

Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 11/100/34. The contractual start date was in April 2013. The final report began editorial review in September 2022 and was accepted for publication in December 2022. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
Copyright © 2023 Stringer et al. This work was produced by Stringer et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This is an Open Access publication distributed under the terms of the Creative Commons Attribution CC BY 4.0 licence, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. See: https://creativecommons.org/licenses/by/4.0/. For attribution the title, original author(s), the publication source – NIHR Journals Library, and the DOI of the publication must be cited.
2023 Stringer et al.
Introduction
Scientific background
Kidney transplants do not last for the natural lifespan of most recipients, 30–40% of patients have their transplant for < 10 years1 and around 3% of prevalent kidney transplants fail annually. 2 This places a large burden on healthcare services. The single biggest cause of transplant failure is immune-mediated injury, the target of which are mismatched donor human leucocyte antigens (HLA).
A validated prognostic biomarker of kidney transplant failure is the appearance of circulating antibodies (Ab) against HLA. 3–8 Patients with HLA Ab have a three-fold greater risk of graft failure compared to those without7,8 and if these are specific for the kidney donor HLA [donor-specific antibodies (DSA)] there is an even higher risk of graft loss compared to those Ab that are not donor-specific (non-DSA). Inappropriately low levels of immunosuppression, either physician-led or due to patient non-adherence, is an important factor allowing the immune-mediated damage to begin and promoting the appearance of the HLA Ab. 9
Rationale for the study
The mechanisms driving graft dysfunction leading to graft failure are most likely complex and although the HLA Ab themselves might be damaging,10 other components including T- and B-lymphocytes may also play a role. 11 Various novel therapies have been tested in small scale, often uncontrolled human studies with some promising results. 12,13 Two randomised controlled trials concluded that B cell depletion with Rituximab was ineffective at preventing graft dysfunction in patients with biopsy-proven chronic antibody-mediated rejection (CAMR)14,15 as did a smaller trial of the anti-IL-6 receptor Ab tocilizumab. 16 However, two small RCTs of the anti-IL-6 monoclonal Ab Clazakizumab have shown benefit17,18 A larger RCT of clazakizumab, with a planned recruitment of 350 patients is underway (https://clinicaltrials.gov/ct2/show/NCT03744910). Several other studies have suggested that optimised oral treatment with tacrolimus (Tac) and mycophenolate mofetil (MMF) can stabilise graft function in small numbers of patients with existing graft dysfunction. 15,19–26 However, to date there have been no large-scale trials testing this strategy by intervening in patients who develop HLA Ab prior to developing graft dysfunction, and none that have assessed if graft failure can be prevented.
Methods
Sections of this report have been reproduced from Dorling et al. 27 under licence CC-BY-2.0.
Sections of this report have been reproduced from Stringer et al. 28 under licence CC-BY-4.0.
Design
OuTSMART was a prospective, open labelled, randomised marker-based strategy (hybrid) trial design, with two arms stratified by biomarker (HLA Ab) status. Recruits were followed up with regular structured visits for at least 32 months (maximum 64 months) and primary endpoint assessed by remote evaluation after approximately 43 months post-randomisation (or post ‘clock reset’ – see below) was achieved by all. The trial design is represented in Figure 1. All eligible patients were screened for HLA Ab before being randomised 1 : 1 into either double-blinded standard care (SC) or unblinded biomarker-led care (BLC). Biomarker stratification generated three groups of recruits in each arm (DSA+, non-DSA+ and HLA Ab-neg). Patients in the blinded SC arm (groups A1, A2 and C in Figure 1) were blind to their biomarker status and remained on baseline immunotherapy throughout, whereas patients in the unblinded (groups B1, B2 and D in Figure 1) knew their HLA Ab status; in this arm, those with HLA Ab were offered ‘intervention’, whereas HLA Ab-negative patients remained on their existing immunotherapy.
FIGURE 1.
Trial design/flow of patients. Legend to figure 1: Randomisation took part in the HLA laboratory, after the first HLA Ab screening round to allow biomarker stratification within the respective arms. All participants assigned to the HLA Ab negative groups in each arm underwent rescreening for HLA Ab every 8 months till month 32. In both arms, recruits changing from HLA Ab negative to positive, at any time after enrolment were asked to complete a further 32 months follow-up (=‘clock reset’), so that the maximum time under structured follow-up for any recruit was 64 months. To maintain blinding, the randomisation system was programmed at each screening round to choose a small random group of HLA Ab-negative recruits from the SC group to complete a further 32 months follow-up. Therefore, a subset of HLA Ab-negative patients had ‘clock reset’ in the blinded SC arm, whereas only HLA Ab+ participants in unblinded BLC had ‘clock reset’.
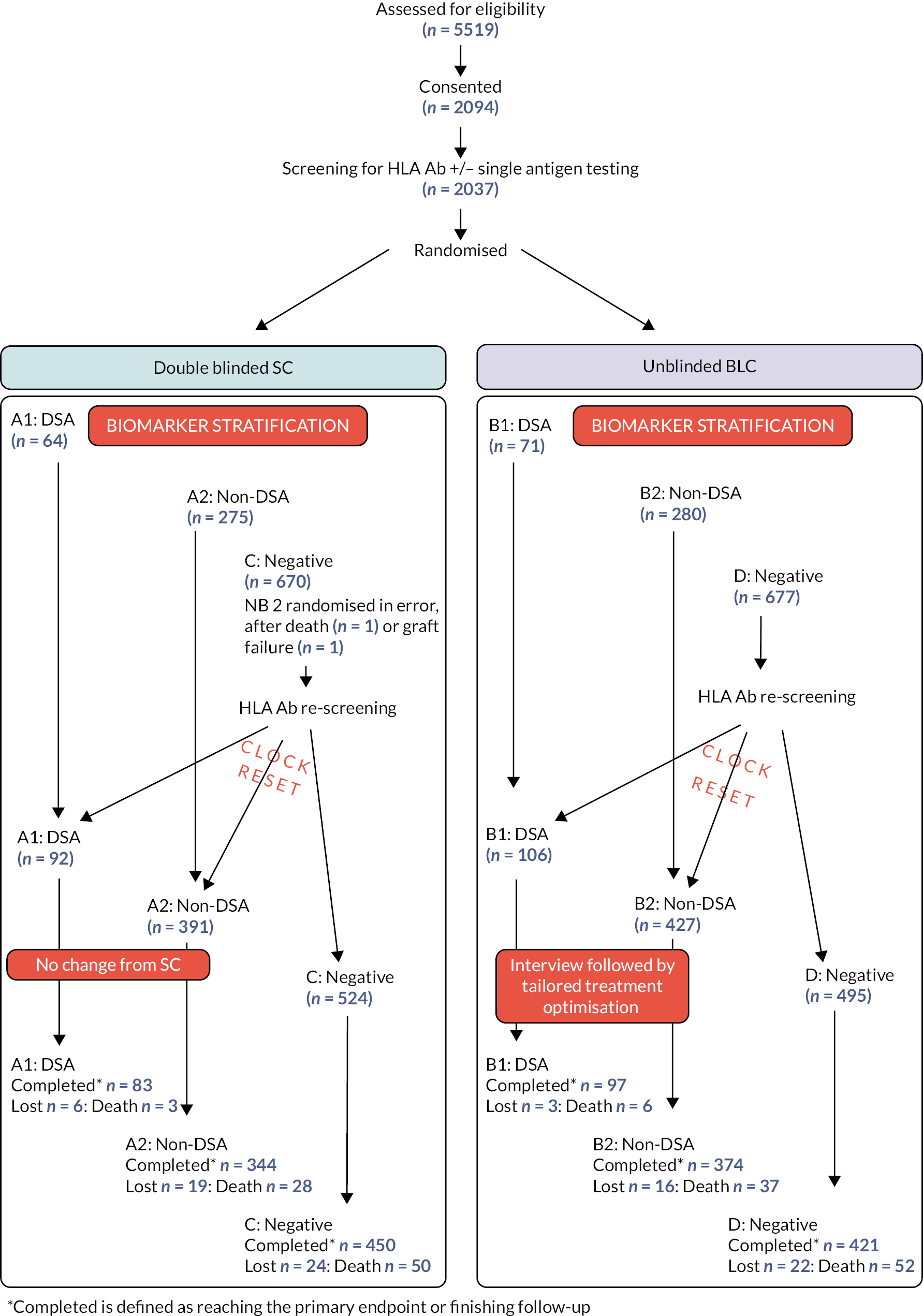
Both groups of HLA Ab-negative recruits had regular Ab status monitoring for the first 32 months. Those patients who became positive during subsequent screening rounds were moved to the appropriate HLA Ab positive groups (DSA+ or non-DSA+) for final data analysis. All patients in groups C and D found to be positive on second or subsequent rounds were intensively followed up for an additional 32 months from the time they become positive (=‘clock reset’): those in the BLC arm were also offered the same ‘intervention’ as those patients who were positive in the first screening round. To maintain blinding in the SC arm, the randomisation system was programmed at each screening round to choose a small random group of HLA Ab-negative recruits from this arm to complete a further 32 months follow-up. Thus the maximum amount of time any single patient remained in intensive follow-up was 64 months. At the end of intensive follow-up, most data collection related to secondary endpoints ceased, but data related to graft failure or death were recorded for all participants up and until the end of the trial (see below), irrespective of the length of follow-up.
Primary objective
Determine the time to graft failure in unblinded BLC HLA Ab+ recruits identified at baseline or within 32 months of randomisation, compared to the control group of blinded SC HLA Ab+ recruits who remain on their established immunotherapy and whose clinicians are not aware of their Ab status. The primary endpoint was to be assessed remotely when approximately 43 months post-randomisation or post-‘clock reset’ had been achieved by all. Graft failure was defined as restarting dialysis or requiring a new transplant. In 2020, because of the impact of the first COVID-19 pandemic, the primary endpoint was redefined as that obtained at the last follow-up prior to 16 March 2020.
Secondary objectives
-
determine the time to graft failure in patients randomised to ‘unblinded’ HLA Ab screening, compared to a control group randomised to ‘blinded’ HLA Ab screening;
-
determine whether ‘treatment’ influences patient survival;
-
determine whether ‘treatment’ influences the development of graft dysfunction as assessed by presence of proteinuria (protein:creatinine ratio > 50 or albumin:creatinine ratio > 35) and change in estimated glomerular filtration rate (eGFR);
-
determine whether ‘treatment’ influences the rates of acute rejection in these groups;
-
determine the adverse effect profiles of ‘treatment’ in this group, in particular whether they are associated with increased risk of infection, malignancy or diabetes mellitus (DM);
-
determine the cost-effectiveness of routine screening for HLA Ab and prolonging transplant survival using this screening/treatment protocol;
-
determine the impact of biomarker screening and ‘treatment’ on the patients’ adherence to drug therapy and their perceptions of risk to the health of the transplant.
For all except (1) and (2), the secondary outcomes were assessed at the end of the intensive follow-up period, which for most was at month 32 but for some was up to 64 months post-enrolment.
Interventions
All Unblinded HLA Ab+ recruits were interviewed by the site principal investigator (PI) and the importance of drug adherence was re-enforced. Changes in drug treatment were tailored to the individual patient, according to compliance, tolerance and achievement of target levels (for Tac). Failure to tolerate one or more of the components of the drug protocol (or refusal to take any of the agents) was not used as a reason for withdrawal from the study.
The ‘optimised treatment’ protocol in the two groups (B1, B2, Figure 1) with HLA Ab was:
-
MMF bd, tds or qds, or enteric-coated mycophenolic acid (MPA) bd, with daily dose determined according to local unit guidelines. The patient was stabilised on the maximum tolerated dose.
-
Tac once daily (od) or bd, according to local unit preference, with dose titrated to achieve 12-hour post-dose levels of 4 μg/L to 8 μg/L (4–8 ng/ml). The patient was stabilised on the maximum tolerated dose that achieves these levels.
-
Prednisolone od starting at 20 mg for two weeks, then reducing by 5 mg od every two weeks down to their previous maintenance dose or 5 mg od, if not previously taking.
After consultation with the Medicines and Healthcare products Regulatory Agency, all these medicines (but no others) were classed as investigational medical products (IMPs) MMF/MPA was used outside of its Marketing Authorisation (which states that it should be used with ciclosporin). However, because it is used so widely in combination of Tac in most units in the United Kingdom (UK), the two were regarded as ‘SC’. Thus, none of the three drugs required labelling in line with annex 13 and all IMPs were managed in the same way as normal that is GP or hospital prescription (as appropriate) and did not require special labelling/accountability/storage, etc.
Setting
Thirteen UK Kidney transplant outpatient clinics.
Participants
Renal transplant recipients > 1 year post-transplantation.
Identification and recruitment
The local transplant clinic database of their prevalent population was used to identify patients meeting the baseline inclusion/exclusion criteria. At the start of the trial, the entire population of transplant clinic attendees who met the eligibility criteria were potentially eligible for recruitment. On subsequent screening rounds, patients who reached 12 months post-transplantation after the start of the trial became eligible. Potentially eligible patients were approached at a routine clinic appointment by the PI or research nurses and given printed and verbal information about the trial. They had the opportunity to return for a second consultation within a few days to give informed consent for recruitment into the study or to do this on their next routine appointment. Alternatively, some eligible patients were sent information about the study through the post, for discussion and consent at their next routine appointment. Following consent, full eligibility criteria were reviewed. This included testing for chronic viral disease (if no such test within last 5 years) or pregnancy (if history suggests possibility of pregnancy).
Randomisation procedure
Prior to randomisation but after consent, site staff registered all recruits online and each assigned a MACRO PIN. Samples from all recruits were sent to the relevant HLA laboratory, along with this PIN and a sample request form containing the other information required for randomisation. HLA laboratory staff performed a screen for HLA Ab and performed single antigen bead testing on positive screening samples to check for the presence of DSA. Once this information was known, the laboratory staff accessed the randomisation system and randomised the patient, using the HLA Ab results and information on the sample form to stratify.
Allocation to SC (blinded) or BLC (unblinded) arms was assigned (1 : 1) by stratified block randomisation with randomly varying block sizes, using a web-based randomisation service provided by the King’s Clinical Trials Unit. Randomisation was stratified by (1) HLA Ab status, to generate three groups within each arm (DSA+, non-DSA+ or HLA Ab-negative), (2) current immunosuppression (to ensure balanced numbers already on Tac or MMF) and (3) site (N = 13). The randomisation allocation was initiated by staff within the five HLA (tissue-typing) laboratories involved in the trial.
Blinding
There was no blinding of arm allocation. In all sites, immediately after randomisation, the PIs and nurses were automatically emailed with information about whether the patient was in the blinded or unblinded groups. If in the unblinded group, the email to the PI contained information about the HLA Ab status. The system told trial staff to enter HLA Ab-negative patients into the subsequent 8 monthly screening rounds.
In blinded patients, HLA Ab status was not fed back to the PIs or trial staff in the emails. All blinded patients had samples taken 8 monthly for HLA Ab screening, though upon sample receipt, HLA laboratory staff used their knowledge of the HLA status to determine those from HLA Ab-negative patients which underwent screening and samples from HLA Ab-positive patients were discarded.
On the second and subsequent HLA Ab screening rounds, the laboratory staff updated the randomisation system within 52 days from the date the rescreen sample was taken. Only the results from patients in the unblinded groups were forwarded to the PI and lab staff, via email. This indicated whether status had changed and triggered the initiation of the treatment protocol in those that had changed from HLA Ab negative to positive. It also indicated that patients who had become HLA Ab+ needed ‘clock reset’ to extend the period of intensive follow-up for a further 32 months.
In the blinded arm, PIs were emailed with a list of patients who required ‘clock reset’ so they got follow-up for a further 32 months. This list contained all the recruits who had become HLA Ab+ on rescreening, but also an equivalent number of recruits who had stayed HLA Ab-; these recruits were randomly chosen by the randomisation system as a mechanism to maintain physician and patient blinding to HLA Ab status within this arm.
There were no blinded study medications in the trial so no emergency code break was required. There were no requests for recruits’ HLA Ab status to be unblinded.
Inclusion criteria
-
sufficient grasp of English to enable written and witnessed informed consent to participate;
-
aged 18–75 years;
-
estimated glomerular filtration rate (eGFR by four variable MDRD) of ≥ 30 ml/min (within the previous 6 months of signing consent or taken at screening if not done in the previous 6 months).
Exclusion criteria29
-
recipient requiring HLA desensitisation to remove Ab for a positive cross match (XM) transplant;
-
recipient known already to have HLA Ab who has received specific intervention for that Ab or for CAMR/chronic rejection;
-
recipient of additional solid organ transplants (e.g. pancreas, heart, etc.);
-
history of malignancy in previous 5 years (excluding non-melanomatous tumours limited to skin);
-
HBsAg+, HepC immunoglobulin G (IgG+) or human immunodeficiency virus (HIV+) recipient (on test performed within previous 5 years);
-
history of acute rejection requiring escalation of immunosuppression in the 6 months prior to screening;
-
patient enrolled in any other studies involving administration of another IMP at time of recruitment;
-
known hypersensitivity to any of the IMPs;
-
known hereditary disorders of carbohydrate metabolism;
-
pregnancy or breastfeeding females (based on verbal history of recipient);
-
pre-menopausal females who refuse to consent to using suitable methods of contraception throughout the trial.
Participant withdrawal
Individual recruits were free to withdraw at any time and the PIs also had the right to withdraw patients from the study drug in the event of inter-current illness, adverse events (AEs), serious adverse events (SAEs), suspected unexpected serious adverse reactions, protocol violations, cure, administrative or other reasons. After every withdrawal from ‘treatment’, efforts were made to obtain permission to continue to collect study-specific data before patients were completely withdrawn from the study. Failure to tolerate one or more components of the ‘treatment’ was not seen as a reason to withdraw an individual participant from the trial.
Significant amendments to study protocol
The complete list of changes over the course of the trial is included in Appendix 1. The following are those judged to have altered the conduct of the trial. All changes were discussed and approved by the Trial Steering Committee or Chairman and, where appropriate, by the Data Monitoring Committee.
In Version 4 (13/5/2013), we clarified that the eGFR measurement on which eligibility was be assessed had to be within 1 month of signing consent, and also clarified the definition of a positive HLA Ab test, which was confusing in the previous protocol versions.
In Version 5 (9/7/2013), the definition of diabetes mellitus was updated to incorporate the WHO definition (use of HbA1c) and the reporting of AEs in this type A trial to the sponsor was clarified.
In Version 7 (7/4/2014), we removed the exclusion criteria ‘history of ongoing or previous infection that would prevent optimisation’ which was being interpreted differently within and across sites. In addition, the gap for the testing of eGFR from within 1 month of signing consent was increased to within the previous 6 months of signing the consent. Finally, the timing of the optimisation process was changed from within 3 months of HLA Ab positivity to ideally within 3 months after positive screening for HLA Ab and allocation to the unblinded treatment arm or as soon as possible thereafter BUT within 8 months of positive screening. This coincided with the realisation that some patients were proving difficult to contact to arrange optimisation and the change was felt to enhance the optimisation process without affecting the outcome of the trial.
In Version 8 (1/7/2014), the time that tissue typing laboratories had to perform the randomisation of patients, was increased from 28 to 56 days post-consent. This was to enhance batching of patient serum for testing, reducing the number of experimental controls and HLA screening beads needed, and therefore the cost of screening.
In Version 10 (11/08/2015), the upper limit for eligibility into the study was increased from 70 to 75 years.
In Version 11 (26/11/2015), the primary objective and endpoint were changed from 3-year graft failure rates in HLA Ab+ patients in the SC versus BLC arms27 to ‘time to graft failure with variable follow-up (with a minimum of 43 months post-randomisation)’. The new primary endpoint was to be assessed remotely from patient notes once 43 months post-randomisation had been achieved by all. This change was required because, after 16 months recruitment, an audit of HLA Ab screening results revealed that the expected 9% prevalence and 3% incidence rates of DSA were actually 5.8% and 1.6%, respectively. 28 All patients already recruited were reconsented to allow this change. This change allowed for a reduction in the number of DSA patients to be recruited, and a significant shortening in the expected study duration while maintaining the power of the study.
Because there was no additional funding for these changes, existing workload was reduced by changing the timing of follow-up visits from 4-monthly to 8-monthly, the end visit for each participant changed from 36 to 32 months, along with the timing of the secondary endpoint assessments. Finally, there was a major reduction in the requirement for SAE reporting to the sponsor.
In Version 12 (1/12/16), we stopped collection of research blood samples and removed the secondary experimental/exploratory ‘scientific’ endpoints. This was required by the funder, who requested that the salary costs associated with the exploratory aspects of the trial be reallocated towards supporting the primary endpoint data collection.
Finally, in Version 14 (08/07/2020), we changed the timing of the collection of the primary endpoint, as a result of the COVID-19 pandemic, in addition to the proposal to included additional sensitivity analyses for the primary endpoint and extension of the study end date.
Statistics methodology
Sample size and power calculations
The primary purpose of the trial was to demonstrate superior outcomes using the defined treatment strategy in BLC recruits, and at the same time demonstrate non-inferior outcomes when the screening strategy is applied to the entire patient population. Time to graft failure was chosen as a clinically relevant primary outcome. As a reference for power calculations, we used the observed failure rates reported by Lachmann et al. 7 for HLA Ab+ and HLA Ab-neg patients. Since Lachman showed that failure rates differed between DSA+ and non-DSA+ patients, sample size calculations were carried out separately for these groups. The estimates of the differences in primary outcome between groups were based on two things; first, the results of preliminary data from patients with CR treated with a similar regime as used here; second, our assessment that large differences in primary outcome would be needed to make the screening programme cost-effective. Our sample size calculations were updated with the change to the protocol in version 11 (see above) and the revised calculations28 are reported here.
Statistical hypotheses
-
Superiority on Biomarker Positive Patients: refer to Figure 1 for groups
-
1.1) Group A1 > group B1: HLA Ab+ patients with DSA, randomised to SC (A1) were hypothesised to show higher graft failure rates than patients randomised to BLC (B1). We then hypothesised that the experimental treatment would bring the failure rate in group B1 down to that of non-DSA patients in SC (A2). Assuming 30% in group A1 should have experienced graft failure by 3 years follow-up (as in7), we expected treatment in group B1 to reduce the rate down to 16%, corresponding to a hazard ratio (HR) of 0.489. The expectation was for 11% and 21% failure among recruits with DSA in group A1 at 1 and 2 years follow-up, respectively, and extrapolating using a HR of 0.489, we expected BLC to reduce these to 5.5% and 10.9%. Using a variable follow-up design assuming an average accrual monthly rate of 3.6 patients per month, and a minimum follow-up of 43 months, recruiting 165 patients with DSA would allow us to observe 23/83 (28%) graft failures under BLC (group B1) and 39/82 (47%) in the SC group (A1). This would provide 80% power and 5% type 1 error for a 2-sided log-rank test.
-
1.2) Group A2 > group B2: HLA Ab+ patients, with non-DSA, randomised to SC (A2) were hypothesised to show higher graft failure rate than patients randomised to BLC (B2). We then hypothesised that the experimental treatment would bring the failure rate in group B2 down to that of HLA Ab-negative patients in SC (C). Assuming 16% with non-DSA in group A2 should have experienced graft failure by 3 years follow-up (as in7), we expected treatment in group B2 to reduce the rate down to 6%, corresponding to a HR of 0.351. The expectation was for 3% and 11% failure among recruits with non-DSA in group A1 at 1 and 2 years follow-up, respectively, and extrapolating using a HR of 0.351, we expected BLC to reduce these to 1.1% and 4.1%. Using a variable follow-up design assuming an average accrual monthly rate of 15.5 patients per month, and a minimum follow-up of 22.4 months, recruiting 296 patients with non-DSA would allow us to observe 8/149 (5.3%) graft failures under BLC (group B2) and 21/147 (14%) in the SC group (A2). This would provide 80% power and 5% type 1 error for a two-sided log-rank test.
-
-
Non-inferiority of all unblinded patients compared to all blinded patients:
-
2.1) Groups A1 + A2 + C ≥ Groups B1 + B2 + D: All patients randomised to unblinded screening were hypothesised to show equal or lower graft failure rates than all patients randomised to blinded screening, irrespective of biomarker status. At the end of the trial, we expected 58% of patients to be in the HLA Ab negative groups, 7% in DSA+ groups and 35% non-DSA+ groups (after dropouts). At the time of planning the trial, based on all the assumptions above, we therefore calculated that the graft failure rate in the whole SC arm would be 13.9%.
-
We established a non-inferiority limit of 5% absolute difference in graft failure rate at 3 years, so that the BLC group would be considered inferior to SC group if they had a graft failure rate of ≥ 18.9%. This corresponded to a HR of 1.4 under the null hypothesis and an HR of 0.63 under the alternative. Therefore, we estimated that recruiting 672 patients to groups C&D (336 per group) with a minimum follow-up of 18.21 months would allow us to observe 22/337 (6.5%) graft failures in the SC group and 32/335 (9.5%) in the BLC group. This would provide 90% power to demonstrate non-inferiority with a one-sided 95% confidence interval (CI) of the HR estimated using a Cox regression model.
Following these calculations, we estimated the number to be screened, based on expected dropout rates, expected screening results and eligibility criteria. We assumed that 6% of initially Ab-neg patients would become Ab+ in each screening round (1/3rd with DSA) and that DSA recruits would comprise 7% of all recruits at the end of the trial. We therefore estimated that we needed to recruit 2357 patients overall to ensure we achieved 165 with DSA. The result of this was that the number of recruits to the other groups was higher than the minimum required as discussed above.
Following 16 months of recruitment, we performed an audit of our assumptions: the observed % of DSA patients (including those from rescreening rounds) was 6.6%. Although the percentage of Ab+ patients at baseline was 35.1%, considerably higher than expected (25–30%), only 5.8% of all patients had DSA at baseline (expected 9%). 300 Ab-neg patients had been rescreened as part of the month-8 screening round, of whom 23 had developed de-novo Ab (7.6% – expected 6%). Five out of the twenty-three had DSA (1.6% of all – expected 2%). Thus, as described above, we redefined the primary endpoint to ‘time to graft failure’ to allow the trial to recruit reduced numbers of DSA+ patients while maintaining power.
Statistical analysis
Statistical analysis was on an intention-to-treat basis. All outcomes were analysed separately within the subgroups of DSA+ and non-DSA+ recruits. Recruits initially HLA Ab-negative who become positive during subsequent screening rounds were moved to the appropriate HLA Ab+ groups (DSA+ or non-DSA+) for analysis. These recruits, who were all followed up for an extra 32 months after the Ab was first discovered, were analysed from the time they became HLA Ab+ in the primary endpoint analyses. In the secondary analysis of time to graft failure in SC versus BLC participant using all recruits, they were analysed from time of randomisation (see below). The statistical analysis plan (SAP) contained detailed descriptions of how we would describe recruit characteristics, broken down by HLA Ab status.
Outcome assumptions and data collection periods
The following treatment effect contrasts for the primary and secondary outcomes were estimated:
-
1a. DSA+ BLC versus DSA+ SC participants (both at randomisation and rescreening);
-
1b. non-DSA+ BLC versus non-DSA+ SC participants (both at randomisation and rescreening);
-
2. all randomised BLC versus SC participants.
For the primary outcome, contrasts 1a and 1b were tested for superiority and contrast 2 was tested for non-inferiority, with non-inferiority concluded if the upper bound of the 95% CI for the HR was less than 1.4. For all secondary outcomes, all contrasts were tested for superiority.
Different comparisons/outcomes used different observation periods. This is outlined in Figure 2.
FIGURE 2.
Diagram to help explain how endpoints were assessed. Figure depicts three recruitment scenarios and explains how different outcomes were assessed, either in relation to recruitment/randomisation or, in the case of recruits turning from HLA Ab-negative to +, in relation to rescreening.
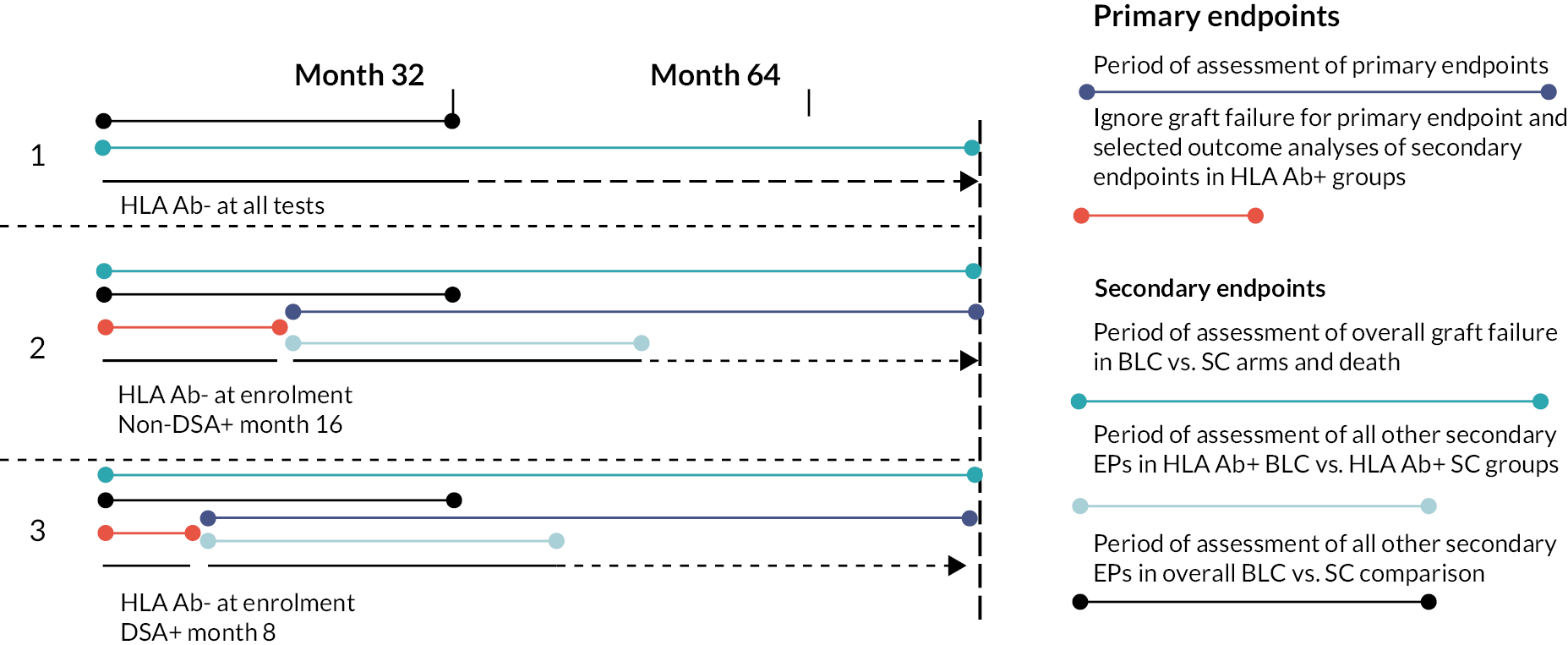
The purpose of these different observation periods for the different comparisons, is that the within DSA+ and within non-DSA+ comparisons aim to estimate the treatment effect of unblinding + optimisation in HLA+ve participants and so include participants at risk from the time they were found to be HLA+ve. The overall unblinded (BLC) versus blinded (SC) comparison aims to estimate the overall effect of the blinding strategy, and so participants time at risk is the time of blinding/unblinding to the HLA result (which is randomisation for all participants).
For the DSA+ and within non-DSA+ comparisons, time at risk started at randomisation for those HLA positive at randomisation and at time of rescreening for those HLA-negative participants who became HLA positive later at rescreening rounds. For the primary outcome, patients’ follow-up time was used up until the pre-COVID-19 collection period. For the overall comparison (statistical hypothesis 2), time at risk started at randomisation for all participants up until the pre-COVID-19 collection period. This was also true for the secondary outcome of death.
For the other secondary outcomes, these were only collected in the intensive data collection period which was from randomisation to 32 months post-randomisation, or in the case of participants who became HLA positive at rescreening rounds, 32 months post-rescreening. Therefore for these secondary outcomes, for the within DSA+ and within non-DSA+ comparisons, time at risk starts at randomisation for those HLA positive at randomisation and at time of rescreening for those HLA-negative participants who became HLA positive later and ends 32 months later. However, for the overall unblinded (BLC) versus blinded (SC) comparison for these other secondary outcomes, time at risk starts at randomisation for all participants and ends 32 months post-randomisation (ignoring any additional follow-up for rescreening HLA-positive participants).
For the primary outcome, the proportional hazards assumption was checked by testing for an interaction between treatment and time or more precisely, testing for a non-zero slope in a generalised linear regression of the scaled Schoenfeld residuals on functions of time (which is equivalent to testing the interaction).
Data analysis plan
The primary analysis used data collected up until 16 March 2020 and analyses were conducted for each of the hypotheses as outlined below. Several sensitivity analyses were also carried out for the primary outcome. These used the same covariates/modelling strategy as the primary analysis unless stated:
-
Excluding site as a covariate: There were a large number of sites, and this was a stratification factor adjusted for the model. However, there were low numbers of participants recruited for some sites such that some estimates for the site covariate were not estimated in the model. An analysis excluding site was carried out to ensure this was not causing instability in treatment effect estimates.
-
A competing risks analysis using competing risk regression, according to the method of Fine and Gray30 (1999), was carried out to examine sensitivity of the results to the competing risk of death. The subhazard ratio for graft failure was estimated.
-
For COVID-19 data: An analysis was carried out using additional follow-up data up until 30 November 2020, which we called the post-COVID-19 time point as these participants’ outcomes may have been affected by the COVID-19 pandemic. The analysis was otherwise exactly the same.
-
Using the primary model for the HLA Ab non-DSA group but restricting it to those participants who were assessed as definite non-DSA (as opposed to non-DSA in the absence of any conclusive evidence of DSA).
-
A sensitivity/per-protocol analysis restricting those in the HLA Ab+ DSA and HLA Ab+ non-DSA groups to those who received the full optimisation protocol (taking MMF, Tac and prednisolone at the visit following the optimisation interview).
Superiority
NB here, hA1(t), hB1(t), etc. represent the graft failure hazard rates in each of the groups.
In order to test superiority for the primary outcome in the BLC (HLA Ab) positive groups (Hypothesis 1.1 and 1.2), we used Cox proportional hazards regression models to estimate the graft failure HR between the BLC and SC groups and test at the 5% level of significance. Results are given as estimates and 95% CIs. Within the model, we adjusted for previous immunosuppression regimen and research site (as these are the randomisation stratification factors) for increased statistical efficiency. We checked the proportional hazards assumption by examining Kaplan–Meier plots and by testing for an interaction between group (BLC or SC) and time to graft failure within the model.
Non-inferiority
In order to test for non-inferiority of the unblinded groups compared to the blinded groups (hypothesis 2.1), we used Cox proportional hazards regression models to estimate the graft failure HR. We adjusted for stratification factors in the model as outlined above and checked the proportional hazards assumption by examining Kaplan–Meier plots and by testing for an interaction between unblended and blinded group and time to graft failure. We concluded non-inferiority if H0 was rejected at 5% significance, and the corresponding upper bound of the 95% CI for the HR excluded the limit δ (HR of 1.4).
All secondary outcomes were analysed comparing BLC versus SC groups within the HLA Ab+ DSA participants and within HLA Ab+ non-DSA participants as well as between unblinded and blinded groups overall, as per the primary outcome analysis. We used similar procedure using Cox proportional hazards regression for the analysis of secondary time-to-event (survival) outcomes. For the secondary outcome of death an additional sensitivity analysis restricting the follow-up time to the first 32 months was carried out (as the original protocol implied that all secondary outcomes will be carried out on the 32 months intensive follow-up period only).
Where numbers allowed, secondary binary outcomes were analysed using logistic regression with adjustment for stratification factors. Where numbers were too small for this, the Z-test or Fisher’s exact was used. For continuous secondary outcomes, linear regression was used (or linear mixed models where accounting for repeated measures), adjusting for baseline values of the outcome and stratification factors. Transformations were considered where data was skewed. Results are given as estimates (odds ratios or differences in proportions) and 95% CIs.
The secondary outcomes of biopsy-proven rejection, infection, malignancy, and diabetes de novo were all analysed using logistic regression, with the outcome as to whether the participant experienced the event (at least once) over the intensive 32-month follow-up period (from randomisation or from rescreening as appropriate). Site was not included as a covariate in these models as small numbers recruited in some sites would lead to perfect prediction and observations being dropped. Baseline immunosuppression was included as a covariate as per the primary outcome model. All participants were included if they had at least one observation post-randomisation (or post-rescreening).
The outcome of proteinuria at month 32 was analysed using a logistic (longitudinal) mixed model, with all observations included between randomisation (or rescreening as appropriate) and month 32 at 4 monthly intervals, although most participants only had data at 8 monthly intervals as frequency of follow up was changed to 8 monthly in Protocol V10 (11 August 2015). Trial arm, time point, an interaction between time point and trial arm and stratification factors were included as covariates. A random intercept was included for participant. Treatment effects at month 32 were estimated using post-estimation commands. All participants were included if they had at least one observation post-randomisation (or post-rescreening).
The outcome of eGFR was analysed using a linear (longitudinal) mixed model, with time points as per the proteinuria model. Trial arm, time point, an interaction between time point and trial arm, baseline eGFR and the stratification factors were included as covariates. A random intercept was included for participant. Treatment effects at month 32 were estimated using post-estimation commands. All participants were included if they had at least one observation post-randomisation or post-rescreening (and so estimates are unbiased under a missing at random assumption as the model uses maximum likelihood).
Statistical considerations
Pre-specified instructions for dealing with missing data (baseline and outcome) are detailed in the SAP (see Supplementary Material). We made no formal adjustment of p-values for multiple testing. An exploratory per-protocol analysis was carried out comparing time to graft failure in only those participants who were optimised to the full treatment protocol in the unblinded arm against all blinded participants, within both the HLA Ab+ DSA and HLA Ab+ non-DSA groups. The proportional hazards assumption was checked for the primary outcome model by testing for an interaction with time. For secondary outcomes, where normally distributed outcomes were assumed, this was checked and transformations considered where departures from normality occurred. Residuals were plotted to check for normality and inspected for outliers.
Changes to the analysis from the SAP following discussion of the results and peer review comments
The following changes were made to the analysis following discussion/review of the results and peer review comments and are not covered in the final SAP (V2.4 09/02/2021):
-
a post-hoc exploratory sensitivity analysis for the primary outcome was carried out (for each of the three comparisons) using only BLC participants taking all three IMPs and with Tac trough levels of between six and eight compared to SC participants;
-
a post-hoc exploratory sensitivity analysis for the primary outcome was carried out (for each of the three comparisons), further adjusting for time of transplant and sex as covariates given chance imbalances between arms for these variables;
-
McNemar tests were carried out comparing whether numbers on immunosuppression medications for BLC HLA+ participants (both DSA and non-DSA) changed from pre-optimisation to the last visit. This was to try to demonstrate that the optimisation intervention did change these participants immunosuppression medications as intended;
-
a post-hoc analysis of the interaction between persisting DSA and time to graft failure in the main analysis was added to test whether those with persisting DSA and those without had different treatment effects;
-
the definition of what was classified as a biopsy-proven rejection was not strictly defined in the SAP or the protocol. This was erroneously taken to be only those participants who showed rejection on the primary pathology for renal biopsies originally. Biopsy-proven rejection is a secondary outcome. This became clear when responding to peer review and the chief investigator (CI) clarified that rejection on secondary pathology should also have been defined as biopsy-proven rejection. This analysis was therefore amended to include these few additional events and the results changed slightly.
Economic evaluation
Aims and methods
The aims of the economic evaluation were to (1) compare health and social care use between both trial arms for HLA Ab + cases, (2) compare health and social care costs between arms and (3) assess the cost-effectiveness of unblinded care compared to blinded care in terms of quality-adjusted life-years (QALYs) accrued. We adopted a health and social care perspective in line with National Institute for Health and Care Excellence (NICE) recommendations. A within-trial analysis was conducted and we focussed on HLA-positive cases with comparisons made between those receiving unblinded care and those with double-blinded care. Two time points provided data for these analyses: the one-year period prior to baseline assessment and the 12-month period prior to 16-month follow-up.
Service use was measured with an adapted version of the Client Service Receipt Inventory. 31 This asked respondents about use of health (primary care and secondary care) and social care service use in the previous 12 months. The number of contacts was recorded or in the case of inpatient care and residential care we asked for the number of days.
Service use was combined with appropriate unit cost information for the year 2019/20 in order to estimate service costs. Unit costs were obtained from the University of Kent’s annual compendium32 and the Department of Health and Social Care (https://www.england.nhs.uk/publication/2019-20-national-cost-collection-data-publication/). For inpatient care, we had data on length of stay and assumed a cost of £500 per day rather than applying a cost for each admission. A list of unit costs used is shown in Table 1. One key cost excluded is the cost of screening itself. It is unknown what this will be in routine practice, and this is addressed in the discussion of the findings.
| Service | Unit | Cost (£s) |
|---|---|---|
| Residential care | Day | 102 |
| Renal inpatient | Day | 500 |
| Intensive care | Day | 1349 |
| Other inpatient | Day | 500 |
| Renal outpatient | Appointment | 135 |
| Other outpatient | Appointment | 135 |
| Day hospital | Visit | 100 |
| A&E | Visit | 182 |
| GP | Appointment | 34 |
| Physiotherapist | Contact | 64 |
| OT | Contact | 85 |
| Speech therapist | Contact | 109 |
| Dietitian | Contact | 92 |
| Nutritionist | Contact | 92 |
| Social worker | Contact | 51 |
| Homecare worker | Contact | 28 |
| Psychologist | Contact | 88 |
| Complementary healthcare | Contact | 58 |
| District nurse | Contact | 43 |
| Psychiatrist | Contact | 135 |
| Counsellor | Contact | 58 |
Quality-adjusted life-years are the outcome measure used in most economic evaluations in the UK. They combine quantity and quality of life, although here we had a time horizon that was restricted to 16 months. The EQ-5D-5L33 was used to derive QALYs and consists of five domains (mobility, self-care, usual activities, pain/discomfort and anxiety/depression). Each domain is scored with an integer from 1 (no problems) to 5 (extreme problems). The health states are then converted into a utility score anchored by 1 (full health) and 0 (death), with negative values indicating health states considered worse than death. The UK crosswalk method was used to derive these values. QALYs were then calculated using the area under the curve method base on a linear change from baseline to 16-month follow-up.
Use of services was compared descriptively between the arms. Total costs and QALYs were compared between arms using a seemingly unrelated regression model to take account of the possibility of correlated errors. In the estimation of cost differences adjustment was made for the baseline costs while for QALYs we adjusted for baseline utility. The model was bootstrapped with 1000 resamples due to the likely skewed cost distribution. The cost difference between arms was divided by the difference in QALYs to produce an incremental cost-effectiveness ratio (ICER). The saved bootstrapped cost and QALY differences were plotted against each other to produce a cost-effectiveness plane and used to derive incremental net benefit values to plot a cost-effectiveness acceptability curve. Discounting was not applied as the time horizon was only 16 months.
Adherence to drug therapy and perceptions of risk to the health of the transplant
Health surveys, consisting of validated psychological measures adapted for this specific health context, were performed at baseline and 12 and 24 months post screening for HLA Ab+, and included the medication adherence report scale (MARS) questionnaire, which consisted of measuring six items on a five-point Likert scale with higher scores representing greater adherence. Most items assessed intentional medication non-adherence; one item measured unintentional non-adherence. MARS was completed for each medication patients received. MARS correlates well with relatively objective measures of adherence in a range of illness contexts, including electronic measures of inhaled corticosteroids for asthma and blood pressure control for hypertension. 34,35 It has also been shown to have good levels of internal consistency, test-retest reliability and construct validity. 35 For Tac, 12-hour trough levels were also compared against the target trough levels (4–8 ng/ml) and a composite adherence scale based on combining MARS scores with trough levels was developed. Concern about the risk of transplant failure was measured using the Brief Illness Perceptions Questionnaire. 36
Analysis of questionnaires was performed separately to the main trial data by the team at University College London. Analyses were based on imputed data: where values were missing for a given survey item, the mean score for that item across all participants was used, providing that a given case had at least 80% complete data for other items on that scale. Mann–Whitney U or chi-squared tests were used to compare scores or proportions across patients in the BLC DSA+ compared to SC DSA+ groups, and BLC non-DSA compared to SC non-DSA groups.
Anti-HLA Ab determination
Serum prepared from 10 mL of blood was used in the commercially available LABScreen tests (One Lambda, Canoga Park, CA through VH Bio, Gateshead, UK), analysed on Luminex equipment (Luminex Corp, Austin, Texas) in the five original sites (Guy’s, Birmingham, Manchester, Leeds and Royal London). All worked to the same standard operating procedure, agreed pre-trial (see Appendix 2). Serum was first analysed using mixed HLA Class I and Class II Ab screening beads, with a positive or negative result assigned based on batch-specific cut-offs designated using validated protocols at the Guys laboratory site. In those patients with positive results, serum was further analysed using single antigen-coated Class I or Class II beads. A positive result was defined as giving a mean fluorescence intensity (MFI) of binding ≥ 2000. Laboratory staff then compared assigned DSA/non-DSA status depending on whether HLA Ab were directed against a mismatched donor HLA antigen. HLA Ab in which it was difficult to label as DSA+ or non-DSA+ (because of insufficient data on donor mismatches, for instance), were categorised as being non-DSA+. Samples with a positive reaction on screening but lacking reactivity with the single antigen beads were considered negative. Screening of all HLA Ab– patients was undertaken every 8 months.
Trial oversight
Trial management group
A trial management group (TMG) was chaired by Professor Anthony Dorling (CI of the study) and consisted of co-applicants of the trial grant, the trial manager, Caroline Murphy (King’s CTU) and Olivia Shaw (Viapath, GSTT Tissue Typing Lab) and members of the research team. The TMG was responsible for decisions on the day-to-day running of the trial. The TMG met quarterly initially, but less frequently as the trial progressed.
Trial Steering Committee
A Trial Steering Committee (TSC) chaired by Professor Christopher Watson (Professor of Transplantation, Cambridge University) was convened in the post-award period. The members were Dr Craig Taylor (HLA Scientist, Addenbrookes Hospital), Professor Sunil Bhandari (Consultant Nephrologist, Hull & York Medical School) and Mr Paul Newton (Representative from the GSTT Kidney Patients Association), Professor Anthony Dorling (CI), the trial manager, Mr Dominic Stringer (Trial Statistician) and two co-applicants of the trial grant. The TSC met every 6 months initially, then annually, according to Terms of Reference drafted prior to recruitment.
Data Monitoring Committee
The Data Monitoring Committee (DMC) was chaired by Dr Nicholas Torpey (Consultant Nephrologist, Addenbrookes Hospital). The DMC remit was to safeguard the interests of trial participants, potential participants, their families, their carers, investigators, and the sponsor; to assess the safety and efficacy of the intervention during the trial and to monitor the trial’s overall conduct and protect its validity and credibility. A DMC charter was drafted prior to recruitment. The members of the DMC were Dr Issy Reading (Independent statistician, University of Southampton), Dr Alan Wong (Trials Pharmacist, Royal Free Hospital) and Dr Vaughan Carter (HLA Scientist, NHS Blood and Transfusion Service). Mr Dominic Stringer (Trial statistician) presented a closed report at each meeting. The DMC met every 6 months initially, then annually, approximately 2 weeks before the TSC.
Patient and public involvement
Kidney transplant patients were involved in the grant application process, first, via their involvement in the local review of projects at GSTT via the TRU Project Board Steering Committee, and second, through a specific meeting with members of the GSTT Kidney Patients Association during the grant application process. At this stage patient involvement led to three significant changes in trial design, including dropping the inclusion of protocol kidney transplant biopsies, reducing the maximum dose of prednisolone used, and because of concerns about the communication of risk associated with the biomarker, recruitment of Professor Rob Horne into the team. The KPA helped Prof Horne with the design of the patient information sheets for the trial and also provided a representative to sit on the TSC. Throughout the conduct of the trial, the KPA were kept updated about how the trial was progressing via their representative on the TSC and through regular updates via the MRC Centre for Transplantation newsletter, annual Clinical Trials Day literature and CI contributions to their quarterly newsletter.
Results
Participants, recruitment and flow
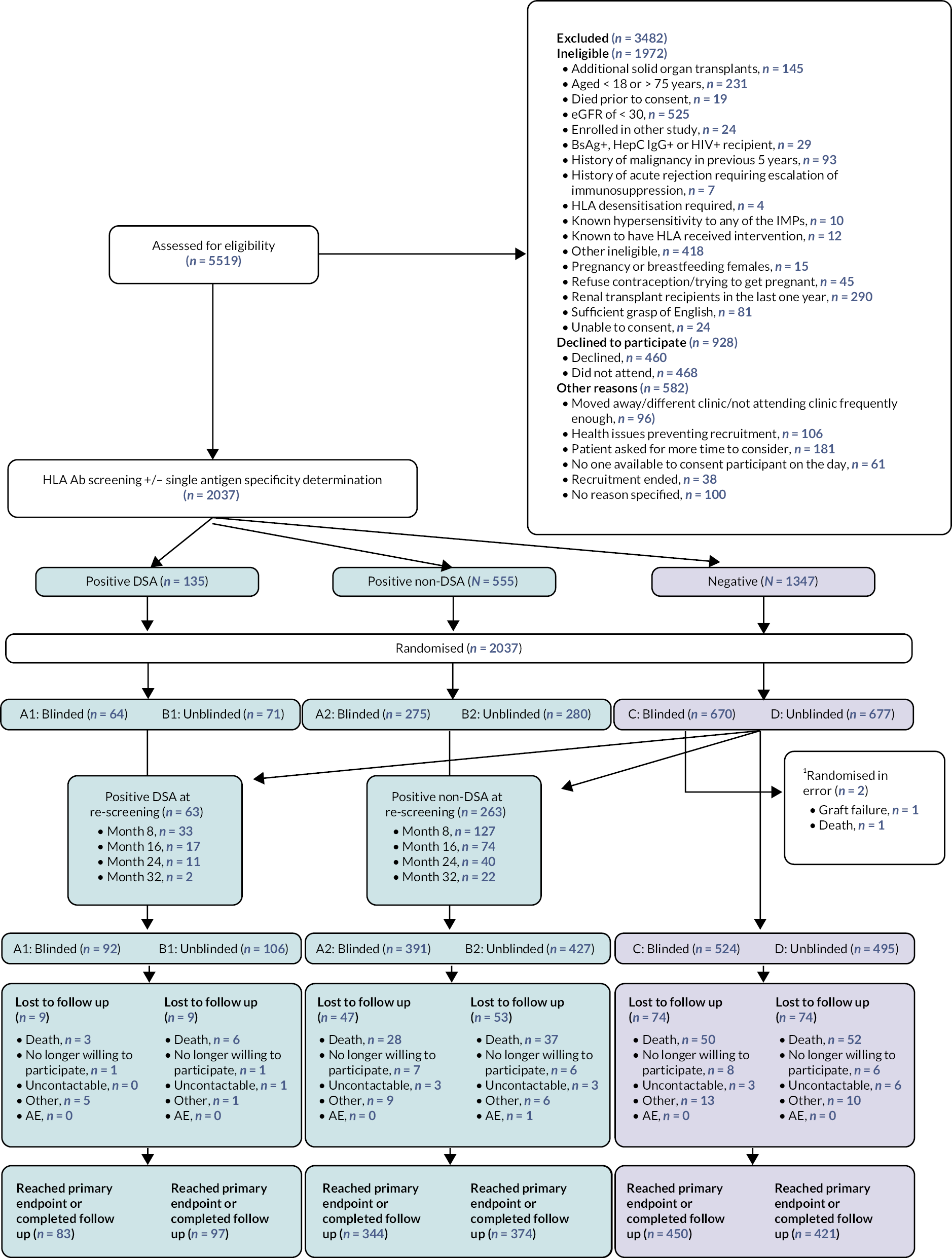
Recruitment was from 13 UK transplant centres and took place between 11 September 2013 and 27 October 2016. During that time, 5519 renal transplant recipients (see Figures 1 and 3) were assessed for eligibility of which 2094 were enrolled after consent. Fifty-seven patients were found to be ineligible after post-consent checks. 2037 were randomised after HLA Ab screening into two arms, each containing three groups based on the HLA Ab screening results; blinded SC (A1, A2 or C), and unblinded BLC (B1, B2 and D). Randomisation broken down by site and year is shown in Table 2. Screening of the HLA Ab-negative groups for HLA Ab finished in June 2017, at which time a further 63 with DSA (28 blinded, 35 unblinded) and 263 non-DSA (116 vs. 147) were identified, leaving 1019 remaining HLA Ab-negative through the course of screening (524 vs. 495). The end of the intensive follow-up period (last person, last visit) occurred 32 months later in March 2020, with the remote primary endpoint collection at a minimum of 43 months post-randomisation, originally scheduled for June 2020 moved to March 2020 because of the pandemic (as described above).
FIGURE 3.
Consort diagram for OuTSMART. 1 Two patients were randomised in error to blinded HLA Ab-negative SC group. These were included from the intention-to-treat analysis. Refer to Supplementary Material for further information.
| Site | 2013 (%) | 2014 (%) | 2015 (%) | 2016 (%) | Total (%) |
|---|---|---|---|---|---|
| St James’s University Hospital, Leeds Teaching Hospitals NHS Trust | 17 (15) | 128 (17) | 92 (18) | 54 (8.5) | 291 (14) |
| The Royal London Hospital, Bart’s Health NHS Trust | 0 (0.0) | 18 (2.3) | 63 (12) | 49 (7.7) | 130 (6.4) |
| Guy’s and St Thomas’ NHS Foundation Trust, London | 100 (86) | 291 (38) | 90 (18) | 48 (7.5) | 529 (26) |
| Manchester Royal Infirmary, Manchester University NHS Foundation Trust | 0 (0.0) | 147 (19) | 98 (19) | 67 (10.5) | 312 (15) |
| Queen Elizabeth Hospital, University Hospitals Birmingham NHS Foundation Trust | 0 (0.0) | 145 (19) | 72 (14) | 0 (0.0) | 217 (11) |
| King’s College Hospital NHS Foundation Trust, London | 0 (0.0) | 38 (4.9) | 32 (6.3) | 73 (11) | 143 (7.0) |
| The York Hospital, York and Scarborough Teaching Hospitals NHS Foundation Trust | 0 (0.0) | 0 (0.0) | 29 (5.7) | 24 (3.8) | 53 (2.6) |
| University Hospitals Coventry and Warwickshire NHS Trust | 0 (0.0) | 3 (0.4) | 35 (6.8) | 15 (2.3) | 53 (2.6) |
| Royal Preston Hospital, Lancashire Teaching Hospitals NHS Foundation Trust | 0 (0.0) | 0 (0.0) | 0 (0.0) | 65 (10) | 65 (3.2) |
| Salford Royal Hospital, Northern Care Alliance NHS Foundation Trust | 0 (0.0) | 0 (0.0) | 0 (0.0) | 52 (8.1) | 52 (2.6) |
| Bradford Royal Infirmary, Bradford Teaching Hospitals NHS Foundation Trust | 0 (0.0) | 0 (0.0) | 0 (0.0) | 48 (7.5) | 48 (2.4) |
| Royal Free Hospital NHS Foundation Trust, London | 0 (0.0) | 0 (0.0) | 0 (0.0) | 125 (20) | 125 (6.1) |
| Epsom and St Helier University Hospitals NHS Foundation Trust | 0 (0.0) | 0 (0.0) | 0 (0.0) | 19 (3.0) | 19 (0.9) |
Of the 90 patients ‘lost’ during the trial, 29 withdrew consent (group A1 n = 1: A2 n = 7: B1 n = 1: B2 n = 6: C n = 8: D n = 6), 16 became uncontactable (group A1 n = 0: A2 n = 3: B1 n = 1: B2 n = 3: C n = 3: D n = 6), 1 was withdrawn for an AE (group B2) and the remaining 44 were withdrawn for reasons listed as ‘other’, but 43 of these were because patients had transferred care to another, non-trial transplant unit and were therefore out of touch (group A1 n = 5: A2 n = 9: B1 n = 1: B2 n = 6: C n = 13: D n = 10).
Protocol violations/randomisation errors
There were two randomisation errors, both participants were randomised to the blinded (SC) arm and were HLA Ab-negative at baseline. One participant had graft failure prior to randomisation and one participant was randomised but was found to have died shortly before randomisation. Randomisation was carried out by lab staff following HLA screening and in error, it was not communicated to the lab staff that these events had occurred prior to randomisation. These participants are excluded from all analyses.
Further, for the primary analysis, one rescreened randomised participant is not included in the DSA group as they were found to have graft failure prior to being rescreened and becoming HLA Ab+ DSA and so were not at risk for the purpose of this analysis. This participant is included in any group/sensitivity analyses where time at risk starts at randomisation for all.
There were several other errors in recording of HLA status in the randomisation system. As per the intention-to-treat principle, these were analysed in the original groups as recorded in the randomisation data and not in the corrected group (as the HLA status as per randomisation data was communicated to the PI if in the unblinded arm, and treatment strategy would have been based on this data). These errors were the following:
-
One participant randomised to the BLC arm and entered as HLA Ab+ DSA at baseline in randomisation system was actually HLA Ab+ definite non-DSA at baseline according to the lab data.
-
One participant randomised to the SC arm and entered as HLA Ab+ DSA at baseline was actually HLA Ab+ definite non-DSA at baseline according to the lab data.
-
One participant in the BLC arm was moved from the HLA Ab-negative to the HLA Ab+ DSA group at rescreening (month 16). However, according to their lab data, their Ab at that time indicated ‘unknown whether DSA’ and should have been allocated to the non-DSA group as per the protocol.
-
One participant randomised to the BLC arm and entered as DSA at baseline actually had Ab at that time indicating ‘unknown whether DSA’ and should have been randomised to the non-DSA group.
-
Two participants randomised to the SC arm who were HLA Ab-negative at baseline were rescreened according to lab data at month 16 and became HLA Ab+ with unknown DSA. However, this was erroneously not entered into the randomisation system (and are considered HLA Ab-negative for the purpose of ITT analysis).
Baseline data
Demographics
There was generally good balance in baseline demographic characteristics between Ab+ and Ab- groups at point of randomisation (see Table 3). The DSA+ unblinded group had a higher proportion of males, longer time from transplant and higher proportion with previous transplants. There was no obvious imbalance in baseline variables after rescreening had finished (see Table 4).
| Characteristic | DSA+ | Non-DSA+ | HLA Ab negative | |||
|---|---|---|---|---|---|---|
| Blinded (SC) | Unblinded (BLC) | Blinded (SC) | Unblinded (BLC) | Blinded (SC) C | Unblinded (BLC) | |
| Group | A1a (N = 64) | B1 (N = 71) | A2 (N = 275) | B2 (N = 280) | (N = 670) | D (N = 677) |
| Age (years) Mean (SD) | 49.5 (12.0) | 47.0 (14.6) | 50.0 (11.9) | 50.6 (12.6) | 50.3 (13.30) | 50.5 (13.2) |
| Male (%) | 66% | 80% | 56% | 59% | 73% | 72% |
| Ethnicity (%) | ||||||
| Asian | 9.4% | 14% | 13% | 13% | 11% | 13% |
| Black | 19% | 14% | 7.6% | 11% | 11% | 9.7% |
| White | 69% | 70% | 76% | 72% | 75% | 75% |
| Mixed | 1.1% | 0% | 1.5% | 1.4% | 0.6% | 0.1% |
| Other | 1.6% | 1.4% | 2.5% | 1.8% | 2.4% | 2.5% |
| Site [N (%)]b | ||||||
| Leeds | 8 (2.7%) | 8 (2.7%) | 41 (14%) | 40 (14%) | 96 (33%) | 98 (34%) |
| Royal London | 6 (4.6%) | 5 (3.8%) | 11 (8.5%) | 12 (9.2%) | 48 (37%) | 48 (37%) |
| Guy’s | 21 (4.0%) | 24 (4.5%) | 69 (13%) | 72 (14%) | 170 (32%) | 173 (33%) |
| Manchester | 8 (2.6%) | 8 (2.6%) | 44 (14%) | 47 (15%) | 103 (33%) | 102 (33%) |
| Birmingham | 3 (1.4%) | 2 (0.9%) | 31 (14%) | 27 (12%) | 77 (36%) | 77 (36%) |
| King’s College Hospital | 6 (4.2%) | 4 (2.8%) | 21 (15%) | 21 (15%) | 44 (31%) | 47 (33%) |
| York | 2 (3.8%) | 2 (3.8%) | 6 (11%) | 7 (13%) | 18 (34%) | 18 (34%) |
| Coventry | 0 (0.0%) | 1 (1.9%) | 6 (11%) | 7 (13%) | 18 (34%) | 21 (40%) |
| Preston | 1 (1.5%) | 4 (6.2%) | 11 (17%) | 8 (12%) | 21 (32%) | 20 (31%) |
| Salford | 1 (1.9%) | 1 (1.9%) | 6 (12%) | 8 (15%) | 19 (37%) | 17 (33%) |
| Bradford | 3 (6.2%) | 5 (10%) | 8 (17%) | 9 (19%) | 12 (25%) | 11 (23%) |
| Royal Free | 5 (4.0%) | 6 (4.8%) | 18 (14%) | 19 (15%) | 38 (30%) | 39 (31%) |
| St Helier | 0 (0.0%) | 1 (5.3%) | 3 (16%) | 3 (16%) | 6 (32%) | 6 (32%) |
| Cause of renal failure [N (%)] | ||||||
| DM | 4 (6.9%) | 2 (3.4%) | 7 (2.9%) | 13 (5.4%) | 38 (6.7%) | 40 (6.8%) |
| GN | 22 (38%) | 19 (33%) | 93 (39%) | 94 (39%) | 216 (38%) | 224 (38%) |
| PKD | 7 (12%) | 9 (16%) | 32 (13%) | 34 (14%) | 105 (19%) | 100 (17%) |
| Hypertension | 6 (10%) | 6 (10%) | 20 (8.3%) | 22 (9.2%) | 43 (7.6%) | 47 (8.0%) |
| Congenital | 7 (12%) | 7 (12%) | 31 (13%) | 22 (9.2%) | 66 (12%) | 47 (8.0%) |
| Obstructive | 8 (14%) | 10 (17%) | 38 (16%) | 34 (14%) | 54 (9.5%) | 80 (14%) |
| Other | 4 (6.9%) | 5 (8.5%) | 19 (7.8%) | 20 (8.3%) | 45 (8.1%) | 46 (7.9%) |
| Previous transplants [N (%)] | ||||||
| 0 | 48 (76%) | 52 (73%) | 193 (71%) | 198 (71%) | 613 (92%) | 633 (94%) |
| 1 | 12 (19%) | 18 (25%) | 71 (26%) | 65 (23%) | 55 (8.2%) | 35 (5.2%) |
| 2 | 3 (4.8%) | 1 (1.4%) | 8 (2.9%) | 13 (4.7%) | 0 (0%) | 5 (0.7%) |
| 3 | 0 (0%) | 0 (0%) | 1 (0.4%) | 3 (1.1%) | 0 (0%) | 0 (0%) |
| Time (years) since Tx Median (IQR) | 6.6 (3.0–12.0) | 9.7 (3.9–14.3) | 5.7 (2.2–10.9) | 4.9 (2.3–11.2) | 5.4 (2.4–9.2) | 5.1 (2.4–9.7) |
| Immunosuppression | ||||||
| CsA [N (%)] | 17 (27%) | 18 (25%) | 49 (18%) | 49 (18%) | 121 (18%) | 120 (18%) |
| Mean dose [mg (SD)] | 170.3 (49.8) | 199.4 (68.5) | 168.6 (65.0) | 168.7 (60.4) | 180.7 (67.9) | 168.7 (63.0) |
| Mean trough level [μg/L (SD)] | 72.3 (34.8) | 80.9 (55.3) | 102.8 (84.8) | 88.6 (56.1) | 100 (71.4) | 109.6 (88.5) |
| Tac [N (%)] | 39 (61%) | 41 (58%) | 205 (75%) | 205 (73%) | 499 (74%) | 501 (74%) |
| Mean dose [mg (SD)] | 6.14 (6.72) | 4.01 (2.24) | 5.08 (3.51) | 5.60 (4.60) | 5.50 (4.12) | 4.89 (3.65) |
| Mean trough level [μg/L (SD)] | 6.49 (2.64) | 5.65 (2.06) | 6.95 (2.93) | 6.86 (2.29) | 6.91 (2.31) | 6.71 (2.47) |
| MMF [N (%)] | 40 (63%) | 41 (58%) | 177 (64%) | 176 (63%) | 460 (69%) | 471 (70%) |
| Mean dose [mg (SD)] | 1156 (476) | 1098 (422) | 1131 (450) | 1117(483) | 1155 (490) | 1136 (466) |
| Aza [N (%)] | 15 (23%) | 19 (27%) | 52 (19%) | 39 (14%) | 90 (13%) | 94 (14%) |
| Mean dose [mg (SD)] | 88.3 (45.2) | 69.7 (33.9) | 76.7 (43.3) | 86.5 (39.3) | 85.3 (34.7) | 85.1 (35.1) |
| Sirolimus [N (%)]c | 2 (3.1%) | 5 (7.0%) | 10 (3.6%) | 4 (1.4%) | 17 (2.5%) | 25 (3.7%) |
| Median dose [mg (SD)] | 2.5 (0.71) | 1.6 (0.55) | 2 (0.82) | 2 (0.82) | 1.65 (0.70) | 2 (0.91) |
| Prednisolone [N (%)] | 37 (58%) | 38 (54%) | 153 (56%) | 154 (55%) | 369 (55%) | 372 (55%) |
| Mean dose [mg (SD)] | 4.97 (1.72) | 4.97 (2.13) | 4.99 (1.45) | 4.99 (1.62) | 5.08 (1.67) | 5.2 (1.62) |
| Taking Tac/MMF/Pred [N (%)] | 13 (20%) | 13 (18%) | 82 (30%) | 70 (25%) | 192 (29%) | 189 (28%) |
| Renal function | ||||||
| Creatinine (μmol/l) [Mean (SD)] | 128.97 (40.32) | 124.96 (37.29) | 123.23 (35.42) | 122.61 (35.81) | 126.17 (38.78) | 126.73 (36.76) |
| eGFR (ml/min/1.73 m2) [Mean (SD)] | 52.31 (15.36) | 56.27 (17.70) | 52.12 (16.54) | 52.89 (16.32) | 53.77 (15.90) | 53.76 (17.26) |
| PCRd (mg/mmol) [Median (IQR)] | 26.50 (15.50–48.25) | 16.50 (10.75–39.25) | 18.00 (8.00-37.25) | 20.00 (9.00–42.50) | 17.00 (9.00–41.25) | 21.00 (10.00–41.00) |
| ACR (mg/mmol) [Median (IQR)] | 1.90 (1.40-1.95) | 5.30 (2.75-7.85) | 2.80 (1.30–6.30) | 7.05 (3.13–15.10) | 3.20 (1.20–9.22) | 3.30 (0.95–10.20) |
| Past medical history [N (%) experienced in that system] | ||||||
| Cardiovascular | 41 (64%) | 43 (61%) | 157 (57%) | 166 (59%) | 406 (61%) | 418 (62%) |
| Respiratory | 9 (14%) | 5 (7%) | 46 (17%) | 51 (18%) | 72 (11%) | 84 (12%) |
| Hepatic | 3 (5%) | 1 (1%) | 3 (1%) | 13 (5%) | 13 (2%) | 32 (5%) |
| Gastrointestinal | 14 (22%) | 9 (13%) | 63 (23%) | 59 (21%) | 123 (18%) | 134 (20%) |
| Genitourinary | 30 (47%) | 33 (46%) | 119 (43%) | 131 (47%) | 269 (40%) | 286 (42%) |
| Endocrine | 24 (38%) | 16 (23%) | 78 (28%) | 91 (33%) | 218 (33%) | 218 (32%) |
| Haematological | 6 (9%) | 3 (4%) | 26 (9%) | 40 (14%) | 80 (12%) | 62 (9%) |
| Musculoskeletal | 23 (36%) | 13 (18%) | 69 (25%) | 83 (30%) | 156 (23%) | 162 (24%) |
| Neoplasia | 4 (6%) | 5 (7%) | 19 (7%) | 31 (11%) | 46 (7%) | 37 (5%) |
| Neurological | 12 (19%) | 6 (8%) | 23 (8%) | 37 (13%) | 66 (10%) | 75 (11%) |
| Psychiatric | 2 (3%) | 3 (4%) | 10 (4%) | 12 (4%) | 20 (3%) | 24 (4%) |
| Immunological | 5 (8%) | 1 (1%) | 17 (6%) | 27 (10%) | 19 (3%) | 31 (5%) |
| Dermatological | 11 (17%) | 11 (15%) | 44 (16%) | 44 (16%) | 85 (13%) | 91 (13%) |
| Allergies | 5 (8%) | 4 (6%) | 33 (12%) | 34 (12%) | 60 (9%) | 76 (11%) |
| Ophthalmological | 6 (9%) | 4 (6%) | 11 (4%) | 23 (8%) | 59 (9%) | 43 (6%) |
| Ear, nose, throat | 6 (9%) | 3 (4%) | 17 (6%) | 20 (7%) | 38 (6%) | 27 (4%) |
| Other | 12 (19%) | 13 (18%) | 56 (20%) | 75 (27%) | 153 (23%) | 132 (20%) |
| Characteristic | DSA+ | Non-DSA+ | No HLA Ab | |||
|---|---|---|---|---|---|---|
| Blinded (SC) | Unblinded (BLC) | Blinded (SC) | Unblinded (BLC) | Blinded (SC) | Unblinded (BLC) | |
| Group | A1b (N = 92) | B1 (N = 106) | A2 (N = 391) | B2 (N = 427) | C (N = 526) | D (N = 495) |
| Age (years) Mean (SD) | 48.1 (13.7) | 46.8 (14.0) | 49.4 (12.7) | 50.3 (12.6) | 51.1 (12.7) | 51.0 (13.3) |
| Male (%) | 72% | 81% | 61% | 59% | 72% | 75% |
| Ethnicity (%) | ||||||
| Asian | 9.9% | 12% | 12% | 14% | 11% | 13% |
| Black | 16% | 12% | 10% | 12% | 9.5% | 8.7% |
| White | 72% | 74% | 74% | 71% | 76% | 76% |
| Mixed | 1.1% | 0% | 1.5% | 0.9% | 0.4% | 0.2% |
| Other | 1.1% | 1.9% | 2.0% | 1.9% | 2.9% | 2.6% |
| Site [N (%)]c | ||||||
| Leeds | 11 (3.8%) | 12 (4.1%) | 70 (24%) | 76 (26%) | 64 (22%) | 58 (20%) |
| Royal London | 8 (6.2%) | 8 (6.2%) | 17 (13%) | 18 (14%) | 40 (31%) | 39 (30%) |
| Guy’s | 32 (6.0%) | 34 (6.4%) | 105 (20%) | 121 (23%) | 123 (23%) | 114 (22%) |
| Manchester | 12 (3.8%) | 9 (2.9%) | 50 (16%) | 54 (17%) | 93 (30%) | 94 (30%) |
| Birmingham | 5 (2.3%) | 12 (5.5%) | 47 (22%) | 42 (19%) | 59 (27%) | 52 (24%) |
| King’s College Hospital | 8 (5.6%) | 5 (3.5%) | 29 (20%) | 28 (20%) | 34 (24%) | 39 (27%) |
| York | 4 (7.5%) | 4 (7.5%) | 9 (17%) | 16 (30%) | 13 (25%) | 7 (13%) |
| Coventry | 0 (0.0%) | 2 (3.8%) | 8 (15%) | 12 (23%) | 16 (30%) | 15 (28%) |
| Preston | 2 (3.1%) | 5 (7.7%) | 13 (20%) | 12 (19%) | 18 (28%) | 15 (23%) |
| Salford | 1 (1.9%) | 1 (1.9%) | 8 (15%) | 8 (15%) | 17 (33%) | 17 (33%) |
| Bradford | 3 (6.2%) | 7 (15%) | 8 (17%) | 12 (25%) | 12 (25%) | 6 (13%) |
| Royal Free | 5 (4.0%) | 6 (4.8%) | 24 (19%) | 22 (18%) | 32 (26%) | 36 (24%) |
| St Helier | 1 (5.3%) | 1 (5.3%) | 3 (16%) | 6 (32%) | 5 (26%) | 3 (16%) |
| Cause of renal failure [N (%)] | ||||||
| DM | 5 (6.0%) | 7 (8.0%) | 17 (5.1%) | 22 (5.9%) | 27 (6.0%) | 26 (6.1%) |
| GN | 28 (34%) | 30 (34%) | 128 (38%) | 147 (40%) | 175 (39%) | 160 (38%) |
| PKD | 10 (12%) | 12 (14%) | 45 (14%) | 54 (15%) | 89 (20%) | 77 (18%) |
| Hypertension | 7 (8.4%) | 7 (8.0%) | 28 (8.4%) | 34 (9.2%) | 34 (7.6%) | 34 (8.0%) |
| Congenital | 13 (16%) | 10 (11%) | 41 (12%) | 34 (9.2%) | 50 (11%) | 32 (7.6%) |
| Obstructive | 12 (15%) | 16 (18%) | 50 (15%) | 48 (13%) | 38 (8.5%) | 60 (14%) |
| Other | 8 (9.6%) | 6 (6.7%) | 25 (7.5%) | 31 (8.4%) | 35 (7.7%) | 34 (7.9%) |
| Previous transplants [N (%)] | ||||||
| 0 | 71 (78%) | 85 (80%) | 301 (77%) | 337 (79%) | 482 (92%) | 461 (94%) |
| 1 | 17 (19%) | 20 (19%) | 79 (20.%) | 73 (17%) | 42 (8%) | 25 (5.1%) |
| 2 | 3 (3.3%) | 1 (0.9%) | 8 (2.1%) | 13 (3.1%) | 0 (0%) | 5 (1.0%) |
| 3 | 0 (0%) | 9 (0%) | 1 (0.3%) | 3 (0.7%) | 0 (0%) | 0 (0%) |
| Time (years) since Tx | ||||||
| Median (IQR) | 5.9 (3.0–11.9) | 6.7 (3.0–12.4) | 5.4 (2.2–9.8) | 5.1 (2.4–10.8) | 5.4 (2.4–9.6) | 5.1 (2.4–9.8) |
| Immunosuppression | ||||||
| CsA [N (%)] | 26 (28%) | 22 (21%) | 69 (18%) | 74 (17%) | 90 (17%) | 89 (18%) |
| Mean dose [mg (SD)] | 187.3 (62.8) | 199.6 (63.6) | 174.4 (62.5) | 160.6 (58.9) | 176.3 (67.8) | 174.7 (62.9) |
| Mean trough level [μg/L (SD)] | 89.3 (56.2) | 80.7 (51.5) | 101.2 (79.8) | 87.3 (52) | 91.9 (52.3) | 116.4 (97.2) |
| Tac [N (%)] | 56 (64%) | 67 (64%) | 296 (76%) | 313 (73%) | 392 (75%) | 366 (74%) |
| Mean dose [mg (SD)] | 6.18 (5.97) | 4.62 (3.33) | 5.14 (3.66) | 5.41 (3.73) | 5.44 (4.13) | 4.70 (3.15) |
| Mean trough level [μg/L (SD)] | 6.56 (2.86) | 5.83 (2.18) | 6.88 (2.74) | 6.68 (2.21) | 6.93 (2.26) | 6.72 (2.52) |
| MMF [N (%)] | 59 (64%) | 62 (59%) | 254 (65%) | 271 (63%) | 361 (69%) | 351 (71%) |
| Mean dose [mg (SD)] | 1165 (482) | 1145 (399) | 1134 (457) | 1112 (472) | 1147 (495) | 1136 (473) |
| Aza [N (%)] | 19 (2.0%) | 26 (25%) | 66 (17%) | 61 (14%) | 71 (13%) | 69 (14%) |
| Mean dose [mg (SD)] | 90.8 (43.5) | 76.9 (32.3) | 78.2 (40.8) | 88.5 (39.4) | 85.2 (33.4) | 83.6 (35.9) |
| Sirolimus [N (%)]d | 2 (2.2%) | 6 (5.7%) | 10 (2.6%) | 6 (1.4%) | 16 (3.0%) | 18 (3.6%) |
| Median dose [mg (SD)] | 2.5 (0.71) | 1.5 (0.55) | 2 (0.82) | 2 (0.89) | 1.62 (0.72) | 2.06 (0.8) |
| Prednisolone [N (%)] | 53 (58%) | 62 (59%) | 210 (54%) | 227 (53%) | 295 (56%) | 274 (55%) |
| Mean dose [mg (SD)] | 5.16 (1.81) | 5.1 (1.87) | 5.01(1.39) | 5.13 (1.53) | 5.11 (1.75) | 5.11 (1.43) |
| Taking Tac/MMF/Pred [N (%)] | 19 (21%) | 24 (23%) | 114 (29%) | 106 (25%) | 152 (29%) | 139 (28%) |
| Renal function | ||||||
| Creatinine (μmol/L) [Mean (SD)] | 129.09 (39.30) | 126.06 (38.25) | 124.08 (35.23) | 121.17 (35.25) | 126.02 (39.71) | 129.07 (36.96) |
| eGFR (ml/min/1.73 m2) [Mean (SD)] | 52.93 (15.23) | 56.16 (18.01) | 52.80 (16.39) | 54.12 (17.30) | 53.59 (15.95) | 52.82 (16.57) |
| PCRe (mg/mmol) [Median (IQR)] | 26.50 (13.75–49.75) | 23.50 (13.00–49.50) | 18.00 (8.00–38.00) | 19.00 (9.00–37.25) | 17.00 (9.00–39.00) | 21.00 (10.00–43.00) |
| ACR (mg/mmol) [Median (IQR)] | 2.00 (1.90–45.60) | 2.30 (0.80–8.00) | 2.80 (1.20–7.70) | 6.40 (2.82–20.10) | 3.20 (1.35–9.22) | 2.55 (0.90–8.75) |
| Past medical history [n (%) experienced in that system] | ||||||
| Cardiovascular | 60 (65%) | 70 (66%) | 221 (57%) | 254 (59%) | 323 (62%) | 303 (61%) |
| Respiratory | 11 (12%) | 9 (8%) | 56 (14%) | 69 (16%) | 60 (11%) | 62 (13%) |
| Hepatic | 3 (3%) | 2 (2%) | 5 (1%) | 17 (4%) | 11 (2%) | 27 (5%) |
| Gastrointestinal | 17 (18%) | 11 (10%) | 81 (21%) | 85 (20%) | 102 (19%) | 106 (21%) |
| Genitourinary | 46 (50%) | 42 (42%) | 169 (43%) | 198 (46%) | 203 (39%) | 207 (42%) |
| Endocrine | 32 (35%) | 34 (32%) | 118 (30%) | 144 (34%) | 170 (32%) | 147 (30%) |
| Haematological | 8 (9%) | 4 (4%) | 36 (9%) | 59 (14%) | 68 (13%) | 42 (8%) |
| Musculoskeletal | 27 (29%) | 25 (24%) | 90 (23%) | 119 (28%) | 131 (25%) | 114 (23%) |
| Neoplasia | 5 (5%) | 6 (6%) | 26 (7%) | 40 (9%) | 38 (7%) | 27 (5%) |
| Neurological | 16 (17%) | 7 (7%) | 33 (8%) | 54 (13%) | 52 (10%) | 57 (12%) |
| Psychiatric | 3 (3%) | 5 (5%) | 11 (3%) | 15 (4%) | 18 (3%) | 19 (4%) |
| Immunological | 5 (5%) | 1 (1%) | 21 (5%) | 37 (9%) | 15 (3%) | 21 (4%) |
| Dermatological | 13 (14%) | 13 (12%) | 55 (14%) | 59 (14%) | 72 (14%) | 74 (15%) |
| Allergies | 8 (9%) | 10 (9%) | 42 (11%) | 51 (12%) | 48 (9%) | 53 (11%) |
| Ophthalmological | 8 (9%) | 7 (7%) | 19 (5%) | 29 (7%) | 49 (9%) | 34 (7%) |
| Ear, nose, throat | 11 (12%) | 3 (3%) | 23 (6%) | 22 (5%) | 27 (5%) | 25 (5%) |
| Other | 20 (22%) | 17 (16%) | 79 (20%) | 98 (23%) | 122 (23%) | 105 (21%) |
HLA Ab status
The HLA Ab status at time of transplant was known for 91% of patients. Of the DSA+, fewer than 25% in either group had HLA Ab at the time of transplantation, indicating that > 75% had developed de novo DSA, whereas 35–40% of the groups with non-DSA HLA Ab, and 7% of the HLA Ab-negative group had HLA Ab at the time of transplantation (see Table 5). Approximately 45% of recruits in each of the DSA+ groups had DSA directed against HLA DQB antigens with a median MFI of 6200–7000, and 15–26% had DSA against HLA A antigens with a median MFI of 3600–4000 (see Table 6). Site investigators were not prevented from asking for routine HLA Ab tests via the normal clinic pathway. 374 patients had their HLA Ab status checked during the trial, including 191 in the blinded care arm. The split by group is illustrated in Table 6. Interestingly, 75–80% of the patients identified at recruitment or on rescreening as having a DSA, and who had DSA status reassessed at the last visit (month 32 post-Ab-detection), had become DSA-negative (see Table 5), with no obvious differences between SC and BLC groups.
| DSA+ | Non-DSA+ | No HLA Ab | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Blinded (SC) A1 | Unblinded (BLC) B1 | Blinded (SC) A2 | Unblinded (BLC) B2 | Blinded (SC) C | Unblinded (BLC) D | |||||||||||||
| Time of Tx | Post-screening | End | Time of Tx | Post-screening | End | Time of Tx | Post-screening | End | Time of Tx | Post-screening | End | Time of Tx | Post-screening | End | Time of Tx | Post-screening | End | |
| Number of Ab+ (%) | 21 (22.8) | 92 (100) | 34 (37*) | 23 (21.7) | 106 (100) | 38 (35.0) | 158 (40.4) | 389 (99.5) | 106 (27.1) | 153 (35.8) | 425 (99.5) | 99 (23.1) | 37 (7) | 2 (0.4) | 9 (1.7) | 33 (6.7) | 0 (0) | 8 (1.6) |
| Definite DSA | - | 91 (98.9) | 18 (19.6) | - | 103 (97.2) | 25 (23.6) | - | 0 (0) | 13 (3.3) | - | 0 (0) | 3 (0.7) | - | 0 (0) | 2 (0.4) | - | 0 (0) | 1 (0.2) |
| Definite non-DSA | - | 1a (1.1) | 13 (14.1) | - | 1a (0.9) | 10 (9.4) | - | 346 (88.5) | 86 (22) | - | 383 (89.7) | 92 (21.5) | - | 0 (0) | 7 (1.3) | - | 0 (0) | 5 (1) |
| Unknown whether DSA | - | 0 (0) | 3 (3.3) | - | 2a (1.8) | 3 (2.8) | - | 43 (11) | 7 (1.8) | - | 42 (9.8) | 4 (0.9) | - | 2b (0.6) | 0 (0) | - | 0 (0) | 2 (0.4) |
| Number of Ab– (%) | 64 (69.6) | 0 (0) | 35 (38) | 72 (67.9) | 0 (0) | 40 (37.7) | 192 (49.1) | 0 (0) | 191 (48.8) | 230 (53.9) | 1c (0.2) | 210 (49.2) | 449 (88.5) | 521 (99) | 437 (83.1) | 431 (87.1) | 489 (98.8) | 402 (81.2) |
| Missing data | 7 (10.5) | 0 (0) | 23 (25) | 11 (10.4) | 0 (0) | 28 (26.4) | 41 (10.5) | 2 (0.5) | 94 (24) | 44 (10.3) | 1 (0.2) | 118 (27.6) | 40 (7.6) | 3 (0.6) | 80 (15.2) | 31 (6.3) | 6 (1.2) | 85 (17.2) |
| Total | 92 (100) | 92 (100) | 92 (100) | 106 (100) | 106 (100) | 106 (100) | 391 (100) | 391 (100) | 391 (100) | 427 (100) | 427 (100) | 427 (100) | 526 (100) | 526 (100) | 526 (100) | 495 (100) | 495 (100) | 495 (100) |
| DSA+ | Non-DSA+ | No HLA Ab- | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Blinded (SC) A1 (N = 92) | Unblinded (BLC) B1 (N = 106) | Blinded (SC) A2 (N = 391) | Unblinded (BLC) B2 (N = 427) | Blinded (SC) C (N = 526) | Unblinded (BLC) D (N = 495) | |||||||
| HLA | Donor MM N (%): *assumed | DSA % [median MFI] | Donor MM N (%) *assumed | DSA % [median MFI] | Donor MM N (%) *assumed |
DSA % [median MFI] | Donor MM N (%) *assumed |
DSA % [median MFI] | Donor MM N (%) *assumed |
DSA % [median MFI] | Donor MM N (%) *assumed |
DSA % [median MFI] |
| A | 78 (85%) *0 | 26% [3998] | 87 (82%) *1 | 15% [3630] | 273 (70%) *0 | 0 | 285 (67%) *0 | 0 | 402 (76%) *0 | 0 | 352 (71%) *0 | 0 |
| B | 84 (91%) *0 | 9.8% [2424] | 93 (88%) *0 | 13% [5990] | 287 (73%) *0 | 0 | 300 (70%) *0 | 0 | 435 (83%) *0 | 0 | 385 (78%) *1 | 0 |
| C | 73 (79%) *1 | 16% [3733] | 80 (76%) *2 | 10% [3401] | 246 (63%) *1 | 0 | 258 (60%) *1 | 0 | 364 (69%) *3 | 0 | 322 (65%) *1 | 0 |
| DRB1 | 65 (71%) *1 | 6.5% [2645] | 89 (84%) *7 | 18% [3155] | 200 (51%) *2 | 0 | 220 (52%) *2 | 0 | 307 (58%) *1 | 0 | 284 (57%) *2 | 0 |
| DRB3 | 16 (17%) *3 | 4.3% [3148] | 17 (16%) *2 | 3.8% [4290] | 38 (9.7%) *0 | 0 | 35 (8.2%) *0 | 0 | 58 (11%) *0 | 0 | 65 (13%) *2 | 0 |
| DRB4 | 17 (19%) *0 | 2.2% [13850] | 25 (24%) *1 | 7.5% [6373] | 42 (11%) *1 | 0 | 49 (12%) *2 | 0 | 58 (11%) *0 | 0 | 82 (17%) *0 | 0 |
| DRB5 | 7 (7.6%) *0 | 1.1% [5326a] | 8 (7.5%) *0 | 0.94% [5568a] | 28 (7.2%) *0 | 0 | 28 (6.6%) *0 | 0 | 50 (9.5%) *0 | 0 | 49 (9.9%) *1 | 0 |
| DQA | 8 (8.7%) *2 | 4.3% [12005] | 9 (8.5%) *6 | 4.7% [12845] | 9 (2.3%) *5 | 0 | 14 (3.3%) *2 | 0 | 10 (1.9%) *2 | 0 | 8 (1.6%) *1 | 0 |
| DQB | 66 (72%) *5 | 44% [6947] | 78 (74%) *5 | 46% [6279] | 161 (41%) *5 | 0 | 189 (44%) *6 | 0 | 281 (53%) *0 | 0 | 265 (54%) *4 | 0 |
| DPB | 11 (12%) *5 | 3.3% [5623] | 9 (8.5%) *4 | 0.94% [5177a] | 31 (7.9%) *15 | 0 | 34 (8%) *15 | 0 | 35 (6.7%) *6 | 0 | 33 (6.7%) *18 | 0 |
| Recruits (%) having HLA Ab test outside trialb | ||||||||||||
| 28 (30%) | 27 (26%) | 75 (19%) | 89 (21%) | 88 (17%) | 67 (14%) | |||||||
Baseline and change in IS during the study
Eighteen per cent of all participants were taking ciclosporin at randomisation, and 15% taking azathioprine. Interestingly, the proportions on ciclosporin or azathioprine were highest in those with DSA compared to those with non-DSA and those who were HLA Ab-negative (see Table 3). The majority of patients were taking Tac (73%) or MMF (67%) at randomisation, though fewer were taking maintenance prednisolone (55%). The proportions on Tac or MMF were lowest in those with DSA, compared to those with non-DSA and those who were HLA Ab-negative (see Table 3). Finally, 27% overall were taking all three drugs. The proportion taking all three drugs was lowest in those with DSA compared to those with non-DSA and those who were HLA Ab-negative (see Table 3). These differences were maintained when considering patients who developed new HLA Ab during the rescreening process. Therefore, the proportions on ciclosporin or azathioprine were still highest in those with DSA after rescreening compared to those with non-DSA and those who were HLA Ab-negative (see Table 4). The proportions on Tac or MMF were still lowest in those with DSA after rescreening, compared to those with non-DSA and those who were HLA Ab-negative (see Table 4). Finally, the proportion taking all three drugs was still lowest in those with DSA after rescreen, compared to those with non-DSA and those who were HLA Ab-negative (see Table 4).
Five hundred twelve of the five hundred thirty-two (97%) HLA Ab+ patients in the BLC arm had an optimisation interview and 33% of the DSA group, and 24% of the non-DSA group underwent steroid boost (see Table 7). The proportion taking all three IMPs increased from 23% immediately post-screening to 54% immediately post-optimisation in the DSA+ BLC group, and from 25% to 44% in the non-DSA+ BLC group. These changes were sustained to the last visit and were statistically significant (see Table 7). There were no discernible differences in demographic characteristics between those optimised according to the full protocol and those not optimised to the full protocol (see Table 8). However, there was significant site variation in the implementation of the full optimisation protocol, with only 6 of 13 sites optimising more than 50% of their BLC HLA Ab+ patients on all three IMPs (see Table 8).
| Characteristic | DSA+ | Non-DSA+ | HLA Ab Negative | |||
|---|---|---|---|---|---|---|
| Blinded (SC) | Unblinded (BLC) | Blinded (SC) | Unblinded (BLC) | Blinded (SC) | Unblinded (BLC) | |
| Group | A1 (N = 92) | B1 (N = 106)d | A2 (N = 391) | B2 (N = 427)e | C (N = 526) | D (N = 495) |
| Had optimisation interview N (%) | 0 (0%) | 102 (96%) | 0 (0%) | 413 (97%) | 0 (0%) | 0 (0%) |
| Taking Tac | ||||||
| Post-screeninga N (%) | 56 (61%) | 68 (64%) | 296 (76%) | 313 (73%) | 392 (75%) | 366 (74%) |
| Mean dose mg (SD) | 6.2 (6) | 4.6 (3.3) | 5.1 (3.7) | 5.4 (4.4) | 5.4 (4.1) | 4.7 (3.2) |
| Mean level (SD) | 6.6 (2.9) | 5.8 (2.2) | 6.9 (2.7) | 6.7 (2.2) | 6.9 (2.3) | 6.7 (2.5) |
| At last visitb N (%) | 58 (63%) | 87 (82%) | 301 (77%) | 355 (85%) | 387 (74%) | 368 (74%) |
| Mean dose mg (SD) | 6.2 (4.4) | 5.2 (3.7) | 4.8 (3.3) | 5.2 (3.7) | 5.1 (3.8) | 4.6 (3.2) |
| Mean level (SD) | 6.6 (2.6) | 6.8 (2.4) | 6.5 (2.3) | 6.7 (2.3) | 6.6 (2.2) | 6.4 (2.0) |
| Taking MMF | ||||||
| Post-screeninga N (%) | 59 (64%) | 62 (59%) | 254 (65%) | 271 (63%) | 361 (69%) | 351 (71%) |
| Mean dose mg (SD) | 1165 (482) | 1145 (399) | 1134 (457) | 1112 (472) | 1147 (495) | 1136 (473) |
| At last visitb N (%) | 59 (64%) | 77 (73%) | 246 (63%) | 305 (72%) | 246 (63%) | 338 (68%) |
| Mean dose mg (SD) | 1178 (470) | 1237 (450) | 1082 (442) | 1149 (457) | 1088 (440) | 1098 (438) |
| Taking prednisolone | ||||||
| Post-screeninga N (%) | 53 (58%) | 62 (59%) | 210 (54%) | 227 (53%) | 295 (56%) | 274 (55%) |
| Mean dose mg (SD) | 5.2 (1.8) | 5.1 (1.9) | 5.0 (1.4) | 5.1 (1.6) | 5.1 (1.8) | 5.1 (1.4) |
| At last visitb N (%) | 55 (60%) | 81 (76%) | 212 (54%) | 268 (63%) | 303 (58%) | 273 (55%) |
| Mean dose mg (SD) | 5.7 (3.7) | 5.3 (2.1) | 5.2 (1.9) | 5.2 (1.8) | 5.7 (4.2) | 5.1 (1.5) |
| Given prednisolone boost N (%) | 0 (0%) | 34 (33%) | 0 (0%) | 101 (24%) | 0 (0%) | 0 (0%) |
| Taking Tac/MMF/Pred N (%) | ||||||
| Post-screening | 19 (21%) | 24 (23%) | 114 (29%) | 106 (25%) | 152 (29%) | 139 (28%) |
| Immediately post-optimisation c | - | 53 (54%) | - | 178 (44%) | - | - |
| At last visit b | 20 (22%) | 51 (48%) | 114 (29%) | 172 (40%) | 142 (27%) | 129 (26%) |
| Optimised to full protocol (N = 231) | Not optimised to full protocol (N = 271) | |
|---|---|---|
| Age (years) Mean (SD) | 47.8 (13.6) | 51.3 (12.3) |
| Male (%) | 66% | 61% |
| Ethnicity (%) | ||
| Asian | 13% | 14% |
| Black | 12% | 11% |
| White | 72% | 73% |
| Mixed | 0.9% | 1% |
| Other | 2.2% | 1.5% |
| Site [N (%)]2 | ||
| Leeds | 14 (16.9%) | 69 (83.1%) |
| Royal London | 11 (44%) | 14 (56%) |
| Guy’s | 97 (65.5%) | 24 (34.5%) |
| Manchester | 17 (29.3%) | 41 (70.7%) |
| Birmingham | 34 (68%) | 16 (32%) |
| King’s College Hospital | 10 (33.3%) | 20 (66.7%) |
| York | 11 (57.9%) | 8 (42.1%) |
| Coventry | 9 (64.3%) | 5 (35.7%) |
| Preston | 10 (58.8%) | 7 (41.2%) |
| Salford | 6 (75%) | 2 (25%) |
| Bradford | 1 (6.2%) | 15 (93.8%) |
| Royal Free | 10 (37%) | 17 (63%) |
| St Helier | 1 (14.3%) | 6 (85.7%) |
| Previous transplants [N (%)] | ||
| 0 | 175 (76%) | 221 (82%) |
| 1 | 47 (20%) | 42 (16%) |
| 2 | 7 (3%) | 7 (2.6%) |
| 3 | 1 (0.4%) | 1 (0.4%) |
| Time (years) since Tx | ||
| Median (IQR) | 4.5 (2.1–9.2) | 7.1 (3.0–12.6) |
| Renal function | ||
| eGFR (ml/min/1.73 m2) [Mean (SD)] | 54.9 (16.76) | 53.91 (17.83) |
| Suffered graft failure [N (%)] | 18 (7.8%) | 16 (5.9%) |
| Past medical history [n (%) experienced in that system] | ||
| Cardiovascular | 150 (65%) | 155 (57%) |
| Respiratory | 30 (13%) | 42 (16%) |
| Hepatic | 8 (3%) | 11 (4%) |
| Gastrointestinal | 39 (17%) | 55 (20%) |
| Genitourinary | 128 (55%) | 103 (38%) |
| Endocrine | 79 (34%) | 89 (33%) |
| Haematological | 32 (14%) | 27 (10%) |
| Musculoskeletal | 55 (24%) | 82 (30%) |
| Neoplasia | 15 (6%) | 30 (11%) |
| Neurological | 32 (14%) | 25 (9%) |
| Psychiatric | 7 (3%) | 11 (4%) |
| Immunological | 21 (9%) | 17 (6%) |
| Dermatological | 24 (10%) | 42 (16%) |
| Allergies | 35 (15%) | 22 (8%) |
| Ophthalmological | 22 (10%) | 14 (5%) |
| Ear, nose, throat | 11 (5%) | 13 (5%) |
| Other | 57 (25%) | 53 (20%) |
Completion of follow-up visits
The majority of participants completed all four of the formal intensive study follow-up visits at months 8, 16, 24 and 32 months. 1.2% were withdrawn, died or reached the primary endpoint prior to the month 8 visit, and 2.5% of the remainder missed this visit. The corresponding figures for month 16 are 3.6% and 1.1%: month 24, 6.4% and 2.8% and month 32, 9.4% and 1.1%.
Primary analysis – time to graft failure in HLA Ab± groups (hypotheses 1a and 1b)
There were 34 graft failures in the blinded SC HLA Ab+ groups (12 DSA+, 22 non-DSA+) compared to 42 in the unblinded BLC HLA Ab+ groups (19 DSA+, 23 non-DSA+), with no evidence that the unblinded BLC strategy is superior to the SC strategy. 95% CIs included the null HR, in both the HLA Ab DSA+ group [HR1.54 (95% CI 0.72 to 3.30)] or non-DSA+ group [HR 0.97 (0.54 to 1.74)] (see Figures 4 and 5, Table 9).
FIGURE 4.
Kaplan–Meier Curves – DSA+ groups. Graph compares time to graft failure in the DSA+ groups. Blue (unbroken) line = patients in unblinded, BLC arm. Black (broken) line = patients in blinded SC arm. The number at risk of graft failure at each time point is shown beneath the graph, followed by (in brackets) the number of graft failures. NB: One HLA-Ab-negative participant in the blinded (SC) group who developed DSA on rescreening was not included in this analysis as the graft failed prior to rescreening, so they were not at risk for the purpose of this analysis.
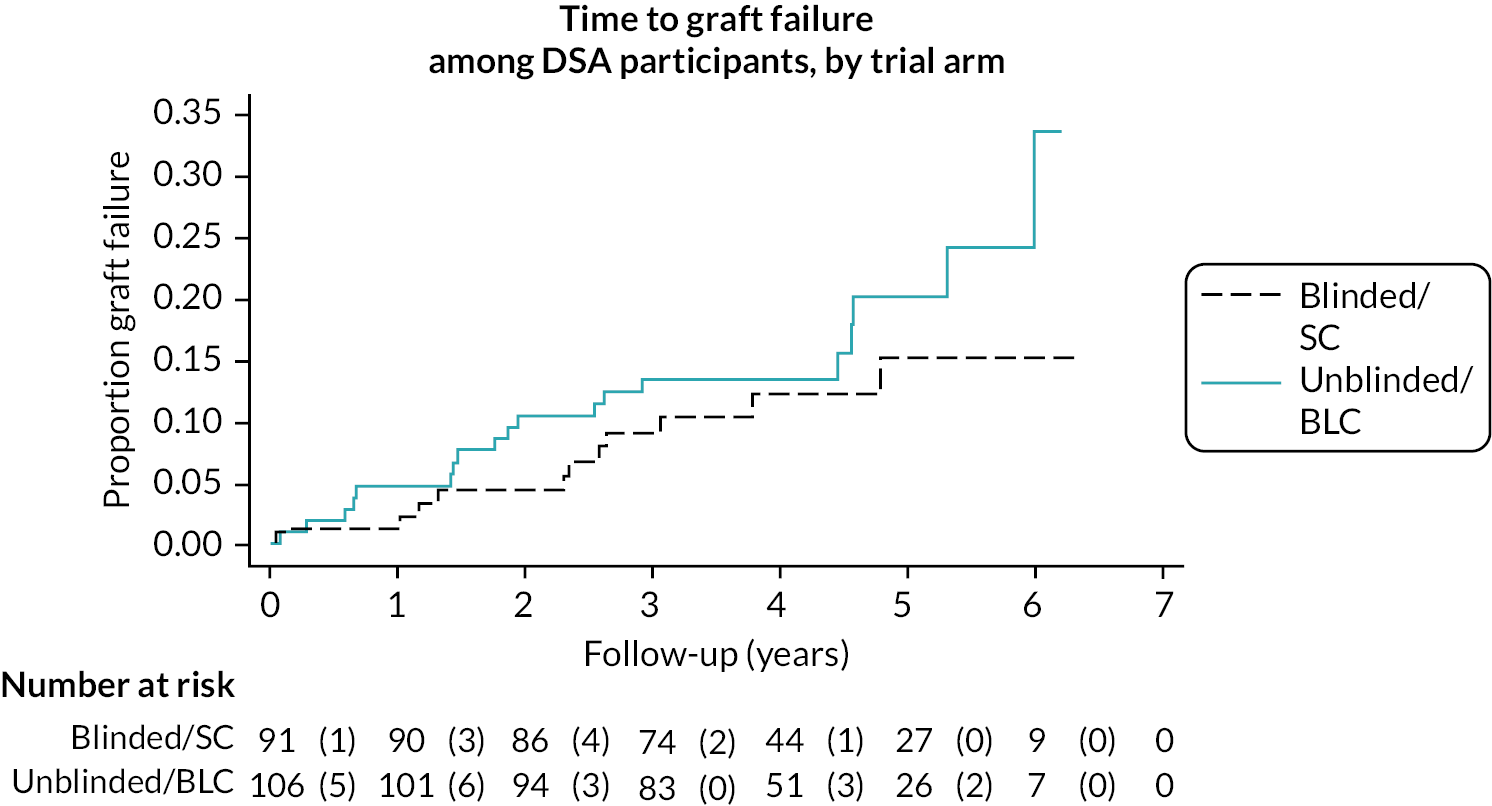
FIGURE 5.
Kaplan–Meier Curves – non-DSA+ groups. Graph compares time to graft failure in the non-DSA+ groups. Blue (unbroken) line = patients in unblinded, BLC arm. Black (broken) line = patients in blinded SC arm. The number at risk of graft failure at each time point is shown beneath the graph, followed by (in brackets) the number of graft failures.
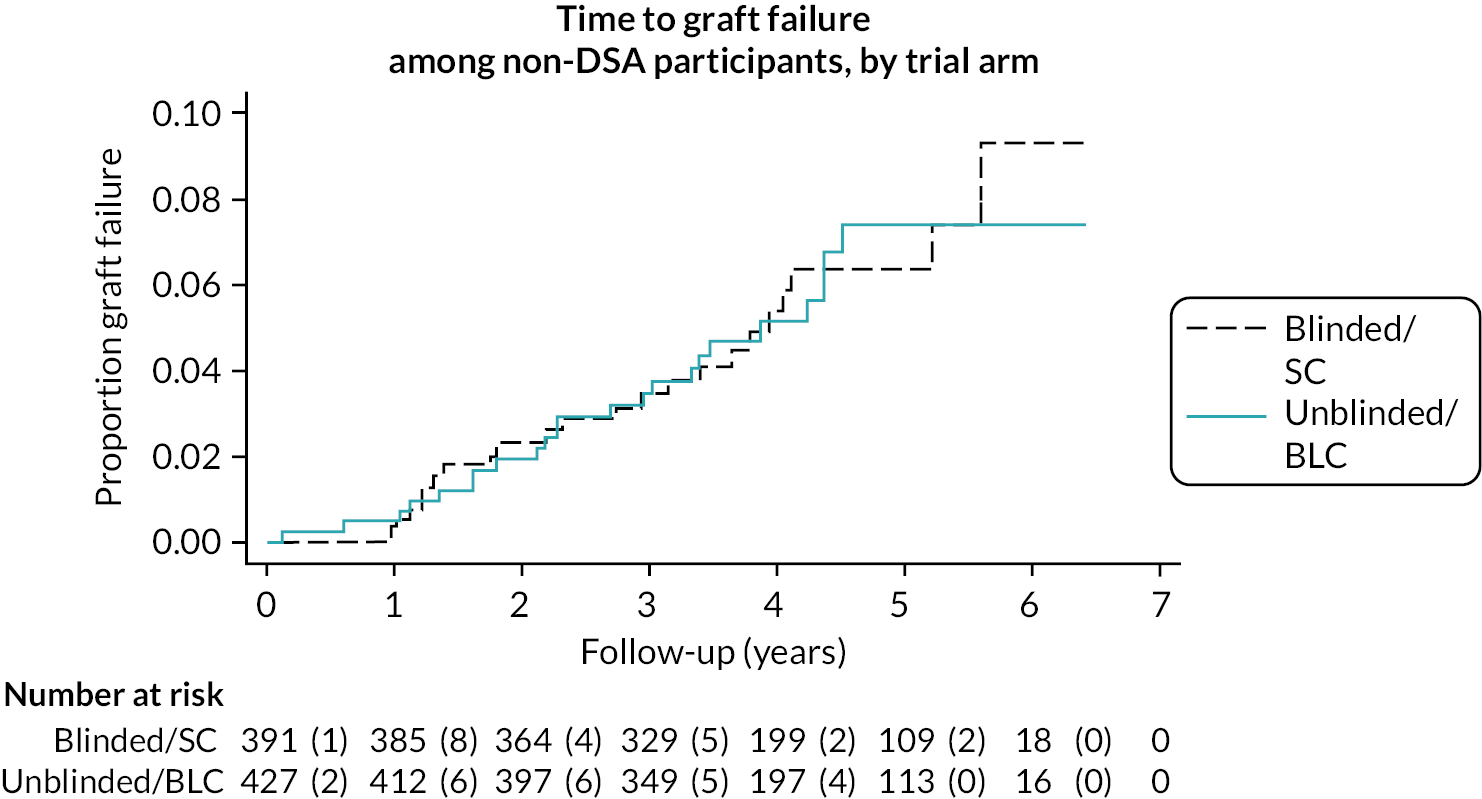
| Group/comparison | Hazard/odds ratioc | 95% CI | p-value |
|---|---|---|---|
| Primary outcome – time to graft failure | |||
| DSA (N = 197a) | 1.54 | 0.72 to 3.30 | 0.27 |
| Non-DSA (N = 818) | 0.97 | 0.54 to 1.74 | 0.91 |
| All participants (N = 2035b) | 1.02 | 0.72 to 1.44 | 0.93 |
| Secondary outcome measures | |||
| Death | |||
| DSA (N = 197) | 2.33 | 0.57 to 9.57 | 0.24 |
| Non-DSA (N = 818) | 1.24 | 0.76 to 2.02 | 0.40 |
| All participants (N = 2035) | 1.14 | 0.85 to 1.54 | 0.38 |
| Biopsy-proven rejection | |||
| DSA (N = 198) | 0.35 | 0.10 to 1.17 | 0.09 |
| Non-DSA (N = 818) | 0.57 | 0.18 to 1.78 | 0.32 |
| All participants (N = 2035) | 0.50 | 0.27 to 0.94 | 0.03 |
| Confirmed infection | |||
| DSA (N = 197) | 1.75 | 0.89 to 3.44 | 0.10 |
| Non-DSA (N = 809) | 1.09 | 0.79 to 1.50 | 0.62 |
| All participants (N = 2010) | 1.08 | 0.88 to 1.33 | 0.46 |
| Malignancy | |||
| DSA (N = 198) | 1.08 | 0.36 to 3.28 | 0.89 |
| Non-DSA (N = 810) | 0.93 | 0.57 to 1.52 | 0.77 |
| All participants (N = 2015) | 0.92 | 0.65 to 1.31 | 0.65 |
| DM | |||
| DSA (N = 198) | 0.99 | 0.19 to 5.21 | 0.99 |
| Non-DSA (N = 818) | 0.56 | 0.25 to 1.26 | 0.16 |
| All (N = 2015) | 0.75 | 0.41 to 1.37 | 0.34 |
| Proteinuria | |||
| DSA (N = 184) | 0.28 | 0.05 to 1.59 | 0.15 |
| Non-DSA (N = 788) | 1.47 | 0.61 to 3.53 | 0.39 |
| All participants (N = 1972) | 0.80 | 0.47 to 1.37 | 0.42 |
| eGFR | Mean difference | ||
| DSA (N = 192) | 0.91 | –2.83 to 4.65 | 0.63 |
| Non-DSA (N = 805) | 0.24 | –1.50 to 1.98 | 0.78 |
| All participants (N = 2015) | –0.46 | –1.98 to 1.05 | 0.55 |
Post-COVID, there were 39 graft failures in the blinded SC HLA Ab+ groups (15 DSA+, 24 non-DSA+) compared to 49 in the unblinded BLC groups (21 DSA+, 28 non-DSA+). Nevertheless, the sensitivity analysis showed no appreciable difference from the primary analysis (see Table 10). In the BLC HLA Ab+ groups, there were 18 graft failures in those who underwent full optimisation according to the protocol, but only 16 in those not optimised according to the protocol and when the former were used in the sensitivity analysis, there was no appreciable difference in the primary outcome. The same is true for all the other planned sensitivity analyses (see Table 10). Post-hoc sensitivity analyses adjusting additionally for factors unbalanced at baseline (sex and time since transplant) or only looking at unblinded BLC recruits that underwent ‘best’ optimisation had no impact on the effect estimates had no impact on the effect estimates (see Table 10).
| Group/comparison | HR | Lower 95% CI | Upper 95% CI | p-value |
|---|---|---|---|---|
| Post-COVID analysis a | ||||
| DSA (N = 197b) | 1.29 | 0.64 | 2.60 | 0.48 |
| Non-DSA (N = 818) | 1.05 | 0.61 | 1.82 | 0.86 |
| All participants (N = 2035c) | 1.03 | 0.74 | 1.42 | 0.88 |
| Excluding site as a covariate | ||||
| DSA (N = 197) | 1.51 | 0.72 | 3.19 | 0.28 |
| Non-DSA (N = 818) | 0.98 | 0.54 | 1.75 | 0.93 |
| All participants (N = 2035) | 1.02 | 0.72 | 1.45 | 0.91 |
| Competing risk of death | ||||
| DSA (N = 197) | 1.53 | 0.70 | 3.35 | 0.29 |
| Non-DSA (N = 818) | 0.96 | 0.53 | 1.74 | 0.90 |
| All participants (N = 2035) | 1.01 | 0.71 | 1.43 | 0.96 |
| Randomisation as time zero d | ||||
| DSA (N = 198) | 1.35 | 0.64 | 2.86 | 0.43 |
| Non-DSA (N = 818) | 0.96 | 0.53 | 1.72 | 0.88 |
| Analysis of only those who underwent IS optimisation (using only BLC participants taking all 3 IMPs)e | ||||
| DSA (N = 145) | 1.17 | 0.44 | 3.14 | 0.75 |
| Non-DSA (N = 569) | 0.96 | 0.44 | 2.10 | 0.91 |
| All participants (N = 1238) | 1.21 | 0.71 | 2.09 | 0.48 |
| Analysis of only those with definite non-DSA | ||||
| DSA (N = 283) | 1.47 | 0.76 | 2.85 | 0.25 |
| Non-DSA (N = 729) | 0.90 | 0.46 | 1.73 | 0.74 |
| Post-hoc sensitivity with sex and time since transplant as additional covariates | ||||
| DSA (N = 197) | 1.60 | 0.73 | 3.49 | 0.24 |
| Non-DSA (N = 818) | 1.02 | 0.56 | 1.85 | 0.96 |
| All participants (N = 2035) | 1.00 | 0.71 | 1.43 | 0.98 |
| Post-hoc sensitivity using only BLC participants taking all 3 IMPs with tac levels 6–8f | ||||
| DSA (N = 118) | 1.23 | 0.30 | 4.98 | 0.77 |
| Non-DSA (N = 496) | 0.70 | 0.23 | 2.12 | 0.53 |
| All participants (N = 1138) | 1.02 | 0.48 | 2.17 | 0.96 |
Secondary outcome analysis
Time to graft failure in all unblinded BLC versus all blinded SC (hypothesis 2)
Overall there were 62 graft failures in the blinded care arm (including 28 HLA Ab-negatives) compared to 64 in the unblinded care arm (including 22 in the HLA Ab-negative groups), providing insufficient evidence for non-inferiority of the unblinded BLC strategy with the upper 95% confidence limit for the HR exceeded the pre-specified threshold of 1.4 (HR 1.02, 95% CI 0.72 to 1.44) (see Figure 6). Time to graft failure in the HLA Ab-negative groups only is shown in Figure 7.
FIGURE 6.
Kaplan–Meier Curves – BLC versus SC. Graph compares time to graft failure in the entire unblinded BLC arm versus blinded SC arm. Blue (unbroken) line = patients in unblinded, BLC arm. Black (broken) line = patients in blinded SC arm. The number at risk of graft failure at each time point is shown beneath the graph, followed by (in brackets) the number of graft failures.
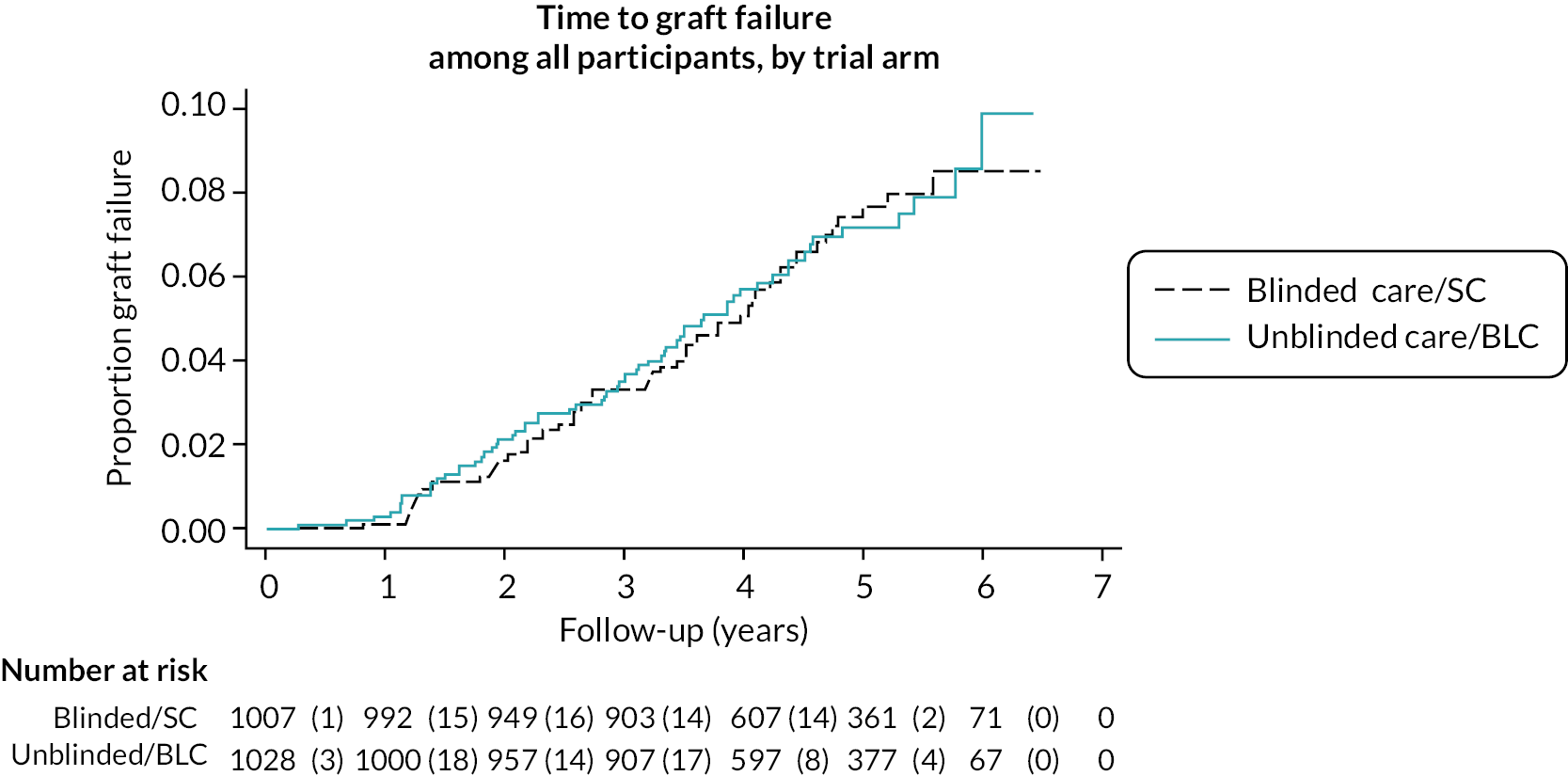
FIGURE 7.
Kaplan–Meier Curves – HLA Ab negatives. Graph compares time to graft failure in the HLA Ab-negative groups. Blue (unbroken) line = patients in unblinded, BLC arm. Black (broken) line = patients in blinded SC arm. The number at risk of graft failure at each time point is shown beneath the graph, followed by (in brackets) the number of graft failures.
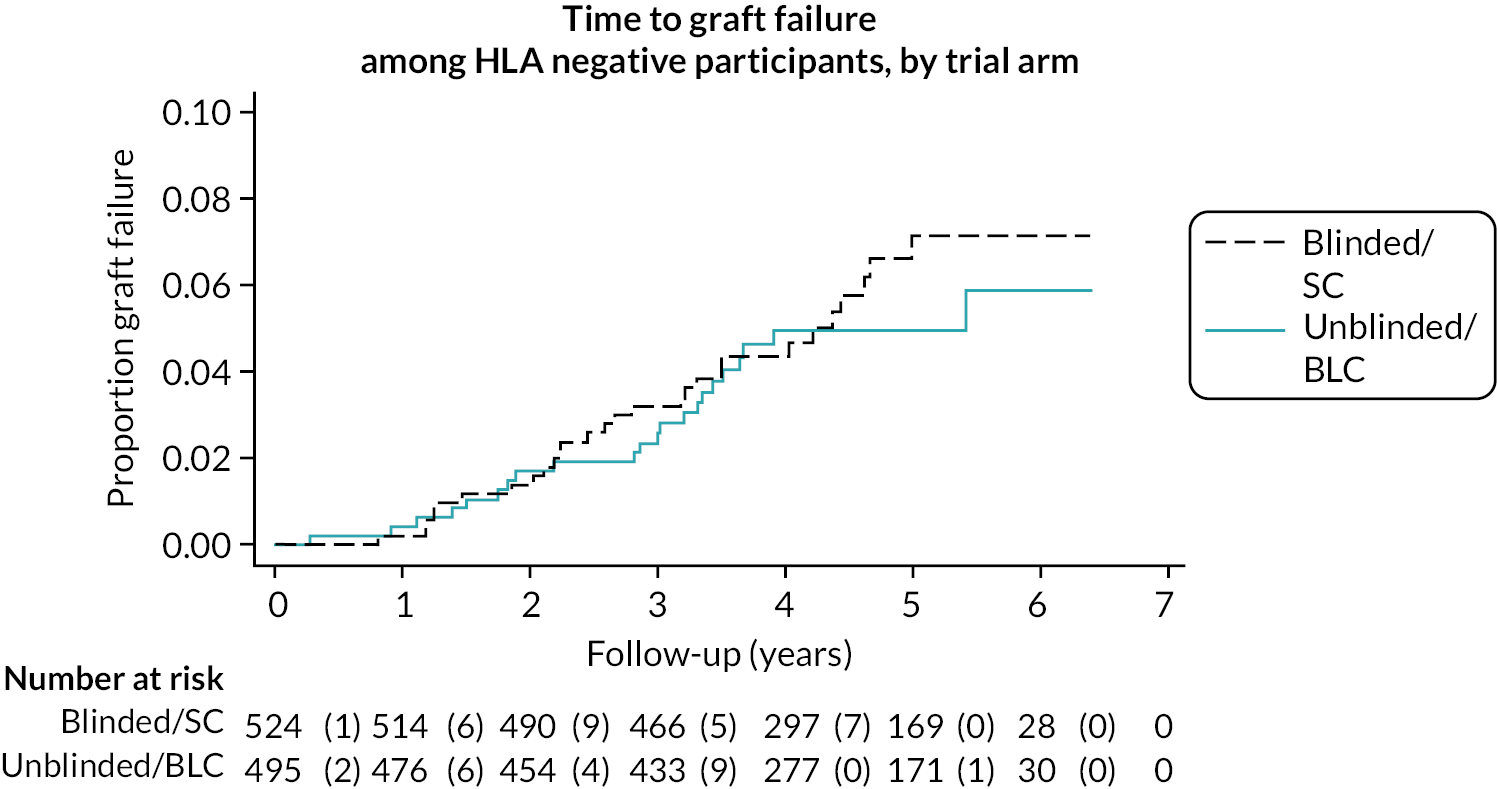
Patient mortality
Survival was 92.7% in the blinded care group and 92.2% in the unblinded care group with no significant differences between arms in any of the specified comparisons (see Table 9).
Biopsy-proven rejection
Forty-seven patients were diagnosed with rejection after a for-cause biopsy, 16 in the unblinded BLC arm (5 DSA+, 6 non-DSA+, 5 Ab-negative) and 31 in the blinded SC arm (11 DSA+, 8 non-DSA+, 12 Ab-negative), though because some recruits had rejection before they developed HLA Ab, and because some clock-reset recruits had rejection after the 32 months post-recruitment period had finished, not all were included in the formal trial analyses (see Table 11). The odds of biopsy-proven rejection were significantly lower in the overall BLC group than in the overall SC group (0.50, 95% CI 0.27 to 0.94; p = 0.03) (see Table 9). The diagnostic features of all biopsies performed in the DSA+ patients are reported in Table 12.
| DSA+ | Non-DSA+ | HLA Ab-negative | |||||
|---|---|---|---|---|---|---|---|
| SC | BLC | SC | BLC | SC | BLC | Total | |
| Biopsy-proven rejection | |||||||
| Total biopsy-proven rejection | 11 | 5 | 8 | 6 | 12 | 5 | 47 |
| Included in formal analysis of rejection in HLA Ab+ groupsa | 9 | 4 | 8 | 5 | N/A | N/A | 26 |
| Included in formal analysis of rejection in overall BLC versus SC comparisonb | 10 | 5 | 7 | 5 | 12 | 5 | 44 |
| Culture/PCR confirmed infections | |||||||
| Total confirmed infections | 21 | 32 | 95 | 109 | 115 | 107 | 479 |
| Included in formal analysis of infection in HLA Ab+ groupsa | 18 | 32 | 92 | 106 | N/A | N/A | 248 |
| Included in formal analysis of infection in overall BLC versus SC comparisonb | 21 | 32 | 87 | 100 | 115 | 107 | 462 |
| Malignancies | |||||||
| Total malignancies | 6 | 10 | 35 | 38 | 36 | 25 | 150 |
| Included in formal analysis of malignancies in HLA Ab+ groupsa | 6 | 8 | 35 | 37 | N/A | N/A | 86 |
| Included in formal analysis of malignancies in overall BLC versus SC comparisonb | 5 | 10 | 29 | 31 | 36 | 25 | 136 |
| DM | |||||||
| Total diabetes | 4 | 3 | 17 | 10 | 7 | 9 | 50 |
| Included in formal analysis of diabetes in HLA Ab+ groupsa | 3 | 3 | 16 | 10 | N/A | N/A | 32 |
| Included in formal analysis of diabetes in overall BLC versus SC comparisonb | 3 | 3 | 15 | 7 | 7 | 9 | 44 |
| BANFF 09 classification | Unblinded BLC DSA+ | Blinded SC DSA+ |
|---|---|---|
| Category 1 normal | 2 | 0 |
| Category 2 ABMR | 2 | 8 |
| C4d deposition only | 0 | 1 |
| Subtype 1 | 0 | 0 |
| Subtype 2 | 0 | 2 |
| Subtype 3 | 1a | 0 |
| Chronic | 1 | 5b |
| Subtype NOS | 0 | 0 |
| Category 3 borderline change | 2 | 3 |
| Category 4 TCMR | 3 | 2 |
| Subtype IA | 1 | 2b |
| Subtype IB | 1 | 0 |
| Subtype IIA | 0 | 0 |
| Subtype IIB | 0 | 0 |
| Subtype III | 1a | 0 |
| Subtype NOS | 0 | 0 |
| Category 5 IFTA without specific cause | 5 | 3 |
| Grade I | 2 | 0 |
| Grade II | 3a | 3 |
| Grade III | 0 | 0 |
| Category 6 | 5c | 6d |
| Insufficient sample | 2 | 0 |
| Total | 21 | 22 |
Other secondary outcome measures
There were no significant differences between groups for any other adverse effect outcome (see Table 9). 231 proven infections were documented during the intensive follow-up period in the blinded SC arm (21 DSA+, 95 non-DSA+, 115 HLA Ab-negative), compared to 248 in the unblinded BLC arm (32 DSA+, 109 non-DSA+, 107 HLA Ab-negative) (see Table 11). Details of specific infections are in Table 13. Seventy-seven malignancies were reported in the blinded SC arm (6 DSA+, 35 non-DSA+, 36 HLA Ab-negative), compared to 73 in the unblinded BLC arm (10 DSA+, 38 non-DSA+, 25 HLA Ab-negative). Details of specific malignancies are given in Table 14. Fifty patients (2.5%) developed de novo DM during the trial, 22 in the BLC arm (3 DSA+, 10 non-DSA+, 9 HLA Ab-negative) and 28 in the SC arm (4 DSA+, 17 non-DSA+ and 7 HLA Ab-negative). For the same reasons as stated above, not all infections, malignancies or case of DM were included in the formal trial analyses (see Table 11). The odds of developing proteinuria in DSA+ BLC group were 0.28 times the odds of developing proteinuria in the DSA+ SC group but the CIs were wide and included the null value. Mean eGFR at month 32 was similar between the DSA BLC group (53.1 SD = 19.8) and DSA SC group (56.1 SD = 22.7) and there was no significant mean difference in eGFR for any of the comparisons (see Table 9).
| DSA+ | Non-DSA+ | No HLA Ab | |||||
|---|---|---|---|---|---|---|---|
| Infections | Blinded (SC) (%) | Unblinded (BLC) (%) | Blinded (SC) (%) | Unblinded (BLC) (%) | Blinded (SC) (%) | Unblinded (BLC) (%) | Total |
| All Infection types | 48 (52) | 65 (61) | 225 (58) | 260 (61) | 276 (53) | 241 (50) | 1115 (55) |
| All Infection types (confirmed by culture or PCR) | 21 (23) | 32 (30) | 95 (25) | 109 (26) | 115 (22) | 107 (22) | 479 (24) |
| Viral | 24 (26) | 30 (28) | 105 (27) | 134 (32) | 120 (23) | 95 (20) | 512 (25) |
| Viral (confirmed) | 4 (4.3) | 5 (4.7) | 19 (4.9) | 22 (5.2) | 25 (4.8) | 22 (4.5) | 97 (4.8) |
| BK | 0 (0.0) | 0 (0.0) | 1 (0.3) | 3 (0.7) | 6 (1.1) | 2 (0.4) | 12 (0.6) |
| CMV | 2 (2.2) | 3 (2.8) | 4 (1.0) | 6 (1.4) | 0 (0.0) | 2 (0.4) | 17 (0.8) |
| EBV | 1 (1.1) | 2 (1.9) | 4 (1.0) | 6 (1.4) | 5 (1.0) | 4 (0.8) | 22 (1.1) |
| Shingles | 1 (1.1) | 1 (0.9) | 1 (0.3) | 1 (0.2) | 5 (1.0) | 3 (0.6) | 12 (0.6) |
| Bacterial | 34 (37) | 46 (43) | 157 (41) | 183 (43) | 197 (38) | 173 (36) | 790 (39) |
| Bacterial (confirmed by culture or PCR) | 19 (21) | 31 (29) | 81 (21) | 93 (22) | 96 (18) | 90 (19) | 412 (20) |
| UTI | 12 (13) | 19 (18) | 64 (17) | 66 (16) | 65 (12) | 59 (12) | 286 (14) |
| Pneumonia | 0 (0.0) | 4 (3.8) | 7 (1.8) | 6 (1.4) | 12 (2.3) | 10 (2.1) | 39 (1.9) |
| TB | 0 (0.0) | 1 (0.9) | 1 (0.3) | 0 (0.0) | 1 (0.2) | 1 (0.2) | 4 (0.2) |
| Fungal | 4 (4.3) | 6 (5.7) | 23 (5.9) | 15 (3.5) | 18 (3.4) | 17 (3.5) | 83 (4.1) |
| Fungal (confirmed by culture or PCR) | 0 (0.0) | 3 (2.8) | 4 (1.0) | 6 (1.4) | 5 (1.0) | 4 (0.8) | 22 (1.1) |
| Pneumocystis jirovecii | 0 (0.0) | 0 (0.0) | 1 (0.3) | 1 (0.2) | 0 (0.0) | 2 (0.4) | 4 (0.2) |
| DSA+ | Non-DSA+ | No HLA Ab | |||||
|---|---|---|---|---|---|---|---|
| Site of malignancy | Blinded (SC) (%) | Unblinded (BLC) (%) | Blinded (SC) (%) | Unblinded (BLC) (%) | Blinded (SC) (%) | Unblinded (BLC) (%) | Total (%) |
| Skin | 3 (3.3) | 6 (5.7) | 23 (5.9) | 23 (5.4) | 24 (4.6) | 15 (3.1) | 94 (4.7) |
| Lymph node | 0 (0.0) | 1 (0.9) | 4 (1.0) | 2 (0.5) | 1 (0.2) | 2 (0.4) | 10 (0.5) |
| Lung | 1 (1.1) | 0 (0.0) | 0 (0.0) | 1 (0.2) | 0 (0.0) | 2 (0.4) | 4 (0.2) |
| Liver | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Breast | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.2) | 0 (0.0) | 0 (0.0) | 1 (0.1) |
| Prostate | 0 (0.0) | 0 (0.0) | 3 (0.8) | 0 (0.0) | 3 (0.6) | 0 (0.0) | 6 (0.3) |
| Stomach | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.2) | 0 (0.0) | 2 (0.4) | 3 (0.1) |
| Colon | 1 (1.1) | 0 (0.0) | 1 (0.2) | 0 (0.0) | 3 (0.6) | 0 (0.0) | 5 (0.3) |
| Cervical/vaginal | 0 (0.0) | 0 (0.0) | 1 (0.3) | 1 (0.2) | 0 (0.0) | 0 (0.0) | 2 (0.1) |
| Bladder | 0 (0.0) | 0 (0.0) | 1 (0.3) | 1 (0.2) | 0 (0.0) | 0 (0.0) | 2 (0.1) |
| Blood | 0 (0.0) | 1 (0.9) | 0 (0.0) | 1 (0.2) | 0 (0.0) | 0 (0.0) | 2 (0.1) |
| Kidney | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (0.2) | 1 (0.2) | 2 (0.1) |
| Tongue/throat/larynx | 1 (1.1) | 1 (0.9) | 3 (0.8) | 2 (0.5) | 0 (0.0) | 0 (0.0) | 7 (0.4) |
| Other | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (0.5) | 3 (0.6) | 2 (0.4) | 7 (0.4) |
| Total | 6 (6.5) | 10 (9.4) | 35 (9.0) | 38 (9.0) | 36 (6.9) | 25 (5.2) | 150 (7.4) |
Health economic analysis
Complete data (i.e. baseline and 16-month costs and EQ-5D-5L) were available for 173 blinded and 189 unblinded cases. The number and percentage of respondents using specific services is shown in Table 15. At baseline, the most commonly used services were renal outpatient, other outpatient, and GP care. Relatively more of the blinded group used other inpatient care. Other services were used by fewer respondents and there were no key differences between arms. At follow-up, similar patterns were evident and again there were few notable differences between the arms.
| Baseline | Follow-up | |||
|---|---|---|---|---|
| Service | Blinded (n = 173) |
Unblinded (n = 189) |
Blinded (n = 173) |
Unblinded (n = 189) |
| Residential care | 3 (1.8) | 3 (1.6) | 1 (0.6) | 1 (0.5) |
| Renal inpatient | 33 (20.6) | 37 (20.0) | 36 (20.9) | 32 (17.0) |
| Intensive care | 9 (5.6) | 5 (2.7) | 2 (1.2) | 3 (1.6) |
| Other inpatient | 30 (18.6) | 20 (11.2) | 26 (15.3) | 31 (16.7) |
| Renal outpatient | 146 (85.4) | 158 (86.3) | 136 (80.0) | 154 (81.9) |
| Other outpatient | 71 (42.8) | 92 (51.1) | 77 (45.3) | 87 (47.3) |
| Day hospital | 4 (2.5) | 5 (2.9) | 4 (2.4) | 7 (3.9) |
| A&E | 33 (20.3) | 36 (20.1) | 38 (22.8) | 36 (20.3) |
| GP | 150 (87.2) | 163 (87.6) | 139 (80.8) | 136 (72.3) |
| Physiotherapist | 27 (16.2) | 27 (15.1) | 18 (10.5) | 35 (19.1) |
| OT | 4 (2.4) | 6 (3.3) | 3 (1.8) | 6 (3.3) |
| Speech therapist | 1 (0.6) | 1 (0.6) | 0 (0) | 2 (1.1) |
| Dietitian | 30 (17.9) | 18 (9.9) | 23 (13.5) | 17 (9.3) |
| Nutritionist | 4 (2.4) | 5 (2.8) | 3 (1.8) | 1 (0.6) |
| Social worker | 6 (3.6) | 3 (1.7) | 1 (0.6) | 4 (2.2) |
| Homecare worker | 2 (1.2) | 4 (2.2) | 1 (0.6) | 3 (1.7) |
| Psychologist | 9 (5.5) | 3 (1.7) | 3 (1.8) | 9 (4.9) |
| Complementary healthcare | 13 (7.9) | 10 (5.7) | 5 (2.9) | 6 (3.3) |
| District nurse | 6 (3.6) | 5 (2.8) | 5 (2.9) | 7 (3.8) |
| Psychiatrist | 4 (2.4) | 2 (1.1) | 0 (0) | 3 (1.7) |
| Counsellor | 7 (4.2) | 5 (2.8) | 11 (6.5) | 12 (6.6) |
The mean number of contacts (or days for residential care and inpatient care) is shown in Table 16. This is for the whole sample and so includes those with zero use. While most services do not differ much between arms, other inpatient care stands out. At baseline this is higher for the blinded group and this reflects the greater proportion of blinded respondents using it. At follow-up there were similar proportions using other inpatient care (as shown in Table 15) but the unblinded group had patients with longest lengths of stay.
| Baseline | Follow-up | |||
|---|---|---|---|---|
| Service | Blinded (n = 173) |
Unblinded (n = 189) |
Blinded (n = 173) |
Unblinded (n = 189) |
| Residential care | 0.14 | 0.16 | 0.03 | 0.02 |
| Renal inpatient | 3.36 | 1.31 | 1.12 | 1.48 |
| Intensive care | 0.18 | 0.07 | 0.02 | 0.02 |
| Other inpatient | 1.34 | 0.69 | 0.45 | 2.65 |
| Renal outpatient | 4.75 | 3.94 | 3.19 | 3.34 |
| Other outpatient | 1.72 | 1.97 | 1.71 | 1.72 |
| Day hospital | 0.04 | 0.03 | 0.04 | 0.05 |
| A&E | 0.29 | 0.35 | 0.29 | 0.33 |
| GP | 2.53 | 3.26 | 2.56 | 2.54 |
| Physiotherapist | 0.64 | 0.54 | 0.43 | 1.12 |
| OT | 0.07 | 0.06 | 0.03 | 0.10 |
| Speech therapist | 0.02 | 0.02 | 0.00 | 0.04 |
| Dietitian | 0.33 | 0.28 | 0.19 | 0.19 |
| Nutritionist | 0.03 | 0.04 | 0.03 | 0.01 |
| Social worker | 0.08 | 0.13 | 0.01 | 0.04 |
| Homecare worker | 0.61 | 2.91 | 0.03 | 0.63 |
| Psychologist | 0.50 | 0.32 | 0.08 | 0.35 |
| Complementary healthcare | 0.43 | 1.12 | 0.24 | 0.76 |
| District nurse | 0.41 | 0.16 | 0.09 | 0.31 |
| Psychiatrist | 0.05 | 0.03 | 0.00 | 0.08 |
| Counsellor | 0.14 | 0.44 | 0.27 | 0.66 |
| Total | 0.14 | 0.16 | 0.03 | 0.02 |
Service costs were highest for inpatient care and outpatient contacts (see Table 17). At baseline, renal and other inpatient costs were substantially higher in the blinded arm. By follow-up, this had switched with the unblinded group having far higher inpatient costs. GP costs were relatively low even though most of the sample used them. Other costs were low and did not differ substantially between arms.
| Baseline | Follow-up | |||
|---|---|---|---|---|
| Service | Blinded (n = 173) |
Unblinded (n = 189) |
Blinded (n = 173) |
Unblinded (n = 189) |
| Residential care | 14 | 16 | 3 | 2 |
| Renal inpatient | 1678 | 654 | 558 | 739 |
| Intensive care | 236 | 88 | 24 | 29 |
| Other inpatient | 671 | 346 | 224 | 1324 |
| Renal outpatient | 641 | 532 | 430 | 451 |
| Other outpatient | 232 | 266 | 230 | 233 |
| Day hospital | 4 | 3 | 4 | 5 |
| A&E | 54 | 64 | 53 | 61 |
| GP | 86 | 111 | 87 | 87 |
| Physiotherapist | 41 | 35 | 28 | 72 |
| OT | 6 | 5 | 3 | 9 |
| Speech therapist | 3 | 2 | 0 | 5 |
| Dietitian | 30 | 26 | 18 | 17 |
| Nutritionist | 3 | 4 | 3 | 1 |
| Social worker | 4 | 7 | 1 | 2 |
| Homecare worker | 17 | 81 | 1 | 18 |
| Psychologist | 43 | 28 | 7 | 31 |
| Complementary healthcare | 25 | 65 | 14 | 44 |
| District nurse | 18 | 7 | 4 | 14 |
| Psychiatrist | 7 | 5 | 0 | 10 |
| Counsellor | 8 | 25 | 16 | 38 |
The overall costs are reported in Table 18 along with the main cost-effectiveness results. At baseline the costs were higher for the blinded group. At follow-up, the costs were higher for the unblinded arm. The regression model showed that the unblinded group had follow-up costs that were £1523 higher than for the blinded group but this was not statistically significant (95% CI, –£49 to £4074).
| Blinded | Unblinded | |
|---|---|---|
| Baseline cost (£s) | 3600 | 2287 |
| Follow-up cost (£s) | 1672 | 3137 |
| Adjusted follow-up cost difference (£s) | 1523 (95% CI, −49 to 4074)a | |
| Baseline utility | 0.7959 | 0.8091 |
| Follow-up utility | 0.7828 | 0.7950 |
| QALYs | 1.0525 | 1.0694 |
| Adjusted follow-up QALY difference | 0.0009 (95% CI, −0.0195 to 0.0237)b | |
| Incremental cost-effectiveness ratio | £1,692,222 per QALY | |
Baseline and follow-up EQ-5D-5L utility scores were similar between groups and with little change over time. The unblinded group had 0.0009 more QALYs than the blinded group and this was not statistically significant (95% CI, −0.019 to 0.024). The ICER showed that for unblinded care to produce one extra QALY, a cost of £1.7 million would be incurred. Uncertainty around the results is shown in Figure 8. The ICER of £1.7 million is far in excess of the threshold used by NICE (£20,000–30,000) and so there is little likelihood of the unblinded option being more cost-effective than blinded care (see Figure 9).
FIGURE 8.
Cost-effectiveness plane. Scatter plot showing that unblinded care is likely to be more expensive, but outcomes are symmetrically distributed around the horizontal axis indicating that unblinded care can result in higher costs with either worse or better outcomes.
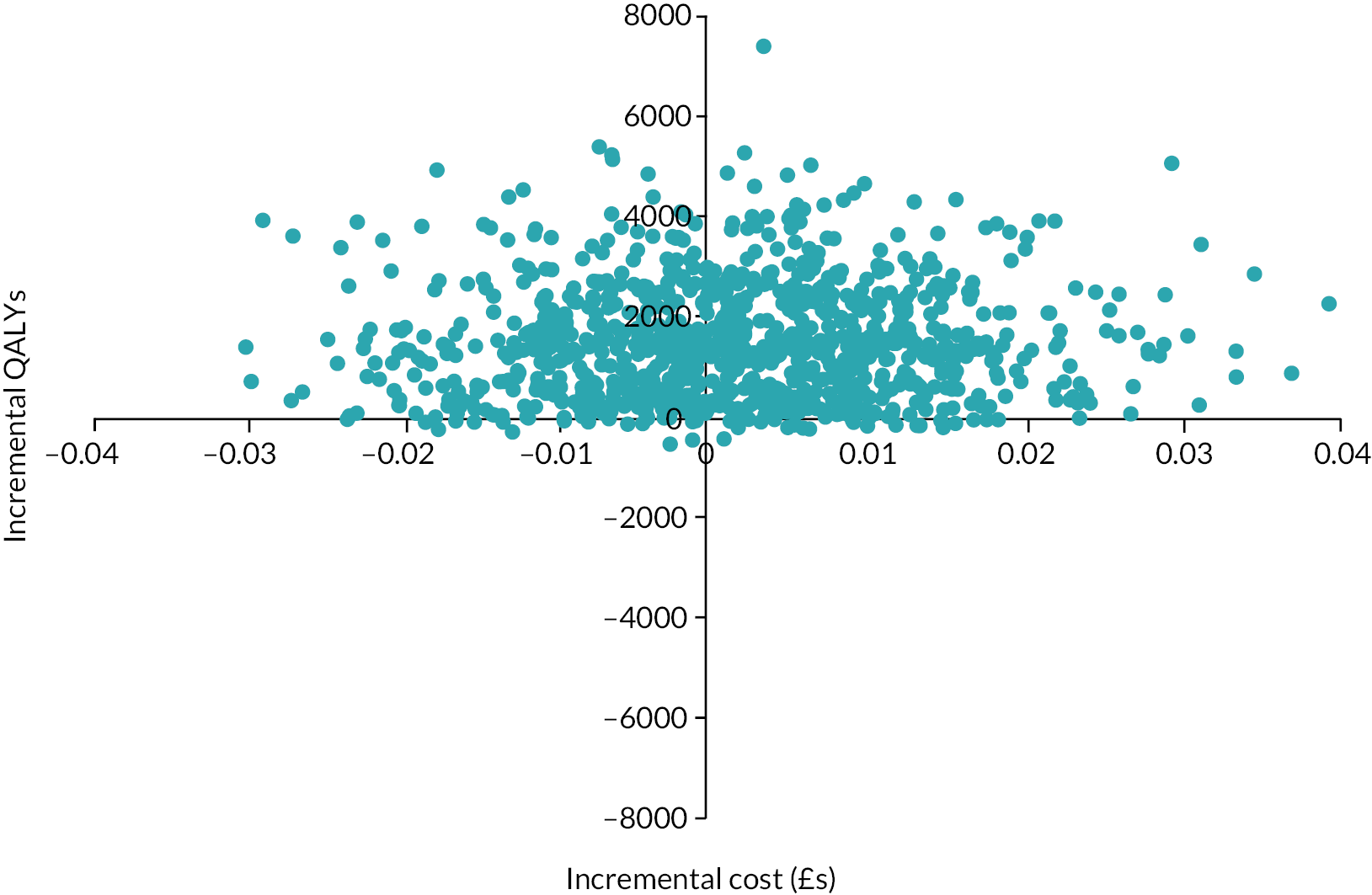
FIGURE 9.
Cost-effectiveness acceptability curve. Graph shows low probability that unblinded care is cost-effective, despite high costs.
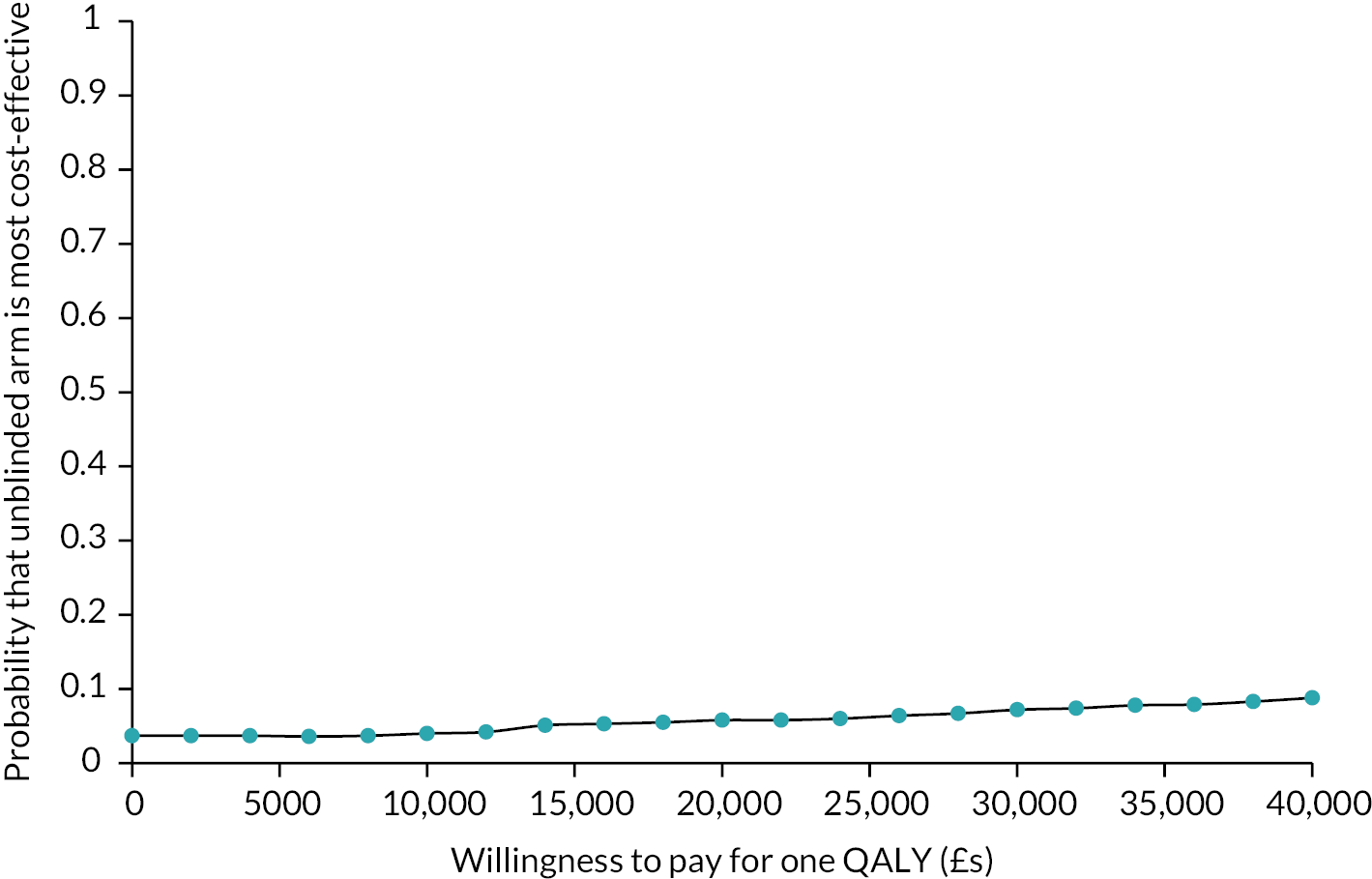
Analysis of adherence and health beliefs
Self-reported adherence, assessed by MARS was no different at any time point for Tac in HLA Ab+ patients in BLC versus SC groups (see Table 19). Assessment of adherence based on Tac levels only (acceptable trough range = 4–8 ng/ml) suggested better adherence at 12 months in the DSA+ BLC group compared to the DSA+ SC group (X2: 5.593 p = .02), though this was lost using a composite score combining MARS with Tac levels. In contrast, self-reported adherence at 12 months was significantly higher in the BLC DSA+ group for both MMF (p = 0.03) and prednisolone (p = 0.04) than in the SC DSA+ group (see Table 19).
| DSA+ | Comparison | Non-DSA+ | Comparison | HLA Ab-Negative | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Unblinded BLC | Blinded SC | Unblinded BLC | Blinded SC | Unblinded BLC | Blinded SC | |||||||||
| n 1 | Mean (SD) |
n 2 | Mean (SD) |
n 3 | Mean (SD) |
n 4 | Mean (SD) |
n 5 | Mean (SD) |
n 6 | Mean (SD) |
|||
| MARS Tac | ||||||||||||||
| T0 | 47 | 4.87 (0.18) | 39 | 4.76 (0.64) | 234 | 4.88 (0.15) | 222 | 4.88 (0.22) | 258 | 4.89 (0.21) | 285 | 4.88 (0.17) | ||
| T12 | 28 | 4.89 (0.16) | 16 | 4.88 (0.14) | p = 0.53 | 125 | 4.86 (0.21) | 100 | 4.89 (0.20) | p = 0.29 | 100 | 4.90 (0.14) | 101 | 4.87 (0.16) |
| T24 | 46 | 4.88 (0.19) | 26 | 4.86 (0.17) | p = 0.57 | 184 | 4.86 (0.22) | 157 | 4.89 (0.13) | p = 0.46 | 195 | 4.88 (0.16) | 203 | 4.87 (0.23) |
| % adherent on Tac trough levels | ||||||||||||||
| T0 | 51 | 88% | 41 | 91% | 260 | 96% | 252 | 97% | 303 | 97% | 321 | 96% | ||
| T12 | 39 | 100% | 19 | 86% | p = 0.02 | 151 | 94% | 130 | 96% | p = 0.79 | 129 | 95% | 139 | 95% |
| T24 | 60 | 92% | 48 | 96% | p = 0.41 | 270 | 95% | 237 | 97% | p = 0.17 | 280 | 95% | 306 | 97% |
| % adherent to Tac on composite adherence measure | ||||||||||||||
| T0 | 37 | 84% | 28 | 78% | 187 | 85% | 185 | 88% | 215 | 88% | 225 | 85% | ||
| T12 | 23 | 82% | 14 | 88% | p = 0.64 | 94 | 79% | 82 | 86% | p = 0.21 | 81 | 87% | 76 | 79% |
| T24 | 35 | 81% | 20 | 80% | p = 0.89 | 142 | 82% | 135 | 91% | p = 0.02 | 161 | 86% | 160 | 84% |
| MARS MMF | ||||||||||||||
| T0 | 40 | 4.89 (0.32) | 39 | 4.76 (0.65) | 212 | 4.89 (0.19) | 190 | 4.88 (0.23) | 259 | 4.90 (0.16) | 255 | 4.88 (0.19) | ||
| T12 | 26 | 4.94 (0.11) | 25 | 4.79 (0.32) | p = 0.03 | 114 | 4.86 (0.24) | 94 | 4.86 (0.28) | p = 0.96 | 94 | 4.91 (0.13) | 103 | 4.88 (0.17) |
| T24 | 44 | 4.85 (0.26) | 30 | 4.87 (0.13) | p = 0.25 | 167 | 4.89 (0.16) | 143 | 4.87 (0.16) | p = 0.13 | 186 | 4.89 (0.13) | 190 | 4.89 (0.17) |
| MARS prednisolone | ||||||||||||||
| T0 | 32 | 4.86 (0.36) | 28 | 4.80 (0.28) | 178 | 4.90 (0.16) | 151 | 4.91 (0.14) | 187 | 4.86 (0.36) | 209 | 4.88 (0.22) | ||
| T12 | 26 | 4.83 (0.40) | 20 | 4.72 (0.34) | p = 0.04 | 97 | 4.87 (0.27) | 68 | 4.93 (0.14) | p = 0.16 | 83 | 4.91 (0.14) | 92 | 4.86 (0.25) |
| T24 | 44 | 4.86 (0.26) | 25 | 4.83 (0.18) | p = 0.06 | 144 | 4.90 (0.20) | 113 | 4.90 (0.13) | p = 0.51 | 138 | 4.90 (0.14) | 154 | 4.87 (0.27) |
| Concern about the risk of transplant failure | ||||||||||||||
| T0 | 73 | 7.27 (2.67) | 67 | 6.75 (3.18) | 338 | 7.38 (2.88) | 306 | 7.30 (2.87) | 380 | 8.0 (2.92) | 411 | 8.01 (3.0) | ||
| T12 | 34 | 6.88 (2.80) | 34 | 6.91 (2.66) | p = 0.98 | 148 | 6.91 (3.06) | 127 | 7.25 (2.87) | p = 0.42 | 139 | 7.71 (2.87) | 146 | 8.13 (2.76) |
| T24 | 62 | 6.97 (2.92) | 42 | 6.64 (3.14) | p = 0.67 | 224 | 7.20 (2.69) | 218 | 6.83 (2.94) | p = 0.24 | 264 | 7.78 (2.89) | 285 | 7.72 (3.01) |
There were no significant differences across any treatment or screening groups on self-reported concern about the risk of transplant failures.
Adverse events
8189 AEs (670 SAEs) were reported, and 1570 patients (77%) experienced at least one AE (see Table 20). Significant differences were observed for five outcomes/codes with HLA Ab+ participants in the unblinded BLC arm being more likely to experience cardiovascular, respiratory, gastrointestinal and GU/renal AEs than HLA Ab+ participants in the blinded SC arm (see Figure 10). These comparisons are not adjusted for multiple testing however, and any AEs of concern are covered by existing secondary outcomes.
| DSA+ | Non-DSA+ | No HLA Ab | |||||
|---|---|---|---|---|---|---|---|
| Body system | Blinded (SC) (%) | Unblinded (BLC) (%) | Blinded (SC) (%) | Unblinded (BLC) (%) | Blinded (SC) (%) | Unblinded (BLC) (%) | Total |
| Allergies | 1 (1.1) | 0 (0.0) | 8 (2.0) | 4 (0.9) | 3 (0.6) | 1 (0.2) | 17 (0.8) |
| Cardiovascular | 4 (4.3) | 15 (14) | 60 (15) | 80 (19) | 66 (13) | 57 (12) | 282 (14) |
| Dermatological | 24 (26) | 27 (26) | 99 (25) | 113 (27) | 101 (19) | 76 (15) | 440 (22) |
| Endocrine | 6 (6.5) | 7 (6.6) | 28 (7.2) | 26 (6.1) | 29 (5.5) | 21 (4.2) | 117 (5.7) |
| Eyes, ear, nose, throat | 19 (21) | 24 (23) | 76 (19) | 89 (21) | 82 (16) | 59 (12) | 349 (17) |
| Gastrointestinal | 29 (32) | 32 (30) | 94 (24) | 128 (30) | 116 (22) | 105 (21) | 504 (25) |
| Genitourinary/renal | 30 (33) | 46 (43) | 141 (36) | 163 (38) | 163 (31) | 130 (26) | 673 (33) |
| Haematological | 7 (7.6) | 7 (6.6) | 25 (6.4) | 29 (6.8) | 35 (6.7) | 26 (5.3) | 129 (6.3) |
| Hepatic | 2 (2.2) | 2 (1.9) | 2 (0.5) | 9 (2.1) | 4 (0.8) | 6 (1.2) | 25 (1.2) |
| Immunological | 3 (3.3) | 1 (0.9) | 13 (3.3) | 7 (1.6) | 9 (1.7) | 4 (0.8) | 37 (1.8) |
| Musculoskeletal | 26 (28) | 32 (30) | 103 (26) | 121 (28) | 134 (26) | 106 (21) | 522 (26) |
| Neoplasia | 2 (2.2) | 1 (0.9) | 16 (4.1) | 17 (4.0) | 9 (1.7) | 9 (1.8) | 54 (2.7) |
| Neurological | 12 (13) | 10 (9.4) | 29 (7.4) | 43 (10) | 31 (5.9) | 30 (6.1) | 155 (7.6) |
| Psychological | 2 (2.2) | 3 (2.8) | 10 (2.6) | 18 (4.2) | 23 (4.4) | 11 (2.2) | 67 (3.3) |
| Respiratory | 37 (40) | 46 (43) | 127 (33) | 176 (41) | 177 (34) | 149 (30) | 712 (35) |
| Other | 34 (37) | 35 (33) | 116 (30) | 157 (37) | 150 (29) | 118 (24) | 610 (30) |
FIGURE 10.
Adverse events recorded in the HLA Ab+ groups. Left panel compares the proportion of patients suffering AEs grouped by body system in blinded (SC: blue circles) and unblinded (BLC: light blue triangles) HLA Ab+ groups. Right panel shows relative risk (95% CI) of developing an AE in the BLC patients, ordered by size of relative risk.
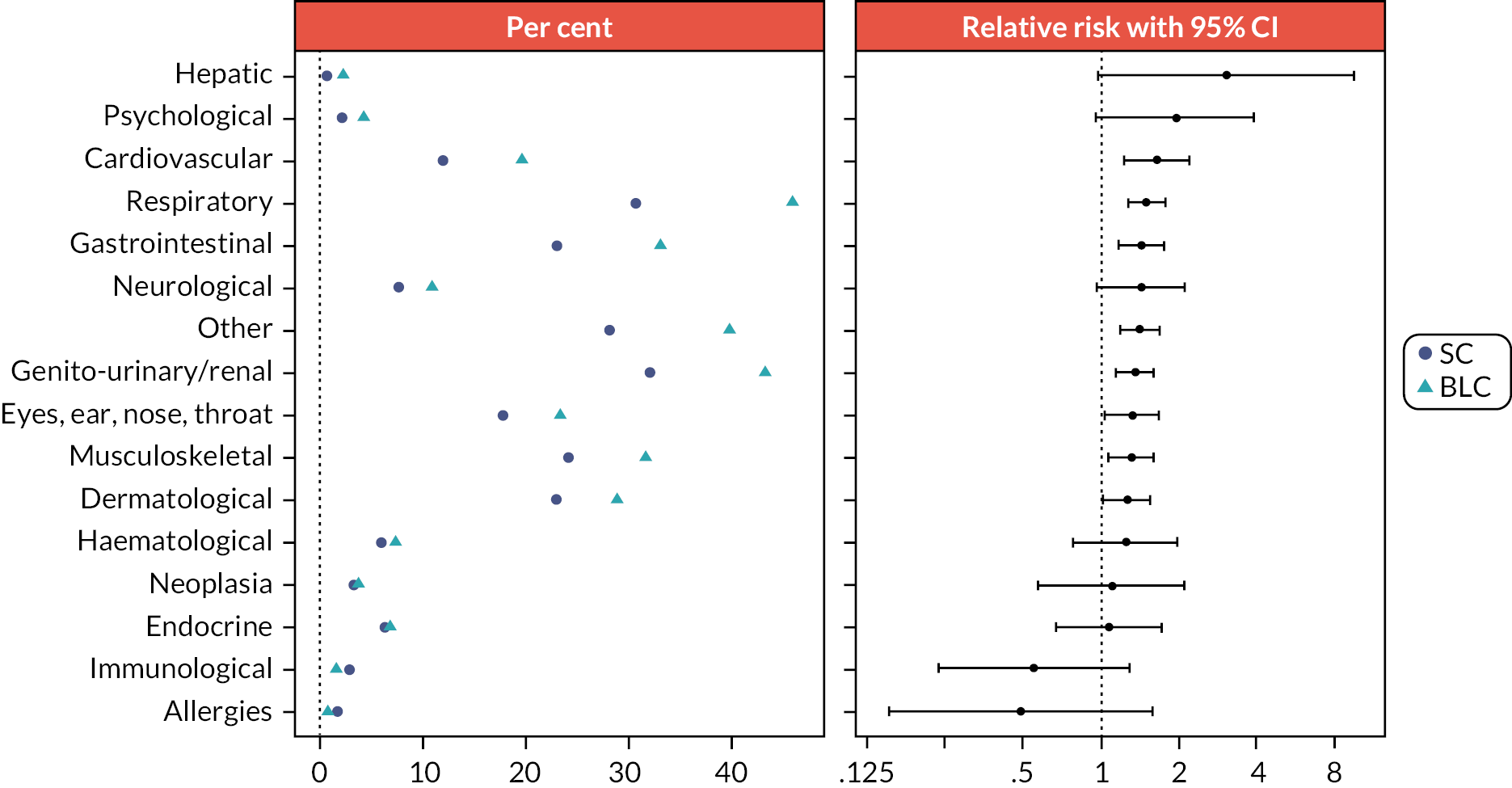
Changes in HLA antibodies
By the end of intensive follow-up, fewer than 2% of the HLA Ab-negative groups became Ab+, more than 50% of the DSA+ participants became HLA Ab-negative, 16–23% lost their DSA but retained non-DSA HLA Ab, and 60–70% of the non-DSA+ participants became Ab-negative. 5.1% of the blinded SC non-DSA+ recruits had developed DSA or possible DSA, compared to 1.6% of the unblinded non-DSA+ participants (see Table 5).
Within the blinded SC group, the same proportion (2/21 [9.5%]) of those with persisting DSA had graft failure as those who became DSA-negative (4/48 [8.3%]). Although in the BLC group, only 2/50 (4%) of the recruits who lost DSA suffered graft failure, compared to 6/28 (21.4%) with persisting DSA, a formal post-hoc analysis of the interaction between persisting DSA and time to graft failure in the main analysis revealed non-significant differences (p = 0.316) in revised HRs. Further analysis of within-group interactions was not undertaken as numbers were small.
Discussion
Equality, diversity and inclusion statement
We have reported the sex and ethnicity of recruits in several tables because these have a well-documented impact on kidney transplant survival and performance. The centres from which we recruited contained very broad and mixed populations drawn from all ethnic backgrounds. The requirement that recruits had a sufficient grasp of English was not felt to disproportionately bias recruitment of any particular group of individuals. All the recruiting centres have well-defined and published equality, diversity and inclusion policies meaning that our research teams comprised of individuals from diverse backgrounds.
Discussion of results
The OuTSMART trial tested the hypothesis that regular screening of kidney transplant recipients for HLA Ab, a validated biomarker for graft failure, followed by an intervention to improve adherence and optimise immunosuppression, would prolong the life of the organ allografts. Using an open-labelled randomised marker-based strategy (hybrid) design, in which all patients were screened, but only half were unblinded to their results and from which only biomarker-positive patients received the intervention, we recruited more than 2000 patients between 2013 and 2016 and followed them until 2020.
As the largest double-blinded study in transplantation to test a stratified medicine approach to post-transplant care, based on HLA Ab status, and the largest RCT to use graft failure as the primary endpoint, there is no ambiguity about how to interpret the results: OuTSMART further validates the prognostic value of DSA, but finds no evidence to support our hypothesis that intervening can prevent graft failure, with little separation in the Kaplan–Meier curves by group and confirmatory 95% CIs for HRs that included the null value. Further, there were no clear signals in favour of biomarker-led care in the HLA Ab+ groups from any of the secondary outcomes, despite indications of improved adherence, especially in DSA+ patients. That said, fewer DSA+ and non-DSA patients in the BLC arm (4 out of 106, and 5 out of 427, respectively) had biopsy-proven rejection than in the SC arm (9 out of 92 and 8 out 391, respectively), though neither of these differences failed to reach statistical significance. Interestingly, fewer HLA Ab-negative patients in the BLC had rejection (5 out of 495) compared to those in the SC arm (12 out of 525), such that there was a statistically significant reduction in biopsy-proven rejection in the whole BLC arm. These data are difficult to explain but they support our signal of improved adherence and potentially suggest that patient awareness of positive or negative risk associated with a prognostic biomarker impacts on behaviour.
Our health economic analyses confirm that renal transplant patients in both arms of the trial had relatively high levels of hospital use at baseline and follow-up. Patterns of care did not differ substantially over time. However, at follow-up, the unblinded BLC group were making more use of inpatient care, with consequently higher costs. QALYs were almost the same for the groups. The incremental cost per QALY for BLC over SC care was substantially greater than the threshold used by NICE. If the costs of screening were included, then this ratio would be even higher indicating that screening is unlikely to be cost-effective. The screening costs were not available at the time of the analysis. However, subsequently these have been reported as approximately £140 per test. It will be clear that this will make no meaningful difference to the findings on cost-effectiveness. The costs of immunotherapy were not included but these amount to only around £100 per patient (with the widespread availability of generics) and so again would not change the findings reported here.
All these data suggest that routine screening for HLA Ab is currently difficult to justify in the absence of any treatment that impacts of transplant survival. These results will impact significantly on how transplant centres around the world organise their post-transplant monitoring and should encourage a global effort to find novel approaches and treatments to prolong allograft survival in the face of DSA.
The validity of HLA Ab as a prognostic biomarker for kidney transplant failure was first demonstrated in retrospective case–control studies showing a higher prevalence of IgG Ab against donor HLA in failed compared to working transplants. 3,4 Later prospective studies reported a higher graft failure rate in those with HLA Ab compared to patients without. 5,6 Lachmann et al.,7 in a cohort of > 1000 patients reported 5-year graft failure rates of 51% for patients with DSA, 30% for patients with non-DSA and 17% for patients with no HLA Ab. This study also established the importance of repeat testing for HLA Ab and demonstrated that the majority of HLA Ab+ patients who were biopsied showed changes consistent with chronic immune-mediated injury. These findings were corroborated by a second study from the Netherlands,8 in which the risk of graft failure with HLA Ab was also shown to be independent of graft dysfunction and proteinuria. These studies provided one of the foundations for OuTSMART.
At the time OuTSMART was conceived, there were no tested strategies for how to intervene in patients with HLA Ab. Multiple trials, reporting since OuTSMART started, have tested agents targeting B cells with rituximab (± IVIg)14,15 or plasma cells with bortezomib and these have failed to show any impact. 37 Agents targeting IL-6 or IL-6 receptor have shown early promise in early phase studies,17 but larger studies assessing their impact are awaited. Other innovative treatments are at earlier stages of assessment. 38
Our hypothesis was that targeting the cells of the immune system rather than the HLA Ab, might prevent graft failure. There were three aspects to this. First, the knowledge that activated T cells associated with development of HLA Ab. 9,11,39–41 Second, the well-described link between immunosuppression reduction, including from non-adherence and development of DSA and graft dysfunction,9,42,43 meant it was logical to try and enhance immunosuppression in this group. Finally, optimised oral immunosuppression to target cell-mediated responses is known to prevent the development of HLA Ab20,21,44 and graft dysfunction,23–25 but has also been shown to stabilise deteriorating function in those with established immune-mediated dysfunction. 15,19,26,45–47
In keeping with previous work, OuTSMART showed that 15–20% of grafts in patients with DSA failed within the period of follow-up (after DSA detection) compared with 7% in the population who stayed consistently HLA Ab-negative. Although in line with recent observations from the Collaborative Transplant Survey (CTS Newsletter 2 : 2020 1st May), this is much lower than we had expected to see based on Lachmann et al. 7 Another important observation from OuTSMART was that patients who developed a non-DSA had a similar time to graft failure as patients without HLA Ab, in contrast to Lachmann’s data, which suggested a graft survival disadvantage associated with non-DSA HLA Ab. 7 A likely explanation for both differences is the different population demographics, most prominently baseline maintenance immunosuppression. For example, the proportion of patients in OuTSMART taking either Tac (73%) or MMF (67%) were double that in Lachmann’s cohort (35% and 33%), reflecting shifts in practice over the last 20 years.
Having screened > 5000 patients, only 37% were randomised, but 25% failed to provide consent, for various reasons, and 34% failed to meet eligibility, which were designed to ensure safe running of the trial and unambiguous interpretation of the results. We are therefore confident our results have generalisability. Several elements of the trial design need further explanation. First, we chose not to exclude patients who were known to be DSA+ (but XM–) at the time of transplantation, and these accounted for ~23% of recruits. Second, the majority of HLA Ab+ patients in all groups had either DSA (~66%) or non-DSA (~68%) at the point of randomisation, and although most of these were de novo Ab and had developed post-transplantation, only a relative minority developed de novo Ab during our rescreening process. Both these were practical compromises, as we calculated that recruiting sufficient numbers of HLA Ab-negative patients to collect enough DSA+ patients from rescreening alone was not feasible. Since patients with DSA that persist > 12 months post-transplantation are known to be at high risk of chronic rejection and graft failure,48,49 and at least one study has shown a similar prognostic significance for persistent non-DSA,48 both these decisions do not compromise the validity of the data. Third, we changed the primary endpoint during the study from graft failure rate over three years, to time to graft failure with minimum follow-up of 43 months. This was because the prevalence and incidence rates of DSA were lower than anticipated with consequent implications for the number of patients needed. 27,28 This change preserved the power of the trial, without affecting the protocol or general modelling strategy. Although the minimum follow-up period was shortened due to the unplanned COVID-19 pandemic, our sensitivity analyses suggested this did not impact on our conclusions. Fourth, in the original design, development of HLA Ab triggered a transplant biopsy to correlate with graft pathology even in the absence of graft dysfunction. This design aspect was removed after a Patient Public Involvement session at which patients raised serious concerns. Fifth, after allocation into HLA Ab+ groups, no further screening was done until the final visit, at which point we were able to retest 70–80%, revealing that only 50% remained DSA+. While we are confident that our testing regimen, which involved a screening test followed by single antigen testing was identifying genuine DSA, these data might indicate heterogeneity within the DSA+ groups not accounted for in our design. However, a formal analysis of interaction between DSA persistence and our primary endpoint indicated non-significant differences on the HRs. Finally, we designed the trial as a ‘real world’ effectiveness study, such that optimisation was tailored to individual patients, according to compliance, tolerance and achievement of target levels (for Tac). This meant that failure to tolerate one or more of the components of the protocol (or refusal to take any of the agents) was not used as a reason for withdrawal from the study. This aspect of the study was regarded as highly relevant and important by patients and PIs, but had the following consequences: all groups had average Tac levels within our target range, only 50% of the unblinded DSA+ group received the ‘steroid boost’ and many in the blinded groups were on immunosuppression that resembled our optimised regimen.
Nevertheless, more than 95% of the unblinded Ab+ group had the intervention interview, the proportion at the end of the trial on Tac, MMF and prednisolone in the unblinded DSA+ group rose from 64% to 82%, 59% to 73%, and 59% to 76%, respectively, whereas proportions in the blinded DSA+ group stayed constant (~60%), and the proportion taking all three IMPs at the end of the trial rose from 23% to 48% in unblinded DSA+ but stayed constant (~22%) in the blinded DSA+ group. In addition, at the end of the trial, the unblinded DSA+ group had the highest average Tac levels and were on the highest average dose of MMF. Moreover, our formal assessments of adherence revealed evidence of significant differences for each of the IMPs, at least at 12 months after the intervention. All these indicate measurable differences related to our intervention. Moreover, these all probably contributed to the lower rates of biopsy-proven rejection in the BLC patients.
There are several methodological limitations that need highlighting. Firstly, within DSA and non-DSA groups, for rescreened participants we defined the start of the time at risk (time zero) for graft failure when they became HLA Ab Positive rather than at time of randomisation. This assumes there is no effect of the blinding/unblinding to the HLA biomarker in the absence of optimised immunosuppression up until that point. However, the overall comparison would seem to support this assumption, given we found no evidence of a treatment effect for the overall unblinding strategy. We also did carry out a sensitivity analysis within the DSA and non-DSA comparisons using time at risk as randomisation for all participants which gave similar results to the main analysis.
We have made certain assumptions as to missing data, the primary analysis assumes a missing at random mechanism (with data censored at loss to follow-up or death). The effect of the intercurrent event of death was assessed with a sensitivity analysis (again which showed similar results). Other than death, the percentage lost to follow-up was very low (4%) and we consider that if the missingness mechanism was ‘missing not at random’, the impact on the results would be quite small. Secondary analyses also assumed a missing at random mechanism. Measures of the secondary outcomes (other than death) are additionally affected by graft failure and death as intercurrent events, for which the data will be subsequently missing. We haven’t directly assessed the impact of these intercurrent events (other than in the competing analysis of graft failure and death) for the secondary outcomes, and so the estimand for the secondary outcomes should be considered as following a treatment policy strategy (i.e. it is the estimated treatment effect in the absence of death or graft failure). Given we did not find evidence of an effect of the intervention on death or graft failure, this approach would seem reasonable. We have not adjusted reported p-values for multiplicity as we have defined a clear single primary outcome,50 and consequently the results on the secondary outcomes should be considered subsidiary and exploratory rather than confirmatory.
Conclusions
In this large, UK multicentre trial we have confirmed that development of DSA (but not non-DSA) is associated with future kidney allograft failure, but with failure rates markedly lower than those reported in cohorts pre-2010. We found no evidence that tailored optimisation of immunosuppression in those with HLA Ab impacts on graft failure, even though patients in our unblinded arm showed higher levels of compliance and lower rates of biopsy-proven rejection.
We conclude that, in the absence of specific and proven interventions to treat DSA, renal transplant recipients on ‘modern era’ immunosuppression regimens most likely do not benefit from regular screening for HLA Ab followed by interventions based on optimising oral treatment. While screening for DSA has clear prognostic value, we need novel strategies to intervene in this group to prevent subsequent graft failure.
Future research
We believe there are several potential areas of future research suggested by these data. The first is understanding of why some people with DSA deteriorate more quickly than others. If we are correct in asserting that improvements in transplant immunosuppression are responsible for why graft failure rates associated with DSA have improved over time, the implication is that better control of adaptive alloimmunity plays a part in preserving graft function. Recent insights into how subpopulations of regulatory T and B cells associate with phenotypes associated with DSA are consistent with this,51,52 and are an exciting area for future research. Understanding whether these insights relate to whether DSA persist or disappear is another area worthy of further research. Work in both these areas might help elucidate the precise pathophysiological contributions that different immune effector mechanisms, beside DSA, make to graft failure. This should all lead to a more rational design of specific therapies that prevent or halt these processes and help preserve graft function. With this in mind, it is very important that promising potential therapies, such as biological agents targeting IL-6 or its receptor, are properly evaluated in well-designed and appropriately powered RCTs.
Acknowledgements/funding
Funding source and their role: The study was indemnified for negligent and non-negligent harm by King’s College London. The study was funded by a National Institute for Health and Care Research (NIHR) Efficacy and Mechanism Evaluation programme grant (ref 11/100/34). VH Bio (Gateshead, UK) provided reagents at cost price. The trial statisticians (DS, R T-T, JP) had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. The corresponding author (AD) made the final decision to submit for publication. This research was funded/supported by the NIHR Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London and/or the NIHR Clinical Research Facility. This study was supported by the United Kingdom Clinical Research Collaboration-registered King’s Clinical Trials Unit at King’s Health Partners, which is partly funded by the NIHR Biomedical Research Centre for Mental Health at South London and Maudsley NHS Foundation Trust and King’s College London and the NIHR Evaluation, Trials and Studies Coordinating Centre. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Contributions of authors
Mr Dominic Stringer (https://orcid.org/0000-0001-5624-1733) (Statistician, Health Informatics) was part of the team of trial statisticians.
Dr Leanne Gardner (https://orcid.org/0000-0002-1233-9613) (Trial Manager, King’s CTU) was the trial manager.
Dr Olivia Shaw (https://orcid.org/0000-0003-4560-0619) (HLA Bioscientist, Guy’s) was part of the team of HLA laboratory principal investigators.
Dr Brendan Clarke (https://orcid.org/0000-0001-7243-1916) (HLA Bioscientist, Leeds) was part of the team of HLA laboratory principal investigators.
Dr David Briggs (https://orcid.org/0000-0002-6796-7086) (HLA Bioscientist, Brimingham) was part of the team of HLA laboratory principal investigators.
Dr Janet Worthington (https://orcid.org/0000-0002-1241-3975) (HLA Bioscientist, Manchester) was part of the team of HLA laboratory principal investigators.
Dr Matthew Buckland (https://orcid.org/0000-0002-5646-4707) (HLA Bioscientist, Barts) was part of the team of HLA laboratory principal investigators.
Dr Rachel Hilton (https://orcid.org/0000-0001-5118-2017) (Consultant Nephrologist, Guy’s) was part of the team of principal investigators at the original recruiting sites.
Dr Michael Picton (https://orcid.org/0000-0002-3066-0638) (Consultant Nephrologist, Manchester) was part of the team of principal investigators at the original recruiting sites.
Dr Raj Thuraisingham (https://orcid.org/0000-0003-2073-9569) (Consultant Nephrologist, Royal London) was part of the team of principal investigators at the original recruiting sites.
Dr Richard Borrows (https://orcid.org/0000-0003-2218-3023) (Consultant Nephrologist, Birmingham) was part of the team of principal investigators at the original recruiting sites.
Dr Richard Baker (https://orcid.org/0000-0002-9260-7664) (Consultant Nephrologist, Leeds) was part of the team of principal investigators at the original recruiting sites.
Ms Rose Tinch-Taylor (https://orcid.org/0000-0003-3861-6364) (Statistician, Health Informatics) was part of the team of trial statisticians.
Professor Robert Horne (https://orcid.org/0000-0002-3068-8438) (Professor of Behavioural Medicine, UCL) led the health beliefs and compliance team.
Professor Paul McCrone (https://orcid.org/0000-0001-7001-4502) (Health Economist, University of Greenwich) was the trial health economist.
Dr Joanna Kelly (https://orcid.org/0000-0002-4389-5284) (Clinial Trials Operations, King’s CTU) was responsible for database design and helped with trial design.
Dr Caroline Murphy (https://orcid.org/0000-0001-7547-8998) (Director of Operations, King’s CTU) was responsible for database design and helped with trial design.
Professor Janet Peacock (https://orcid.org/0000-0002-0310-2518) (Senior Statistician, Health Informatics) was part of the team of trial statisticians.
Professor Anthony Dorling (https://orcid.org/0000-0003-3102-2600) (Professor of Transplant Inflammation and Repair, KCL) was the chief investigator and designed the trial, held the EME grant and wrote the paper.
List of investigators and UK recruiting centres
The following PIs, based in 13 UK centres were: (1) Anthony Dorling (CI), Rachel Hilton (PI) and Olivia Shaw (Lab PI), Guy’s Hospital, Guy’s and St Thomas’ NHS Foundation Trust; (2) Sapna Shah (PI), King’s College Hospital, King’s College Hospital NHS Foundation Trust; (3) Richard Baker (PI) and Brendan Clarke (Lab PI), St James’s University Hospital, Leeds Teaching Hospitals NHS Trust; (4) Michael Picton (PI) and Judith Worthington (Lab PI), Manchester Royal Infirmary, Manchester University NHS Foundation Trust; (5) Raj Thuraisingham (PI) and Matthew Buckland (Lab PI), Royal London Hospital, Bart’s Health NHS Trust; (6) Richard Borrows (PI) and David Briggs (Lab PI), Queen Elizabeth Hospital, University Hospitals Birmingham NHS Foundation Trust; (7) Keith McCullough (PI), York Hospital, York and Scarborough Teaching Hospitals NHS Foundation Trust; (8) Waqar Ayub (PI), University Hospital Coventry, University Hospitals Coventry and Warwickshire NHS Trust; (9) Aimun Ahmed (PI), Royal Preston Hospital, Lancashire Teaching Hospitals NHS Foundation Trust; (10) Janet Hegarty (PI), Salford Royal Hospital, Northern Care Alliance NHS Foundation Trust; (11) John Stoves (PI), Bradford Royal Infirmary, Bradford Teaching Hospitals NHS Foundation Trust; (12) Kin Yee Shiu and Stephen B Walsh (PIs), Royal Free Hospital, Royal Free NHS Foundation Trust; (13) Mysore Phanish (PI), St Helier Hospital, Epsom and St Helier University Hospitals NHS Foundation Trust.
Ethics statement
The initial study protocol was approved on 14 January 2013 and given the reference number 12/LO/1759 by the National Research Ethics Service (NRES Committee London–Hampstead, Skipton House, Ground Floor, NRES/HRA, 80 London Road, London SE1 6LH), who approved all subsequent amendments.
Data-sharing statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, the EME programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, the EME programme or the Department of Health and Social Care.
References
- Lamb KE, Lodhi S, Meier-Kriesche HU. Long-term renal allograft survival in the United States: a critical reappraisal. Am J Transplant 2011;11:450-62. https://doi.org/10.1111/j.1600-6143.2010.03283.x.
- Ravanan R, Udayaraj U, Bakran A, Steenkamp R, Williams AJ, Ansell D. Measures of care in adult renal transplant recipients in the United Kingdom (chapter 11). Nephrol Dial Transplant 2007;22:vii138-54. https://doi.org/10.1093/ndt/gfm334.
- Mizutani K, Terasaki P, Rosen A, Esquenazi V, Miller J, Shih RN, et al. Serial ten-year follow-up of HLA and MICA antibody production prior to kidney graft failure. Am J Transplant 2005;5:2265-72. https://doi.org/10.1111/j.1600-6143.2005.01016.x.
- Lee PC, Zhu L, Terasaki PI, Everly MJ. HLA-specific antibodies developed in the first year posttransplant are predictive of chronic rejection and renal graft loss. Transplantation 2009;88:568-74. https://doi.org/10.1097/TP.0b013e3181b11b72.
- Terasaki PI, Ozawa M. Predictive value of HLA antibodies and serum creatinine in chronic rejection: results of a 2-year prospective trial. Transplantation 2005;80:1194-7. https://doi.org/10.1097/01.tp.0000174338.97313.5a.
- Terasaki PI, Ozawa M, Castro R. Four-year follow-up of a prospective trial of HLA and MICA antibodies on kidney graft survival. Am J Transplant 2007;7:408-15. https://doi.org/10.1111/j.1600-6143.2006.01644.x.
- Lachmann N, Terasaki PI, Budde K, Liefeldt L, Kahl A, Reinke P, et al. Anti-human leukocyte antigen and donor-specific antibodies detected by Luminex posttransplant serve as biomarkers for chronic rejection of renal allografts. Transplantation 2009;87:1505-13. https://doi.org/10.1097/TP.0b013e3181a44206.
- van Timmeren MM, Lems SP, Hepkema BG, Bakker SJ. Anti-human leukocyte antigen antibodies and development of graft failure after renal transplantation. Transplantation 2009;88:1399-400. https://doi.org/10.1097/TP.0b013e3181bc3ef0.
- Wiebe C, Gibson IW, Blydt-Hansen TD, Karpinski M, Ho J, Storsley LJ, et al. Evolution and clinical pathologic correlations of de novo donor-specific HLA antibody post kidney transplant. Am J Transplant 2012;12:1157-67. https://doi.org/10.1111/j.1600-6143.2012.04013.x.
- Colvin RB. Pathology of chronic humoral rejection. Contrib Nephrol 2009;162:75-86. https://doi.org/10.1159/000170814.
- Cherukuri A, Mehta R, Sharma A, Sood P, Zeevi A, Tevar AD, et al. Post-transplant donor specific antibody is associated with poor kidney transplant outcomes only when combined with both T-cell-mediated rejection and non-adherence. Kidney Int 2019;96:202-13. https://doi.org/10.1016/j.kint.2019.01.033.
- Macklin PS, Morris PJ, Knight SR. A systematic review of the use of rituximab for the treatment of antibody-mediated renal transplant rejection. Transplant Rev (Orlando) 2017;31:87-95. https://doi.org/10.1016/j.trre.2017.01.002.
- Choi J, Aubert O, Vo A, Loupy A, Haas M, Puliyanda D, et al. Assessment of tocilizumab (anti-interleukin-6 receptor monoclonal) as a potential treatment for chronic antibody-mediated rejection and transplant glomerulopathy in HLA-sensitized renal allograft recipients. Am J Transplant 2017;17:2381-9. https://doi.org/10.1111/ajt.14228.
- Moreso F, Crespo M, Ruiz JC, Torres A, Gutierrez-Dalmau A, Osuna A, et al. Treatment of chronic antibody mediated rejection with intravenous immunoglobulins and rituximab: A multicenter, prospective, randomized, double-blind clinical trial. Am J Transplant 2018;18:927-35. https://doi.org/10.1111/ajt.14520.
- Shiu KY, Stringer D, McLaughlin L, Shaw O, Brookes P, Burton H, et al. Effect of optimized immunosuppression (including rituximab) on anti-donor alloresponses in patients with chronically rejecting renal allografts. Front Immunol 2020;11. https://doi.org/10.3389/fimmu.2020.00079.
- Chandran S, Leung J, Hu C, Laszik ZG, Tang Q, Vincenti FG. Interleukin-6 blockade with tocilizumab increases Tregs and reduces T effector cytokines in renal graft inflammation: A randomized controlled trial. Am J Transplant 2021;21:2543-54. https://doi.org/10.1111/ajt.16459.
- Doberer K, Duerr M, Halloran PF, Eskandary F, Budde K, Regele H, et al. A randomized clinical trial of anti-IL-6 antibody clazakizumab in late antibody-mediated kidney transplant rejection. J Am Soc Nephrol 2021;32:708-22. https://doi.org/10.1681/ASN.2020071106.
- Jordan SC, Ammerman N, Choi J, Huang E, Najjar R, Peng A, et al. Evaluation of clazakizumab (anti-interleukin-6) in patients with treatment-resistant chronic active antibody-mediated rejection of kidney allografts. Kidney Int Rep 2022;7:720-31. https://doi.org/10.1016/j.ekir.2022.01.1074.
- Theruvath TP, Saidman SL, Mauiyyedi S, Delmonico FL, Williams WW, Tolkoff-Rubin N, et al. Control of antidonor antibody production with tacrolimus and mycophenolate mofetil in renal allograft recipients with chronic rejection. Transplantation 2001;72:77-83. https://doi.org/10.1097/00007890-200107150-00016.
- Lederer SR, Friedrich N, Banas B, Welser G, Albert ED, Sitter T. Effects of mycophenolate mofetil on donor-specific antibody formation in renal transplantation. Clin Transplant 2005;19:168-74. https://doi.org/10.1111/j.1399-0012.2005.00261.x.
- van der Mast BJ, van Besouw NM, Witvliet MD, de Kuiper P, Smak Gregoor P, van Gelder T, et al. Formation of donor-specific human leukocyte antigen antibodies after kidney transplantation: correlation with acute rejection and tapering of immunosuppression. Transplantation 2003;75:871-7. https://doi.org/10.1097/01.TP.0000054840.70526.D0.
- Webster A, Woodroffe RC, Taylor RS, Chapman JR, Craig JC. Tacrolimus versus cyclosporin as primary immunosuppression for kidney transplant recipients. Cochrane Database Syst Rev 2005;2005. https://doi.org/10.1002/14651858.CD003961.pub2.
- Ojo AO, Meier-Kriesche HU, Hanson JA, Leichtman AB, Cibrik D, Magee JC, et al. Mycophenolate mofetil reduces late renal allograft loss independent of acute rejection. Transplantation 2000;69:2405-9. https://doi.org/10.1097/00007890-200006150-00033.
- Meier M, Nitschke M, Weidtmann B, Jabs WJ, Wong W, Suefke S, et al. Slowing the progression of chronic allograft nephropathy by conversion from cyclosporine to tacrolimus: a randomized controlled trial. Transplantation 2006;81:1035-40. https://doi.org/10.1097/01.tp.0000220480.84449.71.
- Meier-Kriesche HU, Merville P, Tedesco-Silva H, Heemann U, Kes P, Haller H, et al. Mycophenolate mofetil initiation in renal transplant patients at different times posttransplantation: the TranCept Switch study. Transplantation 2011;91:984-90. https://doi.org/10.1097/TP.0b013e3182130966.
- Shiu KY, McLaughlin L, Rebollo-Mesa I, Zhao J, Burton H, Douthwaite H, et al. Graft dysfunction in chronic antibody-mediated rejection correlates with B-cell-dependent indirect antidonor alloresponses and autocrine regulation of interferon-gamma production by Th1 cells. Kidney Int 2017;91:477-92. https://doi.org/10.1016/j.kint.2016.10.009.
- Dorling A, Rebollo-Mesa I, Hilton R, Peacock JL, Vaughan R, Gardner L, et al. Can a combined screening/treatment programme prevent premature failure of renal transplants due to chronic rejection in patients with HLA antibodies: study protocol for the multicentre randomised controlled OuTSMART trial. Trials 2014;15. https://doi.org/10.1186/1745-6215-15-30.
- Stringer D, Gardner LM, Peacock JL, Rebollo-Mesa I, Hilton R, Shaw O, et al. Update to the study protocol, including statistical analysis plan, for the multicentre, randomised controlled OuTSMART trial: a combined screening/treatment programme to prevent premature failure of renal transplants due to chronic rejection in patients with HLA antibodies. Trials 2019;20. https://doi.org/10.1186/s13063-019-3602-2.
- European MA. Amsterdam, Netherlands: EMA; 2012.
- Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. Journal of the American Statistical Association 1999;94:496-509. http://dx.doi.org/10.1080/01621459.1999.10474144.
- Beecham J, Knapp M. Measuring Mental Health Needs. London: Gaskell; 2001.
- Curtis L, Burns A. Unit Costs of Health and Social Care 2020. Canterbury: PSSRU, University of Kent; 2020.
- Herdman M, Gudex C, Lloyd A, Janssen M, Kind P, Parkin D, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res 2011;20:1727-36. https://doi.org/10.1007/s11136-011-9903-x.
- Cohen JL, Mann DM, Wisnivesky JP, Home R, Leventhal H, Musumeci-Szabo TJ, et al. Assessing the validity of self-reported medication adherence among inner-city asthmatic adults: the Medication Adherence Report Scale for Asthma. Ann Allergy Asthma Immunol 2009;103:325-31. https://doi.org/10.1016/s1081-1206(10)60532-7.
- Chan AHY, Horne R, Hankins M, Chisari C. The medication adherence report scale: A measurement tool for eliciting patients’ reports of nonadherence. Br J Clin Pharmacol 2020;86:1281-8. https://doi.org/10.1111/bcp.14193.
- Broadbent E, Petrie KJ, Main J, Weinman J. The brief illness perception questionnaire. J Psychosom Res 2006;60:631-7. https://doi.org/10.1016/j.jpsychores.2005.10.020.
- Eskandary F, Regele H, Baumann L, Bond G, Kozakowski N, Wahrmann M, et al. A randomized trial of bortezomib in late antibody-mediated kidney transplant rejection. J Am Soc Nephrol 2018;29:591-605. https://doi.org/10.1681/ASN.2017070818.
- Mayer KA, Budde K, Jilma B, Doberer K, Bohmig GA. Emerging drugs for antibody-mediated rejection after kidney transplantation: a focus on phase II & III trials. Expert Opin Emerg Drugs 2022;27:151-67. https://doi.org/10.1080/14728214.2022.2091131.
- Louis K, Macedo C, Bailly E, Lau L, Ramaswami B, Marrari M, et al. Coordinated circulating t follicular helper and activated b cell responses underlie the onset of antibody-mediated rejection in kidney transplantation. J Am Soc Nephrol 2020;31:2457-74. https://doi.org/10.1681/ASN.2020030320.
- Shiu KY, McLaughlin L, Rebollo-Mesa I, Zhao J, Semik V, Cook HT, et al. B-lymphocytes support and regulate indirect T-cell alloreactivity in individual patients with chronic antibody-mediated rejection. Kidney Int 2015;88:560-8. https://doi.org/10.1038/ki.2015.100.
- Susal C, Slavcev A, Pham L, Zeier M, Morath C. The possible critical role of T-cell help in DSA-mediated graft loss. Transpl Int 2018;31:577-84. https://doi.org/10.1111/tri.13126.
- Sellares J, de Freitas DG, Mengel M, Reeve J, Einecke G, Sis B, et al. Understanding the causes of kidney transplant failure: the dominant role of antibody-mediated rejection and nonadherence. Am J Transplant 2012;12:388-99. https://doi.org/10.1111/j.1600-6143.2011.03840.x.
- Halloran PF, Merino Lopez M, Barreto Pereira A. Identifying subphenotypes of antibody-mediated rejection in kidney transplants. Am J Transplant 2016;16:908-20. https://doi.org/10.1111/ajt.13551.
- Davis S, Wiebe C, Campbell K, Anobile C, Aubrey M, Stites E, et al. Adequate tacrolimus exposure modulates the impact of HLA class II molecular mismatch: a validation study in an American cohort. Am J Transplant 2021;21:322-8. https://doi.org/10.1111/ajt.16290.
- Billing H, Rieger S, Ovens J, Susal C, Melk A, Waldherr R, et al. Successful treatment of chronic antibody-mediated rejection with IVIG and rituximab in pediatric renal transplant recipients. Transplantation 2008;86:1214-21. https://doi.org/10.1097/TP.0b013e3181880b35.
- Fehr T, Rusi B, Fischer A, Hopfer H, Wuthrich RP, Gaspert A. Rituximab and intravenous immunoglobulin treatment of chronic antibody-mediated kidney allograft rejection. Transplantation 2009;87:1837-41. https://doi.org/10.1097/TP.0b013e3181a6bac5.
- Rostaing L, Guilbeau-Frugier C, Fort M, Mekhlati L, Kamar N. Treatment of symptomatic transplant glomerulopathy with rituximab. Transpl Int 2009;22:906-13. https://doi.org/10.1111/j.1432-2277.2009.00896.x.
- Caillard S, Becmeur C, Gautier-Vargas G, Olagne J, Muller C, Cognard N, et al. Pre-existing donor-specific antibodies are detrimental to kidney allograft only when persistent after transplantation. Transpl Int 2017;30:29-40. https://doi.org/10.1111/tri.12864.
- Kimball PM, Baker MA, Wagner MB, King A. Surveillance of alloantibodies after transplantation identifies the risk of chronic rejection. Kidney Int 2011;79:1131-7. https://doi.org/10.1038/ki.2010.556.
- Li G, Taljaard M, Van den Heuvel ER, Levine MA, Cook DJ, Wells GA, et al. An introduction to multiplicity issues in clinical trials: the what, why, when and how. Int J Epidemiol 2017;46:746-55. https://doi.org/10.1093/ije/dyw320.
- Louis K, Fadakar P, Macedo C, Yamada M, Lucas M, Gu X, et al. Concomitant loss of regulatory T and B cells is a distinguishing immune feature of antibody-mediated rejection in kidney transplantation. Kidney Int 2022;101:1003-16. https://doi.org/10.1016/j.kint.2021.12.027.
- Basu S, Dorling A. Regulation of T- and B-cell interactions determines the clinical phenotype associated with donor-specific antibodies. Kidney Int 2022;101:877-9. https://doi.org/10.1016/j.kint.2022.02.020.
Appendix 1
Summary of changes to protocol approved by the ethics committee
All changes were discussed and approved by the Trial Steering Committee or Chairman and, where appropriate, by the Data Monitoring Committee.
The changes made in Version 11 of the protocol reflect the major changes in the design and endpoints that are incorporated into the final version.
Version 2 07/11/12
-
Change to section 3.1 to reflect that MMF was being used outside its marketing authorisation.
Version 3 29/1/2013
-
Changes to sections 2.2.1 and 6.2 relating to assessment of adherence and risk perception. Rather than collecting prescription redemption data (version 1 and 2), we proposed tablet counts on randomly selected patients in addition to the use of iPads or similar tablets to collect the data, prior to electronic transfer to a secure server hosted by University College London. Thirdly, we proposed to pilot the questionnaires and perform quantitative interviews on a small number of participants initially recruited to Guy’s, to inform whether existing standardised questionnaires required change to suit this population.
-
Change to section 7.3 relating to a change in the pharmacovigilance policy of the sponsor to ensure Important Medical Events are recorded as SAEs.
Version 4 13/5/2013
-
Changes to sections 2.2 and 2.3 to clarify that randomisation will be stratified by site.
-
Change to section 4.1 to clarify that the eGFR measurement on which eligibility will be assessed has to be within 1 month of signing consent.
-
Change to section 5.2.1 to clarify the definition of a positive HLA Ab test, which was confusing in the previous protocol versions.
-
Changes to mention of recruitment targets, shifting emphasis away from precise predictions towards a more pragmatic approach that highlights recruitment will stop once minimum numbers required for statistical power have been recruited to each of the individual groups.
Version 5 9/7/2013
-
Change to lab PI at the Royal London Hospital.
-
Change to section 3.1 clarifying the dosing of one the IMPs, Prednisolone.
-
Change to reflect updated WHO definition of DM (addition of HbA1c testing).
-
Change to section 7.3.1, reflecting the fact that certain AEs in this type A trial may not require reporting to the sponsor, but may still require recording in the eCRF.
Version 6 6/12/2013
-
Change to abandon the requirement that recruits be tested for hepatitis B core Ab, as it was hindering collection of samples for scientific analysis. Since core Ab positivity was not a contraindication to optimisation, and testing was not required by King’s College London, as the infectivity of samples from core Ab positive, surface antigen-negative samples is very low, this change was felt not to compromise the trial in any way but would enhance the number of scientific samples obtained at recruitment.
-
Change to allow urine as well as blood testing to rule out pregnancy.
-
Changes relating to recruitment of live donors, to ensure they were tested for HIV and Hepatitis B and C if not tested within the previous 5 years, to increase volume of blood taken to 80 ml and to allow consent to be obtained by non-clinicians be allowed to consent these patients.
Version 7 7/4/2014
-
Changes to maximise and standardise recruitment across sites:
-
Removal of exclusion criteria ‘history of ongoing or previous infection that would prevent optimisation’. This criterion was vague (i.e. did not define which infections were important) and was being interpreted differently within and across sites. As the optimisation for each participant was optimised to that particular individual, immunosuppression could be tailored according to their medical history.
-
Increase in the gap for the testing of eGFR from within 1 month of signing consent to within the previous 6 months of signing the consent. Participants had to have an eGRF≥30 to be eligible for the study, and by increasing the time-period to within the previous 6 months, screening for potential participants can be more efficient as measurements of eGFR from previous renal clinic appointments could be used.
-
-
Removal of need to perform total immunoglobulin testing:
-
This measurement proved to be a difficult and expensive test to perform and was not routinely performed by all hospital laboratories. This test had originally been incorporated into the study to ensure participants were not developing MMF-induced hypogammaglobulinemia. Fortunately, this could still be detected by maintaining requirement to test for IgG, IgM and IgA.
-
Testing and recording of IgG, IgM and IgA moved to every year instead of every four months. Testing for the levels of these immunoglobulins every 12 months was felt to be sufficient by the TSC sufficient for monitoring MMF-induced hypogammaglobulinemia.
-
-
Clarification of and changes to follow-up procedures:
-
Clarification that participants would see a research nurse for all trial-related procedures at follow-up appointments, which will be held at the same time a participant is in routine clinic.
-
Details regarding the fact that only medications being taken at the time of the follow-up would be recorded.
-
Clarification of the timings for questionnaire completion in table 2.2.1.
-
Patients could be given an appointment slip containing a telephone number and/or an email address to contact research nurses if their routine appointments are rescheduled.
-
Clarification of the time windows for follow-up appointments that were allowed without deviation to the protocol.
-
-
Clarification of the optimisation process for participants allocated to the unblinded HLA Ab positive arm:
-
Change of optimisation timing from within 3 months of HLA Ab positivity to ideally within 3 months after positive screening for HLA Ab and allocation to the unblinded treatment arm or as soon as possible thereafter BUT within 8 months of positive screening. This coincided with the realisation that some patients were proving difficult to contact to arrange optimisation and the change was felt to enhance the optimisation process without affecting the outcome of the trial.
-
Clarification of the way that patients will be informed about the group to which they have been allocated. Participants with HLA Ab allocated to the unblinded arm will be told the results of this allocation as soon as possible and invited to undergo optimisation. This can be performed over the phone. Those participants in the blinded groups or in the unblinded HLA Ab negative group will be told the result of their randomisation at their next clinic visit.
-
Clarification that recording details of the optimisation process will be in an Optimisation Log at each site and not in the eCRF.
-
-
Addition of three new sites.
Version 8 1/7/2014
-
Extension to the time that tissue typing laboratories had to perform the randomisation of patients, from 28 to 56 days post consent. This was to optimise batching of patient serum for testing, reducing the number of experimental controls and HLA screening beads needed, and therefore the cost of screening.
-
Clarification, in section 7.1, of when testing of HbA1c should occur.
-
Clarification about tests to be performed to monitor for MMF-induced hypogammaglobulinemia.
-
Changes to allow the use of results from routine clinic blood tests taken up to a week prior to consent, to minimise duplication of tests in sites where it was routine for patients to attend for blood tests prior to their clinic visit.
-
Changes to allow study information to be collected via telephone to minimise time spent with each patient during busy routine clinics.
Version 9 15/10/2014
-
Clarification around timing and need for collection of experimental research samples (laboratory) at point of consent.
-
Further clarification of the time windows for follow-up appointments that were allowed without deviation to the protocol. Changes made to try to ensure collection of three study assessments per year.
-
Change to reduce nurse paperwork: as a Type A trial with a large recruitment target, missing data around sample collection was to be coded in the eCRF but not recorded as a protocol deviation.
-
Change to the PI at one of the sites.
Version 10 11/08/2015
-
Changes to two coinvestigators in the Tissue Typing Laboratories and addition of three new sites.
-
Change to eligibility age range, from 18–70 years to 18–75 years. Originally, we believed that non-transplant-related mortality may be higher in 71–75-year-old age group, however this was reconsidered and felt not to be an issue.
-
Clarification of the inclusion criteria relating to timing of the eGFR used when considering eligibility.
Version 11 26/11/2015
-
Change in primary endpoint
-
The primary endpoint of the trial was changed from ‘graft failure rates over three years’ to ‘time to graft failure with variable follow-up (with a minimum of 43 months post-randomization)’. This change was required to account for the low numbers of DSA-positive participants being recruited to the trial. This change will allow for a reduction in the number of DSA patients to be recruited, and a significant shortening in the expected study duration while maintaining the power of the study. Section 8 on sample size and statistical analyses were changed accordingly. The new primary endpoint was to be assessed remotely from patient notes once 43 months post-randomisation was achieved by all. All patients already recruited were to be reconsented to allow this change.
-
-
Changes to reduce costs associated with extension of the trial
-
Follow-up visits changed from 4-monthly to 8-monthly to reduce nurse workload.
-
End visit for each participant changed from 36 to 32 months.
-
Secondary endpoint assessment changed from 36 to 32 months, except for health economics which was moved from 36 to 16 months.
-
Major reduction of SAE reporting to sponsor incorporated into sections 7.2 and 7.3.
-
-
Change in trial statistician, change in site PIs and addition of a new site.
Version 12 1/12/16
-
Cessation of collection of research blood samples, associated with removal of the secondary experimental ‘scientific’ endpoints. This was required by the funder, who requested that the salary costs associated with the experimental aspects of the trial be reallocated towards supporting the primary endpoint data collection.
Version 13 21/11/18
-
Change to reflect inclusion of albumin:creatinine ratio as a measure of proteinuria in addition to protein:creatinine ratio and clarification that one or the other (not both) are required as one assessment of graft dysfunction.
-
Change to the way that change in eGFR was to be compared between arms.
-
Inclusion of proposed details for how the primary outcome data was to be collected during the period March 2020–June 2020.
-
Inclusion of collection of a final sample for HLA Ab screening from all participants at their final research clinic visit at month 32.
-
Clarification that results from the trial will be presented as estimates and 95% CIs.
-
Clarification that baseline covariates were to be included in the statistical model for the primary outcome.
Version 14 08/07/2020
-
Change to the timing of the collection of the primary endpoint as a result of the COVID-19 pandemic, in addition to the proposal to include additional sensitivity analyses for the primary endpoint and extension of the study end date.
-
Inclusion of a thank you card for all participants who have taken part in the OuTSMART trial.
Appendix 2
SOP relating to HLA Ab determination
| CATEGORY | Tissue typing laboratory work instructions | SOP number: 5 | Version 6.0 (30/09/2019) | ||
|---|---|---|---|---|---|
| TITLE | Detection of HLA Ab in participant samples for OuTSMART study | ||||
| 1.0 | Title | ||||
| Tissue typing for OuTSMART trial project | |||||
| 2.0 | Purpose | ||||
| To describe the procedure for detection of HLA Ab in participant samples for the OuTSMART trial | |||||
| Serum is collected from whole blood by centrifugation and frozen. An aliquot is taken and analysed for the presence of HLA Class I and II Ab. | |||||
| 3.0 | Definitions and abbreviations | ||||
| IgG/PE | Goat anti-human IgG conjugated to phycoerythrin | ||||
| PBS | Phosphate buffer saline | ||||
| NC | Negative control | ||||
| PC | Positive control | ||||
| MFI | Mean fluorescence intensity | ||||
| 4.0 | Equipment and reagents | ||||
| 4.1 | Equipment | ||||
| 4.1.1 | Vacuum manifold and pump | For filter plate method | |||
| 4.1.2 | Orbital mixer | ||||
| 4.1.3 | Luminex analyser | ||||
| 4.1.4 | Benchtop microcentrifuge | ||||
| 4.1.5 | Filter plates | Millipore multiscreen filter plates Cat no: MABVN1250 |
For filter plate method | ||
| 4.1.6 | Precut transparent microplate sealers | Greiner Bio-one Cat no: 676001 Supplied by Jencons-PLS Cat no: 488-097 |
|||
| 4.1.7 | Aluminium foil | ||||
| 4.1.8 | Swinging bucket rotor for 96 well SSP tray (1300g/2600 rpm) | For spin and flick method | |||
| 4.1.9 | Microtube plate V bottom (G&N Laboratory: MA612V96) | For spin and flick method | |||
| 4.1.10 | 96 well low profile SSP tray | For spin and flick method | |||
| 4.2 | Reagents | ||||
| 4.2.1 | Whole blood | Subject source | |||
| 4.2.2 | PBS | MP Biomedicals | LCC CAT no.: 2810305 (Dissolve one tablet in 100 ml distilled water and store at 4°C. Once prepared, discard after 1 month.) |
||
| 4.2.3 | Goat anti-human IgG conjugated to phycoerythrin (freeze dried 100× concentrated and stored 4°C) | OneLambda Cat no: 03LSAB2 |
Reconstituted before use by adding sterile water at least two hours prior to first use. The volume of water to be added is clearly stated on the bottle. The date must be recorded on the side of the bottle, with an expiry date of 6 months post reconstitution date, unless the expiry date provided on the stock is earlier. Once reconstituted the IgG/PE must be stored at 4°C. For use, dilute antihuman IgG/PE 1 : 100 with LABScreen wash buffer that is, 1 part IgG/PE plus 99 parts wash buffer. |
||
| 4.2.4 | FlowPRA Class I and II negative control | VHBio Ltd | 03FLNC (Stored at –80°C. Once defrosted stored at 4°C.) |
||
| 4.2.5 | NIBSC – negative control for FXCM and anti-HLA serology | 09/112 | Reconstituted with 1 ml 0.1% sodium azide and stored at 4°C for up to 1 month. | ||
| 4.2.6 | LABScreen Mixed Class I and II Ab Screening kit – (500 μl) | VHBio Ltd Cat no: 03LSM12 |
Kit must be stored at −80°C. Once defrosted, store kit at 4°C. The date the vial of beads was defrosted, plus the date received in the lab should be recorded on the side of the vial. | ||
| 4.2.7 | LABScreen PRA SA Combi kit (Class I) – (125 μl) |
VHBio Ltd Cat no: 03LS1A04 |
Kit must be stored at −80°C. Once defrosted, store kit at 4°C. The date the vial of beads was defrosted, plus the date received in the lab should be recorded on the side of the vial. | ||
| 4.2.8 | LABScreen PRA SA Class II kit – (125 μl) |
VHBio Ltd Cat no: 03LS2A01 |
Kit must be stored at −80°C. Once defrosted, store kit at 4°C. The date the vial of beads was defrosted, plus the date received in the lab should be recorded on the side of the vial. | ||
| 4.2.9 | 10× Concentrated wash buffer (26 ml). | Provided with screening kit. | This must be diluted 1 : 10 with distilled water prior to use. that is. 1 part wash buffer plus nine parts water. Once diluted label with the Lot number, expiry date and initials, then store at 4°C ready for use. | ||
| 5.0 | Procedures | ||||
| Biological waste should be disposed of according to the current regulations. | |||||
| Note | Safety:
|
||||
| 5.0 | Standard procedure | ||||
| 5.1 | Sample checking and processing | ||||
| 5.1.1 | Samples should arrive in suitably labelled specimen bags. All specimens must be handled over a spill tray. Any soiled paperwork must be discarded in an appropriate waste sack as clinical waste. In this instance, sample details should be manually transcribed onto a clean form, indicating that the original form had to be discarded. | ||||
| 5.1.2 | The details on the sample bottle/ tube should be checked against those on the accompanying request form. Any discrepancies should be noted on the form and identified to a senior member of staff, who will decide on a course of action. If there are discrepancies, details taken from the bottle should be used for data entry. | ||||
| 5.1.3 | Centrifuge clotted blood samples for 5 minutes at 1000 g. | ||||
| 5.1.4 | After centrifugation of the sample, up to 2 ml serum should be transferred to an appropriately labelled serum tube. This transfer should be carried out in such a way to ensure that the serum is transferred to the correct tube. | ||||
| 5.1.5 | Freeze and store serum sample at −20°C until required for testing. | ||||
| Notes |
|
||||
| 5.2 | Procedure for filter plate method | ||||
| 5.2.1 | Remove kit from fridge ensuring the beads and PE remain in the DARK as they are extremely light sensitive. | ||||
| 5.2.2 | Note the Lot number of the kit to be used and ensure that the template has been loaded onto the Luminex software. | ||||
| 5.2.3 | Enter on worksheet the lot numbers and expiry dates of the LABScreen kit, IgG/PE, wash buffer, positive control and negative control. Where appropriate note the date the vial was received and defrosted. | ||||
| 5.2.4 | For screening with LABScreen Mixed kit – the NIBSC negative and the positive control sample should be included. For screening with either the Class I and II single antigen kits – the FlowPRA negative control sample plus a positive control sample should be included. |
||||
| 5.2.5 | Take a new filter plate, or the current ‘in use’ filter plate if enough unused wells are available. Label each well of the plate numerically (corresponding to the serum number on the worksheet) for each serum sample to be screened including positive and negative controls. Labelling must be in the vertical, e.g. sample 1 at A1, sample 2 at B1, sample 3 at C1, etc. | ||||
| 5.2.6 | Using cut-down transparent microplate sealers cover all wells that are not being used for this test. This keeps unused wells clean for future use and ensures a good vacuum when washing with the vacuum manifold. | ||||
| 5.2.7 | For each well to be used pre wet the filter by adding 250 μl of sterile water. Leave for 5 minutes. | ||||
| 5.2.8 | After this time gently aspirate the contents of the wells using the vacuum manifold. | ||||
| 5.2.8.1 | Ensure all tubes are correctly attached to the vacuum pump and the reservoir is empty. | ||||
| 5.2.8.2 | Dampen the top of the manifold by briefly running under the tap, this ensures a good seal for the vacuum. | ||||
| 5.2.8.3 | Place filter plate on top of the manifold and press down. | ||||
| 5.2.8.4 | Turn on the vacuum pump until the contents of the wells have been drawn out of the bottom. | ||||
| 5.2.8.5 | Do not apply excess vacuum as this can damage the filter, and when beads are present cause them to be lost or become trapped in the filter. | ||||
| 5.2.8.6 | Decant contents of reservoir into a slop pot containing 1% Virkon before discarding. | ||||
| 5.2.9 | Prepare the beads by briefly centrifuging the vial at 600-800 g to remove any beads or liquid from the cap or walls of the vial, then thoroughly mix by vortexing for 30 seconds or repeat pipetting to evenly resuspend beads. | ||||
| 5.2.10 | Transfer 3 μl of beads to each of the assigned wells. Addition to the wells must be performed very carefully ensuring the filter is not pierced with the pipette tip. | ||||
| 5.2.11 | Add 12 μl of each serum to the appropriate wells. Mix the well contents using repeat pipetting. Again ensure the filter is not pierced with the pipette tip. | ||||
| 5.2.12 | Cover the plate with the plastic lid provided and wrap in foil to protect from light. | ||||
| 5.2.13 | Incubate for 30 minutes at room temperature (20–24°C) on the orbital mixer, set at 200 rotations per minute. | ||||
| 5.2.14 | Dilute the IgG/PE conjugate. Calculate the amount of conjugate required to add 100 μl to each well plus three wells extra (with each well requiring 1 μl of conjugate diluted in 99 μl of wash buffer). Mix conjugate by pipetting. Cap the tube and wrap completely in foil to protect from the light. Store at room temperature until use. | ||||
| 5.2.15 | After 30 minute incubation, remove the foil and plastic lid from the plate and add 250 μl of wash buffer to each of the wells. | ||||
| 5.2.16 | Gently aspirate the contents of the wells using the vacuum manifold as described in 5.2.8. | ||||
| 5.2.17 | Add 250 μl of wash buffer to each well, and aspirate as described in 5.2.8. | ||||
| 5.2.18 | Repeat step 5.2.17 a further two times to give a total of three washes. | ||||
| 5.2.19 | Add 100 μl of diluted conjugate to each well and cover plate with plastic lid provided and then wrap in foil to protect from light. | ||||
| 5.2.20 | Incubate plate for 30 minutes at room temperature (°C) on the orbital mixer set at 200 rotations per minute. | ||||
| 5.2.21 | Remove plastic lid and add 150 μl of wash buffer. Mix by gently tapping the side of the plate. | ||||
| 5.2.22 | Repeat steps 5.2.16–5.2.18. | ||||
| 5.2.23 | Add 80 μl of room temperature PBS and repeat pipette to mix the well contents. | ||||
| 5.2.24 | The beads are now ready to be analysed. This must be performed within 3 hours to ensure the least chance of obtaining false positive and false negative results. | ||||
| 5.3 | Procedure for ‘Spin and Flick’ method | ||||
| 5.3.1 | Follow steps 5.2.1–5.2.4. | ||||
| 5.3.2 | Prepare the beads by briefly centrifuging the vial at 600–800 g to remove any beads or liquid from the cap or walls of the vial, then thoroughly mix by vortexing for 30 seconds or repeat pipetting to evenly resuspend beads. | ||||
| 5.3.3 | Add 2 µl of LABScreen beads to each test well of a V bottom plate using a multichannel dispenser. | ||||
| 5.3.4 | Add 8 µl of each test serum into the corresponding well and mix. Wrap in foil and incubate for 30 minutes at room temperature (20–24°C) on the orbital mixer. | ||||
| 5.3.5 | Dilute the IgG/PE conjugate. Calculate the amount of conjugate required to add 100 μl to each well plus three wells extra (with each well requiring 1 μl of conjugate diluted in 99 μl of wash buffer). Mix conjugate by pipetting. Cap the tube and wrap completely in foil to protect from the light. Store at room temperature until use. | ||||
| 5.3.6 | Following incubation add 230 µl of diluted (1×) wash buffer to each well of the plate. Cover with tray seal and vortex. Centrifuge at 1300 g for 5 minutes. | ||||
| 5.3.7 | Remove wash buffer from wells of plate by flicking and then blotting on absorbent paper, ensuring the plate is not reinverted between the two actions. | ||||
| 5.3.8 | Repeat steps 5.3.6 and 5.3.7 twice to give a total of 3 washes. | ||||
| 5.3.9 | Add 100 µl of previously diluted PE conjugate to each well. Cover with plate seal and vortex. Wrap in foil and incubate for 30 minutes at room temperature (20–24°C) on the orbital mixer. | ||||
| 5.3.10 | Centrifuge plate at 1300 g for 5 minutes. | ||||
| 5.3.11 | Add 150 µl wash buffer, cover with seal and vortex. Centrifuge at 1300 g for 5 minutes. Repeat wash steps 5.3.6–5.3.7 twice to give a total of 3 washes. | ||||
| 5.3.12 | Add 80 µl of wash buffer to each well and resuspend beads by pipetting up and down. Then transfer the beads to their corresponding positions in a low-profile 96 well PCR tray. The samples are ready for data acquisition. | ||||
| 5.4 | Collecting data using the Luminex analyser | ||||
| 5.4.1 | Set up and calibrate Luminex analyser following local procedure. | ||||
| 5.4.2 | Create Luminex input file following local procedure. | ||||
| 5.4.3 | Load the patient data and create a batch on the Luminex system following local procedure. | ||||
| 5.4.4 | Run plate following local procedure. | ||||
| 5.4.5 | Export raw data for analysis. | ||||
| 5.5 | Analysis of data | ||||
| 5.5.1 | The original hand-signed worksheet should be filed in the research folder. | ||||
| 5.5.2 | Transfer raw data for analysis into HLA Fusion software following local procedure. | ||||
| 5.5.3 | Analysis should be performed using HLA Fusion v2.0 according to local procedure – except for the cut-off values and points detailed below. | ||||
| 5.5.4 | For Class I single antigen analysis ensure that the W6-32 box is ticked. | ||||
| 5.5.5 | For LABScreen mixed screening beads the negative control values should be taken from the NIBSC negative control serum. | ||||
| 5.5.6 | For the single antigen screening beads the FlowPRA negative control values should be used. | ||||
| 5.5.7 | The control values should fit in the following criteria: | ||||
| 5.5.7.1 | The bead count should be greater than 50 for each bead group. | ||||
| 5.5.7.2 | The NC should be greater than 30 and ideally below 500, but should ALWAYS be less than 1000. | ||||
| 5.5.7.3 | The PC should be greater than 1000 and at least twice the NC value. | ||||
| 5.5.7.4 | The PC/NC ratio should be greater than 2. | ||||
| 5.5.7.5 | Any values falling outside these guidelines should be flagged up and discussed with HOS or appropriate before recording results or repeating. | ||||
| 5.5.8 | Samples with an NC value of greater than 1000 should be retested following treatment with Absorbout beads, produced by OneLambda and provided by VH Bio, following the manufacturers’ guidelines. | ||||
| 5.5.9 | For LABScreen mixed analysis a sample should be deemed positive if any Class I bead has a ratio greater than 1.3 and any Class II bead has a ratio greater than 2.5, for the Lot 18 LABScreen mixed bead kit tested using the method described above. For Lot 19 LABScreen mixed bead kits, a sample should be deemed positive if any Class I bead has a ratio greater than 4.0 and when any Class II bead has a ratio greater than 5.5. |
||||
| For Lot 20 LABScreen mixed bead kits, a sample should be deemed positive if any Class I bead has a ratio greater than 1.6 and when any Class II bead has a ratio greater than 4.0. For Lot 22 LABScreen mixed bead kits, a sample should be deemed positive if any Class I bead has a ratio greater than 1.5 and when any Class II bead has a ratio greater than 3.0. |
|||||
| 5.5.10 | Samples tested using the LABScreen single antigen beads will be regarded as positive for the trial if the MFI of any bead is ≥ 2000. If any of the positive beads represent a mismatched donor HLA antigen, this will be assigned as DSA+. The number of DSA with an MFI ≥ 2000 will be recorded to define the Ab ‘burden’ of an individual patient. | ||||
List of abbreviations
- Ab
- antibodies
- ACR
- albumin creatinine ratio
- AE
- adverse event
- BLC
- biomarker-led care
- CAMR
- chronic antibody-mediated rejection
- CI
- confidence interval/chief investigator
- DM
- diabetes mellitus
- DSA
- donor-specific antibody
- eGFR
- estimated glomerular filtration rate
- HIV
- human immunodeficiency virus
- HLA
- human leucocyte antigen
- HR
- hazard ratio
- IgG
- immunoglobulin G
- IMP
- investigational medicinal product
- MARS
- medication adherence report scale
- MFI
- mean fluorescence intensity
- MMF
- mycophenolate mofetil
- MPA
- mycophenolic acid
- NIHR
- National Institute for Health and Care Research
- NICE
- National Institute for Health and Care Excellence
- Non-DSA
- HLA antibodies against non-donor HLA
- od
- once daily
- PCR
- polymerase chain reaction/protein creatinine ratio
- PI
- principal investigator
- QALY
- quality-adjusted life-year
- SAE
- serious adverse event
- SAP
- statistical analysis plan
- SC
- standard care
- Tac
- tacrolimus
- UK
- United Kingdom
- XM
- cross match
Notes
Supplementary material can be found on the NIHR Journals Library report page (https://doi.org/10.3310/KMPT6827).
Supplementary material has been provided by the authors to support the report and any files provided at submission will have been seen by peer reviewers, but not extensively reviewed. Any supplementary material provided at a later stage in the process may not have been peer reviewed.