

Notes
Article history
The research reported in this issue of the journal was funded by the EME programme as project number 12/165/31. The contractual start date was in January 2015. The final report began editorial review in September 2020 and was accepted for publication in July 2021. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The EME editors and production house have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the final report document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
Copyright © 2022 Arndtz et al. This work was produced by Arndtz et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This is an Open Access publication distributed under the terms of the Creative Commons Attribution CC BY 4.0 licence, which permits unrestricted use, distribution, reproduction and adaption in any medium and for any purpose provided that it is properly attributed. See: https://creativecommons.org/licenses/by/4.0/. For attribution the title, original author(s), the publication source – NIHR Journals Library, and the DOI of the publication must be cited.
2022 Arndtz et al.
Chapter 1 Introduction
Scientific background and rationale
End-stage liver disease, regardless of aetiology, is characterised by progressive hepatic fibrosis, culminating in liver cirrhosis and accompanying increased risks of liver cancer, liver failure, portal hypertension and death. Preventing progressive liver fibrosis represents an important area of interest in the development of new drugs suitable for all patients with liver disease. Primary sclerosing cholangitis (PSC) is a prime example of a progressive inflammatory liver disease and is characterised by persistent liver fibrosis and a high unmet need for new therapies.
Primary sclerosing cholangitis
Primary sclerosing cholangitis has a population incidence of 1.3 per 100,000 people annually, with a prevalence of 16.2 per 100,000 people. 1–3 It affects both men and women, with a median age at onset of 41 years,4 and is associated with inflammatory bowel disease (IBD) in 80% of cases. 5 More than 50% of patients require liver transplantation within 10–15 years of symptomatic presentation,6,7 reflecting the failure of medical therapies to have any impact on the clinical outcome. In the UK, for example, PSC is now the leading autoimmune liver disease indication for transplant, despite being the rarest of the autoimmune liver diseases. One barrier to the development of efficacious new medical therapies is the lack of clinically relevant end points, and there is an urgent need to develop appropriate non-invasive surrogate end points to improve clinical trial design. 8 PSC is a progressive immune-mediated biliary disease characterised by bile duct inflammation, fibrosis and accompanying hepatic fibrosis. For patients with elevated alkaline phosphatase (ALP) activity, in particular, progressive disease is predicted, which currently results in a need for liver transplantation in the majority of cases. No current medical therapy has yet been shown to be effective in altering the natural process of the disease. For this reason, patients with PSC with elevated ALP activity were to be recruited to this trial to evaluate the impact of vascular adhesion protein 1 (VAP-1) blockade by BTT1023 in an early-phase trial focused on biochemical efficacy and safety.
Primary sclerosing cholangitis and vascular adhesion protein 1
Vascular adhesion protein 1 is a 170-kDa homodimeric type 2 transmembrane sialoglycoprotein with a short cytoplasmic tail of no known signal sequence, a single transmembrane segment and a large extracellular domain. VAP-1 is constitutively expressed on human hepatic endothelium and supports lymphocyte adhesion and transendothelial migration. Cloning of VAP-1 revealed it to be a copper-dependent semicarbazide-sensitive amine oxidase (SSAO) that catalyses the oxidative deamination of exogenous and endogenous primary amines, resulting in the generation of aldehyde, ammonia and hydrogen peroxide (H2O2). These products activate nuclear factor kappa B (NF-κB)-dependent chemokine secretion and adhesion molecule expression in liver endothelium and may initiate and propagate oxidative stress following the conversion of H2O2 to hydroxyl free radicals. A soluble form of VAP-1 accounts for nearly all of the circulating amine oxidase activity in humans. 9
The progression of PSC to scarring, cirrhosis and hepatobiliary cancer is driven by a chronic inflammatory response and immune cell-mediated destruction of bile ducts. 10 Our research implicates VAP-1 in the inflammation that drives fibrogenesis in liver disease. 11 VAP-1 also acts as an adhesion receptor to support leucocyte recruitment in liver inflammation, a function that is critical in the formation of fibrosis in animal models. 12 Therefore, inhibition of VAP-1 is expected to have an impact on both inflammation and fibrosis. Indeed, treatment with an antibody against VAP-1 prevents fibrosis in murine models of liver injury. 9 Data also show that there are particularly high levels of circulating serum vascular adhesion protein 1 (sVAP-1) in patients with PSC, and there is a strong correlation between sVAP-1/SSAO activity in serum and histological fibrosis scores in patients with fatty liver disease. 9 Based on the strong up-regulation of hepatic VAP-1 reported in PSC patients,9 we hypothesise that levels of sVAP-1/SSAO will correlate with the severity of fibrosis in PSC and will predict patients at risk of progressive disease.
These observations underpin our proposal that VAP-1 has an important role in the progression of liver fibrosis. The BUTEO trial set out to test the hypothesis that inhibiting VAP-1 with a neutralising antibody (BTT1023) will reverse or delay fibrogenesis in patients with PSC. In addition, reliable biomarkers that correlate with fibrosis stage and progression of liver disease are in demand to predict outcome and to stage disease, without the need for invasive liver biopsy. This research will allow us to translate laboratory research into a proof of activity clinical trial that will elucidate the role of VAP-1 in liver fibrosis and its potential as a therapeutic target and biomarker.
BTT1023 (now known as timolumab)
BTT1023 is a fully human monoclonal anti-VAP-1 antibody that blocks the adhesion function of VAP-1, thereby diminishing leucocyte entry into sites of tissue inflammation. BTT1023 will in the future be known as timolumab; however, the original numeric name is used in this manuscript to remain in keeping with the trial protocol. In vivo, blocking VAP-1 function with an anti-mouse anti-VAP-1 antibody significantly alleviates inflammation in mouse models of arthritis and liver fibrosis. 13 BTT1023 appears to be safe and well tolerated in humans. BTT1023 has been given in doses up to 8 mg/kg in patients with rheumatoid arthritis and psoriasis after oral premedication (with cetirizine and ibuprofen), and also appears safe and well tolerated in repeated intravenous (i.v.) dosing. 14 No cytokine release syndrome has been reported. Therefore, this dose appears reasonable as a starting point and is considered to provide appropriate bridging into existing clinical data.
In the well-tolerated arthritis trial,14 patients received five doses at fortnightly intervals, with potentially efficacious trough levels being achieved by day 42 (after three doses). The psoriasis trial used a more rapid loading of the three doses at days 1, 8 and 22, which was also well tolerated (Biotie Therapies, personal communication, 2014). Therefore, we decided to deploy the accelerated dosing scheme (i.e. dosing on days 1 and 8 and biweekly thereafter) in the current trial. 14
BTT1023 is produced in a Chinese hamster ovary cell culture and purified with appropriate methods, including specific viral inactivation and removal procedures. BTT1023 is contained in single-use 100-mg/10-ml glass vials and requires dilution with BTT1023 i.v. infusion diluent (0.9% sodium chloride and 0.02% polysorbate 80 in water for injection) prior to administration by i.v. infusion. The amount of diluent to be added to the BTT1023 concentrate is calculated to consistently provide a total diluted drug product infusion volume of 50 ml. BTT1023 and its diluent are stored at 2–8 °C and the maximum shelf life for the diluted infusion solution is 24 hours when stored at this temperature.
Chapter 2 Methods
Trial design
The BUTEO trial was a two-stage, single-arm, open-label, multicentre hybrid trial of treatment with monoclonal anti-VAP-1 antibody BTT1023 in adult patients with PSC. The sample size was 59 patients, each of whom received up to seven i.v. infusions of BTT1023 over a 78-day treatment period. All patients were followed up until day 120 (42 days after the last administration of treatment). At specified time points during each visit, serum was taken for measurement of circulating levels of BTT1023, as well as anti-drug antibodies and VAP-1 activity, and to provide samples for additional exploratory research. These specified time points included pre dose, during dose and post dose, with some patients also attending 24 hours later for further blood testing. The trial composed two components: (1) a run-in dose confirmation period and (2) a single-arm Simon’s two-stage expansion.
Phase I: dose confirmation
The run-in component of the trial incorporated a conventional 3 + 3 cohort design to confirm the therapeutic dose, with decisions regarding continuation based on toxicity and pharmacokinetic (PK) data (see Report Supplementary Material 1, Figure S1). The trial began with the recruitment of six patients, all of whom received the starting dose of 8 mg/kg. Recruitment was paused while awaiting the results of trough blood serum levels of circulating BTT1023 at visit 7 (i.e. day 50) from all six patients and until the dose-limiting toxicity (DLT) reporting period was completed for each patient [visit 10 (i.e. day 99)].
If results from the first cohorts showed an acceptable DLT rate (see later) and trough levels of BTT1023 met the stipulated success criteria, the trial was to continue into the expansion component (see Phase II: Simon’s two-stage minimax design). Acceptable trough levels were set at 3 µg/ml free-circulating BTT1023 at 8 weeks from the first infusion, which is approximately 100-fold the dissociation constant (Kd) of BTT1023 from VAP-1 and will result in target occupancy of approximately 90%. In the event that the DLT rate was acceptable but the PK values did not meet the success criteria, then the trial was to move into a conventional 3 + 3 cohort design, using escalating doses of BTT1023. In this event, the original cohort of six patients would no longer be evaluated, but a new cohort of three patients would be recruited to receive the newly identified test dose of 12 mg/kg. If there were no DLTs at visit 10 (i.e. day 99), an additional cohort of three patients would be recruited at the new test dose. If the DLT rate remained acceptable but the PK values still did not meet the success criteria, a further 3 + 3 patients would be recruited at the highest dose of 16 mg/kg. If this was found not to result in sufficient blood levels of BTT1023, then the trial would be stopped. If the PK values were found to be too high (e.g. if trough levels consistently exceeded 100 µg/ml), then there was potential to de-escalate the dose in agreement with the Data Monitoring Committee (DMC) and after regulatory approvals. The trial was to be stopped at any stage if patient safety was compromised. Individual patients would receive only one dose level. Once a confirmed dose had been established, the trial was expanded until a total of 37 patients had received treatment with that dose, including those patients who had previously received this dose during the confirmatory period. Those patients not receiving the confirmed dose were not included in the final evaluation.
Figure S1 in Report Supplementary Material 1 shows the trial design.
Phase II: Simon’s two-stage minimax design
The Phase II Simon’s two-stage minimax design centres around the hypothesis test for the trial’s primary outcome. The null hypothesis was that the response rate for PSC patients receiving BTT1023 is < 15% (P0). The alternative hypothesis was that the response rate for PSC patients receiving BTT1023 is > 30% (P1).
A response was considered to be a reduction in inflammation, indicated by reduction in serum ALP activity of ≥ 25% (a comparison of measured ALP activity from baseline to day 99).
The baseline measurements were recorded at pre-infusion visit 3, on day 1 of treatment and on day 99 (i.e. 21 days following last infusion).
The two-stage design incorporated an interim analysis of the accumulating data after 18 patients had received the trial treatment. At this point, the design required that at least 3 of 18 patients had a successful response (i.e. a reduction in ALP activity of ≥ 25%) to allow the trial to continue. If the stage 1 criterion was not met, and fewer than three evaluable patients had a successful response, then the trial was to stop early. However, if the criterion was met, then further patient recruitment was to continue until 37 evaluable patients were recruited. The final design required at least 9 of 37 patients to have a successful response to conclude that the trial treatment successfully met the trial’s primary outcome.
Patients with particularly elevated ALP activity are predicted to be more at risk of progressive disease. Therefore, these patients were selected for trial participation. Six UK academic hospital centres were involved, with four actively recruiting. (Those centres that were actively recruiting were based in Birmingham, Nottingham, Oxford and London, and the centres that failed to recruit were based in Newcastle and Cambridge.) Informed consent was obtained by appropriately trained members of the research team at each site.
A single-arm, rather than placebo-controlled, design was chosen to allow efficient enrolment of patients into the trial because of its intensive nature, as a significant chance of being allocated to the placebo group may have acted as a substantial barrier to enrolment. There was no concurrent control group as the proposed primary end point (ALP activity) was a biochemical measurement and, therefore, not open to subjective bias as a clinical assessment would be.
Given the unpredictable nature of PSC and natural variation of ALP activity, the process of screening for eligibility took place in two stages, with one screening visit 5–7 weeks before beginning infusion and another within 1 week of beginning infusion. Potential participants were eligible for the trial if their ALP activity did not vary by > 25% over this period. During screening, patients underwent routine blood screening and other non-invasive markers of liver fibrosis measured, including Mayo PSC risk score, model for end-stage liver disease (MELD) score, enhanced liver fibrosis (ELF) testing, a FibroScan (Echosens, Paris, France) and magnetic resonance cholangiopancreatography with additional LiverMultiScan® (Perspectum® Ltd, Oxford, UK) imaging. These assessments were repeated during treatment and in the follow-up period to determine if any change had occurred. During all seven treatment visits, patients received premedication with 10 mg of cetirizine plus 400 mg of ibuprofen orally (in the absence of any contraindications) plus 100 mg of i.v. hydrocortisone, 1–2 hours pre infusion (the last for the first three doses only). The first infusion was given over 2 hours, with a 4-hour monitoring period post infusion. Provided that no adverse reactions were seen, the infusion time dropped to 1 hour with an initial 3-hour observation period (for the second dose) and then down to 2 hours’ monitoring post infusion (for all subsequent doses). Safety investigations were completed pre and post infusion, and included haematological/biochemical testing of blood samples, electrocardiography, clinical assessment and physical examination. The full trial schema can be seen as Figure S2 in Report Supplementary Material 1. An aliquot (0.5–1.0 ml) of the BTT1023 infusion solution was taken at the end of every infusion and refrigerated. These samples could be used for analysis of BTT1023 concentration if anomalies in PK data that could be due to errors in BTT1023 preparation were observed.
Trial conduct
The BUTEO trial was a clinician-initiated and clinician-led trial funded by the National Institute for Health Research (NIHR), which receives funds directly from the Department of Health and Social Care. Biotie Therapies Ltd (Helsinki, Finland) supplied BTT1023 free of charge to all individual NHS trusts that were directly treating patients as part of this clinical trial. Payments were made to the individual NHS trusts on a per-patient basis to cover trial running costs. The University of Birmingham (Birmingham, UK) was the trial sponsor and, as such, was responsible for the trial conduct. The trial was conducted under the auspices of the Cancer Research UK Clinical Trials Unit (CRCTU), University of Birmingham, in accordance with its local standard operating procedures. It was managed with a Trial Management Group, together with a Trial Steering Committee (TSC) and an independent DMC. Monitoring was performed by CRCTU. The trial was registered with the European Medicines Agency (as EudraCT 2014-002393-37), NIHR (Portfolio ID 18051) and International Standard Randomised Controlled Trial Number (ISRCTN) Registry (as ISRCTN11233255). The ClinicalTrials.gov identifier was NCT02239211.
Patient and public involvement
A local liver disease patient and public involvement group was run through the, then named, NIHR Birmingham Liver Biomedical Research Unit [Birmingham, UK; URL: www.birmingham.ac.uk/research/activity/mds/centres/liver/BRU/index.aspx (accessed 4 August 2021)] and PSC Support [Manchester, UK; www.pscsupport.org.uk (accessed 4 August 2021)] was involved in the design and review of the trial documents. The panel reviewed and commented on the draft version of the trial protocol, informed consent form and patient information sheet, and views of the panel regarding explanations, clarity and general layout of the document, etc., were implemented where appropriate. The patient groups were also updated with trial progress on a regular basis, which included providing articles for their newsletter and website. PSC Support provided information about the ongoing trial efforts on its website to ensure that patients across the UK are aware and have the opportunity to take part.
The feedback received from the patient group was used to better understand the clinical need, as well as the willingness, of patients to take part in early-stage clinical trials and explain the studies to patients and the public broadly. The trial was also presented to the public through several forums, such as the NIHR Liver Biomedical Research Unit seminar series on current research and open days, to engage public interest. We also used the interactions to help us explain the studies to patients and the public broadly.
Trial aims and objectives
The overall aims of the trial were to:
-
determine the short-term activity and safety of an anti-VAP-1 antibody BTT1023 in patients with PSC
-
confirm the safe and effective dose of BTT1023 in patients with PSC
-
provide insights into the mechanisms of action of VAP-1/SSAO
-
create the framework for subsequent larger-scale interventions in chronic liver disease with the anti-VAP-1 antibody
-
develop soluble VAP-1/SSAO as a biomarker for liver disease by correlating sVAP-1/SSAO levels to liver fibrosis, severity of inflammation and clinical outcome.
Dose-confirmatory phase
The overall aim of the dose-confirmatory phase was to determine a dose of BTT1023 that provides an acceptable level of PK activity and was deemed to be safe, meeting the acceptable DLT level.
This was to be achieved by evaluating:
-
the number of DLTs per dose level of BTT1023
-
PK data per dose level of BTT1023.
Phase II
Primary objectives
-
To determine the activity of the anti-VAP-1 antibody BTT1023 in patients with PSC, as measured by a decrease in ALP activity (i.e. the primary end point), with secondary end points to include various measures of liver injury and fibrosis.
-
To evaluate the safety, effective dosage and tolerability of BTT1023 in patients with PSC.
Secondary objectives
-
To determine the mechanisms of action of BTT1023 through in vivo assessment of VAP-1/SSAO enzyme activity and immune cell function.
-
To evaluate the potential of a novel magnetic resonance imaging (MRI)-based assessment of liver fibrosis and biliary strictures for assessing the therapeutic response in patients with PSC.
-
To assess the use of sVAP-1/SSAO as a biomarker to monitor disease progression in patients with PSC.
Participants
Participants were all patients with a clinical diagnosis of PSC. This was established using recognised eligibility criteria (see Report Supplementary Material 2).
Outcomes
Dose-confirmatory phase
Dose-limiting toxicity definition
-
Any grade 4 or 5 adverse event (AE), as defined by Common Terminology Criteria for Adverse Events (CTCAE) v4.0,15 and considered to be at least possibly related to BTT1023 treatment.
-
Grade 3 cytokine release syndrome considered to be at least possibly related to BTT1023 treatment.
The confirmatory-stage DLT reporting period was from treatment visit 3 up to visit 10 (i.e. the day 99 follow-up visit). The acceptable level of DLTs per dose was one in six patients (≈ 17%).
Pharmacokinetic data
To confirm that the BTT1023 dose was at the required activity level, the trough levels of BTT1023 had to meet the stipulated success criterion of 3 µg/ml free circulating antibody, as measured during the dose-confirmatory stage. If the PK results showed that the levels were too low, then the dose would be escalated. If the PK results showed that the levels were too high, then the dose could be reduced.
Phase II
Primary outcome measure
-
Response at day 99: a reduction in serum ALP activity of ≥ 25% from baseline to day 99.
Secondary outcome measures
-
Safety and tolerability: treatment compliance (including patient withdrawal) and serious adverse event (SAE) and AE frequency.
-
Calculation of any change (improvement or worsening) from baseline to day 99 in:
-
quality-of-life questionnaire scores [e.g. EuroQol-5 Dimensions (EQ-5D), Fatigue Severity Scale (FSS), pruritus visual analogue scale (VAS) score and IBD diaries (if applicable)]
-
tests of liver fibrosis (e.g. ELF and FibroScan)
-
individual markers of liver biochemistry and function [e.g. aspartate transaminase (AST), alanine transaminase (ALT), ALP, gamma-glutamyl transferase (GGT), bilirubin, albumin, international normalised ratio (INR)] and composite risk scores (Mayo PSC risk score and MELD score)
-
LiverMultiScan MRI (as liver MRI is an emerging method for monitoring liver disease and its treatment)
-
sVAP-1/SSAO as a biomarker of liver disease activity across the study period.
-
Exploratory end points
The following exploratory biomarkers of liver fibrosis were collected and analysed:
-
C3M
-
C4M2
-
C5M
-
COL-18N
-
ELM7
-
EL-NE
-
P4NP7S
-
PRO-C3
-
PRO-C5.
The outcome measure was to calculate any change (improvement or worsening) from first treatment (trial visit 3 overall) to day 99 (visit 10).
The following exploratory biomarker ratios were also explored:
-
C3M/PRO-C3
-
C4M2/P4NP7S
-
C5M/PRO-C5.
Experimental assessments
Screening and registration
Data management and monitoring
Data were initially captured on paper (via a case report form) and then entered into the electronic remote data capture (eRDC) systems by the trial data manager and site staff. During the dose expansion, all data were entered directly into the electronic remote data capture by site staff. The CRCTU provided day-to-day support for the site and provided training via the site initiation visits and routine monitoring visits. The principal investigator was responsible for the day-to-day trial conduct at the site. Data quality assurance was maintained through adherence to the sponsor’s standard operating procedures, CRCTU’s standard operating procedures, the trial protocol, the principles of Good Clinical Practice, the Research Governance Framework and Clinical Trial Regulations.
Further details are given in Report Supplementary Material 5.
Statistical methods
Sample size
The overall total sample size needed to accommodate both the dose-confirmatory stage and Phase II of the trial design.
Phase I: dose confirmation stage
The maximum sample size for the Phase I dose confirmation stage was 18 patients. The sample size was based on the classic 3 + 3 design, investigating three fixed dose increments (8, 12 and 16 mg/kg), with no dose skipping.
Phase II: Simon’s two-stage expansion
The sample size for the Phase II Simon’s two-stage expansion was calculated based on a single-arm Simon’s two-stage minimax design,16 with lower and upper acceptability bounds of 15% and 30%, respectively, and error rates of α = 0.10 and β = 0.20. A sample size of 37 patients was required in this stage of the trial; however, to account for patient dropout, estimated to be approximately 10%, the sample size increased by a further four patients. Therefore, the target recruitment was increased to 41 patients for the expansion phase.
In this setting, the interpretation of alpha is the probability of satisfying stage 1 (of the Phase II design) and observing nine or more responses in 37 patients overall when the true response rate is 15%. This is known as the false-positive result (type I error). Beta is the probability of failing to acknowledge activity when the true response rate is 30%, which is known as the type II error. Therefore, the power (1 – β) is the probability of taking an effective treatment forward.
The maximum total number of patients required for the trial overall was 59.
Statistical analysis
The BUTEO trial was an early-phase trial of BTT1023 in immune cell-mediated liver disease, with the rationale being to identify biochemical efficacy (i.e. reduction in ALP activity) and safety in an orphan disease that presently lacks any other effective medical therapy. The trial design, therefore, focused on identifying early biochemical efficacy signals to justify larger-scale randomised controlled trials of a longer duration.
Our primary outcome measure was patient response to treatment at day 99, as measured by a reduction in serum ALP activity of ≥ 25% from baseline to day 99. Our data on stability of ALP activity in PSC suggest that such responses occur very seldom during the natural course of the disease and we can, therefore, reliably assess changes from baseline and response rates for this proof of concept trial to evaluate the therapeutic potential of BTT1023. In addition, we excluded patients in whom ALP values changed significantly naturally by > 25% between the screening visit at 5–7 weeks before infusion and the screening visit within 1 week before infusion.
Secondary outcome measures included safety and tolerability as determined by treatment compliance, patient withdrawal, frequency of SAEs/AEs, change in quality-of-life questionnaires (i.e. EQ-5D, FSS, pruritus VAS score and IBD diaries) and change in quality of BTT1023 efficacy (as determined by tests of liver fibrosis including ELF, FibroScan and liver biochemistry). In addition, liver MRI was an emerging method for monitoring liver disease and its treatment. We evaluated changes in MRI scans pre and post therapy using the LiverMultiScan protocol (or equivalent methodology, at sites where this is possible). Finally, we evaluated changes in levels of sVAP-1/SSAO as a biomarker of liver disease activity across the trial period.
Statistical analyses were conducted on a modified intention-to-treat (mITT) basis, whereby only patients who had received at least one infusion at the confirmed dose of BTT1023 were to be analysed. Descriptive statistics are presented as mean, median, interquartile range (IQR) and minimum and maximum range for numerical variables, with the frequency and percentage given for categorical variables. Statistical methods used are described at the beginning of each relevant analysis section. All analyses were carried out using either Stata® (at least version 14.0 for design and interim analyses) (StataCorp LP, College Station, TX, USA) or R (version 3.6.0 for final analyses) through RStudio (version 1.2.1335) (The R Foundation for Statistical Computing, Vienna, Austria).
Interim analysis
An interim analysis was carried out once 18 patients had been evaluated for the primary outcome (i.e. ALP response). If three or more responses were observed, then the trial would continue. If not, then the trial would cease. If adequate response was seen, then a further 19 patients were to be recruited to obtain the required sample size of 37 patients. Allowing for 10% patient dropout during trial duration, this number could reach a total of 41 patients recruited. The final success criterion chosen maintains power and, in doing so, can result in increased type I error rates (note that the stage 1 criterion is fixed, as the final sample size is unknown at stage 1). These were calculated by the trial biostatistician and independently verified by a CRCTU biostatistician. Patients who could not be evaluated for the primary outcome (e.g. because of withdrawal or lost to follow-up) were treated as non-responders.
Final analysis
Final analyses of the primary outcome were performed when all patients had been followed to day 120 and once the database had been locked. If, overall, there were nine or more responses from 37 evaluable patients, then it could be concluded that the treatment warrants further investigation. Only patients treated at the confirmed dose contributed to the total patient requirement.
Safety
Dose-limiting toxicity
Dose-limiting toxicity is defined as an AE that meets the criteria of grade 3 cytokine release syndrome or the criteria for any other adverse event at grade 4 or 5, as defined in the CTCAE v4.0. Although previous studies have shown no DLTs with BTT1023, toxicity monitoring was ongoing throughout the trial, and any concerns were reported to the trial office within 24 hours of the investigator becoming aware of the event. During the confirmatory stage, the DLT reporting period was defined as the treatment period from visit 3 (first infusion) to visit 10 (i.e. day 99 follow-up visit). An acceptable DLT rate was established for the trial as a maximum of one incident in six patients (≈ 17%). If the DLT rate increased to two or more at any stage during the DLT reporting period [from visit 3 (i.e. day 1) to visit 10 (i.e. day 99)], the trial would have been halted after consultation with the DMC.
Recording and reporting of serious adverse events
The collection and reporting of AEs was in accordance with The Medicines for Human Use (Clinical Trials) Regulations 200417 and its subsequent amendments. The CTCAE v4.0 criteria were used to grade each AE. Any pre-existing conditions were reported in the medical history and were not reported as an AE unless the condition worsened by at least one CTCAE grade during the trial. The reporting period for AEs commenced on the date of consent (visit 1) and continued until the final follow-up visit (visit 11 on day 120) or, alternatively, to 45 days post last infusion if the patient withdrew from the trial prior to completion of all seven trial drug infusions. All trial patients continued to receive standard concomitant clinical care throughout the trial.
The sponsor, appropriate regulatory authority (e.g. Medicines and Healthcare products Regulatory Agency) and the Research Ethics Committee (REC) were informed of all SAEs, as required by current regulations. SAEs judged to have a reasonable causal relationship to the drug were recorded as serious adverse reactions or as suspected unexpected serious adverse reactions, as appropriate, and were reported to the Medicines and Healthcare products Regulatory Agency and the REC within 7 days. The independent DMC also reviewed all SAEs.
In the event that a patient or their partner became pregnant during the SAE reporting period, this was recorded, reported and followed up, subject to required patient/partner approvals.
Chapter 3 Results
The information in the forthcoming analysis was taken on 27 March 2020 unless stated otherwise. Analysis presented is based on the protocol version 5.0 and the statistical analysis plan version 2.0.
Patient withdrawals, discontinuations and deaths
Consolidated Standards of Reporting Trials
The BUTEO trial Consolidated Standards of Reporting Trials (CONSORT) flow diagram is provided in Figure 1.
FIGURE 1.
A CONSORT flow diagram for the BUTEO trial. Note that one patient was registered and then found to be ineligible. Therefore, this patient is counted both as a screen failure and as a patient recruited. a, Interim assessment is based on the first 18 evaluable patients.
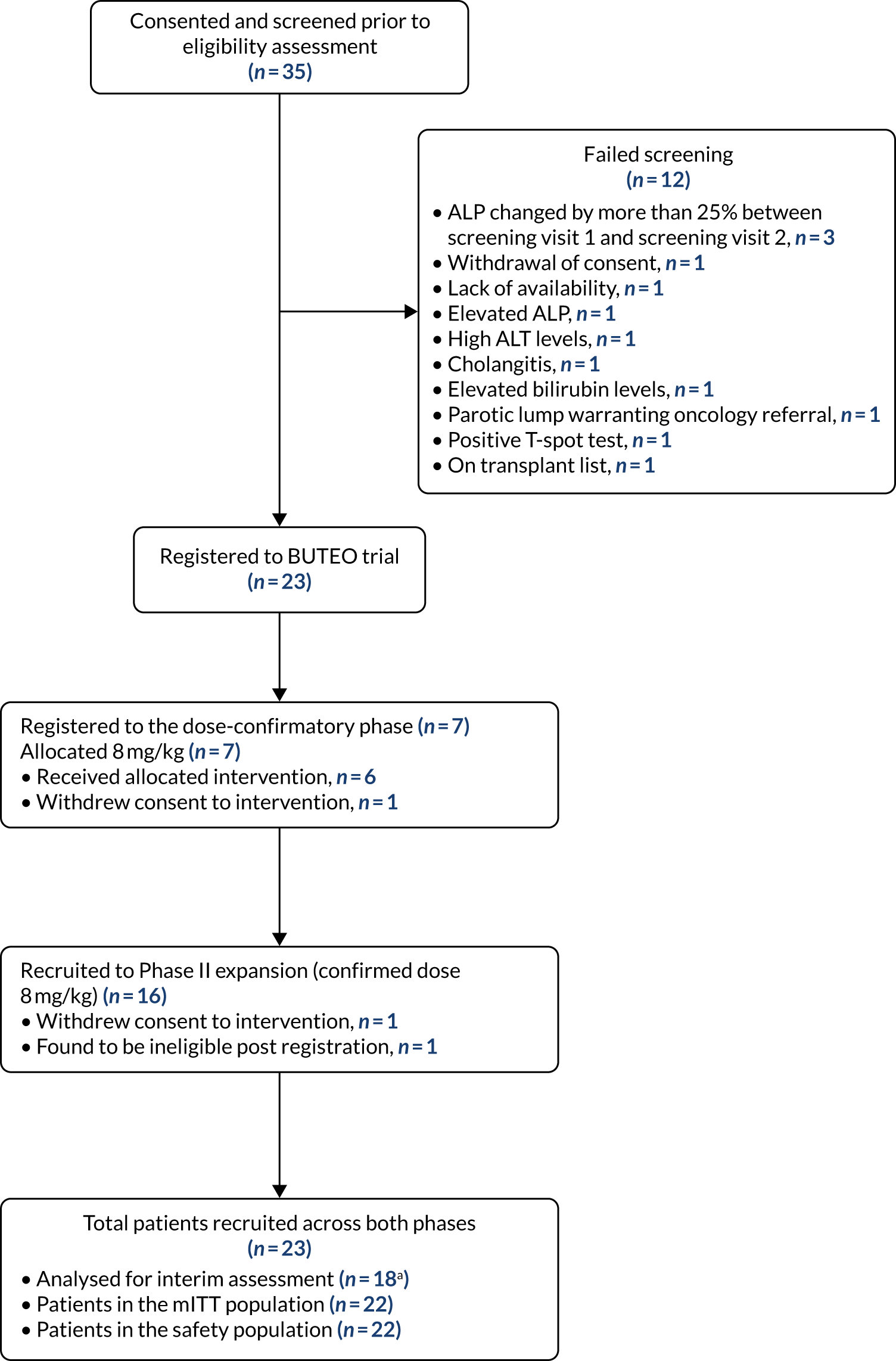
Trial recruitment
Patient information
In the following presentation of baseline patient data, as all patients received the same dose of BTT1023 (i.e. 8 mg/kg), baseline characteristics and medical histories have not been stratified by phase, that is dose-confirmatory phase or Phase II. The characteristics are given for all 23 patients registered, regardless of whether or not they received treatment. This is in contrast to the EudraCT report,18 which listed characteristics for only those patients who received treatment.
Baseline patient characteristics
In the pooled population there was an uneven sex divide, with 82.61% (19/23) of all registered participants being male. Table S8 shows the breakdown of recruitment by sex according to registration centre. In Table S8, it can be seen that, despite the sex imbalance, the John Radcliffe Hospital (Oxford, UK) recruited equal numbers (n = 2) of male and female patients.
The majority of patients (87.0%) identified as Caucasian (n = 20), with the two remaining patients identifying as South Asian (8.7%). Details regarding ethnic identification were missing for a patient who was found to be ineligible post registration. Full details regarding the breakdown of ethnicity by sex can be found in the Additional Material 2 – Patient Information document, Table S9.
Table 1 shows descriptive characteristics regarding patient age, both generally and stratified by sex. From Table 1, it is apparent that the mean age was similar for both males and females (approximately 46 years). However, there is more variability in the age distribution of males, with both a wider range (22–69 years) and a wider IQR (36.0–52.5 years). The observed wider range of age values for males may be attributable to the larger number of male patients registered to the trial. Table S10, Additional Material 2 – Patient Information, shows the number of subjects enrolled per age group, categorised according to EudraCT age categories.
| Summary | Patient sex | Overall | |
|---|---|---|---|
| Male | Female | ||
| n | 19 | 4 | 23 |
| Age (years) | |||
| Mean | 45.11 | 46.00 | 45.26 |
| Median | 46.00 | 47.00 | 46.00 |
| Range | 22.00–69.00 | 39.00–51.00 | 22.00–69.00 |
| IQR | 36.00–52.50 | 42.00–51.00 | 38.00–51.50 |
Table 2 gives a line listing of patient baseline characteristics.
| Patient registration | Age at randomisation (years) | Patient sex | Absolute ALP % change | Dose allocation | Date discontinued | Discontinuation reason | |
|---|---|---|---|---|---|---|---|
| Number | Date | ||||||
| 1 | 7 September 2015 | 22 | Male | 5.80 | 8 mg/kg | ||
| 2 | 3 December 2015 | 50 | Male | 4.70 | 8 mg/kg | 29 January 2016 | Patient withdrew consent to treatment |
| 3 | 7 January 2016 | 61 | Male | 25.20 | 8 mg/kg | ||
| 4 | 8 January 2016 | 43 | Female | 5.00 | 8 mg/kg | ||
| 5 | 8 February 2016 | 38 | Male | 3.30 | 8 mg/kg | ||
| 6 | 19 May 2016 | 52 | Male | 26.58 | 8 mg/kg | ||
| 7 | 8 June 2016 | 51 | Female | 4.55 | 8 mg/kg | ||
| 8 | 2 March 2017 | 69 | Male | 1.60 | 8 mg/kg | ||
| 9 | 8 March 2017 | 63 | Male | 0.00 | 8 mg/kg | ||
| 10 | 23 April 2017 | 27 | Male | 14.40 | 8 mg/kg | 22 April 2017 | Patient found to be ineligible post registration |
| 11 | 8 May 2017 | 34 | Male | 5.50 | 8 mg/kg | 5 September 2017 | Patient withdrew consent to treatment |
| 12 | 10 May 2017 | 53 | Male | 18.40 | 8 mg/kg | ||
| 13 | 10 May 2017 | 39 | Female | 6.70 | 8 mg/kg | ||
| 14 | 4 June 2017 | 46 | Male | 15.30 | 8 mg/kg | ||
| 15 | 20 August 2017 | 51 | Male | 10.00 | 8 mg/kg | ||
| 16 | 4 September 2017 | 38 | Male | 3.90 | 8 mg/kg | ||
| 17 | 24 September 2017 | 23 | Male | 18.40 | 8 mg/kg | ||
| 18 | 5 December 2017 | 38 | Male | 17.20 | 8 mg/kg | ||
| 19 | 29 December 2017 | 51 | Female | 0.40 | 8 mg/kg | ||
| 20 | 23 February 2018 | 29 | Male | 16.50 | 8 mg/kg | ||
| 21 | 2 March 2018 | 67 | Male | 5.00 | 8 mg/kg | ||
| 22 | 17 May 2018 | 50 | Male | 8.00 | 8 mg/kg | ||
| 23 | 18 June 2018 | 46 | Male | 2.00 | 8 mg/kg | ||
Table 2 also shows that patient 6 had an absolute ALP percentage change value that would make him ineligible for the trial. The following deviation was listed: ‘Patient had minor fluctuation in ALP. It was just > 25% and was an oversight and “not” clinically significant’. Following a review by the Trial Management Group, it was decided to allow this individual to participate. For full details on the deviation, see Table S333 in Report Supplementary Material 8.
Patient medical histories
Baseline/screening biochemistry and haematology
Biochemistry
Table 3 shows summary statistics of the biochemistry data at screenings 1 and 2, and the difference between them. The following biochemical data were collected:
-
ALP (IU/l)
-
ALT (IU/l)
-
AST (IU/l)
-
albumin (g/l)
-
bilirubin (direct, µmol/l)
-
bilirubin (indirect, µmol/l)
-
bilirubin (total, µmol/l)
-
GGT (IU/l)
-
sodium (mmol/l)
-
potassium (mmol/l)
-
urea (mmol/l)
-
creatinine (µmol/l)
-
calcium (mmol/l)
-
total protein (g/l)
-
estimated glomerular filtration rate (eGFR) (ml/minute).
| Biochemical test | Test result | Difference | |
|---|---|---|---|
| Screening visit 1 | Screening visit 2 | ||
| ALP (IU/l) | |||
| n/N | 22/23 | 22/23 | 22/23 |
| Mean | 477.64 | 450.77 | –26.86 |
| Median | 406.50 | 346.50 | –16.50 |
| Range | 199.00–1318.00 | 215.00–1075.00 | –243.00 to 118.00 |
| IQR | 295.50–528.00 | 311.25–565.75 | –44.50 to 13.25 |
| ALT (IU/l) | |||
| n/N | 22/23 | 22/23 | 22/23 |
| Mean | 109.77 | 94.68 | –15.09 |
| Median | 84.00 | 73.50 | –10.00 |
| Range | 32.00–303.00 | 33.00–265.00 | –171.00 to 83.00 |
| IQR | 70.25–135.50 | 56.00–107.75 | –26.50 to 1.00 |
| AST (IU/l) | |||
| n/N | 21/23 | 22/23 | 21/23 |
| Mean | 99.24 | 87.77 | –9.00 |
| Median | 79.00 | 74.00 | –5.00 |
| Range | 18.00–357.00 | 24.00–249.00 | –205.00 to 86.00 |
| IQR | 56.00–124.00 | 45.50–104.00 | –13.00 to 6.00 |
| Albumin (g/l) | |||
| n/N | 22/23 | 22/23 | 22/23 |
| Mean | 41.32 | 41.59 | 0.27 |
| Median | 43.00 | 43.00 | 1.00 |
| Range | 29.00–50.00 | 31.00–48.00 | –5.00 to 5.00 |
| IQR | 37.00–44.00 | 38.00–46.00 | –2.00 to 2.00 |
| Direct bilirubin (µmol/l) | |||
| n/N | 19/23 | 19/23 | 17/23 |
| Mean | 12.89 | 15.11 | 3.41 |
| Median | 13.00 | 12.00 | 1.00 |
| Range | 2.00–38.00 | 2.00–44.00 | –1.00 to 21.00 |
| IQR | 5.50–16.50 | 6.50–21.00 | 0.00–4.00 |
| Indirect bilirubin (µmol/l) | |||
| n/N | 18/23 | 17/23 | 15/23 |
| Mean | 7.50 | 9.76 | 1.27 |
| Median | 6.50 | 8.00 | 1.00 |
| Range | 2.00–19.00 | 1.00–25.00 | –6.00 to 13.00 |
| IQR | 5.00–9.75 | 6.00–11.00 | –0.50 to 2.50 |
| Total bilirubin (µmol/l) | |||
| n/N | 22/23 | 22/23 | 22/23 |
| Mean | 21.27 | 24.64 | 3.36 |
| Median | 18.00 | 20.50 | 1.00 |
| Range | 4.00–47.00 | 4.00–57.00 | –5.00 to 30.00 |
| IQR | 13.00–29.00 | 14.25–31.00 | 0.00 to 5.75 |
| GGT (IU/l) | |||
| n/N | 20/23 | 21/23 | 19/23 |
| Mean | 732.85 | 643.24 | –68.00 |
| Median | 447.50 | 398.00 | –16.00 |
| Range | 77.00–2857.00 | 69.00–1918.00 | –939.00 to 114.00 |
| IQR | 297.25–1096.75 | 249.00–1079.00 | –62.50 to 19.50 |
| Sodium (mmol/l) | |||
| n/N | 22/23 | 22/23 | 22/23 |
| Mean | 139.73 | 139.50 | –0.23 |
| Median | 140.00 | 139.00 | 0.00 |
| Range | 135.00–143.00 | 135.00–144.00 | –7.00 to 5.00 |
| IQR | 139.00–140.75 | 139.00–140.75 | –2.00 to 1.00 |
| Potassium (mmol/l) | |||
| n/N | 22/23 | 22/23 | 22/23 |
| Mean | 4.11 | 4.21 | 0.10 |
| Median | 4.10 | 4.10 | 0.10 |
| Range | 3.60–4.60 | 3.50–4.90 | –0.30 to 0.80 |
| IQR | 3.90–4.30 | 3.90–4.50 | –0.08 to 0.18 |
| Urea (mmol/l) | |||
| n/N | 22/23 | 22/23 | 22/23 |
| Mean | 4.38 | 4.64 | 0.26 |
| Median | 4.35 | 4.35 | 0.30 |
| Range | 2.90–6.60 | 3.10–7.00 | –1.40 to 1.60 |
| IQR | 3.50–5.25 | 3.90–5.275 | –0.40 to 0.875 |
| Creatinine (µmol/l) | |||
| n/N | 22/23 | 22/23 | 22/23 |
| Mean | 65.32 | 67.68 | 2.36 |
| Median | 62.00 | 65.50 | 2.00 |
| Range | 36.00–106.00 | 40.00–104.00 | –9.00 to 22.00 |
| IQR | 53.00–77.75 | 58.25–80.25 | –2.75 to 5.75 |
| Calcium (mmol/l) | |||
| n/N | 20/23 | 20/23 | 20/23 |
| Mean | 2.36 | 2.36 | 0.00 |
| Median | 2.36 | 2.34 | 0.01 |
| Range | 2.10 to 2.72 | 2.20 to 2.60 | –0.15 to 0.12 |
| IQR | 2.29–2.44 | 2.29–2.43 | –0.06 to 0.09 |
| Total protein (g/l) | |||
| n/N | 20/23 | 19/23 | 19/23 |
| Mean | 75.80 | 76.84 | 0.95 |
| Median | 73.50 | 75.00 | 2.00 |
| Range | 66.00–93.00 | 64.00–96.00 | –10.00 to 10.00 |
| IQR | 69.50–80.50 | 73.50–80.50 | –1.00 to 3.00 |
| eGFR (ml/minute) | |||
| n/N | 21/23 | 21/23 | 21/23 |
| Mean | 113.44 | 102.70 | –10.75 |
| Median | 96.80 | 90.00 | 0.00 |
| Range | 64.00–204.30 | 65.00–163.00 | –79.00 to 40.10 |
| IQR | 90.00–130.80 | 90.00–116.00 | –11.00 to 2.40 |
Haematology
Table 4 shows summary statistics of the haematological data at screenings 1 and 2, and the difference between them. The following haematological parameters were measured:
-
haemoglobin (g/l)
-
platelets (109/l)
-
red blood cells (1012/l)
-
white blood cells (109/l)
-
haematocrit (l/l)
-
mean cell volume (fl)
-
mean cell haemoglobin (pg)
-
neutrophils (109/l)
-
lymphocytes (109/l)
-
monocytes (109/l)
-
eosinophils (109/l)
-
basophils (109/l)
-
INR
-
activated partial thromboplastin time (APTT) ratio.
| Haematological test | Test result | Difference | |
|---|---|---|---|
| Screening visit 1 | Screening visit 2 | ||
| Haemoglobin (g/l) | |||
| n/N | 20/23 | 22/23 | 20/23 |
| Mean | 136.25 | 140.59 | 3.75 |
| Median | 134.00 | 140.00 | 2.00 |
| Range | 114.00–157.00 | 113.00–163.00 | –12.00 to 23.00 |
| IQR | 130.25–144.25 | 133.50–147.75 | –1.25 to 7.50 |
| Platelets (109/l) | |||
| n/N | 20/23 | 22/23 | 20/23 |
| Mean | 207.25 | 228.23 | 15.45 |
| Median | 205.50 | 230.00 | 14.00 |
| Range | 73.00–375.00 | 86.00–461.00 | –30.00 to 86.00 |
| IQR | 137.00–266.50 | 139.75–291.50 | –1.75 to 28.50 |
| Red blood cells (1012/l) | |||
| n/N | 20/23 | 22/23 | 20/23 |
| Mean | 4.50 | 4.62 | 0.10 |
| Median | 4.51 | 4.62 | 0.06 |
| Range | 3.57–5.35 | 3.51–5.54 | –0.35 to 0.91 |
| IQR | 4.17–4.87 | 4.45–4.86 | –0.11 to 0.22 |
| White blood cells (109/l) | |||
| n/N | 20/23 | 22/23 | 20/23 |
| Mean | 6.11 | 6.93 | 0.76 |
| Median | 5.75 | 6.85 | 0.40 |
| Range | 2.60–10.50 | 3.30–12.20 | –1.70 to 5.40 |
| IQR | 5.00–6.65 | 5.13–8.40 | –0.45 to 1.78 |
| Haematocrit (l/l) | |||
| n/N | 20/23 | 22/23 | 20/23 |
| Mean | 0.41 | 0.42 | 0.01 |
| Median | 0.41 | 0.42 | 0.00 |
| Range | 0.35–0.49 | 0.35–0.49 | –0.03 to 0.07 |
| IQR | 0.39–0.43 | 0.40–0.44 | –0.01 to 0.02 |
| Mean cell volume (fl) | |||
| n/N | 20/23 | 22/23 | 20/23 |
| Mean | 90.91 | 90.90 | 0.02 |
| Median | 91.10 | 91.00 | 0.25 |
| Range | 76.60–100.50 | 75.50–103.20 | –3.00 to 4.00 |
| IQR | 89.53–94.23 | 89.25–93.75 | –1.00 to 0.50 |
| Mean cell haemoglobin (pg) | |||
| n/N | 20/23 | 22/23 | 20/23 |
| Mean | 30.39 | 30.71 | 0.32 |
| Median | 30.90 | 30.75 | 0.25 |
| Range | 24.50–35.50 | 24.00–35.20 | –0.50 to 2.70 |
| IQR | 29.75–31.40 | 29.93–31.83 | –0.13 to 0.43 |
| Neutrophils (109/l) | |||
| n/N | 20/23 | 22/23 | 20/23 |
| Mean | 3.75 | 4.26 | 0.47 |
| Median | 3.60 | 3.90 | 0.10 |
| Range | 1.40–5.90 | 2.10–8.40 | –1.30 to 4.20 |
| IQR | 2.98–4.45 | 3.20–5.43 | –0.43 to 0.93 |
| Lymphocytes (109/l) | |||
| n/N | 20/23 | 22/23 | 20/23 |
| Mean | 1.52 | 1.65 | 0.12 |
| Median | 1.40 | 1.35 | 0.05 |
| Range | 0.60–2.80 | 0.60–2.90 | –0.40 to 0.70 |
| IQR | 1.08–2.00 | 1.13–2.28 | –0.10 to 0.33 |
| Monocytes (109/l) | |||
| n/N | 20/23 | 22/23 | 20/23 |
| Mean | 0.50 | 0.56 | 0.04 |
| Median | 0.50 | 0.55 | 0.00 |
| Range | 0.20–0.90 | 0.20–1.00 | –0.30 to 0.50 |
| IQR | 0.40–0.53 | 0.40–0.70 | –0.03 to 0.13 |
| Eosinophils (109/l) | |||
| n/N | 20/23 | 22/23 | 20/23 |
| Mean | 0.26 | 0.39 | 0.12 |
| Median | 0.20 | 0.20 | 0.00 |
| Range | 0.00–1.20 | 0.00–3.10 | –0.20 to 1.90 |
| IQR | 0.10–0.20 | 0.10–0.40 | 0.00 to 0.10 |
| Basophils (109/l) | |||
| n/N | 20/23 | 22/23 | 20/23 |
| Mean | 0.04 | 0.05 | 0.00 |
| Median | 0.00 | 0.00 | 0.00 |
| Range | 0.00–0.10 | 0.00–0.10 | –0.10 to 0.10 |
| IQR | 0.00–0.10 | 0.00–0.10 | 0.00 to 0.00 |
| INR | |||
| n/N | 22/23 | 22/23 | 22/23 |
| Mean | 1.00 | 1.01 | 0.01 |
| Median | 1.00 | 1.00 | 0.00 |
| Range | 0.90–1.10 | 0.90–1.20 | –0.10 to 0.10 |
| IQR | 1.00–1.00 | 1.00–1.08 | 0.00 to 0.08 |
| APPT ratio | |||
| n/N | 17/23 | 18/23 | 17/23 |
| Mean | 1.14 | 1.12 | –0.02 |
| Median | 1.10 | 1.10 | 0.00 |
| Range | 1.00–1.50 | 1.00–1.40 | –0.10 to 0.20 |
| IQR | 1.00–1.20 | 1.03–1.20 | –0.10 to 0.00 |
Results for dose-confirmatory stage
The BUTEO trial included an initial run-in component to confirm the dose of BTT1023. Data from previous trials of BTT1023 in patients with rheumatoid arthritis or psoriasis supported the use of 8 mg/kg, which was well tolerated. The run-in component of the trial has the potential to incorporate a conventional 3 + 3 cohort design with decision guidelines based on toxicity and PK data.
Owing to the withdrawal, and subsequent replacement, of patient 2 after one dose, seven patients were recruited to the dose confirmation phase. The six remaining evaluable patients all received the scheduled seven infusion treatments. Full details on the withdrawal of patient 2 are given in Report Supplementary Material 6. This was the only patient withdrawal during the dose-confirmatory phase. Measurements of toxicity (assessed by patients) and circulating levels of serum free BTT1023 PK data were recorded to provide evidence that the dose was appropriate or needed to be escalated.
In the dose confirmation phase, all patients were treated at the same dose level of 8 mg/kg.
Toxicity of the treatment was indicated if a patient experienced a DLT, as defined in the trial protocol. An acceptable DLT rate was established for the trial as a maximum of one incident in six patients (≈ 17%). The DLT monitoring period recorded these data from treatment initiation until visit 10 (i.e. day 99). The PK data were obtained from blood samples collected at visit 7 (i.e. day 50 post treatment initiation).
Dose-limiting toxicity evaluation
Version 5.0 of the BUTEO protocol defines a DLT as follows:
. . . an AE that meets the criteria of grade 4 or grade 5 as defined in the Common Terminology Criteria for Adverse Events (CTCAE v4.0) (Protocol Appendix 2). However, if a definite diagnosis of cytokine release syndrome was made, it was classed as a DLT at grade 3 and above, and if considered at least possibly related to BTT1023.
Report Supplementary Material 10, Table S39, lists all grade 3 or higher AEs experienced by all patients during the dose confirmation stage. The table includes grade 3 AEs to ensure that any occurrences of grade 3 cytokine release syndrome were identified.
As evident from Report Supplementary Material 10, Table S39, during the dose-confirmatory phase of the trial, the seven-patient cohort recorded 17 grade 3 AEs and one grade 4 AE. Investigation into the grade 4 AE experienced by patient 2 revealed that this AE was recorded at screening visit 1, prior to any treatment with BTT1023. Consequently, the event was not considered to be a DLT. Moreover, during the dose confirmation phase, there were no instances of confirmed cytokine release syndrome at grade 3 or higher. Therefore, it was concluded that no DLTs were experienced during the dose-confirmatory period.
Pharmacokinetic evaluation of BTT1023
In addition to the incidence of DLTs, the success criteria for transition from the dose confirmation period to the Simon’s two-stage design have been approximated as reaching a trough concentration of 3 µg/ml of free circulating BTT1023 at 8 weeks from first infusion, which is about 100-fold the dissociation constant of BTT1023 from VAP-1 and should result in a target occupancy of approximately 90% with either the starting dose of 8 mg/kg or following dose escalation up to a maximum of 16 mg/kg.
If PK evaluation showed the levels of BTT1023 to be too low, then dose escalation was considered to be appropriate. If the results of PK evaluation showed the levels of BTT1023 to be too high, then dose reduction was required. Blood samples were collected from patients and sent to Biotie Therapies Ltd/Envigo (Bicester, UK) for analysis.
The dose-confirmatory population was taken to be the first six patients who were eligible for PK evaluation 8 weeks from first infusion. As patient 2 discontinued treatment and withdrew from the study after one infusion, this patient was excluded from this analysis. All patients in this cohort were allocated 8 mg/kg. As PK assessments were performed at different time points of the trial visit, pre-infusion values are used.
Report Supplementary Material 10, Table S40, presents descriptive statistics of the circulating BTT1023 levels (µg/ml) recorded at each treatment visit. The laboratory test has a lower limit of quantification (104 ng/ml), and results below this level were set to zero. Moreover, the laboratory test also has an upper limit of quantification. Results above the limit of quantification were treated as missing.
The criterion for continuation to the Simon’s two-stage expansion was for BTT1023 circulating trough levels to reach 3 µg/ml 8 weeks from the first infusion. For the trial to continue to the Simon’s two-stage expansion, circulating levels of BTT1023 8 weeks from infusion trial needed to reach a trough level of 3 µg/ml. As 8 weeks from the first infusion falls between visit 7 (i.e. day 50) and visit 8 (i.e. day 64), the forthcoming analysis presents results at visit 7 and assumes this to be 8 weeks from the first infusion. Summary statistics of circulating levels of BTT1023 at visit 7 pre infusion can be found in Report Supplementary Material 10, Table S40. Table 5 shows patient-specific estimates.
| Patient number | Level of BTT1023 (µg/ml) |
|---|---|
| 1 | 8.47 |
| 3 | 12.70 |
| 4 | 13.10 |
| 5 | 14.70 |
| 6 | 10.40 |
| 7 | 23.60 |
Report Supplementary Material 10, Figure S5, shows a repeated measures plot of circulating BTT1023 levels recorded at each treatment visit in the dose-confirmatory population.
From Report Supplementary Material 10, Figure S5, despite the significant amount of heterogeneity between patients, at visit 7 the circulating levels of BTT1023 exceeded the minimum requirement of 3 µg/ml for all patients in the dose-confirmatory phase. Therefore, the minimum threshold criterion for circulating level was met.
Safety analysis
Adverse event data were recorded for each patient throughout their time on the trial. Any AEs that fulfilled the DLT criteria have been presented in Dose-limiting toxicity evaluation. As the patients from the dose-confirmatory cohort received the final selected dose of BTT1023 for further testing in the expansion part of the trial, the safety data will be presented for the overall total patient cohort (see Safety and toxicity reporting).
Phase II interim assessment results
Once the BTT1023 dose had been confirmed and deemed safe, the trial expanded and moved into Phase II, following a Simon’s minimax two-stage design. As patients in the dose-confirmatory cohort had received the selected dose of 8 mg/kg, these patients were included in the Phase II patient population, constituting part of the sample size. For a type I error rate of α = 0.10 and power of 0.8 (β = 0.8), the Simon’s minimax two-stage design required a total of 37 patients, with 18 patients being evaluable at the interim assessment (i.e. stage I of the trial design). A further 11 patients were recruited. The interim assessment was to determine whether or not there was any evidence of treatment efficacy.
The statistical analysis was carried out on a mITT basis such that only patients who had received at least one infusion of BTT1023 at the confirmed dose were included in the evaluation. Any patient who did not receive any treatment was replaced. Patients who could not be evaluated for the primary outcome (e.g. because of withdrawal or lost to follow-up) were treated as non-responders. A sensitivity analysis was also performed using the per-protocol population (i.e. patients completing the full seven infusions), as scheduled in the trial protocol (details given in Report Supplementary Material 11, Table S63).
Eighteen evaluable patients were required for the interim primary outcome assessment. Patient TNO10 (trial number) was replaced as they were found to be ineligible for the trial post registration and prior to receiving any treatment. Patients TNO2 and TNO11 withdrew from the trial and discontinued treatment, although they remain evaluable as they had received one and six doses of treatment, respectively. Patients TNO 1–9 and TNO 11–19, inclusive, were included in the interim analysis (see Report Supplementary Material 11, Table S63).
Primary outcome measure: response at day 99
The primary outcome of the BUTEO trial was to determine the activity of the anti-VAP-1 antibody BTT1023 in patients with PSC, as measured by a decrease in ALP activity (i.e. the primary end point), with the outcome measure being response to BTT1023 at visit 10 (i.e. day 99). This required patients to exhibit a reduction in serum ALP activity of at least 25% from baseline (i.e. pre-infusion visit 3) to day 99. In an attempt to reduce any between-laboratory analytical variability, blood samples were taken from all patients and processed at a centralised laboratory in Birmingham [see schedule of events in v5.0 of protocol URL: www.journalslibrary.nihr.ac.uk/programmes/eme/1216531/#/documentation (accessed 9 August 2021)]. This ensured that all samples were analysed using the same equipment and procedures. Note that, for the centralised processing, individuals who were treated in Birmingham had their samples recorded on a biochemistry form and may have had their samples recorded on the ALP sample form. For individuals who were not treated in Birmingham, values on the biochemistry form relate to the analysis of the sample performed at the individual treatment centre and values on the ALP sample form relate to records from the centralised processing. Therefore, for individuals treated in Birmingham, data relating to the primary outcome measure were extracted from the biochemistry form. For individuals not treated in Birmingham, data were extracted from the ALP sample form.
Percentage change in ALP was calculated using Equation 1:
Table 6 shows the ALP measurement both at pre-infusion visit 3 and at follow-up visit 10, along with the difference and percentage change in values for those patients constituting the interim data set, ordered according to percentage. In Table 6, patient 19 was missing their follow-up visit 10 measurement. Patient 19 was not treated at the Queen Elizabeth Hospital (Birmingham, UK) and so their sample was to be transported for centralised processing. However, the ALP sample form shows that the ‘sample was never received by the UHB [University Hospitals Birmingham] laboratory’ and that, although tracking was available, it could not be ‘identified where the sample was delivered’. Therefore, as treatment was administered, patient 19 was included in the interim analysis data set, but their follow-up visit 10 value was missing.
| Patient number | Serum ALP activity (IU/l) | Percentage change | ||
|---|---|---|---|---|
| Pre-infusion visit 3 | Follow-up visit 10 | Difference | ||
| 18 | 269 | 154 | –115 | –42.75 |
| 6 | 535 | 374 | –161 | –30.09 |
| 13 | 581 | 474 | –107 | –18.42 |
| 3 | 431 | 356 | –75 | –17.40 |
| 16 | 341 | 294 | –47 | –13.78 |
| 5 | 925 | 812 | –113 | –12.22 |
| 15 | 429 | 417 | –12 | –2.80 |
| 2 | 685 | 673 | –12 | –1.75 |
| 12 | 263 | 261 | –2 | –0.76 |
| 8 | 286 | 289 | 3 | 1.05 |
| 9 | 228 | 232 | 4 | 1.75 |
| 11 | 246 | 251 | 5 | 2.03 |
| 4 | 475 | 531 | 56 | 11.79 |
| 7 | 340 | 386 | 46 | 13.53 |
| 1 | 298 | 353 | 55 | 18.46 |
| 17 | 1069 | 1407 | 338 | 31.62 |
| 14 | 740 | 1192 | 452 | 61.08 |
| 19 | 278 | |||
From Table 6 it can be seen that nine patients (50%) experienced a reduction in ALP activity (i.e. a negative percentage change value), with eight patients (44.44%) experiencing an increase.
In accordance with the Simon’s two-stage design of the trial, for the trial to continue to stage 2, at least three successful responses to BTT1023, out of 18 evaluable responses, were required at the interim assessment. From Table 6, it is immediately apparent that only 2 of the 18 evaluable patients (11.11%) achieved a decrease of ≥ 25%. Following the trial’s interim assessment analyses, and the subsequent DMC and TSC meetings, it was recommended that, despite there being no safety concerns regarding treatment, the trial should be closed because of a lack of efficacy.
Safety data
All AEs and safety reporting can be found in Safety and toxicity reporting.
Overall results based on all recruited patients
A further four patients were recruited to the trial during the interim assessment phase and received the confirmed dose. All data collected from all patients during the trial are analysed together, resulting in a total sample size of 22 patients.
The statistical analysis was carried out on a mITT basis such that only patients who had received at least one infusion of BTT1023 at the confirmed dose were included in the evaluation. Any patient who did not receive any treatment was replaced. Patients who could not be evaluated for the primary outcome (e.g. because of withdrawal or loss to follow-up) were treated as non-responders. A sensitivity analysis was also performed using the per-protocol population (i.e. patients completing the full seven infusions), as scheduled in the trial protocol.
Primary outcome measure: response at day 99
Using definitions given in Primary outcome measure: response at day 99, the percentage change in ALP between visit 3 (pre infusion) and visit 10 (follow-up) can be calculated for the entire study population. Table 7 shows the ALP measurement at both pre-infusion visit 3 and follow-up visit 10, along with the difference and percentage change in values for all evaluable patients, ordered according to percentage.
| Patient number | Serum ALP activity (IU/l) | Percentage change | ||
|---|---|---|---|---|
| Pre-infusion visit 3 | Follow-up visit 10 | Difference | ||
| 18 | 269 | 154 | –115 | –42.75 |
| 6 | 535 | 374 | –161 | –30.09 |
| 13 | 581 | 474 | –107 | –18.42 |
| 3 | 431 | 356 | –75 | –17.40 |
| 16 | 341 | 294 | –47 | –13.78 |
| 5 | 925 | 812 | –113 | –12.22 |
| 20 | 335 | 296 | –39 | –11.64 |
| 15 | 429 | 417 | –12 | –2.80 |
| 2 | 685 | 673 | –12 | –1.75 |
| 12 | 263 | 261 | –2 | –0.76 |
| 8 | 286 | 289 | 3 | 1.05 |
| 22 | 180 | 182 | 2 | 1.11 |
| 9 | 228 | 232 | 4 | 1.75 |
| 23 | 269 | 274 | 5 | 1.86 |
| 11 | 246 | 251 | 5 | 2.03 |
| 21 | 424 | 437 | 13 | 3.07 |
| 4 | 475 | 531 | 56 | 11.79 |
| 7 | 340 | 386 | 46 | 13.53 |
| 1 | 298 | 353 | 55 | 18.46 |
| 17 | 1069 | 1407 | 338 | 31.62 |
| 14 | 740 | 1192 | 452 | 61.08 |
| 19 | 278 | |||
Table 7 shows that there appeared to be a high level of variability in ALP activity, with 10 patients (45.5%) experiencing a decrease in ALP post treatment.
Report Supplementary Material 11, Table S41, shows that only 9.09% of patients achieved the target reduction in serum ALP of 25%. Figure 2 shows a repeated measures plot of all centrally processed serum ALP.
FIGURE 2.
Repeated measures plot of centrally processed ALP activity for the mITT population. A mean locally weighted smoothing (LOESS) trend line is shown in dark blue (thicker line), with uncertainty depicted by the shaded region.
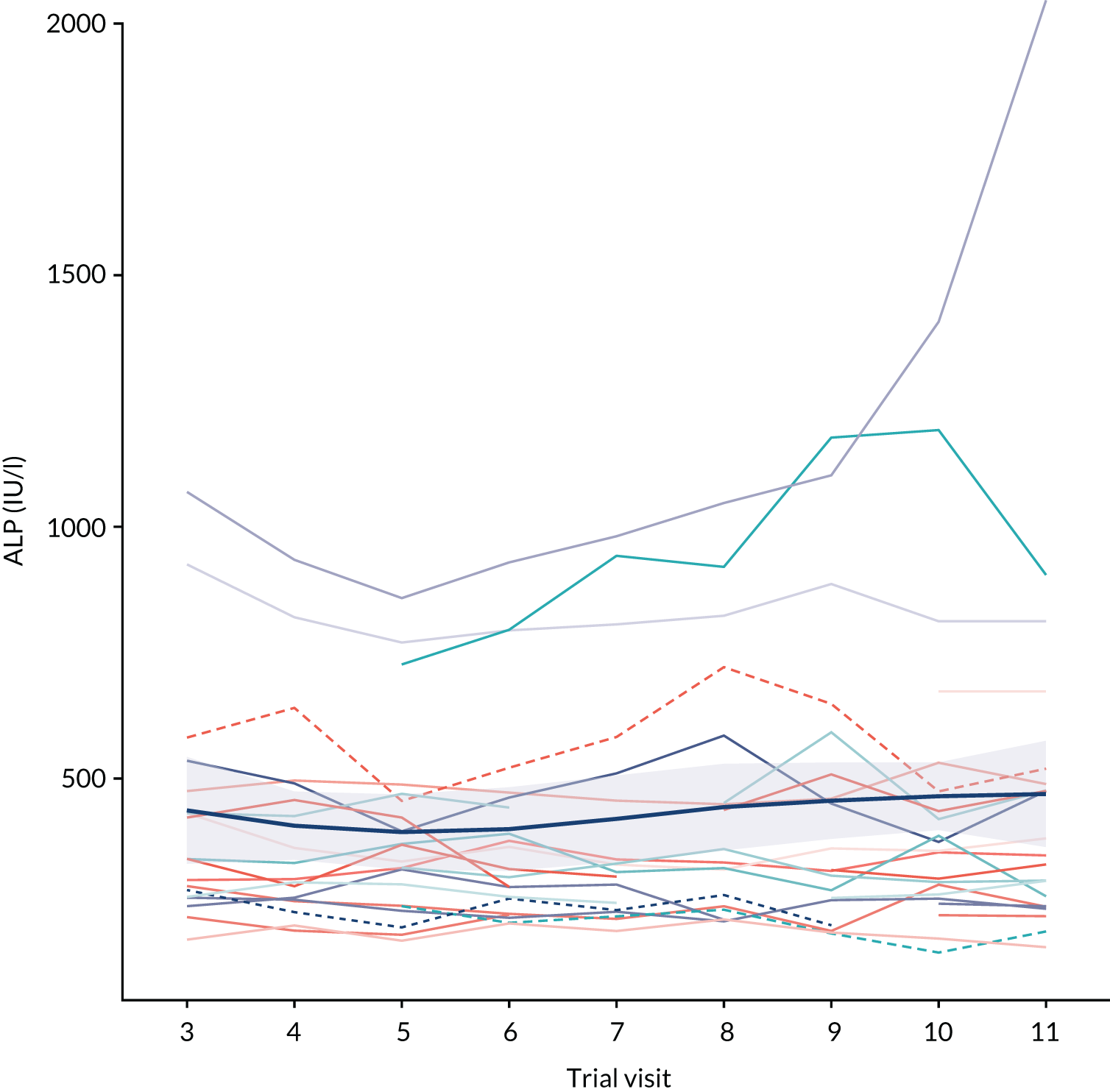
Safety and toxicity reporting
The following section contains AE information pertinent to the safety population of the study. All AEs were graded in accordance with CTCAE v4.0. As per version 2.0 of the statistical analysis plan, the safety population contains all patients who received treatment with BTT1023 at any dose level. Therefore, the safety population included 22 patients.
In total, there were 1133 AEs experienced by 22 patients. Further details regarding the number of patients who experienced AEs can be found in Report Supplementary Material 11, Tables S44 and S45.
All AEs are categorised according to how related to the investigational medicinal product they are on the following scale:
-
unrelated
-
unlikely to be related
-
possibly related
-
probably related
-
definitely related.
Moreover, the following definitions are assumed throughout this section:
-
affected – the number of patients who experienced this event
-
occurrences – the total number of each event reported by those affected
-
related – the number of occurrences that were classified as related to the trial treatment
-
fatal – the number of occurrences for which the outcome was reported as death
-
related and fatal – the number of occurrences that were related to the trial treatment and had death as the outcome.
An event was considered to be related to BTT1023 if it was categorised as at least possibly related.
Report Supplementary Material 11, Table S43, shows the number of AEs experienced by grade and treatment visit and Report Supplementary Material 11, Table S44, shows the number of patients experiencing AEs by grade and treatment visit. Recorded AEs were overwhelmingly either grade 1 (73.02%) or grade 2 (19.49%), with no grade 5 events being reported throughout the trial period. The distribution of AEs by visit was fairly even across all treatment visits. AEs were most commonly reported at screening visit 1 (15.96%). As can be seen in Report Supplementary Material 11, Table S43, eight events were not categorised as happening during a visit. The reason ‘TNO15 missed visit 7, however experienced 8 adverse events on the dates between the dates of visit 6 and visit 8’ was provided. As there was no treatment date that corresponds with the date of these AEs, they were categorised as ‘other’.
Report Supplementary Material 11, Table S45, gives a listing of all AEs that occur in at least one patient, ordered by category and toxicity. The most common toxicities, experienced by 22 patients (100%), were an increase in activity of ALP and GGT, both falling under the CTCAE category of investigations. Moreover, 136 AEs each occurred in only one patient.
Report Supplementary Material 11, Tables S46 and S47, show the overall and patient-level incidence of AEs by toxicity and grade.
Report Supplementary Material 11, Table S48, shows the number of AEs experienced by treatment cycle and relatedness. Generally speaking, AEs were deemed to not be related to BTT1023, with 76.19% being categorised as unrelated and 11.11% being unlikely to be related.
Report Supplementary Material 11, Table S49, gives information on all grade 3 and higher AEs that were deemed to be possibly, probably or definitely related to BTT1023. Report Supplementary Material 11, Table S49, shows that there were 15 instances of a related AE of grade 3 or higher, with only one event being grade 4 and only two events being definitely related. Of the 15 related events, eight were experienced by patient 22.
Report Supplementary Material 11, Table S50, shows the number of AEs of any grade categorised according to CTCAE category by treatment visit. The most common category was investigations, with 802 (70.72%) AEs.
Report Supplementary Material 11, Table S51, shows the duration of AE by CTCAE grade. When the duration of the AE is missing, in all 271 cases this was attributable to the AE being ongoing. Further details regarding which visits these were attributable to are given in Report Supplementary Material 11, Table S52. Moreover, from Report Supplementary Material 11, Table S51, it was apparent that both the mean and median duration of an AE were noticeably longer for grade 4 AEs (mean 90.0 days; median 74 days) than for grade 1–3 AEs, while the range of AE duration was greatest for grade 1 AEs (range 0–2411 days).
Report Supplementary Material 11, Table S52, shows the visits on which the ongoing AEs occurred. Only 23.61% of ongoing AEs occurred during the treatment period (42.44% occurred either pre-screening or at screenings 1 or 2, whereas 33.95% occurred during one of the follow-up visits).
Finally, Report Supplementary Material 11, Table S53, shows the AE outcome by visit. Overwhelmingly, AEs were resolved without sequelae (76.72%), with the second most common outcome being unresolved (23.19%).
Serious adverse events
In total, four SAEs were experienced by four patients, with no one individual experiencing more than one SAE. Table 8 gives a full line listing of all SAEs reported during the BUTEO trial and Report Supplementary Material 11, Table S54, lists the toxicity that the site identified as the AE that prompted the SAE report. The population for the incidence of SAEs was taken to be the safety population.
| TNO | Treatment | Reason | Reason other | Category | Event | Other event | Grade | Relatedness/causality | Other grade | Outcome | Sequelae |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 5 | BTT1023 | Hospitalisation | Allergic reaction to IMP | Non-fatal/life-threatening SUSAR | Infusion-related reaction | NA | 3 | Definitely related | NA | Resolved: no sequelae | |
| 16 | BTT1023 | Hospitalisation | Unrelated SAE | Diarrhoea | Vomiting and gastrointestinal pain | 3 | Unlikely to be related | 2, 1 | Resolved: no sequelae | ||
| 17 | BTT1023 | Other | Patient sent for liver transplant assessment. Principal investigator thought this worthy of reporting because of the unusual schedule of events | Non-fatal/life-threatening SUSAR | Blood bilirubin levels increased | Aspartate aminotransferase increased, ALP activity increased and alanine aminotransferase increased | 4 | Possibly related | 4, 3, 4 | Resolved: with sequelae | Liver transplant |
| 18 | BTT1023 | Other | Medically important event. Patient required urgent outpatient treatment | Unrelated SAE | Colitis | NA | 3 | Unlikely to be related | NA | Resolved: no sequelae |
Secondary outcome measures
In this section, when percentage change calculations were made, the response was calculated as the difference between the first follow-up visit (visit 10) and either screening visit 2 or visit 3 (i.e. the first treatment visit), depending on when each outcome was measured. The stipulation is given at the start of each section.
Calculation of percentage change was always performed with reference to the baseline period, as:
Therefore, a positive percentage change indicates an increase in the measurement between baseline and follow-up, and a negative percentage change indicates a decrease.
Tolerability and treatment compliance
Information pertinent to tolerability (i.e. incidence of AEs and SAEs) can be found in Safety and toxicity reporting and Serious adverse events, above.
In total, 22 patients were registered to the trial and received treatment, thereby, constituting the mITT population. Only one patient (TNO10) registered to the trial did not start trial treatment. The following reason was given: ‘Patient found to be ineligible post registration’.
The BUTEO trial began with an initial dose-confirmatory stage before continuing onto a Simon’s two-stage, single-arm, open-label, multicentre Phase II trial. Report Supplementary Material 11, Table S55, shows the number of patients recruited to each phase of the trial.
If a patient had been fully compliant with the protocol, then they would have attended 11 visits (i.e. screening visits 1 and 2, treatment visits 3–9 and follow-up visits 10 and 11). Report Supplementary Material 11, Table S56, shows the number of patients who complied with the visits per protocol. Report Supplementary Material 11, Table S57, shows descriptive characteristics regarding the number of visits. The reasons for patients not being fully compliant with trial visits were as follows:
-
patient withdrew from study (n = 2, patients 2 and 11)
-
patient missed one treatment cycle, the reason why was unknown (n = 1, patient 15).
From the trial schema, the length of trial treatment from the first screening visit to final follow-up visit was 77 days. Report Supplementary Material 11, Table S58, gives a per-patient listing of length of time on trial treatment and Report Supplementary Material 11, Table S59, gives summary demographics of average length of time on treatment.
Note that in Report Supplementary Material 11, Table S58, the duration of treatment for patient 2 has been hard-coded and manually changed. Patient 2 received the first treatment and immediately withdrew after treatment (because of feelings of unwellness; see Report Supplementary Material 6). When calculating time on treatment, as both the treatment and withdrawal happened on the same day, this comes out as 0 days. To reflect the fact that one cycle of treatment was received, this was hard-coded to be 1. The code for this is given in a note to file in the statistics trial master file.
The dose confirmation phase included the possibility of dose escalation of BTT1023 from 8 mg/kg to potentially 12 or 16 mg/kg, as appropriate, dependent on fulfilment of PK criteria (given in the protocol). Report Supplementary Material 11, Table S60, shows summary information regarding the dose of BTT1023 allocated.
As the minimum trough criterion was met in the first dose explored, no further escalation was required. Therefore, all patients received the same dose.
Report Supplementary Material 11, Table S61, shows the target dose and actual dose received by patient and treatment visit. The target dose was calculated using the treatment allocation. Report Supplementary Material 11, Table S62, then shows how much the allocated treatment deviates from the target dose. This deviation never exceeded 5% in either direction.
Report Supplementary Material 11, Table S63, shows a line listing of per-patient compliance information. In total, there were zero deaths throughout the trial.
Full details on withdrawal and discontinuations are given in Report Supplementary Material 6, Tables S2–S4. During the trial, there were two discontinuations: one patient after one infusion of BTT1023 (patient TNO2) for general feelings of unwellness and one patient after six infusions of BTT1023 (patient TNO11) for toxicity associated with increase in pain.
EuroQol-5 Dimensions questionnaire
The EQ-5D questionnaire comprised two component parts:
-
EuroQol-5 Dimensions, five-level version (EQ-5D-5L), questionnaire.
-
EuroQol Visual Analogue Scale (EQ-VAS).
The results for EQ-5D-5L and EQ-VAS are shown separately.
For each EQ-5D measurement, the following calculations are made: The difference is calculated as:
and the percentage change is calculated as:
EuroQol-5 Dimensions, five-level version
The EQ-5D-5L descriptive system comprises five dimensions (i.e. mobility, self-care, usual activities, pain/discomfort and anxiety/depression). Each dimension has five response levels (i.e. no problems, slight problems, moderate problems, severe problems and unable to/extreme problems). The respondent is asked to indicate their current health state by checking the box next to the most appropriate response level for each of the five dimensions. Responses are coded as single-digit numbers (1–5) expressing the severity level selected in each dimension, with lower numbers equating to better functionality. The digits for the five dimensions can be combined in a five-digit code that describes the respondent’s health state.
In total, there are 3125 possible health states (i.e. 55).
Each health state is then transformed to an index score using England-specific estimates attained from Devlin et al. 19 Transformation of the health state to an index score is country specific, with no global estimates available. As all patients were registered to and treated at hospitals in England, using only the England-specific estimates is not unreasonable. The maximum index score (corresponding to a health state of 11,111, no problems in any dimension) is 1.00, whereas the minimum index score (corresponding to a health state of 55,555, unable to/extreme problems in all dimensions) is –0.285. Therefore, an individual who has health state of 55,555 is considered to be in a state worse than death. For EQ-5D-5L index scores, higher scores equate to better functionality.
Report Supplementary Material 11, Table S64, presents descriptive analyses of the index score at visits 3 and 10 (follow-up), and the difference between them. Report Supplementary Material 11, Table S65, then shows the patient-level absolute difference and percentage change in EQ-5D-5L index scores. Report Supplementary Material 11, Table S66, shows summary measures pooled across the whole mITT population.
Throughout the trial duration, the EQ-5D-5L questionnaire was answered at trial visits 3, 6 and 10. Report Supplementary Material 11, Figure S6, shows a repeated measures plot of the index score with a locally weighted smoothing (LOESS) trend line to help evaluate any potential trend in scores.
Given the extent of heterogeneity surrounding patient-specific intercepts of EQ-5D-5L index score, and the negligible trend in time of the summary smoother line, any formal repeated-measures analysis would be futile.
EuroQol Visual Analogue Scale
The EQ-VAS provides a quantitative measure of the patient’s perception of their overall health. The EQ-VAS records the respondent’s overall current health on a vertical VAS, on which the end points are labelled ‘The best health you can imagine’ and ‘The worst health you can imagine’, corresponding to values of 100 and 0, respectively. An increase in patient-perceived health is, therefore, represented by an increase in VAS score.
As the VAS can take integer values between 0 and 100 only, there is debate as to whether to treat such a measurement as ordinal or continuous. However, as only descriptive analyses with exploratory modelling with no formal testing are being presented, the outcome is considered to be continuous.
Report Supplementary Material 11, Table S67, presents descriptive analyses of the index score at visits 3 and 10 (follow-up), and the difference between them. Report Supplementary Material 11, Table S68, then shows the patient-level absolute difference and percentage change in EQ-5D VAS index scores. Report Supplementary Material 11, Table S69, shows summary measures pooled across the whole mITT population.
Throughout the trial duration, the EQ-5D VAS scale questionnaire was completed at trial visits 3, 6 and 10. Report Supplementary Material 11, Figure S7, shows a repeated-measures plot of the VAS score with a LOESS trend line to help evaluate any potential trend in scores.
Akin to the analysis of EQ-5D-5L, Report Supplementary Material 11, Figure S7, shows there to be an inconsequential trend in the trend line, with patient-level intercepts ranging from 40 to 100. Therefore, no repeated-measures analysis of this outcome was carried out.
Fatigue Severity Scale
The FSS is a method of evaluating the impact of fatigue on a patient through methods of a short questionnaire that requires patients to rate their level of fatigue. The FSS contains nine statements, graded from 1 to 7 based on how accurately the statement reflects the patient’s condition during the past week and the extent to which they agree or disagree that the statement applies to them. A low value (e.g. 1) indicates strong disagreement with the statement, whereas a high value (e.g. 7) indicates strong agreement.
As patients were themselves asked to calculate their score through simple addition, a check was first completed to ensure that the component parts of the questionnaire and the total FSS score were in agreement. No discrepancies were identified.
The following calculations are made: The difference is calculated as:
and the percentage change is calculated as:
Report Supplementary Material 11, Table S70, presents descriptive analyses of the index score at visits 3 and 10 (follow-up), and the difference between them. Report Supplementary Material 11, Table S71, then shows the patient-level absolute difference and percentage change in FSS scores. Report Supplementary Material 11, Table S72, shows summary measures pooled across the whole mITT population.
Report Supplementary Material 11, Figure S8, shows a repeated-measures plot of the FSS score with a LOESS trend line (shown in dark blue, with uncertainty shown by the shaded region) to help evaluate any potential trend in scores.
From Report Supplementary Material 11, Figure S8, it can be seen that there is significant heterogeneity surrounding patient-specific intercepts of FSS, such that repeated-measures analysis would not be fruitful. Therefore, no such analysis was carried out.
Pruritus visual analogue scale
The purpose of the pruritus VAS was to measure the amount of pruritus (itching) that patients experienced while participating in the BUTEO trial. The following reference was given: ‘Mark with a pen on the line below how much you were bothered by itchiness over the past 24 hours. The far left indicates “no itching” and far right indicates ‘intolerable/severe itching’. No itching corresponds to a score of 0.
As only descriptive analyses are to be shown, with no hypothesis testing, the pruritus VAS shall be assumed to be continuous analogous to the analysis of the EQ-5D VAS.
The following calculations are made: The difference is calculated as:
and the percentage change is calculated as:
Report Supplementary Material 11, Table S73, presents descriptive analyses of the index score at visits 3 and 10 (follow-up) and the difference between them. Report Supplementary Material 11, Table S74, shows the patient-level absolute difference and percentage change in pruritus VAS index scores. Report Supplementary Material 11, Table S75, shows summary measures pooled across the whole mITT population.
Report Supplementary Material 11, Figure S9, shows a repeated-measures plot of the pruritus VAS score with trend line to help evaluate any potential trend in scores.
Report Supplementary Material 11, Figure S9, shows that there is no discernible trend in the total pruritus VAS score changes over time and, therefore, as any formal repeated measures analysis would be ineffectual, no such analysis was carried out.
Inflammatory bowel disease diaries
The IBD diary records outcomes on seven sequential days, before visits 2, 6 and 10. It encompasses the following elements:
-
number of stools
-
how often there was blood in the stools
-
abdominal pain or cramps (graded as 0 = good, 1 = average, 2 = poor and 3 = very poor)
-
general well-being (graded as 0 = good, 1 = average, 2 = poor and 3 = very poor).
The IBD diary was completed by 14 patients. All participants who completed the diary had IBD, and all participants who did not complete the diary did not have IBD.
As the diary was completed on seven consecutive days before visit, for the descriptive analyses provided here, the average (either mean or median average) was first calculated for each time point, and then the difference in averages calculated.
The results for each item of the IBD diary are shown separately. Tables are presented showing descriptive analyses of each question at visit 2 (screening) and visit 10 (follow-up) (see Report Supplementary Material 11, Tables S76–S84).
For each question of the IBD diary, the following calculations are made: The difference is calculated as:
and the percentage change is calculated as:
No repeated-measures analysis was undertaken, as there were only 14 patients with IBD. Moreover, there was a high presence of zeros for a lot of questions.
Number of stools
Owing to the positive skew of number of stools, the median average is used. Report Supplementary Material 11, Table S76, shows the summary of the median number of stools per day at visits 2 and 10, and the difference in the median number of stools per day between these two visits. Report Supplementary Material 11, Table S77, shows the average absolute difference and percentage change in median number of stools per day.
Note that there exists one instance where the upper bound cannot be calculated (reported as ‘Inf’), as the median number of stools per day at screening visit 2 was zero. Therefore, Report Supplementary Material 11, Table S78, shows the average absolute difference and percentage change in median number of stools per day (as in Report Supplementary Material 11, Table S77), but omitting any person who had a median of zero stools per day at screening visit 2.
Report Supplementary Material 11, Figure S10, presents a repeated-measure plot of the median number of stools per day for all patients with IBD who completed the IBD diaries. A LOESS trend line has been added to help evaluate any potential trend.
Because the subset of patients who completed the IBD diary is small, and, as there is no discernible trend in the trend line on Report Supplementary Material 11, Figure S10, no repeated-measures analysis has been performed.
Frequency of blood in stool
The median average is again used. Report Supplementary Material 11, Table S79, shows the summary of the median number of blood-containing stools per day at visits 2 and 10, and the difference in the median number of blood-containing stools per day between these two visits. Report Supplementary Material 11, Table S80, shows the average absolute difference and percentage change in median frequency of blood in stools per day.
Owing to the increase propensity for there to be zero frequency of blood in stools per day, many of the percentage change characteristics cannot be calculated.
Report Supplementary Material 11, Figure S11, presents a repeated-measures plot of the median frequency of blood in stools per day for all patients with IBD who completed the IBD diaries.
As the median frequency of blood in stools is zero for all patients at all time points, any repeated-measures analysis would be inappropriate.
Abdominal pain/cramp
As patients were asked about their experience on 7 consecutive days, the median should always fall on an integer, providing meaningful values, given the categorical nature of the data. However, to ensure that this is the case, the summary figures for each individual’s average abdominal pain have been rounded to the nearest integer.
Report Supplementary Material 11, Table S81, shows the summary of the median average abdominal pain in the weeks before visits 2 and 10, and the difference in these figures. Report Supplementary Material 11, Table S82, shows the average absolute difference and percentage change in median average abdominal pain in the weeks preceding visits 2 and 10.
Note that, as many individuals reported their abdominal pain to be ‘good’ (graded 0) at one or both visits, many of the percentage changes cannot be calculated, expressed as either ‘Inf’ (where only screening visit 2 was zero) or ‘NaN’ (where pain at both visits was graded 0).
Report Supplementary Material 11, Figure S12, presents a repeated-measures plot of the median abdominal pain in the week preceding trial visit for all patients with IBD who completed the IBD diaries.
Owing to the categorical nature of the abdominal pain score, no repeated-measures analysis has been performed.
General well-being
The median averages are used here. As the patients are asked about their experience on seven consecutive days, the median should always fall on an integer, giving meaningful values given the categorical nature of the data. However, to ensure this, the summary figures for each individual’s average general well-being have been rounded to the nearest integer.
Report Supplementary Material 11, Table S83, shows the summary of the median average general well-being pain in the weeks before visits 2 and 10, and the difference in these figures. Report Supplementary Material 11, Table S84, shows the average absolute difference and percentage change in median average general well-being in the weeks preceding visits 2 and 10.
Note that, as many individuals reported their abdominal pain to be ‘good’ (graded 0) at one or both visits, many of the percentage changes cannot be calculated, expressed as either ‘Inf’ (where general well-being 2 was graded 0 only at the screening visit) or ‘NaN’ (where it was graded 0 at both visits).
Report Supplementary Material 11, Figure S13, presents a repeated-measures plot of the median general well-being score in the week preceding trial visit for all patients with IBD who completed the IBD diaries.
Owing to the categorical nature of the general well-being score, no repeated-measures analysis has been performed.
Enhanced liver fibrosis
The ELF blood test combines three serum biomarkers (i.e. hyaluronic acid, procollagen III amino-terminal peptide and tissue inhibitor of metalloproteinase 1) to quantify the severity of liver fibrosis.
Enhanced liver fibrosis score
Report Supplementary Material 11, Table S85, presents descriptive analyses of the ELF score at visits 3 and 10, and the difference between them. Report Supplementary Material 11, Table S86, then shows the patient-level absolute difference and percentage change in ELF score. Finally, Report Supplementary Material 11, Table S87, shows summary measures pooled across the whole mITT population.
Report Supplementary Material 11, Figure S14, shows a repeated-measures plot of the ELF score with a LOESS trend line to help evaluate any potential trend in scores.
As the ELF blood test was assessed at only two points, any repeated-measures analysis would be futile.
Enhanced liver fibrosis category
The ELF score can then be categorised according to the following scale:
-
no liver fibrosis to mild liver fibrosis – ELF score < 7.7
-
moderate liver fibrosis – 7.7 ≤ ELF score < 9.8.
-
severe liver fibrosis – ELF score ≥ 9.8.
Report Supplementary Material 11, Table S88, shows a descriptive summary of the ELF category and Report Supplementary Material 11, Table S89, gives a patient line listing of ELF category at each trial visit.
Report Supplementary Material 11, Figure S15, presents a repeated-measures plot of the ELF category.
Owing to the categorical nature of the ELF score, and as it was calculated at only two trial visits, no repeated-measures analyses have been performed.
Enhanced liver fibrosis components
FibroScan kPa
The FibroScan test has two outcome measures: kPa and IQR. The results for these are shown separately in Table 9. For each measurement, the following calculations are made:
and the percentage change is calculated as:
| Measure | FibroScan measurement (kPa) | Difference between measurements at each visit | |
|---|---|---|---|
| Screening visit 2 (N = 22) | Follow-up visit 10 (N = 22) | ||
| Mean | 15.54 | 15.05 | –0.48 |
| Median | 13.55 | 13.20 | 0.20 |
| Range | 3.50 to 38.00 | 7.50 to 32.40 | –15.00 to 5.80 |
| IQR | 7.80 to 19.05 | 9.10 to 17.75 | –3.50 to 3.60 |
Report Supplementary Material 11, Figure S17, shows a repeated-measures plot of the FibroScan measurement, with a LOESS trend line to help evaluate any potential trend in scores.
As FibroScan measurement was undertaken at visits 2 and 10 only, any repeated-measures analysis would be inappropriate and, therefore, has not been carried out.
Interquartile range
Report Supplementary Material 11, Figure S18, shows a repeated-measures plot of the FibroScan measurement IQR, with a LOESS trend line to help evaluate any potential trend in scores.
As FibroScan measurement was undertaken at visits 2 and 10 only, any repeated-measures analysis would be inappropriate and, therefore, has not been carried out.
Liver function tests
The following tests of liver function were carried out:
-
AST
-
ALT
-
ALP
-
GGT
-
albumin
-
direct bilirubin
-
indirect bilirubin
-
INR.
The results for each test of liver function are shown separately. Table 10 shows descriptive analyses of each measurement of liver function at visit 1 (first treatment visit, pre infusion) and at visit 11 (follow-up).
| Test of liver function | Test result | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Visit 1 (screening) | Visit 2 (screening) | Visit 3 (pre dose) | Visit 4 (pre dose) | Visit 5 (pre dose) | Visit 6 (pre dose) | Visit 7 (pre dose) | Visit 8 (pre dose) | Visit 9 (pre dose) | Visit 10 (follow-up) | Visit | |
| AST (IU/l) | |||||||||||
| n | 21 | 22 | 22 | 20 | 19 | 20 | 18 | 19 | 17 | 20 | 19 |
| Mean | 99.24 | 87.77 | 87.45 | 75.20 | 76.00 | 73.35 | 78.33 | 82.79 | 78.82 | 78.25 | 181.53 |
| Median | 79.00 | 74.00 | 71.00 | 63.50 | 80.00 | 57.50 | 63.00 | 67.00 | 72.00 | 73.50 | 82.00 |
| Range | 18.00–357.00 | 24.00–249.00 | 26.00–250.00 | 20.00–146.00 | 21.00–136.00 | 30.00–137.00 | 21.00–170.00 | 28.00–214.00 | 22.00–219.00 | 23.00–230.00 | 25.00–1732.00 |
| IQR | 56.00–124.00 | 45.50–104.00 | 48.25–102.25 | 47.00–102.25 | 42.50–108.00 | 44.00–98.00 | 43.25–100.50 | 46.50–103.50 | 48.00–105.00 | 47.75–94.50 | 49.50–129.50 |
| ALT (IU/l) | |||||||||||
| n | 22 | 22 | 22 | 20 | 21 | 21 | 18 | 19 | 19 | 21 | 22 |
| Mean | 109.77 | 94.68 | 95.14 | 85.80 | 80.81 | 76.38 | 80.44 | 84.21 | 84.42 | 93.33 | 130.86 |
| Median | 84.00 | 73.50 | 71.00 | 73.50 | 73.00 | 68.00 | 66.00 | 68.00 | 74.00 | 72.00 | 84.00 |
| Range | 32.00–303.00 | 33.00–265.00 | 27.00–255.00 | 26.00–227.00 | 29.00–204.00 | 28.00–221.00 | 21.00–214.00 | 26.00–240.00 | 20.00–193.00 | 29.00–249.00 | 19.00–841.00 |
| IQR | 70.25–135.50 | 56.00–107.75 | 60.50–118.50 | 54.25–86.75 | 53.00–97.00 | 47.00–86.00 | 54.25–87.75 | 58.50–101.50 | 59.50–92.50 | 53.00–123.00 | 58.25–112.75 |
| ALP (IU/l) | |||||||||||
| n | 22 | 22 | 22 | 20 | 21 | 21 | 19 | 20 | 20 | 22 | 22 |
| Mean | 477.64 | 450.77 | 443.00 | 403.40 | 397.52 | 402.19 | 412.42 | 446.95 | 453.50 | 456.18 | 479.00 |
| Median | 406.50 | 346.50 | 345.00 | 316.50 | 335.00 | 328.00 | 314.00 | 338.50 | 318.50 | 354.50 | 335.50 |
| Range | 199.00–1318.00 | 215.00–1075.00 | 180.00–1069.00 | 217.00–934.00 | 197.00–858.00 | 215.00–929.00 | 213.00–981.00 | 229.00–1047.00 | 195.00–1177.00 | 142.00–1407.00 | 172.00–2046.00 |
| IQR | 295.50–528.00 | 311.25–565.75 | 288.25–549.25 | 286.25–468.25 | 268.00–467.00 | 264.00–464.00 | 260.50–483.50 | 263.75–497.75 | 252.75–550.75 | 273.25–495.75 | 264.50–516.00 |
| GGT (IU/l) | |||||||||||
| n | 20 | 21 | 22 | 19 | 21 | 21 | 19 | 19 | 19 | 21 | 21 |
| Mean | 732.85 | 643.24 | 609.45 | 515.79 | 608.00 | 556.81 | 444.53 | 590.21 | 549.37 | 583.76 | 630.10 |
| Median | 447.50 | 398.00 | 460.00 | 356.00 | 336.00 | 345.00 | 333.00 | 313.00 | 357.00 | 342.00 | 356.00 |
| Range | 77.00–2857.00 | 69.00–1918.00 | 76.00–1773.00 | 73.00–1694.00 | 59.00–2378.00 | 65.00–2142.00 | 66.00–1402.00 | 108.00–1817.00 | 78.00–2058.00 | 86.00–2032.00 | 74.00–2242.00 |
| IQR | 297.25–1096.75 | 249.00–1079.00 | 245.00–984.75 | 210.00–661.50 | 229.00–951.00 | 237.00–821.00 | 212.00–499.50 | 195.50–997.00 | 197.50–679.00 | 213.00–924.00 | 180.00–872.00 |
| Albumin (g/l) | |||||||||||
| n | 22 | 22 | 22 | 21 | 21 | 21 | 20 | 21 | 20 | 22 | 22 |
| Mean | 41.32 | 41.59 | 40.45 | 39.71 | 39.24 | 39.10 | 38.40 | 38.90 | 38.45 | 40.45 | 40.05 |
| Median | 43.00 | 43.00 | 42.00 | 42.00 | 40.00 | 39.00 | 39.00 | 41.00 | 40.00 | 42.00 | 41.50 |
| Range | 29.00–50.00 | 31.00–48.00 | 30.00–49.00 | 31.00–47.00 | 31.00–47.00 | 29.00–47.00 | 29.00–45.00 | 29.00–44.00 | 26.00–46.00 | 29.00–47.00 | 28.00–48.00 |
| IQR | 37.00–44.00 | 38.00–46.00 | 36.75–43.75 | 35.00–42.00 | 35.00–42.00 | 36.00–43.00 | 35.00–41.25 | 34.00–42.00 | 34.75–42.00 | 36.25–43.00 | 36.50–44.00 |
| Direct bilirubin (µmol/l) | |||||||||||
| n | 19 | 19 | 20 | 20 | 17 | 18 | 17 | 19 | 18 | 21 | 20 |
| Mean | 12.89 | 15.11 | 13.90 | 12.45 | 12.12 | 12.22 | 13.12 | 14.32 | 11.56 | 13.62 | 19.55 |
| Median | 13.00 | 12.00 | 8.50 | 7.50 | 6.00 | 9.50 | 10.00 | 11.00 | 6.00 | 8.00 | 12.00 |
| Range | 2.00–38.00 | 2.00–44.00 | 2.00–35.00 | 2.00–48.00 | 2.00–38.00 | 2.00–35.00 | 2.00–30.00 | 1.00–36.00 | 2.00–32.00 | 3.00–47.00 | 3.00–136.00 |
| IQR | 5.50–16.50 | 6.50–21.00 | 5.00–24.50 | 4.75–17.50 | 4.00–19.00 | 5.25–17.75 | 5.00–22.00 | 5.50–21.50 | 5.00–18.75 | 5.00–17.00 | 4.00–21.50 |
| Indirect bilirubin (µmol/l) | |||||||||||
| n | 18 | 17 | 19 | 19 | 17 | 16 | 15 | 18 | 17 | 19 | 19 |
| Mean | 7.50 | 9.76 | 8.16 | 8.84 | 7.94 | 8.56 | 9.27 | 8.56 | 7.47 | 8.26 | 8.63 |
| Median | 6.50 | 8.00 | 7.00 | 9.00 | 7.00 | 8.00 | 8.00 | 8.50 | 6.00 | 7.00 | 8.00 |
| Range | 2.00–19.00 | 1.00–25.00 | 2.00–23.00 | 1.00–25.00 | 2.00–21.00 | 2.00–16.00 | 2.00–20.00 | 2.00–20.00 | 1.00–23.00 | 1.00–19.00 | 3.00–19.00 |
| IQR | 5.00–9.75 | 6.00–11.00 | 5.00–10.50 | 4.00–11.50 | 5.00–10.00 | 5.00–10.75 | 5.00–14.50 | 5.00–12.50 | 4.00–11.00 | 5.00–11.50 | 4.00–12.50 |
| INR | |||||||||||
| n | 22 | 22 | 22 | 21 | 20 | 21 | 19 | 21 | 20 | 22 | 20 |
| Mean | 1.00 | 1.01 | 1.01 | 1.00 | 1.01 | 1.02 | 1.02 | 1.00 | 1.02 | 1.00 | 1.02 |
| Median | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| Range | 0.90–1.10 | 0.90–1.20 | 0.90–1.20 | 0.90–1.20 | 0.90–1.20 | 0.90–1.20 | 0.90–1.20 | 0.90–1.10 | 0.90–1.20 | 0.80–1.20 | 0.90–1.20 |
| IQR | 1.00–1.00 | 1.00–1.075 | 1.00–1.00 | 1.00–1.00 | 1.00–1.00 | 1.00–1.00 | 1.00–1.00 | 1.00–1.00 | 1.00–1.00 | 1.00–1.075 | 1.00–1.025 |
Descriptive analyses regarding the differences in the liver function tests are also shown.
For each liver function test measurement, the following calculations are made. The difference is calculated as:
and the percentage change is calculated as:
In this section, the measurement data pertinent to tests of liver function originate from blood samples analysed at the patient’s registered local hospital laboratory. This is in contrast to the primary outcome, in that all ALP data came from a centralised laboratory in Birmingham.
Throughout this section, which assesses the change in tests of liver function, the pre-infusion values at visit 3 are used as the baseline value.
Breakdowns of each liver function test are given Report Supplementary Material 11.
Model for end-stage liver disease
Report Supplementary Material 11, Tables S125 and S127, shows descriptive analyses of each measurement of the MELD score at visit 2 (screening) and visit 10 (follow-up). Descriptive analyses regarding the difference in the liver function test measurement are also shown.
The difference is calculated as:
and the percentage change is calculated as:
Note that there exists one instance in which a trial participant has the component parts of the MELD score recorded but not the calculated score.
Report Supplementary Material 11, Figure S22, shows a repeated-measures plot of the MELD score with trend line.
As the MELD score was measured at visits 2 and 10 only, any repeated-measures analysis would be inappropriate and, therefore, has not been carried out.
Mayo primary sclerosing cholangitis risk score
Table 11 and Report Supplementary Material 11, Table S129, show descriptive analyses of each measurement of the Mayo PSC risk score at visits 2 and 10. Descriptive analyses regarding the difference in the liver function test measurement are also shown.
| Measure | Mayo PSC risk score | Difference | |
|---|---|---|---|
| Screening visit 2 (N = 22) | Follow-up visit 10 (N = 20) | ||
| Mean | 0.82 | 0.79 | –0.08 |
| Median | 0.75 | 0.62 | –0.12 |
| Range | 0.03 to 2.68 | 0.02 to 2.84 | –0.58 to 0.40 |
| IQR | 0.25 to 1.14 | 0.28 to 1.12 | –0.19 to 0.08 |
The difference is calculated as:
and the percentage change is calculated as:
For the secondary outcome measure of Mayo PSC score, patient 22 at visit 10 had a Mayo PSC risk score of 24.6981. For all other patients at all other time points, the Mayo PSC risk score falls between 0 and 3. The score for patient 22 at visit 10 is, therefore, excessively high and appears to be an outlier.
After investigation into the case report form and the online calculator used, it was concluded that the units for the component parts differed between the two time points, with alterations needing to be made.
Owing to the length of time that elapsed between the end of patient follow-up and the identification of this data query, no trial staff were still available at the site in question. Therefore, on consultation with the trials team, the value has been changed in the analysis to the correct value by the trial statistician.
Evidence of for the coding of this (including calculation of the correct score) is stored within the statistics trial master file.
Report Supplementary Material 11, Figure S23, shows a repeated-measures plot of the Mayo PSC risk score, with a LOESS trend line to help evaluate any potential patterns in scores over time.
As Mayo PSC risk score was measured at visits 2 and 10 only, any repeated-measures analysis would be inappropriate and, therefore, has not been carried out.
Pharmacokinetic data
All PK data and analysis pertaining to PK data are given in Report Supplementary Material 11.
LiverMultiScan magnetic resonance imaging data
Serum vascular adhesion protein 1/semicarbazide-sensitive amine oxidase liver disease activity biomarker: semicarbazide-sensitive amine oxidase enzyme activity
Semicarbazide-sensitive amine oxidase assays were performed on serum sent to the laboratories at the University of Birmingham, and the laboratories forwarded the results of the completed case report form to the CRCTU. Enzyme activity was assessed at all trial visits [i.e. both screening visits (trial visits 1 and 2), all infusion visits (trial visits 3–9) and both follow-up visits (trial visits 10 and 11)].
Semicarbazide-sensitive amine oxidase monitors the enzyme activity of VAP-1 and is expressed as the pool of H2O2 generated per minute. The difference in SSAO activity between follow-up visit 10 and the first infusion visit (i.e. visit 3) is calculated as:
and the percentage change is calculated as:
In total, SSAO enzyme activity was assessed in only 14 patients.
Figure 3 shows a repeated-measures plot of all SSAO activity at all trial visits in the mITT population. A LOESS trend line has been added to aid identification of any potential trend. Table 12 shows a summary of SSAO enzyme activity over all trial visits. The internal controls (positive control, human serum control and human serum plus VAP control) are also shown.
FIGURE 3.
Repeated-measures plot of SSAO enzyme activity at all trial visits in the mITT population. A LOESS trend line is shown in dark blue (thicker line) with uncertainty depicted by the shaded region.
| Measure | SSAO enzyme activity (pmol H2O2 generated per minute) | POS CTL | HS CTL | HS + VAP CTL | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Visit 1 | Visit 2 | Visit 3 | Visit 4 | Visit 5 | Visit 6 | Visit 7 | Visit 8 | Visit 9 | Visit 10 | Visit 11 | ||||
| Mean | 0.22 | 0.26 | 0.26 | 0.29 | 0.25 | 0.21 | 0.19 | 0.18 | 0.17 | 0.17 | 0.18 | 0.51 | 0.10 | 0.18 |
| Median | 0.21 | 0.22 | 0.23 | 0.24 | 0.22 | 0.19 | 0.17 | 0.14 | 0.13 | 0.10 | 0.08 | 0.53 | 0.10 | 0.18 |
| Range | 0.11–0.42 | 0.13–0.59 | 0.12–0.6 | 0.17–0.76 | 0.14–0.46 | 0.11–0.37 | 0.13–0.38 | 0.09–0.41 | 0.09–0.42 | 0.08–0.72 | 0.03–1.33 | 0.24–0.92 | 0.08–0.12 | 0.13–0.25 |
| IQR | 0.17–0.23 | 0.17–0.31 | 0.16–0.28 | 0.21–0.31 | 0.17–0.31 | 0.16–0.24 | 0.14–0.21 | 0.12–0.20 | 0.11–0.20 | 0.09–0.17 | 0.07–0.10 | 0.41–0.59 | 0.09–0.11 | 0.16–0.19 |
Report Supplementary Material 11, Table S212, presents descriptive analyses of SSAO enzyme activity at visit 3 (i.e. the first treatment visit) and at visit 10 (i.e. the follow-up visit), and the difference between them. Report Supplementary Material 11, Table S213, then shows the patient-level absolute difference and percentage change in SSAO activity. Finally, Report Supplementary Material 11, Table S214, shows summary measures pooled across the whole mITT population.
Figure 3 shows a slight downwards trend over all trial visits and, therefore, the repeated-measures analysis is performed. i is the index patient number and j is the index trial visit number, for j = 1, 2, . . ., 11. Then consider the following mixed-effects model:
The above model was fitted using the lme4 package in R using restricted maximum likelihood to attain the following estimates:
where ai ~ N (0, 0.11632) and εij ~ N (0, 0.0094872).
Report Supplementary Material 11, Figure S49, shows a repeated-measures plot of all SSAO enzyme activity over all trial visits, with a line depicting the fixed effects from the above mixed-effects model.
Report Supplementary Material 11, Figure S50, shows a repeated-measures plot of SSAO enzyme activity as a percentage of the screening 1 value across all trial visits in the mITT population.
Serum vascular adhesion protein 1/semicarbazide-sensitive amine oxidase
The difference in sVAP-1/SSAO between follow-up visit 10 and the first infusion visit (i.e. visit 3) is calculated as:
and the percentage change is calculated as:
Owing to many sVAP-1 samples being below the level of quantification and, therefore, here assumed to be missing, only 14 patients had sVAP-1/SSAO activity assessed at any time point, with only five patients being evaluable for quantification of percentage change between visits 3 and 10.
Report Supplementary Material 11, Figure S51, shows a repeated-measures plot of all SSAO activity at all trial visits in the mITT population. A LOESS trend line has been added to aid identification of any potential trend. Table 13 shows a summary of SSAO enzyme activity over all trial visits.
| Summary | SSAO enzyme activity (pmol H2O2 generated per minute) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Visit 3 (N = 13) | Visit 4 (N = 10) | Visit 5 (N = 10) | Visit 6 (N = 8) | Visit 7 (N = 5) | Visit 8 (N = 3) | Visit 9 (N = 1) | Visit 10 (N = 6) | Visit 11 (N = 11) | |
| Mean | 1492.11 | 5.02 | 26.15 | 12.23 | 9.53 | 419.86 | 4.80 | 104.04 | 1122.75 |
| Median | 1547.45 | 4.07 | 9.70 | 8.88 | 8.13 | 11.38 | 4.80 | 8.33 | 1080.83 |
| Range | 634.60–2303.86 | 1.62–9.97 | 3.21–148.78 | 3.30–29.14 | 3.81–19.64 | 4.85–1243.36 | 4.80–4.80 | 1.96–590.99 | 131.24–2276.19 |
| IQR | 1120.72–1769.55 | 3.77–6.18 | 6.42–18.26 | 6.54–14.1 | 5.03–11.06 | 8.11–627.37 | 4.80–4.80 | 3.74–11.40 | 854.94–1404.30 |
Report Supplementary Material 11, Table S215, presents descriptive analyses of sVAP-1/SSAO activity at visits 3 and 10, and the difference between them. Report Supplementary Material 11, Table S216, then shows the patient-level absolute difference and percentage change in sVAP-1/SSAO activity. Finally, Report Supplementary Material 11, Table S217, shows summary measures pooled across the whole mITT population. Owing to the lack of discernible trend identified and significant heterogeneity present in Report Supplementary Material 11, Figure S51, repeated-measures analysis has not been performed.
Immune cell populations as assessed by flow cytometry
Exploratory analysis
The exploratory biomarkers of fibrosis were analysed by Nordic Bioscience (Herlev, Denmark), with external data uploaded to the database.
Analysis of the exploratory biomarkers and biomarker ratios are given in Report Supplementary Material 11.
Additional analyses
Additional analyses have been performed on the following experimental variables derived from laboratory analyses:
-
total bilirubin (biochemistry)
-
sodium (biochemistry)
-
potassium (biochemistry)
-
urea (biochemistry)
-
creatinine (biochemistry)
-
calcium (biochemistry)
-
total protein (biochemistry)
-
eGFR (biochemistry)
-
haemoglobin (haematology)
-
platelets (haematology)
-
red blood cells (haematology)
-
white blood cells (haematology)
-
haematocrit (haematology)
-
mean cell volume (haematology)
-
mean cell haemoglobin (haematology)
-
neutrophils (haematology)
-
lymphocytes (haematology)
-
monocytes (haematology)
-
eosinophils (haematology)
-
basophils (haematology)
-
APTT ratio (haematology).
Results are presented in Report Supplementary Material 11.
Sensitivity analyses
Sensitivity analyses were performed in the per-protocol population and regarded the primary analysis only. Therefore, using centrally processed samples, the sensitivity analyses evaluate the percentage of patients who completed the treatment as originally allocated, with no dose modifications or missing doses (i.e. patients who received all seven infusions as scheduled in the protocol at the minimum effective dose) and with a clinically meaningful reduction in ALP (a reduction of ≥ 25%). In total, 19 patients were compliant with all treatment visits and, therefore, constituted the per-protocol population. Table 14 shows the number of patients in the per-protocol population achieving a target reduction in ALP.
| ALP reduction of ≥ 25% achieved | Number of patients (%) |
|---|---|
| No | 16 (84.21) |
| Yes | 2 (10.53) |
| Not known | 1 (5.26) |
| Total | 19 (100.00) |
One patient was compliant with all treatment visits but was unevaluable for the sensitivity analysis (see Table 14). This is because the patient was not treated at the Queen Elizabeth Hospital and, therefore, their ALP was sent for central processing. However, the sample was lost and, therefore, the participant was treated as a non-responder, contributing to the denominator only.
Figure 4 shows a repeated measures plot of centrally processed ALP for sensitivity analyses in the per-protocol population.
FIGURE 4.
Repeated-measures plot of centrally processed ALP activity for sensitivity analyses in the per-protocol population.
Concomitant medications
Across all patients, there were 231 concomitant medications recorded. Details regarding concomitant medication usage are given in Report Supplementary Material 9.
Deviations
The following contains deviation information on all patients recruited to the trial (regardless of whether or not they received trial treatment). The sections include the patient who was found to be ineligible post randomisation and withdrew immediately (i.e. patient TNO10).
In total, 144 deviations were reported during the BUTEO trial.
Further details regarding deviations can be found in Report Supplementary Material 8.
Chapter 4 Discussion
Summary of main findings
Primary sclerosing cholangitis is a prime example of a progressive inflammatory liver disease characterised by relentless liver fibrosis. There is a high unmet need for new therapies, as no currently licensed therapy has been shown to alter the natural course of the disease. The progression of PSC to scarring, cirrhosis and hepatobiliary cancer is driven by a chronic inflammatory response and immune cell-mediated destruction of bile ducts. Our research suggests that VAP-1 is heavily implicated as a key driver for fibrogenesis and, as such, it provides a target for the possible slowing, or even reversal, of the liver damage seen in PSC.
The unpredictability of PSC along with its designation as a rare orphan disease poses particular challenges in trial design. ALP activity fluctuates during the natural course of the disease, which limits the usefulness as a primary end point; however, ALP is commonly used as a standard marker of PSC disease activity in the absence of a viable alternative. Therefore, reliable biomarkers that correlate with fibrosis stage and progression of liver disease are in demand to predict outcome and to determine disease stage without the need for invasive liver biopsy. This trial will aid in investigating the role of VAP-1 in liver fibrosis and its potential as a therapeutic target and biomarker.
This unique trial design, incorporating dose-confirmatory and safety stages (based on the traditional 3 + 3 design), followed by a Phase II Simon’s two-stage design, was intended to determine a safe and well-tolerated dose of BTT1023 and the efficacy of this treatment in a new disease group. Screening for this trial commenced on 1 February 2015, with the first patient registered on 10 September 2015. The last patient was recruited on 19 June 2018.
It is clear that this study is limited in its design. The limitations were well recognised at the outset and relate, in large part, to the defined budget with which to test this new agent. In the absence of an unlimited opportunity to test a new agent in large numbers of patients and over a prolonged period, it was necessary to aim to see efficacy in a small cohort over a short period of time. Given the absence of any proven biochemical surrogate of disease activity in PSC, in keeping with multiple prior trials in PSC over the last 20 years, ALP activity was chosen as an end point. 8 This is an inherently difficult end point and one that has been challenged many times. Recent work from our group discusses this in detail. 20
Ethics and dissemination
The trial was performed in accordance with the Recommendations of Guiding Physicians in Biomedical Research Involving Human Subjects,21 adopted by the 48th World Medical Association General Assembly, as well as the Research Governance Framework for Health and Social Care, the applicable UK statutory instruments (which include the Medicines for Human Use Clinical Trials 200417 and subsequent amendments and the Data Protection Act 199822) and guidelines for Good Clinical Practice. The protocol was approved by the REC (reference number 14/EM/1272) (see Report Supplementary Material 12 and 13). The first REC approval date was 6 January 2015, with subsequent amendments on 18 March 2015 (non-substantial), 27 November 2015 (substantial), 16 March 2016 (substantial), 27 March 2018 (substantial) and 31 July 2018 (substantial) (see Report Supplementary Material 14). All active sites obtained local research and development department approval and are up to date with the latest protocol amendment.
Report Supplementary Material 14, Table S334, shows the trial milestones and Report Supplementary Material 14, Table S335, shows a summary of approved protocol versions.
Standard regulations for data-handling and patient confidentiality were followed in accordance with the Data Protection Act 201823 for health and care research and General Data Protection Regulation. 24 During the trial, patients were identified using only their unique trial number, initials and date of birth on the case report form, and this information was also used in correspondence between the trial office and the participating site. Data quality was maintained in accordance with trial-specific guidance and was consistent with the source data. All essential trial documentation and source records will be securely retained for at least 25 years after the end of the trial to allow the results of the trial to be verified. Results will be disseminated via peer-reviewed publication and presentation at international conferences, and additional summaries will be provided to patients and patient support groups.
Strengths
-
The trial had a unique, tailor-made clinical trial design that incorporated a dose-confirmatory stage and a safety stage (based on the traditional 3 + 3 design), followed by a Phase II Simon’s two-stage design.
-
The trial brings the translation of laboratory research into a proof of activity clinical trial.
-
This was an early-phase experimental medicine trial of a novel first-in-class drug in a cohort with chronic disease and a large unmet need for new therapies.
-
The trial aimed to address not just the need for new therapies, but also the need for reliable clinical trial end points and biomarkers for staging and predicting clinical outcomes.
Limitations
-
The trial had only a small cohort because PSC is a rare orphan disease. In addition, clinical unpredictability of the disease made stability for clinical trial inclusion difficult.
-
The duration of the treatment period was too short to demonstrate collective markers of efficacy to justify longer and placebo-controlled trials.
-
There was a limited evidence base for the primary end point of a reduction in ALP activity in the context of antifibrotic agents; however, the trial accepts that there is no alternative surrogate currently available.
-
A translational trial taking laboratory science into patients inevitably brings challenges of identifying biological targets that are relevant in vivo.
Research recommendations
The BUTEO trial was stopped prematurely because of lack of efficacy after stage 1 of the Simon’s two-stage design. Despite this, many things can be learnt from the trial, including the challenge of recruiting across multiple sites in a rare disease, as well as how to choose surrogate markers of treatment efficacy. The lessons learned, to date, reflect an ongoing debate in the literature,8 still unresolved, as regards the choice of efficacy end points and trial duration, depending on proposed new therapy modalities. PSC as a disease is very heterogeneous and, overall, slowly progressive. Despite this, the treatment options for this disease, with it its high rates of morbidity and mortality, remain limited. Clinical trials in the area of PSC have, therefore, as in our case, for some time and to this date, remained challenging. Insufficient data remain to advance new biomarkers, but clear efforts and recommendations for future work include evaluation of large prospective recruited PSC cohorts, including biomarker collection, to aid future trial design. Support for the development of real-world cohorts, followed over time, could provide further insights into short- and long-term outcomes that could be correlated with evaluation of new biomarkers or non-invasive tests. This has the potential to enable more evidence-based approaches to study design, which could result in the ability to study new therapies in small, well-defined cohorts, over limited duration, with greater confidence of reaching clear positive or negative trial findings.
Chapter 5 Conclusion
In an open-label study of patients with PSC treated over a 78-day treatment period with BTT1023 (i.e. an anti-VAP1 monoclonal antibody), no preliminary evidence of disease modification could be demonstrated. Overall, the results showed a lack of efficacy. Only 2 out of 18 patients met the primary end point of a reduction in ALP activity of ≥ 25%. As per the protocol and Simon’s two-stage design of the trial, there needed to be at least three successful evaluable responses to BTT1023 at the interim assessment to continue to stage 2 of the trial.
In two further patients, the percentage change recorded was above 17%. A variety of related secondary biochemical efficacy outcomes were assessed and none was met. These included markers of liver fibrosis. Safety was evaluated in an ongoing manner and no treatment-related effects were evident and no new safety signals for BTT1023 were identified. There were a total of 1133 AEs. Eight-five AEs were severe (≤ grade 3), with 15 of these deemed possibly, probably or definitely related to BTT1023. There were four SAEs, but none was fatal.
No DLTs were reported during the dose confirmation stage of the trial. At the prespecified interim analysis, although no safety concerns were identified, the trial DMC confirmed futility, as evaluable results demonstrated that the study had met the prespecified stopping criteria.
Future research in antifibrotic therapy for patients with PSC will require attention to the ongoing debate regarding the optimal end points for assessing efficacy, as well as consideration of duration of treatment, even in early-phase studies.
Acknowledgements
This trial was sponsored by the University of Birmingham and received a favourable ethics opinion from the National Research Ethics Service Committee East Midlands-Derby (14/EM/1272). Financial support for this trial was provided by the Efficacy and Mechanism Evaluation programme.
The industry partner Acorda Therapeutics, Inc. (formerly known as Biotie Therapies Ltd), funded the study drug and MRI.
The Efficacy and Mechanism Evaluation programme is funded by the Medical Research Council and NIHR, with contributions from the Chief Scientist Office in Scotland, the National Institute for Social Care and Health Research in Wales and the Health and Social Care Research and Development Division Northern Ireland. This research was supported by the NIHR Birmingham Liver Biomedical Research Centre. The authors would like to thank the NIHR, the NIHR Biomedical Research Centre, Biotie Therapies Ltd, the Queen Elizabeth Hospital Liver Patient and Public Involvement Group and the patients involved. In addition, we would like to thank members of the teams at the hospital sites who gave so generously of their time and members of the TSC and the DMC.
The trial would also like to thank the following people: Richard Fox (Senior Trial Statistician) who contributed to the design of the trial, contributed to the statistical analysis plan and supervised the analyses during the trial; Dr Jaspreet Babrah (Senior Trial Co-ordinator) who managed the trial in the final analyses stage; Max Kirvan (Trial Co-ordinator) who managed the trial; and Amisha Desai (Pharmacy Representative) who oversaw trial drug receipt, preparation and dispensing.
Last, we would like to thank all the trial staff at CRCTU who have been involved with the trial over its duration.
Contributions of authors
Katherine Arndtz (https://orcid.org/0000-0002-5049-8893) (Clinical Research Fellow) was a co-investigator at the Birmingham site, assisted with the screening and recruitment of patients, and also supported the principal investigator in their role.
Yung-Yi Chen (https://orcid.org/0000-0003-2999-3715) (Scientist) was responsible for the processing of the blood samples, and ran the enzyme activity assays and flow cytometry on serum and cells isolated from the patients.
Anna Rowe (https://orcid.org/0000-0002-4145-301X) (Trial Management Team Leader) contributed to the development of the trial protocol and oversaw the management of the trial.
Victoria Homer (https://orcid.org/0000-0003-3639-7874) (Trial Statistician) conducted the final analyses.
Amanda Kirkham (https://orcid.org/0000-0002-2593-7970) (Trial Statistician) contributed to the statistical analysis plan and conducted analyses during the trial.
Jessica Douglas-Pugh (https://orcid.org/0000-0003-0189-0933) (Trial Co-ordinator) managed the trial.
Daniel Slade (https://orcid.org/0000-0001-6063-1283) (Senior Trial Statistician) supervised the final analyses.
Douglas Thorburn (https://orcid.org/0000-0002-5610-5448) (Principal Investigator for Royal Free Hospital, London) recruited one patient.
Eleanor Barnes (https://orcid.org/0000-0002-0860-0831) (Principal Investigator for John Radcliffe Hospital, Oxford) recruited four patients.
Guruprasad Aithal (https://orcid.org/0000-0003-3924-4830) (Principal Investigator for Queen’s Medical Centre, Nottingham) recruited five patients.
Philip Newsome (https://orcid.org/0000-0001-6085-3652) (Chief Investigator and Clinical Co-ordinator) evaluated and categorised all relevant (as defined in the protocol) SAEs reported for the trial.
David Smith (https://orcid.org/0000-0003-2135-2884) (Pharmaceutical Company Representative) is a VAP-1 research specialist and was involved in the preclinical and clinical development of the investigational medicinal product.
David Adams (https://orcid.org/0000-0001-6776-0336) (Co-Investigator and Clinical Co-ordinator) developed the trial protocol and helped with evaluation of, and categorisation of, all relevant (as defined in the protocol) SAEs reported for the trial.
Christopher Weston (https://orcid.org/0000-0002-9651-1264) (Scientific Advisor) led the research blood and biomarker analysis, guided the planning of the experiments and provided interpretation of the results.
Gideon Hirschfield (https://orcid.org/0000-0002-6736-2255) (Chief Investigator and Co-Investigator) designed and developed the trial protocol, helped with evaluation, and categorisation, of all relevant (as defined in the protocol) SAEs reported for the trial, and led the study overall.
Publication
Arndtz K, Corrigan M, Rowe A, Kirkham A, Barton D, Fox RP, et al. Investigating the safety and activity of the use of BTT1023 (Timolumab), in the treatment of patients with primary sclerosing cholangitis (BUTEO): a single-arm, two-stage, open-label, multi-centre, phase II clinical trial protocol. BMJ Open 2017;7:e015081.
Data-sharing statement
This trial was a collaboration undertaken with Biotie Therapies Ltd (now part of Acorda Therapeutics, Inc.). A summary of trial results is available on the publicly available EU Clinical Trials Register. For additional data-sharing and accessibility requests, please contact the chief investigator (Philip Newsome) or the lead co-investigator (Gideon Hirschfield) via buteo@trials.bham.ac.uk.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research. The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the MRC, NETSCC, the EME programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the EME programme or the Department of Health and Social Care.
References
- Lindkvist B, Benito de Valle M, Gullberg B, Björnsson E. Incidence and prevalence of primary sclerosing cholangitis in a defined adult population in Sweden. Hepatology 2010;52:571-7. https://doi.org/10.1002/hep.23678.
- Bambha K, Kim WR, Talwalkar J, Torgerson H, Benson JT, Therneau TM, et al. Incidence, clinical spectrum, and outcomes of primary sclerosing cholangitis in a United States community. Gastroenterology 2003;125:1364-9. https://doi.org/10.1016/j.gastro.2003.07.011.
- Molodecky NA, Kareemi H, Parab R, Barkema HW, Quan H, Myers RP, et al. Incidence of primary sclerosing cholangitis: a systematic review and meta-analysis. Hepatology 2011;53:1590-9. https://doi.org/10.1002/hep.24247.
- Bergquist A, Said K, Broomé U. Changes over a 20-year period in the clinical presentation of primary sclerosing cholangitis in Sweden. Scand J Gastroenterol 2007;42:88-93. https://doi.org/10.1080/00365520600787994.
- Loftus EV, Sandborn WJ, Lindor KD, Larusso NF. Interactions between chronic liver disease and inflammatory bowel disease. Inflamm Bowel Dis 1997;3:288-302. https://doi.org/10.1097/00054725-199712000-00007.
- Boonstra K, Weersma RK, van Erpecum KJ, Rauws EA, Spanier BW, Poen AC, et al. Population-based epidemiology, malignancy risk, and outcome of primary sclerosing cholangitis. Hepatology 2013;58:2045-55. https://doi.org/10.1002/hep.26565.
- Broomé U, Olsson R, Lööf L, Bodemar G, Hultcrantz R, Danielsson A, et al. Natural history and prognostic factors in 305 Swedish patients with primary sclerosing cholangitis. Gut 1996;38:610-15. https://doi.org/10.1136/gut.38.4.610.
- Ponsioen CY, Chapman RW, Chazouillères O, Hirschfield GM, Karlsen TH, Lohse AW, et al. Surrogate endpoints for clinical trials in primary sclerosing cholangitis: review and results from an International PSC Study Group consensus process. Hepatology 2016;63:1357-67. https://doi.org/10.1002/hep.28256.
- Weston CJ, Shepherd EL, Claridge LC, Rantakari P, Curbishley SM, Tomlinson JW, et al. Vascular adhesion protein-1 promotes liver inflammation and drives hepatic fibrosis. J Clin Invest 2015;125:501-20. https://doi.org/10.1172/JCI73722.
- Hirschfield GM, Karlsen TH, Lindor KD, Adams DH. Primary sclerosing cholangitis. Lancet 2013;382:1587-99. https://doi.org/10.1016/S0140-6736(13)60096-3.
- Lalor PF, Edwards S, McNab G, Salmi M, Jalkanen S, Adams DH. Vascular adhesion protein-1 mediates adhesion and transmigration of lymphocytes on human hepatic endothelial cells. J Immunol 2002;169:983-92. https://doi.org/10.4049/jimmunol.169.2.983.
- Hsia LT, Ashley N, Ouaret D, Wang LM, Wilding J, Bodmer WF. Myofibroblasts are distinguished from activated skin fibroblasts by the expression of AOC3 and other associated markers. Proc Natl Acad Sci U S A 2016;113:E2162-71. https://doi.org/10.1073/pnas.1603534113.
- Marttila-Ichihara F, Smith DJ, Stolen C, Yegutkin GG, Elima K, Mercier N, et al. Vascular amine oxidases are needed for leukocyte extravasation into inflamed joints in vivo. Arthritis Rheum 2006;54:2852-62. https://doi.org/10.1002/art.22061.
- Biotie Therapies Ltd . BTT12-CD015: A Multiple Ascending Dose Study to Assess the Safety and Pharmacokinetics of Repeated Intravenous Doses of BTT1023 in Patients With Rheumatoid Arthritis – A Double-Blind Randomized Placebo-Controlled Sequential Group Trial 2010. https://clinicaltrials.gov/ct2/show/NCT00851240 (accessed 14 February 2022).
- US Department of Health and Human Services . Common Termonology Criteria for Adverse Events (CTACT) Version 4.0 2009.
- Simon R. Optimal two-stage designs for phase II clinical trials. Control Clin Trials 1989;10:1-10. https://doi.org/10.1016/0197-2456(89)90015-9.
- Great Britain . The Medicines for Human Use (Clinical Trials) Regulations 2004 2004.
- EU Clinical Trials Register . Clinical Trial Results: A Single Arm, Two-Stage, Multi-Centre, Phase II Clinical Trial Investigating the Safety and Activity of the Use of BTT1023, a Human Monoclonal Antibody Targeting Vascular Adhesion Protein (VAP-1), in the Treatment of Patients With Primary Sclerosing Cholangitis (PSC) n.d. www.clinicaltrialsregister.eu/ctr-search/trial/2014-002393-37/results.
- Devlin NJ, Shah KK, Feng Y, Mulhern B, van Hout B. Valuing health-related quality of life: an EQ-5D-5L value set for England. Health Econ 2018;27:7-22. https://doi.org/10.1002/hec.3564.
- Trivedi PJ, Muir AJ, Levy C, Bowlus CL, Manns M, Lu X, et al. Inter- and intra-individual variation, and limited prognostic utility, of serum alkaline phosphatase in a trial of patients with primary sclerosing cholangitis. Clin Gastroenterol Hepatol 2021;19:1248-57. https://doi.org/10.1016/j.cgh.2020.07.032.
- Recommendations of guiding physicians in biomedical research involving human subjects . World Medical Association Declaration of Helsinki. J Med Liban 1994;42:88-9.
- Great Britain . Data Protection Act 1998 1998.
- Great Britain . Data Protection Act 2018 2018.
- GDPR.EU . General Data Protection Regulation n.d. https://gdpr.eu/tag/gdpr/ (accessed 9 August 2021).
List of abbreviations
- AE
- adverse event
- ALP
- alkaline phosphatase
- ALT
- alanine transaminase
- APTT
- activated partial thromboplastin time
- AST
- aspartate transaminase
- CONSORT
- Consolidated Standards of Reporting Trials
- CRCTU
- Cancer Research UK Clinical Trials Unit
- CTCAE
- Common Terminology Criteria for Adverse Events
- DLT
- dose-limiting toxicity
- DMC
- Data Monitoring Committee
- eGFR
- estimate glomerular filtration rate
- ELF
- enhanced liver fibrosis
- EQ-5D
- EuroQol-5 Dimensions
- EQ-5D-5L
- EuroQol-5 Dimensions, five-level version
- EQ-VAS
- EuroQol Visual Analogue Scale
- FSS
- Fatigue Severity Scale
- GGT
- gamma-glutamyl transferase
- IBD
- inflammatory bowel disease
- IMP
- investigational medicinal product
- INR
- international normalised ratio
- IQR
- interquartile range
- i.v.
- intravenous
- LOESS
- locally weighted smoothing
- MELD
- model for end-stage liver disease
- mITT
- modified intention to treat
- MRI
- magnetic resonance imaging
- NIHR
- National Institute for Health Research
- PK
- pharmacokinetic
- PSC
- primary sclerosing cholangitis
- REC
- Research Ethics Committee
- SAE
- serious adverse event
- SSAO
- semicarbazide-sensitive amine oxidase
- sVAP-1
- serum vascular adhesion protein 1
- TNO
- trial number
- TSC
- Trial Steering Committee
- VAP-1
- vascular adhesion protein 1
- VAS
- visual analogue scale
Notes
Supplementary material can be found on the NIHR Journals Library report page (https://doi.org/10.3310/ZPNF4670).
Supplementary material has been provided by the authors to support the report and any files provided at submission will have been seen by peer reviewers, but not extensively reviewed. Any supplementary material provided at a later stage in the process may not have been peer reviewed.