
Notes
Article history
This themed issue of the Health Technology Assessment journal series contains a collection of research commissioned by the NIHR as part of the Department of Health’s (DH) response to the H1N1 swine flu pandemic. The NIHR through the NIHR Evaluation Trials and Studies Coordinating Centre (NETSCC) commissioned a number of research projects looking into the treatment and management of H1N1 influenza. NETSCC managed the pandemic flu research over a very short timescale in two ways. Firstly, it responded to urgent national research priority areas identified by the Scientific Advisory Group in Emergencies (SAGE). Secondly, a call for research proposals to inform policy and patient care in the current influenza pandemic was issued in June 2009. All research proposals went through a process of academic peer review by clinicians and methodologists as well as being reviewed by a specially convened NIHR Flu Commissioning Board.
Declared competing interests of authors
the Health Protection Agency (HPA) receives funding from several vaccine companies for influenza work carried out in the laboratory of Maria Zambon at HPA Centre for Infections. ES has received support to attend scientific meeting from Wyeth Vaccines in 2006, KH has received support to attend international meeting from Sanofi Vaccines in 2006.
Permissions
Copyright statement
© 2010 Queen’s Printer and Controller of HMSO. This journal is a member of and subscribes to the principles of the Committee on Publication Ethics (COPE) (http://www.publicationethics.org/). This journal may be freely reproduced for the purposes of private research and study and may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NETSCC, Health Technology Assessment, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2010 Queen’s Printer and Controller of HMSO
Chapter 1 Background
H1N1 2009 pandemic in the UK
The first cases of H1N1 2009 pandemic influenza in the UK were confirmed in Scotland, on 27 April 2009, in a couple returning from a holiday in Mexico, followed by confirmation of the first case in England on 29 April 2009 in a person who returned on the same flight. The cases that were confirmed in the ensuing month were generally linked to returning travellers from Mexico or the USA or to secondary transmission from such imported cases in schools or other close-contact environments. 1 The subsequent progression of the pandemic in the UK was rapid, particularly in London, with sustained community transmission resulting in a major wave of infection in the summer that was interrupted when schools closed for their annual holidays in mid-July 2009. The second wave of infection in the UK started in September 2009 when children returned to school after their summer holidays, suggesting that transmission of influenza within schools was an important epidemiological determinant of community spread. The UK experience was unlike that of most other European countries, where the first wave of infection occurred in the 2009 autumn/winter season, despite similar exposure to imported cases in the summer months [S Flasche, Health Protection Agency (HPA), 2010, personal communication].
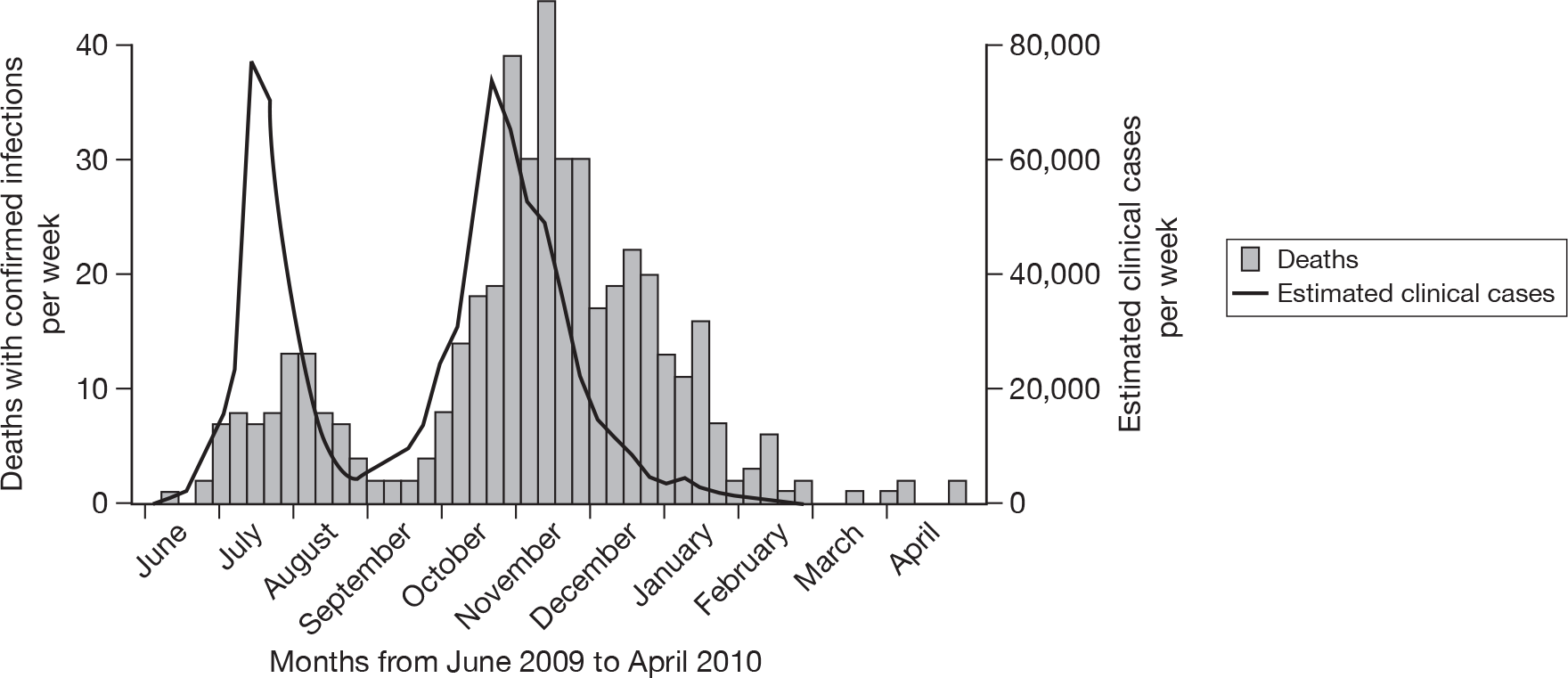
As with seasonal influenza, the number of individuals with an influenza-like illness (ILI) who seek medical care will not be an accurate measure of the number of symptomatic pandemic influenza cases in the population. Not all individuals with ILI consult a doctor and, among those who do, not all will have an illness caused by the pandemic virus. In an attempt to estimate the true number of clinical cases of H1N1 2009 pandemic influenza in England, the HPA devised a statistical model that took account of the estimated proportion of those with an ILI who sought medical care and the proportion of those consulting with ILI who had a confirmed H1N1 2009 infection. 2 The estimated numbers of clinical cases based on this method in the first and second waves of the H1N1 2009 pandemic in England and Wales are shown in Figure 1. These case estimates indicate that the first and second waves were of similar size. However, deaths and hospitalisations in individuals with confirmed H1N1 2009 infection suggest that the second wave was considerably larger than the first. Thus, although these estimated clinical case numbers may provide a good guide as to the progression of the pandemic, they may not give an accurate picture of the relative magnitude of the various waves of infection, possibly due to changes in the propensity to consult among those with an ILI as the pandemic progresses. 3 The inevitable uncertainty in parameters such as the propensity to consult, together with uncertainty about the proportion of H1N1 2009-infected patients who develop ILI, rather than an unrecognised mild or asymptomatic infection, meant that the relationship of these clinical case estimates to the underlying infection rate in the population, and hence the transmission dynamics of this novel influenza virus, was uncertain.
FIGURE 1.
Number of clinical cases of H1N1 2009 in England estimated using the HPA statistical method (as described in the HPA document2) and number of deaths in individuals with confirmed infection in England – June 2009 to April 2010.
Role of seroepidemiology
Rapid understanding of the transmission dynamics of H1N1 2009 was necessary in order to anticipate demands on health-care resources and develop appropriate public health interventions, such as optimal deployment of pandemic strain vaccines. 4 For this, timely seroepidemiological surveys that could provide information on the age-specific incidence of infection as it spreads in the population is essential. The need for serological data was identified as part of the pandemic planning process, particularly in relation to informing the parameterisation of real-time models that could be used to predict the future course of the pandemic and thus assist in contingency planning, although detailed operational plans for achieving timely serum collections were not fully established. Accurate information on the incidence of infection was essential for deriving the true denominator for markers of severity, such as case fatality and hospitalisation rates, and for estimating key transmission parameters for modelling, such as the average number of secondary cases generated from a single index case [known as the reproduction number (R)].
Although H1N1 2009 is genetically and antigenically distinct from current H1N1 seasonal influenza, prior exposure of the population to circulating H1N1 viruses made it likely that there was some cross-reactive immunity, particularly in the older age groups with lifetime experience of seasonal influenza and possible exposure to older H1N1 strains more closely related to H1N1 2009. Serology was essential for assessing the baseline (pre-pandemic) age-specific prevalence of immunity. Knowledge of the baseline immunity was important for understanding the epidemiology of H1N1 2009, in particular whether the lower attack rates that had been documented in the elderly in the early stages of the first wave were likely to be the result of pre-existing cross-protective antibodies.
Measurement of antibodies to the H1N1 2009 virus
Influenza antibody measurements rely on detection of functional antibody, which can be used as a surrogate of protection. This is usually carried out by measurement of haemagglutination inhibiting [haemagglutination inhibition (HI)] or neutralising antibodies, the latter detected using a microneutralisation (MN) assay. Both of these are bioassay techniques, and present more technical challenges than enzyme-linked immunosorbent assay (ELISA) technologies, which are commonly used to determine seroprevalence for infections by human immunodeficiency virus (HIV) or hepatitis virus. HI measures only the proportion of antibody that is directed to the receptor-binding site of viral haemagglutinin (HA) – a major target of neutralising antibodies. In comparison, MN detects a broader range of neutralising antibodies, thus making estimates of immunity based on HI slightly more conservative than those based on MN. For HI, a titre of around 1 : 32 is thought to correlate with protection,5 but there is no accepted correlate of protection for MN titres.
Although HI and MN methods are well established for seasonal influenza, the emergence of a new subtype such as H1N1 2009 in humans poses a number of technical challenges. Immunity to influenza is not only subtype specific, but also strain specific. It is therefore essential to perform any serological analysis with an appropriate representative virus strain for the outbreak. This isolate needs to be genetically and antigenically characterised and grown in suitable quality amounts prior to use in the serological assays. Where such a strain is not immediately available, an antigenically related virus can be identified and used as a substitute in the initial phase of assay development.
Initial classification of the H1N1 2009 virus by the World Health Organization (WHO) as an Advisory Committee on Dangerous Pathogens (ACDP) III (equivalent) pathogen resulted in the requirement for high containment [biosafety level (BSL) 3] conditions for all serological or virological work. This, in turn, meant that the logistics of high-throughput serology could not be achieved. To reduce biosafety hazard and improve laboratory throughput, preparation of reverse genetic (rg) versions of H1N1 2009 that would be classified as BSL2 + pathogens were undertaken. This allowed virus handling under less restrictive conditions. Furthermore, preparation of animal serum standards raised by immunising ferrets with the relevant virus, ancestor and drift strains, were essential for the performance and validation of serological analysis. For the HI test, which depends on the use of red blood cells (RBCs), an appropriate animal source for use in the H1N1 2009 assay also had to be identified.
The time required for each of these key stages in assay development, and the subsequent time needed for assay validation, required a 6- to 8-week period from the end of April, when the first UK viruses were isolated and therefore available to work with, until mid-July 2009 before the first serological testing for this project could begin.
Chapter 2 Study objectives
The objectives of the H1N1 2009 serological surveillance project were twofold, to document:
-
the prevalence of cross-reactive antibodies to H1N1 2009 by age group in the population of England prior to arrival of the pandemic strain virus in the UK
-
the age-specific incidence of infection by month as the pandemic progressed by measuring increases in the proportion of individuals with antibodies to H1N1 2009 by age.
The aim was to generate the baseline prevalence data and monthly incidence estimates as rapidly as possible in order to inform real-time modelling activities. These time constraints inevitably imposed limitations on the number of sera that could be tested each month. It had not been anticipated that there would be differences between geographical regions in the incidence of infection during the pandemic. Thus, while the study was designed to obtain as geographically representative a set of sera as possible, it was not powered to detect differences between regions. Rather the emphasis was placed on measuring incidence within defined age groups, which would be more informative for age-structured, real-time, disease-transmission models.
Chapter 3 Methods
Development of HI and MN assays
Virus isolation and culture of H1N1 2009
Immediately after the notification of a widespread H1N1 2009 outbreak in the USA and Mexico, the Respiratory Virus Unit (RVU) of the HPA identified A/Aragon/R3218/08(H1N1) as a suitable substitute virus for initial assay set-up and validation purposes both for molecular and serological work. The virus, which was classified as a BSL2 + pathogen, was received at the end of April and cultivated in eggs to provide material for positive controls in real-time polymerase chain reaction (RT-PCR) development and also as antigen for the initial stage of serological assay development. The HPA Centre for Emergency Preparedness and Response (CEPR) also received this virus to generate standard ferret sera.
Upon identification of the first clinical cases in the UK (molecularly confirmed by HPA RVU) virus isolation work involving cell culture at BSL3 started from virus-positive clinical swabs taken from these cases. Once isolates from the first UK cases were grown and genetically characterised, a suitable representative was provided to the National Institute for Biological Standards and Control (NIBSC) to attempt generation of an rg virus and preparation of ferret serum standards. NIBSC was also tasked by the WHO with the creation of an rg virus for the A/California/7/09 prototype US strain, which it successfully generated at the end of May 2009 (NIBRG121). The rg version of the first UK isolate (A/England/195/09) became available from NIBSC in the second half of June 2009 (NIBRG122). It was necessary to compare the properties of these and other available viruses to determine the optimum virus to use for large-scale serology studies, a process which is important to ensure that the choice of strain for extensive serology is representative of circulating strains.
Generation of animal sera/antigenic analysis
There is at least a 3-week time delay between the availability of a particular virus strain and availability of a specific ferret standard post-infection serum. A minimum time is required following experimental inoculation of ferrets to develop high-titred antibody. Similar to the strategy for virus cultivation, generation of antisera was performed in stages and a ferret post-infection serum to A/Aragon/R3218/08 became available first (end of May), followed by ferret serum to A/California/7/09 and A/England/195/09. These serum standards and virus prototype strains were used to establish the antigenic relatedness of the various H1N1 2009 isolates that were available at that time from Europe and the USA. In this way, a robust strain selection process was undertaken to ensure optimisation of serological assays used by HPA for seroepidemiological and clinical diagnostic purposes, as well as for the analysis of pandemic vaccine trials.
Strain selection
Table 1 indicates the reactivity of various virus isolates with panels of reference ferret antisera in HI tests. The highest reactivity (titre) is expected in reactions between virus strains and post-infection antisera from ferrets inoculated with the same (homologous) virus. It can be seen that the antigenic reactivity of NIBRG121 and NIBRG122 rg viruses is identical to their respective prototype or parental wild-type strains using a panel of post-infection ferret antisera raised against pandemic, seasonal and older H1N1 strains. This also demonstrates the antigenic relatedness of prototype UK strain to the A/California/4/09 prototype strain and its rg equivalent virus (NIBRG121). Together, these data validate the choice of an rg virus as a suitable alternative to wild-type strains and indicate that sera related to the outbreak in the UK could be analysed with the NIBRG122 virus.
| Viruses | Ferret antisera | |||||
|---|---|---|---|---|---|---|
| A/Aragon/R3218/08 | A/England/195/09 | A/California/7/09 | A/England/117316/86 | A/England/195852/92 | A/Brisbane/59/07 | |
| A/England/195/09 (wt) | 256 | 2048 | 2560 | 512 | 256 | < 8 |
| NIBRG122 (rg) | 256 | 2048 | 2560 | 512 | 256 | < 8 |
| A/California/4/09 (wt) | 256 | 2048 | 2560 | 256 | 256 | < 8 |
| NIBRG121 (rg) | 256 | 2048 | 2560 | 512 | 256 | < 8 |
HI protocol development
Influenza viruses show variation in their capacity to bind RBCs from different species. The ability of HA molecules to bind surface receptors on the RBC depends on which type of sialic acids these receptors contain and what linkage connects the glycan residues. Human viruses agglutinate RBC from chicken/turkey, human and guinea pig, but not from horse. In contrast, avian viruses agglutinate RBC from all four species, whereas the binding of swine viruses depends on whether they have human or avian-like HAs. Assessment of RBC binding of virus strains is a surrogate measure of receptor specificity and knowledge of this property is essential for interpretation of serology, as incorrect use of RBC indicator for the HI bioassay will lead to an underestimate of available antibody, as has been observed in measuring antibody responses to avian virus infection and H5 vaccine responses.
To identify an appropriate source of RBC for the HI assay, empirical investigation of animal RBCs from different species was undertaken to optimise for the A/H1N1 2009 virus. RBCs from chicken, turkey, guinea pig, pig, horse and human (type O) were investigated for adjustment of the HI assay. Turkey RBCs provided the highest sensitivity (Table 2).
| Viruses | RBC species | ||||||
|---|---|---|---|---|---|---|---|
| Chicken | Turkey | Guinea pig | Human type O | Pig | Horse | Duck | |
| A/California/4/09 (wt) | + | ++ | ++ | ++ | – | – | + |
| A/England/195/09 (wt) | – | +++ | ++ | ++ | – | nd | + |
| A/Aragon/R3218/08 (wt) | +++ | ++ | ++ | ++ | – | – | ++ |
Serum validation panels
After development of suitable bioassay protocols, the performance of HI and MN assays was evaluated using archived serum panels. These sera were collected prior to 2009 (i.e. from a population without exposure to swine influenza viruses) and were used to serve in establishing the levels of cross-reactive antibodies in various groups of the population. The panels are described as follows:
-
England age-stratified panels (collected in June 2003 and June 2004) Samples were geographically representative of England and were obtained from residual serum samples from outpatient visits.
-
Influenza-like illness panel (2002/03) These were paired sera from UK patients with ILI, many of whom had serologically confirmed seasonal influenza.
-
Influenza-like illness panel (SARS 2003) These were UK sera collected in 2003 during the SARS (severe acute respiratory illness) incident from patients with suspected SARS infection for which recent seasonal influenza had been excluded.
-
Seasonal Vaccine Study Trivalent inactivated vaccine trial in healthy adult and elderly volunteers (trial performed in southern hemisphere in 2005).
Using the NIBRG122 strain, significant levels of cross-reactive antibody were found in sera without primary exposure to H1N1 2009 virus, using both HI and MN (although analysis was performed with both assays and yielded similar results, we discuss HI only in this section for simplicity). Results of the HI analysis of these four panels are summarised in Table 3. The detection limit for HI is a titre of 1 : 8 (for MN it is 1 : 10).
| Panel | Age group (years) | Sample no. | GMTa | Positive by HI (%) | GMRb | SRCc | |
|---|---|---|---|---|---|---|---|
| Titre ≥ 1 : 8 | Titre ≥ 1 : 32 | ||||||
| England age-stratified panel (2004) | |||||||
| 1–4 | 48 | 4.0 | 0 | 0 | n/a | n/a | |
| 5–10 | 56 | 4.3 | 5.4 | 0 | n/a | n/a | |
| 11–17 | 74 | 4.8 | 6.8 | 5.4 | n/a | n/a | |
| 18–24 | 74 | 4.7 | 8.1 | 2.7 | n/a | n/a | |
| 25–34 | 85 | 4.4 | 5.9 | 3.5 | n/a | n/a | |
| 35–44 | 58 | 4.8 | 10.3 | 3.4 | n/a | n/a | |
| 45–54 | 96 | 5.0 | 10.4 | 6.3 | n/a | n/a | |
| 55–64 | 85 | 5.6 | 12.9 | 7.1 | n/a | n/a | |
| 65–74 | 31 | 5.7 | 22.6 | 9.7 | n/a | n/a | |
| 75–84 | 9 | 10.4 | 44.4 | 11.1 | n/a | n/a | |
| Total | 616 | ||||||
| ILI (2002/03) | |||||||
| Acute | < 55 | 23 | 6.7 | 30.4 | 13.0 | n/a | n/a |
| Convalescent | < 55 | 28 | 8.8 | 32.1 | 21.4 | 1.4 | 4 in 23 pairs |
| Acute | ≥ 55 | 41 | 6.3 | 19.5 | 9.8 | n/a | n/a |
| Convalescent | ≥ 55 | 70 | 5.0 | 11.4 | 4.3 | 0.9 | 1 in 40 pairs |
| Total | 162 | 5 in 63 pairs | |||||
| SRI (2003) | |||||||
| Acute | No information on age group available | 90 | 7.6 | 18.9 | 16.7 | n/a | n/a |
| Convalescent | 74 | 5.5 | 13.5 | 9.5 | 0.7 | 0 in 70 pairs | |
| Total | 164 | 0 in 70 pairs | |||||
| Seasonal vaccine study (2005) | |||||||
| Pre-vaccine | < 55 | 43 | 22.1 | 11.6 | 7.0 | n/a | n/a |
| Post vaccine | < 55 | 43 | 35.4 | 20.9 | 14.0 | 1.2 | 4 in 43 pairs |
| Pre-vaccine | ≥ 55 | 77 | 12.1 | 11.7 | 6.5 | n/a | n/a |
| Post vaccine | ≥ 55 | 77 | 20.6 | 15.6 | 11.7 | 1.2 | 3 in 77 pairs |
| Total | 240 | 7 in 120 pairs | |||||
For the England age-stratified panel, a total of 616 samples collected in July 2004 from four different regions in England were selected to determine the specificity of the HI test. The age range was 0–79 years and 49% were male. Table 3 shows the number of samples by age group, proportions of individuals with HI titres above the cut-off points ≥ 1 : 8 and ≥ 1 : 32, as well as geometric mean titres (GMTs) by age group (information about region was neglected, as regions were not evenly represented in this collection).
We found evidence for cross-reactive antibody in all, except the 1- to 4-year age group. Our analysis suggested that the observed cross-reactivity is age specific, with children under the age of 10 years having little or no cross-reactive antibody, whereas in older adults over the age of 65 years more than 20% of all tested individuals showed titres above the detection limit.
We also observed cross-reactive antibody when analysing sera from subjects with a clinical diagnosis of ILI and severe respiratory illness (SRI) or volunteers from a vaccine trial of seasonal vaccine (which were collected during the 2002–3 season) in 2003 and 2005, respectively. Samples in these panels were grouped into two major age groups (under and above 55 years of age) and, where possible, were analysed as acute/convalescent or pre-/post-vaccination serum pair.
Source of samples for seroepidemiology
The HPA Seroepidemiology Unit (SEU) archive is an opportunistic collection of residual serum samples from routine microbiological testing, submitted voluntarily each year from laboratories throughout the HPA Regional Microbiology Network (RMN) in England. SEU archive sera are stored at the HPA North West (NW) regional laboratory in Manchester, and are anonymised and permanently unlinked from any patient-identifying information, with information only on age, gender, date of collection (if available) and contributing laboratory being retained. The SEU archive has stored over 150,000 samples since collection began in 1986. The HPA has ethical approval (05/Q0505/45) for the collection and use of unlinked and anonymised residual serum samples in cross-sectional antibody prevalence studies for the surveillance of population immunity to vaccine-preventable diseases of public health importance and the collection has been extensively used for this purpose.
Seroepidemiology Unit sera collected in 2008 from eight out of nine RMN regions were available for use in the baseline age-specific serosurvey of antibody to H1N1 2009. However, as the pandemic progressed it became clear that a more rapid and timely method of collection of serum samples was needed to track changes in antibody prevalence to provide estimates for the seroincidence study. Chemical pathology laboratories were identified as potential sources of additional serum samples due to their rapid testing and short sample retention time in comparison with microbiology laboratories. Chemical pathology laboratories were therefore approached at hospitals in each of the RMN regions in August 2009, and requested to provide a monthly contribution of age-stratified residual sera to the SEU archive at the end of each calendar month during the pandemic. Recruitment of these laboratories was helped by the Royal College of Pathologists, whose president made an appeal in the College’s newsletter for collaboration. There was a very positive response from the chemical pathology laboratories, and this, along with the continuing collection from microbiology laboratories, ensured that, for the majority of regions, a regular supply of age-stratified serum samples were obtained rapidly after collection for use in the seroincidence study between August 2009 and April 2010.
Identification of samples from individuals with PCR-confirmed H1N1 2009 infection
To be confident that incidence could be estimated by measuring changes in prevalence of HI or MN antibodies in post-infection human sera taken before and during the pandemic, it was necessary to document the antibody responses in laboratory-confirmed H1N1 2009 cases. This was performed using serum samples opportunistically submitted for serological testing and matching them with a database held at the HPA Centre for Infections (CFI) of laboratory-confirmed cases in England diagnosed by polymerase chain reaction (PCR) detection of H1N1 2009 in clinical respiratory samples. Different sample types were electronically linked using name and date of birth. The request forms submitted with the respiratory samples taken for PCR testing contained the date of sampling during the original illness, and, in some instances, also included the date at onset of symptoms, which was usually a few days earlier than the date the swab was taken. A further database of cases confirmed up to September 2009, generated by local HPA Health Protection Units for infection control purposes, was also available and contained information on date at onset of symptoms for many cases. This second database was also linked with the PCR-positive cases for which a serology sample was available to supplement the information on onset date. Pooling these data sources allowed date of symptom onset to be found for 115 out of 150 (76.7%) serum samples from laboratory-confirmed cases. Intervals were calculated by subtracting date at onset from date of serology sample to give an interval in days and where date of onset was not available using date of PCR sample. Only samples where this final interval was ≥ 0 days were included in the analysis. GMTs and 95% confidence intervals (CIs) for HI and MN by interval since onset or PCR date were calculated for the included samples, together with the percentages with a titre ≥ 1 : 32 by HI or ≥ 1 : 40 by MN, these titres being at least fourfold higher than the starting dilution for each assay.
Statistical methods
Sample size
The study was designed to provide an estimate of the proportion of the population in defined age groups with antibodies against H1N1 2009 before and after the pandemic, and at monthly intervals during the pandemic. The monthly numbers were constrained by the need to generate data quickly and inevitable limitations on laboratory capacity. The aim was to obtain samples spread evenly by age groups (< 5, 5–14, 15–24, 25–44, 45–64, 65–74, 75–79, 80 + years) and regions in the baseline, with each age group having about 200 samples at baseline and post pandemic. The age groups were chosen to match those in the Royal College of General Practitioners (RCGP) influenza surveillance data set with expansion of the 65-and-over age group in order to define with greater precision the relationship between birth cohort and prevalence of pre-existing cross-reactive antibodies to H1N1 2009. A sample size of 1600 (i.e. 200 in each age group) would provide 95% CIs, which were deemed reasonably narrow; for example, for a prevalence of 5%, 95% CI would be 2.9 to 9.0, and for a prevalence of 25% the corresponding 95% CI would be 19.2 to 31.6. This was deemed to provide sufficient precision for meaningful estimation of baseline seroprevalence.
For the monthly incidence estimates, for which there was a testing limit of about 1000 per month, all of those aged 65 + years were collapsed into a single age group. A sample size of 200 per age group would allow prevalence to be estimated with 95% CIs ranging from ± 5% around a prevalence of 10% to ± 7% around a prevalence of 50%. The 95% CIs around various overall incidence estimates based on 1000 samples at each time point are shown in Table 4. Within each age group the precision of the incidence estimate would be much lower.
| p1 (%) | p2 (%) | Cumulative seroincidence (Δ prevalence: p2 – p1), % | 95% CI |
|---|---|---|---|
| 5 | 10 | 5 | 2.7 to 7.3 |
| 10 | 15 | 5 | 2.1 to 7.9 |
| 15 | 20 | 5 | 1.7 to 8.3 |
| 20 | 25 | 5 | 1.3 to 8.7 |
| 5 | 15 | 10 | 7.4 to 12.6 |
| 10 | 20 | 10 | 6.9 to 13.1 |
| 15 | 25 | 10 | 6.5 to 13.5 |
Analysis of baseline seroepidemiology
To investigate whether certain birth cohorts had higher baseline immunity geometric mean HI and MN titres with 95% CIs were calculated by 10-year birth cohorts, as well as the proportions with titres greater or equal to the detection limit and four times the detection limit (i.e. ≥ 1 : 8 and ≥ 1 : 32 for HI and ≥ 1 : 10 and ≥ 1 : 40 for MN) with exact binomial 95% CIs. Results from the HI assay were semiquantitative (in powers of two); the highest titre to which dilutions were made was 1 : 1024. For the MN assay, results were continuous, with an upper dilution of 1 : 320 (a small number of samples were diluted beyond this titre). A multivariable normal errors regression model was also fitted to log(HI) and log(MN) results to investigate the effects of birth cohort/age, sex and region. For all statistical analyses, titres for samples that fell below the detection limit were set to one-half of the detection limit (i.e. four for HI and five for MN), and titres greater or equal to the highest detection limit were set to the maximum detection limit.
Analysis of seroprevalence over time
Seroprevalence over time, based on an HI titre of ≥ 1 : 32, was investigated using all samples taken from August 2009 to April 2010. A logistic regression model was fitted with factors for month, gender, age, region and laboratory type (microbiology or chemical pathology). Interactions were also investigated. This modelling approach was then used to obtain predicted probabilities of a seropositive result by month, age and region (London/outside London).
Estimation of seroincidence from observed changes in seroprevalence
To investigate seroincidence over the course of the pandemic, seroprevalence at three time points [pre-pandemic (2008), post-summer wave and post-autumn wave] was estimated and the change from one time point to another was assumed to reflect seroincidence. The post-summer wave period (weeks 34–37 2009) and autumn wave period (January to April 2010) were defined based on the HPA-estimated case numbers by week and allowing time for seroconversion by HI (at least 3 weeks). An initial descriptive analysis was performed using age-specific reverse cumulative distribution curves of HI titres at the three time points. Age-specific seroincidence was then estimated from baseline to post-summer wave, post-summer wave to post-autumn wave, and baseline to post-autumn wave by calculating the difference in seroprevalence at these time points with 95% CIs. This was carried out using Wilson’s score method, implemented in the Epi library in the r software (The R Foundation for Statistical Computing, Vienna, Austria). Because of the different age composition of the collections, direct age standardisation within the 65-and-over age group, using smaller age groups of 65–74, 75–79 and ≥ 80 years, was required.
Due to time constraints in the laboratory, not all samples could be tested using both HI and MN. Thus, seroincidence was measured using changes in HI from baseline to post-summer wave, and baseline and post-summer wave to post-autumn wave, and by HI and MN from baseline to post-autumn wave. Again due to limited time for testing, the only samples tested that were collected in November and December were from individuals aged under 45 years, in whom changes in incidence were likely to be greatest. Further, some samples could not be tested with MN as there was not enough serum left.
Seroincidence was estimated separately for samples collected in London and samples collected elsewhere. The reason for stratifying by London and elsewhere was that London was one of the regions that experienced a substantial H1N1 2009 wave during the summer. In addition, the demographic structure of the population in London is different to that in other regions, with a higher proportion of individuals in the younger age groups, which, together with the higher population density, may result in a higher R-value for London than elsewhere. As not all regions contributed samples at all time points, a sensitivity analysis was carried out for which seroincidence was estimated for only those regions with samples available from both 2008 (baseline) and 2010 (post-autumn wave).
All statistical analyses were carried out in r version 2.11.1.
Likelihood-based estimation of seroincidence
Estimating the incidence of infection by measuring monthly changes in antibody prevalence from the pre-pandemic baseline has certain limitations. First, the monthly samples are distributed over a 30-day period, during which incidence may be changing, particularly at the height of the wave. Second, the variable time to seroconversion between individuals means that even if all samples were taken on the same date they reflect incidence at different times in the previous weeks. Third, derivation of incidence by comparing prevalence between time points reduces the precision of the estimate for a given sample size and may result in negative point estimates for groups in which incidence is low.
A novel method based on a likelihood approach was therefore developed to overcome some of the limitations of the more conventional statistical method described above. The new method uses information on the distribution of the seroconversion intervals in confirmed cases and the exact date of each serum sample, and allows integration of information on the temporal distribution of cases from clinical surveillance (as coming, for example, from the HPA clinical case estimates2). To link information on distributions of cases from surveillance with the sequential serological samples, we proceeded in three steps:
-
For each age group, we constructed a set of putative H1N1 incidence curves (i.e. number of new infections people per week per age group) by assuming that the proportion of people seeking health care was constant during the epidemic. We thus obtained a set of age-group-specific putative incidence curves, built by multiplying the proportion of cases from surveillance in the considered age group in each week by a constant (as the weekly proportion of cases sums to one, this constant is also the final cumulative incidence in the age group). The inference algorithm aims to assess one parameter per age group.
-
For each of the parameterised incidence curves, using the distribution of intervals from onset of symptoms to seroconversion, we redistributed cases to obtain a seroconversion curve, describing how many people seroconvert daily in each age group. By integrating these cases over time starting from the baseline (2008), for each age group, in turn, we linked the incidence curves with the expected change in antibody prevalence.
-
With the daily value for the antibody prevalence given the final cumulative incidence, we could finally derive a likelihood function for the sequence of serological samples. For this, we assumed that the probability of picking a seropositive sample followed a binomial distribution. We inferred the most likely incidence curve by using maximum-likelihood methods.
To describe the kinetics of seroconversion after infection in step 2, we fitted a scaled Weibull distribution to the subset of samples from confirmed cases (n = 115) with a known date of onset of symptoms. Taking as definition of seroconversion an achievement of a HI titre ≥ 1 : 32, it was possible to derive a likelihood function for a given set of parameters of the scaled Weibull distribution (scaling constant in addition to the two parameters of the Weibull distribution). Assuming that the seroconversion after symptoms follows a Weibull distribution, the probability of observing a positive sample after an interval of n days is equal to the probability of the cumulative Weibull distribution. We derived the parameters of the interval to seroconversion distribution using a maximum-likelihood algorithm.
This method does not require a pre-pandemic baseline (although it uses the information if available) and by using information on date of sample it allows the generation of a continuous curve, describing how incidence is changing over the course of the pandemic. It also removes the possibility of generating negative incidence estimates by sampling error.
This likelihood-based estimation method was applied to the serological data in the age groups (1–4, 5–14, 15–24 and 25–44 years) for which there was a continuous supply of samples throughout the two waves of the pandemic for both London and non-London regions. The temporal distributions of cases by week in the first and second waves based on health-care consultations in London and non-London regions were used to generate potential incidence curves by multiplying them by putative total incidences for the two waves. The most likely incidence given the observed temporal sequence of serological samples with HI titres ≥ 1 : 32 in the given region is then estimated. The CIs were estimated using 10,000 bootstrap samples drawn from the incidence distributions of the different age groups. The baseline seroprevalence was assumed to be the same in and outside London.
Chapter 4 Results
Serological response in confirmed H1N1 2009 cases
A total of 150 serum samples were available, with an HI result from 133 individuals with laboratory-confirmed H1N1 2009 infection identified by the linkage exercise. The date of onset was available for 115 of these (76.7%). For the remaining 35 samples, the date of PCR test was used (the median delay between onset and PCR test among the 115 was 3 days). The age breakdown of the 133 individuals is shown in Table 5. For two individuals in the 45- to 64-year age group, insufficient serum was available to obtain MN titres on two of their sera, although one of these individuals did have another serum included with an MN result for a different time point. Therefore, an MN titre was available for 148 of these samples (i.e. for 132 individuals).
| Age group (years) | n (%) |
|---|---|
| < 5 | 2 (1.5) |
| 5–14 | 22 (16.5) |
| 15–24 | 41 (30.8) |
| 25–44 | 42 (31.6) |
| 45–64 | 24 (18.0) |
| 65–74 | 2 (1.5) |
| Total | 133 (100.0) |
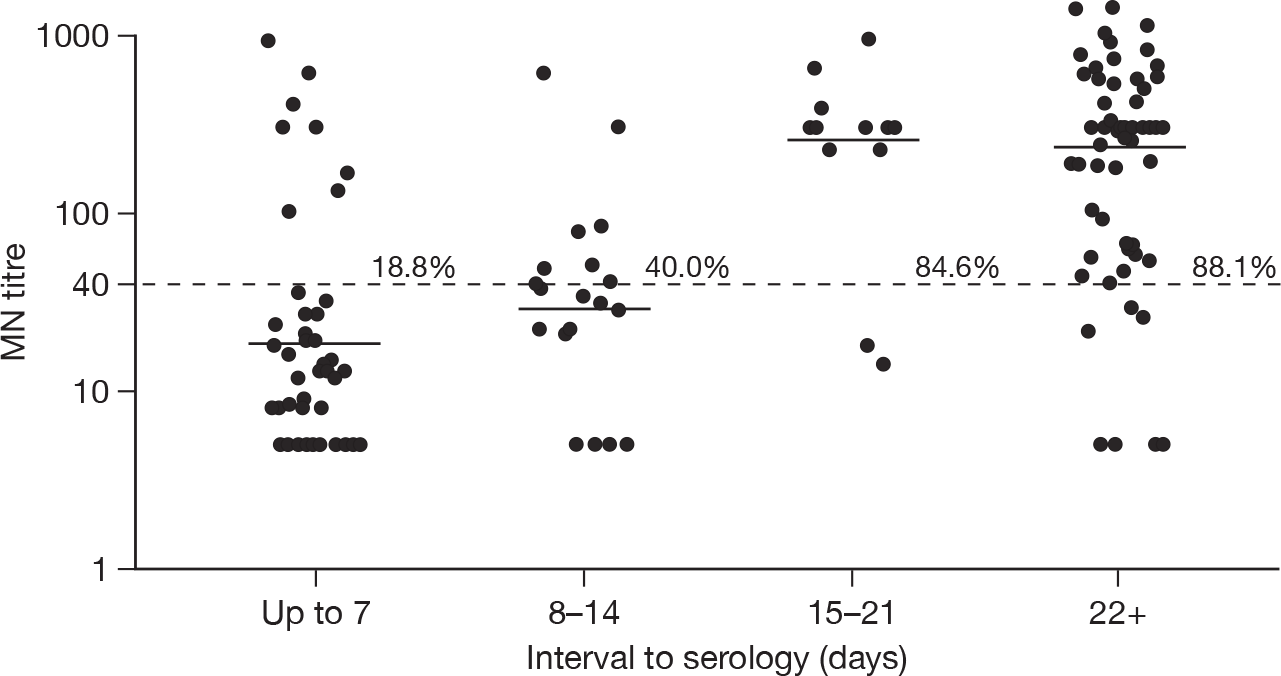
The GMTs and percentages with a titre of four or more times the starting dilution of the assay are shown in Tables 6 and 7 for HI and MN, respectively. Among confirmed cases, there was no evidence of an association of either log(HI) or log(MN) titres with age group once the number of days between onset and serology had been taken into account (F-test p-values comparing linear regression model with and without age were 0.14 for HI and 0.18 for MN). Compared with samples collected at 2–3 weeks after onset, there was a small decline in GMTs for both assays for samples collected at > 3 weeks after onset. This decline was not statistically significant, and there was no decline in the proportion of samples with a titre > 1 : 32. Figures 2 and 3 show the GMTs for HI and MN, respectively, according to delay between onset and sample.
| Onset period | N | n ≥ 1 : 32 (%) | GMT (95% CI) |
|---|---|---|---|
| ≤ 7 days | 49 | 9 (18.4) | 7.9 (5.4 to 11.7) |
| 8–14 days | 20 | 7 (35.0) | 23.0 (9.5 to 56.0) |
| 15–21 days | 13 | 11 (84.6) | 142.4 (61.5 to 330.0) |
| ≥ 22 days | 68 | 59 (86.8) | 125.4 (86.6 to 181.6) |
| Onset period | N | n ≥ 1 : 40 (%) | GMT (95% CI) |
|---|---|---|---|
| ≤ 7 days | 48 | 9 (18.8) | 18.5 (12.0 to 28.7) |
| 8–14 days | 20 | 8 (40.0) | 29.4 (16.3 to 52.9) |
| 15–21 days | 13 | 11 (84.6) | 266.3 (124.0 to 571.9) |
| ≥ 22 days | 67 | 59 (88.1) | 243.9 (163.1 to 364.7) |
FIGURE 2.
Dot plot of HI titres in serum samples from PCR-confirmed cases according to interval from symptom onset or date of PCR test (n = 150). The dashed horizontal line is placed at 1 : 32 (i.e. four times the minimum detection limit); the solid line shows the GMT by interval category. The percentage of samples with a titre that is four or more times the minimum detection limit are also shown.
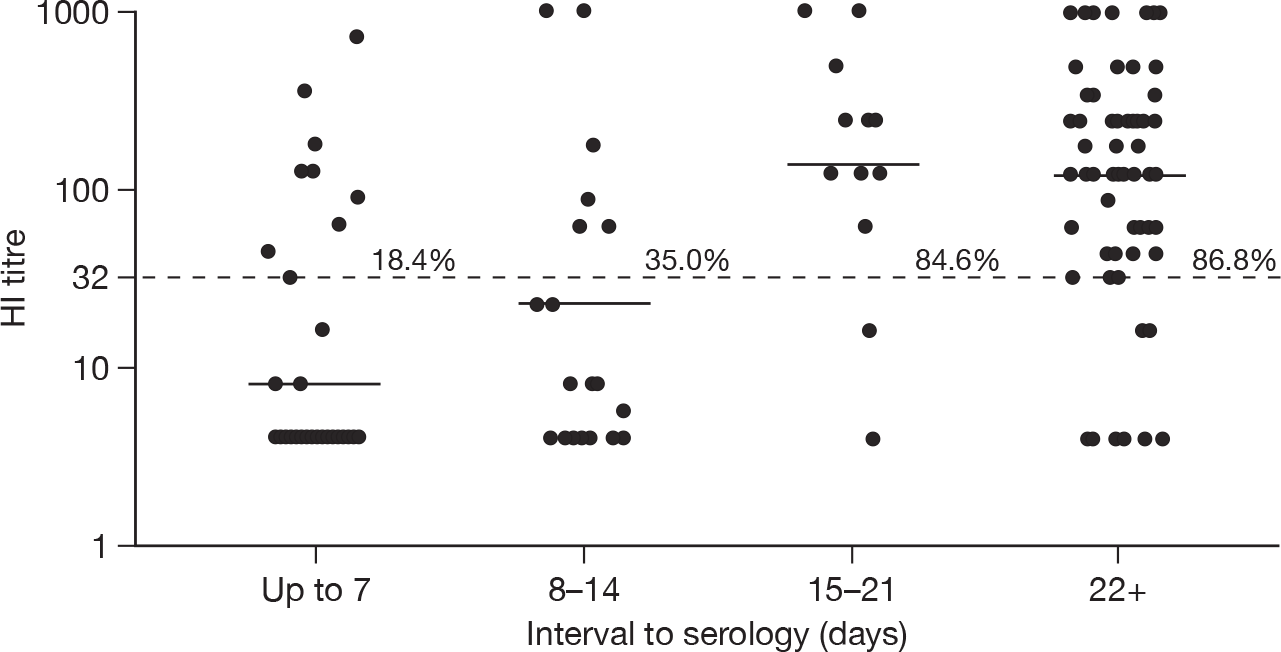
FIGURE 3.
Dot plot of MN titres in PCR-confirmed cases according to interval from symptom onset or date of PCR test (n = 148). The dashed horizontal line is placed at 1 : 40 (i.e. four times the minimum detection limit); the solid line shows the GMT by interval category. The percentage of samples with a titre four or more times the minimum detection limit are also shown.
For each assay there was a small percentage of individuals who failed to develop a measurable response by HI or MN despite having appropriately timed convalescent samples taken. These are shown below in Table 8.
| Interval onset to sample (days) | Titre | Age (years) | |
|---|---|---|---|
| HI | MN | ||
| 24 | < 8 | < 10 | 30.0 |
| 26 | 16 | 27 | 17.4 |
| 30 | 45.2 | 30 | 43.3 |
| 30 | < 8 | < 10 | 30.4 |
| 41 | < 8 | < 10 | 53.8 |
| 49 | 362 | < 10 | 53.7 |
| 67 | < 8 | 22 | 47.8 |
| 69 | < 8 | 56 | 25.2 |
| 69 | < 8 | 95 | 36.8 |
| 70 | < 8 | 45 | 49.2 |
| 87 | 256 | < 10 | 22.2 |
| 128 | 16 | n/a | 54.4 |
Individuals who failed to seroconvert after 21 days on either MN or HI were significantly younger than among the 56 individuals whose sample had been taken more than 21 days after onset but who seroconverted on both assays. The mean age among the non-converters was 32.1 years compared with 27 years among those who did seroconvert on both assays (t test p = 0.03, 95% CI for difference in means 0.4 to 10.0 years).
Pre-pandemic (baseline) seroprevalence
A total of 1403 samples collected between January 2008 and mid-April 2009 from eight different regions in England were selected to determine the baseline seroprofile. All of these were tested by HI and although attempts were made to test 1178 (84.0%) of these samples by MN assay, there was insufficient material for nine samples, leaving 1169 samples with paired HI and MN results. Table 9 shows the number of samples tested by age group and region, showing that all ages and regions are well represented. The age range was 0–87 years and 696 (49.7%) were male. All data items were complete, apart from gender (missing from 3 out of 1403, 0.2%) and sample date (missing from 24 out of 1403, 1.7%); however, sample collection year was available for all 1403 samples.
| Age (years) | Region | Total | |||||||
|---|---|---|---|---|---|---|---|---|---|
| East | London | NE | NW | SE | SW | West Mids | Y&H | ||
| < 5 | 28 | 17 | 31 | 32 | 8 | 31 | 9 | 15 | 171 |
| 5–14 | 25 | 22 | 22 | 18 | 25 | 32 | 21 | 23 | 188 |
| 15–24 | 30 | 12 | 13 | 15 | 11 | 14 | 12 | 13 | 120 |
| 25–44 | 20 | 19 | 20 | 20 | 20 | 20 | 20 | 19 | 158 |
| 45–64 | 36 | 22 | 26 | 24 | 27 | 27 | 28 | 27 | 217 |
| 65–74 | 39 | 26 | 21 | 21 | 21 | 21 | 20 | 21 | 190 |
| 75–79 | 28 | 17 | 26 | 30 | 23 | 25 | 24 | 20 | 193 |
| 80 + | 19 | 13 | 18 | 6 | 25 | 51 | 18 | 16 | 166 |
| Total | 225 | 148 | 177 | 166 | 160 | 221 | 152 | 154 | 1403 |
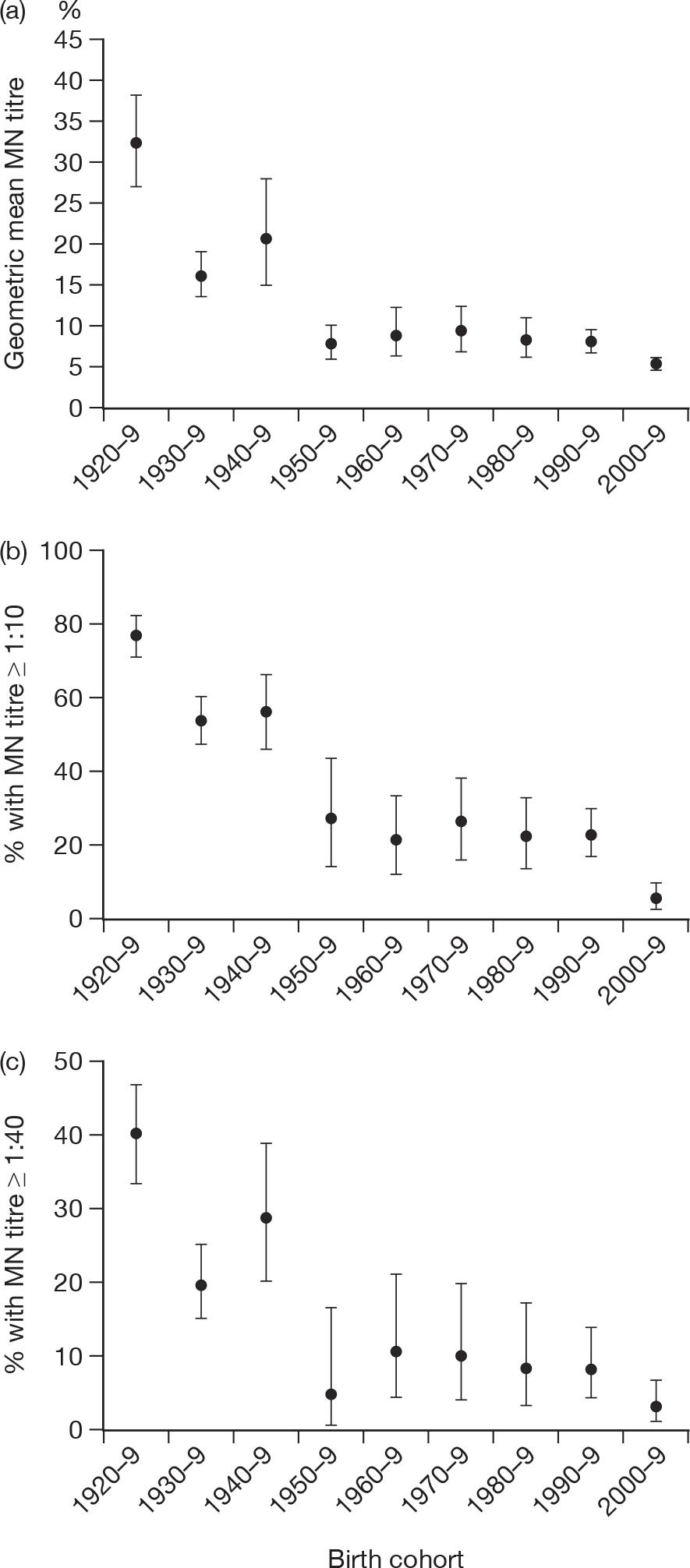
Baseline GMTs as well as proportions ≥ 1 : 8 and ≥ 1 : 32 for HI and ≥ 1 : 10 and ≥ 1 : 40 for MN are shown according to birth cohort in Tables 10 and 11 and Figures 4 and 5. Clear differences can be seen by birth cohort, with those born before 1950 (aged > 60 years) having the highest titres and those born in the last decade (aged < 10 years) having the lowest titres. There was no evidence of a sex difference in baseline log-transformed titres for either HI [likelihood ratio (LR) test p = 0.73] or MN (LR test p = 0.54). There was also no evidence of a regional difference for MN (LR test p = 0.10). For HI there was a regional difference (LR test p < 0.01) that appeared to be due to particularly high titres in the elderly (≥ 65 years) in the West Midlands; if this group [of which 34 out of 62 (54.8%) had titres of ≥ 1 : 32] were excluded then there was no significant regional variation (LR p = 0.08) by HI. MN titres in this age group in the West Midlands were similar to other regions, with 19 out of 62 (31%) having titres of ≥ 1 : 40 compared with 134 out of 458 (29.3%) in other regions (chi-squared test p = 0.98).
| Years | n | GMT (95% CI) | Percentage ≥ 1 : 8 (95% CI) | Percentage ≥ 1 : 32 (95% CI) |
|---|---|---|---|---|
| 1920–9 | 220 | 13.2 (11.1 to 15.8) | 56.8 (50.0 to 63.5) | 28.2 (22.3 to 34.6) |
| 1930–9 | 260 | 9.0 (7.6 to 10.6) | 37.7 (31.8 to 43.9) | 18.5 (13.9 to 23.7) |
| 1940–9 | 158 | 10.4 (8.3 to 13.0) | 42.4 (34.6 to 50.5) | 21.5 (15.4 to 28.8) |
| 1950–9 | 106 | 7.1 (5.7 to 8.7) | 27.4 (19.1 to 36.9) | 10.4 (5.3 to 17.8) |
| 1960–9 | 74 | 5.5 (4.5 to 6.8) | 16.2 (8.7 to 26.6) | 6.8 (2.2 to 15.1) |
| 1970–9 | 79 | 6.0 (4.7 to 7.6) | 16.5 (9.1 to 26.5) | 11.4 (5.3 to 20.5) |
| 1980–9 | 90 | 6.0 (4.9 to 7.2) | 20.0 (12.3 to 29.8) | 11.1 (5.5 to 19.5) |
| 1990–9 | 175 | 5.9 (5.1 to 6.7) | 18.3 (12.9 to 24.8) | 10.3 (6.2 to 15.8) |
| 2000–9 | 241 | 4.4 (4.2 to 4.7) | 4.6 (2.3 to 8.0) | 2.9 (1.2 to 5.9) |
| Years | n | GMT (95% CI) | Percentage ≥ 1 : 10 (95% CI) | Percentage ≥ 1 : 40 (95% CI) |
|---|---|---|---|---|
| 1920–9 | 217 | 32.2 (27.2 to 38.3) | 77.4 (71.3 to 82.8) | 40.1 (33.5 to 46.9) |
| 1930–9 | 243 | 16.2 (13.7 to 19.2) | 54.3 (47.8 to 60.7) | 19.8 (14.9 to 25.3) |
| 1940–9 | 97 | 20.7 (15.2 to 28.2) | 56.7 (46.3 to 66.7) | 28.9 (20.1 to 39) |
| 1950–9 | 40 | 8.0 (6.2 to 10.4) | 27.5 (14.6 to 43.9) | 5.0 (0.6 to 16.9) |
| 1960–9 | 64 | 9.0 (6.5 to 12.4) | 21.9 (12.5 to 34.0) | 10.9 (4.5 to 21.2) |
| 1970–9 | 68 | 9.5 (7.1 to 12.8) | 26.5 (16.5 to 38.6) | 10.3 (4.2 to 20.1) |
| 1980–9 | 80 | 8.6 (6.6 to 11.3) | 22.5 (13.9 to 33.2) | 8.8 (3.6 to 17.2) |
| 1990–9 | 156 | 8.3 (7.1 to 9.8) | 23.1 (16.7 to 30.5) | 8.3 (4.5 to 13.8) |
| 2000–9 | 204 | 5.7 (5.3 to 6.1) | 5.9 (3.1 to 10.0) | 3.4 (1.4 to 6.9) |
FIGURE 4.
Haemagglutination inhibition GMTs and percentage of subjects with titres of ≥ 1 : 8 and ≥ 1 : 32 by birth cohort with 95% CIs.
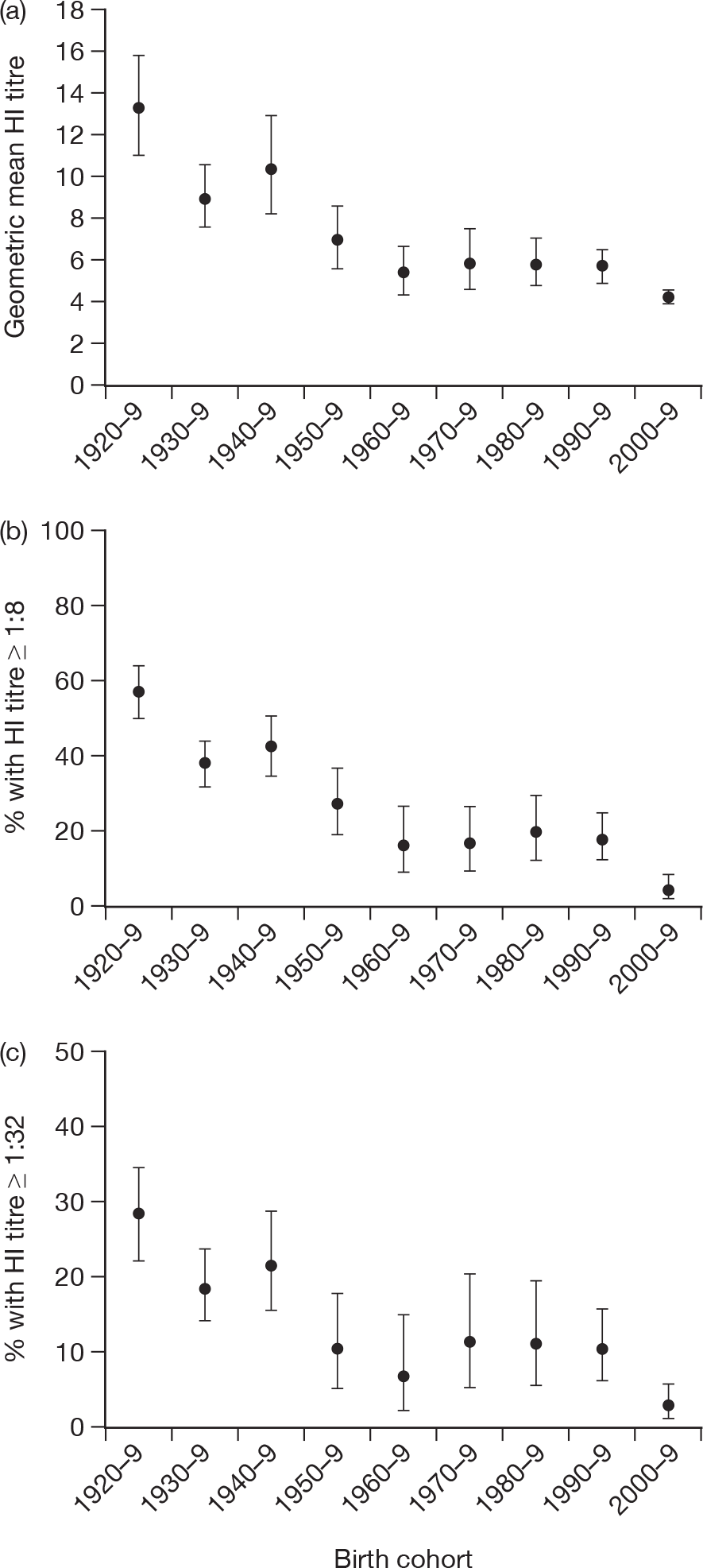
FIGURE 5.
Microneutralisation GMTs and percentage of subjects with titres of ≥ 1 : 10 and ≥ 1 : 40 by birth cohort with 95% CIs.
Seroprevalence from August 2009 to April 2010
A total of 6320 samples collected between 1 August 2009 and 20 April 2010 in eight regions of England were tested by HI [Table 12 – only one sample was tested from Yorkshire and Humber (Y&H)]. Fifty-seven specimens had invalid HI results, leaving 6263 samples for analysis. Of the 6263 samples, 78 (1.2%) had missing information on gender – all other data items were complete.
| Age group (years) | Region | Y&H | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Total | East | East Mids | London | NE | NW | SE | SW | West Mids | ||
| < 5 | 51 | 84 | 209 | 67 | 175 | 10 | 39 | 23 | 0 | 658 |
| 5–14 | 53 | 109 | 198 | 75 | 324 | 30 | 236 | 69 | 0 | 1094 |
| 15–24 | 23 | 67 | 68 | 12 | 296 | 109 | 169 | 44 | 0 | 788 |
| 25–44 | 35 | 146 | 219 | 64 | 297 | 21 | 328 | 183 | 0 | 1293 |
| 45–64 | 45 | 182 | 223 | 52 | 207 | 3 | 305 | 74 | 0 | 1091 |
| 65–74 | 35 | 113 | 125 | 23 | 166 | 14 | 171 | 66 | 1 | 714 |
| 75–79 | 9 | 36 | 39 | 5 | 140 | 13 | 47 | 4 | 0 | 293 |
| 80+ | 3 | 64 | 51 | 22 | 147 | 10 | 88 | 4 | 0 | 389 |
| Total | 254 | 801 | 1132 | 320 | 1752 | 210 | 1383 | 467 | 1 | 6320 |
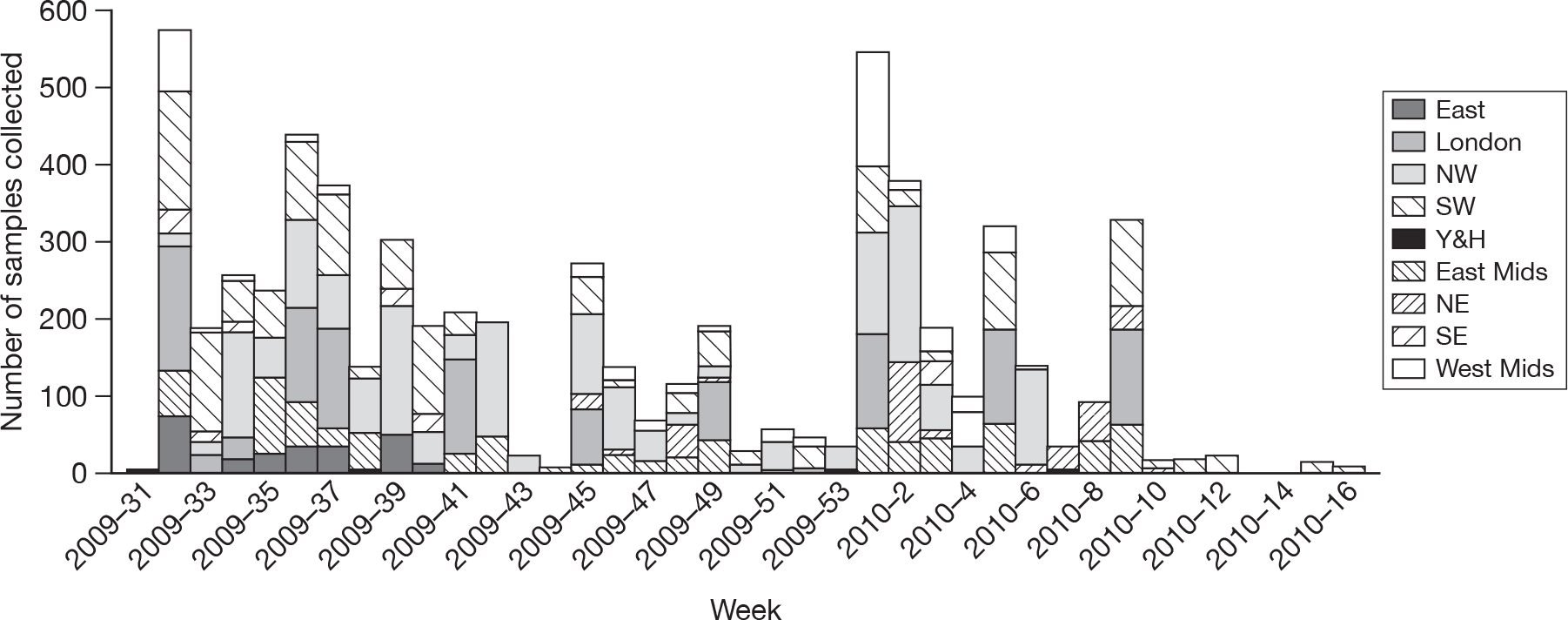
The regions represented across time varied (Figure 6). As shown, some regions [NW, London, South West (SW) and East Midlands (East Mids)] contributed samples throughout the period; others, including East and South East (SE), contributed samples over only a few weeks.
FIGURE 6.
Number of samples collected by region and week – August 2009 to April 2010.
The tables in Appendix 2 summarise the distribution of samples according to age group, region and time period (month).
In a logistic regression model looking at the effect of collecting laboratory type (chemical pathology or microbiology), month, age, region and gender on the odds of having an HI titre of ≥ 1 : 32, no effect of gender was apparent (LR test p = 0.63) so this was not included in further models. Table 13 shows the model with the remaining variables. Seroprevalence clearly increased over time and was highest in those aged 5–14 and 15–24 years. The highest seroprevalence was found in the London region. There was no significant effect of laboratory type, which is important, as the baseline samples came only from microbiology laboratories.
| Linear predictor | n titre ≥ 1 : 32/N (%) | Adjusted OR | LR test p-value |
|---|---|---|---|
| Lab type | |||
| Chemical pathology | 964/4315 (22.3) | 1 | 0.091 |
| Microbiology | 628/1947 (32.3) | 1.17 (0.97 to 1.41) | |
| Month | |||
| August | 168/1248 (13.5) | 1 | < 0.001 |
| September | 168/1317 (12.8) | 0.93 (0.74 to 1.18) | |
| October | 78/525 (14.9) | 0.94 (0.70 to 1.27) | |
| November | 166/625 (26.6) | 1.80 (1.39 to 2.34) | |
| December | 127/322 (39.4) | 3.16 (2.36 to 4.24) | |
| January | 534/1225 (43.6) | 4.62 (3.73 to 5.72) | |
| February | 226/589 (38.4) | 3.91 (3.06 to 4.99) | |
| March | 116/388 (30.0) | 2.32 (1.74 to 3.10) | |
| April | 9/23 (39.1) | 4.00 (1.63 to 9.79) | |
| Age group (years) | |||
| < 5 | 137/635 (21.6) | 1 | < 0.001 |
| 5–14 | 416/1073 (38.8) | 2.63 (2.07 to 3.35) | |
| 15–24 | 226/784 (28.8) | 1.64 (1.26 to 2.14) | |
| 25–44 | 284/1291 (22.0) | 0.94 (0.73 to 1.21) | |
| 45–64 | 195/1086 (18.0) | 0.84 (0.65 to 1.10) | |
| 65 + | 334/1393 (24.0) | 1.32 (1.02 to 1.69) | |
| Region | |||
| North West | 420/1747 (24.0) | 1 | < 0.001 |
| East | 12/254 (4.7) | 0.35 (0.19 to 0.64) | |
| East Midlands | 205/761 (26.9) | 1.17 (0.94 to 1.46) | |
| London | 333/1132 (29.4) | 1.77 (1.45 to 2.17) | |
| North East | 97/320 (30.3) | 0.96 (0.71 to 1.28) | |
| South East | 54/210 (25.7) | 0.90 (0.62 to 1.30) | |
| South West | 292/1371 (21.3) | 1.12 (0.91 to 1.36) | |
| West Midlands | 179/467 (38.3) | 1.59 (1.24 to 2.02) | |
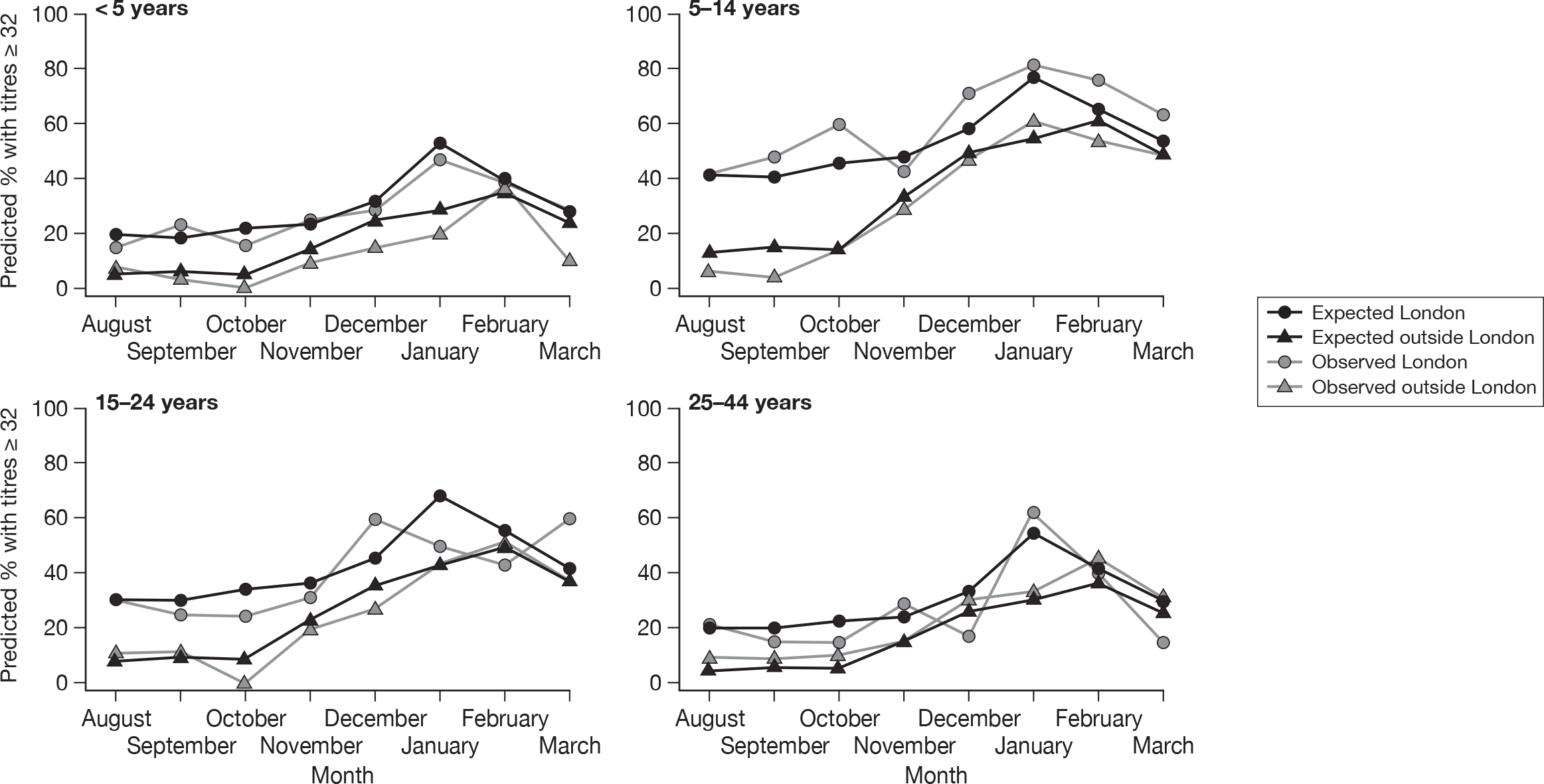
Figure 7 shows observed and predicted seroprevalence of H1N1 HI antibody by age group, region and month for samples from chemical pathology laboratories. These estimates were obtained from a logistic regression model (similar to that presented in Table 13), used to estimate age-specific seroprevalence over time, while taking into account any possible effect of laboratory type. A significant (LR test p < 0.0001) month–region interaction term was also included in this particular model. Note that only age groups up to 25–44 years were included in the model, as there were no samples from persons aged 45 years and above collected in November and December (see Appendix 2). Region was also simplified to London (due to higher incidence in London) and regions outside London. The model was used to predict only the proportion seropositive until March, as there was only a small number of samples collected in April. It is clear the seroprevalence has increased over time and that it was higher in London – particularly in the post-first-wave period (August to September 2009).
FIGURE 7.
Observed and expected (from a logistic regression model) seroprevalence by month, region and age group – August 2009 to March 2010.
The point estimates of the proportion of samples with an HI titre of ≥ 1 : 32 appeared to peak in January then declined. This pattern was not statistically significant in regions outside London (Wald test p-value for March compared with January = 0.21), whereas in London, the odds ratio (OR) for March was significantly lower than in January (0.34, 95% CI 0.17 to 0.67, Wald test p = 0.002).
Estimates of seroincidence after the summer and autumn waves using HI
For assessing changes in seroprevalence between the baseline samples and samples collected after the first and second waves of the pandemic, seroprevalence among the 1403 baseline samples were compared with seroprevalence in 1272 post-first-wave samples and 2225 post-second-wave samples also tested by HI.
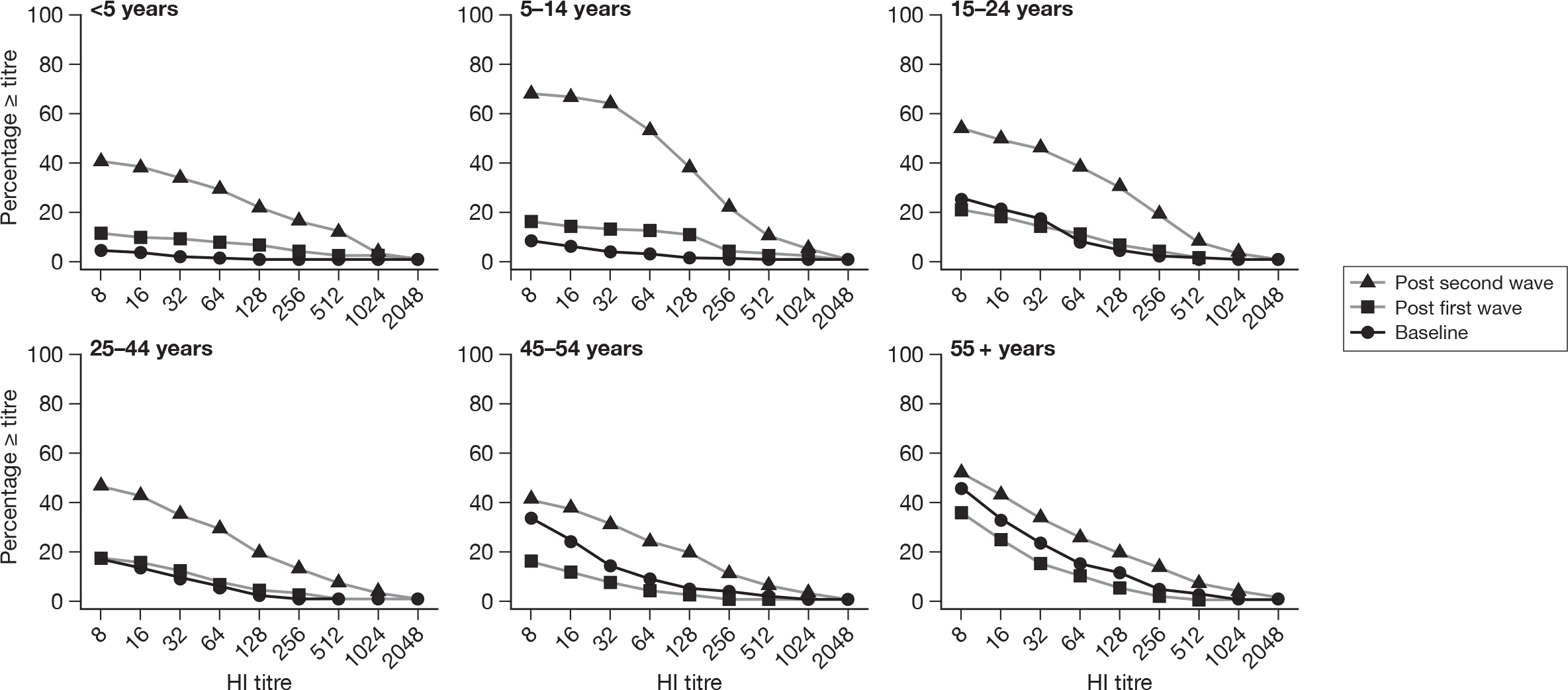
Figure 8 shows reverse cumulative distribution functions of HI titres by age group and time point (pre-pandemic baseline, post first wave and post second wave). The shift in antibody titres between the different time points is clear, particularly the difference in distribution between baseline and post-second-wave samples. The size and timing of this shift in the HI distribution differs according to age group, with the shift occurring later for individuals aged over 15 years. For individuals aged 45 + years the antibody levels in samples collected after the first wave appear to be lower than in the baseline samples.
FIGURE 8.
Reverse cumulative distribution curves by age group and time period.
The seroincidence estimates that equate to the changes between the time periods using the ≥ 1 : 32 cut-off are shown in Table 14. The highest incidence was in the 5- to 14-year age group and the lowest incidence was in the 65+ age group. The younger age groups appear to have higher attack rates in the first wave than older age groups.
| Age group (years) | Baseline (2008) n ≥ 1 : 32/N | Per cent (95% CI) | Post first wave (2009) n ≥ 1 : 32/N | Per cent (95% CI) | Cumulative incidence (95% CI) (2009–baseline) | Post second wave (2010) | Per cent (95% CI) | Cumulative incidence (95% CI) | |
|---|---|---|---|---|---|---|---|---|---|
| 2010–2009 | 2010–baseline | ||||||||
| < 5 | 3/171 | 1.8 (0.6 to 5.0) | 10/109 | 9.2 (5.1 to 16.1) | 7.4 (2.2 to 14.4) | 68/196 | 34.7 (28.4 to 41.6) | 25.5 (16.2 to 33.6) | 32.9 (25.8 to 39.9) |
| 5–14 | 7/188 | 3.7 (1.8 to 7.5) | 24/171 | 14.0 (9.6 to 20.0) | 10.3 (4.5 to 16.6) | 212/324 | 65.4 (60.1 to 70.4) | 51.4 (43.4 to 58.0) | 61.7 (55.2 to 67.0) |
| 15–24 | 21/120 | 17.5 (11.7 to 25.3) | 25/163 | 15.3 (10.6 to 21.7) | –2.2 (–11.3 to 6.4) | 110/240 | 45.8 (39.6 to 52.2) | 30.5 (21.6 to 38.4) | 28.3 (18.4 to 36.9) |
| 25–44 | 14/158 | 8.9 (5.4 to 14.3) | 30/244 | 12.3 (8.7 to 17.0) | 3.4 (–3.1 to 9.3) | 168/470 | 35.7 (31.5 to 40.2) | 23.4 (17.1 to 29.1) | 26.9 (20.0 to 32.5) |
| 45–64 | 31/217 | 14.3 (10.3 to 19.6) | 19/277 | 6.9 (4.4 to 10.5) | –7.4 (–13.2 to –2.0) | 138/435 | 31.7 (27.5 to 36.2) | 24.9 (19.3 to 30.0) | 17.4 (10.7 to 23.5) |
| 65 +a | 128/549 | 23.3 (20.0 to 27.0) | 52/308 | 16.9 (13.9 to 21.5) | –6.4 (–11.7 to –0.8) | 169/560 | 30.2 (26.5 to 34.1) | 13.3 (7.4 to 18.7) | 6.9 (1.7 to 12.0) |
Tables 15 and 16 show these seroincidence estimates stratified by London and elsewhere. When estimating the seroincidence in London the 95% CIs are particularly wide due to the small number of baseline samples collected in London. As there was little evidence of regional variation in the baseline samples the cumulative incidence was also calculated using a baseline of all regions (Table 17). This shows higher incidence in London in the first wave but less clear differences after the second wave.
| Age group (years) | Baseline (2008) n ≥ 1 : 32/N | Per cent (95% CI) | Post first wave (2009) n ≥ 1 : 32/N | Per cent (95% CI) | Cumulative incidence (95% CI) (2009–baseline) | Post second wave (2010) | Per cent (95% CI) | Cumulative incidence (95% CI) | |
|---|---|---|---|---|---|---|---|---|---|
| 2010–2009 | 2010–baseline | ||||||||
| < 5 | 3/154 | 1.9 (0.7 to 5.6) | 4/54 | 7.4 (2.9 to 17.6) | 5.5 (–0.3 to 15.7) | 42/124 | 33.9 (26.1 to 42.6) | 26.5 (13.7 to 36.3) | 31.9 (23.4 to 40.7) |
| 5–14 | 6/166 | 3.6 (1.7 to 7.7) | 11/112 | 9.8 (5.6 to 16.7) | 6.2 (0.3 to 13.4) | 158/251 | 62.9 (56.8 to 68.7) | 53.1 (43.9 to 60.3) | 59.3 (52.0 to 65.4) |
| 15–24 | 21/108 | 19.4 (13.1 to 27.9) | 19/147 | 12.9 (8.4 to 19.3) | –6.5 (–16.1 to 2.5) | 101/222 | 45.5 (39.1 to 52.1) | 32.6 (23.5 to 40.5) | 26.1 (15.4 to 35.2) |
| 25–44 | 12/139 | 8.6 (5.0 to 14.5) | 18/171 | 10.5 (6.8 to 16.0) | 1.9 (–5.1 to 8.5) | 145/411 | 35.3 (30.8 to 40.0) | 24.8 (17.7 to 30.8) | 26.6 (19.3 to 32.6) |
| 45–64 | 29/195 | 14.9 (10.6 to 20.5) | 9/169 | 5.3 (2.8 to 9.8) | –9.5 (–15.7 to –3.3) | 118/362 | 32.6 (28.0 to 37.6) | 27.3 (20.8 to 32.9) | 17.7 (10.4 to 24.3) |
| 65 +a | 112/493 | 22.7 (19.2 to 26.6) | 34/217 | 15.7 (11.4 to 21.1) | –7.0 (–12.8 to –0.6) | 151/486 | 31.1 (27.1 to 35.3) | 15.4 (8.7 to 21.4) | 8.4 (2.8 to 13.8) |
| Age group (years) | Baseline (2008) n ≥ 1 : 32/N | Per cent (95% CI) | Post first wave (2009) n ≥ 1 : 32/N | Per cent (95% CI) | Cumulative incidence (95% CI) (2009–baseline) | Post second wave (2010) | Per cent (95% CI) | Cumulative incidence (95% CI) | |
|---|---|---|---|---|---|---|---|---|---|
| (2010–2009) | (2010–baseline) | ||||||||
| < 5 | 0/17 | 0.0 (0.0 to 18.4) | 6/25 | 24.0 (11.5 to 43.4) | 24.0 (1.7 to 43.4) | 26/72 | 36.1 (26.0 to 47.6) | 12.1 (–9.8 to 29.1) | 36.1 (15.1 to 47.6) |
| 5–14 | 1/22 | 4.5 (0.2 to 21.8) | 12/25 | 48.0 (30.0 to 66.5) | 43.5 (18.5 to 62.3) | 54/73 | 74.0 (62.9 to 82.7) | 26.0 (4.4 to 45.9) | 69.4 (48.9 to 78.9) |
| 15–24 | 0/12 | 0.0 (0.0 to 24.2) | 6/16 | 37.5 (18.5 to 61.4) | 37.5 (6.7 to 61.4) | 9/18 | 50.0 (29.0 to 71.0) | 12.5 (–19.3 to 40.8) | 50.0 (17.9 to 71.0) |
| 25–44 | 2/19 | 10.5 (2.9 to 31.4) | 12/68 | 17.6 (10.4 to 28.4) | 7.1 (–15.0 to 20.3) | 23/59 | 39.0 (27.6 to 51.7) | 21.3 (5.7 to 36.0) | 28.5 (4.7 to 43.3) |
| 45–64 | 2/22 | 9.1 (2.5 to 27.8) | 10/86 | 11.6 (6.4 to 20.1) | 2.5 (–16.9 to 13.3) | 20/73 | 27.4 (18.5 to 38.6) | 15.8 (3.5 to 28.1) | 18.3 (–2.4 to 31.3) |
| 65 +a | 16/56 | 28.6 (18.4 to 41.5) | 17/68 | 25.0 (16.2 to 36.4) | –3.6 (–19.2 to 11.7) | 22/74 | 29.7 (20.5 to 40.9) | 4.7 (–10.0 to 19.0) | 1.2 (–14.7 to 16.3) |
| Age group (years) | Baseline (2008) n ≥ 1 : 32/N | Per cent (95% CI) | Post first wave (2009) n ≥ 1 : 32/N | Per cent (95% CI) | Cumulative incidence (95% CI) (2009–baseline) | Post second wave | Per cent (95% CI) | Cumulative incidence (95% CI) | |
|---|---|---|---|---|---|---|---|---|---|
| 2010–2009 | 2010–baseline | ||||||||
| < 5 | 3/171 | 1.8 (0.6 to 5.0) | 6/25 | 24.0 (11.5 to 43.4) | 22.2 (9.3 to 41.7) | 26/72 | 36.1 (26.0 to 47.6) | 12.1 (–9.8 to 29.1 | 34.4 (23.7 to 46.0) |
| 5–14 | 7/188 | 3.7 (1.8 to 7.5) | 12/25 | 48.0 (30.0 to 66.5) | 44.3 (25.9 to 62.9) | 54/73 | 74.0 (62.9 to 82.7) | 26.0 (4.4 to 45.9) | 70.2 (58.5 to 79.1) |
| 15–24 | 21/120 | 17.5 (11.7 to 25.3) | 6/16 | 37.5 (18.5 to 61.4) | 20.0 (–0.5 to 44.5) | 9/18 | 50.0 (29.0 to 71.0) | 12.5 (–19.3 to 40.8) | 32.5 (10.1 to 54.2) |
| 25–44 | 14/158 | 8.9 (5.4 to 14.3) | 12/68 | 17.6 (10.4 to 28.4) | 8.8 (–0.3 to 20.1) | 23/59 | 39.0 (27.6 to 51.7) | 21.3 (5.7 to 36.0) | 30.1 (17.5 to 43.3) |
| 45–64 | 31/217 | 14.3 (10.3 to 19.6) | 10/86 | 11.6 (6.4 to 20.1) | –2.7 (–10.1 to 6.7) | 20/73 | 27.4 (18.5 to 38.6) | 15.8 (3.5 to 28.1) | 13.1 (2.8 to 25.0) |
| 65 +a | 128/549 | 23.3 (20.0 to 27.0) | 17/68 | 25.0 (16.2 to 36.4) | 1.7 (–7.8 to 13.6) | 22/74 | 29.7 (20.5 to 40.9) | 4.7 (–10.0 to 19.0) | 6.4 (–3.5 to 18.1) |
Weighting these estimates by the regional distribution of the 2009 population6 aged < 15 years living in London on the one hand and in North East (NE), NW, East Mids, SW and SE (the other regions included in the above analysis) on the other, 51.1% (95% CI 46.3 to 55.9) of children aged under 15 years in these regions had seroconverted during the full course of the pandemic. Similar weighted estimates for school-aged children of 5–14 years show that 61.9% (95% CI 56.7 to 67.1) across these regions seroconverted.
To account for changes in the numbers of samples from different regions over time, the analysis of the non-London regions was also restricted to those with samples across all age groups at the baseline and post second wave (Table 18). The only eligible regions were NE, SW and NW. Results can be seen to be very similar to the analysis that included all non-London regions (shown in Table 15).
| Age group (years) | Baseline n ≥ 1 : 32/N | Per cent (95% CI) | 2010 n ≥ 1 : 32/N | Per cent (95% CI) | Cumulative incidence (95% CI) (2010–baseline) |
|---|---|---|---|---|---|
| < 5 | 3/94 | 3.2 (1.1 to 9.0) | 36/98 | 36.7 (27.9 to 46.6) | 33.5 (23.0 to 43.6) |
| 5–14 | 3/72 | 4.2 (1.4,11.5) | 132/213 | 62.0 (55.3 to 68.2) | 57.8 (47.8 to 64.6) |
| 15–24 | 6/42 | 14.3 (6.7 to 27.8) | 68/154 | 44.2 (36.6 to 52.0) | 29.9 (14.3 to 40.8) |
| 25–44 | 1/60 | 1.7 (0.1 to 8.9) | 66/200 | 33.0 (26.9 to 39.8) | 31.3 (21.9 to 38.3) |
| 45–64 | 8/77 | 10.4 (5.4 to 19.2) | 59/220 | 26.8 (21.4 to 33.0) | 16.4 (6.1 to 24.4) |
| 65 +a | 33/219 | 15.1 (10.9 to 20.4) | 86/317 | 27.1 (22.5 to 32.3) | 12.1 (5.0 to 18.7) |
Cumulative seroincidence after the second wave using MN
The MN assay was performed on 1293 out of 2225 (58.1%) samples collected after the second wave (January to April 2010), and valid results were available for 1148 of these. The cumulative seroincidence estimate using a cut-off titre of ≥ 1 : 40 for the MN assay is shown in Table 19 (non-London) and Table 20 (London) and Table 21 (London, with baseline including samples from all regions). Results are similar to those obtained using HI analysis (with a cut-off titre of ≥ 1 : 32).
| Age group (years) | 2008 n ≥ 1 : 40/N | Per cent (95% CI) | 2010 n ≥ 1 : 40/N | Per cent (95% CI) | Cumulative incidence (2010–2008) |
|---|---|---|---|---|---|
| < 5 | 4/126 | 3.2 (1.2 to 7.9) | 38/100 | 38.0 (29.1 to 47.8) | 34.8 (24.8 to 44.8) |
| 5–14 | 6/141 | 4.3 (2.0 to 9.0) | 74/119 | 62.2 (53.2 to 70.4) | 57.9 (47.8 to 66.4) |
| 15–24 | 14/98 | 14.3 (8.7 to 22.6) | 64/149 | 43.0 (35.3 to 51.0) | 28.7 (17.4 to 38.4) |
| 25–44 | 11/119 | 9.2 (5.2 to 15.8) | 46/120 | 38.3 (30.1 to 47.3) | 29.1 (18.6 to 38.9) |
| 45–64 | 13/83 | 15.7 (9.4 to 25.0) | 61/158 | 38.6 (31.4 to 46.4) | 22.9 (11.2 to 32.9) |
| 65 +a | 137/464 | 29.5 (25.6 to 33.8) | 129/345 | 37.4 (32.3 to 42.4) | 7.9 (1.3 to 14.4) |
| Age group (years) | 2008 n ≥ 1 : 40/N | Per cent (95% CI) | 2010n ≥ 1 : 40/N | Per cent (95% CI) | Cumulative incidence (2010–2008) |
|---|---|---|---|---|---|
| < 5 | 0/17 | 0.0 (0.0 to 18.4) | 18/39 | 46.2 (31.6 to 61.4) | 46.2 (22.6 to 61.4) |
| 5–14 | 1/22 | 4.5 (0.2 to 21.8) | 13/20 | 65.0 (43.3 to 81.9) | 60.5 (32.7 to 77.7) |
| 15–24 | 0/12 | 0.0 (0.0 to 24.2) | 3/6 | 50.0 (18.8 to 81.2) | 50.0 (10.5 to 81.2) |
| 25–44 | 1/19 | 5.3 (0.3 to 24.6) | 4/9 | 44.4 (18.9 to 73.3) | 39.2 (7.1 to 68.4) |
| 45–64 | 3/12 | 25.0 (8.9 to 53.2) | 10/32 | 31.2 (18.0 to 48.6) | 6.3 (–25.0 to 29.9) |
| 65 +a | 16/56 | 28.6 (18.4 to 41.5) | 26/51 | 51.0 (37.7 to 64.1) | 22.4 (3.9 to 39.0) |
| Age group (years) | 2008 n ≥ 1 : 40/N | Per cent (95% CI) | 2010 n ≥ 1 : 40/N | Per cent (95% CI) | Cumulative incidence (2010–2008) |
|---|---|---|---|---|---|
| < 5 | 4/143 | 2.8 (1.1 to 7.0) | 18/39 | 46.2 (31.6 to 61.4) | 43.4 (28.2 to 58.7) |
| 5–14 | 7/163 | 4.3 (2.1 to 8.6) | 13/20 | 65.0 (43.3 to 81.9) | 60.7 (38.6 to 77.7) |
| 15–24 | 14/110 | 12.7 (7.7 to 20.2) | 3/6 | 50.0 (18.8 to 81.2) | 37.3 (5.1 to 68.9) |
| 25–44 | 12/138 | 8.7 (5.0 to 14.6) | 4/9 | 44.4 (18.9 to 73.3) | 35.7 (9.5 to 64.9) |
| 45–64 | 16/95 | 16.8 (10.6 to 25.6) | 10/32 | 31.2 (18.0 to 48.6) | 14.4 (–1.5 to 32.8) |
| 65 +a | 153/520 | 29.4 (25.7 to 33.5) | 26/51 | 51.0 (37.7 to 64.1) | 21.6 (7.7 to 35.2) |
Validation of HI and MN cut-offs
To validate the HI cut-off of 1 : 32 and MN cut-off of 1 : 40 the distribution of titres in children (< 15 years of age) was compared between the baseline and the post-pandemic period (January–April 2010). The results are shown in Figure 9.
FIGURE 9.
Distribution of HI and MN titres in children of < 15 years of age in baseline and post-second-wave sera.
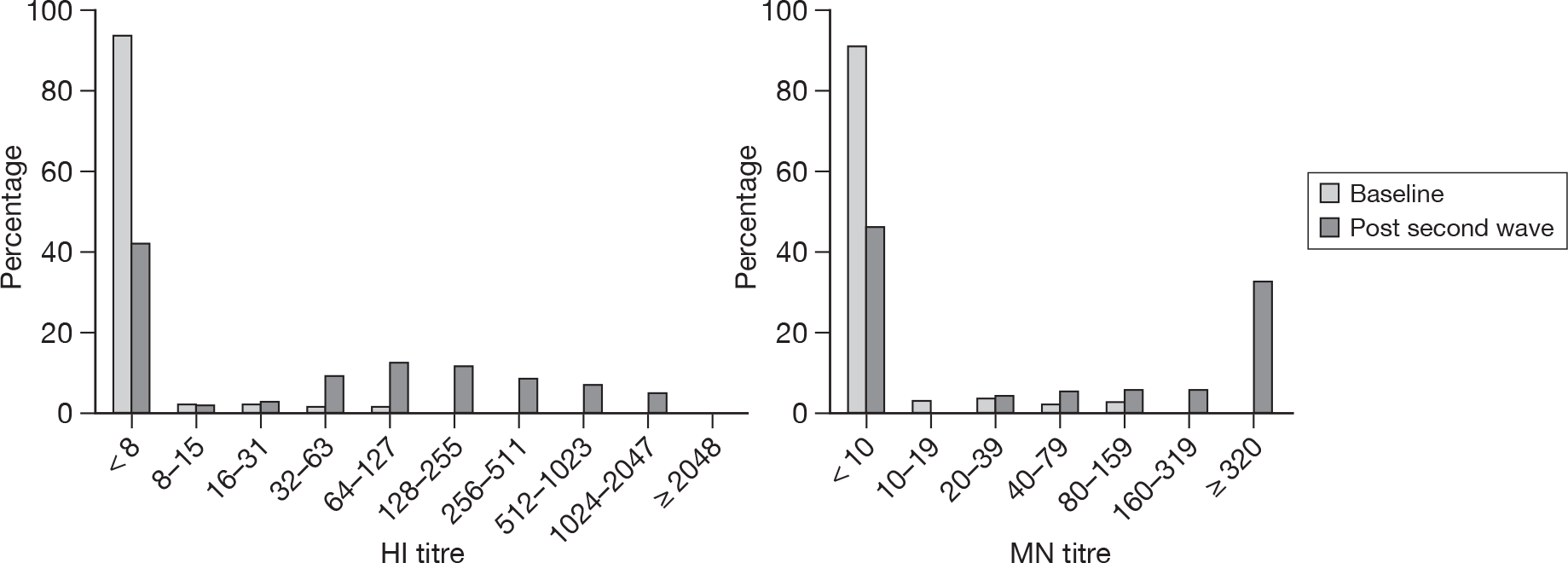
If it is assumed that the baseline period represents a true-negative distribution of titres and that the post-autumn-wave titres represent a mixture of positives and negatives then it can be seen that using HI titres of ≥ 1 : 32 and MN titres of ≥ 1 : 40 to indicate seroconversion will be highly specific, as only a small number of samples from 2008 [10 out of 359 (2.8%) for HI, 11 out of 306 (3.6%) for MN] have titres greater or equal to these cut-off values, and sensitive, as the vast majority of samples with detectable antibody in the post-autumn-wave samples (collected in 2010) have titres greater than these cut-off values [248 out of 268 (92.5%) for HI, 143 out of 156 (91.7%) for MN].
Association between HI and MN results
Pre-pandemic HI/MN correlation
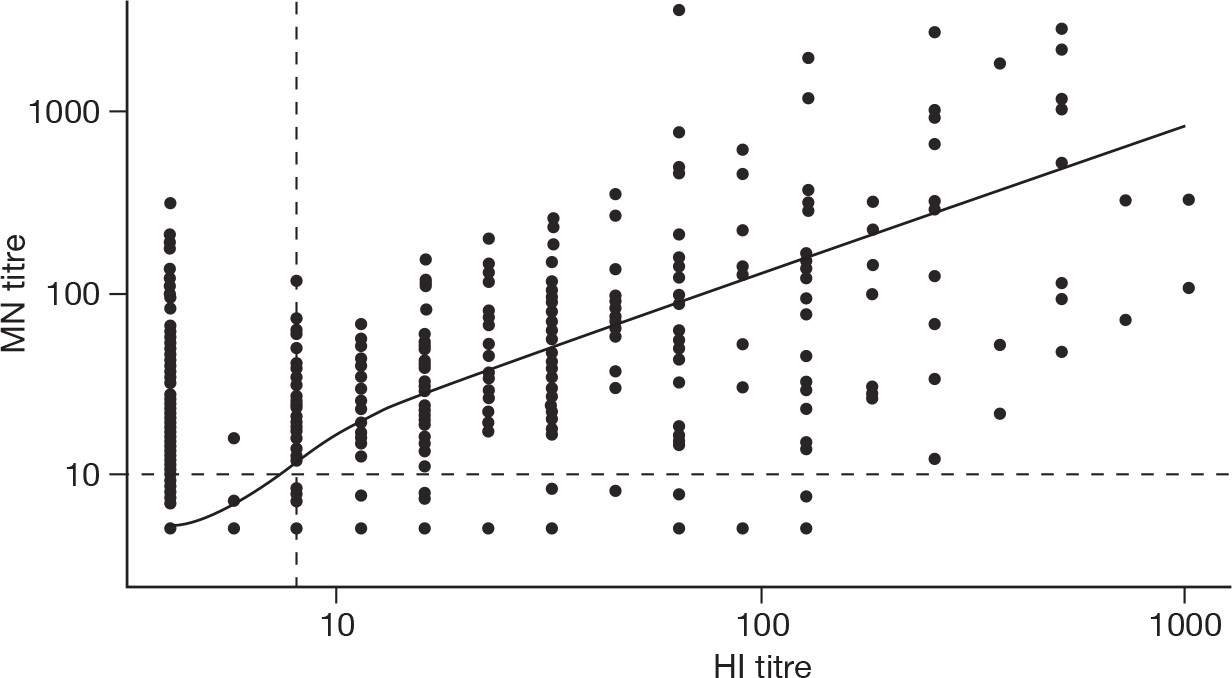
Figure 10 shows MN titres plotted against HI titres with a loess smoother for the 1169 samples tested with both assays. Spearman’s rank correlation r between the HI and MN titres was 0.68. There is therefore some evidence of positive correlation between the assays. Of the 823 samples with HI < 1 : 8, 162 (19.7%) had MN titres of ≥ 1 : 10 and 42 (5.1%) had MN titres of ≥ 1 : 40.
FIGURE 10.
Haemagglutination inhibition vs MN titres, all age groups, for samples collected in 2008. The dashed lines show the minimum detection limit for the two assays (1 : 8 and 1 : 10 for HI and MN, respectively). The solid line is a loess smoother.
Correlation between MN and HI titres in seroincidence samples
Figure 11 shows MN titres plotted against HI titres for the 1148 samples collected between 1 January and 19 April 2010 that had matched valid results on both MN and HI. Note that the highest dilution titrated to was an MN value of 320 so results are censored at this titre.
FIGURE 11.
Haemagglutination inhibition vs MN titres, all age groups, for samples collected January to April 2010. The dashed lines show the minimum detection limit for the two assays (1 : 8 and 1 : 10 for HI and MN, respectively). The solid line is a loess smoother.
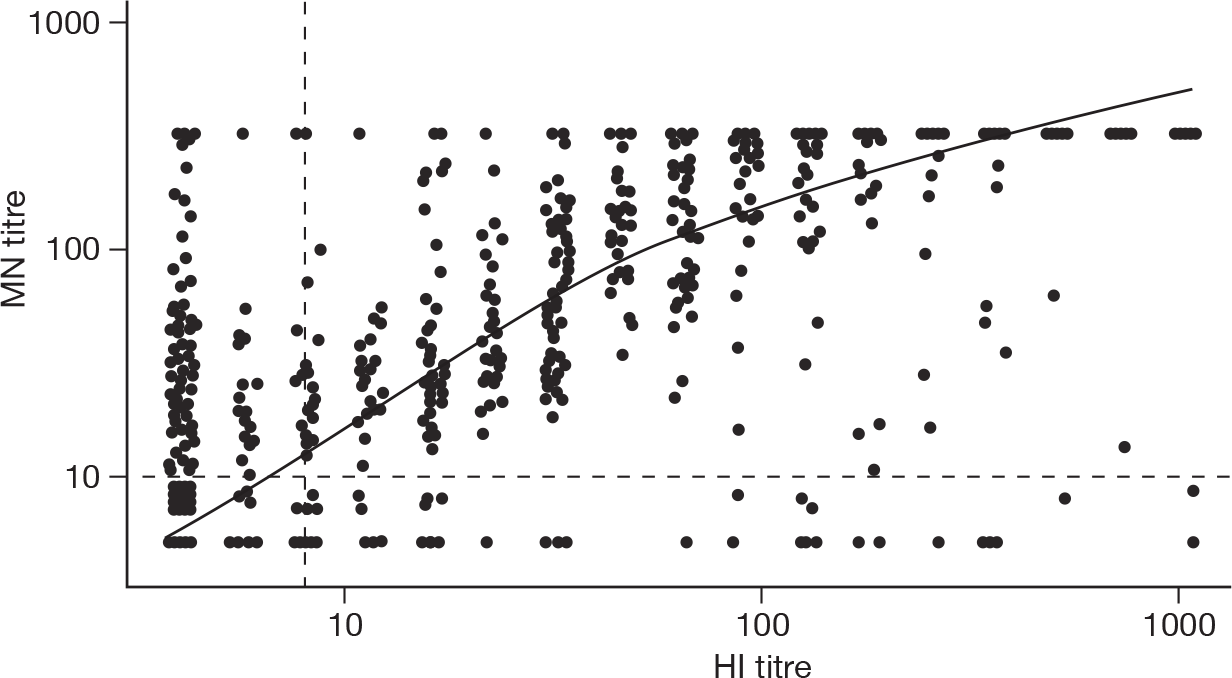
Spearman’s rank correlation r between the HI and MN titres was 0.79. As seen with the baseline samples, a large proportion of those with an HI titre of < 1 : 8 had MN ≥ 1 : 10 (136 out of 1148 samples, 11.8%).
Figure 12 shows the proportion of discordant samples by age group, i.e. the number of samples with detectable antibody on MN but not on HI [MN positive (MN +)/HI negative (HI–) as a proportion of all samples in that age group, and vice versa] in the baseline (Figure 12a) compared with the post-pandemic sera (Figure 12b). For the baseline sera the proportion of samples MN +/HI– increases with age (chi-squared test for trend p < 0.001) but not for samples HI +/MN– (chi-squared test for trend p = 0.30). Similar results were obtained for the post-pandemic sera, with the proportion of samples MN +/HI– increasing with age (chi-squared test for trend p < 0.001) but not for samples not for samples MN–/HI + (chi-squared test for trend p = 0.94.)
FIGURE 12.
(a) Baseline (2008). (b) Post second wave (January to April 2010).
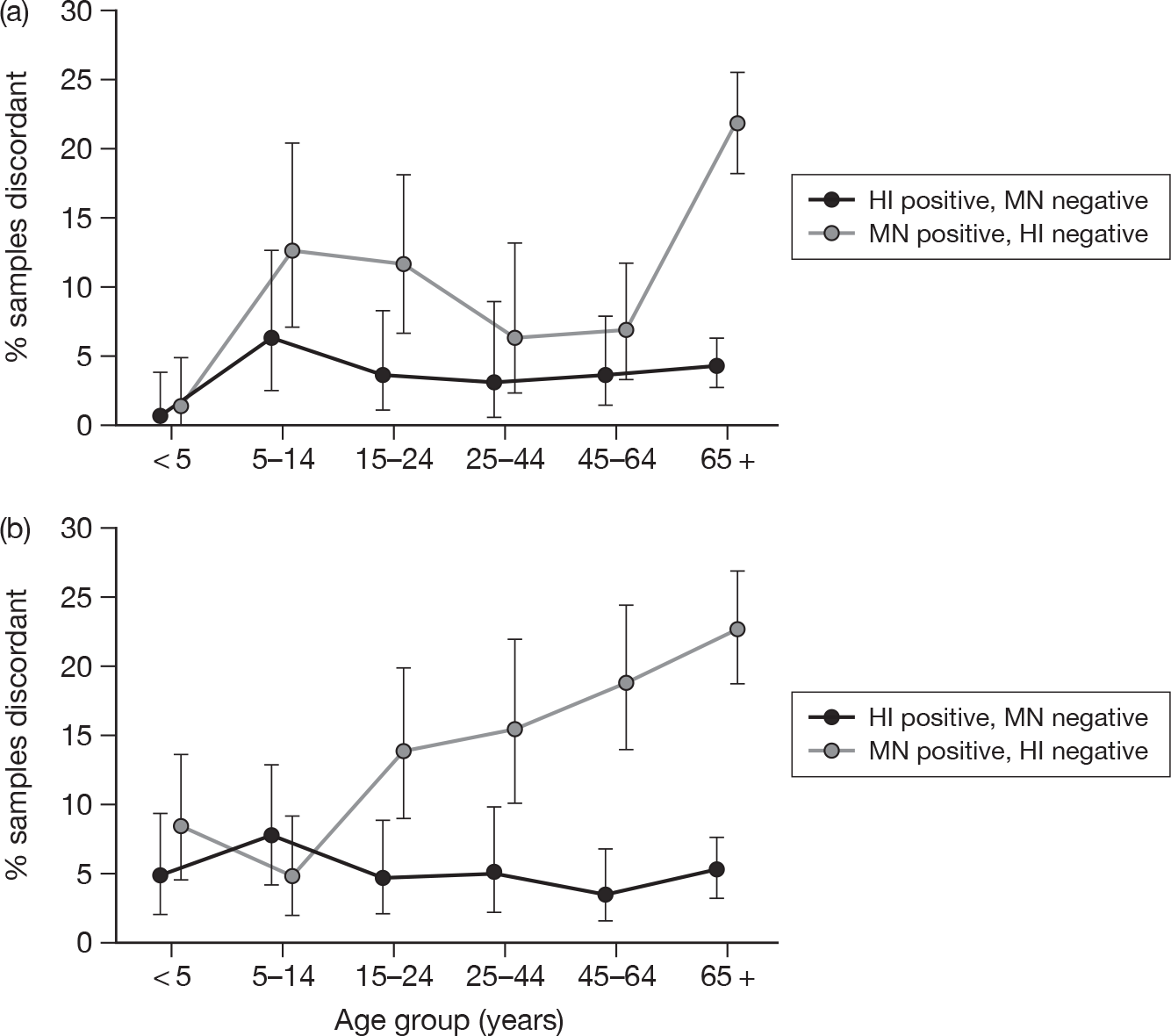
Among younger age groups (< 25 years) the point estimate of the proportion of discrepant samples with detectable antibody by MN but not by HI was smaller in the post-pandemic sera than in the baseline sera.
Incidence estimates using maximum likelihood estimation
The cumulative incidence across the first and second waves as estimated by the likelihood-based method shows a higher incidence in all age groups under 45 years in London than elsewhere (Table 22), consistent with the estimates from the conventional method presented above.
| Age group (years) | Baseline sera | Cumulative incidence | ||
|---|---|---|---|---|
| n ≥ 1 : 32 | n (95% CI) | For London (95% CI) | Outside London (95% CI) | |
| 1–4 | 2/125 | 1.6 (0.4 to 5.6) | 62.7 (48.8 to 76.0) | 42.0 (33.4 to 51.1) |
| 5–14 | 7/188 | 3.7 (1.8 to 7.5) | 82.8 (74.2 to 91.4) | 57.6 (52.1 to 63.0) |
| 15–24 | 21/120 | 17.5 (11.7 to 25.3) | 35.9 (17.7 to 54.0) | 22.6 (17.7 to 28.0) |
| 25–44 | 14/158 | 8.9 (5.4 to 14.3) | 32.2 (21.7 to 43.9) | 23.3 (19.4 to 27.2) |
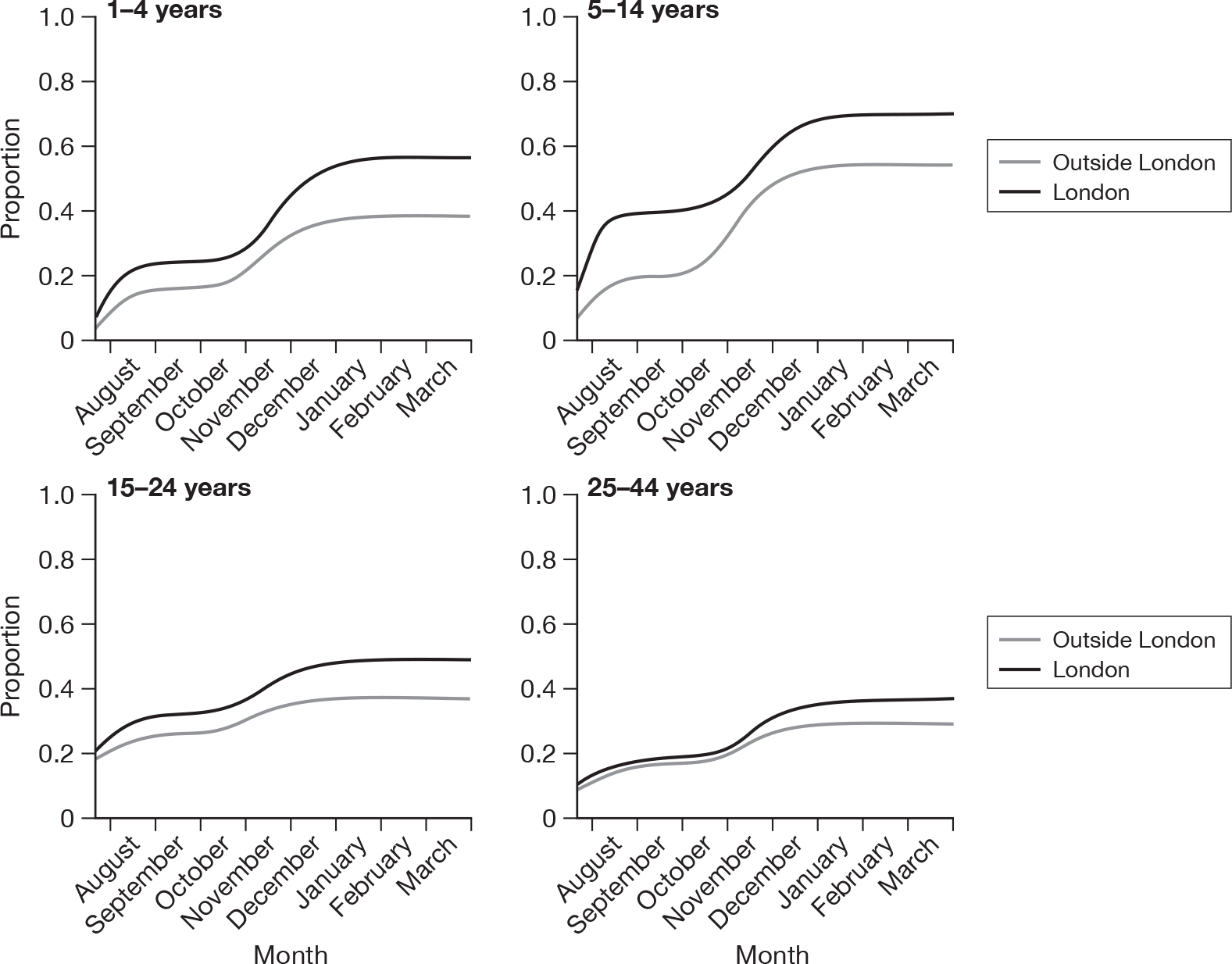
Regarding the timing of the pandemic waves, London had a big first wave among the 5- to 14-year age group, while seroprevalence in the rest of the country increased after the second wave, although cumulative incidence in London remained higher throughout the pandemic in each age group (Figure 13). By the end of the second wave it is estimated that as many as four out of five school-aged children in London had been infected.
FIGURE 13.
Predicted proportion of persons with HI titre ≥ 1 : 32, by age group, in London and elsewhere during the two waves of H1N1 2009 from likelihood estimation method.
Chapter 5 Discussion
Achievement of objectives
The objective of this project was to provide timely information on baseline immunity to the novel H1N1 2009 influenza virus and the incidence of infection as the pandemic progressed in order to improve understanding of its current and future epidemiology. Due to the unavoidable time required to develop and validate appropriate serological assays (a minimum of around 12 weeks from the start of the pandemic), the earliest that serological testing could begin was mid-July 2009.
The first report from the project was made available to the UK Scientific Advisory Group for Emergencies (SAGE), which provided independent advice on the pandemic and its management to government in mid-August 2009. This first report gave the baseline immunity results as measured by HI and preliminary data on the response to infection in confirmed cases. 7 Further serological reports were provided to SAGE in mid-September and October 2009, and revealed the true extent of infection in the first wave in England. A paper reporting the age-specific baseline immunity and incidence of infection in the first wave in England was submitted for publication by late November 2009. 8 The serological data assisted in the parameterisation of the real-time model developed by HPA, which was used to predict the future course of the pandemic and evaluate the likely impact of vaccination. 4 Thus, despite some difficulties in achieving the intended sample quotas each month from all regions and age groups,9,10 the project nevertheless met its overall objective. The excellent collaboration received from chemical pathology laboratories and the continuing participation of microbiology laboratories already contributing to the HPA serological surveillance programme was key in generating the timely incidence estimates.
At the request of the European Centre for Disease Control (ECDC), a guidance document on the conduct of such seroepidemiological studies based on the experience with this National Institute for Health Research (NIHR)/Health Technology Assessment (HTA)-funded project was prepared for the Community Network of Reference Laboratories for Human Influenza in Europe (CNRL) (see Appendix 3).
Interpretation of results
Baseline antibody prevalence
The results of the baseline prevalence survey showed that a substantial proportion of older adults had pre-existing cross-reactive functional antibodies capable of neutralising H1N1 2009, presumably as a result of prior exposure to antigenically related H1N1 influenza viruses circulating in previous decades, or as a result of heterosubtypic antibodies capable of providing broad cross-subtype protection. Examples of antibody that have broad neutralising capability across multiple influenza A subtypes have recently been described. 11–13 During every winter epidemic of influenza, it is estimated that between 10% and 20% of the population are infected with circulating influenza viruses. Virological surveillance in the UK since the 1950s has confirmed the presence of circulating influenza viruses during winter epidemics of ILI. In the twentieth century, the circulating viruses have been influenza A viruses H3N2, H2N2 and H1N1, at various times, or indeed co-circulating. The older an individual, the more influenza seasons and natural infections he/she will have experienced. Increasing time of exposure to influenza A viruses, including H3N2, H2N2 and H1N1, will increase the likelihood of an individual having a repertoire of heterosubtypic antibodies to conserved epitopes of viral proteins. The presence of such antibodies, irrespective of their derivation, is predictive of the probability of protection from infection with H1N1 2009, and is consistent with the lack of observed impact of H1N1 2009 in older age groups.
The prevalence of pre-existing antibodies increased significantly in those born before the 1950s with a further substantial rise in those born before the 1930s. Viruses of the A/H1N1 subtype circulated in humans from 1918 until they were replaced by A/H2N2 influenza, which caused the pandemic of 1957. During this period of circulation, A/H1N1 viruses underwent significant antigenic drift away from their 1918 virus progenitor. 14 In 1977, A/H1N1 influenza viruses from the early 1950s re-emerged in humans, and, until 2009, went through further substantial antigenic evolution in the human host. 15 By contrast, classical H1N1 influenza viruses in swine remained antigenically relatively static until 1998, which has created a substantial antigenic gap between classical swine H1 and human seasonal H1 viruses, to the point that swine became a reservoir of H1 viruses with potential to cause major respiratory outbreaks or even a pandemic in humans as seen in 2009. It is considered, however, that H1N1 in swine in the 1930s arose as a consequence of transmission from humans, or from a common source to the 1918 virus, and that swine viruses from the 1930s are likely to be closely related to viruses circulating in humans at that time.
All adults will therefore have had exposure at some time of their lives to human H1N1 viruses circulating during the last 90 years. Adults born between 1957 and 1977 will not have been exposed to H1N1 viruses as the primary influenza virus encountered in childhood, which may have a bearing on the nature of T-cell memory response and profile of durable subtypic antibody produced. There are no obvious differences, however, in the profile of antibody responses in the cohort of those aged 30–50 years in 2008 using the functional antibody tests described in this report. Elderly adults, i.e. those born in the first decades of the twentieth century, will have encountered 1918-like influenza viruses or close antigenic variants early in life, possibly as a primary infection, and this is likely to be reflected in the measurable antibody response seen to H1N1 2009, which shows closest genetic relationship to the older H1N1 viruses. There are limited data on older H1N1 viruses, derived from study and reconstruction of 1918 viruses, and use of early swine virus isolates from the 1930s. There are no isolates between 1918 and 1930 to help deduce the nature of antigenic variation during this time.
Structural and sequence analysis of H1N1 viruses shows that the HA of pandemic H1N1 2009 virus shares conserved antigenic epitopes with human and swine H1 viruses from the early twentieth century, and that HA from isolates from the 1918 pandemic (A/South Carolina/1/1918) are remarkably close relatives of the pandemic virus, showing only 20% amino acid difference in the antigenic sites, whereas HAs from viruses isolated later, A/Puerto Rico/8/1934 and A/Brisbane/59/2007, differ by 46% and 50%, respectively. 16,17 Recent mouse model experiments have confirmed that prior infection with 1918 H1N1 influenza affords a degree of protection against 2009 pandemic virus antigenic sites on the HA protein of swine lineage influenza viruses that have been preserved from 1918 to the present, and these epitopes may be susceptible to neutralisation by antibodies induced by variants closely related to 1918 influenza. 18 These in vivo experimental data support genetic information about likely relationships between ancestral H1N1 and H1N1 2009.
The proportion of individuals with antibody detectable by MN but not HI (Figure 12a) increased with age and was around 1 : 4 of all those aged 65 + years. As MN measures a larger range of neutralising antibodies than HI, this is consistent with the broad heterosubtypic immunity expected from a lifetime of exposure to influenza A viruses. Comparing the antibody targets of these two tests, HI analysis detects almost exclusively antibodies to the receptor binding site of the HA on the globular head of HA, which is considered to be the main target of neutralising antibodies. MN detects a broader range of neutralising antibodies, which may be directed against more conserved regions of the HA molecule or other viral targets. As a result, MN is a more sensitive and less strain-specific assay than HI,19 although there is generally a good correlation between HI and MN antibody titres for detection of post-infection or vaccination antibody to homologous strains. However, in unexposed individuals, MN is the more sensitive test for detection of cross-reactive antibodies. Individuals showing antibody by MN, but not HI, have been identified frequently (HPA unpublished data). 19,20 While antibody in the baseline is probably caused by previous exposure to seasonal influenza viruses (cross-reactive antibodies, mainly detected by MN, and perhaps longer lasting), antibody levels detected post summer and autumn waves are almost certainly attributable to infection, characterised by recognition of both, strain specific epitopes (detectable by HI, perhaps shorter lasting) as well as conserved epitopes and thus increases the agreement between both functional tests.
The same principles apply also for the older age groups and could explain why we observed the highest levels of discordant results in these groups. The high frequency of positive MN, but negative HI titres indicates the high level of antibody recognising conserved epitopes in these age groups. The agreement between both tests does not increase as a result of the pandemic, as these individuals had a far lower infection rate, indicated by the UK case estimates, numbers of hospitalised/deceased patients and HI data from this study. However, to be more conservative, and as correlates of protection for MN are unknown, we based most of our analysis on HI titres of ≥ 1 : 32. 5
Using the HI correlate of protection of ≥ 1 : 32 suggests that about 25% of adults aged 65 + years should be protected, whereas it may be as high as 67% if any detectable MN antibody is protective. Estimates of the baseline age-specific immunity to H1N1 2009 from transmission models may help determine the likely correlate of protection with each assay, which would be helpful in refining a crude estimate of protection.
Data from our baseline serosurvey are therefore consistent with historical observations and contemporary laboratory experimental work. Cross-reactive antibody titres are highest in birth cohorts that were born during the 1918–57 circulation of H1N1, with the highest titres in those born earlier in that period. However, it is also the case that the oldest age groups will be the most highly vaccinated individuals, consistent with UK national vaccination policy recommending vaccination for all persons over 65 years. A majority of those older than 65 years in our study, birth cohorts from 1910 to 1940, will have received at least one influenza seasonal influenza vaccine, and, more likely, multiple influenza vaccines in the last decade. The vaccines in use in UK vaccination campaigns are all trivalent inactivated vaccines, where the major antigen is the viral HA from influenza A and B strains selected to be representative of circulating viruses. This will include representatives of circulating H1N1 viruses that are antigenically distant from H1N1 2009. There are now reports of prior infection with seasonal influenza A in animal models reducing pandemic virus shedding and transmission. 21 Preliminary data also suggest that seasonal influenza vaccination may confer a degree of cross-protection to other H1N1 strains. 22 In the sample set analysed for this report, seroreactivity to pandemic virus after seasonal influenza vaccination is limited and does not appear to provide significant cross-reactive antibody to H1N1 2009, as illustrated in Table 3. We showed increased seroreactivity to the NIBRG122 virus after receipt of 2005 seasonal vaccine (southern hemisphere) in only 5.8% of all subjects. Similarly, only 7.9% of subjects with clinical diagnosis of ILI prior to the 2009 pandemic showed evidence of seroconversion. Conversely, none of the subjects from the SRI panel seroconverted. Overall, the extent of cross-reactive antibody derived from our own analysis is consistent with work described elsewhere. 20,22 However, the extent to which vaccination in an older population may boost pre-existing antibody and contributes to protection remains to be further explored, and has not been the subject of this work.
Although the data from these assays are useful, and help provide a more detailed explanation of the observed patterns of pandemic attack incidence, they are necessarily pragmatic with a reasonably simple read-out, amenable to high-throughput laboratory work. This provides a population-based approach to analysis of immunity from many different individuals. However, detailed epitope mapping of antibody repertoire at an individual level in older populations is required to deduce the exact reactivity of neutralising antibody reactive in either MN or HI assays for an in-depth understanding of the nature of cross-protection provided in older individuals. This will be fundamental to understanding the pattern of morbidity and mortality seen in all pandemics, where there is sparing of the older adult population, and may have generalisable lessons for vaccine design and the induction of broad cross-reactive immunity. There are different scientific and technical approaches to this, which could include molecular cloning of B cells genes to look at immunoglobulin repertoire23 or the use of phage display to dissect antibody repertoire in individual patients. 24 Both approaches are highly labour intensive, and currently unsuitable for the population-based approach taken here, but are likely to yield important insights in coming years to provide the fundamental detail to explain some of the observations that we and others have made.
Seroincidence estimates
The seroprevalence results for the samples collected after the summer wave in September 2009 were the first to reveal the true extent of H1N1 2009 infection, particularly among school-aged children in London. 8 When compared with the estimated number of clinical cases in this group using the HPA statistical method2 the seroincidence estimates were around 10 times higher. A similar 10-fold scaling factor was estimated from the HPA real-time model. This used the baseline immunity data from the serological survey, together with information on the rate of increase of cases early in the second wave of the pandemic to estimate the age-specific proportion of the population infected in the first wave. 4
Whether the 10-fold difference is due to overestimation of the proportion of individuals with ILI who consulted a health-care professional during the first wave (assumed to be between 20% and 50% in the HPA method) and/or a greater than expected proportion of infections that were asymptomatic or atypical in their clinical presentation cannot be determined on the basis of the serological data alone. In a small household contact study in Hong Kong, of 11 patients with serological evidence of H1N1 2009 infection (fourfold rise in MN titres between acute and convalescent sera) three (27%) had an ILI, although six (55%) had two or more respiratory symptoms. 25 These proportions are similar to those reported for infections with seasonal influenza viruses26 and from a field study in a school in England in May and June 2009 (HPA unpublished data). There is little information, however, on propensity to consult among individuals with ILI in the pandemic in the UK, apart from a small web-based survey that was largely restricted to adults. 27 A large telephone survey in New Zealand suggested that only around 1 in 18 individuals with ILI consulted in the first pandemic wave in that country. 28 This estimate is considerably higher than that used to derive the HPA clinical case estimates.
Our seroincidence estimates show that the second wave of infection that started when the schools re-opened in September 2009 was considerably larger than the first wave that occurred in the summer (Table 14). The relative magnitude of the two waves, as estimated from the serology, is thus more in line with that suggested by the mortality data than the HPA case estimates (Figure 1), which did not take account of likely changes in the propensity to consult over time. There is evidence both from likelihood-based estimates of seroincidence in the second wave and other surveillance data sources3 that the propensity to consult is likely to have been lower in the second than the first wave of infection in the UK.
The difference in magnitude between the two waves varied between regions. In the non-London regions, there was little evidence of infection outside the 5- to 14-year age group in the first wave (estimated seroincidence for this group 6.2%, 95% CI 0.3 to 13.4). In contrast, all age groups outside London showed evidence of infection in the second wave, ranging from 53.1% (95% CI 43.9% to 60.3%) in 5- to 14-year-olds to 15.4% (95% CI 8.7% to 21.4%) in those aged 65 + years (Table 15). The pattern was different in the London region where the first wave was larger than the second (Table 16). The pandemic started earlier in the London region and reached the highest estimated rate of clinical cases/100,000 population of any region in the first wave8 before the national closure of schools took effect and temporarily reduced R to < 1 in all regions. The seroincidence data for the first wave are thus consistent with the regional clinical case estimates using the HPA method. Non-London regions then experienced their major wave of infection when the schools reopened after the summer holidays. The post-second-wave serology results provide some evidence that the final cumulative incidence was higher in London than elsewhere. Given the particular demographic structure of London with a smaller proportion of individuals from the older age groups29 (i.e. those groups with pre-existing immunity), and more contacts due to population density, it can be hypothesised that the higher attack rates in London were due to a bigger R during the two waves. This, together with the likely higher number of imported infections in London than elsewhere, may explain why this region experienced an earlier start of the pandemic and larger overall incidence than other regions.
In addition to regional differences in the timing of the start of the pandemic, there were also differences in timing between age groups, with adults over 24 years in all regions being infected later than those in younger age groups. This is consistent with the key role of children in transmission in the early stages of the pandemic and the social mixing patterns between and within age groups. 30 Clearly, school children are a key target for intervention during a pandemic, especially if the aim is to delay its progression in order to buy time until pandemic strain vaccines become available. The widespread use of antiviral prophylaxis in schools as part of the UK containment policy31 did not appear to be effective in delaying progression of the first wave of infection. While antiviral prophylaxis may have been effective at an individual level1 initial cases in schools were often not identified early enough for prophylaxis to have had a major impact on disease transmission. 32 In contrast, school closures seem extremely effective in reducing transmission as seen by the termination of the first wave when all schools closed for the summer holidays. However, use of national school closure at other unplanned times as a pandemic control measure could result in a considerable economic and social burden. Localised school closures could potentially alleviate the burden on hospital intensive care units that are reaching capacity but a recent modelling study33 shows that, for a range of epidemiologically plausible assumptions, considerable local coordination of school closures would be needed to achieve a substantial reduction in the number of hospitals where capacity is exceeded at the peak of the epidemic. The heterogeneity in demand for intensive care beds means that even widespread school closures are unlikely to have an impact on whether demand will exceed capacity for many hospitals. 33 If school closure is to be used as an intervention strategy in a future pandemic, its deployment may need to be reserved for a more severe strain than the H1N1 2009 virus.
It is not straightforward to disentangle to what the extent the low attack rate in older age groups (and conversely the very high attack rate in younger age groups) is explained by differing social mixing patterns or protective immunity from past infections. There are uncertainties around measuring mixing patterns, partly as a result of their survey origin and partly because it is not clear which type of contacts best describe influenza transmission (i.e. physical or conversational30). There are also uncertainties regarding the interpretation of the high level of baseline cross-reactive antibodies among older persons found in this study in terms of protection against infection and disease. A modelling approach has been used to infer how well mixing patterns derived from the POLYMOD (Improving Public Health Policy in Europe through Modelling and Economic Evaluation of Interventions for the Control of Infectious Diseases) study30 and the observed age-specific immunity profile as described in this study predict the observed pattern of infection. 4 This showed that both factors contribute to reinforce the high attack rate in younger age groups. Some early predictions4 based on scenarios fitted to the HPA clinical case estimates up to October 2009 with a 10-fold scaling factor to derive the number of infections, indicated cumulative attack rates of around 42% in 5- to 14-year-olds and 4% in those aged 65 + years. Even if these numbers need to be revised in the light of the serological data generated in the second wave, these model predictions show that the difference in attack rates between school age children and the elderly can be explained easily by a combination of social mixing patterns and pre-existing immunity.
Study limitations
The serum samples in our study were not obtained as a result of a population screening approach whereby individuals or families are selected at random and asked to provide blood samples for a specific study. While such a method does allow additional clinical and epidemiological information to be obtained with the serum sample, it generally suffers from a low participation rate, which may itself introduce bias and offset the advantages of the random sampling approach. In addition, young children are usually excluded for ethical reasons. For conducting a rapid H1N1 2009 seroincidence survey, the time required to obtain ethics approval and individual patient consent, together with the logistics and costs of such an approach, rendered a population-screening method impractical. The use of serum samples taken from individuals accessing health care for clinical reasons unrelated to a recent illness suggestive of influenza provided a convenient alternative method.
Construction of an HPA annual serum archive from residual aliquots of samples submitted to microbiology laboratories for screening or diagnostic testing has been in operation for over two decades, having been originally established to monitor the impact of the combined measles, mumps and rubella vaccine on age-specific population immunity. 34 The HPA archive has been extensively used for other seroepidemiological studies35 and has proven particularly useful for infections with a high incidence for which exposure is largely age dependent rather than determined by specific behavioural factors (such as for HIV) or associated with particular ethnic groups (such as hepatitis B). Because of the need for rapid generation of incidence data for H1N1 2009, serum collection was extended to chemical pathology laboratories – a new source for seroepidemiological studies. While there were no significant differences in H1N1 seroprevalence between samples from microbiology and chemical pathology laboratories in the logistic regression model (Table 13), patients undergoing regular chemical pathology testing may be more likely to have underlying morbidities (such as chronic cardiac, renal or respiratory conditions) than the general population. This could bias the results if such conditions affect the likelihood of being infected with the H1N1 2009 virus or the likelihood of having been vaccinated with the H1N1 2009 pandemic vaccine. Unless the underlying clinical conditions have a major impact on mixing patterns then bias in the estimates of H1N1 2009 infection through differential exposure would be unlikely. However, these clinical conditions are indications for seasonal and pandemic influenza vaccination, which is a potential source of bias that needs to be considered. As discussed earlier, it is unlikely that prior seasonal influenza vaccination will have generated cross-reactive antibody but the roll-out of pandemic stain vaccine in the UK for high-risk individuals from early November 200936 and for all children under 5 years of age from January 201037 may have biased the analysis, at least in the samples taken from November onwards.
The pandemic vaccine uptake rates by age group in England, derived from database extracts from the 96 general practices enrolled in the RCGP network,38 are shown in Figure 14. Given the relatively high uptake rates in the 65 + age group (26% overall and 41% in high-risk groups), and to a lesser extent in the 45- to 64-year age group (11% overall and 41% in risk groups) some of the increase in the prevalence of H1N1 2009 antibodies in these age groups between the 2008 baseline and the post-second-wave sera is likely to have been due to vaccination, even without selective inclusion of sera from high-risk individuals targeted for vaccination. Vaccine uptake among those aged between 5 and 45 years was much lower overall (< 5%) due to the lower proportion with underlying clinical conditions in these age groups, although coverage in risk groups was relatively high (30%, 19% and 27% for the groups aged 5–14, 15–24 and 25–44 years, respectively). The extent to which vaccination may have contributed to the change in antibody prevalence compared with the baseline in the sera collected from November onwards is therefore difficult to assess. However, even if sera in the 5- to 14-year-olds were exclusively from those in risk groups, the vaccine gave 100% seroconversion, and vaccination only occurred in persons not already infected, the increase in seroprevalence between the first and second wave (51.4%, as shown in Table 14) in this age group is substantially higher than the 30% observed uptake in 5- to 14-year-olds in a risk group. For those aged < 5 years, vaccine was being delivered from January 2010 onwards (while the post-second-wave sera were being taken) and was targeted at all children not just those in risk groups so increases in seroprevalence would be expected from vaccination. It is perhaps surprising therefore that there was evidence of a decline in the proportion with HI titres ≥ 1 : 32 after January in this age group (Figure 7).
FIGURE 14.
Percentage of populations vaccinated with pandemic strain vaccine by age group and week, 2009 and 2010 (data from RCGP).
In estimating cumulative incidence across the two waves of infection, we assumed that HI and MN antibodies developed in response to infection remain at a titre of ≥ 1 : 32 or ≥ 1 : 40, respectively, for around a year, such that that individuals infected early in the first wave (e.g. school-aged children in London in May 2009) would still be seropositive if tested in say March/April 2010. The data on antibody titres in confirmed cases did not allow this assumption to be tested, as there were few samples taken later than 90 days after onset and few overall from children. It is possible that the decline in proportions with HI titres ≥ 1 : 32 in the March 2010 sera, which was statistically significant only in London, may have been the result of waning antibody levels following infection in the first or early in the second wave in this region.
Ideally, to generate information on the serological response in laboratory-confirmed cases of H1N1 2009 infection, a cohort would have been prospectively recruited and those with a PCR-confirmed infection would be followed up with sequential serology samples. Such an approach would be expensive and time consuming and there is no reason to assume that our method of using single serum samples taken from different individuals at various time after confirmation of infection would have produced biased results. The serum samples from confirmed cases that were used to estimate the temporal distribution of the seroconversion interval distribution were obtained from a variety of sources. Many were obtained from the follow-up of the first few hundred confirmed cases and their contacts (the so called FF100 database1). These were actively sought to try and identify seroconversions in contacts of confirmed cases to derive secondary attack rates. Although few paired samples were obtained it is difficult to envisage why those that were obtained from PCR-positive contacts or index cases were somehow biased with respect to the serological response they developed. Other samples were obtained from subjects investigated as part of outbreaks while some were individuals for whom the clinician decided that a serological test for H1N1 2009 was indicated. The distribution for the seroconversion intervals obtained with this approach was consistent with what would be expected for seasonal influenza.
Other limitations of our method of estimating incidence by measuring differences in prevalence between two time points are that it does not take account of the variable time to seroconversion between individuals and the fact that a small proportion of infected individuals do not appear to seroconvert by methods used in this study. It also relies on grouping by calendar month yet incidence may be changing rapidly over this time period. The likelihood-based estimation was developed to overcome some of these limitations and allowed the generation of a continuous cumulative incidence curve by region and age group over the first and second waves. However, to accommodate complexities such as vaccination, or a non-random serum sampling strategy with the potential for oversampling in risk groups for whom vaccination was recommended, or changes in the propensity to consult over time, a more complex set of parameters would need to be estimated. Additionally, the method can incorporate other data sets, such as the vaccine uptake data by age and risk groups or results from vaccine immunogenicity trials. For this, Markov Chain Monte Carlo (MCMC) could be used with the combined likelihood of the different data sets to draw jointly a sample from the parameter space and then the parameters can be evaluated from the sampled distribution. Such a model could also incorporate the effect of waning antibody levels if this is shown to be important using additional data on antibody persistence after infection or vaccination.
Our study did not provide the timely incidence estimates to inform the parameterisation of ‘real-time’ predictive models as originally envisaged, due to the necessary lag time in H1N1 2009 assay development and, to a lesser extent, the collection and testing of the seroincidence samples. However, had the pandemic been with an H5N1 virus for which serological assays had already been developed then generation of baseline data on the age-specific prevalence of cross-reactive antibodies and insight into the incidence of infection would have been obtained more rapidly. Indeed the HPA serum archive has already been used to measure the prevalence of cross-reactive antibodies to the H5N1 virus. Nevertheless the data generated on the prevalence of cross-reactive antibodies to H1N1 2009, which was available prior to the second wave, did assist in the parameterisation of the HPA real-time model used to evaluate the likely impact of vaccination. 4 It also helped validate the scaling factor used in that model to convert the HPA clinical case estimates to infections. 8 For future pandemics, the availability of new serological tools that can detect incident infection in a single sample (e.g. analogous to an Immunoglobulin M test) rather than relying on changes in seroprevalence over time, and the availability of non-invasive techniques, such as oral fluid testing, would greatly facilitate the rapid generation of seroincidence data through a random population-screening approach.
Finally, our study was geographically limited to England. As the pandemic was UK wide, although with some differences in timing and magnitude,39 it would have been informative if a pan-UK study had been undertaken that included Scotland, Wales and Northern Ireland. A proposal for a similar seroepidemiological study was funded in Scotland, for which we shared our protocol and testing standard operating procedures (SOPs), and work is under way to provide data to compare with our study, although is not available at present. Seroepidemiology from Northern Ireland would also have been useful to compare with England, particularly with the different timing and impact of first wave in Northern Ireland. Given the difference between the UK and other European countries in the timing of the first wave of infection, useful epidemiological insights might have been gained if comparable studies had been conducted in those countries. Although serological data are emerging from a range of European Union (EU) countries, the complexity of health systems and variability of studies undertaken makes developing a regional European picture of impact of the pandemic unfeasible at present.
Implications for the NHS
-
The current low levels of susceptibility to the H1N1 2009 virus in the population of England after the second wave, particularly in school-aged children who are the main transmitters of infection, together with the early decline in clinical cases in November 2009, at a time when seasonal influenza is usually increasing, imply that there has been sufficient infection of susceptibles in the population such that a third wave of infection in the 2010–11 influenza season is not to be expected. This interpretation would be consistent with the HPA real-time model that correctly predicted that the second wave would peak in early November 2009 when R fell below ‘1’ due to the exhaustion of susceptibles. 4 The situation could change, however, if there was emergence of an antigenically drifted strain for which antibody generated through infection or vaccination with the H1N1 2009 strain did not provide good cross-protection. Nevertheless, sporadic cases of H1N1 are likely to continue to occur, some of which may arise in particular risk groups and be associated with severe illness. The inclusion of pregnant women in groups recommended for influenza vaccination in the winter of 2010–11 is a recognition of increased risk of severe illness from influenza H1N1 2009, and clinicians should remain alert to the possibility of influenza in this and other risk groups, ensuring early access to antiviral drugs and laboratory confirmation where appropriate.
-
Continued virological surveillance of the strains causing influenza is therefore essential during the 2010–11 season. This requires the ongoing cooperation of NHS colleagues in the enhanced surveillance schemes run by the HPA, whereby patients presenting with suspected influenza in general practice or other health-care facilities are investigated virologically whenever possible, followed by full virus characterisation, ensuring that priority is given to analysis of viruses from severe illness. The use of sentinel hospital trusts for surveillance and weekly reporting of laboratory-confirmed hospitalised cases of influenza H1N1 during winter 2010–11 is intended to provide information about ongoing severe illness.
-
Measurement of the HI and MN titres to any drifted strains in sera generated by infection or vaccination with the H1N1 2009 virus would be essential for the rapid assessment of the potential for a third wave of infection. There is also lack of information on persistence of antibody levels after an influenza infection, especially in children, and thus the degree of protection that can be expected from an infection or vaccination a year ago against the same strain.
-
Opportunities to collaborate with NHS partners to study persistence antibodies after vaccination or infection should therefore be pursued, as well as further investment in pandemic preparation within the NHS to ensure that robust mechanisms for future serosurveillance in different sectors of acute care delivery are in place and can be rapidly activated.
Chapter 6 Research recommendations
The research recommendations are as follows:
-
The authors consider that investment in seroepidemiological studies for seasonal influenza would improve understanding of its epidemiology and the impact of vaccination. Investing in infrastructure for storage and investigation of alternative modalities of collection, such as dried blood spots, would enable more rapid execution of research to inform the management of future epidemics.
-
Collaboration between the devolved administrations in the UK in the preparation of pandemic plans to ensure a common approach to generating comparable seroepidemiologal data.
-
Detailed analysis of surveillance data from H1N1 2009 to ensure legacy systems that can provide information about propensity to consult are developed for use in seasonal influenza.
-
Development of more rapid serological assays that can measure recent infection in a single acute sample and do not require collection of convalescent sera.
-
Further research into key cross-reacting antibodies, their genesis and implications for immunity in older people.
-
Further snapshot of population immunity at regular intervals during the next 5 years to track the waning of immunity to pandemic influenza in the affected ages and investigate the interplay with immunity arising from seasonal circulating viruses.
-
Further development of statistical methods, such as likelihood-based estimation, which can facilitate the rapid interpretation of serological data for real-time model parameterisation.
Acknowledgements
The authors would like to thank the HPA RMN and NHS laboratories that collect samples for the HPA seroepidemiology programme. We also thank the chemical pathology laboratories in the participating regions for their collaboration in the collection of the seroincidence samples and the Royal College of Pathologists and HPA regional microbiologists for their support in recruitment of these laboratories. We are grateful for the technical support provided by staff at (1) the RVU: Janice Baldevarona, Sharleen Braham, Lucy Breakwell, Surita Gangar, Paul Kotzampaltiris and Sejal Morjaria, and (2) at the SEU: Kevin Potts, Lucy Black and Victoria Catterall. We also thank Dr Richard Pebody for use of the serum archive from microbiology laboratories, Dr David Dance for his support in coordinating the sample collection and Dr Douglas Fleming of the RCGP Research and Surveillance Centre for allowing us to use the RCGP database extracts to estimate uptake of pandemic vaccine.
Contribution of authors
Dr Pia Hardelid (Statistician) conducted the statistical analyses of seroprevalence, seroincidence and assay comparisons, and contributed to drafting the methods and results section of the paper.
Nick Andrews (Senior Statistician) helped with the study design, data analysis and drafting the paper.
Dr Katja Hoschler (Clinical Scientist) was responsible for the assay development and validation, as well as performance of serological analysis, and contributed to drafting these sections of the paper.
Elaine Stanford (pre-registration Clinical Scientist) was responsible for the collection and storage of serum samples and demographic data, and contributed to the drafting of this section of the paper.
Dr Marc Baguelin (Mathematical Modeller and Health Economist) carried out the analysis using the likelihood-based method, and helped with the selection of samples for testing and drafting the paper.
Pauline Waight (Senior Scientist) linked the confirmed H1N1 2009 cases and serology databases, managed these databases and contributed to the drafting of these sections of the paper.
Professor Maria Zambon (Director of the CFI) directed the validation and interpretation of laboratory data and contributed to the analysis and drafting of the paper.
Professor Elizabeth Miller (Consultant Epidemiologist) designed the study, submitted the grant application, directed the analysis and contributed to drafting the paper.
All authors commented on, and reviewed, the final paper.
Publication
Miller E, Hoschler K, Hardelid P, Stanford E, Andrews N, Zambon M. Incidence of 2009 pandemic influenza A H1N1 infection in England: a cross-sectional serological study. Lancet 2010;375:1100–8.
Disclaimers
The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the referees for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report. The views expressed in this publication are those of the authors and not necessarily those of the NIHR or the Department of Health.
References
- McLean E, Pebody RG, Campbell C, Chamberland M, Hawkins C, Nguyen-Van-Tam JS, et al. Pandemic (H1N1) 2009 influenza in the UK: clinical and epidemiological findings from the first few hundred (FF100) cases. Epidemiol Infect 2010;138:1531-41.
- Health Protection Agency . Method Used to Estimate New Pandemic (H1N1) Influenza Cases in England in the Week 3 August to 9 August 2009 n.d. www.hpa.org.uk/web/HPAwebFile/HPAweb_C/1250150839845 (accessed 6 September 2010).
- Van Hoek AJ, Miller E. Response to Guest Editorial ‘Influenza Surveillance, the Swine-Flu Pandemic, and the Importance of virology’ n.d. http://clinicalevidence.bmj.com/ceweb/resources/editors-letter-response.jsp (accessed 21 June 2010).
- Baguelin M, Hoek AJ, Jit M, Flasche S, White PJ, Edmunds WJ. Vaccination against pandemic influenza H1N1 v in England: a real-time economic evaluation. Vaccine 2010;28:2370-84.
- Hobson D, Curry RL, Beare AS, Ward-Gardner A. The role of serum haemagglutination-inhibiting antibody in protection against challenge infection with influenza A2 and B viruses. J Hyg (Lond) 1972;70:767-77.
- Office for National Statistics . 2009 Mid-Year Population Estimates n.d. www.nomisweb.co.uk (accessed 13 October 2010).
- Health Protection Agency (HPA) . Centre for Infections (CFI). Update on Assay Development and Preliminary Seroepidemiological Results 2009.
- Miller E, Hoschler K, Hardelid P, Stanford E, Andrews N, Zambon M. Incidence of 2009 pandemic influenza A H1N1 infection in England: a cross-sectional serological study. Lancet 2010;375:1100-8.
- Bird S. Like-with-like comparisons. Lancet 2010;376:684-5.
- Miller E, Hardelid P, Andrews N. Like-with-like comparisons. Authors’ reply. Lancet 2010;376:684-5.
- Chen Y, Qin K, Wu WL, Li G, Zhang J, Du H, et al. Broad cross-protection against H5N1 avian influenza virus infection by means of monoclonal antibodies that map to conserved viral epitopes. J Infect Dis 2009;199:49-58.
- Kashyap AK, Steel J, Oner AF, Dillon MA, Swale RE, Wall KM, et al. Combinatorial antibody libraries from survivors of the Turkish H5N1 avian influenza outbreak reveal virus neutralization strategies. Proc Natl Acad Sci USA 2008;105:5986-91.
- Stropkovská A, Mucha V, Fislová T, Gocník M, Kostolanský F, Varecková E. Broadly cross-reactive monoclonal antibodies against HA2 glycopeptide of Influenza A virus hemagglutinin of H3 subtype reduce replication of influenza A viruses of human and avian origin. Acta Virol 2009;53:15-20.
- Tumpey TM, García-Sastre A, Taubenberger JK, Palese P, Swayne DE, Basler CF. Pathogenicity and immunogenicity of influenza viruses with genes from the 1918 pandemic virus. Proc Natl Acad Sci USA 2004;101:3166-71.
- Hay AJ, Gregory V, Douglas AR, Lin YP. The evolution of human influenza viruses. Philos Trans R Soc Lond B Biol Sci 2001;356:1861-70.
- Xu R, Ekiert DC, Krause JC, Hai R, Crowe JE, Wilson IA. Structural basis of preexisting immunity to the 2009 H1N1 pandemic influenza virus. Science 2010;328:357-60.
- Garten RJ, Davis CT, Russell CA, Shu B, Lindstrom S, Balish A, et al. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 2009;325:197-201.
- Kash JC, Qi L, Dugan VG, Jagger BW, Hrabal RJ, Memoli MJ, et al. Prior infection with classical swine H1N1 influenza viruses is associated with protective immunity to the 2009 pandemic H1N1 virus. Influenza Other Respir Viruses 2010;4:121-7.
- Okuno Y, Tanaka K, Baba K, Maeda A, Kunita N, Ueda S. Rapid focus reduction neutralization test of influenza A and B viruses in microtiter system. J Clin Microbiol 1990;28:1308-13.
- Hancock K, Veguilla V, Lu X, Zhong W, Butler EN, Sun H, et al. Cross-reactive antibody responses to the 2009 pandemic H1N1 influenza virus. N Engl J Med 2009;361:1945-52.
- Laurie KL, Carolan LA, Middleton D, Lowther S, Kelso A, Barr IG. Multiple infections with seasonal influenza A virus induce cross-protective immunity against A(H1N1) pandemic influenza virus in a ferret model. J Infect Dis 2010;202:1011-20.
- Lee VJ, Tay JK, Chen MI, Phoon MC, Xie ML, Wu Y, et al. Inactivated trivalent seasonal influenza vaccine induces limited cross-reactive neutralizing antibody responses against 2009 pandemic and 1934 PR8 H1N1 strains. Vaccine 2010;28:6852-7.
- Wrammert J, Smith K, Miller J, Langley WA, Kokko K, Larsen C, et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 2008;453:667-71.
- Khurana S, Suguitan AL, Rivera Y, Simmons CP, Lanzavecchia A, Sallusto F, et al. Antigenic fingerprinting of H5N1 avian influenza using convalescent sera and monoclonal antibodies reveals potential vaccine and diagnostic targets. PLoS Med 2009;6. 10.1371/journal.pmed.1000049.
- Cowling BJ, Chan KH, Fang VJ, Lau LLH, So HC, Fung ROP, et al. Comparative epidemiology of pandemic and seasonal influenza A in households. N Engl J Med 2010;362:2175-84.
- Carrat F, Vergu E, Ferguson NM, Le Maitre M, Cauchemez S, Leach S, et al. Time lines of infection and disease in human influenza: a review of volunteer challenge studies. Am J Epidemiol 2008;167:775-85.
- London School of Hygiene and Tropical Medicine . Flu Survey n.d. www.flusurvey.org.uk/index.php?option=com_content%26task=view%26id=125%26Itemid=199 (accessed 16 September 2010).
- Baker MG, Wilson N, Huang QS, Paine S, Lopez L, Bandaranayake D, et al. Pandemic influenza A(H1N1)v in New Zealand: the experience from April to August 2009. Euro Surveill 2009;14.
- UK National Statistics n.d. www.statistics.gov.uk/statbase/Product.asp?vlnk=15106 (accessed 25 November 2009).
- Mossong J, Hens N, Jit M, Beutels P, Auranen K, Mikolajczyk R, et al. Social contacts and mixing patterns relevant to the spread of infectious diseases. PLoS Med 2008;5. 10.1371/journal.pmed.0050074.
- Health Protection Agency (HPA) . The Role of the Health Protection Agency in the ‘containment’ Phase During the First Wave of Pandemic Influenza in England in 2009 2010. www.hpa.nhs.uk/web/HPAwebFile/HPAweb_C/1274088320581 (accessed 16 September 2010).
- Smith A, Coles S, Johnson S, Saldana L, Ihekweazu C, O’Moore E. An outbreak of influenza A(H1N1)v in a boarding school in South East England, May–June 2009. Euro Surveill 2009;14.
- House T, Baguelin M, van Hoek AJ, Flasche S, White P, Sadique MZ, et al. Modelling the impact of local reactive school closures on critical care provision during an influenza pandemic. Proc Roy Soc B 2010;276:3239-48.
- Morgan-Capner P, Wright J, Miller CL, Miller E. Surveillance of antibody to measles, mumps and rubella by age. BMJ 1988;297:770-2.
- Osborne K, Gay N, Hesketh L, Morgan-Capner P, Miller E. Ten years of serological surveillance in England and Wales: methods, results, implications and action. Int Journal Epidemiol 2000;29:362-8.
- Chief Medical Officer’s Update, Department of Health (DH) 2009. www.dh.gov.uk/en/Publicationsandstatistics/Lettersandcirculars/Dearcolleagueletters/DH_107169 (accessed 26 April 2010).
- Chief Medical Officer’s Update, Department of Health (DH) n.d. www.nelm.nhs.uk/en/NeLM-Area/News/2009---December/16/CMO-letter-announcing-amendment-of-Pandemrix-license-to-allow-a-one-dose-schedule-in-children-/ (accessed 26 April 2010).
- Royal College of General Practitioners Research and Surveillance Centre . Annual Report 2009 n.d. www.rcgp.org.uk/pdf/ANNUAL%20REPORT%202009%20FINAL.pdf (accessed 13 October 2010).
- Health Protection Agency (HPA) . Epidemiological Report of Pandemic (H1N1) 2009 in the UK n.d. www.hpa.org.uk/web/HPAwebFile/HPAweb_C/1284475321350 (accessed 13 October 2010).
Appendix 1 Protocol for the serological methods
MN
The microneutralisation (MN) will be performed in 96-well format according to previously described protocols [Nicholson KG, Colegate AE, Podda A, Stephenson I, Wood J, Ypma E, et al. Safety and antigenicity of non-adjuvanted and MF59-adjuvanted influenza A/Duck/Singapore/97 (H5N3) vaccine: a randomised trial of two potential vaccines against H5N1 influenza. Lancet 2001;357:1937–43] and standard operating procedures (SOPs) developed at the Respiratory Virus Unit (RVU).
Serum pretreatment
Elimination of complement (e.g. from fetal calf serum in culture medium) by incubation of the sera and appropriate quality control sera (provided and chosen according to test virus by the RVU – usually serum of ferret, sheep or human, with/without neutralisation activity) at + 56 °C/30 minutes. This step will be performed simultaneously for all study samples and control sera.
Microneutralisation test
The analysis with the NIBRG122 virus will be performed. In the early stage of the outbreak the assumption can be made that most sera will show no evidence of antibody to H1N1 2009 virus. Therefore, sera could be screened at a limited dilution range or one dilution only (1 : 10). Only sera that inhibit virus growth at this dilution will be titrated further: a twofold dilution series will be set up for each of the samples and control sera. After addition of a pre-titred virus (usually around 100 × median tissue culture infectious doses (TCID50) per well or 0.1-1 virus particle per cell – input might vary according to the virus used in the assay) neutralisation will be performed by incubation of the virus–serum mixture at room temperature for 1 hour.
As discussed earlier, the dilution range for the study samples might vary depending on the development of the prevalence of antibody in the population over time. We will routinely perform a six-step dilution (covering titres 10–320).
After neutralisation, a suspension of Madin–Darby Canine Kidney (MDCK) cells will be added and the plates will be incubated for 16 hours at 37°C in a CO2 incubator. The remaining infectivity of virus after neutralisation is determined in an enzyme immunoassay (EIA) format using a monoclonal antibody to detect expression of viral nucleoprotein. The amount of nucleoprotein expression is determined photometrically [optical density (OD) 450] using a plate reader.
Reading
An OD reading for each dilution step for each sample will be used to calculate the titre. The titre will be reported as the reciprocal dilution at which 50% of the virus is neutralised (e.g. titre of 100). The MN analysis will be performed in duplicate (in separate runs on 2 days) for each sample.
The two titres for each sample must not differ by more than a twofold serial dilution. In cases, where samples do not fall within this limit, a third analysis is performed and the two closest titres (which must be within a twofold serial dilution) will be reported.
HI
The principle of the haemagglutination inhibition (HI) test is based on the ability of specific anti-influenza antibodies to inhibit haemagglutination of red blood cells (RBCs) by influenza virus HA. The sera to be tested have to be previously treated to eliminate the non-specific inhibitors and the anti-species HAs. The experiment will be performed in accordance to protocols and SOPs established by the RVU.
Serum pretreatment
Elimination of non-specific inhibitors by incubation of the unknown serum samples and quality control sera (serum of ferret or human immunised with influenza virus) with neuraminidase [receptor-destroying enzyme (RDE) II (from Vibrio cholerae, Denka Seiken, Japan): 18 hours/ + 36 °C followed by heat inactivation 1 hour/ + 56 °C].
Preparation will be performed simultaneously for serum obtained pre-vaccination and post vaccination.
Haemagglutination inhibition test
Sera screened in the early phase of the study in a limited dilution range (e.g. 8- to 32-fold dilution) using the NIBRG122 virus. Samples that show activity in this range will be further titrated: eight twofold dilutions, starting at a 1 : 8 dilution of serum sample (or quality control sera) are performed and incubated with the haemagglutinin (HA) antigen suspension [previously titrated to adjust the dilution at eight haemagglutination units (HAUs)/25 µl]. The HA antigen is not added to the well that is dedicated to the RDE quality control.
The mixture is incubated for 1 hour at room temperature and 25 µl of the 0.5% RBC suspension (turkey blood) is added. The reaction is left for 1 hour at room temperature before reading.
Reading
The serum titre is equal to the highest reciprocal dilution, which induces a complete inhibition of haemagglutination. The titre of each quality control serum is close to the previously assigned value (titres have to be within the limits of one serial twofold dilution).
The RBC controls (RBC suspension without antigen) and the RDE controls do not produce any agglutination.
Each serum sample is titrated in duplicate and individual titres will be reported (two for each sample). These must not differ by more than a twofold serial dilution. In cases where samples do not fall within this limit, a third analysis is performed and the two closest titres (which must be within a twofold serial dilution) will be reported. In accordance with laboratory procedure, each sample was tested twice by each assay and the geometric mean titre (GMT) of the two results was used in the analysis. The starting dilution for the HI assay was 1 : 8 and for the MN was 1 : 10.
Reporting
The collaborator(s) will receive results for both assays in form of an excel (Microsoft Corporation, Seattle, WA, USA) table by e-mail.
Appendix 2 No. of seroincidence samples collected by age group, month and region: 2009–10
August 2009
| Age group (years) | Region | Y&H | |||||||
|---|---|---|---|---|---|---|---|---|---|
| East | East Mids | London | NE | NW | SE | SW | West Mids | ||
| < 5 | 26 | 9 | 25 | 0 | 24 | 6 | 19 | 10 | 0 |
| 5–14 | 26 | 22 | 26 | 0 | 30 | 6 | 41 | 15 | 0 |
| 15–24 | 8 | 17 | 10 | 0 | 21 | 24 | 60 | 11 | 0 |
| 25–44 | 18 | 27 | 55 | 0 | 28 | 9 | 74 | 14 | 0 |
| 45–64 | 23 | 38 | 48 | 0 | 31 | 0 | 102 | 25 | 0 |
| 65 + | 24 | 41 | 53 | 0 | 87 | 23 | 101 | 24 | 1 |
September 2009
| Age group (years) | Region | Y&H | |||||||
|---|---|---|---|---|---|---|---|---|---|
| East | East Mids | London | NE | NW | SE | SW | West Mids | ||
| < 5 | 25 | 15 | 25 | 0 | 39 | 1 | 4 | 1 | 0 |
| 5–14 | 27 | 24 | 25 | 0 | 68 | 13 | 26 | 10 | 0 |
| 15–24 | 15 | 6 | 12 | 0 | 83 | 34 | 20 | 9 | 0 |
| 25–44 | 17 | 14 | 53 | 0 | 73 | 0 | 87 | 0 | 0 |
| 45–64 | 22 | 31 | 78 | 0 | 71 | 0 | 86 | 0 | 0 |
| 65 + | 23 | 34 | 62 | 0 | 111 | 12 | 71 | 0 | 0 |
October 2009
| Age group (years) | Region | Y&H | |||||||
|---|---|---|---|---|---|---|---|---|---|
| East | East Mids | London | NE | NW | SE | SW | West Mids | ||
| < 5 | 0 | 9 | 25 | 1 | 29 | 0 | 1 | 0 | 0 |
| 5–14 | 0 | 11 | 25 | 0 | 59 | 0 | 24 | 0 | 0 |
| 15–24 | 0 | 4 | 12 | 0 | 3 | 0 | 9 | 0 | 0 |
| 25–44 | 0 | 9 | 13 | 0 | 21 | 0 | 16 | 0 | 0 |
| 45–64 | 0 | 18 | 24 | 0 | 27 | 0 | 25 | 0 | 0 |
| 65 + | 0 | 20 | 26 | 0 | 74 | 0 | 47 | 0 | 0 |
November 2009
| Age group (years) | Region | Y&H | |||||||
|---|---|---|---|---|---|---|---|---|---|
| East | East Mids | London | NE | NW | SE | SW | West Mids | ||
| < 5 | 0 | 17 | 43 | 24 | 27 | 0 | 1 | 7 | 0 |
| 5–14 | 0 | 16 | 32 | 19 | 69 | 0 | 31 | 19 | 0 |
| 15–24 | 0 | 10 | 5 | 5 | 77 | 0 | 16 | 17 | 0 |
| 25–44 | 0 | 31 | 21 | 21 | 60 | 0 | 35 | 20 | 0 |
| 45–64 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 |
| 65 + | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
December 2009
| Age group (years) | Region | Y&H | |||||||
|---|---|---|---|---|---|---|---|---|---|
| East | East Mids | London | NE | NW | SE | SW | West Mids | ||
| < 5 | 0 | 11 | 19 | 3 | 10 | 0 | 1 | 5 | 0 |
| 5–14 | 0 | 9 | 17 | 8 | 16 | 0 | 31 | 25 | 0 |
| 15–24 | 0 | 11 | 11 | 0 | 10 | 2 | 19 | 7 | 0 |
| 25–44 | 0 | 15 | 18 | 0 | 43 | 0 | 31 | 0 | 0 |
| 45–64 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 65 + | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
January 2010
| Age group (years) | Region | Y&H | |||||||
|---|---|---|---|---|---|---|---|---|---|
| East | East Mids | London | NE | NW | SE | SW | West Mids | ||
| < 5 | 0 | 12 | 23 | 14 | 46 | 3 | 6 | 0 | 0 |
| 5–14 | 0 | 10 | 22 | 23 | 82 | 11 | 31 | 0 | 0 |
| 15–24 | 0 | 15 | 6 | 3 | 94 | 49 | 9 | 0 | 0 |
| 25–44 | 0 | 25 | 21 | 22 | 55 | 12 | 19 | 122 | 0 |
| 45–64 | 0 | 45 | 24 | 25 | 54 | 3 | 24 | 39 | 0 |
| 65 + | 0 | 43 | 25 | 25 | 105 | 2 | 26 | 50 | 0 |
February 2010
| Age group (years) | Region | Y&H | |||||||
|---|---|---|---|---|---|---|---|---|---|
| East | East Mids | London | NE | NW | SE | SW | West Mids | ||
| < 5 | 0 | 8 | 25 | 14 | 0 | 0 | 1 | 0 | 0 |
| 5–14 | 0 | 10 | 26 | 8 | 0 | 0 | 24 | 0 | 0 |
| 15–24 | 0 | 2 | 7 | 3 | 8 | 0 | 14 | 0 | 0 |
| 25–44 | 0 | 21 | 18 | 12 | 17 | 0 | 13 | 27 | 0 |
| 45–64 | 0 | 29 | 25 | 25 | 24 | 0 | 24 | 10 | 0 |
| 65 + | 0 | 41 | 25 | 25 | 76 | 0 | 27 | 0 | 0 |
March 2010
| Age group (years) | Region | Y&H | |||||||
|---|---|---|---|---|---|---|---|---|---|
| East | East Mids | London | NE | NW | SE | SW | West Mids | ||
| < 5 | 0 | 3 | 24 | 11 | 0 | 0 | 2 | 0 | 0 |
| 5–14 | 0 | 7 | 25 | 17 | 0 | 0 | 25 | 0 | 0 |
| 15–24 | 0 | 2 | 5 | 1 | 0 | 0 | 19 | 0 | 0 |
| 25–44 | 0 | 4 | 20 | 9 | 0 | 0 | 47 | 0 | 0 |
| 45–64 | 0 | 16 | 24 | 0 | 0 | 0 | 39 | 0 | 0 |
| 65 + | 0 | 33 | 24 | 0 | 0 | 0 | 31 | 0 | 0 |
April 2010
| Age group (years) | Region | Y&H | |||||||
|---|---|---|---|---|---|---|---|---|---|
| East | East Mids | London | NE | NW | SE | SW | West Mids | ||
| < 5 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 0 | 0 |
| 5–14 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 |
| 15–24 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 0 | 0 |
| 25–44 | 0 | 0 | 0 | 0 | 0 | 0 | 9 | 0 | 0 |
| 45–64 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 0 | 0 |
| 65 + | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 |
Appendix 3 Framework for undertaking seroepidemiological studies at population level, with specific reference to influenza


Background
These notes set out the main considerations when seeking to undertake seroepidemiological studies to provide real-time rapid response to an emerging infection, such as pandemic influenza. Under these circumstances, serological data provide important information, such as levels of cross-protective immunity in different population groups and incidence rates of infection.
A seroepidemiological study can provide information on:
-
age-specific incidence as the disease emerges in the population
-
prevalence of existing cross-protective immunity
-
cumulative prevalence to inform future incidence predictions, for example through disease modelling where seroepidemiological data can be combined with age-specific morbidity data to predict the likely burden of illness and the impact of a novel infection such as pandemic influenza.
These data can be used to complement other descriptive epidemiological data or to inform disease transmission models used to predict the future course of the pandemic.
The requirements are different from those when setting up a seroepidemiological programme, the main focus of which is to monitor levels of immunity to particular diseases within a population to assist in evaluation of the impact of a vaccination programme or to inform the need for other public health interventions. 1,2
For any seroepidemiological study, it is essential that recent, age-stratified baseline sera are available to inform the interpretation of data. Such sera cannot be collected retrospectively so long-term investment by member states is essential to ensure access to population-based stored sera. The absence of recent baseline sera is a significant limiting factor in the interpretation of data for newly emerging infections. For example, knowledge of the baseline prevalence of antibodies to pandemic influenza A/H1N1 in 2009 was essential for understanding the epidemiology and informing vaccine policy, as it indicated that lower clinical attack rates in the elderly were the result of pre-existing cross-reactive antibody, thereby reducing the clinical benefit and cost-effectiveness of delivering vaccine to this group. This required access to sera obtained prior to the pandemic.
It is essential that appropriate personnel (microbiologists, epidemiologists, mathematical modellers and statisticians) are involved in the establishment of seroepidemiological studies from the outset.
When considering region-wide seroepidemiology (e.g. EU wide) it is necessary to form teams with broad representation, and establish a limited network of laboratories or a central laboratory for testing to ensure maximum generalisability. Comparisons between countries may be confounded by differences in assay performance, sensitivity and specificity. 3,4
Populations and sampling methodologies
Serum source
There are two basic methods for obtaining serum samples from populations:
-
Opportunistically available residual samples from routine biochemistry, microbiology or other sources (e.g. blood transfusion service) conducted on outpatient or inpatient populations. While serum samples submitted to diagnostic laboratories may not be entirely representative of the population they are readily accessible and therefore cheap to collect. Special arrangements may be needed for paediatric samples.
-
Establishment of specific studies to achieve sampling of population-based cohorts. This can be sampling of particular groups, for example school children, or targeted sampling [e.g. the Tecumseh study,5 Flu Watch6 or the National Health and Nutrition Examination Survey (NHANES) studies7]. While this is important for evaluating the effect of public health interventions in specific groups, it can be difficult to achieve high levels of compliance and will not always give generalisable results. This method provides the opportunity to collect more detailed demographic data on the participants.
Documentation
The minimum data set for each specimen should be date of birth or age, sex, date of specimen collection, geographic location of specimen collection, and specimen source (e.g. laboratory, hospital, community).
It is usually a condition of ethics approval that samples will be irreversibly unlinked to any possible patient-identifying information to ensure anonymity.
Ethics and approvals
Issues of patient confidentiality and obtaining ethical approval will differ in each member state and may cause significant difficulties in some circumstances. In some member states, implementing an ‘emergency public health response’ or ‘service evaluation’ may invoke an emergency clause that bypasses normal regulatory mechanisms.
Member states should give attention to how to achieve rapid sampling as part of preparedness planning and should establish a rapid ethics approval framework as part of their pandemic plan.
Population
Ideally, sera should be taken from both sick and healthy subgroups in the population to establish levels of cross-protective immunity and levels of seroconversion; however, opportunistic sampling may not permit this.
Emerging infections that have a high clinical attack rate and do not depend on specific behavioural risk factors will cause infection across the population. In these cases, the necessity to sample different populations may not be so critical. It is essential, however, in the event of newly emerging infections (e.g. pandemic influenza, severe acute respiratory syndrome) to ensure the ability to establish seroprevalence in a population ‘baseline sample’ that has been taken or archived prior to the disease emergence.
Immunosuppression and HIV
Samples from patients who are known to be immunosuppressed should be excluded due to difficulties in interpreting these data.
Timing of samples
Samples must be timed appropriately to give interpretable data:
-
Sequential sampling of opportunistic cohorts (e.g. women attending antenatal care) are extremely informative but generalisability to the general population may be limited.
-
Repeated cross-section samples with the same sampling methodology can generate cumulative age-specific prevalence from which infection incidence over time can be derived. 8
Ideally, information should be obtained on whether the sample has been taken post natural infection or post vaccination.
Age stratification
It is essential that serum samples are stratified by age in equal numbers for male and female to provide information about the cross-protective immunity or specific effects in different age groups. All age groups in the population should be represented (e.g. useful age groups include < 5, 5–14, 15–44, 45–64 and ≥ 65 years). It is important to have background information about the age composition of the population to inform interpretation of data, and provide ability to perform predictive analyses.
Geographical representation
It is important to have samples from both urban and rural settings to establish population mixing patterns, and to have information of the population density and population structure, which may vary considerably. This may lead to differences in transmission patterns and, therefore, age-stratified incidence.
Sample size and power calculations
Power calculations need to be undertaken to determine the number of samples required at each time point and in each age group to estimate a change in prevalence according to specified scenarios.
For example, to estimate the incidence of pandemic influenza A/H1N1 in England during the first wave of activity, a serosurvey was performed using samples from prior to the pandemic and then at monthly intervals. The sample consisted of 1600 sera taken prior to the pandemic (200 in each of eight age groups < 1, 1–4, 5–14, 15–44, 45–64, 65–74, 75–79, 80 +) and then 1000 per month, spread across five age groups, with 200 in each (< 5, 5–14,15–44, 45–64, 65 +) with a final post-first-wave cross-sectional survey of 1600 using samples collected May–June 2010.
Using this sample size, the 95% CIs for the estimation of prevalence within each age group and overall are show below (Table 23), for various observed prevalences.
| Prevalence (%) | 95% CIs | |
|---|---|---|
| n = 200 | n = 1000 | |
| 0 | 0.0 to 1.8 | 0.0 to 0.4 |
| 5 | 2.4 to 9.0 | 3.7 to 6.5 |
| 10 | 6.2 to 15.0 | 8.2 to 12.0 |
| 15 | 10.4 to 20.7 | 12.8 to 17.4 |
| 20 | 14.7 to 26.2 | 17.6 to 22.6 |
| 25 | 19.2 to 31.6 | 22.3 to 27.8 |
| 30 | 23.7 to 36.9 | 27.2 to 32.9 |
| 35 | 28.4 to 42.0 | 32.0 to 38.0 |
| 40 | 33.2 to 47.1 | 36.9 to 43.1 |
| 45 | 38.0 to 52.2 | 41.9 to 48.1 |
| 50 | 42.9 to 57.1 | 46.9 to 53.1 |
To estimate incidence from prevalence the difference between the prevalence at two time points can be calculated. When combining all age groups this would give reasonable precision for estimating incidence from prevalence as shown in Table 24 below.
| p1 | p2 | p2–p1 | 95% CI |
|---|---|---|---|
| 5 | 10 | 5 | 2.7 to 7.3 |
| 10 | 15 | 5 | 2.1 to 7.9 |
| 15 | 20 | 5 | 1.7 to 8.3 |
| 20 | 25 | 5 | 1.3 to 8.7 |
| 5 | 15 | 10 | 7.4 to 12.6 |
| 10 | 20 | 10 | 6.9 to 13.1 |
| 15 | 25 | 10 | 6.5 to 13.5 |
Within each age group the precision would be much lower if incidence were calculated this way. However, if incidence (or force of infection) is modelled as a function of time and age then this sample size will still give good precision within age groups.
Assay methodology
Consideration should be given to the choice of assay used and the decision should include epidemiologist, statistician and microbiology involvement. Assays for diagnostic purposes often have different criteria than those selected for seroepidemiology purposes, which do not necessarily require accurate results at the individual patient level.
The magnitude and the kinetics of antibody detection using the chosen methodology need to be understood in relation to the measurements undertaken.
Choice of assay
The choice of assay [e.g. HI, MN, single radial haemolysis (SRH)] for influenza antibodies will depend on the parameters to be measured.
Ideally, the assay chosen should provide a direct correlate of protection. ELISA is not recommended for influenza antibody detection, as it does not measure protective antibody or provide a good measure of disease incidence.
It is important to understand the serological response to infection with the assay methodology used, i.e. the relationship between symptomatic and asymptomatic infection and the relationship between seroconversion and clinical illness.
Statistical analysis
-
Statistical analysis of seroepidemiology data typically provides estimates of age-specific prevalence. If repeat sampling is used this can be used to provide a direct measure of incidence of infection in population.
-
These direct incidence estimates can be compared with those derived from clinical surveillance using direct virus detection tests and modelling to provide an insight into the epidemiology and clinical expression of the infection.
-
Linear regression may be used to estimate the effect of age, sex, geographical origin, gender or other available variables on antibody levels.
-
Reverse cumulative distribution curves can be constructed to compare proportions above various threshold titres.
-
Mixture modelling techniques may be used instead of pre-defined assay cut-off, especially where there is not a clear correlate of protection.
Quality assurance
It is recommended that one or more ‘gold standard’ laboratories are identified at a European level to ensure that country results are comparable. These laboratories would be responsible for the development and distribution of panels of serum specimens, which would include known negatives, acute and convalescent specimens, and specimens from people who have been vaccinated. The panels would be used to establish the sensitivity and specificity of assays.
Use of antibody standards
International antibody standards, such as those produced by the HPA NIBSC are primarily designed for use in serological assays where either population based seroepidemiology is being performed, or where vaccine immunogenicity studies are being undertaken. The significant advantage of a calibrated standard is that it helps to overcome the methodological differences that give rise to significant variation between laboratories in antibody titres. These standards are not designed to be used as ‘run controls’ for routine serological assays. 4
Standard operating procedures
It is recommended that a limited number of laboratories undertake testing, and the number of SOPs is kept to a minimum to limit variability in testing. SOPs are required for sample handling, result validation and result interpretation. Assay reproducibility should be built into existing quality assurance programmes.
Audit and quality assurance
Good practice is required, especially when dealing with large data sets and complex sample collection protocols. Regular monitoring, audit of processes and procedures and data verification is needed to ensure integrity of the process from end to end.
References
- Andrews N, Pebody RG, Berbers G, Blondeau C, Crovari P, Davidkin I, et al. The European Sero-Epidemiology Network: standardizing the enzyme immunoassay results for measles, mumps and rubella. Epidemiol Infect 2000;125:127-41.
- Osborne K, Gay N, Hesketh L, Morgan-Capner P, Miller E. Ten years of serological surveillance in England and Wales: methods, results, implications and action. Int J Epidemiol 2000;29:362-8.
- Monto AS, Napier JA, Metzner HL. The Tecumseh study of respiratory illness. I. Plan of study and observations on syndromes of acute respiratory disease. Am J Epidemiol 1971;94:269-79.
- Stephenson I, Heath A, Major D, Newman RW, Hoschler K, Junzi W, et al. Reproducibility of serologic assays for influenza virus A (H5N1). Emerg Infect Dis 2009;15:1250-9.
- Flu Watch Pandemic Study n.d. URL: https://www.fluwatch.co.uk (accessed 12 May 2010).
- National Health and Nutrition Examination Survey (NHANES) n.d. URL: www.cdc.gov/nchs/nhanes.htm (accessed 12 May 2010).
- Osborne K, Weinberg J, Miller E. The European Sero-Epidemiology Network. Euro Surveill 1997;2:29-31.
- Miller E, Hoschler K, Hardelid P, Stanford E, Andrews N, Zambon M. Incidence of 2009 pandemic influenza A H1N1 infection in England: a cross-sectional serological study. Lancet 2010;375:1100-8.
Appendix 4 Study protocol, version 1 July 2009: no subsequent amendments: Protocol for the assessment of baseline age-specific antibody prevalence and incidence of infection to novel influenza A/H1N1 2009
E Miller,1 NJ Andrews,2 K Hoschler3 and E Stanford4
-
1Immunisation, Hepatitis and Blood Safety Dept, Centre for Infections, Health Protection Agency, London, UK.
-
2Statistics Unit, Centre for Infections, Health Protection Agency, London, UK.
-
3Respiratory Virus Unit, Virus Reference Department, Centre for Infections, Health Protection Agency, London UK.
-
4Vaccine Evaluation/Seroepidemiology Unit, Health Protection Agency, Manchester Royal Infirmary, Manchester, UK.
Background
Knowledge of the baseline prevalence of antibodies to H1N1 2009 is essential for understanding the current and future epidemiology of H1N1 2009. It will inform vaccine policy, as it will indicate whether the lower attack rates so far documented in the elderly in the UK are indeed the result of pre-existing cross-protective antibody, thus reducing the clinical benefit and cost-effectiveness of delivering vaccine to this group. Seroepidemiological data combined with age-specific morbidity data will greatly assist in understanding the spread of this novel virus, and provide valuable information for real-time modellers who will be predicting the likely burden of illness and impact of the pandemic as it progresses during the coming year. The need for such data has been identified as a high priority by the Scientific Advisory Group for Emergencies (SAGE).
The proposal below is designed to generate information on the baseline prevalence of cross-reactive antibodies to the H1N1 2009 virus in England prior to the start of the epidemic and to generate monthly age-specific incidence as the disease progresses during the next year.
Documenting baseline prevalence of antibodies to H1N1 2009 virus in England
Sera taken in 2008 and already stored by the Health Protection Agency (HPA) serological surveillance programme (www.hpa.org.uk/webw/HPAweb&Page&HPAwebAutoListName/Page/1158313434390?p=1158313434390) will be used to document age-stratified prevalence of antibodies to H1N1 2009 prior to the arrival of swine flu in the UK. The sera are collected by 16 laboratories in eight regions in England and are residual aliquots from samples submitted to HPA and NHS laboratories for diagnostic testing or screening and despatched to the Seroepidemiology Unit (SEU) at Manchester. The sera collections have been extensively used for policy-related seroepidemiological studies, and for deriving force of infection estimates for disease modelling purposes (Osborne K, Gay N, Hesketh L, Morgan-Capner P, Miller E. Ten years of serological surveillance in England and Wales: methods, results, implications and action. Int J Epidemiol 2000;29:362–8).
The age groups have been chosen to match those in the Royal College of General Practitioners (RCGP) and Q Flu influenza surveillance data set, with expansion of the 74 + age group to ensure separate groups for 74- to 79-year-olds and those aged 80+ years as follows: < 1, 1–4, 5–14,15–44, 45–64, 65–74, 75–79 and 80 + years. Two hundred samples from each of these groups will be tested.
Demographic data on each sample will be: age in months if under a year, age in years if over, month/year sample taken, gender, collecting laboratory.
Measuring incidence during the evolving epidemic
It is planned to test approximately 1000 serum samples across the age range each month to provide incidence estimates. For this, the principal source of sera will be those from chemical pathology laboratories, as these are usually discarded after 48 hours, whereas the samples from microbiology laboratories are generally retained for longer while testing is completed. This would allow more timely provision of sera for testing. Also sera from the chemical pathology laboratories are not submitted from patients with acute infections and are therefore potentially less biased with respect to risk of influenza. Chemical pathology laboratories on the sites of the existing 16 collecting laboratories will be approached to provide these sera so that they can be despatched together with the microbiological sera using existing procedures.
Collection and documentation of sera
The procedure put in place by the SEU for selection, despatch and documentation of sera is attached in Annex 1. The methods ensure that sera are irreversibly unlinked to any possible patient identifying information. Ensuring that testing is carried out by unlinked anonymous methods is a condition of the ethics approval that has been obtained for the HPA seroepidemiology programme [National Research Ethics Service (NRES) reference no. 05/Q0505/45].
Laboratory testing
Samples will be tested in the Centre for Infections (CFI) Virus Reference Unit (VRU). The HPA SEU will provide the VRU with an excel spreadsheet containing the unique identifier for each sample. The VRU will check the identity of all received samples and feed back mismatches where necessary. All samples will be analysed by microneutralisation (MN) and haemagglutination inhibition (HI) with the NIBRG122 virus [reverse genetic (rg) virus based on A/Engl/195/2009(vH1N1) and A/Puerto Rico/8/34]. Results will be entered onto the excel spreadsheet, which will be returned to the SEU for linking with the basic demographic data available for that sample.
Details of the laboratory methods are attached in Annex 2.
Sample size for swine flu seroprevalence
In order to estimate the incidence of H1N1 2009 during the pandemic, serosurveys will be performed using samples from prior to the pandemic and then monthly through the first wave from August 2009 to May 2010.
The sample size will consist of 1600 sera taken prior to the pandemic (200 in each of eight age groups: <1, 1–4, 5–14, 15–44, 45–64, 65–74, 75–79 and 80 + years) and then 1000 per month, spread across five age groups with 200 in each (< 5, 5–14,15–44, 45–64 and 65 + years) with a final post-first-wave cross-sectional survey of 1600 using samples collected around May–June 2010.
With this sample size the 95% CI for the estimation of prevalence within each age group and overall is shown in Table 1 below for various observed prevalences.
| Prevalence (%) | 95% CIs | |
|---|---|---|
| n = 200 | n = 1000 | |
| 0 | 0.0 to 1.8 | 0.0 to 0.4 |
| 5 | 2.4 to 9.0 | 3.7 to 6.5 |
| 10 | 6.2 to 15.0 | 8.2 to 12.0 |
| 15 | 10.4 to 20.7 | 12.8 to 17.4 |
| 20 | 14.7 to 26.2 | 17.6 to 22.6 |
| 25 | 19.2 to 31.6 | 22.3 to 27.8 |
| 30 | 23.7 to 36.9 | 27.2 to 32.9 |
| 35 | 28.4 to 42.0 | 32.0 to 38.0 |
| 40 | 33.2 to 47.1 | 36.9 to 43.1 |
| 45 | 38.0 to 52.2 | 41.9 to 48.1 |
| 50 | 42.9 to 57.1 | 46.9 to 53.1 |
To estimate incidence from prevalence the difference between the prevalence at two time points can be calculated. When combining all age groups this would give reasonable precision for estimating incidence from prevalence as shown in Table 2 below.
| p1 | p2 | p2–p1 | 95% CI |
|---|---|---|---|
| 5 | 10 | 5 | 2.7 to 7.3 |
| 10 | 15 | 5 | 2.1 to 7.9 |
| 15 | 20 | 5 | 1.7 to 8.3 |
| 20 | 25 | 5 | 1.3 to 8.7 |
| 5 | 15 | 10 | 7.4 to 12.6 |
| 10 | 20 | 10 | 6.9 to 13.1 |
| 15 | 25 | 10 | 6.5 to 13.5 |
Within each age group the precision would be much lower if incidence were calculated this way. However, if incidence (or force of infection) is modelled as a function of time and age then this sample size will still give good precision within age groups.
Data analysis and reporting
Data will be analysed by the CFI Statistics Unit. For the monthly incidence estimates it is intended that testing and analysis of each month’s samples will be completed within a month of receipt of that month’s batch. Tabulations of the proportion positive to H1N1 2009 by test method and age group (< 5, 5–14, 15–44, 45–64 and 65 + years) will be provided regularly to SAGE and the Scientific Pandemic Influenza Sub-Group on Modelling (SPI-M) as appropriate.
Annex 1: Seroepidemiology Unit user guide
Annex 2: laboratory methods
Microneutralisation
The MN will be performed in a 96-well format according to previously described protocols [Nicholson KG, Colegate AE, Podda A, Stephenson I, Wood J, Ypma E, et al. Safety and antigenicity of non-adjuvanted and MF59-adjuvanted influenza A/Duck/Singapore/97 (H5N3) vaccine: a randomised trial of two potential vaccines against H5N1 influenza. Lancet 2001;357:1937–43] and standard operating procedures (SOPs) developed at the Respiratory Virus Unit (RVU).
Serum pretreatment
Elimination of complement (e.g. from fetal calf serum in culture medium) by incubation of the sera and appropriate quality control sera (provided and chosen according to test virus by RVU – usually serum of ferret, sheep or human, with/without neutralisation activity) at + 56 °C/30 minutes. This step will be performed simultaneously for all study samples and control sera.
Microneutralisation test
The analysis with the NIBRG122 virus will be performed. In the early stage of the outbreak the assumption can be made that most sera will show no evidence of antibody to the H1N1 virus. Therefore, sera could be screened at a limited dilution range or one dilution only (1 : 10). Only sera that inhibit virus growth at this dilution will be titrated further: a twofold dilution series will be set up for each of the samples and control sera. After addition of a pre-titred virus (usually around 100 × TCID50 per well or 0.1-1 virus particle per cell – input might vary according to the virus used in the assay) neutralisation will be performed by incubation of the virus–serum mixture at room temperature for 1 hour.
As discussed earlier, the dilution range for the study samples might vary depending on the development of the prevalence of antibody in the population over time. We will routinely perform a six-step dilution (covering titres 20–640), but will determine end point titres for each sample by further titrating those specimen that show titres of > 640.
After neutralisation, a suspension of Madin–Darby Canine Kidney (MDCK) cells will be added and the plates will be incubated for 16 hours at 37 °C in a CO2 incubator. The remaining infectivity of virus after neutralisation is determined in an enzyme immunoassay (EIA) format using a monoclonal antibody to detect expression of viral nucleoprotein. The amount of nucleoprotein expression is determined photometrically [optical density (OD) 450] using a plate reader.
Reading
An OD reading for each dilution step for each sample will be used to calculate the titre. The titre will be reported as the reciprocal dilution at which 50% of the virus is neutralised (e.g. titre of 100). The microneutralisation analysis will be performed in duplicate (in separate runs on 2 days) for each sample.
The two titres for each sample must not differ by more than a twofold serial dilution. In cases, where samples do not fall within this limit, a third analysis is performed and the two closest titres (which must be within a twofold serial dilution) will be reported.
Haemagglutination inhibition
The principle of the HI test is based on the ability of specific anti-influenza antibodies to inhibit haemagglutination of red blood cells (RBCs) by influenza virus haemagglutinin (HA). The sera to be tested have to be previously treated to eliminate the non-specific inhibitors and the anti-species HAs. The experiment will be performed in accordance to protocols and SOPs established by the RVU.
Serum pretreatment
Elimination of non-specific inhibitors by incubation of the unknown serum samples and quality control sera (serum of ferret or human immunised with influenza virus) with neuraminidase [receptor-destroying enzyme (RDE) II: 18 hours/+ 36 °C followed by heat inactivation 1 hour/+ 56 °C]. Preparation will be performed simultaneously for serum obtained pre-vaccination and post vaccination.
Haemagglutination test
Similar to the microneutralisation we will screen sera in the early phase of the study in a limited dilution range (e.g. 8- to 32-fold dilution) using the NIBRG122 virus. Samples which show activity in this range will be further titrated: eight twofold dilutions, starting at a 1 : 8 dilution of serum sample (or quality control sera) are performed and incubated with the HA antigen suspension [previously titrated to adjust the dilution at eight haemagglutination units (HAUs)]/25 µl]. The HA antigen is not added to the well dedicated to the RDE quality control.
The mixture is incubated for 1 hour at room temperature and 25 µl of the 0.5% RBC suspension (turkey blood) are added. The reaction is left for 1 hour at room temperature before reading.
Reading
The serum titre is equal to the highest reciprocal dilution, which induces a complete inhibition of haemagglutination. The titre of each quality control serum is close to the previously assigned value (within one serial twofold dilution limits).
The RBC controls (RBC suspension without antigen) and the RDE controls do not produce any agglutination.
Each serum sample is titrated in duplicate and individual titres will be reported (two for each sample). These must not differ by more than a twofold serial dilution. In cases, where samples do not fall within this limit, a third analysis is performed and the two closest titres (which must be within a twofold serial dilution) will be reported.
List of abbreviations
- ACDP
- Advisory Committee on Dangerous Pathogens
- BSL
- biosafety level
- CEPR
- Centre for Emergency Preparedness and Response
- CFI
- Centre for Infections
- CI
- confidence interval
- CNRL
- Community Network of Reference Laboratories for Human Influenza in Europe
- East Mids
- East Midlands
- ECDC
- European Centre for Disease Control
- EIA
- enzyme immunoassay
- ELISA
- enzyme-linked immunosorbent assay
- EU
- European Union
- GMT
- geometric mean titre
- HA
- haemagglutinin
- HI
- haemagglutination inhibition
- HIV
- human immunodeficiency virus
- HPA
- Health Protection Agency
- HTA
- health technology assessment
- ILI
- influenza-like illness
- LR
- likelihood ratio
- MCMC
- Monte Carlo Markov Chain
- MDCK
- Madin–Darby Canine Kidney
- MN
- microneutralisation
- NE
- North East
- NIBSC
- National Institute for Biological Standards and Control
- NIHR
- National Institute for Health Research
- NW
- North West
- OD
- optical density
- OR
- odds ratio
- PCR
- polymerase chain reaction
- R
- reproduction number
- RBC
- red blood cell
- RCGP
- Royal College of General Practitioners
- RDE
- receptor-destroying enzyme
- rg
- reverse genetic
- RMN
- Regional Microbiology Network
- RT-PCR
- real-time polymerase chain reaction
- RVU
- Respiratory Virus Unit
- SAGE
- Scientific Advisory Group for Emergencies
- SARS
- severe acute respiratory illness
- SE
- South East
- SEU
- Seroepidemiology Unit
- SOP
- standard operating procedure
- SPI-M
- Scientific Pandemic Influenza Sub-Group on Modelling
- SRI
- severe respiratory illness
- SW
- South West
- VRU
- Virus Research Unit
- West Mids
- West Midlands
- WHO
- World Health Organization
- Y&H
- Yorkshire and Humber
All abbreviations that have been used in this report are listed here unless the abbreviation is well known (e.g. NHS), or it has been used only once, or it is a non-standard abbreviation used only in figures/tables/appendices, in which case the abbreviation is defined in the figure legend or in the notes at the end of the table.