

Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 15/39/06. The contractual start date was in June 2017. The draft report began editorial review in March 2021 and was accepted for publication in March 2022. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
Copyright © 2022 Glyn-Jones et al. This work was produced by Glyn-Jones et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This is an Open Access publication distributed under the terms of the Creative Commons Attribution CC BY 4.0 licence, which permits unrestricted use, distribution, reproduction and adaption in any medium and for any purpose provided that it is properly attributed. See: https://creativecommons.org/licenses/by/4.0/. For attribution the title, original author(s), the publication source – NIHR Journals Library, and the DOI of the publication must be cited.
2022 Glyn-Jones et al.
Chapter 1 Introduction
Trial summary
The Managing Avascular Necrosis Treatments: an Interventional Study (MANTIS) trial was, to our knowledge, the first Phase IV UK trial designed to determine the clinical effectiveness and cost-effectiveness of a 12-month course of oral alendronate (70 mg weekly) compared with a matched placebo in reducing the progression of avascular necrosis (AVN) of the hip.
The trial was due to open in a minimum of 10 NHS hospitals, with 280 participants planned to be randomised to one of two treatments:
-
70 mg of alendronate given as an oral tablet weekly for 12 months
-
placebo given as an oral tablet weekly for 12 months.
Background and rationale
Avascular necrosis of the femoral head is an uncommon but devastating condition that affects around 0.001% of the population in developed countries1–3 and occurs when the bone of the hip joint dies, resulting in significant disability in many cases. In the USA, an estimated 10,000–20,000 cases of AVN are diagnosed each year. 3 AVN typically affects younger people, with 45% of patients developing the condition before the age of 30 years. 1 In the early stages of AVN, people often feel pain, followed by a rapid collapse of the joint, which leads to severe immobility and suffering in over 60% of patients. This means that patients often cannot walk comfortably, cannot work and have sleep disturbances. 3,4
The natural history of AVN is well documented; however, its exact pathophysiology is poorly understood. There have been several postulated mechanisms based on whether AVN is classified as post-traumatic or atraumatic. 5 There are also several established risk factors for AVN; glucocorticoid (steroid) therapy and alcoholism are two of the most common associations. Less commonly associated risk factors include sickle cell disease; radiotherapy; cocaine, methadone and heroin use; and Caisson’s disease. 3
Assessing avascular necrosis in the hip
Several radiological and clinical classification systems were proposed. Feasibility work undertaken for the MANTIS trial indicated that standardising the imaging classification used to assess AVN as far as possible across different hospitals and practitioners would produce the best results. Ficat and Arlet, which is based on radiography, magnetic resonance imaging (MRI) changes and clinical symptoms, is the most widely used and clinically relevant classification. 6,7 The early stages (Ficat and Arlet stages 0 and 1) are not detectable on radiographs and rely on MRI scans and clinical assessment. The percentage involvement of the femoral head and the anatomical location of the lesion have been shown to be closely related to progression. Involvement of > 30% of the femoral head, particularly in the weight-bearing area, is correlated with poor outcome and subsequent joint replacement. 5,8 Among patients with untreated Ficat and Arlet stage 1 or 2 disease, 85% progress to femoral head collapse within 2 years and 76% undergo joint replacement within 3 years. 4
Clinician- and patient-based scoring systems are typically used to assess patients with AVN. 4,9–12 Given that many patients require surgery, including total joint replacement, the most appropriate scores are those that are used in these situations. The Oxford Hip Score (OHS) is usually used to assess patients in need of a total hip replacement but has been used previously in patients with AVN and does not seem to be limited by a ceiling effect in this younger patient population,13,14 as their pain is significant and their resultant function very poor. The OHS, therefore, seems to be an acceptable and valid means of assessing AVN. As part of our preparation work, we explored the use of different patient-reported outcome measures [such as the International Hip Outcome Tool (iHOT-33)] with seven patients suffering from femoral head AVN.
Although the Harris Hip Score has been more commonly used in previous studies of AVN,15,16 this is limited in that it is clinician reported and requires that patients be present for the examination. 17
Existing treatments for femoral head avascular necrosis
The early phase of treatment usually involves asking the patient to use walking aids so that their hips are non-weight bearing for 3–7 months, with the aim of preventing hip collapse and controlling symptoms using simple analgesics. However, this is effective in only 15% of patients: most patients become so immobile that their pain can be alleviated only by joint-preserving operations and, eventually, hip replacement.
Joint-preserving operations for AVN have been practised for over 50 years. These either attempt to encourage revascularisation and healing (core decompression or stem cell transplantation) or limit the impact of collapse (osteotomy and bone grafting techniques). Reported success rates vary from 30% to 90% in preventing radiographic progression and relieving pain. 4
Although total hip arthroplasty (THA) is an effective treatment, it is a large operation, with a revision rate of approximately 10 to 20 years. 14,18 In addition, patients who receive joint replacements for AVN are much more likely to require further surgery and be infected postoperatively than those who do not undergo surgery. 18
Five hundred and seventy joint replacements were performed for AVN in England and Wales in 2012,12 at a cost of over £5M. These procedures were predominantly performed in patients under 45 years old. AVN is the primary reason for 25% of hip replacements in patients under 30 years. The outcomes of hip arthroplasty are known to be worse in the English and Welsh population than in other populations in which the same procedure is common. They also present with significantly higher infection, dislocation and 10-year revision rates adding to the longer-term patient, health-care and economic burden of disease. 14,18
Of more promise are pharmaceutical interventions, which provide a potentially cost-effective and lower risk option in comparison with the surgical interventions described above. 9,15–17,19 For example, the cost of oral bisphosphonate therapy is approximately £11 per year in osteoporosis treatment. 20 This is an order of magnitude less than surgical options and other pharmacological therapies. This treatment is also likely to be safer and more acceptable to patients than joint replacement or joint-preserving surgeries.
Bisphosphonates in the treatment of avascular necrosis
At present, there is little evidence for an effective drug treatment that prevents AVN, with very few publications relating to the efficacy of alendronate in the treatment of AVN.
The literature indicates that oral bisphosphonates may be of use in slowing or halting the progression of AVN. 21 Only two randomised controlled trials of bisphosphonates have been published. Chen et al. 16 demonstrated no difference in either clinical/structural appearance or symptoms with an oral bisphosphonate (alendronate) compared with a placebo control over 3 years when looking at the cumulative incidence of THA. By contrast, Lai et al. 10 found that alendronate is much more effective (65% vs. 3% structural progression; p < 0.001) than a placebo control in preventing progression to THA at 2 years; however, this was an open-label trial with unclear randomisation. Agarwala et al. 15 demonstrated a sustained reduction in progression to THA at 10 years in a prospective cohort study of patients receiving alendronate. Despite the potential efficacy of oral alendronate shown in some studies, each trial was limited by critical design issues and the small number of patients included. These trials also had differing inclusion criteria, with glucocorticoid-related AVN, a major cause of the condition, excluded in one trial. 16 Lai et al. 10 allowed surgical intervention in the treatment groups, meaning that the treatment effect of alendronate was difficult to estimate. Yuan et al. 22 performed a low-quality meta-analysis that included five papers of bisphosphonate therapy in femoral head AVN. They did not demonstrate any treatment efficacy and commented that the complications of bisphosphonate use (jaw osteonecrosis and atypical fractures) may outweigh the benefits.
These early results demonstrate that oral bisphosphonates (particularly alendronate) may be of value in the treatment of AVN. Previous feasibility work using patient and public involvement (PPI) groups highlighted that a trial examining the efficacy of bisphosphonates in AVN was needed.
This trial, therefore, aimed to investigate the effect of alendronate plus standard care in the treatment of AVN of the hip.
Chapter 2 Methods
Trial design
The MANTIS trial was designed to be a 66-month project with an initial pilot phase leading into a definitive, multisite, two-arm, parallel-group, placebo-controlled, double-blind, Phase IV randomised controlled superiority trial. Its objective was to evaluate and compare the clinical effectiveness of oral alendronate with a plaebo-matched control for participants with AVN. We planned to assess primary outcomes after 12 (short term) and 36 months (long term), and secondary outcomes at 6, 12, 24 and 36 months.
The trial was preceded by a feasibility study, which identified challenges in identifying patients with AVN of the hip, owing to a low prevalence rate and significant regional variations in at-risk groups and service delivery. To address this concern, the trial was designed with an internal pilot phase, with the intention of identifying hospitals/specialties that would be able to recruit adequate numbers of patients. According to the prespecified go/no-go criteria, the trial would be stopped if key recruitment targets could not be met.
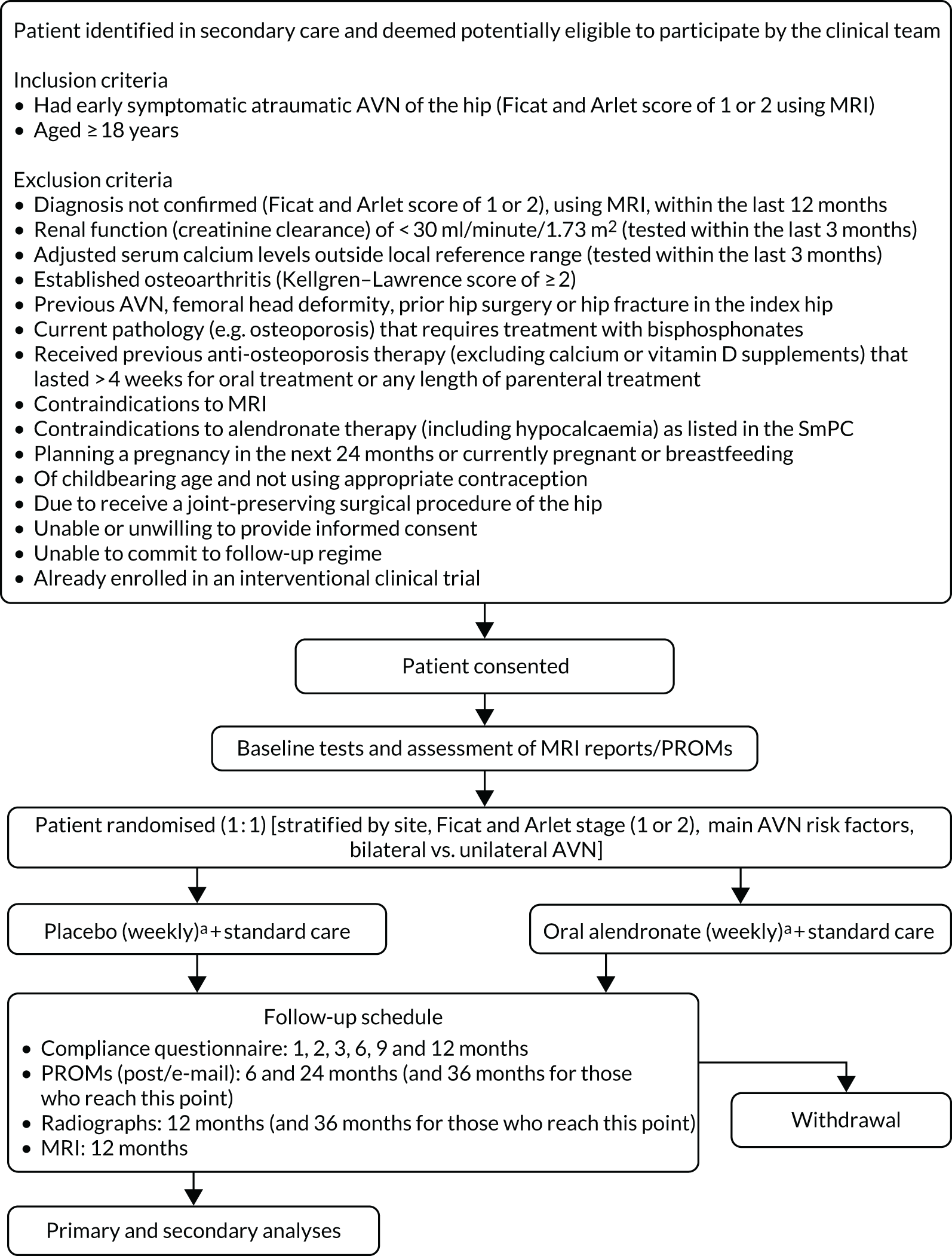
The trial was designed as a placebo-controlled blinded trial with participants, clinicians, pharmacists and the research teams all blinded to the treatment allocation. The trial statistician who set up and monitored the randomisation schedules and prepared the unblinded data for review at independent data monitoring committee meetings was unblinded. Active and placebo oral medication were produced and encapsulated and packaged identically. The trial had two groups (Figure 1):
-
intervention group – active alendronate (70 mg taken as one tablet weekly for 12 months)
-
control group – matched placebo (taken as one tablet weekly for 12 months).
FIGURE 1.
Trial schema. PROM, patient-reported outcome measure; SmPC, summary of product characteristics. a, Treatment must be collected within 14 days of randomisation.
Research governance
The trial was sponsored by the University of Oxford, with regulatory compliance oversight by Oxford Clinical Trials Research Unit (OCTRU) through the Surgical Interventional Trials Unit (SITU).
An ethics application under the Integrated Research Application System (IRAS) ID 230545 was submitted to the South Central – Oxford A Research Ethics Committee on 17 April 2018 and the MANTIS trial received a favourable Research Ethics Committee (REC) opinion on 29 May 2018 (reference number 18/SC/0247) and Health Technology Assessment (HTA) approval on 26 June 2018.
A clinical trial authorisation application under the European Union Drug Regulating Authorities Clinical Trials (EudraCT) number: 2017-002798-21 was submitted to the Medicines and Healthcare products Regulatory Agency (MHRA) on 23 April 2018, updated with further information on 8 June 2018 and the trial gained MHRA approval on 19 June 2018.
To ensure transparency, the trial was registered with the International Standard Randomised Controlled Trial Number (ISRCTN) Registry on 11 June 2018 and was assigned ISRCTN14015902. 23
The trial applied for National Institute for Health and Care Research (NIHR) Clinical Research Network (CRN) Portfolio status and was eligible for this support. The lead supporting network was the Thames Valley and South Midlands CRN.
Global NHS permissions were obtained through the IRAS and local NHS permissions were obtained from each participating NHS trust. A clinical trial site agreement based on the model non-commercial research agreement was signed by each participating NHS trust and the sponsor.
Following NIHR guidelines, a fully independent Data Safety and Monitoring Committee (DSMC) and a 66% independent Trial Steering Committee (TSC) were convened. Both committees met throughout the lifetime of the trial.
Management of the trial
The trial manager (Gemma Greenall) was responsible for day-to-day management of the trial, with support from the data manager (Alistair Gray) and trial statistician (NP). The Trial Management Group (TMG) was responsible for overseeing day-to-day management of the trial and comprised the chief investigator (CI) (SGJ), the lead investigator (Kassim Javaid), the trial statisticians (IR, RK and NP), the trial manager (Gemma Greenall) and health economist (MD).
Design and development of the protocol
Clinicians (including surgeons, rheumatologists and nurses), as well as patient representatives, were invited to discuss the trial protocol. Their feedback was utilised by the applicants when designing and developing the protocol.
Amendments to the trial protocol
Following receipt of a favourable opinion of the trial protocol from the REC on 29 May 2018, no changes were made to the trial protocol – the trial started and ended on version 1.0, dated 10 April 2018.
Amendments to the trial
Following the main REC approval, nine substantial amendments were submitted and received favourable opinion. As the trial was a clinical trial of an investigational medicinal product (IMP), any new sites had to be added as a substantial amendment. The nine substantial amendments were as follows:
-
Change to the investigational medicinal product dossier (IMPD) and updated summary of medicinal product characteristics (SmPC) to be used by the trial (24 August 2018).
-
Addition of one new trial site (Royal Orthopaedic Hospitals NHS Foundation Trust) (12 October 2018).
-
Addition of four new trial sites (Milton Keynes University Hospital NHS Foundation Trust, Taunton and Somerset NHS Foundation Trust, Royal National Orthopaedic Hospital NHS Foundation Trust and St George’s University Hospitals NHS Foundation Trust) (23 January 2019).
-
Addition of two new trial sites (Ashford and St Peter’s Hospitals NHS Foundation Trust and Royal Cornwall Hospitals NHS Trust) (19 March 2019).
-
Addition of two new trial sites (University Hospitals of North Midlands NHS Trust and Royal Free London NHS Foundation Trust) (6 June 2019).
-
Addition of one new trial site (Ayrshire & Arran Health Board) (23 July 2019).
-
Addition of six new trial sites (North Bristol NHS Trust, Maidstone and Tunbridge Wells NHS Trust, South Tyneside and Sunderland NHS Foundation Trust, Sandwell and West Birmingham Hospitals NHS Trust, Cambridge University Hospitals NHS Foundation Trust and Swansea Bay University Health Board) (13 November 2019).
-
Notification of temporary halt of the trial owing to recruitment being substantially below targets set in the original and revised recruitment timelines. Following discussions between the funder and TMG on how to proceed with the trial, it was decided that no further participants would be recruited (5 December 2019).
-
Confirmation that the trial would be closed after the temporary halt. Addition of patient-facing documentation to inform participants of what is happening to the trial, and addition of some patient-facing questionnaires to ask about their experience of the trial (7 February 2020).
Support costs
NHS support and treatment costs were calculated in writing the protocol from health-care resource group (HRG) codes and the patient-level informatics system (PLIS) at the lead site. The PLIS allowed individual treatments, hospital bed occupancy, physiotherapy, outpatients costs and staff costs to be accurately calculated on a patient-level basis.
Routine NHS costs relating to the management of AVN typically consist of the cost of plain radiographs and a MRI scan (to make the diagnosis and stage the disease) and up to eight sessions of outpatient physiotherapy and three outpatient appointments with a clinician within the first year. Each session costs £50 plus £5 for equipment.
We estimated that NHS treatment costs for analgesia would be approximately £5–10 per week for 1 year in patients undergoing observation and investigations during the early stages of AVN (i.e. Ficat and Arlet stages 1 and 2).
NHS treatment costs for bisphosphonate therapy are not currently considered to be routine practice, so all drug costs were costed as a research cost and funded by the grant.
Patient and public involvement
Patient and public involvement was present at all stages of the development and execution of the trial, including during the feasibility and pilot stages. A Research Design Service-appointed PPI representative was involved in the initial trial design and during the development of the patient information sheets.
The Young Adult Hip Group (Nuffield Department of Orthopaedics, Rheumatology and Musculoskeletal Sciences, Oxford, UK. URL: www.ndorms.ox.ac.uk/clinicaltrials/hipyoungadult) was established in conjunction with the local CRN. This was an interest group for patients with conditions such as femoroacetabular impingement (FAI), hip dysplasia, osteoarthritis and AVN, based at the lead site. The trial was developed in conjunction with this group. A member of the group (Peter Lovell) is a co-applicant. In conjunction with advice from INVOLVE (URL: https://involve.org.uk/resources/knowledge-base?gclid=CjwKCAjwu5yYBhAjEiwAKXk_eCs-6b8p7fHnnbTC04L8chUi43tz2KQLZLOBcrIsznSZHHIoub91HRoCLpYQAvD_BwE) and the James Lind Alliance (Southampton, UK), the group advised on research design, developing patient information resources and monitoring screening in outpatient clinics. The co-applicant PPI member sat on the TSC. The Young Adult Hip Group were planning to help disseminate the trial findings through local (hospitals and PPI groups), regional (Thames Valley CRN) and national [National Musculoskeletal Clinical Research Network (Clinical Research Network Coordinating Centre, University of Leeds, Leeds, UK), Arthritis Research UK (Chesterfield, UK) and the James-Lind Alliance] entities at the end of the trial.
During the internal pilot we planned to conduct a focus group discussion with the help of the local NIHR Research Design Service to test the patient acceptability with the use of electronic patient-reported outcomes, the collection of drugs and compliance with drug therapy, and with follow-up and drug delivery method (blister or bottle). As the trial closed prematurely, the focus group discussion did not take place.
Site initiation
At a minimum the trial manager (Gemma Greenall) attended all site initiation visits (SIVs). The CI (SGJ) also attended some of the SIVs. At the SIV a presentation was given that introduced the MANTIS trial – this included the team, the condition being studied and the trial premise and processes. In addition, the management of the MANTIS trial at sites including screening, recruitment, the taking of consent, case report form (CRF) completion, drug management and safety was presented, alongside trial responsibilities and the current status of the trial.
The principal investigator (PI) was always present at these visits and a register was taken of those who attended the visit.
To encourage surgical trainees to become involved in the trial, a MANTIS trial collaborator points scheme was introduced that resulted in certification and the potential to be cited in publications. An additional incentive was the MANTIS trial’s inclusion in the NIHR Associate Principal Investigator Scheme in March 2019, a Royal College of Surgeons-endorsed scheme that aims to engage, recognise and promote trainees involved in NIHR portfolio research, and also to integrate clinical research into routine clinical training.
Investigator site file
The investigator site files (ISFs) consisted of essential documents relating to trial conduct at a specific site (i.e. the location where participant-related trial activities were actually conducted), and were designed to enable both the conduct of the clinical trial and the quality of the data at that site to be evaluated. Among other essential documents, the ISF contained source documents, such as participant screening logs, consent forms, drug accountability records, delegation logs and CVs. Confirmation of receipt of the ISF was requested from sites and then filed centrally.
The ISF was then maintained by the PI at that site; the trial manager (Gemma Greenall) provided instructions on how to maintain the ISF, including the need for direct access to monitors, auditors and inspectors.
Site management
Site monitoring visits
We planned to monitor sites face to face at least once during each site’s recruitment phase, and to undertake triggered monitoring visits if any of the below criteria were met:
-
little or no recruitment for 3 months since site activation
-
poor data quality (deemed appropriate and decided in conjunction with data manager)
-
surgeon non-compliance with IMP administration technique (deemed appropriate and decided in conjunction with CI)
-
persistent errors in the completion of the informed consent form (ICF)
-
a serious breach identified at site
-
any other situation deemed appropriate by the site, trial management team, sponsor and/or CI.
No monitoring visits had been undertaken when the trial closed prematurely.
Inclusion criteria
Each participant in the trial had to meet both of the following criteria to be included in the trial:
-
diagnosed with early symptomatic atraumatic AVN of the hip (Ficat and Arlet stage 1 or 2 using MRI)
-
aged ≥ 18 years.
Exclusion criteria
Otherwise eligible individuals who met any of the following criteria were excluded from the trial:
-
diagnosis (Ficat and Arlet stage 1 or 2) had not been confirmed using MRI within the last 12 months
-
renal function (creatinine clearance) of < 30 ml/minute/1.73 m2 (tested within the last 3 months)
-
adjusted serum calcium levels outside local reference range (tested within the last 3 months)
-
established osteoarthritis (Kellgren–Lawrence score of ≥ 2)
-
previous AVN, femoral head deformity, prior hip surgery or hip fracture in the index hip
-
current pathology (e.g. osteoporosis) requiring treatment with bisphosphonates
-
received previous anti-osteoporosis therapy (excluding calcium or vitamin D supplements) that lasted > 4 weeks for oral treatment or any length of parenteral treatment
-
contraindications to MRI
-
contraindications to alendronate therapy (including hypocalcaemia) as listed in the SmPC
-
planning a pregnancy in the next 24 months or currently pregnant or breastfeeding
-
of childbearing age and not using appropriate contraception
-
due to receive a joint-preserving surgical procedure of the hip
-
unable or unwilling to provide informed consent
-
unable to commit to follow-up regime
-
already enrolled in an interventional clinical trial.
Screening and recruitment
Following attendance at a site initiation meeting, screening and recruitment were commenced at participating sites once the clinical trial site agreement had been signed and all necessary approvals were in place.
Potentially eligible patients were identified and approached by authorised members of staff about taking part in the trial. Patients interested in participating were referred to a research facilitator at recruiting sites, who then assessed whether or not they met the trial inclusion criteria. Information about the trial, which included its purpose, potential risks and benefits, the type of data, and when and how data would be collected during the trial, and who was funding and sponsoring it, was provided to the patient. This information was provided on a site-specific localised patient information sheet (PIS) that included the name and contact details of the local PI. All those approached to participate were able to keep the PIS.
Routinely MRI and radiography images and reports were then screened by the local care team to confirm the diagnosis and the Ficat and Arlet stage. For the diagnosis to be relevant to the trial, radiographs and MRIs had to have been taken no more than 12 months from the point of recruitment.
Before patients were officially recruited into the trial, they needed to have blood samples taken within the previous 3 months to confirm their renal function and adjusted serum calcium and 25(OH)-vitamin D levels. As such blood tests are routinely taken every 3 months in this patient population, all patients should have had these results available for consideration of eligibility.
Following consent, if pregnancy was suspected, participants of childbearing age were asked to take a pregnancy test to confirm their eligibility.
Screening log
To enable full and transparent reporting for the trial, brief details of all patients screened for the trial were recorded at each site. Sites also recorded the date patients were considered for the trial and, for those that were ineligible, the reasons for ineligibility. Those who were considered eligible but declined to participate had their reasons for declining recorded.
Informed consent
Participants had to personally sign and date the latest approved version of the ICF before any trial-specific procedures were performed.
Written and verbal versions of the PIS and ICF detailing the exact nature of the trial, what it involved for the participant, the implications and constraints of the protocol; the known side effects and any risks involved in taking part were presented to participants. Both documents clearly stated that the participant was free to withdraw from the trial at any time and for any reason without prejudice to future care, without affecting their legal rights and with no obligation to provide a reason for withdrawal.
The participant was allowed as much time as needed to consider the information and had the opportunity to question the investigator, their general practitioner (GP) or other independent parties when deciding whether or not they wished to participate in the trial. Written informed consent was obtained by means of a participant-dated signature and dated signature of the person who presented and obtained the informed consent. It was expected that some of the patient population may be difficult to follow up and may not have a fixed address. Therefore, the consent form also asked the participant to consent to their clinical care team contacting their GP for information, which may have helped the clinical care team query missing data. The participant was also made aware of the fact that screening tests and previous tests and scans may need to be reviewed to confirm eligibility, and that images and reports of scans may need to be accessed from their routine appointments by the clinical care team to aid follow-up.
The person who obtained the consent had to be suitably qualified and experienced, and have been authorised to do so by the CI or relevant PI. A copy of the signed ICF was given to the participant, and the original signed ICF was retained at the trial site.
Randomisation and allocation procedure
Randomisation to the interventions was undertaken via the centralised secure web-based randomisation service Registration/Randomisation And Management of Product [RRAMP (OCTRU, Oxford, UK)], run through OCTRU. In the event that sites were unable to randomise patients using RRAMP, they had to contact the central research office, and a member of the trial team was then able to randomise the patient via RRAMP. An emergency backup randomisation system was in place if the central research office was also unable to use RRAMP.
Participants were randomised on a 1 : 1 basis to receive either alendronate or the placebo-matched control. Randomisation was performed using a minimisation algorithm including a random element (p = 0.8) to ensure balanced allocation of participants across the two arms stratified by:
-
randomising site
-
Ficat and Arlet stage (1 or 2)
-
main AVN risk factors (steroid/alcohol/other) obtained from clinical notes
-
bilateral vs. unilateral AVN.
However, the first 28 participants (10% of the expected sample size) were to be randomised using simple randomisation to seed the minimisation algorithm. Owing to the small numbers recruited, the trial did not move out of this seeding stage and so the stratification factors were not taken into account.
The intervention was expected to start within 4 weeks of randomisation.
Treatment groups
A total of 280 participants with AVN of the hip as per the trial inclusion and exclusion criteria were to be randomised to one of two arms to receive one of the following:
-
alendronate
-
placebo-matched control.
Dosing regimen
Both the active substance and the placebo control were manufactured and Qualified Person released for use in the trial by the holder of the Manufacturing and Import Authorisation (IMP) licence. Thereafter, bottles containing 13 weeks' supply of both the active substance and the control were sent to recruiting sites. All IMPs were overencapsulated and the bottles labelled with a MHRA-approved clinical trial label in accordance with Annexe 13 (Investigational Medicinal Products) of the European Union Guidelines on Good Manufacturing Practice. 24 Both the active substance and the placebo control were administered alongside standard care for patients with AVN.
Active substance
Alendronate was used as the active substance in the MANTIS trial. It is an off-white, oval, biconvex, 70-mg tablet, currently used to treat post-menopausal osteoporosis, and to reduce the risk of vertebral and hip fractures. 25
Alendronate is marketed by Accord Healthcare Limited UK [product licence number (PL) 20075/0071] and was given to participants in accordance with the SmPC at the recommended dosage of 70 mg once weekly for the treatment period of the trial (52 weeks).
The effects of alendronate on pregnant and breastfeeding women are not known, as no data are available. However, studies in animals have shown reproductive toxicity; therefore, pregnant and breastfeeding women were not included in the trial and contraception was required for all participants of childbearing age.
Patients also received supplemental calcium and vitamin D if their estimated dietary calcium intake was < 700 mg per day. This was assessed at the point of eligibility screening. Those not requiring calcium supplements were advised to take at least 800 IU of vitamin D3 or D2 per day. Those with a 25(OH)-vitamin D level < 30 nmol/l were recommended treatment as per local vitamin D guidelines. Where no local guidelines existed, we recommended that the Oxfordshire Metabolic Bone Disease Vitamin D guidelines26 be used at participating sites.
Placebo control
A placebo-matched control tablet identical in appearance to the active tablets was manufactured using capsules backfilled with microcrystalline cellulose. Participants were asked to take the placebo treatment in the same way as the active treatment (once per week for the trial treatment period of 52 weeks).
Drug administration
The active substance and the placebo control were both oral tablets to be taken once per week for 52 weeks. Patients received their supply of tablets in bottle form and received a patient card and automated weekly text reminders to take their tablet. The dispensing pharmacy was blinded to the treatment allocation. The tablets were provided to the patient in bottles containing 13 weeks’ worth of treatment, which they needed to collect from the pharmacy within 14 days of randomisation. The patients received the next 13 weeks’ supply at 3-monthly intervals.
To ensure sufficient absorption, the SmPC suggested that alendronate be taken at least 30 minutes before the first food, beverage or medicinal product of the day with plain water only. Therefore, we advised participants that it was preferable to take the tablet first thing in the morning, and take it whole without crushing or chewing it, as doing so could potentially cause oropharyngeal ulcers. Once the alendronate had been taken, patients were advised not to lie down or have the first food of the day for at least 30 minutes.
Data collection
Baseline
Data were collected only once a patient’s eligibility and willingness to participate in the trial had been confirmed and they had given their informed consent. General patient demographics and participant contact details were collected to help enable follow-up assessments. Additional data on the intention for ongoing corticosteroid use, other AVN risk factors [e.g. smoking, drinking, body mass index (BMI) and relevant medical history], whether the participant had unilateral or bilateral involvement, and the current radiographic evidence for the AVN, were collected. The baseline assessment included a questionnaire to be completed by the patient which included the following measures:
-
OHS
-
iHOT-33
-
EuroQol-5 Dimensions, five-level version (EQ-5D-5L)
-
Hospital Anxiety and Depression Scale (HADS).
Subsequent follow-up
Following entry into the trial, participants were to return to hospital, as part of routine clinical care, to undergo radiography (at 1 and 3 years from trial entry) and MRI (at 3 years from trial entry). Images and reports from these routine appointments were to be collected as part of assessing radiological progression. All other assessments were to involve patient-reported outcome questionnaires being sent directly to the participants by the central trial office team in Oxford. Participants could choose whether to complete these electronically (via a link sent in an e-mail) or on paper forms sent out via post with freepost return envelopes.
The follow-up questionnaires were to be sent at 6, 12, 24 and 36 months after entry into the trial.
Compliance with the IMP was planned to be monitored at 1, 2, 3, 6, 9 and 12 months during the 12-month treatment duration for both groups. This was to be conducted via a secure online platform or via post and involved participants completing a short compliance questionnaire.
Reminders to participants with uncompleted or unreturned questionnaires were sent 3 days after the initial due date for the 1- and 2-month questionnaires and 2 weeks following the 3-, 6-, 9-, 12-, 24- and 36-month questionnaires. One further reminder was sent 1 week later. Following this, non-responders were telephoned by a member of the central trial team in Oxford to collect a minimum of the primary outcome (OHS). Participants were then scheduled for their next questionnaires.
An assessment table detailing the timing of assessments and visits is provided in Table 1.
| Assessment | Timing of assessments | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Pre randomisation | Post randomisation (months) | |||||||||
| Screening | Baseline | 1 | 2 | 3 | 6 | 9 | 12 | 24 | 36 | |
| Eligibility check | ✗ | |||||||||
| Demographics | ✗ | |||||||||
| Review screening blood samples (vitamin D levels, calcium levels, renal functioning) | ✗ a | |||||||||
| Informed consent | ✗ | |||||||||
| Pregnancy test | ✗ | |||||||||
| OHS | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||||
| Time to placement on surgical waiting list | ✗ | |||||||||
| iHOT-33 | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||||
| QoL | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||||
| Resource use | ✗ | ✗ | ✗ | ✗ | ✗ | |||||
| MRI reports and images | ✗ a | ✗ a | ||||||||
| Radiography reports and images | ✗ a | ✗ a | ✗ a | |||||||
| Compliance with IMP (both active treatment or placebo) | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||||
| Compliance with prescribed standard care | ✗ | ✗ | ✗ | ✗ | ||||||
Embedded pilot
An internal pilot was planned that would progress to the definitive trial if predefined evaluation criteria regarding recruitment, patient characteristics, equipoise and compliance were met. The pilot trial was to mirror the procedures and logistics to be undertaken in the main definitive trial. We planned for the data that were collected in the pilot trial to contribute to the final analysis.
The pilot aimed to randomise 50 patients or more over a 12-month period from at least 10 sites, with a target recruitment rate of one patient per site per month. If any issues were identified, it was proposed that they be discussed with the TSC; any changes required for the definitive trial were to be discussed with the funder and submitted as amendments for approval.
Evaluation criteria
Recruitment
-
We aimed to determine recruitment rates and referral patterns within different clinical services at each site.
-
We aimed to identify and recruit more sites and estimate recruitment rates at each site.
-
We aimed to work with the staff at recruiting sites to optimise recruitment and identify potential barriers to recruitment (e.g. treatment equipoise).
Patient characteristics
The internal pilot aimed to further refine the inclusion criteria for the trial and identify if any groups (e.g. those with alcohol dependency) were likely to represent barriers to recruitment. The feasibility of including participants with ongoing steroid use in the trial, given that many of these patients would be undergoing treatment for inflammatory joint conditions or undergoing chemo/radiotherapy, was also to be assessed. It was also hoped that monitoring reasons for non-participation would determine if a non-randomised cohort could be appropriately developed alongside the main trial.
Compliance
A weekly bisphosphonate treatment regime was to be used in the trial. Although it was thought that this would be acceptable to most patients, some groups (e.g. those with alcoholism or undergoing chemotherapy) may have faced issues with compliance. The trial team aimed to keep detailed compliance logs along with barriers to compliance as part of the pilot trial. If non-compliance was found to be an issue, then other methods of administration could have been explored.
Evaluation criteria outcomes
The internal pilot trial was set up to consider the following outcomes for the evaluation criteria:
-
recruitment rates
-
number of eligible patients identified and conversion to randomisation
-
feasibility of collecting OHS data
-
quality of data collection for the end points of the main trial.
It was anticipated that the primary and secondary end points for the main trial would not have been analysed separately in the pilot trial.
Outcome measures
Primary outcome measures
The first primary outcome measure, the OHS,27 is a 12-item validated patient-reported outcome measure assessing pain and functional outcomes in the hip. Each item is scored from 0 to 4; total scores are calculated as a sum across the 12 items and range from 0 (severe problems) to 48 (no problems). We planned to measure the OHS at baseline and at 6, 12, 24 and 36 months; the short-term primary outcome time point was at 12 months post randomisation.
The second primary outcome measure was the decision to have a total hip replacement operation within 3 years. This was to be recorded as the date the participant was placed on the surgical waiting list by their surgeon. The decision to have surgery is reached after a series of discussions between the clinical care team and the patient.
Secondary outcome measures
Pain
The OHS at 6, 24 and 36 months would record pain and function throughout the trial.
Hip function
To measure hip function the iHOT-33 questionnaire28 was to be completed at baseline and at 6, 12, 24 and 36 months. This is a validated patient-reported outcome used to measure symptom progression in younger patients who exhibit some ceiling effect with the OHS. The iHOT-33 consists of 33 items and total scores range from 0 (severe problems) to 100 (no problems).
Anxiety and depression
The HADS31 detects states of depression and anxiety in the setting of a hospital medical outpatient clinic. The HADS was to provide an important insight into the overall disease perception and we planned to administer it at baseline and at 6, 12, 24 and 36 months. The HADS consists of two subscales, one for anxiety and one for depression, each scored from 0 (no problems) to 21 (significant problems).
Quality of life
To measure quality of life, the EQ-5D-5L32 was to be administered at baseline and at 6, 12, 24 and 36 months. The EQ-5D-5L is a validated, generalised, health-related quality-of-life questionnaire consisting of five domains related to daily activities with a five-level answer possibility, which is converted into multiattributed utility scores using established algorithms. Utility scores range from –0.594 to 1, with better scores indicating better quality of life and a score of 0 representing a quality of life equivalent to death.
Radiological progression at 1 and 3 years
Both MRI and radiography are conducted as part of routine care for patients with AVN. In addition to the baseline reports and images, the images and reports from routine MRI scans of the index hip using three-dimensional volumetric hip sequences at 1.5 or 3’t were to be obtained at the 3-year assessment for each participant. Images and reports were also to be obtained from routine radiographic examinations of the index hip at 1 and 3 years. During these examinations, anteroposterior (AP) and lateral views of the index hip were to be acquired, and the lateral joint space width of the index hip was to be measured.
Radiological progression was to be measured using the Ficat and Arlet scoring system by comparing the scans obtained during the routine patient assessments with those screened at baseline. The grade of chondral damage was to be assessed using the Kellgren–Lawrence scoring system.
Health-care resource use
Health-care use was to be monitored for the economic analysis at 6, 12, 24 and 36 months. This was to include micro-costing, HRG-based approaches, and detail on costing of components/consumables, health care, rehabilitation, productivity losses and informal care. We had aimed to use the microcosting obtained during the NIHR HTA Clinical Outcomes of Arthroplasty Surgery Trial (COAST) to accurately cost the interventions in any participants who underwent total hip replacement. We had also planned to include the underlying causes of AVN and employment status within the health economic analysis.
As part of the economic evaluation, we had planned to combine within-trial data on resource use, costs and outcomes with an extrapolation model to estimate long-term cost-effectiveness.
Safety monitoring
Alendronate is a licensed medication, with a comprehensive safety profile. Only the adverse events (AEs) detailed in Table 2 were to be recorded on the trial AE form.
| Adverse event | Definition |
|---|---|
| Upper gastrointestinal | Abdominal pain, dyspepsia, constipation, diarrhoea, flatulence, abdominal distension, acid regurgitation, peptic ulcer, nausea, vomiting, gastritis, oesophagitis, oesophageal erosions, melaena, oesophageal ulcer, dysphagia, odynophagia, oesophagitis, oesophageal stricture, oropharyngeal ulceration, upper gastrointestinal perforation, ulcers, bleeding |
| Osteonecrosis of the jaw | 8 weeks of exposed bone in the oral cavity despite usual oral therapy |
| Atypical femoral fracture | Fulfils ASBMR 2013 criteria29 |
| Musculoskeletal pain | Bone, muscle or joint pain unrelated to AVN |
| Hypocalcaemia | Symptoms of cramps and paraesthesiae and a concurrent serum calcium level below the local laboratory reference range |
The listed AEs are known to be expected and related to the trial drug and are detailed in the SmPC. The recording of the listed AEs was to help us assess reasons for non-compliance or withdrawals in this patient population. All AEs were to be recorded on the AE form as soon as the site or central trial office team became aware of them.
The following information was to be recorded on the AE form: description, date of onset, end date and action taken. Follow-up information was to be provided as necessary.
The investigator was to use their clinical judgement to decide whether or not an AE was of sufficient severity to necessitate the participant’s removal from treatment. A participant was able to voluntarily withdraw from treatment because of what he or she perceived as an intolerable AE. In either of these scenarios, the participant was to undergo an end-of-trial assessment and receive appropriate care under medical supervision, until either the symptoms ceased or their condition became stable.
Trial oversight
A DSMC (comprising two medically qualified clinicians and a statistician, all independent) was appointed to safeguard the interests of the trial participants, assess the safety and efficacy of the interventions during the trial, and monitor the overall conduct of the trial, protecting its validity and credibility. The DSMC was independent of the trial investigators and sponsor and adopted a DAta MOnitoring Committees: Lessons, Ethics, Statistics (DAMOCLES)-based charter30 that defined its terms of reference and operation in relation to the oversight of the trial. We planned for this group to meet at least every 12 months over the duration of the trial.
The DSMC also monitored and reviewed accruing data from the internal pilot trial to determine if they would recommend to the TSC that the trial should continue and, if so, whether or not any amendments to the sample size or trial design were required. The DSMC fulfilled this role and, alongside the TSC, concluded that the trial should be closed prematurely.
Throughout the definitive trial, the DSMC would have continued to review accruing data and summaries of those data presented by the alendronate therapy group, and to assess the screening algorithm against the eligibility criteria. It would also have considered emerging evidence from other related trials or research and reviewed any related serious adverse events (SAEs) that were reported.
In addition to a DSMC, a TSC, whose primary function was to act as an oversight body for the trial on behalf of the sponsor and funding body, was appointed. The TSC was chaired by an independent member and considered and acted, as appropriate, upon the recommendations of the DSMC. The TSC also adopted a charter that defined its terms of reference and operation in relation to the oversight of the trial and agreed to meet at least every 12 months over the duration of the trial.
Data management
A Data Management and Sharing Plan was produced for the trial and included reference to confidentiality, access and security arrangements. All data received were processed in accordance with data protection rules. The trial was set up for direct access to be granted to authorised representatives from the sponsor, host institution and the regulatory authorities to permit trial-related monitoring, audits and inspections; however, this was not invoked during the pilot phase of the trial.
All trial data were collected on trial-specific documents, such as questionnaires and CRFs. All trial-specific documents, except for the signed consent form and follow-up contact details, referred to the participant by a unique trial participant number/code and not by name. Participant-identifiable data were stored separately from trial data and in accordance with OCTRU standard operating procedures. All trial data were stored securely in offices accessible only by swipe card by the central co-ordinating team staff in Oxford and authorised personnel.
All data were entered into the trial’s instance of OpenClinica (OpenClinica, LLC, Waltham, MA, USA), which was set up specifically for the trial.
Sample size
Power calculation
The trial required 140 participants per arm (280 in total). This sample size was to provide sufficient power for both co-primary end points of the MANTIS trial.
Short-term end point: the Oxford Hip Score at 12 months
The minimum clinically important difference (MCID) in the OHS is estimated to be 5 points on the scale of 0–48 points. 33 Assuming a common standard deviation (SD) of 10 for the OHS at 12 months, using 90% power and a 5% two-sided significance level, and allowing for a loss to follow-up of 20%, 114 participants were required per arm (228 in total).
Long-term end point: the time to decision that hip replacement is required
The time to the decision that a hip replacement was required was to be measured from randomisation to the date that the participant was placed on the surgical waiting list by their surgeon. This would have allowed us to account for such factors as delays to surgery, or participants not being fit for surgery. It was anticipated that 60% of participants in the control group would require a hip replacement at 36 months post randomisation. This trial was powered to be able to detect an absolute reduction of 20% in the rate of participants requiring a hip replacement in the intervention group (i.e. a reduction to 40% of participants requiring a hip replacement in this trial arm), translating to a hazard ratio of 0.5575.
Using 80% power and a 5% two-sided significance level, and allowing for 20% loss to follow-up assuming that recruitment takes 3 years with an additional 1-year follow-up, 140 participants were required per trial arm (280 in total). The sample size calculation assumed a three-quarters ratio between accrual time and total trial duration.
Sample size calculations were performed in Power Analysis and Sample Size Software (PASS) 11 (NCSS, LLC, Kaysville, UT, USA).
Statistical analyses
Owing to the early halt in trial recruitment, only 21 participants were randomised. Therefore, no formal statistical analyses were planned, as the sample size was too small to provide adequate power for hypothesis testing.
The primary statistical analysis was carried out on the basis of intention to treat (ITT), with all participants being analysed according to their allocated treatment group, irrespective of which treatment they actually received. The analysis was descriptive in nature, and the principal analysis compared results between the intervention and control arms. Numbers and percentages were provided by treatment arm for categorical variables, and numbers, medians and interquartile ranges (IQRs) were provided by treatment arm for continuous variables. For clinical outcomes, means and SDs, and medians and IQRs were provided for both treatment arms.
Chapter 3 Identified challenges
Issues identified for the MANTIS trial
Delayed set-up
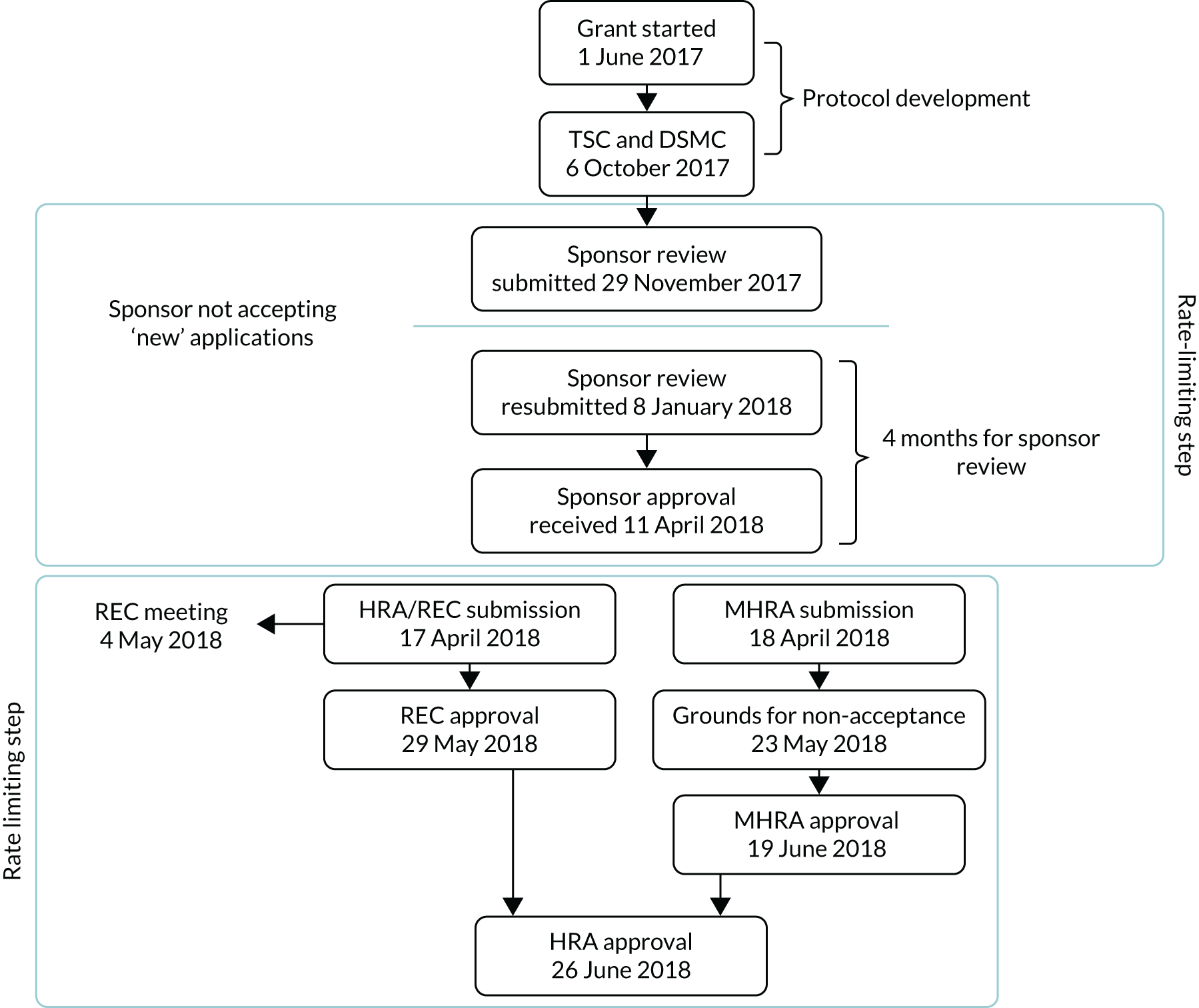
The set-up of the trial was subject to significant delays, which in turn delayed the start of recruitment. These delays have been summarised in Figures 2 and 3.
Overall, set-up resulted in a 14-month delay, with recruitment to the pilot beginning in month 17 rather than month 3. Consequently, the recruitment period was reduced from 36 months to 22 months and the recruitment predictions were remodelled based on estimates from sites. A major contributor to the delay in set-up was our sponsor office’s, which was delayed owing to high local demand and staff shortage; therefore, although it was thought that this review would require only 1 month for completion, it took over 4 months (from 29 November 2018–11 April 2018). In addition, the original submission to the MHRA was not accepted, owing to its operating under the incorrect IMP Product Licence and insufficient details being provided regarding the testing of the over-encapsulated drug product.
FIGURE 2.
Delays experienced with sponsor approval for the MANTIS trial.
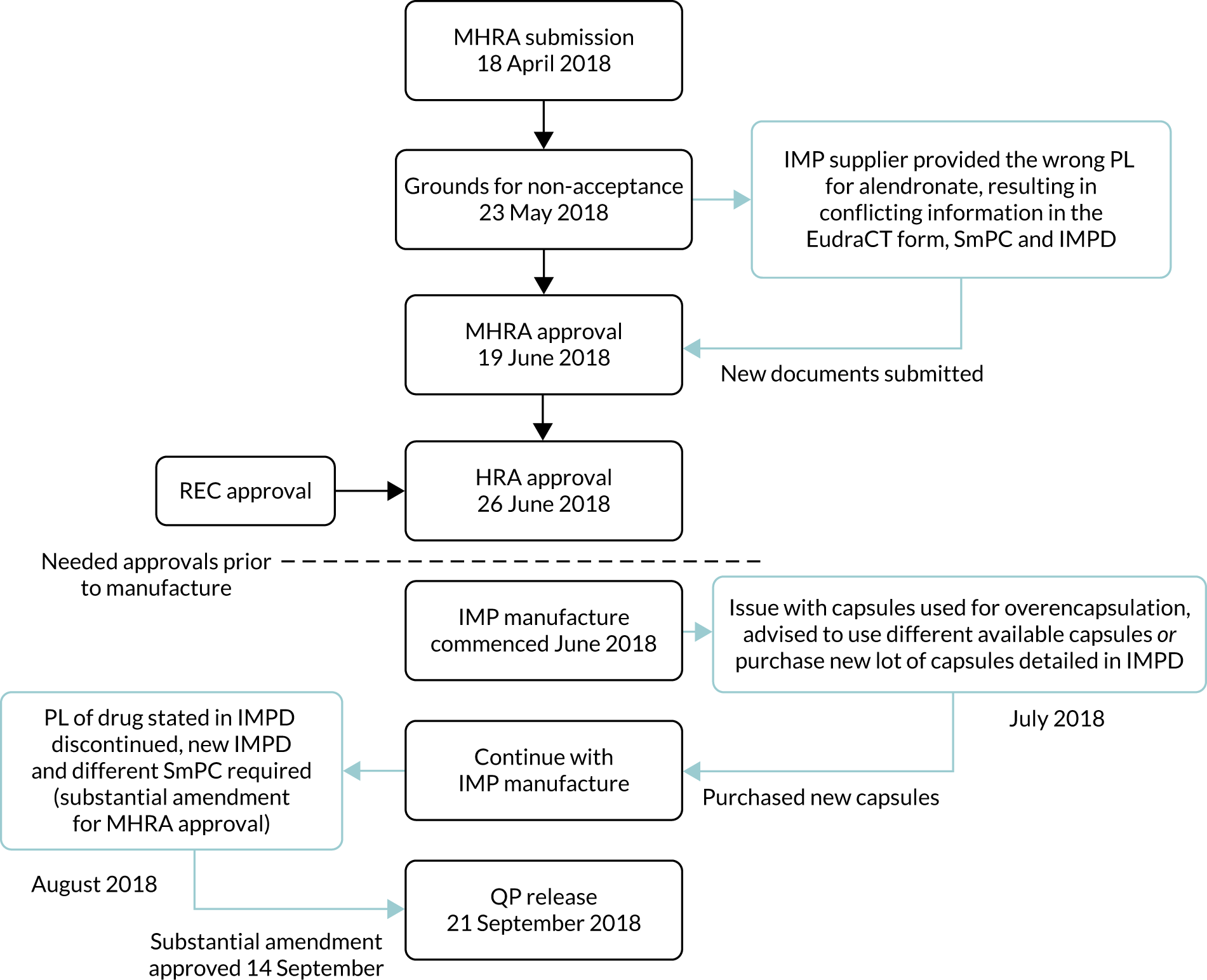
We then encountered delays during the manufacture of the trial IMP; these are specified and detailed in Figure 3.
FIGURE 3.
Delays attributable to the IMP supplier. QP, qualified person.
Poorly defined patient pathway
The patient pathway for the MANTIS trial was complex, with patients identified across multiple specialties including, but not limited to, orthopaedics, haematology, sports medicine, metabolic bone disease, physiotherapy, rheumatology, physiotherapy and oncology. Recruitment predictions for each site were ascertained from hospital databases, a national NIHR CRN feasibility call and business intelligence reports, which may have led to an overestimation, given that these data are usually reflective of the site as a whole, not just one specialty.
In the UK, the patient pathway for AVN is highly variable, both geographically and with respect to the referring specialty. This is a particular challenge given the low disease prevalence. The only means of reliably identifying patients would be the development of a database/registry of hip AVN to detail the current diagnostic pathway, and identify potential solutions for the recruitment of individuals with early-onset disease as well as improvements in clinical care.
When sites were approached regarding the MANTIS trial, site feasibility questionnaires (SFQs) were provided so that the trial team could review predicted recruitment figures and assess the feasibility of local sites. The SFQs encouraged trial teams to think carefully about the number of potentially eligible patients and where they could be identified from (the full patient pathway). The SFQ included the following five questions:
-
How many adult patients with AVN of the hip are seen by your site every year?
-
What information was used to determine this figure?
-
What is your expected recruitment rate for this trial based on the numbers above and the protocol selection criteria (AVN Ficat and Arlet stage 1 or 2)?
-
Which site/department are you associated with?
-
orthopaedic
-
trauma
-
surgical
-
rheumatology
-
oncology
-
transplant
-
renal
-
haematology
-
other (please list).
-
-
How could patients be identified and recruited at your site/department?
On review of the answers provided in the SFQs and information obtained during the pilot phase of the trial, it became apparent that the patient pathway and disease prevalence of the relevant patient population was poorly understood nationally/defined locally. Identifying key patient clinics at recruiting sites was challenging owing to the delay in engaging the relevant clinicians across specialties. Permission to screen other clinics and refer patients to a trial clinician was sought from treating clinicians (who specialised in fields different to those of site PIs) across all sites; however, this was not granted.
The trial team initially recruited only via orthopaedic clinics, but recruitment via this route was slow, and reaching out to clinics for other specialties, including haematology and physiotherapy, resulted in our acquiring new sites and, consequently, improved recruitment rates. Specifically, Milton Keynes University Hospitals NHS Trust was led by a PI who was a haematologist and achieved the highest recruitment rate of all the open MANTIS trial sites.
In addition to this, NIHR National Specialty Groups (musculoskeletal, haematology) were engaged to help identify sites. National Clinical Specialty Groups in haematology and rheumatology were also engaged along with the British Hip Society (London, UK). At the outset of the trial there were no patient/public groups with a specialist interest in AVN of the hip.
Recruitment
In addition to the complex patient pathway, there was a delay between identifying a potential participant and randomisation. This was partly because collaboration was required from the treating clinicians, but also because we required updated imaging and/or blood tests to comply with the eligibility criteria. This was not anticipated at the beginning of the trial, as blood tests and imaging are routinely and regularly conducted in this patient population. However, there are no agreed NHS minimal standards of care for diagnosis and early staging of AVN of the hip. Some sites identified a pool of potential participants who fell outside the inclusion window of 3 months for blood tests and 12 months for imaging and, therefore, had to request updated tests and scans to comply with the eligibility criteria.
At the beginning of the research project, we estimated that AVN had a disease prevalence of approximately 0.001% of the population. As the MANTIS trial was specifically looking at a subset of these patients (i.e. those with early-stage, atraumatic AVN), the number of eligible patients was found to be even lower.
The TMG and TSC also met and discussed the main criteria for ineligibility to determine if it was possible to adjust these. However, they concluded that the ineligibility criteria could not be changed for the following reasons:
-
Patients with advanced disease would not be suitable for this preventative treatment, as advanced disease is usually diagnosed once the hip joint has collapsed. The only suitable treatment for this level of disease is total hip replacement.
-
Bisphosphonates have a long half-life and can metabolise in the bone for over 10 years; therefore, it would be unclear if the treatment effect had been caused by previous treatment or the trial medication.
Capacity at local sites
Various sites that the trial team engaged with early on highlighted capacity issues with pharmacy, radiology and/or the local CRN teams. This remained an ongoing problem with sites already open to recruitment, sites in set-up and sites that expressed an initial interest in taking part in the trial.
Unless we had confirmation of capacity and capability (C&C) across pharmacy, radiology and the local CRN, the MANTIS trial was not feasible at local sites. Sites were slow to set up, primarily owing to a delay in receiving confirmation of C&C within one or more of these departments.
The MANTIS trial was designed to be as pragmatic as possible, with trial tasks coinciding with routine care wherever possible, as in the following examples:
-
In the case of pharmacy, most of the drug management was to be dealt with centrally by the core trial team in Oxford; site departments were required only to dispense the IMP.
-
Radiology had the option to send reports and images to the core trial team for scoring as opposed to completing the CRF locally.
-
All patient follow-up was managed by the core trial team rather than the local CRN (e.g. co-ordination of follow-up questionnaires).
Capacity issues across departments were escalated to a higher level where possible, especially those present within local CRN teams. It is also worth noting that some sites that expressed an initial interest in the trial failed to progress with trial set-up; this was partly due to the discovery that the trial required considerable screening to identify only a small number of patients, and therefore participation in it was regarded as less worthwhile compared with trials that had higher recruitment rates per patient screened (and so could offer greater reimbursement).
Mitigation of issues identified
The trial team mitigated the issues identified in the following ways:
-
Exploring the use of primary care – ‘GP champions’ in two regions across the UK ran searches for patients with AVN to explore whether these patients may be identified in primary care. Unfortunately, both GP champions confirmed that musculoskeletal conditions are poorly coded in primary care, and both regions identified only two potential patients over a 3-year period. It was agreed with the GPs that this would not be a cost-effective way to improve recruitment.
-
Engaging the surgical trainee networks – trainees were engaged through the NIHR Associate PI Scheme and via trainee research collaboratives across the UK. This helped to raise awareness of the trial, encouraged trainees to get involved in research, and resulted in an increased number of patients being screened at sites where trainees were involved.
-
Attending relevant conferences – this again helped us to raise awareness of the trial and engage with potentially interested clinicians. The trial team attended the British Hip Society annual congress in 2018 and 2019, and the British Orthopaedic Association annual congress in 2019, where we engaged with approximately 50 clinicians, some of whom went on to join the trial as members of a recruiting site.
-
Circulating potential patient pathways – as more potential pathways were identified, this information was circulated to existing sites as well as being fed back to potential sites early on in feasibility discussions. Encouraging the engagement between specialties enabled sites to review their recruitment predictions more thoroughly and addressed some capacity issues, as some sites were able to engage more than just the orthopaedic teams.
Further mitigation of issues identified
The trial team researched some additional avenues to be explored at the review with the funder, which took place when recruitment was behind target. These included the following:
-
Advertising the trial through primary care – this would have been in addition to existing posters used in clinic rooms and waiting rooms at recruiting sites. It was thought that this method may have increased recruitment, as patients with early-stage AVN may be unaware of their condition and will sometimes present to the GP only with generalised hip pain. If more patients had been aware of the trial, they may have asked their GP if they could self-refer to a recruiting site for more information.
-
Re-engaging with sites that had been contacted previously – capacity issues may have been resolved since contact was last made and sites may have been more willing to take part in the MANTIS trial owing to the connections that were available through the trainee networks and a better understanding of the patient pathway.
-
Reviewing the per patient payment fee – sites often queried whether or not more money was available to them (to allow them, for example, to cover archiving fees and pay for the time required by research nurses at visits). Although we believed the offered per-patient fee of £100 sufficiently covered the cost to sites (especially when taking into account that the IMP was provided to sites free of charge), the addition of a ‘screening’ fee may have encouraged more sites to take part in the trial. This practice is commonly seen in commercial trials, which often then provide funding for rare disease trials. The trial team could have explored ways that the ‘screening’ fee could be incorporated into the MANTIS trial budget: for example, the extra cost could have been offset by savings made on imaging costs.
However, owing to the early closure of the trial, the trial team did not have the chance to pursue these issues further.
Chapter 4 Results
Explanatory statement
Following the HTA meeting held on 21 November 2019, at which the difficulties in recruiting patients were discussed, it was decided that the MANTIS trial should be closed with immediate effect. Participants were unblinded by the trial team and those in the intervention arm were contacted, and the question of whether they wished to continue to receive alendronate was discussed.
A brief final report is presented in this chapter. No formal statistical tests were performed owing to the small number of participants recruited into the trial.
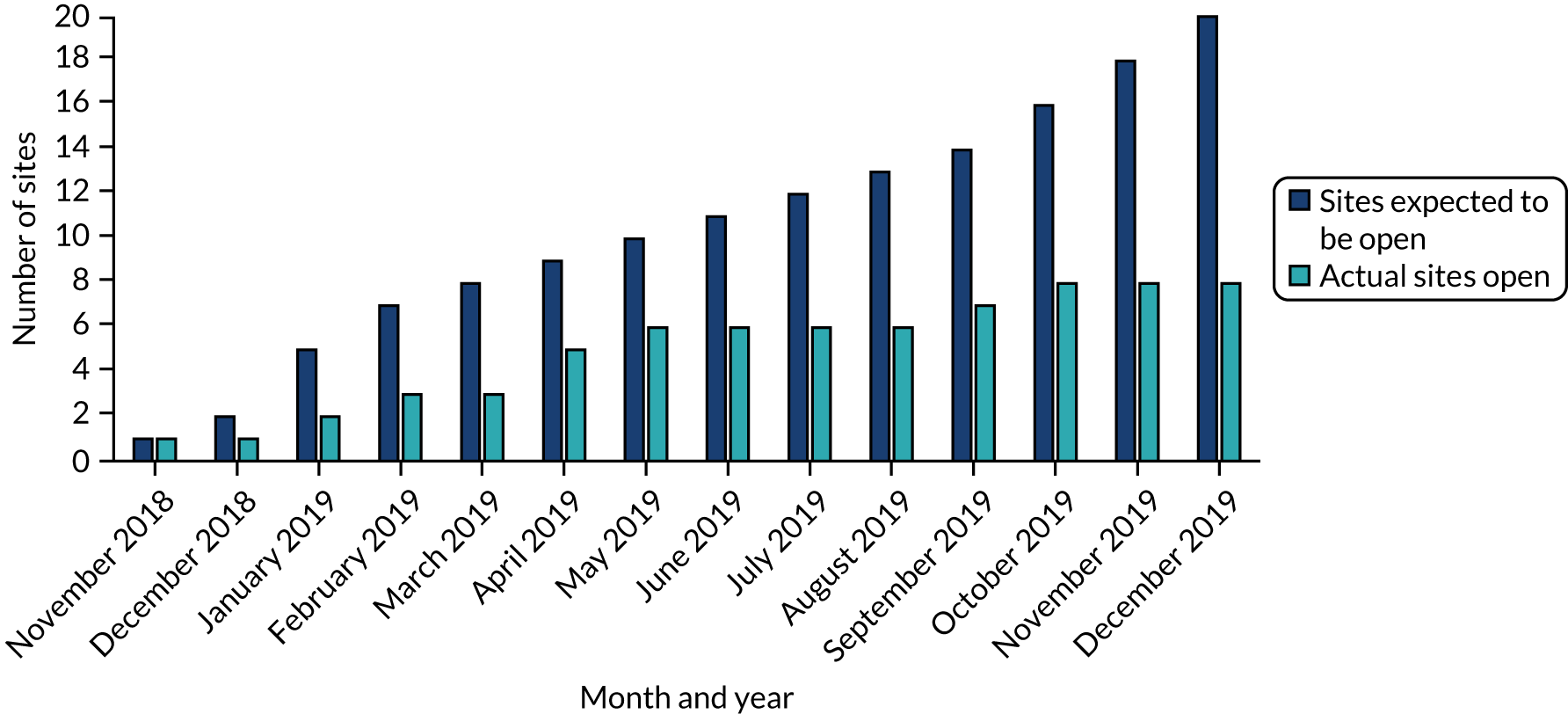
Sites open to recruitment
The trial aimed to open 20 sites. By December 2019, when the trial was shut prematurely, only eight sites had opened to recruitment.
Figure 4 shows the target number of sites compared with the actual number of sites open to recruitment month by month.
FIGURE 4.
Month-by-month target number vs. actual number of sites open to recruitment for the MANTIS trial.
Of the eight open sites, only six recruited at least one participant before the trial was closed to recruitment.
Study participants
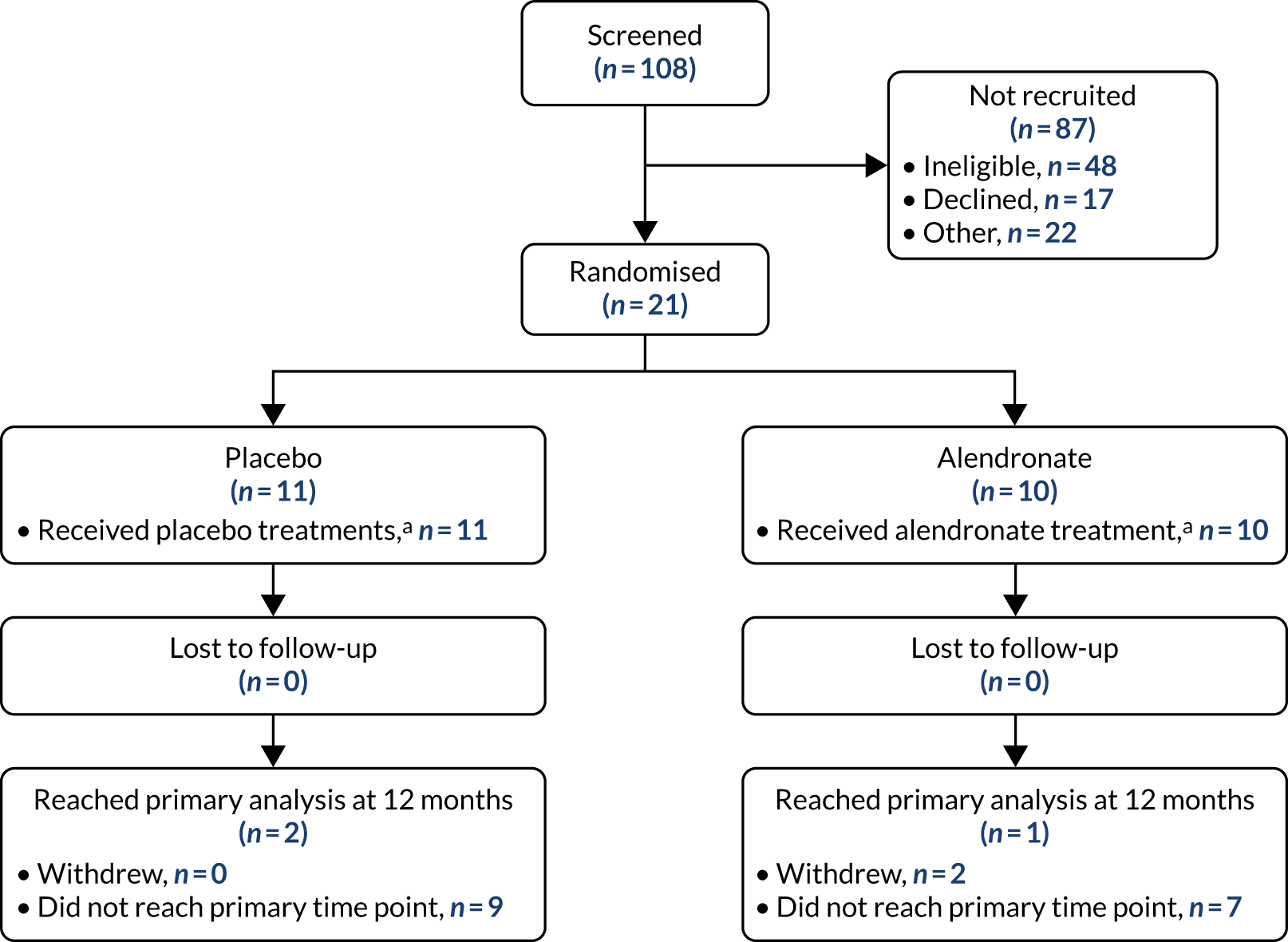
The flow of participants through the trial from screening through randomisation to follow-up is presented in a CONSORT (Consolidated Standards of Reporting Trials) flow diagram (Figure 5). Most randomised participants had not yet reached the primary outcome time point for conversion to hip replacement when the trial was stopped.
FIGURE 5.
Participant flow diagram. a, Patients who received at least one course of treatment.
Recruitment breakdown
Screening and randomisation data, including conversion rates, are summarised by recruitment site in Table 3. Out of 108 patients screened for the MANTIS trial, 48 patients (44%) were deemed ineligible for reasons detailed in Table 4. The most common reason was advanced disease, defined as either a Ficat and Arlet score of > 2 or a Kellgren–Lawrence score of ≥ 2 (indicating osteoarthritis).
| Site | Total screened | Total ineligible | Total declined | Total not randomised with no further informationa | Total randomised | Conversion rate (%)b |
|---|---|---|---|---|---|---|
| Ashford | 1 | 1 | 0 | 0 | 0 | 0 |
| Cornwall | 2 | 0 | 0 | 1 | 1 | 50 |
| Milton Keynes | 15 | 7 | 2 | 0 | 6 | 75 |
| North Midlandsc | 0 | 0 | 0 | 0 | 0 | 0 |
| Oxford | 32 | 16 | 4 | 5 | 7 | 44 |
| Birmingham | 10 | 2 | 5 | 0 | 3 | 38 |
| Stanmore | 27 | 13 | 2 | 11 | 1 | 7 |
| Wrightington | 21 | 9 | 4 | 5 | 3 | 25 |
| Total | 108 | 48 | 17 | 22 | 21 | 35 |
| Site | No evidence of AVN of hip on imaging | Advanced disease | Already on treatment/previous treatment | Contraindication/unable to take treatment | Listed for surgery | Previous fracture | Other | Total |
|---|---|---|---|---|---|---|---|---|
| Ashford | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 |
| Cornwall | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Milton Keynes | 1 | 5 | 0 | 0 | 1 | 0 | 0 | 7 |
| Oxford | 0 | 2 | 5 | 1 | 0 | 0 | 8a | 16 |
| Birmingham | 0 | 1 | 0 | 0 | 0 | 0 | 1b | 2 |
| Stanmore | 4 | 3 | 0 | 1 | 1 | 0 | 4c | 13 |
| Wrightington | 1 | 1 | 2 | 0 | 2 | 3 | 0 | 9 |
| Total | 6 | 12 | 7 | 2 | 5 | 3 | 13 | 48 |
Out of 108 patients screened for the MANTIS trial, 17 declined to take part (17.6%) for reasons detailed in Table 5. Table 6 shows a breakdown of patient recruitment by site, including the numbers recruited at each site for the months in which the trial was open. Table 7 provides more information about the average number of recruits per site per month.
| Site | Language barrier | Treatment preference | Did not respond | Concern over side effects | No reason given | Other reasons | Total |
|---|---|---|---|---|---|---|---|
| Ashford | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Cornwall | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Milton Keynes | 0 | 0 | 0 | 1 | 1 | 0 | 2 |
| Oxford | 0 | 0 | 4 | 0 | 0 | 0 | 4 |
| Birmingham | 2 | 0 | 0 | 0 | 2 | 1a | 5 |
| Stanmore | 0 | 0 | 0 | 1 | 0 | 1b | 2 |
| Wrightington | 0 | 2 | 0 | 0 | 0 | 2c | 4 |
| Total | 2 | 2 | 4 | 2 | 3 | 4 | 17 |
| Site | Date opened | 2018 | 2019 | Total | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| November | December | January | February | March | April | May | June | July | August | September | October | November | |||
| Oxford | 1 November 2018 | 0 | 2 | 1 | 0 | 0 | 2 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 7 |
| Birmingham | 10 January 2019 | – | – | 0 | 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 |
| Wrightington | 7 February 2019 | – | – | – | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 3 |
| Stanmore | 15 April 2019 | – | – | – | – | – | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| Milton Keynes | 16 April 2019 | – | – | – | – | – | 0 | 0 | 0 | 1 | 4 | 0 | 0 | 1 | 6 |
| Cornwall | 10 May 2019 | – | – | – | – | – | – | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| Ashforda | 24 September 2019 | – | – | – | – | – | – | – | – | – | – | 0 | 0 | 0 | 0 |
| North Midlandsb | 10 October 2019 | – | – | – | – | – | – | – | – | – | – | – | 0 | 0 | 0 |
| Total | 0 | 2 | 1 | 0 | 2 | 4 | 1 | 1 | 1 | 5 | 1 | 2 | 1 | 21 | |
| Site | Date opened | Months opena | Screened | Average screened per month | Randomised | Average randomisations per month |
|---|---|---|---|---|---|---|
| Oxford | 1 November 2018 | 12.9 | 32 | 2.48 | 7 | 0.54 |
| Birmingham | 10 January 2019 | 10.6 | 10 | 0.94 | 3 | 0.28 |
| Wrightington | 7 February 2019 | 9.7 | 21 | 2.16 | 3 | 0.31 |
| Stanmore | 15 April 2019 | 7.5 | 27 | 3.60 | 1 | 0.13 |
| Milton Keynes | 16 April 2019 | 7.4 | 15 | 2.03 | 6 | 0.81 |
| Cornwall | 10 May 2019 | 6.7 | 2 | 0.30 | 1 | 0.15 |
| Ashford | 24 September 2019 | 2.2 | 1 | 0.45 | 0 | 0 |
| North Midlands | 10 October 2019 | 1.6 | 0 | 0 | 0 | 0 |
| Total | – | 58.6b | 108 | 1.84 | 21 | 0.36c |
Patient demographics
Tables 8 and 9 show information on the baseline characteristic comparability by allocated treatment arm.
| Characteristic | Placebo, n (%) | Alendronate, n (%) | Total, n (%) |
|---|---|---|---|
| Minimisation factorsa | |||
| Affected hips | |||
| Bilateral | 3 (27.3) | 5 (50.0) | 8 (38.1) |
| Unilateral | 8 (72.7) | 5 (50.0) | 13 (61.9) |
| AVN risk factor | |||
| Alcohol | 1 (9.1) | 2 (20.0) | 3 (14.3) |
| Other | 9 (81.8) | 7 (70.0) | 16 (76.2) |
| Steroid | 1 (9.1) | 1 (10.0) | 2 (9.5) |
| Ficat and Arlet stageb | |||
| 1 | 3 (27.3) | 2 (20.0) | 5 (23.8) |
| 2 | 8 (72.7) | 8 (80.0) | 16 (76.2) |
| Site | |||
| Milton Keynes | 3 (27.3) | 3 (30.0) | 6 (28.6) |
| Oxford | 4 (36.4) | 3 (30.0) | 7 (33.3) |
| Cornwall | 0 (0.0) | 1 (10.0) | 1 (4.8) |
| Stanmore | 1 (9.1) | 0 (0.0) | 1 (4.8) |
| Birmingham | 1 (9.1) | 2 (20.0) | 3 (14.3) |
| Wrightington | 2 (18.2) | 1 (10.0) | 3 (14.3) |
| Baseline characteristics | |||
| Gender | |||
| Female | 4 (36.4) | 1 (10.0) | 5 (23.8) |
| Male | 7 (63.6) | 9 (90.0) | 16 (76.2) |
| Study hip | |||
| Left | 6 (54.5) | 4 (40.0) | 10 (47.6) |
| Right | 3 (27.3) | 4 (40.0) | 7 (33.3) |
| Missing | 2 (18.2) | 2 (20.0) | 4 (19.0) |
| Ethnicity | |||
| Black or black British | 4 (36.4) | 1 (10.0) | 5 (23.8) |
| Indian | 1 (9.1) | 0 (0.0) | 1 (4.8) |
| White British | 5 (45.5) | 8 (80.0) | 13 (61.9) |
| White other | 1 (9.1) | 1 (10.0) | 2 (9.5) |
| Smoking status | |||
| Current smoker | 1 (9.1) | 5 (50.0) | 6 (28.6) |
| Former smoker | 4 (36.4) | 2 (20.0) | 6 (28.6) |
| Never smoked | 6 (54.5) | 3 (30.0) | 9 (42.9) |
| Alcohol frequency | |||
| Two or three times per week | 2 (18.2) | 4 (40.0) | 6 (28.6) |
| Two to four times per month | 1 (9.1) | 0 (0.0) | 1 (4.8) |
| Four or more times per week | 2 (18.2) | 2 (20.0) | 4 (19.0) |
| Monthly or less | 2 (18.2) | 1 (10.0) | 3 (14.3) |
| Never | 4 (36.4) | 3 (30.0) | 7 (33.3) |
| Alcoholic units (week) | |||
| 0–2 | 7 (63.6) | 3 (30.0) | 10 (47.6) |
| 3 or 4 | 3 (27.3) | 3 (30.0) | 6 (28.6) |
| 5 or 6 | 0 (0.0) | 1 (10.0) | 1 (4.8) |
| 7–9 | 0 (0.0) | 1 (10.0) | 1 (4.8) |
| 10 or more | 0 (0.0) | 1 (10.0) | 1 (4.8) |
| Missing | 1 (9.1) | 1 (10.0) | 2 (9.5) |
| Relevant medical history | |||
| No | 8 (72.7) | 3 (30.0) | 11 (52.4) |
| Yesc | 3 (27.3) | 7 (70.0) | 10 (47.6) |
| Concomitant medications (last 6 months) | |||
| No | 1 (9.1) | 1 (10.0) | 2 (9.5) |
| Yesd | 10 (90.9) | 9 (90.0) | 19 (90.5) |
| Kellgren–Lawrence gradeb | |||
| Grade 0 | 4 (36.4) | 3 (30.0) | 7 (33.3) |
| Grade 1 | 5 (45.5) | 3 (30.0) | 8 (38.1) |
| Grade 2 | 1 (9.1) | 1 (10.0) | 2 (9.5) |
| Missing | 1 (9.1) | 3 (30.0) | 4 (19.0) |
| Characteristic | Placebo | Alendronate | Total | ||||||
|---|---|---|---|---|---|---|---|---|---|
| n | Median | IQR | n | median | IQR | n | Median | IQR | |
| BMI (kg/m2) | 10 | 20.8 | 17.5–26.8 | 9 | 20.6 | 17.5–26.8 | 19 | 25.4 | 23.7–32.1 |
| Height (m) | 10 | 1.79 | 1.65–1.87 | 9 | 1.80 | 1.78 1.82 | 19 | 1.79 | 1.72 1.83 |
| Weight (kg) | 10 | 80.4 | 72.0–89.6 | 10 | 94.8 | 87.2–114.0 | 20 | 87.6 | 74.5–98.4 |
| JSW (mm)a | 7 | 4.0 | 3.0–5.0 | 6 | 5.0 | 4.0–6.0 | 13 | 4.0 | 3.5–6.0 |
Treatment compliance
Of the 21 participants randomised, eight (38.1%) completed the full course of treatment as shown in Table 10; four were allocated to the placebo treatment arm and four were allocated to the alendronate treatment arm. Tables 11 and 12 show the information from the self-completed compliance CRF for each treatment arm, where compliance with taking the allocated treatment once per week was high (> 80%), as measured from those responding to the CRF.
| Drug packs issued | Placebo, n (%) | Alendronate, n (%) | Total, n (%) |
|---|---|---|---|
| First | 11 (100.0) | 10 (100.0) | 21 (100.0) |
| Second | 11 (100.0) | 8 (80.0) | 19 (90.5) |
| Third | 8 (72.7) | 7 (70.0) | 15 (71.4) |
| Fourth (full 12-month course) | 4 (36.4) | 4 (40.0) | 8 (38.1) |
| CRF item | Month (n) | |||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 6 | 9 | 12 | |
| Forms returned | 7 | 6 | 5 | 3 | 2 | 1 |
| Tablet once per week | ||||||
| Always (> 80%) | 7 | 6 | 5 | 2 | 2 | 1 |
| Usually (50–80%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Sometimes (< 50%) | 0 | 0 | 0 | 1 | 0 | 0 |
| Never | 0 | 0 | 0 | 0 | 0 | 0 |
| First thing in morning | ||||||
| Always (> 80%) | 6 | 6 | 5 | 1 | 1 | |
| Usually (50–80%) | 0 | 0 | 0 | 1 | 1 | 1 |
| Sometimes (< 50%) | 1 | 0 | 0 | 1 | 0 | 0 |
| Never | 0 | 0 | 0 | 0 | 0 | 0 |
| With water | ||||||
| Always (> 80%) | 6 | 4 | 4 | 3 | 2 | 1 |
| Usually (50–80%) | 0 | 1 | 0 | 0 | 0 | 0 |
| Sometimes (< 50%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Never | 1 | 1 | 1 | 0 | 0 | 0 |
| Swallowed whole | ||||||
| Always (> 80%) | 6 | 5 | 5 | 2 | 1 | 1 |
| Usually (50–80%) | 1 | 1 | 0 | 1 | 1 | 0 |
| Sometimes (< 50%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Never | 0 | 0 | 0 | 0 | 0 | 0 |
| No food or drink | ||||||
| Always (> 80%) | 6 | 5 | 4 | 3 | 1 | 1 |
| Usually (50–80%) | 0 | 0 | 0 | 0 | 1 | 0 |
| Sometimes (< 50%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Never | 1 | 1 | 1 | 0 | 0 | 0 |
| Remained sitting | ||||||
| Always (> 80%) | 3 | 3 | 2 | 2 | 1 | 0 |
| Usually (50–80%) | 2 | 0 | 1 | 0 | 1 | 1 |
| Sometimes (< 50%) | 1 | 3 | 2 | 0 | 0 | 0 |
| Never | 1 | 0 | 0 | 1 | 0 | 0 |
| Worsened heartburn or indigestion | ||||||
| Always (> 80%) | 0 | 1 | 1 | 0 | 0 | 0 |
| Usually (50–80%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Sometimes (< 50%) | 1 | 0 | 0 | 0 | 0 | 0 |
| Never | 6 | 5 | 4 | 3 | 2 | 1 |
| Difficulty swallowing | ||||||
| Always (> 80%) | 1 | 1 | 1 | 0 | 0 | 0 |
| Usually (50–80%) | 0 | 0 | 1 | 0 | 0 | 0 |
| Sometimes (< 50%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Never | 6 | 5 | 3 | 3 | 2 | 1 |
| Chewable calcium supplements | ||||||
| Always (> 80%) | 0 | 1 | 1 | 1 | 0 | 0 |
| Usually (50–80%) | 1 | 0 | 1 | 0 | 0 | 0 |
| Sometimes (< 50%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Never | 6 | 5 | 3 | 2 | 1 | 1 |
| Missing | 0 | 0 | 0 | 0 | 1 | 0 |
| Side effects | ||||||
| No | 4 | 3 | 1 | 1 | 1 | 0 |
| Yes | 1a | 1b | 2c | 1d | 0 | 0 |
| Missing | 2 | 2 | 2 | 1 | 1 | 2 |
| CRF item | Month (n) | |||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 6 | 9 | 12 | |
| Forms returned | 7 | 8 | 7 | 5 | 1 | 1 |
| Tablet once per week | ||||||
| Always (> 80%) | 7 | 8 | 7 | 5 | 1 | 1 |
| Usually (50–80%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Sometimes (< 50%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Never | 0 | 0 | 0 | 0 | 0 | 0 |
| First thing in morning | ||||||
| Always (> 80%) | 6 | 6 | 5 | 3 | 0 | 1 |
| Usually (50–80%) | 0 | 0 | 1 | 1 | 1 | 0 |
| Sometimes (< 50%) | 1 | 1 | 1 | 1 | 0 | 0 |
| Never | 0 | 1 | 0 | 0 | 0 | 0 |
| With water | ||||||
| Always (> 80%) | 4 | 6 | 4 | 2 | 1 | 1 |
| Usually (50–80%) | 1 | 0 | 1 | 1 | 0 | 0 |
| Sometimes (< 50%) | 2 | 2 | 2 | 2 | 0 | 0 |
| Never | 0 | 0 | 0 | 0 | 0 | 0 |
| Swallowed whole | ||||||
| Always (> 80%) | 7 | 8 | 7 | 4 | 1 | 1 |
| Usually (50–80%) | 0 | 0 | 0 | 1 | 0 | 0 |
| Sometimes (< 50%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Never | 0 | 0 | 0 | 0 | 0 | 0 |
| No food or drink | ||||||
| Always (> 80%) | 5 | 7 | 5 | 3 | 1 | 1 |
| Usually (50–80%) | 1 | 1 | 2 | 1 | 0 | 0 |
| Sometimes (< 50%) | 1 | 0 | 0 | 1 | 0 | 0 |
| Never | 0 | 0 | 0 | 0 | 0 | 0 |
| Remained sitting | ||||||
| Always (> 80%) | 6 | 7 | 6 | 4 | 1 | 1 |
| Usually (50–80%) | 0 | 1 | 0 | 0 | 0 | 0 |
| Sometimes (< 50%) | 1 | 0 | 1 | 1 | 0 | 0 |
| Never | 0 | 0 | 0 | 0 | 0 | 0 |
| Worsened heartburn or indigestion | ||||||
| Always (> 80%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Usually (50–80%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Sometimes (< 50%) | 1 | 2 | 2 | 3 | 1 | 0 |
| Never | 6 | 6 | 5 | 2 | 0 | 1 |
| Difficulty swallowing | ||||||
| Always (> 80%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Usually (50–80%) | 0 | 0 | 0 | 1 | 0 | 0 |
| Sometimes (< 50%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Never | 7 | 8 | 7 | 4 | 1 | 1 |
| Chewable calcium supplements | ||||||
| Always (> 80%) | 2 | 2 | 2 | 1 | 0 | 1 |
| Usually (50–80%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Sometimes (< 50%) | 0 | 1 | 0 | 0 | 0 | 0 |
| Never | 5 | 5 | 5 | 4 | 1 | 0 |
| Missing | 3 | 1 | 2 | 0 | 0 | 1 |
| Side effects | ||||||
| No | 1 | 1 | 1 | 1 | 0 | 0 |
| Yes | 4a | 6b | 4c | 4d | 1 | 0 |
| Missing | 5 | 8 | 5 | 5 | 2 | 1 |
Follow-up of patients
The number of follow-up questionnaires containing patient-reported treatment compliance and outcome measures, both expected and received, at 1 and 12 months post randomisation is summarised in Table 13. The compliance rate has been calculated as a percentage of those received out of the total expected. Please see Appendices 1 and 2 for more details on treatment allocation and individual participant details.
Owing to the early closure of the trial, no participants reached the 24 or 36 months post-randomisation time point.
| Questionnaire | Placebo | Alendronate | Total expecteda | Total received, n (%) | ||
|---|---|---|---|---|---|---|
| Expecteda | Received, n (%) | Expecteda | Received, n (%) | |||
| Baselineb | 11 | 11 (100.0) | 10 | 10 (100.0) | 21 | 21 (100.0) |
| 1-month compliance | 11 | 7 (63.6) | 10 | 7 (70.0) | 21 | 14 (66.7) |
| 2-month compliance | 11 | 6 (54.5) | 10 | 8 (80.0) | 21 | 14 (66.7) |
| 3-month compliance | 11 | 5 (45.5) | 9 | 7 (77.8) | 19 | 12 (63.2) |
| 6-month compliance | 9 | 3 (33.3) | 6 | 5 (83.3) | 15 | 8 (53.3) |
| 6-month follow-upc | 9 | 4 (44.4) | 6 | 7 (116.6) | 15 | 11 (73.3) |
| OHS | 9 | 3 (33.3) | 6 | 6 (100.0) | 15 | 9 (60.0) |
| iHot-33 | 9 | 3 (33.3) | 6 | 7 (116.6) | 15 | 10 (66.7) |
| EQ-5D-5L utility | 9 | 4 (44.4) | 6 | 7 (116.6) | 15 | 11 (73.3) |
| EQ-5D-5L VAS | 9 | 4 (44.4) | 6 | 7 (116.6) | 15 | 11 (73.3) |
| HADS | 9 | 4 (44.4) | 6 | 7 (116.6) | 15 | 11 (73.3) |
| Health resource | 9 | 4 (44.4) | 6 | 7 (116.6) | 15 | 11 (73.3) |
| 9-month compliance | 6 | 2 (33.3) | 3 | 1 (33.3) | 9 | 3 (33.3) |
| 12-month compliance | 2 | 1 (50.0) | 1 | 1 (100.0) | 3 | 2 (66.7) |
| 12-month follow-up | 2 | 2 (100.0) | 1 | 1 (100.0) | 3 | 3 (100.0) |
| OHS | 2 | 2 (100.0) | 1 | 1 (100.0) | 3 | 3 (100.0) |
| iHOT-33 | 2 | 2 (100.0) | 1 | 1 (100.0) | 3 | 3 (100.0) |
| EQ-5D-5L utility | 2 | 2 (100.0) | 1 | 1 (100.0) | 3 | 3 (100.0) |
| EQ-5D-5L VAS | 2 | 2 (100.0) | 1 | 1 (100.0) | 3 | 3 (100.0) |
| HADS | 2 | 2 (100.0) | 1 | 1 (100.0) | 3 | 3 (100.0) |
| Health resource | 2 | 2 (100.0) | 1 | 1 (100.0) | 3 | 3 (100.0) |
Withdrawals and protocol deviations
Out of the 21 patients randomised, two withdrew from the trial. The details of both withdrawals are summarised in Table 14. In addition, Table 15 shows information about the two protocol deviations that occured during the trial and the actions taken to address these.
| Allocation | Reason | Site | Time from randomisation to withdrawal (days) | Last CRF |
|---|---|---|---|---|
| Alendronate | Patient had osteoarthritis (not judged to have AVN) | Birmingham | 70 | Compliance (month 2) |
| Alendronate | Participant withdrew consent | Milton Keynes | 93 | Baseline |
| Allocation | Information | Action taken | Site | Date |
|---|---|---|---|---|
| Placebo | Medication was not collected (second drug pack) | Patient was withdrawn from allocated treatment but continued follow-up | Milton Keynes | 20 November 2019 |
| Placebo | No response to the 1-month follow-up; a call was scheduled but was missed owing to staff annual leave | Trial cover plan was implemented | Milton Keynes | 25 December 2019 |
Clinical outcome results
Oxford Hip Scores
Tables 16 and 17 show the OHS for the left and right hip, respectively, summarised by treatment arm at each time point using means and SDs, as well as medians and IQRs.
| Time point | Placebo | Alendronate | Total | ||||||
|---|---|---|---|---|---|---|---|---|---|
| n | Mean (SD) | Median (IQR) | n | Mean (SD) | Median (IQR) | n | Mean (SD) | Median (IQR) | |
| Baseline | 11 | 29.3 (12.5) | 26.0 (19.0–37.0) | 10 | 38.7 (13.3) | 42.0 (31.0–48.0) | 21 | 33.8 (13.4) | 35.0 (24.0–47.0) |
| 6 months | 3 | 31.5 (17.9) | 24.0 (18.5–52.0) | 6 | 39.3 (12.6) | 42.0 (28.0–49.0) | 9 | 36.7 (14.0) | 41.0 (24.0–49.0) |
| 12 months | 2 | 24.5 (0.7) | 24.5 (24.0–25.0) | 1 | 37.0 (–) | 37.0 (–) | 3 | 28.7 (7.2) | 25.0 (24.0–37.0) |
| Time point | Placebo | Alendronate | Total | ||||||
|---|---|---|---|---|---|---|---|---|---|
| n | Mean (SD) | Median (IQR) | n | Mean (SD) | Median (IQR) | n | Mean (SD) | Median (IQR) | |
| Baseline | 11 | 31.2 (14.0) | 30.0 (22.0–43.0) | 10 | 36.0 (13.0) | 36.5 (28.0–45.0) | 21 | 33.5 (13.4) | 35.0 (22.0–43.0) |
| 6 months | 4 | 26.4 (13.1) | 20.0 (19.8–33.0) | 6 | 35.5 (16.4) | 33.5 (23.0–53.0) | 10 | 31.9 (15.1) | 24.0 (20.0–46.0) |
| 12 months | 2 | 13.0 (1.4) | 13.0 (12.0–14.0) | 1 | 13.0 (–) | 13.0 (–) | 3 | 13.0 (1.0) | 13.0 (12.0–14.0) |
iHOT-33 scores
Tables 18 and 19 show the iHOT-33 scores for the left and right hip, respectively, summarised by treatment arm at each time point using means and SDs, as well as medians and IQRs. Both the subscale scores and the total scores are summarised.
| Time point | Placebo | Alendronate | Total | ||||||
|---|---|---|---|---|---|---|---|---|---|
| n | Mean (SD) | Median (IQR) | n | Mean (SD) | Median (IQR) | n | Mean (SD) | Median (IQR) | |
| Section 1: symptoms and functional limitations | |||||||||
| Baseline | 11 | 60.3 (30.1) | 54.0 (27.5–93.9) | 10 | 37.3 (35.9) | 28.3 (3.5–68.9) | 21 | 49.3 (34.2) | 51.6 (19.6–77.4) |
| 6 months | 3 | 55.6 (39.7) | 72.3 (10.3–84.2) | 7 | 37.8 (34.1) | 23.6 (2.6–70.5) | 10 | 43.2 (34.6) | 40.9 (10.3–72.3) |
| 12 months | 2 | 68.5 (22.6) | 68.5 (52.5–84.5) | 1 | 26.1 (–) | 26.1 (–) | 3 | 54.4 (29.3) | 52.5 (26.1–84.5) |
| Section 2: sports and recreational activities | |||||||||
| Baseline | 11 | 50.4 (35.4) | 43.5 (16.7–86) | 10 | 22.3 (34.3) | 8.4 (0.2–21.0) | 21 | 37.0 (36.9) | 21.0 (6.2–68.2) |
| 6 months | 3 | 34.6 (36.8) | 15.4 (11.4–77.0) | 7 | 29.6 (38.4) | 9.0 (0.0–84.2) | 10 | 31.1 (35.9) | 13.4 (6.3–77.0) |
| 12 months | 2 | 43.5 (47.9) | 43.5 (9.6–77.4) | 1 | 5.4 (–) | 5.4 (–) | 3 | 30.8 (40.4) | 9.6 (5.4–77.4) |
| Section 3: job-related concernsa | |||||||||
| Baseline | 8 | 59.1 (34.6) | 72.7 (28.8–83.2) | 5 | 36.4 (38.3) | 21.3 (11.5–43.3) | 13 | 50.4 (36.3) | 43.3 (18.0–82.0) |
| 6 months | 2 | 82.2 (7.8) | 82.2 (76.7–87.8) | 3 | 45.2 (44.8) | 46 (0.0–89.5) | 5 | 60.0 (37.8) | 76.7 (46.0–87.8) |
| 12 months | 2 | 78.3 (14.1) | 78.3 (68.3–88.3) | 1 | 18.0 (–) | 18.0 (–) | 3 | 58.2 (36.2) | 68.3 (18.0–88.3) |
| Section 4: social, emotional and lifestyle concerns | |||||||||
| Baseline | 11 | 47.6 (38.2) | 46.3 (9.2–99.5) | 10 | 26.0 (33.9) | 15.5 (0.0–30.7) | 21 | 37.2 (37.1) | 26.3 (4.8–54.6) |
| 6 months | 3 | 38.6 (48.7) | 11.9 (9.0–94.8) | 7 | 23.4 (34.9) | 6.8 (0.4–48.7) | 10 | 28.0 (37.3) | 9.1 (6.2–48.7) |
| 12 months | 2 | 55.1 (44.3) | 55.1 (23.7–86.4) | 1 | 23.5 (–) | 23.5 (–) | 3 | 44.5 (35.3) | 23.7 (23.5–86.4) |
| Overall score | |||||||||
| Baseline | 11 | 55.7 (30.3) | 50.2 (36.5–94.5) | 10 | 31.3 (33.6) | 24.8 (2.1–44.7) | 21 | 44.1 (33.5) | 37.9 (16.2–64.5) |
| 6 months | 3 | 48.5 (37.1) | 49.2 (11.0–85.2) | 7 | 32.6 (33.2) | 19.7 (1.5–65.4) | 10 | 37.4 (33.2) | 27.9 (11.0–65.4) |
| 12 months | 2 | 62.3 (30.7) | 62.3 (40.6–84.0) | 1 | 21.2 (–) | 21.2 (–) | 3 | 48.6 (32.2) | 40.6 (21.2–84.0) |
| Time point | Placebo | Alendronate | Total | ||||||
|---|---|---|---|---|---|---|---|---|---|
| n | Mean (SD) | Median (IQR) | n | Mean (SD) | Median (IQR) | n | Mean (SD) | Median (IQR) | |
| Section 1: symptoms and functional limitations | |||||||||
| Baseline | 11 | 51.6 (34.1) | 47.3 (18.8–86.9) | 10 | 38.9 (35.0) | 30.6 (6.3–61.6) | 21 | 45.5 (34.2) | 42.6 (17.2–72.3) |
| 6 months | 4 | 63.3 (26.9) | 71.2 (45.2–81.5) | 7 | 40.4 (41.6) | 24.6 (41.0–91.2) | 11 | 48.7 (37.3) | 65.1 (5.1–86.0) |
| 12 months | 2 | 98.4 (1.1) | 98.4 (97.6–99.3) | 1 | 97.9 (–) | 97.9 (–) | 3 | 98.3 (0.9) | 97.9 (97.6–99.3) |
| Section 2: sports and recreational activities | |||||||||
| Baseline | 11 | 47.4 (35.0) | 42.3 (17.5–79.5) | 10 | 26.1 (32.6) | 14.6 (1.5–32.6) | 21 | 37.2 (34.8) | 28.6 (8.0–62.5) |
| 6 months | 4 | 43.7 (27.1) | 38.1 (22.8–64.6) | 7 | 31.5 (34.1) | 23.0 (0.0–73.7) | 11 | 36.0 (30.9) | 26.2 (5.0–73.7) |
| 12 months | 2 | 98.1 (2.7) | 98.1 (96.2–100.0) | 1 | 66.2 (–) | 66.2 (–) | 3 | 87.5 (18.5) | 96.2 (66.2–100.0) |
| Section 3: job-related concernsa | |||||||||
| Baseline | 8 | 57.1 (40.4) | 70.8 (14–91.6) | 6 | 43.5 (39.2) | 38.4 (8.7–75.7) | 14 | 51.3 (39.0) | 57.5 (8.7–83.8) |
| 6 months | 4 | 61.8 (39.8) | 76.7 (39–84.5) | 3 | 37.7 (37.6) | 37.8 (0.0–75.3) | 7 | 51.4 (37.8) | 75.0 (3.0–78.3) |
| 12 months | 2 | 98.3 (1.9) | 98.3 (97.0–99.7) | 1 | 99.0 (–) | 99.0 (–) | 3 | 98.6 (1.4) | 99.0 (97.0–99.7) |
| Section 4: social, emotional and lifestyle concerns | |||||||||
| Baseline | 11 | 48.9 (37.8) | 47.7 (9.2–81.2) | 10 | 34.8 (35.2) | 21.5 (11.0–46.4) | 21 | 42.2 (36.4) | 46.1 (11.0–76.7) |
| 6 months | 4 | 58.9 (44.3) | 57.6 (20.6–97.2) | 7 | 27.6 (31.8) | 25.2 (0.0–39.1) | 11 | 39.0 (38.0) | 25.2 (6.0–90.1) |
| 12 months | 2 | 96.1 (5.5) | 96.1 (92.2–100.0) | 1 | 99.3 (–) | 99.3 (–) | 3 | 97.2 (4.3) | 99.3 (92.2–100.0) |
| Overall scores | |||||||||
| Baseline | 11 | 49.8 (34.7) | 52.8 (15.7–79.9) | 10 | 35.5 (33.7) | 23.9 (7.2–54.9) | 21 | 43.0 (34.2) | 39.6 (14.8–71.9) |
| 6 months | 4 | 58.9 (28.8) | 63.2 (36.3–81.5) | 7 | 34.9 (35.8) | 23.5 (2.9–68.4) | 11 | 43.6 (34.1) | 50.3 (4.6–76.0) |
| 12 months | 2 | 98.0 (2.2) | 98.0 (96.4–99.6) | 1 | 93.0 (–) | 93.0 (–) | 3 | 96.3 (3.3) | 96.4 (93.0–99.6) |
EuroQol-5 Dimensions, five-level version, scores
Table 20 shows the EQ-5D-5L utility and visual analogue scale (VAS) scores summarised by treatment arm at each time point using means and SDs, as well as medians and IQRs.
| Time point | Placebo | Alendronate | Total | ||||||
|---|---|---|---|---|---|---|---|---|---|
| n | Mean (SD) | Median (IQR) | n | Mean (SD) | Median (IQR) | n | Mean (SD) | Median (IQR) | |
| EQ-5D-5L utility score | |||||||||
| Baseline | 10 | 0.47 (0.39) | 0.59 (0.41–0.74) | 10 | 0.23 (0.32) | 0.25 (0.0–0.59) | 20 | 0.36 (0.37) | 0.48 (0.16–0.61) |
| 6 months | 4 | 0.68 (0.12) | 0.73 (0.60–0.75) | 7 | 0.37 (0.36) | 0.53 (0.04–0.60) | 11 | 0.48 (0.33) | 0.56 (0.17–0.74) |
| 12 months | 2 | 0.73 (0.15) | 0.73 (0.62–0.84) | 1 | 0.34 (–) | 0.34 (–) | 3 | 0.60 (0.25) | 0.62 (0.34–0.84) |
| EQ-5D-5L VAS | |||||||||
| Baseline | 11 | 57.8 (32.4) | 60.0 (30.0–95.0) | 10 | 45.5 (26.3) | 47.5 (40.0–50.0) | 21 | 52.0 (29.6) | 50.0 (30.0–75.0) |
| 6 months | 4 | 75.0 (24.8) | 82.5 (57.5–92.5) | 7 | 47.9 (28.6) | 40.0 (15.0–75.0) | 11 | 57.7 (29.4) | 75.0 (35.0–80.0) |
| 12 months | 2 | 87.5 (10.6) | 87.5 (80.0–94.0) | 1 | 40.0 (–) | 40.0 (–) | 3 | 71.7 (28.4) | 80.0 (40.0–95.0) |
Hospital Anxiety and Depression Scale scores
Table 21 shows the HADS scores summarised by treatment arm at each time point using means and SDs, as well as medians and IQRs.
| Time point | Placebo | Alendronate | Total | ||||||
|---|---|---|---|---|---|---|---|---|---|
| n | Mean (SD) | Median (IQR) | n | Mean (SD) | Median (IQR) | n | Mean (SD) | Median (IQR) | |
| Anxiety | |||||||||
| Baseline | 11 | 8.2 (5.4) | 7.0 (3.0–13.0) | 9 | 9.7 (6.1) | 10.0 (4.0–15.0) | 20 | 8.9 (5.6) | 8.5 (3.5–13.0) |
| 6 months | 4 | 5.5 (5.2) | 5.5 (1.0–10.0) | 7 | 10.4 (6.6) | 11.0 (3.0–17.0) | 11 | 8.6 (6.4) | 10.0 (1.0–12.0) |
| 12 months | 2 | 5.5 (7.8) | 5.5 (0.0–11.0) | 1 | 11.0 (–) | 11.0 (–) | 3 | 7.3 (6.4) | 11.0 (0.0–11.0) |
| Depression | |||||||||
| Baseline | 11 | 5.6 (4.2) | 4.0 (2.0–10.0) | 9 | 7.2 (3.4) | 6.0 (5.0–9.0) | 20 | 6.4 (3.9) | 5.0 (3.0–10.0) |
| 6 months | 4 | 5.3 (4.0) | 5.0 (2.0–8.5) | 7 | 9.9 (4.9) | 12.0 (5.0–14.0) | 11 | 8.2 (5.0) | 10.0 (3.0–12.0) |
| 12 months | 2 | 3.5 (4.9) | 3.5 (0.0–7.0) | 1 | 7.0 (–) | 7.0 (–) | 3 | 4.7 (4.0) | 7.0 (0.0–7.0) |
Safety reporting
Details of AEs and SAEs are summarised in Table 22. Among 21 patients, two SAEs were recorded, both of which were considered to be unrelated to the treatment.
| Site | Allocation | Information | Action taken | Relatedness | Time from randomisation to onset (days) |
|---|---|---|---|---|---|
| Adverse events | |||||
| Stanmore | Placebo | Numbness, light euphoric feeling all over. Flatulence. Nausea | Patient advised to eat before taking normal medications | – | 11 |
| Serious adverse events | |||||
| Milton Keynes | Alendronate | Infection of left total hip replacement | Admitted as inpatient for two stage revision of left total hip replacement | Unrelated | 15 |
| Birmingham | Alendronate | Patient complained of bad headaches | Admitted with referral to a neurologist and patient to have eye test. Update: referral came through; appointment due April 2020 | Unlikely to be related | 241 |
Chapter 5 Discussion and conclusions
The MANTIS Trial was designed to determine whether or not a bisphosphonate could modify disease progression in AVN of the hip. Some of the challenges of delivering an interventional trial in a rare condition, such as AVN of the hip, were evident from the pre-trial feasibility work. The trial team therefore sought to introduce a phased study with an initial pilot leading to a main trial, subject to prespecified go/no-go criteria. From the outset, concerns regarding the prevalence of the condition, referral patterns and the variability in geographic distribution meant that identifying high-volume sites was difficult. Recruitment figures were lower than predicted, principally owing to a lower than expected disease prevalence. The CI, in discussion with the funder and trial committees, decided to terminate the study at the end of the internal pilot phase owing to the low recruitment rate, as low recruitment meant that the trial could not be delivered within the funding window of 5 years. Based on recruitment rate data, it is likely that recruitment for the trial would have taken 10 years or more.
Limitations
The small recruitment figures, and the fact that the pilot phase closed prior to the primary outcome being recorded for most participants, makes it very difficult to interpret findings.
Although it is impossible to draw meaningful conclusions from the trial, several observations can be made.
First, we found that the later stages of AVN were those predominantly identified, meaning that many patients classed as Ficat and Arlet stage 3 or above were excluded from the trial. In addition, two-thirds of those included in the trial were classed as having Ficat and Arlet stage 2. This mirrors clinical observations of the condition, which is usually identified at a late stage, when joint-preserving treatments are of no value. These observations are supported by the Non-Arthroplasty Hip Register, which records very few joint-preserving surgical procedures (< 80) per year in early-stage patients. This contrasts starkly with the National Arthroplasty Register, which identifies several hundred joint replacements for late-stage AVN every year. The main barriers to targeting early disease is the fact that symptoms in the early stages of the condition are non-existent or mild. Consequently, even clinicians with a high awareness of the risks may miss early disease, and the few identified cases of early-stage AVN (Ficat and Arlet stage 0 or 1) are often diagnosed during routine screening of the contralateral hip.
Second, we observed that patient-reported outcomes worsen quickly over time in all patients. This further supports the clinical observation that only a small window of opportunity for intervention with joint-preserving treatments in early-stage AVN patients exists, beyond which joint replacement becomes inevitable. Ultimately, this may be the rate-limiting step to treatment.
Last, it seems that the intervention, alendronate, was well tolerated by most participants. The trial team deliberated between different formulations of bisphosphonate while designing the study. Concern was raised regarding the side effects and frequency of dosing in alendronate, but this does not seem to have adversely affected compliance, which was over 80%. However, given the longer rate of onset for oral medication, it remains to be tested whether or not a parenteral therapy would be more effective.
Future work
By the end of the pilot phase, this trial was recruiting small but steady numbers in most sites. Recruitment projections performed at this stage demonstrated that it would take over 10 years to reach the full sample size. In its current form the trial is achievable; however, the cost may be prohibitive. It is likely that many more sites would be required to recruit adequate numbers over a conventional NIHR funding cycle. Our pilot study therefore demonstrates that such a trial is unachievable without a better understanding of the referral pathways and geographic foci of the disease. Such a trial may be feasible in a health-care environment where AVN is more prevalent (e.g. South East Asia).
Early AVN of the hip should be treated as a rare disease in the UK. Although uncommon, the earlier stages of the condition are extremely hard to identify, further reducing its reported prevalence. This makes trials difficult and expensive to conduct, but more importantly, provides a very small window for treatment even if the condition is detected in time. An improved understanding of the condition’s natural history is required, along with an understanding of the geographic locations where the condition is located. During the pilot study, we found it very difficult to identify areas of high disease prevalence. Despite identifying target demographics, we were unable to identify sites with large patient populations who met these criteria. Clinical Specialty Groups and specialist societies engaged extensively in this process; however, they were often unable to provide any useful advice.
This demonstrates the need for a registry or rare disease database for osteonecrosis of the hip. Several databases of rare diseases exist and have been shown to be effective. Many are patient-reported, such as the Rudy Study,34 which provide a repository for patients with rare, long-term conditions. Such a database for osteonecrosis of the hip would help define the population and help clinicians better understand the natural history.
Overall conclusion
The MANTIS trial was terminated at the end of the pilot phase, as it did not meet its go/no-go criteria. The main issues were a low recruitment rate, owing to lower than expected disease prevalence, and difficulties in identifying the condition at an early enough stage.
We would not recommend that a short-term interventional study is conducted on this condition until its prevalence, geographic foci and natural history are better understood. This is likely to be a barrier in most health-care markets.
One means of developing this understanding would be the introduction of a database/registry for AVN of the hip.
Acknowledgements
We wish to thank the NIHR HTA programme for funding this trial. We also wish to thank all of the participants and staff from all of the sites that participated in the trial:
-
Ashford and St Peter’s Hospitals NHS Foundation Trust
-
University Hospitals of North Midlands NHS Trust
-
Ayrshire and Arran NHS Health Board
-
Milton Keynes University Hospital NHS Foundation Trust
-
Oxford University Hospital NHS Foundation Trust
-
The Royal Orthopaedic Hospital NHS Foundation Trust, Birmingham
-
Wrightington, Wigan and Leigh Teaching Hospitals NHS Foundation Trust
-
Royal National Orthopaedic Hospital NHS Trust, Stanmore
-
Royal Cornwall Hospitals NHS Trust, Truro.
Thank you to all of the staff at SITU and OCTRU, Gemma Greenall (Trial Manager), Cushla Cooper (Operational Lead), Heidi Fletcher (Data Manager), Akiko Greshon (Data Support) and Ines Rombach (Statistician), who helped greatly with the set-up and delivery of the trial.
We would also like to thank the external members of the TSC and DSMC for their advice and support during the project.
Trial Steering Committee
Roger Francis (chairperson) (Newcastle University), Sally Hope (Oxford University Hospitals NHS Foundation Trust), Nick Harvey (University of Southampton), Mark Edwards (University of Southampton), Keith Tucker (Chair of Orthopaedic Data Evaluation Panel and Beyond Compliance), Caroline Fairhurst (University of York) and Michael Sheperia (patient representative).
Data Monitoring Committee
Bo Abrahamsen (chairperson) (Holbæk Hospital), John Belcher (Keele University) and Nicola Peel (Sheffield Teaching Hospitals NHS Foundation Trust).
Contributions of authors
Sion Glyn-Jones (https://orcid.org/0000-0002-9130-3167) (Professor of Orthopaedic Surgery and Honorary Consultant Orthopaedic Surgeon) contributed to the design of the trial, the analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
Muhammad K Javaid (https://orcid.org/0000-0001-7985-0048) University Lecturer in Metabolic Bone Disease and Consultant Rheumatologist) contributed to the design of the trial, the analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
David Beard (https://orcid.org/0000-0001-7884-6389) (Director, SITU) contributed to the design of the trial and protocol development, and critically reviewed the manuscript.
Julia Newton (https://orcid.org/0000-0001-7930-299X) (Consultant Rheumatologist) contributed to the design of the trial and protocol development, and critically reviewed the manuscript.
Robert Kerslake (https://orcid.org/0000-0002-3694-2345) (Consultant Musculoskeletal Radiologist) contributed to the design of the trial and protocol development, and critically reviewed the manuscript.
Callum McBryde (https://orcid.org/0000-0002-2500-5313) (Consultant Orthopaedic Surgeon) contributed to the design of the trial and protocol development, and critically reviewed the manuscript.
Tim Board (https://orcid.org/0000-0002-3295-2087) (Consultant Orthopaedic Surgeon) contributed to the design of the trial and protocol development, and critically reviewed the manuscript.
Susan J Dutton (https://orcid.org/0000-0003-4573-5257) (Lead Statistician, OCTRU) contributed to the design of the trial and protocol development, and critically reviewed the manuscript.
Melina Dritsaki (https://orcid.org/0000-0002-1673-3036) (OCTRU Senior Health Economist) contributed to the design of the trial and protocol development, and critically reviewed the manuscript.
Vikas Khanduja (https://orcid.org/0000-0001-9454-3978) (Consultant Orthopaedic Surgeon) contributed to the design of the trial and protocol development, and critically reviewed the manuscript.
Magbor Akanni (https://orcid.org/0000-0003-1864-1319) (Consultant Haemotologist) contributed to and critically reviewed the manuscript.
Shaun Sexton (https://orcid.org/0000-0002-1954-0950) (Consultant Orthopaedic Surgeon) contributed to and critically reviewed the manuscript.
John Skinner (https://orcid.org/0000-0002-1901-9057) (Consultant Orthopaedic Surgeon) contributed to and critically reviewed the manuscript.
Nicholas Peckham (https://orcid.org/0000-0003-1066-0726) (Medical Statistician) contributed to the analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
Ruth Knight (https://orcid.org/0000-0001-6810-2845) (Senior Medical Statistician) contributed to the analysis and interpretation of the data, and drafted and critically reviewed the manuscript.
Ines Rombach (https://orcid.org/0000-0003-3464-3867) (Senior Medical Statistician) contributed to the protocol development and critically reviewed the manuscript.
Loretta Davies (https://orcid.org/0000-0002-4721-356X) (Senior Trial Manager, SITU) drafted and critically reviewed the manuscript.
Vicki Barber (https://orcid.org/0000-0001-9631-3666) (Operations Manager, OCTRU) contributed to the design of the trial, and drafted and critically reviewed the manuscript.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to available anonymised data may be granted following review.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research funded by the National Institute for Health and Care Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, the HTA programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, the HTA programme or the Department of Health and Social Care.
References
- Fukushima W, Fujioka M, Kubo T, Tamakoshi A, Nagai M, Hirota Y. Nationwide epidemiologic survey of idiopathic osteonecrosis of the femoral head. Clin Orthop Relat Res 2010;468:2715-24. https://doi.org/10.1007/s11999-010-1292-x.
- Fukushima W, Ohfuji S, Hirota Y. Epidemiology of idiopathic osteonecrosis of the femoral head in Japan. Clin Calcium 2007;17:843-8.
- Mankin HJ. Nontraumatic necrosis of bone (osteonecrosis). N Engl J Med 1992;326:1473-9. https://doi.org/10.1056/NEJM199205283262206.
- Mont MA, Jones LC, Hungerford DS. Nontraumatic osteonecrosis of the femoral head: ten years later. J Bone Joint Surg Am 2006;88:1117-32.
- Lieberman JR, Engstrom SM, Meneghini RM, SooHoo NF. Which factors influence preservation of the osteonecrotic femoral head?. Clin Orthop Relat Res 2012;470:525-34. https://doi.org/10.1007/s11999-011-2050-4.
- Malizos KN, Karantanas AH, Varitimidis SE, Dailiana ZH, Bargiotas K, Maris T. Osteonecrosis of the femoral head: etiology, imaging and treatment. Eur J Radiol 2007;63:16-28. https://doi.org/10.1016/j.ejrad.2007.03.019.
- Ficat P, Arlet J. Pre-radiologic stage of femur head osteonecrosis: diagnostic and therapeutic possibilities. Rev Chir Orthop Reparatrice Appar Mot 1973;59:26-38.
- Steinberg ME, Bands RE, Parry S, Hoffman E, Chan T, Hartman KM. Does lesion size affect the outcome in avascular necrosis?. Clin Orthop Relat Res 1999;367:262-71. https://doi.org/10.1097/00003086-199910000-00033.
- Agarwala S, Sule A, Pai BU, Joshi VR. Study of alendronate in avascular necrosis of bone. J Assoc Physicians India 2001;49:949-50.
- Lai KA, Shen WJ, Yang CY, Shao CJ, Hsu JT, Lin RM. The use of alendronate to prevent early collapse of the femoral head in patients with nontraumatic osteonecrosis. A randomized clinical study. J Bone Joint Surg Am 2005;87:2155-9. https://doi.org/10.2106/00004623-200510000-00001.
- Steinberg ME, Lai M, Garino JP, Ong A, Wong KL. A comparison between total hip replacement for osteonecrosis and degenerative joint disease. Orthopedics 2008;31. https://doi.org/10.3928/01477447-20080401-35.
- Moriya M, Uchiyama K, Takahira N, Fukushima K, Yamamoto T, Hoshi K, et al. Evaluation of bipolar hemiarthroplasty for the treatment of steroid-induced osteonecrosis of the femoral head. Int Orthop 2012;36:2041-7. https://doi.org/10.1007/s00264-012-1612-8.
- Murray DW, Fitzpatrick R, Rogers K, Pandit H, Beard DJ, Carr AJ, et al. The use of the Oxford hip and knee scores. J Bone Joint Surg Br 2007;89:1010-14. https://doi.org/10.1302/0301-620X.89B8.19424.
- National Joint Registry (NJR) . National Joint Registry for England and Wales: 9th Annual Report 2012 2012.
- Agarwala S, Shah SB. Ten-year follow-up of avascular necrosis of femoral head treated with alendronate for 3 years. J Arthroplasty 2011;26:1128-34. https://doi.org/10.1016/j.arth.2010.11.010.
- Chen CH, Chang JK, Lai KA, Hou SM, Chang CH, Wang GJ. Alendronate in the prevention of collapse of the femoral head in nontraumatic osteonecrosis: a two-year multicenter, prospective, randomized, double-blind, placebo-controlled study. Arthritis Rheum 2012;64:1572-8. https://doi.org/10.1002/art.33498.
- Agarwala S, Jain D, Joshi VR, Sule A. Efficacy of alendronate, a bisphosphonate, in the treatment of AVN of the hip. A prospective open-label study. Rheumatology 2005;44:352-9. https://doi.org/10.1093/rheumatology/keh481.
- Dale H, Hallan G, Hallan G, Espehaug B, Havelin LI, Engesaeter LB. Increasing risk of revision due to deep infection after hip arthroplasty. Acta Orthop 2009;80:639-45. https://doi.org/10.3109/17453670903506658.
- Agarwala S, Sule A, Pai BU, Joshi VR. Alendronate in the treatment of avascular necrosis of the hip. Rheumatology n.d.;41:2002-7. https://doi.org/10.1093/rheumatology/41.3.346-a.
- Joint Formulary Committee . British National Formulary 2017.
- Cardozo JB, Andrade DM, Santiago MB. The use of bisphosphonate in the treatment of avascular necrosis: a systematic review. Clin Rheumatol 2008;27:685-8. https://doi.org/10.1007/s10067-008-0861-9.
- Yuan HF, Guo CA, Yan ZQ. The use of bisphosphonate in the treatment of osteonecrosis of the femoral head: a meta-analysis of randomized control trials. Osteoporos Int 2016;27:295-9. https://doi.org/10.1007/s00198-015-3317-5.
- International Standard Randomised Controlled Trial Number (ISRCTN) Registry . Managing Avascular Necrosis Treatments: An Investigational Study (MANTIS) n.d. www.isrctn.com/ISRCTN14015902 (accessed 20 May 2022).
- European Commission . EudraLex - Volume 4 - Good Manufacturing Practice (GMP) Guidelines n.d. eudralex-volume-4_en.
- Black DM, Cummings SR, Karpf DB, Cauley JA, Thompson DE, Nevitt MC, et al. Randomised trial of effect of alendronate on risk of fracture in women with existing vertebral fractures. Lancet 1996;348:1535-41. https://doi.org/10.1016/S0140-6736(96)07088-2.
- Oxford Clinical Commissioning Group, NHS Formulary . Formulary Chapter 9 – Nutrition and Blood n.d. http://www.oxfordshireformulary.nhs.uk/chaptersSubDetails.asp?FormularySectionID=9%26SubSectionRef=09.06%26SubSectionID=A100.
- Dawson J, Fitzpatrick R, Carr A, Murray. Questionnaire on the perceptions of patients about total hip replacement. J Bone Joint Surg Br 1996;78:185-90. https://doi.org/10.1302/0301-620X.78B2.0780185.
- Mohtadi, Griffin DR, Pedersen ME, Chan D, Safran MR, Parsons N, et al. The development and validation of a self-administered quality-of-life outcome measure for young, active patients with symptomatic hip disease: the International Hip Outcome Tool (iHOT-33). Arthroscopy 2012;28:595-60. https://doi.org/10.1016/j.arthro.2012.03.013.
- Shane E, Burr D, Ebeling PR, Abrahamsen B, Adler RA, Brown TD, et al. Atypical subtrochanteric and diaphyseal femoral fractures: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res 2010;25:2267-94.
- DAMOCLES Study Group . A proposed charter for clinical trial data monitoring committees: helping them to do their job well. Lancet 2005;365:711-22.
- Bjelland I, Dahl AA, Haug TT, Neckelmann D. The validity of the Hospital Anxiety and Depression Scale: an updated literature review. J Psychosom Res 2002;52:69-77. https://doi.org/10.1016/S0022-3999(01)00296-3.
- Herdman M, Gudex C, Lloyd A, Janssen M, Kind P, Parkin D, et al. Development and preliminary testing of the new five-level version of EQ-5D (EQ-5D-5L). Qual Life Res 2011;20:1727-36. https://doi.org/10.1007/s11136-011-9903-x.
- Beard DJ, Harris K, Dawson J, Doll H, Murray DW, Carr AJ, et al. Meaningful changes for the Oxford hip and knee scores after joint replacement surgery. J Clin Epidemiol 2015;68:73-9. https://doi.org/10.1016/j.jclinepi.2014.08.009.
- RUDY Study . RUDY Study n.d. www.rudystudy.org (accessed 17 June 2022).
Appendix 1 List of participants’ relevant medical history, taken at baseline
| Repeat instance | Site | Medical history | Condition ongoing | Allocation |
|---|---|---|---|---|
| 1 | Milton Keynes | Arthritis | Yes | Alendronate |
| 1 | Oxford | Asthma (since childhood) | Yes | Alendronate |
| 2 | Oxford | AVN (2017) | Yes | Alendronate |
| 3 | Oxford | Brain tumour (6 years old) | No | Alendronate |
| 4 | Oxford | Right ankle fracture 2013 – cast | No | Alendronate |
| 1 | Oxford | SS (since birth) | Yes | Placebo |
| 1 | Oxford | Degenerative disease of spine | Yes | Alendronate |
| 2 | Oxford | SS (congenital) | Yes | Alendronate |
| 3 | Oxford | T4 # (injections in the past) | No | Alendronate |
| 1 | Oxford | RTHR (AVN) | No | Placebo |
| 2 | Oxford | Sickle cell anaemia | Yes | Placebo |
| 1 | Cornwall | COPD | Yes | Alendronate |
| 2 | Cornwall | Diverticulitis | Yes | Alendronate |
| 3 | Cornwall | Perianal abscesses | No | Alendronate |
| 4 | Cornwall | Sciatica | Yes | Alendronate |
| 1 | ROH | Fractured right ankle, ORIF (2016) | No | Alendronate |
| 2 | ROH | Psoriatic arthritis right knee | Yes | Alendronate |
| 1 | ROH | Eczema | Yes | Alendronate |
| 1 | Wrightington | Asthma | Yes | Placebo |
| 1 | Wrightington | Hodgkin lymphoma | No | Alendronate |
| 2 | Wrightington | Kidney stones | Yes | Alendronate |
| 3 | Wrightington | Non-Hodgkin lymphoma | No | Alendronate |
| 4 | Wrightington | Road traffic accident | No | Alendronate |
| 5 | Wrightington | Spinal decompression | No | Alendronate |
| 6 | Wrightington | Stem cell transplant | No | Alendronate |
Appendix 2 List of medications taken by participants at baseline
| Repeat instance | Site | Medication | Dose | Frequency | Ongoing | Allocation |
|---|---|---|---|---|---|---|
| 1 | Milton Keynes | Chlordiazepoxide | 20 mg | One tablet every 6 hours | No | Alendronate |
| 2 | Milton Keynes | Thiamine | 100 mg | One tablet thrice daily | Yes | Alendronate |
| 3 | Milton Keynes | Spironolactone | 50 mg | One tablet twice daily | Yes | Alendronate |
| 4 | Milton Keynes | Temazepam | 10 mg | As required | Yes | Alendronate |
| 5 | Milton Keynes | Omeprazole | 20 mg | One tablet once per day | Yes | Alendronate |
| 6 | Milton Keynes | Gabapentin | 100 mg | As required | Yes | Alendronate |
| 1 | Milton Keynes | Naproxen | 500 mg | Two tablets twice per day | Yes | Placebo |
| 1 | Milton Keynes | Ramipril | 20 mg | One tablet once per day | Yes | Alendronate |
| 2 | Milton Keynes | Simvastatin | 20 mg | One tablet once per day | Yes | Alendronate |
| 3 | Milton Keynes | Aspirin | 75 mg | One tablet once per day | Yes | Alendronate |
| 1 | Milton Keynes | Aspirin | NR | NR | NR | Placebo |
| 1 | Milton Keynes | Lansoprazole | 15 mg | One tablet once per day | Yes | Placebo |
| 2 | Milton Keynes | Ramipril | 10 mg | One tablet once per day | Yes | Placebo |
| 3 | Milton Keynes | Atorvastatin, co-codamol | 40/30/500 mg | One tablet once per day when required | Yes | Placebo |
| 4 | Milton Keynes | Metformin | 500 mg | One tablet twice per day | Yes | Placebo |
| 5 | Milton Keynes | Amitriptyline | 25 mg | One tablet per day | Yes | Placebo |
| 6 | Milton Keynes | Naproxen | 500 mg | One tablet once per day | Yes | Placebo |
| 1 | Oxford | MSM, flax seed, omega 3 and collagen | NR | One of each | NR | Alendronate |
| 2 | Oxford | Wheat grass (drink) | NR | One drink and one tablet | NR | Alendronate |
| 3 | Oxford | Inhaler | Two puffs | TT AM, TT night | NR | Alendronate |
| 4 | Oxford | Turmeric, garlic and vitamins B, C, D and K | NR | One of each | NR | Alendronate |
| 5 | Oxford | Calcium, magnesium rosehip and glucosamine | NR | One of each | NR | Alendronate |
| 6 | Oxford | Ventolin | Two puffs | As required | NR | Alendronate |
| 1 | Oxford | Folic acid | 5 mg | One inhalation/puff | Yes | Placebo |
| 2 | Oxford | Paracetamol | 1 g | Up to four times per day | Yes | Placebo |
| 3 | Oxford | Tramadol | 50 mg | As required (up to four times per day) | Yes | Placebo |
| 1 | Oxford | Glucosamine | 500 mg | BD | No | Placebo |
| 2 | Oxford | Floradix | 10 ml | BD | Yes | Placebo |
| 3 | Oxford | Flux D3 | 20,000 IU | Once per week | Yes | Placebo |
| 1 | Oxford | Folic acid | 5 mg | One tablet once per day | NR | Alendronate |
| 2 | Oxford | Steroid injection | NR | NR | NR | Alendronate |
| 3 | Oxford | Penicillin | 250 mg | BD | NR | Alendronate |
| 4 | Oxford | Morphine – Zomorph | 30 mg | BD | NR | Alendronate |
| 5 | Oxford | Vitamin D | NR | Once per day | NR | Alendronate |
| 6 | Oxford | Oramorph | 5 ml | As required | NR | Alendronate |
| 1 | Oxford | Codeine | 30 mg | Twice per day | Yes | Placebo |
| 2 | Oxford | Ramipril | NR | NR | NR | Placebo |
| 3 | Oxford | Bisprolol | NR | NR | NR | Placebo |
| 4 | Oxford | Aspirin | NR | NR | NR | Placebo |
| 1 | Oxford | Ibuprofen | 200 mg | As required | NR | Alendronate |
| 2 | Oxford | Pregablin | 150 mg | BD | Yes | Alendronate |
| 3 | Oxford | Paracetamol | 1 g | 1ds | NR | Alendronate |
| 1 | Oxford | Folic acid | 20 mg | Once per day | Yes | Placebo |
| 2 | Oxford | Penicillin | 25 mg | Once per day | Yes | Placebo |
| 1 | Stanmore | Naproxen | 500 mg | One tablet twice per day | Yes | Placebo |
| 2 | Stanmore | Mirtazapine | 45 mg | One tablet at night | Yes | Placebo |
| 3 | Stanmore | Tramadol | 50 mg | One to two tablets four times per day | Yes | Placebo |
| 4 | Stanmore | Amitriptyline | 25 mg | One to three tablets per night | Yes | Placebo |
| 5 | Stanmore | Codeine | 60 mg | One tablet four times per day | Yes | Placebo |
| 6 | Stanmore | Cetirizine | 10 mg | Once per day | Yes | Placebo |
| 1 | Cornwall | Trimbow | Two ports | BD | Yes | Alendronate |
| 2 | Cornwall | Thiamine | 100 mg | Three times per day | Yes | Alendronate |
| 3 | Cornwall | Morphine sulfate | 30 mg/mlR | BD | Yes | Alendronate |
| 4 | Cornwall | Vitamin B strong | Tablets | Once per day | Yes | Alendronate |
| 5 | Cornwall | Lorazepam | 1 mg | Four times per day | Yes | Alendronate |
| 6 | Cornwall | Lansoprazole | 15 mg | Once per day | Yes | Alendronate |
| 1 | Birmingham | Naproxen | 250 mh | One tablet when required | Yes | Alendronate |
| 2 | Birmingham | Dovonex | 30 mg | Once per day | Yes | Alendronate |
| 3 | Birmingham | Paracetamol | 1 g | Two tablets when required | Yes | Alendronate |
| 4 | Birmingham | Vitamin D + K2 | 3000 iu | Once per day | Yes | Alendronate |
| 5 | Birmingham | Dovobet | 60 mg | Once per day | Yes | Alendronate |
| 6 | Birmingham | Flucidin cream | 10 mg | Once per day | Yes | Alendronate |
| 1 | Birmingham | Dupilumab | 300 mg | Fortnightly | Yes | Alendronate |
| 2 | Birmingham | Presnisolone | 10 mg | Once per day | No | Alendronate |
| 1 | Wrightington | Paracetamol | 1 g | As required | Yes | Placebo |
| 2 | Wrightington | Nytol (diphenhydramine hydrochloride) | 25 mg | NOCTE | Yes | Placebo |
| 1 | Wrightington | Omeprazole | 20 mg | Once per day | Yes | Placebo |
| 2 | Wrightington | Movicol | NR | Two sachets | NR | Placebo |
| 3 | Wrightington | Montelukast | 10 mg | Twice per day | Yes | Placebo |
| 4 | Wrightington | Salbutamol inhaler | 100 µg | Two puffs as required | Yes | Placebo |
| 5 | Wrightington | Fostair inhaler | 100 mcg | Two puffs twice per day | Yes | Placebo |
| 6 | Wrightington | Tramadol | NR | As required | Yes | Placebo |
| 1 | Wrightington | Paracetamol | 100 mg | As required | Yes | Alendronate |
List of abbreviations
- AE
- adverse event
- AP
- anteroposterior
- AVN
- avascular necrosis
- BMI
- body mass index
- C&C
- capability and capacity
- CI
- chief investigator
- CRF
- case report form
- CRN
- Clinical Research Network
- DSMC
- Data Safety and Monitoring Committee
- EQ-5D-5L
- EuroQol-5 Dimensions, five-level version
- EudraCT
- European Union Drug Regulating Authorities Clinical Trials
- GP
- general practitioner
- HADS
- Hospital Anxiety and Depression Scale
- HRG
- health-care resource group
- HTA
- Health Technology Assessment
- ICF
- informed consent form
- iHOT-33
- international Hip Outcome Tool-33
- IMP
- investigational medicinal product
- IMPD
- investigational medicinal product dossier
- IQR
- interquartile range
- IRAS
- Integrated Research Application System
- ISF
- investigator site file
- ITT
- intention to treat
- MANTIS
- Managing Avascular Necrosis Treatments: an Interventional Study
- MHRA
- Medicines and Healthcare products Regulatory Agency
- MRI
- magnetic resonance imaging
- NIHR
- National Institute for Health and Care Research
- OCTRU
- Oxford Clinical Trials Research Unit
- OHS
- Oxford Hip Score
- PI
- principal investigator
- PIS
- participant/patient information sheet
- PL
- product licence number
- PLIS
- patient-level informatics system
- PPI
- patient and public involvement
- REC
- Research Ethics Committee
- RRAMP
- Registration/Randomisation And Management of Product
- SAE
- serious adverse event
- SD
- standard deviation
- SFQ
- site feasibility questionnaire
- SITU
- Surgical Interventions Trials Unit
- SIV
- site initiation visit
- SmPC
- summary of product characteristics
- THA
- total hip arthroplasty
- TMG
- Trial Management Group
- TSC
- Trial Steering Committee