

Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as award number 12/212/15. The contractual start date was in January 2015. The final report began editorial review in October 2022 and was accepted for publication in July 2023. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ manuscript and would like to thank the reviewers for their constructive comments on the final manuscript document. However, they do not accept liability for damages or losses arising from material published in this manuscript.
Permissions
Copyright statement
Copyright © 2024 Iro et al. This work was produced by Iro et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This is an Open Access publication distributed under the terms of the Creative Commons Attribution CC BY 4.0 licence, which permits unrestricted use, distribution, reproduction and adaptation in any medium and for any purpose provided that it is properly attributed. See: https://creativecommons.org/licenses/by/4.0/. For attribution the title, original author(s), the publication source – NIHR Journals Library, and the DOI of the publication must be cited.
2024 Iro et al.
Chapter 1 Introduction
Background
Encephalitis is a rare condition that can result in serious consequences for those affected and their families. Encephalitis can result from an infection of the brain (infectious encephalitis) or from autoantibodies that affect the brain (immune‐mediated encephalitis), or both. 1 Immune‐mediated disorders, such as acute disseminated encephalomyelitis contribute to a significant proportion of cases where no infective cause is identified. 2 Nonetheless, despite routine investigations, no aetiology is found in up to approximately 40–60% of cases of encephalitis. 2,3
Irrespective of the cause, the final common pathway in the pathophysiology in encephalitis is brain inflammation, which leads to changes in neurological function. Therefore, a paradigm for intervention with the greatest presumptive benefit centres on the early attenuation of the extensive inflammation, which is the primary cause of fatality and neurological sequelae and underpins the pathogenesis of most forms of encephalitis.
Direct evidence of efficacy of intravenous immunoglobulin (IVIG) is suggested by the successful outcomes from both its therapeutic and prophylactic use in enteroviral encephalitis in the immunocompromised and in outbreaks in Southeast and East Asia. There is also evidence from case reports that seem to support the use of IVIG in other infectious causes of encephalitis, including infections with West Nile virus, coxsackie viruses and Mycoplasma pneumoniae, where its use has been associated with rapid improvement and reduced morbidity. The authors of one previous study of IVIG in children with Japanese encephalitis (JE) suggest that IVIG may be an effective treatment for JE and other flaviviral encephalitis due to its anti-inflammatory effects, immune augmentation and neutralisation of the JE virus. 4 However, the result of a Cochrane systematic review of IVIG in infective encephalitis was inconclusive due to the risk of bias and quality of evidence of the included studies. 5
Immunotherapy, often in the form of IVIG, also appears to benefit both adults and children with autoimmune encephalitis, resulting in improved outcomes. Further evidence exists to support the benefit of IVIG in various autoimmune neurological conditions that share similar underlying inflammatory mechanisms to encephalitis, including primary inflammatory myopathies, inflammatory neuropathies and multiple sclerosis. Furthermore, given its disease-modifying properties, there is theoretical evidence of benefit from IVIG treatment even in encephalitis patients who appear to have made an initial full recovery since they can still develop persisting symptoms later.
However, in clinical practice the use of IVIG in encephalitis varies. 6 In the immune-mediated forms of encephalitis, IVIG is typically used after inevitable delay (by weeks in some cases) while alternative diagnoses are being excluded, or until a definitive diagnosis is obtained. In other cases, IVIG is used as a last treatment option where clinical improvement is slow; this is usually after several days from hospital admission. Delay in diagnosis and institution of appropriate treatment in encephalitis may contribute to the high rate of morbidity and mortality, prolonged hospitalisation and associated costs from encephalitis. Given the available evidence of a possible beneficial role of IVIG, there is a strong case for the prospective assessment of the potential role of early intervention with IVIG for all children presenting with evidence of inflammatory encephalitis.
Hypothesis
Our hypothesis was that early treatment with IVIG in addition to standard care could improve neurological outcomes in children with encephalitis.
Study objectives
Primary objective
To determine whether early treatment with IVIG improves neurological outcomes at 12 months after randomisation in children with encephalitis, compared with placebo.
Secondary objectives
-
To evaluate whether IVIG is associated with improved clinical outcomes relating to the hospital admission, including duration of ventilation, length of stay on intensive care unit (ICU), and length of hospitalisation.
-
To evaluate whether early IVIG is associated with improved neurological outcomes including motor and behavioural outcomes, and quality of life.
-
To assess whether IVIG has impact on the development of epilepsy in children affected by encephalitis.
-
To evaluate the impact of IVIG on neuropsychological outcomes of patients with encephalitis.
-
To evaluate whether IVIG treatment is associated with improved neuroradiological outcomes.
-
To identify what adverse effects are experienced with IVIG treatment.
-
To evaluate what proportion of recruited children with encephalitis have an autoantibody mediated disease.
Exploratory objectives
-
To correlate neuroimaging findings with primary and secondary outcomes.
-
To correlate clinical and laboratory parameters with neurological outcomes.
-
To compare brain magnetic resonance imaging (MRI) findings with aetiological diagnosis.
-
Analysis of gene expression in whole blood before and after study treatment.
-
Identification of specific DNA sequence and structural genetic variants in patients with encephalitis.
Encephalitis
In this section, we describe the burden of encephalitis, established treatment options and the role that IVIG might have in the treatment of affected patients.
Encephalitis is a syndrome of neurological dysfunction that results from inflammation of the brain parenchyma. The worldwide annual incidence ranges from 3.5 to 7.4 per 100,000, rising to 16 per 100,000 in children, with the highest incidence in infants under 1 year of age. 7 In England, the incidence of childhood encephalitis is 4.02 per 100,000 per year. 8
Pathophysiology
The inflammation that occurs in encephalitis causes the brain parenchyma to swell leading to altered level of consciousness, and often seizures.
Impact of encephalitis
Encephalitis survivors suffer long-term sequelae and around 30–50% of patients fail to make a full recovery, experiencing impairments in concentration, behaviour, speech and memory, and seizures. 9 Affected patients also experience persisting physical, psychological, cognitive, behavioral and social impairments which can be significant. Even those children who appear to have made an initial recovery experience some degree of disability later. 10,11
Across paediatric encephalitis studies, case fatality of 2–16% has been reported,9,12–17 with individual studies reporting an overall long-term morbidity of 30–60%. 11,13,14,16,18–23 In a 10-year Israeli study of 46 children with all-cause encephalitis, 33% of affected children had focal motor deficit at discharge while 15% showed varying levels of cognitive impairment. 10 In the same study, half of affected children had persisting symptoms, including behavioural problems (52%), recurrent headache (22%) and problems with sleep (19%) after a mean follow-up period of 6 years. Children with encephalitis have a significantly higher prevalence of learning disability (20%) compared to the general population (10%), and even those who appear to have made full recovery demonstrate lower intelligence scores when compared with the general population. 10,24 In a prospective study of Malaysian children with JE, 13% of affected children who were of school age never returned to school due to severe physical disabilities from the illness, while 38% had marked deterioration in school performance which resulted in discontinuation of schooling. 14 In an Indian study of JE, 10% of 39 children had Parkinsonian features at discharge and about 30% had residual symptoms at 14 month follow up. 25 A meta-analysis comprising 890 children with infectious encephalitis from 15 studies showed that 10–20% of children with had abnormal behaviour, motor and cognitive impairment at follow-up 1 to 12 years later while a third of patients had developmental delay. 26
Following herpes simplex encephalitis (HSE), 40–65% of affected children develop seizures. 20 In an Australian study of 147 children followed up for a median duration of 7 years, post-encephalitic epilepsy and drug-resistant epilepsy were reported in 31 (21%) and 15 (10%) children respectively. 27
Treatments for encephalitis
Following suspicion of an encephalitis diagnosis, antibiotics and antiviral (aciclovir) treatment are usually commenced and subsequently rationalised based on microbiological results and clinical progress. Aciclovir is widely used for the treatment of confirmed herpes simplex and varicella zoster virus encephalitis. For bacterial infections causing encephalitis, an appropriate antibiotic is used, based on microbiological sensitivity testing. For autoimmune encephalitis, treatment strategies including IV methylprednisolone, plasma exchange and IVIG are commonly used.
Intravenous immunoglobulin
Intravenous immunoglobulin is a blood product made from pooled collections of human plasma collected from thousands of blood donors. The efficacy of IVIG has been demonstrated for a range of neurological conditions. 28 Licensed indications include as replacement therapy for people with primary and secondary antibody deficiency states, Kawasaki disease, haematological conditions (idiopathic immune thrombocytopenic purpura, B‐cell chronic lymphocytic leukaemia) and neurological conditions (multifocal motor neuropathy and chronic demyelinating polyneuropathy). 29 At the time of running the trial, IVIG was sometimes used off‐label for the treatment of children with encephalitis, but has since been commissioned (2021) for use in autoimmune encephalitis. 30
Dosing
The dose of IVIG for each indication varies. 31 For primary and secondary antibody deficiency states, the starting dose is between 0.4 g/kg and 0.6 g/kg of bodyweight with subsequent adjustments made based on clinical outcome. For neurological diseases, two doses of 2 g/kg of bodyweight over 5 days are typically given, 6 weeks apart. For haematological conditions, a dose of 0.8 g/kg to 1 g/kg is used.
Adverse effects
Intravenous immunoglobulin treatment is generally considered safe and well tolerated; however, adverse events (AEs) such as chills, headache, fever, vomiting, allergic reaction, nausea, arthralgia, low blood pressure and low back pain may occur. Rarely, sudden fall in blood pressure, anaphylactic shock, and thromboembolic reactions could occur. Cases of reversible aseptic meningitis and isolated cases of haemolytic anaemia have been observed as well as acute renal failure. Since IVIG is a blood product, there is the risk of transmission of infectious agents such as HIV and viral hepatitis by contaminated products. 32
How IVIG might work
Intravenous immunoglobulin has multiple actions which may operate in concert with each other. For a particular disease, there may be one predominant mechanism of action depending on the underlying disease pathogenesis. The most relevant actions of IVIG include the following:
-
inhibition of complement binding and prevention of membrane attack complex formation33–36
-
regulation of autoantibodies or cytokines by anti‐idiotypic or anticytokine antibodies39,40
-
modulation of T‐cell function and antigen recognition. 43–47
Additional actions include the effect of IVIG on superantigens and enhancement of remyelination. 44 Antiviral functions of IVIG and its potential to inhibit viral infection has been demonstrated in vitro. 48–50
Literature
The literature review was first carried out in PubMed in 2017 and updated in August 2022 (search terms: ‘intravenous immunoglobulin’ or ‘IVIG’ and ‘encephalitis’).
Intravenous immunoglobulin in infectious encephalitis
There are reports of successful outcomes following treatment with IVIG in children with M. pneumoniae encephalitis,51,52 in immunocompromised patients with echovirus meningoencephalitis,53 and in children with JE. 54 Clinical observation following an outbreak of brainstem encephalitis in Catalonia support a beneficial role of IVIG – all but 3 of 34 children who received IVIG showed good clinical response and had no significant sequelae. 55 In a quasi-controlled Indian trial of 83 children with acute encephalitis syndrome complicated by myocarditis, Bhatt et al. demonstrated that who received IVIG treatment demonstrated a borderline significant lower rate of mortality and an improvement in ejection fraction compared with those treated with standard care alone. 56 However, a retrospective review of 35 paediatric patients with confirmed or suspected encephalitis showed similar clinical and neurological outcomes between IVIG treated versus non-treated patients. 57
Intravenous immunoglobulin in autoimmune encephalitis
The largest available evidence suggesting a positive effect of IVIG in anti-N-methyl-D-aspartate receptor (NMDAR) encephalitis is from a Philadelphia multi-institutional observational cohort study in which 577 patients (211 children) were enrolled. 58 Of the 472 patients who underwent first-line immunotherapy (which included any combination of steroids, IVIG and plasmapheresis) or tumour removal, 53% had symptom improvement within 4 weeks; good recovery was observed in 221 patients at 3 months and in 97% of these at 24 months. A retrospective case series of 20 children with N-methyl D aspartate receptor antibody encephalitis (NMDAR-AbE) demonstrated an 85% recovery rate with immunomodulatory treatment including IVIG. 59 A North Indian study of 11 children with NMDAR-AbE reported significant response to steroids and immunoglobulin in 58% of patients. 60 In a prospective Indian study of 15 patients aged 2–64 years with AIE, 67% of patients who were treated with IVIG, steroids or both showed significant improvement, with no further seizures or clinical relapse years at follow-up. 61 A single-arm, open-label study of 41 adult patients with possible autoimmune encephalitis demonstrated a clinical benefit from IVIG with significant improvement in neurological functional outcomes following IVIG treatment. 62 Nosadini et al. 63 carried out a systematic review and meta-analysis of 1550 adults and children from 652 articles with NMDAR-AbE the use of immunotherapy in NMDAR-AbE, which showed that a combination of immunotherapy including IVIG was associated with good functional outcome in affected patients and lack of immunotherapy within the first 30 days of illness onset was associated with poor functional outcomes. Bien and et al. 64 performed a randomised controlled trial (RCT) of 21 patients in which 16 were randomised to either tacrolimus or IVIG and were compared historical untreated controls. They found that immunotreated patients had a longer survival than historical controls. Contrarily, a retrospective case series of 10 children with limbic encephalitis, a form of AIE, failed to show benefit from IVIG treatment,65 and a case series of 10 Taiwanese children with limbic encephalitis showed that all enrolled patients had neuropsychiatric symptoms and 90% developed refractory epilepsy at a mean follow-up of 5.6 years, despite receiving high dose corticosteroid or IVIG treatment. 66
Intravenous immunoglobulin in acute demyelinating encephalomyelitis
Efficacy of IVIG treatment in acute demyelinating encephalomyelitis (ADEM) is suggested by several single case reports and case series. 67–71 In a retrospective case series of 15 children with ADEM admitted to a single institution in Turkey, all 3 children who made poor response with subsequent IVIG treatment. 72 A similar finding was reported in a single-centre Turkish study of 15 children. 73 One study in Japan showed rapid recovery of consciousness and complete clinical improvement in three children with ADEM treated with IVIG. 74 Pradhan et al. reported on four patients with steroid refractory ADEM and also reported quick recovery following IVIG treatment. 75 A prospective observational study of 18 children with ADEM indicated that outcomes were better in children who received IVIG in the first week than those who did so in the second week of the illness. 76 The findings of a single centre Israeli study of 16 children with ADEM suggested a possible beneficial effect from IVIG when either given separately or combined with high dose methylprednisolone. 77 An Australian study demonstrated an improvement in disability scores in 11 children with infectious encephalitis, 8 children with NMDAR antibody encephalitis and 16 children with other immune mediated encephalitis. 78 In the same study, the proportion of children treated with IVIG who had a good outcome at follow-up (mean duration of 52 months) increased from baseline by 88% and 11% for NMDAR antibody encephalitis and other immune-mediated encephalitis respectively.
Summary
Although there is evidence that IVIG benefits some patients with encephalitis, the studies described above have several limitations. Most of the evidence from IVIG use in infectious encephalitis is from single case reports. The largest evidence of IVIG in infectious encephalitis is from a prospective study of children with acute encephalitis complicated by myocarditis, which was a non-randomised study where the study investigators were aware of treatment allocation. 56 In addition, this study was conducted in a developing setting, and it is unclear how the generalisable are the study findings. The evidence from the studies of AIE are compelling. However, the study by Titulaer et al. was not a RCT and enrolled patients received other immunomodulatory treatment alongside IVIG, which makes it impossible to attribute the observed clinical efficacy to IVIG treatment alone. 58 Furthermore, the study by Lee et al. is limited by the small sample size and also lack of a control arm.
The rising use of IVIG in patients with encephalitis in the absence of robust evidence is not without cost implications. In a previously described single-centre study of IVIG use in Australia,79 the total cost of IVIG was US$ 2,595,907 (median $3538/patient, range $544–260,766). In the UK, IVIG costs £18–22 per gram, equivalent to £2100–£3080 per 2 g/kg for a 70-kg patient. 80 Given the high cost associated with IVIG use and its limited availability, and the crucial need to improve outcomes from encephalitis, robust evidence of efficacy from IVIG treatment is imperative.
Chapter 2 Methods
Trial design
ImmunoglobuliN in the Treatment of Encephalitis (IgNiTE) was a randomised, double-blinded, parallel arm, placebo-controlled study to compare early IVIG treatment with placebo in the treatment of childhood encephalitis in individuals aged 6 months to 16 years.
Research governance
This IgNiTE trial was conducted in compliance with the principles of the Declaration of Helsinki (1996), in full conformity with the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Guidelines for Good Clinical Practice (CPMP/ICH/135/95 July 1996), the Research Governance Framework and the Medicines for Human Use (Clinical Trial) Regulations 2004. Ethics approval was granted by the UK National Research Ethics Service (NRES) committee (South Central – Oxford A; REC 14/SC/1416). Clinical trial (CT) authorisation granted via the Medicines and Healthcare Products Regulatory Agency (MHRA) notification scheme (Ref: 21584/0337/001-0001). Written approval from local Research and Development (R&D) departments at each participating site was obtained before recruitment was commenced at each site. This trial was sponsored by the University of Oxford and was adopted on the UK children Research Network portfolio (co-adopted by Infectious Diseases and Neurological) UKCRN ID 18993. Quality assurance strategies were in place to ensure compliance with the CT regulations. The trial was registered for an International Standard Randomised Controlled Trial Number (ISRCTN), which was assigned on 24 June 2015 (ISRCTN 15791925). The study was assigned a European Clinical Trials Database number (2014-002997-35) and was registered with ClinicalTrials.gov (identifier NCT 02308982) on 5 December 2014. The trial protocol was published on 3 November 2016. 81 The study database OpenClinicaTM was designed and delivered by the Oxford Vaccine Group Clinical Trials Unit. A Trial Steering Committee (TSC) was set up to oversee the trial. The committee comprised an independent chairperson, an independent patient and public involvement (PPI) member, two independent clinicians, the chief investigator (CI), co-investigators and trial statisticians. The TSC met regularly to monitor and advise on study progress and conduct. An independent Data Monitoring and Ethics Committee (DMEC) was set up to monitor the main outcome measures and to ensure the safety of trial participants. The committee comprised an independent chairperson, a statistician and an expert clinician. The DMEC met regularly throughout the trial to monitor safety, efficacy and the overall conduct of the study.
The protocol was amended after the trial was terminated early to remove endpoints which could not be derived from the data collected and to update the statistical analysis section; this is reflected in this report.
Funding
The study was funded by the National Institute for Health and Care Research (NIHR) Efficacy and Mechanism Evaluation programme (reference 12/212/15). The investigational medicinal product (IMP), IVIG (Privigen) was provided by CSL Behring. The matching placebo (0.9% saline and 0.1% albumin) was manufactured at the Royal Liverpool and Broadgreen Aseptic Manufacturing Unit under its manufacturing license; funds for this were provided by CSL Behring.
Inclusion and exclusion criteria
The inclusion criteria were adapted from the International Encephalitis Consortium case definition. 82
Inclusion criteria
-
Age 6 weeks to 16 years old.
-
Acute (within 24 hours) or subacute (24 hours to 4 weeks) onset of altered mental state (reduced or altered conscious level, irritability, altered personality or behaviour, lethargy) not attributable to a metabolic cause.
-
At least two of:
-
fever ≥38°C within 72 hours before or after presentation to hospital
-
new or acute onset brain imaging consistent with encephalitis or immune-mediated encephalopathy
-
cerebrospinal fluid (CSF) white cell count (WCC) >4/microlitre
-
generalised or partial seizures not fully attributable to a pre-existing seizure disorde;
-
new-onset focal neurological signs (including movement disorders) for >6 hours
-
EEG abnormality that is consistent with encephalitis and not clearly attributable to another cause.
-
-
Parent/guardian/legal representative consent to the patient participating in the trial.
Exclusion criteria
Children and young people were not eligible to participate if any of the following applied:
-
high clinical suspicion of bacterial meningitis or TB meningitis (e.g. presence of frankly purulent CSF; CSF WCC > 1000/microlitre; bacteria on Gram stain and/or culture)
-
prior receipt of any IVIG product during the index admission
-
traumatic brain injury
-
known metabolic encephalopathy
-
toxic encephalopathy
-
hypertensive encephalopathy/posterior reversible encephalopathy syndrome
-
pre-existing demyelinating disorder; pre-existing antibody-mediated central nervous system disorder; pre-existing CSF diversion
-
ischaemic or haemorrhagic stroke
-
children with a contraindication to IVIG or albumin
-
known hypercoagulable state
-
significant renal impairment defined as glomerular filtration rate of 29 ml/min/1.73 m2 and below (Chronic Kidney Disease Stage 4)
-
known hyperprolinaemia
-
known to be pregnant
-
any other significant disease or disorder which, in the opinion of the investigator, may either put the participants at risk because of participation in the trial or may influence the result of the trial, or the participant’s ability to participate in the trial
-
participants who were being actively followed up in another research trial involving an IMP which had a potential immunomodulatory or neuroprotective effect.
Study procedures
Informed consent
Written informed consent was obtained from all participants before enrolment to the study. Parents and guardians had the opportunity to review the participant information sheet and participant consent form prior to participation. Informed consent was taken by a suitably qualified and experienced medical doctor, as delegated by the CI. Information sheets and assent forms for different age groups were available and where possible, verbal assent and consent from participants was obtained.
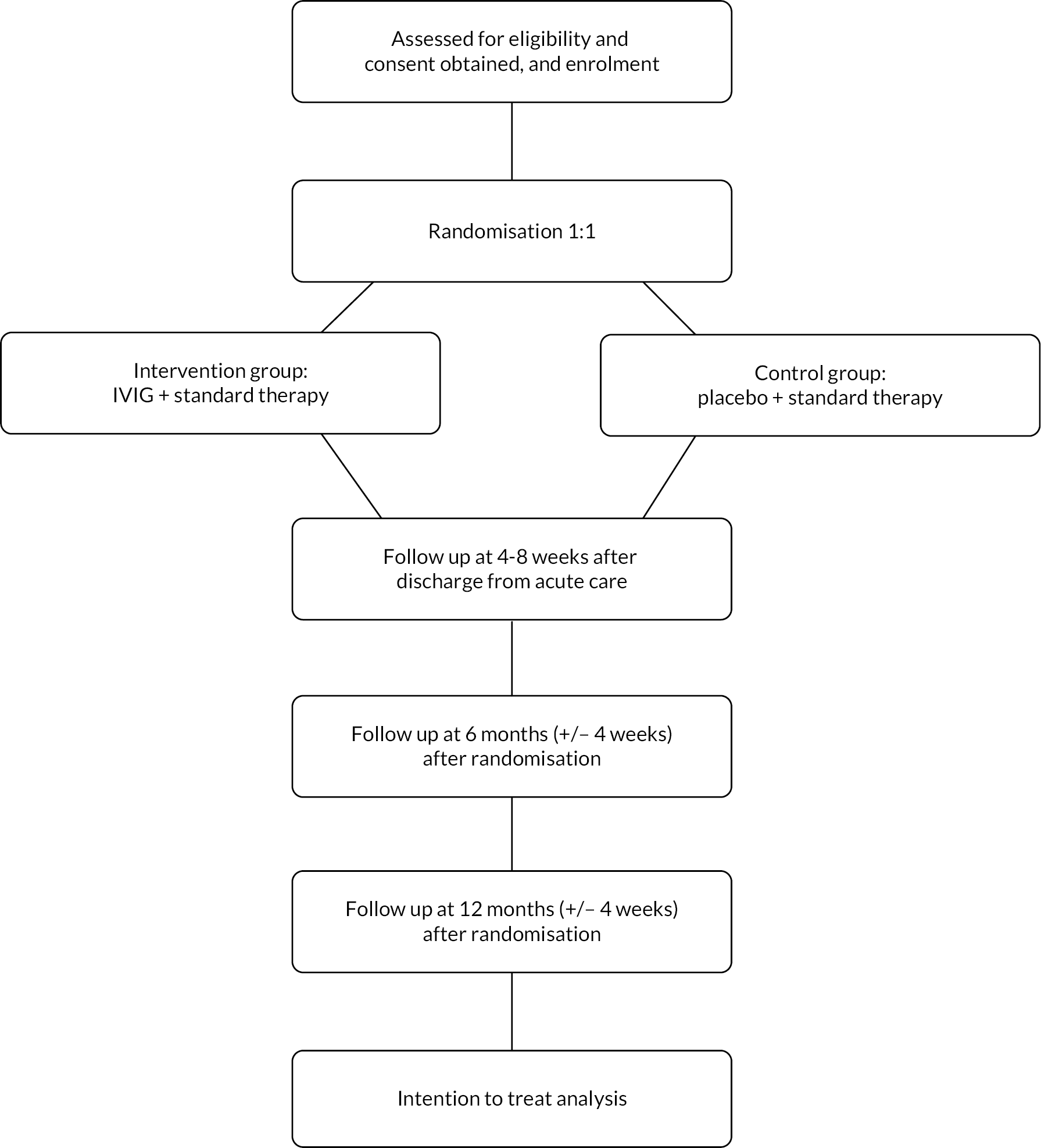
Randomisation and allocation procedure
Randomisation was performed using a secure web-based randomisation system (Sortition®) which was developed by the Clinical Trials Unit in the Nuffield Department of Primary Care Health Sciences, University of Oxford. Randomisation was performed soon as possible after consent was obtained, and within the time window for administration of the first dose of study drug. Allocation sequence was generated by the trial statistician. Stratification variables accounted for at randomisation were age group (<1 year, 1–4 years, 5–9 years, 10–14 years and ≥15 years) and steroid treatment at the time of randomisation, using stratified block randomisation with randomly varying block sizes. Participants were allocated in a 1 : 1 ratio to receive 2 doses (1 g/kg/dose) of either IVIG or matching placebo, in addition to standard care. Confirmation of randomisation was electronically delivered to the investigator performing the randomisation, core members of the central coordinating team, and the independent pharmacy team at each study site.
A rigid blinding process was in place all throughout the study. Participants, their parents/guardians/authorised legal representative, in addition to study staff and clinical staff who were actively involved in the conduct of the study (including recruitment, administration of study treatment, data collection and entry, laboratory analyses) were blind to the treatment allocation through the entire study period. Study monitors who were independent of the study and all site pharmacists were unblinded. The latter was to ensure robust IMP management at each study site. Performance and ascertainment bias were minimised by measures designed to maintain the blinding including identical packaging of IVIG and matched placebo. Dispensing of the correct allocation was performed by the independent pharmacy team who had to be unblinded for this purpose.
Treatment
The IVIG used in the study was Privigen (100 mg/ml solution), with a shelf life of 3 years. The placebo was a mixture of 0.9% saline +0.1 human albumin solution, which had a shelf life of 6 months. The addition of albumin to saline was necessary to make the placebo visually identical to IVIG. Also, similar packaging and labelling were applied to both study treatments. A tear-off label distinguishing both treatments was taken off at the time of dispensing by the unblinded pharmacist at each recruiting site. A dosing guide based on weight band was provided to avoid IMP wastage.
Outcome measures
Primary outcome: Glasgow Outcome Score extended, paediatric version
The primary outcome measure was the Glasgow Outcome Score-Extended, paediatric version (GOS-E Peds) at 12 months after randomisation. The GOS-E Peds is a modified version of the GOS-E, a gold standard for measuring outcomes in adults with traumatic brain injury. The GOS-E Peds provides a developmentally appropriate structured interview necessary to evaluate children across different age groups. Its use has been validated and found to be sensitive to both severity of injury and to recovery over time, at least 6 months after brain injury and has been suggested as useful in guiding treatment in the early phases of recovery from brain injury.
Based on responses provided to the questions contained in the GOS-E Peds, participants were assigned to one of eight categories: 1-Upper Good Recovery, 2-Lower Good Recovery, 3-Upper Moderate Disability, 4-Lower Moderate Disability, 5-Upper Severe Disability, 6-Lower Severe Disability, 7-Vegetative State, and 8-Death. ‘Good recovery’ was defined as a GOS-E Peds score of two or lower and a score of >2 indicated ‘poor recovery’.
Secondary outcomes
Clinical
Secondary clinical outcomes were obtained from routinely collected medical information. These included the need for admission to ICU, invasive ventilation requirement and the length of hospital stay, which was defined as the number of days from admission to a recruiting site to discharge from acute care (i.e. not including days in hospital for neurorehabilitation). At 6 and 12 months after randomisation, information on new diagnosis of epilepsy and need for anti-epileptic treatment since discharge were collected.
Neurological
Secondary neurological outcomes were assessed using various age-appropriate questionnaires and outcome scores, which included the GOS-E Peds (assessed at 6 months after randomisation), Liverpool Outcome Score (LOS), Pediatric Quality of Life Score (PedsQL), Gross Motor Function Classification System (GMFCS), SDQ and Adaptive Behaviors Assessment System, second edition (ABAS-II), all assessed at 4–8 weeks after discharge from acute care and 12 months after randomisation. In addition, a blinded neuropsychology assessment was performed at 12 months after randomisation during which cognitive function was assessed using the following age-appropriate scales: (1) Bayley Scales of Infant and Toddler Development, third edition (1 to 2 years 5 months); (2) Wechsler Preschool Primary Scale of Intelligence IV (2 years 6 months to 5 years 11 months), and (3) Wechsler Intelligence Scale for Children V (6 years to 16 years 11 months).
The LOS is a validated tool for assessing level of disability after encephalitis in infants and children. It was originally designed to assess disease burden following JE and its use has been extended to other forms of encephalitis. For each participant, a total score (sum of scores for all questions) and an outcome score (the lowest score for any single question) were documented. Based on the outcome score only, participants were assigned to one of five outcome categories: 5-Full recovery, 4-Minor sequelae, 3-Moderate sequelae, 2-Severe sequelae, and 1-Death. ‘Good recovery’ was defined as a LOS of 5 and a score of ≤4 indicated ‘poor recovery’.
The PedsQL is a brief measure of health-related quality of life comprised of 23 items assessing quality of life in 4 domains: physical functioning (8 items), emotional functioning (5 items), social functioning (5 items) and school functioning (5 items). Based on the scores in each domain, two summary scores (physical health and psychosocial health summary scores) as well as a total scale score were computed. Total scale scores are presented. A higher total scale score indicates better quality of life.
The GMFCS is an assessment tool based on self-initiated movement and assesses motor function in three areas – walking, sitting and standing. It uses five levels to describe the motor function limitations, taking into consideration age, the use of mobility aids and the quality of movement. The GMFCS is rated from Level 1 (walks without limitations) to Level 5 (transported in a manual wheelchair). Levels 1 and 2 indicate independent mobility. Level 3 indicates ability to move with assistive devices while Levels 4 and 5 indicate significant limitation and dependence on helpers for minor movements. Gross motor function was categorised as mild (Levels 1 and 2), moderate (Level 3) and severe (Levels 4 and 5).
The SDQ is a 25-item questionnaire comprising 5 scales of 5 items each focusing on difficulties relating to emotional functioning, conduct, hyperactivity and interaction with peers. Scale scores and a total difficulties score (generated by summing the scores from all the scales except the prosocial scale) were documented. Based on the total difficulties score, SDQ scores were categorised into four bands: close to average, slightly lower, low and very low, based on a UK community sample. For 2–4-year-old children, the close to average category contains 80%, the slightly raised category contains 12%, the high category contains 4% and the very high category contains 4% of the population. For 4–17-year-old parent-completed questionnaires, the close to average category contains 80%, the slightly raised category contains 10%, the high category contains 5% and the very high category contains 5% of the sampled UK population.
The ABAS-II is an instrument used to evaluate adaptive skills that are important to everyday living and assesses three main domains: (1) Conceptual (summarises performance in the following skill areas – communication, functional academics and self-direction), (2) Social (leisure and social), and (3) Practical (community use, home living, health and safety, self-care). The individual response provided for each skill area question was assigned a score. The total score allocated to each domain was obtained by summing up the skills scores in that domain. Raw scores were converted into composite scores, with a population mean of 100 and a standard deviation of 15, with a lower score signifying worse adaptive behavior. Composite scores were divided into the following categories based on percentiles (%) of the normative population: very superior >130 (≥ 98%); superior 120–129 (91–97%); above average 110–119 (75–90%); average 90–109 (25–74%); below average 80–89 (9–24%); borderline 71–79 (3–8%); extremely low 70 or less (≤2%).
The Bayley Scales of Infant and Toddler Development (BSID-III) is a widely used and validated measure of cognitive functioning which produces three composite scores: cognitive scale, language scale (receptive and expressive) and motor scale (fine and gross). The Wechsler Preschool Primary Scale of Intelligence IV produces scores for: Verbal Comprehension Index (VCI), Visual Spatial Index (VSI), Fluid Reasoning, Working Memory Index (WMI), Processing Speed Index (PSI) and Full-Scale IQ (FSIQ). The Wechsler Intelligence Scale for Children IV assesses general thinking and reasoning skills and is made up of 10 subtests, yielding 4 composite scores (Verbal Comprehension, Perceptual Reasoning Index (PRI), Working Memory, and Processing Speed). The Full-Scale IQ (composite score) is an average of these four scales. Composite standard scores have a mean of 100 and SD of 15. Neurodevelopmental outcome was classified as (1) severe impairment (composite score of <70, >2SD below the mean), (2) mild impairment (score of 70–84, >1SD below the mean) and (3) normal neurodevelopmental (score of ≥85).
Neuroimmunology
Auto-antibody testing was performed by the clinical neuroimmunology service at the Nuffield Department of Clinical Neurosciences, Oxford. Testing was done for antibodies against live neurons, Aquaporin 4, NMDAR, Myelin oligodendrocyte glycoprotein (MOG), leucine-rich, glioma inactivated 1 (LGI1) and Contactin-associated protein-like 2 (CASPR2).
Neuroimaging
Neuroimaging findings were obtained from clinical scans (i.e. scans performed as part of routine clinical care). In addition, an optional follow-up research scan was performed in a subset of participants, where consent was provided. Anonymised research scans provided on a compact disc were analysed by a neuroradiologist at University College London who reported the following:
Initial clinical scan(s):
-
proportion of participants with an abnormal scan
-
distribution of disease – structural and functional anatomy of lesion
-
subset of radiological features (mass effect, hydrocephalus, enhancement, other).
Follow up scan(s):
-
proportion of participants with an abnormal scan
-
lesion resolution/persisting disease
-
presence of new lesions
-
distribution of disease – structural and functional anatomy of lesion
-
subset of radiological features (mass effect, hydrocephalus, enhancement, other).
Mortality
Information on any deaths occurring up to 12 months after randomisation was collected.
Safety
Safety outcomes were obtained throughout the study (see Safety monitoring). In addition, a mandatory full blood count check was performed for all participants 24–48 hours following the second dose of the study treatment to monitor for haemolysis which has previously been described with high concentrations of IVIG treatment. 83
Schedule of visits
A schedule of visits is shown in Appendix 1.
Screening
The purpose of the screening visit was to fully assess the child’s eligibility for the study. Parents/Legal guardians of potential patients were then approached to explain the study and address any queries. The participant information sheet was provided to families who were given sufficient time to decide on whether they wanted their child to participate in the study. Where interest was indicated, written consent was obtained and relevant clinical information was collected. A screening log was maintained throughout the study.
T0: enrolment and randomisation
After obtaining written consent, participants were enrolled to the study and allocated a study number. They were then randomised using the online randomisation system. Following randomisation, electronic confirmation was sent to research staff performing the randomisation, the site Principal Investigator (PI), the site pharmacist and the coordinating team in Oxford. The electronic confirmation specified an allocation number which the site pharmacist used to dispense the study treatment, using an allocation list that was held securely by the pharmacy team and the trial statisticians only.
T1 and T2: IMP administration visits
T1 was the day of administration of the first dose of study treatment. This was as soon as possible after enrolment and within five working days from when a diagnosis of encephalitis was suspected. For participants transferred from a non-participating site during the illness, an additional three working days from admission to the IgNiTE site was allowable if this gave more time for enrolment than five working days from suspicion of a diagnosis of encephalitis. The second dose was administered 24 hours after the first dose (T2).
Each dose of study treatment was based on the participant’s weight (1 g/kg/dose). To avoid wastage a dose-banding guide based on the participant’s weight, which rounded up the total dose of study treatment to be administered to a whole number, was used.
T1 + 24h: research sampling
This visit occurred 24 hours after the first dose of study treatment. Where consent was provided, research specific blood samples were obtained at this time point.
T2 + 24–48 hours
This visit occurred between 24 and 48 hours after receipt of the second dose of study treatment. At this time point, a mandatory full blood count was obtained from all participants to assess for evidence of haemolysis.
T2 + 7 days
This visit occurred 7 days after receipt of the second dose of study treatment. Where consent was provided, research specific blood sample was obtained at this time point.
T3: prior to discharge from medical care
This visit occurred up to 48 hours prior to discharge from medical care. Clinical and laboratory investigation results were collected.
T4: 4–8 weeks after discharge from acute care
At this timepoint, participants completed the study questionnaires.
T5: 6 months after randomisation (+/– 4 weeks)
The GOS-E Peds (secondary outcome) was completed by participants and a research specific MRI scan was performed where consent was provided. Research sampling was also performed at this timepoint, where consent was provided.
T6: 12 months (+/– 4 weeks) after randomisation
Participants were assessed for the primary outcome during a face-to-face visit at this time point. The study questionnaires were completed at this timepoint. A neuropsychology assessment of cognitive function using age-appropriate assessment tools (see section Chapter 2, Secondary outcomes) was performed by a study neuropsychologist who was unaware of the participants’ treatment allocation.
At each study visit time point, eligibility and consent were re-affirmed before any study procedures were performed, relevant clinical information and information regarding serious adverse events (SAEs) were obtained and entered onto an electronic case report form (eCRF).
Collection of research samples and storage
Where specific consent was provided, blood was taken for autoantibody testing, RNA and DNA analysis in line with the study protocol.
Concomitant medication
Details of concomitant medications were collected throughout the study. These included antimicrobials, steroids, anticonvulsants, immunomodulatory treatment (commenced after randomisation), hyperosmolar treatment such as Mannitol and 3% saline, IVIG (as part of routine care and not administered before study treatment), and blood transfusion.
Safety monitoring
Participants were monitored during, and 20 minutes after administration of the study treatment for AEs. The following were reportable in this study:
-
AEs and adverse events of special interest (AESIs) occurring in the first 5 days following receipt of each dose of the study drug
-
SAEs occurring up until 6 months after randomisation
-
serious adverse reactions (SARs) occurring throughout the study period.
An AE was defined as an untoward medical occurrence in a participant that was not necessarily caused by or related to the IMP and was assessed as ‘not related’ or ‘unlikely related’. An adverse reaction (AR) was defined as an untoward and unintended response to the IMP related to any dose administered and was assessed as ‘definitely, likely or possibly related’. An unexpected AR was defined as an AR, the nature and severity of which was not consistent with known information about the IMP. The Summary of Product Characteristics for Privigen was used to assess relatedness of ARs to the study treatment. A SAR was defined as an AE that was both serious and, in the opinion of the reporting Investigator, believed with reasonable probability to be due to one of the trial treatments, based on the information provided. An AESI was defined as any AE of significant scientific, medical, and public interest, relating to an IMP and for which ongoing monitoring and rapid communication by the investigator to the study sponsor could be appropriate.
For each AE, the following information were recorded: description, date of onset and end date, severity and assessment of relatedness to the trial treatment, other suspect drug or device and action taken. The severity was assessed based on the degree to which these affect routine care using the following scale: 1 = mild, 2 = moderate, 3 = severe, 4 = life-threatening, 5 = death. Assessment of causality (i.e. the relationship between an AE and the trial treatment) was performed by a medically qualified investigator at each study site. AEs or ARs that were assessed to be serious (i.e. fatal, life-threatening, resulting in inpatient hospitalisation or prolongation of hospitalisation, resulted in persistent/significant disability or incapacity, or congenital anomaly/birth defect) were reviewed by a delegated medical doctor and reported to the CI, the study sponsor, and CSL Behring within 24 hours after the study team became aware of the event. All AESIs, SAEs and SARS were followed either until resolution, or the event was considered stable. AESIs included the following: (1) anaphylaxis, (2) new onset seizure or abnormal movements not thought to be due to the encephalitis illness, (3) thromboembolism, (4) aseptic meningitis unrelated to the encephalitis illness, (5) acute renal failure, (6) acute haemolysis and (7) other medically significant event as determined by the investigator. The CI and trial manager provided an annual report of all SAEs and SARs (expected and unexpected), which were distributed to the sponsor, and the Research Ethics Committee (REC). The Sponsor reported all SAEs and SARs to the MHRA as part of the annual Drug Safety Update Report. In addition, throughout the duration of the trial, the DMEC reviewed safety data on an ongoing basis to rule out any significant safety concerns.
Data collection and management
Data were collected by clinical staff using paper-based source documents and were subsequently transcribed onto a secure web-based password-protected eCRF, OpenClinicaTM. The paper and electronic data-collection forms were created in accordance with the requirements of the trial protocol. The eCRF was hosted and maintained by the Oxford Vaccine Group Clinical Trials Unit. All participants were identified using a unique trial-specific number and participant-identifiable data were not included in either the paper or eCRF. At the end of the study, the eCRF system was locked and the data were exported for final analysis after data cleaning. All trial data will be stored and archived in line with the Medicines for Human Use (Clinical Trials) Amended Regulations 2006. All study documents will be retained after the completion or discontinuation of the trial for 3 years after the youngest participant turns 18 years.
Management of the study
The trial was coordinated through the Oxford Vaccine Group, a UK Clinical Research Collaboration registered CTs unit working in collaboration with the Primary Care trials unit at the University of Oxford (registration number: 52). The study coordinating team were responsible for the overall management of the trial and comprised the CI, a study lead doctor and nurse, a trial manager and a quality assurance manager. A study team was set up at each recruiting site to oversee the day-to-day running of the trial and publicise the study within the site. Close communication lines were maintained between individual study teams and the coordinating team. In addition, a Trial Management Group was set up to address and discuss clinical queries and the scientific aspects of the study.
The trial master file (TMF) contained all essential documents for the conduct of the trial: approved trial protocols, regulatory approvals, financial and legal documents, the delegation of trial duties log, copies of approved participant information sheets, participant consent forms, screening logs, standard operating procedures, pharmacy/IMP, safety monitoring, etc. The trial manager was responsible for maintaining the TMF.
Monitoring was performed in line with a trial-specific monitoring plan and comprised remote monitoring of the eCRF data and self-monitoring questionnaires completed by each site. A triggered site visit was necessary for sites that either provided a significant proportion of study data or if queries raised during remote monitoring were not resolved. Review of monitoring activity was conducted by a representative of the study sponsor. After each monitoring visit, the trial manager provided a report summarising the documents that had been reviewed and actions required by the study team which was reviewed by the sponsor and CI.
Recruitment and retention
Participant identification and recruitment
Participants were recruited from NHS Hospitals in the UK, as shown in Appendix 2. At each site, potential participants were identified through various routes which included review of medical handover notes, checking the wards for new admissions with a suspected diagnosis of encephalitis, and review of routine clinical investigation results. Following identification of a potentially eligible patient, their clinical presentation was matched against the study inclusion criteria. Parents/guardians of the patients meeting the study criteria were approached by a member of the clinical team to seek their interest in the study. If interest was indicated, a member of the research team approached the family to confirm eligibility and to seek consent. Assent from the participant was also required if they were ≥6 years of age and judged to have sufficient mental capacity to provide this.
Retention of participants
The study team maintained an approachable relationship with participants and provided contact via telephone to discuss any aspect of the study and to remind them of follow-up.
Patient and public involvement
The Encephalitis Society were actively involved in preparing the proposal for this study. The Society agreed that there is the pressing need for improved treatments to address the serious problem of encephalitis, and fully supported the study. A representative of The Encephalitis Society was on the Trial Management Group and provided a patient-centred research perspective to the study design and conduct. The Encephalitis Society was heavily involved in training research nurses and study recruiters. PPI groups were consulted in the development of the essential documents for the study including the participant information sheet and consent forms. Three PPI representatives with previous personal experiences of encephalitis sat on the TSC and contributed to providing overall oversight of the study. Study update meetings were held to which patients previously affected by encephalitis were invited to share their experiences with study teams, thus highlighting the importance of the study. Preliminary blinded results from the study were presented at various national level meetings and conferences. The Plain language summary of this report was reviewed by the Encephalitis Society. The Encephalitis Society will be actively involved in dissemination of the study findings.
Equality, diversity and inclusion
This trial involved individuals with a rare disease; this patient group is often under-served in clinical research. We sought to enrol all eligible children, regardless of their age, gender or ethnicity. The trial was conducted across 21 NHS sites across the UK, serving different local populations. We involved PPI groups in the design and conduct of this study. Children were actively involved in the consent process; children aged ≥6 years and judged to have sufficient mental capacity were required to give assent.
Chapter 3 Statistics
Sample size
At the time of conception of the IgNiTE trial, there were insufficient data from previous studies for sample size calculation. Detection of at least 20% treatment difference from 43% in the ‘good recovery’ rate (i.e. GOS-E Peds score 2 or lower) by 12 months after randomisation was deemed clinically significant. This was based on the results of a large observational study on autoimmune encephalitis by Titulaer et al. 58 A sample size of 308 (154 per group, including an approximate 10% attrition rate) was needed to achieve 90% power (at 5% level of significance, 2-sided) at detecting a difference in the primary outcome between the study groups.
Statistical methods
The analyses were performed on the intention-to-treat (ITT) population; this included all 18 participants who were randomised. In the analysis of the AEs, the population analysed were the 16 participants who received study treatment.
Since only 20% of participants were recruited before the trial was halted, all analyses are descriptive. Hypothesis testing of outcomes has not been conducted, as these analyses would be severely underpowered. Furthermore, no subgroup comparisons or sensitivity analyses were conducted due to the small number of participants.
Descriptive analysis
A Consolidated Standards of Reporting Trials (CONSORT) flow chart was constructed to summarise the flow of participants through the study. Baseline characteristics are summarised by randomised arm to examine balance between the arms at baseline. All outcomes are presented by time point and randomised group, using descriptive statistics. The proportion of participants lost to follow-up or with missing objective are summarised by treatment arm and at each time point. For continuous variables, the mean (normally distributed data) and standard deviation or the median (skewed data) and inter-quartile range (IQR), or range are presented. Binary and categorical variables are presented as counts and percentages.
Statistical software
All analyses were carried out in R version 4.1.1.
Chapter 4 Results
Here we describe the recruitment process and describe results of the primary and secondary outcomes of the study. The exploratory laboratory endpoints were not analysed due to lack of funding to undertake the relevant testing.
Recruitment and participant flow
Recruitment took place between 23 December 2015 and 26 September 2017. During this time, a total of 18 participants were recruited from 21 NHS sites. Appendix 2 shows the recruitment sites and number of participants enrolled from each site. Figure 2 is the CONSORT flow diagram for the trial, which summarises the participant flow through the trial.
FIGURE 2.
CONSORT diagram.

Baseline characteristics
Table 1 summarises the key baseline demographics by treatment arm. The mean age of the participants was 4.09 years (IQR 2.0–11.8), 44% were male, and 89% were of white ethnicity.
| IVIG (n = 10) | Placebo (n = 8) | All (n = 18) | ||
|---|---|---|---|---|
| Age at randomisation (years) | Median (IQR) | 5.55 (1.52–11.8) | 4.09 (2.71–9.64) | 4.09 (2.0–11.8) |
| Sex (%) | Male | 4 (40) | 4 (50) | 8 (44.4) |
| Female | 6 (60) | 4 (50) | 10 (55.6) | |
| Ethnicity (%) | White | 8 (80) | 8 (100) | 16 (88.9) |
| Asian | 1 (10) | 0 (0) | 1 (5.6) | |
| Missing | 1 (10) | 0 (0) | 1 (5.6) | |
| History of immunocompromise (%) | No | 9 (90) | 7 (87.5) | 16 (88.9) |
| Missing | 1 (10) | 1 (12.5) | 2 (11.1) | |
| Previous diagnosis of encephalitis (%) | No | 9 (90) | 7 (87.5) | 16 (88.9) |
| Missing | 1 (10) | 1 (12.5) | 2 (11.1) | |
| History of encephalopathic illness (%) | No | 9 (90) | 7 (87.5) | 16 (88.9) |
| Missing | 1 (10) | 1 (12.5) | 2 (11.1) | |
| Pre-existing diagnosis of epilepsy (%) | No | 9 (90) | 7 (87.5) | 16 (88.9) |
| Missing | 1 (10) | 1 (12.5) | 2 (11.1) |
Ten participants were randomised to IVIG treatment, and eight participants were randomised to the placebo treatment. Baseline characteristics were balanced between the study groups, although there was a tendency towards lower age in the placebo group and the IVIG arm had slightly more females.
Withdrawal from treatment and from the study
The retention rate of the study was high and comparable between both groups at 12 months after randomisation: 80% (IVIG group) versus 75% (placebo group). Two participants (one in each study group) were withdrawn from the study before receipt of the first dose of study treatment; this was due to one participant being transferred urgently to a non-IgNiTE participating hospital and study treatment being unavailable for another participant. One participant in the IVIG group withdrew consent prior to the 12 months after randomisation time point.
Adherence and compliance with treatment
One participant in the placebo group received only one dose of study treatment due to refusal of the second dose.
Loss to follow-up and missing data
At the 12-month visit for primary endpoint, there were two withdrawals from the IVIG group and one withdrawal and one loss to follow up from the placebo group. The median age for these four participants who had missing data at the 12-month visit for primary endpoint was 8.5 years and 3 (75%) were female. The median age for those with no missing data at the same timepoint was 4.1 years, and 50% were female. Due to small numbers, formal comparison of the two groups was not conducted.
All 18 randomised participants were included in the ITT analysis for primary and secondary outcomes, and all 16 participants who received study treatment were included in the safety analysis.
Primary outcome: GOS-E Peds at 12 months
The primary outcome was assessed using the GOS-E Peds, an outcome scale used to assess level of disability, at 12 months after randomisation. The proportion of participants who made good recovery was similar between both study groups, as shown in Table 2.
| IVIG (N = 10) (%) | Placebo (N = 8) (%) | Overall (N = 18) | |
|---|---|---|---|
| GOS-E Peds Scorea | |||
| 1. Upper good recovery | 4 (40) | 4 (50) | 8 (44%) |
| 2. Lower good recovery | 1 (10) | 0 (0) | 1 (6%) |
| 5. Upper severe disability | 1 (10) | 1 (13) | 2 (11%) |
| 6. Lower severe disability | 2 (20) | 1 (13) | 3 (17%) |
| Participants with missing data due to being withdrawn or lost to follow-up | 2 (20) | 2 (25) | 4 (22%) |
Primary analysis
Nine participants [50%; IVIG n = 5 (50%); placebo n = 4 (50%)] made a good recovery, defined as a GOS-E Peds score of <2. Five participants [28%; IVIG n = 3 (30%), placebo n = 2 (25%)] made a poor recovery and four participants [22%; IVIG n = 2 (20%), placebo n = 2 (25%)] did not undergo a GOS-E Peds assessment at 12 months after randomisation.
Secondary outcomes
Clinical outcomes
Table 3 summarises results of the clinical outcomes assessed in the study.
| Outcome | IVIG (N = 10) |
Placebo (N = 8) |
Overall (N = 18) |
|
|---|---|---|---|---|
| During hospital stay | ||||
| Duration of ventilation | Median (IQR) | 2.5 (2.0–3.5) (n = 4) |
2.0 (2.0–3.0) (n = 5) |
2.0 (2.0–3.0) (n = 9) |
| Length of ICU stay | Median (IQR) | 4.0 (3.0–6.0) (n = 5) |
5.0 (2.0–10.0) (n = 5) |
4.5 (3.0–6.8) (n = 10) |
| Length of hospitalisation for acute care | Median (IQR) | 12.0 (8.0–27.0) (n = 9) |
8.0 (6.5–14.0) (n = 7) |
11.0 (7.8–19.5) (n = 16) |
| 6 months post randomisation | ||||
| New diagnosis of epilepsy since discharge | n (%) | 1 (10) | 1 (13) | 2 (11) |
| Anti-epileptic treatment since discharge | n (%) | 1 (10) | 1 (13) | 2 (11) |
| 12 months post randomisation | ||||
| New diagnosis of epilepsy since discharge | n (%) | 0 (0) | 1 (13) | 1 (6) |
| Anti-epileptic treatment since discharge | n (%) | 0 (0) | 0 (0) | 0 (0) |
Duration of ventilation
Ten participants [56%; IVIG n = 5 (50%), placebo = 5 (63%)] were admitted to the ICU during the admission and nine of these (IVIG n = 4, placebo n = 5) required invasive ventilation. The median duration of invasive ventilation was similar between both study groups: 2.5 days (IQR 2–3.5) for the IVIG group versus 2 days (IQR 2–3) for the placebo group.
Length of ICU stay
The median length of stay on ICU did not differ between both study groups and was 4 days (IQR 3–6) for the IVIG group and 5 days (IQR 2–10) for the placebo group.
Length of hospitalisation
The median length of hospitalisation was 12 days (IQR 8–27) for the IVIG group versus 8 days (IQR 6.5–14) for the placebo group.
New diagnosis of epilepsy
At 6 months after randomisation, two participants [11%; IVIG n = 1 (10%), placebo n = 1 (13%)] had a new diagnosis of epilepsy. At 12 months after randomisation, one additional participant in the placebo group (13%) had a new diagnosis of epilepsy versus none of the participants in the IVIG group. Five participants [28%; IVIG n = 2 (20%), placebo n = 3 (38%)] had incomplete data for this outcome.
Neurological outcomes
Table 4 summarises the secondary neurological outcomes.
| Outcome | 4–8 weeks post discharge | 12 months post randomisation | ||
|---|---|---|---|---|
| IVIG (N = 10) | Placebo (N = 8) | IVIG (N = 10) | Placebo (N = 8) | |
| LOS | ||||
| 2. Severe sequelae (%) | 2 (20) | 2 (25) | 2 (20) | 2 (25) |
| 3. Moderate sequelae (%) | 2 (20) | 3 (38) | 1 (10) | 1 (13) |
| 4. Minor sequelae (%) | 1 (10) | 0 (0) | 1 (10) | 1 (13) |
| 5. Full recovery (%) | 3 (30) | 2 (25) | 4 (40) | 2 (25) |
| Missing data due to withdrawal or loss to follow-up of participant (%) | 1 (10) | 1 (13) | 2 (20) | 2 (25) |
| Missing data – assessment not performed (%) | 1 (10) | 0 (0) | 0 | 0 |
| PedsQL | ||||
| Mean (SD) | 77.9 (11.1) | 56.5 (7.8) | 79.9 (21.6) | 63.7 (30.1) |
| Missing data due to withdrawal or loss to follow-up of participant (%) | 1 (10) | 1 (13) | 2 (20) | 2 (25) |
| Missing data – assessment not performed (%) | 4 (40) | 5 (63) | 2 (20) | 4 (50) |
| SDQ | ||||
| Close to average (%) | 4 (40) | 1 (13) | 4 (40) | 1 (13) |
| Slightly raised (%) | 1 (10) | 0 (0) | 1 (10) | 0 (0) |
| Very high (%) | 0 (0) | 1 (13) | 1 (10) | 1 (13) |
| Missing data due to withdrawal or loss to follow-up of participant (%) | 1 (10) | 1 (13) | 2 (20) | 2 (25) |
| Missing data – assessment not performed (%) | 4 (40) | 5 (63) | 2 (20) | 4 (50) |
| ABAS | ||||
| Very superior (%) | 0 (0) | 0 (0) | 1 (10) | 0 (0) |
| Superior (%) | 1 (10) | 0 (0) | 1 (10) | 0 (0) |
| Above average (%) | 1 (10) | 0 (0) | 1 (10) | 0 (0) |
| Average (%) | 2 (20) | 1 (13) | 0 (0) | 1 (13) |
| Below average (%) | 0 (0) | 1 (13) | 1 (10) | 0 (0) |
| Borderline (%) | 1 (10) | 0 (0) | 0 (0) | 0 (0) |
| Extremely low (%) | 1 (10) | 0 (0) | 1 (10) | 1 (13) |
| Missing data due to withdrawal or loss to follow-up of participant (%) | 1 (10) | 1 (13) | 2 (20) | 2 (25) |
| Missing data – assessment not performed (%) | 3 (30) | 5 (63) | 3 (30) | 4 (50) |
| GMFCSa | ||||
| Mild (%) | 5 (50) | 2 (25) | 6 (60) | 1 (13) |
| Severe (%) | 0 (0) | 1 (13) | ||
| Missing data due to withdrawal or loss to follow-up of participant (%) | 1 (10) | 1 (13) | 2 (20) | 2 (25) |
| Missing data – assessment not performed (%) | 4 (40) | 5 (63) | 2 (20) | 4 (50) |
| GOSE-Peds at 6 months post randomisation | ||||
| IVIG (N = 10) | Placebo ( N = 8) | |||
| 1. Upper good recovery (%) | 4 (40) | 4 (50) | ||
| 3. Upper moderate disability (%) | 1 (10) | 1 (13) | ||
| 5. Upper severe disability (%) | 0 (0) | 1 (13) | ||
| 6. Lower severe disability (%) | 3 (30) | 1 (13) | ||
| Missing data due to withdrawal or loss to follow-up of participant (%) | 2 (20) | 1 (13) | ||
| Missing data – assessment not performed (%) | 0 (0) | 0 (0) | ||
Glasgow Outcome Score-Extended, paediatric at 6 months after randomisation
At 6 months after randomisation, four participants in the IVIG group (40%) versus four participants in the placebo group (50%) made a good recovery, while four participants in the IVIG group (40%) versus 3 participants in the placebo group (38%) made poor recovery on the GOS-E Peds assessment. Three participants [17%; IVIG n = 2 (20%), placebo n = 1 (13%)] did not undergo a GOS-E Peds assessment at 6 months after randomisation.
Liverpool Outcome Score
At 4–8 weeks after discharge from acute care, three participants in the IVIG group (30%) versus two participants in the placebo group (25%) made full recovery (i.e. LOS > 4) while five participants in the IVIG group (50%) versus five participants in the placebo group (63%) made a poor recovery, reporting minor to severe sequelae. Three participants [17%; IVIG n = 2 (20%); placebo n = 1 (13%)] did not have LOS data collected at this timepoint.
At 12 months after randomisation, four participants in the IVIG group (40%) versus two participants in the placebo group (25%) made full recovery while four participants in the IVIG group (40%) versus four participants in the placebo group (50%) made poor recovery, reporting minor to severe sequelae. Four participants [22%; IVIG n = 2 (20%); placebo n = 2 (25%)] did not have LOS data collected at this timepoint.
Paediatric quality of life
Paediatric quality of life scores were available for seven participants [39%; IVIG n = 5 (50%), placebo n = 2 (25%)] at 4–8 weeks after discharge from acute care and for eight participants [44%; IVIG n = 6 (60%), placebo n = 2 (25%)] at 12 months post randomisation.
At 4–8 weeks after discharge from acute care, mean PedsQL scores were 77.9 (SD 11.10) for the IVIG group versus 56.5 (SD 7.8) for the placebo group. At 12 months after randomisation, PedsQL scores were 79.9 (SD 21.6) for the IVIG group versus 63.7 (SD 30.1) for the placebo group.
Gross motor function classification system
At 4–8 weeks after discharge from acute care, five participants in the IVIG group (50%) versus two participants in the placebo group (25%) had mild impairment of gross motor functioning. These data were not available for 11 participants [61%; IVIG n = 5 (50%), placebo n = 6 (75%)] at this timepoint.
At 12 months after randomisation, six participants (60%) versus two participants (25%) in the IVIG and placebo groups, respectively, experienced either mild or severe impairment of gross motor function. These data were not available for 10 participants [56%; IVIG n = 4 (40%), placebo n = 6 (75%)] at this timepoint.
Strengths and Difficulty Questionnaire
At 4–8 weeks after discharge from acute care, four participants in the IVIG group (40%) versus one participant in the placebo group (13%) had a score that was close to the average of the normative population, one participant in the IVIG group (10%) and none in the placebo group had a slightly raised score, while no participants in the IVIG group versus one participant in the placebo group (13%) had a very high score. These data were not available for 11 participants [61%; IVIG n = 5 (50%), placebo n = 6 (75%)] at this timepoint.
At 12 months after randomisation, four participants in the IVIG group (40%) versus one participant in the placebo group (13%) had a score that was close to the average for the normative population, one participant in the IVIG group (10%) versus none in the placebo group had a slightly raised score, and one participant each in the IVIG and placebo groups (10% vs. 13% respectively) had a high score when compared with the normative population. These data were not available for 10 participants [56%; IVIG n = 4 (40%), placebo n = 6 (75%)] at this timepoint.
Adaptive Behaviors Assessment System-second edition
At 4–8 weeks after discharge from acute care, four participants in the IVIG group (40%) versus one participant in the placebo group (13%) had a score that was either similar or higher than the average score for the normative population while two participants in the IVIG group (20%) versus one participant in the placebo group (13%) had a score that was lower than the average for the normative population. Ten participants [56%; IVIG n = 4 (40%), placebo n = 6 (75%)] did not have ABAS-II assessment at this timepoint.
At 12 months after randomisation, three participants in the IVIG group (30%) versus one participant in the placebo group (13%) had a score that was similar or higher when compared with the average score for the normative population, while two participants in the IVIG group (20%) versus one participant in the placebo group (13%) had a score that was below the average of the normative population. Eleven participants [61%; IVIG n = 5 (50%), placebo n = 6 (75%)] did not have ABAS-II assessment at this timepoint.
Neuropsychology assessment
The results of neuropsychology assessment at 12 months after randomisation are summarised in Table 5.
| Participant | Age at assessment | Bayley cognitive score | FSIQ | VCI | VSI/PRI | WMI | PSI |
|---|---|---|---|---|---|---|---|
| Placebo arm | |||||||
| 1 | 4y 8m | - | a | a | a | a | |
| 2 | 5y 6m | - | 79 | 95 | 79 | 75 | 71 |
| 3 | 2y 10m | - | a | a | a | a | a |
| 4 | 2y 0m | 110 | - | - | - | - | - |
| 5 | 16y 10m | - | 89 | 99 | 88 | 83 | 94 |
| IVIG arm | |||||||
| 6 | 4y 5m | - | a | a | a | a | a |
| 7 | 9y 2m | - | 104 | 92 | 111 | 107 | 116 |
| 8 | 14y 1m | - | 95 | 102 | 90 | 99 | 91 |
| 9 | 8y 8m | - | 88 | 93 | 96 | 91 | 83 |
| 10 | 2y 2m | 55 | - | - | - | - | - |
| 11 | 3y 9m | - | 65 | 60 | 75 | 72 | - |
| 12 | 2y 1m | - | a | a | a | a | a |
| 13 | 14y 6m | - | 119 | 108 | 110 | 110 | 131 |
Thirteen participants [72%; IVIG n = 8 (80%); placebo n = 5 (63%)] had blinded neuropsychology assessment performed at 12 months after randomisation; four [30%; IVIG n = 2 (25%), placebo n = 2 (40%)] of these participants were unable to complete the full battery of assessments due to attention or behavioural needs. Five participants [28%; IVIG n = 2 (20%), placebo n = 3 (38%)] did not undergo neuropsychology assessment.
The proportion of participants (IVIG vs. placebo) with a score of ≥85 (indicating normal development) for each of the main composite scores were as follows: four (40%) versus one (13%) for FSIQ, four (40%) versus two (25%) for VCI, four (40%) versus one (13%) for VSI, four (40%) versus 0 (0%) for WMI; and three (30%) versus one (13%); for PRI. Two participants (one in each treatment arm) were assessed using the Bayley scale of infant development, one participant (IVIG arm) had severe neurodevelopmental outcome while the other (placebo arm) had a normal neurodevelopmental outcome.
Neuroimaging
Tables 6–8 summarise the neuroimaging results.
At baseline, 5 out of 19 acute scans (26%) were abnormal; 4 of these (80%) showed bilateral changes and one showed unilateral changes. There were nine follow-up scans for eight unique participants of which six (67%) were normal and unchanged from the acute scan while two (25%) scans showed abnormal changes.
| Participant number | Age at time of acute scan | Type of scan | Overall assessment | Laterality of abnormality |
|---|---|---|---|---|
| 1 | 3 years 7 months | MRI | Abnormal | Bilateral |
| 2 | 14 years | MRI | Normal | N/A |
| 3 | 1 year 9 months | MRI | Abnormal | Unilateral (Right) |
| 4 | 13 years | CT scan | Normal | N/A |
| 4 | 13 years | MRI | Abnormal | Bilateral |
| 5 | 8 years 2 months | MRI | Normal | N/A |
| 6 | 15 years 9 months | CT scan | Normal | N/A |
| 6 | 15 years 9 months | MRI | Normal | N/A |
| 7 | 1 year | MRI | Not available | Not available |
| 8 | 2 years 8 months | MRI | Normal | N/A |
| 8 | 2 years 8 months | MRI | Normal | N/A |
| 9 | 4 years 6 months | MRI | Abnormal | Bilateral |
| 10 | 7 years 8 months | CT scan | Normal | N/A |
| 10 | 7 years 8 months | MRI | Abnormal | Bilateral |
| 11 | 7 years 9 months | MRI | Normal | N/A |
| 12 | 1 year | CT scan | Normal | N/A |
| 12 | 1 year | CT scan | Normal | N/A |
| 12 | 1 year | MRI | Normal | N/A |
| 13 | 1 year 1 month | CT scan | Normal | N/A |
| Participant number | TEM | FR | PAR | OCC | INS | BS | CBL | COR | WM | DGM | DGM - BG | DGM-TH | DGM - Other | SUN | SN |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Y | N | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y | Y |
| 3 | N | N | N | N | N | N | N | N | N | Y | N | Y | N | NA | NA |
| 4 | Y | Y | Y | Y | Y | N | N | Y | N | N | N | N | N | N | N |
| 9 | N | Y | Y | Y | N | Y | N | N | Y | N | NA | NA | NA | NA | NA |
| 10 | Y | Y | Y | Y | Y | Y | Y | N | Y | Y | Y | Y | N | NA | NA |
| Participant Number | Somatomotor/sensory | Visual | Auditory | Limbic | Extrapyr system | Radiological pattern (A/V/I) | Mass | Hydro | Enhc |
|---|---|---|---|---|---|---|---|---|---|
| 1 | N | Y | N | Y | Y | V | N | N | Y |
| 3 | N | N | N | N | N | Non-specific | N | N | N |
| 4 | N | N | N | N | N | V | Y | N | N |
| 9 | N | N | N | N | N | V & I | N | N | N |
| 10 | Y | Y | Y | N | N | A & I | N | N | N |
Autoantibody testing
The results of autoantibody testing are shown in Table 9. Two participants (both in the placebo arm) were identified to have specific autoantibodies; one participant was positive for LGI1 antibodies, and one participant was positive for MOG antibodies. Two additional participants (both in the IVIG arm) were positive for IgG to live neurons, but negative for antibodies to the specific antigens tested for.
| Participant number | NEURONS | NR1 (NMDAR-Ab) | MOG-FL IgG1 | AQP4 | LGI1 | CASPR2 |
|---|---|---|---|---|---|---|
| 1 | Negative | Negative | Negative | Negative | Negative | Negative |
| 2 | Negative | Negative | Negative | Negative | Negative | Negative |
| 3 | Negative | Negative | Negative | Negative | Positive | Negative |
| 4 | Positive | Negative | Negative | Negative | Negative | Negative |
| 5 | Negative | Negative | Negative | Negative | Negative | Negative |
| 6 | Positive | Negative | Negative | Negative | Negative | Negative |
| 7 | Negative | Negative | Negative | Negative | Negative | Negative |
| 8 | Negative | Negative | Negative | Negative | Negative | Negative |
| 9 | Negative | Negative | Negative | Negative | Negative | Negative |
| 10 | Negative | Negative | Negative | Negative | Negative | Negative |
| 11 | Negative | Negative | Positive | Negative | Negative | Negative |
| 12 | Negative | Negative | Negative | Negative | Negative | Negative |
Safety reporting
Safety outcomes are summarised in Table 10. One participant in the IVIG group (11%) versus none in the placebo group experienced an AESI within 5 days of receiving the study treatment. The participant developed a fever during infusion of IVIG, which was judged to be unrelated to the study treatment. Ten SAEs were reported in three participants in the placebo group (43%) versus none in the IVIG group. None of the SAEs were judged to be related to the study treatment. None of the participants experienced haemolysis following receipt of two doses of study treatment and there were no deaths reported during the study period.
| Outcome | IVIG (N = 9) | Placebo (N = 7) | |
|---|---|---|---|
| AEs of special interest (AESI) in the first 5 days from each dose | |||
| AESIs reported | No. participants | 1 (11%) | 0 (0%) |
| No. events | 1 | 0 | |
| Relationship to study treatment | Related | 0 (0%) | - |
| Not related | 1 (100%) | - | |
| SAEs up to 6 months post randomisation | |||
| SAEs reported | No. participants | 0 (0%) | 3 (43%) |
| No. events | 0 | 10 | |
| Relationship to study treatment | Related | - | 0 (0%) |
| Not related | - | 10 (100%) | |
| Haemolysis (drop in haemoglobin of >3 g/dl) | |||
| Haemolysis present | Yes (%) | 0 (0) | 0 (0) |
| No (%) | 9 (100) | 4 (57) | |
| Missing data (%) | 0 (0) | 3 (43) | |
Chapter 5 Discussion
Main findings
The IgNiTE trial was the first ever RCT that aimed to evaluate early IVIG treatment in children with encephalitis, irrespective of cause. IgNiTE failed to meet its primary objective due to recruitment difficulties resulting in a small sample size. The study was halted after recruitment of 18 participants due to withdrawal of funding despite proposed alternative strategies to deliver on the study objectives, using a modified protocol. Alternative funding could not be secured; therefore, the trial was ended.
Due to the small sample size, no group comparisons have been conducted and it is impossible to reach any conclusions regarding the efficacy of IVIG in encephalitis. Thus, we comment only on possible trends seen at data analysis. A summary of participant recruitment and flow through the trial is provided by Figure 2.
Results
Primary outcome
Overall, 50% of participants achieved the primary outcome (i.e. GOS-E Peds score of ≤2 indicating a good recovery) while 28% made a poor recovery, experiencing some degree of disability at 12 months after randomisation. The remaining 22% of participants did not undergo assessment at this timepoint. The proportion of participants who made a good recovery at 12 months after randomisation was the same between the study groups.
Secondary outcomes
Clinical outcomes
The duration of ventilation and length of ICU stay were similar between both groups. Participants in the IVIG group had a longer median hospital stay compared with participants in the control group (12 days vs. 8 days). Overall, 17% of participants had a new diagnosis of epilepsy following the encephalitis illness, with a trend towards a higher proportion in the placebo group than the IVIG group.
Disability assessment
Disability was assessed using the GOS-E Peds and LOS questionnaires. A total of 33% of all participants in the study made full recovery (i.e. LOS of > 4) whereas 44% had minor to severe sequelae (LOS ≤ 4) at 12 months after randomisation, and 22% did not have LOS data collected at this timepoint. When assessed using the GOS-E Peds at 6 months, 44% of all participants made a good recovery (GOS-E peds ≤ 2) while 39% experienced moderate to severe disability (GOS-E peds > 2) and 17% did not undergo a GOS-E Peds assessment at this timepoint. The proportion of participants experiencing neurological sequelae based on both the LOS assessment at 12 months and GOS-E Peds assessment at 6 months tended to be higher in the placebo group compared with the IVIG group.
Assessment of quality of life
There was a trend towards higher PedsQL scores (indicating better quality of life) in participants in the IVIG group compared with those in the placebo group.
Assessment of gross motor functioning
The proportion of participants experiencing impairment of gross motor functioning was generally higher in the IVIG group compared with the placebo group; however, this may be influenced by the high number of participants who did not have this assessment at 4–8 weeks or 12 months post-randomisation (61% and 56%, respectively).
Assessment of adaptive skills
There was a tendency towards a higher proportion of participants with adaptive skills scores close to the average for the normative population in the IVIG group compared with the placebo group. The proportion of participants with scores below the average for the normative population was similar for both the IVIG group and placebo group, although similar data were not available for over half of all participants (56%) which may influence these results.
Assessment of cognitive functioning
This was assessed using various age-appropriate cognitive scales. When the main composite outcomes were assessed, there was a trend towards a higher proportion of participants in the IVIG group with normal level of cognition at 12 months compared with those in the placebo group.
Safety
One participant in the IVIG group experienced an AESI but this was deemed unrelated to the study treatment, No SAEs were reported in the IVIG group compared to ten SAEs reported in three participants in the placebo group. No deaths were recorded in both study groups.
Interpretation
The findings demonstrate that the morbidity from encephalitis is significant with 28–45% of participants experiencing some degree of disability at 12 months after the acute presentation. It appears that encephalitis patients treated with IVIG have a better quality of life experience and a lesser degree of disability than patients not treated with IVIG, although we cannot exclude the potential influence of missing assessments on these results. IVIG (Privigen) has been extensively researched in other patient populations and its side effect profile is well known. There were no safety concerns relating to IVIG in this study.
Lessons learned
IgNiTE was conducted to a high standard and all outcomes were ascertained appropriately. Notwithstanding the challenges with recruitment, there are several lessons to be learned that could be applied to future trials.
One of the challenges encountered was significant delays with site opening. For trials of rare conditions like encephalitis, a multicentre design is crucial to optimise recruitment. Given the broad clinical spectrum of encephalitis, a multicentre approach also ensures that the study population is truly representative of the real-world cohort of patients since recruitment from only large hospitals is likely to select only those patients with severe disease. While trial set-up can be straightforward, particularly at large tertiary or quaternary hospitals with a lot of experience in conducting RCTs, it can be a more challenging process for smaller hospitals with limited prior involvement in research and the site set-up process can be slow. It had been envisaged that by 21 months from commencing the IgNiTE trial, all planned 30 sites would have opened to recruitment but the delays with site opening meant that this was not achieved. For some sites, this was due to non-availability of research nurses, lack of pharmacy capacity, changes to PI, and delays in obtaining research and development approval. For future trials, such delays should be anticipated, and contingency plans made such as extending the study recruitment period, to account for such delays.
We observed a lower than anticipated incidence of encephalitis. At the time that IgNiTE was set up, there were minimal data on the incidence of encephalitis in the UK. Based on previously published UK data, we estimated the number of paediatric admissions to be 2 per year in district general hospitals and 10 per year in tertiary paediatric hospitals. However, our experience was different with a significantly lower number of actual numbers recruited when compared with the estimates, even during the autumn/winter months when the natural peak in incidence is to be expected. One reason for this could be the disparity between the entry criteria used in the study and the more pragmatic but less stringent approach to making a clinical diagnosis of encephalitis. A proposal to mitigate the recruitment challenge encountered in IgNiTE, which included revision to the sample size to allow recruitment of a smaller but adequately powered sample size, extension to the recruitment period and an increase to the number of participating sites was unsuccessful. For large full-scale trials of rare conditions like childhood encephalitis, incorporating a pilot phase with clear feasibility objectives, clear analytical plans and explicit stop-go criteria is likely to enhance the success of such trials. Furthermore, different methodologies should be explored when designing trials of rare diseases, as improvements in biomarkers increasingly enable sub-phenotyping of conditions, thus further reducing individual disease sample sizes.
For trials of a condition like encephalitis for which the clinical presentation can be non-specific, in-depth consideration should be given to the choice of an appropriate entry criteria. Having a robust set of entry criteria provides guidance on eligibility (and) provides homogeneity in the cohort recruited. This enables a clearer definition of the target population that would benefit from IMP if a treatment effect is established, but can limit generalisability of the study findings to the wider patient population. The impact that having very stringent entry criteria could have on recruitment should be considered. In IgNiTE, a strict entry set based on the International Encephalitis Consortium case definition was used since this approach automatically selects a population that provides the best opportunity to observe a treatment effect. However, this case definition, which is intended for use in research trials, differs from the diagnostic approach routinely used in clinical practice where a lower threshold is applied. This meant that some patients who met the threshold for a clinician diagnosis of encephalitis diagnosis did not meet the criteria for entry into the IgNiTE trial or did not do so within the timelines for administering the first dose of study treatment and were therefore excluded. Given the challenges with recruitment, one approach could have been to use a less stringent set of entry criteria. Although this approach might have contributed to increasing recruitment, given the non-specific presentation of encephalitis, it would have encouraged enrolment of patients with an alternative diagnosis which could dilute the true effect of the study treatment. Assessing the impact of using a set of inclusion criteria that slightly differs from what is routinely used in clinical practice could be undertaken in the pilot phase to inform decisions for the larger trial.
A further consideration for any future trials investigating treatments for childhood encephalitis is how best to optimise the consent rate. The immediate period following hospital admission and/or diagnosis of encephalitis is a sensitive one for the family and especially if the child is admitted to the ICU. Therefore, a longer period of time is likely to be required by the family to consider information about the study to allow them to reach a decision regarding participation, particularly for trials involving an IMP. While it is crucial that families are given sufficient time to decide regarding enrolling their child to a research trial, where there is a time window for recruitment, as was the case in IgNiTE, delays in reaching a decision could mean that the patient becomes ineligible. Gaining the trust of parents/guardians and providing them with a reasonable degree of reassurance regarding the safety of trial procedures are important to mitigate such delays. Several ways to achieve this include maintaining clarity and consistency in the message provided to parents/guardians about a study. This can be difficult to achieve especially on the ICU, where there is a high level of patient/family contact with multiple health professionals, some of whom may not have adequate knowledge of the trial or may have individual biases. One strategy could be for families to be approached only by key individuals who not only have an established relationship with the family but also have adequate knowledge of the study. Other strategies could include branding the trial with key, easy-to-understand messages, and the use of informed consent videos to provide clarity and consistency for families. Furthermore, introducing the study in stages is likely to limit information overload for families which could impact on their understanding of the trial and decision to participate. An approach could be to provide families with a shorter version of the participant information sheet which highlights the main aspects of the study followed by the full version alongside having adequate clinical and research staff support for any queries or concerns that may arise.
For a time-sensitive trial like IgNiTE, where the study treatment has to be administered within a specific time window, it is important to have a large pool of site staff who are trained in the study procedures available at all times to ensure that eligible patients are approached in a timely fashion, since delays could result in potential participants becoming ineligible for the study. The experience from the IgNiTE trial was that most ward staff nurses did not have good clinical practice (GCP) training and due to the clinical demands and workload in the NHS, it can be challenging to achieve in-depth full GCP training upfront. In IgNiTE, we took a pragmatic approach and provided face-to-face GCP training for site staff through the use of study specific training slides, which meant that ad hoc training could be provided to staff to enable them carry out specific study tasks during their clinical shift. Further strategies that could be considered include the use of training videos or other virtual training platforms so that the clinical staff are able to complete the relevant training at a convenient time. Ultimately, incorporating GCP into routine clinical staff training will undoubtedly be beneficial and will increase the pool of clinical staff that are available to support research trials.
For any RCT, it is crucial that clinicians are in a state of both clinical and personal equipoise. In IgNiTE, some participants who met the inclusion criteria were excluded due to prior receipt or planned administration of IVIG as part of routine care. This observation suggests an increased willingness of clinicians to administer IVIG to children with encephalitis and possibly a perceived lack of equipoise in the use of IVIG in encephalitis. For future trials, appointing trial ambassadors at each site to emphasise the importance of the study, and continued, active engagement of clinical teams are fundamental.
The use of placebo as the control arm in RCTs investigating medicinal products has become commonplace and is necessary to provide a clear picture regarding the efficacy of the treatment being studied. Consideration should be given to the choice of placebo. For IgNiTE, the placebo that was used was a 0.9% saline and 0.1% human albumin mix. Addition of albumin was deemed necessary to make the placebo visually identical to IVIG. However, this resulted in a considerable reduction in shelf life from 3 years for 0.9% saline to 6 months for the final placebo product, so batch manufacturing of the placebo in alignment with the demand from sites was necessary to minimise wastage. However, with the slow recruitment, there was wastage of stock held at the individual sites. For future trials of rare conditions that involve use of an IMP, having a central hub to hold a small number of the study treatment which can be subsequently dispensed to sites depending on demand, could be considered. The success of such strategy will hugely rely on having a robust IMP management process and establishing effective communication lines between study teams.
Strengths and limitations
ImmunoglobuliN in the Treatment of Encephalitis was the first RCT that aimed to evaluate the efficacy of IVIG in children with encephalitis, irrespective of aetiology. The strengths include the expertise of the trial management team, the robust study design, and the use of validated outcome measures. Furthermore, the trial design and oversight were heavily informed by patient and public involvement and there was a strong focus on equality, diversity and inclusion, with children being actively involved in the consent process.
The major limitation of the study was that it was unable to answer the important question regarding the role of IVIG in the treatment of childhood encephalitis, due to not achieving the anticipated sample size.
Implications for health care
Up to half of children with encephalitis experience short to long-term difficulties and there is the unmet need to identify strategies to improve outcomes. There is strong observational evidence to support a beneficial role of IVIG for some forms of encephalitis. However, further research is required to assess the efficacy of IVIG in the context of childhood encephalitis of all causes.
Results of a recent single-arm open-label trial in adults suggest that IVIG improved neurological function in patients with autoimmune encephalitis,62 and a randomised double-blind placebo-controlled trial evaluating the efficacy of IVIG in autoimmune encephalitis in adults in the UK is now under way. 84 However, it is uncertain whether the findings of these adult trials could be reliably translated to autoimmune encephalitis in children, and it the role of early IVIG treatment in infective encephalitis in children remains unanswered.
Recommendations for research
Further studies are needed to determine the precise role of IVIG in encephalitis, and if a treatment effect is established, ascertain the optimum dose, timing of administration and cost-effectiveness. Recommendations on aspects of trial design, conduct and delivery that could be considered for future trials have been highlighted. Consideration should be given to adopting a multinational approach to support timely trial delivery.
Chapter 6 Conclusions
The IgNiTE trial failed to meet recruitment goals therefore the crucial question about the role of IVIG in encephalitis remains unanswered. Given the cost of IVIG and its scarcity, balanced against the significant morbidity that encephalitis places, it is important to answer this question. Future trials are needed to determine the efficacy of IVIG in childhood encephalitis.
Acknowledgements
The study is supported by the NIHR Biomedical Research Centre, which is based at University of Oxford and was coordinated through the Oxford Vaccine Group, University of Oxford. The authors would like to thank all the participants, families, and healthcare professionals for their involvement in the study. The authors would also like to thank CSL Behring for providing the IVIG and funds for manufacturing the placebo.
Contributions of authors
Mildred A Iro (https://orcid.org/0000-0002-9894-6149) (Clinical Research Fellow) was the lead doctor for the trial and prepared the first version, and had input in the design of the trial.
Manish Sadarangani (https://orcid.org/0000-0002-9985-6452) (Paediatric Infectious Diseases and Immunology doctor) is a named investigator on the IgNiTE trial and had input in the design of the trial.
Michael Absoud (https://orcid.org/0000-0002-0577-1897) (Paediatric Neurologist) is a named investigator on the IgNiTE trial and had input in the design of the trial.
Liberty Cantrell (https://orcid.org/0000-0001-5413-7641) (Statistician) conducted analyses for the trial.
Wui K Chong (https://orcid.org/0000-0002-4335-3864) (Neuroradiologist) is a named investigator on the IgNiTE trial.
Christopher Clark (https://orcid.org/0000-0002-8237-6065) (Professor of Neuroimaging) is a named investigator on the IgNiTE trial.
Ava Easton (https://orcid.org/0000-0002-1739-2915) (Chief Executive of The Encephalitis Society) is a named investigator on the IgNiTE trial.
Victoria Gray (https://orcid.org/0000-0002-5828-6369) (Clinical psychologist) is a named investigator on the IgNiTE trial.
Matilda Hill (https://orcid.org/0000-0002-1566-2512) (Paediatric Clinical Research Fellow) prepared subsequent revisions of the manuscript.
Rachael Kneen (https://orcid.org/0000-0002-8729-4122) (Paediatric Neurologist) is a named investigator on the IgNiTE trial and had input in the design of the trial.
Ming Lim (https://orcid.org/0000-0001-7738-8910) (Paediatric Neurologist) is a named investigator on the IgNiTE trial and had input in the design of the trial.
Xinxue Lu (https://orcid.org/0000-0003-1107-0365) (Statistician) conducted analyses for the trial.
Mike Pike (https://orcid.org/0000-0002-9943-3011) (Paediatric Neurologist) conceptualised the trial and is a named investigator on the IgNiTE trial.
Tom Solomon (https://orcid.org/0000-0001-7266-6547) (Professor of Neurology) is a named investigator on the IgNiTE trial and had input in the design of the trial.
Angela Vincent (https://orcid.org/0000-0003-2395-8695) (Professor of Immunology) is a named investigator on the IgNiTE trial.
Louise Willis (https://orcid.org/0000-0003-3115-5964) (Research Nurse) was the lead nurse for the trial.
Ly-Mee Yu (https://orcid.org/0000-0003-0331-7364) (Statistician) is a named investigator on the IgNiTE trial and had input in the design of the trial.
Andrew J Pollard (https://orcid.org/0000-0001-7361-719X) (Professor of Paediatric Infection and Immunity) conceptualised the trial and is a named investigator on the IgNiTE trial.
All authors contributed significantly to the design of the trial, with specific additional contributions from each co-author within their area of expertise. All authors contributed to the manuscript and have approved the final manuscript for publication.
Ignite Study Team
Trial Management Group
Andrew J Pollard (Chief Investigator), Ming Lim, Tom Solomon, Rachel Kneen, Michael Absoud, Mike Pike, Manish Sadarangani, Wui K Chong, Christopher Clarke, Victoria Gray
Trial coordinating team (Oxford)
Mildred A Iro (Clinical Research Fellow), Louise Willis (Research Nurse/Project Manager), David Kerr, Sophie Bradshaw, Svetlana Mica, Simon Kerridge, Emma Plested, Yama Mujadidi
IMP management
Shakeel Herwitker, Mandy Wan
Trial Steering Committee
Claire Cameron, Ming Lim, Adilia Warris, Federico Martinon-Torres, Mike Bale, Sonia Bale, Alan Percival and Ava Easton
Data Monitoring and Ethics Committee
Chris Warlow (Chairperson), Jo Haviland (statistician), David Pace and Simon Nadel
Statistics team
Ly-Mee Yu, Merryn Voysey, Xinxue Liu, Liberty Cantrell
Laboratory team (Oxford)
Sagida Bibi, Amy Beveridge, Amber Thompson, Daniel O’Connor, Sarosh R Irani, Patrick Waters
Neuropsychological assessment support
Lauren Burke, Victoria Gray
Oxford Vaccine Group
Emma Plested, Parvinder Alley
Recruitment sites
| Name of hospital | Principal Investigator |
|---|---|
| Oxford University Hospitals | Andrew J Pollard |
| Great Ormond Street Hospital | Sanjay Bhate |
| Alder Hey Children’s Hospital | Rachel Kneen |
| St George’s Hospital | Paul Heath |
| Barts and the London (Royal London) | Anna Riddell |
| Guy’s and St Thomas’s – Evelina | Ming Lim |
| Sheffield Children’s Hospital | Archana Desurkar |
| NHS Grampian | Elma Stephen |
| Heart of England | Steve Welch |
| Pennine – North Manchester Children’s Hospital | Paddy McMaster |
| Ninewells (Tayside Health Board) | Alice Jollands |
| Royal Hospital for Sick Children, Edinburgh | Jay Shetty |
| University Hospitals Bristol | Kayal Vijayakumar |
| Imperial – St Mary’s Hospital | Leena Mewasingh |
| Nottingham University Hospitals | William Whitehouse |
| Hull Royal Infirmary | Vishal Mehta |
| University Hospitals of North Midland | John Alexander |
| Leeds Teaching Hospital | John Livingston |
| Royal Preston Hospital | Christian de Goede |
| York Teaching Hospital | Dominic Smith |
| Royal Cornwall Hospitals | Andrew Collinson |
Publications
Hill M, Iro M, Sadarangani M, Absoud M, Cantrell L, Chong K, et al.; IgNiTE study team. Intravenous immunoglobulin treatment in childhood encephalitis (IgNiTE): a randomised controlled trial. BMJ Open. 2023 Nov 9;13(11):e072134. https://doi.org/10.1136/bmjopen-2023-072134. PMID: 37945292; PMCID: PMC10649701.
Funding
This award was funded by the National Institute for Health and Care Research (NIHR) Efficacy and Mechanism Evaluation programme (NIHR award ref: 12/212/15) and is published in full in Efficacy and Mechanism Evaluation; Vol. 11, No. 6. See the NIHR Funding and Awards website for further award information. CSL Behring provided the study IMP (IVIG) and funded manufacture of placebo and the supply and distribution of IMP and placebo through an Interlaken Award to the Chief Investigator.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to anonymised data may be granted following review.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that they are stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Ethics statement
This IgNiTE trial was conducted in compliance with the principles of the Declaration of Helsinki (1996), in full conformity with the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Guidelines for Good Clinical Practice (CPMP/ICH/135/95 July 1996), the Research Governance Framework and the Medicines for Human Use (Clinical Trial) Regulations 2004. Ethics approval was granted by the UK National Research Ethics Service (NRES) committee on 29 December 2014 (South Central – Oxford A; REC 14/SC/1416). Clinical trial authorisation granted via the Medicines and Healthcare Products Regulatory Agency (MHRA) notification scheme (Ref: 21584/0337/001-0001). Written approval from local Research and Development (R&D) departments at each participating site was obtained before recruitment was commenced at each site.
Information governance statement
The University of Oxford is committed to handling all personal information in line with the UK Data Protection Act (2018) and the General Data Protection Regulation (EU GDPR) 2016/679. Under the Data Protection legislation, the University of Oxford is the Data Controller, and you can find out more about how we handle personal data, including how to exercise your individual rights and the contact details for our Data Protection Officer here https://compliance.admin.ox.ac.uk/contact.
Department of Health and Social Care disclaimer
This publication presents independent research commissioned by the National Institute for Health and Care Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, MRC, NIHR Coordinating Centre, the EME programme or the Department of Health and Social Care.
This monograph was published based on current knowledge at the time and date of publication. NIHR is committed to being inclusive and will continually monitor best practice and guidance in relation to terminology and language to ensure that we remain relevant to our stakeholders.
Disclaimer
This manuscript presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, the EME programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, the EME programme or the Department of Health and Social Care.
References
- Zuliani L, Graus F, Giometto B, Bien C, Vincent A. Central nervous system neuronal surface antibody associated syndromes: review and guidelines for recognition. J Neurol Neurosurg Psychiatry 2012;83:638-45.
- Granerod J, Ambrose HE, Davies NW, Clewley JP, Walsh AL, Morgan D, et al. UK Health Protection Agency (HPA) Aetiology of Encephalitis Study Group . Causes of encephalitis and differences in their clinical presentations in England: a multicentre, population-based prospective study. Lancet Infect Dis 2010;10:835-44.
- Davison KL, Crowcroft NS, Ramsay ME, Brown DW, Andrews NJ. Viral encephalitis in England, 1989–1998: what did we miss?. Emerg Infect Dis 2003;9:234-40.
- Nightingale S, Rayamajhi A, Bhatta N, Ledger E, Griffiths M, Turtle L, et al. 1636 Intravenous immunoglobulin to treat Japanese encephalitis; a randomised controlled trial in Nepalese children. J Neurol Neurosurg Psychiatry 2012;83.
- Iro MA, Martin NG, Absoud M, Pollard AJ. Intravenous immunoglobulin for the treatment of childhood encephalitis. Cochrane Database Syst Rev 2017;10.
- Kneen R, Michael BD, Menson E, Mehta B, Easton A, Hemingway C, et al. National Encephalitis Guidelines Development and Stakeholder Groups . Management of suspected viral encephalitis in children: Association of British Neurologists and British Paediatric Allergy, Immunology and Infection Group national guidelines. J Infect 2012;64:449-77.
- Thompson C, Kneen R, Riordan A, Kelly D, Pollard AJ. Encephalitis in children. Arch Dis Child 2012;97:150-61.
- Iro MA, Sadarangani M, Goldacre R, Nickless A, Pollard AJ, Goldacre MJ. 30-year trends in admission rates for encephalitis in children in England and effect of improved diagnostics and measles-mumps-rubella vaccination: a population-based observational study. Lancet Infect Dis 2017;17:422-30.
- Mailles A, De Broucker T, Costanzo P, Martinez-Almoyna L, Vaillant V, Stahl JP. Steering Committee and Investigators Group . Long-term outcome of patients presenting with acute infectious encephalitis of various causes in France. Clin Infect Dis 2012;54:1455-64.
- Michaeli O, Kassis I, Shachor-Meyouhas Y, Shahar E, Ravid S. Long-term motor and cognitive outcome of acute encephalitis. Pediatrics 2014;133:e546-52.
- Fowler A, Stödberg T, Eriksson M, Wickström R. Long-term outcomes of acute encephalitis in childhood. Pediatrics 2010;126:e828-35.
- Koskiniemi M, Rautonen J, Lehtokoski-Lehtiniemi E, Vaheri A. Epidemiology of encephalitis in children: a 20-year survey. Ann Neurol 1991;29:492-7.
- Wang IJ, Lee PI, Huang LM, Chen CJ, Chen CL, Lee WT. The correlation between neurological evaluations and neurological outcome in acute encephalitis: a hospital-based study. Eur J Paediatr Neurol 2007;11:63-9.
- Ooi MH, Lewthwaite P, Lai BF, Mohan A, Clear D, Lim L, et al. The epidemiology, clinical features, and long-term prognosis of Japanese encephalitis in central Sarawak, Malaysia, 1997–2005. Clin Infect Dis 2008;47:458-68.
- Britton PN, Khoury L, Booy R, Wood N, Jones CA. Encephalitis in Australian children: contemporary trends in hospitalisation. Arch Dis Child 2016;101:51-6.
- Aygün AD, Kabakuş N, Celik I, Turgut M, Yoldaş T, Gök U, et al. Long-term neurological outcome of acute encephalitis. J Trop Pediatr 2001;47:243-7.
- Bhutto E, Naim M, Ehtesham M, Rehman M, Siddique MA, Jehan I. Prognostic indicators of childhood acute viral encephalitis. J Pak Med Assoc 1999;49:311-6.
- Pillai SC, Hacohen Y, Tantsis E, Prelog K, Merheb V, Kesson A, et al. Infectious and autoantibody-associated encephalitis: clinical features and long-term outcome. Pediatrics 2015;135:e974-84.
- DuBray K, Anglemyer A, LaBeaud AD, Flori H, Bloch K, Joaquin KS, et al. Epidemiology, outcomes and predictors of recovery in childhood encephalitis: a hospital-based study. Pediatr Infect Dis J 2013;32:839-44.
- Elbers JM, Bitnun A, Richardson SE, Ford-Jones EL, Tellier R, Wald RM, et al. A 12-year prospective study of childhood herpes simplex encephalitis: is there a broader spectrum of disease?. Pediatrics 2007;119:e399-407.
- Haglund M, Günther G. Tick-borne encephalitis: pathogenesis, clinical course and long-term follow-up. Vaccine 2003;21:S11-8.
- Ilias A, Galanakis E, Raissaki M, Kalmanti M. Childhood encephalitis in Crete, Greece. J Child Neurol 2006;21:910-2.
- Rismanchi N, Gold JJ, Sattar S, Glaser C, Sheriff H, Proudfoot J, et al. Neurological outcomes after presumed childhood encephalitis. Pediatr Neurol 2015;53:200-6.
- Gau SS, Chang LY, Huang LM, Fan TY, Wu YY, Lin TY. Attention-deficit/hyperactivity-related symptoms among children with enterovirus 71 infection of the central nervous system. Pediatrics 2008;122:e452-8.
- Baruah HC, Biswas D, Patgiri D, Mahanta J. Clinical outcome and neurological sequelae in serologically confirmed cases of Japanese encephalitis patients in Assam, India. Indian Pediatr 2002;39:1143-8.
- Khandaker G, Jung J, Britton PN, King C, Yin JK, Jones CA. Long-term outcomes of infective encephalitis in children: a systematic review and meta-analysis. Dev Med Child Neurol 2016;58:1108-15.
- Pillai SC, Mohammad SS, Hacohen Y, Tantsis E, Prelog K, Barnes EH, et al. Postencephalitic epilepsy and drug-resistant epilepsy after infectious and antibody-associated encephalitis in childhood: clinical and etiologic risk factors. Epilepsia 2016;57:e7-11.
- Hughes RA, Dalakas MC, Cornblath DR, Latov N, Weksler ME, Relkin N. Clinical applications of intravenous immunoglobulins in neurology. Clin Exp Immunol 2009;158:34-42.
- FDA US . Immune Globulin Intravenous (IVIG) Indications 2018 2018. https://www.fda.gov/vaccines-blood-biologics/approved-blood-products/immune-globulin-intravenous-igiv-indications (accessed 31 July 2023).
- NHS-England . Commissioning Criteria Policy for the Use of Therapeutic Immunoglobulin (Ig) England, 2021 2021.
- NHS-England . Updated Commissioning Guidance for the Use of Therapeutic Immunoglobulin (Ig) in Immunology, Haematology, Neurology and Infectious Diseases in England December 2018 2018. https://www.england.nhs.uk/wp-content/uploads/2019/03/PSS9-Immunoglobulin-Commissioning-Guidance-CQUIN-1920.pdf (accessed 31 July 2023).
- Looney RJ, Huggins J. Use of intravenous immunoglobulin G (IVIG). Best Pract Res Clin Haematol 2006;19:3-25.
- Basta M, Illa I, Dalakas MC. Increased in vitro uptake of the complement C3b in the serum of patients with Guillain-Barré syndrome, myasthenia gravis and dermatomyositis. J Neuroimmunol 1996;71:227-9.
- Basta M, Van Goor F, Luccioli S, Billings EM, Vortmeyer AO, Baranyi L, et al. F(ab)‘2-mediated neutralization of C3a and C5a anaphylatoxins: a novel effector function of immunoglobulins. Nat Med 2003;9:431-8.
- Lutz HU, Stammler P, Jelezarova E, Nater M, Späth PJ. High doses of immunoglobulin G attenuate immune aggregate-mediated complement activation by enhancing physiologic cleavage of C3b in C3bn-IgG complexes. Blood 1996;88:184-93.
- Mollnes TE, Høgåsen K, Hoaas BF, Michaelsen TE, Garred P, Harboe M. Inhibition of complement-mediated red cell lysis by immunoglobulins is dependent on the IG isotype and its C1 binding properties. Scand J Immunol 1995;41:449-56.
- Crow AR, Song S, Semple JW, Freedman J, Lazarus AH. A role for IL-1 receptor antagonist or other cytokines in the acute therapeutic effects of IVIg?. Blood 2007;109:155-8.
- Stangel M, Schumacher HC, Ruprecht K, Boegner F, Marx P. Immunoglobulins for intravenous use inhibit TNF alpha cytotoxicity in vitro. Immunol Invest 1997;26:569-78.
- Dietrich G, Kazatchkine MD. Normal immunoglobulin G (IgG) for therapeutic use (intravenous Ig) contain antiidiotypic specificities against an immunodominant, disease-associated, cross-reactive idiotype of human anti-thyroglobulin autoantibodies. J Clin Invest 1990;85:620-5.
- Rossi F, Sultan Y, Kazatchkine MD. Anti-idiotypes against autoantibodies and alloantibodies to VIII:C (anti-haemophilic factor) are present in therapeutic polyspecific normal immunoglobulins. Clin Exp Immunol 1988;74:311-6.
- Dalakas MC. The use of intravenous immunoglobulin in the treatment of autoimmune neuromuscular diseases: evidence-based indications and safety profile. Pharmacol Ther 2004;102:177-93.
- Kazatchkine MD, Kaveri SV. Immunomodulation of autoimmune and inflammatory diseases with intravenous immune globulin. N Engl J Med 2001;345:747-55.
- Caramello P, Canta F, Balbiano R, Lipani F, Ariaudo S, De Agostini M, et al. Role of intravenous immunoglobulin administration in Japanese encephalitis. Clin Infect Dis 2006;43:1620-1.
- Dalakas MC. Mechanism of action of intravenous immunoglobulin and therapeutic considerations in the treatment of autoimmune neurologic diseases. Neurology 1998;51:S2-8.
- Ramakrishna C, Openshaw H, Cantin EM. The case for immunomodulatory approaches in treating HSV encephalitis. Future Virol 2013;8:259-72.
- Tha-In T, Bayry J, Metselaar HJ, Kaveri SV, Kwekkeboom J. Modulation of the cellular immune system by intravenous immunoglobulin. Trends Immunol 2008;29:608-15.
- Trinath J, Hegde P, Sharma M, Maddur MS, Rabin M, Vallat JM, et al. Intravenous immunoglobulin expands regulatory T cells via induction of cyclooxygenase-2-dependent prostaglandin E2 in human dendritic cells. Blood 2013;122:1419-27.
- Frenzel K, Ganepola S, Michel D, Thiel E, Krüger DH, Uharek L, et al. Antiviral function and efficacy of polyvalent immunoglobulin products against CMV isolates in different human cell lines. Med Microbiol Immunol 2012;201:277-86.
- Kishimoto C, Hiraoka Y, Takada H. Effects of immunoglobulin upon murine myocarditis caused by influenza A virus: superiority of intact type to F(ab’)2 type. J Cardiovasc Pharmacol 2004;43:61-7.
- Krause I, Wu R, Sherer Y, Patanik M, Peter JB, Shoenfeld Y. In vitro antiviral and antibacterial activity of commercial intravenous immunoglobulin preparations: a potential role for adjuvant intravenous immunoglobulin therapy in infectious diseases. Transfus Med 2002;12:133-9.
- Saker A, Athman S, Aldosari M, Frayha H. Encephalopathy and axonal neuropathy associated with Mycoplasma pneumoniae infection: response to intravenous immunoglobulin therapy. Child Neurol Open 2016;3.
- Chambert-Loir C, Ouachee M, Collins K, Evrard P, Servais L. Immediate relief of Mycoplasma pneumoniae encephalitis symptoms after intravenous immunoglobulin. Pediatr Neurol 2009;41:375-7.
- Van Maldergem L MF, Ureel D, Jauniaux E, Broeckx W, Vainsel M. Echovirus meningoencephalitis in X‐linked hypogammaglobulinemia. Acta Paediatr Scand 1989;78:325-6.
- Rayamajhi A, Nightingale S, Bhatta NK, Singh R, Kneen R, Ledger E, et al. A preliminary randomized double blind placebo-controlled trial of intravenous immunoglobulin for Japanese encephalitis in Nepal. PLOS ONE 2015;10.
- Casas-Alba D, de Sevilla MF, Valero-Rello A, Fortuny C, Garcia-Garcia JJ, Ortez C, et al. Outbreak of brainstem encephalitis associated with enterovirus-A71 in Catalonia, Spain (2016): a clinical observational study in a children’s reference centre in Catalonia. Clin Microbiol Infect 2017;23:874-81.
- Bhatt GC, Sankar J, Kushwaha KP. Use of intravenous immunoglobulin compared with standard therapy is associated with improved clinical outcomes in children with acute encephalitis syndrome complicated by myocarditis. Pediatr Cardiol 2012;33:1370-6.
- Byun JE, Lee KY. Effectiveness of intravenous immunoglobulin therapy for pediatric viral encephalitis. Annal Child Neurol 2020;28:145-55.
- Titulaer MJ, McCracken L, Gabilondo I, Armangué T, Glaser C, Iizuka T, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013;12:157-65.
- Armangue T, Titulaer MJ, Málaga I, Bataller L, Gabilondo I, Graus F, et al. Spanish Anti-N-methyl-D-Aspartate Receptor (NMDAR) Encephalitis Work Group . Pediatric anti-N-methyl-D-aspartate receptor encephalitis-clinical analysis and novel findings in a series of 20 patients. J Pediatr 2013;162:850.e-856.e2.
- Chakrabarty B, Tripathi M, Gulati S, Yoganathan S, Pandit AK, Sinha A, et al. Pediatric anti-N-methyl-D-aspartate (NMDA) receptor encephalitis: experience of a tertiary care teaching center from North India. J Child Neurol 2014;29:1453-9.
- Pandit AK, Ihtisham K, Garg A, Gulati S, Padma MV, Tripathi M. Autoimmune encephalitis: a potentially reversible cause of status epilepticus, epilepsy, and cognitive decline. Ann Indian Acad Neurol 2013;16:577-84.
- Lee ST, Lee HS, Lee WJ, Cha HA, Kim SH, Shin SY, et al. The safety and efficacy of intravenous immunoglobulin in autoimmune encephalitis. Ann Clin Transl Neurol 2022;9:610-21.
- Nosadini M, Eyre M, Molteni E, Thomas T, Irani SR, Dalmau J, et al. International NMDAR Antibody Encephalitis Consensus Group . Use and safety of immunotherapeutic management of N-methyl-d-aspartate receptor antibody encephalitis: a meta-analysis. JAMA Neurol 2021;78:1333-44.
- Bien CG, Tiemeier H, Sassen R, Kuczaty S, Urbach H, von Lehe M, et al. Rasmussen encephalitis: incidence and course under randomized therapy with tacrolimus or intravenous immunoglobulins. Epilepsia 2013;54:543-50.
- Haberlandt E, Bast T, Ebner A, Holthausen H, Kluger G, Kravljanac R, et al. Limbic encephalitis in children and adolescents. Arch Dis Child 2011;96:186-91.
- Chou IJ, Wang HS, Lin JJ, Kuo CF, Lin KL, Chou ML, et al. CHEESE Study Group . Limbic encephalitis in Taiwanese children and adolescence: a single center study. Pediatr Neonatol 2013;54:246-53.
- Mariotti P, Batocchi AP, Colosimo C, Lo Monaco M, Caggiula M, Colitto F, et al. Multiphasic demyelinating disease involving central and peripheral nervous system in a child. Neurology 2003;60:348-9.
- Assa A, Watemberg N, Bujanover Y, Lerman-Sagie T. Demyelinative brainstem encephalitis responsive to intravenous immunoglobulin therapy. Pediatrics 1999;104:301-3.
- Andersen JB, Rasmussen LH, Herning M, Paerregaard A. Dramatic improvement of severe acute disseminated encephalomyelitis after treatment with intravenous immunoglobulin in a three-year-old boy. Dev Med Child Neurol 2001;43:136-8.
- Straussberg R, Schonfeld T, Weitz R, Karmazyn B, Harel L. Improvement of atypical acute disseminated encephalomyelitis with steroids and intravenous immunoglobulins. Pediatr Neurol 2001;24:139-43.
- Kleiman M, Brunquell P. Acute disseminated encephalomyelitis: response to intravenous immunoglobulin. J Child Neurol 1995;10:481-3.
- İncecik F, Hergüner M, Altunbaşak S. Acute disseminated encephalomyelitis: an evaluation of 15 cases in childhood. Turk J Pediatr 2013;55:253-9.
- Erol I, Ozkale Y, Alkan O, Alehan F. Acute disseminated encephalomyelitis in children and adolescents: a single center experience. Pediatr Neurol 2013;49:266-73.
- Nishikawa M, Hayashi T, Ouchi K, Furukawa S. Intravenous immunoglobulin therapy in acute disseminated encephalomyelitis. Pedatr Neurol 1999;21:583-6.
- Pradhan S, Gupta RP, Shashank S, Pandey N. Intravenous immunoglobulin therapy in acute disseminated encephalomyelitis. J Neurol Sci 1999;165:56-61.
- Sadek AA, Mohamed MA, Abou-Taleb A, Mohammed MI. Pattern and outcome of acute disseminated encephalomyelitis (ADEM) in children: experience in a tertiary center, upper Egypt. Electron Physician 2016;8:2679-85.
- Shahar E, Andraus J, Savitzki D, Pilar G, Zelnik N. Outcome of severe encephalomyelitis in children: effect of high-dose methylprednisolone and immunoglobulins. J Child Neurol 2002;17:810-4.
- Nosadini M, Boniver C, Zuliani L, de Palma L, Cainelli E, Battistella PA, et al. Longitudinal electroencephalographic (EEG) findings in pediatric anti-N-methyl-D-aspartate (anti-NMDA) receptor encephalitis: the Padua experience. J Child Neurol 2015;30:238-45.
- Nosadini M, Mohammad SS, Suppiej A, Sartori S, Dale RC, Group INS. Intravenous immunoglobulin in paediatric neurology: safety, adherence to guidelines, and long-term outcome. Dev Med Child Neurol 2016;58:1180-92.
- Pritchard J, Hughes RAC. Intravenous immunoglobulin-how to use it. Pract Neurol 2001;93.
- Iro MA, Sadarangani M, Absoud M, Chong WK, Clark CA, Easton A, et al. ImmunoglobuliN in the Treatment of Encephalitis (IgNiTE): protocol for a multicentre randomised controlled trial. BMJ Open 2016;6.
- Venkatesan A, Tunkel AR, Bloch KC, Lauring AS, Sejvar J, Bitnun A, et al. International Encephalitis Consortium . Case definitions, diagnostic algorithms, and priorities in encephalitis: consensus statement of the international encephalitis consortium. Clin Infect Dis 2013;57:1114-28.
- Kahwaji J, Barker E, Pepkowitz S, Klapper E, Villicana R, Peng A, et al. Acute hemolysis after high-dose intravenous immunoglobulin therapy in highly HLA sensitized patients. Clin J Am Soc Nephrol 2009;4:1993-7.
- ISRCTN-Registry . Treatment of Autoimmune Encephalitis in Adults With Intravenous Immunoglobulin (ISRCTN69615004) 2020. https://www.isrctn.com/ISRCTN69615004 (accessed 31 July 2023).
Appendix 1 Schedule of visits
| T0 | T1 | T1 + 24h | T2 | T2 + 24-48h | T2 + 7d | T3 | T4 | T5 | T6 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Eligibility assessment | X | |||||||||
| Informed consenta | X | Xb | Xb | Xb | ||||||
| Confirm consent | X | X | X | |||||||
| Demographics | X | |||||||||
| Medical history | X | X | X | X | X | X | ||||
| Obtain relevant clinical information | X | X | X | X | X | X | X | |||
| Enrolment | X | Xc | ||||||||
| Randomisation | X | Xc | ||||||||
| Scavenged samplesd | X | X | X | X | X | X | X | X | ||
| Additional (research) sample if consent obtained) | X | Xc | X | Xc | X | Xe | ||||
| Mandatory full blood count | Xc,f | |||||||||
| Study drug administration and monitoring | X | X | ||||||||
| SAE assessment | X | X | X | X | X | X | Xg | |||
| Completion of research notes and CRFd | X | X | X | X | X | X | X | |||
| Questionnaire completions | X | X | X | |||||||
| Research MRI scan (if consent obtained)h | X | |||||||||
| Neuropsychologist assessment | X |
Appendix 2 Recruiting hospital sites, duration they were open and estimate vs. actual recruitment
| Site | Number of whole months open | Estimated recruitment for number of months open | Actual recruitment |
|---|---|---|---|
| Oxford University Hospitals | 21 | 7–8 | 3 |
| Great Ormond Street Hospital | 19 | 2–3 | 1 |
| Alder Hey Children’s Hospital | 18 | 11–12 | 3 |
| St George’s Hospital | 18 | 4–5 | 1 |
| Barts and the London (Royal London) | 17 | 2–3 | 0 |
| Guy’s and St Thomas’s – Evelina | 16 | 10–11 | 2 |
| Sheffield Children’s Hospital | 16 | 1–2 | 1 |
| NHS Grampian | 16 | 4–5 | 2 |
| Heart of England | 16 | 3–4 | 1 |
| Pennine – North Manchester Children’s Hospital | 15 | 4–5 | 1 |
| Ninewells (Tayside Health Board) | 15 | 0–1 | 0 |
| Royal Hospital for Sick Children, Edinburgh | 14 | 2–3 | 0 |
| University Hospitals Bristol | 13 | 3–4 | 0 |
| Imperial – St Mary’s Hospital | 11 | 5–6 | 1 |
| Nottingham University Hospitals | 11 | 1–2 | 1 |
| Hull Royal Infirmary | 7 | 1–2 | 1 |
| University Hospitals of North Midland | 6 | 1–2 | 0 |
| Leeds Teaching Hospital | 6 | 2–3 | 0 |
| Royal Preston Hospital | 5 | 0–1 | 0 |
| York Teaching Hospital | 4 | 0–1 | 0 |
| Royal Cornwall Hospitals | 2 | 1–2 | 0 |
Appendix 3 Hospital sites intended to be opened, site opening times and date of first recruitment across sites
| No. | Site | SIV date | Date opened | Date of 1st recruit |
|---|---|---|---|---|
| 01 | Oxford University Hospitals | 26 August 2015 | 23 December 2015 | 24 February 2016 |
| 02 | Guy’s and St Thomas’s – Evelina | 6 October 2015 | 19 May 2016 | 30 November 2016 |
| 03 | St George’s Hospital | 12 October 2015 | 22 May 2016 | 27 October 2016 |
| 04 | Alder Hey Children’s Hospital | 16 November 2015 | 30 March 2016 | 19 May 2017 |
| 05 | University Hospitals Bristol | 24 November 2015 | 25 August 2016 | N/A |
| 06 | North Manchester Children’s Hospital | 30 November 2015 | 30 June 2016 | 19 May 2017 |
| 07 | University Hospital Southampton | 10 December 2015 | N/A | N/A |
| 08 | Great Ormond Street Hospital | 14 December 2015 | 1 March 2016 | 21 September 2016 |
| 09 | Barts and the London (Royal London) | 15 January 2016 | 4 May 2016 | N/A |
| 10 | Sheffield Children’s Hospital | 14 January 2016 | 26 May 2016 | 25 May 2017 |
| 11 | NHS Grampian | 24 February 2016 | 7 June 2016 | 24 November 2016 |
| 12 | Ninewells (Tayside Health Board) | 25 February 2016 | 17 June 2016 | N/A |
| 13 | Heart of England | 2 March 2016 | 1 June 2016 | 15 February 2017 |
| 14 | South Tees – James Cook Hospital | 7 March 2016 | N/A | N/A |
| 15 | Birmingham Children’s Hospital | 21 March 2016 | N/A | N/A |
| 16 | Royal Hospital for Sick Children, Edinburgh | 21 April 2016 | 22 July 2016 | N/A |
| 17 | Imperial – St Mary’s Hospital | 20 June 2016 | 11 October 2016 | 13 February 2017 |
| 18 | Cambridge – Addenbrooke’s | 9 August 2016 | N/A | N/A |
| 19 | Nottingham University Hospitals | 1 September 2016 | 7 November 2016 | 22 November 2016 |
| 20 | University Hospitals of North Midland | 19 September 2016 | 9 March 2017 | N/A |
| 21 | Hull Royal Infirmary | 27 October 2016 | 9 February 2017 | 27 February 2017 |
| 22 | Leeds Teaching Hospital | 24 October 2016 | 16 March 2017 | N/A |
| 23 | York Teaching Hospital | 31 October 2016 | 11 May 2017 | N/A |
| 24 | Central Manchester University Hospital | 29 November 2016 | N/A | N/A |
| 25 | Royal Cornwall Hospitals | 15 December 2016 | 18 July 2017 | N/A |
| 26 | Royal Preston Hospital | 26 January 2017 | 13 April 2017 | N/A |
| 27 | Leicester Royal Infirmary | 22 August 2017 | N/A | N/A |
| 28 | Royal Berkshire Hospital | 09 June 2017 | N/A | N/A |
| 29 | Royal Exeter and Devon | 30 June 2017 | N/A | N/A |
List of abbreviations
- ABAS-II
- Adaptive Behaviors Assessment System-Second Edition
- ADEM
- acute demyelinating encephalomyelitis
- AE
- adverse event
- AESI
- adverse event of special interest
- AR
- adverse reaction
- CI
- chief investigator
- CONSORT
- Consolidated Standards of Reporting Trials
- CRF
- case report form
- CSF
- cerebrospinal fluid
- CT
- clinical trials
- FSIQ
- Full-Scale IQ
- GCP
- good clinical practice
- GMFCS
- gross motor function classification system
- GOS-E
- Glasgow Outcome Scale-Extended
- ICH
- international conference of harmonisation
- ICU
- intensive care unit
- ID
- identification
- IgNiTE
- ImmunoglobuliN in the Treatment of Encephalitis
- IMP
- investigational medicinal product
- ITT
- intention-to-treat
- IVIG
- intravenous immunoglobulin
- IQR
- interquartile range
- JE
- Japanese encephalitis
- LOS
- Liverpool Outcome Score
- MHRA
- Medicines and Healthcare products Regulatory Agency
- MRI
- magnetic resonance imaging
- MOG
- myelin oligodendrocyte glycoprotein
- NHS
- National Health Service
- NIHR
- National Institute for Health and Care Research
- NMDAR
- N-methyl-D-aspartate receptor
- NRES
- National Research Ethics Service
- PedsQL
- Paediatric Quality of Life
- PI
- principal investigator
- PRI
- Perceptual Reasoning Index
- PSI
- Processing Speed Index
- RCT
- Randomised controlled trial
- REC
- Research Ethics Committee
- SAE
- serious adverse event
- SAR
- serious adverse reaction
- SDQ
- Strengths and Difficulties Questionnaire
- TMF
- trial master file
- TSC
- Trial Steering Committee
- VCI
- Verbal Comprehension Index
- VSI
- Visual Spatial Index
- WCC
- white cell count
- WMI
- working memory index