

Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 11/47/03. The contractual start date was in March 2013. The final report began editorial review in October 2019 and was accepted for publication in April 2020. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2021. This work was produced by Kehoe et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2021 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction (including scientific background and explanation of rationale)
The health-care costs of Alzheimer’s disease (AD) have previously been estimated to be equivalent to the combined costs of cancer, stroke and heart disease. These costs are also predicted to increase further, with continued increases in life expectancy worldwide set against the current absence of long-term effective therapies. 1–3 Thus, there remains an ongoing and urgent need for the discovery or development of better treatments for people with AD, particularly in the face of a number of failed intervention trials in recent years. 4,5 The aim of these treatments could be to delay the onset or slow the progression of disease, which will extend participants’ quality of life and that of their families and carers and, as a result, reduce the associated health-care burden. There is a striking benefit to be had from a treatment that has even a modest improvement: any treatment that could delay the onset of AD by 5 years could halve its prevalence. 6
Scientific background
There is scientific literature spanning several decades that suggests that hypertension in mid-life7,8 and later life9 and stroke10 increase the risk of dementia. Prior to the commencement of the Reducing pathology in Alzheimer’s Disease through Angiotensin taRgeting (RADAR) trial, we11 and others12 observed that angiotensin II (AngII)-targeting drugs [AngII type 1 receptor antagonists (AT1RAs) and angiotensin I converting enzyme 1 inhibitors (ACE1-Is)] resulted in a lower incidence of AD than other types of antihypertensive drugs. AT1RAs were also significantly more beneficial than ACE1-Is in slowing the rates of progression, hospitalisation and mortality. 11,12 Since the RADAR trial commenced, there has been further observational evidence of the protective role of ACE1-Is and AT1RAs in clinical AD. 13,14 Renin–angiotensin system (RAS)-acting drugs have been found to be associated with lower rates of conversion of mild cognitive impairment (MCI) to AD, as well as less AD-related tau neuropathology in both post-mortem brain tissue and cerebrospinal fluid (CSF) from living AD participants recruited to the Alzheimer’s Disease Neuroimaging Initiative (ADNI),15–17 whereas another analysis of the ADNI cohort showed that one of the earliest likely pathological events in the development of cognitive decline is cerebrovascular dysfunction. 18 However, not all observational studies agree. 19
Although in the vast majority of people with AD the underlying cause remains unclear, there is now acknowledgement of the likely role of vascular dysfunction in the development of AD and dementia;20 however, there remains a lack of understanding as to the possible mechanisms involved. In AD, the loss of acetylcholine and neurons due to the deposition in the brain of amyloid-β (Aβ) peptide and tau pathology is key. 2 As mentioned, cerebrovascular dysfunction is likely to be the earliest pathological event in the development of AD. 18 Such dysfunction [a likely contributor to reduced cerebral blood flow (CBF)], loss of cerebrovascular autoregulation, ischaemia and white matter hyperintensities (WMH) are all pathologies associated with, and are predictive of, loss of cognitive function21–25 and similarly common in AD. 26–28 Previous studies have shown how hypertension is associated with plasma levels of Aβ29 and AD risk. 30 Thus, molecular pathways that can connect the traditional pathological hallmarks of AD with mechanisms involved in blood pressure (BP) regulation and cerebrovascular dysfunction are likely to be strong candidates for intervention in AD, where they may act in addition to or independently of cerebrovascular-mediated pathology.
Angiotensin I converting enzyme 1 (ACE1) and neprilysin, which make AngII, are enzymes that are important to the function of the RAS, and the activity of both enzymes is elevated in the brain of AD patients. 31,32 ACE1 activity is also elevated in the peripheral blood in patients with AD,33 whereas ACE1 and neprilysin degrade Aβ in vitro and in vivo;2 however, variation in the ACE1 gene (ACE1) is associated with lower plasma levels of ACE1 and also with AD risk. 34 The multifunctional peptide AngII is likely to be an important factor in AD, particularly when in excess in the brain,35 because AngII signalling promotes the synthesis of the inflammatory mediator tumour necrosis factor alpha (TNFα),36,37 and has anticholinergic38,39 and antiglutamatergic effects,40 all of which are major sequelae of AD pathology.
Furthermore, AngII has been shown to promote the production of Aβ and phosphorylation of tau at the same amino acid residues that give rise to neurofibrillary tangles, which can be blocked by the AT1RA losartan. 41,42 More recently, we have obtained further evidence of the detrimental effects of AngII in AD. We identified that there is reduced activity of the regulatory renin–angiotensin system (rRAS) pathway that, through the action of the enzyme angiotensin I converting enzyme 2 (ACE2), works to maintain levels of AngII [which is produced by ACE1 in what is now known as the classical renin–angiotensin system (cRAS) pathway], which has been previously reviewed. 20 In these recent studies on post-mortem brain tissue, ACE2 activity was significantly reduced in brain tissue from elderly patients without dementia. In contrast, in AD patients ACE2 reductions were associated with increased Aβ and tau pathology and inversely correlated with ACE1 activity and AngII levels. 35,43 These more recent rRAS findings show very positively that the balance between ACE1 and ACE2 activities, which has a significant bearing on levels of AngII, is highly relevant to the neuropathological basis of AD.
Losartan, an AT1RA, is an effective antihypertensive drug over a wide range of ages. Losartan has been reported to cross the blood–brain barrier (BBB)44 and is the prototype drug developed for the class of AngII-blocking drugs that we, and others, have observed to be associated with reduced incidence of AD. 11,12 Losartan also improves CBF,22 a surrogate marker of cognitive performance in humans,45–47 and limits neuronal damage following ischaemia in stroke rat models. 23 Low-dose losartan (i.e. not reducing BP) has also been shown to reduce pathology and improve cognitive performance in transgenic mouse models of AD. 48 Given its antihypertensive effect, it is also conceivable that it may have a beneficial effect in reducing ischaemia-mediated WMH. 24
The timeliness of the RADAR trial
Since the commencement of the RADAR trial, to the best of our knowledge no clinical trials studying losartan or any related AT1RA drugs as an intervention in AD participants have been reported. The studies that preceded RADAR used losartan (50 mg) and reported modest and non-significant benefits on memory in non-demented hypertensive participants,49,50 which were thought to be independent of BP-lowering effects. 51 Yet, as a hypertension trial, cognition was not the primary outcome and thus was probably underpowered for a more conventional study of cognition. There remain a limited number of systematic reviews that predate the initiation of RADAR and that have assessed the impact of BP-lowering treatments on cognitive decline. 52 McGuinness et al. 52 previously concluded, based on data from four randomised controlled trials (RCTs), that BP reduction was insufficient to prevent dementia and cognitive decline in hypertensive participants with no prior cerebrovascular disease. This was supported by secondary analysis of The ONgoing Telmisartan Alone and in combination with Ramipril Global Endpoint Trial (ONTARGET) and Telmisartan Randomised AssessmeNt Study in ACE iNtolerant subjects with cardiovascular Disease (TRANSCEND) trials, in which neither the ACE1-I (ramipril; Altace®, Aventis, Strasbourg, France) nor another AT1RA (telmisartan; Micardis®, Boerhinger Ingelheim, Ingelheim, Germany) reduced the risk of cognitive decline or the development of any type of dementia in participants with cardiovascular disease or diabetes. 53 A small meta-analysis of hypertension treatment trials by Staessen54 also reported that BP-lowering did not reduce the dementia risk in populations with high cardiovascular morbidity. This finding was supported by a more recent systematic review of previous trials55 and two more recent meta-analyses of several observational longitudinal studies that explored whether or not different antihypertensive medications had any effect on cognitive decline or dementia. 56,57 We previously thought that these studies had limited scope for translation to AD because the study participants were generally younger and were usually recruited exclusively because they had higher cardiovascular burden, and because cognitive outcome measures were not the primary objectives. In addition, the methods of cognitive assessment were less detailed or not as comprehensive as the typical battery of assessments normally used in clinical trials of AD and also rarely used objective outcome measures, such as imaging [e.g. change in various magnetic resonance imaging (MRI) measurements] or other biomarkers. 58,59 We were of the opinion, prior to the commencement of RADAR that these negative findings made a larger-scale (e.g. Phase III) multicentre RCTs of an AT1RA in AD premature, and RADAR was designed to try to further provide supportive evidence. 60
In the intervening years since the commencement of the RADAR trial, no formal clinical trials have tested as to whether or not losartan can change MRI-based measures of AD-related brain atrophy either alone or in combination with a standardised questionnaire-based assessment of cognition in AD participants. Indeed, the use of MRI-based approaches in AD trials has not been commonplace, although there is now growing recognition of their usefulness in trials. 61,62 The protocol of a related, smaller (n = 100), Phase II three-arm US-based trial (NCT00605072), called the Antihypertensives and Vascular, Endothelial and Cognitive function (AVEC) trial, was announced around the time that the RADAR trial commenced. This smaller study recruited hypertensive participants with early (non-AD) cognitive impairment only, and it piloted the comparison of 1-year treatment of participants with candesartan (another AT1RA) with that of lisinopril (an ACE1-I) or hydrochlorothiazide (a diuretic) for their effect on memory and executive function, CBF (measured by transcranial Doppler) and central endothelial function (measured by changes in CBF in response to changes in end-tidal carbon dioxide). 63 This pilot trial later reported encouragingly that the AT1RA candesartan outperformed the ACE1-I (lisinopril) and diuretic (hydrocholorthiazide) in preserving CBF and executive function in these hypertensive participants who had executive dysfunction. 64 In addition, encouraging data, although again not specifically testing the impact that AngII targeting has in AD, were recently announced from the Systolic blood PRessure INtervention Trial Memory and cognition IN Decreased hypertension (SPRINT-MIND) substudy65 of the National Institutes of Health-funded Systolic blood Pressure INtervention Trial (SPRINT). 66 The main SPRINT (NCT01206062) study compared the effectiveness of treating systolic BP, again in a population of hypertensive individuals (without diabetes or a history of stroke or AD) aged > 50 years, to lower than 120 mmHg (compared with the current normotensive threshold of 140 mmHg) on a primary outcome that was a composite of cardiovascular effects as a measure of all-cause mortality. The objective of the SPRINT-MIND substudy was to investigate the impact that the two BP-lowering approaches had on cognitive outcomes, which included the rates of probable dementia (the primary cognitive outcome), or on secondary cognitive outcomes of development of MCI or a composite outcome of the development of MCI and probable dementia. Another objective was to use MRI scanning to measure the impact on levels of small vessel disease in the brain, according to MRI-based measures. 65 Although the main SPRINT trial was stopped early because it was becoming clear that according to its primary outcome measures clinical benefit was evident,66 there was evidence from SPRINT-MIND that lowering BP to below 120 mmHg could reduce rates of MCI and improve the composite secondary cognitive outcome measure. However, there was no significant evidence that the primary cognitive outcome, rate of probable dementia, was reduced. It is very plausible that stopping the main SPRINT trial early, and the impact that this had on follow-up periods in SPRINT-MIND, may have resulted in this aspect of the study being underpowered. 65
It has been reassuring that these findings continue to show that the RADAR trial has been a timely and necessary formal investigation of the therapeutic effect of an available antihypertensive drug in a clinically defined population of AD participants. Meanwhile, the RADAR trial simultaneously allowed for a focused gold-standard way of testing the role of the RAS, particularly that of AngII, in the pathogenesis of AD. The results of RADAR will be the first to be announced in AD that test this hypothesis, to our knowledge. As such, they will provide the first clinical trial data to stand alongside what is still predominantly observational studies, testing the effects of various AT1RAs in reducing rates of MCI and/or rates of progression of dementia, or the effects of AT1RAs on other outcomes proposed to be indicative of age-related or dementia-associated cognitive decline (reviewed by Kehoe20).
Our selection of losartan as the intervention for the RADAR trial was based on several arguments.
First, we chose losartan because of the multifactorial functions described for AngII in the brain. These include being responsible for important physiological processes such as vasoconstriction, and numerous organ- and cell-based functions such as reduction in the release of the neurochemical acetylcholine, the promotion of inflammation and neuronal excitotoxicity,36–40,67 all of which are relevant to the pathogenesis of AD.
Second, selective antagonism of the AngII type 1 receptor by losartan does not inhibit ACE1 activity, which may be important because ACE1 is elevated in AD31,32 but has also been reported to degrade Aβ. 31,68–70 For this reason, we did not wish to interfere with any potentially protective effects that ACE1 activity may have on Aβ pathology. 2 Thus, AT1RAs may be better than ACE1-Is, at least in AD, which has been supported by various pharmacoepidemiological and pre-clinical studies,11–13,15,16,71–73 and they have been demonstrated to have beneficial effects on AD-related pathology and cognitive impairment in murine models of AD. 48
Third, losartan, as the prototype AT1RA, had previously been found to be beneficial to cognitive function (in a 50-mg dose), compared with atenolol, in a small randomised study of their effects on cognitive function in very elderly hypertensive individuals (not in people with AD). 49 It also had the longest use in a clinical setting, thereby providing the most expansive safety data at the time the study commenced. Losartan has been found to be well tolerated and has also been suggested, alongside ACE1-Is, to provide advantages over other antihypertensive medications with respect to quality of life in older people. 74 This longevity in clinical use also offered additional economic advantages over most of the other AT1RAs in that it was already available in a generic form that would, in the event of a successful outcome, provide additional significant economic benefits in future large-scale studies and potential eventual use in a clinical setting.
Finally, given that there was evidence supporting the role of the RAS in AD that may be mediated through both cardiovascular and/or non-cardiovascular molecular mechanisms,49 we opted to test losartan in an AD population that included people with hypertension and those who were normotensive. This was intended to enhance the generalisability of any finding, while examining if any protective mechanisms may be operating independently of or in addition to the BP-lowering effects of these drugs. The decision was also a pragmatic one so that we could recruit from as typical an AD clinical population as possible and hence it would be easier to achieve our sample size. This is an important issue because a considerable proportion of AD participants would be otherwise excluded, depending on whether they had prior exposure to the intervention or related drugs that would make them ineligible, or were excluded because they had no prior hypertension. This intended approach, particularly the intention to recruit people who were normotensive, necessitated important design features in response to initial feedback on the study plan with key stakeholders, as is discussed in Patient and public involvement.
Hypothesis
The RADAR trial tested the hypothesis that blocking AngII signalling using losartan (100 mg) would reduce brain volume loss and, therefore, slow the clinical progression of mild-to-moderate AD.
Study design
This was a two-arm, double-blind, placebo-controlled, multicentre, individually randomised trial comparing the effects of 100 mg of losartan with placebo, preceded by a 1-month open-label phase. The main randomised phase allowed the testing of whether or not 12 months of treatment made any difference to structural brain changes, as measured using a comprehensive neuroimaging protocol involving MRI in both hypertensive and normotensive AD participants.
Duration of treatment and follow-up
We opted for a 12-month period of treatment and follow-up based on previous studies conducted by the ADNI,75–77 which found, using MRI, measurable brain atrophy among AD participants, equivalent to a mean 15.2 ml/year [standard deviation (SD) 8.6 ml/year]. We proposed that a relative between-group difference in atrophy rate of 25% could be considered a clinically meaningful difference that we would detect according to our sample size and power calculations. This relative difference is equivalent to an absolute difference in an annual atrophy rate of at least 3.8 ml/year in total brain volume (TBV).
Patient and public involvement
The study gained helpful input from patient and public involvement (PPI) at several stages of the study. The original design of the RADAR study was modified following consultation with some participants and carers whereby an open-label phase (described below) was introduced to address possible uncertainties that participants who were normotensive may have about taking a BP-lowering drug. Similarly, we sought feedback on the earliest versions of the study information sheets that were to be shared with participants and study partners and these were used at the outset of the study. However, as part of a nested qualitative study that was later conducted to explore other possible obstacles to recruitment,78 these information sheets were revisited and further revisions and simplifications made.
Open-label phase
An open-label phase was introduced as a way of reassuring all participants, particularly normotensive participants, who may have been anxious about taking a BP-lowering drug, as highlighted by participants and carers initially consulted about the intended study. This phase allowed all participants to explore their ability to tolerate the intervention and suitability to be randomised, which would have involved a baseline MRI scan. Participants commenced the open-label phase if they met all of the appropriate criteria, as determined at the initial eligibility and consent visit, and subject to subsequent review of results on bloods taken at the same visit, according to protocol specified ranges. The open-label phase was therefore a 3- to 4-week pre-randomisation phase (Figure 1) that involved participants being on open-label active medication for a maximum of 14 days prior to randomisation.
FIGURE 1.
Open-label phase of the trial.
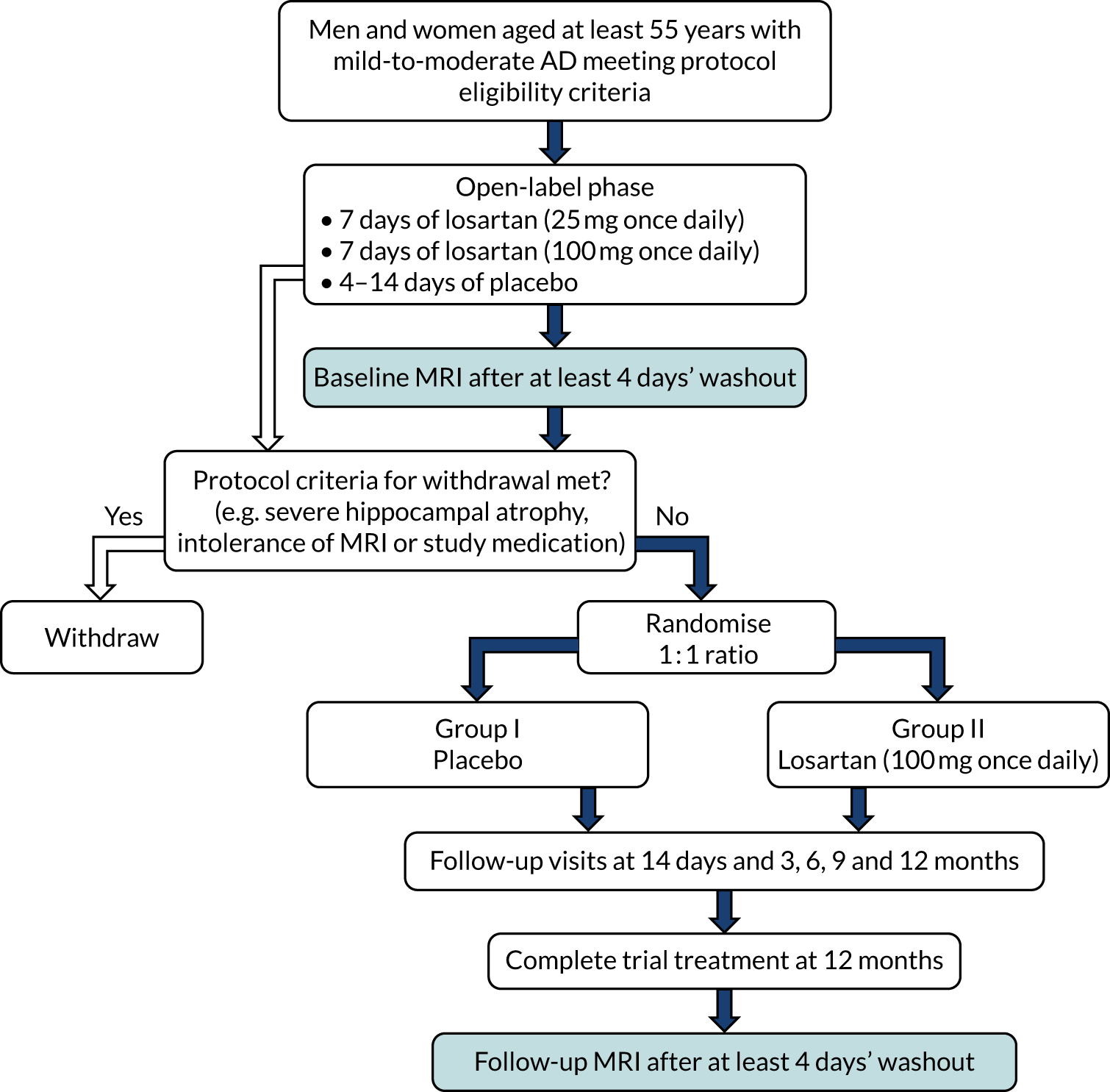
The open-label drug was titrated upwards in participants over a period of 2 weeks, with blood testing carried out to ensure safety, consistent with clinical practice for anyone commencing AT1RAs. The participants were initially prescribed 25 mg of generic losartan (according to brands available locally) to be taken once per day for 7 days and asked to keep a diary of taking the medication and any symptoms that they experienced. They were seen after 7 days, by the research team, had further blood tests taken to ensure safety and asked questions regarding any contraindications up to that point. At this point they were prescribed 100 mg of losartan for a further 7 days and similarly asked to keep a diary of taking the medication and any symptoms that they experienced, which was again reviewed at the end of this period and blood tests taken once more. All participants were given a home use arm-cuff Omron blood pressure monitor (Omron Corporation, Kyoto, Japan) and asked to measure and record their BP daily while they were taking active medication during the 14-day open-label phase. This information was retrieved by the research team member at the visit to review the 7 days on the 100 mg of losartan dose (called the 14-day visit). Each participant was also given an information card with the details of their local research contact in case they needed these. Participants who did not experience or report any contraindications during the 7 days on 100 mg of losartan were prescribed the main study placebo to cover a further 4- to 14-day washout period.
The flexible washout period of 4–14 days was undertaken for several reasons. First, a minimum of 4 days was needed for drug washout. As, according to its original manufacturer, losartan has an estimated half-life of 2 hours and its metabolites have estimated half-lives of up to 8 hours (www.rxlist.com/cozaar-drug.htm), this allowed a generous period of at least five half-lives for elimination of the drug. Second, it allowed sufficient time to review the results of the blood tests taken to ensure safety after participants had taken the maximum dose for 7 days. Third, if the results of these safety bloods taken after the titration of losartan were as expected, there would be sufficient time to schedule a baseline MRI. Fourth, it allowed the assessment of participants’ ability to successfully take the overencapsulated intervention or placebo, which was larger than the normal tablet form, and ensure that there were no tolerability issues with the overencapsulated formulation. Finally, the provision of placebo during the washout period allowed participants to be inculcated into the trial dosage regimen that would continue in the randomised phase.
Throughout the open-label phase, consideration was given to any potential deviations from the BP levels and baseline blood biochemistry measurements taken at the eligibility visit [as assessed by the principal investigator and, if necessary, in further consultation with the RADAR clinical lead (Ian Wilkinson)]. Similarly, any concerns reported by participants or study companions to their local research team were reviewed and discussed with participants.
Randomised phase
After successfully completing the open-label phase, participants were scheduled to undergo baseline MRI (see Primary outcome) and a cognitive assessment battery before being randomised (see Secondary outcomes). Following randomisation, participants underwent an identical upwards titration over 14 days, involving 7 days’ exposure to 25 mg of the intervention or placebo followed by 7 days at the 100-mg dose. Blood tests carried out to ensure safety were again taken on day 14 and more intervention was prescribed to last to the next visit at 3 months post randomisation. Subsequent visits that involved prescribing further allocations of intervention took place at approximately 3-month intervals, at which time drug adherence was also monitored by pill counts, collection of any adverse events (AEs) and conducting repeat assessments (Figure 2). No dose modifications were made after the participant had entered the randomised phase. Participants who began to experience issues that they felt or that were deemed to be related to the intervention were told to stop taking the medication but remained in the study unless they expressed a desire to discontinue.
FIGURE 2.
Participant procedures and data collection after confirmation of eligibility and willingness to participate. Safety bloods include measures of electrolytes, creatinine, and liver function tests according to protocol-defined ranges for inclusion/exclusion. Cognitive assessment includes Alzheimer’s Disease Assessment Scale – Cognitive Subscale (participant), Neuropsychiatric Inventory (companion), Bristol Activities of Daily Living Scale (companion), Dementia Quality of Life (participant) and Dementia Quality of Life-Proxy (companion). Mini Mental State Examination (MMSE) were also compared between eligibility assessment and end of study. Reproduced with permission from Kehoe et al. 79 © 2021 The Author(s). Published by Elsevier Ltd. This is an Open Access article under the CC BY 4.0 license.
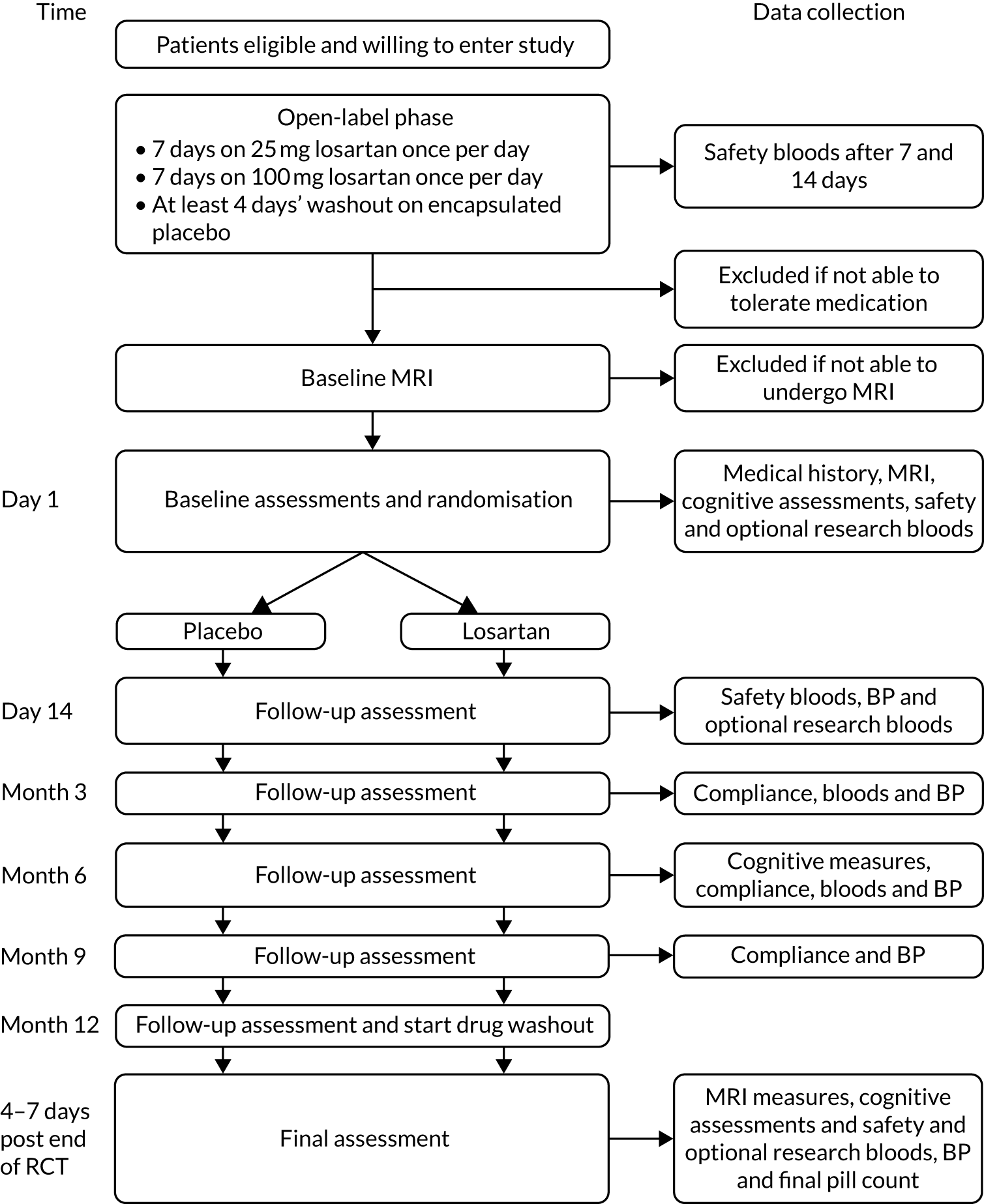
The randomised phase concluded 52 weeks after commencing the first dose of the intervention or placebo. A repeat 4-day washout was initiated and a final MRI was scheduled as close as possible to this as well as the final repeat assessments according to the assessment schedule (see Figure 2).
Throughout the trial period, the trial design was flexible to allow participants’ normal clinical care to continue under the supervision of primary or secondary care providers, as relevant. This included the use of other dementia-related treatments: we aimed for all participants to be on a stable dose of dementia treatments for approximately 3 months prior to study entry but also allowed any naturalistic dose adjustment that may be deemed necessary after entry to the trial.
Study setting
The RADAR trial was designed as a pragmatic multicentre RCT that recruited participants with mild-to-moderate AD from 23 NHS hospital trusts in the UK that routinely diagnose and treat participants with AD. Table 1 lists all the recruitment sites and Figure 3 illustrates their geographical distribution. As some recruitment sites lacked their own MRI facilities, or could not meet the specifications needed for the facilities, patients from these sites underwent MRI at another location (see Table 1). Sites’ eligibility to participate in RADAR was based on whether or not they had prior expertise in recruiting to clinical trials of AD and the capacity to provide MRI facilities to fulfil our neuroimaging protocol.
| Name | Scan site, if different |
|---|---|
| 10 North Bristol NHS Trust | |
| 11 Sheffield Teaching Hospital Trust | |
| 12 2gether NHS Foundation Trust | |
| 13 Oxford Health NHS Foundation Trust | |
| 15 NHS Tayside | |
| 16 NHS Lothian | |
| 17 NHS Borders | 16 NHS Lothian, Edinburgh |
| 18 NHS Grampian | |
| 19 NHS Greater Glasgow and Clyde | |
| 20 North Staffordshire Combined Healthcare NHS Trust | |
| 21 Torbay and South Devon NHS Foundation Trust | |
| 22 Salford Royal NHS Foundation Trust | |
| 23 Norfolk and Suffolk NHS Foundation Trust | |
| 24 Royal Devon and Exeter NHS Foundation Trust | |
| 25 NHS Ayrshire and Arran | 19 NHS Greater Glasgow and Clyde |
| 26 NHS Lanarkshire | 19 NHS Greater Glasgow and Clyde |
| 27 Leeds & York Partnership NHS Trust | |
| 28 NHS Forth Valley | 15 NHS Tayside, Dundee, and 19 NHS Greater Glasgow and Clyde |
| 29 NHS Fife | 15 NHS Tayside, Dundee |
| 30 Newcastle upon Tyne NHS Foundation Trust | |
| 31 Kent and Medway NHS and Social Care Partnership Trust | |
| 32 Midlands Partnership NHS Foundation Trust | Own and 20 North Staffordshire Combined Healthcare NHS Trust |
| 33 Belfast Health and Social Care Trust |
FIGURE 3.
Geographical location of RADAR sites.
Intervention
We used a maximum dose of overencapsulated losartan of 100 mg, which was titrated directly from 25 mg of overencapsulated losartan that was initially given for 7 days and reflected standard clinical practice. This titration approach was undertaken in both the randomised study and the preceding open-label phase already described. The placebo used in this study was similarly overencapsulated and made to match both doses and sourced from St Mary’s Pharmaceutical Unit (SMPU), Cardiff, UK. The maximum dose was chosen with the intention of achieving maximum exposure to the intervention and to ensure the strongest target (AngII type 1 receptor) engagement possible. There were no dose modifications after the participant had entered the randomised phase.
Given the age range of the participant population, we also made provision for the appropriate monitoring of BP. In the open-label phase, the participants themselves took daily measurements of their BP for the duration of the 14-day titration period on generic losartan. BP was also taken at almost every face-to-face meeting between participants and research teams (see Figure 2).
The intervention dose we used was higher than that reported in other studies. These studies used a dose of 50 mg of losartan and observed improvements to cognitive function in very elderly individuals with hypertension and in a secondary analysis in a hypertension study a modest benefit was reported. However, where non-significant benefits on memory were found in hypertensive participants who did not have AD49,50 were thought to be independent of BP-lowering effects.
Ethics approval and research governance
Ethics approval for the study, and approval for all subsequent amendments to the study and study protocol, was given by South East Wales Research Ethics Committee C (MREC, later renamed Wales Research Ethics Committee 2 Cardiff) in February 2013 (reference number 12/WA/033). The clinical trial authorisation was given by the Medicines and Healthcare products Regulatory Agency (MHRA) in July 2013 (reference number 18524/0223/001/0001). The trial was registered with the International Standard Randomised Controlled Trial Number (ISRCTN) ISRCTN93682878 and with the European Union Drug Regulating Authorities Clinical Trials Database (EudraCT) under the reference number 2012-003641-15. Following the introduction of the new Health Regulation Authority the study was adopted on 16 June 2016. The trial was sponsored by North Bristol NHS Trust Research & Innovation (NBT R&I; reference 2625). There were no major changes to the original protocol or the statistical analysis plan during the study and a summary of changes made to the original protocol is given in Table 2.
| Changes to protocol | Date |
|---|---|
| Revisions for clarity, consistency with SOPs and MREC recommendations | 3 June 2013 |
| Addition of information pertaining to pilot imaging procedures | 18 July 2013 |
| Minor clarifications added after review by TSC, DMEC and TMG | 23 July 2013 |
| Change to minimisation details and eligibility criteria | 28 November 2013 |
| Minor corrections to ensure consistency with other documentation | 9 April 2014 |
| Removal of severe hippocampal atrophy as exclusion criterion | 7 October 2014 |
| Removal of residual references to severe hippocampal atrophy as exclusion criterion | 14 November 2014 |
| Change to MMSE inclusion criterion | 3 December 2015 |
| Inclusion of an embedded qualitative component | 3 March 2016 |
| Inclusion of additional prescreening tools, and clarification to consent wording | 27 July 2017 |
| Amendment to the BP range exclusion criterion | 11 October 2017 |
The RADAR trial was overseen by a Trial Steering Committee (TSC) that comprised an independent chairperson and methodologist, external independent experts and lay members, as well as the study sponsor (NBT R&I). The TSC ordinarily met twice per year and more frequently for extraordinary meetings if deemed necessary. The safety and conduct of the study were overseen by an independently chaired Data Monitoring and Ethics Committee (DMEC) that convened every 6 months once data collection commenced and reported to the TSC ahead of each scheduled TSC meeting. The general operations of the trial were overseen by a Trial Management Group (TMG) that convened regularly during the study. The TMG was composed of all chief investigators and all co-applicants of the RADAR study and included the trial manager (TM), other co-opted members of the Dementia Research Centre University College London (DRC-UCL) team or members of the Bristol Randomised Trials Collaboration (BRTC) trials unit at the University of Bristol. The day-to-day administration of the study was conducted by the chief investigator, the TM and the trial administrator, with the support of the sponsor and funder as and when appropriate.
Participants
The trial sought to recruit people aged ≥ 55 years with mild-to-moderate AD diagnosed according to original National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) criteria from NHS hospital trusts across England, Scotland and Northern Ireland where participants with AD are routinely diagnosed and treated. Participants could be male or female, hypertensive or normotensive and could already be taking licensed antidementia treatments.
Inclusion criteria
Women and men diagnosed with mild-to-moderate AD according to original NINCDS-ADRDA criteria80 and who met all of the following criteria were considered eligible:
-
aged ≥ 55 years (to maximise generalisability of the study and avoid exclusion of younger yet otherwise eligible potential participants)
-
had capacity to consent for themselves as judged by a member of the research team with appropriate training and experience
-
had a MMSE score of 15–28 at the consented eligibility assessment
-
scored ≤ 5 on a modified Hachinski Ischaemic Score81
-
had a previous computerised tomography (CT), single-photon emission computed tomography (SPECT) or MRI scan consistent with a diagnosis of AD
-
had a study companion who was willing to participate in the study.
Exclusion criteria
Participants were deemed ineligible if they met any of the following criteria:
-
were already receiving ACE1-Is, AT1RAs, the renin inhibitor aliskiren or potassium-sparing diuretics
-
had known intolerance to, or had experienced renal problems while taking, ACE1-Is or AT1RAs
-
were medically unsuitable for, or unwilling to undergo, MRI
-
had a primary neurodegenerative disease or potential cause of dementia other than AD
-
had a consistent BP (at eligibility visit) of < 115 mmHg systolic and < 70 mmHg diastolic or > 160 mmHg systolic and > 110 mmHg diastolic
-
experienced a fall in BP at the eligibility visit on standing from a seated position of > 20/10 mmHg associated with clinically significant symptoms or a fall > 30/15 mmHg
-
had previously had a cerebrovascular accident and significant residual impairment remained (note that transient ischaemic attack was not automatically a basis for exclusion)
-
had hypertrophic cardiomyopathy or clinically significant aortic valve stenosis
-
had an estimated glomerular filtration rate of < 30 ml/minute/1.73 m2
-
demonstrated evidence of liver disease or significant liver function test (LFT) derangement (aspartate transaminase/alkaline phosphatase/bilirubin more than two times the upper limit of normal) at the eligibility/consent visit
-
had blood levels of potassium > 6.0 mmol/l in a non-haemolysed blood sample
-
had primary neurodegenerative diseases or potential alternative cause of dementia other than AD
-
were female and had not yet reached the menopause (defined as having a period in the previous 12 months) and tested positive for pregnancy, were unwilling to take a pregnancy test prior to trial entry or were unwilling to undertake adequate precautions to prevent pregnancy for the duration of the trial
-
had any severe coincident medical disease, or other factor inhibiting compliance with the study medication or follow-up schedule (e.g. participant unlikely to survive the trial follow-up period because they had a terminal comorbid condition)
-
had participated in a previous Clinical Trial of an Investigational Medicinal Product (CTIMP) within 6 months of RADAR trial entry.
Recruitment procedure
Potential participants were identified at recruitment sites or associated patient identification centres (PICs) from pre-existing lists of patients interested in research or newly referred patients who were receptive to research participation. All sites had scope to recruit from primary care (if sites wanted to use primary care and appropriate arrangements were in place) and from the National Institute for Health Research (NIHR)-funded Join Dementia Research research register portal (see www.joindementiaresearch.nihr.ac.uk/), which was launched after RADAR commenced. Engagement with Join Dementia Research was either by research staff identifying registered potential participants or by registered potential participants contacting local RADAR recruitment centres. Some potential participants contacted the RADAR co-ordinating office or local sites after articles in the national or local press and media. We used social media channels [Twitter (Twitter, Inc., San Francisco, CA, USA) (www.twitter.com/@RadarTrialAD) and Facebook (Facebook, Inc., Menlo Park, CA, USA) (www.facebook.com/RADARTrialAD/)] in concert with various recruitment sites and patient interest channels to reinforce these messages and opportunities to participate, as well as providing information on the RADAR trial website (www.radar-trial.org.uk).
People who made contact directly with recruitment sites, or were referred from the co-ordinating centre, or were identified as a potentially eligible and interested participant on Join Dementia Research were sent a letter of invitation, the initial participant information sheet, the initial companion information sheet, a reply slip and a prepaid envelope. Those with whom the study was discussed face to face were provided with the information packs to read and consider. The reply slip gave options for permission for the potential participant to be contacted by the research team with more information, permission for the research team to access medical records to confirm eligibility to the trial and permission to be contacted by the research team to take part in a telephone eligibility interview. Alternatively, the potential participant could indicate that they did not wish to participate in the trial.
Depending on the response given on the reply slips, further information was sent (for the participant and their companion with details about the MRI procedure) and general practitioners were contacted to find out which medications were currently being taken (to identify if the potential participant was being prescribed medications that would make them ineligible). Researchers telephoned all potential participants to introduce the study formally, to make initial eligibility checks and to arrange the first clinic appointment for consent and screening.
Enhancing site recruitment
During the course of RADAR recruitment, we embedded a qualitative component (before we had recruited 50% of the participants) to further explore the nature of trial site recruitment. This was intended to enhance recruitment and explore the impact of the design and conduct of the trial, and what organisation or training could possibly lead to improvements in recruitment in RADAR or in dementia trials in general. This would not have been possible without effective PPI.
Qualitative interviews were undertaken with a purposeful sample of research nurses and doctors responsible for screening and consent, and trial participants from a range of high- and low-recruiting trial sites to gain insights into barriers to and facilitators of recruitment. With informed consent, interviews were audio-recorded, transcribed and imported into NVivo 10 (QSR International, Warrington, UK) and analysed thematically. 82 Data collection and analysis were conducted in parallel until data saturation was reached. 83 Some of the findings of this nested study resulted in helpful revisions to our participant information packs that were considered to be overly complex and, therefore, were simplified. 78 There were other, more generic, findings identified that were of note and will be discussed later in the overall discussion of findings as they are of relevance not only to the conduct of RADAR but also to clinical trial conduct in general.
Informed consent
Informed, written consent was obtained from every participant and their study companion during the screening visit to confirm their eligibility and before any other data collection took place. As per the study protocol, study participants had to have capacity to consent for themselves. Potential participants were also asked if they wished to consent to donating additional research bloods for future research that may further inform the results of the trial or AD-related research. At this time study participants were asked to identify a legal representative for the purposes of their involvement in the RADAR trial only. This representative was intended to be responsible for deciding whether or not it was in the participant’s best interests to continue in the trial should the participant lose capacity. The legal representative was usually the study companion who had consented to take part. All original signed and dated consent forms were held securely as part of the investigator site file at each of the recruitment sites, with a copy given to the participant, a copy put in the participant’s medical notes and a copy sent to the co-ordinating office for the purpose of central monitoring procedures by the sponsor. The eligibility criteria for the trial included consideration of the results of safety blood samples taken at the screening visit and subject to satisfying inclusion/exclusion criteria. Participants deemed to be ineligible for the study were informed of this and the reasons for it by site staff.
Randomisation
Participants who successfully completed the open-label phase were entered into the main part of the study. A baseline MRI (see Primary outcome and Table 4) was then scheduled and undertaken approximately 4–14 days after completion of taking the active open-label generic losartan. Randomisation was contingent on successful baseline MRI for the primary outcome. Success was adjudicated by our pipeline quality assurance (QA) process provided by our collaborators at the DRC-UCL. Participants in whom a MRI scan was successfully undertaken were then randomised, via an online procedure or pin-access service by telephone hosted by Sealed Envelope (www.sealedenvelope.com), to receive either losartan or placebo in a ratio of 1 : 1 minimised by age and a baseline medial temporal lobe atrophy (MTA) score that encompasses the hippocampus and which was assessed for all sites by team members at the Clinical Research and Imaging Centre (CRIC), University of Bristol (www.bristol.ac.uk/cricbristol/). To expedite the randomisation process, so that it coincided as close as possible with the baseline MRI visit, and alongside the need to commence treatment as soon as possible, we used a subjective rating scale called the Scheltens scale84 of MTA score to gain some assessment of likely disease-associated atrophy and that has good inter-rater reliability. 84 This allowed a rapid provision of some assessment of baseline atrophy, to serve as a minimisation variable to facilitate randomisation. Following randomisation, participants followed the same titration pattern as in the open-label phase, of 25 mg for 7 days and then 100 mg as a maintenance dosage with a 14-day monitoring and dispensing follow-up visit during the randomised phase. If the quality of the scan was insufficient, participants were asked to undergo another scan. If they declined, or the repeat scan(s) were still of insufficient quality (e.g. because the participant did not fully adhere to the MRI protocol), they were deemed to be ineligible for continued inclusion in the study and were informed of this outcome.
Blinding
The study was double blinded, meaning that all participants and study companions, as well as all study personnel (except pharmacists at each site), were blinded to allocation at randomisation. There was scope within the protocol, in the interests of participant safety, for emergency unblinding to occur if a clinician believed that a treatment decision for a participant in certain circumstances could be influenced by the participant receiving losartan. With this in mind, a 24-hour emergency unblinding service was available to all research sites either through each local pharmacy service during working hours and out of hours or through a pharmacy nominated by the RADAR co-ordination team, in which case requests for emergency unblinding were documented by pharmacy staff and logged centrally by the TM. The procedure for any participant for whom emergency unblinding was undertaken was that they would discontinue taking the trial medication but remain part of the study unless they chose to withdraw. Furthermore, in the event of any emergency unblinding being required, the intention was that, where possible, all members of the research team (excluding trial pharmacists) would remain blinded, subject always to clinical need, and reasons for any instances of emergency unblinding would be determined by the TM. Any unblinding that occurred because of a serious adverse event (SAE) was appropriately documented in accordance with the sponsor’s procedures. All participants were given the relevant emergency contact details on their trial participation card when they consented to take part.
Outcome measures and assessments
The full schedule of visits and assessment is summarised in Figure 2. All main assessments (baseline, 6-month and 12-month MRI and face-to-face assessments) were completed by both the participant and their study companion. If consent had been originally given to donate additional research bloods, then these were collected at baseline, 14 days post randomisation and at the end of the study. Wherever possible the researcher arranged to meet the participant for face-to-face assessments and to have follow-up assessments where they felt most comfortable (e.g. at home or at the clinical research centre). Every attempt was made to achieve, as much as possible, consistency of location and time of assessments.
In the case of imaging-related primary and secondary outcome measures, each participant underwent the RADAR scanning protocol, which lasted approximately 30 minutes if all sequences were undertaken (see Primary outcome and Secondary outcomes). Scans and anonymised scan data from all sites were uploaded to a bespoke XNAT platform, and then transferred securely to the DRC-UCL team for QA. Where 12-month MRI differed from baseline in the protocol or positioning sufficiently to adversely affect primary outcomes, a rescan was requested. As previously mentioned, all scans were also independently awarded a visual MTA score, derived from what is more widely known as the Scheltens scale,84 by research staff at the CRIC. This score was intended to inform and expedite the randomisation process.
Primary outcome
The primary outcome was change in brain atrophy, determined by volumetric MRI (vMRI), between baseline and the 12-month follow-up after treatment with losartan. This outcome was measured at a pre-arranged NHS or clinical academic imaging centre. Brain atrophy as measured by vMRI is recognised as a surrogate marker of cognitive decline and AD pathology. 75,76,85–88 Brain atrophy was defined as the absolute difference in TBV between baseline and after 12 months of treatment post randomisation measured using vMRI [T1-weighted magnetisation-prepared rapid-gradient echo (T1-MPRAGE)]. It was chosen as the primary outcome on the assumption that a difference between trial arms of at least 3.8 ml/year would be clinically meaningful.
All MRI was performed at clinical or clinical-academic imaging centres using either 1.5-T or 3-T MRI systems with high-resolution (1-mm isotropic) three-dimensional T1-MPRAGE. The analyses of the magnetisation-prepared rapid-gradient echo (MPRAGE) images were conducted in collaboration with the DRC-UCL. All trial sites piloted the validation of the RADAR MRI protocol under the guidance and support of our DRC-UCL colleagues. The UCL team have developed semiautomated computerised methods to derive brain structure volumes from single-time point MRI and rates of atrophy from serial MRI,89–92 similar to those previously reported for multicentre trials. 93 QA of all scans, and the QA and editing of segmentations, was carried out using Medical Image Display and Analysis Software (MIDAS) version 6.6 (University College London, London, UK). 90 Automated segmentations were performed using brain multi-atlas propagation and segmentation91 for whole brain areas and Similarity and Truth Estimation for Propagated Segmentations (STEPS)94 for hippocampal regions, prior to manual checks and edits if needed. All of the image analysts at DRC-UCL undergo training and regular validation on structure segmentation. Longitudinal change following registration was measured using a DRC-UCL implementation of K-means normalised boundary shift integral (KN-BSI)95 for brains or double-window KN-BSI for the hippocampus. 92,95
Secondary outcomes
A number of secondary outcomes were included in this study (see Table 4):
-
Rates of AD progression as assessed by changes in cognitive assessments, including the 11-item Alzheimer’s Disease Assessment Scale-cognitive subscale (ADAS-Cog),96 the Neuropsychiatry Inventoric97 and the MMSE,98 as well as an examination of change to quality of life [measured using the Dementia Quality of Life Measure (DEMQOL) or the Dementia Quality of Life Measure by proxy (DEMQOL-Proxy)99] and in ability to perform activities of daily living, assessed using the Bristol Activities of Daily Living Scale (BADLS). 100 These secondary outcomes were obtained during a face-to-face assessment by a researcher, who was blinded to the participant’s allocation. The assessments were conducted first at baseline, after successful baseline MRI, and subsequently at the 6-month follow-up visit, and again within 10 days of the 12-month MRI.
-
Change in WMH volume as determined by MRI using T2-weighted fluid-attenuated inversion recovery (T2-FLAIR), possible in only a subset of sites, to explore the effect of the intervention on white matter damage in participants, which exacerbates AD symptoms101 and has been reported to predict 1-year cognitive decline. 24 The WMH volumes were automatically identified and quantified using the longitudinal extension102 of a cross-sectional automated framework103 that performs the analysis on T1- and T2-FLAIR images rigidly aligned to the T1 space, acquired during the same MRI session at baseline and at 1-year follow-up. This framework automatically derives the number of components required to appropriately model the data in a Gaussian mixture model103 that simultaneously accounts for normal and unexpected observations. After convergence of the model, the same model structure is used across time points and candidate lesion voxels are identified based on intensity and anatomical location information. Clusters of selected voxels are then automatically classified as lesion or artefact.
-
In a small subset of sites where MRI facilities were suitable, and data of sufficient quality were available, we also explored the level of change in CBF by arterial spin labelling (ASL) methods. ASL data of sufficient quality were processed using MATLAB® version 2019 (The MathWorks, Inc., Natick, MA, USA) and SPM12 [version 7219 (Statistical Parametric Mapping 12, The Wellcome Centre for Human Neuroimaging, University College London, London, UK)]-based pipeline ExploreASL software verion 1.0.0 (Amsterdam UMC, Amsterdam, the Netherlands)104,105 to generate CBF maps as well as the grey matter spatial coefficient of variation (CoV). Although CBF is proposed to serve as a surrogate marker of cognitive performance,45–47 ASL spatial CoV has been recently proposed as a surrogate global marker of cerebrovascular health. 106,107 These data were acquired at the same time as the baseline and follow-up MRI at 12 months.
-
Change in BP over time is an important indicator (1) that the intervention was biologically active (i.e. observable reductions in BP in the intervention arm would be proof that the medication was active) and (2) of lowered BP, which may also alter the volume of WMH and improve CBF. As such, change to BP over time (visit-to-visit variation) may be a contributing factor to any observed change in cognitive function and thus could serve as an important confounding variable, even though previous studies have suggested that losartan may improve cognitive decline independent of lowered BP. 49–51,74 These data were collected at every visit in the study as described in Tables 3 and 4.
-
Measure of association between the primary MRI measures of atrophy and rate of cognitive decline. Given that atrophy, CBF and WMH volume are all considered proxies in various ways of cognitive decline, as summarised, it will be important to explore levels of association between the various assessment measures of cognitive function, activities of daily living and quality of life described. Examination of these will be important to explore which of the primary and secondary outcomes may be most relevant to use in any future large-scale clinical trials of AD involving losartan or related AT1RAs.
-
Examination of levels of drug compliance and tolerability across the main randomised phase and comparing rates across the active and placebo arms, as well as some examination of any potential differences between participants with hypertension and those who were normotensive. Data to inform this investigation, including AE data and safety bloods taken (with the exception of the 9-month visit, when no safety bloods were taken), were collected throughout the study at each follow-up visit and at the final 12-month face-to-face assessment.
| Visit | Researcher role | Participant role |
|---|---|---|
| Pre-screening phase | ||
| Early eligibility assessment | Gather medication records to verify no potential drug conflicts | Consent on initial reply slip that medical records can be assessed to make sure that there are no conflicts with the study medication |
| Brief telephone assessment | Answer a few brief questions to ensure that eligibility is likely before a face-to-face visit | |
| Screening visit | ||
| Eligibility assessment | MMSEa take baseline bloods for electrolytes, creatinine and LFTs | Give consent of their intention to enter study subject to interview to ascertain eligibility, including blood levels check. Await confirmation to enter open-label phase (within 7 days of blood test) |
| Follow-up telephone call (within 7 days of eligibility assessment) | Feedback blood test results and confirm whether or not participant can proceed. If suitable, arrange for collection of study medication and BP machine | Collection of study medication by participant or companion |
| Open-label phase | ||
| N/A | N/A | Take 25-mg of drug for 7 days, maintain diary of BP check, drug taking and any side effects |
| 7-day visit | Measure sitting and standing BP, take bloods for safety tests,b do pill count, provide next trial drug, record AEs | Take 100-mg dose of drug for 7 days, maintain diary of BP check, drug taking and any side effects |
| 14-day visit | Measure sitting and standing BP, take bloods for safety tests,b do pill count, provide next trial drug, record AEs | Start taking placebo drug for at least 4 days until called for baseline MRI visit |
| Visit | Researcher role | Participant role |
|---|---|---|
| Baseline visit (18–28 days after open-label medication commenced) | MRI to inform randomisation and collect primary outcome measure. At same visit or within 10 days of conducting cognitive assessment;a measure sitting and standing BP, take bloods for safety testsb and optional research samples, provide allocated drug (week 1, 25 mg; week 2, 100 mg), record AEs | Take allocated drug |
| 14 days after randomisation | Measure sitting and standing BP, take bloods for safety tests,b optional research samples, do pill count, provide next trial drug, record AEs | Take allocated drug |
| 3 months after randomisation | Measure sitting and standing BP; take bloods for safety tests,b do pill count, provide next trial drug, record AEs | Take allocated drug |
| 6 months after randomisation | Cognitive assessment,a measure sitting and standing BP, take bloods for safety tests,b do pill count, provide next trial drug, record AEs | Take allocated drug |
| 9 months after randomisation | Measure sitting and standing BP; do pill count, provide next trial drug (no bloods taken at this time), record AEs | Take allocated drug |
| 12 months after randomisation | Contact participant to stop taking trial drug to provide at least 4 study drug-free days (no dose reduction is required) | Stop taking allocated drug |
| End of study (12 months + 4 days after randomisation) | MRI and MMSE.c At same visit or within 10 working days of conducting cognitive assessment,a measure sitting and standing BP, take bloods for safety tests,b record AEs, optional research samples, do final pill count |
Sample size
Our proposed sample size for RADAR was based on previous studies conducted by the ADNI. These studies aim to optimise levels of recruitment to clinical trials of AD that involved MRI as an outcome measure. 75–77 Previous studies have suggested that the 12-month atrophy that is measurable using MRI among AD patients is 15.2 ml/year (SD 8.6 ml/year) and that a relative between-group difference in atrophy rate of 25% is considered a clinically meaningful difference. This is equivalent to an absolute difference in TBV between the trial arms after 12 months of exposure to the intervention of 3.8 ml,108 equivalent to a standardised effect size of 0.44 SDs. We aimed to recruit and randomise a total of 228 participants, which would be sufficient to provide satisfactory primary outcome data on 182 participants for analysis, assuming 20% loss in primary outcome data. This was intended to provide us with 84% power to detect our target difference of at least 3.8 ml/year in 12-month atrophy (therapeutic benefit) with two-sided α = 0.05.
Adherence to intervention
We had defined compliance in the study to be based on participants having taken 80–120% of the intervention, as has been adopted in other studies. 109 Instances of non-compliance by participants were reviewed at each follow-up visit and, where necessary, discussed with the principal investigator or delegated clinician to determine if it was appropriate for the participants in question to discontinue medication. In reality, compliance issues were noted when participants did not meet the lower threshold to be viewed as compliant, rather than the upper limit, because prudent prescribing and the frequency of follow-up visits ensured that participants were prescribed only up to the 100% amount needed between follow-up visits.
Statistical analysis
The analysis and reporting of this trial were undertaken in accordance with Consolidated Standards of Reporting in Trials (CONSORT) guidelines and followed a predefined statistical analysis plan that was agreed with the TSC prior to the completion of data collection. The primary comparative analyses between randomisation arms were conducted on an intention-to-treat basis, without the imputation of missing data, with due emphasis placed on confidence intervals (CIs) for the between-arm comparisons.
Preliminary analyses
Descriptive statistics of demographic and clinical measures were used to summarise the trial population and to compare the randomisation arms at baseline.
Primary analysis
The primary analysis employed linear regression to compare brain volume at 12-month follow-up between arms as randomised, adjusted for baseline brain volume, minimisation variables (age and Scheltens rating score) and recruitment site, to investigate differences in brain atrophy between the intervention and the control arms. The result of the linear regression model is presented as an adjusted difference in means between the intervention and control arms alongside the associated 95% CI and p-value for the comparison.
Secondary analyses
The effect of the intervention on secondary outcomes was investigated using appropriate regression models adjusted for baseline value of the outcome being examined, minimisation variables and recruitment site.
Sensitivity analyses
The sensitivity of the primary analysis to the impact of missing data was explored. Multiple imputation by chained equations (MICE) was used to impute missing primary outcome data. The imputation model included all variables that were part of the primary intention-to-treat analysis, secondary outcomes (from each time point) and baseline variables that were associated with missingness of the primary outcome. Twenty imputed data sets were generated and combined using Rubin’s rules and the primary analysis model was then repeated using the imputed data.
The impact that treatment compliance had on the data was investigated using the allocation-respecting method of complier average causal effects (CACE) via instrumental variable two-stage least squares regression, whereby outcomes of those who ‘complied’ with the intervention are compared with a group of ‘would-be compliers’ in the control arm. Treatment compliance was defined as (self-report of) taking 80–120% of study medication.
The impact that potential outliers had on the data was investigated by repeating the primary analysis model after excluding any participants whose brain volume measurement was > 3 SDs from the mean. The primary analysis was also repeated additionally adjusting for duration of follow-up.
Exploratory analyses
To investigate potential treatment effect modification, the primary analysis model was repeated with the inclusion of a treatment allocation by potential modifier interaction term. The potential treatment effect modifiers investigated were baseline hypertensive status, baseline MMSE, baseline age, duration of AD diagnosis, baseline brain volume and change in systolic BP. The trial was not designed to test interaction effects and, therefore, these analyses should be considered as exploratory in nature and the findings interpreted with due caution.
Chapter 2 Results
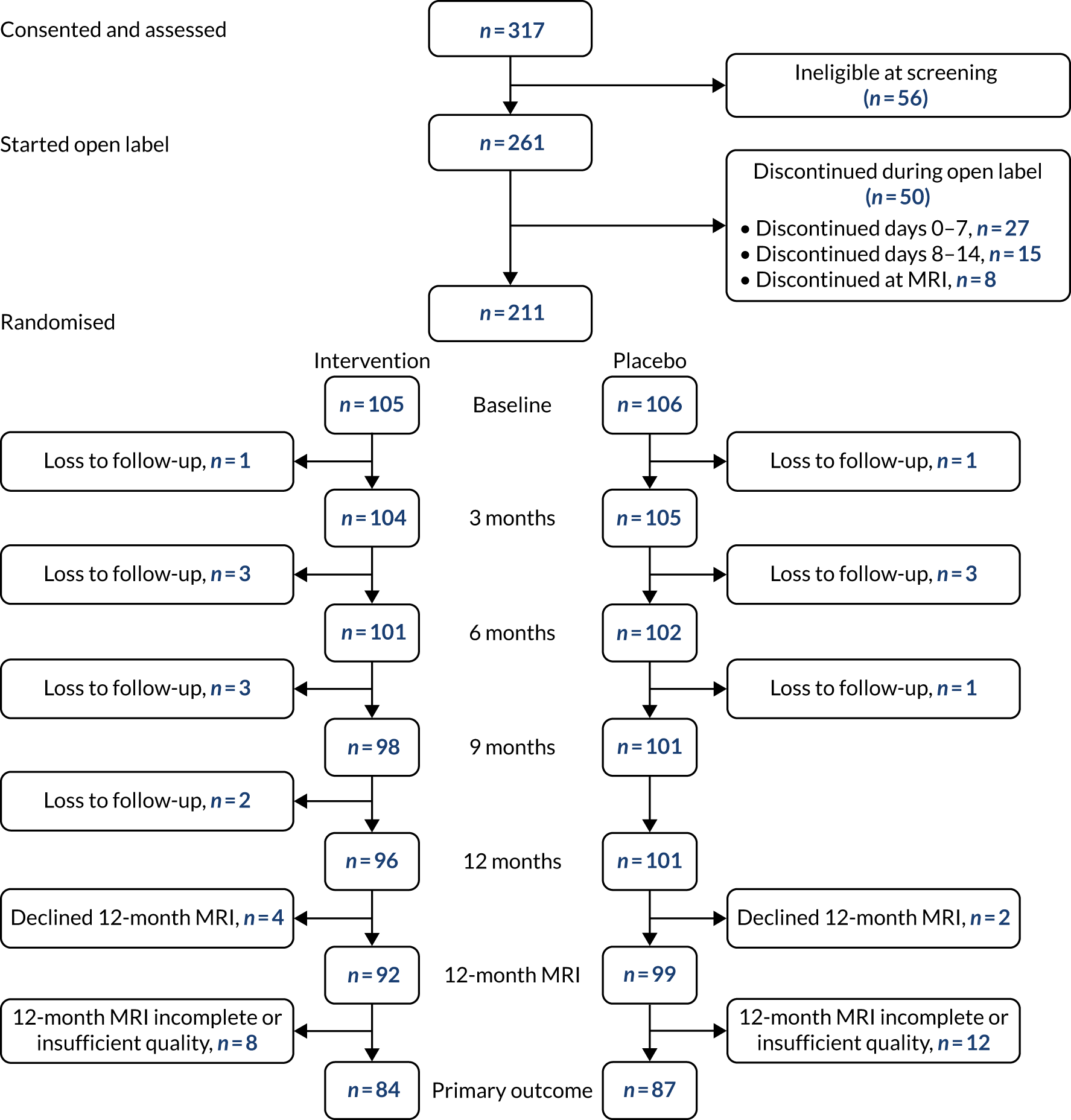
In total, 261 participants, plus 261 study companions, were recruited to the open-label phase of the RADAR trial. Of these, 211 were randomised, with 105 allocated to the intervention group and 106 to the placebo group. Figure 4 presents the CONSORT flow diagram for the trial and summarises participant throughput from eligibility screening, through the open-label phase and randomisation and on to completion of the 3-, 6-, 9- and 12-month follow-ups, as appropriate. The diagram also shows the numbers of participants who did not meet the inclusion criteria.
FIGURE 4.
The RADAR CONSORT flow chart. Reproduced with permission from Kehoe et al. 79 © 2021 The Author(s). Published by Elsevier Ltd. This is an Open Access article under the CC BY 4.0 license.
Baseline characteristics and comparisons
The baseline demographic characteristics of the study sample are summarised in Table 5. This table shows that the randomisation process was very effective in achieving balance between the intervention and placebo groups in standard characteristics of age (including different age strata), gender, ethnicity and the number of years of education. Similarly, there was excellent balance between the intervention and placebo arms in the distribution of people who were hypertensive or normotensive. The same could be said for various readings of their sitting and standing systolic and diastolic BP in the case of both participants who were in the mean normotensive ranges of BP specified in the protocol and those who had hypertension and well-controlled BP.
| Variable | Placebo (N = 106) | Intervention (N = 105) | ||||
|---|---|---|---|---|---|---|
| Categorical | N | n | % | N | n | % |
| Gender (n = male) | 106 | 67 | 64 | 105 | 60 | 57 |
| Ethnicity (n = white) | 106 | 106 | 100 | 105 | 104 | 99 |
| Aged < 70 years | 39 | 37 | 39 | 37 | ||
| Aged 70–79 years | 42 | 40 | 39 | 37 | ||
| Aged > 79 years | 25 | 24 | 27 | 26 | ||
| Hypertensive | 106 | 50 | 47 | 105 | 47 | 45 |
| Continuous | N | Mean/median | SD/[IQR] | N | Mean/median | SD/[IQR] |
| Sitting systolic BP (mmHg) | 85 | 138 | 15 | 85 | 138 | 14 |
| Standing systolic BP (mmHg) | 84 | 134 | 17 | 83 | 137 | 17 |
| Mean systolic BP (mmHg) | 84 | 136 | 15 | 83 | 138 | 13 |
| Sitting diastolic BP (mmHg) | 85 | 78 | 9 | 85 | 77 | 9 |
| Standing diastolic BP (mmHg) | 84 | 79 | 9 | 83 | 80 | 12 |
| Mean diastolic BP (mmHg) | 84 | 78 | 8 | 83 | 79 | 9 |
| Formal education (years) | 106 | 12 | [11–16] | 100 | 12 | [10–16] |
A summary of the baseline characteristics of the study population according to dementia-related outcomes is presented in Table 6. As was evident with the distribution of the general demographic characteristics and general health information, including BP and hypertension status, the randomisation process similarly performed very well in achieving balance between the intervention and placebo arms for the number of years since diagnosis and the status of participants with respect to taking dementia treatments. There was parity between the intervention and placebo arms for various baseline MRI measurements serving as the basis of the primary outcome measure and related to whole-brain structure and volume, as well as measures related to hippocampal volume. Although there was a slight suggestion of imbalance between the trial arms with respect to Scheltens scores of baseline atrophy, our analysis had already included adjustment for this because Scheltens scores were a minimisation variable in our randomisation.
| Characteristic | Placebo (N = 106) | Intervention (N = 105) | ||||
|---|---|---|---|---|---|---|
| n | Mean/median/percentage | SD/[IQR] | n | Mean/median/percentage | SD/[IQR] | |
| Taking antidementia drug | 106 | 105 | ||||
| Yes | 102 | 96% | 100 | 95% | ||
| No | 4 | 4% | 5 | 5% | ||
| Diagnosis (years) | 106 | 1.1 | [0.69–2.43] | 105 | 3.38 | [0.64–2.29] |
| TBV (ml) | 106 | 1036 | 111 | 105 | 1022 | 99 |
| Total intracranial volume (ml) | 106 | 1459 | 146 | 105 | 1440 | 140 |
| LVV (ml) | 106 | 47 | [35–64] | 105 | 48 | [35–69] |
| THV (ml) | 106 | 5.0 | 1.0 | 105 | 5.2 | 0.9 |
| LHV (ml) | 106 | 2.5 | 0.5 | 106 | 2.5 | 0.5 |
| RHV (ml) | 106 | 2.6 | 0.5 | 105 | 2.6 | 0.5 |
| Scheltens score | 106 | 105 | ||||
| Absent/low (0–1) | 62 | 58% | 62 | 59% | ||
| Moderate/severe (2–4) | 44 | 42% | 43 | 41% | ||
| ADAS-Cog score | 104 | 19 | 7 | 103 | 20 | 8 |
| MMSE score | 106 | 22 | 3 | 103 | 22 | 4 |
| NPI score | 106 | 6 | [2–15] | 105 | 8 | [3–18] |
| BADLS score | 106 | 5 | [2–9] | 105 | 7 | [2–13] |
| DEMQOL score | 106 | 96 | [85–102] | 105 | 96 | [87–102] |
| DEMQOL-PROXY score | 106 | 92 | [83–99] | 105 | 91 | [82–100] |
It is also evident from Tables 5 and 6 that there appeared to be excellent balance between trial arms for the various variables needed for the secondary outcomes of the study, including the baseline scores for cognitive assessments, as well as assessments of function and quality of life. These are in addition to the data already mentioned on BP and hypertension status. The imaging data related to subgroups of participants that were used to undertake exploratory analyses of intervention-based change in WMH volume and CBF will be presented and discussed in Secondary outcomes.
Losses to follow-up
We successfully enrolled 211 participants and an equivalent number of study companions (93% of the original recruitment target of 228). Of the 211 subjects recruited, 197 (93% of those enrolled) successfully completed the study, and complete primary outcome data were available for 173 (88% of those randomised). The 7% discontinuation rate was favourable and the total loss of primary outcomes (12%) was just below the 20% we assumed as part of the original power calculations. The impact that recruitment below the original target of 228 participants had on the statistical power of the study was mitigated to some extent by the lower than expected dropout rate and the fact that we secured slightly more primary outcomes than we had assumed. This allowed the study to retain 82% statistical power for our analyses rather than the 84% power that our original assumptions and power calculations for the study were based on.
Adherence to intervention
The study benefited from high levels of adherence to the intervention. This was reflected by a total median adherence for all participants of 96% [interquartile range (IQR) 92–98%], which was almost identical between the two arms of the study [median compliance of 96% (IQR 93–98%) in the control arm, median compliance of 96% (IQR 91–98%) in the intervention arm]. In accordance with our predefined criteria for compliance we determined that 88% of study participants were ‘compliers’. This reflected small differences in the numbers of ‘compliers’ in the control arm (90%) and in the intervention arm (86%).
Primary outcome
The baseline and 12-month vMRI follow-up data for the primary outcome are presented in Table 7. Both the intervention arm and the placebo arm showed some brain atrophy between baseline and the 12-month follow-up. Mean TBV fell from 1022 ml (SD 99 ml) to 1002 ml (SD 98 ml) in the intervention arm and from 1036 ml (SD 111 ml) to 1018 ml (SD 113 ml) in the placebo arm. However, overall the levels of atrophy appeared comparable across both trial arms. When the 12-month cross-sectional brain volumes were analysed, the adjusted mean difference between arms for the total study population (n = 171) with adequate follow-up data amenable to analysis [adjusted for minimisation variables (age and Scheltens score) and baseline brain volume and recruitment centre] was –2.29 ml (95% CI –6.46 ml to 0.89 ml; p = 0.136). This was not significant.
| Primary outcome variable | Placebo | Intervention | Adjusted mean difference (ml)a for n = 171 | 95% CI | p-value | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline (ml) | 12 months (ml) | Baseline (ml) | 12 months (ml) | ||||||||
| n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | ||||
| Brain volume (ml) | 106 | 1036 (111) | 87 | 1018 (113) | 105 | 1022 (99) | 84 | 1002 (98) | –2.29 | –6.46 to 0.89 | 0.136 |
| Brain BSI (ml) | 106 | – | 87 | 19.1 (10.3) | 105 | – | 84 | 20.0 (10.8) | 1.23 | –1.72 to 4.19 | 0.411 |
Further analysis of TBV according to boundary shift interval (BSI), which is more sensitive for the measurement of brain atrophy,89 shows that the mean change in TBV-BSI in the placebo arm (19.1 ml, SD 10.3 ml) was almost identical to that in the intervention arm (20.0 ml, SD 10.8 ml). The adjusted mean difference for TBV-BSI between arms of 1.23 ml (95% CI –1.72 to 4.19 ml) was also non-significant (p = 0.41).
Secondary outcomes
A number of secondary outcomes were investigated:
-
We first investigated rates of AD progression as assessed by changes in cognitive assessments [i.e. the 11-item ADAS-Cog,96 the Neuropsychiatric Inventory (NPI)97 and the MMSE98], as well as an examination of change to quality of life (DEMQOL, DEMQOL-proxy)99 and ability to carry out activities of daily living (BADLS100)]. The associated data for baseline and 6- and 12-month follow-up are presented in Table 8.
| Secondary outcome measure | Placebo | Intervention | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | 6 months | 12 months | Baseline | 6 months | 12 months | |||||||
| n | Mean (SD)/Median [IQR] | n | Mean (SD)/Median [IQR] | n | Mean (SD)/Median [IQR] | n | Mean (SD)/Median [IQR] | n | Mean (SD)/Median [IQR] | n | Mean (SD)/Median [IQR] | |
| ADAS-Cog | 104 | 19 (7) | 98 | 21 (9) | 92 | 24 (12) | 103 | 20 (8) | 96 | 22 (11) | 90 | 23 (12) |
| MMSE | 106 | 22 (3) | – | – | 97 | 19 (6) | 105 | 22 (4) | – | – | 95 | 19 (6) |
| NPI | 106 | 6 [2–15] | 102 | 6 [3–14] | 99 | 8 [3–17] | 105 | 8 [3–18] | 100 | 6 [2–15] | 92 | 8 [3–18] |
| BADLS | 106 | 5 [2–9] | 102 | 6 [3–11] | 100 | 7 [3–14] | 105 | 7 [2–13] | 101 | 8 [3–14] | 94 | 10 [3–17] |
| DEMQOL | 106 | 96 [85–102] | 101 | 95 [83–101] | 95 | 94 [85–101] | 105 | 96 [87–102] | 101 | 96 [89–104] | 91 | 96 [87–105] |
| DEMQOL-proxy | 106 | 92 [83–99] | 101 | 92 [80–98] | 98 | 93 [82–100] | 105 | 91 [82–100] | 99 | 91 [83–99] | 92 | 93 [83–99] |
From Table 8 it appears that, with each successive follow-up visit, all scores worsened in both the intervention and the placebo arms.
It was, therefore, important to examine change over time and so we compared the 12-month assessment scores, conditioned on baseline, for each of the secondary outcome measures. In Table 9 comparisons of the adjusted mean differences between baseline and end of study between the trial arms are summarised. There was little evidence to suggest that losartan afforded any clinical benefit according to the various secondary outcome measures assessed.
| Secondary outcome measure | n | Adjusted mean differencea | 95% CI | p-value |
|---|---|---|---|---|
| ADAS-Cog | 179 | –0.52 | –2.71 to 1.66 | 0.64 |
| MMSE | 192 | –0.33 | –1.43 to 0.78 | 0.56 |
| NPI | 191 | 0.88b | 0.68 to 1.13 | 0.30 |
| BADLS | 194 | 1.00b | 0.83 to 1.21 | 0.98 |
| DEMQOL | 186 | 0.98b | 0.89 to 1.09 | 0.74 |
| DEMQOL-proxy | 190 | 1.43 | –1.43 to 4.28 | 0.33 |
-
We also sought to investigate change in the volume of WMH that was measured in our MRI protocol using T2-FLAIR, given that it may predict a 1-year cognitive decline. 24 This was possible in only a subgroup of the study sample (15 of the 23 sites), which equated to a total of 105 participants split almost equally between the placebo (n = 51) and intervention (n = 54) arms. The data on WMH volume are presented in Table 10 for baseline and 12-month follow-up, and regression analysis shows that there was no evidence of difference longitudinally in the WMH volumes after 12 months of treatment (p = 0.697).
-
In a small subset of sites (9 of the 24 sites) there was scope to undertake the additional MRI protocol sequences to explore the level of change in CBF using ASL. Given that ASL has not been used widely in dementia trials to date and thus far mainly in single-centre formats, the multicentre format in RADAR was also highly exploratory to gain insights to similar approaches in future studies. A summary of all of the scanner and ASL sequences implemented is given in Table 11.
| MRI measure | Placebo | Intervention | Regression analysis,a ratio of geometric means (95% CI) | p-value | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | 12 months | Baseline | 12 months | |||||||
| n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | |||
| WMH | 51 | 7354 (3418–20,019) | 51 | 9793 (4788–20,263) | 54 | 10,910 (2361–24,129) | 54 | 11,992 (2548–24,039) | 0.99b (0.93 to 1.05) | 0.697 |
| ASL sequence type | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 3D GRASE | 3D spiral | 2D EPI | 2D EPI | 3D GRASE | 3D GRASE | 2D EPI | 2D EPI | 2D EPI | |
| Labelling strategy | pCASL | pCASL | pCASL | pCASL | PASL (FAIR QII) | PASL (FAIR QII) | PASL (PICORE Q2 T) | PASL (EPISTAR) | PASL (EPISTAR) |
| Acquisition site | 12.2a | 23 | 11 | 22 | 10 | 31 | 19 | 12.1a | 30 |
| Scanner manufacturer (model name) | Philips Medical Systems (Koninklijke Philips NV, Amsterdam, the Netherlands) (Ingenia) | GE Healthcare (Chicago, IL, USA) (Discovery™ MR 750) | Philips Healthcare (Koninklijke Philips NV, Amsterdam, the Netherlands) (Ingenia) | Philips Medical Systems (Koninklijke Philips NV, Amsterdam, the Netherlands) (Achieva) | Siemens Healthcare GmbH (Erlangen, Germany) (3T Skyra) | Siemens Healthcare GmbH (Erlangen, Germany) (3T Skyra) | Siemens Healthcare GmbH (Erlangen, Germany) (3T Prisma) | Philips Medical Systems (Koninklijke Philips NV, Amsterdam, the Netherlands) (Achieva) | Philips Medical Systems (Koninklijke Philips NV, Amsterdam, the Netherlands) (Achieva) |
| Labelling duration (milliseconds) | 1800 | 1450 | 1650 | 1650 | 700 | 700 | 700 | 440 | 440 |
| Initial PLD (milliseconds) | 2000 | 2025 | 1525 | 1200 | 1320 | 1320 | 1100 | 1360 | 1360 |
| Echo time TE (milliseconds) | 11.5 | 9.9 | 12.4 | 20 | 12 | 12 | 11 | 15 | 15 |
| Repetition time TR (milliseconds) | 6500 | 4739 | 4000 | 4000 | 3500 | 3500 | 3500 | 2619 | 2619 |
| Flip angle | 90° | 111° | 40° | 40° | 130° | 130° | 90° | 90° | 90° |
| Readout module | 3D GRASE, CLEAR (uniformity) | 3D FSE interleaved stack-of-spirals | 2D EPI SENSE 2.5, CLEAR (uniformity) | 2D EPI SENSE 2.2, CLEAR (uniformity) | 3D GRASE | 3D GRASE | 2D FID-EPI | 2D EPI SENSE 2, CLEAR | 2D EPI SENSE 2.3, CLEAR (uniformity) |
| Number of slices | 20 | 30 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Slice thickness (mm) | 6 | 6.0 | 5.25 | 6 | 5.25 | 5.25 | 6.0 | 5 | 5 |
| Acquisition voxel size (mm) | 4.06 × 4.33 × 6.00 | 4.00 × 4.00 × 6.00 | 3.75 × 3.90 × 5.25 | 4.06 × 4.20 × 6.00 | 1.875 × 1.875 × 5.25 | 3.75 × 3.75 × 5.25 | 4.0 × 4.0 × 6.0 | 3.75 × 3.75 × 5.00 | 3.75 × 3.89 × 5.00 |
| Slice gap (mm) | 0 | 0 | 1 | 0.6 | N/A | N/A | N/A | 1 | 1 |
| Background suppression (n pulses) | Yes, n = 4 | Yes, n = 5 | Yes, n = 2 | No | Yes, n = 2 | Yes, n = 2 | No | No | No |
| Acquisition duration (minutes) | 5:10 | 5:37 | 5:28 | 4:08 | 3:51 | 4:47 | 6:06 | 4:37 | 4:37 |
The retrieved ASL data underwent QA assessment and were processed using the ExploreASL software package104,105 to generate quantitative CBF maps. ASL quality control-checked ASL CBF images were then grouped according to the spatial CoV values. Spatial CoV is a ratio of the SD of CBF to the mean of CBF and has been identified as an additional new approach that allow estimates (proxy) measurements of CBF. 106,107
The initial groups were assigned to groups using the following differentiating criteria and stringency towards CBF measurement:
-
predominant CBF contrast: CoV values ≤ 1
-
predominant vascular contrast: 1 < CoV values < 1.5
-
predominantly artefacts CoV ≥ 1.5.
Group 1 data were the most stringent and accurate for the measurement of CBF. Group 2 data, although not as stringent as those in group 1, could serve as a proxy measure for CBF and thus could be used to increase the sample size in addition to group 1 measurements. Group 3 data, because of their largely artefactual nature, were excluded from analyses. All images were then inspected visually by a trained researcher and their categorisation was corrected if needed before further analyses were undertaken.
Owing to the nature of the study design and the intention to measure change over time, we excluded not only data from participants with at least one data set in group 3 (i.e. mainly artefactual), but also data from participants with at least one scan in group 2 (i.e. predominantly vascular contrast) – this participant’s data (even if the other scan was group 1) were assigned to the vascular group.
This approach yielded two data sets:
-
one suitable for CBF perfusion analysis (images with predominant CBF contrast only)
-
one suitable for spatial CoV analysis (images with combinations of predominant CBF contrast and predominant vascular contrast images).
A summary of the distribution of the number of participants per site is presented in Figure 5.
FIGURE 5.
Distribution of ASL data according to RADAR site. Site 12 had two scanners in use throughout the study; however, with different specifications and hence was treated separately.
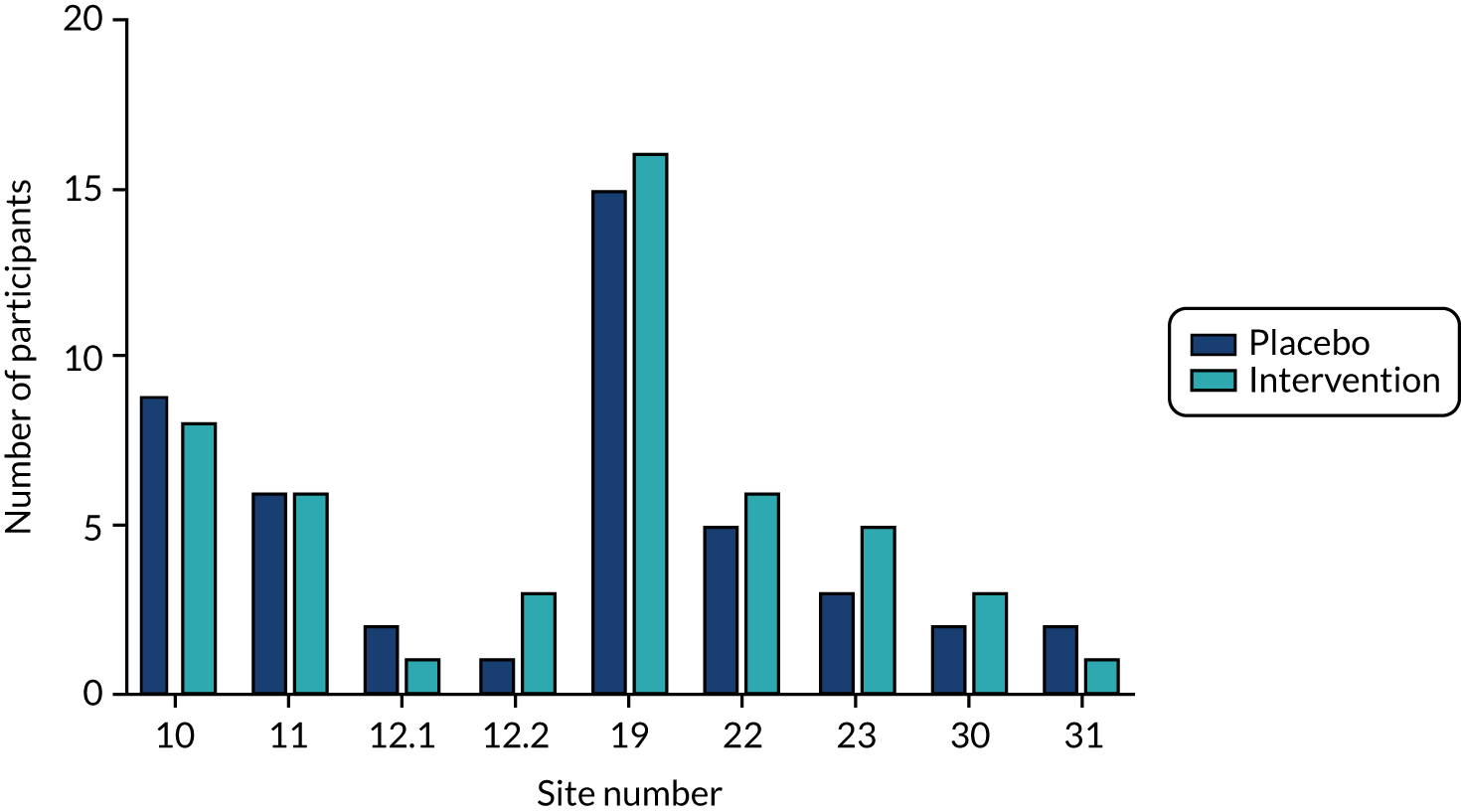
An initial look at the data acquired showed large variation in the interpretation of initial ASL protocols; this varied not only between sites with different MRI scanners, but also between sites with comparable scanners. Following the application of our QA approach, we found that this also had a significant effect on the final numbers of available scans that would be amenable to analysis. These involved group 1 scans (which would provide the greatest specificity for CBF) and group 2 scans (which would provide less specificity for CBF but would allow proxy estimations of arterial transit time).
Given the variability of data, we opted to explore the feasibility of a larger combined analysis for the study by first piloting analysis on data from the subset of sites with the largest number of collected viable ASL scans: these sites were 19 and 10.
Scanning site 19 yielded data from 31 participants (as it served more than one recruitment site). The distribution of scans across treatment arms was relatively equal. Based on the QA, data from only 10 of these 31 participants were amenable to CBF analysis (data set 1), whereas data from 23 individuals fulfilled data set 2 criteria and could be analysed as a proxy estimate for arterial transit time. From this site, data from eight participants were excluded because they were deemed to be predominantly artefactual and thus not amenable to analysis. Similarly, at site 10, which had the second largest number of scans available (n = 17 participants), data from only seven participants were amenable to CBF analysis (data set 1), but data from 15 participants were amenable to the arterial transit time analysis by proxy (data set 2) using the CoV approach.
We were then in a position to explore the differences between treatment arms on an individual site basis both at baseline and at 12-month follow-up. Figure 6 presents the data for site 19 and site 10 (in descending order of the number of scans suitable for CoV analysis). We opted to use the validated106,107 CoV analysis approach on data set 2 scans in each case for these exploratory analyses because it provided more data points and thus greater power than the use of data set 1 scans (Table 12).
FIGURE 6.
Exploratory comparisons of CBF using spatial CoV analysis. (a) Site 19: grey matter spatial CoV distribution from the baseline and 12-month follow-up ASL scans acquired using a 3-T Siemens Prisma with 2D EPI PASL PICORE Q2T; and (b) site 10: grey matter spatial CoV distribution in the baseline and 12-month follow-up ASL scans acquired using a 3-T Siemens Skyra with 3D GRASE pulsed ASL FAIR QII. 2D, two-dimensional; 3D, three-dimensional; EPI, echo-planar imaging; GRASE, gradient and spin echo; FAIR, flow-sensitive alternating inversion recovery; PASL, pulsed arterial spin labelling; PICORE, proximal inversion with control of off-resonance effects.
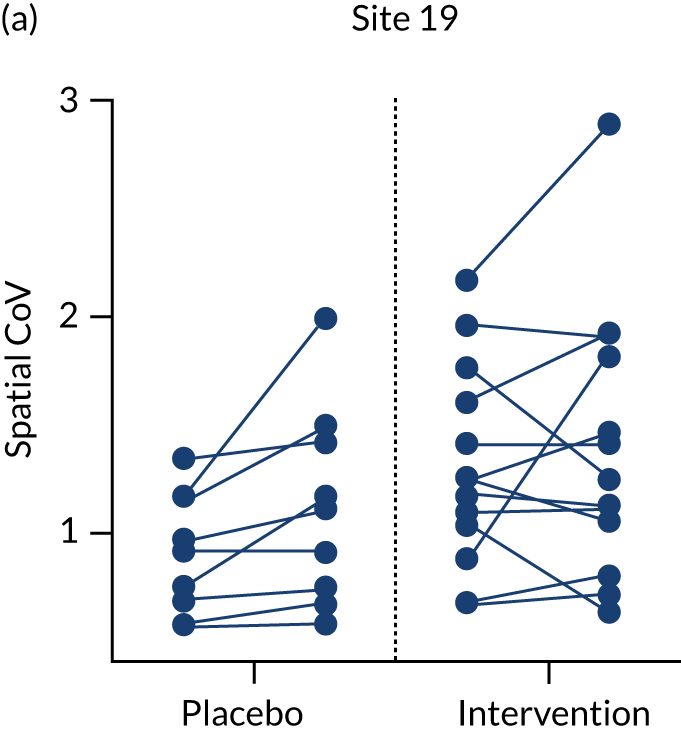
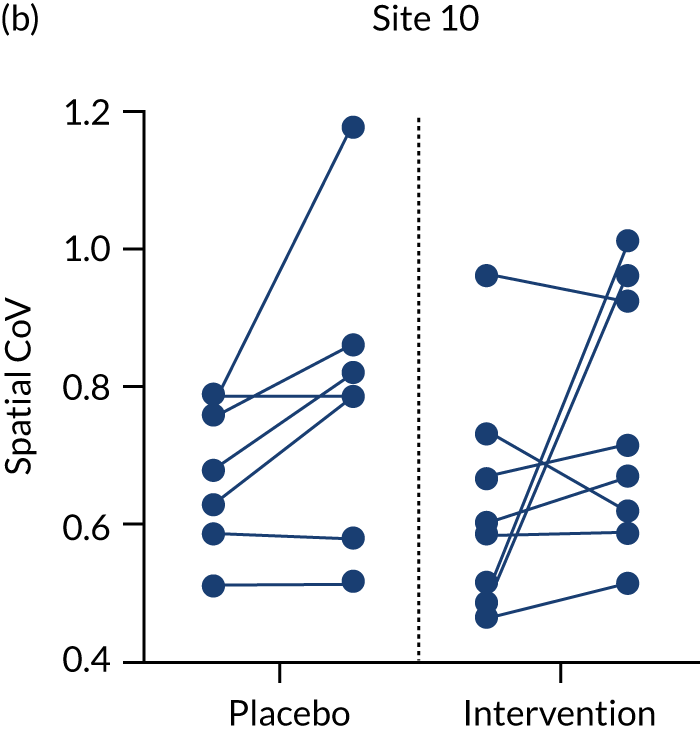
| MRI measure | Placebo | Intervention | ||||||
|---|---|---|---|---|---|---|---|---|
| Baseline | 12 months | Baseline | 12 months | |||||
| n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | |
| CBF (ml/100 g/minute) | 5 | 31.05 (5.47) | 5 | 28.62 (5.41) | 5 | 34.74 (8.64) | 5 | 32.55 (5.29) |
| Spatial CoV | 9 | 0.98 (0.37) | 9 | 1.49 (1.26) | 13 | 1.30 (0.46) | 13 | 1.39 (0.62) |
The information in Figure 6 is presented according to treatment arm and based on data collected during each of the baseline and follow-up visits. Using spatial CoV analyses it would appear that there are no obvious patterns of change between baseline and follow-up in either treatment arm or in either site.
A multiple regression was carried out to test if spatial CoV baseline values and treatment (placebo or intervention) predicted spatial CoV follow-up values for site 19, where the largest number of data survived QA. The results of the regression indicated that the model explained 63.0% of the variance and that the model was significant [F(2,19) = 16.159; p < 0.0005]. It was found that spatial CoV baseline values significantly predicted spatial CoV follow-up values (β1 = 1.085; p < 0.0005), whereas the assignment of treatment did not add statistically significantly to the prediction (β2 = 0.163; p = 0.363).
Similar exploratory comparisons were examined (data not shown) for both sites using the more underpowered data set 1. This showed similar patterns but similarly there were no evident changes between baseline and the 12-month follow-up for either treatment arm but there were fewer participants in each (n = 10 for site 19, five in each arm, and n = 7 for site 10, three in the intervention arm and four in the placebo arm.
The change in BP over time was deemed an important consideration in our study given the normal prescribed purpose of the intervention (i.e. to lower BP in the treatment of hypertension). It was also deemed an important positive control for the intervention to demonstrate (1) that the intervention was biologically active (i.e. observable reductions in BP in the intervention arm would be proof the medication was active) and (2) that variations in BP might be associated with the volume of WMH and cerebrovascular health in general and, thus, CBF. Thus, we were interested in the difference in change in BP over 12 months between trial arms and whether or not this had any influence on the primary and secondary outcomes.
Mean systolic and diastolic BP at baseline and 12-month follow-up are presented in Table 13. As described, the mean BP appeared comparable at baseline between the intervention and placebo arms. A slight reduction in mean systolic BP, but not in diastolic BP, was evident in the intervention arm.
| BP | Baseline, mean (SD) | 12 months, mean (SD) | ||
|---|---|---|---|---|
| Intervention (n = 83) | Placebo (n = 84) | Intervention (n = 95) | Placebo (n = 98) | |
| Systolic | 138 (13) | 136 (15) | 133 (21) | 139 (17) |
| Diastolic | 79 (9) | 79 (8) | 78 (13) | 81 (9) |
Yet on comparison of change in BP over the course of the follow-up period, it is evident that there are statistically significant differences in both systolic and diastolic BP between the intervention and placebo arms (p < 0.001) such that participants in the intervention arm had lower BP over the follow-up period (Table 14).
| BP | n | Adjusted difference in means (95% CI)a | p-value |
|---|---|---|---|
| Systolic | 165 | –6.96 (–10.15 to –3.78) | < 0.001 |
| Diastolic | 165 | –3.59 (–5.29 to –1.89) | < 0.001 |
Given that there were no apparent differences between the intervention and placebo arms of the study at baseline and 12-month follow-up, it was also important to investigate, in line with previous studies,49–51,74 whether or not differences in BP may account for any variation in the primary and secondary outcomes. Correlation analyses showed that there was no strong correlation between systolic or diastolic BP and the primary outcomes, nor was there any evidence of association for any of the assessment-based cognitive, functional or behavioural outcome measures. Although data on the volume of WMH intensities were available for only a subgroup of RADAR participants (45% of randomised participants), we still explored the association with systolic and diastolic BP. Similarly, there was no evidence of any association between volume of WMH and change in systolic and diastolic CBF.
We had also intended to measure the levels of association between the primary outcome of brain atrophy and cognitive decline. This was because brain atrophy has been reported to serve as an objective proxy for cognitive decline in dementia, as previously described. Similarly, it was important to examine the possible association between the rate of brain atrophy and rate of change in our other secondary functional and behavioural outcome measures, predominantly to help inform any future studies where the use of cognitive, functional and behavioural measures would be preferable to using MRI-based outcomes, which, as has been described, can introduce additional complexity and may be more limiting to the identification of recruitment centres.
Table 15 presents the correlations between the primary outcome and the number of secondary cognitive, behavioural and functional outcomes that were assessed. There were variable levels of correlation between the variables presented. The strongest (negative) correlation (–0.56) was between ADAS-Cog scores and MMSE scores. There was also evidence of weak negative correlations between brain volume and ADAS-Cog score (–0.39) and between the functional outcomes DEMQOL-proxy and BADLS (–0.34). By contrast, NPI and BADLS were weakly positively correlated (0.41), as were ADAS-Cog and BADLS (0.36).
| Outcome measure | TBV | TBV-BSI | ADAS-Cog | MMSE | NPI | BADLS | DEMQOL |
|---|---|---|---|---|---|---|---|
| TBV | 1 | ||||||
| TBV-BSI | –0.72 | 1 | |||||
| ADAS-Cog | –0.39 | 0.48 | 1 | ||||
| MMSE | 0.21 | –0.39 | –0.56 | 1 | |||
| NPI | 0 | 0.02 | 0.17 | –0.14 | 1 | ||
| BADLS | –0.2 | 0.23 | 0.36 | –0.24 | 0.41 | 1 | |
| DEMQOL | –0.1 | 0.09 | –0.09 | 0.01 | –0.09 | –0.06 | 1 |
One of our secondary outcome measures was an examination of levels of drug compliance and tolerability across the main randomised phase. As already reported, there were high levels of compliance (88%) across all participants, with compliance only slightly lower in the intervention arm (86%) than in the placebo arm (90%) – a difference that was consistent with chance variability.
We also sought to examine the effect of the intervention with respect to SAEs and AEs reported during the study. There was no significant difference between the number of SAEs reported in the intervention arm (n = 22) and the placebo arm (n = 20). A summary of the recorded SAEs is provided in Appendix 2, Table 24. One SAE was reported in the open-label phase and is also summarised in Appendix 2, Table 24.
A summary of the reported AEs over several follow-up visits from the baseline visit is provided in Table 16. As is apparent from Table 16, there were no obvious or statistically significant differences between the intervention and placebo arms in the numbers of AEs. Indeed, the proportion of participants reporting any AE at 12 months was 6% lower in the intervention arm than in the control arm, although this difference was not significant.
| Adverse event | Follow-up visit | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 14 days | 3 months | 6 months | 9 months | 12 months | ||||||
| n | % | n | % | n | % | n | % | n | % | |
| Control | ||||||||||
| Number entered | 106 | 106 | 101 | 100 | 100 | |||||
| Participants reporting any AE | 32 | 30 | 43 | 41 | 46 | 46 | 41 | 41 | 45 | 45 |
| Intervention | ||||||||||
| Number entered | 104 | 104 | 101 | 99 | 96 | |||||
| Participants reporting any AE | 30 | 29 | 51 | 49 | 46 | 46 | 45 | 45 | 37 | 39 |
Finally, we also explored the results of the primary analysis according to baseline brain volume and according to whether or not participants were taking antidementia drugs. Regarding the analysis to explore the possible interaction between treatment and baseline brain volume, there was no evidence of an interaction (interaction coefficient –0.002, 95% CI 0.039 to 0.036; p = 0.921). Similarly, exploratory analysis of a possible interaction between the primary analysis and taking antidementia drugs revealed no evidence of an interaction (interaction coefficient 8.21, 95% CI –10.18 to 26.59; p = 0.379).
As with all the secondary analyses, the study was powered principally for the primary analyses and so all of these observations should be viewed with appropriate caution.
Chapter 3 Sensitivity analyses
Adherence to intervention
As part of a sensitivity analysis, investigation CACE was undertaken using instrumental variable two-stage least squares regression. In this analysis, outcomes among those who ‘complied’ with the intervention were compared with outcomes among a group of ‘would-be compliers’ in the control arm. As described, treatment compliance was defined as (self-report of) taking 80–120% of study medication. As is evident in Table 17, the CACE analysis showed no differences with the conclusions drawn from the primary analysis.
| Analysis | n | Adjusted difference in means | 95% CI | p-value |
|---|---|---|---|---|
| Primary | 171 | –2.79 | –6.46 to 0.89 | 0.136 |
| CACE | 166 | –3.23 | –7.14 to 0.69 | 0.106 |
Missing data
A thorough investigation of the data showed that approximately 19% of the data or data sets were missing or incomplete, the majority of which related to the data collected from the various assessment tools used to collect some of the secondary outcomes. Reassuringly, there was no evidence that the extent of missingness differed by trial arm (p = 0.70), with 18% missingness in the placebo arm compared with 20% in the intervention arm.
A further exploration of the potential association between baseline characteristics and data missingness was also undertaken. There was no evidence of an association between baseline brain volume (p = 0.76), MMSE (p = 0.12), duration of diagnosis of AD (p = 0.13) and whether or not participants were taking dementia drugs (0.54) with respect to data missingness of the primary outcome. Similarly, there was no evidence of an association between the level of primary outcome data missingness and gender (p = 0.14), whether participants were hypertensive or normotensive (p = 0.63), or their number of years of prior education (p = 0.11). By contrast, we did find evidence of association between participant age and data missingness (p = 0.01) in that older participants were more likely to have missing data.
Multiple imputation using chained equations was used to impute missing data (Table 18). Variables included in the imputation model included all variables in the primary analysis model, baseline variables predictive of missingness and variables associated with the primary outcome. The findings from the analysis on imputed data are consistent with those from the primary analysis model: there was no evidence of a difference in brain volume at 12-month follow-up between the treatment arms.
| Analysis | n | Adjusted difference in means | 95% CI | p-value |
|---|---|---|---|---|
| Primary | 171 | –2.79 | –6.46 to 0.89 | 0.136 |
| MICE | 211 | –1.26 | –5.77 to 3.25 | 0.580 |
Post hoc exploratory analyses
In addition to the primary and secondary outcome analyses that have been described in the protocol, and given the overall lack of evidence in support of the role of losartan as a potential intervention for AD, we considered the potential value of three additional exploratory analyses. These were intended to make use of data gathered to inform the design of future studies of these types of BP-lowering medications in this participant group and in relation to the selection of other alternative outcome measures.
Did pre-existing hypertension affect primary and secondary outcomes?
We first sought to explore further what impact, if any, a prior diagnosis of hypertension may have on the primary outcome and secondary outcomes, given the long-standing recognition of association between mid-life hypertension and increased risk of AD. 20,58 It was therefore considered that participants with pre-existing hypertension, for which they would be receiving treatment at the time of entry to the study, may plausibly have different associations with various outcomes not only at baseline, but also in how they responded over time to the intervention.
The full data are presented in Tables 19 and 20. Table 19 presents the data and regression analyses related to the presence or absence of hypertension on the various imaging data. Table 20 similarly presents the data and regression analyses related to the presence or absence of hypertension on the various cognitive, behavioural and functional outcomes. The regression analyses of these data according to the presence or absence of hypertension at baseline and 12-month follow-up, and according to intervention or placebo, showed that there was no association with any of the MRI-based data on brain volume or hippocampal volume. Similarly, but with the exception of DEMQOL-proxy, there were no significant differences for any of the cognitive (ADAS-Cog and MMSE), behavioural (NPI) or functional (BADLS, DEMQOL) outcome measures. The association with DEMQOL-proxy (interaction coefficient 6.31, 95% CI 0.42 to 12.20; p = 0.04) was unexpected and, given that DEMQOL-proxy is a companion-reported outcome, we cannot discern a reasonable explanation for this association and it most likely represents a type 1 false-positive error.
| Atrophy measure | Placebo, mean (SD) [IQR] | Intervention, mean (SD) [IQR] | Regression analysis | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | 12 months | Baseline | 12 months | Interaction coefficient | 95% CI | p-value | |||||
| Norm | Hyper | Norm | Hyper | Norm | Hyper | Norm | Hyper | ||||
| TBV (ml) | 1048 (109) | 1023 (115) | 1029 (112) | 1009 (114) | 1023 (106) | 1022 (106) | 1044 (97) | 998 (101) | –2.62 | –10.43 to 5.19 | 0.509 |
| LVV (ml) | 44 [34–62] | 50 [38–67] | 49 [38–66] | 54 [42–70] | 48 [38–59] | 50 [31–76] | 51 [40–67] | 55 [38–86] | 1.00 + | 0.97 to 1.04 | 0.795 |
| THV (ml) | 5.1 (1.0) | 5.0 (0.9) | 5.1 (1.0) | 4.7 (0.9) | 5.2 (0.9) | 5.2 (0.9) | 5.0 (0.9) | 5.1 (0.9) | –0.02 | –0.08 to 0.04 | 0.482 |
| LHV (ml) | 2.5 (0.5) | 2.4 (0.5) | 2.5 (0.5) | 2.3 (0.4) | 2.5 (0.5) | 2.5 (0.5) | 2.5 (0.5) | 2.5 (0.5) | –0.02 | –0.05 to 0.02 | 0.397 |
| RHV (ml) | 2.6 (0.5) | 2.5 (0.5) | 2.6 (0.5) | 2.4 (0.5) | 2.6 (0.5) | 2.7 (0.5) | 2.5 (0.5) | 2.6 (0.5) | –0.01 | –0.04 to 0.03 | 0.748 |
| TBV-BSI (ml) | – | – | 19.9 (10.8) | 18.3 (9.8) | – | – | 20.1 (10.9) | 19.8 (10.7) | 1.81 | –4.43 to 8.05 | 0.567 |
| LVV-BSI (ml) | – | – | 5 [3–7] | 5 [3–7] | – | – | 5 [2–8] | 5 [3–7] | 0.24 | –1.87 to 2.36 | 0.819 |
| LHV-BSI (ml) | – | – | 0.11 (0.07) | 0.12 (0.06) | – | – | 0.10 (0.08) | 0.12 (0.08) | 0.01 | –0.03 to 0.06 | 0.604 |
| RHV-BSI (ml) | – | – | 0.11 (0.06) | 0.12 (0.06) | – | – | 0.10 (0.07) | 0.11 (0.08) | < 0.0001 | –0.05 to 0.05 | 0.998 |
| Scheltens score, n (%) | |||||||||||
| 0 | 13 (23) | 7 (14) | 7 (13) | 2 (4) | 13 (22) | 6 (13) | 9 (18) | 5 (13) | 1.31 | 0.28 to 6.25 | 0.732 |
| 1 | 19 (34) | 23 (46) | 15 (29) | 13 (28) | 23 (40) | 20 (43) | 21 (41) | 15 (38) | |||
| 2 | 17 (30) | 15 (30) | 19 (37) | 24 (52) | 8 (14) | 12 (26) | 9 (18) | 12 (30) | |||
| 3 | 7 (13) | 5 (10) | 11 (21) | 7 (15) | 14 (24) | 9 (19) | 12 (24) | 7 (18) | |||
| 4 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (3) | |||
| WMH | 6917 [2791–13,778] | 12,551 [3640–21,071] | 8176 [3029–16,097] | 13,929 [5152–24,833] | 8802 [2805–22,589] | 12,387 [1981–25,837] | 11,036 [3023–23,763] | 13,903 [2126–28,034] | 0.96 + | 0.85 to 1.09 | 0.545 |
| Secondary outcome measure | Placebo, mean (SD) [IQR] | Intervention, mean (SD) [IQR] | Regression analysis | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | 12 months | Baseline | 12 months | Interaction coefficent | 95% CI | p-value | |||||
| Norm | Hyper | Norm | Hyper | Norm | Hyper | Norm | Hyper | ||||
| ADAS-Cog | 20 (7) | 18 (6) | 25 (12) | 23 (11) | 19 (8) | 21 (8) | 22 (12) | 24 (13) | –1.98 | –6.58 to 2.61 | 0.395 |
| MMSE | 22 (3) | 23 (4) | 19 (6) | 20 (5) | 22 (4) | 22 (4) | 19 (6) | 19 (6) | 0.58 | –1.65 to 2.81 | 0.609 |
| NPI | 8 [3–14] | 4 [2–15] | 8 [4–15] | 7 [3–21] | 8 [4–18] | 8 [3–18] | 9 [3–19] | 7 [2–16] | 0.86 + | 0.51 to 1.45 | 0.567 |
| NPI-proxy | 5 [2–10] | 5 [1–9] | 6 [2–9] | 6 [2–14] | 5 [2–10] | 6 [1–9] | 6 [2–10] | 4 [2–7] | 0.78 + | 0.47 to 1.27 | 0.307 |
| BADLS | 5 [3–9] | 5 [2–9] | 7 [3–15] | 7 [4–14] | 4 [1–11] | 9 [3–18] | 9 [3–16] | 12 [3–19] | 0.71 + | 0.48 to 1.04 | 0.075 |
| DEMQOL | 94 [85–103] | 96 [87–101] | 93 [86–100] | 94 [85–102] | 96 [89–102] | 93 [86–102] | 95 [87–105] | 99 [88–107] | 1.12 + | 0.96 to 1.30 | 0.146 |
| DEMQOL-proxy | 90 [82–98] | 95 [84–100] | 92 [82–98] | 95 [85–101] | 93 [86–100] | 90 [75–99] | 93 [83–99] | 94 [83–100] | 6.31 | 0.42 to 12.20 | 0.036 |
Exploring the characteristics of non-randomised participants of the open-label phase
In addition to providing all participants with some reassurance and the opportunity to try the intervention, we used an open-label phase to aid the selection of participants who were most likely to complete the main study. The effectiveness of this is supported by favourable study retention rates in the randomised phase. With this in mind, we wanted to better understand the characteristics of participants who entered the open-label phase but did not go on to the randomised phase, in which, as previously described, there was an excellent balance between the intervention and placebo arms in baseline participant demographics and outcome measures.
Table 21 details the reasons why open-label participants were not randomised. A number of important observations are evident.
| Recorded characteristic | Discontinuation | MRI failure | Total | |
|---|---|---|---|---|
| ≤ 7 days | 8–14 days | |||
| Number of participants | 27 | 15 | 8 | 50 |
| Hypertensive (i.e. taking BP medications) | 5 | 2 | 0 | 7 |
| Normotensive | 22 | 13 | 8 | 43 |
| Discontinuation in relation to intervention | ||||
| Considered unrelated | 3 | 0 | 8 | 11 |
| Considered related or possibly related | 24 | 15 | – | 39 |
| Recorded reasons for discontinuation | ||||
| BP | 17 | 9 | – | 26 |
| Bloods | 2 | 2 | – | 4 |
| Compliance | 1 | 0 | – | 1 |
| AEs | 5 | 3 | – | 8 |
| Other reasons | 2 | 1 | – | 3 |
| MRI | – | – | 8 | 8 |
| Protocol reason | 23 | 11 | 8 | 42 |
| Non-protocol reason | 4 | 4 | – | 8 |
First, over half of participants who withdrew from this phase of the study were in the first 7 days of treatment (at the lower dose of the active medication).
Second, and notable with respect to the relevance of hypertension in this group of participants, was that 86% of the discontinued participants were normotensive; two-thirds of those who withdrew within the first 7 days of treatment and all who withdrew for reasons related to MRI were normotensive.
Third, only 22% of the participants were thought to have discontinued for reasons that were unrelated to losartan. By contrast, the majority of the participants (78%) withdrew for reasons that were considered to be related to the intervention. Among these, there was less certainty that losartan was responsible for discontinuation of six participants, three of whom reported dizziness, confusion and fatigue; one participant reported nausea, one participant had a fall and one participant reported lethargy that continued during and after wash out.
Fourth, of the 50 participants who entered the open-label phase who were not randomised, just over 50% discontinued owing to BP changes that were deemed out of range or clinically significant, and another 20% (n = 8) were not randomised because of either unsuitable or incomplete baseline MRIs; a further 20% discontinued because of reported AEs. Of the eight participants who reported AEs, six are the same six participants whose reasons for discontinuation were less certain to be related to the open-label losartan. Of the remaining two participants, one developed a rash on their lower back and arm that coincided with a drop in their BP and the other developed bradycardia around the same time as their BP dropped. Two participants reported that they wished to discontinue because they viewed the trial to be too problematic (n = 1) or too time-consuming (n = 1), and the remaining participant reported that they felt that the drug was reducing their cognitive function.
Fifth, and finally, 82% of discontinuations were for protocol reasons, and only eight discontinuations were deemed non-protocol related.
Exploration of MRI-based measures of hippocampal atrophy as an alternative outcome measure
Although we used whole-brain atrophy as the primary outcome measure in this study, we were fortunate to be able to extract data on hippocampal atrophy from the relevant scan sequences. Given the potential utility of hippocampal atrophy as an outcome measure for dementia progression in its own right, and that it is measurable with scanning intervals of > 6 months, this study provided a welcome opportunity to explore it in the study population and also explore the relationship between other atrophy-related measures. Indeed, the original development of the visual Scheltens rating scale84 we used in this study as a minimisation variable to expedite randomisation was created because MRI-based methods for measuring hippocampal atrophy were not available at that time.
Table 22 presents a summary of additional measures of hippocampal and ventricular atrophy. We examined total hippocampal volume (THV), as well as left and right hippocampal volumes (LHV and RHV, respectively), at both baseline and 12-month follow-up. We also present change in volume using the BSI method92 applied to the individual hippocampi (LHV-BSI and RHV-BSI). We also explored data regarding lateral ventricle volume (LVV), which we examined at each time point, as well as change over time according to intervention arm (LVV-BSI). We also present these MRI-derived data alongside an exploratory examination of the subjective assessor-derived Scheltens ratings that were captured at baseline to inform randomisation and where these were also captured at 12-month follow-up.
| Placebo | Intervention | Regression analysis, adjusted mean difference (95% CI)a | p-value | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | 12 months | Baseline | 12 months | |||||||
| n | Mean (SD)/median [IQR] | n | Mean (SD)/median [IQR] | n | Mean (SD)/median [IQR] | n | Mean (SD)/median [IQR] | |||
| LVV (ml) | 106 | 47 [35–64] | 86 | 50 [40–67] | 105 | 48 [35–69] | 83 | 51 [40–78] | 0.99b (0.98 to 1.01) | 0.443 |
| LVV-BSI (ml) | – | – | 86 | 5 [3–7] | – | – | 83 | 5 [3–8] | –0.45 (–1.45 to 0.56) | 0.380 |
| LHV (ml) | 106 | 2.5 (0.5) | 87 | 2.4 (0.5) | 105 | 2.5 (0.5) | 84 | 2.5 (0.5) | 0.01 (–0.01 to 0.03) | 0.337 |
| LHV-BSI (ml) | – | – | 87 | 0.11 (0.06) | – | – | 84 | 0.11 (0.08) | –0.01 (–0.03 to 0.01) | 0.385 |
| RHV (ml) | 106 | 2.6 (0.5) | 87 | 2.5 (0.5) | 105 | 2.6 (0.5) | 84 | 2.6 (0.5) | 0.01 (–0.003 to 0.03) | 0.112 |
| RHV-BSI (ml) | – | – | 87 | 0.12 (0.06) | – | – | 84 | 0.11 (0.08) | –0.01 (–0.03 to 0.01) | 0.231 |
| THV (ml) | 106 | 5.0 (1.0) | 87 | 4.9 (0.9) | 105 | 5.2 (0.9) | 84 | 5.0 (0.9) | 0.02 (–0.01 to 0.05) | 0.144 |
| Scheltens score | ||||||||||
| 0 | 20 | 19% | 9 | 9% | 19 | 18% | 14 | 15% | 0.31c (0.14 to 0.68) | 0.004 |
| 1 | 42 | 40% | 28 | 29% | 43 | 41% | 36 | 40% | ||
| 2 | 32 | 30% | 43 | 44% | 20 | 19% | 21 | 23% | ||
| 3 | 12 | 11% | 18 | 18% | 23 | 22% | 19 | 21% | ||
| 4 | 0 | 0% | 0 | 0% | 0 | 0% | 1 | 1% | ||
From Table 22 it is evident that there were no differences according to trial arm for LVV, LHV or RHV at baseline and 12-month follow-up (p = 0.529, p = 0.360 and p = 0.099, respectively). Similarly, the mean change in each of these variables between 12-month follow-up and baseline was also non-significant (LVV-BSI, p = 0.641; LHV-BSI, p = 0.388; and RHV-BSI, p = 0.233). Similarly, there were no significant differences in THV (p = 0.143). These data, therefore, are entirely consistent with the absence of any significant differences recorded for the primary outcome of whole-brain atrophy.
Scheltens scores84 collected during the study are also presented in Table 22. There was a suggestion of a difference between arms whereby the odds of being in a higher (more severe) Scheltens score category at 12 months are lower for participants in the intervention arm than for those in the control arm (p = 0.004). Although this observation, based on the Scheltens rating scale, could be viewed as a possible treatment benefit, it is very important that this subjective rating scale is not overinterpreted when compared with the objective MRI-based measures that we found no effects from. Furthermore, the Scheltens scale rates atrophy of the MTA, which includes the hippocampus, and Scheltens scores not directly comparable with the MRI-based rating of the hippocampus.
Finally, given that both MRI-based measures and Scheltens ratings were focused on assessing levels of atrophy in participants, we thought that it may be helpful to present the correlations between the variables across the entire study population as there were no treatment-based differences. Table 23 summarises the levels of correlation between the MRI-based measures of atrophy and the observer-rated levels of atrophy by Scheltens scale. There was evidence of a strong correlation between THV and both LHV and RHV, whereas there was a weak negative correlation between LVVs and THVs, LHVs and RHVs. Of particular note was the observation that there was negligible correlation between subjective observer-based Scheltens scores and any of the MRI-based measures.
| Atrophy measure | LVV | THV | LHV | RHV | TBV-BSI | LVV-BSI | LHV-BSI | RHV-BSI | S_S |
|---|---|---|---|---|---|---|---|---|---|
| LVV (ml) | 1 | ||||||||
| THV (ml) | –0.40 | 1 | |||||||
| LHV (ml) | –0.36 | 0.87 | 1 | ||||||
| RHV (ml) | –0.31 | 0.86 | 0.50 | 1 | |||||
| TBV-BSI (ml) | 0.79 | –0.47 | –0.45 | –0.37 | 1 | ||||
| LVV-BSI (ml) | 0.98 | –0.40 | –0.35 | –0.32 | 0.80 | 1 | |||
| LHV-BSI (ml) | 0.42 | –0.65 | –0.71 | –0.42 | 0.51 | 0.42 | 1 | ||
| RHV-BSI (ml) | 0.46 | –0.64 | –0.45 | –0.67 | 0.46 | 0.46 | 0.62 | 1 | |
| S_S | 0.11 | 0.05 | 0.02 | 0.07 | 0.12 | 0.12 | –0.06 | –0.03 | 1 |
Chapter 4 Discussion
The RADAR trial was designed to be the first RCT to formally test the angiotensin hypothesis in AD. 20 Central to this hypothesis is the role of AngII, a potent vasoconstrictor known for decades to be implicated in hypertension, acting through the AngII type 1 receptor. 110 AngII levels are in excess in AD,35 as a direct result of excess levels of ACE131,32 or reductions in the activity of ACE235 that have been found in post-mortem brain tissue and compared with brain tissue from non-demented individuals. The relevance of these findings to the pathogenesis of AD is that AngII is a multifunctional peptide with known functions in the synthesis of the inflammatory mediator TNFα,36,37 reduction of acetylcholine release,38,39 promotion of glutamatergic effects,40 and promotion of oxidative stress and mitochondrial dysfunction,111 all of which are major sequelae of AD pathology. Furthermore, AngII has been shown in rodents to promote the production of Aβ and alter the levels of phosphorylation of tau protein, analogous to AD-related changes that give rise to neurofibrillary tangles. 41,42 This has been supported by our own findings of reduced ACE2 activity in post-mortem AD brain tissue that coincided with elevated levels of AngII, was highly correlated with Aβ and tau pathology and was inversely related to ACE1 activity. 35,43
These largely preclinical and ex vivo data have gone some way towards providing a possible mechanistic explanation for findings, spanning several decades, suggesting that hypertension in mid-life7,8 and later life9 and stroke10 all increase risk of dementia. In other words, the association between hypertension and dementia could be related to excess RAS activity in mid-life that contributes to hypertension, whereas the occurrence of dementia later in life derives from what would have been the coincident chronic exposure of the brain to elevated levels of AngII. Alternatively, it may relate to the fact that, although a number of hypertension treatments lower BP effectively, they do not all necessarily lower levels of AngII. Moreover, the more recent finding that cerebrovascular pathology and dysfunction are likely to be the earliest pathological event in the development of AD18 also reinforces this possibility. Similarly, several studies both prior to and since the commencement of the RADAR trial have reported that AngII-targeting drugs [AngII type 1 receptor antagonists (AT1RAs) and ACE1-Is] are associated with a lower incidence of AD than other types of antihypertensive drugs and AT1RAs tend to be more beneficial than ACE1-Is in slowing the rates of progression, hospitalisation and mortality,11–14 although not all studies have found such effects. 19 Similarly, these RAS-acting drugs have also been found to be associated with lower rates of conversion of MCI to AD, as well as less AD-related tau neuropathology in both post-mortem brain tissue and CSF from living AD participants recruited to the ADNI. 15–17
In summary, AngII signalling is a plausible mechanism that can connect the traditional pathological hallmarks of AD with mechanisms involved in BP regulation and cerebrovascular dysfunction, which have been widely reported but poorly understood for decades. As a result, there is now a strong rationale for the use of AngII-targeting drugs as an intervention in AD. The RADAR trial therefore tested the use of the AT1RA losartan, the prototype drug in the AT1RA class, for which there are considerable data on its use as an effective antihypertensive drug for a wide range of ages. Losartan has also been reported to cross the BBB,44 a characteristic that is important in potential treatments for AD. It has also been reported to improve CBF,22 a surrogate marker of cognitive performance in humans. 45–47 Losartan has also been reported to reduce neuronal damage following ischaemia in rat stroke models,23 to inhibit the production of Aβ and AD-related modifications of tau protein in aged rodents41,42 and, in low doses (i.e. having no effect on BP), to reduce AD-related pathology and cognitive decline in transgenic mouse models of AD. 48 It has also been suggested that, given its role in reducing hypertension, losartan may provide a benefit by reducing ischaemia-mediated WMH. 24
General comments on RADAR design and study population
The RADAR trial recruited to 93% of its intended target. However, the use of an open-label phase, preferential participant retention rates and the ability to secure a high proportion of primary outcomes for all completed participants meant that the power of the study to detect our target difference in brain atrophy at 12 months (assumed therapeutic benefit) of at least 3.8 ml/year with a two-sided α = 0.05 was reduced only slightly, by 2% (from 84% to 82%).
Our decision to investigate a 12-month period of treatment and follow-up was based on previous studies conducted by ADNI in which MRI has revealed measurable brain atrophy in this period among AD patients, equivalent to 15.2 ml/year (SD 8.6 ml/year). We had proposed that a relative difference in atrophy between groups of 25%, equivalent to an absolute difference in an annual atrophy rate of 3.8 ml in TBV, would be a clinically meaningful outcome that was measurable. However, it is now worth acknowledging that, since the design and commencement of the RADAR trial in 2012, follow-up periods of 18–24 months have been recommended for investigations where disease modification is being explored and this, therefore, serves as a potential limitation of this study.
Examination of the baseline characteristics of the RADAR study shows very clearly that our randomisation process, which included minimisation variables of age and Scheltens score to attempt good balance in trial arms related to baseline atrophy, was very successful. There were no significant differences in the baseline characteristics of study population in terms of any of the traditional demographic variables or in the baseline primary and secondary outcome measures when examined cross-sectionally.
It was also evident that the level of data missingness was similar in both arms following review at the end of the study. On closer examination it was clear that there were no predictors of the factors that may contribute, such as baseline brain volume, MMSE, duration of diagnosis of AD or whether or not participants were taking dementia drugs (0.539). There were also no differences in data missingness based on the gender or number of years of education of participants, and whether or not they had hypertension did not have any effect. The only variable that was found to be associated with data missingness was participant age (p = 0.01), in that data were more likely to be missing in older participants, arguably not unexpectedly, and the majority of these missing data were related to the assessment-based secondary outcome measures. There may also have been a slight contribution to data missingness from MRI-based outcome measures, because the duration of scans may have been too short, when shortened at the participant’s request, to secure the primary outcome that was taken in the initial MRI sequences. Furthermore, more recruitment sites than anticipated were unable to support the collection of the secondary MRI-based outcome measures related to WMH volume and CBF. However, unless these sites recruited participants who on average were slightly older than those recruited by sites that could support the retrieval of these outcomes, this could have made a small contribution to the levels of data missingness associated with older participants. Our recorded levels of 88% data completeness are comparable to those of another study, NILVAD,112 that recently reported no significant therapeutic benefit of the calcium receptor antagonist nilvadipine in patients with AD. This study also included a single-centre substudy that examined MRI outcome measures similar to those used in the RADAR trial; that study reported 89% data completeness, compared with 88% in the RADAR trial, and, as was found in the RADAR trial, the distribution of missing data was random. 113
It was reassuring to see that there were no significant differences between the intervention and placebo arms of the study in terms of either SAEs or AEs, highlighting the safety of the RADAR trial. This is further supported by comparable retention rates throughout the main study in both treatment arms. These favourable rates are believed to be largely the result of the carefully designed open-label phase; following stakeholder feedback and suggestions, this was undertaken before the randomisation phase. This showed that, in the absence of such an open-label phase, approximately 20% of participants may have entered the RADAR trial and undergone baseline MRI scans only to withdraw within weeks of commencing medication. The open-label phase, therefore, contributed significantly to improving the overall welfare of participants and is likely to have reduced unnecessary participant and study partner burden for a proportion of participants, as well as helping those participants who proceeded to the main study to feel less anxious and more reassured about the possible implications of taking part.
Finally, on a related point regarding safety, only one unblinding request was made during the entire trial, for a participant attending hospital with a chest infection. Few other questions were raised about the need for or possibility of emergency unblinding during the study. This is most likely because the intervention, losartan, is a repurposed drug that is already used, as with a number of related drugs, in clinical practice to treat hypertension and some other cardiac conditions. Thus, in the case of any event necessitating emergency unblinding, it would be reasonable to assume that the patient had been taking losartan and to use existing knowledge about the drug to manage treatment accordingly. Thus, an obvious advantage of using repurposed drugs rather than experimental interventions in a trial is that important safety data can be collected in a relevant patient population, while simultaneously determining the drug’s therapeutic efficacy.
Primary outcome
The primary outcome of the RADAR trial was the measurement, using vMRI, of change in brain atrophy between baseline and the 12-month follow-up after treatment with losartan. The RADAR trial60 was originally designed to provide 84% power, requiring a sample size of 228 participants. The final power was 82% because the number of participants recruited was slightly smaller than originally hoped, but the effects of which were mitigated somewhat by more favourable retention rates, possibly because of the effectiveness of the open-label phase in ensuring the identification of participants who were most likely to tolerate the intervention. The primary outcome, brain atrophy after 12 months’ treatment post randomisation, was chosen on the assumption that an absolute difference in TBV between groups of at least 3.8 ml/year would constitute a clinically meaningful difference. TBV measured using vMRI (T1-MPRAGE) is now widely validated as a surrogate marker of cognitive decline and AD pathology. 75,76,85–88
The analysis showed that there was some progression of brain atrophy in participants with AD in both treatment arms in the RADAR trial. However, there were no significant differences between treatment arms when TBV was analysed using either the 12-month volume cross-sectionally adjusted for baseline or the more sensitive BSI method, showing that losartan afforded no therapeutic benefit following 12 months of treatment. Given the variety of mechanisms reported to be associated with AngII, a number of which are regularly described as significant in the pathogenesis of AD, one may have expected that interference with a target that is a significant contributor to all of these mechanisms may have had a more appreciable effect. These results are therefore surprising and somewhat disappointing and provide yet another example of promising preclinical research and supportive population-based studies failing to translate into RCT evidence of effectiveness. 114 Although loss of original study power was negligble, it remains possible that our powering of the study at 82–84% was on the low side; however, we are reasonably confident that this is unlikely, given the consistently similarly null findings from the secondary analyses.
The RADAR trial is a seminal study from the perspective of formally testing the AT1RA losartan in a formally diagnosed group of people with AD as well as the first use, in a non-commercial study, of vMRI to objectively determine the primary outcome measure. To the best of our knowledge, no other studies with which to compare these results have been conducted to date. Yet these data will provide a sobering benchmark for other small-scale trials that are ongoing. A similarly designed and sized Phase II trial in hypertensive AD patients, SARTAN-AD, which is being undertaken in Canada, commenced after the RADAR trial. It was designed to compare perindopril with telmisartan (https://clinicaltrials.gov/ct2/show/NCT02085265). However, unlike the RADAR trial, this study has failed to recruit even a fraction of the number of participants required because of high rates of prior exposure to drugs related to the intervention, a reliance on the recruitment of only participants with hypertension, and also issues with identifying sufficient numbers of recruitment centres to aid recruitment (Professor Sandra Black, SARTAN-AD chief investigator, University of Toronto, personal communication, 2019). The SARTAN-AD trial is the only study that is close enough to the RADAR trial to allow the results to be compared, particularly given the intention to use vMRI to measure the primary outcome. However, beyond the ongoing concerns of whether or not the study will be successfully completed, its original design will have asked a different research question from the RADAR trial. It was designed to compare the effects of ACE1-Is (perindopril) and AT1RAs (telmisartan) on brain atrophy; however, the restriction of recruitment to people with pre-existing hypertension (unlike the RADAR trial) will limit the generalisability of the SARTAN-AD study. Furthermore, the participants recruited to the SARTAN-AD study include those with prior exposure to related medications and the design of the trial is such that participants could be randomised onto a related drug (ACE1-I or AT1RA). This means that participants in the SARTAN-AD study could have had a prior exposure to the intervention, whereas the RADAR trial was more conservative and recruited only individuals with no previous exposure to the intervention.
The continuation and success of the SARTAN-AD study has important consequences, not only as it will be an additional study to help test the angiotensin hypothesis in AD, but also because it will serve as an important basis of comparison for the RADAR trial. Furthermore, among the other ongoing studies that are exploring the role of other RAS-acting drugs in relation to cognitive impairment, and will be discussed in further detail later, none currently includes investigations of people with AD, thus reinforcing the uniqueness of the RADAR trial.
Only one other study lends itself to some consideration as a comparator with the RADAR trial findings. The RCT NILVAD112 tested the efficacy of the BP-lowering drug nilvadipine in mild-to-moderate AD. Because nilvadipine is a calcium channel blocker and not an AT1RA, the comparison of the studies is imperfect because the drugs work by different mechanisms; however, as both drugs work to lower BP, comparisons in that regard may be worthy of consideration. NILVAD found no any evidence that nilvadipine confers therapeutic value in mild-to-moderate AD. However, NILVAD also included a small substudy, undertaken at a single centre, that included MRI measures of brain (and hippocampal) volume, WMH volume and CBF using similar but not identical approaches to those used in the RADAR trial. It is notable that these data were generated after a follow-up of just 6 months post randomisation and from a group of 44 participants allocated equally to the intervention and placebo arms. 113 Despite the participant group being approximately 20% of that presented in the RADAR trial, the data from NILVAD showed the absence of any therapeutic benefit from BP-lowering with respect to whole-brain volume. In agreement with the RADAR trial findings, this substudy found no evidence to support any benefit from BP-lowering treatment with nilvadipine in relation to either brain volume or the volume of WMH. 113 By contrast, the authors did report that nilvadipine increased hippocampal blood flow, although they did not identify differences in global CBF or CBF in the posterior cingulate cortex, an area they chose to investigate regional differences alongside the hippocampus. 113
Secondary outcomes
A number of secondary outcomes were investigated in the RADAR trial.
We first investigated rates of AD progression, as assessed by changes in various cognitive and behavioural assessments, including the ADAS-Cog,96 the NPI97 and the MMSE. 98 We also explored participant function and quality of life using the BADLS100 as well as examining change in quality of life (DEMQOL, DEMQOL-proxy99). These data were collected at the same time points as the primary outcomes, with an additional collection between these points to explore the short-term benefits.
A cursory examination of all of the assessment scores at each of the three time points (baseline, and 6 and 12 months) showed a worsening of all scores among participants in both the intervention and the placebo arms. However, a comparison of change in scores from baseline to follow-up, similar to the exposure period to the intervention for the primary outcome, showed that there were no significant differences in the secondary outcomes between the intervention arm and the placebo arm that would suggest a therapeutic benefit.
The study also sought to investigate the change in the volume of WMH, which was measured in our MRI protocol using T2-FLAIR, given that it may predict 1-year cognitive decline,24 as well as to explore CBF, using ASL MRI methods in these participants. For ease of discussion and because of their relatedness these will be discussed together. These imaging outcomes and the specific measurement of BP as an additional outcome measure were important, as BP is a key outcome in assessing the efficacy of hypertension treatments and thus may have provided insight into the associations between BP and the secondary MRI measures of cerebrovascular damage and dysfunction.
The levels of data collection for these outcome measures were lower than anticipated across all of the recruitment centres. Indeed, it was known from the outset of the trial that data on WMH and CBF, even if collected at the same level as the primary outcome, would be underpowered to provide any definitive findings and, therefore, these variables were proposed as secondary outcomes.
Data on WMH volume could be collected from only approximately half of the RADAR participants (i.e. from 15 of the 24 sites), reinforcing that these analyses were predominantly exploratory and required appropriate cautious interpretation. The number of participants for whom data were collected was approximately the same in the placebo (n = 51) and intervention arms (n = 54). Comparison of WMH volume at both baseline and 12-month follow-up showed no statistical differences between treatment arms, and regression analysis to measure the change in WMH volume showed that there were no statistically significant differences that may indicate that losartan had any beneficial effect in lowering WMH volume (p = 0.697).
The intended similarly exploratory analysis of CBF was slightly more limited by the number of recruitment sites that could support it. However, the analysis of CBF was also subject to additional limitations posed by the complexities of retrieving data from nine different centres that each used different MRI facilities, different hardware and, as was identified, sometimes adopted different interpretations of the MRI protocols. Although data were collected from a total of 94 participants with similar distribution in the trial arms, it became apparent that there was significant variation in the numbers of scans that could be retrieved that were suitable for analysis once we had applied our QA approaches, both to get indicative results on the potential effect of the intervention and to pilot how data from several centres may be harmonised for analysis.
With these two objectives in mind, we selected data from sites 19 and 10, which between them accounted for almost half of participants from whom data were collected. Our QA of the data from these two centres involved exploration of two approaches. We used either the application of stringent criteria to the data, which were deemed to be of sufficient quality to allow accurate measurement of CBF from the T2-FLAIR scan, or a relatively newer option that uses a spatial CoV approach, which used data to calculate the ratio of the SD of CBF to the mean of CBF in scans. The dual approach was used because the latter is being proposed as a novel robust, but arguably more forgiving, approach when working with data from multicentre studies to allow estimates (proxy) measurements of CBF. 106,107 As is evident from the data generated from pilot studies in the two largest sites in the RADAR trial, the number of scans that were able to be analysed was slightly higher using the CoV approach than when using the more restrictive focus on purely CBF scans; in this study, there were too few CBF scans to allow any meaningful analysis. Moreover, owing to differences in the parameters with which the ASL sequences were run at these two sites, it was not possible to readily combine these data to provide a larger data set that would enable a comparison.
Because of the exploratory nature of CBF as an outcome measure and the complexities of this set of data, exploration of the influence of losartan on CBF following the treatment period was restricted to the CoV data from sites 19 and 10. Unfortunately, all efforts to retrieve data in a usable format from the remaining sites have been unsuccessful at the time of writing. From the CoV data available from sites 19 and 10 there were no obvious differences between treatment arms at either baseline or 12-month follow-up; nor was there any evidence of a difference in mean change in CBF analysed using spatial CoV analysis.
The exploratory data suggest that losartan has no effect on either WMH or CBF in patients with AD following 12 months of treatment, although, given the complexities of the latter data, noted previously, some of these conclusions need to be interpreted cautiously because the study was not designed around the secondary analyses. As mentioned, the recently completed NILVAD trial112 provides some limited basis of comparison for the RADAR trial findings. The NILVAD trial found no evidence to suggest that lowering BP with nilvadipine has any beneficial effect on brain volume; however, it also found no evidence of a nilvadipine-associated change in the volume of WMH. 113 By contrast, the authors reported that nilvadipine had a beneficial effect on increasing hippocampal blood flow, but not on differences in global CBF or CBF in the posterior cingulate cortex, an area that was preselected for investigation of regional blood flow differences, alongside the hippocampus. 113 Again, the fact that this substudy included the measurement of these variables after only 6 months of treatment, compared with 12 months of treatment in the RADAR trial, may have been a limiting factor. Alternatively, it is possible that observations of increased hippocampal blood flow were a true and early but transient phenomenon after participants were randomised to the intervention. However, the fact that there were no similar changes in global or posterior cingulate cortex CBF, and no differences were found in either total brain or hippocampal volume, raises some questions about the impact of the observed increase in hippocampal blood flow on the brain overall. Similarly, the limited numbers of participants for whom data were available and the short follow-up period meant that further analysis of the relationship with cognitive decline, which may have been slightly informative, was not possible.
On balance, it is important to note that the findings of the NILVAD trial are interesting but, given the number of participants, need to be considered with the appropriate levels of caution. The findings of the RADAR trial largely agree with those of the NILVAD substudy and the various MRI outcome measures examined, with the exception of the observed differences in hippocampal blood flow, something that our study did not set out to investigate.
-
It was important to pay due consideration to change in BP over time in the RADAR trial given the normal prescribed purpose of losartan. Interestingly, by virtue of this function, the measurement of BP provided the opportunity to have an important positive control that the intervention was biologically active (i.e. observable reductions in BP in the intervention arm would be proof that the medication was active). The other reason why examination of BP was valuable is that previous research has suggested that variations in BP might be associated with the volume of WMH and CBF, which in turn may affect cognitive performance. 45–47 Yet there have also been some inconsistent findings. 49,50
The findings clearly showed that there was a significant reduction in BP in participants who were randomised to losartan. This equated to a modest drop of approximately 5 mmHg in systolic BP and of 3 mmHg in diastolic BP, although these values are lower than in the NILVAD imaging substudy, which found a drop of in BP of closer to 10 mmHg. 113 Although the RADAR trial has longer follow-up data (12 months vs. 6 months) and a larger study population (193 vs. 44) than the NILVAD trial, in contrast to the main NILVAD trial112 and imaging substudy,113 we found no evidence of an association between reduced BP and change in the primary or secondary outcome measures.
The results of the RADAR and NILVAD trials, being intervention studies in patients with mild-to-moderate AD, can be compared with the findings of the SPRINT-MIND study65 of the main SPRINT (NCT01206062) intervention trial. 66 SPRINT compared the effectiveness of treating systolic BP in a population of hypertensive individuals aged > 50 years (without diabetes or a history of stroke or AD) with the intention of lowering systolic BP to below 120 mmHg, compared with the normal normotensive threshold of 140 mmHg. The sample size in the SPRINT study was an order of magnitude greater (n = 9361 participants randomised) than that of the NILVAD trial (n = 511 randomised) and the RADAR trial (n = 211 randomised). The SPRINT-MIND substudy was similarly larger (n = 2636) and investigated the impact of the two BP-lowering approaches (evenly distributed in the study population) with respect to cognitive outcomes, namely the rate of new cases of probable dementia (the primary cognitive outcome) and the rate of development of MCI, which was investigated along with, or as a composite outcome measure with, the rate of probable dementia.
First, SPRINT-MIND investigated of small-vessel disease in the brain using MRI65 and its association with cognitive outcomes. No such association was observed, although it must be acknowledged that the early stoppage of the main SPRINT trial, because of the earlier than expected observed therapeutic benefit from more aggressive BP-lowering, meant that the planned follow-ups in SPRINT-MIND were curtailed, resulting in fewer data than was originally planned. 65 Yet the lack of observed findings is consistent with the limited findings of both the RADAR trial and the NILVAD trial. The observation from the main SPRINT trial that lowering BP to below 120 mmHg reduced rates of MCI and their composite secondary cognitive outcome measure is not consistent with the findings of the RADAR trial. Yet there are a few notable differences between the studies. First, participants recruited to SPRINT (and SPRINT-MIND), although of comparable lower age to participants in the RADAR trial (50 years in SPRINT and 55 years in the RADAR trial), were hypertensive with no apparent cognitive impairment whereas those in the RADAR trial had mild-to-moderate AD. A similar difference exists between the NILVAD trial and SPRINT. Second, the outcome measures in SPRINT were measures of disease prevention, whereas those in the RADAR trial and the NILVAD trial were measures of reduction in disease progression. Third, the benefits observed in SPRINT were accompanied by a reduction in systolic BP to below the target of 120 mmHg, whereas neither the RADAR trial nor the NILVAD trial achieved such a reduction after 12 months’ treatment [RADAR: mean 133 (SD 21) mmHg; NILVAD: mean 132 (SD 16) mmHg]. This poses the possibility that the reductions in systolic BP achieved in both the RADAR trial and the NILVAD trial may be insufficient to achieve any benefit that may have translated into the chosen primary and secondary outcomes. However, it is also possible that the advanced stage of disease among participants in both the RADAR trial and the NILVAD trial could have negated any potential benefit that may be gained in persons without dementia with no obvious neurodegenerative disease.
In HYVET-COG (Hypertension in the Very Elderly Trial cognitive function assessment), which targeted very elderly people with severe hypertension (systolic BP 160–200 mmHg), the use of indapamide with an optional add-on of the ACE1-I perindopril was compared with placebo. 115 The target BP level in HYVET-COG was a systolic BP of 150 mmHg, higher than both the RADAR trial and the NILVAD trial achieved, but the trial failed to demonstrate any reduction in the incidence of dementia. Like SPRINT-MIND, the full scope of HYVET-COG was limited by the early termination of the main HYVET study because of the observed therapeutic benefit. 115 Yet it may be reasonable to think that the negative findings in HYVET-COG, as in the RADAR trial and the NILVAD trial, may be attributable to insufficient lowering of BP. In the case of the RADAR trial, this would imply that the insufficient BP lowering, rather than the involvement of dysfunctional biochemical systems (i.e. cRAS), could explain the failure to detect any therapeutic benefit of losartan, and the current results cannot exclude this possibility.
Finally, the results of the RADAR trial differ from those of Tedesco et al. , and others, who reported that losartan might confer a beneficial effect on cognitive function. 49–51,74 There are again some notable differences to consider. The Tedesco et al. 50,51 study was smaller (n = 69), involved people with hypertension but not dementia, and used a smaller maximum dose of losartan (50 mg) but for 26 months. We cannot exclude the possibility that the RADAR trial treatment period was too short to demonstrate a similar cognitive benefit in AD participants. The NILVAD study involved treatment for 78 weeks (compared with 52 weeks in the RADAR trial) and, thus, if BP-lowering is an important factor, the longer exposure periods may have benefited participants in both the RADAR trial and the NILVAD trial.
Given that the RADAR trial is one of a few studies to use a combination of MRI measurements that are reported to serve as a proxy for cognitive decline in a population with mild-to-moderate AD, it was also important to explore the potential associations between the primary and secondary outcomes. This examination may help elucidate which outcome measures may be more suitable in future studies. However, as the data from the RADAR trial were null for the primary and secondary outcome measures, it is not possible to determine which outcome measures would be the most informative for a similar study. It was reassuring to see pertinent correlations between MRI-based measures of hippocampal volume and other relevant measures. However, what may have been somewhat surprising is the lack of any strong correlation between the observer-rated measures of hippocampal atrophy (Scheltens scores) and the MRI-based measures of hippocampal atrophy. Any potential issues this may have caused would have been mitigated in our analysis because we adjusted for Scheltens scores as one of our minimisations.
Levels of drug compliance and tolerability were also an important consideration for the RADAR trial. As already described, there were high levels of compliance (88%) across all participants, with compliance only slightly lower (86%) in the intervention arm than in the placebo arm (90%), which sensitivity analyses confirmed was comparable across study arms. Similarly, the numbers of SAEs and AEs were similar in both treatment arms, showing that the intervention was safe to use.
Post hoc exploratory analyses
We considered a small number of additional post hoc exploratory analyses intended to explore questions that the current data may be able to answer to inform future studies.
The first question that could be explored, given that people with and without hypertension were recruited to the RADAR trial, was whether pre-existing hypertension had an effect on the primary and secondary outcomes. This would potentially shed some further light on whether or not any differences may be apparent in the characteristics of people with AD (in a randomised study) in terms of baseline characteristics or how they changed after 12 months of treatment with the BP-lowering agent losartan. The comparability of all the baseline characteristics, as well as the baseline primary and secondary outcomes, suggested that prior hypertension had no bearing on any of the outcome measures in the RADAR trial. In the recent NILVAD substudy that explored similar MRI-based outcomes to those used in the RADAR trial, about 30% of participants enrolled in the study had prior hypertension. However, the authors of the NILVAD study did not comment on the potential effects of prior existence of hypertension and their outcome measures. This is most likely because the arms in the substudy were already relatively small (n = 22 in each arm) and, therefore, meaningful comparisons of the effects with the various MRI-based outcome measures and assessments would have been significantly underpowered. However, given the considerable evidence reported to date that midlife hypertension is thought to increase risk of developing AD,20,58 which the recent findings of the SPRINT-MIND study also support,66 the absence of obvious differences between the treatment and intervention arms is curious. We did not have access to information regarding duration of prior hypertension at the time of writing; the investigators of the SPRINT-MIND study reported on some ongoing analyses of the medication types that participants were taking while trying to achieve the different target BP levels. It was suggested that RAS-acting medications (e.g. ACE1-Is and AT1RAs) appeared to have a greater benefit than other medications, with some indication that AT1RAs (e.g. losartan) offer the most favourable outcome. As yet there has been no publication to support this statement; however, if found to be true and the evidence significant, it would not only provide renewed evidence in support of the legitimacy of investigating losartan in the RADAR trial but also point to the fact that durations of follow-up longer than the 12 months used in the RADAR trial (median follow-up of participants in SPRINT-MIND was 3.14 years) are likely to be needed. 66
Second, the favourable rates of retention of participants, leading to high rates of completion of the primary outcomes and of data completion, were thought to be largely due to the inclusion of an open-label phase as a preceding step to the main randomisation phase. This prompted an examination of the characteristics of those participants who were not randomised, with a view to informing potential revisions to a future study protocol.
There were a number of notable observations from the open-label study. Over half of the participants withdrew within 1 week of entering the study while on the low titration dose for the intervention. Among the 50 participants who withdrew, a surprisingly high proportion (86%) were normotensive. Furthermore, a large number of those people discontinued at the commencement of the study were normotensive participants within the first 7 days (i.e. at the lower dose of the active medication). Yet there was no evidence of unevenness in the randomised phase, showing that our randomisation process worked effectively. Another observation was that almost 20% of the enrolled participants did not proceed to the randomised phase because of an unsuccessful, or lack of, completion of a baseline MRI.
Finally, an exploration of MRI-based measures of hippocampal atrophy was also undertaken as a means to pilot this alternative outcome measure for future trials. As was observed for the primary outcome, there was no evidence to suggest that losartan had a beneficial effect on THV or the volume of individual hippocampi. It was also considered relevant to explore the possible correlation between the MRI-based measures of hippocampal atrophy and rater-based ones (Scheltens score). It was somewhat unexpected to note the poor levels of correlation between these two measures, given that both are intended to measure the same pathological phenomenon.
Conclusions (implications for health care, recommendations for research)
The RADAR trial is the first RCT, to our knowledge, to investigate whether or not the AT1RA losartan has any therapeutic benefit in mild-to-moderate AD and to formally test the angiotensin hypothesis in AD, a hypothesis that is currently gathering increasing levels of support. Furthermore, the RADAR trial is a long overdue study of a BP-lowering intervention in a clinically defined population that numerous observational studies have suggested may benefit from a BP-lowering drug because of decades-old observations of a link between mid-life hypertension and an increased risk of AD. The growing number of preclinical studies that have demonstrated how various means of reducing the synthesis or signalling of AngII reduce rates of cognitive decline and neuropathological hallmarks of AD have continued to support this. Furthermore, there have continued to be some supportive findings, albeit inconsistently, from more recent observational studies. However, the RADAR trial, which used a more objective MRI-based measure as the primary outcomes rather than, as in many studies, assessment-based measures, found no evidence that losartan may provide a therapeutic option for the treatment of AD in the future. This robustly designed study, which had adequate randomisation between treatment arms and favourable participant retention and primary outcome completion rates, showed that losartan did not alter the assumed rate of clinically relevant brain atrophy and unambiguously excluded a reduction in rate of at least 3.8 ml/year. Similarly, there was no significant evidence to support a therapeutic benefit offered by losartan against any of the secondary outcome measures, although this needs to be stated with some caution as the study was not designed around the analysis of these clinical secondary outcomes.
Our study does not exclude the possibility that the duration of treatment, the severity of disease in the participants or the extent to which the drug penetrates the brain are factors that may have resulted in a failure to detect an effect on our primary and secondary outcomes. As described, evidence from the recent SPRINT-MIND study66 showed some reduction in dementia incidence after > 2 years of BP-lowering treatment. However, SPRINT-MIND also examined the therapeutic benefit of aggressive BP-lowering in general, rather than specifically investigating inhibitors of AngII function, or any other individual class of BP-lowering medication. Further analyses of this would be very welcome. Thus, it is not possible to exclude the possibility that more aggressive BP reduction, and for longer, might have had some therapeutic benefit; however, the uniformity of the null findings across all of the primary and secondary outcomes may suggest that AngII inhibition by losartan has no further future as an intervention to be tested.
The findings of the RADAR trial should not serve as a reason for premature termination of investigation of the role of AngII in AD. The evidence in favour of conducting this study was already strong at the time that the RADAR trial commenced, and supportive data have continued to be published in the intervening years. This means that further studies are still required to properly interrogate this hypothesis, as has happened in recent decades to test the therapeutic benefit of stopping or preventing Aβ synthesis, despite several large negative studies. 114 It must be stated that, since the RADAR trial concluded, there has been some renewed interest in the Aβ-lowering intervention aducanumab following subsequent analysis of additional data that became available, after the cut-off point used for a futility analysis that triggered the discontinuation of these trials in early 2019. These data, on a subset of participants, suggested that some participants receiving aducanumab at higher doses and for longer demonstrated reductions in brain Aβ levels, as measured by imaging, and encouraged the commercial owners of aducanumab to seek regulatory marketing approval. However, this development is not without some controversy and scepticism as, despite observable reductions in brain Aβ levels, these reductions were not found to be reflected in separate clinical outcomes also measured in the study, casting some further doubt on the clinical efficacy of the drug. 116,117 Therefore, some of the considerations that relate to the failure of other drugs testing other mechanisms in AD trials may also apply to the RADAR trial, not least the recruitment of participants with a level of disease that may be insurmountable by any intervention by the time clinical symptoms manifest. Indeed, the design of the RADAR trial and its undertaking coincided with growing debate and now acknowledgement that intervention trials may be more likely to be successful in patient populations in the earliest detectable stages of AD, such as those with MCI; longer follow-up periods of at least 18–24 months should be used to try to measure any form of disease modification. 4,5 The recent developments around aducanumab, and in particular the apparent disconnect between evidence of Aβ clearance following longer periods of exposure to the intervention, and in a dose-dependent manner, but in the absence of supportive data around clinical efficacy show once more that there may yet further revisions to perceptions of what may be deemed appropriate follow-up periods in future trials.
Although the findings of the RADAR trial are disappointing with regard to the angiotensin hypothesis of AD, and to patients and their families everywhere hoping for a new treatment, it is possible that the findings of other ongoing related trials will prove to be important in informing whether or not an AngII-inhibiting intervention may have a future role in the treatment and/or lowering the risk of developing AD. Indeed, the recent findings around aducanumab show the importance of having several studies properly test a hypothesis. Aducanumab is still just one in a long succession of compounds that have been tested or are being tested for therapeutic benefit of targeting Aβ. Yet, it is arguably only the first, following a long line of several other failed attempts over several decades,114 to show some promise in a relatively safe manner. With this in mind, it is notable and fortunate that most of the ongoing studies are investigating populations of people who, unlike those in the RADAR trial, have no cognitive impairment but either are at risk of developing dementia [i.e. the HEART study (NCT02471833) and the rrAD study (NCT02913664)] or have MCI [i.e. the CEDAR study (NCT02646982) and the CALIBREX study (NCT01984164)] and thus will provide information on populations of individuals at an earlier stage in the natural history. Moreover, all of these studies use different sartan drugs [rrAD uses losartan, whereas other studies use telmisartan (HEART) or candesartan (CEDAR and CALIBREX)]. Thus, not only will these studies provide information on different outcome measures, but they will do so in various less impaired populations, using a number of other interventions that had similar candidacy to losartan, which was investigated in the RADAR trial. It is therefore conceivable that the contribution of the RADAR trial to the development of any future AngII-inhibiting intervention may be not only in the likely exclusion of losartan as a candidate intervention, but also in providing a robust study design with which to test any other sartan that shows promise in the future.
Acknowledgements
The RADAR study investigators (see Principal investigators) oversaw the participation of their site research team in the RADAR study, and oversaw the recruitment of participants and their involvement throughout the study, in accordance with the trial protocol, with the help of their teams.
Principal investigators
Dr Elizabeth Coulthard (site 10), Dr Kirsty Harkness (site 11), Dr Tarun Kuruvilla (site 12), Dr Rupert McShane (site 13), Dr Peter Connelly (site 15), Professor John Starr (site 16), Dr Gordon Duncan (site 16), Dr Lucy Calvert (site 17), Dr Alasdair Lawrie (site 18), Dr Matthew Sheridan (site 19), Dr Bernard Udeze (site 20), Dr Stephen Pearson (site 21), Dr Tobias Langheinrich (site 22), Dr Andrew Tarbuck (site 23), Dr Suvarna Wagle, (site 23), Dr Joe Butchart (site 24), Dr Ajay Macharouthu (site 25), Dr Andrew Donaldson (site 26), Dr Wendy Neil (site 27), Dr Vivek Pattan (site 28), Dr David Findlay (site 29), Professor Alan Thomas (site 30), Dr Madhusudan Dalvi (site 31), Dr Rashi Negi (site 32) and Dr Bernadette McGuinness (site 33).
The RADAR study investigators would like to give thanks to various important groups of people who made the conduct of this study possible.
We would like to thank every participant and their study companions who consented, for their time, commitment and generosity, which allowed us to undertake this important research study.
We would also like to thank the many members, past and present, of the UCL-DRC team, who played a vital role in the QA review process for all the MRI scans across all the RADAR Centres. Special thanks go to Lloyd Prosser, Maggie Fraser, Laila Ahsan and Jana Kilmova for their ever present help and enthusiasm.
We would like to extend our sincerest thanks and support to all current and previous members of the TSC [Professor Jenny Rusted, University of Sussex (chairperson), Professors Clive Holmes (University of Southampton) and Anthony Bayer (Cardiff University), and Dr Ly-Mee Yu (Oxford University) and Mrs Susan Marshfield, Mrs Aileen Wilson and Mr Bill Mitchell (as our lay members)] for the continuous advice, support and encouragement throughout the conduct of the study. We would similarly like to thank the members of the DMEC [Professor Craig Ritchie, University of Edinburgh (chairperson), Professor Paul Gard (Brighton University), and Dr Sayeed Haque (Birmingham University)] for their time, effort and advice while reviewing data throughout the study.
We would also like to thank each and every member of research teams working to support the RADAR study investigators at every site, including research nurses and allied professionals, local pharmacists, radiographers and other staff related to individual centre involvement in the study as well as members of research and development teams at individual sites for supporting the research teams in adopting the study and for their continued involvement in the study.
Participant centre staff
Site 10, North Bristol NHS Trust (number of randomised participants, 20): H Archer, E Bellavia, S Dillon, L Doran, S Dunn, C Godwin, J Haworth, S Holloway, B Knight, B Owen, V Page, C Pennington, N Rosewell, D Rouse, A Sahni, J Selwood, K Sharma, J Thai, A Wilson and L Wilson.
Site 11, Sheffield Teaching Hospital Trust (number of randomised participants, 13): S Anderson, R Ashek, D Backburn, J Bigley, K Birchall, K Bouakline, J Bray, D Capener, R Clegg, G Cole, A Emery, J Green, C Hammerton, N Hoggard, A Howell, D Jarvis, E King, V Murray, A Patel, T Standering, C Wells and H Young.
Site 12, 2gether NHS Foundation Trust (number of randomised participants, 9): A Ajuili, M Ansell, M Biju, E Bowditch, J Bridge, E Burgess, B Cartwright, A Cox, R Crees, H Dryden, C Dukes, K Hackling-Searle, M Harvey, K Kelly, S Little, M Manju, C Martinez-Clavera, L Moore, D Ogden, E Phillips, G Riley, J Romer, J Spurway, E Tait, L Walker and A Walters.
Site 13, Oxford Health NHS Foundation Trust (number of randomised participants, 18): J Adams, M Akinola, L Benjamin, P Blunden, S Chilcott, M Clarke, D Cooper, C Dransfield, J Fodor, S Gallehawk, A Haddon-Silver, J Hume, O MacDonald, D McConnell, A McIntyre, A Murphy, D Sharma, M Turnbull, Dr Van Der Pott and G Willoughby.
Site 15, NHS Tayside (number of randomised participants, 15): J Anderson, T Chalmers, K Ferguson, D Findlay, N Gandhi, M Hepburn, J Hudson, E Law, A Maxwell, C McCaw, C McDonald, M Semple, T Stewart, Z Tayar, M Tsoumalis and L-A White.
Site 16, NHS Lothian (number of randomised participants, 11): A Anand, G Barclay, C Beveridge, R Buchan, A Cairns, J Carruthers, S Christison, M Cockburn, A Degnaw, S Denham, M Dewar, F Dougherty, G Fraser, G Gentle, E Hall, I Hamilton, J Hartley, C Jardine, J Kerr, L Kesseler, L Killin, M MacRury, G Marwick, D McIntyre, L Melvin, M Murphy, M Parra, N Rangolan, T Russ, E Sandeman, A Shaw, S Simpson and C Sutherland.
Site 17, NHS Borders (number of randomised participants, 7): G Barclay, I Hamilton, B McInally, D McIntyre, M Muir, E Nicol, E Sandman, C Sutherland, S Wiseman and S Wright.
Site 18, NHS Grampian (number of randomised participants, 14): J Adams, F Annison, D Christie, M Christie, P Cooper, N Crouch, E Darling, L Drever, M Hendry, B Jazpal, A Johnson, K Klaasen, M Lloyd, B MacLennan, N Massoud, A McBain, K McClelland-Brookes, C Meneses, C Napier, L Reid, S Scott, C Stewart and B Was.
Site 19, NHS Greater Glasgow and Clyde (number of randomised participants, 11): R Ashworth, E Campbell, E Jackson, C McNeill and V Smith.
Site 20, North Staffordshire Combined Healthcare NHS Trust (number of randomised participants, 3): P Andrews, C Ault, M Booth, D Brockley, S Coates, R Cope, M Dale, G En-Nimr, C Fabian-Ezorizu, L Forrester, T Harrison, C Hudson, S Hussain, L Jackson, K Jones, J Joseph, S Knight, J Lee, K Mason, P May, S Morrey, E Onwuharine, M Slater, K Smith, V Smith, R Sutton, C Tan, R Tarbick and E Williamson.
Site 21, Torbay and South Devon NHS Foundation Trust (number of randomised participants, 9): M Allison, KB Almedilla, E Arbury, H Bearne, C Campbell, M Costelloe, G DeSelincourt, C Dixon, N Donlin, B Finson, J Garfield-Smith, B George, C Good, H Griffin, A Hartford-Brown, A Henderson, K Horan, K Huggett, B Hulance, K-A Kelley, A Kennedy, M Kenny, A Khalil, C Maddison, M Maddock, N Mann, C Manyoni, Dr Marshall, A Mullinger, R Newman, L Paatz, D Pearce, N Portwine, B Reed, F Roger, H Shiels, S Smith, M Stone, S Szago, A Thornton, L Thornton, C Tsane, A Vian and S Wright.
Site 22, Salford Royal NHS Foundation Trust (number of randomised participants, 13): B Blackledge, J Brooke, A Burke, S Corbett, L Craven, AS Francis, L Hankinson, M Howard-Johnes, M Jones, V Parker, N Seferta, E Shields, Dr Skampardoni, M Tayler, N Thomas and B Whitnall.
Site 23, Norfolk and Suffolk NHS Foundation Trust (number of randomised participants, 9): K Clipsham, N Gill, Z Inman, K Kumar, C Rischmiller and C Sheldon.
Site 24, Royal Devon and Exeter NHS Foundation Trust (number of randomised participants, 5): C Browning, T Chapter, A Foden, G Githens-Mazer, A Grice, R James, A Kennedy, T Malone, R Newman, C O’Reilly, J Richards, E Robjohns, S Statton, S Todd, N Walker and F Walters.
Site 25, NHS Ayrshire and Arran (number of randomised participants, 5): D Gilmour, K Greig, J Kerr, L McNeil, J Mitchell, G Scott, S Waltos and M Wilson.
Site 26, NHS Lanarkshire (number of randomised participants, 7): T Baird, K Dempsey, M Fernon, S MacFadyen, C McNeill, R Routh, R Smit and V Smith.
Site 27, Leeds & York Partnership NHS Trust (number of randomised participants, 9): R Asquith, S Bennett, M Dixon, C-J Girling, L Hackney, Z Jawaid, L Kelly, J Kemp, A Locker, D Reynolds, C-B Romain-Hooper and E Sellers.
Site 28, NHS Forth Valley (number of randomised participants, 9): R Ashworth, C Duncan, L Dymock, J Hudson, A Maxwell, E McLennan, C McNeill, B Wong and R Woodward.
Site 29, NHS Fife (number of randomised participants, 2): N Attalla, C Cliff, F Davey, M Simpson and J Tait.
Site 30, Newcastle upon Tyne NHS Foundation Trust (number of randomised participants, 7): R Barber, A Byrne, G Cass, M Chinnasamy, S Edwards, R Gore, H Lynn, D Miller, D O’Donnell, A O’Reilly, H Pilkington, C Price, M Price, L Robson, K Walker and J Wilkinson.
Site 31, Kent and Medway NHS and Social Care Partnership Trust (number of randomised participants, 4): J Coombs, C Cowley, J Dalton, L Elias, J Field, L Hall, A Hammond, H Hedihy, M Howard, B Metelerkamp, P Smith and A Welfare-Wilson.
Site 32, Midlands Partnership NHS Foundation Trust (number of randomised participants, 8): M Ahmed, S Anumanchi, S Bates, R Beckett, M Bodice, E Busby, K Cullinan, P Dolby, E Glaves, E Godwin, L Hamilton, D Handuwalage, S Lavender, A Nash, E Nazir, K Needham, C Silva, A Stuart, R Sutton, A Taylor, L Tudor, R Walsh, C Winkle and V Worsell.
Site 33, Belfast Health and Social Care Trust (number of randomised participants, 3): A Clinton, P Gray and B Wells.
Finally, the RADAR study team would like to thank the BRTC for hosting this study and for its support throughout. We would also like to thank some previous members of the BRTC for their contributions. These include Professor Alan Montgomery, Dr Jo Simons and Hannah Baber for their earlier involvement in setting up the RADAR trial, and Marie Platt, Sam Lamprey, Jo Chambers and Jo Pollitt for providing administrative support during the study. Thanks also go to Dr Taya Thomas and Laura Palmer for help with co-ordination of the retrieval and processing of volunteered biosamples for the study. The RADAR team would also like to thank members past and present in the NIHR project management team and within the NIHR Journals Library for their guidance and help throughout the conduct and completion of the study.
Contributions of authors
Patrick G Kehoe (https://orcid.org/0000-0002-7542-1139) (Professor of Translational Dementia Research) was chief investigator of the trial, led on the drafting and co-ordination of the completion of the report, and oversaw the analysis.
Nicholas Turner (https://orcid.org/0000-0003-1591-6997) (Senior Research Associate in Medical Statistics) was a statistician who undertook the final analysis for the study and also served to support the preparation of data and worked to support the work of the DMEC, and contributed to completion of report.
Beth Howden (https://orcid.org/0000-0001-7824-0803) was the trial manager of the RADAR trial, created the first draft and contributed to the completed report.
Lina Jarutyte (https://orcid.org/0000-0002-7559-9877) (PhD student) was a researcher who undertook some of the ASL analysis and contributed to completion of report.
Shona L Clegg (https://orcid.org/0000-0001-7118-2540) (Imaging Trial Project Manager) was a collaborative researcher who provided guidance and advice in the development of the MRI protocols for the RADAR trial, developed and undertook the training and site validation protocols for all sites with respect to their fulfilment of the MRI protocols and performed a substantive role in QA analysis of scans in the data collection period of the study, and contributed to completion of report.
Ian B Malone (https://orcid.org/0000-0001-7512-7856) (Postdoctoral Research Associate) provided tools and advice for the imaging analysis and contributed to the completed report.
Josephine Barnes (https://orcid.org/0000-0003-3178-025X) (Associate Professor) visually assessed and checked WMH and contributed to the completed report.
Casper Nielsen (https://orcid.org/0000-0002-7112-9595) (Clinical Trials Software Development Manager) provided tools and advice for the imaging analysis and contributed to the completed report.
Carole H Sudre (https://orcid.org/0000-0001-5753-428X) (Research Fellow) processed the longitudinal WMH volumes used and contributed to the report.
Aileen Wilson (https://orcid.org/0000-0001-8692-2273) (Radiographer) undertook the visual assessments of all quality-assured vMRI scans for the entire study at the CRIC Bristol to provide the baseline Scheltens scores for all participants that were used at randomisation as well as at the end of the study.
N Jade Thai (https://orcid.org/0000-0001-6125-7326) (Senior Research Fellow) undertook the visual assessments of all quality-assured vMRI scans for the entire study at the CRIC Bristol to provide the baseline Scheltens scores for all participants that were used at randomisation as well as at the end of the study.
Peter S Blair (https://orcid.org/0000-0002-7832-8087) (Professor of Epidemiology and Statistics and Senior methodologist for the BRTC Unit) was a co-investigator on the study and grant co-applicant, was a senior methodologist and senior statistician for the study, and contributed to completion of report.
Elizabeth J Coulthard (https://orcid.org/0000-0002-0017-9595) (Associate Professor in Dementia Neurology) was a co-investigator on the study, provided clinical expertise and assisted with initial protocol design and application, led one of the recruiting sites in the study, and contributed to completion of report.
J Athene Lane (https://orcid.org/0000-0002-7578-4925) (Professor of Trials Research) was a co-applicant and contributed to the trial conduct and to the completion of the report.
Peter Passmore (https://orcid.org/0000-0003-2858-5509) (Professor of Ageing and Geriatric Medicine) provided clinical expertise and assisted with initial protocol design and application, and contributed to completion of the report.
Jodi Taylor (https://orcid.org/0000-0001-7171-8923) (BRTC Senior Trials Manager) provided oversight for the trial manager and trial administrator and served to support the trial in meeting the sponsor’s requirements in the conduct of the study. She also provided advice and support on the trial protocol.
Henk-Jan Mutsaerts (https://orcid.org/0000-0003-0894-0307) (Research Physician) carried out the ADAS-Cog data analysis, provided supervision and contributed to the drafting and revision of the manuscript.
David L Thomas (https://orcid.org/0000-0003-1491-1641) (Principal MRI Physicist at the Leonard Wolfson Experimental Neurology Centre) was a collaborative researcher who provided guidance and advice on the development of the MRI protocols for the RADAR trial and help and advice particularly in the analysis of the ASL analysis.
Nick C Fox (https://orcid.org/0000-0002-6660-657X) (Professor of Clinical Neurology) was the team lead in the collaboration with UCL for all MRI-related training and scan acquisition and QA analysis, provided clinical expertise and assisted with initial MRI protocol design and grant application.
Ian Wilkinson (https://orcid.org/0000-0001-6598-9399) (Professor of Therapeutics) was a co-investigator on the study and clinical lead for the RADAR trial. He was involved in the design of the study and its funding application and in the overall oversight of the study, as well as the interpretation and completion of the final report.
Yoav Ben-Shlomo (https://orcid.org/0000-0001-6648-3007) (Professor of Clinical Epidemiology) was a co-investigator, helped with the funding application, helped with designing the protocol, oversaw the running of the study, helped with the statistical analysis plan, contributed to the interpretation of the results and modified the initial draft of the report.
Publications
Kehoe PG, Blair PS, Howden B, Thomas DL, Malone IB, Horwood J, et al. The Rationale and Design of the Reducing Pathology in Alzheimer’s Disease through Angiotensin TaRgeting (RADAR) trial. J Alzheimers Dis 2018;61:803–14.
Clement C, Selman LE, Kehoe PG, Howden B, Lane JA, Horwood J. Challenges to and facilitators of recruitment to an alzheimer’s disease clinical trial: a qualitative interview study. J Alzheimers Dis 2019;69:1067–75.
Kehoe PG, Turner N, Howden B, Jarutyte L, Clegg SL, Malone IB, et al. Safety and efficacy of losartan for the reduction of brain atrophy in clinically diagnosed Alzheimer’s disease (the RADAR trial): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol 2021;20:895–906.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to available anonymised data may be granted following review.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, NETSCC, the EME programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the EME programme or the Department of Health and Social Care.
References
- Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, et al. Global prevalence of dementia: a Delphi consensus study. Lancet 2005;366:2112-17. https://doi.org/10.1016/S0140-6736(05)67889-0.
- Kehoe PG, Miners S, Love S. Angiotensins in Alzheimer’s disease – friend or foe?. Trends Neurosci 2009;32:619-28. https://doi.org/10.1016/j.tins.2009.07.006.
- Piau A, Nourhashémi F, Hein C, Caillaud C, Vellas B. Progress in the development of new drugs in Alzheimer’s disease. J Nutr Health Aging 2011;15:45-57. https://doi.org/10.1007/s12603-011-0012-x.
- Cummings J. Lessons learned from Alzheimer disease: clinical trials with negative outcomes. Clin Transl Sci 2018;11:147-52. https://doi.org/10.1111/cts.12491.
- Cummings J, Ritter A, Zhong K. Clinical trials for disease-modifying therapies in Alzheimer’s disease: a primer, lessons learned, and a blueprint for the future. J Alzheimers Dis 2018;64:S3-S22. https://doi.org/10.3233/JAD-179901.
- Gillette Guyonnet S, Abellan Van Kan G, Andrieu S, Aquino JP, Arbus C, Becq JP, . Prevention of progression to dementia in the elderly: rationale and proposal for a health-promoting memory consultation (an IANA Task Force). J Nutr Health Aging 2008;12:520-9. https://doi.org/10.1007/bf02983204.
- Kivipelto M, Helkala EL, Laakso MP, Hänninen T, Hallikainen M, Alhainen K, et al. Midlife vascular risk factors and Alzheimer’s disease in later life: longitudinal, population based study. BMJ 2001;322:1447-51. https://doi.org/10.1136/bmj.322.7300.1447.
- Launer LJ, Ross GW, Petrovitch H, Masaki K, Foley D, White LR, et al. Midlife blood pressure and dementia: the Honolulu-Asia aging study. Neurobiol Aging 2000;21:49-55. https://doi.org/10.1016/S0197-4580(00)00096-8.
- Skoog I, Gustafson D. Update on hypertension and Alzheimer’s disease. Neurol Res 2006;28:605-11. https://doi.org/10.1179/016164106X130506.
- Honig LS, Tang MX, Albert S, Costa R, Luchsinger J, Manly J, et al. Stroke and the risk of Alzheimer disease. Arch Neurol 2003;60:1707-12. https://doi.org/10.1001/archneur.60.12.1707.
- Davies NM, Kehoe PG, Ben-Shlomo Y, Martin RM. Associations of anti-hypertensive treatments with Alzheimer’s disease, vascular dementia, and other dementias. J Alzheimers Dis 2011;26:699-708. https://doi.org/10.3233/JAD-2011-110347.
- Li NC, Lee A, Whitmer RA, Kivipelto M, Lawler E, Kazis LE, et al. Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: prospective cohort analysis. BMJ 2010;340. https://doi.org/10.1136/bmj.b5465.
- Barthold D, Joyce G, Wharton W, Kehoe P, Zissimopoulos J. The association of multiple anti-hypertensive medication classes with Alzheimer’s disease incidence across sex, race, and ethnicity. PLOS ONE 2018;13. https://doi.org/10.1371/journal.pone.0206705.
- Levi Marpillat N, Macquin-Mavier I, Tropeano AI, Bachoud-Levi AC, Maison P. Antihypertensive classes, cognitive decline and incidence of dementia: a network meta-analysis. J Hypertens 2013;31:1073-82. https://doi.org/10.1097/HJH.0b013e3283603f53.
- Hajjar I, Brown L, Mack WJ, Chui H. Impact of Angiotensin receptor blockers on Alzheimer disease neuropathology in a large brain autopsy series. Arch Neurol 2012;69:1632-8. https://doi.org/10.1001/archneurol.2012.1010.
- Hajjar I, Levey A. Association between angiotensin receptor blockers and longitudinal decline in tau in mild cognitive impairment. JAMA Neurol 2015;72:1069-70. https://doi.org/10.1001/jamaneurol.2015.1001.
- Wharton W, Zhao L, Steenland K, Goldstein FC, Schneider JA, Barnes LL, et al. Neurofibrillary tangles and conversion to mild cognitive impairment with certain antihypertensives. J Alzheimers Dis 2019;70:153-61. https://doi.org/10.3233/JAD-190011.
- Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Pérez JM, Evans AC. Alzheimer’s Disease Neuroimaging Initiative. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat Commun 2016;7. https://doi.org/10.1038/ncomms11934.
- Walker VM, Kehoe PG, Martin RM, Davies NM. Repurposing antihypertensive drugs for the prevention of Alzheimer’s disease: a Mendelian randomization study [published online ahead of print July 23 2019]. Int J Epidemiol 2019. https://doi.org/10.1093/ije/dyz155.
- Kehoe PG. The coming of age of the angiotensin hypothesis in Alzheimer’s disease: progress toward disease prevention and treatment?. J Alzheimers Dis 2018;62:1443-66. https://doi.org/10.3233/JAD-171119.
- Hanon O, Latour F, Seux ML, Lenoir H, Forette F, Rigaud AS. REAL.FR Group . Evolution of blood pressure in patients with Alzheimer’s disease: a one year survey of a French Cohort (REAL.FR). J Nutr Health Aging 2005;9:106-11. https://doi.org/10.1016/S0035-3787(05)85355-X.
- Hong KS, Kang DW, Bae HJ, Kim YK, Han MK, Park JM, et al. Effect of cilnidipine vs losartan on cerebral blood flow in hypertensive patients with a history of ischemic stroke: a randomized controlled trial. Acta Neurol Scand 2010;121:51-7. https://doi.org/10.1111/j.1600-0404.2009.01299.x.
- Zhao Z, Tuor UI, Barber P. Chronic treatment with losartan and cerebral ischemic tolerance. J Exp Stroke Transl Med 2009;2:32-40. https://doi.org/10.6030/1939-067X-2.2.32.
- Carmichael O, Schwarz C, Drucker D, Fletcher E, Harvey D, Beckett L, et al. Longitudinal changes in white matter disease and cognition in the first year of the Alzheimer disease neuroimaging initiative. Arch Neurol 2010;67:1370-8. https://doi.org/10.1001/archneurol.2010.284.
- Niwa K, Younkin L, Ebeling C, Turner SK, Westaway D, Younkin S, et al. Abeta 1-40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc Natl Acad Sci US 2000;97:9735-40. https://doi.org/10.1073/pnas.97.17.9735.
- Buée L, Hof PR, Bouras C, Delacourte A, Perl DP, Morrison JH, et al. Pathological alterations of the cerebral microvasculature in Alzheimer’s disease and related dementing disorders. Acta Neuropathol 1994;87:469-80. https://doi.org/10.1007/bf00294173.
- Kalaria RN. Linking cerebrovascular defense mechanisms in brain ageing and Alzheimer’s disease. Neurobiol Aging 2009;30:1512-14. https://doi.org/10.1016/j.neurobiolaging.2007.10.020.
- Miners JS, Baig S, Palmer J, Palmer LE, Kehoe PG, Love S. Abeta-degrading enzymes in Alzheimer’s disease. Brain Pathol 2008;18:240-52. https://doi.org/10.1111/j.1750-3639.2008.00132.x.
- Lambert JC, Dallongeville J, Ellis KA, Schraen-Maschke S, Lui J, Laws S, et al. Association of plasma Aβ peptides with blood pressure in the elderly. PLOS ONE 2011;6. https://doi.org/10.1371/journal.pone.0018536.
- van Oijen M, Hofman A, Soares HD, Koudstaal PJ, Breteler MM. Plasma Abeta(1–40) and Abeta(1–42) and the risk of dementia: a prospective case–cohort study. Lancet Neurol 2006;5:655-60. https://doi.org/10.1016/S1474-4422(06)70501-4.
- Miners JS, Ashby E, Van Helmond Z, Chalmers KA, Palmer LE, Love S, et al. Angiotensin-converting enzyme (ACE) levels and activity in Alzheimer’s disease, and relationship of perivascular ACE-1 to cerebral amyloid angiopathy. Neuropathol Appl Neurobiol 2008;34:181-93. https://doi.org/10.1111/j.1365-2990.2007.00885.x.
- Miners JS, Baig S, Tayler H, Kehoe PG, Love S. Neprilysin and insulin-degrading enzyme levels are increased in Alzheimer disease in relation to disease severity. J Neuropathol Exp Neurol 2009;68:902-14. https://doi.org/10.1097/NEN.0b013e3181afe475.
- Akatsu H, Ogawa N, Kanesaka T, Hori A, Yamamoto T, Matsukawa N, et al. Higher activity of peripheral blood angiotensin-converting enzyme is associated with later-onset of Alzheimer’s disease. J Neurol Sci 2011;300:67-73. https://doi.org/10.1016/j.jns.2010.09.030.
- Lehmann DJ, Cortina-Borja M, Warden DR, Smith AD, Sleegers K, Prince JA, et al. Large meta-analysis establishes the ACE insertion–deletion polymorphism as a marker of Alzheimer’s disease. Am J Epidemiol 2005;162:305-17. https://doi.org/10.1093/aje/kwi202.
- Kehoe PG, Wong S, Al Mulhim N, Palmer LE, Miners JS. Angiotensin-converting enzyme 2 is reduced in Alzheimer’s disease in association with increasing amyloid-β and tau pathology. Alzheimers Res Ther 2016;8. https://doi.org/10.1186/s13195-016-0217-7.
- Benicky J, Sanchez-Lemus E, Honda M, Pang T, Orecna M, Wang J, et al. Angiotensin II AT(1) receptor blockade ameliorates brain inflammation. Neuropsychopharmacology 2011;36:857-70. https://doi.org/10.1038/npp.2010.225.
- Zhou J, Ando H, Macova M, Dou J, Saavedra JM. Angiotensin II AT1 receptor blockade abolishes brain microvascular inflammation and heat shock protein responses in hypertensive rats. J Cereb Blood Flow Metab 2005;25:878-86. https://doi.org/10.1038/sj.jcbfm.9600082.
- Barnes JM, Barnes NM, Costall B, Horovitz ZP, Ironside JW, Naylor RJ, et al. Angiotensin II inhibits acetylcholine release from human temporal cortex: implications for cognition. Brain Res 1990;507:341-3. https://doi.org/10.1016/0006-8993(90)90294-L.
- Barnes JM, Barnes NM, Costall B, Horovitz ZP, Ironside JW, Naylor RJ, et al. Angiotensin II inhibits cortical cholinergic function: implications for cognition. J Cardiovasc Pharmacol 1990;16:234-8. https://doi.org/10.1097/00005344-199008000-00009.
- Rogawski MA, Wenk GL. The neuropharmacological basis for the use of memantine in the treatment of Alzheimer’s disease. CNS Drug Rev 2003;9:275-308. https://doi.org/10.1111/j.1527-3458.2003.tb00254.x.
- Tian M, Zhu D, Xie W, Shi J. Central angiotensin II-induced Alzheimer-like tau phosphorylation in normal rat brains. FEBS Lett 2012;586:3737-45. https://doi.org/10.1016/j.febslet.2012.09.004.
- Zhu D, Shi J, Zhang Y, Wang B, Liu W, Chen Z, et al. Central angiotensin II stimulation promotes β amyloid production in Sprague Dawley rats. PLOS ONE 2011;6. https://doi.org/10.1371/journal.pone.0016037.
- Kehoe PG, Hibbs E, Palmer LE, Miners JS. Angiotensin-III is increased in Alzheimer’s disease in association with amyloid-β and tau pathology. J Alzheimers Dis 2017;58:203-14. https://doi.org/10.3233/JAD-161265.
- Culman J, von Heyer C, Piepenburg B, Rascher W, Unger T. Effects of systemic treatment with irbesartan and losartan on central responses to angiotensin II in conscious, normotensive rats. Eur J Pharmacol 1999;367:255-65. https://doi.org/10.1016/S0014-2999(98)00983-2.
- DeKosky ST, Shih WJ, Schmitt FA, Coupal J, Kirkpatrick C. Assessing utility of single photon emission computed tomography (SPECT) scan in Alzheimer disease: correlation with cognitive severity. Alzheimer Dis Assoc Disord 1990;4:14-23. https://doi.org/10.1097/00002093-199040100-00002.
- Imran MB, Kawashima R, Awata S, Sato K, Kinomura S, Ono S, et al. Tc-99m HMPAO SPECT in the evaluation of Alzheimer’s disease: correlation between neuropsychiatric evaluation and CBF images. J Neurol Neurosurg Psychiatry 1999;66:228-32. https://doi.org/10.1136/jnnp.66.2.228.
- Shiraishi H, Chang CC, Kanno H, Yamamoto I. The relationship between cerebral blood flow and cognitive function in patients with brain insult of various etiology. J Clin Neurosci 2004;11:138-41. https://doi.org/10.1016/j.jocn.2003.02.013.
- Danielyan L, Klein R, Hanson LR, Buadze M, Schwab M, Gleiter CH, et al. Protective effects of intranasal losartan in the APP/PS1 transgenic mouse model of Alzheimer disease. Rejuvenation Res 2010;13:195-201. https://doi.org/10.1089/rej.2009.0944.
- Fogari R, Mugellini A, Zoppi A, Derosa G, Pasotti C, Fogari E, et al. Influence of losartan and atenolol on memory function in very elderly hypertensive patients. J Hum Hypertens 2003;17:781-5. https://doi.org/10.1038/sj.jhh.1001613.
- Tedesco MA, Ratti G, Mennella S, Manzo G, Grieco M, Rainone AC, et al. Comparison of losartan and hydrochlorothiazide on cognitive function and quality of life in hypertensive patients. Am J Hypertens 1999;12:1130-4. https://doi.org/10.1016/S0895-7061(99)00156-9.
- Tedesco MA, Ratti G, Di Salvo G, Natale F. Does the angiotensin II receptor antagonist losartan improve cognitive function?. Drugs Aging 2002;19:723-32. https://doi.org/10.2165/00002512-200219100-00001.
- McGuinness B, Todd S, Passmore AP, Bullock R. Systematic review: blood pressure lowering in patients without prior cerebrovascular disease for prevention of cognitive impairment and dementia. J Neurol Neurosurg Psychiatry 2008;79:4-5. https://doi.org/10.1136/jnnp.2007.118505.
- Anderson C, Teo K, Gao P, Arima H, Dans A, Unger T, et al. Renin–angiotensin system blockade and cognitive function in patients at high risk of cardiovascular disease: analysis of data from the ONTARGET and TRANSCEND studies. Lancet Neurol 2011;10:43-5. https://doi.org/10.1016/S1474-4422(10)70250-7.
- Staessen JA, Thijs L, Richart T, Odili AN, Birkenhäger WH. Placebo-controlled trials of blood pressure-lowering therapies for primary prevention of dementia. Hypertension 2011;57:e6-7. https://doi.org/10.1161/HYPERTENSIONAHA.110.165142.
- van Middelaar T, van Vught LA, van Gool WA, Simons EMF, van den Born BH, Moll van Charante EP, et al. Blood pressure-lowering interventions to prevent dementia: a systematic review and meta-analysis. J Hypertens 2018;36:1780-7. https://doi.org/10.1097/HJH.0000000000001829.
- Peters R, Yasar S, Anderson CS, Andrews S, Antikainen R, Arima H, et al. Investigation of antihypertensive class, dementia, and cognitive decline: A meta-analysis. Neurology 2020;94:e267-e281. https://doi.org/10.1212/WNL.0000000000008732.
- Ding J, Davis-Plourde KL, Sedaghat S, Tully PJ, Wang W, Phillips C, et al. Antihypertensive medications and risk for incident dementia and Alzheimer’s disease: a meta-analysis of individual participant data from prospective cohort studies. Lancet 2020;19:61-70. https://doi.org/10.1016/S1474-4422(19)30393-X.
- Kehoe PG, Passmore PA. The renin-angiotensin system and antihypertensive drugs in Alzheimer’s disease: current standing of the angiotensin hypothesis?. J Alzheimers 2012;30:S251-68. https://doi.org/10.3233/JAD-2012-111376.
- Kehoe PG. Angiotensins and Alzheimer’s disease: a bench to bedside overview. Alzheimers Res Ther 2009;1. https://doi.org/10.1186/alzrt3.
- Kehoe PG, Blair PS, Howden B, Thomas DL, Malone IB, Horwood J, et al. The Rationale and Design of the Reducing Pathology in Alzheimer’s Disease through Angiotensin TaRgeting (RADAR) trial. J Alzheimers Dis 2018;61:803-14. https://doi.org/10.3233/JAD-170101.
- Webster L, Groskreutz D, Grinbergs-Saull A, Howard R, O’Brien JT, Mountain G, et al. Development of a core outcome set for disease modification trials in mild to moderate dementia: a systematic review, patient and public consultation and consensus recommendations. Health Technol Assess 2017;21. https://doi.org/10.3310/hta21260.
- Webster L, Groskreutz D, Grinbergs-Saull A, Howard R, O’Brien JT, Mountain G, et al. Core outcome measures for interventions to prevent or slow the progress of dementia for people living with mild to moderate dementia: systematic review and consensus recommendations. PLOS ONE 2017;12. https://doi.org/10.1371/journal.pone.0179521.
- Hajjar I, Hart M, Milberg W, Novak V, Lipsitz L. The rationale and design of the antihypertensives and vascular, endothelial, and cognitive function (AVEC) trial in elderly hypertensives with early cognitive impairment: role of the renin angiotensin system inhibition. BMC Geriatr 2009;9. https://doi.org/10.1186/1471-2318-9-48.
- Hajjar I, Hart M, Chen YL, Mack W, Novak V, C Chui H, et al. Antihypertensive therapy and cerebral hemodynamics in executive mild cognitive impairment: results of a pilot randomized clinical trial. J Am Geriatr Soc 2013;61:194-201. https://doi.org/10.1111/jgs.12100.
- Williamson JD, Pajewski NM, Auchus AP, Bryan RN, Chelune G, Cheung AK, et al. Effect of Intensive vs standard blood pressure control on probable dementia: a randomized clinical trial. JAMA 2019;321:553-61. https://doi.org/10.1001/jama.2018.21442.
- Williamson JD, Supiano MA, Applegate WB, Berlowitz DR, Campbell RC, Chertow GM, et al. Intensive vs standard blood pressure control and cardiovascular disease outcomes in adults aged ≥ 75 years: a randomized clinical trial. JAMA 2016;315:2673-82. https://doi.org/10.1001/jama.2016.7050.
- Barnes JM, Barnes NM, Costall B, Horovitz ZP, Naylor RJ. Angiotensin II inhibits the release of [3H]acetylcholine from rat entorhinal cortex in vitro. Brain Res 1989;491:136-43. https://doi.org/10.1016/0006-8993(89)90095-4.
- Miners JS, van Helmond Z, Raiker M, Love S, Kehoe PG. ACE variants and association with brain Aβ levels in Alzheimer’s disease. Am J Transl Res 2010;3:73-80.
- Miners S, Ashby E, Baig S, Harrison R, Tayler H, Speedy E, et al. Angiotensin-converting enzyme levels and activity in Alzheimer’s disease: differences in brain and CSF ACE and association with ACE1 genotypes. Am J Transl Res 2009;1:163-77.
- Medvedev AE, Radko SP, Yurinskaya MM, Vinokurov MG, Buneeva OA, Kopylov AT, et al. Neurotoxic effects of Aβ6-42 peptides mimicking putative products formed by the angiotensin converting enzyme. J Alzheimers Dis 2018;66:263-70. https://doi.org/10.3233/JAD-180500.
- Mogi M, Li JM, Tsukuda K, Iwanami J, Min LJ, Sakata A, et al. Telmisartan prevented cognitive decline partly due to PPAR-gamma activation. Biochem Biophys Res Commun 2008;375:446-9. https://doi.org/10.1016/j.bbrc.2008.08.032.
- Wang J, Ho L, Chen L, Zhao Z, Zhao W, Qian X, et al. Valsartan lowers brain beta-amyloid protein levels and improves spatial learning in a mouse model of Alzheimer disease. J Clin Invest 2007;117:3393-402. https://doi.org/10.1172/JCI31547.
- Zou K, Yamaguchi H, Akatsu H, Sakamoto T, Ko M, Mizoguchi K, et al. Angiotensin-converting enzyme converts amyloid beta-protein 1–42 (Abeta(1–42)) to Abeta(1–40), and its inhibition enhances brain Abeta deposition. J Neurosci 2007;27:8628-35. https://doi.org/10.1523/JNEUROSCI.1549-07.2007.
- Fogari R, Zoppi A. Effect of antihypertensive agents on quality of life in the elderly. Drugs Aging 2004;21:377-93. https://doi.org/10.2165/00002512-200421060-00003.
- Fox NC, Ridgway GR, Schott JM. Algorithms, atrophy and Alzheimer’s disease: cautionary tales for clinical trials. Neuroimage 2011;57:15-8. https://doi.org/10.1016/j.neuroimage.2011.01.077.
- Hua X, Gutman B, Boyle CP, Rajagopalan P, Leow AD, Yanovsky I, et al. Accurate measurement of brain changes in longitudinal MRI scans using tensor-based morphometry. Neuroimage 2011;57:5-14. https://doi.org/10.1016/j.neuroimage.2011.01.079.
- Thompson WK, Holland D. Alzheimer’s Disease Neuroimaging Initiative. Bias in tensor based morphometry Stat-ROI measures may result in unrealistic power estimates. Neuroimage 2011;57:1-4. https://doi.org/10.1016/j.neuroimage.2010.11.092.
- Clement C, Selman LE, Kehoe PG, Howden B, Lane JA, Horwood J. Challenges to and facilitators of recruitment to an Alzheimer’s disease clinical trial: a qualitative interview study. J Alzheimers Dis 2019;69:1067-75. https://doi.org/10.3233/JAD-190146.
- Kehoe PG, Turner N, Howden B, Jarutyte L, Clegg SL, Malone IB, et al. Safety and efficacy of losartan for the reduction of brain atrophy in clinically diagnosed Alzheimer’s disease (the RADAR trial): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol 2021;20:895-906. https://doi.org/10.1016/S1474-4422(21)00263-5.
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984;34:939-44. https://doi.org/10.1212/wnl.34.7.939.
- Rosen WG, Terry RD, Fuld PA, Katzman R, Peck A. Pathological verification of ischemic score in differentiation of dementias. Ann Neurol 1980;7:486-8. https://doi.org/10.1002/ana.410070516.
- Braun V, Clarke V. Using thematic analysis in psychology. Qual Res Psychol 2006;3:77-101. https://doi.org/10.1191/1478088706qp063oa.
- Sandelowski M. Sample size in qualitative research. Res Nurs Health 1995;18:179-83. https://doi.org/10.1002/nur.4770180211.
- Scheltens P, Leys D, Barkhof F, Huglo D, Weinstein HC, Vermersch P, et al. Atrophy of medial temporal lobes on MRI in ‘probable’ Alzheimer’s disease and normal ageing: diagnostic value and neuropsychological correlates. J Neurol Neurosurg Psychiatry 1992;55:967-72. https://doi.org/10.1136/jnnp.55.10.967.
- Frisoni GB, Fox NC, Jack CR, Scheltens P, Thompson PM. The clinical use of structural MRI in Alzheimer disease. Nat Rev Neurol 2010;6:67-7. https://doi.org/10.1038/nrneurol.2009.215.
- Chan D, Janssen JC, Whitwell JL, Watt HC, Jenkins R, Frost C, et al. Change in rates of cerebral atrophy over time in early-onset Alzheimer’s disease: longitudinal MRI study. Lancet 2003;362:1121-2. https://doi.org/10.1016/S0140-6736(03)14469-8.
- Jack CR, Bernstein MA, Fox NC, Thompson P, Alexander G, Harvey D, et al. The Alzheimer’s Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging 2008;27:685-91. https://doi.org/10.1002/jmri.21049.
- Fox NC, Scahill RI, Crum WR, Rossor MN. Correlation between rates of brain atrophy and cognitive decline in AD. Neurology 1999;52:1687-9. https://doi.org/10.1212/wnl.52.8.1687.
- Freeborough PA, Fox NC. The boundary shift integral: an accurate and robust measure of cerebral volume changes from registered repeat MRI. IEEE Trans Med Imaging 1997;16:623-9. https://doi.org/10.1109/42.640753.
- Freeborough PA, Fox NC, Kitney RI. Interactive algorithms for the segmentation and quantitation of 3-D MRI brain scans. Comput Methods Programs Biomed 1997;53:15-2. https://doi.org/10.1016/S0169-2607(97)01803-8.
- Leung KK, Barnes J, Modat M, Ridgway GR, Bartlett JW, Fox NC, et al. Alzheimer’s Disease Neuroimaging Initiative. Brain MAPS: an automated, accurate and robust brain extraction technique using a template library. Neuroimage 2011;55:1091-108. https://doi.org/10.1016/j.neuroimage.2010.12.067.
- Leung KK, Barnes J, Ridgway GR, Bartlett JW, Clarkson MJ, Macdonald K, et al. Automated cross-sectional and longitudinal hippocampal volume measurement in mild cognitive impairment and Alzheimer’s disease. Neuroimage 2010;51:1345-59. https://doi.org/10.1016/j.neuroimage.2010.03.018.
- Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med 2014;370:322-33. https://doi.org/10.1056/NEJMoa1304839.
- Jorge Cardoso M, Leung K, Modat M, Keihaninejad S, Cash D, Barnes J, et al. STEPS: Similarity and truth estimation for propagated segmentations and its application to hippocampal segmentation and brain parcelation. Med Image Anal 2013;17:671-84. https://doi.org/10.1016/j.media.2013.02.006.
- Leung KK, Clarkson MJ, Bartlett JW, Clegg S, Jack CR, Weiner MW, et al. Robust atrophy rate measurement in Alzheimer’s disease using multi-site serial MRI: tissue-specific intensity normalization and parameter selection. Neuroimage 2010;50:516-23. https://doi.org/10.1016/j.neuroimage.2009.12.059.
- Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer’s disease. Am J Psychiatry 1984;141:1356-64. https://doi.org/10.1176/ajp.141.11.1356.
- Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA, Gornbein J. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology 1994;44:2308-14. https://doi.org/10.1212/wnl.44.12.2308.
- Folstein MF, Folstein SE, McHugh PR. ‘Mini-mental state’. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189-98. https://doi.org/10.1016/0022-3956(75)90026-6.
- Smith SC, Lamping DL, Banerjee S, Harwood R, Foley B, Smith P, et al. Measurement of health-related quality of life for people with dementia: development of a new instrument (DEMQOL) and an evaluation of current methodology. Health Technol Assess 2005;9. https://doi.org/10.3310/hta9100.
- Bucks RS, Ashworth DL, Wilcock GK, Siegfried K. Assessment of activities of daily living in dementia: development of the Bristol Activities of Daily Living Scale. Age Ageing 1996;25:113-20. https://doi.org/10.1093/ageing/25.2.113.
- Zekry D, Duyckaerts C, Moulias R, Belmin J, Geoffre C, Herrmann F, et al. Degenerative and vascular lesions of the brain have synergistic effects in dementia of the elderly. Acta Neuropathol 2002;103:481-7. https://doi.org/10.1007/s00401-001-0493-5.
- Sudre CH, Cardoso MJ, Ourselin S. Alzheimer’s Disease Neuroimaging Initiative. Longitudinal segmentation of age-related white matter hyperintensities. Med Image Anal 2017;38:50-64. https://doi.org/10.1016/j.media.2017.02.007.
- Sudre CH, Cardoso MJ, Bouvy WH, Biessels GJ, Barnes J, Ourselin S. Bayesian model selection for pathological neuroimaging data applied to white matter lesion segmentation. IEEE Trans Med Imaging 2015;34:2079-102. https://doi.org/10.1109/TMI.2015.2419072.
- Mutsaerts HJMM, Mirza SS, Petr J, Thomas DL, Cash DM, Bocchetta M, et al. Cerebral perfusion changes in presymptomatic genetic frontotemporal dementia: a GENFI study. Brain 2019;142:1108-20. https://doi.org/10.1093/brain/awz039.
- Elvsåshagen T, Mutsaerts HJ, Zak N, Norbom LB, Quraishi SH, Pedersen PØ, et al. Cerebral blood flow changes after a day of wake, sleep, and sleep deprivation. Neuroimage 2019;186:497-509. https://doi.org/10.1016/j.neuroimage.2018.11.032.
- Ibaraki M, Nakamura K, Toyoshima H, Takahashi K, Matsubara K, Umetsu A, et al. Spatial coefficient of variation in pseudo-continuous arterial spin labeling cerebral blood flow images as a hemodynamic measure for cerebrovascular steno-occlusive disease: a comparative 15O positron emission tomography study. J Cereb Blood Flow Metab 2019;39:173-81. https://doi.org/10.1177/0271678X18781667.
- Mutsaerts HJ, Petr J, Václavů L, van Dalen JW, Robertson AD, Caan MW, et al. The spatial coefficient of variation in arterial spin labeling cerebral blood flow images. J Cereb Blood Flow Metab 2017;37:3184-92. https://doi.org/10.1177/0271678X16683690.
- Schott JM, Bartlett JW, Barnes J, Leung KK, Ourselin S, Fox NC. Alzheimer’s Disease Neuroimaging Initiative investigators. Reduced sample sizes for atrophy outcomes in Alzheimer’s disease trials: baseline adjustment. Neurobiol Aging 2010;31. https://doi.org/10.1016/j.neurobiolaging.2010.04.011.
- Jin J, Sklar GE, Min Sen Oh V, Chuen Li S. Factors affecting therapeutic compliance: a review from the patient’s perspective. Ther Clin Risk Manag 2008;4:269-86. https://doi.org/10.2147/TCRM.S1458.
- Kanaide H, Ichiki T, Nishimura J, Hirano K. Cellular mechanism of vasoconstriction induced by angiotensin II: it remains to be determined. Circ Res 2003;93:1015-17. https://doi.org/10.1161/01.RES.0000105920.33926.60.
- Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res 2008;102:488-96. https://doi.org/10.1161/CIRCRESAHA.107.162800.
- Lawlor B, Segurado R, Kennelly S, Olde Rikkert MGM, Howard R, Pasquier F, et al. Nilvadipine in mild to moderate Alzheimer disease: a randomised controlled trial. PLOS Med 2018;15. https://doi.org/10.1371/journal.pmed.1002660.
- de Jong DLK, de Heus RAA, Rijpma A, Donders R, Olde Rikkert MGM, Günther M, et al. Effects of nilvadipine on cerebral blood flow in patients with Alzheimer disease. Hypertension 2019;74:413-20. https://doi.org/10.1161/HYPERTENSIONAHA.119.12892.
- Mehta D, Jackson R, Paul G, Shi J, Sabbagh M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin Investig Drugs 2017;26:735-9. https://doi.org/10.1080/13543784.2017.1323868.
- Peters R, Beckett N, Forette F, Tuomilehto J, Clarke R, Ritchie C, et al. Incident dementia and blood pressure lowering in the Hypertension in the Very Elderly Trial cognitive function assessment (HYVET-COG): a double-blind, placebo controlled trial. Lancet Neurol 2008;7:683-9. https://doi.org/10.1016/S1474-4422(08)70143-1.
- Howard R, Liu KY. Questions EMERGE as Biogen claims aducanumab turnaround. Nat Rev Neurol 2020;16:63-4. https://doi.org/10.1038/s41582-019-0295-9.
- Schneider L. A resurrection of aducanumab for Alzheimer’s disease. Lancet Neurol 2020;19:111-12.
Appendix 1 Details of cognitive assessments, measures of daily life and quality of life
The Mini Mental State Examination (participant)
The MMSE is the most commonly used test for complaints of problems with memory or other mental abilities. The MMSE test can be used by clinicians to help diagnose dementia and to help assess its progression and severity. It consists of a series of questions and tests, each of which scores points if answered correctly. The MMSE tests a number of different mental abilities, including a person’s memory, attention and language. The MMSE is only one part of assessment for dementia. Clinicians will consider a person’s MMSE score alongside their history, symptoms, a physical examination and the results of other tests, possibly including brain scans. The MMSE can also be used to assess changes in a person who has already been diagnosed with dementia. It can help to give an indication of how severe a person’s symptoms are and how quickly their dementia is progressing. It is a 30-point questionnaire that takes 5–10 minutes to be administered by a trained observer. It consists of eight items:
-
orientation to time
-
orientation to place
-
registration
-
attention and calculation
-
recall
-
language
-
repetition
-
complex commands.
The maximum score is 30, with a higher score indicating a lower impairment. Any score of ≥ 24 indicates a normal cognition. Below this, scores can indicate severe (≤ 9 points), moderate (10–18 points) or mild (19–23 points) cognitive impairment.
Alzheimer’s Disease Assessment Scale – Cognitive Subscale (participant)
The Alzheimer’s Disease Assessment Scale takes 30–45 minutes to be administered by a trained observer; it is a standardised assessment of cognitive function and non-cognitive features. The cognitive section of the scale (ADAS-Cog) is the gold standard for measuring change in cognitive function in drug trials. Deterioration of about 10% per year in cognitive tests in participants with AD is regarded as average. The cognitive domains include components of memory, language and praxis, and the non-cognitive features include mood state and behavioural changes. There are 11 main sections testing cognitive function, primarily measuring language and memory. The ADAS-Cog helps evaluate cognition and differentiates between normal cognitive functioning and impaired cognitive functioning. It is especially useful for determining the extent of cognitive decline and can help evaluate which stage of AD a person is in, based on their answers and score. The ADAS-Cog is often used in clinical trials because it can determine incremental improvements or declines in cognitive functioning.
The ADAS-Cog consists of 11 items:
-
word recall task
-
naming objects and fingers
-
following commands
-
constructional praxis
-
ideational praxis
-
orientation
-
word recognition task
-
remembering test directions
-
spoken language
-
comprehension
-
word-finding difficulty.
The test administrator adds up points for each task of the ADAS-Cog for a total score. Total scores range from 0 to 70, with higher scores (≥ 18) indicating greater cognitive impairment. A normal score for someone who does not have AD or another type of dementia is 5.
Dementia Quality of Life (participant)
The DEMQOL assesses quality of life in persons with mild-to-moderate dementia. It is a 28-item interviewer-administered questionnaire that is self-reported by the person with dementia. Responses are on a four-point scale (‘a lot’, ‘quite a bit’, ‘a little’ and ‘not at all’) with a score range of 28–114; a higher score indicates a better health-related quality of life. It takes approximately 10–20 minutes to administer.
Neuropsychiatric Inventory (study companion)
The NPI is the behaviour instrument most widely used in clinical trials of antidementia agents. It may help to distinguish between different causes of dementia; it records severity and frequency separately, and it takes 10 minutes to administer. A screening strategy is used to reduce the length of time that the instrument takes to administer, examining and scoring only those behavioural domains with positive responses to screening questions. Both the frequency (range 1–4) and the severity (range 1–3) of each behaviour are determined. Information for the NPI is obtained from a caregiver familiar with the participant’s behaviour. The NPI assesses 12 behavioural domains common in dementia:
-
hallucinations
-
delusions
-
agitation/aggression
-
dysphoria/depression
-
anxiety
-
irritability
-
disinhibition
-
euphoria
-
apathy
-
aberrant motor behaviour
-
sleep and night-time behaviour change
-
appetite and eating change.
There are two scores for each domain [frequency × severity and caregiver distress (0–5)]. The frequency × severity score range is 12–144, and the caregiver distress score range is 0–60. The higher the total score, the more severe the symptoms (a score of < 20, symptoms are mild; a score of 20–< 50, symptoms are moderate; and a score of ≥ 50, symptoms are severe).
Bristol Activities of Daily Living Scale (study companion)
The BADLS is a 20-item questionnaire designed to measure the ability of someone with dementia to carry out daily activities such as dressing, preparing food and using transport. The scale assesses 20 daily living abilities, each of which has four possible responses (scored 0–3). The score range is 0–60; the higher the total score, the more severe the symptoms.
Dementia Quality of Life-proxy (study companion)
This is the informant version of the DEMQOL and assesses the quality of life in persons with mild-to-moderate dementia. Completed by the main carer, it is a 31-item interviewer-administered questionnaire that is a proxy report of the person with dementia’s DEMQOL report. Responses are on a four-point scale (‘a lot’, ‘quite a bit’, ‘a little’ and ‘not at all’) with a score range of 31–124; a higher score indicates a better health-related quality of life. It takes approximately 10–20 minutes to administer.
Appendix 2 Supplementary adverse event details
| SAE number | Brief summary | Arm |
|---|---|---|
| Cancer | ||
| SAE002 | Participant died of pancreatic cancer | Placebo |
| Cardio-circulatory | ||
| SAE003 | Prolonged syncopal episode | Placebo |
| SAE018 | Fainting and bradycardia | Placebo |
| Dermatological | ||
| SAE030 | Rash around armpits and right side of the abdomen | Intervention |
| Endocrine and metabolic | ||
| SAE013 | Development of diabetes | Intervention |
| Gastrointestinal | ||
| SAE016 | Constipation and prolapse | Placebo |
| SAE033 | Diverticulitis | Placebo |
| SAE027 | Stomach pain and breathing difficulty | Placebo |
| SAE024 | Urinary retention and diarrhoea and vomiting | Intervention |
| Haematological/thrombosis | ||
| SAE040 | Admission 3 days. Right-sided chest pain. Discharged with anticoagulant medication | Placebo |
| Hepatic | ||
| SAE017 | Collapse raised ALT 100 U/l (reference range 0–55 U/l) | Intervention |
| SAE023 | Grossly raised AST serum level | Placebo |
| SAE043 | Safety bloods results outside the safety range | Intervention |
| Infection | ||
| SAE007 | Hospital admission. Sepsis | Intervention |
| SAE019 | Cellulitis left leg | Intervention |
| SAE020 | Cellulitis left leg flare-up | Intervention |
| SAE029 | Recurrent bronchitis | Intervention |
| SAE039 | Dizzy spell, tremor and cold hands. Discharged with antibiotics | Intervention |
| SAE041 | Chest infection | Placebo |
| SAE042 | Lower respiratory tract infection | Intervention |
| Mechanical injury | ||
| SAE006 | Fall | Placebo |
| SAE008 | Fainting | Placebo |
| SAE010 | Participant died. Admitted with femur fracture, dehydration and anaemia. Unrelated to trial | Intervention |
| SAE011 | Fall | Intervention |
| SAE014 | Fainting | Intervention |
| SAE021 | Falls | Intervention |
| SAE035 | Fall | Placebo |
| Neuropsychiatric | ||
| SAE012 | Found slumped on sofa unable to sit up, had visual hallucinations | Placebo |
| SAE004 | Seizures | Intervention |
| SAE026 | Seizure | Placebo |
| SAE032 | Vivid delusions and aggressive outburst | Placebo |
| SAE034 | Temporary admission to dementia assessment unit | Intervention |
| Other | ||
| SAE031 | Ankle swelling (oedema) | Placebo |
| SAE001 | Brain pathology | Intervention |
| SAE005 | Persistent nausea, dizziness and headache | Placebo |
| SAE009 | Mobility problems and unable to get out of bath | Intervention |
| SAE015 | Overdose of dementia medication | Intervention |
| SAE025 | Food poisoning | Intervention |
| SAE028 | Collapse | Intervention |
| SAE036 | Elective surgery (throat biopsy) | Placebo |
| SAE037 | Hospital admission knee pain. Arthritis | Placebo |
| SAE038 | Collapse without loss of consciousness | Placebo |
| Renal | ||
| SAE022 | Increased creatinine | Open label |
Glossary
- Clinical Research and Imaging Centre
- An imaging department based at the University of Bristol that measures all the Scheltens scores on MRI scans.
- Computerised tomography
- An examination using radiography and a computer to create detailed images of the inside of the body. Sometimes referred to as CAT or CT.
- Dementia Research Centre
- A department based at University College London that conducts all of the quality control on MRIs and the data for the primary outcome.
- Efficacy and Mechanism Evaluation programme
- Part of the Medical Research Council and National Institute for Health Research funding for clinical efficacy studies.
- Green light
- The indication that a recruitment site can start recruiting.
- Mini Mental State Examination
- A questionnaire-based tool that can be used to systematically and thoroughly assess mental status. A lower score indicates greater impairment.
- Montreal Cognitive Assessment
- A cognitive screening test designed to assist health professionals in detecting mild cognitive impairment, including Alzheimer’s disease. A lower score indicates greater impairment.
- NiftyFit
- A software package for multiparametric model-fitting of 4D magnetic resonance imaging data.
- Open label
- A type of trial, or segment of a trial period, in which participants are aware of which treatment they are taking.
- Single-photon emission computed tomography
- A type of nuclear imaging test that uses a radioactive substance and a special camera to create 3D pictures.
- XNAT
- The method of securely transferring anonymised scan data from sites to University College London for analysis (www.xnat.org).
List of abbreviations
- Aβ
- amyloid-β
- ACE1
- angiotensin I converting enzyme 1
- ACE1-I
- angiotensin I converting enzyme 1 inhibitor
- ACE2
- angiotensin I converting enzyme 2
- AD
- Alzheimer’s disease
- ADAS-Cog
- Alzheimer’s Disease Assessment Scale – Cognitive Subscale
- ADNI
- Alzheimer’s Disease Neuroimaging Initiative
- AE
- adverse event
- AngII
- angiotensin II
- ASL
- arterial spin labelling
- AT1RA
- angiotensin II type 1 receptor antagonist
- BADLS
- Bristol Activities of Daily Living Scale
- BBB
- blood–brain barrier
- BP
- blood pressure
- BRTC
- Bristol Randomised Trials Collaboration
- BSI
- boundary shift interval
- CACE
- complier average causal effect
- CBF
- cerebral blood flow
- CI
- confidence interval
- CONSORT
- Consolidated Standards of Reporting in Trials
- CoV
- coefficient of variation
- cRAS
- classical renin–angiotensin system
- CRIC
- Clinical Research and Imaging
- CSF
- cerebrospinal fluid
- CT
- computerised tomography
- DEMQOL
- Dementia Quality of Life Measure
- DEMQOL-proxy
- Dementia Quality of Life Measure by proxy
- DMEC
- Data Monitoring and Ethics Committee
- DRC
- Dementia Research Centre
- EudraCT
- European Union Drug Regulating Authorities Clinical Trials Database
- HYVET-COG
- Hypertension in the Very Elderly Trial cognitive function assessment
- IQR
- interquartile range
- ISRCTN
- International Standard Randomised Controlled Trial Register
- KN-BSI
- K-means normalised boundary shift integral
- LFT
- liver function test
- LHV
- left hippocampal volume
- LVV
- lateral ventricle volume
- MCI
- mild cognitive impairment
- MHRA
- Medicines and Healthcare products Regulatory Agency
- MICE
- multiple imputation by chained equations
- MIDAS
- Medical Image Display and Analysis Software
- MMSE
- Mini Mental State Examination
- MPRAGE
- magnetisation-prepared rapid-gradient echo
- MRI
- magnetic resonance imaging
- MTA
- medial temporal lobe atrophy
- NBT R&I
- North Bristol NHS Trust Research and Innovation
- NIHR
- National Institute for Health Research
- NINCDS-ADRDA
- National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association
- NPI
- Neuropsychiatric Inventory
- PIC
- patient identification centre
- PPI
- patient and public involvement
- QA
- quality assurance
- RADAR
- Reducing pathology in Alzheimer’s Disease through Angiotensin targeting
- RAS
- renin–angiotensin system
- RCT
- randomised controlled trial
- RHV
- right hippocampal volume
- rRAS
- regulatory renin–angiotensin system
- SAE
- serious adverse event
- SD
- standard deviation
- SPECT
- single-photon emission computed tomography
- SPRINT
- Systolic blood PRessure INtervention Trial
- SPRINT-MIND
- Systolic blood PRessure INtervention Trial Memory and cognition IN Decreased hypertension
- T1-MPRAGE
- T1-weighted magnetisation-prepared rapid-gradient echo
- T2-FLAIR
- T2-weighted – fluid-attenuated inversion recovery
- TBV
- total brain volume
- THV
- total hippocampal volume
- TM
- trial manager
- TMG
- Trial Management Group
- TNFα
- tumour necrosis factor alpha
- TSC
- Trial Steering Committee
- UCL
- University College London
- vMRI
- volumetric magnetic resonance imaging
- WMH
- white matter hyperintensities