar over time. The interaction between treatment and time was not significant (p = 0.6640). At no time point was the score difference between the active treatment and placebo groups significant. No overall score difference was observed (p = 0.1405) (see Appendix 1, Table 33).
Clinical measurement outcomes
Forced expiratory volume in 1 second
The mean FEV1 value decreased in both the active treatment and placebo groups over time, until 24 months, when the number of individuals became small. The interaction between treatment and time was significant (p = 0.041). At no time point was the difference between the active treatment and placebo groups significant. No overall difference was observed (p = 0.923). The results for the per cent predicted FEV1 are similar, with no overall significant difference (p = 0.321) (see Appendix 1, Tables 34 and 35).
Forced vital capacity
The mean FVC value decreased slightly in both the active treatment and placebo groups over time, until 24 months, when the number of individuals became small (Table 16). The interaction between treatment and time was significant (p = 0.050). At no time point was the difference between the active treatment and placebo groups significant. No overall difference was observed (p = 0.888). The results for the per cent predicted FVC are similar, with no overall difference (p = 0.574); however, there was a significant difference at 36 months (Table 17).
| Time point (months) | Treatment group | Difference (95% CI) | p-value | |||
|---|---|---|---|---|---|---|
| Active treatment | Placebo | |||||
| Mean (SD) | n | Mean (SD) | n | |||
| 3 | 2.28 (0.65) | 120 | 2.27 (0.57) | 134 | –0.004 (–0.20 to 0.19) | 1.00 |
| 6 | 2.23 (0.62) | 105 | 2.26 (0.54) | 112 | –0.02 (–0.23 to 0.18) | 1.00 |
| 12 | 2.26 (0.53) | 63 | 2.23 (0.51) | 77 | –0.02 (–0.23 to 0.20) | 1.00 |
| 18 | 2.22 (0.47) | 39 | 2.14 (0.56) | 50 | –0.03 (–0.26 to 0.20) | 1.00 |
| 24 | 2.34 (0.46) | 19 | 2.14 (0.55) | 26 | 0.11 (–0.16 to 0.38) | 1.00 |
| 30 | 2.17 (0.50) | 11 | 2.20 (0.79) | 9 | –0.21 (–0.55 to 0.13) | 0.693 |
| 36 | 2.41 (0.13) | 2 | 2.04 (0.37) | 4 | 0.47 (–0.11 to 1.05) | 0.209 |
| Overall difference | –0.01 (–0.15 to 0.13) | 0.887 | ||||
| Time point (months) | Treatment group | Difference (95% CI) | p-value | |||
|---|---|---|---|---|---|---|
| Active treatment | Placebo | |||||
| Mean (SD) | n | Mean (SD) | n | |||
| 3 | 56.26 (10.52) | 120 | 55.72 (10.38) | 134 | 0.52 (–2.99 to 4.03) | 1.00 |
| 6 | 55.67 (10.09) | 105 | 55.20 (10.64) | 112 | 0.32 (–3.32 to 3.96) | 1.00 |
| 12 | 54.02 (8.87) | 63 | 53.64 (9.12) | 77 | 0.44 (–3.57 to 4.47) | 1.00 |
| 18 | 52.43 (8.90) | 39 | 51.81 (9.75) | 50 | 0.40 (–4.14 to 4.94) | 1.00 |
| 24 | 56.12 (11.47) | 19 | 51.00 (9.33) | 26 | 4.11 (–1.54 to 9.75) | 0.351 |
| 30 | 55.38 (8.03) | 11 | 52.05 (13.14) | 9 | –3.19 (–10.82 to 4.44) | 1.000 |
| 36 | 69.28 (18.46) | 2 | 47.16 (6.36) | 4 | 17.19 (3.54 to 30.85) | 0.005 |
| Overall difference | 0.68 (–1.69 to 3.05) | 0.574 | ||||
Diffusing capacity of the lung for carbon monoxide
The mean DLCO value increased slightly in both the active treatment and placebo groups over time, until 24 months when the number of individuals became small (Table 18). The interaction between treatment and time was not significant (p = 0.758). At no time point was the difference between the active treatment and placebo groups significant. No overall difference was observed (p = 0.430). The results for the per cent predicted DLCO are similar, with no evidence of a significant difference overall (p = 0.376) (Table 19).
| Time point (months) | Treatment group | Difference (95% CI) | p-value | |||
|---|---|---|---|---|---|---|
| Active treatment | Placebo | |||||
| Mean (SD) | n | Mean (SD) | n | |||
| 3 | 3.32 (1.31) | 82 | 3.65 (1.76) | 99 | –0.24 (–0.87 to 0.40) | 1.00 |
| 6 | 3.47 (1.50) | 77 | 3.77 (1.42) | 73 | –0.17 (–0.84 to 0.51) | 1.00 |
| 12 | 3.49 (1.75) | 50 | 3.71 (1.50) | 60 | –0.11 (–0.88 to 0.65) | 1.00 |
| 18 | 3.60 (2.21) | 26 | 3.55 (1.24) | 29 | –0.13 (–1.13 to 0.86) | 1.00 |
| 24 | 4.18 (2.80) | 16 | 3.67 (1.78) | 19 | 0.32 (–0.88 to 1.53) | 1.00 |
| 30 | 2.50 (1.04) | 7 | 2.89 (1.13) | 6 | –0.83 (–2.71 to 1.04) | 1.00 |
| 36 | 4.79 (1.29) | 2 | 4.40 | 1 | 1.19 (–2.88 to 5.27) | 1.00 |
| Overall difference | –0.15 (–0.53 to 0.23) | 0.431 | ||||
| Time point (months) | Treatment group | Difference (95% CI) | p-value | |||
|---|---|---|---|---|---|---|
| Active treatment | Placebo | |||||
| Mean (SD) | n | Mean (SD) | n | |||
| 3 | 39.21 (14.79) | 82 | 43.21 (19.98) | 99 | –3.10 (–9.70 to 3.49) | 1.00 |
| 6 | 40.89 (17.39) | 77 | 44.33 (14.87) | 73 | –2.59 (–9.72 to 4.54) | 1.00 |
| 12 | 40.22 (17.68) | 50 | 43.17 (16.32) | 60 | –2.06 (–10.27 to 6.15) | 1.00 |
| 18 | 41.06 (25.29) | 26 | 41.36 (14.59) | 29 | –2.06 (–13.08 to 8.96) | 1.00 |
| 24 | 49.35 (34.39) | 16 | 42.48 (18.43) | 19 | 4.23 (–9.27 to 17.74) | 1.00 |
| 30 | 29.82 (11.63) | 7 | 36.53 (14.79) | 6 | –10.16 (–31.57 to 11.26) | 1.00 |
| 36 | 61.72 (26.38) | 2 | 47.90 | 1 | 19.78 (–27.34 to 66.91) | 1.00 |
| Overall difference | –2.22 (–5.95 to 1.52) | 0.244 | ||||
Ancillary analysis
Compliance-adjusted causal effect analysis
Questionnaire outcomes
In addition to the ITT and PP results, a decision was made to include compliance-adjusted causal effect (CACE) results to account for potential biases in the protocol results. Owing to the limitations of this approach, the results are presented for only those outcomes where a linear model could be applied. The questionnaire data were available for approximately 160 individuals at 12 months post randomisation, with more data available in the placebo group than in the active treatment group. For the LCQ total score, the mean score in the active treatment group was 15.37, compared with 14.59 in the placebo group; this mean difference of –0.99 (95% CI –2.54 to 0.57) was not significant (p = 0.215). After accounting for baseline score, the difference decreased slightly to –0.76 (95% CI –2.20 to 0.68; p = 0.300). None of the scores for components of the LCQ was significantly different between the treatment groups, but all of the mean differences were in favour of the active treatment group.
The average CSS was 44.74 in the active treatment group, compared with 49.69 in the placebo group; this difference was not significant (p = 0.198), nor did this change after adjusting for baseline (p = 0.530). The K-BILD questionnaire had average total scores of 50.32 in the active group and 50.74 in the placebo group; this difference was not significant (p = 0.793), nor did this change after adjusting for baseline (p = 0.869). None of the scores for components of the K-BILD questionnaire was significant (see Appendix 1, Table 36).
Lung function outcomes
The lung function CACE analysis is presented in Appendix 1, Table 37. No significant difference was observed in any of the lung function outcomes in the unadjusted or adjusted analyses.
Imputed results
The imputed results are shown in Table 20. These results show no statistically significant difference in any of the comparisons.
| Outcome | Analysis | |||
|---|---|---|---|---|
| Unadjusteda | Adjustedb | |||
| Mean difference (95% CI) | p-value | Mean difference (95% CI) | p-value | |
| LCQ score | ||||
| Total | 0.21 (–13.75 to 14.18) | 0.976 | 0.34 (–13.45 to 14.13) | 0.961 |
| Physical | 0.68 (–7.34 to 8.71) | 0.867 | 0.74 (–7.16 to 8.64) | 0.853 |
| Psychological | –0.19 (–9.92 to 9.54) | 0.970 | –0.16 (–10.00 to 9.67) | 0.974 |
| Social | –0.28 (6.97 to 6.41) | 0.934 | –0.24 (–6.93 to 6.45) | 0.944 |
| K-BILD score | ||||
| Psychological | 1.81 (–3.15 to 6.77) | 0.474 | 2.00 (–2.44 to 6.43) | 0.376 |
| Breathless | 0.78 (–3.97 to 5.52) | 0.748 | –0.16 (–4.24 to 3.91) | 0.937 |
| Chest | –1.74 (–8.47 to 5.00) | 0.612 | –1.48 (–7.80 to 4.85) | 0.646 |
| Total | 0.45 (–2.82 to 3.71) | 0.789 | 0.59 (–2.28 to 3.45) | 0.687 |
| Absolute | ||||
| FVC (l) | –0.00 (–0.14 to 0.14) | 0.990 | –0.01 (–0.11 to 0.08) | 0.760 |
| FEV1 (l) | –0.00 (–0.12 to 0.11) | 0.933 | –0.01 (–0.09 to 0.06) | 0.712 |
| DLCO (mmol/minute/kPa) | 0.23 (–1.36 to 1.81) | 0.780 | 0.19 (–1.40 to 1.78) | 0.813 |
| Per cent predicted | ||||
| FVC | –1.49 (–4.31 to 1.33) | 0.299 | –0.70 (–3.02 to 1.61) | 0.550 |
| FEV1 | –1.56 (–4.56 to 1.44) | 0.307 | –0.21 (–2.68 to 2.25) | 0.865 |
| DLCO | 1.36 (–10.37 to 13.09) | 0.819 | 1.01 (–12.52 to 14.54) | 0.883 |
Biomarker results
Intention-to-treat analysis
The biomarker data were available for 157 patients at baseline. The data are presented in Appendix 1, Table 38; the data were heavily skewed, with extreme values, so a non-parametric testing approach was taken. Overall, the groups were well balanced given the high variability of the measures.
At outcome, differences were observed in CRP levels (p = 0.016), with a small difference in median, but, as seen in Figure 13, the difference was mainly due to the few extreme values in the active treatment group. A significant difference (p = 0.019) was also seen in CA19-9 levels; again, a small difference in median values, but the difference was due to extreme values in the active treatment group, as shown in Figure 14. There was also a difference in the change from baseline in SP-D levels (p < 0.001), as shown in Figure 15; the difference was due to the greater spread of change in placebo group. A significant difference in the change from baseline in CRP levels (p = 0.005) was also observed and was due to some large changes in a few individuals in the active treatment group – this is shown in Figure 16. A significant difference in the change from baseline in CA-125 levels (p = 0.032) was also observed and was due to some large changes in a few individuals in the active treatment group, as shown in Figure 17.
The results are given in Appendix 1, Table 39.
FIGURE 13.
Histogram of CRP levels at 12 months in the ITT sample. (a) Active treatment; and (b) placebo.
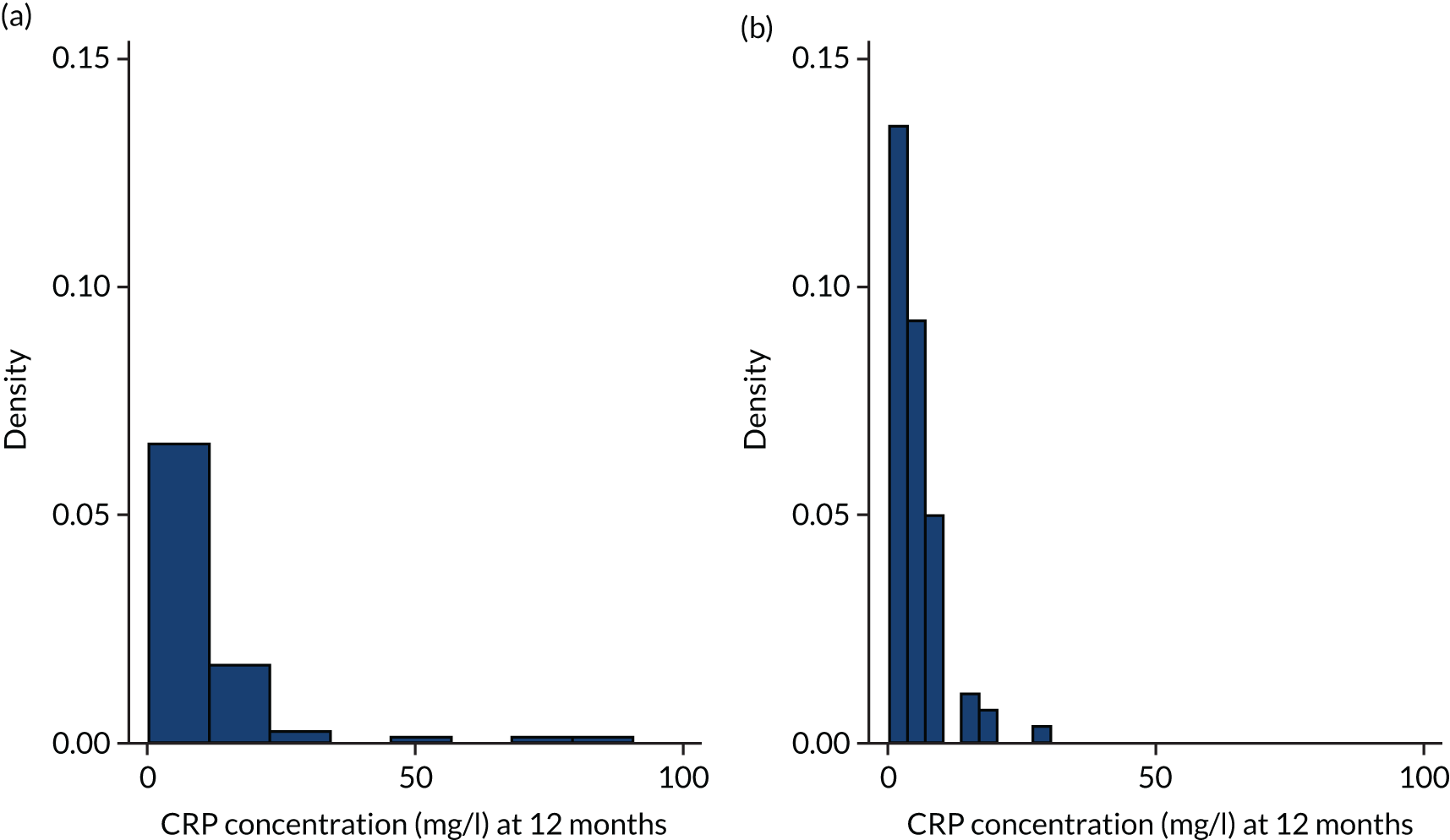
FIGURE 14.
Histogram of CA19-9 levels at 12 months in the ITT sample. (a) Active treatment; and (b) placebo.
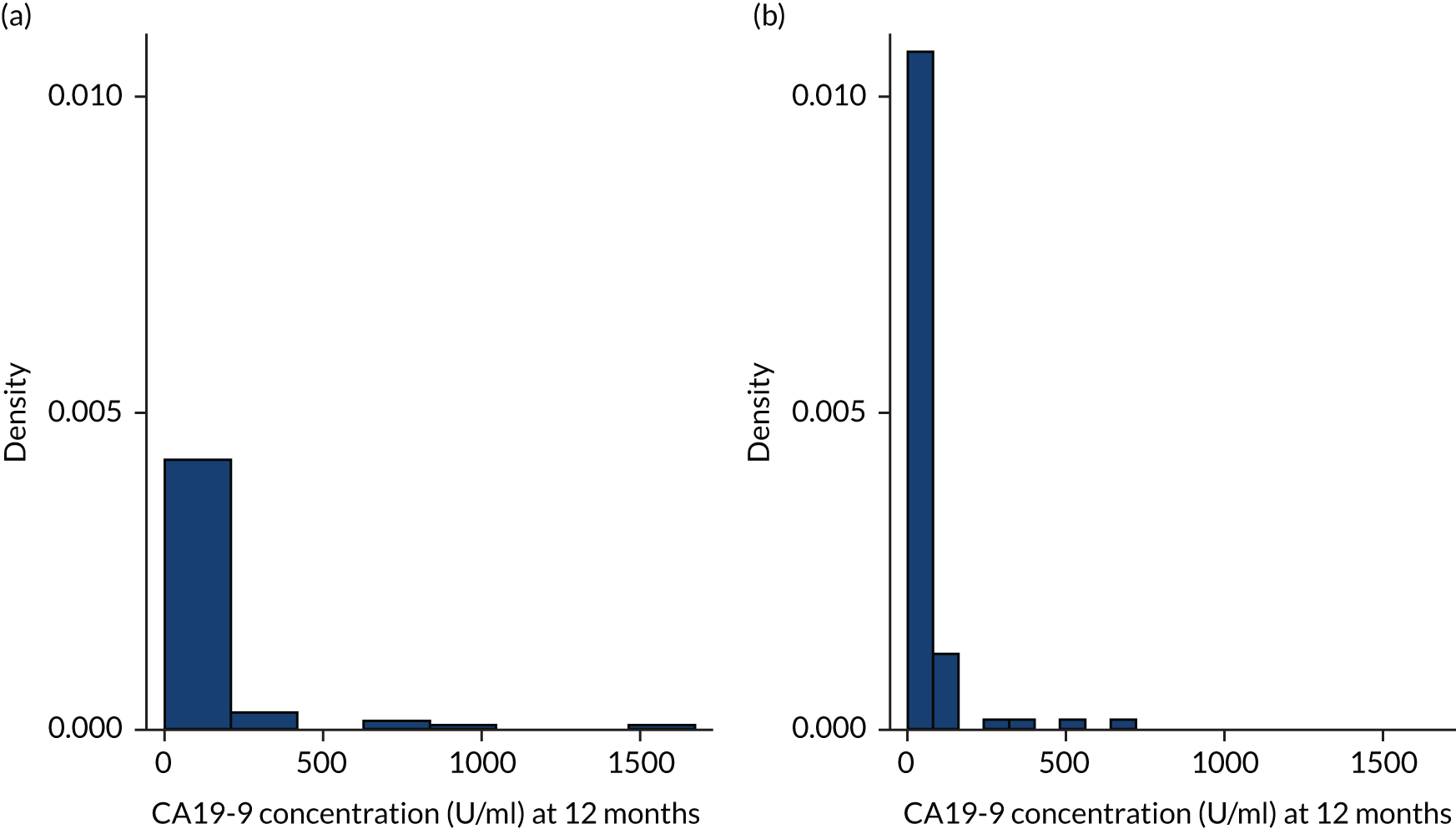
FIGURE 15.
Histogram of change in SP-D levels from baseline to 12 months in the ITT sample. (a) Active treatment; and (b) placebo.
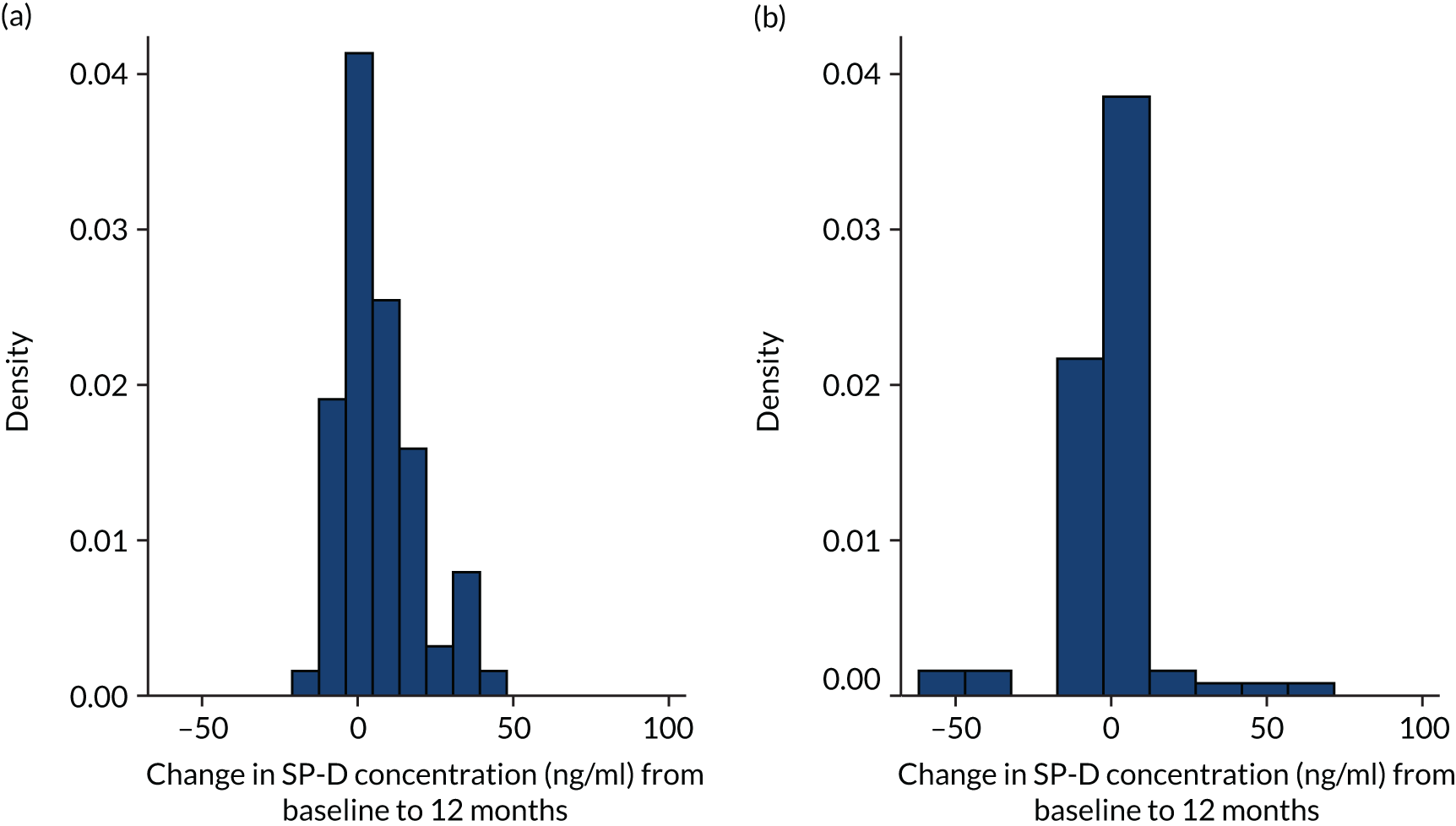
FIGURE 16.
Histogram of change in CRP levels from baseline to 12 months in the ITT sample. (a) Active treatment; and (b) placebo.
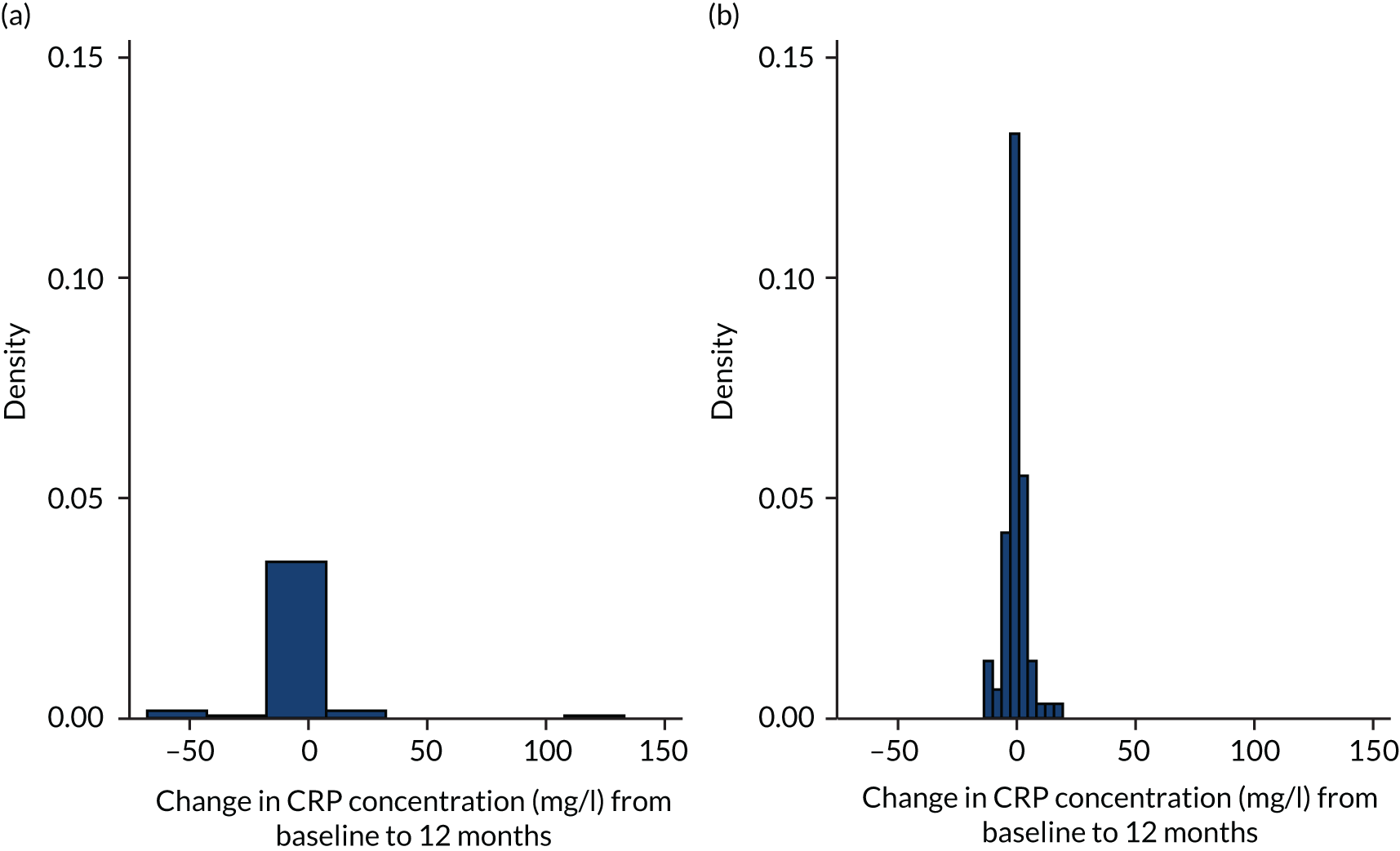
FIGURE 17.
Histogram of change in CA-125 levels from baseline to 12 months in the ITT sample. (a) Active treatment; and (b) placebo.
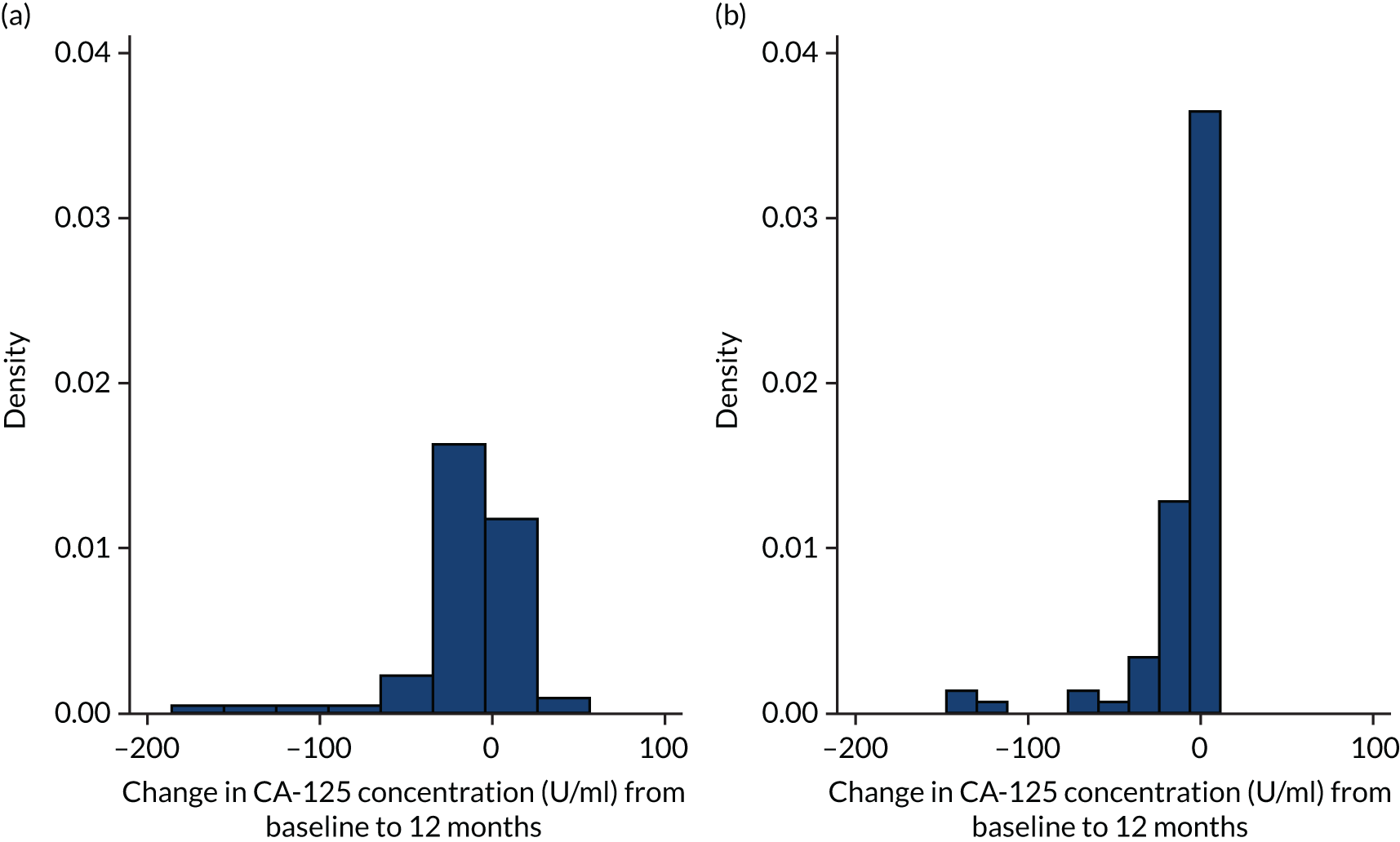
Per-protocol analysis
The biomarker data were available for 108 patients at baseline. The data are presented in Appendix 1, Table 40; the data were heavily skewed, with extreme values, so a non-parametric testing approach was taken. Overall, the treatment groups were well balanced given the high variability of the measures.
At outcome, differences were observed in CRP levels (p < 0.001), with a small difference in medians, but the difference was mainly due to the few extreme values in the active treatment group. A significant difference (p = 0.025) was also seen in CA19-9 levels; again, this was a small difference in median values, but the difference was due to extreme values in the active treatment group. There was also a difference in the change from baseline in SP-D (p < 0.001), CRP (p < 0.001) and CA-125 (p = 0.021) levels. The results are shown in Appendix 1, Table 41.
Modified per-protocol analysis
The biomarker data were available for 108 patients at baseline. The data are presented in Appendix 1, Table 42; the data were heavily skewed, with extreme values, so a non-parametric testing approach was taken. Overall, the treatment groups were well balanced given the high variability of the measures.
At outcome, differences were observed in CRP levels (p < 0.001), with a small difference in medians, but the difference was mainly due to the few extreme values in the active treatment group. A significant difference (p = 0.025) was also seen in CA19-9 levels; again, this was a small difference in median values, but the difference was due to extreme values in the active treatment group. There was also a difference in the change from baseline in SP-D (p < 0.001), CRP (p < 0.001) and CA-125 (p = 0.021) levels and the pro-BNP (p = 0.026) levels. The results are shown in Appendix 1, Table 43.
Safety
Adverse events and serious adverse events
A total of 1336 AEs occurred during the follow-up period: 696 in the active treatment group and 640 in the placebo group. This was roughly equal in both treatment groups. The AEs occurred in 288 individuals: 146 (in 86.4% of individuals randomised) in the active treatment group and 142 (in 82.6% of individuals randomised) in the placebo group. There were 37 SAEs: 20 in the active treatment and 17 in the placebo groups. There were 16 (9.5%) and 12 (6.8%) patients with one or more SAE in the active treatment and placebo groups, respectively.
The classification of AEs is given in Table 21 and differences were seen in three categories, with rates of general disorders, investigations, and metabolism and nutrition disorders all higher in the treatment group than in the placebo group.
| Adverse event | Treatment group | |||
|---|---|---|---|---|
| Active treatment | Placebo | |||
| Total number of events | Number of patients with at least one event, n (%) (N = 169) | Total number of events | Number of patients with at least one event, n (%) (N = 172) | |
| Blood and lymphatic system disorders | 3 | 3 (2) | 3 | 3 (2) |
| Cardiac disorders | 6 | 6 (4) | 4 | 3 (2) |
| Ear and labyrinth disorders | 3 | 2 (1) | 0 | 0 (0) |
| Eye disorders | 5 | 5 (3) | 6 | 5 (3) |
| Gastrointestinal disorders | 216 | 92 (54) | 224 | 81 (47) |
| Nausea | 89 | 53 (31) | 67 | 42 (24) |
| Diarrhoea | 52 | 36 (21) | 84 | 53 (31) |
| Vomiting | 28 | 20 (12) | 20 | 16 (9) |
| Constipation | 11 | 10 (6) | 5 | 5 (3) |
| General disorders and administration site conditions | 36 | 25 (15) | 20 | 17 (10) |
| Fatigue | 15 | 15 (9) | 11 | 10 (6) |
| Chest pain | 8 | 7 (4) | 6 | 5 (3) |
| Oedema peripheral | 5 | 4 (2) | 0 | 0 (0) |
| Immune system disorders | 1 | 1 (1) | 1 | 1 (1) |
| Infections and infestations | 110 | 57 (34) | 127 | 70 (41) |
| Lower respiratory tract infection | 63 | 35 (21) | 66 | 42 (24) |
| Injury, poisoning and procedural complications | 7 | 5 (3) | 10 | 10 (6) |
| Investigations | 44 | 34 (20) | 22 | 16 (9) |
| Weight decrease | 24 | 21 (12) | 16 | 14 (8) |
| Metabolism and nutrition disorders | 57 | 38 (22) | 27 | 19 (11) |
| Decreased appetite | 26 | 18 (11) | 9 | 6 (3) |
| Hyperkalaemia | 24 | 18 (11) | 14 | 11 (6) |
| Musculoskeletal and connective tissue disorders | 21 | 18 (11) | 20 | 14 (8) |
| Neoplasm(s) benign, malignant and unspecified (including cysts and polyps) | 3 | 2 (1) | 1 | 1 (1) |
| Nervous system disorders | 41 | 29 (17) | 32 | 24 (14) |
| Headache | 22 | 16 (9) | 14 | 11 (6) |
| Psychiatric disorders | 5 | 4 (2) | 2 | 2 (1) |
| Renal and urinary disorders | 14 | 12 (7) | 7 | 7 (4) |
| Reproductive system and breast disorders | 0 | 0 (0) | 2 | 2 (1) |
| Respiratory, thoracic and mediastinal disorders | 77 | 46 (27) | 95 | 61 (35) |
| Cough | 27 | 23 (14) | 33 | 30 (17) |
| Dyspnoea | 31 | 25 (15) | 34 | 30 (17) |
| Skin and subcutaneous tissue disorders | 46 | 29 (17) | 30 | 23 (13) |
| Rash | 31 | 23 (14) | 20 | 15 (9) |
| Surgical and medical procedures | 1 | 1 (1) | 2 | 2 (1) |
| Vascular disorders | 0 | 0 (0) | 5 | 3 (2) |
| Total AEs | 696 | 640 | ||
| Number with at least one AE | 146 (86) | 142 (83) | ||
| Number with at least two AEs | 119 (70) | 121 (70) | ||
Blood measures
The summary statistics of the safety blood measures collected at various time points are given in Table 22. Further data on safety blood measures are given in Appendix 1, Tables 44–50. Consistent significant differences were observed at 6 weeks and 3 and 6 months in haemoglobin levels, with mean values approximately 6 g/dl higher in the placebo group than in the active treatment group; red cell count (RCC), with mean values approximately 0.2 × 1012/l higher in the placebo group than in the active treatment group; haematocrit, with mean values approximately 0.01% higher in the placebo group than in the active treatment group; sodium levels, with mean values approximately 2 mmol/l higher in the placebo group than in the active treatment group; creatinine levels, with values approximately 12 µmol/l higher in the active treatment group than in the placebo group; bilirubin levels, with mean values approximately 1.5 µmol/l higher in the placebo group; and alkaline phosphatase levels, with mean values approximately 7 IU/l higher in the active treatment group. At 12 months, there was a significant difference in creatinine levels only. Beyond 12 months, the number of patients is limited and no significant differences are observed.
| Measure | Treatment group | p-value | |||
|---|---|---|---|---|---|
| Active treatment | Placebo | ||||
| Mean (SD) | n | Mean (SD) | n | ||
| White cell count (× 109/l) | 8.85 (2.34) | 68 | 8.44 (1.89) | 87 | 0.23 |
| Haemoglobin (g/dl) | 142.78 (13.22) | 68 | 146.46 (13.97) | 87 | 0.098 |
| RCC (× 1012/l) | 4.66 (0.49) | 67 | 4.78 (0.41) | 86 | 0.088 |
| Mean cell volume (fl) | 92.74 (6.93) | 68 | 92.02 (5.63) | 86 | 0.48 |
| Mean cell haemoglobin (pg) | 30.85 (2.76) | 68 | 30.67 (2.10) | 86 | 0.66 |
| Haematocrit (%) | 0.43 (0.04) | 67 | 0.44 (0.04) | 83 | 0.093 |
| Neutrophils (× 109/l) | 5.99 (1.94) | 67 | 5.65 (1.65) | 86 | 0.24 |
| Lymphocytes (× 109/l) | 1.70 (0.75) | 68 | 1.78 (0.69) | 86 | 0.50 |
| Eosinophils (× 109/l) | 0.29 (0.15) | 68 | 0.25 (0.15) | 86 | 0.11 |
| Basophils(× 109/l) | 0.06 (0.04) | 67 | 0.05 (0.04) | 86 | 0.24 |
| Monocytes (× 109/l) | 0.71 (0.22) | 68 | 0.68 (0.21) | 86 | 0.40 |
| Platelets (× 109/l) | 242.76 (65.36) | 68 | 238.72 (66.98) | 87 | 0.71 |
| Sodium (Na) (mmol/l) | 138.18 (2.79) | 68 | 138.72 (2.70) | 88 | 0.22 |
| Potassium (K) (mmol/l) | 4.40 (0.45) | 68 | 4.37 (0.38) | 88 | 0.61 |
| Urea (mmol/l) | 6.12 (2.38) | 68 | 5.59 (1.72) | 88 | 0.11 |
| Creatinine (µmol/l) | 96.29 (34.67) | 68 | 83.59 (21.08) | 88 | 0.005 |
| Bilirubin, upper limit of normal (µmol/l) | 20.11 (3.11) | 64 | 21.93 (10.71) | 82 | 0.19 |
| Bilirubin (µmol/l) | 8.17 (3.79) | 65 | 9.62 (5.04) | 86 | 0.055 |
| Alanine aminotransferase, upper limit of normal (IU/l) | 48.03 (8.20) | 62 | 48.09 (7.84) | 78 | 0.97 |
| Alanine aminotransferase (IU/l) | 22.87 (12.55) | 68 | 21.27 (10.26) | 85 | 0.39 |
| Alkaline phosphatase (IU/l) | 95.46 (52.95) | 68 | 88.05 (28.96) | 88 | 0.27 |
| Albumin (g/dl) | 39.75 (3.82) | 67 | 39.30 (4.58) | 88 | 0.52 |
| Total protein (g/dl) | 74.00 (6.77) | 56 | 73.17 (6.08) | 75 | 0.46 |
| Globulin (g/dl) | 30.23 (9.90) | 31 | 34.01 (6.53) | 39 | 0.059 |
Microbiology
We obtained 17 sputum samples and one nasal swab in total for all patient visits. Of these, only three grew a relevant microbiological agent on culture: Staphylococcus aureus (n = 1), Haemophilus influenzae (n = 1) and yeasts (n = 1).
Chapter 4 Discussion
Main results
The results of this study have suggested that, for people with moderate to severe IPF, the addition of co-trimoxazole, compared with placebo, does not provide any significant or clinically meaningful benefit in terms of clinical outcomes, disease progression or biomarkers of disease activity. There was no improvement in all-cause mortality, hospitalisation or transplant rates, whether considered together or separately. The findings were the same when the outcomes were restricted to respiratory-related events and to those patients who adhered to high-dose treatment for the duration of the trial. There was a trend to improvement in cough-related quality of life and CSS with co-trimoxazole in the ITT and PP analysis after 1 year of therapy, with statistical significance in CSS when considering data from the duration of the study as per the repeated measure analysis. Co-trimoxazole also improved the chest symptom domain of the K-BILD questionnaire in those patients adhering to the protocol. However, there were no meaningful changes in QALYs or breathlessness scores. Given that adjustments for multiple comparisons were not performed, it is possible that improvements in cough could be due to a type I error. Furthermore, co-trimoxazole did not influence disease progression, as determined by lung function, or exhibit any meaningful change in the serum biomarkers following 12 months of treatment.
Comparison with other studies
Co-trimoxazole
To our knowledge, there have been only two previous trials of prophylactic therapy with co-trimoxazole in people with ILD. 20,21 In a trial of 20 patients with advanced idiopathic fibrotic lung disease, Varney et al. 20 showed an increase in FVC from a median of 1.9 to 2.3 l (95% CI 1.3 to 3.0 l; p = 0.05) and an increase in the shuttle walk test distance from 255 to 355 m (95% CI 200 to 450 m; p = 0.002) after 3 months of treatment. Seven patients had a usual interstitial pneumonia pattern on HRCT and, therefore, would be classified as having IPF; the rest had a HRCT pattern in keeping with a combination of UIP or non-specific interstitial pneumonia, or had unclassifiable fibrotic ILD. Of note is the fact that seven participants in the active treatment group and four in the placebo group were receiving prednisolone at a median dose of 10 mg per day.
The subsequent two trials found that there was no change in FVC measurements with 12 months of co-trimoxazole therapy (from 2.3 to 2.18 l in the TIPAC trial21 and from 2.3 to 2.26 l in the EME-TIPAC trial) and no difference in the change for the placebo and co-trimoxazole treatment arms [0.00 l (95% CI –0.11 to 0.11 l) and –0.01 l (95% CI –0.09 to 0.07 l), respectively]. The lack of benefit identified in the two larger studies is not likely to be caused by the increased variability of measurement as part of a multicentre study. The values were mostly taken from clinical data, as the trials were designed to collect data obtained as part of routine care and, therefore, will have been undertaken to a clinical standard by qualified pulmonary function technologists.
Given the remarkable improvement in these physiological measurements with co-trimoxazole in the initial study, it is possible that the patients were not stable at baseline and co-trimoxazole was treating an unrecognised bacterial lower respiratory tract infection. This is a plausible explanation because clinical stability was not required as part of the entry criteria for the study of Varney et al. 20 and the beneficial effects of FVC were seen after 3 months of treatment, with no further improvement after pulmonary rehabilitation or 1 year of open-label treatment. Interestingly, the shuttle walk test distance did not improve after pulmonary rehabilitation in either the placebo or active treatment groups, which is in contrast to recent systematic review data of the effects of pulmonary rehabilitation in people with IPF. 96
The second trial exhibited a benefit in terms of quality of life and, in the case of those adhering to treatment, all-cause mortality. 21 The symptoms domain score of the SGRQ was reduced in those receiving co-trimoxazole, but increased in the group receiving the placebo, indicating worse quality of life in this group. The difference between the two treatment groups was 6.88 (95% CI 1.7 to 12.06) units when the results were adjusted for baseline, but there was no difference in other domains of the SGRQ or the total SGRQ score. The 6.88-unit difference is larger than the minimum important difference for the SGRQ. 97 The symptom domain of the SGRQ is derived from the first eight questions of this tool, which cover issues relating to cough, sputum, breathlessness, wheeze and attacks of breathlessness. 98 The improvement in these symptoms may reflect the changes seen in the current study in terms of CSS, LCQ and the chest symptom domain of the K-BILD questionnaire in the PP analysis. The symptom domain of the K-BILD questionnaire asks about chest tightness, air hunger and wheeze, and, therefore, is similar to the symptom domain of the SGRQ. This study indicated a significant difference in CSS, a visual analogue scale rating patients’ overall cough severity, after 18 months of treatment, and a significant overall effect on CSS over the duration of the study.
Cough is an important problem for people with IPF. 99 Patients report cough to be particularly bothersome, and cough significantly contributes to the burden of disease. 100 It is an important problem in end-of-life care, but is also present at an early stage of the disease, with one-third of people with IPF having at least one consultation for cough in the year before diagnosis. 101 Unfortunately, it is difficult to manage cough in people with IPF as there are no recognised treatments. 100 The suggestion of improvement in cough quality of life and cough-related symptoms in two separate clinical trials (this trial and the TIPAC trial21) requires further evaluation given the lack of available treatment options.
In the analysis confined to those patients adhering to the treatment and protocol, there was a reduction in deaths and an improvement in QALYs in the TIPAC trial. 21 The reduction in QALYs is likely to reflect mortality, at least in part, because death represents a QALY of zero. Unfortunately, and in contradiction to our hypothesis, we were not able to identify a significant treatment effect, in terms of our composite score of non-elective hospitalisation, all-cause death or transplant rates with co-trimoxazole in people with IPF in the current study.
The discordance between the two results can largely be explained by the change in standard practice between the first and second studies, that is, the discontinuation of immunosuppression and the introduction of antifibrotic therapy. This resulted in a much better survival than in the previous study. 20 In the TIPAC trial,21 the median hospital-free survival in the placebo group was 12.8 months, whereas in the EME-TIPAC trial the median hospital-free survival was 23.3 months.
When the first study was undertaken (i.e. January 2008–December 2010), standard treatment for IPF was immunosuppression with prednisolone, with or without azathioprine or mycophenolate and N-acetylcysteine. However, the results of the Raghu et al. 9 study became available in 2012 and indicated that immunosuppression resulted in higher mortality and hospitalisation rates than no immunosuppression; these findings were considered to be mostly due to respiratory issues, presumably caused by infection and changes in prescribing practices.
In the TIPAC trial,21 60% and 57% of individuals were receiving prednisolone in the placebo and active treatment groups, respectively, with approximately 30% receiving azathioprine and 4% receiving mycophenolate in each group. The majority of those receiving prednisolone were taking a moderate to high dose, with only 13% (placebo group) and 19% (active treatment group) taking < 10 mg per day. This contrasts with the current study, which excluded all those receiving > 10 mg of prednisolone (as part of the entry criteria), and in which only 6% of individuals were taking a low dose (< 10 g/day) of prednisolone.
A systematic review of participants randomised to the placebo group of eight multicentre large-scale randomised controlled trials, totalling 1631 patients with 2067 patient-years’ follow-up, reported that prednisolone therapy results in a 31% increase in all-cause mortality. 25 In a separate analysis,56 two trials totalling 1156 individuals suggested that low-dose prednisolone increased mortality by 54% and a high dose increased mortality more than twofold compared with no prednisolone. To our knowledge, there are no reliable data to evaluate the increase in hospitalisation rates with prednisolone in IPF, but prednisolone had a greater effect on mortality than hospitalisation rates in the TIPAC trial. 21 Prednisolone has been known to result in a 39% increase in pneumonia and a 3.6-fold increase in lower respiratory tract infections. 54 Importantly, prednisolone therapy, particularly at higher doses, is frequently prescribed to patients with severe disease and, therefore, any effect of prednisolone on outcomes from observational data is likely to be an overestimation because of confounding biases. It is clear, therefore, that prednisolone and other immunotherapy increase mortality in people with IPF, possibly because of the increased incidence of respiratory tract infections. Undiagnosed or unrecognised respiratory tract infection may have been prevented or treated with co-trimoxazole in the first study, but not in the current study, resulting in no difference in mortality.
The other major change in prescribing practice was the introduction of antifibrotic therapy. The American Thoracic Society/European Respiratory Society 2011 guidelines8 expressed some caution about using pirfenidone; however, the relevant national boardsfinalised approval of the decision late in 2010, before the results of the CAPACITY study14 were published in May 2011, indicating a reduction in FVC with pirfenidone compared with placebo. Pirfenidone was licensed by the European Medicines Agency on 28 February 2011 for the treatment of adults with mild to moderate IPF and the NICE IPF pirfenidone Technology Appraisal number 282 (TA282) was published in April 2013. 102 The INPULSIS study was published in May 2014103 and the nintedanib Technology Appraisal number 379 (TA379) was published in January 2016. 104 Recruitment into the current trial began in April 2015. Despite the narrow window of eligibility of antifibrotic therapy in the UK (FVC between 50% and 80% predicted), a large proportion of people in the EME-TIPAC trial were eligible for this form of treatment because our entry criteria excluded those with mild disease. Indeed, 75% of people were taking antifibrotic therapy: 137 (40%) patients were taking pirfenidone and 116 (34%) were taking nintedanib.
Although data from randomised controlled trials have not shown a survival benefit for either pirfenidone or nintedanib, there is sufficient evidence to suggest that antifibrotic therapy prolongs life. FVC is considered a good surrogate marker for survival and, therefore, it is reasonable to expect that the improvement in FVC decline with these drugs equates to a survival benefit. Using combined data from the CAPACITY14 and ASCEND74 studies, the relative risk for all-cause mortality in the active treatment groups was half that in the placebo groups (HR 0.52, 95% CI 0.31 to 0.87; p = 0.0107) at 1 year, with maintenance of improved mortality at 120 weeks. 17 In another similar combined analysis using the CAPACITY14 data, an open-label extension study (RECAP105) and people meeting the inclusion criteria for these studies from the Inova Fairfax Hospital database, a survival analysis has shown life expectancy to be 8.72 years (95% CI 7.65 to 10.15 years) with pirfenidone and 6.24 years (95% CI 5.38 to 7.18 years) with standard care. 106 Pirfenidone was estimated to improve life expectancy by 2.47 years (95% CI 1.26 to 4.17 years), which equated to 25% of the expected years of life lost due to IPF. 106 Likewise, a combined post hoc analysis from trials of nintedanib also showed a significant reduction in mortality. It is possible, therefore, that the effects of these drugs will mask any survival benefit from co-trimoxazole. 107
Another difference between the TIPAC trial21 and EME-TIPAC trial is the severity of illness of the patients enrolled; the EME-TIPAC trial recruited people with FVC < 75% predicted. It is known that patients with IPF have greater bacterial burden than healthy individuals,31 but it is not known whether or not disease severity itself influences the microbiological flora. It is known that the treatment response to pirfenidone is similar for people with advanced disease (FVC of < 50%) and those with mild disease. 14,108 Patients not receiving prednisolone in the TIPAC trial21 were recruited only if the PI felt that their disease was declining based on evidence of a reduction in lung function. As a reduction in FVC has been shown to relate to reduced survival,109 this may have also contributed to the higher mortality rates in the TIPAC trial. 21 We do not believe that co-trimoxazole is likely to have been more effective in people with mild disease and that restricting the entry criteria to those with reduced FVC values is responsible for the negative findings of the study; those with more severe disease were likely to benefit from co-trimoxazole, as suggested by the results of a post hoc analysis of the TIPAC trial. 21
In the EME-TIPAC trial, there was an option for people with ARs to high-dose (960 mg twice daily) co-trimoxazole to reduce the dose (to 960 mg three times weekly), whereas, in the TIPAC trial,21 this was not permitted. However, fewer people stepped down to the lower dose (18%) in the EME-TIPAC trial than withdrew from treatment (40%) in the TIPAC trial. 21
In a retrospective review of the Japanese Diagnosis Procedure Combination database,23 293 people with IPF who had an acute exacerbation of IPF, received invasive mechanical ventilation for respiratory failure and were treated with high- (at least 2.88 g/day) or low-dose (480 mg to 2.4 g/day) co-trimoxazole were compared with those who received no treatment. A significant dose-related improvement was seen with co-trimoxazole in terms of survival, with high-dose treatment patients surviving > 100 days and low-dose treatment or no co-trimoxazole patients surviving < 50 days. Like the TIPAC trial,21 but unlike the EME-TIPAC trial, the benefit was seen in people receiving immunosuppression, as all patients were receiving high-dose corticosteroid therapy. This adds weight to the suggestion that co-trimoxazole has a role for people who are immunosuppressed and to the rationale for the discrepancy between the two TIPAC studies. 21
Other antibiotics
Doxycycline may have effects on matrix metalloproteinases110 and has been evaluated in an open-label study that found no physiological improvement, but an improvement in chest X-ray involvement;111 however, there are no data on this drug from placebo-controlled trials. The effects of doxycycline are unknown, but are currently being examined in the Clean-Up IPF study (https://clinicaltrials.gov/ct2/show/NCT02759120; last accessed 20 January 2021). This is a prospective observational study of either co-trimoxazole or doxycycline therapy in over 500 people with IPF over a 12- to 36-month follow-up period, capturing the same outcomes as the EME-TIPAC trial.
Azithromycin attenuates myofibroblast differentiation112 and had beneficial effects in a bleomycin mouse model of pulmonary fibrosis. 113 To our knowledge, there have been no randomised controlled trials with macrolides, but two retrospective studies114,115 have investigated the role of azithromycin in IPF. One was a retrospective review of low-dose macrolide (250 mg three times per week) given to people who had three or more lower respiratory tract infections or courses of antibiotics over the preceding 12 months. The primary end point was hospitalisations, which reduced from 0.29 per patient per year in the 12 months before treatment to 0.08 per patient per year in the 12 months with macrolide treatment. Recurrent chest infections was not an entry criterion for the EME-TIPAC trial, and it is not known if co-trimoxazole had any beneficial effect on this phenotype. Like the TIPAC trial21 and the EME-TIAPC trial, macrolide therapy did not have any effect on the rate of decline of lung function as measured by FVC or DLCO. 114 In another (single-centre) study,115 mortality reduced after the treatment regime changed from quinolones to macrolides for acute exacerbations of IPF. However, as this was not controlled for time of entry into the study, there are likely to be other confounding factors and the findings may be open to question.
Lung microbiota
The results of this study do not support the hypothesis that the lung microbiota influences disease progression and outcomes for people with IPF. This hypothesis is based on two separate moderate- to large-scale studies31,33 using high-throughput DNA sequencing technology based on the highly conserved gene for bacterial 16S rRNA. This technique seeks to detect and identify bacterial species without laboratory culture and is thus able to identify organisms that cannot be grown or are not present in sufficient quantities to be grown. This methodology has altered our perceptions of lung microbiota. Before this culture-independent technique was available, the lungs were considered to be sterile; 16S ribosomal deoxyribonucleic acid (rDNA) detection suggests that this is not the case. Caveats are that the method measures bacterial DNA rather than live bacteria, so it is not possible to determine whether or not the bacteria are viable, let alone medically important. Furthermore, concerns remain that some of the bacteria found enter the sample when it is taken, rather than being present deep in the lung.
The first of these two studies31 was a retrospective analysis of patients from the Royal Brompton Hospital undergoing diagnostic bronchoscopy: 65 patients with IPF were compared with historical samples from healthy individuals (n = 27) and people with COPD (n = 17). Organisms identified from IPF patients were Streptococcus, Prevotella, Fusobacterium and Haemophilus spp., which are also found in healthy individuals. 32 Nonetheless, the bacterial burden, assessed as 16S copies per ml of BALF, was significantly higher in those with IPF than in either the COPD patients or the healthy control patients, with a more than twofold difference between IPF patients and healthy control patients. After splitting the concentrations of 16S copy number/ml into tertiles, it was possible to demonstrate that the bacterial burden was related to survival. Mortality among IPF patients with the highest bacterial load was 4.59-fold higher than among those with the lowest load.
The second analysis33 used a subgroup of the COMET study,116 a multicentre US cohort study, to identify biomarkers of IPF disease progression. Fifty-five patients (five of whom were receiving corticosteroids or azathioprine) were evaluated, and the organisms that had the highest relative abundance were Veillonella and Cronobacter spp. These organisms are also commonly found in healthy lungs. 32 Using a principal component analysis, the presence of Streptococcus spp. and Staphylococcus spp. was associated with worse outcomes (death, acute exacerbation of IPF, transplant reduction in FVC of ≥ 10% or DLCO of ≥ 15%). Statistical modelling showed a significant effect for these bacteria, although smoking history, hypoxaemia (defined as oxygen saturation of < 88%) and gastro-oesophageal reflux disease (GORD) all had a greater effect in the model. Notably, significant burdens of Staphylococcus spp. were found in only 16 people and of streptococci in only eight people with IPF. It may be, as the authors suggest, that these bacteria are involved in the progression of the disease, possibly through the TLR9 pathway;117 however, given their low occurrence, their presence is unlikely to be causative of progression.
It may be that the changes in microbiota represent an association with disease progression rather than causation and, therefore, that antimicrobial pressure on the microbiome will not influence outcomes. In this context, one cause for the increased microbiological burden in IPF could be the presence of GORD. GORD is commonly associated with IPF118 and its presence is related to disease progression. 119 It is possible, therefore, that the greater ‘bacterial burden’ in the lungs of IPF patients than control patients represents a greater incidence of microaspiration, not growth of bacteria in the lung. Furthermore, given that the bronchoscope has to pass through the oropharynx, it is possible that the findings represent contamination from the upper airways. In the COMET study,33 the bronchoscopy technique was not standardised and the nasal route was utilised for some individuals. Neither study evaluated the upper airway microbiota.
An earlier, smaller, study examined the upper and lower respiratory tract microbiota in patients with IIP (n = 5), non-IIP (n = 5) or sarcoidosis (n = 7) and healthy controls (n = 9),30 again using 16S rRNA gene sequencing. The authors did not find significant diversity in the lower airway microbiota in patients with ILD compared with healthy control subjects. In addition, differences between the microbiota of the upper and lower respiratory tracts were found in only 4 out of 26 participants with ILD.
All of the above studies used BAL to sample the lower airways. However, IPF is a disease of the interstitium and evaluation of the lung tissue is required. In a study of explanted lungs,120 the microbiotas of the lung tissue were compared for people with IPF (n = 40), those with cystic fibrosis (n = 5) and healthy controls derived from donated lungs that were not suitable for transplantation (n = 37). In contrast to the studies just outlined, the authors found that the lungs of people with IPF yielded very few 16S rRNA gene sequences, with levels similar to reagent controls, 15-fold lower than those for the cystic fibrosis patients or control lungs. There were differences in the lung microbiota, with ‘skin’ origin taxa (e.g. species of Comamonadaceae, Methylobacterium) more common in IPF patients and ‘oral’ taxa (e.g. species of Prevotella, Streptococcus) more common in control patients. For a small series, the airway microbiota was examined in explanted lungs from people with IPF, showing a higher number of 16S rRNA gene sequences and different taxa from the lung tissue. It is possible, therefore, that airway and lung tissue compartments are separate in IPF, with different microbiota. It is possible that the lungs of IPF patients are ‘walled off’ from the airways as part of the pathogenesis of the disease, compartmentalising bacterial infection. 121
The findings of the current study do not support the view that the bacterial burden is responsible for disease progression in IPF. Co-trimoxazole, as a broad-spectrum antibiotic, should have substantial effects on bacterial load within the lungs. It is widely active against the various anaerobes (Veillonella and Prevotella spp.), haemophili, staphylococci and streptococci variously suggested to have an increased presence in the lungs of IPF patients and does not have a reputation for a swift selection of resistance. 122 Although resistance can occur, it is implausible that it would have been so widespread in these genera as to wholly negate efficacy for a trial population. Indeed, had resistance been the arbiter of outcome, one would have expected some bimodality in the outcome data for the active treatment group, with those patients lacking resistant pathogens faring well and those with resistant pathogens gaining little benefit. However, no evidence of such a pattern was seen.
It should also be emphasised that none of the studies claiming a link between microbiota and progression in IPF has been able to show that a single bacterial agent is more prevalent in the airways of IPF cases than in those of healthy controls and is therefore a likely possible aetiological agent. Rather, the overall flora reported in IPF patients is similar to that seen in healthy control subjects, although with a greater population density. It is inherently unlikely that a mixed population of bacteria would be consistently co-trimoxazole resistant.
Choice of antibiotic
These points link to the further question of ‘Was co-trimoxazole the right antibiotic to trial?’. In context, it should be recalled that the study was initiated to confirm or refute the interesting exploratory findings of the TIPAC trial. 21 When the protocol was written, the prima facie evidence suggested that co-trimoxazole did have a beneficial effect in IPF. In addition, it was unclear whether co-trimoxazole was achieving this through antibacterial or non-antibacterial routes (e.g. through some effect on the immune system). Accordingly, the test drug (co-trimoxazole) and dosing regimen (960 mg twice daily) were kept identical to previous studies.
Given that two large-scale studies of co-trimoxazole (the TIPAC trial21 and the EME-TIPAC trial) have now failed to show a change in FVC or DLCO, which are regarded as key markers of disease activity, and that there were no meaningful changes in any of the blood biomarkers, it seems safe to dismiss the hypothesis that co-trimoxazole has a disease-modifying activity, and, in particular, that it has any non-antibacterial benefit.
There is no reason to suppose that any other antibiotic would have been better in this regard. There remains, however, the possibility that a potential antibacterial benefit was lost owing to widespread resistance, to the selection of resistance or to the involvement of an inherently resistant pathogen. The first two of these possibilities seem unlikely. As already noted, co-trimoxazole is a broad-spectrum combination and, although resistance does arise in the various genera implicated in the IPF lung, there is no evidence to suggest that its prevalence is so large as to entirely negate efficacy in the trial group. To our knowledge, in-therapy selection of resistance has rarely been reported with co-trimoxazole and likewise seems unlikely to have been so frequent as to overwhelm a positive effect. The possibility of an unrecognised co-trimoxazole-resistant pathogen cannot be entirely dismissed, but, to our knowledge, there is no positive evidence to support such a hypothesis.
Among alternative antibiotics, clarithromycin and doxycycline are likely to have similar spectra to co-trimoxazole against the bacteria previously implicated in IPF and there is little obvious reason to suppose that they might achieve better outcomes. Amoxicillin may have greater efficacy against streptococci than co-trimoxazole but would be unreliable against staphylococci, owing to widespread β-lactamase production.
Biomarkers
We assessed a series of serum biomarkers in an attempt to elucidate whether or not co-trimoxazole has a disease-modifying effect in IPF. Specifically, we measured CRP as an acute-phase reactant, as CRP is routinely measured in clinical practice. We found that both treatments resulted in increased CRP concentrations, with a higher increase in the active treatment group. However, the changes were very small and not clinically meaningful.
Notably, the baseline values were low (within normal limits) and, therefore, it is unlikely that this reduction represents a meaningful improvement in inflammation. It should be added that the proven utility of CRP in clinical practice is in the setting of acute infection or inflammation. Its utility in chronic conditions and infections is less established. Thus, for example, long-term (3-months’ treatment) azithromycin reduces CRP in patients with cystic fibrosis123 but short-term (3-week) treatment with doxycycline does not alter CRP in people with stable COPD. 124 The influence of antibiotics on inflammation as assessed by CRP concentrations in stable patients with respiratory disease is, therefore, to our knowledge, not well documented in the current literature.
The concentrations of bronchial epithelium markers, CA19-9 and CA-125 at randomisation were higher in the active treatment group than in the placebo group, and were reduced to a greater extent by active treatment. However, as with CRP, the values at baseline were low; the median values were similar to those found in patients with stable disease. 89 Furthermore, the reduction in concentration was modest and not clinically meaningful.
Given the reported association with IPF and airway neutrophils49,50 and potential mechanisms of co-trimoxazole,39,40 we investigated MPO as a biomarker of neutrophil activity. Unfortunately, we did not find any difference between the MPO concentration with 12 months’ treatment in either the ITT or PP analysis. Likewise, there was no statistically significant difference with TRIAL or osteoprotegerin concentrations. We were not able to show any effect of co-trimoxazole on alveolar epithelial injury or fibroproliferation markers, suggesting that neither co-trimoxazole itself nor its effect on modification of the microbiota influences these key features in the pathogenesis of IPF. 125
Adverse effects
Co-trimoxazole was tolerated well in the study, and the number of AEs was similar in the placebo and active treatment groups. As expected, we found an increase in the number of episodes of hyperkalaemia and rash with co-trimoxazole, although the difference between the two treatment groups was not significant. There were significantly more events of nausea (but not vomiting), but significantly fewer events of diarrhoea, with active treatment than with placebo. However, the findings were due to frequent episodes in a few individuals because there was no significant difference in the number of patients in each group who experienced gastrointestinal ARs. Reassuringly, there were very few episodes of serious hyperkalaemia (one in each group) or rash (one in the active treatment group), and there were no episodes of serious nausea, vomiting or diarrhoea. There were no differences in the levels of any of the safety blood markers other than potassium. Headache was reported more frequently in the active treatment group than in the placebo group. Interestingly, there were fewer episodes of diarrhoea in those taking co-trimoxazole than in those taking placebo and there was a trend towards a reduction in the number of people with diarrhoea in the active treatment group. The reason for this is unclear; although it may be a chance finding, co-trimoxazole may modify the gut flora in a way that reduces diarrhoea. The summary of product characteristics reports diarrhoea as a common side effect of co-trimoxazole.
In the TIPAC trial,21 30% of people in the active treatment group, including one person (concomitantly receiving azathioprine) who had life-threatening neutropenia, and 8% in the placebo group withdrew as a result of adverse effects. There were significantly more people with a rash (15.2% active treatment group vs. 4.7% placebo group), nausea (18.5% vs. 7.0%) and a 10 mmol/l increase in creatinine level (59.3% vs. 12.5%). There was an increase in serum potassium levels in people on co-trimoxazole whether or not they were receiving antikaliuretic drugs, with a small (5.7%) number of people having clinically important hyperkalaemia (> 5.5 mmol/l). 51 In the study by Stegeman et al. ,37 which investigated the role of co-trimoxazole in granulomatosis with polyangiitis, 20% stopped using the drug as a result of ARs over a 3-month period and there was a 17% increase in creatinine levels in the active treatment group.
We introduced the option for patients to step down to a lower dose as the common adverse reactions are dose related, and this may account for the better tolerability in the current study. A larger percentage of people in the co-trimoxazole group (19%) stepped down than in the placebo group (9%); presumably people with ARs due to co-trimoxazole reduced the dose to a tolerable dosing regimen. In this respect, low-dose co-trimoxazole was well tolerated in a study of 116 patients with HIV and P. jirovecii infection. 126 Although 28% had ARs (rash, pruritus and nausea), only 15 withdrew from treatment during a follow-up period of 18.5 months and only 9% were drug intolerant. 126 In a study of 541 children, the rate of adverse drug reactions due to co-trimoxazole (7%) was similar to that with the placebo (6%). 127
Adherence to medication
We showed that the overall adherence was 81% and 86% in the active treatment and placebo groups, respectively, with 72% of patients in both groups complying with treatment by > 80%. In the TIPAC trial,21 overall adherence to the study medication was better, with 96% of patients in the active treatment group and 90% in the placebo group receiving > 80% of the scheduled study drug doses; however, we do not believe that the difference in adherence is responsible for the difference in the findings of the study.
Strengths
The main strength of the EME-TIPAC trial was that it was a large, adequately powered, multicentre, academic clinical trial that used a clinically relevant outcome with high follow-up rates and long-term timescales.
The trial involved 43 centres in the UK, representing one-fifth of the NHS trusts in the UK and nearly all of the specialist centres. We included sites from all of the devolved nations and our recruitment was geographically diverse. We were required to involve specialist centres as the main recruitment sites to ensure that diagnosis followed a MDT meeting. However, we involved sites that were referral centres to specialist centres and managed patients with IPF independently. Although the recruitment numbers were not uniform across all sites, with the 10 highest-recruiting sites responsible for slightly more than half (180 out of 342 patients) of the sample, the recruitment was not dominated by a few large academic centres, with hospitals serving smaller populations also contributing substantially to the study.
The primary outcome was unplanned hospitalisation-free survival, defined as time to death (all causes) and first non-elective hospital admission. These end points have been recommended by the Pulmonary Fibrosis Foundation summit of end points for clinical trials in IPF as the most clinically relevant, as no other end points are either reliable, validated or adequately robust. 52 FVC is frequently utilised as an end point in clinical trials and has been accepted by the US Food and Drug Administration as an appropriate end point for the licensing of medication based on the fact that changes in FVC over time have been repeatedly shown to be highly predictive of mortality. 109 However, it remains a surrogate biomarker of mortality and should be regarded as such. Mortality is clearly meaningful for patients and their relatives. Hospitalisation is also a major clinical event, with the majority of people who are hospitalised with IPF having a poor outcome with high frequency of death. Hospitalisation is financially expensive to health-care providers and has significant social costs to patients. Even proponents of FVC state that ‘all-cause mortality would, indeed be the most clinical meaningful primary endpoint’. 53
Mortality studies in IPF are difficult as they need to recruit several thousand people with mild disease to reliably detect a treatment difference. 53 However, people with more severe disease have poorer outcomes with much higher mortality rates than those with mild disease. 54 We undertook a sensitivity analysis from the TIPAC trial21 to determine the appropriate FVC value for the current study, balancing expected event rates with anticipated recruitment rates. We initially planned to recruit people who had a FVC < 70% predicted but changed this to a FVC of < 75% predicted. This resulted in a marginal improvement in recruitment, with 10 additional patients being randomised into the study, but we do not believe it made a meaningful difference to the event rate.
We aimed to recruit 330 people with IPF; however, as a result of increased initiatives towards the end of the recruitment period, we over-recruited and 342 were randomised into the study. This is much smaller than the recruitment size of multinational pharmaceutically sponsored studies; the ASCEND study randomised 555 patients74 and the INPULSIS studies also randomised more than 500 people103 with IPF. However, to our knowledge, the current study remains the largest study of its type. Furthermore, there was a low withdrawal rate, with only one patient, who was randomised in error, unable to provide data for the primary end point. This was lower than we had anticipated – our sample size calculation had a withdrawal rate of 2%. In addition, patients who wished to stop taking study medication, despite the option to reduce to a low dose, were asked to continue to undergo follow-up assessments. Patients who met the primary end point because of hospitalisation discontinued study medication to reduce their exposure to ARs of the drug, but were also followed up until death.
As we assumed that the hospitalisation-free survival rate in both groups would be 50% higher in the EME-TIPAC trial than in the TIPAC trial,21 we powered the study with the anticipation that there would be 99 events. However, the event rate was noticeably higher than we had anticipated, with 164 events in the all-cause analysis and 82 in the respiratory-related analysis. The study was adequately powered to detect the difference that we were looking for in the primary end point and was nearly adequately powered for the secondary end point.
Another reason that the study delivered to target is that it was designed so that measurements were taken alongside clinical care. This meant that there was a reduced research burden for patients, which may be reflected in the high retention rate, and that lung function measurements were mostly obtained by Association for Respiratory Technology and Physiology-registered pulmonary function technologists. The alignment with clinical and research assessments was difficult to co-ordinate, although we permitted a window of 2 months for each study visit after the 6-month period, as the primary end point was the time to an event and was not dependent on a visit schedule.
We, therefore, conducted an adequately powered study, which recruited to target and had few withdrawals, and our findings are robust. A larger study is unlikely to come to a different conclusion. The treatment effect seen in the TIPAC trial21 was of such magnitude that a mortality study was feasible and appropriate.
Limitations
The main limitation of the study was the inability to evaluate the lung microbiome and the influence that co-trimoxazole had on this measure. We had planned to undertake two bronchoscopy procedures in a subgroup of 50 people, one before and one 3 months after commencing co-trimoxazole administration. Our public and patient advisors and patient acceptability data suggested that this would be feasible. We felt that the risks were low, even in people with moderate disease, and we enrolled centres experienced in undertaking bronchoscopy for clinical and research reasons. However, the recruitment into this substudy was voluntary and, as a result, few people agreed to participate in this. The previous bronchoscopy biomarker and microbiota studies utilised BAL samples taken at a diagnostic bronchoscopy undertaken for clinical need. We could not use samples taken during a diagnostic bronchoscopy as a clinical diagnosis was required before randomisation.
Two patients participated in the substudy and underwent bronchoscopy for the purpose of the trial. Despite increasing the number of sites permitted to undertake bronchoscopy for research and, therefore, the population who may agree, we were unable to increase our sample into this part of the study. In July 2017, the DMC advised against continuing this aspect of the study on the grounds of futility.
We intended that by analysing the microbiota and BAL biomarkers we would understand how co-trimoxazole was exerting its activity. However, given the lack of effect of this treatment, this analysis would not have been helpful. We were unable to determine if or how the microbiota was changed by co-trimoxazole. We could not determine whether or not co-trimoxazole resulted in the emergence of pathological bacteria adversely influencing the outcome in IPF, but given the findings of this study and the mechanism of action of co-trimoxazole, this seems unlikely.
We asked that the results of all microbiological analyses from sputum samples requested for clinical reasons were recorded. However, very few data were captured, and it was not possible to undertake an analysis of the microbiological data. Determining a diagnosis of infection is often difficult in people with IPF, and, given that the data suggesting microbiological association with disease progression were based on non-culture techniques, it is unlikely that sputum samples would have added much to the interpretation of the current findings.
Although data for the primary end point were complete, there were a substantial number of missing data for some of the secondary end points, in particular the questionnaire and lung function data. This was because of both logistical issues regarding booking people into research alongside routine clinic appointments and patient withdrawal from this aspect of the study because of difficulty with transport and mobility. We do not believe that these missing data represented a bias in our results, but they may have weakened the ability to detect a treatment effect. Furthermore, missing data were multiply imputed and subject to sensitivity analysis.
We did not use a central committee to confirm diagnosis prior to entry as is frequently undertaken by commercial studies. This procedure permits some standardisation of diagnosis, but is resource intensive and time-consuming. However, we are confident with the diagnostic accuracy of our sample as all MDT meetings took place with clinical experts in a few specialist centres in the UK. They followed standard national service specification, which is based on international guidelines, and were subject to audit. We employed an independent committee to review whether or not the primary events were respiratory related. This was chaired by an expert in clinical trials and had representation from ILD specialists and experienced general clinicians.
We did not investigate the number of respiratory infection-related events; rather we assessed ‘respiratory-related’ events. Assessing whether or not respiratory infection is present during acute exacerbations or other clinical settings is frequently difficult. Indeed, a recent working group review of acute exacerbation of ILD did not exclude infection as a cause. 128 Our independent review committee reviewed the clinical listings, rather than the chest radiograph or medical notes, which would have been required to make a reliable assessment of the presence or absence of a respiratory tract infection.
We had planned to recruit patients who were within 2 years of diagnosis, so we excluded people with IPF who had stable disease without much evidence of decline or likelihood of meeting an end point. In the TIPAC trial,21 the event rate for unplanned hospitalisation-free survival was 42.5% for those diagnosed within 2 years (one-quarter of randomised patients) versus 35.0% for all patients enrolled in the study. This meant a decrease in unplanned hospitalisation-free survival of about 7%. However, the time of diagnosis proved difficult to define and the time from diagnosis is clearly different from the time of onset of symptoms, which is likely to be more relevant to the time course of the disease. Some patients who were referred had had a clinical diagnosis for a considerable period of time but this was formally confirmed only at the MDT meeting. In addition, to meet the entry criteria of having a FVC of < 75% predicted, patients had to have deteriorated to some extent. Although the condition deteriorates at different rates in different people, stabilisation after an initial deterioration does not usually occur. This entry criterion was, therefore, difficult to clearly establish or monitor, and its strict implementation significantly restricted recruitment rate to the study, leading to its abandonment. We do not believe that this decision significantly influenced the outcome of the study and, in fact, it is likely to have made the study more generalisable.
Although we measured utility, we did not assess costs or undertake an economic analysis. The health economics of co-trimoxazole in IPF have previously been reported, following the TIPAC trial. 21 Given the negative effects of co-trimoxazole, and the potential harms, it is unlikely that a detailed health economic analysis would provide information that would change the conclusions of this study.
Generalisability
The study has good external validity as it recruited from a large number of sites from throughout the UK. Patients were able to continue with their existing treatment for IPF, as long as this was in accordance with current guidelines. There were only a few exclusion criteria, which were required either to ensure patient safety or to ensure that any treatment effect was because of change in IPF. We believe it is highly likely that the EME-TIPAC trial patients were representative of the normal clinical practice in the UK.
We restricted inclusion to the study to people with moderate to severe disease as determined by a FVC of < 75% predicted. We do not believe that the effects of co-trimoxazole are likely to be different according to severity of disease, but those with severe disease have a poorer prognosis and, therefore, this treatment would have been more cost-effective in this group. In addition, had we recruited people with higher lung function, the study would have been prohibitively costly.
Public and patient involvement
The aim of the public and patient involvement in the study was to ensure that the study was relevant for patients, and deliverable from a patient point of view, and that the results of the study were meaningful for patients and shared appropriately. In these respects, the patient and public involvement was effective; however, lessons were learnt during the course of the project.
Patient and public involvement was instrumental to the design of the study. The decision to undertake this study was, in part, kindled by a discussion with patients of the TIAPC trial21 during a research dissemination meeting. The rationale of the study was discussed along with the study measurements and outcomes. An in-depth discussion took place regarding the benefits and risks of bronchoscopy and the patients felt that the majority of patients would agree to this. Eight patients with IPF took part in a semistructured interview about the study. The concerns about bronchoscopy were highlighted by patients, but, overall, the group felt that this outcome was worthwhile. The study was discussed with Cambridge Pulmonary Fibrosis Patient Support group (some of whom took part in the original TIPAC trial). 21 All 40 patients attending the meeting were willing to take part in the study, and 75% were willing to have two consecutive bronchoscopies. Patients were involved with reforming the application following the previous unsuccessful bid. The study was also discussed, in more general terms, with medical staff, members of the public and other patients with IPF. Members of Public and Patient Involvement in Research (https://mcpin.org/resources/service-user-and-carer-groups/east-anglia/norfolk-suffolk-public-and-patient-involvement-in-research-group-ppires-nhs-south-norfolk-clinical-commissioning-group/; accessed 31 March 2020) reviewed a lay summary. They identified jargon and, although they felt that some people would not want a bronchoscopy, they unanimously agreed that the study had the potential to make a difference in the lives of patients with IPF. Perhaps, in retrospect, the results from patient and public involvement regarding the bronchoscopy were biased, as those interested in research were more likely to contribute to the survey and more likely to believe that bronchoscopy would be a successful component of the study than those who were not interested in research. We were reassured by our patient and public involvement that bronchoscopy would be possible, albeit in a subgroup; however, very few people (only two) underwent this procedure in the study.
A patient representative was present on the TSC at the beginning of the study. He helped review the patient-facing material, but withdrew from the study because of illness and was replaced. The second member was helpful in considering different methods of engaging with patients to encourage recruitment. His ideas have formed the basis for a Study Within A Trial (SWAT) to explore patient recruitment alongside a subsequent NIHR Health Technology Assessment (HTA)-funded study. Unfortunately, he developed an acute exacerbation, but still managed to contribute to the running of the study while receiving high-flow oxygen. Sadly, he passed away from IPF during the study. He was not replaced, as the study was already ongoing; however, we had patient and public involvement from Action for Pulmonary Fibrosis, the UK ILD charity. It promoted our study on its website, particularly in the later stages, when we were having difficulty completing the study. We identified a patient and public involvement representative to attend the TSC meetings; although she agreed to take part and was invited to the meetings, she did not attend.
We have learnt from our experience of patient representatives. We have included two patient and public involvement representatives as co-applicants on a subsequent NIHR HTA-funded study. They have had more involvement in the study than the patient and public involvement representatives who joined the previous study at the start-up phase. The NCTU patient and public involvement programme has been fully developed and there are greater resources to support patient and public involvement representatives through studies, including a welcome pack and a NCTU patient and public involvement support lead. In addition, we are better prepared to replace patient and public involvement representatives if they become unwell or unable to support the study. We have also included two patient and public involvement representatives on the TSC.
The results have been shared with members of Action for Pulmonary Fibrosis and patient support groups. They have helped us to write a lay summary and will continue to help with dissemination. This will include a lay summary on the Action for Pulmonary Fibrosis website. While sharing the results with the Norwich patient support group, it became evident that some people were concerned about taking any antibiotics at all. For that reason, we have made it clear that the study was to evaluate prophylactic antibiotic therapy for the purpose of modifying disease progression, not acute antibiotic therapy for respiratory tract or other infections.
Conclusion
This Phase II, double-blind, placebo-controlled, parallel-group, randomised multicentre study evaluated the effects of 960 mg of co-trimoxazole taken twice per day when given as standard care in 354 individuals with moderate to severe IPF over a total exposure time of 394 years. It showed no statistical or clinically meaningful benefit between co-trimoxazole and placebo for total, all-cause or respiratory-related hospitalisations or death. In the prespecified PP analysis and repeated measures analysis, there was an improvement in cough with co-trimoxazole therapy but no change in other patient-reported outcomes, measures of lung function or blood biomarkers.
Implications for clinical practice
Our results suggest that the prophylactic use of co-trimoxazole for the treatment of IPF ought not to be recommended. This study does not make any recommendation regarding the use of co-trimoxazole or other antibiotics in the situation of acute exacerbations or concomitant respiratory infections in IPF.
Recommendations for research
We found a consistent beneficial effect of co-trimoxazole on different measures of cough (CSS, cough quality of life and symptoms of disease-related quality of life) in different analyses. Although it is possible that this is a chance finding, we found similar benefits in the previous TIPAC trial,21 suggesting that these effects are real. A further study to evaluate the effect of co-trimoxazole in terms of cough should be considered.
We examined the effects of sensitivity analysis in terms of adherence to treatment and treatment regime. However, our SAP did not provide provision to undertake exploratory analyses to identify whether or not there are groups of individuals who may benefit from co-trimoxazole (e.g., those with recurrent chest infections, significant traction bronchiectasis or high bacterial burden). Additional studies of antibiotic therapy in those who may benefit most may be warranted.
Although this study rules out a role for co-trimoxazole in unselected individuals with moderate to severe IPF, and it is unlikely that other broad-spectrum antibiotics will be beneficial, we cannot exclude the possibility that other therapies that alter the lung microbiota will improve outcomes in IPF. Other studies of antibiotics, possibly with a more targeted approach, should be considered.
Acknowledgements
We would like to thank all of the patients who took part in the study. We are grateful to all of the staff at the recruitment sites who facilitated the identification, recruitment and follow-up of study patients.
We thank Martin Pond, Anthony Colles and the NCTU data management team for developing and maintaining the electronic CRFs and the study database. We are grateful for the contributions of Mercedes Mills, and also for the contributions of Julie Dawson in her role as sponsor representative.
We are grateful for the members of the TSC and DMC.
We thank Steve Jones from Action for Pulmonary Fibrosis and the two patient representatives.
We would also like to pay our sincere thanks to all of the following PIs and their teams, whose efforts made this research possible.
| Site | PI | Research nurse or trial practitioner |
|---|---|---|
| Norfolk and Norwich University Hospitals NHS Foundation Trust | Andrew Wilson | Susana Robinson |
| Royal Papworth Hospital NHS Foundation Trust | Helen Parfrey | Kane Dorey |
| Royal Brompton and Harefield NHS Trust | Toby Maher | Katie Rhatigan |
| Sheffield Teaching Hospitals NHS Foundation Trust | Stephen Bianchi | Faith Kibutu |
| University Hospital Birmingham NHS Foundation Trust | David Thickett | Diane Griffiths |
| Heart of England NHS Foundation Trust | Gareth Walters | Mary Bellamy |
| University Hospitals of North Midlands NHS Trust | Helen Stone | Loretta Barnett |
| North Bristol NHS Trust | Huzaifa Adamali | Caroline Kilby |
| Cardiff and Vale University Health Board | Ben Hope-Gill | Doria Barbonchielli |
| The Newcastle Upon Tyne Hospitals NHS Foundation Trust | Ian Forrest | Geraldine Jones |
| Gateshead Health NHS Foundation Trust | Robert Allcock | Maureen Armstrong |
| Salford Royal NHS Foundation Trust | Ronan O’Driscoll | Yvonne Rostron |
| University Hospital of South Manchester NHS Foundation Trust | Nazia Chaudhuri | Sukoluhle Moyo |
| Aintree University Hospitals NHS Foundation Trust | Lisa Spencer | Joanne Earley |
| Lancashire Teaching Hospitals NHS Foundation Trust | Yussef Haider | Janet Mills |
| Aberdeen Royal Infirmary | Owen Dempsey | Sandra Steele |
| NHS Greater Glasgow and Clyde | George Chalmers | Helen Banister |
| Oxford University Hospital NHS Foundation Trust | Rachel Hoyles | Debby Nicoll |
| Imperial College Healthcare NHS Trust | Robina Coker | Krystal Johnson |
| NHS Tayside | Andrew Goudie | Heather Loftus |
| Royal Devon and Exeter NHS Foundation Trust | Michael Gibbons | Sinéad Kelly |
| Hull and East Yorkshire Hospitals NHS Trust | Simon Hart | Caroline Wright |
| Nottingham University Hospitals NHS Foundation Trust | Gauri Saini | Lucy Howard |
| Cambridge University Hospitals | Edwin Chilvers | Jacqui Galloway |
| Blackpool, Fylde and Wyre Hospitals NHS Foundation Trust | Thomas Bongers | Melanie Caswell |
| The Shrewsbury and Telford Hospital NHS Trust | Richard Heinink | Heather Button |
| Sherwood Forest Hospitals NHS Foundation Trust | Khaled Amsha | Tracy Brear |
| St George’s University Hospitals NHS Foundation Trust | Raminder Aul | Joyce Kibaru |
| University College London Hospitals NHS Foundation Trust | Joanna Porter | Jagdeep Sahota |
| University Hospitals of Leicester NHS Trust | Felix Woodhead | Nicola Goodman |
| Worcestershire Acute Hospitals NHS Trust | Stephen O’Hickey | Alison Durie |
| Western Health and Social Care Trust | Martin Kelly | Kathryn Ferguson |
| The Royal Wolverhampton NHS Trust | Ahmed Fahim | Lucy Stelfox |
| University Hospital Southampton NHS Foundation Trust | Sophie Fletcher | Rachael Collings |
| University Hospitals of Morecambe Bay NHS Foundation Trust | Timothy Gatheral | Rebecca Jeffery |
| Calderdale and Huddersfield NHS Foundation Trust | Rehan Naseer | Kully Sandhu |
| South Tyneside Foundation Trust | Liz Fuller | Nadia Elkaram |
| NHS Forth Valley | Mark Spears | Anne Todd |
| University Hospitals Coventry and Warwickshire NHS Trust | David Parr | Rhian Hughes |
Role of the funder
The funder, NIHR, had input into the trial design through peer review of the proposal, but did not have a role in data collection, data analysis, data interpretation or the writing of the final report. The corresponding author had access to all of the data and was responsible for the decision to submit the report.
Study website
The study website is as follows: www.uea.ac.uk/eme-tipac (accessed 10 March 2021).
Contributions of authors
Andrew M Wilson (https://orcid.org/0000-0002-5514-4867) (Professor of Respiratory Medicine) was the chief investigator and oversaw the delivery of the study. He contributed to the conception, design and conduct of the trial; the recruitment and follow-up of patients; the interpretation of results; and writing/editing the report
Allan B Clark (https://orcid.org/0000-0003-2965-8941) (Senior Trial Statistician) oversaw the statistical analysis and contributed to the design of the trial. He was responsible for statistical analysis and contributed to the interpretation of results and writing/editing the report
Anthony Cahn (https://orcid.org/0000-0002-7239-0461) (Consultant Physician) provided expertise in clinical trial design and delivery. He contributed to the conception and design of the trial, the interpretation of results and writing/editing the report.
Edwin R Chilvers (https://orcid.org/0000-0002-4230-9677) (Professor of Respiratory Medicine) provided expertise in ILD and lung inflammation. He contributed to the conception and design of the trial, the interpretation of results and writing/editing the report.
William Fraser (https://orcid.org/0000-0003-0556-3358) (Professor of Medicine) oversaw the biochemical analyses. He contributed to the conception and design of the trial, the interpretation of results and writing/editing the report, with particular emphasis on the biochemical analysis.
Matthew Hammond (https://orcid.org/0000-0002-0739-3412) (Clinical Trials Unit Deputy Director) was responsible for the day-to-day management of the trial and contributed to the interpretation of results and writing/editing the report.
David M Livermore (https://orcid.org/0000-0002-9856-3703) (Professor of Medical Microbiology) provided expertise in the microbiological aspects of the study. He contributed to the conception and design of the trial, the interpretation of results and writing/editing the report, with particular emphasis on the microbiological aspects.
Toby M Maher (https://orcid.org/0000-0001-7192-9149) (Professor of Respiratory Medicine) provided expertise in ILD and clinical trial methodology. He contributed to the conception, design and conduct of the trial; the recruitment and follow-up of patients; the interpretation of results; and writing/editing the report.
Helen Parfrey (https://orcid.org/0000-0002-4515-5634) (Consultant Physician) provided expertise in ILD. She contributed to the conception, design and conduct of the trial; the recruitment and follow-up of patients; the interpretation of results; and writing/editing the report.
Ann Marie Swart (https://orcid.org/0000-0002-9359-6995) (Clinical Trials Unit Director) was responsible for the day-to-day management of the trial. She contributed to the interpretation of results and writing/editing the report.
Susan Stirling (https://orcid.org/0000-0001-6663-7846) (Trial Statistician) undertook the statistical analysis. She contributed to the interpretation of results and writing/editing the report.
David Thickett (https://orcid.org/0000-0002-5456-6080) (Professor of Respiratory Medicine) provided expertise in ILD. He contributed to the conception, design and conduct of the trial; the recruitment and follow-up of patients; the interpretation of results; and writing/editing the report.
Moira Whyte (https://orcid.org/0000-0001-7788-1852) (Professor of Respiratory Medicine) provided expertise in ILD. She contributed to the conception and design of the trial, the interpretation of results and writing/editing the report.
Publication
Wilson AM, Clark AB, Cahn T, Chilvers ER, Fraser W, Hammond M, et al. Effect of co-trimoxazole (trimethoprim-sulfamethoxazole) vs. placebo on death, lung transplant, or hospital admission in patients with moderate and severe idiopathic pulmonary fibrosis: the EME-TIPAC randomized clinical trial. JAMA 2020;324:2282–91.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to available anonymised data may be granted following review.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, NETSCC, the EME programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the EME programme or the Department of Health and Social Care.
References
- Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 2018;198:e44-e68. https://doi.org/10.1164/rccm.201807-1255ST.
- Nicholson AG, Colby TV, du Bois RM, Hansell DM, Wells AU. The prognostic significance of the histologic pattern of interstitial pneumonia in patients presenting with the clinical entity of cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med 2000;162:2213-17. https://doi.org/10.1164/ajrccm.162.6.2003049.
- Navaratnam V, Fleming KM, West J, Smith CJP, Jenkins RG, Fogarty A, et al. The rising incidence of idiopathic pulmonary fibrosis in the UK. Thorax 2011;66:462-7. https://doi.org/10.1136/thx.2010.148031.
- Macpherson KJ, Lewsey JD, Jhund PS, Gillies M, Chalmers JW, Redpath A, et al. Trends in incidence and in short term survival following a subarachnoid haemorrhage in Scotland, 1986–2005: a retrospective cohort study. BMC Neurol 2011;11. https://doi.org/10.1186/1471-2377-11-38.
- Navaratnam V, Fogarty AW, Glendening R, McKeever T, Hubbard RB. The increasing secondary care burden of idiopathic pulmonary fibrosis: hospital admission trends in England from 1998 to 2010. Chest 2013;143:1078-84. https://doi.org/10.1378/chest.12-0803.
- Collard HR, Ward AJ, Lanes S, Cortney Hayflinger D, Rosenberg DM, Hunsche E. Burden of illness in idiopathic pulmonary fibrosis. J Med Econ 2012;15:829-35. https://doi.org/10.3111/13696998.2012.680553.
- Lee AS, Mira-Avendano I, Ryu JH, Daniels CE. The burden of idiopathic pulmonary fibrosis: an unmet public health need. Respir Med 2014;108:955-67. https://doi.org/10.1016/j.rmed.2014.03.015.
- Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011;183:788-824. https://doi.org/10.1164/rccm.2009-040GL.
- Raghu G, Anstrom KJ, King TE, Lasky JA, Martinez FJ. Idiopathic Pulmonary Fibrosis Clinical Research Network . Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 2012;366:1968-77. https://doi.org/10.1056/NEJMoa1113354.
- Kubo H, Nakayama K, Yanai M, Suzuki T, Yamaya M, Watanabe M, et al. Anticoagulant therapy for idiopathic pulmonary fibrosis. Chest 2005;128:1475-82. https://doi.org/10.1378/chest.128.3.1475.
- Noth I, Anstrom KJ, Calvert SB, de Andrade J, Flaherty KR, Glazer C, et al. A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2012;186:88-95. https://doi.org/10.1164/rccm.201202-0314OC.
- Demedts M, Behr J, Buhl R, Costabel U, Dekhuijzen R, Jansen HM, et al. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med 2005;353:2229-42. https://doi.org/10.1056/NEJMoa042976.
- Martinez FJ, de Andrade JA, Anstrom KJ, King TE, Raghu G. Idiopathic Pulmonary Fibrosis Clinical Research Network . Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med 2014;370:2093-101. https://doi.org/10.1056/NEJMoa1401739.
- Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011;377:1760-9. https://doi.org/10.1016/S0140-6736(11)60405-4.
- Richeldi L, Costabel U, Selman M, Kim DS, Hansell DM, Nicholson AG, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med 2011;365:1079-87. https://doi.org/10.1056/NEJMoa1103690.
- Raghu G, Rochwerg B, Zhang Y, Garcia CA, Azuma A, Behr J, et al. An Official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med 2015;192:e3-19. https://doi.org/10.1164/rccm.201506-1063ST.
- Nathan SD, Albera C, Bradford WZ, Costabel U, Glaspole I, Glassberg MK, et al. Effect of pirfenidone on mortality: pooled analyses and meta-analyses of clinical trials in idiopathic pulmonary fibrosis. Lancet Respir Med 2017;5:33-41. https://doi.org/10.1016/S2213-2600(16)30326-5.
- National Institute for Health and Care Excellence . Idiopathic Pulmonary Fibrosis in Adults: Diagnosis and Management 2013. www.nice.org.uk/guidance/cg163 (accessed 31 March 2020).
- Skandamis A, Kani C, Markantonis SL, Souliotis K. Systematic review and network meta-analysis of approved medicines for the treatment of idiopathic pulmonary fibrosis. J Drug Assess 2019;8:55-61. https://doi.org/10.1080/21556660.2019.1597726.
- Varney VA, Parnell HM, Salisbury DT, Ratnatheepan S, Tayar RB. A double blind randomised placebo controlled pilot study of oral co-trimoxazole in advanced fibrotic lung disease. Pulm Pharmacol Ther 2008;21:178-87. https://doi.org/10.1016/j.pupt.2007.02.001.
- Shulgina L, Cahn AP, Chilvers ER, Parfrey H, Clark AB, Wilson EC, et al. Treating idiopathic pulmonary fibrosis with the addition of co-trimoxazole: a randomised controlled trial. Thorax 2013;68:155-62. https://doi.org/10.1136/thoraxjnl-2012-202403.
- Wilson EC, Shulgina L, Cahn AP, Chilvers ER, Parfrey H, Clark AB, et al. Treating idiopathic pulmonary fibrosis with the addition of co-trimoxazole: an economic evaluation alongside a randomised controlled trial. Pharmacoeconomics 2014;32:87-99. https://doi.org/10.1007/s40273-013-0112-z.
- Oda K, Yatera K, Fujino Y, Ishimoto H, Nakao H, Hanaka T, et al. Efficacy of concurrent treatments in idiopathic pulmonary fibrosis patients with a rapid progression of respiratory failure: an analysis of a national administrative database in Japan. BMC Pulm Med 2016;16. https://doi.org/10.1186/s12890-016-0253-x.
- Shulgina L, Cahn AP, Chilvers ER, Parfrey H, Clark AB, Wilson EC, et al. Author’s response: co-trimoxazole treatment in idiopathic pulmonary fibrosis. Thorax 2013;68:884-5. https://doi.org/10.1136/thoraxjnl-2013-203765.
- Atkins CP, Loke Y, Wilson AM. S16 Outcomes in idiopathic pulmonary fibrosis: a meta-analysis from placebo controlled trials. Thorax 2013;68. https://doi.org/10.1136/thoraxjnl-2013-204457.23.
- Müllerova H, Chigbo C, Hagan GW, Woodhead MA, Miravitlles M, Davis KJ, et al. The natural history of community-acquired pneumonia in COPD patients: a population database analysis. Respir Med 2012;106:1124-33. https://doi.org/10.1016/j.rmed.2012.04.008.
- Olson AL, Swigris JJ, Raghu G, Brown KK. Seasonal variation: mortality from pulmonary fibrosis is greatest in the winter. Chest 2009;136:16-22. https://doi.org/10.1378/chest.08-0703.
- Richter AG, Stockley RA, Harper L, Thickett DR. Pulmonary infection in Wegener granulomatosis and idiopathic pulmonary fibrosis. Thorax 2009;64:692-7. https://doi.org/10.1136/thx.2008.110445.
- Vidal S, de la Horra C, Martín J, Montes-Cano MA, Rodríguez E, Respaldiza N, et al. Pneumocystis jirovecii colonisation in patients with interstitial lung disease. Clin Microbiol Infect 2006;12:231-5. https://doi.org/10.1111/j.1469-0691.2005.01337.x.
- Garzoni C, Brugger SD, Qi W, Wasmer S, Cusini A, Dumont P, et al. Microbial communities in the respiratory tract of patients with interstitial lung disease. Thorax 2013;68:1150-6. https://doi.org/10.1136/thoraxjnl-2012-202917.
- Molyneaux PL, Cox MJ, Mallia P, Johnston SL, Moffatt MF, Cookson WOC, et al. S38 The role of the respiratory microbiome in idiopathic pulmonary fibrosis. Thorax 2013;68. https://doi.org/10.1136/thoraxjnl-2013-204457.45.
- Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, et al. Disordered microbial communities in asthmatic airways. PLOS ONE 2010;5. https://doi.org/10.1371/journal.pone.0008578.
- Han MKE-D, Zhou Y, Tayob N, Murray S, Flaherty KR, Huffnagle GB, et al. The IPF microbiome: analysis from the COMET study. Am J Respir Crit Care Med 2013;187.
- Savici D, Campbell PA, King TE. Bronchoalveolar macrophages from patients with idiopathic pulmonary fibrosis are unable to kill facultative intracellular bacteria. Am Rev Respir Dis 1989;139:22-7. https://doi.org/10.1164/ajrccm/139.1.22.
- Samara KD, Antoniou KM, Karagiannis K, Margaritopoulos G, Lasithiotaki I, Koutala E, et al. Expression profiles of Toll-like receptors in non-small cell lung cancer and idiopathic pulmonary fibrosis. Int J Oncol 2012;40:1397-404. https://doi.org/10.3892/ijo.2012.1374.
- Hogaboam CM, Trujillo G, Martinez FJ. Aberrant innate immune sensing leads to the rapid progression of idiopathic pulmonary fibrosis. Fibrogenesis Tissue Repair 2012;5. https://doi.org/10.1186/1755-1536-5-S1-S3.
- Stegeman CA, Tervaert JW, de Jong PE, Kallenberg CG. Trimethoprim-sulfamethoxazole (co-trimoxazole) for the prevention of relapses of Wegener’s granulomatosis. Dutch Co-Trimoxazole Wegener Study Group. N Engl J Med 1996;335:16-20. https://doi.org/10.1056/NEJM199607043350103.
- Zycinska K, Wardyn KA, Zielonka TM, Krupa R, Lukas W. Co-trimoxazole and prevention of relapses of PR3-ANCA positive vasculitis with pulmonary involvement. Eur J Med Res 2009;14:265-7. https://doi.org/10.1186/2047-783X-14-S4-265.
- Harvath L, Yancey KB, Katz SI. Selective inhibition of human neutrophil chemotaxis to N-formyl-methionyl-leucyl-phenylalanine by sulfones. J Immunol 1986;137:1305-11.
- Suda T, Suzuki Y, Matsui T, Inoue T, Niide O, Yoshimaru T, et al. Dapsone suppresses human neutrophil superoxide production and elastase release in a calcium-dependent manner. Br J Dermatol 2005;152:887-95. https://doi.org/10.1111/j.1365-2133.2005.06559.x.
- Anderson R, Grabow G, Oosthuizen R, Theron A, Van Rensburg AJ. Effects of sulfamethoxazole and trimethoprim on human neutrophil and lymphocyte functions in vitro: in vivo effects of co-trimoxazole. Antimicrob Agents Chemother 1980;17:322-6. https://doi.org/10.1128/AAC.17.3.322.
- Siegel JP, Remington JS. Effect of antimicrobial agents on chemiluminescence of human polymorphonuclear leukocytes in response to phagocytosis. J Antimicrob Chemother 1982;10:505-15. https://doi.org/10.1093/jac/10.6.505.
- Welch WD, Davis D, Thrupp LD. Effect of antimicrobial agents on human polymorphonuclear leukocyte microbicidal function. Antimicrob Agents Chemother 1981;20:15-20. https://doi.org/10.1128/AAC.20.1.15.
- Cantin AM, North SL, Fells GA, Hubbard RC, Crystal RG. Oxidant-mediated epithelial cell injury in idiopathic pulmonary fibrosis. J Clin Invest 1987;79:1665-73. https://doi.org/10.1172/JCI113005.
- Felton VM, Borok Z, Willis BC. N-acetylcysteine inhibits alveolar epithelial-mesenchymal transition. Am J Physiol Lung Cell Mol Physiol 2009;297:L805-12. https://doi.org/10.1152/ajplung.00009.2009.
- Psathakis K, Mermigkis D, Papatheodorou G, Loukides S, Panagou P, Polychronopoulos V, et al. Exhaled markers of oxidative stress in idiopathic pulmonary fibrosis. Eur J Clin Invest 2006;36:362-7. https://doi.org/10.1111/j.1365-2362.2006.01636.x.
- Behr J, Maier K, Krombach F, Adelmann-Grill BC. Pathogenetic significance of reactive oxygen species in diffuse fibrosing alveolitis. Am Rev Respir Dis 1991;144:146-50. https://doi.org/10.1164/ajrccm/144.1.146.
- Crystal RG, Bitterman PB, Rennard SI, Hance AJ, Keogh BA. Interstitial lung diseases of unknown cause. Disorders characterized by chronic inflammation of the lower respiratory tract (first of two parts). N Engl J Med 1984;310:154-66. https://doi.org/10.1056/NEJM198401193100304.
- Beeh KM, Beier J, Kornmann O, Buhl R. Neutrophilic inflammation in induced sputum of patients with idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis 2003;20:138-43.
- Kinder BW, Brown KK, Schwarz MI, Ix JH, Kervitsky A, King TE. Baseline BAL neutrophilia predicts early mortality in idiopathic pulmonary fibrosis. Chest 2008;133:226-32. https://doi.org/10.1378/chest.07-1948.
- Chan WY, Clark AB, Wilson AM, Loke YK. TIPAC investigators . The effect of co-trimoxazole on serum potassium concentration: safety evaluation of a randomized controlled trial. Br J Clin Pharmacol 2017;83:1808-14. https://doi.org/10.1111/bcp.13263.
- Raghu G, Collard HR, Anstrom KJ, Flaherty KR, Fleming TR, King TE, et al. Idiopathic pulmonary fibrosis: clinically meaningful primary endpoints in phase 3 clinical trials. Am J Respir Crit Care Med 2012;185:1044-8. https://doi.org/10.1164/rccm.201201-0006PP.
- Wells AU, Behr J, Costabel U, Cottin V, Poletti V, Richeldi L, et al. Consensus Group. Hot of the breath: mortality as a primary end-point in IPF treatment trials: the best is the enemy of the good. Thorax 2012;67:938-40. https://doi.org/10.1136/thoraxjnl-2012-202580.
- Atkins CP, Loke YK, Wilson AM. Outcomes in idiopathic pulmonary fibrosis: a meta-analysis from placebo controlled trials. Respir Med 2014;108:376-87. https://doi.org/10.1016/j.rmed.2013.11.007.
- Zisman DA, Schwarz M, Anstrom KJ, Collard HR, Flaherty KR, Hunninghake GW. Idiopathic Pulmonary Fibrosis Clinical Research Network . A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N Engl J Med 2010;363:620-8. https://doi.org/10.1056/NEJMoa1002110.
- du Bois RM, Weycker D, Albera C, Bradford WZ, Costabel U, Kartashov A, et al. Ascertainment of individual risk of mortality for patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011;184:459-66. https://doi.org/10.1164/rccm.201011-1790OC.
- Hammond M, Clark AB, Cahn AP, Chilvers ER, Fraser WD, Livermore DM, et al. The Efficacy and Mechanism Evaluation of Treating Idiopathic Pulmonary fibrosis with the Addition of Co-trimoxazole (EME-TIPAC): study protocol for a randomised controlled trial. Trials 2018;19. https://doi.org/10.1186/s13063-018-2453-6.
- Chan TY. Co-trimoxazole-induced severe haemolysis: the experience of a large general hospital in Hong Kong. Pharmacoepidemiol Drug Saf 1997;6:89-92. https://doi.org/10.1002/(SICI)1099-1557(199703)6:2<89::AID-PDS261>3.0.CO;2-8.
- Patel AS, Siegert RJ, Brignall K, Gordon P, Steer S, Desai SR, et al. The development and validation of the King’s Brief Interstitial Lung Disease (K-BILD) health status questionnaire. Thorax 2012;67:804-10. https://doi.org/10.1136/thoraxjnl-2012-201581.
- Nolan CM, Birring SS, Maddocks M, Maher TM, Patel S, Barker RE, et al. King’s Brief Interstitial Lung Disease questionnaire: responsiveness and minimum clinically important difference. Eur Respir J 2019;54. https://doi.org/10.1183/13993003.00281-2019.
- Sinha A, Patel AS, Siegert RJ, Bajwah S, Maher TM, Renzoni EA, et al. The King’s Brief Interstitial Lung Disease (KBILD) questionnaire: an updated minimal clinically important difference. BMJ Open Respir Res 2019;6. https://doi.org/10.1136/bmjresp-2018-000363.
- Manali ED, Lyberopoulos P, Triantafillidou C, Kolilekas LF, Sotiropoulou C, Milic-Emili J, et al. MRC Chronic Dyspnea Scale: relationships with cardiopulmonary exercise testing and 6-minute walk test in idiopathic pulmonary fibrosis patients: a prospective study. BMC Pulm Med 2010;10. https://doi.org/10.1186/1471-2466-10-32.
- Herdman M, Gudex C, Lloyd A, Janssen M, Kind P, Parkin D, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res 2011;20:1727-36. https://doi.org/10.1007/s11136-011-9903-x.
- Birring SS, Prudon B, Carr AJ, Singh SJ, Morgan MD, Pavord ID. Development of a symptom specific health status measure for patients with chronic cough: Leicester Cough Questionnaire (LCQ). Thorax 2003;58:339-43. https://doi.org/10.1136/thorax.58.4.339.
- Key AL, Holt K, Hamilton A, Smith JA, Earis JE. Objective cough frequency in idiopathic pulmonary fibrosis. Cough 2010;6. https://doi.org/10.1186/1745-9974-6-4.
- Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, et al. Standardisation of spirometry. Eur Respir J 2005;26:319-38. https://doi.org/10.1183/09031936.05.00034805.
- Crapo RO, Morris AH, Gardner RM. Reference spirometric values using techniques and equipment that meet ATS recommendations. Am Rev Respir Dis 1981;123:659-64. https://doi.org/10.1164/arrd.1981.123.6.659.
- Macintyre N, Crapo RO, Viegi G, Johnson DC, van der Grinten CP, Brusasco V, et al. Standardisation of the single-breath determination of carbon monoxide uptake in the lung. Eur Respir J 2005;26:720-35. https://doi.org/10.1183/09031936.05.00034905.
- Quanjer PH. Standardized lung function testing. Bull Europ Physiopathol Respir 1983;19:1-95.
- Ley B, Ryerson CJ, Vittinghoff E, Ryu JH, Tomassetti S, Lee JS, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 2012;156:684-91. https://doi.org/10.7326/0003-4819-156-10-201205150-00004.
- Lee SH, Shim HS, Cho SH, Kim SY, Lee SK, Son JY, et al. Prognostic factors for idiopathic pulmonary fibrosis: clinical, physiologic, pathologic, and molecular aspects. Sarcoidosis Vasc Diffuse Lung Dis 2011;28:102-12.
- Prasse A, Probst C, Bargagli E, Zissel G, Toews GB, Flaherty KR, et al. Serum CC-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2009;179:717-23. https://doi.org/10.1164/rccm.200808-1201OC.
- Neighbors M, Cabanski CR, Ramalingam TR, Sheng XR, Tew GW, Gu C, et al. Prognostic and predictive biomarkers for patients with idiopathic pulmonary fibrosis treated with pirfenidone: post-hoc assessment of the CAPACITY and ASCEND trials. Lancet Respir Med 2018;6:615-26. https://doi.org/10.1016/S2213-2600(18)30185-1.
- King TE, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014;370:2083-92. https://doi.org/10.1056/NEJMoa1402582.
- Xue M, Guo Z, Cai C, Sun B, Wang H. Evaluation of the diagnostic efficacies of serological markers KL-6, SP-A, SP-D, CCL2, and CXCL13 in idiopathic interstitial pneumonia. Respiration 2019;98:534-45. https://doi.org/10.1159/000503689.
- Raghu G, Martinez FJ, Brown KK, Costabel U, Cottin V, Wells AU, et al. CC-chemokine ligand 2 inhibition in idiopathic pulmonary fibrosis: a phase 2 trial of carlumab. Eur Respir J 2015;46:1740-50. https://doi.org/10.1183/13993003.01558-2014.
- McGrath EE, Lawrie A, Marriott HM, Mercer P, Cross SS, Arnold N, et al. Deficiency of tumour necrosis factor-related apoptosis-inducing ligand exacerbates lung injury and fibrosis. Thorax 2012;67:796-803. https://doi.org/10.1136/thoraxjnl-2011-200863.
- Pan T, Nielsen LD, Allen MJ, Shannon KM, Shannon JM, Selman M, et al. Serum SP-D is a marker of lung injury in rats. Am J Physiol Lung Cell Mol Physiol 2002;282:L824-32. https://doi.org/10.1152/ajplung.00421.2000.
- Honda Y, Kuroki Y, Matsuura E, Nagae H, Takahashi H, Akino T, et al. Pulmonary surfactant protein D in sera and bronchoalveolar lavage fluids. Am J Respir Crit Care Med 1995;152:1860-6. https://doi.org/10.1164/ajrccm.152.6.8520747.
- Takahashi H, Fujishima T, Koba H, Murakami S, Kurokawa K, Shibuya Y, et al. Serum surfactant proteins A and D as prognostic factors in idiopathic pulmonary fibrosis and their relationship to disease extent. Am J Respir Crit Care Med 2000;162:1109-14. https://doi.org/10.1164/ajrccm.162.3.9910080.
- Dancer RC, Wood AM, Thickett DR. Metalloproteinases in idiopathic pulmonary fibrosis. Eur Respir J 2011;38:1461-7. https://doi.org/10.1183/09031936.00024711.
- Rosas IO, Richards TJ, Konishi K, Zhang Y, Gibson K, Lokshin AE, et al. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLOS Med 2008;5. https://doi.org/10.1371/journal.pmed.0050093.
- Fujishima S, Shiomi T, Yamashita S, Yogo Y, Nakano Y, Inoue T, et al. Production and activation of matrix metalloproteinase 7 (matrilysin 1) in the lungs of patients with idiopathic pulmonary fibrosis. Arch Pathol Lab Med 2010;134:1136-42. https://doi.org/10.1043/2009-0144-OA.1.
- Song JW, Do KH, Jang SJ, Colby TV, Han S, Kim DS. Blood biomarkers MMP-7 and SP-A: predictors of outcome in idiopathic pulmonary fibrosis. Chest 2013;143:1422-9. https://doi.org/10.1378/chest.11-2735.
- Kakugawa T, Yokota S, Ishimatsu Y, Hayashi T, Nakashima S, Hara S, et al. Serum heat shock protein 47 levels are elevated in acute interstitial pneumonia. BMC Pulm Med 2014;14. https://doi.org/10.1186/1471-2466-14-48.
- Amenomori M, Mukae H, Sakamoto N, Kakugawa T, Hayashi T, Hara A, et al. HSP47 in lung fibroblasts is a predictor of survival in fibrotic nonspecific interstitial pneumonia. Respir Med 2010;104:895-901. https://doi.org/10.1016/j.rmed.2010.01.011.
- Leuchte HH, Neurohr C, Baumgartner R, Holzapfel M, Giehrl W, Vogeser M, et al. Brain natriuretic peptide and exercise capacity in lung fibrosis and pulmonary hypertension. Am J Respir Crit Care Med 2004;170:360-5. https://doi.org/10.1164/rccm.200308-1142OC.
- Song JW, Song JK, Kim DS. Echocardiography and brain natriuretic peptide as prognostic indicators in idiopathic pulmonary fibrosis. Respir Med 2009;103:180-6. https://doi.org/10.1016/j.rmed.2008.11.012.
- Maher TM, Oballa E, Simpson JK, Porte J, Habgood A, Fahy WA, et al. An epithelial biomarker signature for idiopathic pulmonary fibrosis: an analysis from the multicentre PROFILE cohort study. Lancet Respir Med 2017;5:946-55. https://doi.org/10.1016/S2213-2600(17)30430-7.
- Lode H, Schaberg T, Raffenberg M, Mauch H. Diagnostic problems in lower respiratory tract infections. J Antimicrob Chemother 1993;32:29-37. https://doi.org/10.1093/jac/32.suppl_a.29.
- Strumpf IJ, Feld MK, Cornelius MJ, Keogh BA, Crystal RG. Safety of fiberoptic bronchoalveolar lavage in evaluation of interstitial lung disease. Chest 1981;80:268-71. https://doi.org/10.1378/chest.80.3.268.
- Du Rand IA, Blaikley J, Booton R, Chaudhuri N, Gupta V, Khalid S, et al. British Thoracic Society guidelines on diagnostic flexible bronchoscopy in adults: accredited by NICE. Thorax 2013;68:i1-44. https://doi.org/10.1136/thoraxjnl-2013-203618.
- European Medicines Agency . ICH E2A Clinical Safety Data Management: Definitions and Standards for Expedited Reporting n.d. www.ema.europa.eu/en/ich-e2a-clinical-safety-data-management-definitions-standards-expedited-reporting (accessed 16 April 2021).
- Collard HR, Calfee CS, Wolters PJ, Song JW, Hong SB, Brady S, et al. Plasma biomarker profiles in acute exacerbation of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2010;299:L3-7. https://doi.org/10.1152/ajplung.90637.2008.
- Wilson AM, Clark AB, Cahn T, Chilvers ER, Fraser W, Hammond M, et al. Effect of co-trimoxazole (trimethoprim-sulfamethoxazole) vs. placebo on death, lung transplant, or hospital admission in patients with moderate and severe idiopathic pulmonary fibrosis: the EME-TIPAC randomized clinical trial. JAMA 2020;324:2282-91. https://doi.org/10.1001/jama.2020.22960.
- Yu X, Li X, Wang L, Liu R, Xie Y, Li S, et al. Pulmonary rehabilitation for exercise tolerance and quality of life in IPF patients: a systematic review and meta-analysis. Biomed Res Int 2019;2019. https://doi.org/10.1155/2019/8498603.
- Schünemann HJ, Griffith L, Jaeschke R, Goldstein R, Stubbing D, Guyatt GH. Evaluation of the minimal important difference for the feeling thermometer and the St. George’s Respiratory Questionnaire in patients with chronic airflow obstruction. J Clin Epidemiol 2003;56:1170-6. https://doi.org/10.1016/S0895-4356(03)00115-X.
- Jones PW, Quirk FH, Baveystock CM, Littlejohns P. A self-complete measure of health status for chronic airflow limitation. The St. George’s Respiratory Questionnaire. Am Rev Respir Dis 1992;145:1321-7. https://doi.org/10.1164/ajrccm/145.6.1321.
- Ryerson CJ, Abbritti M, Ley B, Elicker BM, Jones KD, Collard HR. Cough predicts prognosis in idiopathic pulmonary fibrosis. Respirology 2011;16:969-75. https://doi.org/10.1111/j.1440-1843.2011.01996.x.
- van Manen MJ, Birring SS, Vancheri C, Cottin V, Renzoni EA, Russell AM, et al. Cough in idiopathic pulmonary fibrosis. Eur Respir Rev 2016;25:278-86. https://doi.org/10.1183/16000617.0090-2015.
- Hewson T, McKeever TM, Gibson JE, Navaratnam V, Hubbard RB, Hutchinson JP. Timing of onset of symptoms in people with idiopathic pulmonary fibrosis. Thorax 2018;73:683-5. https://doi.org/10.1136/thoraxjnl-2017-210177.
- National Institute for Health and Care Excellence . Pirfenidone for Treating Idiopathic Pulmonary Fibrosis: Technology Appraisal Guidance 282 2013. www.nice.org.uk/guidance/ta282 (accessed 19 January 2021).
- Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014;370:2071-82. https://doi.org/10.1056/NEJMoa1402584.
- National Institute for Health and Care Excellence . Nintedanib for Treating Idiopathic Pulmonary Fibrosis: Technology Appraisal Guidance 379 2016. www.nice.org.uk/guidance/ta379 (accessed 19 January 2021).
- Costabel U, Albera C, Lancaster LH, Lin CY, Hormel P, Hulter HN, et al. An open-label study of the long-term safety of pirfenidone in patients with idiopathic pulmonary fibrosis (RECAP). Respiration 2017;94:408-15. https://doi.org/10.1159/000479976.
- Fisher M, Nathan SD, Hill C, Marshall J, Dejonckheere F, Thuresson PO, et al. Predicting life expectancy for pirfenidone in idiopathic pulmonary fibrosis. J Manag Care Spec Pharm 2017;23:17-24. https://doi.org/10.18553/jmcp.2017.23.3-b.s17.
- Lancaster L, Crestani B, Hernandez P, Inoue Y, Wachtlin D, Loaiza L, et al. Safety and survival data in patients with idiopathic pulmonary fibrosis treated with nintedanib: pooled data from six clinical trials. BMJ Open Respir Res 2019;6. https://doi.org/10.1136/bmjresp-2018-000397.
- Nathan SD, Costabel U, Albera C, Behr J, Wuyts WA, Kirchgaessler KU, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis and more advanced lung function impairment. Respir Med 2019;153:44-51. https://doi.org/10.1016/j.rmed.2019.04.016.
- du Bois RM, Weycker D, Albera C, Bradford WZ, Costabel U, Kartashov A, et al. Forced vital capacity in patients with idiopathic pulmonary fibrosis: test properties and minimal clinically important difference. Am J Respir Crit Care Med 2011;184:1382-9. https://doi.org/10.1164/rccm.201105-0840OC.
- Fujita H, Sakamoto N, Ishimatsu Y, Kakugawa T, Hara S, Hara A, et al. Effects of doxycycline on production of growth factors and matrix metalloproteinases in pulmonary fibrosis. Respiration 2011;81:420-30. https://doi.org/10.1159/000324080.
- Bhattacharyya P, Nag S, Bardhan S, Acharya D, Paul R, Dey R, et al. The role of long-term doxycycline in patients of idiopathic pulmonary fibrosis: the results of an open prospective trial. Lung India 2009;26:81-5. https://doi.org/10.4103/0970-2113.53231.
- Tsubouchi K, Araya J, Minagawa S, Hara H, Ichikawa A, Saito N, et al. Azithromycin attenuates myofibroblast differentiation and lung fibrosis development through proteasomal degradation of NOX4. Autophagy 2017;13:1420-34. https://doi.org/10.1080/15548627.2017.1328348.
- Wuyts WA, Willems S, Vos R, Vanaudenaerde BM, De Vleeschauwer SI, Rinaldi M, et al. Azithromycin reduces pulmonary fibrosis in a bleomycin mouse model. Exp Lung Res 2010;36:602-14. https://doi.org/10.3109/01902148.2010.492895.
- Macaluso C, Maritano Furcada J, Alzaher O, Chaube R, Chua F, Wells AU, et al. The potential impact of azithromycin in idiopathic pulmonary fibrosis. Eur Respir J 2019;53. https://doi.org/10.1183/13993003.00628-2018.
- Kawamura K, Ichikado K, Yasuda Y, Anan K, Suga M. Azithromycin for idiopathic acute exacerbation of idiopathic pulmonary fibrosis: a retrospective single-center study. BMC Pulm Med 2017;17. https://doi.org/10.1186/s12890-017-0437-z.
- Naik PK, Bozyk PD, Bentley JK, Popova AP, Birch CM, Wilke CA, et al. Periostin promotes fibrosis and predicts progression in patients with idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2012;303:L1046-56. https://doi.org/10.1152/ajplung.00139.2012.
- Parker D, Prince A. Staphylococcus aureus induces type I IFN signaling in dendritic cells via TLR9. J Immunol 2012;189:4040-6. https://doi.org/10.4049/jimmunol.1201055.
- Raghu G, Freudenberger TD, Yang S, Curtis JR, Spada C, Hayes J, et al. High prevalence of abnormal acid gastro-oesophageal reflux in idiopathic pulmonary fibrosis. Eur Respir J 2006;27:136-42. https://doi.org/10.1183/09031936.06.00037005.
- Savarino E, Carbone R, Marabotto E, Furnari M, Sconfienza L, Ghio M, et al. Gastro-oesophageal reflux and gastric aspiration in idiopathic pulmonary fibrosis patients. Eur Respir J 2013;42:1322-31. https://doi.org/10.1183/09031936.00101212.
- Kitsios GD, Rojas M, Kass DJ, Fitch A, Sembrat JC, Qin S, et al. Microbiome in lung explants of idiopathic pulmonary fibrosis: a case–control study in patients with end-stage fibrosis. Thorax 2018;73:481-4. https://doi.org/10.1136/thoraxjnl-2017-210537.
- Jenkins G. A big beautiful wall against infection. Thorax 2018;73. https://doi.org/10.1136/thoraxjnl-2017-211220.
- Wüst J, Wilkins TD. Susceptibility of anaerobic bacteria to sulfamethoxazole/trimethoprim and routine susceptibility testing. Antimicrob Agents Chemother 1978;14:384-90. https://doi.org/10.1128/AAC.14.3.384.
- Wolter J, Seeney S, Bell S, Bowler S, Masel P, McCormack J. Effect of long term treatment with azithromycin on disease parameters in cystic fibrosis: a randomised trial. Thorax 2002;57:212-16. https://doi.org/10.1136/thorax.57.3.212.
- Prins HJ, Daniels JM, Lindeman JH, Lutter R, Boersma WG. Effects of doxycycline on local and systemic inflammation in stable COPD patients, a randomized clinical trial. Respir Med 2016;110:46-52. https://doi.org/10.1016/j.rmed.2015.10.009.
- Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol 2014;9:157-79. https://doi.org/10.1146/annurev-pathol-012513-104706.
- Ruskin J, LaRiviere M. Low-dose co-trimoxazole for prevention of Pneumocystis carinii pneumonia in human immunodeficiency virus disease. Lancet 1991;337:468-71. https://doi.org/10.1016/0140-6736(91)93402-U.
- Chintu C, Bhat GJ, Walker AS, Mulenga V, Sinyinza F, Lishimpi K, et al. Co-trimoxazole as prophylaxis against opportunistic infections in HIV-infected Zambian children (CHAP): a double-blind randomised placebo-controlled trial. Lancet 2004;364:1865-71. https://doi.org/10.1016/S0140-6736(04)17442-4.
- Collard HR, Ryerson CJ, Corte TJ, Jenkins G, Kondoh Y, Lederer DJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An International Working Group Report. Am J Respir Crit Care Med 2016;194:265-75. https://doi.org/10.1164/rccm.201604-0801CI.
Appendix 1
| Analyte | Assay range | Sensitivity | Unit |
|---|---|---|---|
| CCL18 | 18.8–1200 | 1.77 | pg/ml |
| MCP-1 | 31.2–2000 | 10 | pg/ml |
| TRAIL | 15.6–1000 | 7.87 | pg/ml |
| OPG | 0–20 | 0.07 | pmol/l |
| Pro-BNP | 0.6–4130 | 5 | pmol/l |
| CRP | 0.3–350 | 0.3 | mg/ml |
| CA19–9 | 0.6–10,000 | 0.6 | U/ml |
| CA-125 | 2–3000 | 2.0 | U/ml |
| SP-D | 0.6–40 | 0.094 | ng/ml |
| MMP-7 | 0.2–10 | 0.37 | ng/ml |
| MPO | 0.156–10 | 0.156 | ng/ml |
| NCR ID | Description of NCR |
|---|---|
| 2016_NCR_03 | DMEC not given sufficient AE data to review at initial meeting. Information provided subsequently |
| 2016_NCR_08 | Research nurse sent semi-blinded randomisation e-mail in error |
| 2016_NCR_09 | Patient screening tests completed outside the time window |
| 2017_NCR_01 | Patient did not follow dose reduction regimen as instructed by PI |
| 2017_NCR_07 | Minor IMP temperature excursion not reported within timelines. IMP unaffected |
| 2017_NCR_11 | Local pharmacist attached dispensing labels (containing semi-blinded treatment allocation) to patient-specific trial prescription |
| 2017_NCR_20 | Liver function tests were not performed for patient at screening |
| 2017_NCR_34 | Patient discovered to have not taken folic acid as PP with IMP |
| 2017_NCR_35 | Patient discovered to have not taken folic acid as PP with IMP |
| 2017_NCR_36 | Site did not follow treatment discontinuation rule and instead dispensed 6 months of IMP |
| 2017_NCR_38 | Ineligible patient randomised (ineligibility did not relate to safety) |
| 2018_NCR_011 | Test for folate during screening not completed for two patients who were subsequently enrolled on the trial |
| 2018_NCR_012 | Patient discovered to have not taken correct dose of IMP |
| 2018_NCR_017 | Patient discovered to have not taken folic acid with IMP |
| 2018_NCR_018 | After booking courier to deliver initial IMP supply to the patient, the pharmacy contacted the NCTU to inform them that the pharmacy did not have any stock for the group to which the patient had been allocated |
| 2018_NCR_026 | Site reported that patient was taking IMP incorrectly, at intervals of every other day instead of three times per week. This was identified at a follow-up visit |
| 2019_NCR_018 | Non-emergency unblinding (for patient care) of patient 34001 on 28 July 2018 |
| 2019_NCR_019 | Non-emergency unblinding (for patient care) of patient 42006 on 9 February 2017 |
| 2019_NCR_020 | Non-emergency unblinding (for patient care) of patient 44007 on 8 June 2017 |
| 2019_NCR_024 | Biomarker samples sent to the wrong site by the courier instead of being delivered to the freezers at the UEA. Samples unaffected and redelivered to correct address |
| 2019_NCR_025 | Research blood sample incorrectly processed at site |
| 2019_NCR_027 | Samples were received at UEA with no dry ice and were thawed on receipt and consequently unsuitable for analysis |
| 2019_NCR_028 | The site lost a set of patient samples that were due to be sent back to UEA |
| 2019_NCR_035 | Patient was unblinded (non-emergency) without first informing the chief investigator |
| 2019_NCR_054 | Post data lock, SAP sign off and changes to data set |
| Reason given | Treatment group | Suggested grouping |
|---|---|---|
| ARs made him feel really unwell, nausea and vomiting | Active treatment | Perceived side effects |
| Patient is housebound and unable to come to the hospital for any more visits | Active treatment | Unwilling to continue |
| Does not want to continue in the study | Active treatment | Unwilling to continue |
| He feels he will not be able to comply with taking his medication as instructed | Active treatment | Unwilling to continue |
| Patient did not start study drug because of health personal reasons | Active treatment | Unwilling to continue |
| Patient no longer wishes to participate because of worsening health | Active treatment | Unwilling to continue |
| On active transplant list – patient asked to withdraw from study | Active treatment | Unwilling to continue |
| Patient stopped study medication and now feels unable to continue with extra commitments | Active treatment | Unwilling to continue |
| Patient stopped study medication and now feels unable to continue with extra commitments | Active treatment | Unwilling to continue |
| Patient stopped study medication and now feels unable to continue with extra commitments | Active treatment | Unwilling to continue |
| Patient does not want to be involved and continue in the study | Active treatment | Unwilling to continue |
| Patient has a lot going on at present. He has had a few chest infections recently and is feeling very apprehensive about the future; his wife is unwell at present too | Active treatment | Perceived side effects |
| Patient does not live near to the Newcastle Trust hospital and feels that their general health is failing and no longer wishes to travel for hospital appointments | Active treatment | Unwilling to continue |
| Patient is very frail and lethargic at present | Active treatment | Unwilling to continue |
| Too ill to continue | Active treatment | Unwilling to continue |
| Patient decision because of travelling distance to site | Active treatment | Unwilling to continue |
| Patient has complicated work commitments and does not feel that he would benefit from completing questionnaires and lung function measurements every 3 months | Active treatment | Unwilling to continue |
| Patient felt that symptoms of nausea might be related to IMP/placebo and had a break from study drugs on 19 June 2017. A total of 104 IMP/placebo medications were returned by patient at EOS visit on 20 September 2017 | Active treatment | Perceived side effects |
| Patient is now at a palliative stage | Active treatment | Disease progression |
| Patient said she complained of nausea, vomitting and lack of appetite while on the IMP/placebo | Active treatment | Perceived side effects |
| Deterioration in condition | Active treatment | Disease progression |
| Patient moved to a new house and was very anxious and breathless. Does not want to take part in the study as feels that he has not improved since contracting influenza | Active treatment | Unwilling to continue |
| Too ill at moment to consider continuation | Active treatment | Disease progression |
| Withdrew from study medication as they did not wish to continue with further visits | Active treatment | Unwilling to continue |
| Patient withdrew because of AEs that occurred since starting the study medication. The patient did not wish to have any further visits after the closeout visit was performed | Active treatment | Perceived side effects |
| Having stopped the IMP to allow full recovery from previous AE (23 September 2017) and restarted on 17 October 2017 following return from his holiday and meeting with the PI, the patient reported a return of symptoms. As a result he has requested to withdraw from the trial | Active treatment | Perceived side effects |
| Health is deteriorating and he can no longer come to visit appointments | Active treatment | Disease progression |
| Patient has too much going on at home | Active treatment | Unwilling to continue |
| Too frail to leave the house for visits | Active treatment | Unwilling to continue |
| Too far to travel/deterioration of IPF/on continuous oxygen/frail | Active treatment | Disease progression |
| Patient declined to take part in any further study visits due to starting oxygen therapy and feeling generally unwell | Active treatment | Unwilling to continue |
| Patient is finding it ‘all too much alongside his appointments in Leeds and Huddersfield’ | Active treatment | Unwilling to continue |
| No longer wants to be treated at the hospital for his condition. Would like to just be seen by GP from now on. Reports that hospital appointments are too much for him and has requested all hospital visits to be cancelled | Placebo | Unwilling to continue |
| Deterioration of clinical condition | Placebo | Disease progression |
| Clinical deterioration and inability to tolerate medications | Placebo | Disease progression |
| Recently diagnosed with metastatic prostate cancer and no longer wishes to participate in trial | Placebo | Unwilling to continue |
| Patient is declining in health and is now on oxygen; he feels that he can no longer continue with the study as he is so unwell | Placebo | Disease progression |
| The patient claims travelling to the site is too inconvenient due to time taken to travel (2.5 hours one way) | Placebo | Unwilling to continue |
| Patient is now housebound so is unable to attend visits to the hospital | Placebo | Unwilling to continue |
| Patient’s decision | Placebo | Unwilling to continue |
| Patient stopped study medication and now feels unable to continue with extra commitments | Placebo | Unwilling to continue |
| Discontinued drugs because of diarrhoea and did not wish further input | Placebo | Perceived side effects |
| Withdrawal of consent for treatment by patient | Placebo | Unwilling to continue |
| Patient did not want to continue taking medication or coming to hospital for follow-up as is finding both too much | Placebo | Unwilling to continue |
| Does not want to attend hospital for visits | Placebo | Unwilling to continue |
| Patient stated that she was too unwell to continue on the study as she is now bed/housebound and did not feel that she could carry on with any further visits | Placebo | Disease progression |
| Principal investigator’s decision for patient to withdraw from study | Placebo | Investigator decision |
| Patient was experiencing ARs (as noted on the AE form) from the study drug so has chosen to withdraw | Placebo | Perceived side effects |
| Patient’s choice | Placebo | Unwilling to continue |
| Travelling distance to site | Placebo | Unwilling to continue |
| Patient is at the palliative stages of his lung disease and does not want to continue with clinical trial medication | Placebo | Disease progression |
| Patient is now palliative and is unable to travel to hospital for appointments | Placebo | Disease progression |
| Admitted with community-acquired pneumonia; patient’s choice to withdraw from study | Placebo | Unwilling to continue |
| Patient’s IPF has become worse and they are now struggling to leave the house to attend hospital appointments; does not wish to burden wife with bringing them to appointments; feels that withdrawing from the study would enable them to have one less thing to worry about | Placebo | Disease progression |
| Patient stopped taking study drug on 16 January 2017 as a result of gastrointestinal problems. Patient agreed to attend study closeout visit, but did not wish for biomarkers to be taken and has now withdrawn consent from any further participation in the study | Placebo | Perceived side effects |
| Patient noted no particular benefit – feels that health is much worse and study visits are too much | Placebo | Perceived side effects |
| Feels better since discontinuing study medication. Also finds it difficult to travel to the hospital so is not willing to continue on study and is now off IMP | Placebo | Perceived side effects |
| Patient is too ill to continue; now under hospice care | Placebo | Disease progression |
| Baseline characteristic | Treatment group | |
|---|---|---|
| Active treatment (N = 94) | Placebo (N = 118) | |
| Male patients, n (%) | 82 (87.2) | 111 (94.1) |
| Age in years, mean (SD) | 70.73 (7.30) | 70.73 (6.83) |
| Smoking status, n (%) | ||
| Never smoked | 28 (29.8) | 37 (31.4) |
| Ex-smoker | 66 (70.2) | 80 (67.8) |
| Current smoker | 0 (0.0) | 1 (0.8) |
| Comorbidities, n (%) | ||
| COPD | 2 (2.1) | 2 (1.7) |
| Bronchiectasis | 1 (1.1) | 4 (3.4) |
| Ischaemic heart or angina | 17 (18.1) | 29 (24.6) |
| GORD | 34 (36.2) | 42 (35.6) |
| Diabetes mellitus | 23 (24.5) | 19 (16.1) |
| Osteoporosis | 6 (6.4) | 9 (7.6) |
| Pulmonary hypertension | 6 (6.4) | 4 (3.4) |
| Anxiety or depression | 9 (9.6) | 17 (14.4) |
| Medications, n (%) | ||
| Pirfenidone | 39 (41.5) | 51 (43.2) |
| N-acetylcysteine | 7 (7.4) | 6 (5.1) |
| Other antioxidant | 2 (2.1) | 4 (3.4) |
| Prednisolone | 6 (6.4) | 7 (5.9) |
| Nintedanib | 32 (34.0) | 37 (31.4) |
| Proton pump inhibitor | 46 (48.9) | 48 (40.7) |
| Lung tests | ||
| Absolute value, mean (SD) | ||
| FVC (l) | 2.32 (0.57) | 2.24 (0.53) |
| FEV1 (l) | 1.94 (0.48) | 1.89 (0.43) |
| FEV1/FVC ratio | 0.84 (0.07) | 0.85 (0.11) |
| DLCO (mmol/minute/kPa) | 3.68 (1.79) | 3.77 (1.71) |
| Per cent predicted, mean (SD) | ||
| FVC | 56.66 (9.30) | 54.15 (10.55) |
| FEV1 | 61.69 (9.97) | 59.47 (11.32) |
| DLCO | 43.02 (19.0) | 45.03 (19.59) |
| Minimisation factor, n (%) | ||
| Taking licensed IPF medication | 71 (75.5) | 88 (74.6) |
| Study site, n (%) | ||
| Norwich | 8 (8.5) | 11 (9.3) |
| Papworth | 3 (3.2) | 6 (5.1) |
| Royal Brompton | 3 (3.2) | 8 (6.8) |
| Sheffield | 3 (3.2) | 2 (1.7) |
| Birmingham | 6 (6.4) | 6 (5.1) |
| Heart of England | 2 (2.1) | 2 (1.7) |
| North Midlands | 3 (3.2) | 6 (5.1) |
| Bristol | 6 (6.4) | 3 (2.5) |
| University Hospital Wales | 3 (3.2) | 3 (2.5) |
| Newcastle | 3 (3.2) | 5 (4.2) |
| Gateshead | 2 (2.1) | 2 (1.7) |
| Salford | 2 (2.1) | 1 (0.8) |
| South Manchester | 9 (9.6) | 9 (7.6) |
| Aintree | 6 (6.4) | 7 (5.9) |
| Lancashire | 3 (3.2) | 4 (3.4) |
| Aberdeen | 2 (2.1) | 3 (2.5) |
| Greater Glasgow & Clyde | 1 (1.1) | 1 (0.8) |
| Oxford | 1 (1.1) | 5 (4.2) |
| Imperial College | 0 (0.0) | 3 (2.5) |
| NHS Tayside | 2 (2.1) | 2 (1.7) |
| Royal Devon & Exeter | 1 (1.1) | 1 (0.8) |
| Hull & East Yorkshire | 3 (3.2) | 6 (5.1) |
| Nottingham | 1 (1.1) | 2 (1.7) |
| Cambridge University Hospitals | 1 (1.1) | 1 (0.8) |
| Leicester | 2 (2.1) | 2 (1.7) |
| University College London | 0 (0.0) | 0 (0.0) |
| Blackpool, Fylde and Wyre | 2 (2.1) | 2 (1.7) |
| Shrewsbury & Telford | 0 (0.0) | 0 (0.0) |
| Sherwood Forest Hospitals | 0 (0.0) | 2 (1.7) |
| St George’s University | 1 (1.1) | 2 (1.7) |
| Worcestershire | 0 (0.0) | 0 (0.0) |
| Western Health & Social Care | 2 (2.1) | 3 (2.5) |
| Royal Wolverhampton | 2 (2.1) | 2 (1.7) |
| Southampton | 4 (4.3) | 1 (0.8) |
| Morecambe Bay | 2 (2.1) | 2 (1.7) |
| Calderdale & Huddersfield | 0 (0.0) | 0 (0.0) |
| South Tyneside | 2 (2.1) | 2 (1.7) |
| Forth Valley | 1 (1.1) | 0 (0.0) |
| Coventry | 2 (2.1) | 1 (0.8) |
| Outcome measures | ||
| MRC score, n (%) | ||
| 1: not troubled by breathlessness apart from on strenuous exercise | 4 (4.3) | 4 (3.4) |
| 2: short of breath when hurrying on the level or walking up a slight hill | 42 (44.7) | 58 (49.2) |
| 3: walks slower than most people on the level, stops after 1 mile or so, or stops after 15 minutes walking at own pace | 29 (30.9) | 25 (21.2) |
| 4: stops for breath after walking about 100 yards or after a few minutes on level ground | 14 (14.9) | 21 (17.8) |
| 5: too breathless to leave the house or breathless when undressing | 4 (4.3) | 9 (7.6) |
| MRC score, median (IQR) | 3.00 (2.00–3.00) | 2.00 (2.00–4.00) |
| EQ-5D utility score, mean (SD) | 0.67 (0.18) | 0.69 (0.24) |
| CSS, mean (SD) | 39.05 (25.90) | 41.68 (27.31) |
| LCQ score, mean (SD) | ||
| Total | 15.88 (3.38) | 15.81 (3.73) |
| Physical | 5.20 (1.00) | 5.14 (1.05) |
| Psychological | 5.27 (1.35) | 5.34 (1.49) |
| Social | 5.28 (1.37) | 5.38 (1.40) |
| K-BILD score, mean (SD) | ||
| Psychological | 54.06 (13.80) | 54.79 (16.98) |
| Breathless | 38.13 (14.86) | 38.56 (14.99) |
| Chest | 61.10 (20.87) | 62.01 (21.24) |
| Total | 53.33 (9.60) | 53.39 (10.67) |
| Time point (months) | Treatment group | |||
|---|---|---|---|---|
| Active treatment | Placebo | |||
| Number of patients | Number (%) of dose modifications | Number of patients | Number (%) of dose modifications | |
| 3 | 114 | 19 (16.7) | 137 | 10 (7.3) |
| 6 | 96 | 7 (7.3) | 113 | 6 (5.3) |
| 12 | 61 | 4 (6.6) | 78 | 0 (0.0) |
| 18 | 38 | 1 (2.6) | 46 | 0 (0.0) |
| 24 | 20 | 0 (0.0) | 28 | 0 (0.0) |
| 30 | 10 | 0 (0.0) | 10 | 0 (0.0) |
| 36 | 2 | 0 (0.0) | 3 | 0 (0.0) |
| Any dose change (percentage of patients) | 31 (19) | 16 (9) | ||
| Outcome | Treatment group | Analysis | ||||||
|---|---|---|---|---|---|---|---|---|
| Active treatment | Placebo | Unadjusted | Adjusted | |||||
| n | Mean (SD) | n | Mean (SD) | Mean difference (95% CI) | p-value | Mean difference (95% CI) | p-value | |
| LCQ score | ||||||||
| Total | 37 | 15.67 (3.78) | 44 | 14.13 (4.05) | –1.47 (–3.20 to 0.26) | 0.096 | –1.43 (–2.72 to –0.14) | 0.029 |
| Physical | 37 | 4.99 (1.17) | 45 | 4.62 (1.16) | –0.35 (–0.86 to 0.16) | 0.174 | –0.37 (–0.76 to 0.02) | 0.06 |
| Psychological | 37 | 5.27 (1.33) | 46 | 4.75 (1.56) | –0.48 (–1.11 to 0.16) | 0.141 | –0.54 (–1.04 to –0.05) | 0.03 |
| Social | 37 | 5.41 (1.45) | 46 | 4.92 (1.53) | –0.47 (–1.12 to 0.18) | 0.16 | –0.47 (–0.95 to 0.00) | 0.052 |
| MRC score (percentage of participants), n (%) | ||||||||
| 1 | 0 (0) | 2 (4) | ||||||
| 2 | 18 (47) | 19 (35) | ||||||
| 3 | 9 (24) | 17 (31) | ||||||
| 4 | 9 (24) | 12 (22) | ||||||
| 5 | 2 (5) | 4 (7) | ||||||
| Median (IQR) | 3.00 (2.00–4.00) | 3.00 (2.00–4.00) | 0.6585 | 0.4859 | ||||
| CSS | 43.13 (28.40) | 49.83 (27.20) | 6.33 (–5.31 to 17.98) | 0.287 | 2.31 (–8.25 to 12.87) | 0.668 | ||
| EQ-5D score | 60 | 0.40 (0.37) | 81 | 0.40 (0.37) | –0.03 (–0.14 to 0.09) | 0.663 | –0.01 (–0.12 to 0.10) | 0.836 |
| K-BILD score | ||||||||
| Psychological | 38 | 51.37 (16.16) | 53 | 52.25 (16.74) | 0.97 (–5.98 to 7.92) | 0.784 | –1.01 (–6.9 to 4.87) | 0.735 |
| Breathless | 38 | 35.34 (17.55) | 54 | 35.03 (15.70) | 0.47 (–6.24 to 7.19) | 0.89 | –0.95 (–6.25 to 4.35) | 0.726 |
| Chest | 38 | 60.55 (19.24) | 54 | 54.47 (22.12) | –6.79 (–15.5 to 1.91) | 0.126 | –6.34 (–13.77 to 1.09) | 0.095 |
| Total | 38 | 51.49 (11.67) | 53 | 50.77 (11.21) | –0.6 (–5.39 to 4.18) | 0.805 | –1.39 (–5.3 to 2.51) | 0.485 |
| Outcome | Treatment group | Analysis | ||||||
|---|---|---|---|---|---|---|---|---|
| Active treatment | Placebo | Unadjusted | Adjusted | |||||
| n | Mean (SD) | n | Mean (SD) | Mean difference (95% CI) | p-value | Mean difference (95% CI) | p-value | |
| Absolute | ||||||||
| FVC (l) | 32 | 2.23 (0.51) | 48 | 2.26 (0.53) | 0.03 (–0.21 to 0.26) | 0.83 | 0.06 (–0.05 to 0.17) | 0.30 |
| FEV1 (l) | 32 | 1.84 (0.43) | 48 | 1.89 (0.43) | 0.05 (–0.14 to 0.25) | 0.58 | 0.05 (–0.05 to 0.15) | 0.70 |
| DLCO (mmol/minute/kPa) | 27 | 3.71 (2.19) | 40 | 3.69 (1.47) | 0.01 (–0.86 to 0.87) | 0.99 | 0.36 (–0.43 to 1.14) | 0.37 |
| Per cent predicted | ||||||||
| FVC | 32 | 52.49 (8.49) | 48 | 53.98 (9.59) | 1.51 (–2.55 to 5.57) | 0.47 | 1.16 (–1.49 to 3.82) | 0.39 |
| FEV1 | 32 | 56.16 (9.61) | 48 | 59.20 (11.22) | 3.23 (–1.48 to 7.94) | 0.18 | 1.63 (–1.40 to 4.67) | 0.29 |
| DLCO | 27 | 41.44 (21.27) | 40 | 42.22 (14.83) | 1.27 (–7.40 to 9.93) | 0.77 | 5.30 (–2.96 to 13.57) | 0.21 |
| Time point (months) | Treatment group | Mean difference (95% CI) | p-value (Bonferroni corrected) | |||
|---|---|---|---|---|---|---|
| Active treatment | Placebo | |||||
| Effect size, mean (SD) | n | Effect size, mean (SD) | n | |||
| 3 | 55.10 (17.74) | 122 | 54.11 (17.05) | 142 | –0.78 (–6.44 to 4.88) | 1 |
| 6 | 55.17 (16.97) | 111 | 54.91 (17.37) | 118 | –0.42 (–6.32 to 5.48) | 1 |
| 12 | 49.73 (17.92) | 71 | 51.86 (16.89) | 85 | 1.78 (–4.79 to 8.35) | 1 |
| 18 | 50.84 (15.54) | 43 | 50.70 (18.67) | 52 | 0.24 (–7.41 to 7.89) | 1 |
| 24 | 56.27 (20.29) | 22 | 52.71 (16.85) | 30 | –4.21 (–13.72 to 5.29) | 1 |
| 30 | 54.46 (18.83) | 13 | 52.69 (16.35) | 12 | 3.17 (–9.47 to 15.8) | 1 |
| 36 | 65.40 (14.42) | 2 | 74.22 (21.20) | 4 | 8.84 (–16.61 to 34.29) | 1 |
| Overall difference | –0.11 (–3.8 to 3.59) | 0.954 | ||||
| Time point (months) | Treatment group | Mean difference (95% CI) | p-value (Bonferroni corrected) | |||
|---|---|---|---|---|---|---|
| Active treatment | Placebo | |||||
| Effect size, mean (SD) | n | Effect size, mean (SD) | n | |||
| 3 | 35.34 (17.12) | 123 | 38.71 (16.49) | 142 | 3.63 (–1.89 to 9.15) | 0.54 |
| 6 | 36.28 (16.75) | 112 | 38.98 (15.57) | 118 | 3.11 (–2.61 to 8.84) | 1 |
| 12 | 34.37 (17.42) | 70 | 34.96 (14.55) | 86 | 0.73 (–5.56 to 7.02) | 1 |
| 18 | 32.30 (18.77) | 43 | 36.01 (15.64) | 53 | 3.46 (–3.76 to 10.69) | 1 |
| 24 | 36.75 (23.61) | 23 | 34.98 (15.91) | 30 | –2.85 (–11.64 to 5.93) | 1 |
| 30 | 37.56 (20.94) | 13 | 30.07 (16.89) | 12 | –0.79 (–12.42 to 10.83) | 1 |
| 36 | 63.65 (7.28) | 2 | 44.83 (13.44) | 4 | 1.35 (–21.78 to 24.49) | 1 |
| Overall difference | 2.36 (–1.31 to 6.03) | 0.208 | ||||
| Time point (months) | Treatment group | Mean difference (95% CI) | p-value (Bonferroni corrected) | |||
|---|---|---|---|---|---|---|
| Active treatment | Placebo | |||||
| Effect size, mean (SD) | n | Effect size, mean (SD) | n | |||
| 3 | 59.88 (21.02) | 123 | 60.14 (20.94) | 142 | 0.41 (–6.6 to 7.42) | 1 |
| 6 | 60.74 (22.79) | 112 | 60.46 (20.46) | 117 | 0.54 (–6.81 to 7.89) | 1 |
| 12 | 59.86 (20.26) | 72 | 56.75 (22.82) | 86 | –1.05 (–9.36 to 7.26) | 1 |
| 18 | 57.30 (21.45) | 43 | 52.22 (25.53) | 53 | –1.04 (–10.92 to 8.84) | 1 |
| 24 | 59.59 (26.72) | 23 | 59.72 (16.77) | 30 | 1.97 (–10.46 to 14.4) | 1 |
| 30 | 60.14 (22.48) | 13 | 49.31 (21.86) | 12 | 0.95 (–15.99 to 17.89) | 1 |
| 36 | 74.45 (15.20) | 2 | 66.03 (23.29) | 4 | 8.65 (–26.02 to 43.32) | 1 |
| Overall difference | 0.18 (–4.26 to 4.61) | 0.938 | ||||
| Time point (months) | Treatment group | Mean difference (95% CI) | p-value (Bonferroni corrected) | |||
|---|---|---|---|---|---|---|
| Active treatment | Placebo | |||||
| Mean (SD) | n | Mean (SD) | n | |||
| 3 | 0.61 (0.27) | 131 | 0.63 (0.27) | 153 | 0.04 (–0.05 to 0.14) | 1 |
| 6 | 0.56 (0.31) | 131 | 0.57 (0.31) | 139 | 0.01 (–0.08 to 0.11) | 1 |
| 12 | 0.41 (0.36) | 103 | 0.45 (0.35) | 118 | 0.05 (–0.05 to 0.15) | 1 |
| 18 | 0.29 (0.35) | 85 | 0.36 (0.36) | 96 | 0.08 (–0.03 to 0.18) | 0.397 |
| 24 | 0.23 (0.35) | 69 | 0.27 (0.35) | 74 | 0.06 (–0.05 to 0.18) | 0.935 |
| 30 | 0.20 (0.35) | 43 | 0.18 (0.31) | 38 | 0.08 (–0.06 to 0.22) | 0.815 |
| 36 | 0.06 (0.21) | 23 | 0.14 (0.30) | 21 | 0.06 (–0.11 to 0.23) | 1 |
| Overall difference | 0.05 (–0.01 to 0.11) | 0.116 | ||||
| Time point (months) | Treatment group | Mean difference (95% CI) | p-value | |||
|---|---|---|---|---|---|---|
| Active treatment | Placebo | |||||
| Mean (SD) | n | Mean (SD) | n | |||
| 3 | 1.90 (0.54) | 120 | 1.89 (0.46) | 133 | 0.01 (–0.15 to 0.16) | 1.00 |
| 6 | 1.84 (0.49) | 105 | 1.86 (0.43) | 112 | –0.03 (–0.19 to 0.13) | 1.00 |
| 12 | 1.86 (0.43) | 63 | 1.86 (0.42) | 77 | –0.01 (–0.19 to 0.16) | 1.00 |
| 18 | 1.84 (0.39) | 39 | 1.78 (0.42) | 50 | –0.01 (–0.20 to 0.18) | 1.00 |
| 24 | 1.92 (0.41) | 18 | 1.78 (0.43) | 25 | 0.09 (–0.14 to 0.31) | 1.00 |
| 30 | 1.73 (0.49) | 11 | 1.73 (0.65) | 9 | –0.16 (–0.45 to 0.13) | 0.933 |
| 36 | 2.05 (0.17) | 2 | 1.51 (0.17) | 4 | 0.44 (–0.05 to 0.94) | 0.107 |
| Overall difference | –0.005 (–0.11 to 0.10) | 0.922 | ||||
| Time point (months) | Treatment group | Mean difference (95% CI) | p-value | |||
|---|---|---|---|---|---|---|
| Active treatment | Placebo | |||||
| Mean (SD) | n | Mean (SD) | n | |||
| 3 | 62.04 (18.67) | 120 | 60.43 (11.13) | 133 | 1.82 (–2.39 to 6.03) | 1.00 |
| 6 | 60.17 (9.87) | 10 | 59.04 (11.37) | 112 | 0.51 (–3.89 to 4.91) | 1.00 |
| 12 | 57.83 (9.68) | 63 | 58.15 (10.42) | 77 | 0.81 (–4.18 to 5.79) | 1.00 |
| 18 | 56.31 (10.01) | 39 | 56.35 (9.29) | 50 | 1.23 (–4.52 to 6.97) | 1.00 |
| 24 | 60.31 (12.05) | 18 | 55.13 (9.68) | 25 | 4.83 (–2.63 to 12.29) | 0.572 |
| 30 | 57.44 (11.27) | 11 | 53.24 (14.54) | 9 | –2.44 (–12.60 to 7.72) | 1.00 |
| 36 | 75.37 (17.04) | 2 | 45.97 (4.96) | 4 | 21.53 (3.00 to 40.06) | 0.012 |
| Overall difference | 1.41 (–1.35 to 4.17) | 0.316 | ||||
| Outcome | Treatment group | Analysis | ||||||
|---|---|---|---|---|---|---|---|---|
| Active treatment | Placebo | Unadjusteda | Adjustedb | |||||
| n | Mean (SD) | n | Mean (SD) | Mean difference (95% CI) | p-value | Mean difference (95% CI) | p-value | |
| LCQ score | ||||||||
| Total | 69 | 15.37 (3.99) | 71 | 14.59 (4.00) | –0.99 (–2.54 to 0.57) | 0.215 | –0.76 (–2.20 to 0.68) | 0.300 |
| Physical | 69 | 4.88 (1.22) | 72 | 4.70 (1.16) | –0.23 (–0.72 to 0.26) | 0.353 | –0.16 (–0.67 to 0.36) | 0.550 |
| Psychological | 69 | 5.16 (1.43) | 75 | 4.86 (1.51) | –0.37 (–0.92 to 0.19) | 0.193 | –0.33 (–0.79 to 0.14) | 0.167 |
| Social | 69 | 5.33 (1.49) | 75 | 5.05 (1.49) | –0.35 (–0.87 to 0.17) | 0.182 | –0.30 (–0.78 to 0.18) | 0.216 |
| CSS | 72 | 44.74 (27.01) | 84 | 49.69 (26.68) | 6.70 (–3.50 to 16.90) | 0.198 | 2.97 (–6.30 to 12.23) | 0.530 |
| K-BILD score | ||||||||
| Psychological | 71 | 49.73 (17.92) | 85 | 51.86 (16.89) | 2.64 (–2.89 to 8.19) | 0.348 | 1.92 (–2.12 to 5.97) | 0.352 |
| Breathless | 72 | 34.37 (17.42) | 86 | 34.96 (14.55) | 1.17 (–4.57 to 6.90) | 0.690 | –0.49 (–5.43 to 4.45) | 0.847 |
| Chest | 72 | 59.86 (20.26) | 86 | 56.75 (22.82) | –4.51 (–12.22 to 3.20) | 0.251 | –2.64 (–9.49 to 4.21) | 0.449 |
| Total | 71 | 50.32 (12.26) | 85 | 50.74 (11.20) | 0.52 (–3.39 to 4.44) | 0.793 | 0.26 (–2.83 to 3.34) | 0.869 |
| Outcome | Treatment group | Analysis | ||||||
|---|---|---|---|---|---|---|---|---|
| Active treatment | Placebo | Unadjusteda | Adjustedb | |||||
| n | Mean (SD) | n | Mean (SD) | Mean difference (95% CI) | p-value | Mean difference (95% CI) | p-value | |
| Absolute | ||||||||
| FVC (l) | 48 | 2.21 (0.49) | 50 | 2.27 (0.52) | 0.04 (–0.18 to 0.26) | 0.734 | 0.01 (–0.09 to 0.12) | 0.795 |
| FEV1 (l) | 48 | 1.83 (0.39) | 50 | 1.90 (0.42) | 0.001 (–0.18 to 0.18) | 0.990 | –0.02 (–0.12 to 0.09) | 0.739 |
| DLCO (mmol/minute/kPa) | 39 | 3.37 (1.92) | 40 | 3.69 (1.47) | –0.25 (–1.12 to 0.67) | 0.593 | –0.27 (–1.15 to 0.61) | 0.541 |
| Per cent predicted | ||||||||
| FVC | 48 | 52.75 (8.65) | 50 | 54.02 (9.41) | 0.82 (–2.87 to 4.53) | 0.661 | 0.57 (–2.59 to 3.73) | 0.726 |
| FEV1 | 48 | 56.54 (9.17) | 50 | 59.17 (11.05) | –0.21 (–4.29 to 3.87) | 0.919 | –0.46 (–4.09 to 3.16) | 0.802 |
| DLCO | 39 | 38.47 (19.03) | 40 | 42.22 (14.83) | –3.13 (–12.88 to 6.62) | 0.529 | –3.23 (–12.89 to 6.43) | 0.512 |
| Biomarker | Treatment group, median (IQR) | |
|---|---|---|
| Active treatment (n = 73) | Placebo (n = 84) | |
| MPO (ng/ml) | 242.50 (127.00–346.30) | 181.65 (96.85–338.55) |
| SP-D (ng/ml) | 37.80 (16.20–61.20) | 33.90 (21.00–52.80) |
| MMP-7 (pg/ml) | 8.60 (6.40–13.20) | 8.90 (6.50–11.30) |
| CRP (mg/l) | 3.90 (2.50–6.30) | 3.60 (1.95–6.00) |
| CA-125 (U/ml) | 26.70 (15.80–40.00) | 23.70 (15.80–41.75) |
| CA19-9 (U/ml) | 21.30 (11.40–43.80) | 17.00 (8.20–31.35) |
| Pro-BNP (pg/ml) | 72.70 (45.00–193.70) | 72.60 (40.10–184.80) |
| OPG (pmol/l) | 5.00 (3.60–6.20) | 4.70 (3.30–6.10) |
| CCL18 (pg/ml) | 94,914.00 (77,372.00–120,000.00) | 88,694.00 (70,396.50–125,795.00) |
| TRAIL (pg/ml) | 47.10 (36.40–61.80) | 51.05 (42.40–62.40) |
| MCP1 (pg/ml) | 438.90 (359.80–560.70) | 437.20 (369.15–567.95) |
| Biomarker | Treatment group, median (IQR) | p-value | ||
|---|---|---|---|---|
| Active treatment (n = 73) | Placebo (n = 84) | At 12 months | Change from baseline | |
| MPO (ng/ml) | 201.00 (130.80–324.70) | 184.60 (117.10–296.60) | 0.36 | 1.00 |
| SP-D (ng/ml) | 33.10 (18.10–51.20) | 34.55 (21.90–55.55) | 0.21 | < 0.001 |
| MMP-7 (pg/ml) | 9.90 (7.90–15.00) | 9.45 (6.60–12.80) | 0.23 | 0.52 |
| CRP (mg/l) | 5.10 (2.90–11.60) | 4.25 (2.00–6.90) | 0.016 | 0.005 |
| CA-125 (U/ml) | 36.50 (21.40–61.40) | 29.80 (19.25–46.95) | 0.19 | 0.032 |
| CA19-9 (U/ml) | 24.70 (14.40–69.20) | 19.50 (8.65–39.80) | 0.019 | 0.10 |
| Pro-BNP (pg/ml) | 89.60 (51.70–176.50) | 105.80 (55.55–281.95) | 0.27 | 0.064 |
| OPG (pmol/l) | 5.30 (4.00–6.30) | 4.95 (3.00–6.80) | 0.35 | 0.61 |
| CCL18 (pg/ml) | 100,264.00 (79,783.00–119,274.00) | 99,788.00 (69,781.50–120,000.00) | 0.63 | 0.67 |
| TRAIL (pg/ml) | 42.30 (33.50–53.40) | 45.60 (38.40–57.45) | 0.064 | 0.78 |
| MCP1 (pg/ml) | 493.50 (393.90–622.90) | 453.10 (384.35–552.80) | 0.27 | 0.14 |
| Biomarker | Treatment group, median (IQR) | |
|---|---|---|
| Active treatment (n = 58) | Placebo (n = 50) | |
| MPO (ng/ml) | 229.55 (127.00–337.80) | 174.65 (98.90–298.50) |
| SP-D (ng/ml) | 43.20 (16.20–61.40) | 31.65 (20.50–49.50) |
| MMP-7 (pg/ml) | 8.05 (6.00–12.10) | 8.45 (6.30–11.40) |
| CRP (mg/l) | 3.85 (2.10–6.30) | 2.95 (1.70–5.00) |
| CA-125 (U/ml) | 24.95 (15.00–35.90) | 23.70 (16.40–40.90) |
| CA19-9 (U/ml) | 17.75 (11.40–43.80) | 14.40 (7.50–27.80) |
| Pro-BNP (pg/ml) | 69.85 (45.00–193.70) | 65.10 (34.70–148.20) |
| OPG (pmol/l) | 4.70 (3.20–6.10) | 4.70 (2.70–5.90) |
| CCL18 (pg/ml) | 96,809.50 (77,372.00–117,597.00) | 89,505.00 (59,699.00–126,516.00) |
| TRAIL (pg/ml) | 49.50 (36.70–63.70) | 51.05 (41.30–65.10) |
| MCP1 (pg/ml) | 466.50 (372.40–590.20) | 444.85 (386.70–565.60) |
| Biomarker | Treatment group, median (IQR) | p-value | ||
|---|---|---|---|---|
| Active treatment (n = 73) | Placebo (n = 84) | At 12 months | Change from baseline | |
| MPO (ng/ml) | 197.85 (130.00–306.20) | 182.80 (98.70–284.50) | 0.30 | 0.78 |
| SP-D (ng/ml) | 30.80 (18.00–51.20) | 32.60 (20.70–53.80) | 0.48 | < 0.001 |
| MMP-7 (pg/ml) | 9.25 (7.00–14.00) | 9.75 (7.60–13.20) | 0.97 | 0.61 |
| CRP (mg/l) | 5.25 (2.90–11.80) | 2.55 (1.60–5.80) | < 0.001 | < 0.001 |
| CA-125 (U/ml) | 36.15 (20.80–57.90) | 28.85 (19.10–48.90) | 0.34 | 0.023 |
| CA19-9 (U/ml) | 23.60 (13.80–69.20) | 18.45 (7.80–32.10) | 0.025 | 0.021 |
| Pro-BNP (pg/ml) | 88.35 (47.50–204.30) | 91.40 (43.90–202.00) | 0.91 | 0.19 |
| OPG (pmol/l) | 5.25 (3.80–6.30) | 4.60 (2.90–6.60) | 0.21 | 0.83 |
| CCL18 (pg/ml) | 99,207.00 (79,783.00–119,274.00) | 103,548.00 (58,996.00–120,000.00) | 0.55 | 0.87 |
| TRAIL (pg/ml) | 43.45 (33.70–53.40) | 45.60 (38.50–59.60) | 0.14 | 0.47 |
| MCP1 (pg/ml) | 517.15 (367.10–635.50) | 453.40 (387.30–545.60) | 0.35 | 0.19 |
| Biomarker | Treatment group, median (IQR) | |
|---|---|---|
| Active treatment (n = 38) | Placebo (n = 49) | |
| MPO (ng/ml) | 218.60 (127.00–279.40) | 176.70 (102.70–298.50) |
| SP-D (ng/ml) | 46.75 (25.50–62.40) | 31.80 (20.50–49.50) |
| MMP-7 (pg/ml) | 7.85 (6.00–11.70) | 8.40 (6.30–10.90) |
| CRP (mg/l) | 4.20 (2.00–6.90) | 3.00 (1.70–5.00) |
| CA-125 (U/ml) | 22.50 (14.10–35.90) | 24.10 (16.40–40.90) |
| CA19-9 (U/ml) | 14.50 (9.50–39.30) | 12.50 (7.50–24.60) |
| Pro-BNP (pg/ml) | 63.90 (43.20–104.80) | 67.80 (35.10–148.20) |
| OPG (pmol/l) | 4.40 (3.60–5.70) | 4.70 (3.00–5.90) |
| CCL18 (pg/ml) | 100,774.50 (79,966.00–118,490.00) | 89,752.00 (63,401.00–126,516.00) |
| TRAIL (pg/ml) | 50.10 (38.30–63.70) | 50.80 (41.30–64.60) |
| MCP1 (pg/ml) | 471.05 (402.30–596.80) | 444.60 (386.70–565.60) |
| Biomarker | Treatment group, median (IQR) | p-value | ||
|---|---|---|---|---|
| Active treatment (n = 73) | Placebo (n = 84) | At 12 months | Change from baseline | |
| MPO (ng/ml) | 218.00 (127.20–319.20) | 184.50 (109.30–284.50) | 0.26 | 0.39 |
| SP-D (ng/ml) | 35.55 (18.70–51.20) | 32.70 (20.70–53.80) | 0.74 | < 0.001 |
| MMP-7 (pg/ml) | 9.25 (6.70–12.90) | 9.50 (7.60–13.20) | 0.87 | 1.00 |
| CRP (mg/l) | 5.00 (2.90–12.40) | 2.50 (1.60–5.80) | 0.002 | < 0.001 |
| CA-125 (U/ml) | 40.45 (19.80–57.90) | 28.90 (19.40–48.90) | 0.50 | 0.066 |
| CA19-9 (U/ml) | 21.70 (13.00–69.20) | 17.70 (7.80–29.30) | 0.098 | 0.033 |
| Pro-BNP (pg/ml) | 67.55 (44.00–133.70) | 91.40 (49.60–202.00) | 0.24 | 0.026 |
| OPG (pmol/l) | 5.15 (4.20–6.20) | 4.60 (3.10–6.60) | 0.33 | 0.58 |
| CCL18 (pg/ml) | 101,526.50 (81,487.00–115,539.00) | 104,155.00 (61,200.00–120,000.00) | 0.50 | 0.95 |
| TRAIL (pg/ml) | 44.20 (36.10–53.40) | 45.10 (38.50–57.10) | 0.45 | 0.42 |
| MCP1 (pg/ml) | 538.80 (410.80–635.50) | 454.00 (387.30–545.60) | 0.20 | 0.15 |
| Measure | Treatment group | p-value | |||
|---|---|---|---|---|---|
| Active treatment | Placebo | ||||
| Mean (SD) | n | Mean (SD) | n | ||
| White cell count (× 109/l) | 8.31 (2.23) | 148 | 8.57 (2.39) | 155 | 0.34 |
| Haemoglobin (g/dl) | 141.55 (14.84) | 148 | 147.97 (15.14) | 154 | < 0.001 |
| RCC (× 1012/l) | 4.68 (0.53) | 147 | 4.84 (0.46) | 154 | 0.006 |
| Mean cell volume (fl) | 91.65 (6.24) | 148 | 92.39 (5.40) | 155 | 0.26 |
| Mean cell haemoglobin (pg) | 30.39 (2.36) | 148 | 30.64 (2.05) | 155 | 0.33 |
| Haematocrit (%) | 0.43 (0.04) | 140 | 0.44 (0.04) | 149 | < 0.001 |
| Neutrophils (× 109/l) | 5.40 (1.98) | 148 | 5.63 (2.21) | 155 | 0.35 |
| Lymphocytes (× 109/l) | 1.81 (0.83) | 148 | 1.88 (0.71) | 155 | 0.45 |
| Eosinophils (× 109/l) | 0.35 (0.24) | 146 | 0.30 (0.22) | 155 | 0.056 |
| Basophils (× 109/l) | 0.06 (0.04) | 147 | 0.05 (0.04) | 152 | 0.31 |
| Monocytes (× 109/l) | 0.69 (0.24) | 148 | 0.69 (0.22) | 153 | 0.94 |
| Platelets (× 109/l) | 242.73 (69.17) | 148 | 234.63 (68.99) | 154 | 0.31 |
| Sodium (Na) (mmol/l) | 136.97 (3.17) | 148 | 138.99 (2.83) | 158 | < 0.001 |
| Potassium (K) (mmol/l) | 4.53 (0.38) | 148 | 4.38 (0.40) | 157 | 0.001 |
| Urea (mmol/l) | 5.72 (2.01) | 138 | 5.54 (1.55) | 150 | 0.40 |
| Creatinine (µmol/l) | 94.92 (26.78) | 148 | 83.63 (17.32) | 158 | < 0.001 |
| Bilirubin, upper limit of normal (µmol/l) | 20.62 (2.29) | 138 | 20.59 (2.43) | 147 | 0.89 |
| Bilirubin (µmol/l) | 8.28 (3.57) | 147 | 10.00 (5.24) | 155 | 0.001 |
| Alanine aminotransferase, upper limit of normal (IU/l) | 47.07 (10.50) | 137 | 46.32 (8.45) | 145 | 0.51 |
| Alanine aminotransferase (IU/l) | 28.44 (28.49) | 142 | 21.68 (12.59) | 154 | 0.008 |
| Alkaline phosphatase (IU/l) | 90.25 (31.69) | 146 | 81.22 (24.34) | 156 | 0.006 |
| Albumin (g/dl) | 39.45 (4.00) | 148 | 38.75 (5.60) | 157 | 0.21 |
| Total protein (g/dl) | 73.36 (5.97) | 118 | 73.70 (8.26) | 124 | 0.72 |
| Globulin (g/dl) | 33.36 (7.99) | 66 | 33.70 (9.40) | 76 | 0.82 |
| Measure | Treatment group | p-value | |||
|---|---|---|---|---|---|
| Active treatment | Placebo | ||||
| Mean (SD) | n | Mean (SD) | n | ||
| White cell count (× 109/l) | 8.33 (1.97) | 125 | 8.28 (1.93) | 140 | 0.83 |
| Haemoglobin (g/dl) | 141.86 (14.61) | 125 | 147.63 (13.77) | 140 | 0.001 |
| RCC (× 1012/l) | 4.63 (0.51) | 124 | 4.82 (0.46) | 140 | 0.002 |
| Mean cell volume (fl) | 92.42 (6.35) | 125 | 91.85 (5.04) | 140 | 0.42 |
| Mean cell haemoglobin (pg) | 30.76 (2.29) | 125 | 30.72 (1.93) | 140 | 0.89 |
| Haematocrit (%) | 0.43 (0.04) | 117 | 0.44 (0.04) | 137 | 0.008 |
| Neutrophils (× 109/l) | 5.60 (1.74) | 125 | 5.47 (1.72) | 140 | 0.55 |
| Lymphocytes (× 109/l) | 1.66 (0.68) | 125 | 1.82 (0.70) | 140 | 0.065 |
| Eosinophils (× 109/l) | 0.31 (0.22) | 124 | 0.25 (0.13) | 140 | 0.010 |
| Basophils (× 109/l) | 0.06 (0.04) | 125 | 0.06 (0.04) | 138 | 0.95 |
| Monocytes (× 109/l) | 0.67 (0.21) | 125 | 0.66 (0.21) | 140 | 0.54 |
| Platelets (× 109/l) | 236.86 (60.15) | 125 | 235.21 (67.57) | 140 | 0.83 |
| Sodium (Na) (mmol/l) | 137.55 (3.01) | 126 | 139.09 (2.75) | 141 | < 0.001 |
| Potassium (K) (mmol/l) | 4.44 (0.41) | 126 | 4.29 (0.41) | 141 | 0.005 |
| Urea (mmol/l) | 5.85 (1.93) | 125 | 5.48 (1.52) | 138 | 0.084 |
| Creatinine (µmol/l) | 96.40 (28.19) | 125 | 84.56 (26.86) | 142 | < 0.001 |
| Bilirubin, upper limit of normal (µmol/l) | 20.80 (3.28) | 118 | 20.60 (1.96) | 136 | 0.55 |
| Bilirubin (µmol/l) | 8.37 (4.57) | 124 | 9.67 (4.83) | 139 | 0.026 |
| Alanine aminotransferase, upper limit of normal (IU/l) | 47.92 (8.71) | 119 | 47.98 (7.50) | 133 | 0.95 |
| Alanine aminotransferase (IU/l) | 24.45 (14.06) | 123 | 25.59 (50.52) | 138 | 0.81 |
| Alkaline phosphatase (IU/l) | 88.31 (29.16) | 125 | 81.27 (24.74) | 140 | 0.034 |
| Albumin (g/dl) | 40.10 (4.23) | 126 | 39.82 (4.47) | 141 | 0.61 |
| Total protein (g/dl) | 73.88 (6.71) | 101 | 74.09 (5.17) | 117 | 0.79 |
| Globulin (g/dl) | 33.66 (9.13) | 52 | 33.17 (7.78) | 59 | 0.76 |
| Measure | Treatment group | p-value | |||
|---|---|---|---|---|---|
| Active treatment | Placebo | ||||
| Mean (SD) | n | Mean (SD) | n | ||
| White cell count (× 109/l) | 8.42 (2.27) | 111 | 8.30 (1.93) | 118 | 0.66 |
| Haemoglobin (g/dl) | 141.74 (14.17) | 111 | 147.29 (13.16) | 118 | 0.002 |
| RCC (× 1012/l) | 4.60 (0.49) | 111 | 4.81 (0.44) | 118 | < 0.001 |
| Mean cell volume (fl) | 92.63 (6.10) | 111 | 91.72 (5.44) | 118 | 0.24 |
| Mean cell haemoglobin (pg) | 30.96 (2.42) | 111 | 30.67 (2.01) | 117 | 0.32 |
| Haematocrit (%) | 0.42 (0.04) | 104 | 0.44 (0.04) | 112 | 0.005 |
| Neutrophils (× 109/l) | 5.73 (2.20) | 111 | 5.45 (1.70) | 118 | 0.29 |
| Lymphocytes (× 109/l) | 1.67 (0.69) | 111 | 1.85 (0.69) | 118 | 0.053 |
| Eosinophils (× 109/l) | 0.29 (0.19) | 111 | 0.26 (0.18) | 118 | 0.25 |
| Basophils (× 109/l) | 0.06 (0.04) | 111 | 0.06 (0.04) | 117 | 0.87 |
| Monocytes (× 109/l) | 0.66 (0.22) | 111 | 0.66 (0.20) | 118 | 0.98 |
| Platelets (× 109/l) | 240.86 (63.49) | 111 | 238.39 (68.16) | 118 | 0.78 |
| Sodium (Na) (mmol/l) | 137.77 (2.77) | 111 | 138.93 (2.67) | 119 | 0.001 |
| Potassium (K) (mmol/l) | 4.46 (0.37) | 111 | 4.34 (0.44) | 119 | 0.032 |
| Urea (mmol/l) | 5.86 (2.32) | 111 | 5.63 (1.55) | 119 | 0.36 |
| Creatinine (µmol/l) | 92.32 (25.33) | 111 | 83.33 (19.90) | 119 | 0.003 |
| Bilirubin, upper limit of normal (µmol/l) | 21.62 (10.76) | 105 | 20.37 (2.15) | 114 | 0.23 |
| Bilirubin (µmol/l) | 8.39 (4.12) | 110 | 9.13 (4.56) | 117 | 0.20 |
| Alanine aminotransferase, upper limit of normal (IU/l) | 47.86 (8.00) | 104 | 47.37 (8.02) | 111 | 0.66 |
| Alanine aminotransferase (IU/l) | 23.03 (12.00) | 108 | 20.56 (10.87) | 116 | 0.11 |
| Alkaline phosphatase (IU/l) | 87.48 (28.78) | 111 | 83.12 (24.66) | 119 | 0.22 |
| Albumin (g/dl) | 39.68 (4.20) | 111 | 39.78 (4.22) | 119 | 0.85 |
| Total protein (g/dl) | 73.37 (5.74) | 90 | 74.04 (4.97) | 105 | 0.38 |
| Globulin (g/dl) | 32.51 (9.05) | 44 | 33.72 (6.79) | 53 | 0.46 |
| Measure | Treatment group | p-value | |||
|---|---|---|---|---|---|
| Active treatment | Placebo | ||||
| Mean (SD) | n | Mean (SD) | n | ||
| White cell count (× 109/l) | 8.87 (2.84) | 43 | 8.30 (1.81) | 54 | 0.24 |
| Haemoglobin (g/dl) | 140.49 (13.14) | 43 | 144.87 (14.22) | 54 | 0.12 |
| RCC (× 1012/l) | 4.59 (0.46) | 42 | 4.75 (0.42) | 54 | 0.078 |
| Mean cell volume (fl) | 92.84 (6.66) | 43 | 91.57 (5.40) | 54 | 0.30 |
| Mean cell haemoglobin (pg) | 30.72 (2.52) | 43 | 30.58 (2.15) | 54 | 0.78 |
| Haematocrit (%) | 0.42 (0.04) | 42 | 0.43 (0.04) | 52 | 0.15 |
| Neutrophils (× 109/l) | 6.11 (2.71) | 43 | 5.63 (1.57) | 54 | 0.28 |
| Lymphocytes (× 109/l) | 1.70 (0.78) | 43 | 1.70 (0.74) | 54 | 1.00 |
| Eosinophils (× 109/l) | 0.31 (0.24) | 43 | 0.24 (0.14) | 54 | 0.078 |
| Basophils (× 109/l) | 0.05 (0.04) | 43 | 0.04 (0.04) | 54 | 0.85 |
| Monocytes (× 109/l) | 0.69 (0.22) | 43 | 0.67 (0.20) | 54 | 0.60 |
| Platelets (× 109/l) | 237.77 (67.03) | 43 | 237.81 (76.68) | 53 | 1.00 |
| Sodium (Na) (mmol/l) | 138.58 (2.62) | 43 | 138.61 (2.76) | 54 | 0.96 |
| Potassium (K) (mmol/l) | 4.41 (0.36) | 43 | 4.36 (0.35) | 54 | 0.52 |
| Urea (mmol/l) | 5.58 (2.07) | 43 | 5.85 (1.98) | 54 | 0.51 |
| Creatinine (µmol/l) | 86.88 (23.56) | 43 | 83.76 (20.03) | 54 | 0.48 |
| Bilirubin, upper limit of normal (µmol/l) | 20.23 (2.42) | 39 | 20.56 (2.23) | 50 | 0.51 |
| Bilirubin (µmol/l) | 8.74 (5.35) | 42 | 10.28 (5.66) | 53 | 0.18 |
| Alanine aminotransferase, upper limit of normal (IU/l) | 47.29 (7.80) | 38 | 50.53 (14.12) | 49 | 0.21 |
| Alanine aminotransferase (IU/l) | 24.91 (15.18) | 43 | 22.87 (14.05) | 53 | 0.50 |
| Alkaline phosphatase (IU/l) | 105.95 (83.27) | 42 | 84.96 (21.03) | 54 | 0.078 |
| Albumin (g/dl) | 39.93 (4.02) | 43 | 39.09 (4.31) | 54 | 0.33 |
| Total protein (g/dl) | 72.59 (6.14) | 34 | 73.15 (4.53) | 47 | 0.64 |
| Globulin (g/dl) | 33.16 (10.03) | 16 | 33.21 (8.04) | 25 | 0.99 |
| Measure | Treatment group | p-value | |||
|---|---|---|---|---|---|
| Active treatment | Placebo | ||||
| Mean (SD) | n | Mean (SD) | n | ||
| White cell count (× 109/l) | 8.50 (2.44) | 22 | 8.19 (1.72) | 30 | 0.58 |
| Haemoglobin (g/dl) | 141.17 (14.70) | 22 | 147.17 (12.27) | 30 | 0.11 |
| RCC (× 1012/l) | 4.60 (0.55) | 22 | 4.79 (0.39) | 30 | 0.14 |
| Mean cell volume (fl) | 92.66 (7.40) | 22 | 92.10 (4.55) | 30 | 0.74 |
| Mean cell haemoglobin (pg) | 30.90 (2.96) | 22 | 30.76 (1.93) | 30 | 0.84 |
| Haematocrit (%) | 0.42 (0.05) | 22 | 0.44 (0.03) | 27 | 0.21 |
| Neutrophils (× 109/l) | 5.73 (2.20) | 22 | 5.54 (1.37) | 30 | 0.71 |
| Lymphocytes (× 109/l) | 1.71 (0.63) | 22 | 1.62 (0.61) | 30 | 0.61 |
| Eosinophils (× 109/l) | 0.30 (0.19) | 22 | 0.29 (0.14) | 30 | 0.79 |
| Basophils (× 109/l) | 0.06 (0.03) | 22 | 0.05 (0.03) | 29 | 0.45 |
| Monocytes (× 109/l) | 0.69 (0.23) | 22 | 0.68 (0.18) | 30 | 0.81 |
| Platelets (× 109/l) | 216.30 (51.23) | 22 | 236.53 (65.87) | 30 | 0.23 |
| Sodium (Na) (mmol/l) | 138.43 (3.40) | 22 | 138.63 (2.86) | 30 | 0.82 |
| Potassium (K) (mmol/l) | 4.42 (0.35) | 22 | 4.43 (0.32) | 30 | 0.93 |
| Urea (mmol/l) | 5.63 (2.13) | 22 | 5.93 (2.00) | 30 | 0.61 |
| Creatinine (µmol/l) | 91.55 (28.24) | 21 | 88.77 (22.22) | 30 | 0.69 |
| Bilirubin, upper limit of normal (µmol/l) | 20.27 (1.20) | 21 | 20.68 (0.55) | 28 | 0.12 |
| Bilirubin (µmol/l) | 8.82 (4.45) | 21 | 9.30 (3.58) | 30 | 0.67 |
| Alanine aminotransferase, upper limit of normal (IU/l) | 46.45 (7.42) | 21 | 48.74 (6.92) | 27 | 0.27 |
| Alanine aminotransferase (IU/l) | 22.73 (17.51) | 21 | 19.00 (6.26) | 29 | 0.29 |
| Alkaline phosphatase (IU/l) | 88.00 (32.95) | 21 | 85.43 (22.97) | 30 | 0.74 |
| Albumin (g/dl) | 39.77 (4.37) | 21 | 38.57 (4.22) | 30 | 0.32 |
| Total protein (g/dl) | 71.50 (5.35) | 17 | 74.35 (4.09) | 23 | 0.060 |
| Globulin (g/dl) | 32.80 (4.29) | 9 | 35.05 (8.70) | 12 | 0.47 |
| Measure | Treatment group | p-value | |||
|---|---|---|---|---|---|
| Active treatment | Placebo | ||||
| Mean (SD) | n | Mean (SD) | n | ||
| White cell count (× 109/l) | 7.80 (1.93) | 12 | 8.21 (1.96) | 12 | 0.61 |
| Haemoglobin (g/dl) | 139.17 (14.98) | 12 | 147.25 (8.27) | 12 | 0.12 |
| RCC (× 1012/l) | 4.66 (0.38) | 12 | 4.75 (0.36) | 12 | 0.57 |
| Mean cell volume (fl) | 90.80 (6.40) | 12 | 93.25 (2.86) | 12 | 0.24 |
| Mean cell haemoglobin (pg) | 29.80 (2.06) | 12 | 30.98 (1.63) | 12 | 0.13 |
| Haematocrit (%) | 0.42 (0.04) | 12 | 0.44 (0.02) | 11 | 0.17 |
| Neutrophils (× 109/l) | 5.75 (1.77) | 12 | 5.62 (1.60) | 12 | 0.84 |
| Lymphocytes (× 109/l) | 1.64 (0.77) | 12 | 1.47 (0.70) | 12 | 0.57 |
| Eosinophils (× 109/l) | 0.24 (0.11) | 12 | 0.34 (0.23) | 12 | 0.19 |
| Basophils (× 109/l) | 0.04 (0.03) | 12 | 0.04 (0.03) | 12 | 0.85 |
| Monocytes (× 109/l) | 0.64 (0.19) | 12 | 0.75 (0.16) | 10 | 0.15 |
| Platelets (× 109/l) | 198.42 (48.77) | 12 | 251.18 (70.65) | 11 | 0.048 |
| Sodium (Na) (mmol/l) | 138.85 (2.03) | 13 | 138.00 (2.68) | 11 | 0.39 |
| Potassium (K) (mmol/l) | 4.32 (0.42) | 13 | 4.55 (0.23) | 11 | 0.13 |
| Urea (mmol/l) | 5.13 (1.70) | 13 | 5.12 (1.19) | 11 | 0.98 |
| Creatinine (µmol/l) | 86.08 (30.70) | 13 | 84.82 (16.89) | 11 | 0.90 |
| Bilirubin, upper limit of normal (µmol/l) | 19.54 (4.41) | 13 | 21.08 (1.68) | 12 | 0.27 |
| Bilirubin (µmol/l) | 10.69 (5.14) | 13 | 10.36 (3.88) | 11 | 0.86 |
| Alanine aminotransferase, upper limit of normal (IU/l) | 45.92 (7.63) | 13 | 49.08 (7.51) | 12 | 0.31 |
| Alanine aminotransferase (IU/l) | 18.69 (12.62) | 13 | 19.17 (6.99) | 12 | 0.91 |
| Alkaline phosphatase (IU/l) | 213.15 (420.72) | 13 | 91.17 (23.44) | 12 | 0.33 |
| Albumin (g/dl) | 40.31 (3.82) | 13 | 37.64 (6.09) | 11 | 0.20 |
| Total protein (g/dl) | 74.75 (5.19) | 12 | 75.30 (7.60) | 10 | 0.84 |
| Globulin (g/dl) | 36.80 (6.91) | 5 | 39.50 (6.47) | 6 | 0.52 |
| Measure | Treatment group | p-value | |||
|---|---|---|---|---|---|
| Active treatment | Placebo | ||||
| Mean (SD) | n | Mean (SD) | n | ||
| White cell count (× 109/l) | 6.45 (1.48) | 2 | 7.08 (0.54) | 4 | 0.46 |
| Haemoglobin (g/dl) | 140.50 (2.12) | 2 | 139.50 (7.42) | 4 | 0.87 |
| RCC (× 1012/l) | 4.89 (0.34) | 2 | 4.60 (0.26) | 4 | 0.30 |
| Mean cell volume (fl) | 83.00 (2.83) | 2 | 92.58 (7.81) | 4 | 0.18 |
| Mean cell haemoglobin (pg) | 28.85 (1.63) | 2 | 31.73 (0.95) | 3 | 0.080 |
| Haematocrit (%) | 0.41 (0.01) | 2 | 0.43 (0.03) | 3 | 0.39 |
| Neutrophils (× 109/l) | 3.89 (1.03) | 2 | 5.12 (0.87) | 4 | 0.20 |
| Lymphocytes (× 109/l) | 1.56 (0.02) | 2 | 1.07 (0.59) | 4 | 0.33 |
| Eosinophils (× 109/l) | 0.17 (0.08) | 2 | 0.25 (0.17) | 4 | 0.56 |
| Basophils (× 109/l) | 0.04 (0.01) | 2 | 0.05 (0.04) | 4 | 0.76 |
| Monocytes (× 109/l) | 0.77 (0.31) | 2 | 0.62 (0.20) | 4 | 0.51 |
| Platelets (× 109/l) | 209.00 (46.67) | 2 | 236.50 (40.88) | 4 | 0.50 |
| Sodium (Na) (mmol/l) | 137.00 (1.41) | 2 | 140.75 (2.06) | 4 | 0.087 |
| Potassium (K) (mmol/l) | 4.30 (0.42) | 2 | 4.90 (0.36) | 3 | 0.18 |
| Urea (mmol/l) | 4.00 (1.13) | 2 | 6.73 (1.08) | 4 | 0.045 |
| Creatinine (µmol/l) | 93.00 (25.46) | 2 | 98.75 (19.67) | 4 | 0.77 |
| Bilirubin, upper limit of normal (µmol/l) | 20.00 (0.00) | 2 | 20.50 (0.58) | 4 | 0.31 |
| Bilirubin (µmol/l) | 4.50 (0.71) | 2 | 7.25 (2.06) | 4 | 0.16 |
| Alanine aminotransferase, upper limit of normal (IU/l) | 42.00 (11.31) | 2 | 50.00 (5.00) | 3 | 0.34 |
| Alanine aminotransferase (IU/l) | 15.50 (3.54) | 2 | 15.00 (2.65) | 3 | 0.87 |
| Alkaline phosphatase (IU/l) | 80.00 (8.49) | 2 | 76.50 (12.79) | 4 | 0.75 |
| Albumin (g/dl) | 36.50 (0.71) | 2 | 36.00 (3.16) | 4 | 0.84 |
| Total protein (g/dl) | 76.00 (9.90) | 2 | 71.50 (2.12) | 2 | 0.59 |
| Globulin (g/dl) | 39.50 (10.61) | 2 | 37.50 (2.12) | 2 | 0.82 |
List of abbreviations
- AE
- adverse event
- AR
- adverse reaction
- BAL
- bronchoalveolar lavage
- BALF
- bronchoalveolar lavage fluid
- CA-125
- cancer antigen 125
- CA19-9
- carbohydrate antigen 19-9
- CACE
- compliance-adjusted causal effect
- CCL18
- chemokine ligand 18
- CI
- confidence interval
- CONSORT
- Consolidated Standards of Reporting Trials
- COPD
- chronic obstructive pulmonary disease
- CRF
- case report form
- CRP
- C-reactive protein
- CSM
- central statistical monitoring
- CSS
- Cough Symptom Score
- CV
- coefficient of variation
- DLCO
- diffusing capacity of the lung for carbon monoxide
- DMC
- Data Monitoring Committee
- DNA
- deoxyribonucleic acid
- EC
- European Commission
- ECLIA
- electrochemiluminescence immunoassay
- ELISA
- enzyme-linked immunosorbent assay
- EME-TIPAC
- Efficacy and Mechanism Evaluation of Treating Idiopathic Pulmonary Fibrosis with the addition of Co-trimoxazole
- EQ-5D
- EuroQol-5 Dimensions
- EQ-5D-5L
- EuroQol-5 Dimensions, five-level version
- EQC
- external quality control
- FEV1
- forced expiratory volume in 1 second
- FVC
- forced vital capacity
- G6PD
- glucose-6-phosphate dehydrogenase
- GCP
- good clinical practice
- GORD
- gastro-oesophageal reflux disease
- GP
- general practitioner
- GSST PMU
- Guy’s and St Thomas’ NHS Foundation Trust Pharmacy Manufacturing Unit
- HIV
- human immunodeficiency virus
- HR
- hazard ratio
- HRCT
- high-resolution computerised tomography
- HSP47
- heat shock protein 47
- HTA
- Health Technology Assessment
- ICH
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use
- IIP
- idiopathic interstitial pneumonia
- ILD
- interstitial lung disease
- IMP
- investigational medicinal product
- IPF
- idiopathic pulmonary fibrosis
- IQC
- internal quality control
- IQR
- interquartile range
- ISF
- investigator site file
- ISRCTN
- International Standard Randomised Controlled Trial Number
- ITT
- intention to treat
- K-BILD
- King’s Brief Interstitial Lung Disease
- LCQ
- Leicester Cough Questionnaire
- MCP-1
- monocyte chemotactic protein 1
- MDT
- multidisciplinary team
- MedDRA
- Medical Dictionary for Regulatory Activities
- MHRA
- Medicines and Healthcare products Regulatory Agency
- MMP-7
- matrix metalloproteinase 7
- MPO
- myeloperoxidase
- mPP
- modified per protocol
- MRC
- Medical Research Council
- NCTU
- Norwich Clinical Trials Unit
- NICE
- National Institute for Health and Care Excellence
- NIHR
- National Institute for Health Research
- OPG
- osteoprotegerin
- PI
- principal investigator
- PP
- per protocol
- pro-BNP
- pro-brain natriuretic protein
- PSF
- pharmacy site file
- QALY
- quality-adjusted life-year
- QMMP
- quality management and monitoring plan
- RCC
- red cell count
- rRNA
- ribosomal ribonucleic acid
- SAE
- serious adverse event
- SAP
- statistical analysis plan
- SAR
- serious adverse reaction
- SD
- standard deviation
- SGRQ
- St George’s Respiratory Questionnaire
- SP-D
- surfactant protein D
- SUSAR
- suspected unexpected serious adverse reaction
- TIPAC
- Treating Idiopathic Pulmonary Fibrosis with the Addition of Co-trimoxazole
- TLR
- toll-like receptor
- TMG
- Trial Management Group
- TRAIL
- tumour necrosis factor-related apoptosis-inducing ligand
- TSC
- Trial Steering Committee
- UIP
- usual interstitial pneumonia
- ULN
- upper limit of normal
- ULOQ
- upper limit of quantitation