
Notes
Article history
This themed issue of the Health Technology Assessment journal series contains a collection of research commissioned by the NIHR as part of the Department of Health’s (DH) response to the H1N1 swine flu pandemic. The NIHR through the NIHR Evaluation Trials and Studies Coordinating Centre (NETSCC) commissioned a number of research projects looking into the treatment and management of H1N1 influenza. NETSCC managed the pandemic flu research over a very short timescale in two ways. Firstly, it responded to urgent national research priority areas identified by the Scientific Advisory Group in Emergencies (SAGE). Secondly, a call for research proposals to inform policy and patient care in the current influenza pandemic was issued in June 2009. All research proposals went through a process of academic peer review by clinicians and methodologists as well as being reviewed by a specially convened NIHR Flu Commissioning Board.
Declared competing interests of authors
KGN has been an ad hoc consultant to GlaxoSmithKline (GSK), Merck and Novartis. He has received funding to speak at meetings organised by Novartis, Baxter, Berna Biotech, Esteves, and the European Scientific Working Group on Influenza, and H5 vaccines from Novartis to support a Medical Research Council-funded research project, and H1N1 vaccines from Baxter AG and GSK to support this National Institute for Health Research-funded research project. A colleague in KGN’s department has received research funding from Roche. JS N-V-T has received funding to attend influenza-related meetings, ad hoc lecture and consultancy fees, and research funding from several influenza antiviral drug and vaccine manufacturers (including both GSK and Baxter AG), and is a former employee of SmithKline Beecham plc (now GlaxoSmithKline), Roche Products Ltd and Sanofi–Pasteur MSD. MZ and KH have been investigators of clinical trials sponsored by Novartis, Baxter, Sanofi–Pasteur and CSL Australia Ltd. KH has been sponsored by Sanofi–Pasteur to take part and speak at one international meeting. RCR has been an investigator of clinical trials sponsored by Novartis Vaccines and Sanofi–Pasteur. WSL is in receipt of an unrestricted educational grant towards study of pneumococcal pneumonia from Wyeth, UK. KRA has acted as a paid consultant to the health-care industry generally (for the provision of advice and short courses), but specifically has not advised either Baxter or GSK in relation to either vaccine, or any other body with regard to influenza vaccination policy. TWC has been an investigator of clinical trials sponsored by Novartis and Roche.
Permissions
Copyright statement
© 2010 Queen’s Printer and Controller of HMSO. This journal is a member of and subscribes to the principles of the Committee on Publication Ethics (COPE) (http://www.publicationethics.org/). This journal may be freely reproduced for the purposes of private research and study and may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NETSCC, Health Technology Assessment, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2010 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction and background
Background
Introduction
Influenza viruses are unique among respiratory viruses with respect to their segmented genome, antigenic diversity, seasonality in the northern and southern hemispheres, and economic and social impacts. In the northern hemisphere, influenza is characterised by the occurrence of annual outbreaks during winter and by worldwide pandemics that have occurred at 11- to 52-year intervals during the past 300 years. 1
Influenza is a highly contagious, acute febrile respiratory infection caused by the influenza virus. Influenza viruses cause seasonal epidemics and, very occasionally, global pandemics. The word pandemic (from the Greek pan meaning ‘all’ and demos meaning ‘people’) describes an epidemic that affects the whole population. Typically, several waves of infection, occurring over a few years, are needed before most of the world’s population is affected by pandemic influenza. Worldwide pandemics of influenza may occur following the emergence of a ‘new’ subtype of influenza A.
Past experiences indicate that there is no regularity to pandemics and no reliable basis for predicting when and where they might arise. During the twentieth century, pandemics occurred at relatively long and unpredictable intervals of 9–39 years during 1918 (H1N1), 1957 (H2N2), 1968 (H3N2) and, to a lesser extent, in 1977 (H1N1). In 1957, the H2N2 virus completely replaced the previous H1N1 virus and in 1968 the H3N2 virus replaced it in turn. The re-emergence of H1N1 virus in 1977 did not cause a ‘true’ pandemic, as many people born before 1957 were partially immune. Moreover, in 1977 the H1N1 virus did not replace H3N2 virus. Since 1968, both H1N1 and H3N2 subtypes have cocirculated, with influenza B causing ‘interpandemic’ or ‘seasonal’ outbreaks in humans most winters in the northern hemisphere.
Influenza vaccines have been available for over 60 years. Extensive experience during this long period has demonstrated their safety and efficacy. In populations that are at risk of severe complications, vaccination cuts hospital admissions and deaths. Vaccination is thus the cornerstone of influenza prevention.
The influenza virus
Influenza is an orthomyxovirus, comprising a lipid membrane surrounding a matrix protein shell and a core consisting of ribonucleic acid–nucleoprotein complexes. There are three types of influenza virus – influenza types A, B and C – which differ in their core proteins. Influenza types A and B are responsible for nearly all influenza-associated clinical illnesses. Influenza type A is responsible for pandemic influenza.
Type A influenza viruses are divided into subtypes, depending on antigenic differences between two surface glycoproteins, specifically the haemagglutinin (HA) and neuraminidase (NA). The HA facilitates the entry of the virus into cells of the respiratory epithelium, while the NA facilitates the release of newly produced viral particles (virions) from infected cells. Sixteen distinct HAs and nine different NAs are known. Influenza type A viruses of all HA and NA antigenic subtypes have been recovered from aquatic birds, whereas only a few have been isolated from other animal species – mostly humans, pigs and birds – indicating that aquatic birds are the natural reservoir of influenza A viruses. Three subtypes of influenza A – H1N1, H2N2 and H3N2 – have formed stable lineages in man during the last century; the introduction of each subtype was associated with pandemic influenza – H1N1 in 1918, H2N2 in 1957, H3N2 in 1968, and, to a lesser extent, H1N1 in 1977.
Nomenclature
Each strain of influenza is described on the basis of type (i.e. whether influenza type A, B or C), the original host of origin, the place of origin, the strain number (as designated by the laboratory that first grew the virus), the year of isolation, and, for influenza A viruses, the subtypes of the HA and NA. While the host of origin is recorded for animal viruses, it is not recorded for human strains. Thus:
-
Influenza A/California/7/2009 (H1N1) was isolated from human beings in CA, USA, strain number 7, in 2009, with HA subtype 1 and NA subtype 1.
-
Influenza A/turkey/Turkey/1/2005 (H5N1) was isolated from a turkey in Turkey, strain number 1, in 2005, with HA subtype 5 and NA subtype 1. The H5 subtype has been further divided into virus clades that differ antigenically, genetically and in geographic distribution.
-
Influenza B/Malaysia/2506/2004, the recommended influenza B strain included in vaccines for the 2006–7 season, was isolated in Malaysia, strain number 2506, in 2004. This particular strain is a descendant of the influenza B/Victoria/2/1987 lineage. While influenza B strains have not been subtyped (like influenza A), they have evolved into two antigenically dissimilar lineages (B/Victoria/2/1987 and B/Yamagata/16/88).
Antigenic shift and drift of the surface haemagglutinin and neuraminidase glycoproteins
The antigenic variability of influenza viruses arises from two distinct mechanisms: ‘antigenic drift’ and ‘antigenic shift’.
Antigenic drift reflects a gradual evolution of a virus subtype in response to immune pressure. The HA and NA are the virion surface antigens that are associated with humoral immunity. With antigenic drift, new strains of influenza evolve that are antigenically related to those circulating during preceding epidemics. Antigenic drift arises from gene mutations in the amino acid sequences of genes that encode the HA (and NA). This process leads to a new strain of influenza within the same subtype of influenza A and also occurs with influenza B viruses. Antigenic drift is associated with annual (or ‘seasonal’ or ‘interpandemic’) outbreaks of influenza, as the new strain is able to infect people who had developed immunity to ancestral strains. Because of antigenic drift, the World Health Organization (WHO) reviews the composition of interpandemic vaccines twice annually.
Antigenic shift occurs when an entirely new subtype of influenza A is introduced into the population, causing human–human transmission, community-wide infections and disease. Antigenic shift occurs when a novel HA, and possibly NA, is introduced into humans from the avian reservoir. This could occur directly (e.g. from poultry) or indirectly (e.g. from pigs), which can be infected with human, avian and porcine influenza. Antigenic shift probably occurred in 1918, when an avian H1N1 subtype adapted to man. It also occurred in 1957 (when the H1N1 subtype was replaced by an H2N2 virus) and in 1968 (when the H2N2 subtype was replaced by an H3N2 virus) when the genomes of the circulating human viruses were mixed with those of avian origin by genetic reassortment. This gene shuffling (genetic reassortment) is possible when a susceptible host is coinfected with influenza virus from different animal species. Pandemic influenza occurred with the antigenic shifts of 1957 and 1968 because populations across the world had little or no immunity to the new strains. Pandemics normally cause considerable morbidity and mortality.
In 1977, an outbreak of A/USSR/90/1977 (H1N1) occurred; antigenic variants of this virus have cocirculated with influenza A H3N2 viruses ever since. This virus was reintroduced into the global population two decades after the 1957 pandemic, when the influenza A H2N2 subtype replaced the H1N1 subtype. The A/USSR/90/1977 virus was close antigenically to one that circulated in the early 1950s and may have been accidentally introduced into the community. 2
Reasons for decline and emergence of dominant subtypes is unclear, although it seems likely that during interpandemic intervals, population immunity reaches a point where the prevalent strain loses its capacity for further drift capable of eluding host defences.
Pandemic definition
Historically, a pandemic is considered imminent or said to exist when the following apply:
-
Antigenic shift occurs, i.e. the emergence in humans of a new HA subtype of influenza A that is serologically distinct from viruses circulating in humans for many preceding years and could not have arisen from earlier viruses by mutation.
-
A ‘high’ proportion of the population lacks immunity to the new virus, i.e. no or low antibody titres to the HA of the novel virus detected in major segments of the population.
-
The new virus spreads from person to person, causing disease.
-
The new virus spreads rapidly beyond the community in which it was first identified.
The WHO has defined phases in the evolution of an influenza pandemic, with the goal of facilitating a stepwise escalation to preparedness planning and response leading up to the declaration of the onset of a pandemic. The WHO phases were first published in 1999 and were updated by WHO in April 20053 and 2009. 4 In keeping with the historical definition of pandemic influenza, the 1999 and 2005 WHO documents relate pandemic influenza to the introduction of a new influenza virus subtype. In the 2009 revision,4 WHO refers to the occurrence of human infections with an animal or human–animal reassortant virus rather than a ‘new’ subtype of influenza. Additionally, the phase description refers to ‘community level outbreaks’ with no mention of immunity to antigenically similar viruses that circulated previously. In the 2009 revision, Phase 6, (i.e. pandemic influenza) is defined by community level outbreaks of a ‘new’ virus in at least two different WHO regions. Pandemic H1N1 influenza was declared by the Director General of WHO on 11 June 2009.
Seasonality
Interpandemic influenza
While influenza transmission in the tropics and subtropics may extend throughout most of the year with increased activity during monsoon or wet seasons, outbreaks in temperate zones exhibit marked seasonality, occurring during the ‘winter’ months from October to April in the northern hemisphere, and from May to September in the southern hemisphere. In the northern hemisphere, influenza virus may be recovered sporadically during the summer, but summertime outbreaks of influenza are unusual. The UK influenza season typically occurs between December and March. It usually begins abruptly, peaks nationally within 2–3 weeks and lasts for about 5–7 weeks. Successive or overlapping waves of infection by different subtypes of influenza A (i.e. H1N1 and H3N2) or by influenza A and B may result in a more prolonged period of disease activity. Summertime outbreaks of interpandemic influenza that are similar in scale to winter outbreaks do not occur in temperate regions.
Pandemic influenza
A feature of pandemic influenza is successive waves of infection that may occur within several months of one another, or may be separated by a year or more. One of these waves may occur during the summer or autumn.
-
1918–19 In England, Wales and other European countries, the 1918 pandemic spread in three rapidly recurring waves within an ∼9-month interval. 5 British soldiers were first struck by the pandemic in France in April 1918, but the first wave of the infection began in England on 23 June 1918. 6 In the USA, the first wave began in March 1918. This ‘spring’ wave hit mainland Europe in May and June. By July and August it was waning, but was rapidly followed by an ‘autumn’ wave that began in France during August and spread throughout Europe during September and October. The final wave occurred during the early months of 1919. 7
-
1957–8 The 1957–8 pandemic originated in the Yunan Province of China in February 1957. Infection spread to Europe in June,8 laboratory reports of influenza in public health and other laboratories in the UK increased during July and August and peaked in late September. 9 In the north of England, claims for sickness benefit peaked during late September. As judged by claims for sickness benefit, the outbreak began and was more prevalent in the north than in the south. There was little or no evidence of a second wave in the north and west of the country, but there was a definite increase in excess sickness claims in the south and east, the second peak occurring approximately 10 weeks after the initial peak (i.e. during the final week of 1957) in association with excess mortality. Influenza deaths in England and Wales peaked during the third week of October 1957 and second week of January 1958. Deaths from all causes were higher in the September and December quarters of 1957 than in the same period in previous years back to 1950. Comparisons of the deaths during these periods provided a crude estimate of 33,431 for the toll in deaths for the influenza epidemic of 1957. 10
-
1968–70 The epicentre of the influenza A Hong Kong/1968 pandemic was in Kweichow Province of China. The virus was isolated in Hong Kong on 17 July 1968 and the first wave peaked in Hong Kong during the last week of August 1968. 11 Despite a number of virus seedings into Japan, an epidemic of A/Hong Kong/68 virus did not occur there until October 1968; spread was gradual and sporadic, in contrast with an influenza B epidemic that affected the whole country during the same period, and also in contrast with the 1957–8 Asian influenza epidemic. 12 In the UK, the first outbreak was identified in a residential school on 24 September 1968. The attack rate was low (9%), and until the end of 1968 only a few scattered outbreaks in residential facilities were reported by January and February 1969. 13 Nationally, claims for sickness benefit climbed steeply during the second week of January 1969, and eventually peaked during the first week of March 1969. The RCGP consultation rates for ‘clinical influenza’ increased gradually from the beginning of 1969 and peaked at ∼150 per 100,000 population during the first 2 weeks of March. The numbers of laboratory confirmed cases began to increase in the last week of December 1968, climbed to a peak of 161 cases in the week ending 31 January 1969, fell for 2 weeks, and then was maintained at a level of 126–161 per week until early April 1969. Weekly deaths assigned to influenza and influenza pneumonia during the first 3 months of 1969 was less than one-quarter of the deaths during the corresponding period of the previous year. The first wave of the pandemic was associated with no sudden or excess demand on either general medical practitioners or hospital services. While the rest of Europe also experienced a mild first wave, the experience in the USA was different; some 30%–40% of the population was affected, schools had 50% absenteeism and > 56,000 deaths were attributed to the outbreak. In the UK, the influenza A/Hong Kong/68 pandemic had its maximum impact during a second wave of infection that occurred during the winter of 1969–70. 14
Manifestations and burden of seasonal influenza
Seasonal influenza A and B affect about 10%–20% of the population each year. 15 In the USA, localised outbreaks are typically of 4–12 weeks’ duration. 16 The spectrum of influenza is broad, ranging from asymptomatic infection in about half, through an acute respiratory illness with or without systemic features; upper respiratory complications including sinusitis and otitis media; lower respiratory complications, including acute bronchitis, croup, asthma bronchiolitis and pneumonia; multisystem complications affecting the cardiovascular system, brain, liver, muscle and kidneys; to death, most commonly due to cardiopulmonary complications. 17
Despite the importance of influenza infection as a cause of morbidity and mortality, very few data exist from which estimates of the influenza disease burden, for the purposes of health economic studies, can be made. The difficulty in obtaining accurate information arises from a variety of sources: many episodes of illness may not come to medical attention; a specific diagnosis of influenza is frequently not sought; the disease is not reportable; outbreaks and epidemics may occur only in some areas or regions at different times; and many of the hospitalisations or deaths actually due to influenza may be attributed to other causes. Moreover, consultation rates, clinical practices and hospitalisation rates for influenzal illness may differ from country to country, and influenza epidemics can vary in magnitude and severity from one year to the next.
Recognised manifestations of influenza and influenza-like illness (ILI) include the use of over-the-counter relief medication, bed-days and restricted-activity days,18–20 school and workplace absenteeism,21–23 medical consultations for influenza and its complications,24–28 and hospitalisation and excess deaths. 28–30
Influenzal complications and death rates are not uniform across age bands. Most deaths occur in those aged > 75 years, and the risk of death is elevated considerably by the presence of certain chronic medical conditions, particularly respiratory and cardiac disorders, and by residential care.
Mortality from past pandemics
The H1N1 pandemic of 1918–19 was the most devastating in history, with a total mortality of 40–50 million. 31 In the USA, it killed 550,000 people, representing approximately 0.5% of the population. In Scotland, 1 in 200–300 of the population died. In England and Wales there were 200,000 deaths, and by December 1918, an estimated 4.9 million excess deaths (about 2% of the whole population) occurred in British India, the vast majority occurring within the space of 2 months. During 1918–19, morbidity and mortality were unusually high in young otherwise healthy adults. During the 1918–19 pandemic, mortality varied by ethnicity within certain countries (e.g. New Zealand and the USA), presumably reflecting differences that might affect the risk of infection and severity of illness, for example overcrowding, nutritional status, pregnancy, comorbidity and access to medical care. 32 Mortality increased during the second pandemic wave in comparison with the first wave. Whereas pneumonia developed in 3% of patients during the first wave, it occurred in 18% during the second. The fatality rate among US army personnel in the USA increased from 0.2% during the first wave to 4.2% during the second, and in the US army in France, the case–fatality rate increased from 0.3% during the first wave to 4.4% during the second. The reasons for this are not known. It has been suggested that with adaptation to man, the virus may become more virulent.
The mortality during the ‘Asian’ H2N2 influenza pandemic in 1957 was moderate in comparison to that seen during 1918–19, with an estimated 2 million deaths globally. 31 In England and Wales, mortality was estimated at 33,000 deaths. In the USA, 80,000 deaths were attributed to influenza during the 1957–8 and 1960 epidemics, with nearly one-half occurring in the first 3 months of the 1957–8 epidemic. During the ‘Hong Kong’ H3N2 pandemic of 1968, the global mortality was estimated at around 1 million deaths,31 while in the USA it was estimated at around 30,000 deaths. In Britain, mortality was also estimated at around 30,000 deaths.
The pandemics in 1957 and 1968 affected all ages, with the greatest excess mortality occurring in the elderly and in people of all ages with underlying medical conditions. The re-emergence of H1N1 virus in 1977 mostly affected young people and the outbreak was benign in comparison with the episodes in 1957 and 1968.
Swine influenza in humans
Influenza as a disease of pigs was first described during 1918 when outbreaks of respiratory disease occurred simultaneously in humans and swine herds living and working in close proximity. Pigs are thought to have an important role in interspecies transmission, as they possess receptors in their respiratory tract capable of binding both avian and human influenza.
Occasional isolation of swine influenza viruses from humans with respiratory illness has confirmed that sporadic human infection can occur. 33 Generally, cases have been limited to laboratory workers or those with occupational swine exposure. However, a pandemic alert was raised in 1976 when swine H1N1 caused an outbreak of respiratory illness with one fatality among 13 soldiers at a military base in Fort Dix, NJ, USA. 34 No exposure to pigs was found and seroepidemiological investigation identified up to 230 further soldiers had been infected, suggesting human–human transmission. Mass vaccination of the US public was initiated and halted amid reports of adverse vaccine reactions, media scepticism and the lack of pandemic activity. 35
Emergence of the H1N1 pandemic in 2009
During the spring of 2009, a novel influenza A/H1N1 virus of swine origin was isolated from cases of human infection and acute respiratory illness in Mexico. 36,37 In April 2009, near the end of the usual influenza season in the northern hemisphere, the first two cases of swine origin H1N1 influenza virus were identified in the USA. 38 The US Centers for Disease Control (CDC) confirmed that these cases were caused by a genetically similar swine virus that had not been previously identified in the USA. Clusters of severe pneumonia were first recognised in Mexico in mid-April 2009. On 23 April 2009, 18 of the Mexican cases were laboratory confirmed in Canada as swine origin influenza A/H1N1; a further five cases in California and Texas were confirmed as swine origin influenza A/H1N1 on 24 April 2009.
Genetic analysis of the strains showed that they were derived from a new reassortment of six gene segments from the known triple reassortant swine virus, and two gene segments (NA and matrix protein) from the Eurasian influenza A/H1N1 swine virus lineage. 39 After initially spreading among persons in the USA and Canada, the virus spread globally, and by the time WHO declared a pandemic on 11 June 2009,40 a total of 74 countries and territories had reported laboratory-confirmed infections, with evidence of community spread in more than one WHO region.
Characteristics of the 2009 H1N1 pandemic virus
The HA of the 2009 A/H1N1 pandemic virus is antigenically distinct from recent seasonal human H1N1 viruses. Antibodies to seasonal H1N1 virus do not protect against the pandemic H1N1 virus. 41 Antigenically, the 2009 A/H1N1 viruses are homogeneous and are most similar to classical swine A/H1N1 viruses, as well as to North American-lineage-triple-reassortant A/H1N1 viruses that have circulated in swine over the past 10 years in the USA and that have occasionally infected humans. 41 Sequence analysis of pandemic A/H1N1 viruses show that they were genetically homogeneous. Importantly, serological studies have shown that pandemic A/H1N1 viruses are antigenically homogeneous and similar to the A/California/7/2009 (H1N1) virus that has been selected for vaccine production. 42
Preliminary studies in the USA have shown that crossreactive microneutralisation (MN) antibody titres of ≥ 160 to A/California/2009 H1N1 were detected in 6% of adults aged 18–40 years, 9% of adults 18–64 years and 33% of adults aged 60 years and older. 43
Effectively all isolates are susceptible to NA inhibitors, but are resistant to M2 inhibitors (which inhibit the ion-channel function of the M2 protein which is integral in the viral envelope of the influenza A virus, e.g. amantadine). Oseltamivir-resistant virus has been identified in 20 countries in four WHO regions. As of 3 February 2010, a total of 225 oseltamivir-resistant cases had been reported worldwide. All these oseltamivir-resistant isolates have the same mutation in the NA gene (H275Y), conferring resistance to oseltamivir, but not to zanamivir. 44 Most cases have been sporadic, and, although three clusters have been described – two in severely immunocompromised patients – there is no evidence that oseltamivir-resistant pandemic virus has spread in the community. Of the 142 cases of oseltamivir-resistant pandemic influenza virus for which data are available, 56 (40%) occurred in severely immunocompromised patients, 54 (38%) were associated with the treatment of influenza, 16 (11%) with chemoprophylaxis45 and 16 (11%) had no known association with antiviral use. 44
Seasonal influenza vaccines
Inactivated influenza virus vaccines represent the mainstay of efforts to prevent influenza and its complications. Current licensed seasonal vaccines are produced from virus grown in eggs or cell culture systems and consist of either whole-virion (WV), detergent-treated ‘split-product’ or purified HA and NA (subunit) surface antigen formulations.
Vaccine efficacy of 70%–95% in healthy adults is obtained when there is a good match between the vaccine and the circulating strains. 46 Seasonal vaccines have reduced efficacy against antigenically drifted viruses and are considered ineffective against unrelated subtypes.
The use of mammalian cell lines, notably Vero cells and Madin–Darby Canine Kidney (MDCK) cells, to grow influenza virus are approved substrates for production of licensed trivalent seasonal vaccines that may allow for increased vaccine production at short notice to meet unexpected demand.
As vaccine responses are generally lower in elderly subjects, efforts to improve immunogenicity have been investigated. The addition of MF59, a squalene-containing, oil-in-water emulsion adjuvant, was found to increase postvaccination antibody titres and seroconversion rates (SCRs) in elderly and immunocompromised subjects. 47 MF59-adjuvanted seasonal influenza vaccines have been licensed for clinical use since 1997. More recently, two other squalene-containing oil-in-water adjuvants have been developed by GlaxoSmithKline (GSK) and Sanofi Pasteur. The GSK AS03A oil-in-water adjuvant has been extensively evaluated in association with H5N1 antigens.
Global manufacturing capacity for pandemic influenza vaccine
Seasonal influenza vaccines are given at doses of 15 µg of HA per virus strain. The global human population (August 2010) is estimated at 6.86 billion. 48 The annual global vaccine manufacturing capacity for trivalent seasonal influenza vaccines was 852 million doses in May 2009. 49 Assuming that the yield of the 2009 H1N1 antigen is comparable to that for seasonal virus strains, the present manufacturing capacity equates to 2.56 billion doses of monovalent H1N1 vaccine containing 15 µg of HA per dose. This would be enough for only 2.56 billion people if two doses containing 7.5 µg of HA were immunogenic, but could protect more people if one dose was sufficient. Pandemic H1N1 vaccines will be supplied over a period of 6 months or more, emphasising the importance of dose-sparing formulations and regimens to protect as many vulnerable people as possible.
Experience with pandemic and mock pandemic vaccines since the 1970s
Historically, influenza vaccines were first developed as ‘whole-virion’ formulations. During the late 1970s, WV vaccines were replaced by ‘split’ and highly purified ‘surface antigen’ formulations that caused fewer local and systemic reactions than WV vaccines, but are equally immunogenic when given to primed (i.e. had been infected or vaccinated with an antigenically similar influenza A virus, of the same subtype, previously) individuals as ‘seasonal’ or ‘interpandemic’ vaccine. But as outlined below, WV vaccines were found to be more immunogenic in people who were unprimed (i.e. were unlikely to have been infected or vaccinated with an antigenically similar influenza A virus, of the same subtype, previously).
Experience with H1N1 vaccines during the 1970s
Experience in unprimed individuals with vaccines produced from Hsw1N1 viruses (A/New Jersey/8/76) or H1N1 viruses (A/USSR/90/77) indicated that high concentrations of antigen (> 50 µg of HA) were needed in a single vaccine dose to generate haemagglutination inhibition (HI) titres that met the current European licensing criteria. In a two-dose schedule, HI titres of ≥ 40 could be achieved with two doses containing 5 µg of HA. Overall, WV vaccines were more immunogenic than split or subunit vaccines. The split and surface antigen vaccine formulations were notably less immunogenic than WV vaccine when given to children, both during 1976 when influenza A/New Jersey/76 (H1N1) posed a pandemic threat50 and during 1977 when A/USSR/77 (H1N1) virus re-emerged. 51
Immunogenicity of plain (i.e. non-adjuvanted split and subunit influenza vaccines) mock pandemic influenza vaccines
As outlined below, neither split nor subunit vaccine formulations of H5, H7 and H9 avian influenza satisfy all three CHMP licensing criteria when given at doses of up to 90 µg HA.
Treanor et a.l52,53 showed that neither two 90-µg doses of plain (i.e. non-adjuvanted) recombinant, baculovirus-expressed, H5 HA nor two 90-µg doses of egg-grown, plain, inactivated, subvirion influenza A/Vietnam/1203/2004 (H5N1) vaccine satisfied the CHMP regulatory criteria. Nicholson et a.l54 showed that two doses of 7.5-, 15- and 30-µg formulations of plain A/Duck/Singapore/97 (H5N3) surface antigen vaccine failed to meet the CHMP criteria.
Bresson et a.l55 showed that two doses of three 7.5- to 30-µg HA formulations of plain, split-virus, A/Vietnam/1194/2004 (H5N1) vaccine satisfied the CHMP criterion for a greater than 2.5-fold increase in antibody titre, but 47% vaccinees failed to achieve protective levels of antibody after a second dose. Nolan et a.l56 evaluated two doses of split A/Vietnam/1194/2004 (H5N1) vaccine containing 7.5–45 µg of HA with and without an alum adjuvant. All formulations met the CHMP criterion for a > 2.5-fold increase in HI antibody titres after the second dose, but not the criterion for > 70% of participants achieving seroprotection.
Keitel et a.l57 evaluated subvirion inactivated influenza A/H5N1 vaccine containing 3.75, 7.5, 15 or 45 µg of HA. Dose-related increases in antibody responses were noted after both vaccinations, but no formulation attained the CHMP criteria.
Stephenson et a.l58 evaluated two 7.5-, 15- and 30-µg doses of plain, subunit, influenza A/Hong Kong/1073/99 (H9N2) vaccines. The CHMP criterion for a > 2.5-fold increase in HI antibody titres was met after the second dose, but 86% vaccinees failed to attain protective levels of antibody. Cox et a.l59 evaluated two doses of split H7N1 virus vaccine containing 12 or 24 µg HA. Neither formulation fulfilled the CHMP licensing criteria.
Immunogenicity of whole-virion vaccines and vaccines adjuvanted with oil-in-water emulsions (mock pandemic influenza vaccines)
Whole-virion vaccines and vaccines adjuvanted with oil-in-water emulsions are more immunogenic in man than split and subunit vaccines. 54,58,60–67
Lin et a.l68 showed that a two-dose regimen of an aluminium hydroxide-adjuvanted whole-virion A/Vietnam/1194/2004 (H5N1) vaccine containing 10 µg of HA met all CHMP regulatory requirements for annual licensing of seasonal influenza vaccine. Ehrlich et al. 60 evaluated WV A/Vietnam/1203/2004 (H5N1) vaccine (manufactured by Baxter Healthcare) at doses of 3.75, 7.5, 15 or 30 g of HA with an alum adjuvant, and 7.5 or 15 µg without an adjuvant. Maximum responses to the vaccine strain were obtained with formulations without an alum adjuvant. When assessed by SRH, the 7.5-µg dose met all three CHMP licensing criteria. Two criteria were met when antibodies were measured by HI. The vaccine also induced a neutralising immune response against clade 2 and 3 strains, and results without an alum adjuvant elicited significantly higher immune responses than those with an alum adjuvant.
A Phase I randomised trial of subunit and whole-virion A/Hong Kong/1073/99 (H9N2) vaccine, given in two doses containing doses of 7.5, 15 or 30 µg of HA, revealed the presence of crossreacting antibodies in participants born before 1969 who were older than 32 years58 – this finding is comparable to the recent observation of an age-related presence of crossreacting antibodies to A/California/2009 (H1N1) in the USA. In participants older than 32 years, one dose of WV or subunit vaccine evoked antibody responses associated with protection. However, in people aged 32 years or younger, WV vaccine produced a significantly higher probability of seroconversion than with subunit virus for this age group. 58
Nicholson et al. 54 evaluated two doses of subunit A/Duck/Singapore/97 (H5N3) vaccine containing 3.75, 7.5 and 15 µg of HA with and without MF59 oil-in-water adjuvant. In this Phase I randomised trial, the GMTs of antibody and SCRs were significantly higher with MF59 adjuvanted vaccine. After the second injection, all MF59-adjuvanted vaccine doses met all three CHMP licensing criteria. Further studies showed improved antibody persistence with MF59 containing vaccine, improved immune responses to other clades of H5 virus, and significantly higher antibody responses on boosting. 61–64
Leroux-Roels et al. 65 evaluated A/Vietnam/1194/2004 (H5N1) vaccine manufactured by GSK at doses of 3.8, 7.5, 15 and 30 μg HA with and without its proprietary AS03A adjuvant. The adjuvanted formulations were significantly more immunogenic than the non-adjuvanted formulations at all antigen doses. At the lowest antigenic dose, immune responses for the adjuvanted vaccine against the vaccine strain met or exceeded all the US Food and Drug Administration (FDA) and CHMP licensure criteria. Further research showed broad cross-clade immune responses at the lowest antigen dose (3.8 µg) with adjuvant, but no cross-clade response in the non-adjuvanted group. 66,67
European licensing criteria for seasonal and pandemic vaccines
During the late 1970s, influenza vaccines were poorly standardised. Subsequently, improved methods of measuring vaccine potency and ensuring vaccine standardisation were introduced, and in Europe, by criteria for licensure of seasonal,69 and, latterly, pandemic vaccines. 70
As specified in the EU Committee for Medicinal Products for Human Use (CHMP), ‘Guideline on dossier structure and content for pandemic influenza vaccine marketing authorisation application’,70 a pandemic candidate vaccine should at least be able to elicit sufficient immunological responses to meet and preferably exceed all three of the current standards set for existing vaccines in unprimed adults or elderly subjects as specified for seasonal vaccines. 69
These include assessments of the mean geometric increase in antibody titre (the seroconversion factor), the number of seroconversions or significant increases in antibody and the seroprotection rate (i.e. the proportion attaining ‘protective’ levels of antibody). The criteria are based on the HI assay or single radial haemolysis (SRH). Both assays have been established as surrogates for protection. The CHMP guidelines stipulate that vaccines should be tested in adults (18–60 years) and elderly (> 60 years), in groups of > 50 subjects, and attain the following:
Adults (18–60 years):
-
seroconversions/or significant rises (i.e. a fourfold increase in postvaccination titre) by > 40%
-
mean-fold increase in geometric mean titre (GMT) postvaccination > 2.5
-
significant levels of antibody (i.e. having post-vaccination HI titres ≥ 1 : 40) in > 70%.
Elderly (> 60 years):
-
seroconversions/or significant rises (i.e. a fourfold increase in postvaccination titre) by > 30%
-
mean-fold increase in GMT > 2
-
significant levels of antibody (i.e. having postvaccination HI titres ≥ 1 : 40) in > 60%.
Reproducibility of the serology assays for influenza
For research purposes and vaccine licensure, influenza vaccines are evaluated by clinical trials that assess immunogenicity by the presence of serum antibody. Collaborative studies have shown that the serology assays are highly variable between laboratories – with variability between laboratories for HI assays varying by up to 32-fold. 71–73 This leads to difficulties in interpreting results from different manufacturers. At the fifth WHO Meeting on Evaluation of Pandemic Influenza Prototype Vaccines in Clinical Trials, 12–13 February 2009, WHO highlighted the need for standardised assays and internationally accepted antiserum standards. 74
Relationship between antibody and protection
Based on observations at the Medical Research Council Common Cold Unit,75 an HI titre of 1 : 40 is generally accepted to be associated with a 50% reduction in the risk of illness in a susceptible population and is referred to as the 50% protective titre (50% PT). An HI titre of 1 : 40 is a required target for vaccine licensure. The research conducted by Hobson et al. 75 included virus challenge studies, or vaccine field studies, and involved 1032 subjects exposed to influenza A and B. Hobson et a.l75 concluded that the 50% PT ranged from 1/18 to 1/36. Recently, Coudeville et al. 76 developed a model that estimates the level of clinical protection against influenza at any HI titre. The source data were derived from a systematic literature review that identified 15 studies, representing a total of 5899 adult subjects and 1304 influenza cases. Significant relationships between HI titre and clinical protection against influenza were observed in all tested models, irrespective of the virus type and strain. The 50% PT obtained with this model was 1 : 29. The relationship is not exact, and a titre of 1 : 40 could be associated with protection of < 60% to > 80%. Other antigens, for example the NA and M2 protein, may also be associated with protection. There is no correlation of protection for MN antibodies.
The pandemic H1N1 vaccines purchased by the government
During 2009, the UK Department of Health (DH) purchased pandemic H1N1 vaccines from Baxter Healthcare and GSK. The Baxter vaccine (trade name Celvapan) is a plain (i.e. non-adjuvanted), monovalent, WV, Vero cell-grown, influenza A/H1N1 vaccine, containing 7.5 µg of HA per 0.5 ml dose. The GSK vaccine (trade name Pandemrix) is a monovalent AS03A-adjuvanted, split-product, egg-grown, influenza A/H1N1 vaccine, containing 3.75 µg of HA and oil-in-water AS03A adjuvant (composed of squalene, DL-α-tocopherol and polysorbate 80). To ensure that protection is provided as rapidly as possible, it is imperative that vaccine is used efficiently.
This multicentre study examines the adverse events (AEs) and immune responses of both vaccines in young, middle-aged and elderly adults.
Chapter 2 Study objectives and methods
Study design
The study was an observer-blind, age-stratified, multicentre, parallel-group randomised controlled trial (RCT) comparing the immunogenicity and short-term reactogenicity of two scheduled doses of WV or AS03A oil-in-water adjuvanted, split-virion 2009 H1N1 vaccine in healthy adults.
Figure 1 summarises the study method, and Table 1 shows the time and event schedule.
FIGURE 1.
Study method.
| Events | Study visit | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
| Days after the first vaccination: | ||||||||
| Window (days) | ||||||||
| 0 | (± 1) | (± 2) | (± 2) | (± 2) | (± 3) | (± 3) | (± 10) | |
| Study day | ||||||||
| 0 | 7 | 14 | 21 | 28 | 35 | 42 | 180 | |
| Informed consent | × | |||||||
| Inclusion/exclusion criteria | × | × | ||||||
| Medical/medication history | × | |||||||
| Pregnancy test | × | × | ||||||
| Blood sample – antibody studies | × | × | × | × | × | × | × | × |
| Vaccination | × | × | ||||||
| Thermometer/diary card | × | × | ||||||
| Diary card training | × | |||||||
| Diary card returned/review | × | × | ||||||
| Reminder regarding unsolicited events | × | × | × | × | × | × | × | |
| AEs monitoring | × | × | × | × | × | × | × | |
| Termination of study | × | |||||||
A list of the case record forms used in the study is shown in Appendix 1.
Study objectives
This RCT was designed to compare the immunogenicity in adults of one and two doses of the two vaccines purchased by the government in response to the 2009 H1N1 pandemic. Each vaccine was assessed in its licensed formulation, to reflect the antibody response and levels of protection that are likely to occur in adults in the general population up to 6 months after vaccination.
Specific trial objectives were as follows.
Primary
To evaluate the immunogenicity of Baxter cell-culture, non-adjuvanted, WV H1N1 vaccine, and GSK AS03A-adjuvanted, split-virion H1N1 vaccine, with respect to CHMP and FDA licensing criteria. 69,70,77
Secondary
-
To identify whether one or two doses of vaccine are required to satisfy the licensing criteria.
-
To examine the short-term reactogenicity of the vaccines.
-
To examine the kinetics of the antibody responses to vaccination.
-
To examine persistence of antibody at 6 months.
-
And, if appropriate, (i.e. an antigenic drift variant emerges prior to the 2010–11 influenza season), to evaluate the breadth of the antibody response to the antigenic variant.
Vaccines
The 2009 H1N1 vaccines used were licensed products available in the UK: Celvapan™ (Baxter) and Pandemrix™ (GSK). The HA content of each vaccine was determined by single radial immunodiffusion. NA content is not standardised and is unknown. Vaccines were stored at 4°C until use.
Celvapan™, the non-adjuvanted, WV vaccine, was manufactured by Baxter AG (Vienna, Austria). The seed virus was egg-derived, wild-type A/California/7/2009 (H1N1). Vaccine was prepared using a serum-free Vero-cell culture system, without antibiotics, and was formulated with 7.5 µg of formaldehyde- and UV-inactivated H1N1 HA per 0.5-ml dose. Vaccine suspension containing trometamol, sodium chloride, water and polysorbate 80 was presented without thiomersal in 5-ml multidose vials; 0.5 ml of suspension was drawn into a single syringe for injection. Opened vials were used within 3 hours.
Pandemrix™, the adjuvanted split-virion vaccine, was manufactured by GSK (GSK Biologicals, Dresden, Germany). The vaccine virus [New York Medical College (NYMC) X-179A] was generated from the A/California/7/2009 strain, and supplied by the US CDC. The seed virus was propagated on hens’ eggs and harvested virus was split using standard processes for interpandemic (Fluarix®, GSK Biologicals, Dresden, Germany) vaccine production. Vaccine was supplied as two multidose vials: H1N1 antigen with thiomersal and AS03A-adjuvant emulsion (GSK Biologicals, Rixensart, Belgium). The final formulation was prepared immediately before administration by mixing equal 0.25-ml volumes of antigen and AS03A-adjuvant to give a 0.5-ml injection containing 3.75 µg of H1 HA, 10.69 mg of squalene, 11.86 mg of DL-α-tocopherol and 4.86 mg of polysorbate 80.
Outcomes
The primary outcome measure was vaccine immunogenicity using CHMP and FDA licensing criteria (Appendix 2). 69,70,77 The immunogenicity of the two-dose schedule of the two influenza 2009 H1N1 vaccines was assessed by HI assay, according to standard methods,78–80 at the Centre for Infections, Health Protection Agency, London, UK with egg-grown NIBRG-121 virus, generated from A/California/7/2009 and A/PR/8/34 strains using reverse genetics, as the test antigen [National Institute for Biological Standards and Control (NIBSC) UK].
The three immunogenicity end points were:
-
the seroprotection rate – i.e. the proportion of subjects with HI titres of ≥ 1 : 40,
-
the SCR – i.e. the proportion of subjects with either seroconversion or significant increase in HA titre (i.e. prevaccination HI titre ≤ 1 : 8 and a postvaccination titre ≥ 1 : 40; or a prevaccination titre ≥ 1 : 8 and an increase in the titre by fourfold or more), and
-
the mean fold titre elevation – i.e. the factor increase in the geometric mean HI titre, pre-vaccination and postvaccination.
Immunogenicity end points were assessed by HI on day 0 (before vaccination), and at 21 and 42 days later. The kinetics of the HI antibody response, measured 7 and 14 days after each vaccination, and the persistence of antibody, measured 6 months after the first vaccination, were also assessed using the above three immunogenicity end points. The breadth of the antibody response, as assessed by antibody responses to antigenic variants of the pandemic H1N1 virus, was a further planned end point. This end point was not assessed due to the failure of antigenic drift variants of the pandemic H1N1 virus to emerge during the study.
Immunogenicity was also assessed by MN assay, but there are no CHMP or FDA licensing criteria to assess vaccines by MN. Accordingly, immunogenicity end points by MN were:
-
the proportion of subjects with MN titres of ≥ 1 : 40, and
-
the GMT.
Immunogenicity end points were assessed by MN on day 0 (before vaccination), and at 21 and 42 days later. The kinetics of the MN antibody response, measured 7 and 14 days after each vaccination was also assessed using the above two immunogenicity end points.
Subjects and recruitment
This observer-blind, multicentre study was undertaken at three study sites in the English East Midlands, mostly in teaching hospital settings in Leicester (Leicester Royal Infirmary), Nottingham (Nottingham City Hospital) and Sheffield (Royal Hallamshire Hospital). Some elderly subjects were recruited in surgeries following invitations from general practitioners (GPs) in Newbold Verdon, Leicestershire, UK. The study population included healthy male and female adults, or adults with stable chronic medical conditions. We recruited six groups of male and female adults, who were stratified by age (18–44, 45–64 and 65 years and older).
Potential participants were identified from several sources in each study centre, including medical students, nursing and medical staff, and staff and students at universities within each city. They were given an information leaflet and had an opportunity to discuss the study with a member of the research team. Training was given to research teams about the project and research governance. A member of the research team saw each participant to discuss the study. Consent was sought at the screening visit.
Inclusion criteria
The same inclusion and exclusion criteria were used in each age group and in each centre. Adults who fulfilled all of the following inclusion criteria were eligible:
-
mentally competent adults who give signed informed consent after receiving a detailed explanation of the study protocol
-
clinically healthy, male or female volunteers aged 18 years of age and older, including those aged 65 years and over, and those with stable, high-risk medical conditions; ‘stable’ is defined as having no medical consultations for an exacerbation or worsening of any chronic medical condition during the preceding 8 weeks, and maintenance on a stable drug regimen for at least 2 weeks prior to study entry, as assessed by the medical history
-
those who understand and comply with all study procedures and can complete study diaries
-
those who can be contacted and are available for all study visits
-
women using secure contraceptive precautions: either (1) the oral contraceptive pill or (2) condom/barrier contraception, or (3) their partner has had a vasectomy or (4) they have been surgically sterilised or (5) are postmenopausal (defined as at least 2 years since the last menstrual period).
Exclusion criteria
-
Unable to lead an independent life either physically or mentally.
-
Pregnancy or lactation.
-
Refusal to use reliable contraception (women of reproductive age) during days 0–42 of the study.
-
Laboratory-confirmed infection with H1N1 pandemic influenza.
-
Treatment with oseltamivir or zanamivir for ILI since May 2009.
-
Oseltamivir or zanamivir treatment of a household member for ILI since May 2009.
-
Laboratory-confirmed pandemic H1N1 infection in a household member.
-
Received another investigational vaccine or medicinal product during the preceding 4 weeks.
-
Unwilling to refuse participation in another study during days 0–42 of the study.
-
Clinically significant concurrent illness or unstable medical condition including malignancy, progressive renal or hepatic pathology, chronic obstructive pulmonary disease requiring oxygen therapy, and any active neurological disorder.
-
Systemic antibiotic or antiviral therapy during the preceding 7 days (chronic antibiotic therapy for prevention of urinary tract infections is acceptable).
-
A temperature ≥ 38°C within 3 days of vaccination.
-
Acute illness at the time of vaccination (note: minor infections without fever or systemic upset are not contraindications/exclusion criteria).
-
Known or suspected impairment/alteration of immune function, including:
-
– treatment with oral immunosuppressive drugs or other drugs listed in section 8 of the British National Formulary (BNF), or chloroquine, gold or penicillamine or other drugs listed in section 10.1.3 of the BNF to suppress a chronic disease process (note: long-term, inhaled steroids for asthma management is acceptable)
-
– treatment with immunostimulants or interferon
-
– treatment with an immunoglobulin preparation, blood products and/or plasma derivatives within 3 months of the study
-
– is at high risk of developing immunocompromising condition
-
– radiotherapy or chemotherapy within 6 months of the study.
-
-
Planned surgery during days 0–42 of the study.
-
Regularly drink > 40 units of alcohol weekly.
-
Drug abuse (recreational or prescribed, known or suspected).
-
Conditions that might complicate interpretation of the study results.
-
Previous anaphylaxis or serious reactions to vaccines, hypersensitivity (other than anaphylaxis) to influenza viral protein or to any component of the study vaccines, products containing mercury, egg and chicken protein, ovalbumin, formaldehyde, gentamicin sulphate, sodium deoxycholate or benzonase.
-
History of any neurological symptoms and signs following administration of any vaccine.
-
Actual or planned receipt of another vaccine, excluding seasonal influenza vaccine, during the period 3 weeks before to 3 weeks after vaccination on days 0 and 21.
Study procedures
Table 1 summarises the time and event schedule.
Having sought consent, the screening assessment was completed by a clinical investigator or study nurse. The inclusion/exclusion criteria were reviewed to ensure that the participant was eligible. The assessment included demographic details, review of medical history, medication (including the use of analgesia or antipyretic medications before vaccination, seasonal vaccination against influenza, previous vaccination against H5 or H9 avian influenza, and the occurrence of ILI since May 2009). Female participants of child-bearing potential were required to have a negative urine pregnancy test in order to be included in the study and to agree to use adequate contraception throughout its duration. A 10-ml blood sample was collected at baseline before vaccination and oral temperature was recorded before vaccination.
The first vaccine dose was administered according to the randomisation list by intramuscular (IM) injection into the deltoid muscle of the non-dominant arm. Subjects were observed for 30 minutes after vaccination and any local or systemic reactions were recorded. Subjects were instructed how to evaluate and record local reactions and were given a diary card and thermometer. Over the next 7 days, subjects recorded – in self-completed diaries – the severity of solicited local (pain, bruising, erythema and swelling) and systemic symptoms (chills, malaise, muscle aches, nausea and headache), oral temperature and use of analgesic medications.
A 10-ml blood sample was collected 7 days after vaccination to measure HI and MN antibody responses and the first diary card was reviewed and collected. Further 10-ml blood samples for HI and MN antibody titrations were collected 14 and 21 days after the first vaccination. Female participants of child-bearing potential were required to have a negative urine pregnancy test immediately before administration of the second dose of vaccine that was administered 21 days after the first. The oral temperature was measured before the second dose that was of the same type and antigen content as the first dose, and was administered by IM injection into the deltoid muscle of the non-dominant arm. Subjects were observed for 30 minutes after vaccination and the oral temperature and any local or systemic reactions were recorded in a second diary card. Over the next 7 days, subjects recorded – in self-completed diaries – the severity of solicited local and systemic symptoms as before. Blood samples (10 ml) for HI and MN antibody titrations were collected 7, 14 and 21 days after the second vaccination. The second diary card was reviewed and collected 7 days after the second vaccination.
A final blood sample was collected 180 days after the first vaccination.
Participant withdrawal criteria
According to the judgement of the lead clinician, participants could be withdrawn from the study if they were prescribed systemic steroids, other immunosuppressive agents, blood or plasma derivates, including immunoglobulin, and non-study vaccines (with the exception of postexposure vaccinations in a medical emergency, e.g. hepatitis, rabies and tetanus) during the study. Additionally, the investigator could withdraw a subject if, in his/her clinical judgement, it was in the best interest of the subject, for example following occurrence of convulsions or any other neurological disturbances after vaccination, hypersensitivity to the investigational vaccine and other suspected side effects that could compromise the subject’s well-being or if the subject could not comply with the protocol. If a participant discontinued the study prematurely (i.e. before completion of the protocol), the primary reason for discontinuation was recorded when given. In all cases the investigator ensured that the participant received medical follow-up as necessary. Withdrawn participants were not replaced.
Randomisation
Randomisation was organised by the trial statistician at the University of Leicester, UK. Participants were stratified by age (ages 18–44, 45–64 and 65 years and older) and trial centre (Leicester, Nottingham and Sheffield), and randomised to WV vaccine (Baxter) or AS03A-adjuvanted vaccine (GSK) in a 1 : 1 ratio using randomly permuted block sizes of two, four and six, generated using the ralloc procedure within stata (version 11; StataCorp LP, College Station, TX, USA). Each centre was provided with a randomisation list. The statistician at Leicester University generated and provided the chief investigator three sealed ‘randomisation’ lists for each centre with a list for each age band. The statistician also provided individually numbered randomisation envelopes for the three sites for all three age groups.
Subjects were assigned a five-digit subject number. The first digit identified the study site (1 for Leicester, 2 for Nottingham, 3 for Sheffield); the second digit reflected the age of the subjects on day 0 (1 for 18–44 years, 5 for 45–64 years, and 9 for 65 years and older). The unblinded study nurse/doctor opened the individual randomisation envelopes in sequence and allocated the vaccine as per the slip in the envelope. To maintain blinding, volunteers were told to look away, both during preparation and administration of the vaccine.
Grading of events after vaccination
Symptoms were graded as: ‘none’, ‘mild’ (if they did not interfere with normal activities), ‘moderate’ (if they interfered with normal activities) and ‘severe’ (if they prevented engagement in daily activities and necessitated medical attention). Serious adverse reactions were any reaction requiring medical attention during the study period. Solicited local reactions were considered to be vaccine related, whereas the investigator assessed the causality of solicited systemic and unsolicited AEs.
A serious adverse event (SAE) was any untoward medical occurrence that:
-
resulted in death
-
was life-threatening (i.e. the subject was, in the opinion of the investigator, at immediate risk of death from the event as it occurred)
-
required inpatient hospitalisation
-
resulted in persistent or significant disability/incapacity (i.e. caused a substantial disruption of a person’s ability to conduct normal life functions)
-
resulted in a congenital anomaly/birth defect
-
required intervention to prevent permanent impairment or damage, or
-
an important and significant medical event that may not have been immediately life threatening or resulting in death or hospitalisation but, based upon appropriate medical judgement, may have jeopardised the subject or may have required intervention to prevent one of the other outcomes listed above.
Sample size
The primary aim of the trial is to establish whether the GSK and Baxter pandemic H1N1 vaccines satisfy CHMP and FDA licensing criteria (Appendix 2), and, if so, compare them in terms of immunogenicity (for each vaccine/age group and each vaccine type). The sample size is in line with standard practice. The protocols for seasonal EU vaccine clinical trials and the criteria for assessment have been standardised within the EU. They stipulate that trials should be carried out with groups of at least 50 subjects. We planned to recruit 60 subjects per group, allowing for up to 17% dropout. With 180 subjects per arm (60 in each age group), the study had over 95% power to detect an overall difference in seroprotection/seroconversion of 20% at the 5% level of statistical significance, and 80% power to detect an overall difference of 15%, each assuming a baseline of 50%.
Blinding
Symptom diary cards were completed by study participants who were blind to the randomisation group. To enable blinding to be achieved, research nurses who were tasked with vaccine administration in each centre played no other role in the study. All other research staff were blinded to randomisation. Participants were instructed to look away during the preparation and administration of vaccine. Aliquots of sera were labelled with participant’s trial code number and day of collection. The titration of sera for HI and MN antibodies was undertaken by laboratory staff who were blind to the randomisation group and demographic details. The trial code was broken only when all data queries were resolved and day 0–42 antibody titrations were completed.
Statistical methods
The three immunogenicity end points were: the proportion of subjects with HI titres of 1 : 40, the proportion of subjects with either seroconversion or significant increase in titre, and the factor increase in the GMT. Secondary end points included the frequency, duration and intensity of postvaccination reactions (solicited and unsolicited for 7 days) and the incidence of SAEs during the study period.
Geometric mean titres and 95% confidence interval (CI) for each visit were computed by taking the exponential (log10) of the mean and of the lower and upper limits of the 95% CI of the log10-transformed titres. GMTs were compared between each pair of vaccine groups by means of one-way analysis of variance (ANOVA) on the log10-transformed titres. The proportions of subjects in whom seroconversion (prevaccination HI titre ≤ 1 : 8 and a postvaccination titre ≥ 1 : 40; or a prevaccination titre ≥ 1 : 8 and an increase in the titre by fourfold or more) or seroprotection (HI titre of ≥ 1 : 40) was achieved were compared between each group by two-sided Fisher’s exact test. The role of age, centre, vaccine type and receipt of seasonal vaccination was explored by multiple logistic regression analysis for GMT, and multiple logistic regression for seroprotection. A stepwise model selection procedure (with a significance criterion of 0.05) was used to identify important factors, with potential interactions between these factors and vaccine type being assessed using likelihood ratio tests. Age-related trends in the proportion of subjects with seroconversion or seroprotection were computed by chi-squared test for trend, while age-related trends in GMTs were examined using linear regression. Analyses for the immunogenicity end points stratified patients into those who had antibodies present at baseline, those that did not, and a combined population. The combined population was considered as analogous to an ITT analysis/population, as in practice antibody status will not be known at the time of vaccination.
For solicited and unsolicited reactions, the percentages of subjects (point estimates and 95% CIs) were based on the frequency and severity of reported responses after vaccination. Exact (Clopper–Pearson) CIs are reported for all proportional end points. We used a two-sided Fisher’s exact test to compare proportions between vaccine groups, with no adjustments for multiple testing; values of ≤ 0.05 were considered to indicate statistical significance.
All analyses were performed using stata (version 11).
Laboratory tests
Antibody responses were detected by MN and HI assays, according to standard methods78–80 at the Centre for Infections, HPA, London, UK, with egg-grown NIBRG-121 virus, generated from A/California/7/2009 and A/PR/8/34 strains using reverse genetics, as the test antigen. NIBRG-121 virus was prepared and provided by the NIBSC, UK. The gene segments encoding the HA and NA were derived from the influenza A/California/7/2009 strain, with the remaining genes taken from the influenza A/PR/8/34 virus. The antigen for both assays was propagated in 11-day-old embryonated hens’ eggs at 37°C and harvested 3 days after infection.
Serum samples were tested with the use of 1 : 2 serial dilutions. For HI assays, sera were tested at an initial dilution of 1 : 8, and those that were negative were assigned a titre of 1 : 4. Samples were analysed to determine absolute end point titres, and the final dilution was 1 : 65,536. For MN assays, sera were tested blind at an initial dilution of 1 : 10, and those that were negative were assigned a titre of 1 : 5. The final dilution was 1 : 320, and samples for which the end point titres were greater were assigned a value of 1 : 640.
Four positive and two negative laboratory control sera were included in each run of both the HI and MN assays. All four positive control sera contained high titres of antibody to NIBRG-121 virus, both by HI and MN. In addition, the international H1N1 standard antibody (supplied by the NIBSC, UK), was tested on five occasions by HI and three by MN.
Ethical arrangements and research governance
The Multicentre Research Ethics Committee (MREC) approval for the study was given by Royal Free Hospital and Medical School Research Ethics Committee on 16 September 2009. Site-specific approval was obtained from the following:
-
Leicester, Leicestershire and Rutland Primary Care Research Office.
-
University Hospitals of Leicester NHS Trust, Directorate of Research and Development.
-
NHS Derby City Research and Development Office.
-
Derbyshire County NHS Primary Care Trust Research and Development Office.
-
Nottinghamshire County Teaching PCT Research and Evaluation.
-
Sheffield Teaching Hospitals Research and Development Office.
-
Regulatory approval was granted by the Medicines and Healthcare Products Regulatory Agency (MHRA) and the trial was conducted in accordance with the International Conference on Harmonisation – Good Clinical Practice (ICHGCP). 81
Summary of any changes to the project protocol
One amendment was made to the protocol and implemented following regulatory approval. We amended the exclusion criteria allowing receipt of seasonal influenza vaccine both before and during the study. Due to delays in getting the study under way in Nottingham and Sheffield, additional subjects were recruited above the target of 40 per age group in Leicester. The primary outcome was assessed separately in groups with and without antibody at baseline, and then subsequently for the whole study population.
Chapter 3 Results
Study recruitment
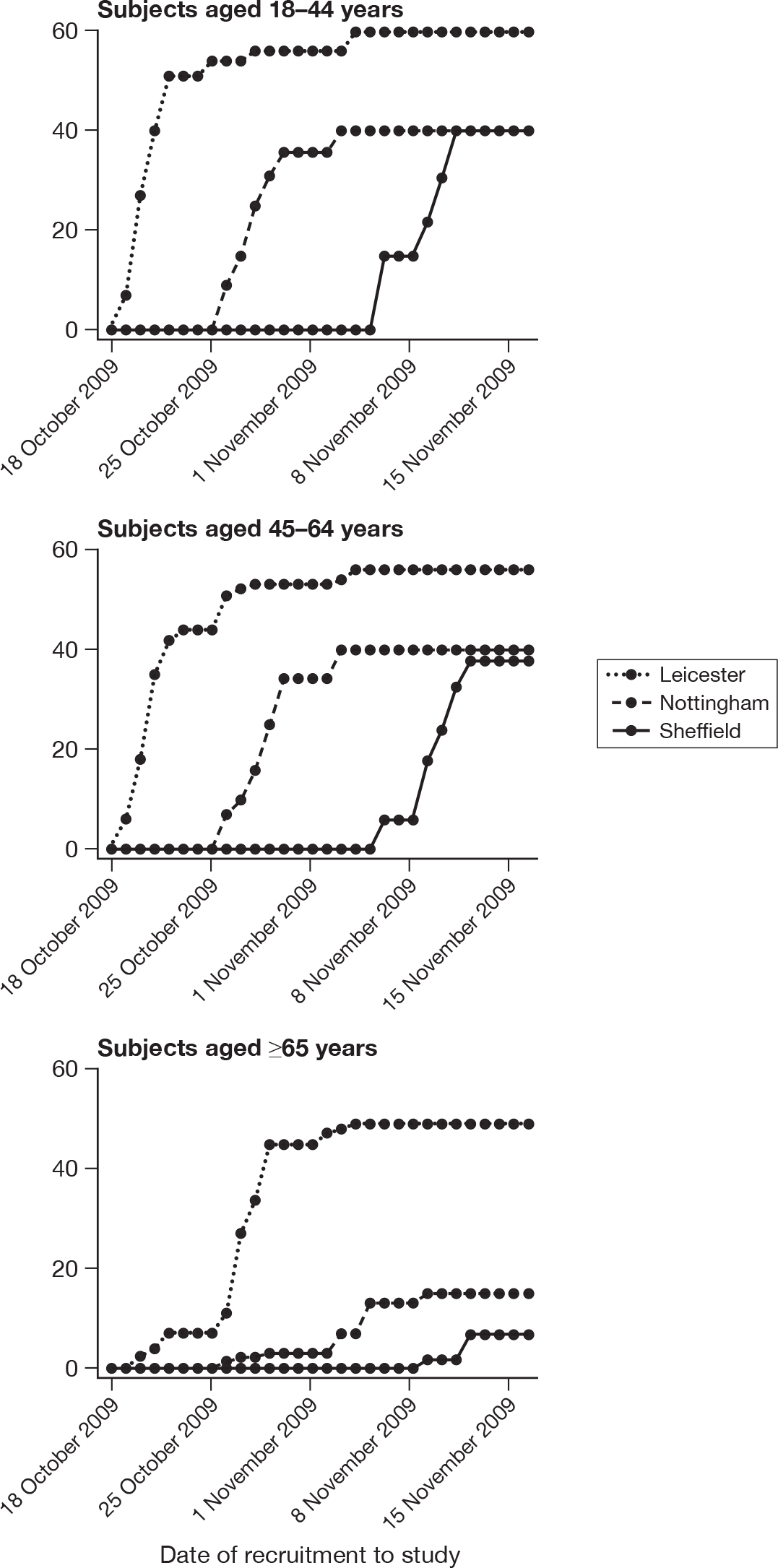
Between 19 October 2009 and 12 November 2009, 347 participants were enrolled and received the first vaccine in age groups of ≥ 18–44 years (n = 140), ≥ 45–64 years (n = 136) and ≥ 65 years (n = 71) years. One hundred and seventy-two (49.6%) participants were randomised to receive WV vaccine and 175 (50.4%) received adjuvanted vaccine. Weekly cumulative recruitment by age and by centre is shown in Figure 2. One hundred and sixty-five (47.6%) participants were enrolled in Leicester, 95 (27.3%) were enrolled in Nottingham and 86 (24.8%) were enrolled in Sheffield.
FIGURE 2.
Weekly cumulative recruitment by centre and by age.
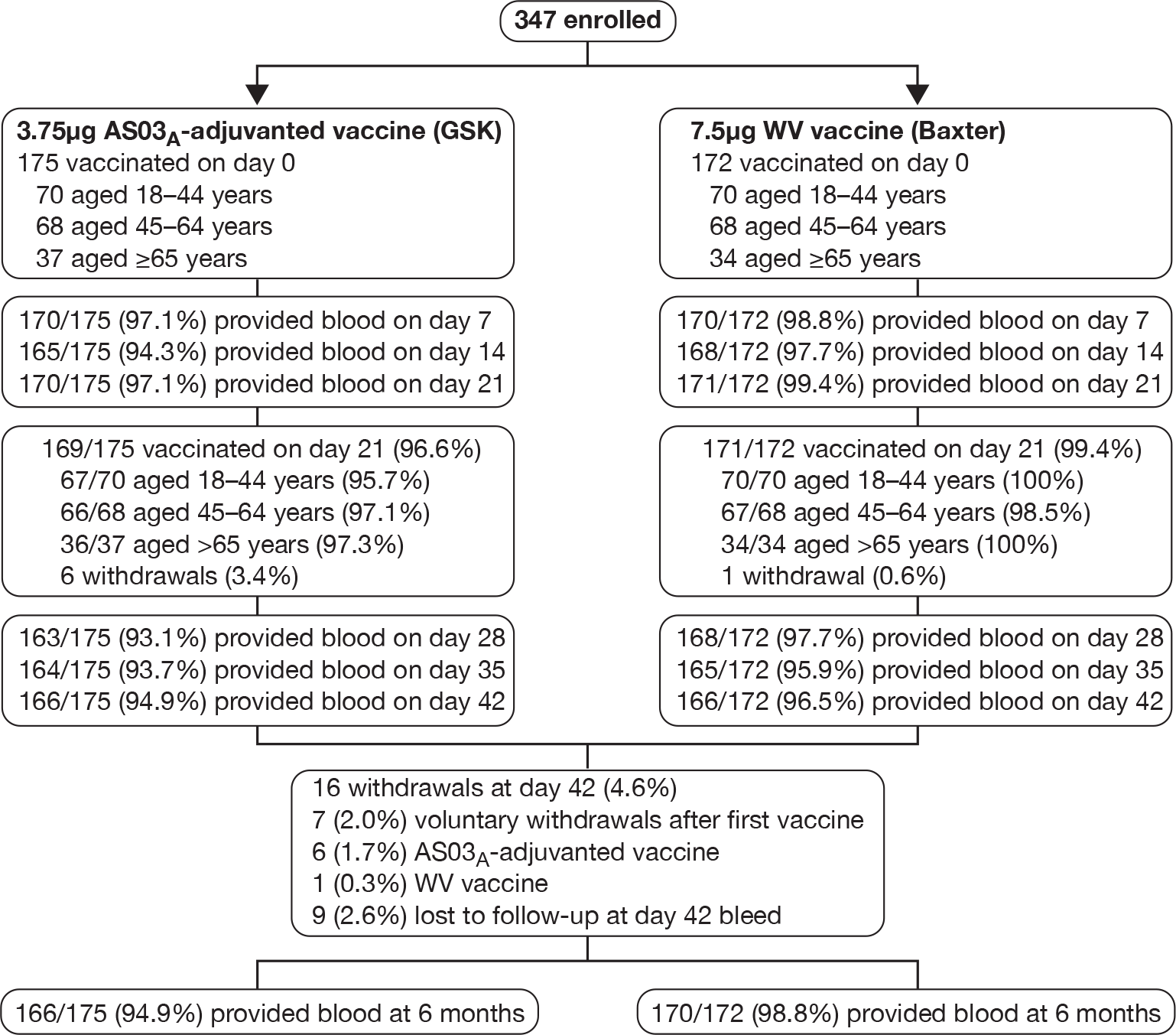
Study compliance
Figure 3 shows the participant flow through the trial. Both vaccine doses were given in 340 subjects (98%). There were seven (2%) withdrawals after the first vaccine: three each from the 18- to 44-year (all adjuvanted) and 45- to 64-year groups (one WV and two adjuvanted), and one from the ≥ 65-year group. There was no significant difference in the withdrawal rates during the 3 weeks after the first vaccination with either the Baxter or the GSK vaccine (p = 0.0713). Data from 680 (99%) of 687 issued diary cards were returned. Sera was obtained from 340 (98.0%), 333 (96%), 341 (98.3%), 330 (95.1%), 328 (94.5%) and 331 (95.4%) subjects on days 7, 14, 21, 28, 35 and 42, respectively. Three hundred and forty-six and 345 subjects were included in the safety and immunogenicity analyses.
FIGURE 3.
Study profile. Note: seven cases withdrew after the first vaccination for the following reasons: (1) pressure of work; (2) breast carcinoma identified during routine mammography; (3) spray-back of uncertain amount of vaccine during the first injection; (4) failure to meet inclusion/exclusion criteria when the second injection was due; and (5–7) no reason specified. Examination of the symptom records for the seven subjects who withdrew after the first vaccination revealed no severe local or systemic reactions to the first vaccination or any fever. One recipient of adjuvanted vaccine stayed at home during the 24-hour period after vaccination.
Study population
Randomisation groups were well matched at baseline with regard to demography and previous receipt of seasonal influenza vaccine (Table 2).The median age was 49 years (range 18–83 years), 62.5% were female, 92.5% were white and 50.7% had previously received influenza vaccine – 34.4% during 2008. Overall, seasonal influenza vaccine uptake during 2008 increased with age (p < 0.0001, chi-squared test for linear trend).
| Characteristic | WV vaccine | AS03A-adjuvanted split-virion vaccine | All subjects (N = 347) | ||||
|---|---|---|---|---|---|---|---|
| 18–44 years (n = 70) | 45–64 years (n = 68) | ≥ 65 years (n = 34) | 18–44 years (n = 70) | 45–64 years (n = 68) | ≥ 65 years (n = 37) | ||
| Age: years | |||||||
| Median | 29 | 52.5 | 71 | 27 | 53 | 71 | 49 |
| Range | 19–44 | 45–64 | 65–81 | 18–44 | 45–64 | 65–83 | 18–83 |
| Sex: no. (%) | |||||||
| Female | 45 (64.3) | 41 (60.3) | 19 (55.9) | 50 (71.4) | 47 (69.1) | 15 (40.5) | 217 (62.5) |
| Male | 25 (35.7) | 27 (39.7) | 15 (44.1) | 20 (28.6) | 21 (30.9) | 22 (59.5) | 130 (37.5) |
| Race: no. (%) | |||||||
| White | 60 (85.7) | 66 (97.1) | 33 (97.1) | 60 (85.7) | 65 (95.6) | 37 (100) | 321 (92.5) |
| Other | 10 (14.3) | 2 (2.9) | 1 (2.9) | 10 (14.3) | 3 (4.4) | 0 (0.0) | 26 (7.5) |
| Previous receipt of seasonal influenza vaccinea | 16 (22.9) | 36 (52.9) | 34 (100) | 17 (24.3) | 37 (54.4) | 36 (97.3) | 176 (50.7) |
| Received 2008–9 seasonal influenza vaccine | 8 (11.4) | 28a (41.8) | 28 (82.4) | 4 (5.7) | 22 (32.3) | 29 (78.4) | 119a (34.4) |
Prevaccination antibody was detected by HI (titre ≥ 1 : 8) and MN (titre ≥ 1 : 10) in 44 (12.7%) and 103 (29.7%) of subjects, respectively; this was inversely related to age, but not statistically significant (HI, p = 0.2; MN, p = 0.2), and not by previous receipt of 2008–9 seasonal vaccine for HI (p = 0.3), but was for MN (p = 0.002). GMTs differed between centres for the 18- to 44-year group (HI, p = 0.001; MN, p = 0.02), but not for 45- to 64-year age group or those aged 65 years and over. Baseline GMTs did not differ between the WV vaccine and adjuvanted vaccine groups (HI, p = 0.7; MN, p = 0.4). Immunogenicity was assessed in populations without antibody at baseline, subjects with antibody at baseline, and all subjects regardless of the presence of baseline antibody.
Subjects without baseline HI antibody
Appendix 3 shows the results of HI assays on days 0, 7, 14, 21, 28, 35 and 42 in subjects without baseline HI antibody. There was an age–response relationship regarding seroprotection rates (≥ 1 : 40) for adjuvanted (p < 0.002)) and WV vaccine (p < 0.0025, each visit), and GMTs for adjuvanted (all p < 0.0001) and WV vaccine (all p < 0.0001). Vaccine type and subject age, but not receipt of 2008–9 seasonal vaccine, were independent predictors of the response by HI on day 21 (both p < 0.0001) and day 42 (both p < 0.001). However, after adjusting for age, vaccine type remained statistically significant on both days 21 (p < 0.001) and 42 (p < 0.001), and there was no evidence of an interaction between vaccine type and age at either days 21 (p = 0.2) or 42 (p = 0.9).
Subjects with baseline HI antibody
Appendix 4 shows the results of HI assays on days 0, 7, 14, 21, 28, 35 and 42 in subjects with baseline HI antibody. There was an age-response relationship regarding GMTs for adjuvanted vaccine on days 7 (p = 0.02), 14 (p = 0.02), 21 (p = 0.04), 28 (p = 0.003), 35 (p = 0.005) and 42 (p = 0.003), but not for WV vaccine (all p > 0.05). After adjustment for age, adjuvanted vaccine was superior to WV vaccine (day 7, p = 0.007; day 14, p = 0.005; day 21, p = 0.001; day 28, p = 0.01; day 35, p = 0.01; and day 42, p = 0.02). No further multiple linear or logistic regression was performed for those subjects with baseline antibodies due to the relatively small numbers of patients.
All subjects, regardless of baseline HI antibody
Appendix 5 shows the results of HI assays on days 0, 7, 14, 21, 28, 35 and 42 in all subjects regardless of baseline HI antibody. There was an age–response relationship regarding GMTs for adjuvanted (p < 0.0001, all visits) and WV vaccine (p < 0.0001, all visits). After adjustment for age, adjuvanted vaccine was superior to WV vaccine (p < 0.0001, all visits). Stepwise multiple linear regression identified only centre in addition to vaccine type and age group as being important on days 21 (p = 0.04) and 42 (p = 0.004), although after adjustment for both centre and age group, adjuvanted vaccine was superior to WV vaccine (p < 0.0001, both days) and there was very little or no evidence of a vaccine–factor interaction either on day 21 (p = 0.04, centre; p = 0.2, age group) or day 42 (p = 0.1, centre; p = 0.9, age group).
Subjects without baseline MN antibody
Appendix 6 shows the results of MN assays on days 0, 7, 14, 21, 28, 35 and 42 in subjects without baseline MN antibody. There were significant age–response relationships regarding the development of postvaccination titres of 1 : 40 or more on day 7 (p < 0.0001), day 14 (p < 0.0001) and day 21 (p ≤ 0.0008) for adjuvanted vaccine, and at any postvaccination visit for WV vaccine (all p < 0.0001). There were also significant age–response relationships regarding GMTs, at any postvaccination visit, for both adjuvanted vaccine (all p < 0.0001) and WV vaccine (day 7, p = 0.02; days 14, 21, 28, 35 and 42, all p < 0.0001).
At day 21 for ‘seroprotection’, stepwise logistic regression identified only age group as being a significant factor as well as vaccine group, and, after adjustment for this, adjuvanted vaccine continued to yield a greater seroprotection rate compared with WV vaccine [odds ratio (OR) 5.83, 95% CI 2.89 to 11.77, p < 0.001]. At day 42 only previous seasonal vaccination in 2008–9 was identified as being statistically significant in addition to vaccine group (p < 0.001). However, after adjustment for this, adjuvanted vaccine continued to yield a greater seroprotection rate compared with WV vaccine (OR 12.75, 95% CI 3.68 to 44.16, p < 0.001) and there was no evidence of an interaction between vaccine type and previous vaccination (p = 0.8). In terms of GMTs at day 21, stepwise multiple linear regression identified age group, previous season vaccination and centre as important factors, although after adjustment for all of these, adjuvanted vaccine continued to be superior to WV (p < 0.001). However, there was evidence of a centre–vaccine type interaction (p = 0.002) and, to a lesser extent, previous vaccination (p = 0.04) and age group (p = 0.03). At day 42, age group and previous vaccination in 2008–9 appeared to be important in addition to vaccine type, with an inverse relationship with age and those individuals who had had a previous vaccination having lower GMTs. However, adjustment for these factors did not alter the statistical significance of vaccine type (p < 0.0001), while there was little evidence of factor–vaccine type interactions (p = 0.05, age group; p = 0.06, previous vaccination), nor was there evidence of an interaction between age group and previous vaccination (p = 0.9).
Subjects with baseline MN antibody
Appendix 7 shows the results of MN assays on days 0, 7, 14, 21, 28, 35 and 42 in subjects with baseline MN antibody. There was an age–response relationship for both adjuvanted vaccine (day 7, p = 0.002; day 14, p = 0.001, day 21, p < 0.001; day 28, p = 0.003; day 35, p = 0.02; and day 42, p = 0.01) and WV vaccine (p < 0.001, all visits). After adjustment for age, adjuvanted vaccine was superior to WV vaccine on days 28 (p = 0.02), 35 (p = 0.002) and 42 (p = 0.002). No multiple further linear or logistic regression was performed for those subjects with baseline MN antibodies due to the relatively small numbers of subjects.
All subjects, regardless of baseline MN antibody
Appendix 8 shows the results of MN assays on days 0, 7, 14, 21, 28, 35 and 42 in all subjects regardless of baseline HI antibody. GMTs at all visits decreased with increasing age, for adjuvanted vaccine (p < 0.001, all visits), and WV vaccine (p ≤ 0.001, all visits). Adjuvanted vaccine GMTs were greater compared with WV vaccine at all visits (p < 0.001). Adjustment of the effect of age group did not alter the effect of vaccine type (p < 0.001, all visits). In terms of ‘seroprotection’, adjuvanted vaccine produced greater seroprotection rates compared with WV vaccine at all visits (p < 0.001), while there was a decreasing effect of age for both adjuvanted and WV vaccine at all visits (p < 0.0001), except for adjuvanted vaccine at days 28 (p = 0.08), 35 (p = 0.03) and 42 (p = 0.08). At day 21, OR for adjuvanted compared with WV vaccine was 3.36 (95% CI 1.92 to 5.87, p < 0.001) and at day 42 OR was 10.11 (95% CI 3.49 to 29.26, p < 0.001). Adjustment for the effect of age group did not alter the comparative effect estimates (p < 0.001, all visits) and there was little evidence for an age group–vaccine type interaction (p > 0.1, all visits). At day 21, the adjusted OR was 4.11 (95% CI 2.25 to 7.48) and at day 42 it was 11.71 (95% CI 3.97 to 34.49). At day 21, stepwise regression did not identify any factor other than age group that had an effect on either GMTs or seroprotection. At day 42 previous seasonal influenza vaccination during the 2008–9 season was identified as a potentially important factor for both GMTs (p = 0.03) and seroprotection (p = 0.05), but there was little evidence of an interaction between previous vaccination and the effect of vaccine type for either GMTs (p = 0.03) or seroprotection (p = 0.6).
Primary outcome
The CHMP and FDA immunogenicity criteria were assessed separately by vaccine group and by age in: (1) subjects without HI antibody at baseline; (2) subjects with HI antibody at baseline; and (3) the whole study population, i.e. including subjects with and without HI antibody at baseline. CHMP and FDA immunogenicity criteria were assessed 21 days after each vaccination.
The Committee for Medicinal Products for Human Use criteria in participants without baseline HI antibody
Table 3 shows the response to vaccination in relation to CHMP criteria in participants without baseline HI antibody. On day 21, WV vaccine met two out of three CHMP criteria (seroconversions and factor increase in GMT) in vaccinees aged 18–44 years and all age groups combined, and one of the three criteria (factor increase in GMT) in the 45- to 64-year-olds and over-65-year-olds. All three criteria were satisfied 21 days after the first dose of adjuvanted vaccine in 18- to 44-year-olds and 45- to 64-year-olds, and all age groups combined (Table 3). Adjuvanted vaccine satisfied two out of three criteria (seroconversions and factor increase in GMT) in the over-65-year-olds after one dose of vaccine. Twenty-one days after the second dose (i.e. on day 42), WV vaccine met two out of three criteria (seroconversions and factor increase in GMT) in each age group and all age groups combined. All three CHMP criteria were satisfied 21 days after adjuvanted vaccine in each age group and all age groups combined (see Table 3).
| WV vaccine | AS03A-adjuvanted split-virion vaccine | |||||||
|---|---|---|---|---|---|---|---|---|
| 18–44 years | 45–64 years | ≥ 65 years | All subjects | 18–44 years | 45–64 years | ≥ 65 years | All subjects | |
| Participants without baseline antibody as measured on HI assay | ||||||||
| Baseline | ||||||||
| No. of subjects | 58 | 62 | 30 | 150 | 60 | 59 | 34 | 153 |
| GMT value (95% CI) | 4.1 (4.0 to 4.2) | 4.0 (4.0 to 4.1) | 4.2 (3.9 to 4.4) | 4.1 (4.0 to 4.1) | 4.1 (4.0 to 4.2) | 4.0 (4.0 to 4.0) | 4.1 (3.9 to 4.3) | 4.1 (4.0 to 4.1) |
| After first dose: day 21 | ||||||||
| No. of subjects | 58 | 61 | 30 | 149 | 57 | 57 | 34 | 148 |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 65.5 (51.9 to 77.5) | 36.1 (24.2 to 49.4) | 30.0 (14.7 to 49.4) | 46.3 (38.1 to 54.7) | 93.0 (83.0 to 98.1) | 73.7 (60.3 to 84.5) | 50.0 (32.4 to 67.6) | 75.7 (68.0 to 82.4) |
| Seroconversions: % (95% CI) | 65.5 (51.9 to 77.5) | 36.1 (24.2 to 49.4) | 30.0 (14.7 to 49.4) | 46.3 (38.1 to 54.7) | 93.0 (83.0 to 98.1) | 73.7 (60.3 to 84.5) | 50.0 (32.4 to 67.6) | 75.7 (68.0 to 82.4) |
| Factor increase in GMT: value (95% CI) | 18.3 (11.5 to 29.2) | 4.8 (2.9 to 8.0) | 4.2 (2.6 to 7.0) | 7.9 (5.8 to 10.8) | 90.0 (61.6 to 131.5) | 22.4 (14.1 to 35.5) | 8.7 (4.7 to 16.0) | 30.8 (22.7 to 41.6) |
| After first dose: day 42 | ||||||||
| No. of subjects | 56 | 61 | 29 | 146 | 55 | 56 | 34 | 145 |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 67.9 (54.0 to 79.7) | 41.0 (28.6 to 54.3) | 34.5 (17.9 to 54.3) | 50.0 (41.6 to 58.4) | 100 (93.5 to 100) | 89.3 (78.1 to 96.0) | 76.5 (58.8 to 89.3) | 90.3 (84.3 to 94.6) |
| Seroconversions: % (95% CI) | 67.9 (54.0 to 79.7) | 41.0 (28.6 to 54.3) | 34.5 (17.9 to 54.3) | 50.0 (41.6 to 58.4) | 100 (93.5 to 100) | 89.3 (78.1 to 96.0) | 76.5 (58.8 to 89.3) | 90.3 (84.3 to 94.6) |
| Factor increase in GMT: value (95% CI) | 23.0 (14.3 to 36.8) | 6.2 (3.9 to 9.8) | 3.7 (2.2 to 6.1) | 9.2 (6.8 to 12.5) | 123.3 (98.4 to 154.4) | 32.8 (22.6 to 47.7) | 18.1 (11.7 to 27.9) | 47.1 (37.4 to 59.4) |
The US Food and Drug Administration criteria in participants without baseline HI antibody
Table 3 shows the response to vaccination in relation to FDA criteria in participants without baseline HI antibody.
On day 21, WV vaccine met one of two end points (i.e. exceeding the lower bound of the two-sided 95% CI for the percentage of subjects with seroconversions) in vaccinees aged 18–44 years. Neither end point was met on day 21 in participants aged 45–64 years, ≥ 65 years, and all age groups combined after WV vaccine.
Adjuvanted vaccine met both end points (seroconversions and seroprotection) on day 21 in vaccinees aged 18–44 years, and one end point (i.e. exceeding the lower bound of the two-sided 95% CI for the percentage of subjects with seroconversions) in those aged 45–64 years and ≥ 65 years, and all age groups combined.
After the second dose (i.e. on day 42), WV vaccine met one FDA end point (exceeding the lower bound of the two-sided 95% CI for the percentage of subjects with seroconversions) in vaccinees aged 18–44 years, and all age groups combined. Both FDA end points were met on day 42 after adjuvanted vaccine in participants aged 18–44 years, 45–64 years, and all age groups combined; the seroconversion end point was met in subjects aged ≥ 65 years.
Participants with baseline HI antibody
Table 4 shows the baseline HI antibody levels in 44 participants with baseline HI antibody. At baseline, among recipients of WV vaccine, HI titres were ≥ 1 : 40 among 11 out of 12 (91.7%) 18- to 44-year-olds, four out of six (66.7%) 45- to 64-year-olds, and one out of four (25.0%) elderly participants. Among recipients of AS03A-adjuvanted vaccine, baseline HI titres were ≥ 1 : 40 among 8 out of 10 (80.0%) 18- to 44-year-olds, six out of nine (66.7%) 45- to 64-year-olds, and all three (100%) over-65-year-olds.
| WV vaccine | AS03A-adjuvanted split-virion vaccine | |||||||
|---|---|---|---|---|---|---|---|---|
| 18–44 years | 45–64 years | ≥ 65 years | All subjects | 18–44 years | 45–64 years | ≥ 65 years | All subjects | |
| Participants with baseline antibody as measured on HI assay | ||||||||
| Baseline | ||||||||
| No. of subjects | 12 | 6 | 4 | 22 | 10 | 9 | 3 | 22 |
| GMT value (95% CI) | 80.7 (59.7 to 109.1) | 67.7 (20.3 to 226.0) | 22.6 (7.5 to 68.2) | 61.0 (41.7 to 89.2) | 137.3 (54.0 to 349.1) | 57.3 (32.2 to 102.0) | 71.7 (19.1 to 268.7) | 87.9 (54.6 to 141.4) |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 91.7 (61.5 to 99.8) | 66.7 (22.3 to 95.7) | 25.0 (0.6 to 80.6) | 72.7 (49.8 to 89.3) | 80.0 (44.4 to 97.5) | 66.7 (29.9 to 92.5) | 100 (29.2 to 100) | 77.3 (54.6 to 92.2) |
| After first dose: day 21 | ||||||||
| No. of subjects | 12 | 6 | 4 | 22 | 10 | 9 | 3 | 22 |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 100 (73.5 to 100) | 66.7 (22.3 to 95.7) | 50.0 (6.8 to 93.2) | 81.8 (59.7 to 94.8) | 100 (69.2 to 100) | 100 (66.4 to 100) | 66.7 (9.4 to 99.2) | 95.5 (77.2 to 99.9) |
| Seroconversions: % (95% CI) | 50.0 (21.1 to 78.9) | 33.3 (4.3 to 77.7) | 25.0 (0.6 to 80.6) | 40.9 (20.7 to 63.6) | 60.0 (26.2 to 87.8) | 77.8 (40.0 to 97.2) | 66.7 (9.4 to 99.2) | 68.2 (45.1 to 86.1) |
| Factor increase in GMT: value (95% CI) | 3.1 (1.9 to 4.9) | 2.8 (1.4 to 5.9) | 3.1 (0.1 to 117.4) | 3.0 (1.9 to 4.9) | 6.1 (2.3 to 16.1) | 10.4 (4.4 to 24.9) | 2.8 (0.1 to 62.7) | 6.8 (3.9 to 12.0) |
| After first dose: day 42 | ||||||||
| No. of subjects | 11 | 5 | 4 | 20 | 9 | 9 | 3 | 21 |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 100 (71.5 to 100) | 80.0 (28.4 to 99.5) | 50.0 (6.8 to 93.2) | 85.0 (62.1 to 96.8) | 100 (66.4 to 100) | 100 (66.4 to 100) | 66.7 (9.4 to 99.2) | 95.2 (76.2 to 99.9) |
| Seroconversions: % (95% CI) | 54.5 (23.4 to 83.3) | 40.0 (5.3 to 85.3) | 25.0 (0.6 to 80.6) | 45.0 (23.1 to 68.5) | 55.6 (21.2 to 86.3) | 77.8 (40.0 to 97.2) | 33.3 (0.8 to 90.6) | 61.9 (38.4 to 81.9) |
| Factor increase in GMT value (95% CI) | 3.9 (2.1 to 7.0) | 3.3 (1.7 to 6.3) | 3.1 (0.1 to 117.4) | 3.5 (2.1 to 6.1) | 6.35 (2.1 to 19.3) | 8.0 (2.7 to 23.8) | 1.8 (0.2 to 15.5) | 5.8 (3.1 to 11.1) |
The Committee for Medicinal Products for Human Use criteria in participants with baseline HI antibody
Table 4 shows the response to vaccination in relation to the CHMP criteria. On day 21, WV vaccine met all three CHMP criteria in participants aged 18–44 years and all age groups combined, and one of three criteria (factor increase in GMT) in the 45- to 64-year-olds and the elderly. Adjuvanted vaccine met all three CHMP criteria in all three age groups and all age groups combined.
On day 42, WV vaccine met all three CHMP criteria in participants aged 18–44 years and all age groups combined, two of three criteria (seroprotection and factor increase in GMT) in the 45- to 64-year-olds, and one of three criteria (factor increase in GMT) in the elderly. Adjuvanted vaccine met all three CHMP criteria in participants aged 18–44 and 45–64 years and all age groups combined; it met two of three criteria (seroprotection and seroconversions) in the elderly.
The US Food and Drug Administration criteria in participants with baseline HI antibody
Table 4 shows the response to vaccination in relation to the FDA criteria. The 95% CIs were wide due to the small numbers of subjects in each group. On day 21, WV vaccine met one end point (seroprotection) in 18- to 44-year-olds. Adjuvanted vaccine met both end points (seroconversions and seroprotection) in all age groups combined. On day 42, both vaccines met one end point (seroprotection) – WV vaccine in the 18- to 44-year-olds, and adjuvanted vaccine in all age groups combined.
Participants with and without baseline HI antibody
Table 5 shows the baseline HI antibody levels in all 347 participants with and without baseline HI antibody. At baseline, in the WV vaccine arm, HI titres were ≥ 1 : 40 among 11 of 70 (15.7%) 18- to 44-year-olds, 4 out of 68 (5.9%) 45- to 64-year-olds and 1 out of 34 (2.9%) over-65-year-olds. In the adjuvanted vaccine arm, baseline HI titres were ≥ 1 : 40 among 8 out of 70 (11.4%) of 18- to 44-year-olds, 6 out of 68 (8.8%) 45- to 64-year-olds, and 3 out of 37 (8.1%) over-65-year-olds.
| WV vaccine | AS03A-adjuvanted split-virion vaccine | |||||||
|---|---|---|---|---|---|---|---|---|
| 18–44 years | 45–64 years | ≥ 65 years | All subjects | 18–44 years | 45–64 years | ≥ 65 years | All subjects | |
| Participants with and without baseline antibody as measured on HI assay | ||||||||
| Baseline | ||||||||
| No. of subjects | 70 | 68 | 34 | 172 | 70 | 68 | 37 | 175 |
| GMT value (95% CI) | 6.8 (5.2 to 9.0) | 5.2 (4.2 to 6.4) | 5.1 (4.1 to 6.3) | 5.8 (5.0 to 6.7) | 6.8 (4.9 to 9.3) | 5.7 (4.5 to 7.2) | 5.2 (4.0 to 6.8) | 6.0 (5.1 to 7.0) |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 15.7 (8.1 to 26.4) | 5.9 (1.6 to 14.4) | 2.9 (0.1 to 15.3) | 9.3 (5.4 to 14.7) | 11.4 (5.1 to 21.3) | 8.8 (3.3 to 18.2) | 8.1 (1.7 to 21.9) | 9.7 (5.8 to 15.1) |
| After first dose: day 21 | ||||||||
| No. of subjects | 70 | 67 | 34 | 171 | 67 | 66 | 37 | 170 |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 71.4 (59.4 to 81.6) | 38.8 (27.1 to 51.5) | 32.4 (17.4 to 50.5) | 50.9 (43.1 to 58.6) | 94.0 (85.4 to 98.4) | 77.3 (65.3 to 86.7) | 51.4 (34.4 to 68.1) | 78.2 (71.3 to 84.2) |
| Seroconversions: % (95% CI) | 62.9 (50.5 to 74.1) | 35.8 (24.5 to 48.5) | 29.4 (15.1 to 47.5) | 45.6 (38.0 to 53.4) | 94.0 (85.4 to 98.4) | 74.2 (62.0 to 84.2) | 51.4 (34.4 to 68.1) | 74.7 (67.5 to 91.0) |
| Factor increase in GMT value (95% CI) | 13.5 (8.9 to 20.6) | 4.6 (2.9 to 7.3) | 4.1 (2.5 to 6.7) | 7.0 (5.3 to 9.2) | 60.1 (39.6 to 91.3) | 20.2 (13.3 to 30.5) | 7.9 (4.4 to 14.1) | 25.3 (19.1 to 33.6) |
| After first dose: day 42 | ||||||||
| No. of subjects | 67 | 66 | 33 | 166 | 64 | 65 | 37 | 166 |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 73.1 (60.9 to 83.2) | 43.9 (31.7 to 56.7) | 36.4 (20.4 to 54.9) | 54.2 (46.3 to 62.0) | 100 (94.4 to 100) | 90.8 (81.0 to 96.5) | 75.7 (58.8 to 88.2) | 91.0 (85.5 to 94.9) |
| Seroconversions: % (95% CI) | 65.7 (53.1 to 76.8) | 40.9 (29.0 to 53.7) | 33.3 (18.0 to 51.8) | 49.4 (41.6 to 57.3) | 93.8 (84.8 to 98.3) | 87.7 (77.2 to 94.5) | 72.9 (55.9 to 86.2) | 86.7 (80.6 to 91.5) |
| Factor increase in GMT value (95% CI) | 17.1 (11.1 to 26.4) | 5.9 (3.9 to 9.0) | 3.6 (2.2 to 6.0) | 8.2 (6.2 to 10.8) | 81.2 (57.4 to 115.0) | 27.0 (18.7 to 38.9) | 15.0 (9.5 to 23.7) | 36.2 (28.5 to 46.0) |
The Committee for Medicinal Products for Human Use criteria in participants with and without baseline HI antibody
Table 5 shows the response to vaccination in relation to CHMP criteria. On day 21, WV vaccine met all three CHMP criteria in 18- to 44-year-olds, two CHMP criteria in all age groups combined (seroconversions and factor increase in GMT), and one (factor increase in GMT) in 45- to 64-year-olds and over-65-year-olds. On day 21, adjuvanted vaccine met three criteria in 18- to 44-year-olds and 45- to 64-year-olds and all age groups combined (Table 5), and two criteria (seroconversions and factor increase in GMT) in the over-65-year-olds.
On day 42, WV vaccine met all three CHMP criteria in vaccinees aged 18–44 years, and two criteria (seroconversions and factor increase in GMT) in 45- to 64-year-olds, over-65-year-olds, and all age groups combined (Table 5). Adjuvanted vaccine satisfied all three criteria in each age group and all age groups combined.
The US Food and Drug Administration criteria in participants with and without baseline HI antibody
Table 5 shows the response to vaccination in relation to the FDA criteria. On day 21, WV vaccine met one end point (seroprotection) in vaccinees aged 18–44 years. Adjuvanted vaccine met both end points (seroconversions and seroprotection) in 18- to 44-year-olds and all groups combined, and the seroconversion target in the 45- to 64-year-olds, and over-65-year-olds. On day 42, WV vaccines met the seroconversion end point in 18- to 44-year-olds and all age groups combined. Adjuvanted vaccine met both end points in 18- to 44-year-olds, 44- to 64-year-olds, and all age groups combined, and the seroconversion end point in the over-65-year-olds.
Secondary outcomes
Number of doses of vaccine required to satisfy The Committee for Medicinal Products for Human Use licensing criteria
Table 6 summarises the immunogenicity data, by age, after each dose of WV and adjuvanted vaccine in relation to attainment of CHMP criteria:
| Age (years) | No. of CHMP criteria satisfied by the first vaccine dose in groups ± baseline antibody | No. of CHMP criteria satisfied by the second vaccine dose in groups ± baseline antibody | ||||
|---|---|---|---|---|---|---|
| No baseline HI antibody | Baseline HI antibody | All subjects | No baseline HI antibody | Baseline HI antibody | All subjects | |
| WV vaccine | ||||||
| 18–44 | 2a,b | 3a–c | 3a–c | 2a,b | 3a–c | 3a–c |
| 45–64 | 1b | 1b | 1b | 2a,b | 2b,c | 2a,b |
| ≥ 65 | 1b | 1b | 1b | 2a,b | 1b | 2a,b |
| All age groups | 2a,b | 3a–c | 2a,b | 2a,b | 3a–c | 2a,b |
| AS03A-adjuvanted vaccine | ||||||
| 18–44 | 3a–c | 3a–c | 3a–c | 3a–c | 3a–c | 3a–c |
| 45–64 | 3a–c | 3a–c | 3a–c | 3a–c | 3a–c | 3a–c |
| ≥ 65 | 2a,b | 3a–c | 2a,b | 3a–c | 2a,c | 3a–c |
| All age groups | 3a–c | 3a–c | 3a–c | 3a–c | 3a–c | 3a–c |
Whole-virion vaccine
18- to 44-year-olds
-
All three CHMP criteria were met after one dose of WV vaccine in subjects with baseline HI antibody, and in all subjects (i.e. with and without baseline HI antibody).
-
Two CHMP criteria were met after one and two doses of WV vaccine in subjects without baseline HI antibody. WV vaccine missed the seroprotection target in subjects without baseline HI antibody by about 4% and 2% after one and two doses, respectively (see Table 3).
45- to 64-year-olds
-
Two CHMP criteria were met after two doses of WV vaccine in subjects without HI baseline antibody, subjects with baseline HI antibody, and in all subjects.
-
One CHMP criterion was met after one dose of WV vaccine in subjects without baseline HI antibody, subjects with baseline HI antibody, and in all subjects.
Over-65-year-olds
-
Two CHMP criteria were met after two doses of WV vaccine in subjects without baseline HI antibody and in all subjects.
-
One CHMP criterion was met after one dose of WV vaccine in subjects without baseline HI antibody, subjects with baseline HI antibody, and in all subjects, and after two doses in subjects with baseline HI antibody.
All age groups
-
All three CHMP criteria were met after one dose of WV vaccine in subjects with baseline HI antibody.
-
Two CHMP criteria were met after one dose of WV vaccine in subjects without baseline HI antibody, and in all subjects.
AS03A-adjuvanted vaccine
18- to 44-year-olds
-
All three CHMP criteria were met after one dose of adjuvanted vaccine in subjects without baseline antibody, subjects with baseline HI antibody, and in all subjects.
45- to 64-year-olds
-
All three CHMP criteria were met after one dose of adjuvanted vaccine in subjects without baseline antibody, subjects with baseline HI antibody, and in all subjects.
Over-65-year-olds
-
All three CHMP criteria were met after one dose of adjuvanted vaccine in subjects with baseline HI antibody, and two doses in subjects without baseline HI antibody, and in all subjects.
-
Two CHMP criteria were met after one dose of adjuvanted vaccine in subjects without baseline HI antibody and in all subjects.
All age groups
-
All three CHMP criteria were met after one dose of adjuvanted vaccine in subjects without baseline HI antibody, subjects with baseline antibody, and in all subjects.
Short-term reactogenicity
Solicited reactions during the first 7 days following any vaccine dose are shown in Table 7. Local reactions of pain, erythema and swelling, and systemic reactions of muscle aches, chills and malaise occurred more frequently after adjuvanted than WV vaccine. The occurrence of reactions after adjuvanted vaccine generally diminished with age. The frequency or severity of reactions did not increase after the second dose of either vaccine (Appendix 9, Tables 9 and 10). Self-reported reactions were graded mostly as mild or moderate and generally resolved within 72 hours.
| WV vaccine | AS03A-adjuvanted split–virion vaccine | |||||||
|---|---|---|---|---|---|---|---|---|
| 18–44 years (n = 70) | 45–64 years (n = 68) | ≥ 65 years (n = 34) | All subjects (n = 172) | 18–44 years (n = 70) | 45–64 years (n = 67) | ≥ 65 years (n = 37) | All subjects (n = 174) | |
| Local reaction [% (95% CI)] | ||||||||
| Pain at injection siteb | ||||||||
| None | 63 (51 to 73) | 75 (63 to 84) | 82 (66 to 92) | 72 (64 to 78) | 10 (5 to 20) | 24 (15 to 36) | 54 (38 to 69) | 25 (19 to 32) |
| Mild | 31 (22 to 43) | 22 (14 to 33) | 15 (6 to 31) | 24 (19 to 31) | 56 (44 to 67) | 55 (43 to 67) | 43 (28 to 59) | 53 (45 to 60) |
| Moderate | 6 (2 to 14) | 3 (1 to 11) | 3 (0 to 18) | 4 (2 to 8) | 30 (20 to 42) | 21 (13 to 32) | 3 (0 to 17) | 21 (15 to 27) |
| Severe | 0 | 0 | 0 | 0 | 4 (1 to 13) | 0 | 0 | 2 (1 to 5) |
| Redness diameterb | ||||||||
| 0 mm | 84 (74 to 91) | 90 (80 to 95) | 88 (72 to 96) | 87 (81 to 91) | 57 (45 to 68) | 68 (56 to 78) | 81 (65 to 91) | 66 (59 to 73) |
| 1–4 mm | 14 (8 to 25) | 4 (1 to 13) | 9 (3 to 24) | 9 (6 to 15) | 24 (16 to 36) | 12 (6 to 22) | 8 (3 to 22) | 16 (11 to 22) |
| ≥ 5 mm | 1 (0 to 10) | 6 (2 to 15) | 3 (0 to 18) | 3 (2 to 8) | 19 (11 to 29) | 21 (13 to 32) | 11 (4 to 26) | 18 (13 to 24) |
| Swelling diameterb | ||||||||
| 0 mm | 91 (82 to 96) | 97 (89 to 99) | 97 (82 to 100) | 95 (90 to 97) | 61 (50 to 72) | 71 (59 to 80) | 81 (65 to 89) | 69 (62 to 76) |
| 1–4 mm | 7 (3 to 16) | 0 | 3 (0 to 18) | 3 (2 to 8) | 20 (12 to 31) | 15 (8 to 25) | 8 (3 to 22) | 15 (11 to 22) |
| ≥ 5 mm | 1 (0 to 10) | 3 (1 to 11) | 0 | 2 (1 to 5) | 19 (11 to 29) | 15 (8 to 25) | 11 (4 to 26) | 15 (11 to 22) |
| Bruising diameter | ||||||||
| 0 mm | 90 (80 to 95) | 96 (87 to 99) | 91 (76 to 97) | 92 (87 to 96) | 84 (74 to 91) | 87 (76 to 93) | 100 | 89 (83 to 93) |
| 1–4 mm | 4 (1 to 13) | 0 | 0 | 2 (1 to 5) | 9 (4 to 18) | 6 (2 to 15) | 0 | 6 (3 to 10) |
| ≥ 5 mm | 6 (2 to 14) | 4 (1 to 13) | 9 (3 to 24) | 6 (3 to 10) | 7 (3 to 16) | 7 (3 to 17) | 0 | 6 (3 to 10) |
| Systemic reaction | ||||||||
| Muscle achesc | ||||||||
| None | 73 (61 to 82) | 78 (67 to 86) | 76 (59 to 88) | 76 (69 to 81) | 41 (30 to 53) | 51 (39 to 63) | 70 (54 to 83) | 51 (44 to 59) |
| Mild | 14 (8 to 25) | 16 (9 to 27) | 21 (10 to 37) | 16 (11 to 23) | 30 (20 to 42) | 30 (20 to 42) | 27 (15 to 43) | 29 (23 to 37) |
| Moderate | 13 (7 to 23) | 6 (2 to 15) | 3 (0 to 18) | 8 (5 to 13) | 27 (18 to 39) | 19 (12 to 31) | 3 (0 to 17) | 19 (14 to 26) |
| Severe | 0 | 0 | 0 | 0 | 1 (0 to 10) | 0 | 0 | 1 (0 to 4) |
| Chillsc | ||||||||
| None | 96 (87 to 99) | 90 (80 to 95) | 85 (69 to 94) | 91 (86 to 95) | 69 (57 to 78) | 76 (64 to 85) | 84 (68 to 93) | 75 (68 to 81) |
| Mild | 3 (1 to 11) | 9 (4 to 18) | 9 (3 to 24) | 6 (4 to 11) | 9 (4 to 18) | 15 (8 to 26) | 11 (4 to 26) | 11 (8 to 17) |
| Moderate | 1 (0 to 10) | 1 (0 to 10) | 6 (1 to 21) | 2 (1 to 6) | 20 (12 to 31) | 9 (4 to 19) | 3 (0 to 17) | 12 (8 to 18) |
| Severe | 0 | 0 | 0 | 0 | 3 (1 to 11) | 0 | 3 (0 to 17) | 2 (1 to 5) |
| Malaised | ||||||||
| None | 89 (79 to 94) | 79 (68 to 87) | 88 (72 to 96) | 85 (79 to 90) | 69 (57 to 78) | 72 (60 to 81) | 95 (81 to 99) | 75 (68 to 81) |
| Mild | 7 (3 to 16) | 13 (7 to 24) | 6 (1 to 21) | 9 (6 to 15) | 24 (16 to 36) | 16 (9 to 27) | 5 (1 to 19) | 17 (12 to 24) |
| Moderate | 4 (1 to 13) | 7 (3 to 17) | 6 (1 to 21) | 6 (3 to 10) | 7 (3 to 16) | 12 (6 to 22) | 0 | 7 (4 to 12) |
| Severe | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Headache | ||||||||
| None | 66 (54 to 76) | 51 (40 to 63) | 71 (53 to 83) | 61 (54 to 68) | 43 (32 to 55) | 54 (42 to 65) | 86 (71 to 94) | 56 (49 to 64) |
| Mild | 23 (14 to 34) | 31 (21 to 43) | 18 (8 to 34) | 25 (19 to 32) | 34 (24 to 36) | 28 (19 to 40) | 8 (3 to 22) | 26 (20 to 34) |
| Moderate | 10 (5 to 20) | 16 (9 to 27) | 12 (4 to 28) | 13 (9 to 19) | 23 (14 to 34) | 15 (8 to 26) | 5 (1 to 19) | 16 (11 to 22) |
| Severe | 1 (0 to 10) | 1 (0 to 10) | 0 | 1 (0 to 5) | 0 | 3 (1 to 11) | 0 | 1 (0 to 5) |
| Nausea | ||||||||
| None | 83 (72 to 90) | 90 (80 to 95) | 85 (69 to 94) | 86 (80 to 90) | 73 (61 to 82) | 82 (71 to 90) | 92 (78 to 97) | 80 (74 to 86) |
| Mild | 10 (5 to 20) | 6 (2 to 15) | 9 (3 to 24) | 8 (5 to 13) | 19 (11 to 29) | 15 (8 to 26) | 8 (3 to 22) | 15 (10 to 21) |
| Moderate | 7 (3 to 16) | 4 (1 to 13) | 3 (0 to 18) | 5 (3 to 10) | 9 (4 to 18) | 3 (1 to 11) | 0 | 5 (2 to 9) |
| Severe | 0 | 0 | 3 (0 to 18) | 1 (0 to 4) | 0 | 0 | 0 | 0 |
| Fever: temperature ≥ 38°C | 3 (1 to 11) | 1 (0 to 10) | 3 (0 to 18) | 2 (1 to 6) | 4 (1 to 13) | 3 (1 to 11) | 0 | 3 (1 to 7) |
| Use of analgesic | 30 (20 to 42) | 18 (10 to 29) | 6 (1 to 21) | 20 (15 to 27) | 39 (28 to 50) | 24 (15 to 35) | 5 (1 to 19) | 26 (20 to 33) |
The most frequent solicited local event was pain, reported by 28% and 76% of subjects after either dose of WV or adjuvanted vaccine, respectively (OR 7.71, 95% CI 4.48 to 13.24, p < 0.0001). Three subjects (2%) reported severe local pain after adjuvanted vaccine.
The most common systemic event was myalgia, reported by 24% and 49% of subjects after either dose of WV or adjuvanted vaccine (OR 2.99, 95% CI 1.86 to 4.80, p < 0.0001). Headache on days 1 or 2 was reported by 19% and 30% of subjects after WV or adjuvanted vaccine, respectively (p = 0.03). Analgesic use was reported by 20% and 25% of subjects after WV or adjuvanted vaccine, respectively (p = 0.1). Fever of ≥ 38.0°C was reported by two subjects after WV vaccine and three after adjuvanted vaccine. Severe reactions were reported by three subjects (1.7%; 95% CI 0.4 to 5.0) after WV, and by six (3.4%; 95% CI 1.3 to 7.3) after adjuvanted vaccine. Absenteeism on day 1 was reported by one and nine subjects after WV and adjuvanted vaccine, respectively (p = 0.01).
Unsolicited AEs were reported by 33.1% (26.2% to 40.7%) and 23.6% (17.5% to 30.6%) of subjects after WV or adjuvanted vaccine (Appendix 10, Tables 11 and 12). In total, 13.5% and 36.5% of unsolicited AEs were considered to be vaccine related after WV vaccine and adjuvanted vaccine, respectively. The most frequent unsolicited events were sore throat, cough and symptomatic colds. Two serious AEs occurred during the course of the study: one subject in Leicester was withdrawn following detection of breast malignancy by routine mammography performed during the study; and a second subject in Sheffield was withdrawn following the diagnosis of oesophageal cancer.
Kinetics of the HI antibody response to vaccination in subjects without baseline HI antibody
Seroprotection in subjects with no baseline HI antibody
There was an age–response relationship regarding seroprotection rates (≥ 1 : 40) and GMTs for adjuvanted (p < 0.002) and WV vaccine (p < 0.0025, each visit), with the most robust response in 18- to 44-year-olds. Figure 4 shows the percentage of participants stratified by age with HI titres of ≥ 1 : 40 (seroprotection) at weekly intervals after vaccination.
FIGURE 4.
Percentage of subjects without baseline HI antibody with seroprotection (HI titres of ≥ 1 : 40) at 7-day intervals after vaccination on days 0 and 21 (see Appendix 3 for 95% CIs). (a) Age 18–44 years. (b) Age 45–64 years. (c) Age ≥ 65 years. (d) All age groups combined.
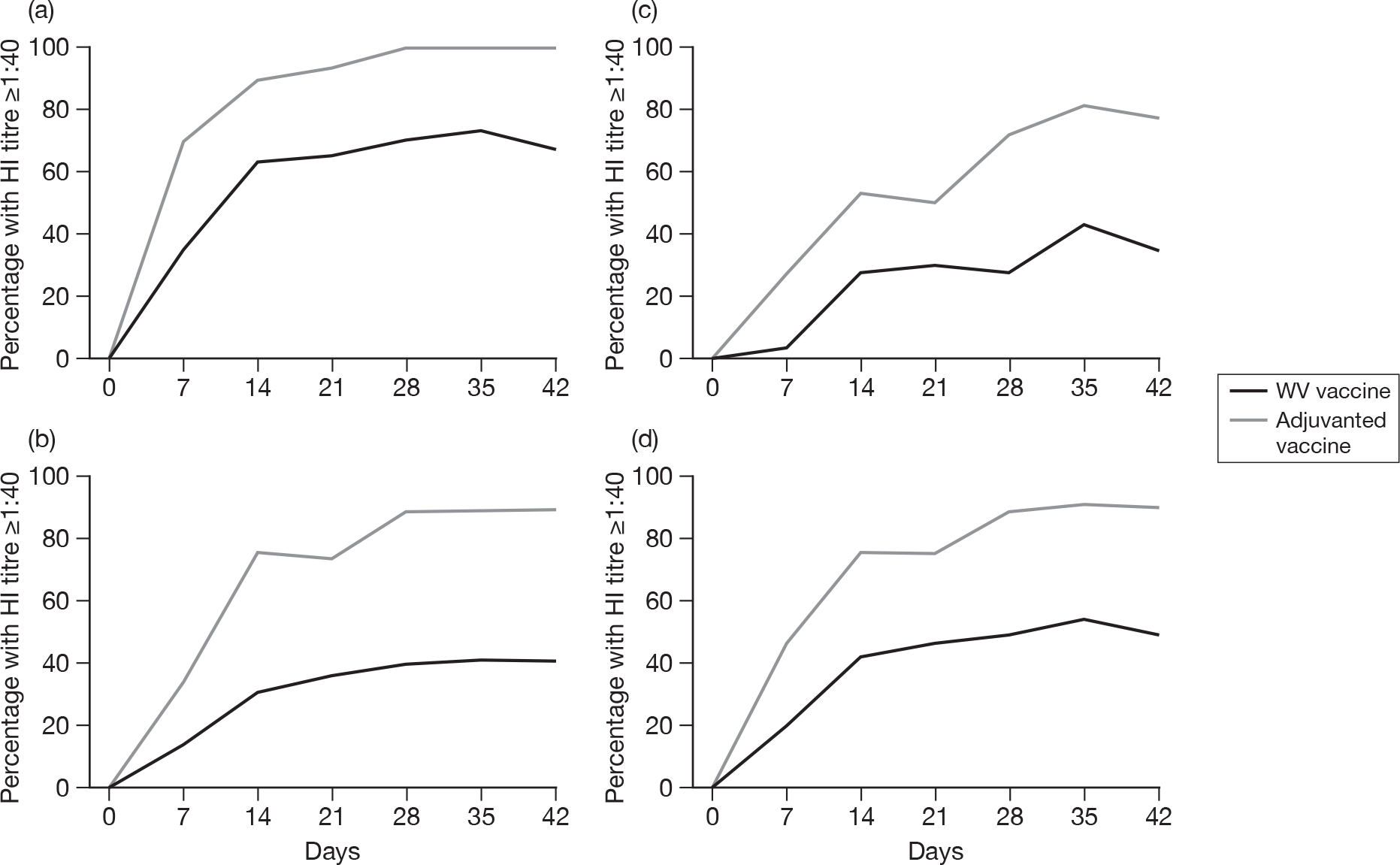
Seroprotection was attained more rapidly with adjuvanted vaccine than WV vaccine, with more subjects in each age group attaining HI titres of ≥ 1 : 40 (p < 0.001 each visit). Seroprotection occurred in 46.6% (95% CI 38.4 to 55.0), 76.4% (95% CI 68.6 to 83.1), and 75.7% (95% CI 68.0 to 82.4) of subjects of all ages on days 7, 14 and 21, respectively, after the first dose of adjuvanted vaccine, and in 19.6% (95% CI 13.5 to 26.9), 42.5% (95% CI 34.3 to 50.9), and 46.3% (95% CI 38.1 to 54.7), respectively, after WV vaccine. On days 28, 35 and 42, seroprotection occurred in 89.4% (95% CI 83.1 to 93.9), 91.6% (95% CI 85.8 to 95.6), and 90.3% (95% CI 84.3 to 94.6) of all subjects, respectively, after the second dose of adjuvanted vaccine, and in 49.3% (95% CI 41.0 to 57.7), 54.5% (95% CI 46.0 to 62.8) and 50.0% (95% CI 41.6 to 58.4), respectively, after WV vaccine.
GMT responses in subjects with no baseline HI antibody
Figure 5 shows the GMT HI titres of participants stratified by age at weekly intervals after vaccination. Among all age groups GMTs and mean-fold titre elevations were higher after adjuvanted than WV vaccine (p < 0.001 each visit). After the first vaccine dose, peak GMTs occurred on day 14 in each age group given adjuvanted vaccine and on day 21 in each age group given WV vaccine.
FIGURE 5.
Haemagglutination inhibition GMTs in subjects without baseline HI antibody before vaccination and 7-day intervals after vaccination on days 0 and 21 (see Appendix 3 for 95% CIs). (a) Age 18–44 years. (b) Age 45–64 years. (c) Age ≥ 65 years. (d) All age groups combined.

Kinetics of the HI antibody response to vaccination in subjects with baseline HI antibody
Seroprotection and GMTs in subjects with baseline HI antibody
Figure 6a shows the percentage of all participants with baseline HI antibody with seroprotection (HI titres of ≥ 1 : 40) before vaccination and at weekly intervals afterwards. There was no difference in prevaccination titres or seroprotection rates in subjects given adjuvanted or WV vaccine. Higher seroprotection rates were observed after adjuvanted than WV vaccine on day 14 (100%, 95% CI 83.9 to 100; 81.8%, 95% CI 59.7 to 94.8, p = 0.04). Peak seroprotection rates were observed after a single dose of adjuvanted vaccine on day 14, and after two doses of WV vaccine on day 28 (90.9%, 95% CI 70.8 to 98.9).
FIGURE 6.
Seroprotection rates (percentage with HI titres ≥ 1 : 40) and HI GMTs before vaccination and at 7-day intervals after vaccination (days 0 and 21) in subjects with baseline HI antibody (see Appendix 4 for 95% CIs). (a) All age groups combined. (b) All age groups combined.
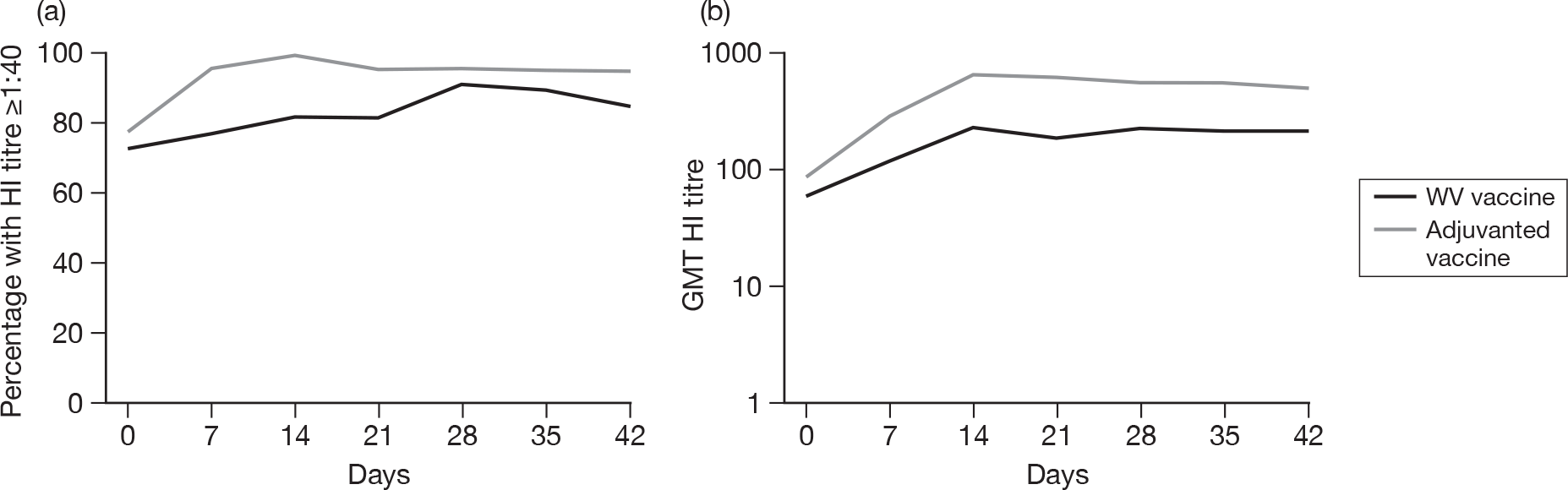
Figure 6b shows the GMT HI titres of all participants with baseline HI antibody before vaccination and at weekly intervals afterwards. There was no difference in prevaccination GMTs in subjects given adjuvanted or WV vaccine. GMTs were higher after adjuvanted than WV vaccine subsequently after vaccination (p ≤ 0.02, each occasion), and peak GMTs occurred 14 days after the first dose of either vaccine.
Kinetics of the HI antibody response to vaccination in subjects with and without baseline HI antibody
Seroprotection in subjects with and without baseline HI antibody
There was an age–response relationship regarding seroprotection rates (≥ 1 : 40) and GMTs for adjuvanted (p = <0.0005) and WV vaccine (p = <0.0005, each visit), with the most robust response occurring in 18- to -44-year-olds. Figure 7 shows the percentage of participants stratified by age with HI titres of ≥ 1 : 40 (seroprotection) at weekly intervals after vaccination.
FIGURE 7.
Percentage of subjects with and without baseline HI antibody with seroprotection (HI titres of ≥ 1 : 40) at 7 day intervals after vaccination on days 0 and 21 (see Appendix 5 for 95% CIs). (a) Age 18–44 years. (b) Age 45–64 years. (c) Age ≥ 65 years. (d) All age groups combined.
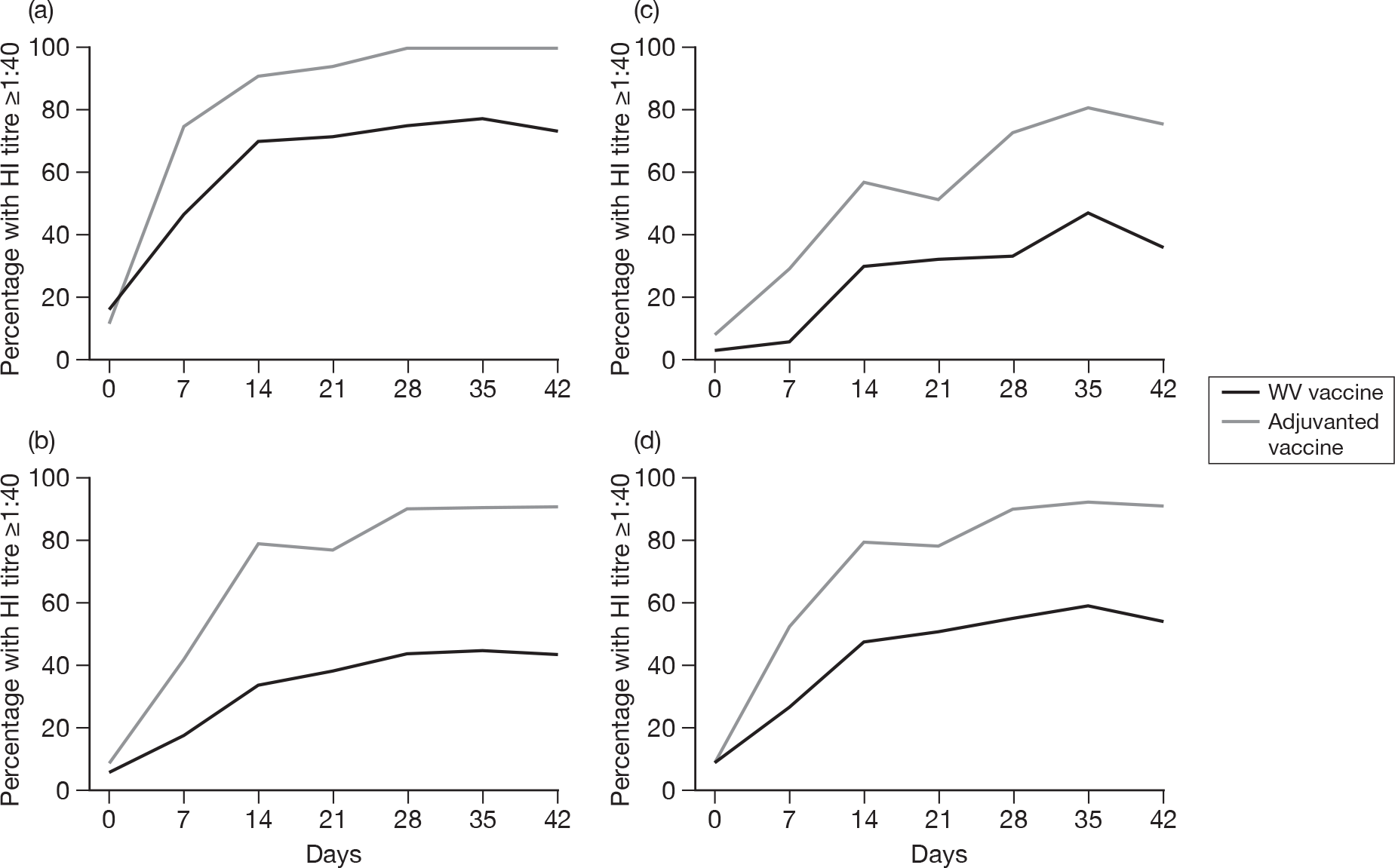
Seroprotection was attained more rapidly with adjuvanted vaccine than WV vaccine, with more subjects in each age group attaining HI titres of ≥ 1 : 40 (18–44 years, p < 0.004, all visits; 45–64 years, p < 0.002, all visits; ≥ 65 years, day 7, p = 0.01; day 14, p = 0.03; day 21, p = 0.2; day 28, p = 0.002; day 35, p = 0.006; day 42, p = 0.002). Seroprotection occurred in 52.9% (95% CI 45.2 to 60.6), 79.4 (95% CI 72.4 to 85.3), and 78.2 (95% CI 71.3 to 84.2) of subjects of all ages on days 7, 14 and 21, respectively, after the first dose of adjuvanted vaccine, and in 27.1% (95% CI 20.5 to 34.4), 47.6% (95% CI 39.9 to 55.5) and 50.9% (95% CI 43.1 to 58.6), respectively, after WV vaccine. On days 28, 35, and 42 seroprotection occurred in 90.2% (95% CI 84.6 to 94.3), 92.1% (95% CI 86.8 to 95.7) and 91.0% (95% CI 85.5 to 94.9) of all subjects, respectively, after the second dose of adjuvanted vaccine, and in 54.8% (95% CI 46.9 to 62.4), 58.8% (95% CI 50.9 to 66.4) and 54.2% (95% CI 46.3 to 62.0), respectively, after WV vaccine. The age-adjusted ORs for adjuvanted compared with WV vaccine, in terms of seroprotection, at 21 and 42 days were 4.42 (95% CI 2.63 to 7.44, p < 0.001) and 11.21 (95% CI 5.80 to 21.64, p < 0.001), respectively.
GMTs responses in subjects with and without baseline HI antibody
Figure 8 shows the GMT HI titres of participants stratified by age at weekly intervals after vaccination. Among all age groups GMTs and mean-fold titre elevations were higher after adjuvanted than WV vaccine (18–44 years, p < 0.0001, all visits; 45–64 years, p = 0.002, all visits; ≥ 65 years, days 28, 35 and 42, p < 0.001). After the first vaccine dose, peak GMTs occurred on day 14 in each age group given adjuvanted vaccine, and on day 14 in the over-65-year-olds, and day 21 in the other age groups given WV vaccine.
FIGURE 8.
Haemagglutination inhibition GMTs in subjects with and without baseline HI antibody before vaccination and 7-day intervals after vaccination on days 0 and 21 (see Appendix 5 for 95% CIs). (a) Age 18–44 years. (b) Age 45–64 years. (c) Age ≥ 65 years. (d) All age groups combined.
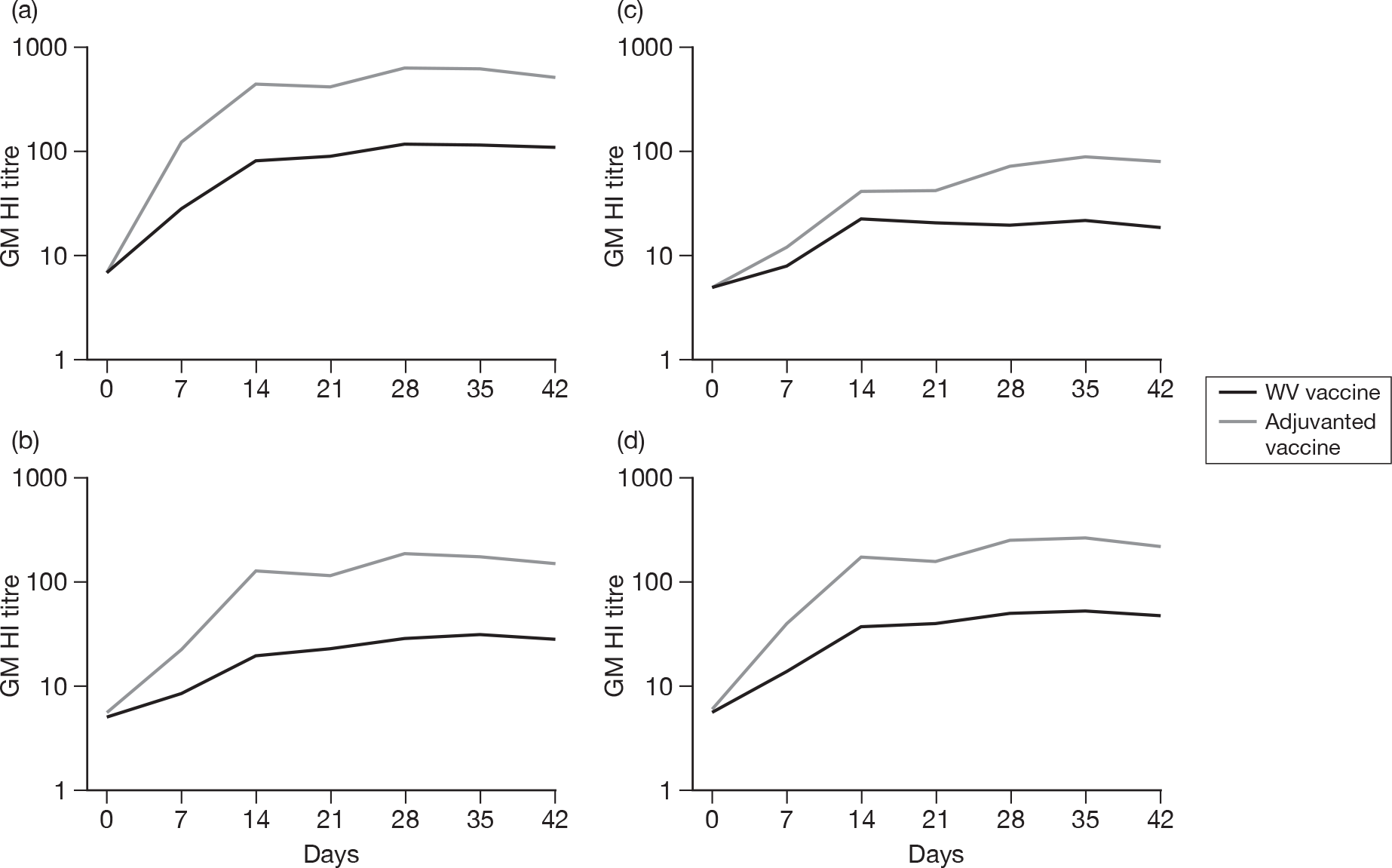
Kinetics of the MN antibody response to vaccination in subjects without baseline MN antibody
Attainment of MN antibody titres of ≥1 : 40
There was an age–response relationship regarding the development of postvaccination titres of > 1 : 40 and GMTs on days 7, 14 and 21 after the first dose of adjuvanted (all p ≤ 0.0008) and WV vaccine (all p < 0.0001), with the most robust response in 18- to 44-year-olds.
Figure 9 shows the percentage of participants without baseline MN antibody, stratified by age, who develop MN titres of ≥ 1 : 40 at 7-day intervals after vaccination. MN antibody titres of ≥ 1 : 40 generally occurred sooner with adjuvanted vaccine than WV vaccine. Among subjects aged 18–44 years, 45–64 years, and all age groups combined, more subjects attained a titre of 1 : 40 or more after a single dose of adjuvanted vaccine than WV vaccine (18–44 years: 98.0% and 80.9%, respectively, p = 0.008; 45–64 years: 85.7% and 44.2%, respectively, p < 0.0001; all subjects: 84.2% and 56.8%, respectively, p < 0.0001); the trend in the older age group was non-significant (52.2% and 31.6%, respectively, p = 0.2).
FIGURE 9.
Percentage of subjects without baseline MN antibody with MN titres of ≥ 1 : 40 at 7-day intervals after vaccination on days 0 and 21 (see Appendix 6 for 95% CIs). (a) Age 18–44 years. (b) Age 45–64 years. (c) Age ≥ 65 years. (d) All age groups combined.
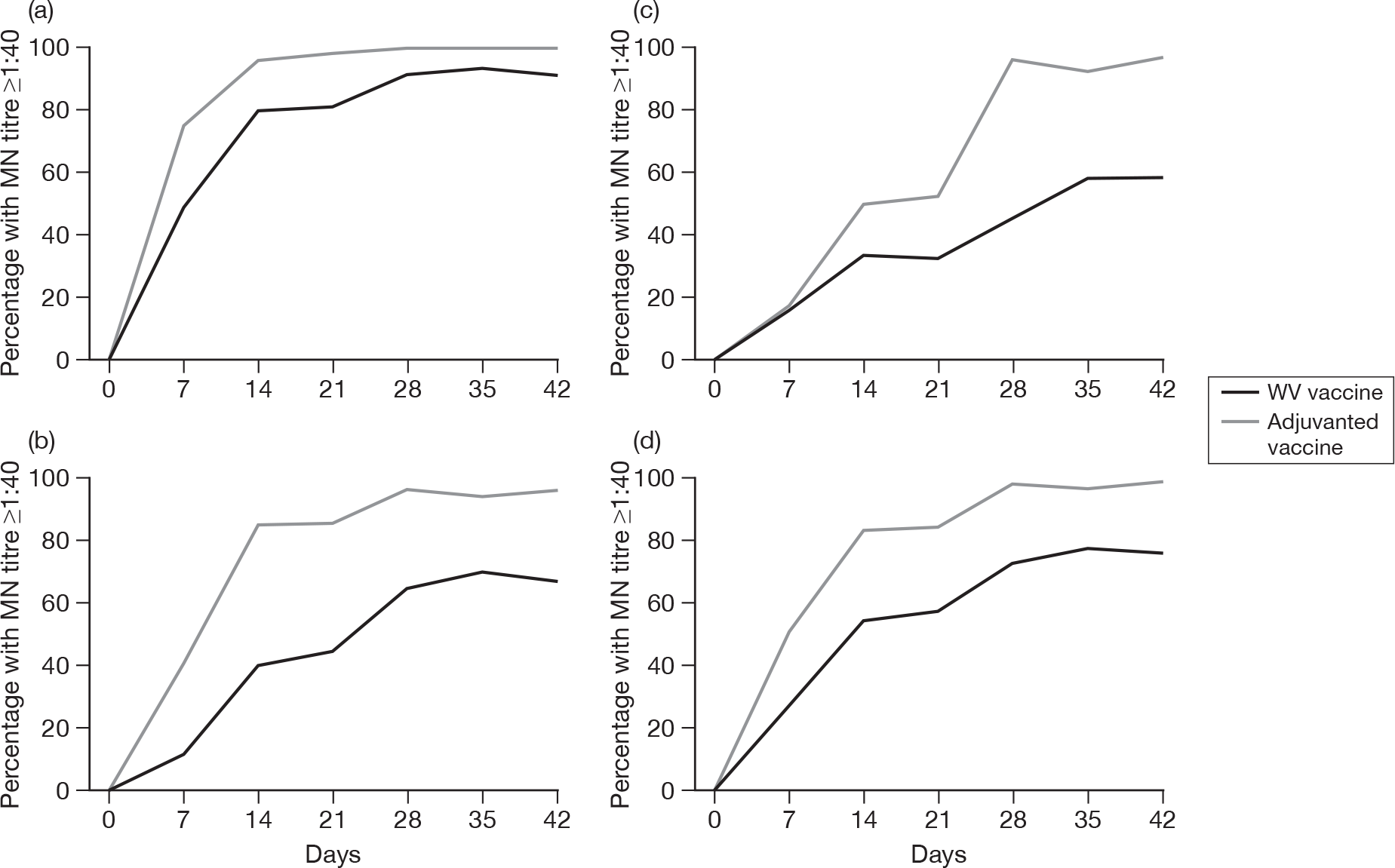
Microneutralisation antibody titres of ≥ 1 : 40 occurred in 50.8% (95% CI 41.6 to 60.1), 82.9% (95% CI 74.8 to 89.2) and 84.2% (95% CI 76.4 to 90.2) of subjects of all ages on days 7, 14 and 21, respectively, after the first dose of adjuvanted vaccine, and in 27.4% (95% CI 19.5 to 36.4), 54.3% (95% CI 44.8 to 63.6) and 56.8% (95% CI 47.3 to 65.9), respectively, after WV vaccine. Titres of ≥ 1 : 40 occurred in 97.3% (95% CI 92.4 to 99.4), 95.8% (95% CI 90.4 to 98.6) and 97.5% (95% CI 92.8 to 99.5) of subjects of all ages on days 28, 35 and 42, respectively, after the second dose of adjuvanted vaccine, and in 72.2% (95% CI 63.0 to 80.1), 77.2% (95% CI 68.4 to 84.5) and 75.0% (95% CI 66.1 to 82.6), respectively, after WV vaccine.
GMTs responses in subjects with no baseline MN antibody
Figure 10 shows the GMT MN titres of participants stratified by age at weekly intervals after vaccination. GMTs were higher after adjuvanted than WV vaccine (p < 0.0001 each visit). Among subjects aged 18–44 years, 45–64 years and all age groups combined, the GMTs were higher at each postvaccination visit after adjuvanted vaccine than after WV vaccine (18–44 years: day 7, p = 0.0007, day 14, p = 0.002, day 21, p = 0.0002, day 28, p = 0.0003, day 35, p = 0.0006, day 42, p < 0.0001; 45–64 years: day 7, p = 0.0026, days 14, 21, 28, 35 and 42, all p < 0.0001; all subjects: all postvaccination visits, p < 0.0001); in the older age group, the trend was non-significant on day 7 (p = 0.4712) and day 14 (p = 0.0869), but significant on day 21 (p = 0.03), day 28 (p = 0.0004), day 35 (p = 0.0002) and day 42 (p = 0.003).
FIGURE 10.
Microneutralisation GMTs in subjects without baseline MN antibody before vaccination and 7-day intervals after vaccination on days 0 and 21 (see Appendix 6 for 95% CIs). (a) Age 18–44 years. (b) Age 45–64 years. (c) Age ≥ 65 years. (d) All age groups combined.
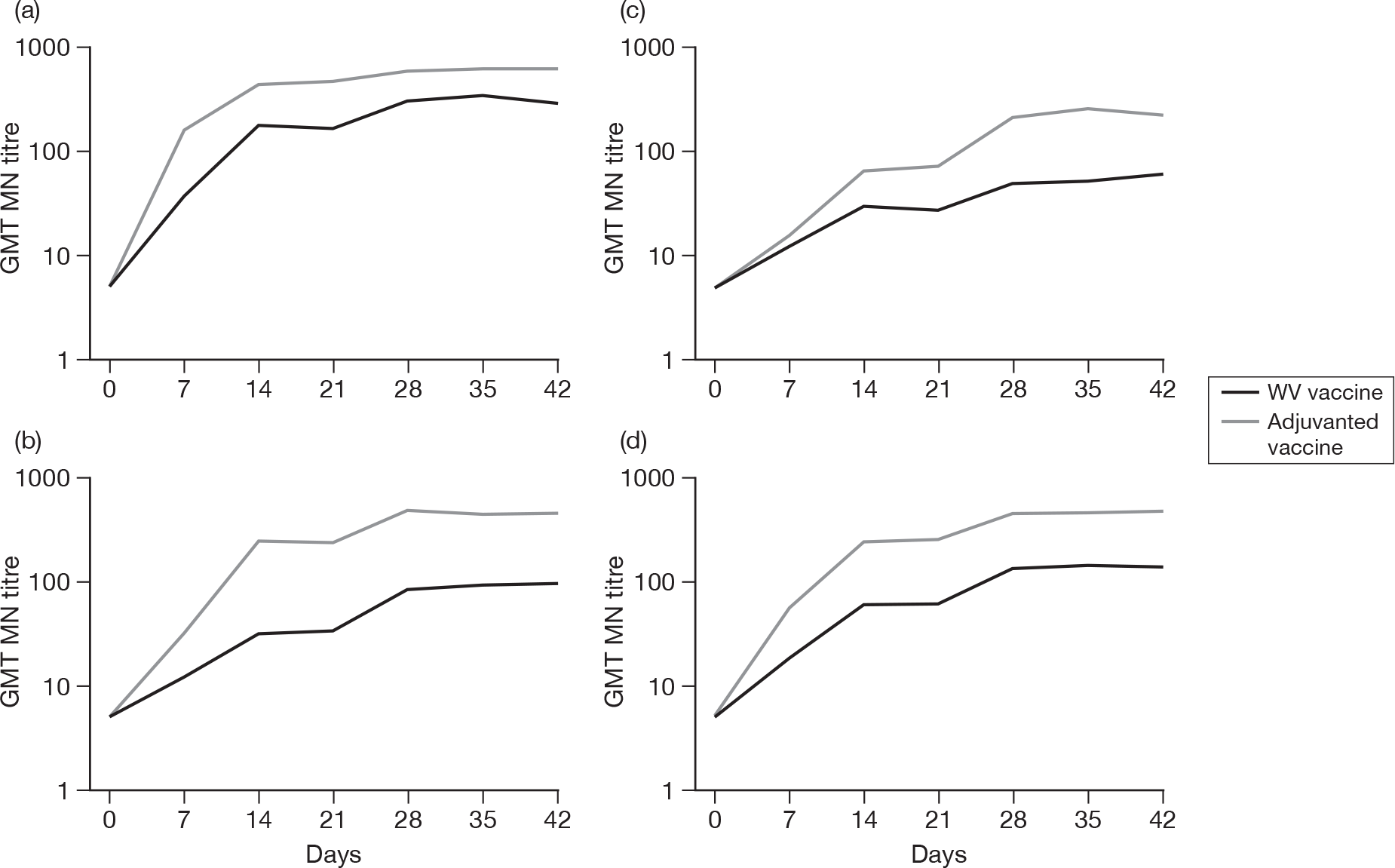
Kinetics of the MN antibody response to vaccination in subjects with baseline MN antibody
Figure 11a shows the percentage of all subjects with baseline MN antibody titres of ≥ 1 : 40 before vaccination and at weekly intervals afterwards, and Figure 11b shows the corresponding GMTs. Baseline GMTs and the percentage of subjects with MN titres of ≥ 1 : 40 were similar in the groups given adjuvanted vaccine and WV vaccine [all age groups: GMTs, 1 : 83 (95% CI 1 : 54 to 1 : 127) and 1 : 67 (95% CI 1 : 44.5 to 1 : 102.2), respectively (p = 0.5); percentage of subjects with MN titres of 1 : 40 or more, 50.0% (95% CI 35.5 to 64.5) and 62.3% (95% CI 47.9 to 75.2), respectively (p = 0.2)].
FIGURE 11.
Percentage of subjects with MN titres ≥ 1 : 40 and MN GMTs before vaccination and at 7-day intervals after vaccination (days 0 and 21) in subjects with baseline MN antibody (see Appendix 7 for 95% CIs). (a) All age groups combined. (b) All age groups combined.
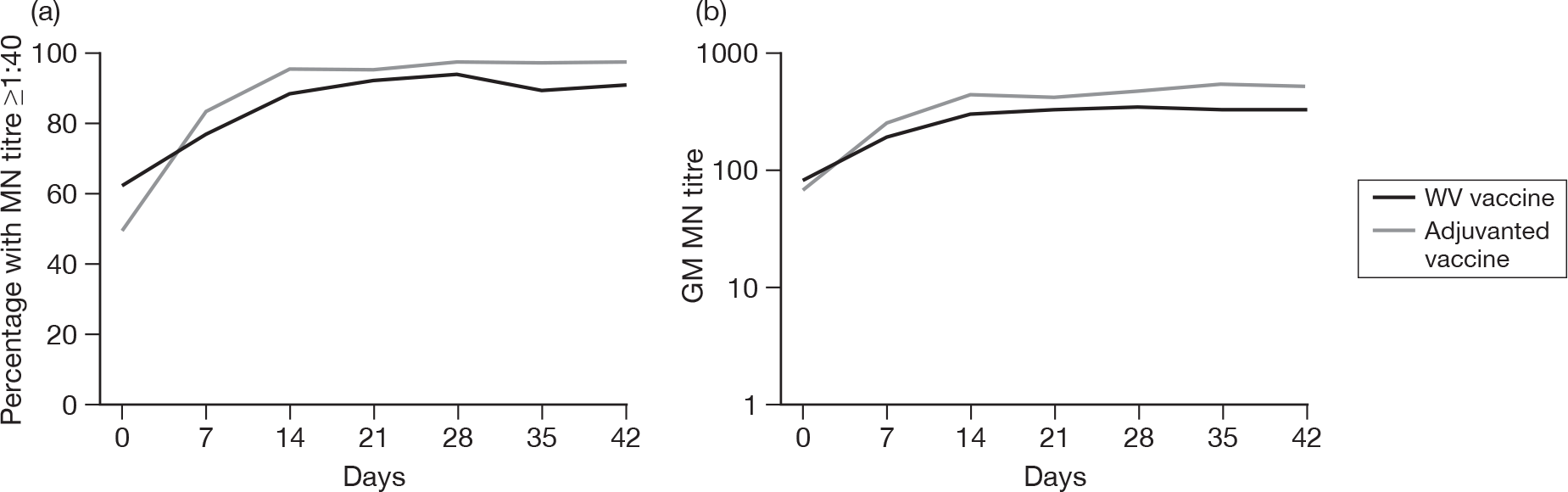
Among all subjects with baseline MN antibody, MN titres of ≥ 1 : 40 occurred at similar rates in both vaccine groups on each occasion. Peak rates for the occurrence of MN titres of ≥ 1 : 40 occurred in both vaccine groups on day 21 after the first dose of adjuvanted vaccine (96.0%, 95% CI 86.3 to 99.5) and WV vaccine (92.5%, 95% CI 81.8 to 97.9), and on day 28 after the second dose of adjuvanted vaccine (98%, 95% CI 89.4 to 99.9) and WV vaccine (94.2%, 95% CI 84.1 to 98.8). The GMTs peaked on day 14 after the first dose of adjuvanted vaccine and on day 21 after the first dose of WV vaccine.
Kinetics of the MN antibody response to vaccination in subjects with and without baseline MN antibody
Attainment of MN antibody titres of ≥ 1 : 40
In all subjects, regardless of baseline antibody status on MN, GMTs at all visits decreased with increasing age, for adjuvanted vaccine (p < 0.001, all visits), and WV vaccine (p ≤ 0.001, all visits). Adjustment of the effect of age group did not alter the effect of vaccine type (p < 0.001, all visits). In terms of seroprotection, adjuvanted vaccine produced greater seroprotection rates compared with WV vaccine at all visits (p < 0.001), while there was a decreasing effect of age for both AS03A-adjuvanted and WV vaccine at all visits (p < 0.0001) except for adjuvanted at days 28 (p = 0.08), 35 (p = 0.03) and 42 (p = 0.08).
Figure 12 shows the percentage of participants with and without baseline MN antibody, stratified by age, who develop MN titres of ≥ 1 : 40 at weekly intervals after vaccination. Among subjects aged 18–44 years, 45–64 years, and all age groups combined, more subjects attained a titre of 1 : 40 or more after a single dose of adjuvanted vaccine than WV vaccine (18–44 years, 98.5% and 87.1%, respectively, p = 0.01; 45–64 years, 89.4% and 56.7%, respectively, p < 0.0001; all subjects, 87.6% and 67.8% respectively, p < 0.0001); the trend in the older age group was non-significant (64.9% and 50.0%, respectively, p = 0.2).
FIGURE 12.
Percentage of subjects with and without baseline MN antibody with MN titres of > 1 : 40 at 7-day intervals after vaccination on days 0 and 21 (see Appendix 8 for 95% CIs). (a) Age 18–44 years. (b) Age 45–64 years. (c) Age ≥ 65 years. (d) All age groups combined.
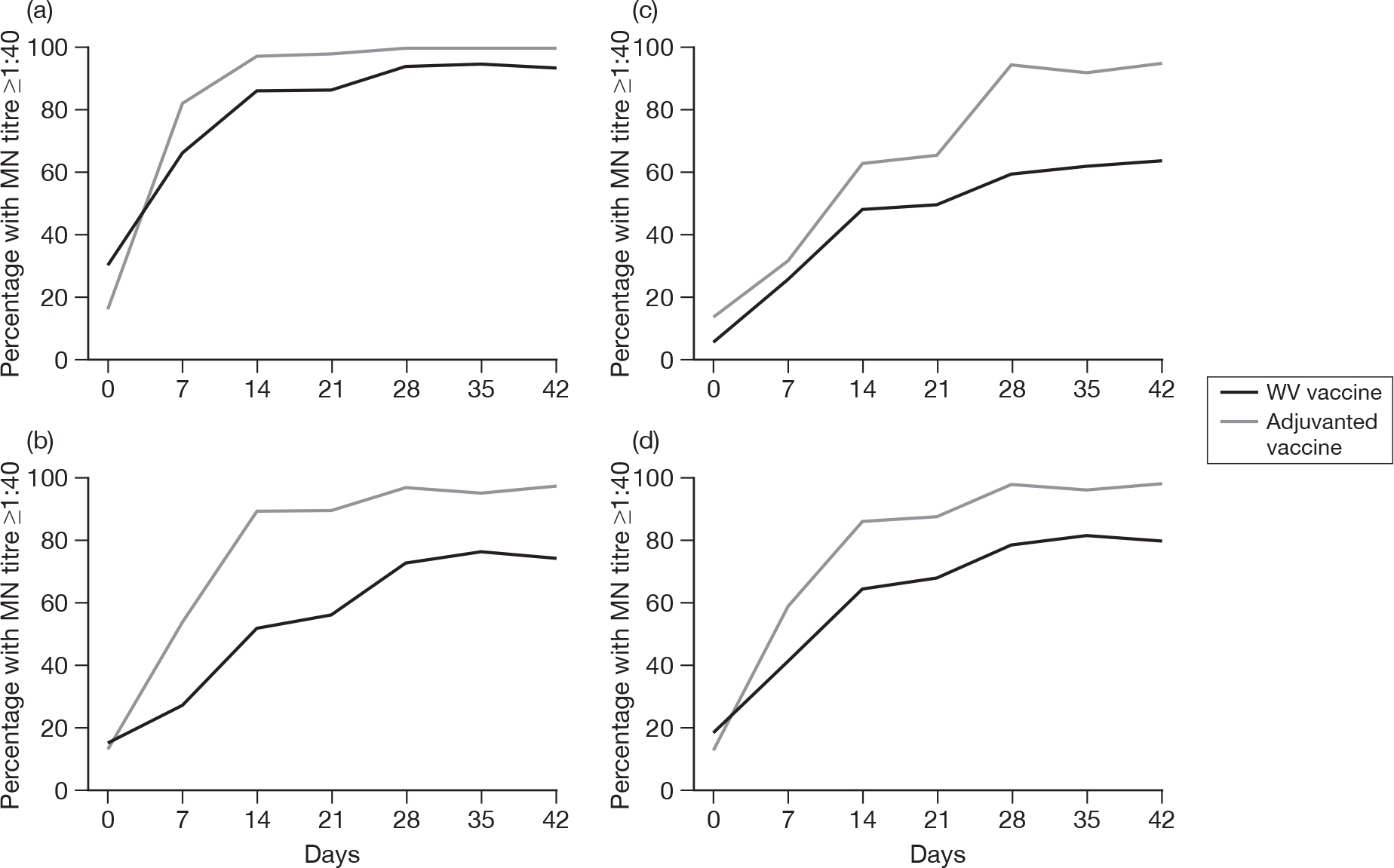
Microneutralisation antibody titres of ≥ 1 : 40 occurred in 60.6% (95% CI 52.8 to 68.0), 86.7% (95% CI 80.5 to 91.5) and 87.6% (95% CI 81.7 to 92.2) of subjects of all ages on days 7, 14 and 21, respectively, after the first dose of adjuvanted vaccine, and in 42.9% (95% CI 35.4 to 50.7), 64.9% (95% CI 57.2 to 72.1) and 67.8% (95% CI 60.3 to 74.8), respectively, after WV vaccine. Titres of ≥ 1 : 40 occurred in 97.5% (95% CI 93.8 to 99.3), 96.4% (95% CI 92.3 to 98.7) and 97.6% (95% CI 94.0 to 99.3) of subjects of all ages on days 28, 35 and 42, respectively, after the second dose of adjuvanted vaccine and in 79.0% (95% CI 72.1 to 84.9), 81.2% (95% CI 74.4 to 86.9) and 80.1% (95% CI 73.2 to 85.9), respectively, after WV vaccine.
GMTs responses in subjects with and without baseline MN antibody
Figure 13 shows the GMT MN titres of participants stratified by age at weekly intervals after vaccination. Adjuvanted vaccine GMTs were greater than WV vaccine GMTs at all visits (across all ages) after vaccination (p < 0.0003). Adjuvanted vaccine GMTs were greater than WV vaccine at all visits in the 18- to 44-year and 45- to 64-year age groups (p < 0.01 and p < 0.004, respectively) and at days 28, 35 and 42 (all p < 0.001) for the ≥ 65-year age group.
FIGURE 13.
Microneutralisation GMTs in subjects with and without baseline MN antibody before vaccination and 7-day intervals after vaccination on days 0 and 21 (see Appendix 8 for 95% CIs). (a) Age 18–44 years. (b) Age 45–64 years. (c) Age ≥ 65 years. (d) All age groups combined.
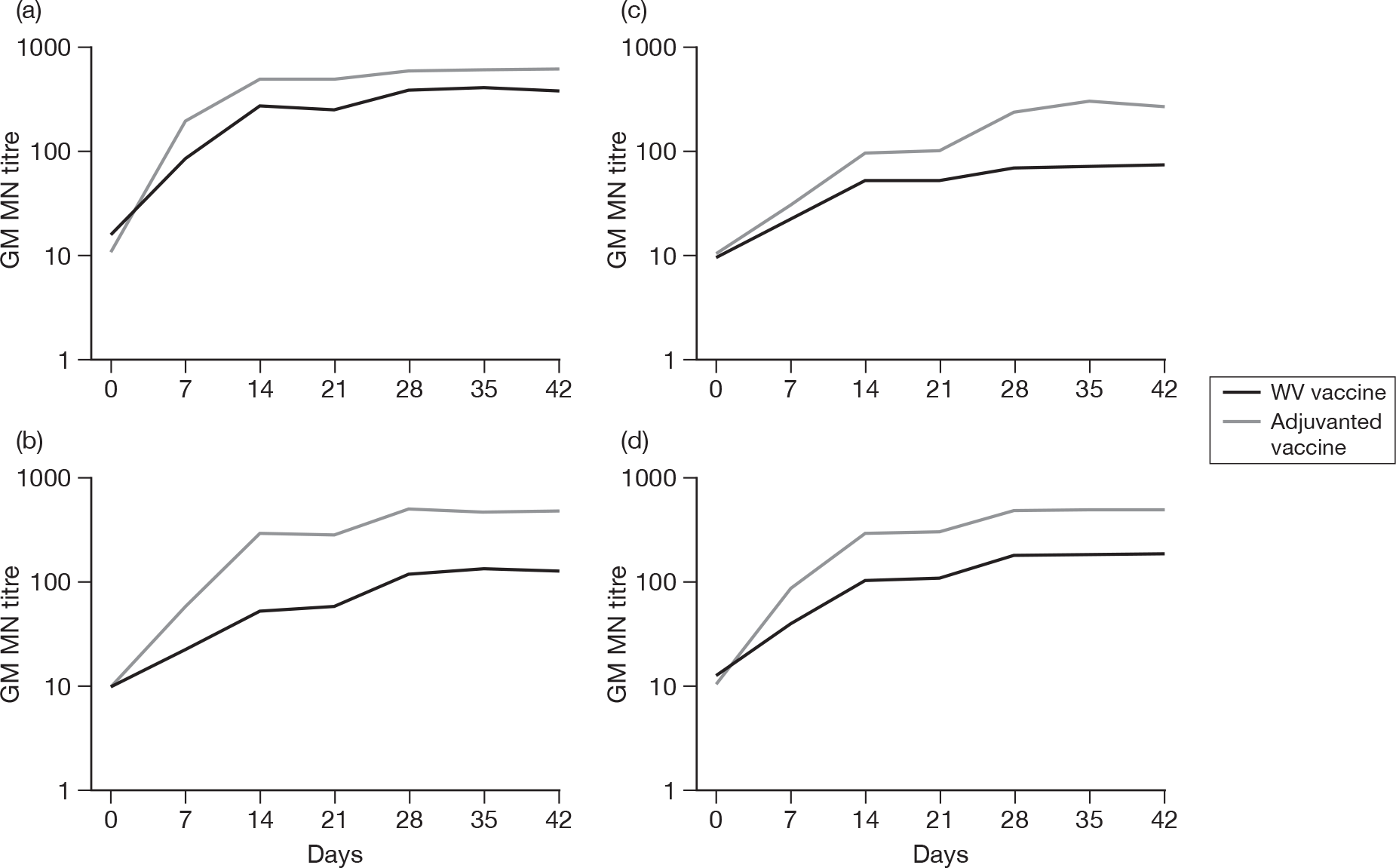
Persistence of antibody
HI antibody titres 6 months after vaccination
Only the HI assay was performed at 6 months and 336 (97%) of the 347 vaccinated at baseline had outcome data at this time point: 165 out of 175 (95%) in the adjuvanted vaccine arm and 170 out of 172 (98%) in the WV vaccine arm.
Table 8 shows the presence of HI antibody 6 months after the first vaccination in subjects without baseline antibody, those with baseline HI antibody, and all subjects with and without baseline antibody. The 6-month results are shown as the percentage of participants with seroprotection (HI titres of ≥ 1 : 40) HI GMTs and the factor increase in GMT compared with baseline.
| Immunogenicity end point | WV vaccine | AS03A-adjuvanted split-virion vaccine | ||||||
|---|---|---|---|---|---|---|---|---|
| 18–44 years | 45–64 years | ≥ 65 years | All subjects | 18–44 years | 45–64 years | ≥ 65 years | All subjects | |
| No baseline HI antibody | ||||||||
| No. of subjects | 58 | 61 | 30 | 149 | 55 | 57 | 34 | 146 |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 74.1 (61.0 to 84.7) | 50. 8 (37.7 to 63.9) | 30. 0 (14.7 to 49.4) | 55.7 (47.4 to 63.8) | 98.2 (90.3 to 100) | 79.0 (66.1 to 88.6) | 55.9 (37.9 to 72.8) | 80.8 (73.5 to 86.9) |
| GMT value (95% CI) | 85.3 (58.4 to 124.6) | 37.1 (25.2 to 54.5) | 18.4 (12.3 to 27.5) | 44.5 (34.9 to 56.9) | 202.8 (158.5 to 259.5) | 86.2 (64.9 to 114.6) | 49.1 (31.3 to 77.0) | 104.4 (85.7 to 127.1) |
| Factor increase in GMT value (95% CI) | 20.9 (14.3 to 30.5) | 9.2 (6.3 to 13.5) | 4.4 (3.0 to 6.6) | 10.9 (8.6 to 14.0) | 49.4 (38.8 to 63.0) | 21.6 (16.2 to 28.6) | 11.9 (7.7 to 18.4) | 25.7 (21.1 to 31.2) |
| Baseline HI antibody | ||||||||
| No. of subjects | 11 | 6 | 4 | 21 | 8 | 9 | 3 | 20 |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 100 (71.5 to 100) | 100 (54.1 to 100) | 25.0 (0.6 to 80.6) | 85.7 (63.7 to 97.0) | 100 (63.1 to 100) | 100 (66.4 to 100) | 66.7 (9.4 to 99.2) | 95.0 (75.1 to 99.9) |
| GMT value (95% CI) | 272.7 (171.5 to 433.4) | 203.2 (75.2 to 548.9) | 45.3 (6.7 to 305.7) | 178.1 (109.1 to 290.7) | 449.6 (308.3 to 655.7) | 287.4 (188.6 to 437.9) | 90.5 (6.8 to 1197.8) | 289.0 (200.5 to 416.6) |
| Factor increase in GMT value (95% CI) | 3.5 (2.1 to 6.0) | 3.0 (1.4 to 6.5) | 2.0 (0.8 to 4.9) | 3.0 (2.2 to 4.2) | 3.0 (1.0 to 9.2) | 5.0 (2.4 to 10.6) | 1.3 (0.3 to 4.7) | 3.3 (1.9 to 5.7) |
| With and without baseline HI antibody | ||||||||
| No. of subjects | 69 | 67 | 34 | 170 | 63 | 66 | 37 | 166 |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 78.3 (66.7 to 87.3) | 55.2 (42.6 to 67.4) | 29.4 (15.1 to 47.5) | 59.4 (51.6 to 66.9) | 98.4 (91.5 to 100) | 81.8 (70.4 to 90.2) | 56.8 (39.5 to 72.9) | 82.5 (75.9 to 88.0) |
| GMT value (95% CI) | 102.6 (73.1 to 144.1) | 43.2 (29.7 to 62.8) | 20.4 (13.8 to 30.2) | 52.8 (41.9 to 66.6) | 224.3 (178.6 to 281.8) | 101.6 (77.6 to 133.0) | 51.6 (33.8 to 78.8) | 118.0 (98.1 to 141.9) |
| Factor increase in GMT value (95% CI) | 15.8 (11.0 to 22.6) | 8.3 (5.8 to 11.9) | 4.0 (2.8 to 5.8) | 9.3 (7.5 to 11.7) | 34.6 (24.7 to 48.5) | 17.7 (13.3 to 23.5) | 9.9 (6.3 to 15.6) | 20.0 (16.3 to 24.7) |
Subjects without haemagglutination inhibition antibodies at baseline
In subjects without antibodies at baseline, seroprotection was significantly increased at 6 months on adjuvanted vaccine (80.8%, 95% CI 73.5% to 86.9%) compared with WV vaccine (55.7%, 95% CI 47.4% to 63.8%) (OR 3.35, 95% CI 1.98 to 5.65, p < 0.001). There was a statistically significant decreasing effect of seroprotection with age group for both adjuvanted vaccine (p < 0.001) and WV vaccine (p < 0.001). However, after adjusting for the effect of age adjuvanted vaccine conferred greater seroprotection than WV vaccine (OR 4.32, 95% CI 2.42 to 7.74, p < 0.001) and there was no evidence of an interaction between the effect of vaccine and age group (p = 0.2). In addition, stepwise logistic regression identified previous seasonal influenza vaccine in the 2008–9 season (p < 0.001) and white ethnic origin (p = 0.03) as having a significant effect on seroprotection. However, after adjusting for both of these factors, in addition to age group, adjuvanted vaccine still conferred greater seroprotection than WV vaccine (OR 4.21, 95% CI 2.30 to 7.70, p < 0.001) and there was no evidence of an interaction between the effect of vaccine and either factor (p = 0.5, previous vaccine; p = 0.9, ethnicity).
Geometric mean titres were significantly higher in the adjuvanted vaccine arm (1 : 104) than in the WV vaccine arm (1 : 45) (p < 0.0001). As with seroprotection, there was a statistically significant decreasing effect on GMTs with age group for both adjuvanted vaccine (p < 0.001) and WV vaccine (p < 0.001). However, after adjusting for the effect of age, adjuvanted vaccine had higher GMTs than WV vaccine (p < 0.001) and there was no evidence of an interaction between the effect of vaccine and age group (p = 0.9). In addition, stepwise multiple linear regression identified previous seasonal influenza vaccine in the 2008–9 season (p = 0.01) and centre (p = 0.04) as having a significant effect on GMT at 6 months. However, after adjusting for both of these factors, in addition to age group, adjuvanted vaccine still had a significantly greater GMT than WV vaccine (p < 0.001), and there was no evidence of an interaction between the effect of vaccine and either factor (p = 0.2, previous vaccine; p = 0.4, centre).
Subjects with haemagglutination inhibition antibodies at baseline
In subjects with antibodies at baseline, there was not a statistically significant difference in seroprotection rates between adjuvanted vaccine (95.0%, 95% CI 75.1% to 99.9%) compared with WV vaccine (85.7%, 95% CI 63.7 to 97.0) (OR 3.17, 95% CI 0.30 to 33.31, p = 0.3). There was a decreasing effect of seroprotection with age group for both adjuvanted vaccine (p = 0.07) and WV vaccine (p = 0.002), although it was statistically significant for only the latter, and due to small numbers of observations a logistic regression model could not be applied.
Geometric mean titres were higher in the adjuvanted vaccine arm (1 : 289) compared with the WV vaccine arm (1 : 178), although not significantly so (p = 0.1). There was a statistically significant decreasing effect on GMTs with age group for both adjuvanted vaccine (p = 0.002) and WV vaccine (p = 0.005). However, after adjusting for the effect of age, adjuvanted vaccine still had higher GMTs than WV vaccine (p = 0.04) and there was no evidence of an interaction between the effect of vaccine and age group (p = 0.8). Stepwise multiple linear regression did not identify any other baseline factors other than age group that significantly were associated with GMT.
All subjects (i.e. with and without haemagglutination inhibition antibodies at baseline)
In all subjects, ignoring baseline antibody status, seroprotection was significantly increased at 6 months on adjuvanted vaccine (82.5%, 95% CI 75.9 to 88.0) compared with WV vaccine (59.4%, 95% CI 51.6 to 66.9) (OR 3.23, 95% CI 1.95 to 5.34, p < 0.001). There was a statistically significant decreasing effect of seroprotection with age group for both adjuvanted vaccine (p < 0.001) and WV vaccine (p < 0.001). However, after adjusting for the effect of age, adjuvanted vaccine conferred greater seroprotection than WV vaccine (OR 4.29, 95% CI 2.43 to 7.56, p < 0.001) and there was no evidence of an interaction between the effect of vaccine and age group (p = 0.2). In addition, as with those subjects who did not have antibodies at baseline, stepwise logistic regression identified previous seasonal influenza vaccine in the 2008–9 season (p = 0.002) and white ethnic origin (p = 0.04) as having a significant effect on seroprotection. However, after adjusting for both of these factors, in addition to age group, adjuvanted vaccine still conferred greater seroprotection than WV vaccine (OR 4.35, 95% CI 2.43 to 7.78, p < 0.001), and there was no evidence of an interaction between the effect of vaccine and either factor (p = 0.3, previous vaccine; p = 0.9, ethnicity).
Geometric mean titres were significantly higher in the adjuvanted vaccine arm (1 : 118) than in the WV vaccine arm (1 : 53) (p < 0.0001). As with seroprotection, there was a statistically significant decreasing effect on GMTs with age group for both adjuvanted vaccine (p < 0.001) and WV vaccine (p < 0.001). However, after adjusting for the effect of age, adjuvanted vaccine gave higher GMTs than WV vaccine (p < 0.001) and there was no evidence of an interaction between the effect of vaccine and age group (p = 0.9). Stepwise multiple linear regression did not identify any other baseline factors other than age group that significantly were associated with GMT.
Breadth of the antibody response
No antigenic variants emerged during the course of the study and the effect of vaccine type on cross-protection against a new variant was not studied.
Chapter 4 Discussion
Our study compares two licensed pandemic H1N1 vaccines and finds significant differences in tolerability and immunogenicity. As comparison of available vaccine options is limited by the variability and poor standardisation of influenza serological assays, our results highlight the importance of comparing formulations within a single study to identify optimal regimens, and have implications for control of both pandemic and seasonal influenza.
The first wave of H1N1 pandemic influenza had already passed when the study began and the second was under way. Through the study exclusion criteria we endeavoured to avoid subjects who were infected prior to immunisation. Baseline HI and MN antibody to the pandemic strain was present in 14% and 31% of subjects, respectively, suggesting that most participants were immunologically naive. The highest prevalence of baseline HI and MN antibodies and the highest geometric mean HI and MN antibody titres were found in the younger participants, which is in keeping with higher illness rates in young adults during the first pandemic wave. We assessed vaccine immunogenicity separately in groups with and without baseline antibodies, and in all subjects regardless of baseline antibody, to examine vaccine tolerability and immunogenicity in each population.
The randomisation groups were well matched at baseline and there were no significant differences in baseline GMTs between WV and adjuvanted vaccine groups for any age group, either by HI or MN assay. While two trial centres failed to recruit the target number of elderly subjects (≥ 65 years), compliance with the study protocol was excellent. Overall, 98% of subjects were given both doses of vaccine and sera were obtained from 94.5%–98.3% of subjects on days 7 to 42. Outcome data were available at 6 months for 95% of subjects in the adjuvanted vaccine arm and 98% in the WV vaccine arm. In general, the findings by HI and MN at all time points were comparable, with similar age-related trends occurring with both assays, indicating that they are robust despite the shortfall in elderly participants. Overall, adjuvanted vaccine was more immunogenic than WV vaccine by HI and MN. However, the adjuvanted vaccine was associated with greater reactogenicity than the WV vaccine, including more frequent absenteeism. Nonetheless, pharmacovigilance has not identified significant safety concerns associated with any 2009 H1N1 vaccine formulations. 82,83
While seasonal influenza vaccines are required to satisfy only one of three CHMP criteria, the CHMP guidelines69,70 indicate that pandemic influenza vaccines should at least be able to elicit sufficient immunological responses to meet all three of the current standards set for seasonal vaccines in adults of working age and the elderly. CHMP set lower targets for subjects aged > 60 years than those aged between 18 and 60 years. In this study we used the CHMP criteria for 18- to 60-year-olds to evaluate adjuvanted and WV pandemic vaccines in subjects aged 18–44 and 45–64 years, and the CHMP criteria for over 60-year-olds for subjects aged ≥ 65 years.
Antibody responses to influenza HA are more robust in primed than unprimed populations. Given increased prevalence of crossreactive antibodies against the 2009 pandemic strain in people born before 1950,84,85 and the relative sparing of adults older than 60 years of age from 2009 H1N1 pandemic influenza,39,86,87 we anticipated that some over-65-year-olds in our study would behave as a primed population with a rapid and high-titred antibody response to a single dose of vaccine. Accordingly, the profound age-related decline in antibody responses throughout the study (which was manifest by lower MN and HI GMTs, seroprotection rates and seroconversions, and occurred in both arms across the three age groups, with the lowest antibody responses in the over-65-year-olds) was greater than expected.
The benefit of oil-in-water emulsion adjuvants is best seen when subjects are considered immunologically naive, such as with seasonal vaccine in infants88 or with avian strains. 54,65,89 In our study, the greatest vaccine response was seen among 18- to -44-year-olds, who have lower levels of crossreacting antibodies to 2009 H1N1 virus than older subjects and have suffered the highest attack rates after children. The immune-stimulatory benefit of AS03A-adjuvanted vaccine declined with age, and in those ≥ 65 years, a single dose of adjuvanted vaccine failed to meet the three CHMP criteria. The performance of AS03A-adjuvanted H1N1 2009 vaccine in younger adults is consistent with the benefit of MF59-adjuvanted seasonal vaccine over non-adjuvanted vaccine observed in young children, compared with a modest improvement in primed adults. 88–90
As assessed by attainment of CHMP criteria 21 days after vaccination, a single dose of adjuvanted split-virion vaccine was more immunogenic than WV vaccine. Among subjects with and without baseline HI antibody, one dose of adjuvanted vaccine met all three criteria in 18- to 44-year-olds, 45- to 64-year-olds and all age groups combined, and two of three criteria in the over-65-year-olds. WV vaccine met all three CHMP criteria in 18- to 44-year-olds, two criteria in all age groups combined, and one in 45- to 64-year-olds and over-65-year-olds. Twenty-one days after the second dose, the adjuvanted vaccine met all three criteria in all three age groups and all age groups combined. WV vaccine met all three CHMP criteria in 18- to 44-year-olds and two criteria in the older age groups and all age groups combined.
The principal difference between the two vaccines was in the attainment of ‘seroprotective’ HI titres of 1 : 40 or more. Regardless of baseline antibodies, HI titres of 1 : 40 or more were seen on day 21 after one dose of vaccine in 23% more 18- to 44-year-olds (94.0% vs 71.4%, respectively), 38.5% more 45- to -64-year-olds (77.3% vs 38.8%, respectively), 19% more over-65-year-olds (51.4% vs 32.4%) and 27% more people of all ages (78.2% vs 50.9%) after adjuvanted vaccine than WV vaccine. Similarly, 21 days after the second vaccination, HI titres of 1 : 40 or more were seen in 27% more 18- to -44-year-olds (100% vs 73.1%), 47% more 45- to -64-year-olds (90.8% vs 43.9%), 39% more over-65-year-olds (75.7% vs 36.4%) and 37% more people of all ages (91.0% vs 54.2%) after adjuvanted vaccine than WV vaccine. As each dose of WV vaccine contains twice as much antigen as adjuvanted vaccine, 100 doses of adjuvanted vaccine contains the same amount of viral HA as 50 doses of WV vaccine and could attain seroprotection in around 78 of 100 people of all ages, compared with around 25 of 50 recipients of WV vaccine. Thus, about three times as many people could attain seroprotective levels of antibody after adjuvanted vaccine than WV vaccine using the same amount of antigen. However, the relationship between HI titres and protection is imprecise, varying from one study to another, either due to the imprecision of the HI assay or differences between strains and subtypes of influenza. Nonetheless, the observed antigen-sparing property of AS03A adjuvant could be crucially important when vaccines are first prepared in response to pandemic influenza and are limited in supply.
The HI assay is used extensively in the assessment of immunity to influenza and in vaccine licensing, but HI does not measure the full repertoire of antibodies that may be important in protection. To date there are no correlates of protection using the MN assay and CHMP has no licensing criteria based on MN. Only 191 of the 10 published studies of 2009 pandemic H1N1 vaccine reported results of MN tests. 91–100 In people with and without baseline antibodies, titres of 1 in 40 or more were seen 21 days after the first dose of WV vaccine in HI and MN assays, respectively, in 71.4% and 87.1% of 18- to -44-year-olds, 38.8% and 56.7% of 45- to -64-year-olds, 32.4% and 50.0% of ≥ 65-year-olds, and 50.9% and 67.8% of all age groups, and 21 days after the second dose of WV vaccine in 73.1% and 94.0% of 18- to -44-year-olds, 43.9% and 74.2% of 45- to -64-year-olds, 36.4% and 63.6% of ≥ 65-year-olds, and 54.2% and 80.1% of all age groups. Similarly, titres of 1 in 40 or more were seen 21 days after the first dose of adjuvanted vaccine in HI and MN assays, respectively, in 94.0% and 98.5% of 18- to -44-year-olds, 77.3% and 89.4% of 45- to -64-year-olds, 51.4% and 64.9% of ≥ 65-year-olds, and 78.2% and 87.6% of all age groups, and 21 days after the second dose in 100% and 100% of 18- to -44-year-olds, 90.8% and 97.0% of 45- to -64-year-olds, 75.7% and 94.6% of ≥ 65-year-olds, and 91.0% and 97.6% of all age groups. Thus, in our study, MN and HI discerned comparable differences between adjuvanted and WV vaccines, but the numbers attaining a titre of 1 : 40 were higher by MN than HI. This is unsurprising as HI measures antibodies directed towards the receptor binding portion of the HA, whereas MN identifies a broader range of antibody, potentially to other antigens. On five of seven occasions when a vaccine failed to attain the CHMP criteria for seroprotection by HI, it also failed to attain a titre of 1 : 40 or more by MN.
Our study and a similar study in children,91 reveal heightened immunogenicity of AS03A-adjuvanted vaccine in comparison with WV vaccine by HI and MN. Waddington etal. 91 did not measure antibodies after the first vaccine dose or assess vaccine immunogenicity using CHMP criteria. The HI and MN antibody assays in the paediatric study were carried out in the same laboratory as in our study. After two doses, the adjuvanted vaccine was associated with significantly higher percentages of children with HI titres ≥ 1 : 32 (99.3% vs 78.2%) and MN titres ≥ 1 : 40 (99.3% vs 88.5%).
We are disappointed by the performance of the WV vaccine in our study in comparison with that of single doses of various pandemic H1N1 vaccines in other publications, notably in relation to 70% target for seroprotection. 92,94,95,97–99 The failure of one dose of AS03A-adjuvanted vaccine in the over-65-year-olds (primed and unprimed) to meet the target for seroprotection was unexpected. 95,97,99,100 In our study, both vaccines were transported and stored using appropriate temperature monitoring, as were the samples for HI and MN. Multidose vials of WV vaccine were discarded within 3 hours of opening. The WV vaccine was prepared using an egg-derived wild-type A/California/7/2009 (H1N1) virus grown in a Vero-cell culture system and the HI assay was carried out with egg-grown NIBRG-121 virus, generated from A/California/7/2009 (H1N1) virus and influenza A/PR/8/34 strains using reverse genetics as test antigen. The adjuvanted vaccine was prepared using the NYMC X-179A virus generated from the same A/California/7/2009 (H1N1) virus as used in the WV vaccine. According to unpublished findings of the same type of WV vaccine used in this study [data presented by Baxter to the European Medicines Agency (EMA)], seroprotection rates on HI assay increased significantly, by about 10%, when homologous Vero cell-derived H1N1 antigen was used in HI assay rather than egg-derived antigen. Serology assays are highly variable between laboratories, with variability for HI assays varying by up to 32-fold between laboratories. 71–73 Such variability creates difficulties in interpreting results from different manufacturers, and in February 2009 the WHO highlighted the need for standardised assays and internationally accepted anti-serum standards. 74 In our study, sera were tested blind, in duplicate, with four positive and two negative laboratory control sera included in each run of HI and MN. In addition, the international H1N1 standard antibody was tested on five occasions during the analyses. Our observations underscore the need for further comparative studies of vaccines, for greater harmonisation of assays and standards between laboratories, and for independent work on the role of egg-derived versus cell culture-derived antigen in assays of vaccine propagated on Vero cells.
Our study is the first to consider the kinetics of HI or MN antibody response to pandemic H1N1 vaccine. Seroprotection in all subjects regardless of baseline antibody was attained on HI more rapidly with adjuvanted vaccine, occurring on days 7, 14 and 21 in 53%, 79% and 78% of participants, respectively, and in 27%, 48% and 51% after WV vaccine. Similarly, MN titres of 1 : 40 or more were attained more rapidly with adjuvanted vaccine, occurring in 61%, 87% and 87% of subjects on days 7, 14 and 21, and in 43%, 65% and 68%, respectively, after WV vaccine. Similarly, seroprotection in subjects without baseline antibody was attained on HI more rapidly with adjuvanted vaccine, occurring on days 7, 14 and 21 in 47%, 76% and 76% of participants, respectively, and in 20%, 42.5% and 46% after WV vaccine. Similarly, MN titres of 1 : 40 or more were attained more rapidly with adjuvanted vaccine, occurring in 51%, 83% and 84% of subjects on days 7, 14 and 21, and in 27%, 54% and 57%, respectively, after WV vaccine. These more rapid early antibody responses to a single dose of adjuvanted vaccine could be crucial among immunologically naive essential key workers and high-risk populations, in whom rapid protection is essential.
In general, a second dose of either study vaccine failed to overcome the age-related decline in antibody titres (i.e. with achievement of antibody titres to levels seen in younger subjects). In all subjects, regardless of baseline antibody status, a second dose of AS03A-adjuvanted vaccine boosted seroprotection rates from day 21 levels from 77% to 91% among 45- to 64-year-olds, and from 51% to 81% among older adults. In contrast, a single dose of WV vaccine induced seroprotection rates on day 21 of 39% and 32% among 45- to -64-year-olds and older adults, with the second dose failing to attain seroprotection rates of 50%. As the overall case–fatality rate among hospitalised patients with 2009 H1N1 infection is highest among ≥ 50-year-olds, a two-dose vaccine schedule with adjuvanted vaccine in these subjects should be considered. But overall, our experience indicates that health benefits from 2009 H1N1 vaccine will be greater using a one-dose regimen rather than a two-dose regimen in half as many people.
Our study is the first to evaluate antibody persistence at 6 months after vaccination. Ignoring baseline antibody status, seroprotection at 6 months was significantly greater on adjuvanted vaccine than WV vaccine, as in all previous samples. Comparison of results at 6 months in participants with and without baseline antibody with those on day 42 showed that the HI seroprotection rate after adjuvanted vaccine fell modestly (83% vs 91%) and the GMT fell approximately twofold to 1/118. After WV vaccine, the HI seroprotection rate (59%) at 6 months was similar to the rate observed on day 42 (54%), as was the GMT (1/53). HI antibodies were well maintained by subjects who were HI antibody negative at baseline – titres of 1 in 40 or more were found after adjuvanted vaccine in 98%, 79% and 56% in subjects aged 18–44 years, 45–64 years, and ≥ 65 years, respectively, and 74%, 51% and 30%, respectively, after WV vaccine. The nature of influenza is unpredictable, but continued circulation of 2009 H1N1-like viruses is likely, so the durable immunity from vaccination last winter should be beneficial.
Our study focused on immunogenicity and did not evaluate vaccine efficacy or effectiveness. It is unclear whether the observed differences in immunogenicity from use of adjuvanted vaccine compared with WV vaccine would ultimately lead to greater protection against pandemic influenza and its complications. Given current knowledge of the relationship between HI antibody levels and protection,76 it seems likely that the improved immunogenicity associated with adjuvanted vaccine would provide more social, public health, and economic benefits than WV vaccine.
Hitherto, the evaluation of existing and potential vaccine candidates has been limited by single vaccine studies, and the variability of laboratory assays and immunogenicity end points. International antibody standards reduce variability in serological testing78 but, as exemplified by this study, head-to-head trials are essential to identify differences between vaccine approaches.
Chapter 5 Conclusions
Our data indicate that both vaccines are generally well tolerated, with adjuvanted vaccine causing more local and systemic reactions than WV vaccine. Neither the frequency nor severity of reactions increased after the second dose of either vaccine. The most frequent local AE was pain, which was reported three times more often after adjuvanted vaccine than WV vaccine by three-quarters of those given adjuvanted vaccine. About 1 in 50 people given adjuvanted vaccine experienced severe local pain.
The most common systemic effect after either vaccine was myalgia, which occurred twice as often after either dose of adjuvanted vaccine than WV vaccine in around 50% of people. Headache occurring shortly after vaccination was also reported more often after adjuvanted vaccine than WV vaccine. Analgesia was taken by comparable numbers of people given adjuvanted and WV vaccines, but absenteeism was higher after adjuvanted vaccine.
Overall, the majority of reactions were graded as mild or moderate and the extremely high uptake of the second dose was in keeping with the self-reported grading. However, the participants in this study were recruited mostly in a tertiary health-care setting, and can be expected to be more knowledgeable of influenza and its complications, better motivated, and more accepting of a second dose than the general population. It is conceivable that the higher incidence of local and systemic reactions with adjuvanted vaccine might affect uptake of a second dose, should this be required to confront a future pandemic.
A striking feature of this pandemic has been the excellent antibody response of vaccinees in this and other studies to a single dose of H1N1 vaccine. Our experience and that of others indicate that health benefits will be greater using a one-dose regimen rather than a two-dose regimen in half as many people.
Generally, two doses of avian H5, H7 and H9 vaccines are required to produce responses that meet the EMA CHMP criteria. However, with vaccine manufacturers using their own assays and in the absence of standardisation of assays, it has been difficult to compare results obtained by different laboratories and manufacturers. Similarly, because of social and demographic differences between populations that participate in vaccine studies, it has been difficult to compare acceptability across studies. Our head-to-head evaluation reveals real benefits over studies undertaken by individual manufacturers.
An important finding in this study was the demonstration of an antibody response as early as day 7 after the first vaccination of people without baseline antibody. By day 7, HI antibody titres of 1 : 40 or more develop after WV vaccine in about 20% of people of all ages. By MN, antibody titres of 1 : 40 or more are found after WV vaccine in almost 30% of people of all ages. Adjuvanted vaccine increases the early antibody response to about 50% by day 7 by HI and MN. Antibody assessments on days 14 and 21 show a continuing advantage of adjuvanted vaccine over WV vaccine – with higher GMTs and seroprotection, both by HI and MN. A more rapid antibody response also occurs in all subjects, i.e. ignoring baseline antibody status. We conclude that adjuvanted vaccine is more able than WV vaccine to elicit an early antibody response and in doing so offers greater public health benefits.
We used the CHMP criteria to compare the immunogenicity of the two vaccines. CHMP criteria measure antibodies directed against the HA that correlate with protection. However, the HI assay does not measure all antibodies that reduce infectivity and the relationship between HI antibody and protection is imprecise. To date, MN antibodies have not been correlated with protection and HI is used by regulators to evaluate vaccines, although there is a clear recognition that the CHMP criteria are somewhat arbitrary and may need refining. EMA anticipates that pandemic vaccines should be able to elicit sufficient immunological responses to meet all three of the current standards set for seasonal influenza vaccines. In all subjects, i.e. with and without baseline antibody, one dose of adjuvanted vaccine met all three CHMP criteria in all vaccinees and WV vaccine met two – also in response to the second dose. Thus adjuvanted pandemic vaccine is more immunogenic than WV vaccine and the use of adjuvant reduces the dosage of antigen needed and is therefore dose sparing, as has been demonstrated in studies of H5 vaccines, and pandemic H1N1 vaccine in children. 88
Trials of other pandemic H1N1 vaccines that differ in antigen content, use of adjuvant and method of production, together with our study, show that the CHMP standard for seroprotection is met commonly by one dose. However, one dose of WV vaccine elicited only modest levels of seroprotection in the over 45-year-olds, moreover there was a profound age effect with both vaccines eliciting lower HI and MN antibody responses with increasing age. Overall, our experience indicates that health benefits from 2009 H1N1 vaccine are greater using a one dose regimen rather than a two-dose regimen in half as many people. However, regardless of baseline antibody, a second dose of AS03A-adjuvanted vaccine boosted day 21 seroprotection rates from 51% to 81% among subjects aged 65 years and older, suggesting that a two-dose vaccine schedule with adjuvanted vaccine in these subjects should be considered.
Trials of avian influenza vaccines show that antibody levels wane over time and are boosted by further vaccination. Antibody titres in our study were lower at 6 months after WV vaccine than adjuvanted vaccine with persistence of the age-related effect. At 6 months, seroprotection after adjuvanted vaccine (83%) exceeded the CHMP target. It was well maintained (59%) after WV vaccine, and while an effect of clinical or subclinical infection in both groups cannot be excluded, the available information suggests the possibility of durable immunity resulting from the national immunisation programme for pandemic vaccine last winter.
There has been little or no antigenic drift since the onset of the pandemic so it was not possible to compare the breadth of the immune response. The size of the study precludes any comments on the occurrence of rare events, such as Guillain–Barré syndrome. The results of the clinical trial support the decision to use adjuvanted vaccine in preference to WV vaccine during the national immunisation programme, despite the higher incidence of local and systemic reactions.
Recommendations for future research
Based on our findings, we make the following recommendations for future research studies on:
Understanding individual immunological repertoire
-
The breadth of the antibody response We recommend work to identify whether there is an advantage of adjuvanted H1N1 vaccine over WV vaccine in generating broader antibody responses to drift variants that might emerge over time.
-
Further work on vaccine adjuvants Oil-in-water adjuvants such as AS03A improve antibody responses considerably compared with conventional unadjuvanted vaccines and WV vaccine, particularly in unprimed subjects. In our study AS03A did not reverse the age-related decline in immune response to the level attained in younger adults. It is unclear whether this goal can ever be achieved. It is also unclear whether the reactogenicity and immunostimulatory effects of oil-in-water adjuvants can be disentangled.
-
Basic research on immunogenicity to different HA subtypes A striking feature of this pandemic was the excellent antibody responses of vaccinees to single doses of unadjuvanted vaccine. This experience contrasts with that observed with H5, H7 and H9 vaccines for reasons that are unclear. Possibilities include a priming effect of previous infection or genuine differences in immunogenicity relating to structural or other differences in HAs. A better understanding of the underlying immune response mechanism(s) is required.
Clinical vaccine studies
-
Further comparative studies of novel vaccines There has been an understandable reluctance of vaccine manufacturers to support comparative studies such as ours. Head-to-head studies provide the best means of identifying optimal strategies to confront a future pandemic and to optimise the choice of seasonal vaccine formulations, especially in young children and the elderly where immunogenicity is known to be problematic. These should be built into the procurement process for pandemic vaccines. Meanwhile, we recommend that comparative studies of vaccine immunogenicity and efficacy be carried out using new and conventional vaccine formulations of vaccines for seasonal influenza in young immunologically naive children – they remain at high risk from interpandemic influenza and may act as a surrogate for pandemic influenza.
-
Comparative studies of vaccine effectiveness Our study focused on immunogenicity and did not evaluate vaccine efficacy or effectiveness. It is unclear whether the observed differences in immunogenicity ultimately led to better protection from use of adjuvanted vaccine in practice. We recommend that when different types of vaccine are procured to confront a future pandemic, that consideration be given to observational studies that assess product-specific vaccine benefits, including potential reductions in complications, hospitalisations and mortality.
-
Age-related differences in vaccine efficacy The elderly are particularly vulnerable to severe influenza and its complications, and have relatively poor responses to potent vaccines. Immunosenescence may be critically important, but other factors may contribute. The biological basis for the age-related decline in response to influenza vaccine requires a better understanding in order to derive strategies for mitigation.
-
Health economic modelling Our study identified differences in reactogenicity to the two vaccines with a higher absenteeism rate during the 24 hours after vaccination to adjuvanted vaccine. It is conceivable that the observed differences in immunogenicity might lead to different levels of protection and socioeconomic benefits. The cost-effectiveness of different vaccines and vaccine strategies will be influenced by the availability of vaccine in relation to the course of the pandemic, vaccine procurement and distribution costs, vaccine uptake and wastage, the safety profile of vaccines, the attack rate and severity of the pandemic, and the level and duration of protection from vaccination among other factors. Health economic analyses of these various factors could help inform future vaccine strategies.
Improvements in technical parameters
-
Microneutralisation as a surrogate of protection Antibodies measured by HI correlate with protection but the analyses that established the relationship were carried out many years ago during a different virological era using poorly standardised HI tests. The response to vaccination is increasingly being undertaken by MN. It is conceivable that antibodies measured by a standardised MN assay may provide a better correlate of protection than those measured by HI. We recommend that MN be included in future trials of vaccine efficacy. Consideration should also be given to the study design, as antibody titres wane after vaccination.
-
Role of egg-derived versus cell culture-derived virus in antibody assays As noted above, it is conceivable that cell culture-derived virus may be more relevant in measuring antibodies to vaccines grown on mammalian cells than virus that is propagated in eggs. Manufacturers and bodies, such as NIBSC, should explore this further.
Acknowledgments
Thanks are due to Dr Iain Stephenson for his support in the organisation and conduct of the trial and to colleagues in the University Hospitals of Leicester (UHL) Research and Development Department, specifically Carolyn Maloney, for their support and guidance. Thanks are also due to Nicola McMaster and Rachael Wilmott for the provision of local approval in Nottingham, and colleagues for the provision of approval in Sheffield. Thanks are also due to Dr Michael Pegg, Chairman and Rosemary Brown, Committee Co-ordinator, for help in expediting ethical approval, and Elaine Godfrey and Martyn Ward for accelerating the approval process at the MHRA.
In addition, thanks are also due to colleagues in the National Institute for Health Research (NIHR) Centres in Leicester [Chris Cannaby, Research Management and Governance (RM&G) Manager], Clare O’Neill (RM&G Manager), Nottingham (Penny Scardifield) and Sheffield (Alison Mortimer, Jenny Powell and Ramila Patel) for their support and guidance throughout the study.
We are especially grateful to Neil Formica, GSK, and Adrian Kilcoyne, Baxter for their invaluable help in providing vaccine for the study and the supporting materials for regulatory approval.
The invaluable contributions of Ann Walkden, Phayre Parkinson, Adam Lewszuk, Hilary Pateman, Sharon Holling (Research Nurses) and Suzanne Verster (Administrator) to data collection and day-to-day conduct of the study in Leicester are gratefully acknowledged. Similarly, the invaluable contributions of Sonia Greenwood, Thomas Bewick, Emily Jarvis, Gemma Thompson, Maria Benitez and Raquel Velos to data collection and day-to-day conduct of the study in Nottingham are gratefully acknowledged.
The contribution of Tiffany Jones from the UHL Communications Department in helping to inform staff of the study is gratefully acknowledged. We also thank University Hospital Nottingham Communications Team in helping to inform staff, WRVS University Hospital Nottingham, Women’s Institute, Age Concern and BBC Radio Nottingham for their interest and help in advertising the study, and Rachel Holt, the staff of the Pulmonary Function Laboratory, and the Nottingham Respiratory Biomedical Research Unit for use of facilities during the study.
We thank Neena Vadher, Trial Pharmacist, University Hospitals of Leicester NHS Trust, and David Lovett, Principal Pharmacist, Leicester Royal Infirmary, for support in transferring vaccines to Sheffield and Nottingham, and for their invaluable contribution to the conduct of the trial. We also thank pharmacy staff in Nottingham (Sheila Hodgson and Rachel Payne) and Sheffield.
We are grateful for excellent technical support and assistance from individuals in the respiratory virus laboratory at the Centre for Infections, Health Protection Agency, in particular Janice Baldevarona, Lucy Breakwell, Dipa Lackman, Surita Gangar and Ruth Reith.
Finally, we would like to thank all the study participants whose time and support made this study possible.
KRA is supported as a NIHR Senior Investigator (NI-SI-0508-10061).
Contributions of authors
KG Nicholson (Professor of Infectious Diseases, UHL, Chief investigator) led on development of the proposal and to the design, management, volunteer recruitment, data collection and data analysis, and wrote the report.
KR Abrams (Professor of Medical Statistics) contributed to the development of the proposal and to its design, led on the statistical analyses, and contributed to the writing and editing of this report.
S Batham (Trial Manager) contributed to management, volunteer recruitment, data collection and editing of the report.
TW Clark (Research Registrar) contributed to management, volunteer recruitment, data collection and editing of the report.
K Hoschler (Clinical Scientist) led on the laboratory analyses and editing of the report.
WS Lim (Consultant Respiratory Physician,) acted as Principal Investigator in Nottingham and contributed to the design, management, volunteer recruitment, data collection and editing of the report.
M Medina (Research Associate) contributed to data collection, handling of laboratory specimens and editing of the report.
JS Nguyen-Van-Tam (Professor of Health Protection, University of Nottingham) contributed to the development of proposal, its design and management, and editing of the report.
RC Read (Professor of Infectious Diseases) acted as Principal Investigator in Sheffield and contributed to the design, management, volunteer recruitment, data collection and editing of the report.
FC Warren (statistical expert) contributed to the statistical analyses.
M Zambon (Director, Centre for Infections, Health Protection Agency) contributed to the development of proposal, its design and management, and editing of the report.
All authors contributed to and approved this report.
Disclaimers
The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the referees for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report. The views expressed in this publication are those of the authors and not necessarily those of the NIHR or the Department of Health.
References
- Potter CW, Nicholson KG, Webster RG, Hay AJ. Textbook of influenza. Oxford: Blackwell; 1998.
- Kendal A P, Noble GR, Skehel JJ, Dowdle WR. Antigenic similarity of influenza A (H1N1) viruses from epidemics in 1977–1978 to ‘Scandinavian’ strains in epidemics of 1950–1951. Virology 1978:89-6.
- World Health Organization . WHO Global Influenza Preparedness Plan 2005. www.who.int/csr/resources/publications/influenza/WHO_CDS_CSR_GIP_2005_5.pdf (accessed 13 August 2010).
- World Health Organization . Pandemic Influenza Preparedness and Response 2009. www.who.int/csr/disease/influenza/PIPGuidance09.pdf (accessed 13 August 2010).
- Morens DM, Fauci AS. The 1918 influenza pandemic: insights for the 21st century. J Inf Dis 2007;195:1018-28.
- Adams DL. Putting Pandemics into Perspective: England and the Flu, 1889–1919 2008. http://kuscholarworks.ku.edu/dspace/bitstream/1808/4432/1/Adams_ku_0099D_10045_DATA_1.pdf (accessed 18 December 2010).
- Reid A. The effects of the 1918–19 influenza pandemic on infant and child health in Derbyshire. Med Hist 2005;49:29-54.
- Potter CW. A history of influenza. J Appl Microbiol 2001;91:572-9.
- McDonald JC. Asian influenza in Great Britain 1957–58. Proc R Soc Med 1958;51:1016-18.
- Anonymous . Reports on Public Health and Medical Subjects 1960.
- Chang WK. National influenza experience in Hong Kong, 1968. Bull WHO 1969;41:349-51.
- Fukumi H. Summary report on Hong Kong influenza in Japan. Bull WHO 1969;41:3-9.
- Roden AT. National experience with Hong Kong influenza in the United Kingdom, 1968–69. Bull WHO 1969;41:375-80.
- Fleming DM, Elliot AJ. Lessons from 40 years’ surveillance of influenza in England and Wales. Epidemiol Infect 2008;136:866-75.
- Nguyen-Van-Tam JS, Nicholson KG, Webster RG, Hay AJ. Textbook of influenza. London: Blackwell Science; 1998.
- Nichol KL, Mendelman PM, Mallon KP, Jackson LA, Gorse GJ, Belshe RB, et al. Effectiveness of live, attenuated intranasal influenza virus vaccine in healthy, working adults. JAMA 1999;282:137-44.
- Nicholson KG, Nicholson KG, Webster RG, Hay AJ. Textbook of influenza. London: Blackwell Science; 1998.
- Smith AP, Thomas M, Brockman P, Kent J, Nicholson KG. Effect of influenza B infection on human performance. BMJ 1993;306:760-1.
- Sullivan KM, Monto AS, Longini IM. Estimates of the US health impact of influenza. Am J Publ Hlth 1993;83:1712-16.
- Nicholson KG, Kent J, Hammersley V, Cancio E. Acute viral infections of upper respiratory tract in elderly people living in the community: comparative, prospective, population based study of disease burden. BMJ 1997;315:1060-4.
- Yassi A, Kettner J, Hammond G, Cheang M, McGill M. Effectiveness and cost–benefit of an influenza vaccination program for health care workers. Can J Inf Dis 1991;2:101-8.
- Kumpulainen V, Makela M. Influenza vaccination among healthy employees: a cost–benefit analysis. Scand J Inf Dis 1997;29:181-5.
- Keech M, Scott AJ, Ryan PJ. The impact of influenza and influenza-like illness on productivity and healthcare resource utilization in a working population. Occ Med 1998;48:85-90.
- Monto AS, Sullivan KM. Acute respiratory illness in the community. Frequency of illness and the agents involved. Epidemiol Infect n.d.:110-60.
- Vutuc C, Kunze M. Influenza: incidence and costs of inpatient treatment. MMW Fortschr Med 1993;111:508-9.
- Connolly AM, Salmon RL, Williams DH. What are the complications of influenza and can they be prevented? Experience from the 1989 epidemic of H3N2 influenza A in general practice. BMJ 1993;306:1452-4.
- Fleming DM. The impact of three influenza epidemics on primary care in England and Wales. PharmacoEconomics 1996;9:38-45.
- Neuzil KM, Reed GW, Mitchel EF, Griffin MR. Influenza-associated morbidity and mortality in young and middle-aged women. JAMA 1999;281:901-7.
- Ahmed AH, Nicholson KG, Nguyen-Van-Tam JS, Pearson JCG. Effectiveness of influenza vaccine in reducing hospital admissions during the 1989-90 epidemic. Epidemiol Infect 1997;18:27-33.
- Colquhoun AJ, Nicholson KG, Botha JL, Raymond NT. Effectiveness of influenza vaccine in reducing hospital admissions in people with diabetes. Epidemiol Infect 1997;119:335-41.
- World Health Organization . Ten Concerns If Avian Influenza Becomes a Pandemic 2005. www.who.int/csr/disease/influenza/pandemic10things/en/ (accessed 16 August 2010).
- Rice GW, Palmer E. Pandemic influenza in Japan, mortality patterns and official responses. J Jap Stud 1993;19:389-420.
- Myers KP, Olsen CW, Gray GC. Cases of swine influenza in humans a review of the literature. Clin Infect Dis 2007;44:1084-8.
- Hodder RA, Gaydos JC, Allen RG, Top FH, Nowosiwsky T, Russell PK, et al. Influenza A/swine at Fort Dix, NJ (Jan–Feb 1976), III. Extent of Spread and Duration of the Outbreak. J Infect Dis 1977;136:369-75.
- Sencer DJ, Millar JD. Reflections on the 1976 swine flu vaccination program. Emerg Infect Dis 2006;12:29-33.
- Echevarria-Zuno S, Mejia-Arangure JM, Mar-Obeso AJ, Grajales-Muñiz C, Robles- Pérez E, González-León M, et al. Infection and death from influenza A H1N1 virus in Mexico: a retrospective analysis. Lancet 2009;374:2072-9.
- Perez-Padilla R, de la Rosa-Zamboni D, Ponce de Leon S, Hernandez M, Quiñones-Falconi F, Bautista E, et al. Pneumonia and respiratory failure from swine-origin influenza A (H1N1) in Mexico. N Engl J Med 2009;361:680-9.
- Ginsberg M, Hopkins J, Maroufi A, Dunne G, Sunega DR, Giessick J, et al. Swine influenza A (H1N1) infection in two children: Southern California, March–April 2009. Morb Mortal Wkly Rep 2009;58:400-2.
- Dawood FS, Jain S, Lyn Finelli PH, Shaw MW, Lindstrom S, Garten RJ, et al. Novel Swine-Origin Influenza A (H1N1) Virus Investigation Team. Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N Engl J Med 2009;360:2605-15.
- Chan M. World Now at the Start of 2009 Influenza Pandemic n.d. www.who.int/mediacentre/news/statements/2009/h1n1_pandemic_phase6_20090611/en/index.html (accessed 16 August 2010).
- Garten RJ, Davis CT, Russell CA, Shu B, Lindstrom S, Balish A, et al. Antigenic and genetic characteristics of swine-origin 2009 A (H1N1) influenza viruses circulating in humans. Science 2009;325:197-201.
- Anonymous . Recommended composition of influenza virus vaccines for use in the 2010 influenza season (southern hemisphere winter). Wkly Epidemiol Rec 2009;84:421-31.
- Katz J, Hancock K, Veguilla V, Zhong W, Lu XH, Sun H. Serum cross-reactive antibody response to a novel influenza A (H1N1) virus after vaccination with seasonal influenza vaccine. Morb Mortal Wkly Rep 2009;58:521-4. www.cdc.gov/mmwr/preview/mmwrhtml/mm5819a1.htm (accessed 16 August 2010).
- Update on oseltamivir-resistant pandemic A (H1N1) 2009 influenza virus: January 2010. Wkly Epidemiol Rec 2010;85:37-40.
- Baz M, Abed Y, Papenburg J, Bouhy X, Hamelin M-E, Boivin G. Emergence of oseltamivir-resistant pandemic H1N1 virus during prophylaxis. N Engl J Med 2009;361:2296-7.
- Meiklejohn G, Eickhoff TC, Graves P. Antigenic drift and efficacy of influenza virus vaccines, 1976–1977. J Infect Dis 1978;138:618-24.
- De Donato S, Granoff D, Minutello M, Lecchi G, Faccini M, Agnello M, et al. Safety and immunogenicity of MF59-adjuvanted influenza vaccine in the elderly. Vaccine 1999;17:3094-101.
- US Census Bureau n.d. www.census.gov/ipc/www/popclockworld.html (accessed 16 August 2010).
- Kieny M-P. WHO Briefing on H1N1 Vaccines 2009. www.who.int/vaccine_research/H1N1vaccines.pdf (accessed 16 August 2010).
- Lermann SJ. Reactogenicity and immunogenicity of monovalent A/New Jersey/76 influenza virus vaccines in children. J Infect Dis 1977:136-70.
- Nicholson KG, Tyrrell DAJ, Harrison, Potter CW, Jennings. R, Clark A, . Clinical studies of monovalent whole virus and subunit A/USSR/77 (H1N1) vaccine: serological responses and clinical reactions. J Biol Stand 1979;7:123-36.
- Treanor JJ, Wilkinson BE, Masseoud F, Hu-Primmer J, Battaglia R, O’Brien D, et al. Safety and immunogenicity of a recombinant hemagglutinin vaccine for H5 influenza in humans. Vaccine 2001;19:1732-7.
- Treanor JJ, Campbell JD, Zangwill KM, Rowe T, Wolff M, . Safety and immunogenicity of an inactivated subvirion influenza A (H5N1) vaccine. N Engl J Med 2006;354:1343-51.
- Nicholson KG, Colegate AE, Podda A, Stephenson I, Wood J, Ypma E, et al. Safety and antigenicity of non-adjuvanted and MF59-adjuvanted influenza A/Duck/Singapore/97 (H5N3) vaccine: a randomised trial of two potential vaccines against H5N1 influenza. Lancet 2001;357:1937-43.
- Bresson J-L, Perronne C, Launay O, Gerdil C, Saville M, Wood J, et al. Safety and immunogenicity of an inactivated split-virion influenza A/Vietnam/1194/2004 (H5N1) vaccine: phase I randomised trial. Lancet 2006;367:1657-64.
- Nolan TM, Richmond PC, Skeljo MV, Pearce G, Hartel G, Formica NT, et al. Phase I and II randomised trials of the safety and immunogenicity of a prototype adjuvanted inactivated split-virus influenza A (H5N1) vaccine in healthy adults. Vaccine 2008;26:4160-7.
- Keitel WA, Campbell JD, Treanor JJ, Walter EB, Patel SM, He F, et al. Safety and immunogenicity of an inactivated influenza A/H5N1 vaccine given with or without aluminum hydroxide to healthy adults: results of a phase I–II randomized clinical trial. J Infect Dis 2008;198:1309-16.
- Stephenson I, Nicholson KG, Glück R, Mischler R, Newman RW, Palache AM, et al. Safety and antigenicity of whole virus and subunit influenza A/Hong Kong/1073/99 (H9N2) vaccine in healthy adults: phase I randomised trial. Lancet 2003;362:1959-66.
- Cox RJ, Madhun AS, Hauge S, Sjursen H, Major D, Kuhne M, et al. A phase I clinical trial of a PER.C6 cell grown influenza H7 virus vaccine. Vaccine 2009;27:1889-97.
- Ehrlich HJ, Müller M, Oh HML, Tambyah PA, Joukhadar C, Montomoli E, et al. A clinical trial of whole-virus H5N1 vaccine derived from cell culture. N Engl J Med 2008;358:2573-84.
- Stephenson I, Nicholson KG, Colegate A, Podda A, Wood J, Ypma E, et al. Boosting immunity to influenza H5N1 with MF59-adjuvanted H5N3 A/Duck/Singapore/97 vaccine in a primed human population. Vaccine 2003;21:1687-93.
- Stephenson I, Bugarini R, Nicholson KG, Podda A, Wood JM, Zambon MC, et al. Cross-reactivity to highly pathogenic avian influenza H5N1 viruses after vaccination with nonadjuvanted and MF59-adjuvanted influenza A/Duck/Singapore/97 (H5N3) vaccine: a potential priming strategy. J Infect Dis 2005;191:1210-15.
- Stephenson I, Nicholson KG, Hoschler K, Zambon MC, Hancock K, DeVos J, et al. Antigenically distinct MF59-adjuvanted vaccine to boost immunity to H5N1. N Engl J Med 2008;359:1631-3.
- Galli G, Hancock K, Hoschler K, DeVos J, Praus M, Bardelli M, et al. Fast rise of broadly cross-reactive antibodies after boosting long-lived human memory B cells primed by an MF59 adjuvanted prepandemic vaccine. Proc Natl Acad Sci USA 2009;106:7962-7.
- Leroux-Roels I, Borkowski A, Vanwolleghem T, Dramé M, Clement F, Hons E, et al. Antigen sparing and cross-reactive immunity with an adjuvanted rH5N1 prototype pandemic influenza vaccine: a randomised controlled trial. Lancet 2007;370:580-9.
- Leroux-Roels I, Bernhard R, Gérard P, Dramé M, Hanon E, Leroux-Roels G, et al. Broad Clade 2 cross-reactive immunity induced by an adjuvanted clade 1 rH5N1 pandemic influenza vaccine. PLoS One 2008;3.
- Levie K, Leroux-Roels I, Hoppenbrouwers K. An adjuvanted, low-dose, pandemic influenza A (H5N1) vaccine candidate is safe, immunogenic, and induces cross-reactive immune responses in healthy adults. J Infect Dis 2008;198:642-9.
- Lin J, Zhang J, Dong X, Fang H, Chen J, Su N, et al. Safety and immunogenicity of an inactivated adjuvanted whole-virion influenza A (H5N1) vaccine: a phase I randomised controlled trial. Lancet 2006;368:991-7.
- European Committee for Proprietary Medicinal Products . Note for Guidance on Harmonization of Requirements for Influenza Vaccines 1997. www.emea.europa.eu/pdfs/human/bwp/021496en.pdf (accessed 16 August 2010).
- Committee for Proprietary Medicinal Products . Guideline on Dossier Structure and Content for Pandemic Vaccine Marketing Authorisation 2004. http://archives.who.int/prioritymeds/report/append/62EMEAguidelines.pdf (accessed 16 August 2010).
- Stephenson I, Das RG, Wood JM, Katz JM. Comparison of neutralising antibody assays for detection of antibody to influenza A/H3N2 viruses: an international collaborative study. Vaccine 2007;25:4056-63.
- Wood JM, Gaines-Das RE, Taylor J, Chakraverty P. Comparison of influenza serological techniques by international collaborative study. Vaccine 1994;12:167-74.
- Wood J. Update on Development of International Standards for H5N1 Antibodies n.d. www.who.int/vaccine_research/diseases/influenza/120209_John_Wood.pdf (accessed 16 August 2010).
- n.d. www.who.int/vaccine_research/diseases/influenza/meeting_120209/en/ (accessed 16 August 2009).
- Hobson D, Curry RL, Beare AS, Ward-Gardner A. The role of serum haemagglutination-inhibiting antibody in protection against challenge infection with influenza A2 and B viruses. J Hyg (Lond) 1972;70:767-77.
- Coudeville L, Bailleux F, Riche B, Megas F, Andre P, Ecochard R. Relationship between haemagglutination inhibiting antibody titres and clinical protection against influenza: development and application of a Bayesian random-effects model. BMC Med Res Methodol 2010;10. www.biomedcentral.com/1471-2288/10/18.
- US Department of Health and Human Services, Food and Drug Administration . Guidance for Industry: Clinical Data Needed to Support the Licensure of Pandemic Influenza Vaccines 2007. www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/Vaccines/ucm091985.pdf (accessed 16 August 2009).
- Stephenson I, Heath A, Major D, Newman RW, Hoschler K, Junzi W, et al. Reproducibility of serologic assays for influenza virus A (H5N1). Emerg Infect Dis 2009;15:1252-9.
- Rowe T, Abernathy RA, Hu-Primmer J, Thompson WW, Lu X, Lim W, et al. Detection of antibody to avian influenza A (H5N1) virus in human serum by using a combination of serologic assays. J Clin Microbiol 1999;37:937-43.
- Ellis JS, Zambon MC. Molecular investigation of an outbreak of influenza in the United Kingdom. Eur J Epidemiol 1997;13:369-72.
- International Conference on Harmonisation/Good Clinical Practice . International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use 1996. www.scribd.com/doc/14677530/ICH-GCP-Guidelines (accessed 18 December 2010).
- Bautista E, Chotpitayasunondh T, Gao Z, Harper SA, Shaw M, Uyeki TM, et al. Writing Committee of the WHO Consultation on Clinical Aspects of Pandemic (H1N1) 2009 Influenza. Clinical Aspects of Pandemic 2009 Influenza A (H1N1) Virus Infection. N Engl J Med 2010;362:1708-19.
- European Medicines Agency . Twenty-Second Pandemic Pharmacovigilance Update 2010. www.ema.europa.eu/influenza/updates.html (accessed 17 September 2010).
- Hancock K, Veguilla V, Xiuhua L, Zhong W, Butler EN, Sun H, et al. Cross-reactive antibody responses to the 2009 pandemic H1N1 influenza virus. N Engl J Med 2009;361:1945-52.
- Miller E, Hoschler K, Hardelid P, Stanford E, Andrews N, Zambon M. Incidence of 2009 pandemic influenza A H1N1 infection in England: a cross-sectional serological study. Lancet 2010;375:1100-8.
- Reed C, Angulo FJ, Swerdlow DL, Lipsitch M, Meltzer MI, Jernigan D, et al. Estimates of the prevalence of pandemic (H1N1) 2009, United States, April–July 2009. Emerg Infect Dis 2009;15:2004-7.
- Donaldson LJ, Rutter PD, Ellis BM, Greaves FEC, Mytton OT, Pebody RG, et al. Mortality from pandemic A/H1N1 2009 influenza in England: public health surveillance study. BMJ 2009;339.
- Vesikari T, Pellegrini M, Karvonen A, . Enhanced immunogenicity of seasonal influenza vaccines in young children using MF59 adjuvant. Pediatr Infect Dis 2009;28:563-71.
- Atmar RL, Keitel WA, Patel SM, Katz JM, She D, El Sahly H, et al. Safety and immunogenicity of nonadjuvanted and MF59-adjuvanted influenza H9N2 vaccine preparations. Clin Infect Dis 2006;43:1135-42.
- Frey S, Poland G, Percell S, Podda A, . Comparison of the safety, tolerability and immunogenicity of MF59-adjuvanted and non-adjuvanted influenza vaccine in non-elderly adults. Vaccine 2003;21:4234-37.
- Waddington CS, Oeser C, Reiner A, John T, Wilkins S, Casey M, et al. Safety and immunogenicity of AS03B adjuvanted split virion versus non-adjuvanted whole virion H1N1 influenza vaccine in UK children aged 6 months-12 years: open label, randomised, parallel group, multicentre study. BMJ 2010;340. 10.1136/bmj.c2649.
- Arguedas A, Soley C, Lindert K. Responses to 2009 H1N1 vaccine in children 3 to 17 years of age. N Engl J Med 2010;362:370-2.
- Clark TW, Pareek M, Hoschler K, . Trial of 2009 influenza A (H1N1) monovalent MF59-adjuvanted vaccine. N Engl J Med 2009;361:2424-35.
- Greenberg ME, Lai MH, Hartel GF, . Response to a monovalent 2009 influenza A (H1N1) vaccine. N Engl J Med 2009;361:2405-13.
- Liang X-F, Wang H-Q, Wang J-Z, Fang H-H, Wu J, Zhu F-C, et al. Safety and immunogenicity of 2009 pandemic influenza A H1N1 vaccines in China: a multicentre, double-blind, randomised, placebo-controlled trial. Lancet 2010;375:56-6.
- Nolan T, McVernon J, Skeljo M, Richmond P, Wadia U, Lambert S, et al. Immunogenicity of a Monovalent 2009 influenza A(H1N1) vaccine in infants and children: a randomized trial. JAMA 2009;303:37-46.
- Plennevaux E, Sheldon E, Blatter M, Reeves-Hochq MK, Denis M. Immune response after a single vaccination against 2009 influenza A H1N1 in USA: a preliminary report of two randomised controlled phase 2 trials. Lancet 2010;375:41-8.
- Roman F, Vaman T, Gerlach B, Markendorf A, Gillard P, Devaster JM. Immunogenicity and safety in adults of one dose of influenza A H1N1v 2009 vaccine formulated with and without AS03A-adjuvant: Preliminary report of an observer-blind, randomised trial. Vaccine 2010;28:1740-5.
- Vajo Z, Tamas F, Sinka L, Jankovics I. Safety and immunogenicity of a 2009 pandemic influenza A H1N1 vaccine when administered alone or simultaneously with the seasonal influenza vaccine for the 2009-10 influenza season: a multicentre, randomised controlled trial. Lancet 2010;375:49-55.
- Zhu FC, Wang H, Fang HH, Wang JG, Lin XJ, Liang XF, et al. A novel influenza A (H1N1) vaccine in various age groups. N Engl J Med 2009;361:2414-23.
Appendix 1 List of case record forms
The following case record forms were used in the study:
-
poster advertisement
-
information letter
-
volunteer information sheet
-
volunteer consent form (consent to take part in the study)
-
randomisation
-
letter to GPs informing them that their patient has agreed to participate in the study
-
diary card for first immunisation
-
diary card for second immunisation
-
case report form (CRF).
Copies of the CRFs are available from Professor Karl Nicholson, e-mail: kgn2@le.ac.uk
Appendix 2 The Committee for Medicinal Products for Human Use and the US Food and Drug Administration licensing criteria for pandemic vaccines
The Committee for Medicinal Products for Human Use licensing criteria
The following information concerning immunogenicity criteria is taken from the CPMP document, ‘Note for guidance on harmonisation of requirements for influenza vaccines’ and the CHMP documents, ‘Guideline on dossier structure and content for pandemic influenza vaccine marketing authorisation application’. 69,70
However, with no other criteria to suggest at present, it is anticipated that mock-up vaccines should at least be able to elicit sufficient immunological responses to meet all three of the current standards set for existing vaccines in adults or older adults in CPMP/BWP/214/96. In addition, neutralising antibodies should be measured, preferably at one or a few selected reference centres. 70
Requirements in CPMP/BWP/214/96
2.3 Trial procedure
-
Just prior to vaccination a 10-ml venous blood sample shall be taken from each trial subject, for baseline titration of circulating anti-HA antibodies.
-
Immediately thereafter, each subject shall receive one dose of vaccine (0.5 ml) by IM or subcutaneous injection into the upper arm. The injection shall be given into the opposite arm from which the blood was drawn.
-
Approximately 3 weeks after vaccination, a 10-ml blood sample shall be taken from each subject. Sera shall be separated and stored at –20°C; samples shall be kept at the disposal of the control laboratories for epidemiological studies and possible further antibody titration.
-
In the event of intercurrent infection, nasal and/or pharyngeal swabs shall be collected, in order to allow diagnosis of either influenza or another viral respiratory infection …
2.5 Antibody titration
All sera shall be assayed for anti-HA antibody against the prototype strains by HI (Palmer et al. 1975) or SRH (Schild et al. 1975, Aymard et al. 1980) tests. Positive and negative sera as well as reference preparations may be obtained from a reference laboratory.
2.6 Interpretation of results and statistics
Antibody titrations shall be done in duplicate; pre-vacciniation and postvaccination sera shall be titrated simultaneously.
The titre assigned to each sample shall be the geometric mean of two independent determinations:
-
For the purpose of calculation, and HI result < 10 (= undetectable) shall be expressed as 5, and any negative SRH result shall be expressed as 4 mm2.
-
In HI tests, seroconversion corresponds to:
-
– negative prevaccination serum/post-vaccination serum ≥ 40
-
– a significant increase in antibody titre, i.e. at least a fourfold increase in titre.
-
-
In SRH tests, seroconversion corresponds to:
-
– negative prevaccination serum/postvaccination serum ≥ 25mm2
-
– a significant increase in antibody titre, i.e. at least a 50% increase in area.
-
-
Statistical parameters to be determined:
-
– geometric mean of prevaccination serum anti-HA antibody titres
-
– increase in the geometric mean of antibody titre
-
– number of seroconversions
-
– proportion of subjects with a titre of antibodies before vaccination
-
– proportion of subjects with a titre of antibodies after vaccination.
-
-
Clinical tolerance – frequency, mean time of appearance and duration of all local and general side effects shall be calculated.
Interpretation of results should take into account the route of administration and any recent history of influenza immunisation or infection…
3.1 Serological data
-
The following serological assessments should be considered for each strain in adult subjects, aged between 18 and 60 years, and at least one of the assessments should meet the indicated requirements:
-
– number of seroconversions or significant increase in anti-HA antibody titre > 40%
-
– mean geometric increase > 2.5
-
– the proportion of subjects achieving an HI titre ≥ 40 or SRH titre ≥ 25mm2 should be > 70%.
-
-
The following serological assessments should be considered for each strain in adult subjects, aged over 60 years, and at least one of the assessments should meet the indicated requirements:
-
– number of seroconversions or significant increase in anti-haemagglutinin antibody titre > 30%
-
– mean geometric increase > 2.0
-
– the proportion of subjects achieving an HI titre ≥ 40 or SRH titre ≥ 25mm2 should be > 60%.
-
The US Food and Drug Administration licensing criteria
The following information concerning immunogenicity criteria is taken from the FDA document ‘Guidance for Industry: Clinical Data Needed to Support the Licensure of Pandemic Influenza Vaccines’. 77
Approval of a pandemic influenza vaccine for manufacturers of a US licensed seasonal inactivated influenza vaccine where the process for manufacturing the pandemic influenza vaccine is the same All submissions for the initial licensure of a pandemic influenza vaccine should be submitted as biologics license applications (BLAs), which will provide for a trade name and labelling specific to the pandemic vaccine. For sponsors with existing licensed seasonal inactivated influenza vaccines who intend to file a BLA for a pandemic influenza vaccine that utilises the same manufacturing process, we would expect that the BLA would reference the original BLA, including the non-clinical and chemistry, manufacturing and controls (CMC) data in their original BLA. The GMTs at pre-vaccination and postvaccination should also be included.
-
Immunogenicity
-
– Data to support the selected dose and regimen should be based on the evaluation of immune responses elicited by the vaccine. The HI antibody assay has been used to assess vaccine activity and may be appropriate for the evaluation of the pandemic influenza vaccine. Appropriate end points may include: (1) the percentage of subjects achieving an HI antibody titre ≥ 1 : 40 and (2) rates of seroconversion, defined as the percentage of subjects with either a pre-vaccination HI titre of < 1:10 and a postvaccination HI titre of ≥ 1 : 40 or a pre-vaccination HI titre of ≥ 1 : 10 and a minimum fourfold rise in postvaccination HI antibody titre. In a prepandemic setting it is likely that most subjects will not have been exposed to the pandemic influenza viral antigen(s). Therefore, it is possible that vaccinated subjects may reach both suggested end points. Thus, for studies enrolling subjects who are immunologically naive to the pandemic antigen, one HI antibody assay end point, such as the percentage of subjects achieving an HI antibody titre ≥ 1 : 40, may be considered. Point estimates and the two-sided 95% CIs of these evaluations should be provided with the BLA. The GMTs at pre-vaccination and postvaccination should also be included.
-
– Considerable variability can be introduced into the laboratory assay used to measure HI antibodies as a result of a number of factors including differences in viral strains and red blood cell types, and the presence of non-specific inhibitors in the assay medium. Thus, suitable controls and assay validation are important for interpreting HI antibody results. It is also recommended that adequate serum sample volumes be obtained and stored for possible later use in confirmatory or comparative assay studies, if needed.
-
– Other end points and the corresponding immunological assays, such as the MN assay, might also be used to support the approval of a pandemic influenza vaccine BLA (ref. 18). Sponsors are encouraged to discuss their proposals with the Center for Biologics Evaluation and Research early in development …
-
Accelerated approval of a pandemic influenza vaccine manufactured by a process not US licensed The following may be used as a guide in developing end points that would support accelerated approval of pandemic influenza vaccines.
-
For adults < 65 years of age and for the paediatric population:
-
– The lower bound of the two-sided 95% CI for the percentage of subjects achieving seroconversion for HI antibody should meet or exceed 40%.
-
– The lower bound of the two-sided 95% CI for the percentage of subjects achieving an HI antibody titre ≥ 1 : 40 should meet or exceed 70%.
-
-
For adults ≥ 65 years of age:
-
– The lower bound of the two-sided 95% CI for the percentage of subjects achieving seroconversion for HI antibody should meet or exceed 30%.
-
– The lower bound of the two-sided 95% CI for the percentage of subjects achieving an HI antibody titre of ≥ 1 : 40 should meet or exceed 60%.
-
Appendix 3 Immune responses after the first and second dose of H1N1 vaccine in subjects without baseline antibody, as measured on haemagglutination inhibition
| Immunogenicity end point | WV vaccine | AS03A-adjuvanted split-virion vaccine | ||||||
|---|---|---|---|---|---|---|---|---|
| 18–44 years | 45–64 years | ≥ 65 years | All subjects | 18–44 years | 45–64 years | ≥ 65 years | All subjects | |
| Baseline | ||||||||
| No. of subjects | 58 | 62 | 30 | 150 | 60 | 59 | 34 | 153 |
| GMT value (95% CI) | 4.1 (4.0 to 4.2) | 4.0 (4.0 to 4.1) | 4.2 (3.9 to 4.4) | 4.1 (4.0 to 4.1) | 4.1 (4.0 to 4.2) | 4.0 (4.0 to 4.0) | 4.1 (3.9 to 4.3) | 4.1 (4.0 to 4.1) |
| After first dose | ||||||||
| Day 7 | ||||||||
| No. of subjects | 57 | 61 | 30 | 148 | 58 | 56 | 34 | 148 |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 35.1 (22.9 to 48.9) | 13.1 (5.8 to 24.2) | 3.3 (0.1 to 17.2) | 19.6 (13.5 to 26.9) | 70.7 (57.3 to 81.9) | 33.9 (21.8 to 47.8) | 26.5 (12.9 to 44.4) | 46.6 (38.4 to 55.0) |
| GMT value (95% CI) | 19.5 (12.2 to 31.1) | 6.9 (4.8 to 10.0) | 6.2 (4.6 to 8.4) | 10.1 (7.8 to 13.0) | 100.4 (57.9 to 174.0) | 15.1 (9.9 to 23.0) | 10.8 (6.6 to 17.7) | 29.3 (21.1 to 40.7) |
| Factor increase in GMT value (95% CI) | 4.8 (3.0 to 7.6) | 1.7 (1.2 to 2.5) | 1.5 (1.1 to 2.0) | 2.5 (1.9 to 3.2) | 24.5 (14.2 to 42.3) | 3.8 (2.5 to 5.8) | 2.6 (1.6 to 4.2) | 7.2 (5.2 to 10.0) |
| Day 14 | ||||||||
| No. of subjects | 55 | 62 | 28 | 146 | 57 | 55 | 32 | 144 |
| Subjects with HI titre ≥ 1 : 40%: (95% CI) | 63.6 (49.6 to 76.2) | 30.7 (19.6 to 43.7) | 27.6 (12.7 to 47.2) | 42.5 (34.3 to 50.9) | 89.5 (78.5 to 96.0) | 76.4 (63.0 to 86.8) | 53.1 (34.7 to 70.9) | 76.4 (68.6 to 83.1) |
| GMT value (95% CI) | 64.0 (38.7 to 106.0) | 17.0 (10.5 to 27.5) | 17.0 (10.3 to 28.1) | 28.0 (20.6 to 38.1) | 403.9 (270.0 to 604.5) | 104.1 (63.7 to 170.2) | 37.7 (19.8 to 71.9) | 142.1 (103.4 to 195.2) |
| Factor increase in GMT value (95% CI) | 15.9 (9.6 to 26.3) | 4.2 (2.6 to 6.8) | 4.1 (2.5 to 6.7) | 6.9 (5.1 to 9.4) | 98.6 (66.0 to 147.2) | 26.0 (15.9 to 42.5) | 9.1 (4.8 to 17.1) | 34.9 (25.5 to 47.9) |
| Day 21 | ||||||||
| No. of subjects | 58 | 61 | 30 | 149 | 57 | 57 | 34 | 148 |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 65.5 (51.9 to 77.5) | 36.1 (24.2 to 49.4) | 30.0 (14.7 to 49.4) | 46.3 (38.1 to 54.7) | 93.0 (83.0 to 98.1) | 73.7 (60.3 to 84.5) | 50.0 (32.4 to 67.6) | 75.7 (68.0 to 82.4) |
| GMT value (95% CI) | 74.8 (46.7 to 119.6) | 19.5 (11.7 to 32.3) | 17.6 (10.6 to 29.1) | 32.2 (23.7 to 43.8) | 368.7 (251.7 to 540.1) | 89.5 (56.4 to 141.8) | 35.9 (19.2 to 67.0) | 125.1 (92.3 to 169.7) |
| Factor increase in GMT value (95% CI) | 18.3 (11.5 to 29.2) | 4.8 (2.9 to 8.0) | 4.2 (2.6 to 7.0) | 7.9 (5.8 to 10.8) | 90.0 (61.6 to 131.5) | 22.4 (14.1 to 35.5) | 8.7 (4.7 to 16.0) | 30.8 (22.7 to 41.6) |
| Day 28 | ||||||||
| No. of subjects | 57 | 60 | 29 | 146 | 54 | 54 | 33 | 141 |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 70.2 (56.6 to 81.6) | 40 (27.6 to 53.5) | 27.6 (12.7 to 47.2) | 49.3 (41.0 to 57.7) | 100 (93.4 to 100) | 88.9 (77.4 to 95.8) | 72.7 (54.5 to 86.7) | 89.4 (83.1 to 93.9) |
| GMT value (95% CI) | 97.6 (61.5 to 155.0) | 24.9 (15.5 to 40.0) | 15.2 (9.0 to 25.4) | 38.4 (28.3 to 52.2) | 597.3 (469.5 to 759.9) | 165.4 (114.5 to 238.8) | 66.8 (42.8 to 104.2) | 218.7 (171.9 to 278.2) |
| Factor increase in GMT value (95% CI) | 23.9 (15.1 to 38.0) | 6.2 (3.9 to 9.9) | 3.7 (2.2 to 6.1) | 9.4 (7.0 to 12.8) | 145.5 (115.3 to 183.8) | 41.3 (28.6 to 59.7) | 16.1 (10.5 to 24.9) | 53.7 (42.3 to 68.2) |
| Day 35 | ||||||||
| No. of subjects | 57 | 58 | 30 | 145 | 54 | 56 | 33 | 143 |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 73.7 (60.3 to 84.5) | 41.4 (28.6 to 55.1) | 43.3 (25.5 to 62.6) | 54.5 (46.0 to 62.8) | 100 (93.4 to 100) | 89.3 (78.1 to 96.0) | 81.8 (64.5 to 93.0) | 91.6 (85.8 to 95.6) |
| GMT value (95% CI) | 97.5 (62.6 to 151.8) | 27.8 (17.3 to 44.6) | 18.2 (10.7 to 30.9) | 41.7 (31.0 to 56.1) | 597.3 (478.5 to 745.6) | 157.0 (110.6 to 222.8) | 83.2 (53.2 to 130.2) | 224.6 (178.7 to 282.2) |
| Factor increase in GMT value (95% CI) | 23.9 (15.3 to 37.2) | 6.9 (4.3 to 11.0) | 4.4 (2.6 to 7.4) | 10.2 (7.6 to 13.8) | 145.5 (117.2 to 180.8) | 39.2 (27.7 to 55.7) | 20.1 (13.0 to 31.1) | 55.2 (44.0 to 69.2) |
| Day 42 | ||||||||
| No. of subjects | 56 | 61 | 29 | 146 | 55 | 56 | 34 | 145 |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 67.9 (54.0 to 79.7) | 41.0 (28.6 to 54.3) | 34.5 (17.9 to 54.3) | 50.0 (41.6 to 58.4) | 100 (93.5 to 100) | 89.3 (78.1 to 96.0) | 76.5 (58.8 to 89.3) | 90.3 (84.3 to 94.6) |
| GMT value (95% CI) | 93.6 (58.2 to 150.7) | 25.0 (15.8 to 39.4) | 15.2 (9.1 to 25.5) | 37.6 (27.8 to 50.8) | 505.6 (402.6 to 635.1) | 131.3 (90.4 to 190.7) | 74.6 (47.7 to 116.7) | 191.8 (151.9 to 242.0) |
| Factor increase in GMT value (95% CI) | 23.0 (14.3 to 36.8) | 6.2 (3.9 to 9.8) | 3.7 (2.2 to 6.1) | 9.2 (6.8 to 12.5) | 123.3 (98.4 to 154.4) | 32.8 (22.6 to 47.7) | 18.1 (11.7 to 27.9) | 47.1 (37.4 to 59.4) |
Appendix 4 Immune responses after the first and second dose of H1N1 vaccine in subjects with baseline antibody, as measured on haemagglutination inhibition
| Immunogenicity end point | WV vaccine | AS03A-adjuvanted split-virion vaccine | ||||||
|---|---|---|---|---|---|---|---|---|
| 18–44 years | 45–64 years | ≥ 65 years | All subjects | 18–44 years | 45–64 years | ≥ 65 years | All subjects | |
| Baseline | ||||||||
| No. of subjects | 12 | 6 | 4 | 22 | 10 | 9 | 3 | 22 |
| GMT value (95% CI) | 80.7 (59.7 to 109.1) | 67.7 (20.3 to 226.0) | 22.6 (7.5 to 68.2) | 61.0 (41.7 to 89.3) | 137.3 (54.0 to 349.1) | 57.3 (32.2 to 102.0) | 71.7 (19.1 to 268.7) | 87.9 (54.6 to 141.4) |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 91.7 (61.5 to 99.8) | 66.7 (22.3 to 95.7) | 25.0 (0.6 to 80.6) | 72.7 (49.8 to 89.3) | 80.0 (44.4 to 97.5) | 66.7 (29.9 to 92.5) | 100 (29.2 to 100) | 77.3 (54.6 to 92.2) |
| After first dose | ||||||||
| Day 7 | ||||||||
| No. of subjects | 12 | 6 | 4 | 22 | 10 | 9 | 3 | 22 |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 100 (73.5 to 100) | 66.7 (22.3 to 95.7) | 25.0 (0.6 to 80.6) | 77.3 (54.6 to 92.2) | 100 (69.2 to 100) | 100 (66.4 to 100) | 66.7 (9.4 to 99.2) | 95.5 (77.2 to 99.9) |
| Subjects with seroconversion: % (95% CI) | 25.0 (5.5 to 57.2) | 0 (0.0 to 45.9) | 25.0 (0.6 to 80.6) | 18.2 (5.2 to 40.3) | 20.0 (2.5 to 55.6) | 66.7 (29.9 to 92.5) | 0.0 (0.0 to 70.8) | 36.4 (17.2 to 59.3) |
| GMT value (95% CI) | 170.9 (115.0 to 254.0) | 96.0 (21.5 to 429.3) | 58.9 (1.4 to 2566.4) | 120.3 (67.8 to 213.6) | 415.9 (216.5 to 798.8) | 335.2 (170.5 to 659.2) | 71.8 (8.2 to 627.0) | 299.7 (189.4 to 474.3) |
| Factor increase in GMT value (95% CI) | 2.1 (1.4 to 3.2) | 1.4 (0.9 to 2.3) | 2.6 (0.2 to 37.8) | 2.0 (1.4 to 2.9) | 3.0 (1.2 to 7.8) | 5.9 (2.1 to 16.7) | 1.0 (0.4 to 2.4) | 3.4 (1.9 to 6.2) |
| Day 14 | ||||||||
| No. of subjects | 12 | 6 | 4 | 22 | 10 | 8 | 3 | |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 100 (73.5 to 100) | 66.7 (22.3 to 95.7) | 50.0 (6.8 to 93.2) | 81.8 (59.7 to 94.8) | 100 (69.2 to 100) | 100 (63.1 to 100) | 100 (29.2 to 100) | 100 (83.9 to 100) |
| Subjects with seroconversion: % (95% CI) | 58.3 (27.7 to 84.8) | 33.3 (4.3 to 77.7) | 50.0 (6.8 to 93.2) | 50.0 (28.2 to 71.8) | 70.0 (34.8 to 93.3) | 75.0 (34.9 to 96.8) | 66.7 (9.4 to 99.2) | 71.4 (47.8 to 88.7) |
| GMT value (95% CI) | 279.3 (189.5 to 411.7) | 203.2 (42.2 to 978.0) | 181.0 (2.1 to 15617.2) | 236.7 (128.7 to 435.1) | 922.9 (533.1 to 1597.7) | 724.0 (389.7 to 1345.3) | 180.7 (8.0 to 4058.9) | 666.5 (430.2 to 1032.7) |
| Factor increase in GMT value (95% CI) | 3.5 (2.2 to 5.5) | 3.0 (1.3 to 7.0) | 8.0 (0.2 to 261.7) | 3.9 (2.4 to 6.4) | 6.7 (2.6 to 17.5) | 11.8 (3.6 to 38.7) | 2.5 (0.4 to 18.4) | 7.2 (3.9 to 13.4) |
| Day 21 | ||||||||
| No. of subjects | 12 | 6 | 4 | 22 | 10 | 9 | 3 | 22 |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 100 (73.5 to 100) | 66.7 (22.3 to 95.7) | 50.0 (6.8 to 93.2) | 81.8 (59.7 to 94.8) | 100 (69.2 to 100) | 100 (66.4 to 100) | 66.7 (9.4 to 99.2) | 95.5 (77.2 to 99.9) |
| Subjects with seroconversion: % (95% CI) | 50.0 (21.1 to 78.9) | 33.3 (4.3 to 77.7) | 25.0 (0.6 to 80.6) | 40.9 (20.7 to 63.6) | 60.0 (26.2 to 87.8) | 77.8 (40.0 to 97.2) | 66.7 (9.4 to 99.2) | 68.2 (45.1 to 86.1) |
| GMT value (95% CI) | 284.9 (170.3 to 364.0) | 191.8 (41.4 to 888.4) | 69.7 (0.7 to 7249.2) | 183.9 (96.1 to 352.1) | 831.7 (452.1 to 1530.0) | 597.2 (368.7 to 967.4) | 203.2 (3.8 to 10835.9) | 599.3 (391.4 to 917.7) |
| Factor increase in GMT value (95% CI) | 3.1 (1.9 to 4.9) | 2.8 (1.4 to 5.9) | 3.08 (0.1 to 117.4) | 3.01 (1.9 to 4.9) | 6.1 (2.3 to 16.1) | 10.4 (4.4 to 24.9) | 2.8 (0.1 to 62.7) | 6.8 (3.9 to 12.0) |
| Day 28 | ||||||||
| No. of subjects | 12 | 6 | 4 | 22 | 10 | 9 | 3 | 22 |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 100 (73.5 to 100) | 83.3 (35.9 to 99.6) | 75.0 (19.4 to 99.4) | 90.9 (70.8 to 98.9) | 100 (69.2 to 100) | 100 (66.4 to 100) | 66.7 (9.4 to 99.2) | 95.4 (77.2 to 99.9) |
| Subjects with seroconversion: % (95% CI) | 50.0 (21.1 to 78.9) | 33.3 (4.3 to 77.7) | 75.0 (19.4 to 99.4) | 50.0 (28.2 to 71.8) | 60.0 (26.2 to 87.8) | 77.8 (40.0 to 97.2) | 33.3 (0.8 to 90.6) | 63.6 (40.7 to 82.8) |
| GMT value (95% CI) | 313.4 (207.1 to 474.1) | 180.9 (45.4 to 719.9) | 107.6 (0.7 to 16698.4) | 222.1 (114.2 to 431.9) | 922.9 (476.2 to 1788.5) | 512.0 (322.7 to 812.2) | 128.0 (6.5 to 2526.0) | 554.0 (349.8 to 877.1) |
| Factor increase in GMT value (95% CI) | 3.9 (2.3 to 6.7) | 2.7 (1.6 to 4.6) | 4.8 (0.1 to 277.2) | 3.6 (2.1 to 6.2) | 6.7 (2.5 to 18.3) | 8.9 (3.5 to 23.1) | 1.8 (0.2 to 15.5) | 6.3 (3.5 to 11.4) |
| Day 35 | ||||||||
| No. of subjects | 10 | 6 | 4 | 20 | 9 | 9 | 3 | 21 |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 100 (69.2 to 100) | 83.3 (35.9 to 99.6) | 75.0 (19.4 to 99.4) | 90.0 (68.3 to 98.8) | 100 (66.4 to 100) | 100 (66.4 to 100) | 66.7 (9.4 to 99.2) | 95.2 (76.2 to 99.9) |
| Subjects with seroconversion: % (95% CI) | 60.0 (26.2 to 87.8) | 33.3 (4.3 to 77.7) | 50.0 (6.8 to 93.2) | 50.0 (27.2 to 72.8) | 66.7 (29.9 to 92.5) | 77.8 (40.0 to 97.2) | 33.3 (0.8 to 90.6) | 66.7 (43.0 to 85.4) |
| GMT value (95% CI) | 315.2 (199.8 to 497.1) | 191.8 (51.8 to 710.5) | 90.4 (0.7 to 11308.6) | 211.5 (104.1 to 429.8) | 877.8 (514.3 to 1498.3) | 553.0 (292.8 to 1044.2) | 128.0 (6.5 to 2526.0) | 546.9 (344.7 to 867.8) |
| Factor increase in GMT value (95% CI) | 4.14 (2.24 to 7.6) | 2.8 (1.8 to 4.5) | 4.0 (0.1 to 187.2) | 3.7 (2.1 to 6.3) | 6.9 (2.4 to 19.3) | 9.7 (3.1 to 30.1) | 1.8 (0.2 to 15.5) | 6.6 (3.5 to 12.5) |
| Day 42 | ||||||||
| No. of subjects | 11 | 5 | 4 | 20 | 9 | 9 | 4 | 21 |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 100 (71.5 to 100) | 80.0 (28.4 to 99.5) | 50.0 (6.8 to 93.2) | 85.0 (62.1 to 96.8) | 100 (66.4 to 100) | 100 (66.4 to 100) | 66.7 (9.4 to 99.2) | 95.2 (76.2 to 99.9) |
| Subjects with seroconversion: % (95% CI) | 54.5 (23.4 to 83.3) | 40.0 (5.3 to 85.3) | 25.0 (0.6 to 80.6) | 45.0 (23.1 to 68.5) | 55.6 (21.2 to 86.3) | 77.8 (40.0 to 97.2) | 33.3 (0.8 to 90.6) | 61.9 (38.4 to 81.9) |
| GMT value (95% CI) | 299.7 (199.2 to 450.7) | 256.0 (52.7 to 1244.2) | 69.7 (0.7 to 7249.2) | 215.2 (107.2 to 432.2) | 812.7 (454.8 to 1452.5) | 456.1 (259.2 to 802.6) | 128.0 (6.5 to 2526.0) | 487.3 (310.8 to 764.0) |
| Factor increase in GMT value (95% CI) | 3.9 (2.1 to 7.0) | 3.3 (1.7 to 6.3) | 3.1 (0.1 to 117.4) | 3.5 (2.1 to 6.1) | 6.35 (2.1 to 19.3) | 8.0 (2.7 to 23.8) | 1.8 (0.2 to 15.5) | 5.8 (3.1 to 11.1) |
Appendix 5 Immune responses after the first and second dose of H1N1 vaccine in subjects with and without baseline antibody, as measured on haemagglutination inhibition
| Immunogenicity end point | WV vaccine | AS03A–adjuvanted split–virion vaccine | ||||||
|---|---|---|---|---|---|---|---|---|
| 18–44 years | 45–64 years | ≥ 65 years | All subjects | 18–44 years | 45–64 years | ≥ 65 years | All subjects | |
| Baseline | ||||||||
| No. of subjects | 70 | 68 | 34 | 172 | 70 | 68 | 37 | 175 |
| GMT value (95% CI) | 6.8 (5.2 to 9.0) | 5.2 (4.2 to 6.4) | 5.1 (4.1 to 6.3) | 5.8 (5.0 to 6.7) | 6.8 (4.9 to 9.3) | 5.7 (4.5 to 7.2) | 5.2 (4.0 to 6.8) | 6.0 (5.1 to 7.0) |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 15.7 (8.1 to 26.4) | 5.9 (1.6 to 14.4) | 2.9 (0.1 to 15.3) | 9.3 (5.4 to 14.7) | 11.43 (5.1 to 21.3) | 8.8 (3.3 to 18.2) | 8.1 (1.7 to 21.9) | 9.7 (5.8 to 15.1) |
| After first dose | ||||||||
| Day 7 | ||||||||
| No. of subjects | 69 | 67 | 34 | 170 | 68 | 65 | 37 | 170 |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 46.4 (34.3 to 58.8) | 17.9 (9.6 to 29.2) | 5.9 (0.7 to 19.7) | 27.1 (20.5 to 34.4) | 75.0 (63.0 to 84.7) | 43.1 (30.9 to 56.0) | 29.7 (15.9 to 47.0) | 52.9 (45.2 to 60.6) |
| Subjects with seroconversion: % (95% CI) | 33.3 (22.4 to 45.7) | 11.9 (5.3 to 22.2) | 5.9 (0.7 to 19.7) | 19.4 (13.8 to 26.2) | 63.2 (50.7 to 74.6) | 38.5 (26.7 to 51.4) | 24.3 (11.8 to 41.2) | 45.3 (37.7 to 53.1) |
| GMT value (95% CI) | 28.4 (18.3 to 44.0) | 8.8 (5.9 to 13.0) | 8.1 (5.1 to 12.6) | 13.9 (10.7 to 18.1) | 123.7 (75.8 to 201.9) | 23.1 (14.6 to 36.6) | 12.6 (7.7 to 20.5) | 39.6 (28.9 to 54.2) |
| Factor increase in GMT value (95% CI) | 4.1 (2.8 to 6.1) | 1.7 (1.2 to 2.4) | 1.6 (1.2 to 2.2) | 2.4 (1.9 to 3.0) | 18.0 (10.8 to 30.1) | 4.0 (2.7 to 5.9) | 2.4 (1.5 to 3.8) | 6.5 (4.9 to 8.8) |
| Day 14 | ||||||||
| No. of subjects | 67 | 68 | 33 | 168 | 67 | 63 | 35 | 165 |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 70.2 (57.7 to 80.7) | 33.8 (22.8 to 46.3) | 30.3 (15.6 to 48.7) | 47.6 (39.9 to 55.5) | 91.0 (81.5 to 96.6) | 79.4 (67.3 to 88.5) | 57.1 (39.4 to 73.7) | 79.4 (72.4 to 85.3) |
| Subjects with seroconversion: % (95% CI) | 62.7 (50.0 to 74.2) | 30.9 (20.2 to 43.3) | 30.3 (15.6 to 48.7) | 43.5 (35.8 to 51.3) | 86.6 (76.0 to 93.7) | 76.2 (63.8 to 86.0) | 54.3 (36.6 to 71.2) | 75.8 (68.5 to 82.1) |
| GMT value (95% CI) | 83.4 (53.8 to 129.2) | 21.1 (13.0 to 34.2) | 22.7 (12.5 to 41.40 | 37.0 (27.5 to 49.9) | 456.9 (320.1 to 652.1) | 133.2 (83.9 to 211.4) | 43.1 (23.3 to 79.8) | 173.0 (129.1 to 231.7) |
| Factor increase in GMT value (95% CI) | 12.1 (7.8 to 18.8) | 4.1 (2.6 to 6.4) | 4.5 (2.7 to 7.3) | 6.4 (4.9 to 8.4) | 66.0 (42.9 to 101.5) | 23.5 (15.0 to 36.9) | 8.2 (4.5 to 14.8) | 28.6 (21.3 to 38.4) |
| Day 21 | ||||||||
| No. of subjects | 70 | 67 | 34 | 171 | 67 | 66 | 37 | 170 |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 71.4 (59.4 to 81.6) | 38.8 (27.1 to 51.5) | 32.4 (17.4 to 50.5) | 50.9 (43.1 to 58.6) | 94.0 (85.4 to 98.4) | 77.3 (65.3 to 86.7) | 51.4 (34.4 to 68.1) | 78.2 (71.3 to 84.2) |
| Subjects with seroconversion: % (95% CI) | 62.9 (50.5 to 74.1) | 35.8 (24.5 to 48.5) | 29.4 (15.1 to 47.5) | 45.6 (38.0 to 53.4) | 88.1 (77.8 to 94.7) | 74.2 (62.0 to 84.2) | 51.4 (34.4 to 68.1) | 74.7 (67.5 to 91.0) |
| GMT value (95% CI) | 91.9 (61.2 to 138.0) | 23.9 (14.5 to 39.3) | 20.7 (11.8 to 36.2) | 40.3 (30.1 to 54.0) | 416.3 (296.4 to 584.7) | 115.9 (75.3 to 178.4) | 41.3 (22.5 to 75.8) | 153.2 (115.7 to 203.0) |
| Factor increase in GMT value (95% CI) | 13.5 (8.9 to 20.6) | 4.6 (2.9 to 7.3) | 4.1 (2.5 to 6.7) | 7.0 (5.3 to 9.2) | 60.1 (39.6 to 91.3) | 20.2 (13.3 to 30.5) | 7.9 (4.4 to 14.1) | 25.3 (19.1 to 33.6) |
| Day 28 | ||||||||
| No. of subjects | 69 | 66 | 33 | 168 | 64 | 63 | 36 | 163 |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 75.4 (63.5 to 85.0) | 43.9 (31.7 to 56.7) | 33.3 (18.0 to 51.8) | 54.8 (46.9 to 62.4) | 100 (94.4 to 100) | 90.5 (80.4 to 96.4) | 72.2 (54.8 to 85.8) | 90.2 (84.6 to 94.3) |
| Subjects with seroconversion: % (95% CI) | 66.7 (54.3 to 77.6) | 39.4 (27.6 to 52.2) | 33.3 (18.0 to 51.8) | 49.4 (41.6 to 57.2) | 93.8 (84.8 to 98.3) | 87.3 (76.5 to 94.4) | 69.4 (51.9 to 83.7) | 85.9 (79.6 to 90.8) |
| GMT value (95% CI) | 119.6 (80.2 to 178.3) | 29.8 (18.8 to 47.3) | 19.2 (10.4 to 35.5) | 48.4 (36.1 to 64.8) | 639.3 (511.2 to 799.6) | 194.3 (139.2 to 271.3) | 70.5 (46.2 to 107.5) | 247.9 (198.7 to 309.4) |
| Factor increase in GMT value (95% CI) | 17.5 (11.5 to 26.6) | 5.7 (3.7 to 8.8) | 3.8 (2.2 to 6.4) | 8.3 (6.3 to 11.0) | 90.0 (62.4 to 130.0) | 33.2 (23.2 to 47.6) | 13.4 (8.6 to 21.1) | 40.2 (31.4 to 51.5) |
| Day 35 | ||||||||
| No. of subjects | 67 | 64 | 34 | 165 | 63 | 65 | 36 | 164 |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 77.6 (65.8 to 86.9) | 45.3 (32.8 to 58.3) | 47.1 (29.8 to 64.9) | 58.8 (50.9 to 66.4) | 100 (94.3 to 100) | 90.8 (81.0 to 96.5) | 80.6 (64.0 to 91.8) | 92.1 (86.8 to 95.7) |
| Subjects with seroconversion: % (95% CI) | 71.6 (59.3 to 82.0) | 40.6 (28.5 to 53.6) | 44.1 (27.2 to 62.1) | 53.9 (46.0 to 61.7) | 95.2 (86.7 to 99.0) | 87.7 (77.2 to 94.5) | 77.8 (60.8 to 89.9) | 88.4 (82.5 to 92.9) |
| GMT value (95% CI) | 116.1 (78.4 to 172.0) | 33.3 (21.0 to 52.7) | 22.0 (12.2 to 39.8) | 50.8 (38.2 to 67.4) | 631.0 (515.5 to 772.5) | 186.9 (134.7 to 259.3) | 86.3 (56.6 to 131.5) | 251.7 (203.7 to 310.9) |
| Factor increase in GMT value (95% CI) | 18.4 (12.2 to 27.8) | 6.3 (4.1 to 9.8) | 4.3 (2.6 to 7.3) | 9.0 (6.9 to 11.9) | 94.1 (66.3 to 133.5) | 32.3 (22.8 to 45.8) | 16.4 (10.3 to 26.2) | 42.0 (33.1 to 53.3) |
| Day 42 | ||||||||
| No. of subjects | 67 | 66 | 33 | 166 | 64 | 65 | 37 | 166 |
| Subjects with HI titre ≥ 1 : 40: % (95% CI) | 73.1 (60.9 to 83.2) | 43.9 (31.7 to 56.7) | 36.4 (20.4 to 54.9) | 54.2 (46.3 to 62.0) | 100 (94.4 to 100) | 90.8 (81.0 to 96.5) | 75.7 (58.8 to 88.2) | 91.0 (85.5 to 94.9) |
| Subjects with seroconversion: % (95% CI) | 65.7 (53.1 to 76.8) | 40.9 (29.0 to 53.7) | 33.3 (18.0 to 51.8) | 49.4 (41.6 to 57.3) | 93.8 (84.8 to 98.3) | 87.7 (77.2 to 94.5) | 72.9 (55.9 to 86.2) | 86.7 (80.6 to 91.5) |
| GMT value (95% CI) | 113(74.9 to 171.4) | 29.8(18.9 to 46.9) | 18.3(10.3 to 32.7) | 46.4(34.7 to 62.0) | 540.5(438.0 to 667.1) | 156.0(110.7 to 220.0) | 77.9(51.0 to 119.0) | 215.8(174.0 to 267.6) |
| Factor increase in GMT value: 95% CI | 17.1 (11.1 to 26.4) | 5.9 (3.9 to 9.0) | 3.6 (2.2 to 6.0) | 8.2 (6.2 to 10.8) | 81.2 (57.4 to 115.0) | 27.0 (18.7 to 38.9) | 15.0 (9.5 to 23.7) | 36.2 (28.5 to 46.0) |
Appendix 6 Immune responses after the first and second dose of H1N1 vaccine in subjects without baseline antibody, as measured on microneutralisation
| Immunogenicity end point | WV vaccine | AS03A–adjuvanted split–virion vaccine | ||||||
|---|---|---|---|---|---|---|---|---|
| 18–44 years | 45–64 years | ≥ 65 years | All subjects | 18–44 years | 45–64 years | ≥ 65 years | All subjects | |
| Baseline | ||||||||
| No. of subjects | 47 | 53 | 19 | 119 | 51 | 51 | 23 | 125 |
| GMT value (95% CI) | 5.0 (5.0 to 5.0) | 5.0 (5.0 to 5.0) | 5.0 (5.0 to 5.0) | 5.0 (5.0 to 5.0) | 5.0 (5.0 to 5.0) | 5.0 (5.0 to 5.0) | 5.0 (5.0 to 5.0) | 5.0 (5.0 to 5.0) |
| After first dose | ||||||||
| Day 7 | ||||||||
| No. of subjects | 46 | 52 | 19 | 117 | 49 | 48 | 23 | 120 |
| Subjects with MN titre ≥ 1 : 40: % (95% CI) | 50.0 (34.9 to 65.1) | 11.5 (4.4 to 23.4) | 15.8 (3.4 to 39.6) | 27.4 (19.5 to 36.4) | 75.5 (61.1 to 86.7) | 41.7 (27.6 to 56.8) | 17.4 (5.0 to 38.8) | 50.8 (41.6 to 60.1) |
| GMT value (95% CI) | 39.0 (21.0 to 72.4) | 12.2 (8.2 to 18.2) | 12.6 (7.6 to 20.7) | 19.4 (14.1 to 26.7) | 170.5 (97.5 to 298.3) | 32.3 (18.9 to 55.0) | 15.8 (8.8 to 28.5) | 55.6 (38.5 to 80.3) |
| Day 14 | ||||||||
| No. of subjects | 45 | 53 | 18 | 116 | 48 | 47 | 22 | 117 |
| Subjects with MN titre ≥ 1 : 40: % (95% CI) | 80.0 (65.4 to 90.4) | 39.6 (26.5 to 54.0) | 33.3 (13.3 to 59.0) | 54.3 (44.8 to 63.6) | 95.8 (85.7 to 99.5) | 85.1 (71.7 to 93.8) | 50.0 (28.2 to 71.8) | 82.9 (74.8 to 89.2) |
| GMT value (95% CI) | 185.5 (113.3 to 303.6) | 32.4 (19.1 to 55.1) | 29.7 (11.6 to 75.9) | 62.9 (43.6 to 90.8) | 439.9 (343.0 to 564.2) | 243.9 (158.0 to 376.5) | 64.1 (27.4 to 149.8) | 241.7 (182.9 to 319.2) |
| Day 21 | ||||||||
| No. of subjects | 47 | 52 | 19 | 118 | 48 | 49 | 23 | 120 |
| Subjects with MN titre ≥ 1 : 40: % (95% CI) | 80.9 (66.7 to 90.9) | 44.2 (30.5 to 58.7) | 31.6 (12.6 to 56.6) | 56.8 (47.3 to 65.9) | 97.9 (88.9 to 99.9) | 85.7 (72.8 to 94.1) | 52.2 (30.6 to 73.2) | 84.2 (76.4 to 90.2) |
| GMT value (95% CI) | 169.0 (104.8 to 272.7) | 34.9 (20.4 to 59.5) | 27.3 (11.6 to 64.6) | 62.9 (44.1 to 89.7) | 467.3 (375.3 to 581.8) | 241.6 (161.4 to 361.6) | 72.4 (37.0 to 141.6) | 249.7 (194.4 to 320.8) |
| Day 28 | ||||||||
| No. of subjects | 46 | 51 | 18 | 115 | 45 | 46 | 22 | 113 |
| Subjects with MN titre ≥ 1 : 40: % (95% CI) | 91.3 (79.2 to 97.6) | 64.7 (50.1 to 77.6) | 44.4 (21.5 to 69.2) | 72.2 (63.0 to 80.1) | 100 (92.1 to 100) | 95.7 (85.2 to 99.5) | 95.5 (77.2 to 99.9) | 97.3 (92.4 to 99.4) |
| GMT value (95% CI) | 313.5 (222.7 to 441.1) | 85.8 (53.3 to 138.0) | 49.4 (23.1 to 105.7) | 132.1 (97.7 to 178.6) | 611.1 (556.8 to 670.7) | 483.9 (388.9 to 602.2) | 213.7 (134.6 to 339.1) | 452.9 (391.6 to 523.7) |
| Day 35 | ||||||||
| No. of subjects | 46 | 49 | 19 | 114 | 46 | 49 | 23 | 118 |
| Subjects with MN titre ≥ 1 : 40: % (95% CI) | 93.5 (82.1 to 98.6) | 69.4 (54.6 to 81.7) | 57.9 (33.5 to 79.7) | 77.2 (68.4 to 84.5) | 100 (92.3 to 100) | 93.9 (83.1 to 98.7) | 91.3 (72.0 to 98.9) | 95.8 (90.4 to 98.6) |
| GMT value (95% CI) | 350.7 (250.0 to 492.1) | 95.7 (58.0 to 157.9) | 52.8 (24.1 to 115.6) | 146.3 (107.3 to 199.6) | 640.0 (640.0 to 640.0) | 444.0 (344.0 to 573.1) | 258.1 (169.2 to 393.6) | 460.6 (399.4 to 531.3) |
| Day 42 | ||||||||
| No. of subjects | 45 | 52 | 19 | 116 | 47 | 49 | 23 | 119 |
| Subjects with MN titre ≥ 1 : 40: % (95% CI) | 91.1 (78.8 to 97.5) | 67.3 (52.9 to 79.7) | 57.9 (33.5 to 79.7) | 75.0 (66.1 to 82.6) | 100 (92.5 to 100) | 95.9 (86.0 to 99.5) | 95.7 (78.1 to 99.9) | 97.5 (92.8 to 99.5) |
| GMT value (95% CI) | 303.0 (212.2 to 432.7) | 98.2 (62.7 to 153.8) | 60.6 (28.2 to 130.2) | 140.5 (105.1 to 187.7) | 640.0 (640.0 to 640.0) | 460.2 (358.6 to 590.5) | 217.5 (129.2 to 366.1) | 453.5 (388.4 to 529.6) |
Appendix 7 Immune responses after the first and second dose of H1N1 vaccine in subjects with baseline antibody, as measured on microneutralisation
| Immunogenicity End Point | WV vaccine | AS03A-adjuvanted split-virion vaccine | ||||||
|---|---|---|---|---|---|---|---|---|
| 18–44 years | 45–64 years | ≥ 65 years | All subjects | 18–44 years | 45–64 years | ≥ 65 years | All subjects | |
| Baseline | ||||||||
| No. of subjects | 23 | 15 | 15 | 53 | 19 | 17 | 14 | 50 |
| GMT value (95% CI) | 186.5 (107.7 to 323.0) | 86.9 (36.5 to 207.0) | 23.1 (12.9 to 41.4) | 83.2 (54.2 to 127.6) | 103.5 (49.7 to 215.5) | 67.8 (31.8 to 144.3) | 36.8 (18.8 to 72.2) | 67.1 (44.5 to 101.2) |
| Subjects with MN titre ≥ 1 : 40: % (95% CI) | 91.3 (72.0 to 98.9) | 66.7 (38.4 to 88.2) | 13.3 (1.7 to 40.5) | 62.3 (47.9 to 75.2) | 57.9 (33.5 to 79.7) | 52.9 (27.8 to 77.0) | 35.7 (12.8 to 64.9) | 50.0 (35.5 to 64.5) |
| After first dose | ||||||||
| Day 7 | ||||||||
| No. of subjects | 23 | 15 | 15 | 53 | 19 | 17 | 14 | 50 |
| Subjects with MN titre ≥ 1 : 40: % (95% CI) | 100 (85.2 to 100) | 80.0 (51.9 to 95.7) | 40.0 (16.3 to 67.7) | 77.4 (63.8 to 87.7) | 100 (82.4 to 100) | 88.2 (63.6 to 98.5) | 57.1 (28.9 to 82.3) | 84.0 (70.9 to 92.8) |
| Subjects with seroconversion: % (95% CI) | 43.5 (23.2 to 65.5) | 20.0 (4.3 to 48.1) | 20.0 (4.3 to 48.1) | 30.2 (18.3 to 44.3) | 47.4 (24.4 to 71.1) | 52.9 (27.8 to 77.0) | 8.6 (8.4 to 58.1) | 44.0 (30.0 to 58.7) |
| GMT value (95% CI) | 511.7 (409.5 to 639.4) | 180.7 (76.5 to 426.8) | 48.0 (24.0 to 96.0) | 195.1 (130.0 to 292.6) | 433.4 (307.8 to 610.3) | 327.1 (157.5 to 679.3) | 98.1 (39.9 to 241.0) | 259.8 (176.0 to 383.6) |
| Day 14 | ||||||||
| No. of subjects | 22 | 15 | 15 | 52 | 19 | 16 | 13 | 48 |
| Subjects with MN titre ≥ 1 : 40: % (95% CI) | 100 (84.6 to 100) | 93.3 (68.1 to 99.8) | 66.7 (38.4 to 88.2) | 88.5 (76.6 to 95.6) | 100 (82.4 to 100) | 100 (79.4 to 100) | 84.6 (54.6 to 98.1) | 95.8 (85.7 to 99.5) |
| Subjects with seroconversion: % (95% CI) | 45.5 (24.4 to 67.8) | 33.3 (11.8 to 61.6) | 40.0 (16.3 to 67.7) | 40.4 (27.0 to 54.9) | 57.9 (33.5 to 79.7) | 68.8 (41.3 to 89.0) | 61.5 (31.6 to 86.1) | 62.5 (47.4 to 76.0) |
| GMT value (95% CI) | 640.0 (640.0 to 640.0) | 317.1 (174.1 to 577.5) | 101.2 (40.1 to 255.9) | 307.0 (213.9 to 440.8) | 640.0 (640.0 to 640.0) | 523.6 (384.2 to 713.6) | 216.9 (96.7 to 486.7) | 446.5 (346.1 to 576.1) |
| Day 21 | ||||||||
| No. of subjects | 23 | 15 | 15 | 53 | 19 | 17 | 14 | 50 |
| Subjects with MN titre ≥ 1 : 40: % (95% CI) | 100 (85.2 to 100) | 100 (78.2 to 100) | 73.3 (44.9 to 92.2) | 92.5 (81.8 to 97.9) | 100 (82.4 to 100) | 100 (80.5 to 100) | 85.7 (57.2 to 98.2) | 96.0 (86.3 to 99.5) |
| Subjects with seroconversion: % (95% CI) | 47.8 (26.8 to 69.4) | 40.0 (16.3 to 67.7) | 53.3 (26.6 to 78.7) | 47.2 (33.3 to 61.4) | 57.9 (33.5 to 79.7) | 64.7 (38.3 to 85.8) | 50.0 (23.0 to 77.0) | 58.0 (43.2 to 71.8) |
| GMT value (95% CI) | 594.0 (532.8 to 662.3) | 383.2 (230.2 to 637.7) | 116.2 (49.3 to 273.9) | 330.7 (239.4 to 456.7) | 640.0 (640.0 to 640.0) | 493.8 (353.4 to 689.8) | 185.7 (82.6 to 417.1) | 414.4 (315.1 to 544.8) |
| Day 28 | ||||||||
| No. of subjects | 23 | 15 | 14 | 52 | 19 | 17 | 14 | 50 |
| Subjects with MN titre ≥ 1 : 40: % (95% CI) | 100 (85.2 to 100) | 100 (78.2 to 100) | 78.6 (49 to 2 to 95.3) | 94.2 (84.1 to 98.8) | 100 (82.4 to 100) | 100 (80.5 to 100) | 92.9 (66.1 to 99.8) | 98.0 (89.4 to 99.9) |
| Subjects with seroconversion: % (95% CI) | 47.8 (26.8 to 69.4) | 46.7 (21.3 to 73.4) | 57.1 (28.9 to 82.3) | 50.0 (35.8 to 64.2) | 57.9 (33.5 to 79.7) | 70.6 (44.0 to 89.7) | 71.4 (41.9 to 91.6) | 66.0 (51.2 to 78.8) |
| GMT value (95% CI) | 640.0 (640.0 to 640.0) | 397.6 (252.9 to 625.3) | 102.2 (42.3 to 247.4) | 340.5 (246.0 to 471.3) | 622.6 (587.5 to 659.7) | 568.4 (476.8 to 677.7) | 282.4 (138.2 to 577.0) | 483.7 (390.6 to 599.1) |
| Day 35 | ||||||||
| No. of subjects | 21 | 15 | 15 | 51 | 17 | 17 | 13 | 47 |
| Subjects with MN titre ≥ 1 : 40: % (95% CI) | 100 (83.9 to 100) | 100 (78.2 to 100) | 66.7 (38.4 to 88.2) | 90.2 (78.6 to 96.7) | 100 (80.5 to 100) | 100 (80.5 to 100) | 92.3 (64.0 to 99.8) | 97.9 (88.7-99.9) |
| Subjects with seroconversion: % (95% CI) | 52.4 (29.8 to 74.3) | 53.3 (26.6 to 78.7) | 46.7 (21.3 to 73.4) | (51.0) (36.6 to 65.2) | 58.8 (32.9 to 81.6) | 70.6 (44.0 to 89.7) | 76.9 (46.2 to 95.0) | 68.1 (52.9 to 80.9) |
| GMT value (95% CI) | 604.5 (536.6 to 680.9) | 453.9 (301.2 to 684.0) | 96.6 (41.7 to 223.8) | 324.0 (230.6 to 455.3) | 640.0 (640.0 to 640.0) | 607.9 (545.1 to 677.9) | 371.8 (196.5 to 703.7) | 540.6 (453.1 to 645.0) |
| Day 42 | ||||||||
| No. of subjects | 22 | 14 | 14 | 50 | 17 | 17 | 14 | 48 |
| Subjects with MN titre ≥ 1 : 40: % (95% CI) | 100 (84.6 to 100) | 100 (76.8 to 100) | 71.4 (41.9 to 91.6) | 92.0 (80.8 to 97.8) | 100 (80.5 to 100) | 100 (80.5 to 100) | 92.9 (66.1 to 99.8) | 97.9 (88.9 to 99.9) |
| Subjects with seroconversion: % (95% CI) | 100 (84.6 to 100) | 100.0 (76.8 to 100) | 71.4 (41.9 to 91.6) | 92.0 (80.8 to 97.8) | 100 (80.5 to 100) | 100 (80.5 to 100) | 92.9 (66.1 to 99.8) | 97.9 (88.9 to 99.9) |
| GMT value (95% CI) | 608.4 (547.6 to 676.0) | 423.3 (253.4 to 707.1) | 89.6 (36.4 to 220.9) | 321.5 (225.6 to 458.1) | 640.0 (640.0 to 640.0) | 582.7 (508.4 to 667.8) | 359.0 (194.2 to 663.5) | 523.0 (435.4 to 628.2) |
Appendix 8 Immune responses after the first and second dose of H1N1 vaccine in subjects with and without baseline antibody, as measured on microneutralisation
| Immunogenicity end point | WV vaccine | AS03A–adjuvanted split–virion vaccine | ||||||
|---|---|---|---|---|---|---|---|---|
| 18–44 years | 45–64 years | ≥ 65 years | All subjects | 18–44 years | 45–64 years | ≥ 65 years | All subjects | |
| Baseline | ||||||||
| No. of subjects | 70 | 68 | 34 | 172 | 70 | 68 | 37 | 175 |
| GMT value (95% CI) | 16.4 (10.5 to 25.6) | 9.4 (6.7 to 13.1) | 9.8 (6.8 to 14.1) | 11.9 (9.4 to 15.0) | 11.4 (7.8 to 16.5) | 9.6 (6.9 to 13.3) | 10.6 (7.1 to 15.9) | 10.5 (8.5 to 12.9) |
| Subjects with MN titre ≥ 1 : 40: % (95% CI) | 30.0 (19.6 to 42.1) | 14.7 (7.3 to 25.4) | 5.9 (0.7 to 19.7) | 19.2 (13.6 to 25.9) | 15.7 (8.1 to 26.4) | 13.2 (6.2 to 23.6) | 13.5 (4.5 to 28.8) | 14.3 (9.5 to 20.4) |
| After first dose | ||||||||
| Day 7 | ||||||||
| No. of subjects | 69 | 67 | 34 | 170 | 68 | 65 | 37 | 170 |
| Subjects with MN titre ≥ 1 : 40: % (95% CI) | 66.7 (54.3 to 77.6) | 26.9 (16.8 to 39.1) | 26.5 (12.9 to 44.4) | 42.9 (35.4 to 50.7) | 82.4 (71.2 to 90.5) | 53.8 (41.0 to 66.3) | 32.4 (18.0 to 49.8) | 60.6 (52.8 to 68.0) |
| Subjects with seroconversion: % (95% CI) | 47.8 (35.6 to 60.2) | 13.4 (6.3 to 24.0) | 17.6 (6.8 to 34.5) | 28.2 (21.6 to 35.6) | 67.6 (55.2 to 78.5) | 44.6 (32.3 to 57.5) | 21.6 (9.8 to 38.2) | 48.8 (41.1 to 56.6) |
| GMT value (95% CI) | 91.9 (55.3 to 152.7) | 22.4 (14.3 to 35.0) | 22.7 (14.4 to 35.8) | 39.8 (29.5 to 53.7) | 221.3 (145.2 to 337.2) | 59.1 (35.9 to 97.2) | 31.6 (18.1 to 55.2) | 87.4 (64.7 to 118.2) |
| Day 14 | ||||||||
| No. of subjects | 67 | 68 | 33 | 168 | 67 | 63 | 35 | 165 |
| Subjects with MN titre ≥ 1 : 40: % (95% CI) | 86.6 (76.0 to 93.7) | 51.5 (39.0 to 63.8) | 48.5 (30.8 to 66.5) | 64.9 (57.2 to 72.1) | 97.0 (89.6 to 99.6) | 88.9 (78.4 to 95.4) | 62.9 (44.9 to 78.5) | 86.7 (80.5 to 91.5) |
| Subjects with seroconversion: % (95% CI) | 68.7 (56.2 to 79.4) | 38.2 (26.7 to 50.8) | 36.4 (20.4 to 54.9) | 50.0 (42.2 to 57.8) | 85.1 (74.3 to 92.6) | 81.0 (69.1 to 89.8) | 54.3 (36.6 to 71.2) | 77.0 (69.8 to 83.2) |
| GMT value (95% CI) | 278.6 (195.0 to 397.9) | 53.6 (33.0 to 87.1) | 51.9 (26.7 to 100.7) | 102.8 (76.4 to 138.3) | 489.2 (408.2 to 586.5) | 296.1 (210.9 to 415.8) | 100.8 (54.3 to 187.3) | 288.9 (233.3 to 357.8) |
| Day 21 | ||||||||
| No. of subjects | 70 | 67 | 34 | 171 | 67 | 66 | 37 | 170 |
| Subjects with MN titre ≥ 1 : 40: % (95% CI) | 87.1 (77.0 to 93.9) | 56.7 (44.0 to 68.8) | 50.0 (32.4 to 67.6) | 67.8 (60.3 to 74.8) | 98.5 (92.0 to 100) | 89.4 (79.4 to 95.6) | 64.9 (47.5 to 79.8) | 87.6 (81.7 to 92.2) |
| Subjects with seroconversion: % (95% CI) | 70.0 (57.9 to 80.4) | 43.3 (31.2 to 56.0) | 41.2 (24.6 to 59.3) | 53.8 (46.0 to 61.4) | 86.6 (76.0 to 93.7) | 80.3 (68.7 to 89.1) | 51.4 (34.4 to 68.1) | 76.5 (69.4 to 82.6) |
| GMT value (95% CI) | 255.5 (180.2 to 362.2) | 59.7 (36.5 to 97.4) | 51.8 (27.5 to 97.4) | 105.2 (78.9 to 140.2) | 510.9 (435.7 to 599.1) | 290.4 (211.6 to 398.6) | 103.4 (61.7 to 173.2) | 289.8 (238.2 to 352.5) |
| Day 28 | ||||||||
| No. of subjects | 69 | 66 | 32 | 167 | 64 | 63 | 36 | 163 |
| Subjects with MN titre ≥ 1 : 40: % (95% CI) | 94.2 (85.8 to 98.4) | 72.7 (60.4 to 83.0) | 59.4 (40.6 to 6.3) | 79.0 (72.1 to 84.9) | 100 (94.4 to 100) | 96.8 (89.0 to 99.6) | 94.4 (81.3 to 99.3) | 97.5 (93.8 to 99.3) |
| Subjects with seroconversion: % (95% CI) | 76.8 (65.1 to 86.1) | 60.6 (47.8 to 72.4) | 50.0 (31.9 to 68.1) | 65.3 (57.5 to 72.5) | 87.5 (76.8 to 94.4) | 88.9 (78.4 to 95.4) | 86.1 (70.5 to 95.3) | 87.7 (81.7 to 92.3) |
| GMT value (95% CI) | 397.7 (313.1 to 505.1) | 121.5 (80.8 to 182.9) | 67.9 (38.9 to 118.7) | 177.4 (139.7 to 25.2) | 614.5 (574.8 to 656.9) | 505.4 (428.5 to 596.0) | 238.1 (163.4 to 347.1) | 462.1 (410.3 to 520.6) |
| Day 35 | ||||||||
| No. of subjects | 67 | 64 | 34 | 165 | 63 | 66 | 36 | 165 |
| Subjects with MN titre ≥ 1 : 40: % (95% CI) | 95.5 (87.5 to 99.1) | 76.6 (64.3 to 86.2) | 61.8 (43.6 to 77.8) | 81.2 (74.4 to 86.9) | 100 (94.3 to 100) | 95.5 (87.3 to 99.1) | 91.7 (77.5 to 98.2) | 96.4 (92.3 to 98.7) |
| Subjects with seroconversion: % (95% CI) | 80.6 (69.1 to 89.2) | 65.6 (52.7 to 77.1) | 52.9 (35.1 to 70.2) | 69.1 (61.4 to 76.0) | 88.9 (78.4 to 95.4) | 87.9 (77.5 to 94.6) | 86.1 (70.5 to 95.3) | 87.9 (81.9 to 92.4) |
| GMT value (95% CI) | 416.0 (327.1 to 529.1) | 137.8 (90.2 to 210.5) | 68.9 (39.6 to 119.8) | 187.1 (146.6 to 238.6) | 640.0 (640.0 to 640.0) | 481.4 (397.1 to 583.6) | 294.4 (209.5 to 413.9) | 482.1 (430.5 to 540.0) |
| Day 42 | ||||||||
| No. of subjects | 67 | 66 | 33 | 166 | 64 | 66 | 37 | 167 |
| Subjects with MN titre ≥ 1 : 40: % (95% CI) | 94.0 (85.4 to 98.3) | 74.2 (62.0 to 84.2) | 63.6 (45.1 to 79.6) | 80.1 (73.2 to 85.9) | 100 (94.4 to 100) | 97.0 (89.5 to 99.6) | 94.6 (81.8 to 99.3) | 97.6 (94.0 to 99.3) |
| Subjects with seroconversion: % (95% CI) | 32.8 (21.8 to 45.4) | 21.2 (12.1 to 33.0) | 30.3 (15.6 to 48.7) | 27.7 (21.1 to 35.2) | 26.6 (16.3 to 39.1) | 25.8 (15.8 to 38.0) | 35.1 (20.2 to 52.5) | 28.1 (21.5 to 35.6) |
| GMT value (95% CI) | 380.9 (296.2 to 489.9) | 133.9 (90.4 to 198.3) | 71.5 (41.1 to 124.6) | 180.3 (142.6 to 227.9) | 640.0 (640.0 to 640.0) | 489.0 (405.2 to 590.2) | 262.9 (178.3 to 387.6) | 472.5 (418.3 to 533.7) |
Appendix 9 Solicited local and systemic adverse effects after each vaccine dose
| Effect | WV vaccine | AS03A-adjuvanted split-virion vaccine | ||||||
|---|---|---|---|---|---|---|---|---|
| 18–44 years (n = 70) | 45–64 years (n = 68) | ≥ 65 years (n = 34) | All subjects (n = 172) | 18–44 years (n = 70) | 45–64 years (n = 67) | ≥ 65 years (n = 37) | All subjects (n = 174) | |
| Local reaction [% (95% CI)] | ||||||||
| Pain at injection siteb | ||||||||
| None | 73 (61 to 82) | 85 (75 to 92) | 82 (66 to 92) | 80 (73 to 85) | 14 (8 to 25) | 34 (24 to 46) | 62 (46 to 76) | 32 (26 to 40) |
| Mild | 24 (16 to 36) | 15 (8 to 25) | 15 (6 to 31) | 19 (13 to 25) | 56 (44 to 67) | 49 (37 to 61) | 38 (24 to 54) | 49 (42 to 57) |
| Moderate | 3 (1 to 11) | 0 | 3 (0 to 18) | 2 (1 to 5) | 29 (19 to 40) | 16 (9 to 27) | 0 | 18 (13 to 24) |
| Severe | 0 | 0 | 0 | 0 | 1 (0 to 10) | 0 | 0 | 1 (0 to 4) |
| Redness diameterb | ||||||||
| 0 mm | 89 (79 to 94) | 1 (82 to 96) | 88 (72 to 96) | 90 (84 to 93) | 71 (59 to 81) | 81 (69 to 88) | 89 (74 to 96) | 79 (72 to 84) |
| 1–4 mm | 10 (5 to 20) | 3 (1 to 11) | 9 (3 to 24) | 7 (4 to 12) | 17 (10 to 28) | 6 (2 to 15) | 11 (4 to 26) | 12 (8 to 17) |
| ≥ 5 mm | 1 (0 to 10) | 6 (2 to 15) | 3 (0 to 18) | 3 (2 to 8) | 12 (6 to 22) | 13 (7 to 24) | 0 | 10 (6 to 15) |
| Swelling diameterb | ||||||||
| 0 mm | 96 (87 to 99) | 97 (89 to 99) | 97 (82 to 100) | 97 (92 to 98) | 76 (64 to 84) | 84 (73 to 91) | 92 (78 to 97) | 82 (76 to 87) |
| 1–4 mm | 3 (1 to 11) | 0 | 3 (0 to 18) | 2 (1 to 5) | 10 (5 to 20) | 4 (1 to 13) | 5 (1 to 19) | 7 (4 to 12) |
| ≥ 5 mm | 1 (0 to 10) | 3 (1 to 11) | 0 | 2 (1 to 5) | 14 (8 to 25) | 12 (6 to 22) | 3 (0 to 17) | 11 (7 to 17) |
| Bruising diameter | ||||||||
| 0 mm | 94 (86 to 98) | 100 | 91 (76 to 97) | 96 (92 to 98) | 90 (80 to 95) | 94 (85 to 98) | 100 | 94 (89 to 96) |
| 1–4 mm | 3 (1 to 11) | 0 | 0 | 1 (0 to 5) | 6 (2 to 15) | 3 (1 to 11) | 0 | 3 (2 to 8) |
| ≥ 5 mm | 3 (1 to 11) | 0 | 9 (3 to 24) | 3 (1 to 7) | 4 (1 to 13) | 3 (1 to 11) | 0 | 3 (1 to 7) |
| Systemic reaction [% (95% CI)] | ||||||||
| Muscle achesc | ||||||||
| None | 86 (75 to 92) | 82 (71 to 90) | 88 (72 to 96) | 85 (79 to 90) | 56 (44 to 67) | 61 (49 to 72) | 78 (62 to 89) | 63 (55 to 70) |
| Mild | 9 (4 to 18) | 15 (8 to 25) | 9 (3 to 24) | 11 (7 to 17) | 23 (14 to 34) | 27 (18 to 39) | 19 (9 to 35) | 24 (18 to 30) |
| Moderate | 6 (2 to 14) | 3 (1 to 11) | 3 (0 to 18) | 4 (2 to 8) | 21 (13 to 33) | 12 (6 to 22) | 3 (0 to 17) | 14 (9 to 20) |
| Severe | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Chillsc | ||||||||
| None | 99 (90 to 100) | 93 (83 to 97) | 94 (79 to 99) | 95 (91 to 98) | 80 (69 to 88) | 82 (71 to 90) | 86 (71 to 94) | 82 (76 to 87) |
| Mild | 1 (0 to 10) | 6 (2 to 15) | 0 | 3 (1 to 7) | 10 (5 to 20) | 12 (6 to 22) | 8 (3 to 22) | 10 (7 to 16) |
| Moderate | 0 | 1 (0 to 10) | 6 (1 to 21) | 2 (1 to 5) | 10 (5 to 20) | 6 (2 to 15) | 3 (0 to 17) | 7 (4 to 12) |
| Severe | 0 | 0 | 0 | 0 | 0 | 0 | 3 (0 to 17) | 1 (0 to 4) |
| Malaised | ||||||||
| None | 93 (84 to 97) | 87 (76 to 93) | 91 (76 to 97) | 90 (85 to 94) | 79 (68 to 87) | 79 (68 to 87) | 100 | 82 (76 to 87) |
| Mild | 7 (3 to 16) | 9 (4 to 18) | 3 (0 to 18) | 7 (4 to 12) | 20 (12 to 31) | 13 (7 to 24) | 0 | 13 (9 to 19) |
| Moderate | 0 | 4 (1 to 13) | 6 (1 to 21) | 3(1 to 7) | 4 (1 to 13) | 7 (3 to 17) | 0 | 5 (2 to 9) |
| Severe | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Headache | ||||||||
| None | 79 (67 to 87) | 68 (56 to 78) | 82 (66 to 92) | 75 (68 to 81) | 59 (47 to 70) | 69 (57 to 79) | 92 (78 to 97) | 70 (62 to 76) |
| Mild | 14 (8 to 25) | 19 (11 to 30) | 12 (4 to 28) | 16 (11 to 22) | 26 (17 to 37) | 21 (13 to 32) | 5 (1 to 19) | 20 (14 to 26) |
| Moderate | 7 (3 to 16) | 13 (7 to 24) | 6 (1 to 21) | 9 (6 to 15) | 16 (9 to 26) | 9 (4 to 19) | 3 (0 to 17) | 10 (7 to 16) |
| Severe | 0 | 0 | 0 | 0 | 0 | 1 (0 to 10) | 0 | 1 (0 to 4) |
| Nausea | ||||||||
| None | 96 (87 to 99) | 94 (85 to 98) | 94 (79 to 99) | 95 (90 to 97) | 86 (75 to 92) | 87 (76 to 93)) | 97 (83 to 100) | 89 (83 to 92) |
| Mild | 1 (0 to 10) | 4 (1 to 13) | 3 (0 to 18) | 3 (1 to 7) | 10 (5 to 20) | 12 (6 to 22) | 3 (0 to 17) | 9 (6 to 15) |
| Moderate | 3 (1 to 11) | 1 (0 to 10) | 3 (0 to 18) | 2 (1 to 6) | 4 (1 to 13) | 1 (0 to 10) | 0 | 2 (1 to 6) |
| Severe | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Fever – temperature ≥ 38°C | 3 (1 to 11) | 0 | 3 (0 to 18) | 2 (1 to 5) | 0 | 1 (0 to 10) | 0 | 1 (0 to 4) |
| Use of analgesic | 21 (13 to 33) | 0 | 0 | 21 (13 to 33) | 24 (16 to 36) | 0 | 0 | 24 (16 to 36) |
| Effect | WV vaccine | AS03A-adjuvanted split-virion vaccine | ||||||
|---|---|---|---|---|---|---|---|---|
| 18–44 years (n = 70) | 45–64 years (n = 68) | ≥ 65 years (n = 34) | All subjects (n = 172) | 18–44 years (n = 70) | 45–64 years (n = 67) | ≥ 65 years (n = 37) | All subjects (n = 174) | |
| Local reaction [% (95% CI)] | ||||||||
| Pain at injection siteb | ||||||||
| None | 72 (61 to 82) | 80 (68 to 88) | 94 (79 to 99) | 80 (73 to 85) | 27 (18 to 39) | 39 (28 to 51) | 69 (53 to 82) | 41 (34 to 49) |
| Mild | 22 (14 to 33) | 17 (10 to 28) | 3 (0 to 18) | 16 (11 to 22) | 55 (42 to 66) | 52 (39 to 64) | 28 (16 to 44) | 48 (40 to 55) |
| Moderate | 6 (2 to 15) | 3 (1 to 12) | 3 (0 to 18) | 4 (2 to 9) | 14 (7 to 24) | 9 (4 to 19) | 3 (0 to 17) | 10 (6 to 15) |
| Severe | 0 | 0 | 0 | 0 | 5 (1 to 13) | 0 | 0 | 2 (1 to 5) |
| Redness diameterb | ||||||||
| 0 mm | 91 (82 to 96) | 92 (83 to 97) | 100 | 93 (89 to 96) | 73 (61 to 82) | 77 (65 to 85) | 86 (71 to 94) | 77 (70 to 83) |
| 1–4 mm | 9 (4 to 18) | 2 (0 to 10) | 0 | 4 (2 to 9) | 18 (11 to 29) | 8 (3 to 18) | 3 (0 to 17) | 11 (7 to 17) |
| ≥ 5 mm | 0 | 6 (2 to 15) | 0 | 2 (1 to 6) | 9 (4 to 19) | 16 (9 to 27) | 11 (4 to 26) | 12 (8 to 18) |
| Swelling diameterb | ||||||||
| 0 mm | 94 (85 to 98) | 98 (90 to 100) | 100 | 97 (93 to 99) | 71 (59 to 81) | 80 (68 to 88) | 83 (67 to 92) | 77 (70 to 83) |
| 1–4 mm | 6 (2 to 15) | 0 | 0 | 2 (1 to 6) | 17 (9 to 28) | 11 (5 to 21) | 6 (1 to 20) | 12 (8 to 18) |
| ≥ 5 mm | 0 | 2 (0 to 10) | 0 | 1 (0 to 4) | 12 (6 to 22) | 9 (4 to 19) | 11 (4 to 26) | 11 (7 to 17) |
| Bruising diameter | ||||||||
| 0 mm | 96 (87 to 99) | 95 (87 to 99) | 100 | 96 (92 to 98) | 92 (83 to 87) | 94 (84 to 98) | 100 | 95 (90 to 97) |
| 1–4 mm | 1 (0 to 10) | 0 | 0 | 1 (0 to 4) | 5 (1 to 13) | 3 (1 to 12) | 0 | 3 (1 to 7) |
| ≥ 5 mm | 3 (1 to 11) | 5 (1 to 13) | 0 | 3 (1 to 7) | 3 (1 to 11) | 3 (1 to 12) | 0 | 2 (1 to 6) |
| Systemic reaction [% (95% CI)] | ||||||||
| Muscle achesc | ||||||||
| None | 77 (65 to 85) | 88 (77 to 94) | 85 (69 to 94) | 83 (76 to 88) | 61 (48 to 72) | 70 (58 to 80) | 78 (61 to 89) | 68 (61 to 75) |
| Mild | 13 (7 to 23) | 9 (4 to 19) | 15 (6 to 31) | 12 (8 to 18) | 27 (18 to 39) | 19 (11 to 30) | 22 (11 to 39) | 23 (17 to 30) |
| Moderate | 10 (5 to 20) | 3 (1 to 2) | 0 | 5 (3 to 10) | 11 (5 to 21) | 11 (5 to 21) | 0 | 8 (5 to 14) |
| Severe | 0 | 0 | 0 | 0 | 2 (0 to 10) | 0 | 0 | 1 (0 to 4) |
| Chillsc | ||||||||
| None | 97 (89 to 99) | 95 (87 to 99) | 91 (76 to 97) | 95 (91 to 98) | 77 (66 to 86) | 88 (77 to 94) | 97 (83 to 100) | 86 (79 to 90) |
| Mild | 1 (0 to 10) | 5 (1 to 13) | 9 (3 to 24) | 4 (2 to 9) | 9 (4 to 19) | 9 (4 to 19) | 3 (0 to 17) | 8 (5 to 13) |
| Moderate | 1 (0 to 10) | 0 | 0 | 1 (0 to 4) | 11 (5 to 21) | 3 (1 to 12) | 0 | 5 (3 to 10) |
| Severe | 0 | 0 | 0 | 0 | 3 (1 to 11) | 0 | 0 | 1 (0 to 5) |
| Malaised | ||||||||
| None | 93 (84 to 97) | 88 (77 to 94) | 97 (82 to 100) | 92 (86 to 95) | 80 (69 to 88) | 84 (73 to 91) | 94 (80 to 99) | 85 (79 to 90) |
| Mild | 3 (1 to 11) | 9 (4 to 19) | 3 (0 to 18) | 5 (3 to 10) | 17 (9 to 28) | 11 (5 to 21) | 6 (1 to 20) | 12 (8 to 18) |
| Moderate | 4 (1 to 13) | 3 (1 to 12) | 0 | 3 (1 to 7) | 3 (1 to 11) | 5 (2 to 14) | 0 | 3 (1 to 7) |
| Severe | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Headache | ||||||||
| None | 77 (65 to 85) | 71 (59 to 81) | 82 (66 to 92) | 76 (68 to 82) | 65 (53 to 76) | 67 (55 to 78) | 92 (77 to 97) | 72 (64 to 78) |
| Mild | 19 (11 to 30) | 22 (13 to 33) | 12 (4 to 28) | 18 (13 to 25) | 23 (14 to 34) | 23 (15 to 35) | 6 (1 to 20) | 19 (14 to 26) |
| Moderate | 3 (1 to 11) | 6 (2 to 15) | 6 (1 to 21) | 5 (2 to 9) | 12 (6 to 22) | 8 (3 to 18) | 3 (0 to 17) | 8 (5 to 14) |
| Severe | 1 (0 to 10) | 2 (0 to 10) | 0 | 1 (0 to 5) | 0 | 2 (0 to 10) | 0 | 1 (0 to 4) |
| Nausea | ||||||||
| None | 86 (75 to 92) | 92 (83 to 97) | 91 (76 to 97) | 89 (84 to 93) | 82 (71 to 89) | 92 (82 to 97) | 94 (80 to 99) | 89 (83 to 93) |
| Mild | 10 (5 to 20) | 5 (1 to 13) | 6 (1 to 21) | 7 (4 to 12) | 12 (6 to 22) | 6 (2 to 16) | 6 (1 to 20) | 8 (5 to 14) |
| Moderate | 4 (1 to 13) | 3 (1 to 12) | 0 | 3 (1 to 7) | 6 (2 to 15) | 2 (0 to 10) | 0 | 3 (1 to 7) |
| Severe | 0 | 0 | 3 (0 to 18) | 1 (0 to 4) | 0 | 0 | 0 | 0 |
| Fever – temperature ≥ 38°C | 0 | 2 (0 to 10) | 0 | 1 (0 to 4) | 5 (2 to 14) | 2 (0 to 11) | 0 | 2 (1 to 6) |
| Use of analgesic | 10 (5 to 20) | 18 (10 to 29) | 6 (1 to 21) | 12 (8 to 18) | 24 (15 to 35) | 24 (15 to 35) | 3 (0 to 17) | 19 (14 to 26) |
Appendix 10 Unsolicited adverse events
| Percentage | n | |
|---|---|---|
| People with any unsolicited event | 33.1 | 57 |
| No. of unsolicited events | 89 | |
| Events possibly related to vaccinationa | 13.5 | 12 |
| General disorders and administration site conditions | ||
| ILI | 1.2 | 2a |
| Lethargy | 0.6 | 1a |
| Injection site pruritus | 0.6 | 1a |
| Nervous system disorders | ||
| Paraesthesiae | 1.2 | 2a |
| Migraine | 1.2 | 2 |
| Motion sickness | 0.6 | 1 |
| Musculoskeletal disorders | ||
| Musculoskeletal pain | 4.1 | 7 |
| Arthralgia | 1.2 | 2 |
| Pain in extremity | 0.6 | 1 |
| Discomfort in extremity | 0.6 | 1a |
| Gastrointestinal disorders | ||
| Diarrhoea | 1.2 | 2 |
| Epigastric discomfort | 0.6 | 1 |
| Dyspepsia | 0.6 | 1 |
| Skin and subcutaneous conditions: | ||
| Rash | 1.7 | 3a |
| Folliculitis | 0.6 | 1 |
| Respiratory disorders: | ||
| Cough | 5.2 | 9 |
| Rhinorrhoea | 2.3 | 4 |
| Sneezing | 0.6 | 1 |
| Nasal stuffiness | 0.6 | 1 |
| Nasal irritation | 0.6 | 1 |
| Chest congestion | 0.6 | 1 |
| Ear and labyrinth disorders | ||
| Vertigo | 0.6 | 1a |
| Ear pain | 0.6 | 1 |
| Metabolism and nutrition disorders | ||
| Decreased appetite | 0.6 | 1 |
| Psychiatric disorders | ||
| Insomnia | 0.6 | 1 |
| Confusional state | 0.6 | 1a |
| Depression | 0.6 | 1 |
| Infections | ||
| Pharyngitis | 11.6 | 20 |
| Common cold | 5.2 | 9 |
| Sinusitis | 1.7 | 3 |
| Toothache | 1.2 | 2 |
| Herpes labialis | 0.6 | 1 |
| Injury and procedural disorders | ||
| Wound infection | 0.6 | 1 |
| Outer ear injury | 0.6 | 1 |
| Obstetric and gynaecological | ||
| Dysmenorrhoea | 0.6 | 1 |
| Percentage | n | |
|---|---|---|
| People with any unsolicited event | 23.6 | 41 |
| No. unsolicited events | 52 | |
| Events possibly related to vaccinationa | 36.5 | 19 |
| General disorders and administration site conditions | ||
| ILI | 0.6 | 1a |
| Lethargy | 0.6 | 1a |
| Injection site pruritus | 0.6 | 1a |
| Injection site stiffness | 0.6 | 1a |
| Rigors | 0.6 | 1a |
| Poor sleep due to administration site discomfort | 0.6 | 1a |
| Nervous system disorders | ||
| Paraesthesiae | 1.1 | 2a |
| Migraine | 1.1 | 2 |
| Dizziness | 1.7 | 3a |
| Musculoskeletal disorders | ||
| Back pain | 1.1 | 2a |
| Musculoskeletal pain | 1.1 | 2a |
| Pain in extremity | 0.6 | 1 |
| Gastrointestinal disorders | ||
| Abdominal pain | 0.6 | 1 |
| Abdominal discomfort | 0.6 | 1 |
| Dyspepsia | 0.6 | 1 |
| Skin and subcutaneous conditions: | ||
| Pruritic rash | 1.1 | 2a |
| Rash | 0.6 | 1 |
| Pruritus | 0.6 | 1a |
| Eczema | 0.6 | 1 |
| Respiratory disorders | ||
| Cough | 2.9 | 5 |
| Productive cough | 1.1 | 2 |
| Rhinorrhoea | 1.1 | 2 |
| Dyspnoea | 0.6 | 1 |
| Tachypnoea | 0.6 | 1 |
| Infections | ||
| Pharyngitis | 3.4 | 6 |
| Common cold | 1.7 | 3 |
| Surgical site abscess | 0.6 | 1 |
| Toothache | 0.6 | 1 |
| Injury and procedural disorders | ||
| Back injury | 1.1 | 2 |
| Blood and lymphatic system disorders | ||
| Axillary tenderness | 0.6 | 1a |
| Lymphadenopathy | 0.6 | 1 |
Appendix 11 Protocol
Title
A randomised, partially observer-blind, multicentre, head-to-head comparison of a two-dose regimen of Baxter and GSK H1N1 pandemic vaccines, administered 21 days apart
Short title: Head-to-head study of influenza H1N1 vaccines in adults
EudractCT number: 2009-015743-16
CLRN research number 31843
Funder’s number: 09/93/01
Investigator’s research number: NIHRH1
ISRCTN: ISRCTN92328241
Version: 3.0
| Sponsor | University Hospitals of Leicester NHS Trust |
| Trust Headquarters | |
| Gwendolen Road | |
| Leicester LE5 4PW | |
| Chief investigator | Professor Karl G Nicholson |
| Infectious Diseases Unit | |
| Leicester Royal Infirmary | |
| Infirmary Square | |
| Leicester LE1 5WW | |
| Principal investigator (Leicester) | |
|---|---|
| Dr Iain Stephenson | Infectious Diseases Unit |
| HEFCE Senior Lecturer & Hon Consultant In Infectious Diseases | Level 6, Windsor Building |
| Leicester Royal Infirmary | |
| Infirmary Square | |
| Leicester LEI 5WW | |
| Principal investigator (Nottingham) | |
| Dr Wei Shen Lim | David Evans Building, Nottingham University Hospitals |
| Consultant Respiratory Physician | City Hospital Campus |
| Nottingham NG8 2NE | |
| Principal investigator (Sheffield) | |
| Professor Robert Read | Division of Genomic Medicine |
| Professor of Infectious Diseases | Sheffield University Medical School |
| 10 Beech Hill Road | |
| Sheffield S10 2RX | |
| Co-investigator | |
| Professor Keith Abrams | Department of Health Sciences |
| Professor of Medical Statistics | University of Leicester |
| 2nd Floor, Adrian Building | |
| University Road | |
| Leicester LE1 7RH | |
| Co-investigator | |
| Professor Jonathan Nguyen-Van-Tam | Room A40d Clinical Sciences Building |
| Professor of Health Protection | City Hospital |
| Nottingham NG5 1PB | |
| Co-investigator | |
| Professor Maria Zambon | Health Protection Agency, Centre for Infections |
| Director, HPA Centre for Infections | Colindale, 61 Colindale Avenue |
| London NW9 5EQ | |
| Trial manager | |
| Mrs Sally Batham | Infectious Diseases Unit |
| Senior Research Nurse | Level 6, Windsor Building |
| Leicester Royal Infirmary | |
| Infirmary Square | |
| Leicester LEI 5WW | |
Study synopsis
Title of study
A randomised, partially observer-blind, multicentre, head-to-head comparison of a two-dose regimen of Baxter and GSK H1N1 pandemic vaccines, administered 21 days apart.
Objectives
Primary
-
To evaluate the immunogenicity of Baxter cell-culture, non-adjuvanted, whole-virion H1N1 vaccine, and GlaxoSmithKline (GSK) AS03-adjuvanted, split H1N1 vaccine with respect to the EU Committee of Human Medicinal Products (CHMP) and the US Food and Drug Administration (FDA) licensing criteria.
Secondary
-
To identify whether one or two doses of vaccine are required to satisfy the licensing criteria.
-
To examine the short-term reactogenicity of the vaccines.
-
To examine the kinetics of the antibody responses to vaccination.
-
To examine persistence of antibody at 6 months.
and, if appropriate (i.e. an antigenic drift variant emerges prior to the 2010–11 influenza season):
-
To evaluate the breadth of the antibody response to the antigenic variant.
-
To assess cellular responses to influenza haemagglutinin (HA) before and after vaccination in one subset of 18- to -44-year-old subjects.
Design
An observer-blind, multicentre study in which six groups of 60 male and female adults stratified by age (18–44, 45–64, and 65 years and older) will be randomly allocated to receive two 7.5-µg HA doses of cell culture plain (i.e. non-adjuvanted) whole-virion A/California/2009 (H1N1) vaccine, or two doses of AS03-adjuvanted influenza A/California/2009 (H1N1) split-virus vaccine containing 3.75 µg of HA by intramuscular (IM) injection. A second dose of the same vaccine containing the same quantity of antigen as in the first dose will be administered 21 days later. Subjects will be observed for local and systemic reactions for 30 minutes after each immunisation and will be monitored for any reactions and other AEs for 7 days after each immunisation.
Blood for immunogenicity studies will be obtained at day 0 (pre-immunisation), day 7 (± 1 day), day 14 (± 2 days), day 21 (± 2 days), day 28 (± 2 days), day 35 (± 3 days), day 42 (± 3 days) and day 180 (± 10 days). Blood for cellular assays will be taken on day 0, day 21 (± 2 days) and day 42 (± 3 days).
Immunogenicity to influenza viruses will be evaluated by haemagglutination inhibition (HI), virus neutralisation (MN), and possibly single radial haemolysis (SRH) responses. Cellular assays will be by ELISPOT to influenza HA.
Duration
Approximately 6 months per subject. Subjects will screened to ensure entry criteria are met and then vaccinated. After a second dose of vaccine on day 21, subjects will be followed up for an additional 159 days.
Start date
The study is planned to commence early September 2009.
Setting
This multicentre study will be conducted in University Hospitals of Leicester, Nottingham, and Sheffield, and possibly in GP surgeries in Leicestershire, Nottinghamshire, Yorkshire and Derbyshire.
Study schedule: flow diagram
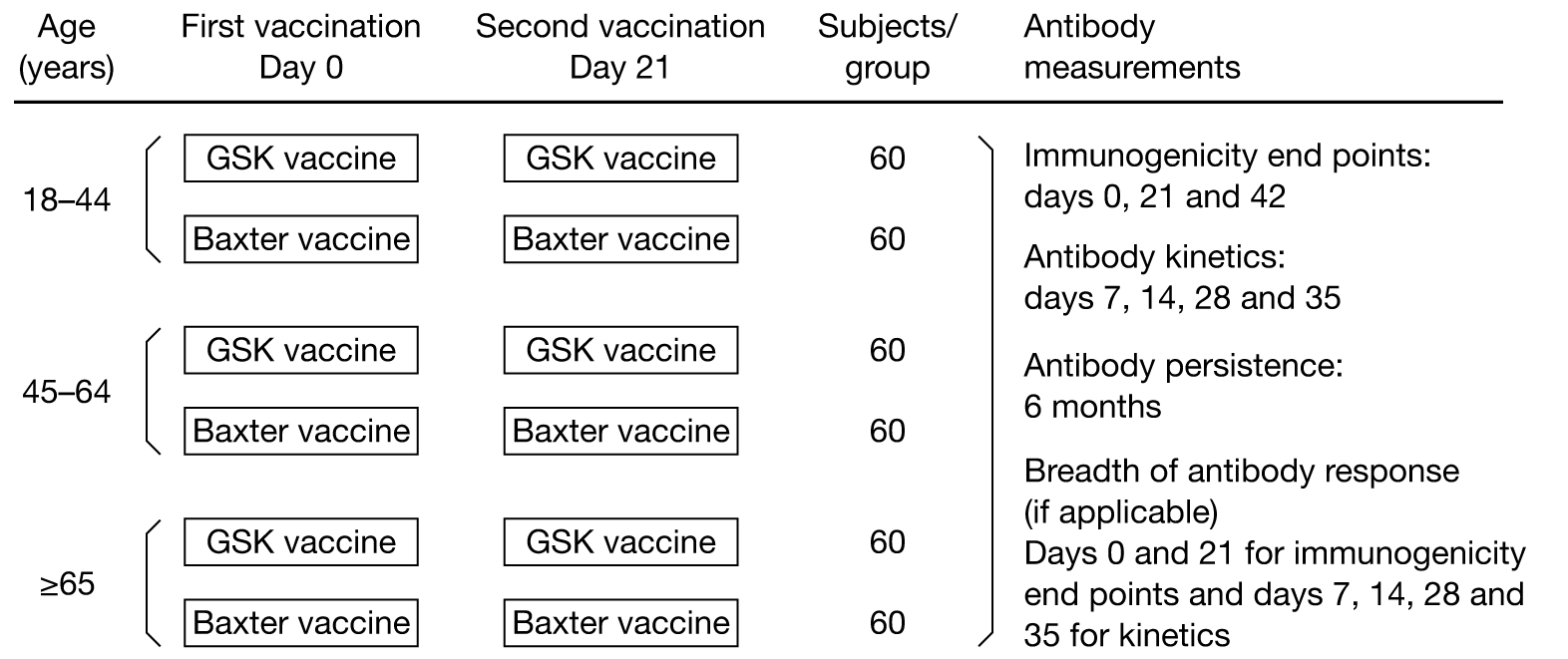
Time and events table
| Study visit | ||||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
| Days after the first vaccination: | ||||||||
| Window (days) | ||||||||
| 0 | (± 1) | (± 2) | (± 2) | (± 2) | (± 3) | (± 3) | (± 10) | |
| Study day | ||||||||
| 0 | 7 | 14 | 21 | 28 | 35 | 42 | 180 | |
| Informed consent | × | |||||||
| Inclusion/exclusion criteria | × | × | ||||||
| Medical/medication history | × | |||||||
| Pregnancy test | × | × | ||||||
| Blood sample – antibody studies | × | × | × | × | × | × | × | × |
| Vaccination | × | × | ||||||
| Thermometer/diary card | × | × | ||||||
| Diary card training | × | |||||||
| Diary card returned/review | × | × | ||||||
| Reminder regarding unsolicited events | × | × | × | × | × | × | × | |
| AEs monitoring | × | × | × | × | × | × | × | |
| Termination of study | × | |||||||
Number of subjects
| Vaccine | Age | Vaccine dose (µg) | Total | |
|---|---|---|---|---|
| 3.75 | 7.5 | |||
| GSK | 18–44 | 60 | – | 60 |
| 45–64 | 60 | – | 60 | |
| ≥ 65 | 60 | – | 60 | |
| Baxter | 18–44 | – | 60 | 60 |
| 45–64 | – | 60 | 60 | |
| ≥ 65 | – | 60 | 60 | |
| Total | 180 | 180 | 360 | |
Inclusion criteria
-
Mentally competent adults, who have signed an informed consent form after having received a detailed explanation of the study protocol.
-
Clinically healthy, male or female volunteers aged 18 years of age and older, including the over-65-year-olds, and those with stable high-risk medical conditions. (Note: ‘Stable’ is defined as having no medical consultations for an exacerbation or worsening of any chronic medical condition during the preceding 8 weeks, and having been maintained on a stable drug regimen for at least 2 weeks prior to study entry as assessed by the medical history.)
-
Individuals who are able to understand and comply with all study procedures and to complete study diaries.
-
Individuals who can be contacted and are available for all study visits.
-
Females should be using secure contraceptive precautions including (1) the oral contraceptive pill or (2) condom/barrier contraception, or (3) partner has had a vasectomy; (4) be surgically sterilised; or (5) postmenopausal (defined as at least 2 years since the last menstrual period).
Exclusion criteria
-
Subjects who are unable to lead an independent life either physically or mentally.
-
Women should not be pregnant or lactating.
-
Women who refuse to use a reliable contraceptive method on days 0–42 of the study;
-
Confirmed H1N1 infection, as determined by laboratory tests.
-
Have received oseltamivir or zanamivir for influenza-like illness (ILI) since May 2009.
-
Have a household member who had confirmed H1N1 infection, as determined by laboratory tests, and/or received oseltamivir or zanamivir for ILI since May 2009.
-
Receipt of another investigational agent (vaccine or medicinal product) in the preceding 4 weeks.
-
Unwilling to refuse participation in another study during days 0–42 of the study.
-
Any clinically significant concurrent illness or unstable medical condition including: malignant tumours, acute or progressive renal or hepatic pathology, chronic obstructive pulmonary disease requiring oxygen therapy, and any active neurological disorder.
-
Individuals who have had acute respiratory pathology or infections requiring systemic antibiotic or antiviral therapy during the preceding 7 days (chronic antibiotic therapy for prevention of urinary tract infections is acceptable).
-
Subjects who had a temperature ≥ 38°C within 3 days of vaccination.
-
Any acute illness at the time of vaccination. (Note: minor infections without fever or systemic upset are not contraindications/exclusion criteria.)
-
Subjects with known or suspected impairment/alteration of immune function, including:
-
receipt of oral immunosuppressive drugs or other drugs listed in section 8 of the British National Formulary (BNF) or chloroquine, gold or penicillamine or other drugs listed in section 10.1.3 of the BNF to suppress a chronic disease process (note: long-term, inhaled steroids for asthma management is acceptable.)
-
receipt of immunostimulants or interferon
-
receipt of an immunoglobulin preparation, blood products, and/or plasma derivatives within 3 months of the study
-
anyone at high risk of developing immunocompromising condition
-
received radiotherapy or chemotherapy during the 6 months preceding the study.
-
-
Subjects for whom surgery is planned during days 0–42 of the study.
-
Regularly drink more than 40 units of alcohol weekly.
-
Known or suspected drug abuse (recreational or prescribed).
-
Individuals who, in the opinion of the investigator, have conditions that might complicate interpretation of the study results.
-
Subjects with a history of anaphylaxis or serious reactions to vaccines; known hypersensitivity (other than anaphylactic reaction) to influenza viral protein, to any component of the study vaccines, to products containing mercury and to residues (egg and chicken protein, ovalbumin, formaldehyde, gentamicin sulphate, sodium deoxycholate and benzonase).
-
Subjects with a history of any neurological symptoms and signs, or anaphylactic shock following administration of any vaccine.
-
Actual or planned receipt of another vaccine, excluding seasonal influenza vaccine, during the period 3 weeks before to 3 weeks after vaccination on days 0 and 21.
Test vaccines, antigen content, dosage regimen, route of administration
All subjects will be allocated either two doses of Baxter vaccine (i.e. cell-culture, non-adjuvanted, whole-virion influenza A/California/2009 (H1N1) vaccine, containing 7.5 µg of HA) or two doses of GSK vaccine (i.e. egg-grown, AS03-adjuvanted, split-virus influenza A/California/2009 (H1N1) vaccine, containing 3.75 µg of HA), administered 21 days apart, by IM injection into the deltoid muscle of, preferably, the non-dominant arm.
Concomitant vaccines/medications
There are no concomitant vaccines or medication.
Assessments
Study entry
-
Inclusion/exclusion criteria.
-
Medical history.
-
Demography.
During the study
-
Exclusion criteria.
-
Solicited and unsolicited events (local and systemic symptoms).
-
Immune responses to influenza virus.
Analysis
-
Baseline demographic data, including age, sex, ethnicity, previous influenza vaccination (past three seasons), ILI (since May 2009), and pre-vaccination antibody to the vaccine strain.
-
Solicited and unsolicited events, including local and systemic symptoms, relief medication, and absence from work due to any AEs.
-
Measures of immunogenicity (see below).
Measures of immunogenicity
Immunogenicity will be measured by HI, neutralising antibody (MN) and possibly SRH antibody responses to influenza H1N1 at each visit.
The principal objective of the study is to evaluate the immunogenicity of each dose of Baxter cell-culture, non-adjuvanted, whole-virion H1N1 vaccine, and GSK AS03-adjuvanted, split H1N1 vaccine with respect to CHMP and FDA licensing criteria, i.e. 21 days after the first and second doses.
Immunogenicity will be assessed in terms of the ‘magnitude’ and ‘kinetics’ of the antibody response, and, when appropriate, the ‘breadth’ of the antibody response:
-
magnitude i.e. measurement of the antibody titres (to the vaccine strain) to one and two 0.5-ml IM doses of Baxter and GSK vaccines [i.e. by comparing (1) mean geometric increases (ratio of day 21 GMT–day 0 GMT and ratio of day 42 GMT–day 0 GMT, by age group and all age groups combined); (2) the SCR, or significant increases in titre; and (3) the seroprotection rate]
-
kinetics i.e. application of the above immunogenicity criteria 7 and 14 days after each dose, after each vaccine type, in each age group, and in all age groups combined
-
breadth i.e. application of the above immunogenicity criteria 21 days after each dose, after each vaccine type, in each age group, and in all age groups combined, to any antigenic drift variant that emerges prior to the 2010–11 influenza season.
Serology
Serum samples will be assessed by means of HI, MN and, possibly, SRH tests. HI and MN assays will be performed at the Health Protection Agency Centre for Infections, Enteric, Respiratory & Neurological Virus Laboratory, London, UK. SRH tests may be done at the National Institute for Biological Standards and Control.
Cellular assays
Cellular assays (T-cell ELISPOT) will be used to assess responses before and after vaccination. This will be performed at Imperial College, London.
Statistical hypothesis
The aim of the trial is to establish whether GSK and Baxter vaccines satisfy all three Committee for Proprietary Medicinal Products (CPMP) criteria, and, if so, compare them in terms of immunogenicity (for each vaccine/age group and each vaccine type). The sample/group size is in line with standard practice. The protocols for seasonal European Union (EU) vaccine clinical trials and the criteria for assessment have been standardised within the EU. They stipulate that trials should be done with groups of at least 50 subjects. We will recruit 60 per group, allowing for up to 17% dropout.
Interim/preliminary analyses
To provide the Department of Health (DH) with information as rapidly as possible with the goal of informing DH vaccination strategy, the following interim analyses of data from this study are planned:
-
Solicited/unsolicited events (local and systemic symptoms, relief medication, and absence from work due to any AEs) during:
-
days 0–6 (first vaccination)
-
days 0–21 (first vaccination)
-
days 21–27 (second vaccination)
-
days 21–41 (second vaccination).
-
-
HI and MN antibody titres on days:
-
days 0, 7, 14, 21 (first tranche of sera measuring antibodies before and after first injection)
-
days 21, 28, 35, 42 (second tranche of sera measuring antibodies before and after second injection).
-
List of abbreviations and definitions of terms
Abbreviations
- AE
- adverse event
- AP
- (statistical) analysis plan
- CCA
- chick cell agglutination
- CI
- confidence interval
- CPMP
- Committee for Proprietary Medicinal Products
- CRF
- case report form
- EC
- ethics committee
- EMEA
- European Agency for the Evaluation of Medicinal Products
- GCP
- good clinical practice
- GMA
- geometric mean area
- GMR
- geometric mean ratio
- GMT
- geometric mean titre
- HA
- haemagglutinin
- HI
- haemagglutination inhibition
- ICH
- International Conference on Harmonisation
- ICF
- informed consent form
- IM
- intramuscular
- ITT
- intention to treat
- IUD
- intrauterine device
- LSLV
- last subject last visit
- MedDRA
- Medical Dictionary for Regulatory Activities
- MHRA
- Medicines and Healthcare Products Regulatory Agency
- MRC CDVIP
- Medical Research Council Committee for the Development of Vaccines and Immunisation Procedures
- MN
- microneutralisation
- NA
- neuraminidase
- SA
- surface antigen
- SAE
- serious adverse event
- SOP
- standard operating procedure
- SP
- split product
- SRH
- single radial haemolysis
- SUSAR
- Suspected Unexpected Serious Adverse Reaction
- UHL
- University Hospitals of Leicester
- WHO
- World Health Organization
- WV
- whole virion
Definition of terms
- Adverse event
- An adverse event (AE) is any untoward medical occurrence in a patient or clinical investigation subject administered a pharmaceutical product at any dose that does not necessarily have to have a causal relationship with this treatment. An AE can, therefore, be any unfavourable and unintended sign (including an abnormal laboratory finding, for example), symptom or disease temporally associated with the use of an investigational product, whether or not considered related to the investigational product. This definition includes intercurrent illnesses or injuries and exacerbations of pre-existing conditions.
- Concomitant medication
- All prescription medications being taken by the subjects on entry to the study and all prescription medications given in addition to the study vaccine during 21 days after each vaccination are to be regarded as concomitant medication.
- End of trial
- The end of trial corresponds with the last visit of the last subject undergoing the trial (LSLV, last subject last visit).
- Local and systemic reactions
- Selected local and systemic AEs are routinely monitored in vaccine clinical trials as indicators of vaccine reactogenicity. It is recognised that each of these events, and particularly those of a systemic nature, may, under some circumstances, in any individual subject, have a cause that is unrelated to the study vaccine. However, as a matter of convenience, and in accordance with common clinical practice, all such events occurring within 6 days after immunisation are herein termed ‘local and systemic reactions’.
- Month, day
- Study months are based upon 30-day cycles. The study day refers to the number of days after enrolment, with the day of first vaccination being designated ‘day 0’.
- Serious adverse event
- Any experience or reaction that suggests a significant hazard, contraindication, side effect or precaution. These events include any experience that is fatal or life-threatening, requires or prolongs inpatient hospitalisation, is permanently disabling, leads to congenital abnormality, requires intervention to prevent permanent impairment or damage, or is important and significant medical event that, based upon appropriate medical judgement, may jeopardise the subject.
- Stable medical condition
- Is defined as having no medical consultations for an exacerbation or worsening of any chronic medical condition during the preceding 8 weeks and have been maintained on a stable drug regimen for at least 2 weeks prior to study entry as assessed by the medical history.
- Study monitor
- The study monitor is the sponsor’s designated representative responsible for managing, supervising and monitoring the overall conduct of the trial.
Ethics
Approval of study protocol
This protocol and any accompanying material provided to the patient (such as patient information sheets or descriptions of the study used to obtain informed consent) will be submitted for expedited Integrated Research Application System (IRAS) ethical approval for projects on pandemic influenza by the principal investigator. Approval will be obtained before starting the study, and will be documented in a letter to the investigator specifying the date on which the committee met and granted approval for the study and the protocol identification (title, version, date).
The ethics committee (EC) should also be asked for a written statement regarding the composition of the committee and should comply with Good Clinical Practice (GCP) and the applicable regulatory requirement(s). The trial will not be initiated until appropriate EC approval of the protocol and informed consent document. In addition, all documents will be submitted to other authorities [e.g. Medicines and Healthcare Products Regulatory Agency (MHRA)] in compliance with local jurisdictions.
Prior to enrolment, the sponsor and the investigator must exchange written confirmation that their ethical and legal responsibilities have been observed. The EC and, if applicable, other authorities, must be informed of protocol amendments in accordance with local legal requirements. Appropriate reports on the progress of the study will be made to the EC and the sponsor by the investigator in accordance with applicable governmental regulations and in agreement with policy established by the sponsor.
Any modifications made to the protocol after receipt of the EC approval must be submitted by the investigator to the EC in accordance with local procedures.
Ethical conduct and good clinical practice
This trial will be conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki, and that are consistent with GCPs and the applicable regulatory requirement(s) for the country in which the trial is conducted, GCP according to International Conference on Harmonisation (ICH) guidelines, and applicable standard operating procedures (SOPs). Specifically, this trial is based on adequately performed laboratory procedures; the trial will be conducted under a protocol reviewed and approved by an EC, the trial will be conducted by scientifically and medically qualified persons; the benefits of the study are in proportion to the risks; the rights and welfare of the subjects will be respected; the physicians conducting the trial do not find the hazards to outweigh the potential benefits; each subject, or where applicable, each subject’s legally acceptable representative(s), will give his or her written informed consent before any protocol-driven tests or evaluations are performed. A copy of the ICH GCP guidelines and of the Declaration of Helsinki (version 1996) will be included in the investigator’s study file.
Informed consent of subject and confidentiality
Informed consent
The investigator is responsible to obtain informed consent in adherence to GCP and according to applicable regulations prior to entering the subject into the trial.
The information about the trial must be given orally and in an understandable form. Written information about the trial will also be provided. In addition to the explanation of the trial and the subject’s legal rights the information should comprise that access to original medical records and processing of coded personal information must be authorised. The informed consent discussion must be conducted by a person who is qualified according to applicable local regulations. The subject should have the opportunity to inquire about details of the trial and to consider participation.
The informed consent form (ICF) must be signed and dated by the subject and must be countersigned by the person who conducted the informed consent discussion (according to local laws and GCP).
If a person is unable to read or write, oral consent in the presence of an impartial witness is possible, if this is permitted by local legislation. In this case, the witness is to be present during the meeting in which the significance of the informed consent will be orally explained. After the informed consent discussion and after the subject has orally consented to participate in the clinical trial the witness should sign and personally date the consent form to attest that information concerning the clinical trial and the subject’s rights was accurately explained to, and apparently understood by the subject and that informed consent was freely given.
The investigator will provide a copy of the signed informed consent to the subject, and will maintain the original in the investigator’s study file.
The written informed consent form and any other written information to be provided to subjects should be revised whenever important new information becomes available that may be relevant to the subject’s consent. Any revised written informed consent form, and written information should receive EC’s approval before use.
The subject should be informed in a timely manner if new information becomes available that may affect the decision to participate in the clinical trial. The communication of this information should be documented.
Subject confidentiality
Subject names will not be supplied to the sponsor (University Hospitals of Leicester NHS Trust). Only the subject numbers and subject identification codes will be recorded in the case report form (CRF), and if a subject’s name appears on any other document, it will be obliterated before a copy of the document is supplied to the sponsor. Study findings stored on a computer will be subject to local data protection laws. The subject, or where applicable, the subject’s legally acceptable representative, will be informed that representatives of the sponsor, EC, or regulatory authorities may inspect their medical records to verify the information collected, and that all personal information made available for inspection will be handled in strictest confidence.
The investigator or designee will maintain a personal list of subject numbers and subject identification codes to enable records to be found at a later date.
Indemnity
This study is being undertaken in response to pandemic H1N1 influenza following a call for scientific proposals to help inform national strategy/policy. This study is being done with vaccine purchased by the Department of Health to help protect the population from pandemic influenza. The investigators and the Sponsor (UHL NHS Trust) and others who facilitate the study will be indemnified by the DH of England and Wales against non-negligent harm (in accordance with applicable laws and regulations, against financial loss resulting from personal injury and/or other damages), which may arise as a consequence of the administration of Baxter and GSK H1N1 vaccines used in this study. This indemnity is applicable to subjects vaccinated in this study in health-care settings (University Hospitals of Leicester, Nottingham, and Sheffield, and general practice facilities) in Leicestershire, Nottinghamshire, Yorkshire, and Derbyshire.
Investigators
The trial will be administered and monitored by employees or representatives of the UHL NHS Trust, University Hospitals of Nottingham NHS Trust, and University Hospitals of Sheffield NHS Trust. The Principal Investigators in Leicester (Dr Iain Stephenson), Nottingham (Dr Weishen Lim) and Sheffield (Professor Robert Read), together with the Chief Investigator (Professor Karl Nicholson), will be responsible for the timely reporting of SAEs.
The investigators and study nurses undertaking the trial either hold, or will hold (when appointed), appointments, or honorary appointments, with the UHL NHS Trust, University Hospitals of Nottingham NHS Trust, or University Hospitals of Sheffield NHS Trust.
Study monitors will monitor the sites on a periodic basis and perform verification of source documentation for volunteers. The principal investigator at each study site will be readily available to provide appropriate medical expertise on trial-related medical questions. The sponsors and investigators responsibilities as regards reporting SAEs and suspected unexpected serious adverse events (SUSARs) will be in accordance with the European Directive 2001/20/EC.
Background and rationale
Influenza virus diversity
Influenza A viruses are antigenically distinguished and classified by subtypes of HA and neuraminidase (NA) with 16 HA and nine NA subtypes identified within the natural reservoir of aquatic birds. The HA and NA surface antigens of influenza A virus are responsible for virus attachment and release from host cell receptors and are targeted by host antibodies. The HA is the major component of influenza vaccines and is subject to mutations resulting in antigenic drift that enables the virus to escape immune recognition.
In the northern hemisphere influenza is characterised by the occurrence of annual outbreaks during winter and worldwide pandemics, which have occurred at 11- to 52-year intervals during the past 300 years. 1 Pandemics inflict huge socioeconomic costs. The 1918–19 pandemic caused an estimated 40–100 million deaths globally. Pandemic influenza results from the emergence of an influenza A virus possessing a ‘new’ HA (antigenic ‘shift’) to which the population possesses little or no immunity and which is capable of spreading with a high attack rate in all parts of the world. Despite this viral diversity, only three HAs and two NAs have established human lineages during the last 100 years. In 1918 (Spanish flu: A/H1N1), 1957 (Asian flu: A/H2N2), 1968 (Hong Kong flu: A/H3N2) and 1977 (A/H1N1) strains emerged to cause widespread human infections.
Reasons for decline and emergence of dominant subtypes is unclear, although it seems likely that during interpandemic intervals, population immunity broadens to a point where the prevalent strain loses its capacity for further drift capable of eluding host defences.
Swine influenza in humans
Influenza as a disease of pigs was first described during 1918 when outbreaks of respiratory disease occurred simultaneously in humans and swine-herds living and working in close proximity. Pigs are thought to have an important role in interspecies transmission as they possess receptors in their respiratory tract that are capable of binding both avian and human influenza. Consequently, they have been proposed as a possible mixing vessel in which novel reassortant viruses of pandemic potential may be generated. Occasional isolation of swine influenza viruses from humans with respiratory illness has confirmed that sporadic human infection can occur. 2 Generally, cases have been limited to laboratory workers or those with occupational swine exposure. However, a pandemic alert was raised in 1976 when swine H1N1 caused an outbreak of respiratory illness with one fatality among 13 soldiers at a military base in Fort Dix, New Jersey, USA. 3 No exposure to pigs was found and seroepidemiological investigation identified up to 230 further soldiers had been infected suggesting human-to-human transmission. Mass vaccination of the US public was initiated and halted amid reports of adverse vaccine reactions, media scepticism and the lack of pandemic activity. 4
Pandemic A/H1N1 emergence in 2009
In April 2009, near the end of the usual influenza season in the northern hemisphere, the first two cases of swine origin H1N1 influenza virus were identified in the USA. 5 The CDC confirmed that these cases were caused by a genetically similar swine virus that had not been previously identified in the USA. Genetic analysis of the strains showed that they were derived from a new reassortment of six gene segments from the known triple reassortant swine virus, and two gene segments (NA and matrix protein) from the Eurasian influenza A/H1N1 swine virus lineage. 6 Effectively all isolates are susceptible to neuraminidase inhibitors, but resistant to M2 inhibitors. The World Health Organization (WHO) raised its pandemic alert level to level 6 on June 11 2009,7 reflecting community level outbreaks of novel H1N1 infection in at least two different WHO regions and the onset of a new pandemic. H1N1 virus has reached over 170 countries and territories worldwide as of August 6 2009, causing at least 177457 cases and 1462 deaths. 8 In the UK, the first pandemic wave is waning, but a substantial increase in cases of novel H1N1 infection is expected following the re-opening of schools in September.
Burden of H1N1 disease, hospitalisations and admissions to ITU
As of 7 August , analysis of the distribution by age of 8974 individual case reports of influenza A/H1N1v infection in 27 EU/EEA countries reveal that the age-distribution of non-hospitalised cases of H1N1 influenza is highest in the 10- to 19-year age group, followed by 0- to 9-year and 20- to 29-year age groups. The age-distribution of hospitalised cases is significantly higher in the 20- to 29-year age group than in the under 20-year-olds. 9 Estimates of transmissibility (Ro 1.4–1.6) are significantly higher than observed in seasonal influenza and comparable to previous pandemics. 10 In England, admission rates have been highest in the under-5-year-olds, but life-threatening complications requiring admissions to intensive treatment unit (ITU) increase with age and presence of ‘high-risk’ comorbidity. It is unclear whether the higher prevalence of infection in the under-30-year-olds is related primarily to social mixing, crossreacting antibodies that are more prevalent with increasing age, or both.
Preliminary studies in the USA have shown that crossreactive MN antibody titres of ≥ 160 to A/California/2009 H1N1 were detected in 6% of adults aged 18–40 years, 9% of adults aged 18–64 years, and 33% of adults aged 60 years and older. 11 After vaccination with seasonal vaccine, 7% of adults aged 18–40 years, 25% of adults aged 18–64 years, and 43% of adults aged 60 years and older had postvaccination titres of ≥ 160.
Infection with the current influenza H1 pandemic virus is mostly mild, with no increase in mortality above threshold national statistics in the USA, despite widespread infection. The observed age distribution is unusual and different from seasonal influenza, being skewed towards younger age groups, resulting in deaths and ITU admissions in people who are normally affected less frequently.
Currently, it is difficult to be certain about the impact of novel H1N1 infection on hospitalisation rates. A rate of 11% has been observed in the USA, but this is likely to be an overestimate due to the mild nature of the disease in many cases, and differences in clinical practice. An overall rate for Europe is around 5–6%, but this too may be an overestimate due to the practice in some countries of admitting patients for isolation to control spread, rather than for severity of illness. In the UK, the observed rate has been around 1–2%.
Current seasonal influenza vaccines
Inactivated influenza virus vaccines represent the mainstay of efforts to prevent influenza and its complications. Current licensed vaccines are produced from virus grown in eggs or cell culture systems and consist of either whole-virion, detergent-treated ‘split-product’, or purified HA and NA (subunit) surface antigen formulations.
Vaccine efficacy of 70–95% in healthy adults is obtained when there is a good match between the vaccine and the circulating strains. 12 They display reduced efficacy against antigenically drifted viruses and are considered ineffective against unrelated subtypes.
The use of mammalian cell lines, notably Vero and Madin–Darby Canine Kidney (MDCK) cells, to grow influenza virus are approved substrates for production of licensed trivalent seasonal vaccines which may allow for increased vaccine production at short notice to meet unexpected demand.
As vaccine responses are generally lower in elderly subjects, efforts to improve immunogenicity have been investigated. The addition of MF59, a squalene-containing oil-in-water emulsion adjuvant, has been shown to increase postvaccination antibody titres and SCRs in elderly and immunocompromised subjects. 13 MF59-adjuvanted seasonal influenza vaccines have been licensed for clinical use since 1997. More recently, two other squalene-containing oil-in-water adjuvants have been developed by GSK and Sanofi. The GSK AS03 oil-in-water adjuvant has been extensively evaluated in association with H5N1 antigens.
Global manufacturing capacity for pandemic influenza vaccine
Seasonal influenza vaccines are given at doses of 15 µg of HA per virus strain. The global human population is estimated at 6.77 billion. 14 The annual global vaccine manufacturing capacity for trivalent seasonal influenza vaccines was 852 million doses in May 2009. 15 Assuming that the yield of H1N1(v) antigen is comparable to that for seasonal virus strains, the present manufacturing capacity equates to 2.56 billion doses of monovalent H1N1v vaccine containing 15 µg of HA per dose. This would be enough for only 2.56 billion people if two doses containing 7.5 µg of HA were immunogenic, but could protect more people if one dose was sufficient in older people. Pandemic H1N1v vaccines will be supplied over a period of 6 months or more, so it is essential that dose-sparing formulations and regimens are identified and deployed rapidly to protect as many vulnerable people as possible.
Experience of pandemic and mock pandemic vaccines since the 1970s
Historically, influenza vaccines were first developed as ‘whole-virion’ formulations. During the late 1970s, whole-virion vaccines were replaced by ‘split’, and highly purified ‘surface antigen’ formulations that caused fewer local and systemic reactions than whole-virion vaccine, but are equally immunogenic when given to primed individuals as ‘seasonal’ or ‘interpandemic’ vaccine.
Experience with H1N1 vaccines during the 1970s
Experience in unprimed individuals with vaccines produced from Hsw1N1 viruses (A/New Jersey/8/76) or H1N1 viruses (A/USSR/90/77) indicated that high concentrations of antigen (> 50 µg of HA) were needed in a single vaccine dose to generate HI titres that met the current European licensing criteria. In a two-dose schedule, HI titres of ≥ 40 could be achieved with two doses containing 5 µg of HA. Overall, whole-virion vaccines were more immunogenic than split or subunit vaccines. The split and surface antigen vaccine formulations were notably less immunogenic than whole-virion vaccine when given to children, both during 1976 when influenza A/New Jersey/76 (H1N1) posed a pandemic threat,16 and during 1977 when A/USSR/77 (H1N1) virus re-emerged. 17
European licensing criteria
During the late 1970s, influenza vaccines were poorly standardised. Subsequently, improved methods of measuring vaccine potency and ensuring vaccine standardisation were introduced, and in Europe, by criteria for licensure of seasonal,18 and, latterly, pandemic vaccines. 19
As specified in the CHMP ‘Guidelines on dossier structure and content for pandemic influenza vaccine marketing authorisation application’,19 it is anticipated that a pandemic candidate vaccine should at least be able to elicit sufficient immunological responses to meet and preferably exceed all three of the current standards set for existing vaccines in unprimed adults or elderly subjects as specified for seasonal vaccines. 18
These include assessments of the mean geometric increase in antibody titre (the seroconversion factor), the number of seroconversions or significant increases in antibody, and the seroprotection rate (i.e. the proportion attaining ‘protective’ levels of antibody). The criteria are based on the HI assay or SRH. Both assays have been established as surrogates for protection.
The CHMP guidelines stipulate that vaccines should be tested in adults (18–60 years) and elderly (> 60 years), in groups of > 50 subjects, and attain the following:
-
Adults (18–60 years):
-
– seroconversions/or significant rises (i.e. a fourfold increase in postvaccination titre) by > 40%
-
– mean-fold increase in GMT post vaccination > 2.5
-
– significant levels of antibody (i.e. having postvaccination HI titres > 1 : 40) in > 70%.
-
-
Elderly (> 60 years):
-
– seroconversions/or significant rises (i.e. a fourfold increase in postvaccination titre) by > 30%
-
– mean-fold increase in GMT >2
-
– significant levels of antibody (i.e. having postvaccination HI titres > 1 : 40) in > 60%.
-
Immunogenicity of plain (i.e. non-adjuvanted split and subunit avian influenza vaccines)
As outlined below, neither split nor subunit vaccine formulations of H5, H7 and H9 avian influenza satisfy all three CHMP licensing criteria when given at doses of up to 90 µg of HA.
Treanor et al. 20 showed that neither two 90-µg doses of plain (i.e. non-adjuvanted) recombinant, baculovirus-expressed, H5 HA nor two 90-µg doses of egg-grown, plain, inactivated, subvirion influenza A/Vietnam/1203/2004 (H5N1) vaccine satisfied the CHMP regulatory criteria. 21 Nicholson et al. 22 showed that two doses of all three 7.5- to 30-µg formulations of plain A/Duck/Singapore/97 (H5N3) surface antigen vaccine failed to meet the CHMP criteria.
Bresson et al. 23 showed that two doses of all three 7.5- to 30-µg HA formulations of plain, split virus, A/Vietnam/1194/2004 (H5N1) vaccine satisfied the CHMP criterion for a greater than 2.5-fold increase in antibody titre, but 47% vaccinees failed to achieve protective levels of antibody after a second dose. Nolan et al. 24 evaluated two doses of split A/Vietnam/1194/2004 (H5N1) vaccine containing 7.5–45 µg of HA with and without alum adjuvant. All formulations met the CPMP criterion for a greater than 2.5-fold increase in HI antibody titres after the second dose, but not the criterion for greater than 70% of participants achieving seroprotection.
Keitel et al. 25 evaluated subvirion inactivated influenza A/H5N1 vaccine containing 3.75, 7.5, 15 or 45 μg of HA. Dose-related increases in antibody responses were noted after both vaccinations, but no formulation attained the CHMP criteria. 25
Stephenson et al. 26 evaluated two 7.5-, 15- and 30-µg doses of plain, subunit, influenza A/Hong Kong/1073/99 (H9N2) vaccines in people before and after their 32nd birthday. The CHMP criterion for a greater than 2.5-fold increase in HI antibody titres was met after the second dose, but 86% vaccinees failed to attain protective levels of antibody. Cox et al. 27 evaluated two doses of split H7N1 virus vaccine containing 12 or 24 µg of HA. Neither formulation fulfilled the CHMP licensing criteria.
Immunogenicity of whole-virion vaccines and vaccines adjuvanted with oil-in-water emulsions
Whole-virion vaccines and vaccines adjuvanted with oil-in-water emulsion are more immunogenic in man than split and subunit vaccines. 22,26,28–35
Lin et al. 36 showed that a two-dose regimen of an aluminium hydroxide adjuvanted whole-virion A/Vietnam/1194/2004 (H5N1) vaccine containing 10 µg of HA met all CHMP regulatory requirements for annual licensing of seasonal influenza vaccine. Ehrlich et al. 37 evaluated whole-virion A/Vietnam/1203/2004 (H5N1) vaccine (manufactured by Baxter Healthcare) at doses of 3.75, 7.5, 15 or 30 µg of HA with alum adjuvant, and 7.5 or 15 µg without adjuvant. Maximum responses to the vaccine strain were obtained with formulations without alum adjuvant. When assessed by SRH, the 7.5-µg dose met all three CHMP licensing criteria. Two criteria were met when antibodies were measured by HI. 37 The vaccine also induced a neutralising immune response against clade 2 and 3 strains, and results without alum adjuvant elicited significantly higher immune responses than those with alum.
A Phase I randomised trial of subunit and whole-virion A/Hong Kong/1073/99 (H9N2) vaccine, given in two doses containing doses of 7.5, 15 or 30 µg of HA, revealed the presence of crossreacting antibodies in participants born before 1969 who were older than 32 years38 – this finding is comparable to the recent observation of an age-related presence of crossreacting antibodies to A/California/2009 (H1N1v) in the USA. In participants older than 32 years, one dose of whole-virion or subunit vaccine evoked antibody responses associated with protection. However, in people aged 32 years or younger, whole-virion vaccine produced a significantly higher probability of seroconversion than with subunit virus for this age group. 38
Nicholson et al. 22 evaluated two doses of subunit A/Duck/Singapore/97 (H5N3) vaccine containing 3.75, 7.5 and 15 µg of HA with and without MF59 oil-in-water adjuvant. In this Phase I randomised trial, the GMTs of antibody, and SCRs, were significantly higher with MF59 adjuvanted vaccine. After the second injection, all MF59-adjuvanted vaccine doses met all three CHMP licensing criteria. Further studies showed improved antibody persistence with MF59 containing vaccine, improved immune responses to other clades of H5 virus, and significantly higher antibody responses on boosting. 29–32
Leroux-Roels et al. 33 evaluated A/Vietnam/1194/2004 (H5N1) vaccine manufactured by GlaxoSmithKline at doses of 3.8, 7.5, 15 and 30 µg of HA with and without its proprietary AS03 adjuvant. The adjuvanted formulations were significantly more immunogenic than the non-adjuvanted formulations at all antigen doses. At the lowest antigenic dose, immune responses for the adjuvanted vaccine against the vaccine strain met or exceeded all FDA and CHMP licensure criteria. Further research showed broad cross-clade immune responses at the lowest antigen dose (3.8 µg) with adjuvant, but no cross-clade response in the non-adjuvanted group. 34,35
Reproducibility of the serology assays for influenza
For research purposes and vaccine licensure, influenza vaccines are evaluated by clinical trials that assess immunogenicity by the presence of serum antibody. Collaborative studies have shown that the serology assays are highly variable between laboratories – with variability between laboratories for HI assays varying by up to 32-fold. 39–41 This leads to difficulties in interpreting results from different manufacturers. At the 5th WHO Meeting on Evaluation of Pandemic Influenza Prototype Vaccines in Clinical Trials, 12–13 February 2009, WHO highlighted the need for standardised assays and internationally accepted antiserum standards. 42
The H1N1v vaccines purchased by the government
The UK DH has purchased pandemic vaccines from Baxter Healthcare and GSK. The Baxter vaccine (trade name ‘celvapan’) is a plain (i.e. non-adjuvanted), whole-virion, Vero-cell-grown, influenza A/H1N1v pandemic formulation, containing 7.5 µg of HA per 0.5-ml dose. The GSK vaccine (trade name ‘pandemrix’) is an AS03-adjuvanted, split-product, egg-grown, influenza A/H1N1v formulation, containing 3.75 µg of HA and oil-in-water AS03 adjuvant composed of squalene, DL-α-tocopherol and polysorbate 80. The expectation is that the initial limited supplies of both vaccines will be prioritised for those deemed to a greatest risk, but eventually everyone will have access to either vaccine. To ensure that protection is provided as rapidly as possible, it is imperative that vaccine is used efficiently.
Conclusions
The available evidence indicates that whole-virion vaccines, and split and subunit vaccines that contain oil-in-water adjuvant, are more immunogenic for avian H5, H7 and H9 HAs, and A/New Jersey/76 (HSw1N1) and A/USSR/77 (H1N1) than split and surface antigen vaccines without adjuvant. They also offer the potential for broadened antibody responses that could be critical in the event of antigenic drift during the course of the pandemic. Vaccine is likely to be in short supply (vaccine production takes time and is subject to various rate-limiting factors) and demand will be high worldwide. The available antigen will therefore need to be given optimally with consideration being given to the logistics of vaccine administration, immune responses, and the frequency and nature of adverse clinical reactions.
There have been no head-to-head comparisons of avian vaccines manufactured by Baxter Healthcare and GSK. Due to variability of serological assays, there are uncertainties as to whether one preparation offers advantages over the other in terms of dose sparing in ‘older’ people, and significantly more ‘seroconversions’ in the young.
Study objectives
Primary
-
To evaluate the immunogenicity of Baxter cell-culture, non-adjuvanted, whole-virion H1N1 vaccine, and GSK AS03-adjuvanted, split H1N1 vaccine with respect to the CHMP and FDA licensing criteria.
Secondary
-
To identify whether one or two doses of vaccine are required to satisfy the licensing criteria.
-
To examine the short-term reactogenicity of the vaccines.
-
To examine the kinetics of the antibody responses to vaccination.
-
To examine persistence of antibody at 6 months.
And, if appropriate (i.e. an antigenic drift variant emerges prior to the 2010–11 influenza season):
-
To evaluate the breadth of the antibody response to the antigenic variant.
-
To evaluate cellular responses before and after vaccination.
Overall study design
Overall study design
This observer-blind, multicentre study will be performed at three study sites in England (Leicester, Nottingham and Sheffield) in a study population of healthy male and female adults, or adults with stable chronic medical conditions. Six groups of 60 male and female adults will be stratified by age (18–44, 45–64 and 65 years and older):
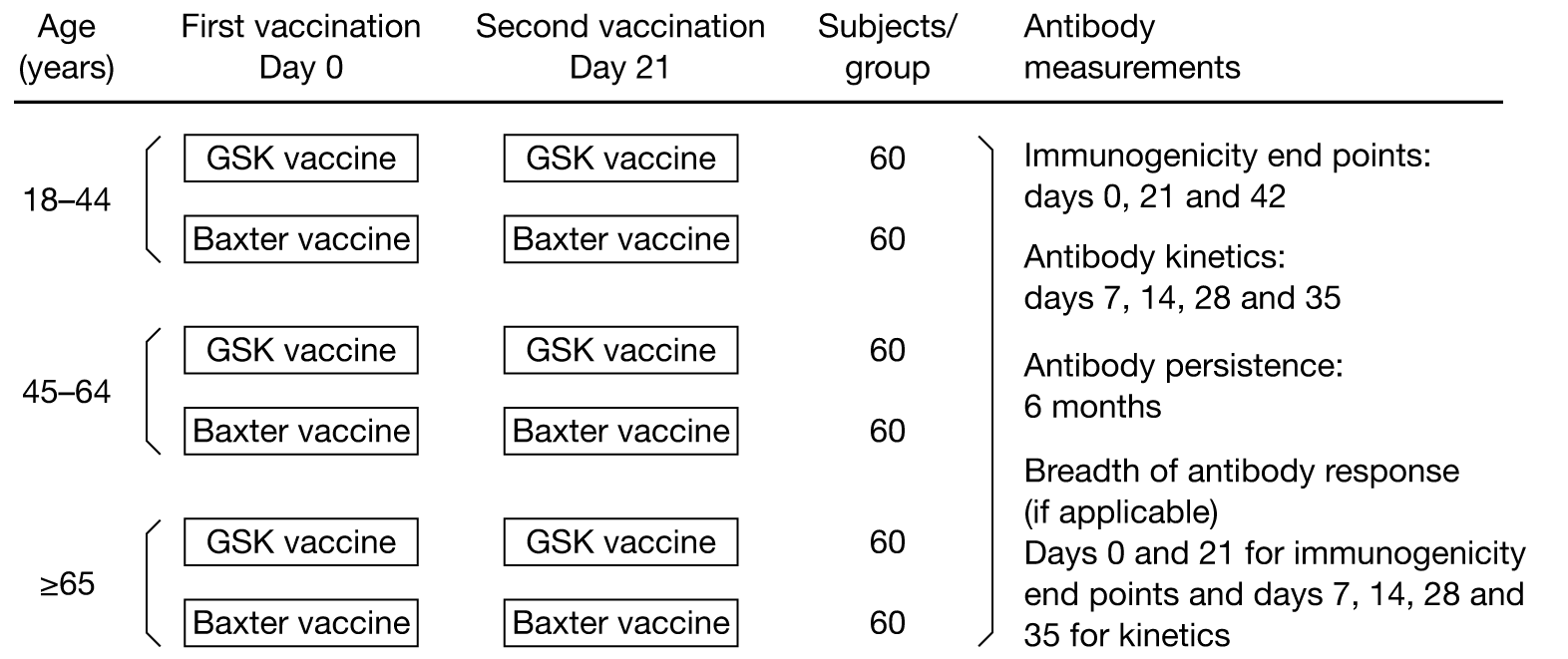
At least 360 subjects will be randomly allocated to receive two 7.5-µg HA doses of cell culture plain (i.e. non-adjuvanted) whole-virion A/California/2009 (H1N1) vaccine or two doses of AS03-adjuvanted influenza A/California/2009 (H1N1) split virus vaccine containing 3.75 µg of HA by IM injection. A second dose of the same vaccine containing the same quantity of antigen as in the first dose will be administered by the same route 21 days later. Subjects will be observed for local and systemic reactions for 30 minutes after each immunisation and will be monitored for any reactions and other AEs for 7 days after each immunisation.
Blood for immunogenicity studies will be obtained at day 0 (pre-immunisation), day 7 (± 1 day), day 14 (± 2 days), day 21 (± 2 days), day 28 (± 2 days), day 35 (± 3 days), day 42 (± 3 days) and day 180 (± 10 days). Blood for cellular responses will be taken on day 0, day 21 (± 2) and day 42 (± 3).
Time and events table
| Study visit | ||||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
| Days after the first vaccination: | ||||||||
| Window (days) | ||||||||
| 0 | (± 1) | (± 2) | (± 2) | (± 2) | (± 3) | (± 3) | (± 10) | |
| Study day | ||||||||
| 0 | 7 | 14 | 21 | 28 | 35 | 42 | 180 | |
| Informed consent | × | |||||||
| Inclusion/exclusion criteria | × | × | ||||||
| Medical/medication history | ||||||||
| Pregnancy test | × | × | ||||||
| Blood sample – antibody studies | × | × | × | × | × | × | × | × |
| Vaccination | × | × | ||||||
| Thermometer/diary card | × | |||||||
| Diary card training | × | |||||||
| Diary card returned/review | × | × | ||||||
| Reminder regarding unsolicited events | × | × | × | × | × | × | × | |
| AEs monitoring | × | × | × | × | × | × | × | |
| Termination of study | × | |||||||
Immunogenicity to influenza viruses will be evaluated by HI, MN, and possibly SRH responses.
Planned duration of the study
-
Expected enrolment interval: approximately 2 weeks.
-
Duration of individual subject’s participation: 6 months.
-
Total duration of study: approximately 7 months.
-
End of trial: corresponds to the last visit of the LSLV.
Subjects will be screened and consented to ensure entry criteria are met and then vaccinated. After a second dose of vaccine on 21 days later, subjects will be followed up for an additional 159 days.
Premature discontinuation of the study
The sponsor (UHL Trust), or the Chief Investigator (following consultation with the DH/National Institute for Health Research (NIHR) – the funder) has the right to discontinue this study at any time. If the clinical study is prematurely terminated, the Chief Investigator is to promptly inform the study subjects and should assure appropriate follow-up for the subjects. If the study is prematurely terminated, all procedures and requirements pertaining to archiving of documents will be observed.
Discussion of overall study design
This study was designed to evaluate immunogenicity of Baxter cell-culture, non-adjuvanted, whole-virion H1N1 vaccine, and GSK AS03-adjuvanted, split H1N1 vaccine with respect to the CHMP and FDA licensing criteria, and occurrence of local and systemic symptoms and signs following vaccine administration in adult and elderly people who have never previously been vaccinated with pandemic H1N1 influenza vaccine. The exclusion criteria reduce the likelihood of prior pandemic H1N1 infection among vaccinees, but the timing of the study cannot avoid this possibility. Moreover, it is possible that a further outbreak of pandemic H1N1 infection may occur during the first 42 days of the study. Nonetheless, this study design was considered best to evaluate the pandemic H1N1 vaccines purchased by the government. The need for the study was discussed by advisors to the DH. The study proposal was reviewed anonymously as part of the NIHR funding process.
Start date
The study is planned to commence early September 2009.
Study setting
This multicentre study will be conducted in University Hospitals of Leicester, Nottingham and Sheffield, and possibly in GP surgeries in Leicestershire, Nottinghamshire, Yorkshire and Derbyshire.
Study population
Initial approach
We will approach staff and health-care workers in university and health-care settings in Leicester, Nottingham and Sheffield either directly (i.e. personal contact) or through written information by post, e-mail or poster advertising. We may also request GPs to contact potentially suitable patients, either in writing or verbally, asking whether they would be prepared to learn more about the study with a view to participation. We may advertise the study through news items or advertisements on the local radio or in local newspapers. Respondents will be enrolled after they receive a detailed explanation of the study protocol and providing they meet inclusion and exclusion criteria and give signed informed consent. We will approach staff in frontline areas (acute admissions unit, acute wards, intensive care, bone marrow units) before non-health-care workers are enrolled.
Inclusion criteria
-
Mentally competent adults, who have signed an informed consent form after having received a detailed explanation of the study protocol.
-
Clinically healthy, male or female volunteers aged 18 years of age and older, including the over-65-year-olds, and those with stable high-risk medical conditions. (Note: ‘Stable’ is defined as having no medical consultations for an exacerbation or worsening of any chronic medical condition during the preceding 8 weeks and have been maintained on a stable drug regimen for at least 2 weeks prior to study entry as assessed by the medical history.)
-
Are able to understand and comply with all study procedures and to complete study diaries.
-
Individuals who can be contacted and are available for all study visits.
-
Females using secure contraceptive precautions including (1) the oral contraceptive pill or (2) condom/barrier contraception, or (3) partner has had a vasectomy, (4) be surgically sterilised or (5) post-menopausal (defined as at least 2 years since the last menstrual period).
Exclusion criteria
-
Subjects who are unable to lead an independent life either physically or mentally.
-
Women should not be pregnant or lactating.
-
Women who refuse to use a reliable contraceptive method on days 0–42 of the study.
-
Confirmed H1N1 infection, as determined by laboratory tests.
-
Have received oseltamivir or zanamivir for ILI since May 2009.
-
Have a household member who had confirmed H1N1 infection, as determined by laboratory tests, and/or received oseltamivir or zanamivir for ILI since May 2009.
-
Receipt of another investigational agent (vaccine or medicinal product) in the preceding 4 weeks.
-
Unwilling to refuse participation in another study during days 0–42 of the study.
-
Any clinically significant concurrent illness or unstable medical condition including: malignant tumours, acute or progressive renal or hepatic pathology, chronic obstructive pulmonary disease requiring oxygen therapy, and any active neurological disorder.
-
Individuals who have had acute respiratory pathology or infections requiring systemic antibiotic or antiviral therapy during the preceding 7 days (chronic antibiotic therapy for prevention of urinary tract infections is acceptable).
-
Subjects who had a temperature ≥ 38°C within 3 days of vaccination.
-
Any acute illness at the time of vaccination. (Note: minor infections without fever or systemic upset are not contraindications/exclusion criteria.)
-
Subjects with known or suspected impairment/alteration of immune function, including:
-
receipt of oral immunosuppressive drugs or other drugs listed in section 8 of the BNF or chloroquine, gold or penicillamine or other drugs listed in section 10.1.3 of the BNF to suppress a chronic disease process (Note: long-term, inhaled steroids for asthma management is acceptable.)
-
receipt of immunostimulants or interferon
-
receipt of an immunoglobulin preparation, blood products, and/or plasma derivatives within 3 months of the study
-
anyone at high risk of developing immunocompromising condition
-
received radiotherapy or chemotherapy during the 6 months preceding the study.
-
-
Subjects for whom surgery is planned during days 0–42 of the study.
-
Regularly drink more than 40 units of alcohol weekly.
-
Known or suspected drug abuse (recreational or prescribed).
-
Individuals who, in the opinion of the investigator, have conditions that might complicate interpretation of the study results.
-
Subjects with a history of anaphylaxis or serious reactions to vaccines; known hypersensitivity (other than anaphylactic reaction) to influenza viral protein, to any component of the study vaccines, to products containing mercury and to residues (egg and chicken protein, ovalbumin, formaldehyde, gentamicin sulphate, sodium deoxycholate and benzonase).
-
Subjects with a history of any neurological symptoms and signs, or anaphylactic shock following administration of any vaccine.
-
Actual or planned receipt of another vaccine, excluding seasonal influenza vaccine, during the period 3 weeks before to 3 weeks after vaccination on days 0 and 21.
Prior and concomitant treatment
During this trial medication prescribed to the subject prior to the start of the study will not be collected. All prescription medication (except minerals and vitamins), including non-study vaccines, being taken by the subjects on entry to the study and all prescription medication given in addition to the study vaccine during this clinical trial are to be regarded as concomitant medication and must be documented on the Concomitant Medications CRF.
In consideration of the overlapping northern hemisphere influenza vaccination campaign, the use of seasonal flu vaccines during the period 3 weeks before to 3 weeks after vaccination on days 0 and 21 is an exclusion criterion.
All subjects may continue therapy for chronic medical conditions provided that they are not listed in section 8 of the BNF or include chloroquine, gold or penicillamine or other drugs listed in section 10.1.3 of the BNF to suppress a chronic disease process. Subjects with chronic medical conditions must be maintained on a stable regimen for at least 2 weeks prior to study entry as assessed by the medical history.
Use of other medication including over-the-counter products should be discouraged during the study. Investigational drugs are prohibited during the course of the study.
The following concomitant treatments are discouraged and, if used, might lead to a major protocol violation and result in withdrawal of the subject from the study according to the medical judgement of the lead physician (see Exclusion criteria and Removal of subjects from therapy or assessments, above):
-
systemic steroids
-
other immunosuppressive agents
-
blood or plasma derivates, including immunoglobulin
-
non-study vaccines (with the exception of postexposure vaccinations in a medical emergency, e.g. hepatitis, rabies, tetanus) within 2 weeks.
Removal of subjects from therapy or assessments
The subject, or where applicable, the subject’s legally acceptable representative(s) can withdraw consent for participation in the study at any time without prejudice. The investigator can withdraw a subject if, in his/her clinical judgement, it is in the best interest of the subject or if the subject cannot comply with the protocol.
In addition, a subject may not be eligible for subsequent immunisation or may be discontinued from the study following occurrence of:
-
convulsions or any other neurological disturbances after vaccination
-
hypersensitivity to the investigational vaccine
-
other suspected side effects that could compromise the subject’s well-being.
Any subject who, despite the requirement for adequate contraception, becomes pregnant during the trial will not receive further immunisation. The site should maintain contact with the pregnant subject, and obtain pregnancy outcome information. It should be noted that pregnant women are at substantially increased risk from pandemic H1N1 influenza. The WHO and the Advisory Committee on Immunization Practices (ACIP) recommend the administration of pandemic H1N1 vaccine to pregnant women. It is likely that the DH will also recommend that pregnant women should receive pandemic H1N1 vaccine.
The subject will be followed up after withdrawal, the cause of which will be recorded in detail on the Study Termination CRF, and where appropriate, on the AEs and/or Concomitant Medications CRF. Where the withdrawal of a subject resulted from an AE, this will be documented in accordance with the procedures in Documentation/reporting of AEs and other clinical events, below.
Whenever possible, the tests and evaluations listed for the termination visit will be carried out.
Withdrawn subjects will not be replaced.
All subjects who have received investigational vaccines should be included in clinical events assessments, and all who provided pre-immunisation and postimmunisation blood samples at the scheduled times should be included in the immunogenicity assessments.
Stopping/pausing rule
There are no predetermined stopping rules other than those described above in Removal of subjects from therapy or assessments.
Vaccines
Vaccines
All subjects in this study will be randomised to receive two doses of the two pandemic influenza vaccines purchased by the government to confront the current H1N1 pandemic. Specifically the vaccine are:
-
Baxter, plain (i.e. non-adjuvanted), whole-virion, Vero cell-grown, influenza A/California/2009 (H1N1) pandemic vaccine, containing 7.5 µg of viral HA per 0.5-ml dose (given the trade name, Celvapan).
-
GlaxoSmithKline, AS03-adjuvanted, split-product, egg-grown, influenza A/California/2009 (H1N1) pandemic vaccine, containing 3.75 µg of viral HA and AS03 adjuvant composed of squalene, DL-α-tocopherol and polysorbate 80 (given the trade name Pandemrix).
The vaccines will be supplied by the DH as part of its initial consignment of vaccine from each manufacturer. It will be labelled, packaged and supplied with a package information leaflet exactly as procured from the manufacturers for the government’s national pandemic influenza vaccine administration programme.
Vaccine labelling, storage and packaging
All vaccine supplies must be stored between +2 °C to +8 °C, protected from light. The vaccine must not be frozen. Vaccines that have not been stored according to manufacturer’s instructions must not be used. In the event that the vaccine cannot be used, the vaccine must be replaced with fresh stock. The DH will supply the investigational H1N1 vaccine. The investigator (or pharmacist) will make an inventory and acknowledge receipt of all shipments of study vaccine.
Vaccine administration
The Principal Investigators at each study site will be responsible for the administration of the vaccine to subjects enrolled into the study according to the procedures stipulated in this study protocol. All vaccines will only be administered by personnel who are qualified to perform that function under applicable local laws and regulations for the specific study site.
The vaccine should be allowed to reach room temperature before use. The vaccine must be gently shaken and visually inspected before use. The vaccination site should be disinfected with a skin disinfectant (e.g. 70% alcohol). Before vaccination, the skin must be dry. DO NOT inject intravascularly or subcutaneously.
Precautions to be observed in administering study vaccine:
-
Study vaccines should not be administered to individuals with known hypersensitivity to any component of the vaccine.
-
An axillary temperature ≥ 38°C or serious active infection (with fever and systemic symptoms) are reasons for delaying vaccination.
-
Standard immunisation practices should be observed and care should be taken to administer the injection intramuscularly. As with all injectable vaccines, appropriate medical treatment and supervision should be readily available in case of rare anaphylactic reactions following administration of the study vaccine. Epinephrine 1 : 1000 and chlorphenamine (or equivalent adrenaline and antihistamine agents) should be available in case of any anaphylactic reactions. Care must be taken to ensure the vaccine is not injected into a blood vessel.
Administration of GSK Pandemrix vaccine
Nature and contents of container
Pandemrix is supplied in multidose vials (type I glass) containing 2.5 ml of vaccine (antigen) suspension (10 × 0.25-ml doses) with a stopper (butyl rubber) and vials (type I glass) of 2.5 ml of adjuvant (emulsion) (10 × 0.25-ml doses) with a stopper (butyl rubber).
Prior to administration, the two components should be mixed. The volume after mixing one vial of suspension (2.5 ml) with one vial of emulsion (2.5 ml) corresponds to 10 doses of vaccine (5 ml).
Instructions for mixing and administration of the vaccine
-
Before mixing the two components, the emulsion and suspension should be allowed to reach room temperature, shaken and inspected visually for any foreign particulate matter and/or abnormal physical appearance. In the event of either being observed, discard the vaccine.
-
The vaccine is mixed by withdrawing the contents of the vial containing the emulsion (vial B) by means of a syringe and by adding it to the vial containing the suspension (vial A).
-
After the addition of the emulsion to the suspension, the mixture should be well shaken. The mixed vaccine is a whitish emulsion. In the event of other variation being observed, discard the vaccine.
-
The volume of Pandemrix (5 ml) after mixing corresponds to 10 doses of vaccine.
-
The vial should be shaken prior to each administration.
-
Each vaccine dose of 0.5 ml is withdrawn into a syringe for injection.
-
The needle used for withdrawal must be replaced by a needle suitable for intramuscular injection.
Administration of Baxter Celvapan vaccine
Celvapan is supplied in multidose vials (type I glass) of 5 ml suspension (10 × 0.5 ml doses) with a stopper (bromobutyl rubber). The vaccine should be allowed to reach room temperature before use. Shake before use. Each vaccine dose of 0.5 ml is withdrawn into a syringe for injection.
Method of assigning subjects to Baxter and GSK vaccine groups
It would be desirable if vaccinees could be assigned Baxter and GSK vaccines at the same time, i.e. both vaccines arrived in each centre before regulatory approval (ethics and MHRA) is obtained. This may not occur, so procedures will be put in place to meet the following scenarios:
-
Both vaccines arrive at each centre prior to regulatory approval.
-
Only one vaccine arrives at each centre prior to regulatory approval.
Both vaccines arrive at each centre prior to regulatory approval We will use a block randomisation scheme to ensure that balance between vaccines is maintained and all volunteers are randomly allocated to groups. Subjects who meet the study admission criteria will be enrolled into the study and will be assigned a 5 digit subject number:
-
The first digit identifies the study site (1 for Leicester, 2 for Nottingham, 3 for Sheffield).
-
The second digit reflects the age of the subjects on day 0 (1 for 18–44 years, 5 for 45–64 years, and 9 for 65 years and older).
-
The following three digits identify the subject within the site and will be assigned sequentially, starting with 001 corresponding to the first subject enrolled within each age band.
Thus each centre will have three randomisation lists, each list corresponding to each age band.
Each centre will continue recruiting volunteers within an age band until the tally for the centres reaches a total of 120 subjects for that age band.
Volunteers in each age band will be randomised by a computer-generated randomisation code (1 : 1 proportions in block size(s) that will be determined by the statistician to ensure balance across groups). The randomisation code for each age band will be stored in individual, sequentially numbered envelopes, specific for each centre, and will be opened by the nurse with responsibility for vaccine administration. The type of vaccine for administration will be printed on an adhesive label. The label will be peeled from its backing and entered into an appropriate space in the Vaccine Log, ensuring that the instruction and vaccine that is administered actually agree (see below).
Only one study nurse/doctor will be responsible for vaccine administration in each centre. This study nurse/doctor will be unblinded with respect to the type of vaccine administered; he/she will play no other role in the study. To maintain blinding, volunteers will be told to look away, both during preparation and administration of the vaccine. It is essential that volunteers do not see the syringe that is used or whether the vaccine is translucent or cloudy. The ‘vaccine’ study nurse/doctor will document in the CRF: (1) the time of vaccine administration, and (2) the site of vaccine administration (left or right – vaccine will normally be given into the non-dominant arm). The ‘vaccine’ study nurse/doctor will record the volunteer’s trial number, together with the vaccine that was administered (the adhesive label in the envelope) in the ‘vaccine log’. The study nurse will check that the instruction in the envelope corresponds with the type of vaccine that is administered before and after each injection. He/she must inform the Principal Investigator immediately if the wrong vaccine was administered.
Only one vaccine arrives at each centre prior to regulatory approval This scenario is the one most likely to occur. It will be implemented if all regulatory approval is in place, and only one of the two trial vaccines is available in each centre, and the second vaccine will not arrive within 5 days of arrival of the first vaccine. Subjects who meet the study admission criteria will be enrolled into the study and will be assigned a 5 digit subject number:
-
As before, the statistician would generate three ‘randomisation’ lists for each centre, with a list for each age band.
-
As before, the first digit identifies the study site (1 for Leicester, 2 for Nottingham, 3 for Sheffield).
-
As before, the second digit reflects the age of the subjects on day 0 (1 for 18–44 years, 5 for 45–64 years; and 9 for 65 years and older);
-
As before, the next three digits identify the subject within the site.
However, the distribution of the numbers will be randomised, with the first 60 numbers corresponding to the first vaccine to arrive (vaccine 1), and the second 60 numbers corresponding to the second vaccine (vaccine 2).
As an example, the first person to be vaccinated in Leicester could have the number ‘10054’, i.e. with ‘1’ corresponding to Leicester, ‘0’ reflecting that he/she is aged 18–44 years, and ‘054’ being the volunteer’s unique trial number.
Each centre will continue recruiting volunteers within an age band until the tally for the centres reaches a total of 60 subjects for that age band for vaccine 1. The same process will then be carried out with vaccine 2, when it arrives.
Depending on the interval between arrival of GSK and Baxter vaccines, we will endeavour to ship sera from some volunteers who received vaccine 2 in the first tranche of sera that was sent for analysis. The laboratory staff will not know when vaccine 2 arrived and was first given, so will not know whether sera were collected from recipients of GSK or Baxter vaccine.
Vaccinees will not be told which vaccine they receive. They will be instructed to look away, both during preparation and administration of the vaccine. It is essential that volunteers do not see the syringe that is used or whether the vaccine is translucent or cloudy.
As before, only one study nurse/doctor will be responsible for vaccine administration in each centre. This study nurse/doctor will be unblinded with respect to the type of vaccine administered; he/she will play no other role in the study.
The ‘vaccine’ study nurse/doctor will record the volunteer’s trial number, together with the vaccine that was administered (the adhesive label in the envelope) in the Vaccine Log.
Adherence to randomisation
Vaccine will be given according to the randomisation list. Subjects will not be able to choose between GSK or Baxter vaccines.
Code break
Should both vaccines arrive at each centre prior to regulatory approval and GSK and Baxter vaccines be allocated randomly, the Principal Investigators will be provided with a sealed envelope containing individual code-break envelopes for each subject. These would be opened in a medical emergency only should a SAE occur, defined as: requiring medical intervention; frank myonecrosis; ulceration, superinfection or phlebitis at the injection site; extreme pain or tenderness with complete limitation of use of arm; or severe intractable headache requiring repeated narcotic treatment. In the event of such reactions, the investigators will notify the Sponsor immediately and document the event in the CRF.
Vaccination compliance
The site Principal Investigator will be responsible for adequate and accurate accounting of vaccine usage. The investigator or designee will administer the study vaccines only to individuals included in this study following the procedures set out in this study protocol. The date and time of vaccinations will be recorded. The investigator or delegate will track vaccines received, used and wasted and will retain all unused or expired products until it has been established that all accountability records are correct. Thereafter, all unused vaccines will be returned to the DH or destroyed at the investigational site. An overall summary of vaccines supplied, received, wasted, used and returned, re-assayed or destroyed will be prepared at the conclusion of the study.
Study procedures
Clinical procedures
Informed consent must be obtained from the subject, or where applicable, the subject’s legally acceptable representative(s) prior to the performance of any trial specific tests or evaluations, i.e. any unusual or non-routine procedures that involve risk, however trivial, to the subject.
The following procedures will be done during visits 1–8:
Visit 1: day 0: screening and first vaccination
-
Subjects will be enrolled providing they meet inclusion and exclusion criteria and give fully informed, written consent.
-
For females of childbearing potential, perform a urine pregnancy test.
-
If the subject meets all the inclusion criteria and none of the exclusion criteria, assign a study subject number relevant to the subject’s age and study centre (for further information see Method of assigning subjects to Baxter and GSK vaccine groups, above).
-
Obtain basic demography, including age, sex, ethnicity, previous influenza vaccination (past three seasons) and ILI (since May 2009).
-
Obtain and record significant medical history.
-
Record any current medications taken. Verify if the subject has taken an analgesic/antipyretic medication on day 0 prior to study vaccination. Document this information on the subject’s appropriate CRF.
-
Collect 10 ml of clotted blood pre-immunisation for baseline antibodies. Process blood and store serum for serology assays as described in Processing of samples for serology, below. If willing an additional 60 ml of blood will be taken for cellular assays.
-
Record oral temperature.
-
The vaccine administrator will give vaccine according to the randomisation list (if GSK and Baxter vaccines are both available) by IM injection in the deltoid muscle of the upper non-dominant arm.
-
Examine the site of injection of the vaccine for local reactions at the end of 30 minutes. These findings and any systemic reactions will be recorded on the appropriate CRF page.
-
Instruct each subject in the evaluation of local and systemic reactions (e.g. how to measure the maximum diameter of induration and erythema in millimetres at the injection site and how to record temperature with a thermometer). As a guide for subsequent evaluations, enter the findings from the 30-minute postinjection evaluation onto the subject diary card.
-
Give the subject diary card and thermometer for immunisation reactions to subjects and instructions for its completion. Tell subjects to:
-
complete the diary at approximately the same time each day;
-
notify study personnel immediately if the subject experiences a SAE. SAEs are defined in Definition of terms (above) and Adverse events, SUSARs and SAEs (below).
-
return the diary card to the site on the day 7 visit.
-
-
Schedule the day 7 visit.
Visit 2: day 7 (± 1): serology and diary card review
The following activities will be carried out on day 7 and recorded in the CRF:
-
Collect and review the first Immunisation Diary Card, including new medication/analgesia/antipyretics taken during the preceding week (concomitant medication).
-
Remind subject to notify study personnel immediately if the subject experiences a SAE.
-
Collect 10 ml of clotted blood for serology. Process blood and store serum.
-
Schedule the day 14 visit.
Visit 3: day 14 (± 2): serology
The following activities will be carried out on day 14 and recorded in the CRF:
-
Collect 10 ml of clotted blood for serology; process blood and store serum.
-
Remind subject to notify study personnel immediately if the subject experiences a SAE.
-
Schedule the day 21 visit.
Visit 4: day 21 (± 2): second vaccination
The following activities will be carried out on day 21 and recorded in the CRF:
-
Collect 10 ml of clotted blood for serology. Process blood and store serum. If willing and participated in cellular assay study, 60 ml will be obtained.
-
For females of childbearing potential, perform a urine pregnancy test.
-
Check if the subject has taken an analgesic/antipyretic medication on day 21 prior to study vaccination. Document this information on the subject’s appropriate CRF.
-
Record oral temperature.
-
The vaccine administrator will give the same type of vaccine as before by IM injection in the deltoid muscle of the upper non-dominant arm.
-
Examine the site of injection of the vaccine for local reactions at the end of 30 minutes. These findings and any systemic reactions will be recorded on the appropriate CRF page.
-
Remind each subject in the evaluation of local and systemic reactions (e.g. how to measure the maximum diameter of induration and erythema in millimetres at the injection site and how to record temperature with a thermometer).
-
Give the second diary card. Tell subjects to:
-
complete the diary at approximately the same time each day and to return it at the next visit
-
notify study personnel immediately if the subject experiences a SAE; SAEs are defined in Definition of terms (above) and Adverse events, SUSARs and SAEs (below).
-
return the diary card to the site on the day 28 visit.
-
-
Schedule the day 28 visit.
Visit 5: day 28 (± 2): serology and diary card review
The following activities will be carried out on day 28 and recorded in the CRF:
-
Collect and review the second Immunisation Diary Card, including new medication/analgesia/antipyretics taken during the preceding week (concomitant medication).
-
Remind subject to notify study personnel immediately if the subject experiences a SAE.
-
Collect 10 ml of clotted blood for serology; process blood and store serum.
-
Schedule the day 35 visit.
Visit 6: day 35 (± 3): serology
The following activities will be carried out on day 35 and recorded in the CRF:
-
Collect 10 ml of clotted blood for serology; process blood and store serum.
-
Remind subject to notify study personnel immediately if the subject experiences a SAE.
-
Schedule the day 42 visit.
Visit 7: day 42 (± 3): serology
The following activities will be carried out on day 42 and recorded in the CRF:
-
Collect 10 ml clotted blood for serology. Process blood and store serum. If willing and participated in cellular study, an additional 60mls of blood will be obtained.
-
Remind subject to notify study personnel immediately if the subject experiences a SAE.
-
Schedule the day 180 visit.
Visit 8: day 180 (± 10): study termination
The following activities will be carried out on day 180 and recorded in the CRF:
-
Collect 10 ml of clotted blood for serology; process blood and store serum.
-
Ensure completion of all study termination CRFs.
Processing of samples for serology and cellular responses
At least 10 ml of serum should be available for immunogenicity assays (HI, MN and, possibly, SRH). On each of the scheduled days requiring serum samples, the clotted blood will be stored at 2°C to 8°C and centrifuged within 24 hours. Sera will be stored in triplicate in cryovials, with all cryovials labelled with the volunteer’s trial code number, and day of collection. Samples should be stored frozen (below –14°C) until shipment. Blood for cellular assays will be obtained in a subset of 18- to 44-year-olds. Blood will be collected in Na-heparin tubes and stored at room temperature before centrifuging on a sucrose gradient to remove peripheral white blood cells. Cells will be stored at –80°C until processing.
Diary cards
Diary cards will be issued to all subjects instructing them to record their temperature and local and systemic symptoms at 6 and 24 hours after vaccination, and then daily for a total of 7 days, or longer should symptoms persist.
Information will be sought concerning the presence or absence of: redness at the injection site; local itching; local swelling; ulceration at the injection site; local pain; warm feeling at the injection site; tenderness to touch at the injection site; limitation of the use of the arm; headache; nausea; dizziness; diminished appetite; breathlessness; cough; coryza; wheeze; skin rash; generalised itching; fatigue; ‘other’ symptoms, use of antipyretic/relief medication and any changes in medication.
Subjects will score the severity of symptoms and effect on daily activities ranging from 0 (symptom absent), 1 (symptom occurred but not often or severe enough to cause inconvenience), 2 (symptom occurred often and severe enough to interfere with daily activities and requires no medical intervention) to 3 (symptom occurred often, severe enough to markedly interfere with daily activities; requires medical intervention).
Subjects will also be asked to document any unsolicited symptoms.
Subjects will be instructed that in the event of any SAE, he/she must notify the investigator immediately and be reviewed clinically. SAEs are defined in Definition of terms (above) and Adverse events, SUSARs and SAEs (below).
Local and systemic reactions
The occurrence of selected indicators of reactogenicity (listed below), which by definition, can only occur up to 6 days post vaccination, will be recorded on the Local and Systemic Reactions CRF rather than the AEs CRF:
-
local reactions ecchymosis, erythema, induration, swelling and pain at injection site
-
systemic reactions chills, malaise, myalgia, arthralgia, nausea, headache, sweating and fatigue
-
other indicators of reactogenicity stayed at home due to reactions, oral temperature and use of analgesic/antipyretic medication.
AEs, SUSARS and SAEs
An adverse event (AE) is defined as any untoward medical occurrence in a patient or clinical investigation subject administered a pharmaceutical product at any dose that does not necessarily have to have a causal relationship with this treatment. An AE can be, therefore, any unfavourable and unintended sign (including an abnormal laboratory finding, for example), symptom or disease temporally associated with the use of an investigational product, whether or not considered related to the investigational product. This definition includes intercurrent illnesses or injuries and exacerbation of pre-existing conditions.
All AEs will be monitored until resolution or, if the AE becomes chronic, a cause identified. If an AE is unresolved at the conclusion of the study, a clinical assessment will be made by the investigator and Medical Monitor whether continued follow-up of the AE is warranted.
The severity of events reported on the AEs CRF will be determined by the investigator as:
-
mild transient effect with no limitation in normal daily activity
-
moderate some limitation in normal daily activity, but no medical attention needed
-
severe unable to perform normal daily activity and requiring medical attention.
The relationship of the study treatment to an AE will be determined by the investigator based on the following definitions:
-
Not related The AE is not related if exposure to the investigational vaccine has not occurred or the occurrence of the AE is not reasonably related in time or the AE is considered unlikely to be related to use of the investigational vaccine, i.e. there are no facts (evidence) or arguments to suggest a causal relationship.
-
Possibly related The administration of the investigational vaccine and AE are considered reasonably related in time and the AE could be explained by causes other than exposure to the investigational vaccine.
-
Probably related Exposure to the investigational vaccine and AE are reasonably related in time and the investigational vaccine is more likely than other causes to be responsible for the AE or is the most likely cause of the AE.
The relationship of the study treatment to an AE will be determined by the Principal Investigator.
A suspected unexpected serious adverse event (SUSAR) is one that is not listed in the current Summary of Product Characteristics or the Investigator’s Brochure or an event that is by nature more specific or more severe than a listed event.
A serious adverse event (SAE) is defined as any untoward medical occurrence that at any dose:
-
results in death
-
is life-threatening (i.e. the subject was, in the opinion of the investigator, at immediate risk of death from the event as it occurred); it does not refer to an event which hypothetically might have caused death if it were more severe
-
requires or prolongs inpatient hospitalisation
-
results in persistent or significant disability/incapacity (i.e. the event causes a substantial disruption of a person’s ability to conduct normal life functions)
-
results in a congenital anomaly/birth defect
-
requires intervention to prevent permanent impairment or damage
-
is an important and significant medical event that may not be immediately life-threatening or resulting in death or hospitalisation but, based upon appropriate medical judgement, may jeopardise the patient/subject or may require intervention to prevent one of the other outcomes listed above.
Note: A ‘possible vaccine failure’ should be reported as a serious AE only if it resulted in an infectious disease which should have been prevented by the vaccine implied.
Adverse events that do not fall into these categories are defined as non-serious. It should be noted that a severe AE need not be serious in nature and that a SAE need not, by definition, be severe.
In addition, a pre-existing event or condition that results in hospitalisation should be recorded on the medical history CRF. If the onset of an event occurred before the subject entered the trial (e.g. any preplanned hospitalisation for conditions like cosmetic treatments or for non-emergency routine visits for a pre-existing condition), the hospitalisation would not lead to an AE being classified as serious unless, in the view of the investigator, hospitalisation was prolonged as a result of participation in the clinical trial or was necessary due to a worsening of the pre-existing condition.
Documentation/reporting of adverse and other clinical events
All study subjects will be observed for at least 30 minutes after a vaccination for evidence of immediate reactions in general and in particular for symptoms of allergic phenomena (such as rashes, itching or other allergic manifestations). Each subject, or where applicable, the subject’s legally acceptable representative(s) will be instructed to complete a diary card for 7 days following each administration, to describe local and systemic reactions and other selected indicators of reactogenicity. If a local and systemic reaction or fever (derived from measured oral temperatures ≥ 38.0°C) continues beyond the 7-day period after a vaccination, it will also be recorded on the AEs CRF. If the subject recovers on the last day, then this fact will be recorded on the Local and Systemic Reaction CRF. All AEs must be reported and documented. The period of observation for AEs extends from the time the subject receives vaccination through until 3 weeks after vaccination.
All AEs necessitating a physician’s visit or consultation and/or leading to premature study discontinuation and all SAEs will be collected throughout the entire study and data will be reconciled at study termination.
All AEs, regardless of severity, will be monitored by the investigator until resolution. All subjects experiencing AEs – whether considered associated with the use of the study vaccine or not – must be monitored until symptoms subside and any abnormal laboratory values have returned to baseline, or until there is a satisfactory explanation for the changes observed, or until death, in which case a full pathologist’s report should be supplied, if possible. All findings must be reported on an AEs CRF and on the Serious Adverse Event form, if necessary, which is part of the investigator’s study file. All findings in subjects experiencing AEs must be reported also in the subject’s medical records.
In addition, any event resulting in a subject’s withdrawal from subsequent vaccinations or from follow-up should be reported according to the protocol instructions.
All SAEs that occur during the course of the trial, whether considered to be associated with the study vaccination or not, have to be reported within 24 hours by telephone or fax to either of the following:
-
Study Sponsor Mrs Carolyn Maloney, Research and Development, Leicester General Hospital, Gwendolen Road, Leicester LE4 5PW.
-
Chief Investigator Professor Karl Nicholson, Infectious Diseases Unit, Leicester Royal Infirmary, Leicester.
For trial-related emergencies out of office hours please contact the Principal Investigators at each site:
-
Principal Investigator, Leicester Dr Iain Stephenson, Infectious Diseases Unit, Leicester Royal Infirmary, Leicester.
-
Principal Investigator, Nottingham Professor Jonathan Nguyen-Van-Tam, Room A40d Clinical Sciences Building, City Hospital, Nottingham NG5 1PB.
-
Principal Investigator, Sheffield Professor Robert Read, Royal Hallamshire Hospital, Sheffield.
As far as possible, all points raised on the ‘Serious Adverse Event’ form need to be addressed and faxed immediately to the Study Monitor. The original must be retained by the investigator. The event must also be documented on the Adverse Events CRF. After receipt of the initial report, the Study Monitor/Sponsor will review the information and contact the investigator if it is necessary to obtain further information for assessment of the event.
Any medication or other therapeutic measures used to treat the event will be recorded on the appropriate CRF(s) in addition to the outcome of the AE. Any serious adverse reaction must be reported to the EC in a timely manner, according to local regulations. Adequate documentation will be provided to the sponsor showing that the EC has been properly notified. The sponsor must also comply with the applicable regulatory requirement(s) relating to the reporting of unexpected serious and non-serious adverse drug reactions to the regulatory authority(ies) and the EC.
Poststudy events
Any AE occurring at any time outside the observation period or after the end of the study and considered to be caused by the study vaccine – and therefore a possible adverse drug reaction – must be reported to the sponsor.
Halting criteria
Any serious adverse reaction will be reported to the sponsor and EC in accordance with the European Directive 2001/20/EC. If any SAE is considered as probably related to the study vaccine, this will be considered by the Chief Investigator, study co-applicants, sponsor and DH representatives, who will make a collective decision to continue or halt the study.
Study monitoring/auditing
Investigators and/or their study staff will be trained at the latest during the initiation meeting. Monitoring and auditing procedures will be followed in order to comply with GCP guidelines and to ensure validity of the study data. During each monitoring visit source data verification will be performed by qualified staff.
Monitoring
The clinical study sites will be monitored by regular site visits and telephone calls to the investigator by qualified staff representing the Sponsor. By frequent communication, the site monitor will ensure that the study is conducted according to the protocol. CRFs and all original data collected at the site should be available for review during monitoring visits. During these visits, the site monitor should review drug accountability records and might review document retention including the Investigator’s Study File. Additionally, the site monitor should check that clinical study procedures are observed and discuss any problems with the investigators.
Source data verification
Inspection and examination of CRFs and source documents (all original recordings, medical records) – giving due consideration to data protection and medical confidentiality – will be undertaken by representatives of the Sponsor.
All data not recorded directly on the CRFs, as defined in Documentation of study findings of this study protocol, below, will be verified by checking CRF entries against source documents in order to ensure that the data have been completely and accurately reported as required by the study protocol.
Source data verification will be performed and recorded following the sponsor’s SOP. The subject or the subject’s legally acceptable representative must also allow access to the subject’s medical records, if required. Each subject, or the subject’s legally acceptable representative, will be informed of this prior to the start of the study.
During or after the clinical study, the regulatory authorities, the EC and/or representatives of the sponsor may request access to all source documents, CRFs and other study documentation for on-site audit or inspection.
Documentation of study findings
All study data must be entered into the CRFs by the investigator who will sign and date the entries. If the investigator authorises other persons in his/her staff to make entries on the CRF, the names, positions, signatures and initials must be supplied to the sponsor.
The following data may be reported directly on the CRFs and are considered to be source data:
-
medical history
-
vaccination time, concomitant medication
-
study termination, and
-
comments.
CRFs must be completed during/after each study visit.
A reasonable explanation must be given by the investigator for all missing data.
If corrections are made to entries in the CRF by the investigator or designates, the words or figures must be crossed through, leaving the initial entry legible. The correction must then be dated and initialled. Incorrect entries must not be covered with correcting fluid, obliterated or made illegible in any way. If further corrections are made after review and signature by the investigator, he/she must confirm and endorse the changes by signing and dating the study termination CRF again.
As part of the conduct of the trial, the sponsor may have questions about the CRF data. These questions will be documented using Data Clarification Forms (DCFs). The investigator will file each of the DCFs for the trial.
Record retention
Investigators must retain all study records according to applicable regulations in a secure and safe facility. The investigator must notify the sponsor of any change in the location, disposition or custody of the study files.
Upon completion of the study, all study documents will be collated and stored by the Chief Investigator.
The CHMP requires retention for the maximum period of time permitted by the institution, but not less than 15 years. It is the responsibility of the sponsor to inform the investigator/institution as to when these documents no longer need to be retained.
Data protection
The sponsor respects the subjects’ rights to privacy and will ensure the confidentiality of their medical information in accordance with all applicable laws and regulations. The sponsor as Data Controller according to the European Directive on the protection of individuals with regard to the processing of personal data and on the free movement of such data [95/46/EC] confirms herewith compliance to Directive 95/46/EC in all stages of Data Management.
Changes in the conduct of the study or planned analysis
Planned changes in the conduct of the study will be described in protocol amendments; changes in the planned analysis will be described in the clinical study report. An amendment is a written description of change(s) to, or formal clarification of, a study protocol. The EC must be informed of all amendments and if necessary prior review and documented approval/favourable opinion must be sought for ethical aspects. Approval must also be obtained from the authorities, if necessary. Such amendment will be agreed upon by the sponsor, the investigator, the EC and authorities, if necessary, prior to implementation.
Analysis
Blood sampling windows
Blood samples taken in the following time windows will be evaluable:
-
Pre-immunisation:
-
– day 0.
-
-
Post immunisation:
-
– day 7 (window: days 6–8)
-
– day 14 (window: days 12–16)
-
– day 21 (window: days 19–23)
-
– day 28 (window: days 26–30)
-
– day 35 (window: days 32–38)
-
– day 42 (window: days 39–45)
-
– day 180 (window: days 170–190).
-
Statistical methods, sample size, and analyses
Sample size
The aim of the trial is to establish whether GSK and Baxter vaccines satisfy all three CPMP criteria, and if so compare them in terms of immunogenicity (for each vaccine/age group and each vaccine type). The sample size is in line with standard practice. The protocols for seasonal EU vaccine clinical trials and the criteria for assessment have been standardised within the EU. They stipulate that trials should be done with groups of at least 50 subjects. We will recruit 60 per group, allowing for up to 17% dropout. This will enable for example the trial to detect a 10–20% difference in seroprotection and SCRs at the 5% significance level with 80% power.
Definition of populations to be analysed
Definition of populations to be analysed:
All enrolled population
-
All subjects in the enrolled population.
Full analysis set, immunogenicity
-
All subjects in the enrolled population who:
-
– actually receive at least one dose of study vaccination, and provide at least one evaluable serum sample both before and after baseline.
-
Per protocol (PP) population, Immunogenicity
-
All subjects in the enrolled population who:
-
– receive all the relevant doses of vaccine correctly, and provide evaluable serum samples at the relevant time points and have no major protocol violation as defined prior to statistical analysis.
-
A major deviation is defined as a protocol deviation that is considered to have a significant impact on the immunogenicity result of the subject.
AEs population
-
All subjects who received at least one dose of the study vaccine who:
-
– provide postbaseline AEs data.
-
Analysis of demographic and baseline characteristics
Descriptive statistics (mean, standard deviation, median, minimum and maximum) for age, together with distributions of subjects by sex and ethnic origin, previous influenza vaccination (during the past three seasons), recent ILI in the patient (since May 2009) and presence of prevaccination antibody to the vaccine strain will be summarised overall, for each vaccine group, and by age group.
Analysis of immunogenicity criteria
Blood samples for immunogenicity assays will be collected on days 0 (prevaccination), 7, 14, 21, 28, 35, 42 and 180.
Antibody response will be evaluated by HI, neutralisation and, possibly, SRH in all subjects.
Haemagglutination inhibition and/or MN antibody titres need to be assessed rapidly and so will be analysed in three tranches:
-
first tranche of sera measuring antibodies before and after first injection: days 0, 7, 14 and 21
-
second tranche of sera measuring antibodies before and after second injection: days 21, 28, 35 and 42.
-
third tranche of sera measuring persistence of antibodies: days 0, 42 and 180.
Haemagglutination inhibition and MN assays will be performed at the Health Protection Agency Centre for Infections, Enteric, Respiratory & Neurological Virus Laboratory, London, UK. SRH tests may be done at the National Institute for Biological Standards and Control. Antibody titrations will be done in duplicate. Pre- and postvaccination samples from each person will be titrated simultaneously, as specified in the CHMP guidance. 5 The titre assigned will be the geometric mean of two independent determinations.
Cellular assays will be performed at Imperial College, London, by means of the T-cell ELISPOT assay. Antigen will be influenza HA (swine H1). There are no recognised correlates of protection for influenza.
Immunogenicity objectives
Primary immunogenicity objective
The primary immunogenicity objective is to evaluate the immunogenicity of Baxter cell-culture, non-adjuvanted, whole-virion H1N1 vaccine, and GSK AS03-adjuvanted, split H1N1 vaccine with respect to CHMP and FDA licensing criteria.
These criteria for adults aged between 18 and 60 years are, for sera collected ‘approximately 3 weeks after vaccination’:
-
number of seroconversions or significant increase in anti-HA antibody titres > 40%
-
mean geometric increase > 2.5
-
the proportion of subjects achieving an HI titre of ≥ 40 or SRH titre of 25 mm2 should be > 70%.
For adults aged over 60 years these criteria are:
-
number of seroconversions or significant increase in anti-HA antibody titres > 30%
-
mean geometric increase > 2.0
-
the proportion of subjects achieving an HI titre of ≥ 40 or SRH titre of 25 mm2 should be > 60%.
The measures of immunogenicity will be calculated as:
Geometric mean titre (GMT) or geometric mean area (GMA) For each vaccine/age group and each vaccine type, least squares GMTs for HI and MN data (GMAs for SRH data), associated 95% confidence interval (CI) and median, minimal and maximal titre value will be determined for each visit, i.e. days 0, 7, 14, 21, 28, 35, 42 and 180.
Geometric mean ratio (GMR) (increase) For each vaccine/age group and each vaccine type, the least squares GMRs will be calculated for the HI, MN and SRH results for the following time points of the study: day 7/day 0, day 14/day 0, day 21/day 0, day 28/day 0, day 35/day 0, day 42/day 0, as well as the associated 95% CIs and the median, minimal and maximal n-fold increase. Statistical methods used to analyse GMRs will be identical to those described above for GMTs (GMAs).
Seroconversions – percentages of subjects with seroconversion (or significant increase in HI titre) The number and proportion of subjects achieving seroconversion or significant increase in HI titres or SRH area from pre-immunisation to each visit after first immunisation will be tabulated for each vaccine/age group and each age group.
-
Seroconversion is defined as negative pre-vaccination serum (< 10 for HI, < 4 for SRH)/positive post-vaccination titre (≥ 40 for HI, area ≥ 25 mm2 for SRH).
-
Significant increase in antibody titre/area is defined as at least a fourfold increase in HI or a 50% increase in area from non-negative pre-vaccination serum ( 10 for HI, ≥ 4 for SRH).
Seroprotection – percentages of subjects achieving each of the following thresholds, inverse HI titre ≥ 40, SRH area ≥ 25 mm2 The number and proportion of subjects achieving each threshold at each visit will be tabulated for each vaccine/age group and each vaccine type.
All statistical analyses for HI and SRH will be performed on the logarithmically (base 10) transformed values. Titres below the limit of detection for assays will be set to 1 in 5 for HI and 4 mm2 for SRH for the purposes of analysis. Original values will be presented in all listings.
The above immunogenicity criteria will be applied to sera collected 21 days after completion of vaccination, i.e. 21 days after the second vaccine dose. For pandemic vaccines, all three criteria should be met.
Should both GSK and Baxter vaccines satisfy all three CPMP criteria, we will compare the immunogenicity of the two vaccines (for each vaccine/age group and each vaccine type) in terms of:
-
geometric mean titres 21 days after each vaccination
-
geometric mean ratio increases 21 days after each vaccination
-
seroprotection rates 21 days after each vaccination
-
SCRs 21 days after each vaccination.
Comparisons will be made using parametric or non-parametric tests as appropriate for continuous outcomes, and Pearson’s chi-squared test or Fisher’s exact test, where appropriate, for categorical outcomes. Further analyses using (generalised) linear models will explore the effect of baseline covariates on the outcomes. For populations (1) and (2), analyses using multiple imputation to allow for missing data will also be undertaken as a sensitivity analysis. No formal adjustment will be made for multiple testing but associated p-values will be interpreted cautiously.
Secondary immunogenicity measures
The secondary immunogenicity measures are:
-
To identify whether one or two doses of vaccine are required to satisfy the licensing criteria We will assess whether either GSK or Baxter vaccine is able to meet all three Committee for Proprietary Medicinal Products (CPMP) criteria 21 days after the first vaccine dose. This will be assessed for each vaccine/age group, and each vaccine type.
-
To examine the kinetics of the antibody responses to vaccination This will be measured in terms of the ability of GSK or Baxter vaccine to meet any one, or all three, of the CPMP criteria 7 and 14 days after the first and second doses – for each vaccine/age group, and each vaccine type.
-
To examine persistence of antibody at 6 months This will be assessed in terms of the proportion of vaccinees who have inverse HI antibody titres of ≥ 40, SRH areas of ≥ 25 mm2, and inverse MN antibody titres of ≥ 40 and ≥ 80. Antibody persistence will be assessed for each vaccine/age group, and each vaccine type;
-
To evaluate the breadth of the antibody response to the antigenic variant (if appropriate, i.e. an antigenic drift variant emerges prior to the 2010–11 influenza season) This will be done using the new antigenic variant as test antigen in HI, MN and, possibly, by SRH tests by comparing the above primary immunogenicity measures on days 21 and 42 for each vaccine, by age group, and all age groups combined.
As with the primary immunogenicity outcomes comparisons will be made using parametric or non-parametric tests as appropriate for continuous outcomes, and Pearson’s chi-squared test or Fisher’s exact test where appropriate for categorical outcomes. Further analyses using (generalised) linear models will explore the effect of baseline covariates on the outcomes. For populations (1) and (2), analyses using multiple imputation to allow for missing data will also be undertaken as a sensitivity analysis. No formal adjustment will be made for multiple testing but associated p-values will be interpreted cautiously.
Exploratory analyses
The possible effect of previous seasonal influenza vaccination, age, presence of detectable levels of antibody before vaccination, as well as vaccine type – and any interactions will be explored using (generalised) linear models.
If one vaccine is available significantly before the other, then the trial will adopt a sequential allocation procedure switching to randomisation when the second one becomes available. If such a situation arises then the comparisons outlined above in Primary immunogenicity objective, will be non-randomised ones and potentially subject to bias (for example due to temporal effects). In this situation the use of date (time) of vaccination will also be used as an explanatory covariate in (generalised) linear models in order to explore the potential impact of this on the trial outcomes.
Analysis of local and systemic symptoms and AEs
For the purpose of data partitioning and analysis, the study will be partitioned into the following broad time intervals to compare short-term reactogenicity to vaccination:
-
vaccination to 48 hours
-
48 hours to 7 days, and
-
beyond 7 days.
Analysis will focus on comparisons of the incidence and severity of local and systemic reactions as indicated by the presence of specific symptoms, symptom scores and temperature elevations.
Local and systemic symptoms
The incidences of local and systemic reactions following vaccination will be summarised by vaccine group.
The occurrence of selected indicators of reactogenicity (listed below), which by definition, can only occur up to 6 days post vaccination, will be recorded on the Local and Systemic Reactions Diary CRF rather than the Adverse Events CRF.
-
local reactions ecchymosis, erythema, induration, swelling and pain at injection site
-
systemic reactions chills, malaise, myalgia, arthralgia, nausea, headache, sweating and fatigue
-
other indicators of reactogenicity stayed at home due to reactions, oral temperature, and use of analgesic/antipyretic medication.
If a reaction occurs more than once for a subject, the reaction will be classified according to the highest occurring severity.
All study subjects will be observed for at least 30 minutes after a vaccination for evidence of immediate reactions in general and in particular for symptoms of allergic phenomena (such as rashes, itching or other allergic manifestations).
Each subject will be instructed to complete a diary card for 7 days following each vaccination, to describe local and systemic reactions and other selected indicators of reactogenicity. If a local and systemic reaction or fever (derived from measured oral temperatures ≥ 38.0°C) continues beyond the 7-day period after a vaccination, it will also be recorded on the Adverse Events CRF. If the subject recovers on the last day, then this fact will be recorded on the Local and Systemic Reaction CRF.
We will summarise the occurrence of solicited local and systemic symptoms (point estimates and 95% CI) by vaccine type in terms of incidence, intensity, and relation to vaccination. We will use the two-sided Fisher’s exact tests to compare groups where appropriate.
Frequencies and percentages (together with 95% CIs) of subjects experiencing each reaction will be presented for each symptom severity. Summary tables showing the occurrence of any local or systemic reaction overall and at each time point will also be presented.
The severity of local reactions, including injection-site ecchymosis, erythema, swelling, and induration, will be categorised as none, 1 to ≤ 25 mm, 26 to ≤ 50 mm and > 50 mm.
The severity of pain and systemic reactions will be categorised as ‘none’, ‘mild’ (transient with no limitation in normal daily activity), ‘moderate’ (some limitation in normal daily activity) and ‘severe’ (unable to perform normal daily activity).
Distribution of body temperature, staying at home due to vaccine reaction and the use of analgesic/antipyretic medication occurring during 7 days after each vaccination will be tabulated. Fever will be defined as a temperature of ≥ 38.0°C, and severe fever as ≥ 40.0°C. Oral temperature will be categorised as < 38°C, 38°C to < 39°C, 39°C to < 40°C, and ≥ 40°C.
All postvaccination reactions (local and systemic), use of relief medication and absence from work due to any AEs will be summarised as none versus any.
For the local and systemic reaction safety variables, differences among the groups after vaccination will be analysed by using Pearson’s chi-squared test or Fisher’s exact test where appropriate.
Adverse events
The reporting and documentation of unsolicited events throughout the study is described above (AEs, SUSARs and SAEs; Documentation/reporting of adverse and other clinical events).
The original verbatim terms used by investigators to identify AEs in the CRFs will be mapped to preferred terms using the MedDRA dictionary. The AEs will then be grouped by MedDRA preferred terms into frequency tables according to system organ class. All reported AEs, as well as AEs at least possibly related to study vaccine, will be summarised according to system organ class and preferred term within system organ class. These summaries will be presented by vaccination group. When an AE occurs more than once for a subject, the maximal severity and strongest relationship to the vaccine group will be counted.
Additionally, three separate summaries will be produced: (1) SAEs; (2) AEs that are possibly or probably related to vaccine; and (3) AEs that are unrelated to vaccine. Data listings of all AEs will be provided by subject. In addition, a listing of subjects withdrawn from the study because of an AE will be presented.
Interim/preliminary analyses
To provide the DH with information as rapidly as possible, with the goal of informing DH vaccination strategy, the following interim analyses of data from this study are planned:
-
Solicited/unsolicited events (local and systemic symptoms, relief medication, and absence from work due to any AEs) during:
-
– days 0–6 (first vaccination)
-
– days 0–21 (first vaccination)
-
– days 21–27 (second vaccination)
-
– days 21–41 (second vaccination).
-
-
HI and MN antibody titres on days:
-
– days 0, 7, 14, 21 (first tranche of sera measuring antibodies before and after first injection)
-
– days 21, 28, 35, 42 (second tranche of sera measuring antibodies before and after second injection).
-
References
- Potter CW, Nicholson KG, Webster RG, Hay AJ. Textbook of influenza. Oxford: Blackwell; 1998.
- Myers KP, Olsen CW, Gray GC. Cases of swine influenza in humans a review of the literature. Clin Infect Dis 2007;44:1084-8.
- Hodder RA, Gaydos JC, Allen RG, Top FH, Nowosiwsky T, Russell PK, et al. Influenza A/swine at Fort Dix, NJ (Jan–Feb 1976), III. Extent of Spread and Duration of the Outbreak. J Infect Dis 1977;136:369-75.
- Sencer DJ, Millar JD. Reflections on the 1976 swine flu vaccination program. Emerg Infect Dis 2006;12:29-33.
- Centers for Disease Control and Prevention (CDC) . Swine influenza A (H1N1) infection in two children: Southern California, March–April 2009. Morb Mortal Wkly Rep 2009;58:400-2.
- Novel Swine-Origin Influenza A (H1N1) Virus Investigation Team . Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N Engl J Med 2009;360:2605-15.
- Chan M. World Now at the Start of 2009 Influenza Pandemic n.d. URL: www.who.int/mediacentre/news/statements/2009/h1n1_pandemic_phase6_20090611/en/index.html (accessed 14 August 2009).
- World Health Organization (WHO) . Pandemic (H1N1) 2009: Update 61 n.d. URL: www.who.int/csr/don/2009_08_12/en/index.html (accessed 14 August 2009).
- ECDC Surveillance report . Pandemic (H1N1) 2009: Analysis of Individual Cases Reports in EU and EAA Countries 2009. URL: www.ecdc.europa.eu/en/healthtopics/Documents/090810_Influenza_A(H1N1)_Analysis_of_individual_data_EU_EEA-EFTA.pdf.
- Fraser C, Donnelly CA, Cauchemez S, . Pandemic potential of a strain of influenza A (H1N1): Early findings. Science n.d.:324-61.
- Centers for Disease Control and Prevention (CDC) . Serum cross-reactive antibody response to a novel influenza A (H1N1) virus after vaccination with seasonal influenza vaccine. Morb Mortal Wkly Rep 2009;58:521-4. URL: www.cdc.gov/mmwr/preview/mmwrhtml/mm5819a1.htm.
- Meiklejohn G, Eickhoff TC, Graves P. Antigenic drift and efficacy of influenza virus vaccines, 1976–1977. J Infect Dis 1978;138:618-24.
- De Donato S, Granoff D, Minutello M, Lecchi G, Faccini M, Agnello M, et al. Safety and immunogenicity of MF59-adjuvanted influenza vaccine in the elderly. Vaccine 1999;17:3094-101.
- US Census Bureau n.d. URL: www.census.gov/ipc/www/popclockworld.html.
- Kieny M-P. WHO Briefing on H1N1 Vaccines 2009.
- Lermann SJ. Reactogenicity and immunogenicity of monovalent A/New Jersey/76 influenza virus vaccines in children. J Infect Dis 1977:136-70.
- Nicholson KG, Tyrrell DAJ, Harrison, Potter CW, Jennings. R, Clark A, . Clinical studies of monovalent whole virus and subunit A/USSR/77 (H1N1) vaccine: serological responses and clinical reactions. J Biol Stand 1979;7:123-36.
- European Committee for Proprietary Medicinal Products . Note for Guidance on Harmonization of Requirements for Influenza Vaccines 1997. URL: www.emea.europa.eu/pdfs/human/bwp/021496en.pdf (accessed 15 July 2009).
- Committee for Proprietary Medicinal Products (CPMP) . Guideline on Dossier Structure and Content for Pandemic Vaccine Marketing Authorisation 2004. URL: http://archives.who.int/prioritymeds/report/append/62EMEAguidelines.pdf (accessed 15 July 2009).
- Treanor JJ, Wilkinson BE, Masseoud F, Hu-Primmer J, Battaglia R, O’Brien D, et al. Safety and immunogenicity of a recombinant hemagglutinin vaccine for H5 influenza in humans. Vaccine 2001;19:1732-37.
- Treanor JJ, Campbell JD, Zangwill KM, Rowe T, Wolff M, . Safety and immunogenicity of an inactivated subvirion influenza A (H5N1) vaccine. N Engl J Med 2006;354:1343-51.
- Stephenson I, Nicholson KG, Colegate AE, Podda A, Wood J, Ypma E, et al. Safety and antigenicity of non-adjuvanted and MF59-adjuvanted influenza A/Duck/Singapore/97 (H5N3) vaccine: a randomised trial of two potential vaccines against H5N1 influenza. Lancet 2001;357:1937-43.
- Bresson J-L, Perronne C, Launay O, Gerdil C, Saville M, Wood J, et al. Safety and immunogenicity of an inactivated split-virion influenza A/Vietnam/1194/2004 (H5N1) vaccine: phase I randomised trial. Lancet 2006;367:1657-64.
- Nolan TM, Richmond PC, Skeljo MV, Pearce G, Hartel G, Formica NT, et al. Phase I and II randomised trials of the safety and immunogenicity of a prototype adjuvanted inactivated split-virus influenza A (H5N1) vaccine in healthy adults. Vaccine 2008;26:4160-7.
- Keitel WA, Campbell JD, Treanor JJ, Walter EB, Patel SM, He F, et al. Safety and immunogenicity of an inactivated influenza A/H5N1 vaccine given with or without aluminum hydroxide to healthy adults: results of a phase I–II randomized clinical trial. J Infect Dis 2008;198:1309-16.
- Stephenson I, Nicholson KG, Glück R, Mischler R, Newman RW, Palache AM, et al. Safety and antigenicity of whole virus and subunit influenza A/Hong Kong/1073/99 (H9N2) vaccine in healthy adults: phase I randomised trial. Lancet 2003;362:1959-66.
- Cox RJ, Madhun AS, Hauge S, Sjursen H, Major D, Kuhne M, et al. A phase I clinical trial of a PER.C6 cell grown influenza H7 virus vaccine. Vaccine 2009;27:1889-97.
- Ehrlich HJ, Müller M, Oh HML, Tambyah PA, Joukhadar C, Montomoli E, et al. A clinical trial of whole-virus H5N1 vaccine derived from cell culture. N Engl J Med 2008;358:2573-84.
- Stephenson I, Nicholson KG, Colegate A, Podda A, Wood J, . Boosting immunity to influenza H5N1 with MF59-adjuvanted H5N3 A/Duck/Singapore/97 vaccine in a primed human population. Vaccine 2003;21:1687-93.
- Stephenson I, Bugarini R, Nicholson KG, Podda A, Wood JM, Zambon MC, et al. Cross-reactivity to highly pathogenic avian influenza H5N1 viruses after vaccination with nonadjuvanted and MF59-adjuvanted influenza A/Duck/Singapore/(7 (H5N3) vaccine: a potential priming strategy. J Infect Dis 2005;191:1210-15.
- Stephenson I, Nicholson KG, Hoschler K, Zambon MC, Hancock K, DeVos J, et al. Antigenically distinct MF59-adjuvanted vaccine to boost immunity to H5N1. N Engl J Med 2008;359:1631-3.
- Galli G, Hancock K, Hoschler K, DeVos J, Praus M, Bardelli M, et al. Fast rise of broadly cross-reactive antibodies after boosting long-lived human memory B cells primed by an MF59 adjuvanted prepandemic vaccine. Proc Natl Acad Sci USA 2009;106:7962-7.
- Leroux-Roels I, Borkowski A, Vanwolleghem T, Dramé M, Clement F, Hons E, et al. Antigen sparing and cross-reactive immunity with an adjuvanted rH5N1 prototype pandemic influenza vaccine: a randomised controlled trial. Lancet 2007;370:580-9.
- Leroux-Roels I, Bernhard R, Gérard P, Dramé M, Hanon E, Leroux-Roels G, et al. Broad Clade 2 cross-reactive immunity induced by an adjuvanted clade 1 rH5N1 pandemic influenza vaccine. PLoS One 2008;3.
- Levie K, Leroux-Roels I, Hoppenbrouwers K. An adjuvanted, low-dose, pandemic influenza A (H5N1) vaccine candidate is safe, immunogenic, and induces cross-reactive immune responses in healthy adults. J Infect Dis 2008;198:642-9.
- Lin J, Zhang J, Dong X, Fang H, Chen J, Su N, et al. Safety and immunogenicity of an inactivated adjuvanted whole-virion influenza A (H5N1) vaccine: a phase I randomised controlled trial. Lancet 2006;368:991-7.
- Ehrlich HJ, Müller M, Oh HML, Tambyah PA, Joukhadar C, Montomoli E, et al. A clinical trial of whole-virus H5N1 vaccine derived from cell culture. N Engl J Med 2008;358:2573-84.
- Stephenson I, Nicholson KG, Glück R, Mischler R, Newman RW, Palache AM, et al. Safety and antigenicity of whole virus and subunit influenza A/Hong Kong/1073/99 (H9N2) vaccine in healthy adults: phase I randomised trial. Lancet 2003;362:1959-66.
- Stephenson I, Das RG, Wood JM, Katz JM. Comparison of neutralising antibody assays for detection of antibody to influenza A/H3N2 viruses: an international collaborative study. Vaccine 2007;25:4056-63.
- Wood JM, Gaines-Das RE, Taylor J, Chakraverty P. Comparison of influenza serological techniques by international collaborative study. Vaccine 1994;12:167-74.
- Wood J. Update on Development of International Standards for H5N1 Antibodies n.d.
- n.d. URL: www.who.int/vaccine_research/diseases/influenza/meeting_120209/en/ (accessed July 2009).
List of abbreviations
- AE
- adverse event
- ANOVA
- analysis of variance
- BNF
- British National Formulary
- BWP
- Biotechnology Working Party
- CDC
- Centers for Disease Control
- CHMP
- Committee for Medicinal Products for Human Use
- CI
- confidence interval
- CPMP
- Committee for Proprietary Medicinal Products
- CRF
- case report form
- DH
- Department of Health
- EMA
- European Medicines Agency
- FDA
- the US Food and Drug Administration
- GMT
- geometric mean titre
- GSK
- GlaxoSmithKline
- HA
- haemagglutinin
- HI
- haemagglutination inhibition
- ICHGCP
- International Conference on Harmonisation – Good Clinical Practice
- ILI
- influenza-like illness
- IM
- intramuscular
- MHRA
- Medicines and Healthcare Products Regulatory Agency
- MN
- microneutralisation
- MREC
- Multicentre Research Ethics Committee
- NA
- neuraminidase
- NIBSC
- National Institute for Biological Standards and Control
- NYMC
- New York Medical College
- OR
- odds ratio
- PT
- protective titre
- RCT
- randomised controlled trial
- SAE
- serious adverse event
- SCR
- seroconversion rate
- SRH
- single radial haemolysis
- UHL
- University Hospitals of Leicester
- WHO
- World Health Organization
- WV
- whole virion
All abbreviations that have been used in this report are listed here unless the abbreviation is well known (e.g. NHS), or it has been used only once, or it is a non-standard abbreviation used only in figures/tables/appendices, in which case the abbreviation is defined in the figure legend or in the notes at the end of the table.