
Notes
Article history
This themed issue of the Health Technology Assessment journal series contains a collection of research commissioned by the NIHR as part of the Department of Health’s (DH) response to the H1N1 swine flu pandemic. The NIHR through the NIHR Evaluation Trials and Studies Coordinating Centre (NETSCC) commissioned a number of research projects looking into the treatment and management of H1N1 influenza. NETSCC managed the pandemic flu research over a very short timescale in two ways. Firstly, it responded to urgent national research priority areas identified by the Scientific Advisory Group in Emergencies (SAGE). Secondly, a call for research proposals to inform policy and patient care in the current influenza pandemic was issued in June 2009. All research proposals went through a process of academic peer review by clinicians and methodologists as well as being reviewed by a specially convened NIHR Flu Commissioning Board.
Declared competing interests of authors
JSNVT has received funding to attend influenza-related meetings, for lecture and consultancy fees and for research from several influenza antiviral drug and vaccine manufacturers, including GlaxoSmithKline (GSK) and Baxter AG, which supplied pandemic vaccine to the UK. He is a former employee of SmithKline Beecham plc (now GSK), Roche Products Ltd and Sanofi Pasteur MSD. GSK funding is anticipated for the secondary objective of this study. RLP has received funding from Sanofi Pasteur to attend two influenza-related meetings. GSK funding is anticipated for the secondary objective of this study. KGN has received H5 avian influenza vaccines from Novartis and A/H1N1v pandemic influenza vaccines from GSK and Baxter AG to facilitate trials funded by the Medical Research Council and the National Institute for Health Research. He has received consultancy fees from Novartis and GSK, and lecture fees from Baxter. IS has received funding to attend international scientific meetings (Novartis Vaccines), for lecture and consultancy fees (Novartis, GSK and Baxter) and for research from several pharmaceutical companies, including Novartis Vaccines, GSK, Baxter Vaccines and Hoffman-La Roche. MZ received a Interscience Conference on Antimicrobial Agents and Chemotherapy/Imedex keynote speaker honorarium. KH received funding from Sanofi Pasteur MSD for one scientific meeting attendance. JE has received consultancy fees from GSK and performed paid work for the Department of Health, England.
Permissions
Copyright statement
© 2010 Queen’s Printer and Controller of HMSO. This journal is a member of and subscribes to the principles of the Committee on Publication Ethics (COPE) (http://www.publicationethics.org/). This journal may be freely reproduced for the purposes of private research and study and may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NETSCC, Health Technology Assessment, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2010 Queen’s Printer and Controller of HMSO
Objective
The primary objective was to determine the proportion of babies who acquired passive immunity to A/H1N1v, born to mothers who accepted vaccination as part of the national vaccination programme while pregnant (during the second and/or third trimesters) against the novel A/H1N1v influenza virus (exposed group) compared with unvaccinated (unexposed) mothers.
Design
An observational study at three sites in the UK. The purpose was to determine if mothers immunised against A/H1N1v during the pandemic vaccination period transferred that immunity to their child in utero.
Setting
Three sites in the UK [Queen’s Medical Centre, Nottingham; City Hospital, Nottingham (both forming University Hospitals Nottingham), and Leicester Royal Infirmary (part of University Hospitals Leicester)].
Participants
All pregnant women in the second and third trimester presenting at the NHS hospitals above to deliver were eligible to participate in the study. Women were included regardless of age, social class, ethnicity, gravida and parity status, past and current medical history (including current medications), ethnicity, mode of delivery and pregnancy outcome (live/stillbirth).
Interventions
At enrolment, participants provided written consent and completed a questionnaire. At parturition, venous cord blood was obtained for serological antibody analysis. Serological analysis was undertaken by the Respiratory Virus Unit (RVU), Health Protection Agency (HPA) Centre for Infections, London.
Main outcome measures
The primary end point in the study was the serological results of the cord blood samples for immunity to A/H1N1v. Regarding a suitable threshold for the determination of a serological response consistent with clinical protection, this issue is somewhat complex for pandemic influenza. The European Medicines Agency (EMEA) Committee for Human Medicinal Products (CHMP) judges that a haemagglutination inhibition (HI) titre of 1 : 40 is an acceptable threshold. However, this level was set in the context of licensing plain trivalent seasonal vaccine, where a titre of 1 : 40 is but one of several related immunogenicity criteria, and supported by paired sera capable of demonstrating a fourfold rise in antibody titre in response to vaccination. The current study mainly investigated the effects of an AS03-adjuvanted monovalent vaccine, and it was not possible to obtain paired sera where the initial sample was taken before vaccination (in vaccinated subjects). Of possibly greater relevance is the fact that it has been established from the study of early outbreaks of pandemic influenza in secondary schools in the UK (HPA, unpublished observations) that an HI antibody titre of 1 : 32 seems to be the threshold for a humoral response to ‘wild-type’ A/H1N1v infection. On that basis, a threshold of 1 : 32 is at least as appropriate as one of 1 : 40, especially in unvaccinated individuals. Given the difficulties that would accrue by applying thresholds of 1 : 32 in unvaccinated patients and 1 : 40 in vaccinated patients, we have therefore applied a threshold of 1 : 32 and 1 : 40, to increase the robustness of our findings. Differences arising are described. A microneutralisation (MN) titre of 1 : 40 may be also used, although it is not part of the CHMP criteria for vaccine licensure. Nonetheless, we utilised this analysis as a secondary end point, based on a conservative threshold of 1 : 60.
Results
Reverse cumulative distribution percentage curves for haemagglutinin dilution and MN titres demonstrate background immunity in babies of unvaccinated mothers of 25%–30%. Humoral immunity in babies of vaccinated mothers was present in 80% of the group. The difference in positive immunity between the babies of unvaccinated and vaccinated mothers was statistically significant (chi-squared test, p < 0.001).
Conclusions
Our findings reveal a highly significant difference in HI titres between babies born to mothers vaccinated with pandemic-specific vaccine against A/H1N1v during the 2009–10 pandemic period. The subjects recruited were comparable from a baseline perspective and thus do not represent different groups that otherwise could have introduced bias into the study. Continued circulation of 2009 A/H1N1-like viruses is uncertain, but is possible as seasonal influenza in years to come. It is possible that future seasonal waves may display increased virulence. Given the adverse outcomes experienced for a small proportion of pregnant women during the influenza pandemic of 2009–10, this study provides useful evidence to support vaccination in pregnancy to protect both the mother and baby.
Funding
The National Institute for Health Research Health Technology Assessment programme.
Chapter 1 Introduction
Influenza has long been known to cause a higher level of complications (including hospitalisations) and death in particular high-risk groups, such as the elderly and those with underlying comorbidities, for instance cardiopulmonary disease. Although less widely known, pregnant women also fall into this high-risk category. Evidence suggests that this effect can be seen with seasonal influenza,1–4 but is far more evident with pandemic influenza, the most notable observations arising from the 1957 A/H2N2 pandemic, during the second and third trimesters of pregnancy. 5–7 In addition, adverse effects of influenza on perinatal and early neonatal outcomes have also been observed. 8–10 The epidemiological profile of the A/H1N1v influenza virus that emerged in 2009 was different from normal seasonal influenza, in that working-age adults and children suffered higher rates of complications (including hospitalisations) than the elderly. The effect was noted to be most pronounced in individuals with underlying comorbidities and during pregnancy, in whom clear signals regarding the relationship between the premorbid state and influenza illness severity were observed, despite being based on small data sets. 11–15 In parallel with this, there is increasing evidence also suggesting that young children of < 2 years of age are at greater risk of developing complications and death from influenza than at any other time in childhood, and the rate of hospitalisation in this age group (due to seasonal influenza) broadly equals that seen in working-age adults with underlying high-risk conditions. 16
Contemporary management of paediatric influenza cases is complicated, in terms of policy and practice, by the fact that both neuraminidase inhibitors [oseltamivir (Tamiflu®, Roche) and zanamivir (Relenza®, GlaxoSmithKline, GSK)] are unlicensed for use in children of < 12 months and 5 years, respectively, and, if required, have to be given off label, if at all, in children who are < 12 months (zanamivir is an oral inhalational drug that would be almost impossible to administer in its marketed form). Furthermore, the licensing of novel A/H1N1 vaccines, utilised from October 2009 onwards, excluded children of < 6 months of age. 17–20
Seasonal influenza vaccination, until now, has not been routinely recommended for pregnant women in the UK. It has been in use in the USA in some pregnant women in all trimesters since 2004 because of the perceived risk–benefit profile. 2,21 However, the take-up is low (published figures indicate approximately 14%–16%), suggesting that its benefits are not widely appreciated by pregnant women or health professionals. 2,22,23
The predisposition of the new A/H1N1v influenza virus to cause severe illness in pregnant women led to recommendations for them to be vaccinated in the UK and elsewhere. 11,12,15
Two pharmaceutical companies were contracted by the Department of Health (DH) to provide vaccine for the UK – GSK and Baxter AG. Both originally involved a two-dose strategy; however, the GSK vaccine used a dose-sparing adjuvant alongside split-virion antigen, and subsequent guidance called for a single dose of vaccine in the UK, whereas the Baxter AG product was a Vero cell-grown, ‘wild-type’, whole cell product that necessitated a two-dose approach. 17–19,24 In the UK, the GSK product (Pandemrix™) was the preferred vaccine for pregnant women because of the data on rapidity of immune response with just one dose. 17
Effect of maternal vaccination and acquired (vertical/passive) immunity in children
Influenza vaccination in pregnancy offers benefit to the mother by reducing the risk of infection and resultant complications. It has also been established that immunisation in pregnancy with trivalent, unadjuvanted, seasonal influenza vaccine does provide vertical immunity to the child through the cord blood. 2,25–28 However, the clinical impact of the finding is less clear with some studies indicating benefit and others not. 16,26–28
The immunity offered by monovalent, new variant influenza vaccine, with or without adjuvant, has not yet been established. This study was designed to help answer this question.
Rationale for the proposed study
Given the emergent risk profile of the A/H1N1v pandemic virus, it could have been assumed that pregnant women would readily choose to be vaccinated. However, perception and response to threats and assessment of risk do not necessarily align in terms of human behaviour. Research has suggested that people tend to overestimate the likelihood and impact of rare events, and underestimate for more common situations. 29,30 The public response to the measles, mumps and rubella (MMR) vaccination scare, and its possible link to autism and Crohn’s disease, is an example. Despite substantial and sound evidence to the contrary, many parents chose to refuse MMR vaccination for their children on the basis of a theoretical association that has been dismissed by most scientists and policy-makers, therefore exposing them to the risk of serious disease from measles, mumps or rubella. 29,31,32 Another example of a similar response is that to whooping cough vaccine in the 1970s. 33,34 Whether the low take-up of seasonal influenza vaccine in pregnant women in the USA is due to similar anxiety is uncertain. 35,36 Nonetheless, policy-makers will be concerned to ensure maximum uptake of vaccination for future pandemic situations. Evidence to support the approach may help encourage women to come forward for immunisation. If data were available, which revealed that vaccination of pregnant women appeared to confer meaningful protection against A/H1N1v influenza to their babies after birth, this would enable messages to pregnant women of potential vaccinees to be shifted from ‘evidence that you are likely to benefit and no evidence that your baby will be harmed’ to ‘evidence that you are likely to benefit and further evidence that your baby will also benefit’. 36
The study was therefore designed to assess the immunity conferred to infants of mothers who had been vaccinated against A/H1N1v influenza by obtaining venous umbilical cord blood samples at delivery and submitting them for serological analysis and comparing with those of unvaccinated mothers in the same birth cohort.
Chapter 2 Methods
The study was designed by the chief investigators (RP/JVT) in conjunction with the co-researchers. RP was responsible for managing data resulting from the study, analysis and drafting the manuscript. The data and analyses were fully accessible and interpreted by all authors, who individually, and collectively, vouch for their accuracy and completeness. The Leicester, Northamptonshire and Rutland Research Ethics Committee 1 (LNRREC1) and participating centres approved the study (09/H0406/107). The University of Nottingham was the sponsor for the study. The study was funded by the UK DH (National Institute for Health Research, NIHR).
Research objectives
The primary objective was to determine the proportion of babies who acquired passive immunity to A/H1N1v born to mothers who accepted vaccination as part of the national vaccination programme37 while pregnant (during the second and/or third trimesters) against the novel A/H1N1v influenza virus (exposed group) compared with unvaccinated (unexposed) mothers.
(A secondary objective was to record and investigate influenza-like illness during winter 2009–10/spring–summer 2010 in the babies of mothers who took part in the study; however, this was funded separately from the primary objective and is ongoing and therefore is not the subject of this report.)
Babies recruited to the study are being followed for 5 years by a ‘flag’ applied to the Office for National Statistics (ONS) records.
Study design
During November 2009–March 2010 the researchers conducted a prospective, observational study at three sites in the UK [Queen’s Medical Centre, Nottingham; City Hospital, Nottingham (both forming University Hospitals Nottingham); and Leicester Royal Infirmary (part of University Hospitals Leicester)] in accordance with the principles of the Declaration of Helsinki and UK regulatory requirements. 38,39
The purpose was to determine if mothers immunised against A/H1N1v during the pandemic vaccination period transferred that immunity to their child in utero.
Pregnant women presenting for delivery at one of the sites listed above were screened for eligibility and provided informed consent.
Eligibility
Inclusion criteria
Subject to the exclusions listed below, all pregnant women in the second and third trimester presenting at the NHS hospitals above to deliver were eligible to participate in the study.
Women were included regardless of age, social class, ethnicity, gravida and parity status, past and current medical history (including current medications), ethnicity, mode of delivery and pregnancy outcome (live birth/stillbirth).
Exclusion criteria
The main exclusion criteria were pregnant women who were still in their first trimester and women delivering before the age of fetal viability (23 weeks and 6 days’ gestation). 40
Other exclusion criteria were:
-
incapacity to provide informed consent for participation
-
refusal (including refusal to agree to both primary and secondary objectives)
-
women who were prisoners
-
inability to take cord blood samples, for example cord blood needed for other clinical purpose so none available for the study
-
involvement in another study entailing clinical interventions
-
women who did not routinely live in the East Midlands.
Study procedures
The researchers enrolled subjects in two groups – those vaccinated during pregnancy and those not [vaccinated (exposed)/unvaccinated (unexposed)]. This study was not randomised because prior vaccination status ascertainment of participants was required to determine eligibility. Members of the usual care team (midwives/obstetricians) recruited the participants after admission at an appropriate point in their clinical care prior to delivery.
Definition of vaccination status [vaccinated (exposed)/unvaccinated (unexposed)]
Women approached for enrolment were asked if they had been vaccinated against ‘pandemic flu’ (A/H1N1v). If the woman had been vaccinated then the date and batch number of the vaccine was asked for, as at the study design point it was considered that this detail would probably be entered into the woman’s personal handheld pregnancy record. However, the researchers also recognised that this detail might be missing and therefore obtained express consent at enrolment to clarify missing details with the woman’s general practitioner. These details of date and batch of vaccine were mostly missing from the enrolment records and therefore were subsequently checked retrospectively.
Unvaccinated (unexposed) women
Unvaccinated (unexposed) women were defined as those who reported that they had not been vaccinated against A/H1N1v.
Vaccinated (exposed) women
Vaccinated (exposed) women were those who reported having been vaccinated against A/H1N1v.
Definition of vaccination exposure date
The vaccination schedule adopted in England is outlined in detail later in this document. Nonetheless, two different types of vaccine from two manufacturers were available, adopting dissimilar vaccination regimens. Pandemrix™ was given as a single dose in pregnancy, whereas Celvapan™ adopted a two-dose schedule. Therefore, the exposure date was taken as the date of vaccination recorded for Pandemrix™ and the latest date of vaccination recorded for Celvapan™. 17,37,41
Lot/batch numbers were used to identify which vaccine had been used. In the case of Pandemrix™, the numbers recorded from vaccine/diluents or package were taken as robust evidence of type given. At analysis stage, it became clear that only Pandemrix™ appeared to have been used. Therefore, the date of vaccination was taken as the one provided by the recruit’s general practitioner.
Enrolment questionnaire
At enrolment, participants provided written consent to take part in the study (primary and secondary objective) and completed a questionnaire (see Appendix 3). This included details of any past or current medical history that might have influenced the decision of the subject to accept or refuse vaccination and therefore could have biased the results. These included cardiovascular disease, respiratory disease, renal disease, liver disease, diabetes (gestational or pre-existing), immunosuppression and hypertension of pregnancy/pre-eclampsia. Likewise, details of past obstetric history were also elicited.
Demographic details (to allow follow-up) were recorded separately from clinical details but were linked using a unique (study-specific) identifier (pseudoanonymised). Although a history of prior infection with A/H1N1v might have provided the research with some additional insights, it was not sought from participants for the following reasons.
-
Research evidence indicates that for both seasonal influenza and A/H1N1v some people acquire the infection and seroconvert asymptomatically. Asking for a history of infection would therefore miss substantial numbers.
-
Requesting participant reports of influenza-like illness would not be sufficiently sensitive or specific to determine prior infection. 42,43
At parturition, cord blood was obtained for serological antibody analysis. Venous cord blood was obtained by the delivering midwife/clinician from the umbilical cord (vein) after delivery was complete, and was placed in a serum sample collection tube. The samples were pseudoanonymised using the study-specific identifier and the test request only, so that laboratory staff were blinded to the vaccination status of the donor (recruiting staff were not required to add additional details). The laboratory request form gave no details of the vaccination status of the mother. Samples were spun down and serum was separated from red cells. The samples were then stored at –20 °C until analysis. Samples were transported in two batches to the Respiratory Virus Unit (RVU), Health Protection Agency (HPA) Centre for Infections, London, for serological analysis. Both batches contained samples from vaccinated and unvaccinated subjects.
Vaccine was not provided as part of this study.
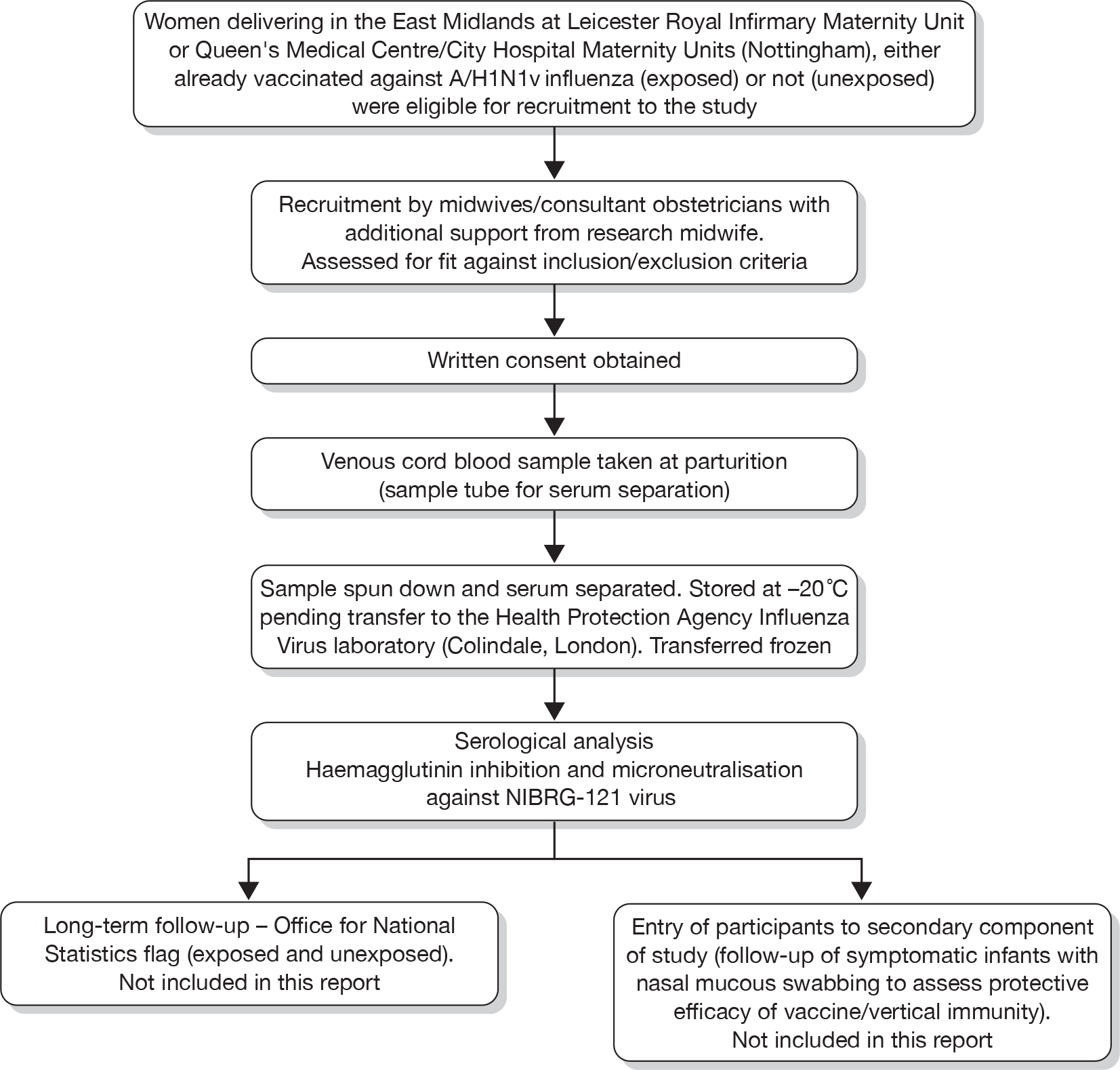
The study participant recruitment and involvement is set out diagrammatically in Figure 1, below.
FIGURE 1.
Study participant flow diagram.
Outcome measures
Primary end point
The primary end point in the study was the serological results of the cord blood samples for immunity to A/H1N1v. The issue of a suitable threshold for the determination of a serological response consistent with clinical protection is somewhat complex for pandemic influenza. The European Medicines Agency (EMEA) Committee for Human Medicinal Products (CHMP) judges that a haemagglutination inhibition (HI) titre of ≥ 1 : 40 is an acceptable threshold. However, this level was set in the context of licensing plain trivalent seasonal vaccine, where a titre of ≥ 1 : 40 is but one of several related immunogenicity criteria, and is supported by paired sera capable of demonstrating a fourfold rise in antibody titre in response to vaccination. 44 The current study mainly considered the effects of an AS03-adjuvanted monovalent vaccine. It was not possible to obtain paired sera where the initial sample would have been taken before vaccination (in vaccinated subjects).
Of possibly greater relevance is the fact that it has been established from the study of early outbreaks of pandemic influenza in secondary schools in the UK (HPA, unpublished observations) that a HI antibody of ≥ 1 : 32 seems to be the threshold for a humoral response to wild-type A/H1N1v infection. 45 On this basis, a threshold of 1 : 32 is at least as appropriate as one of 1 : 40, especially in unvaccinated individuals.
Given the difficulties that would accrue by applying a threshold of ≥ 1 : 32 in unvaccinated patients and ≥ 1 : 40 in vaccinated patients, we have therefore applied a threshold of ≥ 1 : 32 and ≥ 1 : 40 to increase the robustness of our findings. Differences arising are described.
Microneutralisation titre of ≥ 1 : 40 may be also used, although not part of the CHMP criteria for vaccine licensure. Nonetheless, we utilised this analysis as a secondary end point, based on a conservative threshold of ≥ 1 : 60.
Changes to protocol
The original submitted protocol (version 9) is appended (see Appendix 1). It was reviewed by the LNRREC1 on 2 October 2009. Minor modifications to some of the supporting documentation to the study were required prior to approval (e.g. advertising poster and patient information sheet). Final approval was received on 20 October 2009.
Minor amendments were submitted and accepted on 22 and 29 October 2009, 3 November 2009 and 14 April 2010. These related to small changes in supporting documentation and swab collection, and were approved without further ethical review.
A substantial amendment was submitted on 13 November 2009, requesting the involvement of an additional partner (for objective 2) and therefore a change to protocol (version 12) (see Appendix 2).
Vaccines
This study did not provide vaccination as part of the investigation. Recruits had either been immunised already [vaccinated (exposed)] or had not been offered/declined vaccination [unvaccinated (unexposed)]. Those who had been vaccinated had accepted immunisation as part of the DH Pandemic Influenza A/H1N1v programme. 37,41 The 2009 A/H1N1v vaccines used in the national programme were pandemic-specific licensed products: Celvapan™ (Baxter AG) and Pandemrix™ (GSK).
Celvapan™ is a non-adjuvanted, whole-virion vaccine, manufactured by Baxter AG (Vienna, Austria), based on wild-type A/California/07/2009 (H1N1). 18
Pandemrix™ is an adjuvanted split-virion vaccine, manufactured by GSK (GSK Biologicals, Dresden, Germany) based on a reverse genetic virus derived from an A/California/7/2009 strain. 19
Laboratory assays for serological analysis
The cord blood samples were submitted for serological antibody analysis46 using HI and microneutralisation (MN),47,48 according to standard methods at the HPA Centre for Infections, London, with egg-grown NIBRG121 virus, generated from A/California/7/2009 strain, using reverse genetics, as the test antigen (National Institute for Biological Standards and Controls, UK).
Serum samples were tested with the use of 1 : 2 serial dilutions for HI. For MN assays, sera were tested at an initial dilution of 1 : 10, and those that were negative were assigned a titre of 1 : 5. The final dilution was 1 : 320, and samples for which the end-point titres were greater were assigned a value of 1 : 640. Blinded specimens were tested, in duplicate, and the geometric mean values of these duplicates were used in analyses.
In more detail, samples collected at each study site were centrifuged, separated into two aliquots and tested in parallel.
The principle of the HI test was based on the ability of specific anti-influenza antibodies to inhibit haemagglutination of red blood cells (RBCs) by influenza virus haemagglutinin. The sera to be tested were treated to eliminate the non-specific inhibitors and the anti-species haemagglutinins prior to testing. The analysis of the samples was performed in accordance to protocols and standard operating procedures (SOPs) developed with the RVU, HPA Centre for Infections, London.
Elimination of non-specific inhibitors was achieved by incubation of the study serum samples and quality control sera (serum of ferret or human immunised with influenza virus) with neuraminidase from Vibrio cholerae [receptor-destroying enzyme (RDE II), Denka Seiken Co. Ltd, Tokyo, Japan], according to the manufacturer’s instructions: 18 hours/+ 36 °C followed by heat inactivation 1 hour/+ 56 °C). All batched samples were prepared simultaneously.
For the HI analysis with the NIBRG121 virus, samples and controls were titrated in an eight-step twofold dilution series (covering titres 8–1024) and incubated with the haemagglutinin antigen (HA) suspension [previously titrated to adjust the dilution at 4 HAUs (haemagglutination units)/25 µl, 50% end point]. The HA antigen was not added to the well that was dedicated to the RDE quality control.
The mixture was incubated for 1 hour at room temperature and 25 µl of the 0.5% RBC suspension (turkey blood) was added. The reaction was left for half an hour at room temperature before reading.
The serum titre is equal to the highest reciprocal dilution that induces a complete inhibition of haemagglutination. The titre of each quality control serum is close to the previously assigned value (within one serial twofold dilution limits). The RBC controls (RBC suspension without antigen) and the RDE controls do not produce any agglutination. Each serum sample is titrated in duplicate, and individual titres were reported (two for each sample).
The MN was performed using a 96-well format, according to previously described protocols and SOPs developed with the RVU, HPA Centre for Infections, London.
Elimination of complement (e.g. from fetal calf serum in culture medium) was achieved by incubation of study sera and appropriate quality control sera (provided and chosen according to test virus by the RVU – usually serum of ferret, sheep or human, with/without neutralisation activity) at + 56 °C/30 minutes. This step was performed simultaneously for all study samples and control sera.
The MN analysis with the NIBRG121 virus was performed as follows: a six-step twofold dilution series (covering titres 20–640) was set up for each of the samples and control sera. After addition of a pretitred virus (100 × TCID50 per well or 0.1-1 virus particle per cell) neutralisation was performed by incubation of the virus/serum mixture at room temperature for 1 hour. After neutralisation, a suspension of Madin–Darby Canine Kidney (MDCK) cells was added and the plates incubated for 16 hours at 37 °C in a carbon dioxide incubator. The remaining infectivity of virus after neutralisation was determined in an enzyme immunoassay (EIA) format using a monoclonal antibody (mAb) to detect expression of viral nucleoprotein. The amount of nucleoprotein expression was determined photometrically [optical density (OD) reading = OD450] using a plate reader.
An OD reading for each dilution step for each sample was used to calculate the titre. The titre was reported as the reciprocal dilution at which 50% of the virus was neutralised (e.g. titre of 100). The MN analysis was performed in duplicate (in separate runs on 2 days) for each sample.
Statistical analysis
The researchers used a conservative power calculation for this study based upon an estimated 20% seroconversion rate from wild-type infection in unvaccinated women. The researchers also estimated conservatively that seroconversion in vaccinated women would be 50%, although evidence suggests that it was more likely to be 70% after two doses. Thus, a power calculation based on 20% versus 50% was very conservative, and a more optimistic comparison would have been 10% versus 70%.
Based on 20% versus 50%, with 80% statistical power and 5% significance (two-tailed statistics), 38 subjects per group were required – total 76. However, anticipating that two-thirds of women would accept vaccine, the ratio of unvaccinated–vaccinated subjects was predicted to be 0.5. Allowing for this imbalance, the researchers chose a total study size of 89 subjects (59 vaccinated, 30 unvaccinated). Assuming a total of 89 subjects and more optimistic estimates of 10% versus 70%, the study would have had 100% power to detect such a difference. Nonetheless, to allow for possible losses during analysis or inadequate specimens the researchers planned to recruit 100 study subjects.
The researchers used the following statistical approaches [using statistical software package stata 11 (StataCorp LP, College Station, TX, USA) for analysis]. 49
Primary objective of study
-
Characteristics of vaccinated and unvaccinated mothers.
-
Assessing vertical transmission of immunity to A/H1N1v influenza virus in vaccinated and unvaccinated mothers using cord-blood analyses:
-
– Exposure variable vaccinated/unvaccinated mother (binary variable).
-
– Outcome variable binary variable ‘immune (yes/no)’, based on either threshold level of antibodies in cord blood.
-
The immunogenicity end point was the proportion of subjects with HI titres of ≥ 1 : 32 and ≥ 1 : 40 and MN titre of ≥ 1 : 60. 46–48 In normal serological determination, paired samples looking for a rise in titre would also be utilised; however, this was not possible in this study, as cord blood is clearly available only once and reflects the immune status as the result of vaccination (or possible wild-type infection) occurring some time before. Paired samples would therefore have been meaningless. Geometric mean titres were calculated from the sample duplicate analysis results and were compared between babies of vaccinated mothers and babies of unvaccinated mothers. The proportions of subjects in whom seroconversion (HI titres ≥ 1 : 32 and ≥ 1 : 40 and MN ≥ 1 : 60) was achieved were compared between each group using a chi-squared test.
Additionally, the difference in immunity between offspring of vaccinated and unvaccinated mothers and subgroup analysis according to health status and prior obstetric history and other biologically relevant covariates were described.
Chapter 3 Results
The researchers enrolled 117 subjects in two groups – those vaccinated during pregnancy and those not [vaccinated (exposed)/unvaccinated (unexposed)] between 18 November 2009 and 20 March 2010 (presented diagrammatically in Figures 2 and 3).
FIGURE 2.
Results study flow diagram (with numbers recruited and final samples received).
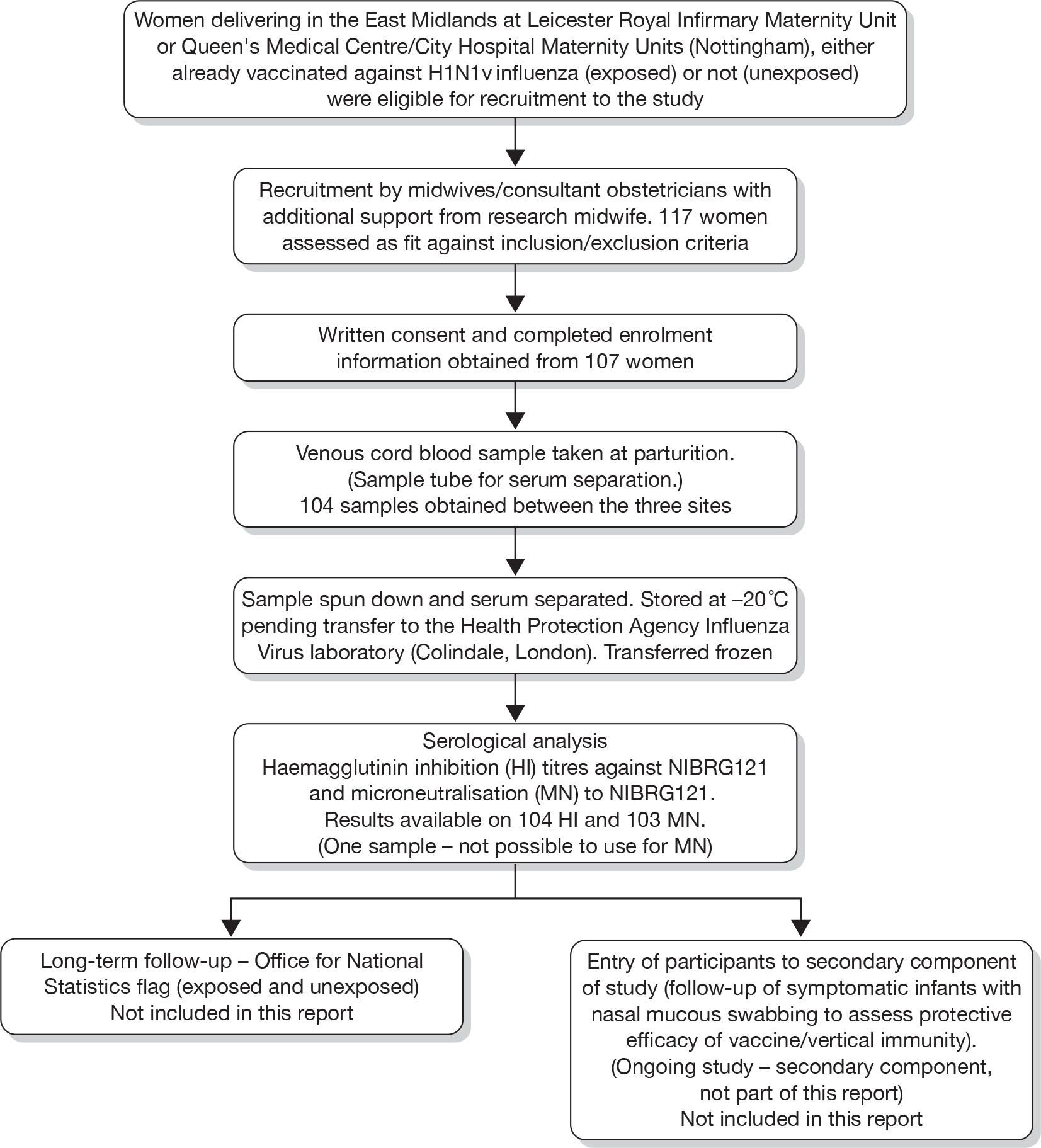
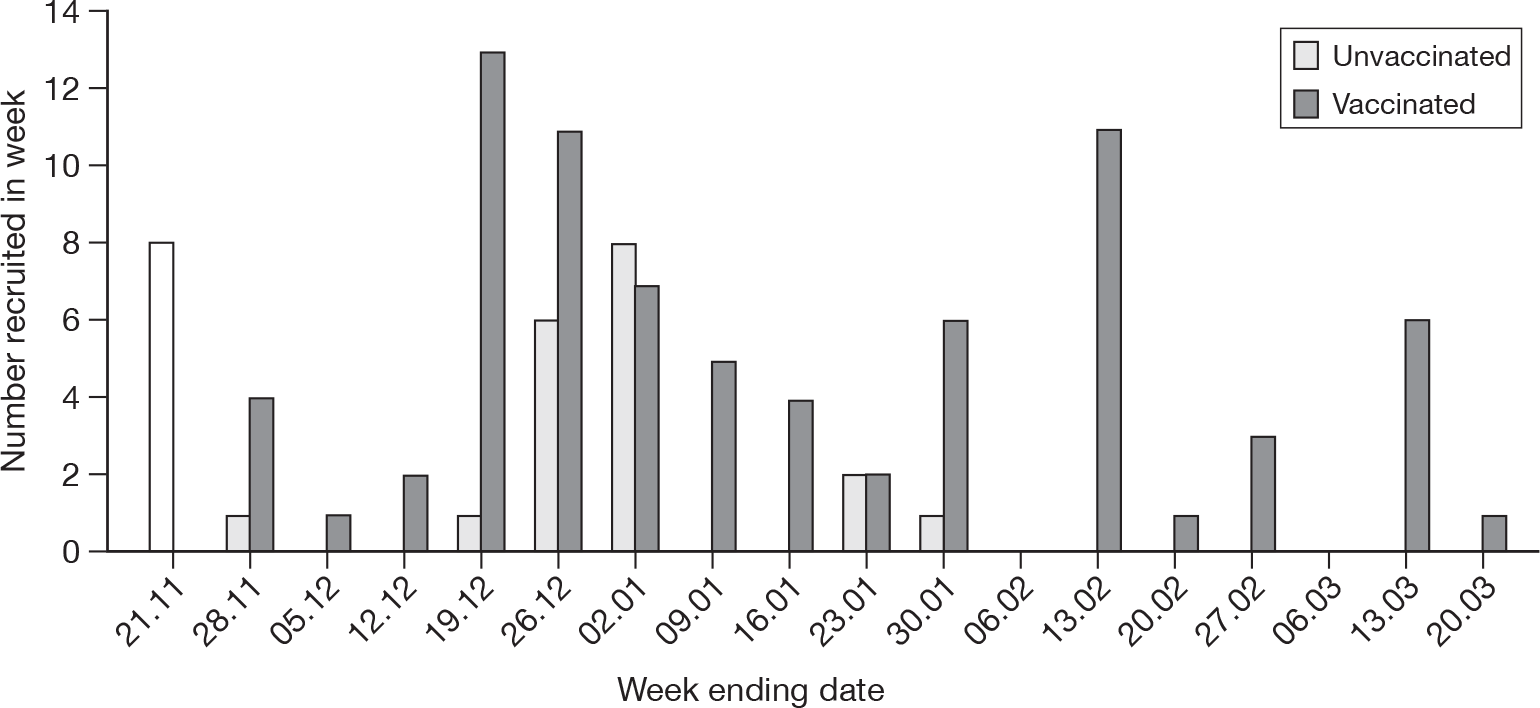
FIGURE 3.
Recruitment curves (week ending date) – vaccinated and unvaccinated mothers.
A total of 10 enrolees were subsequently excluded, as, although a sample was taken and analysed, no clinical data were returned. An additional three cases were also excluded, as, although recruited, no sample was obtained at delivery. In total, therefore, clinical and sample data on 104 subjects were obtained. One sample was suitable only for HI analysis, as strong haemolysis prevented MN titres from being assessed.
The researchers were concerned that the lag for recruiting vaccinated mothers could introduce a bias by increasing the duration for exposure to wild-type A/H1N1v, and therefore giving them a higher serological immune profile than from the vaccine alone (see Table 1, data markeda,b,c). However, this is considered unlikely, as, first, the period after the end of January 2010 was when circulating A/H1N1v had dropped, and, second, the mothers (except for one who delivered 2 days after vaccination) delivering in this period had been already been vaccinated for a considerable period (32–108 days, mean 74 days, median 78 days). Additionally, Figure 4, below, shows the recruitment curve of the unvaccinated women, and the date of vaccination plus 10 days (to allow for seroconversion) for the vaccinated recruits. Figure 4 demonstrates that the vaccinated women recruited largely had less time to have exposure to wild-type infection before vaccination than the unvaccinated group, and, therefore, if anything, any bias would be towards reducing the effect observed. This has been confirmed statistically (date of serological immunity of vaccinated women significantly earlier than unvaccinated women delivered – non-parametric Mann–Whitney U-test, p = 0.021).
| Characteristics | Vaccinated: n = 77 (74%) | Unvaccinated: n = 27 (26%) | p-value | |
|---|---|---|---|---|
| Age of mother, years, mean (95% CI) | 31 (29–32) | 29 (26–31) | 0.141 | |
| Ethnicity | White | 67 (89) | 25 (93) | 0.625 |
| Other | 8 (11) | 2 (7) | ||
| Gravida | One | 35 (46) | 10 (37) | 0.417 |
| Two or more | 41 (54) | 17 (63) | ||
| Parity | Zero | 45 (59) | 16 (59) | 0.996 |
| One or more | 31 (41) | 11 (41) | ||
| Any past medical history | None | 67 (87) | 22 (81) | 0.481 |
| One or more | 10 (13) | 5 (19) | ||
| Any past obstetric history | None | 50 (65) | 16 (59) | 0.598 |
| One or more | 27 (35) | 11 (41) | ||
| Mode of delivery | Normal, assisted, elective or caesarean unspecified | 67 (88) | 22 (88) | 0.983 |
| Emergency caesarean section | 9 (12) | 3 (12) | ||
| Sex of baby | Male | 38 (50) | 16 (59) | 0.408 |
| Female | 38 (50) | 11 (41) | ||
| Weight of baby (g) | 3365 (3223–3507) | 3600 (3400–3799) | 0.085 | |
| Gestational age of baby (weeks) | 39.3 (38.9–39.7) | 40.1 (39.5–40.7) | 0.061 (< 37 weeks vs > 37 weeks) | |
| Under-5-year-olds at home | 0 | 43 (57) | 17 (63) | 0.610 |
| One or more | 32 (43) | 10 (37) | ||
| Estimated due date | 17 November 2009 to 10 April 2010 | 5 November 2009 to 6 February 2010 | < 0.001a | |
| Actual delivery date | 25 November 2009 to 12 March 2010 | 18 November 2009 to 26 January 2010 | 0.001b | |
| Estimated date of serological conversion (vaccinated mothers) vs date of delivery of unvaccinated mothers (last possible date of seroconversion to wild-type infection) | 11 November 2009 to 23 February 2010 | 18 November 2009 to 26 January 2010 | 0.021c | |
| Any current medication | Zero | 62 (82) | 24 (89) | 0.379 |
| One or more | 14 (18) | 3 (11) | ||
| No. of smokers in household | Zero | 54 (82) | 16 (70) | 0.217 |
| One or more | 12 (18) | (30) |
FIGURE 4.
Recruitment curve – unvaccinated mothers and vaccination dates + 10 days curve of mothers recruited who had already been vaccinated (week ending date).

Table 1 illustrates the underlying characteristics of vaccinated and unvaccinated mothers and indicates that between the groups there was no systematic difference that could have introduced a source of error into the study other than recruitment date. However, as highlighted, the vaccinated group had less time for exposure to wild-type infection to achieve natural seroconversion than the unvaccinated group, and, therefore, this would tend to have narrowed the degree of effect seen, as the women who had not been vaccinated would have had a longer opportunity to acquire natural immunity.
Table 2 illustrates the results of the HI titres. Using the cut-off of ≥ 1 : 32 and separately ≥ 1 : 40 dilution, the results were categorised into immune/not immune (based on the geometric mean of the tests conducted on each sample obtained). Statistical analysis (chi-squared test) indicates a highly significant statistical difference between the numbers of immune babies born to vaccinated mothers compared with those who were not.
| Vaccinated (all) | Vaccinated (> 10 days from vaccination to delivery) | Vaccinated cohort delivered before 24 January 2010 | Unvaccinated (all) | Unvaccinated, delivered before 24 January 2010 | p-value | |
|---|---|---|---|---|---|---|
| Immune status NIBRG121 HI titres, n = 104 (%) | ||||||
| Proportion of samples with an immune titre ≥ 1 : 32 | 63/77 (82) | – | – | 8/27 (30) | – | < 0.001 |
| – | 57/67 (85) | – | 8/27 (30) | – | < 0.001 | |
| – | – | 40/49 (82) | – | 8/26 (31) | < 0.001 | |
| Proportion of samples with an immune titre ≥ 1 : 40 | 58/77 (75) | – | – | 5/27 (19) | – | < 0.001 |
| – | 53/67 (79) | – | 5/27 (19) | – | < 0.001 | |
| – | – | 36/49 (74) | – | 5/26 (19) | < 0.001 | |
| Immune status NIBRG121 MN titres, n = 103 (%) | ||||||
| Proportion of samples with an immune titre ≥ 1 : 60 | 64/76 (84) | – | 42/49 (86) | 7/27 (26) | – | < 0.001 |
| – | 57/66 (86) | – | 7/27 (26) | – | < 0.001 | |
| – | – | 42/49 (65) | – | 7/26 (27) | < 0.001 | |
Additionally, MN titres were performed. Although MN titres do not indicate immunity, they can provide additional confirmation/validation of the HI titres. The results of these are also tabulated below in Table 2.
The immune status analysis was repeated including the results from the 10 samples where no clinical details were returned, treating this group as vaccinated and unvaccinated in turn. The results remained highly significantly different (p < 0.001 for all thresholds used).
The kappa statistic agreement between NIBRG121 HI titres (1 : 32 threshold) and MN titres was 0.89 (95% agreement) (p < 0.001). This provides additional support to the use of MN titres to validate HI titres.
Table 3, below, indicates that there was a difference in the recruitment profile between the hospital centres and this was significant.
| Vaccine | |||
|---|---|---|---|
| PandemrixTM: n (%) | 72/76 (95) | ||
| Unknown: n (%) | 4/76 (5)a | ||
| Recruitment site | |||
| Vaccinated | Unvaccinated | p-value | |
| Nottingham University Hospitals: n (%) | 53/77 (69) | 12/27 (44) | p = 0.024 |
| University Hospitals Leicester: n (%) | 24/77 (31) | 15/27 (56) | |
Table 2 also indicates the number of vaccinated mothers who had been immunised with each vaccine type. As can be seen, where researchers were able to obtain data, none of the recruits appears to have been immunised with Celvapan™.
Subgroup analysis
Limited subgroup analysis, first using a simple model and then multivariate logistic regression, examined the effect of other independent variables that could have had a biologically plausible effect on transfer/acquisition of immunity. The model utilised immune status (yes or no) by HI and MN titres (separately) as the dependent variable, with vaccination status (yes or no) as the independent variable. Other covariates include past obstetric history (any), past medical history (any), gravida and parity status. (Full details of covariates used are set out in Tables 4–7.) Odds ratios were calculated and 95% confidence intervals (CIs) around these results were also determined. Where the CI did not include 1 or the p-value was < 0.05, the result was concluded to be significant.
| Variable | Unadjusted OR for immunity, cut-off ≥ 1 : 32 | CIs | p-value | Unadjusted OR for immunity, cut-off ≥ 1 : 40 | 95% CI | p-value |
|---|---|---|---|---|---|---|
| Vaccinated | 10.69 | 3.90 to 29.31 | < 0.001 | 13.43 | 4.47 to 40.38 | < 0.001 |
| Covariate adjusted for | Adjusted OR for immunity, cut-off ≥ 1 : 32 | CIs | p-value | Adjusted OR for immunity, cut-off ≥ 1 : 40 | 95% CI | p-value |
|
Ethnicity (n = 102) (Caucasian or other) |
10.37 | 3.77 to 28.53 | 0.935 | 13.08 | 4.33 to 39.48 | 0.788 |
|
Any past obstetric history (n = 104) |
10.63 | 3.87 to 29.24 | 0.499 | 13.38 | 4.45 to 40.24 | 0.856 |
|
Any past medical history (n = 104) |
10.65 | 3.85 to 29.46 | 0.260 | 13.35 | 4.42 to 40.34 | 0.332 |
|
Gravida status (n = 103) (One/two or more) |
10.43 | 3.79 to 28.66 | 0.812 | 13.23 | 4.38 to 39.93 | 0.962 |
|
Parity status (n = 103) (None/one or more) |
10.54 | 3.84 to 28.95 | 0.787 | 13.21 | 4.39 to 39.73 | 0.891 |
|
Under-5-year-olds in household (n = 102) (None/one or more) |
11.26 | 4.06 to 31.25 | 0.769 | 13.85 | 4.58 to 41.91 | 0.690 |
|
Sex of baby (n = 103) |
10.39 | 3.78 to 28.56 | 0.718 | 13.37 | 4.42 to 40.47 | 0.801 |
|
Birth weight (g) (n = 99) |
13.92 | 4.62 to 41.95 | 0.391 | 21.67 | 5.76 to 81.47 | 0.515 |
|
Mode of delivery (n = 101) (Normal, assisted, elective caesarean section/caesarean section unspecified or emergency caesarean) |
11.65 | 4.05 to 33.53 | 0.371 | 12.02 | 3.96 to 36.46 | 0.800 |
|
Gestation (n = 103) (< 37 weeks or ≥ 37 weeks) |
10.89 | 3.87 to 30.66 | 0.755 | 14.03 | 4.57 to 43.06 | 0.541 |
| Maternal age at delivery (n = 103) | 13.47 | 4.49 to 40.38 | 0.091 | 14.12 | 4.57 to 43.57 | 0.496 |
|
Smokers at home (n = 89) (Zero, n, one or more, n) |
14.18 | 4.42 to 45.51 | 0.455 | 14.33 | 4.23 to 48.58 | 0.410 |
| On any current medication (n = 103) | 11.17 | 3.99 to 31.29 | 0.387 | 13.74 | 4.52 to 41.81 | 0.509 |
| Variable | Unadjusted OR for immunity | 95% CI | p-value |
|---|---|---|---|
| Vaccinated | 15.24 | 5.29 to 43.93 | 0.001 |
| Covariate adjusted for | Adjusted OR for immunity | 95% CI | p-value |
|
Ethnicity (n = 101) (Caucasian or other) |
14.86 | 5.13 to 43.04 | 0.848 |
|
Any past obstetric history (n = 103) |
15.16 | 5.26 to 43.74 | 0.755 |
|
Any past medical history (n = 103) |
15.10 | 5.23 to 43.59 | 0.607 |
|
Gravida status (n = 102) (One/two or more) |
16.49 | 5.51 to 49.37 | 0.305 |
|
Parity status (n = 102) (Zero/one or more) |
15.17 | 5.24 to 43.97 | 0.621 |
|
Under-5-year-olds in household (n = 101) (Zero/one or more) |
16.70 | 5.63 to 49.55 | 0.284 |
|
Sex of baby (n = 102) |
14.98 | 5.18 to 43.33 | 0.972 |
|
Birth weight (g) (n = 98) |
20.79 | 6.37 to 67.79 | 0.172 |
|
Mode of delivery (n = 100) (Normal, assisted, elective caesarean section/caesarean section unspecified or emergency caesarean) |
17.27 | 5.63 to 52.96 | 0.307 |
|
Gestation (n = 102) (< 37 weeks or ≥ 37 weeks) |
14.29 | 4.87 to 41.94 | 0.672 |
|
Maternal age at delivery (n = 102) |
18.27 | 5.84 to 57.13 | 0.192 |
|
Smokers at home (n = 88) (Zero, n or one or more, n) |
20.04 | 5.93 to 67.71 | 0.904 |
|
On any current medication (n = 102) |
15.37 | 5.27 to 44.85 | 0.707 |
| Immune titre ≥ 1 : 32 | Immune titre ≥ 1 : 40 | ||||||
|---|---|---|---|---|---|---|---|
| Adjusted ORa | p-value | 95% CI | Adjusted ORa | p-value | 95% CI | ||
| Vaccinated | 22.17 | 0.001 | 5.81 | 84.59 | 22.76 | < 0.001 | 5.57 to 93.02 |
| Gravida (two or more) | 0.87 | 0.867 | 0.16 | 4.68 | 1.41 | 0.679 | 0.28 to 7.02 |
| Parity (one or more) | 0.99 | 0.994 | 0.05 | 19.65 | 0.47 | 0.568 | 0.04 to 6.23 |
| Number of under-5year-olds in household (one or more) | 2.07 | 0.587 | 0.15 | 28.56 | 2.48 | 0.411 | 0.29 to 21.60 |
| Mode of delivery (emergency section) | 0.46 | 0.321 | 0.10 | 2.12 | 0.87 | 0.849 | 0.20 to 3.74 |
| Gestational age (37 weeks) | 1.80 | 0.577 | 0.23 | 14.12 | 2.20 | 0.400 | 0.35 to 13.74 |
| Any past medical history (one or more) | 0.62 | 0.543 | 0.13 | 2.91 | 0.63 | 0.540 | 0.15 to 2.75 |
| Baby weight (g) | 1.00 | 0.288 | 1.00 | 1.00 | 1.00 | 0.307 | 1.00 to 1.00 |
| Maternal age at delivery (years) | 0.96 | 0.379 | 0.86 | 1.06 | 1.01 | 0.864 | 0.92 to 1.10 |
| Immune titre ≥ 1 : 32 | Immune titre ≥ 1 : 40 | ||||||
|---|---|---|---|---|---|---|---|
| Adjusted ORa | p-value | 95% CI | Adjusted ORa | p-value | 95% CI | ||
| Vaccinated | 20.22 | < 0.001 | 5.23 | 78.10 | 21.03 | < 0.001 | 5.08 to 87.11 |
| Duration of exposure (days)b | 0.99 | 0.645 | 0.96 | 1.02 | 1.00 | 0.732 | 0.96 to 1.03 |
| Gravida (two or more) | 0.82 | 0.814 | 0.15 | 4.48 | 1.35 | 0.721 | 0.26 to 6.86 |
| Parity (one or more | 1.18 | 0.919 | 0.05 | 26.15 | 0.53 | 0.643 | 0.04 to 7.77 |
| Number of under-5-year-olds in household (one or more) | 1.85 | 0.652 | 0.13 | 27.19 | 2.35 | 0.446 | 0.26 to 21.07 |
| Mode of delivery (emergency section) | 0.50 | 0.367 | 0.11 | 2.28 | 0.92 | 0.916 | 0.21 to 4.04 |
| Gestational age (≥ 37 weeks) | 1.75 | 0.600 | 0.22 | 14.11 | 2.10 | 0.430 | 0.33 to 13.16 |
| Any past medical history (one or more) | 0.60 | 0.516 | 0.13 | 2.84 | 0.62 | 0.528 | 0.14 to 2.72 |
| Baby weight (g) | 1.00 | 0.272 | 1.00 | 1.00 | 1.00 | 0.315 | 1.00 to 1.00 |
| Maternal age at delivery (years) | 0.95 | 0.344 | 0.85 | 1.06 | 1.00 | 0.914 | 0.92 to 1.10 |
| Adjusted ORa | p-value | 95% CI | |
|---|---|---|---|
| Vaccinated | 48.95 | < 0.001 | 9.64 to 248.72 |
| Gravida (two or more) | 2.20 | 0.432 | 0.31 to 15.78 |
| Parity (one or more) | 0.72 | 0.851 | 0.02 to 21.87 |
| Number of under-5-year-olds in household (one or more) | 3.26 | 0.436 | 0.17 to 63.88 |
| Mode of delivery (emergency section) | 0.43 | 0.308 | 0.08 to 2.18 |
| Gestational age (37 weeks) | 1.31 | 0.835 | 0.11 to 16.28 |
| Any past medical history (one or more) | 1.00 | 0.998 | 0.17 to 6.02 |
| Baby weight (g) | 1.00 | 0.174 | 1.00 to 1.00 |
| Maternal age at delivery (years) | 0.95 | 0.355 | 0.84 to 1.06 |
| Adjusted ORa | p-value | 95% CI | |
|---|---|---|---|
| Vaccinated | 45.33 | < 0.001 | 8.82 to 232.85 |
| Duration of exposure (days)b | 0.99 | 0.740 | 0.96 to 1.03 |
| Gravida (two or more) | 2.13 | 0.451 | 0.30 to 15.12 |
| Parity (one or more | 0.82 | 0.912 | 0.03 to 27.47 |
| Number of under under-5-year-olds in household (one or more) | 2.98 | 0.480 | 0.14 to 62.17 |
| Mode of delivery (emergency section) | 0.46 | 0.357 | 0.09 to 2.40 |
| Gestational age (≥ 37 weeks) | 1.24 | 0.870 | 0.10 to 15.87 |
| Any Past Medical History (one or more) | 0.97 | 0.973 | 0.16 to 5.84 |
| Baby weight (g) | 1.00 | 0.169 | 1.00 to 1.00 |
| Maternal age at delivery (years) | 0.94 | 0.354 | 0.83 to 1.07 |
The results of the logistic regression models are tabulated (Tables 4 and 5) and show that immune status was not additionally affected by any of the covariates. The logistic regression calculations also indicate that vaccination in the mother was strongly predictive of the baby being immune, with odds ratios of approximately 10, 1 : 32 threshold, 13, 1 : 40 threshold for HI titres, and 15 for MN titres. These odds ratios had highly significant p-values; however, the CIs were wide, indicating that the precision of this estimate could have been more accurate with a larger sample size.
Multivariate models
In the absence of any of the covariates reaching significance in the bivariate models, the covariates included in the multivariate model (Tables 6a and 7a) were on an a priori basis, where the researchers considered that they could have a biological bearing on immune status:
-
Gravida, parity and number of under-5-year-olds in the household Children are efficient spreaders of influenza and therefore more children in the household might increase exposure opportunity for natural immunity to occur.
-
Mode of delivery On the basis that emergency section may indicate a problem with fetal health and therefore immune acquisition.
-
Maternal past medical history On the basis that underlying medical illness could affect immune response/make it more likely to be vaccinated.
-
Gestational age and baby weight On the basis that premature babies may have not received as much intrauterine immune transfer/lighter babies may reflect prematurity/poorly functioning placenta.
-
Maternal age Placental function.
[Table 6-7]
Further analysis was undertaken to determine if duration of exposure to A/H1N1v could have had a bearing on the findings. The first possible exposure date was taken as 26 April 2009,50 when the first suspected (confirmed the following day) case of A/H1N1v was announced in the UK. The results are shown in Tables 6b and 7b, respectively, and demonstrate that duration of exposure was not a significant determinant of immune status.
The geometric mean titres for the HI and MN have been also plotted on reverse cumulative distribution percentage curves for babies of vaccinated and unvaccinated mothers. These are shown in Figures 5 and 6. These figures demonstrate the different proportions that are immune between the vaccinated and unvaccinated groups.
FIGURE 5.
Reverse cumulative distribution curves – haemagglutinin titres.
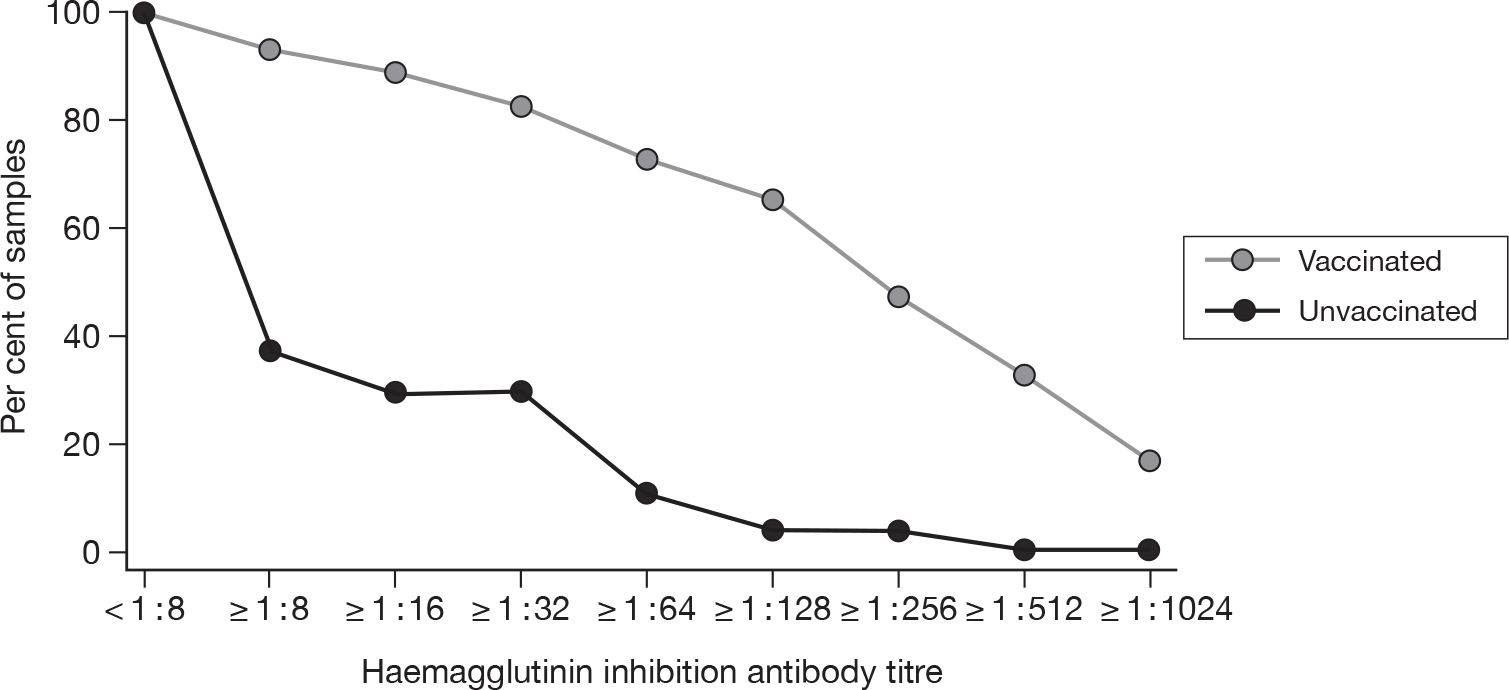
FIGURE 6.
Reverse cumulative distribution curves – microneutralisation titres.
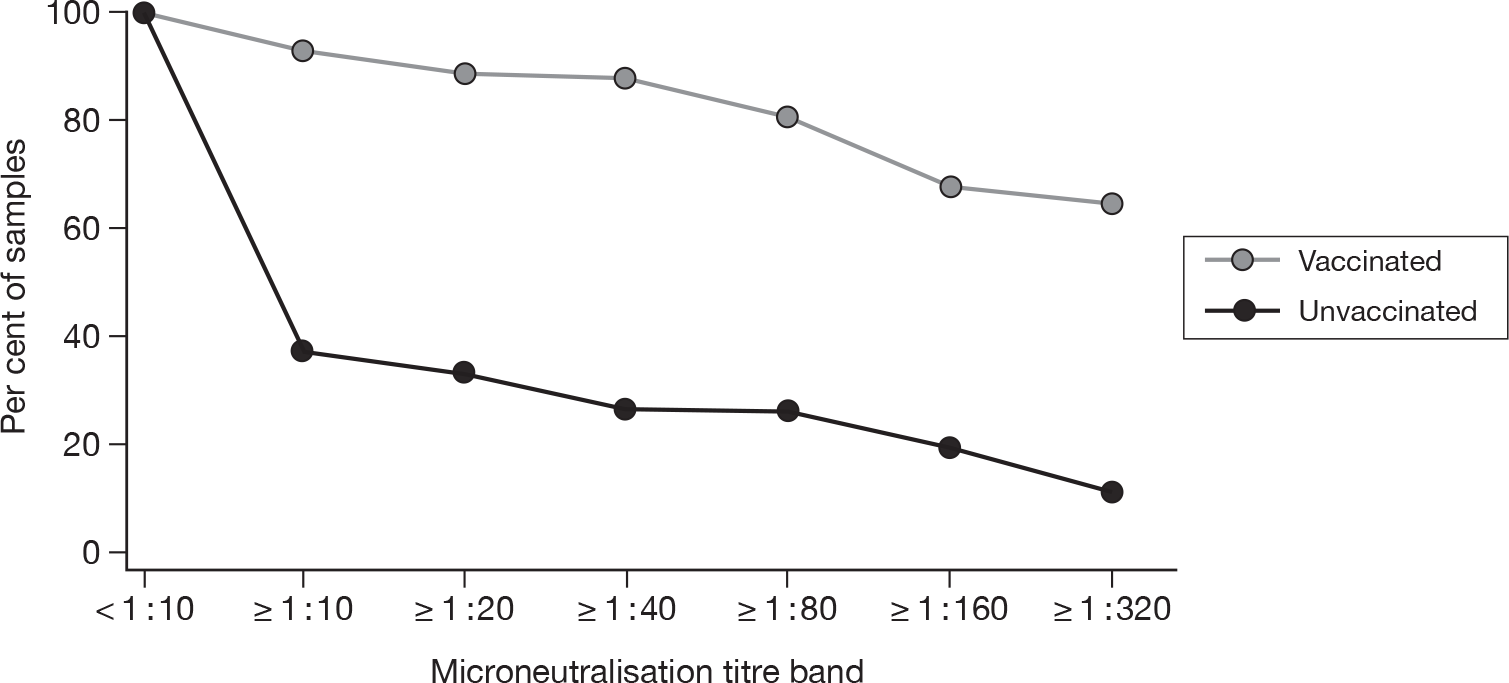
Figure 5 demonstrates that utilising HI titre estimates of immunity, approximately 80% of babies from vaccinated mothers had serological evidence of immunity. This falls to 30% or 20% (dependent on the immune threshold used) of babies born to unvaccinated mothers and is consistent with the prevalence of natural immunity gained during the pandemic. MN titres provide additional corroboration of this difference in immunity between the two groups and demonstrate similar distribution curves (Figure 6).
Chapter 4 Discussion
The influenza A/H1N1v pandemic in 2009–10 was very mild in terms of its overall impact, even although the initial data emanating from Mexico suggested a more serious picture. Roughly 1% of patients with clinical illness required hospital care and the case fatality rate was no more than 0.1%. 51–53 While most people contracting pandemic influenza suffered a short, self-limiting illness, smaller subsets, including pregnant women, suffered a higher risk of developing serious illness or death. 11–13 Subsequent reports from the USA, England and elsewhere indicate that pregnant women were at substantially (statistically significant) greater risk of requiring admission to hospital with symptomatic infection from A/H1N1v than the general population (four times higher), up to one-fifth of those admitted needed intensive-level care, and were also over-represented in the mortality data (5% of all deaths). 11,12,15,54 Early diagnosis and antiviral drug treatment were also important prognostic determinants. 13,55 Although associated comorbidities further increased the risks, nonetheless there were some otherwise premorbidly completely healthy people who were adversely affected. 15,51
Despite advances in vaccine production and novel centralised European procedures for rapid licensure of pandemic vaccines,56 vaccines were not available for widespread use until mid-October 2009, approximately 5.5 months after the pandemic began to emerge, and 4 months after World Health Organization Phase 6 (pandemic) was declared. 17,37,41,57 The UK experienced a major pandemic wave in spring–summer 2009, which peaked in late July. 45,51 Thus, by the time vaccine was available, the cumulative serological attack rate in the UK was approximately equal to the prevalence of immunity found in unvaccinated mothers recruited in this study. 58
The cumulative attack rate, however, masks considerable differences between age groups and regions. A serological prevalence survey undertaken with samples from before and after the outbreak45 has shown that prior to the pandemic the presence of neutralising antibodies to A/H1N1v was overall 14.5%, but with a marked age gradation (increasing with seniority). By September 2009, the prevalence of samples with a HI titre above 1 : 32 had increased across all age ranges (except 45- to 65-year-olds), but particularly in children and adults below the age of 45 years. For example, in older children and young adults (aged 15–24 years) the prevalence of serological immunity had increased from 17.5% at baseline to 38.1% and in adults (aged 25–49 and 25–44 years) from 9.8% to 15.1%. 45
Attack rates varied across the country during the pandemic, with London and the West Midlands having the highest disease burden. The East Midlands (where this study was based) had lower attack rates that were more in line with other parts of England, and therefore it can probably be safely assumed that the baseline prevalence of serological immunity at commencement of vaccination (and also recruitment to the study for unvaccinated mothers) might have been lower in the region than the cumulative national estimates would otherwise suggest. 52
This study was based on a relatively simple concept of obtaining immediately postpartum venous cord blood from mothers who were both vaccinated and unvaccinated. We did not influence whether vaccination took place or not. In the UK, the AS03-adjuvanted vaccine (Pandemrix™, GSK Biologicals)17,37,41 was the recommended product of choice in most women, based on the availability of data suggesting rapid seroconversion after one dose, and the need to protect pregnant women as quickly as possible prior to a potential UK second wave. Although we included all women in our primary analysis, a restricted analysis based on only those women with an interval of at least 10 days between vaccination and parturition (those women with time to seroconvert) revealed almost identical results. 59–61 We assumed that cord venous blood is an acceptable proxy measure of the degree of humoral immunity transmitted from mother to child. The primary outcome measure was based on a single HI antibody titre because obtaining paired sera would not have been possible. Thus, we were able to analyse only HI antibody titres, as opposed to fourfold rises or seroconversion rates. Although CHMP specifies an HI antibody threshold of ≥ 1 : 40 as a correlate of clinical protection for seasonal vaccination, we used a titre of ≥ 1 : 32 based on information from pandemic outbreaks (see Chapter 2). We also performed a secondary analysis based on MN because, although less well defined in terms of agreed thresholds for clinical protection, the approach is considered important for corroboration of HI antibody data. 46–48
To avoid the risk of introducing bias into the serological analysis of samples, the laboratory staff were blinded to the vaccination status of the subject to which the sample related (no details on vaccination status were included on the request form, which had been prelabelled with the unique identifier and test required to obviate recruiting staff inadvertently revealing the status of the subject). However, the separation of sample and subject identity meant that for 10 samples the researchers could not assign the results to either the vaccinated or unvaccinated group, as it was impossible to trace them because their recruitment details were not returned. Likewise, in the case of three subjects no cord sample was collected. These 13 subjects therefore had to be excluded from the study. This might have introduced a source of bias; however, this is considered unlikely. Further analysis of immune status including the 10 samples for which a result was available, but for which no clinical data was presented, treating them, in turn, as if from babies of vaccinated/unvaccinated mothers, respectively, remained highly significant, whichever immune threshold measure was used, regardless of whether these samples were treated as being from babies of vaccinated or unvaccinated mothers.
The collected samples were stored, frozen, at the respective study sites and then sent in mixed batches for analysis at the HPA laboratory, which would have reduced the risk of deducing the vaccination status. Nonetheless, despite not being asked for, some recruiting staff/local laboratory staff at the research site entered the date of sample on to some forms (48 out of 104). To address the potential for inadvertent unblinding of laboratory staff which could have led to unintentional bias occurring, the researchers re-ran the analyses, using only samples for which no date of collection was noted (and therefore no possible deduction of vaccine status by date). The results remained highly significant.
Overall, our results suggest that vaccination with one dose of AS03-adjuvanted monovalent A/H1N1v vaccine is likely to provide considerable protection to the newborn child via humoral antibody transmitted in utero from a vaccinated mother from the time of birth. The difference between vaccinated and unvaccinated mothers was highly significant, irrespective of whether a HI titre threshold of ≥ 1 : 32 or ≥ 1 : 40 was used. However, our data do not provide information on the duration of antibody persistence. A study by Zaman et al. 27 reported that immunisation of mothers with trivalent seasonal influenza vaccine provided protection for 6 months to the infant. Maternal passive immunity transferred to the infant is thought to persist for between 3 and 12 months. 25,62–65 However, it is possible that protection may last longer. In this regard, reduction in disease severity may be as important as absolute prevention of infection. Investigation of transmission of maternal tetanus antibodies (when vaccinated in pregnancy) showed continued highly sensitised responses at 13 months. 65 To investigate if a similar effect is seen with A/H1N1v we have therefore obtained separate funding to follow up the children of the recruited mothers from this study to explore the longer-term protective effect of the passive transfer of immunity. Nevertheless, based on the available data from other studies it seems likely that protection via passively transferred antibodies would persist for at least 6 months, until the child is old enough to receive active vaccination. 25,63–65
In the East Midlands, the vaccination programme against A/H1N1v in pregnant women took time to become established. Recruitment of unvaccinated women was easy, but for the first few weeks after the national immunisation programme began there were few women presenting for delivery who had been vaccinated. The comment made by many of the women presenting for delivery was that they were near the end of their pregnancy and had chosen to take the risk of not having the vaccine, i.e. their personal risk–benefit assessment may have mitigated against vaccine acceptance. We therefore deliberately slowed the pace of recruitment of unvaccinated women so that they were contemporaneous with vaccinated women and therefore exposed to similar background circulating influenza in the community, otherwise this could have introduced an unintentional bias into the results. If bias has been introduced whereby dates of delivery in unvaccinated women were generally earlier than in vaccinated women, any differences in antibody titres might have been accentuated and the degree of protection in vaccinated mothers might be, in part, wrongly ascribed to the effect of the vaccine rather than the effect of recent infection. Nonetheless, despite slowing the recruitment of unvaccinated women, the recruitment curve does demonstrate an apparent differential between vaccinated and unvaccinated mothers. The researchers were concerned that the later recruitment of vaccinated women could have accounted for the difference in immunity found, and therefore undertook additional analyses to refute this possibility. First, the baseline differences between the two groups were not significant for the other variables examined (other than date of recruitment) (see Table 1). This provides reassurance that there was not some systematic difference between vaccinated and unvaccinated subjects (including factors that might be associated with vaccination acceptance). Second, as can be seen from Table 2, dropping the recruits enrolled after 23 January 2010 to avoid this possible confounder (tail of vaccinated recruits) has no effect on the significance. Likewise, women who delivered shortly after vaccination could have biased the results because of insufficient time to seroconvert. Again, excluding these from the analyses had no effect on the significance determined. Additionally, the researchers calculated the estimated date of seroconversion in vaccinated women and compared this to the last possible date of seroconversion in unvaccinated women (date of recruitment) and also determined the interval between the first possible date of exposure to A/H1N1v (26 April 2009 – first case reported in the UK – confirmed positive on 27 April 2009) and these two dates for the two groups. The vaccinated women were found to have been exposed to wild-type infection for significantly less time than the unvaccinated women and therefore would not have achieved their enhanced immune status from longer exposure. Also, adjustment for the interval between first possible exposure and seroconversion (vaccinated/unvaccinated) in the logistic regression model did not change the findings.
Additionally, the epidemic curve (HPA, published data) provides further corroborative evidence that differential exposure to wild-type A/H1N1 was unlikely to be a significant confounder. The epidemic curve indicates that at the point recruitment started, incident cases had declined substantially (the peak having occurred in approximately week 30). Although the number of new cases increased again in the autumn, the curve was flattened but prolonged, with a peak of only one-third of that observed in the summer and at the point recruitment started – this secondary wave was declining. Therefore, it would be expected that seroconversion due to exposure to natural infection (if it occurred) would have occurred substantially before recruitment for both groups and would therefore have led to similar proportions of mothers being immune/not immune in vaccinated and unvaccinated mothers at baseline, which was not found in practice.
Our study revealed a statistically significant difference in vaccine uptake by centre (lower in Leicester). This is interesting in its own right and may reflect different cultural attitudes to vaccination [Leicester City has a large, non-white population (> 35%)]. Attitudes to vaccination (in pregnancy) by cultural background therefore warrant further research. Although the difference in vaccine uptake could have led to a bias in exposure to the wild-type virus and therefore serological immune titres, Leicester City was affected more adversely during the outbreak than Nottingham and therefore would probably have had a higher background prevalence of immunity prior to enrolment. This would have had the effect of narrowing the HI titres between vaccinated and unvaccinated women and therefore would, if anything, have reduced the significance. Therefore, given that a robust significance was still determined, it is not viewed as material.
The researchers had concerns that it would not be possible to obtain sufficient cord blood to enable robust serological analysis. This, however, was not a problem in practice. Nonetheless, usual practice in assessing serological immunity is to observe a rise in immune titres. As previously commented, this was not possible. This could have created difficulties for interpreting the different results between the two groups had they not been so starkly different.
Despite extensive media promotion, take up of the A/H1N1v-specific vaccine was below that hoped for, leaving potentially vulnerable individuals at increased risk of infection, who did, in some cases, go on to experience serious morbidity and mortality. 66 It is therefore vital to be able to persuade high-risk groups to accept vaccination, even when the overall risk to the wider population seems low. Faced with a similar situation in future, the results of this study will help policy-makers, providers, individual clinicians and patients to make informed decisions based on evidence as to the merits of influenza vaccination during pregnancy and an outbreak situation. Pregnancy rightly remains a period where patients and their health-care advisors are concerned to avoid pharmaceutical interventions unless there is a strong case that the benefit to the mother and unborn child exceeds the risks. This scientific investigation shows robust evidence of benefit to the mother and, furthermore, demonstrates support for national policy that vaccination will also provide immune protection to the baby and therefore may help to persuade pregnant mothers in the future to accept both seasonal and pandemic-specific influenza vaccination.
Chapter 5 Conclusions
Our findings reveal a highly significant difference in HI titres from venous cord samples obtained from babies born to mothers vaccinated with pandemic-specific vaccine against A/H1N1v during the 2009–10 pandemic period compared with those from mothers who were not vaccinated. The subjects recruited were comparable from a baseline perspective and thus do not represent different groups that otherwise could have introduced bias into the study.
Continued circulation of 2009 A/H1N1-like viruses is uncertain but is possible as seasonal influenza in years to come. 67–70 It is possible that future seasonal waves may display increased virulence. Given the adverse outcomes experienced for a small proportion of pregnant women during the influenza pandemic of 2009–10, this study provides useful evidence to support vaccination in pregnancy to protect both the mother and baby.
Implications for health care, recommendations for research
The results of this investigation indicate definite transfer of immunity for A/H1N1v from a vaccinated mother to her unborn child, and the proportion provided with this immunity is far greater than those who have acquired natural immunity. This provides support to the policy decision during the influenza pandemic of 2009–10 to vaccinate pregnant women and for future such events. 17,37,41 Although seasonal influenza vaccination for pregnant women in the USA has been recommended for some years, this was the first time that influenza vaccination during pregnancy (seasonal or pandemic specific) had occurred in the UK. 26 The results should therefore also provide additional support to making the seasonal vaccination routine for pregnant women in the future in the UK.
The secondary component of this study – determining whether the babies of vaccinated women would experience less influenza/influenza-like illness over the long term is still ongoing and will report at a later date.
Acknowledgements
The researchers extend their thanks to the mothers and their babies who agreed to take part in this study.
We are grateful to the administrative assistance of the NIHR, the LNRREC1, the Division of Epidemiology and Public Health at Nottingham University, Trent Comprehensive Local Research network, the Nottingham University Hospitals NHS Trust, the Leicester University Hospitals NHS Trust, the University of Nottingham Research Innovation Services and the HPA at the Centre for Infections, Colindale, London.
We would like to extend particular thanks to Sheila O Malley, Carl Edwards, Nichola Goddard, Mark Van Veen, Angela Shone and Sharon Figgens.
This study was funded by the NIHR Health Technology Assessment Programme and was supported by the HPA (salary support for JVT/RP/MZ/KH).
Contributions of authors
Professor Jonathan Nguyen-Van-Tam (Professor of Health Protection) contributed to study design, data interpretation and was Chief Investigator.
Dr Richard Puleston (Associate Professor of Health Protection) contributed to study design, data collection, patient enrolment, data analysis and interpretation, as well as being responsible for project management, including coordination across sites, study logistics, data management and preparation of regulatory submissions. He drafted the study protocol and this report, which were both reviewed by all authors.
Professor Maria Zambon contributed to study design and serological analysis.
Dr Katja Hoschler provided serological analysis.
Dr George Bugg contributed to study design and participant recruitment.
Professor Justin Konje contributed to study design and participant recruitment.
Professor James Thornton contributed to study design and participant recruitment.
Dr Iain Stephenson contributed to study design and participant recruitment.
Professor Karl G. Nicholson contributed to study design.
Dr Puja Myles contributed to study design and statistical support.
Mrs Joanne Enstone provided additional project assistance.
Mrs Glenda Augustine provided additional project assistance.
Mrs Yvette Davis assisted with participant recruitment.
Mrs Sharon Figgens provided study administration support.
Disclaimers
The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the referees for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report. The views expressed in this publication are those of the authors and not necessarily those of the NIHR or the Department of Health.
References
- Nicholson KG, Snacken R, Palache AM. Influenza immunization policies in Europe and the United States. Vaccine 1995;13:365-9.
- Mak TK, Mangtani P, Leese J, Watson JM, Pfeifer D. Influenza vaccination in pregnancy: current evidence and selected national policies. Lancet Infect Dis 2008;8:44-52.
- Mullooly JP, Barker WH, Nolan TFJ. Risk of acute respiratory disease among pregnant women during influenza A epidemics. Public Health Rep 1986;101:205-11.
- Neuzil KM, Reed GW, Mitchel EF, Simonsen L, Griffith MR. Impact of influenza on acute cardiopulmonary hospitalizations in pregnant women. Am J Epidemiol 1998;148:1094-102.
- Glezen WP. Emerging infections: pandemic influenza. Epidemiol Rev 1996;18:64-76.
- Ward P, Small I, Smith J, Suter P, Dutkowski R. Oseltamivir (Tamiflu®) and its potential for use in the event of an influenza pandemic. J Antimicrob Chemother 2005;55:5-21.
- Larsen JW. Influenza and pregnancy. Clinical Obstet Gynecol 1982;25:599-604.
- Hardy JM, Azarowicz EN, Mannini A, Medearis DN, Cooke RE. The effect of Asian influenza on the outcome of pregnancy, Baltimore, 1957–1958. Am J Public Health 1961;51:1182-8.
- Sagrera X, Ginovart G, Raspall F, Rabella N, Sala P, Sierra M, et al. Outbreaks of influenza A virus infection in neonatal intensive care units. Pediatr Infect Dis J 2002;21:196-200.
- Neuzil KM, Dupont WD, Wright PF, Edwards KM. Efficacy of inactivated and cold-adapted vaccines against influenza A infection, 1985 to 1990: the pediatric experience. Pediatr Infect Dis J 2001;20:733-40.
- Jain S, Kamimoto L, Bramley AM, Schmitz AM, Benoit SR, Louie J, et al. Hospitalized Patients with 2009 H1N1 influenza in the United States, April–June 2009. N Engl J Med 2009;361:1935-44.
- Jamieson DJ, Honein MA, Rasmussen SA, Williams JL, Swerdlow DL, Biggerstaff MS, et al. H1N1 2009 influenza virus infection during pregnancy in the USA. Lancet 2009;374:451-8.
- Scriven J, Mcewen R, Mistry S, Green C, Osman H, Bailey M. Swine flu: a Birmingham experience. Clin Med 2009;9:534-8.
- Kumar A, Zarychanski R, Pinto R, Cook DJ, Marshall J, Lacroix J, et al. Critically ill patients with 2009 influenza A(H1N1) infection in Canada. JAMA 2009:302-9.
- Louie JK, Acosta M, Jamieson DJ, Honein MA. Severe 2009 H1N1 influenza in pregnant and postpartum women in California. N Engl J Med 2010;362:27-35.
- Teo SSS, Nguyen-Van-Tam JS, Booy R. Influenza burden of illness, diagnosis, treatment, and prevention: what is the evidence in children and where are the gaps?. Arch Dis Child 2005;90:532-6.
- Salisbury DM. CMO letter announcing amendment of Pandemrix® licence to allow a one-dose schedule in children. London: CMO’s Update, Department of Health (DH); 2009.
- European Medicines Agency (EMEA) . Celvapan: Summary of Product Characteristics 2009.
- European Medicines Agency (EMEA) . Pandemrix: Product Information As Approved by the CHMP on 22 April 2010, Pending Endorsement by the European Commission, Pandemrix: Summary of Product Characteristics 2008.
- Ramet J, Weil-Olivier C, Sedlak W. Influenza vaccination: the paediatric perspective. Vaccine 2007;25:780-7.
- van Essen GA, Palache AM, Forleo E, Fedson DS. Influenza vaccination in 2000: recommendations and vaccine use in 50 developed and rapidly developing countries. Vaccine 2003;21:1780-5.
- Lu PJ, Bridges CB, Euler GL, Singleton JA. Influenza vaccination of recommended adult populations, US, 1989–2005. Vaccine 2008;26:1786-93.
- Fiore AE, Shay DK, Haber P, Iskander JK, Uyeki TM, Mootrey G. Prevention and control of influenza. Recommendations of the Advisory Committee on Immunization Practices (ACIP), 2007. MMWR Recomm Rep 2007;56:1-54.
- Cook S. European agency approves swine flu vaccines for licensing. BMJ 2009;339.
- Englund JA, Mbawuike IN, Hammill H, Holleman MC, Baxter BD, Glezen WP. Maternal immunization with influenza or tetanus toxoid vaccine for passive antibody protection in young infants. J Infect Dis 1993;168:647-56.
- Sumaya CV, Gibbs RS. Immunization of pregnant women with influenza A/New Jersey/76 virus vaccine: reactogenicity and immunogenicity in mother and infant. J Infect Dis 1979;140:141-6.
- Zaman K, Roy E, Arifeen SE, Rahman M, Raqib R, Wilson E, et al. Effectiveness of maternal influenza immunization in mothers and infants. N Engl J Med 2008;359:1555-64.
- Munoz FM, Greisinger AJ, Wehmanen OA, . Safety and Effectiveness of Influenza Vaccine in Pregnant Women and Their Infants n.d.
- Bellaby P. Communication and miscommunication of risk: understanding UK parents attitudes to combined MMR vaccination. BMJ 2003;327:725-8.
- Weinstein ND, Nicolich M. Correct and incorrect interpretations of correlations between risk perceptions and risk behaviors. Health Psychol 1993;12:235-45.
- Offit PA, Coffin SE. Communicating science to the public: MMR vaccine and autism. Vaccine 2003;22:1-6.
- Bauch CT, Earn DJD. Vaccination and the theory of games. Proc Natl Acad Sci USA 2004;101:13391-4.
- Kulenkampff M, Schwartzman JS, Wilson J. Neurological complications of pertussis inoculation. Arch Dis Child 1974;49:46-9.
- Bedford H, Elliman D. Concerns about immunisation. BMJ 2000;320:240-3.
- Munoz FM, Greisinger AJ, Wehmanen OA, Mouzoon ME, Hoyle JC, Smith FA, et al. Safety of influenza vaccination during pregnancy. Am J Obstet Gynecol 2005;192:1098-106.
- Esposito S, Tremolati E, Bellasio M, Chiarelli G, Marchisio P, Tiso B, et al. Attitudes and knowledge regarding influenza vaccination among hospital health workers caring for women and children. Vaccine 2007;25:5283-9.
- Donaldson L, Beasely C, Ridge K. The H1N1 swine flu vaccination programme 2009–2010. London: Department of Health; 2009.
- World Medical Association . WMA Declaration of Helsinki – Ethical Principles for Medical Research Involving Human Subjects 2008.
- Medical Research Council (MRC) . Guidelines for Good Clinical Practice in Clinical Trials 1998.
- Allen MC, Donohue PK, Dusman AE. Racial differences in temporal changes in newborn viability and survival by gestational age. Paediatr Perinat Epidemiol 2000;14:152-8.
- Salisbury DM. H1N1 swine flu vaccination programme: information materials and vaccine schedule information. London: Department of Health; 2009.
- Lee V, Yap J, Tay JK, Barr I, Gao Q, Ho HJ. Seroconversion and asymptomatic infections during oseltamivir prophylaxis against influenza A H1N1 2009. BMC Infect Dis 2010;10.
- McCaw JM, Wood JG, McCaw CT, McVernon J. Impact of emerging antiviral drug resistance on influenza containment and spread: influence of subclinical infection and strategic use of a stockpile containing one or two drugs. PLoS ONE 2008;3.
- Hobson D, Curry RL, Beare AS, Ward-Gardner A. The role of serum haemagglutination-inhibiting antibody in protection against challenge infection with influenza A2 and B viruses. J Hyg (Lond) 1972;70:767-77.
- Miller E, Hoschler K, Hardelid P, Stanford E, Andrews N, Zambon M, et al. Incidence of 2009 pandemic influenza A H1N1 infection in England: a cross-sectional serological study. Lancet 2010;375:1100-108.
- de Jong JC, Palache AM, Beyer WEP, Rimmelzwaan GF, Boon ACM, Osterhaus ADME. Haemagglutination-inhibiting antibody to influenza virus. Dev Biol 2003;115:63-7.
- Aymard-Henry M, Coleman MT, Dowdle WR, Laver WG, Schild GC, Webster RG. Influenzavirus neuraminidase and neuraminidase-inhibition test procedures. Bull World Health Organ 1973;48:199-202.
- Frank AL, Puck J, Hughes BJ, Cate TR. Microneutralization test for influenza A and B and parainfluenza 1 and 2 viruses that uses continuous cell lines and fresh serum enhancement. J Clin Microbiol 1980;12:426-32.
- STATA/SE/11 2010.
- UK Monitoring Swine Flu Outbreak n.d. http://news.bbc.co.uk/1/hi/8018887.stm (accessed 27 September 2010).
- Pebody RG, McLean E, Zhao H, Cleary P, Bracebridge S, Foster K. Pandemic Influenza A (H1N1) 2009 and mortality in the United Kingdom: risk factors for death, April 2009 to March 2010. Eurosurveillance 2010;15.
- Health Protection Agency (HPA) . Pandemic (H1N1) 2009 in England: An Overview of Initial Epidemiological Findings and Implications for the Second Wave 2009.
- Dunning J, Openshaw PJM. Impact of the 2009 influenza pandemic. Thorax 2010;65:471-2.
- Girard MP, Tam JS, Assossou OM, Kieny MP. The 2009 A (H1N1) influenza virus pandemic: a review. Vaccine 2010;28:4895-902.
- Siston AM, Rasmussen SA, Honein MA, Fry AM, Seib K, Callaghan WM, et al. Pandemic 2009 Influenza A(H1N1) virus illness among pregnant women in the United States. JAMA 2010;303:1517-25.
- European Vaccine Manufacturers (EVM) . Influenza Pandemic Preparedness – EVM Proposal for an Action Plan to Ensure Availability of Effective Vaccines in the Event of an Influenza Pandemic 2006.
- Zarocostas J. World Health Organization declares A(H1N1) influenza pandemic. BMJ 2009;338.
- Health Protection Agency . Pandemic (HINI) 2009 in England: An Overview of Initial Epidemiological Findings and Implications for the Second Wave. Version 4 2009. www.hpa.org.uk/web/HPAwebFile/HPAweb_C/1258560552857 (accessed 22 November 2010).
- Gross PA, Russo C, Dran S, Cataruozolo P, Munk G, Lancey SC. Time to earliest peak serum antibody response to influenza vaccine in the elderly. Clin Diagn Lab Immun 1997;4:491-2.
- Rastogi S, Gross PA, Bonelli J, Dran S, Levandowski RA, Russo C. Time to peak serum antibody response to influenza vaccine. Clin Diagn Lab Immun 1995;2:120-1.
- Department of Health (DH) . In Influenza. Immunisation Against Infectious Disease ‘ The Green Book’ 2007.
- Englund J, Glezen WP, Piedra PA. Maternal immunization against viral disease. Vaccine 1993;16:1456-63.
- Reuman PD, Ayoub EM, Small PA. Protection of infants from infection with influenza A virus by transplacentally acquired antibody. J Infect Dis 1980;142:844-9.
- Englund, JA, Mbawuike IN, Hammill H, Holleman MC, Baxter BD, Glezen WP. Maternal immunization with influenza or tetanus toxoid vaccine for passive antibody protection in young infants. J Infect Dis 1993;168:647-56.
- Gill TJ, Repetti CF, Metlay LA, Rabin BS, Taylor FH, Thompson DS, et al. Transplacental immunization of the human fetus to tetanus by immunization of the mother. J Clin Invest 1983;72:987-96.
- Department of Health (DH) . Pandemic H1N1 Vaccine Uptake Figures for England by SHA and PCT 2010.
- Zimmer SM, Burke DS. Historical perspective – emergence of influenza A (H1N1) Viruses. N Engl J Med 2009;361:279-85.
- Sullivan SJ, Jacobson RM, Dowdle WR, Poland GA. 2009 H1N1 Influenza. Mayo Clin Proc 2009;85:64-76.
- FLU.GOV . Protection Against 2009 H1N1 To Be Included in 2010–2011 Seasonal Flu Vaccine 2010. www.flu.gov/news/blogs/blog20100222.html (accessed 22 November 2010).
- World Health Organization (WHO) . Recommended Viruses for Influenza Vaccines for Use in the 2010–2011 Northern Hemisphere Influenza Season 2010.
Appendix 1 Original protocol
Title: Observational study to investigate vertically acquired passive immunity in babies of mothers vaccinated against H1N1v (swine influenza) during pregnancy (draft 9, 22 September 2009)
Short title: Vertically acquired immunity in babies born to mothers vaccinated against H1N1 v.swine
Acronym: Mummy flu
Study registration: www.clinicaltrials.gov
Study sponsor: University of Nottingham
Funding source: National Institute for Health Research (NIHR)
Study/study personnel and contact details
Sponsor: University of Nottingham
Contact name: Mr Roger Brooks, Deputy Director of Research Innovation Services, Research Innovation Services, King’s Meadow Campus, Lenton Lane, Nottingham, NG7 2NR
Chief Investigator: Professor Jonathan Nguyen-Van-Tam (medical expert)
Co-investigators: Dr Richard Puleston, Associate Professor of Health Protection, University of Nottingham
Professor Jim Thornton, Professor of Obstetrics and Gynaecology, University of Nottingham

Dr George Bugg, Consultant obstetrician and Gynaecologist, Nottingham University Hospitals
Professor Karl Nicholson, Professor of Infectious Diseases, University of Leicester
Dr Iain Stephenson, Senior Lecturer in Infectious Diseases, University of Leicester
Professor Maria Zambon, Director of Centre for Infections, Health Protection Agency
Professor Justin Konje, Professor of Obstetrics and Gynaecology, University of Leicester
Mrs Joanne Enstone, Research Coordinator, University of Nottingham
Study/Study Statistician: Dr Puja Myles
Study/Study Coordinating Centre: University of Nottingham, Clinical Sciences Building, City Hospital, Nottingham, NG5 1PB
Project/Study Manager: Dr Richard Puleston, Associate Professor of Health Protection
Synopsis
| Title | Observational study to investigate vertically acquired passive immunity in babies of mothers vaccinated against H1N1v (swine influenza) during pregnancy |
|---|---|
| Acronym | Mummy flu |
| Short title | Vertically acquired immunity in babies born to mothers vaccinated against H1N1 v.swine |
| Chief Investigator | Professor Jonathan Nguyen-Van-Tam |
| Objectives |
Primary objective To determine the proportion of babies who have acquired passive immunity to A/H1N1v (swine influenza) born to mothers who accept vaccination as part of the national vaccination programme whilst pregnant (during the second and/or third trimesters) against the novel A/H1N1 swine influenza virus (exposed group), compared with unvaccinated (unexposed) mothers Secondary objective To record and investigate influenza-like illness during winter 2009–10 in the babies of mothers who take part in the study and to flag for long-term follow-up babies born to mothers who take part in the study |
| Study configuration | Observational study |
| Setting | Primary/secondary care – obstetrics and paediatrics |
| Sample size estimate | 59 vaccinated and 30 unvaccinated |
| No. of participants | 100 (additional numbers to allow for losses) |
| Eligibility criteria |
Eligible pregnant women will be normally resident in the East Midlands and will be in the second or third trimester of pregnancy and will deliver during the study period at Nottingham University Hospitals (Queen’s Medical Centre/City Hospital) or Leicester Royal Infirmary, University Hospital Leicester, and intend to remain in the region for the remainder of the research period Exposure will be defined as any pregnant woman normally resident in the East Midlands, who is in the second or third trimester of her pregnancy and is booked to, and delivers at, one of the three hospitals listed above at any time in the study period, and has been immunised as part of the national vaccination programme against H1N1 swine (one or two doses and no minimum interval between delivery and first injection) Non-exposed will be those pregnant women fitting the same criteria as cases, except not having been previously vaccinated against H1N1 swine either because they have declined it or have not yet been offered immunisation In both the vaccinated/unvaccinated, a pregnancy resulting in live or stillbirth will be eligible Prior medical conditions, medication, obstetric history – gravida and parity status, age and previous confirmed or possible H1N1 swine influenza infection will not affect eligibility but will be recorded, as will ethnicity Due to the logistical difficulties of ensuring antenatal recruitment leads to sampling at birth, women will instead be recruited at parturition. It is likely that the focus of recruitment will be on women in the later stages of pregnancy, as it is probable they will be prioritised for vaccination ahead of those earlier on and thus will form the majority of labours presenting. However, this will not preclude the participation of women who deliver early – i.e. earlier in third trimester or second trimester For the secondary objective of the study, all live born babies born to mothers enrolled in the study will be eligible |
| Description of interventions |
There will be no direct intervention to the mother. The study does not involve administering vaccine to the participants. Those exposed will be those who have already accepted immunisation as part of the national vaccination programme; the unexposed will be those who have declined or not yet been offered it The primary intervention will be obtaining a sample of cord blood at delivery for serological assessment of immune status of the baby Additional secondary interventions will be to: |
| Duration of study |
Commencement as soon as the Department of Health (DH) releases and starts vaccinating women against H1N1 swine flu – anticipated October 2009. This will continue until mid-December 2009. The further swabbing follow-up will be until 31 March 2010 (or 31 July 2010 if additional funding can be obtained). ONS flags will also be applied to the child(s) and mother Therefore, the cord blood sampling part will last approximately 2 months, the follow-up swabbing up to 5–6 months |
| Randomisation and blinding | None required or possible for recruitment. However, the samples will be blinded to the laboratory conducting the serological/PCR analysis |
| Outcome measures |
Primary efficacy variable The study will use relevant measures of seroconversion (from cord blood samples). All cord blood serology samples will be analysed by microneutralisation (MN) and haemagglutination inhibition (HI) with the NIBRG121 virus [reverse genetics virus based on A/California/7/2009 (vH1N1) and A/Puerto Rico/8/34] Secondary efficacy variable Proportion of neonates presenting with laboratory-confirmed influenza infection, as detected by validated PCR for A/H1N1v swine |
| Statistical methods |
Primary objective Analysis – descriptive:Secondary objective The statistical approaches to be used will either be: Cohort analysis involving:Alternatively, a nested case–control study (cases = influenza babies, controls = non-influenza babies) may be required, and analysed using logistic regression |
Study background information and rationale
Influenza has long been known to cause a higher level of complications (including hospitalisations) and death in particular high-risk groups, such as the elderly and those with underlying comorbidities, such as cardiopulmonary disease. Although less widely known, pregnant women also fall into this high-risk category. Evidence suggests that this effect can be seen with seasonal influenza,1,2,24–26 but is far more evident with pandemic influenza, the most notable observations arising from the 1957 A/H2N2 pandemic, during the second and third trimesters of pregnancy. 3–12 In addition, adverse effects of influenza on perinatal and early neonatal outcomes have also been observed. 2,13,14 The epidemiological profile of the emergent H1N1v swine influenza virus suggests that it is behaving differently from normal seasonal influenza, in that working age adults and children appear to be suffering higher rates of complications (including hospitalisations) than the elderly. The effect has been noted to be most pronounced in individuals with underlying comorbidities and during pregnancy where clear signals regarding the relationship between the premorbid state and influenza illness severity have already been observed, despite being based on small data sets. 15 There is increasing evidence that also suggests that young children of < 2 years of age are at greater risk of developing complications and death from influenza than at any other time in childhood, and the rate of hospitalisation in this age group (due to seasonal influenza) broadly equals that seen in working age adults with underlying high-risk conditions. 16
In relation to the present pandemic, the implications of the above to paediatric management are complicated in terms of policy and practice by the fact that both neuraminidase inhibitors [oseltamivir (Tamiflu®, Roche) and zanamivir (Relenza®, GlaxoSmithKline, GSK)] are unlicensed in children younger than 12 months and 5 years, respectively, and will need to be given off label, if at all, in children < 12 months old (zanamivir is an oral inhalational drug that would be almost impossible to administer in its marketed form). Furthermore, novel A/H1N1 vaccines under development and likely to be deployed in the UK from October 2009 onwards will not be licensed in children of this age group.
Seasonal influenza vaccination has not, until now, been routinely recommended for pregnant women in the UK. It has been in use in the USA in pregnant women in the second and third trimester for approximately 4 years because of the perceived risk–benefit profile. However, the take-up is low (published figures indicate approximately 13%), suggesting that its benefits are not widely appreciated by pregnant women or health professionals.
Vaccination against H1N1 swine is being developed currently, and preliminary stocks are likely to be delivered towards the end of August 2009, although the national vaccination programme is not anticipated to start until October. By the end of the year, the government expects that it will have taken delivery of 60 million doses of inactivated H1N1 swine influenza vaccine approximating to enough for half of the UK population, with more to follow over the ensuing 6 months for the remainder of the population.
Given the apparent predisposition of the new H1N1 swine influenza virus to cause severe illness in pregnant women, vaccination will be recommended for them and they will be prioritised for immunisation, with vaccination most likely to be offered from October 2009.
Two pharmaceutical companies have been contracted to provide vaccine – GSK and Baxter AG. Both will involve a two-dose strategy; however, the GSK vaccine will use a dose-sparing adjuvant alongside split-virion antigen, whilst the Baxter AG product is a Vero cell-grown, wild-type, whole cell product. For this reason the vaccines will not be interchangeable. It is not yet clear whether pregnant women will be prioritised to the plain Baxter AG vaccine or will also be offered the adjuvanted GSK version, or both.
The risk of influenza complications increases as pregnancy progresses, with women in the third trimester being at greater risk than in the second trimester, with the first-trimester group being at least risk. For this reason, women in the third trimester will probably be selected for vaccination first. Women in the first trimester will probably not be offered vaccination under national policy.
Effect of maternal vaccination and acquired (vertical/passive) immunity in children
Influenza vaccination in pregnancy offers benefit to the mother by reducing the risk of infection and the resultant complications. It has also been established that immunisation in pregnancy with trivalent, unadjuvanted, seasonal influenza vaccine does provide vertical immunity to the child through the cord blood. 17–19,22,24 However, the clinical impact of the finding is less clear, with some studies indicating benefit and others not. 16–19 The same is not true of a monovalent, two-dose schedule, new variant influenza vaccine with or without adjuvant, where the scientific evidence for vertical transmission of passive immunity, although likely, has not yet been established.
Rationale for the proposed study
Given the current risk profile of the emergent pandemic virus, it could be assumed that pregnant women would readily choose to be vaccinated. However, perception and response to threats and assessment of risk do not necessarily align in terms of human behaviour. Research has suggested that people tend to overestimate the likelihood and impact of rare events and underestimate for more common situations. 27,28 The public response to the measles, mumps and rubella (MMR) vaccination scare, and its possible link to autism and Crohn’s disease, is an example. Despite substantial and sound evidence to the contrary, many parents chose to refuse MMR vaccination for their children on the basis of a theoretical association that has by most scientists and policy-makers been dismissed, therefore exposing them to the risk of serious disease from measles, mumps or rubella. Another example of a similar response is that to whooping cough vaccine in the 1970s. Whether the low take-up of seasonal influenza vaccine in pregnant women in the USA is due to similar anxiety is uncertain; however, policy-makers will be concerned to ensure maximum uptake of the H1N1 swine flu vaccination. Therefore, evidence to support the approach may help encourage women to come forward for immunisation. If data were available which revealed that vaccination of pregnant women appeared to confer meaningful protection against swine influenza to their babies after birth, this would enable messages to pregnant women of potential vaccinees to be shifted from ‘evidence that you are likely to benefit and no evidence that your baby will be harmed’ to ‘evidence that you are likely to benefit and further evidence that your baby will also benefit’.
The researchers propose to assess the immunity conferred to infants of mothers who have been vaccinated against H1N1 swine influenza by obtaining umbilical cord blood samples at delivery and submitting them for serological analysis, comparing with unvaccinated mothers in the same birth cohort.
Additionally, the study also plans to assess the efficacy of vaccination of the mother at prevention influenza/respiratory illness in the infant. This will be by following the child up for a period after birth and taking nasal swabs to look for influenza/other respiratory viruses and comparing the rate of infection in those of mothers who were vaccinated versus those who were not.
Study objectives and purpose
Purpose
To improve policy implementation by providing clarity on the degree of protection transferred from the vaccinated mother to infant and to enable clinicians to provide pregnant patients with accurate information with which they can make an informed decision over whether to accept immunisation or not, and to allow public health messages to be strengthened.
FIGURE 1.
Observational study to investigate vertically acquired passive immunity in babies of mothers vaccinated against H1N1v (swine influenza) during pregnancy.
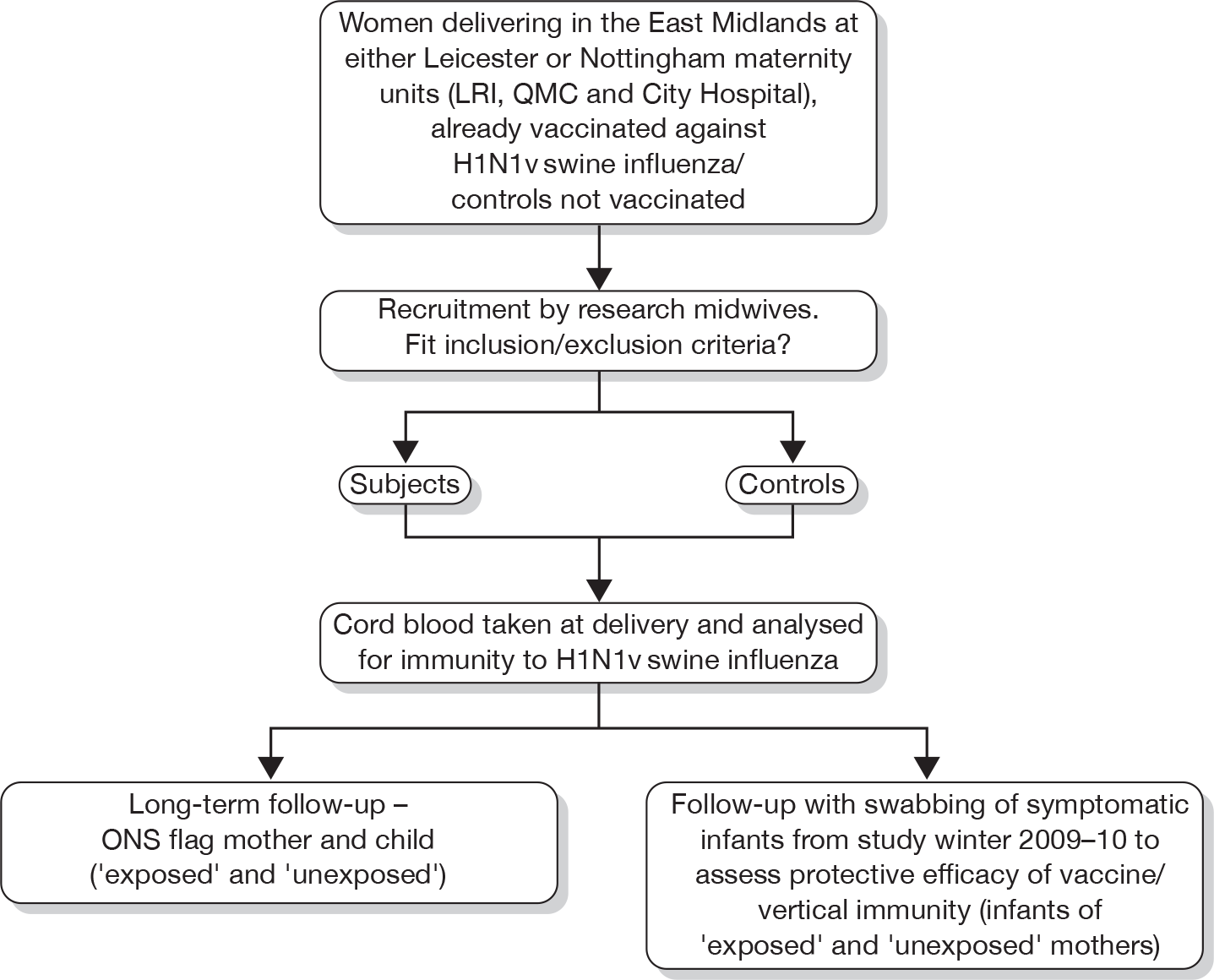
Objectives
Primary objective To determine the proportion of babies who have acquired passive immunity to A/H1N1v (swine influenza) born to mothers who accept vaccination as part of the national vaccination programme whilst pregnant (during the second and/or third trimesters) against the novel A/H1N1 swine influenza virus compared with unvaccinated mothers.
Secondary objective To record and investigate influenza-like illness during winter 2009–10 and also to flag for long-term follow-up of babies born to mothers who take part in the study.
Study design
Study configuration
Observational cohort study across three centres [Queen’s Medical Centre and City Hospital, Nottingham, and Leicester Royal Infirmary (University Hospital Leicester)].
Full randomisation is not possible within this study, as the participants will/will not have previously been vaccinated as part of the national swine influenza vaccination programme.
Recruitment will be at delivery. Although the researchers would prefer to recruit prior to delivery, they recognise that this has real practical difficulties, especially ensuring that those consented antenatally are sampled at delivery. Therefore, it is considered that recruitment will have to occur at partition. For these reasons it is probable that the majority of participants will be women at term; however, those in the earlier stages of pregnancy (second or third trimester) and presenting in premature labour will be eligible for recruitment.
For the secondary objective (protective effectiveness of the vertical immunity provided to the baby from the maternal H1N1 swine vaccine against acquisition of A/H1N1 swine), subject to the availability of additional funding, the mother will be contacted after birth by the research midwife and provided with nasal swabs to obtain samples if the child becomes symptomatic with a influenza-like illness. The mother/parents will be provided with an information sheet including diagrams on how to do this and will also be trained by demonstration in taking these. With the participant’s consent, intermittent follow-up calls will be made to the mother over the period to 31 March 2010 to reinforce and remind her about swabbing if the child becomes ill. (If further funding can be obtained this may be extended to 31 July 2010.)
With maternal consent a flag will be applied to the ONS record of the mother and child (from both the vaccinated and unvaccinated groups).
Statistical analysis will be undertaken in two stages as a minimum: firstly when all the cord samples (anticipated December 2009) have been obtained and secondly when the nasal swabbing sampling is complete (spring 2010).
Primary end point
The primary end point in the study will be the serological results of the cord blood samples for immunity to H1N1 swine and will be determined by measures of seroconversion as specified by the European Medicines Agency (EMEA) – Committee for Human Medicinal Products (CHMP). These criteria are formally accepted to be measures of seroconversion consistent with clinical protection against seasonal influenza in adults, and are used routinely for testing the immunogenicity of influenza vaccines in adults (HI titre ≥ 40 or single radial haemolysis > 25 mm2). MN titre ≥ 40 may be also used although is not part of EMEA assessment.
(The extent to which these criteria are relevant to pandemic vaccines and to babies could both be debated but at present there are no widely accepted alternative measures and these criteria are being used for the licensure of pandemic vaccines. They are therefore appropriate for this study.)
Secondary end point
Viral PCR from nasal swab samples from symptomatic babies born to the vaccinated and unvaccinated mothers. Proportion of neonates presenting with laboratory confirmed influenza infection as detected by validated PCR for A/H1N1v swine.
Safety end points
Primary outcome safety end point – as such because there is no specific intervention being undertaken to the mother or child for the assessment of vertical immunity transmission; there are no identifiable risk factors to the recruits. However, there is a safety end point to consider in relation to this outcome as follows:
-
If a major issue with the vaccine itself becomes apparent either externally or through the study then there may be a case for recruiting additional vaccinated mothers to help clarify the situation, but this would be subject to obtaining additional funds. It is important to re-emphasise here that the investigation will not be administering vaccine and that other monitoring procedures will be in place for the national vaccination programme, but it is possible the study may also uncover an issue that would warrant flagging to the appropriate authorities.
For the secondary end point (efficacy at preventing respiratory illness in babies) there will again be a possible safety end point as follows:
-
A problem with the mother taking nasal swab samples from her baby; however, this is thought unlikely because there is established precedent in other studies for using this method. 29 Another possible safety end point is if the study uncovers another health problem with the vaccinated group. Although the national programme will be monitored, it is possible that there could be effects that might be identified through the study. If this were the case then it might be appropriate to expand the study to help answer the problem.
Stopping rules and discontinuation
Discontinuation criteria will be:
-
Recruitment of sufficient subjects to meet power requirements for primary and secondary objectives.
-
Date deadlines set (31 December 2009 for primary objective and 31 March 2010 for secondary objective – unless additional funding can extend this component to 31 July 2010 to allow longer follow-up) or if the national programme of vaccination is delayed then similar time intervals would be used but shifted further back. Additionally, termination of study objective 2, will depend on current level of circulating influenza. If below the baseline activity for the time of year (sustained) this may lead to cessation; alternatively, if still higher than normal at 31 March 2010 or further outcome clarity is required then further funding may be sought to allow an extension to the investigation.
-
National vaccination programme ceases (for whatever reason), leading to a loss of sufficient vaccinated participants.
Randomisation and blinding
As such, because the exposed participants to be used in the study will be those who have already been vaccinated against H1N1 swine and the unexposed participants controls will be those who have not, it is not possible to randomise at this primary selection level. However, it will be important to avoid the introduction of bias at this stage. It is possible that those accepting vaccination (exposed) as part of the national programme may represent a different group from those who do not/who have not yet been offered it (unexposed). For this reason, data at the point of recruitment on age, ethnicity, socioeconomic status, gravida, parity and pre-existing medical history will be sought to allow further analysis/enable potential confounders to be adjusted for. This should help to reduce the effect of any biases.
For obvious reasons the subjects cannot be blinded, as they will know if they have been vaccinated or not and, therefore, likewise it will not be possible to blind the midwife recruiting/delivering the baby and taking the cord blood sample for the primary objective. Additionally for the secondary objective it will not be possible to blind the mother for nasal sampling. However, the cord blood serological analysis and nasal swab PCR determination will be blinded to the laboratory by use of unique code allocation to sampling. The results will then have to be unblinded at this point in order to allow statistical analysis. The Study Statistician will not have any direct contact will any of the participants.
Unique codes will be applied to the sample request forms and media with no other identifying data so that the laboratory will be blinded. The code will be linked to a separate participant study record that will be sealed and stored securely and will be accessible only to the study team after receipt of the result in order to allow linkage and statistical analysis. (See maintenance of codes below for further detail.)
Enrolment will be coordinated by research midwives at the respective delivery units as previously listed above, with support from the usual care teams (midwife/obstetrician).
Unblinding
Unblinding of the cord sampling/nasal swab PCR results will occur once they are received to allow linkage to the participant study record, entry into the database and subsequent statistical analysis. The researchers will statistically analyse the results as the investigation proceeds to provide interim analysis.
Study management
Given the relatively small scale of this study and the speed with which results are required, a single project manager will control the study. However, there is a Trial Steering Committee (TSC) composed of the study principle applicant and co-applicants. They will initially meet monthly or more frequently as required.
Trial Steering Committee composition
The TSC will comprise the Chief (Principal) Investigator, the co-investigators and the Study Statistician (as listed earlier). Additionally, the study group will endeavour to engage a research consultant neonatologist and general practitioner (GP) to the TSC.
Trial Steering Committee terms of reference
-
To monitor and supervise the progress of the study towards its interim and overall objectives.
-
To review at regular intervals relevant information from other sources (e.g. related studies).
-
To consider the recommendations of the Ethics Committee.
-
In the light of 1, 2 and 3 above, to inform the NIHR Board on the progress of the investigation.
-
To advise the NIHR on publicity and the presentation of all aspects of the study.
Duration of the study and participant involvement
Pregnant women will be recruited to the study in the autumn of 2009. The government has announced that the H1N1 swine flu-specific vaccination will be released in October 2009. Pregnant women will be one of the early groups to be targeted. The researchers anticipate that recruitment will commence in earnest from then. Recruited pregnant women will therefore be enrolled on the study until the end of the project (31 March 2010). The duration of participation for each pregnant woman will be up to 4–6 months.
For the secondary objective, assessing the efficacy of maternal vaccination in preventing respiratory illness in the infant, the researchers expect that the first babies born to vaccinated mothers will be in early November 2009, continuing until late December 2009. The babies will then remain enrolled on the study until 31 March 2010 – a period of up to 4–5 months.
Enrolment of the pregnant mothers will commence as soon as pregnant women start to be offered vaccination. At this time, this is assumed to be mid-October 2009 but may be subject to further delays. Enrolment will continue until sufficient subjects have been recruited or 31 December 2009, whichever is the sooner.
The overall study will close on the 31 March 2010 unless additional funding enables extension until 31 July 2010. The cord blood sampling part will close on 31 December 2009. Total study duration is expected to be approximately 6 months (unless additional funding can be found, in which case it may be extended to 31 July 2010).
End of the study
The end of the study will be date based and is to be 31 March 2009 (subject to additional funding caveat as above).
Selection and withdrawal of participants
Recruitment
The study setting will be at the antenatal clinics/obstetric units of the three hospitals listed previously (Queen’s Medical Centre and City Hospital, Nottingham, and Leicester Royal Infirmary). These are all teaching hospitals. They have been chosen for the following reasons:
-
large delivery numbers per year (in excess of 20,000 deliveries per year combined)
-
established research bases for obstetrics and influenza/infectious diseases at each site
-
close proximity to the researchers.
Clearly, this study is focusing on pregnant women and their babies, so all other groups will be excluded, i.e. men, older persons and older children. Pregnant women will be eligible to take part regardless of ethnicity or socioeconomic status.
Participants will be recruited from the delivery units of the above hospitals. The potential participant will be approached by a member of the patient’s usual care midwifery team for possible recruitment to the study. Information about the study will be on display in relevant clinical areas and advertised as widely as possible so that pregnant women are aware of the research before recruitment. If the patient is interested in partaking then the usual care midwife in the relevant unit will then go through the details of the investigation with the potential participant of all aspects pertaining to participation in the study, including obtaining consent.
All of the maternity units have experience of recruiting women to studies during labour and this has been found to be an acceptable time to obtain consent. One of the key midwifery roles in labour is to act as the woman’s advocate. The usual care midwives are therefore very well qualified to decide when or if it is appropriate to approach a woman during her labour, depending on her level of distress and the possibility of clinical complications. The maternity unit will be paid for each cord sample taken but the money will not be paid directly to the usual care midwives – it will be used to improve general staff facilities in the unit.
The researchers have considered carefully whether pregnant women should be recruited antenatally, prior to parturition. However, logistically, it will be not possible to ensure that at delivery the samples of consented mothers are taken. For this reason, it is viewed that women will have to be enrolled at delivery.
Research midwives at each site will help to facilitate participation and coordinate the practical aspects of specimen collection.
If needed, the usual hospital interpreter and translator services will be available to assist with discussion of the study. The participant information sheets and consent forms will be available printed in other languages as far as is reasonably practical. It will be explained to the potential participant that entry into the study is entirely voluntary and that her treatment and care will not be affected by her decision. It will also be explained that she can withdraw at any time. In the event of her withdrawal it will be explained that the data collected so far will be retained.
Inclusion criteria
All pregnant women in the second and third trimester will be eligible to participate in the study, if they present in labour; however, due to the short timescales required by the study commissioners to obtain results, the largest group recruited is likely to be those at term.
Women will be included regardless of age (up to 39 years 364 days), social class, ethnicity, gravida and parity status, past and current medical history (including current medications), ethnicity mode of delivery and pregnancy outcome (live/stillbirth). However, all of these parameters will be noted for each participant to allow further analysis later.
The researchers have thought carefully about whether to exclude women who had underlying health conditions that might predispose them towards being vaccinated, as it is possible that their condition may alter the development of an appropriate immune response (e.g. if on immunosuppressants/renal impairment). However, the conclusion reached was that the information from this group was particularly important and therefore should be included, but to avoid the confounder effects adjustment at analysis may be required.
Exclusion criteria
Primary objective:
The main exclusion criterion is pregnant women in the first trimester/women delivering before the age of fetal viability (23 weeks and 6 days’ gestation).
Other exclusion criteria will be:
-
incapacity to provide informed consent for participation refusal
-
prisoners
-
inability to take cord blood samples (e.g. cord blood needed for other clinical purpose, so none available for the study)
-
involvement in another study entailing clinical interventions
-
women who do not routinely live in the East Midlands
-
assisted conception
-
age > 40 years.
Secondary objective:
-
Inability to measure the outcome – this may be relevant if the woman is not normally resident in the UK or is moving abroad shortly after the child’s birth or is homeless and of no fixed abode. Compliance here may also be an issue. It may be that the possible participant may have such difficulty understanding the requirements of the secondary objective that it will not be practically possible to include her and her child.
-
Refusal by the participant to agree to both parts of the study.
Expected duration of participant participation
Mothers, up to 6 months in total, and their babies, up to 5 months, depending on date of delivery (unless funding allows extension).
Removal of participants
Participants may have to be removed for the secondary objective if they cannot be contacted; however, this would only be accepted as a loss to follow-up if two reminder letters fail to re-engage the participant.
During the enrolment period, where pregnant mothers are being recruited for cord blood sampling at delivery and subsequent follow-up of the baby, if the participant withdraws consent the researchers will seek to replace the lost individual if the number recruited to that date is insufficient to meet the power requirements of the study. After the enrolment period, if a mother subsequently declines to partake in nasal sampling then it will not be possible to replace that individual, as there will be no corresponding cord blood sample to provide comparison (exposed/unexposed to vaccine).
Abrupt termination from the study will not have safety implications for the participant.
Participants may be withdrawn from the study either at their own request or at the discretion of the Principal Investigator.
Informed consent
All participants will provide valid, written, informed consent. The Informed Consent Form will be signed and dated by the participant before she enters the study. The usual care midwife or team/research midwife will explain the details of the study and provide a Participant Information Sheet, ensuring that the participant has sufficient time to consider participating or not before obtaining consent if she wishes to partake. The midwife will answer any questions that the participant has concerning study participation. Consent will be obtained from the potential participant at the point of enrolment. Informed consent will be obtained from each participant before she undergoes any interventions (including history-taking and cord blood sampling) related to the study. One copy of this will be kept by the participant, one will be kept by the Investigator (and stored with the participant’s study record), and a third will be retained in the patient’s hospital records.
Consent will be obtained through face-to-face discussion with the potential participant.
The consent form will be kept with the case report form. Should there be a need later to amend the study (unlikely unless the study commissioner requests a subsequent modification) then each participant will be approached individually and reconsented by the research midwife or other member of the research team. This will be by written informed consent, which will again be stored in the same way as the original consent.
Explicit consent will be sought for both the primary and secondary objective participation, and the use and retention of the relevant data.
At the consent stage it will be made clear to the participant that the cord sample will be destroyed at the end of the study and will not be used for other scientific investigations. Participants will also be advised at this point that the sample will also not be available for subsequent use for clinical reasons, such as paternity determination or stem cell treatment of the baby if later required (as the sample collection and storage would not be appropriate to meet this need) and therefore if cord blood storage for this purpose was desired it would be for the participant to discuss and agree with the usual care team.
Study regimen
Vaccinated (exposed) and unvaccinated (unexposed) participants will be handled in exactly the same way throughout the investigation. The only difference between the two groups will be their H1N1 swine influenza vaccination status prior to entry into the study. Exposed women will have been vaccinated through the national programme and unexposed women will not.
At recruitment, after consent has been obtained, the midwife will take and record basic demographic details, including name, address, postcode, telephone number(s), GP and ethnicity (which will be collected by self-defined method using a standardised form). All of the identifiable demographic data will be separated from the data to be collected specific to the research. A unique identifying number will enable subsequent linkage if later necessary.
As part of the specific data required for the research, a proforma questionnaire covering relevant personal history will be completed. This will include the following:
-
estimated due date (based on scan or last menstrual period date) and actual date of delivery
-
gravida and parity status
-
previous obstetric history
-
vaccination date(s) and batch number (to identify which vaccine given – Baxter AG or GSK); this will be obtained from the patient’s handheld pregnancy record
-
previous and current medical history specifically looking for risk factors that may lead to priority for vaccination/alter immune response; this will include:
-
– cardiovascular disease
-
– respiratory disease
-
– renal disease
-
– liver disease
-
– diabetes (gestational or pre-existing)
-
– immune compromise
-
– pre-eclampsia
-
-
current prescribed medication
-
ethnicity
-
number of children in the household (in particular the number under 5 years of age) – (which may be different from the parity status, e.g. if in a multioccupancy household)
-
smokers in household.
Usually, these answers will all be obtained when the woman presents to the maternity unit at parturition. If clinical or other requirements dictate otherwise the questionnaire may have to be completed after delivery.
At delivery a cord blood sample will be obtained as per standard procedures. This will be taken by the attending midwife/obstetrician as appropriate. There will be no specific invasive intervention on the mother or child at this stage.
The mode of delivery will be noted (e.g. spontaneous vaginal or elective or emergency caesarean), as will the gestational age and outcome – live birth, complicated live birth (e.g. neonatal intensive care unit/special care baby unit admission) or stillbirth.
Depending on the length of stay of the mother and baby in hospital, the mother will be followed up by the usual care/research midwife in hospital or at home to provide training for the nasal swabbing secondary objective of the study. For example, for those mothers staying in hospital for < 24 hours it may be necessary to follow them up at home.
At consenting, the participant will be advised that they will be requested to take the nasal swab only once trained in the simple procedure (with minimal risk) of taking the swab and are comfortable with doing so. The swabs will only be for collecting a sample of mucous discharge at the exterior nares (babies and young children excrete the virus in high titre therefore sampling from inside the nose will not be necessary unlike in adults).
At the point where the training is delivered, the mother will be reminded what this component of the study is trying to establish, when swabbing will be appropriate and the packaging and delivery of the sample. The training (by demonstration) will be supplemented with an information leaflet with accompanying diagram.
To optimise compliance the mother will be regularly followed up until the end of the investigation (31 March 2010). This will be arranged as follows.
Follow-up support telephone calls:
-
during the first week after discharge from hospital/birth whichever is the sooner
-
4–6 weeks post delivery
-
10–12 weeks (and 16 and 20 weeks if born early in the study) postnatally.
(With possible further calls at 4- to 6-week intervals if the study is extended to 31 July 2010.)
At each of the follow-up points there will be a structured format to the call; this will include a general health enquiry of the mother and infant, including whether a swab has been taken in the intervening period, the baby or mother has required any medical care or assessment, medication or other treatments including routine vaccinations, a reminder of when and how to take the swab, current feeding method for baby (e.g. breast or bottle), change in the occupancy of the household (especially the under 5s) and child-care arrangements (if relevant) for the infant and how to get advice regarding the study if needed.
The mother will be informed at the outset of recruitment that the results of the cord blood sample and any nasal swabs taken will not be communicated to the participant or the GP because they will not affect the clinical management of the participant or child.
Compliance
Compliance with meeting the primary objective is unlikely to be a problem from a patient perspective. The research midwife coordinating on site will be based on the maternity unit as much as practicable in order to optimise enrolment and sample collection.
During the secondary objective phase (nasal swabbing), compliance will be promoted through the regular telephone contact of the participants and direct enquiry at that time whether the infant has had any respiratory infection in the intervening period and if so if they have been swabbed. Where there is a mismatch, the history of a respiratory infection without a swab having been taken will be noted and the mother reminded on the importance of obtaining a swab under these circumstances.
Criteria for terminating the study
The investigation may cease at any of the proposed centres if the adherence to the study protocol is unacceptable or the trust requests it. However, these are not anticipated to be likely and steps to correct problems would be identified first.
There are unlikely to be any specific safety issues with the cord blood sampling that would lead to complete study termination. (See below in the section on the transport and storage of the tissues re. the safe handling of the samples.) Likewise, this is also unlikely with the nasal swab sampling component, as there is established precedent from other studies for this method. Additionally, the mother/parents will be taught how to sample safely.
Possible external reasons for ceasing the whole investigation might include major and sustained business continuity problems at the relevant hospitals/investigating units, either as a direct consequence of the influenza pandemic or other issues. New information from another study source globally may render the need for the investigation unnecessary, unforeseen fund withdrawal and serology or PCR analysis problem may also lead to the investigation being terminated early. Additionally, if the vaccination programme were terminated this would prevent acquisition of vaccinated mothers and would necessitate ceasing the investigation. If, however, nationally, a safety issue with the vaccine were raised, it is likely that the study would need to be expanded – subject to additional funding availability.
Transport and storage of the tissues
Samples taken for the study will be transported as follows.
Cord blood samples
Cord blood samples will be collected in appropriate sample containers. These will be subsequently collected by the research midwife on each site and packaged according to usual handling standards into appropriate containers designed for the safe transport of biological specimens (safe potential biohazard sample handling). Each sample and the accompanying request form will be labelled with the participant’s unique code only. As described elsewhere in this document, the unique code can be linked to the participant’s study record to allow linkage of the results but maintain anonymisation until then. Once packaged the samples will be transported (by post) to the Health Protection Agency (HPA) Centre for Infections, London, for analysis.
Nasal swab samples
Nasal dry swab samples will be collected in appropriate sample containers. These will be subsequently packaged into appropriate containers designed for the safe transport of biological specimens by the participant (having been previously shown how to do this). Each sample and the accompanying request form will be labelled with the participant’s unique code only. As described elsewhere in this document, the unique code can be linked to the participant’s study record to allow linkage of the results but maintain anonymisation until then. The samples will be transported (by post) to the Leicester Royal Infirmary, Clinical Microbiology Department, for suspension into viral transport medium and subsequent molecular analysis.
At the conclusion of the study, the cord blood and nasal swab samples will be destroyed.
Laboratory analyses
Cord blood serology
All cord blood serology samples will be analysed (by MN and HI) with the NIBRG121 virus [reverse genetics virus based on A/California/7/2009 (vH1N1) and A/Puerto Rico/8/34]. Samples collected at each study site will be centrifuged and separated into two aliquots. Samples will be tested in parallel.
The MN will be performed in 96-well format according to previously described protocols and standard operating procedures (SOPs) developed with the Respiratory Virus Unit (RVU), HPA Centre for Infections.
Elimination of complement (e.g. from fetal calf serum in culture medium) will be achieved by incubation of study sera and appropriate quality control sera (provided and chosen according to test virus by RVU, usually serum of ferret, sheep or human, with/without neutralisation activity) at + 56 °C/30 minutes. This step will be performed simultaneously for all study samples and control sera.
The MN analysis with the NIBRG121 virus will be performed as follows: a six-step twofold dilution series (covering titres 20–640) will be set up for each of the samples and control sera. After addition of a pretitred virus [usually around 100 × median tissue culture infectious dose (TCID50) per well or 0.1-1 virus particle per cell] neutralisation will be performed by incubation of the virus/serum mixture at room temperature for 1 hour. After neutralisation, a suspension of Madin–Darby Canine Kidney (MDCK) cells will be added and the plates will be incubated for 16 hours at 37 °C in a CO2 incubator. The remaining infectivity of virus after neutralisation is determined in an EIA format using a monoclonal antibody to detect expression of viral nucleoprotein. The amount of nucleoprotein expression is determined photometrically [optical density (OD) reading = OD450] using a plate reader.
An OD reading for each dilution step for each sample will be used to calculate the titre. The titre will be reported as the reciprocal dilution at which 50% of the virus is neutralised (e.g. titre of 100). The MN analysis will be performed in duplicate (in separate runs on 2 days) for each sample. The two titres for each sample must not differ by more than a twofold serial dilution. In cases, where samples do not fall within this limit, a third analysis is performed and the two closest titres (which must be within a twofold serial dilution) will be reported.
The principle of the HI test is based on the ability of specific anti-influenza antibodies to inhibit haemagglutination of red blood cells (RBCs) by influenza virus haemagglutinin. The sera to be tested have to be previously treated to eliminate the non-specific inhibitors and the anti-species haemagglutinins. The experiment will be performed in accordance to protocols and SOPs established by RVU.
Elimination of non-specific inhibitors will be achieved by incubation of unknown serum samples and quality control sera (serum of ferret or human immunised with influenza virus) with neuraminidase [receptor-destroying enzyme (RDE) II: 18 hours/+ 36 °C followed by heat inactivation 1 hour/+ 56 °C]. All samples – sera pre-vaccination, post vaccination and controls – will be prepared simultaneously.
For the HI analysis with the NIBRG121 virus, samples and controls will be titrated in an eight-step twofold dilution series (covering titres 8–1024) and incubated with the haemagglutination antigen suspension (previously titrated to adjust the dilution at 4 haemagglutination units/25 µl; 50% end point). The haemagglutination antigen is not added to the well dedicated to the RDE quality control.
The mixture is incubated for 1 hour at room temperature and 25 µl of the 0.5% RBC suspension (turkey blood) are added. The reaction is left for half an hour at room temperature before reading.
The serum titre is equal to the highest reciprocal dilution, which induces a complete inhibition of haemagglutination. The titre of each quality control serum is close to the previously assigned value (within one serial twofold dilution limits). The RBC controls (RBC suspension without antigen) and the RDE controls do not produce any agglutination. Each serum sample is titrated in duplicate and individual titres will be reported (two for each sample). These must not differ by more than a twofold serial dilution. In cases, where samples do not fall within this limit, a third analysis is performed and the two closest titres (which must be within a twofold serial dilution) will be reported.
Nasal swab PCR
Nucleic acid extraction and real-time PCR (RT-PCR) will be performed by research staff at the Molecular Diagnostics Laboratory of the Leicester Royal Infirmary Microbiology Department, according to the UK National Standard Methods VSOP 25 (RT quadriplex PCR for the detection of influenza) and VSOP 29 [swine-lineage influenza A (H1)-specific fast RT-PCR]. Equipment available for automated nucleic acid extraction includes the Corbett Xtractor Gene and the Qiagen Qiasymphony. RT-PCR analysis will be performed using the Corbett Rotor-Gene. The Leicester Microbiology Department is under the Pathology Directorate of the University Hospitals of Leicester/Leicester Royal Infirmary. Regular participation in performance evaluations, such as those of the National External Quality Assessment Service (NEQAS) and HPA, are the SOP to maintain laboratory accreditation.
Statistics
Methods
The overall programme manager, Dr Richard Puleston, Associate Professor of Health Protection at the University of Nottingham, will be responsible for the evaluation and analysis of the findings of the study. He will be assisted in this task by the statistical expertise provided by Dr Puja Myles and other statistical experts from the same unit. stata 11 will be the primary statistical analysis software, with additional use of Microsoft excel (Microsoft Corporation, Seattle, WA, USA) and other graphing/statistical software as appropriate.
It is likely the researchers will use the following approaches.
Primary objective of study
Assessing vertical transmission of immunity to swine influenza virus in vaccinated and unvaccinated mothers using cord blood analyses:
-
Exposure variable vaccinated/unvaccinated mother (binary variable).
-
Outcome variable binary variable immune (yes/no) based on either threshold level of antibodies in cord blood; or immune status as a categorical variable with a range of values signifying no/low immunity, moderate immunity, high immunity.
Analysis – descriptive:
-
Characteristics of vaccinated and unvaccinated mothers (mean, range, standard deviation, t-test or percentage, chi-squared test).
-
Proportion of babies immune in vaccinated and unvaccinated mothers.
-
Percentage difference in immunity between offspring of vaccinated and unvaccinated mothers with 95% CI (should not include 0 to indicate a statistically significant difference in immunity) and p-values (chi-squared test).
-
For the interim analysis the researchers will compute headline figures only.
-
For the full analysis the researchers will explore the proportions and percentage differences in subgroups of vaccinated and unvaccinated mothers.
-
Subgroup analysis – among vaccinated mothers, is there a difference in vertical transmission of immunity by underlying health status? (Per cent difference.)
Secondary objective of study
Analytical investigation to assess statistical association between:
[Note: two possible exposure variables: maternal vaccination status (yes/no), immune status of child at birth (yes/no)]
-
maternal vaccination status and subsequent development of influenza-like/respiratory illness in babies
-
immune status at birth and subsequent development of influenza-like/respiratory illness in babies.
With respect to the outcome, the researchers will count only the first H1N1 swine influenza episode for each child and censor from study once a positive H1N1 swine influenza diagnosis has been obtained.
Adjustment for covariates may also be used (e.g. number of children in household).
The statistical approaches to be used will either be:
-
Cohort analysis involving:
-
– Kaplan–Meier survival curves to demonstrate differences in event (influenza-like/respiratory illness) in babies over the study period by exposure status.
-
– Cox regression analysis – ‘hazard’ or risk of developing influenza-like/respiratory illness in vaccinated group compared with unvaccinated group (hazard ratio, 95% CI and p-values).
-
-
Alternatively, a nested case–control study (cases = influenza babies, controls = non-influenza babies) may be required, and analysed using logistic regression with results expressed as odds ratio, 95% CI and p-values. This would also allow the analysis to control for other covariates of interest.
(Note: cannot compute rates or rate differences using Poisson regression because no denominators.)
These will be specified in more detail in the statistical analysis plan, which will be developed prior to the unblinding of the results of the cord blood samples. Any changes to the statistical methods used will be documented in the study report.
Interim findings will be assessed as previously set out to review efficacy of immune transfer (objective one) and protection against respiratory infection (objective two). In order to achieve this, the results available at that stage will have to be unblinded. Analysis at both interim and full investigation results will be by the same team. Because of the urgent need for results the interim findings will be made available to the main strategy group and also the DH, which may lead to changes in the conduct of the study if appropriate.
Sample size and justification
The researchers have used a conservative power calculation based upon an estimated 20% seroconversion rate in unvaccinated women; in reality it may be closer to 10% as the cumulative clinical attack rate in the UK is still very low (probably < 2%) and so the true serological attack rate is probably < 4%. In addition underlying immunity in the age group 15–44 years (women of child-bearing age) is considered to be low. We have similarly estimated conservatively that seroconversion in vaccinated women will be 50%; in fact it is more likely to be 70% after two doses. Thus a power calculation based on 20% versus 50% is very conservative, and a more optimistic comparison would be 10% versus 70%.
Based on 20% versus 50%, with 80% statistical power and 5% significance (two-tailed statistics), 38 subjects per group are required – total 76. However, we anticipate that two-thirds of women will accept vaccine, so the ratio of unvaccinated–vaccinated subjects will be 0.5; allowing for this imbalance a total study size of 89 subjects (59 vaccinated 30 unvaccinated) would be needed. Assuming a total of 89 subjects and more optimistic estimates of 10% versus 70%, the study would have 100% power to detect such a difference. To allow for possible losses during analysis or inadequate specimens, the researchers plan to recruit a maximum of 100 study subjects.
Assessment of efficacy
Primary efficacy end points will as has previously been indicated be based on the internationally recognised standard for immunity to influenza. Measures of seroconversion as specified by the EMEA–CHMP will be used. These criteria are formally accepted to be measures of seroconversion consistent with clinical protection against seasonal influenza in adults, and are used routinely for testing the immunogenicity of influenza vaccines in adults (HI titre ≥ 40 or single radial haemolysis > 25 mm2). MN tests may be used to assess responses to pandemic influenza but there are no recognised correlates of protection. Subjects achieving MN titres of ≥ 40 are may be considered as an immunogenicity end point. The extent to which these criteria are relevant to pandemic vaccines and to babies could both be debated, but at present there are no widely accepted alternative measures and these criteria are being used for the licensure of pandemic vaccines. They are therefore appropriate for this study.
Laboratory-confirmed influenza in symptomatic neonates will be measured by detection of H1N1 swine by validated PCR from clinical nasal samples.
The efficacy parameter will be the difference in means for both the primary and secondary efficacy end points.
Procedures for missing, unused and spurious data
Missing data may fall into two categories: direct results from the primary and secondary objectives or indirect supporting information, such as date of birth or GP details.
Where the former is missing, every effort will be taken to track down what might be the problem, such as sample not actually taken through to result not being communicated. Where resolvable, the data will then be collated with the other findings. Where data items appear spurious, for example an implausible serological conversion level, clarification will be sought with the testing laboratory to ascertain its veracity and, if necessary, request requantification, although the laboratory will have its own quality control procedures that should detect issues of this nature before reporting.
Where supporting data are missing the research midwives will endeavour to complete the data item either directly from the participant (e.g. number of children under 5 years in the household) or, if more appropriate (e.g. details of the delivery), from the clinical record.
Where data items remain missing despite these efforts, they will be treated as missing and will be noted as such in the analysis.
Definition of populations analysed
Interim analysis set, primary objective Analysis will be ongoing through the study. The relevance of the results as the study progresses will be based on the probability of obtaining such results for the number known.
Interim analysis set, secondary objective This will occur only if the national influenza activity on 31 January 2010 is above the normal baseline for the time of year.
Full analysis set, primary objective All individuals for whom a cord blood sample has been received at the close of recruitment and their subsequent delivery.
Full analysis set, secondary objective All infants for whom a nasal swab has been received by the termination date of the study, 31 March (July – if extended) 2010, or the end of the pandemic, whichever is the sooner.
Efficacy will be assessed on the interim and full analysis set. Ineligible participants will be excluded before recruitment. However, if a participant becomes ineligible through withdrawal of consent then the research record for that person will be marked as such and noted at the time of analysis.
Adverse events
For the purposes of the study, the researchers consider that, although unlikely, there may be the possibility of some adverse events occurring. These may include issues becoming apparent as a result of vaccination. Although administering vaccination is not part of the investigation, it is possible that, in the course of the study, issues in the vaccinated case group may become apparent such as a(n):
-
exacerbation of a pre-existing illness
-
increase in frequency or intensity of a pre-existing episodic event or condition
-
condition detected or diagnosed after vaccine administration, even though it may have been present prior to the start of the study
-
continuous persistent disease or symptoms present at baseline that worsen following the start of the study.
At this stage it is not obvious what these may be, and, as the study is not geared to be looking for these, they are unlikely to present in this way.
Issues as a result of parental swabbing of infant’s nose:
-
exacerbation of a pre-existing illness
-
increase in frequency or intensity of a pre-existing episodic event or condition
-
condition detected or diagnosed after nasal swab taking, even though it may have been present prior to the start of the study
-
continuous persistent disease or symptoms present at baseline that worsen following the start of the study.
Taking a nasal swab is unlikely to have any impact on any of these possibilities. The only conceivable problems could be causing minor trauma to the nose if done incorrectly or exacerbating respiratory upset of the child. These risks will be minimised by providing the mother/parents with careful instruction on self-sampling. Additionally, there is established precedent for this type of parental sampling in studies on infants.
A serious adverse event (SAE) is any adverse event occurring following study mandated procedures that results in any of the following outcomes:
-
death
-
a life-threatening adverse event
-
inpatient hospitalisation or prolongation of existing hospitalisation
-
a disability/incapacity.
As a direct result of the study, it is not considered that any of these will occur as the interventions are not of the nature that could cause these.
Reporting of adverse events
Participants will be asked to contact the study site immediately in the event of any actual or perceived SAE. All adverse events will be recorded and closely monitored until resolution, stabilisation or until it has been shown that the study intervention is not the cause. The Principal (Chief) Investigator shall be informed immediately of any SAEs and shall determine seriousness and causality in conjunction with any treating medical practitioners.
Participant removal from the study due to adverse events
Any participant who experiences an adverse event may be withdrawn from the study at the discretion of the Principal Investigator.
Ethical and regulatory aspects
Ethics committee and regulatory approvals
The study will not be initiated before the protocol, Informed Consent Forms and participant and general information sheets have received approval/favourable opinion from the Research Ethics Committee (REC), and the respective NHS Research and Development (R&D) department. Should a protocol amendment be made that requires REC approval, the changes in the protocol will not be instituted until the amendment and revised Informed Consent Forms and participant and GP information sheets (if appropriate) have been reviewed and received approval/favourable opinion from the REC and R&D departments. A protocol amendment intended to eliminate an apparent immediate hazard to participants may be implemented immediately, providing that R&D and the REC are notified as soon as possible and an approval is requested. Minor protocol amendments only for logistical or administrative changes may be implemented immediately, and the REC will be informed.
The study will be conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki, 1996, and the DH Research Governance Framework for Health and Social Care, 2005.
Informed consent and participant information
The process for obtaining participant informed consent or assent and parent/guardian informed consent will be in accordance with the REC guidance, and good clinical practice (GCP) and any other regulatory requirements that might be introduced. The investigator or their nominee (usual/research midwife or obstetrician) and the participant or other legally authorised representative shall both sign and date the Informed Consent Form before the person can participate in the study.
The participant will receive a copy of the signed and dated forms and the original will be retained in the Study Master File. A second copy will be filed in the participant’s medical notes, and a signed and dated note made in the notes that informed consent was obtained for the investigation.
The decision regarding participation in the study is entirely voluntary. The investigator or their nominee shall emphasise to them that consent regarding study participation may be withdrawn at any time without penalty or affecting the quality or quantity of their future medical care, or loss of benefits to which the participant is otherwise entitled. No study-specific interventions will be done before informed consent has been obtained.
The Principal Investigator will inform the participant of any relevant information that becomes available during the course of the study, and will discuss with them, whether they wish to continue with the study. If applicable they will be asked to sign revised consent forms.
If the Informed Consent Form is amended during the study, the investigator shall follow all applicable regulatory requirements pertaining to approval of the amended Informed Consent Form by the REC and use of the amended form (including for ongoing participants).
Records
Case report forms
Each participant will be assigned a unique study identity code number [one for the mother and different one(s) for the infant(s), allocated at recruitment], for use on case report forms (CRFs), other study documents including request forms and the electronic database. The documents and database will also use their initials (of first and last names separated by a hyphen or a middle name initial when available) and date of birth (dd/mm/yy).
Case report forms will be treated as confidential documents and held securely in accordance with regulations. The investigator will make a separate confidential record of the participant’s name, date of birth, local hospital number or NHS number, and unique study identity code number (the Study Recruitment Log), to permit identification of all participants enrolled in the investigation to allow results linkage.
Case report form access will be restricted to those personnel approved by the Principal Investigator and recorded on the ‘Study Delegation Log’.
All paper forms will be filled in using black ballpoint pen. Errors shall be lined out, but not obliterated, using correction fluid, and the correction inserted, initialled and dated.
The Principal Investigator shall sign a declaration ensuring accuracy of data recorded in the CRF.
Source documents
Source documents shall be filed at the investigator’s site and may include but are not limited to, consent forms, case report forms/study records and laboratory results. Only investigative staff as listed on the Study Delegation Log shall have access to study documentation other than for regulatory requirements.
Direct access to source data/documents
The CRF and all source documents, including progress notes and copies of laboratory and medical test results, shall made be available at all times for review by the Principal Investigator, Sponsor’s designee and inspection by relevant regulatory authorities.
Data protection
All study staff and investigators will endeavour to protect the rights of the study’s participants to privacy and informed consent, and will adhere to the Data Protection Act, 1998. The CRF will only collect the minimum required information for the purposes of the investigation. CRFs will be held securely, in a locked room or locked cupboard or cabinet. Access to the information will be limited to the study staff and investigators and relevant regulatory authorities (see above). Computer-held data, including the study database, will be held securely and password protected. All data will be stored on a secure dedicated server. Access will be restricted by user identifiers and passwords (encrypted using a one-way encryption method).
Information about the study in the participant’s medical records/hospital notes will be treated confidentially in the same way as all other confidential medical information.
Electronic data will be backed up every 24 hours to both local and remote media in encrypted format.
Quality assurance and audit
Insurance and indemnity
Insurance and indemnity for study participants and study staff is covered within the NHS indemnity arrangements for clinical negligence claims in the NHS, issued under cover of Health Service Guidelines (96)48. There are no special compensation arrangements, but study participants may have recourse through the NHS complaints procedures.
The University of Nottingham has taken out an insurance policy to provide indemnity in the event of a successful litigious claim for proven non-negligent harm.
Study conduct
The study conduct will be subject to systems audit of the Study Master File for inclusion of essential documents, permissions to conduct the study, Study Delegation Log, curricula vitae of investigative staff and training received, local document control procedures, consent procedures and recruitment logs, adherence to procedures defined in the protocol (e.g. inclusion/exclusion criteria, timeliness of visits) and adverse event recording and reporting.
The Study Coordinator, or, where required, a nominated designee of the Sponsor, shall carry out a site systems audit at least once during the course of the study and an audit report shall be made to the TSC.
Study data
Monitoring of study data will include confirmation of informed consent, source data verification, data storage and data transfer procedures, local quality control checks and procedures, back-up and disaster recovery of any local databases and validation of data manipulation. The Study Manager or suitable deputy, or where required, a nominated designee of the Sponsor, shall carry out monitoring of study data as an ongoing activity.
Entries on CRFs will be verified by inspection against the source data. A sample of CRFs (10%) will be checked on a regular basis for verification of all entries made. In addition the subsequent capture of the data on the study database will be checked. Where corrections are required these will carry a full audit trail and justification.
Study data and evidence of monitoring and systems audits will be made available for inspection by the regulatory authority as required.
Record retention and archiving
In compliance with the International Conference on Harmonisation (ICH)/GCP guidelines, regulations and in accordance with the University of Nottingham Research Code of Conduct, the Principal Investigator will maintain all records and documents regarding the conduct of the study. These will be retained for at least 7 years or for longer if required. If the responsible investigator is no longer able to maintain the study records, a second person will be nominated to take over this responsibility.
The Study Master File and study documents held by the Principle Investigator on behalf of the Sponsor shall be finally archived at secure archive facilities at the University of Nottingham. This archive shall include all study databases and associated meta-data encryption codes.
Discontinuation of the study by the sponsor
The Sponsor reserves the right to discontinue this study at any time for failure to meet expected enrolment goals, for safety or any other administrative reasons. The Sponsor shall take advice from the TSC as appropriate in making this decision.
Statement of confidentiality
Individual participant medical information obtained as a result of this study are considered confidential and disclosure to third parties is prohibited with the exceptions noted above.
Participant confidentiality will be further ensured by utilising unique identification code numbers in the computer files.
Such medical information may be given to the participant’s usual care team and all appropriate medical personnel responsible for the participant’s welfare.
Data generated as a result of this investigation will be available for inspection on request by the participating physicians, the University of Nottingham representatives, the REC, local R&D departments and the regulatory authorities.
Publication and dissemination policy
The results of the study are expected to be of importance in the management of the current influenza pandemic. As the primary client it is expected that the DH will be informed of the results by means of reports prior to any formal publication. These reports may be required to influence policy recommendations and therefore may be delivered at several stages to report on the primary and secondary outcomes, respectively, as the results are obtained. The results on the primary objective – vertical immunity is expected before the year end, 2009 – therefore will be made available to the DH at that time. The secondary viral swabbing follow-up of symptomatic babies will not be complete until spring 2010 and thus a report will be provided to DH then unless data suggest that an earlier statement is warranted.
It is possible that the influenza pandemic may raise considerable public disquiet again in autumn 2009 to spring 2010, including with respect to the increased risk of complications in pregnant women. Therefore, it is possible that there may be media interest in the findings of the study. The researchers/their academic institutions will therefore liaise closely with the DH/NIHR on appropriate handling of media interest.
Formal academic publication will be sought in a minimum of two stages, firstly to report on the vertical immune transmission and subsequently on the protection conferred (baby swabbing element). These will be in winter 2009/early 2010 and summer/autumn 2010, respectively.
Conferences and other professional meetings may also be utilised to disseminate the findings.
Patient confidentiality and anonymity will be maintained through any reporting/publication of the study results.
User and public involvement
Due to the tight time scales involved in obtaining results for the study commissioner, it has not been/will not be possible to engage the public in the design of this investigation.
Study finances
Funding source
This study is funded by the NIHR.
Participant stipends and payments
Participants will not be paid to participate in this study.
Signature pages
Signatories to protocol.
Chief Investigator:
(Name) ________________________________________________
Signature: ______________________________________________
Date: __________________________________________________
Co-investigator:
(Name) ________________________________________________
Signature: ______________________________________________
Date: __________________________________________________
Study Statistician:
(Name) ________________________________________________
Signature: ______________________________________________
Date: __________________________________________________
Appendix 2 Final protocol
Observational study to investigate vertically acquired passive immunity in babies of mothers vaccinated against H1N1v (swine influenza) during pregnancy (final – version 12, 13 November 2009)
Short title: Vertically acquired immunity in babies born to mothers vaccinated against H1N1 v.swine
Acronym: Mummy flu
Study registration: www.clinicaltrials.gov
Study sponsor: University of Nottingham
Funding source: National Institute for Health Research (NIHR)
Study/study personnel and contact details
Sponsor: University of Nottingham
Contact name: Mr Roger Brooks, Deputy Director of Research Innovation Services, Research Innovation Services, King’s Meadow Campus, Lenton Lane, Nottingham, NG7 2NR
Chief Investigator: Professor Jonathan Nguyen-Van-Tam (medical expert)
Co-investigators: Dr Richard Puleston, Associate Professor of Health Protection, University of Nottingham
Professor Jim Thornton, Professor of Obstetrics and Gynaecology, University of Nottingham

Dr George Bugg, Consultant obstetrician and Gynaecologist, Nottingham University Hospitals
Professor Karl Nicholson, Professor of Infectious Diseases, University of Leicester
Dr Iain Stephenson, Senior Lecturer in Infectious Diseases, University of Leicester
Professor Maria Zambon, Director of Centre for Infections, Health Protection Agency
Professor Justin Konje, Professor of Obstetrics and Gynaecology, University of Leicester
Mrs Joanne Enstone, Research Coordinator, University of Nottingham
Study/Study Statistician: Dr Puja Myles
Study/Study Coordinating Centre: University of Nottingham, Clinical Sciences Building, City Hospital, Nottingham, NG5 1PB
Project/Study Manager: Dr Richard Puleston, Associate Professor of Health Protection
Synopsis
| Title | Observational study to investigate vertically acquired passive immunity in babies of mothers vaccinated against H1N1v (swine influenza) during pregnancy |
|---|---|
| Acronym | Mummy flu |
| Short title | Vertically acquired immunity in babies born to mothers vaccinated against H1N1 v.swine |
| Chief Investigator | Professor Jonathan Nguyen-Van-Tam |
| Objectives |
Primary objective To determine the proportion of babies who have acquired passive immunity to A/H1N1v (swine influenza) born to mothers who accept vaccination as part of the national vaccination programme whilst pregnant (during the second and/or third trimesters) against the novel A/H1N1 swine influenza virus (exposed group), compared with unvaccinated (unexposed) mothers Secondary objective To record and investigate influenza-like illness during winter 2009–10 in the babies of mothers who take part in the study and to flag for long-term follow-up babies born to mothers who take part in the study |
| Study configuration | Observational study |
| Setting | Primary/secondary care – obstetrics and paediatrics |
| Sample size estimate | 59 vaccinated and 30 unvaccinated |
| No. of participants | 100 (additional numbers to allow for losses) |
| Eligibility criteria |
Eligible pregnant women will be normally resident in the East Midlands and will be in the second or third trimester of pregnancy and will deliver during the study period at Nottingham University Hospitals (Queen’s Medical Centre/City Hospital) or Leicester Royal Infirmary, University Hospital Leicester, and intend to remain in the region for the remainder of the research period Exposure will be defined as any pregnant woman normally resident in the East Midlands, who is in the second or third trimester of her pregnancy and is booked to, and delivers at, one of the three hospitals listed above at any time in the study period, and has been immunised as part of the national vaccination programme against H1N1 swine (one or two doses and no minimum interval between delivery and first injection) Non-exposed will be those pregnant women fitting the same criteria as cases, except not having been previously vaccinated against H1N1 swine either because they have declined it or have not yet been offered immunisation In both the vaccinated/unvaccinated, a pregnancy resulting in live or stillbirth will be eligible Prior medical conditions, medication, obstetric history – gravida and parity status, age and previous confirmed or possible H1N1 swine influenza infection will not affect eligibility but will be recorded, as will ethnicity Due to the logistical difficulties of ensuring antenatal recruitment leads to sampling at birth, women will instead be recruited at parturition. It is likely that the focus of recruitment will be on women in the later stages of pregnancy as it is probable they will be prioritised for vaccination ahead of those earlier on and thus will form the majority of labours presenting. However, this will not preclude the participation of women who deliver early – i.e. in earlier third trimester or second trimester For the secondary objective of the study, all live born babies born to mothers enrolled in the study will be eligible |
| Description of interventions |
There will be no direct intervention to the mother. The study does not involve administering vaccine to the participants. Those exposed will be those who have already accepted immunisation as part of the national vaccination programme, the unexposed will be those who have declined or not yet been offered it The primary intervention will be obtaining a sample of cord blood at delivery for serological assessment of immune status of the baby Additional secondary interventions will be to: |
| Duration of study |
Commencement as soon as the Department of Health (DH) releases and starts vaccinating women against H1N1 swine – anticipated October 2009. This will continue until mid-December 2009. The further swabbing follow-up will be until 31 July 2010. ONS flags will also be applied to the child(s) and mother Therefore, the cord blood sampling part will last approximately 2 months, the follow-up swabbing up to 9 months |
| Randomisation and blinding | None required or possible for recruitment. However, the samples will be blinded to the laboratory conducting the serological/PCR analysis |
| Outcome measures |
Primary efficacy variable The study will use relevant measures of seroconversion (from cord blood samples). All cord blood serology samples will be analysed by microneutralisation (MN) and haemagglutination inhibition (HI) with the NIBRG121 virus (reverse genetics virus based on A/California/7/2009 (vH1N1) and A/Puerto Rico/8/34) Secondary efficacy variable Proportion of neonates presenting with laboratory-confirmed influenza infection as detected by validated PCR for A/H1N1v swine |
| Statistical methods |
Primary objective Analysis – descriptive:Secondary objective The statistical approaches to be used will either be Cohort analysis involving:Alternatively, a nested case–control study (cases = influenza babies, controls = non-influenza babies) may be required, and analysed using logistic regression |
Study background information and rationale
Influenza has long been known to cause a higher level of complications (including hospitalisations) and death in particular high-risk groups, such as the elderly and those with underlying comorbidities, such as cardiopulmonary disease. Although less widely known, pregnant women also fall into this high-risk category. Evidence suggests that this effect can be seen with seasonal influenza1,2,24–26 but is far more evident with pandemic influenza, the most notable observations arising from the 1957 A/H2N2 pandemic, during the second and third trimesters of pregnancy. 3–12 In addition, adverse effects of influenza on perinatal and early neonatal outcomes have also been observed. 2,13,14 The epidemiological profile of the emergent H1N1v swine influenza virus suggests that it is behaving differently from normal seasonal influenza in that working age adults and children appear to be suffering higher rates of complications (including hospitalisations) than the elderly. The effect has been noted to be most pronounced in individuals with underlying comorbidities and during pregnancy where clear signals regarding the relationship between the premorbid state and influenza illness severity have already been observed, despite being based on small data sets. 15 There is increasing evidence that also suggests that young children of < 2 years of age are at greater risk of developing complications and death from influenza than at any other time in childhood, and the rate of hospitalisation in this age group (due to seasonal influenza) broadly equals that seen in working age adults with underlying high-risk conditions. 16
In relation to the present pandemic, the implications of the above to paediatric management are complicated in terms of policy and practice by the fact that both neuraminidase inhibitors [oseltamivir (Tamiflu®, Roche) and zanamivir (Relenza®, GlaxoSmithKline, GSK)] are unlicensed in children younger than 12 months and 5 years, respectively, and will need to be given off label, if at all, in children < 12 months old (zanamivir is an oral inhalational drug that would be almost impossible to administer in its marketed form). Furthermore, novel A/H1N1 vaccines under development and likely to be deployed in the UK from October 2009 onwards will not be licensed in children of this age group.
Seasonal influenza vaccination has not until now been routinely recommended for pregnant women in the UK. It has been in use in the USA in pregnant women in the second and third trimester for approximately 4 years because of the perceived risk–benefit profile. However, the take-up is low (published figures indicate approximately 13%), suggesting that its benefits are not widely appreciated by pregnant women or health professionals.
Vaccination against H1N1 swine is being developed currently and preliminary stocks are likely to be delivered towards the end of August 2009, although the national vaccination programme is not anticipated to start until October. By the end of the year, the government expects that it will have taken delivery of 60 million doses of inactivated H1N1 swine influenza vaccine, approximating to enough for half of the UK population, with more to follow over the ensuing 6 months for the remainder of the population.
Given the apparent predisposition of the new H1N1 swine influenza virus to cause severe illness in pregnant women, vaccination will be recommended for them and they will be prioritised for immunisation, with vaccination most likely to be offered from October 2009.
Two pharmaceutical companies have been contracted to provide vaccine – GSK and Baxter AG. Both will involve a two-dose strategy; however, the GSK vaccine will use a dose-sparing adjuvant alongside split-virion antigen, whilst the Baxter AG product is a Vero-cell grown, wild-type, whole cell product. For this reason the vaccines will not be interchangeable. It is not yet clear whether pregnant women will be prioritised to the plain Baxter AG vaccine or will also be offered the adjuvanted GSK version or both.
The risk of influenza complications increases as pregnancy progresses, with women in the third trimester being at greater risk than in the second trimester, with the first trimester group being at least risk. For this reason, women in the third trimester will probably be selected for vaccination first. Women in the first trimester will probably not be offered vaccination under national policy.
Effect of maternal vaccination and acquired (vertical/passive) immunity in children
Influenza vaccination in pregnancy offers benefit to the mother by reducing the risk of infection and the resultant complications. It has also been established that immunisation in pregnancy with trivalent, unadjuvanted, seasonal influenza vaccine does provide vertical immunity to the child through the cord blood. 17–19,22,24 However, the clinical impact of the finding is less clear, with some studies indicating benefit and others not. 16–19 The same is not true of a monovalent, two-dose schedule, new variant influenza vaccine with or without adjuvant, where the scientific evidence for vertical transmission of passive immunity, although likely, has not yet been established.
Rationale for the proposed study
Given the current risk profile of the emergent pandemic virus, it could be assumed that pregnant women would readily choose to be vaccinated. However, perception and response to threats and assessment of risk do not necessarily align in terms of human behaviour. Research has suggested that people tend to overestimate the likelihood and impact of rare events and underestimate for more common situations. 27,28 The public response to the measles, mumps and rubella (MMR) vaccination scare and its possible link to autism and Crohn’s disease is an example. Despite substantial and sound evidence to the contrary, many parents chose to refuse MMR vaccination for their children on the basis of a theoretical association that has by most scientists and policy-makers been dismissed, therefore exposing them to the risk of serious disease from measles, mumps or rubella. Another example of a similar response is that to whooping cough vaccine in the 1970s. Whether the low take-up of seasonal influenza vaccine in pregnant women in America is due to similar anxiety is uncertain; however, policy-makers will be concerned to ensure maximum uptake of the H1N1 swine flu vaccination. Therefore, evidence to support the approach may help encourage women to come forward for immunisation. If data were available which revealed that vaccination of pregnant women appeared to confer meaningful protection against swine influenza to their babies after birth, this would enable messages to pregnant women of potential vaccinees to be shifted from ‘evidence that you are likely to benefit and no evidence that your baby will be harmed’ to ‘evidence that you are likely to benefit and further evidence that your baby will also benefit’.
The researchers propose to assess the immunity conferred to infants of mothers who have been vaccinated against H1N1 swine influenza by obtaining umbilical cord blood samples at delivery and submitting them for serological analysis, comparing with unvaccinated mothers in the same birth cohort.
Additionally, the study also plans to assess the efficacy of vaccination of the mother at prevention influenza/respiratory illness in the infant. This will be by following the child up for a period after birth and taking nasal swabs to look for influenza/other respiratory viruses and comparing the rate of infection in those of mothers who were vaccinated versus those who were not.
Study objectives and purpose
Purpose
To improve policy implementation by providing clarity on the degree of protection transferred from the vaccinated mother to infant and to enable clinicians to provide pregnant patients with accurate information with which they can make an informed decision over whether to accept immunisation or not, and to allow public health messages to be strengthened.
FIGURE 1.
Observational study to investigate vertically acquired passive immunity in babies of mothers vaccinated against H1N1v (swine influenza) during pregnancy.
Objectives
Primary objective To determine the proportion of babies who have acquired passive immunity to A/H1N1v (swine influenza) born to mothers who accept vaccination as part of the national vaccination programme whilst pregnant (during the second and/or third trimesters) against the novel A/H1N1 swine influenza virus (exposed group), compared with unvaccinated (unexposed) mothers.
Secondary objective To record and investigate influenza-like illness during winter 2009–10 and also to flag for long-term follow-up of babies born to mothers who take part in the study.
Study design
Study configuration
Observational cohort study across three centres [Queen’s Medical Centre and City Hospital, Nottingham, and Leicester Royal Infirmary (University Hospital Leicester)].
Full randomisation is not possible within this study, as the participants will have previously been vaccinated (exposed group) as part of the national swine influenza vaccination programme or not (in the case of the unexposed).
Recruitment will be at delivery. Although the researchers would prefer to recruit prior to delivery, they recognise that this has real practical difficulties, especially ensuring that those consented antenatally are sampled at delivery. Therefore, it is considered that recruitment will have to occur at parturition. For these reasons it is probable that the majority of participants will be women at term; however, those in the earlier stages of pregnancy (second or third trimester) and presenting in premature labour will be eligible for recruitment.
For the secondary objective (protective effectiveness of the vertical immunity provided to the baby from the maternal H1N1 swine vaccine against acquisition of A/H1N1 swine), the mother will be contacted after birth by the research midwife and provided with nasal swabs to obtain samples if the child becomes symptomatic with a influenza-like illness. The mother/parents will be provided with an information sheet including diagrams on how to do this and will also be trained by demonstration in taking these. With the participant’s consent, intermittent follow-up calls will be made to the mother over the period to 31 July 2010 to reinforce and remind her about swabbing if the child becomes ill.
With maternal consent a flag will be applied to the ONS record of the mother and child (from both the vaccinated and unvaccinated groups).
Statistical analysis will be undertaken in two stages as a minimum – firstly when all the cord samples (anticipated December 2009) have been obtained and secondly when the nasal swabbing sampling is complete (spring 2010).
Primary end point
The primary end point in the study will be the serological results of the cord blood samples for immunity to H1N1 swine and will be determined by measures of seroconversion as specified by the European Medicines Agency (EMEA) – Committee for Human Medicinal Products (CHMP). These criteria are formally accepted to be measures of seroconversion consistent with clinical protection against seasonal influenza in adults, and are used routinely for testing the immunogenicity of influenza vaccines in adults (HI titre ≥ 40 or single radial haemolysis > 25 mm2). MN titre ≥ 40 may be also used although is not part of EMEA assessment.
(The extent to which these criteria are relevant to pandemic vaccines and to babies could both be debated but at present there are no widely accepted alternative measures and these criteria are being used for the licensure of pandemic vaccines. They are therefore appropriate for this study.)
Secondary end point
Viral PCR from nasal swab samples from symptomatic babies born to the vaccinated and unvaccinated mothers. Proportion of neonates presenting with laboratory-confirmed influenza infection as detected by validated PCR for A/H1N1v swine.
Safety end points
Primary outcome safety end point: as such because there is no specific intervention being undertaken to the mother or child for the assessment of vertical immunity transmission, there are no identifiable risk factors to the recruits. However, there is a safety end point to consider in relation to this outcome as follows.
-
If a major issue with the vaccine itself becomes apparent either externally or through the study then there may be a case for recruiting additional vaccinated mothers to help clarify the situation, but this would be subject to obtaining additional funds. It is important to re-emphasise here that the investigation will not be administering vaccine and that other monitoring procedures will be in place for the national vaccination programme, but it is possible the study may also uncover an issue that would warrant flagging to the appropriate authorities.
For the secondary end point (efficacy at preventing respiratory illness in babies) there will again be a possible safety end point as follows:
-
A problem with the mother taking nasal swab samples from her baby, however this is thought unlikely because there is established precedent in other studies for using this method. 29 Another possible safety end point is if the study uncovers another health problem with the vaccinated group. Although the national programme will be monitored, it is possible that there could be effects that might be identified through the study. If this were the case, then it might be appropriate to expand the study to help answer the problem.
Stopping rules and discontinuation
Discontinuation criteria will be:
-
Recruitment of sufficient subjects to meet power requirements for primary and secondary objectives.
-
Date deadlines set – 31 December 2009 for primary objective (subject to full recruitment which if incomplete may necessitate continuing recruitment into early 2010) and 31 July 2010 for secondary objective or if the national programme of vaccination is delayed then similar time intervals would be used but shifted further back. Additionally, termination of study objective 2 will depend on current level of circulating influenza. If below the baseline activity for the time of year (sustained), this may lead to cessation; alternatively, if still higher than normal at 31 March 2010 or further outcome clarity is required then further funding may be sought to allow an extension to the investigation.
-
National vaccination programme ceases (for whatever reason) leading to a loss of sufficient vaccinated participants.
Randomisation and blinding
As such, because the ‘exposed’ participants to be used in the study will be those who have already been vaccinated against H1N1 swine and the ‘unexposed’ participants controls will be those who have not, it is not possible to randomise at this primary selection level. However, it will be important to avoid the introduction of bias at this stage. It is possible that those accepting vaccination (exposed) as part of the national programme may represent a different group from those who do not/who have not yet been offered it (unexposed). For this reason, data at the point of recruitment on age, ethnicity, socioeconomic status, gravida, parity and pre-existing medical history will be sought to allow further analysis/enable potential confounders to be adjusted for. This should help to reduce the effect of any biases.
For obvious reasons the subjects cannot be blinded as they will know if they have been vaccinated or not and therefore likewise it will not be possible to blind the midwife recruiting/delivering the baby and taking the cord blood sample for the primary objective. Additionally for the secondary objective it will not be possible to blind the mother for nasal sampling. However, the cord blood serological analysis and nasal swab PCR determination will be blinded to the laboratory by use of unique code allocation to sampling. The results will then have to be unblinded at this point in order to allow statistical analysis. The Study Statistician will not have any direct contact will any of the participants.
Unique codes will be applied to the sample request forms and media with no other identifying data so that the laboratory will be blinded. The code will be linked to a separate participant study record that will be sealed and stored securely, and will be accessible to the study team only after receipt of the result in order to allow linkage and statistical analysis. (See maintenance of codes below for further detail.)
Enrolment will be coordinated by research midwives at the respective delivery units as previously listed above, with support from the usual care teams (midwife/obstetrician).
Unblinding
Unblinding of the cord sampling/nasal swab PCR results will occur once they are received to allow linkage to the participant study record, entry into the database and subsequent statistical analysis. The researchers will statistically analyse the results as the investigation proceeds to provide interim analysis.
Study management
Given the relatively small scale of this study and the speed with which results are required, a single project manager will control the study. However, there is a Trial Steering Committee (TSC) composed of the study principle applicant and co-applicants. They will initially meet monthly or more frequently as required.
Trial Steering Committee composition
The TSC will comprise the Chief (Principal) Investigator, the co-investigators and the Study Statistician (as listed earlier). Additionally, the study group will endeavour to engage a research consultant neonatologist and general practitioner (GP) to the TSC.
Trial Steering Committee terms of reference
-
To monitor and supervise the progress of the study towards its interim and overall objectives.
-
To review at regular intervals relevant information from other sources (e.g. related studies).
-
To consider the recommendations of the Ethics Committee.
-
In the light of 1, 2 and 3 above, to inform the NIHR Board and GSK on the progress of the investigation.
-
To advise the NIHR/GSK on publicity and the presentation of all aspects of the study.
Duration of the study and participant involvement
Pregnant women will be recruited to the study in the autumn of 2009. The government has announced that the H1N1 swine flu specific vaccination will be released in October 2009. Pregnant women will be one of the early groups to be targeted. The researchers anticipate that recruitment will commence in earnest from then. Recruited pregnant women will therefore be enrolled on the study until the end of the project (31 July 2010). The duration of participation for each pregnant woman will be up to 7–9 months.
For the secondary objective, assessing the efficacy of maternal vaccination in preventing respiratory illness in the infant, the researchers expect that the first babies born to vaccinated mothers will be in early November 2009, continuing until late December 2009. The babies will then remain enrolled on the study until 31 July 2010 – a period of up to 9 months.
Enrolment of the pregnant mothers will commence as soon as pregnant women start to be offered vaccination. At this time, this is assumed to be mid-October 2009, but may be subject to further delays, enrolment will continue until sufficient subjects have been recruited.
The overall study will close on 31 July 2010. The cord blood sampling part will close on 31 December 2009, provided that sufficient participants have been recruited by that date. If not recruitment may need to continue until early 2010. Total study duration is expected to be approximately 9 months.
End of the study
The end of the study will be date based and is to be 31 July 2010.
Selection and withdrawal of participants
Recruitment
The study setting will be at the antenatal clinics/obstetric units of the three hospitals listed previously (Queen’s Medical Centre and City Hospital, Nottingham, and Leicester Royal Infirmary). These are all teaching hospitals. They have been chosen for the following reasons:
-
large delivery numbers per year (in excess of 20,000 deliveries per year combined)
-
established research bases for obstetrics and influenza/infectious diseases at each site
-
close proximity to the researchers.
Clearly, this study is focusing on pregnant women and their babies, so all other groups will be excluded, i.e. men, older persons and older children. Pregnant women will be eligible to take part regardless of ethnicity or socioeconomic status.
Participants will be recruited from the delivery units of the above hospitals. The potential participant will be approached by a member of the patient’s usual care midwifery team for possible recruitment to the study. Information about the study will be on display in relevant clinical areas and advertised as widely as possible, so that pregnant women are aware of the research before recruitment. If the patient is interested in partaking then the usual care midwife in the relevant unit will then go through the details of the investigation with the potential participant of all aspects pertaining to participation in the study, including obtaining consent.
All of the maternity units have experience of recruiting women to studies during labour and this has been found to be an acceptable time to obtain consent. One of the key midwifery roles in labour is to act as the woman’s advocate. The usual care midwives are therefore very well qualified to decide when or if it is appropriate to approach a woman during her labour, depending on her level of distress and the possibility of clinical complications. The maternity unit will be paid for each cord sample taken but the money will not be paid directly to the usual care midwives – it will be used to improve general staff facilities in the unit.
The researchers have considered carefully whether pregnant women should be recruited antenatally, prior to parturition. However, logistically, it will be not possible to ensure that at delivery the samples of consented mothers are taken. For this reason, it is viewed that women will have to be enrolled at delivery.
Research midwives at each site will help to facilitate participation and coordinate the practical aspects of specimen collection.
If needed, the usual hospital interpreter and translator services will be available to assist with discussion of the study. The participant information sheets and consent forms will be available printed in other languages as far as is reasonably practical. It will be explained to the potential participant that entry into the study is entirely voluntary and that her treatment and care will not be affected by her decision. It will also be explained that she can withdraw at any time. In the event of her withdrawal it will be explained that the data collected so far will be retained.
Inclusion criteria
All pregnant women in the second and third trimester will be eligible to participate in the study if they present in labour; however, due to the short time scales required by the study commissioners to obtain results, the largest group recruited is likely to be those at term.
Women will be included regardless of age, social class, ethnicity, gravida and parity status, past and current medical history (including current medications), ethnicity mode of delivery and pregnancy outcome (live/stillbirth). However, all of these parameters will be noted for each participant to allow further analysis later.
The researchers have thought carefully about whether to exclude women who had underlying health conditions that might predispose them towards being vaccinated, as it is possible that their condition may alter the development of an appropriate immune response (e.g. if on immunosuppressants/renal impairment). However, the conclusion reached was that the information from this group was particularly important and therefore should be included, but to avoid the confounder effects adjustment at analysis may be required.
Exclusion criteria
Primary objective:
The main exclusion criterion is pregnant women in the first trimester/women delivering before the age of fetal viability (23 weeks and 6 days’ gestation).
Other exclusion criteria will be:
-
incapacity to provide informed consent for participation refusal
-
prisoners
-
inability to take cord blood samples (e.g. cord blood needed for other clinical purpose, so none available for the study)
-
involvement in another study entailing clinical interventions
-
women who do not routinely live in the East Midlands.
Secondary objective:
-
Inability to measure the outcome – this may be relevant if the woman is not normally resident in the UK or is moving abroad shortly after the child’s birth or is homeless and of no fixed abode. Compliance here may also be an issue. It may be that the possible participant may have such difficulty understanding the requirements of the secondary objective that it will not be practically possible to include her and her child.
-
Refusal by the participant to agree to both parts of the study.
Expected duration of participant participation
Mothers up to 6 months in total, and their babies up to 5 months, depending on date of delivery (unless funding allows extension).
Removal of participants
Participants may have to be removed for the secondary objective if they cannot be contacted; however, this would only be accepted as a loss to follow-up if two reminder letters fail to re-engage the participant.
During the enrolment period, where pregnant mothers are being recruited for cord blood sampling at delivery and subsequent follow-up of the baby, if the participant withdraws consent, the researchers will seek to replace the lost individual if the number recruited to that date is insufficient to meet the power requirements of the study. After the enrolment period, if a mother subsequently declines to partake in nasal sampling then it will not be possible to replace that individual, as there will be no corresponding cord blood sample to provide comparison (exposed/unexposed to vaccine).
Abrupt termination from the study will not have safety implications to the participant.
Participants may be withdrawn from the study either at their own request or at the discretion of the Principal Investigator.
Informed consent
All participants will provide valid, written, informed consent. The Informed Consent Form will be signed and dated by the participant before they enter the study. The usual care midwife or team/research midwife will explain the details of the study and provide a Participant Information Sheet, ensuring that the participant has sufficient time to consider participating or not before obtaining consent if she wishes to partake. The midwife will answer any questions that the participant has concerning study participation. Consent will be obtained from the potential participant at the point of enrolment. Informed consent will be obtained from each participant before they undergo any interventions (including history-taking and cord blood sampling) related to the study. One copy of this will be kept by the participant, one will be kept by the Investigator (and stored with the participant’s study record), and a third will be retained in the patient’s hospital records.
Consent will be obtained through face-to face discussion with the potential participant.
The consent form will be kept with the case report form. Should there be a need later to amend the study (unlikely unless the study commissioner requests a subsequent modification) then each participant will be approached individually and reconsented by the research midwife or other member of the research team. This will be by written informed consent, which will again be stored in the same way as the original consent.
Explicit consent will be sought for both the primary and secondary objective participation, and the use and retention of the relevant data.
At the consent stage it will be made clear to the participant that the cord sample/nasal swab samples will, at the end of the study, be transferred to the University of Nottingham study bank, where they will be stored anonymously for use in other research, but that there will be no possible linkage to the individual participant. Participants will also be advised at this point that the sample will also not be available for subsequent use for clinical reasons, such as paternity determination or stem cell treatment of the baby if later required (as the sample collection and storage would not be appropriate to meet this need) and therefore if cord blood storage for this purpose was desired it would be for the participant to discuss and agree with the usual care team.
Study regimen
Vaccinated (exposed) and unvaccinated (unexposed) participants will be handled in exactly the same way throughout the investigation. The only difference between the two groups will be their H1N1 swine influenza vaccination status prior to entry into the study. Exposed women will have been vaccinated through the national programme and unexposed women will not.
At recruitment, after consent has been obtained the midwife will take and record basic demographic details, including name, address, postcode, telephone number(s), GP and ethnicity (which will be collected by self-defined method using a standardised form). All of the identifiable demographic data will be separated from the data to be collected specific to the research. A unique identifying number will enable subsequent linkage if later necessary.
As part of the specific data required for the research a proforma questionnaire covering relevant personal history will be completed. This will include the following:
-
estimated due date (based on scan or last menstrual period date) and actual date of delivery
-
gravida and parity status
-
previous obstetric history
-
vaccination date(s) and batch number (to identify which vaccine given – Baxter AG or GSK); this will be obtained from the patient’s handheld pregnancy record
-
previous and current medical history specifically looking for risk factors that may lead to priority for vaccination/alter immune response; this will include:
-
– cardiovascular disease
-
– respiratory disease
-
– renal disease
-
– liver disease
-
– diabetes (gestational or pre-existing)
-
– immune compromise
-
– pre-eclampsia
-
-
current prescribed medication
-
ethnicity
-
number of children in the household (in particular the number under 5 years of age) – (which may be different from the parity status, e.g. if in a multi-occupancy household)
-
smokers in household.
Usually, these answers will all be obtained when the woman presents to the maternity unit at parturition. If clinical or other requirements dictate otherwise, the questionnaire may have to be completed after delivery.
At delivery a cord blood sample will be obtained as per standard procedures. This will be taken by the attending midwife/obstetrician as appropriate. There will be no specific invasive intervention on the mother or child at this stage.
The mode of delivery will be noted (e.g. spontaneous vaginal or elective or emergency caesarean), as will the gestational age and outcome – live birth, complicated live birth (e.g. neonatal intensive care unit/special care baby unit admission) or stillbirth.
Depending on the length of stay of the mother and baby in hospital, the mother will be followed up by the usual care/research midwife in hospital or at home to provide training for the nasal swabbing secondary objective of the study. For example, for those mothers staying in hospital for < 24 hours, it may be necessary to follow them up at home.
At consenting, the participant will be advised that they will be requested to take the nasal swab only once trained in the simple procedure (with minimal risk) of taking the swab and are comfortable with doing so. The swabs will only be for collecting a sample of mucous discharge at the exterior nares (babies and young children excrete the virus in high titre, therefore sampling from inside the nose will not be necessary unlike in adults).
At the point where the training is delivered, the mother will be reminded what this component of the study is trying to establish, when swabbing will be appropriate and the packaging and delivery of the sample. The training (by demonstration) will be supplemented with an information leaflet with accompanying diagram.
To optimise compliance the mother will be regularly followed up until the end of the investigation (31 July 2010). This will be arranged as follows.
Follow-up support telephone calls:
-
during the first week after discharge from hospital/birth, whichever is the sooner
-
4–6 weeks post delivery
-
10–12 weeks (and 16 and 20 weeks if born early in the study) postnatally.
(With further calls at 4- to 6-week intervals with the study being extended to 31 July 2010.)
At each of the follow-up points there will be a structured format to the call: this will include a general health enquiry of the mother and infant, including whether a swab has been taken in the intervening period, the baby or mother has required any medical care or assessment, medication or other treatments including routine vaccinations, a reminder of when and how to take the swab, current feeding method for baby (e.g. breast or bottle), change in the occupancy of the household (especially the under 5-year-olds) and child-care arrangements (if relevant) for the infant and how to get advice regarding the study if needed.
The mother will be informed at the outset of recruitment that the results of the cord blood sample and any nasal swabs taken will not be communicated to the participant or the GP because they will not affect the clinical management of the participant or child.
Compliance
Compliance with meeting the primary objective is unlikely to be a problem from a patient perspective. The research midwife coordinating on site will be based on the maternity unit as much as practicable in order to optimise enrolment and sample collection.
During the secondary objective phase (nasal swabbing), compliance will be promoted through the regular telephone contact of the participants and direct enquiry at that time whether the infant has had any respiratory infection in the intervening period and if so if they have been swabbed. Where there is a mismatch, the history of a respiratory infection without a swab having been taken will be noted and the mother reminded on the importance of obtaining a swab under these circumstances.
Criteria for terminating the study
The investigation may cease at any of the proposed centres if the adherence to the study protocol is unacceptable or the trust requests it. However, these are not anticipated to be likely and steps to correct problems would be identified first.
There are unlikely to be any specific safety issues with the cord blood sampling that would lead to complete study termination. (See below in the section on the transport and storage of the tissues re. the safe handling of the samples.) Likewise, this is also unlikely with the nasal swab sampling component, as there is established precedent from other studies for this method. Additionally, the mother/parents will be taught how to sample safely.
Possible external reasons for ceasing the whole investigation might include major and sustained business continuity problems at the relevant hospitals/investigating units, either as a direct consequence of the influenza pandemic or other issues. New information from another study source globally may render the need for the investigation unnecessary, unforeseen fund withdrawal and serology or PCR analysis problem may also lead to the investigation being terminated early. Additionally, if the vaccination programme were terminated this would prevent acquisition of vaccinated mothers and would necessitate ceasing the investigation. If, however, nationally, a safety issue with the vaccine were raised, it is likely that the study would need to be expanded – subject to additional funding availability.
Transport and storage of the tissues
Samples taken for the study will be transported as follows.
Cord blood samples
Cord blood samples will be collected in appropriate sample containers. These will be subsequently collected by the research midwife on each site and packaged according to usual handling standards into appropriate containers designed for the safe transport of biological specimens (safe potential biohazard sample handling). Each sample and the accompanying request form will be labelled with the participant’s unique code only. As described elsewhere in this document, the unique code can be linked to the participant’s study record to allow linkage of the results but maintain anonymisation until then. Once packaged the samples will be transported (by post) to the Health Protection Agency (HPA) Centre for Infections, London, for analysis.
Nasal swab samples
Nasal dry swab samples will be collected in appropriate sample containers. These will be subsequently packaged into appropriate containers designed for the safe transport of biological specimens by the participant (having been previously shown how to do this). Each sample and the accompanying request form will be labelled with the participant’s unique code only. As described elsewhere in this document, the unique code can be linked to the participant’s study record to allow linkage of the results but maintain anonymisation until then. The samples will be transported (by post) to the HPA or Leicester Royal Infirmary, Clinical Microbiology Department, for suspension into viral transport medium and subsequent molecular analysis.
At the conclusion of the study, the cord blood and nasal swab samples will be transferred to the University of Nottingham Tissue Bank for anonymous storage for other researchers to use.
Laboratory analyses
Cord blood serology
All cord blood serology samples will be analysed by MN and HI with the NIBRG121 virus [reverse genetics virus based on A/California/7/2009 (vH1N1) and A/Puerto Rico/8/34]. Samples collected at each study site will be centrifuged and separated into two aliquots. Samples will be tested in parallel.
The MN will be performed in 96-well format according to previously described protocols and standard operating procedures (SOPs) developed with the Respiratory Virus Unit (RVU), HPA Centre for Infections.
Elimination of complement (e.g. from fetal calf serum in culture medium) will be achieved by incubation of study sera and appropriate quality control sera (provided and chosen according to test virus by RVU: usually serum of ferret, sheep or human, with/without neutralisation activity) at + 56 °C/30 minutes. This step will be performed simultaneously for all study samples and control sera.
The MN analysis with the NIBRG121 virus will be performed as follows: a six-step twofold dilution series (covering titres 20–640) will be set up for each of the samples and control sera. After addition of a pretitred virus [usually around 100 × median tissue culture infectious dose (TCID50) per well or 0.1-1 virus particle per cell] neutralisation will be performed by incubation of the virus/serum mixture at room temperature for 1 hour. After neutralisation, a suspension of Madin–Darby Canine Kidney (MDCK) cells will be added and the plates will be incubated for 16 hours at 37 °C in a CO2 incubator. The remaining infectivity of virus after neutralisation is determined in an EIA format using a monoclonal antibody to detect expression of viral nucleoprotein. The amount of nucleoprotein expression is determined photometrically (optical density = OD450) using a plate reader.
An OD reading for each dilution step for each sample will be used to calculate the titre. The titre will be reported as the reciprocal dilution at which 50% of the virus is neutralised (e.g. titre of 100). The MN analysis will be performed in duplicate (in separate runs on 2 days) for each sample. The two titres for each sample must not differ by more than a twofold serial dilution. In cases, where samples do not fall within this limit, a third analysis is performed and the two closest titres (which must be within a twofold serial dilution) will be reported.
The principle of the HI test is based on the ability of specific anti-influenza antibodies to inhibit haemagglutination of red blood cells (RBCs) by influenza virus haemagglutinin. The sera to be tested have to be previously treated to eliminate the non-specific inhibitors and the anti-species haemagglutination antigens. The experiment will be performed in accordance to protocols and SOPs established by RVU.
Elimination of non-specific inhibitors will be achieved by incubation of unknown serum samples and quality control sera (serum of ferret or human immunised with influenza virus) with neuraminidase [receptor-destroying enzyme (RDE) II: 18 hours/+36 °C followed by heat-inactivation 1 hour/+ 56 °C]. All samples – sera pre-vaccination and post vaccination and controls will be prepared simultaneously.
For the HI analysis with the NIBRG121 virus, samples and controls will be titrated in an eight-step twofold dilution series (covering titres 8–1024) and incubated with the HA antigen suspension (previously titrated to adjust the dilution at 4 haemagglutination units/25 µl, 50% end point). The HA antigen is not added to the well dedicated to the RDE quality control.
The mixture is incubated for 1 hour at room temperature and 25 µl of the 0.5% RBC suspension (turkey blood) are added. The reaction is left for half an hour at room temperature before reading.
The serum titre is equal to the highest reciprocal dilution, which induces a complete inhibition of haemagglutination. The titre of each quality control serum is close to the previously assigned value (within one serial twofold dilution limits). The RBC controls (RBC suspension without antigen) and the RDE controls do not produce any agglutination. Each serum sample is titrated in duplicate and individual titres will be reported (two for each sample). These must not differ by more than a twofold serial dilution. In cases, where samples do not fall within this limit, a third analysis is performed and the two closest titres (which must be within a twofold serial dilution) will be reported.
Nasal swab PCR
Nucleic acid extraction and real-time PCR (RT-PCR) will be performed by research staff at the Molecular Diagnostics Laboratory of the Leicester Royal Infirmary Microbiology Department, according to the UK National Standard Methods VSOP 25 (RT quadriplex PCR for the detection of influenza) and VSOP 29 (Swine-lineage Influenza A(H1)-specific fast RT-PCR). Equipment available for automated nucleic acid extraction includes the Corbett Xtractor Gene and the Qiagen Qiasymphony. RT-PCR analysis will be performed using the Corbett Rotor-Gene. The Leicester Microbiology Department is under the Pathology Directorate of the University Hospitals of Leicester/Leicester Royal Infirmary. Regular participation in performance evaluations, such as the National External Quality Assessment Service (NEQAS) and HPA, are SOP to maintain laboratory accreditation.
Statistics
Methods
The overall programme manager, Dr Richard Puleston, Associate Professor of Health Protection at the University of Nottingham, will be responsible for the evaluation and analysis of the findings of the study. He will be assisted in this task by the statistical expertise provided by Dr Puja Myles and other statistical experts from the same unit. stata 11 will be the primary statistical analysis software, with additional use of Microsoft excel and other graphing/statistical software as appropriate (e.g. spss).
It is likely the researchers will use the following approaches.
Primary objective of study
Assessing vertical transmission of immunity to swine influenza virus in vaccinated and unvaccinated mothers using cord blood analyses:
-
Exposure variable vaccinated/unvaccinated mother (binary variable).
-
Outcome variable binary variable ‘immune (yes/no)’ based on either threshold level of antibodies in cord blood; or immune status as a categorical variable with a range of values signifying ‘no/low immunity, moderate immunity, high immunity’.
Analysis – descriptive:
-
Characteristics of vaccinated and unvaccinated mothers (mean, range, standard deviation, t-test or percentage, chi-squared test).
-
Proportion of babies immune in vaccinated and unvaccinated mothers.
-
Percentage difference in immunity between offspring of vaccinated and unvaccinated mothers with 95% CI (should not include 0 to indicate a statistically significant difference in immunity) and p-values (chi-squared test).
-
For the interim analysis the researchers will compute headline figures only.
-
For the full analysis the researchers will explore the proportions and percentage differences in subgroups of vaccinated and unvaccinated mothers.
-
Subgroup analysis – among vaccinated mothers, is there a difference in vertical transmission of immunity by underlying health status? (Per cent difference.)
Secondary objective of study
Analytical investigation to assess statistical association between:
[Note: two possible exposure variables: maternal vaccination status (yes/no), immune status of child at birth (yes/no)]
-
maternal vaccination status and subsequent development of influenza-like/respiratory illness in babies
-
immune status at birth and subsequent development of influenza-like/respiratory illness in babies.
With respect to the outcome, the researchers will count first only the first H1N1 swine influenza episode for each child and censor from study once a positive H1N1 swine influenza diagnosis has been obtained.
Adjustment for covariates may also be used (e.g. number of children in household).
The statistical approaches to be used will either be:
-
Cohort analysis involving:
-
– Kaplan–Meier survival curves to demonstrate differences in event (influenza-like/respiratory illness) in babies over the study period by exposure status.
-
– Cox regression analysis: ‘hazard’ or risk of developing influenza-like/respiratory illness in vaccinated (exposed) group compared with unvaccinated (unexposed) group (hazard ratio, 95% CI and p-values).
-
-
Alternatively, a nested case–control study (cases = influenza babies, controls = non-influenza babies) may be required, and analysed using logistic regression with results expressed as odds ratio, 95% CI and p-values. This would also allow the analysis to control for other covariates of interest.
(Note: cannot compute rates or rate differences using Poisson regression because no denominators.)
These will be specified in more detail in the statistical analysis plan, which will be developed prior to the unblinding of the results of the cord blood samples. Any changes to the statistical methods used will be documented in the study report.
Interim findings will be assessed as previously set out to review efficacy of immune transfer (objective one) and protection against respiratory infection (objective two). In order to achieve this, the results available at that stage will have to be unblinded. Analysis at both interim and full investigation results will be by the same team. Because of the urgent need for results the interim findings will be made available to the main strategy group and also the DH, which may lead to changes in the conduct of the study if appropriate.
GlaxoSmithKline may require access to the anonymised data set for their own scientific purposes. Participants will be specifically asked to provide consent for this.
Sample size and justification
The researchers have used a conservative power calculation based upon an estimated 20% seroconversion rate in unvaccinated women; in reality it may be closer to 10% as the cumulative clinical attack rate in the UK is still very low (probably < 2%) and so the true serological attack rate is probably < 4%. In addition underlying immunity in the age group 15–44 years (women of child-bearing age) is considered to be low. We have similarly estimated conservatively that seroconversion in vaccinated women will be 50%; in fact it is more likely to be 70% after two doses. Thus a power calculation based on 20% versus 50% is very conservative, and a more optimistic comparison would be 10% versus 70%.
Based on 20% versus 50%, with 80% statistical power, and 5% significance (two-tailed statistics), 38 subjects per group are required – total 76. However we anticipate that two-thirds of women will accept vaccine, so the ratio of unvaccinated–vaccinated subjects will be 0.5; allowing for this imbalance, a total study size of 89 subjects (59 vaccinated 30 unvaccinated) would be needed. Assuming a total of 89 subjects and more optimistic estimates of 10% versus 70%, the study would have 100% power to detect such a difference. To allow for possible losses during analysis or inadequate specimens, the researchers plan to recruit a maximum of 100 study subjects.
Assessment of efficacy
Primary efficacy end points will, as has previously been indicated, be based on the internationally recognised standard for immunity to influenza. Measures of seroconversion as specified by the EMEA–CHMP will be used. These criteria are formally accepted to be measures of seroconversion consistent with clinical protection against seasonal influenza in adults, and are used routinely for testing the immunogenicity of influenza vaccines in adults (HI titre ≥ 40 or single radial haemolysis > 25 mm2). MN tests may be used to assess responses to pandemic influenza but there are no recognised correlates of protection. Subjects achieving MN titres of ≥ 40 are may be considered as an immunogenicity end point. The extent to which these criteria are relevant to pandemic vaccines and to babies could both be debated but at present there are no widely accepted alternative measures, and these criteria are being used for the licensure of pandemic vaccines. They are therefore appropriate for this study.
Laboratory-confirmed influenza in symptomatic neonates will be measured by detection of H1N1 swine by validated PCR from clinical nasal samples.
The efficacy parameter will be difference in means for both the primary and secondary efficacy end points.
Procedures for missing, unused and spurious data
Missing data may fall into two categories: direct results from the primary and secondary objectives or indirect supporting information, such as date of birth or GP details.
Where the former is missing, every effort will be taken to track down what might be the problem, such as sample not actually taken through to result not being communicated. Where resolvable, the data will then be collated with the other findings. Where data items appear spurious, for example an implausible serological conversion level, clarification will be sought with the testing laboratory will be made to ascertain its veracity and, if necessary, request requantification, although the laboratory will have its own quality control procedures that should detect issues of this nature before reporting.
Where supporting data are missing the research midwives will endeavour to complete the data item either directly from the participant (e.g. number of children under 5 years in the household) or if more appropriate (e.g. details of the delivery) from the clinical record.
Where data items remain missing despite these efforts they will be treated as missing and will be noted as such in the analysis.
Definition of populations analysed
Interim analysis set, primary objective Analysis will be ongoing through the study. The relevance of the results as the study progresses will be based on the probability of obtaining such results for the number known.
Interim analysis set, secondary objective This will occur only if the national influenza activity on 31 January 2010 is above the normal baseline for the time of year.
Full analysis set, primary objective All individuals for whom a cord blood sample has been received at the close of recruitment and their subsequent delivery.
Full analysis set, secondary objective All infants for whom a nasal swab has been received by the termination date of the study, 31 July 2010, or the end of the pandemic, whichever is the sooner.
Efficacy will be assessed on the interim and full analysis set. Ineligible participants will be excluded before recruitment. However, if a participant becomes ineligible through withdrawal of consent then the research record for that person will be marked as such and noted at the time of analysis.
Adverse events
For the purposes of the study, the researchers consider that although unlikely there may be the possibility of some adverse events occurring. These may include:
Issues becoming apparent as a result of vaccination. Although administering vaccination is not part of the investigation, it is possible that in the course of the study, issues in the vaccinated case group may become apparent such as a(n):
-
exacerbation of a pre-existing illness
-
increase in frequency or intensity of a pre-existing episodic event or condition
-
condition detected or diagnosed after vaccine administration even though it may have been present prior to the start of the study
-
continuous persistent disease or symptoms present at baseline that worsen following the start of the study.
At this stage it is not obvious what these may be, and as the study is not geared to be looking for these they are unlikely to present in this way.
Issues as a result of parental swabbing of infant’s nose:
-
exacerbation of a pre-existing illness
-
increase in frequency or intensity of a pre-existing episodic event or condition
-
condition detected or diagnosed after nasal swab taking even though it may have been present prior to the start of the study
-
continuous persistent disease or symptoms present at baseline that worsen following the start of the study.
Taking a nasal swab is unlikely to have any impact on any of these possibilities. The only conceivable problems could be causing minor trauma to the nose if done incorrectly, or exacerbating respiratory upset of the child. These risks will be minimised by providing the mother/parents with careful instruction on self sampling. Additionally, there is established precedent for this type of parental sampling in studies on infants.
A serious adverse event (SAE) is any adverse event occurring following study mandated procedures that results in any of the following outcomes:
-
death
-
a life-threatening adverse event
-
inpatient hospitalisation or prolongation of existing hospitalisation
-
a disability/incapacity.
As a direct result of the study, it is not considered that any of these will occur as the interventions are not of the nature that could cause these.
Reporting of adverse events
Participants will be asked to contact the study site immediately in the event of any actual or perceived SAE. All adverse events will be recorded and closely monitored until resolution, stabilisation, or until it has been shown that the study intervention is not the cause. The Principal (Chief) Investigator shall be informed immediately of any SAEs and shall determine seriousness and causality in conjunction with any treating medical practitioners.
Participant removal from the study due to adverse events
Any participant who experiences an adverse event may be withdrawn from the study at the discretion of the Principal Investigator.
Ethical and regulatory aspects
Ethics committee and regulatory approvals
The study will not be initiated before the protocol, Informed Consent Forms and participant and GP information sheets have received approval/favourable opinion from the Research Ethics Committee (REC), and the respective NHS Research and Development (R&D) department. Should a protocol amendment be made that requires REC approval, the changes in the protocol will not be instituted until the amendment and revised Informed Consent Forms and participant and GP information sheets (if appropriate) have been reviewed and received approval/favourable opinion from the REC and R&D departments. A protocol amendment intended to eliminate an apparent immediate hazard to participants may be implemented immediately providing that R&D and REC are notified as soon as possible and an approval is requested. Minor protocol amendments only for logistical or administrative changes may be implemented immediately, and the REC will be informed.
The study will be conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki, 1996, and the DH Research Governance Framework for Health and Social Care, 2005.
Informed consent and participant information
The process for obtaining participant informed consent or assent and parent/guardian informed consent will be in accordance with the REC guidance, and good clinical practice (GCP) and any other regulatory requirements that might be introduced. The investigator or their nominee (usual/research midwife or obstetrician) and the participant or other legally authorised representative shall both sign and date the Informed Consent Form before the person can participate in the study.
The participant will receive a copy of the signed and dated forms and the original will be retained in the Study Master File. A second copy will be filed in the participant’s medical notes and a signed and dated note made in the notes that informed consent was obtained for the investigation.
The decision regarding participation in the study is entirely voluntary. The investigator or their nominee shall emphasise to them that consent regarding study participation may be withdrawn at any time without penalty or affecting the quality or quantity of their future medical care, or loss of benefits to which the participant is otherwise entitled. No study-specific interventions will be done before informed consent has been obtained.
The Principal Investigator will inform the participant of any relevant information that becomes available during the course of the study, and will discuss with them, whether they wish to continue with the study. If applicable they will be asked to sign revised consent forms.
If the Informed Consent Form is amended during the study, the investigator shall follow all applicable regulatory requirements pertaining to approval of the amended Informed Consent Form by the REC and use of the amended form (including for ongoing participants).
Records
Case report forms
Each participant will be assigned a unique study identity code number [one for the mother and different one(s) for the infant(s), allocated at recruitment], for use on case report forms (CRFs), other study documents including request forms and the electronic database. The documents and database will also use their initials (of first and last names separated by a hyphen or a middle name initial when available) and date of birth (dd/mm/yy).
Case report forms will be treated as confidential documents and held securely in accordance with regulations. The investigator will make a separate confidential record of the participant’s name, date of birth, local hospital number or NHS number, and unique study identity code number (the Study Recruitment Log), to permit identification of all participants enrolled in the investigation to allow results linkage.
Case report forms access will be restricted to those personnel approved by the Principal Investigator and recorded on the ‘Study Delegation Log’.
All paper forms will be filled in using black ballpoint pen. Errors shall be lined out but not obliterated by using correction fluid and the correction inserted, initialled and dated.
The Principal Investigator shall sign a declaration ensuring accuracy of data recorded in the CRF.
Source documents
Source documents shall be filed at the investigator’s site and may include, but are not limited to, consent forms, case report forms/study records and laboratory results. Only investigative staff as listed on the Study Delegation Log shall have access to study documentation other than for regulatory requirements.
Direct access to source data/documents
The CRF and all source documents, including progress notes and copies of laboratory and medical test results, shall made be available at all times for review by the Principal Investigator, Sponsor’s designee and inspection by relevant regulatory authorities.
GlaxoSmithKline will not have direct access to identifiable data; they will be provided only with anonymised data.
Data protection
All study staff and investigators will endeavour to protect the rights of the study’s participants to privacy and informed consent, and will adhere to the Data Protection Act, 1998. The CRF will only collect the minimum required information for the purposes of the investigation. CRFs will be held securely, in a locked room, or locked cupboard or cabinet. Access to the information will be limited to the study staff and investigators and relevant regulatory authorities (see above). Computer-held data including the study database will be held securely and password protected. All data will be stored on a secure dedicated server. Access will be restricted by user identifiers and passwords (encrypted using a one-way encryption method).
Information about the study in the participant’s medical records/hospital notes will be treated confidentially in the same way as all other confidential medical information.
Electronic data will be backed up every 24 hours to both local and remote media in encrypted format.
Quality assurance and audit
Insurance and indemnity
Insurance and indemnity for study participants and study staff is covered within the NHS indemnity arrangements for clinical negligence claims in the NHS, issued under cover of Health Service Guidelines (96)48. There are no special compensation arrangements, but study participants may have recourse through the NHS complaints procedures.
The University of Nottingham has taken out an insurance policy to provide indemnity in the event of a successful litigious claim for proven non-negligent harm.
Study conduct
The study conduct will be subject to systems audit of the Study Master File for inclusion of essential documents, permissions to conduct the study, Study Delegation Log, curricula vitae of investigative staff and training received, local document control procedures, consent procedures and recruitment logs, adherence to procedures defined in the protocol (e.g. inclusion/exclusion criteria, timeliness of visits) and adverse event recording and reporting.
The Study Coordinator or, where required, a nominated designee of the Sponsor, shall carry out a site systems audit at least once during the course of the study and an audit report shall be made to the TSC.
Study data
Monitoring of study data will include confirmation of informed consent, source data verification, data storage and data transfer procedures, local quality control checks and procedures, back-up and disaster recovery of any local databases, and validation of data manipulation. The Study Manager or suitable deputy, or where required, a nominated designee of the Sponsor, shall carry out monitoring of study data as an ongoing activity.
Entries on CRFs will be verified by inspection against the source data. A sample of CRFs (10%) will be checked on a regular basis for verification of all entries made. In addition, the subsequent capture of the data on the study database will be checked. Where corrections are required these will carry a full audit trail and justification.
Study data and evidence of monitoring and systems audits will be made available for inspection by the regulatory authority as required.
Record retention and archiving
In compliance with the International Conference on Harmonisation (ICH)/GCP guidelines, regulations and in accordance with the University of Nottingham Research Code of Conduct, the Principal Investigator will maintain all records and documents regarding the conduct of the study. These will be retained for at least 7 years or for longer if required. If the responsible investigator is no longer able to maintain the study records, a second person will be nominated to take over this responsibility.
The Study Master File and study documents held by the Principle Investigator on behalf of the Sponsor shall be finally archived at secure archive facilities at the University of Nottingham. This archive shall include all study databases and associated meta-data encryption codes.
Discontinuation of the study by the sponsor
The Sponsor reserves the right to discontinue this study at any time for failure to meet expected enrolment goals, for safety or any other administrative reasons. The Sponsor shall take advice from the TSC as appropriate in making this decision.
Statement of confidentiality
Individual participant medical information obtained as a result of this study are considered confidential and disclosure to third parties is prohibited with the exceptions noted above.
Participant confidentiality will be further ensured by utilising unique identification code numbers in the computer files.
Such medical information may be given to the participant’s usual care team and all appropriate medical personnel responsible for the participant’s welfare.
Data generated as a result of this investigation will be available for inspection on request by the participating physicians, the University of Nottingham representatives, the REC, local R&D departments and the regulatory authorities.
Publication and dissemination policy
The results of the study are expected to be of importance in the management of the current influenza pandemic. As the primary and secondary clients it is expected that the DH and GSK will be informed of the results by means of reports prior to any formal publication. These reports may be required to influence policy recommendations and therefore may be delivered at several stages to report on the primary and secondary outcomes, respectively, as the results are obtained. The results on the primary objective – vertical immunity is expected before the year end, 2009, and therefore will be made available to the DH/GSK at that time. The secondary viral swabbing follow-up of symptomatic babies will not be complete until spring 2010 and thus a report will be provided to DH/GSK then unless data suggests that an earlier statement is warranted.
It is possible that the influenza pandemic may raise considerable public disquiet again in the autumn 2009 to spring 2010, including with respect to the increased risk of complications in pregnant women. Therefore, it is possible that there may be media interest in the findings of the study. The researchers/their academic institutions will therefore liaise closely with the DH/NIHR and GSK on appropriate handling of media interest.
Formal academic publication will be sought in a minimum of two stages: firstly to report on the vertical immune transmission and subsequently on the protection conferred (baby swabbing element). These will be in winter 2009/early 2010 and summer/autumn 2010, respectively.
Conferences and other professional meetings may also be utilised to disseminate the findings.
Patient confidentiality and anonymity will be maintained through any reporting/publication of the study results.
User and public involvement
Due to the tight time scales involved in obtaining results for the study commissioner, it has not been/will not be possible to engage the public in the design of this investigation.
Study finances
Funding source
This study is funded by the NIHR. The secondary objective will also be supported by a grant from GSK subject to contractual agreements being completed.
Participant stipends and payments
Participants will not be paid to participate in this study,
Signature pages
Signatories to protocol.
Chief Investigator:
(Name) ________________________________________________
Signature: ______________________________________________
Date: __________________________________________________
Co-investigator:
(Name) ________________________________________________
Signature: ______________________________________________
Date: __________________________________________________
Study Statistician:
(Name) ________________________________________________
Signature: ______________________________________________
Date: __________________________________________________
Appendix 3 Participant questionnaire
List of abbreviations
- A/H1N1v
- newly emergent pandemic influenza A/H1N1v (variant)
- CHMP
- Committee for Human Medicinal Products
- CI
- confidence interval
- CRF
- case report form
- DH
- Department of Health
- EIA
- enzyme immunoassay
- EMEA
- European Medicines Agency
- GCP
- good clinical practice
- GSK
- GlaxoSmithKline
- HA
- haemagglutinin
- HAU
- haemagglutination unit
- HI
- haemagglutinin inhibition
- HPA
- Health Protection Agency
- LNRREC1
- Leicestershire, Northamptonshire and Rutland Research Ethics Committee 1
- mAb
- monoclonal antibody
- MDCK
- Madin–Darby Canine Kidney
- MMR
- measles, mumps and rubella
- MN
- microneutralisation
- NIBRG121
- Reassortant vaccine virus produced using reverse genetics technology, from an A/California/7/2009(H1N1)v virus by the National Institute for Biological Standards and Control
- NIHR
- National Institute for Health Research
- OD
- optical density
- ONS
- Office for National Statistics
- PCR
- polymerase chain reaction
- R&D
- Research and Development
- RBC
- red blood cell
- RDE
- receptor-destroying enzyme
- REC
- Research Ethics Committee
- RVU
- Respiratory Virus Unit
- SAE
- serious adverse event
- SOP
- standard operating procedure
- TCID50
- median tissue culture infectious dose
- TSC
- Trial Steering Committee
All abbreviations that have been used in this report are listed here unless the abbreviation is well known (e.g. NHS), or it has been used only once, or it is a non-standard abbreviation used only in figures/tables/appendices, in which case the abbreviation is defined in the figure legend or in the notes at the end of the table.