

Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 13/50/17. The contractual start date was in November 2015. The final report began editorial review in December 2020 and was accepted for publication in June 2021. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
Copyright © 2022 Cro et al. This work was produced by Cro et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This is an Open Access publication distributed under the terms of the Creative Commons Attribution CC BY 4.0 licence, which permits unrestricted use, distribution, reproduction and adaption in any medium and for any purpose provided that it is properly attributed. See: https://creativecommons.org/licenses/by/4.0/. For attribution the title, original author(s), the publication source – NIHR Journals Library, and the DOI of the publication must be cited.
2022 Cro et al.
Chapter 1 Introduction
Material throughout the report has been adapted from the trial protocol by Cornelius et al. 1 © The Author(s). 2018 Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (https://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
Parts of this report are also based on Cro et al. 2 © 2021 British Association of Dermatologists.
Scientific background
Psoriasis is a common condition (estimated prevalence of 2% in the UK) that is known to have an impact on quality of life at a level comparable to other major diseases, including chronic heart disease and cancer. 3,4 Pustular forms of psoriasis are characterised by painful, intensely inflamed red skin studded with sheets of monomorphic, sterile, neutrophilic pustules. These pustules may be chronic. Pustular psoriasis is typically localised and involves the hands and feet [known as acral pustular psoriasis (APP)], although it can also occur more rarely as generalised, episodic and potentially life-threatening [generalised pustular psoriasis (GPP)]. 5,6 Some individuals may experience both forms throughout their life.
Although pustular psoriasis constitutes < 10% of all cases of psoriasis, it often ranks the highest of all psoriasis phenotypic variants in terms of symptoms (itch, pain and functional impairment, causing limited mobility and interference with daily living tasks and work). 7–9 Ultimately, the consequential impact is immense and equivalent to that of psychiatric illness and other major medical diseases. 10,11
Over the past decade, significant investment in novel therapies and the advent of biological therapies have revolutionised the treatment and management of plaque-type psoriasis. This has been primarily driven by scientific investigations of underlying genetic and immunological disease pathways. 12 By contrast, the treatment options for pustular psoriasis are currently profoundly limited. Super-potent (topical) corticosteroids, phototherapy, oral treatments [e.g. acitretin (Neotigason®, Teva UK Ltd, Harlow, UK), methotrexate (Hospira UK Ltd, Maidenhead, UK) and ciclosporin (Capimune®, Mylan, Canonsburg, PA, USA)] and targeted biologic therapies (notably tumour necrosis factor antagonists) are all used, although the evidence for their benefit is poor. 13 There is, therefore, a very significant unmet need in this patient group.
Rationale for the study
Recent evidence indicates that the molecular pathways underlying pustular psoriasis are distinct (from that observed with plaque-type disease) and involve the interleukin (IL) 36/IL-1 axis. Research has identified functionally relevant IL36RN mutations in both GPP and APP. 14–16 IL36RN encodes the IL-36 receptor antagonist IL-36Ra (this is an IL-1 family member that antagonises the pro-inflammatory activity of IL-36 cytokines). Disease mutations disrupt the inhibitory function of IL-36Ra, causing enhanced production of downstream inflammatory cytokines (including IL-1). 15,16 Indeed, in individuals with IL36RN mutations, IL-1 production has been shown to be significantly upregulated in response to IL-36 stimulation. 15 Furthermore, IL-1 is a cytokine that is known to sustain the inflammatory responses initiated by skin keratinocytes. 17
Interleukin 1 antagonists have previously shown therapeutic benefits in the treatment of IL-1-mediated diseases (many of which feature neutrophilic infiltration of the skin). 18 Furthermore, there has been research that suggests a key pathogenic role for IL-1 in pustular forms of psoriasis. 19,20
The model IL-1 antagonist proposed for the study was anakinra [Sobi (Swedish Orphan Biovitrum AB), Stockholm, Sweden]. Anakinra is an IL-1 receptor antagonist that is licensed for the treatment of rheumatoid arthritis and, during the timeline of this trial, periodic fever syndromes and Still’s disease. Anakinra was selected in preference of other licensed IL-1 antagonists for several reasons. It uniquely blocks the activity of both IL-1a and IL-1b. 18 Financially, it has the lowest drug acquisition cost (and this is of relevance to the NHS should anakinra show efficacy) and we had access to fully funded trial drugs through the manufacturer Sobi. Anakinra also possesses a rapid onset of action and an established safety profile (with > 70,000 patient-years’ exposure). Furthermore, there is early evidence of therapeutic benefit in participants with pustular psoriasis. 21
Hypothesis
We hypothesised that an IL-1 blockade would deliver therapeutic benefits in pustular forms of psoriasis. Therefore, this project aimed to investigate the clinical efficacy of an IL-1 blockade in palmoplantar pustulosis (PPP) (the most common form of pustular psoriasis) using the model IL-1 antagonist, anakinra, in a randomised, placebo-controlled trial with a two-staged adaptive design, followed by an open-label extension (OLE).
Study objectives
Primary objective
The primary objective of the study was to determine the efficacy of anakinra (compared with placebo) in the treatment of adults with PPP. The primary end point was change in disease activity at 8 weeks, adjusted for baseline, measured using the Palmoplantar Pustulosis Psoriasis Area Severity Index (PP-PASI).
Secondary objectives
-
Determine the treatment group difference in fresh pustule count, adjusted for baseline.
-
Determine the treatment group difference in total pustule count, adjusted for baseline.
-
Determine the time to response of PPP (defined as a 75% reduction in fresh pustule count compared with baseline) and relapse rate (defined as return to baseline fresh pustule count) with anakinra compared with placebo.
-
Determine the proportion of randomised participants who achieved clearance of PPP with anakinra compared with placebo by 8 weeks.
-
Determine the treatment effect on the development of a disease flare (> 50% deterioration in PP-PASI score compared with baseline) at 8 weeks.
-
Determine any treatment effect of anakinra in pustular psoriasis at non-acral sites as measured by change in percentage area of involvement at 8 weeks compared with baseline.
-
Determine any treatment effect of anakinra in plaque-type psoriasis (if present) measured using the Psoriasis Area Severity Index (PASI) at 8 weeks compared with baseline.
-
Determine the impact of anakinra on participants’ symptoms and quality of life compared with placebo at 8 weeks, adjusted for baseline, as assessed using the Palmoplantar Quality of Life (PP-QoL) instrument, Dermatology Life Quality Index (DLQI), Participants Global Assessment (PGA) and EuroQol-5 Dimensions, three-level version (EQ-5D-3L).
-
Determine the proportion of randomised participants who found the treatment acceptable or ‘worthwhile’.
-
Determine the proportion of randomised participants who adhered to treatment.
-
Determine whether or not there are any treatment group differences in episodes of serious infections, as defined by any infection leading to death or hospital admission or requiring intravenous antibiotics.
-
Determine whether or not there are any treatment group differences in neutropenia (neutrophil count of ≤ 1.0 × 109/l on at least one occasion).
-
Collect data on the adverse event (AE) profile and adverse reactions induced by anakinra compared with placebo to evaluate the safety and tolerability of anakinra in the treatment of PPP.
Exploratory objectives (mechanistic studies)
-
To validate the hypothesis that abnormal IL-1 signalling is a key driver in the pathogenesis of pustular psoriasis.
-
To determine the genetic status of individuals who responded to treatment as a preliminary step for future pharmacogenetic studies by comparing the genotypes of responders with those of non-responders.
-
To characterise the immune phenotype of all subjects entering the trial to establish whether or not the disease was associated with alterations in the number or activation status of IL-1-producing cells.
-
To collect mechanistic sample data sets on participants with pustular psoriasis for studies investigating disease pathogenesis [pustular psoriasis – elucidating underlying mechanisms (PLUM)].
Open-label extension objectives
The primary objective of the OLE was to boost recruitment; it was introduced part-way through the trial when funding for the required additional anakinra IMP was secured. In addition, we also obtained the following:
-
observational data on disease activity on anakinra [measured using the PP-PASI, fresh pustule count, total pustule count, Palmoplantar Pustulosis – Investigators’ Global Assessment (PPP-IGA) and PASI] over an initial 8-week treatment period for individuals originally prescribed placebo who chose to continue into the open-label component
-
observational data on disease activity on anakinra (measured using the PP-PASI, fresh pustule count, total pustule count, PPP-IGA scores and PASI scores) over a second 8-week treatment period for individuals originally prescribed anakinra who chose to continue into the open-label component
-
additional safety data following 8 weeks of anakinra treatment and also at 90 days post last dose of anakinra for individuals originally prescribed placebo
-
longer-term safety data on anakinra for individuals originally prescribed anakinra in the double-blind study period.
Chapter 2 Methods
Design
This was a Phase IV, two-stage (stages 1 and 2), double-blind, randomised, placebo-controlled trial with an adaptive element followed by an OLE. 1 Participant data from both stages were included in the main stage 2 analysis.
Stage 1 compared treatment groups to ensure that there was sufficient efficacy and safety to progress to stage 2. The pre-planned interim analysis for stage 1 occurred after 24 participants had been randomised and followed up for 8 weeks. A decision to embark on stage 2 was made using stop/go efficacy criteria. Fresh pustule counts and PP-PASI scores at week 8 were compared between treatment groups to assess efficacy. If, at the end of stage 1, the placebo group did as well as, or better than, the anakinra group on both of the two outcomes, then the study would be stopped. However, because the anakinra group did better than the placebo group for at least one outcome, the study proceeded (to stage 2).
Furthermore, the primary outcome for stage 2 was chosen at the end of stage 1. The two candidate primary outcomes assessed were the fresh pustule count (across palms and soles) and the PP-PASI score. These were recorded at baseline and at weeks 1, 4, 8 and 12. To determine the efficacy of anakinra for PPP compared with placebo, the primary end point for stage 2 was prespecified to be the change in disease activity at 8 weeks (adjusted for baseline) measured using fresh pustule count (the default primary outcome) unless the PP-PASI score was judged to be more reliable and discriminating.
Stage 2 commenced with the PP-PASI score designated as the primary outcome (see Chapter 3, Stage 1). Stage 2 included the randomisation of a further 40 participants (64 in total). 1,22
Interventions
Participants were randomised (1 : 1) to receive (100 mg per day) either anakinra or placebo for 8 weeks, which was self-administered daily as a subcutaneous injection.
Participants who opted to take part in the OLE received a further 8 weeks of anakinra (100 mg per day) treatment, which was self-administered daily as a subcutaneous injection. The OLE was optional, and was offered to all participants who completed the 8-week treatment period and the 12-week follow-up visit.
Setting
The trial was set in 16 hospitals across England, Scotland and Wales.
Participants
Given that GPP is rare, episodic and potentially life-threatening, including GPP participants in a trial setting would have been difficult and potentially unethical (with the placebo group). Thus, the population designated for the study was participants with PPP. This chronic, localised form of pustular psoriasis involves the hands and/or feet, and is associated with significant disability. It is the most common form of pustular psoriasis, making recruitment feasible, and typically features chronic development of pustules so that we would expect to capture any treatment effect within the 8-week treatment period.
All participants were adults (aged ≥ 18 years) with a diagnosis of PPP that had been made by a trained dermatologist, with a disease duration of > 6 months and disease of sufficient impact and severity to require systemic therapy. To be randomised into the study, at the baseline visit participants had to exhibit at least moderate disease on the PPP-IGA, with evidence of active pustulation on palms and/or soles.
Women who were pregnant, breastfeeding or of child-bearing age and not on adequate contraception and men planning conception were excluded from taking part in the trial.
The specific inclusion criteria and exclusion criteria for the double-blind, placebo-controlled trial and the OLE are detailed below.
Identification and recruitment
Potentially eligible participants were identified by the following four methods:
-
In clinic at participating sites. Potentially eligible participants were identified in clinics and were approached directly by a member of the trial team and/or clinical care team, who explained the study to the patient and provided them with the patient information leaflet. Participants were then given as much time as they required (and at least 24 hours) to read the information leaflet and come to a decision about their participation.
-
Searching existing local health-care/medical databases at participating sites. Once sites were opened, local study teams identified potentially eligible participants through searching local clinic and pharmacy lists, electronic patient records, referral lists and letters, research databases and other lists (as appropriate). Potential participants were then contacted by their consultant and the research team (by letter, e-mail or telephone call), were invited to participate the study and were provided with the patient information leaflet.
-
Self-referral. Potential study participants identified themselves after becoming aware of the study. The study website (http://apricot-trial.com/; accessed 28 February 2020) included a specific page (which was taken down following the end of recruitment) on which participants could register through an interactive web-based patient recruitment questionnaire (see Appendix 1, Figure 19) for more information. These results were automatically sent to the trial manager and were used as the first line of eligibility screening. The trial manager/research nurse contacted potentially eligible patients by telephone and e-mail and invited them to participate (and provided them with the patient information leaflet if this had not already been downloaded by the patient from the trial website). If the patient remained interested in participating, then, with their consent, their contact details were provided to the trial team geographically closest to them to arrange a formal research consultation.
-
Participant identification centres (PICs). Potential study participants were identified at PICs following clinic visits or review of local clinic and pharmacy lists, electronic patient records, referral lists and letters, research databases and other lists. They were then contacted by their direct clinical care team (usually by letter, e-mail, telephone call or in person) and then invited to self-refer on the trial website (as detailed above) or (with their agreement) referred directly to the team at their chosen trial site for further information regarding participation.
Randomisation procedure
The randomisation service for the study was provided by the King’s Clinical Trials Unit (CTU). Following written consent at the screening visit, each participant was registered on the MACRO electronic case report form system version 4 (InferMed Macro) that generated a unique patient identification number (PIN). This unique PIN was then recorded on all source data worksheets and was used to identify the participants throughout the study.
At the baseline visit, randomisation occurred via a bespoke web-based randomisation system hosted by the King’s CTU (https://cturandomisation.iop.kcl.ac.uk/APRICOT/Login.aspx?ReturnUrl%20=%20%2fAPRICOT; accessed 1 September 2020). Authorised site staff were allocated a username and password for the randomisation system by the trial manager. An authorised staff member (typically the principal investigator or research nurse) logged into the randomisation system and entered in the patient’s details, including the unique study PIN.
Once a participant was randomised, the system automatically generated e-mails to key study staff. For example, an e-mail was sent to the local site pharmacy to alert the staff to a participant’s treatment group (treatment 1 or 2). Additional blinded and unblinded e-mails were generated from the randomisation system to notify key trial site staff (e.g. the chief investigator and trial manager) depending on their role in the study.
The randomisation sequence was generated using blocked randomisation, stratified by centre.
Blinding
Investigational medical product
Participants, investigators, co-investigators, research nurses, clinical trial co-ordinators and clinical trial practitioners were blind to the IMP allocation for the duration of the trial.
Each randomised participant was provided with a card giving the code-break telephone numbers and emergency contact details.
Emergency code-break services were provided by ESMS Global (London, UK): a 24-hour cover service. To support patient safety, emergency unblinding could be performed according to strict criteria.
In the event of an emergency code break, ESMS Global was to notify the King’s Health Partners Clinical Trials Office of any emergency code break requests received, irrespective of outcome. The King’s Health Partners Clinical Trials Office clinical research associate would then inform the chief investigator and relevant principal investigator of the instance of unblinding. This would then be recorded so that the study statistician could be informed at the analysis stage of the trial.
Skin assessments
The active trial medication is known to cause injection site reactions in the majority of participants. If such reactions were apparent during study skin assessments, this could have led to inadvertent unblinding. 23 To avoid this, primary outcome assessments of fresh pustule count and PP-PASI score were carried out by an independent assessor blind to study treatment (a member of the study team trained in the assessment protocol but independent of the rest of the trial). At study visits, the independent blinded assessor was introduced as such to the participant by the clinical research team and had sight of the participant’s hands and feet only (injection site reactions occur at the site of administration, which is generally the abdomen/thighs). The independent blinded assessors were also instructed not to speak to the participant to maintain blinding.
Once the relevant outcome measures were assessed, the independent blinded assessor was instructed to leave the consulting room and the treating physician or research nurse then conducted the rest of the study visit (and the protocol-mandated procedures).
A second assessment of the PP-PASI score and PPP-IGA was also conducted by the treating physician or research nurse at each study visit.
Site staff were instructed that, whenever possible, the independent blinded assessor for a particular participant should be the same throughout the study.
During stage 1, fresh pustule counts were also assessed by a central blinded assessor using photography (prespecified views of palms and soles at baseline and weeks 1 and 8 of treatment; see Appendix 8, Figures 21 and 22).
Inclusion criteria for the double-blind treatment stage, placebo-controlled study
-
Adults (aged ≥ 18 years) with a diagnosis of PPP made by a trained dermatologist, with disease of sufficient impact and severity to require systemic therapy.
-
Disease of duration of > 6 months and not responding to an adequate trial of topical therapy, including very potent corticosteroids.
-
Evidence of active pustulation on palms and/or soles to ensure sufficient baseline disease activity to detect efficacy.
-
At least moderate disease on the PPP-IGA.
-
In the case of women of child-bearing potential, being on adequate contraception (see Appendix 2, Contraception guidelines, for guidance) and not pregnant or breastfeeding.
-
Written informed consent to participate.
Exclusion criteria for the double-blind treatment stage, placebo-controlled study
-
Previous treatment with anakinra or other IL-1 antagonists.
-
A history of recurrent bacterial, fungal or viral infections that, in the opinion of the principal investigator, presented a risk to the patient.
-
Evidence of active infection or latent tuberculosis (TB), human immunodeficiency virus (HIV) positivity or hepatitis B or C seropositivity.
-
A history of malignancy of any organ system (other than treated, localised non-melanoma skin cancer), treated or untreated, within the past 5 years.
-
Use of therapies with potential or known efficacy in psoriasis during or within the following specified time frame before treatment initiation (week 0, visit 1):
-
very potent topical corticosteroids within 2 weeks
-
topical treatment that is likely to impact signs and symptoms of psoriasis (e.g. corticosteroids, vitamin D analogues, calcineurin inhibitors, retinoids, keratolytics, coal tar, urea) within 2 weeks
-
methotrexate, ciclosporin, acitretin and alitretinoin (Toctino®, Stiefel, Brentford, UK) within 4 weeks
-
phototherapy or psoralen and ultraviolet A radiation (PUVA) within 4 weeks
-
etanercept (Enbrel®, Pfizer, New York, NY, USA) or adalimumab (Humira®; AbbVie Inc., Chicago, IL, USA) within 4 weeks
-
infliximab (Remicade®, Janssen Biotech Inc., Horsham, PA, USA) or ustekinumab (Stelara®, Janssen Biotech Inc.) or secukinumab (Cosentyx®, Novartis Pharmaceuticals UK Ltd, London, UK) within 3 months
-
other tumour necrosis factor (TNF) antagonists within 3 months
-
other immunosuppressive or immunomodulatory therapy within 30 days or five half-lives prior to treatment initiation, whichever was longer
-
any other investigational drugs within 30 days (or 3 months for investigational monoclonal antibodies) or five half-lives prior to treatment initiation, whichever was longer.
-
-
Moderate renal impairment (defined as creatinine clearance of < 50 ml/minute).
-
Neutropenia (defined as neutrophil count of < 1.5 × 109/l).
-
Thrombocytopenia (defined as platelet count of < 150 × 109/l).
-
Known moderate hepatic disease and/or raised hepatic transaminases [alanine aminotransferase (ALT) or aspartate aminotransferase (AST)], more than two times the upper limit of normal (ULN), at baseline. Participants who failed this screening criterion could still be considered following review by a hepatologist and confirmed expert opinion that study entry was clinically appropriate.
-
Live vaccinations within 3 months prior to the start of study medication, during the trial and up to 3 months following the last dose.
-
In the case of women, pregnancy, breastfeeding or being of child-bearing age and not on adequate contraception and, in the case of men, planning conception.
-
Poorly controlled diabetes mellitus, cardiovascular disease, asthma and concomitant therapy that may interact with anakinra (e.g. phenytoin or warfarin) or any condition in which, in the opinion of the investigator, anakinra would a present risk to the patient.
-
Unable to give written informed consent.
-
Unable to comply with the study visit schedule.
-
Diagnosis (or historic diagnosis) of either childhood- or adult-onset Still’s disease.
Inclusion criteria for the open-label extension
-
Participation in the double-blind placebo-controlled study.
-
Completion past visit 4 (week 8) of the double-blind placebo-controlled study.
-
In the case of women of child-bearing potential, being on adequate contraception (see Appendix 2, Contraception guidelines for guidance) and not pregnant or breastfeeding.
-
Written informed consent to participate.
Exclusion criteria for the open-label extension
-
A history of recurrent bacterial, fungal or viral infections that, in the opinion of the principal investigator, presented a risk to the patient.
-
Evidence of active infection or latent TB or of HIV positivity or hepatitis B or C seropositivity (required only for participants who were beyond visit 5, the double-blind treatment stage, of the placebo-controlled study).
-
A history of malignancy of any organ system (other than treated, localised non-melanoma skin cancer), treated or untreated, within the past 5 years.
-
Use of therapies with potential or known efficacy in psoriasis during or within the following specified time frame before treatment initiation (visit OLE 1):
-
methotrexate, ciclosporin, acitretin or alitretinoin within 4 weeks
-
phototherapy or PUVA within 4 weeks
-
etanercept or adalimumab within 4 weeks
-
infliximab, ustekinumab or secukinumab within 3 months
-
other TNF antagonists within 3 months
-
other immunosuppressive or immunomodulatory therapy within 30 days or 5 half-lives prior to treatment initiation, whichever was longer
-
any other investigational drugs within 30 days (or 3 months for investigational monoclonal antibodies) or 5 half-lives prior to treatment initiation, whichever was longer.
-
-
Moderate renal impairment (defined as creatinine clearance of < 50 ml/minute).
-
Neutropenia (defined as neutrophil count of < 1.5 × 109/l).
-
Thrombocytopenia (defined as platelet count of < 150 × 109/l).
-
Known moderate hepatic disease and/or raised hepatic transaminases (ALT/AST), more than two times the ULN, at baseline. Participants who failed this screening criterion could still be considered following review by a hepatologist and confirmed expert opinion that study entry was clinically appropriate.
-
Live vaccinations within 3 months prior to the start of study medication, during the trial and up to 3 months following the last dose.
-
In the case of women, pregnancy, breastfeeding or being of child-bearing age and not on adequate contraception or, in the case of men, planning conception.
-
Poorly controlled diabetes mellitus, cardiovascular disease and asthma, and concomitant therapy that may interact with anakinra (e.g. phenytoin or warfarin) or any condition in which, in the opinion of the investigator, anakinra would present a risk to the patient.
-
Inability to give written informed consent.
-
Inability to comply with the study visit schedule.
-
Having been previously invited to have the OLE therapy and declined.
-
Diagnosis (or historic diagnosis) of either childhood-onset or adult-onset Still’s disease.
Concomitant medication, prohibited medication and rescue therapy information
The list of concomitant medication, prohibited medication and rescue therapy for the double-blind RCT is presented in Appendix 3, Table 36. The list of prohibited medication for the OLE is presented in Appendix 3, Table 37.
Participant pathway (trial procedures)
The participant pathway consisted of four periods: a screening period, a treatment period, a follow-up period and an optional OLE.
The overall study flow is detailed in Figure 1 and the detailed visit schedule is listed in Appendix 2, Tables 33–35.
FIGURE 1.
Trial flow chart. ITT, intention to treat.
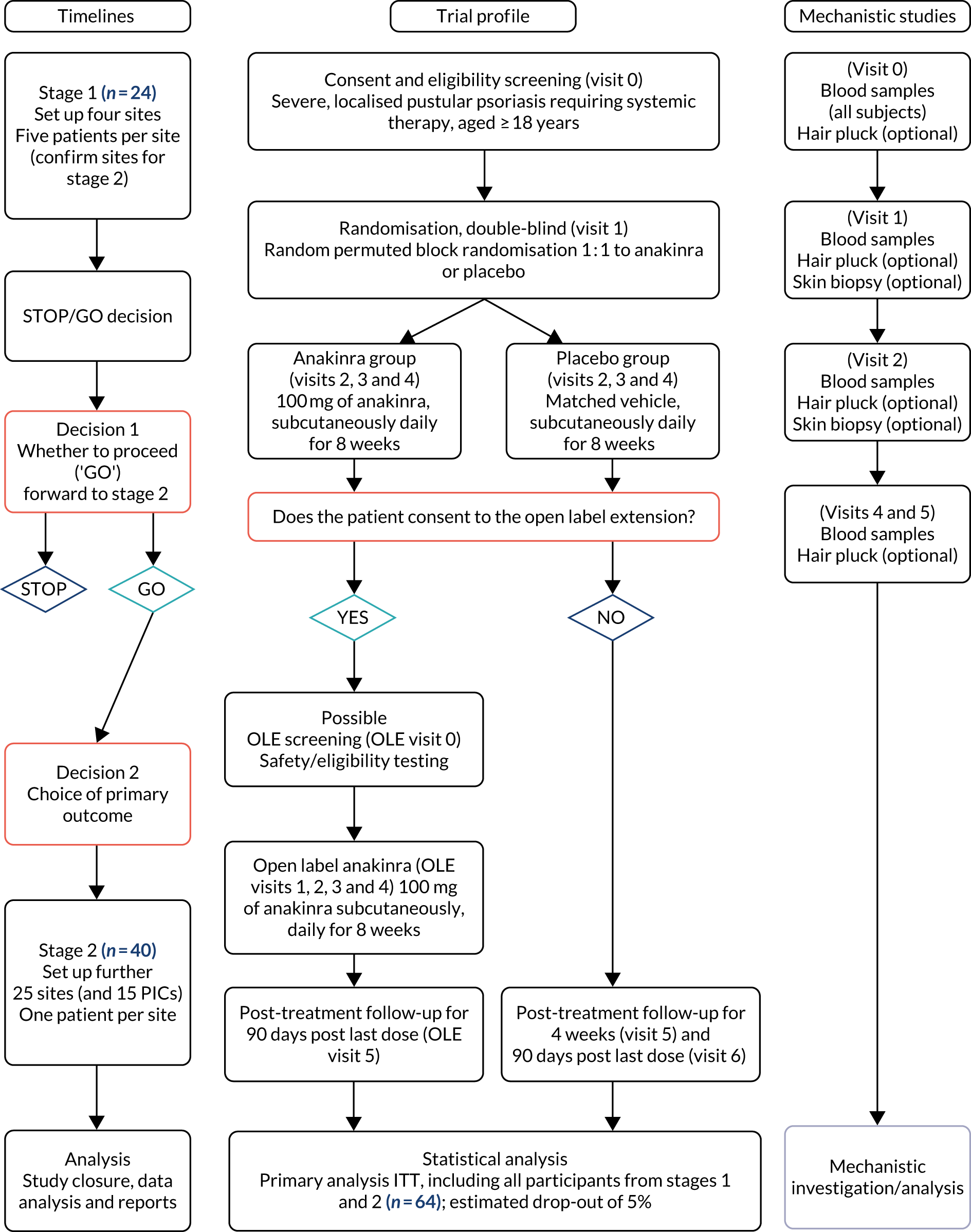
The screening period, that is the period between the screening visit (visit 0) and baseline (visit 1), was a minimum of 5 days and a maximum of 3 months, and was used to assess eligibility and to taper off prohibited medicines (as part of the washout period for the study). Participants who failed the screening period (did not satisfy one or more eligibility criteria) had the option to be re-screened if clinically appropriate.
The treatment period (visits 1–4) was 8 weeks. At the start of the treatment period, eligible participants were randomised to receive the intervention (as described above).
The follow-ups (visits 5 and 6) at week 12 and 90 days post last treatment date were used to assess disease relapse off study treatment, to follow up any AEs that had been previously reported and to plan for post-treatment management of the participants’ condition.
If a participant decided to take part in the optional 8-week OLE, there were two possible pathways:
-
Participants who decided to take part in the OLE before or at the week 12 follow-up visit (visit 5) were to begin their 8-week OLE period directly after the week 12 follow-up (i.e. their OLE baseline visit could be on the same day as the week 12 follow-up visit). Their final follow-up visit would take place 90 days after their last dose of anakinra.
-
Participants who were beyond the week 20 follow-up visit (visit 6) may have been receiving another treatment for their PPP when they decided to take part in the OLE. These participants were required to have an OLE screening visit, a possible washout period (as per the study protocol) and an OLE baseline visit arranged once the required washout period was completed. A final follow-up visit was then conducted 90 days after the last dose of anakinra.
To achieve the exploratory objectives (mechanistic studies), all participants were invited to provide biological samples for use in exploratory laboratory tests. These were blood samples taken at visit 0, and longitudinal blood samples were taken at visits 1, 2, 4 and 5.
In addition, participants were invited to provide microbiopsy samples from the skin on the lateral edge of the base of their feet or palms prior to treatment initiation at baseline (visit 1) and then at visit 2 (approximately 1 week later). These samples were used to understand the underlying pathogenesis of pustular psoriasis and the mechanism by which anakinra may work, and to identify potential biomarkers of response.
Participant withdrawal
Participants had the right to withdraw from the study at any time and for any reason. The principal investigator also had the right to withdraw participants from the study drug in the event of intercurrent illness, AEs, serious adverse events (SAEs), suspected unexpected serious adverse reactions (SUSARs) or protocol violation, or for administrative reasons or other pertinent reasons.
Participants had to discontinue the investigational product (and non-investigational product at the discretion of the investigator) in the event of any of the following:
-
withdrawal of informed consent (if the participant decided to withdraw for any reason)
-
the occurrence of any clinical AE, laboratory abnormality or intercurrent illness that, in the opinion of the investigator, indicated that continued participation in the study was not in the best interest of the participant
-
the need, in the principal investigator’s opinion, to administer concomitant medication not permitted by the trial protocol
-
pregnancy (in which case the chief investigator was notified immediately).
If a participant decided to withdraw from the trial, all efforts were made to report the reason for withdrawal as thoroughly as possible and participants were encouraged to provide follow-up data for the remaining trial visits, but at a minimum were asked for outcome data and safety data (AE records) at week 8 and at 90 days post last dose follow-up. They were also asked if they were willing to provide trial-specific clinical data (i.e. outcome measures) and/or samples for mechanistic study, as per the remaining trial schedule. All data and samples collected up to the date of withdrawal were retained.
Safety bloods should have been taken as per the trial schedule for all participants and/or as considered appropriate by the principal investigator.
Significant amendments to the study protocol
For a summary of amendments, see Appendix 4, Table 38.
Removal of visits from the study schedule
In April 2017 (as part of substantial amendment 4), the week 2 and week 6 visits were removed from the study protocol (originally visits 3 and 5, respectively). The study visits were renumbered accordingly (as required) throughout the protocol to accommodate this change.
The visit schedule was amended to decrease visits (while still maintaining the necessary safety assessments) to make the study more enticing to potential participants (by making it less burdensome to them). These changes were made in response to recruiting clinician feedback and informal participant feedback from the initial recruits.
The week 1 visit (visit 2) stayed in place to ensure that an early set of outcome measures were still collected, with some of the procedures that were previously undertaken at the week 2 visit being undertaken at the week 1 visit (visit 2).
At the time of this change, only 11 participants had been recruited. The analysis model was flexible enough to accommodate the change and the data from participants who had already provided the week 2 outcomes could be included in all analyses, substituted for week 1.
These protocol changes had no impact on the mechanistic work.
Open-label extension introduction
An OLE was added to the study in July 2019 (as part of substantial amendment 11). The primary purpose of the OLE was to enhance recruitment to the randomised, double-blind, placebo-controlled study, so that all participants had the potential opportunity to access anakinra. The OLE was added when agreement and funding for the required additional anakinra IMP were secured from the trial drug manufacturer, Sobi.
To retain the integrity of the primary randomised, double-blind, placebo-controlled study, only participants who had completed the 8-week treatment period schedule, as well as the week 12 follow-up visit, could take part in an optional 8-week period of anakinra treatment as an OLE to the trial.
To ensure equality of access, all participants who had already participated in APRICOT (Anakinra for Pustular Psoriasis: Response in a Controlled Trial), were currently taking part in APRICOT or were considering taking part in APRICOT were made aware of the 8-week OLE therapy and the criteria for enrolment. The only exceptions were the seven participants from the Manchester site, who were not offered the OLE because of a lack of research capacity, and one participant from the Guy’s Hospital site who wished to defer entry because of concerns about risk associated with travel to the study site during the COVID-19 pandemic [this participant has since been offered study anakinra IMP outside the context of the OLE for 8 weeks, with permission from the Medicines and Healthcare products Regulatory Agency (MHRA)].
Inclusion/exclusion criteria amendments
Thrombocytopenia
New safety information in the summary of product characteristics (SmPC) led to the decision to add exclusion criterion viii [with thrombocytopenia (defined as platelet count of < 150 × 109/l)] and this formed part of substantial amendment 4 to the protocol in April 2017.
The requirements for reporting and temporary treatment discontinuation were also amended following consultation with the study collaborator expert, who recommended that a platelet count of < 75 × 109/l should be reported as an important medical event and trigger a temporary halt in IMP treatment.
Latex allergy
During stage 1 of the study, latex was removed from the IMP containers and packaging and, thus, all trial stock was confirmed as being latex free. Therefore, the original exclusion criterion xii [latex allergy (inner needle cover of pre-filled syringe contains natural rubber)] was removed as part of substantial amendment 4 to the protocol in April 2017.
Still’s disease
Following an update to the information in the SmPC, it was found that there were reports of cases of macrophage activation syndrome (MAS) in Kineret-treated participants with Still’s disease. It must be noted that a causal relationship between Kineret® (Sobi Inc., Stockholm, Sweden) and MAS has not (yet) been established.
Following discussion within the Data Monitoring Committee (DMC), the study team opted to treat this finding with extreme caution and explicitly excluded participants with Still’s disease from the trial (submitted as substantial amendment 12 in September 2019 to update the protocol accordingly). Thus, exclusion criterion xv [diagnosis (or historic diagnosis) of either childhood-onset or adult-onset Still’s disease] for the double-blind, placebo-controlled study and the OLE were added as part of this amendment. This is a rare condition, and no participants with this condition entered into the trial.
Statistics methodology
Sample size
The overall sample size for APRICOT was calculated prior to the completion of stage 1 of the study, at which time the primary outcome of the main trial analysis was unknown. 24
The sample size was calculated using a standardised effect size. A large effect size of 0.9 standard deviations (SDs) was selected to be the minimum important difference to detect because of the cost of the drug and high patient burden arising from the requirement for participants to adhere to daily self-administered subcutaneous injection treatment. In addition, larger effect sizes have been reported with oral retinoids (historically etretinate and now acitretin), a recommended systemic intervention for pustular psoriasis. 25–27
To achieve 90% power with a 5% significance level for the detection of a difference of 0.9 SDs, a sample size of 27 participants per group was required. To allow for a (conservative) approximate 15% withdrawal rate, 32 participants per group (n = 64 in total) were required for the study. In APRICOT, the observed SD for the baseline PP-PASI score (n = 64) was 10.5; therefore, 0.9 SDs was approximately equivalent to a change of 9.5 in the PP-PASI score.
The sample size for stage 1 was based on the correct ordering of group means. A high probability of continuing (‘go’) was needed if there was a true (conservative) difference in means between the groups of 0.5 SDs in favour of the treatment group. With 20 participants (n = 10 per group), assuming a real difference of 0.5 SDs, the probability that the mean for the treatment group would be correctly ordered (i.e. the treatment mean is greater than the placebo mean) was 0.85. If two outcomes were assessed, each with an expected difference of 0.5 SDs, then the overall probability of failing to ‘go’ was (1 – 0.85)3 = 0.0225, that is less than 3 in 100. There was, therefore, a minimal chance of failing to continue if the treatment really was beneficial. If there was no treatment benefit, the probability of not progressing to the next stage was 0.25 based solely on these rules. Stage 1 did not involve statistical tests. To ensure that 10 participants contributed to each group, it was planned that the interim stage 1 analysis would be carried out after 24 participants had been randomised and followed up.
Statistical analysis
General statistical principles
The analysis was conducted subgroup blind (i.e. as group A vs. group B) in accordance with the APRICOT statistical analysis plans,22,24 which were finalised prior to database lock. The main analysis was based on the intention-to-treat (ITT) principle, that is all participants with at least one follow-up were analysed in the group to which they were randomised regardless of subsequent treatment received. The use of a longitudinal model for the primary analysis meant that a minimal number of participants would be excluded. Every effort was made to obtain all follow-up data for all participants, including those who stopped treatment.
The safety set population consisted of all participants who received at least one dose of the assigned IMP intervention and was used in the analysis to describe AEs.
All regression analyses included adjustment for centre because this was a stratification factor in the randomisation. The inclusion of this adjustment was necessary in the analysis to maintain the correct type I error rate. 28,29
Estimates are presented with 95% confidence intervals and p-values. A p-value of < 0.05 was interpreted as statistically significant for the primary outcome. All analyses were conducted using Stata® version 15.1 (StataCorp LP, College Station, TX, USA).
Stage 1 analysis22
At the end of stage 1, the baseline-adjusted mean treatment group differences in the fresh pustule count and PP-PASI score, averaged across follow-up visits, were calculated using a linear regression model. These results informed the decision to progress to stage 2. The trial continued to stage 2 if the treatment group did, on average, better than the placebo group on at least one measure. The primary outcome for stage 2 was selected based on an assessment of reliability and distributional properties for two candidate outcomes: the fresh pustule count and the PP-PASI score. The reliability of the fresh pustule count was assessed by examining the agreement between the assessments made at the site and those assessed centrally based on photographs. Agreement was formally assessed using the Bland–Altman method and the intraclass correlation coefficient (ICC) was calculated using a mixed-effects analysis of variance (ANOVA) with a random intercept for patient and rater. 30,31 The closer the ICC value was to 1, the better the level of consistency. The reliability of the PP-PASI was assessed by examining the agreement between assessments made at the site by two independent assessors using the same methods outlined above. Distribution properties for each candidate outcome were assessed using standardised mean differences and histograms by treatment group.
Stage 2 analysis24
A Consolidated Standards of Reporting Trials (CONSORT) flow chart32 was constructed to summarise the participant flow through the study. Baseline characteristics were summarised by treatment group to examine the balance between the groups at baseline. Treatment adherence, reasons for withdrawal and use of rescue medication, prohibited therapy and other topical treatments were summarised by treatment group. All primary and secondary outcomes were also summarised by time point and treatment group. Continuous variables were summarised using the mean (SD) where approximately normally distributed and the median [interquartile range (IQR)] where skewed. Categorical variables were summarised by frequency and percentage.
The primary analysis was based on the ITT principle and estimated the effect of the treatment policy. 33 A linear (Gaussian) mixed-effects model including PP-PASI data from weeks 1, 4 and 8 was utilised to obtain an estimate of the mean treatment group difference in PP-PASI scores at week 8. The model included random intercepts for participant and centre and fixed effects for study visit, treatment group, study visit by treatment group interaction and baseline PP-PASI scores. An unstructured covariance matrix was used to model the covariance structure, as it allows for all variances and covariances to be distinct, and the model was fitted with restricted maximum likelihood. The mean difference in the week 8 PP-PASI scores, adjusted for baseline, between the two treatment groups formed the focal point of the primary outcome analysis. The main conclusion of the trial was, therefore, based on this (week 8) analysis time point. However, treatment effects at weeks 1 and 4 were also calculated and reported.
In accordance with the ITT principle, all participants who provided data from at least one follow-up visit (at weeks 1, 4 or 8) were included in the primary analysis model as randomised. All missing response values were assumed to be missing at random (MAR) (i.e. the probability that the response is missing does not depend on the value of the response after allowing for the observed variables).
A pre-planned sensitivity analysis was performed to explore the impact of departures from the main MAR analysis assumption and potential missing not at random (MNAR) mechanisms on the trial results using multiple imputation (MI) and a pattern mixture approach. 34,35
Four pre-planned supplementary analyses targeted alternative treatment estimands for the trial’s primary outcome:
-
Supplementary analysis that estimated the treatment effect if rescue therapy was not available. Data post initiation of rescue therapy were set as missing and MI was used to explore the impact of a worse outcome post initiation on rescue therapy on trial results. The primary analysis model was retained for use in the analysis, following MI.
-
Supplementary analysis that estimated the treatment effect if rescue therapy and prohibited therapy were not available. Data post initiation of rescue therapy and prohibited medication were set as missing, and MI was used to explore the impact of a worse outcome post initiation on rescue therapy on the trial results. The primary analysis model was retained for use in the analysis, following MI.
-
Supplementary analysis that estimated the treatment effect if all topical therapy was not available. Data during the use of topical therapy were set as missing and MI was used to explore the impact of observing on-treatment behaviour (MAR) in the absence on topical therapy on the trial results. The primary analysis model was retained for use in the analysis, following MI.
-
Supplementary analysis to estimate the complier-average causal effect (CACE). The CACE preserves the benefits of randomisation and compares the average outcome of the compliers in the treatment group with the average outcome of the comparable group of ‘would-be compliers’ in the placebo group. To identify the CACE it is assumed that (1) members of the placebo group have the same probability of non-compliance as members of the intervention group and (2) being offered the treatment, that is randomisation itself, has no effect on outcome. We estimated the CACE using a two-stage least squares instrumental variable regression for the primary end point. Here, we initially defined a ‘complier’ as anyone who had received > 50% of the total number of planned injections (at any time point). Randomisation was used as an instrumental variable for treatment received, with adjustment for baseline PP-PASI scores (excluding centre from the analysis). We also calculated the CACE by defining a complier as, alternatively, anyone receiving 60–90% of the total number of planned injections.
Secondary outcome statistical analysis
Continuous secondary outcomes were analysed using the same modelling approach as specified above for the primary outcome. Binary outcomes were analysed using mixed logistic regression models and ordered categorical outcomes using mixed ordered logistic models. Similar to the primary analysis model, the models for secondary outcomes included participant and centre as a random intercept and fixed effects for time, time by treatment group interaction and baseline value of the outcome.
Kaplan–Meier curves were plotted for time to response and time to relapse outcomes. Given that outcomes were observed at a relatively few discrete time intervals (weeks 4, 8 and 12), complementary log–log models were fitted to estimate the treatment effect for the time-to-event outcomes, as this is an analysis model suitable for discrete survival time data. The time-to-event models included a fixed effect for treatment group and a random intercept for centre (stratification variable).
Adverse event analysis
Data concerning AEs were collected during study visits from reports of testimony from study participants, clinical observations, clinical examinations and blood tests.
Local clinicians rated the relationship of each AE to the study medication as none/unlikely/possible/likely/definite. From this classification, adverse reactions were the subset of non-serious AEs considered to have a possible/likely/definite relationship with the study medication. Serious adverse reactions (SARs) consisted of the subset of SAEs considered to have a possible/likely/definite relationship with the study medication. Furthermore, if an event was considered related to the study IMP, local clinicians also rated whether or not the reaction was unexpected.
All AEs were coded using terms referencing the Medical Dictionary for Regulatory Activities (MedDRA) at the ‘preferred terms’ level. These were also summarised by MedDRA system organ class and intensity (when subjectively assessed by local clinicians as mild/moderate/severe).
Adverse events were tabulated by treatment group for both the number of events and the number of participants with each type of event. AEs were also listed individually by MedDRA preferred term level and intensity (subjectively assessed by local clinical investigators as mild/moderate/severe) and summarised by MedDRA system organ class level. To identify the events with the strongest evidence for between-group difference, we constructed a volcano plot, in which the difference between treatment groups in the risk of non-serious AEs and reactions, by MedDRA system organ class, was plotted against the p-value from a Fisher’s exact test. 36 To further aid interpretation, AEs were also summarised visually in a dot plot, which displayed the proportions of individuals experiencing each type of event, by group, and the relative difference with 95% confidence intervals (CIs). The number of events related to an infection was also tabulated.
Exploratory analysis
A longitudinal analysis was undertaken using a linear (Gaussian) mixed model to determine the treatment difference in PP-PASI scores at week 12. The analysis model was the same as in the primary analysis, but included additional data at week 12. The treatment effect for PP-PASI scores at week 12 was estimated and reported with a 95% CI. Given that it was hypothesised that palmar disease may respond more quickly to anakinra than plantar disease, pre-planned exploratory analysis separately estimated the efficacy of anakinra on (1) disease activity at week 8, measured using fresh pustule count on the palms, adjusted for baseline, compared with placebo, and (2) disease activity at week 8, measured using fresh pustule count on the soles, adjusted for baseline, compared with placebo. For each of the palms and soles fresh pustule count, a linear mixed-effects model was used, which included fixed effects for treatment group, time (weeks 1, 4 and 8), treatment group by time interaction and baseline value of the associated outcome. A random intercept for participant and centre was also included in each of the models.
Post hoc analysis
The treatment group difference in ≥ 50% improvement in PP-PASI (PP-PASI 50) scores and ≥ 75% improvement in PP-PASI (PP-PASI 75) scores at week 8 was assessed using a mixed-logistic binary model that included centre as a random intercept and fixed effects for treatment group and baseline PP-PASI scores. We also examined the treatment group difference in the PP-PASI pustule subscale scores at week 8, separately for palms and soles, using a mixed-ordered logistic model that included participant and centre as a random intercept and fixed effects for time, time by treatment group interaction and baseline PP-PASI pustule subscale scores. For each participant and region (i.e. palm or sole), the maximum severity pustule rating across the left or right component of the region was utilised in analyses.
Mechanistic samples
Genetic analyses, including whole-exome sequencing, bulk ribonucleic acid (RNA) sequencing, pathway enrichment analyses and upstream regulator analysis, were used on mechanistic samples obtained during the trial to investigate the pathogenic involvement of IL-1 in PPP.
Open-label extension analysis
The number of participants who entered the OLE was summarised by original randomised treatment group. Baseline characteristics of all participants in the original double-blind period were descriptively compared against those of the participants entering the OLE period.
In the OLE, some participants continued their medication (some following a 4-week break and some with a longer break) and some participants started the medication for the first time. For this reason, it was not possible to undertake a randomised comparison for this extended follow-up period. Therefore, this was treated as an observational intervention period.
For the population of participants who continued into the OLE stage, descriptive statistics were presented for the open-label outcomes recorded at the OLE baseline visit and 8 weeks after OLE treatment initiation (fresh pustule count, total pustule count, PP-PASI scores, PPP-IGA, clearance on PPP-IGA and PASI) by original randomised treatment.
The week 8 outcomes of the participants originally randomised to the active group from the double-blind part of the trial were combined with the week 8 outcomes of participants originally randomised to the placebo group from the OLE to form a first-time exposure group. Descriptive statistics were presented for the first-time exposure group.
Adverse events were recorded for all participants in the OLE until the final follow-up visit.
No statistical testing was performed because of the open-label study design and because some participants commenced OLE anakinra treatment immediately following the week 12 visit (of the randomised double-blind placebo-controlled study), whereas others had previously completed the full double-blind trial schedule.
Trial oversight
Trial Management Group
The Trial Management Group (TMG) was chaired by Professor Catherine Smith (chief investigator of the study) and consisted of the co-applicants of the trial grant, a patient representative (Helen McAteer, Chief Executive of the Psoriasis Association) and the trial manager, and was responsible for decisions on the day-to-day running of the trial. The TMG provided the forum and mechanism through which the opinion of the central co-ordinating team (at Guy’s Hospital) and co-applicant on study matters was sought and discussed. The TMG met monthly and reported to the Trial Steering Committee (TSC) and the DMC.
Trial Steering Committee
The TSC comprised an independent chairperson (Professor Edel O’Toole, Queen Mary University of London), two independent members [Professor Hervé Bachelez, Consultant Dermatologist (with internationally recognised clinical and academic expertise in pustular forms of psoriasis), University Paris Diderot/Saint-Louis Hospital; and Dr Stephen Kelly, Consultant Rheumatologist, Barts Health NHS Trust], an independent patient representative (Mr David Britten), the chief investigator of the study (Professor Catherine Smith) and the trial statistician (Dr Victoria Cornelius, Imperial Clinical Trials Unit).
The TSC met as required and was the main decision-making body for the study. It had overall responsibility for scientific strategy and direction while also providing supervision and advice to study members.
Data Monitoring Committee
The DMC was chaired by an independent chairperson [Professor Deborah Symmons, Consultant Rheumatologist (who provided pharmacovigilance expertise in rheumatological interventions including anakinra), University of Manchester] and was responsible for monitoring evidence for treatment harm. The DMC also included an independent member (Dr Mike Ardern-Jones, University of Southampton), an independent statistician (Professor Simon Skene, University of Surrey), the chief investigator of the study (Professor Catherine Smith), the trial statisticians (Dr Suzie Cro, Imperial Clinical Trials Unit, and Dr Victoria Cornelius, Imperial Clinical Trials Unit) and the APRICOT trial manager. The DMC also collated data reports and reviewed all decisions pertaining to the safety aspects of the study.
The DMC met on initiation of the project and at specific study milestones thereafter, with the opportunity to convene extraordinary meetings to discuss SAEs if necessary.
Patient and public involvement
Patient and public involvement (PPI) has been central to the APRICOT study throughout its course. In this section, we summarise the different ways in which participants and the public have been involved and have had an impact on the study. A detailed description is provided in Appendix 7.
Aim
We convened patient and public partners, including people with pustular psoriasis and representatives from the Psoriasis Association (Northampton, UK), to provide input and support into all aspects of the study, including study design and ethics issues, patient support materials and questionnaires, delivery, results interpretation and communication of study outcomes.
Methods
From the outset (pre-funding preparation), Helen McAteer, Chief Executive of the Psoriasis Association, partnered with the study group as a co-applicant to ensure that we effectively engaged with participants and the public in the design, implementation, evaluation and communication of programme of research. She was also a member of the TSC and the TMG. When the outline application was being made, one-to-one discussions were held with participants (n = 3, two with APP requiring systemic therapy and personal experience of participating in placebo-controlled RCTs) to seek their advice on the study design and outcome measures. For the development of the full application, a formal Patient and Lay members Group (PLAG) meeting was held that consisted of one patient with APP, one patient with GPP, one patient with psoriasis, a NICE psoriasis guideline committee member, Helen McAteer, the Biomedical Research Centre PPI co-ordinator and (the chief investigator) Catherine Smith. A patient representative (David Britten) was part of the TSC and regularly attended and actively participated in these meetings to provide guidance and support to the APRICOT study. The APRICOT study was regularly mentioned at all of the PPI events held by the St John’s Institute of Dermatology (at Guy’s and St Thomas’ NHS Foundation Trust), and in Manchester and Newcastle. During these events, participants were asked for their feedback and suggestions about the trial experience (for themselves) and how it could potentially be improved. These findings were fed back to the central co-ordinating team for consideration by the TMG/TSC.
Impact of patient and public involvement
The study design involved a RCT with a placebo. The discussions held with participants about the outline application led to the decision to limit the trial treatment duration to 8 weeks and to extend the scope of the patient-orientated outcome measures for the study to include the pustular psoriasis-specific quality of life. The PLAG meeting shaped the trial design and led to amendments (e.g. the inclusion of rescue topical corticosteroid) that were applied to the full application prior to formal submission. With respect to samples for mechanistic studies, the PLAG considered and approved the planned sampling strategy, including skin biopsies.
During the trial, the removal of some visits in substantial amendment 4 (see Appendix 4) was heavily informed by PPI to help to boost study recruitment. Patient feedback suggested that some were concerned about missing out on treatment if they were in the placebo group, and this became a concern that was instrumental in devising and implementing the OLE to help to boost study recruitment. Furthermore, in response to feedback from the TSC patient representative, various sites and potential participants, staff at study sites were encouraged to ensure that it was made clear to all potential and actual participants that travel expenses would be reimbursed in full. This was important given that a number of participants had to travel significant distances to attend study visits.
Discussion and conclusions
The PPI in the APRICOT study enabled the study to recruit to target. The input gained from the PPI was important in designing the study and in shaping the trial once running. PPI was also crucial in the promotion of the trial, which ultimately generated study awareness and helped to enhance recruitment.
Reflections/critical perspective
The Psoriasis Association participated regularly in discussions about recruitment strategies and was extremely helpful in advertising the APRICOT study via social media, its magazine and its website. It helped to guide participants, directing any queries to the study website and e-mail address (where interested parties could self-refer). It also helped with the review and amendment of study materials. The PPI could have been enhanced by including more than one formal lay patient representative in the study infrastructure to more accurately reflect the PPP patient population and to enable more diverse conversations and guidance during the trial. A recent PPI event on psoriasis held during the COVID-19 pandemic over Zoom (Zoom Video Communications, San Jose, CA, USA) had more than 120 attendees; the question and answer format worked well, suggesting that virtual formats may be efficient and cost-effective ways of engaging a wider audience that would appeal to participants. We have opted to use this format to disseminate the findings once published and will also utilise the Psoriasis Association and its social medial channels to facilitate this.
Chapter 3 Results
Recruitment and participant flow
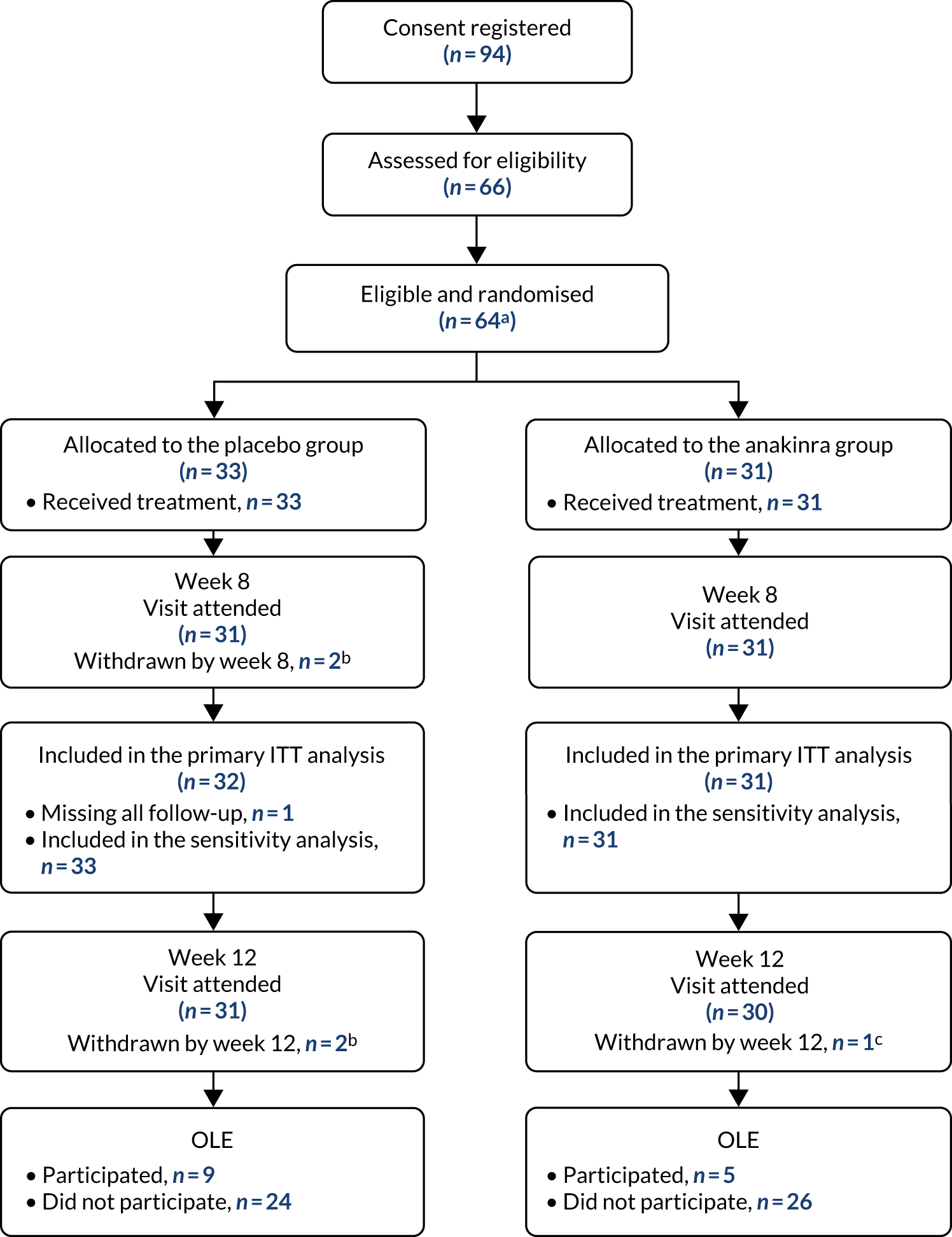
Between October 2016 and January 2020, a total of 64 eligible participants were enrolled: 33 were randomly allocated to the placebo group and 31 to the anakinra group (see Appendix 6, Tables 40 and 41, and Figure 20). An additional two consenting participants were randomised in error and never received any treatment and are excluded from all analysis. Figure 2 is the CONSORT flow chart for the trial, which summarises the participant flow through the trial. A total of 64 participants received treatment (placebo group, n = 33; anakinra group, n = 31), of whom 62 (placebo group, n = 31; anakinra group, n = 31) attended the week 8 visit and 61 attended the week 12 visit (placebo group, n = 31; anakinra group, n = 30). A total of 14 participants entered the OLE (placebo group, n = 9; anakinra group, n = 5).
FIGURE 2.
The CONSORT flow chart. a, An additional two participants were randomised, making a total of 66 randomised; however, these two participants were randomised in error, as they were ineligible. They were not offered treatment and were immediately withdrawn and excluded from all analysis. b, One participant withdrew in week 1. One participant was lost to follow-up and withdrawn post week 4. c, One participant withdrew at week 8. Note: numbers withdrawn from the trial are cumulative.
Stage 1
Recruitment began in October 2016 and the pre-planned interim stage 1 analysis was performed in January 2018 after the first 24 participants had been followed up for 8 weeks (placebo group, n = 13; anakinra group, n = 11). The unadjusted PP-PASI score averaged over weeks 1–8 was 16.2 points (SD 11.1 points) in the placebo group and 12.9 points (SD 7.9 points) in the anakinra group. The baseline-adjusted treatment group difference in PP-PASI scores averaged over weeks 1–8 was –1.2 points (95% CI –5.5 to 3.1 points), and the point estimate was in favour of anakinra. The unadjusted fresh pustule count averaged over weeks 1–8 was 47.2 points (SD 59.4 points) in the placebo group and 61.6 points (SD 76.9 points) in the anakinra group. The baseline-adjusted treatment group difference in the fresh pustule count averaged over weeks 1–8 was 16.5 points (95% CI –51.0 to 49.6 points), and the point estimate was in favour of placebo. Given that one outcome was in favour of anakinra (PP-PASI score), the trial met the criteria to progress to stage 2.
The mean difference in agreement between the fresh pustule count assessed at sites and the fresh pustule count assessed centrally using photography was 27 pustules (95% CI –131 to 185 pustules). The ICC was used as a measure of agreement for fresh pustule count between the site assessor and the photographic central assessor, and was found to be 0.13 pustules. The mean difference in agreement in PP-PASI scores between the first and the second site assessors was –0.56 points (95% CI –5.9 to 4.8 points). The ICC for fresh pustule count between two independent site assessors was 0.73 pustules. Overall, the DMC decided unanimously to recommend PP-PASI score as the primary outcome variable for stage 2 because this outcome was judged to be more reliable than fresh pustule count; the agreement (ICC) between the two PP-PASI independent assessors was considerably higher (0.73 points) than the agreement between the site and the central photo fresh pustule count assessments (0.13 points). Additional results from stage 1, including the unadjusted standardised mean differences between the treatment groups for the stage 1 outcomes by time point, can be found in Appendix 9, Table 42, and Figures 23 and 24.
Baseline characteristics
Table 1 summarises baseline demographics by randomised group. The baseline characteristics of the placebo and anakinra treatment groups were generally well matched, including demographics and the severity and impact of participants’ disease.
| Baseline demographic | Treatment group | Total (N = 64) | |
|---|---|---|---|
| Placebo (N = 33) | Anakinra (N = 31) | ||
| Age (years), mean (SD) | 51.7 (13.6) | 49.9 (11.9) | 50.8 (12.7) |
| Sex, n (%) | |||
| Male | 6 (18) | 4 (13) | 10 (16) |
| Female | 27 (82) | 27 (87) | 54 (84) |
| Ethnicity, n (%) | |||
| White | 31 (94) | 28 (90) | 59 (92) |
| Asian/Asian British | 1 (3) | 1 (3) | 2 (3) |
| Black/black British | 0 (0) | 1 (3) | 1 (2) |
| Chinese/Japanese/Korean/Indochinese | 0 (0) | 1 (3) | 1 (2) |
| Other | 1 (3) | 0 (0) | 1 (2) |
| Smoker, n (%) | |||
| Current smoker | 19 (58) | 16 (52) | 35 (55) |
| Ex-smoker | 9 (27) | 12 (39) | 21 (33) |
| Non-smoker | 5 (15) | 3 (10) | 8 (13) |
| PP-PASI score | |||
| Mean (SD) | 18.0a (10.4) | 17.5 (10.8) | 17.8 (10.5) |
| Median (IQR) | 15.9 (10.4–21.3) | 15.4 (11.7–20.7) | 15.6 (10.6–21.0) |
| Fresh pustule count (palms and soles) | |||
| Mean (SD) | 36.1 (33.1) | 39.8† (46.3) | 37.9 (39.6) |
| Median (IQR) | 28.0 (18.0–45.0) | 25.5 (11.0–58.0) | 27.0 (15.0–49.0) |
| Fresh pustule count (soles) | |||
| Mean (SD) | 25.9 (23.4) | 29.6a (43.2) | 27.7 (34.1) |
| Median (IQR) | 23.0 (4.0–36.0) | 15.0 (5.0–37.0) | 19.0 (4.0–37.0) |
| Fresh pustule count (palms) | |||
| Mean (SD) | 10.2 (19.2) | 10.2a (16.5) | 10.2 (17.8) |
| Median (IQR) | 2.0 (0.0–13.0) | 2.5 (0.0–13.0) | 2.0 (0.0–13.0) |
| Total pustule count (palms and soles) | |||
| Mean (SD) | 116.9 (96.4) | 154.3a (198.7) | 134.7 (153.7) |
| Median (IQR) | 97.0 (45.0–169.0) | 89.0 (45.0–157.0) | 95.0 (45.0–169.0) |
| PPP-IGA,b n (%) | |||
| Moderate | 16 (48) | 16 (52) | 32 (50) |
| Severe | 17 (52) | 15 (48) | 32 (50) |
| Participant global assessment, n (%) | |||
| Almost clear | 0 (0) | 2 (6) | 2 (3) |
| Mild | 3 (9) | 3 (10) | 6 (9) |
| Moderate | 14 (42) | 14 (45) | 28 (44) |
| Severe | 13 (39) | 7 (23) | 20 (31) |
| Very severe | 3 (9) | 5 (16) | 8 (13) |
| DLQI | |||
| Mean (SD) | 13.9 (7.2) | 15.1 (7.0) | 14.5 (7.1) |
| PASIc | |||
| Mean (SD) | 2.1 (5.4) | 1.1 (1.6) | 1.6 (4.1) |
| Median (IQR) | 0.0 (0.0–1.8) | 0.2 (0.0–1.6) | 0.0 (0.0–1.6) |
| PP-QoL | |||
| Mean (SD) | 46.4 (13.8) | 45.5 (14.8) | 46.0 (14.2) |
| EQ-5D utility score | |||
| Mean (SD) | 0.37 (0.43) | 0.47 (0.35) | 0.42 (0.40) |
| Median (IQR) | 0.62 (0.09–0.73) | 0.62 (0.16–0.73) | 0.62 (0.09–0.73) |
| EQ-5D-VAS score | |||
| Mean (SD) | 57.7 (27.7) | 68.4d (18.3) | 62.5 (24.4) |
| Median (IQR) | 65.0 (45.0–80.0) | 75.0 (55.0–80.0) | 70.0 (50.0–80.0) |
The mean age of the participants was 50.8 years; 84% were female and 92% were of white ethnicity. Current smokers made up 55% of participants and ex-smokers made up 22%. The mean disease severity at baseline, as measured by the PP-PASI, was 17.8 points (SD 10.5 points). The median fresh pustule count including both the palms and soles was 27.0 pustules (IQR 15.0–49.0 pustules) and total pustule count across the palms and soles was 9.0 pustules (IQR 45.0–169.0 pustules).
Withdrawals from treatment and from the study
During the trial, a total of 11 participants (17%) withdrew from study treatment: six from the placebo group and five from the anakinra group (Table 2).
| Time of and reason for withdrawala | Number of withdrawals (%) | Total (N = 64) | |
|---|---|---|---|
| Placebo group (N = 33) | Anakinra group (N = 31) | ||
| Point of treatment discontinuation | |||
| Baseline (n = 64) | 0 (0) | 0 (0) | 0 (0) |
| Week 1 (n = 63) | 1 (3) | 0 (0) | 1 (2) |
| Week 2 (n = 63) | 0 (0) | 1 (3) | 1 (2) |
| Week 3 (n = 63) | 0 (0) | 0 (0) | 0 (0) |
| Week 4 (n = 63) | 3 (9) | 3 (10) | 6 (9) |
| Week 5 (n = 63) | 1 (3) | 0 (0) | 1 (2) |
| Week 6 (n = 62) | 1 (3) | 1 (3) | 2 (3) |
| Week 7 (n = 62) | 0 (0) | 0 (0) | 0 (0) |
| Week 8 (n = 62) | 0 (0) | 0 (0) | 0 (0) |
| Reason for permanent trial treatment discontinuation | |||
| AE | 1b (3) | 4c (13) | 5 (8) |
| Withdrawal of consent | 2 (6) | 1 (3) | 3 (5) |
| Lack of response | 2 (6) | 0 (0) | 2 (3) |
| Condition worsening wants other treatment | 1 (3) | 0 (0) | 1 (2) |
| Total (n = 64) | 6 (18) | 5 (16) | 11 (17) |
Retention in the study was high (see Figure 2). Only three participants (5%) who withdrew from treatment also withdrew entirely from the study. One participant who withdrew from treatment in the placebo group prior to the end of week 1 did not attend any further follow-up appointments and was withdrawn from the study because of non-compliance with the visit schedule. Two further participants who withdrew from treatment early continued in the trial immediately following treatment cessation, but were later withdrawn: one placebo participant was withdrawn post week 4 prior to week 8 due to loss to follow-up and one anakinra participant was withdrawn at the week 8 visit prior to week 12 due to a wish to start other therapies.
Temporary treatment discontinuations, after which trial treatment was recommenced, occurred more frequently in the anakinra group than in the placebo group. There was a total of nine participants recorded to have temporarily discontinued treatment, with 3 out of 33 (9%) in the placebo group and 6 out of 31 (19%) in the anakinra group (Table 3). Temporary discontinuations were mainly as a result of AEs, except for one discontinuation in the anakinra group from an individual who wanted to start other treatments because their condition was worsening. The larger number of discontinuations in the anakinra group was driven by injection site reactions. The temporary treatment numbers include one participant in the placebo group and one in the anakinra group who later permanently withdrew from treatment and who are also included above.
| Time of and reason for temporary treatment discontinuationa | Number of discontinuations (%) | Total (N = 64) | |
|---|---|---|---|
| Placebo group (N = 33) | Anakinra group (N = 33) (N = 31) | ||
| First point of treatment discontinuation | |||
| Baseline (n = 64) | 0 (0) | 0 (0) | 0 (0) |
| Week 1 (n = 63) | 1b (3) | 1c (3) | 2 (3) |
| Week 2 (n = 63) | 0 (0) | 2d (6) | 2 (3) |
| Week 3 (n = 63) | 1 (3) | 2e,f (6) | 3 (5) |
| Week 4 (n = 63) | 0 (0) | 0 (0) | 0 (0) |
| Week 5 (n = 63) | 0 (0) | 0 (0) | 0 (0) |
| Week 6 (n = 62) | 1 (3) | 1g (3) | 2 (3) |
| Week 7 (n = 62) | 0 (0) | 0 (0) | 0 (0) |
| Week 8 (n = 62) | 0 (0) | 0 (0) | 0 (0) |
| Reason | |||
| AE | 3h (9) | 5i (16) | 8 (13) |
| Condition worsening wants other treatment | 0 (0) | 1 (3) | 1 (2) |
| Total | 3 (9) | 6 (19) | 9 (14) |
Adherence to treatment
Adherence to trial treatment was recorded by using the responses to daily text messages [from a short message service (SMS)], self-reporting from participants using a paper trial diary (issued at the baseline visit and checked at each study visit) and verbal self-recall at study visits. For those who used the SMS service (see Table 4), a daily SMS was sent out to enquire about whether or not the participant had administered their dose that day and required a response of ‘yes’. Unfortunately, an operational incident (which was discovered in January 2019 and was rectified during mid-March 2019) meant that the SMS service was not utilised by all participants (six participants were affected during this period). After excluding the known treatment withdrawals using SMS (Table 4), out of a maximum of seven injections per week, the placebo group reported, on average, 5.7 injections per week at both week 1 and week 8, and the anakinra group reported, on average, 5.8 injections per week at week 1 and 5.4 injections per week at week 8.
Table 4 also summarises the average number of injections received weekly, as self-reported at each clinical visit; these adherence data were self-reported using either a paper trial diary or verbal self-recall: it was not possible to separate the two methods of measurement. For those with self-reported adherence data (see Table 4), after excluding withdrawals, an average of 6.5 injections was reported per week in week 1 and 6.4 in week 8 in the placebo group, compared with 6.9 at week 1 and 6.7 at week 8 in the anakinra group. When combining the SMS and self-recalled adherence data (taking the mean weekly adherence where both measurements were reported per participant), reported adherence was similar in both groups, with the average number of injections received per week being 6.3 at week 1 and 6.2 at week 8 in the placebo group and 6.7 at week 1 and 6.4 at week 8 (after excluding withdrawals) in the anakinra group. However, when including the withdrawals data from the participants who were known to receive no injections following treatment withdrawal or during temporary withdrawal periods to get an overall picture of adherence per week (Table 5), the adherence was a little lower in the placebo group than in the anakinra group: an average of 6.1 injections per week at week 1 and 4.8 injections per week at week 8 in the placebo group, compared with 6.7 at week 1 and 5.3 at week 8 in the anakinra group.
| Treatment perioda | Number of doses per week | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SMS datab | Self-reported datac | Mean of SMS and self-reported data | ||||||||||
| Placebo group | Anakinra group | Placebo group | Anakinra group | Placebo group | Anakinra group | |||||||
| n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | |
| Week 1 (N = 63) | 10 | 5.7 (1.8) | 13 | 5.8 (1.4) | 28 | 6.5 (1.5) | 29 | 6.9 (0.3) | 29 | 6.3 (1.6) | 29 | 6.7 (0.6) |
| Week 2 (N = 60) | 12 | 6.3 (1.2) | 11 | 6.5 (0.9) | 27 | 6.4 (1.8) | 27 | 6.9 (0.6) | 28 | 6.4 (1.5) | 27 | 6.8 (0.6) |
| Week 3 (N = 57) | 12 | 6.5 (0.7) | 10 | 6.5 (0.7) | 26 | 6.6 (1.4) | 24 | 6.8 (0.5) | 28 | 6.6 (1.4) | 24 | 6.8 (0.5) |
| Week 4 (N = 58) | 12 | 6.3 (1.8) | 13 | 6.2 (0.8) | 26 | 6.6 (1.5) | 25 | 6.9 (0.4) | 28 | 6.5 (1.5) | 25 | 6.7 (0.5) |
| Week 5 (N = 53) | 12 | 6.3 (1.4) | 13 | 6.5 (0.9) | 24 | 6.7 (1.4) | 23 | 7.0 (0.0) | 26 | 6.5 (1.4) | 23 | 6.9 (0.3) |
| Week 6 (N = 51) | 12 | 6.3 (1.2) | 12 | 5.9 (1.8) | 22 | 6.5 (1.5) | 22 | 7.0 (0.2) | 24 | 6.4 (1.5) | 22 | 6.7 (0.7) |
| Week 7 (N = 50) | 12 | 5.8 (2.0) | 12 | 6.1 (1.4) | 22 | 6.5 (1.6) | 23 | 7.0 (0.2) | 24 | 6.4 (1.7) | 23 | 6.7 (0.6) |
| Week 8 (N = 51) | 12 | 5.7 (1.9) | 12 | 5.4 (2.0) | 22 | 6.4 (1.8) | 24 | 6.7 (1.0) | 24 | 6.2 (1.8) | 24 | 6.4 (1.2) |
| Treatment period (N = 64) | Number of doses per week (mean of SMSa and self-reported datab) | |||
|---|---|---|---|---|
| Placebo group (N = 33)c | Anakinra group (N = 31)d | |||
| n | Mean (SD) | n | Mean (SD) | |
| Week 1 | 30 | 6.1 (1.9) | 29 | 6.7 (0.6) |
| Week 2 | 30 | 5.9 (2.2) | 29 | 6.7 (0.8) |
| Week 3 | 30 | 6.2 (1.9) | 29 | 5.9 (2.1) |
| Week 4 | 30 | 6.2 (2.1) | 29 | 5.9 (2.1) |
| Week 5 | 30 | 5.7 (2.6) | 29 | 5.7 (2.5) |
| Week 6 | 31 | 5.1 (2.9) | 29 | 5.3 (2.7) |
| Week 7 | 31 | 4.9 (3.1) | 29 | 5.5 (2.6) |
| Week 8 | 31 | 4.8 (3.1) | 29 | 5.3 (2.7) |
Given that the number of participants with data on adherence available varied by week, we calculated an overall level of adherence per participant to further explore the overall levels of adherence across the 8-week treatment period. For each participant, using the obtained weekly data on adherence, an overall measure of total adherence was calculated as the proportion of injections received relative to the total number of injections planned over the 8-week treatment period (total planned = 8 × 7 = 56). See Table 14 for a summary of overall compliance levels based on receiving 50–90% of the planned injections. At least 50% of the total planned injections were received by 79% of the placebo group and 81% of the anakinra group. At least 90% of the total planned injections were received by 61% of the placebo group and 48% of the anakinra group.
Non-randomised treatment use
Rescue therapies are listed in Appendix 3, Rescue therapy. Over the 8-week treatment period, eight participants in the placebo group and 11 in the anakinra group were initiated on rescue therapy. After the treatment period, an additional three participants in the placebo group and one in the anakinra group were initiated on rescue therapy prior to week 12 (Table 6). The proportion starting rescue medication was similar in both groups, but more participants in the anakinra group than in the placebo group received this earlier.
| Details of rescue therapy | Number of participants (%) | Total (N = 64) | |
|---|---|---|---|
| Placebo group (N = 33) | Anakinra group (N = 31) | ||
| Time of rescue medication initiation | |||
| Baseline | 0 (0) | 2 (6) | 2 (3) |
| Prior to week 1 visit | 0 (0) | 0 (0) | 0 (0) |
| Prior to week 4 visit | 3 (9) | 8 (26) | 11 (17) |
| Prior to week 8 visit | 5 (15) | 1 (3) | 6 (9) |
| Prior to week 12 visit | 3 (9) | 1 (3) | 4 (6) |
| Type of rescue medication initiated | |||
| Moderately potent corticosteroid | |||
| Clobetasone butyrate (Eumovate®, GlaxoSmithKline, London, UK) | 1 (3) | 0 (0) | 1 (2) |
| Potent corticosteroid | |||
| Betamethasone valerate (Betnovate®, GlaxoSmithKline UK)a | 5 (15) | 5 (16) | 10 (16) |
| Betamethasone dipropionate and salicylic acid (Diprosalic™, Organon Pharma UK Ltd, London, UK) | 1 (3) | 0 (0) | 1 (2) |
| Mometasone furoate (Elocon®, Organon Pharma UK Ltd) | 5 (15) | 8 (26) | 13 (20) |
| Total number of participantsb | 11 (33) | 12 (39) | 23 (36) |
Over the 8-week treatment period, three (9%) participants in the placebo group and three (10%) in the anakinra group were initiated on prohibited therapy. After the treatment period, an additional two participants in the placebo group and two in the anakinra group were initiated on prohibited therapy prior to week 12 (Table 7).
| Details of prohibited medication initiation | Number of participants (%) | Total (N = 64) | |
|---|---|---|---|
| Placebo group (N = 33) | Anakinra group (N = 31) | ||
| Time of prohibited medication initiation | |||
| Baseline | 0 (0) | 0 (0) | 0 (0) |
| Prior to week 1 visit | 0 (0) | 0 (0) | 0 (0) |
| Prior to week 4 visit | 0 (0) | 0 (0) | 0 (0) |
| Prior to week 8 visit | 3 (9)a | 3 (10)b | 6 (9) |
| Prior to week 12 visit | 2 (6) | 2 (6) | 4 (6) |
| Type of prohibited medication initiated | |||
| Topical super-potent corticosteroid | |||
| Clobetasol propionate | 3 (9) | 4 (13) | 7 (11) |
| Systemic therapy | |||
| Acitretin | 2 (6) | 2 (6) | 4 (6) |
| Ciclosporin | 0 (0) | 1 (3) | 1 (2) |
| Prednisolone | 1 (3) | 0 (0) | 1 (2) |
| Total number of participantsc | 5 (15) | 5 (16) | 10 (16) |
A larger number of ‘other topical treatments’ (excluding topical rescue therapy and prohibited treatments) were used by participants in the anakinra group (n = 12) than by participants in the placebo group (n = 7) over the 8-week treatment period (Table 8). Following the treatment period, an additional one (3%) participant in the placebo group and one (3%) in the anakinra group were recorded as having initiated other topical therapy prior to week 12. A summary of the other topical treatments used is given in Table 8. The use of emollients may not have been accurately reported because it was captured by self-recall on a concomitant medication form. It is likely that the use of emollients reported is an underestimate. Furthermore, the site of use for all other topical treatments, as site of PPP or otherwise (e.g. for injection site reactions), cannot be distinguished for these reported uses. For simplicity, we have included all of the topical treatment uses reported on the concomitant medication data form. Therefore, these data on other topical treatments should be interpreted cautiously.
| Details of initiation of other topical therapy | Number of participants (%) | Total (N = 64) | |
|---|---|---|---|
| Placebo group (N = 33) | Anakinra group (N = 31) | ||
| Time of initiation of other topical therapy | |||
| Baseline | 1 (3) | 1 (3) | 2 (3) |
| Prior to week 1 visit | 0 (0) | 1 (3) | 1 (2) |
| Prior to week 4 visit | 4 (12) | 8 (26) | 12 (19) |
| Prior to week 8 visit | 2 (6) | 2 (6) | 4 (6) |
| Prior to week 12 visit | 1 (3) | 1 (3) | 2 (3) |
| Type of topical therapy initiated | |||
| Antiseptics, antibiotics or antifungals | |||
| Naseptin | 1 (3) | 0 (0) | 1 (2) |
| Nystatin | 0 (0) | 1 (3) | 1 (2) |
| Mild corticosteroids | |||
| Hydrocortisone | 1 (3) | 7 (23) | 8 (13) |
| Clobetasone 17-butyrate [with oxytetracycline and nystatin (Trimovate™)] | 0 (0) | 1 (3) | 1 (2) |
| Potent corticosteroids | |||
| Betamethasone valerate | 1 (3) | 2 (6) | 3 (5) |
| Emollients | |||
| Cetraban | 1 (3) | 1 (3) | 2 (3) |
| Dermol | 1 (3) | 1 (3) | 2 (3) |
| Doublebase | 0 (0) | 1 (3) | 1 (2) |
| E45 | 1 (1) | 0 (0) | 1 (2) |
| Epaderm | 3 (9) | 2 (6) | 5 (8) |
| Totala | 8 (24) | 13 (42) | 21 (33) |
Loss to follow-up and missing data
Loss to follow-up was low (see Figure 2). Appendix 10, Table 43, summarises the completeness of the primary PP-PASI scores outcome. Only four participants had missing primary end-point data at week 8 (6%), which is lower than the 15% factored into the sample size calculation. This meant that a total of 60 (94%) participants provided week 8 PP-PASI (primary outcome) data.
Primary outcome: Palmoplantar Pustulosis Psoriasis Area Severity Index
Figures 3 and 4 display the individual participant PP-PASI profiles over time by treatment group. Figure 5 and Table 9 summarise the mean PP-PASI score outcome by time point and treatment group, with the unadjusted mean treatment group differences. In both treatment groups, the mean PP-PASI score was lower at week 8 than at baseline. The unadjusted mean difference in PP-PASI scores at week 8 for those in the anakinra group compared with those in the placebo group was –1.4 points (95% CI –6.0 to 3.2 points), for which the point estimate was in favour of anakinra.
FIGURE 3.
Placebo participant PP-PASI profiles over time. The raw PP-PASI values for each participant are plotted on the y-axis against the time point on the x-axis.
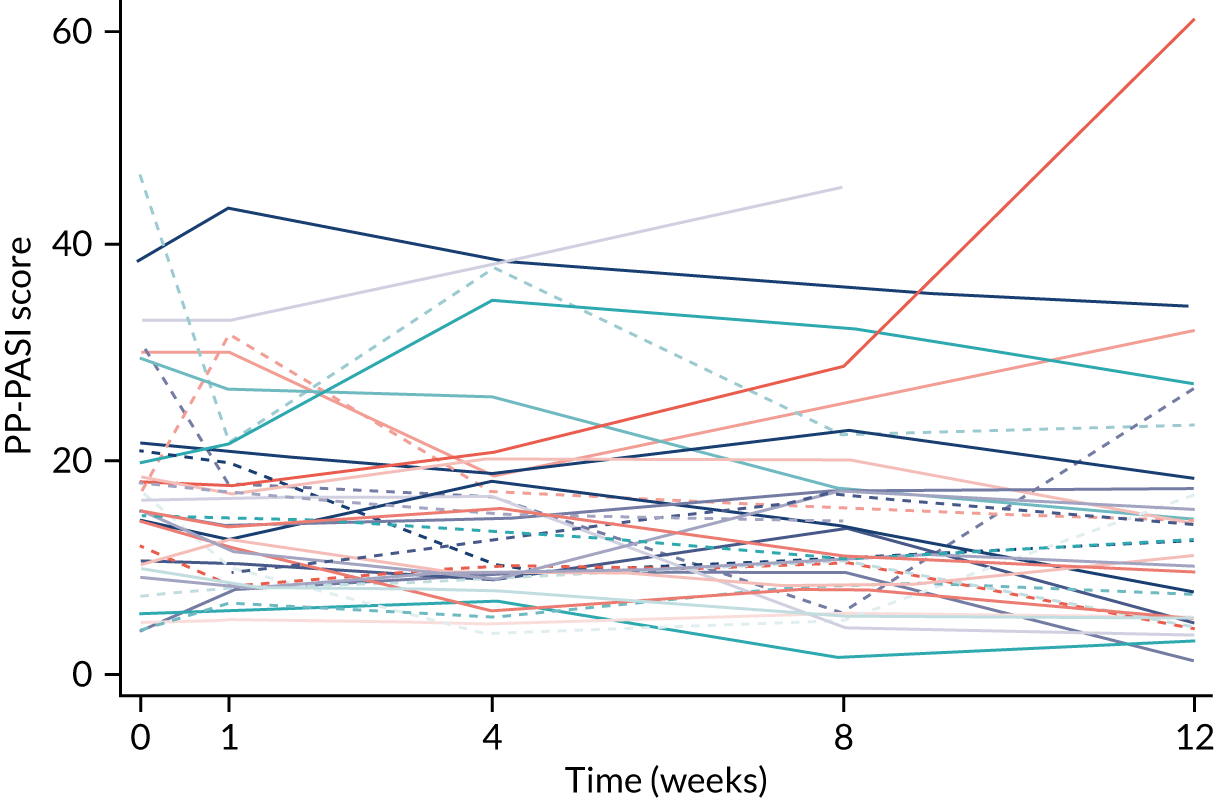
FIGURE 4.
Anakinra participant PP-PASI profiles over time. The raw PP-PASI values for each participant are plotted on the y-axis against the time point on the x-axis.
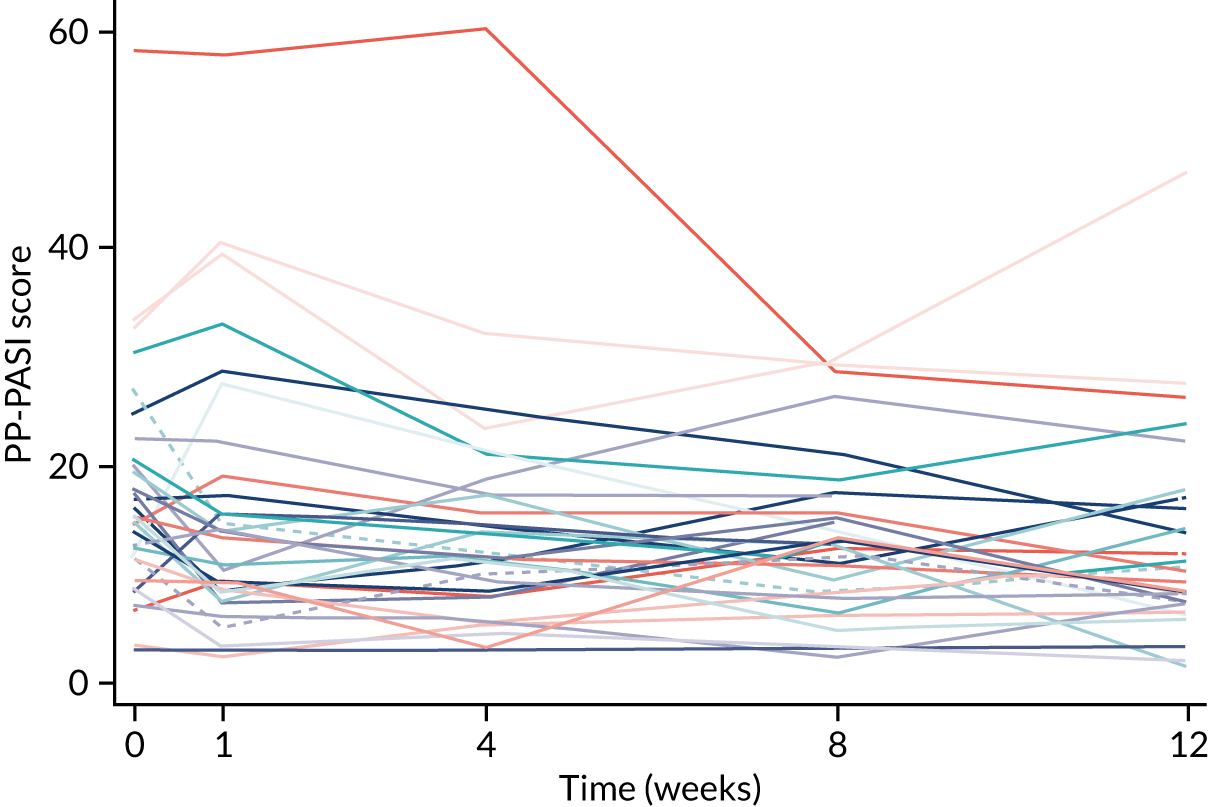
FIGURE 5.
The PP-PASI over the 12-week follow-up period. The unadjusted mean PP-PASI is plotted on the y-axis, against the time point on the x-axis for each treatment group. The error bars represent 95% CIs for the unadjusted treatment group means.
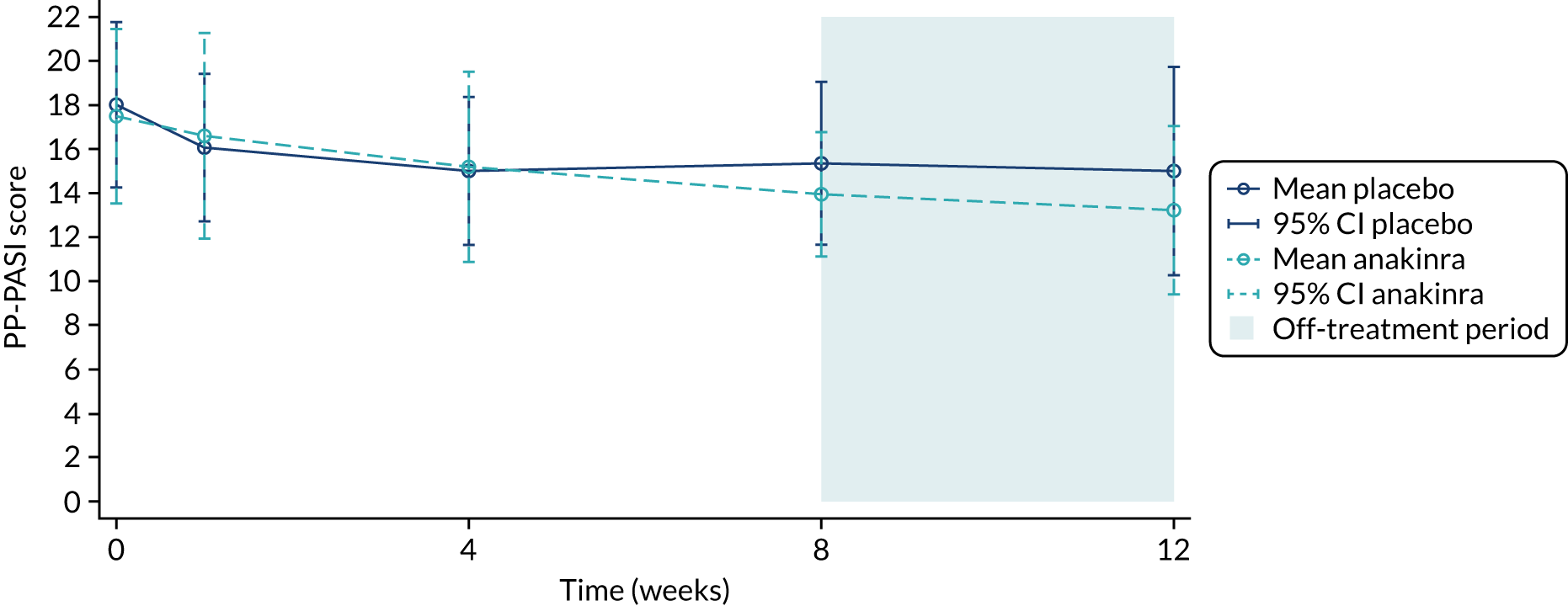
| Time point | Placebo group (N = 33) | Anakinra group (N = 31) | Total (n) | Unadjusted mean difference (anakinra – placebo) (95% CI) | ||
|---|---|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | |||
| Baseline | 32 | 18.0 (10.4) | 31 | 17.5 (10.8) | 63 | – |
| Week 1 | 31 | 16.1 (9.1) | 30 | 16.6 (12.5) | 61 | 0.5 (–5.1 to 6.1) |
| Week 4 | 31 | 15.0 (9.1) | 28 | 15.2 (11.1) | 59 | 0.2 (–5.1 to 5.5) |
| Week 8 | 31 | 15.4 (10.1) | 29 | 13.9 (7.4) | 60 | –1.4 (–6.0 to 3.2) |
| Week 12 | 29 | 15.0 (12.4) | 27 | 13.2 (9.7) | 56 | –1.8 (–7.8 to 4.2) |
Primary analysis
The primary analysis included a total of 62 participants who had at least one post-baseline follow-up (placebo group, n = 32; anakinra group, n = 30). A mixed linear regression model, including a random intercept for participant and centre, was used to adjust the week 8 treatment group difference for baseline PP-PASI scores. The adjusted mean treatment group difference in the week 8 PP-PASI scores for those in the anakinra group compared with those in the placebo group was –1.65 points (95% CI –4.77 to 1.47 points; p = 0.300). The adjusted mean treatment group difference in the week 4 PP-PASI scores for those in the anakinra group compared with those in the placebo group was –0.72 points (95% CI –3.86 to 2.43 points) and the adjusted mean treatment group difference in the week 1 PP-PASI scores for those in the anakinra group compared with those in the placebo group was 0.22 points (95% CI –2.88 to 3.31 points) (see Appendix 10, Figures 25 and 26).
Sensitivity analysis
A sensitivity analysis was conducted to explore the impact of the missing data on the primary PP-PASI scores outcome. In each treatment group, one participant was missing PP-PASI follow-up data over weeks 1–8; in addition, in each treatment group, one participant was missing week 8 data only and one participant in the placebo group and two in the anakinra group were missing interim week 4 follow-up data (see Appendix 10, Table 43). The primary analysis assumes that the missing responses are MAR conditional on the variables included in the model: treatment group, observed PP-PASI scores at baseline, weeks 1, 4 and 8, and centre. The sensitivity analysis explored the robustness of the primary analysis results to various MNAR assumptions.
The estimated intervention effect (the baseline adjusted mean treatment group difference in PP-PASI scores at week 8) did not change when it was assumed that the rate of change of the PP-PASI score between the observed and the unobserved cases for each week unobserved was 0.039 to 0.39 points worse (a higher PP-PASI score outcome) than that predicted under MAR (Table 10), corresponding to outcomes that were worse by 0–100% of the unadjusted mean rate of change in the PP-PASI scores observed over 8 weeks.
| Analysis | Mean treatment group difference in week 8 PP-PASI scores (points), anakinra – placebo (95% CI) | p-value |
|---|---|---|
| Primary analysis | ||
| MAR using mixed model (n = 62) | –1.65 (–4.77 to 1.47) | 0.300 |
| MAR using MI (n = 64) | –1.66 (–5.11 to 1.79) | 0.346 |
| MNARa sensitivity analysis (n = 64) | ||
| MNAR using MI – MAR + 10% × δ | –1.66 (–5.11 to 1.80) | 0.347 |
| MNAR using MI – MAR + 20% × δ | –1.66 (–5.11 to 1.80) | 0.348 |
| MNAR using MI – MAR + 30% × δ | –1.65 (–5.11 to 1.80) | 0.348 |
| MNAR using MI – MAR + 40% × δ | –1.65 (–5.11 to 1.81) | 0.349 |
| MNAR using MI – MAR + 50% × δ | –1.65 (–5.11 to 1.81) | 0.350 |
| MNAR using MI – MAR + 60% × δ | –1.65 (–5.11 to 1.81) | 0.350 |
| MNAR using MI – MAR + 70% × δ | –1.65 (–5.11 to 1.82) | 0.351 |
| MNAR using MI – MAR + 80% × δ | –1.65 (–5.11 to 1.82) | 0.352 |
| MNAR using MI – MAR + 90% × δ | –1.65 (–5.11 to 1.82) | 0.352 |
| MNAR using MI – MAR + 100% × δ | –1.64 (–5.11 to 1.83) | 0.353 |
| MNARa in placebo group (n = 64) | ||
| MNAR using MI – MAR + 10% × δ | –1.67 (–5.13 to 1.78) | 0.343 |
| MNAR using MI – MAR + 20% × δ | –1.69 (–5.14 to 1.77) | 0.339 |
| MNAR using MI – MAR + 30% × δ | –1.70 (–5.16 to 1.76) | 0.335 |
| MNAR using MI – MAR + 40% × δ | –1.71 (–5.17 to 1.74) | 0.331 |
| MNAR using MI – MAR + 50% × δ | –1.73 (–5.19 to 1.73) | 0.328 |
| MNAR using MI – MAR + 60% × δ | –1.74 (–5.20 to 1.72) | 0.324 |
| MNAR using MI – MAR + 70% × δ | –1.75 (–5.22 to 1.71) | 0.321 |
| MNAR using MI – MAR + 80% × δ | –1.77 (–5.23 to 1.70) | 0.318 |
| MNAR using MI – MAR + 90% × δ | –1.78 (–5.25 to 1.69) | 0.314 |
| mnar using mi – mar + 100% × δ | –1.78 (–5.26 to 1.70) | 0.315 |
| MNARa in anakinra group (n = 64) | ||
| MNAR using MI – MAR + 10% × δ | –1.64 (–5.10 to 1.81) | 0.351 |
| MNAR using MI – MAR + 20% × δ | –1.63 (–5.08 to 1.82) | 0.355 |
| MNAR using MI – MAR + 30% × δ | –1.61 (–5.07 to 1.84) | 0.360 |
| MNAR using MI – MAR + 40% × δ | –1.60 (–5.05 to 1.86) | 0.365 |
| MNAR using MI – MAR + 50% × δ | –1.58 (–5.04 to 1.87) | 0.369 |
| MNAR using MI – MAR + 60% × δ | –1.57 (–5.02 to 1.89) | 0.374 |
| MNAR using MI – MAR + 70% × δ | –1.55 (–5.01 to 1.90) | 0.378 |
| MNAR using MI – MAR + 80% × δ | –1.54 (–4.99 to 1.92) | 0.382 |
| MNAR using MI – MAR + 90% × δ | –1.52 (–4.98 to 1.93) | 0.387 |
| MNAR using MI – MAR + 100% × δ | –1.51 (–4.96 to 1.95) | 0.392 |
Supplementary analysis
Treatment effect if no rescue therapy was used
As summarised in Table 6, a total of 19 participants, (placebo group, n = 8; anakinra group, n = 11) were initiated on rescue therapy some time prior to week 8. Data collection continued post rescue initiation for all 19 rescued participants, and all recorded data post rescue initiation were used in the primary analysis, in keeping with the ITT principle. Appendix 10, Table 44, summarises the proportions of observed data included in the primary analysis by rescued status.
To estimate the treatment effect in those not receiving rescue therapy, an analysis was performed in which all data collected after starting rescue therapy were set to be missing. We then examined a series of different assumptions as to what the data could have looked like in the absence of rescue therapy use. First, an assumption of MAR was made for post-rescued data, which provided an estimate of the treatment effect under the assumption that those rescued would have had data similar to those who were not rescued, in the absence of rescue initiation. This treatment effect was slightly larger than the main ITT analysis, being –2.30 (95% CI –7.48 to 2.87) compared with the ITT analysis of –1.65 (95% CI –4.77 to 1.47). Subsequently, MNAR assumptions were made and these assumed that participants would have had progressively worse outcomes had they not used rescue medication relative to those observed (not rescued). Compared with the main ITT effect, the results under the MNAR assumption also found a slightly larger treatment effect for those in the anakinra group than for those in the placebo group on the baseline-adjusted week 8 PP-PASI scores (Table 11).
| Analysis | Mean treatment group difference in week 8 PP-PASI scores (points), anakinra – placebo (95% CI) | p-value |
|---|---|---|
| MAR | ||
| MAR using MI (n = 64) | –2.30 (–7.48 to 2.87) | 0.381 |
| MNARa (n = 64) | ||
| MNAR using MI – MAR + 10% × δ | –2.26 (–7.43 to 2.92) | 0.390 |
| MNAR using MI – MAR + 20% × δ | –2.21 (–7.39 to 2.96) | 0.400 |
| MNAR using MI – MAR + 30% × δ | –2.17 (–7.34 to 3.10) | 0.410 |
| MNAR using MI – MAR + 40% × δ | –2.12 (–7.29 to 3.06) | 0.422 |
| MNAR using MI – MAR + 50% × δ | –2.07 (–7.25 to 3.11) | 0.431 |
Treatment effect if no rescue or prohibited therapy was used
As summarised in Table 7, six participants, three in each group, were initiated on prohibited therapy some time prior to week 8 and post week 4. This included three participants who were also initiated on rescue treatment prior to week 8, (placebo group, n = 2; anakinra group, n = 1) and three participants who were not previously initiated on rescue treatment, (placebo group, n = 1; anakinra group, n = 2). Of the six participants who were started on prohibited treatments, five (placebo group, n = 3; anakinra group, n = 2) had follow-up data at week 8 that were recorded post initiation of prohibited therapy and included in the primary analysis following the ITT principle. Appendix 10, Table 45, summarises the proportions of data included in the primary analysis by use of rescue or prohibited therapy compared with the use of neither therapy.
As with the prohibitive medication analysis above, to estimate the treatment effect in the absence of use of rescue or prohibitive therapy, we set all data collected post initiation of rescue therapy or prohibited therapy to be missing and examined a series of assumptions as to what the data could look like in the absence of rescue or prohibited therapy. First, an assumption of MAR was made for post-rescue/prohibited data, which provided an estimate of the treatment effect under the assumption that those rescued or started on prohibited therapy would have had data similar to those who were not rescued or started on prohibited therapy. This effect (–2.09, 95% CI –8.47 to 4.29) was, on average, larger than the main ITT analysis (–1.65, 95% CI –4.77 to 1.47). Subsequently, MNAR assumptions were made that assumed that progressively worse outcomes would have been observed in the absence of rescue or prohibited therapy initiation, relative to those observed. In these MNAR analyses, the treatment effects were also slightly larger than the main ITT effect (Table 12).
| Analysis | Mean treatment group difference in week 8 PP-PASI score (points), anakinra – placebo (95% CI) | p-value |
|---|---|---|
| MAR | ||
| MAR using MI (n = 64) | –2.09 (–8.47 to 4.29) | 0.518 |
| MNARa (n = 64) | ||
| MNAR using MI – MAR + 10% × δ | –2.04 (–8.42 to 4.35) | 0.528 |
| MNAR using MI – MAR + 20% × δ | –1.99 (–8.37 to 4.40) | 0.539 |
| MNAR using MI – MAR + 30% × δ | –1.93 (–8.32 to 4.46) | 0.551 |
| MNAR using MI – MAR + 40% × δ | –1.88 (–8.27 to 4.51) | 0.562 |
| MNAR using MI – MAR + 50% × δ | –1.83 (–8.22 to 4.56) | 0.572 |
Treatment effect in the absence of topical treatment
Table 8 summarises the time point of initiation of other topical treatments (excluding topical prohibited and rescue therapies). In the following analyses, data during the use of the specified topical treatment were set to be missing and assumed to be MAR conditional on treatment group, baseline PP-PASI scores and observed PP-PASI scores until the time of treatment initiation. Analyses provided an estimate of the treatment effect in the absence of the stated topical therapy, under the assumption that participants whose data were set to be missing during topical therapy usage would have had a similar outcome to those observed with the same history and profile in the absence of topical therapy (Table 13). For simplicity, we have included all of the topical treatment uses reported on the concomitant medication data form and, as acknowledged above, site of use for other topical treatments was unknown. Therefore, the results of these analyses should be interpreted with caution. The treatment effect in the absence of all topical therapy was much smaller than the main ITT effect.
| Supplementary analysis | Mean treatment group difference in week 8 PP-PASI scores (points), anakinra – placebo (95% CI) | p-value |
|---|---|---|
| Data during topical treatment (rescue/prohibited or other topical) set to be missing and assumed to be MAR (where missing stop date assumed topical use ongoing) | 0.30 (–3.24 to 3.85) | 0.866 |
| Data during topical treatment (rescue/prohibited or other topical) set to be missing and assumed to be MAR (where missing stop date assume topical use at closest visit only) | –0.47 (–3.77 to 2.82) | 0.779 |
| Data post rescue/prohibited initiation and only during other topical treatment set to be missing (where missing stop date assumed other topical use ongoing) | 0.08 (–3.64 to 3.80) | 0.967 |
| Data post rescue/prohibited initiation and only during other topical treatment set to be missing (where missing stop date assumed topical use at closest visit only) | –1.02 (–4.63 to 2.59) | 0.580 |
Complier-average causal effect
For each participant, the proportion of injections used relative to the number of injections prescribed (total planned: 8 × 7 = 56) was calculated based on the self-reported data obtained via SMS and at follow-up visits. Compliance was defined as receiving at least 50–90% of planned injections and is summarised in Table 14.
| Compliancea | Number of participants (%) | |
|---|---|---|
| Placebo group | Anakinra group | |
| ≥ 50% of injections | 26 (79) | 25 (81) |
| ≥ 60% of injections | 24 (73) | 24 (77) |
| ≥ 70% of injections | 22 (67) | 24 (77) |
| ≥ 80% of injections | 22 (67) | 23 (74) |
| ≥ 90% of injections | 20 (61) | 15 (48) |
The CACE was initially estimated for all participants who had data at baseline and week 8 follow-up (n = 60). The CACE estimate for a treatment complier, defined as an individual receiving ≥ 50% of injections, was –2.30 (95% CI –6.54 to 1.93; p = 0.287). For a complier defined as an individual receiving ≥ 90% of injections, the CACE was –3.80 (95% CI –10.76 to 3.16; p = 0.285), indicating a larger average treatment effect for individuals with greater levels of compliance (Table 15).
Subsequently, MI was employed to handle missing outcome data (under the assumption of MAR) and the CACE was estimated for all participants who (1) had data at baseline and at least one follow-up over week 8, so the included set of participants would correspond with the primary analysis (n = 62), and (2) had been randomised (n = 64). The CACE estimates in these unplanned sensitivity analyses were very similar to the complete case CACE (see Table 15). When individuals missing compliance status were excluded from the analysis (rather than assumed to be non-compliant in accordance with the APRICOT statistical analysis plan), the results similarly did not vary (results not shown).
| Analysis | Mean treatment group difference in week 8 PP-PASI scores (points), anakinra – placebo (95% CI) | p-value |
|---|---|---|
| Primary analysis (N = 62) | ||
| Mixed model (ITT)a | –1.65 (–4.77 to 1.47) | 0.300 |
| CACE (n = 60, complete case) | ||
| ≥ 50% of injections | –2.30 (–6.54 to 1.93) | 0.287 |
| ≥ 60% of injections | –2.30 (–6.54 to 1.93) | 0.287 |
| ≥ 70% of injections | –2.30 (–6.54 to 1.93) | 0.287 |
| ≥ 80% of injections | –2.41 (–6.85 to 2.04) | 0.289 |
| ≥ 90% of injections | –3.80 (–10.76 to 3.16) | 0.285 |
| CACE (n = 62, MI post hoc sensitivity) | ||
| ≥ 50% of injections | –1.94 (–6.16 to 2.29) | 0.369 |
| ≥ 60% of injections | –2.02 (–6.43 to 2.39) | 0.369 |
| ≥ 70% of injections | –2.02 (–6.43 to 2.39) | 0.369 |
| ≥ 80% of injections | –2.11 (–6.74 to 2.51) | 0.370 |
| ≥ 90% of injections | –3.33 (–10.56 to 3.90) | 0.366 |
| CACE (n = 64, MI post hoc sensitivity) | ||
| ≥ 50% of injections | –2.18 (–6.58 to 2.22) | 0.331 |
| ≥ 60% of injections | –2.27 (–6.85 to 2.31) | 0.331 |
| ≥ 70% of injections | –2.27 (–6.85 to 2.31) | 0.331 |
| ≥ 80% of injections | –2.37 (–7.15 to 2.42) | 0.332 |
| ≥ 90% of injections | –3.62 (–10.90 to 3.65) | 0.329 |
Exploratory analysis for the Palmoplantar Pustulosis Psoriasis Area Severity Index
A total of 63 participants, (placebo group, n = 32; anakinra group, n = 31) were included in the analysis of the primary outcome up to 12 weeks. The adjusted mean treatment group difference in the week 12 PP-PASI scores for those in the anakinra group compared with those in the placebo group was –2.42 points (95% CI –5.97 to 1.13 points; p = 0.182). The results are presented in Figure 6.
FIGURE 6.
The PP-PASI scores over the 12-week follow-up period by treatment group: mixed-model estimates. The baseline adjusted mean PP-PASI score is plotted on the y-axis against the time point on the x-axis for each treatment group. The error bars represent 95% CIs for the adjusted treatment group means, estimated from the primary linear mixed model with week 12 data added in.
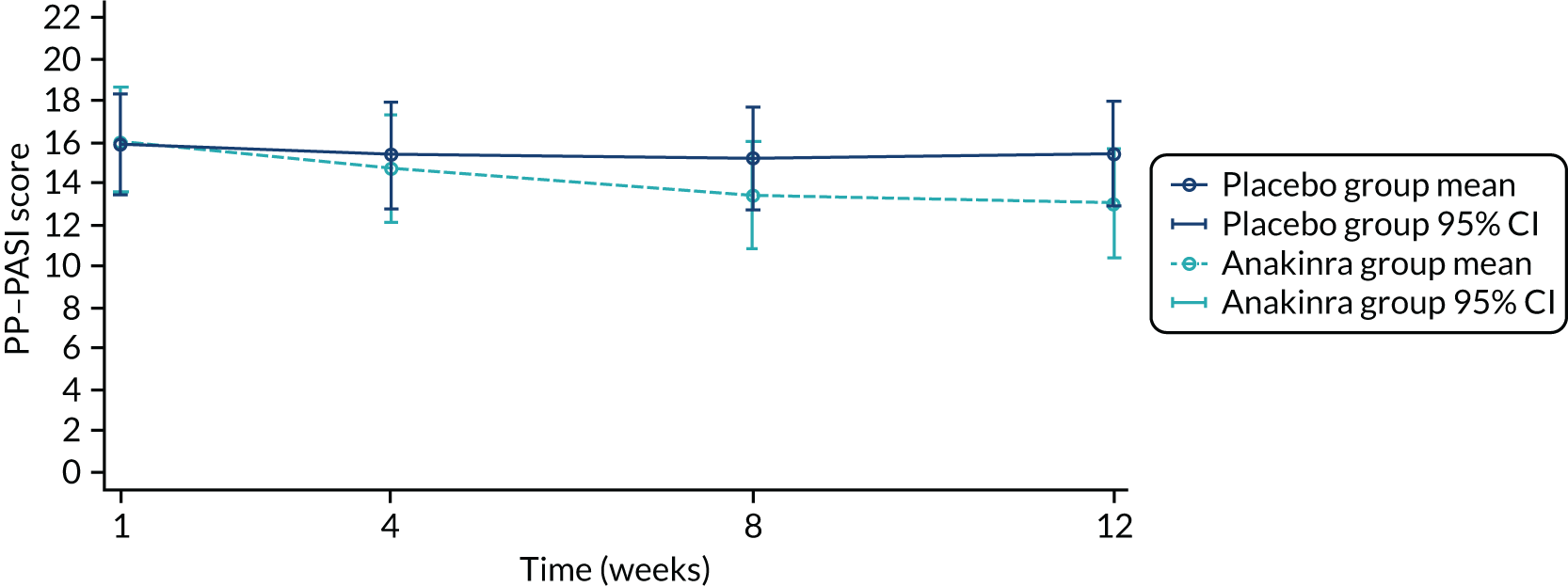
Secondary outcomes
Secondary investigator-assessed outcomes
Fresh pustule count (palms and soles)
The mean fresh pustule counts by visit are shown in Figure 7 and are summarised in Table 16. A total of 61 participants, (placebo group, n = 32; anakinra group, n = 29) were included in the analysis of the fresh pustule count. One individual in the anakinra group [identification (ID) = 100029] had a baseline fresh pustule count of 799 and it was known that their pustule counts were measured incorrectly; therefore, this participant was excluded from the analysis. Sensitivity analysis is performed below including this participant. The adjusted mean difference in the week 8 fresh pustule count for those in the placebo group compared with those in the anakinra group was 2.94 pustules (95% CI –26.44 to 32.33 pustules; p = 0.844), where the point estimate is in favour of the placebo group. The adjusted mean difference in the week 4 fresh pustule count for those in the anakinra group compared with those in the placebo group was 9.98 pustules (95% CI –19.40 to 39.36 pustules), and the adjusted mean difference in the week 1 fresh pustule count for those in the anakinra group compared with those in the placebo group was –2.41 pustules (95% CI –31.76 to 26.94 pustules).
FIGURE 7.
Fresh pustule count over the 12-week follow-up period. The unadjusted mean fresh pustule count is plotted on the y-axis against the time point on the x-axis for each treatment group. The error bars represent 95% CIs for the unadjusted treatment group means.
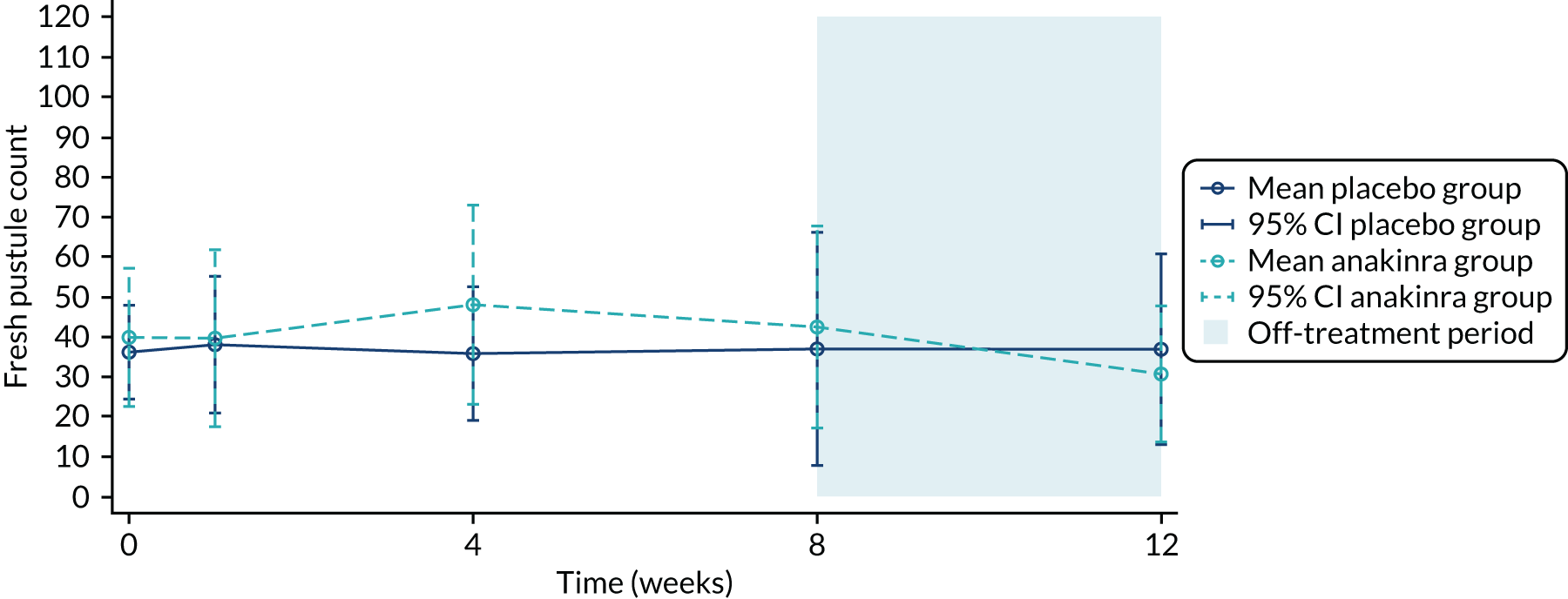
| Time point | Placebo group (N = 33) | Anakinra group (N = 31) | Total (N) | Unadjusted mean difference, anakinra – placebo (95% CI) | ||
|---|---|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | |||
| Baseline | 33 | 36.1 (33.1) | 30 | 39.8a (46.3) | 63 | – |
| Week 1 | 30 | 38.0 (45.8) | 29 | 39.6 (58.2) | 59 | 1.6 (–25.6 to 28.8) |
| Week 4 | 31 | 35.7 (45.5) | 28 | 48.0 (64.3) | 59 | 12.2 (–16.6 to 41) |
| Week 8 | 31 | 36.9 (79.5) | 28 | 42.4 (65.1) | 59 | 5.5 (–32.6 to 43.6) |
| Week 12 | 29 | 36.8 (62.7) | 27 | 30.6 (43.0) | 56 | –6.2 (–35.2 to 22.8) |
In the sensitivity analysis, which included the individual from the anakinra group who had an outlying pustule count at baseline (n = 799), the adjusted mean treatment group difference in the week 8 fresh pustule count for those in the anakinra group compared with those in the placebo group (baseline) was 3.08 pustules (95% CI –27.8 to 34.0 pustules; p = 0.845; n = 62). The adjusted mean difference in the week 4 fresh pustule count for those in the anakinra group compared with those in the placebo group was 2.99 pustules (95% CI –27.9 to 33.9 pustules) and the adjusted mean difference in the week 1 fresh pustule count for those in the anakinra group compared with those in the placebo group was 4.59 pustules (95% CI –26.29 to 35.5 pustules; p = 0.771).
Fresh pustule count: exploratory analysis by palms and soles separately
Fresh pustule counts on the palms are summarised in Table 17. A total of 62 participants, (placebo group, n = 32; anakinra group, n = 30) were included in the analysis of the fresh pustule count on the palms. The adjusted mean treatment group difference in the week 8 fresh pustule count of the palms for those in the anakinra group compared with those in the placebo group was 4.07 pustules (95% CI –5.78 to 13.92 pustules; p = 0.418), where the point estimate was in favour of the placebo group.
| Time point | Placebo group (N = 33) | Anakinra group (N = 31) | Total (N) | Unadjusted mean difference, anakinra – placebo (95% CI) | ||
|---|---|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | |||
| Baseline | 33 | 10.2 (19.2) | 30 | 10.2a (16.5) | 63 | – |
| Week 1 | 31 | 11.1 (29.5) | 29 | 10.7 (16.3) | 60 | –0.4 (–12.8 to 12.0) |
| Week 4 | 31 | 8.9 (20.9) | 28 | 11.7 (25.7) | 59 | 2.8 (–9.4 to 15.0) |
| Week 8 | 31 | 7.0 (14.7) | 29 | 10.8 (19.2) | 60 | 3.9 (–4.9 to 12.7) |
| Week 12 | 29 | 8.5 (21.8) | 27 | 9.6 (22.4) | 56 | 1.1 (–10.7 to 13.0) |
In the post hoc sensitivity analysis, which included the one individual from the anakinra group who had an outlying pustule count on the palms (ID 100029, n = 209), the adjusted mean treatment group difference in the week 8 fresh pustule count of the palms for those in the anakinra group compared with those in the placebo group was 1.83 pustules (95% CI –8.88 to 12.54 pustules; p = 0.738).
Fresh pustule counts on the soles are summarised in Table 18. A total of 62 participants (placebo group, n = 32; anakinra group, n = 30) were included in the analysis of the fresh pustule count on the soles.
| Time point | Placebo group (N = 33) | Anakinra group (N = 31) | Total (N) | Unadjusted mean difference, anakinra – placebo (95% CI) | ||
|---|---|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | |||
| Baseline | 33 | 25.9 (23.4) | 30 | 29.6a (43.2) | 63 | – |
| Week 1 | 31 | 26.2 (34.4) | 29 | 28.9 (50.2) | 60 | 2.7 (–19.4 to 24.9) |
| Week 4 | 31 | 26.8 (38.1) | 28 | 36.3 (61.1) | 59 | 9.4 (–16.9 to 35.7) |
| Week 8 | 31 | 29.9 (69.1) | 28 | 31.4 (61.2) | 59 | 1.5 (–32.7 to 35.7) |
| Week 12 | 29 | 28.3 (46.0) | 28 | 20.3 (36.0) | 57 | –8.1 (–30.1 to 13.9) |
The adjusted mean treatment group difference in the week 8 fresh pustule count of the soles for those in the anakinra group compared with those in the placebo group was –1.42 pustules (95% CI –27.33 to 24.48 pustules; p = 0.914).
In the post hoc sensitivity analysis, which included the one individual from the anakinra group who had an outlying pustule count on the soles (ID = 100029), the adjusted mean treatment group difference in the week 8 fresh pustule count of the soles for those in the anakinra group compared with those in the placebo group was –0.02 pustules (95% CI –26.7 to 26.69 pustules; p = 0.999).
Total pustule count
The mean total pustule count scores by visit are shown in Figure 8 and are summarised in Table 19. A total of 61 participants (placebo group, n = 32; anakinra group, n = 29) were included in the analysis of the total pustule count. The adjusted mean difference in the week 8 total pustule count for those in the anakinra group compared with those in the placebo group was –30.08 pustules (95% CI –83.20 to 23.05 pustules; p = 0.267), where the point estimate was in favour of the anakinra group.
FIGURE 8.
Total pustule count over the 12-week follow-up period. The unadjusted mean total pustule count is plotted on the y-axis against the time point on the x-axis for each treatment group. The error bars represent 95% CIs for the unadjusted treatment group means.
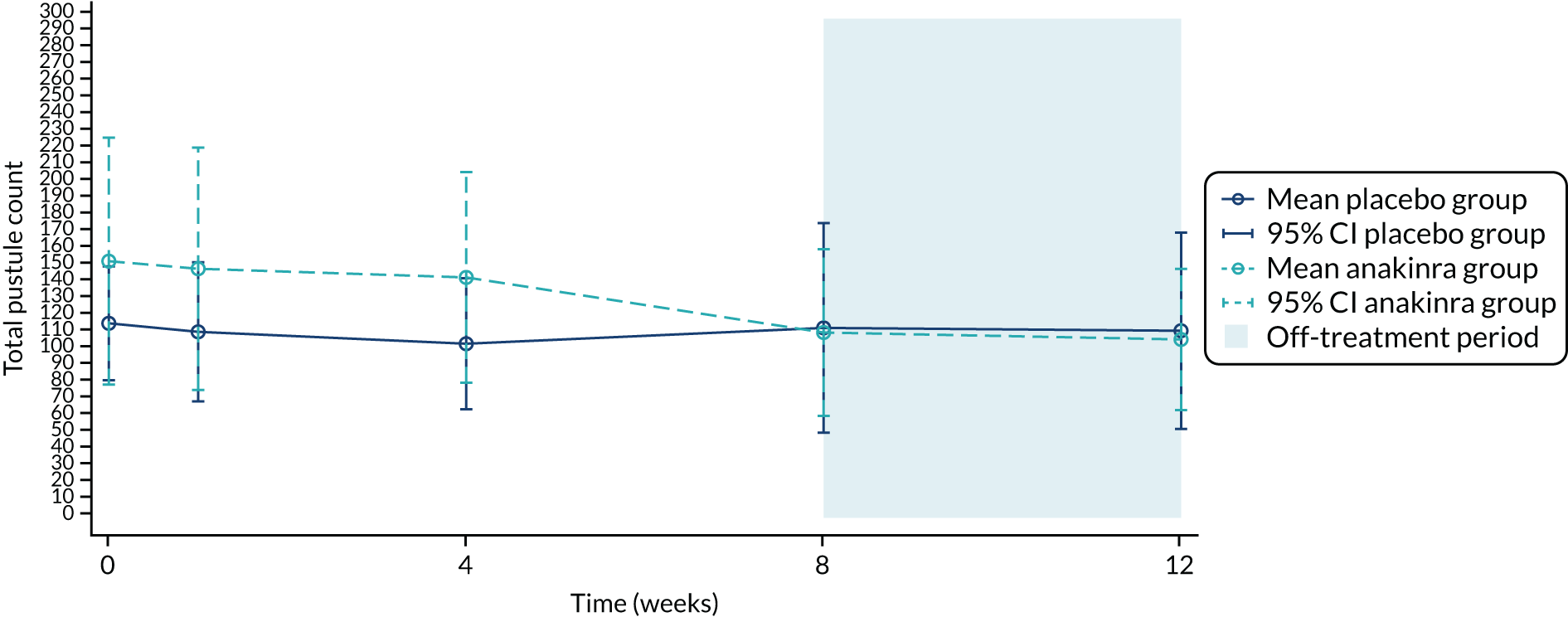
| Time point | Placebo group (N = 33) | Anakinra group (N = 31) | Total (N) | Unadjusted mean difference, anakinra – placebo (95% CI) | ||
|---|---|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | |||
| Baseline | 33 | 116.9 (96.4) | 30 | 154.3a (198.7) | 63 | – |
| Week 1 | 30 | 111.8 (112.1) | 29 | 149.7 (191.5) | 59 | 37.9 (–43.5 to 119.4) |
| Week 4 | 31 | 104.6 (107.3) | 28 | 144.5 (163.3) | 59 | 39.9 (–31.5 to 111.2) |
| Week 8 | 31 | 114.2 (171.8) | 28 | 111.4 (129.3) | 59 | –2.8 (–82.7 to 77.2) |
| Week 12 | 29 | 112.4 (155.0) | 27 | 107.2 (107.3) | 56 | –5.2 (–77.2 to 66.7) |
The adjusted mean difference in the week 4 total pustule count for those in the anakinra group compared with those in the placebo group was 15.82 pustules (95% CI –37.26 to 68.89 pustules). The adjusted mean difference in the week 1 total pustule count for those in the anakinra group compared with those in the placebo group was 11.06 pustules (95% CI –41.98 to 64.10 pustules).
In post hoc sensitivity analysis, which included the one individual from the anakinra group who had an outlying total pustule count of 1186 at baseline (ID = 100029), the adjusted mean treatment group difference in the week 8 total pustule count for those in the anakinra group compared with those in the placebo group was –27.98 pustules (95% CI –84.51 to 28.54 pustules; p = 0.332). The adjusted mean difference in the week 4 total pustule count for those in the anakinra group compared with those in the placebo group was 1.73 pustules (95% CI –54.73 to 58.19 pustules). The adjusted mean difference in the week 1 total pustule count for those in the anakinra group compared with those in the placebo group was 23.59 pustules (95% CI –32.85 to 80.04 pustules).
Palmoplantar Pustulosis – Investigator’s Global Assessment
The PPP-IGA ratings over the 12-week follow-up period are summarised in Table 20. A total of 63 participants (placebo group, n = 32; anakinra group, n = 31) were included in the analysis of the PPP-IGA. There was no evidence for statistical superiority in the odds of a higher PPP-IGA rating between the treatment groups at week 8 (OR 0.54, 95% CI 0.13 to 2.19; p = 0.384), for which the point estimate favoured the anakinra group. There was also no evidence for statistical superiority in the odds of a higher PPP-IGA rating between the treatment groups at week 4 (OR 0.64, 95% CI 0.17 to 2.44) and week 1 (OR 1.26, 95% CI 0.33 to 4.78). No participants achieved a clear rating on the PPP-IGA at week 8: 0 out of 28 (0%) in the placebo group compared with 0 out of 30 (0%) in the anakinra group.
| PPP-IGA rating | Placebo group | Anakinra group |
|---|---|---|
| Baseline | ||
| Mild, n (%) | 4 (12) | 4 (13) |
| Moderate, n (%) | 16 (48) | 17 (55) |
| Severe, n (%) | 13 (39) | 10 (32) |
| Median (IQR) | 3.0 (3.0–4.0) | 3.0 (3.0–4.0) |
| Week 1 | ||
| Mild, n (%) | 4 (13) | 6 (20) |
| Moderate, n (%) | 20 (65) | 14 (47) |
| Severe, n (%) | 7 (23) | 10 (33) |
| Median (IQR) | 3.0 (3.0–3.0) | 3.0 (3.0–4.0) |
| Week 4 | ||
| Almost clear, n (%) | 0 (0) | 1 (4) |
| Mild, n (%) | 7 (23) | 6 (21) |
| Moderate, n (%) | 18 (58) | 17 (61) |
| Severe, n (%) | 6 (19) | 4 (14) |
| Median (IQR) | 3.0 (3.0–3.0) | 3.0 (2.5–3.0) |
| Week 8 | ||
| Almost clear, n (%) | 2 (7) | 1 (3) |
| Mild, n (%) | 4 (14) | 6 (20) |
| Moderate, n (%) | 12 (43) | 17 (57) |
| Severe, n (%) | 10 (36) | 6 (20) |
| Median (IQR) | 3.0 (3.0–4.0) | 3.0 (3.0–3.0) |
| Week 12 | ||
| Almost clear, n (%) | 3 (10) | 0 (0) |
| Mild, n (%) | 7 (24) | 9 (33) |
| Moderate, n (%) | 12 (41) | 13 (48) |
| Severe, n (%) | 7 (24) | 5 (19) |
| Median (IQR) | 3.0 (2.0–3.0) | 3.0 (2.0–3.0) |
Time to response (75% reduction in fresh pustule count)
A total of 28 participants achieved a 75% reduction in fresh pustule count compared with their baseline count during the 12-week follow-up period: 15 out of 31 (48%) in the placebo group and 13 out of 30 (43%) in the anakinra group (Figure 9). A total of 61 participants (placebo group, n = 31; anakinra group, n = 30) were included in the analysis of the time to response. There was no evidence of statistical superiority of anakinra in the time to response between the treatments (HR 0.58, 95% CI 0.22 to 1.50; p = 0.263), although the point estimate favoured the anakinra group.
FIGURE 9.
Time to response by treatment group.
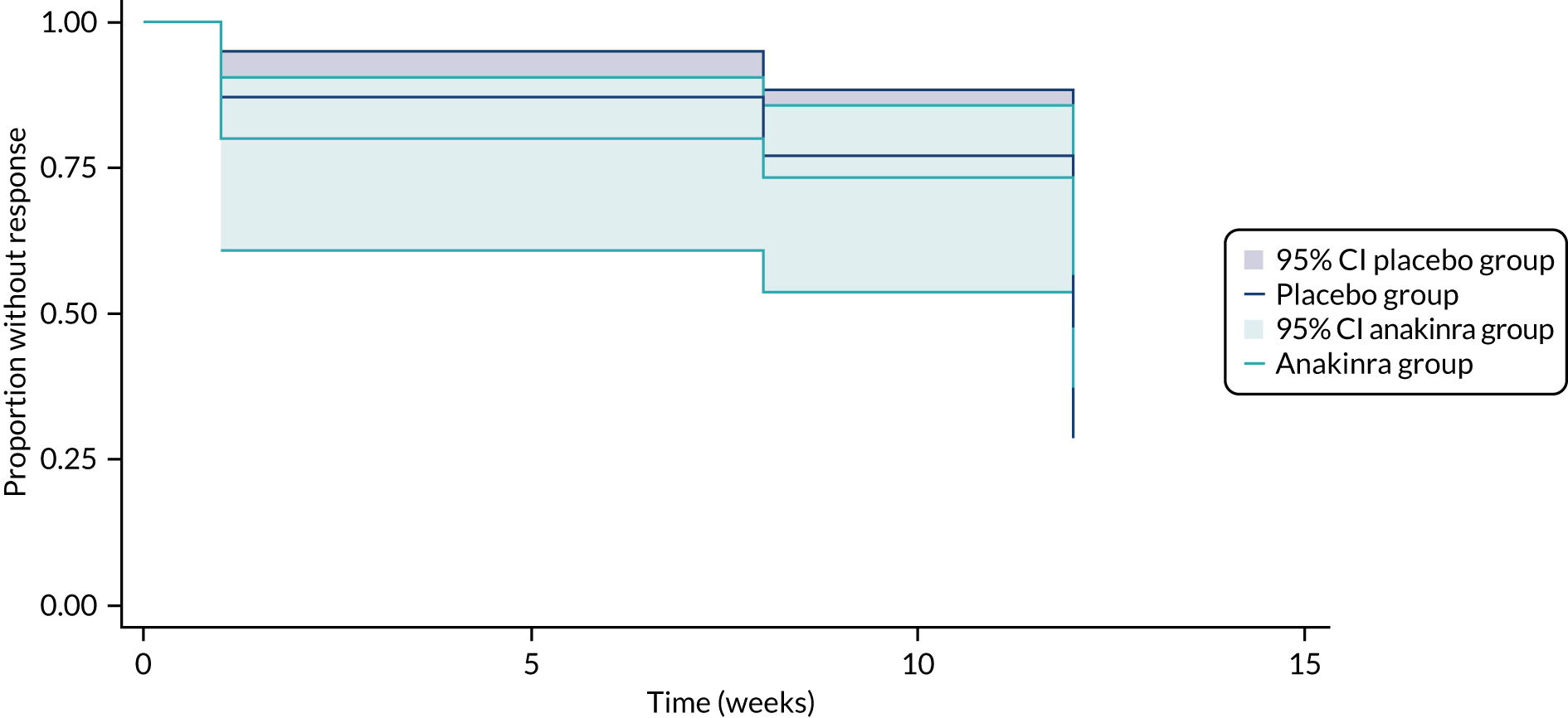
Time to relapse (return to baseline fresh pustule count)
A total of 39 participants returned to their baseline fresh pustule count during the 12-week follow-up period: 19 out of 31 (61%) in the placebo group and 20 out of 30 (67%) in the anakinra group (Figure 10). A total of 61 participants (placebo group, n = 31; anakinra group, n = 30) were included in the analysis of the time to relapse. The time to relapse between the treatment was found to be similar (HR 0.94, 95% CI 0.50 to 1.78; p = 0.853).
FIGURE 10.
Time to relapse by treatment group.
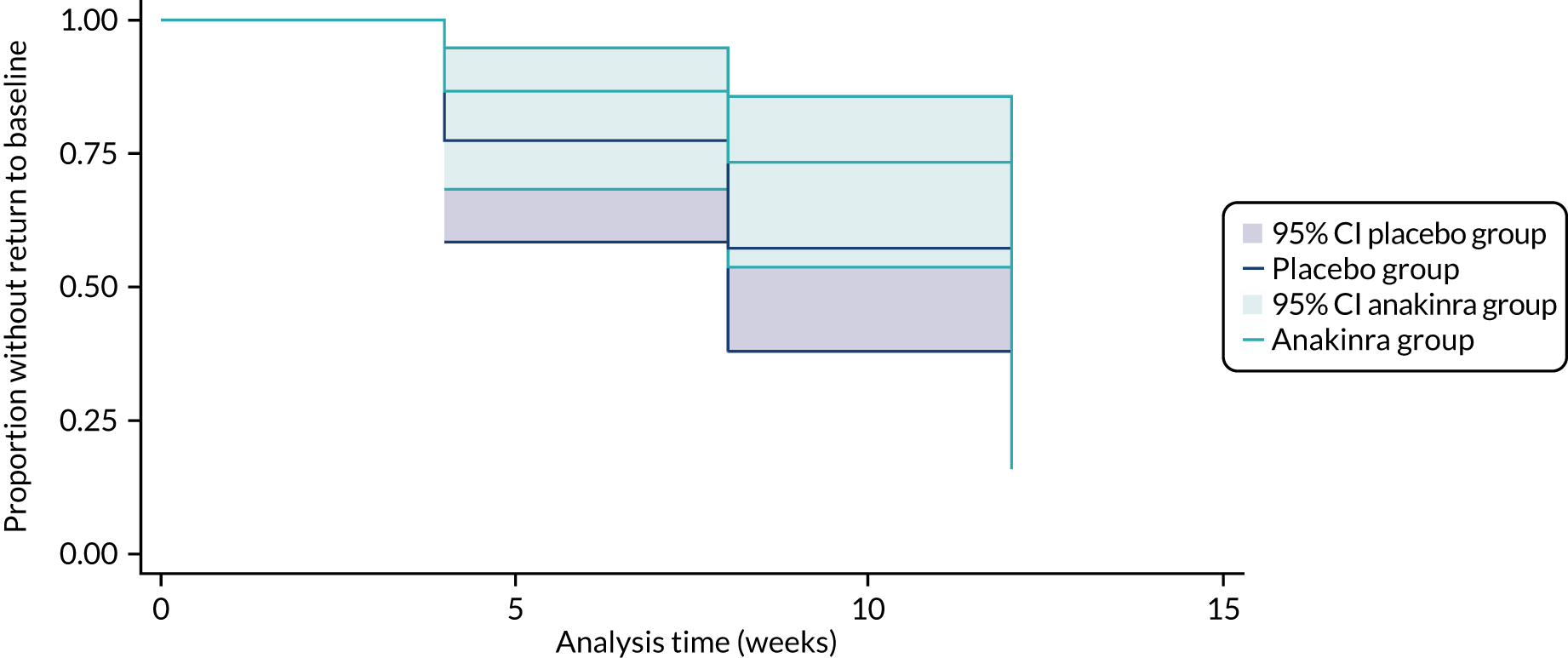
Development of disease flare
A disease flare is defined as a > 50% deterioration in PP-PASI score compared with baseline. The denominator for the analysis of development of disease flare includes the number of participants followed up over the full treatment period, that is up to week 8. Four participants in the placebo group experienced disease flare compared with two in the anakinra group [12.9% vs. 6.9%, unadjusted difference in proportions –6.0% (95% CI –20.98% to 8.97%)]. A mixed-logistic regression model was used to adjust the treatment group difference for baseline PP-PASI scores and centre. There was no evidence of statistical superiority in the odds of disease flare for anakinra relative to placebo (OR 0.55, 95% CI 0.08 to 3.71; p = 0.542), with the point estimate in favour of anakinra (i.e. those receiving the anakinra treatment are less likely to develop disease flare).
Plaque type psoriasis
The mean PASI scores by visit are shown in Figure 11 and summarised in Table 21. A total of 38 participants (placebo group, n = 20; anakinra group, n = 18) had plaque psoriasis evident at one or more follow-up visits and were included in the analysis of the PASI. The adjusted mean difference in the week 8 PASI scores for those in the anakinra group compared with those in the placebo group was –0.41 (95% CI –0.96 to 0.15; p = 0.151), for which the point estimate was in favour of the anakinra group. The adjusted mean difference in the week 4 PASI scores for those in the anakinra group compared with those in the placebo group was –0.08 (95% CI –0.61 to 0.45).
FIGURE 11.
The PASI scores over the 12-week follow-up period. The unadjusted mean PASI scores are plotted on the y-axis against the time point on the x-axis for each treatment group. The error bars represent 95% CIs for the unadjusted treatment group means.
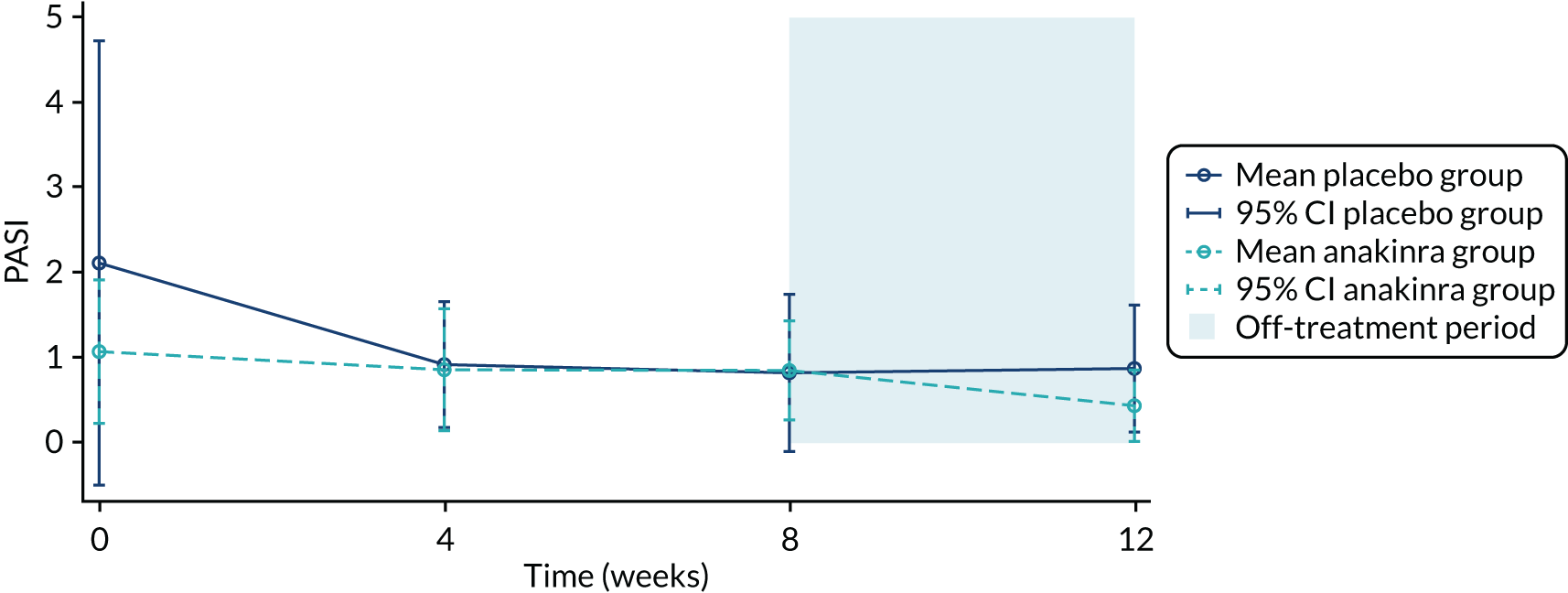
| Time point | Placebo group (N = 33) | Anakinra group (N = 31) | Total (N) | Unadjusted mean difference, anakinra – placebo (95% CI) | ||
|---|---|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | |||
| Baseline | 19 | 2.1 (5.4) | 16 | 1.1 (1.6) | 35 | – |
| Week 4 | 18 | 0.9 (1.5) | 18 | 0.9 (1.4) | 36 | –0.1 (–1.1 to 0.9) |
| Week 8 | 16 | 0.8 (1.7) | 15 | 0.9 (1.1) | 31 | 0.0 (–1.0 to 1.1) |
| Week 12 | 17 | 0.9 (1.5) | 18 | 0.4 (0.8) | 35 | –0.4 (–1.2 to 0.4) |
Palmoplantar Pustulosis Psoriasis Area Severity Index-50 and Palmoplantar Pustulosis Psoriasis Area Severity Index-75
In the post hoc analysis, the treatment group difference in PP-PASI-50 and PP-PASI-75 scores at week 8 was explored because these two outcomes were reported in two recently published randomised controlled trials investigating interventions in PPP. 37,38
Five participants in the placebo group experienced PP-PASI-50, compared with six in the anakinra group (16.1% vs. 20.7%; unadjusted difference in proportions –4.6%, 95% CI –15.1% to 24.2%). There was no evidence for statistical superiority in the odds of PP-PASI-50 for those in the anakinra group compared with those in the placebo group (OR 1.68, 95% CI 0.35 to 8.19; p = 0.520), for which the point estimate was in favour of the anakinra group. One placebo group participant experienced PP-PASI-75, compared with zero participants in the anakinra group (3.2% vs. 0.0%; unadjusted difference in proportions –3.2%, 95% CI –9.4% to 3.0%). Owing to the small numbers of events, it was not possible to adjust the treatment effect on PP-PASI-75 by baseline PP-PASI score and centre, as a mixed-logistic regression model did not converge.
Palmoplantar Pustulosis Area Severity Index pustule subscales
In the post hoc analysis, we explored the treatment group difference in the PP-PASI pustule subscale, separately for palms and soles, at week 8 (Table 22). There was no evidence for statistical superiority in the odds of a higher PP-PASI pustule rating on the palms across treatment groups for those in the anakinra group compared with those in the placebo group (OR 2.51, 95% CI 0.56 to 11.28; p = 0.231), for which the point estimate was in favour of placebo. There was no difference in the odds of a higher PP-PASI pustule rating on the soles across treatment groups for those in the anakinra group than for those in the placebo group (OR 1.63, 95% CI 0.49 to 5.46; p = 0.426), for which the point estimate was similarly in favour of placebo (a more severe pustule subscale rating for those in the anakinra group relative to those in the placebo group).
| PP-PASI pustule subscale rating | Number of participants (%) | |
|---|---|---|
| Placebo group | Anakinra group | |
| Palm (worst pustule subscale across left and right palm) | ||
| Baseline | ||
| None | 12 (36) | 10 (32) |
| Slight | 9 (27) | 9 (29) |
| Moderate | 7 (21) | 10 (32) |
| Severe | 1 (3) | 2 (6) |
| Very severe | 4 (12) | 0 (0) |
| Week 8 | ||
| None | 14 (45) | 11 (37) |
| Slight | 10 (32) | 9 (30) |
| Moderate | 5 (16) | 8 (27) |
| Severe | 2 (6) | 2 (7) |
| Very severe | 0 (0) | 0 (0) |
| Sole (worst pustule subscale across left and right sole) | ||
| Baseline | ||
| None | 3 (9) | 4 (13) |
| Slight | 5 (15) | 2 (6) |
| Moderate | 5 (15) | 9 (29) |
| Severe | 12 (36) | 13 (42) |
| Very severe | 8 (24) | 3 (10) |
| Week 8 | ||
| None | 3 (10) | 2 (7) |
| Slight | 6 (195) | 8 (28) |
| Moderate | 11 (35) | 8 (28) |
| Severe | 9 (29) | 9 (31) |
| Very severe | 2 (6) | 2 (7) |
Secondary participant-assessed outcomes
Participants’ global assessment
Participants’ global assessments over the 12-week follow-up period are summarised in Table 23.
| Participant’s global assessment | Placebo group (N = 33) | Anakinra group (N = 31) |
|---|---|---|
| Baseline | ||
| Clear, n (%) | 0 (0) | 0 (0) |
| Nearly clear, n (%) | 0 (0) | 2 (6) |
| Mild, n (%) | 3 (9) | 3 (10) |
| Moderate, n (%) | 14 (42) | 14 (45) |
| Severe, n (%) | 13 (39) | 7 (23) |
| Very severe, n (%) | 3 (9) | 5 (16) |
| Median (IQR) | 3.0 (3.0–4.0) | 3.0 (3.0–4.0) |
| Week 1 | ||
| Clear, n (%) | 0 (0) | 0 (0) |
| Nearly clear, n (%) | 0 (0) | 0 (0) |
| Mild, n (%) | 5 (16) | 7 (23) |
| Moderate, n (%) | 12 (38) | 14 (45) |
| Severe, n (%) | 11 (34) | 8 (26) |
| Very severe, n (%) | 4 (13) | 2 (6) |
| Median (IQR) | 3.0 (3.0–4.0) | 3.0 (3.0–4.0) |
| Week 4 | ||
| Clear, n (%) | 0 (0) | 0 (0) |
| Nearly clear, n (%) | 1 (3) | 0 (0) |
| Mild, n (%) | 7 (22) | 6 (20) |
| Moderate, n (%) | 9 (28) | 14 (47) |
| Severe, n (%) | 11 (34) | 8 (27) |
| Very severe, n (%) | 4 (13) | 2 (7) |
| Median (IQR) | 3.0 (2.5–4.0) | 3.0 (3.0–4.0) |
| Week 8 | ||
| Clear, n (%) | 1 (3) | 0 (0) |
| Nearly clear, n (%) | 3 (10) | 3 (10) |
| Mild, n (%) | 4 (13) | 5 (16) |
| Moderate, n (%) | 11 (37) | 11 (35) |
| Severe, n (%) | 10 (33) | 10 (32) |
| Very severe, n (%) | 1 (3) | 2 (6) |
| Median (IQR) | 3.0 (2.0–4.0) | 3.0 (2.0–4.0) |
| Week 12 | ||
| Clear, n (%) | 0 (0) | 0 (0) |
| Nearly clear, n (%) | 4 (13) | 2 (7) |
| Mild, n (%) | 5 (17) | 7 (25) |
| Moderate, n (%) | 12 (40) | 10 (36) |
| Severe, n (%) | 7 (23) | 7 (25) |
| Very severe, n (%) | 2 (7) | 2 (7) |
| Median (IQR) | 3.0 (3.0–4.0) | 3.0 (3.0–4.0) |
A total of 63 participants (placebo group, n = 32; anakinra group, n = 31) were included in the analysis of the PGA. There was no evidence for statistical superiority in the odds of a higher PPP-IGA rating between the treatment groups at week 8 (OR 1.39, 95% CI 0.41 to 4.70; p = 0.597), for which the point estimate was in favour of placebo. There was also no evidence for statistical superiority in the odds of a higher PPP-IGA rating between the treatment groups at week 4 (OR 0.74, 95% CI 0.22 to 2.50) or week 1 (OR 0.57, 95% CI 0.18 to 1.87).
Palmoplantar Quality of Life Instrument
The mean Palmoplantar Quality of Life (PP-QoL) scores by visit are shown in Figure 12 and are summarised in Table 24. A total of 62 participants (placebo group, n = 31; anakinra group, n = 31) were included in the analysis of the PP-QoL. The adjusted mean difference in the week 8 PP-QoL scores for those in the anakinra group compared with those in the placebo group was 1.27 (95% CI –3.04 to 5.57; p = 0.564), for which the point estimate was in favour of the placebo group.
FIGURE 12.
The PP-QoL scores over the 12-week follow-up period. The unadjusted mean PP-QoL scores are plotted on the y-axis against the time point on the x-axis for each treatment group. The error bars represent 95% CIs for the unadjusted treatment group means.
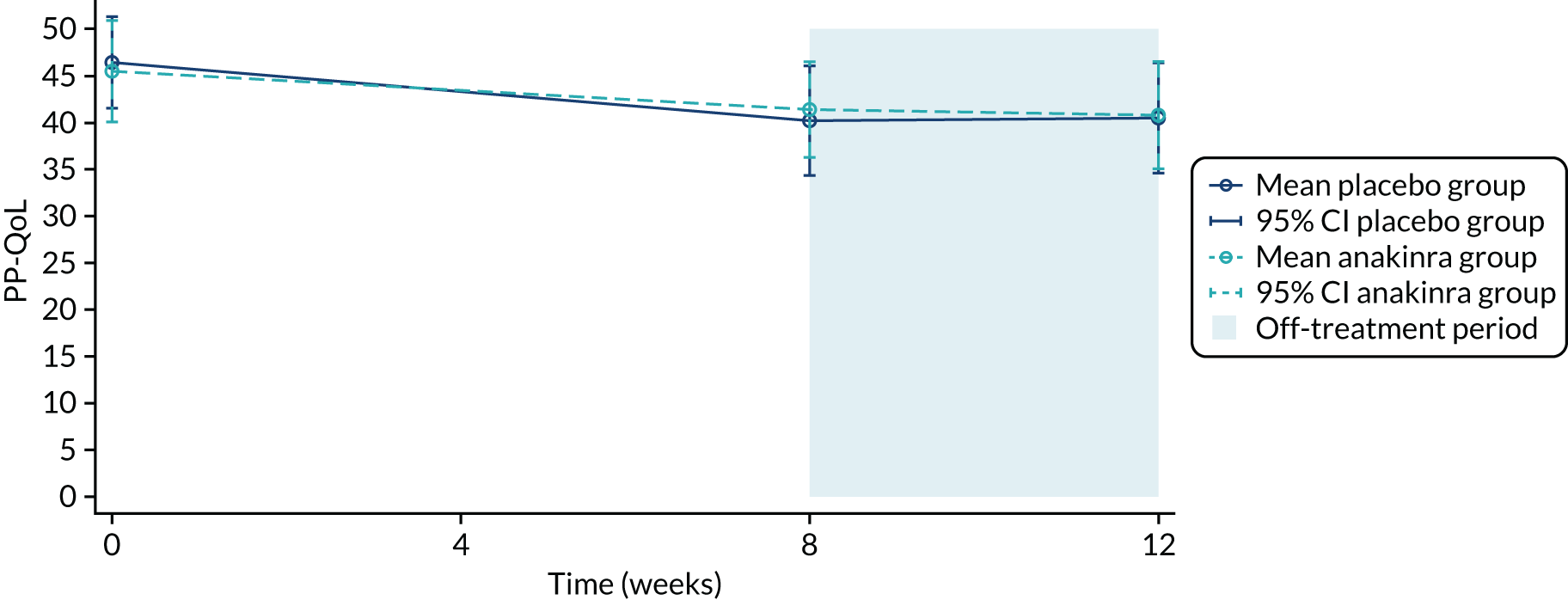
| Time point | Placebo group (N = 33) | Anakinra group (N = 31) | Total (N) | Unadjusted mean difference, anakinra – placebo (95% CI) | ||
|---|---|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | |||
| Baseline | 33 | 46.4 (13.8) | 31 | 45.5 (14.8) | 64 | – |
| Week 8 | 31 | 40.2 (16.0) | 31 | 41.4 (13.9) | 62 | 1.2 (–6.4 to 8.8) |
| Week 12 | 29 | 40.5 (15.5) | 28 | 40.8 (14.8) | 57 | 0.3 (–7.7 to 8.3) |
Dermatology Life Quality Index
The mean DLQI scores by visit are shown in Figure 13 and are summarised in Table 25. A total of 62 participants (placebo group, n = 31; anakinra group, n = 31) were included in the analysis of the DLQI scores. The adjusted mean difference in the week 8 DLQI scores for those in the anakinra group compared with those in the placebo group was 0.52 (95% CI –2.04 to 3.07; p = 0.692), for which the point estimate was in favour of the placebo group.
FIGURE 13.
Dermatology Life Quality Index scores over the 12-week follow-up period. The unadjusted mean DLQI scores are plotted on the y-axis against the time point on the x-axis for each treatment group. The error bars represent 95% CIs for the unadjusted treatment group means.
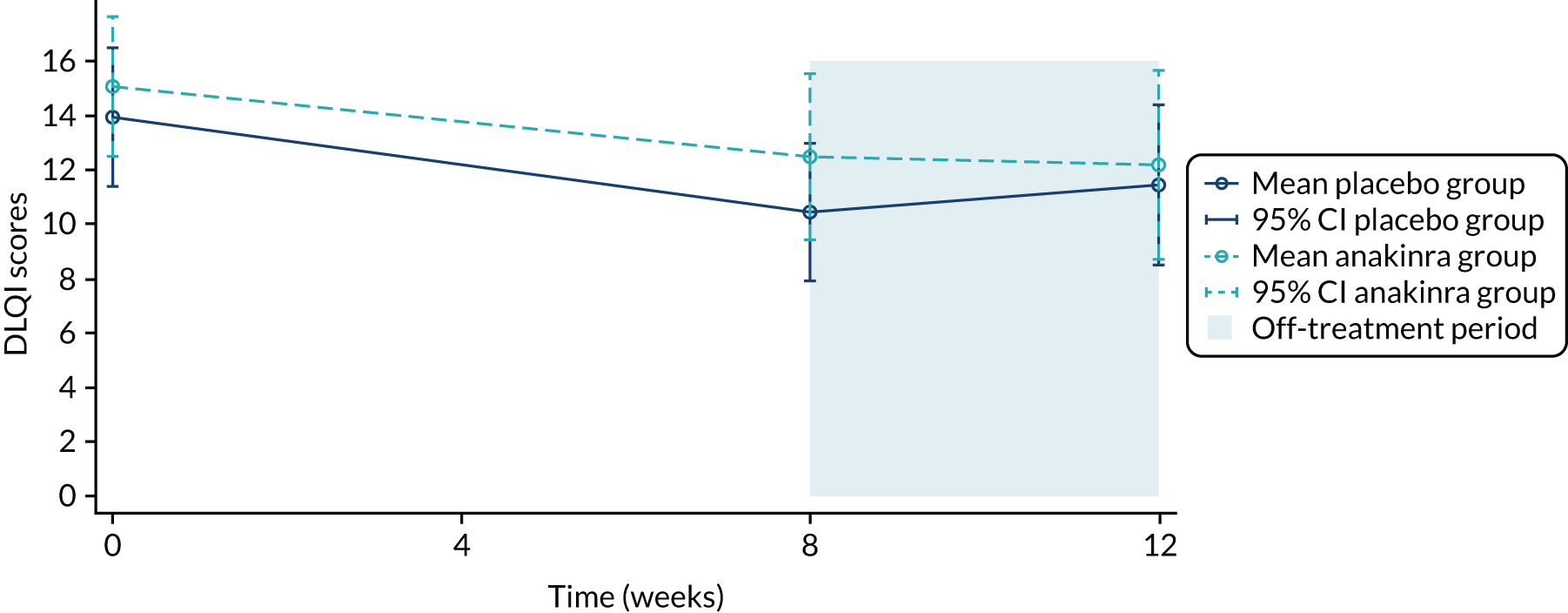
| Time point | Placebo group (N = 33) | Anakinra group (N = 31) | Total (N) | Unadjusted mean difference, anakinra – placebo (95% CI) | ||
|---|---|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | |||
| Baseline | 33 | 13.9 (7.2) | 31 | 15.1 (7.0) | 64 | – |
| Week 8 | 31 | 10.5 (6.9) | 31 | 12.5 (8.3) | 62 | 2.0 (–1.9 to 5.9) |
| Week 12 | 29 | 11.4 (7.7) | 27 | 12.2 (8.8) | 56 | 0.7 (–3.7 to 5.2) |
EuroQol-5 Dimensions, three-level version, utility index
The mean EuroQol-5 Dimensions, three-level version (EQ-5D-3L), scores by visit are shown in Figure 14 and are summarised in Table 26. A total of 62 participants (placebo group, n = 31; anakinra group, n = 31) were included in the analysis of the EQ-5D-3L utility index scores. A mixed-linear regression model was used to adjust the treatment group difference for baseline EQ-5D-3L score and centre. The adjusted mean difference in the week 8 EQ-5D-3L utility index score for those in the anakinra group compared with those in the placebo group was –0.09 (95%CI –0.23 to 0.06; p = 0.227), for which the point estimate was slightly in favour of the placebo group.
FIGURE 14.
The EQ-5D-3L utility index scores over the 12-week follow-up period. The unadjusted mean EQ-5D-3L scores are plotted on the y-axis against the time point on the x-axis for each treatment group. The error bars represent 95% CIs for the unadjusted treatment group means.
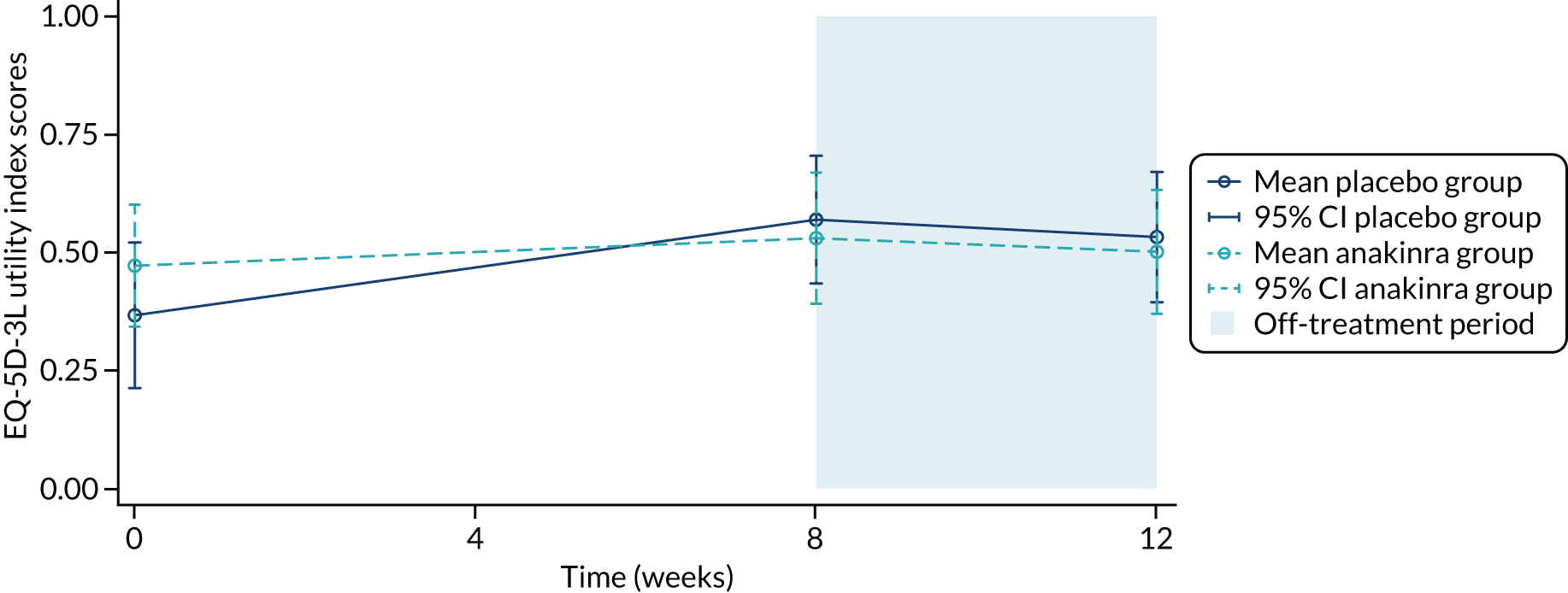
| Time point | Placebo group (N = 33) | Anakinra group (N = 31) | Total (N) | Unadjusted mean difference, anakinra – placebo (95% CI) | ||
|---|---|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | |||
| Baseline | 33 | 0.4 (0.4) | 31 | 0.5 (0.4) | 64 | – |
| Week 8 | 31 | 0.6 (0.4) | 31 | 0.5 (0.4) | 62 | 0.0 (–0.2 to 0.2) |
| Week 12 | 30 | 0.5 (0.4) | 29 | 0.5 (0.3) | 59 | 0.0 (–0.2 to 0.2) |
Treatment acceptability
The number of participants who strongly agreed that the treatment was worthwhile was larger in the anakinra group (n = 12/29, 41%) than in the placebo group (n = 4/14, 14%) (Table 27). The mean difference in the proportion strongly agreeing that the treatment was worthwhile for those in the anakinra group compared with those in the placebo group was 27.1% (95% CI 5.0% to 49.2%).
| Level of agreement with the statement ‘the treatment was worthwhile’ | Number of participants (%) | |
|---|---|---|
| Placebo group | Anakinra group | |
| Strongly agree | 4 (14) | 12 (41) |
| Agree | 8 (29) | 7 (24) |
| Neither agree nor disagree | 7 (25) | 6 (21) |
| Disagree | 5 (18) | 3 (10) |
| Strongly disagree | 4 (14) | 1 (3) |
| Total | 28 | 29 |
Figures 15 and 16 display the PP-PASI profiles over time for the participants who strongly agreed that the treatment was worthwhile by treatment group, which can be compared with the PP-PASI profiles across all trial participants in Figure 3. One participant who received anakinra, who strongly agreed that the treatment was worthwhile, had the highest baseline PP-PASI score across the trial of 58 points and saw the largest decrease in PP-PASI score to 28.8 points at week 8.
FIGURE 15.
The PP-PASI profiles for the placebo group participants who strongly agreed that the treatment was worthwhile.
FIGURE 16.
The PP-PASI profiles for the anakinra group participants who strongly agreed that the treatment was worthwhile.
Table 28 summarises the overall levels of compliance with the 8-week treatment (proportion of total planned injections received) by treatment group, and strong agreement that the treatment was worthwhile. Among those in the anakinra group receiving at least 90% of the planned total injections over the 8-week treatment period, there was a higher proportion of participants who strongly agreed that the treatment was worthwhile (67%) than those who did not (37%), indicating a potential relationship between treatment adherence and acceptability.
| Compliancea | Number of participants (%) | |||
|---|---|---|---|---|
| Placebo group | Anakinra group | |||
| Strongly agreed | Did not strongly agree | Strongly agreed | Did not strongly agree | |
| ≥ 50% of injections | 3 (75) | 23 (82) | 9 (75) | 16 (84) |
| ≥ 60% of injections | 3 (75) | 21 (75) | 9 (75) | 15 (79) |
| ≥ 70% of injections | 2 (50) | 20 (71) | 9 (75) | 15 (79) |
| ≥ 80% of injections | 2 (50) | 20 (71) | 9 (75) | 14 (74) |
| ≥ 90% of injections | 2 (50) | 18 (64) | 8 (67) | 7 (37) |
Safety outcomes
Serious infection and neutropenia
No participants experienced a serious infection (placebo group, n = 0/33, 0%; anakinra group, n = 0/31, 0%). Similarly, no participants experienced neutropenia (neutrophil count < 1.0 × 109/l: placebo group, n = 0/33, 0%; anakinra group, n = 0/31, 0%).
Pregnancy
One trial participant became pregnant (despite following the protocol regarding contraception) (a pregnancy test carried out at the week 8 visit was positive). The baby was born healthy at full term.
Safety monitoring
Table 29 summarises the types of AEs by treatment group. A full listing of the non-SAEs and reactions by MedDRA-preferred term and treatment group is given in Appendix 5, Table 39. Figures 17 and 18 summarise the non-SAEs by MedDRA system organ class. There was a notably larger number of participants experiencing general disorders and administration site conditions in the anakinra group than in the placebo group (anakinra group, n = 23, 74%; placebo group, n = 4, 12%).
| Event | Placebo group | Anakinra group | Total | |||
|---|---|---|---|---|---|---|
| Participants (n) | Events (n) | Participants (n) | Events (n) | Participants (n) | Events (n) | |
| Total non-serious AEs | 26 | 84 | 29 | 114 | 55 | 198 |
| AE | 24 | 52 | 24 | 66 | 48 | 118 |
| AR | 10 | 30 | 26 | 48 | 36 | 78 |
| Unexpected adverse reaction (subset of adverse reaction) | 2 | 3b | 2 | 2c | 4 | 5 |
| Unclassifiablea | 1 | 2 | 0 | 0 | 1 | 2 |
| Total SAEs | 0 | 0 | 0 | 0 | 0 | 0 |
| SAE | 0 | 0 | 0 | 0 | 0 | 0 |
| SAR | 0 | 0 | 0 | 0 | 0 | 0 |
| SUSAR (subset of SAR) | 0 | 0 | 0 | 0 | 0 | 0 |
| Total | 26 | 84 | 29 | 114 | 55 | 198 |
FIGURE 17.
Adverse events and reactions by MedDRA System Organ Class. This figure displays the proportions of individuals experiencing each type of event by treatment group on the left-hand side, the relative treatment group difference expressed as relative risk with 95% CI in the middle and numbers of participants experiencing each event and event totals on the right-hand side.
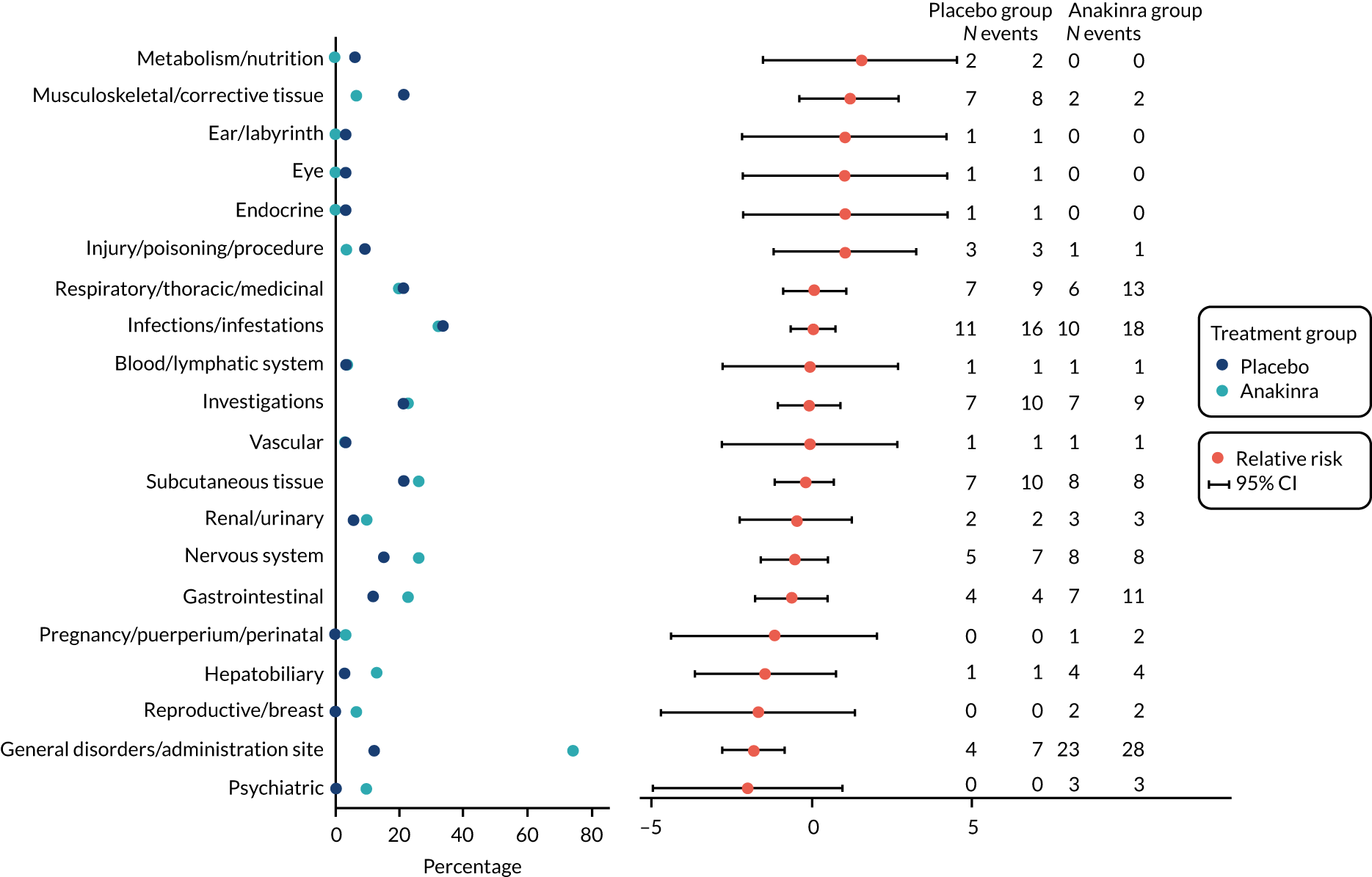
FIGURE 18.
Volcano plot of AEs and reactions by MedDRA System Order Class. In the volcano plot, the x-axis represents the difference in proportions of participants experiencing each category of AE between the treatment groups (placebo and anakinra). Risk difference of < 0 favours placebo. The y-axis represents the p-value from a Fisher’s exact test on a negative log-scale; smaller p-values are situated higher up the y-axis. The centre of each circle indicates the co-ordinates for a particular category of AE and the size of the circle is proportional to the total number of events for both treatment arms combined. AE categories have been labelled where p < 0.2.
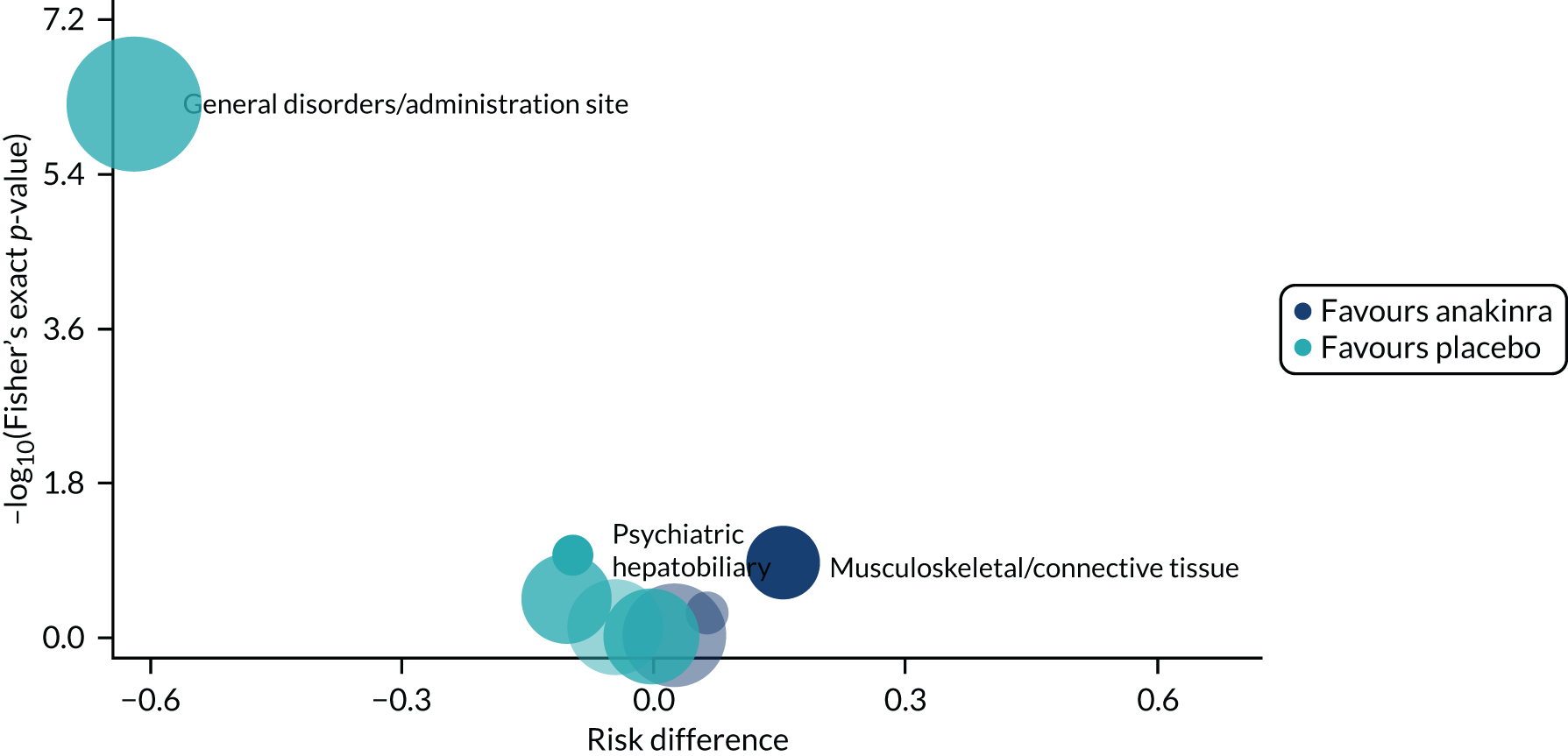
Examining the events at the preferred term level reveals that the difference is the result of a larger number of injection site reactions in the anakinra group (see Appendix 5, Table 39); there were 20 injection site reactions among 19 participants (61%) in the anakinra group compared with one injection site reaction (3%) in the placebo group. Injection site reactions led to temporary treatment interruption in 5 out of 31 (16%) of the anakinra group participants (see Table 3); 3 out of 31 (10%) of the anakinra group participants stopped treatment permanently because of injection site reactions (including one participant who had previously temporarily stopped because of these reactions; see Table 2).
Table 30 summarises the number of AEs that related to an infection by treatment group. The total number of events relating to an infection was larger in the anakinra group (37 events) than in the placebo group (23 events). The number of participants who were prescribed medication for an AE relating to an infection was similar across the groups (anakinra group, n = 11; placebo group, n = 9). Table 31 summarises the blood assessments. At week 8, as expected, there was a fall in the total white cell count, driven by a fall in the neutrophil count (which in two participants was > 50% from baseline) and platelet counts in the anakinra group, but no participants experienced a clinically significant change. This is consistent with the known effects on full blood count. Collectively, this AE profile in people with PPP is comparable to that reported for licensed indications, such as rheumatoid arthritis.
| Event type | Total number of events related to infection | Prescribed medication | ||||
|---|---|---|---|---|---|---|
| Events (n) | Participants (n) | |||||
| Placebo group | Anakinra group | Placebo group | Anakinra group | Placebo group | Anakinra group | |
| AE | 14 | 24 | 6 | 9 | 6 | 8 |
| AR | 9 | 13 | 4 | 5 | 3 | 5 |
| Unclassified (non-serious) | 0 | 0 | 0 | 0 | 0 | 0 |
| SAE | 0 | 0 | 0 | 0 | 0 | 0 |
| SAR | 0 | 0 | 0 | 0 | 0 | 0 |
| Total | 23 | 37 | 10 | 14 | 9 | 11 |
| Time | Placebo group | Anakinra group | Total (n) | Unadjusted mean difference, anakinra – placebo (95% CI) | Adjusteda mean difference, anakinra – placebo (95% CI) | p-value | ||
|---|---|---|---|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | |||||
| Neutrophil count (× 109/l) | ||||||||
| Baseline | 32 | 5.0 (1.7) | 29 | 4.9 (1.5) | 61 | |||
| Week 8 | 30 | 5.1 (1.7) | 31 | 4.3 (2.3) | 61 | –0.8 (–1.8 to 0.3) | ||
| Week 8 change | 30 | 0.2 (1.3) | 29 | –0.7 (2.1) | 59 | –0.9 (–1.8 to 0.0) | –0.9 (–1.7 to 0.01) | 0.053 |
| Total white cell count (× 109/l) | ||||||||
| Baseline | 32 | 8.3 (2.0) | 29 | 8.0 (2.1) | 61 | |||
| Week 8 | 30 | 8.4 (2.6) | 31 | 7.4 (2.6) | 61 | –1.0 (–2.3 to 0.3) | ||
| Week 8 change | 30 | 0.3 (1.7) | 29 | –0.7 (2.2) | 59 | –1.0 (–2.0 to 0.1) | –1.0 (–2.01 to 0.00) | 0.051 |
| Haemoglobin (g/l) | ||||||||
| Baseline | 32 | 139.7 (8.9) | 29 | 137.7 (10.3) | 61 | |||
| Week 8 | 30 | 138.4 (7.1) | 31 | 140.5 (9.2) | 61 | 2.1 (–2.2 to 6.3) | ||
| Week 8 change | 30 | –0.1 (5.1) | 29 | 2.2 (6.4) | 59 | 2.3 (–0.7 to 5.3) | 2.0 (–0.6 to 4.7) | 0.129 |
| Platelets (× 109/l) | ||||||||
| Baseline | 32 | 283.8 (74.3) | 29 | 272.3 (65.6) | 61 | |||
| Week 8 | 30 | 279.9 (68.5) | 31 | 254.1 (57.8) | 61 | –25.8 (–58.2 to 6.7) | ||
| Week 8 change | 30 | 2.0 (27.8) | 29 | –22.2 (33.8) | 59 | –24.2 (–40.3 to –8.1) | –25.3 (–39.6 to –11.1) | < 0.001 |
| CRP (mg/l) | ||||||||
| Baseline | 26 | 5.0 (5.7) | 27 | 6.2 (7.6) | 53 | |||
| Week 8 | 9 | 3.2 (2.4) | 8 | 3.1 (2.2) | 17 | –0.1 (–2.5 to 2.3) | ||
| Week 8 change | 9 | –1.4 (2.0) | 7 | 0.0 (0.6) | 16 | 1.4 (–0.2 to 3.1) | 1.05 (–0.5 to 2.6) | 0.174 |
Open-label extension
A total of 14 out of 64 (22%) participants entered the optional OLE: nine placebo and five anakinra participants. Appendix 11, Table 46, summarises the baseline characteristics of all participants in the original double-blind period against those of participants entering the OLE period. Table 32 summarises the outcomes from the 8-week OLE, including by first-time exposure period. Participants entering the OLE had improved disease severity relative to baseline and required no washout period. Only eight participants provided 8-week follow-up data. On average, investigator-assessed outcome improved over the 8-week open-label period; however, these results should be interpreted with caution because there was no control in this phase and the results are based on a small self-selecting group of participants. Furthermore, because the OLE started part-way through the overall trial (in July 2019, when 40 participants had already completed the RCT component), many had already been moved onto other treatments and did not want to participate.
| Time point | First time exposure | Total | Open-label exposurea (N = 14) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RCT: anakinra group (N = 5) | OLE: placebo group (N = 9) | |||||||||||
| n | Mean (SD) | 95% CI | n | Mean (SD) | 95% CI | n | Mean (SD) | 95% CI | n | Mean (SD) | 95% CI | |
| PP-PASI score (points) | ||||||||||||
| Baseline | 5 | 18.6 (7.9) | 8.8 to 28.3 | 8 | 15.0 (11.3) | 5.5 to 24.4 | 13 | 16.4 (9.9) | 10.3 to 22.4 | 13 | 13.1 (9.8) | 7.2 to 19.0 |
| Week 8 | 5 | 10.5 (7.6) | 1.0 to 20.0 | 6 | 11.0 (9.4) | 1.2 to 20.8 | 11 | 10.8 (8.2) | 5.3 to 16.3 | 8 | 10.0 (8.2) | 3.2 to 16.9 |
| Week 8 change | 5 | –8.0 (6.8) | –16.5 to 0.38 | 6 | –5.2 (9.1) | –14.8 to 4.3 | 11 | –6.5 (7.9) | –11.8 to –1.2 | 8 | –5.4 (7.9) | –12.0 to 1.2 |
| Fresh pustule count | ||||||||||||
| Baseline | 5 | 28.0 (19.7) | 3.5 to 52.5 | 9 | 40.7 (57.6) | –3.6 to 84.9 | 14 | 36.1 (46.9) | 9.1 to 63.2 | 14 | 35.4 (47.1) | 8.1 to 62.6 |
| Week 8 | 5 | 25.2 (19.0) | 1.6 to 48.8 | 6 | 6.0 (9.6) | –4.0 to 16.0 | 11 | 14.7 (17.1) | 3.3 to 26.2 | 8 | 5.4 (8.4) | –1.6 to 12.4 |
| Week 8 change | 5 | –2.8 (33.1) | –43.9 to 38.3 | 6 | –15.2 (37.8) | –54.8 to 24.5 | 11 | –9.5 (34.5) | –32.8 to 13.7 | 8 | –18.1 (33.0) | –45.7 to 9.5 |
| Total pustule count | ||||||||||||
| Baseline | 5 | 86.6 (39.0) | 38.2 to 135.0 | 9 | 102.1 (118.0) | 11.4 to 192.8 | 14 | 96.6 (95.4) | 41.5 to 151.6 | 14 | 102.4 (100.5) | 44.3 to 160.4 |
| Week 8 | 5 | 69.2 (28.2) | 34.2 to 104.2 | 6 | 29.7 (26.0) | 2.4 to 56.9 | 11 | 47.6 (32.9) | 25.5 to 69.7 | 8 | 29.9 (22.0) | 11.5 to 48.3 |
| Week 8 change | 5 | –17.4 (47.0) | –75.8 to 41.0 | 6 | –20.7 (47.5) | –70.5 to 29.2 | 11 | –19.2 (44.9) | –49.3 to 11.0 | 8 | –34.8 (62.1) | –86.7 to 17.2 |
| PPP-IGA baseline, n (%) | N = 5 | N = 9 | N = 14 | N = 14 | ||||||||
| Clear | 0 (0) | 0 (0) | 0 (0) | 0 (0) | ||||||||
| Nearly clear | 0 (0) | 0 (0) | 0 (0) | 0 (0) | ||||||||
| Mild | 1 (20) | 1 (11) | 2 (14) | 3 (21) | ||||||||
| Moderate | 4 (80) | 5 (56) | 9 (64) | 8 (57) | ||||||||
| Severe | 0 (0) | 3 (33) | 3 (21) | 3 (21) | ||||||||
| PPP-IGA week 8, n (%) | N = 5 | N = 6 | N = 11 | N = 7 | ||||||||
| Clear | 0 (0) | 0 (0) | 0 (0) | 0 (0) | ||||||||
| Nearly clear | 0 (0) | 1 (17) | 1 (9) | 0 (0) | ||||||||
| Mild | 3 (60) | 1 (17) | 4 (36) | 1 (14) | ||||||||
| Moderate | 2 (40) | 3 (50) | 5 (45) | 2 (29) | ||||||||
| Severe | 0 (0) | 1 (17) | 1 (9) | 3 (43) | ||||||||
| Serious infection | 0 (0) | 0 (0) | 0 (0) | 1 (14) | ||||||||
| Neutropenia | 0 (0) | 0 (0) | 0 (0) | 0 (0) | ||||||||
A total of 26 non-serious AEs were recorded over the OLE (see Appendix 11, Table 47). A total of five injection site reactions occurred among five participants (5/14, 36%) in the OLE.
Exploratory objectives: mechanistic studies
Abnormal IL-1 signalling in the pathogenesis of pustular psoriasis
Whole-exome sequencing was carried out on the mechanistic samples from trial participants to explore the possibility that abnormal IL-1 activity contributes to disease onset and to investigate whether or not affected individuals harbour mutations in IL-1-related genes.
There was a view to query a set of 14 genes. These were selected because they encode key components of the IL-1 receptor complex (IL1R1, IL1R12, IL1RAP, IL1RA and IL1RN), as well as proteins that regulate IL-1 processing and signalling (LPIN2, MEFV, MVK, NLRP1, NLRP3, NLRP12, NLRC4, PSTPIP1 and MVK).
The COVID-19 pandemic disrupted the transfer of samples from recruiting centres and, thus, data could be generated for only 16 participants. This was compensated for by sequencing 83 unrelated cases ascertained through the PLUM consortium (see Mechanistic sample data set collection from participants with pustular psoriasis for studies investigating disease pathogenesis for more information). Thus, the analysis included a total of 99 PPP participants.
The analysis of these data sets revealed 12 rare and deleterious alleles, affecting eight IL-1-related genes. The only recurrent change was found in IL1R1 (encoding a subunit of the IL-1 receptor), for which a p.Gly398Arg substitution was observed in three participants from the PLUM cohort. Of note, the frequency of this allele among affected individuals was higher than that observed in a publicly available control data set (Avon Longitudinal Study of Parent and Children39) (1.5% vs. 0.1%; p = 0.004).
Comparing the genotypes of responders and non-responders to determine the genetic status of individuals who responded to treatment as a preliminary step for future pharmacogenetic studies
Two trial participants carried rare damaging alleles in IL-1-related genes. One harboured a p.Gly121Val substitution in NLRC4 (encoding a key component of one the inflammasomes responsible for IL-1 processing). They were randomised to the anakinra group of the trial and their PP-PASI score dropped from 9 points (moderate on PGA) at baseline to 3.6 points (almost clear on PGA) at week 8. The second participant showed a p.Arg277Cys variant in IL1RAP (encoding the accessory subunit of the IL-1 receptor). They received the placebo treatment and the severity of their symptoms worsened over the course of the trial (their PP-PASI score increased from 18 points at baseline to 29 points at week 8).
Characterising the immune phenotype of all trial participants
This was carried out to establish whether the disease was associated with alterations in the number or activation status of IL-1-producing cells. Bulk RNA sequencing was used to characterise the immune phenotype of trial participants and investigate the role of IL-1 in propagating abnormal inflammatory responses.
Whole-blood (nine unrelated cases vs. four healthy controls) and skin samples (eight perilesional, three lesional and seven control biopsies) obtained before treatment initiation were analysed.
The analysis of the blood samples uncovered 109 genes that were differentially expressed in cases compared with controls [log2(fold change) > 0.5 or log2(fold change) < –0.5; false discovery rate (FDR) < 0.05].
Pathway enrichment analyses showed that genes involved in phagosome formation were marginally over-represented among those that were upregulated in affected individuals (nominal p-value for over-representation = 4.7 × 10–4; FDR taking into account multiple testing = 0.10).
The analysis of lesional compared with perilesional samples identified 984 differentially expressed genes (DEGs). As anticipated, pathways related to innate signalling and granulocyte infiltration were enriched among DEGs (e.g. IL-8 signalling, IL-6 signalling, granulocyte adhesion and diapedesis; FDR < 10–4 for all). In keeping with these observations, an upstream regulator analysis showed a very significant enrichment of innate cytokines among the molecules that drive the expression of upregulated genes (e.g. oncostatin M, TNF, IL-1b; FDR < 10–25 for all).
The comparison of perilesional with control skin identified 531 DEGs, revealing an unexpected over-representation of pathways related to T-cell activation (e.g. CD28 signalling in T helper cells, iCOS-iCOSL signalling in T helper cells, Th1 and Th2 activation pathway; FDR< 10–8 for all). An analysis of upstream drivers confirmed the involvement of Th1 and Th2 cytokines (IFN-γ, IL-4, IL-15; FDR< 10–15 for all), while also providing evidence for IL-1β activity (FDR < 10–13).
Mechanistic sample data set collection from participants with pustular psoriasis for studies investigating disease pathogenesis
To leverage the recruitment drive underlying the trial, a sister study was set up to collect samples for research purposes. The PLUM (Pustular psoriasis, eLucidating Underlying Mechanisms) study was approved by the Health Research Authority on 7 March 2017. The study has been adopted by 29 recruiting centres, enabling the recruitment of over 370 participants, and is still actively recruiting (at the time of writing this report).
All participants were phenotyped on a standardised case report form and donated blood samples for genetic analyses.
At the time of writing this report, from APRICOT, the study team have obtained 89 DNA samples, 313 plasma samples, 296 RNA samples, 33 lesional skin biopsies and 21 non-lesional skin biopsies. From PLUM, the study team have obtained 309 DNA samples, 78 plasma samples, two serum samples, 91 RNA samples, one lesional skin biopsy and six non-lesional skin biopsies.
Chapter 4 Discussion
Summary findings
This trial assessed the use of an IL-1 receptor antagonist, anakinra, in adult participants with PPP using a novel two-stage adaptive trial design that enabled confirmation of primary outcome within the trial and opportunity for early stopping. In the randomised double-blind phase, there was no evidence of treatment benefit after 8 weeks of anakinra compared with placebo on the primary PP-PASI outcome. Similarly, there was no evidence of statistical superiority of anakinra on the secondary objective of investigator-assessed outcomes or participant-assessed outcomes. In keeping with the known safety profile of anakinra, neutrophil and platelet counts were smaller following treatment, but the difference did not reach clinical significance; there were no SAEs, but there were a larger number of injection site reactions with anakinra relative to placebo.
Interpretation and clinical relevance
Failing to demonstrate efficacy in a trial may be because of a number of factors. First, the study was underpowered. This study size was small (n = 64) and established to detect a large effect size of 0.9 SDs. At baseline, the observed SD for the PP-PASI (n = 64) was 10.5 points; therefore, according to post hoc calculations, 0.9 SDs is approximately equivalent to a change of 9.5 points in the PP-PASI. A standardised effect size was chosen because this was calculated prior to the conformation of the primary outcome for stage 2 and made with input from clinicians and participants. Thus, estimates for some of the secondary outcomes lacked precision. However, we had high follow-up rates and a robust primary outcome demonstrating no benefit, which was supported by secondary outcomes.
Second, the lack of treatment effect may be because of inadequate drug exposure: dose and/or duration. With respect to dose, the (100-mg daily subcutaneous injection) dose of anakinra used in this trial was the same as that used for other licensed indications in adults. Anakinra is licensed for use to treat rheumatoid arthritis and Still's disease (specialist use only) in the UK. 40 Nevertheless, a daily self-administered subcutaneous injection that is often associated with injection site reactions places a significant burden on participants and adherence was variable across participants. Approximately 80% of the anakinra group received ≥ 50% of the total planned daily doses over the 8-week treatment period, but just under half of the participants in the anakinra group received ≥ 90% of the total planned. Notably, the CACE, which estimates the causal effect of treatment for the population of eligible participants who would be able to comply with the treatment schedule, indicated that, compared with the primary ITT effect of –1.65, the treatment effect for an individual who could adhere to at least 90% of total planned injections was just over double, at –3.80, although this finding did not reach statistical significance. The 8-week duration of treatment was selected as being long enough to see an effect on pustules, the expected main target for anakinra, and, based on PPI feedback, the maximum reasonable duration for a placebo-controlled trial requiring daily injections. Nevertheless, the anakinra observed treatment effect (as measured by the PP-PASI) was maintained and slightly increased at 12 weeks (albeit remaining insignificant). Individual participant PP-PASI profiles in the anakinra group over 12 weeks show improvements beyond week 8.
In addition, two randomised trials investigating the effectiveness of guselkumab (a mAb directed against the IL-23 subunit p19) and secukinumab (mAb directed against IL-17) in PPP published during our trial support the notion that treatment duration may be relevant because the treatment benefits for both therapeutic agents, although modest, improved consistently through to 52 weeks (primary outcomes at 16 weeks). 37,38 Thus, taken together (along with the notable finding that a greater proportion of participants in the anakinra group than in the placebo group strongly agreed that the treatment was worthwhile), it seems possible that for people to be able to adhere to treatment, and, if treatment had been given for longer, there may be some treatment benefit with anakinra. If this is the case, have we missed a clinically relevant treatment benefit?
Overall, across primary and secondary outcomes there was no evidence for statistical superiority of anakinra at 8 weeks in the ITT analyses. No clinical differences in usage of rescue or prohibited treatments were observed between the treatment groups, and the analysis to estimate the treatment effect if rescue and prohibited treatment had not been used did not suggest that this was likely to have influenced the observed results. Although the CACE was suggestive that poor adherence may have contributed to the observed lack of efficacy, adherence rates are likely to be even lower in routine clinical practice. Thus, we can confidently conclude from this trial that there is no evidence for clinically relevant benefit with 8 weeks of anakinra for those with PPP. Based on the results of mechanistic studies, it is also reasonable to conclude that genetically determined IL-1 upregulation is not a major disease driver in this condition.
Strengths and limitations
Methodologically this trial has made a number of novel contributions that are potentially generalisable beyond this specific indication. First, as proof-of-concept data and safety information were limited in this rare disease because of the small PPP population, a novel two-stage adaptive trial design with prespecified progression criteria was adopted. 1 This allowed the trial to stop after 24 participants if the results of the interim analysis flagged a concern for safety or if there was no signal for efficacy. A conventional approach to the design of the interim stage would have resulted in a prohibitively large sample size. Second, as there were no validated outcomes to measure disease change for PPP, this design enabled the most reliable outcome with the best distribution properties out of two prespecified candidate outcomes to be used as the primary outcome for the main trial analysis. The PP-PASI was unanimously selected by the independent DMC to be the primary trial outcome following an assessment of reliability and its distributional properties in comparison with the fresh pustule count. There was a large degree of variability in fresh pustule count values between independent site and central assessors from photographs and much less in PP-PASI outcomes between two independent site assessors. Selecting PP-PASI as the primary outcome was supported by the main analysis findings in which the uncertainty around the fresh pustule count treatment effect remained (with wide 95% CIs).
In addition to providing evidence on the efficacy and safety of anakinra, this trial also provides important data on the natural history of PPP and change in disease severity over time. Notably, improvements in outcomes over time were observed in both treatment groups during the trial, a trend observed in other recent placebo-controlled trials of targeted interventions in PPP. 37,38
This might be in part because of a selection bias towards less severe or unstable participants entering the trial: the study was placebo controlled with no guaranteed access to anakinra for the majority of the recruitment period, as well as stipulated washout periods for all interventions with potential or known effectiveness in PPP. The trial cohort was predominantly female, white and current/ex-smokers. However, this is consistent with the observed clinical bias in those with PPP. PPP shows a marked sex bias, with women accounting for 60–90% of affected individuals and is associated with smoking, with up to 90% of participants self-identifying as smokers at the time of diagnosis. 41 Higher prevalence rates have also been reported in white people than in other ethnic groups. 42
Currently validated outcome measures in PPP are lacking, and validation of end points is required. The obtained trial data include measurements of pustule counts, PP-PASI, Investigator Global Assessment and various patient-reported outcome measures. As a result, APRICOT has also provided a valuable source data that will be used in future research to validate outcome measures in PPP. On a related note, it was intriguing that a greater proportion of participants in the anakinra group than in the placebo group strongly agreed that the treatment was worthwhile. We did identify a higher rate of compliance among those who strongly agreed in the anakinra group relative to those who did not strongly agree. But this difference raises the question as to whether or not there are any outcomes that were not measured, such as pain or fatigue, that would be important to explore further in future research.
The results of the OLE should be interpreted cautiously. The OLE was added to the trial primarily to aid recruitment into the trial. Only around 20% of total participants optionally entered this phase of the trial. A greater proportion of participants entering this phase were from the placebo group (27%) relative to the anakinra group (16%). However, overall there was a highly self-selective group entering this phase of the trial, without washout. Moreover, 8-week follow-up data were obtained for only just over half of those entering this phase.
Mechanistic findings
Mechanistic studies provided evidence for potential involvement of IL-1 pathways in a subset of affected individuals. For instance, whole-exome sequencing identified a small number of PPP cases with mutations in IL-1-related genes.
However, because of the small size of the data set and the small number of responders in the trial, it was not possible to draw any definitive conclusions about the genotypes of responders and non-responders.
Bulk RNA sequencing highlighted important differences between the activity of IL-1 in perilesional and lesional skin. Although the immune landscape of the former was dominated by Th1 and Th2 cell activation, the inflammatory milieu of the lesional skin was mostly characterised by neutrophil infiltration. In this context, there was strong evidence for IL-1β activity in lesional skin, whereas other cytokines were dominant in perilesional samples. This suggests that IL-1β is a likely driver of skin pustulation but may be less important for the maintenance of chronic inflammation.
Given that some participants agreed to provide skin samples after treatment initiation, further experiments could be carried out to monitor the expression of IL-1 signature genes at week 1. Differences between responders and non-responders could then be investigated in the light of compliance data.
Finally, the mechanistic studies were accompanied by the recruitment of the PLUM cohort. To our knowledge, this is one of the largest and best-characterised data sets to be held in a single research centre. It has enabled and established a productive partnership with the European Rare And Severe Psoriasis Network (ERASPEN), leading to the publication of two research papers. 41,43 It has also enabled the identification of a novel disease gene that has been described in a recent article. 44
Since the inception of this trial, IL-36 per se has been further validated as a potential therapeutic target in pustular psoriasis, with the first proof-of-concept phase 1 study of the IL-36 receptor antagonist monoclonal antibody BI655130 showing efficacy in GPP in a series of seven participants. 45 As there is only limited in vitro evidence to suggest that an IL-1 blockade may abrogate IL-36 signalling, the fact that we have not demonstrated therapeutic efficacy with anakinra does not preclude IL-36 being of pathogenic importance in localised forms of pustular psoriasis such as PPP. 46
However, although clinical trials have shown that an IL-36 blockade ameliorates the symptoms of GPP, limited efficacy in PPP has recently been shown. 45,47,48 Equally, the biological therapies, particularly those targeting the canonical IL-23/IL-17 pathway, which deliver such impressive clearance rates in plaque psoriasis show only modest benefit, with two recent randomised controlled trials37,38 reporting data for secukinumab and guselkumab, respectively. Thus, our findings alongside the recent published findings in IL-36 suggest a need to determine other drug targets. There are two explanations: (1) both are targeting the wrong pathway or (2) poor drug exposure may be contributing to poor outcomes.
Palmoplantar pustulosis remains an area of high unmet need. We recommend that further research is conducted to (1) identify new drug targets, (2) determine the contributory role of drug exposure (including pharmacokinetics and adherence) and (3) validate outcome measures in PPP.
Conclusion
There was no evidence for clinically relevant benefit with 8 weeks of anakinra in treating PPP and by inference that IL-1 blockade over 8 weeks is not a useful intervention.
Acknowledgements
General acknowledgements
First and foremost, we would like to thank the participants who participated in APRICOT and made this study possible. They all enthusiastically engaged in the research process, were incredibly charitable with their time and in many cases generously donated mechanistic samples.
A particular thanks to Rosemary Wilson and Angela Pushpa-Rajah, who previously managed the study and were central figures in the trial’s development and operation, respectively.
Our sincere gratitude to all the independent members of the TSC (Professor Edel O’Toole, Professor Hervé Bachelez and Dr Stephen Kelly) and the DMC (Professor Deborah Symmons, Dr Mike Ardern-Jones and Professor Simon Skene), who all provided their time and expertise in helping to construct the study and then made sure that it was run with integrity by helping to interpret all the findings throughout the whole duration of the trial.
A special thanks to Mr David Britten, who was our patient representative on the TSC and who throughout the whole study offered invaluable insight in participant outreach and retention, contributed to vital discussions on strategy and helped to form various trial-related materials. Thanks also to Giselle Folloni, who was a passionate advocate for the study on social media.
An additional thanks to Emma Gray and Aysar Al-Rawi, who were the study clinical research associates and provided a comprehensive data monitoring service and general study support.
Thank you to Sobi: its generous support enabled the study to go ahead and for the OLE to be successfully implemented. Sobi kindly provided anakinra and matched placebo for the study.
Our gratitude also extends to Ange Cape and the Guy’s Hospital Pharmacy Manufacturing Unit for processing, labelling and co-ordinating the distribution of the study IMP.
Thank you to Caroline Murphy and the King’s Clinical Trials Unit staff for their assistance with developing the electronic case report form and for their support in delivering the trial.
Finally, a general thanks goes out to everyone whose efforts and support in the conduct and implementation of APRICOT allowed us to successfully complete the study and, consequently, this final report.
The APRICOT Team acknowledges the support of the National Institute for Health Research Clinical Research Network (NIHR CRN).
The study was developed with support from the UK Dermatology Clinical Trials Network (UK DCTN). The UK DCTN is grateful to the British Association of Dermatologists and the University of Nottingham for financial support of the network.
This study was supported by the UK Clinical Research Collaboration-registered King’s Clinical Trials Unit at King’s Health Partners, which is part-funded by the NIHR Biomedical Research Centre for Mental Health at South London and Maudsley NHS Foundation Trust and King’s College London and the NIHR Evaluation, Trials and Studies Co-ordinating Centre.
Thank you to the BADBIR pharmacovigilance team for providing MedDRA coding for AEs. MedDRA® terminology is the international medical terminology developed under the auspices of the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). MedDRA® trademark is registered by IFPMA on behalf of ICH.
Collaborators
Site principal investigators and participating sites
Dr Davide Altamura (Broomfield Hospital).
Dr Suzannah August (Poole Hospital, University Hospitals Dorset NHS Foundation Trust).
Dr Gabrielle Becher (West Glasgow Ambulatory Care Hospital).
Dr Giles Dunnill (Bristol Royal Infirmary).
Dr Adam Ferguson (University Hospitals of Derby and Burton NHS Foundation Trust).
Dr Sharizan Ghaffar (Ninewells Hospital & Medical School).
Dr John Ingram (University Hospital of Wales).
Dr Svetlana Kavakleiva (Royal Lancaster Infirmary).
Dr Effie Ladoyanni (Russells Hall Hospital).
Dr Joyce Leman (Queen Margaret Hospital and Victoria Hospital).
Dr Abby Macbeth (Norfolk and Norwich University Hospitals NHS Foundation Trust).
Dr Areti Makrygeorgou (West Glasgow Ambulatory Care Hospital).
Dr Richard Parslew (Liverpool University Hospitals NHS Foundation Trust).
Professor Vincent Piguet (University Hospital of Wales).
Dr Andrew Pink (Guy’s and St Thomas’ NHS Foundation Trust).
Professor Nick Reynolds (Royal Victoria Infirmary, Newcastle upon Tyne NHS Foundation Trust).
Dr Ashish Sharma (Nottingham Circle).
Dr Catriona Sinclaire (Broomfield Hospital).
Professor Catherine Smith (Guy’s and St Thomas’ NHS Foundation Trust).
Dr Roberto Verdolini (The Princess Alexandra Hospital NHS Trust).
Dr Rachel Wachsmuth (Royal Devon and Exeter NHS Foundation Trust).
Dr Marc Wallace (Addenbrooke’s Hospital).
Professor Richard Warren (Salford Royal NHS Foundation Trust).
Professor Andrew Wright (Bradford Teaching Hospitals NHS Foundation Trust).
Clinical trial co-ordinators/co-investigators/research nurses at the collaborating sites
Addenbrooke’s Hospital: Dr Douglas Maslin.
Bradford Teaching Hospitals NHS Foundation Trust: Carol Denniss, Annette Essex and Jennifer Ott.
Bristol Royal Infirmary: Natasha Beck, Andrew Harris, Dr Debbie Shipley, Vicky Taylor and Jebin Tomy.
Broomfield Hospital: Stacey Cotterell, Victoria Mead, Susan Smolen and Joanne Topliffe.
Guy’s and St Thomas’ NHS Foundation Trust: Aisling Clery, John Gregory, Louise Griffiths, Ewa Kislowska, Freya Meynell, Elizabeth Osuji, Linda Reid-Amoruso, Emmanuel Toni, Dr Wedad Abdelrahman and Dr Richard Woolf.
Liverpool University Hospitals NHS Foundation Trust: Sarah Aldwinckle and Tessa Garland.
Ninewells Hospital & Medical School: Paul Johnston and Janice Rowland.
Norfolk and Norwich University Hospitals NHS Foundation Trust: Catherine Haughton, Dr Jocelyn Keshet-Price, Dr Priya Patel, Gill Pout and David Tomlinson.
Nottingham Circle: Dr Sowjanya Ayyalaraju and Grace Miller.
Poole Hospital University Hospitals Dorset NHS Foundation Trust: Maxine Ashton and Charlotte Barclay.
Queen Margaret Hospital and Victoria Hospital: Anna Gill.
Royal Devon and Exeter NHS Foundation Trust: Dr Peter Acheson and Rob James.
Royal Lancaster Infirmary: Jackie Toomey.
Royal Victoria Infirmary: Ross Farley, Panos Maniatis and Sara Wilkinson.
Russells Hall Hospital: Tracy Henn, Dr Kayalvizhi Mohankumar, Wara Mudunge and Dr John Talaganis.
Salford Royal NHS Foundation Trust: Jean Bastrilles, Marie Durkin, Jackie Evans, Dr Durga Viswesvariah and Caroline White.
The Princess Alexandra Hospital NHS Trust: Ervin Shpuza.
University Hospitals of Derby and Burton NHS Foundation Trust: Dr Joelle Dobson and Dr Jasmine Mann.
University Hospital of Wales: Vianne Britten, Delyth Braim, Bev Gambles, Dr Flora Kiss, Bethan Morse and Dr Jui Vyas.
West Glasgow Ambulatory Care Hospital: Christina MacNeil and Anne Thorrat.
Participant identification sites and staff
Kings College Hospital: Dr Aisling Ryan.
Queen Elizabeth Hospital: Dr Anna Chapman and Dr Kavitha Sundararaj.
Kingston Hospital: Dr Nisha Arujuna and Dr Abigail Fogo.
St George’s University Hospitals NHS Foundation Trust: Dr Alya Abdul-Wahab, Charlotte Fleming and Ruth Lamb.
South Tees Hospitals NHS Foundation Trust: Dr Jaskiran Azad and Jacqueline Dodds.
Portsmouth Hospitals NHS Trust: Sonia Baryschpolec, Dr Hywel Cooper and Dr Alexa Shipman.
Contributions of authors
Suzie Cro (https://orcid.org/0000-0002-6113-1173) (Research Fellow and Clinical Trials Statistician, Study Statistician) designed the statistical analysis plan; carried out the statistical analyses for the DMC and the final stage 2 analysis; contributed to interpretation of the data; was a member of the TMG and TSC; drafted the report; and reviewed the full report.
Victoria Cornelius (https://orcid.org/0000-0002-0080-1065) (Reader in Medical Statistics and Head of Statistics and Trial Methodology, Study Statistician) designed the novel adaptive study and statistical analysis plan; contributed to the grant application, the interpretation of data and the report; obtained grant funding; carried out the statistical analyses for stage 1; was a member of the TMG and TSC; and reviewed the full report.
Francesca Capon (https://orcid.org/0000-0003-2432-5793) (Reader in Inflammation Genetics, Head of Laboratory) contributed to the grant application, was the head of the central laboratory facility; was the scientific lead; was responsible for design, delivery and interpretation with mechanistic studies; edited reports for publication; and drafted relevant parts of this report.
Jonathan Barker (https://orcid.org/0000-0002-9030-183X) (Professor of Medical Dermatology and Honorary Consultant Dermatologist, Senior Co-investigator) contributed to the grant application and recruitment and provided clinical guidance and expertise with critical input into the mechanistic and trial findings.
David Burden (https://orcid.org/0000-0001-7395-9931) (Consultant Dermatologist) contributed to the grant application; provided clinical guidance and expertise, operational leadership and important contributions to the conception and design of the trial and subsequent acquisition of data; and assisted with the interpretation of findings and critical appraisal of all related publications (including this report).
Christopher Griffiths (https://orcid.org/0000-0001-5371-4427) (Foundation Professor of Dermatology) contributed to the grant application; provided clinical guidance and expertise, operational leadership and important contributions to the conception and design of the trial and subsequent acquisition of data; and assisted with the interpretation of findings and critical appraisal of all related publications (including this report).
Helen Jane Lachmann (https://orcid.org/0000-0001-8378-2498) (Professor in Medicine and Honorary Consultant Nephrologist) contributed to the grant application and to the design and conduct of the study throughout the duration of the trial, and provided clinical guidance and expertise, particularly with the use of anakinra.
Helen McAteer (https://orcid.org/0000-0002-2887-5533) (Chief Executive at Psoriasis Association, Patient Representative) contributed to the grant application and to the design and conduct of the study, including the development of patient-facing literature and material, participation in TMG meetings and communication and dissemination of study progress and results; was the lead PPI representative for the trial and provided guidance.
Prakash Patel (https://orcid.org/0000-0001-9971-0130) (Clinical Trials Manager) was the trial manager for the project; was involved with the implementation, oversight and management of the study; was a member of the DMC and TMG; edited reports for publication; and was responsible for co-authoring and reviewing this report.
Andrew Pink (https://orcid.org/0000-0001-5151-5539) (Consultant Dermatologist, Senior Coinvestigator) was a principal investigator for the study, provided clinical guidance and expertise, and contributed to recruitment.
Nick Reynolds (https://orcid.org/0000-0002-6484-825X) (Professor Dermatology and Consultant Dermatologist) contributed to the grant application and to recruitment; was a principal investigator for the study; provided clinical guidance, expertise, operational leadership, including with mechanistic studies and important contributions to the conception and design of the trial and subsequent acquisition of data; and assisted with the interpretation of findings and critical appraisal of all related publications (including this report).
Richard Warren (https://orcid.org/0000-0002-2918-6481) (Professor of Dermatology and Therapeutics and Honorary Consultant Dermatologist) contributed to the grant application and to recruitment; was a principal investigator for the study; provided clinical guidance, expertise, operational leadership, and important contributions to the conception and design of the trial and subsequent acquisition of data; was a member of the TMG; and assisted with the interpretation of findings and critical appraisal of all related publications (including this report).
Catherine Smith (https://orcid.org/0000-0001-9918-1144) (Professor of Dermatology and Therapeutics and Consultant Dermatologist, Chief Investigator) co-conceived the project and led with the grant application, contributed widely to the design and conduct of the trial including recruitment and ongoing development, was responsible for the overall management and oversight of the study throughout its duration, reviewed data analyses, provided clinical guidance and expertise and a critical review of all reports for publication, and co-authored this report.
Publications
Cornelius V, Wilson R, Cro S, Barker J, Burden D, Griffiths C, et al. A small population, randomised, placebo-controlled trial to determine the efficacy of anakinra in the treatment of pustular psoriasis: study protocol for the APRICOT trial. Trials 2018;19:465.
Cro S, Smith C, Wilson R, Cornelius V. Treatment of pustular psoriasis with anakinra: a statistical analysis plan for stage 1 of an adaptive two-staged randomised placebo-controlled trial. Trials 2018;19:534.
Cro S, Patel P, Barker J, Burden D, Griffiths C, Lachmann H, et al. A randomised placebo controlled trial of anakinra for treating pustular psoriasis: statistical analysis plan for stage two of the APRICOT trial. Trials 2020;21:158.
Twelves S, Mostafa A, Dand N, Burri E, Farkas K, Wilson R, et al. Clinical and genetic differences between pustular psoriasis subtypes. J Allergy Clin Immunol 2019;143:1021–6.
Cro S, Cornelius VR, Pink AE, Wilson R, Pushpa-Rajah A, Patel P, et al. Anakinra for palmoplantar pustulosis: results from a randomized, double-blind, multicentre, two-staged, adaptive placebo-controlled trial (APRICOT). Br J Dermatol 2022;186:245–56.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, NETSCC, the EME programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the EME programme or the Department of Health and Social Care.
References
- Cornelius V, Wilson R, Cro S, Barker J, Burden D, Griffiths CEM, et al. A small population, randomised, placebo-controlled trial to determine the efficacy of anakinra in the treatment of pustular psoriasis: study protocol for the APRICOT trial. Trials 2018;19. https://doi.org/10.1186/s13063-018-2841-y.
- Cro S, Cornelius VR, Pink AE, Wilson R, Pushpa-Rajah A, Patel P, et al. Anakinra for palmoplantar pustulosis: results from a randomized, double-blind, multicentre, two-staged, adaptive placebo-controlled trial (APRICOT). Br J Dermatol 2022;186:245-56. https://doi.org/10.1111/bjd.20653.
- NHS UK . Psoriasis – Overview 2018. www.nhs.uk/conditions/psoriasis/ (accessed 1 May 2020).
- Bhutani T, Patel T, Koo B, Nguyen T, Hong J, Koo J. A prospective, interventional assessment of psoriasis quality of life using a nonskin-specific validated instrument that allows comparison with other major medical conditions. J Am Acad Dermatol 2013;69:e79-88. https://doi.org/10.1016/j.jaad.2012.10.009.
- Griffiths C, Christophers E, Barker J, Chalmers R, Chimenti S, Krueger G, et al. A classification of psoriasis vulgaris according to phenotype. Br J Dermatol 2007;156:258-62. https://doi.org/10.1111/j.1365-2133.2006.07675.x.
- Navarini AA, Burden AD, Capon F, Mrowietz U, Puig L, Köks S, et al. European consensus statement on phenotypes of pustular psoriasis. J Eur Acad Dermatol Venereol 2017;31:1792-9. https://doi.org/10.1111/jdv.14386.
- Menter A, Gottlieb A, Feldman S, Voorhees A, Leonardi C, Gordon K, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 1. Overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol 2008;58:826-50. https://doi.org/10.1016/j.jaad.2008.02.039.
- Sampogna F, Gisondi P, Melchi CF, Amerio P, Girolomoni G, Abeni D. Prevalence of symptoms experienced by patients with different clinical types of psoriasis. Br J Dermatol 2004;151:594-9. https://doi.org/10.1111/j.1365-2133.2004.06093.x.
- Farley E, Masrour S, McKey J, Menter A. Palmoplantar psoriasis: a phenotypical and clinical review with introduction of a new quality-of-life assessment tool. J Am Acad Dermatol 2009;60:1024-31. https://doi.org/10.1016/j.jaad.2008.11.910.
- Sampogna F, Tabolli S, Söderfeldt B, Axtelius B, Aparo U, Abeni D. Measuring quality of life of patients with different clinical types of psoriasis using the SF-36. Br J Dermatol 2006;154:844-9. https://doi.org/10.1111/j.1365-2133.2005.07071.x.
- Rapp S, Feldman S, Exum M, Fleischer A, Reboussin D. Psoriasis causes as much disability as other major medical diseases. J Am Acad Dermatol 1999;41:401-7. https://doi.org/10.1016/S0190-9622(99)70112-X.
- Tsoi LC, Spain SL, Knight J, Ellinghaus E, Stuart PE, Capon F, et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet 2012;44:1341-8. https://doi.org/10.1038/ng.2467.
- Obeid G, Do G, Kirby L, Hughes C, Sbidian E, Le Cleach L. Interventions for chronic palmoplantar pustulosis. Cochrane Database Syst Rev 2020;1. https://doi.org/10.1002/14651858.CD011628.pub2.
- Li M, Han J, Lu Z, Li H, Zhu K, Cheng R, et al. Prevalent and rare mutations in IL-36RN gene in Chinese patients with generalized pustular psoriasis and psoriasis vulgaris. J Invest Dermatol 2013;133:2637-9. https://doi.org/10.1038/jid.2013.267.
- Onoufriadis A, Simpson MA, Pink AE, Di Meglio P, Smith CH, Pullabhatla V, et al. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am J Hum Genet 2011;89:432-7. https://doi.org/10.1016/j.ajhg.2011.07.022.
- Marrakchi S, Guigue P, Renshaw BR, Puel A, Pei XY, Fraitag S, et al. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N Engl J Med 2011;365:620-8. https://doi.org/10.1056/NEJMoa1013068.
- Albanesi C, Madonna S, Gisondi P, Girolomoni G. The interplay between keratinocytes and immune cells in the pathogenesis of psoriasis. Front Immunol 2018;9. https://doi.org/10.3389/fimmu.2018.01549.
- Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov 2012;11:633-52. https://doi.org/10.1038/nrd3800.
- Johnston A, Xing X, Wolterink L, Barnes DH, Yin Z, Reingold L, et al. IL-1 and IL-36 are dominant cytokines in generalized pustular psoriasis. J Allergy Clin Immunol 2017;140:109-20.
- Tauber M, Viguier M, Alimova E, Petit A, Lioté F, Smahi A, et al. Partial clinical response to anakinra in severe palmoplantar pustular psoriasis. Br J Dermatol 2014;171:646-9. https://doi.org/10.1111/bjd.13012.
- Viguier M, Guigue P, Pagès C, Smahi A, Bachelez H. Successful treatment of generalized pustular psoriasis with the interleukin-1-receptor antagonist anakinra: lack of correlation with IL1RN mutations. Ann Intern Med 2010;153:66-7. https://doi.org/10.7326/0003-4819-153-1-201007060-00030.
- Cro S, Smith C, Wilson R, Cornelius V. Treatment of pustular psoriasis with anakinra: a statistical analysis plan for stage 1 of an adaptive two-staged randomised placebo-controlled trial. Trials 2018;19. https://doi.org/10.1186/s13063-018-2914-y.
- Electronic Medicines Compendium . Kineret 100 Mg Solution for Injection in a Pre-Filled Syringe 2020. www.medicines.org.uk/emc/product/559/smpc#UNDESIRABLE_EFFECTS (accessed 2 December 2020).
- Cro S, Patel P, Barker J, Burden DA, Griffiths CEM, Lachmann HJ, et al. A randomised placebo controlled trial of anakinra for treating pustular psoriasis: statistical analysis plan for stage two of the APRICOT trial. Trials 2020;21. https://doi.org/10.1186/s13063-020-4103-z.
- National Institute for Health and Care Excellence . Systemic Non-Biological Therapy for Psoriasis 2017. https://pathways.nice.org.uk/pathways/psoriasis#path=view%3A/pathways/psoriasis/systemic-non-biological-therapy-for-psoriasis.xml&content=view-node%3Anodes-drug-choice (accessed 2 December 2020).
- Marsland A, Chalmers R, Hollis S, Leonardi-Bee J, Griffiths C. Interventions for chronic palmoplantar pustulosis. Cochrane Database Syst Rev 2006;1. https://doi.org/10.1002/14651858.CD001433.pub2.
- Lassus A, Geiger JM. Acitretin and etretinate in the treatment of palmoplantar pustulosis: a double-blind comparative trial. Br J Dermatol 1988;119:755-9. https://doi.org/10.1111/j.1365-2133.1988.tb03499.x.
- Kahan BC, Morris TP. Reporting and analysis of trials using stratified randomisation in leading medical journals: review and reanalysis. BMJ 2012;345. https://doi.org/10.1136/bmj.e5840.
- Kahan B, Morris TP. Analysis of multicentre trials with continuous outcomes: when and how should we account for centre effects?. Stat Med 2013;32:1136-49. https://doi.org/10.1002/sim.5667.
- Martin B, Altman D. Agreement between methods of measurement with multiple observations per individual. J Biopharm Stat 2007;17:571-82. https://doi.org/10.1080/10543400701329422.
- McGraw K, Wong S. Forming inferences about some intraclass correlation coefficients. Psychol Methods 1996;1:30-46. https://doi.org/10.1037/1082-989X.1.1.30.
- Moher D, Hopewell S, Schulz KF, Montori V, Gøtzsche PC, Devereaux PJ, et al. CONSORT 2010 explanation and elaboration: updated guidelines for reporting parallel group randomised trials. BMJ 2010;23. https://doi.org/10.1136/bmj.c869.
- European Medicines Agency . ICH E9 (R1) Addendum on Estimands and Sensitivity Analysis in Clinical Trials to the Guideline on Statistical Principles for Clinical Trials 2017. www.ema.europa.eu/en/documents/scientific-guideline/draft-ich-e9-r1-addendum-estimands-sensitivity-analysis-clinical-trials-guideline-statistical_en.pdf (accessed 2 December 2020).
- Carpenter J, Kenward M. Missing Data in Randomised Controlled Trials: A Practical Guide. Birmingham: Health Technology Assessment Methodology Programme; 2007.
- White IR, Horton NJ, Carpenter J, Pocock SJ. Strategy for intention to treat analysis in randomised trials with missing outcome data. BMJ 2011;342. https://doi.org/10.1136/bmj.d40.
- Zink RC, Wolfinger RD, Mann G. Summarizing the incidence of adverse events using volcano plots and time intervals. Clin Trials 2013;10:398-406. https://doi.org/10.1177/1740774513485311.
- Terui T, Kobayashi S, Okubo Y, Murakami M, Hirose K, Kubo H. Efficacy and safety of guselkumab, an anti-interleukin 23 monoclonal antibody, for palmoplantar pustulosis: a randomized clinical trial. JAMA Dermatol 2018;154:309-16. https://doi.org/10.1001/jamadermatol.2017.5937.
- Mrowietz U, Bachelez H, Burden AD, Rissler M, Sieder C, Orsenigo R, et al. Secukinumab for moderate-to-severe palmoplantar pustular psoriasis: results of the 2PRECISE study. J Am Acad Dermatol 2019;80:1344-52. https://doi.org/10.1016/j.jaad.2019.01.066.
- Fraser A, Macdonald-Wallis C, Tilling K, Boyd A, Golding J, Davey Smith G, et al. Cohort profile: the Avon Longitudinal Study of Parents and Children: ALSPAC mothers cohort. Int J Epidemiol 2013;42:97-110. https://doi.org/10.1093/ije/dys066.
- Joint Formulary Committee . British National Formulary [online] n.d. https://bnf.nice.org.uk/drug/anakinra.html#preTreatmentScreeningInformation.
- Benzian-Olsson N, Dand N, Chaloner C, Bata-Csorgo Z, Borroni R, Burden AD, et al. Association of clinical and demographic factors with the severity of palmoplantar pustulosis. JAMA Dermatol 2020;156:1216-22. https://doi.org/10.1001/jamadermatol.2020.3275.
- Parisi R, Iskandar IYK, Kontopantelis E, Augustin M, Griffiths CEM, Ashcroft DM. National, regional, and worldwide epidemiology of psoriasis: systematic analysis and modelling study. BMJ 2020;369. https://doi.org/10.1136/bmj.m1590.
- Twelves S, Mostafa A, Dand N, Burri E, Farkas K, Wilson R, et al. Clinical and genetic differences between pustular psoriasis subtypes. J Allergy Clin Immunol 2019;143:1021-6. https://doi.org/10.1016/j.jaci.2018.06.038.
- Vergnano M, Mockenhaupt M, Benzian-Olsson N, Paulmann M, Grys K, Mahil SK, et al. Loss-of-function myeloperoxidase mutations are associated with increased neutrophil counts and pustular skin disease. Am J Hum Genet 2020;107:539-43. https://doi.org/10.1016/j.ajhg.2020.06.020.
- Bachelez H, Choon SE, Marrakchi S, Burden AD, Tsai TF, Morita A, et al. Inhibition of the interleukin-36 pathway for the treatment of generalized pustular psoriasis. N Engl J Med 2019;380:981-3. https://doi.org/10.1056/NEJMc1811317.
- Hudock KM, Collins MS, Imbrogno M, Snowball J, Kramer EL, Brewington JJ, et al. Neutrophil extracellular traps activate IL-8 and IL-1 expression in human bronchial epithelia. Am J Physiol Lung Cell Mol Physiol 2020;319:L137-L147. https://doi.org/10.1152/ajplung.00144.2019.
- Mrowietz U, Burden AD, Pinter A, Reich K, Schäkel K, Baum P, et al. Spesolimab, an anti-interleukin-36 receptor antibody, in patients with palmoplantar pustulosis: results of a phase IIa, multicenter, double-blind, randomized, placebo-controlled pilot study. Dermatol Ther 2021;11:571-85. https://doi.org/10.1007/s13555-021-00504-0.
- AnaptysBio, Inc . AnaptysBio Reports Imsidolimab POPLAR Phase 2 Clinical Trial in Moderate-to-Severe Palmoplantar Pustulosis (PPP) Did Not Meet Primary Endpoint 2021. https://ir.anaptysbio.com/news-releases/news-release-details/anaptysbio-reports-imsidolimab-poplar-phase-2-clinical-trial (accessed 17 March 2021).
- National Institute for Health and Care Excellence . Psoriasis: Assessment and Management 2017. www.nice.org.uk/guidance/cg153 (accessed 7 December 2020).
Appendix 1 Recruitment materials
FIGURE 19.
Self-referral from website.
Appendix 2 Study information
Contraception guidelines
Women of child-bearing potential were eligible to participate in the study following confirmation of agreement to remain abstinent during the period of IMP dosing and for at least 4 weeks after the last dose. Abstinence was acceptable if it was in line with the preferred and usual lifestyle of the patient. Alternatively, confirmation of the use of single or combined contraceptive methods that resulted in a failure rate of < 1% per year during the IMP dosing and for at least 4 weeks after the last dose of study treatment was also acceptable.
Examples of contraceptive methods with a failure rate of < 1% per year include tubal ligation, male sterilisation, hormonal implants, combined contraceptives (oral/injection) and certain intrauterine devices. Alternatively, two methods (e.g. two barrier methods, such as a condom and a cervical cap) may be used to achieve a failure rate of < 1%. Barrier methods must always be supplemented with use of spermicide.
| Allowed visit window: ± 3 days | Screening | Treatment period | Follow-up | Safety follow-up | |||
|---|---|---|---|---|---|---|---|
| Visit 0 | Visit 1 | Visit 2 | Visit 3 | Visit 4 | Visit 5a | Visit 6 | |
| Baseline | Week 1 | Week 4 | Week 8 | Week 12 | Week 20 | ||
| Study enrolment | Treatment initiation | Treatment end | Study end | ||||
| Informed consent | ✗ | ||||||
| Randomisation | ✗ | ||||||
| Medical history | ✗ | ✗ | |||||
| Physical examination | ✗ | ||||||
| Vital signs | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| Fresh pustule countb | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| Total pustule countb | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| PP-PASIb (× 2) | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| PPP–IGAb (× 2) | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| PASI (plaque psoriasis only) | ✗ | ✗ | ✗ | ✗ | ✗ | ||
| BSA | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| PGA | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| PP-QoLb | ✗ | ✗ | ✗ | ||||
| DLQI | ✗ | ✗ | ✗ | ||||
| EQ-5D-3L | ✗ | ✗ | ✗ | ||||
| SMS/text compliance | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| Acceptability questionnaire | ✗ | ||||||
| Photography | ✗ | ✗ | ✗ | ✗ | |||
| Chest X-ray | ✗ | ||||||
| TBSpot.TBc | ✗ | ||||||
| HIV, HBV and HCV | ✗ | ||||||
| Safety bloodsd,e | ✗f | ✗f | ✗ | ✗ | ✗ | ✗ | |
| bHCG (blood)g | ✗ | ✗ | ✗ | ||||
| Exploratory laboratory tests (see Table 35) | ✗ | ✗ | ✗ | ✗ | ✗ | ||
| Urine analysis (dipstix) | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| Prescribing and dispensing trial IMP | ✗ | ✗ | ✗ | ||||
| Concomitant meds | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| AE monitoring | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |
| Allowed visit window: ± 3 days | Screeninga | Treatment period | Safety follow-up | |||
|---|---|---|---|---|---|---|
| Visit OLE 0a | Visit OLE 1 | Visit OLE 2 | Visit OLE 3 | Visit OLE 4 | Visit OLE 5 | |
| Baseline | Week 1 | Week 4 | Week 8 | Week 20 | ||
| Treatment initiation | Treatment end | Study end | ||||
| Informed consent | ✗a | |||||
| Eligibility review | ✗a | ✗ | ||||
| Physical examination | ✗a | |||||
| Check washout period | ✗a | ✗ | ||||
| Vital signs | ✗a | ✗ | ✗ | ✗ | ✗ | |
| Fresh pustule count | ✗ | ✗ | ||||
| Total pustule count | ✗ | ✗ | ||||
| PP-PASI | ✗ | ✗ | ||||
| PPP–IGA | ✗ | ✗ | ||||
| PASI (plaque psoriasis only) | ✗ | ✗ | ||||
| Safety bloodsb,c | ✗a,d | ✗d | ✗ | ✗ | ✗ | |
| TBSpot.TBe | ✗a | |||||
| HIV, HBV and HCV | ✗a | |||||
| bHCG (blood)f | ✗a | ✗ | ||||
| Urine analysis (dipstix) | ✗a | ✗ | ✗ | ✗ | ✗ | |
| Prescribing and dispensing anakinra | ✗ | |||||
| Concomitant medications | ✗a | ✗ | ✗ | ✗ | ✗ | ✗ |
| AE monitoring | ✗a | ✗ | ✗ | ✗ | ✗ | ✗ |
| Laboratory test | Screening | Treatment period | Follow-up | Safety follow-up | |||
|---|---|---|---|---|---|---|---|
| Visit 0 | Visit 1 | Visit 2 | Visit 3 | Visit4 | Visit 5 | Visit 6 | |
| Baseline | Week 1 | Week 4 | Week 8 | Week 12 | Week 20 | ||
| Study enrolment | Treatment initiation | Treatment end | Study end | ||||
| DNAa (1 × 10 ml) | ✗ | ||||||
| RNA isolation (1 × 3 ml)b | ✗ | ✗ | ✗ | ✗ | ✗ | ||
| Immune phenotyping (1 × 25 ml)b | ✗ | ||||||
| Plasma (1 × 5 ml)b | ✗ | ✗ | ✗ | ✗ | ✗ | ||
| Skin microbiopsy (optional),b,c unaffected skin | ✗ | ||||||
| Skin microbiopsy (optional),b,c affected skin | ✗ | ✗ | |||||
| Hair plucks (optional)b | ✗ | ✗ | ✗ | ✗ | ✗ | ||
Appendix 3 Concomitant medication, prohibited medication and rescue therapy information
Topical therapy
Emollient therapy was permitted throughout the trial.
For injection sites
To treat the common side effect of an injection site reaction, the use of topical mild corticosteroid (e.g. hydrocortisone up to 2.5%) or antihistamine cream/ointment was permitted.
For plaque psoriasis
Use of emollients was recommended as the first-line intervention, but mild–moderate topical corticosteroids were permitted at the discretion of the investigator as a second-line intervention for plaques at sites other than the hands and feet. Gloves should have been worn for application.
For palmoplantar pustulosis
Rescue therapy
Investigator-directed ‘rescue’ medication in the form of a potent corticosteroid (e.g. mometasone furoate, betamethasone valerate ointment or cream) once per day to affected areas of PPP could be dispensed if necessary to provide substantial symptomatic relief. Rescue medication could be prescribed as part of normal clinical care, and the volume prescribed recorded at study visits to evaluate any potential confounding effect of topical corticosteroid use.
Systemic therapy
Any concomitant treatments for other indications that are not listed in the prohibited medication section should have been at a stable dose for at least 4 weeks before the first study treatment administration. Dose adjustments of these treatments should have been avoided during the study.
Prohibited medication for the initial double-blind treatment stage
Any therapy likely to have efficacy in PPP or psoriasis or to compound the potential immunosuppressive effects of anakinra was prohibited and stipulated washout periods should have been adhered to. If treatment with any of the prohibited treatments was essential, the patient should have notified the study team and they should have been withdrawn from the trial.
| Rule | Therapy |
|---|---|
| Prohibited |
Very potent topical corticosteroids (e.g. Dermovate) Any topical treatment that is likely to have an impact on signs and symptoms of PPP (e.g. corticosteroids, vitamin D analogues, calcineurin inhibitors, retinoids, keratolytics, tar and urea) Phototherapy or PUVA Methotrexate, cyclosporine, acitretin, alitretinoin and fumaric acid esters Etanercept or adalimumab Infliximab, ustekinumab and secukinumab Other TNF antagonists Other systemic immunosuppressive therapy Other investigational monoclonal antibody Other investigational drugs |
| Allowable topical therapy |
Emollients Topical hydrocortisone and antihistamine for injection site reactions Mild topical corticosteroids for the treatment of psoriasis at sites other than hands and feet, applied with gloves |
| Allowable therapy | Oral antihistamine for injection site reactions |
| ‘Rescue’ topical therapy | Potent corticosteroid od. To be dispensed only by the study team, at the investigator’s discretion. Amounts prescribed to be recorded |
Prohibited medication for the open-label extension
Stipulated washout periods should have been adhered to. Concomitant topical treatment (only) was allowed only during the OLE stage. If treatment with any of the prohibited systemic treatments (as indicated in Table 37) was essential, the patient should have notified the study team and they should have been withdrawn from the trial and anakinra should have been discontinued.
| Rule | Therapy |
|---|---|
| Prohibited |
Phototherapy or PUVA Methotrexate, cyclosporine, acitretin, alitretinoin and fumaric acid esters Etanercept or adalimumab Infliximab, ustekinumab and secukinumab Other TNF antagonists Other systemic immunosuppressive therapy Other investigational monoclonal antibody Other investigational drugs |
| Allowable topical therapy |
Emollients Topical hydrocortisone, antihistamine for injection site reactions Mild topical corticosteroids for the treatment of psoriasis at sites other than hands and feet, applied with gloves Very potent topical corticosteroids (e.g. dermovate) These topical treatments: corticosteroids, vitamin D analogues, calcineurin inhibitors, retinoids, keratolytics, tar and urea |
| Allowable therapy | Oral antihistamine for injection site reactions |
Appendix 4 Amendments and extensions summary
Please note that, in addition to protocol updates, other study documents (e.g. patient information sheets, informed consent forms, GP letters and posters) were often also amended to reflect the study changes and submitted for regulatory body approval as part of each amendment. In addition, small typographical and grammatical corrections were also often carried out to the study documents and submitted for regulatory body approval as part of each amendment.
| Amendment | Regulatory body approval date | Brief description of amendment |
|---|---|---|
| Substantial | ||
| 1 | 16 May 2016 |
|
| 2 | 12 October 2016 |
|
| 3 | 20 December 2016 |
|
| 4 | 21 June 2017 |
|
| 5 | 7 August 2017 |
|
| 6 | 5 December 2017 |
|
| 7 | 19 June 2018 |
|
| 8 | 27 June 2018 |
|
| 9 | 15 August 2018 |
|
| 10 | 3 October 2018 |
|
| 11 | 18 June 2019 |
|
| 12 | 17 October 2019 |
|
| Non-substantial | ||
| 1 | 22 February 2017 |
|
| 2 | Not applicable |
|
| 3 | 11 October 2018 |
|
| 4 | Not applicable |
|
| 5 | 27 December 2019 |
|
| 6 | 16 March 2020 |
|
| 7 | 18 May 2020 |
|
The funder approved a 16-month no-cost extension to the study in November 2018.
Following the start of the COVID-19 pandemic, the funder also approved a 3-month no-cost extension to the study in June 2020.
Appendix 5 Adverse events listing
| AE term | Placebo group events (n) | Anakinra group events (n) | Total events (n) | Placebo group participants (n) | Anakinra group participants (n) | Total participants (n) |
|---|---|---|---|---|---|---|
| Abdominal discomfort | 1 | 0 | 1 | 1 | 0 | 1 |
| Abdominal pain lower | 0 | 1 | 1 | 0 | 1 | 1 |
| Arthralgia | 2 | 1 | 3 | 2 | 1 | 3 |
| Back injury | 1 | 0 | 1 | 1 | 0 | 1 |
| Biopsy (skin) | 0 | 1 | 1 | 0 | 1 | 1 |
| Blood creatinine increased | 1 | 0 | 1 | 1 | 0 | 1 |
| Blood pressure increased | 0 | 1 | 1 | 0 | 1 | 1 |
| C-reactive protein increased | 1 | 1 | 2 | 1 | 1 | 2 |
| Catarrh | 1 | 0 | 1 | 1 | 0 | 1 |
| Cellulitis | 1 | 0 | 1 | 1 | 0 | 1 |
| Constipation | 0 | 1 | 1 | 0 | 1 | 1 |
| Contusion | 2 | 1 | 3 | 2 | 1 | 3 |
| Cough | 2 | 5 | 7 | 2 | 4 | 6 |
| Cystitis | 1 | 0 | 1 | 1 | 0 | 1 |
| DNA antibody positive | 1 | 0 | 1 | 1 | 0 | 1 |
| Decreased appetite | 1 | 0 | 1 | 1 | 0 | 1 |
| Depressed mood | 0 | 3 | 3 | 0 | 3 | 3 |
| Dermatitis | 1 | 0 | 1 | 1 | 0 | 1 |
| Diabetes mellitus | 1 | 0 | 1 | 1 | 0 | 1 |
| Diarrhoea | 0 | 5 | 5 | 0 | 5 | 5 |
| Dizziness | 0 | 1 | 1 | 0 | 1 | 1 |
| Ear pain | 1 | 0 | 1 | 1 | 0 | 1 |
| Eosinophilia | 0 | 1 | 1 | 0 | 1 | 1 |
| Epistaxis | 2 | 0 | 2 | 1 | 0 | 1 |
| Flushing | 1 | 0 | 1 | 1 | 0 | 1 |
| Folliculitis | 1 | 2 | 3 | 1 | 1 | 2 |
| Gestational diabetes | 0 | 1 | 1 | 0 | 1 | 1 |
| Glomerular filtration rate decreased | 1 | 0 | 1 | 1 | 0 | 1 |
| Glucose urine present | 1 | 0 | 1 | 1 | 0 | 1 |
| Haematuria | 1 | 2 | 3 | 1 | 2 | 3 |
| Head injury | 0 | 1 | 1 | 0 | 1 | 1 |
| Headache | 4 | 6 | 10 | 2 | 6 | 8 |
| Hepatitis B antibody positive | 0 | 1 | 1 | 0 | 1 | 1 |
| Hepatotoxicity | 1 | 4 | 5 | 1 | 4 | 5 |
| Hyperkalaemia | 1 | 0 | 1 | 1 | 0 | 1 |
| Hypertension | 0 | 1 | 1 | 0 | 1 | 1 |
| Influenza | 1 | 0 | 1 | 1 | 0 | 1 |
| Influenza-like illness | 1 | 0 | 1 | 1 | 0 | 1 |
| Injection site discomfort | 0 | 1 | 1 | 0 | 1 | 1 |
| Injection site erythema | 1 | 2 | 3 | 1 | 2 | 3 |
| Injection site pain | 0 | 1 | 1 | 0 | 1 | 1 |
| Injection site pruritus | 1 | 0 | 1 | 1 | 0 | 1 |
| Injection site rash | 0 | 1 | 1 | 0 | 1 | 1 |
| Injection site reaction | 1 | 20 | 21 | 1 | 19 | 20 |
| Injection site swelling | 1 | 2 | 3 | 1 | 2 | 3 |
| Lethargy | 1 | 0 | 1 | 1 | 0 | 1 |
| Lower respiratory tract infection | 3 | 3 | 6 | 3 | 3 | 6 |
| Lymphadenopathy | 1 | 0 | 1 | 1 | 0 | 1 |
| Malaise | 2 | 0 | 2 | 1 | 0 | 1 |
| Mean cell volume increased | 1 | 0 | 1 | 1 | 0 | 1 |
| Menorrhagia | 0 | 1 | 1 | 0 | 1 | 1 |
| Metrorrhagia | 0 | 1 | 1 | 0 | 1 | 1 |
| Migraine | 2 | 0 | 2 | 2 | 0 | 2 |
| Monocyte count increased | 1 | 0 | 1 | 1 | 0 | 1 |
| Myalgia | 1 | 0 | 1 | 1 | 0 | 1 |
| Nasopharyngitis | 3 | 5 | 8 | 3 | 4 | 7 |
| Nausea | 2 | 2 | 4 | 2 | 2 | 4 |
| Neuralgia | 0 | 1 | 1 | 0 | 1 | 1 |
| Neutrophil count increased | 0 | 1 | 1 | 0 | 1 | 1 |
| Oedema peripheral | 0 | 1 | 1 | 0 | 1 | 1 |
| Oropharyngeal pain | 1 | 3 | 4 | 1 | 3 | 4 |
| Osteoporosis | 1 | 0 | 1 | 1 | 0 | 1 |
| Pain in extremity | 1 | 1 | 2 | 1 | 1 | 2 |
| Pain of skin | 0 | 1 | 1 | 0 | 1 | 1 |
| Pharyngeal oedema | 1 | 0 | 1 | 1 | 0 | 1 |
| Post-procedural infection | 1 | 0 | 1 | 1 | 0 | 1 |
| Pregnancy | 0 | 1 | 1 | 0 | 1 | 1 |
| Proteinuria | 0 | 1 | 1 | 0 | 1 | 1 |
| Pruritus | 0 | 1 | 1 | 0 | 1 | 1 |
| Psoriasis | 2 | 3 | 5 | 2 | 3 | 5 |
| Psoriatic arthropathy | 1 | 0 | 1 | 1 | 0 | 1 |
| Pustular psoriasis | 2 | 2 | 4 | 2 | 2 | 4 |
| Pyuria | 0 | 1 | 1 | 0 | 1 | 1 |
| Rash (macular) | 1 | 0 | 1 | 1 | 0 | 1 |
| Rash (papular) | 1 | 0 | 1 | 1 | 0 | 1 |
| Rhinitis | 1 | 1 | 2 | 1 | 1 | 2 |
| Rhinitis allergic | 0 | 1 | 1 | 0 | 1 | 1 |
| Rhinorrhoea | 0 | 1 | 1 | 0 | 1 | 1 |
| Sinusitis | 1 | 2 | 3 | 1 | 2 | 3 |
| Skin infection | 2 | 1 | 3 | 2 | 1 | 3 |
| Skin irritation | 1 | 0 | 1 | 1 | 0 | 1 |
| Skin lesion | 1 | 0 | 1 | 1 | 0 | 1 |
| Synovial cyst | 1 | 0 | 1 | 1 | 0 | 1 |
| Synovitis | 1 | 0 | 1 | 1 | 0 | 1 |
| Tonsillitis | 0 | 1 | 1 | 0 | 1 | 1 |
| Toothache | 1 | 0 | 1 | 1 | 0 | 1 |
| Transaminases increased | 0 | 1 | 1 | 0 | 1 | 1 |
| Upper respiratory tract infection | 0 | 1 | 1 | 0 | 1 | 1 |
| Urinary tract infection | 3 | 4 | 7 | 3 | 4 | 7 |
| Urine analysis abnormal | 0 | 1 | 1 | 0 | 1 | 1 |
| Viral infection | 2 | 0 | 2 | 2 | 0 | 2 |
| Visual acuity reduced | 1 | 0 | 1 | 1 | 0 | 1 |
| Vomiting | 0 | 2 | 2 | 0 | 2 | 2 |
| White blood cell count increased | 0 | 1 | 1 | 0 | 1 | 1 |
| White blood cells urine positive | 2 | 0 | 2 | 2 | 0 | 2 |
| Urine analysis abnormal | 1 | 1 | 2 | 1 | 1 | 2 |
Appendix 6 Participant recruitment and randomisation
| Site name | Date opened | Number of participants randomised, n (%) |
|---|---|---|
| Guy’s and St Thomas’ NHS Foundation Trust | 9 August 2016 | 21 (33) |
| Salford Royal NHS Foundation Trust | 20 October 2016 | 7 (11) |
| Royal Victoria Infirmary | 20 October 2016 | 4 (6) |
| University Hospital of Wales | 3 February 2017 | 4 (6) |
| Ninewells Hospital & Medical School | 3 April 2017 | 1 (2) |
| Liverpool University Hospitals NHS Foundation Trust | 12 June 2017 | 5 (8) |
| Bradford Teaching Hospitals NHS Foundation Trust | 28 June 2017 | 1 (2) |
| Royal Lancaster Infirmary | 15 August 2017 | 1 (2) |
| Russells Hall Hospital | 29 August 2017 | 1 (2) |
| Bristol Royal Infirmary | 6 September 2017 | 5 (8) |
| Addenbrooke’s Hospital | 3 January 2018 | 0 (0) |
| Poole Hospital NHS Foundation Trust University Hospitals Dorset | 3 January 2018 | 2 (3) |
| The Princess Alexandra Hospital NHS Trust | 23 May 2018 | 0 (0) |
| Norfolk and Norwich University Hospitals NHS Foundation Trust | 5 June 2018 | 4 (6) |
| University Hospitals of Derby and Burton NHS Foundation Trust | 15 August 2018 | 2 (3) |
| Royal Devon and Exeter NHS Foundation Trust | 5 November 2018 | 2 (3) |
| Nottingham Circle | 20 November 2018 | 0 (0) |
| Broomfield Hospital | 27 November 2018 | 2 (3) |
| West Glasgow Ambulatory Care Hospital | 7 December 2018 | 2 (3) |
| Queen Margaret Hospital and Victoria Hospital | 23 April 2019 | 0 (0) |
| Total randomised | 66 | |
FIGURE 20.
Planned vs. actual randomisation.
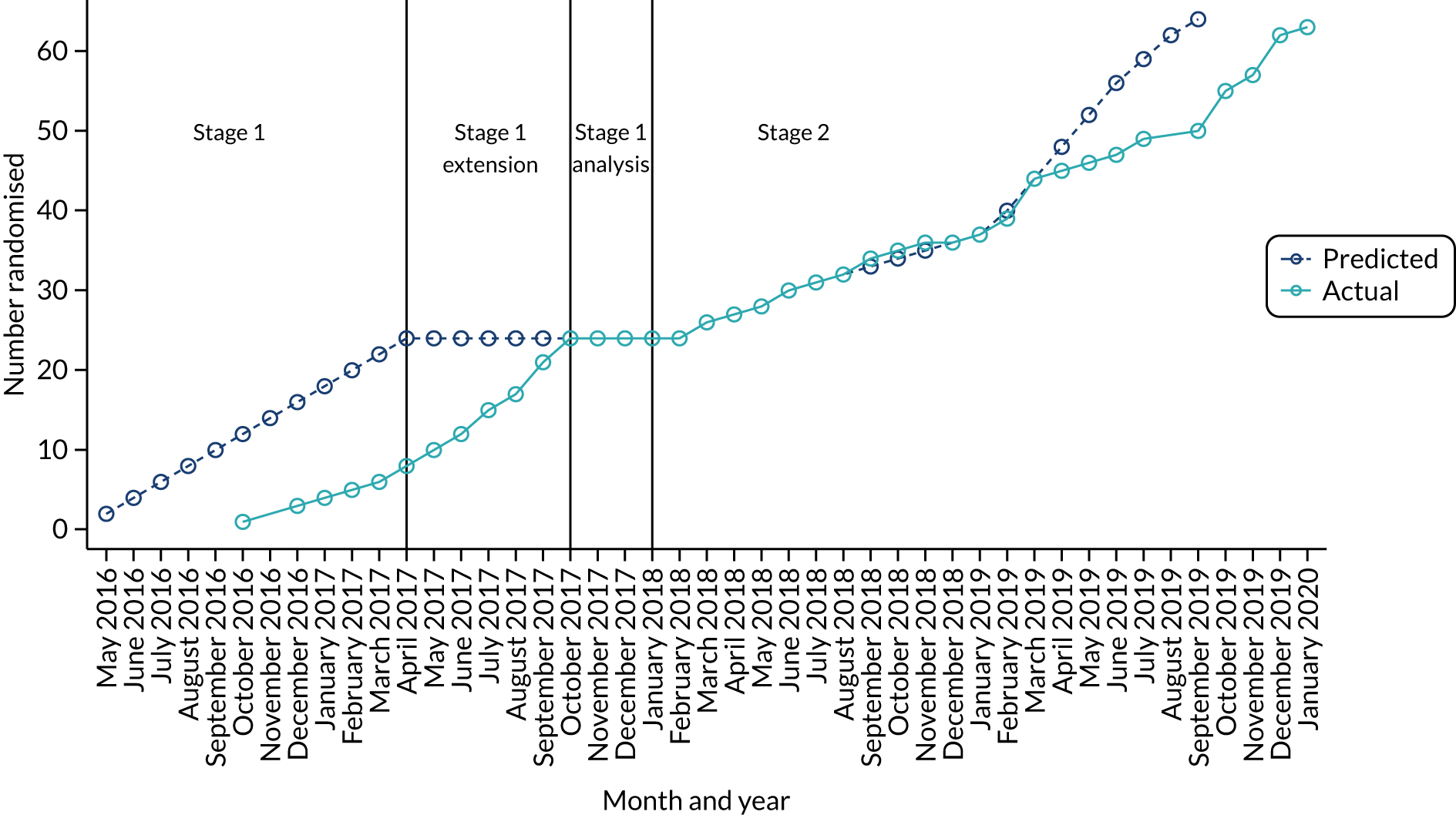
| Site name | Number of participants identified (n) | Percentage of participants randomised from those identified | Number of participants randomised, n (%) |
|---|---|---|---|
| Guy’s and St Thomas’ NHS Foundation Trust | 94 | 22 | 21 (33) |
| Salford Royal NHS Foundation Trust | 51 | 14 | 7 (11) |
| Royal Victoria Infirmary | 17 | 24 | 4 (6) |
| University Hospital of Wales | 40 | 10 | 4 (6) |
| Ninewells Hospital & Medical School | 11 | 9 | 1 (2) |
| Liverpool University Hospitals NHS Foundation Trust | 22 | 23 | 5 (8) |
| Bradford Teaching Hospitals NHS Foundation Trust | 22 | 5 | 1 (2) |
| Royal Lancaster Infirmary | 1 | 100 | 1 (2) |
| Russells Hall Hospital | 10 | 10 | 1 (2) |
| Bristol Royal Infirmary | 18 | 28 | 5 (8) |
| Addenbrooke’s Hospital | 9 | 0 | 0 (0) |
| Poole Hospital NHS Foundation Trust University Hospitals Dorset | 3 | 67 | 2 (3) |
| The Princess Alexandra Hospital NHS Trust | 4 | 0 | 0 (0) |
| Norfolk and Norwich University Hospitals NHS Foundation Trust | 27 | 15 | 4 (6) |
| University Hospitals of Derby and Burton NHS Foundation Trust | 25 | 8 | 2 (3) |
| Royal Devon and Exeter NHS Foundation Trust | 2 | 100 | 2 (3) |
| Nottingham Circle | 4 | 0 | 0 (0) |
| Broomfield Hospital | 8 | 25 | 2 (3) |
| West Glasgow Ambulatory Care Hospital | 5 | 40 | 2 (3) |
| Queen Margaret Hospital and Victoria Hospital | 1 | 0 | 0 (0) |
| Total | 374 | 17 | 64 |
Screening data are not consistently recorded across sites. Therefore, the reported total number of participants identified for screening is an underestimate of true number of screened participants.
Appendix 7 Patient and public involvement
Introduction
Pustular psoriasis had been identified as an area of unmet need during the development of the NICE guidelines49 on the management of psoriasis, which had substantial input from participants and the public, with the lack of effective/safe interventions in pustular psoriasis being highlighted as a research gap. In addition, the Psoriasis Association, the largest and most important UK patient organisation for people with psoriasis, had also made a major commitment to this area by funding two PhD (Doctor of Philosophy) studentships at King’s College London investigating disease pathogenesis. The study group, therefore, invited Helen McAteer, Chief Executive of the Psoriasis Association, to partner with the study group to ensure that we effectively engaged with participants and the public in the design, implementation, evaluation and communication of programme of research.
Aim
To develop a trial PPI infrastructure that would:
-
advise on study design and ethics issues
-
develop patient support materials and questionnaires
-
facilitate shared learning and reflection from the study
-
advise on the best methods to disseminate research outputs and review articles for publication in the lay press.
Methods
Qualitative feedback through the Patient and Lay Members Group
When the outline application was being made, one-to-one discussions were held with participants (n = 3, two with APP requiring systemic therapy, and personal experience as study participants in a placebo-controlled randomised controlled trial) for their advice on the study design and outcome measures.
For the development of the full application, a formal PLAG meeting was held that consisted of one patient with APP, one patient with GPP, one patient with psoriasis, a NICE psoriasis guideline committee member, Helen McAteer, the Biomedical Research Centre PPI co-ordinator and (the chief investigator) Professor Catherine Smith.
Patient representation in trial committees
Helen McAteer was enlisted as a co-applicant to the study to support study design, for ethics issue consultation and to help with recruitment using social/web-based media. She was also a member of the Trial Steering Committee (TSC) and the monthly Trial Management Group (TMG).
A patient representative (David Britten) was part of the TSC and regularly attended and actively participated in these meetings to provide guidance and support to APRICOT.
Social media communications
During the study, Giselle Folloni, a very active PPP patient based in Italy, who is very active on the PPP community on Facebook (Facebook, Inc., Menlo Park, CA, USA; www.facebook.com), became an advocate for the study. They regularly received study updates and newsletters and promoted the study on Facebook.
Patient and public involvement events
The APRICOT study was regularly mentioned at all of the PPI events held by the St John’s Institute of Dermatology (at Guy’s and St Thomas’ NHS Foundation Trust), Manchester and Newcastle. During these events, participants were asked for their feedback and suggestions about the trial experience (for themselves) and how it could be potentially improved. These findings were fed back to the central co-ordinating team for consideration by the TMG/TSC.
Impact of patient and public involvement on the study results
Trial design and development
The discussions held with participants for the outline application motivated the decision to limit the trial treatment duration to 8 weeks and to also extend the scope of the patient-orientated outcome measures for the study, including the pustular psoriasis-specific QoL.
The PLAG meeting shaped the trial design and led to amendments (e.g. the inclusion of a rescue topical corticosteroid) that were applied to the full application prior to formal submission. With respect to samples for mechanistic studies, the PLAG considered and approved the planned sampling strategy, including skin biopsies.
Helen McAteer was very influential in the study design and also provided ethics guidance for the amendments that were made to the trial that affected participants.
Trial delivery
The Psoriasis Association participated regularly in discussions about recruitment strategies and were extremely helpful in advertising APRICOT via social media, its magazine and its website. It helped to guide participants, directing any queries to the study website and e-mail address (where interested parties could self-refer). It also helped with the review and amendment of study materials. David Britten (the TSC patient representative) was very active throughout the whole study and offered meaningful insight into participant outreach and retention. He also contributed to vital discussions on strategy and study design and helped to form various trial-related materials. For example, for the submission of substantial amendment 12 (to add a new exclusion criterion relating to Still’s disease), his input and feedback was used to gauge the scope of changes needed for the protocol and participant information leaflet.
The PPP patient (Giselle Folloni) in Italy was an immense promoter and supporter of the trial, and their Facebook posts led PPP participants to the study website and raised an awareness of both APRICOT and PLUM in the PPP community.
The study design involved a randomised controlled trial with a placebo. Patient feedback suggested that some were concerned about missing out on treatment if they were in the placebo group and this became a concern that was instrumental in devising and implementing the OLE to help boost study recruitment.
Furthermore, in response to feedback from the TSC patient representative, various sites and potential participants, study sites were encouraged to ensure that it was made clear to all potential and actual participants that travel expenses would be reimbursed in full. This was important given that a number of participants had to travel significant distances to attend study visits.
Trial results and dissemination
Helen McAteer has co-authored a number of related publications and has critically appraised this report. We have drafted a results communication plan including a PPI event. This is an ongoing project to be delivered over 2021/22.
Discussion and conclusions
The PPI experience in APRICOT was astoundingly excellent and decisive, and enabled the study to recruit to target.
The input gained from the PPI was important in designing the study and also in shaping the trial once running. Fundamental changes to the trial design (e.g. the removal of some visits in substantial amendment 4 and the addition of the OLE as part of substantial amendment 12) were heavily informed by PPI. The same can be said for development of the study-related literature and patient-facing documentation and the amendments made to them throughout the study.
Patient and public involvement was also crucial in the promotion of the trial, which ultimately generated study awareness and helped to enhance recruitment.
Beyond the study, Helen McAteer and the Psoriasis Association have provided useful guidance on how to disseminate the study results and logistical support in doing so.
Reflections/critical perspective
The PPI network had a completely positive effect on the study and there were no negative factors of the involvement of the individuals and their conduct.
One possible suggestion that could have enhanced the PPI would be to seek to include more than one formal lay patient representative to the study infrastructure to reflect the PPP patient population more accurately and to enable more diverse conversations and guidance. A recent PPI event held during the pandemic over Zoom (Zoom Video Communications, San Jose, CA, USA) on psoriasis had more than 120 attendees; the question and answer format worked well, suggesting that virtual formats may be an efficient, and cost-effective, way of engaging a wider audience that would appeal to participants. We are exploring this format for our PPI results dissemination strategy.
Appendix 8 Study photography
FIGURE 21.
Study photograph taken of one participant’s soles.
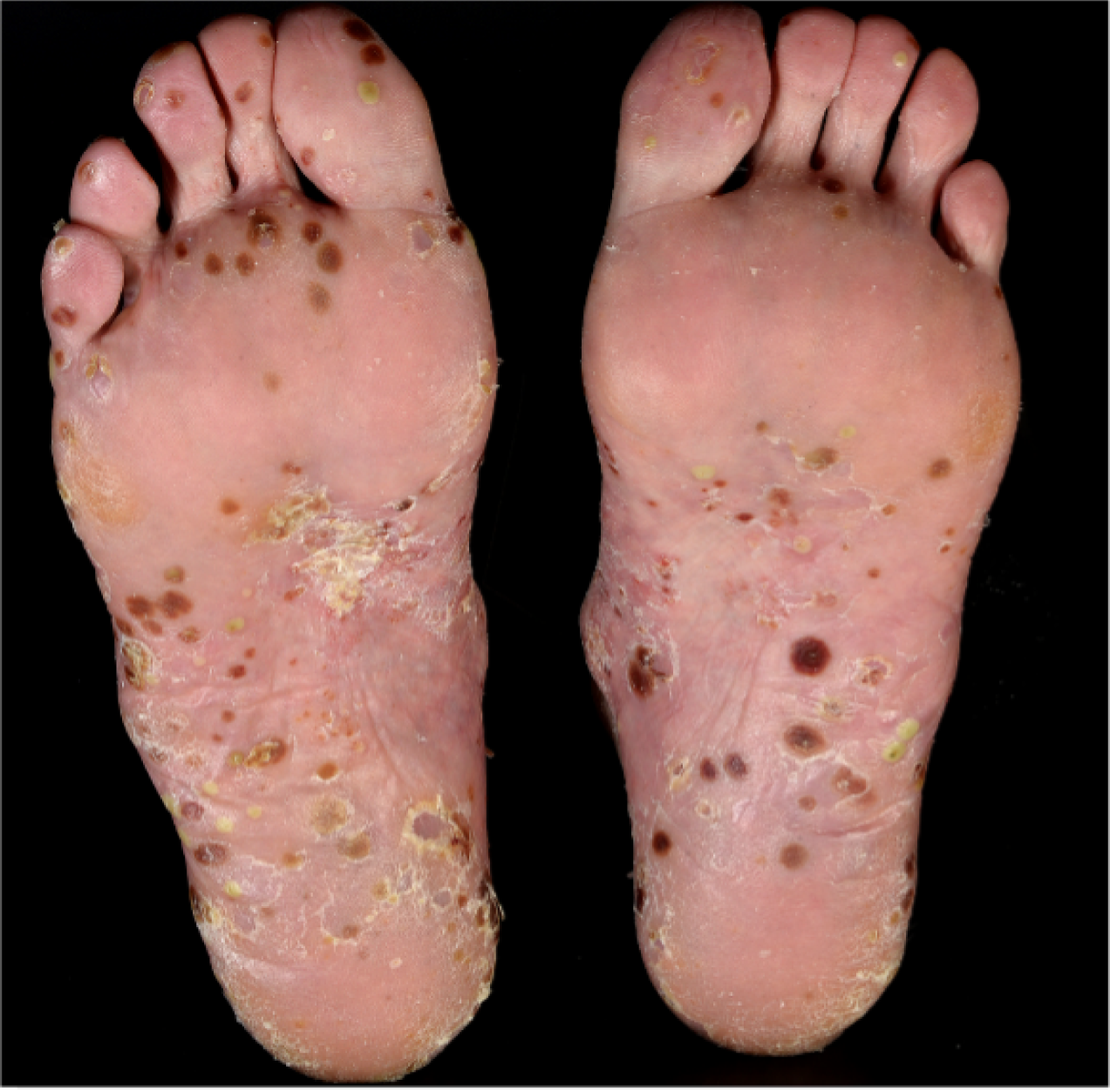
FIGURE 22.
Study photograph taken of participant’s palms.
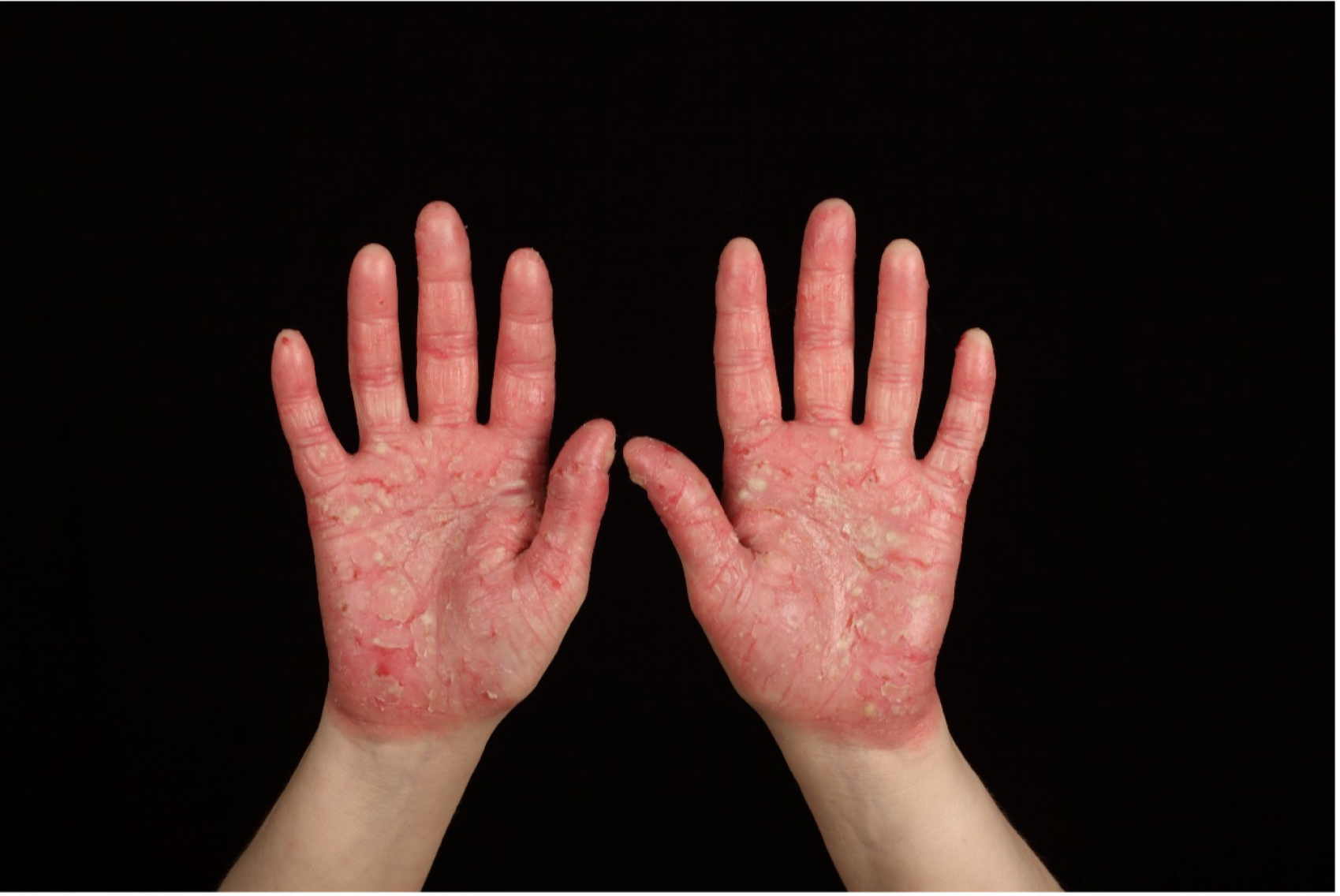
Appendix 9 Additional stage 1 results
| Outcome | Week | Total (N) | Placebo (n) | Anakinra (n) | SMD (95% CI) |
|---|---|---|---|---|---|
| Fresh pustule count (site assessed) | Baseline | 23 | 13 | 10 | 0.06 (–0.77 to 0.88) |
| 1 | 22 | 13 | 9 | –0.18 (–1.03 to 0.67) | |
| 4 | 20 | 12 | 8 | –0.53 (–1.43 to 0.39) | |
| 8 a | 23 | 13 | 10 | –0.11 (–0.93 to 0.72) | |
| 12 | 16 | 9 | 7 | –0.04 (–1.02 to 0.95) | |
| Fresh pustule count (central photographic assessor) | Baseline | 24 | 13 | 11 | 0.39 (–0.43 to 1.19) |
| 1 | 24 | 13 | 11 | 0.37 (–0.45 to 1.18) | |
| 8 a | 22 | 12 | 10 | 0.06 (–0.78 to 0.90) | |
| PP-PASI (first site assessor) | Baseline | 24 | 13 | 11 | 0.24 (–0.57 to 1.05) |
| 1 | 23 | 13 | 10 | –0.25 (–1.07 to 0.59) | |
| 4 | 21 | 12 | 9 | 0.44 (–0.44 to 1.31) | |
| 8 a | 23 | 12 | 11 | 0.41 (–0.42 to 1.23) | |
| 12 | 17 | 9 | 8 | 0.1 (–0.86 to 1.05) | |
| PP-PASI (second site assessor) | Baseline | 24 | 13 | 11 | 0.32 (–0.49 to 1.12) |
| 1 | 23 | 13 | 10 | 0.18 (–0.64 to 1.01) | |
| 4 | 23 | 13 | 10 | 0.48 (–0.36 to 1.31) | |
| 8 a | 24 | 13 | 11 | 0.14 (–0.67 to 0.94) | |
| 12 | 17 | 9 | 8 | 0.08 (–0.87 to 1.04) |
FIGURE 23.
Agreement between site assessor and photographic central assessment for fresh pustule count. The mean difference in the fresh pustule count between the site assessment and the central assessment from photographs is plotted on the y-axis against the average of the two paired measures for individual participant measures recorded at baseline and weeks 1, 4, 8 and 12. The dark-blue horizontal line indicates the mean difference. The black horizontal lines indicate the region within which 95% of the differences fall.
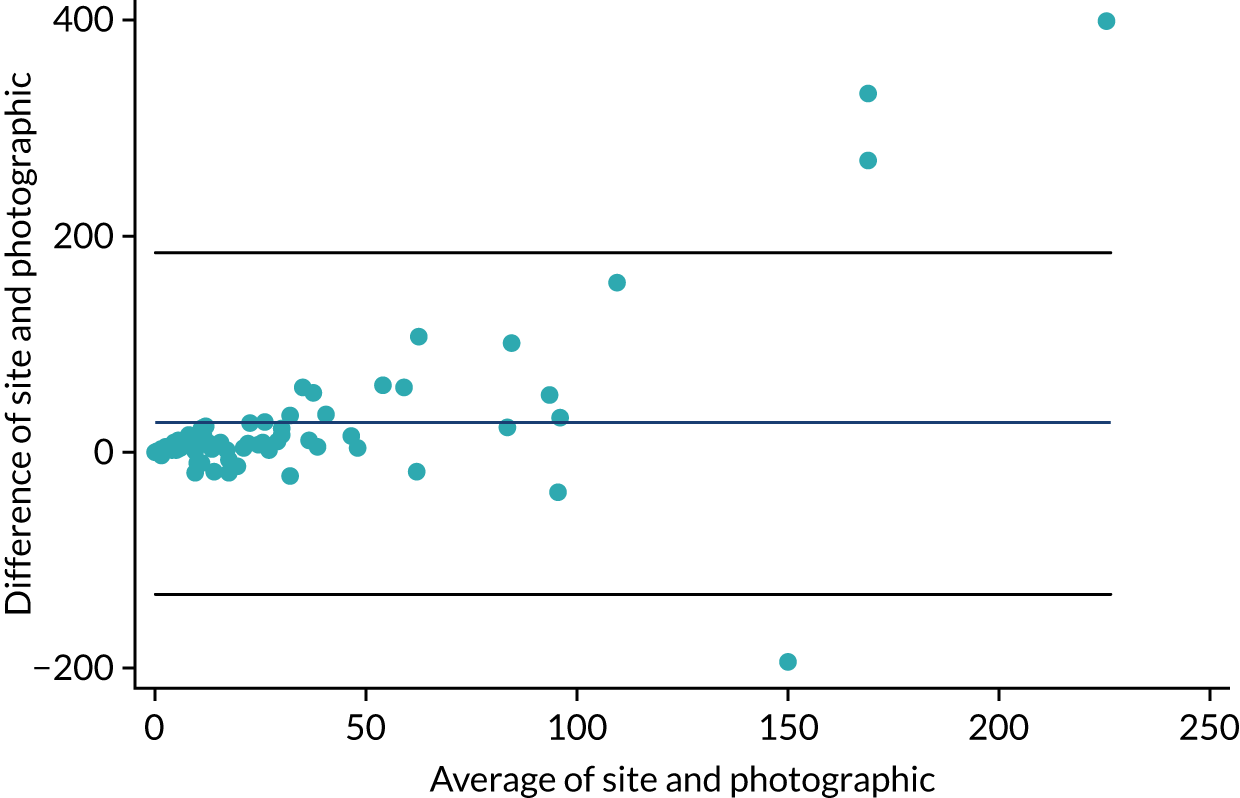
FIGURE 24.
Agreement between site assessor 1 and site assessor 2 for PP-PASI scores. The mean difference in the PP-PASI scores between the two paired site assessments is plotted on the y-axis against the average of the two paired measures for individual participant measures recorded at baseline and weeks 1, 4, 8 and 12. The dark-blue horizontal line indicates the mean difference. The black horizontal lines indicate the region within which 95% of the differences fall.
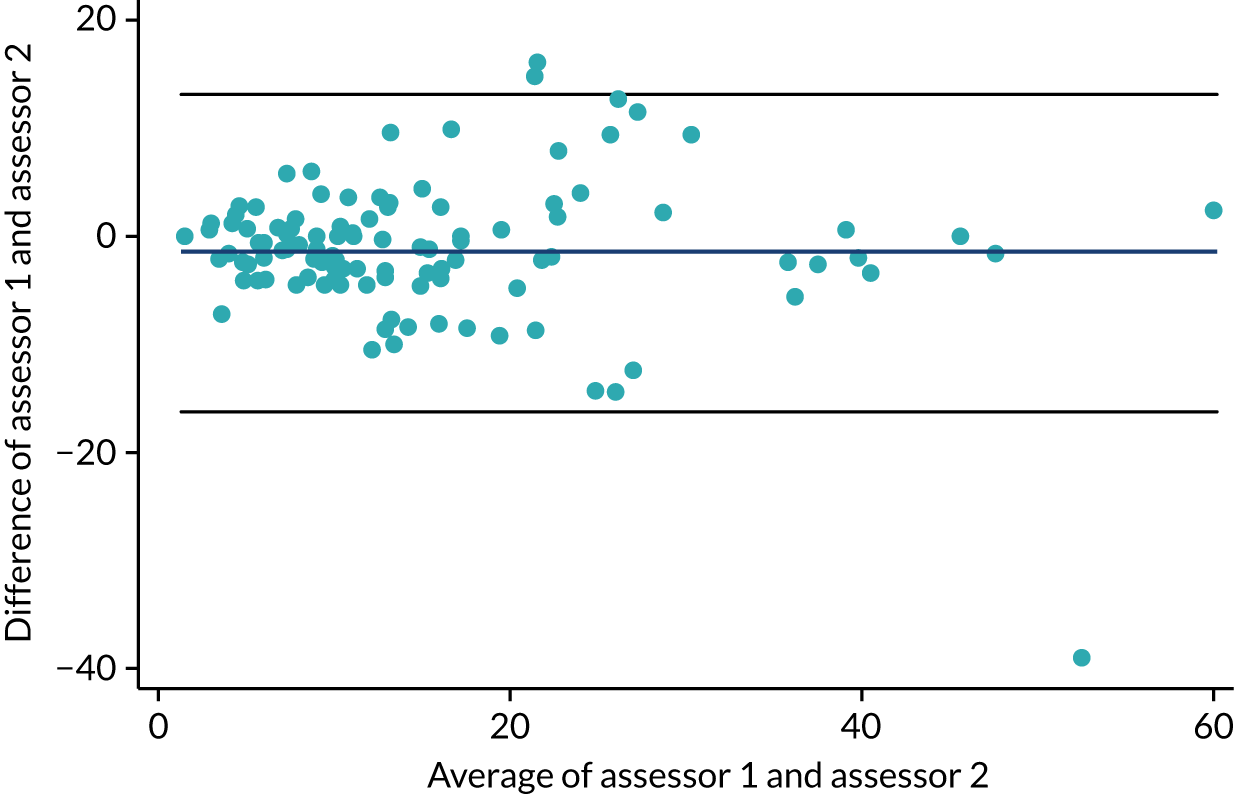
Appendix 10 Additional stage 2 results
| Missing time point | Treatment group, n (%) | Total (N = 64) | |
|---|---|---|---|
| Placebo (N = 33) | Anakinra (N = 31) | ||
| Baseline | 1 (3) | 0 (0) | 1 (2) |
| Week 1 | 2 (6) | 1 (3) | 3 (5) |
| Week 4 | 2 (6) | 3 (10) | 5 (8) |
| Week 8 | 2 (6) | 2 (6) | 4 (6) |
| Week 12 | 4 (12) | 4 (13) | 8 (13) |
FIGURE 25.
The PP-PASI scores over the 8-week follow-up period by treatment group: mixed-model estimates by treatment group. The baseline adjusted mean PP-PASI score is plotted on the y-axis, against the time point on the x-axis for each treatment group. The error bars represent 95% CIs for the adjusted treatment group means, estimated from the primary linear mixed model.
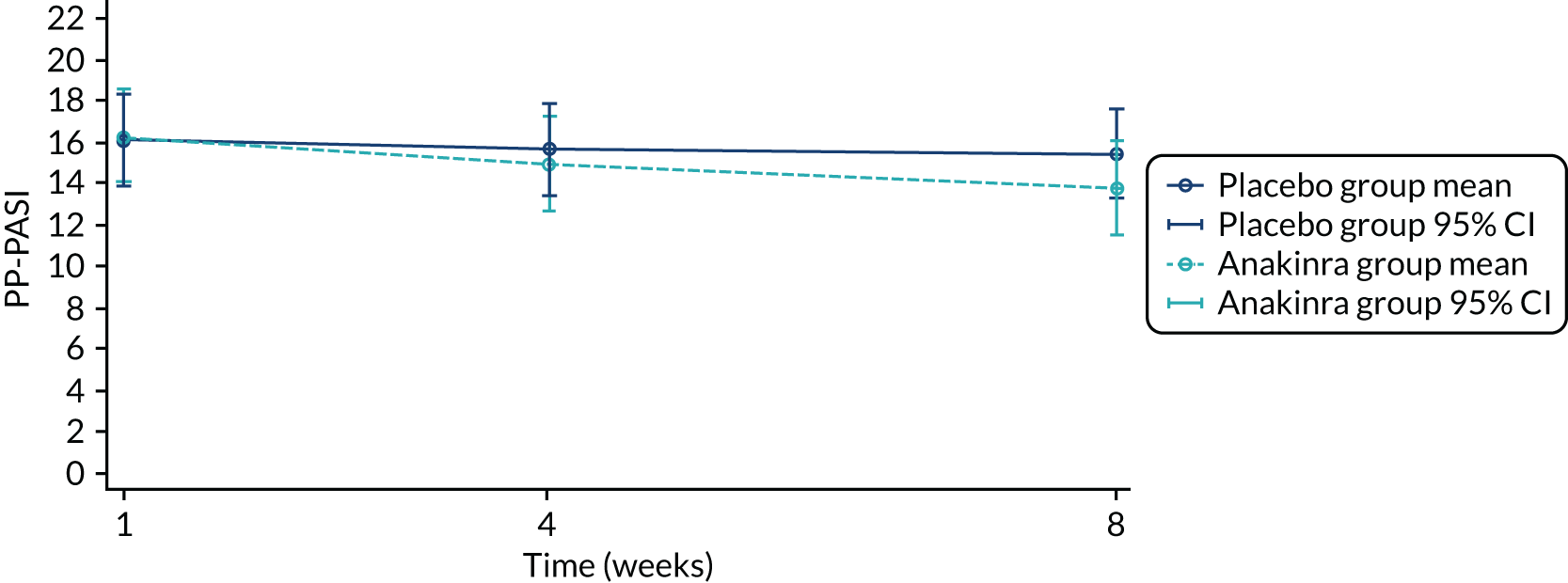
FIGURE 26.
The PP-PASI scores over the 8-week follow-up period by treatment group: mixed-model estimates for treatment group difference. The baseline adjusted mean treatment group differences (anakinra – placebo) in PP-PASI scores is plotted on the y-axis against the time point on the x-axis. The error bars represent 95% CIs for the adjusted treatment group differences, estimated from the primary linear mixed model.
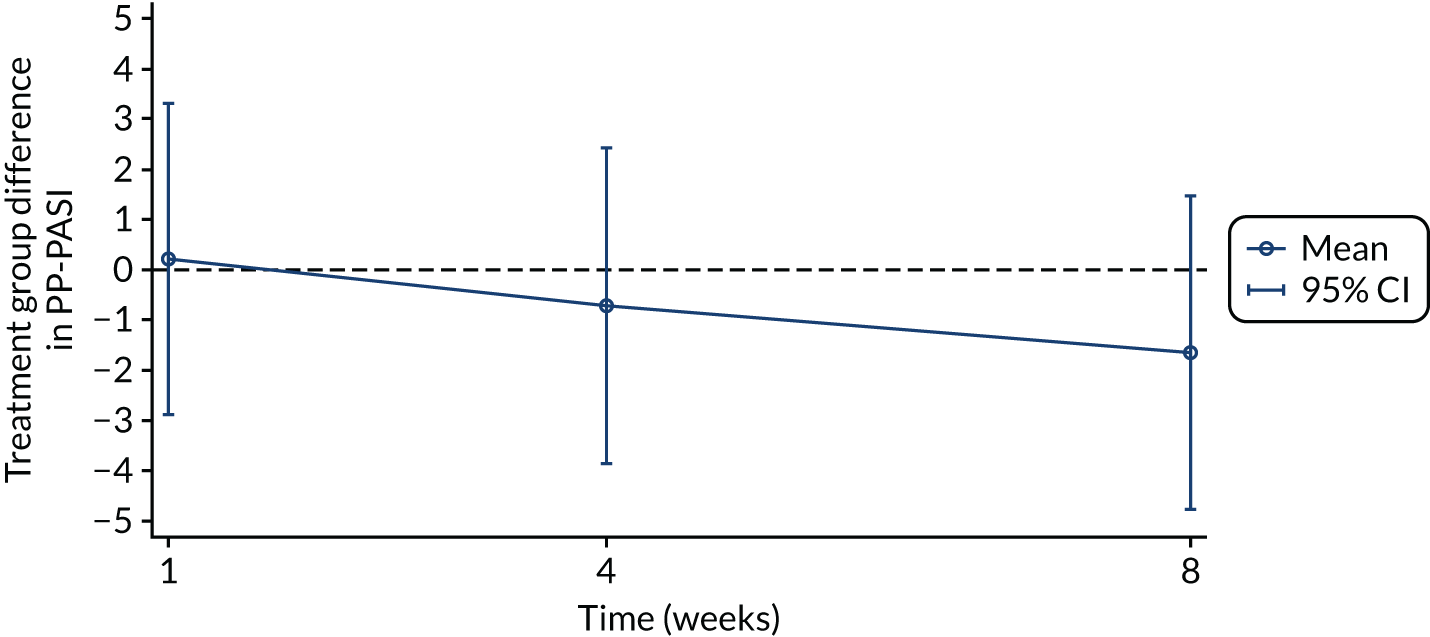
| Data | Treatment group, n (%) | |||
|---|---|---|---|---|
| Placebo (N = 31) | Anakinra (N = 30) | |||
| No rescue | Rescue | No rescue | Rescue | |
| Week 1 PP-PASI score | 31 (100) | 0 (0) | 28 (93) | 2 (7) |
| Week 4 PP-PASI score | 28 (90) | 3 (10) | 19 (68) | 9 (32) |
| Week 8 PP-PASI score | 24 (77) | 7 (23) | 19 (66) | 10 (34) |
| Summary | No rescue | At least one PP-PASI score included post rescue | No rescue | At least one PP-PASI score included post rescue |
| Total in primary PP-PASI score analysis | 24 (75) | 8 (25) | 19 (63) | 11 (37) |
| Data | Treatment group, n (%) | |||
|---|---|---|---|---|
| Placebo (N = 31) | Anakinra (N = 30) | |||
| No rescue or prohibited | Rescue or prohibited | No rescue or prohibited | Rescue or prohibited | |
| Week 1 PP-PASI score | 31 (100) | 0 (0) | 28 (93) | 2 (7) |
| Week 4 PP-PASI score | 28 (90) | 3 (10) | 19 (68) | 9 (32) |
| Week 8 PP-PASI score | 23 (74) | 8 (26) | 17 (59) | 12 (41) |
| Summary | No rescue or prohibited | At least one PP-PASI score included post rescue or prohibited | Not rescue or prohibited | At least one PP-PASI score included post rescue or prohibited |
| Total in primary PP-PASI analysis | 23 (72) | 9 (28) | 17 (57) | 13 (43) |
Appendix 11 Additional open-label extension results
| Characteristic | Individuals not entering OLE (N = 50) | Individuals entering OLE (N = 14) | ||||
|---|---|---|---|---|---|---|
| Placebo (N = 24) | Anakinra (N = 26) | Total (N = 50) | Placebo (N = 9) | Anakinra (N = 5) | Total (N = 14) | |
| Age (years) | ||||||
| Mean (SD) | 51.9 (14.4) | 50.1 (11.3) | 50.9 (12.7) | 51.3 (11.9) | 49.1 (16.6) | 50.5 (13.2) |
| Sex, n (%) | ||||||
| Male | 5 (21) | 2 (8) | 7 (14) | 1 (11) | 2 (40) | 3 (21) |
| Female | 19 (79) | 24 (92) | 43 (86) | 8 (89) | 3 (60) | 11 (79) |
| Ethnicity, n (%) | ||||||
| White | 22 (92) | 23 (88) | 45 (90) | 9 (100) | 5 (100) | 14 (100) |
| Asian or Asian British | 1 (4) | 1 (4) | 2 (4) | 0 (0) | 0 (0) | 0 (0) |
| Black or Black British | 0 (0) | 1 (4) | 1 (2) | 0 (0) | 0 (0) | 0 (0) |
| Chinese, Japanese, Korean, Indochinese | 0 (0) | 1 (4) | 1 (2) | 0 (0) | 0 (0) | 0 (0) |
| Other | 1 (4) | 0 (0) | 1 (2) | 0 (0) | 0 (0) | 0 (0) |
| Smoking status, n (%) | ||||||
| Current smoker | 13 (54) | 13 (50) | 26 (52) | 6 (67) | 3 (60) | 9 (64) |
| Ex-smoker | 7 (29) | 12 (46) | 19 (38) | 2 (22) | 0 (0) | 2 (14) |
| Non-smoker | 4 (17) | 1 (4) | 5 (10) | 1 (11) | 2 (40) | 3 (21) |
| PP-PASI score | ||||||
| Mean (SD) | 19.0 (9.4)a | 17.3 (11.4) | 18.1 (10.4) | 15.5 (12.8) | 18.6 (7.9) | 16.6 (11.1) |
| Median (IQR) | 17.1 (11.9–29.6) | 14.9 (11.7–20.2) | 15.4 (11.7–21.0) | 14.4 (7.4–17.9) | 17.9 (15.6–24.8) | 15.9 (7.4–18.0) |
| Fresh pustule count (palms and soles) | ||||||
| Mean (SD) | 35.6 (25.3) | 71.3 (156.3) | 54.1 (114.4) | 37.4 (50.3) | 28.0 (19.7) | 34.1 (41.2) |
| Median (IQR) | 29.5 (18.5–48.0) | 27.0 (11.0–63.0) | 28.5 (18.0–53.0) | 21.0 (18.0–34.0) | 21.0 (15.0–37.0) | 21.0 (15.0–37.0) |
| Fresh pustule count (palms) | ||||||
| Mean (SD) | 27.5 (23.5) | 52.7 (118.8) | 40.6 (87.3) | 21.8 (24.1) | 22.0 (18.0) | 21.9 (21.4) |
| Median (IQR) | 27.5 (7.5–38.0) | 17.0 (5.0–41.0) | 20.5 (7.0–39.0) | 21.0 (4.0–34.0) | 15.0 (9.0–37.0) | 18.0 (4.0–36.0) |
| Fresh pustule count (soles) | ||||||
| Mean (SD) | 8.1 (13.3) | 18.6 (42.6) | 13.6 (32.2) | 15.7 (30.2) | 6.0 (6.3) | 12.2 (24.5) |
| Median (IQR) | 1.5 (0.0–12.5) | 2.5 (0.0–21.0) | 2.0 (0.0–15.0) | 4.0 (0.0–14.0) | 5.0 (0.0–12.0) | 4.5 (0.0–13.0) |
| Total pustule count (palms and soles) | ||||||
| Mean (SD) | 118.8 (83.3) | 207.0 (290.4) | 164.7 (219.7) | 111.9 (131.0) | 86.6 (39.0) | 102.9 (105.8) |
| Median (IQR) | 99.5 (52.0–178.5) | 101.5 (45.0–279.0) | 99.5 (45.0–192.0) | 57.0 (33.0–105.0) | 83.0 (82.0–120.0) | 82.5 (33.0–120.0) |
| PPP-IGA scores, n (%) | ||||||
| Clear | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Almost clear | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Mild | 2 (8) | 3 (12) | 5 (10) | 2 (22) | 1 (20) | 3 (21) |
| Moderate | 12 (50) | 13 (50) | 25 (50) | 4 (44) | 4 (80) | 8 (57) |
| Severe | 10 (42) | 10 (38) | 20 (40) | 3 (33) | 0 (0) | 3 (21) |
| PGA scores, n (%) | ||||||
| Clear | 0 (0) | 2 (8) | 2 (4) | 0 (0) | 0 (0) | 0 (0) |
| Almost clear | 2 (8) | 3 (12) | 5 (10) | 0 (0) | 0 (0) | 0 (0) |
| Mild | 9 (38) | 10 (38) | 19 (38) | 1 (11) | 0 (0) | 1 (7) |
| Moderate | 11 (46) | 6 (23) | 17 (34) | 5 (56) | 4 (80) | 9 (64) |
| Severe | 2 (8) | 5 (19) | 7 (14) | 2 (22) | 1 (20) | 3 (21) |
| Very severe | 0 (0) | 2 (8) | 2 (4) | 0 (0) | 0 (0) | 0 (0) |
| DLQI scores | ||||||
| Mean (SD) | 14.5 (6.7) | 15.7 (7.1) | 15.2 (6.9) | 12.3 (8.5) | 11.6 (6.1) | 12.1 (7.5) |
| PP-QoL | ||||||
| Mean (SD) | 47.2 (11.9) | 46.3 (14.7) | 46.7 (13.3) | 44.3 (18.6) | 41.2 (16.3) | 43.2 (17.3) |
| AE term | Total events (n) |
|---|---|
| Abdominal pain | 1 |
| Arthralgia | 2 |
| Cystitis | 1 |
| Eczema | 1 |
| Facial bones fracture | 1 |
| Fall | 1 |
| Glucose urine present | 1 |
| Haemoptysis | 1 |
| Hepatotoxicity | 2 |
| Injection site reaction | 5 |
| Migraine | 2 |
| Nasopharyngitis | 1 |
| Osteoporosis | 1 |
| Psoriasis | 1 |
| Swelling | 1 |
| Viral infection | 1 |
| Vomiting | 2 |
| Urine analysis abnormal | 1 |
| Total | 26 |
Glossary
- Acral pustular psoriasis
- Forms of pustular psoriasis affecting the hands and/or feet (acrodermatitis of Hallopeau and palmoplantar pustulosis are forms of acral pustular psoriasis) (as defined by the European Rare and Severe Psoriasis Expert Network).
- Acrodermatitis continua of Hallopeau
- Primary, persistent (> 3 months), sterile, macroscopically visible pustules affecting the nail apparatus (as defined by the European Rare and Severe Psoriasis Expert Network).
- Compliance-average causal effect
- A measure of effect among those complying with anakinra treatment compared with those receiving the placebo.
- Estimand
- A precise definition of the treatment effect reflecting a clinical question that is the target of estimation.
- Generalised pustular psoriasis
- Primary, sterile, macroscopically visible epidermal pustules on non-acral skin (excluding cases in which pustulation is restricted to psoriatic plaques) with/without plaque psoriasis, with/without systemic inflammation, or relapsing (more than one episode) or persistent (> 3 months) (as defined by the European Rare and Severe Psoriasis Expert Network).
- Palmoplantar pustulosis
- Primary, persistent (> 3 months), sterile, macroscopically visible epidermal pustules on palms and/or soles (as defined by the European Rare and Severe Psoriasis Expert Network).
- Treatment policy estimand
- A measure of effect of deciding to prescribe anakinra compared with deciding to prescribe placebo (regardless of treatment adherence).
List of abbreviations
- AE
- adverse event
- ALT
- alanine aminotransferase
- APP
- acral pustular psoriasis
- APRICOT
- Anakinra for Pustular Psoriasis: Response in a Controlled Trial
- AST
- aspartate aminotransferase
- CACE
- complier-average causal effect
- CI
- confidence interval
- CONSORT
- Consolidated Standards of Reporting Trials
- CTU
- clinical trials unit
- DEG
- differentially expressed gene
- DLQI
- Dermatology Life Quality Index
- DMC
- Data Monitoring Committee
- EQ-5D-3L
- EuroQol-5 Dimensions, three-level version
- GPP
- generalised pustular psoriasis
- FDR
- false discovery rate
- HIV
- human immunodeficiency virus
- ICC
- intraclass correlation coefficient
- ID
- identification
- IL
- interleukin
- IMP
- investigational medical product
- IQR
- interquartile range
- ITT
- intention to treat
- MAR
- missing at random
- MAS
- macrophage activation syndrome
- MedDRA
- Medical Dictionary for Regulatory Activities
- MI
- multiple imputation
- MNAR
- missing not at random
- OLE
- open-label extension
- PASI
- Psoriasis Area Severity Index
- PGA
- Participants Global Assessment
- PIC
- participant information centre
- PIN
- patient identification number
- PLAG
- patient and lay members group
- PLUM
- Pustular Psoriasis – Elucidating Underlying Mechanisms
- PPI
- patient and public involvement
- PP-QoL
- Palmoplantar Quality of Life
- PPP
- palmoplantar pustulosis
- PP-PASI
- Palmoplantar Pustulosis Psoriasis Area Severity Index
- PP-PASI 50
- ≥ 50% improvement in PP-PASI
- PP-PASI 75
- ≥ 75% improvement in PP-PASI
- PPP-IGA
- Palmoplantar Pustulosis – Investigator’s Global Assessment
- PUVA
- psoralen and ultraviolet A radiation
- RNA
- ribonucleic acid
- SAE
- serious adverse event
- SD
- standard deviation
- SmPC
- summary of product characteristics
- SMS
- short message service
- Sobi
- Swedish Orphan Biovitrum AB
- SUSAR
- suspected unexpected serious adverse reaction
- TB
- tuberculosis
- TMG
- trial management group
- TNF
- tumour necrosis factor
- TSC
- Trial Steering Committee
- ULN
- upper limit of normal