

Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 14/152/14. The contractual start date was in October 2015. The final report began editorial review in May 2022 and was accepted for publication in December 2022. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
Copyright © 2024 Crombie et al. This work was produced by Crombie et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This is an Open Access publication distributed under the terms of the Creative Commons Attribution CC BY 4.0 licence, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. See: https://creativecommons.org/licenses/by/4.0/. For attribution the title, original author(s), the publication source – NIHR Journals Library, and the DOI of the publication must be cited.
2024 Crombie et al.
Chapter 1 Introduction
Background
For many years, trauma care has been predominantly based upon offering casualties with traumatic injuries basic treatment on scene to then safely allow transfer to hospital for further and definitive care. In the last two decades however, the emphasis in civilian practice has started to change in the direction of delivering more advanced interventions to patients while still in the pre-hospital phase. This shift, intended to provide earlier physiological stability and prevent so-called secondary damage occurring, has been driven in part by lessons learned in conflict by military medical systems, and includes advanced haemorrhage control and blood product-based resuscitation. 1–3
The introduction of blood component resuscitation during military casualty retrieval initially produced encouraging results with reports of reduced mortality amongst recipients receiving red blood cells (RBCs) and pre-thawed plasma. 4–6 However, a subsequent systematic review and meta-analysis of studies investigating outcomes following pre-hospital transfusion across both military and civilian practice found only modest advantages in those with moderate severity of injury. 7 In more recent years, two large randomised trials examining the use of pre-hospital plasma produced differing results – one trial favoured the use of plasma8 and one trial was stopped early due to futility. 9 There is therefore a lack of consistent and reliable data, confounded by the wide variety of systems in which studies have been conducted. In 2020, a review of pre-hospital transfusion of RBCs was unable to demonstrate a survival benefit, again highlighted the absence of randomised controlled trials (RCTs) and recommended further studies incorporating the use of individualised transfusion criteria and the use of plasma. 10
Extrapolating results from military trauma-based studies into civilian practice is not straightforward. The mechanisms and severity of injury sustained in conflict are rarely replicated in civilian practice, the patient demographic of active combatants is comparatively narrow and the medical infrastructure in dedicated field hospitals is different to many civilian emergency departments (EDs).
The provision of blood products as early treatment of major haemorrhage may seem logical, but it is also not without complication. Stored citrated blood can present a significant metabolic burden when administered rapidly and has the potential to cause further disruption to coagulation and myocardial function, especially in trauma. 11 There are also considerations around the provision of blood products including the demand for ‘universal’ blood products, regulatory compliance and secure cold-chain governance to avoid unnecessary wastage of products. 12
The lack of robust evidence surrounding the administration of pre-hospital RBCs or plasma in civilian practice, coupled with the challenges this poses to the transfusion community, make a prospective RCT important if further developments in this area of practice are to be justifiable. 13
We present the results of resuscitation with pre-hospital blood products (PHBP) (RePHILL) – a prospective multi-centre RCT which investigates the hypothesis that the administration of pre-hospital RBCs and lyophilised plasma (LyoPlas) would improve tissue perfusion as measured by the clearance of lactate and/or reduce mortality in patients demonstrating shock secondary to traumatic haemorrhage compared to resuscitation with crystalloid infusion. 14
Chapter 2 Methods
Study design
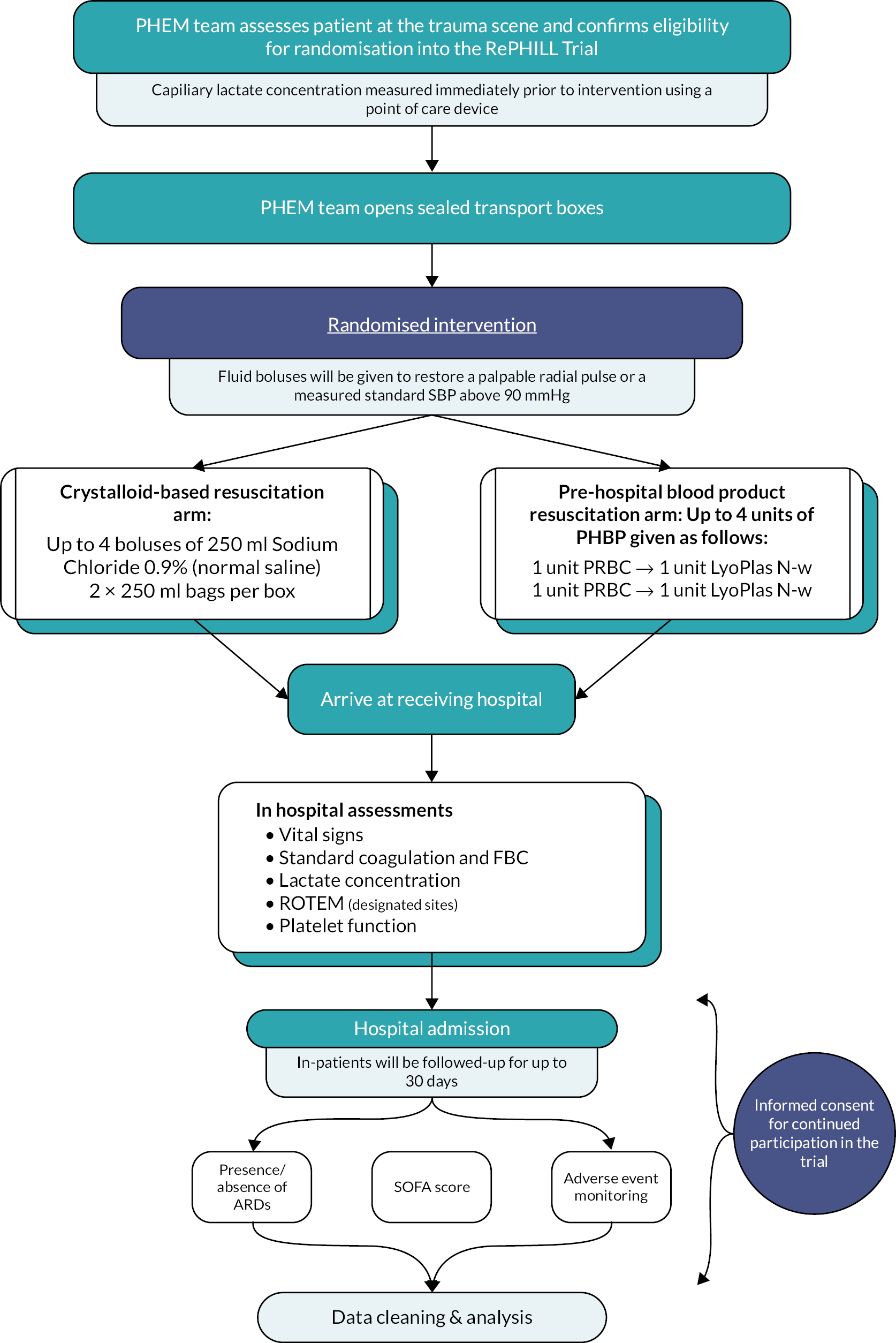
We conducted a multi-centred two-arm (1 : 1) open-labelled phase 3 RCT of pre-hospital blood product administration versus standard care for patients following traumatic haemorrhage. Research Ethics Committee approval was granted prior to study initiation (South Central – Oxford C, ref: 15/SC/0691). The trial was registered with the International Standard Randomised Controlled Trial Number (ISRCTN62326938) as well as with the European Union Drug Regulating Authorities Clinical Trials Database (registration: 2015-001401-13) and the Medicines and Healthcare products Regulatory Agency (Clinical Trials Authorisation: 16719/0228/001-0001). The trial was run in accordance with Good Clinical Practice requirements. Reporting is undertaken according to the Consolidated Standards of Reporting Trials (CONSORT) guidance. 15 A trial protocol has been published previously. 14 Figure 1 presents a summary of the trial protocol.
FIGURE 1.
RePHILL trial protocol summary.
Internal pilot
The RePHILL trial included an internal pilot. The purpose of the internal pilot was to assess the trial logistics to determine if it is both feasible and practical to carry on and recruit into the trial. It was intended that the pilot would be run at multiple sites to validate the multi-centre aspects of the trial.
The trial progression criteria were defined as:
-
minimum of 25 participants recruited across at least two active sites;
-
in participants recruited to the trial intervention arm, at least one unit of RBC and one unit of LyoPlas delivered to at least 80% of participants before reaching hospital;
-
at least 90% complete data capture;
-
Data Monitoring and Ethics Committee (DMEC) reports no safety concerns, which would prohibit continuation to main trial.
Changes to trial protocol
There were no major changes made to the study design. Table 1 lists the key amendments to the trial protocol.
| Amendment date | Protocol version number | Summary of amendment |
|---|---|---|
| 18 July 2016 | 1.1 | Changes to the chief and PIs Addition of new sites |
| 21 September 2016 | 1.1 | Change of PI |
| 2.0 | Administrative updates to TMG (formal change to CI requested as part of SA1) Updates to members of the oversight committees Clarification on the primary outcome (see note 1) Update to exclusion criteria (see note 2) Update to include delivery of interventions by the intraosseous route Clarification of the informed consent process Clarification on the randomisation and enrolment process Update to the schedule of events Clarification on adverse event (AE) reporting Clarification on data collection Statistical updates Clarification on monitoring requirements Removal of participating sites Change of PIs |
|
| 6 February 2017 to 1 February 2019 (covering 11 amendments) | 2.0 | Change of PIs and addition of sites |
| 18 February 2019 | 3.0 | Removal of participating sites table RePHILL Trial Protocol version 3.0, 8 April 2019 EudraCT Number: 2015-001401-13 Page 4 of 58 Update to secondary outcomes (see note 3) Update to who will assess and confirm eligibility (see note 4) Update to the exclusion criteria (see note 2) Removal of NHS digital, long-term follow-up Clarification to trial procedures on scene Clarification of informed consent procedure Update on blood sampling Removal of blood sampling for future analysis Update to pharmacovigilance reporting requirements Update to categorisation of causality table Update to data protection regulations Update to end of trial definition |
Eligibility criteria
Eligibility was assessed by the Pre-Hospital Emergency Medical (PHEM) team (doctor and/or paramedic). The principal investigator (PI) for intervention delivery sites (IDS) was always a medically qualified doctor, and they were responsible for maintaining oversight of the confirmation of eligibility process.
Inclusion criteria
Patients were eligible for inclusion in RePHILL if:
-
they had suffered a traumatic injury;
-
the PHEM team attended;
-
hypotension [systolic blood pressure (SBP) <90 mmHg or absence of palpable radial pulse] believed to be due to traumatic haemorrhage.
Exclusion criteria
Patients were excluded if:
-
children (known or apparently aged <16 years);
-
blood administered on scene, prior to arrival of the RePHILL PHEM team;
-
traumatic cardiac arrest where (1) the arrest occurred prior to arrival of the PHEM team and/or (2) the primary cause is not hypovolaemia;
-
refusal of blood product administration; known Jehovah’s Witness;
-
pregnancy (known or apparent);
-
isolated head injury without evidence of external haemorrhage;
-
known prisoners in the custody of HM Prison and Probation Service.
Clinician training
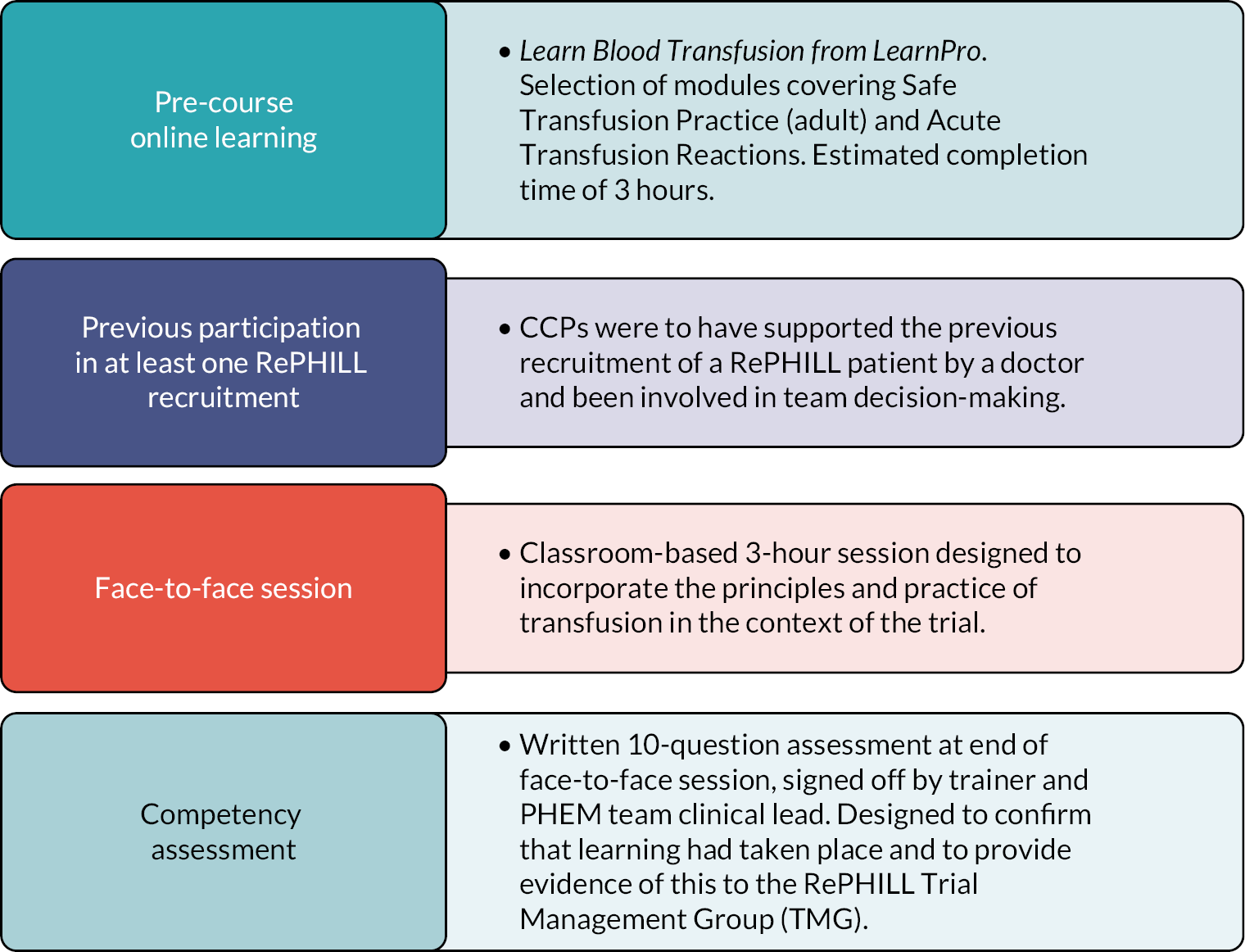
All clinical staff who participated in the trial completed online Good Clinical Practice and protocol training. At the start of the trial, authorisation for the administration of the interventions was limited to doctors working with the PHEM teams. However, this led to missed recruitments by paramedic-only crews. With agreement from relevant parties, the protocol was amended in 2019 and developed a training programme to ensure the safe administration of blood products by paramedics in the context of the trial, that is non-medical authorisation (NMA).
A bespoke training to enable NMA of blood was developed. The training programme comprised four elements as outlined in Figure 2. The programme was successful delivered to three critical care paramedics who cascaded the training to 14 colleagues.
FIGURE 2.
Non-medical authorisation programme.
Informed consent
Prospective informed consent for the participation in the trial was not feasible due to the nature of the injuries and incapacitation that was anticipated to fulfil eligibility criteria. Patients were enrolled in the trial in accordance with Good Clinical Practice, the Declaration of Helsinki, the Clinical Trials Regulations 2004 and the Human Tissue Act 2004. After enrolment, consent for participants to remain in the trial was sought as early as feasible and appropriate when capacity was regained. At the time of enrolment, consent was sought from a professional legal representative shortly after arrival at the receiving hospital. Further consent was sought from a personal or legal representative (such as a close relative or friend). Information regarding the trial was on display in locations likely to be visited by relatives, with a brief summary of the trial and contact details for further information. Some patients may wish to avoid blood product transfusion due to prior held beliefs (such as the Jehovah’s Witness community). In such circumstances, an advance medical directive would have been sought (as per usual practice in emergency resuscitation).
Although an eligibility criterion was that participants should be aged 16 or over, due to the nature of the trial it was not always possible to confirm the age of the participant before recruitment. If a participant was recruited and later found to be under the age of 16, child assent was sought alongside consent from a parent or guardian.
Study setting
The trial was situated within NHS England major trauma network. PHEM teams were recruited to become IDS. At the start of the research, it was anticipated recruitment would take place in six regional air ambulances services. During the set-up of the trial, two air ambulance services withdrew (Dorset and Somerset Air Ambulance and Essex and Herts Air Ambulance) due to loss of equipoise. A further air ambulance service briefly joined the trial (Yorkshire Air Ambulance) but subsequently withdrew prior to recruitment starting due to loss of equipoise.
The following air ambulances participated in the trial:
-
East Anglian Air Ambulance, Norwich, UK (www.eaaa.org.uk, accessed 7 February 2022)
-
Magpas Air Ambulance, Huntingdon, UK (www.magpas.org.uk, accessed 7 February 2022)
-
Midlands Air Ambulance and MERIT, West Midlands Ambulance Service NHS Trust, West Midlands, UK (www.midlandsairambulance.com and https://wmas.nhs.uk, accessed 7 February 2022)
-
The Air Ambulance Service, Warwickshire (https://theairambulanceservice.org.uk, accessed 7 February 2022)
The hospital sites which served as receiving hospitals and participating blood banks and pharmacies are listed in Appendix 1.
Randomisation
Sequence generation
Randomisation was provided by a computer-generated programme at the Birmingham Clinical Trials Unit (BCTU), with a 1 : 1 ratio of experimental intervention and control. The randomisation procedure was stratified by IDS to account for variation in trauma care and type of trauma between sites.
Allocation concealment
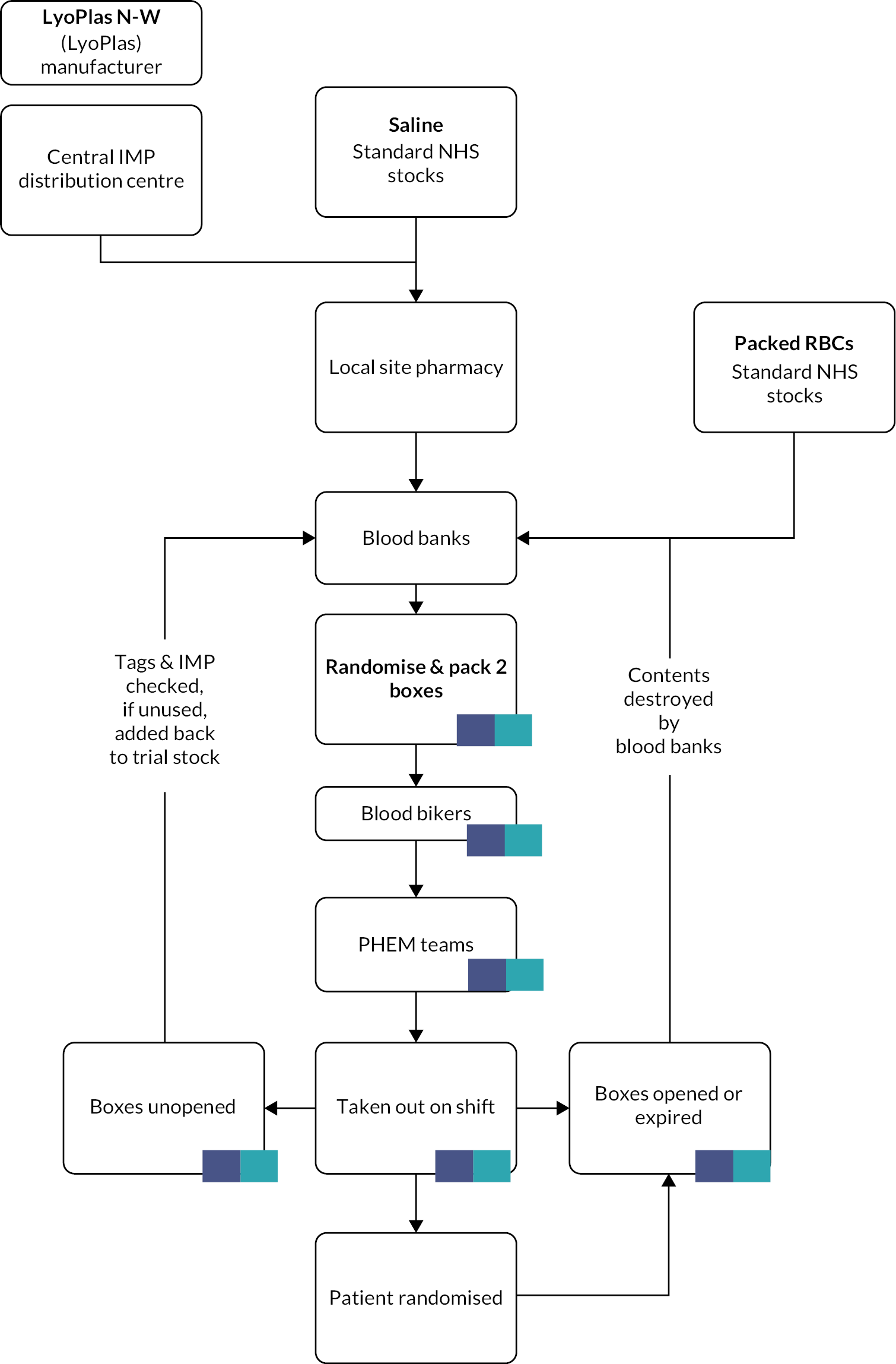
Blood banks were supplied with pre-printed ‘treatment box number’ labels. A registered user at the blood bank requested a treatment allocation from the BCTU and received a treatment box number and treatment arm allocation. The allocated trial intervention was placed into transport boxes affixing the correct labels. These transport boxes were then issued as a pair, one marked red (containing either 2 units of RBC or 2 bags of 250 ml 0.9% saline) and one marked yellow (containing either 2 units of LP or 2 bags of 250 ml 0.9% saline). These sealed boxes were then dispatched to the PHEM base where they were taken to the scene of any pre-hospital trauma patient(s). The weight and appearance of the boxes were the same between trial arms. Those assessing eligibility were blinded to the trial intervention before enrolling patients into the trial. No formal assessment of the success of allocation concealment was undertaken.
Interventions
The experimental intervention was a combination of universal donor (blood group O) RBCs and LyoPlas. LyoPlas was used as the plasma product for this trial (License PEI.H.03075.01.1, Germany) in accordance with MHRA approval for import and use as an investigational medicinal product (IMP). The control intervention was 0.9% saline, which was the most commonly delivered non-blood product fluid at the time of trial design. 3 Participants received up to four boluses of the assigned intervention in order to achieve SBP ≥90mmHg or a palpable radial pulse. For the experimental intervention, this was delivered as alternating units of RBC and LyoPlas until a total of 2 RBC and 2 LyoPlas were delivered. For the control intervention, up to four boluses of 250 ml 0.9% saline were delivered. All boluses were administered using fluid warming devices. If any further fluid resuscitation was required in either interventional arm of the trial, this was given in further boluses of 250 ml 0.9% saline and recorded on the PHEM case report form.
Storage and delivery
Lyophilised plasma should be stored between 2 and 25°C and RBC between 2 and 6°C. As LyoPlas is very difficult to reconstitute when cold, it was necessary to store it separately from the RBC. The trial tested and validated two insulated box systems for this purpose – ORCA boxes (Intelsius, Elvington, UK) and Credo boxes (Pelican BioThermal, Leighton Buzzard, UK). The latter boxes were preferred by air ambulances due to their smaller size and weight. The insulated boxes were preconditioned for a minimum of 24 hours in a freezer below –25°C, then removed 30 minutes before use to allow them to reach the required operating temperature before packing. Each transport container also had a temperature data logger, the choice of which was determined by the local blood bank. Sealed treatment boxes were transferred from blood banks to air ambulance/ambulances bases by volunteers from the Blood Bikes charity (www.bloodbikes.org.uk, accessed 9 February 2022). The intervention pathway is shown in Figure 3.
FIGURE 3.
IMP management.
Outcomes
Primary outcome
The primary outcome was a composite measure consisting of episode mortality and lactate clearance defined as a failure to achieve lactate clearance ≥20% per hour in the first 2 hours from randomisation.
Therefore, if the participant experiences either:
-
Episode mortality, or
-
A failure to achieve lactate clearance ≥20% per hour in the first 2 hours after randomisation,
they will be considered to have experienced the primary outcome. If they have survived to the point of exiting the trial through discharge from acute care and have experienced lactate clearance ≥20% per hour in the first 2 hours after randomisation, they will be considered to not have experienced the primary outcome.
Rationale for the primary outcome
RePHILL was designed to assess the superiority of pre-hospital blood and plasma transfusion over the existing standard of care at the time, which was 0.9% saline. We hypothesised that early blood would accelerate the reversal of hypovolaemic shock, improve coagulopathy and thereby improve episode mortality.
The absence of a core outcome set for trauma trials creates a challenge for investigators each time they plan and develop a trial. The trial team carefully considered using mortality as the sole and primary outcome for the study but were concerned that mortality was too a blunt a tool to assess the effectiveness of the intervention as a certain proportion of patients recruited would either be too well or too sick to benefit from the treatment. Furthermore, sample size estimates to detect small but important differences in mortality were prohibitively large (several thousand participants).
After a review of the literature and further discussion with the funder and co-investigators, it was agreed to use lactate clearance as (1) it could be easily measured; (2) it is related to tissue perfusion, a target specifically related to the intervention; and (3) it has been shown to be prognostically related to mortality. 16
During the peer review process and contracting with the funder, we were required to include mortality and thus create a composite outcome.
The investigators appreciate that interpretation of a composite outcome is challenging, particularly when the clinical importance of the outcomes is different. We attempt to mitigate this through reporting the individual components of the primary outcome separately in accordance with recommended practice for composite outcomes.
Definitions of components of primary outcome
Episode mortality was defined as those participants who die during the study between the time of injury/recruitment and discharge from the primary receiving facility to non-acute care (this includes participants who die on scene). The date of discharge from acute care and date of death are recorded on the exit form. Any deaths occurring after the date of discharge from acute care are not considered to be cases of episode mortality.
Lactate clearance was calculated according to the baseline capillary lactate (recorded immediately prior to intervention delivery), and measured using a point of care lactate device. The value, date, and time of the 2-hour post-randomisation lactate concentration were defined as the value, date and time of either:
-
the capillary lactate concentration taken if the participant has not reached hospital within 2 hours of randomisation; or
-
the venous lactate concentration taken in ED if the participant has reached hospital within 2 hours of randomisation; or
-
the arterial lactate concentration taken in ED if the participant has reached hospital within 2 hours of randomisation and venous access is not available.
The lactate concentration value available from (1) to (3) above which is closest to the 2 hours from randomisation time point will be used as the 2-hour lactate concentration value regardless of method of collection.
Using the following notation:
| Lac 0 | = | Randomisation capillary lactate concentration |
| Lac h | = | 2-hour post-randomisation lactate concentration |
| T 0 | = | Date and time of Lac0 |
| T h | = | Date and time of Lach |
| Interval | = | Th–T0 (in minutes) |
The interval time is given by:
| Time between 2-hour post-randomisation lactate concentration and randomisation lactate (minutes) | = | Date and time of the 2-hour post-randomisation lactate concentration (Th) | - | Date and time of the randomisation capillary lactate concentration (T0) |
Lactate clearance, expressed as a percentage per hour (%/h), is calculated using the formula:
A normal lactate is taken to be ≤2.2 mmol/L. Achieving ≥20% per hour lactate clearance is defined as follows in participants whose:
-
Lac0 is >2.2 mmol/L and whose Lach demonstrates lactate clearance of ≥20% per hour; or
-
Lach is >2.2 mmol/L, but whose Lach is ≤2.2 mmol/L, regardless of the magnitude of the change; or
-
Lac0 and Lach are both ≤2.2 mmol/L, regardless of the magnitude and direction of any difference.
All the above will be counted as participants achieving ≥20% per hour lactate clearance.
The above can be summarised in Table 2.
| Lac0 (mmol/L) | Lach (mmol/L) | Required lactate clearance |
|---|---|---|
| >2.2 | >2.2 | ≥20% per hour |
| >2.2 | ≤2.2 | Not applicable |
| ≤2.2 | ≤2.2 | Not applicable |
Achieving <20% per hour lactate clearance is defined as follows in participants:
-
Whose Lac0 is >2.2 mmol/L and whose Lach demonstrates lactate clearance of <20% per hour; or
-
Who die prior to interval sampling (e.g. before the Lach measurement is taken at Th). For this we require the date and time of death from the exit form to determine if the participant died within two hours and 30 minutes of randomisation.
The Table 3 below summarises what is considered an event (failure to achieve lactate clearance).
| Lac0 (mmol/L) | Lach (mmol/L) | Lactate clearance <20% per hour | Lactate clearance ≥20% per hour |
|---|---|---|---|
| >2.2 | >2.2 | Failure to clear (event) | Achieves clearance |
| >2.2 | ≤2.2 | Achieves clearance | Achieves clearance |
| ≤2.2 | ≤2.2 | Achieves clearance | Achieves clearance |
| Dies prior to interval sampling and within 2.5 hours of randomisation | Failure to clear (event) | ||
There are instances where the lactate value is too high for a value to be reported, that is the value is out of range (OOR) of the detection level of the test. In these cases, the lactate measurement is recorded on the database as ‘too high to be recorded’. In these instances, at database lock prior to analysis, a review of these lactate values was undertaken by two statisticians blind to treatment allocation to assess whether the participant cleared their lactate or not. For example, if randomisation lactate is OOR, but the two- hour randomisation lactate is ≤2.2, then as per the table above the participant would be considered to have achieved clearance; or if both the randomisation and two- hour lactates are OOR, then the participant will be considered to have failed to clear. If unable to determine, then the lactate component of the primary outcome was considered missing.
Secondary outcomes
The secondary outcomes comprised:
-
individual components of the primary outcome;
-
all-cause mortality within 3 hours of randomisation;
-
pre-hospital time and type and volume of fluid;
-
vital signs (SBP, heart rate, capillary oxygen saturation);
-
(venous) lactate concentration;
-
haemoglobin concentration on ED arrival;
-
trauma-induced coagulopathy [defined as International Normalised Ratio (INR) >1.5];
-
coagulation measured viscoelastically by rotational thromboelastometry (ROTEM)III;
-
platelet function using multiple electrode impedance aggregometry (MultiPlate)III;
-
total blood product receipt;
-
acute respiratory distress syndrome;
-
transfusion-related complications;
-
organ failure-free days.
Justification of the secondary outcomes
The secondary outcomes were selected to assess whether PHBP improved blood pressure, heart rate and capillary oxygenation on ED arrival, prolonged on-scene time, reduced pre-hospital fluid and in-hospital transfusion requirements, reduced trauma-induced coagulopathy and preserved platelet function. The study also set out to monitor transfusion-related complications, including acute respiratory distress syndrome and other adverse events.
Definitions of organ failure-free days
The presence of organ failure is defined as any Sequential Organ Failure Assessment (SOFA) component score of ≥3. 3,17 Organ failure will be assumed to be absent if the participant is discharged from hospital and will be assumed to be present if the participant has died.
Sample size calculation
There were no prior published analyses using our composite primary outcome. However, there is considerable evidence from observational data for the survival benefit of RBC and plasma in civilian and military studies. Through consultation with those with expertise in pre-hospital trauma resuscitation, we chose an absolute reduction of 10% in the proportion of patients having one of the components of our composite primary outcome as a clinically meaningful effect size. Therefore, using this difference between proportions (two-sided Fisher’s exact test) with 80% power, and a type 1 error rate of 5% (i.e. α = 0.05), we calculated a requirement for 219 participants in each arm of the trial (438 participants in total in our 1 : 1 design). A target of 490 patients was set in order to account for 10% loss to follow-up rate.
The interim analysis for the DMC meeting in May 2018 reported the results on the 192 participants recruited by 20 April 2018. A pooled event rate of 65% experiencing either episode mortality or lactate clearance <20%/h in the two hours post-randomisation was observed in these participants. This observed rate did not correspond with the pooled event rate of 15% assumed in the original sample size calculations. On the DMC’s recommendations, this issue was discussed with the Trial Steering Committee (TSC) in October 2018. The TSC recommended that the power calculations be framed in terms of a relative risk rather than an absolute risk, with the original target sample size of 490 unchanged.
Assuming the pooled event rate remains at 65% and allowing for a 10% loss to follow-up rate, 490 participants will provide 80% power to detect a relative risk ratio of 0.82 (i.e. from 71.7% in the standard care group to 58.3% in the group receiving PHBP) using the method of difference between proportions (two-sided Fisher’s exact test), and a type 1 error rate of 5% (i.e. α = 0.05). This estimated relative risk ratio is consistent with the relative risk ratios of 1.54 and 0.70 reported in two recent pre-hospital RCTs using plasma in one of the treatment arms. 8,9
Statistical analysis
All analyses followed a statistical analysis plan which was finalised prior to database lock (see Report Supplementary Material 1). All analyses were undertaken in SAS v9.4.
Primary analyses
The intention-to-treat principle (i.e. analysis according to the randomisation schedule irrespective of treatment received) informed all primary analyses of the primary and secondary outcomes. Participants who withdrew from the study were non-assessable. A model-based approach to analysis was used with IDS included as a fixed effect covariate in the model. Treatment effects are presented with two-sided 95% confidence intervals (CIs). No adjustment for multiple comparisons was made. The risk ratio and absolute risk difference are adjusted for IDS. In the primary analyses of the primary outcome, all lactate clearance measurements are included regardless of whether they were inside the two hour ± 30 minute window. The 0.9% saline treatment arm is the reference level.
The study reports both the relative effect and absolute effect for binary outcomes (e.g. primary outcome and the individual components) as recommended by CONSORT. Binary outcomes were analysed using log-binomial regression models to obtain adjusted relative risks along with 95% CIs. A relative risk <1 favoured the RBC/LyoPlas group. Adjusted risk differences along with 95% CIs were estimated using a binomial regression model with identity link. A risk difference <0 favoured the RBC/LyoPlas group. Continuous data were analysed using linear regression models to obtain adjusted mean differences between groups along with 95% CIs.
Bayesian analysis
We planned a priori a Bayesian analysis of the primary outcome and its individual components using non-informative, sceptical and informative priors.
Subgroup analysis
We also planned a priori various exploratory subgroup analyses according to: IDS, mode of transport (air vs. ground), initial lactate concentration (≤2.2 mmol/L vs. >2.2 mmol/L), time to hospital arrival from injury (≤1 hour vs. >1 hour), mode of injury (blunt vs. penetrating vs. crush), volume of pre-hospital fluid given (total intervention (4 boluses vs. <4 boluses), age (<50 years, 50–70 years, >70 years), presence of head injury, compressible haemorrhage, prior history of anticoagulant/antiplatelet use (anticoagulant or antiplatelet medication vs. no anticoagulant or antiplatelet medication) and cardiac arrest (arrested vs. not arrested).
Sensitivity analyses
Sensitivity analyses were undertaken for the primary outcome:
-
An analysis in which the risk ratio and absolute risk difference for the composite outcome were both adjusted for IDS and the following prognostic variables: age, capillary lactate, cardiac arrest and Glasgow Coma Score (GCS) at randomisation.
-
A per-protocol analysis, the per-protocol population comprised those who received one or more dose of the randomised intervention/control, unless there was a clinical justification for withholding it.
-
An analysis in which only lactate concentrations recorded within specified time windows were included.
-
A multiple imputation analysis to assess the effect of missing response.
Post-hoc analyses
Post-hoc analyses comprised a comparison of total transfusion volume (i.e. pre-hospital and hospital transfusion), the influence of injury severity score and transport time from scene to hospital (post-hoc subgroup analyses), and an additional per-protocol analysis in which the per-protocol population was defined as those who received one or more units of the randomised intervention.
We also modelled different scenarios had the trial achieved the intended sample size.
Interim analyses and stopping guidelines
An independent DMEC was established to oversee the safety of trial participants. The DMEC met prior to the trial opening and again once the first 25 patients had been entered into the study. All data discussed remained confidential except to members of the DMEC and the trial statisticians performing the analyses. The DMEC then met on an annual basis throughout the trial. Interim analyses were provided to the DMEC, as well as any new evidence from other sources that may have shown that one treatment was definitely more, or less, effective than the other. The trial would be stopped if the DMEC had advised the TSC that any of the randomised comparisons in the trial had provided both (1) ‘proof beyond reasonable doubt’ that for all, or for some, types of patient one particular treatment is definitely indicated or definitely contraindicated in terms of a net difference in the major end-points, and (2) evidence that might reasonably be expected to influence the patient management of many clinicians who are already aware of the other main trial results. The trial statistician performed all of the interim and final analyses. Analyses were verified and reviewed by an independent statistician, and the statistical analysis report was reviewed by the senior statistician on the study.
Chapter 3 Results
Internal pilot
There were delays in starting the trial due to changes to the regulatory requirements, change of chief investigator and difficulty in procuring LyoPlas for use in the trial. The trial opened to recruitment at the Midlands Air Ambulance and West Midlands Ambulance sites on 29 November 2016, with the first patient being recruited on 6 December 2016. By 31 May 2017:
-
Twenty-six participants had been randomised across the two active sites.
-
100% of patients randomised to the PHBP arm have received at least one unit of RBC and one unit of LyoPlas.
-
A review of 22 completed case report forms confirmed 100% data completion for the primary outcome and 93% data for the secondary outcome.
-
The TSC and DMC reviewed trial progress and recommended to the funder that the trial be continued.
The funder confirmed successful completion of the internal pilot and approved progression to the main trial.
Recruitment
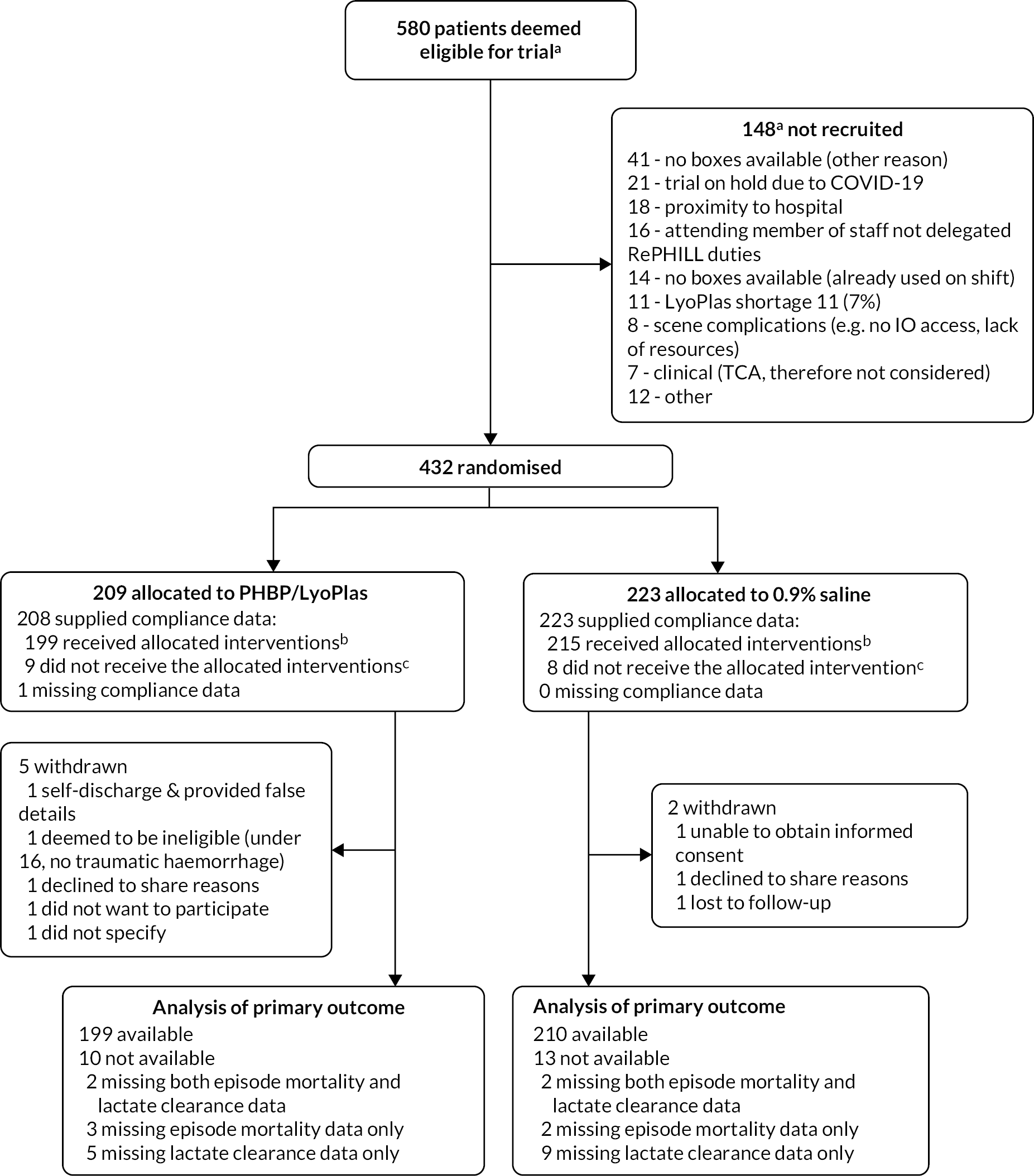
Following successful completion of the pilot, the trial extended to the three remaining additional sites. Participant recruitment continued through to 1 January 2021. The trial was closed on 2 January 2021 prior to achieving the intended sample size due to the ongoing impact of the COVID-19 pandemic. The decision to close the trial was made by the TSC and sponsor, without any knowledge of the data or results of interim analyses. During the 49 months of trial recruitment, at least 580 patients were assessed for eligibility, with 432 (74%) deemed eligible and recruited into the trial (see Figure 4). The flow of participants and reasons for non-recruitment are summarised in Figure 5. Recruitment was approximately equal between both trial arms with 209 (46%) participants allocated to receive RBC/LyoPlas and 223 (54%) allocated to receive 0.9% saline.
FIGURE 4.
Recruitment. Thick red line denotes observed recruitment. Aqua line denotes the revised target recruitment still based on a total of 490. Dashed blue line denotes projected recruitment based on rate observed up to the previous DMEC meeting in January 2020. Lighter aqua shaded region denotes period during which no LyoPlas was available to be supplied to the trial. Dashed black line denotes the scheduled end of trial recruitment. Darker aqua shaded region denotes period during which recruitment was paused due to COVID-19. A further breakdown of recruitment (by site) is provided in Appendix 2.
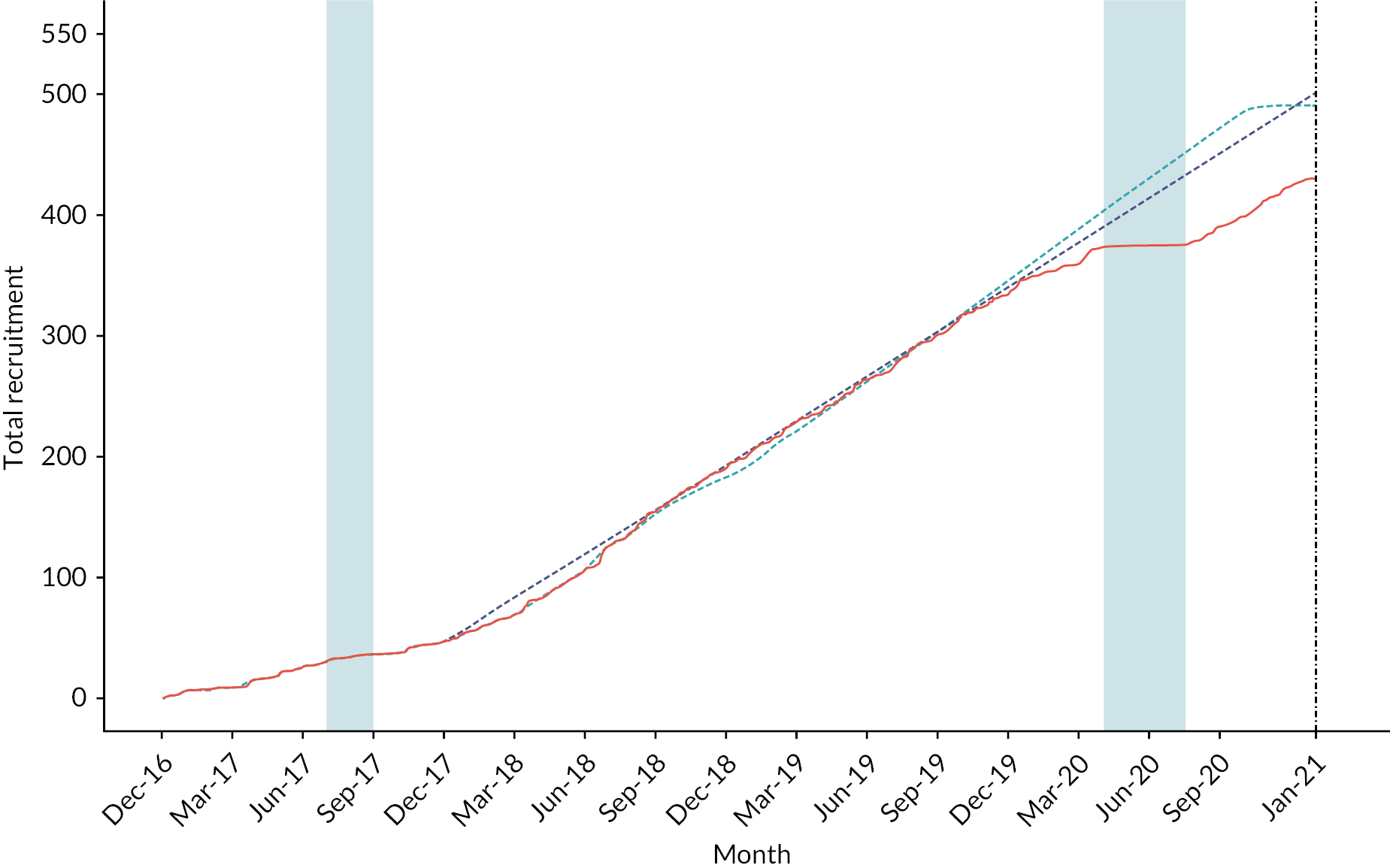
FIGURE 5.
CONSORT flow diagram. abased on screening lists provided by each IDS. IO = intraosseous, TCA = traumatic cardiac arrest. ballocated interventions are, unless clinically justified: the administration of at least one unit of RBC and one unit of LyoPlas in the PHBP/LyoPlas arm of the trial, and the administration of at least one bolus of fluid in the 0.9% saline arm of the trial. creasons for participants not receiving any units of allocated intervention (with no clinical justification) were: 9 due to equipment absence/failure (e.g. of giving sets or lactate monitors), 1 due to complex scene conditions, 1 due to decision to stop resuscitation, 1 due to non-trial saline already being administered to patient and 5 gave no reason.
All participants were followed up until trial exit, with data collection ending at the first occurrence of: withdrawal, acute care discharge, death or at 30 days follow‐up. Episode mortality data was collected up to discharge from the acute care setting, which may be >30 days. The median duration of study follow-up was 8 days [interquartile range (IQR) to 34] across all 432 participants. The median follow-ups for each treatment group were 9 days (IQR 1 to 34) for participants in the RBC/LyoPlas group and 7 days (IQR 0 to 31) for participants in the 0.9% saline group.
The allocation of participants between treatment arms exhibited a slight imbalance, with 14 more participants allocated to receive 0.9% saline than to receive RBC/LyoPlas. Across all 8188 treatment boxes issued by the blood banks, 4094 contained 0.9% saline and 4094 contained RBC/LyoPlas. Treatment boxes issued by blood banks were split 50:50 between RBC/LyoPlas and 0.9% saline in each IDS. Although the proportion of issued treatment boxes used by participants did vary by IDS, from 2.8% to 11.5%, there was no systematic imbalance by study arm. The pre-hospital teams were unaware of the treatment allocation prior to enrolling a participant, so we assume that the imbalance in the number of participants in each arm is down to random chance.
Baseline demographic and clinical characteristics
Baseline demographic and clinical characteristics by treatment arm are presented in Table 4. Participants were well-balanced across baseline characteristics, with most participants being male (82%), of white ethnicity (62%) and with a median age of 38 years (IQR 26 to 58). Participants could record more than one injury mechanism and the most commonly recorded was road traffic collision (62%), with stabbing (16%) and falls (14%) being the other main mechanisms of injury. Other mechanisms of injury were recorded by 9% of participants and comprised 13 laceration injuries, 6 pedestrian incidents with trains, 4 agricultural incidents, 4 industrial accidents and 5 other injuries.
| RBC/LyoPlas (n = 209) | 0.9% saline (n = 223) | All (n = 432) | |
|---|---|---|---|
| Stratification variable | |||
| IDS | |||
| Site 1 | 68 (32%) | 64 (29%) | 132 (31%) |
| Site 2 | 37 (18%) | 41 (18%) | 78 (18%) |
| Site 3 | 60 (29%) | 61 (27%) | 121 (28%) |
| Site 4 | 44 (21%) | 57 (26%) | 101 (23%) |
| Demographic and other baseline variables | |||
| Sex | ( n = 208) | ( n = 223) | ( n = 431) |
| Male | 170 (82%) | 183 (82%) | 353 (82%) |
| Age (n) | ( n = 196) | ( n = 211) | ( n = 407) |
| Median (IQR) | 38 (27, 56.5) | 39 (24, 59) | 38 (26, 58) |
| Ethnic group a | ( n = 166) | ( n = 168) | ( n = 334) |
| White | 104 (63%) | 104 (62%) | 208 (62%) |
| Black | 2 (1%) | 3 (2%) | 5 (1.5%) |
| Mixed | 4 (2%) | 5 (3%) | 9 (3%) |
| Asian | 8 (5%) | 8 (5%) | 16 (5%) |
| Other | 1 (1%) | 4 (2%) | 5 (1.5%) |
| Not known/provided | 47 (28%) | 44 (26%) | 91 (27%) |
| Injury details | |||
| Injury mechanism b | |||
| RTC | 130 (62%) | 139 (62%) | 269 (62%) |
| Stabbing | 33 (16%) | 35 (16%) | 68 (16%) |
| Fall | 26 (12%) | 35 (16%) | 61 (14%) |
| Gunshot | 4 (2%) | 4 (2%) | 8 (2%) |
| Burn | 0 (0%) | 1 (0.4%) | 1 (0.2%) |
| Inhalation | 1 (0.5%) | 0 (0%) | 1 (0.2%) |
| Otherc | 19 (9%) | 22 (10%) | 41 (9%) |
| Injury characteristics | |||
| Concomitant head injuryd | 29/60 (48%) | 32/68 (47%) | 61/128 (48%) |
| Compressible haemorrhage | 50/208 (24%) | 49/223 (22%) | 99/431 (23%) |
| Non-compressible haemorrhage | 171/208 (82%) | 186/223 (83%) | 357/431 (83%) |
| Traumatic cardiac arreste | 21/151 (14%) | 20/175 (11%) | 41/326 (13%) |
| Blunt force trauma | 162/208 (78%) | 178/223 (80%) | 340/431 (79%) |
| Penetrating trauma | 47/208 (23%) | 48/223 (22%) | 95/431 (22%) |
| Crush trauma | 6/208 (3%) | 2/223 (1%) | 8/431 (2%) |
| Suspected at time of injury | |||
| Alcohol | |||
| Yes | 30 (14%) | 32 (14%) | 62 (14%) |
| No | 175 (84%) | 186 (83%) | 361 (84%) |
| Other illicit substances | |||
| Yes | 13 (6%) | 12 (5%) | 25 (6%) |
| No | 187 (89%) | 201 (90%) | 388 (90%) |
| Pre-hospital timeline | |||
| Time from 999 call to arrival on scene (mins) Median (IQR) (n) | 26 (19–36) (209) | 27 (19–37) (223) | 26 (19–37) (432) |
| Time from arrival on scene to administration of first intervention (mins) Median (IQR) (n) | 22 (15–33) (201) | 21 (13–35) (209) | 22 (15–34) (410) |
| On-scene vital signs | |||
| Heart Rate (bpm)f Mean (SD, n) | 115 (31, 185) | 109 (33, 198) | 112 (32, 383) |
| SBP (mmHg)f Mean (SD, n) | 73 (16, 128) | 73 (20, 148) | 73 (18, 276) |
| DBP (mmHg)f Mean (SD, n) | 47 (13, 125) | 46 (16, 147) | 46 (15, 272) |
| Respiratory rate (/min)f Mean (SD, n) | 24 (9, 172) | 23 (11, 186) | 24 (10, 358) |
| Oxygen saturation (%)f Mean (SD, n) | 92 (8, 131) | 91 (9, 144) | 92 (9, 275) |
| GCS Median (IQR) (n) | 8 (3–14) (209) | 6 (3–14) (222) | 7 (3–14) (431) |
| Capillary lactate concentration (mmol/L) Mean (SD, n) | 9.1 (4.4, 199) | 9.2 (5.0, 207) | 9.2 (4.7, 406) |
| Medical history g | |||
| ISSh Median (IQR) (n) | 36 (24.5–49) (148) | 36 (25–50) (152) | 36 (25–50) (300) |
| NISSh Median (IQR) (n) | 43 (34–57) (144) | 48 (34–57) (148) | 43 (34–57) (292) |
| Comorbidities | |||
| Yes | 18/172 (10%) | 20/170 (12%) | 38/342 (11%) |
| No | 154/172 (90%) | 149/170 (87.5%) | 303/342 (88.75%) |
| Anticoagulant medication | |||
| Yes | 16/172 (9%) | 24/170 (14%) | 40/342 (12%) |
| No | 125/172 (73%) | 105/170 (62%) | 230/342 (67%) |
| Unknown | 30/172 (17.5%) | 41/170 (24%) | 71/342 (20.75%) |
| Antiplatelet medication | |||
| Yes | 3/172 (2%) | 3/170 (2%) | 6/342 (1.75%) |
| No | 138/172 (80%) | 127/170 (75%) | 265/342 (78%) |
| Unknown | 30/172 (17.5%) | 40/170 (23%) | 70/342 (20%) |
| Concomitant treatments | |||
| Tranexamic acid | 182 (87%) | 206 (92%) | 388 (90%) |
| Fluid volume given prior to intervention (ml) Mean (SD) | 422 (499) | 437 (482) | 430 (490) |
| Mode of transport | |||
| Air | 80 (38%) | 86 (39%) | 166 (38%) |
| Ground | 129 (62%) | 137 (61%) | 266 (62%) |
Injury characteristics were similar across treatment arms. The presence of a concurrent brain injury was only collected in the last 16 months of the trial and, during that period, around half of the participants had a concurrent brain injury (48%). Participants could experience both compressible and non-compressible haemorrhages, with non-compressible haemorrhages being the most commonly recorded (83%). Traumatic cardiac arrest, defined as those with a heart rate of 0 and blood pressure of 0, was experienced by 13% of participants providing on-scene heart rate and blood pressure measurements. Blunt force trauma injuries were recorded in 79% of participants, with penetrating trauma injuries recorded in 22% of participants. Acute excessive consumption or alcoholism was suspected in 14% of participants, and the presence of other illicit substances was suspected in 6% of participants. On-scene vital signs were very similar between both groups, with participants exhibiting a mean [(standard deviation (SD)] blood pressure of 73 (18)/46 (15) mmHg, a mean (SD) heart rate of 112 (32) bpm, a mean (SD) respiratory rate of 23.8 (10.1) per minute, a mean (SD) oxygen saturation of 92% (9%) and a median (IQR) GCS of 7 (3, 14). Capillary lactate concentration was very similar across both groups, with a mean (SD) of 9.15 (4.69) mmol/L across all participants.
Both measures of injury severity, injury severity score (ISS) and new injury severity score (NISS), are recorded only on those participants that were Trauma Audit Research Network (TARN) eligible. This excludes participants who died prior to arrival at the ED, so cannot strictly be defined as a baseline characteristic. For the 300 participants providing injury severity scores, the median ISS was 36 (IQR 25 to 50) and median NISS was 43 (IQR 34 to 57). The pre-hospital teams enrolling RePHILL patients attended scene via road ambulance for 62% of participants. They arrived on scene at a median of 26 minutes (IQR 19 to 37) after the time of injury, approximated via the time of the 999 call, and administered the first bolus of trial intervention at a median of 22 minutes (IQR 15 to 34) later.
Mode of transport
There was variability between IDS in the mode of transport the pre-hospital teams enrolling RePHILL patients used to attend the scene (see Table 3). Over all participants, 62% were attended via road ambulance, but this proportion varies between 48% and 71% across the four IDS.
The distribution of mode of transport is summarised by IDS and treatment group in Table 5.
| Site | Mode of transport | RBC/LyoPlas (%) | 0.9% saline (%) | Total (%) |
|---|---|---|---|---|
| Site 1 | Air | 19/68 (28) | 19/64 (30) | 38/132 (29) |
| Ground | 49/68 (72) | 45/64 (70) | 94/132 (71) | |
| Site 2 | Air | 17/37 (46) | 18/41 (44) | 35/78 (45) |
| Ground | 20/37 (54) | 23/41 (56) | 43/78 (55) | |
| Site 3 | Air | 23/60 (38) | 17/61 (28) | 40/121 (33) |
| Ground | 37/60 (62) | 44/61 (72) | 81/121 (67) | |
| Site 4 | Air | 21/44 (48) | 32/57 (56) | 53/101 (52) |
| Ground | 23/44 (52) | 25/57 (44) | 48/101 (48) |
Pre-hospital timeline
The pre-hospital timeline (see Table 6) displays the time, in minutes, between the key trial events that occurred prior to arrival at hospital for each treatment group. The timings are very similar across the two treatment groups, with little evidence that administering boluses of RBC and LyoPlas, compared to 0.9% saline, delayed arrival at ED. The time from opening the treatment box to arrival at ED was a median of 32 minutes (IQR 21 to 45) for participants in the RBC/LyoPlas group and a median of 30 minutes (IQR 20 to 45) for participants in the 0.9% saline group.
| Pre-hospital event | 999 call | On-scene attendance | Randomisation capillary lactate | Treatment box opening | Left scenea | Arrival at EDb |
|---|---|---|---|---|---|---|
| 999 call | 26 (19, 36) (n = 209) |
46 (35, 60) (n = 203) |
47 (35, 62) (n = 209) |
57.5 (41, 76) (n = 62) |
83 (63, 109) (n = 169) |
|
| On-scene attendance | 27 (19, 37) (n = 223) |
18 (11, 28) (n = 203) |
19 (11, 30) (n = 209) |
31 (22, 45) (n = 62) |
57 (40, 78) (n = 169) |
|
| Randomisation capillary lactate | 48.5 (34, 65) (n = 214) |
17.5 (10, 30) (n = 214) |
0 (0, 1) (n = 203) |
4 (0, 19) (n = 59) |
33 (22, 47) (n = 165) |
|
| Treatment box opening | 48 (34, 65) (n = 223) |
19 (10, 31) (n = 223) |
1 (0, 2) (n = 214) |
3 (–2, 15) (n = 62) |
32 (21, 45) (n = 169) |
|
| Left scene a | 66 (42, 87.5) (n = 68) |
34.5 (19, 45) (n = 68) |
10 (–3, 24.5) (n = 64) |
10.5 (–3, 24.5) (n = 68) |
26 (17, 36) (n = 53) |
|
| Arrival at EDb | 85 (66, 111) (n = 171) |
57 (41, 74) (n = 171) |
31 (21, 45) (n = 167) |
30 (20, 45) (n = 171) |
26.5 (18, 36.5) (n = 56) |
Primary outcomes
The composite primary outcome of episode mortality and/or failure to clear lactate occurred in 128/199 (64%) of participants in the RBC/LyoPlas group, and in 136/210 (65%) of participants in the saline 0.9% group (see Table 5). After adjusting for IDS, allocation to treatment with RBC/LyoPlas was observed across the 432 study participants to have a 1% higher relative risk of episode mortality and/or failure to clear lactate than allocation to treatment with 0.9% saline with a 95% two-sided compatibility interval of 0.88 to 1.17, indicating a moderate range of plausible true treatment effects. The degree of evidence against the null hypothesis that the treatments are interchangeable is p = 0.86. After adjusting for IDS, allocation to treatment with RBC/LyoPlas was observed across the 432 study participants to have a 0.025% lower absolute risk of episode mortality and/or failure to clear lactate than allocation to treatment with 0.9% saline with a 95% two-sided compatibility interval of −9% to 9%, indicating a moderate range of plausible true treatment effects. The degree of evidence against the null hypothesis that the treatments are interchangeable is p = 0.996.
The qualifying events for the primary outcome in each treatment group are given in Table 7. Due to participant drop-out and missing data, 23 participants did not provide primary outcome data. For the 199 participants providing a primary outcome in the RBC/LyoPlas group, the most common outcome was survival and clearance of lactate (36%), 29% of participants experienced episode mortality and failed to clear their lactate, 20% survived but failed to clear their lactate and 15% cleared their lactate but experienced episode mortality. For the 210 participants providing a primary outcome in the 0.9% saline group, the most common outcome was experiencing episode mortality and failure to clear their lactate (36%), 35% of participants survived and cleared their lactate, 18% survived but failed to clear their lactate and 11% cleared their lactate but experienced episode mortality.
| RBC/LyoPlas (%) | 0.9% saline | Total | |
|---|---|---|---|
| Qualifying event, n (%) | |||
| Both episode mortality and failure to clear lactate | 58/209 (28) | 76/223 (34) | 134/432 (31) |
| Episode mortality alone | 30/209 (14) | 23/223 (10) | 53/432 (12) |
| Failure to clear lactate alone | 40/209 (19) | 37/223 (17) | 77/432 (18) |
| Alive and cleared lactate | 71/209 (34) | 74/223 (33) | 145/432 (34) |
| Missing/not available | 10/209 (5) | 13/223 (6) | 23/432 (5) |
The individual components of the primary outcome were analysed separately (see Table 8). Episode mortality occurred in 88/203 (43%) of participants in the RBC/LyoPlas group, and in 99/218 (45%) of participants in the saline 0.9% group (see Table 8). Analyses, adjusted for IDS, yielded an estimated risk ratio of 0.97 (95% CI 0.78 to 1.20; p-value: 0.75) and an estimated risk difference of −3% (95% CI: −12% to 7%; p-value: 0.57). Failure to clear lactate occurred in 98/196 (50%) of participants in the RBC/LyoPlas group, and in 113/206 (55%) of participants in the saline 0.9% group (see Table 8). Analyses, adjusted for IDS, yielded an estimated risk ratio of 0.94 (95% CI 0.78 to 1.13; p-value: 0.52) and an estimated risk difference of −5% (95% CI −14% to 5%; p-value: 0.33). The estimates of treatment effects for each individual component produced relatively wide CIs including both benefits and harms. The degree of evidence against the null hypothesis that the treatments are interchangeable ranged from 0.33 to 0.75 across these analyses.
| RBC/LyoPlas (%) | 0.9% saline (%) | Adjusted risk ratio (95% CI) | Adjusted average difference (95% CI) | |
|---|---|---|---|---|
| Primary outcome | ||||
| Episode mortality and/or failure to clear lactate | 128/199 (64) | 136/210 (65) | 1.01 (0.88 to 1.17)a; P = 0.86 | −0.025% (−9% to 9%)b; P = 0.996 |
| Episode mortality | 88/203 (43) | 99/218 (45) | 0.97 (0.78 to 1.20)a; P = 0.75 | −3% (−12% to 7%)b; P = 0.57 |
| Failure to clear lactate | 98/196 (50) | 113/206 (55) | 0.94 (0.78 to 1.13)a; P = 0.52 | −5% (−14% to 5%)b; P = 0.33 |
Table 6 summarises which of the primary outcome components were met by the 432 participants: the 409 who recorded a primary outcome, and the 23 who did not provide information to determine a primary outcome.
Secondary outcomes
The secondary outcomes are summarised in Tables 9–13. Selected secondary outcomes are summarised in Table 9. The volume of post intervention fluids was similar between treatment groups [adjusted mean difference −34 ml (95% CI –101 to 32)]. There was little evidence of any meaningful difference in the time taken to arrive at ED from either the time of injury [recorded as the time of the 999 call, adjusted mean difference 0.60 minutes (95% CI –6.14 to 7.35)] or from the time of randomisation [recorded as the time the first treatment box was opened, adjusted mean difference 3.03 minutes (95% CI –1.40 to 7.46)]. Vital signs recorded at ED arrival were similar across treatment groups [adjusted mean differences for heart rate −0.80 (95% CI –5.83 to 4.23) beats per minute, SBP −1.19 (95% CI –8.19 to 5.82) mmHg, diastolic blood pressure 2.26 (95% CI –3.77 to 8.29) mmHg, respiratory rate 0.59 (95% CI –0.79 to 1.97) per minute and oxygen saturation 0.48 (95% CI –0.86 to 1.82)]. Laboratory results showed that lactate concentration on ED arrival was similar across groups [adjusted mean difference −0.08 (95% CI −0.97 to 0.82) mmol/L]. Although INR was only collected on 158 participants, the proportion with an INR >1.5 was similar across treatment groups [adjusted risk ratio 0.91 (95% CI 0.44 to 1.90)]. Haemoglobin concentration on ED arrival was significantly higher in the RBC/LyoPlas group compared to the 0.9% saline group [adjusted mean difference 15 (95% CI 10 to 19) g/L]. A post-hoc analysis found that blood product use from admission to hospital through to 24 hours later was higher in the RBC/LyoPlas group than in the 0.9% saline group [adjusted mean difference of 1.80 (95% CI 0.58 to 3.01) units of RBCs and 1.54 (95% CI 0.57 to 2.50) units of plasma]. Measures of mortality within 3 hours of injury and within 30 days of mortality were both slightly lower in the RBC/LyoPlas group. Analyses, adjusted for IDS, yielded an estimated risk ratio of 0.75 (95% CI 0.50 to 1.13) and an estimated risk difference of −7% (95% CI –15% to 1%) for 3-hour mortality. For 30-day mortality, adjusted analyses produced an estimated risk ratio of 0.94 (95% CI 0.76 to 1.17) and an estimated risk difference of −4% (95% CI –13% to 6%).
| RBC/LyoPlas | 0.9% saline | Adjusted risk ratio (95% CI) | Adjusted average difference (95% CI) | |
|---|---|---|---|---|
| Secondary outcomes | ||||
| Post intervention fluids (ml) | 123 (310), 207 | 160 (389), 221 | −34 (−101 to 32)a; P = 0.31 | |
| Time to ED arrival (mins) | ||||
| From 999 call | 90 (35), 169 | 91 (35), 171 | - | 0.60 (–6.14 to 7.35)a; P = 0.86 |
| From randomisation | 37 (22), 169 | 35 (22), 171 | - | 3.03 (−1.40 to 7.46)a; P = 0.18 |
| Vital signs at ED arrival | ||||
| Heart rate (bpm) | 107 (29), 157 | 105 (24), 154 | - | −0.80 (−5.83 to 4.23)b; P = 0.76 |
| SBP (mmHg) | 114 (27), 111 | 114 (29), 124 | - | −1.19 (−8.19 to 5.82)b; P = 0.74 |
| Diastolic blood pressure (mmHg) | 75 (24), 107 | 72 (24), 123 | - | 2.26 (−3.77 to 8.29)b; P = 0.46 |
| Respiratory rate (/min) | 20 (6.5), 128 | 19 (5.6), 126 | - | 0.59 (−0.79 to 1.97)b; P = 0.40 |
| Oxygen saturation (%) | 97 (5.2), 105 | 97 (5.2), 114 | - | 0.48 (−0.86 to 1.82)b; P = 0.48 |
| Laboratory results (ED arrival) | ||||
| Lactate concentration (mmol/L) | 7.04 (4.50) | 6.93 (4.58) | - | −0.08 (−0.97 to 0.82)b; P = 0.87 |
| INR >1.5 | 12/84 (14%) | 12/74 (16%) | 0.91 (0.44 to 1.90)c; P = 0.80 | |
| Haemoglobin (g/L) | 133 (19), 154 | 118 (23), 152 | - | 15 (10 to 19)a; P < 0.0001 |
| Total blood product up to 24 hours after ED arrival | ||||
| RBC | 6.34 (7.09), 209 | 4.41 (6.17), 223 | - | 1.80 (0.58 to 3.01)a; P = 0.0037 |
| Plasma | 5.04 (5.56), 209 | 3.37 (5.04), 223 | - | 1.54 (0.57 to 2.50)a; P = 0.0018 |
| Death | ||||
| Within 3 hours | 32/197 (16%) | 46/208 (22%) | 0.75 (0.50 to 1.13)c; P = 0.17 | −7% (−15% to 1%)d; P = 0.083 |
| Within 30 days | 86/204 (42%) | 99/219 (45%) | 0.94 (0.76 to 1.17)c; P = 0.59 | −4% (−13% to 6%)d; P = 0.44 |
| Within 24 hours | 47/197 (24%) | 65/207 (31%) | 0.77 (0.56 to 1.06)c; P = 0.10 | −8% (−17% to 0.05%)d; P = 0.066 |
Pre-hospital fluid (type and volume) and vital signs are summarised in Table 10. The proportion of participants receiving fluids given prior to intervention, and the volumes of fluids received, were similar between both groups [adjusted risk ratio 0.95 (95% CI 0.83 to 1.07) and adjusted mean difference −17 (−108 to 74) ml]. The proportion of participants receiving fluids given after intervention, and the volumes of fluids received, were also similar between both groups [adjusted risk ratio 0.84 (95% CI 0.58 to 1.21) and adjusted mean difference −34 (−101 to 32) ml]. Vital signs were recorded at regular intervals from arrival on scene to 24 hours after arrival at hospital. Vital signs are only summarised for participants who were still alive at the time of assessments (i.e. no values of 0 are imputed for participants known to have died at a previous time point). All vital signs were broadly similar across both treatment groups at each time point. Mean heart rate values appeared to stabilise at around 86–90 beats per minute two hours after participants arrived in hospital. SBP increased from a mean of 73 mmHg on scene to around 110–117 mmHg at all in-hospital assessments. Diastolic blood pressure increased from a mean of 46 mmHg on scene to a mean of 73 mmHg when arriving at hospital, before reducing to a mean of 62 mmHg 12 hours after arriving at hospital. Respiratory rate was slightly elevated on scene with a mean of 23 per minute, but this reduced to an average rate between 18 and 20 per minute after arrival at hospital. Oxygen saturation was relatively low on scene with a mean of 92%, but this increased to a mean saturation greater than 97% once participants arrived in hospital. Lactate concentration was similar across both arms at each time point with values decreasing from arrival at hospital to 2 hours after arrival at hospital. At 2 and 6 hours after arrival at hospital, the proportion of participants with an INR >1.5 was similar across treatment groups, however given the low incidence of participants with an INR >1.5 there is considerable uncertainty associated with these estimates. Calcium concentration on ED arrival was similar across treatment groups [adjusted mean difference −0.03 (95% CI −0.12 to 0.05) mmol/L].
| Outcome | RBC/LyoPlas | 0.9% saline | Adjusted risk ratio (95% CI) | Adjusted average difference (95% CI) |
|---|---|---|---|---|
| Pre-hospital fluid type and volume | ||||
| Fluids given prior to intervention | 142/209 68%) | 159/223 (71%) | 0.95 (0.83 to 1.07)a; P = 0.40 | |
| Salineb | 140/209 (67%) | 159/223 (71%) | ||
| Hartmann’sb | 1/209 (0.5%) | 2/223 (1%) | ||
| Otherb | 7/209 (3%) | 4/223 (2%) | ||
| Volume given prior to intervention | 422 (499), 209 | 437 (482), 223 | −17 (−108 to 74)c; 0.71 | |
| Fluids given after intervention | 40/207 (19%) | 52/221 (24%) | 0.84 (0.58 to 1.21)a; P = 0.35 | |
| Salineb | 33/207 (16%) | 39/221 (17%) | ||
| Hartmann’sb | 3/207 (1%) | 6/221 (3%) | ||
| Otherb | 5/207 (2%) | 15/221 (7%) | ||
| Volume given after intervention | 123 (310), 207 | 160 (389), 221 | −34 (−101 to 32)c; P = 0.31 |
|
| Vital signs | ||||
| Heart rate (bpm) | ||||
| On scene | 115 (31),185 | 109 (33), 198 | 5.83 (−0.61 to 12.27)c; P = 0.076 | |
| ED arrival | 107 (29), 157 | 105 (24), 154 | −0.80 (−5.83 to 4.23)d; P = 0.76 | |
| 2 hrs after ED arrival | 95 (22), 147 | 91 (22), 147 | 3.80 (−1.09 to 8.70)d; P = 0.13 | |
| 6 hrs after ED arrival | 88 (21), 148 | 86 (21), 137 | 2.57 (−2.34 to 7.49)d; P = 0.31 | |
| 12 hrs after ED arrival | 90 (21), 149 | 89 (23), 139 | 1.23 (−3.81 to 6.28)d; P = 0.63 | |
| 24 hrs after ED arrival | 90 (20), 144 | 90 (22), 134 | −1.05 (−5.94 to 3.84)d; P = 0.67 | |
| SBP (mmHg) | ||||
| On scene | 73 (16), 128 | 73 (20), 148 | −0.05 (v4.23 to 4.14)c; P = 0.98 | |
| ED arrival | 114 (27), 111 | 114 (29), 124 | −1.19 (−8.19 to 5.82)d; P = 0.74 | |
| 2 hrs after ED arrival | 114 (24), 113 | 115 (21), 121 | 0.04 (−5.75 to 5.83)d; P = 0.99 | |
| 6 hrs after ED arrival | 109 (21), 116 | 114 (23), 117 | −5.22 (−10.87 to 0.43)d; P = 0.070 | |
| 12 hrs after ED arrival | 113 (22), 110 | 115 (24), 118 | −2.27 (−8.23 to 3.69)d; P = 0.45 | |
| 24 hrs after ED arrival | 114 (20), 109 | 117 (21), 114 | −3.24 (−8.59 to 2.12)d; P = 0.24 |
|
| Diastolic blood pressure (mmHg) | ||||
| On scene | 47 (13), 125 | 46 (16), 147 | 0.77 (−2.70 to 4.24)c; P = 0.66 | |
| ED arrival | 75 (24), 107 | 72 (24), 123 | 2.26 (−3.77 to 8.29)d; P = 0.46 | |
| 2 hrs after ED arrival | 67 (17), 111 | 65 (15), 119 | 2.07 (−1.97 to 6.12)d; P = 0.31 | |
| 6 hrs after ED arrival | 64 (15), 114 | 67 (15), 117 | −2.76 (−6.57 to 1.04)d; P = 0.15 | |
| 12 hrs after ED arrival | 62 (13), 108 | 62 (13), 118 | −0.36 (−3.60 to 2.88)d; P = 0.83 | |
| 24 hrs after ED arrival | 61 (14), 107 | 62 (12), 114 | −1.44 (−4.73 to 1.84)d; P = 0.36 | |
| Respiratory rate (/min) | ||||
| On scene | 24 (9.5), 172 | 23 (10.6), 191 | 0.98 (−1.10 to 3.05)c; P = 0.36 | |
| ED arrival | 20 (6.5), 128 | 19 (5.6), 126 | 0.59 (–0.79 to 1.97)d; P = 0.40 | |
| 2 hrs after ED arrival | 19 (4.8), 121 | 19 (4.7), 123 | 0.45 (−0.72 to 1.62)d; P = 0.45 | |
| 6 hrs after ED arrival | 19 (6.3), 133 | 18 (4.1), 129 | 0.62 (−0.66 to 1.91)d; P = 0.34 | |
| 12 hrs after ED arrival | 19 (5.2), 140 | 18 (3.8), 133 | 0.49 (−0.59 to 1.58)d; P = 0.37 | |
| 24 hrs after ED arrival | 18 (4.11), 140 | 18 (3.7), 129 | 0.38 (−0.56 to 1.31)d; P = 0.43 | |
| Oxygen saturation (%) | ||||
| On scene | 92 (7.6),131 | 91 (9.3), 144 | 0.92 (−1.10 to 2.94)c; P = 0.37 | |
| ED arrival | 97 (5.2), 105 | 97 (5.2), 114 | 0.48 (−0.86 to 1.82)d; P = 0.48 | |
| 2 hrs after ED arrival | 98 (3.9), 104 | 98 (4.9), 108 | 0.03 (−1.14 to 1.20)d; P = 0.96 | |
| 6 hrs after ED arrival | 98 (4.4), 109 | 98 (6.0), 103 | 0.48 (−0.94 to 1.90)d; P = 0.51 | |
| 12 hrs after ED arrival | 97 (6.9), 108 | 98 (3.9), 102 | −0.38 (−1.91 to 1.15)d; P = 0.63 | |
| 24 hrs after ED arrival | 97 (2.6), 105 | 98 (2.4), 96 | −0.02 (−0.70 to 0.65)d; P = 0.95 | |
Total blood product receipt was recorded at 6, 12 and 24 hours after arrival at hospital and is summarised in Table 12. The mean number of units of RBCs used at each time point was slightly higher in participants in the RBC/LyoPlas group compared to the 0.9% saline group. Similarly, the mean number of units of plasma used at each time point was also slightly higher in the RBC/LyoPlas group compared to the 0.9% saline group. Participants on the RBC/LyoPlas arm received a higher volume of crystalloid than participants on the 0.9% saline arm [adjusted mean difference at 12 hours after hospital arrival 628 (95% CI 211 to 1034) ml, adjusted mean difference at 24 hours after hospital arrival 708 (95% CI 180 to 1236) ml]. The number of bags of cryoprecipitate and platelets was similar across all three time points. The volume of colloid was lower in participants in the RBC/LyoPlas group compared to the 0.9% saline group, but the high variability of colloid volume means there is considerable uncertainty associated with these adjusted mean differences.
| Outcome | RBC/LyoPlas | 0.9% saline | Adjusted risk ratio (95% CI) | Adjusted average difference (95% CI) |
|---|---|---|---|---|
| Laboratory results | ||||
| Lactate concentration (mmol/L) | ||||
| 2 hrs post-randomisation based on time | 5.42 (4.45) (n = 168) |
5.78 (4.68) (n = 169) |
−0.37 (−1.28 to 0.53)a; P = 0.42 | |
| 2 hrs post-randomisation based on CRF | 4.91 (4.14) (n = 153) |
5.40 (4.41) (n = 152) |
−0.34 (−1.24 to 0.55)a; P = 0.46 | |
| Arrival at ED | 7.04 (4.50) (n = 157) |
6.93 (4.58) (n = 161) |
−0.08 (−0.97 to 0.82)a; P = 0.87 | |
| 2 hrs after ED arrival | 4.45 (3.57) (n = 134) |
4.46 (3.33) (n = 138) |
−0.07 (−0.84 to 0.70)a; P = 0.86 | |
| INR >1.5 | ||||
| ED arrival | 12/84 (14%) | 12/74 (16%) | 0.91 (0.44 to 1.90)b; P = 0.80 | |
| 2 hrs after ED arrival | 1/27 (4%) | 4/29 (14%) | 0.27 (0.03 to 2.25)c; P = 0.23 | |
| 6 hrs after ED arrival | 3/48 (6%) | 3/46 (7%) | 0.81 (0.17 to 3.88)b; P = 0.79 | |
| Haemoglobin (g/L) arrival at ED | 133 (19), 154 | 118 (23), 152 | 15 (10 to 19)d; P = < 0.0001 |
|
| Calcium (mmol/L) arrival at ED | 1.21 (0.42), 152 | 1.24 (0.37), 156 | −0.03 (−0.12 to 0.05)d; P = 0.44 | |
| ROTEM | ||||
| EXTEM | ||||
| A05 (mm) | 35.8 (9.9), 32 | 33.2 (11.9), 23 | 2.61 (−3.07 to 8.29)d; P = 0.37 | |
| CFT (seconds) | 107 [84.5, 131.5], 32 | 110 [79, 145], 22 | −3 (−36 to 30)e; P = 0.86 | |
| MCF (mm) | 55.7 (12.4),32 | 54.9 (6.01), 20 | 0.64 (−5.10 to 6.37)d; P = 0.83 | |
| CT (seconds) | 78 [73, 107], 33 | 78 [69, 122], 3 | 3 (−22 to 28)c; P = 0.81 | |
| α angle (degree) | 70 [66, 73], 28 | 71 [65, 74],23 | −1 (−6 to 4)c; P = 0.67 | |
| Ly30 (%) | 100 [100, 100], 32 | 100 [100, 100], 23 | 0 (−0 to 0)e; - | |
| Ly60 (%) | 99.5 [99, 100], 22 | 98.5 [97, 100], 18 | 1 (−0.29 to 2.29)c; P = 0.13 | |
| FIBTEM | ||||
| A05 (mm) | 8.73 (3.78), 30 | 5.86 (2.71), 22 | 2.89 (1.06 to 4.71)d; P = 0.0020 | |
| CFT (seconds) | 76 (-), 1 | - | - | |
| MCF (mm) | 12.0 (9.6), 29 | 7.85 (3.34), 20 | 4.21 (−0.13 to 8.54)d; P = 0.057 | |
| CT (seconds) | 73 [67, 101], 31 | 84 [70, 121], 22 | −9 (−33 to 15)c; P = 0.46 | |
| α angle (degree) | 63 (7.3), 15 | 60 (9.0), 7 | 2.51 (−3.90 to 8.93)d; P = 0.44 | |
| Ly30 (%) | 100 [100, 100], 29 | 100 [100, 100], 22 | 0 (–0 to 0)e; - | |
| Ly60 (%) | 100 [100, 100], 21 | 100 [100, 100], 17 | 0 (0 to 0)e; - | |
| Multiplate | ||||
| TRAP | 93.4 (49.8), 21 | 77.6 (44.2), 10 | 11.0 (−24.2 to 46.2)d; P = 0.54 | |
| ADP | 53.5 (40.4), 22 | 42.8 (24.6), 10 | 6.36 (−19.7 to 32.4)d; P = 0.63 | |
| ASPI | 66.2 (41.8), 21 | 51.4 (36.5), 10 | 12.8 (−17.1 to 42.7)d; P = 0.40 | |
| Outcome | RBC/LyoPlas | 0.9% saline | Adjusted average difference (95% CI)a |
|---|---|---|---|
| Total blood product receipt | |||
| RBCs (units) | |||
| 6 hrs after arrival at ED | 5.61 (5.92), 132 | 5.31 (5.84), 137 | 0.09 (−1.27 to 1.45); P = 0.89 |
| 12 hrs after arrival at ED | 6.03 (7.62), 144 | 5.26 (6.08), 143 | 0.55 (−1.00 to 2.10); P = 0.49 |
| 24 hrs after arrival at ED | 5.63 (6.14), 139 | 5.31 (6.33), 134 | 0.18 (−1.22 to 1.59); P = 0.80 |
| Plasma (units) | |||
| 6 hrs after arrival at ED | 4.31 (4.68), 143 | 3.97 (4.75), 144 | 0.16 (−0.90 to 1.22); P = 0.77 |
| 12 hrs after arrival at ED | 4.72 (5.69), 144 | 4.26 (5.17), 143 | 0.30 (−0.92 to 1.51); P = 0.63 |
| 24 hrs after arrival at ED | 4.50 (4.76), 139 | 4.31 (5.40), 134 | 0.12 (–1.03 to 1.26); P = 0.84 |
| Crystalloid (volume) | |||
| 6 hrs after arrival at ED | 1417 (1610), 142 | 1037 (1175), 144 | 382 (61 to 702); P = 0.020 |
| 12 hrs after arrival at ED | 2388 (2031), 143 | 1782 (1550), 143 | 628 (221 to 1034); P = 0.0025 |
| 24 hrs after arrival at ED | 3620 (2479), 139 | 2947 (2115), 134 | 708 (180 to 1236); P = 0.0086 |
| Cryoprecipitate (bags) | |||
| 6 hrs after arrival at ED | 0.66 (1.23), 143 | 0.64 (1.38), 144 | 0.001 (−0.30 to 0.30); P = 0.99 |
| 12 hrs after arrival at ED | 0.89 (1.73), 144 | 0.80 (1.65), 143 | 0.05 (−0.33 to 0.43); P = 0.79 |
| 24 hrs after arrival at ED | 0.96 (2.03), 139 | 0.88 (2.00), 134 | 0.06 (−0.41 to 0.52); P = 0.82 |
| Platelets (bags) | |||
| 6 hrs after arrival at ED | 0.54 (0.97), 143 | 0.42 (0.87), 144 | 0.10 (−0.11 to 0.31); P = 0.37 |
| 12 hrs after arrival at ED | 0.63 (1.14), 144 | 0.55 (1.02), 143 | 0.06 (−0.18 to 0.31); P = 0.62 |
| 24 hrs after arrival at ED | 0.71 (1.19), 139 | 0.67 (1.31), 134 | 0.02 (−0.27 to 0.31); P = 0.90 |
| Colloid (volume) | |||
| 6 hrs after arrival at ED | 28 (155), 142 | 83 (317), 144 | −55 (−113 to 3); P = 0.062 |
| 12 hrs after arrival at ED | 31 (144), 143 | 128 (499), 142 | −98 (−183 to 13); P = 0.024 |
| 24 hrs after arrival at ED | 105 (368), 138 | 197 (701), 134 | −89 (−221 to 43); P = 0.18 |
Further secondary outcomes are summarised in Table 13. Acute respiratory distress syndrome (ARDS) was recorded in 9/142 (6%) participants in the RBC/LyoPlas group and 3/129 (2%) in the saline 0.9% group, adjusted relative risk 2.71 (95% CI 0.75 to 9.81). The rate of transfusion-related complications in the first 24 hours after arrival in hospital was similar across the two treatment groups [11/148 (7%) in the RBC/LyoPlas group and 9/137 (7%) in the 0.9% saline group, adjusted risk ratio 1.05 (95% CI 0.46 to 2.42)]. The number of organ failure-free days experienced by participants was similar across treatment groups [mean 12.19 days (SD 13.0) in the RBC/LyoPlas group and 12.1 days (SD 13.1) in the 0.9% saline group, adjusted mean difference 0.86 (95% CI −1.64 to 3.36) days].
| Outcome | RBC/LyoPlas | 0.9% saline | Adjusted risk ratio (95% CI) | Adjusted average difference (95% CI) |
|---|---|---|---|---|
| ARDS | 9/142 (6%) | 3/129 (2%) | 2.71 (0.75 to 9.81)a; P = 0.13 | |
| Transfusion-related complications (in first 24 hours in ED) | 11/148 (7%) | 9/137 (7%) | 1.05 (0.46 to 2.42)a; P = 0.90 | |
| Organ failure-free days b | 12.9 (13.0), 202 | 12.1 (13.1), 212 | 0.86 (−1.64 to 3.36)c; P = 0.50 | |
| All-cause mortality ≤ 3 hrs of randomisation | ||||
| Using time of death only | 6/171 (4%) | 6/168 (4%) | 0.94 (0.32 to 2.82)a; P = 0.92 | −0.001 (−0.04 to 0.04)d; P = 0.98 |
Laboratory results, ROTEM and Multiplate are summarised in Table 11. Coagulation measured viscoelastically by rotational thromboelastometry (ROTEM©) and platelet function using multiple electrode impedance aggregometry (MultiPlate) data was only collected on participants from selected receiving hospitals. The measurements taken for both sets of EXTEM and FIBTEM tests were similar across treatment group, suggesting that participants in each treatment arm exhibited equivalent coagulation measured viscoelastically profiles. Multiplate data were only collected on 32 participants, and the uncertainty associated with these small sample sizes makes it hard to reach any meaningful conclusions other than that there was an absence of evidence of any meaningful difference between treatment arms.
Exploratory outcomes are summarised in Table 14. Both the length of stay in ITU and in hospital, recorded from admission to hospital up to a maximum of 30 days, were similar between treatment groups [adjusted mean differences of −0.40 days (95% CI −2.84 to 2.04) and −1.67 days (95% CI −4.31 to 0.97) respectively]. The occurrence of organ failure on at least one day during hospital stay was assessed for each organ system. Around two-thirds of participants experienced organ failure on at least one day in their respiratory, neurological or cardiovascular systems. One in four participants experienced organ failure on at least one day in their renal system, one in eight participants experienced organ failure on at least one day in their coagulation system, and one in 13 experienced organ failure on at least one day in their liver system. Of the 406 participants providing a response, nearly all (97%) received an initial dose of TXA, of the 307 participants providing a response, over two-thirds (69%) received a second dose. Rates of surgery in the first 24 hours following admission to hospital were similar in both treatment groups, with the highest rates reported up to 6 hours after arrival at hospital.
| Outcome | RBC/LyoPlas | 0.9% saline | Adjusted risk ratio (95% CI) | Adjusted average difference (95% CI) |
|---|---|---|---|---|
| ITU length of stay (up to day 30) | (n = 142) | (n = 130) | ||
| Up to discharge from ITU | 10.5 (10.2) | 10.9 (10.4) | −0.40 (−2.84 to 2.04)a | |
| Hospital length of stay (up to day 30) | ||||
| Up to discharge from hospital | 17.9 (11.4) | 19.7 (10.8) | −1.67 (−4.31 to 0.97)a | |
| Any organ failure by system during hospital stay (up to day 30) [SOFA ≥3] | ||||
| Respiratory | 83/118 (70%) | 68/113 (60%) | 1.16 (0.96 to 1.40)b | |
| Neurological | 89/139 (64%) | 74/130 (57%) | 1.13 (0.93 to 1.37)b | |
| Cardiovascular | 95/138 (69%) | 80/126 (63%) | 1.09 (0.91 to 1.29)b | |
| Liver | 13/130 (10%) | 6/122 (5%) | 2.09 (0.82 to 5.35)b | |
| Coagulation | 12/135 (9%) | 19/127 (15%) | 0.58 (0.29 to 1.14)b | |
| Renal | 32/136 (24%) | 33/126 (26%) | 0.92 (0.61 to 1.40)b | |
| Use of tranexamic acid | ||||
| First TXA dose received | 186/193 (96%) | 208/213 (98%) | 0.99 (0.98 to 1.01)c | |
| Second TXA dose received | ||||
| On arrival at ED | 85/168 (51%) | 68/168 (40%) | 1.24 (0.97 to 1.56)b | |
| 2 hrs after arrival at ED | 54/127 (43%) | 60/139 (43%) | 1.00 (0.76 to 1.31)b | |
| 6 hrs after arrival at ED | 52/116 (45%) | 50/110 (45%) | 1.00 (0.77 to 1.32)b | |
| Any second dose received | 111/157 (71%) | 102/150 (68%) | 1.02 (0.89 to 1.17)b | |
| Surgery | ||||
| 2 hrs after arrival at ED | 51/162 (31%) | 45/166 (27%) | 1.08 (0.78 to 1.49)b | |
| Between 2 and 6 hrs after arrival at ED | 60/155 (39%) | 49/151 (32%) | 1.10 (0.83 to 1.46)b | |
| Between 6 and 12 hrs after arrival at ED | 39/152 (26%) | 25/148 (17%) | 1.54 (0.98 to 2.42)b | |
| Between 12 and 24 hrs after arrival at ED | 32/148 (21%) | 31/137 (23%) | 0.96 (0.62 to 1.49)b | |
Exploratory Bayesian analyses
We decided a priori to include a Bayesian analysis of the primary outcome and its individual components. The rationale for doing so was to directly estimate the probability of a clinically meaningful treatment effect, which is a measurement of direct interest to clinicians. 18
Following the DMEC and TSC meetings in May and October 2018, the power calculations were reframed in terms of relative risk rather than absolute risk while maintaining the original target sample size of 490. Based on the observed pooled event rate in May 2018 (65%), and allowing for a 10% loss to follow-up rate, 490 participants would provide 80% power to detect a relative risk ratio of 0.82. This effect size was used to inform the sceptical and information prior distributions in the exploratory Bayesian analyses.
Bayesian models were fitted using three different prior distributions: a non-informative prior, a sceptical prior such that the probability of observing a treatment effect at least as large as a relative risk ratio of 0.82 is <5% and an informative prior reflecting current knowledge. For each set of the priors, Table 15 provides summary statistics (the mean value, and upper and lower 2.5% quantiles) of the primary outcome event rates in each treatment group. Posterior probabilities of primary outcome rates by treatment group were estimated for Bayesian analyses using each of the three priors, encompassing varying assumptions of benefit from RBC/LyoPlas.
| Prior | Treatment group | Summary statistics for prior distributions: assumed event rate of composite primary outcome | ||
|---|---|---|---|---|
| 2.5% | Mean, % | 97.5% | ||
| Non-informative | 0.9% saline | 2.5 | 50 | 97.5 |
| RBC/LyoPlas | 2.5 | 50 | 97.5 | |
| Sceptical | 0.9% saline | 55.7 | 66.7 | 76.8 |
| RBC/LyoPlas | 55.7 | 66.7 | 76.8 | |
| Informative | 0.9% saline | 40 | 70 | 93.2 |
| RBC/LyoPlas | 19.4 | 60 | 92.5 | |
Bayesian analysis of the composite primary outcome
For each set of priors, the median risk ratios and associated 95% higher posterior density intervals for the primary outcome are presented in Table 16, along with the posterior probabilities that the risk ratio is <1, 0.8 and 0.7. The results are extremely similar across all three prior specifications, with posterior risk ratios of 1.01 (95% HDI 0.88 to 1.16) estimated for each. These estimates match the frequentist estimate of the adjusted risk ratio: 1.01 (95% CI 0.88 to 1.17). For each analysis the posterior probability that the risk ratio of experiencing either episode mortality or failure to clear lactate was lower in the RBC/LyoPlas group than in the 0.9% saline group which was 44%. The probability that the risk ratio of experiencing either episode mortality or failure to clear lactate was at least 20% lower (risk ratio of <0.8) in the RBC/LyoPlas group than in the 0.9% saline group which was 0.1%.
| Composite outcome | RBC/LyoPlas (N = 209) | 0.9% saline (N = 223) | Priors | Median risk ratio | 95% HDI | Probability of risk ratio | ||
|---|---|---|---|---|---|---|---|---|
| < 1.0, % | < 0.8, % | < 0.7, % | ||||||
| Yes | 128 (64%) | 136 (65%) | Non-informative | 1.01a | (0.88–1.16)a | 43.5 | 0.1 | 0 |
| No | 71 (36%) | 74 (35%) | Sceptical | 1.01a | (0.87–1.16)a | 44 | 0.1 | 0 |
| Missing | 10 | 13 | Informative | 1.01a | (0.87–1.16)a | 44 | 0.1 | 0 |
For each set of priors, the median absolute risk differences and associated 95% higher posterior density intervals for the primary outcome are presented in Table 17, along with the posterior probabilities that the absolute risk difference is less than 0, −10% and −20%. The results are consistent across all three prior specifications, with posterior absolute risk differences ranging from 0.6% (95% HDI −7% to 8%) to −0.4% (HDI −9% to 8%). These estimates align with the frequentist estimate of the adjusted absolute risk differences: −0.025% (95% CI −9% to 9%). Across the Bayesian analyses the posterior probability that the absolute risk of experiencing either episode mortality or failure to clear lactate was lower in the RBC/LyoPlas group than in the 0.9% saline group ranged from 44.1% to 53.4%. The probability that the absolute risk of experiencing either episode mortality or failure to clear lactate was at least 10 percentage points lower in the RBC/LyoPlas group than in the 0.9% saline group which ranged from 0.3% to 1.6%.
| Composite Outcome | RBC/LyoPlas (N = 209) | 0.9% saline (N = 223) | Priors | Median absolute risk difference | 95% HDI | Probability of absolute risk difference | ||
|---|---|---|---|---|---|---|---|---|
| < 0.0, % | < −0.1, % | < −0.2, % | ||||||
| Yes | 128 (64%) | 136 (65%) | Non-informative | 0.002a | (−0.09 to 0.09)a | 48.2 | 1.3 | 0.007 |
| No | 71 (36%) | 74 (35%) | Sceptical | 0.006a | (−0.07 to 0.08)a | 44.1 | 0.3 | 0 |
| Missing | 10 | 13 | Informative | −0.004a | (−0.09 to 0.08)a | 53.4 | 1.6 | 0 |
Bayesian analysis of episode mortality
For each set of priors, the median risk ratios and associated 95% higher posterior density intervals for episode mortality are presented in Table 18, along with the posterior probabilities that the risk ratio is <1, 0.8 and 0.7. As for the primary outcome analysis, the results are extremely similar across all three prior specifications, with posterior risk ratios for episode mortality of 0.97 (95% HDI 0.77 to 1.17) or 0.97 (95% HDI 0.77 to 1.18) estimated for each. These estimates are consistent with the frequentist estimate of the adjusted risk ratio: 0.97 (95% CI 0.78 to 1.20). For each analysis, the posterior probability that the risk ratio of experiencing episode mortality was lower in the RBC/LyoPlas group than in the 0.9% saline group was between 63% and 64%. The probability that the risk ratio of experiencing episode mortality was at least 20% lower (risk ratio of <0.8) in the RBC/LyoPlas group than in the 0.9% saline group <5% in all three analyses.
| Episode mortality | RBC/LyoPlas (N = 209) | 0.9% saline (N = 223) | Priors | Median risk ratio | 95% HDI | Probability of risk ratio | ||
|---|---|---|---|---|---|---|---|---|
| < 1.0, % | < 0.8, % | < 0.7, % | ||||||
| Yes | 88 (43%) | 99 (45%) | Non-informative | 0.97a | (0.77–1.18)a | 63.1 | 4.2 | 0.2 |
| No | 115 (57%) | 119 (55%) | Sceptical | 0.97a | (0.77–1.17)a | 63.6 | 4.6 | 0.2 |
| Missing | 6 | 5 | Informative | 0.97a | (0.77–1.17)a | 64.0 | 4.6 | 0.2 |
For each set of priors, the median absolute risk differences and associated 95% higher posterior density intervals for episode mortality are presented in Table 19, along with the posterior probabilities that the absolute risk difference is <0, −10% and −20%. The results are consistent across all three prior specifications, with posterior absolute risk differences ranging from −5% (95% HDI −12% to 3%) to −3% (HDI −12% to 6%). These estimates align with the frequentist estimate of the adjusted absolute risk differences: −3% (95% CI −12% to 7%). Across the Bayesian analyses, the posterior probability that the absolute risk of experiencing episode mortality was lower in the RBC/LyoPlas group than in the 0.9% saline group ranged from 71.2% to 88.2%. The probability that the absolute risk of experiencing episode mortality was at least 10 percentage points lower in the RBC/LyoPlas group than in the 0.9% saline group ranged from 5.9% to 9.5%.
| Episode mortality | RBC/LyoPlas (N = 209) | 0.9% saline (N = 223) | Priors | Median absolute risk difference | 95% HDI | Probability of absolute risk difference | ||
|---|---|---|---|---|---|---|---|---|
| < 0.0, % | < −0.1, % | < −0.2, % | ||||||
| Yes | 88 (43%) | 99 (45%) | Non-informative | −0.03a | (−0.12 to 0.06)a | 71.2 | 5.9 | 0.009 |
| No | 115 (57%) | 119 (55%) | Sceptical | −0.05a | (−0.12 to 0.03)a | 88.2 | 8.4 | 0 |
| Missing | 6 | 5 | Informative | −0.04a | (−0.13 to 0.05)a | 80.7 | 9.5 | 0.013 |
Bayesian analysis of failure to clear lactate
For each set of priors, the median risk ratios and associated 95% higher posterior density intervals for failure to clear lactate are presented in Table 20, along with the posterior probabilities that the risk ratio is <1, 0.8 and 0.7. The results are similar across all three prior specifications, with posterior risk ratios of either 0.94 (95% HDI 0.78 to 1.13) or 0.94 (95% HDI 0.77 to 1.12) estimated for each. These estimates align well with the frequentist estimate of the adjusted risk ratio: 0.94 (95% CI 0.78 to 1.13). Across all three analyses, the posterior probability that the risk ratio of failure to clear lactate was lower in the RBC/LyoPlas group than in the 0.9% saline group ranged from 73.5% to 74.5%. The probability that the risk ratio of failure to clear lactate was at least 20% lower (risk ratio of <0.8) in the RBC/LyoPlas group than in the 0.9% saline group ranged from 4.3% to 4.5%.
| Failure to clear lactate | RBC/LyoPlas (N = 209) | 0.9% saline (N = 223) | Priors | Median risk ratio | 95% HDI | Probability of risk ratio | ||
|---|---|---|---|---|---|---|---|---|
| < 1.0, % | < 0.8, % | <0.7, % | ||||||
| Yes | 98 (50%) | 113 (55%) | Non-informative | 0.94a | (0.78–1.13)a | 73.5 | 4.3 | 0.09 |
| No | 98 (50%) | 93 (45%) | Sceptical | 0.94a | (0.77–1.12)a | 74.4 | 4.4 | 0.14 |
| Missing | 13 | 17 | Informative | 0.94a | (0.77–1.12)a | 74.5 | 4.5 | 0.12 |
For each set of priors, the median absolute risk differences and associated 95% higher posterior density intervals for failure to clear lactate are presented in Table 21, along with the posterior probabilities that the absolute risk difference is <0, −10% and −20%. The results are consistent across all three prior specifications, with posterior absolute risk differences ranging from −4% (95% HDI −13% to 5%) to −5% (HDI −14% to 4%). These estimates align closely with the frequentist estimate of the adjusted absolute risk differences: −5% (95% CI −14% to 5%). Across the Bayesian analyses, the posterior probability that the absolute risk of failure to clear lactate was lower in the RBC/LyoPlas group than in the 0.9% saline group ranged from 81.3% to 86.9%. The probability that the absolute risk of failure to clear lactate was at least 10 percentage points lower in the RBC/LyoPlas group than in the 0.9% saline group ranged from 5.6% to 11.5%.
| Failure to clear lactate | RBC/LyoPlas (N = 209) | 0.9% saline (N = 223) | Priors | Median absolute risk difference | 95% HDI | Probability of absolute risk difference | ||
|---|---|---|---|---|---|---|---|---|
| < 0.0, % | < –0.1, % | < –0.2, % | ||||||
| Yes | 98 (50%) | 113 (55%) | Non-informative | −0.04a | (−0.13 to 0.05)a | 81.3 | 11.5 | 0.05 |
| No | 98 (50%) | 93 (45%) | Sceptical | −0.04a | (−0.11 to 0.04)a | 84.6 | 5.6 | 0 |
| Missing | 13 | 17 | Informative | −0.05a | (−0.14 to 0.04)a | 86.9 | 15.3 | 0.07 |
Sensitivity analyses
Sensitivity analyses were undertaken for the primary outcome and each component of the primary outcome: episode mortality and failure to clear lactate. For each sensitivity analysis, the adjusted risk ratio and adjusted risk differences are reported.
Sensitivity analyses for primary outcome
The results of the sensitivity analyses for the primary outcome are presented in Table 22. The results from the original analyses are included in the first row for reference. All of the results of the sensitivity analyses are consistent with the results from the original analyses. There is little evidence that missing data influenced the results of the primary outcome analysis. Restricting the analysis to the participants providing lactate measurements within the allowable time window did not change the interpretation of the trial results. Restricting the analysis to the participants who adhered to their allocated treatment, using both definitions, did not change the trial results. Accounting for other covariates in the analysis reduced the analysis population, but produced estimates of treatment effect that were consistent with those from the primary analysis.
| RBC/LyoPlas | 0.9% saline | Sensitivity analysis | Adjusted risk ratio (95% CI); p-value | Adjusted risk difference (95% CI); p-value |
|---|---|---|---|---|
| 128/199 (64%) | 136/210 (65%) | Original analysis | 1.01 (0.88 to 1.17)a; P = 0.86 | −0.00025 (−0.09 to 0.09)b; P = 0.996 |
| 77/130 (59%) | 87/151 (58%) | Further covariate adjustment | 1.02 (0.95 to 1.08)c; P = 0.63 | 0.02 (−0.10 to 0.13)d; P = 0.79 |
| 125/192 (65%) | 130/203 (64%) | Per-protocol analysis | 1.04 (0.90 to 1.20)a; P = 0.63 | 0.014 (−0.08 to 0.11)b; P = 0.76 |
| 125/196 (64%) | 126/198 (64%) | Secondary per-protocol analysis | 1.03 (0.89 to 1.19)a; P = 0.68 | 0.012 (−0.08 to 0.11)b; P = 0.80 |
| 95/151 (65%) | 103/153 (67%) | Timing of lactate Concentration | 0.99 (0.85 to 1.17)a; P = 0.94 | −0.014 (−0.12 to 0.09)b; P = 0.80 |
| - | - | Missing responses | (0.87 to 1.17)e; P = 0.90 | −0.001 (−0.094 to 0.091)f; P = 0.98 |
Sensitivity analyses for episode mortality
The results of the sensitivity analyses for episode mortality are presented in Table 23. The results from the original analyses are included in the first row for reference. As for the primary outcome analyses, all of the results of the sensitivity analyses are consistent with the results from the original analyses. Imputing missing data returned treatment estimates that were nearly identical to those in the original analysis. Restricting the analysis to the participants that adhered to their allocated treatment, using both definitions, produced estimates of treatment effect that were even closer to the null values, but the overall interpretation of the episode mortality results did not change. Accounting for other covariates in the analysis reduced the analysis population and produced estimates of episode mortality that were slightly higher in the RBC/LyoPlas treatment arm compared to the 0.9% saline arm, but the uncertainty associated with these estimates meant that the results were consistent with those from the original analysis.
| RBC/LyoPlas | 0.9% saline | Sensitivity analysis | Adjusted risk ratio (95% CI); p-value |
Adjusted risk difference (95% CI); p-value |
|---|---|---|---|---|
| 88/203 (43%) | 99/218 (45%) | Original analysis | 0.97 (0.78 to 1.20)a; P = 0.75 | −0.03 (−0.12 to 0.07)b; P = 0.57 |
| 50/131 (38%) | 55/152 (36%) | Further covariate adjustment | 1.02 (0.96 to 1.09)a; P = 0.50 | 0.01 (−0.10 to 0.12)c; P = 0.86 |
| 85/194 (44%) | 94/210 (45%) | Per-protocol analysis | 0.99 (0.80 to 1.23)a; P = 0.93 | −0.02 (−0.11 to 0.08)b; P = 0.73 |
| 85/197 (44%) | 91/204 (45%) | Secondary per-protocol analysis | 0.98 (0.79 to 1.22)a; P = 0.86 | −0.02 (−0.12 to 0.07)b; P = 0.65 |
| - | - | Missing responses | 0.96 (0.78 to 1.19)d; P = 0.73 | −0.03 (−0.12 to 0.07)e; P = 0.56 |
Sensitivity analyses for failure to clear lactate
The results of the sensitivity analyses for failure to clear lactate are presented in Table 24. The results from the original analyses are included in the first row for reference. As for the primary outcome and episode mortality analyses, all of the results of the sensitivity analyses are consistent with the results from the original analyses. The missing data analyses produced results that are extremely similar to the results of the original analysis. Restricting the analysis to participants providing lactate measurements made little difference to the results and did not change their interpretation. Restricting the analysis to the participants that adhered to their allocated treatment, using both definitions, produced estimates of treatment effect that were even closer to the null values, but the overall interpretation of the failure to clear lactate results did not change. Accounting for other covariates in the analysis reduced the analysis population and produced lower estimated incidences of failure to clear lactate in each group, but still produced estimates of treatment effect that were consistent with those from the primary analysis.
| RBC/LyoPlas | 0.9% saline | Sensitivity analysis | Adjusted risk ratio (95% CI); p-value | Adjusted risk difference (95% CI); p-value |
|---|---|---|---|---|
| 98/196 (50%) | 113/198 (55%) | Original analysis | 0.94 (0.78 to 1.13)a; P = 0.52 | −0.05 (−0.14 to 0.05)b; P = 0.35 |
| 55/131 (42%) | 70/148 (47%) | Further covariate adjustment | 0.96 (0.89 to 1.03)a; P = 0.29 | −0.06 (−0.17 to 0.06)c; P = 0.33 |
| 98/190 (52%) | 109/200 (55%) | Per-protocol analysis | 0.97 (0.81 to 1.17)a; P = 0.75 | −0.03 (−0.13 to 0.07)b; P = 0.56 |
| 98/193 (52%) | 105/195 (55%) | Secondary per-protocol analysis | 0.97 (0.80 to 1.16)a; P = 0.71 | −0.03 (−0.13 to 0.07)b; P = 0.50 |
| 74/149 (50%) | 85/150 (57%) | Timing of lactate concentration | 0.92 (0.74 to 1.14)a; P = 0.45 | −0.06 (−0.17 to 0.05)a; P = 0.29 |
| - | - | Missing responses | 0.94 (0.78 to 1.13)d; P = 0.50 | –0.03 (−0.12 to 0.07)e; P = 0.56 |
Subgroup analyses
Subgroup analyses were conducted for the composite primary outcome and for episode mortality.
Subgroups for primary outcome
The results of the pre-specified subgroup analyses for the composite primary outcome are presented in Table 25. There was no compelling evidence of any subgroup effect for the primary outcome. Although the incidence rates of the primary outcome varied by IDS, and there was some difference in rates across treatments arms within an IDS, these differences were not enough to conclude that there was a significant difference in treatment effect between IDS. There was little evidence that the estimated risk ratios for the composite primary outcome varied by IDS.
| Subgroup description | RBC/LyoPlas (N = 199) (%) | 0.9% saline (N = 210) | Adjusted risk ratio (95% CI) | p-value for interaction |
|---|---|---|---|---|
| IDS | ||||
| Site 1 | 49/67 (73) | 43/63 (68) | 1.07 (0.86 to 1.34)a | 0.28 |
| Site 2 | 26/33 (79) | 24/37 (65) | 1.21 (0.90 to 1.63)a | |
| Site 3 | 31/58 (53) | 36/57 (63) | 0.85 (0.62 to 1.16)a | |
| Site 4 | 22/41 (54) | 33/53 (62) | 0.86 (0.61 to 1.23)a | |
| Mode of transport | ||||
| Air | 49/76 (64) | 53/80 (66) | 0.98 (0.79 to 1.23)a | 0.74 |
| Ground | 79/123 (64) | 83/130 (64) | 1.03 (0.86 to 1.24)a | |
| Initial lactate concentration | ||||
| ≤2.2 mmol/L | 3/3 (100) | 1/1 (100) | 1.05 (0.99 to 1.13)b | 0.36 |
| >2.2 mmol/L | 120/190 (63) | 126/199 (63) | 1.00 (0.94 to 1.06)b | |
| Cardiac arrest | ||||
| Yes | 20/21 (95) | 20/20 (100) | 0.96 (0.91 to 1.01)b | 0.32 |
| No | 70/123 (57) | 82/147 (56) | 1.01 (0.93 to 1.09)b | |
| Time to ED from injury | ||||
| ≤1 hour | 19/34 (56) | 15/30 (50) | 1.18 (0.74 to 1.86)a | 0.47 |
| >1 hour | 74/128 (58) | 80/135 (59) | 0.98 (0.80 to 1.19)a | |
| Mode of injury | ||||
| Blunt | 101/147 (69) | 112/163 (69) | 1.00 (0.94 to 1.06)b | 0.83 |
| Penetrating | 24/43 (56) | 23/43 (53) | 1.02 (0.89 to 1.17)b | |
| Crush | 0/2 (0) | 0/0 (-) | - | |
| Multiple modes | 3/7 (43) | 1/4 (25) | 1.11 (0.73 to 1.67)b | |
| Volume of pre-hospital fluid given | ||||
| 4 units | 55/80 (69) | 56/73 (77) | 0.93 (0.77, 1.13)a | 0.35 |
| <4 units | 73/118 (62) | 80/137 (58) | 1.06 (0.87 to 1.29)a | |
| Age | ||||
| <50 years | 72/124 (58) | 74/124 (60) | 0.99 (0.81 to 1.22)a | 0.58 |
| 50 – 70 years | 30/44 (68) | 35/56 (63) | 1.09 (0.82 to 1.45)a | |
| >70 years | 13/18 (72) | 16/19 (84) | 0.87 (0.60 to 1.25)a | |
| Head injury c | ||||
| Positive | 2/2 (100) | 5/6 (83) | 1.06 (0.90 to 1.24)b | 0.97 |
| Negative | 38/58 (66) | 33/58 (57) | 1.06 (0.95 to 1.18)b | |
| Compressible haemorrhage | ||||
| Compressible haemorrhage | 20/37 (54) | 18/35 (51) | 1.07 (0.69 to 1.65)a | 0.27 |
| Non-compressible haemorrhage | 98/149 (66) | 113/164 (69) | 0.97 (0.83 to 1.14)a | |
| Both types of haemorrhage | 10/13 (77) | 5/11 (45) | 1.67 (0.82 to 3.39)a | |
| Pre-morbid drug history d | ||||
| Anticoagulant/antiplatelet medication | 11/17 (65) | 10/24 (42) | 1.16 (0.96 to 1.42)b | 0.42 |
| No anticoagulant/antiplatelet medication | 59/120 (49) | 49/99 (49) | 1.00 (0.92 to 1.10)b | |
| Unknown anticoagulant/antiplatelet medication | 27/29 (93) | 34/41 (83) | 1.04 (0.96 to 1.13)b | |
| Age of blood products e | ||||
| <8 days | 11/14 (79) | - | ||
| ≥8 days | 113/179 (63) | - | ||
The results of two additional post-hoc subgroup analyses are presented in Table 26. One subgroup analysis examined the association between the primary outcome and band of injury severity score (NISS < 16 vs. 16 to 30 vs. > 30) and the other examined the association between the primary outcome and transport time from scene to hospital (<20 minutes vs. ≥20 minutes). As with the pre-specified subgroup analyses, there was no compelling evidence of any subgroup effect for the primary outcome in these post-hoc analyses.
| Subgroup description | RBC/LyoPlas (N = 50) (%) | 0.9% saline (N = 54) (%) | Adjusted risk ratio (95% CI) | p-value for interaction |
|---|---|---|---|---|
| NISS band | ||||
| <16 | 1/7 (14) | 1/7 (14) | 0.95 (0.07 to 12.30)a | 0.82 |
| 16 to 30 | 11/26 (42) | 8/24 (33) | 1.32 (0.64, 2.70)a | |
| > 30 | 68/107 (64) | 69/111 (62) | 1.04 (0.86, 1.27)a | |
| Transport time: Time to ED from leaving scene | ||||
| <20 minutes | 12/17 (71) | 8/15 (53) | 1.12 (0.91, 1.37)b | 0.64 |
| ≥20 minutes | 20/33 (61) | 22/39 (56) | 1.05 (0.91, 1.22)b | |
Subgroups for episode mortality
The results of the pre-specified subgroup analyses for episode mortality are presented in Table 27. There was no strong evidence of any subgroup effect for episode mortality, although two subgroup analyses did suggest a possible subgroup effect. Similar to the findings for the primary outcome, the incidence rates of episode mortality varied by IDS, and there was some difference in rates across treatments arms within an IDS. Estimated treatment effects varied from 0.55 (95% CI 0.31 to 0.99) to 1.42 (95% CI 0.89 to 2.27) by IDS, but these differences were not large enough to conclude that there was a significant difference in treatment effect between IDS. The subgroup analysis examining premorbid drug history suggests that participants receiving either anticoagulant or antiplatelet medication may experience a higher rate of episode mortality if allocated to RBC/LyoPlas compared to 0.9% saline. This treatment effect is higher than in participants known to be receiving neither anticoagulant nor antiplatelet medication and in participants with unknown history of anticoagulant or antiplatelet medication. However, this analysis is based on small numbers of episode mortality events in the participants receiving either anticoagulant or antiplatelet medication, and the initial regression model experiences convergence problems. Hence, this result should be interpreted with caution.
| Subgroup description | RBC/LyoPlas (N = 199) (%) | 0.9% saline (N = 210) (%) | Adjusted risk ratio (95% CI) | p-value for interaction |
|---|---|---|---|---|
| IDS | ||||
| Site 1 | 33/67 (49) | 30/64 (47) | 1.05 (0.74 to 1.50)a | 0.07 |
| Site 2 | 21/36 (58) | 16/39 (41) | 1.42 (0.89 to 2.27)a | |
| Site 3 | 23/58 (40) | 27/60 (45) | 0.88 (0.58 to 1.35)a | |
| Site 4 | 11/42 (26) | 26/55 (47) | 0.55 (0.31 to 0.99)a | |
| Mode of transport | ||||
| Air | 40/77 (52) | 41/84 (49) | 1.07 (0.79 to 1.44)a | 0.41 |
| Ground | 48/126 (38) | 58/134 (43) | 0.89 (0.67 to 1.20)a | |
| Initial lactate concentration | ||||
| ≤2.2 mmol/L | 1/4 (25) | 0/1 (0) | 1.30 (0.93 to 1.83)b | 0.35 |
| >2.2 mmol/L | 82/190 (43) | 90/201 (45) | 0.99 (0.92 to 1.06)b | |
| Cardiac arrest | ||||
| Yes | 20/21 (95) | 20/20 (100) | 0.97 (0.92 to 1.02)b | 0.49 |
| No | 43/126 (34) | 50/151 (33) | 1.01 (0.93 to 1.09)b | |
| Time to ED from injury | ||||
| ≤1 hour | 9/33 (27) | 11/30 (37) | 0.75 (0.36 to 1.55)a | 0.45 |
| >1 hour | 46/133 (35) | 47/140 (34) | 1.02 (0.74 to 1.41)a | |
| Mode of injury | ||||
| Blunt | 73/151 (48) | 79/169 (47) | 1.01 (0.93 to 1.08)b | 0.14 |
| Penetrating | 13/43 (30) | 20/44 (45) | 0.90 (0.78 to 1.04)b | |
| Crush | 0/2 (0) | 0/0 (-) | - | |
| Multiple modes | 2/7 (29) | 0/5 (0) | 1.26 (0.98 to 1.62)b | |
| Volume of pre-hospital fluid given | ||||
| 4 units | 41/80 (51) | 42/76 (55) | 0.95 (0.71 to 1.28)a | 0.96 |
| <4 units | 47/122 (39) | 57/142 (40) | 0.96 (0.71 to 1.30)a | |
| Age | ||||
| <50 years | 41/124 (33) | 54/129 (42) | 0.80 (0.58 to 1.10)a | 0.29 |
| 50–70 years | 21/46 (46) | 21/56 (38) | 1.24 (0.78 to 1.96)a | |
| >70 years | 13/20 (65) | 13/22 (59) | 1.05 (0.64 to 1.72)a | |
| Head injuryc | ||||
| Positive | 2/2 (100) | 2/6 (33) | 1.46 (1.10 to 1.93)b | 0.13 |
| Negative | 26/58 (45) | 22/62 (35) | 1.06 (0.94 to 1.21)b | |
| Compressible haemorrhage | ||||
| Compressible haemorrhage | 9/37 (24) | 13/37 (35) | 0.72 (0.35 to 1.47)a | 0.08 |
| Non-compressible haemorrhage | 72/153 (47) | 84/169 (50) | 0.95 (0.76 to 1.19)a | |
| Both types of haemorrhage | 7/13 (54) | 2/12 (17) | 3.32 (0.85 to 12.94)a | |
| Pre-morbid drug history d | ||||
| Anticoagulant/antiplatelet medication | 7/19 (37) | 0/26 (0) | 1.36 (1.16 to 1.60)b | 0.01 |
| No anticoagulant/antiplatelet medication | 26/121 (21) | 26/102 (25) | 0.97 (0.88 to 1.06)b | |
| Unknown anticoagulant/antiplatelet medication | 25/29 (86) | 30/41 (73) | 1.07 (0.97 to 1.19)b | |
| Age of blood products e | ||||
| <8 days | 6/14 (43) | - | ||
| ≥8 days | 78/182 (43) | - | ||
Safety outcomes
The rates of complications and adverse events by treatment group are reported in Table 28. There was only one reported transfusion-related acute lung injury, and rates of thromboembolism were similar across the treatment groups. There was the suspicion or clinical evidence of infection in 65% of participants who completed at least one in-hospital daily assessment. This rate was similar across both treatment groups. There were only two serious adverse events recorded in the RePHILL trial. There were no required dose reductions during the trial, and no participants needed to have their trial treatment discontinued for drug-related toxicity. None of the deaths in the RePHILL study were related to the trial treatments.
| RBC/LyoPlas (n = 142) | 0.9% saline (n = 130) | |
|---|---|---|
| Transfusion-related acute lung injury | ||
| Yes | 0/142 (0%) | 1/130 (1%) |
| No | 142/142 (100%) | 128/130 (99%) |
| Missing | 0/140 | 1/130 |
| Thromboembolism | ||
| Yes | 17/142 (12%) | 11/130 (8%) |
| No | 125/142 (88%) | 118/130 (91%) |
| Missing | 0/142 (0%) | 1/130 (1%) |
| Type of thromboembolisma | ||
| Deep vein thrombosis | 3/142 (2%) | 3/130 (2%) |
| Pulmonary embolism | 9/142 (6%) | 8/130 (6%) |
| Stroke | 3/142 (2%) | 0/130 (0%) |
| Other | 2/142 (1%) | 3/130 (2%) |
| Suspicion or clinical evidence of infection | (n = 142) | (n = 130) |
| Yes (%) | 92/142 (65%) | 83/130 (64%) |
| No (%) | 50/142 (35%) | 47/130 (36%) |
| Missing | 0/142 (0%) | 0/130 (0%) |
| Type of infectiona | ||
| Intra-abdominal | 26/142 (18%) | 15/130 (12%) |
| Meningitis | 3/142 (2%) | 1/130 (1%) |
| Respiratory | 61/142 (43%) | 59/130 (45%) |
| Urinary tract infection | 5/142 (4%) | 10/130 (8%) |
| Soft tissue | 35/142 (25%) | 20/130 (15%) |
| Indwelling device | 16/142 (11%) | 13/130 (10%) |
| Blood-borne | 8/142 (6%) | 7/130 (5%) |
| Other | 46/142 (32%) | 40/130 (31%) |
Blood product wastage
RBC and LyoPlas wastage was measured at two of the trial blood banks. In total, 2496 RBC units were issued, 171 units were administered to patients, 2325 units were returned to blood bank stock and 53 units required disposal. This equates to a wastage rate of 2.12%. Amongst 2496 units of LyoPlas that were issued, 31 were wasted, equating to a rate of 1.24%.
Good clinical practice of compliance statement
The trial was run in accordance with the requirements of Good Clinical Practice. No serious breaches occurred during the conduct of the trial.
Chapter 4 Discussion
The RePHILL prospective multi-centre randomised controlled superiority trial recruited 432 patients with trauma-related haemorrhagic shock in a civilian setting. For the primary outcome, a composite of episode mortality and lactate clearance, the trial did not demonstrate that pre-hospital RBC/LyoPlas resuscitation was superior to 0.9% sodium chloride. Participants’ vital signs (heart rate, blood pressures, respiratory rate, oxygen saturations), markers of shock (lactate) and coagulopathy were similar between groups at hospital arrival. Participants randomised to the RBC/LyoPlas group had a higher haemoglobin on admission to hospital and received cumulatively more blood products in total than those in the control group.
Although the point estimates for the individual components of the primary outcome and some other secondary outcomes (early survival) are consistent with a benefit from allocation to RBC/LyoPlas, the CIs are wide and include the possibility of both benefits and harms. The Bayesian exploratory analysis allowed the trial to examine the composite primary outcome and its individual components across a range of prior beliefs about the effectiveness of blood/LyoPlas. For the composite primary outcome, the probability that RBC/LyoPlas was superior (i.e. an absolute risk difference >0) compared to 0.9% saline was between 44.1 and 53.%. For episode mortality, the probability was 71.2 to 88.2% and for lactate clearance 81.3 to 86.9%. However, the 95% credible intervals crossed zero, included the possibility for benefit as well as harm.
The use of PHBP for haemorrhagic shock following major trauma has been driven by the desire to deliver earlier in the patient pathway interventions traditionally reserved until hospital arrival. Research in both military and civilian settings suggests that early transfusion improves survival although the evidence from randomised trials remains inconclusive. 10 Trials involving pre-hospital plasma have produced conflicting results. The Pre-hospital Air Medical Plasma (PAMPer) clinical trial showed a nearly 30% relative reduction in mortality with plasma transfusion in the pre-hospital environment (30 day mortality 23.2% vs. 33.0%). 8 In contrast the Control of Major Bleeding After Trauma Trial (COMBAT) trial which randomised adults with haemorrhagic shock to pre-hospital plasma or saline was stopped after enrolment of 144/150 participants due to futility (28 day mortality was 15% in the plasma group vs. 10% in the control group). The Pre-hospital Administration of Lyophilized Plasma for Post-traumatic Coagulopathy Treatment (PREHO-PLYO) trial randomised 150 participants with major trauma to 4 LyoPlas units or 0.9% saline. The study found no difference in the primary outcome [INR on arrival at the hospital or in 30-day survival (17.6% plasma group vs. 15.2%), OR 1.20 (0.43 to 3.37)]. 19
It is worth considering why the present study did not find clear benefit from PHBP. First, the study took place in a civilian setting within an established major trauma network. Pre-hospital critical care is provided at the scene of the incident by specialist doctors and critical care paramedics, who already bring forward some of the advanced life support treatments traditionally delayed until hospital arrival. Second, the national trauma network has been configured to facilitate transfer to a major trauma centre within 60 minutes of injury. 20 Although the overall time from scene to arrival at the ED was on average median 57 (IQR 40 to 78) minutes, a third of participants with recorded transport times (from scene to ED) had an interval of less than 20 minutes. Whilst our subgroup analysis did not find evidence of an interaction based on transport time, a larger, post-hoc analysis of the PAMPer and COMBAT trials reported that the survival benefit from pre-hospital transfusion was limited to patients who had transport times of less than 20 minutes. 21 Third, due to the logistical challenges created by the limited post-thaw shelf life of fresh frozen plasma at the time of the study, RePHILL used freeze-dried (LyoPlas). This decision was based on evidence that LyoPlas has similar or improved biological efficacy relative to fresh frozen plasma. 22,23 However, a secondary, non-randomised analysis of the PAMPer trial showed that compared with crystalloid only resuscitation, resuscitation with fresh frozen plasma in combination with red cells reduced 30 day mortality (37% vs. 26%, P = 0.05). 24 The authors however highlight that their findings could be limited by residual confounding. Further research is required to determine if the apparent differences were due to the use of LyoPlas or fresh frozen plasma. Fourthly, RePHILL used a red cell to plasma ratio of 1 : 1 consistent with UK national guidelines for major haemorrhage. 20 Whether different ratios, the addition of platelets, coagulation factors or administration of whole blood would have made a difference remains to be determined by future research. Finally, the population of civilian participants enrolled in RePHILL were older, more severely injured and had a higher proportion of blunt traumatic injuries than in observational military studies which have suggested benefit from pre-hospital blood transfusion. 25,26 Although the subgroup analysis according to injury severity scores (<16, 16 to 30 or ≥ 30) did not find evidence of interaction, it remains possible that some patients were too well or too sick to benefit from treatment with blood and plasma.
Part of the hypothesis for the putative benefits of transfusing red cells relate to early optimisation of oxygen delivery to the tissues. Guidelines recommend red cell transfusion when the level of haemoglobin falls below 70–90 g/L, except in older patients and those with traumatic brain injury who may benefit from higher concentrations of Hb. 27 Despite observing higher haemoglobin levels on admission to hospital [133 (SD 19) in the RBC/LyoPlas group compared with 118 (SD 23) in the saline group], RePHILL did not find evidence that pre-hospital transfusion improved oxygen delivery as measured by lactate level or the rate of lactate clearance. This may in part be explained by only a small proportion of participants (9/152, 6%) in the 0.9% saline group arriving at hospital with a haemoglobin <80, highlighting the difficulty in identifying patients in the pre-hospital setting who are anaemic at the time of assessment.
Blood and blood components are a scarce resource. Substantial efforts are therefore put into the supply chain to minimise wastage. This is particularly important when using O negative blood as demand for O negative RBCs is 12% in the UK, but this group makes up only 8% of the population. 28 Data from the National Blood and Transplant Service during the conduct of the trial indicates that blood product wastage occurred in 2.5% of units issued for red cells (4.7% for group O negative blood). 29 Given the environmental challenges inherent with the delivery of care in the pre-hospital environment, when extending transfusion operations into that setting it is important to take care to minimise wastage. In a prospective observational study, in the context of a quality improvement initiative, the London Air Ambulance explored blood wastage related to the supply of whole blood. 30 Over a two-year period, which involved four Plan-Do-Study-Act cycles, mean weekly wastage reduced from 8.36 units (70%) to 3.19 units (27%). Although not directly comparable (as the shelf life for whole blood is 14 days compared to 35 days for RBCs), the low wastage seen in the RePHILL trial (2%) provides assurance that the use of pre-hospital RBCs does not lead to substantial wastage. However, the finding that those in the pre-hospital transfusion group not only received earlier blood but also had higher total blood product usage, without definitive evidence of benefit, suggests the need for more careful assessment of transfusion requirements in hospital in those who received pre-hospital blood.
Equality, diversity and inclusion
Given the unpredictable nature of life-threatening haemorrhagic shock and the need for emergency treatment, it was not possible for us to take active steps to enrich the diversity of the population of patients recruited. However, the population enrolled broadly reflected the ethnicity characteristics of England and Wales (www.ethnicity-facts-figures.service.gov.uk/uk-population-by-ethnicity/national-and-regional-populations/population-of-england-and-wales/latest, accessed 6 July 2022). The study protocol excluded people with a known objection to blood or blood product transfusion in order to respect the individual’s rights to accept or refuse treatment. We also excluded women who were known or suspected to be pregnant due to the potential greater transfusion risks in this population. Finally, we excluded prisoners due to the perceived difficulty of obtaining follow-up data for this population. In subsequent research, we have shown that it is feasible to obtain survival outcomes data for this group of patients through the Office for National Statistics. 31
Strengths and limitations
RePHILL is the first RCT to evaluate the clinical effectiveness of pre-hospital blood transfusion. The trial overcame substantial regulatory, supply and logistical challenges. It is the first to engage multiple air ambulance charities in a randomised clinical trial of an investigational medicinal product. The case mix of participants enrolled were typical of UK civilian major trauma. The injury severity scores confirm that the group of participants enrolled had, as anticipated, severe injuries. These findings support the generalisability of the research findings to the UK civilian trauma population.
Alongside these strengths, the trial also has limitations. First, we recruited only 88% of our planned sample size due to the impact of COVID-19 and the withdrawal of several air ambulance partners. Given the similarity of the primary and secondary outcomes between the intervention and control group, although it is possible that not recruiting the full sample size may have led to a type 2 error, we consider it unlikely that it would have led to a substantively different finding. This position is further supported by simulations examining the effect of large increases in sample sizes which would not have materially altered the findings for the primary outcome. Whether a larger trial would have influenced the secondary outcomes, remains to be determined in future studies.
COVID-19 had a global impact on clinical trials leading to thousands of clinical trials closing prior to reaching the intended sample size. 32 The CONSORT and SPIRIT Extension for RCTs Revised in Extenuating Circumstances (CONSERVE) statement provides guidance to help improve the transparency, quality and completeness of reporting. 33 Adopting that framework in the context of RePHILL can be summarised as:
-
Extenuating circumstance: (1) COVID-19 led to an inability to maintain safe oversight for the trial due to a combination sickness and redeployment of clinical and research staff. (2) National lockdowns reduced the incidence of major trauma.
-
Impact: (1) Sponsor paused trial recruitment (March 2020 through to July 2020). (2) Fewer cases of major trauma leading to lower recruitment rates. Together these meant that the study fell short of reaching the intended sample size.
-
Mitigations: Recruitment window extended; addition of Bayesian analysis.
-
These are important modifications as they reduced the statistical power of the study.
Although multi-centre RCTs are considered the gold standard of evidence, it takes considerable time to design and recruit sufficient numbers. Other pre-hospital transfusion trials have faced challenges – with the Control of Major Bleeding After Trauma – COMBAT stopped prematurely due to futility9 and the Pre-Hospital Use of Plasma for Traumatic Hemorrhage – PUPTH due to insufficient recruitment. 34 The withdrawal of several air ambulance services prior to the start of the trial also adversely affected delivering the trial on time and target. The clinical and commercial pressures for early adoption (www.londonsairambulance.org.uk/news-and-stories/charity/uks-first-air-ambulance-to-carry-blood-on-board, accessed 25 February 2022)35,36 and loss of equipoise37 meant that early evaluations of pre-hospital blood were limited to observational studies38,39 and reduced the pool willing to participate in a randomised evaluation. Future randomised studies will need to carefully consider the pressures for early adoption inherent in this sector and work carefully with air ambulance partners to agree future research priorities.
Research in trauma-related haemorrhagic shock is limited by the absence of a core outcome set. A systematic review examining outcome measures used in clinical research evaluating pre-hospital blood component transfusion in traumatically injured bleeding patients identified over 212 outcome measures across 34 studies,40 highlighting significant heterogeneity in outcomes across previous trials. A composite primary outcome reflecting the efficacy of initial resuscitation (lactate clearance) and overall effectiveness (death) was chosen after careful consideration by the trial investigators, funders and patient and public involvement groups. Despite the empirical attractiveness to this composite outcome, the direction of effects observed in RePHILL for mortality −3% (−12% to 7%) and improved lactate clearance −5% (−14% to 5%) do not appear to have been additive when combined as a composite outcome [i.e. negligible difference seen in the composite outcome −0.025% (−9% to 9%)]. This could be explained by the most seriously injured patients, at highest risk of early death, would also have high lactate loads. An effective treatment that averted early death could be mitigated by producing a survivor with impaired lactate clearance. This suggests investigators should be cautious about its use in future studies.
Given the nature of the intervention and the setting for administration, it was not possible to mask treatment allocation to those delivering the intervention. It is possible that this may have led to performance bias during both the pre-hospital and ED treatment of enrolled patients. The key trial outcomes were objective measurements and thus it was unlikely that they were influenced by knowledge of treatment assignment. More subjective measures (e.g. transfusion reactions, adverse events) may have been more susceptible to detection bias.
Future research
There remains an urgent need to develop evidence-based interventions to improve outcomes from haemorrhagic shock secondary to acute traumatic injuries.
A key challenge is to identify the population of patients most likely to benefit from pre-hospital interventions. 41 The RePHILL trial recruited a population of severely injured patients characterised by high injury severity scores and overall high episode mortality. 42 By contrast, the COMBAT trial recruited less severely injured patients (and had a short time between intervention and hospital arrival) and similarly did not find evidence of benefit from up to two units of thawed plasma. 9 The PAMPer trial,8 the only trial to find a benefit from pre-hospital transfusion (of up to two units of plasma) recruited a group of moderately injured patients who were on average 42 minutes from away from definitive hospital-delivered treatments. This suggests there may be an optimal set of inclusion/exclusion criteria for recruitment to future trials which avoids recruiting those unlikely to benefit from having either catastrophic injury (e.g. traumatic cardiac arrest) or least severe injuries who are likely to survive irrespective of pre-hospital transfusions. However, there remains a key challenge to identify this population of patients at the roadside. Whilst some post-hoc analyses (e.g. longer pre-hospital transport time, CT evidence of brain injury) suggest there may be subgroups with better outcomes, none of the a priori subgroup analyses across the three trials, which can be assessed at the point of injury, demonstrated evidence of benefit. Further research is required to identify the characteristics of patients most likely to benefit from pre-hospital transfusion.
The discordant findings from the four prior randomised transfusion trials highlight the need for further research to identify the optimal intervention(s) in trauma-related haemorrhagic shock. 8,9,19,42,43 Consideration should be given to both the components transfused, and the sequence and dose. Such research should be informed by the findings (once published) of the Pre-hospital Plasma or Red Blood Cell Transfusion Strategy in Major Bleeding (PRIEST; www.clinicaltrials.gov/ct2/show/NCT04879485, accessed 28 September 2022). 44 The role of whole blood, which reduces the need for transfusing individual components, showed promise in a single-centre pilot randomised trial45 and is being explored in the Type O Whole Blood and Assessment of Age During Pre-hospital Resuscitation Trial (TOWAR; https://clinicaltrials.gov/ct2/show/NCT04684719, accessed 28 September 2022)46 and Study of Whole Blood In Frontline Trauma (SWIFT; www.nhsbt.nhs.uk/clinical-trials-unit/current-trials-and-studies/swift/trial, accessed 28 September 2022). 47 Noting that early treatment is critical for best outcomes in traumatic haemorrhage,48,49 future trials will need to consider how to enable interventions to be delivered earlier in the patient pathway.
A systematic review of outcome measures used in clinical research evaluating pre-hospital blood component transfusion patients with trauma-related haemorrhagic shock identified substantial heterogeneity in the outcomes reported. Many outcomes focused on evidence of clinical effectiveness with limited insights provided into safety and complications. 40 The absence of a core outcome set for pre-hospital transfusion trials presents a significant challenge for researchers seeking to identify the optimal outcomes for individual trials and to facilitate meta-analyses between trials. The Core Outcomes Set for Cardiac Arrest (COSCA) includes survival to hospital discharge (or 30 days), survival with a favourable neurological outcome and health-related quality of life. 50 While survival to and beyond hospital discharge may be desirable for large effectiveness trials, any treatment effect from pre-hospital transfusion will likely be diluted due to severity of underlying injuries and treatments administered after hospital arrival. Future trials that wish to demonstrate evidence of efficacy may need to focus on outcomes earlier in the patient journey such as survival to hospital admission or within the first 24 hours of injury. Further research and consensus work is needed to define the optimal outcomes set for efficacy and effectiveness trials, what constitutes the minimal clinically important differences for these outcomes and what the optimal experimental designs are to answer the specific research questions. 18
Chapter 5 Conclusions
This multi-centre, allocation concealed, open-label, parallel group RCT in participants with trauma-related haemorrhagic shock did not demonstrate that pre-hospital RBC/LyoPlas was superior to 0.9% saline for the composite outcome of episode mortality and/or lactate clearance.
Acknowledgements
Contributions of authors
Nicholas Crombie (https://orcid.org/0000-0003-3740-5530) (Consultant Trauma Anaesthetist) conceived, designed and obtained funding for the study, supervised the research, wrote the original draft and reviewed and approved the draft.
Heidi A Doughty (https://orcid.org/0000-0002-5444-6222) (Consultant in Transfusion Medicine) conceived, designed and obtained funding for the study, supervised blood and LyoPlas supplies, wrote the original draft and reviewed and approved the draft.
Jonathan RB Bishop (https://orcid.org/0000-0003-1789-5886) (Senior Medical Statistician) conceived, designed and obtained funding for the study, was responsible for data curation, formal analysis, data verification and data visualisation, wrote the original draft and reviewed and approved the draft.
Amisha Desai (https://orcid.org/0000-0002-7305-8401) (Advanced Clinical Pharmacist) supervised investigational medicinal product management, and reviewed and approved the draft.
Emily F Dixon (https://orcid.org/0000-0003-3660-5354) (Trial Manager) co-ordinated the study and reviewed and approved the draft.
James M Hancox (https://orcid.org/0000-0002-5797-0898) (Critical Care Paramedic) supervised patient recruitment in collaboration with sites and reviewed and approved the draft.
Mike J Herbert (Blood Transfusion Discipline Lead) supervised blood and LyoPlas supplies and reviewed and approved the draft.
Caroline Leech (https://orcid.org/0000-0002-5301-9798) (Consultant in Emergency Medicine and Pre-hospital Emergency Medicine) managed resources at sites and supervised patient recruitment and reviewed and approved the draft.
Simon J Lewis (https://orcid.org/0000-0002-2350-1436) (Consultant in Emergency Medicine and Pre-hospital Emergency Medicine) managed resources at sites and supervised patient recruitment and reviewed and approved the draft.
Mark R Nash (https://orcid.org/0000-0002-3793-4937) (Consultant Anaesthetist) managed resources at sites and supervised patient recruitment and reviewed and approved the draft.
David N Naumann (https://orcid.org/0000-0003-2243-2325) (Military Surgeon, Trauma Fellow) conceived, designed and obtained funding for the study and wrote the original draft.
Karen Piper (https://orcid.org/0000-0001-5087-7052) (Programme Manager: NIHR Surgical Reconstruction and Microbiology Research Centre) who played a substantial role in the conception and design of the study and obtained funding for the study.
Gemma Slinn (https://orcid.org/0000-0003-1096-4148) (Trials Management Team Leader) managed the set-up and oversaw the operational delivery of the study at Birmingham Clinical Trials Unit, and reviewed and approved the draft.
Hazel Smith (https://orcid.org/0000-0002-7652-7699) (Research Paramedic) supervised patient recruitment in collaboration with sites, wrote the original draft, and reviewed and approved the draft.
Iain M Smith (https://orcid.org/0000-0001-5941-410X) (Specialty Registrar in General Surgery) conceived, designed and obtained funding for the study and reviewed and approved the draft.
Rebekah K Wale (https://orcid.org/0000-0003-1343-7538) (Senior Trial Manager) co-ordinated the study and reviewed and approved the draft.
Alistair Wilson (https://orcid.org/0000-0001-8042-1654) (Consultant Trauma Surgeon) managed resources at sites and supervised patient recruitment and reviewed and approved the draft.
Aisling Crombie (https://orcid.org/0000-0003-4739-9613) (Deputy Director of Nursing and Quality) played a substantial role in the conception and design of the study and obtained funding for the study.
Mark Midwinter (https://orcid.org/0000-0003-1836-7137) (Professor of Clinical Anatomy, General and Trauma Surgeon) played a substantial role in the conception and design of the study and obtained funding for the study.
Natalie Ives (https://orcid.org/0000-0002-1664-7541) (Reader in Clinical Trials) conceived, designed and obtained funding for the study, was responsible for data curation, formal analysis and data visualisation, supervised the research, wrote the original draft and reviewed and approved the draft.
Gavin D Perkins (https://orcid.org/0000-0003-3027-7548) (Professor in Critical Care Medicine) conceived, designed and obtained funding for the study, supervised the research, wrote the original draft and reviewed and approved the draft.
We thank the patients and their relatives who supported the trial, principal investigators and other research staff at ambulance services, receiving hospitals, air ambulance charities, participating blood banks and pharmacy staff, the blood bikers, Birmingham Clinical Trials Unit and Sponsors office, the Trial Steering Committee, and the Data Monitoring and Ethics Committee. This project is funded by the Efficacy and Mechanism Evaluation Programme (study reference 14/152/14) of the National Institute for Health and Care Research (NIHR).
Ethics statement
The study was approved by South Central Research Ethics Committee (15/SC/0691) on 15 December 2015.
Data-sharing statement
Requests for access to data from the RePHILL trial should be addressed to the corresponding author at rephill@trials.bham.ac.uk. The individual participant data collected during the trial (including the data dictionary) will be available, after de-identification, with no end date. All proposals requesting data access will need to specify how the data will be used, and all proposals will need the approval of the trial management group before data release.
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, the EME programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, the EME programme or the Department of Health and Social Care.
References
- CRASH-2 Trial Collaborators . Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet 2010;376:23-32.
- Holcomb JB, del Junco DJ, Fox EE, Wade CE, Cohen MJ, Schreiber MA, et al. PROMMTT Study Group . The prospective, observational, multicenter, major trauma transfusion (PROMMTT) study comparative effectiveness of a time-varying treatment with competing risks. JAMA Surg 2013;148:127-36.
- Vincent JL, Moreno R, Takala J, Willatts S, De Mendonça A, Bruining H, et al. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure: on behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med 1996;22:707-10.
- Morrison JJ, Oh J, DuBose JJ, O’Reilly DJ, Russell RJ, Blackbourne LH, et al. En-route care capability from point of injury impacts mortality after severe wartime injury. Ann Surg 2013;257:330-4.
- Bailey JA, Morrison JJ, Rasmussen TE. Military trauma system in Afghanistan: lessons for civil systems?. Curr Opin Crit Care 2013;19:569-77.
- O’Reilly DJ, Morrison JJ, Jansen JO, Apodaca AN, Rasmussen TE, Midwinter MJ. Prehospital blood transfusion in the en route management of severe combat trauma: a matched cohort study. J Trauma Acute Care 2014;77:S114-20.
- Smith IM, James RH, Dretzke J, Midwinter MJ. Prehospital blood product resuscitation for trauma: a systematic review. Shock 2016;46:3-16.
- Sperry JL, Guyette FX, Brown JB, Adams PW. Prehospital plasma during air medical transport in trauma patients at risk for hemorrhagic shock. New Engl J Med 2018;379:315-26.
- Moore HB, Moore EE, Chapman MP, McVaney K, Bryskiewicz G, Blechar R, et al. Plasma-first resuscitation to treat haemorrhagic shock during emergency ground transportation in an urban area: a randomised trial. Lancet 2018;392:283-91.
- van Turenhout EC, Bossers SM, Loer SA, Giannakopoulos GF, Schwarte LA, Schober P. Prehospital transfusion of red blood cells. Part 2: a systematic review of treatment effects on outcomes. Transfusion Med 2020;30:106-33.
- DeBot M, Sauaia A, Schaid T, Moore EE. Trauma-induced hypocalcemia. Transfusion 2022;62:274-80.
- Jenkins D, Stubbs J, Williams S, Berns K, Zielinski M, Strandenes G, et al. Implementation and execution of civilian remote damage control resuscitation programs. Shock 2014;41:84-9.
- Jenkins DH, Rappold JF, Badloe JF, Berséus O, Blackbourne L, Brohi KH, et al. Trauma hemostasis and oxygenation research position paper on remote damage control resuscitation: definitions, current practice, and knowledge gaps. Shock 2014;41:3-12.
- Smith IM, Crombie N, Bishop JR, . RePHILL: protocol for a randomised controlled trial of pre-hospital blood product resuscitation for trauma. Transfusion Med 2018;28:346-56.
- Turner L, Shamseer L, Altman DG, Weeks L, Peters J, Kober T, et al. Consolidated standards of reporting trials (CONSORT) and the completeness of reporting of randomised controlled trials (RCTs) published in medical journals. Cochr Datab Syst Rev 2012;11.
- Régnier MA, Raux M, Le Manach Y, Asencio Y, Gaillard J, Devilliers C, et al. Prognostic significance of blood lactate and lactate clearance in trauma patients. Anesthesiology 2012;117:1276-88.
- Chase JG, Pretty CG, Pfeifer L, Shaw GM, Preiser J-C, Le Compte AJ, et al. Organ failure and tight glycemic control in the SPRINT study. Crit Care 2010;14.
- Yarnell CJ, Darryl A, Baldwin MR, Brodie D, Fan E, Ferguson ND, et al. Clinical trials in critical care: can a Bayesian approach enhance clinical and scientific decision making?. Lancet Respir Med 2021;9:207-16.
- Jost D, Lemoine L, Lemoine F, Derkenne C, Beaume S, Lanoë V, et al. Prehospital lyophilized plasma transfusion for trauma-induced coagulopathy in patients at risk for hemorrhagic shock: a randomized clinical trial. JAMA Netw Open 2022;5.
- National Institute for Health and Care Excellence . Major Trauma: Assessment and Initial Management NICE Guideline 2016. www.nice.org.uk/guidance/ng39 (accessed 21 December 2021).
- Pusateri AE, Moore EE, Moore HB, Le TD, Guyette FX, Chapman MP, et al. Association of prehospital plasma transfusion with survival in trauma patients with hemorrhagic shock when transport times are longer than 20 minutes: a post hoc analysis of the PAMPer and COMBAT clinical trials. JAMA Surg 2020;155.
- Bux J, Dickhorner D, Scheel E. Quality of freeze-dried (lyophilized) quarantined single-donor plasma. Transfusion 2013;53:3203-9.
- Garrigue D, Godier A, Glacet A, Labreuche J, Kipnis E, Paris C, et al. French lyophilized plasma versus fresh frozen plasma for the initial management of trauma-induced coagulopathy: a randomized open-label trial. J Thromb Haemost 2018;16:481-9.
- Guyette FX, Sperry JL, Peitzman AB, Billiar TR, Daley BJ, Miller RS, et al. Prehospital blood product and crystalloid resuscitation in the severely injured patient: a secondary analysis of the prehospital air medical plasma trial. Ann Surg 2021;273:358-64.
- Shackelford SA, del Junco DJ, Powell-Dunford N, Mazuchowski EL, Howard JT, Kotwal RS, et al. Association of prehospital blood product transfusion during medical evacuation of combat casualties in Afghanistan with acute and 30-day survival. JAMA: J Am Med Assoc 2017;318:1581-91.
- O’Reilly DJ, Morrison JJ, Jansen JO, Apodaca AN, Rasmussen TE, Midwinter MJ. Prehospital blood transfusion in the en route management of severe combat trauma: a matched cohort study. J Trauma Acute Care Surg 2014;77:S114-20.
- Spahn DR, Bouillon B, Cerny V, Duranteau J, Filipescu D, Hunt BJ, et al. The European guideline on management of major bleeding and coagulopathy following trauma: fifth edition. Critical Care 2019;23.
- Foukaneli D, Bassey S, Bend M. Survey of group O D negative red cell use: NHS Blood and Transplant 2018. https://nhsbtdbe.blob.core.windows.net/umbraco-assets-corp/17159/o-neg-survey-2018-final2.pdf (accessed 21 December 2021).
- NHS Blood and Transplant . NHSBT Hospital Blood Supply Chain 2017 18 Annual Report 2018.
- McCullagh J, Proudlove N, Tucker H, Davies J. Making every drop count: reducing wastage of a novel blood component for transfusion of trauma patients. BMJ Open Qual 2021;10.
- Perkins GD, Ji C, Deakin CD, Quinn T, Nolan JP, Scomparin C, et al. PARAMEDIC2 Collaborators . A randomized trial of epinephrine in out-of-hospital cardiac arrest. N Engl J Med 2018;379:711-21.
- US Department of Health and Human Services Food and Drug Administration . Conduct of Clinical Trials of Medical Products During the COVID-19 Public Health Emergency. Guidance for Industry, Investigators, and Institutional Review Boards n.d. www.fda.gov/media/136238/download (accessed 21 December 2021).
- Orkin AM, Gill PJ, Ghersi D, Campbell L, Sugarman J, Emsley R, et al. CONSERVE Group . Guidelines for reporting trial protocols and completed trials modified due to the COVID-19 pandemic and other extenuating circumstances: the CONSERVE 2021 statement. JAMA 2021;326:257-65.
- Reynolds PS, Michael MJ, Cochran ED, Wegelin JA, Spiess BD. Prehospital use of plasma in traumatic hemorrhage (The PUPTH Trial): study protocol for a randomised controlled trial. Trials 2015;16.
- London’s Air Ambulance Charity . UK’S First Air Ambulance to Carry Blood on Board 2013. www.londonsairambulance.org.uk/news-and-stories/charity/uks-first-air-ambulance-to-carry-blood-on-board (accessed 25 February 2022).
- O’Neill J, Jarman H, Greenhalgh R, Russell M, Lyon R. Pre-hospital Blood Transfusions on a UK Air Ambulance Service: The First Ten Months. Kent: Kent, Surrey and Sussex Helicopter Emergency Medical Service; 2014.
- Thies KC, Truhlář A, Keene D, Hinkelbein J, Rützler K, Brazzi L, et al. Pre-hospital blood transfusion: an ESA survey of European practice. Scand J Trauma Resusc Emerg Med 2020;28.
- Griggs JE, Jeyanathan J, Joy M, Russell MQ, Durge N, Bootland D, et al. Kent, Surrey and Sussex Air Ambulance Trust . Mortality of civilian patients with suspected traumatic haemorrhage receiving pre-hospital transfusion of packed red blood cells compared to pre-hospital crystalloid. Scand J Trauma Resusc Emerg Med 2018;26.
- Rehn M, Weaver AE, Eshelby S, Røislien J, Lockey DJ. Pre-hospital transfusion of red blood cells in civilian trauma patients. Transfus Med 2018;28:277-83.
- Tucker H, Avery P, Brohi K, Davenport R, Griggs J, Weaver A, et al. Outcome measures used in clinical research evaluating prehospital blood component transfusion in traumatically injured bleeding patients: a systematic review. J Trauma Acute Care Surg 2021;91:1018-24.
- Yazer MH, Cap AP, Glassberg E, Green L, Holcomb JP, Khan MA, et al. Toward a more complete understanding of who will benefit from prehospital transfusion. Transfusion 2022;62:1671-9.
- Crombie N, Doughty HA, Bishop JRB, Desai A, Dixon EF, Hancox JM, et al. Resuscitation with blood products in patients with trauma-related haemorrhagic shock receiving prehospital care (RePHILL): a multicentre, open-label, randomised, controlled, phase 3 trial. Lancet Haematol 2022;9:250-61.
- Chapman MP, Moore EE, Chin TL, Ghasabyan A, Chandler J, Stringham J, et al. Initial experience with a randomized clinical trial of plasma-based resuscitation in the field for traumatic hemorrhagic shock. Shock 2015;44:63-70.
- Prehospital Transfusion Strategy in Bleeding Patients 2021. www.clinicaltrials.gov/ct2/show/NCT04879485 (accessed 28 September 2022).
- Cotton BA, Podbielski J, Camp E, Welch T, del Junco D, Bai Y, et al. A randomized controlled pilot trial of modified whole blood versus component therapy in severely injured patients requiring large volume transfusions. Ann Surg 2013;258:527-33.
- Type O Whole Blood and Assessment of Age During Prehospital Resuscitation Trial 2020. https://clinicaltrials.gov/ct2/show/NCT04684719 (accessed 28 September 2022).
- Study of Whole Blood In Frontline Trauma n.d. www.nhsbt.nhs.uk/clinical-trials-unit/current-trials-and-studies/swift/ (accessed 21 December 2021).
- Woolley T, Thomson P, Kirkman E, Reed R, Ausset S, Beckett A, et al. Trauma Hemostasis and Oxygenation Research Network position paper on the role of hypotensive resuscitation as part of remote damage control resuscitation. J Trauma Acute Care Surg 2018;84:S3-1.
- Shakur H, Roberts I, Afolabi A, Brohi K, Coats T, Dewan Y, et al. CRASH-2 collaborators . The importance of early treatment with tranexamic acid in bleeding trauma patients: an exploratory analysis of the CRASH-2 randomised controlled trial. Lancet 2011;377:1096-101.
- Haywood K, Whitehead L, Nadkarni VM, Felix A, Beesems S, Böttiger BW, et al. COSCA (Core Outcome Set for Cardiac Arrest) in adults: an advisory statement from the International Liaison Committee on Resuscitation. Resuscitation 2018;127:147-63.
Appendix 1 The RePHILL Collaborative Group
Trial Management Group
Prof. Gavin Perkins, Dr Nicholas Crombie, Iain Smith, Dr Heidi Doughty, Dr David Naumann, Hazel Smith, Dr Margaret Grant, Gemma Slinn, Dr Rebekah Wale, Emily Dixon, Dr Karen Piper, Deborah Papoola, Amisha Desai, Natalie Ives, Dr Jon Bishop
Data Monitoring Committee
Prof. John Nicholl (Chair), Dr Jan Jansen, Prof. Fiona Lecky
Trial Steering Committee
Prof. Ian Roberts (Chair), Prof. John Holcomb, Dr Simon Stanworth, Prof. Jason Smith, Prof. Timothy Coats, Andrew Cox, Timothy Marshall
Blood Banks and Pharmacies
Mike Herbert, Mindy Sahota (New Cross Hospital); Camran Khan, Zeeshan Parvez (Worcester Acute Hospital); Tina Taylor, Julie Nortcote, Mojid Khan (University Hospitals Coventry and Warwickshire); Claire Newsam, Katherine Philpott, Lynne Whitehead (Addenbrooke’s Hospital); Debbie Asher, Gail Healey (Norfolk and Norwich University Hospital)
Blood Bikers
Midland Freewheelers, Warwickshire and Solihull Blood Bikers
Air Ambulance Sites (IDS)
Alistair Wilson (PI), Pam Chrispin (PI), Richard Hinson (East Anglian Air Ambulance); Caroline Leech (PI), Mark Beasley, Sam Cooper (The Air Ambulance Service); Mark Nash (PI) (West Midlands Ambulance Service); Simon Lewis (PI), Oliver Robinson, Rod Mackenzie (Magpas Air Ambulance)
Receiving Hospital Sites
David Yeo (PI; Queen Elizabeth Hospital); Caroline Leech (PI; University Hospitals Coventry and Warwickshire); Tom James (University Hospitals of North Midlands); Jason Kendall (PI), Johannes von Vopelius-Feldt (PI) (Southmead Hospital Bristol); Christopher Gough (PI) (Queen’s Medical Centre Nottingham); Gary Mills (PI; Northern General Hospital Sheffield); Aquib Hafeez (PI; Oxford University Hospitals); Sarah Hazleman (PI; Addenbrooke’s Hospital); Francoise Sheppard (PI; Norfolk and Norwich University Hospital); Ben Bloom (PI; Royal London Hospital)
Appendix 2 Recruitment by site
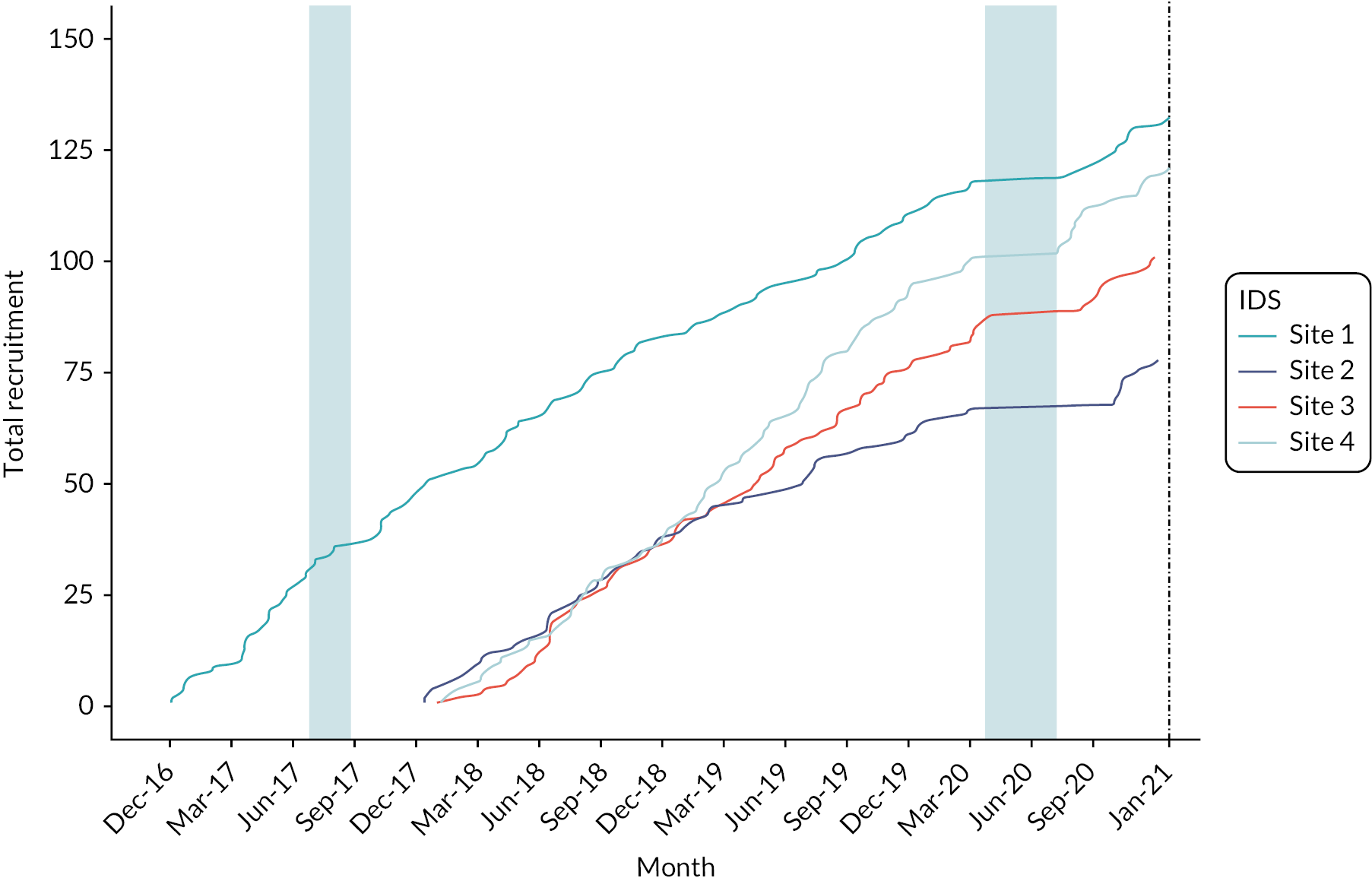
List of abbreviations
- CI
- confidence interval
- CONSORT
- Consolidated Standards of Reporting Trials
- DMEC
- Data Monitoring and Ethics Committee
- ED
- emergency department
- IDS
- intervention delivery site
- INR
- international normalised ratio
- IQR
- interquartile range
- LyoPlas
- lyophilised plasma
- PHBP
- pre-hospital blood products
- PHEM
- pre-hospital emergency medicine
- PI
- principal investigator
- PPI
- patient and public involvement
- RBC
- red blood cells
- RCT
- randomised controlled trial
- SBP
- systolic blood pressure
- SD
- standard deviation
- TAAS
- The Air Ambulance Service
- TSC
- Trial Steering Committee
Notes
Supplementary material can be found on the NIHR Journals Library report page (https://doi.org/10.3310/TDNB9214).
Supplementary material has been provided by the authors to support the report and any files provided at submission will have been seen by peer reviewers, but not extensively reviewed. Any supplementary material provided at a later stage in the process may not have been peer reviewed.