

Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as award number 12/205/56. The contractual start date was in April 2015. The draft manuscript began editorial review in June 2022 and was accepted for publication in June 2023. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ manuscript and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this article.
Permissions
Copyright statement
Copyright © 2024 Pieles et al. This work was produced by Pieles et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This is an Open Access publication distributed under the terms of the Creative Commons Attribution CC BY 4.0 licence, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. See: https://creativecommons.org/licenses/by/4.0/. For attribution the title, original author(s), the publication source – NIHR Journals Library, and the DOI of the publication must be cited.
2024 Pieles et al.
Chapter 1 Introduction
Background
Barth syndrome is a very rare, life-threatening, X-linked recessive genetic disease that almost exclusively affects young males and is caused by abnormal lipids in mitochondria. 1 The tafazzin gene (TAZ) encodes tafazzin, a phospholipid acyltransferase. Tafazzin transfers unsaturated fatty acids with acyl chains from phospholipids to monolysocardiolipin (MLCL) and regulates the remodelling and maturation of cardiolipin,2,3 a phospholipid located exclusively in the inner mitochondrial membrane. Mature cardiolipin (CLm) is essential in the mitochondrial membrane for maintaining mitochondrial membrane potential and the structural integrity of electron transport chain complexes and mitochondrial cristae architecture. It plays a role in apoptosis, autophagy, cell cycle regulation, and iron–sulphur cluster biosynthesis. 2
Cardiolipin is a major constituent of inner mitochondrial membranes, and therefore the major effects of the aberrant gene are on the muscular tissues that are most reliant on energy production. The cardiac and skeletal manifestations of Barth syndrome are thought to be the result of impaired formation of respiratory chain super-complexes and, specifically in myocardial tissue, the cardio-specific loss of succinate dehydrogenase. 4 TAZ mutations result in reduced formation of CLm, predominantly tetralinoleoyl-cardiolipin (L4-CL), and an increase in intermediate species of MLCLs, resulting in a marked and pathognomonic increase in the ratio of MLCL:L4-CL. 5 This forms the basis for a highly sensitive and specific biochemical test for Barth syndrome. 1,6,7
The effects of the aberrant gene on muscular tissues result in infantile cardiomyopathy (including stillbirth) and lifelong severe exercise intolerance, lethargy and fatigue. 1,8 Low neutrophil numbers (neutropenia), poor feeding and growth delay are less intuitive but common features. 1
Barth syndrome is characterised by many gene mutations; more than 120 have been identified in only 200 affected persons. 1,9 There is profound phenotypic variability between affected patients within families and in different families, both in severity of the cardinal problems (e.g. cardiomyopathy, neutropenia) and the number of different organs that are significantly affected. To date, there has been no definitive proof of any genotype–phenotype correlation in Barth syndrome.
Twenty-six males and one female were currently alive with the disease in the UK when the trial was designed. The disease carries many risks and problems for its sufferers, as well as major health-care costs. Almost one-third (30%) of living UK patients have undergone cardiac transplantation. Several patients who have died had previously undergone cardiac transplantation due to cardiac rejection or post-transplant lymphoproliferative disease. Other patients have deteriorated too fast to undergo transplantation or died of complications of ventricular assist devices or extracorporeal membrane oxygenation while awaiting transplantation. In the 5 years before this study started, four babies/infants had been candidates for cardiac transplantation, requiring cumulatively approximately 20 months of intensive care bed stays. Sadly, only one underwent successful transplantation due to factors including shortage of donor hearts. Even those who have not received transplants can develop life-threatening heart rhythm disturbances.
Neutropenia is another life-threatening issue due to the associated risk of serious bacterial infection. As a result, two-thirds of UK patients are treated with chronic subcutaneous injection therapy with granulocyte colony-stimulating factor (G-CSF), an expensive medication whose administration is distressing for patients. 10 Furthermore, the management of neutropenia in this disease is challenging since patients have highly variable neutrophil counts, which prevents administration of a consistent daily dose and requires repeated blood counts and physician oversight. Patients often have neutropenia intermittently even when receiving G-CSF.
Exercise intolerance, lethargy and fatigue are universal, interfering with daily life, schoolwork and play and often necessitating the use of wheelchairs. Similarly, patients cannot hold down strenuous or demanding jobs. Many patients have major feeding problems from the neonatal period through into adult life and require long-term supplemental feeding via gastrostomies.
There can be rapid deterioration during periods of stable health even when under expert medical care. Ventricular arrhythmia (tachycardia or fibrillation) affects 10% of adolescents and can cause sudden cardiac death at any stage of childhood, including the neonatal period. 3 These seemingly random acute crises are not predictable by genotype or recent medical history, causing chronic anxiety in affected families and creating a need for cardiac resuscitation training.
There are no specific treatments for Barth syndrome other than supportive care for acute symptoms. Children continue to die from this disease despite best conventional therapy. Supplements such as coenzyme Q, carnitine and antioxidants are frequently used to treat other mitochondrial diseases but have proven ineffective in Barth syndrome,1 and there is no evidence that standard medications for cardiomyopathy ameliorate long-term poor outcomes. Treatment involves a multidisciplinary approach, with options currently limited to treatment of intercurrent bacterial infections, bone marrow support, physiotherapy, nutritional support, management of heart failure and arrhythmia, and consideration of heart transplantation. The societal and psychological impacts of the condition mean that children struggle with education and adults struggle to retain work. These facts highlight the need for disease-specific therapy for this group of patients to prevent morbidity, mortality, psychological distress and disruption of quality of life (QoL), and potentially easing strain on healthcare resources.
A number of potential treatments have been proposed for Barth syndrome, with some showing promise. Experiments using lymphoblasts from patients with Barth syndrome show that treatment with either bezafibrate or resveratrol can partially normalise the deranged cardiolipin ratio,11 suggesting these drugs’ potential as specific therapies. This may be important, since a subset of patients with Barth syndrome has been described who have cardiolipin ratios intermediate between those of typical patients and normal individuals; these patients lack neutropenia and tend to have good exercise tolerance. 12
Resveratrol is a naturally occurring food supplement available from nutraceutical companies. It affects energy metabolism and mitochondrial function and has a short half-life in blood. The short half-life may explain its lack of consistent clinical efficacy in a range of mammalian and human conditions. 13 Bezafibrate is an agonist of peroxisome proliferator-activated receptors (PPARs), nuclear receptor proteins that play essential roles as transcription factors in metabolic regulation. By contrast with resveratrol, it is licensed and well established as a lipid-lowering agent in adults and children, with a good safety record in long-term use. 14 It has been proposed as a potential treatment for metabolic mitochondrial disorders15,16 and has previously been used in muscle disease resulting from mitochondrial problems.
Encouragingly, bezafibrate has also been shown to improve left ventricular (LV) function at supraphysiological doses in a TAZ knockdown mouse model of the disease. 17 This research showed attenuation of cardiac dysfunction and significant amelioration of impaired exercise capacity. 17 Treatment with bezafibrate resulted in upregulation of genes involved in metabolism of fatty acids, ketone bodies, and glucose; metabolism of proteins; mitochondrial protein transport; RNA metabolism; gene expression; DNA repair; chromatin organisation; immune system; and organelle biogenesis and maintenance. The effect was shown in cardiomyopathy induced both by adrenergic (isoproterenol) stress and by chronic administration in unstressed knockout mice. In the latter condition, it prevented the cardiac deterioration typically seen by 7 months of age. 17 Surprisingly, in contradiction to the previous cellular evidence, this cardiac benefit was not accompanied by improvement of the cardiolipin ratio; it is postulated that the drug works by increasing mitochondrial biogenesis (mitochondrial numbers) in cardiomyocytes through a role in activating PPARs, rather than ameliorating the abnormal lipid chemistry. Bezafibrate has also been shown during a 4-month period at a clinically relevant dose to protect cardiac LV systolic function and, in combination with everyday voluntary running, to significantly ameliorate the impaired exercise capacity in TAZ knockdown mice. 18
Rationale
Disease-specific therapy is now required in order to prevent morbidity, mortality, psychological distress and disruption of QoL in patients with Barth syndrome and their families. In addition to the direct impact this may have on affected patients, it has the potential to produce major savings for the UK NHS. In response to a themed call for research on treatments for very rare diseases, we proposed a randomised controlled trial (RCT) to investigate the potential risks and benefits of bezafibrate treatment in this population, to provide high-quality evidence about the effectiveness of bezafibrate in patients with Barth syndrome.
The UK’s NHS is uniquely well positioned to explore therapy in patients with Barth syndrome, having the world’s highest density of diagnosed patients and the world’s only national multidisciplinary service. The NHS Specialised Services Barth Syndrome Service (BSS) currently cares for 26 boys and one girl from England, Scotland and Wales (from approximately 200 diagnosed worldwide). 1 When the trial was being set up, 20 of these were above 6 years of age and candidates for the trial.
Bezafibrate is a lipid-lowering drug approved by the European Medicines Agency with established use in adults and children and a good safety record in long-term use. 14 These characteristics made it an excellent candidate for investigation. Bezafibrate has also been reported to significantly ameliorate a mitochondrial myopathy (CPT2 deficiency) in adults19 as well as giving promising results in a range of animal/human cellular models of mitochondrial disease.
Although bezafibrate and resveratrol had equivalent effects in the mouse model/cells, resveratrol improved the cardiolipin ratio significantly more in human Barth syndrome fibroblasts (Dr Mindong Ren, New York University, 2016, personal communication). However, resveratrol has been included in the trial for comparative in vitro studies only due to concerns that its use could aggravate pre-existing neutropenia and that its half-life in blood is extremely short.
Several of the laboratory assays and clinical investigations proposed in this study [cardiolipin profiling, cardiac magnetic resonance spectroscopy (MRS) and electron microscopic evaluation of mitochondrial morphology] were also considered potentially important beyond the scope of this randomised trial for assessing associations between genotype and phenotype and for future evaluations of therapies in Barth syndrome. Patients with unexplained conditions very closely allied to Barth syndrome are known to this team. The technical expertise developed here may allow better investigation of these patients and those with other unexplained conditions such as idiopathic neutropenia or cardiomyopathy.
Aims and objectives
Aim
To determine whether bezafibrate (or resveratrol in vitro) increases mitochondrial biogenesis and potentially modifies the cellular ratio of MLCL to L4-CL, ameliorating disease phenotype in those living with the disease, without significant side effects at clinically effective doses.
This study also aimed to look at many aspects of the practicality and robustness of study design when evaluating a treatment for a very rare disease.
Objectives
Specific objectives were to:
-
Estimate the effect on clinical, biochemical and QoL outcome measures of bezafibrate compared to placebo in Barth syndrome (see Chapters 4, 5, and 6).
-
Investigate whether clinical improvements parallel in vitro changes in cardiolipin ratio/profile and mitochondrial morphology in each participant’s cells when exposed to bezafibrate in laboratory culture (see Chapter 8).
-
Investigate whether clinical improvements and culture findings with bezafibrate parallel in vitro changes in participants’ cells when exposed to resveratrol in laboratory culture (see Chapter 8).
-
Describe the most feasible methods and standardised outcome measures to optimise the conduct of future trials and evaluations in Barth syndrome (see Chapter 11).
-
Describe features of the research infrastructure which optimised recruitment, retention and communication with families and people with Barth syndrome (see Chapters 7, 10 and 11).
-
Describe the perceptions of participants and their families of research and any important potential barriers to participation (see Chapter 7).
Chapter 2 Methods
Study design
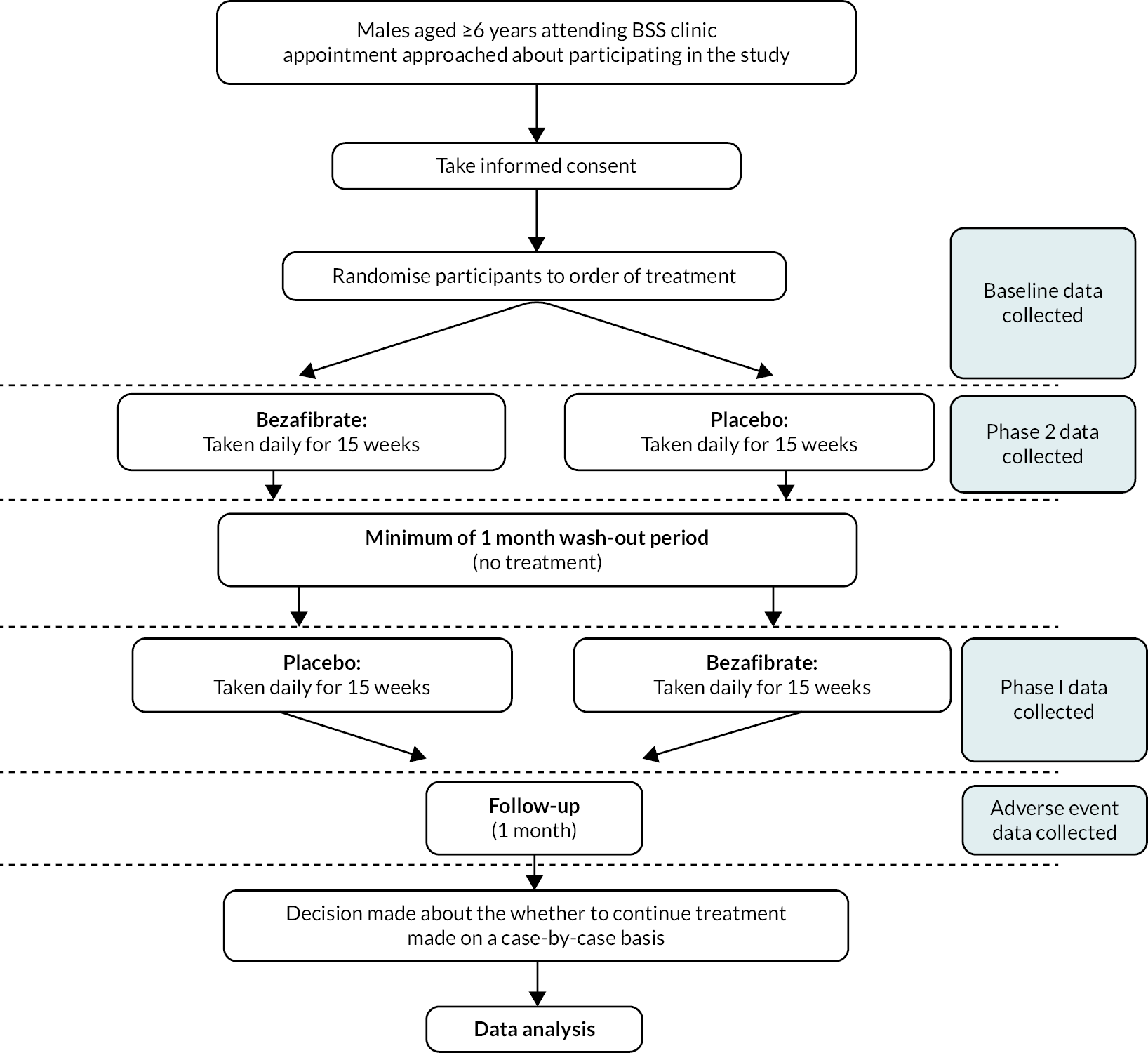
The CARDIOlipin MANipulation trial (CARDIOMAN) was a double-blind, placebo-controlled crossover randomised trial, registered as International Standard Randomized Controlled Trial Number ISRCTN58006579. Treatment was given in two 15-week phases with a minimum of 1 month’s washout period between these when no treatment was given. Participants were followed up for 1 month after the end of the second treatment phase (Figure 1).
FIGURE 1.
Diagram showing the crossover design of the CARDIOMAN trial.
Changes to study design after commencement of the study
One substantial amendment was approved after recruitment commenced. The following changes to the protocol were through this amendment:
-
Removal (retrospectively) of references to an interim analysis during the washout period, which was deemed inappropriate by the Trial Steering and Data Monitoring and Safety Committees (TSC and DMSC).
-
Removal of near-infrared spectroscopy as a secondary outcome because data generated during the first study visits demonstrated that it was too unstable to be useful.
-
Removal of the requirement for all monthly safety blood test results to be available for the Chief/Principal Investigator to make a satisfactory assessment of safety. This was requested by the clinical team (and approved) because monthly safety blood tests were performed at participants’ local healthcare facilities (general practitioner, local hospital, school) as per the usual arrangements for the participant. We observed variation in the results obtained, despite clear requests for the tests required as part of the protocol. For example, one centre’s ‘U and Es’ test protocol did not include urea in the results, so urea results were frequently missing for one participant.
-
Clarification of the formula to be used for paediatric estimated glomerular filtration rate (eGFR) results, and the upper age limit when it should be applied, due to it becoming apparent that biochemistry laboratories did not routinely report paediatric eGFR results, because of no formula having been validated in children. Therefore, eGFR results for the paediatric participants were calculated by the Chief/Principal Investigator during each visit and for the monthly safety blood assessments.
-
The duration for each treatment phase was shortened from 16 weeks to 15 weeks. This decision was made when scheduling the required three clinic trial visits for each participant, to avoid the final visits being too close to the Christmas holiday period, which might otherwise have reduced attendance.
-
One of the measurements used to assess cardiac function was changed from shortening fraction to 2-D strain by speckle tracking echocardiography, as 2-D strain has recently been shown to be a more load-independent and more sensitive measure of myocardial performance and ventricular function in inherited cardiomyopathies. 20
-
MRS of skeletal muscle only was carried out as a measurement at the last assessment. The reason for this was that this very novel methodology was not ready for use earlier. The delay in availability of MRS was discussed with the National Institute for Health and Care Research (NIHR) and we were requested to include it when it became available to provide information about its potential.
Participants
Eligibility criteria
Parts of this section have been reproduced from Dabner et al. 21 This is an open-access article distributed under the terms of the Creative Commons Attribution Licence (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work, first published in JMIR Research Protocols, is properly cited.
The reference population was males with a confirmed diagnosis of Barth syndrome.
The study population was males aged ≥ 6 years in the UK, with a confirmed diagnosis of Barth syndrome with the following inclusion/exclusion criteria applied. Males under 6 years of age were ineligible due to the difficulty in obtaining data on the primary outcome through bicycle ergometry in young children.
Participants were eligible to enter the study if ALL the following applied:
-
male aged ≥ 6 years
-
clinical diagnosis of Barth syndrome with characteristic abnormality of the L4-CL/ MLCL ratio plus identified mutation in TAZ
-
under the care of the NHS BSS
-
stable cardiac condition
-
able to swallow trial investigational medicinal product (IMP; bezafibrate or placebo tablets) manufactured for the study, of a similar size to ibuprofen caplets.
Participants were ineligible to enter the study if ANY of the following applied:
-
known hypersensitivity to bezafibrate, to any component of the product or to other fibrates
-
known photoallergic or phototoxic reactions to fibrates
-
hepatic dysfunction and/or liver function test (LFT) with a result greater than twice the normal level
-
a shortening fraction of < 25% in the cardiac chamber (or a significant drop in shortening fraction in the previous year)
-
documented atrial or ventricular arrhythmia (atrial/ventricular tachycardia or atrial/ventricular fibrillation) that has not been stabilised by treatment
-
renal impairment (creatinine clearance < 90 ml/min)
-
pre-existing known gallbladder disease
-
recent unspecified significant deterioration in general health
-
prisoners and adults lacking capacity to provide informed consent.
There are reports of rhabdomyolysis occurring in patients treated with a combination of bezafibrate and statins (see Safety criteria for dose escalation and continuation of study drug). 22 This is relevant because several males eligible for the trial had previously undergone cardiac transplantation and were maintained on the statin pravastatin. For patients who wished to participate in the trial, this issue was discussed with their respective local cardiology teams, who agreed for their statin medication to be ceased during the trial period.
Settings
The study visits took place at the Bristol Clinical Research and Imaging Centre (CRIC), which is part of the University Hospitals Bristol and Weston NHS Foundation Trust (UHBW). This hospital is a tertiary care NHS Trust in the South West United Kingdom. The NHS Specialised Services BSS, which is a national and centralised service, commissions this NHS Trust to provide the service.
Patients attending BSS clinics undergo extensive multidisciplinary review as part of their usual care, including consultations with a dietitian, cardiologist, psychologist, metabolic clinicians and the clinical nurse specialist. Due to the volume and nature of the study assessments, it was not possible to accommodate these during BSS clinics. In addition, the BSS clinics were (at the time of trial set-up) run biannually, although patients attended annually, and the study protocol required three study visits within 9 months. Therefore, the only way to complete the study assessments was to arrange separate research-specific clinics.
Conduct (screening and consent process)
Potential participants were identified through the BSS. Potential recruits were well known to clinicians in the study team before recruitment due to the clinicians’ involvement in the patients’ clinical care and the small number of patients with the disease. Once the eligibility criteria had been defined, eligible patients could be identified without traditional screening methods being required. During the trial set-up, the funder also requested formal written expressions of interest from potential participants or their families to determine whether there were sufficient participants to proceed; a minimum number of 10 participants expressing interest was required. Therefore, the study team were aware of individuals’ interest in joining the trial before recruiting them.
The number of assessments for the research and the availability of research personnel meant that only four patients could be seen in a clinic spanning 2 days. The number of participants was limited to 12 due to constraints on scheduling clinics (see Patient and public involvement).
Approximately 1 month before recruitment was due to commence, age-appropriate patient information leaflets (PILs) (see Report Supplementary Material 1) were sent out to potential participants and their families known to be eligible and who had previously expressed an interest in participating in the trial. Research nurses then telephoned potential participants/families to confirm their interest in the trial and to arrange a research clinic appointment if appropriate.
Participants and their families attended a baseline research clinic (clinic one of three), during which eligibility was confirmed. Clinicians discussed the study with the participants and their families and answered any questions. If the participant and the family remained willing to take part, written informed consent was completed. For participants under the age of 16, written informed consent was provided by the parents/guardians. Participants aged 16 or over provided consent for themselves.
Recruitment took place between March and April 2019. Each research clinic was on a Friday and Saturday (to facilitate attendance and availability of research personnel without compromising other NHS services). Participants were required to attend both days to accommodate the volume of study assessments. Four participants attended each clinic, and we carefully considered friendships between participants or their families, who often knew each other, when scheduling clinics. By ensuring that some participants attended the same clinics together longitudinally throughout the study, we were able to enhance the social aspect of the trial, promoting retention.
Clinics two and three (end of phase 1 and end of phase 2) were conducted during July and November/December 2019.
Trial interventions
All participants received 15 weeks of the intervention (bezafibrate) and 15 weeks of placebo. Each participant was randomised to receive either bezafibrate in phase 1 and placebo in phase 2, or the reverse order. All participants had a minimum of one calendar month’s washout period between phases, when no treatment was given.
The study intervention was prescribed once at the start of the study and again at the end of phase 1, for phase 2. The prescription only specified the trial IMP to ensure that all investigators, members of the research team (apart from pharmacy) and participants remained blind to treatment allocation throughout the trial. The order was determined by the randomisation list provided to the UHBW Pharmacy Trials Unit (PTU) which had been generated in advance by the Bristol Trials Centre. Participants were dispensed a supply of 15 weeks’ bezafibrate or placebo on each occasion. All dispensing was managed by the UHBW PTU. Participants remained on an initial loading dose until instructed to increase the dose after a satisfactory assessment of safety by the Chief/Principal Investigator.
Bezafibrate tablets (100 mg) and placebo were manufactured for the trial by Mawdsley-Brooks and Co. Ltd., (Salford, UK) who were also responsible for packaging, labelling and ‘Qualified Persons’ approval and release of the drug for the trial. The final containers were stored and drug accountability managed by the UHBW PTU in accordance with their standard operating procedures.
Intervention: Bezafibrate taken orally in tablet formulation.
-
Children aged 6–9 years: commenced on 100 mg once daily for the first 4 weeks and if well tolerated, increased to 100 mg twice daily for the remaining 11-week treatment period.
-
Children aged 10–17 years: commenced on 200 mg once daily for the first 4 weeks and if well tolerated increased to 200 mg twice daily for the remaining 11-week treatment period.
-
Adults (≥ 18 years): 200 mg twice daily.
Current licensed indications of bezafibrate are for the treatment of severe hypertriglyceridaemia with or without low high-density lipoprotein cholesterol and mixed hyperlipidaemia when a statin is contraindicated or not tolerated. Bezafibrate has a half-life of 1–2 hours in the body. 23
Placebo: Tablet formulation with no active substance taken orally. The placebo was visually identical and was as similar as possible in taste and smell to the intervention. No discernible difference in taste or smell was expressed by any participant.
Participants who changed prescribing age category during the trial due to a birthday stayed on the same dose regimen that they started at the beginning of the trial.
The following concomitant medication guidelines were followed throughout (maintaining blinding), as advised in the summary of product characteristics (SmPC) (see Report Supplementary Material 2):
-
‘Care is required in administering bezafibrate to patients taking coumarin-type anticoagulants, the action of which may be potentiated. The dosage of anticoagulant should be reduced by up to 50% and readjusted by monitoring blood coagulation.’
-
‘As bezafibrate improves glucose utilisation the action of antidiabetic medication, including insulin, may be potentiated and should be considered.’
-
‘Should combined therapy with an ion-exchange resin be considered necessary, there should be an interval of 2 hours between the intake of the resin and bezafibrate, otherwise the absorption of bezafibrate may be impaired.’
-
‘In isolated cases, a pronounced though reversible impairment of renal function (accompanied by a corresponding increase in serum creatinine level) has been reported in organ transplant patients receiving immuno-suppressant therapy and concomitant bezafibrate. Accordingly, renal function should be closely monitored in these patients and, in the event of relevant significant changes in laboratory parameters, bezafibrate should if necessary be discontinued. (NB creatinine will be measured at 2 weeks after commencing study medication in both phases of the study in participants who have received an organ transplant).’
-
‘MAO-inhibitors (with hepatotoxic potential) should not be administered together with bezafibrate.’
-
‘Interaction between 3-hydroxy-3-methyl-glutaryl-CoA reductase inhibitors and fibrates may vary in nature and intensity depending on the combination of the administered drugs. A pharmacodynamic interaction between these two classes of drugs may, in some cases, also contribute to an increase in the risk of myopathy for specific dose recommendations of statins. Refer also to the SmPC of the relevant product.’
Participants continued to be prescribed G-CSF as clinically indicated since there are no known drug interactions with this treatment. If a patient was shown to have a neutrophil count < 0.5 × 109/l, either in their monthly monitoring counts or if they presented with signs and symptoms of infection and required additional blood tests as part of their normal care, G-CSF therapy was initiated or dose/frequency of dosing was reviewed and potentially increased in line with routine care of the participant. Subsequent weekly full blood counts were requested on days after G-CSF therapy for the next 2 weeks (as this is the routine way of monitoring neutrophil counts in these patients). If the patient had three consecutive blood counts of < 0.5 × 109/l, the protocol specified that administration of the intervention should be terminated.
Once participants completed the study treatment, their medical care reverted to the standard care received from the BSS. However, if bezafibrate had shown a beneficial effect on individual participants, provision would have been made for them to continue receiving bezafibrate, on a case-by-case basis.
Treatment decisions
Safety criteria for dose escalation and continuation of study drug
Bezafibrate is a widely used and well tolerated medication in the general population, but it had not previously been tested in patients with Barth syndrome. We used a conservative dosing approach for paediatric participants in the trial (see Trial Interventions). After the first month of each treatment phase, paediatric participants increased their dose upon a satisfactory assessment of safety by the Chief Investigator. This assessment included review of results from monthly safety blood tests (including eGFR results) and reports of adverse events (AEs) experienced by the participant.
The monthly safety blood tests included: absolute neutrophil count, routine renal and LFTs, plasma triglycerides/total cholesterol/low density lipoprotein cholesterol and creatine phosphokinase. The latter was included because bezafibrate can induce rhabdomyolysis, although this risk is most significant in those being additionally treated with statins. 22 An additional blood test for creatinine was scheduled to be carried out at 2 weeks after the initiation of study drug in both phases in participants who had had an organ transplant. However, in practice, we found that it was not possible to arrange the 2-week test and obtain the results from the local providers in a timely fashion, so that it was sufficiently separate from the monthly blood samples. Therefore, the 2-week creatinine sample and initial monthly blood test were combined. The study research nurses facilitated the arrangement of these tests with the participants/families and their local provider and obtained the results from the place of testing.
For participants aged < 17 years, the eGFR was calculated by the Chief Investigator using the following formula: eGFR (ml/min/1.73 m2) = creatinine (µmol/l)/height (cm) × 40. 24 Older participants were classed as ‘adults’ for the purposes of the eGFR calculations, which were calculated by the Chief Investigator using the local UHBW laboratory formula in discussion with expert renal clinicians at the trial site.
The study research nurses had regular contact with participants to collect AE data: after the first week of starting treatment and then monthly thereafter until the end of follow-up. Patients were also asked to report by telephone any unexpected symptoms or hospital visits/admissions to the BSS clinical and nursing team or the research team. If participants required help outside of normal service hours, they were able to contact the on-call cardiologist at Bristol Royal Hospital for Children who in turn could contact one of the clinicians allied to the BSS.
Usual care
Throughout the trial, participants continued with their usual clinical care as part of the BSS, including any concomitant medication not contraindicated by bezafibrate.
Outcomes
Primary outcome
The primary outcome measure was peak oxygen consumption on bicycle ergometry (i.e. peak VO2). This outcome is strongly associated with activity intolerance and may correlate with subjective fatiguability, which we believed when designing the study to be the most important determinant of QoL in these patients. Peak VO2 was assessed at baseline and in the final week of each treatment phase.
Secondary outcomes
-
MLCL/L4-CL ratio and cardiolipin profile in blood cells
-
phosphocreatine (PCr)/ATP ratio in cardiac muscle on 31P MRS
-
skeletal muscle oxidative function/on 31P MRS
-
QoL assessed using age-appropriate Pediatric Quality of Life Inventory (PedsQL™) questionnaires
-
absolute neutrophil count
-
amino acid expression (serum arginine and cysteine levels)
-
cardiac function [left ventricular ejection fraction (LVEF) and 2-D strain]
-
mitochondrial size in lymphocytes
-
mitochondrial number in lymphocytes
-
mitochondrial area in lymphocytes
-
mitochondrial area as proportion of cytoplasm area
-
mitochondrial function and cristae organisation in lymphocytes/neutrophils
-
arrhythmia profile from 12-lead electrocardiogram (ECG) at rest and during exercise (for potential rhythm abnormalities).
In addition to the data collected from the clinical trial, we integrated qualitative research methods to explore participants’ and families’ experiences of the different interventions.
Assessments
The following assessments took place at baseline and during the final week of therapy at the end of each treatment phase, so that patients were still receiving the study medication at the time of testing but had maximum cumulative exposure to bezafibrate (or placebo).
Clinical outcomes
Peak oxygen consumption
Peak VO2 was assessed using bicycle ergometry on an electronically braked echocardiography exercise eBike (GE Healthcare eBike EL, GE Medical Systems Information Technologies GmbH, Freiburg, Germany). This stress exercise test was conducted simultaneously with echocardiography, as described below. A step protocol was used, with each participant starting at 0 W and the intensity increasing by 25 W every 3 minutes (modified McMaster protocol) as described by our group previously. 25 The 3-minute steps were used to obtain detailed ‘steady-state’ information for each exercise stage and to facilitate echocardiographic image acquisition. Participants were encouraged to cycle at 60 r.p.m. throughout and were told to cycle until completion or to the point of exhaustion, whichever came earlier. The exercise test was terminated when the participant was unable to sustain a cadence of 60 ± 5 r.p.m.
Oxygen consumption was measured using a portable metabolic ‘cart’ (Metalyzer 3B Cortex, Biophysik, Leipzig, Germany). The device was calibrated using a known reference gas before testing and a 3-l calibration syringe was used to calibrate the turbine volume transducer (Hans Rudolph, Kansas City, MO, USA). The gas cart provided breath-by-breath gas exchange analysis, averaged to 10-second time intervals. The highest 10-second average VO2 represented that individual’s peak VO2. Average VO2 for each stage was calculated as the mean of the last 30 seconds of data for each 3-minute interval. The Borg Scale was used to measure the rating of perceived exertion at the end of each 3-minute interval, with the participants asked to subjectively indicate the grading (scale out of 10) relevant to them at that point in the test.
Functional transthoracic echocardiography
Parts of this section have been reproduced from Pieles et al. 26 This article is licensed under a Creative Commons Attribution 4.0 International Licence, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The below text includes minor changes to the original text.
Functional transthoracic echocardiography at rest was performed by a sonographer or paediatric cardiologist following the guideline of the American Society of Echocardiography27 and using the following hardware: Canon (Canon Medical Systems LTD, Crawley, UK). LV systolic function was assessed during rest and exercise using short axis and long axis fractional shortening, pulse wave–tissue Doppler imaging-derived myocardial systolic velocities and 2-D myocardial strain analysis (speckle tracking). For longitudinal and radial strain analysis, 2-D ultrasound images of the left ventricle were obtained in apical four-chamber and parasternal short axis views. Images for strain analysis were obtained at a rate of 40–90 frames per minute during rest and exercise, and raw data were stored as a DICOM file. Dedicated software (Vitrea, Canon Medical Systems Ltd.) was used to generate strain values. Diastolic LV function was assessed using pulse wave–tissue Doppler imaging-derived myocardial diastolic velocities and pulse wave Doppler mitral valve inflow velocities.
Before exercise stress testing, participants underwent a full structural and functional resting (baseline) echocardiogram following international paediatric guidelines. 27,28 Echocardiographic measurements and analysis were performed using a Canon i900 ultrasound machine and a 2.0–4.8 MHz transducer and UltraExtend v.3.2 software (Canon Medical Systems, Japan). LV diameters were measured from 2-D echocardiography in the parasternal short axis view at the base of the left ventricle. Ejection fraction (EF) was calculated using the Simpson 2-D biplane method. A parasternal short axis and left or right ventricle-focused apical four-chamber view were captured for 2-D strain analysis. Three cardiac cycles were acquired at rates of 60–100 frames per second, and analysis was performed on one manually selected cardiac cycle. The endocardial borders were manually contoured at end-systole with the range of interest adjusted to include the whole myocardium. Mean peak systolic longitudinal and circumferential strain were defined as the maximal deformation value of a segment during systole in the endocardial segment and are represented as percentages; mean peak systolic strain rate was defined as the maximal rate of deformation of a segment in systole over time and is expressed per second. 29 Circumferential peak systolic strain was measured at the base of the left ventricle. Mean values for circumferential and longitudinal strain were calculated for each stage only if good tracking was obtained in a minimum of four segments. Image acquisition and offline analysis were performed by investigators experienced in paediatric echocardiography. Exercise stress echocardiography and 2-D strain analysis were performed using the same protocol and by the same internationally accredited operator, as described by our research group previously. 25 Briefly, focused echocardiography was performed for 2-D strain analysis during the free breathing exercise 60 seconds into each exercise stage at baseline (rest), 0 (unloaded pedalling), 50, 100, 150 W and during recovery at 2 and 6 minutes after the end of the exercise. The gas exchange threshold, representing the break point in breath-by-breath values of carbon dioxide uptake and oxygen uptake, was expressed as a percentage of VO2 peak. Myocardial reserve was defined as the difference in 2-D mean peak systolic strain between baseline and each exercise stage up to 150 W; strain values were not calculated at work rates higher than 150 W to ensure sufficient image quality and frame rate for reliable strain analysis. Only images with high frame rates of 60–100 frames per second were used, to ensure capture of sufficient frames for 2-D strain analysis at higher heart rates. A minimum of three cardiac cycles were recorded to capture at least one cardiac cycle in expiration, to obtain best image quality, which was confirmed visually and then used to perform strain analysis. The method has been described in detail by our group previously. 25,26
Cardiac magnetic resonance imaging
Cardiac magnetic resonance imaging (MRI) was performed on participants using the following standard clinical cardiomyopathy protocol and sequences on a Siemens 3Tesla Skyra scanner (Siemens Medical Systems, Erlangen, Germany):
-
HASTE™ axial dark blood axial (free breathing so paediatric participants do not get tired)
-
FISP™ coronal bright blood (free breathing so paediatric participants do not get tired)
-
Steady-state free precession (SSFP) four chamber – breath hold
-
SSFP two chamber – breath hold
-
SSFP stack of short axis – slice thickness 8 mm, no gap – breath hold
-
Native T1 mapping four chamber – breath hold
-
Native T1 mapping in all short axis view, no gadolinium (copy the cine short axis) – breath hold.
The acquisition time was between 30 minutes and 1 hour. If participants became tired during the scan, step 4 was omitted.
Continuous 12-lead ECGs were collected during exercise and recovery to assess participants’ arrhythmia profiles. Electrodes were connected to a 12-lead standard ECG monitor linked to the metabolic exercise cart with integrated ECG software (Metalyzer 3B Cortex, Biophysik, Leipzig, Germany).
Magnetic resonance spectroscopy
Participants underwent high-energy phosphate MRS (31P MRS) on a Siemens 3T Skyra (pTX-MNO) whole-body MRI system located in the CRIC facility, part of University of Bristol (UoB). This was performed only at a single time point at the end of the study. An in-house-built 11-cm diameter loop coil attached to a Stark Contrast 31P TX/RX interface unit was used. The coil included a 1.5-cm diameter central calibration sphere containing a solution of phenylphosphonic acid in ethanol and a set of cod liver oil fiducial markers, both in the plane of the loop coil.
Magnetic resonance spectroscopy acquisition for cardiac muscle
The participants were asked to lie supine head-first in the scanner with knees flexed, resting on a wedge support. The 31P coil was empirically positioned on the left side of the chest and the position subsequently refined by reference to proton images (acquired using the built-in body coil) to lie just below the mitral valve level of the heart. Localised B0 shimming over the region of interest was also performed using the body coil. The spectroscopic acquisition employed depth-resolved spectroscopy, a sequence with a slice-selective excitation (TR = 1 s, BW = 2000 Hz, SL = 20 mm). Three averaged spectra were obtained, one for each of the rest (4 minutes), exercise (5 minutes) and recovery (6 minutes) phases. The exercise took the form of alternating flexion/extension of legs with resistance bands placed around the calves and light lead weights on the ankles. The 31P transmitter voltage was in each case set to a fixed value of 80 V to approximate a 90-degree flip angle at the depth of interest. In addition, a series of non-localised inversion-recovery free induction decay acquisitions were performed at a range of transmitter voltages to facilitate subsequent estimation of the effective flip angle based on measurements of the phenylphosphonic acid 31P signal from the calibration sphere in the plane of the coil.
The spectra at each of the three time points were analysed in the time domain using a nonlinear least-squares algorithm (AMARES) from within the Oxford spectroscopy analysis package OXSA30,31 to determine the areas under the PCr, β-adenosine triphosphate (ATP) and 2,3-diphosphoglycerate (2,3-DPG) peaks; and subsequently the PCr/ATP peak ratios.
Magnetic resonance spectroscopy acquisition for skeletal muscle
The participants were positioned prone, head-first inside the scanner and the 31P coil placed and secured on the back of the thigh. Hamstring muscle bioenergetics were measured at rest and during and after exercise. The exercise again took the form of repetitive leg flexion/extension with a resistance band placed around the calf and lead ankle weight. A non-localised free induction decay acquisition was used to acquire the 31P spectra. Initially, several acquisitions were made using different transmitter voltages to determine a suitable value to be used for the main acquisition. The main acquisition consisted of a dynamic series of spectra (BW = 1500Hz) obtained with a repetition time of 2 seconds for 1–2 minutes of rest, 5 minutes of exercise and 6 minutes of recovery, making a total of 360–390 spectra.
The spectra in the time series were each independently fitted in the time domain using the non-linear least-squares algorithm AMARES in the OXSA software package30,31 to determine the peak areas and chemical shifts of PCr, ATP and inorganic phosphate (Pi) as a function of time. A mono-exponential relaxation curve was subsequently fitted to the PCr peak areas during the recovery phase. The time constant for this recovery (together with estimates of intracellular pH from the chemical shifts of the Pi peak) was then used to derive measures of mitochondrial function.
In vivo biochemical outcomes
Cardiolipin ratio and profile
The Pierce bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific, Altrincham, UK) was used to measure total protein concentrations for all samples, following the manufacturer’s instructions. All protein assays were carried out in triplicate. A standard curve from 25 μg/ml to 2 mg/ml was prepared using the N,O-bis(trimethylsilyl)acetamide (BSA) standard (2 mg/ml in 0.9% saline containing 0.05% sodium azide) and diluting with distilled water.
The assay kit contained two pre-prepared reagents: A – sodium carbonate, sodium bicarbonate, BCA and sodium tartrate in 0.1 M sodium hydroxide; and B – 4% cupric sulphate. To make the working reagent, 1 ml of BCA reagent B was added to 50 ml of BCA reagent A. Standard and sample protein solutions were then added to the BCA working reagent (50 μl of sample to 1 ml of reagent) and incubated at 37°C for 30 minutes. After incubation, the samples were allowed to cool to room temperature before the protein concentration was measured on a DeNovix DS-11 (DeNovix Inc., Wilmington, DE, USA) spectrophotometer at 562 nm.
For the lipid extraction, an aliquot with an equivalent volume for 20 μg of protein was extracted. The cell sample was disrupted by sonication for 5 minutes, then centrifuged at 16,000 × g for 30 seconds. The supernatant was discarded and 10 μl of chloroform added to the pellet, which was vortexed to extract the lipids. The matrix solution of 9-aminoacridine in 2-propanol/acetonitrile (60/40, v/v) at a concentration of 10 mg/ml was used in all cases. A 10-μl aliquot of the matrix solution was added to the chloroform lipid-extracted pellet and the sample mixed. A 0.4-mM solution of tetra-myristoyl cardiolipin (14 : 0) (Avanti Polar Lipids, Alabaster, AL, USA) in chloroform was prepared and used as an internal standard. A 0.5-μl aliquot of the internal standard solution was added to the lipid extract–matrix mixture and this solution was centrifuged at 16,000 × g for 30 seconds.
Matrix-assisted laser desorption ionisation (MALDI) mass spectroscopy was used to analyse the lipid extract. The lipid extract–matrix solution was spotted onto the polished steel MALDI target in droplets of 0.35 μl, allowed to air dry, and analysed. MALDI–time of flight mass spectra of intact lipids were acquired on a Bruker Ultraflex mass spectrometer (Bruker Daltonics, Bremen, Germany). Instrument performance and calibration was routinely checked against a known mixture of peptides in the mass range 900–3600 Da (AB Sciex Calmix 2, AB Sciex UK Ltd, Warrington, UK). The calibrant solution was prepared as recommended by the manufacturer using α-cyano-4-hydroxycinnamic acid as a matrix in acetonitrile/water (7: 3, v/v) with 0.1% trifluoroacetic acid as an additive.
Absolute neutrophil count
Neutrophil counts were determined by the UHBW laboratory according to their standard laboratory protocol.
Amino acid expression
Blood levels of arginine and cysteine were determined by the UHBW laboratory according to their standard laboratory protocol.
Electron microscopy (mitochondrial tests)
Lymphocytes were isolated from study participants within 3 hours of venepuncture, followed by preparation for electron microscopy by high-pressure freezing and freeze substitution. Sections were stored at room temperature until use. Cells from sections were viewed on a FEI Tecnai 12 (Fei UK Limited, Altrincham, UK) electron microscope, and images of lymphocytes captured for analysis. Where possible, images were captured and stored from 20–25 individual lymphocytes on a single section for each participant at each time point. Cells damaged by the preparation process were not analysed. Images of cells were viewed, and measurements of size and area were performed using either ImageJ (National Institutes of Health, Bethesda, MD, USA) or Adobe® Photoshop® (Adobe Systems Incorporated, San Jose, CA, USA) software. The following measurements were made:
-
Number of mitochondria per lymphocyte.
-
Area of each mitochondrion.
-
Total area of mitochondria per lymphocyte.
-
Area of mitochondria as a proportion of the cytoplasm in each lymphocyte. The area of the nucleus was subtracted from the cell area to give the area of the cytoplasm.
The cristae organisation within the mitochondria was analysed by eye, making note of those where (1) there were few cristae, (2) cristae were short, or (3) cristae protruded randomly from the membrane.
Mitochondrial content and membrane potential
Following peripheral blood mononuclear cell and lymphocyte isolation, 1.5 × 105 cells were resuspended in 50 μl of serum-free RPMI medium and incubated for 20 minutes at 37˚C. Equal volumes of media containing either tetramethylrhodamine methyl ester perchlorate (TMRE) (final concentration 25 nM) or Mitotracker™ Green FM (final concentration 5 nM) were added and incubated for 20 minutes at 37°C. An additional sample per donor was stained with TMRE after receiving a 10-minute pretreatment of carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) (final concentration 6.6 µM) to act as a negative staining control. Following incubation cells were washed in wash buffer (phosphate-buffered saline, 5 mM ethylenediaminetetraacetic acid and 0.5% BSA) and data were recorded using by a BD X20 (Becton Dickinson Biosciences, Wokingham, UK) Fortessa flow cytometer. Data were analysed using FlowJo (FlowJo, LLC, Ashland, OR, USA). Following doublet discrimination, monocytes were identified within peripheral blood mononuclear cell samples using forward and side scatter. Mitotracker was analysed by median fluorescent intensity (MFI), TMRE was analysed by TMRE MFI – TMRE + FCCP MFI.
In vitro drug-treated biochemical outcomes
Lymphoblasts were cultured in complete medium (RPMI-1640 supplemented with 10% fetal calf serum, 50 IU/ml penicillin, 0.1 mg/ml streptomycin and 4 mM L-glutamine) with the addition of dimethyl sulfoxide and either 400 µM of bezafibrate or 40 µM resveratrol at 37°C in 5% CO2 for 48 hours.
The cells were then analysed by electron microscopy for the physical measurements of the lymphocytes and mitochondria. Cells were also prepared and sent for cardiolipin profiling.
Quality of life
Participants’ QoL was assessed using the core and multidimensional fatigue scales of the PedsQL questionnaires. 32 Each scale contained the following age-appropriate forms given to the participants: young child (aged 5–7 years), child (aged 8–12 years), teen (aged 13–18 years) and young adult (aged 18–12 years). Parents/guardians were also asked to assess their child’s QoL during the study using the PedsQL parent/guardian questionnaires. All questionnaires were self-administered and completed during the research clinics.
Patient experience
Parents of younger patients (< 18 years), and patients aged > 14 years (with consent of the patients or parents), were invited to take part in semistructured one-to-one interviews during research clinics at the end of each treatment phase. The first interview (at the end of the first treatment phase) lasted approximately 20 minutes and explored the parents’/participants’ experience of the intervention. The second interview (at the end of the second treatment phase) lasted approximately 40 minutes and explored the parents’/participants’ experience of the second intervention and their perception of participating in the trial as a whole. Interviews with participants and parents/guardians were digitally recorded with their consent and transcribed verbatim. Data from interviews were analysed using framework analysis methodology with the aid of the NVivo (QSR International, Warrington, UK) data management software package.
Topic guides (see Report Supplementary Material 3) were developed for both phases of the qualitative study based on discussions with the study team and grant co-applicants, which included social scientists and medical specialists.
The topic guide for phase 1 included experience of the trial up to the end of the first phase of the trial including: family, schooling, occupation and so on; background diagnosis and symptoms of Barth syndrome; previous and current treatment for Barth syndrome; living with Barth syndrome; QoL; social, physical and psychological impact of Barth syndrome; support; experience of the past 4 months in the trial; adherence and issues with medication; and engagement with medical practitioners.
The topic guide for phase 2 explored experiences of the trial since the last interview, which included: symptoms of Barth syndrome; experience of the trial; QoL; social, physical and psychological impact of Barth syndrome; washout period; and adherence and issues with medication.
Adverse events
Serious adverse event (SAE) and AE data were collected from participants from the time of consent until 1 month after the final treatment phase was completed. Participants’ general practitioners were notified of their participation, with a request to inform the research team of any suspected AEs or reactions.
A research nurse asked questions about AEs at each research clinic visit and during monthly phone calls with the participants/families, including during the washout period. All AEs were recorded. SAEs were reviewed by the Chief Investigator, who was blind to the random allocation of order of administration of bezafibrate and placebo. He made the decision whether the SAE was related to bezafibrate.
All unexpected SAEs and suspected serious adverse reactions (i.e. serious reactions expected of bezafibrate) were reviewed by the Chief Investigator and by the Chair of the DMSC. A list of all anticipated (due to the disease), expected and unexpected AEs and SAEs was presented to the DMSC after the completion of the first phase of treatment. The DMSC then made recommendations about the continuation of the trial to an independent TSC. Unexpected SAEs which were judged to be causally related to the intervention were subject to expedited reporting to the Research Ethics Committee (REC), Medicines and Healthcare Products Regulatory Agency (MHRA) and DMSC.
Sample size
A total of 20 males aged between 6 and 24 years attended the NHS National BSS at the time of recruitment. We anticipated that 12–15 of them would elect to take part.
The sample size was dictated by the number of eligible boys willing to take part. The primary analysis estimated the difference in mean peak VO2 (see Primary outcome) between placebo and bezafibrate phases, assuming a two-tailed alpha of 0.05. For illustration, a sample size of 12 participants allowed the trial to detect a difference of 0.90 (within subject) standard deviations (SDs) with 80% power, or 1.05 SDs with 90% power.
Randomisation
The random allocations of order of administration of bezafibrate and placebo were generated before starting the study by an independent statistician in the Bristol Trials Centre [Clinical Trials and Evaluation Unit (CTEU)], using blocks of undisclosed size. Allocations were generated by computer and concealed from all clinical and research personnel. The sequence was appended to a list of consecutive study IDs and provided to the PTU.
After a participant had provided informed consent and eligibility had been confirmed, the participant was assigned to the next consecutive study ID. The study physician prescribed the study IMP and the pharmacy dispensed the appropriate intervention according to the participant’s study ID and the randomised sequence provided.
Blinding
No one (no participant, investigator or any other member of the research team), apart from the pharmacist dispensing the IMP prescription, knew the order in which bezafibrate and placebo were administered to a participant.
The bezafibrate and placebo tablets did not have a particularly strong or unusual smell or taste and were visually identical. Therefore, inadvertent unblinding due to the characteristics of the IMP was not anticipated.
The PIL and discussions with the participants and their families during the informed consent process explained the uncertainty around the potential beneficial effects of bezafibrate over a placebo. Therefore, in the event of inadvertent unblinding, the participant should not have had a strong expectation that one or other method would lead to a more favourable result. Participants were told before giving consent that they would not be told which treatment they would receive in each phase.
Data collection and follow-up schedule
Case report forms for the study are available at the NIHR Library study webpage (see Report Supplementary Material 4). Collection of the required data at scheduled points of contact is shown in the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) diagram in Table 1. Data were collected face to face (i.e. clinic appointments in Bristol) at scheduled time points −t1, 0, t4 and t8. Scheduled time points t1, t2, t3, washout, t5, t6, t7 and t9 represent telephone calls.
| STUDY PERIOD | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Enrolment | Allocation | Post-allocation | Close-out | ||||||||||
| TIME POINT | −t 1 a | 0a | t 1 b | t 2 b | t 3 b | t 4 a | Washout ~ | t 5 b | t 6 b | t 7 b | t 8 a | T 9 b | |
| ENROLMENT: | |||||||||||||
| Eligibility confirmation | X | ||||||||||||
| Informed consent | X | ||||||||||||
| Allocation | X | ||||||||||||
| INTERVENTIONS: | |||||||||||||
| Drug prescribing | X | X | |||||||||||
| Drug dispensing | X | X | |||||||||||
| Bezafibrate |

|

|
|||||||||||
| Placebo |

|

|
|||||||||||
| ASSESSMENTS: | |||||||||||||
| Medical history | X | ||||||||||||
| Height and weight | X | X | X | ||||||||||
| Clinical examinationc | X | X | X | ||||||||||
| Bicycle ergometry (exercise bike test) | Peak oxygen consumption | X | X | X | |||||||||
| Tissue Doppler studies | X | X | X | ||||||||||
| Echocardiogram (at rest and during exercise)d | X | X | X | ||||||||||
| 12-lead ECG at rest and during exercise (during echocardiography) | X | X | X | ||||||||||
| Blood sample (20–30 ml total)e | Transformed lymphoblast line for in vitro incubation with bezafibrate and resveratrol | X | |||||||||||
| FBC, absolute neutrophil count, urea/electrolytes, LFTs, CK, plasma arginine/cysteine, full lipid profile (total cholesterol, high-density lipoprotein, triglycerides), brain natriuretic peptide | X | X | X | ||||||||||
| Mitochondrial assessmente | X | X | X | ||||||||||
| Blood sample (safety assessment) performed locally to patient: FBC including absolute neutrophil count, routine renal tests and LFTs, plasma triglyceride/total cholesterol/ low-density lipoprotein-cholesterol and CK, creatinine |
X | X | X | X | X | X | X | ||||||
| Cardiac/skeletal muscle MRI/MRS scanf | X | X | X | ||||||||||
| PedsQL | X | X | X | ||||||||||
| AEs | X | X | X | X | X | X | X | X | X | X | |||
| Qualitative interview | X | X | |||||||||||
Analysis methods
Planned statistical analyses
The statistical analysis plan (SAP; see Report Supplementary Material 5) for the study prespecified that:
-
All summaries and analyses of the primary and secondary outcomes would be conducted according to the intention-to-treat (ITT) principle. The ITT population consisted of all participants and periods, according to the order of administration of bezafibrate and placebo specified by the randomised allocation.
-
In addition to the ITT analysis, a complier average casual effect analysis would be considered for the primary outcome if a considerable number of participants had major protocol deviations (> 20% of participants).
-
The safety population consisted of all randomised participants, classified according to the treatment received in each period, who received at least one dose of IMP.
We intended that all participants withdrawn from the trial, and those found to be ineligible post randomisation, would continue to be followed until the end of the study (without taking any IMP), unless they withdrew full consent for further follow-up data to be collected. In such instances, all data collected up to the time of withdrawal would be used in the analysis, unless consent to use the collected data was withdrawn.
The placebo group was designated as the reference category. All treatment effects therefore represent the effect of bezafibrate in comparison with placebo. All applicable statistical tests were two-sided and were performed using a 5% significance level, except for tests for interactions that were performed using a 10% significance level; 95% confidence intervals (CIs) were used unless otherwise stated. No formal adjustment was made for multiple testing, but consideration was given to the number of statistical tests performed and the consistency, magnitude and direction of treatment estimates for different outcomes when interpreting the results.
All percentages reported are calculated with respect to the total number of participants with data available, with any missing data described in footnotes. For categorical and binary data, all percentages have been rounded to at most one decimal place. Continuous measures (means and CIs) are summarised to one more decimal place than the number of decimal places to which the raw data were collected. p-values > 0.001 are summarised to two significant figures, and those < 0.001 are reported as < 0.001.
Outcome data are summarised descriptively using means and SDs [or medians and interquartile ranges (IQRs) depending on the distribution] or counts and percentages (where appropriate) by treatment, period and overall. The treatment effect for the primary outcome was analysed using mixed linear regression models, adjusting for period as a fixed effect (exploring interactions where necessary) and participants as random effects. Nested models were compared using likelihood ratio tests. Model assumptions were tested using standard methods (e.g. residual plots), and the effect of carry-over was estimated by including treatment order in the model. Table 2 shows other continuously scaled secondary outcomes for which the SAP prespecified that treatment effects were to be estimated and tested, using the same methods as used for the primary outcome.
| Outcome |
|---|
| Rest VO2 |
| MLCL/L4-CL ratio |
| QoL |
| Absolute neutrophil count |
| Plasma arginine level |
| Plasma cysteine level |
| Cardiac function, echocardiography: left ventricular biplane ejection fraction (LVEFecho%) |
| Cardiac function, echocardiography: peak systolic mean longitudinal strain |
| Cardiac function, echocardiography: LV peak systolic mean circumferential strain |
| Cardiac function, echocardiography: diastolic ratio (MV E/LV E’ cm/s) |
| Cardiac function, MRI: LVEF at rest (LVEFMRI%) |
| Cardiac function, MRI: right ventricular biplane ejection fraction (RVEFMRI%) |
Qualitative research
All the interviews were audio-recorded and transcribed verbatim. A sample of transcripts representing both time points were read and re-read by two experienced qualitative researchers to familiarise themselves with the data. The qualitative researchers then discussed their overall impressions of each data set and how the data should be analysed. It was agreed that both data sets should be analysed thematically,33 as this would enable comparisons to be made both within and across the data sets (boys with Barth syndrome and parents). The sampled transcripts were independently coded and a coding frame developed and verified for the entire data set. Once each coding frame was finalised, all the transcripts were imported into NVivo 12 and electronically coded. Each data set was analysed separately before comparisons were made between participation at each phase of the trial.
Coding development
At each phase, there were codes that were specific to participants/families’ experiences of participation in the trial regarding acceptability and feasibility. These codes included: reason for trial participation, experience of trial, treatment adherence, perceived benefits of participation in the trial, symptoms of Barth syndrome, communication with health professionals, trial assessment and monitoring, and trial conduct.
There were also codes considered to be specific to the wider ‘lived experience’ of Barth syndrome and the impact it has on individuals and families. These codes included: living with Barth syndrome, treatment of Barth syndrome, child development and growth, communication and role of the multidisciplinary team, management and coping strategies, social functioning and integration, view of self, schooling and Barth syndrome, social and psychological support for Barth syndrome, perceived stigma and normalisation.
Biobank samples
Due to the valuable nature of samples obtained from patients with this rare disease, we specified at the outset that the samples collected in the trial would be retained for use in other future research. When the REC was notified that the study had closed, the samples were transferred to Dr Allison Blair at the UoB under a separate research ethics approval. All participants consented for their samples to be used in future research.
Adherence monitoring
Adherence to study treatment was assessed using two methods:
-
During monthly follow-up telephone calls, participants (or the carers of young children) were asked whether any doses had been missed and, if yes, the number of doses missed was recorded.
-
Participants were asked to return all their study medication bottles at the end of each treatment phase. Unused tablets were counted and compared to the expected number of tablets to be returned. The proportion of tablets taken by each participant during each treatment phase was then calculated. All tablets were assumed to be taken if the bottle was empty. Participants were classified as adherent if they took at least 70% of their tablets.
Emergency unmasking
Members of the participant’s healthcare team were able to request unmasking of the study medication in the event of a SAE, if they considered that the information would alter the management of the SAE. Participants/their families were given a card to carry at all times. This card described instructions for the attending doctor on how to request unmasking. Arrangements were made for CTEU Bristol to facilitate unblinding during office hours (but with the UHBW Pharmacy communicating with the person requesting unblinding, to keep the CTEU staff blind) and the UHBW Pharmacy department to be contacted directly outside of office hours. The on-call pharmacist had access to the unblinded list of randomised order allocations. In the event of unblinding being requested, the following information had to be documented: person requesting unblinding, reason for the request, the time and date of the request, and the person who performed unblinding.
Patient and public involvement
Design of the research
Doctors and scientists in Bristol have collaborated closely with people and families affected by Barth syndrome since the 1990s, both nationally and internationally, often via the UK and US Barth syndrome charities Barth Syndrome Trust (BST, UK) and Barth Syndrome Foundation (BSF, USA). Patients with Barth syndrome and their families were involved in the trial design from an early stage in its conception. This was done by a combination of focus groups, face-to-face interviews and telephone discussions with adult and adolescent sufferers of the disease, their parents, and officers of the BST.
The major feedback from these sessions was: (1) for the trial to limit as much as possible the numbers of routine blood tests and detailed assessment points, to optimise schooling, employment and QoL; and (2) to ensure that the young people taking part in the study felt able and prepared to take the study medications.
Management of the research
The mother of a boy who died from Barth syndrome some years ago kindly agreed to become a member of the TSC. The first TSC meeting was held in February 2019, shortly before the trial commenced, and she provided much helpful insight and advice during this meeting.
Mrs Michaela Damin, chair of the BST, who is also a co-applicant of the study, provided invaluable input and advice throughout the trial and helped us to liaise with the participants and their families.
Dissemination of research findings
We planned to hold a social gathering for the trial participants after the results become available so that we could feed back the information from the trial to the participants and their families in person and thank them for their participation. We planned to send a written lay summary of the results to all patients/families in the UK with Barth syndrome.
Chapter 3 Results: trial cohort
Screened patients
Patient screening for this trial did not take place in the usual way because the entire population of patients with Barth syndrome was under the care of the BSS. The population is also very stable, with few incident cases, so that clinicians involved in the study knew from the outset which patients were potential candidates for the trial and their clinical status. The UK Barth syndrome community was also aware of the trial from its early stages, so clinicians had some idea of the interest in the trial of individuals and their families before they were formally approached by the study team.
Twenty boys with Barth syndrome were under the care of the BSS and of a suitable age to enter the trial. The funder had specified a stop/go criterion of at least 10 participants and requested that written expressions of interest be obtained from potential participants to ensure the viability of the trial. Three potential participants were not approached to express an interest for the following reasons: one patient was known not to swallow tablets (and was not expected to be able to do so in the future), one patient had learning difficulties, and one was known not to be interested in participating because of resolution of cardiomyopathy and lack of disease symptoms, except for occasional neutropenia, and had previously indicated that taking time off work would be difficult.
Seventeen patients were approached for an expression of interest, of whom four indicated that they did not want to participate. Over time, a further two patients were unable to take part. One patient declined due to his concerns about his clinical status, and another was unable to swallow tablets after a period of training to help him achieve this. However, one patient who previously indicated that they were not interested in the trial became interested and eligible after successfully completing a period of pill-swallowing training. Therefore, 12 patients are shown at the top level of the Consolidated Standards of Reporting Trials (CONSORT) diagram (Figure 2).
FIGURE 2.
Trial CONSORT diagram.
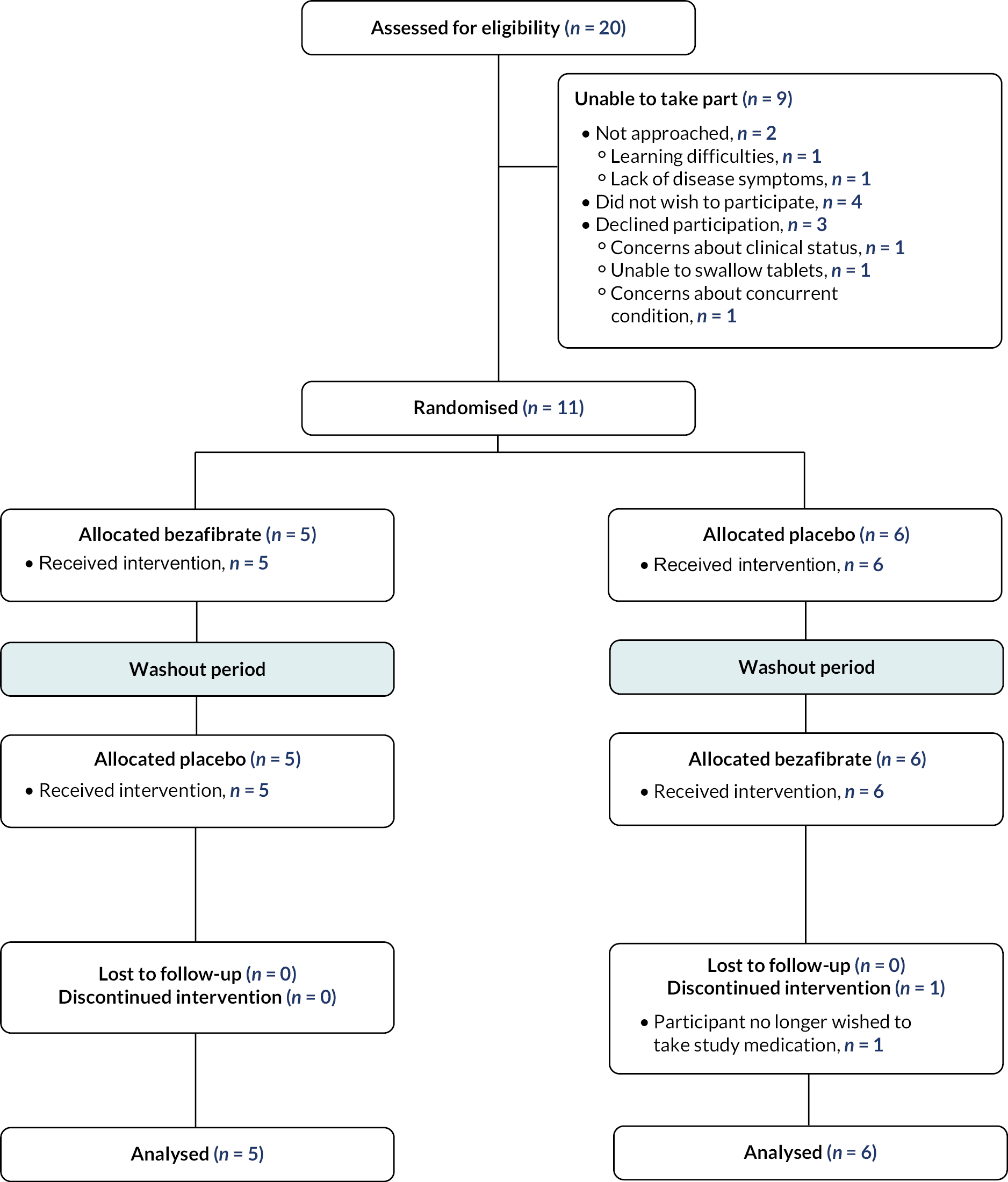
Age-appropriate PILs were sent to these 12 patients and arrangements made for them to attend the research clinics. However, one patient declined to participate the day before his scheduled attendance at the final baseline research clinic due to concerns about a concurrent medical condition.
Randomised patients
Eleven participants were randomised. The first participant was randomised on 29 March 2019 and the final participant randomised on 12 April 2019. Follow-up for the trial was completed on 6 January 2020. The flow of participants in the trial is shown in the CONSORT diagram (see Figure 2).
Recruitment to the trial
When the trial was conceived, we anticipated that 12–15 patients would be recruited into the trial. This was based on the number of patients aged 6 years and above being managed in the BSS who were expected to be able to swallow tablets and willing to take part. The minimum of 10 participants who expressed willingness to participate before randomisation required by the funder for the trial to proceed was achieved (see Figure 2).
The UK population of people with Barth syndrome is widely dispersed across the country. Participation required attending a 2-day clinic in Bristol three times within a year. Therefore, we reimbursed travel expenses and arranged local hotel accommodation for those wishing to take part. The volume and length of study assessments required clinics to be conducted over 2 days. We arranged clinics on Fridays and Saturdays – that is, 1 day falling on a weekend – to minimise absences from work or school. Clinics could not be run entirely at weekends due to local hospital health and safety and working conditions policies. For example, non-urgent cardiac MRIs could not take place outside of normal working hours.
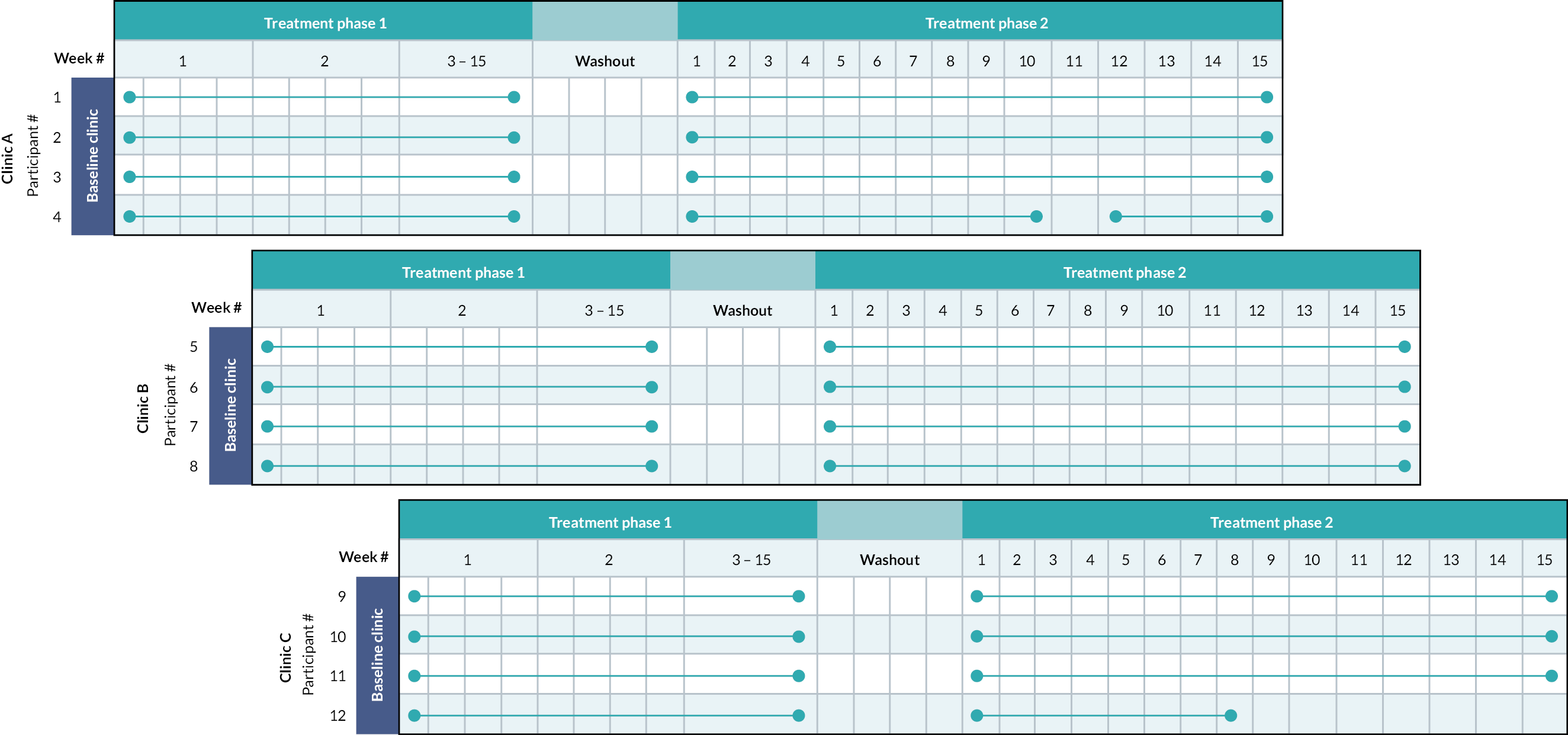
The volume and length of study assessments also limited the number of participants we could see in one clinic (four per clinic). As 12 patients were scheduled to take part, we therefore arranged clinics over three consecutive Fridays/Saturdays for the baseline, end of phase 1 and end of phase 2 assessments (Figure 3).
FIGURE 3.
Participants’ time on treatment diagram.
Participant withdrawals
One participant withdrew from taking the study medication during the second treatment phase due to an AE (diarrhoea). However, the parents did not withdraw their child from the study as a whole and they attended the end of treatment phase research clinics and completed all the study assessments. Thus, all participants attended all three research clinics.
Major protocol deviations
There were two instances requiring corrective and preventative action.
The parents of one 13-year-old participant increased the dose of IMP too early in the second phase. The participant was assigned to bezafibrate in the second phase, although this was not known at the time (and allocation was not unmasked). The participant experienced vomiting for one night some days before the error was identified. No other untoward effects were observed or identified from safety blood tests.
The second instance concerned calculation of eGFR results. For adult participants, but not children, an eGFR result was provided on a laboratory readout (calculated from the creatinine level in a blood sample). At the point of recruitment, when the eligibility criteria checklist was completed, a blood test was undertaken (or reviewed if a recent test was available) and the laboratory readout made available. Because there were no eGFR laboratory readouts available for the paediatric patients, the eGFR results were calculated at the baseline clinic by the clinical team. The Chief Investigator (or delegate) assessed whether all participants were eligible and all blood results were satisfactory. However, these eGFR results were not documented in source data and, therefore, there was no documentary evidence that the eGFR results were calculated to establish eligibility. After the baseline clinics, the research nurses recalculated the eGFR values for the paediatric patients so that they could be documented in the case report forms. On advice from a renal consultant, the nurses were advised to use a particular formula to calculate eGFR rates for the paediatric patients: (height in cm/creatinine in mg/dl) × 40. The nurses used the paediatric formula for a 17.5-year-old participant, which showed an eGFR < 90. However, there is uncertainty about the upper age limit for which paediatric formulae should be used; commonly recommended age limits range from 16 to 18 years of age. During the clinic, the eGFR for the participant was calculated using the UKidney website calculator (https://ukidney.com/nephrology-resources/egfr-calculator), which incorporates patient age in the formula. The UKidney calculator is a widely used formula for calculating eGFR in adults. Based on the result of 111 ml/min, the patient was considered eligible and recruited. We did not specify in the protocol how eGFRs for paediatric patients should be calculated. In addition, formulae for eGFR calculations are not validated in children (hence why laboratories do not calculate eGFRs using any formula). There was also uncertainty about whether a formula implemented locally for paediatric patients, or a widely used online calculator, should be used for a patient aged 17.5 years. The issue was identified after all the patients had been recruited and had their eligibility assessed, so we were unable to make any changes to the process of checking and documenting the eGFR results during CARDIOMAN recruitment.
Emergency unblinding was not required. One participant experienced multiple episodes of diarrhoea during phase 2, but unblinding was not requested. Instead, the Chief Investigator decided to stop the participant’s IMP temporarily and restart it after 2 weeks. Restarting was considered justifiable because the participant had by this time undergone all indicated investigations with normal results and the symptoms had settled. The participant was closely monitored after restarting IMP and did not experience any recurrence of the symptoms. Another participant withdrew from taking the study IMP during the second treatment phase due to non-serious AEs; his allocation was not unblinded and he completed the final study assessments as scheduled.
Finally, the first group of four participants stayed on their allocated IMP for one extra week at the end of phase 1 because the exercise equipment failed. They returned the following week to complete the exercise assessments.
Adherence to the investigational medicinal product regimen
Adherence was defined as taking > 70% of the expected number of pills prescribed. Participants returned used bottles at their next study visits (or posted empty bottles back to the study team if they forgot to bring them to the study visits) and remaining pills were counted. For bottles not returned by participants (lost bottles), the assumption was made that no pills were remaining; that is, the participant had taken all the pills and discarded the bottle.
The numbers of participants who were adherent during each phase of the study, according to whether placebo or bezafibrate was being taken at the time, are shown in Table 3. There was no suggestion that non-adherence (18% overall) was worse when participants were taking bezafibrate than placebo. The participant who was non-adherent in phase 1 (when taking bezafibrate) was also non-adherent in phase 2 (when taking placebo). One participant who was non-adherent in phase 2 (when taking bezafibrate) stopped IMP permanently. Another participant who was non-adherent in phase 2 (when taking placebo) had concurrent illness during the study, which may explain his non-adherence. The participant who stopped temporarily in phase 2 was still adherent overall with respect to the > 70% criterion.
| Prescription | Adherence when taking bezafibrate (n = 11) (%) | Adherence when taking placebo (n = 11) (%) |
|---|---|---|
| Phase 1 | 1/5 (20) | 0/6 (0) |
| Phase 2 | 1/6 (17) | 2/5 (40) |
| Overall | 2/11 (18) | 2/11 (18) |
Participant characteristics and data at baseline
Participants’ baseline characteristics are shown in Table 4. In addition, none of the participants had fainted or experienced palpitations, chest pain or tightness, or an unexpected decrease in exercise capacity of fitness level in the preceding 4 months. One participant in each group had experienced dizziness in the preceding 4 months.
| Characteristic | Randomised to bezafibrate first (n = 5) | Randomised to placebo first (n = 6) | Overall (n = 11) | |
|---|---|---|---|---|
| Median age in years at randomisation (IQR) | 18 (17–21) | 13 (10–14) | 14 (10–27) | |
| Ethnicity | White | 5 | 5 | 10 |
| Mixed | 0 | 1 | 1 | |
| Height in cm | 173 (170, 173) | 140 (131, 145) | 145 (118, 175) | |
| Weight in kg | 78 (52, 91) | 34 (29, 51) | 51 (19, 98) | |
| Systolic blood pressure, mmHg | 120.0 (81.0, 128.0) | 94.5 (83.0, 122.0) | 98 (81, 128) | |
| Diastolic blood pressure, mmHg | 60.0 (51.0, 79.0) | 59.5 (48.0, 76.0) | 60 (48, 79) | |
| Heart rate, b.p.m. | 92.0 (84.0, 101.0) | 93.0 (88.0, 102.0) | 92 (84, 102) | |
| Respiratory rate | 17.0 (16.0, 19.0) | 19.0 (17.0, 25.0) | 18 (16, 25) | |
| Oxygen saturation | 97.0 (90.0, 99.0) | 97.0 (96.0, 98.0) | 97 (90, 99) | |
| Bicep skinfold, mm | 5.4 (3.0, 11.2) | 9.9 (6.3, 18.8) | 9 (3, 19) | |
| Tricep skinfold, mm | 11.0 (8.6, 21.0) | 12.2 (6.3, 21.0) | 12 (6, 21) | |
| Subscapula skinfold, mm | 13.4 (4.4, 25.0) | 10.8 (8.0, 20.0) | 11 (4, 25) | |
| Suprailiac skinfold, mm | 9.2 (5.2, 23.0) | 12.1 (5.2, 25.0) | 12 (5, 25) | |
| Normal heart sounds | 5 (100.0%) | 6 (100.0%) | 11 (100.0%) | |
| Regular pulses | 5 (100.0%) | 6 (100.0%) | 11 (100.0%) | |
| No signs of heart failure | 5 (100.0%) | 5 (83.3%) | 10 (90.9%) | |
| Previous cardiac transplant | 1 (20.0%) | 1 (16.7%) | 2 (18.2%) | |
Additional treatments administered as usual care
Participants were often prescribed several other medications at baseline as part of their usual care. Two of five participants allocated to bezafibrate and five of six participants allocated to placebo in phase 1 were taking cardiac medications. One of five participants allocated to bezafibrate and three of six participants allocated to placebo in phase 1 were taking general supportive-care medications. Concurrent cardiac and other medications at baseline by participant are shown in Table 5. Two participants (study IDs 6 and 9) were not taking any additional medication at baseline.
| Study ID | Sequence | Drug | Dose | Units | Frequency |
|---|---|---|---|---|---|
| 1 | Bezafibrate → placebo | Supportive care: omeprazole | 20 | mg | o.d. |
| Supportive care: sertraline | 200 | mg | o.d. | ||
| Transplant-related: prednisolone | 2 | mg | o.d. | ||
| Transplant-related: sirolimus | 2 | g | o.d. | ||
| Infection prophylaxis: cotrimoxazole | 960 | mg | alternate daily | ||
| Other medication: allopurinol | 10 | mg | o.d. | ||
| Other medication: dihydrocodeine | 90 | mg | twice a week | ||
| Other medication: oral morphine | 20 | mg | p.r.n. | ||
| 2 | Placebo → bezafibrate | Cardiac: carvedilol | 6.25 | mg | b.d. |
| Cardiac: enalapril | 7.5 | mg | b.d. | ||
| Infection prophylaxis: G-CSF | 0.3 | µg | twice weekly | ||
| Infection prophylaxis: penicillin | 250 | mg | b.d. | ||
| 3 | Bezafibrate → placebo | Cardiac: carvedilol | 6.25 | mg | b.d. |
| Cardiac: digoxin | 62.5 | µg | b.d. | ||
| Cardiac: enalapril | 5 | mg | b.d. | ||
| Infection prophylaxis: G-CSF | 0.4 | ml | twice weekly | ||
| Infection prophylaxis: penicillin | 250 | mg | b.d. | ||
| 4 | Placebo → bezafibrate | Cardiac: carvedilol | 6.25 | mg | b.d. |
| Cardiac: furosemide | 10 | mg | o.d. | ||
| Cardiac: lisinopril | 5 | mg | o.d. | ||
| Cardiac: spironolactone | 25 | mg | o.d. | ||
| Supportive care: omeprazole | 10 | mg | o.d. | ||
| Infection prophylaxis: G-CSF | 263 | µg | three times per week | ||
| Infection prophylaxis: azithromycin | 160 | mg | three times per week | ||
| 5 | Placebo → bezafibrate |
Cardiac: enalapril | 5 | mg | b.d. |
| Cardiac: furosemide | 10 | mg | o.d. | ||
| Cardiac: spironolactone | 12.5 | mg | o.d. | ||
| 7 | Placebo → bezafibrate | Cardiac: captopril | 12.5 | mg | t.d.s. |
| Supportive care: folic acid | 2.5 | mg | o.d. | ||
| Infection prophylaxis: cotrimoxazole | 7.5 | ml | o.d. | ||
| Infection prophylaxis: G-CSF | 158 | µg | three times per week | ||
| Other medication: ferrous sulphate | 200 | mg | o.d. | ||
| 8 | Bezafibrate → placebo | Cardiac: bisoprolol | 3.75 | mg | o.d. |
| Cardiac: enalapril | 7.5 | mg | o.d. | ||
| Cardiac: spironolactone | 12.5 | mg | o.d. | ||
| 10 | Placebo → bezafibrate | Supportive care: glycozade | 10 | g | ×6 |
| Transplant-related: mycophenolate mofetil | 0.1 | g | b.d. | ||
| Transplant-related | 1.2 | mg | b.d. | ||
| Infection prophylaxis: G-CSF | 0.25 | ml | three times per week | ||
| Other medication: cefalexin | nr | nr | nr | ||
| 11 | Placebo → bezafibrate | Cardiac: carvedilol | 3.125 | µg | b.d. |
| Cardiac: lisinopril | 2.5 | mg | o.d. | ||
| Cardiac: spironolactone | 25 | mg | b.d. | ||
| Infection prophylaxis: G-CSF | 52.5 | µg | twice per week | ||
| Infection prophylaxis: penicillin | 125 | mg | b.d. |
Chapter 4 Results: clinical outcomes
Peak VO2
Figure 4 shows a box plot of peak VO2 by treatment. Table 6 summarises the peak VO2 levels by treatment and period, respectively.
FIGURE 4.
Box plot of peak VO2 by treatment.
| Baseline | Bezafibrate | Placebo | |
|---|---|---|---|
| Baseline | First period | Second period | |
| N | 11 | 11 | 11 |
| Mean (SD) | 18.20 (5.90) | 16.93 (5.58) | 17.51 (6.28) |
| Median (IQR) | 16.0 (13.0–25.1) | 15.9 (11.9–22.3) | 14.5 (12.4–22.1) |
| N | 11 | 11 | 11 |
| Mean (SD) | 18.20 (5.90) | 16.81 (6.26) | 17.63 (5.59) |
| Median (IQR) | 16.0 (13.0–25.1) | 16.3 (11.6–22.1) | 15.5 (13.7–22.3) |
The results for the comparison between bezafibrate and placebo, adjusting for period as a fixed effect and participants as random effects, are shown in Table 7. There was no statistically significant difference between bezafibrate and placebo. Peak VO2 was 0.66 ml/kg/min lower (95% CI −2.34 to 1.03 ml/kg/min; p = 0.43) when participants were taking bezafibrate than placebo. Measurements taken in the second period were similar to the first period (0.88 ml/kg/min, 95% CI −0.81 to 2.56 ml/kg/min; p = 0.29).
| Effect | Level | Estimate (95% CI), ml/kg/min | p-value |
|---|---|---|---|
| Treatment effect | Bezafibrate vs. placebo | −0.66 (−2.34 to 1.03) | 0.4266 |
| Period effect | Second vs. first period | 0.88 (−0.81 to 2.56) | 0.2905 |
Pre-exercise VO2
At each clinic, VO2 was also measured before exercising for the peak VO2 measurement. Figure 5 is a box plot of baseline pre-exercise VO2 by treatment, and Table 8 shows the mean baseline pre-exercise VO2 by treatment and sequence, and overall. The results of the multilevel model to determine the effect of bezafibrate in comparison with placebo are shown in Table 9.
FIGURE 5.
Box plot for pre-exercise VO2 levels by treatment.
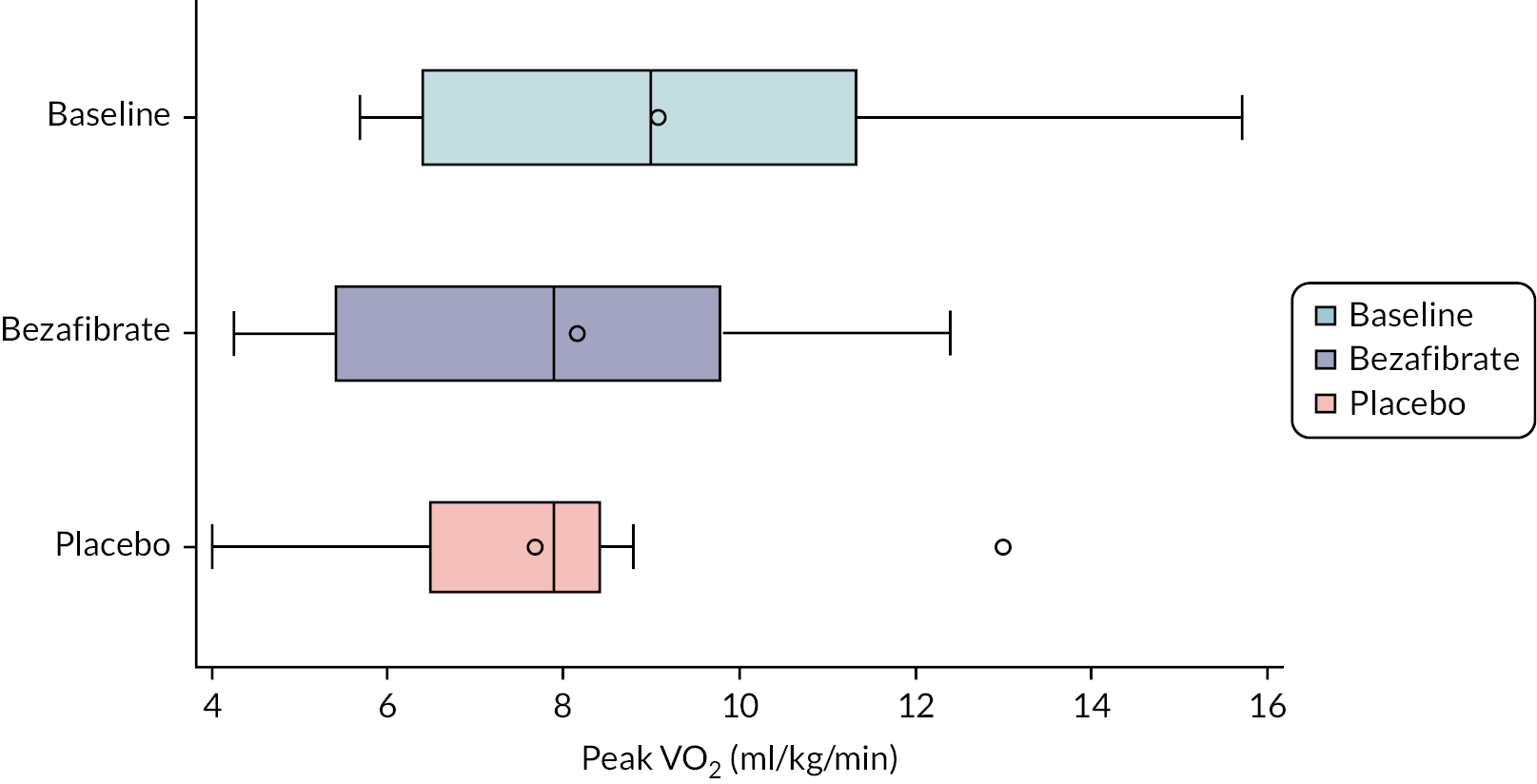
| Baseline (ml/kg/min) | Bezafibrate (ml/kg/min) | Placebo (ml/kg/min) | |
|---|---|---|---|
| Baseline | First period | Second period | |
| N | 11 | 11 | 11 |
| Mean (SD) | 9.10 (3.13) | 8.16 (2.65) | 7.69 (2.26) |
| Median (IQR) | 9.0 (6.4–11.3) | 7.9 (5.4–9.8) | 7.9 (6.5–8.4) |
| N | 11 | 11 | 11 |
| Mean (SD) | 9.10 (3.13) | 7.52 (2.44) | 8.34 (2.44) |
| Median (IQR) | 9.0 (6.4–11.3) | 7.9 (5.4–8.8) | 7.9 (6.5–9.8) |
| Effect | Level | Estimate (95% CI), ml/kg/min | p-value |
|---|---|---|---|
| Treatment effect | Bezafibrate vs. placebo | 0.40 (−0.90 to 1.70) | 0.5285 |
| Period effect | Second vs. first period | 0.78 (−0.51 to 2.08) | 0.2216 |
The effect of bezafibrate compared to placebo on pre-exercise VO2 was also estimated, as for peak VO2. There was no statistically significant effect of bezafibrate (see Table 9). Peak VO2 was 0.40 ml/kg/min lower (95% CI −0.90 to 1.60 ml/kg/min; p = 0.53) with bezafibrate compared to placebo. Measurements taken in the second period were similar to the first period (0.78 ml/kg/min, 95% CI −0.51 to 2.08 ml/kg/min; p = 0.22). There was no evidence of a difference in effect depending on the sequence (p = 0.95) and no evidence of an interaction between treatment and period (p = 0.76).
Cardiac function
Cardiac function was measured in several ways. Secondary outcomes for which bezafibrate and placebo measurements were formally compared were:
-
echocardiography: LVEFecho% at rest
-
echocardiography: LV peak systolic mean longitudinal strain at rest, peak exercise and after 2 minutes’ recovery
-
echocardiography: LV peak systolic mean circumferential strain at rest, peak exercise and after 2 minutes’ recovery
-
MRI: LVEFMRI% at rest
-
MRI: RVEFMRI% at rest.
There were missing values for most parameters in each period due to suboptimal and varying echocardiography windows in individual patients. The numbers of observations for each summary statistic are included in the tables. Echocardiography parameters were not collected at the first baseline clinic after 2 minutes’ recovery.
Ventricular ejection fraction
Ventricular EF was measured at rest both by echocardiography (LVEFecho%) and by MRI (LVEFMRI% and RVEFMRI%). Figure 6 and Table 10 summarise these measurements, by allocation.
FIGURE 6.
Box plots of LVEF% and RVEF% parameters at rest. Top: LVEFecho%. Middle: LVEFMRI%. Bottom: RVEFMRI%.
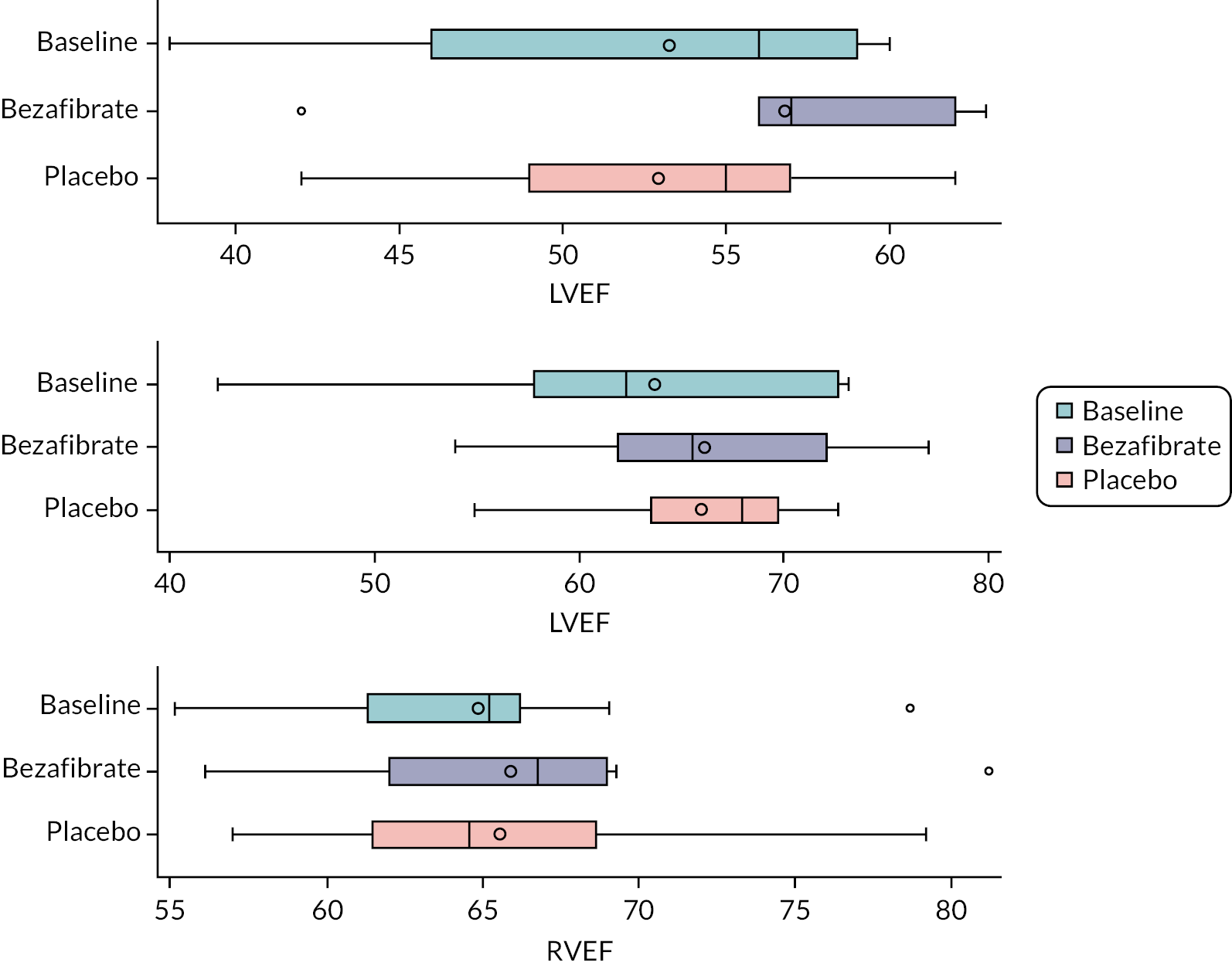
| Baseline | Bezafibrate | Placebo | |
|---|---|---|---|
| LVEFecho% | |||
| N | 11 | 11 | 11 |
| Mean (SD) | 53.27 (7.43) | 56.82 (5.60) | 52.91 (6.19) |
| Median (IQR) | 56.0 (46.0–59.0) | 57.0 (56.0–62.0) | 55.0 (49.0–57.0) |
| LVEFMRI% | |||
| N | 11 | 10 | 10 |
| Mean (SD) | 63.68 (9.28) | 66.11 (6.82) | 65.96 (5.54) |
| Median (IQR) | 62.3 (57.8–72.7) | 65.5 (61.9–72.1) | 68.0 (63.5–69.7) |
| RVEFMRI% | |||
| N | 11 | 10 | 10 |
| Mean (SD) | 64.85 (6.17) | 65.87 (7.27) | 65.55 (6.13) |
| Median (IQR) | 65.2 (61.3–66.2) | 66.8 (62.0–69.0) | 64.6 (61.5–68.6) |
The results of fitting regression models to compare bezafibrate and placebo for these parameters are summarised in Table 11. For LVEFecho% there was a borderline statistically significant effect of bezafibrate in comparison with placebo (3.72, 95% CI −0.26 to 7.69; p = 0.065). There was no evidence of a period effect (p = 0.2788). For LVEFMRI% there was no evidence of a difference between bezafibrate and placebo (0.15, 95% CI −3.21 to 3.51; p = 0.926). There was similarly no evidence of a difference between bezafibrate and placebo for RVEFMRI% (0.32, 95% CI −2.32 to 2.96; p = 0.801). There was no evidence of a period effect for either model (p = 0.436 and p = 0.552).
| Factor | Factor level | Estimate (95% CI) | p-value |
|---|---|---|---|
| LVEFecho% | |||
| Treatment effect | Bezafibrate vs. placebo | 3.72 (−0.26 to 7.69) | 0.065 |
| Period effect | Second vs. first period | 2.12 (−1.86 to 6.09) | 0.279 |
| LVEFMRI% | |||
| Treatment effect | Bezafibrate | 0.15 (−3.21 to 3.51) | 0.926 |
| Period effect | Second vs. first period | 1.27 (−2.09 to 4.63) | 0.436 |
| RVEFMRI% | |||
| Treatment effect | Bezafibrate | 0.32 (−2.32 to 2.96) | 0.801 |
| Period effect | Second vs. first period | 0.76 (−1.88 to 3.40) | 0.552 |
Other echocardiography outcomes
Figures 7–9 and Tables 12–14 summarise longitudinal and circumferential strain and diastolic ratio at rest, peak exercise and after 2 minutes’ recovery, by allocation. By consensus, more negative strain values are considered to indicate better heart function.
FIGURE 7.
Box plots of longitudinal and circumferential strain and diastolic ratio at rest.
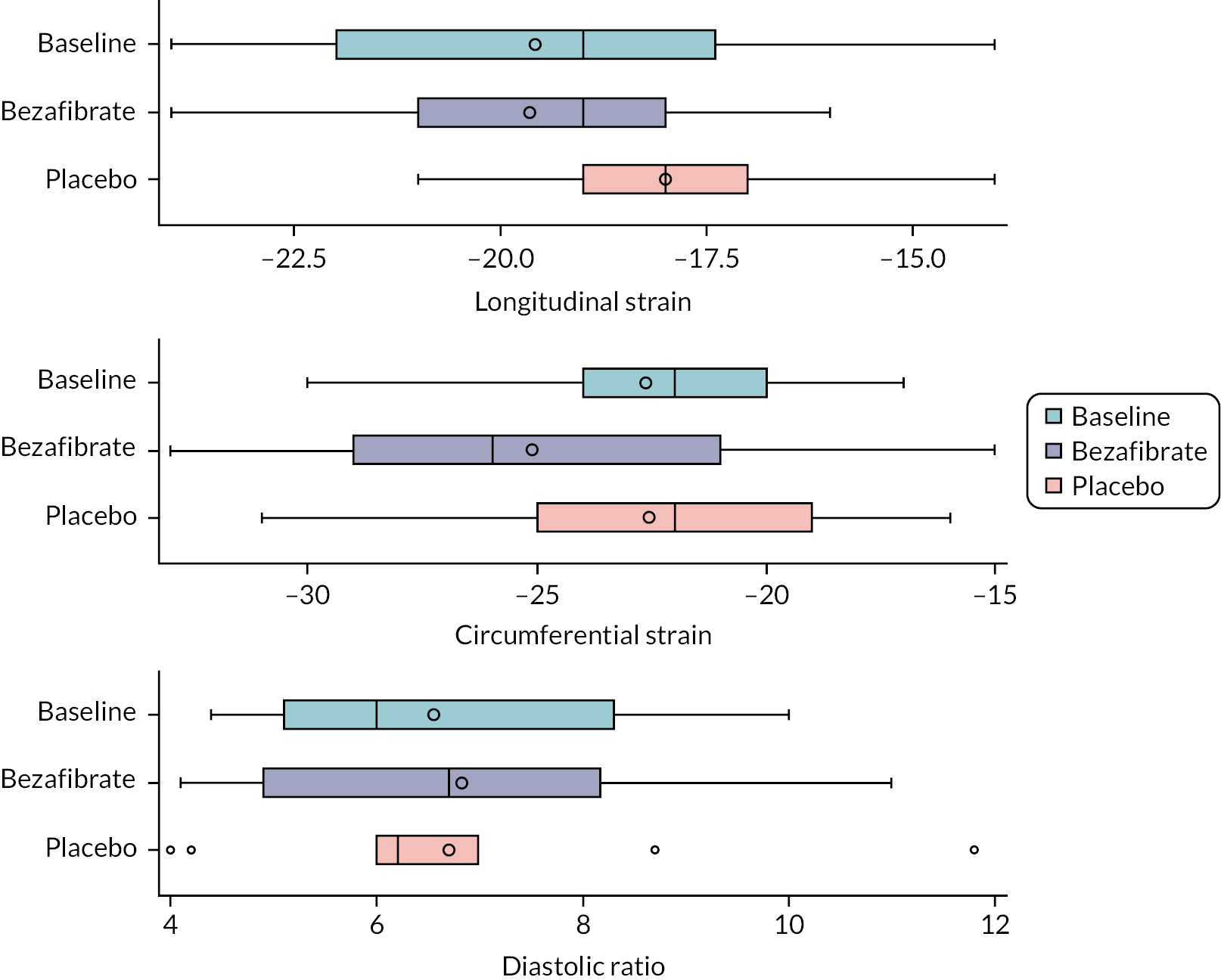
FIGURE 8.
Box plots of longitudinal and circumferential strain and diastolic ratio at peak exercise.
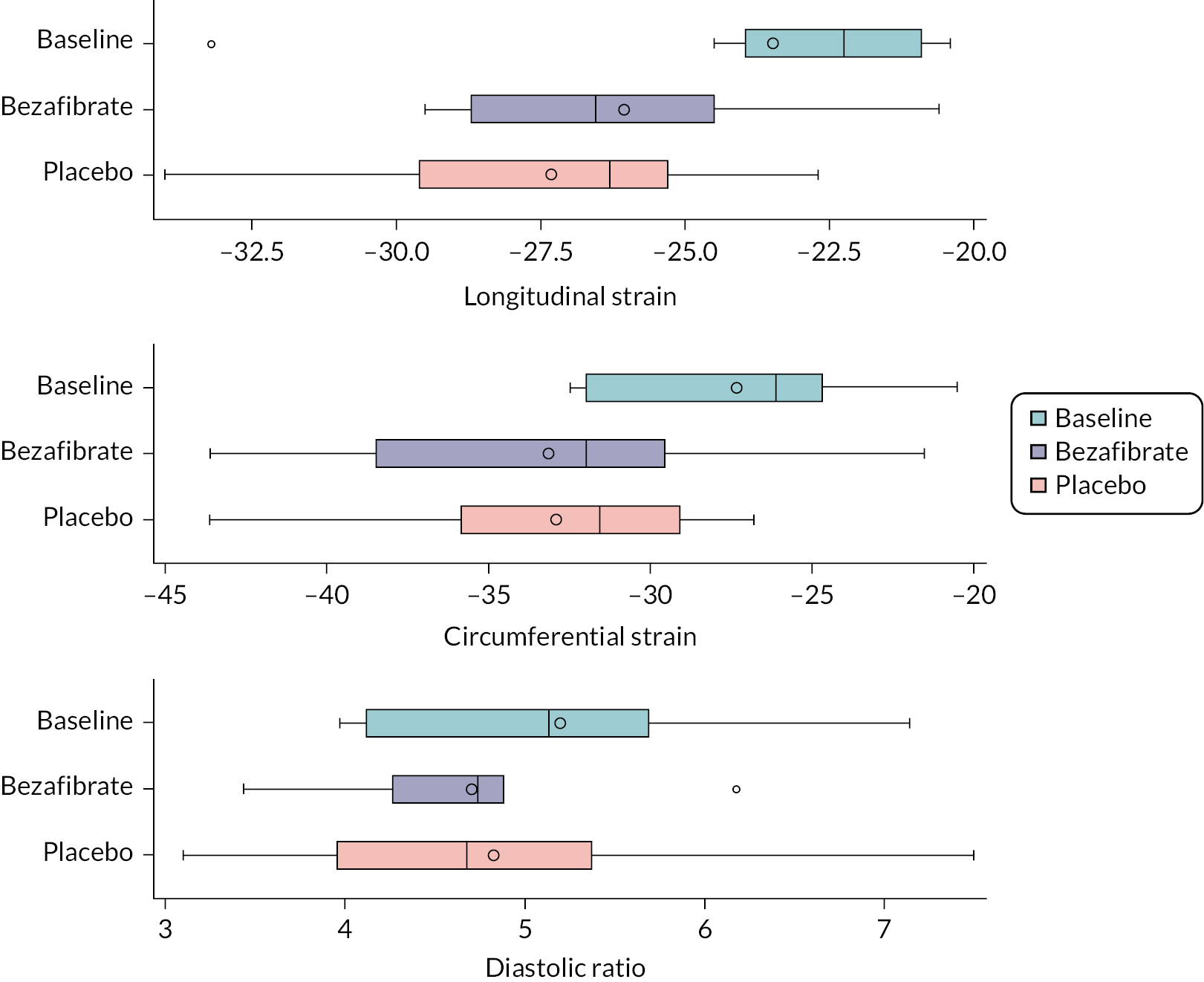
FIGURE 9.
Box plots of longitudinal and circumferential strain and diastolic ratio after 2 minutes’ recovery.
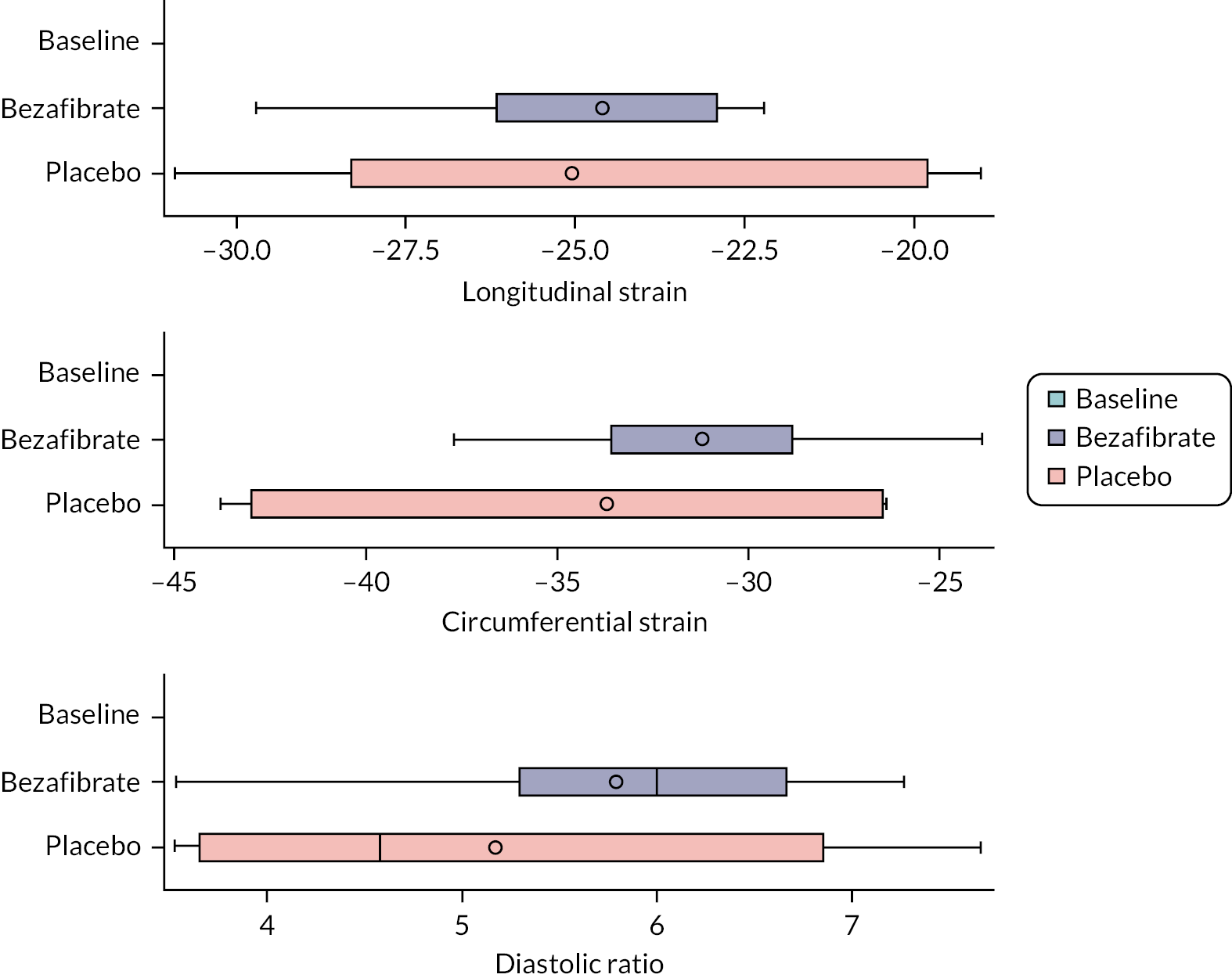
| Baseline | Bezafibrate | Placebo | |
|---|---|---|---|
| LV systolic longitudinal strain | |||
| N | 11 | 11 | 11 |
| Mean (SD) | −19.58 (3.05) | −19.64 (2.11) | −18.00 (2.05) |
| Median (IQR) | −19.0 (−22.0 to −17.4) | −19.0 (−21.0 to −18.0) | −18.0 (−19.0 to −17.0) |
| LV systolic circumferential strain | |||
| N | 11 | 11 | 11 |
| Mean (SD) | −22.64 (3.44) | −25.09 (5.15) | −22.55 (4.32) |
| Median (IQR) | −22.0 (−24.0 to −20.0) | −26.0 (−29.0 to −21.0) | −22.0 (−25.0 to −19.0) |
| Diastolic ratio: MV E/LV E’ | |||
| N | 11 | 11 | 10 |
| Mean (SD) | 6.55 (1.87) | 6.82 (2.26) | 6.70 (2.24) |
| Median (IQR) | 6.0 (5.1–8.3) | 6.7 (4.9–8.2) | 6.2 (6.0–7.0) |
| Baseline | Bezafibrate | Placebo | |
|---|---|---|---|
| LV systolic longitudinal strain | |||
| N | 8 | 6 | 9 |
| Mean (SD) | −23.48 (4.17) | −26.07 (3.46) | −27.32 (3.39) |
| Median (IQR) | −22.3 (−24.0 to −20.9) | −26.6 (−28.7 to −24.5) | −26.3 (−29.6 to −25.3) |
| LV systolic circumferential strain | |||
| N | 8 | 8 | 8 |
| Mean (SD) | −27.34 (4.40) | −33.14 (7.26) | −32.93 (5.49) |
| Median (IQR) | −26.2 (−32.0 to −24.7) | −32.0 (−38.5 to −29.6) | −31.6 (−35.9 to −29.1) |
| Diastolic ratio: MV E/LV E’ | |||
| N | 6 | 6 | 8 |
| Mean (SD) | 5.20 (1.21) | 4.71 (0.90) | 4.83 (1.35) |
| Median (IQR) | 5.1 (4.1–5.7) | 4.7 (4.3–4.9) | 4.7 (4.0–5.4) |
| Baseline | Bezafibrate | Placebo | |
|---|---|---|---|
| LV systolic longitudinal strain | |||
| N | 0 | 8 | 7 |
| Mean (SD) | – | −24.60 (2.57) | −25.04 (4.57) |
| Median (IQR) | – | −23.4 (−26.2 to −22.9) | −26.1 (−28.3 to −19.8) |
| LV systolic circumferential strain | |||
| N | 0 | 8 | 7 |
| Mean (SD) | – | −31.21 (4.28) | −33.70 (7.20) |
| Median (IQR) | – | −31.6 (−33.6 to −28.9) | −31.6 (−43.0 to −26.5) |
| Diastolic ratio: MV E/LV E’ | |||
| N | 0 | 9 | 8 |
| Mean (SD) | – | 5.79 (1.21) | 5.17 (1.73) |
| Median (IQR) | – | 6.0 (5.3–6.7) | 4.6 (3.7–6.9) |
Measurements for longitudinal and circumferential strain and diastolic ratio were modelled at rest, peak exercise and after 2 minutes’ recovery to compare bezafibrate and placebo. The results are summarised in Tables 15–17. The difference in longitudinal strain at rest between bezafibrate and placebo (see Table 15) was statistically significant (−1.67, 95% CI −3.11 to −0.22; p = 0.026). There was no evidence of a period effect (p = 0.635). Similarly, the difference in circumferential strain at rest between bezafibrate and placebo was statistically significant (−2.72, 95% CI −5.03 to −0.40; p = 0.024), with no evidence of a period effect (p = 0.105). There was no difference in diastolic ratio between bezafibrate and placebo [1.01 (i.e. a 1% increase in diastolic ratio for bezafibrate), 95% CI 21% reduction to 30% increase; p = 0.931] and no period effect (p = 0.727).
| Factor | Factor level | Estimate (95% CI) | p-value |
|---|---|---|---|
| Longitudinal strain | |||
| Treatment effect | Bezafibrate vs. placebo | −1.67 (−3.11 to −0.22) | 0.026 |
| Period effect | Second vs. first period | 0.33 (−1.11 to 1.78) | 0.635 |
| Circumferential strain | |||
| Treatment effect | Bezafibrate | −2.72 (−5.03 to −0.40) | 0.024 |
| Period effect | Second vs. first period | 1.88 (−0.43 to 4.20) | 0.105 |
| Diastolic ratio a | |||
| Treatment effect | Bezafibrate | 1.01 (0.79 to 1.30) | 0.931 |
| Period effect | Second vs. first period | 0.96 (0.75 to 1.23) | 0.727 |
There were no differences in longitudinal strain, circumferential strain or diastolic ratio at peak exercise between bezafibrate and placebo (see Table 16): longitudinal strain (2.02, 95% CI −2.08 to 6.11; p = 0.305); circumferential strain (0.28, 95% CI −5.34 to 5.91; p = 0.915); diastolic ratio (0.99, 1% decrease in diastolic ratio for bezafibrate, 95% CI 23% decrease to 27% increase; p = 0.948). There were also no period effects but a sequence effect for diastolic ratio: participants who received placebo first had on average a 27% increase in diastolic ratio compared to those who received bezafibrate first.
| Factor | Factor level | Estimate (95% CI) | p-value |
|---|---|---|---|
| Longitudinal strain | |||
| Treatment effect | Bezafibrate vs. placebo | 2.02 (−2.08 to 6.11) | 0.305 |
| Period effect | Second vs. first period | 3.77 (−0.24 to 7.77) | 0.063 |
| Circumferential strain | |||
| Treatment effect | Bezafibrate | 0.28 (−5.34 to 5.91) | 0.915 |
| Period effect | Second vs. first period | 1.98 (−3.64 to 7.61) | 0.460 |
| Diastolic ratio a | |||
| Treatment effect | Bezafibrate | 0.99 (0.77 to 1.27) | 0.948 |
| Period effect | Second vs. first period | 1.06 (0.83 to 1.36) | 0.603 |
| Sequence effect (period 1 – period 2) | Placebo → bezafibrate vs. Bezafibrate → placebo | 1.27 (1.03 to 1.56) | 0.031 |
There were no differences in longitudinal strain, circumferential strain or diastolic ratio after 2 minutes’ recovery between bezafibrate and placebo (Table 17): longitudinal strain (0.30, 95% CI −3.84 to 4.43; p = 0.866); circumferential strain (1.88, 95% CI −4.24 to 7.99; p = 0.484); diastolic ratio (1.10, 10% increase in diastolic ratio for bezafibrate, 95% CI 12% decrease to 37% increase; p = 0.344). There was no evidence of a period effect for any of the measures.
| Factor | Factor level | Estimate (95% CI) | p-value |
|---|---|---|---|
| Longitudinal strain | |||
| Treatment effect | Bezafibrate vs. placebo | 0.30 (−3.84 to 4.43) | 0.866 |
| Period effect | Second vs. first period | −2.03 (−6.16 to 2.11) | 0.275 |
| Circumferential strain | |||
| Treatment effect | Bezafibrate | 1.88 (−4.24 to 7.99) | 0.484 |
| Period effect | Second vs. first period | −1.74 (−7.86 to 4.37) | 0.514 |
| Diastolic ratio a | |||
| Treatment effect | Bezafibrate | 1.10 (0.88 to 1.37) | 0.344 |
| Period effect | Second vs. first period | 0.89 (0.71 to 1.10) | 0.226 |
Arrhythmia profile
Arrhythmia profile was assessed using at rest and during exercise at the end of each period. All participants had data for all three time points, and all participants had sinus rhythm at rest and during exercise for all three time points.
Metabolic function (magnetic resonance spectroscopy)
Magnetic resonance spectroscopy was only carried out at the end of the second period for 10 participants (one participant had missing MRS data).
Phosphocreatine/adenosine triphosphate ratio in cardiac muscle
The PCr/ATP ratio was not collected due to problems with collecting MRS data, so this outcome could not be analysed.
Skeletal muscle oxidative function
Figure 10 and Table 18 summarise the Tau and Qmax by treatment for the second period.
FIGURE 10.
Box plots of MRS skeletal muscle oxidative function in the second period by allocation.
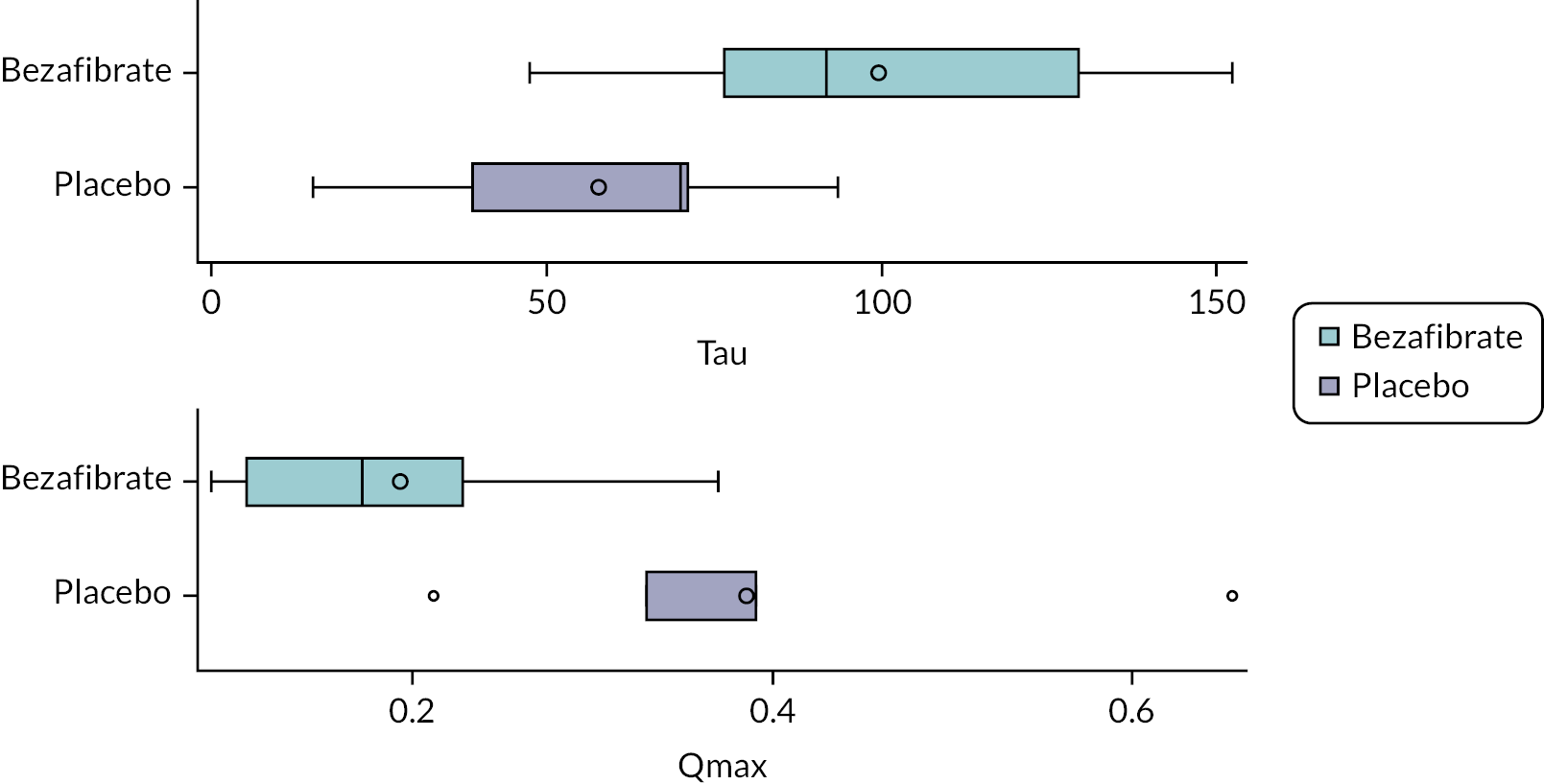
| Bezafibrate | Placebo | |
|---|---|---|
| Tau | ||
| N | 5 | 5 |
| Mean (SD) | 99.47 (41.79) | 57.71 (30.80) |
| Median (IQR) | 91.8 (76.4–129.4) | 69.9 (38.9–71.1) |
| Q max | ||
| N | 5 | 5 |
| Mean (SD) | 0.19 (0.11) | 0.39 (0.16) |
| Median (IQR) | 0.2 (0.1–0.2) | 0.3 (0.3–0.4) |
Chapter 5 Results: in vivo blood samples, laboratory outcomes
Monolysocardiolipin/tetralinoleoyl-cardiolipin ratio
The MLCL/L4-CL ratio was calculated from MLCL and CLm data collected as part of a panel of cardiolipin data points. In addition, the MLCL + CLi/CLm ratio was also calculated post hoc. Each participant had at least one replicate (median of 2) for each period, hence varying numbers of observations in the tables below. Figures 11 and 12 and Table 19 summarise the distributions of MLCL/L4-CL ratio by allocation and period.
FIGURE 11.
Box plot of MLCL/L4-CL ratio by allocation.
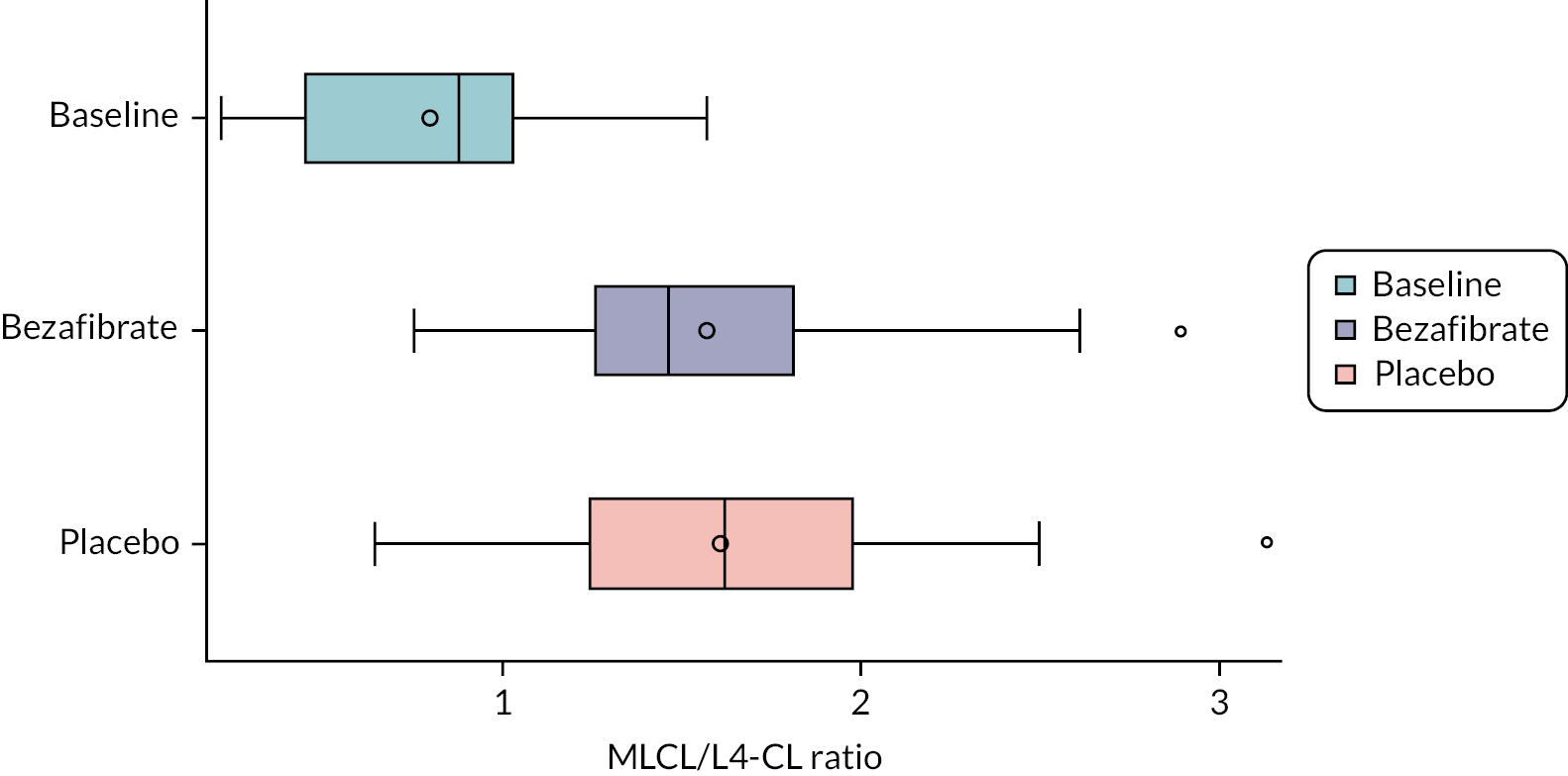
FIGURE 12.
Box plot of MLCL/L4-CL ratio by period.
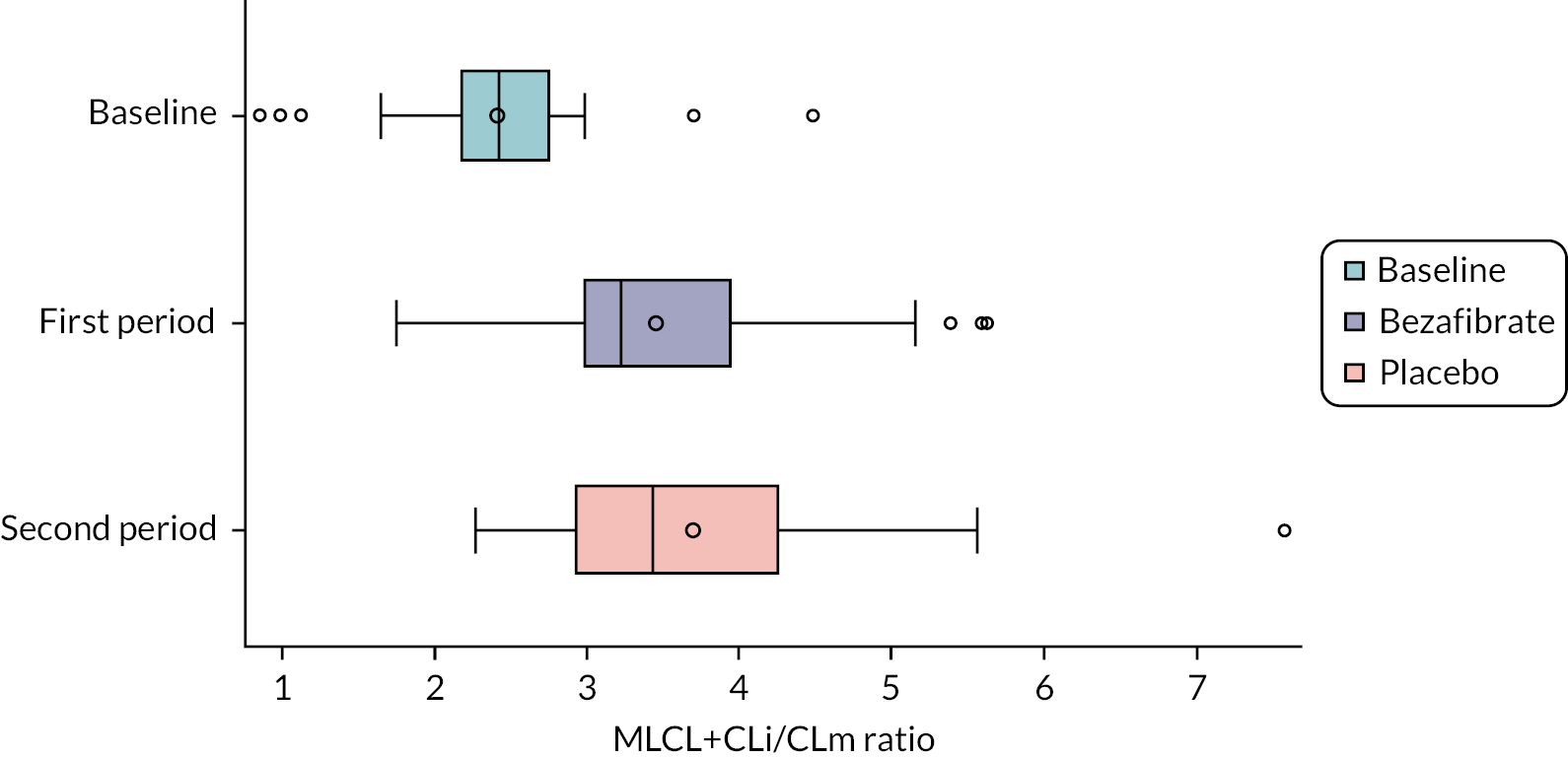
| Baseline | Bezafibrate | Placebo | |
|---|---|---|---|
| Baseline | First period | Second period | |
| N | 18 | 38 | 37 |
| Mean (SD) | 0.797 (0.370) | 1.567 (0.501) | 1.604 (0.493) |
| Median (IQR) | 0.877 (0.445–1.027) | 1.461 (1.258–1.816) | 1.618 (1.242–1.974) |
| N | 18 | 36 | 39 |
| Mean (SD) | 0.797 (0.370) | 1.472 (0.523) | 1.691 (0.447) |
| Median (IQR) | 0.877 (0.445–1.027) | 1.369 (1.162–1.692) | 1.641 (1.371–2.001) |
Monolysocardiolipin/L4-CL ratio was modelled on the log base 10 scale (due to the non-normal distribution and to aid interpretation) to formally compare bezafibrate and placebo. Effect estimates for this outcome are therefore geometric mean ratios. The results of the model are summarised in Table 20. There was no significant difference in the MLCL/L4-CL ratio between bezafibrate and placebo (5% reduction in MLCL/L4-CL ratio for bezafibrate compared with placebo, 95% CI 19% reduction to 11% increase; p = 0.53). However, there was evidence of a period effect, with the MLCL/L4-CL ratio estimated to be 20% higher in the second period compared to the first period (95% CI 3% increase to 40% increase; p = 0.02).
| Factor | Factor level | Estimatea (95% CI) | p-value |
|---|---|---|---|
| Treatment effect | Bezafibrate vs. placebo | 0.95 (0.81 to 1.11) | 0.531 |
| Period effect | Second vs. first period | 1.20 (1.03 to 1.40) | 0.024 |
Absolute neutrophil count
All participants had absolute neutrophil counts collected for all periods. Figure 13 shows absolute neutrophil counts by allocation and Table 21 summarises counts by allocation and period.
FIGURE 13.
Box plot of absolute neutrophil counts by treatment.
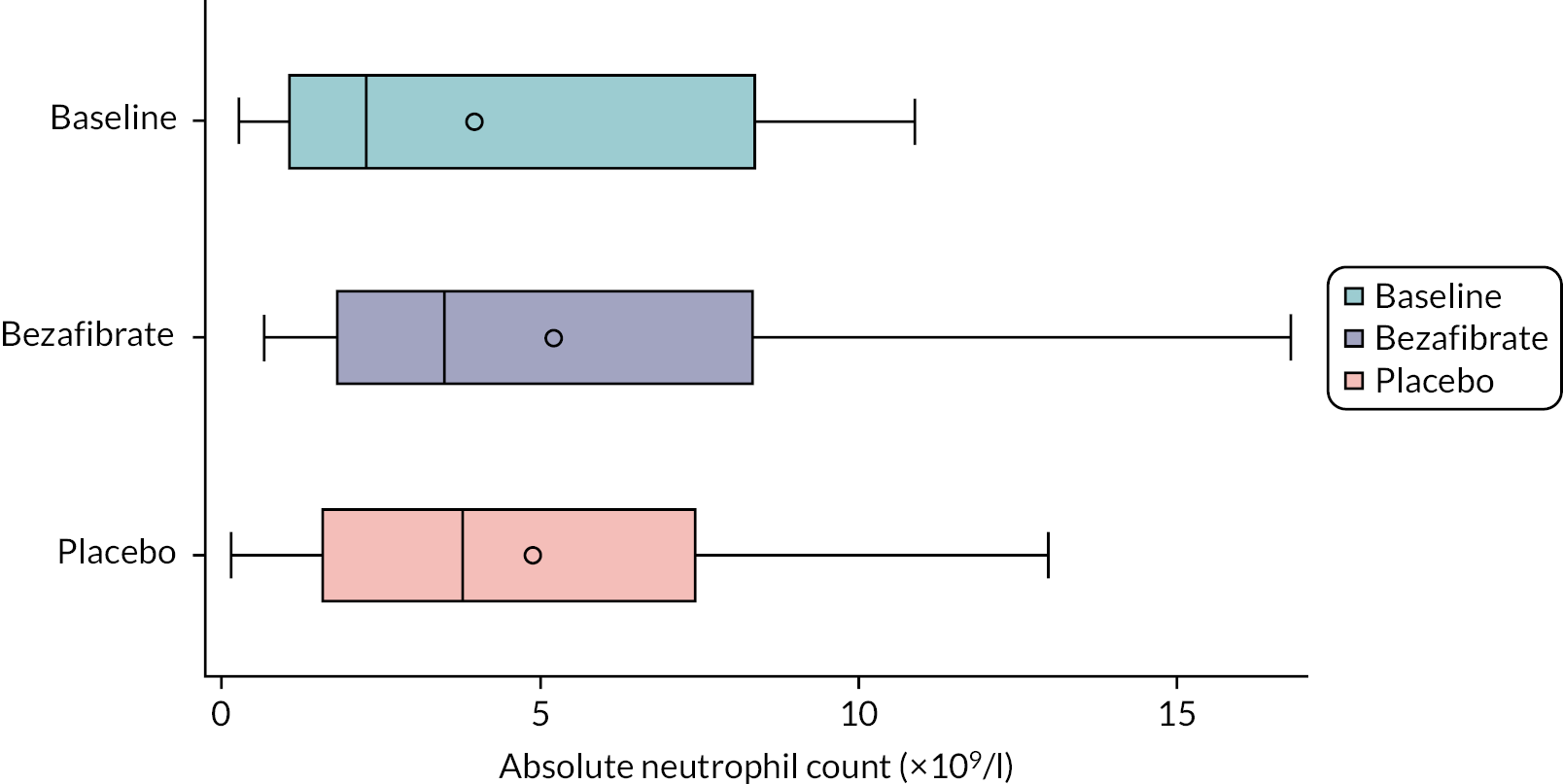
| Baseline | Bezafibrate | Placebo | |
|---|---|---|---|
| Baseline | First period | Second period | |
| N | 11 | 11 | 11 |
| Mean (SD), ×109/l | 3.95 (3.83) | 5.21 (5.10) | 4.89 (4.37) |
| Median (IQR), ×109/l | 2.3 (1.1–8.4) | 3.5 (1.8–8.3) | 3.8 (1.6–7.4) |
| N | 11 | 11 | 11 |
| Mean (SD), ×109/l | 3.95 (3.83) | 3.92 (3.70) | 6.19 (5.35) |
| Median (IQR), ×109/l | 2.3 (1.1–8.4) | 3.5 (1.0–5.2) | 3.8 (1.9–11.6) |
The model comparing absolute neutrophil count between bezafibrate and placebo is shown in Table 22. There was no evidence of a difference between bezafibrate and placebo (0.11 ×109/l, 95% CI −2.07 to 2.30 × 109/l; p = 0.915). However, there was a statistically significant difference between periods with the second period having a higher average count (2.26 × 109/l, 95% CI 0.08 to 4.45 × 109/l; p = 0.043).
| Factor | Level | Estimate (95% CI) | p-value |
|---|---|---|---|
| Treatment effect | Bezafibrate vs. placebo | 0.11 (−2.07 to 2.30) | 0.915 |
| Period effect | Second vs. first period | 2.26 (0.08 to 4.45) | 0.043 |
Amino acid levels
Amino acid expression was assessed by measuring plasma arginine and plasma cysteine levels. All participants had both plasma values collected for all periods. Figure 14 shows these levels by allocation and Table 23 summarises them by allocation and period.
FIGURE 14.
Box plots of arginine and plasma levels by allocation.
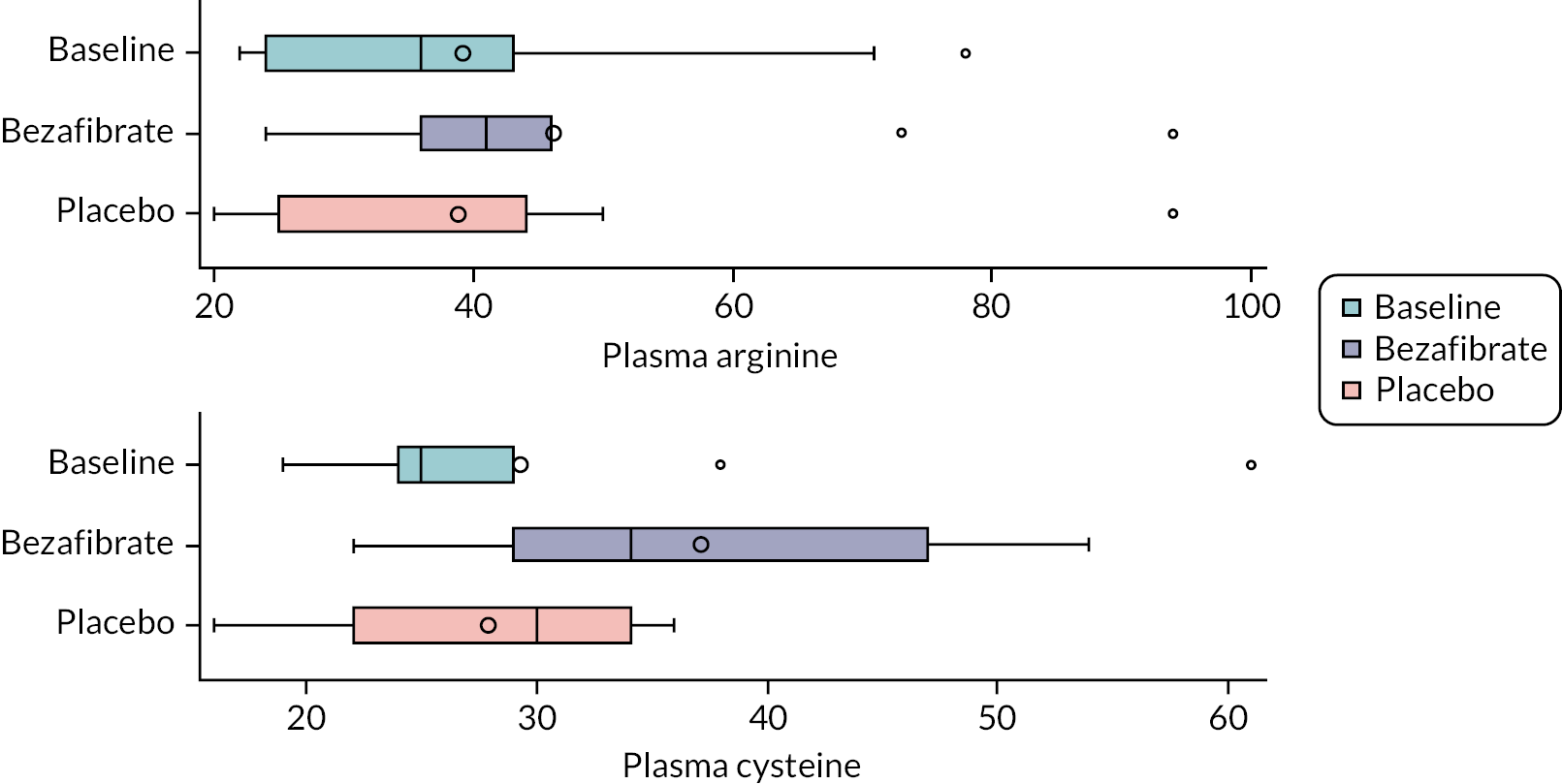
| Baseline | Bezafibrate | Placebo | |
|---|---|---|---|
| Baseline | First period | Second period | |
| Plasma arginine | |||
| N | 11 | 11 | 11 |
| Mean (SD), microMol/L | 39.18 (19.02) | 46.09 (20.09) | 38.82 (20.78) |
| Median (IQR), microMol/L | 36.0 (24.0–43.0) | 41.0 (36.0–46.0) | 31.0 (25.0–44.0) |
| Plasma cysteine | |||
| N | 11 | 11 | 11 |
| Mean (SD), microMol/L | 29.27 (11.66) | 37.18 (11.28) | 27.91 (6.80) |
| Median (IQR), microMol/L | 25.0 (24.0–29.0) | 34.0 (29.0–47.0) | 30.0 (22.0–34.0) |
| Plasma arginine | |||
| N | 11 | 11 | 11 |
| Mean (SD), microMol/L | 39.18 (19.02) | 48.09 (26.54) | 36.82 (9.50) |
| Median (IQR), microMol/L | 36.0 (24.0–43.0) | 40.0 (30.0–73.0) | 41.0 (26.0–43.0) |
| Plasma cysteine | |||
| N | 11 | 11 | 11 |
| Mean (SD), microMol/L | 29.27 (11.66) | 32.82 (11.05) | 32.27 (9.92) |
| Median (IQR), microMol/L | 25.0 (24.0–29.0) | 34.0 (25.0–41.0) | 30.0 (25.0–34.0) |
Regression models compared arginine and cysteine levels on log scales between bezafibrate and placebo (Table 24). There was no difference in plasma arginine level between bezafibrate and placebo (24% increase with bezafibrate, 95% CI 10% reduction to 72% increase; p = 0.180) and no evidence of a period effect (p = 0.228). However, there was strong evidence of a difference in plasma cysteine level between bezafibrate and placebo (32% increase with bezafibrate, 95% CI 8% to 62%; p = 0.001). There was no evidence of a period effect (p = 0.813).
| Factor | Level | Estimate (95% CI)a | p-value |
|---|---|---|---|
| Plasma arginine | |||
| Treatment effect | Bezafibrate vs. placebo | 1.24 (0.90 to 1.72) | 0.180 |
| Period effect | Second vs. first period | 0.82 (0.59 to 1.14) | 0.228 |
| Plasma cysteine | |||
| Treatment effect | Bezafibrate vs. placebo | 1.32 (1.08 to 1.62) | 0.001 |
| Period effect | Second vs. first period | 0.98 (0.80 to 1.20) | 0.813 |
Lymphocyte mitochondria measurements
Lymphocyte mitochondria were characterised in several ways: mitochondrial size, number and total area of mitochondria per lymphocyte, area of mitochondria as proportion of cytoplasm, and mitochondrial content (Mitotracker) and mitochondrial membrane potential (TMRE) in lymphocytes. Each patient had multiple lymphocytes (~20) assessed at each visit, and each lymphocyte had four or five mitochondria. As specified by the SAP, mitochondria measurements are descriptive and not formally compared. These outcomes are summarised in Tables 25 and 26 and Figure 15. They were assessed as averages for varying numbers of mitochondria and lymphocytes per participant and time; therefore denominators vary to some extent.
| Baseline | Bezafibrate | Placebo | |
|---|---|---|---|
| Mitochondrial size | |||
| N | 883 | 1145 | 1097 |
| Mean (SD) | 0.159 (0.114) | 0.153 (0.111) | 0.150 (0.103) |
| Median (IQR) | 0.126 (0.084–0.194) | 0.127 (0.079–0.192) | 0.126 (0.086–0.181) |
| Mitochondrial number | |||
| N | 220 | 215 | 234 |
| Mean (SD) | 4.01 (3.23) | 5.33 (4.07) | 4.70 (3.12) |
| Median (IQR) | 3.0 (2.0–6.0) | 5.0 (2.0–7.0) | 4.0 (2.0–6.0) |
| Total area of mitochondria per lymphocyte | |||
| N | 220 | 215 | 234 |
| Mean (SD) | 0.636 (0.550) | 0.815 (0.636) | 0.705 (0.475) |
| Median (IQR) | 0.527 (0.205–0.971) | 0.675 (0.343–1.189) | 0.618 (0.289–1.016) |
| Area of mitochondria as proportion of cytoplasm | |||
| N | 220 | 215 | 234 |
| Mean (SD) | 4.958 (3.846) | 5.950 (4.100) | 5.709 (3.464) |
| Median (IQR) | 4.224 (2.067–7.589) | 5.070 (2.954–8.415) | 5.348 (2.860–8.139) |
| Outcome | Baseline | Bezafibrate | Placebo |
|---|---|---|---|
| Mitotracker Green FM MFI in monocytes | |||
| N | 7a | 11 | 11 |
| Mean (SD) | 135.8 (53.6) | 491.4 (214.9) | 302.7 (157.6) |
| Median (IQR) | 117.0 (89.9–198.0) | 373.0 (335.0–733.0) | 273.0 (216.0–385.0) |
| FITC MFI in lymphocytes using Mitotracker by allocation | |||
| N | 4b | 11 | 9c |
| Mean (SD) | 140.3 (51.8) | 711.6 (557.6) | 645.3 (407.8) |
| Median (IQR) | 123.0 (109.5–171.0) | 699.0 (261.0–1053.0) | 506.0 (371.0–916.0) |
| [TMRE MFI] – [TMRE + FCCP MFI] in monocytes | |||
| N | 7d | 11 | 11 |
| Mean (SD) | 11,129 (11,446) | 7861 (9019) | 14,025 (15,108) |
| Median (IQR) | 11,792 (320–18,617) | 3182 (1361–12,957) | 10,465 (117–26,148) |
| [TMRE MFI] – [TMRE + FCCP MFI] in lymphocytes | |||
| Mean (SD) | 1376 (2053) | 7385 (10,576) | 19,463 (16,561.2) |
| Median (IQR) | 561 (66–2687) | 1607 (622–12,031) | 17,942 (1719–35,096) |
| N | 4e | 11 | 9f |
FIGURE 15.
Box plot of mitochondrial content (Mitotracker) and mitochondrial membrane potential (TMRE) by allocation.
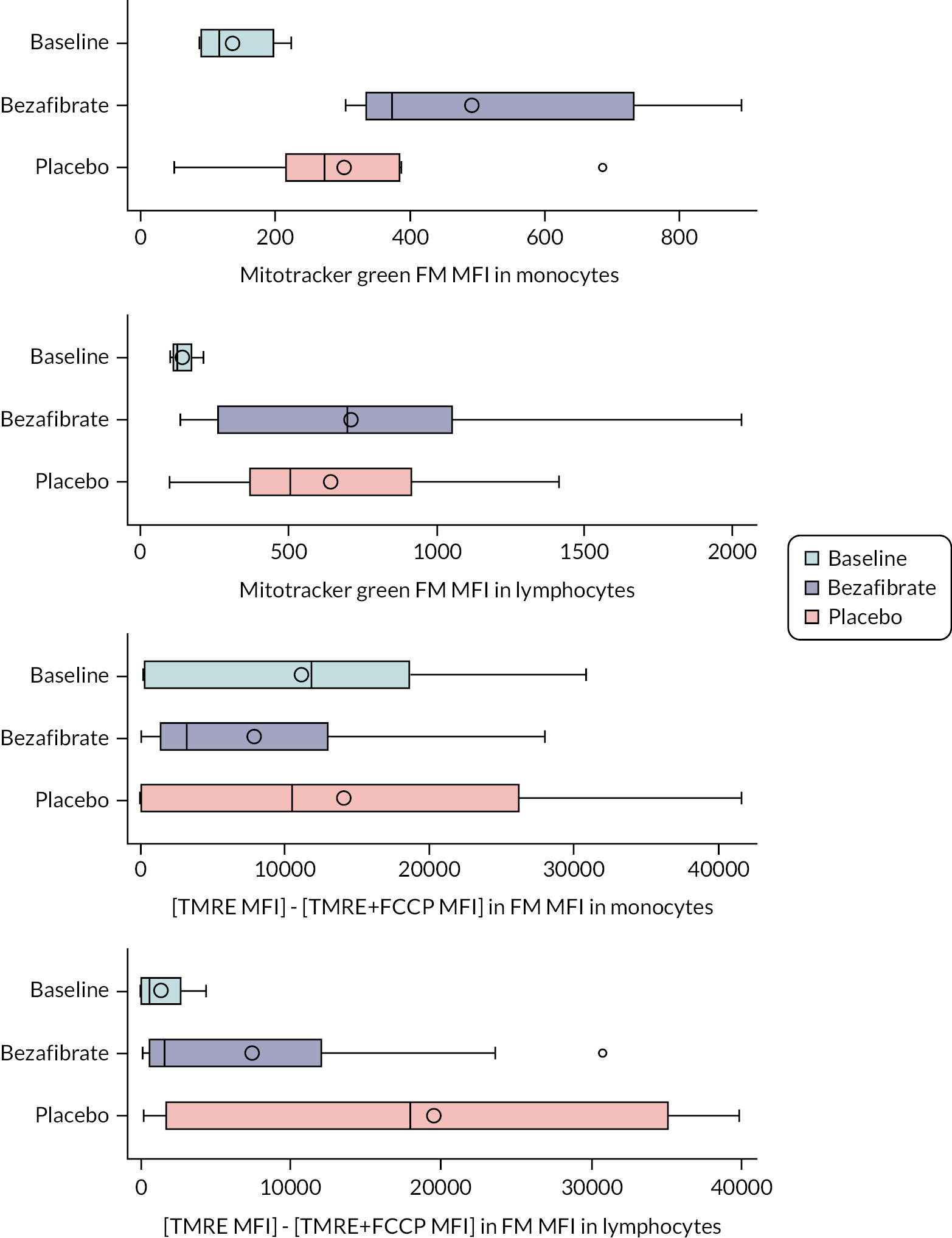
Chapter 6 Results: quality of life
Quality of life
Scores were calculated according to the PedsQL scoring manual. The young child report used a three-point scoring scale (0: not at all, 2: sometimes, 4: a lot), and others used a Likert five-point scoring scale (0: never, 1: almost never, 2: sometimes, 3: often, 4: almost always) where answers to the questions are transformed as follows: 0 = 100, 1 = 75, 2 = 50, 3 = 25, 4 = 0. When high numbers were worse outcomes in the questionnaire, this transformation made higher transformed numbers better outcomes (on a 0–100 scale).
Figure 16 and Table 27 summarise QoL scores by allocation. Table 28 presents the model results for the effect of treatment on core and fatigue scores respectively. Bezafibrate had no effect on core (−3.41, 95% CI −8.67 to 1.86; p = 0.192) or fatigue (1.71, 95% CI −6.28 to 9.71; p = 0.659) QoL scores for patients. There was no evidence of a period effect for either model.
FIGURE 16.
Box plots of QoL scores for the core and fatigue domains for patients and parents by allocation.
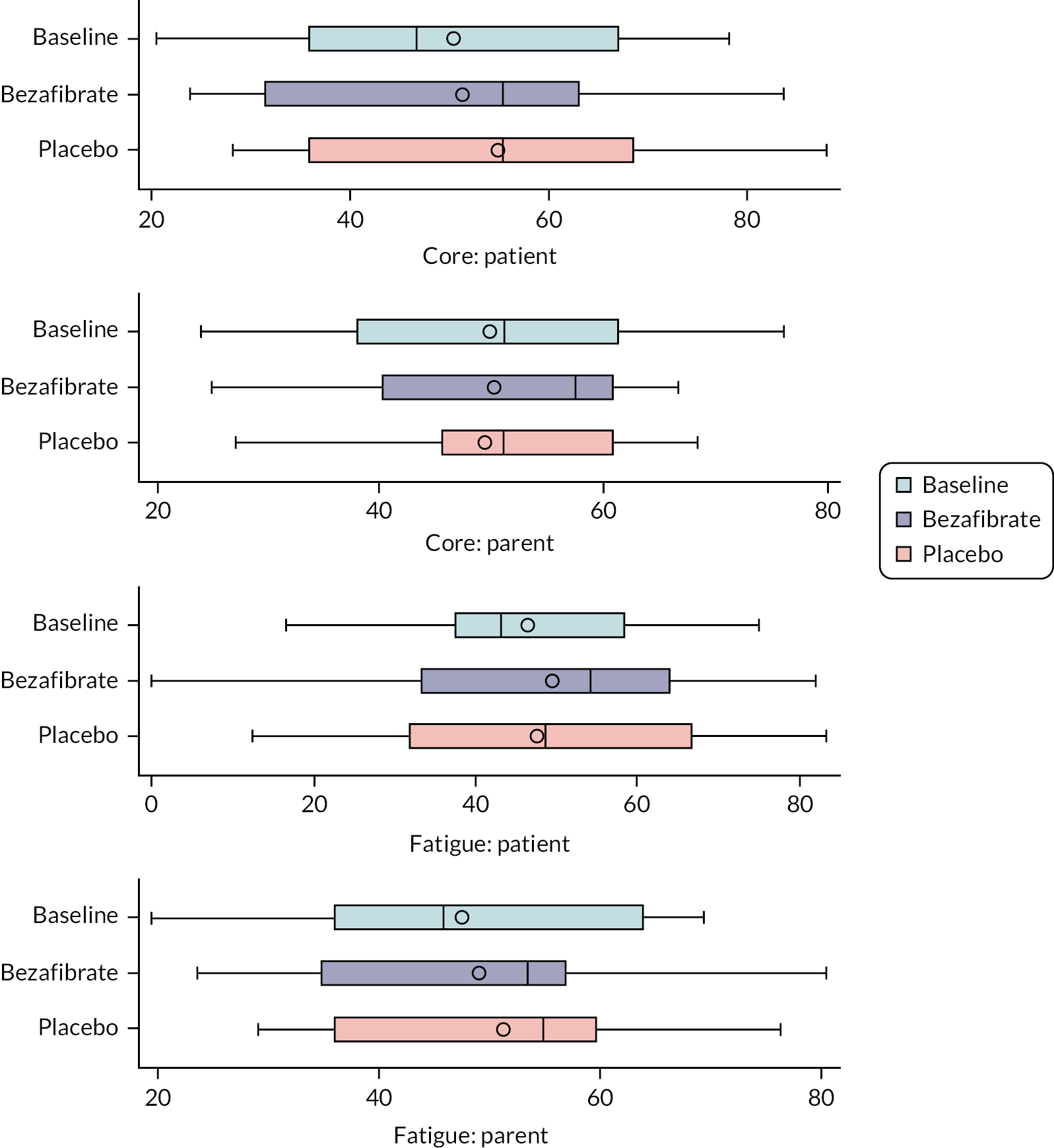
| Baseline | Bezafibrate | Placebo | |
|---|---|---|---|
| Core: patient scores | |||
| N | 11 | 11 | 11 |
| Mean (SD) | 50.5 (20.1) | 51.4(20.2) | 54.8 (18.6) |
| Median (range) | 46.8 (20.5–78.3) | 55.4 (23.9–83.7) | 55.4 (28.3–88.0) |
| IQR | 35.9–67.0 | 31.5–63.0 | 35.9–68.5 |
| Core: parent scores a | |||
| N | 10 | 10 | 10 |
| Mean (SD) | 49.8 (17.6) | 50.1 (15.3) | 49.3 (15.9) |
| Median (range) | 51.1 (23.9–76.1) | 57.6 (25.0–66.7) | 51.1 (19.6–68.5) |
| IQR | 38.0–61.4 | 40.2–60.9 | 45.7–60.9 |
| Fatigue: patient scores | |||
| N | 11 | 11 | 11 |
| Mean (SD) | 46.4 (16.6) | 49.5 (22.6) | 47.6 (21.0) |
| Median (range) | 43.1 (16.7–75.0) | 54.2 (0.0–81.9) | 48.6 (12.5–83.3) |
| IQR | 37.5–58.3 | 33.3–63.9 | 31.9–66.7 |
| Fatigue: parent scores a | |||
| N | 10 | 10 | 10 |
| Mean (SD) | 47.5 (16.5) | 49.028 (16.8) | 51.3 (15.1) |
| Median (range) | 45.8 (19.4–69.4) | 53.5 (23.6–80.6) | 55.0 (29.2–76.4) |
| IQR | 36.1–63.9 | 34.7–56.9 | 36.1–59.7 |
| Factor | Factor level | Estimate (95% CI) | p-value |
|---|---|---|---|
| Core domain | |||
| Treatment effect | Bezafibrate vs. placebo | −3.41 (−8.67 to 1.86) | 0.192 |
| Period effect | Second vs. first period | −0.58 (−5.85 to 4.69) | 0.820 |
| Fatigue domain | |||
| Treatment effect | Bezafibrate vs. placebo | 1.71 (−6.28 to 9.71) | 0.659 |
| Period effect | Second vs. first period | 1.99 (−6.00 to 9.98) | 0.6082 |
Chapter 7 Results: patient experience/qualitative research
Face-to-face semistructured interviews were conducted with all 11 participants (Table 29). The interviews were conducted at two time points: one in the final week of each treatment phase. All interviews were conducted while families were attending the CRIC centre for clinical assessment for the study. The interviews took place between July and December 2019.
| ID | Phase 1 – month (interviewed with) | Phase 2 – month (interviewed with) | Total interviews |
|---|---|---|---|
| 01 | July (alone) | November (alone) | 2 |
| 02 | July (mother/father) | November (father) | 2 |
| 03 | July (alone) | November (alone) | 2 |
| 04 | July (with mother) | November (with mother) | 2 |
| 05 | July (alone) | November (with father) | 2 |
| 06 | July (alone) | November (alone) | 2 |
| 07 | July (mother/father) | November (with mother) | 2 |
| 08 | July (with mother/father) | November (with father) | 2 |
| 09 | July (with father) | December (with father) | 2 |
| 10 | July (with mother) | December (with mother) | 2 |
| 11 | July (with mother) | November (with mother) | 2 |
| Total interviewees | 4 interviewed alone, 8 with parents | 3 interviewed alone, 9 with parents | 22 |
Participants aged under 14 years at time of study were interviewed with at least one parent, and participants above 14 years were interviewed alone or with a parent if preferred.
Emerging themes
Themes were generated for the participants’ experience of the trial; for example, acceptability and feasibility, impact of growth and development on trial conduct and outcome, adherence to trial treatment, perceived symptoms of Barth syndrome, and communication with the trial team. There were also overarching themes that had implications for participation in the trial but were also related to individuals’ and families’ lived experience of Barth syndrome (Figure 17). These themes represent the context of living with Barth syndrome and the extent to which participants were able to commit to the demands of the trial and adhere to the treatment. These themes include perceived symptoms of Barth syndrome, family and social support, existing treatment for Barth syndrome management and coping strategies, social functioning and integration, schooling and Barth syndrome, employment opportunities and Barth syndrome, social comparison and peers.
FIGURE 17.
Breakdown of emerging themes relating to the lived experience of Barth syndrome and participation in trial.
Phase 1 emerging themes
-
Reason for trial participation.
-
Adherence to trial treatment.
-
Acceptability and conduct of trial.
-
Communication with trial team.
-
Physical assessment and monitoring.
-
Travel and accommodation.
-
Perceived benefits of trial.
Phase 2 emerging themes
-
Adherence to trial treatment.
-
Acceptability and conduct of trial.
-
Communication with trial team.
-
Physical assessment and monitoring.
-
Travel and accommodation.
-
Perceived efficacy of trial.
-
Impact of growth and development.
It should be noted that this document will report on the themes relating to participation in the trial and the overarching themes that are pertinent to families’ participation in the trial. Themes relating to the lived experience of Barth syndrome are documented in a separate report.
Reasons for participation in trial
Participants were asked for their reasons for participation in the trial. In addition to seeking a therapeutic benefit from the trial drug in the management of Barth syndrome, most participants were involved for altruistic reasons and wanted to give something back to the medical community that had supported them and their families:
I do my part to help myself and to help all the others who are in the same situation. Because it’s not a nice situation, but it is manageable and it becomes more manageable the more things like this we do. So, this is why we do them.
03, phase 2
Participation in the trial was also seen as a positive action for those reaching adulthood and taking steps towards responsibility for their health:
It’s just helped me learn to take charge of my health and stuff, because usually, I just go in with my parents, but I was at that stage where I just need to learn to be more independent and take more control, so the trial’s helped a lot in doing that. It’s just helped a lot really, just helped me with my independence and just learning to do things on my own …
06, phase 2
However, some parents were concerned that they had consented to their child being subjected to more blood tests and monitoring than they already receive, and this was associated with guilt:
So, no, the trial overall, apart from, obviously, the time frames that it takes to do it, I’d have to say my emotion was, ‘Oh my God I’ve said we would be involved in this, and I’ve therefore put him forward and he needs to have as many bloods done as this, and this is horrendous’, but again we’ve kind of ironed that out. So, kind of guilt-free now, you know. The rest of it we kind of look forward to seeing whether it’s something that can be implemented long-term.
011, phase 1, mother
Expectations and perceived benefits of the trial
Overall, there was a desire to be realistic, with a perception that there would unlikely be a measurable benefit from the treatment and that any perceived benefit would be a bonus. There was also acknowledgement of the idiosyncratic nature of Barth syndrome suggesting that not all participants would necessarily gain the same benefit from bezafibrate:
I mean it may work absolutely fantastically for one person and that’s amazing. If it works for one person, it’s done its job. But unfortunately, it may not do it for someone else. But I think the fact that we’re trying it and the fact we’ve been given the opportunity to try it, we’d be stupid not to. I mean I know there are quite a lot of people that have either turned down the trial for whatever reason it may be but I honestly think if there is a chance to actually …
01, phase 1
Surprisingly, few participants/families expressed concerns regarding potential risks or side effects of bezafibrate, despite it not being specifically developed for managing Barth syndrome:
With this trial, me and his mother wanted to be part of it, but you always have in the back of your mind the risks involved. Of course, with this drug, a lot of those risks were immediately taken away because it’s not an experimental drug, it’s just the way the drug is being used. It might be slightly different to what it was intended for. We would still want to be involved in that, but if it was an experimental drug and there were still, shall we say, more inherent risks and after effects, we would have to listen much more to what the boys had to say about that. If either of them said, ‘No, I don’t fancy that,’ we’d have to obviously take that into consideration.
05, phase 2, father
Initially, there was a low expectation that participation would yield a positive outcome for symptoms. However, for some, participation became an ‘emotional investment’ that needed to be met with a change in subjective symptoms or other objective measures of outcome:
I’m genuinely actually looking forward to the next month to see what happens. It’s like you’ve researched, ‘This has happened’, but when you’re actually living the research study, it is a far more emotional investment. Personally, I started going, ‘If it works or if it doesn’t, it’s just another tablet’. But now I’ve got to this point, I’m there going, ‘OK, now I want to know symptoms. I want to know results. I want to see correlations’.
01, phase 1
Some participants also articulated their understanding of the putative mechanism of action of bezafibrate:
I believe it is. I think the science of it [bezafibrate] is it lowers lipids in the blood to … and it makes your mitochondria work a little easier because of it and … Well, I’ve been on it for – well, I’ve been on a drug; whether it’s the placebo, or not, I don’t know, but I haven’t felt any difference. I haven’t had any improvements. I haven’t had anything go wrong. It’s just been very stale. The same, same.
03, phase 1
Some parents expressed that a perceived benefit from their sons’ participation in the trial might be tempered by seasonal variation and increased risk of infection. However, there was also an assumption that the efficacy of the trial medication would manifest itself through observable physical signs:
It’s hard to say because we started it as he was coming out of the winter phase. So, over winter he’s very tired, because he picks up everything. He had a lot of time off before Christmas because he was poorly. Then, it’s the summer stage, so it’s hard to say, but he is always going, ‘Look at my muscles,’ he’s always done that. We never distract him, but whether it’s true or not, obviously the tests will tell and we’ll figure it out. He is coming into his summer stage where he does have a lot more energy, anyway.
04, phase 1, mother
This extract is taken from Searle et al. 34 This is an open-access article distributed under the terms of the Creative Commons CC-BY licence, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Parents also appreciated that the participation in the trial gave them opportunities to meet with other families and discuss their concerns regarding Barth syndrome:
I think unless the families build up their own networks between them, obviously this is an essential hub and everything else and I understand the whole purpose of it is not to become a social event, but it’s useful for some of the feedback because when they do meet up once a year and have these gatherings a lot of parents then talk to each other and actually share common problems with their children, where they’re able to discuss and say, ‘Oh, we were told to try this’ and it’s learning from learning, second-degree learning, that is passed on from the team here. So those things are always useful. It does feel slightly detached, but that’s not to say that when we’re here the care isn’t great.
08, phase 1, father
Adherence with trial treatment
Participants generally reported high levels of adherence with the trial treatment. The tablet was considered easy to swallow and was not an issue with regard to its size and taste:
Been fine, and actually he’s always been able to swallow the tablets because he takes them anyway, so we’ve not had an issue with the size of the tablet as well, just to point that out, or taste or anything like that.
08, phase 1, mother
In fact, it was not seen as an additional burden as most participants were taking other medications routinely. During the interviews, there were few reported side effects. One parent reported a rash which could be attributed to other causes:
Yes, I mean from the actual trial itself, I think it’s been pretty straightforward. We haven’t missed a dose or anything. There’s no real pressure to think, ‘Oh my God, we’ve got this extra thing to do’, because we have to administer medicines morning and evening anyway so it’s just another box. There hasn’t been an impact on there at all. I think the first week, he developed a rash. We were also away on holiday, so it was trying to attribute whether that was to do with the sun cream, whether it was to do with the sun, the weather, the seawater, or the trial. So that was the only little bump in the road. Other than that, it was plain sailing.
08, phase 1, father
Most parents of younger participants adopted a ‘no-nonsense’ policy towards medication adherence and this was something entrenched in family life:
He’s very adherent with medication because since he was a tiny baby, we’ve had a no-nonsense policy … He’s taken every dose of this trial all the way through, including the dose where he managed to drop a tablet down the side of the bed and we actually hunted out the tablet. So, we didn’t give it to him, but we hunted it out, so we didn’t lose it. He’s very good with medicine.
010, phase 2, mother
Yes, there’s four of your regular pills and then the two trial pills on top. And then you can stick them – organise them – for the week and just take them, and it makes absolutely no difference taking them, at his age, and they’re so small. It’s not a problem.
02, phase 2, mother
Only one family reported making errors in administering medication:
We prepare it and he just takes it and it wasn’t because of him. What happened was I picked up the wrong bottle and it said on it, the first ones were, ‘Take two once a day’, and when I was preparing the next lot, I think I must have picked up the old bottle and only prepared, for that week, just morning meds, not morning and afternoon meds. So, I think it’s about eight days.
07, phase 1, mother
Older boys were given the responsibility to take medication, although they were prompted by parents:
As I say, the wife should be here rather than me I suppose but, yes, she asks him regularly, I ask him regularly, he always says yes. I think he has missed a few, and I think he said he’s missed about four or five. Apart from that, yes, I believe him, he’s got no reason to lie I don’t think.
09, phase 1, father
Finally, one participant stated that he found it harder to adhere to medication since he had started to work nights:
Well, it’s been, sort of, hard because the first lot of medication I took, I wasn’t working or anything, but the second lot, I started working, so it’s been hard. Obviously, I used to sleep during the day but now, I work nights, so I’m asleep in the day anyway. But if I wasn’t taking any tablets, I’d be able to work or, like, keep up with it.
09, phase 2
Perceived efficacy of trial treatment
Most participants were in clinical equipoise about their treatment status during phase 1 of the trial but were anticipating noticing a difference once they had completed phase 2:
Well when I first started taking it, well I actually thought it was like maybe like helping, but I wasn’t sure if that was my mind playing tricks or if it actually was helping. But further along I’m thinking I’m not really sure if it is. So yeah, I guess I’ll know if I take the other tablets, see if that will make me feel any different again.
09, phase 1
Due to the clinical nature of Barth syndrome and the close social network associated with the condition, it was inevitable that participants would discuss their perceptions of treatment with each other and potentially raise their expectations for therapeutic benefit. However, an awareness of the extent of individual differences was also acknowledged:
But I know, speaking to some of the others, that they feel it’s had an effect, which is brilliant. But I think it is going to be a bit like Barth syndrome. It does affect everyone individually. You can’t categorise it.
01, phase 1
Although some participants were not able to subjectively determine a therapeutic benefit, some were curious to know whether they had fared better on other quantitative measures such as QoL:
I mean, honestly, from the first stage to the washout period there was almost no change, and then back to this set that I’m on now. Also very little change. It’s not a medication that I’m on and I feel like, ‘Wow. This is amazing’, but there might be and I am very curious to see if there are readings that can be taken that are slightly better, or if results I’ve given in quality of life tests would be better … If I’m honest, no. I don’t feel any different, one from the other, to not being on it either.
03, phase 2
Subjectively, participants were not able to identify a treatment benefit following either phase of the trial; however, some parents observed behavioural differences in their sons in the first phase of the trial:
What we have noticed – and this, again, just could be pure coincidence – that his appetite, I think, has improved, definitely, I think, but it was already improving. It’s just continued. So there haven’t been any negatives that we can see so … We were at the [other hospital] last week and they told us that his heart had made quite a significant improvement from the last scan a couple of months ago, so that’s the only thing I can say in terms of figures and actually reporting lines.
08, phase 1, mother
I know when he first started he was taking the dogs for a walk, obviously the weather’s been really hot lately so we haven’t been taking the dogs out, so I don’t know. In my opinion he is still lethargic. I don’t want to say he doesn’t do enough because that might be unfair to say, obviously I’m not with him 24 hours a day, but he’s lying around a lot, constantly tired. I know when he first started taking it after a while he said, ‘I think I’m on the real stuff here.’
09, phase 1, father
Health-wise, we haven’t noticed any tiredness or extra tiredness. He’s got low energy levels anyway, but we haven’t noticed any dip in that. We don’t think the drugs, or the placebo, whichever it may be this time, has affected him in that way. No upset stomachs, nothing like that.
05, phase 2, father
One participant reported feeling and looking stronger due to being in the trial. However, at 14 years of age, it is difficult to determine whether this was a result of natural growth and development. Also, this participant did not distinguish between experiences at both phases of the trial.
I think since I’ve been on the drugs it’s kind of changed me as I am. Like, from the beginning to now, it’s just like changed me, the experience and that … because I’ve never really done anything like this before. And the drugs, I feel like the drugs have changed me as well … Because like, the first one I felt stronger. And I looked more stronger.
04, phase 2
What we’ve been told is that in Barth boys, they can have very rapid growth spurts over quite a short period of time, which can have problems with the skeletal muscular system as well because the bones are growing but the ligaments and things aren’t growing as quickly.
02, phase 1, mother
Some participants referred to experiencing side effects that dissipated in the washout period:
Again, she’s [nurse?] seen what we presume are side effects of the drug at the first stage when I was feeling run down and lethargic, and all of that. But she said that was the only thing that she’s witnessed during the first – before the washout. ‘You were really, really, really ill. Wash out. Now you’re fine.’ But she said, ‘Other than that, you know, behaviour-wise, you haven’t been any different.’
01, phase 2
One parent alluded to signs that their son may have received active treatment due to him experiencing a rash – although this happened in both treatment phases:
I kind of think he possibly started on it [active treatment] and now he’s on the placebo. That’s what my wife thinks, just because of the rash. And when he goes onto a new medication he always – usually breaks out with something. But then he would have done it with the other one as well, because it still contains something in there, there’s taste flavours, there’s other things as well, components in the ingredients. So, personally, I don’t know. I was probably leaning towards the second time, only because, when we did phone up about something when we were in [XXXX], there seemed to be more urgency in getting back to us and more concern, than there was the first time, when he had the rash. However, as this is a blind test and no one knows, that doesn’t really mean anything because the actual person maybe was just a bit more diligent than they were the first time around, or maybe they had less work on and were able to react a little bit quicker.
08, phase 2, father
There were extraneous activities within the trial cohort that could potentially skew the results of the laboratory aerobic tests such as the bike and treadmill. For example, one participant had been referred to an exercise referral scheme by his general practitioner, and his father had noticed an increased capacity in heart and lung function:
Yes. I have noticed a difference and he’s not out of breath as much as he was. As I say, [Name], who takes him has kept it – he was on an exercise referral scheme and, obviously, because he knows [Name] needs it more, because of his problems. He’s got him on it longer than he normally would.
06, phase 1, father
Another participant had received cardiovascular training advice within the context of gym membership:
No weights or anything like that, but he’s worked out a routine for him for cardiovascular where he’s working his body but not putting too much strain on it and it’s beginning to reap benefits now, so if we can get that much off again, I think you’d be where you need to be then.
05, phase 2, father
Finally, one parent suggested that there was no ‘normality’ due to the impact of Barth syndrome, which makes it difficult to evaluate any benefits of participation in a trial:
And I think one of the community teams that once came to the house a few years ago, we were running through a whole load of things, and she did say, ‘This is your norm, but it’s not the norm.’ And there are just things that you don’t think of. Like, for example, having this interview now and you’re asking certain questions and so on. And I’m sure, if I had this particular survey e-mailed in a week’s time, I’d have something completely different to say. It’s whatever is constant in your head and you’re trying to, again, distinguish and break out this is a relation of maybe CARDIOMAN, this is a relation of Barth, this is just him being him, this is what normal teenagers or pre-teenagers go through.
08, phase 2, father
Acceptability and conduct of trial
All participants and families were accepting of the trial procedures, and the trial was generally considered to be well conducted during both phases. Participants and families also praised the professional attitude of staff:
I would say solely from the trial, and not any side effects of the drug or anything like that; solely based on all of this, I don’t think we could ask for more, literally. They have been extremely accommodating. They go out of their way to make sure we get our monthly phone calls, to make sure … Have there been any changes? You know, even if it’s a drug change, for example, then I know they need to know this, this, this, this, this. Dates. They’re on it. They’re absolutely on it. And what’s nice as well is like, I was saying to [Name], you come into a hospital environment, ‘Oh, I’ve got to go and have these tests. Oh, no. I’m getting my blood …’ But it doesn’t actually feel like you’re coming to a hospital appointment.
01, phase 2
However, some families experienced logistical issues with laboratory and blood tests:
Yes, so you’re under a bit of pressure, but there are 11 boys. If one boy doesn’t go through, the trial is through. Then, one blood came back … The timing has to be right. You get your bloods sorted out and then it’s shown … It’s important to do the timing right for the trial. The lab did what they were told. They sent the blood results to here. Here said, ‘Oh, the lab screwed up,’ but, actually, they hadn’t sent the right information totally for one of those blood things. That was true because I talked about it with [Name] and that. That sort of thing, but generally, the staff are full-on professional, like the blood takers here. The staff numbers are high, relative to how many are coming, so it’s like we’re spoilt. It’s a well-run thing. It’s very well run.
02, phase 1, mother
The same family felt that it would have been more efficient if the blood results for the trial had been shared with the BSS in Bristol:
We get a blood test during the trial in our local hospitals, to get your numbers of specific things in your blood [neutrophils]. And then the hospital e-mails them back to the trial people. But it would have been handier if those blood results were put to our – there’s a team in Bristol, that runs all the Barth kids, there’s a specialist service. And it would have been good if they passed on those results to the – there were some people coming, like me, and they’re going, ‘Do you know what my last neutrophil result was for last month and the month before?’ It would be handy, because it’s nice to know it when you’re at home.
02, phase 2, father
The trial, as usual, runs really smoothly. It’s fine. The people are lovely. The testing seems intelligent and efficient and I never feel like I’m like, ‘Why am I here?’ It’s concise. It’s MRI, bloods, exercise. Yes, it’s really clean, which is nice. Kind of amazing, considering it’s the first of its kind in the UK, full stop.
03, phase 2
Another parent suggested to his son that his confidence may have been affected due to wearing an oxygen mask and the sight of wires while being monitored on the bike:
[Parent to child]: Were you scared? When you’re doing it on the bike in there, you have all the wires and stuff and the oxygen mask. Was it the fact of everything being new and you feeling perhaps a bit scared? Sometimes if you’re scared or you’re not as confident as you could be, that can affect how you do things physically. It’s like football, you’ve got a footballer, trains day in, day out, he can do it, but when it comes to playing in front of 60,000 people, it’s a different matter.
05, phase 2, father
Some of the boys found the MRI scanner uncomfortable, noisy and claustrophobic, which was a source of distress for them:
Too warm in there. When it starts, I’m alright at first, but when it starts to get warmer – I get irritated and I start moving about and then … come out. It’s supposed to be about 20 minutes.
04, phase 2
I would say, especially with children like, who’ve got anxiety issues that, particularly on the first session, to make sure that they’re OK. Because he went in the MRI on his own. Because I wasn’t – I didn’t – I think that panicked him more than not being – he was more or less in floods of tears when he came out.
04, phase 2, mother
Because you feel like you’ve got to relax, and then you hear this little tick and then you just hear a really loud noise all of a sudden, and it feels like it’s getting louder and louder each time. And then it stops, and then it starts again.
08, phase 2
Communication and the Barth Syndrome Service
The wider aspects of living with Barth syndrome and communication with specialists both within and outside of the BSS were explored with the trial families.
We’ve got consultants in [place name]. We’ve got a paediatrician who is a local person and we see him at least once or twice a year. We have also seen a cardiologist, who’s just retired, so I think they’re thinking that we won’t now see a cardiologist in [place name]. We’ll just come for their annual clinics here and have a full cardiac screening here, which I’m not that happy about because it’s quite nice to know that someone is checking regularly for any changes, especially during this period of puberty and rapid growth.
02, phase 1, father
Although families were aware of the range of professionals involved in their child’s care in the UK, there was an appreciation of the close-knit Barth syndrome community from which they could also seek advice:
And there are some psychologists, and a haematologist, and a cardiologist. There’s everybody we could ever need. Every kind of ologist you could think of. Our community is very, very [laughter] close. Like, we all know each other. There are certain people we know. You could name any … because it’s just a very rare disease. There are like 200 of us known, only. And there are certain people you could just go – and it’s like, you could just say – you could pull out any name randomly and if it’s an affected individual, she’s like, ‘Oh, yes. That person lives over there in America,’ wherever they are, and it’s just … We’re very close, and very tight. We’re like a family, and it’s really nice.
03, phase 1
The qualitative interviews explored patient-centred effects on QoL (e.g. burdens and limitations that acute or chronic symptoms impose on daily lives). During both phases of the trial, participants were asked about any perceived differences in their QoL and whether they felt they had experienced benefits. Symptom experience was defined as disease-related symptoms and experiences that can be best assessed by and/or noticed only by the patient; therefore symptom experience is subjective. Although patients did not report any relief of symptoms that could be directly attributed to trial participation and/or active treatment, some parents reported subtle changes in their son’s daily functioning, symptoms or signs of Barth syndrome, such as increased stamina or improved muscle tone, that the patients had not reported and/or been subjectively aware of. However, these observed differences in well-being may not be directly attributable to the trial or the IMP being taken at the time and could be a function of extraneous and contextual variables such as seasonal variation. Furthermore, one of the major and potentially confounding issues regarding the therapeutic benefit of the trial treatment is the fact that most of the boys were reporting the psychological and emotional impact of living with Barth syndrome but are still developing physically and emotionally. Some of the boys were also engaging in physical activities that may have served to increase cardiovascular capacity and muscle development in the trial period. Finally, from a psychosocial perspective, the data suggest that participation in the trial served families well with regard to social support and was an opportunity for the boys to sustain friendships, socialise and share experiences of living with Barth syndrome and potential treatments.
Although the data have highlighted issues with needle phobia, MRI scanning and bicycle ergometry (due to short stature), none of the participants conveyed that such issues were a barrier to participation in the trial. Most participants and families found strategies for overcoming phobic experiences, and none reported any long-term issues in this context.
All patients were either in full-time education or work and, if living with their families, were not dependent on earning an income. Such dependency on families served to ensure retention in the study and good levels of adherence with medication. In fact, living with a child with Barth syndrome meant that families had to be highly organised about medication adherence, observing symptoms, and employing strategies for their daily functioning. For some families, this level of organisation placed restrictions on their type and location of employment, choice of school and impact on siblings.
Families’ experience of the professional staff (of both the BSS and the Clinical Trials Unit) and the facilities of the CRIC offered them a highly organised and effective infrastructure that optimised recruitment, retention and communication. Participants and families were pleased to be included in the trial and viewed their participation as a means of ‘giving back’ to the medical community. No participants withdrew from the trial and it was not perceived to be too onerous for families, even for those travelling long distances for study visits. Participating families found communicating with study staff easy.
Participation in the research gave both individuals and families a sense of responsibility for their condition. This sense of responsibility was partly driven by an appreciation of the BSS and its staff expertise and was displayed through their deference to the medical community.
Reflexivity statement
The qualitative researchers have extensive experience of illness and engagement with health services. However, the primary qualitative researcher (AS) was the interviewer and primary data analyst but had no prior experience of Barth syndrome or working within the BSS. Similarly, the second data analyst (GH) was also an experienced qualitative researcher with no prior experience of Barth syndrome or its treatment.
Chapter 8 Results: in vitro drug-treated biochemical outcomes
The MLCL/L4-CL ratio in vitro was calculated in triplicate from samples of cells incubated with control, bezafibrate or resveratrol. Figure 18 shows box plots summarising medians and IQRs for MLCL/L4-CL ratios by condition of incubation (i.e. control, bezafibrate or resveratrol) and shows the effects of incubating cells with bezafibrate or resveratrol compared to the control. Each drug reduced the MLCL/L4-CL ratio, but by only a small amount in relation to the ratio found in people without Barth syndrome.
FIGURE 18.
MLCL + CLi/CLm by in vitro drug application.
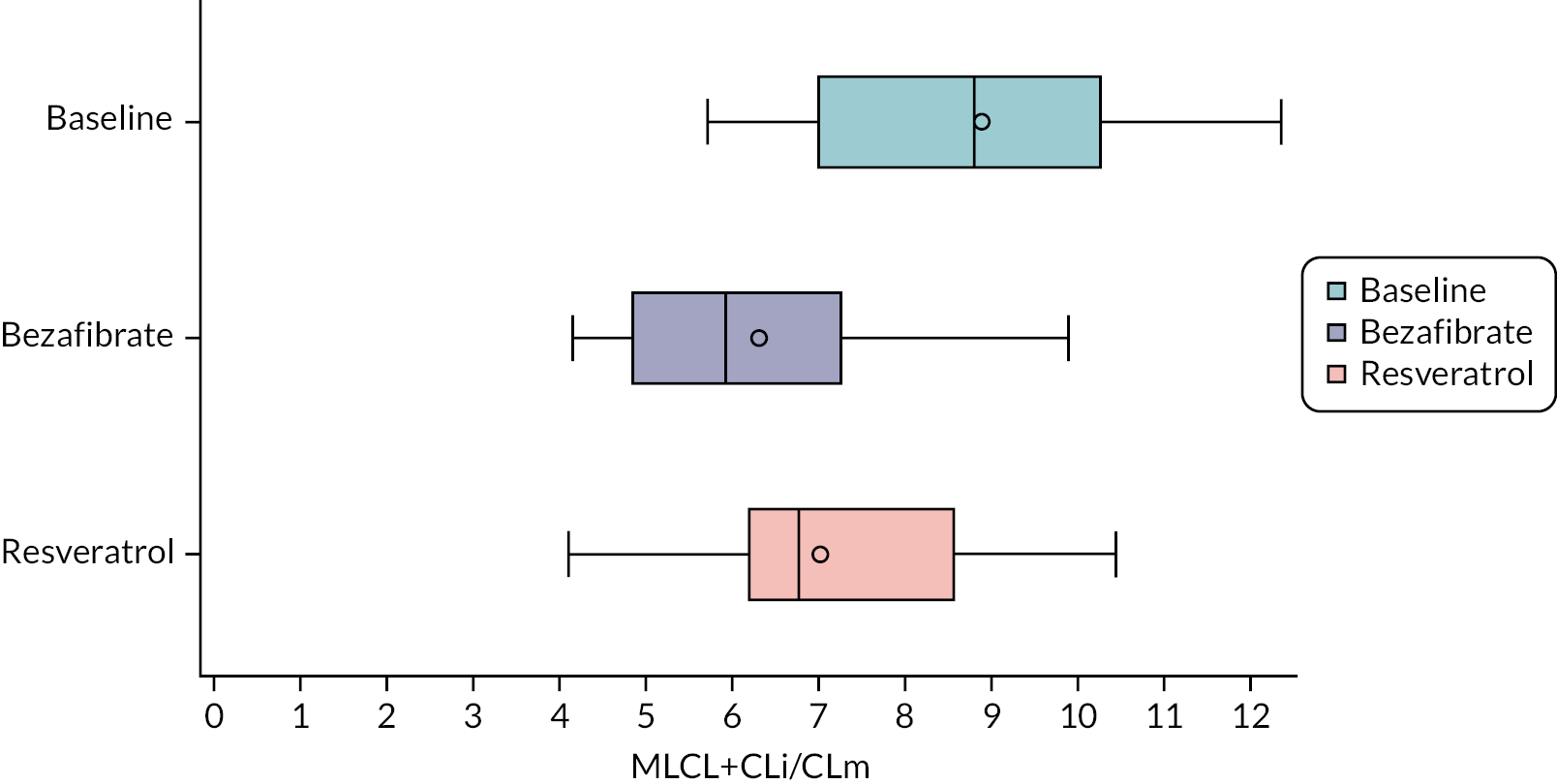
Profiles of MLCL and CLi and CLm in individual participants are shown in Figures 19 and 20. These show that both drugs appear to have reduced the MLCL/L4-CL ratio, largely by increasing intermediate cardiolipin species, and more so with resveratrol than bezafibrate. Ratios were nevertheless profoundly abnormal in comparison to patients with intermediate Barth phenotypes. 12
FIGURE 19.
Proportion of %MLCL, %CLi and %CLm by patient from in vitro drug applications.
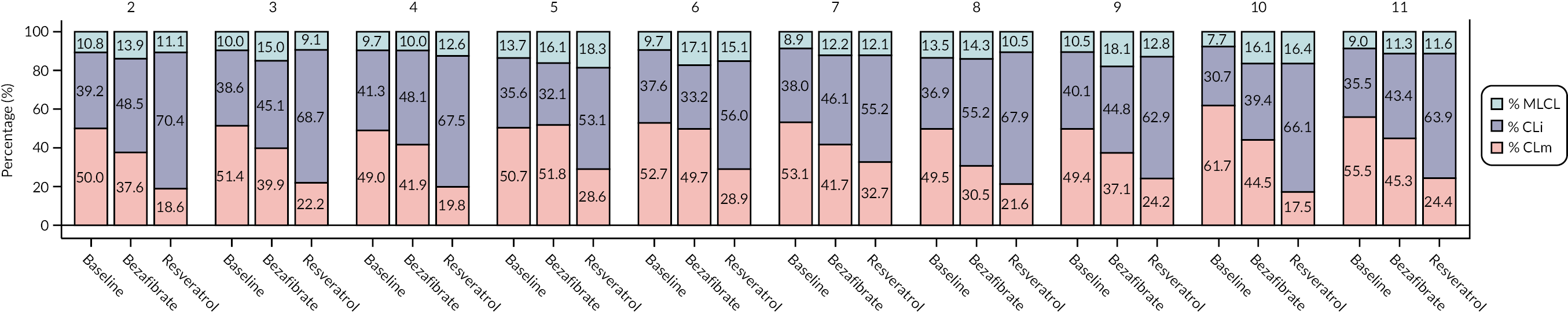
FIGURE 20.
Proportion of %MLCL, %CLi and %CLm for all patients grouped by baseline and in vitro drug applications.
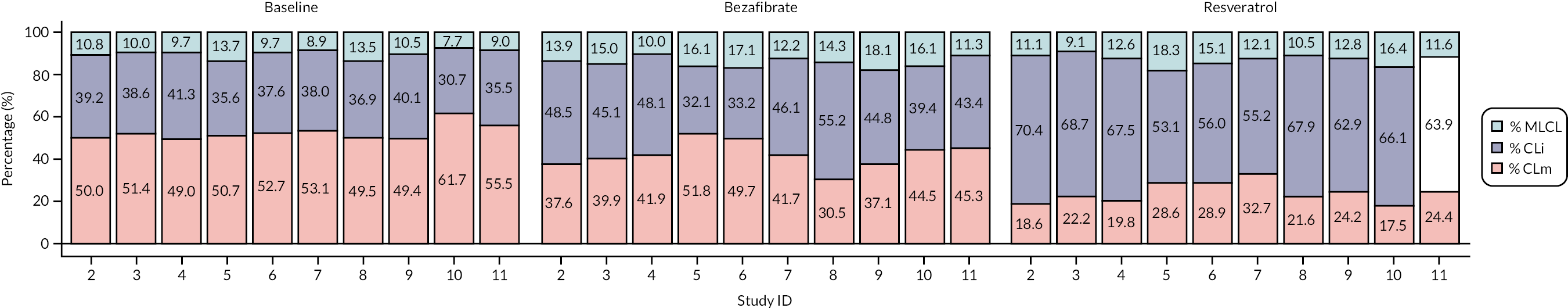
Mitochondrial studies were not completed on cells incubated with bezafibrate or resveratrol.
Chapter 9 Safety
Safety is summarised by SAEs and AEs reported during the trial.
Two participants experienced one or more SAE while taking bezafibrate or during the washout after taking bezafibrate, and one participant while taking placebo (Table 30). Seven participants experienced (non-serious) AEs while taking bezafibrate, and one participant while taking placebo (Table 31).
| Study ID | Allocation | Expectednessa | Description | Specification | Date of onset | Relatednessb | Classification |
|---|---|---|---|---|---|---|---|
| 1 | Bezafibrate | Expected event of bezafibrate | Muscle cramp | 4 March 2019 | Possibly related | Resulted in persistent or significant disability/incapacity | |
| 1 | Bezafibrate | Disease-related anticipated event | Neutropenia | Neutropenic chest infection | 11 March 2019 | Unlikely to be related | Required hospitalisation |
| 1 | Bezafibrate | Unexpected AE | Chest infection | 11 March 2019 | Unlikely to be related | Required hospitalisation | |
| 1 | Bezafibrate | Unexpected AE | Bladder control loss | 8 June 2019 | Unlikely to be related | Required hospitalisation | |
| 4 | Bezafibrate | Expected event of bezafibrate | Diarrhoea and vomiting | 17 September 2019 | Probably related | Required hospitalisation | |
| 4 | Bezafibrate | Expected event of bezafibrate | Diarrhoea | 3 October 2019 | Probably related | Required hospitalisation | |
| 1 | Washout period | Unexpected AE | Pyrexia of unknown origin | 8 August 2019 | Unlikely to be related | Required hospitalisation | |
| 1 | Placebo | Unexpected AE | Viral illness | 19 November 2019 | Unlikely to be related | Required hospitalisation | |
| 8 | Placebo | Unexpected AE | Tonsilitis | 20 November 2019 | Not related | Required hospitalisation |
| Allocation | Expectednessa | Event description | Number of events |
|---|---|---|---|
| Bezafibrate | Expected event of bezafibrate | Stomach cramps | 1 |
| Bezafibrate | Expected event of bezafibrate | Nausea | 1 |
| Bezafibrate | Unexpected AEs | Temperature | 1 |
| Bezafibrate | Unexpected AEs | Vomiting | 2 |
| Bezafibrate | Unexpected AEs | Abdominal pain | 1 |
| Bezafibrate | Expected event of bezafibrate | Raised creatinine | 1 |
| Bezafibrate | Expected event of bezafibrate | Raised CK | 1 |
| Bezafibrate | Expected event of bezafibrate | Leg muscle cramps | 1 |
| Bezafibrate | Unexpected AEs | Achilles tendon injury/pain | 1 |
| Bezafibrate | Expected event of bezafibrate | Vomiting | 2 |
| Bezafibrate | Unexpected AEs | Pulled hamstring | 1 |
| Bezafibrate | Disease-related anticipated event | Fatigue | 1 |
| Placebo | Disease-related anticipated event | Fatigue | 1 |
| Bezafibrate | Expected event of bezafibrate | Diarrhoea | 1 |
| Placebo | Disease-related anticipated event | Diarrhoea | 1 |
| Placebo | Disease-related anticipated event | Neutropenia | 2 |
| Bezafibrate | Expected event of bezafibrate | Cheek rash | 1 |
| Bezafibrate | Unexpected AEs | Cold/puffy eyes | 1 |
| Bezafibrate | Unexpected AEs | Pedal verruca | 1 |
| Bezafibrate | Unexpected AEs | Urine infection | 1 |
| Bezafibrate | Expected event of bezafibrate | Abdominal pain/discomfort | 1 |
| Bezafibrate | Expected event of bezafibrate | Legs aching | 1 |
| Bezafibrate | Expected event of bezafibrate | Gastrointestinal symptoms | 1 |
| Placebo | Unexpected AEs | Nose bleed | 1 |
Chapter 10 Patient and public involvement during the trial
Mrs Damin, chair of the BST (now Barth Syndrome UK), was a co-applicant on the funding application for the study and has made significant contributions to both the design and facilitation of the research. During the set-up period, she assisted the research team with drafting and editing the PILs, to ensure clarity of understanding for potential participants and their families. This was challenging due to the number of different versions of the PIL (adult participant, parent/guardian, 11- to 15-year-old and under-11-year-old participants), and particularly with trying to communicate the complexity of the trial to younger participants. As is the case with all paediatric trials, it was challenging to strike the right balance of information so that children can be well informed of the requirements of the trial and what is involved for them, while also not overwhelming them with too much information beyond their current capacity to understand. Having the PILs reviewed by Mrs Damin, with her understanding of the potential participants involved, helped greatly to achieve the right balance.
Before starting the trial, many patients with Barth syndrome underwent rigorous pill-swallowing training with the clinical psychologist and other members of the BSS team. During the training, patients learned to swallow pieces of food with different textures in incrementally larger sizes, until they were able to swallow hard-textured ‘Tic-Tac’ style pieces, similar in size to many commercially available medications. ‘Pill Glide’ was also used during pill-swallowing training as an aid to help patients swallow tablets. Pill Glide is a commercially available flavoured lubricant that is sprayed into the mouth, coating the tablet and making it easier to swallow.
Mrs Damin talked with the Barth syndrome patients and their families to help engage them with this training, not only for potential trial purposes but also as a skill in its own right, as it would benefit all patients with Barth syndrome in terms of their future health care. Several CARDIOMAN participants who, before the trial started, were anxious about their ability to swallow tablets, proceeded to successfully participate in the trial due to the pill-swallowing training. Therefore, the background training conducted during the usual care pathway of the patients was instrumental to conducting the trial successfully.
Mrs Damin’s knowledge of the relationships between participants helped us to arrange the clinics so that participants could attend according to their preferred social groups – that is, friends attending the same research clinic – which helped to make the clinics a more enjoyable experience for the participants and their families and may have helped with study retention.
Ms Liz Stobart-Hook, whose son previously died from Barth syndrome, participated as a lay member of the TSC. During the joint DMSC/TSC meeting held in February 2019, she advised on aspects of the trial where family views were needed.
One participant made a documentary film about his and the other participants’ experience of taking part in the trial, including interviews with the research team, participants and their families. The film will be shown to the UK Barth syndrome community during one of BST’s regular events and at the international Barth syndrome conference in the USA, subject to acceptance. He intended to show the video documentary at the end-of-study celebration. Unfortunately, the end-of-study celebration was postponed due to the COVID-19 pandemic (particularly because Barth patients are a clinically vulnerable group) and is yet to take place.
We sent a summary of the results to all participants and their families, as planned, and made this available more widely to patients outside the UK through the BST and BSF websites. Trial participants are rarely made aware of the processes required to analyse results and ensure they are robust enough to publish. Some larger (usually multicentre) trials communicate information about timelines for trial results to their participants via periodic newsletters, but this does not often explain the processes involved; for example, obtaining and collating data from multiple sources, resolving data queries, locking the database, carrying out statistical analyses and writing up the findings. To help explain to the CARDIOMAN participants the steps required after their participation ends and before study results become available (and therefore why the trial results would not be available for some time), the study team created an educational infographic (see project document 3 or see Report Supplementary Material 6). Mrs Damin was involved in reviewing the infographic, advised on amendments and distributed the final version to the participants and their families. The infographic has been well received by the Barth syndrome participants and their families and by colleagues in the research community – so much so that it has been adapted for other trials. This initiative fits with the national directive of the NIHR communications working group to improve overall communications and engagement with trial participants throughout the life of a trial.
Chapter 11 Discussion
Main findings: challenges
Process challenges
Several significant challenges were experienced when setting up the trial, including logistical arrangements, issues related to the study design, and IMP formulation.
The trial was required by the funder to recruit a minimum of 10 participants, which meant that clinicians providing the BSS needed to encourage trial participation within the Barth syndrome community without unduly persuading or coercing patients and their families to take part. The UK patients with Barth syndrome, and their families, were aware of the study from the early stages of development and, fortunately, are highly engaged with their care and related research. However, this also gave rise to an unusual situation where, in theory, the refusal of a small number of individuals could stop the entire study despite the potential benefits to the group. Rarely does an individual’s decision carry so much weight and risk disrupting the potential benefit of a piece of research for an entire group. In the case of Barth syndrome, for which there are no current treatment options other than symptomatic care, failure to proceed with the trial would likely spell the end of future investigations into potential treatments and the hopes that many patients and families have of finding a treatment option.
The funder requested formal expressions of interest to determine whether the trial should proceed. The research team remained anxious about the future of the trial, since they were aware that potential participants considering any trial can change their minds due to changing health and life circumstances. Ultimately, the interest of patients matters (in terms of numbers) only at the point at which they consent to enter a trial. While the research team understood that assurances (as far as possible) were required to gauge the viability of a trial, none of the researchers had experienced a previous requirement to obtain formal interest from potential participants before they were recruited. Since the UK governance framework does not recognise this situation, the researchers sought advice from the Chair of the NHS REC that reviewed and approved our trial, asking whether we could contact potential participants to obtain expressions of interest. They agreed that this would be possible as a form of ‘Patient and Public Involvement’ and did not require approval from the REC.
Funders of future RCTs investigating treatments for rare diseases will need to reconcile the risk of not recruiting enough participants with the need for a greater knowledge and understanding of the disease in question. If, as the Department of Health review showed,35 barriers to diagnosis and treatment in rare diseases are a result of limited knowledge of the disease, it is unclear at what financial or methodological threshold research should not be pursued because it is not considered viable. These thresholds and the rationale behind them need to be transparent and clear from the outset, and the expectations of the potential study population managed appropriately throughout the process. To fund and set up a trial – raising the hopes of those with the disease and their families – only to disband it if not enough participants are recruited (which could be a matter of one or two patients declining to take part) seems at best inefficient and, at worst, unfair to patients willing to take part, even if the recruitment numbers are not as expected. The cost of setting up a RCT in a rare disease population is not cheaper because there are fewer participants, and the balance between value for money versus the health-care needs of patients with rare diseases needs to be weighed carefully.
Another condition of funding (contested at the time by the applicants) was the requirement for an interim analysis during the washout period, to estimate the efficacy of bezafibrate. The rationale was that, if bezafibrate showed no effect (or less than what would be expected by chance), the trial would not continue to the second treatment phase – despite the interim analysis necessarily being between (rather than within) groups with markedly less precision. During the ethical approval process, the REC rejected this requirement but agreed instead for the decision to be devolved to the DMSC for the study. Difficulties in finding independent committee members with relevant expertise in rare diseases delayed the first DMSC meeting until shortly before recruitment started, leaving uncertainty during the set-up period about whether an interim analysis would be required and difficulty in planning dates for research clinics. If an interim analysis was required, a longer washout period would be needed to accommodate the time to carry out the interim analysis, yet the clinic facilities needed for the research had to be booked well in advance. We planned the clinics around a likely washout period of 1 month, which required a very short recruitment period, so that the clinics could occur as close in time as possible to allow data for the first period to be collected, and entered and analysed, the interim analysis conducted and reported, the DMSC to have met and the funder to have implemented any recommendation, before the first recruited participants were due to enter the second phase. An amendment to the protocol was made to allow the washout period to be extended, but there was no guarantee that the clinic facilities could be rebooked if research clinic dates had to be rescheduled. In the event, both oversight TSC and DMSC unanimously rejected performing an interim analysis.
Participants and their families were all of school or working age. To increase the likelihood of uptake and retention, we tried to minimise absences from school/work by arranging the clinics so that at least 1 day fell on a weekend. We were unable to conduct the clinics solely on weekends due to health and safety restrictions on working practices of some of the support departments involved; for example, the MRI scans were required to be conducted during normal working hours. Each ‘clinic’ was arranged over 2 days due to the number of assessments required, and to avoid too much physical activity for participants in a single day. The availability of the facilities and personnel dictated that our clinics were arranged over three consecutive weeks (Fridays and Saturdays). These arrangements also needed to be juggled with finding times during the year that minimised disruption to patients and families (e.g. during summer holidays and at Christmas) and avoiding (as far as possible) the winter season, when viral illnesses often adversely affect the health of Barth patients. All the staff involved also had to be available for the clinics, including NHS staff who were not researchers who needed to fit the research clinics around existing clinical duties. The calendars of 22 personnel across three different organisations and 11 different departments had to be aligned, which was only possible due to the extraordinary goodwill of all those involved.
A key requirement was finding a formulation of bezafibrate that was acceptable to the target population, to enable us to recruit the minimum of 10 patients. Many patients had difficulties in swallowing; few could swallow tablets reliably, and several Barth syndrome patients were fed via nasogastric tubes. Our original intention was to use a liquid formulation of bezafibrate/placebo, but stability testing showed the formulation to be unstable after only 2 weeks and so unpalatable as to ensure unblinding would occur. Therefore, tablets were the only alternative. The smallest dose in commercially available bezafibrate tablets was 200 mg, but the study protocol required 100 mg to be prescribed for younger children. We explored methods such as cutting marketed bezafibrate tablets in half (to achieve the 100-mg dose) and over-encapsulating the cut tablets, but the width of the tablet determined the length and size of the encapsulating shell. The width of an existing 200-mg bezafibrate tablet resulted in a very large capsule (approximately antibiotic-sized) that the younger children would be unlikely to be able to swallow. Therefore, we investigated whether 100-mg tablets of bezafibrate/placebo could be manufactured.
We contacted several ‘specials’ manufacturing companies (both private companies and NHS pharmacy manufacturing units) in accordance with NHS competitive procurement policies. Out of those, only two private companies were able to provide quotations for our requirements and were willing, in principle, to accept the contract. The estimated cost of one supplier was prohibitive. Other manufacturers may have been able to supply to our requirements, but a strategic search was difficult. The NIHR could help researchers in similar situations by compiling information about UK specials manufacturers and their capabilities; this would streamline the identification of appropriate manufacturers, obtaining quotations and placing a contract.
After securing agreement in principle from the funder to proceed, the contracting process began. This took over 12 months because each part of the contracting process was undertaken only after the previous one had been completed, instead of different parts being conducted in parallel. Had we been aware that multiple contractual documents were required, we might have been able to expedite the process, but the manufacturer did not explain the process to us at the outset. Completion of the contract with the drug manufacturer also required that the contract variation with the funder be completed first. Without this, the study sponsor would have been required to bear the financial risk of the drug manufacture.
The number of bezafibrate/placebo tablets ordered was estimated based on the age-dependent dosing in the protocol. However, extensive delays during set-up meant that the ages of the children had increased from when the order was estimated to the time when recruitment started; more tablets for the older ages were required. Fortunately, the minimum order of 12,000 tablets provided ample IMP to cover this change, but it is recommended that researchers and/or PTUs have a protocol/standard operating procedure in place for assessing and checking work orders, independently of the person initiating the calculation, with special consideration for the effect of potential set-up delays on age-dependent dosing. The volume of IMP when manufacturing a special may not markedly influence the cost of a contract if the drug involved is inexpensive; in such situations, we recommend researchers consider ordering a much larger volume than needed providing that the IMP expiry date covers the likely duration of the study (including some delay).
Outcomes challenges
It is tempting to collect as many data as possible from participants with rare diseases, accepting that having more data per participant does not add additional power. This is due partly to having fewer opportunities to collect research data from participants with rare diseases, and partly to a perceived pressure to demonstrate that the study is providing value for money. CARDIOMAN was not immune to this temptation, with 14 outcomes in the protocol, many requiring highly specialised equipment for assessment. Although the value of secondary outcomes may be higher in a population with a rare disease, as in any trial it is important for researchers to weigh carefully the insights gained from the additional information against the burden for patients and the resource implications.
Patients attending the BSS clinics already undergo extensive multidisciplinary review, including consultations with a dietitian, cardiologist, psychologist, metabolic clinicians and the clinical nurse specialist, so it was not possible to accommodate the volume of trial assessments into the patients’ usual care pathway. In addition, the BSS clinics were (at the time of trial set-up) run biannually, with patients usually attending annually; by contrast, the study protocol required three study visits within 9 months. Consequently, the only way to complete the study assessments was to arrange separate research clinics.
We also had to consider the exercise intolerance and fatigue experienced by the participants. It was not practicable for participants to move between different areas of the hospital to undergo the various study assessments (including in some buildings situated on steep hills), but some of the assessments required the use of static equipment in different places to the clinic areas (e.g. MRI scanner and bicycle ergometer). Fortunately, the CRIC had clinic rooms (and other facilities such as a recreational room for participants and their families) in the same building as the static equipment. Without this facility, it might not have been possible to conduct the trial. Having a dedicated area in which to conduct our research was central to its successful delivery.
The small sample sizes inevitable in rare disease research put greater pressure on research teams to recruit and retain participants, because data from each participant carry greater weight than in studies with large sample sizes. Therefore, a greater degree of effort and problem-solving is required to ensure that logistical arrangements accommodate patients’ needs. This requirement for the research to be more accommodating of patients’ needs and wishes may be unfamiliar to some trial managers and other researchers. In studies with large populations of potential participants, if the study arrangements do not suit a participant and they decline to take part, researchers can wait for the next available and willing participant. We have demonstrated that it is possible to create the research infrastructure to successfully deliver a RCT in this rare disease population.
As CARDIOMAN was funded by the NIHR Efficacy and Mechanisms Evaluation programme, one purpose of the study was to evaluate potential disease mechanisms of Barth syndrome. We included in our investigations an evaluation of the PCr/ATP ratio in cardiac muscle and oxidative function in skeletal muscle using 31P MRS. 31P MRS technology is not yet widely used in the UK – only two other centres in the UK have this facility – and was not available at our centre. Therefore, to obtain the data for these outcomes, we first had to implement the use of 31P MRS technology at our centre, a major undertaking which was not well understood at the outset.
The use of 31P MRS technology required a third-party electromagnetic coil to be integrated in the locally available MRI scanner. Commercially available coils are available but the cost was prohibitive (> £25,000). Instead, we approached the Faculty of Engineering at the UoB to manufacture the coil. They agreed and were able to design and build the coil at a significantly reduced cost. In addition to the construction of the coil, several other governance processes and administrative checks were required before it could be used on study participants, including: (1) obtaining the sequence code for the MRI scanner (for the scanner to recognise the third-party device), (2) obtaining approvals from the manufacturer to use the coil in the MRI scanner, (3) obtaining approval from the CRIC research facility, (4) testing on ‘phantom’ objects (i.e. inanimate objects containing phosphorus solution) and (5) testing the coil on healthy volunteers before using it on study participants. The latter involved a separate ethics committee approval.
These processes took approximately 3 years to complete, very much longer than anticipated, for several reasons. There was no precedent at our centre for using 31P MRS technology for cardiac and skeletal muscle. The unfamiliarity of our requirements meant that the staff involved (engineer, medical physicist, clinicians, radiographer and administrative support teams) were learning the technical requirements while ensuring due diligence, which did not make the process straightforward. This challenge was compounded by limited national expertise to call upon. Staff at the University of Oxford kindly agreed to assist with some of the technical aspects of the 31P MRS, without which we would have been unlikely to be able to proceed. Therefore, to complete the MRS investigations, we were largely reliant on individuals and teams that operated externally to the funded research team, both within the same organisation and outside it, and we were not able to implement the 31P MRS assessments until the last visit of the study. The CARDIOMAN research team had no option but to accommodate delays from the coil manufacturing and implementation process.
Current organisational arrangements do not seek to impose contracts on departments within the same institution that are, effectively, acting as suppliers. This makes it very difficult to impose a deadline as there is no penalty (for the supplying department) if the deadline is missed. Academic relationships are forged and informal arrangements agreed, but individuals in such departments have their own competing priorities and there is no accountability mechanism, apart from a threat to reputation. Research teams are subject to external contractual deadlines to deliver their projects (e.g. with the NIHR), often with financial penalties or reputational risk if the team fails to deliver, but this accountability, although communicated to those involved, may be difficult to cascade through the ‘supply chain’ without increasing the contractual burden. Imposing contracts may also disadvantage a study when there is a limited choice of collaborators to meet a research need.
At the outset, we understood the potential benefits of the 31P MRS technology but were not fully aware of the challenges in setting up the facility locally. It is important that researchers are cautious when committing to exploratory work in clinical research on mechanisms study. The use of exploratory techniques (or novel technology) may help researchers gain new information and understanding about a disease, but unexpected delays may arise which limit the potential benefit.
Main findings: study results
The main findings of the study were:
-
No clinically important or statistically significant improvement in peak VO2 (the primary outcome) when participants were taking bezafibrate compared to placebo. This was also true for VO2 measured before exercising.
-
No statistically significant improvements in the core or fatigue QoL domains.
-
Inconsistent findings for LVEF; there was a borderline improvement when assessed by echocardiography but no difference when assessed by cardiac MRI.
-
Inconsistent findings for strain on echocardiography; longitudinal and circumferential strain measurements were better at rest when participants were taking bezafibrate compared to placebo, but not at peak exercise. There was a borderline improvement when assessed by echocardiography but no difference when assessed by cardiac MRI.
-
Blood samples from participants were tested directly for outcomes: amino acid and cysteine levels were significantly higher after bezafibrate treatment; there was a trend to improved arginine levels; but no significant change in neutrophil numbers. There was no significant change in cardiolipin ratio in participants’ blood after bezafibrate treatment.
-
With respect to Epstein–Barr virus (EBV)-transformed lymphoblasts incubated in vitro with bezafibrate, resveratrol or placebo, improvements in cardiolipin ratio were seen with either bezafibrate or resveratrol, consistent with the previous laboratory findings which underpinned this trial. 11 This included a more marked beneficial effect with resveratrol than bezafibrate. Mitochondrial measurements were not successful due to a variety of technical challenges relating to electron microscopy and COVID laboratory shutdown.
-
There were no significant period effects except for the MLCL/L4-CL ratio, which showed a 20% increase in the second compared to the first period, and neutrophil count in blood samples, which was also higher in the second period.
We recruited 42% of all known patients with Barth syndrome in the UK and about 70% of eligible patients. Therefore, we regard these findings as applicable to anyone who would have been eligible. Several exclusion criteria relate to patients with contraindications to bezafibrate or swallowing tablets, and the findings cannot be applied to these patients.
Volume of oxygen peak as primary outcome was carefully chosen as the most reliable parameter in the scientific literature of functional capacity and exercise function. The lack of a treatment effect on VO2 peak can be taken as a reliable true outcome here, as VO2 peak is an extremely well validated parameter in these settings. A disadvantage of VO2 peak as a global parameter of exercise capacity is, however, that its value is a conglomerate of many physiological components such as cardiac function, cell metabolism and respiratory function, and mild treatment effects on any one of these can be masked by non-effects in the other physiological systems.
Echocardiographic functional parameters as secondary outcomes were chosen based on their proven track record in cardiac RCTs in adults, as fewer data are available from paediatric trials. Possible treatment effects on systolic function by echocardiography (EF and resting 2-D strain) were observed, but the increase in EF was not confirmed by cardiac MRI, the gold standard measurement method. EF measured by echocardiography, while reproducible, is more operator-dependent than that acquired by cardiac MRI. However, changes in 2-D strain could indicate a subclinical effect on systolic LV function, as 2-D strain has been shown to be more sensitive than EF and to register mild differences in cardiac function that do not translate into clinically measurable differences. 36 Hence, it is unsurprising that health-related QoL did not differ.
Quantitative exercise echocardiography was a novel echocardiographic assessment used in the trial. This method of assessment was previously piloted and validated by our research group. 25 It was included in the trial to complement the exercise assessment by VO2 peak. No differences between treatment and placebo were found. No other cardiac assessments showed significant changes.
With respect to the laboratory aspects of the study, bezafibrate therapy was originally proposed in response to a series of observations. Ren et al reported that co-culture of either TAZ knockdown mouse fibroblasts or human Barth syndrome fibroblasts with either bezafibrate or resveratrol improved the ratio of MLCL to mature L4-CL in these cells,11 ameliorating the grossly elevated MLCL/L4-CL ratio so pathognomonic of Barth syndrome. This finding added to an intriguing observation that some patients with Barth syndrome appear to have an ameliorated phenotype with absence of neutropenia and mild or absent cardiac or skeletal myopathy. 12 Such patients are characterised by better L4-CL levels (in some cases even overlapping those seen in normal controls), although they still have grossly elevated MLCL levels. For these reasons, it was hypothesised that treatment with either bezafibrate or resveratrol could improve the aberrant cardiolipin ratio, pushing treated patients towards an ameliorated phenotype and hence reducing symptoms of their disease.
Work published after we proposed this trial17 confirmed these in vitro suggestions of benefit in a TAZ knockdown mouse model. These animals have an inducible form of Barth syndrome which develops progressively after introducing doxycycline into their diet as neonates while also administering chronic isoproterenol infusions to accelerate development of a cardiac phenotype. LV function was shown by echocardiography to be depressed by 4.5 months of age. Bezafibrate intake over a 2-month period substantially ameliorated this development of LV dysfunction. However, in contradiction to the in vitro experiments described above, improvement in systolic function was accompanied by a reduction of L4-CL content and an increase of MLCL levels in cardiac muscle; that is, worsening of the aberrant cardiolipin ratio. Accompanying mitochondrial studies suggested that the beneficial effect of bezafibrate resulted from increased mitochondrial biogenesis – that is, increased mitochondrial numbers – rather than amelioration of cardiolipin chemistry.
Several laboratory secondary outcome measures were assessed in CARDIOMAN. The principal aim was to quantify improvement of cardiolipin ratios in response to therapy with bezafibrate but also, in the event that patients varied in their response, to see whether laboratory outcomes changed in a way consistent with in vivo observations. The ultimate hope was that in vitro testing might enable prediction of an individual’s response to therapy. Cardiolipin ratio was therefore measured at baseline and at the end of each treatment phase. Lymphocytes were also EBV-immortalised to produce cell lines which were then treated with bezafibrate or resveratrol to look for evidence of differential effect of these two drugs. Since so much of the pathology of this disease is thought to be mitochondrial in origin, we investigated a range of mitochondrial parameters by either electron microscopy (seeking evidence of increased mitochondrial biogenesis) or mitochondrial function testing. Absolute neutrophil counts were also investigated to look for amelioration by bezafibrate therapy. Finally, there is some evidence that specific amino acid levels are reduced in Barth syndrome – notably serum arginine, but also cysteine levels37 – and hence these were assayed.
No significant changes were found in any of these parameters except amino acid levels. There was no evidence at all for an improvement in the cardiolipin ratio in participants’ blood (taken at the end of each treatment phase) after bezafibrate treatment. When participants’ cells were treated in the laboratory with bezafibrate or resveratrol, some improvements in cardiolipin ratio were seen (confirming the original findings of Dr Mindong Ren, New York University, 2016, personal communication) but not to the levels seen in patients with the ameliorated phenotype. Although analysed in blood rather than the cardiac cells examined in the live murine studies, these CARDIOMAN findings are analogous in finding no improvement of the cardiolipin ratio in vivo. However, importantly and in contrast to the murine studies, we did not find significant changes in mitochondrial size, number or function after in vivo drug treatment to support increased mitochondrial biogenesis.
The electron microscopic and mitochondrial assessments proved problematic in a variety of ways which would discourage us from suggesting them as more routine investigations in Barth syndrome patients or as outcomes in future trials (see Reporting Equality, Diversity and Inclusion). Those cell lines that could be analysed showed a mixture of normal and abnormal mitochondria, including some large with empty spaces, some with disordered cristae and an onion-like mitochondrion as previously reported in EBV-transformed Barth syndrome B-cell lines. 38 Intriguingly, these were not seen in freshly isolated lymphocytes either at baseline or after drug treatment phases, a discrepancy previously noted in this laboratory (Dr A Bowron, PhD thesis, University of Bristol). 37 The majority of mitochondria from all study patients were also comparable in size with those seen in control samples in previous work in this area39 (SJ Groves, 2021, personal communication).
Amino acid levels did improve. Cysteine levels were significantly higher after bezafibrate treatment, and there was a trend to improved arginine levels. However, these observations must be treated with caution, since ideally amino acid levels should only be assayed in blood taken after a 4-hour fast and it was not deemed safe or considerate to insist on this in children who were undergoing a wide range of tests, including exercise testing. Recent meals or snacks could therefore have contributed to these rises. There was no significant change in neutrophil numbers.
It is important to view these CARDIOMAN results in the context of a highly comparable US trial of a daily subcutaneous injectable drug called elamipretide. This is a cell-permeable peptide that targets the inner mitochondrial membrane, where it binds to cardiolipin via electrostatic and hydrophobic interactions. It is proposed to sustain the cristae network and remediate bioenergetic dysfunction.
Elamipretide therapy was investigated in a Phase II randomised, double-blind, placebo-controlled crossover trial (Clinicaltrials.gov NCT03098797) to evaluate the safety, tolerability, and efficacy. 40 Enrolment began in July 2017, and 12 patients aged 12 years and over were recruited. Patients were randomised to elamipretide or placebo for 12 weeks, followed by a 4-week washout and then 12 weeks on the opposite arm. Ten subjects continued to a secondary open-label extension study, with eight subjects reaching 36 weeks (i.e. 48 weeks of therapy in total). Primary outcomes were 6-minute walk test (6MWT) and improvement on a Barth syndrome symptom assessment scale (BTHS-SA); there was no significant change in either by the end of the primary study. However, after 36 weeks of open-label extension there were significant improvements in 6MWT (+95.9 m; p = 0.024) and BTHS-SA (−2.1 points; p = 0.031) and a mean 42% improvement in grip strength as assessed by hand-held dynamometry (p = 0.001). There were also significant improvements in secondary endpoints including knee extensor strength, patient global impression of symptoms, and some cardiac parameters. Despite these positive findings in the open-label extension study, the US Food and Drug Administration (FDA) refused a new drug application from Stealth BioTherapeutics, the manufacturer of elamipretide, in October 2021.
There are several areas of relevance here to CARDIOMAN. Firstly, no change in MLCL/L4-CL ratio was seen with elamipretide despite strong evidence of benefit in response to prolonged therapy. That and the in vivo mouse bezafibrate trial therefore demonstrate that improvements in clinical parameters are not accompanied by improvement in the MLCL/L4-CL ratio in blood or cardiac tissue respectively. Thus our underlying hypothesis that improving the cardiolipin ratio to shift patients towards an intermediate phenotype appears flawed.
Arguably the more important consideration is the duration of the trial. CARDIOMAN patients received approximately half the therapeutic dose of bezafibrate for the first month of the trial to check tolerance. The duration of therapy at full dose was a further 3 months, hence very comparable to the elamipretide trial. No significant effects were seen with 12 weeks of elamipretide: 6MWT compared with placebo was −0.8 m (p = 0.97) and BTHS-SA total fatigue score compared with placebo (+0.06; p = 0.89). However, improvements were apparent for both primary outcome measures after the first 12 weeks of open-label extension therapy. Mean improvement on the 6MWT was 60.5 m (16% increase; paired t-test, p = 0.02) and BTHS-SA total fatigue score was −1.6 points (average 19% reduction/improvement; paired t-test, p = 0.03). Similarly, hand-held dynamometry showed a 30% improvement (p = 0.003) during this second 12 weeks of therapy. Each of these areas of improvement further increased over the 36-week open-label extension.
Would the same have been observed with bezafibrate in continued use, especially since some echocardiographic parameters suggested improvement after 4 months? Only a second bezafibrate study with a duration of at least 6 months, and by preference longer, could fully answer this. Further trials need to consider these findings when choosing the duration of treatment period and the dose of medication; for example, the safety tolerance may need to be lowered.
Patient and public involvement
Mrs Damin, Chairperson of the Barth Syndrome UK charity, was extensively involved in the conception and design stages of this project, including critical appraisal of the trial protocol and assisting with writing and editing the PILs. Mrs Damin also discussed with the research team the pros and cons of different formulations of trial medication and helped plan the tablet-swallowing training sessions. She was instrumental in keeping open channels of communication with patients and families about progress with the trial and explaining the importance of new drug development for management of both existing and future patients.
In addition, Bristol Trials Centre liaised closely with Mrs Damin to arrange clinic dates with the prospective families and optimise the groups of individuals in each clinic to increase the likelihood of engagement and retention. The insight and information regarding the dynamics involved with this patient group was invaluable for making the practical arrangements for the clinic dates. Mrs Damin also helped to ‘prime’ the families for some of the tests they would experience as part of the trial by providing them with links to video clips and other (non-trial-related) information that showed patients and their families what to expect; for example, videos of an MRI machine and what to expect when having an MRI scan.
After completion of the trial, we held a teleconference inviting all participants, their families, and the wider Barth syndrome community to a presentation of the results. This event was advertised through both the UK and US Barth syndrome charities to ensure that as many interested people as possible could attend. Many attendees were from the US-based research community, and they expressed their admiration and gratitude for our achievement of conducting a trial in this patient group.
Overall, we had an exceptionally high level of engagement with everyone in the Barth syndrome community, in both the UK and the USA. Mrs Damin’s (unofficial) role of liaising between the research team and the UK patient group has been critical in ensuring that we could recruit to the trial. The type of liaison work that Mrs Damin undertook is not included in the remit of the research team (e.g. trial managers or research nurses would not be allowed to contact potential participants before they consented to provide information to ‘prime’ participants for research tests, or get involved in activities that form part of usual care). Having a central figure (in our case a co-applicant) who was familiar with the patient group and who could liaise between the research team and patients has been extremely helpful and important for engagement, and ensured communication continued even during times of substantial delays and difficulties. Without her taking on this role when we were experiencing delays, we would have risked losing the interest of some potential participants, which would have jeopardised the entire trial. Therefore, we recommend that future trials of treatments for very rare diseases try to identify a person with a similar role/function to Mrs Damin’s in the design, planning and implementation of the trial.
Similarly, we were fortunate to receive additional technical support from others in the Barth community, such as Todd Cade in the USA with respect to MRS exercise testing, and others outside of the Barth community; for example, Oxford MRS experts.
The tight-knit nature of the international Barth syndrome community is evidenced by help received from the BSF. The BSF awarded funding at a critical time to enable the clinics to go ahead, and Erik Lontok, Director of Research at the BSF, has been keenly interested in progress of the trial and encouraged us at every step, including facilitating webinars with Mrs Damin and offering help with reviewing lay summary results.
Reporting equality, diversity and inclusion
Due to the very small patient population affected by the condition, we were restricted in our ability to include participants from diverse backgrounds, as the cohort was dictated by the demographic profile of patients with Barth syndrome. However, by the nature of conducting research in very rare diseases, we were required to include and recruit as many of the population as possible to reach a minimum number of participants to make the study viable. The BSS is also a UK-wide specialised service run centrally through UHBW, so potential participants were already widely geographically dispersed across the UK. We also ensured that participants and their families were reimbursed for their travel and accommodation expenses, and that refreshments were provided throughout their time in the research clinics, so as not to preclude participation on the basis of financial ability.
The included patient cohort was representative of the Barth patient population in terms of spectrum and severity of disease, treatment modalities, and also gender and age. It must be noted that the youngest patient age range was not represented, as these patients did not meet the inclusion criteria.
There was a wide range of experience and expertise across the research team members. This was partly due to the study encompassing a wide range of outcomes, so expertise came from across a multitude of disciplines, such as exercise physiology, electrical engineering, cellular medicine, medical physics, trial methodology, and cardiology. Some members of staff who had less research experience were also included in conducting parts of the research, enabling them to gain valuable insight into conducting high-quality RCTs. Development opportunities were provided for the sonographers and exercise physiology students, and a NIHR clinical fellow was also involved in helping with the MRS outcomes.
Strengths and limitations
There are several strengths of the trial. Despite the small sample size, randomisation and placebo blinding minimised the risk of bias. The crossover design maximised the power afforded by the small eligible population precision by comparing the effects of bezafibrate and placebo within participants, an achievement largely attributable to the commitment of participants and their families. Many crossover trials are compromised by withdrawals between periods of study; the fact that all participants attended both assessment clinics in this trial is both a strength and an achievement for all concerned. Other strengths include the completeness of the data, good adherence, and tolerance of the IMP.
The trial was also subject to several limitations. These include:
-
The sample size achieved satisfied the funder’s condition for progressing but was very small. We could not have done anything different to increase the sample size. This limitation could only be addressed by an international collaboration (see Future research).
-
Interpretation of effects estimated for many secondary outcomes was especially challenging. We made no adjustment for multiple testing.
-
It may be that a longer treatment duration or a higher bezafibrate dose might have produced better outcomes. The drug dose was chosen to ensure the safety of participants; we achieved this, as evidenced by only one boy stopping the drug 8 weeks into treatment due to gastrointestinal symptoms. The duration of each period was necessitated by the funder’s request to reduce the cost of the study; we had originally specified a 6-month period. The trial of elamipretide, like CARDIOMAN, also did not observe a statistically significant improvement in the primary outcome (6MWT) after 12 weeks, but some participants reported major improvements in an uncontrolled open-label extension study. 40 Only by repeating the trial with a longer duration of treatment can the risk of bias due to using a subjective outcome in an open-label extension be excluded.
-
Bezafibrate may have induced adverse reactions in some participants, inadvertently causing unblinding. Most non-serious AEs arose when participants were taking bezafibrate. However, none of the participants or their families expressed a strong feeling during either treatment phase about whether they were receiving bezafibrate or placebo.
-
The group of participants randomised to receive bezafibrate first and placebo second was older than the group randomised to the opposite order. Other characteristics associated with age differed in the same direction. The crossover design controlled for potential confounding by participants’ characteristics, but these could explain period effects.
-
The electron microscopic and mitochondrial assessments proved problematic in a variety of ways. It proved difficult to obtain enough blood for all tests (see Table 26). Although in general the high-pressure freezing and freeze substitution produced good-quality, clear images, issues with preparation and staining of samples led to insufficient cells being analysed in a minority of cases. Where possible, only intact cells were analysed, but on occasion cells with slight tears had to be accepted for analysis.
-
It was not possible to complete the electron microscopic work on the immortalised cell lines. There were problems with some samples, there being either no cells present in the electron microscopic section or cells that were unfit for analysis. Repeats were attempted for a few samples but without success. The costs had exceeded the original budget for this element of the study by this point, and further work was also prohibited by the COVID-19 pandemic. Thirdly, lymphocyte cell numbers were limited in some samples, providing insufficient samples for some of the mitochondrial function measurements, as isolated lymphocytes were prioritised for the electron microscopic work. Assessment of cristae organisation is subjective and the assumption was made that the relatively small numbers of cells analysed were representative of all cells. Although stringent care was taken to avoid overgrowth of cell lines and exhaustion of media/nutrients, it is likely that such lines contain more aged or apoptotic cells than fresh peripheral blood samples. Potentially it is these senescent cells that display abnormal mitochondria.
-
Measurement of mitochondrial content and membrane potential by flow cytometry (Mitotracker FM, TMRE) unfortunately proved highly variable, with no clear pattern of response to treatment. Technical limitations, such as variability in time of sample acquisition, sample preparation, dye preparation and staining, may have led to variability in either Mitotracker or TMRE intensity between samples acquired on different days. Future improvements may include using a standardised control sample in every staining to which samples can be normalised, thus gaining a picture of mitochondrial health between samples taken weeks apart.
-
The finding that amino acid levels improved – significantly so for cysteine, and with a trend to improvement for arginine – must be treated with caution, since ideally amino acid levels should only be assayed in blood taken after a 4-hour fast. It was not deemed safe or considerate to insist on this in children who were undergoing a wide range of tests, including exercise testing. Recent meals or snacks could therefore have contributed to rises in amino acids.
-
Exercise imaging by echocardiography is particularly dependent on obtaining good imaging windows. In many participants, images were suboptimal.
-
Due to the COVID-19 restrictions on social gatherings, coupled with the participants’ need to shield throughout the pandemic, we have so far been unable to hold a dissemination meeting with participants.
Lessons for the future
The trial attempted to use state-of-the-art assessment and diagnostic tools, such as exercise echocardiography and muscle MRS. Whereas the former has been validated and introduced into clinical care by our research group, MRS in children is still an exploratory method in the clinical research setting, and protocols need to be better validated in larger numbers of children. The duration in the scanner was felt to be too long for some participants, yet is unavoidable at present. In summary, this methodology is very promising as it represents a potential objective functional outcome and is the only clinical investigation that can detect metabolic changes in muscle. However, more research on its implementation is needed before it is appropriate for inclusion as an outcome in future RCTs in paediatric populations.
It was difficult to get reproducible results for cardiolipin profiling. The methods are appropriate for diagnosis because the Barth cardiolipin profile is so extremely different from a normal profile. However, we now feel that cardiolipin ratio is not a good outcome measure. In patients with Barth syndrome, numerous different species of cardiolipin are present, whereas samples from normal humans have relatively few clearly defined peaks.
Cellular investigations also proved not to be fit for purpose. Electron microscopy is resource-intensive and expensive, and we were unable to image cells reproducibly despite expert technical input. Our experience leads us to recommend that these measurements are not considered as routine investigations in patients with Barth syndrome or as outcomes in future trials.
The elamipretide trial included a wider range of functional outcomes; for example, grip strength, 6MWT. 40 The latter was the primary outcome almost certainly because this was the outcome that the FDA was prepared to recognise with respect to a marketing authorisation. This is a reasonable outcome if an intervention can be effectively blinded, but there is a serious risk of bias if not – for example, in an open-label extension study – due to the confounding effect of a participant’s effort. We would recommend peak VO2 as the primary outcome in a future trial because it is less susceptible to bias; oxygen consumption is monitored dynamically, allowing an operator to distinguish peak exercise from submaximal effort. An international collaboration on a future trial may require the choice of primary outcome to be discussed with the FDA.
Future research
Evidence from this study and the murine bezafibrate trial performed after its conception suggest that manipulation of cardiolipin chemistry may not be achievable. We found no evidence for mitochondrial biogenesis in blood cells, although strong evidence was found in mouse cardiac muscle, paralleling evidence of cardiac benefit. However, given the findings of the trial, we do not think that further evaluation of bezafibrate as a treatment for Barth syndrome – for example, for a longer duration or at a higher dose – is warranted. More importantly, we also believe that there would be no appetite for further evaluation of bezafibrate among the Barth syndrome population and their families.
With respect to future trials of an alternative intervention, we think there is a need to carry out an international trial over a longer period. During the set-up of the trial, there was interest from the USA in running a parallel trial, but bezafibrate was not licensed there so this was not possible. We also now have a better idea of what endpoints are readily achievable as primary and secondary outcomes. Our experience of MRS was sufficient to enthuse us about the possibilities of the technology in future, but its lack of availability to a standard protocol is currently a barrier.
International non-commercial clinical trials of IMPs can be difficult to conduct for organisational and regulatory reasons. Running parallel trials (with separate sponsors and governance) may solve some issues, but there can still be major challenges and costs associated with manufacturing/importing identical IMPs and obtaining regulatory approval in different jurisdictions. A further major challenge is presented by the excellent degree of national and international cohesion and exchange of information through social media within this community of patients and families. The fact that we saw improvement in neither our primary outcome nor QoL after use of bezafibrate has been communicated through this network; hence our belief that the Barth community would not consider a longer trial of bezafibrate.
Although elamipretide has not been used in children below 12 years of age, statistically significant beneficial effects have been reported in the open-label extension study and sustained over as long as 3.5 years. 40 These observations arguably make elamipretide a stronger candidate for an international trial. If the manufacturer of elamipretide were to initiate a future commercial trial, the UK should seek to join it. Commercial sponsorship would help to overcome the barriers of governance and IMP manufacturing/distribution described above.
Chapter 12 Conclusion
The CARDIOMAN randomised controlled crossover trial did not show a significant treatment effect of bezafibrate for the primary outcome, peak VO2. Some secondary outcomes – such as systolic function measured by echocardiography, and amino acid levels – provided some evidence of benefit. Nevertheless, the trial produced important insights about trial methods and conduct in the context of a very rare paediatric disease that should inform future studies. These include the advantages and disadvantages of different outcomes, the intricacies of scheduling clinics with extensive multidisciplinary clinical assessments when using a crossover design, and recognising and enabling the importance of patient involvement throughout the trial to promote retention and adherence. The qualitative interviews found that the study was acceptable to participants and the burden of participating not too onerous. Adherence to the study medication was also good, with parents reporting few difficulties with pill swallowing.
Additional information
Contributions of authors
Guido Pieles (https://orcid.org/0000-0003-1203-688X) (Consultant Paediatric Cardiologist and Chief Investigator) contributed to the design of the study, obtained funding for the study, conducted some of the study assessments, contributed to the interpretation of the results, and contributed to the drafting and editing of the report.
Colin Steward (https://orcid.org/0000-0001-6291-0710) (Emeritus Professor of Paediatric Stem Cell Transplantation, laboratory lead and previous Chief Investigator), led the conception of the study and its design, obtained funding for the study, contributed to the interpretation of the results, led the drafting of the introduction and laboratory outcomes, and contributed to drafting the rest of the report.
Lucy Dabner (https://orcid.org/0000-0001-7269-1945) (Trial management, Bristol Trials Centre) was responsible for the set-up and day-to-day management of the study, coordinated the specials manufacture of bezafibrate and placebo, coordinated the research clinics, led the drafting of the methods, patient and public involvement and challenges sections, and contributed to drafting the rest of the report.
Laura Collett (https://orcid.org/0000-0002-6681-2146) (Statistician, Bristol Trials Centre) conducted the statistical analysis, contributed to the interpretation of results, and led the drafting of the SAP.
Lucy Culliford (https://orcid.org/0000-0002-9255-6617) (Senior Research Fellow in Trial Management, Bristol Trials Centre) contributed to the design of the study and advised on trial management throughout the study.
Karen Sheehan (https://orcid.org/0000-0003-1606-8184) (Research Sister, UHBW) contributed to the study design, recruitment and follow-up of participants and data acquisition, and led the research nurse team conducting the study clinics.
Lucy Ellis (https://orcid.org/0000-0001-8179-5172) (Senior Research Associate in Clinical Trials, Bristol Trials Centre) contributed to the day-to-day management of the study and data acquisition.
Michaela Damin (https://orcid.org/0000-0003-0776-5914) (Barth Syndrome UK) contributed to the study design, obtained funding for the study, and liaised with participants, their families and the wider Barth syndrome community during the trial.
Eva Sammut (https://orcid.org/0000-0002-4198-6860) (NIHR Clinical Fellow, Cardiology) contributed to MRS design and data acquisition, and contributed to drafting the report.
Nuno Duarte (https://orcid.org/0000-0003-3136-0079) (Sonographer, UHBW) contributed to data acquisition of the echocardiograms.
Owen Burgess (https://orcid.org/0000-0002-2321-6838) (Sonographer, UHBW) contributed to data acquisition of the echocardiograms.
Curtis Wadey (https://orcid.org/0000-0003-3275-7975) (PhD student, University of Exeter) contributed to data acquisition of the metabolic exercise testing.
Craig Williams (https://orcid.org/0000-0002-1740-6248) (Professor and Director of the Children’s Health and Exercise Research Centre, University of Exeter) contributed to the design of the exercise assessments and to data acquisition of the metabolic exercise testing and interpretation of results.
John Crosby (https://orcid.org/0000-0002-8389-7102) (Senior Lecturer, School of Chemistry, UoB) conducted the cardiolipin assessments and contributed to the drafting the corresponding methods and results sections of the report and the interpretation of the results.
Sarah Groves (https://orcid.org/0000-0001-8801-5749) (Laboratory technician, UoB) prepared the study samples for laboratory analysis, conducted the laboratory tests (data acquisition), supervised and coordinated the management of laboratory samples, and contributed to the drafting of the methods section of the report.
Aidan Searle (https://orcid.org/0000-0001-9860-3253) (Qualitative researcher, UoB) contributed to the conduct and management of the qualitative study and the interpretation of the results, and drafted the corresponding methods and results sections of the report.
Borko Amulic (https://orcid.org/0000-0002-8518-8393) (Senior Research Fellow, UoB) contributed to the analysis of some laboratory assessments and their interpretation for the report.
Chris Rice (https://orcid.org/0000-0002-8158-4022) (Senior Research Associate, UoB) contributed to the analysis of some laboratory assessments and their interpretation for the report.
Chiara Bucciarelli-Ducci (https://orcid.org/0000-0002-2515-0852) (Consultant Cardiologist, UHBW) contributed to the design, obtained funding for the study, and carried out or supervised analysis of the cardiac MRI assessments.
Andrew Ness (https://orcid.org/0000-0003-3548-9523) (Emeritus Professor of Epidemiology, UoB) contributed to the initial study conception and the study design, and obtained funding for the study.
Julian Hamilton-Shield (https://orcid.org/0000-0003-2601-7575) (Professor in Diabetes and Metabolic Endocrinology, UoB) contributed to the initial study conception and the study design, and obtained funding for the study.
Chris A Rogers (https://orcid.org/0000-0002-9624-2615) (Professor of Medical Statistics, UoB) contributed to the design of the study, and was responsible for oversight of the statistical analyses and SAP.
Barnaby C Reeves (https://orcid.org/0000-0002-5101-9487) (Professor of Health Services Research, UoB) contributed to the design of the study, obtained funding for the study, and contributed to the interpretation of the results and drafting of the final report.
Acknowledgements
The trial could not have been completed successfully without the help of Kathleen Selway and Julie Madden, both research nurses with the paediatric cardiac surgery research team at UHBW. They worked tirelessly throughout the recruitment and follow-up periods of the trial, including (but not limited to) welcoming participants and their families to clinics, conducting study assessments, arranging safety blood tests, making follow-up telephone calls, and collecting study data on Case Report Forms. Kathleen Selway was also involved in the planning stages of the trial and helped with the clinic arrangements. The trial team would also like to express their thanks and gratitude to the members of the oversight committees for their help, advice and guidance throughout the trial.
Trial Steering Committee: list of independent members
| Name | Position | Role in TSC |
|---|---|---|
| Professor Tim Barrett | Leonard Parsons Professor of Paediatrics, University of Birmingham | Chair |
| Dr Paul Clift | Consultant Congenital Cardiologist | Member |
| Professor Rob Wynn | Consultant Paediatric Haematologist and Director of Paediatric BMT Programme | Member |
| Mrs Elizabeth Stobart-Hook | Patient and public representative | Member |
Data monitoring and safety committee: list of independent members
| Name | Position | Role in DMSC |
|---|---|---|
| Professor Stephen Evans | Professor of Pharmacoepidemiology | Chair |
| Professor John Gregory | Professor of Paediatric Endocrinology | Member |
| Dr Jacob Simmonds | Consultant Cardiologist and Transplant Physician | Member |
Data-sharing statement
Anonymised individual patient data (baseline, intervention, outcome data and AEs) will be made available for secondary research, conditional on assurance from the secondary researcher that the proposed use of the data is compliant with the UK Policy Framework for Health and Social Care Research and MRC Policy on Data Preservation and Sharing regarding scientific quality, ethical requirements and value for money. All data requests should be submitted to btc-mailbox@bristol.ac.uk for consideration, and access may be granted following review.
Data will only be made available after publication of the primary results. Only data from patients who have consented for their data to be shared with other researchers will be provided.
Ethics statement
The trial was given a favourable ethical opinion (reference IRAS 1703710) by the UK (South West – Central Bristol) National Research Ethics Service Committee on 12 November 2015. The trial was also approved by the MHRA (reference EUDRACT 2015-001382-10).
Information governance statement
All information collected is held securely and treated in accordance with the Regulation (EU) 2016/679 (the ‘General Data Protection Regulation’ or ‘UK GDPR’) and the Data Protection Act 2018. Under the Data Protection legislation, both the University Hospitals Bristol and Weston NHS Foundation Trust (UHBW) and the University of Bristol (UoB) are controllers of personal data for the purposes of the Data Protection Act 2018 and the General Data Protection Regulation. You can find out more about how we handle personal data, including how to exercise your individual rights and the contact details for our Data Protection Officers here: https://www.uhbw.nhs.uk/p/how-we-use-your-data/what-we-do-with-your-information and https://www.bristol.ac.uk/secretary/data-protection/policy/research-participant-fair-processing-notice/).
Disclosure of interests
Full disclosure of interests: Completed ICMJE forms for all authors, including all related interests, are available in the toolkit on the NIHR Journals Library report publication page at https://doi.org/10.3310/JDBC7982.
Primary conflicts of interest: Guido Pieles is lead researcher in a contractual research partnership between the University of Bristol and Canon Medical Systems UK. He is vice chair of UK MHRA CHM Paediatric Medicine Expert Advisory Group (PMEAG) and COVID vaccine external adviser to JCVI, CHM and UK HSA. He is chair of the UK HSA WG on COVID vaccine-associated myocarditis and chairing author of the UK HSA guideline on this topic. Colin Steward reports voluntary membership of the Scientific and Medical Advisory Board, Barth Syndrome Foundation, USA. Chris A Rogers reports membership of a Clinical Trials Unit funded by the National Institute for Health and Care Research. She also reports membership of the Health Technology Assessment Funding Committee Policy Group (formerly CSG) and the Health Technology Assessment Commissioning Committee. She has no other competing interests. Barnaby C Reeves reports former membership of: the Health Technology Assessment Commissioning Board (from January 2012 to 31 March 2016); the Health Technology Assessment Efficient Study Designs Board (October–December 2014); HTA Commissioning Sub-Board (EOI) from 1 April 2016 to 31 March 2017; HTA IP Methods Group from 7 December 2016 (end date unknown); and SRP – Cochrane Programme Grant Funding Meeting (dates unknown). He also reports: current membership of the Health Technology Assessment Interventional Procedures Committee B Methods Group and Systematic Reviews Programme Advisory Group (Systematic Reviews National Institute for Health and Care Research Cochrane Incentive Awards and Systematic Review Advisory Group); and membership of a CTU funded by the NIHR. He has no other competing interests.
Publications
Dabner L, Pieles GE, Steward CG, Hamilton-Shield JP, Ness AR, Rogers CA, et al. Treatment of Barth Syndrome by Cardiolipin Manipulation (CARDIOMAN) with bezafibrate: protocol for a randomized placebo-controlled pilot trial conducted in the Nationally Commissioned Barth Syndrome Service. JMIR Res Protoc 2021;10(5):e22533. https://doi.org/10.2196/22533
Searle A, Herbert G, Dabner L, Steward CG, Damin M, Pieles G. Self-regulation in Barth syndrome: a qualitative perspective of adolescents, adults and parents in the UK. Orphanet J Rare Dis 2021;16:404. https://doi.org/10.1186/s13023-021-02027-5
Disclaimers
This article presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, the EME programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, the EME programme or the Department of Health and Social Care.
References
- Clarke SL, Bowron A, Gonzalez IL, Groves SJ, Newbury-Ecob R, Clayton N, et al. Barth syndrome. Orphanet J Rare Dis 2013;8.
- Schlame M. Cardiolipin remodeling and the function of tafazzin. Biochim Biophys Acta 2013;1831:582-8.
- Basu Ball W, Neff JK, Gohil VM. The role of nonbilayer phospholipids in mitochondrial structure and function. FEBS Lett 2018;592:1273-90.
- Dudek J, Cheng I-F, Chowdhury A, Wozny K, Balleininger M, Reinhold R, et al. Cardiac-specific succinate dehydrogenase deficiency in Barth syndrome. EMBO Mol Med 2016;8:139-54.
- van Werkhoven MA, Thorburn DR, Gedeon AK, Pitt JJ. Monolysocardiolipin in cultured fibroblasts is a sensitive and specific marker for Barth syndrome. J Lipid Res 2006;47:2346-51.
- Kulik W, van Lenthe H, Stet FS, Houtkooper RH, Kemp H, Stone JE, et al. Bloodspot assay using HPLC–tandem mass spectrometry for detection of Barth syndrome. Clin Chem 2008;54:371-8.
- Houtkooper RH, Rodenburg RJ, Thiels C, van Lenthe H, Stet F, Poll-The BT, et al. Cardiolipin and monolysocardiolipin analysis in fibroblasts, lymphocytes, and tissues using high-performance liquid chromatography–mass spectrometry as a diagnostic test for Barth syndrome. Anal Biochem 2009;387:230-7.
- Storch EA, Keeley M, Merlo LJ, St Amant JB, Jacob M, Storch JF, et al. Psychosocial Functioning in Youth with Barth Syndrome. Child Health Care 2009;38:137-56.
- Human Tafazzin Gene Variants Database n.d. https://www.barthsyndrome.org/research/tafazzindatabase.html (accessed 9 June 2023).
- Steward CG, Groves SJ, Taylor CT, Maisenbacher MK, Versluys B, Newbury-Ecob RA, et al. Neutropenia in Barth syndrome: characteristics, risks, and management. Curr Opin Hematol 2019;26:6-15.
- Xu Y, Phoon CKL, Berno B, D’Souza K, Hoedt E, Zhang G, et al. Loss of protein association causes cardiolipin degradation in Barth syndrome. Nat Chem Biol 2016;12:641-7.
- Bowron A, Honeychurch J, Williams M, Tsai-Goodman B, Clayton N, Jones L, et al. Barth syndrome without tetralinoleoyl cardiolipin deficiency: a possible ameliorated phenotype. J Inherit Metab Dis 2015;38:279-86.
- Bhullar KS, Hubbard BP. Lifespan and healthspan extension by resveratrol. Biochim Biophys Acta 2015;1852:1209-18.
- Goldenberg I, Boyko V, Tennenbaum A, Tanne D, Behar S, Guetta V. Long-term benefit of high-density lipoprotein cholesterol-raising therapy with bezafibrate: 16-year mortality follow-up of the bezafibrate infarction prevention trial. Arch Intern Med 2009;169:508-14.
- Monk JP, Todd PA. Bezafibrate. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in hyperlipidaemia. Drugs 1987;33:539-76.
- Tenenbaum A, Fisman EZ. Balanced pan-PPAR activator bezafibrate in combination with statin: comprehensive lipids control and diabetes prevention?. Cardiovasc Diabetol 2012;11.
- Huang Y, Powers C, Moore V, Schafer C, Ren M, Phoon CKL, et al. The PPAR pan-agonist bezafibrate ameliorates cardiomyopathy in a mouse model of Barth syndrome. Orphanet J Rare Dis 2017;12.
- Schafer C, Moore V, Dasgupta N, Javadov S, James JF, Glukhov AI, et al. The effects of PPAR stimulation on cardiac metabolic pathways in Barth syndrome mice. Front Pharmacol 2018;9.
- Bonnefont JP, Bastin J, Laforêt P, Aubey F, Mogenet A, Romano S, et al. Long-term follow-up of bezafibrate treatment in patients with the myopathic form of carnitine palmitoyltransferase 2 deficiency. Clin Pharmacol Ther 2010;88:101-8.
- Dorobantu DM, Wadey CA, Amir NH, Stuart AG, Williams CA, Pieles GE. The role of speckle tracking echocardiography in the evaluation of common inherited cardiomyopathies in children and adolescents: a systematic review. Diagnostics (Basel) 2021;11.
- Dabner L, Pieles GE, Steward CG, Hamilton-Shield JP, Ness AR, Rogers CA, et al. Treatment of Barth Syndrome by Cardiolipin Manipulation (CARDIOMAN) With Bezafibrate: protocol for a randomized placebo-controlled pilot trial conducted in the Nationally Commissioned Barth Syndrome Service. JMIR Res Protoc 2021;10. www.researchprotocols.org/2021/5/e22533/ (accessed 29 March 2023).
- Graham DJ, Staffa JA, Shatin D, Andrade SE, Schech SD, La Grenade L, et al. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA 2004;292:2585-90.
- Bezafibrate n.d. https://go.drugbank.com/drugs/DB01393 (accessed 29 March 2023).
- Schwartz GJ, Work DF. Measurement and estimation of GFR in children and adolescents. Clin J Am Soc Nephrol 2009;4:1832-43.
- Pieles GE, Gowing L, Forsey J, Ramanujam P, Miller F, Stuart AG, et al. The relationship between biventricular myocardial performance and metabolic parameters during incremental exercise and recovery in healthy adolescents. Am J Physiol Heart Circ Physiol 2015;309:H2067-76.
- Pieles GE, Gowing L, Ryding D, Perry D, McNally SR, Stuart AG, et al. Characterisation of LV myocardial exercise function by 2-D strain deformation imaging in elite adolescent footballers. Eur J Appl Physiol 2021;121:239-50.
- Lopez L, Colan SD, Frommelt PC, Ensing GJ, Kendall K, Younoszai AK, et al. Recommendations for quantification methods during the performance of a pediatric echocardiogram: a report from the pediatric measurements writing group of the American Society of Echocardiography Pediatric and Congenital Heart Disease Council. J Am Soc Echocardiogr 2010;23:465-95.
- Lai WW, Geva T, Shirali GS, Frommelt PC, Humes RA, Brook MM, et al. Task Force of the Pediatric Council of the American Society of Echocardiography . Guidelines and standards for performance of a pediatric echocardiogram: a report from the Task Force of the Pediatric Council of the American Society of Echocardiography. J Am Soc Echocardiogr 2006;19:1413-30.
- Voigt JU. Cardiac resynchronization therapy responders can be better identified by specific signatures in myocardial function. Eur Heart J Cardiovasc Imaging 2016;17:132-3.
- Purvis LAB, Clarke WT, Biasiolli L, Valkovič L, Robson MD, Rodgers CT. OXSA: an open-source magnetic resonance spectroscopy analysis toolbox in MATLAB. PLoS One 2017;12.
- OXSA (Oxford Spectroscopy Analysis) Toolbox n.d. https://github.com/oxsatoolbox/oxsa (accessed 29 March 2023).
- Alderfer MA, Marsac ML. Encyclopedia of Behavioral Medicine. New York, NY: Springer New York; 2013.
- Braun V, Clark V. Using thematic analysis in psychology. Qual Res Psychol 2006;3:77-101.
- Searle A, Herbert G, Dabner L, Steward CG, Damin M, Pieles G. Self-regulation in Barth syndrome: a qualitative perspective of adolescents, adults and parents in the UK. Orphanet J Rare Dis 2021;16.
- NIHR Themed Call: Very Rare Diseases 2012. https://www.nihr.ac.uk/documents/themed-call-very-rare-diseases/24064 (accessed 29 March 2023).
- Dragulescu A, Mertens LL. Developments in echocardiographic techniques for the evaluation of ventricular function in children. Arch Cardiovasc Dis 2010;103:603-14.
- Vernon HJ, Sandlers Y, McClellan R, Kelley RI. Clinical laboratory studies in Barth syndrome. Mol Genet Metab 2014;112:143-7.
- Acehan D, Xu Y, Stokes DL, Schlame M. Comparison of lymphoblast mitochondria from normal subjects and patients with Barth syndrome using electron microscopic tomography. Lab Invest 2007;87:40-8.
- Groves SJ. Personal Communication n.d.
- Reid Thompson W, Hornby B, Manuel R, Bradley E, Laux J, Carr J, et al. A phase 2/3 randomized clinical trial followed by an open-label extension to evaluate the effectiveness of elamipretide in Barth syndrome, a genetic disorder of mitochondrial cardiolipin metabolism. Genet Med 2021;23:471-8.
List of abbreviations
- 6MWT
- 6-minute walk test
- AE
- adverse event
- ATP
- adenosine triphosphate
- BSF
- Barth Syndrome Foundation
- BSS
- Barth Syndrome Service
- BST
- Barth Syndrome Trust
- BTHS-SA
- Barth syndrome symptom assessment scale
- CARDIOMAN
- CARDIOlipin MANipulation trial
- CI
- confidence interval
- CLi
- immature cardiolipin
- CLm
- mature cardiolipin
- CONSORT
- Consolidated Standards of Reporting Trials
- CRIC
- Clinical Research and Imaging Centre
- CTEU
- Clinical Trials and Evaluation Unit
- DMSC
- Data Monitoring and Safety Committee
- EBV
- Epstein–Barr virus
- ECG
- electrocardiogram
- EF
- ejection fraction
- eGFR
- estimated glomerular filtration rate
- FCCP
- carbonyl cyanide-p-trifluoromethoxyphenylhydrazone
- FDA
- US Food and Drug Administration
- G-CSF
- granulocyte colony-stimulating factor
- IMP
- investigational medicinal product
- IQR
- interquartile range
- ITT
- intention-to-treat
- L4-CL
- tetralinoleoyl-cardiolipin
- LFT
- liver function test
- LV
- left ventricular
- LVEF
- left ventricular ejection fraction
- MFI
- median fluorescent intensity
- MHRA
- Medicines and Healthcare Products Regulatory Agency
- MLCL
- monolysocardiolipin
- MRC
- Medical Research Council
- MRI
- magnetic resonance imaging
- MRS
- magnetic resonance spectroscopy
- NIHR
- National Institute for Health and Care Research
- PCr
- phosphocreatine
- PedsQL™
- Pediatric Quality of Life Inventory
- PIL
- patient information leaflet
- PTU
- pharmacy trials unit
- QoL
- quality of life
- RCT
- randomised controlled trial
- REC
- research ethics committee
- RVEF
- right ventricular ejection fraction
- SAE
- serious adverse event
- SAP
- statistical analysis plan
- SD
- standard deviation
- SmPC
- Summary of Product Characteristics
- TAZ
- tafazzin gene
- TMRE
- tetramethylrhodamine methyl ester perchlorate
- TSC
- Trial Steering Committee
- UHBW
- University Hospitals Bristol and Weston NHS Foundation Trust
- UoB
- University of Bristol
- VO2
- volume of oxygen
Notes
-
Patient information leaflets for: children < 11 years, children 11-15 years, adults (≥ 16 years), and parents/guardians
Supplementary material can be found on the NIHR Journals Library report page (https://doi.org/10.3310/JDBC7982).
Supplementary material has been provided by the authors to support the report and any files provided at submission will have been seen by peer reviewers, but not extensively reviewed. Any supplementary material provided at a later stage in the process may not have been peer reviewed.