

Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 14/02/17. The contractual start date was in February 2016. The final report began editorial review in February 2020 and was accepted for publication in June 2021. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
Copyright © 2021 Cooke et al. This work was produced by Cooke et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This is an Open Access publication distributed under the terms of the Creative Commons Attribution CC BY 4.0 licence, which permits unrestricted use, distribution, reproduction and adaption in any medium and for any purpose provided that it is properly attributed. See: https://creativecommons.org/licenses/by/4.0/. For attribution the title, original author(s), the publication source – NIHR Journals Library, and the DOI of the publication must be cited.
2021 Cooke et al.
Chapter 1 Introduction
Background
Hepatitis C virus (HCV) is a major challenge in the UK both for the individual with the virus and for public health. In 2013, an estimated 215,000–265,000 individuals were living with HCV infection in the UK. 1,2 Those who are chronically infected are at risk of severe liver diseases (e.g. cirrhosis, liver failure and hepatocellular carcinoma). Progression to end-stage liver disease is more rapid in those who have other medical conditions, particularly co-infection with human immunodeficiency virus (HIV). Treatment of infected individuals has the additional potential to reduce ongoing transmission through needle use and sexual contact, and from mother to child.
Morbidity and mortality from HCV have been an increasing challenge to the NHS. In 2013, health-care costs related to HCV were estimated to be £82.7M per year and productivity losses £184M–367M per year. 3 HCV-related hospital admissions rose from 612 in 1998 to 2268 in 2011. HCV-related deaths rose from 98 in 1996 to 381 in 2011. The proportion of liver transplants undertaken because of HCV rose steadily from 10% in 1996 to 18% in 2011.
Viral genotype remains a key factor in determining the preferred treatment options for HCV in the NHS. Globally, genotype 1 is the most common, accounting for approximately 46% of all infections, which is very similar to the estimated rate in the UK. 4 As the most common genotype in most well-resourced health economies, particularly the USA, genotype 1 was the greatest focus of the initial development of new oral drugs. However, other genotypes also make a substantial contribution to the HCV burden in the UK.
Curative treatments have been available for HCV for some time. However, in early 2015, standard treatment for HCV infection still involved long courses (i.e. 24–48 weeks) of relatively toxic therapy (pegylated interferon alpha plus ribavirin), with a modest chance of cure (40–50%). The nature of therapy remained a major barrier to the uptake of treatment and, hence, control of the epidemic. Since 2015, a new generation of well-tolerated oral direct-acting antivirals (DAAs) has transformed HCV treatment, with the potential to cure HCV in most patients after 8–12 weeks of therapy. All HCV-infected adults with mild disease could, in theory, be cured with these regimens, substantially reducing future morbidity and mortality. For genotype 1, the first two interferon-free combination regimens approved in the NHS were:
-
a ritonavir-boosted triple combination of paritaprevir/ritonavir, ombitasvir (Viekirax®; AbbVie, Chicago, IL, USA) and dasabuvir (Exviera®; AbbVie, Chicago, IL, USA)
-
sofosbuvir and ledipasvir (Harvoni®; Gilead Sciences, Inc., Foster City, CA, USA).
The ritonavir-boosted combination of paritaprevir, ritonavir and ombitasvir (without dasabuvir) has also been approved for genotype 4 infection. Other regimens that are active against more, or even all, viral genotypes have also been approved or have been submitted for approval.
The new treatments for HCV offer the potential for curative therapy for the individual and the opportunity to break transmission pathways, leading to the real possibility of eliminating the HCV epidemic in the UK. A recent systematic review showed a clear benefit of HCV cure in improving health outcomes across a range of clinical settings,5 and there is no evidence to suggest that this differs according to the means used to achieve cure. However, initial costs for treatment were very high, at approximately £3000 per week, placing strain on limited health budgets. 6 Beyond costs, licensed durations of therapy with DAAs, at 8–12 weeks, although significantly shorter, and therefore more tolerable than those of previous interferon-based therapies7, remain challenging for many patients with HCV, whose chaotic lifestyles are a barrier to adhering to treatment. Shorter courses of treatment would potentially increase access to treatment for difficult-to-reach groups and could have an impact on onward transmission.
The rapid development of treatment options for HCV has led to an ambitious World Health Organization strategy to eliminate viral hepatitis as a global public health threat by 2030. 8 This includes a target of treating 80% of those chronically infected with HCV, many of whom will be patients who will still find it challenging to complete a full treatment course of 8–12 weeks. Such patients will become an increasingly important part of clinical practice as treatment coverage expands to reach marginalised groups.
Shorter treatment courses of licensed therapies are one clear mechanism to increase coverage into these groups, including those with active illicit drug use, as they should increase adherence. 9,10 Shortened courses of licensed therapy may have sufficiently high efficacy in acute or recent HCV infection for them to be recommended routinely. 11 However, limited data are available in chronic infection to identify which patients might be able to achieve high cure rates with shorter durations of therapy. In particular, in two small Phase II studies, short-duration treatment in all patients, without risk stratification, achieved cure rates of only 20–40% after a fixed 4-week treatment course and of 57–95% after a fixed 6-week treatment course. 12,13 Furthermore, few of the combinations or durations that have been trialled have subsequently been licensed for use. In the case of licensed therapies, recommendations to shorten therapy to 8 weeks (rather than 12 weeks) are based on baseline viral load (VL) (< 6,000,000 IU/ml)14 and subgenotype,15 but no criteria for recommending < 8 weeks’ therapy have been tested or validated in those with chronic infection. Nevertheless, the very high cure rates (i.e. > 95%) on standard 8- to 12-week treatment courses make it clear that many patients are being prescribed much more medication than they require to be cured, resulting in unnecessary inconvenience and costs.
When a clinician initiates treatment in the knowledge that there is a high risk that the patient may not complete therapy, or aims to use an ultrashort course of therapy for the same reason, an important concern is that virological failure may be accompanied by emerging resistance, which could then compromise future treatment options. However, less resistance could also theoretically emerge with shorter courses of treatment. Ribavirin, a generically available guanosine analogue, was widely used to increase cure rates with previous pegylated interferon-based therapies, and there is some evidence that it may improve rates of virological cure with shorter treatment courses. 16 There is also the possibility that it might reduce the rate at which resistance emerges in those failing treatment when added to short-course therapy. 17 However, these hypotheses have not been tested in a randomised trial.
Rationale
The very high cure rates achieved with 12- and 8-week DAA regimens raise the question ‘What is the minimum duration of treatment that can achieve cure in the majority of patients?’. 18 As above, minimising (effective) treatment duration is important for ensuring the widest and most equitable access to curative therapy across all patients (particularly those who will struggle to take medicine and are likely to require support) for the same fixed budget, and for minimising toxicity. However, as observed with previous interferon-based therapies, it is likely that response to DAA treatment will depend on individual-level characteristics, offering the opportunity to stratify short-course treatment. The best-studied biomarker for stratifying treatment duration is plasma HCV ribonucleic acid (RNA) VL. Based on mean baseline VL levels and decline from VL baseline levels in studies to date, and assuming, at most, a modest negative correlation between initial values and rates of decline, a ‘sliding scale’ of 4–7 weeks’ combination DAA treatment (where the precise duration of treatment depends on the individual’s pre-therapy HCV VL) should reduce virus levels to < 1 copy in the whole person. The question addressed in this trial is whether or not such an HCV VL-stratified DAA duration (13–50% shorter, i.e. 50–83% of the original length) followed by retreatment for those failing initial treatment gives similar cure rates to longer fixed-duration (8-week) therapy followed by retreatment in individuals with mild chronic HCV disease. Biomarker-stratified variable-duration ultrashort-course treatment would enable more patients to be cured within the same overall budget, and would also have benefits in terms of less potential toxicity and regimens that are easier to adhere to for patients. This is particularly important for HCV, as a substantial minority of those infected come from disadvantaged populations (e.g. drug users, homeless persons and prisoners).
In addition, although ribavirin was an essential component of previous interferon-based treatments, its role in DAA regimens is less clear, potentially providing minimal additional benefit when added to more potent regimens. However, it is cheap and has less toxicity when given for short duration, and modest benefits could allow shorter DAA regimens to be used more effectively. Therefore, the trial also tested whether or not the addition of ribavirin is beneficial in short-course treatment, using a partial factorial design in those randomised to therapy that is shorter than the full licensed duration. First-line treatment choice was in line with recommended NHS options.
Ombitasvir/paritaprevir/ritonavir with or without dasabuvir
All participants randomised to variable ultrashort strategy (VUS) DAA treatment with the DAA combination of ombitasvir, paritaprevir and ritonavir with or without dasabuvir were additionally factorially randomised to adjunctive ribavirin (or not). The rationale for the factorial randomisation in both groups (i.e. fixed and variable duration) was that the ‘8-week’ treatment arm still represents a shorter duration than standard of care for this combination.
Glecaprevir/pibrentasvir
All participants randomised to VUS DAA treatment with the DAA combination of glecaprevir/pibrentasvir were additionally factorially randomised to adjunctive ribavirin (or not). However, participants randomised to the 8-week treatment were not additionally randomised to adjunctive ribavirin (or not), as 8 weeks of this combination without ribavirin is the licensed standard-of-care indication for mild HCV.
At the time the trial was designed, two licensed combination therapies were available for patients infected with HCV genotype 1: (1) 12 weeks’ ombitasvir/paritaprevir/dasabuvir/ritonavir and (2) 8–12 weeks’ sofosbuvir and ledipasvir. In the trial, ombitasvir/paritaprevir/dasabuvir/ritonavir was used as first-line treatment (with two alternative shortened treatment durations; see above), and sofosbuvir and ledipasvir as retreatment (as a 12-week course with ribavirin). A priori, it is reasonable to assume that the ordering of the two main combination treatments in first-line treatment compared with retreatment would be similar, although, to the best of our knowledge, no studies to date have addressed the question of whether or not regimen sequencing has an impact on performance in terms of overall cure from biomarker-stratified shortened first-line treatment plus retreatment. Other new combinations, including those active against other genotypes, were licensed during the course of the trial. To enable data to be generated on other genotypes, trial patients could alternatively be treated with:
-
ombitasvir/paritaprevir/ritonavir first line, they were infected with HCV genotype 4 (as this combination, without dasabuvir, is licensed for the treatment of infection with HCV genotype 4, but not HCV genotype 1)
-
glecaprevir/pibrentasvir first line, they were infected with HCV genotype 1a/1b or 4 (8-week standard course licensed in both of these genotypes).
All trial patients continued to receive sofosbuvir and ledipasvir as retreatment (as a 12-week standard course with ribavirin).
The provision of retreatment within the trial will generate important data to inform strategic use of DAAs in treatment pathways for the NHS. The scientific knowledge generated is likely to be generalisable to other new HCV DAAs. In addition, the mechanistic insights gained [in collaboration with the STOP-HCV-1 (Stratified Treatment OPtimisation for HCV-1) consortium, involving most of the leading HCV scientists in the UK] into the role of initial VL declines and viral quasi-species, human polymorphisms and immune responses will inform the development and evaluation of further treatment strategies (e.g. tailoring treatment duration based on on-treatment responses), ultimately improving outcomes across the NHS.
Objectives
The overarching aim is to evaluate the efficacy of biomarker-stratified treatment of HCV infection and of adjunctive ribavirin with combination DAAs. This would allow the identification of patients with minimal fibrosis and chronic HCV infection who can be offered a high probability of cure with shortened courses of interferon-free all-oral DAA regimens. Such stratification will reduce the cost per cure and improve access for those unable to adhere to 8–12 weeks of treatment.
The primary objectives of the STOP-HCV-1 trial were to test:
-
whether or not a biomarker-stratified short-course first-line treatment (with variable duration of between 4 and 7 weeks determined by patient baseline VL) followed by 12 weeks of retreatment for those failing therapy is non-inferior to a fixed-duration, 8-week first-line treatment followed by 12 weeks of retreatment for those failing therapy, in terms of overall HCV cure in patients with minimal fibrosis and chronic genotype 1 or 4 HCV infection
-
the benefits and risks of adding adjunctive ribavirin to 4–8 weeks’ first-line therapy for HCV infection.
The different strategies above will be tested using ombitasvir/paritaprevir/dasabuvir/ritonavir (genotype 1), ombitasvir/paritaprevir/ritonavir (genotype 4) or glecaprevir/pibrentasvir (genotypes 1 and 4) as first-line treatment, and sofosbuvir/ledipasvir/ribavirin as retreatment (genotypes 1 and 4).
The secondary objectives were as follows:
-
To test whether or not 4–7 weeks’ first-line biomarker-stratified treatment is non-inferior to 8 weeks’ fixed-duration first-line treatment in mild HCV infection (i.e. excluding retreatment responses).
-
To test whether or not retreatment with 12 weeks of an alternative combination regimen, given after detecting virological failure on first-line treatment, still achieves cure in the majority of the small proportion of patients failing short-course first-line treatment.
-
To explore whether or not factors other than baseline HCV VL influence and, therefore, could better predict the response to (1) short-course DAA treatment and (2) retreatment. Factors explored will include viral factors (such as minority resistance variants and viral diversity, and initial virological response), host factors {such as age, body mass index (BMI) and human genetic variation [notably, interleukin (IL) 28 polymorphisms]} and immune factors (such as immune phenotyping before and after treatment initiation). Mechanistic work will be embedded in the Medical Research Council (MRC) HCV Stratified Medicine Consortium (to be reported separately).
-
To validate the performance of a novel point-of-care device for detecting IL-28B polymorphism (to be reported separately).
Chapter 2 Methods
Trial design
The STOP-HCV-1 trial was an open-label randomised controlled trial that tested biomarker-stratified short-course first-line and retreatment DAA oral treatment regimens to cure mild chronic HCV disease. Patients were allocated 1 : 1 using a factorial design to each of:
-
open-label variable ultrashort treatment compared with fixed-duration first-line treatment (1 : 1)
-
open-label adjunctive ribavirin or not (1 : 1). (Note that patients receiving glecaprevir/pibrentasvir and randomised to fixed 8 weeks’ first-line treatment were excluded.)
All patients received first-line ombitasvir/paritaprevir/(dasabuvir)/ritonavir or glecaprevir/pibrentasvir (based on genotype and local availability of the different regimens) and sofosbuvir/ledipasvir/ribavirin retreatment as necessary.
Participants
All participants met the trial-specified inclusion and exclusion criteria detailed below. Written informed consent was obtained from all participants after they received an explanation of the aims, methods, benefits and potential hazards of the trial and before any trial-specific procedures were performed or any blood was taken for the trial.
Inclusion criteria
-
Aged ≥ 18 years.
-
Infected with HCV genotype 1a/1b or 4 with access to first-line treatment appropriate for their genotype [ombitasvir/paritaprevir/(dasabuvir)/ritonavir or glecaprevir/pibrentasvir].
-
At least one episode of detectable viraemia in the 6 months prior to randomisation (by quantitative HCV RNA, qualitative assay or HCV genotype), with no intervening undetectable results.
-
Plasma HCV VL greater than lower limit of quantification (LLOQ) at screening.
-
No evidence of significant liver fibrosis resulting from any aetiology [defined as FibroScan® (Echosens, Paris, France) score of ≤ 7.1 kPa, equivalent to F0–F1,19 within 180 days prior to planned randomisation or biopsy consistent with mild fibrosis (i.e. Ishak score ≤ 2/6) within 180 days prior to planned randomisation].
-
BMI ≥ 18 kg/m2.
-
Laboratory tests: platelets ≥ 60 × 109/l, haemoglobin > 12 g/dl (male) or > 11 g/dl (female), creatinine clearance (estimated using Cockcroft–Gault) ≥ 60 ml/minute and an international normalised ratio of < 1.5.
-
Screening HCV VL < 10,000,000 IU/ml.
-
Written informed consent obtained from the patient.
If patients were infected with HIV, then an additional eligibility criterion was:
-
on antiretrovirals with a HIV VL of < 50 copies/ml for > 24 weeks at the screening visit.
Exclusion criteria
-
Previous DAA exposure for this infection. (Previous treatment with pegylated interferon and/or ribavirin allowed and successful previous treatments with therapy allowed.)
-
Lactating, pregnant, planning to become pregnant or not willing to use effective contraception during the study and for 4 months after last dose of the study medication (female patients only).
-
Currently taking ethinyloestradiol-containing medicinal products, such as those contained in most combined oral contraceptives or contraceptive vaginal rings (female patients only).
-
Planning pregnancy with female partner or not willing to use effective contraception during the study and for 7 months after last dose of the study medication (male patients only).
-
Malignancy within 5 years prior to screening.
-
Any condition that, in the judgement of the investigator, might limit the patient’s life expectancy.
-
Currently receiving medication known to interact with study medication.
-
Disorder that may cause ongoing liver disease, including, but not limited to, active hepatitis B virus and ongoing alcohol misuse.
-
Any disorder that, in the opinion of the investigator, may have a significant negative impact on the ability of the patient to adhere to the trial regimen.
-
Use of other investigational products within 60 days of screening.
-
Known hypersensitivity to any active ingredient and/or excipients of the study medicines, namely microcrystalline cellulose, lactose monohydrate, croscarmellose sodium, magnesium stearate, gelatine, shellac, propylene glycol, polyethylene glycol, ammonium hydroxide, pregelatinised maize starch, sodium starch glycolate (type A), maize starch, hypromellose, talc, ethylcellulose aqueous dispersion, triacetin, copovidone, colloidal anhydrous silica, vitamin E (tocopherol) polyethlyene glycol succinate, sodium stearyl fumarate, polyvinyl alcohol, macrogol 3350, sunset yellow FCF aluminium lake (E110), colouring agent (E132), titanium dioxide (E171), yellow iron oxide (E172), red iron oxide (E172) and black iron oxide (E172)
-
History of severe pre-existing cardiac disease, including unstable or uncontrolled cardiac disease, in the previous 6 months.
-
Haemoglobinopathies (e.g. thalassaemia and sickle-cell anaemia).
Trial setting
Participants were recruited from 14 UK NHS hospital trusts:
-
Singleton Hospital, Swansea Bay University Health Board
-
University Hospitals of Leicester NHS Trust
-
Imperial College NHS Trust
-
St George’s Healthcare NHS Foundation Trust
-
Royal Free London NHS Foundation Trust
-
Nottingham University Hospitals NHS Trust
-
Royal Surrey County Hospital NHS Foundation Trust
-
Brighton and Sussex University Hospitals NHS Trust
-
John Radcliffe Hospital, Oxford University Hospitals NHS Foundation Trust
-
Glasgow Royal Infirmary, NHS Greater Glasgow and Clyde
-
Newcastle Freeman Hospital, Newcastle Upon Tyne Hospitals NHS Foundation Trust
-
Chelsea and Westminster Hospital NHS Foundation Trust
-
Central and North West London NHS Foundation Trust
-
Sheffield Teaching Hospitals NHS Foundation Trust.
The main criterion for selecting participating hospitals was that they had the potential to recruit the required number of chronic (> 6 months) HCV genotype 1a/1b- and 4-infected participants within the agreed recruitment period. This was established by the use of a trial-specific site survey. Sites also needed to meet the following criteria:
-
no competing studies that would have an impact on the ability to enrol quickly to the trial
-
turnaround of no more than 7 days for HCV VL test results
-
ability to provide 24-hour cover for trial patients
-
local governance approval likely to take < 3 months.
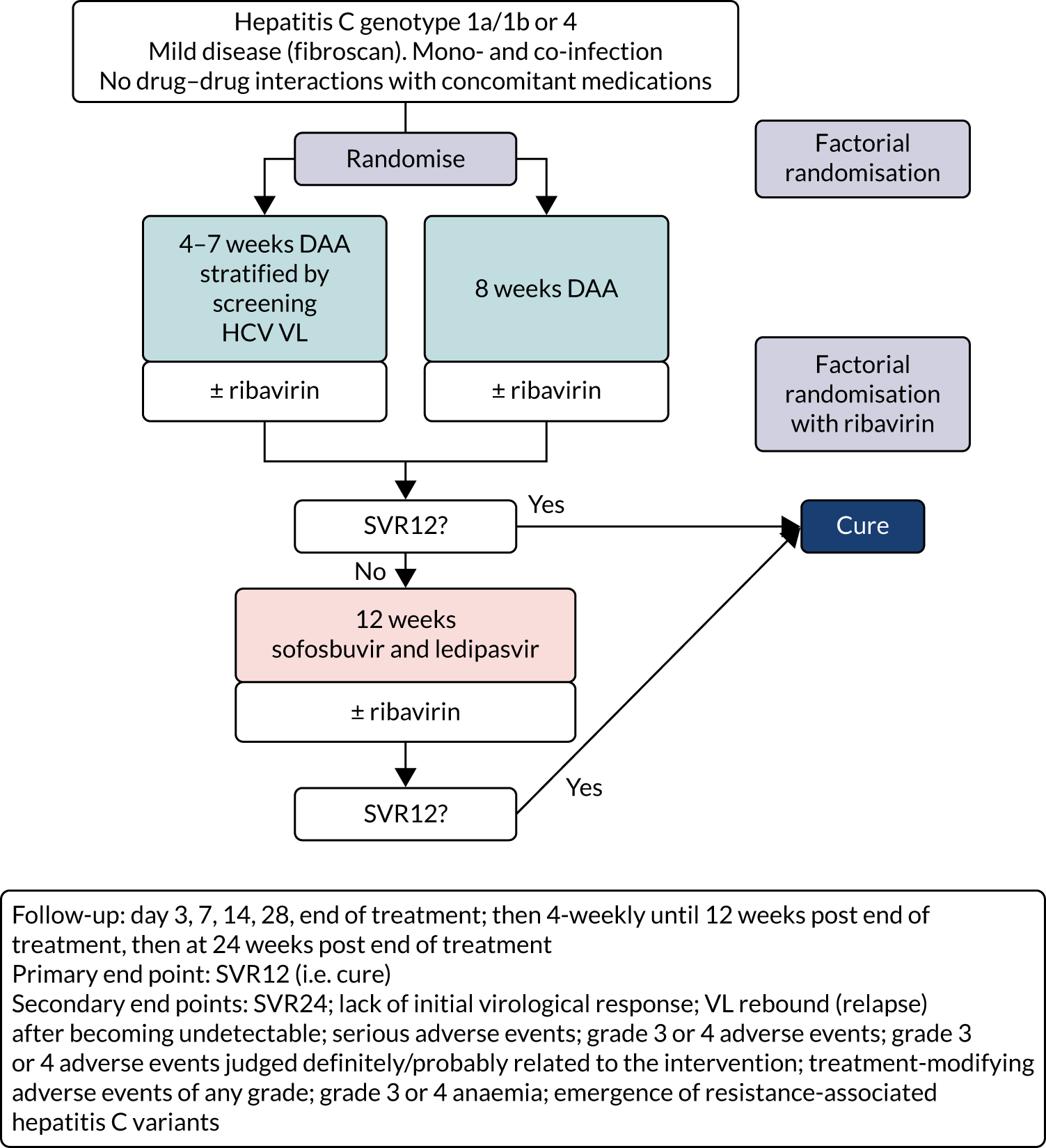
The overall trial design is summarised in Figure 1.
FIGURE 1.
Trial schema. Note that, as above, the ribavirin randomisation was a partial factorial in those randomised to a course shorter than the full licensed duration of therapy (i.e. the vast majority of patients recruited to the trial; see Chapter 3).
Patient and public involvement
The trial was developed with the Hepatitis C Trust (London, UK) and, in particular, Rachel Halford (who succeeded Charles Gore), who was one of two patient and public involvement representatives on the Trial Steering Committee. The Hepatitis C Trust advised on the design of the interventions, in particular the determination of the duration of variable-course therapy by baseline HCV VL, the acceptability of the follow-up schedule and assessments and the information provided to patients. The Hepatitis C Trust is helping to disseminate the trial’s results beyond the academic and health-care professional community to other patient groups.
Trial intervention: duration of treatment
All patients were randomised to VUS (intervention) or fixed 8-week (control) initial treatment. In protocol versions 1.0–4.0 inclusive, the intervention duration was between 4 and 7 weeks’ first-line treatment, on a sliding scale determined by the screening HCV VL. The proposed stratification rule was determined from the mean and standard deviation (SD) baseline VL, and the mean estimated declines, from previous trials (mean screening VL ≈ 6.25 log10 IU/ml, SD 0.4 log10 IU/ml; mean estimated decline 2.15 log10 IU/ml per week). Together, these could be used to estimate the duration of treatment needed to reduce levels to ≈ 1 copy in the whole body at end of treatment (< 0.0001 IU/ml), including a conservative assumption of a moderate negative correlation between baseline and decline in VL, as no data are available on this parameter.
This biomarker-stratified treatment duration was implemented as a specific number of days of first-line treatment based on the screening VL, as shown in Table 1. (Note that the declines are linear on a log-scale and so the absolute value in IU/ml does not increase linearly across the categories in this table.) Based on recent trials, it was expected that ≈ 15% of recruited patients (with screening HCV VL of < 10,000,000 IU/ml) would receive the minimum treatment and ≈ 5% of the maximum treatment.
| From HCV VL (IU/ml) | To HCV VL (IU/ml) | Days if randomised before 1 April 2017 (VUS1) | Days if randomised after 1 April 2017 (VUS2) |
|---|---|---|---|
| LLOQ | 50,000 | 28 | 28 |
| 50,001 | 65,000 | 28 | 29 |
| 65,001 | 82,500 | 28 | 30 |
| 82,501 | 110,000 | 28 | 31 |
| 100,001 | 140,000 | 28 | 32 |
| 150,001 | 180,000 | 28 | 33 |
| 175,001 | 235,000 | 28 | 34 |
| 225,001 | 300,000 | 28 | 35 |
| 300,001 | 400,000 | 29 | 36 |
| 400,001 | 500,000 | 30 | 37 |
| 500,001 | 550,000 | 30 | 38 |
| 550,001 | 650,000 | 31 | 38 |
| 650,001 | 750,000 | 31 | 39 |
| 750,001 | 850,000 | 32 | 39 |
| 850,001 | 1,100,000 | 32 | 40 |
| 1,100,001 | 1,300,000 | 33 | 41 |
| 1,300,001 | 1,450,000 | 34 | 41 |
| 1,450,001 | 1,700,000 | 34 | 42 |
| 1,700,001 | 1,850,000 | 35 | 42 |
| 1,850,001 | 2,200,000 | 35 | 43 |
| 2,200,001 | 2,400,000 | 36 | 43 |
| 2,400,001 | 2,850,000 | 36 | 44 |
| 2,850,001 | 3,150,000 | 37 | 44 |
| 3,150,001 | 3,600,000 | 37 | 45 |
| 3,600,001 | 4,100,000 | 38 | 45 |
| 4,050,001 | 4,550,000 | 38 | 46 |
| 4,550,001 | 5,250,000 | 39 | 46 |
| 5,250,001 | 5,700,000 | 39 | 47 |
| 5,700,001 | 6,800,000 | 40 | 47 |
| 6,800,001 | 7,100,000 | 40 | 48 |
| 7,100,001 | 8,800,000 | 41 | 48 |
| 8,800,001 | Upwards | 42 | 49 |
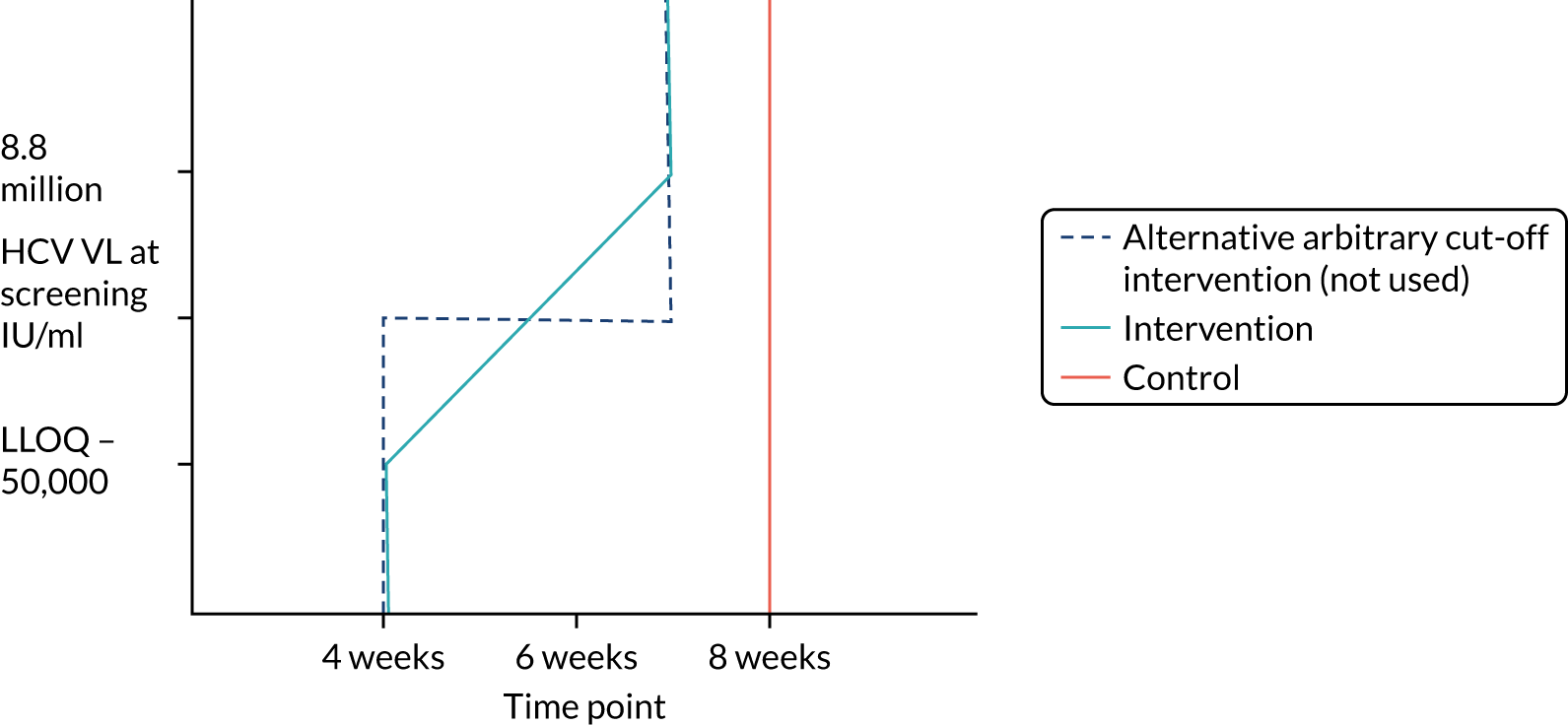
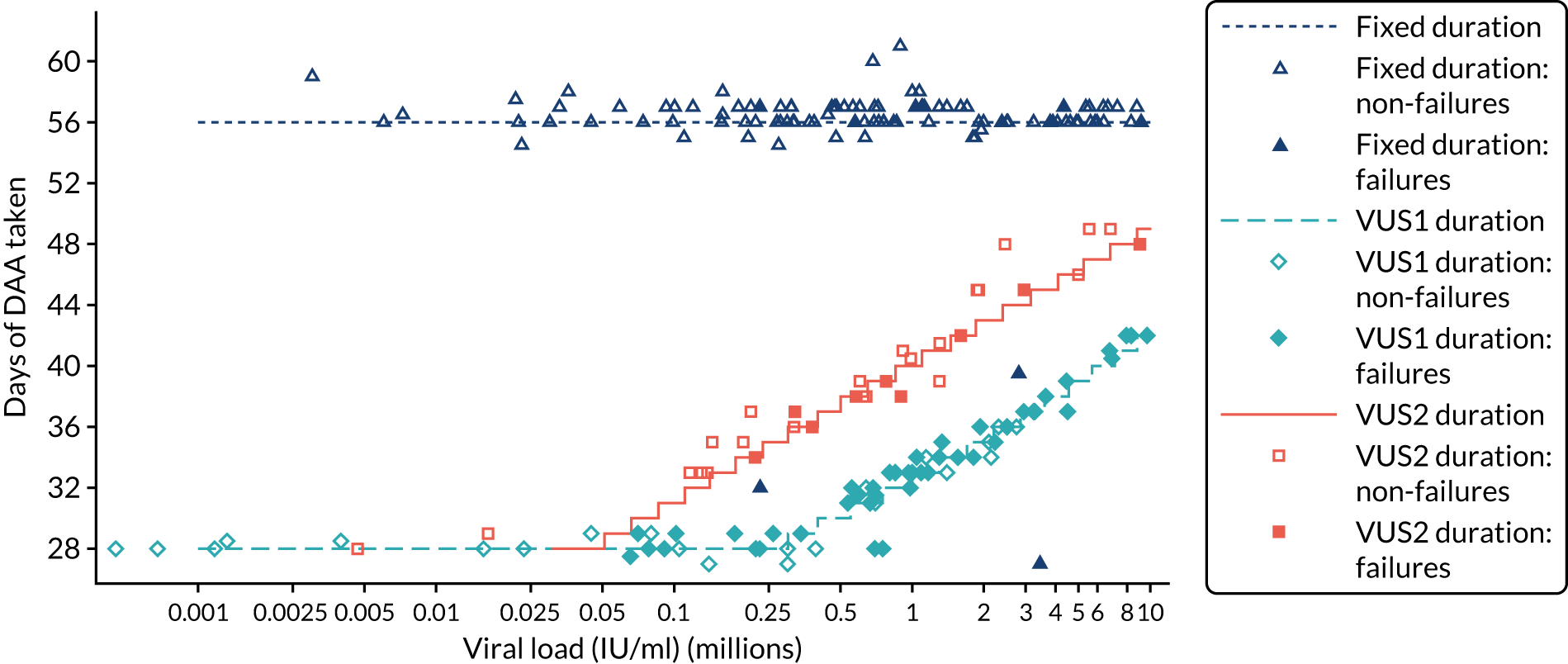
Protocol versions 1.0–4.0 prespecified that the biomarker-stratified duration would be adapted if the upper limit of the 99.9% confidence interval (CI) for the risk difference between variable and fixed duration was < 65%. The adaptation criterion was met at the second meeting of the Data Monitoring Committee (DMC) and the decision was taken by the DMC to change the potential DAA treatment length for variable-duration participants from 4–6 weeks (VUS1) to 4–7 weeks (VUS2), implemented in protocol version 5.0, and illustrated as the solid blue line on Figure 2. An alternative ‘cut-off point’ (shown as dashed blue line in Figure 2) would require a single threshold HCV VL to be chosen and reflects biological variation less well. All participants randomised from 1 April 2017 were treated under VUS2.
FIGURE 2.
Biomarker-stratified variable ultrashort first-line treatment duration from protocol version 5.0 onwards (VUS2).
All patients were prescribed the first 4 weeks of first-line therapy at randomisation and the remaining first-line treatment (as per their randomised group) was provided at the week 4 visit. All patients were offered an optional patient diary card personalised with their specific combination regimen [tablets once daily (OD)/bis in die (twice a day) (b.i.d.)] and treatment duration to help them record pill taking. Any doses missed during the treatment course were to be taken at the end of the prescribed course.
Choice of 8-week first-line fixed-duration control group
Both the ombitasvir/paritaprevir/(dasabuvir)/ritonavir and the sofosbuvir and ledipasvir combinations are licensed as 12-week treatments for the cure of HCV. However, several trials comparing fixed shorter durations had promising results, such that the vast majority of patients are still likely to achieve cure with 8 weeks’ treatment. Glecaprevir/pibrentasvir is licensed as an 8-week treatment without ribavirin. In the trial, therefore, the duration of first-line treatment was fixed at 8 weeks in the control group. All patients who did not achieve cure with 8 weeks’ treatment received retreatment with 12 weeks’ sofosbuvir/ledipasvir/ribavirin in accordance with the protocol, such that their overall probability of being cured within the trial was extremely high.
Trial intervention: drug regimens
The trial allowed three possible first-line drug combinations with which participants could be treated, depending on their genotype and local availability:
-
Viekirax (ombitasvir/paritaprevir/ritonavir) and Exviera (dasabuvir) for genotype 1a/1b
-
Viekirax (ombitasvir/paritaprevir/ritonavir) for genotype 4
-
Maviret® (AbbVie, Chicago, IL, USA) (glecaprevir/pibrentasvir) for genotypes 1a/1b and 4.
The licensed duration of Viekirax, with or without Exviera, was a 12-week treatment course and so the fixed-duration arm represented a shorter than standard course. The licensed duration of Maviret was an 8-week treatment course and so the fixed-duration arm represented the standard course. Viekirax without Exviera was added in protocol amendment 5 and Maviret in protocol amendment 6.0 (see Appendix 2). In practice, very few patients in the trial received these regimens (see Chapter 3).
With all three possible first-line treatments, participants randomised to the VUS arm were also randomised to receive or not receive ribavirin. In the case of participants randomised to the 8-week fixed-duration arm, those taking Viekirax, with or without Exviera, were also randomised to receive or not to receive ribavirin. Participants taking Maviret who were randomised to 8 weeks’ fixed-duration treatment did not receive ribavirin because this 8-week course was already the licensed duration.
Participants in whom first-line treatment failed were offered 12 weeks’ Harvoni (sofosbuvir and ledipasvir) with ribavirin, regardless of their initial DAA regimen.
Ombitasvir/paritaprevir/(dasabuvir)/ritonavir
Ombitasvir/paritaprevir/(dasabuvir)/ritonavir is a triple combination of three DAAs active against HCV genotype 1a/1b and 4 manufactured by AbbVie, namely ombitasvir/paritaprevir/ritonavir (12.5 mg/75 mg/50 mg) co-formulated film-coated tablets OD (total daily dosage: 25 mg/150 mg/100 mg) plus one dasabuvir 250-mg tablet b.i.d. (total daily dosage: 500 mg). Dosing was orally and b.i.d.:
-
morning – two tablets of ombitasvir 12.5 mg/paritaprevir 75 mg/ritonavir 50 mg plus one 250-mg tablet of dasabuvir with food without regard to fat or calorie intake
-
evening – one 250-mg tablet of dasabuvir with food without regard to fat or calorie intake.
Patients with HCV genotype 4 took only ombitasvir/paritaprevir/ritonavir, following the licensing indication.
Patients were instructed that, if vomiting occurred within 6 hours of dosing, then an additional dose of trial drug should be taken. If vomiting occurred > 6 hours after dosing, then no further dose was needed. If a dose of trial drug was missed, then the prescribed dose could be taken within 6 hours. If > 6 hours had passed since the drug was usually taken then the missed dose should not have been taken and the patient should have taken the next dose as per the usual dosing schedule. Patients should have been instructed not to take a double dose. Any doses missed during the treatment course should have been taken at the end of the prescribed course.
Dose modifications and interruptions of ombitasvir/paritaprevir/(dasabuvir)/ritonavir were primarily considered for hepatic impairment. If a patient developed symptomatic hepatitis, or remained asymptomatic but with alanine aminotransferase (ALT) > 10 × upper limit of normal (ULN) and the investigator believed that this could possibly be related to the drug, all HCV drugs were to be ceased. Re-challenge was not to occur until the case had been discussed with the trial team. It was recommended that asymptomatic patients experiencing five or more ULN elevations of ALT be monitored more closely with weekly ALT testing until resolution. In a pooled analysis of ombitasvir/paritaprevir/(dasabuvir)/ritonavir taken with or without ribavirin, 1% of patients experienced elevations of ALT > 5 × ULN [Viekerax, summary of product characteristics (SmPC)]. Most occurred early (mean time 20 days after start of treatment, range 8–57 days), were asymptomatic and resolved without any dose interruption. The strongest association was with being female on ethinyloestradiol-containing contraception and, therefore, the co-administration of contraception containing this form of hormone was contraindicated in the trial. Other oestrogens, such as oestradiol or conjugated oestrogens, were not associated with liver enzyme elevations.
No dose adjustment of Viekirax with or without dasabuvir was required for patients with mild, moderate or severe renal impairment.
In early-phase studies, the highest single dose administered to healthy volunteers was 400 mg in the case of paritaprevir (with 100 mg of ritonavir), and 350 mg in the case of ombitasvir. No adverse events (AEs) were observed, although transient elevations of bilirubin were seen. As per the SmPC in the case of overdose, the patient was to be observed for any AE and symptomatic treatment of any AE initiated. The highest documented single dose of dasabuvir administered to healthy volunteers was 2 g. No study drug-related adverse reactions or clinically significant laboratory abnormalities were observed. In case of overdose, it was recommended that the patient be monitored for any signs or symptoms of adverse reactions or effects and appropriate symptomatic treatment be instituted immediately.
Sofosbuvir/ledipasvir (Harvoni)
The fixed-dose combination of sofosbuvir (400 mg)/ledipasvir (90 mg) was taken OD. Patients were instructed to swallow the tablet whole with or without food. As the film-coated tablet has a bitter taste, it was recommended that it not be chewed or crushed. Patients were instructed that, if vomiting occurred within 5 hours of dosing, then an additional tablet of the trial drug should be taken. If vomiting occurred > 5 hours after dosing, then no further dose was needed. If a dose was missed and it was within 18 hours of the normal time, then patients were instructed to take the tablet as soon as possible and then take the next dose at the usual time. If it was after 18 hours, then patients were instructed to wait and take the next dose at the usual time. Patients were instructed not to take a double dose. Any doses missed during the treatment course should have been taken at the end of the prescribed course.
No dose adjustment of sofosbuvir and ledipasvir was required for patients with mild, moderate or severe hepatic impairment [Child–Pugh–Turcotte (CPT) class A, B or C] or with mild or moderate renal impairment. The safety of sofosbuvir and ledipasvir has not been assessed in patients with severe renal impairment (estimated creatinine clearance < 30 ml/minute/1.73 m2) or end-stage renal disease requiring haemodialysis.
No details on overdoses were provided in the SmPC. Cases of overdose were, therefore, discussed on a case-by-case basis with the trial team.
Glecaprevir/pibrentasvir (Maviret)
The fixed-dose combination of glecaprevir/pibrentasvir is a pangenotypic DAA regimen manufactured by AbbVie. Each film-coated tablet contains 100 mg of glecaprevir and 40 mg of pibrentasvir (total daily dosage of three tablets is 300 mg of glecaprevir and 120 mg of pibrentasvir, taken OD). Patients were instructed to swallow tablets whole with food and not to chew, crush or break the tablets, as this may alter the bioavailability of the agents. Patients were instructed that, if vomiting occurred within 3 hours of dosing, then an additional tablet of the trial drug should be taken. If vomiting occurred > 3 hours after dosing, then no further dose was needed. If a dose was missed and it was within 18 hours of the normal time, then patients were instructed to take the tablet as soon as possible and then take the next dose at the usual time. If it was after 18 hours, then patients were instructed to wait and take the next dose at the usual time. Patients were instructed not to take a double dose. Any doses missed during the treatment course were to be taken at the end of the prescribed course.
No dose adjustment of glecaprevir/pibrentasvir was required in patients with mild hepatic impairment (CPT class A). Glecaprevir/pibrentasvir is not recommended in patients with moderate hepatic impairment (CPT class B) and is contraindicated in patients with severe hepatic impairment (CPT class C). As only patients with mild disease were eligible for the trial, no dose adjustment was necessary. No dose adjustment of glecaprevir/pibrentasvir was required in patients with any degree of renal impairment, including patients on dialysis.
The highest documented doses administered to healthy volunteers was 1200 mg OD for 7 days for glecaprevir and 600 mg OD for 10 days for pibrentasvir. Asymptomatic serum ALT elevations (> 5 × ULN) were observed in 1 out of 70 healthy subjects following multiple doses of glecaprevir (700 or 800 mg) OD for ≥ 7 days. In case of overdose, patients were to be monitored for any signs and symptoms of toxicities, and appropriate symptomatic treatment initiated immediately. All cases of suspected overdose were to be discussed with the trial team.
Ribavirin
Ribavirin film-coated tablets (or hard capsules) contain either 200 or 400 mg of ribavirin per tablet. The standard dose is weight based (Table 2). Ribavirin is administered orally each day in two divided doses (morning and evening) with food. The tablets/capsules should not be chewed or crushed. Patients were instructed that, if vomiting occurred within 6 hours of dosing, then an additional dose of trial drug should be taken. If vomiting occurred > 6 hours after dosing, then no further dose was needed. If a dose was missed, then the prescribed dose could be taken within 6 hours. If > 6 hours had passed since the drug was usually taken then the missed dose should not be taken and the patient should take the next dose as per the usual dosing schedule. Patients were instructed not to take a double dose. Any doses missed during the treatment course were to be taken at the end of the prescribed course.
| Weight-based daily ribavirin dose | Number of 200-mg ribavirin tablets |
|---|---|
| Body weight < 75 kg: 1000 mg | Five 200-mg tablets (two in the morning and three in the evening) |
| Body weight ≥ 75 kg: 1200 mg | Six 200-mg tablets (three in the morning and three in the evening) |
Table 3 provides guidelines for dose modifications and discontinuation based on the patient’s haemoglobin concentration and cardiac status. These were to be applied for ribavirin used as either first-line treatment or retreatment. If ribavirin was withheld because of either a laboratory abnormality or a clinical manifestation, an attempt could be made to restart ribavirin at 600 mg daily and further increase the dose to 800 mg daily. However, it was not recommended that ribavirin be increased to the originally assigned dose (of 1000–1200 mg daily). Intensive monitoring of haemoglobin concentrations, with corrective action as necessary, was employed throughout the treatment period.
| Laboratory parameter | Reduce ribavirin dose to 600 mg/day if . . . | Discontinue ribavirin if . . . |
|---|---|---|
| Haemoglobin in patients with no cardiac disease | < 10 g/dl | < 8.5 g/dl |
| Haemoglobin in patients with history of stable cardiac disease | ≥ 2 g/dl decrease in haemoglobin during any 4-week treatment period | < 12 g/dl despite 4 weeks at reduced dose |
Based on pharmacokinetic modelling and simulation, dose reductions are recommended in patients with significant renal impairment [i.e. creatinine clearance (Cockcroft–Gault) (CrCl) < 50 ml/minute] (Table 4). These adjusted doses were expected to provide ribavirin plasma exposures comparable to those achieved in patients with normal renal function receiving the standard dose. Most of the recommended doses were derived from pharmacokinetic modelling and simulation and have not been studied in clinical trials. Although patients with CrCl < 60 ml/minute were not eligible for the trial, other patients may develop renal impairment during the trial, in which case doses should be adjusted as below. Furthermore, it was possible that those needing retreatment could have developed renal impairment and this was checked before commencing retreatment. Ribavirin was to be initiated, or continued if renal impairment developed while on therapy, with extreme caution in those with CrCl < 50 ml/minute.
| CrCl | Ribavirin dose (daily) |
|---|---|
| 30–50 ml/minute | Alternating doses (200 and 400 mg every other day) |
| < 30 ml/minute | 200 mg daily |
| Haemodialysis | 200 mg daily |
No cases of overdose of ribavirin had been reported in clinical trials. Hypocalcaemia and hypomagnesaemia have been observed in persons administered dosages greater than four times the maximal recommended dosages. In many of these instances, ribavirin was administered intravenously. Owing to the large volume of distribution of ribavirin, significant amounts of ribavirin are not effectively removed by haemodialysis.
Randomisation
Randomisation was performed via a computer-generated program at the STOP-HCV-1 Co-ordinating Centre [MRC Clinical Trials Unit (CTU) at University College London (UCL), London, UK]. Patients were allocated 1 : 1 using a factorial design to each of:
-
biomarker-stratified VUS compared with fixed-duration treatment
-
adjunctive ribavirin or not (a partial factorial in those randomised to a shorter course than the full licensed duration of therapy).
Randomisation was stratified by study centre, HCV genotype and study drug regimen using a minimisation algorithm, incorporating a probabilistic element securely into the online trial database. Randomisation determined the duration of first-line therapy rather than the choice of DAAs, which was prespecified by the investigator before randomisation based on local availability.
Allocation concealment mechanism
Each allocation was generated within the trial database only at the point of randomisation and after it was confirmed that the participant was eligible and was to be randomised. Allocations were generated using minimisation with a probabilistic element and so there was no predetermined allocation sequence to conceal. To further conceal the potential allocation, study centres were not informed of the randomisation strata.
Implementation
On the day of randomisation, participant eligibility was checked at sites and the data confirming eligibility were entered onto a case report form and sent to MRC CTU. The data were entered into the database at MRC CTU and, again, checked for eligibility. Once the participant had been confirmed as eligible, the database would perform randomisation using the computer-generated program. Sites were then informed of the allocation and length of DAA treatment required for the participant.
Blinding
All randomisations were open label and, therefore, there were no unblinding procedures. It would have been infeasible to blind the durations of five different drugs for variable durations from 4 to 8 weeks. Given the lack of blinding, the primary outcome was based on an objective laboratory biomarker (HCV VL).
Assessment and follow-up
All participants were followed by the site teams for 24 weeks after the end of first-line treatment or retreatment (where applicable) for evaluation of virological response and toxicity. Participants on first-line therapy had clinical assessments on days 3, 7, 10, 14 and 28 and at the end of therapy (EOT) (where EOT was not day 28), followed by weeks 2, 4, 8, 12 and 24 after EOT. All outcome measures below were assessed at these clinic visits. All patients failing treatment were retreated as soon as practicable after failure was identified. The schedule of assessment is in Appendix 3.
The trial closed in August 2018, after which time no further recruitment was possible.
Outcomes
Primary outcome measure: biomarker-stratified duration comparison
The primary outcome for the biomarker-stratified duration comparison is the proportion of patients in each randomised group who achieve sustained virological response (persistently undetectable) 12 weeks after end of therapy (SVR12) following first-line treatment (‘first-line SVR12’) and retreatment (where necessary) [i.e. SVR12 across the treatment/retreatment pathway (‘overall SVR12’)].
Sustained virological response (persistently undetectable) (SVR) was defined as undetectable plasma (HCV VL < LLOQ) measured 12 weeks after the EOT (i.e. first-line treatment with or without retreatment) and without failure, defined as:
-
two consecutive measurements of HCV VL greater than the LLOQ (taken at least 1 week apart) after two consecutive visits with HCV VL less than the LLOQ, at any time, with the latter confirmatory measurement also being > 2000 IU/ml
-
two consecutive measurements of HCV VL (taken at least 1 week apart) that are > 1 log10 increase above HCV VL nadir on treatment and > 2000 IU/ml, at any time.
Therefore, for patients who did not fail on first-line treatment (and were not retreated), this was SVR12 after first-line treatment. For those who failed on first-line treatment and start retreatment, this was SVR12 after retreatment. Any patient in whom first-line treatment failed and who chose not to be retreated was counted as a failure.
For the vast majority of patients with SVR12, their HCV VL 8 weeks post EOT was also undetectable (i.e. SVR12 unconfirmed). Any patient whose HCV VL was greater than the LLOQ for the first time 12 weeks post EOT had a second test performed at least 1 week later to confirm failure. Such patients were conservatively assumed to not have achieved SVR12, regardless of the value of the confirmatory test, but continued to be followed closely.
Many studies have shown very strong associations between SVR12 and sustained virological response (persistently undetectable) 24 weeks after end of therapy (SVR24). The latter was used historically to define cure. SVR12 is now the accepted outcome measure for regulatory trials. 20 Durable SVR (at either 12 or 24 weeks) has been shown across many studies to have long-term benefits on clinical outcomes, including all-cause mortality, progression of liver disease and hepatocellular carcinoma. 5
The primary end point of the trial was the overall cure rate after first-line treatment and retreatment, specifically to address the question as to whether or not failing on a shorter duration of treatment ultimately affects overall chance of cure, or whether or not the percentage of patients cured with shorter treatment can make it more cost-effective to give everyone shorter courses initially and then retreat those who do not achieve cure. This was a non-inferiority comparison. The specific first-line cure rates are not critical to answering either of these questions.
Primary outcome measure: ribavirin comparison
For the ribavirin comparison, the primary outcome was the proportion of patients in each randomised group who achieved SVR12 following first-line treatment, assessed 12 weeks after EOT (i.e. first-line SVR12).
The reason for focusing on first-line cure for the ribavirin comparison is the hypothesis that adjunctive ribavirin is superior (i.e. it will increase cure rates) and that retreatment will be successful in curing all patients who fail first-line treatment. Therefore, the primary interest was in the impact of ribavirin on first-line cure.
Secondary outcome measures
Secondary outcomes for all randomised comparisons (where not the primary outcome measure) were:
-
proportion of patients achieving SVR12 following first-line therapy (stratified duration comparison)
-
proportion of patients achieving SVR12 overall following first-line treatment plus retreatment therapy (ribavirin comparison)
-
sustained virological response 24 weeks after completion of all therapy (overall SVR24)
-
sustained virological response 24 weeks after completion of first-line therapy only (first-line SVR24)
-
proportion of patients with primary first-line treatment failure (confirmed > 1 log10 increase from HCV VL nadir on treatment and > 2000 IU/ml)
-
VL rebound (i.e. HCV VL greater than the LLOQ) after two consecutive visits with HCV VL less than the LLOQ, with the latter confirmatory measurement also being > 2000 IU/ml (on first-line therapy and after stopping first-line therapy)
-
proportion of patients with detectable HCV VL 4 weeks after randomisation
-
proportion of patients with one or more serious adverse events (SAEs)
-
proportion of patients with one or more severe (grade 3/4) AEs
-
proportion of patients with one or more grade 3/4 AEs judged definitely/probably related to one or more study medications
-
proportion of patients requiring any change to study medication because of AEs
-
proportion of patients with grade 3/4 anaemia
-
proportion of patients with emergent resistance-associated substitutions (RASs)
-
overall total treatment cost and treatment cost per cure
-
sensitivity and specificity of Epistem’s (Epistem Ltd, Manchester UK) diagnostic platform for detecting presence of IL-28B T allele.
Adverse events were graded using the toxicity gradings in the Division of AIDS (DAIDS) Table21 for grading the severity of adult and paediatric AEs.
Hepatitis C virus genome sequences were generated using next-generation sequencing with probe enrichment in a single laboratory, as previously described. 22 Briefly, RNA was extracted from 500 µl of plasma using the NucliSENS® magnetic extraction system (bioMérieux, Basingstoke, UK). Libraries were prepared using the NEBNext® Ultra Directional RNA Library Prep kit for Illumina (New England BioLabs Inc., Hitchin, UK) and quantified before pooling into equimolar proportions. A 500-ng aliquot of the pooled library was enriched using the xGen Lockdown protocol from Integrated DNA Technologies (Coralville, IA, USA). Enriched pools were reamplified (12 cycles), repurified and normalised using quantitative polymerase chain reaction before a single run with 150-basepaired-end reads was performed using the Illumina MiSeq system (v2 chemistry, San Diego, CA, USA).
Methods to protect against bias included the use of a ‘failure’ primary outcome measure that is based on a routine laboratory test assayed without knowledge of randomisation and, therefore, not subject to clinical opinion. The test (HCV VL) is widely used in clinical practice and all centres in the trial use laboratories that participate in external quality assurance programmes. Randomisation was stratified by centre. Therefore, even if there were very small differences between laboratories, these would not bias the randomised comparison. All patients followed the same visit schedule after EOT, ensuring that measurement frequency was identical.
Sample size
The trial was originally powered to demonstrate:
-
non-inferiority of biomarker-stratified variable ultrashort 4- to 7-week first-line treatment followed by 12 weeks’ retreatment compared with fixed 8-week first-line treatment with the same retreatment
-
superiority of adjunctive ribavirin in first-line treatment.
The primary end point for the non-inferiority comparison is overall SVR12 after first-line treatment and retreatment (where necessary), which was estimated at 98% for the control group, regardless of first-line combination, given the very high cure rates achieved with the 12-week ribavirin-containing regimens that will be used for retreatment and the limited impact of prior DAAs treatment on response to subsequent regimens.
As an example, from previous trials,23 we can assume 96% and 84% SVR12 with 8 weeks’ ombitasvir/paritaprevir/dasabuvir/ritonavir in patients with genotypes 1b and 1a, respectively. With a 1 : 2 ratio of presenting cases (reflecting UK prevalence24), the cure rate in the control first-line group would be 88% prior to retreatment. Conservatively assuming a cure rate of retreatment of 85% to allow for potential role of mutations, particularly in the NS5A gene, would lead to an overall 98% cure rate in the control group. However, similar overall 98% cure rates could be achieved with lower first-line treatment and higher retreatment cure rates (e.g. 65% first-line treatment and 94% retreatment) or higher first-line treatment and lower retreatment cure rates (e.g. 95% first-line treatment and 60% retreatment). Although first-line cure rates may be slightly lower or higher with 8 weeks of different first-line combinations (in particular, first-line cure rates might be expected to be higher with 8 weeks’ glecaprevir/pibrentasvir, as this is the licensed indication, albeit without much real-world experience to date), in practice, it is unlikely that an overall cure rate of 98% from first-line treatment plus retreatment can be exceeded, and so the control group rate of 98% is reasonable across different first-line regimens.
Assuming a 98% cure rate overall for the control group, for a 4% non-inferiority margin, 80% power, a one-sided test and an alpha of 0.025, the required sample size for the biomarker-stratified duration comparison is 408 patients, allowing for 5% early withdrawal.
The 4% non-inferiority margin is arbitrary, but ensures that overall cure rates in the biomarker-stratified short-course group would be well over 90% if the trial were to declare non-inferiority. Furthermore, even small genuine reductions in overall cure rate with short-course treatment substantially decrease the trial’s power to demonstrate non-inferiority [i.e. a reduction of 52%, 25% and 11% of short-course treatment (from 80%) genuinely achieves overall cure rates that are 1%, 2% or 3% lower, respectively].
If non-inferiority is not demonstrated, a total of 408 patients is likely to provide reasonable power to investigate other predictors of cure, such as presence of viral quasispecies, including resistance, age (related to immune health), IL-28 polymorphisms and BMI.
The calculation of sample size for the fixed and duration non-inferiority comparison is conducted under the null hypothesis for the ribavirin superiority comparison (i.e. no effect). Given its partial factorial nature, estimates of the effect of adjunctive ribavirin are determined from generalised linear models, which include terms to reflect the randomisations and the specific first-line DAA regimen received. Therefore, patients randomised to 8 weeks’ glecaprevir/pibrentasvir effectively do not contribute to this comparison. When protocol version 6.0 was approved, we estimated that this would be approximately 25% of patients (n = 102). In practice, only two patients received this regimen in the trial (see Chapter 3). A total of 306 patients randomised to adjunctive ribavirin (or not) provides 75–85% power to identify a 10% improvement in first-line cure rate associated with adjunctive ribavirin for first-line cure rates of 83–86% without ribavirin and of 93–96% with ribavirin (two-sided alpha = 0.05), and > 80% power to identify a 15% improvement in first-line cure rate associated with adjunctive ribavirin for first-line cure rates of 60–80%, allowing for 5% early withdrawal, as above.
Interim monitoring and analyses
The protocol prespecified that the DMC would meet approximately 6-monthly, and four 6-monthly meetings took place. The protocol prespecified an adaptation in the case that the upper limit of the 99.9% CI for the risk difference between variable and fixed duration was < 65%. The adaptation criterion was met at the second DMC meeting on 10 April 2017 and the decision was taken by the DMC to change the potential DAA treatment length for variable-duration participants from 4–6 weeks (VUS1) to 4–7 weeks (VUS2).
Statistical methods
Analyses followed the principle of intention to treat, including all follow-up, regardless of changes to treatment. The statistical analysis plan (SAP) prespecified that any patient who was randomised in error (defined as the realisation that the patient should not have been randomised before taking study drug and not ever taking study drug) and, hence, not followed up would be excluded.
A patient was formally considered as lost to follow-up (LTFU) if they had not been seen at the final EOT plus 24 weeks visit within a –6- to +12-week window. If a patient was LTFU before the visit at which an outcome was measured (EOT plus 12 weeks for SVR12 and EOT plus 24 weeks for SVR24), then the following methods were prespecified in the SAP, but not in the protocol, to be used to determine the patient’s outcome:
-
If the missing HCV VL was between two undetectable measurements, then it was assumed to be undetectable.
-
HCV VL results from local practice were sought for patients with missing SVR12 and SVR24 outcomes. If the local result was undetectable, then the patient was assumed to be undetectable at EOT plus 12 weeks/EOT plus 24 weeks. (One patient had a detectable local VL, but as they had no confirmatory subsequent VL they were not considered to have failed and were also not counted as a cure.)
After following these methods, the percentage of patients without an outcome was < 10% and so the analysis was restricted to complete cases (as prespecified in the SAP).
Primary analyses of outcomes restricted to first-line therapy were stratified by first-line DAA strategy in place [VUS1 (before 1 April 2017) or VUS2 (after 1 April 2017)] as a main effect, and as an interaction with randomised group (fixed duration vs. variable duration), where the p-value for the interaction term was < 0.05. For analyses of SVR12 after first-line therapy only, analysis was also performed separately within the VUS1/VUS2 strata. Primary analyses of outcomes, including retreatment, were unstratified, reflecting the overall strategy comparison and because no patients failed after receiving retreatment. Primary analyses were not stratified by centre, given the large number of centres with small numbers of patients recruited.
Primary analysis of the primary end point included all randomised participants other than those considered randomised in error (following the SAP) and for whom no VL data could be obtained. A per-protocol analysis (prespecified in the protocol) included patients receiving > 90% and < 110% of the prescribed duration of first-line treatment and where the difference between screening and enrolment HCV VL values would have led to a difference of ≤ 2 days in allocated duration of DAAs had they been allocated to the variable-duration group. Secondary analyses were conducted considering all LTFU patients as failures and all LTFU patients as cured. An additional secondary analysis excluding reinfections identified by genome sequencing, which was prespecified in the protocol, was not performed as no reinfections were identified.
For the primary analysis, a risk difference and 95% CI were obtained from a binomial regression on the risk difference scale using a generalised linear model. Kaplan–Meier plots and Cox proportional hazard models were used for analyses of time until failure (any type). Secondary analyses of primary treatment failure and VL rebound (i.e. the components of overall treatment failure) used competing risks methods (e.g. cumulative incidence plots and subhazard ratios) to account for the possibility that the patient would experience the other type of failure. Binomial generalised estimating equations with an independent working correlation were used to analyse the percentage of patients with undetectable HCV VL at each time point.
Safety outcomes were analysed using chi-squared p-values, and Cox proportional hazard models were also used. To assess the change in laboratory values over time (other than for HCV VL), generalised estimating equations (normal distribution) with an independent correlation structure adjusted for baseline values were used. Sensitivity analyses of changes in laboratory values used alternative error structures and mixed-effects models, but these provided results similar to those of the primary analysis.
Baseline values of laboratory test results were those taken closest to randomisation. No laboratory test results taken after randomisation were used for baseline. HCV VL was log10 transformed for analysis as a continuous variable. Other continuous measures were transformed using Box–Cox transformations when there were gross (p < 0.0001) deviations from normality, as assessed using the Shapiro–Wilk test. Analyses of measurements at a given point in follow-up used the closest available measurement to that time point in evenly spaced windows. If a visit fell in two visit windows, then it was classed as belonging to the later window, except where this led to no visits within in the first window and two within the second, in which case it was classed as belonging to the first visit window.
Subgroup analyses were conducted to assess the consistency of effects across different participant characteristics. All subgroup analyses were adjusted for the interaction between VUS strategy and duration randomisation because it was highly significant in the primary analysis. For the duration comparison, interaction tests within binomial models on the risk difference scale were used for subgroup analyses. For the ribavirin comparison, owing to non-convergence of the models, p-values were obtained from marginal effects after logistic regression for subgroups. Heterogeneity p-values for IL-28B polymorphisms considering CC/CT/TT genotype as an ordinal factor were obtained from ordered logistic regression. Continuous factors were categorised into terciles, as well as using fractional polynomial models. Heterogeneity p-values could not be estimated for all subgroups because of small numbers or perfect prediction. No formal adjustment for multiple testing was made for subgroup analyses.
Protocol changes
The trial was approved by Cambridge South Research Ethics Committee (reference 15/EE/0435) and the Medicines and Healthcare products Regulatory Agency. See Appendix 2 for changes to the protocol.
Chapter 3 Results
Recruitment and participant allocation
Recruitment opened on 17 March 2016, with the first participant randomised on 18 March 2016, and closed on 31 August 2018, with the last participant randomised on 28 August 2018. In total, 217 individuals were screened for entry to the trial and 204 participants were randomised to variable ultrashort treatment of 4–7 weeks (n = 102) or DAA treatment for a fixed duration of 8 weeks (n = 102), and to receive adjunctive ribavirin (n = 101) or not (n = 103) (Figure 3). The most common reason for not randomising a participant was that their screening HCV VL was too high (n = 4). Two participants were not eligible because they had not been infected with HCV for ≥ 6 months. Other reasons for ineligibility were infection with HCV of a genotype other than 1a/1b or 4, a FibroScan result that was too high, having been previously exposed to DAAs for the current infection, having low estimated CrCl, receiving contraindicated medication, not using effective contraception, not attending randomisation visit and moving away during screening (n = 1 for each).
FIGURE 3.
Participant flow diagram. Note that individuals can have more than one reason for exclusion.
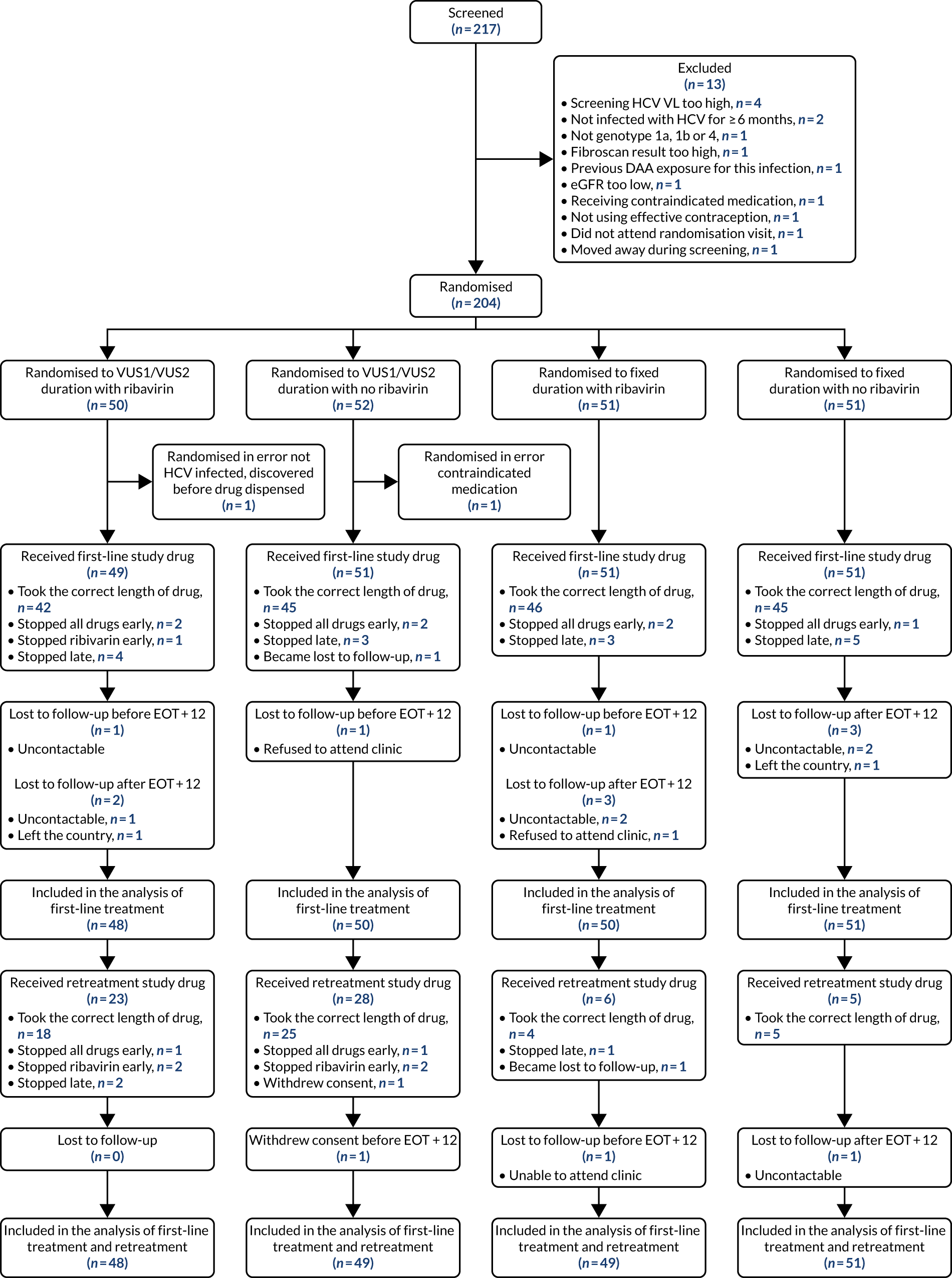
Two participants were randomised in error, defined as never intended to be randomised (e.g. data entry error, not infected with HCV, study drug never being dispensed), with the error realised and notified immediately. Therefore, these two participants were excluded from all analyses, as prespecified in the SAP. One of these participants was receiving contraindicated medication. The other was confirmed to not be infected with HCV after randomisation but prior to drugs being dispensed.
Recruitment
The trial stopped recruiting after randomising 204 participants because of slow recruitment and the lack of available patients at participating centres. Follow-up continued until the last participant’s last visit (24 weeks after EOT) on 4 April 2019.
Baseline characteristics
Sixty-two (31%) participants were female; participants’ median age was 45 [interquartile range (IQR) 37–53] years and their median FibroScan score was 4.9 (IQR 4.2–5.8) kPa (Table 5). Sixty-eight (34%) participants were co-infected with HIV.
| Characteristic | Total (N = 202) | Treatment arm | |||
|---|---|---|---|---|---|
| VUS duration (N = 100) | Fixed duration (N = 102) | Ribavirin (N = 100) | No ribavirin (N = 102) | ||
| Randomised under first protocol (VUS1), n (%) | 136 (67) | 68 (68) | 68 (67) | 68 (68) | 68 (67) |
| Age (years), median (IQR) | 45.5 (37.5–53.0) | 45.2 (38.8–51.6) | 46.3 (36.6–54.1) | 46.1 (36.7–52.4) | 44.8 (37.7–54.1) |
| Female at birth, n (%) | 62 (31) | 28 (28) | 34 (33) | 34 (34) | 28 (27) |
| BMI (kg/m2), median (IQR) | 24.9 (22.2–27.2) | 24.9 (22.6–26.7) | 24.9 (21.8–27.7) | 23.7 (21.7–26.5) | 25.8 (23.3–27.6) |
| White ethnicity, n (%) | 176 (87) | 89 (89) | 87 (85) | 89 (89) | 87 (85) |
| Screening HCV VL (IU/ml), median (IQR) | 711,423 (218,776–1,995,262) | 790,664 (214,388–1,917,731) | 687,916 (220,000–2,381,846) | 700,272 (169,717–2,071,064) | 750,523 (275,000–1,949,844) |
| Enrolment HCV VL (IU/ml) (n = 199), median (IQR) | 741,946 (249,097–1,872,136) | 801,000 (251,188–1,500,000) | 614,047 (248,000–2,238,721) | 657,858 (178,842–1,500,000) | 801,000 (385,595–2,200,000) |
| HCV genotype/subgenotype, n (%) | |||||
| 1a | 166 (82) | 82 (82) | 84 (82) | 84 (84) | 82 (80) |
| 1b | 34 (17) | 17 (17) | 17 (17) | 16 (16) | 18 (18) |
| 4 | 2 (1) | 1 (1) | 1 (1) | 0 | 2 (2) |
| HIV co-infected, n (%) | 68 (34) | 32 (32) | 36 (35) | 35 (35) | 33 (32) |
| FibroScan result (kPa), median (IQR) | 4.9 (4.2–5.8) | 5.0 (4.3–5.9) | 4.8 (4.1–5.5) | 4.8 (4.4–5.8) | 4.9 (4.1–5.9) |
| Haemoglobin (g/dl), median (IQR) | 14.7 (14.0–15.6) | 14.8 (14.1–15.6) | 14.7 (13.8–15.6) | 14.7 (13.8–15.6) | 14.8 (14.0–15.7) |
| ALT (IU/l), median (IQR) | 52 (34–87) | 50 (34–90) | 54 (34–87) | 51 (35–89) | 54 (31–87) |
| AST (IU/l) (n = 189), median (IQR) | 38 (30–57) | 38 (29–57) | 38 (31–58) | 39 (31–55) | 38 (29–58) |
| ALP (IU/l), median (IQR) | 72 (59–91) | 71 (59–87) | 75 (59–94) | 76 (61–95) | 69 (58–85) |
| CrCl (ml/minute), median (IQR) | 109 (93–131) | 109 (94–126) | 109 (92–138) | 107 (92–126) | 110 (93–133) |
| Total bilirubin (μmol/l), median (IQR) | 9 (6–12) | 8 (6–11) | 9 (6–12) | 9 (6–12) | 9 (6–12) |
| IL-28B genotype, n (%)a | |||||
| CC | 60 (30) | 32 (32) | 28 (27) | 29 (29) | 31 (30) |
| CT | 106 (52) | 51 (51) | 55 (54) | 56 (56) | 50 (49) |
| TT | 27 (13) | 14 (14) | 13 (13) | 11 (11) | 16 (16) |
| No result | 9 (4) | 3 (3) | 6 (6) | 4 (4) | 5 (5) |
| RAS to any prescribed first-line drug (N = 188), n (%) | 27 (14) | 10 (11) | 17 (18) | 16 (17) | 11 (12) |
| Current/recent alcoholism/alcohol abuse, n (%) | 13 (6) | 5 (5) | 8 (8) | 7 (7) | 6 (6) |
| Current/recent illicit substance abuse, n (%) | 64 (32) | 31 (31) | 33 (32) | 28 (28) | 26 (25) |
| Ever spontaneously cleared and reinfected, n (%) | 6 (3) | 4 (4) | 2 (2) | 2 (2) | 4 (4) |
| Ever successfully treated with interferon and/or ribavirin and reinfected, n (%) | 10 (5) | 5 (5) | 5 (5) | 5 (5) | 5 (5) |
| Previously unsuccessfully treated with interferon and/or ribavirin, n (%) | 24 (12) | 12 (12) | 12 (12) | 11 (11) | 13 (13) |
| Intolerant relapser, n (%) | 6 (26) | 3 (25) | 3 (27) | 1 (10) | 5 (38) |
| Relapser after full treatment, n (%) | 7 (30) | 5 (42) | 2 (18) | 4 (40) | 3 (23) |
| Non-responder, n (%) | 8 (35) | 3 (25) | 5 (45) | 5 (50) | 3 (23) |
| Breakthrough on treatment, n (%) | 2 (9) | 1 (8) | 1 (9) | 0 | 2 (15) |
| Modes of HCV infection, n (%) | |||||
| No known risk factor (n = 197) | 18 (9) | 7 (7) | 11 (11) | 9 (9) | 9 (9) |
| Injecting drug use (n = 200) | 99 (50) | 51 (51) | 48 (48) | 50 (51) | 49 (49) |
| Blood/blood products (n = 197) | 11 (6) | 7 (7) | 4 (4) | 6 (6) | 5 (5) |
| Perinatal exposure (n = 197) | 4 (2) | 0 | 4 (4) | 3 (3) | 1 (1) |
| Known HCV-positive sexual partner (n = 197) | 21 (11) | 11 (11) | 10 (10) | 8 (8) | 13 (13) |
| Born abroad (n = 197) | 27 (14) | 12 (12) | 15 (15) | 14 (14) | 13 (13) |
| High-risk sexual partner (n = 201) | 71 (35) | 37 (37) | 34 (34) | 35 (35) | 36 (36) |
| Tattoo (n = 197) | 27 (14) | 14 (14) | 19 (19) | 14 (14) | 19 (19) |
| Health-care exposure (n = 197) | 19 (10) | 8 (8) | 11 (11) | 11 (11) | 8 (8) |
| Other (n = 196) | 20 (10) | 11 (11) | 9 (9) | 10 (10) | 10 (10) |
| Treated with paritaprevir/ombitasvir/dasabuvir/ritonavir, n (%) | 198 (98) | 98 (98) | 100 (98) | 100 (100) | 98 (96) |
| Treated with paritaprevir/ombitasvir/ritonavir, n (%) | 2 (1) | 1 (1) | 1 (1) | 0 | 2 (2) |
| Treated with glecaprevir/pibrentasvir/ritonavir, n (%) | 2 (1) | 1 (1) | 1 (1) | 0 | 2 (2) |
Median screening VL was 711,423 (IQR 218,776–1,995,262) IU/ml and median enrolment VL was slightly higher, at 741,946 (IQR 249,097–1,872,136) IU/ml (Lin’s concordance coefficient 0.84), in samples taken a median of 19 (IQR 13–33) days apart. A total of 166 (82%) patients were infected with genotype 1a, 34 (17%) were infected with genotype 1b and two (1%) were infected with genotype 4. Sixty (30%) patients had the CC genotype of the IL-28B gene, 106 (52%) had the CT genotype and 27 (13%) had the TT genotype, with the genotype unknown in nine (4%) participants. Of the 188 participants with baseline sequencing available for post-trial analysis, 27 (14%) had a RAS to any prescribed first-line drug.
Twenty-four (12%) participants had previously been treated unsuccessfully with interferon and ribavirin for their current infection, 10 (5%) had been successfully treated for a previous infection and three (2%) had spontaneously cleared a previous infection. The most common causes of HCV infection were injecting drug use (n = 99, 50%) and having a high-risk sexual partner (n = 71, 35%). Sixty-four (32%) participants had current or recent illicit substance abuse and 13 (6%) had current or recent alcohol abuse.
All but four (2%) participants received ombitasvir/paritaprevir/ritonavir plus dasabuvir as their first-line treatment (see Table 5).
Follow-up and treatment received
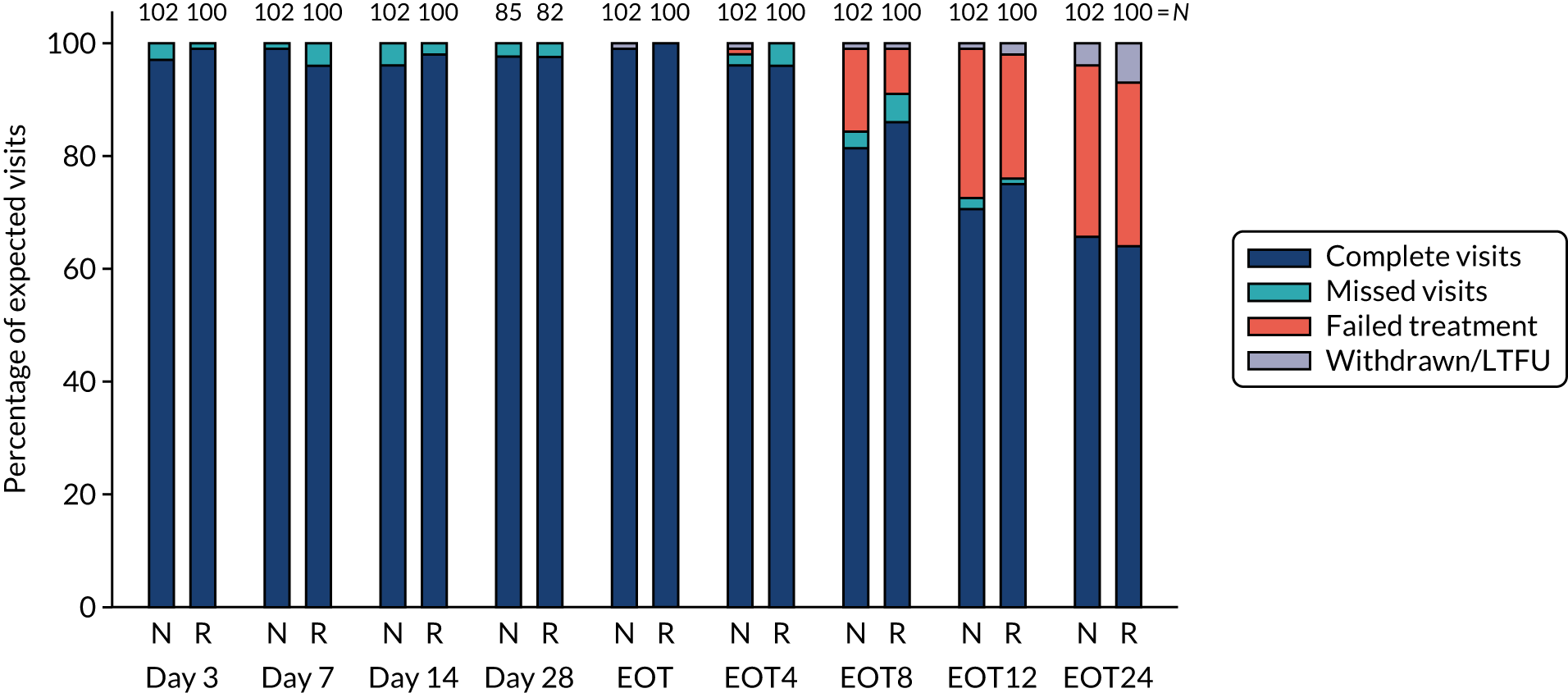
Overall, first-line follow-up was very good, with only a small number of visits missed [four (2%) at day 3, five (2%) at day 7, six (3%) at day 14, four (2%; n = 167 expected) at day 28, none at EOT, six (3%) at EOT plus 4 weeks, eight (4%) at EOT plus 8 weeks and three (1%) at EOT plus 12 weeks] (Figures 4 and 5). In total, 13 (6%) participants were LTFU and one participant withdrew consent (Table 6). Eleven (5%) participants were LTFU during first-line treatment, but eight of these participants missed the last visit only (i.e. EOT plus 24 weeks) and attended the visit at which the primary outcome was measured (i.e. EOT plus 12 weeks). In the case of the other three participants who were LTFU while on first-line treatment, the last visit was in one case on day 28, in one case at EOT plus 4 weeks and in one case at EOT plus 8 weeks. Follow-up for retreatment was similarly good, with three (5%) of the visits missed at week 2, one (2%) visit missed at week 4, two (3%) visits missed at week 8, two (3%) visits missed at EOT, four visits (6%) visits missed at EOT plus 4 weeks, 10 (16%) visits missed at EOT plus 8 weeks and three (5%) visits missed at EOT plus 12 weeks (Figure 6). During the retreatment phase, one participant withdrew consent (last visit at week 4) and two participants became LTFU (last visits at week 2 and EOT plus 12 weeks).
FIGURE 4.
First-line follow-up by duration randomisation.
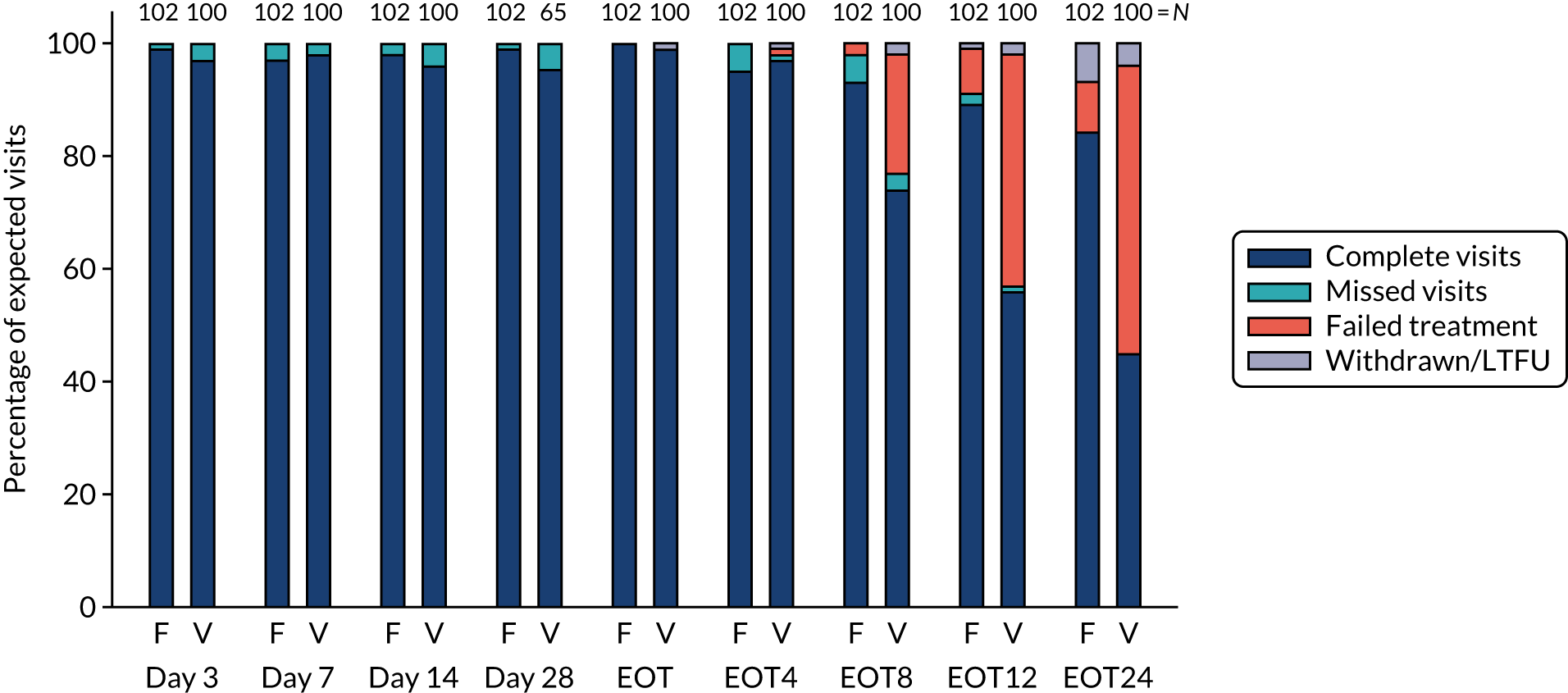
FIGURE 5.
First-line follow-up by ribavirin randomisation.
| Follow-up | Treatment arm | Total (N = 202) | |||
|---|---|---|---|---|---|
| Varying duration with ribavirin (N = 49) | Varying duration with no ribavirin (N = 51) | Fixed duration with ribavirin (N = 51) | Fixed duration with no ribavirin (N = 51) | ||
| Median (IQR) [range] weeks from randomisation to last visit | 30 (29–53) [8–82] | 47 (29–53) [4–62] | 32 (32–34) [32–67] | 32 (32–33) [20–80] | 32 (30–50) [4–82] |
| Died, n | 0 | 0 | 0 | 0 | 0 |
| Withdrew consent, n (%) | 0 | 1 (2) | 0 | 0 | 1 (< 1) |
| LTFU (did not withdraw consent), n (%) | 3 (6) | 1 (2) | 5 (10) | 4 (8) | 13 (6) |
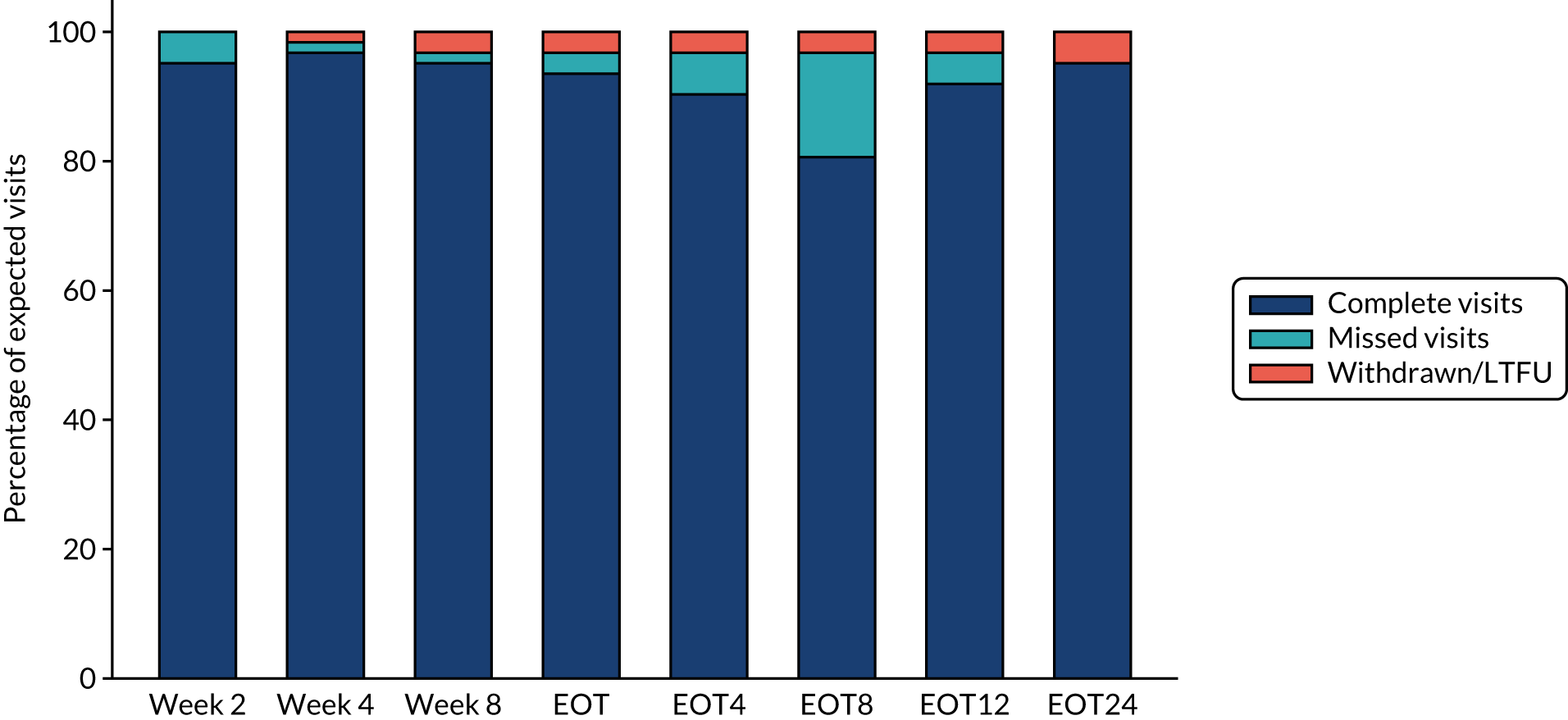
FIGURE 6.
Retreatment follow-up.
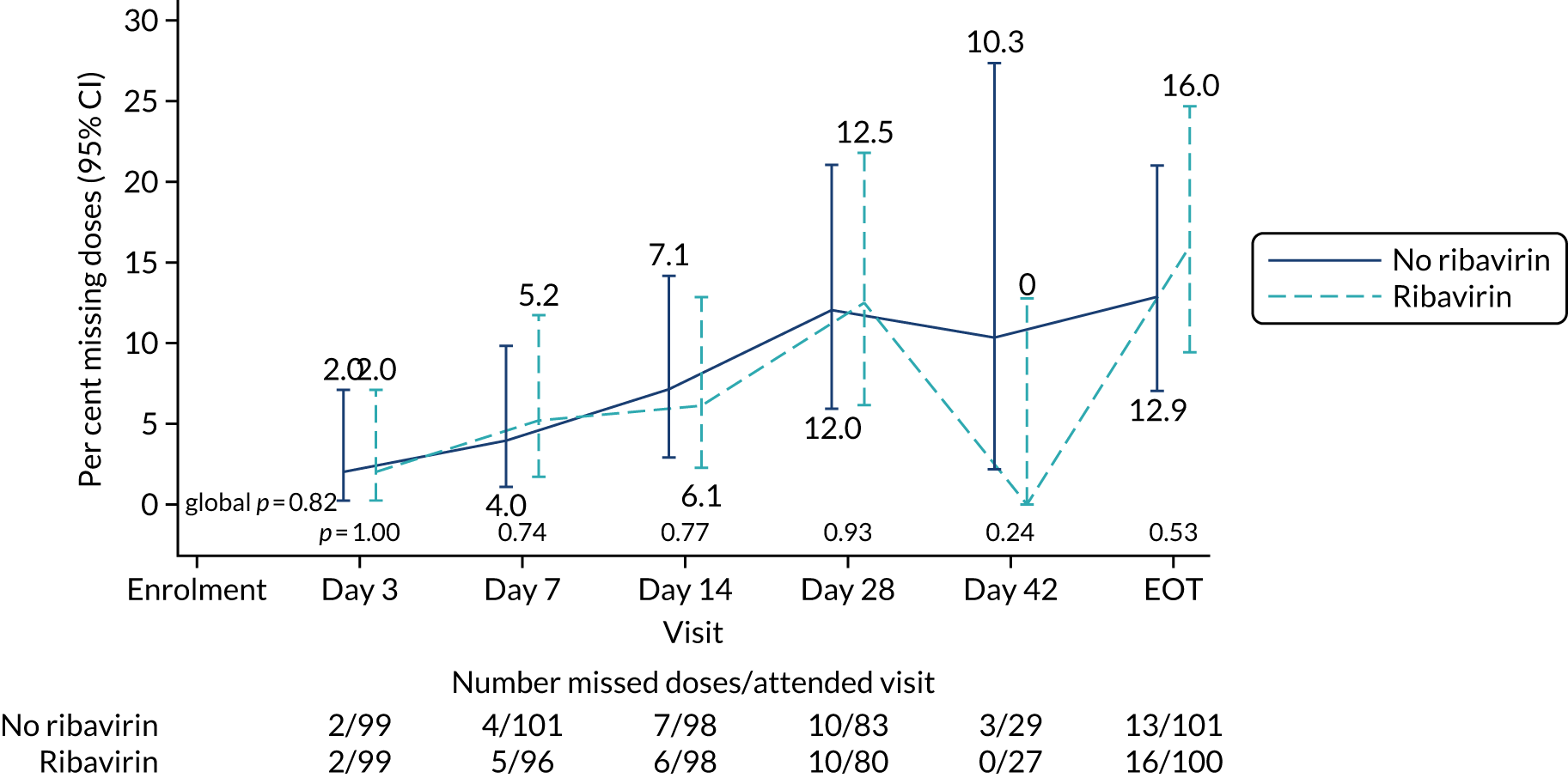
The mean number of days of first-line DAA treatment was 56 (SD 4.2) in the fixed-duration arm and 35 (SD 5.7) in the VUS arm, with those randomised to VUS1 taking 32 (SD 4.2) days and those randomised to VUS2 taking 39 (SD 5.6) days (Figure 7). One participant was LTFU before completing first-line treatment, seven stopped DAAs early, one stopped ribavirin early and seven stopped late (Table 7). Those participants stopping late were not prescribed more than their allocated duration (the difference resulted from taking any missed doses at the end of treatment, as instructed) (see Chapter 2). Among those participants stopping early, two chose to do so, a further two stopped because of AEs, two missed doses and did not take these at the end of treatment, one lost 4 days’ worth of drugs and one had drug supply issues. Fifty-five (28%) participants reported missing at least one first-line dose, with 40 (20%) participants reporting missing a dose only once. The percentage of participants reporting a missed dose increased with time on treatment, rising to 29 (14%) participants reporting a missed dose at their EOT visit (Figures 8 and 9).
FIGURE 7.
Randomised and actual treatment duration by failure at SVR24.
| Adherence | Treatment arm, n (%) | Total (N = 198), n (%) | |||
|---|---|---|---|---|---|
| Varying duration (N = 98) | Fixed duration (N = 100) | With ribavirin (N = 100) | Without ribavirin (N = 98) | ||
| Any missed doses reported | 23 (23) | 32 (32) | 29 (29) | 26 (27) | 55 (28) |
| Number of forms reporting missed doses | |||||
| 0 | 75 (77) | 68 (68) | 71 (71) | 72 (73) | 143 (72) |
| 1 | 20 (20) | 20 (20) | 22 (22) | 18 (18) | 40 (20) |
| 2 | 1 (1) | 8 (8) | 5 (5) | 4 (4) | 9 (5) |
| 3–5 | 2 (2) | 4 (4) | 2 (2) | 4 (4) | 6 (3) |
| LTFU or withdrew consent before EOT | 1 (7) | 0 | 0 | 1 (1) | 1 (1) |
| Stopped DAA before EOT | 4 (4) | 3 (3) | 4 (4) | 4 (4) | 7 (4) |
| Stopped ribavirin only early before EOT | 1 (1) | 0 | 1 (1) | 1 (1) | 1 (1) |
| Stopped late | 7 (7) | 8 (8) | 7 (7) | 7 (7) | 15 (8) |
| Reasons for stopping early (% stopped early) | |||||
| Participant choice | 0 | 2 (67) | 2 (40) | 0 | 2 (25) |
| AEa | 2 (40) | 0 | 2 (40) | 0 | 2 (25) |
| Other | 3 (60) | 1 (33) | 1 (20) | 3 (100) | 4 (50) |
| Reasons for dose or frequency change (% changes) | |||||
| AEb | 1 (100) | 1 (100) | 2 (100) | 0 | 2 (100) |
FIGURE 8.
Reported first-line missed doses by duration randomisation.
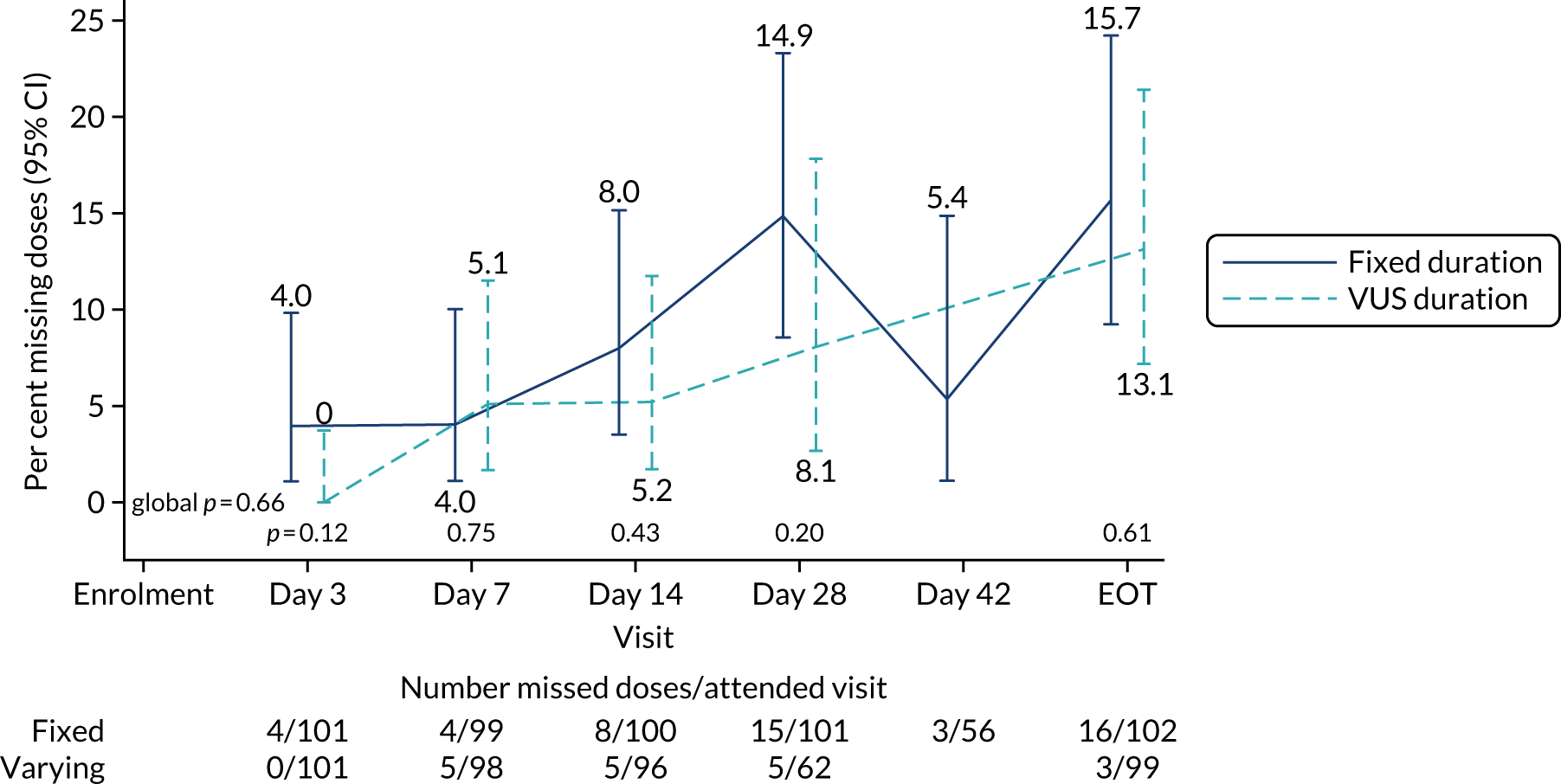
FIGURE 9.
Reported first-line missed doses by ribavirin randomisation.
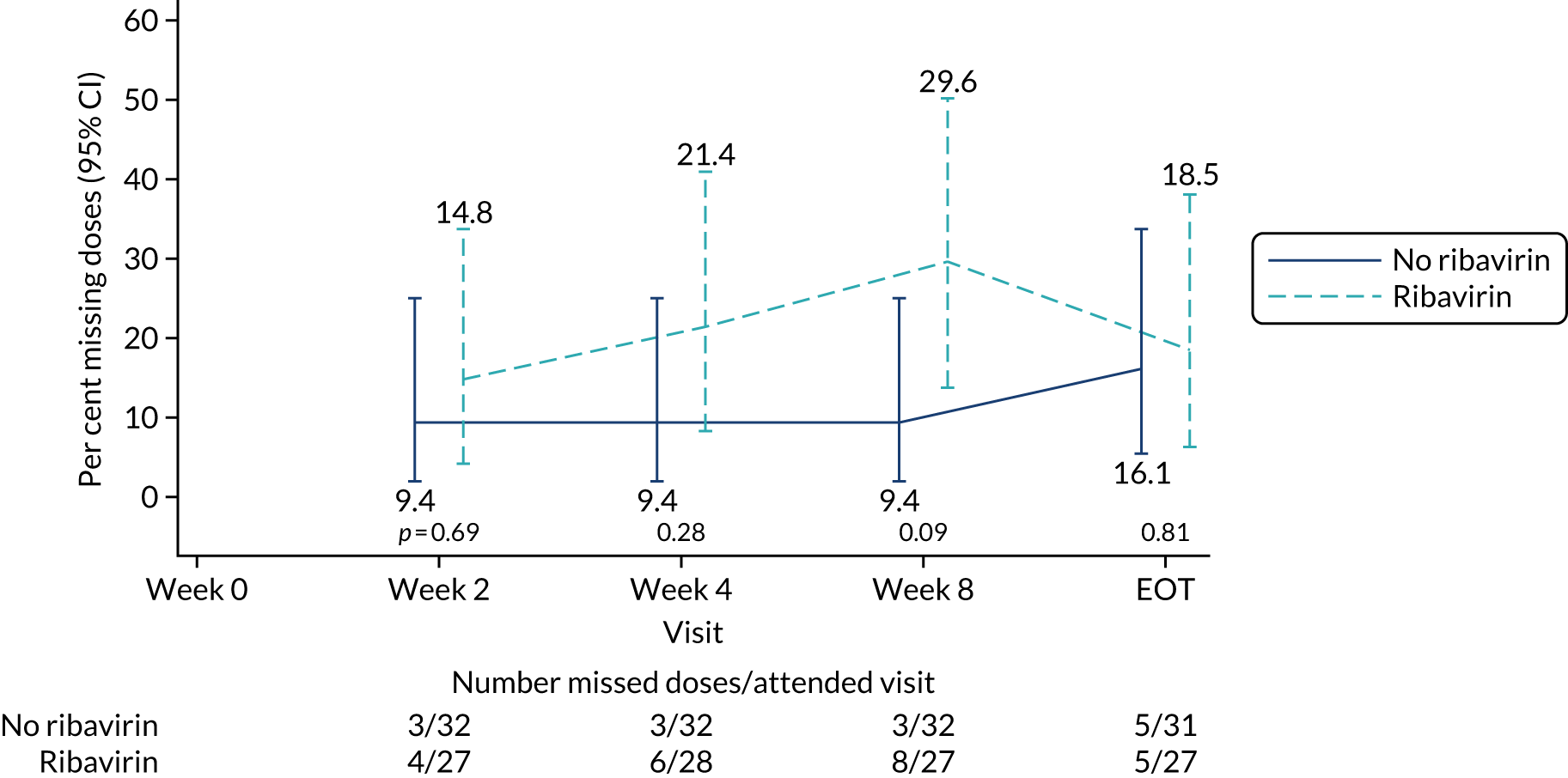
The mean days of first-line plus retreatment DAAs was 64 (SD 24.3) in the fixed-duration arm and 77 (SD 42.8) in the VUS arm, with those randomised to VUS1 taking 85 (SD 43.2) days and those randomised to VUS2 taking 63 (SD 36.8) days. One participant was LTFU, one withdrew consent before completing retreatment, two stopped all retreatment early, three stopped ribavirin early and three stopped late (Table 8). Of those participants stopping early, two did so because of AEs and three missed doses and did not take these at the end of treatment. Twenty-four (39%) participants reported missing at least one retreatment dose, with 17 (27%) participants reporting missing a dose only once (Figures 10 and 11).
| Retreatment adherence | Total (N = 62), n (%) |
|---|---|
| Any missed doses reported | 24 (39) |
| Number of missed doses reported | |
| 0 | 38 (61) |
| 1 | 17 (27) |
| 2 | 4 (6) |
| 4 | 3 (5) |
| LTFU or withdrew consent before EOT | 2 (3) |
| Stopped all treatment before EOT | 2 (3) |
| Stopped ribavirin only before EOT | 3 (5) |
| Stopped late | 3 (5) |
| Reasons for stopping early (% stopped early) | |
| AEa | 2 (40) |
| Other | 3 (60) |
| Reasons for dose or frequency change (% changes) | |
| AEb | 5 (63) |
| Other | 3 (38) |
FIGURE 10.
Reported retreatment missed doses by duration randomisation.
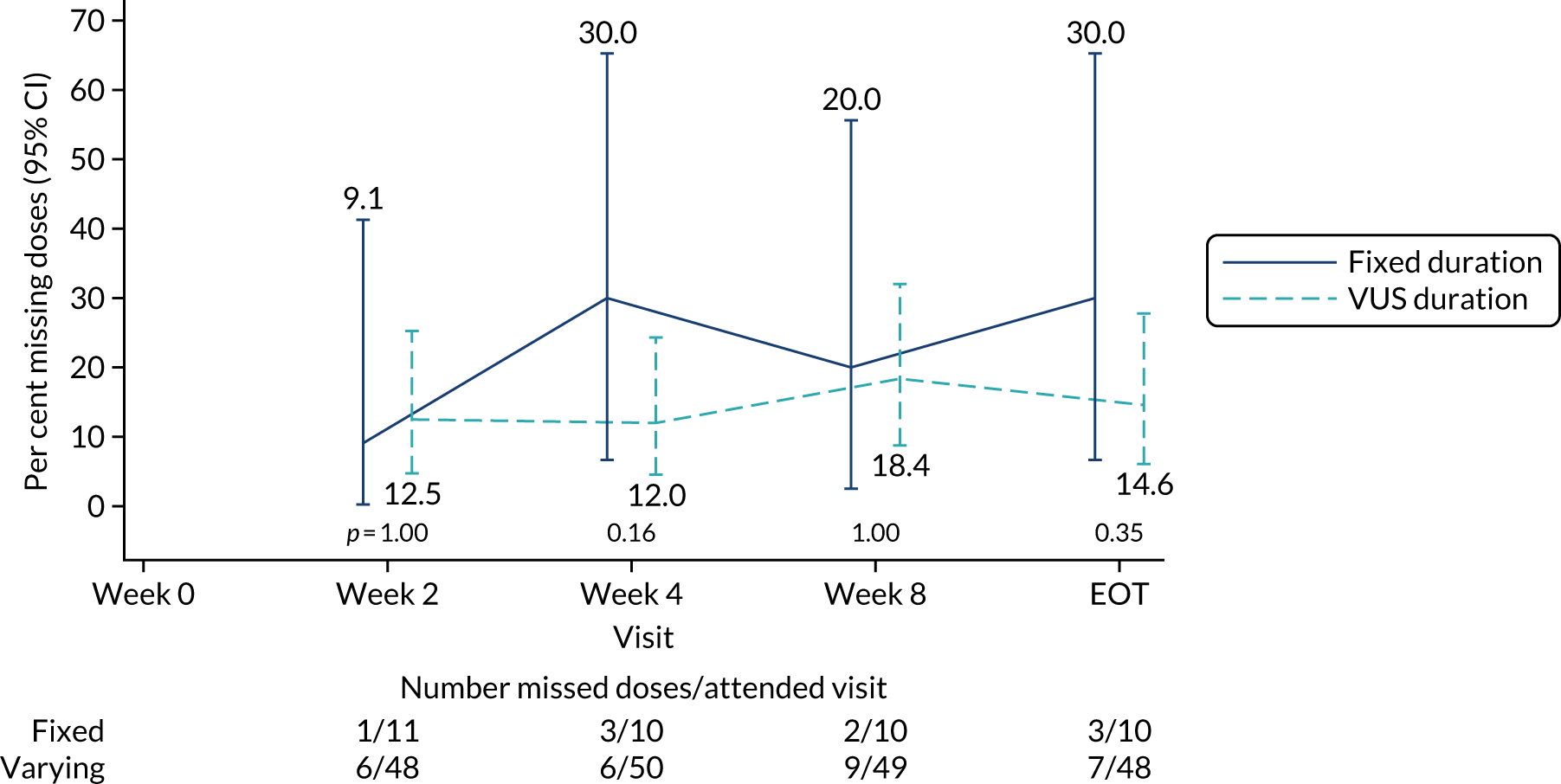
FIGURE 11.
Reported retreatment missed doses by ribavirin randomisation.
Numbers analysed
Although some participants were LTFU before the time point at which some outcomes were measured, it was possible to ascertain many of these outcomes through routine medical records when participants had returned to clinic for their usual care. Specifically, in total, one participant withdrew consent (at retreatment week 4) and a further 13 (6%) participants were LTFU (first-line treatment, n = 1; post first-line EOT, n = 9; on retreatment, n = 2; post retreatment EOT, n = 1). However, HCV VL results were available from medical notes for most of those who did not withdraw consent, meaning that first-line SVR12 and SVR24 could not be ascertained for only three (1%) and six (3%) participants, respectively, and overall (i.e. first-line treatment plus retreatment) SVR12 and SVR24 for only five (2%) and eight (4%) participants, respectively (excluded from the corresponding analyses following the SAP; see Chapter 2).
All analysis was by original assignment groups. The primary analysis was by intention to treat, with a secondary per-protocol analysis for SVR12. The per-protocol population was defined as those receiving first-line treatment for > 90% and < 110% of the prescribed duration based on prescription and temporary/permanent discontinuation and in whom the difference in HCV VL values between screening and enrolment would have led to a difference in allocated duration of DAAs of ≤ 2 days had participants been allocated to the VUS group. Although the median difference in VL between screening and enrolment was 0.01 (IQR –0.19–0.21) log10 IU/ml, the absolute differences were greater (Figure 12), leading to 57 (28%) participants being excluded from the per-protocol analysis because the difference in duration of treatment with DAAs would have been ≥ 3 days had duration been determined by the enrolment rather than the screening VL [31 (23%) VUS1 participants vs. 26 (39%) VUS2 participants because the second strategy received more drug overall] (see Figure 7). In total, 70 participants (70%) randomised to VUS compared with 72 (71%) participants randomised to fixed duration, and 68 (68%) participants randomised to ribavirin compared with 74 (73%) participants not randomised to ribavirin, were included in the per-protocol population (Table 9).
FIGURE 12.
Hepatitis C virus VL at screening and enrolment (all patients) (a) by assay and (b) by duration randomisation and failure status.
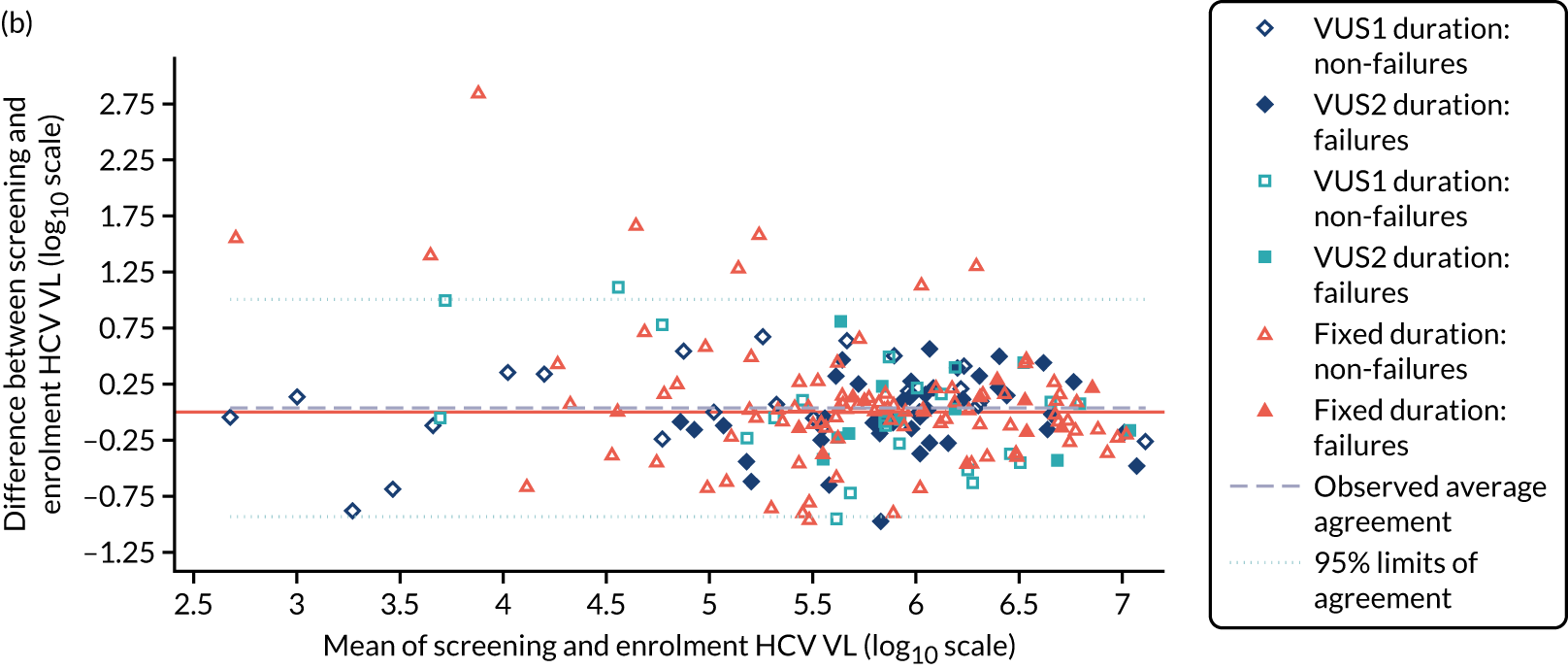
| Randomisation | Treatment arm, n (%) | Total, n (%) | |
|---|---|---|---|
| Varying duration | Fixed duration | ||
| Randomised | 100 | 102 | 202 |
| Included in per-protocol population | 70 (70) | 72 (71) | 142 (70) |
| Reasons for exclusiona | |||
| Failing VL criteria (first DAA strategy) | 14 | 17 | 31 |
| Failing VL criteria (second DAA strategy) | 15 | 11 | 26 |
| Missed doses | 0 | 2 | 2 |
| Stopping early | 2 | 3 | 5 |
| With ribavirin | Without ribavirin | ||
| Randomised | 100 | 102 | 202 |
| Included in per-protocol population | 68 (68) | 74 (73) | 142 (70) |
| Retreatment | |||
| Starting retreatment | 62 | ||
| In per-protocol population | 43 (69) | ||
Outcomes and estimation
SVR12
All participants (197/197) achieved SVR12 after first-line treatment and any retreatment (Table 10), with a difference of 0% (95% CI –3.8% to 3.7%), within the prespecified 4% non-inferiority margin. Ninety-one per cent (91/101; 95% CI 86% to 97%) of those randomised to the 8 weeks’ fixed-duration treatment achieved SVR12 after first-line treatment, compared with 48% (47/98; 95% CI 39% to 57%) of those randomised to VUS (a difference of –43%, 95% CI –54% to –32%; p < 0.001) (Figure 13). The proportion of participants who achieved SVR12 after first-line treatment was significantly higher in the group randomised to VUS2 (72%, 23/43) than in the group randomised to VUS1 (36%, 24/66; p = 0.001) (interaction between duration randomisation and strategy p = 0.001). There was evidence of differences in rates of SVR12 after first-line treatment between those randomised to ribavirin (68%, 68/98) and those not (72%, 70/101; p = 0.48 adjusting for interaction between duration randomisation and strategy). There was no evidence of and interaction between ribavirin and duration randomisations overall (heterogeneity p = 0.16, adjusted for the interaction between duration randomisation and strategy) or in the variable-duration group, where SVR12 was 52% (25/48; 95% CI 37% to 67%) with ribavirin compared with 44% (22/50; 95% CI 30% to 59%) without ribavirin (difference 8%, 95% CI –10% to 27%; p = 0.38).
| Outcome | Treatment arm, n (%, 95% CI) | Total, n (%, 95% CI) | Risk difference (95% CI); p-value | |
|---|---|---|---|---|
| Varying duration | Fixed duration | |||
| Randomised | 100 | 102 | 202 | |
| Primary outcome evaluable | 97 | 100 | 197 | |
| Primary outcome: SVR12 – first-line treatment or retreatment | 97 (100, 96 to 100) | 100 (100, 96 to 100) | 197 (100, 98 to 100) | 0 (–0.038 to 0.037) |
| SVR12: first-line treatment evaluable | 98 | 101 | 199 | |
| SVR12: first-line treatment only | 47 (48, 39 to 57) | 92 (91, 86 to 97) | 139 (70, 64 to 75) | –0.43 (–0.54 to –0.32); p < 0.001a |
| Received VUS1 | 66 | 67 | 133 | |
| SVR12: first-line treatment only | 24 (36, 25 to 48) | 62 (93, 86 to 99) | 86 (65, 58 to 71) | –0.56 (–0.69 to –0.43); p < 0.001 |
| Received VUS2 | 32 | 34 | 66 | |
| SVR12: first-line treatment only | 23 (72, 56 to 87) | 30 (88, 77 to 99) | 53 (80, 71 to 90) | –0.16 (–0.35 to 0.03); p = 0.09 |
| Ribavirin | No ribavirin | |||
| Randomised | 100 | 102 | 202 | |
| Primary outcome evaluable | 97 | 100 | 197 | |
| SVR12: first-line treatment or retreatment | 97 (100, 96 to 100) | 100 (100, 96 to 100) | 197 (100, 98 to 100) | 0 (–0.038 to 0.037) |
| SVR12: first-line treatment evaluable | 98 | 101 | 199 | |
| Primary outcome: SVR12 – first-line treatment only | 69 (68, 61 to 76) | 70 (72, 65 to 78) | 139 (70, 65 to 75) | –0.03 (–0.13 to 0.06); p = 0.48b |
FIGURE 13.
Overall SVR12 and first-line SVR12 by randomised groups and strategy (VUS1/VUS2). Fixed, overall SVR12 for 8-week therapy; fixed1, fixed duration when VUS duration received VUS1; fixed2, fixed duration when VUS duration received VUS2; FL, first line; RT, retreatment.
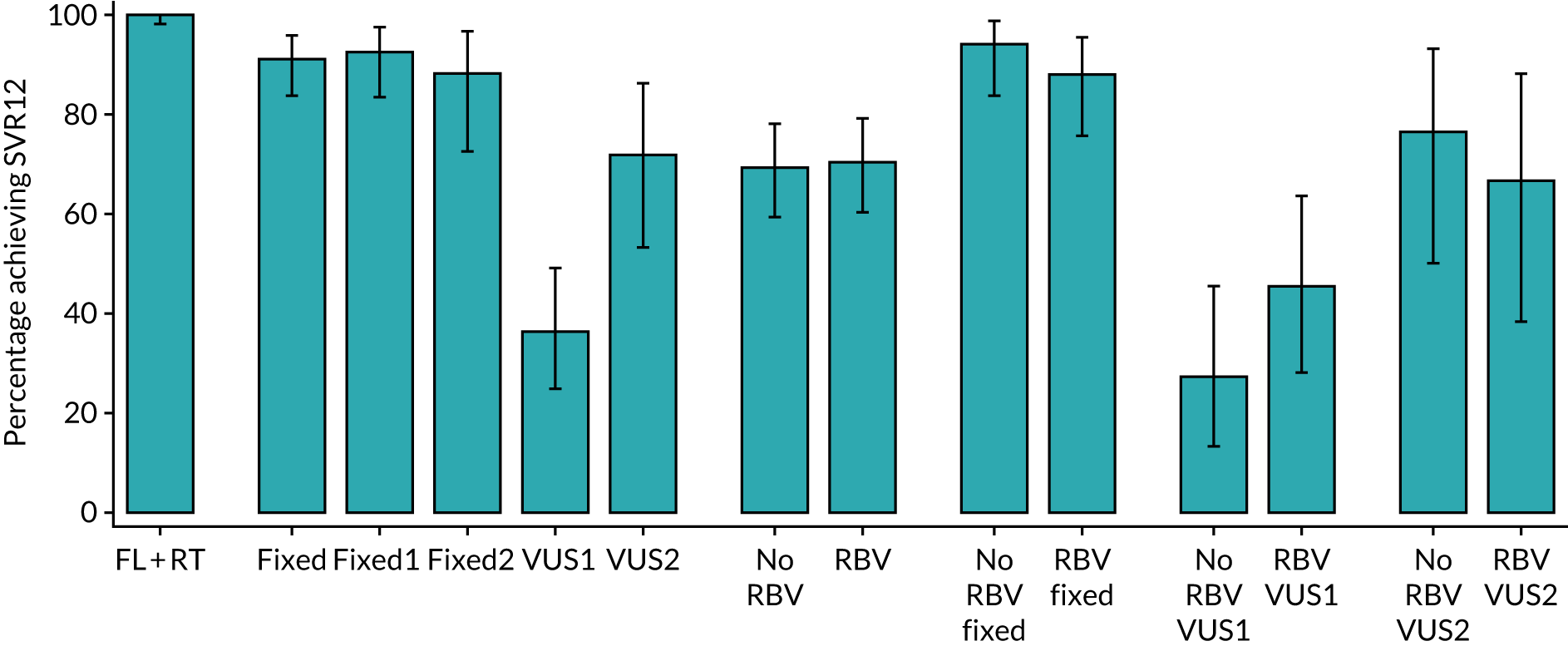
Subgroup analysis (prespecified)
Sixteen subgroups were prespecified in the protocol and SAP. Differences between duration randomisation groups in rates of first-line SVR12 were significantly smaller among participants in whom the virus was suppressed at days 7 and 14 than among those in whom the virus was not suppressed (heterogeneity p = 0.02 and p = 0.03, respectively) (Figures 14 and 15). The effect of the virus being suppressed at day 3 could not be formally tested because all participants in whom VL was already suppressed by day 3 achieved SVR12 on first-line treatment, but the difference in SVR12 among participants in whom VL was not suppressed was substantially larger (0% VUS vs. 40% fixed duration). There was also a trend for difference in SVR12 on first-line treatment between the fixed- and variable-duration groups to be larger for those with baseline RAS than for those without (heterogeneity p = 0.051). Although the evidence was weak, the difference in rate of SVR12 on first-line treatment between fixed- and variable-duration groups was numerically much larger in the case of those who had previously been unsuccessfully treated with interferon than among those who had not (heterogeneity p = 0.13). No subgroup favoured variable duration.
FIGURE 14.
Subgroup analysis by duration comparison. D, day.
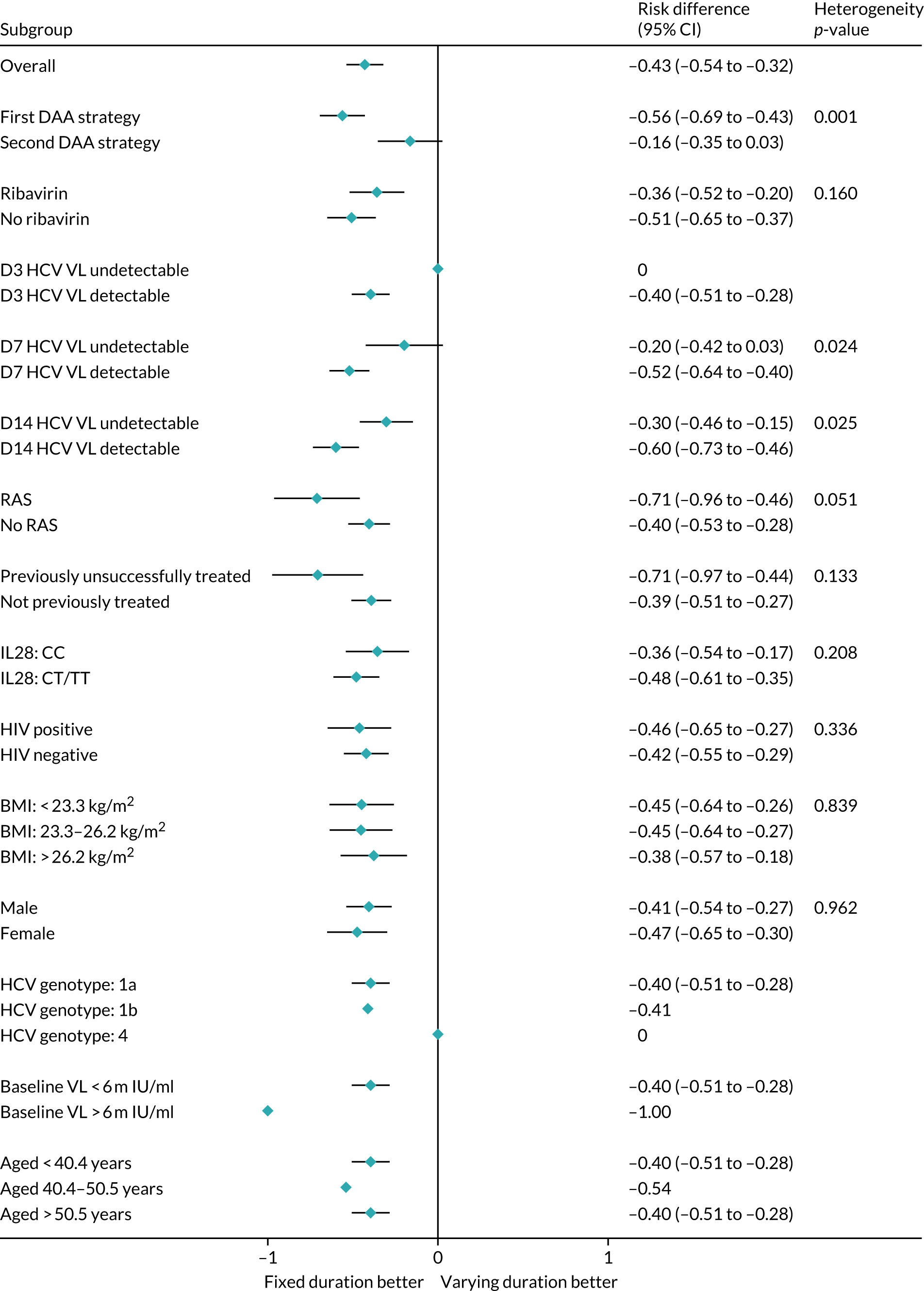
FIGURE 15.
Variation in the difference in the rate of first-line SVR12 between fixed- and variable-duration groups by key subgroups for fixed duration vs. varying duration. Note that solid bars represent the first subgroup (detectable VL on the various days shown, no previous unsuccessful treatment and no baseline resistance to drugs taken as first-line treatment) and empty bars the second subgroup (undetectable VL on the various days shown, previous unsuccessful treatment and baseline resistance to drugs taken as first-line treatment). p-values are heterogeneity p-values comparing the difference between fixed and VUS1/2 strategies across the two subgroups. A heterogeneity p-value for day 3 VL cannot be estimated because of perfect prediction. D, day.
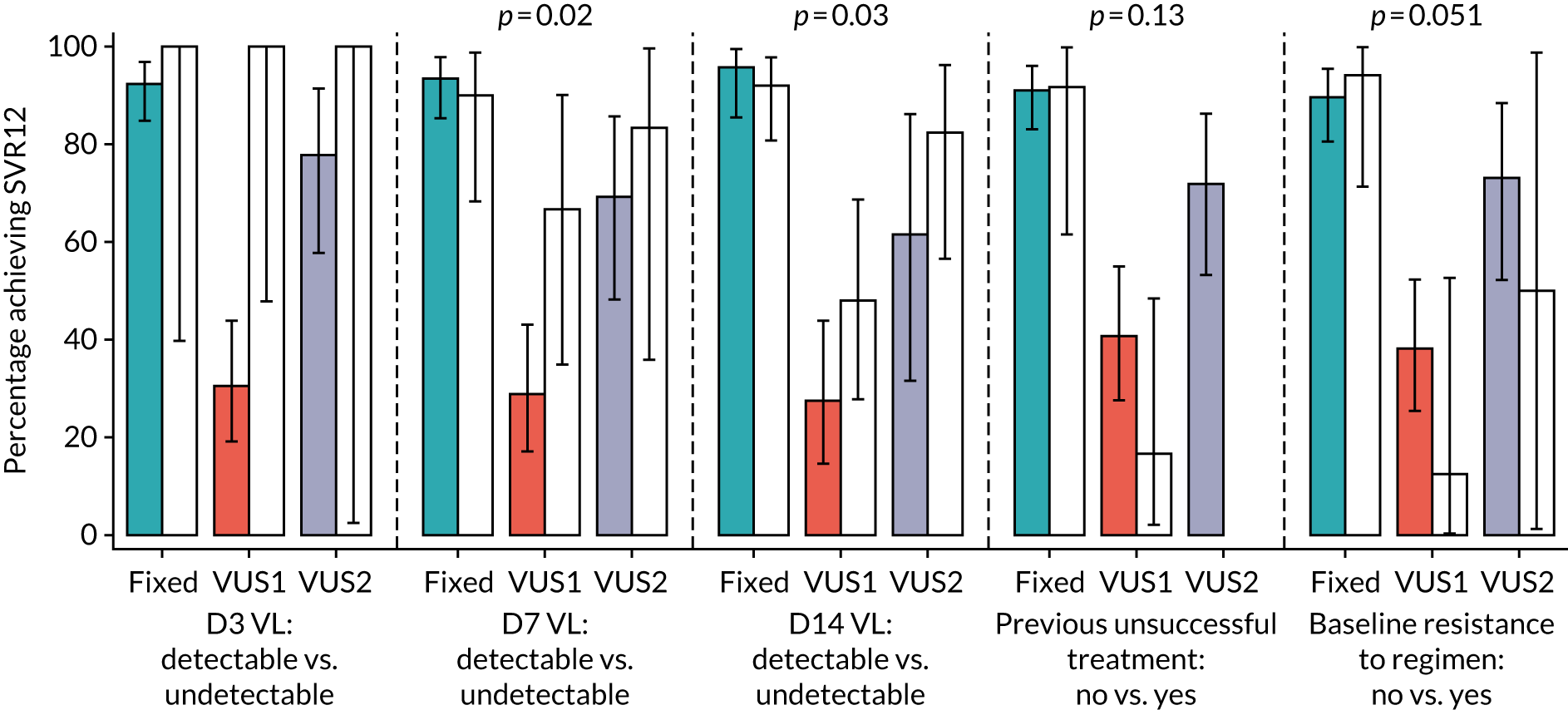
There was no evidence of any variation in the (lack of) effect of ribavirin across these subgroups (heterogeneity p ≥ 0.16) (Figure 16).
FIGURE 16.
Subgroup analysis by ribavirin comparison. D, day.
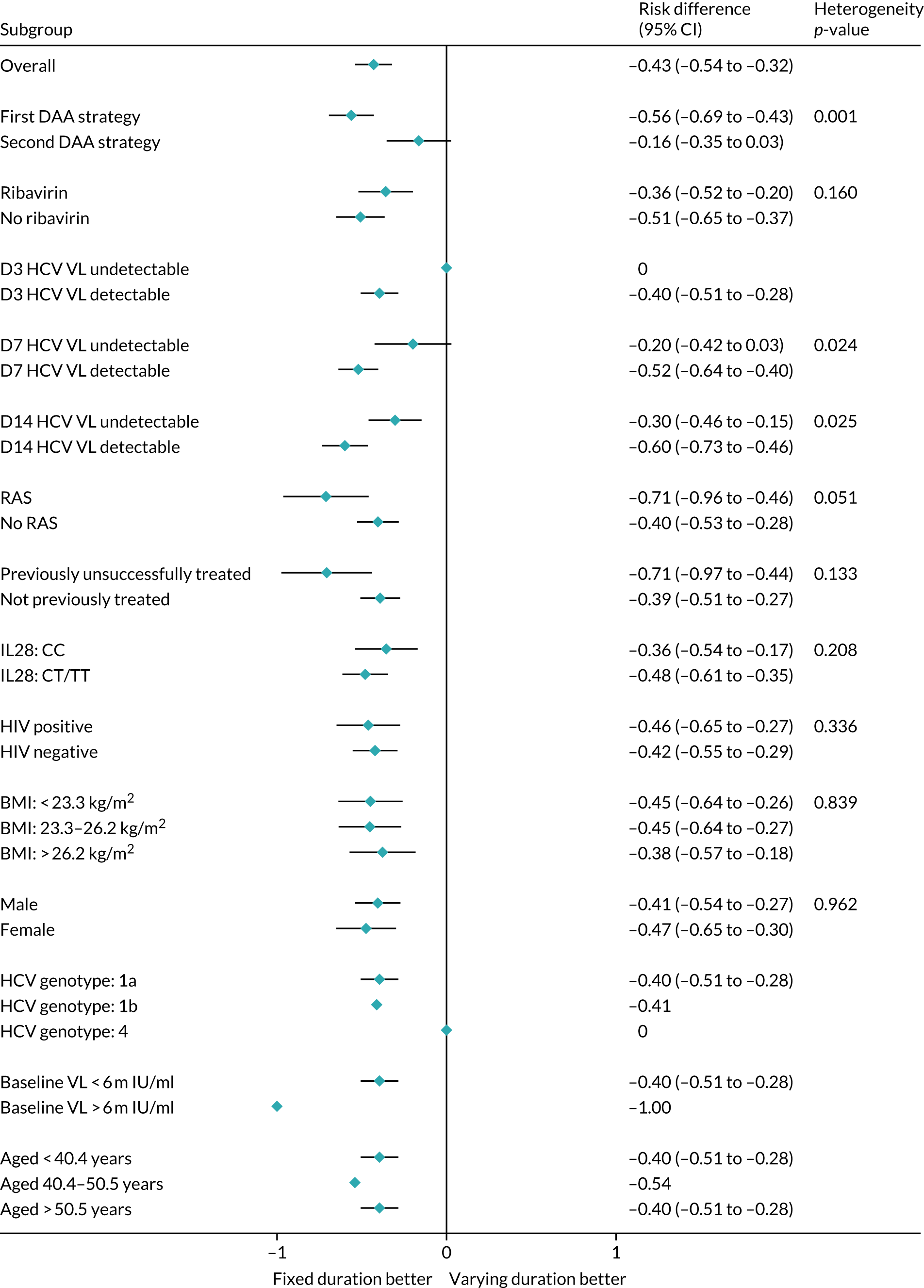
Considering the time when individuals first became undetectable (rather than the prespecified subgroup analyses according to whether they were detectable or undetectable at specific time points), all 10 individuals who became undetectable at day 3 of treatment achieved first-line SVR12 regardless of treatment duration [as did 31 of 38 (82%) participants who were first undetectable at day 7] (Figures 15 and 17).
FIGURE 17.
First-line SVR12 by time first suppressed. Note that figure does not include eight patients who never had VL suppression. In addition, for patients allocated 28–31 days, their EOT visit is also their 28-day visit and they are included in each group. D, day.
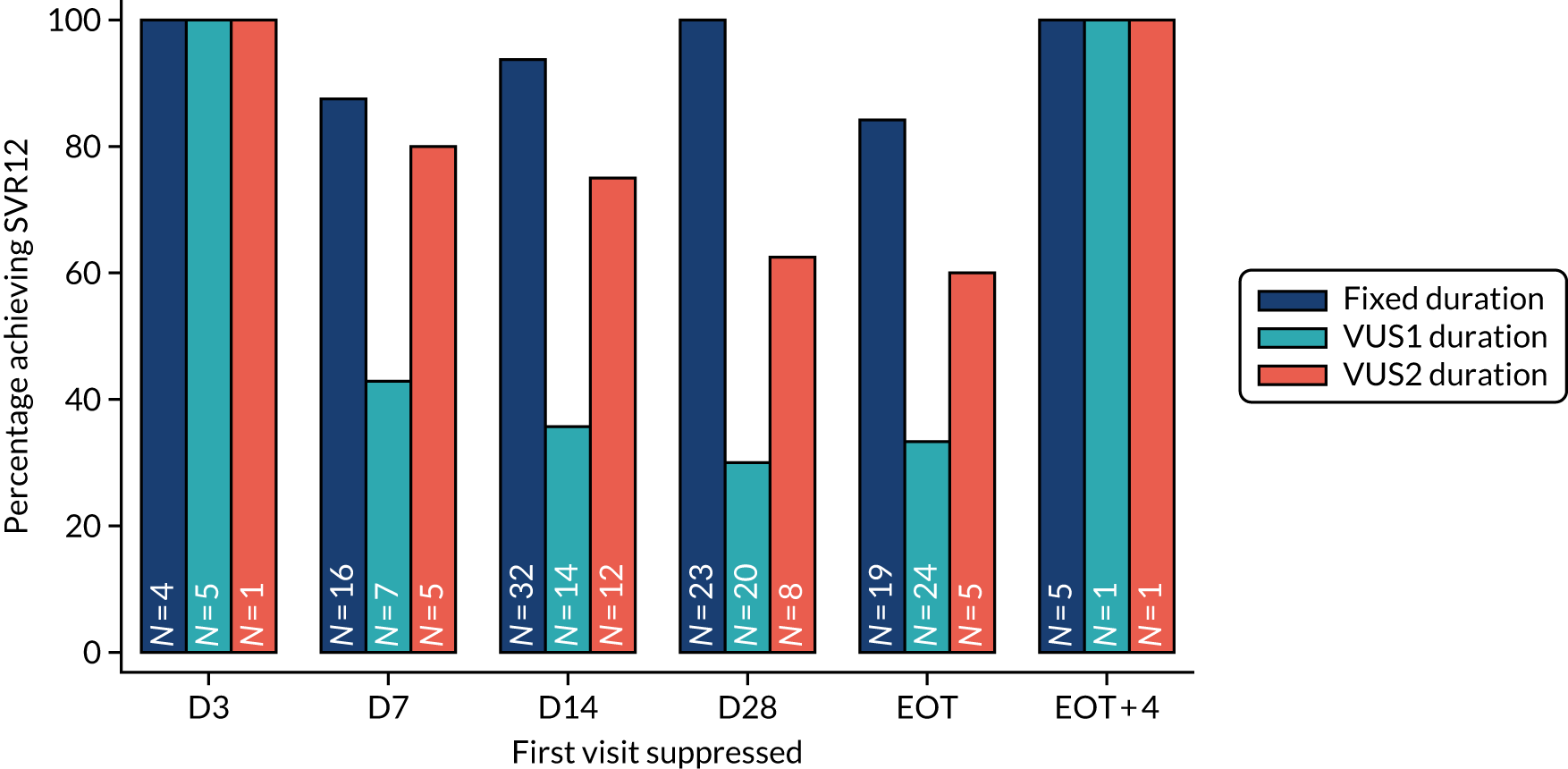
SVR24
All participants achieved SVR24 after first-line treatment or retreatment (194/194; risk difference 0%, 95% CI –3.8% to 3.8%) (Table 11). After first-line treatment only, 89% (88/99; 95% CI 83% to 95%) of participants randomised to fixed duration achieved SVR24, compared with 47% (46/97; 95% CI 38% to 56%) of participants randomised to VUS1/2, that is a difference of –42% (95% CI –53% to 31%; p < 0.001). There was no evidence of differences between the groups randomised to or not randomised to ribavirin for SVR24 after first-line treatment (p = 0.87).
| Outcome | Treatment arm, n (%, 95% CI) | Total, n (%, 95% CI) | Risk difference (95% CI); p-value | |
|---|---|---|---|---|
| Varying duration | Fixed duration | |||
| Randomised | 100 | 102 | 202 | |
| Outcome evaluable | 96 | 98 | 194 | |
| SVR24: first-line treatment or retreatment | 96 (100, 96 to 100) | 98 (100, 96 to 100) | 194 (100, 98 to 100) | 0 (–0.038 to 0.038) |
| Reached EOT plus 24 weeks | 97 | 99 | 196 | |
| SVR24: first-line treatment only | 46 (47, 38 to 56) | 88 (89, 83 to 95) | 134 (68, 63 to 74) | –0.42 (–0.53 to –0.31); p < 0.001a |
| Ribavirin | No ribavirin | |||
| Randomised | 100 | 102 | 202 | |
| Outcome evaluable | 96 | 98 | 194 | |
| SVR24: first-line treatment or retreatment | 96 (100, 96 to 100) | 98 (100, 96 to 100) | 194 (100, 98 to 100) | 0 (–0.038 to 0.038) |
| Reached EOT plus 24 weeks | 97 | 99 | 196 | |
| Primary outcome: SVR24 – first-line treatment only | 68 (69, 61 to 76) | 66 (68, 60 to 76) | 134 (68, 63 to 74) | 0.01 (–0.09 to 0.11); p = 0.87b |
Hepatitis C virus viral load rebound and primary first-line treatment failure
In total, 41 (20%) participants had HCV VL rebound, defined as having a confirmed HCV VL greater than the LLOQ after two consecutive visits with HCV VL less than the LLOQ, with the latter confirmatory result also > 2000 IU/ml. Six (6%) participants in the fixed-duration group and 35 (35%) participants in the variable-duration group experienced rebound (a difference of 29%, 95% CI 18% to 39%; p < 0.001) (Figure 18). There was no evidence of a difference in the percentage of participants experiencing rebound between those randomised to and those not randomised to ribavirin (19% vs. 22%, respectively; p = 0.59) (Figure 19).
FIGURE 18.
Cumulative incidence plot of HCV VL rebound by duration randomisation.
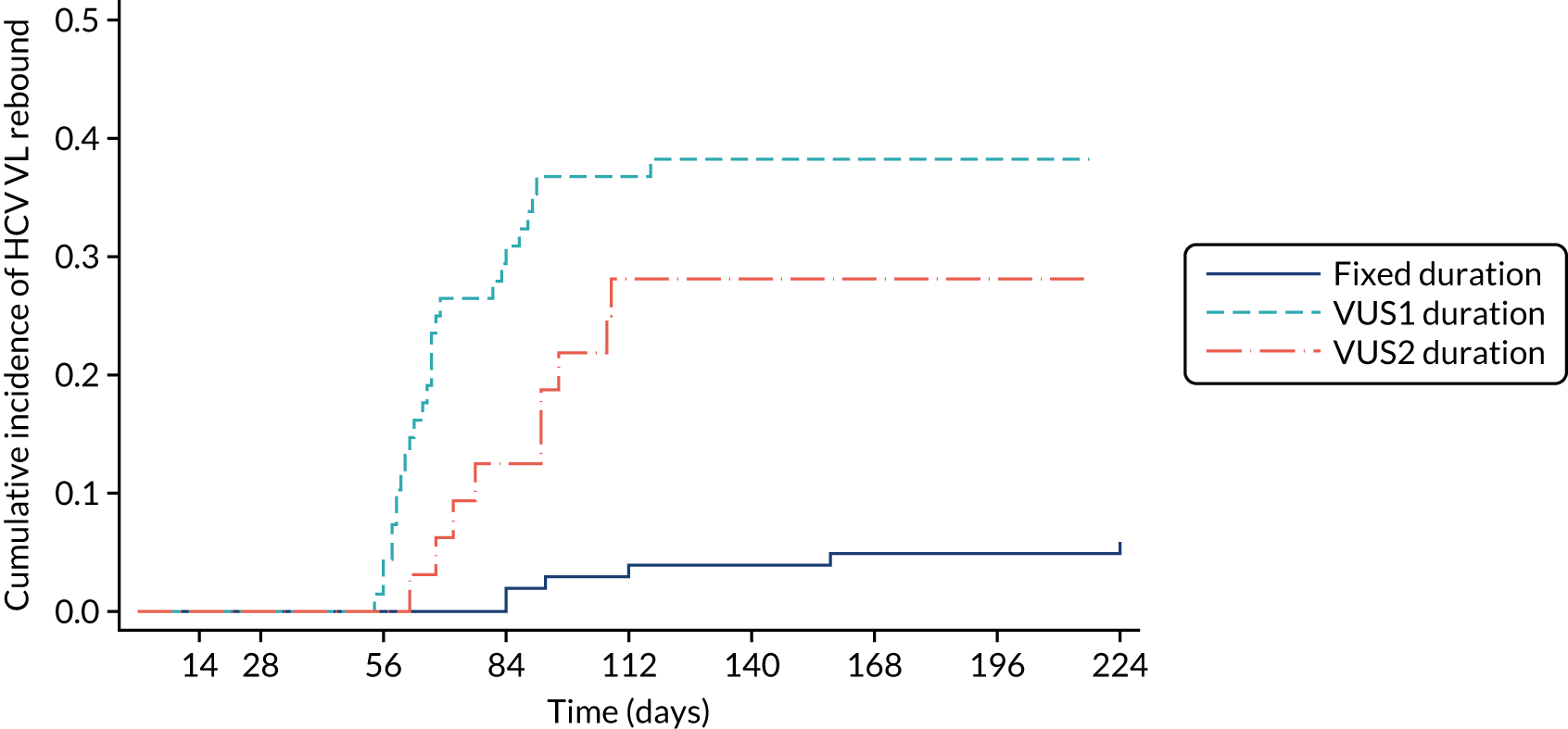
FIGURE 19.
Cumulative incidence plot of HCV VL rebound by ribavirin randomisation.
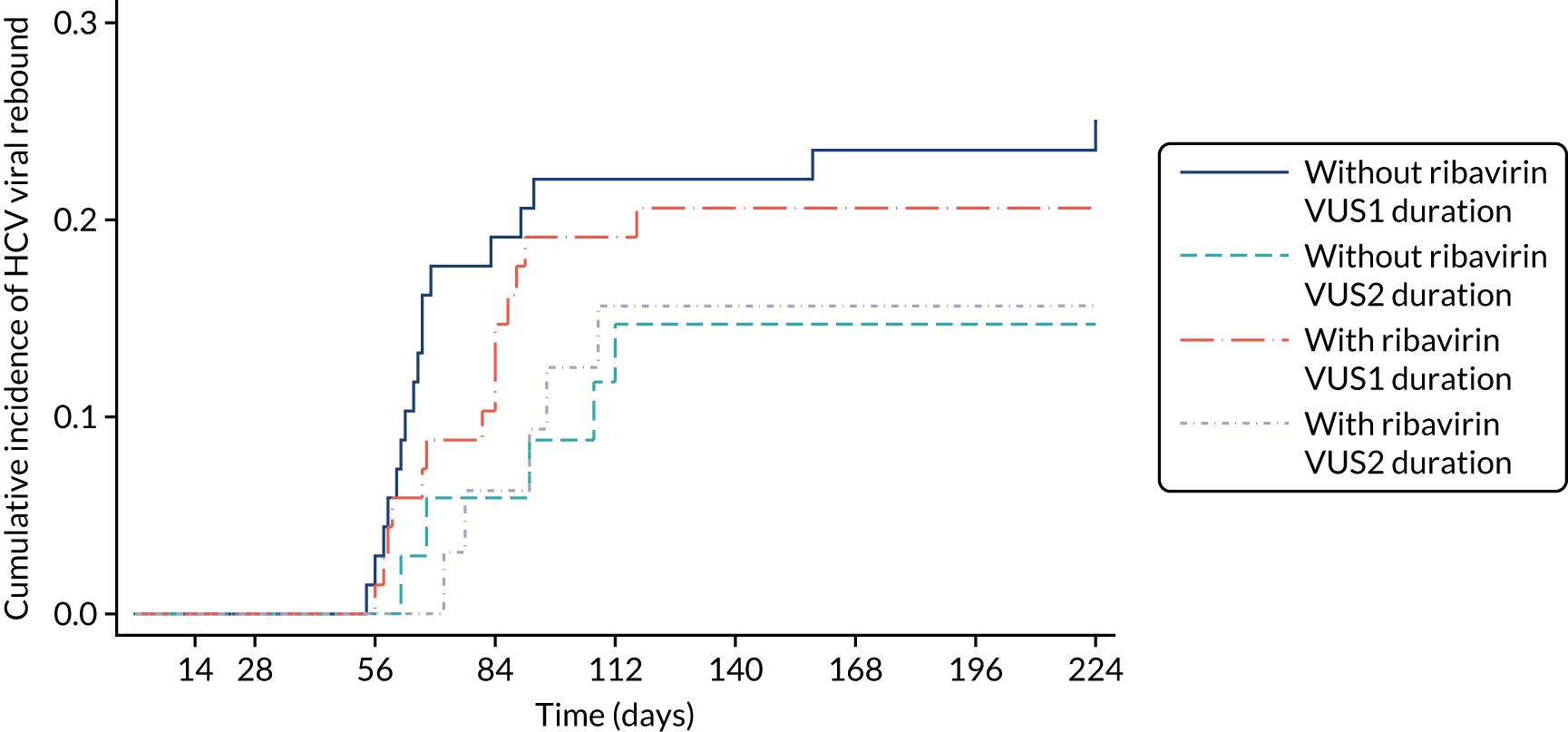
Twenty-one (10%) participants had primary first-line treatment failure, defined as having a confirmed > 1 log10 increase from HCV VL nadir on treatment and > 2000 IU/ml (Figure 20). Participants randomised to variable duration were significantly more likely to experience primary failure than those randomised to fixed duration, with an estimated difference of 10% (95% CI 2% to 18%; p = 0.008). There was no evidence of differences in percentages with primary first-line treatment failure between those randomised to receive or not receive ribavirin (10% vs. 11%, respectively; p = 0.83) (Figures 21 and 22).
FIGURE 20.
Cumulative incidence plot of primary first-line treatment failure by duration randomisation.
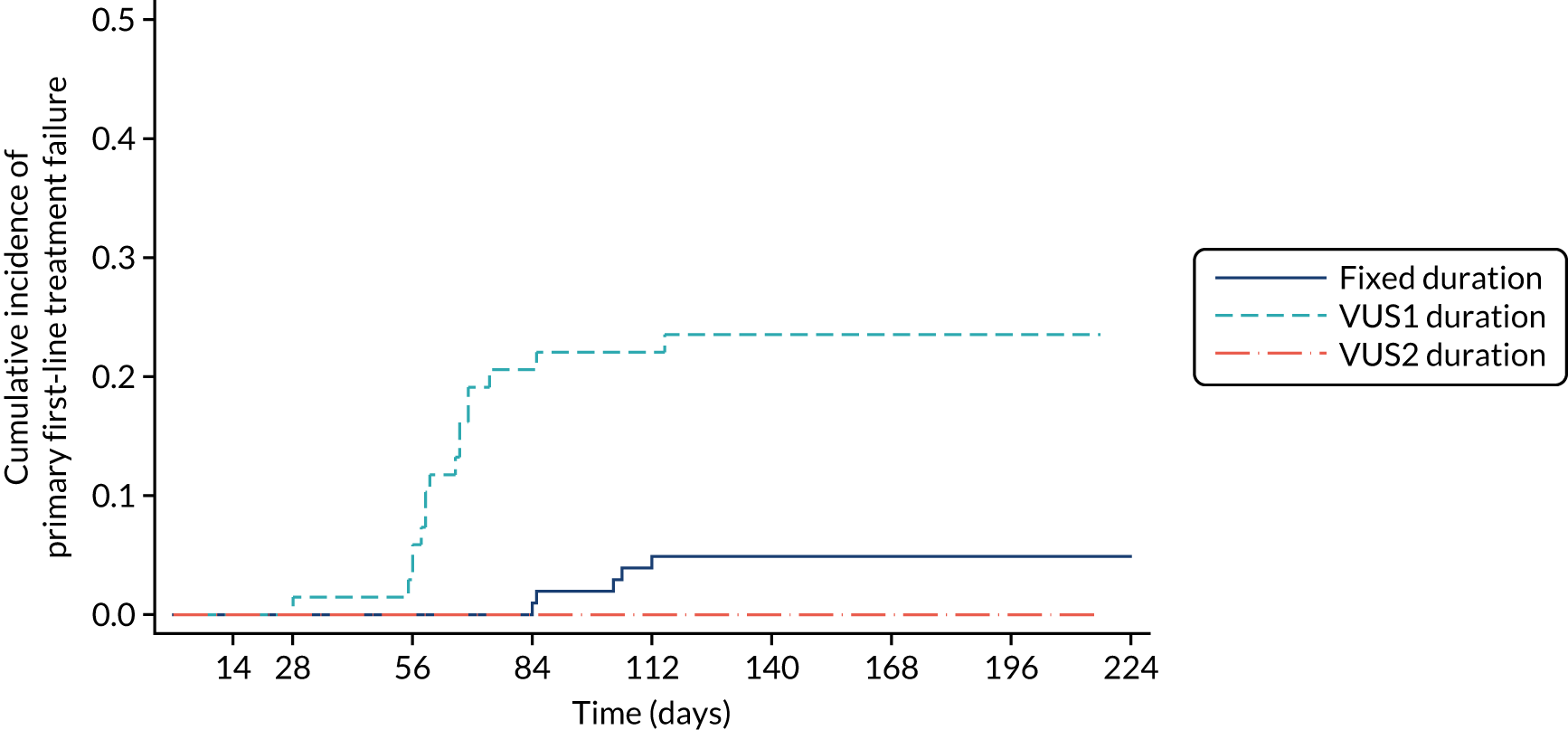
FIGURE 21.
Cumulative incidence plot of primary first-line treatment failure by ribavirin randomisation.
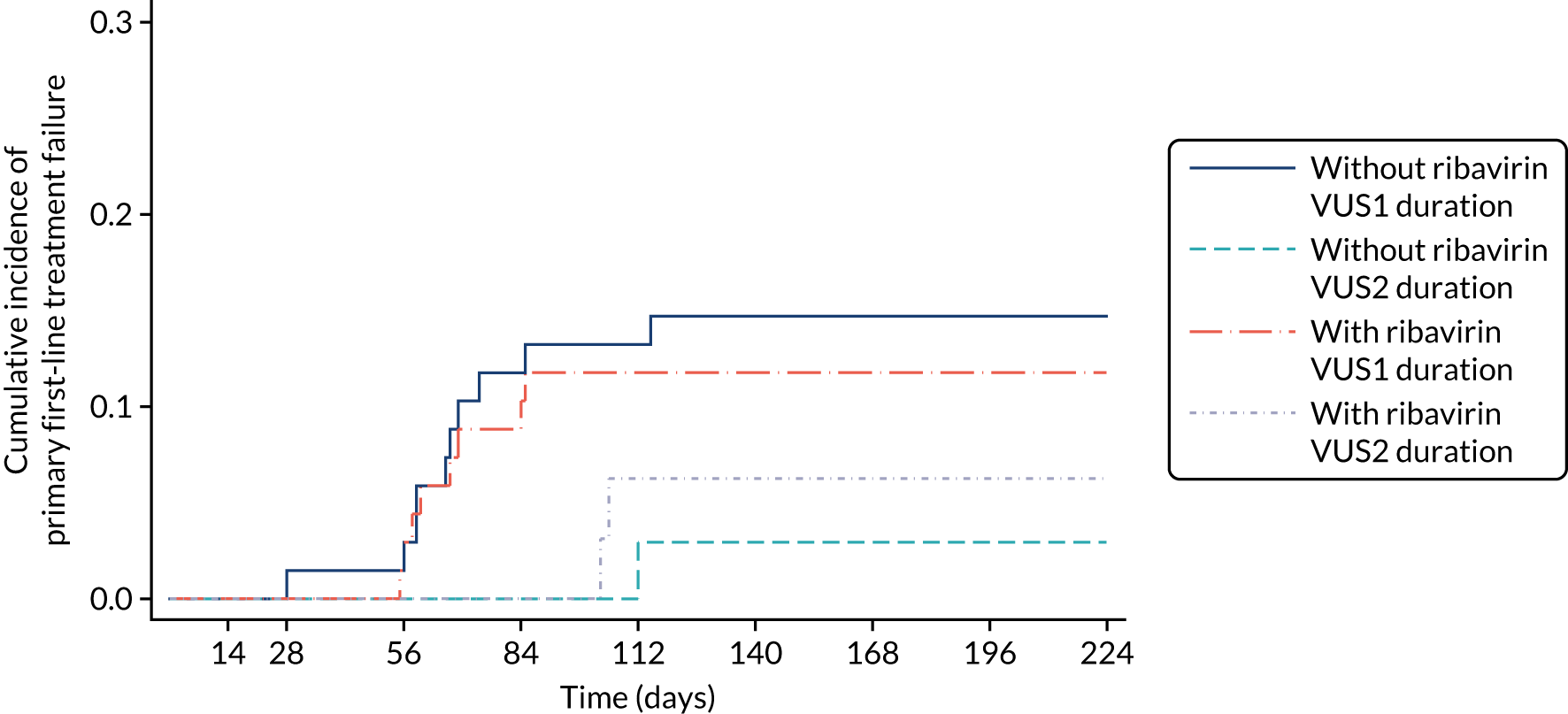
FIGURE 22.
Resistance at baseline (all tested participants) and at first-line treatment failure (failures only) classified by (a) first-line drugs and (b) retreatment drugs. p-values are relate to comparisons of resistance to (a) first-line drugs and (b) retreatment drugs with resistance to any other DAA between ribavirin groups. Dark-blue bars represent resistance to drugs received as (a) first-line treatment and (b) retreatment and light-blue bars represent resistance to all DAAs, including those not used in the trial.
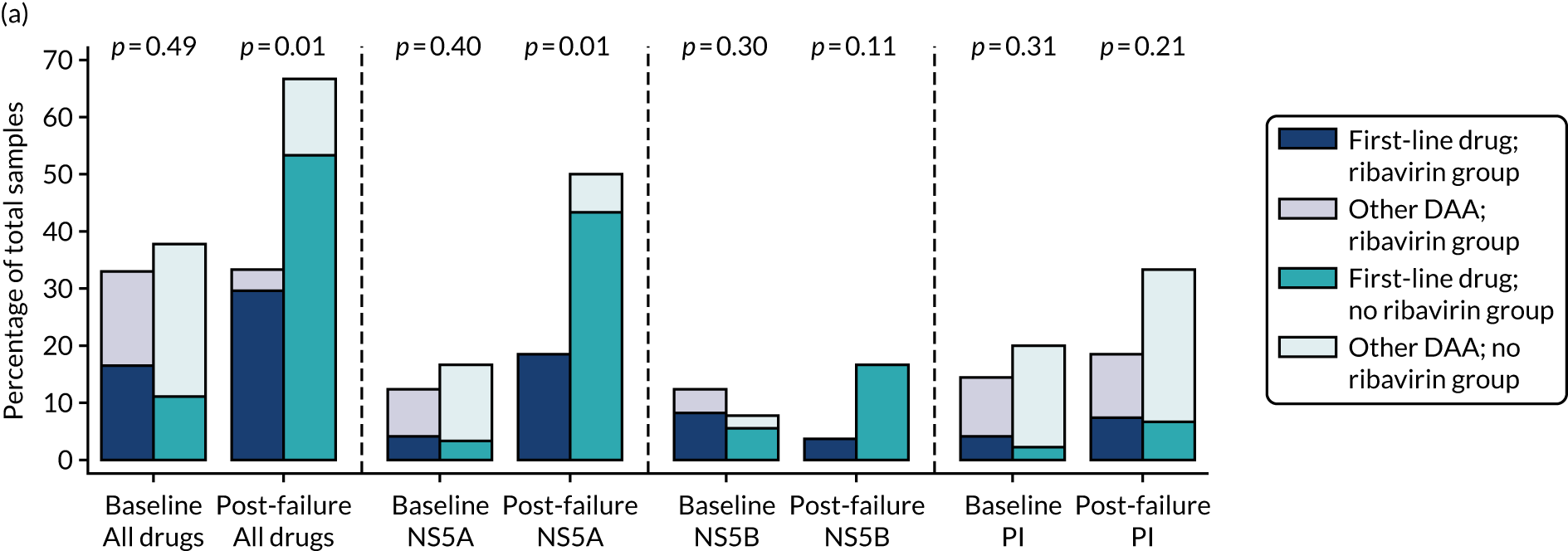
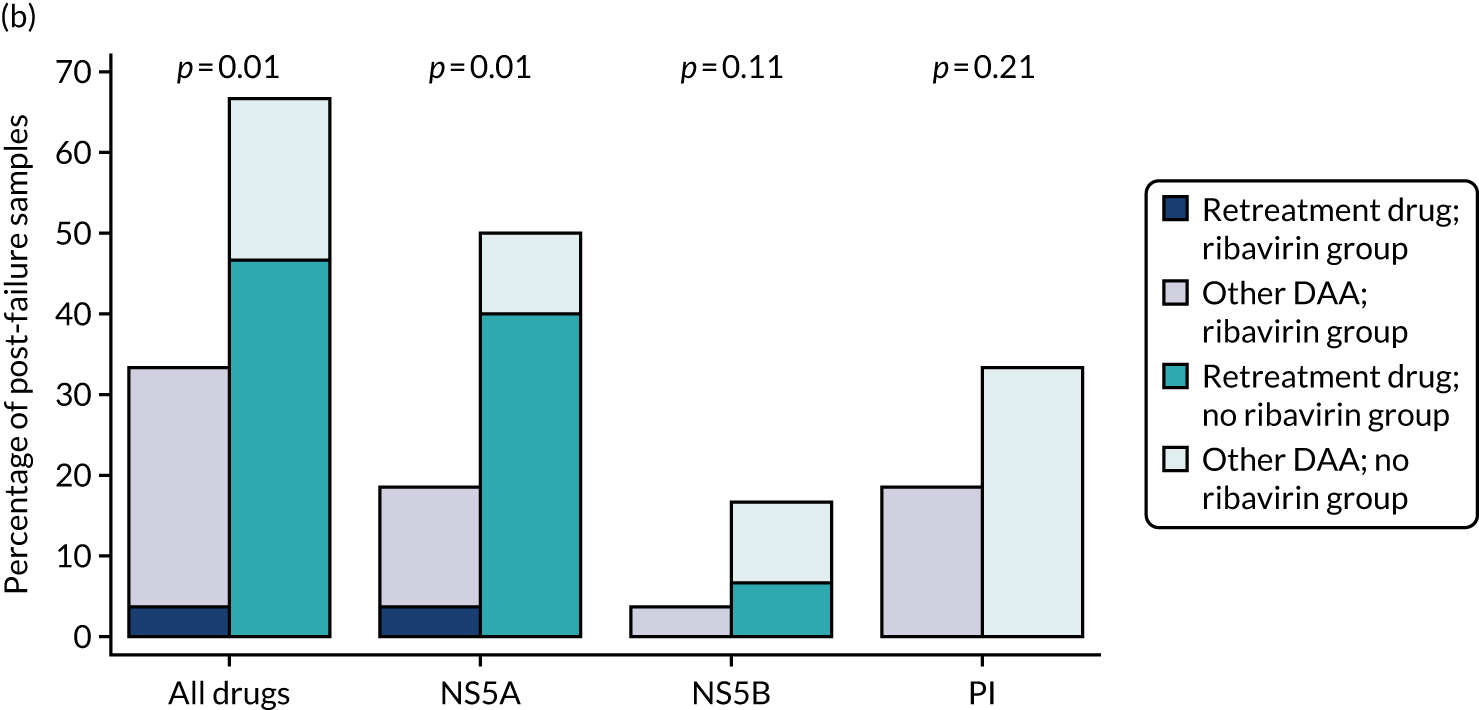
Detectable viral load 4 weeks after randomisation
Overall, 21% (41/197) of participants had a detectable VL at day 28. There was no evidence of differences between fixed- and variable-duration groups or between the ribavirin group and no-ribavirin group (p = 0.08 and p = 0.26, respectively) (Table 12).
| Outcome | Treatment arm, n (%) | Total, n (%) | Risk difference (95% CI); p-value | |
|---|---|---|---|---|
| Varying duration | Fixed duration | |||
| Randomised | 100 | 102 | 202 | |
| HCV VL 4 weeks after randomisation | 96 | 101 | 197 | |
| Detectable VL | 15 (16) | 26 (26) | 41 (21) | –0.10 (–0.21 to 0.01); p = 0.08 |
| Ribavirin | No ribavirin | |||
| Randomised | 100 | 102 | 202 | |
| HCV VL 4 weeks after randomisation | 97 | 100 | 197 | |
| Detectable VL | 17 (18) | 24 (24) | 41 (21) | –0.06 (–0.18 to 0.05); p = 0.26 |
Emergent resistance-associated substitutions to first-line treatment
Sixty-two (31%) participants met the criteria for failure on first-line treatment [fixed-duration group, 11/102 (11%); variable-duration group, 51/100 (51%), p < 0.0001; ribavirin group, 29 (29%); no-ribavirin group, 33 (32%), p = 0.68]. More failures occurred with VUS1 (62%, 42/68) than with VUS2 (28%, 9/32) (p = 0.002; interaction with fixed vs. variable strategy p = 0.003). Twenty-five per cent (14/56) of participants developed a new RAS (not present at baseline) to at least one of their prescribed first-line drugs (Table 13). Within paired samples available at baseline and failure, there was no evidence of difference in development of emergent RASs between those randomised to variable and fixed durations (24% vs. 30%, respectively; p = 0.77). However, those randomised to ribavirin were significantly less likely to develop new RASs (11% vs. 38% of those not randomised to ribavirin, a difference of –27%, 95% CI –48% to –6%; p = 0.01), with significantly lower rates of resistance to any DAA and to non-structural protein 5A (NS5A) inhibitors (both p = 0.01).
| Outcome | Treatment arm | Total | Risk difference (95% CI); p-value | |
|---|---|---|---|---|
| Varying duration | Fixed duration | |||
| Randomised, n | 100 | 102 | 202 | |
| Sequenced data after failure/total failures, n/N | 46/51 | 10/11 | 56/62 | |
| Emergent resistance-associated variant to first-line drugs, n (%) | 11 (24) | 3 (30) | 14 (25) | –0.05 (–0.36 to 0.27); p = 0.78 |
| Ribavirin | No ribavirin | |||
| Randomised, n | 100 | 102 | 202 | |
| Sequenced data after failure, n/N | 27/29 | 29/33 | 56/62 | |
| Emergent resistance-associated variant to first-line drugs, n (%) | 3 (11) | 11 (38) | 14 (25) | –0.27 (–0.48 to –0.06); p = 0.01 |
Ancillary analyses
Secondary analysis of SVR12: per-protocol analysis (prespecified)
Rates of both first-line SVR12 and SVR12 after first-line treatment and any retreatment in the per-protocol population were similar to those in all participants (Table 14). All per-protocol participants achieved SVR12 after first-line treatment and any retreatment (140/140), with a difference of 0% (95% CI –5% to 5%). The rate of first-line SVR12 was significantly lower among participants randomised to variable-duration treatment than among those randomised to fixed-duration treatment, with a difference of –43% (95% CI –54% to –32%; p < 0.001). There was no evidence of differences in the rate of first-line SVR12 between those randomised to ribavirin and those not (p = 0.93).
| Outcome | Treatment arm, n (%, 95% CI) | Total, n (%, 95% CI) | Risk difference (95% CI); p-value | |
|---|---|---|---|---|
| Varying duration | Fixed duration | |||
| Randomised | 70 | 72 | 142 | |
| Primary outcome evaluable | 69 | 71 | 140 | |
| Primary outcome: SVR12 – first-line treatment or retreatment | 69 (100, 95 to 100) | 71 (100, 95 to 100) | 140 (100, 97 to 100) | 0 (–0.05 to 0.05) |
| SVR12: first-line treatment evaluable | 69 | 71 | 140 | |
| SVR12: first-line treatment only | 32 (47, 36 to 59) | 66 (93, 87 to 99) | 98 (70, 64 to 76) | –0.46 (–0.59 to –0.33); p < 0.001a |
| Ribavirin | No ribavirin | |||
| Randomised | 68 | 74 | 142 | |
| Primary outcome evaluable | 66 | 74 | 140 | |
| SVR12: first-line treatment or retreatment | 66 (100, 95 to 100) | 74 (100, 95 to 100) | 140 (100, 97 to 100) | 0 (–0.06 to 0.05) |
| SVR12: first-line treatment evaluable | 66 | 74 | 140 | |
| SVR12: first-line treatment only | 48 (70, 61 to 78) | 50 (70, 62 to 78) | 98 (70, 64 to 76) | –0.00 (–0.11 to 0.10); p = 0.93b |
Secondary analysis of SVR12: all missing SVR12 particpants are considered failures (prespecified)
When the three and five missing participants (achieving first-line SVR12 and SVR12 after first-line treament and any retreatment, respectively) are considered failures (i.e. the worst-case scenario), the results are largely similar to those of the complete-case analysis (Table 15). For both the duration randomisation and ribavirin randomisation, 98% (197/202; 95% CI 95% to 100%) of participants achieved SVR12 after first-line treatment and any retreatment, giving a difference for both comparisons of –1% (95% CI –5% to 3%; p = 0.64). The rate of first-line SVR12 was significantly lower in the variable-duration group than in the fixed-duration group, with a difference of –43% (95% CI –54% to –32%; p < 0.001), but there was no evidence of differences between those receiving ribavirin and those not (p = 0.37).
| Outcome | Treatment arm, n (%, 95% CI) | Total, n (%, 95% CI) | Risk difference (95% CI); p-value | |
|---|---|---|---|---|
| Varying duration | Fixed duration | |||
| Randomised | 100 | 102 | 202 | |
| Primary outcome evaluable | 100 | 102 | 202 | |
| Primary outcome: SVR12 – first-line treatment or retreatment | 97 (97, 94 to 100) | 100 (98, 95 to 100) | 197 (98, 95 to 100) | –0.01 (–0.05 to 0.03); p = 0.64 |
| SVR12: first-line treatment evaluable | 100 | 102 | 202 | |
| SVR12: first-line treatment only | 47 (47, 38 to 56) | 92 (90, 84 to 96) | 139 (69, 63 to 74) | –0.43 (–0.54 to –0.32); p < 0.001a |
| Ribavirin | No ribavirin | |||
| Randomised | 100 | 102 | 202 | |
| Primary outcome evaluable | 100 | 102 | 202 | |
| SVR12: first-line treatment or retreatment | 97 (97, 94 to 100) | 100 (98, 95 to 100) | 197 (98, 95 to 100) | –0.01 (–0.05 to 0.03); p = 0.64 |
| SVR12: first-line treatment evaluable | 100 | 102 | 202 | |
| Primary outcome: SVR12 – first-line treatment only | 69 (67, 60 to 75) | 70 (71, 65 to 78) | 139 (69, 64 to 75) | –0.04 (–0.14 to 0.05); p = 0.37b |
Secondary analysis of SVR12: all missing participants are considered cured (prespecified)
When the three and five missing participants (achieving first-line SVR12 and SVR12 after first-line treament and any retreatment, respectively) are considered cures (i.e. the best-case scenario), the results are very similar to those of the complete-case analysis (Table 16). All participants achieved SVR12 after first-line treatment and any retreatment (202/202), with a difference of 0% (95% CI –3.7% to 3.6%), within the prespecified 4% non-inferiority margin. The rate of first-line SVR12 was significantly lower in the variable-duration group than in the fixed-duration group, with a difference of –42% (95% CI –53% to –31%; p < 0.001), but there was no evidence of differences between those receicing ribavirin and those not (p = 0.48).
| Outcome | Treatment arm, n (%, 95% CI) | Total, n (%, 95% CI) | Risk difference (95% CI); p-value | |
|---|---|---|---|---|
| Varying duration | Fixed duration | |||
| Randomised | 100 | 102 | 202 | |
| Primary outcome evaluable | 100 | 102 | 202 | |
| Primary outcome: SVR12 – first-line treatment or retreatment | 100 (100, 96 to 100) | 102 (100, 96 to 100) | 202 (100, 98 to 100) | 0 (–0.037 to 0.036) |
| SVR12: first-line treatment evaluable | 100 | 102 | 202 | |
| SVR12: first-line treatment only | 49 (49, 40 to 59) | 93 (91, 86 to 97) | 142 (70, 65 to 76) | –0.42 (–0.53 to –0.31); p < 0.001a |
| Ribavirin | No ribavirin | |||
| Randomised | 100 | 102 | 202 | |
| Primary outcome evaluable | 100 | 102 | 202 | |
| SVR12: first-line treatment or retreatment | 100 (100, 96 to 100) | 102 (100, 96 to 100) | 202 (100, 98 to 100) | 0 (–0.037 to 0.036) |
| SVR12: first-line treatment evaluable | 100 | 102 | 202 | |
| Primary outcome: SVR12 – first-line treatment only | 71 (69, 62 to 76) | 71 (72, 65 to 79) | 142 (71, 65 to 76) | –0.03 (–0.12 to 0.06); p = 0.48b |
Failures
Overall, 62 participants failed on first-line treatment (Table 17), all of whom started retreatment a median 2.9 weeks after meeting the failure criteria. Only one participant failed while on treatment (at EOT, 28 days DAA) (Figure 23). The majority of VUS1 participants failed at EOT plus 4 weeks, with smaller numbers failing at EOT plus 8 weeks and EOT plus 12 weeks. By contrast, more VUS2 participants failed at EOT plus 8 weeks than at EOT plus 4 weeks (comparing timing of failure between VUS1 and VUS2, p = 0.08; variable duration vs. fixed duration, p = 0.07; VUS1 vs. fixed duration, p = 0.03). Two participants, both randomised to 8 weeks’ fixed-duration treatment without ribavirin, failed after achieving SVR12. However, there was no evidence of differences in failure VLs (VUS1, median 158,073 IU/ml; VUS2, median 89,125 IU/ml; fixed-duration group, median 346,737 IU/ml; VUS2 vs. VUS1, p = 0.41; VUS2 vs. fixed-duration group, p = 0.21) (see Figure 23).
| Outcome | Treatment arm | Total (N = 202) | |||
|---|---|---|---|---|---|
| Varying duration, ribavirin (N = 49) | Varying duration, no ribavirin (N = 51) | Fixed duration, ribavirin (N = 51) | Fixed duration, no ribavirin (N = 51) | ||
| Meeting failure criteria, n (%) | 23 (47) | 28 (55) | 6 (12) | 5 (10) | 62 (31) |
| Started retreatment, n (%) | 23 (100) | 28 (100) | 6 (100) | 5 (100) | 62 (100) |
| Median (IQR) [range] weeks from meeting criteria to retreatment | 3.3 (2.0–6.9) [0.9–32.7] | 2.6 (2.1–3.2) [1.3–10.1] | 4.6 (2.6–8.1) [2.6–14.0] | 3.1 (2.1–4.9) [1.9–7.0] | 2.9 (2.0–4.4) [0.9–32.7] |
FIGURE 23.
First-line failure. (a) Timing of first-line treatment failure and (b) HCV VL at first-line treatment failure. Note that HCV VL shown is the first, not the confirmatory, VL. No failures were considered reinfections after genome sequencing.
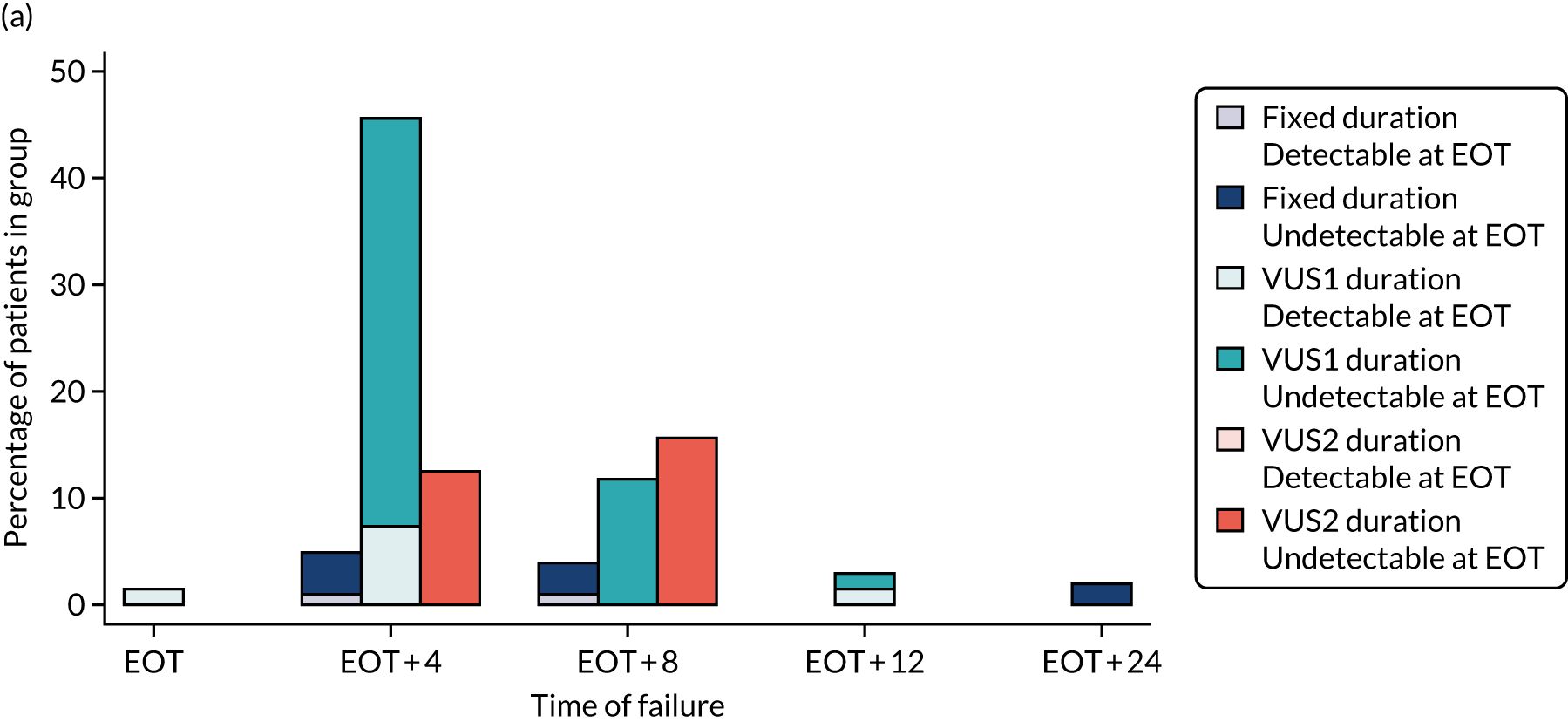
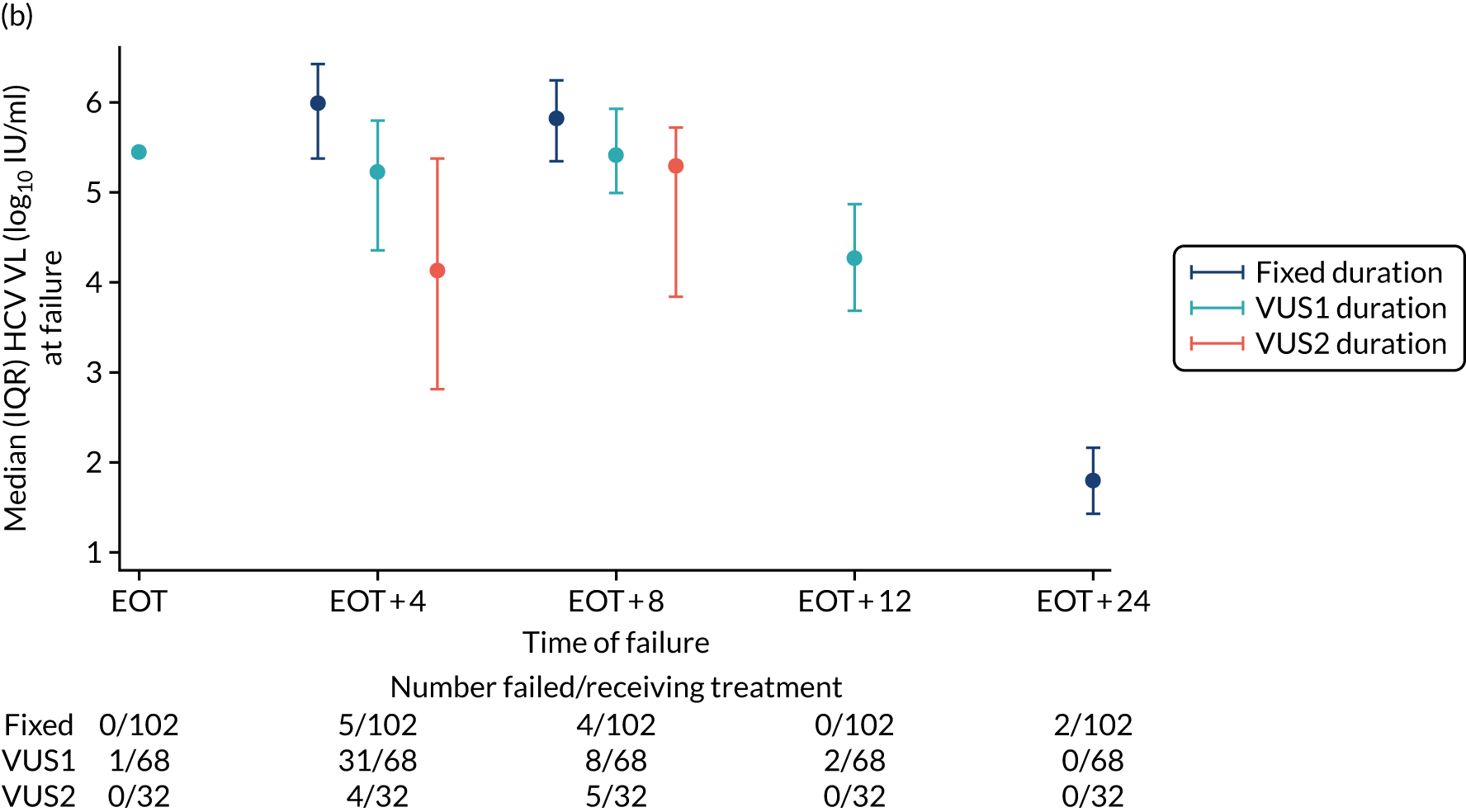
Changes in viral load and viral load suppression (prespecified analysis)
There was no evidence of differences in the mean HCV VL according to either type of randomisation (duration or ribavirin) from screening up to day 14 (Figures 24 and 25). There was also no evidence of difference in the percentage of patients with known undetectable VLs between those randomised to variable-duration treatment and those randomised to fixed-duration treatment up to day 28 (p = 0.13) (Figure 26). After EOT, the percentage of patients with an undetectable VL was significantly lower in the group randomised to VUS1 than in those randomised to fixed-duration treatment, but there was no evidence of a difference between those randomised to VUS2 and those randomised to fixed-duration treatment (p < 0.001 and p = 0.53, respectively). There was no evidence of difference between the percentages with undetectable VL at any time point between ribavirin randomisation groups (global p = 0.82) (Figure 27).
FIGURE 24.
Mean HCV VL on first-line treatment by duration randomisation.
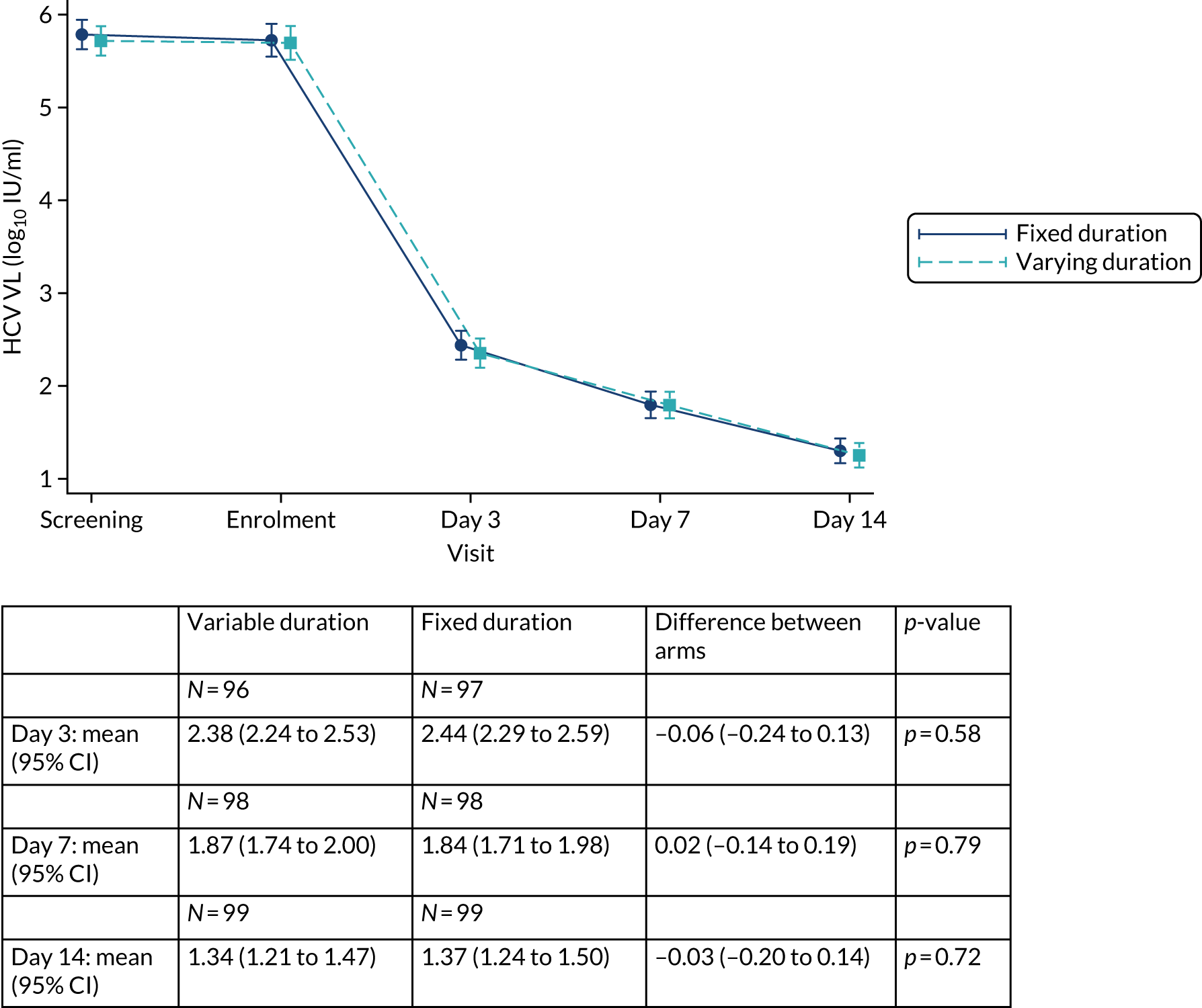
FIGURE 25.
Mean HCV VL on first-line treatment by ribavirin randomisation.
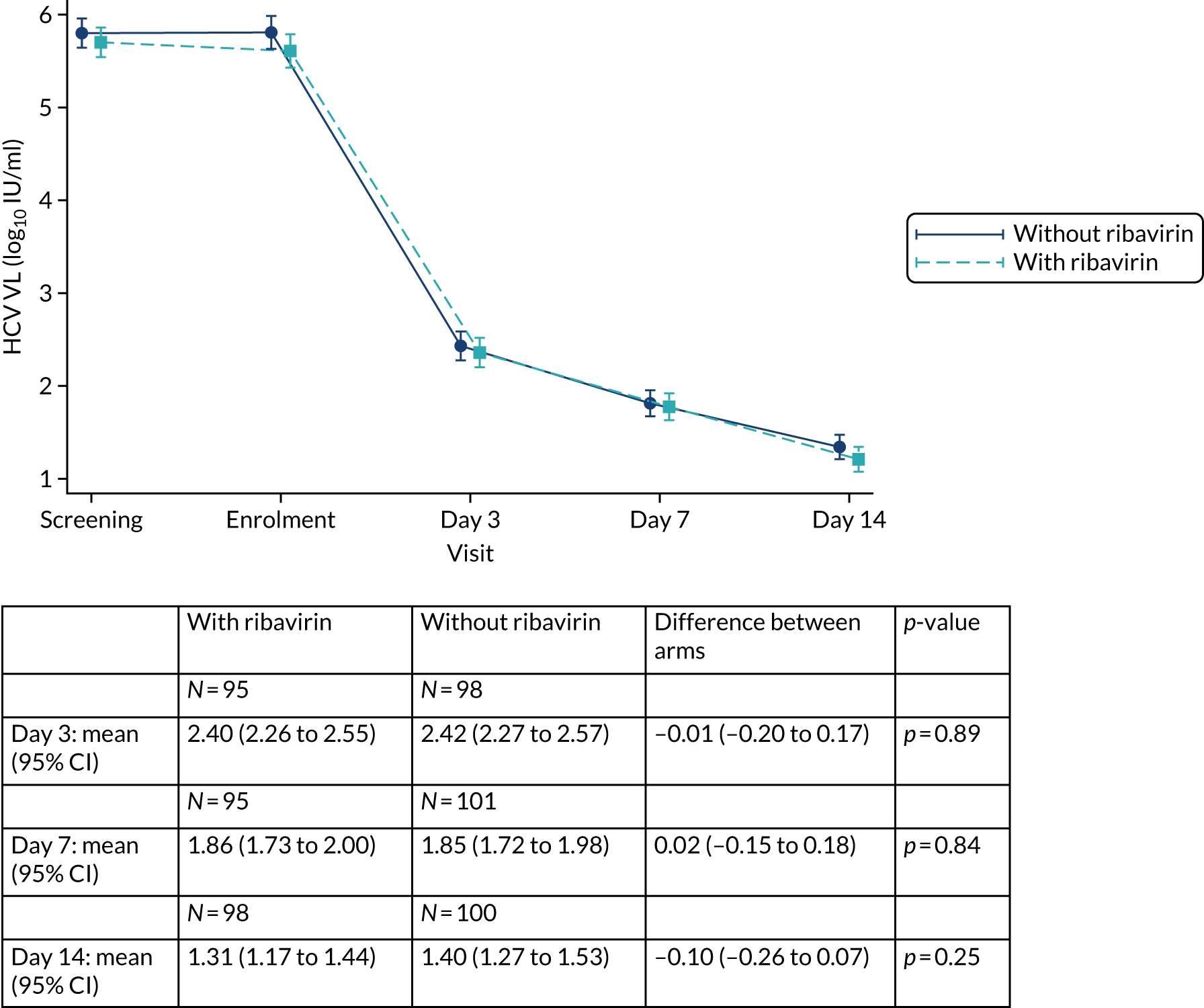
FIGURE 26.
Hepatitis C virus VL suppression by duration randomisation.
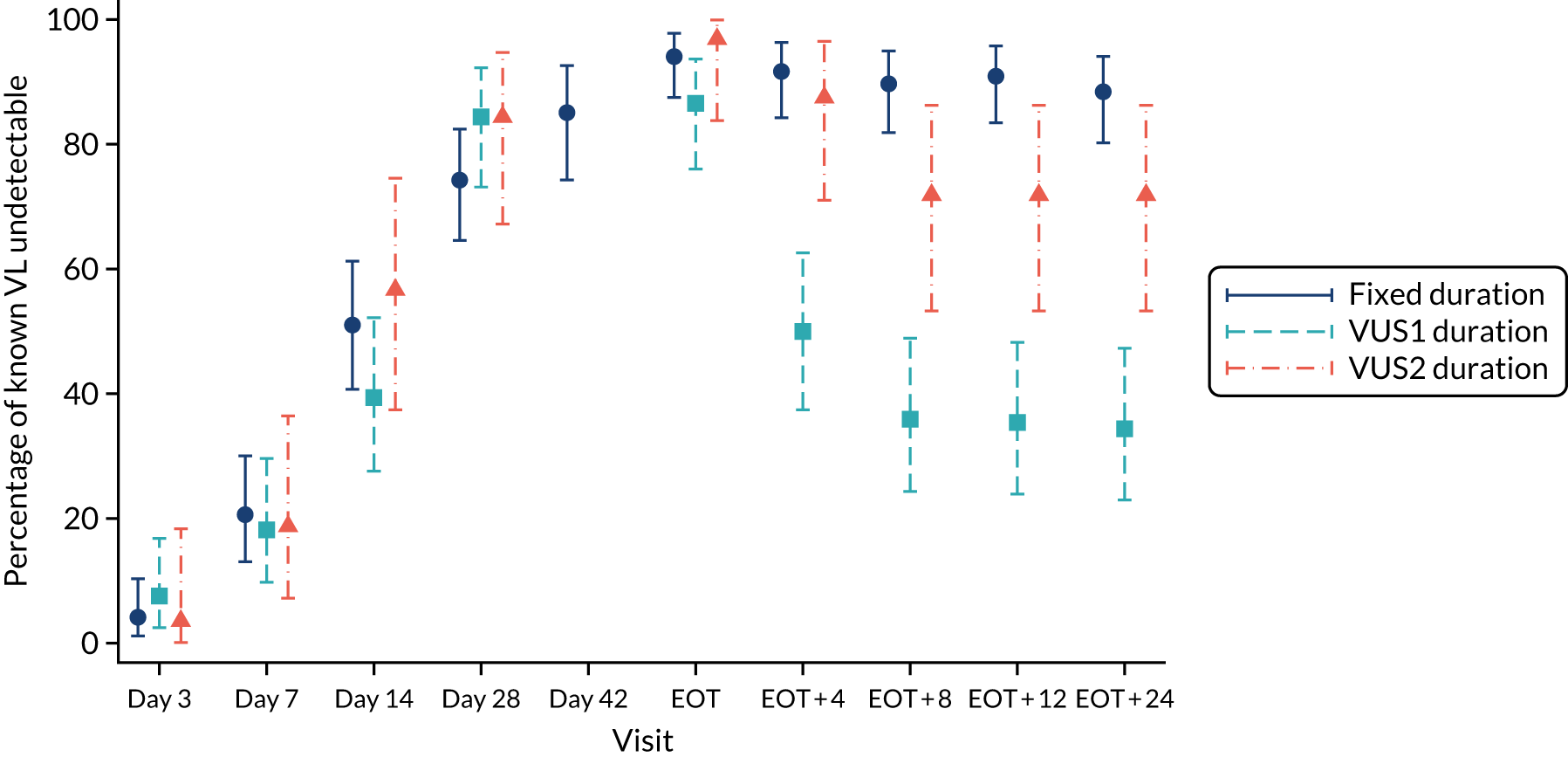
FIGURE 27.
Hepatitis C virus VL suppression on first-line treatment by ribavirin randomisation.
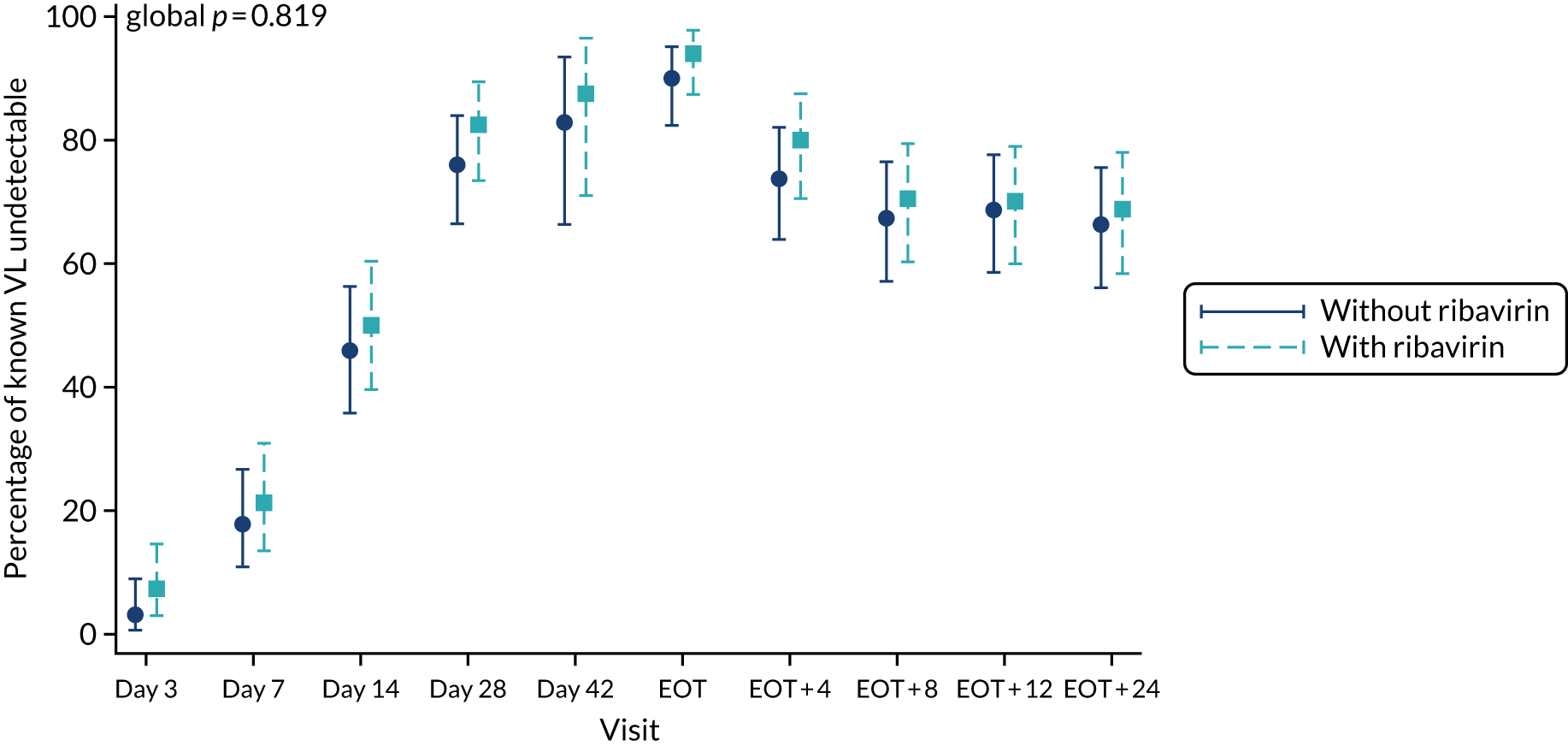
Adverse events (harms)
Ten SAEs occurred during the trial, five in each of the duration groups (Table 18) and five in each of the ribavirin groups (Table 19). None was related to study drug (Table 20). Two SAEs were life-threatening, nine necessitated or prolonged hospitalisation and one was another important medication condition. There were 21 grade 3 or 4 AEs in 14 participants, four of which were probably or definitely related to first-line drugs and eight of which were probably or definitely related to retreatment drugs. Sixteen AEs led to changes in study drugs. There was a trend towards more first-line drug changes in those randomised to ribavirin (p = 0.06) and more retreatment drug changes in those randomised to VUS treatment (p = 0.06), but there was no evidence of differences in the number of AEs between groups according to either randomisation. Three grade 3 or 4 anaemias were observed and all occurred in participants randomised to VUS treatment and ribavirin (p = 0.12).
| AE | Treatment arm | Total | p-value | |
|---|---|---|---|---|
| Varying duration | Fixed duration | |||
| Number randomised | 100 | 102 | 202 | |
| Median follow-up (IQR) [range] in weeks | 49 (29–54) [4–82] | 32 (32–33) [16–80] | 32 (31–50) [4–82] | |
| SAEs, n (%) | 5 (5) | 5 (5) | 10 (5) | p = 1.00a |
| SAE criteria, n (%) | ||||
| Life-threatening | 1 (1) | 1 (1) | 2 (1) | |
| Required or prolonged hospitalisation | 5 (5) | 4 (4) | 9 (4) | |
| Other important medical condition | 0 | 1 (1) | 1 (< 1) | |
| Relationship to trial drug, n (% of SAEs) | ||||
| Unlikely | 2 (40) | 2 (40) | 4 (40) | |
| Not related | 3 (60) | 3 (60) | 6 (60) | |
| Severe AEs, n (%) | 9 (9) | 5 (5) | 14 (7) | p = 0.28b |
| Relationship to trial drug, n (% of severe AEs) | ||||
| Definitely | 8 (50) | 0 | 8 (38) | |
| Probably | 2 (13) | 2 (40) | 4 (19) | |
| Possibly | 3 (19) | 0 | 3 (14) | |
| Unlikely | 0 | 1 (20) | 1 (5) | |
| Not related | 3 (19) | 2 (40) | 5 (24) | |
| AEs probably/definitely related to first-line drugs, n (%) | 3 (3) | 1 (1) | 4 (2) | p = 0.37 |
| AEs probably/definitely related to retreatment drugs, n (%) | 3 (3) | 1 (1) | 4 (2) | p = 0.37 |
| First-line drug changes due to AEs, n (%) | 3 (3) | 1 (1) | 4 (2) | p = 0.37 |
| Retreatment drug changes due to AEs, n (%) | 6 (6) | 1 (1) | 7 (3) | p = 0.06 |
| Grade 3/4 anaemia, n (%) | 3 (3) | 0 | 3 (1) | p = 0.12 |
| SAE | Treatment arm | Total | p-value | |
|---|---|---|---|---|
| With ribavirin | Without ribavirin | |||
| Number randomised | 100 | 102 | 202 | |
| Median follow-up (IQR) [range] in weeks | 32 (30–50) [8–82] | 32 (32–49) [4–80] | 32 (31–50) [4–82] | |
| SAEs, n (%) | 5 (5) | 5 (5) | 10 (5) | p = 1.00a |
| SAE criteria, n (%) | ||||
| Life-threatening | 1 (1) | 1 (1) | 2 (1) | |
| Required or prolonged hospitalisation | 5 (5) | 4 (4) | 9 (4) | |
| Other important medical condition | 0 | 1 (1) | 1 (< 1) | |
| Relationship to ribavirin, n (% of SAEs) | ||||
| Unlikely | 2 (40) | 2 (40) | 4 (40) | |
| Not related | 3 (60) | 3 (60) | 6 (60) | |
| Severe AEs, n (%) | 9 (9) | 5 (5) | 14 (7) | p = 0.28b |
| Relationship to trial drug, n (% of severe AEs) | ||||
| Definitely | 8 (53) | 0 | 8 (38) | |
| Probably | 1 (7) | 3 (50) | 4 (19) | |
| Possibly | 3 (20) | 0 | 3 (14) | |
| Unlikely | 1 (7) | 0 | 1 (5) | |
| Not related | 2 (13) | 3 (50) | 5 (24) | |
| AEs probably/definitely related to first-line drugs, n (%) | 3 (3) | 1 (1) | 4 (2) | p = 0.37 |
| AEs probably/definitely related to retreatment drugs, n (%) | 2 (2) | 2 (2) | 4 (2) | p = 1.00 |
| First-line drug changes due to AEs, n (%) | 4 (4) | 0 | 4 (2) | p = 0.06 |
| Retreatment drug changes due to AEs, n (%) | 4 (4) | 3 (3) | 7 (3) | p = 0.72 |
| Grade 3/4 anaemia | 3 (3) | 0 | 3 (1) | p = 0.12 |
| Event | Treatment arm (n) | Total events (n) | |||
|---|---|---|---|---|---|
| Variable duration, ribavirin | Variable duration, no ribavirin | Fixed duration, ribavirin | Fixed duration, no ribavirin | ||
| SAE | |||||
| Accidental drug overdosea | 1 | 0 | 0 | 0 | 1 |
| Acute appendicitis | 0 | 0 | 0 | 1 | 1 |
| Adenocarcinoma in lower third of oesophagusa | 0 | 0 | 0 | 1 | 1 |
| Burn to foot: degree unknown | 1 | 0 | 0 | 0 | 1 |
| Liver abscess | 1 | 0 | 0 | 0 | 1 |
| Lower respiratory tract infection: pneumonia | 1 | 0 | 0 | 0 | 1 |
| Musculoskeletal pain in chest radiating to left arm | 0 | 0 | 0 | 1 | 1 |
| Pericarditis | 1 | 0 | 0 | 0 | 1 |
| Right epididymo-orchitis | 0 | 0 | 0 | 1 | 1 |
| Urinary sepsis | 0 | 0 | 0 | 1 | 1 |
| Severe AE | |||||
| Abscess leg | 1 | 0 | 0 | 0 | 1 |
| Alcohol intoxication acute | 0 | 0 | 0 | 1 | 1 |
| Anaemia | 2 | 0 | 0 | 0 | 2 |
| Cellulitis of leg | 0 | 1 | 1 | 0 | 2 |
| Concentration loss | 1 | 0 | 0 | 0 | 1 |
| Haemoglobin low | 1 | 0 | 0 | 0 | 1 |
| Hyperbilirubinaemia | 0 | 1 | 0 | 0 | 1 |
| Inguinal hernia | 1 | 0 | 0 | 0 | 1 |
| Insomnia | 3 | 0 | 0 | 0 | 3 |
| Jaundice | 0 | 0 | 0 | 1 | 1 |
| Lethargy | 1 | 0 | 0 | 0 | 1 |
| Low mood | 1 | 0 | 0 | 0 | 1 |
| Mouth sores | 1 | 0 | 0 | 0 | 1 |
| Pyelonephritis | 0 | 0 | 1 | 0 | 1 |
| Suicidal ideation | 1 | 0 | 0 | 0 | 1 |
| Syncope | 0 | 0 | 0 | 1 | 1 |
| Tinnitus | 1 | 0 | 0 | 0 | 1 |
| AEs probably/definitely related to trial drugs | |||||
| Anaemia | 2 | 0 | 0 | 0 | 2 |
| Concentration loss | 1 | 0 | 0 | 0 | 1 |
| Haemoglobin low | 1 | 0 | 0 | 0 | 1 |
| Hyperbilirubinaemia | 0 | 1 | 0 | 0 | 1 |
| Insomnia | 3 | 0 | 0 | 0 | 3 |
| Jaundice | 0 | 0 | 0 | 1 | 1 |
| Lethargy | 1 | 0 | 0 | 0 | 1 |
| Low mood | 1 | 0 | 0 | 0 | 1 |
| Syncope | 0 | 0 | 0 | 1 | 1 |
| Drug changes due to AEs | |||||
| Anaemia | 3 | 3 | 0 | 0 | 6 |
| Concentration loss | 1 | 0 | 0 | 0 | 1 |
| Haemoglobin low | 1 | 0 | 1 | 0 | 2 |
| Hair loss | 0 | 0 | 1 | 0 | 1 |
| Hyperbilirubinaemia | 0 | 1 | 0 | 0 | 1 |
| Insomnia | 1 | 0 | 0 | 0 | 1 |
| Lethargy | 1 | 0 | 0 | 0 | 1 |
| Low mood | 1 | 0 | 0 | 0 | 1 |
| Mouth ulcer | 1 | 0 | 0 | 0 | 1 |
| Mouth sores | 1 | 0 | 0 | 0 | 1 |
There was no evidence of differences in levels of haemoglobin, alkaline phosphatase (ALP) or bilirubin or in CrCl between the duration randomisation groups at any time point (p > 0.10); however, there were differences at EOT and EOT plus 12 weeks in ALT level (each p = 0.02) and from day 28 in aspartate aminotransferase (AST) level (p < 0.01) (Figure 28). In the case of the ribavirin randomisation, there was no evidence of a difference between randomised groups in ALT, AST or ALP level or in CrCl at any time point (p > 0.11) (Figure 29). For those randomised to ribavirin, there was a significant decrease in haemoglobin and a significant increase in bilirubin while on treatment (p < 0.001); however, levels had returned to normal by EOT plus 12 weeks.
FIGURE 28.
Change in laboratory parameters by duration randomisation: (a) haemoglobin; (b) ALT; (c) AST; (d) ALP; (e) CrCl; and (f) bilirubin. RT, retreatment; W, week.
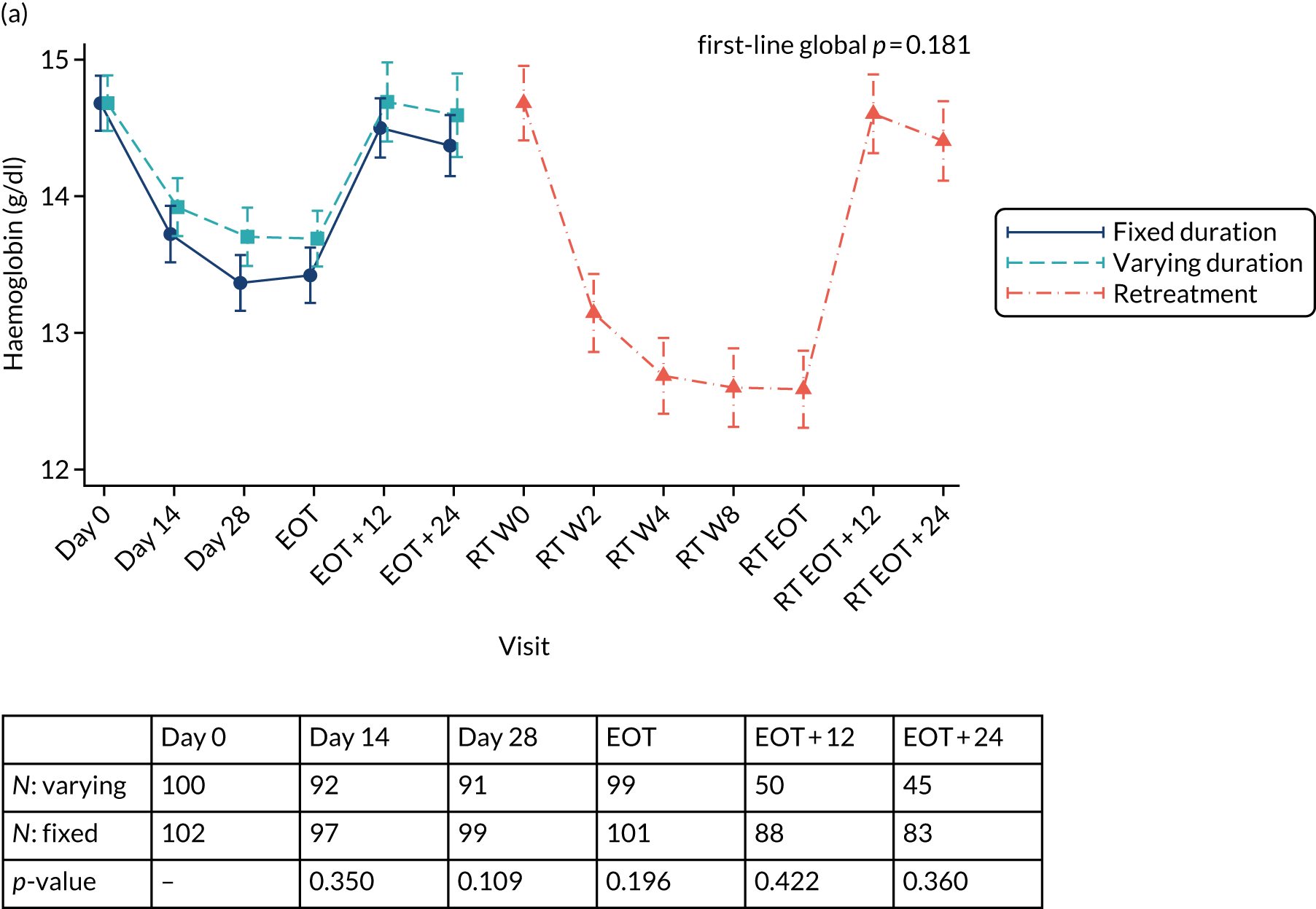
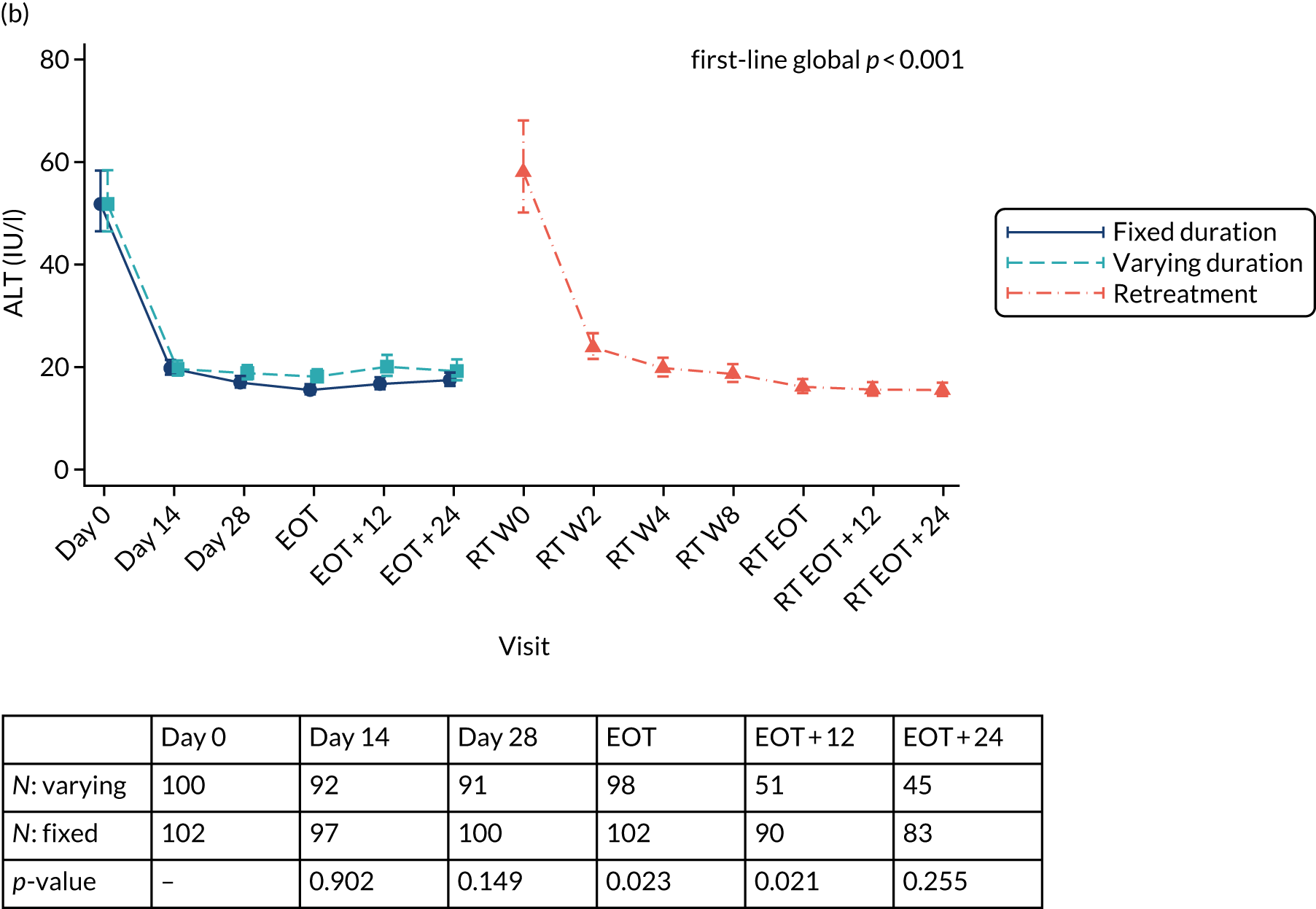
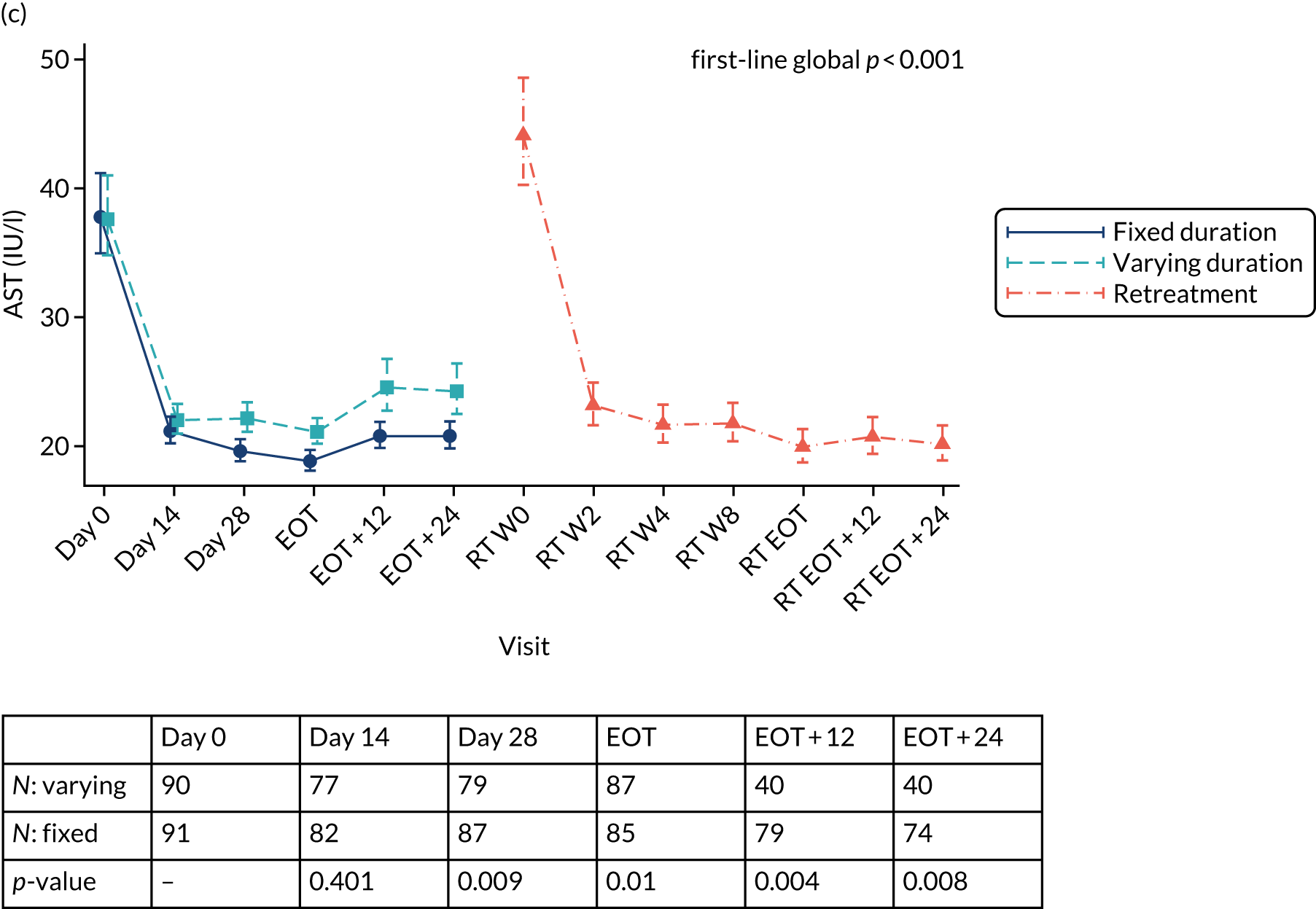
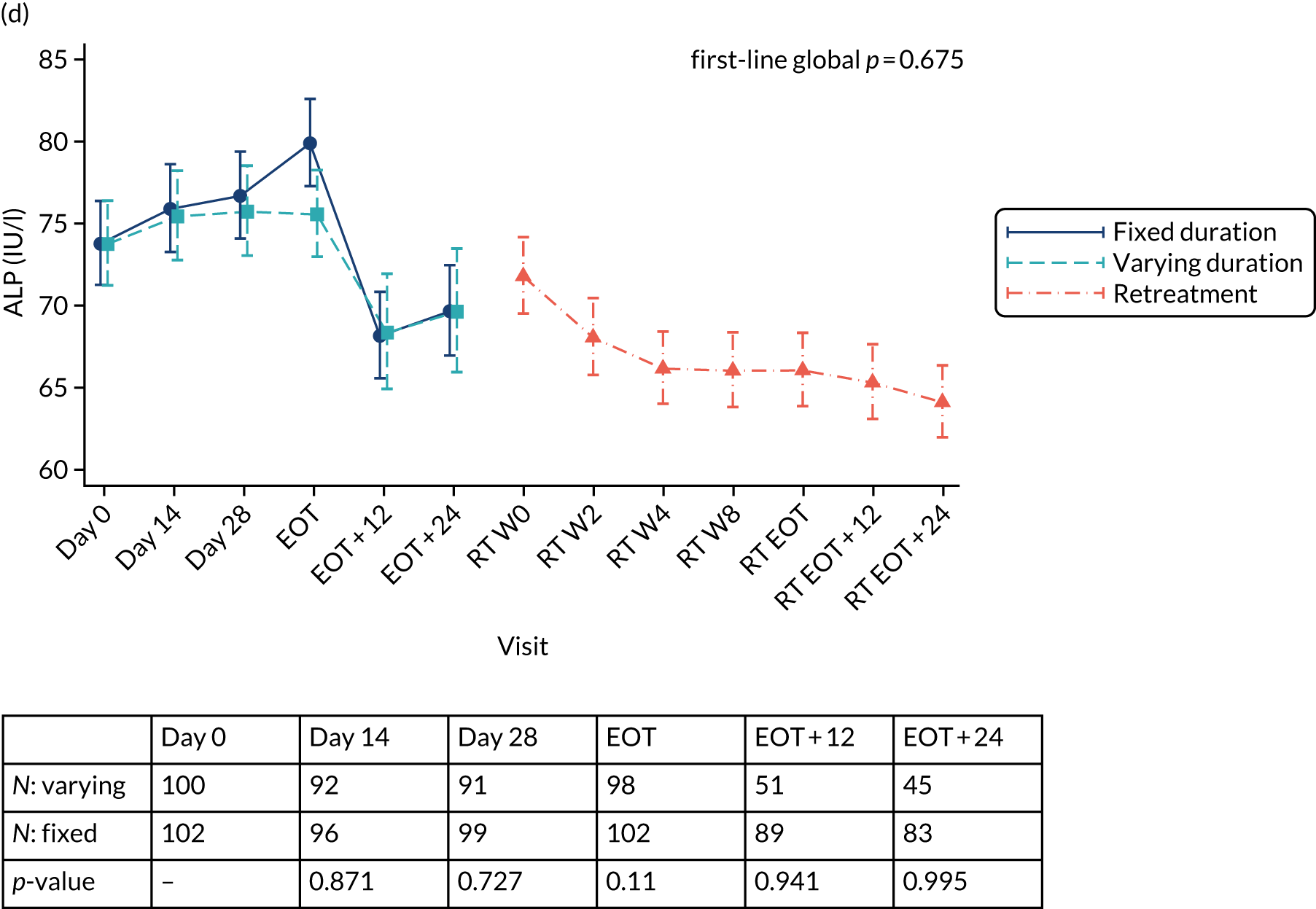
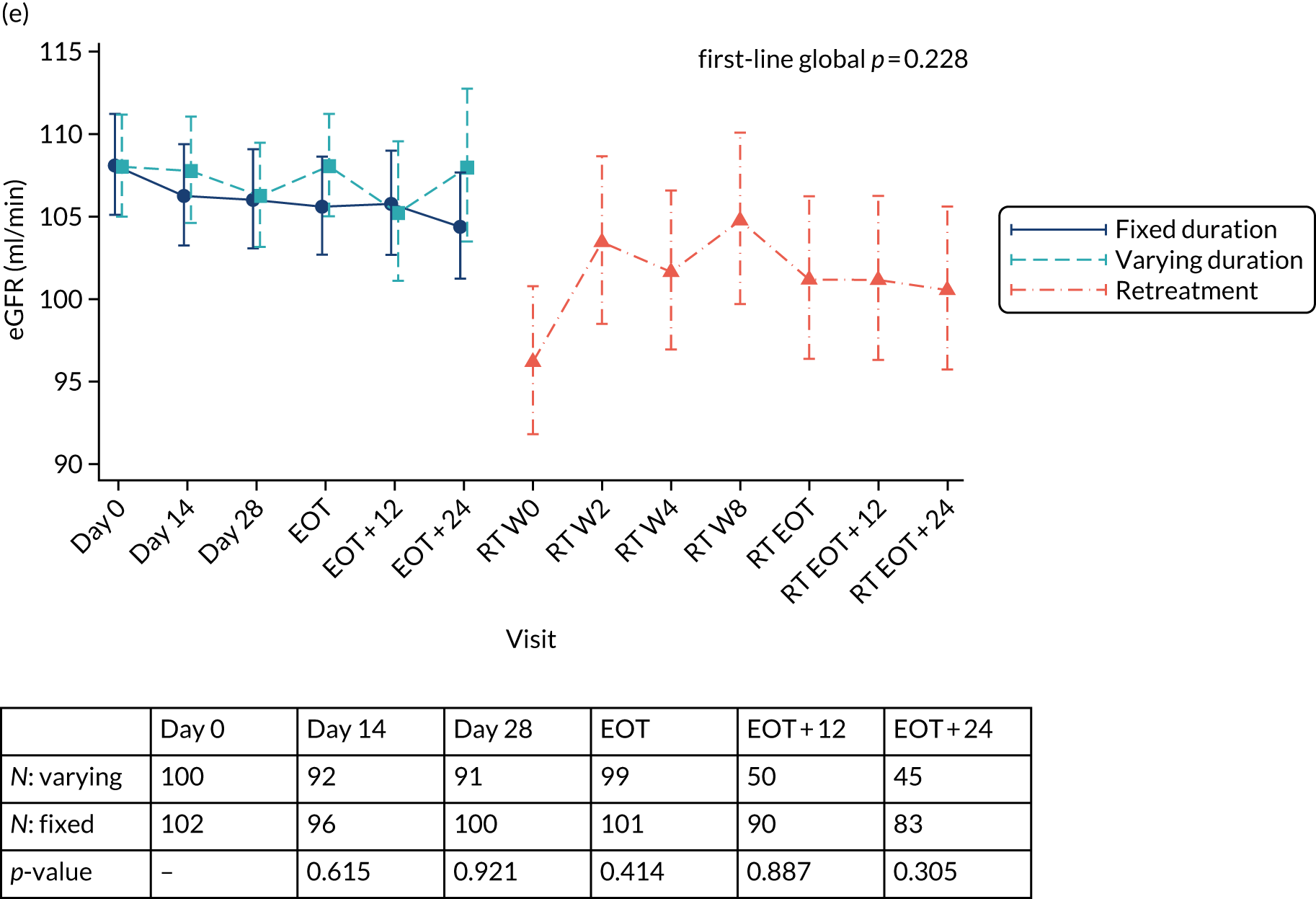
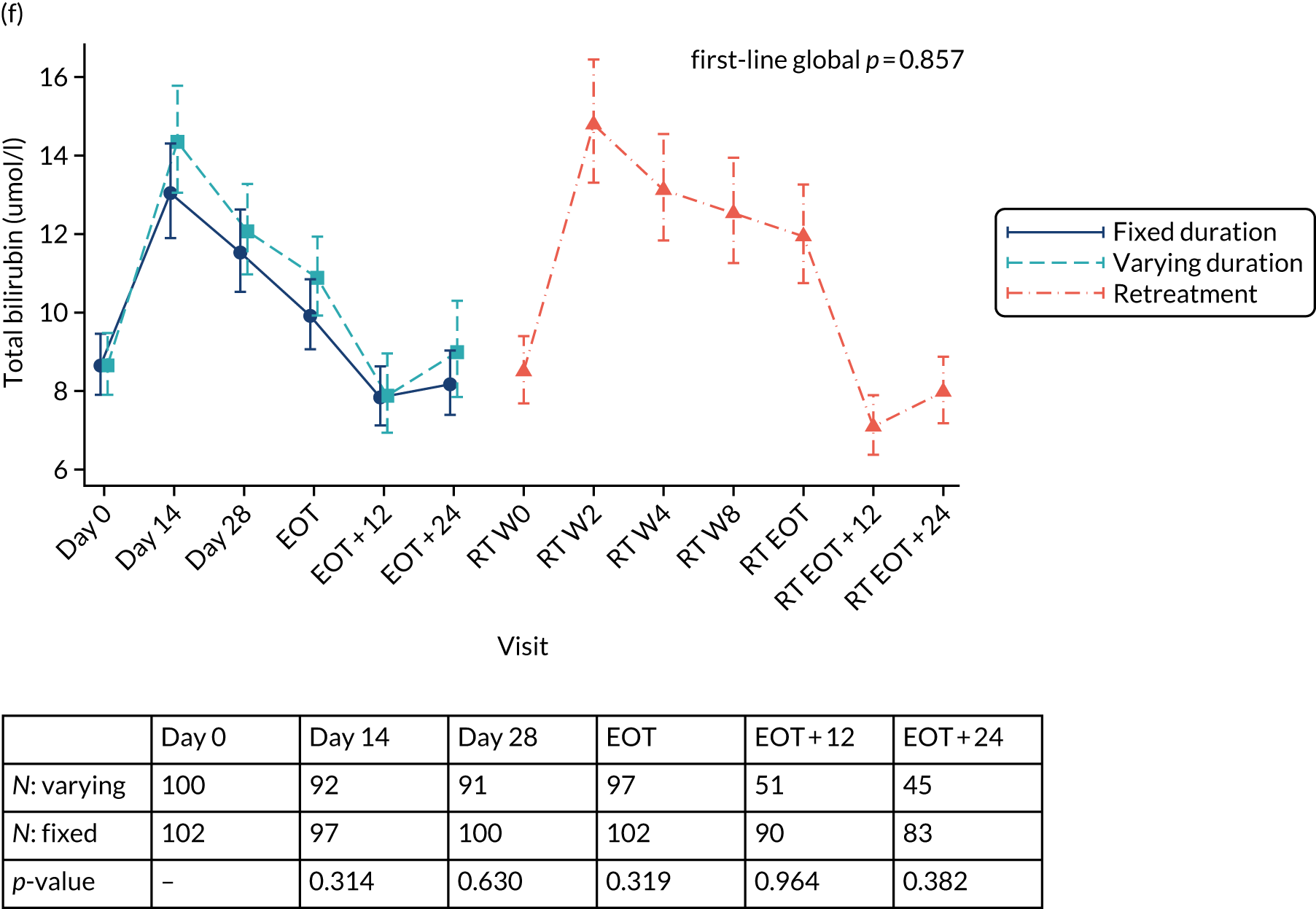
FIGURE 29.
Change in laboratory parameters by ribavirin randomisation: (a) haemoglobin; (b) ALT; (c) AST; (d) ALP; (e) CrCl; and (f) bilirubin. RT, retreatment; W, week.
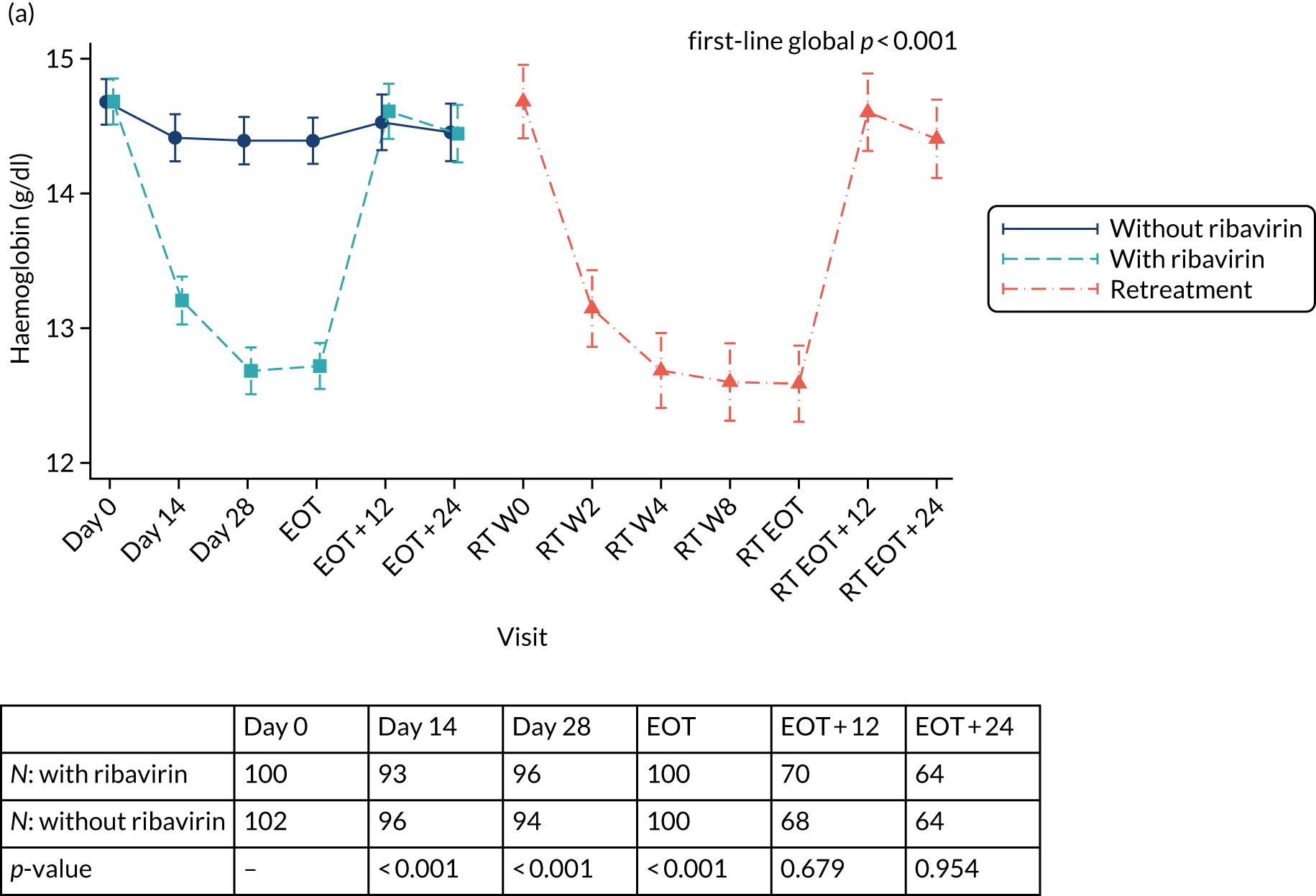
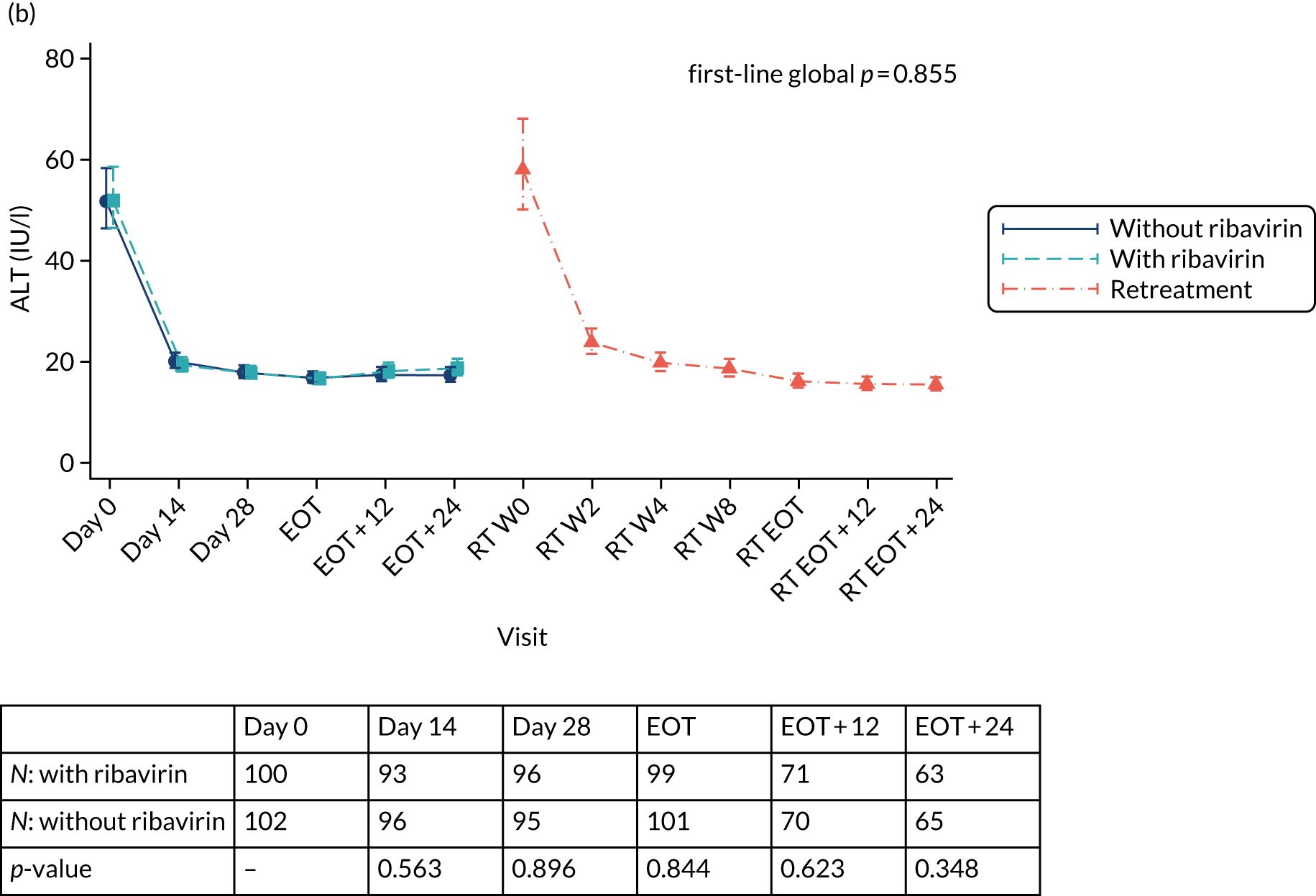
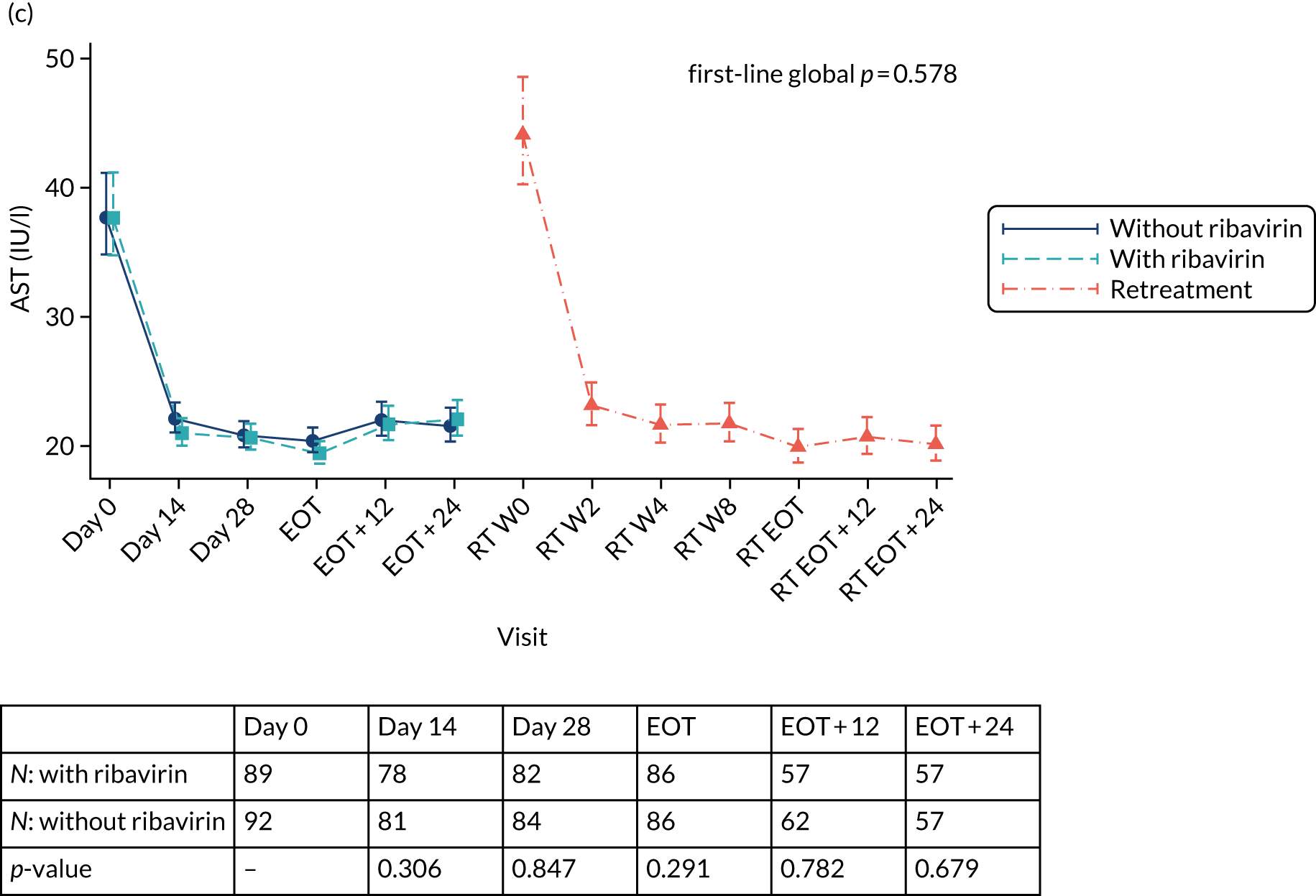
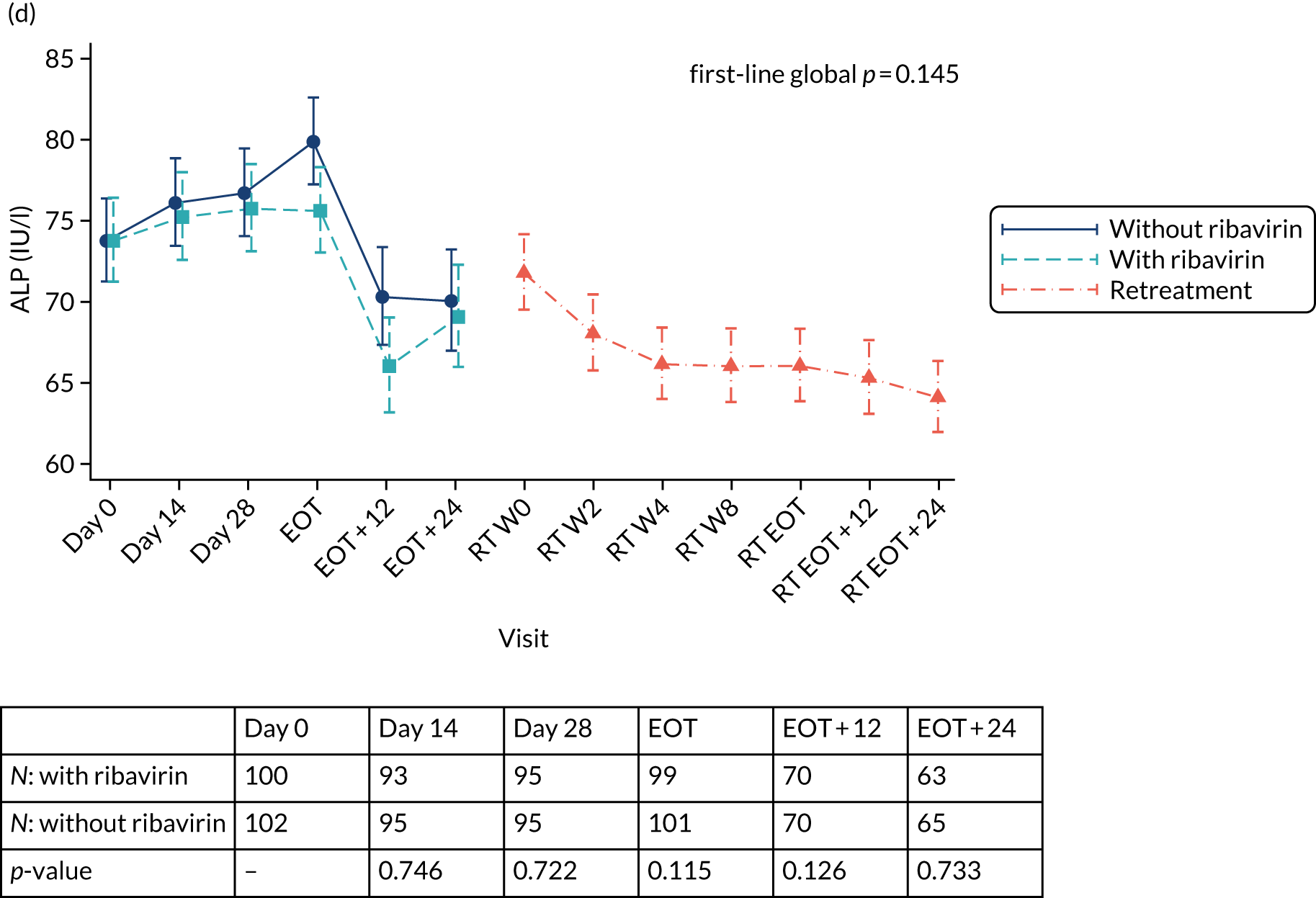
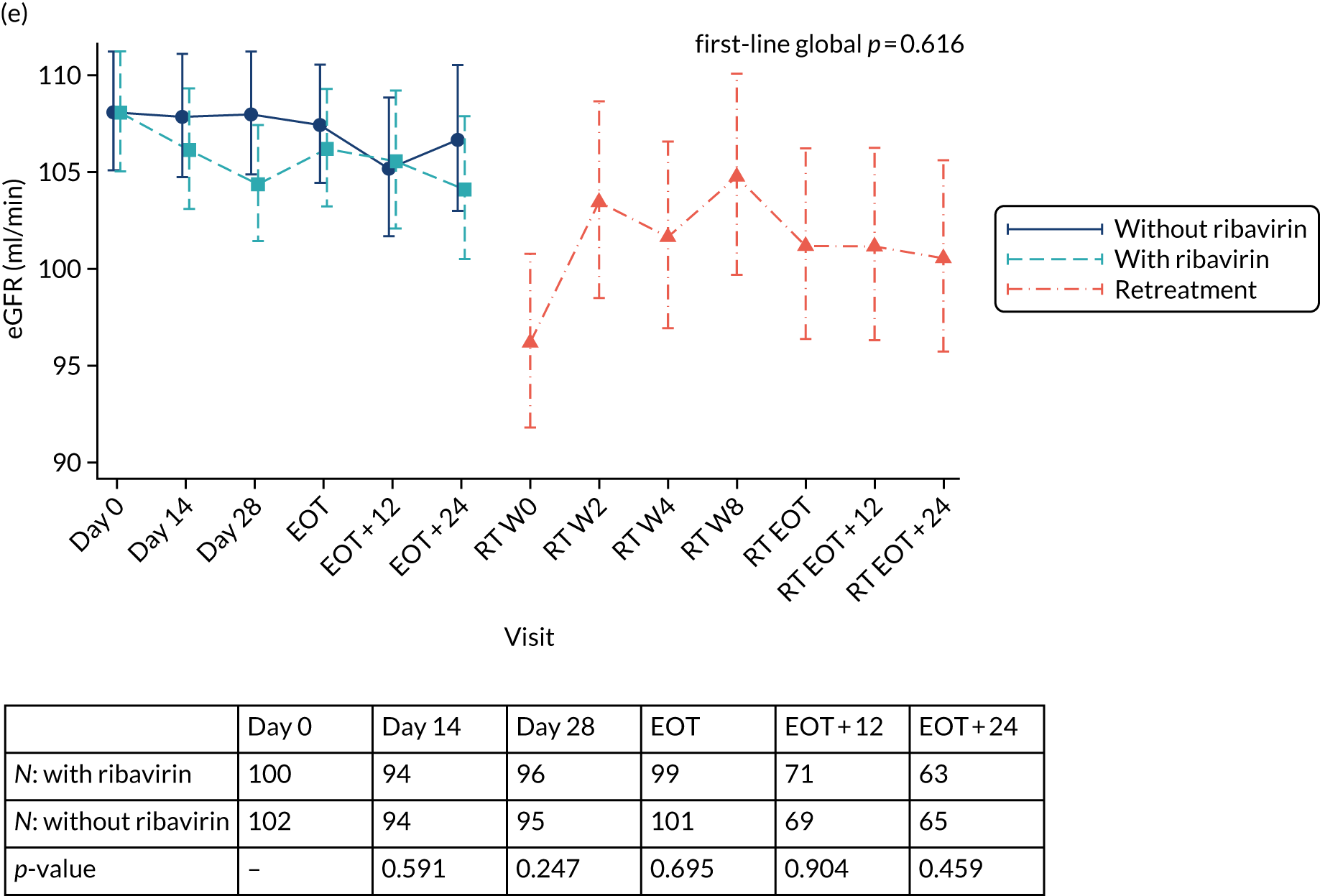
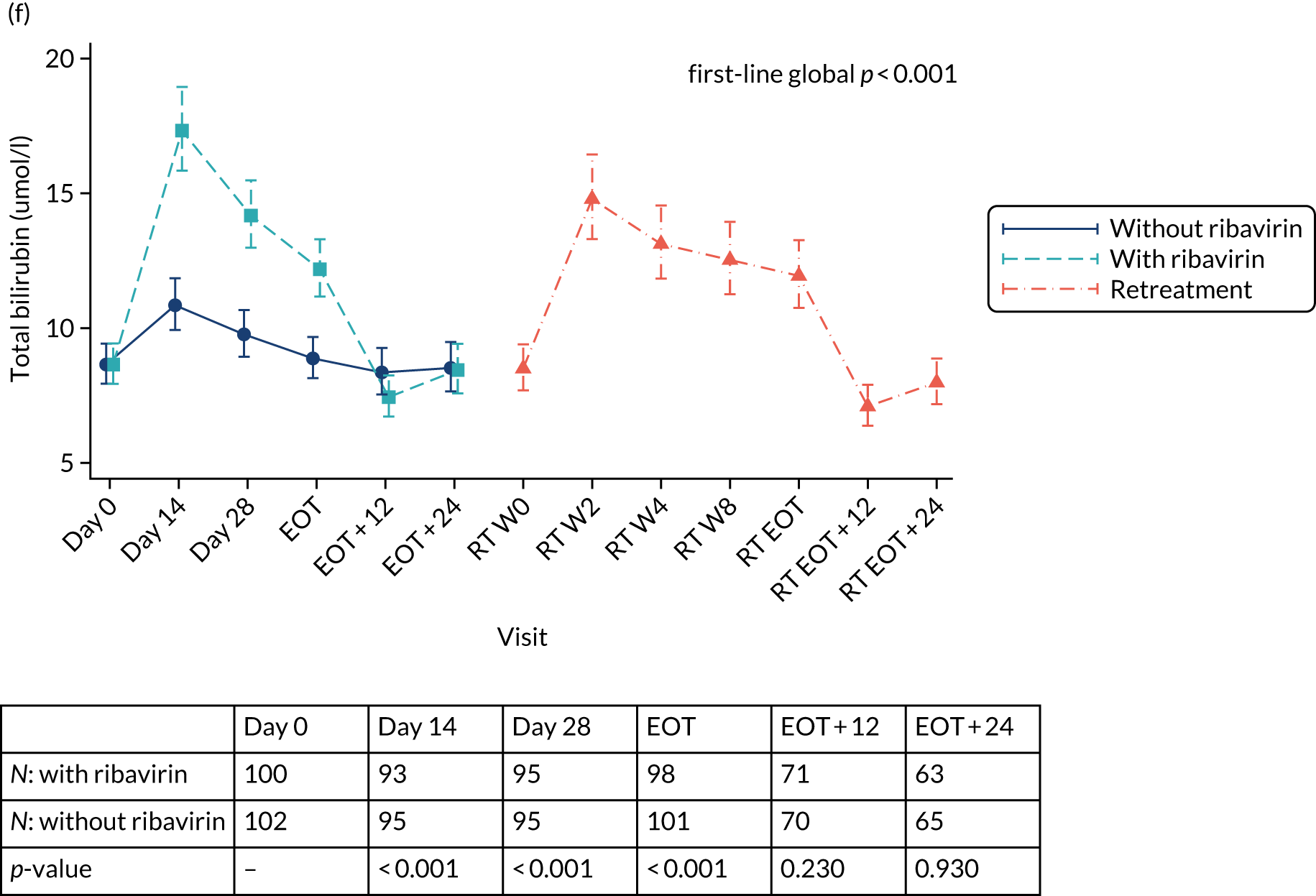
Chapter 4 Discussion
Interpretation
The main question addressed by the trial was whether or not a strategy of using shorter, and personalised (based on screening HCV VL), first-line DAA treatment followed by immediate retreatment with standard 12-week treatment courses in everyone not achieving initial cure, overall, can achieve the same rates of SVR12 as standard fixed-duration first-line treatment courses, potentially while using less drug overall. To the best of our knowledge, this is one of the very few, large, strategic, post-licensing trials in this disease area.
Overall, we found non-inferiority of strategies using first-line ultrashort treatment durations, with both groups achieving 100% SVR12 rate after retreatment. Although the initial shortening strategy (i.e. VUS1) was able to cure only 36% of participants after first-line treatment, strikingly, a relatively small increase in ultrashort treatment duration (from a mean of 32 days to 39 days) led to SVR12 rates doubling from 36% to 72%. Interestingly, the rate of SVR12 achieved by the 8-week fixed-duration strategy (91%) was not higher than in previous Phase II trials of shorter treatment courses,23 even though the trial inclusion criteria limited VL to under 6,000,000 IU/ml, which would have been expected to reduce the risk of failure. The first-line cure rates achieved even with VUS2 are not sufficiently high to routinely recommend such a strategy for stable patients able to adhere to 8–12 weeks’ therapy, particularly as overall mean treatment duration with VUS2 was very similar to the overall mean treatment duration with the fixed 8-week duration. However, the findings do suggest that a high proportion of patients can be cured with, on average, approximately 60% of the licensed duration of first-line therapy with the agents used in the trial. Further work is ongoing to identify predictors of cure on VUS1 and VUS2.
Declining adherence to DAA therapy as treatment continues has been demonstrated previously, with patients suggesting ‘feeling as if the treatment is working’ as a reason for decreasing adherence. 10 Adherence is often considered to be better in trials than in real-world situations, but we found similar decreases in adherence with time on first-line treatment, with 28% of participants reporting any missed first-line doses. Adherence to retreatment was even poorer than adherence to first-line treatment. However, the fact that retreatment resulted in 100% SVR12 despite this again illustrates the likely overtreatment of the majority of individuals by standard courses. SVR12 was 72% with VUS2 of mean duration 39 days (despite 28% of participants reporting missing any first-line doses), suggesting that intermittent non-adherence may be less important than overall adherence during weeks 4–8 of first-line treatment. These findings emphasise the importance of supporting adherence after week 4 of therapy to ensure good cure rates in hard-to-reach populations. 14,25
In practice, one important concern about starting treatment in a patient considered unlikely to complete an 8- to 12-week course is the risk of virological failure with emergent resistance that might compromise retreatment options. This is particularly the case when, as in this trial, retreatment does not include a protease inhibitor (in contrast to licensed retreatment options). In our population, shorter first-line treatment strategies did not reduce participants’ ultimate ability to achieve SVR12. The trial used a first-line regimen that is active only against specific genotypes; however, there is no obvious reason why this would not also be the case with pangenotypic first-line treatment regimens. Retreatment with a 12-week course of sofosbuvir, ledipasvir and ribavirin achieved 100% cure rates, a finding that is reassuring from an ethics perspective in terms of future trials of short-course or personalised therapy, but also suggests that it may be a reasonable retreatment option for patients failing therapy more generally, at least in some situations, for example where access to licensed retreatment options, such as sofosbuvir/velpatasvir/voxilaprevir, remains limited.
Ribavirin is generally considered to have a relatively poor side-effect profile, with anaemia and fatigue, in particular, being common. The very high efficacy and low AE rates achieved with DAAs7,16 have, therefore, limited ribavirin’s role in treatment recommendations in the DAA era. However, some patients may still benefit from ribavirin,17 with some preliminary work suggesting that adding ribavirin to short-course therapy does increase SVR12 rates26 and possibly also reduces the rate of emergent resistance in patients failing therapy. 27 Here, adjunctive ribavirin was actually well tolerated in first-line treatment and retreatment, with only 2–4% of participants experiencing AEs related to trial drugs or causing changes to trial drugs, including ribavirin, despite careful elicitation of AEs at scheduled visits.
However, across both the fixed 8-week and the variable ultrashort randomised groups, there was no evidence of improvements in SVR12 with adjunctive ribavirin. In participants randomised to 4–7 weeks’ first-line therapy, SVR12 rates were 8% higher in those randomised to ribavirin, but this result did not reach statistical significance. However, for the first time in a randomised comparison, we found that the emergence of resistance was significantly lower in those failing therapy (12% with ribavirin vs. 38% without). Further work to characterise the impact of ribavirin on different resistance variants and the mechanism of action of ribavirin is ongoing. However, it is important to note that the success of 12 weeks’ retreatment was unaffected by emergent resistance. The main role for adjunctive ribavirin may, therefore, be to reduce the potential for compromise of subsequent retreatment in patients considered at high risk of not completing standard courses of therapy.
Tailoring the duration of therapy based on initial treatment response, so-called response-guided therapy, was standard for interferon-based treatment courses that were prolonged and associated with substantial toxicity. 28 The very high viral suppression rates after 2 weeks of DAA therapy mean that response-guided approaches are not currently recommended, and there are limited data supporting such a strategy in routine practice. 29,30 For the first time, we show that very early response to treatment, namely achieving VL suppression by day 3, or even day 7, may help predict success of shortened courses of licensed therapy. Relatively few patients achieved this early suppression (approximately 5% and 20%, respectively) and early monitoring will be impractical for many outpatient settings. However, where patients are treated in a supervised setting, for example prisoners or as inpatients, such an approach may help guide management and should be investigated further. 30,31
In settings where treatment costs are proportional to duration of therapy, shortened therapy and retreatment could theoretically offer a more cost-effective approach to treatment than standard duration therapy for all. A prior modelling study comparing fixed-duration (8-week) therapy with shortened therapy (4 or 6 weeks, fixed duration) found that the 8-week fixed strategy was most likely to be cost-effective unless SVR12 rates reached 77% for 6 weeks’ therapy. 32 In our trial, a strategy with VUS1 actually used more drug (a mean of 85 days, including retreatment) and so is very unlikely to be cost-effective, although VUS2 (a mean of 63 days, including retreatment) may be in some circumstances, depending on local costs for treatment. Whether or not such an approach is viable will depend on the local health system context.
Limitations
The main trial limitation is that the trial fell short of its original recruitment target for complex reasons relating to public sector commissioning for drug treatment. However, the higher than anticipated success of retreatment (predicted to be 85% but was actually 100%) meant that the trial was still able to demonstrate non-inferiority according to its prespecified margin. Most patients were randomised to a treatment that is now not widely used and so the relevance to other treatment courses will need to be established prospectively.
Generalisability
A major strength of the trial is that it did recruit a range of participants, including around one-third of participants who were HIV/HCV co-infected and in whom we found no evidence of differential response to variable-duration strategies or to ribavirin. Around one-third of participants were also female.
This trial was designed to test strategies for treatment, rather than specific regimens. Almost all recruitment took place when the combination of ombitasvir, paritaprevir, dasabuvir and ritonavir (Viekirax) was a preferred first-line treatment in the UK NHS. This combination still remains a recommended option in the NHS, but the degree to which our findings can be generalised to other combinations with broader genotype coverage is unknown. The similar declines in HCV VL and increases in suppression seen with this and with other DAA combinations suggest that it is plausible that the relationship between duration of therapy and SVR12 is similar for other combinations approved for 12-week treatment courses in patients with mild disease.
The trial did require a stable population able to adhere to a follow-up schedule with significantly more visits than regular standard of care. Despite the fact that around one-third of participants reported actively using recreational drugs, visit non-attendance was low and self-reported adherence reasonably high.
Overall evidence
Overall, we found that unsuccessful short-course first-line therapy did not compromise retreatment with sofosbuvir/ledipasvir/ribavirin, with 100% SVR12 achieved overall and evidence of non-inferiority compared with a fixed 8-week treatment duration plus retreatment. SVR12 rates were significantly increased when ultrashort treatment varied between 4 and 7 weeks rather than between 4 and 6 weeks. We found no evidence of ribavirin significantly improving first-line SVR12, but it significantly reduced resistance emergence in those failing first-line treatment.
Chapter 5 Conclusions
Implications for health care
Currently, clinical decision-making is complex when there are concerns that a patient will be unable to complete a full course of treatment. Our findings suggest that shortened courses of treatment (of 4–7 weeks) are able to achieve cure in a majority of patients in these settings. Furthermore, where it is feasible to measure early virological response, this may have a role in identifying a group of participants who will be cured with shorter courses of therapy. More importantly, with the combination used (which included a protease inhibitor in first-line therapy), any resistance that developed did not appear to compromise retreatment and the addition of ribavirin significantly reduced the emergence of resistance in treatment failure.
Recommendations for future research
Improve individual prediction of outcome of short-course treatment
Ongoing work from this trial and other cohorts will seek to identify predictors of which patients can be successfully cured with short-course therapy before they start therapy. Such an approach is likely to include a combination of host, viral and clinical features (factors that can identify patients likely to be cured). Greater confidence in prediction will help staff and patients, save drug costs and help improve access to those who struggle to engage with health-care services.
Test the outcomes of short-course treatment in real-world settings
Increasingly, treatment is being started in patients who are unlikely to complete 8–12 weeks of therapy. Understanding the outcomes for such patients in real-world settings is important to identify what support might need to be in place to ensure that they can be cured of infection.
Understand how ribavirin reduces the emergence of resistance
The mechanism of action of ribavirin remains the subject of debate, and comparative analysis of full viral sequence from this study should shed light on how ribavirin works, with relevance not just to HCV but other infections that it is used to treat.
Validate prospectively response-guided therapy
Although VL was suppressed in only a small number of patients after 3 days of therapy, all achieved cure. Prospective validation of this approach is needed before it can be widely recommended.
Identify optimum retreatment strategies
Retreatment SVR12 rates of 100% were achieved in this study with sofosbuvir/ledipasvir/ribavirin. This drug combination is much cheaper than the National Institute for Health and Care Excellence-approved retreatment option of sofosbuvir/velpatasvir/voxilaprevir and further work is needed to understand the preferred options for retreatment, particularly where access to sofosbuvir/velpatasvir/voxilaprevir is limited.
Acknowledgements
We thank all the participants and staff from all the centres participating in the STOP-HCV-1 trial (listed in Appendix 1) and NHS England specialist commissioning.
Virological analysis was supported by the MRC Stratified Medicine Consortium (MR/K01532X/1 – STOP-HCV Consortium) and core funding from the Wellcome Trust Centre for Human Genetics provided by the Wellcome Trust (reference 090532/Z/09/Z). The MRC CTU at UCL is supported by funding from the MRC (reference MC_UU_12023/22).
STOP-HCV-1 co-applicants
Professor Graham Cooke, Professor Ann Sarah Walker, Professor Ellie Barnes, Professor Sarah Pett, Dr Stephen Ryder, Dr Daniel Forton, Professor Graham Foster, Kosh Agarwal, Ashley Brown, Dr Richard Riley and Mr Charles Gore (patient representative).
STOP-HCV-1 Trial Steering Committee
Professor Sir Ian Weller (chairperson), Dr Collette Smith, Dr Patrick Ingiliz, Mr Robert James, Ms Rachel Halford, Professor Graham Cooke, Professor Ann Sarah Walker and Professor Ellie Barnes.
STOP-HCV-1 Trial Management Group
Professor Graham Cooke, Professor Ann Sarah Walker, Fleur Hudson, Ellie Barnes, Dr Stephen Ryder, Katie Topping, Emma Hudson, Leanne McCabe, Aminata Sy, Emily Dennis, Cara Purvis, Dr Christopher Jones, Professor Sarah Pett, Helen Ainscough, Ania Spurdens, Charlotte Russell and Deborah Alawode.
Data Monitoring Committee
Professor David Lalloo (chairperson), Professor John Dillon, Professor Tim Peto, Professor Katherine Fielding and Dr Oscar Bortolami.
Contributions of authors
Graham S Cooke (https://orcid.org/0000-0001-6475-5056) was the chief investigator, he conceived, designed and managed the trial, was a site principal investigator and was responsible for participant recruitment and data collection, and co-wrote the first draft of the report.
Sarah Pett (https://orcid.org/0000-0002-4851-4727) conceived, designed and managed the trial, and reviewed and provided comment on the report.
Leanne McCabe (https://orcid.org/0000-0002-6509-8999) performed statistical analysis, and reviewed and provided comment on the report.
Christopher Jones (https://orcid.org/0000-0002-6301-2282) reviewed and provided comment on the report.
Richard Gilson (https://orcid.org/0000-0002-9572-193X) was a site principal investigator and was responsible for participant recruitment and data collection, and reviewed and provided comment on the report.
Sumita Verma (https://orcid.org/0000-0001-7021-8409) was a site principal investigator and was responsible for participant recruitment and data collection, and reviewed and provided comment on the report.
Stephen D Ryder (https://orcid.org/0000-0001-9649-4444) was a site principal investigator and was responsible for participant recruitment and data collection, and reviewed and provided comment on the report.
Jane D Collier (https://orcid.org/0000-0002-7046-1713) was a site principal investigator and was responsible for participant recruitment and data collection, and reviewed and provided comment on the report.
Stephen T Barclay (https://orcid.org/0000-0003-0415-785X) was a site principal investigator and was responsible for participant recruitment and data collection, and reviewed and provided comment on the report.
Aftab Ala (https://orcid.org/0000-0001-6257-3160) was a site principal investigator and was responsible for participant recruitment and data collection, and reviewed and provided comment on the report.
Sanjay Bhagani (https://orcid.org/0000-0003-2557-4337) was a site principal investigator and was responsible for participant recruitment and data collection, and reviewed and provided comment on the report.
Mark Nelson (https://orcid.org/0000-0002-5944-6096) was a site principal investigator and was responsible for participant recruitment and data collection, and reviewed and provided comment on the report.
Chin Lye Ch’Ng (https://orcid.org/0000-0003-3179-9313) was a site principal investigator and was responsible for participant recruitment and data collection, and reviewed and provided comment on the report.
Benjamin Stone (https://orcid.org/0000-0003-4999-2250) was a site principal investigator and was responsible for participant recruitment and data collection, and reviewed and provided comment on the report.
Martin Wiselka (https://orcid.org/0000-0002-4587-5826) was a site principal investigator and was responsible for participant recruitment and data collection, and reviewed and provided comment on the report.
Daniel Forton (https://orcid.org/0000-0002-0903-9169) was a site principal investigator and was responsible for participant recruitment and data collection, and reviewed and provided comment on the report.
Stuart McPherson (https://orcid.org/0000-0002-5638-2453) was a site principal investigator and was responsible for participant recruitment and data collection, and reviewed and provided comment on the report.
Rachel Halford (https://orcid.org/0000-0003-4569-2824) reviewed and provided comment on the report.
Dung Nguyen (https://orcid.org/0000-0001-9390-9990) performed sequencing and bioinformatic analyses, and reviewed and provided comment on the report.
David Smith (https://orcid.org/0000-0001-7778-7137) performed sequencing and bioinformatic analyses, and reviewed and provided comment on the report.
M Azim Ansari (https://orcid.org/0000-0003-2790-8353) performed sequencing and bioinformatic analyses, and reviewed and provided comment on the report.
Helen Ainscough (https://orcid.org/0000-0003-2912-2661) reviewed and provided comment on the report.
Emily Dennis (https://orcid.org/0000-0002-9634-7767) managed the trial, and reviewed and provided comment on the report.
Fleur Hudson (https://orcid.org/0000-0001-5891-7514) managed the trial, and reviewed and provided comment on the report.
Eleanor J Barnes (https://orcid.org/0000-0002-0860-0831) conceived and designed the trial, and reviewed and provided comment on the report.
Ann Sarah Walker (https://orcid.org/0000-0002-0412-8509) conceived, designed and managed the trial, and co-wrote the first draft of the report.
Data-sharing statement
The MRC CTU at UCL supports data-sharing where appropriate. Requests should be made to the corresponding author.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, NETSCC, the EME programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the EME programme or the Department of Health and Social Care.
References
- Hepatitis C Trust . Confronting the Silent Epidemic: A Critical Review of Hepatitis C Management in the UK 2013.
- Public Health England . Hepatitis C in the UK 2013.
- Patruni B, Nolte E. Hepatitis C: A Projection of Healthcare and Economic Burden in the UK. Santa Monica, CA: RAND Corporation; 2013.
- Messina JP, Humphreys I, Flaxman A, Brown A, Cooke GS, Pybus OG, et al. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology 2014;61:77-8. https://doi.org/10.1002/hep.27259.
- Hill A, Saleem J, Simmons B, Cooke GS. Effects of Sustained Virological Response on the Risk of Liver Transplant, Hepatocellular Carcinoma, Death and Re-Infection: Meta-Analysis of 129 Studies in 34,563 Patients With Hepatitis C Infection n.d.
- Hill A, Cooke G. Medicine. Hepatitis C can be cured globally, but at what cost?. Science 2014;345:141-2. https://doi.org/10.1126/science.1257737.
- AASLD-IDSA HCVGuidance Panel . Hepatitis C guidance 2018 update: AASLD-IDSA recommendations for testing, managing, and treating hepatitis C virus infection. Clin Infect Dis 2018;67:1477-92. https://doi.org/10.1093/cid/ciy585.
- World Health Organization (WHO) . Global Health Sector Strategy on Viral Hepatitis 2016.
- Younossi ZM, Stepanova M, Henry L, Nader F, Younossi Y, Hunt S. Adherence to treatment of chronic hepatitis C: from interferon containing regimens to interferon and ribavirin free regimens. Medicine 2016;95. https://doi.org/10.1097/MD.0000000000004151.
- Petersen T, Townsend K, Gordon LA, Sidharthan S, Silk R, Nelson A, et al. High adherence to all-oral directly acting antiviral HCV therapy among an inner-city patient population in a phase 2a study. Hepatol Int 2016;10:310-19. https://doi.org/10.1007/s12072–015–9680–7.
- Martinello M, Matthews GV. Management of acute HCV in the era of direct-acting antivirals: implications for elimination. Lancet Gastroenterol Hepatol 2019;4:256-7. https://doi.org/10.1016/S2468-1253(19)30001-9.
- El Sherif O, Afhdal N, Curry M. No one size fits all-shortening duration of therapy with direct-acting antivirals for hepatitis C genotype 1 infection. J Viral Hepat 2017;24:808-13. https://doi.org/10.1111/jvh.12734.
- Jones CR, Flower BF, Barber E, Simmons B, Cooke GS. Treatment optimisation for hepatitis C in the era of combination direct-acting antiviral therapy: a systematic review and meta-analysis. Wellcome Open Res 2019;4. https://doi.org/10.12688/wellcomeopenres.15411.1.
- Gilead Sciences . Harvoni US Full Prescribing Information 2017. www.accessdata.fda.gov/drugsatfda_docs/label/2014/205834s000lbl.pdf (accessed 15 June 2021).
- Welzel TM, Asselah T, Dumas EO, Zeuzem S, Shaw D, Hazzan R, et al. Ombitasvir, paritaprevir, and ritonavir plus dasabuvir for 8 weeks in previously untreated patients with hepatitis C virus genotype 1b infection without cirrhosis (GARNET): a single-arm, open-label, phase 3b trial. Lancet Gastroenterol Hepatol 2017;2:494-500. https://doi.org/10.1016/S2468-1253(17)30071-7.
- EASL . EASL recommendations on treatment of hepatitis C. J Hepatol 2018;69:461-51. https://doi.org/10.1016/j.jhep.2018.03.026.
- Feld JJ, Jacobson IM, Sulkowski MS, Poordad F, Tatsch F, Pawlotsky JM. Ribavirin revisited in the era of direct-acting antiviral therapy for hepatitis C virus infection. Liver Int 2017;37:5-18. https://doi.org/10.1111/liv.13212.
- Emmanuel B, Wilson EM, O’Brien TR, Kottilil S, Lau G. Shortening the duration of therapy for chronic hepatitis C infection. Lancet Gastroenterol Hepatol 2017;2:832-6. https://doi.org/10.1016/S2468-1253(17)30053-5.
- Nitta Y, Kawabe N, Hashimoto S, Harata M, Komura N, Kobayashi K, et al. Liver stiffness measured by transient elastography correlates with fibrosis area in liver biopsy in patients with chronic hepatitis C. Hepatol Res 2009;39:675-84. https://doi.org/10.1111/j.1872–034X.2009.00500.x.
- Chen J, Florian J, Carter W, Fleischer RD, Hammerstrom TS, Jadhav PR, et al. Earlier sustained virologic response end points for regulatory approval and dose selection of hepatitis C therapies. Gastroenterology 2013;144:1450-5.e2. https://doi.org/10.1053/j.gastro.2013.02.039.
- DAIDS Regulatory Support Center . Division of AIDS (DAIDS) Table n.d. https://rsc.niaid.nih.gov/clinical-research-sites/daids-adverse-event-grading-tables (accessed 23 July 2021).
- Thomson E, Ip CL, Badhan A, Christiansen MT, Adamson W, Ansari MA, et al. Comparison of next-generation sequencing technologies for comprehensive assessment of full-length hepatitis C viral genomes. J Clin Microbiol 2016;54:2470-84. https://doi.org/10.1128/JCM.00330–16.
- Kowdley KV, Lawitz E, Poordad F, Cohen DE, Nelson DR, Zeuzem S, et al. Phase 2b trial of interferon-free therapy for hepatitis C virus genotype 1. N Engl J Med 2014;370:222-32. https://doi.org/10.1056/NEJMoa1306227.
- Harris KA, Gilham C, Mortimer PP, Teo CG. The most prevalent hepatitis C virus genotypes in England and Wales are 3a and 1a. J Med Virol 1999;58:127-31. https://doi.org/10.1002/(SICI)1096-9071(199906)58:2<127::AID-JMV5>3.0.CO;2-K.
- Electronic Medicines Compendium . Harvoni 90 mg 400 mg Film-Coated Tablets 2019. www.medicines.org.uk/emc/product/3461 (accessed 15 June 2021).
- Madsen LW, Ovrehus A, Gerstoft J, Weis N, Berfod TS, Laurson A, et al. 4 Week Treatment for Hepatitis C – A Randomised Controlled Trial (4RIBC). Vienna: EASL; 2019.
- Galli A, Mens H, Gottwein JM, Gerstoft J, Bukh J. Antiviral effect of ribavirin against HCV associated with increased frequency of G-to-A and C-to-U transitions in infectious cell culture model. Sci Rep 2018;8. https://doi.org/10.1038/s41598–018–22620–2.
- Di Martino V, Richou C, Cervoni JP, Sanchez-Tapias JM, Jensen DM, Mangia A, et al. Response-guided peg-interferon plus ribavirin treatment duration in chronic hepatitis C: meta-analyses of randomized, controlled trials and implications for the future. Hepatology 2011;54:789-800. https://doi.org/10.1002/hep.24480.
- Yakoot M, Abdo AM, Abdel-Rehim S, Helmy S. Response tailored protocol versus the fixed 12 weeks course of dual sofosbuvir/daclatasvir treatment in Egyptian patients with chronic hepatitis C genotype-4 infection: a randomized, open-label, non-inferiority trial. EBioMedicine 2017;21:182-7. https://doi.org/10.1016/j.ebiom.2017.05.011.
- Lau G, Benhamou Y, Chen G, Li J, Shao Q, Ji D, et al. Efficacy and safety of 3-week response-guided triple direct-acting antiviral therapy for chronic hepatitis C infection: a phase 2, open-label, proof-of-concept study. Lancet Gastroenterol Hepatol 2016;1:97-104. https://doi.org/10.1016/S2468–1253(16)30015–2.
- Dahari H, Canini L, Graw F, Uprichard SL, Araújo ES, Penaranda G, et al. HCV kinetic and modeling analyses indicate similar time to cure among sofosbuvir combination regimens with daclatasvir, simeprevir or ledipasvir. J Hepatol 2016;64:1232-9. https://doi.org/10.1016/j.jhep.2016.02.022.
- Fawsitt CG, Vickerman P, Cooke G, Welton NJ. STOP-HCV Consortium . A cost-effectiveness analysis of shortened direct-acting antiviral treatment in genotype 1 noncirrhotic treatment-naive patients with chronic hepatitis C virus. Value Health 2019;22:693-70. https://doi.org/10.1016/j.jval.2018.12.011.
- Ware J, Kosinski M, Keller SD. A 12-Item Short-Form Health Survey: construction of scales and preliminary tests of reliability and validity. Med Care 1996;34:220-33. https://doi.org/10.1097/00005650–199603000–00003.
- Revicki DA, Chan K, Gevirtz F. Discriminant validity of the Medical Outcomes Study cognitive function scale in HIV disease patients. Qual Life Res 1998;7:551-9. https://doi.org/10.1023/a:1008866122441.
Appendix 1 Members of the STOP-HCV trial team
Participating centres
Brighton, Royal Sussex County Hospital
Sumita Verma, Royal Sussex County Hospital, Brighton, UK
Allison Leslie, Royal Sussex County Hospital, Brighton, UK
Maggie Cole, Royal Sussex County Hospital, Brighton, UK
Wezwei Li, Royal Sussex County Hospital, Brighton, UK
Jane Lyttle, Royal Sussex County Hospital, Brighton, UK
Heidi Barkhordar, Royal Sussex County Hospital, Brighton, UK
Mary Flowerdew, Royal Sussex County Hospital, Brighton, UK
Michelle Villanueva, Royal Sussex County Hospital, Brighton, UK
Samantha Aplin, Royal Sussex County Hospital, Brighton, UK
Lisa Furnival, Royal Sussex County Hospital, Brighton, UK
Raquel Nsuesha Akieme, Royal Sussex County Hospital, Brighton, UK
Lorraine Shah-Goodwin, Royal Sussex County Hospital, Brighton, UK
Vicky Kennard, Royal Sussex County Hospital, Brighton, UK
Lucia Macken, Royal Sussex County Hospital, Brighton, UK
Caroline McPherson, Royal Sussex County Hospital, Brighton, UK
Memory Kamhuka, Royal Sussex County Hospital, Brighton, UK
Mel Smith, Royal Sussex County Hospital, Brighton, UK
Michelle Powell, Royal Sussex County Hospital, Brighton, UK
John Bell, Royal Sussex County Hospital, Brighton, UK
Onyinyechi Uwasomba, Royal Sussex County Hospital, Brighton, UK
Dominika Wlazly, Royal Sussex County Hospital, Brighton, UK
Abraham Gihawi, Royal Sussex County Hospital, Brighton, UK
Rhea Savvidov Strudwick, Royal Sussex County Hospital, Brighton, UK
Paul Frattaroli, Royal Sussex County Hospital, Brighton, UK
Ahmed Hashim, Royal Sussex County Hospital, Brighton, UK
Zhengmai He, Royal Sussex County Hospital, Brighton, UK
Christine Laycock, Royal Sussex County Hospital, Brighton, UK
Sephora Shaw, Royal Sussex County Hospital, Brighton, UK
Hannah Heron, Royal Sussex County Hospital, Brighton, UK
Tenesa Sargent, Royal Sussex County Hospital, Brighton, UK
Edinburgh, Western General Hospital
Clifford Leen, Western General Hospital, Edinburgh, UK
Sheila Morris, Western General Hospital, Edinburgh, UK
Amy Shepherd, Western General Hospital, Edinburgh, UK
Aileen Cairns, Western General Hospital, Edinburgh, UK
Hazel Rae, Western General Hospital, Edinburgh, UK
Ruaridh Buchan, Western General Hospital, Edinburgh, UK
Glasgow, Glasgow Royal Infirmary
Stephen Barclay, Glasgow Royal Infirmary, Glasgow, UK
Dominic Rimmer, Glasgow Royal Infirmary, Glasgow, UK
Janet Johnstone, Glasgow Royal Infirmary, Glasgow, UK
John Alexander, Glasgow Royal Infirmary, Glasgow, UK
Joan Blevings, Glasgow Royal Infirmary, Glasgow, UK
Neil Lachlan, Glasgow Royal Infirmary, Glasgow, UK
Ewan Forrest, Glasgow Royal Infirmary, Glasgow, UK
Sharon Grant, Glasgow Royal Infirmary, Glasgow, UK
Susanne Cathcart, Glasgow Royal Infirmary, Glasgow, UK
Dario Salutous, Glasgow Royal Infirmary, Glasgow, UK
Guildford, Royal Surrey County Hospital
Aftab Ala, Royal Surrey County Hospital, Guildford, UK
Shahrzad Shahmehri, Royal Surrey County Hospital, Guildford, UK
Rebecca Wills, Royal Surrey County Hospital, Guildford, UK
Fiona Butler, Royal Surrey County Hospital, Guildford, UK
Laura Gordon, Royal Surrey County Hospital, Guildford, UK
Jenneke Schmit, Royal Surrey County Hospital, Guildford, UK
Tineke Edmunds, Royal Surrey County Hospital, Guildford, UK
Catherine Medcalf, Royal Surrey County Hospital, Guildford, UK
Veronica Davies, Royal Surrey County Hospital, Guildford, UK
Louise Gallagher, Royal Surrey County Hospital, Guildford, UK
Clare Kelly, Royal Surrey County Hospital, Guildford, UK
Cassila Peixoto, Royal Surrey County Hospital, Guildford, UK
Sanja Mathew, Royal Surrey County Hospital, Guildford, UK
Federica Regis, Royal Surrey County Hospital, Guildford, UK
Lee Moore, Royal Surrey County Hospital, Guildford, UK
Victoria Westall, Royal Surrey County Hospital, Guildford, UK
Max Wong, Royal Surrey County Hospital, Guildford, UK
Nichola Wakeford, Royal Surrey County Hospital, Guildford, UK
Sally Bradfeld, Royal Surrey County Hospital, Guildford, UK
Marinos Pericleous, Royal Surrey County Hospital, Guildford, UK
Leicester, Leicester Royal Infirmary
Martin Wiselka, Leicester Royal Infirmary, Leicester, UK
Adam Lewszuk, Leicester Royal Infirmary, Leicester, UK
Valerie Renals, Leicester Royal Infirmary, Leicester, UK
Kate Ellis, Leicester Royal Infirmary, Leicester, UK
Karl Hastings, Leicester Royal Infirmary, Leicester, UK
Paul Underwood, Leicester Royal Infirmary, Leicester, UK
Natasha Patel, Leicester Royal Infirmary, Leicester, UK
London, Chelsea and Westminster Hospital
Mark Nelson, Chelsea and Westminster Hospital, London, UK
Carina Bautista, Chelsea and Westminster Hospital, London, UK
Mini Thankachen, Chelsea and Westminster Hospital, London, UK
Kathryn McCormick, Chelsea and Westminster Hospital, London, UK
Helen Morgan, Chelsea and Westminster Hospital, London, UK
Francesca Ferretti, Chelsea and Westminster Hospital, London, UK
Daniel Bradshaw, Chelsea and Westminster Hospital, London, UK
Nicole Pagani, Chelsea and Westminster Hospital, London, UK
Maria Elena Noval, Chelsea and Westminster Hospital, London, UK
Maddalena Cerrone, Chelsea and Westminster Hospital, London, UK
London, Mortimer Market Centre
Richard Gilson, Mortimer Market Centre, London, UK
Sarah Pett, Mortimer Market Centre, London, UK
Nicola Stewart, Mortimer Market Centre, London, UK
Holly Wing, Mortimer Market Centre, London, UK
Marzia Fiorino, Mortimer Market Centre, London, UK
Diarmuid Nugent, Mortimer Market Centre, London, UK
Binta Sultan, Mortimer Market Centre, London, UK
Kristian Warnes, Mortimer Market Centre, London, UK
Erica Pool, Mortimer Market Centre, London, UK
Ana Milinkovic, Mortimer Market Centre, London, UK
Asma Ashraf, Mortimer Market Centre, London, UK
Jack Brophy, Mortimer Market Centre, London, UK
Lewis Haddow, Mortimer Market Centre, London, UK
Hinal Lukha, Mortimer Market Centre, London, UK
June Minton, Mortimer Market Centre, London, UK
Indrajit Ghosh, Mortimer Market Centre, London, UK
Nina Bason, Mortimer Market Centre, London, UK
London, Royal Free Hospital
Sanjay Bhagani, Royal Free Hospital, London, UK
Anne Carroll, Royal Free Hospital, London, UK
Glykeria Pakou, Royal Free Hospital, London, UK
Filippo Ferro, Royal Free Hospital, London, UK
Jonathan Edwards, Royal Free Hospital, London, UK
Nnenna Ngwu, Royal Free Hospital, London, UK
Alice Nightingale, Royal Free Hospital, London, UK
Thomas Fernandez, Royal Free Hospital, London, UK
Eric Witele, Royal Free Hospital, London, UK
Vivian Wu, Royal Free Hospital, London, UK
Janet North, Royal Free Hospital, London, UK
Mathew Wright, Royal Free Hospital, London, UK
Fatema Essaji, Royal Free Hospital, London, UK
Parizade Raymode, Royal Free Hospital, London, UK
Tabitha Mahungu, Royal Free Hospital, London, UK
Stephanie Harris, Royal Free Hospital, London, UK
Nomusa Mpofu, Royal Free Hospital, London, UK
Conor Bowman, Royal Free Hospital, London, UK
Alison Rodger, Royal Free Hospital, London, UK
Sabina Melander, Royal Free Hospital, London, UK
Aarti Nandani, Royal Free Hospital, London, UK
London, St George’s Hospital
Daniel Forton, St George’s Hospital, London, UK
Hannah Rine, St George’s Hospital, London, UK
Nick Tatman, St George’s Hospital, London, UK
Beverly Edwards, St George’s Hospital, London, UK
Alice Dainty, St George’s Hospital, London, UK
Helen Webb, St George’s Hospital, London, UK
Nia Al-Samarrai, St George’s Hospital, London, UK
Anet Soubieres, St George’s Hospital, London, UK
London, St Mary’s Hospital
Graham Cooke, St Mary’s Hospital, London, UK
Christopher Jones, St Mary’s Hospital, London, UK
Wilbert Ayap, St Mary’s Hospital, London, UK
Chi-man Wong, St Mary’s Hospital, London, UK
Ajerico Del Rosario, St Mary’s Hospital, London, UK
Michael Wood, St Mary’s Hospital, London, UK
Ken Legg, St Mary’s Hospital, London, UK
Claire Petersen, St Mary’s Hospital, London, UK
Heather Lewis, St Mary’s Hospital, London, UK
Andrew Lovell, St Mary’s Hospital, London, UK
Maryam Khan, St Mary’s Hospital, London, UK
Newcastle upon Tyne, Freeman Hospital
Stuart McPherson, Freeman Hospital, Newcastle upon Tyne, UK
Lorna Brownlee, Freeman Hospital, Newcastle upon Tyne, UK
Sarah Hogg, Freeman Hospital, Newcastle upon Tyne, UK
Maureen Foreman, Freeman Hospital, Newcastle upon Tyne, UK
Afnaan Nadeem, Freeman Hospital, Newcastle upon Tyne, UK
Steven Masson, Freeman Hospital, Newcastle upon Tyne, UK
Jack Oliver, Freeman Hospital, Newcastle upon Tyne, UK
Jennifer Gallagher, Freeman Hospital, Newcastle upon Tyne, UK
Shion Gosrani, Freeman Hospital, Newcastle upon Tyne, UK
Helen Dixon, Freeman Hospital, Newcastle upon Tyne, UK
Wendy Nichol, Freeman Hospital, Newcastle upon Tyne, UK
Penny Bradley, Freeman Hospital, Newcastle upon Tyne, UK
Rachael Forbes, Freeman Hospital, Newcastle upon Tyne, UK
Hannah Stevenson, Freeman Hospital, Newcastle upon Tyne, UK
Heather Russell, Freeman Hospital, Newcastle upon Tyne, UK
Phillipa Thwaites, Freeman Hospital, Newcastle upon Tyne, UK
Jade Davison, Freeman Hospital, Newcastle upon Tyne, UK
Nottingham, Queens Medical Centre
Stephen Ryder, Queens Medical Centre, Nottingham, UK
Juliette Verheyden, Queens Medical Centre, Nottingham, UK
Zoe Rose, Queens Medical Centre, Nottingham, UK
Jack Squires, Queens Medical Centre, Nottingham, UK
Sheila Hodgson, Queens Medical Centre, Nottingham, UK
Bernie Cook, Queens Medical Centre, Nottingham, UK
Sally Hodgkinson, Queens Medical Centre, Nottingham, UK
Paul Douglas, Queens Medical Centre, Nottingham, UK
Victoria Scott-South, Queens Medical Centre, Nottingham, UK
Elizabeth Keddie Gray, Queens Medical Centre, Nottingham, UK
David Nash, Queens Medical Centre, Nottingham, UK
Robert Scott, Queens Medical Centre, Nottingham, UK
G Aithal, Queens Medical Centre, Nottingham, UK
J Chalmers, Queens Medical Centre, Nottingham, UK
W Fateen, Queens Medical Centre, Nottingham, UK
L James, Queens Medical Centre, Nottingham, UK
M Barnes, Queens Medical Centre, Nottingham, UK
Varinder Ryan, Queens Medical Centre, Nottingham, UK
B Reynolds, Queens Medical Centre, Nottingham, UK
R Harris, Queens Medical Centre, Nottingham, UK
P Thiagarayan, Queens Medical Centre, Nottingham, UK
Martin James, Queens Medical Centre, Nottingham, UK
Susan Congreave, Queens Medical Centre, Nottingham, UK
Oxford, John Radcliffe Hospital
Jane Collier, John Radcliffe Hospital, Oxford, UK
Denise O’Donnell, John Radcliffe Hospital, Oxford, UK
Lizzie Stafford, John Radcliffe Hospital, Oxford, UK
Louise Holland, John Radcliffe Hospital, Oxford, UK
Ellie Barnes, John Radcliffe Hospital, Oxford, UK
Nellia Sande, John Radcliffe Hospital, Oxford, UK
Christine Tsang, John Radcliffe Hospital, Oxford, UK
Wendy Chau, John Radcliffe Hospital, Oxford, UK
Mark Ainsworth, John Radcliffe Hospital, Oxford, UK
Anna Simpson, John Radcliffe Hospital, Oxford, UK
Katerina Klimova, John Radcliffe Hospital, Oxford, UK
Paola Cicconi, John Radcliffe Hospital, Oxford, UK
Adele Melton, John Radcliffe Hospital, Oxford, UK
Oliver Duncan, John Radcliffe Hospital, Oxford, UK
Evelyn Chan, John Radcliffe Hospital, Oxford, UK
Forzia Mushtaq, John Radcliffe Hospital, Oxford, UK
Toby Cade, John Radcliffe Hospital, Oxford, UK
Sheffield, Royal Hallamshire Hospital
Ben Stone, Royal Hallamshire Hospital, Sheffield, UK
Dr Christopher Durojaiye, Royal Hallamshire Hospital, Sheffield, UK
Sarah Berry, Royal Hallamshire Hospital, Sheffield, UK
Deborah Allen, Royal Hallamshire Hospital, Sheffield, UK
Meena Chopra, Royal Hallamshire Hospital, Sheffield, UK
Carol Jaques, Royal Hallamshire Hospital, Sheffield, UK
Tracey Jackson, Royal Hallamshire Hospital, Sheffield, UK
Helen Bowler, Royal Hallamshire Hospital, Sheffield, UK
Lynsey Jones, Royal Hallamshire Hospital, Sheffield, UK
Lynne Smart, Royal Hallamshire Hospital, Sheffield, UK
Sophia Fell, Royal Hallamshire Hospital, Sheffield, UK
Esme Marshall, Royal Hallamshire Hospital, Sheffield, UK
Dr Katherine Cartwright, Royal Hallamshire Hospital, Sheffield, UK
Dr Alicia Vedio, Royal Hallamshire Hospital, Sheffield, UK
Susan Thompson, Royal Hallamshire Hospital, Sheffield, UK
Dr Paul Collini, Royal Hallamshire Hospital, Sheffield, UK
Cecilia Tawiah, Royal Hallamshire Hospital, Sheffield, UK
Swansea, Singleton Hospital
Chin Lye Ch’Ng, Singleton Hospital, Swansea, UK
Caradog Thomas, Singleton Hospital, Swansea, UK
Sue Thomson, Singleton Hospital, Swansea, UK
Lynda Connor, Singleton Hospital, Swansea, UK
Glyn Gainard, Singleton Hospital, Swansea, UK
Euan Pratt, Singleton Hospital, Swansea, UK
Keith Baldwin, Singleton Hospital, Swansea, UK
Paul John, Singleton Hospital, Swansea, UK
Helen Thompson-Jones, Singleton Hospital, Swansea, UK
Sally Kneath, Singleton Hospital, Swansea, UK
Trial co-ordination and oversight
Medical Research Council Clinical Trials Unit at the University College London
Ann Sarah Walker, Medical Research Council Clinical Trials Unit at the University College London, London, UK
Fleur Hudson, Medical Research Council Clinical Trials Unit at the University College London, London, UK
Emily Dennis, Medical Research Council Clinical Trials Unit at the University College London, London, UK
Cara Purvis, Medical Research Council Clinical Trials Unit at the University College London, London, UK
Leanne McCabe, Medical Research Council Clinical Trials Unit at the University College London, London, UK
Sarah Pett, Medical Research Council Clinical Trials Unit at the University College London, London, UK
Helen Ainscough, Medical Research Council Clinical Trials Unit at the University College London, London, UK
Ania Spurdens, Medical Research Council Clinical Trials Unit at the University College London, London, UK
Deborah Alawode, Medical Research Council Clinical Trials Unit at the University College London, London, UK
Aminata Sy, Medical Research Council Clinical Trials Unit at the University College London, London, UK
Nafisah Atako, Medical Research Council Clinical Trials Unit at the University College London, London, UK
Charlotte Russell, Medical Research Council Clinical Trials Unit at the University College London, London, UK
Will Everett, Medical Research Council Clinical Trials Unit at the University College London, London, UK
Data management systems
Mary Rauchenberger
Chiara Borg
Appendix 2 Protocol changes
| Submission | Date of submission | Details | Date approval received | ||
|---|---|---|---|---|---|
| REC | MHRA | HRA | |||
| Initial application | 3 November 2015 | Initial application | Response required | Response required | Prior to HRA formation |
| Response | 15 December 2015 | Response to questions | 29 December 2015 | 31 December 2015 | Prior to HRA formation |
| Substantial amendment 1 | 21 January 2016 | Protocol v2.0 and addition of new sites | 10 February 2016 | 1 March 2016 | Prior to HRA formation |
| Substantial amendment 2 | 26 January 2016 | Change in site name: Glasgow | 29 January 2016 | n/a | Prior to HRA formation |
| Substantial amendment 3 | 13 April 2016 | Protocol v3.0 and PIS update | 29 April 2016 | 18 May 2016 | 10 June 2016 |
| Initial HRA submission | 6 May 2017 | Initial submission following HRA set up | n/a | n/a | 22 June 2016 |
| Non-substantial amendment | 5 September 2017 | Diary card update | n/a | n/a | 7 September 2016 |
| Substantial amendment 4 | 3 October 2016 | Addition of new site: Edinburgh | 5 October 2016 | n/a | 5 October 2016 |
| Substantial amendment 5 | 26 October 2016 | Protocol v4.0 and PIS update | 7 November 2016 | 30 November 2016 | 2 December 2016 |
| Substantial amendment 6 | 9 March 2017 | Change in PI at Imperial College Healthcare NHS Trust | 15 March 2017 | n/a | 15 March 2017 |
| Substantial amendment 7 | 28 April 2017 | Protocol v5.0 and PIS update (change in variable-duration intervention) | 8 May 2017 | 9 May 2017 | 6 June 2017 |
| Substantial amendment 8 | 19 June 2017 | Updated patient diary cards | 26 June 2017 | n/a | 27 June 2017 |
| Substantial amendment 9 | 17 August 2017 | Protocol v6.0 and PIS update (addition of genotype 4 patients and two further initial DAA regimens) | 19 October 2017 | 22 September 2017 | 27 October 2017 |
| Non-substantial amendment | 27 November 2017 | Recruitment extension | n/a | n/a | 28 November 2017 |
| Substantial amendment 10 | 22 February 2018 | Protocol V7.0 and PIS update | 27 March 2018 | 29 March 2018 | 11 April 2018 |
Appendix 3 Trial assessment schedules
| First-line treatment real-time tests | Screeninga | Days post randomisationb | EOT | Week post EOT | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 3 | 7 | 14 | 28 | 4 | 8 | 12 | 24 | |||
| Control: 8 weeks’ treatment | DAA | [DAA] | [DAA] | [DAA] | DAA | (See retreatment schedule below for any treatment after first-line EOT) | |||||
| Intervention maximum: 7 weeks’ treatment | DAA | [DAA] | [DAA] | [DAA] | DAA | ||||||
| Intervention minimum: 4 weeks’ treatment | DAA | [DAA] | [DAA] | [DAA] | DAA | ||||||
| Eligibility assessment | ✗ | ||||||||||
| Patient information sheet and consent | ✗ | ||||||||||
| Randomisation | ✗ | ||||||||||
| Clinical assessmentc | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| Self-reported adherence | ✗ | ✗ | ✗ | ✗ | ✗ | ||||||
| FibroScan or biopsyd | (✗) | ||||||||||
| Weight (kg) | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||||
| Height (m) | ✗ | ||||||||||
| Urine pregnancy test if child-bearing potential | ✗ | ✗ | ✗ | ✗ | ✗ | ||||||
| Quality of lifee | ✗ | ✗ | ✗ | ||||||||
| EDTA blood for haematologyf,g,h (5 ml) | (✗) | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||||
| Clotted blood for biochemistryg,h,i (5 ml) | (✗) | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||||
| Coagulation markers (2.5 ml) | (✗) | ||||||||||
| Real-time HCV VLg,h (10 ml) | (✗) | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| Point-of-care IL-28 polymorphism test (Epistem)j,k | ✗ | ||||||||||
| Total blood draw (ml) for real-time tests | – | 20 | 10 | 10 | 20 | 22.5 | 22.5 | 10 | 10 | 22.5 | 22.5 |
| If HIV-infected | |||||||||||
| HIV VL (9 ml) | (✗) | ✗ | ✗ | ||||||||
| Additional CD4 cell countl | (✗) | (✗) | (✗) | ||||||||
| Retreatment | Start of retreatment(0)a | Weeks from start of retreatment | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 2 | 4 | 8 | 12 (EOT) | 16 (EOT plus 4 weeks) | 20 (EOT plus 8 weeks) | 24 (EOT plus 12 weeks) | 36 (EOT plus 24 weeks) | ||
| 12 week’s treatment | DAA | [DAA] | DAA | DAA | |||||
| Clinical assessmentb | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| Self-reported adherence | ✗ | ✗ | ✗ | ✗ | |||||
| Weight (kg) | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||
| Urine pregnancy test if child-bearing potential | ✗ | ✗ | ✗ | ✗ | ✗ | ||||
| Quality of lifec | ✗ | ✗ | ✗ | ||||||
| EDTA blood for haematologyd,e,f (5 ml) | (✗) | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||
| Clotted blood for biochemistrye,f,g (5 ml) | (✗) | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||
| Coagulation markerse (2.5 ml) | (✗) | ||||||||
| Real-time HCV VLe,f (10 ml) | (✗) | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| Storage: sites processing all samples locally | |||||||||
| EDTA plasma for storagee,f (10 ml blood) | (✗) | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| Total blood draw (ml) if storing locally | 32.5 | 30 | 32.5 | 30 | 32.5 | 20 | 20 | 32.5 | 32.5 |
| EDTA plasma for Glasgowe,f (10 ml blood) | (✗) | ✗h | |||||||
| Total blood draw (ml) if not storing locally | 32.5 | 20 | 22.5 | 20 | 22.5 | 10 | 10 | 32.5 | 22.5 |
| Remnant plasma obtainable from local service laboratory on request from study teami | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||
| If HIV-infected | |||||||||
| HIV VL (9 ml) | ✗ | ✗ | ✗ | ||||||
| Additional CD4 cell count | (✗) | (✗) | (✗) | ||||||
Appendix 4 Trial recruitment graph
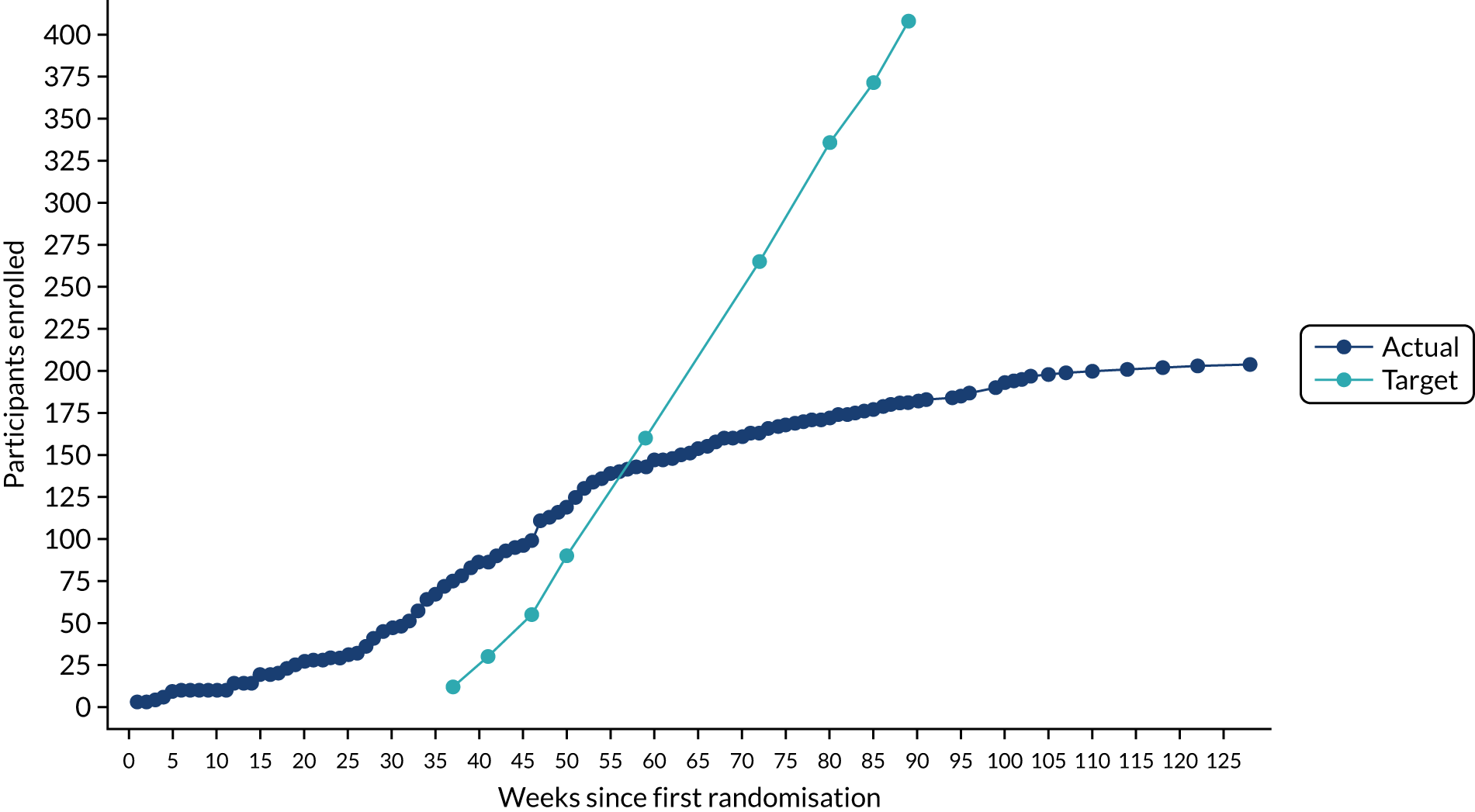
List of abbreviations
- AE
- adverse event
- ALP
- alkaline phosphatase
- ALT
- alanine aminotransferase
- AST
- aspartate aminotransferase
- b.i.d.
- bis in die (twice a day)
- BMI
- body mass index
- CI
- confidence interval
- CPT
- Child–Pugh–Turcotte
- CrCl
- creatinine clearance (Cockcroft–Gault)
- CTU
- Clinical Trials Unit
- DAA
- direct-acting antiviral
- DMC
- Data Monitoring Committee
- EOT
- end of therapy
- HCV
- hepatitis C virus
- HIV
- human immunodeficiency virus
- IL
- interleukin
- IQR
- interquartile range
- LLOQ
- lower limit of quantification
- LTFU
- lost to follow-up
- MRC
- Medical Research Council
- NS5A
- non-structural protein 5A
- OD
- once daily
- RAS
- resistance-associated substitution
- RNA
- ribonucleic acid
- SAE
- serious adverse event
- SAP
- statistical analysis plan
- SD
- standard deviation
- SmPC
- summary of product characteristics
- STOP-HCV-1
- Stratified Treatment OPtimisation for HCV-1
- SVR
- sustained virological response (persistently undetectable)
- SVR12
- sustained virological response (persistently undetectable) 12 weeks after end of therapy
- SVR24
- sustained virological response (persistently undetectable) 24 weeks after end of therapy
- UCL
- University College London
- ULN
- upper limit of normal
- VL
- viral load
- VUS
- variable ultrashort strategy