
Notes
Article history
The research reported in this issue of the journal was commissioned and funded by the HTA programme on behalf of NICE as project number 06/60/01. The protocol was agreed in June 2007. The assessment report began editorial review in January 2008 and was accepted for publication in May 2010. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the referees for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
none
Permissions
Copyright statement
© 2011 Queen’s Printer and Controller of HMSO. This journal is a member of and subscribes to the principles of the Committee on Publication Ethics (COPE) (http://www.publicationethics.org/). This journal may be freely reproduced for the purposes of private research and study and may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NETSCC, Health Technology Assessment, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2011 Queen’s Printer and Controller of HMSO
Chapter 1 Background
Description of health problem
Description of Crohn’s disease
Inflammatory bowel disease (IBD) refers to a group of chronic intestinal diseases characterised by inflammation of the gastrointestinal mucosa. The most common types of IBD are ulcerative colitis and Crohn’s disease (CD). CD can affect any part of the gastrointestinal tract, from mouth to anus, but most commonly the terminal ileum (35%) or the ileocaecal region (40%) are affected. 8
The main symptoms of CD are dependent on disease location and include chronic or nocturnal diarrhoea, abdominal pain, anal lesions, rectal bleeding and weight loss. Clinical signs include pallor, cachexia, abdominal mass or tenderness, perianal fissures, fistulas or abscesses. Systemic symptoms include malaise, anorexia or fever. 8–10 Extraintestinal symptoms related to intestinal inflammation include spondyloarthritis, cutaneous manifestations or ocular inflammation. 10 In children, growth failure may be the primary manifestation of CD. 11
CD can be defined using the Vienna classification (see Disease classification), i.e. by location, disease behaviour (inflammatory, stricturing, penetrating) and age at diagnosis. 12 Stricturing disease refers to the narrowing of the bowel which can lead to bowel obstruction, while penetrating (or fistulising) disease refers to the creation of abnormal passageways (fistulas) between the bowel and other structures such as the skin. Inflammatory disease (non-stricturing, non-penetrating) causes inflammation without any strictures or fistulas.
Approximately 40%–50% of patients present with ileocolonic disease at the time of diagnosis, approximately 30% have isolated small bowel disease and approximately another 30% have pure colonic disease. It is estimated that only 10%–15% of patients have a change in disease localisation in the 10 years after diagnosis. 13 Disease behaviour at diagnosis is inflammatory (non-stricturing and non-penetrating) in 70% of patients, stricturing in 17% and penetrating (fistulas, abscesses or both) in 13% of patients. 14
Where the ileum and colon are affected, this is usually complicated by intestinal obstruction, inflammatory mass or abscess. Where disease is limited to the colon, patients commonly present with rectal bleeding, perianal complications and extraintestinal complications involving the skin or joints. Gastric and duodenal manifestations include nausea and vomiting, epigastric pain or gastric outlet obstruction. 15
Common complications are strictures, fistulas and perianal disease. Fistulas can develop between loops of bowel adjacent to the bladder, vagina or skin. Perianal disease comprises fissures, fistulas and abscesses, and perianal manifestations may precede the onset of bowel symptoms. 8,15 Symptomatic perianal disease requiring therapy occurs in around 35% of CD patients. 16 CD may also be complicated by sequelae related to malabsorption such as anaemia or metabolic bone disease. 15 Rare complications include acute dilatation, perforation and massive haemorrhage, especially when the disease affects the colon.
CD is characterised by recurring flares alternating with periods of remission. Most patients take medication for a large period of their life because if they stop they might experience a disease flare, but some drugs are tapered off during periods of remission, then if a patient experiences a flare he or she then returns to therapy. 13
Aetiology
The aetiology of CD remains unknown. It is generally accepted that the disease is a response to environmental triggers (infection, drugs or other agents) in genetically susceptible individuals. 9 Smoking has been shown to be a risk factor in CD, with suggestions that smokers are more than twice as likely to develop the disease. 17 Areas under investigation to identify pathogenic mechanisms include epidemiology (e.g. diet, drugs, water supply), the gut–environmental interface (e.g. work on luminal bacteria), the inflammatory process (e.g. cell signalling pathways) and genetics (e.g. studies on gene expression). 9 Exacerbating factors include intercurrent infections, smoking and the use of non-steroidal anti-inflammatory drugs, while the issue of stress initiating or exacerbating CD remains controversial. 15
Diagnosis
Recent efforts have focused on discovery of biomarkers that may eventually lead to the development of specific diagnostic tests for CD,18 but as yet no definitive diagnostic test exists for CD. Overlapping features with other IBDs, a potentially insidious onset, and the heterogeneity of manifestations and/or presentation without gastrointestinal symptoms can make diagnosis difficult. 15 Diseases with symptoms in common with CD include infectious diarrhoea, small bowel lymphoma, ulcerative colitis, appendicitis, coeliac disease and irritable bowel syndrome. A detailed clinical history, a physical examination, laboratory tests and endoscopic evaluation are necessary to make an accurate diagnosis. A diagnosis of IBD should be contemplated in patients presenting with chronic (bloody or non-bloody) diarrhoea, particularly nocturnal diarrhoea and/or weight loss, abdominal pain, fever or extraintestinal manifestations. Family history of the disease should be considered. Signs of volume depletion, ulceration of the oral mucosa, perianal lesions or abdominal tenderness may be observed on physical examination. Laboratory tests should rule out infection and look for markers of IBD such as low serum albumin level or vitamin B12 deficiency. Imaging studies of the bowel may be helpful; abdominal radiography may reveal mucosal oedema or dilated loops of small bowel or colon consistent with either inflammation or obstruction. On endoscopy, CD is characterised by deep, linear ulcerations that can occur as segmental areas of mucosal involvement separated by areas of normal intervening mucosa (‘skip lesions’). Biopsy findings usually demonstrate transmural inflammation. 19
CD may be unsuspected and incorrectly diagnosed in the elderly, with as many as 60% of patients being misdiagnosed initially compared with a misdiagnosis rate of only 15% in younger people. The delay in diagnosis has been calculated as 6.4 years after onset of symptoms in older patients compared with 2.4 years in younger individuals. 20
Disease classification
CD is a heterogeneous condition with a variety of clinical manifestations and presentations. The ‘Vienna classification’ introduced a schema to categorise the disease according to three important elements: age at diagnosis (A), location of the disease (L) and disease behaviour (B). The Vienna classification, summarised in Table 1, was revised in 2005 and this modified version, termed the Montreal classification,21 expanded the number of categories within each of the three elements as shown in Table 1.
| Classification element | Vienna | Montreal | |
|---|---|---|---|
| AGE at diagnosis (years) (A) | A1 < 40 | A1 < 16 | |
| A2 > 40 | A2 17–40 | ||
| A3 > 40 | |||
| LOCATION of disease (L) | L1 Terminal ileum | L1 Terminal ileum | L1 + L4 Terminal ileum + upper GI |
| L2 Colon | L2 Colon | L2 + L4 Colon + upper GI | |
| L3 Ileocolon | L3 Ileocolon | L3 + L4 Ileocolon + upper GI | |
| L4 Upper GI | L4 Upper GI | ||
| BEHAVIOUR of disease (B) | B1 Non-stricturing non-penetrating | B1 Non-stricturing non-penetrating | B1p Non-stricturing non-penetrating + perianal |
| B2 Stricturing | B2 Stricturing | B2p Stricturing + perianal | |
| B3 Penetrating | B3 Penetrating | B3p Penetrating + perianal | |
The categorisation of CD using these classification systems has allowed descriptions of the progression/natural history of the disease and raised the possibility of identifying genes or environmental factors (including treatments) that may be associated with particular features of the disease or with the rapidity of progression from one category to another. For example, using the Vienna classification and retrospective analysis of case notes for a cohort of 290 patients with up to 25 years of follow-up, Louis et al. 22 found that location of disease was relatively stable with only 15.9% of patients exhibiting a change over a decade while disease behaviour was more labile, changing for 45.9% of patients in a decade. Similarly Cosnes et al. 23 reported that over 20 years most patients progressed to penetrating or stricturing disease, that initial location of disease was a determinant of disease behaviour, and that year-by-year disease activity was poorly influenced by previous behaviour of the CD.
Natural history
The disease location of CD is fairly stable; however, the behaviour of the disease can vary substantially during its course. The disease changes from non-stricturing to either stricturing (in 27% of patients) or penetrating disease (in 29% of patients). 14 After the first year of diagnosis, 10%–30% of patients have an exacerbation, 15%–25% have low activity and 55%–65% are in remission; 13%–20% have a chronic active course of disease activity, 67%–73% have a chronic intermittent course and only 10%–13% remain in remission for several years. 14 Most patients with CD will require surgery within 20 years. 14 The lifetime risk for developing fistulas has been reported to be between 20% and 40%. Perianal fistulas are most common, followed by entero-enteric, with many patients developing a fistula at or before diagnosis of CD. 24 CD is associated with an increased risk of colonic carcinoma and the overall mortality is slightly higher than that of the overall population. 9
A Danish study25 of an inception cohort of 373 CD patients found the following disease activity distributions: 80% of patients had high activity at diagnosis, decreasing to an almost stable value of 30% in the following 25 years; a constant 15% of patients overall had low activity; and around 55% could expect to be in remission each year. Individual patients however changed from year to year between relapse and remission. The study further found that over a 10-year period 20%–30% of patients could expect to go into remission each year. There was a slight indication of the disease ‘burning out’, as late in the disease course (more than 15 years post diagnosis) slightly more patients (29%) changed from activity to remission than the 14% who changed from remission to activity. A separate analysis of 171 patients followed for at least 7 years after diagnosis found that, between years 3 and 7, 25% of patients had active disease every year, 22% were in remission and 53% changed between years in remission and years with relapse. 25 This disease course was independent of initial treatment, age, sex, localisation and symptoms at diagnosis or time from onset to diagnosis. With regard to hospital admissions, 83% were admitted during the year of diagnosis; this decreased during the following 5 years to a constant 20% each year.
A US modelling study26 examined a retrospective cohort and estimated a future life expectancy of 46.4 years for a representative CD patient aged 28.1 years at the time of diagnosis. The projected clinical course consisted of 11.1 years in remission (with no medication), 0.51 years of requiring surgery, 18.9 years in post-surgery remission (no medication), 12.7 years of receiving aminosalicylate or a similar medication, and disease severe enough to require corticosteroids or immunosuppressives lasted 3.2 years. This was based on a sample of 174 patients and on treatment practices used between 1970 and 1993, which may have changed over the course of the study.
A Norwegian study12 which followed up 221 CD patients prospectively for 5 years found that during the observation period 28% had undergone surgery. At the time of the 5-year visit 54% used sulfasalazine and 5-aminosalicylic acid, 25% used oral glucocorticosteroids and 13% used azathioprine. There were 16% who had symptoms that interfered with everyday activities and 72% had taken oral glucocorticosteroids at some point during the 5 years.
These cohort studies and the models based on them indicate that the clinical course estimates will vary depending on a variety of characteristics of the patients within the cohort.
Incidence and prevalence
CD can occur at any age, but manifests itself mainly during late adolescence or early adulthood. Peak onset is between 15 and 30 years of age. 27,28 The incidence in younger years is higher in women than in men. 27,29 There is some inconsistency regarding differences in prevalence between women and men overall, with some studies finding a higher prevalence in women, and some finding no difference. 29 There is an increased prevalence among first- and second-degree relatives, suggesting the involvement of genetic factors. 28 CD may also present later in life (sixth and seventh decades) when there tends to be more colonic involvement and disease manifestations may be less severe. 20
The extent of CD varies across the world and is most common in developed countries, with the UK having one of the highest rates. It was previously thought that IBD occurred less frequently among ethnic minorities. However, studies of migrant populations have shown that ethnic and racial differences are more likely to be attributable to lifestyle and environmental influences than true genetic differences. Similar rates of IBD have been found in African-Caribbean and white children and adults in the UK. 17 No association between CD and social class was found in a UK prevalence study; it has been suggested that this is attributable to exposure to risk factors becoming more similar across social classes. 29
In regions with a high prevalence of CD, the incidence increased between the 1950s and 1980s, and stabilised after that, which can be explained by an increased availability of gastroenterology units and increased awareness of the disease. 28,30 Some studies suggest that there is still an upward trend, which may be due to continued variations in environmental risk factors. 29 Increases in less developed countries have recently been noted, and it has been suggested that this is a result of changes in lifestyle (e.g. more exposure to smoking, changes in diet). 28
Table 2 shows the incidence and prevalence of CD in the UK, taken from studies published from 2000 onwards. The incidence ranges from 3.8 to 10 per 100,000 per year and the prevalence ranges from 50 to 375 per 100,000. For children, the British Paediatric Surveillance Unit found an estimated incidence of 5.3 per 100,000 per year. 11 Differences in incidence and prevalence estimates may result from the way data are gathered, changes in disease awareness and diagnosis over time, or changes in disease risk factors. There is no national CD database that could be used to determine numbers of CD patients.
| Study | Population/sample | Incidence CD (adults) | Prevalence CD (adults) |
|---|---|---|---|
| Carter et al., 20049 | Review by the British Society of Gastroenterology (based on several studies, no details on sample size) | 5–10/100,000 per year | 50–100/100,000 |
| Ehlin et al., 200329 | The 1970 British Cohort study and the 1958 National Child Development Study (1-week national birth cohorts); total sample population of 22,680 (70% of target population) | NR |
1970 cohort at age 30 years: 375/100,000 (95% CI 262 to 488) 1958 cohort at age 30 years: 211/100,000 (95% CI 127 to 295) 1958 cohort at age 42 years: 325/100,000 (95% CI 221 to 430) |
| Rubin et al., 200030 | Systematic search of GP records in North England (based on a population of 135,723) | 8.3/100,000 per year (95% CI 7.5 to 20.3) | 144.8/100,000 (95% CI 124.8 to 168.8) |
| NACC31 | UK (no details on sample) | 5–10/100,000 per year | 100/100,000 |
| Shivananda et al., 199632 | Multicentre study of 20 centres across Europe during 1991–3, one of these in Leicester (total sample size unclear) |
Non-immigrants: 3.8/100,000 per year (95% CI 0.7 to 6.9) Immigrants: 5.6/100,000 per year (95% CI 0.0 to 12.5) All aged 15–64 years |
NR |
| Stone et al., 200333 | Fifteen general practices recruited through the Trent Focus Collaborative Research Network, UK (based on a population of 86,801) | NR | 130/100,000 (95% CI 107 to 157) |
| Yapp et al., 200034 | Information from clinical records, the department of pathology database and a questionnaire sent to local family practitioners in the city of Cardiff (total sample size unclear) | 5.6/100,000 per year (95% CI 4.4 to 6.8) | NR |
Impact of health problem
Significance for patients in terms of ill health
The impact on patients and society is high, as patients are often diagnosed at a young age and ill health may be lifelong. Medical treatments can cause secondary health problems and surgery can result in complications such as impotence or intestinal failure. Patients can find symptoms embarrassing and humiliating, and may have difficulties in gaining employment or insurance. Younger people in particular may have psychological problems and growth failure or retarded sexual development. Approximately 75% of patients are fully capable of work 1 year after diagnosis and 15% of patients are unable to work after 5–10 years of disease. 9 Similarly, a Danish study25 found that, except for the year of diagnosis, 75%–80% of patients were fully capable of work each year, 9%–16% were incapable and 9%–11% only partly capable; after 15 years, 15% of patients obtained a disablement pension. The National Association of Colitis and Crohn’s Disease (NACC) website35 states that most sufferers can be maintained in remission for most of the time and are able to lead a full working life; however, some with severe disease do not achieve their educational and career potential.
Information sheets produced by the NACC35 relating to the most frequently asked questions to the NACC helpline cover the following issues: difficulties finding insurance companies that will provide life cover, mortgage protection, or travel, critical illness or health insurance (when offered, insurance can be more expensive than if they did not have CD); managing bloating and wind; managing diarrhoea; concerns for young people (particularly focusing on emotional aspects such as embarrassment, body image, anxiety); and supporting someone with CD.
A prospective cohort study36 of health-related quality of life (HRQoL) in 231 patients with CD found that patients’ main worries (in decreasing order of magnitude of concern) related to ‘having an ostomy bag’, ‘uncertain nature of disease’, ‘energy level’, ‘having surgery’, ‘pain and suffering’, ‘eating normally’, ‘feelings about my body’ and ‘effects of medication’. Other concerns related to loss of bowel control, career/finances, sexual relationships, body/self-image, being a burden to others, developing cancer or dying early. Quality of life (QoL), as measured in this study by the Short Form (36) Health Survey questionnaire (SF-36), was lower for CD patients than for the general population (the SF-36 measures various aspects of physical and mental functioning). Factors having a negative impact on QoL were active disease, hospitalisation, receiving steroids, having colonic disease and surgery.
A discussion with a patient representative, who has also worked for the NACC helpline, highlighted the following issues of particular concern to patients who contact the helpline (Denise Cann, NACC, 2007, personal communication):
-
difficulty in coping with unpredictability of disease (particularly where patients have been in remission) and a lack of control over it
-
difficulty in gaining employment or staying employed
-
difficulty in finding insurance
-
impact on family and social life
-
impact on relationships, sexual activity and pregnancy
-
embarrassing nature of disease, e.g. flatulence, need to frequently use toilets because of diarrhoea, incontinence
-
distressing symptoms such as rectovaginal fistulas where faeces can be passed through the vagina
-
coping with the general tiredness, malaise and lack of energy
-
coping with side effects of treatments
-
fear that (new) treatment may not work
-
coping with depression
-
difficulty particularly for children and teenagers to cope emotionally
-
costs: drug and continence prescription charges, cost of many sets of clothing/linen, trips to hospital, loss of earnings.
Significance for NHS
A UK study from 200437 calculated the cost of CD. The setting was an NHS university hospital with a target population of around 330,000. Table 3 lists the costs for different patient groups.
| Patient group | Mean cost for 6 monthsa |
|---|---|
| All CD patients (with complete 6-month follow-up ‘prevalent’ cases) | £1652 (95% CI £1221 to £2239) |
| Ambulatory group | £516 (95% CI £452 to £618) |
| Patients hospitalised during study period | £6923 (95% CI £5415 to £8919)b |
| Quiescent disease | £275 (95% CI £235 to £319) |
| Ambulatory patients suffering disease exacerbation (‘flare’) | £578 (95% CI £431 to £701) |
| Hospitalised patients | £5444 (95% CI £3894 to £9242)b |
| New ‘incident’ cases | £2662 (95% CI £1006 to £5866) |
Costs comprised all secondary care costs, including drugs, tests (e.g. endoscopy, laboratory tests), in- and outpatient services and surgery. Cost estimates also included all associated costs such as staff salaries, pharmacy services and other miscellaneous costs. Costs did not include visits to a GP, but these were estimated separately and amounted to < £30 per patient per 6 months. The median number of days lost from household and recreational activities in 6 months were 20 [interquartile range (IQR) 9–60]. Fifty per cent of employed patients had some loss of employment days, with a median loss of earnings of £299 (IQR £119–597). Mean out-of-pocket expenses were £66 (range £0–750) and included travel and over-the-counter medication. No patient in this cohort received infliximab or another tumour necrosis factor (TNF) inhibitor (anti-TNF-α antibody).
The contribution of different items and services to the overall cost of CD in all patients was as follows (estimated from Figure 1 in Bassi et al. 37): 37% surgery, 24% inpatient costs, 11% outpatient costs, 11% tests (laboratory tests, X-ray, endoscopy) and 17% drugs.
Six-month resource use in ambulatory and hospitalised CD patients is shown in Table 4 (adapted from Table 2 in Bassi et al. 37). There were a total of 260 bed-days for CD within the 6-month period, 196 surgical bed-days and 12 days of intensive care bed occupancy.
| Parameter (per 6 months) | Ambulatory CD patients (n = 130) Mean (range) |
Hospitalised CD patients (n = 28) Mean (range) |
|---|---|---|
| Outpatient services (visits) | ||
| IBD related | 2.2 (0–7) | 2.9 (0–8) |
| Extraintestinal | 1.25 (1–3) | – |
| Dietitian | 0.07 (0–3) | 0.1 (0–1) |
| Stoma nurse | – | 0.03 (0–1) |
| Laboratory testsa | 7.6 (0–28) | 35.3 (9–66) |
| Radiology | ||
| Plain X-ray | 0.07 (0–1) | 1.4 (0–4) |
| Barium enema | 0.01 (0–1) | 0.07 (0–1) |
| Barium follow-through | 0.1 (0–1) | 0.30 (0–2) |
| Ultrasound abdomen | 0.02 (0–1) | 0.18 (0–1) |
| CT abdomen/pelvis | 0.01 (0–1) | 0.01 (0–1) |
| MRI abdomen/pelvis | – | 0.07 (0–1) |
| White blood cell scan | 0.01 (0–1) | 0.07 (0–1) |
| DEXA scan | 0.07 (0–1) | – |
| Fistulogram | 0.01 (0–1) | – |
| Endoscopy | ||
| OGD | 0.15 (0–1) | 0.11 (0–1) |
| Sigmoidoscopy | 0.05 (0–2) | 0.18 (0–1) |
| Colonoscopy | 0.1 (0–1) | 0.3 (0–3) |
| Hospital admission | NA | |
| Number of admissions | – | 1.1 (1–2) |
| Length of each admission (days) | – | 14 (4–40) |
Measurement of disease severity in adults
Working definitions of disease severity have been developed by the Practice Parameters Committee of the American College of Gastroenterology,10 and are:
-
Mild–moderate disease:
Mild-moderate Crohn’s disease applies to ambulatory patients able to tolerate oral alimentation without manifestations of dehydration, toxicity (high fevers, rigors, prostration), abdominal tenderness, painful mass, obstruction, or > 10% weight loss.
-
Moderate–severe disease:
Moderate–severe disease applies to patients who have failed to respond to treatment for mild–moderate disease or those with more prominent symptoms of fever, significant weight loss, abdominal pain or tenderness, intermittent nausea or vomiting (without obstructive findings), or significant anaemia.
-
Severe–fulminant disease:
Severe–fulminant disease refers to patients with persisting symptoms despite the introduction of steroids as outpatients, or individuals presenting with high fever, persistent vomiting, evidence of intestinal obstruction, rebound tenderness, cachexia, or evidence of an abscess.
-
Remission:
Remission refers to patients who are asymptomatic or without inflammatory sequelae and includes patients who have responded to acute medical intervention or have undergone surgical resection without gross evidence of residual disease. Patients requiring steroids to maintain well-being are considered to be ‘steroid dependent’ and are usually not considered to be ‘in remission’.
The severity of CD is difficult to assess, and a global measure encompassing clinical, endoscopic, biochemical and pathological features is not available. 38 The most widely used disease activity measures include the Crohn’s Disease Activity Index (CDAI), the Harvey–Bradshaw Index (HBI) or Simple Index, a simplified version of the CDAI, and the Perianal Disease Activity Index (PDAI). A commonly used HRQoL measure is the Inflammatory Bowel Disease Questionnaire (IBDQ). Other measures include the Crohn’s Disease Endoscopic Index of Severity (CDEIS).
The CDAI was developed in the 1970s as there was a need for a single index to assess disease severity. Variables measured include number of liquid stools, abdominal pain, general well-being, extraintestinal complications, use of antidiarrhoeal drugs, abdominal mass, haematocrit and body weight. Scores range from 0 to approximately 600, with higher scores corresponding to more severe disease (see Appendix 1 for full description of the index and the scoring system used). Values of below 150 are suggestive of quiescent disease (remission) and values above 450 are associated with very severe disease. 39 Severe disease is thought to be above 300. Some investigators, however, have arbitrarily labelled CDAI scores of 150–219 as mildly active disease and scores of 220–450 as moderately active disease. 38
The CDAI has been criticised for having limitations. It does not cover aspects of QoL, such as psychological, social, sexual and occupational functioning. A patient with a low CDAI score may still be severely limited by the disease in those areas. 40 Substantial variability exists when different observers review the same case histories and calculate the CDAI score, although this can be reduced after discussion and education about the terminology. The calculation is based in part on a daily diary kept by the patient for 7 days before the evaluation. In practice, some investigators and study co-ordinators assist the patient to retrospectively complete the diary at the time of an evaluation visit; there is no information on the prevalence of this practice. The CDAI score may be low in patients whose primary symptom is drainage of enterocutaneous fistulas, presumably because the presence of an actively draining fistula contributes only 20 points to the score. The CDAI is therefore not an appropriate instrument for assessing the activity of draining abdominal or perianal enterocutaneous fistulas. The CDAI has been criticised for giving too much weight to ‘general well-being’ and ‘intensity of abdominal pain’, as these are relatively subjective items. However, these aspects of disease are important to patients. 41
Clinical studies have variously defined a clinical response as a decrease in CDAI of 50, 60, 70 or 100 points. The US Food and Drug Administration (FDA) and the European Medicines Agency (EMEA) suggested in 2000 that a clinically meaningful decrease in the CDAI score is a decrease of 100 points. 41
The HBI is a modified/simplified version of the adult CDAI. It uses a single day’s reading for diary entries and excludes three variables (body weight, haematocrit and use of drugs for diarrhoea). Code values are added together rather than summing the products of code values and coefficients (see Appendix 1). Scores range from 0 to 20, with higher scores corresponding to worse disease. The CDAI can be predicted reasonably well from the HBI. 42 Other instruments derived from the CDAI are: the Cape Town Index, which includes parameters on subjective symptoms, physician clinical findings and laboratory data; the three-variable version of the CDAI used for survey research; and the van Hees Index, which includes laboratory parameters, sex (male or female) and seven clinical features and excludes subjective, patient-related items such as well-being and pain. 40
The PDAI was developed to account for the morbidity and impairment of QoL of patients with perianal disease, and to evaluate the effectiveness of perianal disease treatment. Variables include discharge, pain/restriction of activities, restriction of sexual activity, type of perianal disease (including number of fistulas) and degree of induration. Scores range from 0 to 20. 16
The reliance on traditional disease activity measures (such as the CDAI) to measure treatment effectiveness fails to take into account the impaired QoL experienced by CD patients. The IBDQ is an HRQoL measure. It is a 32-item questionnaire and evaluates general activities of daily living, intestinal function, social performance, personal interactions and emotional status. Four-dimensional scores cluster items under bowel function, emotional function, systemic function and social function. Scores range from 32 to 224. 43
The CDEIS was developed to take into account endoscopic data, such as lesion severity, when assessing severity of the disease. Variables include the presence or absence of deep or superficial ulceration in various segments of the intestinal tract, the surface involved (in cm), the surface ulcerated (in cm) and presence of ulcerated stenosis. Scores range from 0 to 30. 44
Measurement of disease severity in children
The paediatric CDAI is a multi-item measure of severity that includes linear growth and places less emphasis on subjectively reported symptoms and more on laboratory parameters of intestinal inflammation than the adult CDAI. It includes 11 variables: weight, height, abdominal mass, perirectal disease, extraintestinal manifestation, haematocrit, erythrocyte sedimentation rate, albumin, abdominal pain, number of liquid stools and general well-being. Scores range from 0 to 100: ≤ 10 indicates inactive disease, 11–30 mild disease and > 30 moderate-to-severe disease. 45,46
Current service provision
CD treatment includes nutrition, drugs and surgery. Nutrition includes complete elemental diets and nutritional supplements. Drug treatments can include aminosalicylates (mesalazine and sulfasalazine) and corticosteroids (prednisolone, budesonide, intravenous (i.v.) hydrocortisone and methylprednisolone). Licensed drugs are being used in unlicensed indications for chronically active CD, including immunomodulators (azathioprine, mercaptopurine and methotrexate) and the antibiotic metronidazole. 47 Cytokine modulators (also known as biologics) such as adalimumab and infliximab are licensed for severe active CD. Use of infliximab is subject to the National Institute for Health and Clinical Excellence (NICE) guidance (see below). Adalimumab is discussed in the next section (see Description of technology under assessment). Surgery is not curative and is used to manage symptoms. In patients with fistulas, treatment can include use of setons (devices to keep fistulas open and allow drainage) and surgery. At least 50% of CD patients require surgical treatment in the first 10 years of disease and around 70%–80% require surgery within their lifetime. 9
The NICE guidance on the current use of infliximab in CD is as follows (Technology Appraisal Guidance No. 40):1
1.1. Infliximab is recommended for the treatment of patients with severe Crohn’s disease who fulfil all three of the following criteria:
Patients who have severe active Crohn’s disease. These patients will already be in very poor general health with weight loss and sometimes fever, severe abdominal pain and usually frequent (3–4 or more) diarrhoeal stools daily. They may or may not be developing new fistulas or have extra-intestinal manifestations of the disease. This clinical definition normally corresponds to a Crohn’s Disease Activity Index (CDAI) score of 300 or more and a Harvey–Bradshaw Index of 8/9 or above.
Patients whose condition has proved to be refractory to treatment with immuno-modulating drugs (e.g. azathioprine or 6-mercaptopurine, methotrexate) and corticosteroids, or who have been intolerant of, or experienced toxicity from, these treatments.
Patients for whom surgery is inappropriate (e.g. because of diffuse disease and/or a risk of short bowel syndrome).
1.2. Treatment can be repeated for those patients who match the above criteria and have responded to the initial treatment course, but then relapsed. A decision about whether or not to re-administer infliximab after the first course or subsequently should be made only after discussion with the patient who has been fully informed of the potential risks and benefits of repeated therapy (episodic treatment).
1.3. Infliximab should be prescribed by a gastroenterologist experienced in the management of Crohn’s disease.
1.4. Infliximab is not recommended for patients with fistulising Crohn’s disease who do not have the other criteria for severe active Crohn’s disease as detailed in section 1.1.
For current conventional treatment, the recommendations below are taken from the UK guidelines for the management of IBD in adults from 20049 (see Appendix 2 for full details on medical management of CD). In this guideline, treatment options are complex and depend on the severity of disease, whether first-line treatments have failed, side effects, stage/type of disease (active, in remission, chronic, fistulising). Also, some treatment may be adjunctive.
For patients with active, ileal/ileocolonic/colonic disease, options include aminosalicylates (e.g. mesalazine), corticosteroids (e.g. prednisolone), antibiotics (e.g. metronidazole), immunosuppressants (e.g. azathioprine), nutritional therapy and surgery. Patients with fistulising and perianal disease can be treated with antibiotics or immunosuppressants; infliximab where CD is severe and active, and fistulas are refractory to other treatment; nutritional therapy; and surgery.
The efficacy of treatment for maintenance of remission depends on how remission was achieved (medically or surgically), on risk of relapse and on the site of disease. In addition to smoking cessation (one of the most important factors in maintaining remission), aminosalicylates, immunosuppressants or antimetabolites (e.g. methotrexate) can be used. Infliximab can be used for up to 44 weeks as part of a treatment strategy including immunomodulation. Corticosteroids are not effective for the maintenance of remission, although some patients appear steroid dependent. Immunomodulation should be tried as first-line treatment in steroid-dependent patients; infliximab should be reserved for patients with moderate-to-severe CD who are refractory or intolerant of treatment with steroids, mesalazine, azathioprine/mercaptopurine and methotrexate, and where surgery is considered inappropriate. It has been estimated that around 2% of patients have severe, drug-refractory disease, but this is based on a Markov model rather than on cohort data. 48
In children, enteral nutrition is used as primary therapy for active CD by the majority of paediatric gastroenterologists in the UK. 11
An audit49 carried out in collaboration between the British Society of Gastroenterology, the Royal College of Physicians, the Association of Coloproctology of Great Britain and Ireland and the NACC found marked variation in the resources and quality of care: They found that:
-
44% of sites did not have an IBD nurse specialist
-
there was poor provision of dietetic services
-
there was a lack of adequate toilet provision in hospitals
-
fewer than one-fifth of hospitals were able to refer patients directly for psychological support
-
42% of patients with IBD had a stool sample sent for culture
-
52% of CD patients were weighed
-
37% of CD patients seen by a dietitian
-
many patients with CD were receiving inappropriately prolonged course of steroids
-
there was inadequate prophylactic bone protection therapy for patients on systemic steroids and inadequate screening for osteoporosis
-
there was infrequent participation in clinical research into IBD in the UK.
Description of technology under assessment
Adalimumab and infliximab are TNF inhibitors (anti-TNF-α antibodies). TNF-α is a cytokine, a small protein molecule acting as a cell messenger and involved in inflammatory conditions. It is a key mediator of the inflammation associated with CD and can be detected in diseased areas of the bowel wall, and in blood and faeces of patients with the disease. 50 Both adalimumab and infliximab are manufactured antibodies that bind to and inhibit TNF-α thus reducing the inflammatory response. They belong to the pharmacotherapeutic group of selective immunosuppressive agents. 51 The term ‘biologics’ is also applied to these drugs as their production depends on cells that have been genetically engineered to produce a specific protein.
Adalimumab (Humira®, Abbott Laboratories, Abbott Park, IL, USA) is a recombinant, fully human monoclonal antibody expressed in Chinese hamster ovary cells. It binds specifically to TNF and neutralises its biological function. Adalimumab is available as Humira 40 mg solution; each 0.8-ml single-dose vial contains 40 mg of adalimumab. It is administered by subcutaneous injection. Treatment with adalimumab should be initiated and supervised by specialist physicians experienced in the treatment of CD. After training, patients may self-inject with adalimumab, with medical follow-up as necessary. Adalimumab is also licensed for use in rheumatoid arthritis, psoriatic arthritis and ankylosing spondylitis. 52
The licence indication for CD detailed in the Summary of Product Characteristics (SPC)52 is as follows:
Humira is indicated for treatment of severe, active Crohn’s disease, in patients who have not responded despite a full and adequate course of therapy with a corticosteroid and/or an immunosuppressant; or who are intolerant to or have medical contraindications for such therapies. For induction treatment, Humira should be given in combination with cortiocosteroids. Humira can be given as monotherapy in case of intolerance to corticosteroids or when continued treatment with corticosteroids is inappropriate.
The recommended Humira induction dose regimen for adult patients with severe Crohn’s disease is 80 mg at week 0 followed by 40 mg at week 2. In case there is a need for a more rapid response to therapy, the regimen 160 mg at week 0 (dose can be administered as four injections in one day or as two injections per day for two consecutive days), 80 mg at week 2, can be used with the awareness that the risk for adverse events is higher during induction.
After induction treatment, the recommended dose is 40 mg every other week via subcutaneous injection. Alternatively, if a patient has stopped Humira and signs and symptoms of disease recur, Humira may be re-administered. There is little experience from re-administration after more than 8 weeks since the previous dose. During maintenance treatment, corticosteroids may be tapered in accordance with clinical practice guidelines. Some patients who experience decrease in their response may benefit from an increase in dose intensity to 40 mg Humira every week.
Infliximab [Remicade®, Schering-Plough (formerly Schering-Plough Ltd., since 2009 Merck & Co., Kenilworth, NJ, USA)] is a chimaeric human–murine monoclonal antibody manufactured from a recombinant cell line. It binds with high affinity to soluble and transmembrane forms of TNF thus inhibiting the functional activity of TNF. Infliximab is available as Remicade 100 mg powder for concentrate for solution for infusion; each vial contains 100 mg of infliximab. Treatment with infliximab should be initiated and supervised by specialist physicians experienced in the treatment of CD. Infliximab is administered intravenously over a 2-hour period. Infusions should be administered by qualified health-care professionals trained to detect infusion-related issues; patients should be observed for at least 1–2 hours post infusion for acute infusion-related reactions, and emergency equipment (such as adrenaline) must be available. Patients may be pre-treated in order to avoid infusion-related reaction, particularly where these have occurred previously. Infliximab is also licensed for use in rheumatoid arthritis, ulcerative colitis, ankylosing spondylitis, psoriatic arthritis and psoriasis.
The licence indication for CD detailed in the SPC53 is as follows:
Adult Crohn’s disease: Remicade is indicated for:
treatment of severe, active Crohn’s disease, in patients who have not responded despite a full and adequate course of therapy with a corticosteroid and/or an immunosuppressant; or who are intolerant to or have medical contraindications for such therapies
treatment of fistulising, active Crohn’s disease, in patients who have not responded despite a full and adequate course of therapy with conventional treatment (including antibiotics, drainage and immunosuppressive therapy).
Paediatric Crohn’s disease:
Treatment of severe, active Crohn’s disease, in paediatric patients aged 6 to 17 years, who have not responded to conventional therapy including a corticosteroid, an immunomodulator and primary nutrition therapy; or who are intolerant to or have contraindications for such therapies. Remicade has been studied only in combination with conventional immunosuppressive therapy.
Severe, active Crohn’s disease:
5 mg/kg given as an intravenous infusion over a 2-hour period. Available data do not support further infliximab treatment, in patients not responding within 2 weeks to the initial infusion. In responding patients, the alternative strategies for continued treatment are:
maintenance: additional infusions of 5 mg/kg at 2 and 6 weeks after the initial dose, followed by infusions every 8 weeks or
readministration: infusion of 5 mg/kg if signs and symptoms of the disease recur.
Fistulising, active Crohn’s disease:
An initial 5 mg/kg infusion given over a 2-hour period is to be followed with additional 5 mg/kg infusion doses at 2 and 6 weeks after the first infusion. If a patient does not respond after these three doses, no additional treatment with infliximab should be given.
In responding patients, the strategies for continued treatment are:
additional infusions of 5 mg/kg every 8 weeks or
readministration if signs and symptoms of the disease recur followed by infusions of 5 mg/kg every 8 weeks.
In Crohn’s disease, experience with readministration if signs and symptoms of disease recur is limited and comparative data on the benefit/risk of the alternative strategies for continued treatment are lacking.
Crohn’s disease (6 to 17 years):
5 mg/kg given as an intravenous infusion over a 2-hour period followed by additional 5 mg/kg infusion doses at 2 and 6 weeks after the first infusion, then every 8 weeks thereafter. Some patients may require a shorter dosing interval to maintain clinical benefit, while for others a longer dosing interval may be sufficient. Available data do not support further infliximab treatment in paediatric patients not responding within the first 10 weeks of treatment.
As outlined in the licence indications, patients eligible for treatment with anti-TNF therapy are adults or children with severe, active (or fistulising CD) who have not responded to and/or are intolerant to conventional treatment. There is no standard definition for what constitutes severe CD. NICE guidance defines severe as a score of > 300 on the CDAI or 8–9 on the HBI. 1 The group that developed the CDAI defines values of 150 and below as quiescent disease and values above 450 as extremely severe disease; no intermediate cut-off point is given for severe disease. 39 The NICE scope for the current appraisal states that the population of interest consists of patients with moderate-to-severe CD; there is no standard definition of what constitutes moderate-to-severe. Trials have described patients with a CDAI of 220–400 (or 450) as having moderate-to-severe CD. 57
Adverse events with anti-TNF treatment
A number of adverse events (AEs) have been associated with anti-TNF therapy and have been reported for infliximab and adalimumab. As the immune response is suppressed, infections may be more likely to occur. These include tuberculosis, other bacterial infections including sepsis and pneumonia, fungal infections and opportunistic infections such as pneumocystosis or cytomegalovirus infection. Cases of reactivation of hepatitis B infection have been observed, as have rare cases of jaundice and hepatitis, optic neuritis and onset or exacerbation of demyelinating disorders including multiple sclerosis. A deficiency of TNF may result in the initiation of an autoimmune process, and the occurrence of lupus-like syndrome has been observed. There is the possibility of an increased risk of lymphoma or other malignancies, worsening of heart failure or of AEs of the haematological system (e.g. cytopenias). Infliximab has been associated with acute, infusion-related reactions (including anaphylactic shock) and delayed hypersensitivity reactions. Injection site reactions are common with adalimumab. Common AEs for both infliximab and adalimumab are upper respiratory infections (such as sinus infections), headache, rash, nausea and stomach pains. The development of anti-TNF antibodies may be associated with a decrease in efficacy and predispose the patient to an additional risk of recurrent delayed or acute allergic reactions. 52–56
This report will consider the following patient groups (where information is available): adults with moderate-to-severe active CD intolerant or resistant to conventional treatment; children with moderate-to-severe active CD intolerant or resistant to conventional treatment; and adults with fistulising CD intolerant or resistant to conventional treatment. Where possible, patients with severe (rather than moderate-to-severe) CD will be considered as this is in line with the licence indication.
Degree of diffusion
There is no up-to-date evidence available on the degree of diffusion of adalimumab and infliximab for CD treatment in the UK. The only evidence that is available from routinely collected data is for the total number of adalimumab and infliximab prescriptions for all conditions.
Chapter 2 Definition of the decision problem
The main aims of the report were:
-
To update a previous Technology Assessment Report (TAR)5 on the effectiveness and cost-effectiveness of infliximab in adults with moderate-to-severe CD or fistulising CD who are refractory to or intolerant of conventional treatment.
-
To review the evidence on the clinical effectiveness and cost-effectiveness of infliximab in children with moderate-to-severe CD who are refractory to or intolerant of conventional treatment.
-
To review the evidence on the clinical effectiveness and cost-effectiveness of a further anti-TNF-α antibody, adalimumab, in adults with moderate-to-severe CD who are refractory to or intolerant of conventional treatment.
-
To investigate whether there is evidence for greater clinical effectiveness or cost-effectiveness for either adalimumab or infliximab.
Decision problem
Interventions
Adalimumab and infliximab are drugs for use in patients with severe active CD or fistulising active CD (infliximab) who have not responded to conventional treatment or who have experienced toxicity from these treatments. There has been a distinction made between induction treatment and maintenance treatment, but it is unclear where the boundary lies between these for the interventional drugs. Similarly there has been a distinction between ‘episodic’ treatment, i.e. treatment when a disease flare starts (or at a clinician’s discretion), and maintenance treatment, where patients are treated at regular (scheduled) intervals with the intention of keeping them in remission, but it is unclear where the boundary lies between these treatment strategies. It would be useful to know the most effective dosing regimen for each of the drugs.
Comparators
Conventional treatment includes no treatment, dietary intervention, drug treatment with aminosalicylates, methotrexate, corticosteroids (prednisolone, budesonide and hydrocortisone), azathioprine or metronidazole or surgical intervention.
Given that licences for both drugs are for use only when conventional treatment has failed, it is unlikely that randomised controlled trials (RCTs) would compare the drugs to conventional treatment. Instead, the most likely comparator will be no treatment or placebo, but where patients in all trial arms continue to receive elements of conventional therapy. Another relevant comparator may be a different dosing regimen of the same drug.
For comparisons between both drugs under review, head-to-head comparisons within the same trial would be the ideal scenario. It is important to note that, because of earlier licensing, infliximab could be viewed either as the intervention of interest in some of the RCTs or as part of conventional treatment in others. It would also be useful to establish the effectiveness of both drugs compared with non-drug treatments such as surgery or nutrition, particularly in children.
Population and relevant subgroups
Infliximab is licensed for use in adults and children with severe active CD or in adults with fistulising disease who are intolerant or resistant to treatment. Adalimumab is licensed for severe active CD; current information does not indicate whether this is in adults only.
There is no standard definition for what constitutes severe CD. NICE guidance defines severe as a score of > 300 on the CDAI or 8–9 on the HBI. 1 The group that developed the CDAI defines values of ≤ 150 as quiescent disease and values > 450 as extremely severe disease; no intermediate cut-off point is given for severe disease. 39
The NICE scope for the current appraisal stated that the population of interest consists of patients with ‘moderate-to-severe’ CD. There is no standard definition of what constitutes ‘moderate-to-severe’, but RCTs have described patients with a CDAI of 220–400 as having moderate-to-severe CD. 57 Note that this assessment report is therefore investigating treatments outside their licence indications. The main thrust of the work should be to investigate the clinical effectiveness of treatments in patients with a CDAI score of 300 or more. However, it is unlikely that any RCTs have included only these CD patients. Therefore, the options are:
-
To look only at subgroups of patients in RCTs with a CDAI score of ≥ 300. This is unlikely to be a valid comparison unless the RCT stratified patients by being more or less than CDAI 300.
-
To widen the inclusion criteria of the assessment report to include RCTs where CD patients had lower CDAI scores. 58
It may be that there is a different effectiveness of the interventions in CD patients with CDAI scores of > 220 compared with > 300.
Most work on measurement of CD has been carried out in adult patients. Where a child has CD, it is unclear how this would be consistently categorised as severe CD or moderate-to-severe CD. Although there is a children’s version of CDAI – Paediatric Crohn’s Disease Activity Index (PCDAI) – it is unclear how well this measure is validated and how it relates to CDAI cut-off points.
It could be important to look at populations of patients who have failed either infliximab or adalimumab therapy to determine if unresponsiveness to a particular drug is a persistent state and whether unresponsiveness to one drug can be linked to similar unresponsiveness to the other. Finally, it is unclear exactly how resistance to treatment is measured or how long a treatment trial would go on for before a patient would be categorised as being resistant or responsive to treatment.
Outcomes
Key factors are the clinical effectiveness of both drugs particularly in terms of enhancing patient QoL, maintenance of remission, delaying disease progression and prolonging survival. More specifically, outcomes could include overall survival, progression-free survival, HRQoL, disease activity (remission, response, relapse, changes in disease activity indices, number of fistulas for fistulising disease), maintenance of response to treatment over time, need for surgery, need for an ostomy, hospitalisation rates, need for steroid treatment, dropout rates from TNF-α treatment and adverse effects of treatment. It is unclear how outcomes such as mucosal healing would impact on clinical outcomes such as QoL.
Where disease severity and effect of treatment is measured by CDAI or PCDAI scores, it is uncertain how large a change in CDAI score constitutes a clinically significant change and whether this would be the same change for more severe CD as for less severe CD.
Trials in patients with fistulising disease will measure fistula closure but it is uncertain whether this is a good measure of effectiveness as abscesses can form if the fistula is no longer patent; so abscess occurrence may be a better outcome measure. Other clinical outcomes could include abscess formation rates and seton use (if reported).
Overall aims and objectives of assessment
The overall decision problem is ‘What is the cost-effectiveness of adalimumab and infliximab in the management of moderate-to-severe CD in the UK NHS?’. Ideally, this analysis would be based on head-to-head comparisons. In the absence of these, this decision problem is operationalised as a number of complementary cost-effectiveness analyses (depending on availability of data):
-
What is the expected incremental cost-effectiveness ratio (ICER) for infliximab therapy (induction or episodic/clinician discretion or scheduled maintenance) compared with standard care (SC) in the management of moderate-to-severe CD?
-
What is the expected ICER for adalimumab therapy (induction or episodic/clinician discretion or scheduled maintenance) compared with SC in the management of moderate-to-severe CD?
-
What is the expected ICER for one dosing regimen of infliximab therapy compared with another dosing regimen of infliximab in the management of moderate-to-severe CD?
-
What is the expected ICER for one dosing regimen of adalimumab therapy compared with another dosing regimen of adalimumab therapy in the management of moderate-to-severe CD?
-
What is the expected ICER for (different dosing regimens of) infliximab therapy compared with (different dosing regimens of) adalimumab therapy in the management of moderate-to-severe CD?
This report contains reference to confidential information provided as part of the NICE appraisal process. This information has been removed from the report and the results, discussions and conclusions of the report do not include the confidential information. These sections are clearly marked in the report.
Chapter 3 Assessment of clinical effectiveness
Methods for reviewing clinical effectiveness
Search strategy
The search strategy was designed to update that undertaken for the previous technology assessment of the clinical effectiveness and cost-effectiveness of infliximab in adults with moderate-to-severe CD5 and to encompass the new anti-TNF therapies identified for review. A search was undertaken to find existing good quality systematic reviews in order to document the evidence base to date. Searches for primary studies were restricted to RCTs. The following sources were searched for relevant primary studies:
-
bibliographic databases: Cochrane Library [Cochrane Central Register of Controlled Trials (CENTRAL)] 2007, Issue 2; MEDLINE (Ovid) 2000 to May/June 2007; MEDLINE In-Process & Other Non-Indexed Citations (Ovid) 4 June 2007 and 26 June 2007; EMBASE (Ovid) 2000 to May/June 2007. Searches were based on index and text words that encompass the condition: CD and the interventions: adalimumab, certolizumab pegol, infliximab and natalizumab. [Natalizumab and certolizumab pegol were originally part of this technology appraisal so were included in the searches. They were subsequently dropped from the report after completion of searches (see Protocol modification).] Where it was appropriate, a methodological ‘filter’ was applied to identify RCTs
-
EMEA, FDA and other relevant websites
-
citations of relevant studies
-
contact with experts
-
research registries of ongoing trials including National Research Register 2007, Issue 2, Current Controlled Trials and ClinicalTrials.gov
-
submissions from industry
-
hand search of conference abstracts in 2006 and 2007: British Society of Gastroenterology, Digestive Disease Week, United European Gastroenterology Meeting, European Crohn’s and Colitis Organisation, Federation of Clinical Immunology Societies.
Searches were not limited by language. Full search strategies can be found in Appendix 3.
Inclusion and exclusion criteria
Only studies meeting the following inclusion criteria were included:
-
Study design: RCTs (study designs other than RCTs were excluded).
-
Population: adults (≥ 18 years) and children (6–17 years) with moderate-to-severe, active CD intolerant or resistant to conventional treatment; adults (≥ 18 years) with fistulising CD resistant to conventional treatment. ‘Moderate-to-severe’ disease includes patients with an average CDAI score of ≥ 220 or those who are described by trial authors as having moderate-to-severe disease.
-
Intervention: adalimumab or infliximab (any dosage/treatment regimen).
-
Comparator: conventional treatment without TNF-α inhibitors including no treatment, placebo, dietary intervention, drug treatment with aminosalicylates, methotrexate, corticosteroids (prednisolone, budesonide and hydrocortisone), azathioprine, metronidazole or surgical intervention. Adalimumab and infliximab compared with each other. Different dosage or treatment regimens of the same drug.
-
Outcomes: at least one of the following: overall survival, progression-free survival, HRQoL, disease activity (remission, response, relapse, changes in disease activity indices, number of fistulas for fistulising disease), need for surgery, hospitalisation rates and adverse effects of treatment.
-
Trials that looked at both induction and maintenance of remission were included.
Based on the above inclusion/exclusion criteria, study selection was made independently by two reviewers. Discrepancies were resolved by discussion, with involvement of a third reviewer when necessary. All discrepancies were resolved in this way.
Data extraction strategy
Information on study characteristics, study quality and results for each trial was extracted by one reviewer and checked by a second reviewer. Four reviewers were involved in data extraction. A standardised data extraction form was used, based on the form designed for the previous TAR on infliximab. 5 The data extraction template can be found in Appendix 4. Where necessary the template was adapted to accommodate details relevant to a specific trial. Where required, information was extracted from graphs as follows (see Appendix 5): the graph was scanned into a word document, overlaid with an appropriate template with graph gridlines, and printed and enlarged to A3 size, and information was extracted using the gridline template. To reduce error in this procedure, extracted information was checked by comparing graph readings with any available values in the report text and/or by redrawing the graph using the extracted data and comparing this with the original (see Appendix 5 for examples). Data extraction discrepancies were resolved by discussion, with involvement of a third reviewer when necessary. All discrepancies were resolved in this way.
Quality assessment strategy
Quality assessment was based on the published papers only and note was taken that absence of a quality criterion may be due to lack of reporting rather than actual poor methodological quality. Authors were not contacted for further information. Quality assessment was descriptive, a quality scoring system was not used. The quality criteria assessed were based on guidelines suggested by the Cochrane Collaboration, inviting consideration of threats arising from selection, performance, attrition and detection biases. Individual checklist items were: randomisation, concealment, blinding, comparability of groups, follow-up of trial participants, handling of missing data [intention-to-treat (ITT) analysis], power calculation and selective reporting (see Appendix 4 for checklist). Study quality was assessed by one reviewer and checked by a second. Discrepancies were resolved by discussion, with involvement of a third reviewer when necessary. All discrepancies were resolved in this way.
Handling of manufacturer and other submissions
The main industry submissions (including appendices) were checked for additional relevant trials and additional clinical effectiveness data for included trials. Because editorial constraints meant the results available in published accounts of the trials were necessarily selective, information in the submitted Clinical Study Reports was sourced as required for purposes of balance and completeness. It was not possible to systematically review all such additional information submitted owing to the volume of the submissions [e.g. more than 38,000 pages for the clinical study report of ACCENT (A Crohn’s disease Clinical trial Evaluating inflixmab in a New long-term Treatment regimen) I,2,3 more than 5000 pages for the Clinical Study Report of Targan et al. ,57 both included studies]. No references to specific sections of the clinical study reports were made in the main industry submissions. [Please note that the clinical study reports for the CLASSIC (CLinical assessment of Adalimumab Safety and efficacy Studied as Induction therapy in Crohn’s disease), CHARM (Crohn’s Trial of the Fully Human Antibody Adalimumab for Remission Maintenance) and GAIN (Gauging Adalimumab efficacy in Infliximab Nonresponders) RCTs that were received from the manufacturers of adalimumab started on section 4 and had no page numbers or tables of contents. Also some of the appendices were missing, particularly ones referred to in the text as having all of the raw results in tables. Therefore it is unclear whether some pages are missing from the middle of these reports or not and potentially the most useful appendices were not supplied.] For details on how the submitted economic models were assessed see Chapter 4, Critique of the submission on infliximab by Schering-Plough.
Analysis strategy
The clinical effectiveness section of this report mainly focuses on the results from RCTs and/or RCT trial arms in which the drugs were administered within the limits of their current respective licence indication (see Appendix 6). Results of trials are organised and reported in four categories:
-
induction trials in adult populations predominantly or wholly constituted of non-fistulising patients
-
maintenance trials in adult populations predominantly or wholly constituted of non-fistulising CD patients
-
trials in populations wholly constituted of patients with fistulising CD
-
trials in paediatric patients.
Results are reported within these four categories on a trial-by-trial basis except with regard to AEs and side effects which were considered simultaneously across all included trials across both drugs. Most outcome results are presented in forest plots so as to provide an overview of the quantitative spread of effect sizes. These are accompanied with brief narrative commentary. In some instances outcome results are tabulated. Both placebo and intervention rates and both risk difference and risk ratio effect sizes are presented for most outcomes in Tabulation of included studies. The confidence intervals (CIs) quoted were not adjusted for repeated measures.
The clinical heterogeneity of trials, or the existence of only a single trial, precluded pooling of data in meta-analysis. The feasibility of undertaking indirect comparison analysis was considered in-depth in order to assess the relative effectiveness of different drugs because there were no RCTs directly comparing both drugs included in this technology appraisal. However, indirect comparisons were not done because of the variation in placebo effect sizes in the RCTs (induction trials), the lack of similarity in the apparently common comparator (i.e. placebo arm maintenance trials), and the reporting of subgroup results only at follow-up (i.e. variously defined responders only) in many of the RCTs.
Protocol modification
The protocol originally encompassed assessment of the clinical effectiveness and cost-effectiveness of infliximab, adalimumab, certolizumab pegol and natalizumab within their licensed indications for moderate-to-severe CD. At the time of producing the protocol, certolizumab pegol and natalizumab were not licensed for CD, but imminent licensing was anticipated by the commissioners of this report (NICE). After the start of the review process it became clear that neither drug would achieve a licence within the time frame required for this technology assessment and consequently both drugs were dropped from the review. This occurred after completion of the search strategy. As of November 2010 these drugs remain unlicensed for CD.
Results
Quantity of research available
Eleven relevant trials were identified,2,3,45,46,57,58,62–67 some supported by multiple publications. Figure 1 details the trial identification process.
FIGURE 1.
Study identification process.
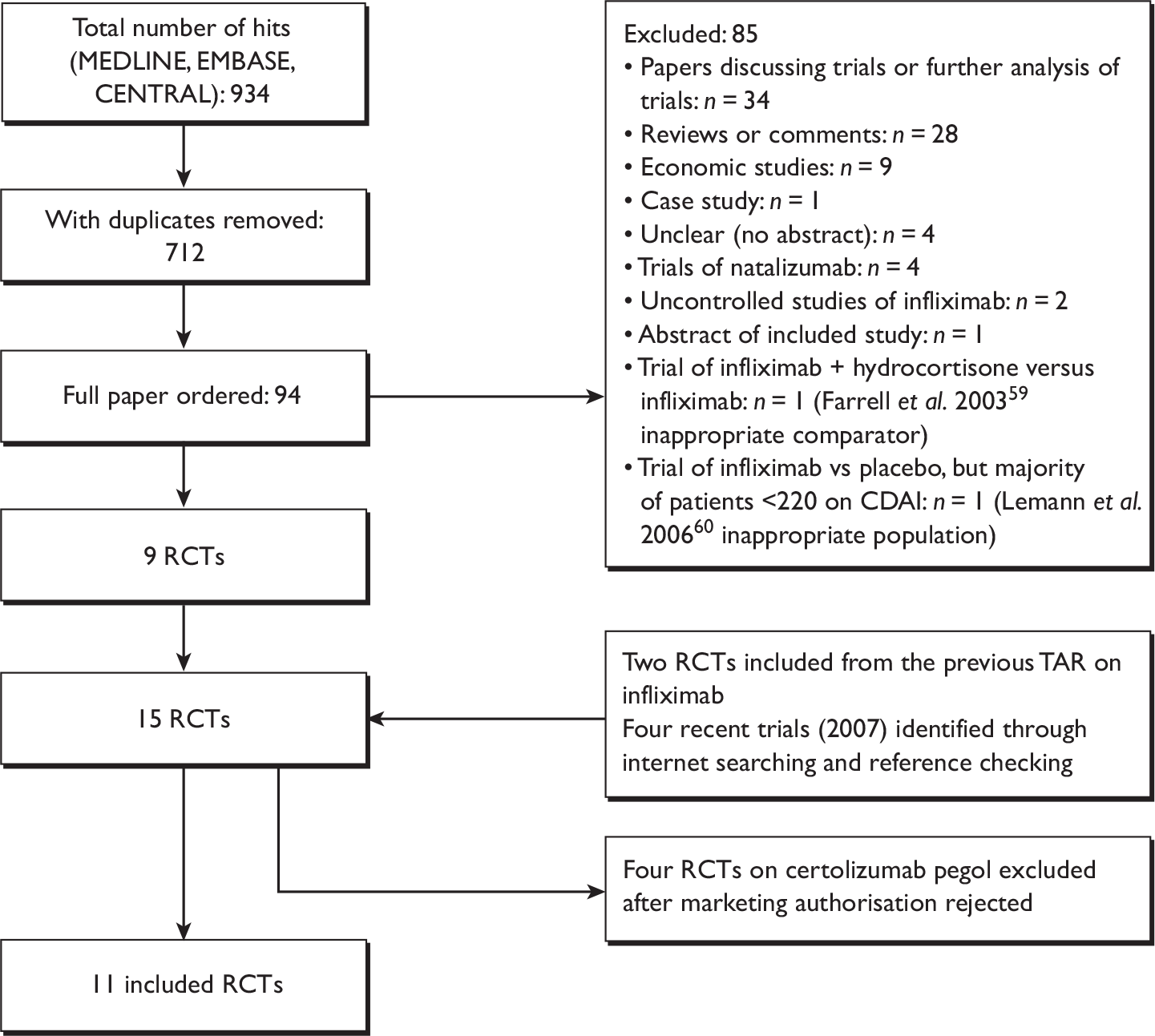
At the time of writing of this report, 11 hard copies of ordered publications were still outstanding or not available; none of these are likely to contain new trial data (see Appendix 7 for details of publications).
Eleven RCTs were included in total. 2,3,45,46,57,58,62–67 Seven trials meeting the inclusion criteria were identified through the main database searches. 2,3,45,46,58,63,65,67 Two additional studies57,62 from the previous TAR on infliximab were included,5 as were two trials from 2007 which had been published after the search cut-off date. 64,66
Searching through the main industry submissions from both manufacturers did not yield any additional RCTs. The search for conference abstracts yielded no further relevant trials. An abstract of the study by Hommes et al. 61 was identified, which is referred to in Chapter 5, Other relevant factors. This study did not meet the criterion of a population of CD patients who are resistant or intolerant to conventional treatment.
The search for ongoing trials yielded four potentially relevant RCTs, all of adalimumab (see Appendix 8). All were at the recruitment stage (or not yet recruiting) at the time the information was verified by the respective manufacturers. Two were trials (induction and maintenance) of adalimumab in Japanese patients with moderate-to-severe CD. Two multicentre trials of adalimumab were in patients with moderate-to-severe ileocolonic CD and in children with moderate-to-severe CD respectively. Two ongoing trials of infliximab were identified, but did not meet the inclusion criteria as they compared either infliximab with infliximab plus methotrexate or infliximab with infliximab plus azathioprine. No ongoing trials of head-to-head comparisons of adalimumab and infliximab were identified. No preliminary reports of any of these ongoing trials were identified in the manufacturer submissions.
Tabulation of included studies
All of the included RCTs recruited patients having ‘moderate-to-severe CD’ defined according to CDAI scores of between 220 and 450, or 220 and 400; it is therefore likely that they do not reflect the intended licensed population of severe active CD (i.e. CDAI score of more than 300).
The included studies encompassed two trial designs, induction therapy and maintenance therapy, in any of three populations: adults predominantly or wholly non-fistulising, fistulising adults and children. Table 5 gives an overview of the included studies with reference to trial design and recruited patient population.
| Type of trial | Drug | Population | ||
|---|---|---|---|---|
| Wholly or predominantly non-fistulising adults | Fistulising adults | Children | ||
| Induction | Infliximab | aTargan et al., 199757 | Present et al., 199962 | Baldassano et al., 200346 |
| Adalimumab |
CLASSIC I Hanauer et al. , 200663 GAIN Sandborn et al. , 200764 |
No trials identified | No trials identified | |
| Maintenance | Infliximab |
Rutgeerts et al. , 199958 |
ACCENT II; Sands et al., 200465 | REACH; Hyams et al., 200745 |
| Adalimumab |
CLASSIC II; Sandborn et al. , 200766 CHARM; Colombel et al. , 200767 |
No trials identified | No trials identified | |
Of the 11 included RCTs,2,3,45,46,57,58,62–67 nine compared infliximab or adalimumab with placebo. 2,3,57,58,62–67 Two RCTs compared different doses of infliximab only and these were both in children. 45,46 Two RCTs of infliximab were in patients with fistulising disease. 62,65 Both induction and maintenance trials were identified for both drugs. All RCTs were multicentre studies conducted mainly in North America and Europe. No RCTs of head-to-head comparisons of adalimumab and infliximab were identified. No RCTs of adalimumab in children were identified. Based on the information in the published papers, all RCTs were either industry sponsored or in part industry sponsored, had participants from industry involved in study design or manuscript writing, or had one or more authors with industry involvement.
In the induction trials, patients received short-duration anti-TNF or placebo to see if a favourable clinical response was induced. In the maintenance trials, all patients received short-term induction therapy with anti-TNF and then continued with longer term anti-TNF or placebo. In the maintenance trials most published results reported only the follow-up of patients who initially responded to the induction therapy, and results for ‘non-responders’ were generally not provided.
The most widely reported outcomes were based on CDAI scores (see Appendix 1 for details). Although group mean or median CDAI scores were usually recorded at various times of follow-up, the variance of these scores was incompletely reported and trials emphasised binary outcome measures derived by dichotomising CDAI scores. Three such binary measures were used:
-
response 70: defined as a reduction of 70 or more in CDAI score relative to baseline
-
response 100: defined as a reduction of 100 or more in CDAI score relative to baseline
-
remission: defined as a CDAI score of less than 150.
The definitions of the binary measures given above were often qualified by stipulation of additional criteria usually including no requirement for a change in concomitant medication because of worsening clinical condition and no requirement for surgery.
This section describes the results about the effectiveness of the anti-TNF interventions. The results reviewed were taken mainly from publications. When judged necessary for purposes of completeness and balance, information in the unpublished industry trial reports was also sourced.
There are four sections in the clinical effectiveness results: induction treatment in adults (predominantly non-fistulising), maintenance in adults (predominantly non-fistulising), treatments in adult patients exclusively with fistulising CD, and paediatric CD (≤ 18 years old). Within each section infliximab is reported before adalimumab and the earliest trial publication date first. Each of the four sections are organised for each trial as follows:
-
description of intervention used in the trial and other unusual points about the trial design
-
report of outcomes organised as A, response 70; B, response 100; C, remission; D, other outcomes; and E, other considerations, in the first two sections, primary and secondary outcomes in the last two sections
-
quality assessment
-
summary for that trial (in box).
Adverse events and side effects are considered simultaneously across all included trials for both drugs at the end of the clinical effectiveness section (see Adverse events), just before the discussion of clinical effectiveness (see Discussion of results and assessment of effectiveness).
Induction trials in adult populations (wholly or predominantly non-fistulising)
Induction trials are patients who were not receiving anti-TNF therapy at the time of randomisation. Three trials were identified. 57,63,64 One, Targan et al. ,57 compared infliximab with placebo. A further publication, D’Haens et al. ,68 reported on a subgroup from Targan et al. 57 and so will not be further discussed. Two trials compared adalimumab with placebo (CLASSIC I63 and GAIN64). Apart from the subgroup study the trials recruited patients who had initial CDAI scores between 220 and 450. The outcomes reported are summarised in Table 6 and trial details are summarised in Table 7.
| % with remission | % with response 100 | % with response 70 | CDAI score | IBDQ score | Other outcomes | |
|---|---|---|---|---|---|---|
| Infliximab | ||||||
| Targan et al., 199757 | ✓ | ✗ | ✓ | ✓ | ✓ | CRPc |
| Adalimumab | ||||||
| CLASSIC I63 | ✓ | ✓ | ✓ | ✓ | ✓ | CRPc |
| GAIN64 | ✓ | ✓ | ✓ | ✓ | ✓ | CRPc, improvement in draining fistulas, fistula remission at week 4 (in subgroup) |
| Studya |
Study weeks n |
Population: severity of CD (baseline CDAI and IBDQ if stated) |
Intestinal areas affected | Main concomitant medication, % not on any medication | Previous/concomitant treatment with anti-TNF inhibitors | Intervention and comparator (dosing regimen) |
|---|---|---|---|---|---|---|
|
Targan et al. , 199757 Infliximab |
4b 108 |
Moderate-to-severe, CDAI 220–450 Eligible if receiving mesalamine or oral corticosteroids or mercaptopurine or azathioprine Mean baseline CDAI (SD): 288 ± 54) placebo, 312 ±56), 318 ±59), 307 ±50) infliximab groups Mean baseline IBDQ (SD): 128 ±29) placebo, 122 (29), 116 ±23), 118 ±28) infliximab groups |
Mainly ileum/colon, also colon only, some ileum only |
Aminosalicylates or corticosteroids, also mercaptopurine or azathioprine % not on medication (if any) not stated |
Exclusion criterion: previous treatment with monoclonal antibodies | One 2-hour i.v. infusion of: 5 mg/kg, 10 mg/kg or 20 mg/kg infliximab or of placebo |
|
Hanauer et al. , 200663 CLASSIC I Adalimumab |
4 299 |
Moderate-to-severe, CDAI 220–450 Mean baseline CDAI (SD): placebo 296 (60); adalimumab groups 299 (57); 301 (61); 295 (52) Median baseline IBDQ (range): placebo, 131 (52–200); adalimumab groups 129 (81–218); 128 (63–200); 127 (37–192). |
Mainly ileum and colon |
Aminosalicylates, also corticosteroids, immunosuppressives, and few on antibiotics % not on medication (if any) not stated |
Exclusion criterion: infliximab or other anti-TNF therapy |
Subcutaneous infusion at weeks 0 and 2: 40 mg/20 mg, 80 mg/40 mg or 160 mg/80 mg adalimumab at week 0 and 2 respectively. Placebo at weeks 0 and 2 |
|
Sandborn et al. , 200764 GAIN Adalimumab |
4 325 |
Moderate-to-severe, CDAI 220–450 Mean baseline CDAI (SD): placebo 313 (66); adalimumab 313 (58) Mean baseline IBDQ (SD): 124 (28) placebo, 120 (27) adalimumab |
Mainly ileum or colon, some rectum, perianal or anus or gastro-duodenal |
Corticosteroids or immunosuppressives, also oral aminosalicylates % not on medication (if any) not stated |
Patients must have been treated with infliximab and either lost response or been intolerant; excluded patients with primary non-response to infliximab | Subcutaneous injections 160 mg adalimumab at week 0 and 80 mg at week 2 or placebo at weeks 0 and 2 |
Targan et al., 199757 (infliximab)
This RCT had four arms. 57 Patients were randomised to a single i.v. infusion of placebo (n = 25) or of infliximab at 5 mg/kg (n = 27), 10 mg/kg (n = 28) or 20 mg/kg (n = 28). Disease status (remission, response 70 and CDAI score) was monitored at baseline and at weeks 2 and 4 after infusion. The 4-week blinded phase was followed by an open-label phase with a further 12 weeks of follow-up. The primary outcome measure was defined as a response 70 at week 4 with no change in any concomitant medication.
A, response 70
Response 70 at week 4 was the primary outcome. Results for response 70 at weeks 2 and 4 are summarised in Figure 2. For response 70 at week 4 there was a statistically significant difference in favour of the infliximab groups (combined) compared with placebo (p < 0.001). The percentage of placebo patients achieving response 70 was ≤ 16% at both time points and for infliximab groups at week 4, and was between 50% and 81% depending on dose regimen. Point estimates of percentage response were associated with considerable uncertainty. The rate of response 70 at week 4 for the combined infliximab groups was 61% (95% CI 51% to 71%). At week 4 the risk difference (infliximab–placebo) was between 0.34 and 0.65, and risk ratio (infliximab/placebo) was between 3.1 and 5.1 depending on dose. Both risk difference and risk ratio at week 4 reached statistical significance in favour of intervention.
FIGURE 2.
Response 70 rates in Targan et al. 57 At week 4 risk difference p < 0.001, p = 0.0045, p < 0.001, for 5, 10 and 20 mg/kg dose regimens respectively. At week 4 risk ratio p < 0.001, p = 0.022, p < 0.004 for 5, 10 and 20 mg/kg dose regimens respectively. LCI, lower confidence interval; RD, risk difference; RR, risk ratio; UCI, upper confidence interval.
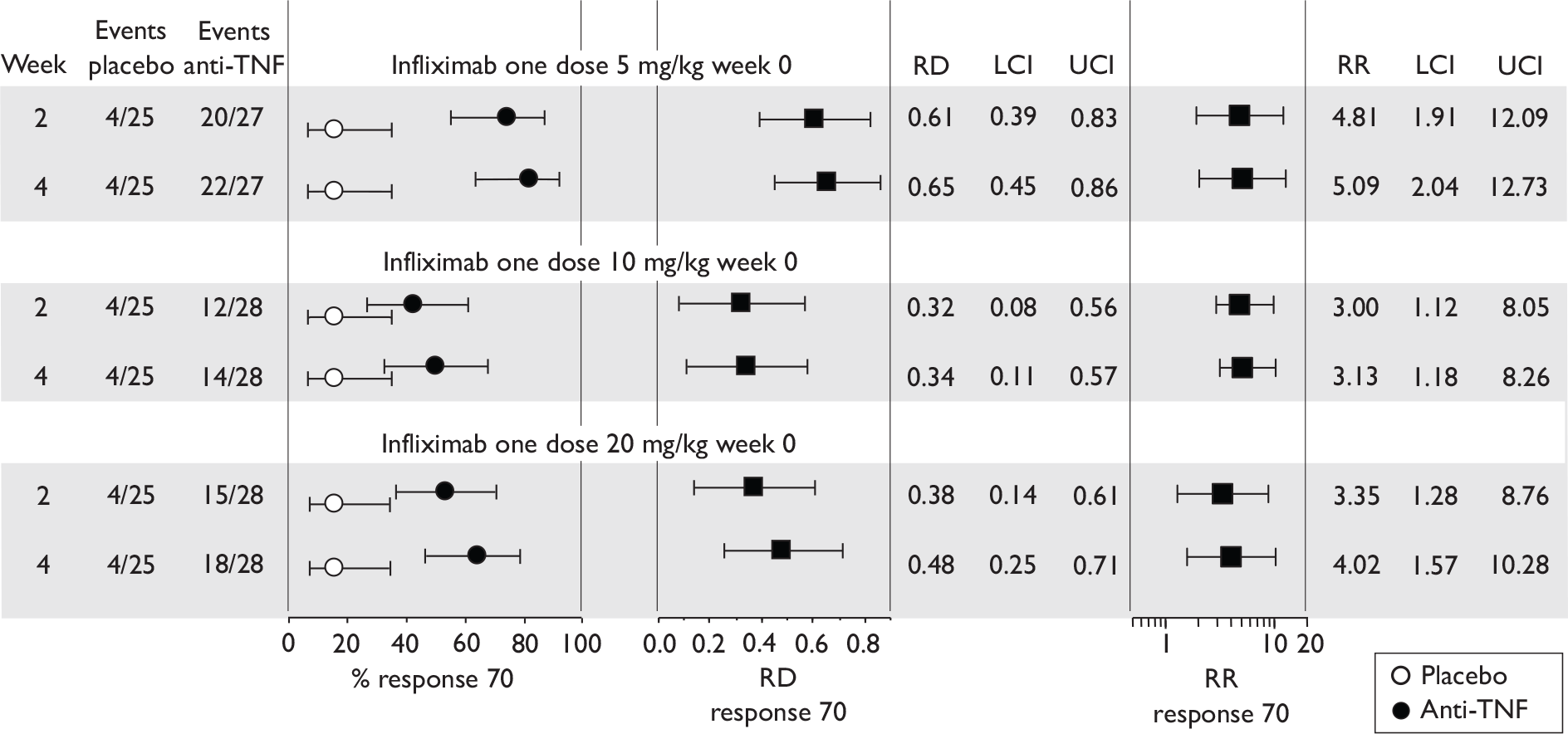
Table 8 summarises the comparison between different dose regimens for response 70 at week 4. The low-dose regimen (5 mg/kg) appeared more effective than the 10 mg/kg regimen (p = 0.009). The difference between dose regimens for other comparisons did not reach statistical significance.
| Dose comparison | Risk difference | Lower CI | Upper CI |
|---|---|---|---|
| 5 mg/kg vs 10 mg/kg | 0.315 | 0.079 | 0.551 |
| 5 mg/kg vs 20 mg/kg | 0.172 | –0.058 | 0.402 |
| 10 mg/kg vs 20 mg/kg | –0.143 | –0.399 | 0.114 |
B, response 100 was not reported
C, remission
Figure 3 summarises remission rates. At 4 weeks, between 25% and 48% of patients in the infliximab groups were in remission, depending on dose, but only one placebo patient achieved remission.
FIGURE 3.
Remission rates in Targan et al. 57 At week 4 risk difference p < 0.001, p = 0.0206 and p = 0.0206, for 5, 10 and 20 mg/kg dose regimens respectively. At week 4 risk ratio p = 0.013, p = 0.076 and p = 0.076 for 5, 10 and 20 mg/kg dose regimens respectively. LCI, lower confidence interval; RD, risk difference; RR, risk ratio; UCI, upper confidence interval.
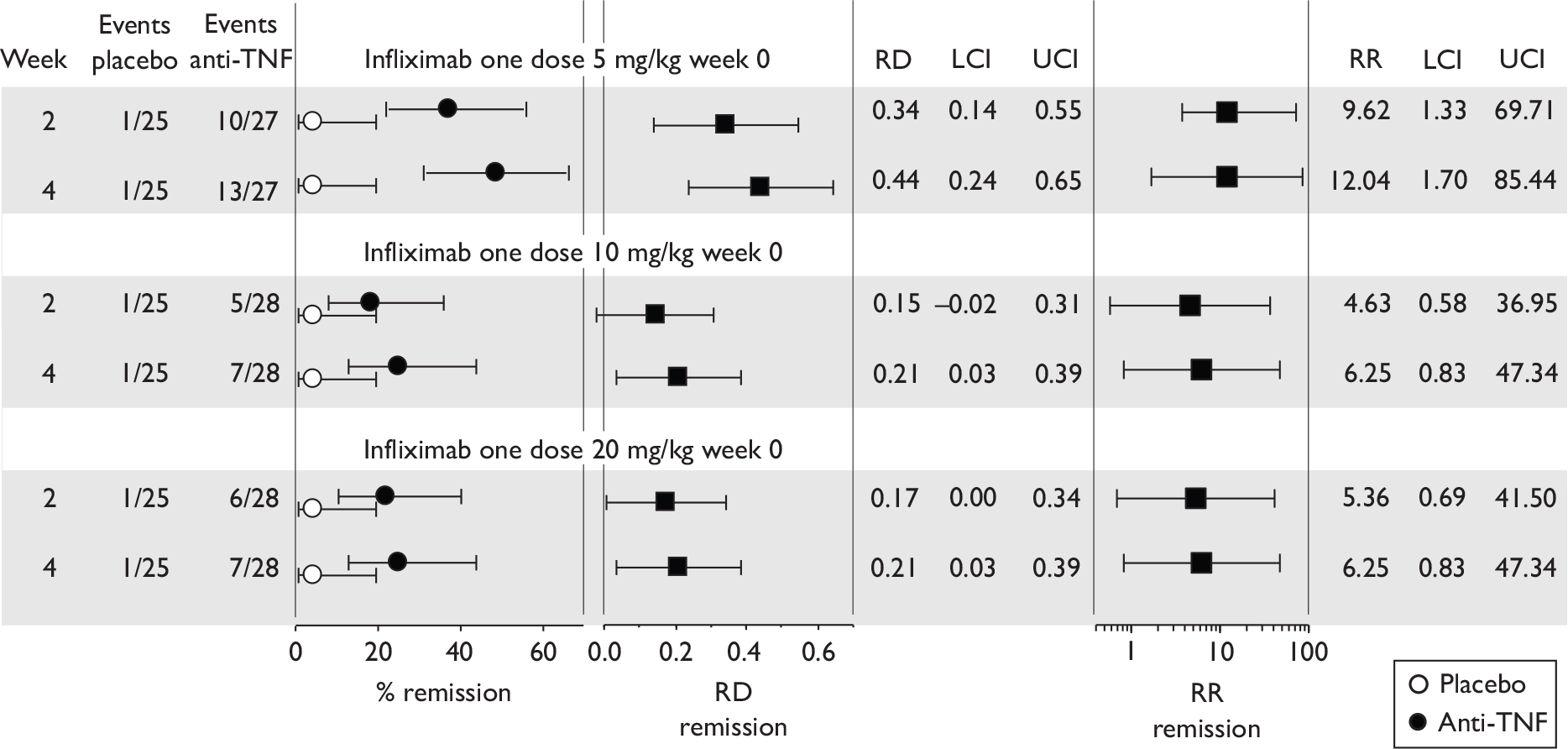
There was a discrepancy between remission rates published in Targan et al. 57 and rates presented in the manufacturer’s submission. The latter for the 5 mg/kg group at week 4 were placebo rate 4% (1/24), infliximab rate 0% (0/24). These remission rates generate a negative risk difference (infliximab–placebo) at week 4 (–0.04). CIs for risk ratios (infliximab/placebo) in the manufacturer’s submission were described as ‘unadjusted’, but were unexpectedly narrow compared with those calculated using standard software packages or using the standard error of ln (risk ratio) given by:69 ([ei]–2 + [ep]–2 – [Ti]–2 – [Tp]–2)0.6, where ei, and ep, are the number of patients with the outcome in the intervention and placebo arms respectively, and Ti and Tp are total number of patients in the intervention and placebo arms respectively. (This discrepancy in CIs applies to CDAI-based binary risk ratios for all trials in the infliximab industry submission.)
At week 4 there were 54/83 (65%) responders (response 70) to infliximab (combined dose groups); by 12 weeks (see E, open-label phase below) there were 34 responders (41%). At week 4, 27/83 (33%) patients given infliximab had gained remission and at 12 weeks 20 patients (24%) were in remission.
D, other outcomes
At week 4 favourable responses to treatment were reported for CDAI scores, for QoL scores (IBDQ), and for C-reactive protein (CRP) levels. The results reported are summarised in Table 9.
| Placebo n = 25 | 5 mg/kg n = 27 | 10 mg/kg n = 28 | 20 mg/kg n = 28 | All infliximab groups n = 83 | |
|---|---|---|---|---|---|
| Score on CDAI | |||||
| Baseline | 288 ± 54 | 312 ± 56 | 318 ± 59 | 307 ± 50 | 312 ± 55 |
| 4 weeks | 211 ± 82 | 166 ± 76a | 226 ± 115b | 211 ± 107a | 201 ± 103a |
| Score on IBDQ | |||||
| Baseline | 128 ± 29 | 122 ± 29 | 116 ± 23 | 118 ± 28 | 118 ±27 |
| 4 weeks | 133 ± 28 | 168 ± 36a | 146 ± 41c | 149 ± 35d | 154 ± 38e |
| CRP (mg/l)f | |||||
| Baseline | 12.8 + 13.9 | 22.1 + 23.6 | 23.2 + 34.2 | 22.4 ± 23.9 | 22.5 ± 21.4 |
| 4 weeks | 14.8 ± 18.6 | 5. 1 ± 9.3g | 12.1 ± 18.6 | 6.9 ± 11.6a | 8.3 ± 1.39a |
Figure 4 shows the mean difference in IBDQ score (infliximab–placebo) at week 4. Mean difference reached statistical significance only for patients who received the low-dose regimen.
FIGURE 4.
Mean IBDQ scores and mean difference at baseline and week 4 of Targan et al. 57 NS, not significant.
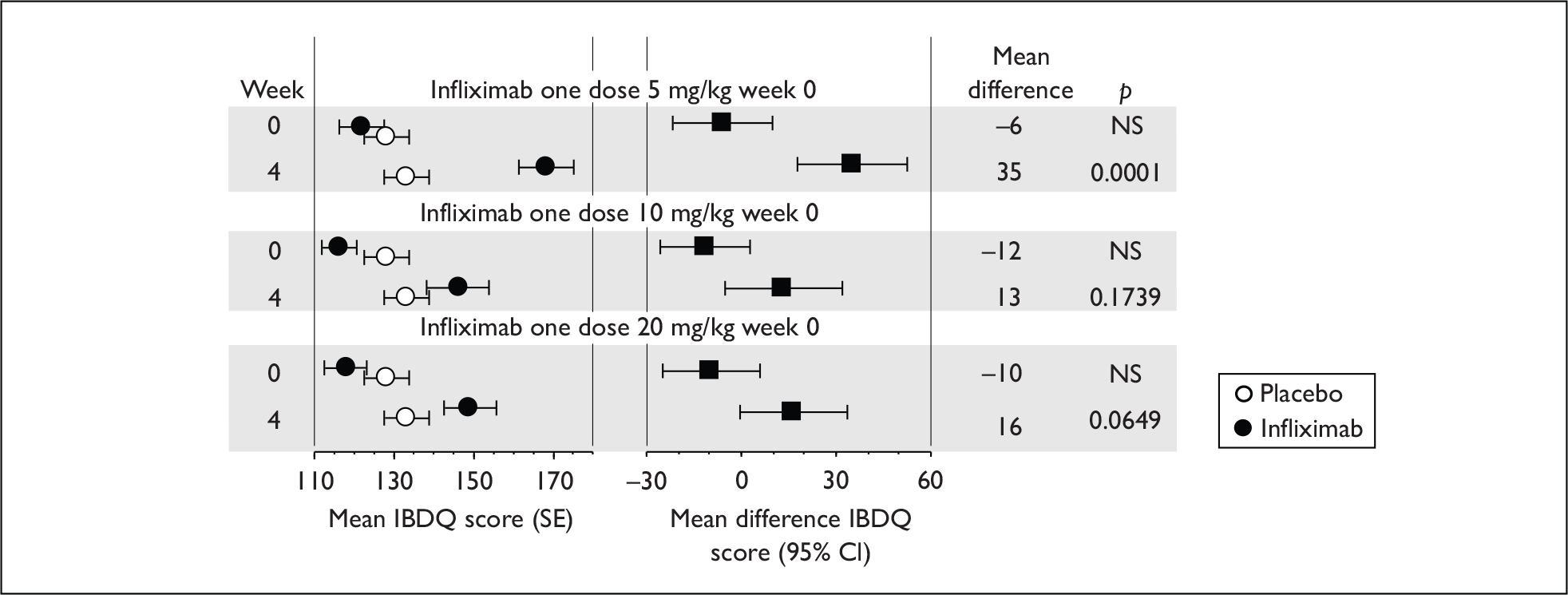
E, other considerations – open-label phase
In the open-label phase of the trial, extending by at least 12 weeks from week 4, non-responder patients at week 4 were eligible for a 10 mg/kg infusion of infliximab. The distribution of this second infusion among the patient groups is summarised in Table 10. Of the original 25 placebo group patients, 19 non-responders received infliximab; 29 non-responder patients who had received a first dose of infliximab received the second dose. Table 10 lists the percentage of the patients (not responsive at week 4) in each group who subsequently achieved response 70 at follow-up weeks 4, 8 and 12 after the second infusion.
| Original randomisation group (n) | Number receiving and not receiving second infusion (%) | Response 70 at times after second infusion (non-responders at week 4 after first infusion) | |||
|---|---|---|---|---|---|
| Did not receive | Received | Week 4 | Week 8 | Week 12 | |
| Placebo (25) | 6 (24) | 19 (76) | 11/19 (58%) | 13/19 (68%) | 10/19 (53%) |
| 5 mg/kg group (27) | 21 (78) | 6 (22) | 2/6 (33%) | 3/6 (50%) | 1/6 (17%) |
| 10 mg/kg group (28) | 13 (46) | 15 (54) | 6/15 (40%) | 5/15 (33%) | 5/15 (33%) |
| 20 mg/kg group (28) | 20 (71) | 8 (29) | 2/8 (25%) | 4/8 (50%) | 2/8 (25%) |
| Combined infliximab groups | 8/29 (28%) | ||||
| All groups (108) | 60 (56) | 48 (44) | 21/48 (44%) | 25/48 (52%) | 18/48 (38%) |
Of patients unresponsive to the first dose of infliximab, 28% (8/29) responded by week 12 following the second dose, compared with 53% (10/19) of patients whose second infusion was their first exposure to active intervention. During this open-label phase there was a lack of a true placebo control group and the results therefore only suggest that some patients poorly responsive to an initial infusion may respond subsequently on receipt of further infusion. Whether a 10 mg/kg second dose represents the most appropriate dose regimen for this second-dose strategy is unknown.
Quality assessment (based on published report)
Randomisation, allocation concealment, and blinding (up to week 4) were all adequate. Baseline characteristics were similar between groups except for CRP levels and for the proportion of patients with ileal involvement. Placebo CRP level {mean 12.8 [standard deviation (SD) 13.9]} was substantially lower than that for the active intervention groups [mean (SD): 22.1 (23.6), 23.2 (34.2) and 22.4 (23.9) for the 5 mg/kg, 10 mg/kg and 20 mg/kg groups respectively]. The potential impact on results of the imbalanced CRP levels is difficult to determine. Follow-up appeared almost complete. The original study protocol did not specify the use of ITT analysis, but the publication stated that patients were analysed according to assignment. A power calculation was conducted; this assumed a 30% response in the placebo group presumably reflecting the authors’ assessment of placebo rates reported in other CD trials. The actual placebo response rate observed was less than half this value (16%) and was low compared with other similar trials. The low placebo rate and imbalance of placebo CRP level may indicate an atypical placebo population possibly stemming from the small sample size of the group (n = 25).
CLASSIC I63 (adalimumab)
In this trial,63 patients (n = 299) were randomised to two subcutaneous injections 2 weeks apart of either placebo (n = 74) or adalimumab at dose regimens of 40 mg then 20 mg (n = 74), at 80 mg then 40 mg (n = 75), or at 160 mg then 80 mg (n = 76). Patients were excluded if they had previously received any anti-TNF treatment. At baseline 11% of patients had fistulas. Outcomes were monitored at weeks 1, 2 and 4 after the first injection. The primary outcome was defined as the proportion of patients in remission at week 4 in the two high-dose adalimumab groups versus the placebo group (tested using chi-squared test).
A single i.v. infusion of infliximab (5, 10 or 20 mg/kg) was more effective than placebo at delivering a clinical response (a reduction of ≥ 70 points in CDAI score) at week 4 of follow-up (p < 0.005 for risk differences and p < 0.022 for risk ratios). Estimates of the percentage of patients responding to infliximab were associated with considerable uncertainty, and at 4 weeks ranged between 50% and 80% depending on dose. Of the dose regimens used, the lowest appeared to be the most effective, suggesting the possibility that the most appropriate dose could be less than the lowest used in the trial (5 mg/kg). A proportion of patients (∼30%) not responsive at week 4 did respond subsequently when given a second dose of infliximab (10 mg/kg); although it is likely this ‘second-dose’ response required active intervention, this was not properly demonstrated because the trial lacked a true placebo comparator after week 4. The most effective dose regimen for a ‘second-dose’ response was uncertain. After week 4 nearly all trial participants had received active intervention, and inferences about the relation of outcomes to infliximab were obscured. The Targan et al. 57 trial was completed more than a decade ago and no further induction trial of infliximab in this population has been conducted, so the uncertainties described above remain to be addressed.
A, response 70
At week 4 for the less robust measure of a clinical improvement by more than 70 points in CDAI score from baseline (response 70), a statistically significant result was observed for both risk difference and risk ratio for all three dose regimens (results are summarised in Figure 5).
FIGURE 5.
Rates of response 70 in CLASSIC I. 63 At week 4 for risk difference p = 0.029, p = 0.005 and p = 0.004 for 40/20, 80/40 and 160/80 dose regimens, At week 4 for risk ratio p = 0.0357, p = 0.0088 and p = 0.0073 for 40/20, 80/40 and 160/80 dose regimens. LCI, lower confidence interval; RD, risk difference; RR, risk ratio; UCI, upper confidence interval.
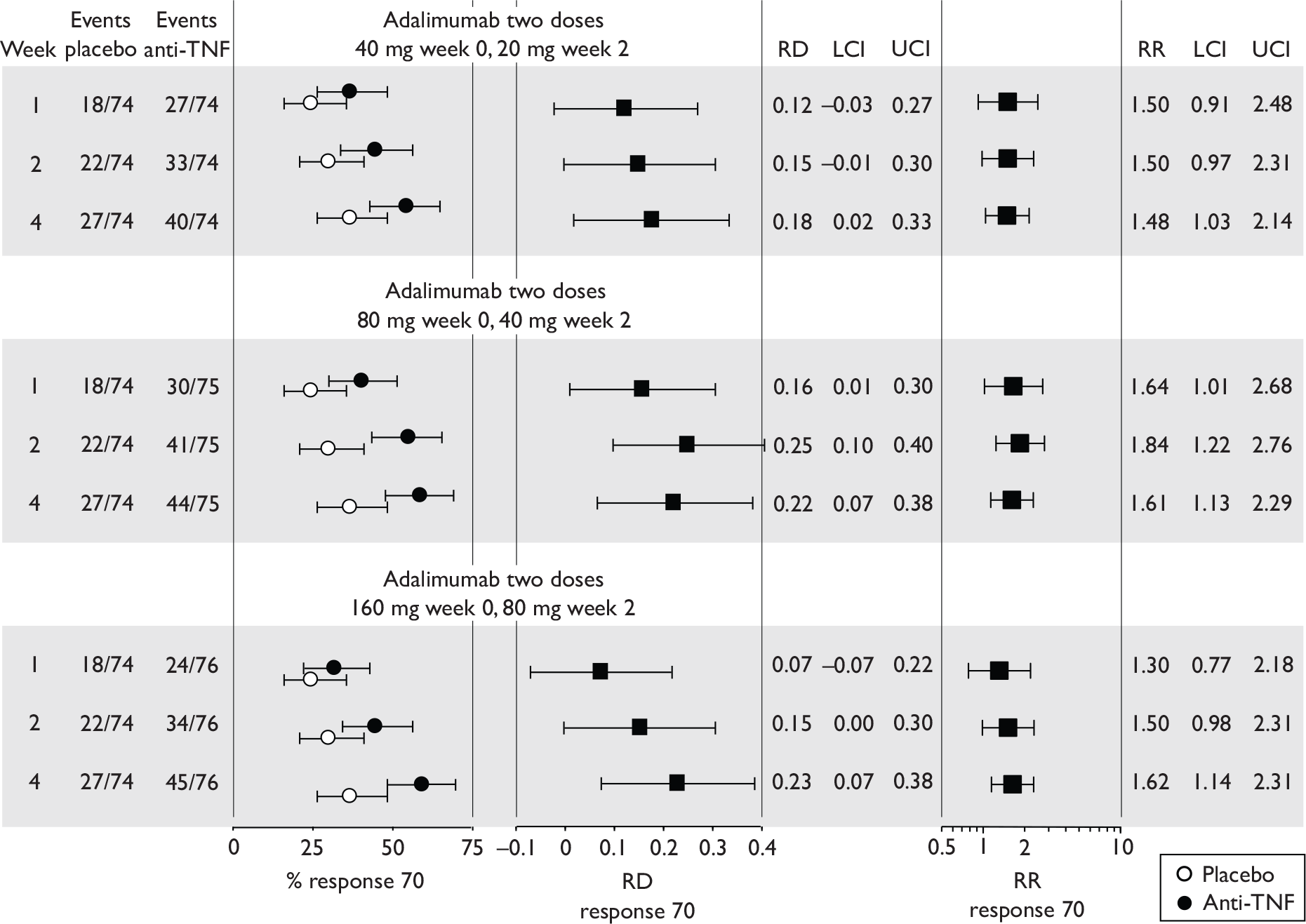
B, response 100
At week 4 the risk difference for response 100 (intervention–placebo) reached statistical significance only for the highest dose regimen while risk ratio (intervention/placebo) reached statistical significance for the two higher dose regimen groups. The results for response 100 are summarised in Figure 6.
FIGURE 6.
Rates of response 100 in CLASSIC I. 63 At week 4 for risk difference p = 0.279, p = 0.060 and p = 0.0015 for 40/20, 80/40 and 160/80 dose regimens respectively. At week 4 for risk ratio p = 0.284, p = 0.0682 and p = 0.0036 for 40/20, 80/40 and 160/80 dose regimens respectively. LCI, lower confidence interval; RD, risk difference; RR, risk ratio; UCI, upper confidence interval.
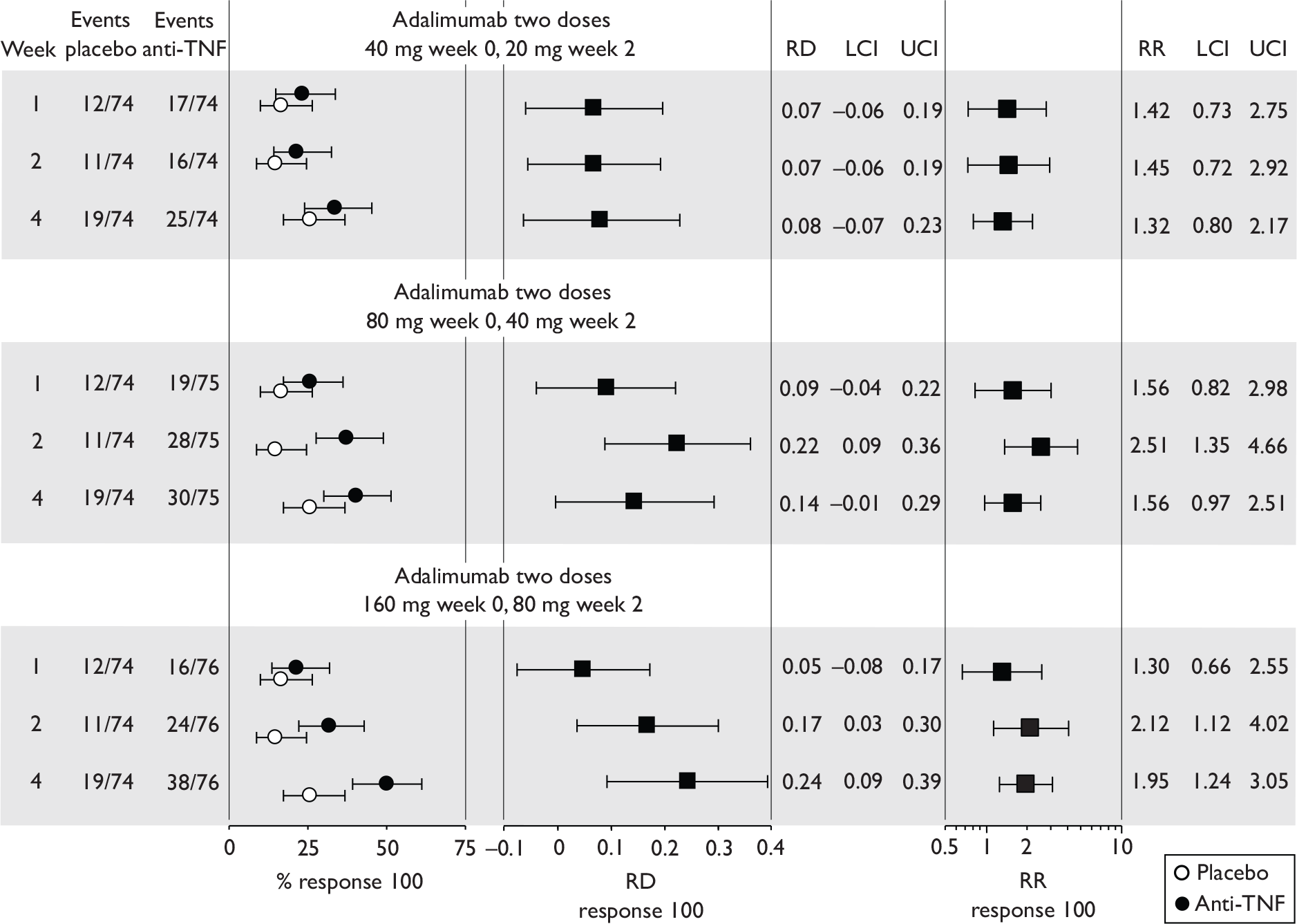
C, remission rates
Remission rates were the primary outcome in this RCT. For remission rates there was a statistically significant difference in favour of the two high-dose adalimumab regimens relative to placebo for the proportion of patients in remission at (45/151 vs 9/74; p = 0.004). At week 4 the risk difference (intervention–placebo) and risk ratio (intervention/placebo) reached statistical significance only in the highest dose regimen group. Remission rates are summarised in Figure 7.
FIGURE 7.
CLASSIC I remission rates. 63 At week 4 risk difference p = 0.354, p = 0.057 and p = 0.0005 for 40/20, 80/40 and 160/80 dose regimens. At week 4 risk ratio p = 0.359, p = 0.0691 and p = 0.0021 for 40/20, 80/40 and 160/80 dose regimens respectively. LCI, lower confidence interval; RD, risk difference; RR, risk ratio; UCI, upper confidence interval.
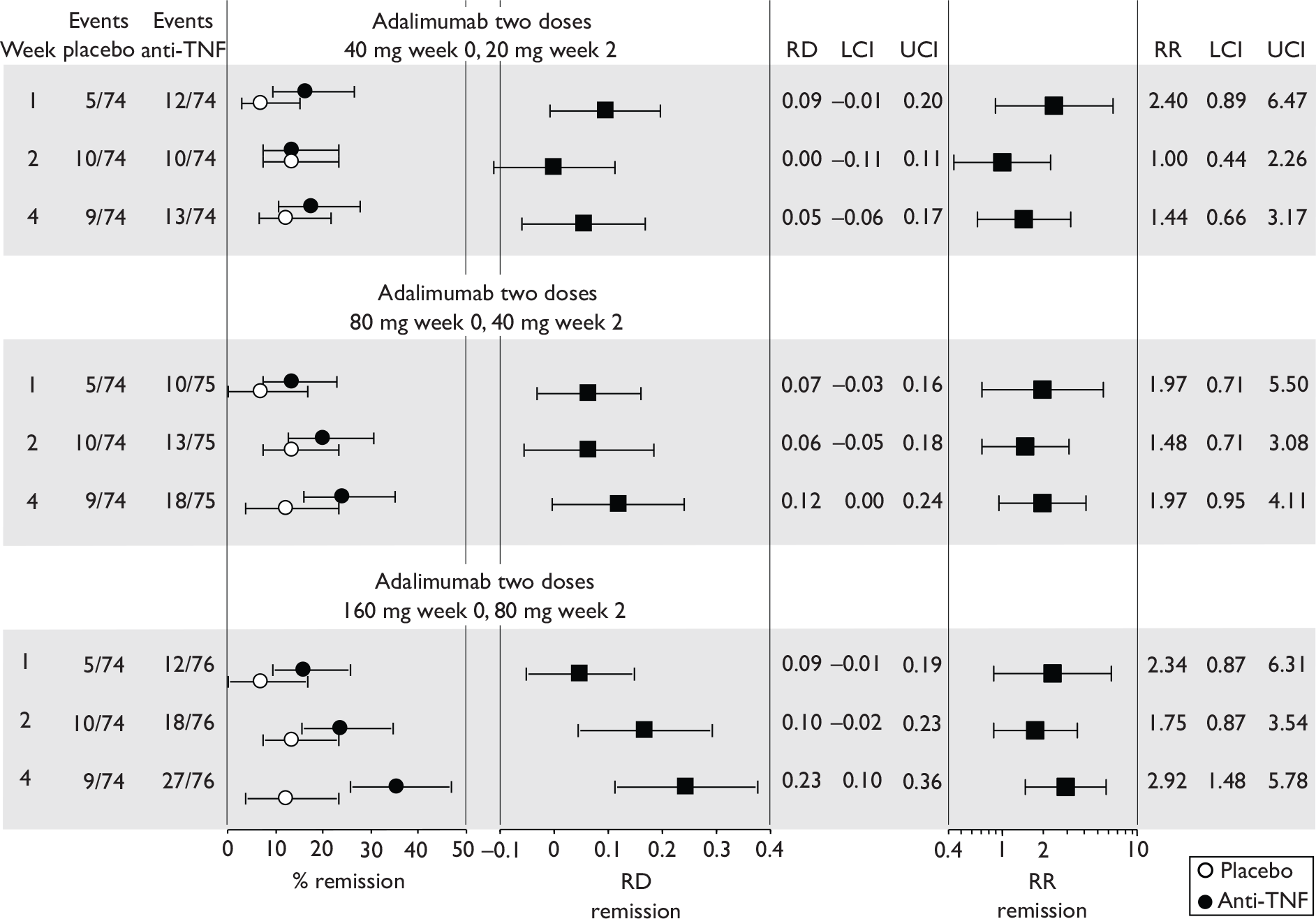
For each of the three CDAI-based binary outcome measures there was an apparent linear dose response trend with greater effectiveness for higher dose.
D, other outcomes
At week 4, favourable responses to treatment were reported for CDAI scores, for QOL scores (IBDQ), and for CRP levels. The results reported are summarised in Table 11.
| Placebo (n = 74) | 40/20 (n = 74) | 80/40 (n = 75) | 160/80 (n = 76) | |
|---|---|---|---|---|
| Score on CDAI: mean (SD) | ||||
| Baseline | 296 (60) | 299 (57) | 301 (61) | 295 (52) |
| 4 weeks | 240 (NR) | 228 (NR) | 210 (NR)a | 193 (NR)b |
| Score on IBDQ median (range) | ||||
| Baseline | 131 (52–200) | 129 (81–218) | 128 (63–200) | 127 (37–192) |
| 4 weeks | 147 (NR) | 147 (NR) | 158 (NR)c | 158 (NR)c |
| CRP (mg/l) median (range)d | ||||
| Baseline | 0.9 (0–17.3) | 0.9 (0–11.3) | 0.9 (0–14.9) | 0.7 (0–9.3) |
| 4 weeks | 0.8 (0–9.3) | 0.3 (0–8.6)e | 0.4 (0–34.0)f | 0.2 (0–4.6)g |
E, other considerations – subgroup analyses
Logistic regression failed to show a relationship between baseline CRP levels or concomitant immunosuppressive therapy and remission rates at week 4 with placebo or adalimumab.
For the small subgroup of patients with fistulas (11%), no significant differences were observed between placebo and intervention with regard to fistula improvement or remission.
Quality assessment (based on published report)
Randomisation, allocation concealment and blinding were adequate. Baseline characteristics were reasonably well balanced between groups. There were no losses to follow-up, and withdrawals were limited to 5%. Efficacy estimates appear to have been calculated using ITT analysis, but this was not stated explicitly. A power calculation was conducted; this assumed 20% and 45% remission rates in the placebo and intervention arms respectively (the observed placebo rate in the trial was about 12%). Last observation carried forward was used for analysis of IBDQ scores, but the number of missing data was not stated.
Two subcutaneous injections of adalimumab given 2 weeks apart at 40 mg then 20 mg, at 80 mg then 40 mg, or at 160 mg then 80 mg, were more effective than placebo at achieving remission (CDAI score < 150) at week 4 after the first injection (p = 0.004 for the two high-dose regimens combined vs placebo). The percentage of placebo-treated patients gaining remission at week 4 was ∼12% compared with between ∼18% and ∼36% for adalimumab-treated patients depending on dose regimen received. Point estimates of response 70 rates, response 100 rates and remission rates were associated with considerable uncertainty, but for all three outcome measures a trend was evident for higher doses to be more effective. At week 4 of follow-up, risk differences (intervention–placebo) and risk ratios (intervention/placebo) for the highest dose regimen reached statistical significance in favour of adalimumab for all three outcomes. Subgroup analyses failed to identify any baseline characteristics associated with a better response to active intervention relative to placebo.
GAIN64 (adalimumab)
In this trial,64 325 patients were randomised to two subcutaneous injections 2 weeks apart of either placebo (n = 166) or adalimumab at a dose regimen of 160 mg then 80 mg (n = 159). To be included patients had to have been previously exposed to infliximab treatment and found to be intolerant (n = 190), unresponsive (n = 164), or intolerant and unresponsive (n = 40). The primary response was defined as the proportion of patients in remission at week 4 after the first injection.
A, response 70; B, response 100; and C, remission
The primary outcome was remission rates. The remission rate at week 4 was 7% in the placebo group and 21% in the adalimumab group (p < 0.001). This result and those for the secondary outcomes as reported are summarised in Table 12. The CDAI-based binary response outcome measures reported are summarised graphically in Figure 8. At weeks 2 and 4, risk differences (adalimumab–placebo) and risk ratios (adalimumab/placebo) were in favour of the intervention and reached statistical significance.
| Placebo (n = 159) | Adalimumab 160/80 (n = 164) | Difference (95% CI) (adalimumab–placebo) | p a | |
|---|---|---|---|---|
| Remission (rate; %)b | ||||
| Week 1 | 4% | 6% | 2.7% (–2.0 to 7.4) | (CiC information has been removed) |
| Week 2 | 6% | 21% | 14.7% (7.2 to 22) | (CiC information has been removed) |
| Week 4 | 7% | 21% | 14.2% (6.7 to 21.6) | (CiC information has been removed) |
| Response 70 (rate; %) | ||||
| Week 1 | 21% | 35% | 14.1% (4.5 to 23.7) | 0.004 |
| Week 2 | 33% | 52% | 19.7% (9.1 to 30.1) | (CiC information has been removed) |
| Week 4 | 34% | 52% | 17.8% (7.3 to 28.4) | (CiC information has been removed) |
| Response 100 (rate; %) | ||||
| Week 1 | 12% | 20% | 7.4% (–0.5 to 15.4) | (CiC information has been removed) |
| Week 2 | 18% | 37% | 18.4% (8.9 to 27.9) | (CiC information has been removed) |
| Week 4 | 25% | 38% | 13.7% (3.7 to 23.7) | (CiC information has been removed) |
| CDAI: mean (SD) | ||||
| Baseline | 313 (66) | 313 (58) | 0 | |
| Week 1 | 287 (NR) | 264 (NR) | –23 | |
| Week 2 | 281 (NR) | 232 (NR) | –49 | |
| Week 4 | 264 (NR) | 226 (NR) | –38 | |
| IBDQ score: mean (SD) | ||||
| Baseline | 124 (28) | 120 (27) | + 4 | |
| Week 4 | 139 (NR) | 150 (NR) | + 11 | < 0.001 |
| CRP: median (range) mg/ld | ||||
| Baseline | 7.0 (0–235) | 9.0 (0–115) | + 2 | |
| Week 4 | 7.0 | 5.0 | –2 | |
| Change from baseline | 0 | 4 | 4 | Significant |
FIGURE 8.
Response 70, response 100 and remission rates reported in GAIN. 64 At week 4 risk difference p = 0.001, p = 0.007 and p = 0.0002 for response 70, response 100 and remission respectively. At week 4 risk ratio p = 0.0014, p = 0.009 and p = 0.0006 for response 70, response 100 and remission respectively. LCI, lower confidence interval; RD, risk difference; RR, risk ratio; UCI, upper confidence interval.
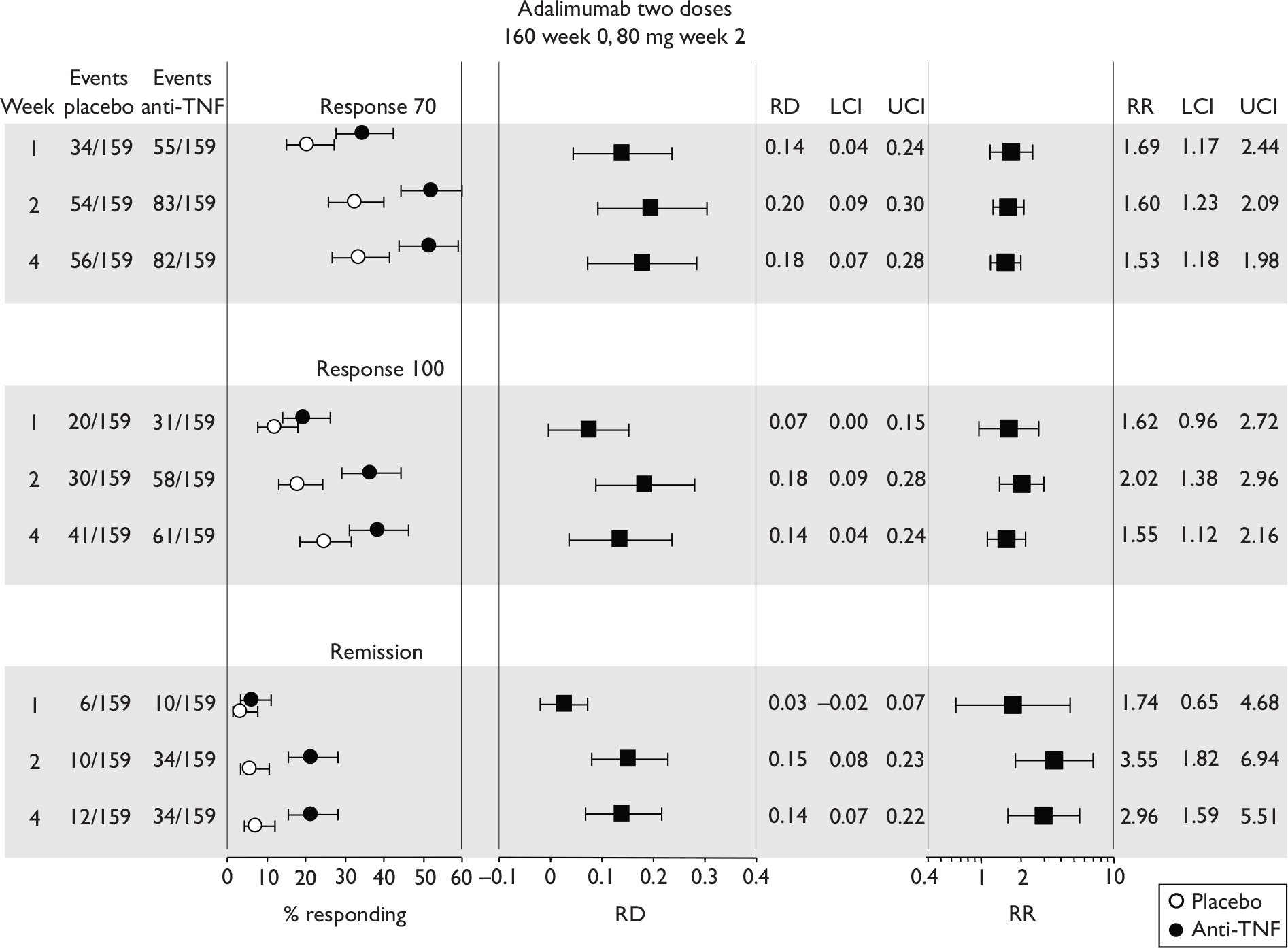
D, other outcomes
Results for these are also shown in Table 12. Mean CDAI scores reduced from baseline to a greater extent with adalimumab than with placebo (at week 4, p < 0.001 for mean change from baseline). At week 4 the improvements from baseline in IBDQ scores were 30 and 15 for the adalimumab and the placebo groups respectively. CRP levels at week 4 relative to baseline were more normalised in the intervention than in the placebo group. The change from baseline comparing adalimumab with placebo reached statistical significance in favour of adalimumab.
E, other considerations – subgroup analyses
The primary outcome (remission at week 4) was reported for subgroups of patients defined according to: previous response or intolerance to infliximab; receiving or not receiving immunosuppressive agents at baseline; receiving or not receiving corticosteroids at baseline; or having a negative or positive test for antibodies to infliximab. Risk difference was in favour of adalimumab relative to placebo for all subgroups.
A small proportion of patients (14%, n = 45) had draining fistulas or perianal fistulas at baseline. Rates of fistula improvement and remission were similar between placebo and adalimumab groups.
Quality assessment (based on published report)
Randomisation, allocation concealment and blinding were adequate. Baseline characteristics were well balanced between groups. There were no losses to follow-up, and withdrawals were limited to 4%. Efficacy estimates appear to have been calculated using ITT analysis for remission and response outcomes. For continuous variables such as IBDQ, last observation was carried forward; the number of missing data for IBDQ was small (eight patients). A power calculation was conducted; this assumed 20% and 35% remission rates in the placebo and intervention arms respectively (the observed rates at week 4 in the trial were 7% and 21% respectively).
Two subcutaneous injections of 160 mg and then 80 mg of adalimumab given 2 weeks apart were more effective than injections of placebo at achieving remission (CDAI score < 150) at week 4 after the first injection (p < 0.001). The percentage of placebo-treated patients gaining remission at week 4 was 7% (95% CI 4% to 12%) compared with 21% (95% CI 14% to 27%) for adalimumab-treated patients. At weeks 2 and 4 of follow-up, risk differences (intervention–placebo) and risk ratios (intervention/placebo) reached statistical significance in favour of adalimumab for remission, response 70 and response 100. A statistically significant difference in favour of adalimumab versus placebo was observed for change in IBDQ score at week 4 relative to baseline.
Pooling and indirect comparison
The two adalimumab trials differed with respect to their populations: CLASSIC I63 excluded patients if they had previously received any anti-TNF treatment while the GAIN64 trial recruited only patients who had previously experienced infliximab treatment but had proved intolerant or unresponsive; because of these clear population differences results from the two trials were not pooled. The existence of only a single induction trial for infliximab in this population precluded pooling.
No head-to-head induction trial of infliximab versus adalimumab has been conducted. A possible approach to compare effectiveness of the two drugs is by indirect comparison using trials with a ‘common’ comparator (e.g. placebo). The Targan et al. population,57 in contrast to that in GAIN,64 was naive to anti-TNF therapy and therefore indirect comparison between these trials was not judged productive. The placebo rates for remission and response 70 in Targan et al. 57 were low compared with those in the adalimumab trials and are indicative of likely differences between the potentially ‘common’ comparator groups possibly stemming from the very small sample size of the placebo group in the Targan et al. 57 trial. Because of the likely difference in target placebo populations, indirect comparison was judged more likely to be misleading than informative. It is relevant that neither industry submission undertook an indirect comparison between these induction trials. One way clinical heterogeneity may be expressed is in different response rates in placebo groups. Although CDAI scores at baseline may be similar between trials, this could mask considerable clinical heterogeneity because CDAI is a summary score and patients can achieve the same score yet may have problems with quite different aspects of their disease.
Maintenance trials in adults (wholly or predominantly non-fistulising)
These are trials in which all patients receive short-term induction therapy with anti-TNF and then proceed to longer term treatment with either placebo or anti-TNF. The predominant aim of these trials was to investigate whether anti-TNF was superior to placebo in maintaining any favourable clinical response observed from induction therapy. As no true placebo comparator existed during the induction therapy it is not possible to determine how much of the favourable clinical response seen from induction was actually attributable to active intervention. This complicates interpretation of results.
Four trials were identified, two with infliximab [Rutgeerts et al. 58 and ACCENT I (Hanauer et al. 3 and Rutgeerts et al. 2)] and two with adalimumab [CLASSIC II (Sandborn et al. 66) and CHARM (Colombel et al. 67)]. These studies were characterised by distinct differences in induction regimens.
The Rutgeerts et al. 58 trial was an extension of the Targan et al. 57 infliximab induction trial. Patients eligible had received variably one or two previous infusions of placebo or of infliximab at doses of 5, 10 or 20 mg/kg. Patients with a response 70 were then eligible for the trial. The induction regimen of participants in this trial was variable and not clearly defined, making it difficult to identify the precise target population involved.
Similarly to Rutgeerts et al. ,58 the CLASSIC II66 trial was an extension of a previously conducted induction trial, namely the CLASSIC I63 study of adalimumab. Patients eligible for CLASSIC II were required to be in remission (CDAI < 150) at week 4 of CLASSIC I and also 4 weeks later. These patients may have received two subcutaneous injections 2 weeks apart of various doses of adalimumab (40 mg then 20 mg, 80 mg then 40 mg, or 160 mg then 80 mg) or of placebo.
The ACCENT I2,3 (infliximab) and CHARM67 (adalimumab) trials were free-standing maintenance trials with more straight forward induction regimens. In ACCENT I patients received a single induction infusion of 5 mg/kg infliximab. In CHARM patients received subcutaneous induction injections of 160 mg of adalimumab and of 80 mg of adalimumab 2 weeks apart.
The main study and population characteristics are shown in Table 13. The main outcome measures described in the published reports of the four trials are summarised in Table 14.
| Studya Drug |
Weeks n |
Population: severity of CD Baseline CDAI and IBDQ if stated |
Areas affected | Main concomitant medicationb | Previous anti-TNF therapy | Intervention and comparator (dosing regimen) |
|---|---|---|---|---|---|---|
|
Infliximab |
48 73 |
Moderate-to-severe, CDAI 220–400 ‘treatment resistant’ Median CDAI: placebo 305; infliximab 310 Median IBDQ: placebo 121; infliximab 111 |
Mainly ileum and colon or colon only, some ileum only | Corticosteroids or immunosuppressive agents ‘allowed’, non-responders to aminosalicylates ‘eligible’ | Excluded if had received monoclonal antibodies prior to Targan et al. study57 | Variable treatment with infliximab or placebo in previous RCT then re-randomisation to placebo or infliximab (10 mg/kg/kg i.v.) at 8-week intervals |
|
Hanauer et al. , 20023 and Rutgeerts et al. , 20042 – ACCENT I2,3 Infliximab |
54 573 |
Moderate-to-severe, CDAI 220–450 CDAI median (IQR): placebo 292 (256–341); infliximab 303 (268–346) and 297 (256–346) IBDQ median (IQR): placebo 126 (110–144); infliximab 126 (109–146) and 131 (109–152) |
Mainly ileum/colon, also colon only or ileum only; some gastroduodenum | Corticosteroids, immunosuppressives, oral aminosalicylates | Excluded if previously treated with any anti-TNF agent |
All receive 5 mg/kg infliximab i.v.; then seven additional infusions (weeks 2, 6, then every 8 weeks) of either placebo or infliximab (5 mg/kg or 10 mg/kg) (Note both infliximab groups received 5 mg/kg at weeks 2 and 6) |
|
Sandborn et al. , 200766 – dCLASSIC II66 Adalimumab |
56 55 |
All patients in remission week 0 (week 4 CLASSIC I63) Baseline corresponds to CLASSIC I63 week 4 CDAI mean (SD): Placebo 107 (62); adalimumab 106 (33) and 88 (50) IBDQ median (range): placebo 191 (138–224); adalimumab 188 (128–213) and 200 (138–216) |
No further details As CLASSIC I63 |
Mainly oral aminosalicylates or corticosteroids, some immunosuppressive agents | Unclear if all previously received adalimumab or if patients in remission after placebo were included | Subcutaneous infusion 40-mg adalimumab from weeks 4–55, weekly or e.o.w. Not stated if placebo weekly or e.o.w. |
|
Colombel et al. , 200767 – CHARM67 Adalimumab |
56 778 |
Moderately-to-severely active CD, CDAI 220–450 CDAI mean (SD)e: 313.1 (62.0) IBDQ median (range)e: 122.0 (44–205) |
Mainly ileum or colon, few gastroduodenal or other (not stated) | Corticosteroids, immunosuppressive agents, oral aminosalicylates |
424 (49.6%) previously exposed to anti-TNF (must not have exhibited an initial non-response) |
All received adalimumab 80 mg subcutaneously, then 40 mg at week 2; randomisation at week 4, then 40-mg adalimumab, weekly or e.o.w. Not stated if placebo weekly or e.o.w. |
| % of patients in remission (CDAI score < 150) | % achieving 100-point response on CDAI | % achieving 70-point response on CDAI | CDAI score (mean or median) | IBDQ score (mean or median) | Additional outcomes | |
|---|---|---|---|---|---|---|
| Infliximab | ||||||
| Rutgeerts et al., 199958 | ✓ | ✗ | ✓ | ✓ | ✓ | Median CRPc. Time to loss of response |
| ACCENT I2,3 | ✓ | ✗ | ✓ | ✓ | ✓ | Patients with CD-related intra-abdominal surgery; CD-related hospitalisations; patients discontinuing and remaining free from corticosteroids; mucosal healing (subgroup) |
| Adalimumab | ||||||
| CLASSIC II66 | ✓ | ✓ | ✓ | ✗ | ✓ | Median CRPc, % of patients discontinuing steroids without loss of remission |
| CHARM67 | ✓ | ✓ | ✓ | ✓ | ✓ | % of patients in remission at week 4 who were also in remission at week 56; median time in remission; corticosteroid free remission; fistula response |
Rutgeerts et al., 199958 (infliximab)
The Rutgeerts et al. 58 trial was an extension of the Targan et al. 57 infliximab induction trial and included 73 of the original 108 patients. Targan et al. 57 consisted of a 4-week comparison between placebo and one dose of infliximab in three arms (5 mg/kg, 10 mg/kg or 20 mg/kg). This was followed after a maximum of 2 weeks by an open-label phase with 12 weeks of follow-up that started with the option of a 10 mg/kg dose of infliximab for week 4 non-responder patients. To be eligible to enrol in Rutgeerts et al. 58 the Targan et al. week 4 responder patients needed to achieve a response 70 at week 8, and the week 4 non-responder patients needed to achieve a response 70 at week 8 after the open-label option of a 10 mg/kg infusion of infliximab. Four weeks after qualifying (week 8 after induction infliximab or 8 weeks after open-label infliximab) the eligible patients were randomised to i.v. infusion of placebo or 10 mg/kg infliximab (designated week 12 of maintenance phase) and a further three infusions at 8-week intervals (a total of four infusions after becoming eligible to participate; administered weeks 12, 20, 28 and 36). Follow-up continued to week 48.
The induction regimen in this study was variable between patients in duration and in exposure to infliximab. In consequence, induction was ill-defined and the distinction between the induction regimen and maintenance regimen was also unclear. The eligible patients could have received any of the following possible infusions of infliximab: no infliximab (placebo), one 5 mg/kg infusion, one 10 mg/kg infusion or one 20 mg/kg infusion; a second infusion of 10 mg/kg could be given (to any patients) at week 4 if there was no response. Four patients received no infliximab (placebo and no second 10 mg/kg dose as a response was achieved). How closely the trial induction phase corresponds to the licence indication is uncertain.
A, response 70
No primary outcome measure was identified. The response 70 results presented (summarised in Figure 9) referred to point prevalence at assessment time points and do not necessarily indicate maintenance of individual patient response. At week 8 > 90% of patients had a response 70 (CDAI reduced by > 70 points relative to baseline in Targan et al. 57). At week 12 (randomisation week) this had diminished to about 75% and by week 48 had further diminished to 33% in the placebo group and 57% in the infliximab group (p = 0.038 for risk difference and p = 0.054 for risk ratio). Point estimates were associated with considerable uncertainty. The authors stated that of patients with response 70 at the last infusion (week 36), 62% of the infliximab group and 37% of the placebo group maintained their response for the 8 weeks to week 44 (p = 0.16).
FIGURE 9.
Response 70 rates in Rutgeerts et al. 58 At weeks 24 and 48 risk difference p = 0.094 and p = 0.038 respectively. At weeks 24 and 48 risk ratio p = 0.108 and p = 0.054 respectively. LCI, lower confidence interval; RD, risk difference; RR, risk ratio; UCI, upper confidence interval.
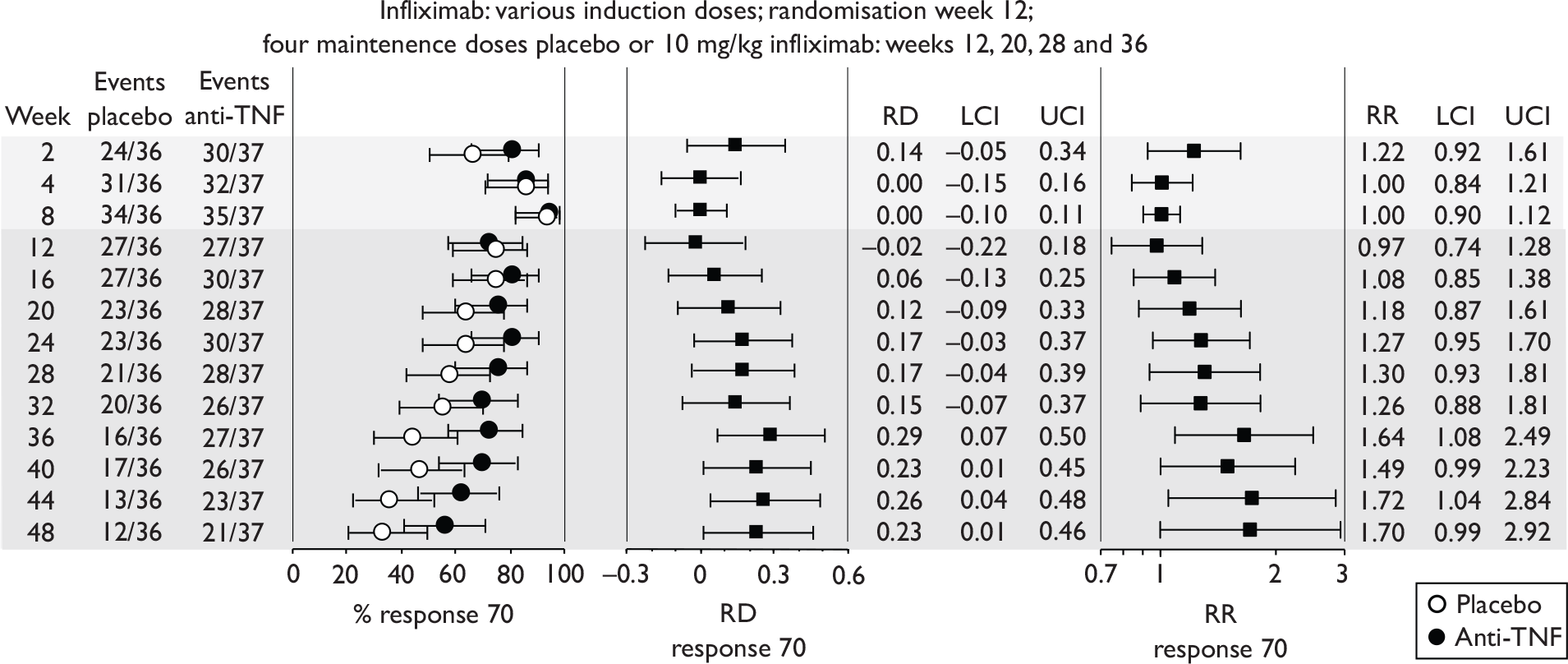
B, response 100
This outcome was not reported.
C, remission
The point prevalence of remission at different follow-up weeks was reported (results are summarised in Figure 10). Point estimates were associated with considerable uncertainty. At randomisation (week 12) ∼38% of patients were in remission in the infliximab group; this increased to ∼ 60% during weeks 16–40. The corresponding values for the placebo group were ∼ 44% (week 12) and 35% (weeks 16–40). Risk difference (infliximab–placebo) and risk ratio (infliximab/placebo) just reached statistical significance (p < 0.05) at most time points for weeks 16–40.
FIGURE 10.
Remission rates in Rutgeerts et al. 58 LCI, lower confidence interval; RD, risk difference; RR, risk ratio; UCI, upper confidence interval.
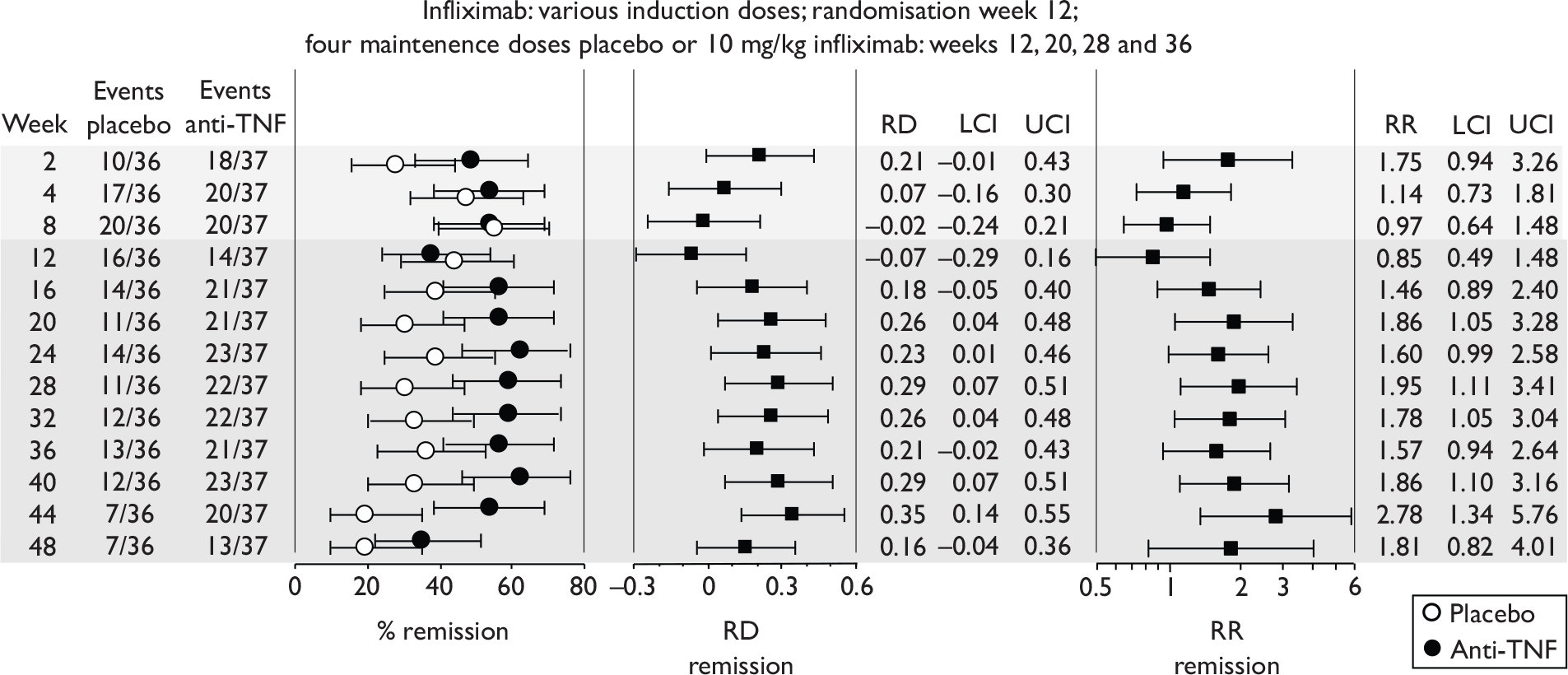
D, other outcomes
Time to loss of response for patients achieving a response at ‘any time’ during follow-up after randomisation was reported. The criteria for loss of response were not explicit. Over 48 weeks it is possible for a patient to enter a response state on several occasions. The publication did not make clear which occasion(s) were used in the analysis, or how and if double counting was avoided. The log rank test for difference between placebo and infliximab groups just failed to reach statistical significance (p = 0.057).
Median CDAI score, median IBDQ score and median CRP concentrations were reported, but range of values and statistical analyses for these outcomes were not presented. The results were in favour of infliximab relative to placebo with greater reduction in CDAI scores, larger increases in IBDQ scores and more ‘normalisation’ of CRP concentrations. The results published are summarised in Figure 11.
FIGURE 11.
Median CDAI, IBDQ and CRP levels reported in Rutgeerts et al. 58 Data taken from published graphs and redrawn. Where necessary the authors carried last observation forward.
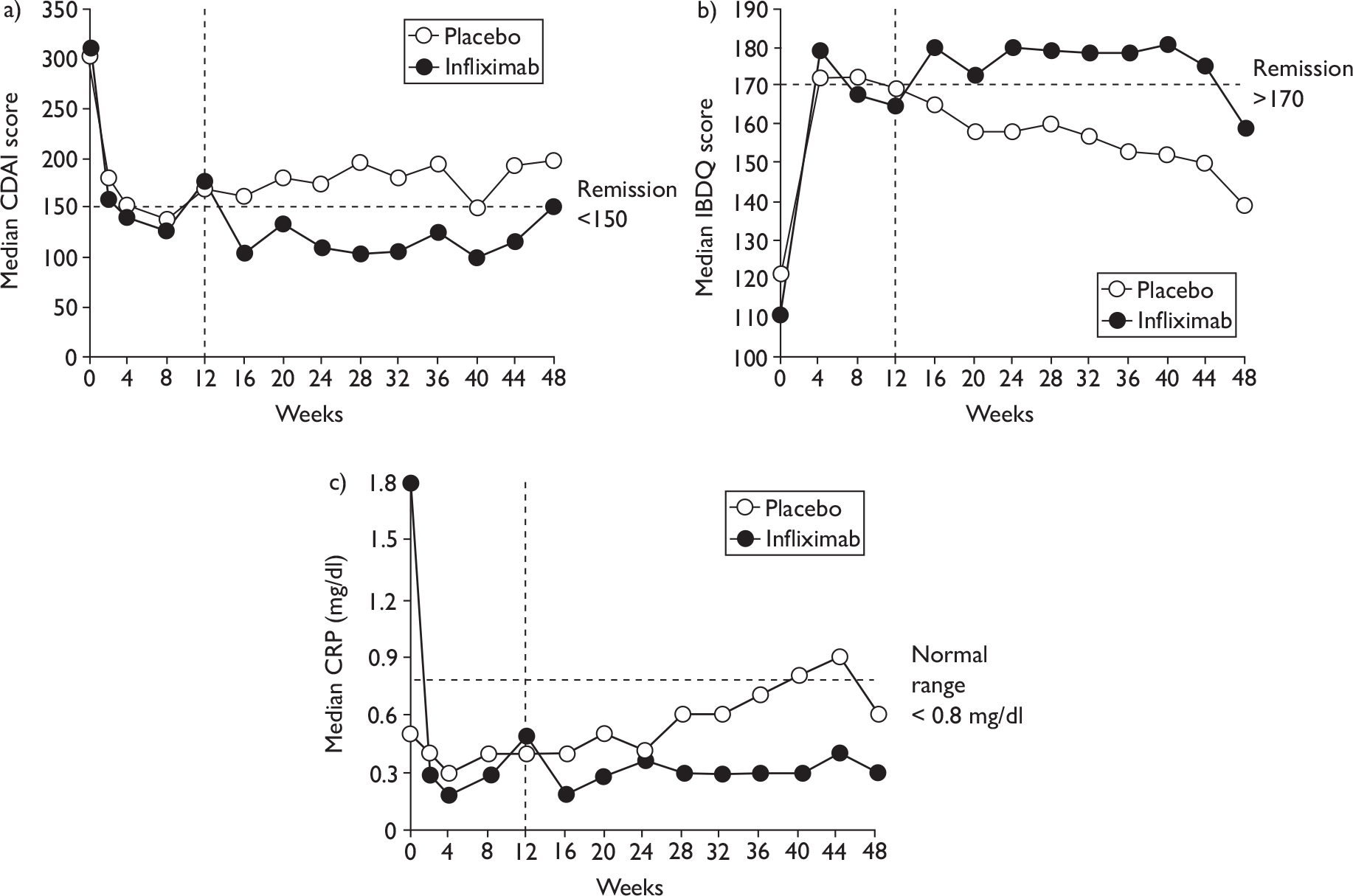
Quality assessment (based on published report)
Randomisation, allocation concealment and blinding were adequate. Baseline values for those characteristics reported were evenly balanced, but values for CRP, which was not balanced in the original Targan et al. trial,57 were unclear. Analysis of response 70 and remission rates was by ITT; the results presented were point prevalence values at various follow-up times, and they therefore represent maintenance of response at the group level only and not maintenance by individual patients. For continuous outcomes, last observation was carried forward where necessary, but the number of missing data was not reported. No primary outcome was identified and no power calculation was described; the combined trials appear to have been powered only for the induction analysis of Targan et al. 57 (at week 4 of that study). The maintenance part of the study was probably underpowered. About 33% of patients withdrew.
The study recruited patients from among responders (CDAI score reduced by 70 points) following on from the Targan et al. trial57 and the resulting induction phase varied between patients in both duration and dose regimen. Subsequent maintenance treatment with infliximab (four infusions of 10 mg/kg at 8-week intervals) generated a greater proportion of patients with a response 70 and with remission than did treatment with placebo. Point prevalence estimates for these outcomes were associated with considerable uncertainty. The trial left unanswered how well a clinical response is sustained at the individual patient level.
ACCENT I2,3 (infliximab)
This was a free-standing maintenance trial (i.e. newly started). 2,3 There were 580 eligible patients (CDAI range 220–400), of whom 573 received a single induction infusion of 5 mg/kg infliximab. Two weeks later patients were randomised to placebo, to 5 mg/kg infliximab at weeks 2 and 6 and then every 8 weeks to week 54, or to 5 mg/kg at weeks 2 and 6 and then 10 mg/kg infliximab every 8 weeks to week 54 (these groups are here termed 5 mg/kg and 10 mg/kg groups respectively). At week 2 (randomisation week) patients were classified as responders (335/573, 58.5%) or non-responders (238/573, 41.5%) depending on whether they achieved a response 70 (a reduction of > 70 points in CDAI score at week 2 relative to baseline). At week 14 patients who initially responded but then worsened were eligible to cross over to treatment with increased dosage of infliximab; this crossover treatment for the placebo group was termed ‘episodic treatment’. The results for responders were published in 2002 (Hanauer et al. 3) and patients who crossed over to increased dosage after week 14 for most of these analyses were considered as treatment failures.
Effectiveness results published for responders only in 2002 (Hanauer et al. 3) are reviewed below, and results for all patients, irrespective of responder status at week 2 and published in 2004 (Rutgeerts et al. 2), are considered in the following section.
ACCENT I: results for responders
Of the 335 responders (58.5% of those who had received an induction dose of 5 mg/kg infliximab), 110 were randomised to placebo, 113 to the 5 mg/kg infliximab group and 112 to the 10 mg/kg infliximab group.
A, response 70
The published results for responders3 included graphical presentation of point prevalence of response 70 at weeks 30 and 54. These results are summarised in Table 15. A statistically significant difference in rates in favour of infliximab versus placebo was reported for both infliximab groups at weeks 30 and 54. The manufacturer’s submission provided point prevalence rates for response 70 for all assessment visit weeks from 2 to 54. These results are summarised in Figure 12.
| Dose regimen (n) | Week 30b | Week 54b | ||
|---|---|---|---|---|
| Response 70 (%)b | p c | Response 70 (%)b | p c | |
| Placebo (110) | 27% | NA | 16% | NA |
| 5 mg/kg group (113) | 51% | 0.0002 | 38% | 0.0001 |
| 10 mg/kg group (112) | 58% | < 0.0001 | 47% | 0.0001 |
Point estimates were associated with appreciable uncertainty. Week 2 response rates of ∼90% had diminished in all groups by week 54 to 15% in the placebo group and 38% and 47% in the 5-mgkg and 10 mg/kg infliximab groups respectively. Risk differences (infliximab–placebo) remained fairly constant from week 14 onwards. Risk differences and risk ratios (infliximab/placebo) reached statistical significance in favour of infliximab at all visit times from week 10 to week 54. It is unclear why week 2 response rates were less than 100%; it is possible some patients with a 70-point CDAI reduction from baseline nevertheless required surgery or a change in concomitant medication for worsening of clinical condition. After week 2, decline of response occurred in both placebo and intervention groups, then after week 10 risk differences remained similar [e.g. for the 5 mg/kg arm risk differences (infliximab–placebo) remained similar after week 14 as follows: at weeks 10, 14, 22, 30, 38, 46 and 54 risk differences were 0.14, 0.23, 0.26, 0.24, 0.21, 0.23 and 0.23 respectively]. This suggested that most benefit of infliximab was delivered in the first 10–12 weeks of the trial.
B, response 100
This outcome was not reported.
C, remission rates
Remission was a coprimary outcome. The results published for remission at week 30 and week 54 are summarised in Table 16. For this outcome patients who worsened and crossed over to ‘episodic treatment’ (allowed from week 14 onwards) were counted as treatment failures (i.e. as no longer in remission). The results reported measured the point prevalence of remission for each group at week 30 and did not require maintenance of response from week 2 to week 30 at the patient level. A statistically significant greater proportion of patients were in remission at weeks 30 and 54 in the infliximab groups than in the placebo group. At week 30 the risk differences (infliximab–placebo) were 18% and 25% for the 5 mg/kg and 10 mg/kg groups respectively and the corresponding numbers needed to treat (NNT) (30 weeks) were 5.66 and 4. Note this NNT estimate does not include non-responders who had been administered induction infliximab. The point prevalence of remission had diminished somewhat by week 54.
| Dose regimen (n) | Week 30 | Week 54 | |||
|---|---|---|---|---|---|
| Remission: % (95% CIb) (number) | Odds ratio (95% CI) intervention/placebo | p c | Remission (%)d | p c | |
| Placebo (110) | 21% (14% to 29%) (23) | NR | NA | 14% | NA |
| 5 mg/kg group (113) | 39% (30% to 48%) (44) | NR | 0.003 | 28% | 0.007 |
| 10 mg/kg group (112) | 45% (36% to 55%) (50) | NR | 0.002 | 38% | < 0.0001 |
| 5 mg/kg and 10 mg/kg groups combined (225) | 42% (36% to 48%) (94) | 2.7 (1.6 to 4.6) | NR | 33% | |
The unpublished Industry Trial Report for ACCENT I2,3 provided information regarding the maintenance of remission at the individual patient level for weeks 14–54. The percentages were slightly discrepant with those in the published report as indicated in Table 17.
| % in remission at all visits from weeks 14 to 54 | |||
|---|---|---|---|
| Placebo | 5 mg/kg group | 10 mg/kg group | |
| Published report | 11% | 25% | 33% |
| Trial report | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) |
The manufacturer’s submission and the Industry Trial Report provided commercial-in-confidence (CiC) point prevalence rates for remission for all assessment visits from weeks 2 to 54. These results are summarised in Figure 13.
FIGURE 13.
Remission rates for responders throughout follow-up in ACCENT I. 2,3 At week 30 risk difference p = 0.0027 and p < 0.0001 for 5 mg/kg and 10 mg/kg groups respectively. At week 30 risk ratio p = 0.0047 and p = 0.00025 for 5 mg/kg and 10 mg/kg groups respectively. LCI, lower confidence interval; RD, risk difference; RR, risk ratio; UCI, upper confidence interval.
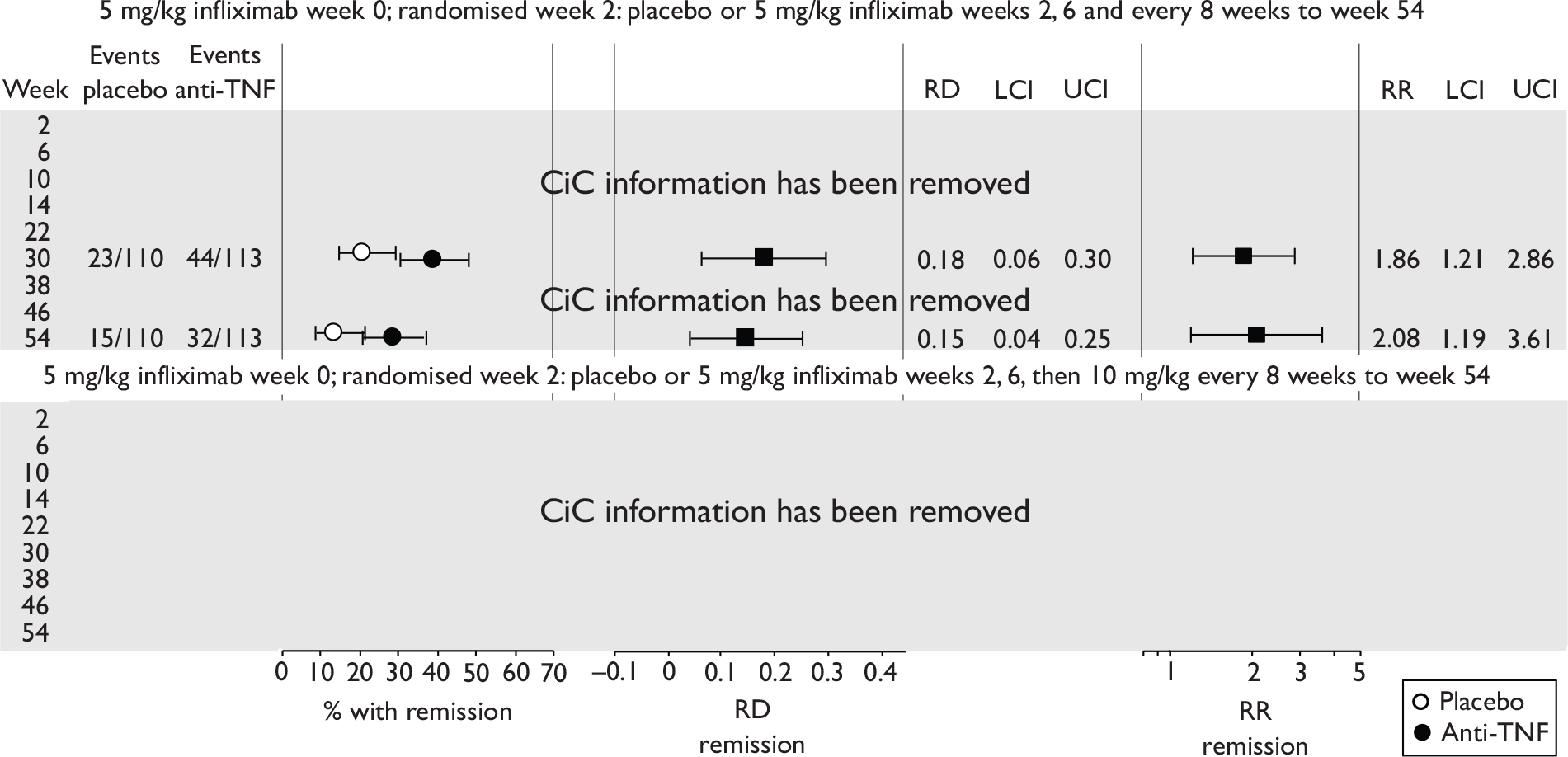
Point estimates were associated with appreciable uncertainty (CiC information has been removed). From week 10, remission rates diminished in all groups and risk difference (infliximab–placebo) diminished or remained fairly constant; risk differences and risk ratios (infliximab/placebo) reached statistical significance at all visit times from week 10 onwards. It is evident that loss of remission was continuous after weeks 6–10 of follow-up and that the advantage of intervention over placebo was mostly gained by about weeks 6–10, the phase of the study during which dose frequency was greatest. Thereafter decline of response was about the same for both placebo and intervention groups despite continued infliximab every 8 weeks in the treatment arms; for example, for the 5 mg/kg arm risk differences (infliximab–placebo) remained similar after week 14 as follows: at weeks 10, 14, 22, 30, 38, 46 and 54 risk differences were 0.15, 0.21, 0.20, 0.18, 0.15, 0.15 and 0.15 respectively.
D, other outcomes
The primary outcome in ACCENT I2,3 was identified as time to loss of response. (Note: a protocol amendment added the proportion of responder patients in remission at week 30 as a coprimary outcome, which has been reported above.) Loss of response was defined as a CDAI of ≥ 175, a CDAI increased by ≥ 35% and a CDAI increased by ≥ 70 points relative to the qualifying value for a response on at least two consecutive assessments, or requirement for change in medication or requirement for surgery. Assessments were scheduled at weeks 0, 2, 6, 10, 14 and then every 8 weeks to week 54. With this definition of loss of response, it is possible for an individual responder to no longer qualify as achieving a response 70 status, but counter-intuitively nevertheless to not have lost response. (For example, an individual with a CDAI of 221 at enrolment would qualify as a responder at week 2 with a CDAI score reduced by 71 points to 150. If this patient’s CDAI subsequently rose to 170 he or she would no longer be in a response 70 but would nevertheless not have lost response because the increase in score from week 2 was < 70 points, < 35% of week 2 score and below a score of 175.) For this primary outcome, patients in the active intervention arms had significantly longer time to loss of response than patients given placebo (p = 0.0002, log rank test). The median times to loss of response are summarised in Table 18.
| Dose regimen (n) | Median time (weeks) to loss of response | IQR (weeks) |
|---|---|---|
| Placebo (110) | 19 | 10 to 45 |
| 5 mg/kg group (113) | 38 | 15 to > 54 |
| 10 mg/kg group (112) | > 54 | 21 to > 54 |
| 5 mg/kg and 10 mg/kg groups combined (225) | 46 | 17 to > 54 |
Published effectiveness results for responders included median CDAI scores and median IBDQ scores. These are summarised in Table 19. For missing values of CDAI and IBDQ, the nearest observation was carried forward. CDAI scores and IBDQ scores diminished and increased respectively to a greater extent in the infliximab groups than in the placebo group. The IQRs for median values during follow-up were not reported.
| Week | CDAI: mediana | IBDQ: mediana | ||||||
|---|---|---|---|---|---|---|---|---|
| Placebo (n = 110) | 5 mg/kg (n = 113) | 10 mg/kg (n = 112) | p | Placebo (n = 110) | 5 mg/kg (n = 113) | 10 mg/kg (n = 112) | p | |
| 0 | 290 | 305 | 305 | NSb,c | 129 | 128 | 130 | NSb,c |
| 2 | 157 | 155 | 152 | 0.01b 0.04c | 173 | 169 | 173 | NR |
| 6 | 159 | 138 | 140 | < 0.0001b < 0.002c | 165 | 174 | 161 | NR |
| 10 | 165 | 131 | 127 | < 0.0001b,c | 160 | 170 | 169 | NSb,c |
| 14 | 197 | 145 | 125 | < 0.0001b,c | 155 | 167 | 172 | 0.05b 0.0076c |
| 22 | 217 | 163 | 135 | < 0.0001b,c | 142 | 164 | 169 | 0.013b < 0.0001 c |
| 30 | 225 | 172 | 150 | < 0.0001b,c | 144 | 162 | 167 | 0.015b 0.001c |
| 38 | 238 | 214 | 140 | < 0.0001b,c | 137 | 151 | 170 | 0.015b < 0.0001 c |
| 46 | 235 | 200 | 142 | < 0.0001b,c | 135 | 144 | 169 | 0.06b < 0.0001c |
| 54 | 238 | 192 | 152 | < 0.0001b,c | 136 | 150 | 167 | 0.015b < 0.0001 c |
The manufacturer’s submission provided information about QoL measures (SF-36). The SF-36 scores were reported separately for mental and physical components for weeks 30 and 54 of the trial, and mean improvement from baseline was reported. SDs of values were provided. The results are summarised in Table 20. Change from baseline for SF-36 physical component reached statistical significance in favour of infliximab at both weeks 30 and 54.
| SF-36 component | Group | SF-36 score | Mean improvement from baseline | |||
|---|---|---|---|---|---|---|
| Baseline | Week 30 | Week 54 | Week 30 | Week 54 | ||
| Physical Component | Infliximab | 33.0 ± 8.5 | 40.4 ± 11.3 | 39.2 ± 11.9 | 7.3 ± 10.3 | 6.1 ± 10.8 |
| Placebo | 33.9 ± 8.8 | 37.2 ± 11.3 | 36.5 ± 11.0 | 3.1 ± 9.5 | 2.5 ± 9.0 | |
| p = 0.002 | p = 0.014 | |||||
| Mental Component | Infliximab | 38.8 ± 11.3 | 43.2 ± 11.4 | 43.9 ± 12.2 | 4.6 ± 12.7 | 5.1 ± 12.8 |
| Placebo | 39.8 ± 11.3 | 42.8 ± 12.0 | 42.1 ± 12.0 | 2.9 ± 11.2 | 2.0 ± 10.9 | |
| p = 0.348 | p = 0.072 | |||||
Median daily steroid dose was reduced by week 14 in all groups and then remained constant. The reduction in the infliximab groups was greater than that for the placebo group. The odds ratio for discontinuation of steroid use (infliximab/placebo) at week 54 was 4.2 (95% CI 1.5 to 11.5).
E, other considerations – subgroup analysis of remission rate in severe CD patients
The manufacturer’s submission for infliximab provided CiC information about the proportion of responder patients who initially had severe disease (defined as a baseline CDAI score > 300) and who achieved remission status during follow-up. Results presented referred to patients classified as having severe disease who were randomised to the 5 mg/kg infliximab group [n = 63/113 (56%)] and placebo group [n = 48/110 (44%)]. No information was provided regarding patients with severe disease among non-responders. The remission rates in placebo and 5 mg/kg infliximab arms and the risk difference for this subgroup of patients are shown in Figure 14. Remission rates were slightly poorer in this more severe CDAI group than for all responders, but a similar pattern was shown during follow-up, in that most of the advantage from the intervention was achieved with the first three doses (early phase). Thereafter, remission decayed at approximately similar rates in the two arms even though patients in the intervention arm received further doses of infliximab and risk differences decreased from week 14 onwards.
ACCENT I2,3 (responders): quality assessment (based on published report of Hanauer et al.3)
Randomisation, allocation concealment and blinding were adequate. Baseline characteristics were only reported for all patients (i.e. for all responders and for all non-responders). It was therefore not possible to judge if baseline characteristics were evenly balanced between the three arms of responders that were analysed for effectiveness outcomes. Similarly the number of patients who withdrew was reported for all enrolled patients and it was not possible to determine how many responders discontinued their randomised treatment. Where necessary, the nearest or last observation was carried forward for continuous outcomes but the number of missing data was not reported. A power calculation was conducted and based on the primary outcome of loss of response. The definition of loss of response was complex and did not correspond to a failure to maintain a response 70 status, and its clinical meaning was difficult to gauge.
ACCENT I: results for all patients (Rutgeerts et al.2) (infliximab)
The results for all 573 patients who received an induction dose in ACCENT I2,3 were presented by Rutgeerts et al. 2 in a paper published 2 years after that, describing results for responders only. Separate results for non-responders have not been published. The 573 patients were 335 responders and 238 non-responders (defined according to whether a 70-point reduction in CDAI score was attained by week 2 after the induction infusion).
Randomisation at week 2 resulted in allocation of 188 patients to the placebo group, 192 to the 5 mg/kg group and 193 to the 10 mg/kg group.
The authors stated ‘the primary objective of the analysis was to examine the difference in efficacy between episodic and scheduled treatment strategies with infliximab under conditions that simulate clinical practice’. For this purpose the patients in the original placebo group were designated as receiving ‘episodic strategy’, and those in the infliximab groups as receiving a ‘5 mg/kg scheduled strategy’ and a ’10 mg/kg scheduled strategy’ respectively. From week 14 onwards patients who had shown a response to infliximab therapy at any time but then worsened were eligible to cross over to ‘active episodic treatment as needed with infliximab 5, 10, and 15 mg/kg for patients originally assigned to episodic, 5 mg/kg scheduled, and 10 mg/kg scheduled treatment strategies respectively’. This description is confusing as it clearly states that active episodic treatment is given in both episodic and scheduled strategies, which renders a comparison of episodic and scheduled strategies problematic. The publication designates the start of episodic treatment to be week 14 (see Appendix 9 for patient flow through the trial).
Of the 573 patients (with baseline CDAI 220–400), 58.5% (335) achieved response 70 2 weeks after a single induction infusion of 5 mg/kg infliximab. These patients were designated ‘responders’. It is unclear if the three trial arms of randomised responders were well balanced at baseline. Of responders, (CiC information has been removed)% were in remission (CDAI < 150) at week 2. This represented (CiC information has been removed)% of the original 573 patients. The proportion of responders with remission had declined by week 30 to 23% (95% CI 14% to 29%) for those who only received placebo after induction and to 39% (95% CI 30.% to 48%) for those who received four infusions of 5 mg/kg infliximab (at weeks 2, 6, 14 and 22) and to 42% (95% CI 36% to 55%) for those who received four infusions consisting of 5 mg/kg at weeks 2 and 6 and 10 mg/kg at weeks 14 and 22. Risk differences (infliximab–placebo) and risk ratios (infliximab/placebo) for remission at week 30 reached statistical significance in favour of infliximab for both infliximab groups. By week 54 the percentage of patients in remission had diminished further in all three groups. Most of the advantage of intervention relative to placebo was achieved by weeks 10–14; thereafter risk differences remained fairly stable. A similar pattern of results was observed for response 70. Published information regarding maintenance of remission at the patient level (as distinct from group level) was meagre. Between weeks 14 and 54, 11% of placebo patients retained remission at all six study visits; the corresponding values were 25% and 33% respectively for 5 mg/kg and 10 mg/kg infliximab groups. Somewhat lower values of (CiC information has been removed)%, (CiC information has been removed)% and (CiC information has been removed)% respectively were quoted in the Industry Trial Report. Results favouring infliximab over placebo were reported for several other outcomes including median CDAI scores and median IBDQ scores. These measures required last or nearest observation carried forward in order to allow for missing data.
The treatment regimens received before week 14 in each of the randomised groups were as follows:
-
placebo/‘episodic group’: 5 mg/kg infliximab week 0, placebo weeks 2 and 6
-
5 mg/kg group ‘scheduled strategy’: 5 mg/kg infliximab weeks 0, 2 and 6
-
10 mg/kg group ‘scheduled strategy’: 5 mg/kg infliximab weeks 0, 2 and 6.
Treatment to week 14 was therefore similar for the two infliximab ‘scheduled strategy’ groups and was determined according to randomisation. From week 14, crossover to an increase in infliximab dosage was allowed in all three trial arms for patients whose CD worsened. The criteria for worsening were ‘an increase CDAI of ≥ 70 points from the qualifying score with a total score of at least 175, an increase in CDAI of 35% or more from baseline value, or the introduction of new treatment for active Crohn’s disease’. From week 14 onwards it was possible for patients in different arms to be receiving identical infliximab treatment; for example, a placebo patient might cross over at week 14 to receive 5 mg/kg and this corresponds to treatment received by a 5 mg/kg ‘scheduled strategy’ patient who did not cross over. This complicates the interpretation of any comparisons between groups.
A, response 70
No primary outcome was identified. Analyses were according to randomised group irrespective of crossover after week 14 to different treatment regimen, and comparisons were drawn between the ‘episodic group’ and the two ‘scheduled strategy’ groups. The results for response 70 for all patients in ACCENT I are summarised in Figure 15. By week 14, statistically significant differences in CD status were evident between placebo group and intervention groups (p-values for risk differences and risk ratios are shown in Table 21). Risk differences and risk ratios for comparison between ‘episodic’ and ‘scheduled’ strategies after week 14 were in favour of ‘scheduled strategies’ but failed to reach statistical significance at most time points. Interpretation of these differences is problematic.
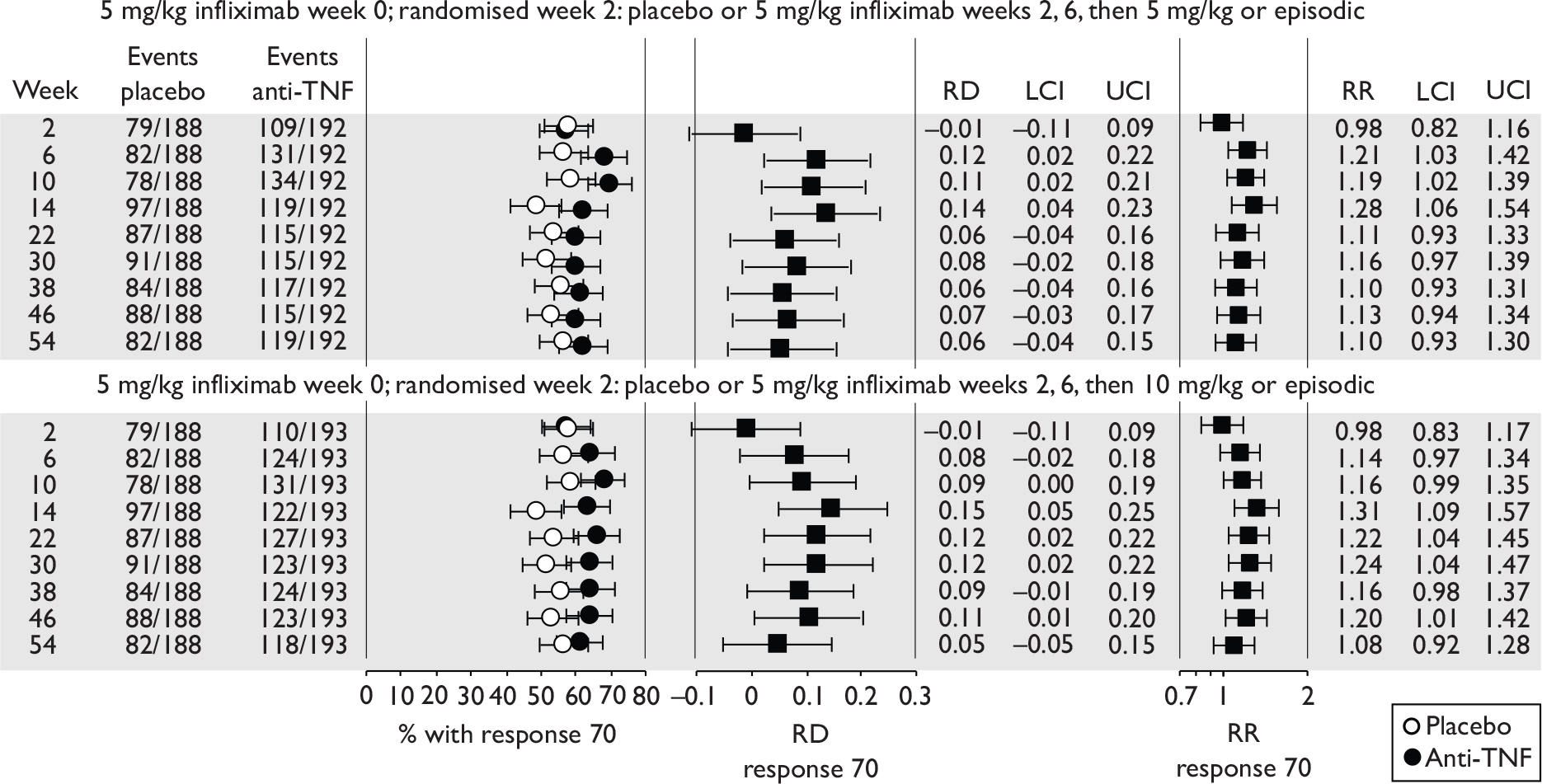
| Risk difference (placebo–active intervention) | Risk ratio (placebo/active intervention) | |||
|---|---|---|---|---|
| vs 5 mg/kg group | vs 10 mg/kg group | vs 5 mg/kg group | vs 10 mg/kg group | |
| p | 0.00725 | 0.00326 | 0.00865 | 0.00418 |
B, response 100
This outcome was not reported.
C, remission rates
Figure 16 summarises the published results for rates of remission at clinic visits to end of follow-up (week 54). Week 14 remission rates were greater in the two ‘scheduled treatment’ arms (37.5% in the 5 mg/kg group and 43% in the 10 mg/kg group) than in the ‘episodic’ group (25.5%). p-values for week 14 comparisons between placebo and intervention groups are shown in Table 22.
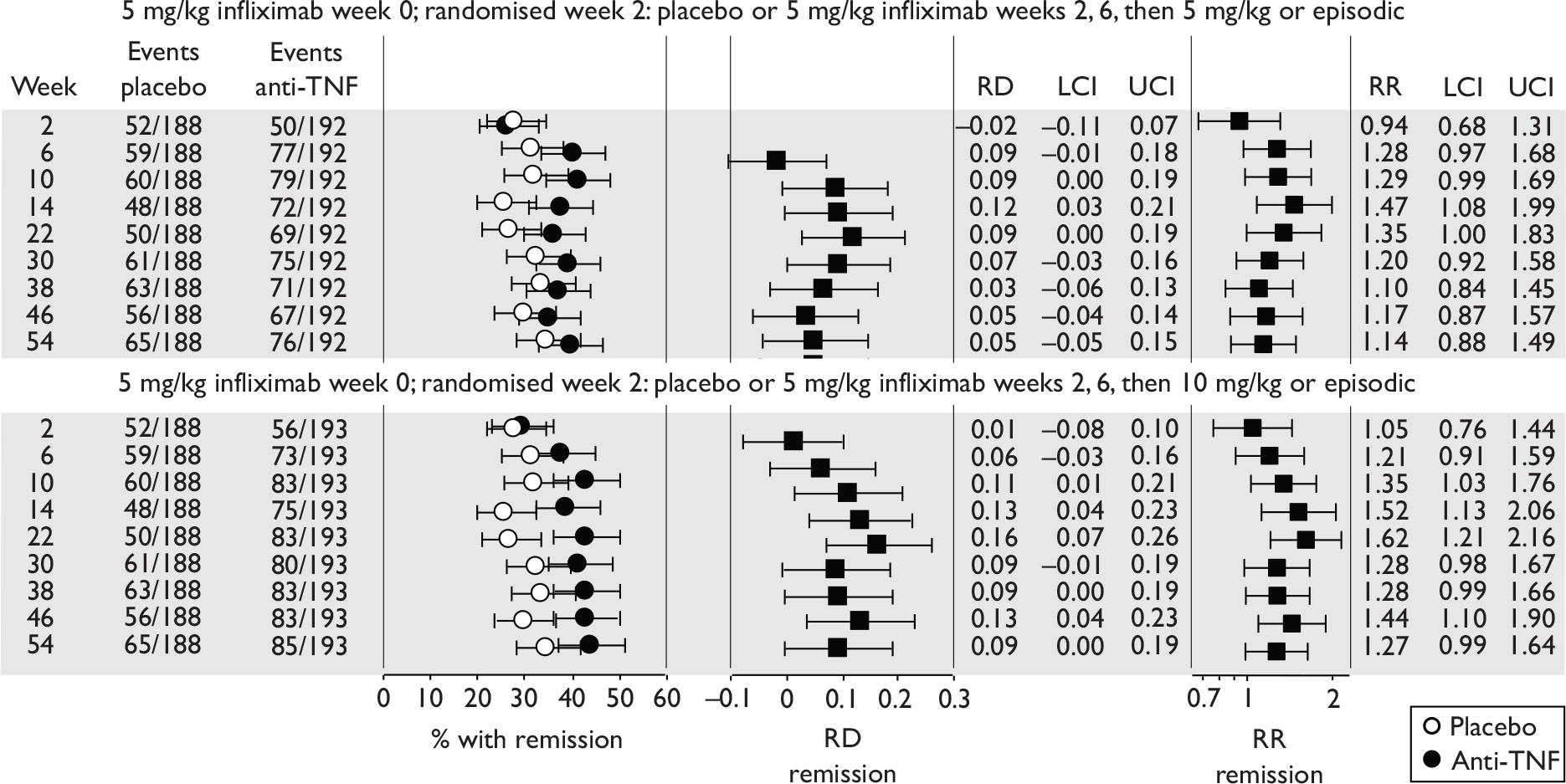
| Risk difference (placebo–active intervention) | Risk ratio (placebo/active intervention) | |||
|---|---|---|---|---|
| vs 5 mg/kg group | vs 10 mg/kg group | vs 5 mg/kg group | vs 10 mg/kg group | |
| p | 0.0113 | 0.0049 | 0.0135 | 0.0063 |
Treatment regimens up to week 14 were strictly pre-specified and designed to examine effectiveness for maintenance of the induced response. After week 14 treatment regimens became variable (termed ‘episodic’ by the authors). It is clear that by week 14 the CD status of patients in the placebo/‘episodic’ arm had departed from that of patients in the two ‘scheduled strategy’ arms; this means that at baseline (week 14) for the comparison of ‘episodic’ with ‘scheduled strategies’, the groups were imbalanced. Comparisons between ‘episodic’ and ‘scheduled’ strategies after week 14 are not randomised comparisons. For a randomised comparison of the two strategies patients should have been rerandomised at week 14. Such rerandomisation was precisely the study design adopted by Menter et al. 70 when comparing continuous with intermittent treatment strategies with infliximab in psoriasis.
Risk differences and risk ratios for comparison between ‘episodic’ and ‘scheduled’ strategies after week 14 were in favour of ‘scheduled strategies’, but failed to reach statistical significance at nearly all time points. Interpretation of these differences is problematic because, as described above, the comparisons are not between properly randomised groups and because patients in all groups were allowed the option of ‘episodic’ treatment.
D, other outcomes
Median CDAI score and the proportion of patients with IBDQ score > 170 were reported and are summarised in Table 23 and presented graphically in Appendix 10. By week 14, statistically significant differences in CDAI median scores were evident between the placebo group and the intervention groups. Differences were less pronounced after week 14, especially for the placebo versus 5 mg/kg comparison. The percentage of patients with IBDQ score > 170 did not differ significantly between placebo and 5 mg/kg groups, but after week 14 favoured the 10 mg/kg group relative to placebo.
| Week | CDAI: mediana | % patients with IBDQ score > 170a | ||||||
|---|---|---|---|---|---|---|---|---|
| Placebo (n = 188) |
5 mg/kg (n = 193) |
10 mg/kg (n = 192) |
p | Placebo (n = 188) |
5 mg/kg (n = 193) |
10 mg/kg (n = 192) |
p | |
| 0 | 292 | 303 | 297 | NSb,c | 4.8 | 5.2 | 8.3 | NSb,c |
| 2 | 197.5 | 205 | 195 | NSb,c | 35.6 | 32.3 | 35.8 | NSb,c |
| 6 | 205 | 180 | 180 | NSb,c | 33.5 | 41.7 | 38.3 | NSb,c |
| 10 | 187.5 | 170 | 167.5 | < 0.05b,c | 35.1 | 41.7 | 39.9 | NSb,c |
| 14 | 225 | 185 | 182.5 | < 0.05b,c | 29.8 | 38.0 | 40.9 | NSb< 0.05c |
| 22 | 212.5 | 185 | 167.5 | < 0.05b,c | 29.3 | 37.0 | 44.0 | NSb < 0.05c |
| 30 | 212.5 | 180 | 177.5 | NSb < 0.05c | 33.5 | 39.6 | 44.0 | NSb < 0.05c |
| 38 | 200 | 187.5 | 170 | NSb < 0.05c | 35.1 | 34.9 | 47.7 | NSb < 0.05c |
| 46 | 205 | 190 | 175 | NSb < 0.05c | 33.5 | 35.4 | 48.7 | NSb < 0.05c |
| 54 | 205 | 185 | 170 | NSb < 0.05c | 35.1 | 37.5 | 46.1 | NSb < 0.05c |
The manufacturer’s submission provided information regarding CD-related hospitalisation rates and rates for intra-abdominal surgery. These rates and the relative risk for the 5 mg/kg ‘scheduled maintenance’ group relative to the ‘episodic’ are summarised in Table 24. The results for mucosal healing observed for a small subgroup of patients (n = 58) at European study centres who underwent endoscopy examination are also tabulated. The interpretation of the comparisons is problematical for the reasons already described, in particular after week 14. The extent to which avoidance of hospitalisation and abdominal surgery might depend on the administration of active intervention is not measurable because no true control (placebo) group existed after that time.
| Endoscopy | Hospitalisationsa | Abdominal surgery | ||||
|---|---|---|---|---|---|---|
| Mucosal healing at week 54 | p b | n/N (%) | Relative risk (95% CI) | n/N | Relative risk (95% CI) | |
| Placebo | 4/22 | NA | 71/188 (38%) | NA | 14/188 | NA |
| 5 mg/kg group | 8/19 | 0.093 | 5/193 | 0.348c (0.128 to 0.947) | ||
| 10 mg/kg group | 8/17 | 0.053 | 6/192 | |||
| Combined 5- and 10 mg/kg groups | 16/36 | 0.041 | 86/305 (23%) | 0.591b,c (0.455 to 0.768) | 11/385 | 0.373 (0.173 to 0.806) |
Quality assessment of ACCENT I2,3 (all patients): (based on published report of Rutgeerts et al.2)
Randomisation, allocation concealment and blinding were adequate. Baseline characteristics at week 0 were well balanced. Where necessary, the nearest or last observation was carried forward for continuous outcomes but the number of missing data was not reported. No power calculation was conducted for the analysis of all patients. The number of patients who withdrew was reported except for patients who crossed over to a 15 mg/kg dose regimen from a 10 mg/kg regimen. The proportion of patients who withdrew before the end of the trial was substantial.
It must be questionable whether the ‘episodic’ (placebo) arm did ‘simulate clinical practice’ as stated to be an objective of the study. Patients in this arm of the study received one dose of 5 mg/kg infliximab at week 0, followed by an interim period of > 3 months with no active infliximab therapy before the ‘episodic’ use of infliximab according to worsening disease (for patients ‘who had responded at any time to infliximab therapy’). There is little evidence to support the idea that this resembles clinical practice. The scheduled strategy is difficult to define as it did not follow a prescribed programme of treatment as might be anticipated by the term ‘scheduled strategy’, but encompassed ‘episodic’ treatment in the same manner as the ‘episodic’ arm.
Because of the large numbers of patients who withdrew from treatment and crossed over to dose escalations, the actual treatments received in the three different trial arms are difficult to define. Figure 17 summarises the progression of patients through the trial with respect to withdrawal from treatment and crossover to increased dose of infliximab.
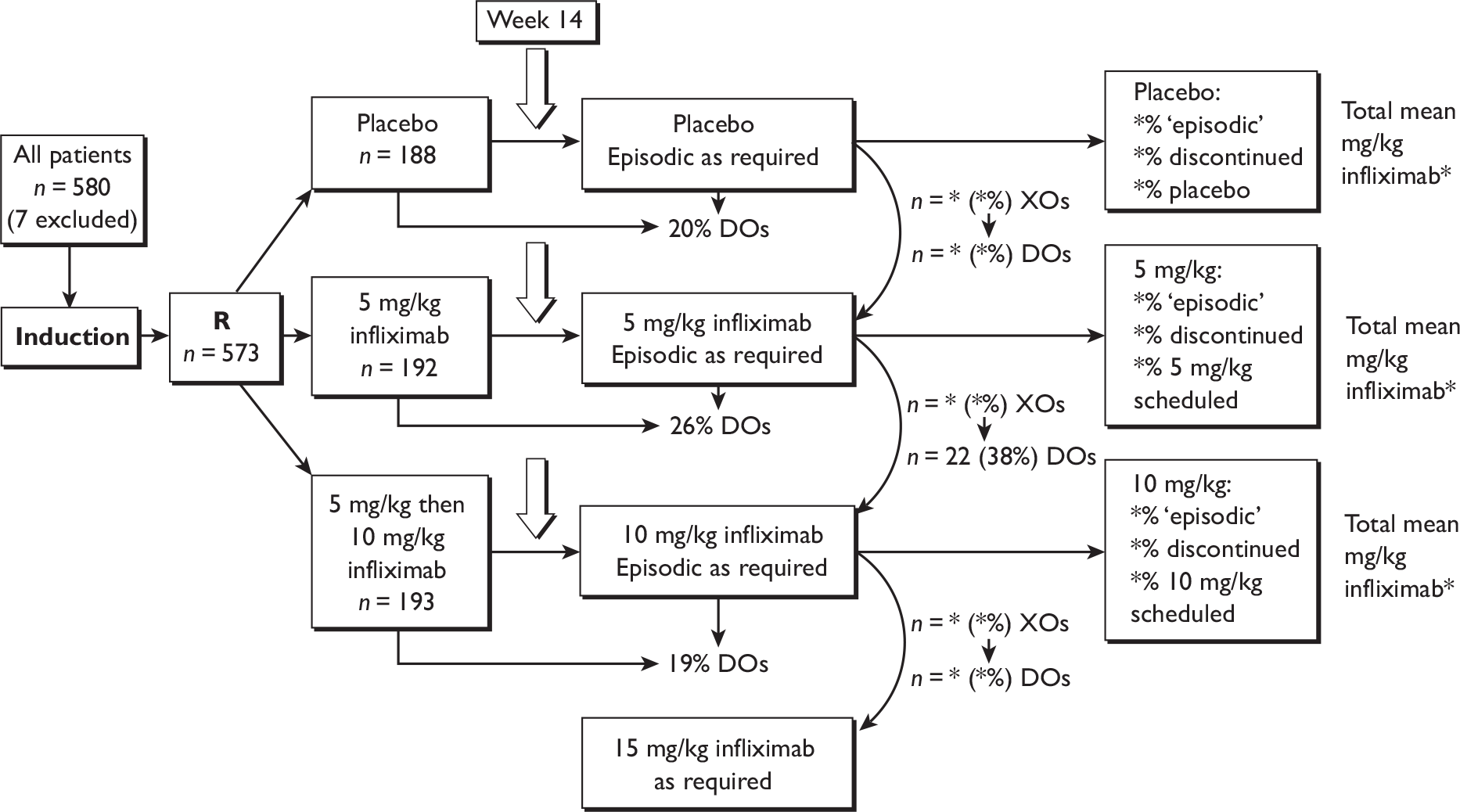
Over a period of 1 year, about a quarter of patients withdrew from treatment, and of those allocated active intervention at randomisation only about half completed the trial receiving the treatment regimen to which they had been allocated at randomisation.
The authors’ stated primary objective ‘…was to examine the difference in efficacy between episodic and scheduled treatment strategies with infliximab’. 2 They concluded that the scheduled treatment strategy was superior to episodic treatment. Unfortunately, the comparisons were compromised by strong biases introduced as a result of the study design. These biases are explained below:
-
Crossover to increased infliximab was allowed for patients ‘who had responded at any time to infliximab therapy’ and subsequently worsened. In the placebo group (‘episodic strategy’), 78 of 188 patients (41%) were classified at week 2 as non-responders and received no further infliximab to week 14; these patients were unlikely to become responsive and therefore to qualify for crossover to active intervention. In contrast to this group the week 2 non-responders in the ‘scheduled strategy’ arms received additional doses of infliximab (5 mg/kg) at both weeks 2 and 6, boosting their opportunity to ‘respond at any time’ to infliximab. The greater opportunity to respond at any time in the ‘scheduled strategy’ arms represents a strong bias in their favour in any subsequent comparison with the episodic arm. Relative to the scheduled strategy this resulted in a substantial proportion of patients in the episodic arm being denied access to active therapy. This is reflected in the very large difference between arms in their exposure to infliximab stated to be 3 and 5 times greater in the two scheduled strategy arms than in the episodic arm.
-
Episodic treatment was introduced at week 14 of the trial, but by this time the CD status of patients in the placebo ‘episodic’ arm was significantly inferior to that in the scheduled strategy arms in terms of several efficacy measures. This advantage for the scheduled strategy arms is reflected in increases not seen in the placebo group from week 2 in the response 70 rates and at weeks 6 and 10 in the remission rates. The result is a bias in favour of scheduled strategy for any comparison between strategies at times after week 14. Essentially, the compared arms were unbalanced at the start of the compared strategies (week 14).
Two infusions of 5 mg/kg infliximab at weeks 2 and 6 after a single induction infusion of 5 mg/kg were better than placebo infusions at generating remission and response 70. At week 14, risk differences (infliximab–placebo) and risk ratios (infliximab/placebo) were in favour of infliximab and reached statistical significance (p < 0.02 for remission, p < 0.01 for response 70).
At week 14, ‘episodic’ treatment was introduced and subsequent comparisons were made between the original placebo arm (designated ‘episodic treatment strategy’) and original infliximab arms (termed ‘scheduled treatment strategies’). Because of bias strongly in favour of scheduled strategy groups, the post 14-week comparisons were not valid estimates of the relative effectiveness of strategies. Biases identified arose from: (a) reduced opportunity for crossover to active therapy for patients in the episodic group compared with the scheduled groups; and (b) gross imbalance in disease status at the start of the strategies (week 14).
Difficulties in interpreting post 14-week comparisons between groups were compounded by the very high rate of withdrawal from treatment and the use of ‘episodic’ treatment in all three arms of the trial so that the distinction between episodic and scheduled strategies was obscured except for the fact that the original infliximab groups were allowed larger dosages of active intervention.
CLASSIC II66 (adalimumab)
The CLASSIC II66 trial was an extension of the previously conducted adalimumab induction trial, CLASSIC I,63 which had enrolled 299 patients. To be eligible for CLASSIC II, patients were required to be in remission (CDAI < 150) at week 4 of CLASSIC I and also 4 weeks later (equivalent to week 8 of CLASSIC I and designated week 4 of CLASSIC II). These patients may have received two subcutaneous injections 2 weeks apart of various doses of adalimumab (40 mg then 20 mg, 80 mg then 40 mg, or 160 mg then 80 mg) or two injections of placebo. Fifty-five eligible patients entered CLASSIC II, this means about 12 patients did not retain remission from weeks 4–8 of CLASSIC I or declined to participate. The 55 patients were randomised at week 4 of CLASSIC II to receive placebo (n = 18) or 40 mg of adalimumab every other week (e.o.w.) (n = 19) or 40 mg of adalimumab weekly (n = 18) from weeks 4 to 54. Thus CLASSIC II analysed only strong responders from the CLASSIC I trial.
For the purposes of the ‘primary efficacy analysis’, patients who had continued non-response defined as ‘a decrease in CDAI ≤ 70 points vs. Week-0 value in CLASSIC I’ were considered treatment failures and became eligible for open-label treatment. This means patients in remission at start of CLASSIC II became treatment failures if they ceased to qualify as response 70 responders relative to their baseline CDAI score in CLASSIC I. In addition, patients who flared during CLASSIC II follow-up were also counted as treatment failures and were eligible for open-label treatment. CD flare was defined as an increase of ≥ 70 points above the week 4 CLASSIC II value (which by definition was < 150) AND a CDAI score > 150 (no longer in remission). Thus a patient in remission at week 4 (CLASSIC II) with a CDAI score of 149 would need to move to a CDAI of at least 219 to be classified as having experienced flare. For this patient a score of 218 would not count as a flare but could count as treatment failure if his or her week 0 CLASSIC I CDAI score had been < 288 (for reference the mean baseline CDAI score at week 0 for 299 CLASSIC I patients was 298).
A, response 70 and B, response 100
Response 100 and response 70 rates throughout follow-up were among the secondary outcome measures of efficacy. Results reported for responses 100 and 70 are summarised in Figure 18. The placebo rates were high for these less rigorous measures of effectiveness, and the risk differences (adalimumab–placebo) and risk ratios (adalimumab/placebo) failed to reach statistical significance at most time points.
FIGURE 18.
Response 100 (upper panel) and response 70 (lower panel) rates in CLASSIC II. 66 LCI, lower confidence interval; RD, risk difference; RR, risk ratio; UCI, upper confidence interval.
C, remission
The primary outcome was the proportion of patients in remission at week 56 in each arm of the randomised cohort. Remission throughout follow-up was among the secondary outcome measures of efficacy. For the primary outcome, 10 patients (18%) withdrew before week 56 (five from placebo and five from adalimumab). These were counted as remission failures for the primary analysis. Remission rates at week 56 are summarised in Table 25. Remission rates during the trial are summarised in Figure 19.
| Dose regimen (n ) | Number in remission (%; 95% CI) | p a |
|---|---|---|
| Placebo (18) | 8 (44%; 25 to 66) | NA |
| 40-mg adalimumab e.o.w. (19) | 15 (79%; 57 to 91) | < 0.05 |
| 40-mg adalimumab weekly (18) | 15 (83%; 61 to 94) | < 0.05 |
| Open-label [(CiC information has been removed)b] | (CiC information has been removed)c [(CiC information has been removed)%] |
FIGURE 19.
Remission rates in CLASSIC II. 66 LCI, lower confidence interval; RD, risk difference; RR, risk ratio; UCI, upper confidence interval.
Point estimates of remission rate during the trial were associated with considerable uncertainty, reflecting the small number of patients in the trial. The fact that rates rose and fell during follow-up indicated the values reported referred to point prevalence. Nearly half of patients in the placebo group were in remission at week 56 despite not receiving active intervention from 2 weeks prior to randomisation onwards. Risk differences (intervention–placebo) and risk ratios (intervention/placebo) were in favour of intervention at all follow-up times and reached statistical significance at several time points.
D, other outcomes
The results published for continuous measures are summarised in Table 26. These measures involved last observation carried forward to allow for missing values. The amount of missing values was not published but was available (CiC) in the unpublished Industry Trial Report. For week 56 changes in favour of adalimumab relative to placebo were reported for mean IBDQ and CDAI scores.
| Placebo (n = 18) | Adalimumab 40 mg e.o.w. (n = 19) |
Adalimumab 40 mg weekly (n = 18) |
p | |
|---|---|---|---|---|
| Mean IBDQ scorea | ||||
| Week 0b | 187.5 | 181 | 191.5 | |
| Week 4 | 188.5 | 187 | 191 | |
| Week 8 | 178 | 181 | 187 | |
| Week 12 | 170.5 | 182.5 | 189 | |
| Week 16 | 172.5 | 181 | 182 | |
| Week 20 | 170.5 | 177 | 186.5 | |
| Week 24 | 167.6 | 176.3 | 192.2 | < 0.005c,d |
| Week 32 | 166.5 | 182 | 192 | < 0.05c < 0.005d |
| Week 40 | 167 | 179 | 188 | < 0.005c,d |
| Week 48 | 163.5 | 178 | 183.5 | |
| Week 56 | 162.4 | 178.4 | 185.6 | |
| CDAI: mean change (95% CI) from baseline in CLASSIC Ie | ||||
| Week 56 | –119.6 (–74 to –65.1) | –158 (–202 to –99.8) | –197.7 (–248 to –147) | < 0.005c,d |
| CRP concentration mg/dl: median (range) [levels of CRP < 0.88 mg/dl are considered normal] | ||||
| 24 | 0.5 (0 to 1.2) | 0.4 (0 to 1.9) | 0.1 (0 to 1.6) | NR |
| 56 | 0.4 (0 to 0.9) | 0.3 (0 to 2.8) | 0.3 (0 to 1.2) | NR |
At the start of CLASSIC II,66 49% of patients were receiving systemic steroids or budesonide; seven of the placebo group, seven of the e.o.w. adalimumab group, and eight of the weekly adalimumab group. Using the last observation carried forward it was reported that by week 56 the number who had discontinued steroids was four in both the placebo and e.o.w. adalimumab groups, and seven in the weekly adalimumab group.
E, other considerations – open-label study
Most patients from CLASSIC I63 who did not qualify for CLASSIC II66 participated in an open-label study in parallel with CLASSIC II. The results reported were not randomised comparisons and are outwith the inclusion criteria for this report.
Quality assessment (based on published report)
Randomisation, allocation concealment and blinding were adequate. Baseline characteristics at week 0 were well balanced. The study was powered for the primary outcome (remission at week 4) of CLASSIC I, and no further power calculation was conducted for CLASSIC II. The number of patients who withdrew was reported; 5 of 18 placebo patients withdrew and 5 of 37 patients given adalimumab withdrew. There were 32 patients (58%) who completed to 56 weeks of double-blind follow-up. The last observation was carried forward as necessary for continuous outcomes, but the number of missing data was not reported.
The trial population (n = 55) was recruited from responders in the previous CLASSIC I63 adalimumab induction trial (n = 299). Only responders with a strong response (remission for at least a month) were selected; they had received various induction dose regimens.
Maintenance injections of 40 mg of adalimumab administered weekly or e.o.w. generated a statistically significant greater proportion of patients in remission at week 56 than did placebo (frequency of administration not published). About half of the placebo group and 81% of those who received infliximab were in remission at week 56. Point estimates of response rates were associated with considerable uncertainty due to the small size of the trial. There were no statistically significant differences in effectiveness between e.o.w. and weekly adalimumab regimens.
CHARM67 (adalimumab)
This was a free-standing maintenance trial (i.e. newly started). 67 There were 854 enrolled patients (CDAI range 220–400), of whom 130 (15.2%) had fistulas at screening and baseline. An induction regimen consisting of an 80-mg injection of adalimumab at week 0 and a 40-mg injection 2 weeks later was followed by randomisation of 778 patients at week 4 to one of three arms as follows: placebo to week 56 (n = 261), 40 mg adalimumab e.o.w. to week 56 (n = 260) and 40 mg adalimumab weekly to week 56 (n = 257). There were 76 (8.9%) withdrawals prior to randomisation. Assessment visits were planned for weeks 0, 2, 4, 6, 8, 12, 16, 20, 26, 32, 40, 48, 56 and 60.
At week 4 patients were classified as responders or non-responders. Responders had to have a reduction of ≥ 70 CDAI points relative to baseline. Of the 854 patients given the induction regimen, 499 (58%) were categorised as responders and were the focus of the published effectiveness results. This population was different to that followed up in the other adalimumab maintenance trial, CLASSIC II,66 in that the latter were on average better responders, having achieved remission from induction. The numbers of responders randomised to the three trial arms of CHARM were 170 to placebo, 172 to adalimumab e.o.w. and 157 to adalimumab weekly.
The coprimary outcome measures were designated: the percentage of week 4 responders who achieved remission at weeks 26 and 56. Pre-specified secondary outcomes included (1) percentage achieving response 70 and response 100 at weeks 26 and 56; (2) change in IBDQ score from baseline at weeks 26 and 56; (3) percentage achieving clinical remission at weeks 26 and 56 who were able to discontinue corticosteroid use; (4) percentage achieving clinical remission at weeks 26 and 56 who were able to discontinue steroids for ≥ 90 days; (5) percentage of patients with fistula remission (closure of all fistulas that were draining at screening and baseline visits); and (6) median time in clinical remission among randomised responders achieving remission. Post hoc analyses examined subgroup responses and sustainability of response.
At or after week 12, patients with disease flare (an increase of ≥ 70 CDAI points from the score at week 4 and a CDAI score > 220) or sustained non-response (CDAI score not reduced by ≥ 70 points from week 0) were eligible to cross over to 40-mg adalimumab e.o.w. which could be escalated to 40-mg weekly for patients with continued non-response or recurrent flare. For the primary effectiveness outcome (responders), any patients who crossed over were counted as remission failures.
A, response 70 and B, response 100
The published response 70 and response 100 rates at weeks 26 and 56 are summarised in Table 27. Rates reached statistical significance in favour of adalimumab for both dose regimens at both time points.
| Dose regimen (n) | Number with response 100 (%; 95% CI) | p a | |
|---|---|---|---|
| Week 26 | Week 56 | ||
| Placebo (170) | 45 (26.5%; 20 to 34) | 28 (16.5%; 12 to 23) | NA |
| 40-mg adalimumab e.o.w. (172) | 89 (52%; 44 to59) | 71 (41%; 34 to 49) | < 0.001 |
| 40-mg adalimumab weekly (157) | 82 (52%; 44 to 60) | 75 (48%; 40 to 56) | < 0.001 |
| Number with response 70 (%; 95% CI) | |||
| Week 26 | Week 56 | ||
| Placebo (170) | 48 (28%; 22 to 35) | 30 (18%; 13 to 24) | NA |
| 40-mg adalimumab e.o.w. (172) | 93 (54%; 47 to 61) | 74 (43%; 36 to 50) | < 0.001 |
| 40-mg adalimumab weekly (157) | 88 (56%; 48 to 63) | 77 (49%; 41 to 57) | < 0.001 |
The unpublished Industry Trial Report for CHARM provided (CiC) values for response 70 at time points for all assessment visits. These are summarised in Figure 20. (CiC information has been removed.)
FIGURE 20.
Response 70 rates among responders in CHARM. 67 LCI, lower confidence interval; RD, risk difference; RR, risk ratio; UCI, upper confidence interval.
Similar CiC results were observed for response 100 and are summarised in Figure 21. From week 8 onwards (CiC information has been removed).
FIGURE 21.
Rates of response 100 among responders in CHARM. 67 LCI, lower confidence interval; RD, risk difference; RR, risk ratio; UCI, upper confidence interval.
C, remission
The primary outcome was the proportion of patients in remission at weeks 26 and 56. The results are summarised in Table 28. The difference between adalimumab groups and placebo reached statistical significance in favour of adalimumab for both dose regimens.
| Dose regimen (n) | Number in remission (%; 95% CI) | p a | |
|---|---|---|---|
| Week 26 | Week 56 | ||
| Placebo (170) | 29 (17%; 12 to 23) | 20 (12%; 8 to 13) | NA |
| 40-mg adalimumab e.o.w. (172) | 68 (40%; 32 to 47) | 62 (36%; 29 to 43) | < 0.001 |
| 40-mg adalimumab weekly (157) | 73 (47%; 39 to 54) | 65 (41%; 34 to 49) | < 0.001 |
The secondary outcomes of remission rates for each follow-up visit to week 56 are summarised in Figure 22. Risk differences (adalimumab–placebo) and risk ratios (adalimumab/placebo) reached statistical significance in favour of adalimumab at all time points after week 6. Rates of remission in the adalimumab e.o.w. arm diminished through follow-up. From weeks 12–16 onwards, risk differences remained stable so that most benefit of the intervention appeared to be delivered in the first quarter of the trial. The rates reported were group point prevalence values and do not reflect maintenance of remission at the patient level. The difference in rates between the two adalimumab regimens at week 56 was not significant (risk difference p = 0.32, risk ratio p = 0.32).
FIGURE 22.
Remission rates reported during follow-up in CHARM. 67 At week 56 risk difference and risk ratio for both regimens of adalimumab versus placebo, p < 0.0001. LCI, lower confidence interval; RD, risk difference; RR, risk ratio; UCI, upper confidence interval.
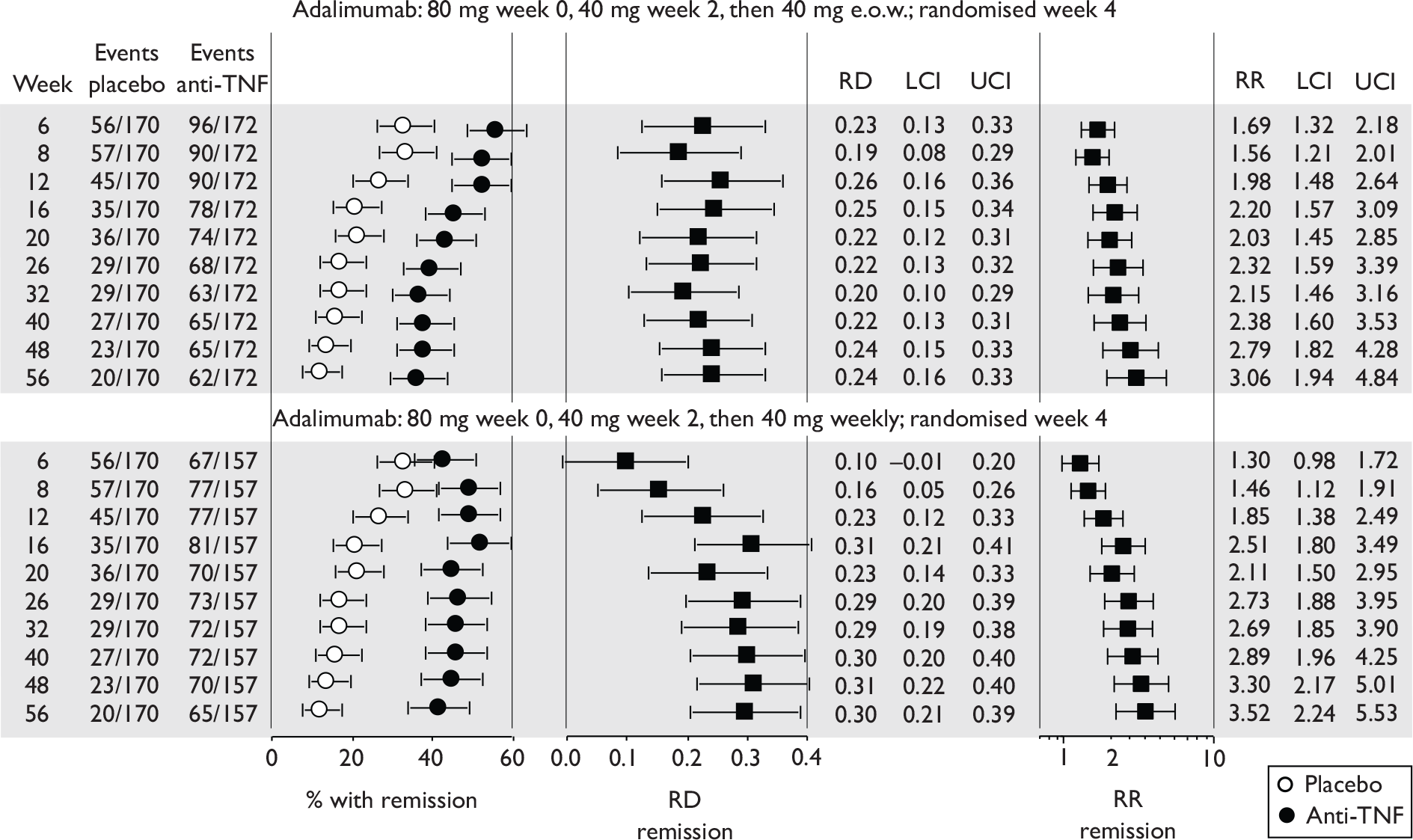
Patient level maintenance of remission was published for weeks 26–56. In the adalimumab arms, 81% of patients in remission at week 26 sustained remission to week 56; this represented 114 patients and 27% of all those randomised to adalimumab. For patients randomised to placebo, 48% of those in remission at week 26 sustained remission to week 56. This represented 14 patients and 5% of all those randomised to placebo. The median time in clinical remission that started at any time was 127 days for the placebo group, 378 days for the adalimumab e.o.w. group and > 392 days for the adalimumab weekly group (p = 0.002 and p < 0.001 vs placebo respectively). Over 56 weeks it was possible for a patient to enter a remission state on several occasions. The publication did not make clear which occasion(s) were used in the analysis or how and if double counting was avoided.
D, other outcomes
Published mean CDAI and IBDQ scores are summarised in Table 29. No variance information was provided. After week 12, CDAI and IBDQ scores for patients who crossed over to increased adalimumab doses were included in the calculation of group mean scores although this was not made explicit. Mean CDAI scores decreased and mean IBDQ scores increased, to a greater degree respectively in the adalimumab groups than the placebo group. Given that a true placebo group did not exist after week 14, the results thereafter are difficult to interpret. The last observation was carried forward; the proportion of patients evaluated at week 56 (CiC information has been removed).
| Week | CDAI:meana | IBDQ mediana | ||||||
|---|---|---|---|---|---|---|---|---|
| Placebo (n = 170) |
40-mg adalimumab e.o.w. (n = 172) | 40-mg adalimumab weekly (n = 157) | p | Placebo (n = 170) |
40-mg adalimumab e.o.w. (n = 172) | 40-mg adalimumab weekly (n = 157) | p | |
| 0 | 318 | 317 | 310 | NR | 125 | 128 | 123 | NR |
| 2 | 215 | 200 | 203 | NR | NR | NR | NR | NR |
| 4 | 170 | 153 | 162 | NR | 166.6 | 174 | 165 | NR |
| 6 | 178 | 150 | 162 | NR | NR | NR | NR | NR |
| 8 | 183 | 147 | 155 | NR | NR | NR | NR | NR |
| 12 | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | NR | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | NR |
| 16 | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | NR | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | NR |
| 20 | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | NR | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | NR |
| 26 | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | NR | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | NR |
| 32 | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | NR | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | NR |
| 40 | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | NR | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | NR |
| 48 | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | NR | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | NR |
| 56 | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | NR | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | NR |
From week 8 the responder patients who were receiving steroids at baseline could begin reducing steroid use (presumably at the physician’s discretion). This involved 66 placebo patients, 58 and 74 patients respectively in the adalimumab e.o.w. and adalimumab weekly groups. The percentage of these patients who were in remission at week 26 and who had discontinued steroids was 3% (2/66) in the placebo group, and 34% (20/58) and 30% (22/74) in the adalimumab e.o.w. and weekly groups respectively. Corresponding percentages at week 56 were 6%, 29% and 23% respectively. The percentage who were in remission at week 26 and who were steroid free for at least 90 days was 3% in the placebo group and 19% and 15% in the adalimumab e.o.w. and weekly groups respectively. Corresponding percentages at week 56 were 5%, 29% and 20% respectively.
Details on hospitalisation rates from the CHARM trial67 were reported in the industry submission, referenced to published abstracts by Wu et al. 71 and by Feagan et al. 72 The latter abstract reports the hospitalisation rates in the placebo arm and the combined adalimumab arms, which were 22.4% and 14.0% respectively. The 56-week actuarial CD-related hospital admission rates for the placebo and for the combined adalimumab arms were 13.9% and 5.9% respectively. A difference in relative risk was apparent at 2 weeks after randomisation, and placebo patients had 4.5 times the risk of hospitalisation at month 3 as adalimumab patients. Wu et al. 71 used a Cox proportional hazard regression model and found that lower CDAI scores were associated with a decreased risk of hospitalisation and CD-related hospitalisation. Simulated 1-year rates indicated that a 70-point reduction on the CDAI throughout the follow-up period reduced all-cause hospitalisation risk by 28.3% and CD-related hospitalisation by 36.5% at year end. Further simulations indicated that remission was associated with a 43.7% decrease in the 1-year risk of all-cause hospitalisation and a 60.3% decrease in CD-related hospitalisation.
E, other considerations – subgroup analyses and crossover issues
Outcomes for patients with draining fistulas are included in the next section.
The manufacturer’s submission to NICE provided weeks 26 and 56 results for placebo and e.o.w. adalimumab group patients who had severe disease at baseline (CDAI > 300). Results for all severe patients and for severe week 4 responders were provided allowing calculation of results for non-responders with severe CD (Table 30). There were 96 severe CD patients in both placebo and e.o.w. adalimumab groups. Rates of remission, response 70 and response 100 are summarised in Figure 23. Remission rates at week 56 in adalimumab and placebo arms were 35% and 10% respectively; higher rates were recorded for the less stringent response 70 and 100 outcomes. These rates were similar to those reported for all week 4 responders (within 5%; listed in Table 30). The rates in the e.o.w. arm for week 4 non-responders with severe CD were about half those for week 4 responders with severe CD.
| Outcome | Week | All week 4 responders | Severe CD responders | All severe CD patients | Severe CD non-responders | |||
|---|---|---|---|---|---|---|---|---|
| Placebo (n = 170) | e.o.w. anti-TNF (n = 157) | Placebo (n = 96) | e.o.w. anti-TNF (n = 96) | Placebo (n = 149) | e.o.w. anti-TNF (n = 135) | e.o.w. anti-TNF (n = 39) | ||
| Remission | 26 | 17% | 40% | 14% | 36% | 11% | 30% | 15% |
| 56 | 11.8% | 36% | 9% | 33% | 8% | 27% | 8% | |
| Response 100 | 26 | 26.5% | 52% | 28% | 56% | 21% | 47% | 26% |
| 56 | 16.5% | 44% | 17% | 44% | 13% | 36% | 18% | |
| Response 70 | 26 | 28% | 54% | 29% | 58% | 23% | 47% | 28% |
| 56 | 17.6% | 43% | 18% | 45% | 13% | 36% | 18% | |
FIGURE 23.
Response and remission rates for severe disease responders in CHARM. 67 LCI, lower confidence interval; RD, risk difference; RR, risk ratio; UCI, upper confidence interval.
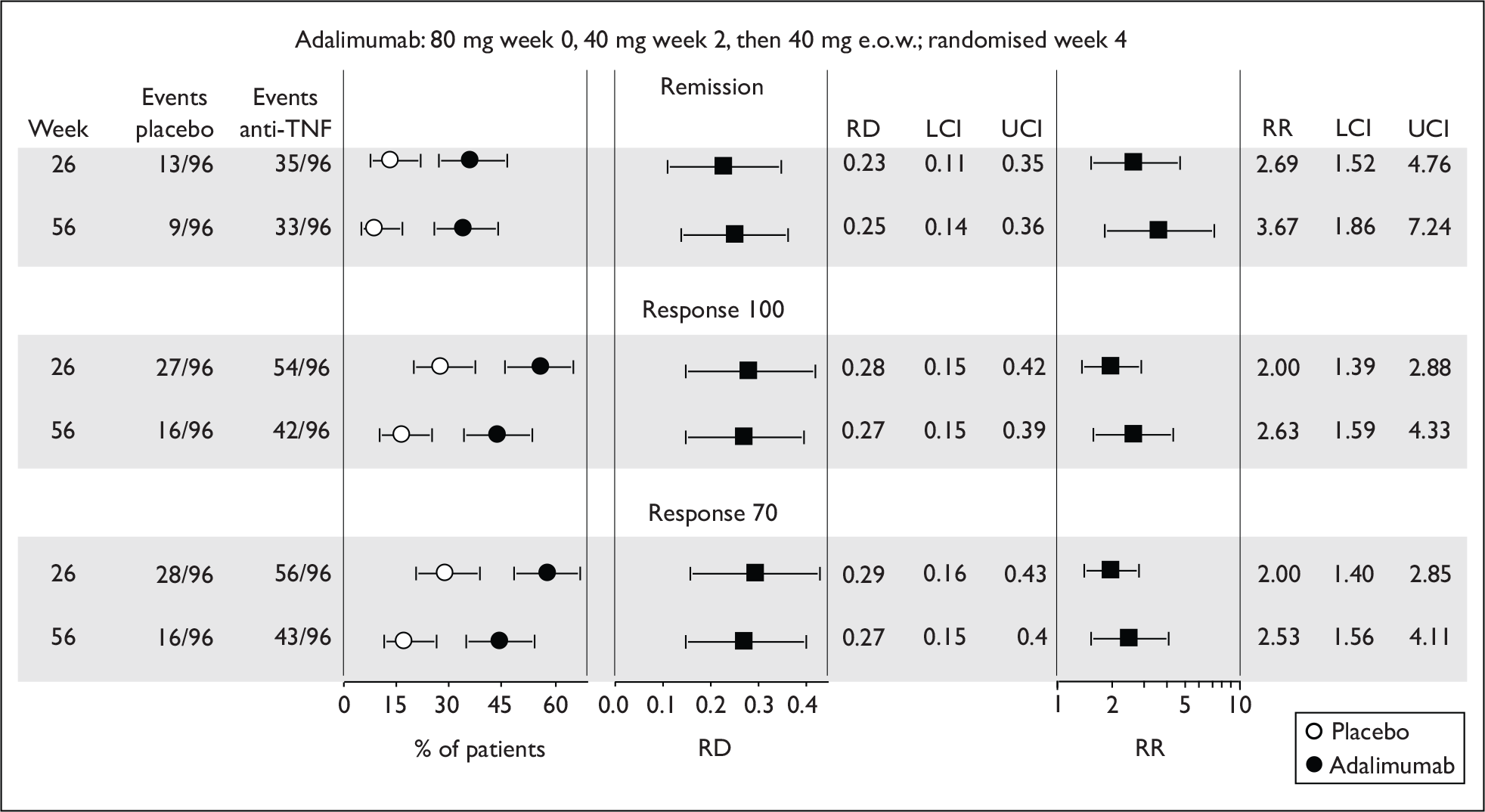
Other post hoc subgroup analyses
Several post hoc analyses explored the effectiveness of adalimumab among subgroups of patients defined according to various criteria including: baseline CRP level > or < 1 mg/ml; concomitant treatment with or without immunosuppressant medication; and previous experience of anti-TNF therapy or no previous experience. No statistically significant subgroup differences in adalimumab effectiveness were observed.
Premature withdrawal from treatment and crossover due to worsening disease
The published information about withdrawal from treatment and crossover to open-label therapy was difficult to disentangle. The Industry Trial Report provided fuller detail. Of 499 responders 29% (144) withdrew prematurely: (CiC information has been removed)% of the placebo group [(CiC information has been removed)], (CiC information has been removed)% of the e.o.w. adalimumab group [(CiC information has been removed)] and (CiC information has been removed)% of the weekly adalimumab group [(CiC information has been removed)]. The Industry Trial Report stated the overall premature discontinuation rate among all patients was (CiC information has been removed)% [(CiC information has been removed)], with 79 of these occurring before randomisation. Among all 788 randomised patients, withdrawals during the randomised phase were (CiC information has been removed) [(CiC information has been removed)%] in placebo group, (CiC information has been removed) [(CiC information has been removed)%] in the adalimumab e.o.w. group and (CiC information has been removed) ((CiC information has been removed)%) in the adalimumab weekly group, giving an overall rate of (CiC information has been removed)% [(CiC information has been removed)/(CiC information has been removed)] slightly (CiC information has been removed) than for responders only. Of patients randomised to adalimumab maintenance therapy, the rate of premature withdrawal was the (CiC information has been removed).
Crossover to open-label treatment after week 12 involved (CiC information has been removed) [(CiC information has been removed)%] of patients randomised to placebo, (CiC information has been removed) [(CiC information has been removed)%] of those randomised to adalimumab e.o.w. and (CiC information has been removed)/(CiC information has been removed) [(CiC information has been removed)] of those randomised to adalimumab weekly. These numbers represented patients experiencing worsening disease by flare or discontinued response. Transfer to open-label for patients in the weekly adalimumab group involved continuation of the same dose regimen (as crossover was described as ‘…switched to open-label treatment with 40-mg adalimumab eow… escalated to 40-mg weekly for those with continued non-response or recurrent flare’. After crossover ‘…continued non-response with open-label 40-mg weekly dosage resulted in withdrawal’;67 however, there was no published information about how long the state of flare or non-response was allowed to continue before withdrawal was implemented. The number of responder patients who crossed over to open-label was not published. The Industry Trial Report allowed calculation of crossovers and withdrawals among all randomised patients; this information is summarised in Figure 24.
FIGURE 24.
Withdrawals from treatment and crossovers for flare or non-response in CHARM. 67 *There was a discrepancy concerning one patient in the values for the adalimumab weekly group. (CiC information has been removed.)
Quality assessment (based on published report)
Randomisation, allocation concealment and blinding were adequate. Baseline characteristics were reported only for all patients, for all responders and for all non-responders, not for each of the trial arms. It was therefore not possible to judge if baseline characteristics were evenly balanced between the three arms of responders (placebo and e.o.w. or weekly adalimumab groups) that were analysed for effectiveness outcomes. The frequency of placebo injections was not documented. Information about patients who withdrew was reported. After week 12, patients with disease flare or non-response were allowed to cross over to the open-label treatment. It was difficult to determine how many responders and how many randomised patients in each group crossed over to open-label treatment. There was no statement defining how long after crossover flare or non-response was allowed to continue before withdrawal was implemented. Where necessary, the nearest or last observation was carried forward for continuous outcomes, but this was not stated explicitly and the number of missing data was not reported. A power calculation was conducted and based on the primary analysis of 4-week responders achieving remission at weeks 26 and 56.
The published text stated ‘…secondary efficacy analyses were conducted for all treated patients, including both randomised responder and randomised non-responder groups (all randomised patients who failed to achieve a clinical response at week 4)’. 67 Although this might be technically correct, in the sense that analyses were conducted, it is misleading because the results of these analyses were not reported, with the single exception of data on healing of fistulas for a subgroup of patients with fistulas at baseline and screening.
Seven hundred and seventy-eight patients given induction injections of 80 mg and 40 mg of adalimumab separated by 2 weeks were randomised at week 4 to maintenance therapy with placebo or 40-mg adalimumab e.o.w. or weekly. Only results for responders were published. Responders were defined as patients who at week 4 had a CDAI score reduced by ≥ 70 points from baseline. At weeks 26 and 56 there were significantly more responder patients in remission in the e.o.w. and weekly adalimumab groups than the placebo group, 40% and 47% respectively versus 17% at week 26, and 36% and 41% respectively versus 12% at week 56 (p < 0.001 for adalimumab vs placebo). The risk difference (adalimumab–placebo) for remission reached statistical significance in favour of adalimumab from week 8 onwards and remained stable from about week 12 or 16 to the end of follow-up (week 56), indicating that most of the benefit from active intervention was delivered during the first quarter to third of the trial.
The proportion of responders (response 70) had diminished to < 50% in all groups by week 56. Response 70 rates diminished (CiC information has been removed) delivered during the first part of the trial. Premature withdrawal from randomised treatments (adalimumab and placebo) was (CiC information has been removed)%; withdrawal rate from active intervention (adalimumab) was (CiC information has been removed) responders (CiC information has been removed)% (CiC information has been removed) non-responders [(CiC information has been removed)%]. Amongst the whole trial population randomised to adalimumab maintenance therapy, (CiC information has been removed)% crossed over to open-label treatment due to flare or non-response. The distribution of crossovers between responders and non-responders was unclear.
Pooling and indirect comparisons
The two adalimumab trials, CLASSIC II66 and CHARM,67 differed fundamentally with respect to populations analysed for outcome results. CLASSIC II66 reported results for responders who had achieved remission, whereas the responders in CHARM67 had achieved only the less stringent response of a 70-point reduction in CDAI score. It would be inappropriate to combine the results from these two trials. It is relevant that the manufacturer’s submission for adalimumab did not adopt a pooling approach. In a 2008 Cochrane review73 the authors stated ‘the two studies evaluating adalimumab were evaluated separately due to heterogeneity among the two trials (i.e. CLASSIC II and CHARM)’. Surprisingly, the results section of the review provided pooled results for remission (random effects model), and a further different pooled result (which may have been fixed effects) was presented in the discussion. On contacting the authors regarding these inconsistencies, we were informed that the review would be amended and the modified version, with no pooled results, is now available in the Cochrane Library.
The two infliximab trials, Rutgeerts et al. 58 (extension from Targan et al. 57) and ACCENT I,2,3 both employed a 10 mg/kg infliximab maintenance therapy arm and both reported results for responders based on a CDAI score reduced from baseline by ≥ 70 points. Therefore there is potential for pooling results. However, the pre-maintenance ‘induction’ phases of the two trials were very different, so the populations analysed for maintenance outcomes were likely to be quite different at the start of maintenance. Responders in ACCENT I2,3 were selected 2 weeks after a single exposure to a 5 mg/kg dose of infliximab. In contrast, responders in Rutgeerts et al. 58 were selected between 8 and 12 weeks after their first exposure to infliximab and were required to have a response 70 lasting 4 weeks. A further considerable difference between the responders in the two trials was the degree of exposure to infliximab prior to their selection as responders; in ACCENT I2,3 responders were defined after a single 5 mg/kg exposure, whereas Rutgeerts et al. ’s58 responders could have been exposed to any of the following: one 5 mg/kg, one 10 mg/kg, one 20 mg/kg, one 5 mg/kg and one 10 mg/kg, two 10 mg/kg, one 20 mg/kg and one 10 mg/kg, or no infliximab. The cumulative effect of these differences in the responder population (up to sixfold difference in exposure, different requirement in duration of response 70, and between four- and sixfold difference in duration of induction phase) is that the populations were unlikely to be sufficiently similar for the pooling of results to be informative.
Indirect comparison between the placebo-controlled maintenance trials, so as to gain an estimate of relative effectiveness of the two anti-TNF agents, was not undertaken for this non-fistulising adult population. Indirect comparison requires that trials for different interventions of interest share a common comparator arm (‘exchangeability’, see Glenny et al. 74). For the maintenance trials, the differences between ‘placebo’ groups were numerous and not easily quantifiable; different induction drugs were administered on differing numbers of occasions for different periods of time, followed by selection of responders by differing criteria representing different proportions of the randomised populations. The basis of indirect comparison depends on strict comparability of the trial arms common to the compared trials (in this case the placebo arms). In these circumstances indirect comparison would be misleading and unjustified. It is noteworthy that neither of the manufacturers’ submissions performed formal indirect comparison based on these trials.
Trials recruiting patients with fistulas
Two trials, Present et al. 62 – an induction trial – and ACCENT II65 – a maintenance trial – compared infliximab with placebo for adults with fistulising CD. There were no trials of adalimumab that enrolled only from this patient group. In these two trials all patients had one or more fistulas at the time of randomisation, and the main outcome measures focused on the status of fistulas during follow-up. The outcomes measured are listed in Table 31 and the main trial characteristics summarised in Table 32. For reference purposes this section also includes fistula status results for the small subgroups of adult patients who had fistulas in other trials.
| % achieving 70-point response on CDAI | CDAI score (mean or median) | IBDQ score (mean or median) | PDAI score (mean or median) | Response: reduction of 50% or more of draining fistula | Complete response: absence of draining fistula | Additional outcomes | |
|---|---|---|---|---|---|---|---|
| Infliximab | |||||||
| Present et al., 199962 | ✗ | ✓ | ✗ | ✓ |
✓ (at two or more consecutive visits) |
✓ (at two or more consecutive visits) |
Time to beginning of response; duration of response |
| ACCENT II65 |
✓ (subgroup only) |
✓ | ✓ | ✗ |
✓ time until loss of response (at consecutive visits > 4 weeks apart) |
✓ | Subsequent response among previous non-responders, response rate in patients who lost response and crossed over |
| Studya Drug |
Study weeks (n) |
Population: severity of CD (baseline PDAI, CDAI and IBDQ if stated) |
Intestinal areas affected; fistula: n and location |
Main concomitant medicationb | Previous/concomitant treatment with anti-TNF inhibitors | Intervention and comparator (dosing regimen) |
|---|---|---|---|---|---|---|
|
Present et al. , 199962 Infliximab |
18 94 |
≥ 1 draining abdominal or perianal fistula of ≥ 3 months’ duration Mean baseline PDAI (IQR): 9 (7–10.5) placebo; 8 (7–10), 10 (8–12) infliximab groups Mean baseline CDAI (SD): 193 (92) placebo; 184 (98), 185 (97) infliximab groups (IBDQ not stated) |
Mainly ileum and colon, also ileum only and colon only Fistula: 1, 45%; > 1, 55%; mainly perianal fistula, a few abdominal |
Aminosalicylates (mainly), also mercaptopurine or azathioprine, corticosteroids and antibiotics | Exclusion criterion: Infliximab within 3 months of study; no further details |
Intravenous infusions of placebo, 5 mg/kg infliximab or 10 mg/kg infliximab at weeks 0, 2 and 6 Study visits at least every 21 days; total follow-up not stated |
|
Sands et al. , 200465 – ACCENT II Infliximab |
54 282 |
≥ 1 draining abdominal or perianal fistula of ≥ 3 months’ duration PDAI scores not stated CDAI at baseline: 60% ≥ 150, 33% ≥ 220 Median baseline IBDQ (IQR) (responders): 168 (145–193) placebo; 155 (135–187) infliximab; 161 [136–176 (non-responders) |
Mainly ileum and colon, also ileum only and colon only Fistula: 1, 44%; > 1, 56%; mainly perianal fistula, some abdominal or recto-vaginal |
Aminosalicylates (mainly), also mercaptopurine or azathioprine, corticosteroids and antibiotics, few methotrexate Previous medication: mercaptopurine/azathioprine (mainly) and antibiotics, some ciclosporin, tacrolimus or methotrexate |
Exclusion criterion: previously treated with infliximab | Intravenous infusions of 5 mg/kg infliximab at weeks 0, 2 and 6 for all patients; at week 14 responders and non-responders randomised to placebo or 5 mg/kg infliximab at weeks 14, 22, 30, 38 and 46 |
Present et al., 199962 (infliximab)
Present et al. 62 was a small study that randomised 31 patients to placebo, 31 and 32 patients respectively to 5 mg/kg or 10 mg/kg infliximab infused at weeks 0, 2 and 6. Follow-up extended to at least week 18 with assessment visits every 4 weeks from week 2 onwards.
Primary outcome
The primary outcome was a > 50% reduction in the number of draining fistulas relative to baseline evaluated by physical evaluation and observed over at least two consecutive study visits at any time during the trial. Secondary outcomes included: complete absence of draining fistula observed over at least 4 weeks (i.e. across at least two consecutive study visits) at any time during the study, time to beginning of response and duration of response. Changes in CDAI and PDAI scores were reported for some patients.
The results for the primary outcome (for 50% reduction in draining fistula occurring at any time over at least two consecutive clinic visits and for complete absence of draining fistula over two consecutive clinic visits) are summarised in Figure 25. For both these outcomes, infliximab at both dose regimens was more effective than placebo (p = 0.002 and p = 0.02 for 5- and 10 mg/kg regimens respectively). The point estimates for response rates were associated with substantial uncertainty because of the small group size; for the combined infliximab groups the response rate was 62% (95% CI 50% to 73%) compared with 26% (95% CI 14% to 43%) for the placebo group (p < 0.001). For those with a response, the median time to response was 6 weeks in the placebo group and 2 weeks in the infliximab groups (Table 33).
FIGURE 25.
Rates and risk differences for a 50% reduction and absence of draining fistulas. LCI, lower confidence interval; RD, risk difference; RR, risk ratio; UCI, upper confidence interval.
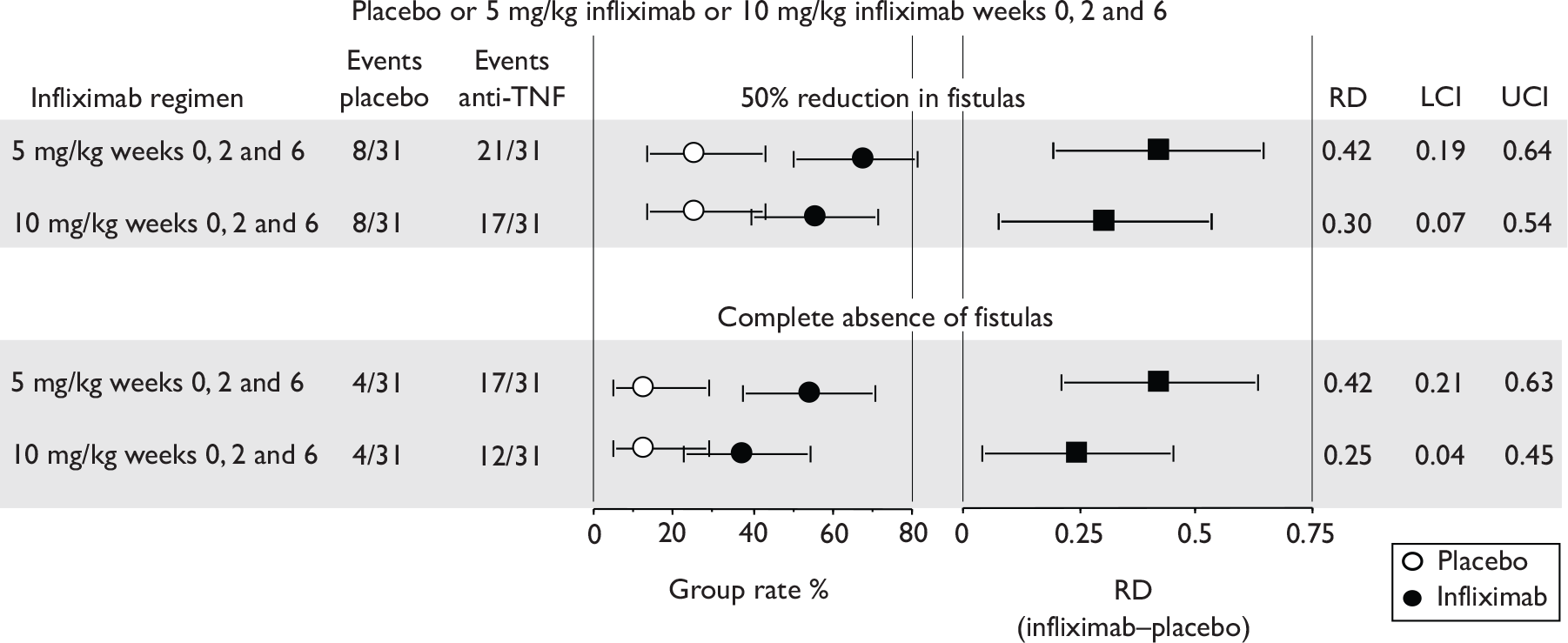
| Length of time to the beginning of a responsea (days: median, IQR) | ||||
|---|---|---|---|---|
| Placebo (n = 8) | 5 mg/kg (n = 21) | 10 mg/kg (n = 18) | 5 or 10 mg/kg (n = 39) | |
| Median (days) | 42 | 14 | 14 | 14 |
| IQR | 15–72 | 14–42 | 14–42 | 14–42 |
| p vs placebo | NA | NR | NR | NR |
Response and complete response
The median duration of response (defined as the maximum period during which the patient experienced a 50% reduction in draining fistulas) was approximately 3 months. For infliximab patients, 29/63 (46%; 95% CI 34% to 58%) experienced complete absence of draining fistulas for at least two consecutive clinic visits compared with 4/31 (13%; 95% CI 5% to 29%) of patients in the placebo group (p < 0.001).
The median CDAI and PDAI scores reported for baseline and weeks 2 and 18 of follow-up are summarised in Table 34. By week 2, statistically significantly better (i.e. lower) scores were found for the infliximab groups than for the placebo group. The statistical significance of the difference between groups had weakened or disappeared by week 18. Not all patients contributed data for the analyses (i.e. this was not an ITT analysis).
| Outcome | Placebo (n = 25) | Infliximab 5 mg/kg Weeks 0, 2 and 6; (n = 27) |
Infliximab 5 mg/kg Weeks 0, 2 and 6; (n = 27) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Week | Median | IQR | Median | IQR | p a | Median | IQR | p a | |
| CDAI scoreb | 0 | 162 | 126–265 | 163 | 99–284 | 0.71 | 203 | 112–254 | 0.66 |
| 2 | 171 | 114–252 | 108 | 83–203 | 0.04 | 111 | 89–164 | 0.06 | |
| 18 | 160 | 72–206 | 104 | 47–177 | 0.23 | 123 | 58–175 | 0.32 | |
| PDAI scoreb | 0 | 9 | 7–10.5 | 8 | 7–10 | 0.69 | 10.0 | 8.0–12.0 | 0.31 |
| 2 | 8 | 6–10 | 6 | 3–7 | 0.02 | 6.0 | 4.0–8.0 | 0.04 | |
| 18 | 7 | 4–9 | 4 | 1–7 | 0.05 | 5.0 | 3.0–8.0 | 0.14 | |
Quality assessment based on published report62
Randomisation and blinding were adequate and allocation concealment was likely to have been adequate. Baseline characteristics were generally well balanced although there was a greater proportion of patients in the infliximab groups that had undergone previous segmental resections than in the placebo group. Draining fistulas of < 3 months’ duration were excluded from the primary analysis. However, the number or frequency of these fistulas was not reported, and it was unclear if these were also excluded from the secondary outcome of a complete absence of a draining fistula. Total follow-up time for the primary outcome was unclear. No power calculation was performed. Last observation was carried forward for CDAI and PDAI analyses, but the number of missing data was unclear. There were only six premature withdrawals from treatment, four from the placebo group and one patient from each of the infliximab groups.
Patients with one or more draining fistula of more than 3 months’ duration, and an unreported number of fistulas of < 3 months’ duration, were randomised to placebo or 5 mg/kg infliximab or 10 mg/kg infliximab, by i.v. infusion at weeks 0, 2 and 6. More patients in the infliximab groups than in the placebo group achieved the primary outcome defined as: a reduction in the number of 3-month duration draining fistulas present at baseline by at least 50% lasting for at least two consecutive clinic visits. The percentage of patients responding to infliximab was 62% (95% CI 50% to 73%) compared with 26% (95% CI 14% to 43%) for the placebo group (p < 0.002). The median time to response was 2 weeks for infliximab groups and 6 weeks for placebo group. The duration of response was the same for both groups (median about 12 weeks).
More patients in the infliximab groups than in the placebo group achieved the secondary outcome of absence of draining fistula lasting for at least two consecutive clinic visits. The percentage of patients responding to infliximab for this outcome was 46% (95% CI 34% to 58%) compared with 13% (95% CI 5% to 29%) for the placebo group (p < 0.001).
ACCENT II65 (infliximab)
This was a maintenance trial that recruited 306 patients who had one or more fistulas of at least 3 months’ standing. 65 Of the 306 enrolled patients, 282 were assessed for ‘response’ at week 14 after administration of infusions at weeks 0, 2 and 6 of 5 mg/kg infliximab. ‘Responders’ were defined as those patients with at least 50% reduction in draining fistulas relative to baseline, observed at both weeks 10 and 14. Sixty-nine per cent (195) of patients were classified as responders. Both responders and non-responders were randomised to placebo (96 responders, 43 non-responders) or to 5 mg/kg infliximab (99 responders, 44 non-responders), which were administered at weeks 14, 22, 30, 38 and 46. Assessment visits were scheduled at weeks 0, 2, 6, 10, 14, 22, 30, 38, 46 and 54. After week 22, patients losing response could cross over to 5 mg/kg infliximab from placebo and from 5 mg/kg infliximab to 10 mg/kg infliximab. The fistula status outcome measures were:
-
loss of response – defined as a recrudescence of draining fistula, a change in therapy, a need for surgery, dropout due to lack of efficacy, or a worsening of luminal disease activity
-
response – defined as 50% reduction from baseline in draining fistula observed at consecutive visits 4 or more weeks apart
-
complete absence of draining fistula.
Primary outcome
The primary outcome was designated as time to loss of response in responders. The results are summarised in Figure 26.
FIGURE 26.
Time to loss of response by responders in ACCENT II. 65 Data taken from published graph and redrawn.
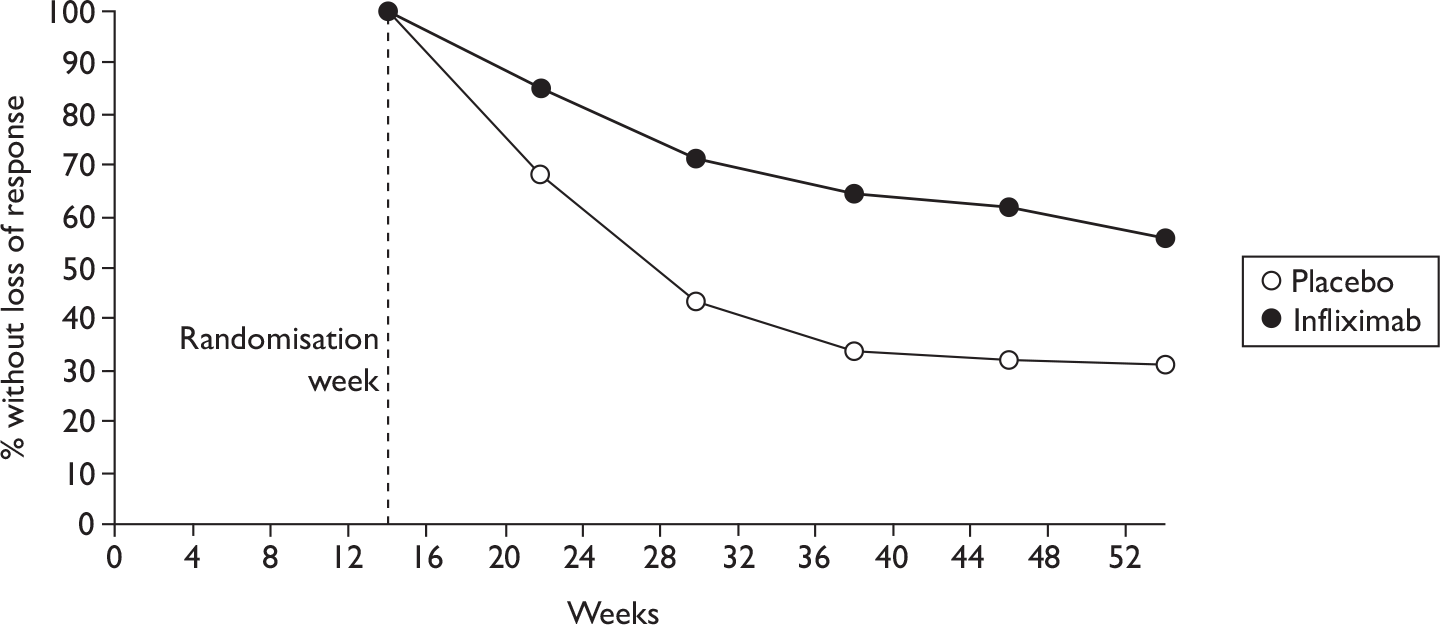
The median time to loss of response after randomisation was 14 weeks in the placebo group and > 40 weeks in the infliximab group (p < 0.001 by log rank test). In the infliximab group, 42% of responders lost response, and in the placebo group, 62% lost response. The main reasons for loss of response in the primary outcome were: change in treatment (38% of placebo, 25% of infliximab) or recrudescence of fistula (22% placebo, 16% infliximab).
Response and complete response
At 30 weeks, 33% and 64% of the placebo and infliximab groups respectively had a response (50% reduction in draining fistula from baseline for at least two consecutive visits), and at week 54 the corresponding percentages had diminished to 23% and 46% respectively (p = 0.001). The manufacturer’s submission to NICE contained CiC information for additional weeks of follow-up. These are summarised in Figure 27. Prior to randomisation, except at week 2, the rates were about equal as would be expected, given that all responder patients received identical induction therapy up to week 14 and baseline characteristics were well balanced. At week 2, a surprising difference between groups was observed with higher rates for the patients subsequently randomised to infliximab. At week 14, the placebo group did not receive infliximab. After randomisation at week 14, response rates diminished in both groups. From week 22 the risk difference (infliximab–placebo) reached statistical significance in favour of infliximab, after week 30 risk differences diminished indicating that most benefit for maintenance of response from active intervention was delivered between weeks 14 and 30. By week 30 the intervention group had received two more infusions of infliximab than placebo patients.
FIGURE 27.
Rates and risk differences for ≥ 50% reduction of draining fistulas in ACCENT II65 responders. LCI, lower confidence interval; RD, risk difference; RR, risk ratio; UCI, upper confidence interval.
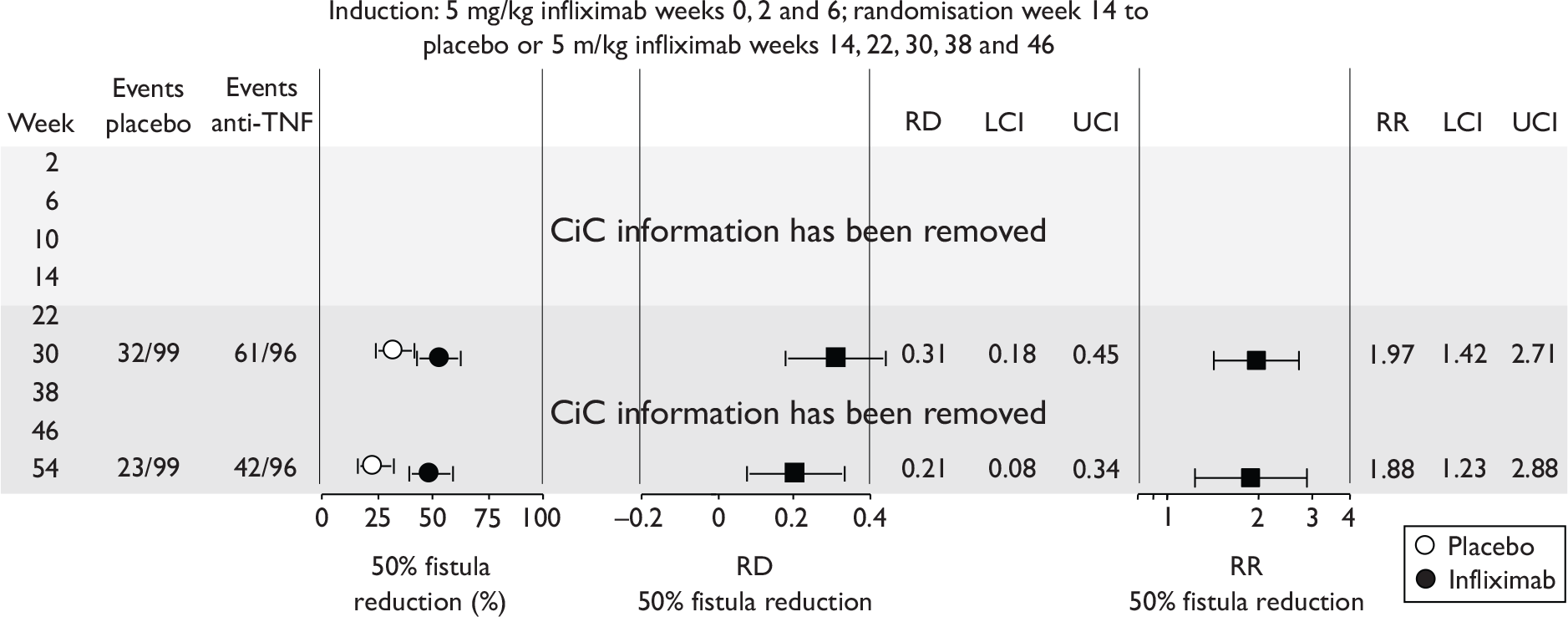
Responders with loss of response during the post-randomisation phase were allowed to cross over after week 22 to an increased dose of infliximab. The renewed response rate in these crossover patients was reported as 25/41 (61%) in the placebo group (crossed over to 5 mg/kg dose) and 12/21 (57%) in the intervention group (crossed over to 10 mg/kg dose). However, Figure 1 of the ACCENT II65 published report shows 50 crossovers from placebo and 28 from 5 mg/kg infliximab.
The published report provided information about the rates of ‘complete response’ among responders. 65 A complete response was defined as a complete absence of draining fistulas. The definition for a response required ≥ 50% reduction in fistulas for at least 4 weeks, a ‘complete response’ differed in that no minimum duration was specified. It was unclear, but likely, that this definition applied only to draining fistulas of at least 3 months’ standing at baseline. The frequency of draining fistulas at baseline that were of less than 3 months’ standing was not reported. The results for a complete response are summarised in Figure 28.
FIGURE 28.
Rates, risk difference and risk ratio for complete response among responders in ACCENT II. 65 LCI, lower confidence interval; RD, risk difference; RR, risk ratio; UCI, upper confidence interval.
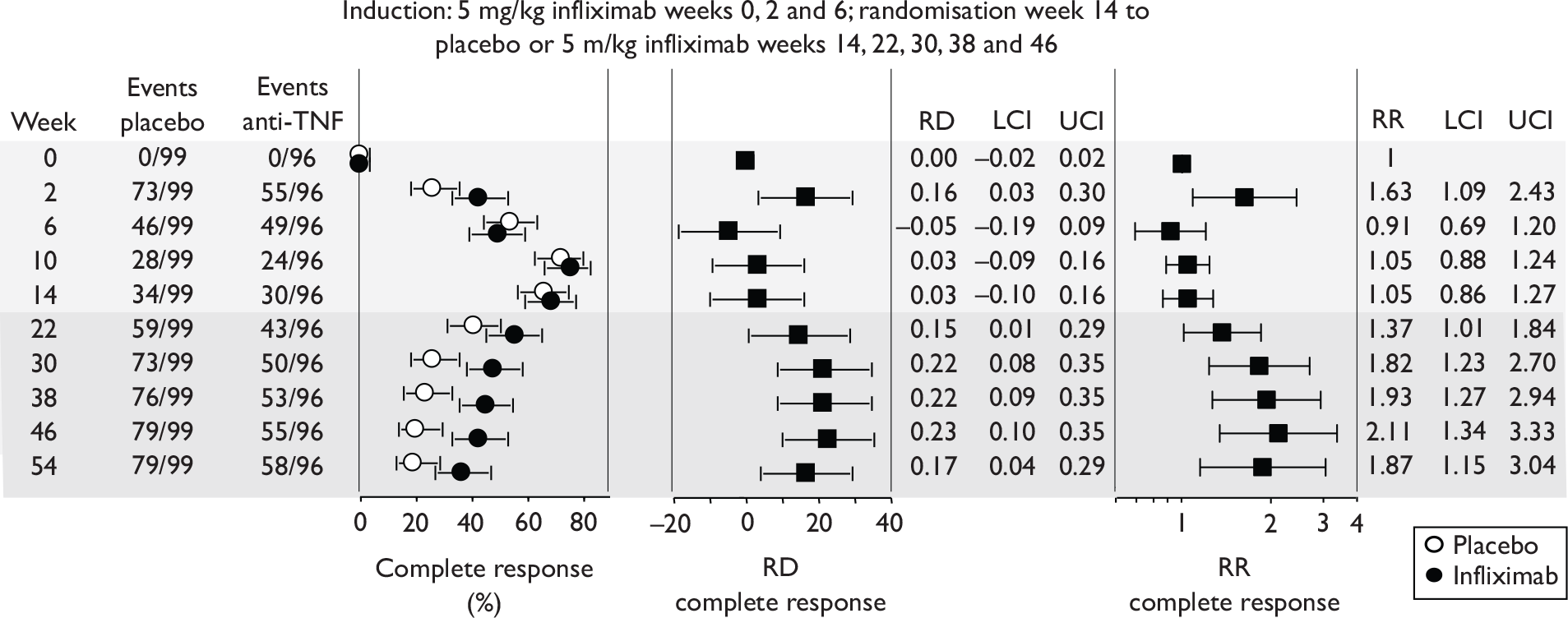
At week 2, after only a single dose of infliximab, 66% (128/195) of patients already had a ‘complete response’. Unexpectedly, more patients who were subsequently randomised to infliximab had a complete response than those subsequently randomised to placebo (p = 0.014). By 14 weeks, 66% and 69% of responder patients who were randomised to placebo and infliximab respectively had a complete response. The rate of complete response in responders diminished in both groups after week 14. From week 22 the risk difference (infliximab–placebo) reached statistical significance in favour of infliximab and from week 30 remained stable, indicating that most benefit in maintenance of response from active intervention was delivered between weeks 14 and 30.
The rate for a complete response among all enrolled patients at week 14 was reported to be 48% (147/306); this generated a 75% [147/(99 + 96)] complete response for responders at week 14 which according to Figure 2B in the ACCENT II65 published report corresponded to week 10 rather than to week 14.
Hospitalisations and major surgery
The manufacturer’s submission to NICE presented results for major surgery and for hospitalisation for all patients in ACCENT II65 whether or not they crossed over. For this purpose the placebo arm was termed ‘episodic treatment’ and the infliximab arm ‘scheduled treatment’. A 2.4-fold lower rate was reported for the scheduled treatment group. There were two important differences between these treatments. Firstly, patients in the ‘episodic’ arm experienced a 4-month mandatory withdrawal of active intervention (from weeks 6 to 22) not experienced by patients in scheduled treatment. Secondly, after week 22 the ‘episodic’ group patients were restricted to 5 mg/kg infliximab at episodes of worsening disease, whereas the ‘scheduled treatment’ group were able to receive 10 mg/kg. Restricted access to treatment (weeks 6–22) and restricted dosage represent biases likely to favour the ‘scheduled treatment’ group for any comparisons after week 6. Furthermore the ‘episodic treatment’ procedure was unlikely to reflect how an episodic strategy might be implemented in real world clinical practice, both with respect to the 4-month gap in active intervention and with regard to restriction of dose. Because of bias in the comparisons made and the probable dissimilarity between the trial episodic treatment and likely clinical practice, it was considered here that the hospitalisation rate for the ‘episodic’ treatment and the comparison with the scheduled treatment were very approximate guides.
The considerations described above also apply to the values reported for the percentages of patients requiring major surgery (13% and 2% in ‘episodic’ and scheduled treatment arms respectively).
Other outcomes reported for ACCENT II
The ACCENT II65 published report presented the median decrease from baseline in CDAI score at weeks 30 and 54 for all patients. Improvements in median CDAI were statistically significantly greater for the infliximab group (p = 0.004). Median increases from baseline in IBDQ scores at weeks 30 and 54 were also significantly greater for the infliximab group than for the placebo group. Baseline scores for all patients by group were not provided and baseline balance was therefore uncertain. In the case of missing values, the last observations were carried forward for the CDAI and IBDQ outcomes. The results are summarised in Table 35.
| Week | Infliximab (n = 139) | Placebo (n = 143) | p infliximab vs placebo |
|---|---|---|---|
| Median decrease in CDAI score from baseline | |||
| 30 | 42 | 16 | 0.004 |
| 54 | 40 | 15 | 0.004 |
| Median increase in IBDQ score from baseline | |||
| 30 | 14 | 4 | 0.002 |
| 54 | 10 | 5 | 0.03 |
Further results for the ACCENT II65 trial were presented in two separate papers. 75,76 One reported a post hoc analysis of the subgroup of responder patients with rectovaginal fistulas (11 received placebo and 14 received the 5 mg/kg dose regimen of infliximab);75 the other paper performed a post hoc analysis on incidence of abscess development in patients responding to infliximab with closure of fistulas. 76 The first of these papers was too underpowered for firm conclusions to be drawn. In ACCENT II,65 crossover to an increased dose of infliximab was allowed for all randomised groups (including placebo) from week 22 onwards; this resulted in the mean dose of infliximab in the placebo group (quoted as 20 mg/kg) being approximately half that of the intervention groups (quoted as 40 mg/kg). The post hoc analysis for abscess development compared these two groups and reported no statistically significant difference in rates (15% vs 19%; p = 0.526).
Quality assessment (based on published report)
Randomisation and blinding were adequate and allocation concealment likely to be so. Baseline characteristics for responders were well balanced between placebo and infliximab arms; however, for the all-patient comparisons between infliximab and placebo arms (e.g. of change in IBDQ and CDAI scores relative to baseline) it was not possible to ascertain if groups were balanced at baseline. There was a lack of clarity in the methods section so that it was difficult to determine if the sentence ‘…data for patients who crossed over from placebo to infliximab were censored before crossover occurred…’ referred to the survival analysis of loss of response. If it did, the reason for different handling of crossovers in the compared groups is difficult to interpret. The number of patients who withdrew prematurely was unclear except for discontinuation for AEs. No power calculation was undertaken. The last observation was carried forward as necessary for continuous outcomes, but the number of missing data was not reported.
After induction infusions of 5 mg/kg infliximab at weeks 0, 2 and 6, 64% of enrolled patients were classified as responders. Responders were defined as patients experiencing at both weeks 10 and 14 a ≥ 50% reduction in the number of draining fistulas that were present at baseline of at least 3 months’ standing.
After week 14, the median time to loss of response by responder patients was greater for patients randomised to continued infliximab treatment of 5 mg/kg at 8-week intervals than for those randomised to placebo (p < 0.001). More responder patients randomised to infliximab at week 14 experienced a response (closure of ≥ 50% of draining fistula for at least 4 weeks) than did responder patients randomised to placebo, and from week 22 the risk difference (infliximab–placebo) reached statistical significance in favour of infliximab. After week 14, response rates diminished in both groups. From week 30, risk differences diminished indicating that most benefit from infliximab was delivered between weeks 14 and 30.
Other trials reporting on subgroups of adults with fistulas
Two other trials reported on effectiveness of anti-TNF therapy for closure of fistulas – the GAIN induction trial of adalimumab64 and the CHARM maintenance trial of adalimumab. 67 In the GAIN trial,64 at the end of follow-up (week 4) similar rates of fistula improvement were recorded for adalimumab and placebo groups (3/20 and 5/25 respectively). The CHARM trial67 reported a measure termed ‘fistula remission’ for the subgroup of trial patients who had fistula at screening and baseline. Fistula remission was defined as the percentage of patients with closure of all fistulas that were draining at screening and at baseline (separated by 2 weeks). Fistula remission was observed for 30% (21/70) and 13% (6/47) of combined adalimumab groups and placebo group respectively at week 26 and for 33% (23/70) and 13% (6/47) respectively at week 56.
Paediatric Crohn’s disease trials
Patients in these trials were ≤ 18 years of age. Two trials, Baldassano et al. 46 and REACH [A randomized, multicenter, open-label study to evaluate the safety and efficacy of anti-TNFα chimeric monoclonal antibody (infliximab, Remicade®) in pediatric subjects with moderate-to-severe Crohn’s disease] (Hyams et al. 45), looked at the effectiveness of different doses of infliximab in paediatric CD patients. There was no placebo arm in either trial. There were no trials of adalimumab in children. The outcomes measured are shown in Table 36 and the study characteristics are summarised in Table 37.
| % of patients in remission (PCDAI score < 10) | % achieving response | PCDAI score (mean or median) | Additional outcomes | |
|---|---|---|---|---|
| Infliximab | ||||
| Baldassano et al. 200346 | ✓ |
✓ Decrease in CDAI of ≥ 70 points OR ≥ 10 points on PCDAI |
✓ | Endoscopic lesion severity score (in consenting patients) |
| REACH45 |
✓ (or CDAI < 150) |
✓ Decrease in PCDAI of ≥ 15 points and total PCDAI score < 30 |
✓ | IMPACT III score; % discontinuing corticosteroids; change in height status (subgroup); clinical response following crossover |
| Study Sponsor |
Country | Study length/size | Population: severity of CD (baseline CDAI and IBDQ if stated) |
Intestinal areas affected | Main concomitant medication, % not on any medication | Previous/concomitant treatment with anti-TNF inhibitors | Intervention and comparator (dosing regimen) |
|---|---|---|---|---|---|---|---|
|
Baldassano et al. , 200346 Supported by Centocor |
Multicentre (USA, Europe) |
12 weeks n = 21 |
Moderate-to-severe active disease despite previous treatment, PCDAI ≥ 30 or modified CDAI ≥ 200 Median PCDAI 56, 45 and 41 infliximab groups (no placebo group), median modified CDAI score 455, 317 and 312 (IBDQ not stated) |
Mainly ileum and colon, also colon only and gastroduodenal |
Mainly aminosalicylates and corticosteroids, also mercaptopurine or azathioprine, antibiotics, few methotrexate % not on medication (if any) not stated |
No details |
Single intravenous infusion of 1 mg/kg infliximab, 5 mg/kg infliximab or 10 mg/kg infliximab over at least 2 hours at week 0 (No placebo group) |
|
Hyams et al. , 2007 – REACH45 Supported by Centocor |
Multicentre (USA, Canada, Europe) |
54 weeks n = 103 |
Moderate-to-severe CD PCDAI > 30 at baseline Mean baseline PCDAI (SD) 42.1 (9.2) and 40.1 (6.8) (infliximab groups, no placebo group) (no other baseline measures) |
Mainly colon and/or ileum, also upper tract |
Mainly mercaptopurine or azathioprine, also aminosalicylates and corticosteroids, few methotrexate % not on medication (if any) not stated |
Exclusion criteria: previously treated with infliximab or other anti-TNF agent | Three i.v. infusions as induction therapy with infliximab 5 mg/kg (weeks 0, 2 and 6) followed by: five infusions of maintenance therapy with infliximab 5 mg/kg administered at weeks 14, 22, 30, 38 and 46 or three infusions with infliximab 5 mg/kg at weeks 18, 30 and 42 |
Baldassano et al., 200346 (infliximab)
The small trial of Baldassano et al. 46 examined whether a single dose of infliximab induced a response in paediatric patients. Patients were randomised to a 1 mg/kg (n = 6), 5 mg/kg (n = 7) or 10 mg/kg (n = 8) infusion. Patients were followed up to week 12. The primary outcomes were improvements from baseline in PCDAI and modified CDAI score. Other outcomes were the percentage of patients responding and the percentage in remission.
Table 38 shows the median percentage improvement in PCDAI score at various follow-up times relative to baseline. No clear pattern relating to follow-up time or dose regimen was apparent. To what extent improvement in scores resulted from infliximab treatment is impossible to determine because of lack of an appropriate placebo control group.
| Week | Median % improvement from baseline in PCDAI score | ||
|---|---|---|---|
| Infliximab dose | |||
| 1 mg/kg | 5 mg/kg | 10 mg/kg | |
| 1 | 47 | 37 | 35 |
| 2 | 40 | 65 | 53 |
| 4 | 27 | 57 | 28 |
| 8 | 32 | 28 | 64 |
| 12 | 27 | 13 | 40 |
Response and remission results are summarised in Figure 29. All estimates were associated with great uncertainty due to the small number of participants. The proportion of patients in response approached 100% after 1 week in all groups and then tended to decline during follow-up. There was little difference between the groups. How much of the response was intervention dependent cannot be determined because of the lack of an appropriate placebo control group that did not receive infliximab. For remission, no clear pattern relating to dose or length of follow-up was apparent. Again, because of the lack of an inactive control it is impossible to determine the contribution of infliximab to the observed results.
FIGURE 29.
Response and remission rates reported in Baldassano et al. 46 (results as reported, not ITT). Response was defined as at least a 10-point reduction in PCDAI or at least a 70-point reduction in modified CDAI score; remission was defined as a PCDAI score < 10 or a modified CDAI of < 150.
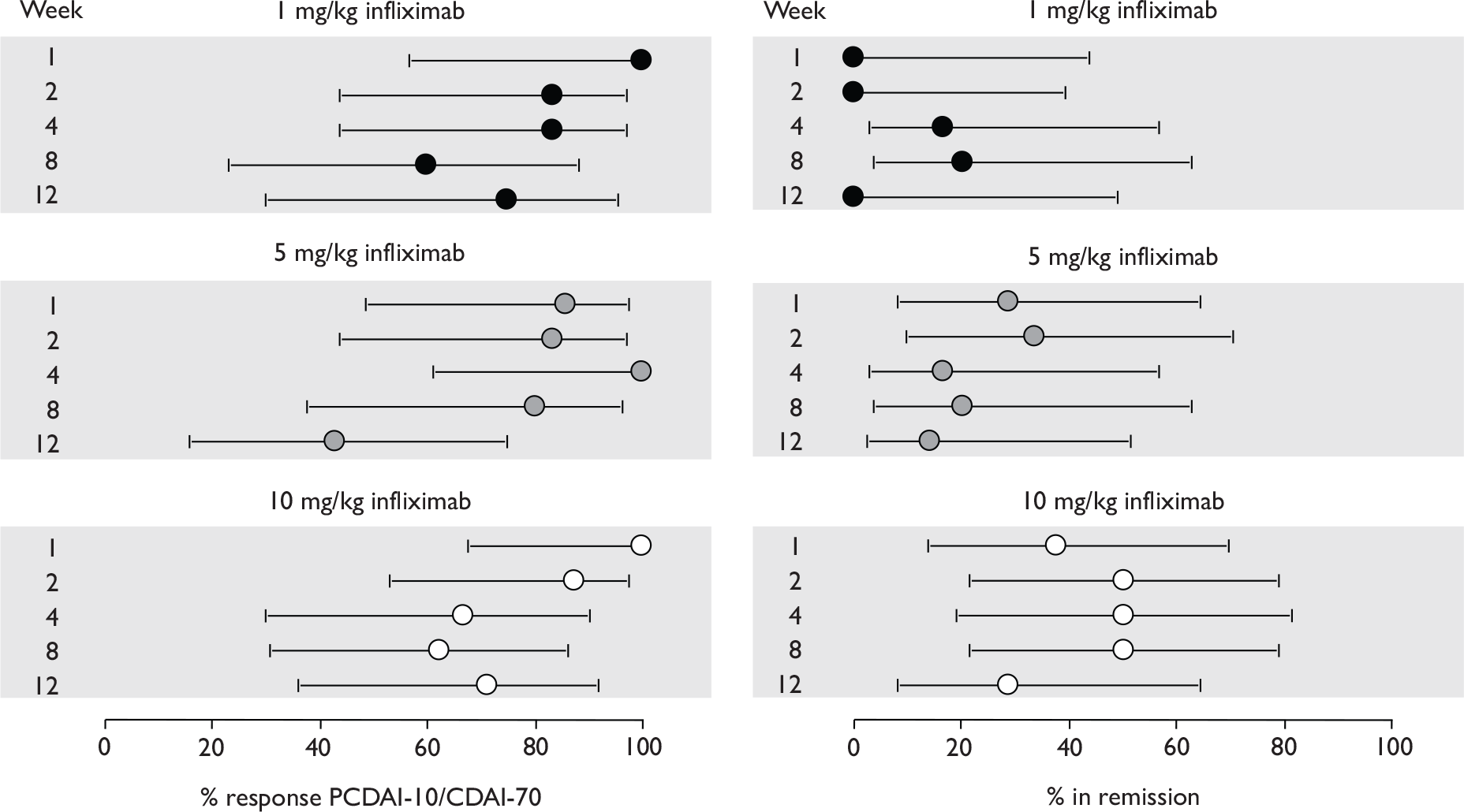
Randomisation and blinding were adequate and allocation concealment likely to be so. With such small numbers in each group it is not surprising that some baseline characteristics were imbalanced; notably, the 10 mg/kg group consisted almost exclusively of boys and the baseline CDAI score was substantially higher for the 1 mg/kg group than for the 5 mg/kg and 10 mg/kg groups. The number of patients completing the trial was reported to be 90%. No power calculation was done and analyses did not appear to be ITT.
An induction infusion of 1, 5 or 10 mg/kg infliximab improved PDAI scores relative to baseline. Induction increased the proportion of patients in response (40%–100% depending on dose and follow-up time) and in remission (0%–50% depending on dose and follow-up time). The study was underpowered, so these effectiveness estimates were associated with great uncertainty; no clear pattern was evident relating outcomes to dose regimen. The lack of a placebo control group renders interpretation of results problematic.
REACH45 (infliximab)
The REACH trial45 was called an ‘induction and maintenance’ study. Patients received induction doses of 5 mg/kg infliximab at weeks 0, 2 and 6. Responders were defined as those who reduced baseline PCDAI by at least 15 points and had a score of ≤ 30 at week 10. Responders (only) at week 10 were randomised to either five further doses of 5 mg/kg every 8 weeks delivered at weeks 14, 22, 30, 38 and 46 or three further doses delivered every 12 weeks at weeks 18, 30 and 42. Of 112 patients entering the induction phase, 103 were classified as responders and 99 were analysed. The lack of a placebo control group not receiving infliximab means that it is difficult to determine to what extent maintenance of response after induction was attributable to infliximab intervention. No primary outcome was identified. Response and remission results were reported for weeks 30 and 54 and weeks 10, 30 and 54 respectively. These are summarised in Figure 30. The differences between the two dose regimens for both response and remission at weeks 30 and 54 reached statistical significance (p < 0.05) in favour of the more frequent dose regimen.
FIGURE 30.
Post-induction response and remission rates for responders in the REACH trial. 45 Response defined as decrease in PCDAI of ≥ 15 points from baseline and total ≯ 30. Remission defined as a PCDAI ≤ 10 points.
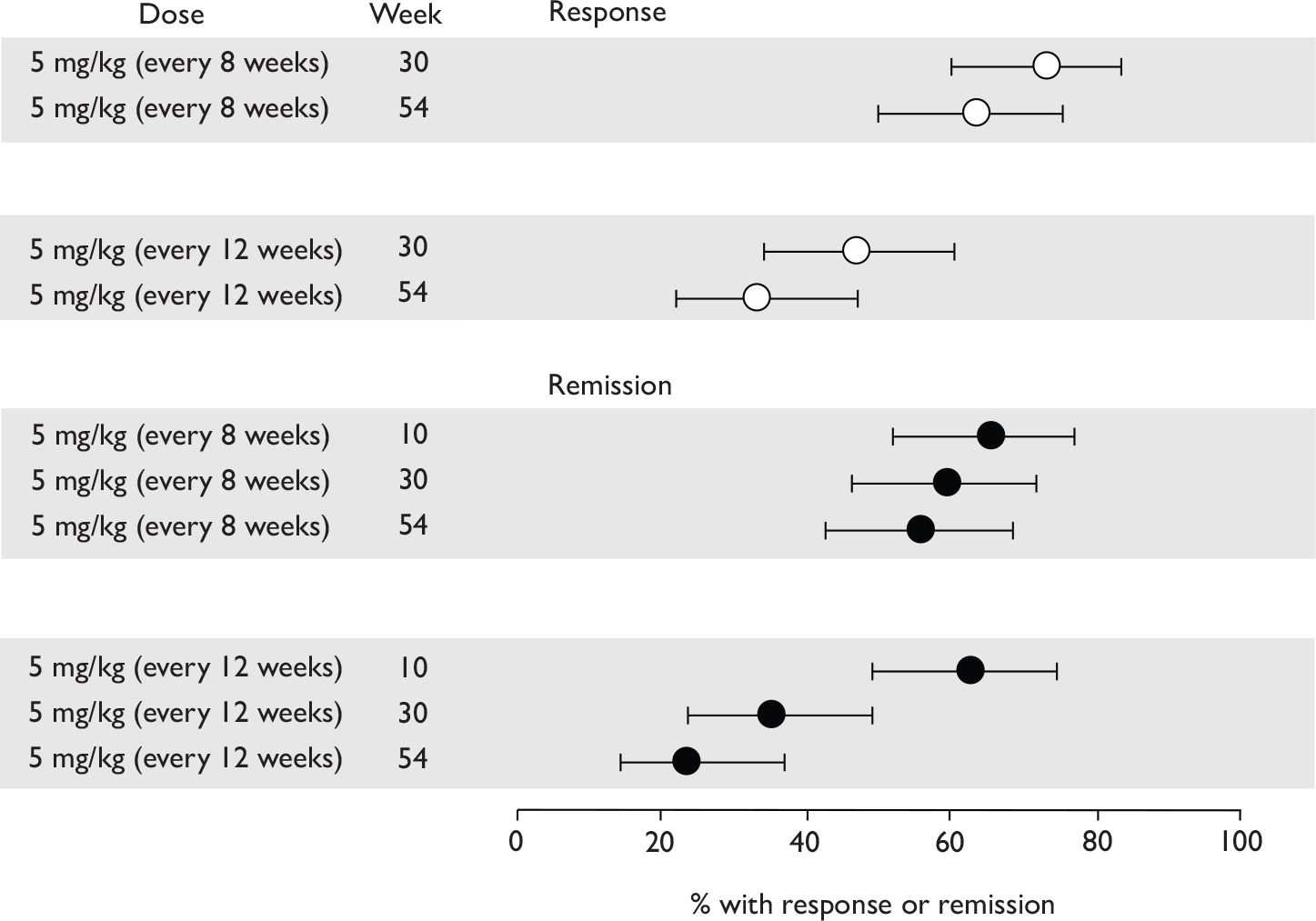
The REACH publication45 also reported changes from baseline (mean and SD) in PCDAI score, IMPACT III score (a QoL measure; scores range from 35 to 175 with higher scores representing better QoL) and daily corticosteroid use. Last observation was carried forward where values were missing. Information was provided for all ‘responders’ (i.e. the two trial arms combined) or separately for the two different treatment groups, at weeks 10, 30 and 54. The results are summarised in Table 39. The ‘all responders results’ do not represent a randomised comparison but rather a ‘before versus after treatment’ comparison for a subgroup of patients (responders) who were selected because they exhibited a favourable response. Given that a ‘no-treatment control group’ was not included in this trial, the analyses do not provide robust quantitative information about the effectiveness of infliximab for paediatric patients, and the favourable changes reported are difficult to interpret as an indeterminate proportion of the effects observed may have been infliximab independent.
| Group | Week 10 | Week 30 | Week 54 | n |
|---|---|---|---|---|
| PCDAI: mean (SD) decrease from baseline [improvement]a | ||||
| Doses every 8 weeks | –33.2 | NR | NR | 52 |
| Doses every 12 weeks | –29.4 | NR | NR | 51 |
| Groups combined | –31 (10) | –25.5 (16) | –27 (16) | 103 |
| IMPACT III: mean (SD) increase from baseline [improvement]a | ||||
| Doses every 8 weeks | NR | 24.7 | 26.5 | NR |
| Doses every 12 weeks | NR | 18.3 | 22.5 | NR |
| Groups combined | 23.9 (16) | 21 (18) | 24 (17) | 76 |
| Corticosteroid dose (mg/kg): mean (SD) decrease from baseline [improvement]a | ||||
| Doses every 8 weeks | NR | NR | NR | 52 |
| Doses every 12 weeks | NR | NR | NR | 51 |
| Groups combined | 0.3 (0.4) | 0.39 (0.4) | 0.3 (0.59) | 103 |
| Patients’ corticosteroid use: n discontinued of N users (%) | ||||
| Doses every 8 weeks | 12 of 24 at baseline (50%) | NR | 10 of 12 at week 10 (83%) | NA |
| Doses every 12 weeks | 3 of 12 at baseline (25%) | NR | 5 of 9 at week 10 (56%) | NA |
| Groups combined | 15 of 36 at baseline (42%) | NR | 15 of 21 at week 10 (71%) | NA |
After week 10 responder patients were allowed to cross over to increased infliximab for worsening disease state. The increases in infliximab allowed included transfer from infusions every 12 weeks to every 8 weeks and increase in infusion dose from 5 mg/kg to 10 mg/kg. The proportion of patients who crossed over was 40%. The number of responder patients who withdrew prematurely was reported as 22 (21%), but it was unclear if this included withdrawals of patients who had crossed over to increased infliximab.
Randomisation was adequate and allocation concealment likely to be so. This was an open-label study with no blinding. 45 Baseline characteristics were well balanced except for steroid use. The number of patients withdrawing was reported, but it was not clear if this also included crossover patients who later withdrew. A power calculation was done and analyses were ITT.
A 10-week induction phase with infusions of 5 mg/kg infliximab at weeks 0, 2 and 6 was followed by randomisation of responders at week 10 to further 5 mg/kg infusions every 8 or 12 weeks. At week 10, 88% of enrolled patients were classified as responders. Response rates for responders diminished to less than 50% by week 54. The difference between dose regimens reached statistical significance in favour of the 8-weekly infliximab dose regimen. About 40% of patients crossed over to increased infliximab because of worsening disease status. About 20% of patients withdrew from treatment prematurely.
Results in non-responders
Published results for maintenance trials focused on early responders (determined at week 2, or week 4 in the two large trials, ACCENT I2,3 and CHARM67). It is important to attempt to determine if such a subgroup analysis can be justified.
The question of whether results were published for non-responders is summarised in Table 40. Out of all of the maintenance trials, only two trials (ACCENT I2,3 and II65) published results including initial non-responders. Additional information was obtained from the industry submission for results in responders and non-responders from the CHARM67 trial (see Appendix 11). Table 40 also details whether non-responders were randomised.
| Study | Were non-responders randomised to maintenance treatment? | Were results reported for both responders and non-responders separately (RCT only)? |
|---|---|---|
|
CLASSIC II66 (adalimumab) |
No Only those patients (from CLASSIC I63) in remission at week 0 and 4 eligible for randomisation; those not in remission at week 0 or no longer in remission at week 4 entered an open-label cohort |
No Results reported for patients not eligible for randomisation who entered open-label cohort |
|
(infliximab) |
Yes Responders and non-responders randomised |
No Results for ALL patients (responders + non-responders) reported in Rutgeerts et al. ;2 results for responders only reported in Hanauer et al. 3 Industry submission: subgroup analysis of mucosal healing and CDEIS scores in responders and non-responders together; hospitalisation and surgery reported for responders and non-responders together |
|
Rutgeerts et al. , 199958 (infliximab) |
No Responders from Targan et al. 57 RCT eligible Initial non-responders in Targan et al. 57 given an additional 8 weeks of open-label treatment during which they could respond and still be eligible for maintenance treatment Unclear if any responders drawn from placebo group of RCT |
No No non-responders included in RCT Proportion of non-responders at week 4 (Targan et al. 57 RCT) subsequently responding during open-label treatment unclear |
|
ACCENT II65 (infliximab, fistulising) |
Yes Non-responders randomised for secondary analysis |
Yes Results reported for response |
|
REACH45 (infliximab, paediatric) |
No Only responders randomised (no placebo control) |
No No non-responders included in RCT (no placebo control) |
|
CHARM67 (adalimumab) |
Yes Non-responders randomised for secondary analysis (randomisation stratified by responder status) |
No Stated that secondary efficacy analyses were conducted for total population, but results presented only for fistula closure, which relates to a subgroup of patients (15% of patients have fistula) Industry submission: present results (remission, CDAI decrease > 70, CDAI decrease > 100, IBDQ score) for responders and for patients with CDAI > 300 The trial report submitted to NICE contained information on non-responders |
ACCENT I2,3 (infliximab)
Results for responders and non-responders were not published separately nor presented in the manufacturer’s submission. However, by subtracting CiC information for responders from published information for all patients, it is possible in theory to calculate the response and remission rates for non-responders. Results for responders and for all patients were available in publications or CiC information in the industry submission for the following outcomes in the ACCENT I2,3 trial:
-
Median CDAI scores at numerous follow-up times. These were published in separate papers for responders only and for all patients. 3 No indication of variance was given, so robust analysis was not possible.
-
IBDQ scores. These were recorded but reported differently in the two publications2,3 (median scores for responders and a dichotomised outcome for ‘all’ patients); this information cannot be used for estimation of non-responder results. 3
-
Remission and response 70 for responder patients at multiple follow-up times. The manufacturer’s submission on infliximab provided CiC results for remission and response 70 for responder patients at multiple follow-up times for ACCENT I. 2,3 Results for these outcomes for all patients were available in the public domain. It was possible to calculate the outcome for non-responder patients randomised to placebo or infliximab (5 mg/kg) by appropriate subtraction of responder rates from all-patient rates. Unfortunately, in practice this was meaningful for only the first 14 weeks of the trial because after week 14, patients who crossed over to increased infliximab dosage regimen on exacerbation of their disease contributed to the numbers achieving outcomes in the all-patient results but were discounted in the analyses for responders only.
The combined lack of complete long-term results, and the introduction of crossover to different treatments at week 14 of the trial, made it difficult to determine the rates of response of ‘non-responders’ in the ACCENT I2,3 trial, and renders problematic the interpretation of these rates when the limited available data allows their calculation. Appendix 11 provides the results calculated for non-responders in ACCENT I. 2,3
ACCENT II65 (infliximab)
Limited results for responders and non-responders were reported separately for this trial65 that investigated patients with fistulas. The response rate among initial non-responders was 7/44 (16%) in the placebo group and 9/43 (21%) in the infliximab group (p = 0.6). A response was defined as a reduction of at least 50% from baseline in the number of draining fistulas at consecutive visits 4 or more weeks apart. The time point for this result was not stated and it is unclear whether these were patients who ever had a response during the 54-week trial. There are no details on whether these response rates were maintained. It is difficult to compare these results with those of the initial responders as the trial looked at the maintenance of response in initial responders rather than induction of response.
Adverse events
This section includes in-licence and non-licence trial results so as to include all relevant evidence. All studies reported AEs. There were six malignancies among 573 patients followed for 54 weeks in ACCENT I. 2 The most serious AEs, and/or those thought potentially to be associated with anti-TNF therapy have been tabulated. In Table 41 trials are combined and the number of patients with selected AEs listed for treatment and placebo groups. Where there were several treatment groups, these have been combined. AEs occurring during induction or open-label periods of maintenance trials are listed separately according to availability of information (CHARM67 and CLASSIC II66). There were differences in how trialists reported or grouped together AEs (see notes to Table 42). Where an event was not reported it is possible this was because the event did not occur. Excluding trials from the total count where the event did not occur may lead to an overestimation of the frequency of an AE. Where patients experienced more than one type of AE within a category (e.g. infusion reactions), they will have been counted more than once.
| Event (drug) | Treatment RCT data only |
Placebo RCT data only |
Induction or open-label phase only (CHARM,67 CLASSIC II66) |
|---|---|---|---|
| Deaths (both) | 0.18% (3/1673) | 0.11% (1/918) | NA |
| Deaths (adalimumab) | 0% (0/938) | 0% (0/519) | 0.09% (1/1075) |
| Deaths (infliximab) | 0.41% (3/735) | 0.25% (1/399) | NA |
| AEs leading to withdrawal or discontinuation of treatment (both) | 2.45% (43/1756) | 6.36% (60/943) | NA |
| AEs leading to withdrawal or discontinuation of adalimumab treatment | 3.84% (36/938) | 8.29% (43/519) | 8.65% (93/1075) |
| AEs leading to withdrawal or discontinuation of infliximab treatment | 0.86% (7/818) | 4.01% (17/424) | NA |
| Serious infections (both) | 2.73% (47/1719) | 3.42 (31/907) | NA |
| Serious infections (adalimumab) | 1.71% (16/938) | 2.50% (13/519) | 1.77% (19/1075) |
| Serious infections (infliximab) | 3.97% (31/781) | 4.64% (18/388) | NA |
| TB (both) | 0.23% (3/1323) | 0% (0/707) | NA |
| TB (adalimumab) | 0.21% (2/938) | 0% (0/519) | 0% (0/1075) |
| TB (infliximab) | 0.26% (1/385) | 0% (0/188) | NA |
| Cancer (both) | 0.25% (4/1610) | 0.56% (5/887) | NA |
| Cancer (adalimumab) | 0% (0/938) | 0.39% (2/519) | 0% (0/1075) |
| Cancer (infliximab) | 0.60% (4/672) | 0.82% (3/368) | NA |
| Lupus (-like syndrome) (both) | 0.29% (3/1018) | 0% (0/513) | NA |
| Lupus (-like syndrome) (adalimumab) | 0% (0/421) | 0% (0/258) | 0% (0/221) |
| Lupus (-like syndrome) (infliximab) | 0.50% (3/597) | 0% (0/255) | NA |
| Demyelinating disorders (both) | 0% (0/666) | 0% (0/279) | NA |
| Demyelinating disorders (adalimumab) | 0% (0/554) | 0% (0/279) | 0.09% (1/1075) |
| Demyelinating disorders (infliximab) | 0% (0/112) | NR | NA |
| All infusion reactions (both) | 16.43% (292/1777) | 8.59% (81/943) | NA |
| All infusion reactions (adalimumab) | 17.48% (164/938) | 7.71% (40/519) | 12.74% (137/1075) |
| All infusion reactions (infliximab) | 15.26% (128/839) | 9.67% (41/424) | NA |
| Anaphylactic reaction (both) | 1.79% (2/112) (possible reactions) | NR | NA |
| Anaphylactic reaction (adalimumab) | NR | NR | NR |
| Anaphylactic reaction (infliximab) | 1.79% (2/112) (possible reactions) | NR | NA |
| Study | Infliximab | Adalimumab | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Baldassano et al., 200346 | Hyams et al., 200745 REACH | Present et al., 199962 | Sands et al., 200465 ACCENT II | Targan et al., 199757 | Hanauer et al., 20023 ACCENT I | Rutgeerts et al., 199958 | Hanauer et al., 200663 CLASSIC I | Sandborn et al., 200764 GAIN | Colombel et al., 200767 CHARM | Colombel et al., 200767 CHARM | Sandborn et al., 200766 CLASSIC II | Sandborn et al., 200766 CLASSIC II | |
| Study duration (weeks) | 12 weeks (I, P) | 54 weeks (M, P) | 18 weeks (I, F) | 54 weeks (M, F) | 4–16 weeks (I)a | 54 weeks (M) | 48 weeks (M) | 4 weeks (I) | 4 weeks (I) | 4-week induction period (N = 854) | 56 weeks (M) (N = 778) | 56 weeks (M) | Open-label cohort |
| Deaths – Rx | NR | 0/112 | 0/63 | 0/138 | NR | 3/385 | 0/37 | 0/225 | 0/159 | 1/854 | 0/517 | 0/37 | 0/221 |
| Deaths – placebo | NA | NA | 0/31 | 0/144 | NR | 0/188 | 1/36 | 0/74 | 0/166 | NA | 0/261 | 0/18 | NA |
| AEs leading to withdrawal or discontinuation of treatment – Rx | NR | 12/112 | 1/63 | 5/138 |
2/83 Unclear |
45/385 | 6/37 | 2/225 | 2/159 | 54/854 | 30/517 | 2/37 | 39/221 |
| AEs leading to withdrawal or discontinuation of treatment – placebo | NA | NA | 0/31 | 12/144 |
0/25 Unclear |
5/188 | 0/36 | 2/74 | 4/166 | NA | 35/261 | 2/18 | NA |
| Serious infections – Rx | NR | 9/112 | 3/63 | 4/138 | 1/83 | 14/385 | NR | 2/225 | 0/159 | 10/854 | 14/517 | 0/37 | 9/221 |
| Serious infections – placebo | NA | NA | 0/31 | 9/144 | 1/25 | 8/188 | NR | 0/74 | 4/166 | NA | 9/261 | 0/18 | NA |
| TB – Rx | NR | NR | NR | NR | NR | 1/385 | NR | 0/225 | 0/159 | 0/854 | 2/517 | 0/37 | 0/221 |
| TB – placebo | NA | NA | NR | NR | NR | 0/188 | NR | 0/74 | 0/166 | NA | 0/261 | 0/18 | NA |
| Cancer – Rx | NR | 0/112 | NR | 0/138 | NR | 4/385 | 0/37 |
0/225 Lymphoma |
0/159 | 0/854 | 0/517 |
0/37 Lymphoma |
0/221 Lymphoma |
| Cancer – placebo | NA | NA | NR | 0/144 | NR | 2/188 | 1/36 |
0/74 Lymphoma |
0/166 | NA | 1/261 |
1/18 Lymphoma |
NA |
| Lupus (-like syndrome) – Rx | NR | 0/112 | 0/63 | NR | NR | 2/385 | 1/37 | 0/225 | 0/159 | NR | NR | 0/37 | 0/221 |
| Lupus (-like syndrome) – placebo | NA | NA | 0/31 | NR | NR | 0/188 | 0/36 | 0/74 | 0/166 | NA | NR | 0/18 | NA |
| Demyelinating disorders – Rx | NR | 0/112 | NR | NR | NR | NR | NR | NR | NR | 1/854 | 0/517 | 0/37 | 0/221 |
| Demyelinating disorders – placebo | NA | NA | NR | NR | NR | NR | NR | NR | NR | NA | 0/261 | 0/18 | NA |
| All infusion reactions – Rx | 0/21 | 19/112 | 4/63 | 22/138 | 2/83 | 80/385 | 1/37 | 66/225 | 17/159 |
111/ 854 Injection-site reaction |
80/517 Injection-site reaction | 1/37 | 26/221 |
| All infusion reactions – placebo | NA | NA | 0/31 | 24/144 | 0/25 | 17/188 | 0/36 | 12/74 | 17/166 | NA |
9/261 Injection-site reaction |
2/18 | NA |
| Anaphylactic reaction – Rx | NR |
2/112 Possible reaction |
NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR |
| Anaphylactic reaction – placebo | NA | NA | NR | NR | NR | NR | NR | NR | NR | NA | NR | NR | NA |
Adverse events leading to withdrawal included worsening of CD, infection or obstruction. Serious infections included sepsis, colitis, abscess and pneumonia. Injection site reactions included burning, rash, pain, bruising or irritation, while i.v. infusion reactions included pruritus, chest pain, flushing, dizziness, dyspnoea, injection site irritation and nausea. Very few deaths were reported.
Little difference was found between treatment and placebo groups for the selected AEs. The only cases of tuberculosis and lupus-like syndrome occurred in the treatment groups. AEs leading to withdrawal were slightly higher in the placebo groups and infusion reactions slightly higher in the treatment groups.
Table 42 lists AEs according to trial. It appears that for reporting of AEs, the placebo groups of the maintenance trials also included patients who crossed over to a treatment group. For ACCENT I,2,3 ACCENT II,65 CHARM67 and CLASSIC II,66 crossover was specified as an option for those patients who had a non-response or experienced a disease flare. There were no details regarding potential crossovers from placebo to treatment in Rutgeerts et al. 58 (n = 73). See section on quality for details on number of crossovers from placebo groups (see Quality assessment sections and Appendix 12).
Crossover to treatment may have had an effect on the types and numbers of AEs reported in the placebo groups; for example an increase of those types of AEs associated with the treatment (e.g. infection) and/or an underestimate of AEs associated with no treatment (e.g. worsening of CD). It should be noted that in the maintenance trials, all patients (including those subsequently randomised to placebo) initially received the study drug during the induction phase; the effects of this may have carried over into the placebo phase of the RCT.
None of the maintenance trials reported AEs for patients according to whether they had ever or never received the treatment during the RCT phase of the study. As some of the AEs reported are very rare, it is possible that any differences between treatment and placebo groups are due to chance.
Development of antibodies
This section describes all included studies. Table 43 lists numbers of patients developing antibodies to anti-TNF agents, nuclear antibodies and antibodies to double-stranded deoxyribonucleic acid (DNA). Most (10/11) studies reported the development of antibodies to the respective anti-TNF agent;2,3,45,46,57,58,62–66 four studies2,3,45,65,66 reported anti-nuclear antibodies and eight studies2,3,45,46,57,58,62,65,66 reported anti-double-stranded DNA antibodies.
| % evaluated Abs | Patients with Abs to anti-TNF agent | Patients with anti-nuclear Abs | Patients with Abs to double-stranded DNA | |
|---|---|---|---|---|
| Adalimumab | ||||
|
Hanauer et al. , 2006 CLASSIC I63 INDUCTION |
NR |
PLAC: 1/74 (1.4%) +ve (assumed n) ADA: 1/225 (0.4%) +ve (assumed n) |
NR | NR |
|
Sandborn et al. , 2007 GAIN64 INDUCTION |
Appears to be measured in all |
PLAC: 0/166 (0%) +ve ADA: 0/159 (0%) +ve Presence of measurable ADA precluded determination of Abs in most patients treated with ADA |
NR | NR |
|
Sandborn et al. , 2007 CLASSIC II66 MAINTENANCE |
269/276 (97.5%; includes 221 from open-label cohort) for anti-ADA Abs 185/276 (67.0%; includes 221 from open-label cohort) for anti-nuclear and anti-DNA Abs |
7/269 (2.6%) +ve All groups including open-label cohort |
36/185 (19.5%) +ve At baseline and/or at final visit |
33/185 (17.8%) +ve At baseline and/or at final visit |
|
Colombel et al. , 2007 CHARM67 MAINTENANCE |
Not measured | Not measured | Not measured | Not measured |
| Infliximab | ||||
|
Baldassano et al. , 200346 INDUCTION CHILDREN |
21/21 (100%) NB no PLAC group |
0/21 (0%) +ve | NR | 0/21 (0%) +ve |
|
Hyams et al. , 2007 REACH45 MAINTENANCE CHILDREN |
105/112 (93.8%) for Abs to INF 91/112 (81.3%) for anti-nuclear Abs 99/112 (88.4%) for anti-DNA Abs Note that no PLAC group; includes patients who were not randomised to maintenance therapy |
3/105 (2.9%) +ve 21/105 (20.0%) –ve 81/105 (77.1%) inconclusive sample (detectable INF concentration) |
23/91 (25.3%) +ve | 7/99 (7.1%) +ve |
|
Present et al. , 199962 INDUCTION FISTULISING |
92/94 (97.9%) for Abs to INF Unclear for anti-DNA Abs (appears all in INF groups only) |
3/92 (3.3%) +ve 13/92 (14.1%) inconclusive sample INF and PLAC groups |
NR |
8/63 (12.7%) +ve INF groups only |
|
Sands et al. , 2004 ACCENT II65 MAINTENANCE FISTULISING |
258/282 (91.5%) for Abs to INF 254/282 (90.1%) for anti-nuclear Abs 243/282 (86.2%) for anti-DNA Abs |
44/258 (17.1%) +ve 80/258 (31.0%) –ve 134/258 (51.9%) inconclusive sample Not detailed by group |
INF: 56/122 (45.9%) +ve PLAC: 24/132 (18.2%) +ve |
INF: 27/116 (23.3%) +ve PLAC: 8/127 (6.3%) +ve |
|
Targan et al. , 199757 INDUCTION |
101/108 (93.5%) for Abs to INF 98/108 (90.1%) for anti-DNA Abs Note that samples include post-RCT open-label patients |
6/101 (5.9%) +ve who received INF blinded or as open label Note that in 2/3 INF patients, INF was still detectable and may have interfered with assay |
NR | 3/98 (3.1%) +ve who received INF blinded or as open label |
|
Hanauer et al. , 2002 and Rutgeerts et al. , 2004 ACCENT I2,3 MAINTENANCE |
442/573 (77.1%) for Abs to INF Note that number with Abs reported according to actual treatment received bearing in mind that patients crossed over |
PLAC: 41/442 (9.3%) +ve INF: 23/442 (5.2%) +ve In 46% of patients INF still detectable therefore inconclusive |
PLAC: 63/180 (35%) +ve INF groups: 363/648 (56%) +ve |
PLAC: 19/173 (11%) +ve INF groups: 123/362 (34%) +ve |
|
Rutgeerts et al. ,58 1999 MAINTENANCE |
71/73 (97%) |
7/47 total (14.9%) +ve PLAC: 5/NR INF: 2/NR 24/71 (33.8%) inconclusive as measurable INF in sample |
NR |
2/47 total (4.3%) +ve PLAC: 0/ND INF: 2/ND |
Five induction trials reported the proportion of patients with antibodies to an anti-TNF agent;46,57,62–64 these ranged from 0% to 6% (adalimumab: 0%, 1.3%; infliximab: 0%, 3.3%, 6%). All reported antibody development either for the intervention group only, or split by placebo and intervention group, except Present et al. ,62 which reported antibodies for placebo and intervention group together. Targan et al. 57 included patients from the post-RCT open-label extension. This was also the longest follow-up study among induction trials (16 weeks) and had the highest level of antibodies (6%).
Five maintenance trials reported antibodies to an anti-TNF agent;2,3,45,58,65,66 these ranged from 2.6% to 17% (infliximab: 2.9% to 17%; adalimumab: 2.6%). All patients were exposed to anti-TNF during induction. A patient’s subsequent exposure was variable according to randomisation group and crossover to active intervention or escalated dosage regimen. Three studies reported antibodies for the intervention and placebo groups together (ACCENT II,65 Rutgeerts et al. 58 and CLASSIC II66). The majority of patients in CLASSIC II66 came from the open-label cohort component of the study. The lowest antibody levels occurred in CLASSIC II66 (adalimumab); the other large adalimumab maintenance trial (CHARM67) did not measure antibodies.
Seven studies listed the proportion of inconclusive samples,2,3,45,57,58,62,64,65 which were generally high and ranged from 14% to ‘most’ patients. These samples had detectable concentrations of anti-TNF agent, which could compete for the detection of antibodies to the anti-TNF agent in the immunoassay used, and would therefore not give a valid result. It is unclear whether the overall percentages of antibodies to the anti-TNF agent would have been different if they could have been measured in all patients.
As with the AEs described above, it should be noted that patients in the placebo groups of the maintenance trials would have all received the treatment as part of induction and may also have crossed over to a treatment group during later stages of the trials.
The proportions of anti-nuclear antibodies were variable: 25% in REACH45 (infliximab), 18%/46% [active treatment (anti-TNF) group (Rx)/placebo] in ACCENT II65 (infliximab), 35%/56% (Rx/placebo) in ACCENT I2,3 (infliximab) and 19% in CLASSIC II66 (adalimumab).
Antibodies to double-stranded DNA were measured in three infliximab induction trials46,57,62 (range 0%–13%) and four maintenance trials2,3,45,58,65 (range 4%–34%); only one adalimumab trial (CLASSIC II,66 19%) measured this parameter.
Given the proportion of missing data (inconclusive samples), the varying numbers of patients receiving treatment in different trials (those who crossed over) and the relatively small number of trials, it is not possible to conclude that one of the interventions is more or less likely to result in the development of antibodies to the anti-TNF agent. Whether different types of assays were used for the detection of antibodies or whether there were differences in the number of frequency of assessments, which could have led to differences between studies or drugs, was not investigated.
Based on the results for all patients (responders and non-responders) from the ACCENT I trial,2 it appeared that scheduled treatment led to the formation of fewer antibodies to infliximab than ‘episodic’ treatment (28% in placebo/episodic treatment arm, 9% in 5 mg/kg scheduled arm and 6% in 10 mg/kg scheduled arm). It should be noted that the comparison between ‘episodic’ and scheduled treatment is not a randomised one (see Quality assessmentof ACCENT 1). Given that the ‘episodic’ group included patients who crossed over from the scheduled treatment groups and the fact that 46% of total samples were inconclusive, it is unclear how robust these results are.
Safety issues; rare serious adverse events
Information extracted from the RCTs included for review of clinical effectiveness provides little long-term evidence about safety. Anti-TNF therapies have now been licensed for multiple indications and data about rare serious AEs have gradually accumulated. In this section the relevant safety issues following from these data are briefly reviewed.
Bongartz et al. 77 meta-analysed rates of malignancy and of serious infection reported in placebo controlled RCTs of infliximab and adalimumab in rheumatoid arthritis. Information in published papers and from the US FDA website was used for the analysis. Odds ratios (anti-TNF versus placebo) for malignancy and infection were 3.3 (95% CI 1.2 to 9.1) and 2 (95% CI 1.2 to 3.1) respectively, and the numbers needed to harm were 154 and 59 respectively over a treatment period of 3–12 months. Higher drug doses were associated with greater risk. Similar results were reported in a meta-analysis conducted by Shoor. 78
Tumour necrosis factor-α has an important role in the host immune response to Mycobacterium tuberculosis and in the immunopathology of tuberculosis. 79 Patients to be treated with anti-TNF agents should be screened for tuberculosis before starting anti-TNF therapy, they should be monitored for tuberculosis during therapy and those with latent tuberculosis should be appropriately treated prior to initiation of anti-TNF therapy. A 2007 publication (Raval et al. 79) detailed 130 infliximab-associated cases of tuberculosis spontaneously reported to the US FDA between 1 November 2001 and 30 May 2006. In 45% of cases there was extrapulmonary disease. In a subset of 67 cases notified after the addition of a tuberculosis warning to the boxed medication it was noted that in six instances no test had been performed and that of 47 tuberculin skin tests performed, 34 gave a negative result. The false-negative rate was unknown. These results emphasise the requirement for vigilance by physicians caring for patients treated or about to be treated with anti-TNF therapies.
Ramos-Casals et al. 80 identified 233 cases of autoimmune disease apparently associated with anti-TNF therapies. Of these, 17 occurred in CD patients. Anti-TNF agents infliximab, adalimumab and etanercept were associated with various autoimmune manifestations including lupus, vasculitis and interstitial lung diseases. Overall incidence rates or rates for individual anti-TNF agents are unknown.
Elevated TNF-α is associated with heart failure and its level is correlated with severity of heart failure. Case reports (n = 47) reviewed by Kwon et al. 81 indicate that anti-TNF therapy might trigger new onset heart failure in a subset of patients and might exacerbate the condition of some patients. The SPCs carry warning of this potential risk.
Treatment with monoclonal antibodies has been associated with potentially fatal induction of progressive multifocal leuco-encephalopathy. A 2008 systematic review of primary data by Socal et al. 82 identified 29 cases most of which (n = 23) were associated with rituximab therapy which depletes the B-cell population. The single instance associated with anti-TNF treatment was reported for a 74-year-old woman given etanercept for rheumatoid arthritis. 83
Discussion of results and assessment of effectiveness
Patient heterogeneity
Patient heterogeneity may affect results across different trials. The inclusion criteria of the trials specified a CDAI score between 220 and 400 or 450. The inclusion of patients already at a CDAI level close to remission could have improved the remission rates found. However, if patients already had a low CDAI count, achieving a reduction of 70 or 100 points would have been harder to achieve. The opposite would be more likely to be true for patients with very high initial CDAI scores. Therefore, it is unlikely that the initial wide spread of CDAI scores would have much impact on the results unless there were more patients at one end of the spread than the other. Mean CDAI scores at entry did not vary greatly between trials, so it appears unlikely that patient populations taken as a whole differed substantially between trials with respect to this parameter. Nevertheless populations probably did differ between trials as the placebo rates were heterogeneous. The corollary is that CDAI is not necessarily a reliable indicator of the seriousness of disease or of its likely progression. The CDAI score is a summary score and patients can achieve the same score yet have problems with very different aspects of their disease. Similarly, if a patient had a reduction of 70 points, that could be achieved in a variety of different ways. It is also uncertain whether a reduction of 70 or 100 points means the same in terms of reduction of disease severity for patients starting at different ends of the severity spectrum.
Cohort studies (e.g. Munkholm et al. 25) demonstrate that most CD patients, at some time in their disease history, experience ‘highly active disease’ and that they cycle between highly active and quiescent periods of varying durations. Whether CD is severely debilitating for an individual depends to some extent on the frequency with which the episodes of highly active disease are repeated. Cohort studies show that this varies between patients. For these reasons a patient’s CDAI score at a particular time, such as at recruitment into a trial, is not a good indicator for the likely duration of that level of disease activity or of the likely subsequent recurrence of highly active disease.
The licence indications for infliximab and adalimumab specify ‘severe’ CD but do not define how severe disease may be determined. It has been assumed that this is a CDAI score of ≥ 300. Trials have recruited patients having ‘moderate-to-severe CD’ defined according to CDAI scores of between 220 and 450, or 220 and 400; it is therefore unclear to what extent these populations fully reflect the intended licensed population.
Induction trials – placebo rates
CD is a chronic relapsing and remitting disease. Induction trials selected patients in relapse. On average, irrespective of treatment, relapsed patients will tend to improve, i.e. remit with time (their CDAI scores will reduce as they regress to the mean). This tendency would be reflected in relatively high rates of improvement in placebo groups in placebo-controlled trials and also in variation in these rates dependent on the relapse–remission cycling characteristics of each of the patients enrolled in the different trials.
The rates of response (reduction in CDAI of 70 or 100 points) and of remission in the placebo arms of the included induction trials varied from trial to trial and in some trials reached high levels (see Appendix 13 for details). Except in the Targan et al. 57 trial of infliximab, by week 4 one-third or more of placebo patients had already achieved the least stringent measure of improvement (response 70). Similarly, at least 20%–25% achieved response 100 by week 4. Varied and high rates of placebo response have previously been documented for many CD intervention trials (Su et al. 84). For dichotomous outcomes, variable placebo rates can profoundly influence effect size values such as risk difference and risk ratio. Thus placebo and intervention rates in two trials of 10% and 20% respectively in one and 30% and 40% respectively in the other generate identical outcome measures for risk difference (0.1 or 10%) but considerably different measures for risk ratio (2.0 and 1.3 respectively). For this reason, both placebo and intervention rates and both risk difference and risk ratio effect sizes have been presented in this report for most outcomes in the results section. The CIs quoted were not adjusted for repeated measures.
These high and varied placebo rates probably result from three influences: the tendency for CDAI scores to regress to the mean; a placebo effect; and possibly the effect of concomitant treatments allowed in the trials. The variation in placebo rates makes comparisons between trials problematic and indicates that CDAI scores alone are unlikely to be good prognostic indicators. Although recruited populations in the trials conformed to similar ranges, means or medians of CDAI score, they are likely to be clinically dissimilar.
Induction trials – effect sizes
By week 4 all induction trials, except for CLASSIC I63 at the lower dose level for adalimumab (80/40 mg/kg weeks 0 and 2), exhibited statistically significant effect sizes for anti-TNF relative to placebo for remission and response, irrespective of whether these were measured in terms of risk difference or risk ratio. The trial of infliximab by Targan et al. 57 was remarkable in that the effect sizes observed were much greater than those seen in the other trials; placebo rates were notably lower in Targan et al. 57 than in any other trials. Targan et al. 57 was the earliest anti-TNF induction RCT and was a relatively small trial, so the point estimates of effectiveness were associated with more uncertainty than was the case for the larger induction trial of adalimumab (GAIN64). Since the publication of Targan et al. ,57 no infliximab induction trial has been reported that can provide confirmatory evidence for the large effect size point estimates from the Targan et al. 57 trial. The response 70 rate at 4 weeks in the intervention arm of Targan et al. 57 was 81%. In the induction phase of the ACCENT I2,3 maintenance trial of infliximab, the response 70 rate at week 4 was considerably less at 59%. ACCENT I2,3 patients were administered the same dose at week 0 and patient baseline characteristics were similar to Targan et al. ,57 e.g. very similar CDAI and IBDQ scores. The contribution of infliximab to the initial 59% response 70 rate in ACCENT I2,3 cannot be gauged because of lack of an appropriate control group.
The follow-up in the published adalimumab trial reports was to 4 weeks only, and there is no reliable evidence on the effectiveness of induction with adalimumab beyond this time period. Targan et al. 57 provided data on infliximab for some patients up to 16 weeks (4 weeks’ induction + 12 weeks’ open label).
Maintenance trials – general comments on trial design
The maintenance trials conformed to what have been called ‘adaptive’ trial designs. The main features of such designs have been reviewed by Chang and Chow. 85 ACCENT I,2,3 CHARM67 and CLASSIC II66 trials had adaptive trial design of the type described as ‘drop-the-loser’ with in some cases ‘adaptive treatment switching’. 85 An inherent problem of ‘drop-the-loser’ design is that groups that are dropped may contain valuable information regarding the response to treatment under study. A further problem concerns how such studies should be powered; whether for the interim analysis at the point when ‘losers’ are dropped, or for the final analysis involving winners only. With treatment switching come problems of identifying the target population for the therapy of interest and a precise definition of the therapy provided. Treatment switching can lead to a change to a different hypothesis being tested. Chang and Chow state ‘From a statistical point of view adaptations to trial and or statistical procedures could (i) introduce bias/variation to data collection, (ii) result in a shift in location and scale of the target population, (iii) lead to inconsistency between the hypothesis being tested and the corresponding statistical tests’. 85 In summary, these trials are susceptible to difficulties of analysis and interpretation.
Maintenance trials in adult populations wholly or predominantly of non-fistulising patients
For each drug, one large maintenance trial has been published that employed within-licence treatment regimens: the CHARM67 trial (adalimumab) and the ACCENT I2,3 trial (infliximab). The interpretation of results from the maintenance trials was hampered by the nature of the trial designs, most of which allowed for scheduled crossovers into other treatment arms (or to ‘open-label treatment’). This led to a proportion of patients in the placebo arms of the trials receiving variable amounts of drug. In order to comply with an ITT analysis, these patients (and those who withdrew) were mainly counted as treatment failures for binary outcomes such as remission or response. Not all trials clearly defined the handling of missing data or data for patients who crossed over. Where there were missing continuous data, the last observation carried forward was used in ACCENT I2,3 but not in CHARM,67 the effect of which on results is unclear.
There were particular concerns over the ACCENT I trial (Rutgeerts et al. 2 publication), as its stated aim of comparing episodic with scheduled treatment is misleading as no patients were randomised to an episodic treatment arm. Proper comparison of two strategies has been implemented by Breban et al. 86 who randomised patients with ankylosing spondylitis to induction with infliximab followed by continuous treatment or followed by treatment adapted to symptom recurrence. Similarly, Menter et al. 70 conducted an unbiased comparison of continuous and intermittent infliximab strategies for psoriasis by re-randomising at the start of the compared strategies (week 14) patients who had initially been randomised (week 0) to different infliximab induction regimens. There were also uncertainties regarding the impact of methods for handling of missing data in the analysis including both responders and non-responders.
Responder/non-responder subgroups
The interpretation of the maintenance trials was further complicated by the fact that a subgroup of patients (responders) were selected for analysis or randomisation at varying time points after an induction period during which all patients received the study drug. For both of the large maintenance trials of within-licence treatment regimens (CHARM67 – adalimumab; ACCENT I2,3 – infliximab) the published effectiveness results all focused on the ‘responders’ subgroup. Separate results for non-responders were not reported (see Table 40), although both CHARM67 and ACCENT I2,3 randomised both responder and non-responder patients. The definition of responders differed somewhat between the two trials. Furthermore, the induction phases used in both trials differed with respect to duration and number of induction doses administered. The consequence of these considerations is that attempting any comparisons of effectiveness between the trials is very problematic. The proportion of patients categorised as responders in each of these trials was 64% for CHARM67 and 58% for ACCENT I. 2,3
It is known from trials where results were also reported for (randomised) non-responders that initial non-responders can still respond later, so it is unclear which patients this subgroup of responders actually represents in clinical practice. It is possible that a subgroup of responders chosen at a different time point would have led to different results. There is no published evidence or information in the manufacturers’ submissions to show that compared with non-responders, responders benefit more from the treatment (compared with placebo). The selection of responders at different time points in different trials also hampers any comparisons between the trials.
Reporting effectiveness results for a subgroup but not for all randomised patients (or not for all patients who commenced treatment) appears at odds with usual practice. For example, in placebo-controlled randomised trials of anti-TNF agents (infliximab, adalimumab and etanercept) for the treatment of rheumatoid arthritis, results for all patients have been analysed and presented. 87 Dichotomising patients into responders and non-responders makes clinical sense only if a ‘response’ at the time of the dichotomisation is a good prognostic tool for identifying those patients most likely to benefit from maintenance of treatment. In order to find this out, the comparison of results for responders and non-responders is required, which, unfortunately, is the precise analysis that was not undertaken in these trials. Thus there is no evidence available to indicate that subgrouping patients in the ways described is a useful practice. The usefulness of the results reported for responders only is therefore questionable.
The ACCENT I2,3 trial dichotomised patients according to their response at 2 weeks after the induction infusion of infliximab. The decision to do this may have been derived from previous research. The 1997 induction study of Targan et al. 57 provided data up to 4 weeks after a single infusion of infliximab at 5 mg/kg. This study reported that the mean CDAI score in the placebo group remained constant from weeks 2 to 4, while the risk differences (infliximab vs placebo) for remission (score < 150 points) and for a 70-point reduction in CDAI score increased from 0.37 to 0.44 and from 0.62 to 0.65 respectively. Placebo rates for these outcomes remained constant from weeks 2 to 4. Although the study was small and the point estimates were associated with considerable uncertainty, these results imply that some patients not responding at 2 weeks do in fact go on to respond at a later time. In ACCENT I2,3 (CiC information has been removed).
In the absence of appropriate analyses it appears that dichotomising patients as early as 2 weeks after a single infliximab infusion is probably premature and does not appear to be a clinically meaningful procedure.
In the CHARM trial67 the time chosen for categorisation into responders and non-responders (at week 4) was not based on efficacy data but was ‘based on pharmacokinetic model estimates for when maximal drug concentrations should be present’. CiC results were available for non-responder patients at weeks 12, 26 and 56, so that it was possible to calculate the response rates among all randomised patients. The risk difference and risk ratio results for remission, response 100 and response 70 are summarised in Figures 31 and 32 respectively. (CiC information has been removed.)
FIGURE 31.
(CiC information has been removed.)
FIGURE 32.
(CiC information has been removed.)
The two large maintenance trials (CHARM67 and ACCENT I2,3) provided evidence that for the subgroups of patients defined as ‘responders’, anti-TNF therapy was more beneficial than placebo with respect to the proportions of patients exhibiting remission or response (70 or 100). Rates at multiple follow-up times extending to week 56 for CHARM67 (remission rates) were reported in the published paper. For the ACCENT I2,3 trial, results for weeks 30 and 54 (response 70 and remission) only were published, but CiC information for multiple time points was provided. The higher rates of response for intervention versus placebo might lead to the conclusion that extended therapy over the prolonged follow-up is beneficial and/or necessary for maintenance of response. However, examination of all the available information indicates that nearly all benefit observed for intervention over placebo was generated early on and that risk differences thereafter remained relatively stable or decreased. These results imply that prolonged treatment after the initial benefit has been attained is uneconomical and, as anti-TNF agents are associated with significant health risks, may be clinically ill-advised. The dose regimens required to attain this early benefit are likely to be different for adalimumab and infliximab.
The published results for the CHARM67 trial graph ‘% patients maintaining remission’ (Figure 2B in CHARM67 publication) versus follow-up time depicted increased rates of remission following after decreased remission rates, demonstrating that patients who achieved remission at late follow-up times are counted as ‘maintaining remission’ and that in fact, the point prevalence of remission is represented in the graph rather than maintenance of individual patients in remission. If this is the case, a late time point (e.g. 30 or 54 weeks) value reported does not necessarily inform about maintenance of response during follow-up as it is possible that those registered as ‘in response’ may only have achieved this status just prior to the time point reported. It was unclear, but appeared possible, that point prevalence of response was the statistic reported in the ACCENT I2,3 published reports; however, (CiC information has been removed).
The most appropriate way to determine the ability of anti-TNF agents to maintain response in patients who were defined as responders is time-to-event analysis with statistical comparison using a log rank test. For ACCENT I,2,3 median time to treatment failure was 19 weeks and 38 weeks for placebo and 5 mg/kg infliximab groups respectively (p = 0.002); however, the definition of treatment failure used in this analysis was complex, did not correspond to a loss of response 70 status, and its clinical impact was difficult to gauge. The CHARM67 trial (adalimumab) reported median duration of remission for those responders who achieved remission starting at any time during follow-up. The median times were 127 days for the placebo group, 378 days for the 40 mg/kg adalimumab e.o.w. group and > 392 days for the 40 mg/kg weekly dosage regimen group.
Trials recruiting patients with draining fistula
One induction trial provided evidence that infliximab promotes fistula closure to a greater extent than placebo. The ACCENT II65 trial of infliximab maintenance treatment (IMT) focused on responders (69% of patients receiving induction doses). IMT promoted closure of fistulas to a statistically significantly greater extent than did placebo. There was evidence that a reduction of dose frequency from every 4 weeks to every 8 weeks was associated with poorer maintenance of fistula closure. Limited evidence from the CHARM67 trial suggested that adalimumab may also promote fistula closure.
It is possible that fistula closure may not necessarily be a desirable outcome as it may result in increased development of abscesses. A post hoc analysis of patients participating in the ACCENT II65 trial found no significant difference in abscess incidence between two groups receiving different mean dosages of infliximab. Interpretation of these results is problematic because results for the most appropriate comparison (placebo vs infliximab) were not available.
Trials recruiting paediatric patients
Two trials of infliximab, one induction and the other maintenance, reported on the treatment of paediatric patients. Unfortunately, in these trials all patients received infliximab, and no reliable inferences regarding the effectiveness of the intervention were possible because the spontaneous response rates in the population were unknown. The more frequent of the two dosage regimens used in the REACH45 trial (5 mg/kg every 8 or 12 weeks) resulted in statistically significant greater rates of response and remission, a dose response relationship that likely implies beneficial effect of infliximab relative to placebo or standard treatment, but a placebo-controlled trial would have provided far stronger evidence of effectiveness.
Differences in effectiveness of anti-TNF agents; indirect comparisons
No head-to-head trials were found that compared the effectiveness of adalimumab and infliximab. However, the existence of placebo-controlled induction and maintenance trials for both drugs means that adjusted indirect comparisons of effectiveness were theoretically feasible using methods described by Glenny et al. 74
The indirect comparison of trials was hampered in this case by a number of factors. One of these was differing placebo rates found for induction trials and unknown or uncertain placebo rates in maintenance trials (because all patients receive active intervention early in the trial). Patients with CD can experience spontaneous clinical improvement without treatment. Su et al. 84 conducted a meta-analysis of CD trials looking at placebo rates for remission and response (based on the CDAI). The authors found substantial heterogeneity between placebo rates and found that these were in the main attributable to follow-up duration, number of follow-up visits and CDAI score at entry to the trial (see Appendix 13). Because of the variation in placebo rates in the induction trials, indirect comparison was not made.
Indirect comparison of effectiveness using maintenance trials was judged unlikely to deliver valid results. For responders, the placebo arms of compared trials were not truly comparable because the groups had received different anti-TNF induction drugs on differing numbers of occasions and for different periods of time; furthermore, the ‘responder’ groups consisted of different proportions of the randomised populations according to differing criteria. For all patients’ results again placebo groups were not truly similar between trials and, additionally, availability of results for all patients in the adalimumab trials was limited (see Appendix 11); furthermore, the permitted crossover of variable proportions of placebo group patients to active intervention at weeks 12 and 14 of the CHARM67 and ACCENT I2,3 trials would render analyses unreliable.
Adverse events
The large number of crossovers in the trials made the comparison of AE rates between treatment and placebo arms difficult, as many patients in the placebo arms will have received some study drug. In addition, the maintenance trials either gave an induction bolus of the drug at the start of the trial then randomised to treatment or placebo, or enrolled patients from a previous induction trial. Similarly, it is difficult to tell what the true rates for the development of antibodies are for each of the drugs, again due to crossovers and induction doses. It was not within the remit of this project to examine the test accuracy of antibody determination used in the trials. Increased risk of malignancies and of infection is evidenced from published information about anti-TNF agents used in all their indications. Vigilance with respect to tuberculosis and patients with potential or suspected heart failure should be mandatory.
Summary of effectiveness results
There were no included RCTs with severe CD patients only. They all included moderate-to-severe CD.
-
The general pattern of results is similar for the two drugs.
-
There is a good initial, clinically significant, improvement for the majority of patients when given induction treatment with infliximab or adalimumab. The short duration induction trials demonstrated that the majority of CD patients with moderate-to-severe disease gained clinical benefit from a single i.v. infusion of infliximab (5 or 10 mg/kg) or two subcutaneous injections of adalimumab (80 mg and 40 mg or 160 mg and 80 mg) separated by 2 weeks. Published estimates for the proportion benefiting depended on the measure of clinical response employed and were associated with considerable uncertainty (e.g. 95% lower CI to upper CI ranged from 13% to 66% and 16% to 47% for remission at week 4 from infliximab and adalimumab respectively depending on dose and trial). Obtaining a valid estimate of effectiveness for the two drugs and for their relative efficacy was plagued with difficulties contingent on the small number of trials, their small size, differences between the populations examined, and uncertainties concerning the most appropriate induction regimen to be used and the imprecision of trial results.
-
Although there exists a core of responders of indeterminate size who maintain an anti-TNF dependent response, in general the initial good response is not well maintained with extended treatment. This is evidenced in three ways:
-
The percentage of patients in response (or with remission) fades away after the first weeks or so of maintenance therapy.
-
Large numbers of patients drop out of treatment. In ACCENT I,2,3 34% of patients dropped out, some before dose escalation, some after; in the active treatment arms, 25% withdrew in ACCENT I2,3 and (CiC information has been removed)% in CHARM. 67 Among responders in CHARM,67 about (CiC information has been removed)% withdrew from active treatment.
-
Large numbers of patients required dose escalation and/or transfer to open label (CHARM67) because of worsening disease. In ACCENT I,2,3 68% in the 5 mg/kg arm required dose escalation, as did 49% of those in the higher dose arm. In CHARM67 about 30% in the adalimumab arms crossed over to escalated dose or open-label therapy.
-
These results indicate that during extended treatment an appreciable proportion of patients decide there is an unsatisfactory balance between the actual benefit of anti-TNF and its perceived benefits. The withdrawal rates in these trials are not similar to other monoclonal antibody interventions and contrast with the > 90% compliance over 52 weeks observed for i.v. weekly infused eculizumab. 88 The high requirement for dose escalation reflects efforts to resuscitate a fading response; the continuing dropout rate after escalation shows that these efforts meet with limited success.
Conclusions
Evidence from at least one induction and one maintenance trial for each drug administered within the licensed dose regimen demonstrates that for selected patients, relative to SC, these anti-TNF agents (infliximab and adalimumab) deliver statistically significant benefits of disease remission and improvement based on CDAI binary measures. Remission, response 70 and response 100 rates measured in maintenance trials indicate that for ‘responders’ nearly all benefit is achieved in about the first 12 weeks of treatment. Thereafter risk differences (anti-TNF minus placebo) remain relatively stable. These results imply that a short burst of treatment is likely to be more clinically effective and cost-effective than prolonged treatment and that after about 12 weeks the likelihood the intervention will be clinically effective and cost-effective will steadily diminish as treatment is extended unless other favourable outcomes additional to those based on CDAI measures are delivered later than 10–12 weeks.
The recruitment of patients who may not have failed alternative treatments together with the selective reporting of outcomes for early responders in the maintenance trials means it is difficult to gauge the effectiveness of these drugs in maintaining favourable outcome among the whole patient population with moderate-to-severe CD who are resistant to other treatments. Because of inappropriate study designs, heterogeneity of patients, incomplete and/or selective reporting of outcomes and lack of head-to-head trials, no convincing objective evidence was available to indicate whether one drug was superior to another either in respect to effectiveness or to safety.
Chapter 4 Assessment of cost-effectiveness
Systematic review of existing cost-effectiveness evidence
Introduction
This chapter explores the published cost-effectiveness literature on the costs and benefits of TNF-α inhibitors. Within the UK, the licensed anti-TNF treatments are adalimumab and infliximab. The following section goes on to describe the results of a systematic literature review of these treatments for CD.
When assessing the economic impact of CD, costs can be divided into direct costs and indirect costs. Direct costs refer to the costs of an intervention itself and include the value of all resources consumed in the provision of an intervention, including all side effects occurring as a result of treatment, and all future health-care expenditures contingent on either the intervention or side effects. Direct costs include all goods, services and other resources used, both within and outside of the health-care sector. Health-care costs include all medication, diagnostic tests, supplies, staff and medical facility costs. Costs outside health care can include the costs to the patients and to other public agencies. Indirect costs include those resources consumed that are not directly paid for by any party. As the ability of patients to work is related to general health and the time spent in treatment, indirect costs include productivity gains and losses. Indirect costs also include the productivity costs of unpaid carers including family members.
In CD, the perspective taken may have a significant impact on the costs associated with the disease and the overall conclusions drawn from the evidence. Several perspectives can be adopted and the NICE reference case recommends concentrating only on the direct health-care and public social service (PSS) costs. A researcher may also wish to consider a societal perspective that includes direct and indirect costs to all parties as a sensitivity analysis in an economic evaluation.
Methods for reviewing cost-effectiveness
Search strategy
A comprehensive literature search on the cost and cost-effectiveness of adalimumab, infliximab, certolizumab pegol and natalizumab for the treatment of CD from a UK perspective was conducted. Natalizumab and certolizumab pegol were originally part of this technology appraisal so were included in the searches; they were subsequently dropped from the appraisal after completion of searches.
Studies on costs, QoL, cost-effectiveness and modelling were identified from the following sources:
-
bibliographic databases: MEDLINE (Ovid) 2000 to May/June 2007, EMBASE (Ovid) 2000 to May/June 2007, Cochrane Library [NHS Economic Evaluation Database and DARE (Database of Abstracts of Reviews of Effects)] 2007 Issue 2, and HEED (Health Economic Evaluations Database) (June 2007)
-
industry submissions
-
internet sites of national economic units.
Searches were not limited by language. Full search strategies can be found in Appendix 14.
In addition, searches for cohort studies of infliximab for CD and also clinical guidelines for CD were undertaken for background information for the decision analytic model. Full details can be found in Appendix 14.
Inclusion and exclusion criteria
Only studies meeting the following criteria were included:
-
Study design: fully published economic evaluations only (abstracts without full publication were excluded).
-
Population: patients with CD (adults or children).
-
Intervention: adalimumab or infliximab (any dosage/treatment regimen).
-
Comparator: conventional treatment without TNF-α inhibitors including no treatment, placebo, dietary intervention, drug treatment with aminosalicylates, methotrexate, corticosteroids (prednisolone, budesonide and hydrocortisone), azathioprine, metronidazole or surgical intervention.
-
Outcomes: cost–utility, cost-effectiveness.
Inclusion, quality assessment and data extraction strategies
Two reviewers independently reviewed studies for inclusion using title and abstract. Disagreements were resolved by discussion. After this initial sift, full papers were obtained and assessed for inclusion. All studies were quality assessed using a standard checklist (Drummond and Jefferson89) by two independent reviewers. If a substantial part of the economic evaluation was missing because of the material being commercially in confidence, formal quality assessment methods were not used. Data extraction of included studies was performed by one reviewer, extracted data was then checked by a second reviewer.
Results of systematic review of existing cost-effectiveness studies
Inclusion and exclusion of studies
Using the search strategy and previous knowledge of the literature, an initial 814 papers were identified. Initial sifting identified 64 articles for further investigation. These articles identified seven further papers that could have provided relevant information for economic modelling but were not included in the systematic review as they were not cost-effectiveness studies.
Quantity and quality of included studies
Only four full papers met the review criteria and were subsequently reviewed. These were Arseneau et al. 90 from the USA, Jaisson-Hot et al. 7 from France, Marshall et al. 6 from Canada and Clarke et al. 5 from the UK. A further two papers were available in abstract form only. Given the difficulty of extracting reliable information from this format, these were not formally reviewed. Several excluded papers provided either the costs or benefits of treatment but not both.
None of the four papers declared any conflicts of interest. Two of the four papers were peer-reviewed published works by independent researchers,7,90 one was commissioned by the Canadian Collaborating Centre for Health Technology Assessment (CCHTA),6 and one was a Health Technology Assessment (HTA) report from the UK5 commissioned by NICE. Given the restrictions on CiC information in HTA reports, the UK HTA was not quality assessed using the checklist.
Of the three quality-assessed studies, the CCHTA report6 scored highly in comparison to the remaining two papers, and was both clearly written and transparent. These remaining two papers7,90 omitted several key features (including an incremental analysis of all comparators), and resource usage was outlined in cost terms only.
Characteristics of economic studies
All four studies conducted cost–utility analyses of infliximab and were reported in a total of five papers. 5–7,90,91 The HTA study5 considered non-fistulising and fistulising disease, Marshall et al. 6 and Jaisson-Hot et al. 7 considered non-fistulising disease only, and Arseneau et al. 90 considered fistulising disease only. No published economic studies were found for adalimumab in CD.
Within economic analyses of infliximab in CD, a lack of relevant observational data has led to the widespread use of information from a relevant study conducted in Olmstead County, and in particular the model constructed in Silverstein et al. 26 Using 24 years of data, Silverstein et al. 26 constructed a Markov model of the course of CD to calculate the excess lifetime costs of the disease. The model considered seven states (remission, mild disease, drug-responsive severe disease, drug-dependent severe disease, drug-refractory severe disease, surgery and postsurgical remission) plus death. This model was not an economic evaluation, but has been highly influential in the modelling carried out in non-fistulising CD.
Gregor et al. 92 elicited HRQoL values from 180 consecutive Canadian CD patients. This evaluation provided both standard gamble and time trade-off data for hypothetical chronically active CD, acute disease and remitted states, and also by the patient’s own health and CDAI status. This information was also used in a number of the economic models reviewed here.
Non-fistulising disease
Within the published models, the comparator treatment strategies comprised surgery and medical treatment,7 placebo5 or usual care6 in populations that were resistant/non-responsive to standard therapy. Only one model was UK-based,5 with the others based in France7 and Canada. 6 The French model7 was lifetime-based, with the Canadian model6 taking a 1-year time frame. The time frame in the UK model5 was unclear, with the treatment considered up to three retreatments within a single year, but stated that the time frame of treatment was ‘1 or more years’.
The French,7 Canadian6 and UK5 models used the Olmstead County data when estimating transition probabilities. The French7 and Canadian6 models used this data to model states where infliximab was not used (in the French case, following surgery, and in the Canadian model, from baseline). In the modified industry model in the UK HTA report,5 these Olmstead data were used to define post-remission health states for the infliximab arm. Clark et al. 5 noted that the use of this information was likely to lead to bias if used to populate a Markov model that moved CD patients responding to treatment into a remitted state. They noted that the prognosis of those in a remitted state following disease flare and infliximab treatment was likely to differ from those who had not experienced a disease flare in the observational cohort.
Both the French7 and Canadian6 models used a third-party payer perspective and, while not described, the UK HTA report5 likely used an NHS/PSS perspective in line with the NICE reference case. 93
Fistulising disease
In fistulising disease, the comparator treatment strategies comprised placebo (Clark et al. 5) or the combination of 6-mercaptopurine/metronidazole (6MP/met) and/or infliximab in different regimens (Arseneau et al. 90). Both studies (one UK, Clark et al. ;5 one US, Arseneau et al. 90) used a 1-year time frame. The US model used a third-party payer perspective but the UK model was unclear on this point but again probably took an NHS/PSS perspective. A lack of existing clinical data beyond 18 weeks required Arseneau et al. 90 to make strong assumptions about both the effectiveness of infliximab as second-line and reinfusion therapies, and the longer term chances of fistula recurrence.
The four models considered the cost-effectiveness of infliximab treatment for 70-kg adult CD patients (Clark et al. 5 fistulising and non-fistulising, Arseneau et al. 90 and Marshall et al. 6). In the remaining study the assumed weight of the Markov cohort was unclear. 7
Calculation of cost data
With the exception of Marshall et al. 6 (non-fistulising Canadian model) and Arseneau et al. 90 (fistulising US model), the assessed models reported costs and resource usage poorly. None of the remaining models reported resource use separately from costs, and in many cases the costs of individual items was not given. In the UK HTA model,5 the source of the cost data was not given. Expert opinion was used to estimate resource use items in two models (Jaisson-Hot et al. 7 and Marshall et al. 6).
As the models were typically of 1 year’s duration, there was no discounting. In the single French analysis7 of a longer duration (lifetime), a discount rate of 5% was used. (While the US analysis90 is only 1-year in duration, it claimed to use a discount rate of 3%. It was not clear how this discounting was calculated.)
Health outcomes and data sources
Effectiveness in non-fistulising disease
In the UK model,5 many of the clinical data were removed for confidentiality reasons. Effectiveness in the model was based on two scenarios where the effectiveness of infliximab was either aggregated across doses (Scenario 1) or based on the 5 mg/kg dose (Scenario 2). Scenario 1 gave lower effectiveness estimates and was used in the company submission. Values for both scenarios were given in summary tables. The French model7 calculated effectiveness data from published evidence (Targan et al. 57) and expert opinion, but details were unclear. The Canadian model6 used effectiveness data from the Targan et al. 57 and Rutgeerts et al. 58 trials.
Effectiveness in fistulising disease
The US model by Arseneau et al. 90 converted pooled data from 12 studies to calculate transition probabilities. The model assessed benefits through fistula improvement rather than closure, so that the improved state included both complete closure and symptomatic improvement. While acknowledging that clinically relevant end points were a subject of debate, the authors acknowledge that this choice of definition was likely to increase the effectiveness of treatment and may have biased estimates.
The UK HTA model5 used data from the Present et al. 62 study for fistulising disease, but no details of precise estimates were given.
Utility estimates
Utility estimates were based on the study by Gregor et al. 92 in the three non-fistulising models. As the Gregor et al. estimates did not include fistulising states, separate estimates were used in the models for fistulising disease. The US fistulising model by Arseneau et al. 90 used standard gamble utilities from 32 CD patients and 20 healthy volunteers, while Clark’s (UK) modified industry model5 used an unpublished algorithm based on CDAI and PDAI scores from the Gregor et al. 92 data.
Cost-effectiveness results
The comparison of cost-effectiveness results across studies is always problematical. For comparison purposes, the methods used were to transform cost estimates based on purchasing power parities94 (as appropriate) and reflate according to all-item UK retail price index figures95 to provide estimates in 2006 pound sterling where possible. Where the base year for costs was not given, figures could not be reflated and the original stated values are used here.
Differences in comparators, methods, data and the non-disclosure or removal of pertinent information prevent reliable interpretation of the results of such comparisons. In these results, caution should be taken in the interpretation, as ICERs relate to the cost of increasingly more effective treatments while cost-effectiveness ratios may be compared with a common comparator. The former is preferred as it allows assessment of the marginal costs and effectiveness of treatment.
In only two6,90 of the models were total costs and effectiveness data given for all the compared strategies. In only one6 of the models was it possible to calculate ICERs. Back-calculation of figures was avoided as this may have introduced errors, while transforming provided figures is hazardous given that they are not displayed to sufficient precision.
In non-fistulising disease, the UK model5 compared single and ‘episodic’ treatment with placebo. Against placebo, episodic treatment (defined as a single 5 mg/kg dose plus up to three 5 mg/kg retreatments within a single year) was estimated to have an ICER of £62,016 per quality-adjusted life-year (QALY) when using effectiveness data from the 5 mg/kg dose group (base year not given). Treatment with a single dose of infliximab (no episodic reinfusions) was found to be less cost-effective.
The French model7 estimated the cost-effectiveness against usual care only. As neither total nor incremental QALY figures were given (and back-calculating is not reliable), incremental figures could not be calculated. Against usual care, ‘episodic’ treatment and maintenance treatments of infliximab were estimated to have cost-effectiveness ratios of €63,700 and €784,057 per QALY (base year not given) respectively.
The converted results from the Canadian model6 suggested an ICER of £105,900 per QALY for single dose versus usual care, £280,600 per QALY for ‘episodic’ versus single-dose infliximab, and £407,000 per QALY for maintenance versus ‘episodic’ treatment with infliximab.
In fistulising disease, the UK model5 suggested a cost-effectiveness ratio of £102,000 (base year unclear) for initial infliximab treatment versus placebo. In the US model90 (converted to 2005 pounds sterling), only cost-effectiveness ratios could be calculated as the outcomes figures were not given with sufficient precision. Against the comparator treatment of 6MP/met, the interventions had cost-effectiveness ratios of £274,100 per QALY (infliximab, with 6MP/met as second-line treatment), £278,000 per QALY (infliximab, with infliximab reinfusions as second-line treatment) and £290,770 per QALY (6MP/met + episodic infliximab reinfusion).
Sensitivity analysis
The reporting of sensitivity analyses was variable, with probabilistic sensitivity analysis conducted in only the Canadian model. 6 This analysis suggested that usual care was favoured up to a threshold of approximately £105,000 per QALY, with a single dose of infliximab favoured between this figure and approximately £251,000 per QALY in non-fistulising disease (converted figures). While this study suggested that the rate of surgical admissions for drug-refractory treatment had little effect on cost-effectiveness, it was sensitive to the variations in the cost of infliximab. With a sufficiently large price reduction it suggested that infliximab treatment may have become cost-effective.
In the UK study5 for non-fistulising disease, the one-way sensitivity analyses conducted on utility, duration of response and the rate of averted surgeries did not result in any ICER below £40,000 per QALY (base year not given).
In fistulising disease, the UK model5 varied the rate of success in retreatment and reclosure of fistulas alongside the level of cost offset owing to averted surgeries. In no case did this produce a ratio below £80,000 per QALY. In the other fistulising model (USA),90 all ICERs remained above £79,000 per QALY (converted figures) regardless of the changes made in one-way sensitivity analyses other than in the price of infliximab. Only where the price of infliximab was reduced to £160 per dose (a reduction of 90% from the modelled price) did the ICER for ‘episodic’ reinfusion fall marginally below £30,000 per QALY (converted figures).
Author conclusions
All of the studies considered were conducted by non-industry authors, with the Canadian6 and UK5 studies commissioned by national HTA bodies. The remaining studies7,90 did not disclose any industry affiliation.
The results of all non-fistulising studies suggested that infliximab was not necessarily cost-effective over the usual range for thresholds. The study by Jaisson-Hot et al. 7 (France) suggested that ‘episodic’ infliximab treatment could possibly have been cost-effective, but that maintenance treatment may not have justified the increased costs required. The study by Clark et al. 5 (UK) suggested that the cost-effectiveness was relatively insensitive to changes in the key assumptions in their model but that the key criterion for the cost-effectiveness of ‘episodic’ treatment would have been the duration of benefit.
The study by Marshall et al. 6 (Canada) was limited by the use of non-Canadian data, the need to convert utility data to populate estimated states, the use of expert opinion to inform resource usage and the lack of longer term clinical data. They noted that while CD may severely impact morbidity and affect productivity, there was no detailed information available on productivity losses to make allowances for this. They justified the relatively short time frame in his model with the lack of clinical data to populate a longer term model.
In fistulising disease, Clark et al. 5(UK) stated that the cost-effectiveness ratios were high under even the most favourable assumptions for retreatment and closure in the industry model. The study by Arseneau et al. 90 (USA) suggested that the high cost-effectiveness ratio for infliximab was due to both the high incremental cost of infliximab and a similar effectiveness to 6MP/met treatment. They acknowledged the difficulties with the ‘fistula improvement’ state and noted that infliximab may have been more effective if it had promoted closure rather than merely symptomatic improvement. The availability of only 18-week data was also acknowledged as a difficulty.
Conclusions
There have been no published studies on the cost-effectiveness of adalimumab alone or in comparison to infliximab. Given the lack of comparison between both alternative treatments considered here, it was not possible to infer the relative cost-effectiveness of treatments from existing evidence. Also, while the indirect productivity costs of non-treatment may be appreciable in CD, these costs were not included in the cost-effectiveness studies owing to a lack of evidence as to their magnitude.
All four studies5–7,90 reviewed were conducted by non-industry authors, with the Canadian6 and UK5 studies commissioned by national HTA bodies. The remaining studies7,90 did not disclose any industry affiliation. Taken together, the papers suggested that single use or ‘episodic’ treatment with infliximab has a relatively high cost-effectiveness ratio for both non-fistulising and fistulising disease. Maintenance therapy was considered only for non-fistulising disease and this is partly due to its potentially prophylactic role in this disease group. Both models considering maintenance infliximab therapy suggested that it would have a particularly high cost-effectiveness ratio relative to both SC and ‘episodic’ infliximab treatment.
Full details for included studies of study characteristics, models used, costs and resources, efficacy data, total costs and outcomes, sensitivity analyses and author conclusions can be found in Appendix 15.
Critique of the submission on infliximab by Schering-Plough
Model structure and inputs
The economic component of the Schering-Plough submission96 took the form of three cost–utility analyses from the perspective of the health service provider. The model structures were informed by the structure of ACCENT I2,3 and included the ‘episodic’ treatment over which concern has been previously expressed (see Glossary). The term ‘infliximab clinical discretion’ (ICD) has been used in place of ‘infliximab episodic treatment’ here.
The three models in the Schering-Plough submission were: a version considering cost-effectiveness of IMT compared with ICD and SC without infliximab among patients with severe active CD (CDAI scores 220–400) in England and Wales (MODEL A); a second version comparing IMT against SC in fistulising CD (MODEL B); and a third model considering paediatric CD patients (MODEL C).
Infliximab maintenance treatment consisted of 5 mg/kg infliximab at week 0, 5 mg/kg infliximab at weeks 2 and 6 and every 8 weeks thereafter. Those receiving ICD received an induction dose of 5 mg/kg infliximab at week 0 and thereafter 5 mg/kg infliximab according to clinical discretion. Those receiving SC received a placebo infusion at weeks 2 and 6 and every 8 weeks thereafter.
MODELS A–C were primarily based on data from two recent trials, ACCENT I2,3 and ACCENT II. 65 Further trial data came from Targan et al. ,57 Present et al. 62 and REACH. 45 ACCENT I2,3 was designed to compare a single 5 mg/kg infusion of infliximab followed by maintenance or a placebo for patients with CD. Participants were recruited from across North America, Europe and Israel. Participants must have had CD for > 3 months and a CDAI score of between 220 and 400. All participants were given 5 mg/kg infliximab at week 0. At week 2, whether participants were responders or not, they were randomly assigned to one of the following three groups:
-
Group I: placebo infusion at weeks 2 and 6, and every 8 weeks thereafter to week 46 (n = 188).
-
Group II: 5 mg/kg infliximab at weeks 2 and 6, and every 8 weeks thereafter to week 46 (n = 192).
-
Group III: 5 mg/kg infliximab at weeks 2 and 6, followed by 10 mg/kg every 8 weeks thereafter to week 46 (n = 193).
Response was defined as a decrease in CDAI score of ≥ 70 points and a minimum 25% reduction in total CDAI score. After week 14, patients who initially responded but experienced exacerbation of symptoms could cross over to 5, 10 or 15 mg of infliximab on an ‘as needed’ or ‘episodic’ basis.
The ACCENT II65 trial compared long-term treatment regimens for patients with fistulising CD. Participants all had CD with single or multiple draining fistulas and were recruited from across North America, Europe and Israel. All participants were given 5-mg infliximab at weeks 0, 2 and 6. At week 14, all patients, regardless of whether they were responders, were randomly assigned to one of the following two groups:
-
placebo infusion at weeks 14, 22, 30, 38 and 46, and follow-up at week 54 (n = 99)
-
5 mg infliximab at weeks 14, 22, 30, 38 and 46, and follow-up at week 54 (n = 96).
In this trial, response was defined as a reduction of at least 50% from baseline in the number of draining fistulas at consecutive visits 4 weeks apart. After week 22, patients in the placebo group who experienced a loss of response could crossover to IMT of 5 mg/kg infliximab.
MODEL A used a Markov model to simulate the progression of patients and to calculate the cost per QALY of the infliximab treatment over a 5-year period. For severe active CD, the model states were active, remission, death, non-responding active (patients who failed to respond either initially by week 2 or discontinued treatment in the second week due to loss of response), surgery, post-surgery remission and post-surgery complications. MODEL B (fistulising) replicated this but expanded the active state to: active + fistula closure, active + fistula, remission + fistula closure and remission + fistula. In the severe active model, patients stayed in the active state (on treatment) for the first 2 weeks before movement to other states. In contrast, patients stayed in the active state for the first 14 weeks within the fistulising model, as assessment of these patients in ACCENT II65 occurred at this point. Transition probabilities for the active state were based on ACCENT II65 and Present et al. 62 trial results. The transition probabilities for the ‘on treatment’ health states were estimated from the Targan et al. 57 and ACCENT I2,3 studies; while the transition probabilities for the ‘off treatment’ health states were estimated from the literature. MODEL C (paediatric) mirrored those of the severe active model with transition probabilities based on data from the Targan et al. ,57 ACCENT I2,3 and REACH45 studies.
The probability of surgery and post-surgery states were obtained from a variety of sources (Marshall et al. ,6 Wolters et al. 97 and Jess et al. 98). The authors assumed an equivalent surgery rate (64%) in all three models (severe active, fistulising and paediatric). Post-surgery complications were estimated from Marshall et al. ,6 which showed no significant differences between groups with and without infliximab prior to surgery, so a weighted average was used. Recurrence rates were based on those from Wolters et al. ;97 while the study contained data from nine European countries this did not include the UK, so expert opinion was sought to confirm the similarity of the estimates with the UK.
The methods for the estimation of quantities and unit costs were, in the main, comprehensively described. The cost of hospitalisation and assessments used data taken from Jewell8 (a UK study). This retrospective observational study (n = 205) compared resource use 6 months pre- and 6 months post infliximab. The pre-infliximab figures were used for SC. Data on post-surgery health states (post-surgery remission and post-surgery complications) were not available so resource use was estimated by an expert panel (consisting of UK gastroenterologists) and the average estimates taken. The cost of immunomodulators was excluded from the analysis on the basis that the efficacy of the treatment would not have been affected (the authors did a post hoc analysis of ACCENT I2,3 and II65 trials that indicated that there was no significant difference in infliximab treatment effect with or without immunomodulators). AEs were assumed to be included in the infusion and hospitalisation costs. The cost of infliximab infusions was estimated using an average adult body weight of 60 kg, which the authors stated was based on previous guidance from NICE. A cost of administering infliximab of £96 was suggested by the authors but was incompletely referenced. The closest match found99 suggests a cost of £257.50 per infusion, which was considerably higher than suggested by the manufacturer.
Health-state utilities were based on a number of different data sources. For severe active CD, health-state preferences for pre surgery were taken from a Spanish study [n = 201 CD patient responses to EuroQoL-5 Dimensions (EQ-5D) and converted into utilities using UK tariffs]. 100,101 Surgery and post-surgery preferences were based on data from a secondary care database of patients in Cardiff and The Vale of Glamorgan, Wales. 34 No information was available for complications post surgery, and a utility value of 0.4 was assigned. The authors justified the value given in terms of complications that would lead to significant hospitalisation.
The transitional probabilities were subject to sensitivity analyses with the exception of post-surgery health states because no treatment effect was assumed beyond surgery. One-way sensitivity analyses was conducted on patient weight, time horizon, discount rate, baseline age and infliximab administration cost, and the resultant cost per QALY was reported.
Model results
The results of the analysis are shown in Table 44.
| Time | Treatment | Mean costs (£) | Difference (£) | Mean QALYs | Difference | ICERs (£ per QALY) |
|---|---|---|---|---|---|---|
| Severe active CD | ||||||
| 5 years | IMT | 31,040 | 4831 | 2.145 | 0.186 | 25,903 |
| SC | 26,209 | 1.959 | ||||
| 5 years | ICD | 25,501 | –708 | 2.133 | 0.174 | ICD dominates |
| SC | 26,209 | 1.959 | ||||
| 5 years | IMT | 31,040 | 5539 | 2.145 | 0.011 | 457,386 |
| ICD | 25,501 | 2.133 | ||||
| Fistulising | ||||||
| 5 years | IMT | 36,626 | 6049 | 2.449 | 0.202 | 30,005 |
| SC | 30,577 | 2.247 | ||||
| Paediatric | ||||||
| 5 years | IMT | 33,504 | 5833 | 2.566 | 0.420 | 13,891 |
| SC | 27,672 | 2.146 | ||||
The results in MODEL A were analysed using both a probabilistic sensitivity analysis (which contained flaws) and a univariate sensitivity analysis [patient weight 70 kg (vial sharing) or 80 kg; time horizon at 2 years or at lifetime; discount rate changes; baseline ages of 30 and 60 years vs 45 years; increased administration costs]. In MODEL A the dominance of ICD over SC remained in all univariate analyses except patient weight 80 kg where it remained highly cost-effective. While the industry submission concluded that maintenance was cost-effective against SC, the relevant comparison where ICD is feasible is between maintenance and ICD. Here, the ICER for maintenance treatment over ICD remains above £400,000 in all analyses.
Critique of the industry submissions for infliximab
The design of the cost–utility analysis is in line with what would be expected for this type of submission but the results are limited by a number of factors. While the comparators appear to be justified, the analysis comparing both SC and infliximab maintenance to ICD care is hampered by the definition of ‘episodic care’ used for the ICD comparison. Although episodic care was described as treatment ‘as required’, no further details were given.
The analysis relied heavily, but not exclusively, on the ACCENT I2,3 and II65 trials. The information used in the economic analysis did not compare treatments consistently across those who were potential responders and non-responders to anti-TNF treatment. This is likely to overestimate the effectiveness of IMT in the industry submission. In addition, the costs of those who did not respond do not appear to have been included, giving lower estimated costs. Both scenarios produce a lower cost per QALY.
In line with the other industry submissions, the primary comparison is with SC. The rationale for this comparator is that the majority of patients eligible for biological treatments in England and Wales still receive SC. While the authors cite market research as evidence of this, unfortunately the information cited is not in the public domain. Also, the ACCENT I2,3 and II65 trials were conducted outside the UK, so it is not possible to determine how ‘standard care’ in the trials compares to that in the UK.
Throughout the submission, a CDAI score of 220–400 was used to indicate severe active CD. While there is no formal quantification of the level at which moderate CD becomes severe active, 220 is lower than has been used in a number of other studies, which makes comparison difficult.
Evaluating MODEL A: active CD (220 < CDAI < 400)
Clinical effectiveness of the comparators in the economic model
The economic model compared maintenance (IMT), ICD and SC. As the details of ICD treatment are unclear, it is not possible to satisfactorily verify or interpret the model. In particular, the description of ICD treatment neither guarantees episodic care nor precludes the use of maintenance treatment. Thus, it both is extremely broad in definition and does not guarantee that clinically identical individuals would receive the same treatment. ICD is limited in the degree to which it represents an identifiable treatment strategy.
Much of the model was based on the ACCENT I2,3 trial. This trial distinguished between those who did and did not respond by week 2 on both the placebo maintenance and infliximab maintenance arms (of which the economic analysis considers only the 5 mg/kg dosage). The placebo arm in the ACCENT I2,3 trial included treatment with (1a) placebo maintenance treatment and (1b) ICD at 5 mg/kg for those not responding to 1a. The infliximab arm in the ACCENT I2,3 trial included treatment with (2a) maintenance at 5 mg/kg and (2b) ICD at 10 mg/kg treatment for those not responding to (2a).
Hanauer et al. 3 compared the outcomes for week 2 responders (1a) with treatment (2a), with those crossing to (1b) and (2b) considered to be treatment failures. Rutgeerts et al. 2 attempted to compare outcomes for both week 2 responders and non-responders in their initial and crossover treatment, attempting to compare (1a) plus (1b) with (2a) plus (2b). The economic model attempted to compare (1a) plus (1b) (as ICD) against only (2a) (as infliximab maintenance).
Confidential clinical information in the industry submission suggested that, however constituted, ICD provides very similar clinical outcomes to maintenance therapy. The clinical study report included data on how many patients retained a response to treatment at week 30. Among week 2 responders, 51% of those receiving IMT retained a response, as against (CiC information has been removed) of those receiving SC (placebo) at week 30 (week 54 clinical study report). The ICD arm is based on those failing on placebo treatment receiving infliximab at 5 mg/kg. All those receiving infliximab on ICD would have been considered failures and on this basis, it would have been expected that ICD would have the same effectiveness in retaining a response as placebo treatment.
A clearer comparison is available using the week 30 clinical study report that includes data for week 2 responders and non-responders, those who crossed over, and those who received protocol-prohibited medication changes or surgery. At 30 weeks, (CiC information has been removed) of patients receiving ICD maintained a clinical response to treatment. As it would be expected that non-responders would have poorer outcomes than responders, these results may have underestimated the effectiveness of ICD for week 2 responders. Given this, it would not be surprising for ICD to match maintenance therapy in health outcomes achieved, and any advantage for maintenance over ICD is likely to be very minor. As maintenance therapy patients received infliximab regularly, whereas ICD patients received infliximab according to clinician discretion (at the same dosage), ICD would be less expensive than maintenance. It appears that IMT is very unlikely to be cost-effective against ICD.
Use of trial data in MODEL A
Although the treatment scenarios were well presented, there were some limitations which were primarily associated with the sample characteristics of the studies used. The active CD treatment strategies estimated were based on ACCENT I2,3 data together with data from another smaller study – Targan et al. . 57 The Targan et al. 57 study was used for transitions between week 0 and week 2, with transitions following week 2 estimated using ACCENT I. 2,3 The use of Targan et al. 57 data is questionable as they come from a smaller trial preferred in place of a larger trial which provided less positive results (ACCENT I2,3).
The economic model also appeared to use two different populations in active CD, with both the SC (placebo) and ICD arms using week 2 responders and non-responders and the maintenance arm using week 2 responders only. If week 2 responders do indeed have a better response than week 2 non-responders, then this is likely to bias the comparisons in the economic model. Given the data above, this bias may account for any positive effect found for infliximab maintenance against ICD.
Assessment group revisions to MODEL A
The cost of drug infusions was estimated using an average adult body weight of 60 kg, which the authors state was based on previous guidance from NICE. 1 This is likely to have underestimated the cost per QALY. The Targan et al. 57 trial recorded mean body weights of between 68 and 74 kg (for different treatment groups), while for ACCENT I2,3 these are recorded only in the (confidential) clinical study reports (CiC information has been removed). ACCENT I aside, there were four larger trials (Targan et al. ,57 CLASSIC I,63 GAIN64 and CHARM67) in the clinical effectiveness section in adults (where n > 100) that gave mean weight of included patients – approximately 71.5 kg.
A weight of 60 kg exactly corresponds to the use of three 100-ml vials of infliximab, and the model therefore assumed no wastage. A revised model was constructed by the assessment group. In this model a weight of 70 kg was used, which remains conservative given that no wastage is incorporated into the model. The price of infliximab within the model was also increased to reflect the cost as published in the British National Formulary (£419.73). 102 In the revised model, administration costs were taken to be half of a day case – H26 (Day Case Rheumatology) – in line with a 2006 HTA report,99 which was £293.67 when adjusted to 2006 prices using the Personal Social Services Research Unit (PSSRU) NHS Pay and Prices Index. 103 The cost-effectiveness of ICD, IMT and SC was calculated using a probabilistic sensitivity analysis (1000 iterations) with cost-effectiveness acceptability curves (CEACs) calculated (Figure 33).
FIGURE 33.
Cost-effectiveness acceptability curve for active CD.
The conclusions of the MODEL A as revised by the evidence review group differ from those of the industry MODEL A, a result of these input changes and correction of errors in the industry sensitivity analysis. For threshold levels between £0 and £2466 per QALY, placebo had the highest chance of being cost-effective. For threshold levels between £2466 and approximately £481,000 per QALY, ICD treatment had the highest chance of being cost-effective. Infliximab treatment according to clinical discretion appears to be cost-effective at thresholds of £20,000–30,000 per QALY, although this is contingent on a series of caveats, including the ill-defined nature of the ‘episodic’ intervention itself.
Evaluating MODEL B: fistulising CD
Use of trial data in MODEL B
For fistulising CD, the industry submission stated that evidence from the ACCENT I2,3 trial suggested that maintenance infliximab therapy may bring significant QALY gains related to improved QoL (as opposed to improved life expectancy) in adults with fistulising CD. However, it is not possible to ascertain from the submission or from published papers of the trial whether the sample included adults with fistulising CD. The submission did however report evidence presented in the ACCENT II65 trial (fistulising CD patients) that showed a significantly longer time to loss of response for infliximab maintenance than for placebo maintenance and significantly improved CDAI scores for the infliximab group versus the placebo group.
The fistulising CD strategies were based on responders only. The ACCENT II65 trial showed that 69% of the sample were responders after the induction period. There was, however, no placebo comparison during this period so it is not possible to determine the proportion of patients who would have gone into remission without infliximab. While the proportion of responders was higher than the ACCENT I2,3 trial, the definition of a responder differs and the time at which they were assessed as a responder/non-responder was much later (14 weeks rather than 2 weeks). This highlights the arbitrary nature of the time point chosen to identify responders and the impact it may have had on the results.
The treatment strategy was modelled on the Present et al. 62 trial (0–14 weeks) and the ACCENT II65 trial (14–54 weeks). The Present et al. 62 trial, like the Targan et al. 57 trial, had relatively small numbers in each arm (31–32). The ACCENT II65 scheduled maintenance arm excluded those who switched to 10-mg ‘episodic’ treatment after week 22. Again it is not clear how SC in the UK compares with that in the Present et al. 62 trial (recruitment was in the USA and Europe) and the ACCENT II65 trial.
Assessment group revisions to MODEL B
As with severe active CD, the cost of drug infusions was estimated using an average adult body weight of 60 kg which the authors state was based on previous guidance from NICE. 1 Questions remain about the suitability of this figure given that it is lower than the values found in clinical trials on which the analyses are based. As above, a figure of 70 kg was used for the average weight of patients, which changed both the cost of infliximab and administration costs to a more accurate figure.
The health-state utilities within MODEL B were based on two different sources:
-
A Spanish study (n = 201 CD patients) measuring EQ-5D then converted into utilities using UK tariffs which assumed that the preferences measured are concordant with UK patient preferences. 100,101
-
A secondary care database of patients in Cardiff and The Vale of Glamorgan, Wales, measuring surgery and post-surgery preferences. This was based on a small sample size and looked at surgery (< 2 months after surgery, n = 17) and remission post surgery (i.e. > 2 months with no recurrence/complication n = 21). 34
Despite specific utility estimates being available for the fistulising CD model, they were not used because they are generally higher than the utilities found in the Spanish study, which was used to provide values in the severe active CD model. The authors stated that the estimates were not in accordance with the NICE reference case93 because they were taken from CD patients and healthy individuals (n = 32 and n = 20 respectively). The utilities allocated assumed that patients with fistula closure had identical utilities to patients without fistulas. For all other fistula states 0.15 was deducted from utility estimates, the authors gave no explanation for this figure but did include the variable in the sensitivity analysis. While the assessment group notes this as an issue, no changes were made to MODEL B on this point.
The model was rerun using the revised weight and correcting the probabilistic sensitivity analysis for calculation errors. The CEAC for maintenance versus SC suggests that maintenance care was more likely to be cost-effective against SC as the threshold value increased. At £20,000 and £30,000 per QALY, infliximab treatment was found to be cost-effective 32.5% and 48.1% of the time respectively. Following the weight adjustment and recalculation of the CEAC, the curve shifted downwards. Now, at £20,000 and £30,000 per QALY infliximab treatment was found to be cost-effective only 6.3% and 19.1% of the time respectively (Figure 34).
FIGURE 34.
Cost-effectiveness acceptability curve for fistulising CD (IMT versus SC).
Evaluating MODEL C: paediatric Crohn’s disease
The economic model provided with the industry submission contained both circular references and broken links. While broken links may be repaired with the provision of the files containing the information on which the model is based, the file was not available. Any analysis conducted on this model would necessarily be based on such computational guesswork and would not withstand scrutiny. Although they had received opportunities to do so, Schering-Plough provided a working version of its paediatric model only at a sufficiently late stage that it was not possible to verify either the model structure or its assumptions. At this stage, three variants of a model outlined were provided and a cursory examination was sufficient to discover important – and within the available time frame – unresolvable flaws. It was therefore not possible to provide a report of the models provided beyond the brief summary of the original (unverified) submission that appears below.
Use of trial data in MODEL C
For paediatric CD, the submission states that evidence from the ACCENT I2,3 trial ‘suggested that maintenance infliximab therapy may bring significant QALY gains, related to improved QoL (as opposed to improved life expectancy) in adult and paediatric patients with CD’. While the sample characteristics provided in the trial summary do not show whether paediatric CD patients were included in ACCENT I,2,3 a paper in The Lancet3 reporting on the ACCENT I2,3 trial gave the patient age range as 18–76 years, suggesting no paediatric patients were included. It is difficult to tell whether the authors’ statement is based on data from these subgroups or previous evidence.
Paediatric CD strategies were based on data from Targan et al. ,57 REACH45 and ACCENT I. 2,3 For both the scheduled maintenance and SC arms, the model was based on the Targan et al. 57 trial response rates at week 2. The Targan et al. 57 study was not a paediatric study and no age range was given (the mean ages that were given at baseline of those receiving 20 mg and 10 mg of chimeric monoclonal antibody cA2 were 36 and 39.3 years respectively). As there was a small sample in Targan et al. 57 it is unlikely that a subsample of paediatric patients was used. The transitions of these Targan et al. 57 study responders were estimated using data from the REACH45 trial (at between 2 and 54 weeks). The REACH45 trial was a paediatric study that compared scheduled maintenance at 8 weeks versus every 12 weeks. While the study assessed response only at week 10, the response rate was particularly high (99/112) compared with the ACCENT I2,3 and II65 trials. However, the REACH45 study did not have a placebo arm so it is not possible to determine the proportion of patients who would have been classified as responders without infliximab.
The comparison of the two treatment strategies used in the model is inappropriate. The SC arm was based on ACCENT I2,3 data from week 2 onwards. Like the Targan et al. 57 study, ACCENT I2,3 is not a paediatric study; thus the paediatric SC treatment strategy is based only on adult study data; paediatric data are used only in the infliximab scheduled maintenance arm. As with the adult comparisons, the paediatric model was based on an optimistic assumption of 40-kg weight. Of the paediatric studies used in the analysis, the mean weights recorded were between 45 and 55 kg and 42 and 48 kg.
Discussion of the Schering-Plough submission
The Schering-Plough submission included three submodels considering (1) active CD for the CDAI range covered in the ACCENT I2,3 trial including both moderate and more severe forms of CD, (2) fistulising CD and (3) paediatric CD. The industry submissions contained errors, some of which were resolved by the manufacturer following the consultation period on the draft HTA report. Others could not be corrected, such as the selective use of responders in only the infliximab maintenance arm in the active CD model.
For active CD, the corrected models suggested that infliximab treatment (ICD) could be cost-effective, while maintenance care was unlikely to be cost-effective even at several times the normal threshold values. The lack of detail on what constitutes ICD or ‘episodic’ treatment is unhelpful. To the degree that they can be investigated, the models provided by Schering-Plough mostly meet the standards in the NICE reference case93 (Table 45). There remain issues regarding the selection of studies, the use of data within the selected studies and some inputs used in the modelling. The utility values used in one model rely on a small sample, but are broadly in line with the reference case. 93
| Element of health technology assessment | Principles | |
|---|---|---|
| Defining the decision problem | The scope developed by the Institute | Yes |
| Comparator | Alternative therapies routinely used in the NHS | Yes |
| Perspective on costs | NHS and PSS | NHS only |
| Perspective on outcomes | All health effects on individuals | Yes |
| Type of economic evaluation | Cost-effectiveness analysis | Yes |
| Time horizon | Sufficient to reflect any differences in costs or outcomes between the technologies compared | Yes |
| Synthesis of evidence on outcomes | Based on a systematic review | Partial; doubt remains on selection of studies |
| Measure of health benefits | QALYs | Yes |
| Description of health states for calculation of QALYs | Health states described using a standardised and validated generic instrument | Yes |
| Method of preference elicitation for health-state valuation | Choice-based method, for example, time trade-off, standard gamble (not rating scale) | Yes |
| Source of preference data | Representative sample of the public | Partial; one source of particularly small sample size |
| Discount rate | An annual rate of 3.5% on both costs and health effects | Yes |
| Equity position | An additional QALY has the same weight regardless of the other characteristics of the individuals receiving the health benefit | Yes |
| Modelling methods | Structural assumptions and data inputs clearly documented and justified | Partial; assumptions made not justified |
| Probabilistic sensitivity analysis should be conducted | Yes |
Critique of the submission on adalimumab by Abbott
Introduction to the evaluation
An economic analysis was conducted for Abbott for their submission4 to NICE by Analysis Group. The submission comprised two economic models – one comparing the cost-effectiveness of adalimumab as a maintenance therapy against SC and one comparing the cost-effectiveness of adalimumab and infliximab as maintenance therapies. This latter model will be relevant only where both adalimumab and infliximab have been first justified as maintenance therapies versus SC. Where one or both maintenance therapies are not cost-effective versus SC, this comparison provides no information to decision-makers.
This evaluation therefore begins by concentrating on the former model assessing the cost-effectiveness of adalimumab as a maintenance therapy. The model considers both fistulising and non-fistulising forms of CD together, and comprises a printed economic submission and accompanying working model in excel. The economic model contained several assumptions that were not fully explained or justified at its initial submission. Abbott took the opportunity in responding to the draft assessment report to clarify some of these issues raised and their model incorporates elements of the health economic critique to their earlier version.
Model inputs and structure
The stated aim of the company’s primary submission was to produce a comparison of lifetime maintenance on adalimumab versus SC. The adalimumab arm of the models was based on data up to week 56 in the CHARM67 trial, which were then extrapolated to also produce a lifetime analysis by assuming that all those responding at week 56 would continue to respond for the remainder of their lives. A regression based on the CLASSIC I63 trial was used to provide SC outcomes for the CHARM67 arm. The company’s comments received regarding the modified model are acknowledged below.
All patients enrolled in the CHARM67 trial had baseline CDAI scores between 220 and 450. In this trial, all patients were given open-label 80 mg at week 0, 40 mg at week 2 and then randomised blind at week 4 to a placebo, adalimumab 40 mg e.o.w. or adalimumab 40 mg weekly. After week 12, those who did not respond to randomised treatment (defined as a drop of less than 70 points in CDAI) were switched to open-label adalimumab 40 mg e.o.w., as were those ‘responders’ who experienced a treatment flare after week 12. Those not responding to open-label adalimumab 40 mg e.o.w. were switched to adalimumab 40 mg weekly. Those not responding to 40 mg weekly were returned to SC.
In the CLASSIC I63 trial, patients had a baseline CDAI between 220 and 450 and had had no previous exposure to any anti-TNF therapy. There were 299 individuals who were randomised to either placebo (n = 74) or adalimumab induction regimens in weeks 0 and 2 of 40 mg and 20 mg (n = 74), 80 mg and 40 mg (n = 75), or 160 mg and 80 mg (n = 76) respectively. All patients were followed for 4 weeks, and the primary end point was the proportion with a CDAI score < 150 in week 4.
The industry submission compared the cost–utility of the 40 mg adalimumab e.o.w. strategy versus SC. As the standard arm of the CHARM67 trial began with adalimumab induction at 80 mg in week 0 and 40 mg in week 2, this did not provide suitable estimates for either the cost or the effectiveness of SC. As the placebo arm in the CLASSIC I63 trial received no adalimumab, the economic submission used these data to predict health states in the SC arm.
The models in the industry submission were based on both the 56 weeks of the CHARM67 trial and an extrapolation to give a lifetime model. The 56-week model included no discounting, the longer model used a discount rate of 3.5% for both costs and benefits. The lifetime model assumed that health remains constant across the group (in terms of the profile of health states) from week 56 to death and assumed a baseline age of 37 years (in line with the average age for CHARM67), with life expectancy of 66 years. There was no mortality between years 37 and 66 due to treatment, CD or other causes.
The model structure was based around four health states defined as remission (CDAI < 150), moderate (150 ≤ CDAI < 300), severe (300 ≤ CDAI < 450) and very severe (CDAI ≥ 450). Patients enrolled in the CHARM67 trial had baseline CDAI scores between 220 and 450, and fell in only the moderate and severe categories at baseline. Utility data were based on these health states. Costs were calculated based both on trial arms (for anti-TNF medication costs) and on the time spent in each of these disease states (for hospitalisation costs and all other costs). Overall costs and QALY benefits for the CHARM67 trial were calculated for the baseline moderate and severe groups (150 ≤ CDAI < 450) and the baseline severe subgroup (300 ≤ CDAI < 450).
Univariate sensitivity analyses were conducted that modified the method of imputing states for those leaving the trial, incorporated indirect costs and made several other changes to the cost assumptions. Using details in the revised industry model for adalimumab versus SC, it was possible to replicate the multivariate sensitivity analysis provided in the revised model.
Estimates of standard care outcomes
The relationship between CDAI-based health states and prognostic factors was estimated using the CLASSIC I63 trial using an ordered probit regression that predicted the chances of an individual falling into each of the four states (remission, moderate, severe and very severe). Variables were included for baseline CDAI, previous anti-TNF exposure, corticosteroid use, fistulising disease, and time and treatment dummy. Health states for the first 4 weeks in the SC arm were found by applying this regression to the clinical factors observed in the CHARM67 40-mg e.o.w. arm. It was assumed that the proportion of people in each health state would remain constant from week 4 onwards. Although patients previously receiving such treatment were excluded from the CLASSIC I63 trial, the previous use of anti-TNF treatment appeared as a predictor in the ordered probit regression. It is unclear how this effect was estimated.
Estimates of adalimumab maintenance outcomes
The adalimumab outcomes were estimated using data derived from the CHARM67 trial data. Within the CHARM67 trial, 778 patients were randomised but 854 patients were enrolled at week 0 in order to achieve this sample. The 76 patients [(CiC information has been removed) with CDAI ≥ 300] who withdrew prior to week 4 did so for a variety of reasons, including AEs (45), lack of efficacy (13) and, in one case, death. (This death was judged not to be related to the use of adalimumab by the CHARM trial investigators. 67)
The revised industry model incorporated the costs of these non-randomised individuals by including them within the adalimumab modelling arm. Given that the 260 individuals receiving adalimumab comprised approximately one-third of the randomised CHARM cohort, it was assumed that one-third of the non-randomised individuals would have been randomised to this arm. The individuals who were not expected to receive a standard adalimumab course were modelled as if they were SC patients but each incurred an additional £974 in medication costs.
Those expected to be able to receive a standard adalimumab course (i.e. those randomised at 4 weeks) were modelled as the 40-mg e.o.w. arm from the CHARM67 trial, based on randomisation at 4 weeks. CHARM67 randomisation was stratified by 4-week response (reduction in CDAI of 70 points from baseline). At 12 weeks, those not responding (reduction in CDAI of 70 points from baseline) could be shifted to open-label treatment and leave the randomised study. Figure 35 shows the comparison between the randomised data at 4 weeks and the observational groups defined at 12 weeks. Of the 260 individuals, only (CiC information has been removed) received their scheduled treatment at week 56.
FIGURE 35.
Clinical data: CHARM67 evidence versus that used in the economic model. (CiC information has been removed.)
Those patients removed from the trial at 12 weeks were referred to in the industry submission as ‘deleted non-responders’ in the diagram shown in Figure 36 (reproduced as non-CiC) from the economic submission. Missing individuals were those who discontinued from the trial for other reasons, including disease flares and protocol violations.
The economic model included a mixture of those responding and not responding at 4 weeks. The results in the economic model therefore differ from the coprimary clinical end points of the trial, which concerned week 4 randomised responders only.
In the main analysis, the economic model used the last value carried forward (LVCF) to impute missing values. As a sensitivity test, the model included results where the course of the patient’s disease reverted back to the state the patient was in prior to adalimumab therapy (‘simulated placebo’).
Health-state costs and utility estimates
Each health state was linked to an expected number of hospitalisations using a Poisson regression model based on a variety of clinical and background characteristics. This was used to construct patient-level predictions of hospitalisation events per year. The unit cost was estimated using published UK data (Bassi et al. 37) and inflated using PSSRU figures to produce a cost per hospitalisation of £7441 in 2006 pound sterling. The CHARM67 trial did not record CD-related surgeries, and so this hospitalisation factor incorporated the cost of surgery. (Note that the submission was inconsistent whether the hospitalisation figure applied per year or over the 56 weeks of CHARM. 67)
Other non-hospitalisation disease costs (excluding anti-TNF medication) were estimated using Bassi et al. 37 Bassi et al. used a seven-state classification for CD states similar to Silverstein et al. 26 The model assumed that ‘very severe’ corresponded to ‘indicated for surgery’ in Bassi et al. ,37 with ‘severe’, ‘moderate’ and ‘remission’ corresponding to ‘severe, drug-refractory’, ‘mild disease’ and ‘remitted’ states. Estimated non-hospitalisation disease costs are given in Table 46.
| Health state | CDAI score | Non-hospitalisation disease costsa | Utility |
|---|---|---|---|
| Remission | < 150 | 8.45 | 0.859 |
| Moderate | 150 ≤ CDAI < 300 | 23.66 | 0.795 |
| Severe | 300 ≤ CDAI < 450 | 43.11 | 0.693 |
| Very severe | ≥ 450 | 78.55 | 0.433 |
Table 46 also displays utility estimates for the four health states that were based on a reanalysis of previously published primary standard gamble data (Gregor et al. 92). These estimates were based on 180 consecutive Canadian patients presenting with CD between December 1995 and December 1996.
Adalimumab cost estimates
The cost per 40-mg adalimumab dose was assumed to be £357.50, with one dose necessary every 2 weeks per patient after an initial three-dose induction in the first 4 weeks. No administration costs were included.
Results of the adalimumab industry submission
The results reported here refer to the adalimumab industry submission produced in response to comments in the draft assessment report. The industry submission 56-week model suggested that for baseline moderate and severe patients (150 ≤ CDAI < 450), the incremental cost of adalimumab 40-mg e.o.w. treatment was £2496 for an incremental increase of 0.0823 QALYs (Table 47). The estimated ICER was £30,319. For patients with severe disease (300 ≤ CDAI < 450), the incremental costs and benefits were estimated at £1254 and 0.1045, giving an ICER of £11,998 per QALY.
| Moderate and severe | Severe only | |||||
|---|---|---|---|---|---|---|
| Adalimumab | Standard care | Difference | Adalimumab | Standard care | Difference | |
| QALYs | 0.8566 | 0.7743 | 0.0823 | 0.8384 | 0.7339 | 0.1045 |
| Drug costs | £6533 | £0 | £7119 | £0 | ||
| Health-state related costs | £1249 | £2049 | £1429 | £2407 | ||
| Hospitalisation | £2028 | £5265 | £2598 | £7485 | ||
| NHS costs | £9810 | £7315 | £2496 | £11,146 | £9892 | £1254 |
| ICER | £30,319 | £11,998 | ||||
In the original submission, the univariate sensitivity analysis suggested that for patients with severe CD, adalimumab was close to or below £20,000 per QALY for a variety of different assumptions. When considering both moderate and severe CD together, the baseline assumption was close to £30,000 per QALY and typically exceeded this whenever any adverse change was made to the model assumptions. While the industry submission included an induction regimen at 160 mg and 80 mg at weeks 0 and 2, it should be noted that this was not used in the CHARM67 trial and the results will differ if it is associated with higher AEs. This did not change significantly in the second model.
The second industry submission model assumed that parameters for hospital and health-state related costs were distributed according to gamma distributions. Utilities for the remission state were assumed to be distributed according to a beta distribution, with constant ratios between all four utility values (Table 48). When estimating uncertainty in adalimumab outcomes, a Dirichlet distribution was used based on the time spent in each state by the 260 patients within CHARM. 67
| Type | Health state | Type | A | B |
|---|---|---|---|---|
| Hospitalisation costs (£) | Gamma | 6.25 | 1190.56 | |
| Health-state related costs (£) | Remission | Gamma | 6.25 | 1.35 |
| Moderate | Gamma | 6.25 | 3.79 | |
| Severe | Gamma | 6.25 | 6.90 | |
| Very severe | Gamma | 6.25 | 12.57 | |
| Utilities | Remission | Beta | 3.5280 | 0.5791 |
In the second industry model (based on 1000 samples), adalimumab treatment was estimated to be cost-effective below £20,000 per QALY in 62.8% of samples, and below £30,000 per QALY in 80.9% of samples. Figure 37 is based on 5000 samples and gives similar results (61% and 79%, respectively).
FIGURE 37.
Cost-effectiveness acceptability curve for severe disease (LVCF method).
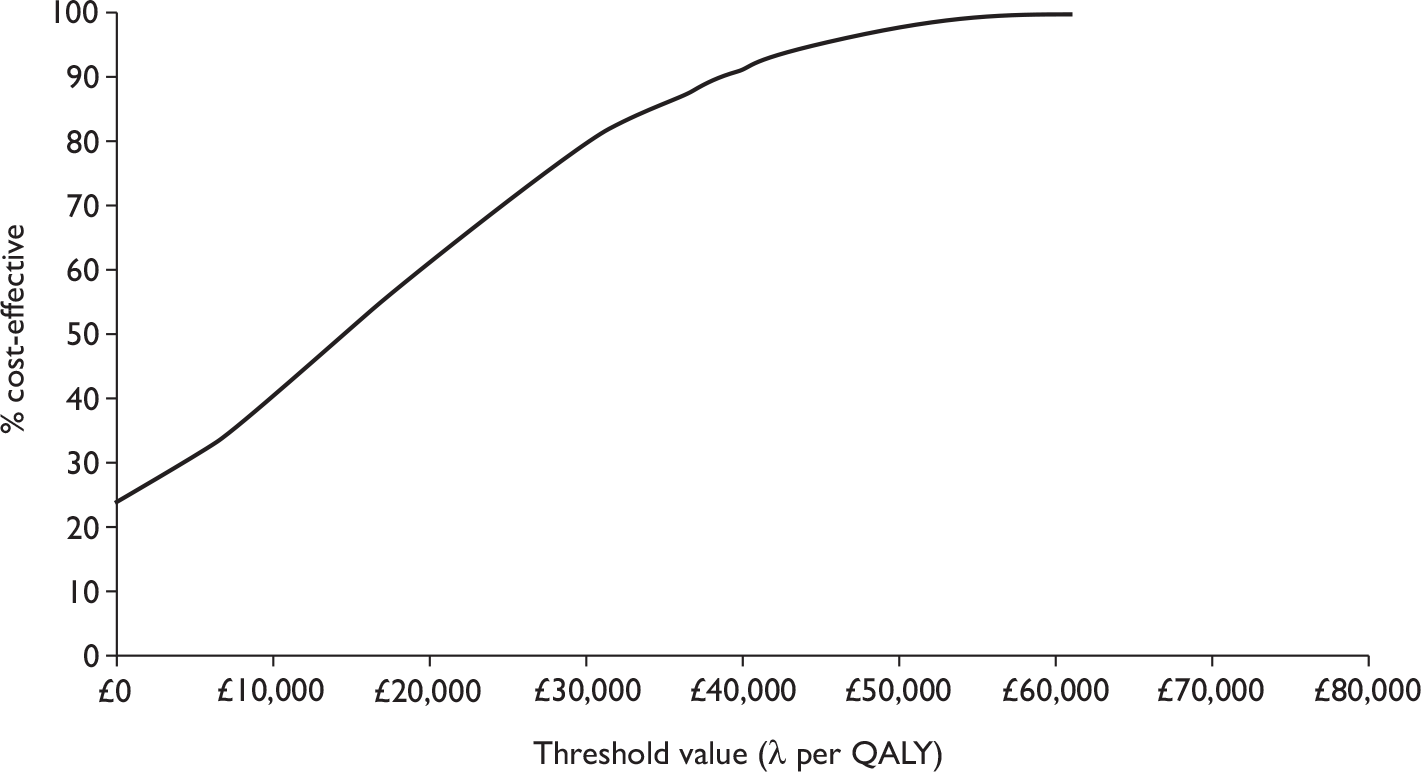
Evaluating the industry submission for adalimumab maintenance versus standard care
The inputs to the industry model of adalimumab maintenance were modified to investigate the robustness of the model. The revised model used the ‘simulated placebo’ method of imputing missing values. Those leaving the CHARM67 trial did so because of disease flare or other issues requiring protocol violating treatments, and so their health may have been poorer than an ‘equivalent’ simulated SC outcome (which represented expected health at 4 weeks). The ‘simulated placebo’ assumption is more neutral with respect to the prognosis of those leaving blinded CHARM67 treatment than the LVCF used in the industry model.
Aside from a preference for ‘the simulated placebo’, the major differences between the second industry model and the revised industry model are the assumptions made regarding the use of adalimumab beyond the study period. In both the first and second industry models it was assumed that all those receiving adalimumab at 56 weeks would continue to do so for their entire lives. In the revised model, the rate of withdrawal from adalimumab maintenance post 56 weeks was increased from zero to that of the CHARM 40-mg e.o.w. arm. Outcomes for patients with moderate CD were also inferred as a separate subgroup where this was possible. Unless otherwise indicated, the model description here is kept as in the industry submission.
The industry model’s use of CHARM data
The clinical end points of the CHARM67 trial related to week 4 responders (a reduction in CDAI of 70 points from baseline), and all published data referred to this group. This causes difficulties in interpreting the data, as terms were duplicated with few caveats. Where published data and the industry clinical submission referred to responders and non-responders, they did so based on a comparison of baseline and 4-week data (randomisation); the economic submission appeared to define this split using baseline versus week 12 data. However, the remission rates for moderate and severe patients do appear to be broadly in line with an all-comers analysis of the trial.
The CHARM67 trial randomised patients at 4 weeks to one of three (blinded) arms: placebo care, adalimumab 40 mg e.o.w. and adalimumab 40 mg weekly. This blinded treatment stage in the trial was maintained for 12 weeks for all randomised patients. Those who did not achieve a sufficient improvement in health at 12 weeks were termed ‘non-responders’ in the economic model, which appeared to define this as a reduction of < 70 points in CDAI from baseline. It appears that week 12 non-responders were moved to open-label 40-mg e.o.w. treatment, as were week 12 responders who experienced disease flares after 12 weeks. Those receiving open-label e.o.w. treatment could subsequently move to weekly treatment as required, and then subsequently to SC following persistent non-response.
Outcomes beyond 56 weeks
The initial industry submission did not adequately describe the assumptions used in constructing its economic model. This was particularly problematic when considering lifetime costs and effects, as this extrapolated data at the end of CHARM trial67 for an additional 37 years. The industry models suggested an average expected adalimumab 40-mg e.o.w. use of 13.3 vials per year after year 1, which was consistent with the numbers receiving e.o.w. treatment at 56 weeks. However, with an approximately constant number of people leaving the trial’s adalimumab arm from week 7 onwards within CHARM, it could be predicted when the last individual would cease to receive adalimumab on this until-flare maintenance regimen. With the limited data made available from the economic model, it was predicted that the last dose of adalimumab corresponding to the blinded treatment on CHARM67 would have occurred in week 189. A lifetime model was not necessary here as – under the assumptions of the placebo method of imputing lost values – the SC and adalimumab model arms would have been identical after 4 years, therefore a 4-year time frame would have sufficed.
In analysing this information, the assessment group used a ‘best guess’ interpretation of the industry model in which only blinded treatment was costed within the model. Subsequent communication verified that the industry model included all treatments received, including open-label 40-mg e.o.w. and weekly regimens. This highlights some unresolved issues within the data and apparent difficulties with adherence. On a period-by-period basis (e.g. weeks 12–16, weeks 16–20, etc.), those on a 40 mg weekly dosage receive only (CiC information has been removed)% of the total dosage received by those on a 40-mg e.o.w. dosage, which is lower than the 200% that would be expected. If all patients who received treatment had the indicated dose, then on a period-by-period basis after week 12 it appears that only (CiC information has been removed)% of patients prescribed 40 mg e.o.w. and (CiC information has been removed)% of patients prescribed 40 mg weekly went on to receive it. This suggested that many individuals appeared to miss scheduled treatment on 40 mg e.o.w., and that those shifted on to 40 mg weekly were even less likely to actually receive it. This may indicate issues in the tolerability of adalimumab and its long-term acceptability for patients.
The accurate analysis of long-term adalimumab usage relies, at the very least, on better data about both the longer term acceptability of treatment to those still ‘on’ treatment, and the assessment of how many patients are now formally off treatment. It is possible to infer vastly different profiles of future usage (and hence benefit) from the CHARM data. Given CiC data provided by Abbott in the consultation process, little confidence can be placed in either the original or revised model beyond the 56 weeks of CHARM. For this reason, outcomes beyond a 1-year time frame are not considered further here. A lifetime model for adalimumab usage is warranted, and as the evidence base improves it may be possible to analyse such issues in greater depth.
Separating moderate and severe subgroups
At randomisation (week 4), the CHARM67 40-mg e.o.w. arm included 125 patients with moderate CD (CDAI between 150 and 300) and 135 with severe CD (CDAI between 300 and 450). The industry submission provided the expected frequency of health states, adalimumab use and hospitalisations for both moderate and severe, and severe only groups. This allowed separate outcomes to be inferred for those with moderate disease within the 56 weeks of the CHARM model. These are reported below.
Results of revised adalimumab model
The industry submission predicted an ICER of £11,998 per QALY at 56 weeks for those with a baseline CDAI at or above 300. With the preferred ‘placebo method’ of imputing missing data, this rose to £30,964 per QALY. The industry submission did not predict an ICER for the moderate subgroup. In the estimates presented here, it was found that treatment was far less cost-effective than for the severe subgroup, and above £100,000 per QALY using the ‘placebo method’ of imputing missing data.
In the 56-week model it appears that treatment for severe patients is likely to approach £30,000 per QALY under more conservative assumptions, but will fall below £20,000 per QALY under optimistic assumptions. For moderate patients, even optimistic assumptions appear to give relatively large ICERs for adalimumab treatment.
The numbers in Table 49 suggest that treatment of those with severe disease (300 ≤ CDAI < 450) will be cost-effective under optimistic (LVCF) assumptions, and marginally over £30,000 per QALY with the preferred and more conservative assumptions. There is less ambiguity surrounding the treatment of those with moderate disease (150 ≤ CDAI < 300). Even under optimistic assumptions, the smaller additional health benefit of 0.0589 comes at a higher incremental cost, leading to an ICER above £60,000 per QALY.
| Severe patients only | Moderate patients only | |||||
|---|---|---|---|---|---|---|
| Adalimumab | Standard care | Difference | Adalimumab | Standard care | Difference | |
| Values imputed using LVCF – optimistic estimate | ||||||
| QALYs | 0.8384 | 0.7339 | 0.1045 | 0.8769 | 0.8180 | 0.0589 |
| Drug costs | £7119 | £0 | £6029 | £0 | ||
| Health-state related costs | £1429 | £2407 | £1046 | £1663 | ||
| Hospitalisation | £2598 | £7485 | £1465 | £2868 | ||
| NHS costs | £11,146 | £9892 | £1254 | £8540 | £4531 | £4009 |
| ICER | £11,998 | £68,065 | ||||
| Values imputed using simulated placebo – pessimistic estimate | ||||||
| QALYs | 0.8225 | 0.7339 | 0.0886 | 0.8605 | 0.8180 | 0.0425 |
| Drug | £7119 | £0 | £6029 | £0 | ||
| Health-state related costs | £1565 | £2407 | £1205 | £1663 | ||
| Hospitalisation | £3952 | £7485 | £2099 | £2868 | ||
| NHS costs | £12,636 | £9892 | £2744 | £9333 | £4531 | £4802 |
| ICER | £30,964 | £113,008 | ||||
Figure 38 shows the CEAC for the treatment of severe disease where the (conservative) simulated placebo method is used. Across 5000 samples, 24.4% of ICERs fell below £20,000 per QALY and 44.1% fell below £30,000 per QALY. These figures compare to 61.2% and 79.1% respectively using LVCF.
FIGURE 38.
Cost-effectiveness acceptability curve for severe disease (placebo method).
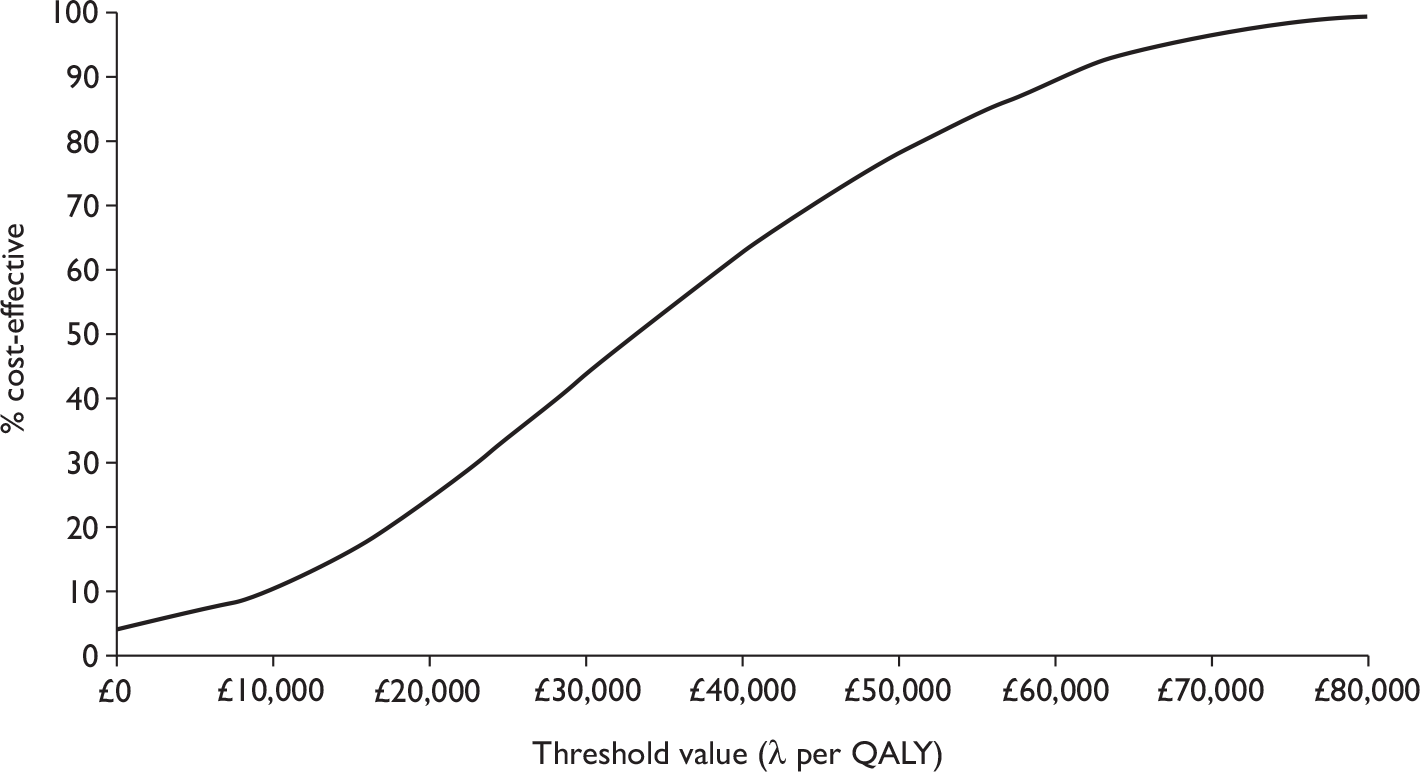
For moderate disease, Figure 39 shows the CEAC for the optimistic case (LVCF). Here, only 1.4% of samples fall below £20,000 per QALY and only 7.9% fall below £30,000 per QALY. Under the pessimistic assumption (simulated placebo) these figures fall to 0.04% and 2.0% respectively.
FIGURE 39.
Cost-effectiveness acceptability curve for moderate disease (LVCF method).
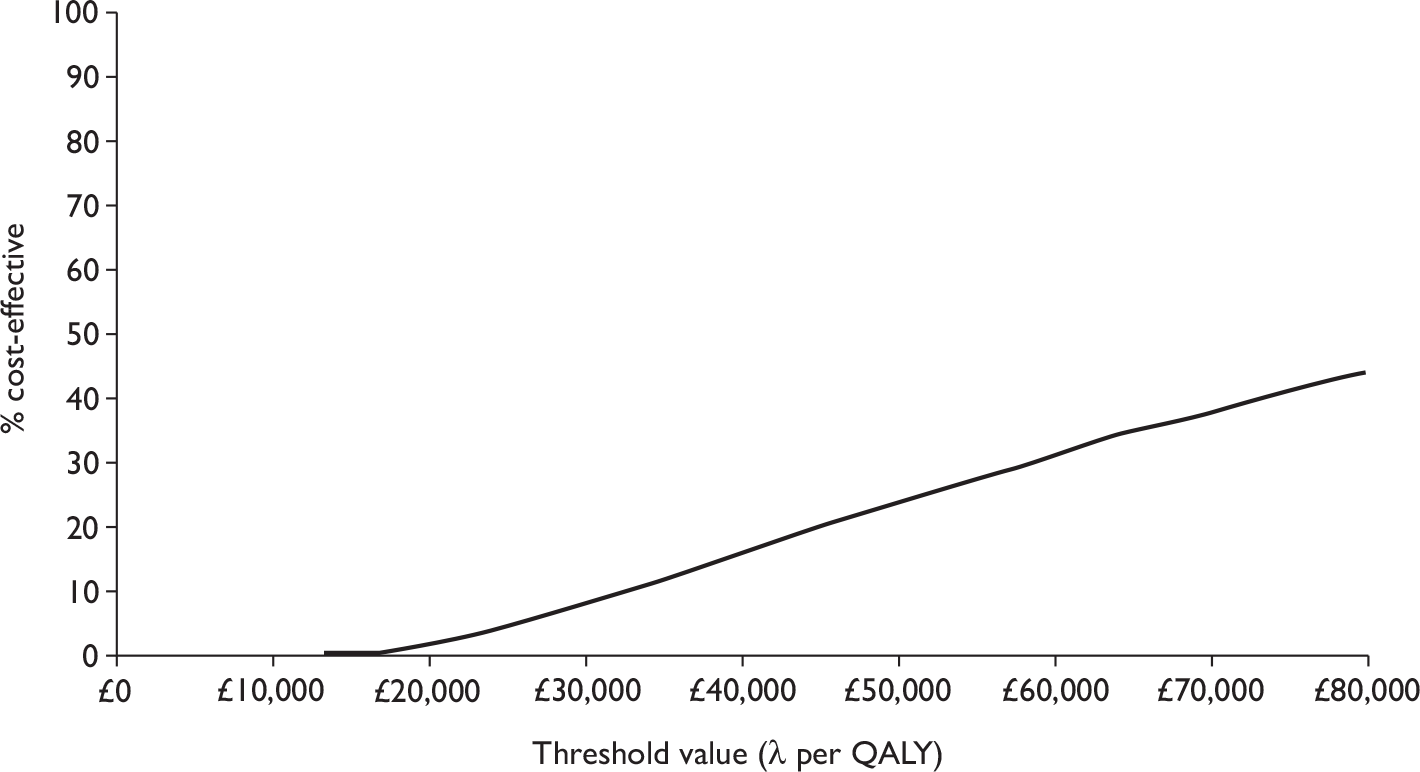
Discussion of adalimumab industry submission
Neither the analysis of the 56-week CHARM67 trial data nor the lifetime adalimumab economic model used the modified ITT analysis on which the major clinical findings of the published CHARM67 trial were based. In reviewing the economic evidence, there are concerns over the comparators used in the adalimumab model. Given the structure of the CHARM67 trial, SC could have been compared with an induction only dose of adalimumab, an until-flare maintenance regimen (based on blinded treatment in CHARM67) and a lifetime regimen (based on blinded and open treatment in CHARM67). As the results for an induction only regime appear as the ‘standard care’ arm of the CHARM67 trial, it should have been included in the economic submission.
Previous use of other anti-TNF therapy is an important predictor of response to adalimumab that is not addressed within the industry submission. In the CHARM67 trial, patients with previous experience of anti-TNF therapy were excluded only if they had no clinical response to the therapy, or had used it in the past 12 weeks. Fifty per cent of all CHARM67 patients had had prior exposure to anti-TNF therapy. As the clinical response was superior in those who had not previously received anti-TNF treatment, it is highly likely that cost-effectiveness would be superior in this group. Given that the 56-week revised model suggested a relatively high cost-effectiveness ratio even for the severe subgroup, this may be an important consideration.
A lack of clarity over the source and interpretation of data has hampered the analysis of the economic submission. Overall, the economic model met most of the requirements of the NICE reference case93 (Table 50), but crucial elements of the model could not be verified. The analysis here has attempted to address concerns over the methodology and interpretation of the economic model. It appears that the cost-effectiveness ratio for moderate CD patients is particularly high at 56 weeks, and it is not at all clear that this figure will approach cost-effectiveness even over a longer period of time. The cost-effectiveness of adalimumab treatment is far more favourable for patients with severe CD.
| Element of health technology assessment | Principles | Met requirements of NICE reference case |
|---|---|---|
| Defining the decision problem | The scope developed by NICE | Yes |
| Comparator | Alternative therapies routinely used in the NHS | Partial. Not all relevant comparators are used for the adalimumab |
| Perspective on costs | NHS and PSS | NHS only |
| Perspective on outcomes | All health effects on individuals | Yes |
| Type of economic evaluation | Cost-effectiveness analysis | Yes |
| Time horizon | Sufficient to reflect any differences in costs or outcomes between the technologies compared | Not applicable, given limits of evidence based |
| Synthesis of evidence on outcomes | Based on a systematic review | Partial. Details unclear and not necessarily reproducible |
| Measure of health benefits | QALYs | Yes |
| Description of health states for calculation of QALYs | Health states described using a standardised and validated generic instrument | Yes |
| Method of preference elicitation for health-state valuation | Choice-based method, for example, time trade-off, standard gamble (not rating scale) | Yes |
| Source of preference data | Representative sample of the public | No. Patient values used |
| Discount rate | An annual rate of 3.5% on both costs and health effects | Yes |
| Equity position | An additional QALY has the same weight regardless of the other characteristics of the individuals receiving the health benefit | Yes |
| Modelling methods | Structural assumptions and data inputs clearly documented and justified | No |
| Probabilistic sensitivity analysis should be conducted | Yes |
Industry submission comparison of adalimumab and infliximab maintenance therapies
The Abbott submission4 also included a cost model comparing adalimumab and infliximab maintenance regimens. The stated aim of the maintenance comparison was to compare adalimumab against infliximab on the basis that infliximab is the alternative most likely to be displaced by the prescription of adalimumab. However, neither adalimumab maintenance nor infliximab maintenance would be the most appropriate comparator in such an analysis. Owing to a lack of trial results comparing these treatments directly, the comparison is secondary in nature.
The infliximab comparator used appears to combine those who were judged to be responders and non-responders on the 5 mg/kg arm of ACCENT I2,3 using the Rutgeerts et al. 2 data including both 5 mg/kg standard dosage and 10 mg/kg ‘as needed’ dosage. The adalimumab comparator used adalimumab maintenance at 80-mg/40-mg induction with 40-mg dosage e.o.w. The adalimumab outcomes were found using a weighted sample from the CHARM67 trial for those with CDAI between 220 and 400 (in line with ACCENT I2,3) and with weights derived so that the same gender distribution, median age and CDAI quartile figures (lower quartile, median, upper quartile) held across the infliximab and modified adalimumab groups. Those with CDAI above 400 were excluded from the analysis for comparability with the ACCENT I2,3 trial. Those who had previously used anti-TNF treatments were not excluded from the adalimumab group, although this was an exclusion criterion in ACCENT I. 2,3 Missing data for both comparators were inferred using LVCF.
The adalimumab arm costs were found by assuming that all those receiving adalimumab and not responding at week 12 would have continued to receive it, which led to higher costs than in the main model. The infliximab arm drug costs were assumed to include an average wastage of 0.5 vials per infusion. Hospitalisation costs were estimated using the Poisson regression for the adalimumab group and observed hospitalisations (plus inferred data from the adalimumab group) in the infliximab group. The model used an excess hospitalisation of 0.098 per infliximab patient per 56 weeks.
As the infliximab data used remission/non-remission rather than the four health states of the main adalimumab model, the health status based costs were estimated using the frequencies reported in Bassi et al. 37 A cost of £38.48 per week was calculated for non-remission costs, and the cost of remitted patients per week was assumed to be £8.45. Overall, the model suggested an excess cost for infliximab patients of £4414 over 56 weeks, of which the majority was due to medication costs (£3526).
While this model also attempted to compare health outcomes, no summary quantitative figures were provided on which to base a cost–consequences analysis, cost-effectiveness or a cost–utility analysis. Using the proportion of patients in remission (partially inferred using LVCF assumptions), it was claimed that adalimumab led to a higher proportion of patients in remission from week 6 onwards. In conjunction with the cost findings, the model claimed that adalimumab maintenance dominated infliximab maintenance.
This model is reported but not analysed in depth. The model compared adalimumab maintenance against infliximab maintenance without comparing either against a standard treatment. As a comparison of non-standard treatments, it fell outside the scope of the assessment. Furthermore, given that both adalimumab and infliximab maintenance appeared to have ICERs far outside the suggested ranges, the results of this model are of little practical relevance to the decision problem faced.
It was also noted that the infliximab regimen modelled here included the 10 mg/kg dosage only. Given the uncertainty relating to which treatments were actually received by patients in ACCENT I,2,3 on what basis these treatments were received, and to what degree the treatments received would be legitimate in a NHS context, it would be difficult to place any confidence in this model.
Independent economic assessment
Introduction
The overall decision problem for this appraisal was ‘What is the cost-effectiveness of anti-TNFs in the management of moderate-to-severe CD in the UK NHS?’. In order to undertake cost-effectiveness analyses to address this decision problem it was necessary to (a) define moderate and severe CD; (b) specify the specific roles for anti-TNFs in the management of CD that were to be evaluated; and (c) specify the patient groups for whom cost-effectiveness would be assessed. The first part of this section addresses these questions. The second part describes the de novo cost-effectiveness model developed to answer the decision problem, and provides base-case results. A series of sensitivity analyses to explore the impact of uncertainty in key model parameters on the estimates of cost-effectiveness were then undertaken. The section finishes with a discussion of the implications of the results of this model.
Disease severity can be expressed in terms of current status or life course. It can be measured using a wide range of indices including frequency of symptoms, severity of symptoms, biochemical activity levels and intensity of treatment required. Available evidence does not provide a strong basis for differentiating CD severity in terms of life course. Munkholm et al. 25 reported that ‘The clinical course of CD differs markedly over time, with ever-relapsing cases, to a quiescent course with remission for several years, interrupted by years with relapse’. No predictive factors have been found for the subsequent course with regard to age, sex, extent of disease at diagnosis and treatment in the year of diagnosis.
The current severity of CD is difficult to assess, and a global measure encompassing clinical, endoscopic, biochemical and pathological features is not available. 39 The most widely used disease activity measures include the CDAI, the HBI or Simple Index, a simplified version of the CDAI, and the PDAI. A commonly used HRQoL measure is the IBDQ. Other measures include the CDEIS.
The CDAI is the measure used most extensively in the anti-TNF clinical trials. It measures current disease severity using a recall period of the previous 7 days. Variables captured in the measure include number of liquid stools, abdominal pain, general well-being, extraintestinal complications, use of antidiarrhoeal drugs, abdominal mass, haematocrit and body weight. Scores range from 0 to 600. Values of < 150 are suggestive of quiescent disease (remission) and values > 450 are associated with very severe disease. 40 Some investigators have arbitrarily labelled CDAI scores of 150–219 as mildly active disease and scores of 220–450 as moderately active disease. 39
Given that the anti-TNF trials use CDAI to measure disease severity and to determine response, the cost-effectiveness analysis used the following definitions of disease severity:
-
severe disease: CDAI > 300
-
moderate disease: 220 < CDAI ≤ 300.
It should be noted that in line with the decision problem and the use of CDAI in the trials, this definition says nothing about the frequency of relapse. A patient who has been in remission for 5 years and relapses with moderate disease refractory to standard therapy is as much a target for treatment with anti-TNFs as a patient who has had two relapses in the last 12 months, with moderate disease that is refractory to standard therapy.
The scope for this appraisal identified three categories of use of anti-TNFs in the management of CD: induction, episodic and maintenance. There is some uncertainty as to the precise definition of each of these categories.
Maintenance therapy is perhaps the most straightforward to define. It can be described as the chronic use of anti-TNF therapy to maintain remission in patients who have responded (and continued to respond) to anti-TNF therapy when in relapse. In maintenance therapy, the key challenges in arriving at a working definition are:
-
What is the criterion for defining a patient as a responder? Is it the achievement of remission (achieving a CDAI score below a defined threshold) or improvement (a defined minimum fall in their CDAI score)?
-
In those currently receiving treatment, is non-response identified by looking at their response over a period of time, or their response after a certain number of treatments?
Defining ‘episodic’ treatment is less straightforward (see Glossary). Seven different working definitions of episodic treatment were identified in the literature and submissions to this appraisal. In the previous appraisal of anti-TNFs in the management of CD, episodic treatment was defined as giving up to three additional courses of treatment when a patient experienced a disease relapse if that patient initially responded to anti-TNF therapy. The relapse could have occurred once in several years or much more frequently. The key uncertainties in this definition are the same as with maintenance therapy above.
Induction treatment is the use of anti-TNF therapy with the aim to achieve remission. It is not straightforward to draw a distinction between repeated use of anti-TNF as induction on the one hand and episodic therapy as described above. Induction therapy may merely be the initial application of anti-TNF to a patient in relapse which establishes his or her responder status prior to the subsequent provision of ‘episodic’ or maintenance therapy. To consider the cost-effectiveness of induction therapy divorced from its value in informing future decisions on ‘episodic’ or maintenance therapy would be clinically unrealistic and would produce an inaccurate estimate of its cost-effectiveness. As with ‘episodic’ and maintenance treatment, the definition of response and the identification of responders are important issues.
Given the problems with assessing the cost-effectiveness of induction therapy in isolation, one-off induction therapy has not been modelled. Instead, the cost-effectiveness of ‘induction’ as a series of individual induction treatments received when a patient falls into relapse that is non-responsive (or given past treatment expected to be unresponsive) to standard therapy was examined. Thus a repeated reinduction treatment was considered here. Each individual induction treatment was composed of an appropriate dosage regimen based on clinical trial information. Those who did not respond within 8 weeks of induction (i.e. after induction doses at 0, 2 and 6 weeks for infliximab and 0 and 2 weeks for adalimumab) were deemed non-responders and transferred to SC. In maintenance, treatment occurred every 8 weeks for infliximab and every 2 weeks for adalimumab. On relapse, doses increased to match the higher induction regime. For infliximab, treatment occurred at relapse, relapse + 2 weeks and relapse + 6 weeks. For adalimumab, treatment occurred at relapse (at the higher induction dose), then relapse + 2 weeks, relapse + 4 weeks and relapse + 6 weeks at the standard dose in line with maintenance treatment. Remission status following this reinduction dose was assessed at relapse + 8 weeks, and subsequent treatment was determined by whether or not remission was achieved: maintenance treatment continued where remission was achieved by 8 weeks; treatment reverted to SC where remission was not achieved at 8 weeks.
There are a number of alternatives to defining responder status. Within the trials, responders were defined in two distinct ways: (1) patients whose CDAI improved by a pre-specified amount following administration of anti-TNF; and (2) patients who achieved remission following administration of anti-TNF. For the purpose of economic evaluation the first approach to defining a responder was problematic as it said nothing about the relative improvement in health for any given reduction in CDAI score. Both the health gain associated with any given improvement in the CDAI and the value attached to the health gain would depend upon the pre-treatment CDAI. Defining responders using a pre-specified improvement in CDAI does not differentiate between patients for whom treatment controls the disease and patients for whom treatment merely reduces the severity of the symptoms. Thus it was not possible to ascribe a robust utility value for the health of responders defined in this way. By contrast, patients in remission (CDAI < 150) will typically have few CD-specific symptoms and this is a health state for which it would be possible to ascribe a robust utility value. For this reason, response was defined as achieving remission following anti-TNF therapy. This method of defining responders may bias results against treatment in patients with severe disease for whom achieving an absolute reduction in CDAI score may be more difficult. By using an absolute score for response rate it is not possible to take account of all possible health benefits that a patient may gain from a large change in relative response. Yet, as stated above, there is no possible way of determining the health gain associated with a particular relative reduction in CDAI score given the data available. So even when considering the possible bias this approach might introduce in to the model, it is still the only methodologically appropriate way of valuing health states.
The scope for this appraisal identified a number of patient groups for whom the NICE Appraisals Committee would be interested in obtaining specific estimates of the cost-effectiveness of anti-TNF therapy: adults; children; severe CD; moderate CD; fistulising; and non-fistulising.
The randomised placebo-controlled trial evidence for the anti-TNFs did not include paediatric trials, even though infliximab does have a licence for use in the paediatric population. In the absence of estimates of effectiveness that can be used to model the magnitude of effect for anti-TNFs compared with SC, robust modelling of the cost-effectiveness of anti-TNF therapy in a paediatric population was not possible. However, to assist the committee in its deliberations; a scenario analysis using the adult models was presented, where paediatric administration and drug costs were substituted for the adult costs. This was equivalent to assuming that treatment was equally effective in paediatric and adult populations and that all other costs of treatments and utilities gained remained the same.
Separate models are presented for patients with moderate and severe disease, as the value of the health gain associated with remission will be systematically different for these two patient groups, as would be the likely costs of managing relapse.
While the trials of anti-TNFs differentiated between fistulising and non-fistulising disease, it was not possible to identify a long-term usual care cohort study for fistulising patients. In the absence of this evidence, the trial-based evaluations submitted by the manufacturers provided the best available estimates of the likely cost-effectiveness of treatment in the fistulising population. However, it is important to emphasise the difference in the characteristics of the trial populations and the characteristics of the population who are the focus of the decision problem for this appraisal as defined in the NICE scope – i.e. all patients with moderate-to-severe disease that is refractory to standard treatment.
The trial populations were patients in relapse. However, frequency of relapse was not an inclusion criterion for treatment in the decision problem for this appraisal. Patients who relapse more frequently have more capacity to benefit from an effective treatment and therefore, assuming effectiveness is not lower in patients who relapse frequently, treatment would be more cost-effective in those patients than in the population defined for this appraisal.
In summary, de novo cost-effectiveness analyses for adults with moderate-to-severe CD were presented, where response was defined as remission within 8 weeks. The objective of the cost-effectiveness analysis was to estimate the incremental cost per QALY between (a) SC, (b) induction therapy and (c) maintenance therapy for moderate and for severe disease.
Model structure
The NICE methods guide recommends that models of chronic diseases normally adopt a lifetime horizon because exacerbations in chronic diseases usually relate to reduced life expectancy. 93 Analysing a cohort of patients followed from 1993–4 to 2003–4, Wolters et al. 97 reported that there was no significant CD-specific mortality, with the age at diagnosis being the only significant association with mortality. As none of the clinical trials provided evidence of an impact upon mortality, it was reasonable to assume there was no differential mortality rate and, therefore, a lifetime horizon would not add meaningfully to the precision of the cost-effectiveness estimate. The time horizon for the Markov model was 1 year (with sensitivity analyses of 5, 10 and 20 years) and the cycle duration was 4 weeks, i.e. the model had 13 cycles and did not include mortality. All models were constructed and analysed in treeage pro 2008 (TreeAge Software Inc., Williamstown, MA, USA). Both the induction and maintenance model started with a cohort of patients suffering from an SC-refractory relapse.
In the absence of time-dependent transition probabilities for the natural history of the cohort with moderate-to-severe CD at onset, a natural history cohort that reflects the average transition rate over time was used. If the appropriate data had been available, the preferred model structure would have been a time-dependent state transition model, allowing the possibility to capture more accurately the performance of the therapies in the early phase of the disease, where relapse rates are notably higher, as in the short-duration RCT data provided by the manufacturers. In the absence of such data a balance must be struck between the potential underestimate of treatment in the early stages of the disease and the overestimate of the value of maintenance therapy in longer established disease.
Conceptually, the cost-effectiveness model developed was an expanded version of a four-state Markov model. At any time and on any given treatment, a patient was in remission, in relapse, undergoing surgery or in post-surgery remission. For ease of identification, each state is identified by a prefix showing the type of treatment received be it SC, maintenance (MNT – in this chapter only when referring to the Markov model) or induction (IND – in this chapter only when referring to the Markov model). For example, those who received SC therefore occupied states SC remission, SC relapse, SC surgery or SC post-surgery remission within the Markov model. Figure 40 shows the basic structure of the SC model.
FIGURE 40.
Schematic of SC arm of de novo cost-effectiveness model. Incr, incremental; Init, initial; rwd, reward.
The models for induction and maintenance treatments were conceptually similar: each involved one set of states for anti-TNF treatment and another for SC treatments (where necessary). For IND there were three treatment-related states – remission and surgery-related (IND remission, IND surgery, IND post-surgery remission). There were also two relapse states, one for each 4-week period in which response was assessed (IND relapse and IND relapse 2). Those failing to respond to treatment after 8 weeks of continued relapse (i.e. passing through both IND relapse and then IND Relapse 2) transited to the SC states (SC remission, SC relapse, SC surgery, SC post-surgery remission). In order to correctly assign anti-TNF costs, this occurred through a temporary transition state which was identical to SC relapse in its transition probabilities and utility and which only differed in its costs. Maintenance treatment used the same structure as induction.
Transition probabilities for the SC states were based on the Silverstein et al. 26 cohort and the derivation process is detailed in Standard care transition probabilities. Transition probabilities for the IND model assigned a treatment effect by using relapse to remission probabilities from RCT evidence. Transition probabilities for the MNT model assigned the treatment effect by using the this same relapse to remission data and, by inference, a lower remission to relapse rate. Details of the derivation of IND and MNT models are given in Induction transition probabilities and Maintenance transition probabilities below.
Standard care transition probabilities
The population of interest for this appraisal was an inclusive one, rather than the tightly defined populations often found in clinical trials. Therefore, it was important to identify evidence from a population cohort that included patients who were resistant to standard therapy. It was also important the cohort reported data from the time before the advent of biological therapy.
The frequently cited study by Silverstein et al. ,26 as mentioned above, met these criteria. This study reported a 2-monthly transition matrix estimated from 20 years’ follow-up of an inception cohort of 174 patients. Patients were characterised as being in one of eight states: remission, mild, drug-responsive, drug-dependent, drug-refractory, surgery, post-surgery remission and death. Figure 41 shows a schematic of Silverstein et al. ’s26 Markov model.
FIGURE 41.
Schematic of Silverstein et al. ’s26 clinical classification. Incr, incremental; Init, initial; rwd, reward.
As the model did not include death as a state, this had to be removed from the Silverstein et al. 26 matrix when estimating transition probabilities. A relationship existed between the SC remission and SC relapse states and the Silverstein et al. 26 states for mild disease (mild) and active disease (drug-dependent, drug-responsive, drug-refractory). Within Silverstein et al. ,26 patients were effectively managed with non anti-TNF treatments in both the drug-dependent and drug-responsive states; these states were therefore combined to form the SC remission state. In contrast, in the drug-refractory state, the patient was not effectively managed by treatment and was not responding to SC. It was this group who were candidates for anti-TNF treatment and hence this group was used to form the SC relapse state. The process for removing mild and death states from the Silverstein et al. 26 model was slightly more complex and appears below. The four-state matrix: remission, relapse, surgery and post-surgery remission was derived using the following steps.
Begin with the Silverstein et al. 26 transition matrix. Reprinted with permission from Elsevier.
| Silverstein et al.26 original – 2-month transition probabilities | ||||||||
|---|---|---|---|---|---|---|---|---|
| Remission | Mild | Drug-responsive | Drug-dependent | Drug-refractory | Surgery | Post-surgery | Death | |
| Remission | 0.89688 | 0.07016 | 0.00939 | 0.00639 | 0.00363 | 0.00793 | 0.00395 | 0.00167 |
| Mild | 0.05751 | 0.90952 | 0.00829 | 0.00619 | 0.00968 | 0.00585 | 0.00281 | 0.00015 |
| Drug-responsive | 0.25261 | 0.2217 | 0.41262 | 0.02563 | 0.00817 | 0.04569 | 0.02733 | 0.00626 |
| Drug-dependent | 0.05274 | 0.03484 | 0.00193 | 0.88626 | 0.00592 | 0.01071 | 0.00543 | 0.00217 |
| Drug-refractory | 0.06174 | 0.05888 | 0.00392 | 0.02599 | 0.74207 | 0.06435 | 0.03466 | 0.00839 |
| Surgery | 0.00657 | 0.06906 | 0.00801 | 0.03421 | 0.02397 | 0.33714 | 0.52022 | 0.00082 |
| Post-surgery | 0.00054 | 0.00849 | 0.001 | 0.00152 | 0.00096 | 0.00436 | 0.98126 | 0.00187 |
| Death | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Step 1: Removing death. It was supposed that death from all states was equally likely. The chance of death in each (t0) state was divided by six and this was added to the six non-mild, non-death states.
| Step 1 – Identify and assign increase in probabilities following the removal of the death state Uses original Silverstein et al.26 matrix, without mild or dead |
||||||||
|---|---|---|---|---|---|---|---|---|
| Remission | Mild | Drug-responsive | Drug-dependent | Drug-refractory | Surgery | Post-surgery | Death | |
| Remission | 0.00028 | – | 0.00028 | 0.00028 | 0.00028 | 0.00028 | 0.00028 | – |
| Mild | – | – | – | – | – | – | – | – |
| Drug-responsive | 0.00104 | – | 0.00104 | 0.00104 | 0.00104 | 0.00104 | 0.00104 | – |
| Drug-dependent | 0.00036 | – | 0.00036 | 0.00036 | 0.00036 | 0.00036 | 0.00036 | – |
| Drug-refractory | 0.00140 | – | 0.00140 | 0.00140 | 0.00140 | 0.00140 | 0.00140 | – |
| Surgery | 0.00014 | – | 0.00014 | 0.00014 | 0.00014 | 0.00014 | 0.00014 | – |
| Post-surgery | 0.00031 | – | 0.00031 | 0.00031 | 0.00031 | 0.00031 | 0.00031 | – |
| Death | – | – | – | – | – | – | – | – |
Step 2: Removing the mild state. A more complex process was used for the mild state. Here, the issue was not now where one entered the state from, but where one would exit to after leaving mild.
-
2.1. Each of the non-death transition probabilities out of mild were deflated (subtract?) by the total chance of leaving the mild state (0.090). These probabilities for the exit state for mild were: remission, 0.636; drug-responsive, 0.092; drug-dependent, 0.068; drug-refractory, 0.107; surgery, 0.065; and post-surgery recovery, 0.031.
-
2.2. These exit probabilities were multiplied by the chance of entering mild from each of the other initial health states (t0) and used to distribute the probabilities. Here, the chance of a person in remission remaining in remission increased by 0.070 (the chance of leaving for mild) × 0.636 (the chance of moving from mild to remission).
-
2.3. The initial Silverstein et al. 26 transition probabilities were increased by the probabilities in (2.1) and (2.2).
| Step 2 – Identify and assign increase in probabilities following the removal of the death state, and apply both increases | ||||||||
|---|---|---|---|---|---|---|---|---|
| 2.1 Identify transitions out of mild from Silverstein et al.26 matrix excluding death | ||||||||
| Remission | Mild | Drug-responsive | Drug-dependent | Drug-refractory | Surgery | Post-surgery | Death | |
| 1. Mild | 0.05751 | 0.90952 | 0.00829 | 0.00619 | 0.00968 | 0.00585 | 0.00281 | – |
| 2. Mild (excluding mild → mild) | 0.63667 | – | 0.09177 | 0.06853 | 0.10716 | 0.06476 | 0.03111 | – |
| 2.2 Identify increase in probability using row 2 above, the row-specific transition to mild in Silverstein et al.26 | ||||||||
| Remission | Mild | Drug-responsive | Drug-dependent | Drug-refractory | Surgery | Post-surgery | Death | |
| Remission | 0.04467 | – | 0.00644 | 0.00481 | 0.00752 | 0.00454 | 0.00218 | – |
| Mild | – | – | – | – | – | – | – | – |
| Drug-responsive | 0.14115 | – | 0.02035 | 0.01519 | 0.02376 | 0.01436 | 0.00690 | – |
| Drug-dependent | 0.02218 | – | 0.00320 | 0.00239 | 0.00373 | 0.00226 | 0.00108 | – |
| Drug-refractory | 0.03749 | – | 0.00540 | 0.00403 | 0.00631 | 0.00381 | 0.00183 | – |
| Surgery | 0.04397 | – | 0.00634 | 0.00473 | 0.00740 | 0.00447 | 0.00215 | – |
| Post-surgery | 0.00541 | – | 0.00078 | 0.00058 | 0.00091 | 0.00055 | 0.00026 | – |
| Death | – | – | – | – | – | – | – | – |
| 2.3 Identify modified Silverstein et al.26 matrix using increases from Step 1 and Step 2.2 matrices | ||||||||
| Remission | Mild | Drug-responsive | Drug-dependent | Drug-refractory | Surgery | Post-surgery | Death | |
| Remission | 0.94183 | – | 0.01611 | 0.01148 | 0.01143 | 0.01275 | 0.00641 | – |
| Mild | – | – | – | – | – | – | – | – |
| Drug-responsive | 0.39480 | – | 0.43401 | 0.04187 | 0.03297 | 0.06109 | 0.03527 | – |
| Drug-dependent | 0.07528 | – | 0.00549 | 0.88901 | 0.01002 | 0.01333 | 0.00688 | – |
| Drug-refractory | 0.10063 | – | 0.01072 | 0.03142 | 0.74978 | 0.06956 | 0.03789 | – |
| Surgery | 0.05067 | – | 0.01448 | 0.03908 | 0.03151 | 0.34175 | 0.52250 | – |
| Post-surgery | 0.00626 | – | 0.00209 | 0.00241 | 0.00218 | 0.00522 | 0.98184 | – |
| Death | – | – | – | – | – | – | – | – |
Step 3. The drug-responsive state has Markov probabilities summing to 1.00001 due to rounding error in the original paper. These have now been corrected.
| Step 3 – Correct for summation error in drug-responsive (no changes to five decimal places in matrix) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Remission | Mild | Drug-responsive | Drug-dependent | Drug-refractory | Surgery | Post-surgery | Death | |
| Remission | 0.94183 | – | 0.01611 | 0.01148 | 0.01143 | 0.01275 | 0.00641 | – |
| Mild | – | – | – | – | – | – | – | – |
| Drug-responsive | 0.39480 | – | 0.43401 | 0.04187 | 0.03297 | 0.06109 | 0.03527 | – |
| Drug-dependent | 0.07528 | – | 0.00549 | 0.88901 | 0.01002 | 0.01333 | 0.00688 | – |
| Drug-refractory | 0.10063 | – | 0.01072 | 0.03142 | 0.74978 | 0.06956 | 0.03789 | – |
| Surgery | 0.05067 | – | 0.01448 | 0.03908 | 0.03151 | 0.34175 | 0.52250 | – |
| Post-surgery | 0.00626 | – | 0.00209 | 0.00241 | 0.00218 | 0.00522 | 0.98184 | – |
| Death | – | – | – | – | – | – | – | – |
Step 4. Steps 1–3 produce a matrix in six states. The states remission, drug-responsive, and drug-dependent were then combined into a single remission state.
-
4.1. The chance of being in any one of these states was assessed using figures in Silverstein et al. 26 Of the three states, there was an 89.1% chance of being in remission, a 2.1% chance of being in a drug-responsive state, and an 8.8% chance of being in a drug-dependent state.
-
4.2. The chance of remaining in the (broader) remission state was calculated as the average of the chance of moving to any of the three earlier states from the three earlier states, weighted by the 89.1%, 2.1% and 8.8% from (4.1).
-
4.3. The chance of moving from this (broader) remission state to a relapse, surgical or post-surgery state was also taken as a similar weighted average.
-
4.4. The chance of transiting to the (broader) remission state was calculated as the sum of the probabilities of the earlier states comprising the (broader) remission state.
| Step 4 – Combine states for remission, drug-responsive and drug-dependent into a single state | ||||||||
|---|---|---|---|---|---|---|---|---|
| 4.1 Identify chances of being in each state from Silverstein et al.26 | ||||||||
| Remission | Mild | Drug-responsive | Drug-dependent | Drug-refractory | Surgery | Post-surgery | Death | |
| Time spent in states (total person-years) | 737.38 | 471.35 | 17.53 | 72.50 | 37.37 | 26.95 | 570.08 | – |
| Selected states (total person-years) | 737.38 | – | 17.53 | 72.50 | – | – | – | |
| Probability of being in state | 89.12% | – | 2.12% | 8.76% | – | – | – | – |
| Identify total chances of remaining in any remission-type state | ||||||||
| Remission | Mild | Drug-responsive | Drug-dependent | Drug-refractory | Surgery | Post-surgery | Death | |
| Remission | 0.96941 | – | – | – | 0.01143 | 0.01275 | 0.00641 | – |
| Mild | – | – | – | – | – | – | – | – |
| Drug-responsive | 0.87067 | – | – | – | 0.03297 | 0.06109 | 0.03527 | – |
| Drug-dependent | 0.96978 | – | – | – | 0.01002 | 0.01333 | 0.00688 | – |
| Drug-refractory | 0.14277 | – | – | – | 0.74978 | 0.06956 | 0.03789 | – |
| Surgery | 0.10424 | – | – | – | 0.03151 | 0.34175 | 0.52250 | – |
| Post surgery | 0.01076 | – | – | – | 0.00218 | 0.00522 | 0.98184 | – |
| Death | – | – | – | – | – | – | – | – |
| 4.2, 4.3, 4.4 Apply chances of being in each state from Silverstein et al.26 to weight outcomes | ||||||||
| Remission | Mild | Drug-responsive | Drug-dependent | Drug-refractory | Surgery | Post-surgery | Death | |
| Remission | 0.96735 | – | – | – | 0.01176 | 0.01383 | 0.00706 | – |
| Mild | – | – | – | – | – | – | – | – |
| Drug-responsive | – | – | – | – | – | – | – | – |
| Drug-dependent | – | – | – | – | – | – | – | – |
| Drug-refractory | 0.14277 | – | – | – | 0.74978 | 0.06956 | 0.03789 | – |
| Surgery | 0.10424 | – | – | – | 0.03151 | 0.34175 | 0.52250 | – |
| Post-surgery | 0.01076 | – | – | – | 0.00218 | 0.00522 | 0.98184 | – |
| Death | – | – | – | – | – | – | – | – |
Step 5. This gave a matrix in four states (remission, relapse, surgery and post-surgery remission) for 2-monthly cycles. This was modified to form a 4-week transition matrix by halving the figures off the main diagonal and setting the diagonal entries to one minus the remaining values in each row.
| Step 5 – Transform to 1-month transition probabilities | ||||||||
|---|---|---|---|---|---|---|---|---|
| Remission | Mild | Drug-responsive | Drug-dependent | Drug-refractory | Surgery | Post-surgery | Death | |
| Remission | 0.98368 | – | – | – | 0.00588 | 0.00691 | 0.00353 | – |
| Mild | – | – | – | – | – | – | – | – |
| Drug-responsive | – | – | – | – | – | – | – | – |
| Drug-dependent | – | – | – | – | – | – | – | – |
| Drug-refractory | 0.07139 | – | – | – | 0.87489 | 0.03478 | 0.01894 | – |
| Surgery | 0.05212 | – | – | – | 0.01575 | 0.67087 | 0.26125 | – |
| Post-surgery | 0.00538 | – | – | – | 0.00109 | 0.00261 | 0.99092 | – |
| Death | – | – | – | – | – | – | – | – |
Step 6. Reformat the table and add evidence review group health-state labels to give the final transition matrix used in the evidence review group analysis.
| Step 6 – Apply labels and reformat | ||||
|---|---|---|---|---|
| SC remission | SC relapse | SC surgery | SC post-surgery remission | |
| SC remission | 0.98368 | 0.00588 | 0.00691 | 0.00353 |
| SC relapse | 0.07139 | 0.87489 | 0.03478 | 0.01894 |
| SC surgery | 0.05212 | 0.01575 | 0.67087 | 0.26125 |
| SC post-surgery remission | 0.00538 | 0.00109 | 0.00261 | 0.99092 |
Steps 1–5 created the transition matrix in Table 51, which was used to model SC in both the main SC model and the submodels for maintenance and induction care where patients transit to SC.
| SC remission | SC relapse | SC surgery | SC post-surgery remission | |
|---|---|---|---|---|
| SC remission | 0.9837 | 0.0059 | 0.0069 | 0.0035 |
| SC relapse | 0.0713 | 0.8749 | 0.0348 | 0.0189 |
| SC surgery | 0.0521 | 0.0158 | 0.6709 | 0.2613 |
| SC post-surgery remission | 0.0054 | 0.0011 | 0.0026 | 0.9909 |
These probabilities were used for both severe and moderate disease. The clinical course framework described above did not differentiate between these two states and it was not clear how a mild/moderate division could have been placed upon the active disease patients reported in Silverstein et al. 26 The implicit assumption was that SC treatments were equally likely to achieve remission in moderate and severe disease.
Similar to the Silverstein et al. 26 analysis, the matrix included transitions to post-surgery remission from relapse and remission states. These transitions were most likely to be an artefact of the maximum likelihood method used to estimate the Silverstein et al. 26 transition matrix. Silverstein et al. 26 did not report complication rates from surgery and thus it was not included as a state in the model here. As the number of these types of transitions were small, it was not considered here to have substantially weakened the Silverstein et al. 26 study as the preferred basis for modelling SC.
Induction transition probabilities
The main difference between the transition matrices for SC and IND was the probability of transiting from relapse to remission. In the SC model, there was a 7.13% chance of a person starting the period in the SC remission state moving to SC relapse at the end of one cycle.
Effectiveness for infliximab and adalimumab treatment were derived from ACCENT I2,3 and CHARM,67 respectively. Neither trial reported 4-week outcomes, and 6-week outcomes were used here as highly favourable estimates of efficacy. At 6 weeks, 63/113 patients in the infliximab 5 mg/kg groups (see Figure 13) and 96/172 patients in the adalimumab 40-mg e.o.w. groups (see Figure 22) were in remission. For both groups, this 6-week effectiveness represented a peak efficacy which fell thereafter. Distributions based on these figures were used to estimate the transition probability from IND relapse to IND remission.
Table 52 shows the IND transition matrix, where the response rate (RESP in the table) is the estimated transition probability for the respective anti-TNF treatment. All individuals started in IND relapse, and those who were to remain in relapse moved to the second relapse state (IND relapse 2). Those who were to remain in relapse for a third 4-week period were deemed non-responders and entered the transitional state. In this state, a patient received SC and had transition probabilities corresponding to the SC relapse state. Following this, the patient remained solely in SC (the SC states).
| IND remission | IND relapse | IND relapse 2 | Transitional | IND surgery | IND post-surgery remission | SC remission | SC relapse | SC surgery | SC post- surgical remission | |
|---|---|---|---|---|---|---|---|---|---|---|
| IND remission | 0.9837 | 0.0059 | 0.0069 | 0.0035 | ||||||
| IND relapse | RESP | # | 0.0348 | 0.0189 | ||||||
| IND relapse 2 | RESP | # | 0.0348 | 0.0189 | ||||||
| Transitional | 0.0713 | 0.8749 | 0.0348 | 0.0189 | ||||||
| IND surgery | 0.0521 | 0.0158 | 0.6709 | 0.2613 | ||||||
| IND post-surgery remission | 0.0054 | 0.0011 | 0.0026 | # | ||||||
| SC remission | SC transition matrix – Table 51 | |||||||||
| SC relapse | ||||||||||
| SC surgery | ||||||||||
| SC post-surgery remission | ||||||||||
In the probabilistic sensitivity analysis, the response variables (ACCENT_response, CHARM_response) were modelled using the beta distributions, using the values for the effectiveness estimates outlined in Table 53.
| Anti-TNF | Source | Mean | Variable and distribution |
|---|---|---|---|
| Infliximab | Peak (6 weeks) effectiveness from ACCENT I 5 mg/kg arm Figure 13 | 55.7% |
ACCENT_response Beta; α = 63, β = 50 |
| Adalimumab | Peak (6 weeks) effectiveness from CHARM 40-mg e.o.w. arm Figure 22 | 55.8% |
CHARM_response Beta; α = 96, β = 76 |
Figure 42 shows the effectiveness of infliximab per period under the highly favourable estimates used here, with Figure 43 showing the equivalent figures for adalimumab. The mean value for both distributions was similar, with slightly less uncertainty in the CHARM67 data.
FIGURE 42.
Infliximab effectiveness (n = 113).
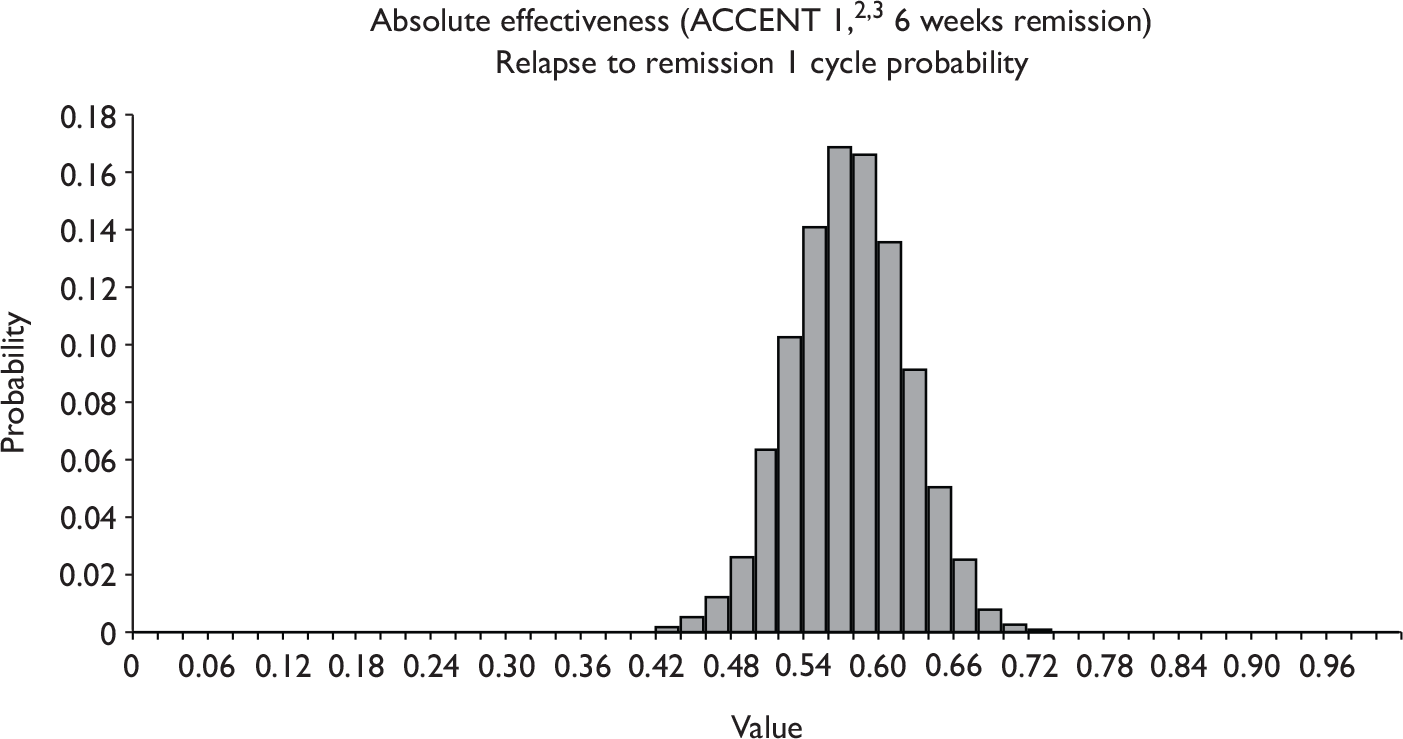
FIGURE 43.
Adalimumab effectiveness (n = 172).
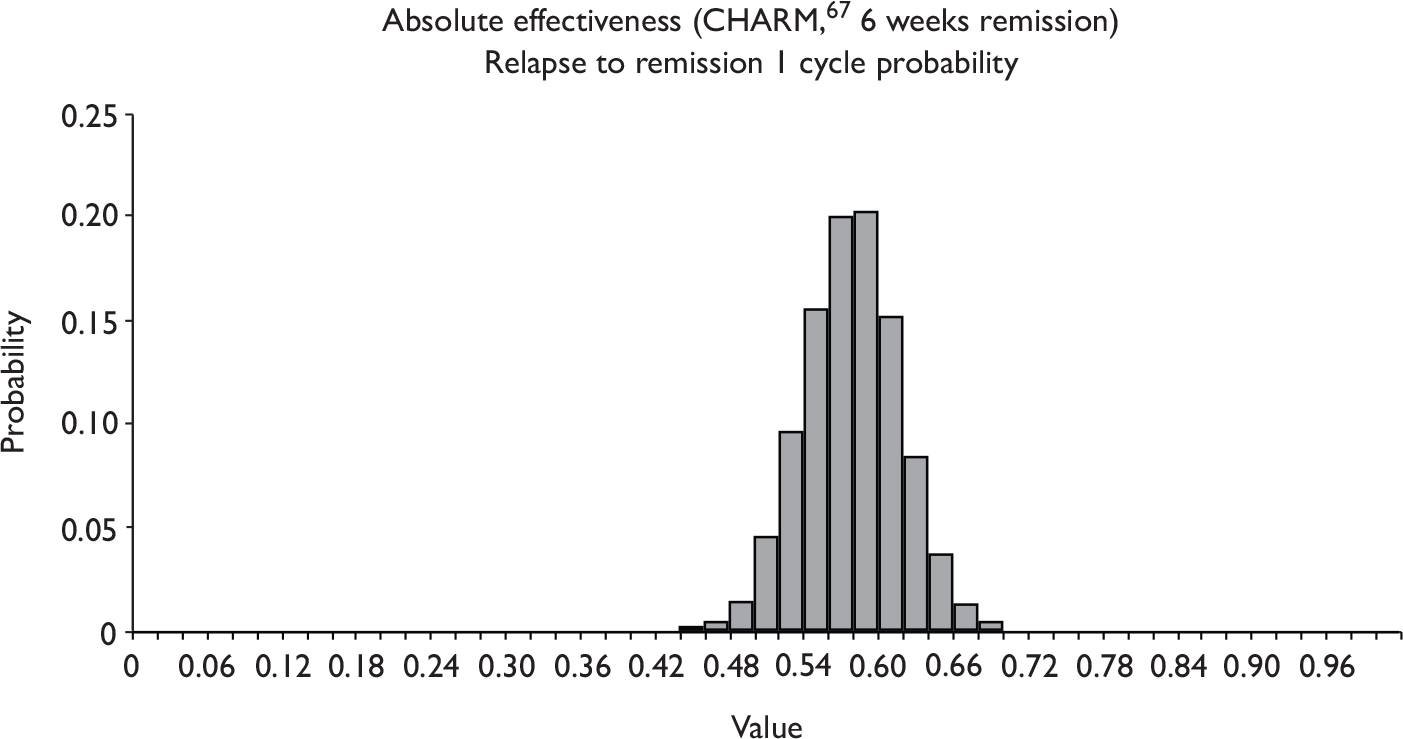
Maintenance transition probabilities
Given that the initial doses in trials were identical between induction and maintenance treatment, it would be expected that maintenance would have had the same initial (and subsequent) effectiveness in increasing the relapse to remission rate as induction treatment. Maintenance treatment was expected to have an additional effect, however, in reducing the probability of an individual relapsing from a remitted state. Ideally, RCT evidence from ACCENT I2,3 and CHARM67 would have included separate remission to relapse and relapse to remission rates. In the absence of this information the inverse of the relapse to remission rate was applied to the remission to relapse SC transition probability. Thus, as anti-TNF maintenance therapy was assumed to give about eight times the chance of entering remission from relapse (56% against 7%), the model assumed that there was one-eighth the chance of relapsing among patients in remission. These values changed depending on the precise value of the response variables (ACCENT_response and CHARM_response) in each iteration of the model. Thus, if the response variable was RESP, then the relative effectiveness in obtaining remission was RESP/0.0713, and the expected MNT remission to MNT relapse rate was 0.0059/(RESP/0.0713).
So here, the incremental benefit of maintenance treatment over induction treatment was expected to be a reduction in the probability of relapse from remission states (MNT remission and MNT post-surgery remission). While the chance of relapse from MNT remission was expected to be very small (0.0059 per period) in the base-case analysis, this baseline probability was increased in one-way sensitivity analyses reported below. Barring labelling, the MNT transition matrix (Table 54) was identical to the IND transition matrix except in two cells: the probability of relapse from remission and the probability of relapse from post-surgery remission.
| MNT remission | MNT relapse | MNT relapse 2 | Transitional | MNT surgery | MNT post-surgery remission | SC remission | SC relapse | SC surgery | SC post- surgical remission | |
|---|---|---|---|---|---|---|---|---|---|---|
| MNT remission | 0.9837 | Rel 1 | 0.0069 | 0.0035 | ||||||
| MNT relapse | RESP | # | 0.0348 | 0.0189 | ||||||
| MNT relapse 2 | RESP | # | 0.0348 | 0.0189 | ||||||
| Transitional | 0.0713 | 0.8749 | 0.0348 | 0.0189 | ||||||
| MNT surgery | 0.0521 | 0.0158 | 0.6709 | 0.2613 | ||||||
| MNT post-surgery remission | 0.0054 | Rel 2 | 0.0026 | # | ||||||
| SC remission | SC transition matrix – Table 51 | |||||||||
| SC relapse | ||||||||||
| SC surgery | ||||||||||
| SC post-surgery remission | ||||||||||
Utilities
Table 55 shows the utility distributions associated with each modelled state. Most utilities were derived from a widely cited study of HRQoL in CD. 78 While this study did not meet the reference case specification in the NICE methods guide,93 in the absence of an alternative study that did meet these criteria, it had the desirable characteristics of providing values derived from a choice-based method (time trade-off), being a well-conducted study and providing utility values for differing severities of disease – the type of data required for this analysis. The only states where utility values were not available concerned surgery, and in the absence of published estimates it was assumed that the average utility for individuals in the major surgery state would be equivalent to EQ-5D state 22222 with utility weight of 0.516.
| Type | States | Variable and distribution |
|---|---|---|
| All remission states | SC remission, IND remission, MNT remission, SC post-surgery remission, IND post-surgery remission, MNT post-surgery remission |
utility_remission Normal; mean = 0.073, SD = 0.00801 |
| Relapse states (severe disease) | SC relapse, IND relapse, IND relapse 2, MNT relapse, MNT relapse 2, transitional |
utility_severe_relapse Normal; mean = 0.056, SD = 0.0012 |
| Relapse states (moderate disease) | SC relapse, IND relapse, IND relapse 2, MNT relapse, MNT relapse 2, transitional |
utility_moderate_relapse Normal; mean = 0.068, SD = 0.0012 |
| Surgery states | SC surgery, IND surgery, MNT surgery |
utility_surgery Normal; mean = 0.039, SD = 0.0012 |
It is possible that the choice of method used to value health states will affect the results of the analysis and increase uncertainty in the results. In this instance, the time trade-off method was selected as this has some methodological consistency with the EQ-5D where EQ-5D results are not available. The difference in the health-state utilities obtained by different methods is a well-known phenomenon and is not, as far as we are aware, a function of the clinical condition under consideration. Rather it reflects methodological uncertainty, not parameter uncertainty. Inflating parameter uncertainty to reflect methodological uncertainty would be methodologically inappropriate and so is not done here. This is not recommended in either the 2004104 or 200893 edition of the NICE methods guide. Indeed, we are not aware of any guide to good practice in economic evaluation that has recommended this, nor are we aware of any methodological paper describing how it might be done if it were deemed to be a desirable thing to do.
Utility values per cycle were calculated by taking the annual utility value and dividing by the number of cycles (13) run by the model. The mean values reported in Table 55 show the per cycle utility associated with each health state along with the distribution used to capture the uncertainty in these estimates. Although in some cases these values may appear high when taken at face value, it is useful to consider how these should be interpreted. In a standard gamble context, respondents giving this value would accept a one in four chance of death for a procedure that returned them to full health. In a time trade-off context, given a life expectancy of 10 years they would give up slightly over 2.5 years in order to be returned to full health. When looked at from this perspective, these values are not necessarily extremely high or of questionable validity but rather reflect patient views on how this illness, which is not life-threatening, affects their day-to-day life.
Anti-TNF costs
In order to identify the initial (and reinduction) cost of anti-TNF treatments it was necessary to consider the induction drug regimen under each anti-TNF for both induction and maintenance treatments. Where relapse occurred at week 0:
-
For infliximab treatment, induction (infliximab IND) involved a loading dose comprising treatment at 0, 2 and 6 weeks at 5 mg/kg (a loading dose) irrespective of response status at 4 weeks.
-
Infliximab maintenance treatment (infliximab MNT) also involved the same treatment at 0, 2 and 6 weeks, but with additional doses at weeks 14 and 20 and every subsequent 8 weeks for those entering remission (except in the case of subsequent relapse).
-
Adalimumab induction (adalimumab IND) involved a loading dose of 80 mg at week 0 and 40 mg at week 2, with no further treatment.
-
Adalimumab maintenance (adalimumab MNT) involved the induction loading doses at weeks 0 and 2, with additional doses of 40 mg at weeks 4 and 6 regardless of response at week 4 (i.e. in either MNT remission or MNT relapse 2).
Infliximab administration costs were taken from a previous HTA report,99 at £257.50 per treatment. For a single infliximab treatment, total costs were £1936.42 (assuming four 100-mg vials at £419.73 plus administration costs of £257.50). This was received three times in induction, so infliximab IND costs £5809.26 per relapse set against the IND relapse state. There were no anti-TNF costs levied in any other state within the infliximab IND arm.
For infliximab maintenance, each standard dose after induction provided treatment for 8 weeks, so that the standard cost (post induction, pre relapse) was therefore £968.21 each 4 weeks (0.5 × £1936.42). Following induction (or reinduction given relapse), the first treatment post induction occurred at 14 weeks, so that treatment following relapse could be thought of as purchasing 14 weeks’ worth of treatment. This was 10 weeks beyond what would normally be purchased in a 4-week period. Hence, the cost of relapse for maintenance was reduced by the equivalent of 2.5 4-weekly charges. MNT relapse infliximab costs were then £3388.74 [£5809.26 – (2.5 × £968.21)]. MNT remission, MNT post-surgery remission and MNT relapse 2 were all allocated the standard 4-week costs, while it was assumed that no infliximab was received in a 4-week period where surgery occurred. If a patient failed to respond within 8 weeks of treatment for a relapse, he or she entered the transitional state. Here, 8 weeks’ worth of relapse costs have been previously paid, leaving 6 weeks of unallocated treatment costs (£1452.32) which were set against the transitional state before entering the normal SC states.
For an adalimumab induction arm, three 40-mg doses were assumed to be given, at a cost of £357.50 per dose, so that IND relapse cost £1072.50. No administration costs were included in this value, and no state other than IND relapse incurred an anti-TNF cost. For an adalimumab maintenance arm, all induction doses were incurred within 4 weeks, so that MNT relapse also incurred £1072.50 in anti-TNF costs. MNT relapse 2, MNT remission and MNT post-surgery remission all required two 40-mg doses of adalimumab (£715). Transitional, MNT surgery, and all SC states involved no anti-TNF costs.
Total health-state costs
The model was developed from an NHS/PSS perspective, as per the NICE reference case. 93 Direct NHS costs were modelled as the sum of anti-TNF costs and type-specific health-state costs; no costs related to PSS were identified as part of the modelling process. Where possible, these health-state costs were taken from the NHS Reference Cost database 2005–6. 105 The reference costs for surgery were modelled as the cost of inpatient IBD interventions, while moderate and severe relapse costs were modelled as the cost of IBD outpatient major and intermediate interventions. Post-surgery remission costs were based on outpatient surgical gastrointestinal follow-up. Relapse costs were based on a gastrointestinal admission to hospital. Remission costs were modelled using Bassi et al. 37 and indexed using the PSSRU NHS Pay and Prices Index. 103 The Bassi et al. 37 cost for quiescent CD was used as the cost for the remission state.
Distributions for the probabilistic sensitivity analysis were found by identifying the mean and SD of each state. For the database-derived costs, an SD was inferred from the IQR assuming normality. From here, gamma distributions were derived using parameters . Mean costs and probabilistic sensitivity analysis distributions for each health state are given in Table 56.
| Type | States | Mean cost (£) | Variable and distribution |
|---|---|---|---|
| Remission states | SC remission, IND remission, MNT remission, | 52 |
cost_remission Gamma; α = 182.43, λ = 3.51 |
| Relapse states (severe disease) | SC relapse, IND relapse, IND relapse 2, MNT relapse, MNT relapse 2, transitional | 1489 |
cost_severe_relapse Gamma; α = 1406.01, λ = 0.944 |
| Relapse states (moderate disease) | SC relapse, IND relapse, IND relapse 2, MNT relapse, MNT relapse 2, transitional | 474 |
cost_moderate_relapse Gamma; α = 1826.81, λ = 3.85 |
| Surgery states | SC surgery, IND surgery, MNT surgery | 4592 |
cost_surgery Gamma; α = 1232.50, λ = 0.26 |
| Post-surgery states | SC post-surgery remission, IND post-surgery remission, MNT post-surgery remission | 72 |
cost_surgical_remission Gamma; α = 349.74, λ = 4.86 |
Overall, total costs for each state were defined by drug costs (fixed) plus the costs for the corresponding SC state (drawn from the relevant distribution). Non-hospitalisation medical costs were not included in the model except in the administration of infliximab. Table 57 presents mean total costs for each state to the nearest pound for IND and MNT states, with equivalent costs for SC states given in Table 56 (note: anti-TNF costs are zero).
| Infliximab | Adalimumab | |||||
|---|---|---|---|---|---|---|
| State costs | Drug costs | Total costs | State costs | Drug costs | Total costs | |
| IND remission | 52 | 0 | 52 | 52 | 0 | 52 |
| IND relapse (severe disease) | 1489 | 5809 | 7298 | 1489 | 1073 | 2562 |
| IND relapse 2 (severe disease) | 1489 | 0 | 1489 | 1489 | 0 | 1489 |
| Transitional (severe disease) | 1489 | 0 | 1489 | 1489 | 0 | 1489 |
| IND relapse (moderate disease) | 474 | 5809 | 6283 | 474 | 1073 | 1547 |
| IND relapse 2 (moderate disease) | 474 | 0 | 474 | 474 | 0 | 474 |
| Transitional (moderate disease) | 474 | 0 | 474 | 474 | 0 | 474 |
| IND surgery | 4592 | 0 | 4592 | 4592 | 0 | 4592 |
| IND post-surgery remission | 72 | 0 | 72 | 72 | 0 | 72 |
| MNT remission | 52 | 968 | 1020 | 52 | 715 | 767 |
| MNT relapse (severe disease) | 1489 | 3389 | 4878 | 1489 | 715 | 2204 |
| MNT relapse 2 (severe disease) | 1489 | 968 | 2457 | 1489 | 715 | 2204 |
| Transitional (severe disease) | 1489 | 1452 | 2941 | 1489 | 0 | 1489 |
| MNT relapse (moderate disease) | 474 | 3389 | 3863 | 474 | 715 | 1189 |
| MNT relapse 2 (moderate disease) | 474 | 968 | 1442 | 474 | 715 | 1189 |
| Transitional (moderate disease) | 474 | 1452 | 1926 | 474 | 0 | 474 |
| MNT surgery | 4592 | 0 | 4592 | 4592 | 0 | 4592 |
| MNT post-surgery remission | 72 | 968 | 1040 | 72 | 715 | 787 |
Probabilistic sensitivity analysis
Monte Carlo simulation was used to estimate the expected mean costs and effects for SC and each intervention. Each analysis used 10,000 simulations. For each cost-effectiveness analysis the following variables were included in the multivariate probabilistic sensitivity analysis:
-
utilities in remission, relapse, surgery and post-surgery remission (see Table 53)
-
effectiveness of anti-TNF therapy (see Table 55)
-
direct health-care costs in remission, relapse, surgery and surgical remission (see Table 56).
The probabilistic sensitivity analysis was partial and did not include uncertainties in either the Silverstein et al. 26 transition matrix or anti-TNF costs. Additional one-way sensitivity analyses are detailed in Sensitivity analyses. CEACs were calculated for induction and maintenance therapies in moderate and severe disease.
Results
Table 58 gives the mean costs and QALYs and expected cost-effectiveness ratios and ICERs for each intervention in induction and maintenance therapy for those suffering from moderate and severe CD.
| Costs (£) | QALYs | CER vs SC | ICERs | |||
|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | |||
| Severe disease | ||||||
| SC | 13,415 | 278 | 0.8119 | 0.0455 | – | Dominated |
| Infliximab IND | 12,051 | 488 | 0.8943 | 0.8193 | Dominates | Baseline |
| Infliximab MNT | 19,143 | 187 | 0.8957 | 0.0813 | £68,315 per QALY | £5.03M per QALY |
| SC | 13,421 | 283 | 0.8118 | 0.0457 | – | Dominated |
| Adalimumab IND | 7053 | 410 | 0.8942 | 0.0816 | Dominates | Baseline |
| Adalimumab MNT | 14,047 | 197 | 0.8956 | 0.0823 | £7749 per QALY | £4.98M per QALY |
| Moderate disease | ||||||
| SC | 6615 | 117 | 0.8926 | 0.0454 | – | Baseline |
| Infliximab IND | 9573 | 202 | 0.9240 | 0.0813 | £94,321 per QALY | £94,321 per QALY |
| Infliximab MNT | 16,751 | 146 | 0.9245 | 0.0819 | £317,991 per QALY | £13.9M per QALY |
| SC | 6615 | 117 | 0.8922 | 0.0459 | – | Dominated |
| Adalimumab IND | 4583 | 175 | 0.9231 | 0.0818 | Dominates | Baseline |
| Adalimumab MNT | 11,657 | 95 | 0.9236 | 0.0824 | £160,079 per QALY | £13.9M per QALY |
Cost-effectiveness acceptability curves
For patients with severe disease, infliximab induction treatment was found to be cost-effective relative to maintenance treatment and SC in over 99% of cases at all points up to £100,000 per QALY. Likewise, adalimumab induction treatment was found to be cost-effective relative to maintenance treatment and SC for thresholds up to £100,000 per QALY. Given that these diagrams are relatively uninformative, they are not displayed. Figures 44 and 45 show the CEACs for induction and maintenance therapies for infliximab and adalimumab respectively in patients with moderate disease.
FIGURE 44.
Cost-effectiveness acceptability curve for infliximab in moderate disease. Note: the MNT infliximab line runs along the x-axis, as at no stage on the scale of the figure is the likelihood of this treatment being cost-effective > 0.
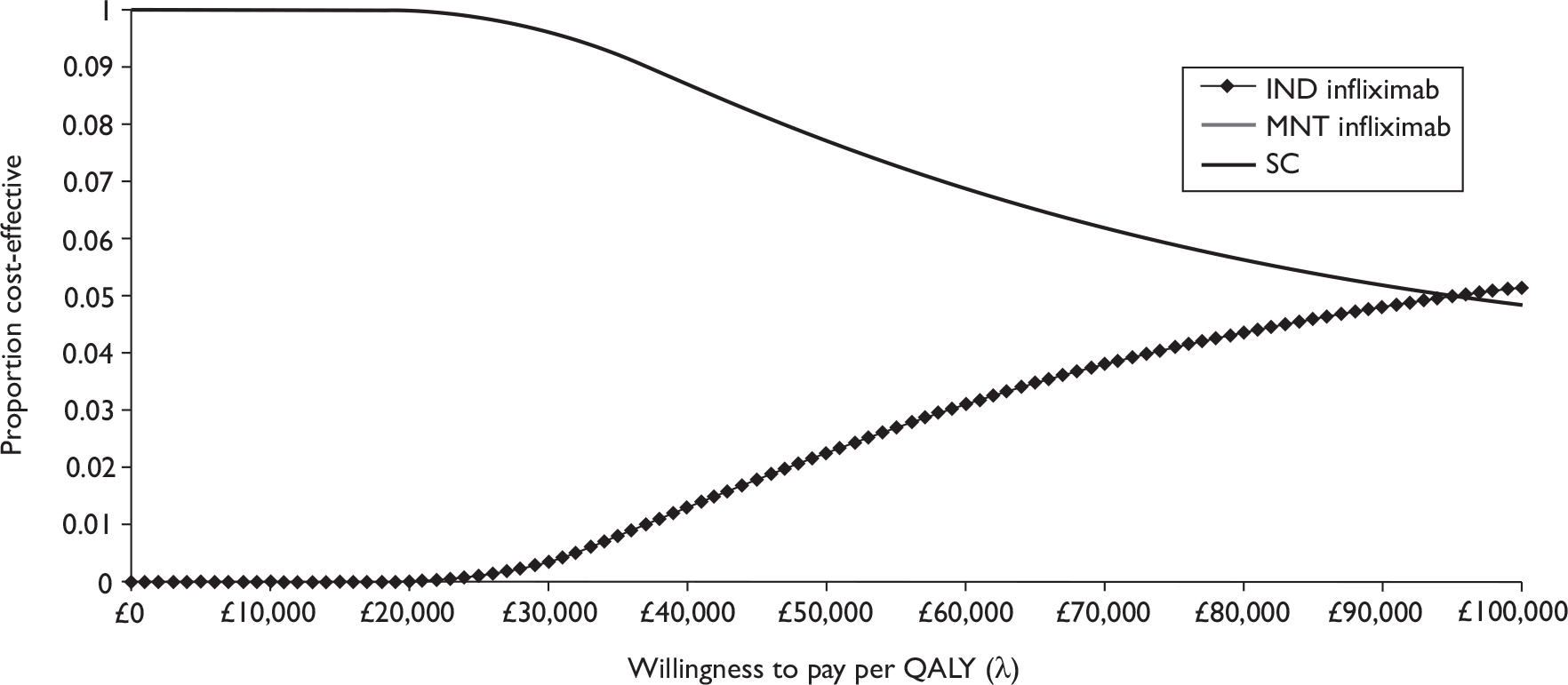
FIGURE 45.
Cost-effectiveness acceptability curve for adalimumab in moderate disease. Note: the MNT adalimumab line runs along the x-axis, as at no stage on the scale of the figure is the likelihood of this treatment being cost-effective > 0.
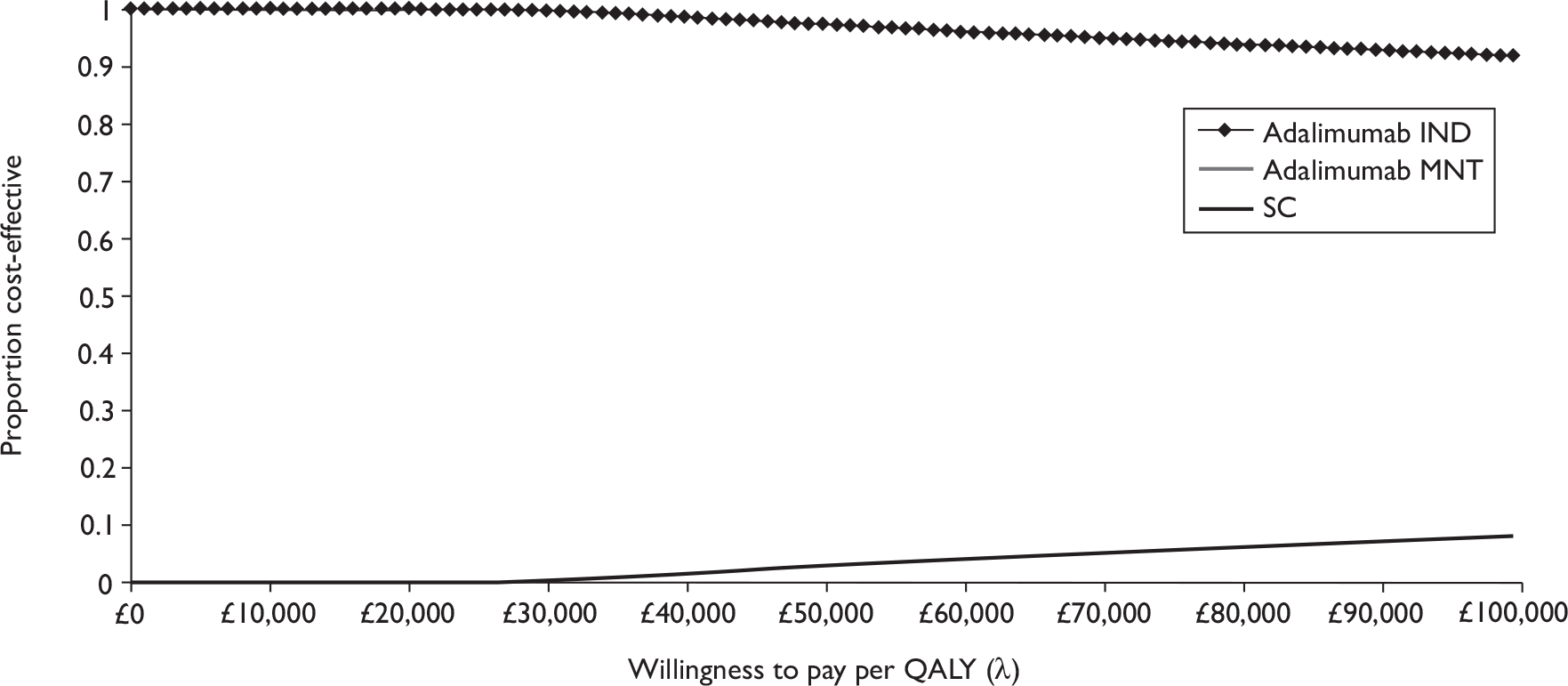
Sensitivity analyses
The de novo cost-effectiveness model used probabilistic sensitivity analysis to characterise the uncertainty in the clinical and cost data. This is a widely accepted method for addressing uncertainty in decision analysis modelling and is the preferred method according to the NICE reference case. 93 In some cases, insufficient information was available to estimate the uncertainty surrounding a parameter value, therefore a series of scenario analyses were also undertaken in order to explore the consequences for the estimates of cost-effectiveness of changes in these values.
Stakeholders who commented on previous draft versions of this report noted that there were aspects of the model that they wished to see changed and, where appropriate, these changes have been made. Given that the focus of the NICE technology appraisal was on severe active CD (because of the licence indication of the drugs) and the cost-effectiveness of treatment in moderate disease is typically poor, these analyses concentrate on severe disease only. The results of all scenarios analyses and other suggested changes are given in the text of the following sections.
The base case was modified in several ways. The SC transition matrix was modified to allow the following:
-
incorporation of different relapse rates from remission states
-
consideration of the effect of ‘implausible’ transitions in the SC matrix
-
provision of alternative transitions from the surgical states.
A further (fourth) analysis extended the time horizon of analysis up to 20 years. The fifth set of analyses modified the effectiveness estimates used in the base case. In the final set of analyses, infliximab dosages were adjusted according to patient weight in order to provide an analysis that may be relevant to a paediatric population, albeit with caveats as to the applicability of adult data to this population.
Following consultation on the draft report, another request was to consider an analysis of the cost-effectiveness of the anti-TNF agents based on the calculation of dosage using body surface area instead of weight. However, clinical expert advice suggested that there was little evidence to suggest that dose scaling based on body surface area is likely to have an impact on the effectiveness of the treatment (Professor C Twelves, Cancer Research UK Leeds, 2008, personal communication). Moreover, because the clinical evidence that was available was based on doses calculated based on weight, there was no suggestion as to what the differential effectiveness would be, making such an analysis speculative at best and misleading at worst. Finally, as the cost-effectiveness estimates were based on a per vial cost, minor adjustments to the dose required would have been unlikely to shift those categories of treatments that were not cost-effective to being cost-effective, for the reasons discussed previously in the report.
Changes to the standard care matrix: relapse rates
The first set of sensitivity tests related to relapse rates in SC and, by extension, the other transition matrices. In the baseline case, there was a relatively small chance of relapse once remission was achieved. In ‘standard’ remission there was a 0.59% chance of relapse per 4-week period (SC remission to SC relapse) and a 1.6% chance of leaving remission for any reason. Following surgery, there was a 0.11% chance of relapse per 4-week period (SC post-surgery remission to SC relapse) and a 0.81% chance of leaving remission for any reason. These values characterise the risk of an average patient leaving remission within the Silverstein et al. 26 cohort.
For groups that had a higher risk of relapse than the Silverstein et al. 26 cohort, the transition matrices derived above will not necessarily characterise this risk. As the risk of relapse increases, the time a successfully treated patient spent in remission drops, therefore the general efficacy of anti-TNF treatments against SC would have been expected to fall. As relapse risk increased, the health benefits of maintenance over induction treatments would also have been expected to increase, as the benefit of maintenance was a proportionate reduction in this relapse risk – the higher the risk, the greater would be the absolute benefit of maintenance over induction.
Two different analyses were conducted in order to assess the importance of relapse risk. In the first, the SC remission to SC relapse rate (‘sc_relapse’) was increased. In the second, sc_relapse was changed and the post-surgery remission rate (SC post-surgery remission to SC relapse, ‘ps_relapse’) was also changed by the same proportion as sc_relapse (ps_relapse = sc_relapse × 0.0059/0.0011). Changes were made to all the relapse probabilities in all three transition matrices (SC, IND, MNT) for each anti-TNF to explore the impact of such changes.
Supposing that the level of relapse from standard remission was 10 times its original level of 0.59% (0.0059), if sc_relapse had a value = 0.0590, the average length of time in remission would drop from 4.68 years to 1.07 years. Table 59 presents the cost-effectiveness of anti-TNFs in this analysis. Here, both infliximab and adalimumab induction therapy remained cost-effective versus SC, and neither maintenance therapy was generally cost-effective versus induction/reinduction therapy.
| Costs (£) | QALYs | CER vs SC | ICERs | |||
|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | |||
| SC | 14,271 | 304 | 0.8020 | 0.0414 | – | Baseline |
| Infliximab IND | 15,492 | 405 | 0.8795 | 0.0751 | £15,755 per QALY | £15,755 per QALY |
| Infliximab MNT | 19,498 | 218 | 0.8920 | 0.0806 | £58,078 per QALY | £320,480 per QALY |
| SC | 14,268 | 300 | 0.8019 | 0.0416 | – | Dominated |
| Adalimumab IND | 8714 | 429 | 0.8799 | 0.0758 | Dominates SC | Baseline |
| Adalimumab MNT | 14,291 | 218 | 0.8925 | 0.0814 | £254 per QALY | £442,619 per QALY |
Neither of the maintenance treatments were cost-effective. However, at higher relapse rates – and shorter periods of expected remission – maintenance treatment may become cost-effective. Table 60 shows the predicted cost-effectiveness of infliximab treatment as relapse rates were increased. The figures shown correspond to the final column of the tables above, identifying both the baseline treatment and the incremental cost-effectiveness of alternative treatments. For treatments that were dominated (directly or through extended dominance), the ICERs are not given but a line displayed.
| Relapse rate (SC) | Remission length | ICERs (£ per QALY) | |||||
|---|---|---|---|---|---|---|---|
| sc_relapse only changing | Both relapse rates changing | ||||||
| SC | Infliximab IND | Infliximab MNT | Infliximab SC | Infliximab IND | Infliximab MNT | ||
| 0.025 | 2.13 years | – | Baseline | 1,039,584 | – | Baseline | 1,017,400 |
| 0.05 | 1.24 years | Baseline | 10,041 | 412,633 | Baseline | 9955 | 399,734 |
| 0.075 | 314 days | Baseline | 24,776 | 204,973 | Baseline | 24,550 | 193,601 |
| 0.1 | 240 days | Baseline | 39,264 | 101,100 | Baseline | 38,821 | 91,641 |
| 0.125 | 193 days | Baseline | – | 49,892 | Baseline | – | 47,232 |
| 0.15 | 161 days | Baseline | – | 48,137 | Baseline | – | 44,936 |
| 0.175 | 137 days | Baseline | – | 46,702 | Baseline | – | 43,119 |
| 0.2 | 119 days | Baseline | – | 45,587 | Baseline | – | 41,629 |
| 0.225 | 105 days | Baseline | – | 44,793 | Baseline | – | 40,380 |
| 0.25 | 94 days | Baseline | – | 44,195 | Baseline | – | 39,414 |
| 0.275 | 84 days | Baseline | – | 43,812 | Baseline | – | 38,587 |
| 0.3 | 76 days | Baseline | – | 43,621 | Baseline | – | 37,986 |
| 0.325 | 69 days | Baseline | – | 43,481 | Baseline | – | 37,518 |
| 0.35 | 64 days | Baseline | – | 43,503 | Baseline | – | 37,135 |
| 0.375 | 59 days | Baseline | – | 43,634 | Baseline | – | 36,859 |
| 0.4 | 54 days | Baseline | – | 43,866 | Baseline | – | 36,713 |
| 0.425 | 50 days | Baseline | – | 44,148 | Baseline | – | 36,587 |
| 0.45 | 47 days | Baseline | – | 44,559 | Baseline | – | 36,542 |
| 0.475 | 44 days | Baseline | – | 44,967 | Baseline | – | 36,571 |
| 0.5 | 41 days | Baseline | – | 45,408 | Baseline | – | 36,668 |
For groups at relatively little risk of relapse, infliximab induction/reinduction appeared to be a cost-effective strategy. The additional benefit of maintenance was not cost-effective in these cases (ICER > £30,000/QALY).
Standard care became the most cost-effective treatment at the relapse rate between 0.075 and 0.100 (average remission lengths of 314 and 240 days). As the relapse rate increased further, obtaining QALYs with maintenance treatment became less expensive than with induction treatment. However, in none of the cases considered here did the cost-effectiveness ratio for infliximab maintenance fall below £30,000 per QALY. At very high risks of relapse, the ICERs for maintenance treatment even appeared to increase with the relapse rates, suggesting that even higher relapse rates were unlikely to provide clear cost-effectiveness.
In contrast, the similar efficacy and lower costs of adalimumab suggested a lesser role for SC. At all relapse rates, adalimumab IND dominated SC in all cases (up to sc_relapse = 0.500) regardless of whether or not the post-surgery remission rates were also modified. As a result, the relevant comparison was always between induction/reinduction treatment and maintenance treatment. Here, higher relapse rates placed increasing prominence on maintenance over induction treatment.
Table 61 presents the ICERs for the adalimumab treatment options. From standard relapse rates to high relapse rates, the adalimumab induction/reinduction treatment was cost-effective. Above this standard rate, maintenance treatment was first cost-effective and then dominated induction/reinduction. However, the relapse rate cut-off for such treatment appeared very high, and depended only slightly on whether post-surgery relapse rates also increased. Maintenance treatment may have been cost-effective for those who typically suffered severe relapses within 10 or 11 weeks. However, this group was expected to comprise only a very small proportion of CD patients.
| Relapse rate (SC) | Remission length | ICERs (£ per QALY) | |||||
|---|---|---|---|---|---|---|---|
| sc_relapse only changing | Both relapse rates changing | ||||||
| SC | Adalimumab IND | Adalimumab MNT | SC | Adalimumab IND | Adalimumab MNT | ||
| 0.025 | 2.13 years | – | Baseline | 1,133,765 | – | Baseline | 1,112,324 |
| 0.05 | 1.24 years | – | Baseline | 532,789 | – | Baseline | 520,769 |
| 0.075 | 314 days | – | Baseline | 333,906 | – | Baseline | 322,790 |
| 0.1 | 240 days | – | Baseline | 234,236 | – | Baseline | 224,302 |
| 0.125 | 193 days | – | Baseline | 174,035 | – | Baseline | 165,702 |
| 0.15 | 161 days | – | Baseline | 134,504 | – | Baseline | 122,261 |
| 0.175 | 137 days | – | Baseline | 106,350 | – | Baseline | 98,714 |
| 0.2 | 119 days | – | Baseline | 85,136 | – | Baseline | 77,959 |
| 0.225 | 105 days | – | Baseline | 68,862 | – | Baseline | 61,774 |
| 0.25 | 94 days | – | Baseline | 55,883 | – | Baseline | 48,849 |
| 0.275 | 84 days | – | Baseline | 45,445 | – | Baseline | 38,320 |
| 0.3 | 76 days | – | Baseline | 36,710 | – | Baseline | 29,663 |
| 0.325 | 69 days | – | Baseline | 29,397 | – | Baseline | 22,308 |
| 0.35 | 64 days | – | Baseline | 23,149 | – | Baseline | 16,007 |
| 0.375 | 59 days | – | Baseline | 17,807 | – | Baseline | 10,564 |
| 0.4 | 54 days | – | Baseline | 13,164 | – | Baseline | 5852 |
| 0.425 | 50 days | – | Baseline | 9121 | – | Baseline | 1692 |
| 0.45 | 47 days | – | Baseline | 5543 | – | Baseline | Dominates |
| 0.475 | 44 days | – | Baseline | 2385 | – | Baseline | Dominates |
| 0.5 | 41 days | – | – | Dominates | – | Baseline | Dominates |
Changes to the standard care matrix: removal of implausible transitions
The SC transition matrix presented in Table 51 included four transitions to remission states that could be considered implausible. Those who have not had surgery (SC remission and SC relapse) would not transit to a post-surgery remission state; equally, those who have had surgery (SC surgery and SC post-surgery remission) would not move to a non-surgical remission state. The analyses were rerun with the SC matrix revised so that these transitions were no longer possible and any probability was reassigned to the expected remission state. For example, the SC surgery state originally had a 0.0521 probability of moving to the SC remission state and a 0.2613 chance of moving to the SC post-surgery remission state. Here, there was a 0.3134 chance (0.0521 + 0.2613) of moving to any remission state. Table 62 shows the results of removing the implausible transitions (here there is no chance of moving to SC remission from SC surgery) and presents a modified SC matrix with a 0.3134 chance of moving to the (expected) SC post-surgery remission state.
| SC remission | SC relapse | SC surgery | SC post-surgery remission | |
|---|---|---|---|---|
| SC remission | 0.9872 | 0.0059 | 0.0069 | 0 |
| SC relapse | 0.0902 | 0.8749 | 0.0348 | 0 |
| SC surgery | 0 | 0.0158 | 0.6709 | 0.3134 |
| SC post-surgery remission | 0 | 0.0011 | 0.0026 | 0.9963 |
Since the IND (see Table 52) and MNT matrices (see Table 54) were based on the SC matrix, similar changes were also made to these. The revised model was run, and results are given in Table 63. The general results were extremely similar to the base case (see Table 58), with both induction/reinduction treatments dominating SC. Maintenance treatments cost slightly less per QALY against SC (infliximab £71,000 vs £68,000; adalimumab £8200 vs £7700), and slightly more per QALY against induction (infliximab £4.96M vs £5.03M; adalimumab £4.58M vs £4.98M). As such, any impacts of the implausible transitions in the base-case analysis were very minor.
| Costs (£) | QALYs | CER vs SC | ICERs | |||
|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | |||
| SC | 13,450 | 285 | 0.8129 | 0.0457 | – | Dominated |
| Infliximab IND | 12,261 | 504 | 0.8925 | 0.0809 | Dominates SC | Baseline |
| Infliximab MNT | 19,209 | 192 | 0.8939 | 0.0815 | £71,099 per QALY | £4.96M per QALY |
| SC | 13,450 | 285 | 0.8129 | 0.0457 | – | Dominated |
| Adalimumab IND | 7269 | 427 | 0.8947 | 0.0809 | Dominates SC | Baseline |
| Adalimumab MNT | 14,137 | 204 | 0.8962 | 0.0816 | £8247 per QALY | £4.58M per QALY |
Changes to the standard care matrix: surgical assumptions
During consultation on the draft report there was discussion about the most appropriate way to incorporate estimates of the probability of having a second surgery following an unsuccessful first surgery into the model. It was argued that the method used in the initial analysis (and for the base-case analysis in this report) overestimated the number of patients who would require repeat surgery. The information as reported in Silverstein et al. 26 was based on the probability of requiring repeat surgery within 2 months of the initial surgery. To obtain an estimate of this probability over a 4-week cycle, half of the 2-month value was used to arrive at the base-case values; this method likely led to an overestimate of the number of patients who had repeat surgery over the 1-year model time horizon. In the sensitivity analysis below, the 2-month value is used. This method was likely to result in an underestimate in the number of patients requiring surgery.
As can be seen from Table 64 when compared with the base case, the largest effect was on the SC arm. This was to be expected – more patients in the SC arm underwent surgery than in any of the treatment arms; reducing the second surgery rate like this lowered the overall cost of the SC arm by more than it lowered the overall cost of the treatment arms.
| Severe disease | Costs (£) | QALYs | CER vs SC | ICERs | ||
|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | |||
| SC | 12,070 | 274 | 0.8233 | 0.0482 | – | Dominated |
| Infliximab IND | 11,270 | 443 | 0.9004 | 0.08330 | Dominates | Baseline |
| Infliximab MNT | 17,809 | 150 | 0.9018 | 0.0836 | £73,108 per QALY | £4.7M per QALY |
| SC | 12,068 | 269 | 0.8215 | 0.0483 | – | Dominated |
| Adalimumab IND | 6299 | 368 | 0.8991 | 0.0843 | Dominates | Baseline |
| Adalimumab MNT | 12,876 | 161 | 0.9004 | 0.0840 | £10,240 per QALY | £5.1M per QALY |
Modelling assumptions: changes to the time horizon
The longer term cost-effectiveness of anti-TNFs was considered and estimates of cost-effectiveness at 5, 10 and 20 years are reported in Table 65. Reasons for not adopting a lifetime horizon for this model are discussed in Model structure. Estimates of effectiveness in these scenarios have not been changed as no reliable evidence was available to show the effectiveness of either drug at any of the longer term time horizons. As a consequence, these results must be treated with caution. It should also be remembered that no evidence was found to suggest either the direction of change or the magnitude if it were decided to alter the estimates of effectiveness. These results are illustrative only and should be assumed to be sufficiently uncertain as to be unreliable estimates of cost-effectiveness over the time frames modelled.
| Costs (£) | QALYs | CER vs SC | ICERs | |||
|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | |||
| 5 years | ||||||
| SC | 25,631 | 469 | 4.5157 | 0.4277 | – | Dominated |
| Infliximab IND | 22,162 | 759 | 4.6443 | 0.4793 | Dominates SC | Baseline |
| Infliximab MNT | 69,069 | 1490 | 4.6545 | 0.4838 | £1.6M per QALY | £4.6M per QALY |
| SC | 25,670 | 470 | 4.5202 | 0.4217 | – | Dominated |
| Adalimumab IND | 16,214 | 684 | 4.6374 | 0.4728 | Dominates SC | Baseline |
| Adalimumab MNT | 53,009 | 804 | 4.6476 | 0.4772 | £214,592 per QALY | £3.6M per QALY |
| 10 years | ||||||
| SC | 36,822 | 662 | 9.1791 | 0.9364 | – | Dominated |
| Infliximab IND | 33,144 | 942 | 9.3285 | 0.9958 | Dominates SC | Baseline |
| Infliximab MNT | 129,951 | 3807 | 9.3500 | 0.9963 | £544,933 per QALY | £4.5M per QALY |
| SC | 36,823 | 656 | 9.2126 | 0.9287 | – | Dominated |
| Adalimumab IND | 26,273 | 914 | 9.3453 | 0.9875 | Dominates SC | Baseline |
| Adalimumab MNT | 100,338 | 2156 | 9.3653 | 0.9962 | £415,946 per QALY | £3.7M per QALY |
| 20 years | ||||||
| SC | 58,481 | 1066 | 18.5560 | 1.9365 | – | Dominated |
| Infliximab IND | 53,991 | 1374 | 18.6430 | 2.0012 | Dominates SC | Baseline |
| Infliximab MNT | 250,391 | 8584 | 18.7536 | 2.0259 | £971,204 per QALY | £1.8M per QALY |
| SC | 58,486 | 1071 | 18.5585 | 1.9242 | – | Dominated |
| Adalimumab IND | 45,716 | 1377 | 18.7180 | 1.9956 | Dominates SC | Baseline |
| Adalimumab MNT | 193,840 | 4928 | 18.7566 | 2.0124 | £683,261 per QALY | £3.8M per QALY |
Modelling assumptions: changes to the effectiveness estimates
In the baseline model, peak clinical effectiveness values (56% remission) were applied every 4 weeks which was highly favourable to the estimated cost-effectiveness of anti-TNF treatments. In this modification, the 4-week effectiveness was reduced in order to provide the peak effectiveness (56%) over 8 weeks. As an example, in order to obtain a 56% effectiveness over 8 weeks, approximately a 34% chance of remission every 4 weeks would be required. Here, 34% of patients achieved remission at 4 weeks (66% remained in relapse) and a further 22% (34% × 66%) achieved remission by 8 weeks. Table 66 presents the revised cost-effectiveness from this sensitivity analysis.
| Severe disease | Costs (£) | QALYs | CER vs SC | ICERs | ||
|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | |||
| SC | 13,417 | 278 | 0.8113 | 0.0460 | – | Baseline |
| Infliximab IND | 16,040 | 544 | 0.8481 | 0.0623 | £71,315 per QALY | £71,315 per QALY |
| Infliximab MNT | 20,285 | 242 | 0.8491 | 0.0628 | £181,475 per QALY | £3.9M per QALY |
| SC | 13,416 | 279 | 0.8123 | 0.0461 | – | Dominated |
| Adalimumab IND | 9862 | 460 | 0.8637 | 0.0686 | Dominates | Baseline |
| Adalimumab MNT | 15,144 | 235 | 0.8650 | 0.0692 | £32,759 per QALY | £4.1M per QALY |
The effect of these changes highlights the importance of uncertainty in the effectiveness estimates, and particularly in the case of infliximab. In the baseline case, infliximab induction/reinduction treatment dominated SC; in this case, such treatment was no longer cost-effective at a standard £30,000 per QALY threshold. The impact of this assumption on adalimumab was far less critical, as adalimumab IND continued to dominate SC. In neither case would maintenance treatment be justified on cost-effectiveness grounds.
In the absence of specific clinical trial data on the impact of maintenance on relapse probabilities, the model assumed that anti-TNF maintenance impacted on relapse probabilities to approximately the same degree as anti-TNF treatment affected the probability of entering remission. Consequently, where anti-TNF maintenance therapy was assumed to give about eight times the chance of entering remission from relapse, the model assumed there was one-eighth the chance of relapsing among patients in remission.
This assumption was tested by assuming that relapse under maintenance was one-quarter, one-eighth (approximate baseline) or one-sixteenth of the relapse rate under SC and induction (post-surgery relapse was also changed by the same general amount.) In the standard case, the impact of this assumption was very limited given the low chance of relapse. For the purposes of comparison, higher relapse rates (sc_relapse = 0.05, ps_relapse = 0.05 × 0.0011/0.0059 = 0.0093) were therefore used, in order to allow the impact of this assumption to be assessed.
The scenarios in Table 67 suggest that there may have been some potential for maintenance to be somewhat more cost-effective by modifying these maintenance assumptions, e.g. from £399,000 to £330,000 per QALY in infliximab treatment as the relapse rate moved from one-eighth of the standard rate to one-sixteenth. Here, halving the numbers relapsing per period made some, though relatively little, difference to the cost-effectiveness of treatment. However, doubling those relapsing per period (from one-eight to one-quarter of the standard rate) made a much larger difference – here the infliximab ICER moved from £399,000 to £537,000 per QALY.
| Costs (£) | QALYs | CER vs SC | ICERs | |||
|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | |||
| MNT relapse = ¼ cs_relapse | ||||||
| SC | 14,207 | 301 | 0.8028 | 0.0419 | – | Baseline |
| Infliximab IND | 15,010 | 424 | 0.8824 | 0.0766 | £10,126 per QALY | £10,126 per QALY |
| Infliximab MNT | 19,687 | 195 | 0.8911 | 0.0808 | £62,061 per QALY | £537,241 per QALY |
| SC | 14,207 | 301 | 0.8028 | 0.0419 | – | Dominates |
| Adalimumab IND | 8492 | 435 | 0.8824 | 0.0765 | Dominates | Baseline |
| Adalimumab MNT | 14,401 | 212 | 0.8918 | 0.0806 | £2,180 per QALY | £628,617 per QALY |
| MNT relapse = ⅛ cs_relapse | ||||||
| SC | 14,207 | 301 | 0.8028 | 0.0419 | – | Baseline |
| Infliximab IND | 15,010 | 424 | 0.8824 | 0.0766 | £10,126 per QALY | £10,126 per QALY |
| Infliximab MNT | 19,438 | 198 | 0.8954 | 0.0820 | £57,674 per QALY | £398,649 per QALY |
| SC | 14,207 | 301 | 0.8028 | 0.0419 | – | Dominates |
| Adalimumab IND | 8492 | 435 | 0.8824 | 0.0765 | Dominates | Baseline |
| Adalimumab MNT | 14,253 | 210 | 0.8935 | 0.812 | £507 per QALY | £519,000 per QALY |
| MNT relapse = 116 cs_relapse | ||||||
| SC | 14,207 | 301 | 0.8028 | 0.0419 | – | Baseline |
| Infliximab IND | 15,010 | 424 | 0.8824 | 0.0766 | £10,126 per QALY | £10,126 per QALY |
| Infliximab MNT | 19,311 | 200 | 0.8940 | 0.0819 | £55,119 per QALY | £330,615 per QALY |
| SC | 14,207 | 301 | 0.8028 | 0.0419 | – | Dominates |
| Adalimumab IND | 8492 | 435 | 0.8824 | 0.0765 | Dominates | Baseline |
| Adalimumab MNT | 14,180 | 207 | 0.8938 | 0.0799 | Dominates | £498,947 per QALY |
While the results of the analysis were sensitive to the assumptions made regarding maintenance efficacy, higher efficacy values are unlikely to affect the ICER much, but lower values may have a much larger effect.
Paediatric Crohn’s disease threshold analysis
The review of clinical effectiveness evidence reported found no randomised, placebo-controlled evidence on the effectiveness of infliximab in paediatric CD. An executable model to estimate the cost-effectiveness of infliximab was provided by the manufacturers. However, programming errors in that model prevented validation of the results. A corrected version of the model was eventually provided, but was received outside the accepted time frame for new evidence to be considered as part of the assessment.
A threshold analysis using the de novo cost-effectiveness model based on the adult population effectiveness estimates was undertaken to obtain the estimated required effectiveness of infliximab in paediatric patients. This analysis must be interpreted with caution as it is often neither straightforward nor advisable to extrapolate the results of research in adults to a paediatric population. It would be a mistake to uncritically accept the notion that research results that apply to adults are applicable to children. In the case of anti-TNF therapy in particular, it is important to consider how the effectiveness of the drugs might differ in a paediatric population, whether or not the same or similar AEs can be expected, the differences in costs of the treatments including both drug costs and the requirement for specialist paediatric services, and finally the potentially different value attached to different aspects of HRQoL in children when compared with adults.
The costs associated with treating children may differ from the costs of treating adults. This may be due to the different costs associated with the drug itself or related to some other factor such as the setting in which care takes place. In these circumstances, given that infliximab doses are according to weight, the costs of treatment may be expected to be lower if a linear relationship between dose and effect is assumed (leaving aside the issue of whether the dose-response relationship holds for the paediatric population as it is assumed to do for the adult population). However, the cost of the drug is only a single factor in establishing the total cost of care for the paediatric population. Often children may need to be seen in specialist paediatric settings, which will attract costs different to those that apply in adult clinics. Generally it would be expected that treatment of paediatric patients will be different to that of adults. An estimate of this cost for the threshold analysis has been made, but given the paucity of available evidence (e.g. on required drug dose, on the model and location of care) the estimate should be viewed with caution.
Owing to a lack of specific evidence, the assumption in this threshold analysis has been made that the utility weights assigned to children for all states in the model were the same as for adults in the same state. But research was clear that it was not necessarily an appropriate assumption. In assessing the HRQoL of children, due consideration was required relating to the domains of life considered important by children (Rosenbaum and Saigal106), to the relevant stage of their physiological and mental development (Harris and Butterworth107), and to the social context in which they find themselves (Matza et al. 108) which impinges on their social roles (e.g. including aspects of life related their dependence and autonomy) (Fox-Rushby and Parker109). Those domains of life that were considered important in an adult population may not necessarily have been appropriate indicators of QoL in children (Petrou and Henderson110). It should be clear then that to apply adult utility values to an analysis of paediatric patients was a suboptimal approach to the problem, though the information available permitted no other course of action. After taking into consideration the above arguments, it was difficult to reach a precise conclusion about the effectiveness of infliximab for the treatment of paediatric CD.
In this sensitivity analysis, two key changes to the base case were made. Firstly, it was assumed that the average weight of the paediatric patients would be less than that of the adult patients. Secondly, a change to the administration cost of the drug was made which assumed that this will take place in paediatric gastroenterology service, with a cost of £232. 111 Table 68 shows the results from analysis of a paediatric population where the weight of the patients was assumed to be between 40 kg and < 60 kg and between 20 kg and < 40 kg. The average weight of the children in the evidence supplied by the manufacturer varied, with a mean of 49.1 kg in Baldassano et al. 46 and 43.8 kg in REACH. 45 This analysis was conducted on a per vial basis, as once a vial is opened it must be used or discarded and this represents the true cost to the NHS.
| Costs (£) | QALYs | CER vs SC | ICERs | |||
|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | |||
| 40–59 kg (three vials) | ||||||
| SC | 13,418 | 278 | 0.8130 | 0.0462 | – | Dominated |
| Infliximab IND | 10,652 | 497 | 0.8960 | 0.0824 | Dominates | Baseline |
| Infliximab MNT | 16,918 | 268 | 0.8974 | 0.0830 | £41,469 per QALY | £4.5M per QALY |
| 20–39 kg (two vials) | ||||||
| SC | 13,418 | 280 | 0.8112 | 0.0458 | – | Dominated |
| Infliximab IND | 9317 | 490 | 0.8930 | 0.0819 | Dominates | Baseline |
| Infliximab MNT | 14,813 | 340 | 0.8944 | 0.0825 | £16,767 per QALY | £3.9M per QALY |
It is clear in the analysis presented in Table 68 that induction therapy with infliximab for patients with severe disease was the only option that may be cost-effective where two or three vials were used. In neither weight range case is maintenance treatment cost-effective.
In the absence of valid estimates of effectiveness in children, a threshold analysis of those scenarios where the initial analysis showed treatments to be cost-ineffective for children with weight between 40 kg and < 60 kg was undertaken. When maintenance therapy is compared with induction therapy the ICER was sufficiently high to preclude the possibility of the former being cost-effective relative to the latter at any estimate of effectiveness. Maintenance therapy in severe disease may be cost-effective compared with SC at the same threshold if the average benefit were equal to 0.93 QALYs.
This threshold analysis demonstrated two things. Firstly, there is the possibility that treatments that were not cost-effective in the base case could become cost-effective if the costs of treatment were significantly lower. Secondly, it highlighted the uncertainty inherent in the model estimates of effectiveness (and in turn the total cost of treatment), given that small changes in estimates of effectiveness can lead to large swings in the estimated cost-effectiveness of treatment.
Discussion
The analyses described in this chapter indicate that infliximab is not likely to be cost-effective, according to the criteria laid out in the NICE Guide to the methods of technology appraisal,93 in the management of moderate CD. While adalimumab may be cost-effective, there is uncertainty regarding the ICER value. Neither of these therapies is likely to be cost-effective as maintenance therapy for moderate or severe disease. Both treatments are highly cost-effective, with no meaningful uncertainty, as induction therapy in severe disease.
The estimates of cost-effectiveness in maintenance therapy must be viewed as exploratory. This is because of the shortness of the randomised placebo-controlled period of the maintenance trials for these drugs. Essentially these do not provide evidence of the magnitude of effect compared with usual care owing to allowing patients to cross over to ‘episodic’ treatment relatively quickly after trial commencement. The evidence required to model the cost-effectiveness was the proportion of patients transiting between remission and relapse, and between relapse and remission, with and without treatment at regular time points. Given the absence of this evidence, it has therefore been necessary to postulate a maintenance effect based upon what has been reported in the trials. The implicit assumption for estimating the effect is that anti-TNFs interfere with the underlying biochemical process that causes relapses and that the effectiveness is equivalent whether or not the process has led to symptomatic relapse and whether the patient is in remission or relapse. The Silverstein et al. 26 matrix suggested that 82% of patients in remission were expected to be maintained in remission each year on SC. On average, then, the capacity for additional benefit from anti-TNF maintenance therapy is small, approximately 0.045 QALYs per annum for patients with severe disease. Against this background, it is unlikely that maintenance therapy has sufficient scope for generating health gain to justify its use at current prices.
Another key decision in estimating the cost-effectiveness was to use the Silverstein et al. 26 cohort to model usual care for all interventions, and use the Targan et al. 57 trial to provide an estimate of absolute effect, but not relative effect. The use of the Silverstein et al. 26 data model has some inherent limitations. Expert opinion suggested that the surgery rates were higher than would be seen in practice. Also, the relapse rates were much lower than was likely prevailing in clinical trials. The Silverstein et al. model was based upon long-term data on a substantial sample of patients and there was reasonable evidence that, for many patients, the longer term disease course is one of decreasing activity. Thus it would be expected that a model based upon long-term data would predict lower event rates than one based upon shorter term data from patients with highly active disease. However, the scope of the appraisal did not specify a disease activity inclusion criterion, only disease severity and treatment responsiveness criteria. In contrast, the trials applied recent disease activity inclusion criteria. On this basis it was concluded that the Silverstein et al. model was more appropriate given the scope of the appraisal than the clinical trial data, while accepting its limitations.
Having chosen to use the Silverstein et al. 26 model, the analysis made the assumption that the drug-responsive and drug-dependent states were both effectively managed on standard therapy, and therefore the two states were combined into a single state within the model. This might mean that the costs of the remission state were underestimated as the drug-dependent patients would have been on maintenance therapy to retain a remission status, while drug-responsive patients would have been managed with induction therapy. The effect of this will be to overstate the cost saving associated with achieving remission and thereby improve the cost-effectiveness of anti-TNFs because of their higher remission rate.
The relative effect seen in the Targan et al. 57 trial was an outlier due to the very low rate of remission at 4 weeks in the control arm. However, the absolute magnitude of effect was consistent with the remission rate seen in the pre-randomisation phase of the ACCENT I2,3 trial of infliximab.
An important difference between this model and others was the use of a 1-year time horizon and the exclusion of death from the model. Silverstein et al. 26 reported a small risk of death in each state. The mean risk of death varied between states, i.e. between 0.00015 and 0.00839. The greatest risks were for the ‘drug-responsive’ and ‘drug-refractory’ states (0.00626 and 0.00839 respectively). If these results had been used as the basis for incorporating mortality into the model and therefore adopting a lifetime horizon, the effectiveness of the drug in inducing remission would have produced an apparent mortality gain for treatment of approximately 0.00213 per additional remissions created – i.e. slightly over two lives in a cohort of 1000 patients. As the total QALYs produced by induction therapy were in the region of seven QALYs per year, this would have completely swamped the direct effectiveness, and made the treatments appear highly cost-effective, even though the evidence did not support a causal link between status in the Silverstein et al. 26 framework and mortality, and there was no direct evidence of a mortality benefit from anti-TNF treatment.
The analysis of uncertainty was probabilistic but not comprehensively so. Notably, the SC transition probabilities were entered into the model deterministically. The effect of this was to understate the uncertainty regarding the costs and outcomes from standard therapy, and, by extension, the uncertainty around the cost-effectiveness of the anti-TNFs in both induction/episodic and maintenance therapy. The primary effect of incorporating additional uncertainty was to increase the model’s prediction of expected value of subsequent research.
It was useful to consider briefly the implications on the cost-effectiveness of treatment for patients who were outside the four-vial analysis owing to their weight. The model effectively assumed that all patients were likely to fall somewhere between 60 kg and < 80 kg, when the clinical reality was that many patients would be either above or below this range. There was no evidence to suggest that weight had a direct effect on the efficacy of treatment – an assumption supported by a weight-based dosing regimen. Therefore the only difference that should have been expected would be in the costs of the treatment – the smaller the patient, the fewer vials required, the lower the cost, and vice versa. From an empirical standpoint, the paediatric sensitivity analysis can be extrapolated (see Table 68) to test how changes in the number of vials required affected the cost-effectiveness. It is worth noting that the administration costs in the paediatric analysis were slightly lower than in the adult analysis, but this did not matter greatly for the purposes of this argument.
From that analysis, it was clear that there was only one change to the relative cost-effectiveness when compared with the base case. In this case, when only two vials were required, the use of infliximab for patients with moderate disease could be considered cost-effective, whereas in the base case it would not be. Based on the three-vial paediatric analysis, the decisions would likely be the same as in the base case. Mindful that a very small number of adults with CD may have a weight of less than 40 kg, it seemed unlikely that treatment for patients who fell below the average weight would be cost-effective. Similarly, given the assumption that effectiveness was unaltered by weight and that costs would only increase for patients with a weight above 80 kg, it followed that the treatment was unlikely to be cost-effective for patients above the average weight.
The empirical evidence was only one of two parts of the argument. The second point at issue was whether or not it would be appropriate to discriminate against patients based on their weight when this was unrelated to the effectiveness of the treatment but changed the cost of treatment and therefore the relative cost-effectiveness of the treatment. This form of subgroup discrimination is not permitted by the NICE reference case. 93 Therefore it would have been inappropriate to suggest that one group of people with weight less than some critical level received treatment while those with weight above this level were denied it, where there is no demonstrable difference in health outcomes as a result of the difference in weight.
A further important consideration was the focus of this analysis on the cost-effectiveness of these treatments in the induction of remission. The trials reported response rates for remission, and CDAI response 70 and response 100. CDAI response rates cannot be converted into improvements in health without knowing the baseline CDAI for each patient. Although Gregor et al. 92 demonstrated a linear relationship between CDAI scores and utility values, they also found that the relationship was only modestly correlated, with increasing levels of uncertainty in utility scores as CDAI score increased. It was not considered possible here, therefore, to attach a utility gain to a 70- or 100-point gain on the CDAI without knowledge of the pre-treatment CDAI status.
As discussed above, it was chosen to construct a model based upon health state (remission, relapse, requiring surgery and remission following surgery) rather than CDAI score. This decision was guided by the desire to quantify the cost-effectiveness of treatments in producing health rather than their cost-effectiveness in shifting the clinical pathway. The results from the Gregor et al. 92 study suggested that when patients were grouped as per the Silverstein et al. 26 framework, the differences in mean HRQoL were extremely small, and much smaller than those used in the structure of health states adopted in this analysis.
A simple model of CD was constructed which focused on the cost-effectiveness of anti-TNF therapies in achieving or maintaining remission. The assumptions made here regarding cost of care and utilities gains from treatment favoured the anti-TNFs over usual care. The analyses drew out the much larger health benefit for patients with severe disease compared with moderate disease and how this fed through to ICERs that were likely to be acceptable for severe disease but not moderate disease. The analysis also highlighted the important variations in effectiveness and cost between the therapies. Perhaps most importantly, the analysis reflected the assumption that a substantial number of patients would achieve remission under SC and that the incidence of relapse among those in remission was such that maintenance therapy would have to be much less costly for it to be a cost-effective option.
Budget impact assessment
The NICE guidance on infliximab from 20021 estimated that 31,000 patients in England and 1800 in Wales had CD, that 2% had very severe disease and that between 1050 and 4200 patients would have been eligible for treatment. These estimates were made in the absence of good-quality CD prevalence studies. There is now more information on the UK prevalence of CD but not as much on the typical spread of severity.
It was estimated from the incidence/prevalence section in this report that the prevalence of CD in the UK is approximately 150 per 100,000 but could be between 50 and 400 per 100,000. The incidence of new cases of CD has been estimated to be approximately 5 per 100,000 per year but could be between 3.8 and 10 per 100,000 per year. The incidence and prevalence estimates from both industry submissions are shown in Table 69.
| Incidence | Prevalence | |
|---|---|---|
| Adalimumab submission4 | 10/100,000 per year used in budget impact section (derived from NICE guidance) |
50–100/100,000 ‘however this is likely to be an underestimation’ 62.5/100,000 used in budget impact section (derived from NICE guidance) |
| Infliximab submission96 | 14/100,000 per year | 50–100/100,000, 145/100,000 |
These incidence and prevalence estimates are for all CD patients rather than those with moderate-to-severe CD or severe CD. A large cross-sectional survey of CD patients with CDAI scores used to indicate the percentage with mild, moderate and severe CD was not found.
In a UK study37 of 172 CD patients attending a university hospital during a 6-month period, 7% were in remission, 33% had mild disease, 42% had severe disease, 8% had surgery and 10% were in post-surgery remission. Severity was judged by treatments being used rather than on CDAI score so severe CD patients were those being treated with corticosteroids or immunosuppressive regimens.
In a Canadian QoL study,92 180 consecutive CD patients referred to a tertiary care hospital had CDAI scores measured. The overall mean CDAI score was 182 (95% CI 166 to 199). There were 52 patients classified as ‘chronically active therapy resistant’ with mean CDAI of 246 (95% CI 220 to 272), 34 patients classified as ‘chronically active therapy responsive’ with CDAI 72 (95% CI 60 to 84), 45 patients classified as ‘acute disease exacerbation’ with CDAI 249 (95% CI 217 to 281) and 49 patients in remission with CDAI 129 (95% CI 110 to 148). This equates to 54% with severe disease (CDAI > 220) and 46% with mild disease (CDAI < 220). These 46% of patients with mild disease would also be categorised as in remission (CDAI < 150).
In a regional cohort of 373 CD patients from Denmark, in the first year 80% had highly active disease (defined as more than four stools daily, blood or pus daily, severe or daily abdominal pains and systemic symptoms such as fever or weight loss). 25 In the second year 40% had high activity, 22% had low activity and 38% were in remission. In subsequent years the proportions were approximately 30%, 20% and 50% respectively.
From the three studies mentioned above it can be estimated that approximately 40% of CD patients will have moderate-to-severe disease and may be considered eligible for treatment according to the inclusion criteria for the RCTs.
In the Olmstead County cohort study of 174 CD patients,26 follow-up information for up to 10 years was used in a Markov model to estimate the probability of future clinical course. From this it was estimated that 1.77% of CD patients might be in a severe, drug-refractory disease state. As this was based on a model, it may be much less reliable than actual cohort study results.
With a prevalence of 150/100,000 and a total population of approximately 50 million in England and 3 million in Wales, there would be approximately 79,500 CD patients – 75,000 in England and 4500 in Wales. If 40% had moderate-to-severe disease, this would be 31,800 CD patients. There is no information on the proportion of patients with severe CD as defined by CDAI > 300 within the moderate-to-severe category. However, it is noticeable that the mean CDAI score for all of the induction trials included in the clinical effectiveness review was approximately 300. These RCTs included patients described as having moderate-to-severe CD. 57,63,64 From this it can be estimated that if there is a roughly normal distribution, approximately 50% of patients with moderate-to-severe CD will have a CDAI score of more than 300 (Table 70).
| CD severity | Number in England and Wales | Percentage of total CD population |
|---|---|---|
| All CD | 79,500 | 100% |
| Moderate-to-severe CD | 31,800 | 40% |
| Severe CD | 15,900 | 20% |
| Severe and drug-resistant CD (estimate from a Silverstein et al.26 study Markov model only) | 1590 | 2%a |
The cost of treatment with the new interventions (induction and maintenance) for adults (non-fistulising CD) with both drugs is shown in Table 71. This includes the cost of administration in hospital or clinic in the case of infliximab. The administration cost would include the presence of a health professional during the 2 hours of infusion and for a period of time afterwards. As there is a (small) risk of acute allergic reactions, emergency equipment should be available. No administration costs were given for adalimumab on the grounds that it can be given subcutaneously. However, training must be given before this can occur which will incur a cost.
| From industry submission | Induction | Maintenance for 1 year | |
|---|---|---|---|
| Adalimumab | Cost per 40-mg vial – £357.50, no administration cost given | 80 mg at week 0 then 40 mg at week 2 (two doses) = £1072.50 (+ administration) | 40 mg e.o.w. (26 doses) = £9295 (+ admin) |
| Infliximab |
Cost per 100-mg vial – £419.73. Total cost per infusion – £1355.19 (Assumes 60-kg person so at 5 mg/kg would need three vials, plus administration cost of £96) |
One dose at weeks 0, 2 and 6 (three doses) = £4065.57 | 5 mg/kg every 8 weeks (6.5 doses) = £8808.74 |
For infliximab, the estimated three vials per person is likely to be an underestimate, as the mean weight of patients from the four large trials included in the clinical effectiveness review that gave this information (CHARM,67 CLASSIC I,63 GAIN,64 Targan et al. 57) suggested that the mean weight of CD patients was approximately 71.5 kg so a dose of 5 mg/kg would require four vials per person. Also, it was unclear how the administration cost of £96 taken from an HTA report on psoriatic arthritis (Woolacott et al. 99) was actually derived. In that HTA report, the annual administration cost for treatment (every 8 weeks so – 6.5 treatments) was estimated to be £1673.75, which equates to £257.50 per treatment. Taking these revised costs into account would give the induction dose estimate as £5809.26 {[(4 × 419.73) + 257.50] × 3} and the annual maintenance cost (not including the initial induction dose) estimate as £12,586.73 {[(4 × 419.73) + 257.50] ×6.5} gives the cost per treatment, of which 6.5 are required per year.
If 31,800 CD patients in England and Wales with moderate-to-severe CD received treatment, this equates to a total budget impact for both drugs that can be seen in Table 72. If only CD patients with a CDAI score > 300 are treated (a much more likely scenario), this equates to a total budget impact for both drugs that can be seen in Table 73. The current NICE guidance on infliximab states that it should be used in patients with severe active CD whose condition is refractory to other treatment, who are intolerant to or experience toxicity from these treatments, and for whom surgery is inappropriate. 112 It is unclear how many people would be in this category so the precise budget impact if the current NICE guidance is maintained is unclear. The estimates in Tables 72 and 73 will be an overestimation (compare with Table 74).
| Induction | Maintenance for 1 year | |
|---|---|---|
| Adalimumab from industry submission | £34,105,500 | £295,581,000 |
| Infliximab from industry submission | £129,285,126 | £280,117,932 |
| Infliximab from recalculation | £184,734,468 | £400,258,014 |
| Induction | Maintenance for 1 year | |
|---|---|---|
| Adalimumab from industry submission | £17,052,750 | £147,790,500 |
| Infliximab from industry submission | £64,642,563 | £140,058,966 |
| Infliximab from recalculation | £92,367,234 | £200,129,007 |
| Induction | Maintenance for 1 year | |
|---|---|---|
| Adalimumab from industry submission | £1,705,275 | £14,779,050 |
| Infliximab from industry submission | £6,464,256 | £14,005,897 |
| Infliximab from recalculation | £9,236,723 | £20,012,901 |
Fistulising disease occurs in 17%–43% of people with CD (ACCENT II65). In two trials of moderate-to-severe CD that also gave details on fistulising patients, the proportions were 14% (GAIN64) and 15% (CHARM67). Therefore it is possible that more people with fistulas have mild CD as measured by CDAI scores. If approximately 30% of all CD patients (23,850 in England and Wales) have fistulas then the estimated budget impact is shown in Table 75. Note that the prevalence used in these estimates does not include children. It is estimated that the incidence of CD in children is 5.3 per 100,000 per year (Jenkins11) and that 20%–30% of all new cases of CD are in people aged < 20 years (infliximab industry submission96).
| Induction | Maintenance for 1 year | |
|---|---|---|
| Adalimumab from industry submission | £25,579,125 | £221,685,750 |
| Infliximab from industry submission | £96,963,844 | £210,088,449 |
| Infliximab from recalculation | £138,550,851 | £300,193,510 |
Estimations of the budget impact do not include changes to potential costs arising from two situations. The first is the need for retreatment for patients who initially respond to treatment and then relapse. These patients will require a second induction dose of treatment, increasing the likely cost of treatment, resulting in a likely underestimate of the total budget impact. The budget impact estimates also do not account for people who are responding to treatment but choose to discontinue treatment. This will lead to an overestimate of the cost of the total budget impact.
To put the above calculations into perspective, the total NHS drug bill for 2004–5 was £9,965,000,000. 113 The mean annual cost of treating CD per patient (data collection in 2000, when infliximab was not being widely used) was approximately £3300 (see Table 3), so if 31,800 patients were treated at that time this would have amounted to a cost of approximately £105,067,000.
As a comparison, the industry submission for adalimumab used the 2002 NICE guidance1 on infliximab to estimate that there would be a prevalence of 27,811, of whom 1112 would be eligible for treatment with adalimumab. Combined with the incidence estimates for CD they estimated that 1287 CD patients would be eligible for adalimumab treatment in 2007, rising to 2000 patients in 2011. This would cost £11,971,784 in 2007, rising to £18,604,165 in 2011. They compared this with the budget impact of treating these patients with infliximab of £19,211,660 in 2007 to £29,854,950 in 2011.
The industry submission for infliximab estimated that the total cost of infliximab to the NHS per year would be £24,165,283 in the first year, rising to £38,916,321 in the fifth year. This assumed that 2744 people would be eligible for treatment in the first year, rising to 4419 people in the fifth year. They estimated that 28% of all patients with CD would be eligible for treatment with infliximab.
Mortality rates
No excess mortality rates with adalimumab or infliximab were found in any of the RCTs included in the clinical effectiveness review. However, there are reports in the medical press of relatively high rates of serious AEs with disease-modifying antirheumatic drugs. In a report of the serious adverse drug events reported to the US FDA between 1998 and 2005, infliximab was the seventh most frequently suspected drug for deaths and the third most frequently suspected drug for disability and other serious outcomes. Adalimumab was also listed as having 2389 serious adverse drug events. 114 It is not known how many people were taking these drugs.
In the UK, the drug analysis prints compiled from suspected adverse drug reactions are reported through the Yellow Card Scheme. Fatal reactions reported up to 26 May 2006 are summarised in Table 76. The highest number of deaths was due to infections but it is surprising that the category of diseases of the circulatory system, particularly including myocardial infarctions, was relatively high for both adalimumab and infliximab. Tuberculosis was not linked to many deaths. It is known that the Yellow Card Scheme tends to have an under-reporting of AEs. It has been calculated that £50,390,200 was spent on infliximab in 2006 (for all indications). 115 Given that infliximab costs £419.73 per vial, this would suggest that the NHS used 120,052 vials in 2006. If three vials were used per person, ∼40,000 people would have received infliximab, suggesting a very approximate overall mortality rate of 0.5%. It is unclear from this information whether there is an excess mortality in patients receiving infliximab. There is no information on the number of people taking infliximab for CD or the mortality rate in this patient group.
| Cause of death | Number of reported deaths | |
|---|---|---|
| Adalimumab | Infliximab | |
| Infections (not TB) | 31 | 70 |
| TB | 2 | 6 |
| Neoplasms | 9 | 28 |
| Mental and behavioural disorders | 0 | 1 |
| Diseases of the circulatory system | 31 | 40 |
| Diseases of the respiratory system | 9 | 32 |
| Diseases of the digestive system | 0 | 4 |
| Death/sudden death | 11 | 23 |
| Other | 3 | 10 |
| Total fatal outcome | 96 | 214 |
| Total number of reports | 693 | 1949 |
Chapter 5 Discussion
Statement of principal findings
Clinical effectiveness review
Eleven RCTs were identified that had at least one study arm that included some participants within the UK licensed indication for adalimumab or infliximab. 2,3,45,46,57,58,62–67 The results from these are summarised below. Results for other trial arms that used adalimumab and infliximab outside licence dose regimens are presented in Appendix 10.
For adalimumab, two induction trials (CLASSIC I63 and GAIN64) and two maintenance trials (CLASSIC II66 and CHARM67) in adults with moderate-to-severe CD were identified.
For infliximab, one induction trial (Targan et al. 57) and two maintenance trials in adults with moderate-to-severe CD (ACCENT I2,3 and Rutgeerts et al. 58), one induction (Present et al. 62) and one maintenance trial (ACCENT II65) in adults with fistulising CD, and one induction (Baldassano et al. 46) and one maintenance trial (REACH45) in children with moderate-to-severe CD were identified.
All were placebo-controlled trials, with the exception of the paediatric trials which compared different doses of infliximab, and there were no head-to-head comparisons of the two drugs.
There was no information on the relative effectiveness of treatments depending on ethnicity or other similar groups.
There were concerns regarding the trial design and study quality, particularly for the maintenance trials. These concerns related to the division of patients into subgroups (responders and non-responders) at different time points, the high proportions of scheduled crossovers resulting in the lack of a true placebo group, uncertainties regarding the handling and number of missing binary and continuous data, and the occasional dose escalation beyond licensed indication.
Particular concerns related to the ACCENT I2,3 trial. The comparison between ‘episodic’ and ‘scheduled’ treatment described in the publication by Rutgeerts et al. 3 is not a valid comparison. The ‘placebo’ arm changed to ‘episodic treatment’ after 14 weeks and the scheduled maintenance arm participants could switch to episodic increased treatment. There was no randomisation to episodic and scheduled maintenance arms at the beginning of the trial.
Statistically significant effect sizes in favour of anti-TNF therapy compared with placebo were found in all induction trials by week 4 for both CDAI response rates and remission (for CLASSIC I63 this was only at the highest induction dose regimen of adalimumab); effect sizes in Targan et al. 57 (infliximab) were greater than those for adalimumab but were associated with greater uncertainty.
High and varied placebo response rates observed in the induction trials are thought to result from a tendency of CDAI scores to regress to the mean, from a placebo effect and possibly from differences in concomitant treatment in the trials.
There was statistically significant evidence from both large maintenance trials (CHARM,67 adalimumab and ACCENT I,3 infliximab2) that for the subgroups defined as ‘responders’, anti-TNF therapy was more beneficial than placebo with respect to remission or response rates at reported follow-up times. However, it appeared that point prevalence rather than sustained response (remission) was reported and so the results represented group rather than individual response (remission) and did not inform on persistence of the response (remission) state in the individual.
Indirect comparisons between adalimumab and infliximab were not made because they were judged unlikely to be valid because of the heterogeneity between the trials caused by variation in placebo rates, the apparently arbitrary selection of responders only in the maintenance trials and the varied definition of responder status.
The practice of dichotomising patients into responders and non-responders was considered to be clinically useful only if ‘responders’ were more likely to benefit from maintenance of treatment. There was no evidence available from the identified trials to confirm or refute this.
There was evidence from both the induction and maintenance trials that infliximab promotes fistula closure to a greater extent than placebo (which was statistically significant for maintenance treatment). However, it is possible that fistula closure may not always be the most desirable outcome as it may result in increased development of abscesses.
In the paediatric infliximab trials, no reliable quantitative estimate of the effectiveness of infliximab was possible as the placebo or SC response rates were not measured. In REACH45 there was an improved response with higher dosage of infliximab which implied a possible beneficial effect of infliximab, should a comparison ever be made with a control with zero dosage of infliximab.
Patient-related QoL was measured by the IBDQ in seven trials (induction and maintenance). 2,3,57,58,63,64,66,67 Overall there was a beneficial effect (statistically significant at some time points) of anti-TNF therapy, shown by greater improvement (or less deterioration over time) in IBDQ scores in the treatment arms.
Cost-effectiveness review
A review and quality assessment of existing published literature on cost-effectiveness identified four papers for inclusion into the review. 5–7,90 All concerned infliximab; no published studies on the cost-effectiveness of adalimumab were identified.
The four published infliximab cost-effectiveness studies were all independently funded and the results suggested that single use or ‘episodic’ treatment (various definitions) with infliximab had a relatively high cost-effectiveness ratio for both non-fistulising and fistulising disease (all above £50,000/QALY for non-fistulising disease and all above £100,000/QALY for fistulising disease).
The results of both industry submissions (adalimumab and infliximab) typically showed ICERs of under £30,000 for both anti-TNFs versus SC.
For the adalimumab industry submission model there was a lack of clarity over the source and interpretation of data used in the industry model, and key elements of the model could not be verified. Corrected results for both severe CD and moderate and severe (combined) CD were substantially higher than in the industry submitted model; in the severe subgroup of patients the corrected ICER approached cost-effectiveness (at a threshold of £30,000).
For infliximab, errors were identified in the industry model (active CD), some of which could not be corrected. The revised model was suggestive of infliximab being cost-effective for ‘episodic’ (clinician discretion) treatment, though the exact nature of this intervention remained unclear. Scheduled maintenance treatment with infliximab was unlikely to be cost-effective. The industry model for fistulising CD revised here also suggested that infliximab was unlikely to be cost-effective. No functioning model was provided for paediatric CD within the time frame of the project, so no conclusions could be made from the reported findings.
De novo economic model
A simple Markov model was developed from the NHS/PSS perspective to estimate the incremental cost per QALY for both drugs compared with SC in (a) induction/episodic therapy (as defined for the purposes of the economic model) for moderate and severe disease; and (b) maintenance therapy for moderate and severe disease. The model had a 1-year time horizon and was constructed and analysed in treeage pro 2008.
The findings were that for induction/episodic treatment, both adalimumab and infliximab were cost-effective (dominant relative to SC) in the management of severe CD and that adalimumab (but not infliximab) was cost-effective for moderate CD, according to the criteria laid out in the NICE Guide to the methods of technology appraisal. 93 Neither drug was cost-effective as maintenance therapy for moderate or severe disease.
Budget impact assessment
A simple budget impact assessment was conducted using information from prevalence data and the industry submissions. It suggested that the total cost to the NHS in England and Wales for induction in severe disease only could range between £17M and £92M and for maintenance for 1 year between £140M and £200M. These totals would be less only if those CD patients whose condition is refractory to other treatment, who are intolerant to or experience toxicity from these treatments and for whom surgery is inappropriate were treated. It is unclear how many people would be in this category, so the precise budget impact if the current NICE guidance is maintained was unclear.
Strengths and limitations of the assessment
-
Well-established systematic review techniques were used for this technology assessment, which lends considerable strength to its validity and reliability.
-
Searches for RCTs were conducted systematically. Using a sensitive search strategy was likely to have identified all of the relevant evidence; checking industry submissions did not yield additional RCTs.
-
Both the licence indications (for adalimumab and infliximab) and current NICE guidance112 on infliximab specify the use of the drugs in ‘severe’ CD but the NICE scope for this work specified ‘moderate-to-severe’ CD. The identified induction RCTs (or induction phases of maintenance RCTs) included patients with moderate-to-severe CD or a CDAI score between 220 and 400 or 450. This means that none of the included trials matched the NICE guidance or licence indications with reference to the severity of CD. Subgroup results for patients with an initial CDAI score of ≥ 300 have been presented here if they were available from the trials. However, none of the trials were planned for this specific subgroup so did not stratify by whether patients were above or below the 300 CDAI threshold. Furthermore, there were no consensus guidelines in the literature on what CDAI score constitutes ‘severe’ CD. Both the licence indications and the NICE guidance specify that adalimumab and infliximab should be used in patients who are resistant and/or intolerant to conventional treatment. While many or most of the patients in the included studies were likely to meet this criterion, some may not have done. Only one study (Rutgeerts et al. 58 in infliximab) had an inclusion criterion that patients should be treatment resistant.
-
Considerable efforts were made to try to understand the flow of patients through the trials. Several of the included trials had very complicated structures where patients could take several different pathways with different treatments, and one of these has been diagrammed to illustrate patient flow as clearly as possible (see Appendix 9).
-
The assessment of relative effectiveness of adalimumab and infliximab was limited by the fact that no head-to-head comparisons were available. A formal indirect comparison was inappropriate owing to clinical heterogeneity between trials, indicated by variation in placebo rates, and the variable subgroup selection of responders and non-responders.
-
For dichotomous outcomes, variable placebo rates can influence the effect size values depending on the outcome measure used. In order to gain accurate estimates of effect sizes, both placebo and intervention rates and both risk differences and risk ratios are presented in the clinical effectiveness section (see Chapter 3).
-
Trial designs for the maintenance trials were unusual, so trial quality and any potential impact on the validity of results were investigated in detail.
-
The systematic appraisal of both the published papers and industry models facilitated a comprehensive review of the cost-effectiveness evidence in this area. However, the evidence is limited; only four published economic studies met the review inclusion criteria,5–7,90 of which all considered infliximab, and none considered adalimumab. One paper was not quality assessed because of a lack of detail,5 and the remaining three papers were of variable quality. 6,7,90
-
The assessments of the industry models were hampered by inconsistent use of data, lack of clarity over the source and interpretation of data and, in one case, unclear details of treatment, which meant that it was not possible to satisfactorily verify or interpret the model.
-
The strength of the new economic model presented here is its simple and transparent structure and inputs. However, CD is a very complex disease so it could be argued that the simple model presented here does not take account of all of the nuances of the disease. On the other hand, the more complicated a model becomes, the harder it is to establish accurate inputs to populate the model. Given that there was much uncertainty around a number of model parameters, not least the effectiveness estimates and relapse rates, on balance it was felt to be more appropriate to have a simpler model.
Uncertainties
All of the included trials in the clinical effectiveness review were funded by the relevant drug companies. It is uncertain whether independently funded research in this area would yield different results; it may, however, have much more simple designs, which would aid interpretation of the results considerably.
CD is a lifelong condition with sometimes relatively long cycles of relapse and remission. The trials were mostly of 1 year’s duration or less. It is uncertain whether the effect of the drugs would gradually wear off over time, and whether this might be associated with an increase in antibodies to the drug.
The way the included trials were conducted and reported has provided considerable uncertainty as to the effectiveness of the drugs. Aspects of this are discussed in detail in Chapter 3, Discussion of results and assessment of effectiveness, and, for the maintenance trials, include:
-
How the relatively large proportions who crossed over or were lost to follow-up were counted.
-
The use of point prevalence, rather than number of patients remaining in remission or as responders.
-
Different or unclear handling of missing binary and continuous data.
-
The division of patients into subgroups of responders and non-responders at different time points.
One area of considerable uncertainty is regarding the division of patients as ‘responders’ and ‘non-responders’ on the basis of initial response to a single dose or up to three or four doses only. Where trials did give maintenance treatment to ‘non-responders’, separate results have not been published. It may be that in the ‘non-responder’ group there are CD patients who will respond to treatment but take longer to do so. The finding regarding the division of patients into responders and non-responders at specific time points has implications for the licence indications. The current licence indication for infliximab mentions that if patients have not responded to induction treatment within 2 weeks, there is no evidence to support further treatment. No evidence was identified to support this statement, so it is unclear whether this part of the licence indication is evidence-based. It may be that the some of the so-called non-responders are taking longer to respond because of drug–drug interactions that have not yet been evaluated.
The patients included in most of the trials had varying levels of severity of CD. They were mostly described as having moderate-to-severe CD or a CDAI score between 220 and 400 or 450. The trials were all multicentre and there is no indication whether patients from different countries had different mean levels of severity. Patients in the USA may have been enrolled at a level of less severity than UK or European patients because of the different health systems in the different countries. Also there is no information on the ethnic group of participants. There is no information on whether the drugs were found to be more effective in one country than another or in one ethnic group than another. Therefore it is unclear how generalisable the results of these trials are to the UK.
Applicability to individual patients is also uncertain. Although patients within the categories of ‘moderate-to-severe’ CD and fistulising CD may appear to be fairly homogeneous populations, this is unlikely to be the case in practice. Owing to the variable nature of the disease, these are actually likely to be very heterogeneous populations in terms of manifestation of disease, severity of disease, treatment (including surgical) history or concomitant medications, and impact of disease on patients’ lives. Therefore the effect of a drug on a specific type of patient is also unclear, and it is not known if there are subgroups of patients who would benefit more or less from these drugs.
The main outcome measures used in the trials are based on the CDAI, which may not be an adequate measure for capturing clinically meaningful changes in disease severity (see Chapter 3, Discussion of results and assessment of effectiveness for further detail) or capturing aspects of QoL such as psychological, social and occupational functioning. The disease-specific QoL measure IBDQ was reported in a number of trials but a generic QoL measure such as EQ-5D may have been more useful.
There was very little information from any of the included trials about hospitalisation rates. This is a key cost driver in the industry economic models and the economic model presented here. Also, some hospitalisations are for relatively minor procedures, such as fistula drainage in someone who is relatively well, and can just be an overnight stay whereas others are because patients are seriously ill and have to stay in hospital for weeks. Therefore simple counts of hospitalisations will not take into account all relevant information.
There was considerable uncertainty as to whether treatment might affect mortality rates.
The comparison of AE rates was affected by the design of the maintenance trials, as all patients initially received the study drug before being randomised to drug or placebo and, additionally, patients in most maintenance trials had the opportunity to cross over from placebo to drug treatment if specified criteria were met. Therefore, there is uncertainty around AEs due to the study drug.
The uncertainties in the clinical data (as outlined above) have complicated the economic analysis. It is difficult to define comparators where the details of treatment are uncertain. In such cases, the interpretation of economic models within the published papers becomes problematical.
The published economic models relied heavily on a small body of data, primarily 24 years of data from Olmstead County, USA. A Markov analysis of these data has been widely used. Similarly, in part, the industry models relied on data from small samples.
Both the published cost-effectiveness studies and the industry submission models lacked long-term data.
The analyses within all of the economic models typically used a Markov model. Markov models assume zero memory; how long a patient has been in a health state and how they got there may impact on resources. This could be important in a CD patient group.
Other relevant factors
It was outside the remit of this assessment to look at the effectiveness of adalimumab or infliximab as first-line or ‘top-down’ therapy. It has been suggested that there may be advantages to this approach in terms of avoiding complications such as surgery and hospitalisations. 61,116
Chapter 6 Conclusions
Implications for service provision
Adalimumab and infliximab gave statistically significant effect sizes in favour of anti-TNF therapy compared with placebo in all induction trials for moderate-to-severe CD patients [between 6% and 24% (adalimumab) and 21% and 44% (infliximab) more patients in remission with anti-TNFs than with placebo]. There was statistically significant evidence from one large maintenance trial for adalimumab and one large maintenance trial for infliximab that, for the subgroups defined as ‘responders’, anti-TNF therapy was more beneficial than placebo with respect to remission or response rates at reported follow-up times [between 24% and 29% (adalimumab) and 14% and 24% (infliximab) more patients achieved remission with anti-TNFs].
The findings of the economic model were that for induction, both adalimumab and infliximab were cost-effective (dominant relative to SC) in the management of severe CD and adalimumab was cost-effective for moderate CD (dominant relative to SC), according to the criteria laid out in the NICE Guide to the methods of technology appraisal. 93 Induction therapy with infliximab was not cost-effective for moderate CD (ICER of £94,321). Neither drug was cost-effective as maintenance therapy for moderate or severe disease (ICERs around £14M and £5M respectively for both drugs).
The cost-effectiveness analysis highlights important variations in effectiveness and cost between the two therapies. Perhaps most importantly, the analysis reflects the fact that a substantial number of patients will achieve remission under SC and that the incidence of relapse among those in remission is such that maintenance therapy with the anti-TNF drugs assessed here would have to be much less costly for it to be a cost-effective option.
Suggested research priorities
Independently funded RCT research on effectiveness of treatment
-
If the licence indication for both drugs remains for patients with severe active CD who are resistant and/or intolerant to other CD treatments, trials for anti-TNF drugs should be conducted in these patients separately.
-
In order to take into account natural fluctuations in relapse and remission in CD, any future trials should be conducted for a period of at least 1 year.
-
Any future trials in children should include a placebo (SC) arm, as there is currently no evidence of the benefit of anti-TNF therapy compared with SC.
-
As there is currently no evidence that the subgroup of ‘responders’ is more likely to benefit than the whole group of eligible CD patients (‘responders’ and ‘non-responders’), any future maintenance trials should be undertaken in the whole patient group; subgroup analysis of ‘responders and ‘non-responders’ can be undertaken as part of the analysis, providing the trial has sufficiently high patient numbers.
-
The potential benefit of ‘episodic’ treatment (treatment as required/deemed clinically necessary) compared with scheduled treatment should be investigated in an appropriately designed RCT, randomised to three treatment arms (placebo or SC, ‘episodic’ treatment and scheduled treatment) after appropriate induction.
-
CD is a relapsing and remitting condition. Each individual will have episodes of varying length and severity and periods of remission of varying length and mildness. Some patients will go into remission without the use of additional drug treatment. Therefore it is vital that this is taken into account when planning RCTs to assess accurately the added benefit of a particular drug treatment.
-
There should be no scheduled crossovers in RCTs as this means that there is no true placebo arm and results become difficult to interpret, particularly where high proportions of patients cross over. Where patients need to use alternative treatment during the course of a trial, they should be considered as withdrawals.
-
Any future trials should measure QoL (using a generic QoL measure, e.g. EQ-5D) and should also record number and type of hospitalisations (including length of stay in hospital), as this information is important when considering the cost-effectiveness of treatments. If CDAI continues to be used as the main outcome measure there needs to be much more work on how this translates to the effect of the disease on the person. Does a change of 50 points from 150 to 200 have a similar magnitude of impact as a change from 350 to 400?
-
Reporting of trial results needs to be clear, with results reported for all patients, and responders and non-responders separately if appropriate, and numbers of withdrawals at each time point clearly stated.
Research into the natural history of Crohn’s disease
There is currently little information on the natural history of the disease in individual patients. A cohort study following individual patients over several years would provide information on mortality rates, and the length of time patients have mild, moderate or severe disease or are in remission; this information in turn would facilitate the interpretation of trial results. This cohort study must include a variety of patient attributes including ethnicity.
Acknowledgements
Dr Tariq Iqbal and Dr Ralph Boulton (University Hospital Birmingham) for providing clinical advice, Natalie Rowles for administration, Denise Cann (NACC) for advice on the patient perspective and Dr Fujian Song for internal peer review.
Contributions of authors
The protocol was written by J Dretzke, C Meads and M Connock, with comments from all coauthors. Literature searching was carried out by A Fry-Smith. Study selection, data extraction and quality assessment clinical effectiveness were carried out by J Dretzke, M Connock, J Czeczot and C Meads. M Connock performed the clinical effectiveness analysis, with comments from all coauthors. Systematic review cost-effectiveness was carried out by R Edlin, C Hulme and C McCabe. R Edlin, C Hulme and C McCabe did the appraisal of industry models with comments from all coauthors. C McCabe, R Edlin and J Round ran the de novo economic model. J Dretzke, R Edlin, J Round, M Connock, C Meads, C Hulme and C McCabe wrote this report, with contributions from all coauthors. C Meads was the project manager.
About ‘home unit’
The West Midlands Health Technology Assessment Collaboration (WMHTAC) is an organisation involving several universities and academic groups who collaboratively undertake research synthesis to produce health technology assessments. Most of the members are based in the Department of Public Health & Epidemiology, University of Birmingham, Birmingham, UK; however, other members are drawn from a wide field of expertise including economists and mathematical modellers from the Health Economics Facility, University of Birmingham, Birmingham, UK.
The WMHTAC produce systematic reviews, health technology assessments and economic evaluations for the NHS R&D HTA programme (NCCHTA), NICE, and for the health service in the West Midlands. The WMHTAC also undertakes methodological research on research synthesis, and provides training in systematic reviews and health technology assessment.
Disclaimers
The views expressed in this publication are those of the authors and not necessarily those of the HTA programme or the Department of Health.
References
- National Institute for Health and Clinical Excellence . Guidance on the Use of Infliximab for Crohn’s Disease. Technology Appraisal No. 40 2002. www.nice.org.uk/nicemedia/pdf/NiceCROHNS40GUIDANCE.pdf (accessed October 2007).
- Rutgeerts P, Feagan BG, Lichtenstein GR, Mayer LF, Schreiber S, Colombel JF, et al. Comparison of scheduled and episodic treatment strategies of infliximab in Crohn’s disease. Gastroenterology 2004;126:402-13.
- Hanauer SB, Feagan BG, Lichtenstein GR, Mayer LF, Schreiber S, Colombel JF, et al. Maintenance infliximab for Crohn’s disease: The ACCENT I randomised trial. Lancet 2002;359:1541-9.
- Abbott Laboratories Ltd . Adalimumab (Humira®). Submission to the National Institute for Health and Clinical Excellence 2007:1-98.
- Clark W, Raftery J, Song F, Barton P, Cummins C, Fry-Smith A, et al. Systematic review and economic evaluation of the effectiveness of infliximab for the treatment of Crohn’s disease. Health Technol Assess 2003;7.
- Marshall J, Blackhouse G, Goeree R, Brazier N, Irvine E, Faulkner L, et al. Infliximab for the Treatment of Crohn’s Disease: A Systematic Review and cost–utility Analysis. Technology Report No 24 2002.
- Jaisson-Hot I, Flourie B, Descos L, Colin C. Management for severe Crohn’s disease: a lifetime cost-utility analysis. Int J Technol Assess Health Care 2004;20:274-9.
- Jewell D. Crohn’s disease. Medicine 1998;26:87-92.
- Carter MJ, Lobo AJ, Travis SP. on behalf of the IBD section of the British Society of Gastroenterology . Guidelines for the management of inflammatory bowel disease in adults. Gut 2004;53:V1-V16.
- Hanauer SB, Sandborn W. Management of Crohn’s disease in adults. Am J Gastroenterology 2001;96:635-43.
- Jenkins HR. Inflammatory bowel disease. Arch Dis Child 2001;85:435-7.
- Henriksen M, Jahnsen J, Lygren I, Aadland E, Schulz T, Vatn MH, et al. Clinical course in Crohn’s disease: results of a five-year population-based follow-up study (the IBSEN study). Scand J Gastroenterol 2007;42:602-10.
- Vermeire S, Van Assche G, Rutgeerts P. Review article: altering the natural history of Crohn’s disease-evidence for and against current therapies. Aliment Pharmacol Ther 2006;25:3-12.
- Baumgart DC, Sandborn WJ. Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet 2007;369:1641-57.
- Hanauer SB, Meyers S. Managment of Crohn’s disease in adults. Am J Gastroenterol 1997;92:559-65.
- Irvine EJ. Usual therapy improves perianal Crohn’s disease as measured by a new Disease Activity Index. J Clin Gastroenterol 1995;20:27-32.
- Loftus EV. Clinical epidemiology of inflammatory bowel disease: incidence, prevalence, and environmental influences. Gastroenterology 2004;126:1504-17.
- Meuwis MA, Fillet M, Chapelle JP, Malaise M, Louis E, Merville MP. New biomarkers of Crohn’s disease: serum biomarkers and development of diagnostic tools. Expert Rev Mol Diagn 2008;8:327-37.
- Knigge KL. Inflammatory bowel disease. Clin Cornerstone 2002;4:49-60.
- Gurudu S, Fiocchi C, Katz JA. Inflammatory bowel disease. Best Pract Res Clin Gastroenterol 2002;16:77-90.
- Silverberg MS, Satsangi J, Ahmad T, Arnott ID, Bernstein CN, Brant SR, et al. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: Report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. Can J Gastroenterol 2005;19:5-36.
- Louis E, Collard A, Oger AF, Degroote E, Aboul Nasr El Yafi FA, Belaiche J. Behaviour of Crohn’s disease according to the Vienna classification: changing pattern over the course of the disease. Gut 2001;49:777-82.
- Cosnes J, Cattan S, Blain A, Beaugerie L, Carbonnel F, Parc R, et al. Long-term evolution of disease behavior of Crohn’s disease. Inflamm Bowel Dis 2002;8:244-50.
- Felley C, Mottet C, Juillerat P, Froehlich F, Burnand B, Vader JP, et al. Fistulizing Crohn’s disease. Digestion 2005;71:26-8.
- Munkholm P, Langholtz E, Davidsen M, Binder V. Disease activity courses in a regional cohort of Crohn’s disease patients. Scand J Gastroenterol 1995;30:699-706.
- Silverstein MD, Loftus EV, Sandborn WJ, Tremaine WJ, Feagan BG, Nietert PJ, et al. Clinical course and costs of care for Crohn’s disease: Markov model analysis of a population-based cohort. Gastroenterology 1999;117:49-57.
- Russel MGVM. Changes in the incidence of inflammatory bowel disease: what does it mean?. Eur J Int Med 2000;11:191-6.
- Hanauer SB. Inflammatory bowel disease: epidemiology, pathogenesis, and therapeutic opportunities. Inflamm Bowel Dis 2006;12:S3-9.
- Ehlin AGC, Montgomery SM, Ekbom A, Pounder RE, Wakefield AJ. Prevalence of gastrointestinal diseases in two British national birth cohorts. Gut 2003;52:1117-21.
- Rubin GP, Hungin APS, Kelly PJ, Ling J. Inflammatory bowel disease: epidemiology and management in an English general practice population. Aliment Pharmacol Ther 2000;14:1553-9.
- National Association for Colitis and Crohn’s Disease . Inflammatory Bowel Disease Basics 2004. http://nacc.org.uk/content/ibd.asp (accessed 25 April 2007).
- Shivananda J, Lennard-Jones J, Logan R, Fear N, Price A, Carpenter L, et al. Incidence of inflammatory bowel disease across Europe: is there a difference between north and south? Results of the European collaborative study on inflammatory bowel disease (EC-IBD). Gut 1996;39:690-7.
- Stone MA, Mayberry JF, Baker R. Prevalence and management of inflammatory bowel disease: a cross-sectional study from central England. Eur J Gastroenterol Hepatol 2003;15:1275-80.
- Yapp TR, Stenson R, Thomas GAO, Lawrie BW, Williams GT, Hawthorne AB. Crohn’s disease incidence in Cardiff from 1930: an update for 1991–1995. Eur J Gastroenterol Hepatol 2000;12:907-11.
- National Association for Colitis and Crohn’s Disease . Information Sheets and Drug Treatment Information 2004. www.nacc.org.uk/content/services/infoSheets.asp (accessed 7 November 2007).
- Blondel-Kucharski F, Chircop C, Marquis P, Cortot A, Baron F, Gendre J, et al. Health-related quality of life in crohn’s disease: a prospective longitudinal study in 231 patients. Am J Gastroenterol 2001;96:2915-20.
- Bassi A, Dodd S, Williamson P, Bodger K. Cost of illness of inflammatory bowel disease in the UK: a single centre retrospective study. Gut 2004;53:1471-8.
- Sostegni R, Daperno M, Scaglione N, Lavagna A, Rocca R, Pera A. Review article: Crohn’s disease: monitoring disease activity. Aliment Pharmacol Ther 2003;17:11-7.
- Best WR, Becktel JM, Singleton JW, Kern F. Development of a Crohn’s Disease Activity Index. National Co-operative Crohn’s disease study. Gastroenterology 1976;70:439-44.
- Yoshida EM. The Crohn’s Disease Activity Index, its derivatives and the Inflammatory Bowel Disease Questionnaire: a review of instruments to assess Crohn’s disease. Can J Gastroenterol 1999;13:65-73.
- Sandborn WJ, Feagan BG, Hanauer SB, Lochs H, Lofberg R, Modigliani R, et al. A review of activity indices and efficacy endpoints for clinical trials of medical therapy in adults with Crohn’s disease. Gastroenterology 2002;122:512-30.
- Best WR. Predicting the Crohn’s Disease Activity Index from the Harvey–Bradshaw Index. Inflamm Bowel Dis 2006;12:304-10.
- Irvine EJ, Feagan BG, Rochon J, Archambault A, Fedorak RN, Groll A, et al. Quality of Life: a valid and reliable measure of therapeutic efficacy in the treatment of inflammatory bowel disease. Gastroenterology 1994;106:287-96.
- Mary JY, Modigliani R. Development and validation of an endoscopic index for the severity for Crohn’s disease: a prospective multicentre study. Gut 1989;30:983-9.
- Hyams J, Crandall W, Kugathasan S, Griffiths A, Olson A, Johanns J, et al. Induction and maintenance infliximab therapy for the treatment of moderate-to-severe Crohn’s disease in children. Gastroenterology 2007;132:863-73.
- Baldassano R, Braegger CP, Escher JC, DeWoody K, Hendricks DF, Keenan GF, et al. Infliximab (REMICADE) therapy in the treatment of pediatric Crohn’s disease. Am J Gastroenterol 2003;98:833-8.
- British National Formulary. London: British Medical Association and Royal Pharmaceutical Society of Great Britain; 2008.
- Loftus EV, Schoenfeld P, Sandborn WJ. The epidemiology and natural history of Crohn’s disease in population-based patient cohorts from North America: a systematic review. Aliment Pharmacol Ther 2002;16:51-60.
- Royal College of Physicians . UK IBD Audit 2006. National Results for the Organisation &Amp; Process of IBD Care in the UK 2006. www.rcplondon.ac.uk/clinical-standards/ceeu/Current-work/IBD/Pages/1st-Round.aspx (accessed November 2010).
- Schreiber S, Rutgeerts P, Fedorak RN, Khaliq-Kareemi M, Kamm MA, Boivin M, et al. A randomized, placebo-controlled trial of certolizumab pegol (CDP870) for treatment of Crohn’s disease. Gastroenterology 2005;129:807-18.
- Calabrese L. The yin and yang of tumor necrosis factor inhibitors. Cleve Clin J Med 2006;73:251-6.
- European Medicines Agency . Summary of Product Characteristics (Humira) n.d. www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000481/WC500050870.pdf (accessed November 2010).
- European Medicines Agency . Summary of Product Characteristics (Remicade) n.d. www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000240/WC500050888.pdf (accessed November 2010).
- Centocor I. Medication Guide Remicade 2007. www.remicade.com/remicade/assets/Med_Guide.pdf (accessed 10 October 2007).
- Abbott Laboratories . Important Safety Information Humira 2007. www.humira.com (accessed 10 October 2007).
- Han PD, Cohen RD. Managing immunogenic responses to infliximab. Drugs 2004;64:1767-77.
- Targan S, Hanauer SB, van Deventer SJH, Mayer L, Present DH, Braakman T, et al. A short term study of chimeric monoclonal antibody cA2 to tumour necrosis factor alpha for Crohn’s disease. N Engl J Med 1997;337:1029-35.
- Rutgeerts P, D’Haens G, Targan S, Vasiliauskas E, Hanauer SB, Present DH, et al. Efficacy and safety of retreatment with anti-tumor necrosis factor antibody (infliximab) to maintain remission in Crohn’s Disease. Gastroenterology 1999;117:761-9.
- Farrell RJ, Alsahli M, Jeen Y-T, Falchuk KR, Peppercorn MA, Michetti P. Intravenous hydrocortisone premedication reduces antibodies to infliximab in Crohn’s disease: A randomized controlled trial. Gastroenterology 2003;124:917-24.
- Lemann M, Mary J, Duclos B, Veyrac M, Dupas J, Delchier JC, et al. Infliximab Plus azathioprine for steroid-dependent Crohn’s disease patients: a randomized placebo-controlled trial. Gastroenterology 2006;130:1054-61.
- Hommes DW, Baert F, Van Assche G, Caenepeel F, Vergauwe P, Tuynman H, et al. Management of recent onset Crohn’s disease: a controlled, randomised trial comparing step-up and top-down therapy (Abstract). Gastroenterology 2005;129.
- Present DH, Rutgeerts P, Targan S, Hanauer SB, Mayer L, van Hogezand Rea. Infliximab for the treatment of fistulae in patients with Crohn’s disease. N Engl J Med 1999;340:1398-405.
- Hanauer SB, Sandborn WJ, Rutgeerts P, Fedorak RN, Lukas M, MacIntosh D, et al. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn’s disease: The CLASSIC-I trial. Gastroenterology 2006;130:323-32.
- Sandborn WJ, Rutgeerts P, Enns R, Hanauer SB, Colombel JF, Panaccione R, et al. Adalimumab induction therapy for Crohn disease previously treated with infliximab: a randomized trial. Ann Intern Med 2007;146:829-38.
- Sands BE, Anderson FH, Bernstein CN, Chey WY, Feagan BG, Fedorak RN, et al. Infliximab maintenance therapy for fistulizing Crohn’s Disease. N Engl J Med 2004;350:876-85.
- Sandborn WJ, Hanauer SB, Rutgeerts PJ, Fedorak RN, Lukas M, Macintosh DG, et al. Adalimumab for maintenance treatment of Crohn’s Disease: Results of the CLASSIC II Trial. Gut 2007;56:1232-9.
- Colombel J, Sandborn WJ, Rutgeerts P, Enns R, Hanauer SB, Panaccione R, et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn’s Disease: The CHARM Trial. Gastroenterology 2007;132:52-65.
- D’Haens G, van Deventer S, van Hogezand R, Chalmers D, Kothe C, Baert F. Endoscopic and histological healing with infliximab anti-tumour necrosis factor antibodies in Crohn’s disease. Gastroenterology 1999;116:1029-34.
- Egger M, Davey Smith G, Altman DG. Systematic reviews in health care; meta-analysis in context. London: BMJ Books; 2001.
- Menter A, Feldman SR, Weinstein GD, Papp K, Evans R, Guzzo C, et al. A randomized comparison of continuous vs. intermittent infliximab maintenance regimens over 1 year in the treatment of moderate-to-severe plaque psoriasis. J Am Acad Dermatol 2007;56:15-31.
- Wu EQ, Yu A, Atanasov P, Tang J, Pollack P, Lomax K, et al. Lower Disease Activity and Clinical Remission Are Associated With Reduced Hospitalisation Risk in Crohn’s Disease n.d.
- Feagan B, Panaccione R, Sandborn W, D’Haens G, Schreiber S, Rutgeerts P, et al. An Evaluation of Adalimumab on the Risk of Hospitalisation in Patients With Crohn’s Disease, Data from CHARM n.d.
- Behm BW, Bickston SJ. Tumor necrosis factor-alpha antibody for maintenance of remission in Crohn’s disease. Cochrane Database Syst Rev 2008;1.
- Glenny AM, Altman DG, Song F, Sakarovitch C, Deeks JJ, D’Amico R. Indirect comparisons of competing interventions. Health Technol Assess 2005;9.
- Sands BE, Blank MA, Patel K, Van Deventer SJ. Long-term treatment of rectovaginal fistulas in Crohn’s disease: Response to infliximab in the ACCENT II study. Clin Gastroenterol Hepatol 2004;2:912-20.
- Sands BE, Blank MA, Diamond RH, Barrett JP, Van Deventer SJ. Maintenance infliximab does not result in increased abscess development in fistulizing Crohn’s disease: Results from the ACCENT II study. Aliment Pharmacol Ther 2006;23:1127-36.
- Bongartz T, Sutton AJ, Sweeting MJ, Buchan I, Matteson EL, Montori V. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA 2006;295:2275-85.
- Shoor S. Review: anti-tumor necrosis factor antibody therapy for rheumatoid arthritis increases risk for serious infection and malignancy. ACP J Club 2006;145.
- Raval A, khavan-Toyserkani G, Brinker A, Avigan M. Brief communication: characteristics of spontaneous cases of tuberculosis associated with infliximab. Ann Intern Med 2007;147:699-702.
- Ramos-Casals M, Brito-Zeron P, Munoz S, Soria N, Galiana D, Bertolaccini L, et al. Autoimmune diseases induced by TNF-targeted therapies: analysis of 233 cases. Medicine (Baltimore) 2007;86:242-51.
- Kwon HJ, Cote TR, Cuffe MS, Kramer JM, Braun MM. Case reports of heart failure after therapy with a tumor necrosis factor antagonist. Ann Intern Med 2003;138:807-11.
- Socal MP, Vargas AP, Laporte EA, Costa AF, Picon PD. Progressive Multifocal Leuko-Encephalopathy Induced by Monoclonal Antibodies- a Systematic Review: Preliminary Data n.d.
- Yamomoto M, Takahashi H, Wakasugi H, Sukawa Y, Saito M, Suzuki C, et al. Leukoencephalopathy during administration of etanercept for refractory rheumatoid arthritis. Mod Rheumatol 2007;17:72-4.
- Su C, Lichtenstein G, Krok K, Brensinger CM, Lewis JD. A meta-analysis of the placebo rates of remission and response in clinical trials of active Crohn’s disease. Gastroenterology 2004;126:1257-69.
- Chang M, Chow S-C. Adaptive design methods in clinical trials – a review. Orphanet J Rare Dis 2008;3:11-36.
- Breban M, Ravaud P, Claudepierre P, Baron G, Henry YD, Hudry C, et al. Maintenance of infliximab treatment in ankylosing spondylitis: results of a one-year randomized controlled trial comparing systematic versus on-demand treatment. Arthritis Rheum 2008;58:88-97.
- Chen Y-F, Jobanputra P, Barton P, Jowett S, Bryan S, Clark W, et al. A systematic review of the effectiveness of adalimumab, etanercept and infliximab for the treatment of rheumatoid arthritis in adults and an economic evaluation of their cost-effectiveness. Health Technol Assess 2006;10.
- Brodsky RA, Young NS, Antonioli E, Risitano AM, Schrezenmeier H, Schubert J, et al. Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Blood 2008;111:1840-7.
- Drummond MF, Jefferson T. Guidelines for authors and peer reviewers of economic submissions to the BMJ. BMJ 1996;313:275-83.
- Arseneau KO, Cohn SM, Cominelli F, Connors AFJ. Cost-utility of initial medical management for Crohn’s disease perianal fistulae. Gastroenterology 2001;120:1640-56.
- Marshall JK, Blackhouse G, Goeree R, Brazier N, Irvine EJ, O’Brien BJ. Clinical and economic assessment: infliximab for the treatment of Crohn’s disease. Technology overview no. 8. Ottawa: Canadian Coordinating Office for Health Technology Assessment; 2002.
- Gregor J, McDonald JWD, Klar N, Wall R, Atkinson K, Lamba B. An evaluation of utility measure in Crohn’s disease. Inflamm Bowel Dis 1997;3:265-76.
- National Institute of Health and Clinical Excellence . Guide to the Methods of Technology Appraisal 2008.
- Organisation for economic co-operation and development . Purchasing Power Parities (PPP) n.d. www.oecd.org/std/ppp (accessed November 2007).
- Office for National Statistics . Retail Prices Index: Annual Index Numbers of Retail Prices 1948-2009 (RPI) (RPIX) n.d. www.statistics.gov.uk/statbase/tsdataset.asp?vlnk=7172%26More=N%26All=Y (accessed November 2007).
- Schering-Plough Ltd . Remicade® in the Treatment of Crohn’s Disease in the United Kingdom. Submission to the National Institute for Health and Clinical Excellence 2007:1-67.
- Wolters FL, Russel MG, Sijbrandij J, Schouten L, Odes S, Riis L, et al. Crohn’s disease: increased mortality 10 years after diagnosis in a Europe-wide population based cohort. Gut 2006;55:510-18.
- Jess T, Riis L, Vind I, Winther KV, Borg S, Binder V, et al. Changes in clinical characteristics, course and prognosis of inflammatory bowel disease during the last 5 decades: a population-based study from Copenhagen, Denmark. Inflamm Bowel Dis 2007;13:481-9.
- Woolacott N, Bravo Vergel Y, Hawkins N, Kainth A, Khadjesari Z, Misso K, et al. Etanercept and infliximab for the treatment of psoriatic arthritis: a systematic review and economic evaluation. Health Technol Assess 2006;10.
- Casellas F, Arenas JI, Baudet JS, Fabregas S, Garcia N, Gelabert J, et al. Impairment of health-related quality of life in patients with inflammatory bowel disease: a Spanish multicenter study. Inflamm Bowel Dis 2005;11:488-96.
- Dolan P, Gudex C, Kind P, Williams A. A social tariff of EuroQol: results from a UK general population survey. Centre for Health Economics discussion paper 138; 1995.
- British Medical Association and Royal Pharmaceutical Society of Great Britain . British National Formulary 2007.
- Curtis L, Netten A. Unit costs of of health and social care. Canterbury: Personal Social Services Research Unit; 2006.
- National Institute of Health and Clinical Excellence . Guide to the Methods of Technology Appraisal 2004.
- Department of Health . NHS Reference Costs 2005–06 2006. www.dh.gov.uk/en/publicationsandstatistics/Publications/PublicationsPolicyAndGuidance/DH_062884 (accessed June 2008).
- Rosenbaum PL, Saigal S, Spilker B. Quality of life and pharmacoeconomics in clinical trials. Philadelphia: Lippincott-Raven; 1996.
- Harris M, Buttterworth G. Developmental psychology: A student’s handbook. Hove: Psychology Press; 2002.
- Matza LS, Swensen AR, Flood EM, Secnik K, Leidy NK. Assessment of health-related quality of life in children: a review of conceptual, methodological, and regulatory issues. Value Health 2004;7:79-92.
- Fox-Rusby J, Parker M. Culture and the measurement of health-related quality of life. Eur Rev Appl Psychol 1995;45:257-63.
- Petrou S, Henderson J. Preference-based approaches to measuring the benefits of perinatal care. Birth 2003;30:217-26.
- Department of Health . NHS Reference Costs 2006–07 n.d. www.dh.gov.uk/en/Publicationsandstatistics/Publications/PublicationsPolicyAndGuidance/DH_082571 (accessed June 2008).
- National Institute of Health and Clinical Excellence . Use of Tumour Necrosis Factor Alpha (TNF A) Inhibitors (adalimumab, and Infliximab [review]) for Crohn’s Disease (review of TA40) 2010. http://guidance.nice.org.uk/TA187 (accessed November 2010).
- TheyWorkForYou.com . NHS: Drugs. Health. Written Answers and Statements, 12 July 2007 2007. www.theyworkforyou.com/wrans/?id=2007-07-12b.149489.h%26s=NHS+drug+budget+2004+2005+2006-01-01. 2008-01-01#g149489.r0 (accessed 29 October 2007).
- Moore TJ, Cohen MR, Furberg CD. Serious adverse drug events reported to the Food and Drug Administration, 1998–2005. Arch Intern Med 2007;167:1752-9.
- The Information Centre . Hospital Prescribing, 2006: England. Statistical Bulletin IC 2007:1-24.
- Hanauer SB. Turning traditional treatment strategies on their heads: Current evidence for ‘step-up’ versus ‘top-down’. Rev Gastroenterol Disord n.d.;7:S17-S22.
- Schreiber S, Khaliq-Kareemi M, Lawrance IC, Ostergaard Thomsen O, Hanauer SB, McColm J, et al. Maintenance therapy with certolizumab pegol for Crohn’s disease. N Engl J Med 2007;357:239-50.
- Winter TA, Wright J, Ghosh S, Jahnsen J, Innes A, Round P. Intravenous CDP870, a PEGylated Fab’ fragment of a humanized antitumour necrosis factor antibody, in patients with moderate-to-severe Crohn’s disease: an exploratory study. Aliment Pharmacol Ther 2004;20:1337-46.
Appendix 1 Calculation of Crohn’s Disease Activity Index (adapted from Best et al.39)
| Variable | Description | Scoring | Multiplier |
|---|---|---|---|
| Number of liquid stools | Sum of 7 days | ×2 | |
| Abdominal pain | Sum of 7 days’ ratings |
0 = none 1 = mild 2 = moderate 3 = severe |
×5 |
| General well-being | Sum of 7 days’ ratings |
0 = generally well 1 = slightly under par 2 = poor 3 = very poor 4 = terrible |
×7 |
| Extraintestinal complications | Number of complications listed | Arthritis/arthralgia, iritis/uveitis, erythema nodosum, pyoderma gangrenosum, aphtous stomatitis, anal fissure/fistula/abscess, fever > 37.8 °C | ×20 |
| Antidiarrhoeal drugs | Use in the previous 7 days |
0 = no 1 = yes |
×30 |
| Abdominal mass |
0 = no 2 = questionable 5 = definite |
×10 | |
| Haematocrit | Expected–observed haematocrit |
Men: 47 observed Women: 42 observed |
×6 |
| Body weight | Ideal/observed ratio | [1 – (ideal/observed)] × 100 | ×1 (not < –10) |
Appendix 2 Guidelines on the medical management of Crohn’s disease
From: Carter et al.9 on behalf of the British Society of Gastroenterology
The severity of CD is more difficult to assess than ulcerative colitis. The general principles are to consider the site (ileal, ileocolic, colonic, other), pattern (inflammatory, stricturing, fistulising) and activity of the disease before treatment decisions are made in conjunction with the patient.
An alternative explanation for symptoms other than active disease should be considered (such as bacterial overgrowth, bile salt malabsorption, fibrotic strictures, dysmotility, gall stones) and disease activity confirmed (usually by CRP or erythrocyte sedimentation rate) before starting steroids. Individuals with CD have many investigations over their lifetime, and imaging (colonoscopy, small bowel radiology) should not be repeated unless it will alter management or a surgical decision depends on the result.
1.1 Active ileal/ileocolonic/colonic disease
Patients should be encouraged to participate actively in the decision to treat with high dose aminosalicylates, different corticosteroids, nutritional therapy, antibiotics, new biological agents, or surgery. Infliximab is considered in section 1.5.
In mild ileocolonic CD, high-dose mesalazine (4 g/day) may be sufficient initial therapy (grade A).
For patients with moderate to severe disease, or those with mild to moderate ileocolonic CD that has failed to respond to oral mesalazine, oral corticosteroids such as prednisolone 40-mg daily is appropriate (grade A).
Prednisolone should be reduced gradually according to severity and patient response, generally over 8 weeks. More rapid reduction is associated with early relapse (grade C).
Budesonide 9-mg daily is appropriate for patients with isolated ileo-caecal disease with moderate disease activity, but marginally less effective than prednisolone (grade A).
Intravenous steroids (hydrocortisone 400 mg/day or methylprednisolone 60 mg/day) are appropriate for patients with severe disease (grade B). Concomitant IV metronidazole is often advisable, because it may be difficult to distinguish between active disease and a septic complication.
Elemental or polymeric diets are less effective than corticosteroids, but may be used to induce remission in selected patients with active CD who have a contraindication to corticosteroid therapy, or who would themselves prefer to avoid such therapy (grade A).
Elemental or polymeric diets are appropriate adjunctive therapy (grade C).
Total parenteral nutrition is appropriate adjunctive therapy in complex, fistulising disease (grade B).
Sulphasalazine 4-g daily is effective for active colonic disease, but cannot be recommended as first line therapy in view of a high incidence of side effects. It may be appropriate in selected patients (grade A).
Metronidazole 10–20 mg/kg/day, although effective, is not usually recommended as first line therapy for CD in view of the potential for side effects (grade A). It has a role in selected patients with colonic or treatment-resistant disease, or those who wish to avoid steroids.
Topical mesalazine may be effective in left-sided colonic CD of mild to moderate activity (grade B).
Azathioprine 1.5–2.5 mg/kg/day or mercaptopurine 0.75–1.50 mg/kg/day may be used in active CD as adjunctive therapy and as a steroid sparing agent. However, its slow onset of action precludes its use as a sole therapy (grade A).
Infliximab 5 mg/kg is effective (grade A), but is best avoided in patients with obstructive symptoms (see section 1.5).
Surgery should be considered for those who have failed medical therapy, and may be appropriate as primary therapy in patients with limited ileal or ileo-caecal disease (grade C).
Recommendations
1.1.1 Initial treatment of active ileal or ileocolonic CD with high dose mesalazine, corticosteroids, nutritional therapy or surgery should be tailored to the severity of disease and take the views of the patient into account.
1.1.2 There is insufficient evidence to recommend the use of other agents outside trials or specialist centres.
1.2 Fistulising and perianal disease
Active perianal disease or fistulas are often associated with active CD elsewhere in the gastrointestinal tract. The initial aim should be to treat active disease and sepsis. For more complex, fistulising disease, the approach involves defining the anatomy, supporting nutrition and potential surgery. For perianal disease, magnetic resonance imaging and examination under anaesthetic are particularly helpful.
Metronidazole 400-mg three times daily (grade A) and/or ciprofloxacin 500-mg twice daily (grade B) are appropriate first line treatments for simple perianal fistulas.
Azathioprine 1.5–2.5 mg/kg/day or mercaptopurine 0.75–1.50 mg/kg/day are potentially effective for simple perianal fistulas or enterocutaneous fistulas where distal obstruction and abscess have been excluded (grade A).
Infliximab (three infusions of 5 mg/kg at 0, 2 and 6 weeks) should be reserved for patients whose perianal or enterocutaneous fistulas are refractory to other treatment, and should be used as part of a strategy that includes immunomodulation and surgery (grade A).
Surgery (see section 7 of the guidelines), including seton drainage, fistulectomy and the use of advancement flaps, is appropriate for persistent or complex fistulas in combination with medical treatment (grade C).
Elemental diets or parenteral nutrition have a role as adjunctive therapy, but not as sole therapy (grade B).
There is insufficient evidence to recommend other agents outside clinical trials or specialist centres.
Recommendation
1.2.1 Controlled therapeutic trials combining medical and surgical therapy in perianal CD should be conducted.
1.3 Other sites
The same general principles apply, although there are no RCTs in the treatment of gastroduodenal or diffuse small bowel disease.
Oral CD. This is best managed in conjunction with a specialist in oral medicine. Topical steroids, topical tacrolimus, intralesional steroid injections, enteral nutrition and infliximab may have a role in management but there are no RCTs.
Gastroduodenal disease. Symptoms are often relieved by proton pump inhibitors. Surgery is difficult and may be complicated by fistulation.
Diffuse small bowel disease. Stricture dilatation or strictureplasty with or without triamcinolone injection should be considered. Nutritional support before and after surgery is usually essential. Other approaches, including the combination of infliximab with surgery for residual strictures, are evolving.
1.4 Maintenance of remission
The efficacy of drug therapy appears to depend on whether remission was achieved with medical or surgical therapy, on the risk of relapse and on the site of disease. Smoking cessation is probably the most important factor in maintaining remission.
To reduce the risk of relapse in CD, all smokers should be strongly advised to stop (grade A), with help (counselling, nicotine patches or substitutes) offered to achieve this.
Mesalazine has limited benefit and is ineffective at doses < 2 g/day or for those who have needed steroids to induce remission (grade A).
Azathioprine 1.5–2.5 mg/kg/day and mercaptopurine 0.75–1.5 mg/kg are effective, but reserved as second line therapy because of potential toxicity (grade A).
Methotrexate (15- to 25-mg intramuscular weekly) is effective for patients whose active disease has responded to intramuscular methotrexate (grade A). It is appropriate for those intolerant of, or who have failed, azathioprine/mercaptopurine therapy (grade B) once potential toxicity and other options, including surgery, have been discussed with the patient. Folic acid 5 mg once a week, taken 3 days after methotrexate, may reduce side effects. Subcutaneous or oral therapy may be effective (grade B).
Infliximab is effective at a dose of 5–10 mg/kg every 8 weeks in patients who have responded to an initial infusion 12 weeks earlier, for up to 44 weeks (grade A). It is best used as part of treatment strategy including immunomodulation once other options, including surgery, have been discussed with the patient (grade B).
Sulphasalazine cannot be recommended (grade A).
Corticosteroids, including budesonide, are not effective (grade A), although some patients have chronic active disease who appear steroid dependent (below).
Recommendations
1.4.1 Patients with CD who smoke should be offered help to stop.
1.4.2 Immunomodulation with azathioprine, mercaptopurine or methotrexate is usually appropriate if patients relapse more than once per year as steroids are withdrawn.
1.5 Chronic active and steroid-dependent disease
Long-term treatment with steroids is undesirable. Patients who have a poor response to steroids can be divided into steroid refractory and steroid dependent. Steroid-refractory disease may be defined as active disease in spite of an adequate dose and duration of prednisolone (20 mg/day for 2 weeks) and steroid dependence as a relapse when the steroid dose is reduced below 20 mg/day, or within 6 weeks of stopping steroids. Such patients should be considered for treatment with immunomodulators if surgery is not an immediate consideration.
Azathioprine 1.5–2.5 mg/kg/day, or mercaptopurine 0.75–1.25 mg/kg/day are the first line agents of choice for steroid-dependent disease (grade A).
Monitoring the full blood count to detect neutropenia is advisable, although there is no evidence that this is effective because profound neutropenia and sepsis can develop rapidly. The full blood count is best checked within 4 weeks of starting therapy and every 6–12 weeks thereafter, although may be done more frequently. Routine measurement of thiopurine methyltransferase activity before treatment, which may identify some (but not all) patients at risk of neutropenia, cannot yet be recommended but is debated. Large published series report safe use of azathioprine without thiopurine methyltransferase assay.
Methotrexate intramuscular 25-mg weekly for up to 16 weeks followed by 15-mg weekly is effective for chronic active disease (grade A). Oral dosing is effective for many patients (grade B).
Infliximab (5 mg/kg) should be reserved for patients with moderate to severe CD who are refractory to or intolerant of treatment with steroids, mesalazine, azathioprine/mercaptopurine and methotrexate, and for whom surgery is considered inappropriate (grade A).
Recommendation
1.5.1 Immunomodulation with azathioprine, mercaptopurine or methotrexate should be tried if steroids cannot be withdrawn without deterioration in disease activity.
Appendix 3 Search strategy clinical effectiveness
Clinical effectiveness searches
Note: certolizumab pegol and natalizumab were originally part of this appraisal; they were subsequently excluded after searching had been completed.
Source – MEDLINE (Ovid), 1950 to week 4 May 2007
-
(adalimumab or humira).mp., (540)
-
(certolizumab or cimzia).mp. (19)
-
(infliximab or remicade).mp. (3096)
-
(natalizumab or tysabri).mp.(208)
-
or/1-4 (3473)
-
Crohn Disease/ (21,624)
-
crohn$.mp. (25,626)
-
or/6-7 (25,626)
-
5 and 8 (1046)
-
randomized controlled trial.pt. (235,561)
-
controlled clinical trial.pt. (74,973)
-
randomized controlled trials.sh. (48,808)
-
random allocation.sh. (57,966)
-
double blind method.sh. (91,410)
-
single blind method.sh. (10,959)
-
or/10-15 (399,453)
-
(animals not human).sh. (4,090,275)
-
16 not 17 (365,869)
-
clinical trial.pt. (436,028)
-
exp clinical trials/ (191,534)
-
(clin$adj25 trial$).ti,ab. (130,375)
-
((singl$or doubl$or trebl$or tripl$) adj25 (blind$or mask$)).ti,ab. (90,759)
-
placebo$.ti,ab. (102,414)
-
random$.ti,ab. (372,182)
-
placebos.sh. (26,175)
-
research design.sh. (47,543)
-
or/19-26 (846,379)
-
27 not 17 (743,134)
-
28 not 18 (394,326)
-
18 or 29 (760,195)
-
9 and 30 (276)
-
limit 31 to yr=“2000 - 2007” (258)
Source – MEDLINE (Ovid), 1950 to week 2 June 2007*
-
ca2.mp. (105,839)
-
d2e7.mp. (23)
-
cdp870.mp. (26)
-
pha-738144.mp. (0)
-
pha 738144.mp. (0)
-
(anti adj2 4 integrin).mp. (45)
-
anti alpha4 integrin.mp. (49)
-
anti alpha 4 integrin.mp. (32)
-
or/1-8 (105,978)
-
crohn disease/ (21,691)
-
crohn$.mp. (25,715)
-
or/10-11 (25,715)
-
9 and 12 (66)
-
randomized controlled trial.pt. (236,980)
-
controlled clinical trial.pt. (75,195)
-
randomized controlled trials.sh. (49,205)
-
random allocation.sh. (58,180)
-
double blind method.sh. (91,776)
-
single blind method.sh. (11,028)
-
or/14-19 (401,708)
-
(animals not human).sh. (4,106,179)
-
20 not 21 (367,711)
-
clinical trial.pt. (436,884)
-
exp clinical trials/(192,444)
-
(clin$adj25 trial$).ti,ab. (131,452)
-
((singl$or doubl$or trebl$or tripl$) adj25 (blind$or mask$)).ti,ab. (91,157)
-
placebo$.ti,ab. (102,972)
-
random$.ti,ab. (374,725)
-
placebos.sh. (26,255)
-
research design.sh. (47,827)
-
or/23-30 (851,045)
-
not 21 (747,002)
-
32 not 22 (396,637)
-
22 or 33 (764,348)
-
13 and 34 (26)
-
limit 35 to yr=“2000 - 2007” (22)
*Additional search to account for alternative terminology used for the drugs.
Source – EMBASE (Ovid), 1980 to week 22 2007
-
(adalimumab or humira).mp. (2036)
-
(certolizumab or cimzia).mp., (230)
-
(infliximab or remicade).mp., (7811)
-
(natalizumab or tysabri).mp. (843)
-
or/1-4 (8685)
-
Crohn Disease/ (20,817)
-
crohn$.mp. (23,756)
-
or/6-7 (23,756)
-
5 and 8 (2554)
-
limit 9 to “treatment (2 or more terms min difference)” (506)
-
limit 10 to yr=“2000 - 2007” (492)
Source – EMBASE (Ovid), 1980 to week 25* 2007
-
ca2.mp. (115,879)
-
d2e7.mp. (65)
-
cdp870.mp. (16)
-
pha-738144.mp. (1)
-
pha 738144.mp. (1)
-
(anti adj2 4 integrin).mp. (9)
-
anti alpha4 integrin.mp. (37)
-
anti alpha 4 integrin.mp. (2)
-
or/1-8 (116,001)
-
crohn$.mp. (23,876)
-
crohn disease/ (20,928)
-
or/10-11 (23,876)
-
9 and 12 (72)
-
limit 13 to (“treatment (2 or more terms min difference)” and yr=“2000 - 2007”) (17)
*Additional search to account for alternative terminology used for the drugs.
Cochrane Library (CENTRAL), 2007, issue 2
-
#1 adalimumab OR humira
-
#2 certolizumab OR cimzia
-
#3 infliximab OR remicade
-
#4 natalizumab OR tysabri
-
#5 (#1 OR #2 OR #3 OR #4)
-
#6 crohn*
-
#7 MeSH descriptor Crohn Disease explode all trees
-
#8 (#6 OR #7)
-
#9 (#5 AND #8)
Cochrane Library (CENTRAL), 2007, issue 2*
-
#1 ca2
-
#2 d2e7
-
#3 cdp870
-
#4 pha-738144
-
#5 pha next 738144
-
#6 antegren
-
#7 integrin
-
#8 (#1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7)
-
#9 crohn*
-
#10 MeSH descriptor Crohn Disease explode all trees
-
#11 (#9 OR #10)
-
#12 (#8 AND #11)
*Additional search to account for alternative terminology used for the drugs.
Source – MEDLINE (Ovid) In-Process & Other Non-Indexed Citations, 4 June 2007
-
(adalimumab or humira).mp (71)
-
(certolizumab or cimzia).mp (10)
-
(infliximab or remicade).mp (209)
-
(natalizumab or tysabri).mp. (14)
-
or/1-4 (249)
-
crohn$.mp. (549)
-
5 and 6 (77)
-
limit 7 to yr=“2000 - 2007” (76)
Source – MEDLINE (Ovid) In-Process & Other Non-Indexed Citations, 26 June 2007*
-
ca2.mp. (1007)
-
d2e7.mp. (0)
-
cdp870.mp. (0)
-
pha-738144.mp. (0)
-
pha 738144.mp. (0)
-
(anti adj2 4 integrin).mp. (0)
-
anti alpha4 integrin.mp. (2)
-
anti alpha 4 integrin.mp. (0)
-
or/1-8 (1009)
-
crohn$.mp. (602)
-
9 and 10 (0)
*Additional search to account for alternative terminology used for the drugs.
Ongoing studies
Source – National Research Register (2007 Issue 2)
See above Cochrane Library clinical effectiveness search strategy.
Sources – Current Controlled Trials and ClinicalTrials.gov
Search terms: adalimumab OR humira; certolizumab OR cimzia; infliximab OR remicade; natalizumab OR tysabri; ca2 OR d2e7; cdp870 OR pha-738144; pha 738144 OR anti 4 integrin; anti alpha4 integrin OR anti alpha 4 integrin
References were selected where they also included CD.
Appendix 4 Data extraction form
Reviewer: Date:
Study author, year: Reference: Geographical location of the study
| Baseline characteristics | Placebo n = | Drug 1 n = | Drug 2 n = | Drug 3 n = |
|---|---|---|---|---|
| Mean age ± SD | ||||
| Sex | ||||
| Ethnicity | ||||
| Mean weight (kg) ± SD | ||||
| Mean height ± SD | ||||
| Number smokers | ||||
| Mean duration of Crohn’s disease (years) ± SD | ||||
|
Intestinal area involved Ileum only Colon only Ileum/colon Jejunal only Perianal only Other |
||||
| % with fistulas | ||||
|
Where all with fistulising disease: Number of (draining) fistulas Location of fistulas Mean PDAI score |
||||
| Previous surgery for Crohn’s | ||||
| Mean baseline CDAI ± SD | ||||
| Mean baseline IBDQ median (range) | ||||
| Other disease activity index or measure of disease severity (e.g. Harvey–Bradshaw) | ||||
| Mean C-reactive protein ± SD | ||||
|
Previous or concurrent biologic agent (state which) (% of patients previously received/receiving agent, % naive) |
||||
| Other concurrent medication | ||||
| Corticosteroids (e.g. prednisone or budesonide) state which | ||||
| Immunosuppressive agent (e.g. mercaptopurine, methotrexate, azathioprine) state which | ||||
| Oral aminosalicylate | ||||
| Antibiotic | ||||
| Other: specify |
Notes: (identify any statistically significant differences)
Study design/methodology – see flow chart
List all outcomes:
Do not extract data on laboratory parameters
Outcomes: state which type of analysis (e.g. efficacy, ITT, safety, etc.)
Outcome 1)
| Placebo n = | Drug 1 n = | Drug 2 n = | Drug 3 n = | |
|---|---|---|---|---|
| Baseline | ||||
| First time point | ||||
| p-value vs placebo | ||||
| Second time point | ||||
| p-value vs placebo | ||||
| Third time point | ||||
| p-value vs placebo |
List number of patients for each study arm at each time point
Repeat table for all relevant outcomes
Subgroup analyses (if applicable):
Safety
| Adverse event | Placebo n = | Drug 1 n = | Drug 2 n = | Drug 3 n = | |
|---|---|---|---|---|---|
| Average follow-up | |||||
| Any adverse event (%) | |||||
| Death | |||||
| Adverse event leading to withdrawal | |||||
| GI | Nausea | ||||
| Vomiting | |||||
| Abdominal pain | |||||
| CNS | Headache | ||||
| Pain | |||||
| Fatigue | |||||
| Infection | URTI | ||||
| Other infection | |||||
| Serious infection | |||||
| TB | |||||
| Haematological | |||||
| Cardiovascular | Chest pain | ||||
| Hypotension | |||||
| Hypertension | |||||
| Heart failure | |||||
| Skin | Pruritus | ||||
|
Injection site reaction (give details) |
|||||
| Hypersensitivity | Acute | ||||
| Delayed | |||||
| Respiratory | Dyspnoea | ||||
| MS | MS or symptoms of MS (e.g. demyelination) | ||||
| Bone marrow | |||||
| Other | Myalgia | ||||
| Fever | |||||
| Abscess | |||||
| Antibodies to DNA | |||||
| Human anti-TNF agent | |||||
| Lupus arthritis | |||||
| AE during or within 2 hours of infusion | |||||
| Other | |||||
| Other | |||||
Inclusion/exclusion criteria
Inclusion criteria:
| Age/sex |
| Duration of CD |
| Severity of CD |
| Surgical history |
| Concurrent treatment (non biologics) |
| Concurrent treatment (biologics) |
| Previous treatment (non biologics) |
| Previous treatment (biologics) |
| Concurrent disease |
| Female patients of child bearing potential included? |
Exclusion criteria:
| Concurrent treatment (non biologics) |
| Concurrent treatment (biologics) |
| Previous treatment (non biologics) |
| Previous treatment (biologics) |
| Previous/imminent surgery |
| Concurrent disease |
Are patients within UK licence in terms of severity of disease and resistance/intolerance to conventional treatment?
Follow-up of patients through trial
| Number of patients enrolled: | |||||
| Number of patients excluded (state main reasons) | |||||
| Number of patients randomised: | |||||
| Number of patients at each time point and reasons for withdrawal | |||||
| Placebo | Drug 1 | Drug 2 | Drug 3 | Drug 4 | |
|---|---|---|---|---|---|
| Time point 1 | |||||
| Time point 2 | |||||
| Time point 3 | |||||
| Time point 4 | |||||
| Number completed | |||||
Duration of study:
Number of infusions:
(how administered/where administered)
Number of assessments:
Additional notes on trial design (if applicable):
Funding source:
Quality assessment
| Randomisation | Details on method of randomisation | |
| If described, was the method adequate? | ||
| Concealment | Details of method of allocation concealment | |
| If described, was the method adequate? | ||
| Blinding | Details on placebo (indistinguishable from intervention?) | |
| Details of blinding: patients | ||
| Details of blinding: study investigators | ||
| Details of blinding: study co-ordinators | ||
| Details of blinding: data analysts | ||
| Details of blinding: other | ||
| Comparability of groups |
Were groups comparable at baseline? For a) baseline scores For b) demographics |
|
|
Were groups treated the same throughout the trial, with the exception of the intervention? For a) assessments For b) other care |
||
| Analysis | Were all trial participants accounted for throughout trial? | |
|
Was loss to follow-up >20%? (state actual loss to follow-up for each time point) |
||
| Was it stated that an ITT analysis was performed? | ||
|
ITT: data from all assessments used regardless of compliance with allocated treatment Sensitivity analysis should be performed where assessment data missing |
Was an ITT analysis performed for all relevant outcomes (according to the reported data), or was a sensitivity analysis performed? If other analysis (e.g. including open-label patients, describe) |
|
| Was a sample size calculation performed? | ||
| Was there any selective reporting of outcome measures? |
Description of which patients were included in which analysis: (primary, secondary, efficacy, ITT, open label, safety, etc.)
Appendix 5 Extraction of data from published graphs
Scans of published graphs were overlayed with a grid, printed, enlarged to A3 and then used to extract data. The data were used to redraw the graph and compare with the original. Examples are shown below.
Scan of published graph with grid overlay
(CiC information has been removed.)
Scan of published graph with grid overlay
(CiC information has been removed.)
Scans of published graphs overlayed with graphs redrawn using data extracted from grid-overlayed originals
(CiC information has been removed.)
Appendix 6 Consistency of trials with licence indications
| Licence indication | Study | Population/study characteristics |
|---|---|---|
| Adalimumab | ||
|
Hanauer et al. , 2006 CLASSIC I63 INDUCTION |
|
|
Sandborn et al. , 2007 GAIN64 INDUCTION |
|
|
|
Colombel et al. , 2007 CHARM67 MAINTENANCE |
|
|
|
Sandborn et al. , 2007 CLASSIC II66 MAINTENANCE |
|
|
| Infliximab – adults | ||
|
Targan et al. , 199757 INDUCTION |
|
|
Hanauer et al. , 20023 and Rutgeerts et al. , 20042 MAINTENANCE |
|
|
|
Rutgeerts et al. , 199958 (follow-on from Targan et al. 57 trial) MAINTENANCE |
|
|
| Infliximab – fistulising CD | ||
|
Present et al. , 199962 INDUCTION |
|
|
Sands et al. , 2004 ACCENT II65 MAINTENANCE |
|
|
| Infliximab – children | ||
|
Baldassano et al. , 200346 INDUCTION |
|
|
Hyams et al. , 2007 REACH45 MAINTENANCE |
|
|
Appendix 7 Publications not obtained
Infliximab. A last resort for Crohn’s disease after failure of steroids and azathioprine. Prescrire Int 2000;9:163–5.
Abramowitz L. Treatments of anoperineal localized Crohn’s disease. [French]. Acta Endoscopica 2005;35:748–50.
Bayes M, Rabasseda X, Prous JR. Gateways to clinical trials: November 2006. Methods Find Exp Clin Pharmacol 2006;28:657–78.
Bayes M, Rabasseda X, Prous JR. Gateways to clinical trials: January/February 2007. Methods Find Exp Clin Pharmacol 2007;29:53–71.
Dotan I, Yeshurun D, Hallak A, Horowitz N, Tiomny E, Reif S, et al. [Treatment of Crohn’s disease with anti TNF alpha antibodies--the experience in the Tel Aviv Medical Center]. [Hebrew]. Harefuah 2001;140:289–93.
Escher JC, van-den BG, Kate FT, te VA, van DS. Mucosal healing and down-regulation of inflammation with anti-tumor necrosis factor a (Infliximab) in children with refractory Crohn’s disease. J Pediatr Gastroenterol Nutr 2000;31:S19.
Isaacs KL. Adalimumab induction therapy in Crohn disease. Evid Base Gastroenterol 2006;7:67–8.
Koltun WA. A Paradigm for the Management of Complex Perineal Crohn’s Disease in the Anti-TNF Era. Semin Colon Rectal Surg 2006;17:61–7.
Mahadevan U. TNF-alpha antagonists: Benefits beyond remission. Rev Gastroenterol Disord 2007;7(Suppl. 1):S13–S19.
Mealy NE, Bayes M. Treatment of gastrointestinal disorders: Certolizumab pegol. Drugs Future 2005;30:600–1.
Schreiber S, Rutgeerts P, Fedorak RN, Khaliq-Kareemi M, Kamm MA, Boivin M, et al. A randomized, placebo-controlled trial of certolizumab pegol (CDP870) for treatment of Crohn’s disease. Gastroenterology 2005;129:807–18. [Erratum published in Gastroenterology 2005;129:1808.]
Appendix 8 Ongoing trials
| Study/source | Country | Study design | Population | Treatment | Trial start/likely completion |
|---|---|---|---|---|---|
|
Study M04–729 NCT00445939 Clinicaltrials.gov Information provided by Abbott |
Japan | Multicentre, randomised, double-blind, placebo-controlled study of adalimumab for the induction of clinical remission in Japanese subjects with CD | Japanese subjects with CD, CDAI score of ≥ 220 and ≤ 450; if previously received infliximab, subjects who discontinued owing to a loss of response or intolerance | Adalimumab | Study start March 2007; recruitment stage (information verified March 2007) |
|
Study M06–837 NCT00445432 Clinicaltrials.gov Information provided by Abbott |
Japan | Multicentre, randomised, double-blind, placebo-controlled study of adalimumab for the maintenance of clinical remission in Japanese subjects with CD | Japanese subjects with CD enrolled in and completed study M04–729 | Adalimumab | Study start March 2007; recruitment stage (information verified March 2007) |
|
Study M05–769 NCT00348283 Clinicaltrials.gov Information provided by Abbott |
Multicentre (USA, Canada, Europe) | Multicentre, randomised, double-blind, placebo-controlled study of the human anti-TNF monoclonal antibody adalimumab endoscopy trial to evaluate the effects on mucosal healing in subjects with CD involving the colon | Patients with moderate-to-severe ileocolonic CD | Adalimumab | Study start August 2006; recruitment stage (information verified April 2007) |
|
Study M06–806 NCT00409682 Clinicaltrials.gov Information provided by Abbott |
Multicentre (USA, Canada, Europe) | Multicentre, randomised, double-blind, placebo-controlled study to evaluate the safety, efficacy and pharmacokinetics of the human anti-TNF monoclonal antibody adalimumab in paediatric subjects with moderate-to-severe CD | Children aged 6–17 years with moderate-to-severe CD | Adalimumab | Study start March 2007; recruitment stage or not yet recruiting (information verified April 2007) |
| Study/source | Country | Study design | Population | Treatment | Trial start/likely completion |
|---|---|---|---|---|---|
|
RP0401 NCT00132899 Information provided by Robarts Research Institute, Schering-Plough |
Canada | A phase III randomised, placebo-controlled, double-blind, parallel group, multicentre study to evaluate the safety and efficacy of infliximab with methotrexate for the long-term treatment of CD | Patients with symptoms that are persistent enough to require corticosteroid therapy | Infliximab (vs infliximab plus methotrexate) | Study start December 2005, expected completion December 2007 (information verified December 2005) |
|
CR004804 NCT00094458 Information provided by Centocor, Inc., Schering-Plough |
Multicentre (USA, Canada, Europe) | Multicentre, randomised, double-blind, active controlled trial comparing Remicade (Infliximab) and Remicade plus azathioprine in the treatment of patients with CD naive to both immunomodulators and biologic therapy (SONIC trial) | Patients with CDAI score of > 220 to < 450 | Infliximab (vs infliximab plus azathioprine) | Study start March 2005; recruitment stage or no longer recruiting (information verified May 2007) |
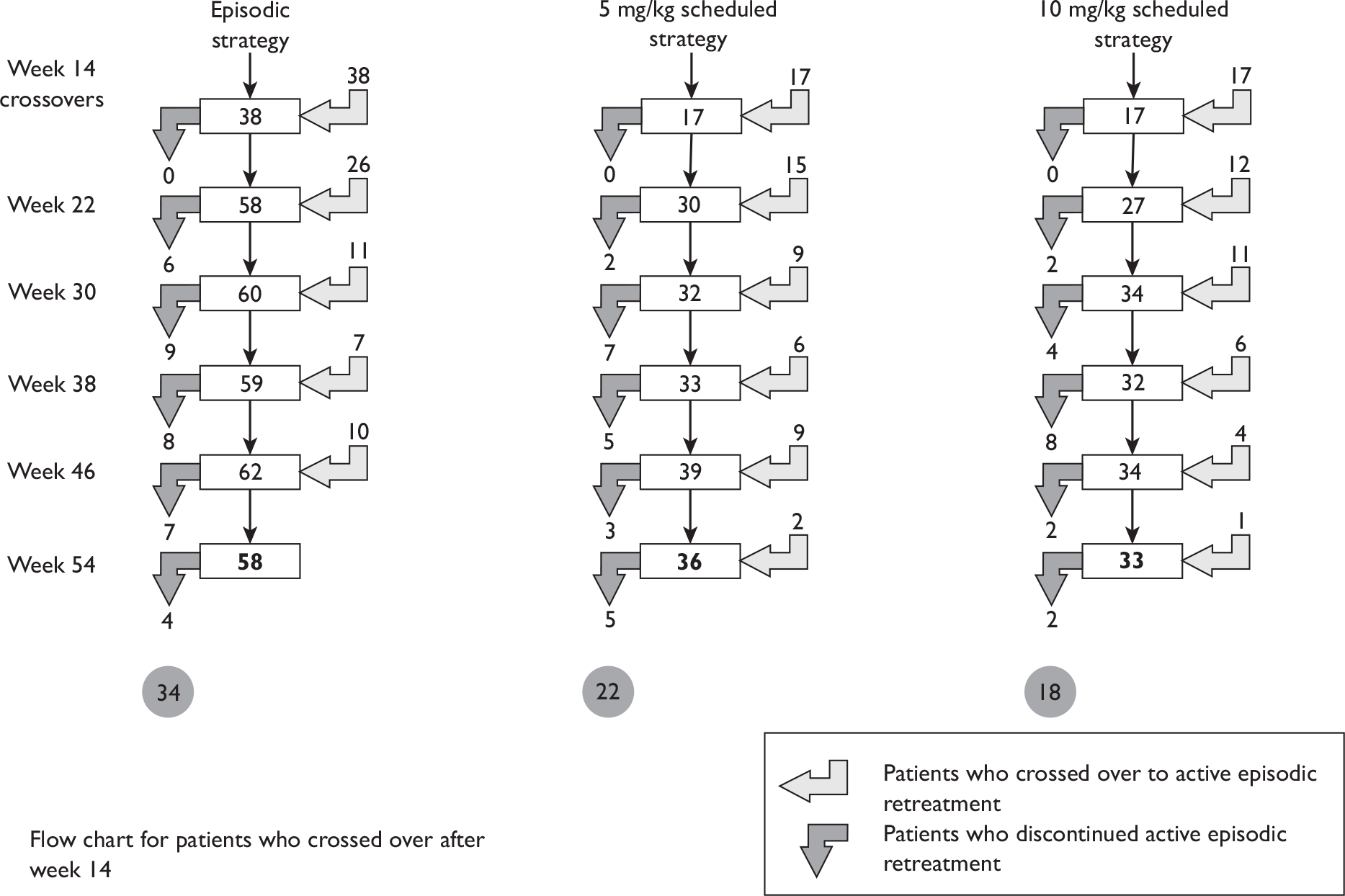
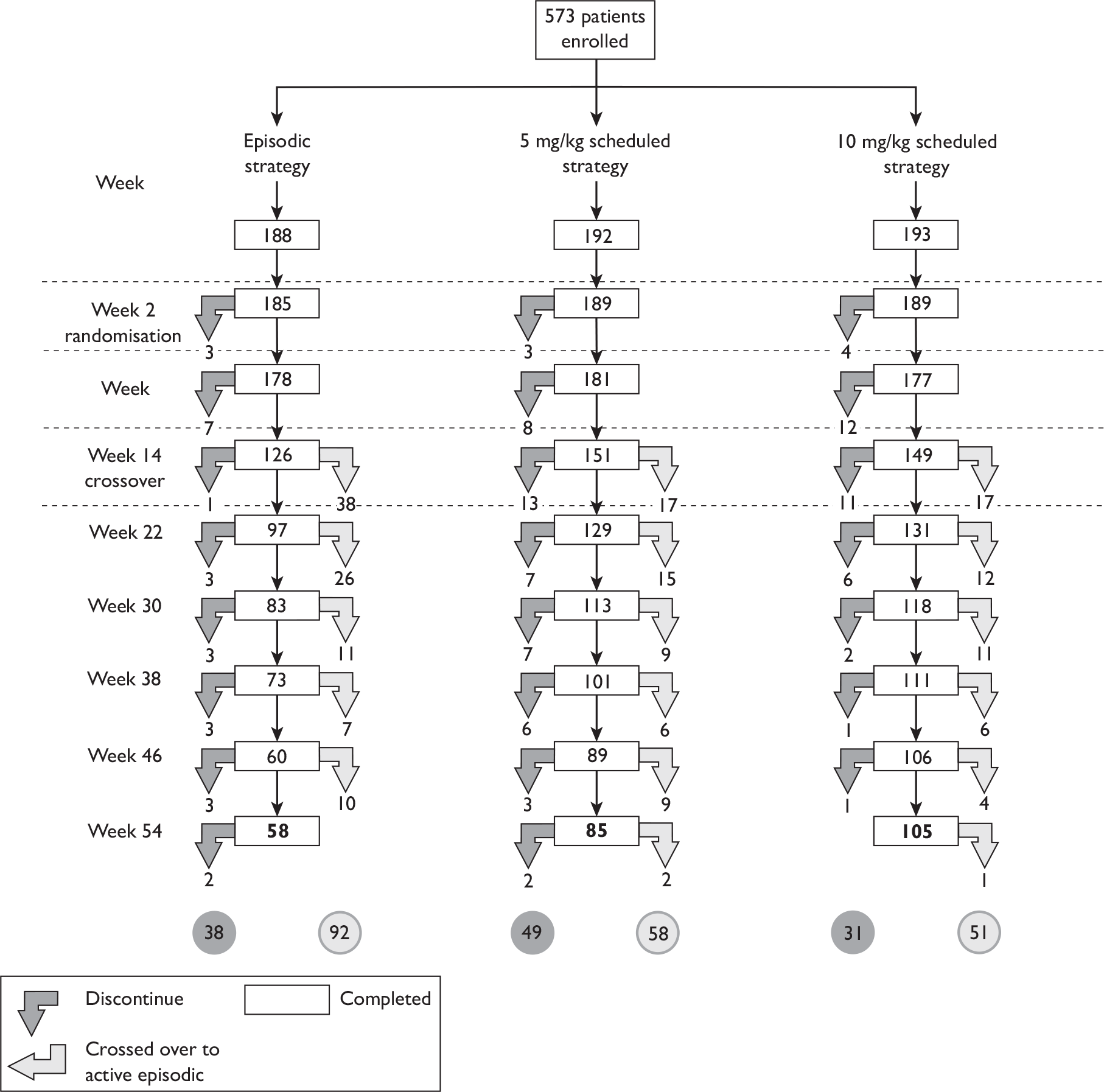
Appendix 10 Results for all included studies irrespective of licence indication
This appendix presents the results from the included trials by outcome measure in the form of forest plots.
Induction trials
FIGURE 46.
Induction trials – risk ratio of remission. Ada, adalimumab; Inflix, infliximab; RR, risk ratio.
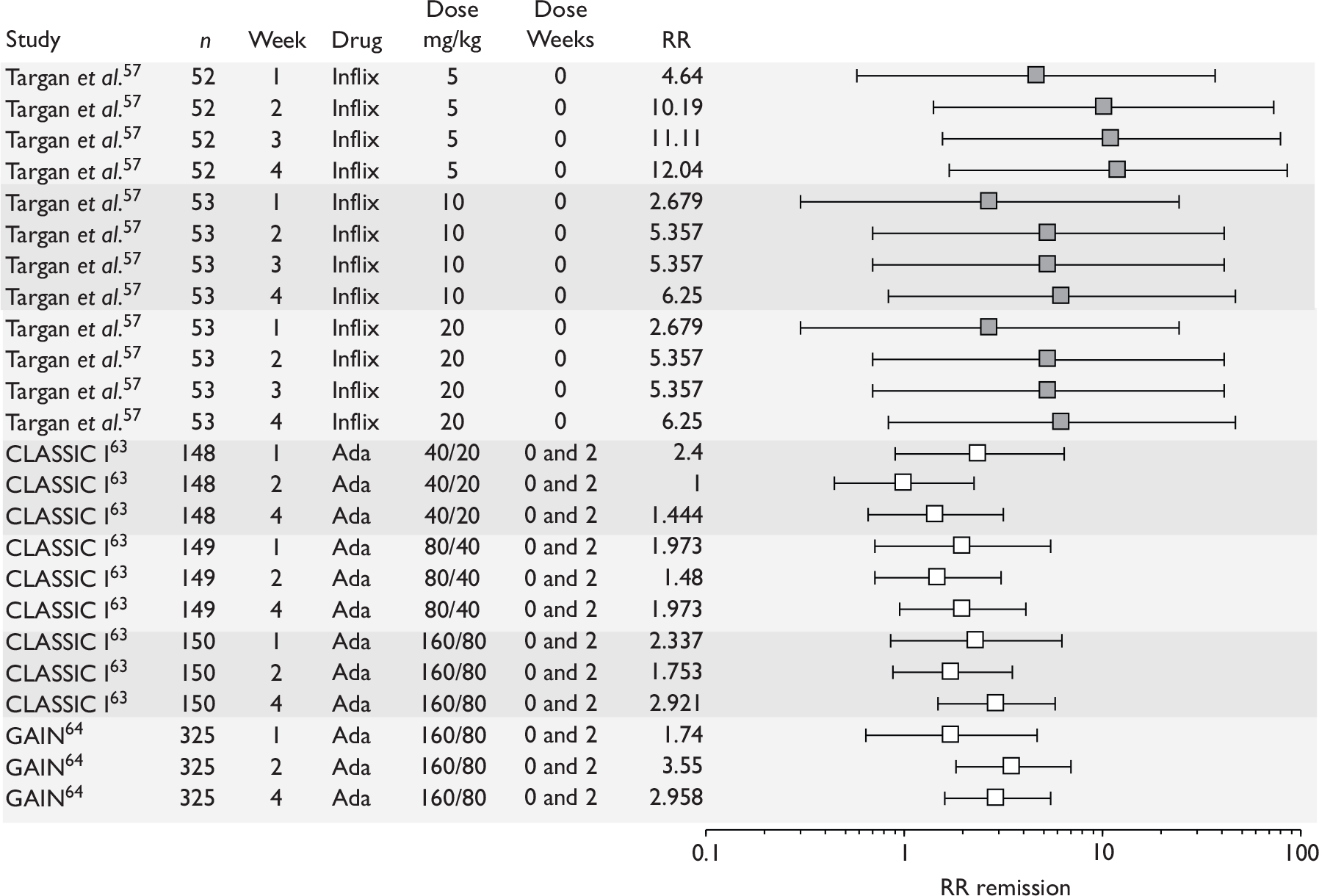
FIGURE 47.
Induction trials – risk difference of remission. Ada, adalimumab; Inflix, infliximab; RD, risk difference.
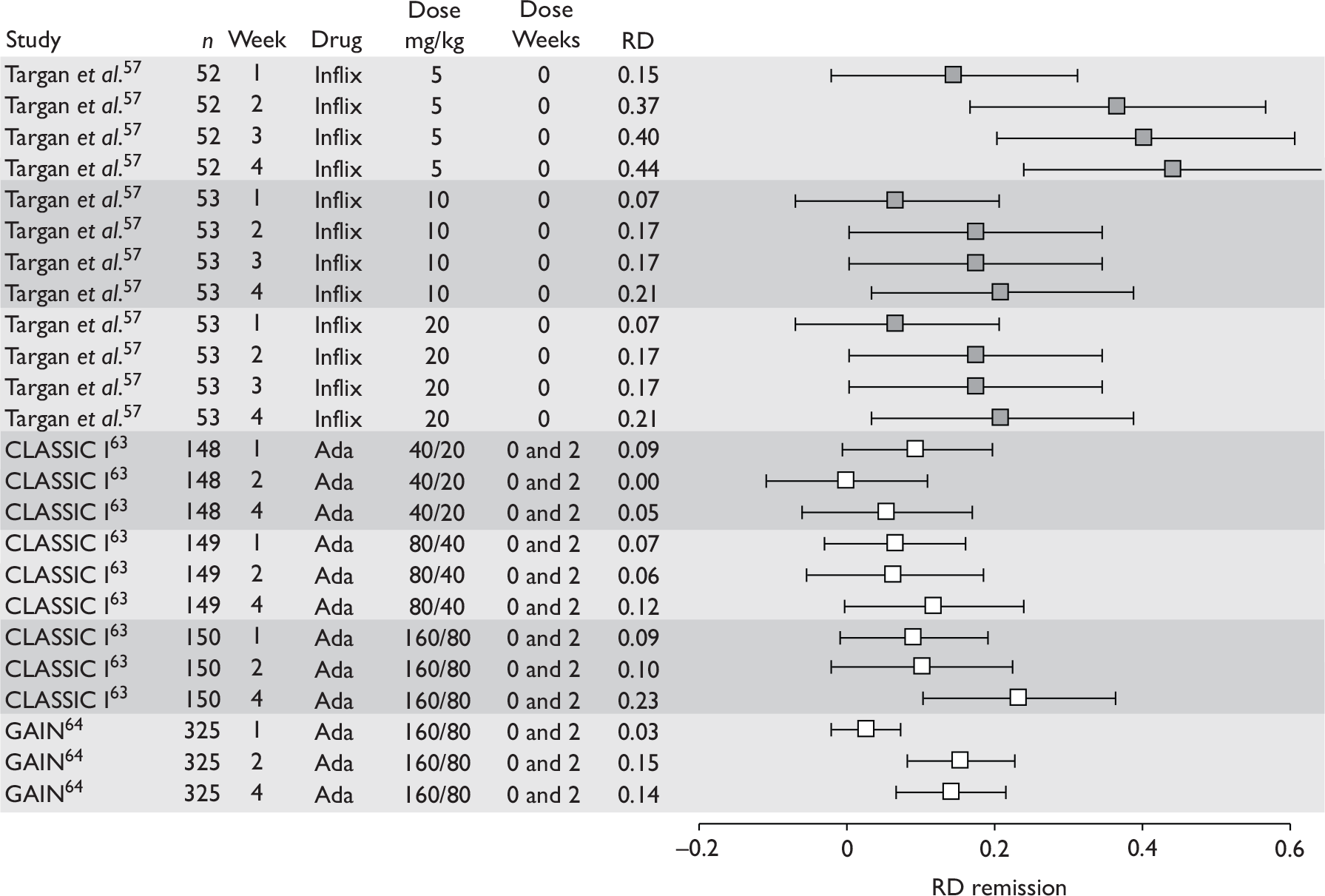
FIGURE 48.
Induction trials – risk ratio of response 100. Adal, adalimumab; RR, risk ratio.
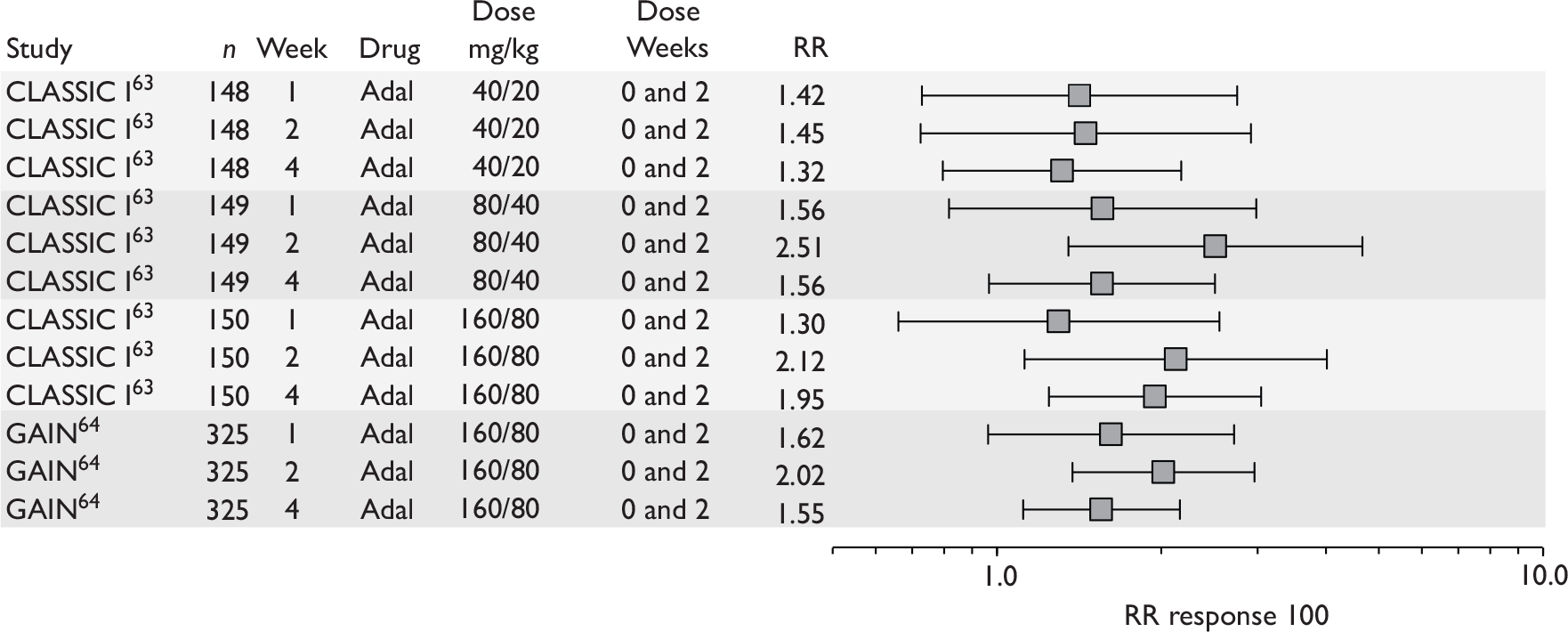
FIGURE 49.
Induction trials – risk difference of response 100. Adal, adalimumab; RD, risk difference.
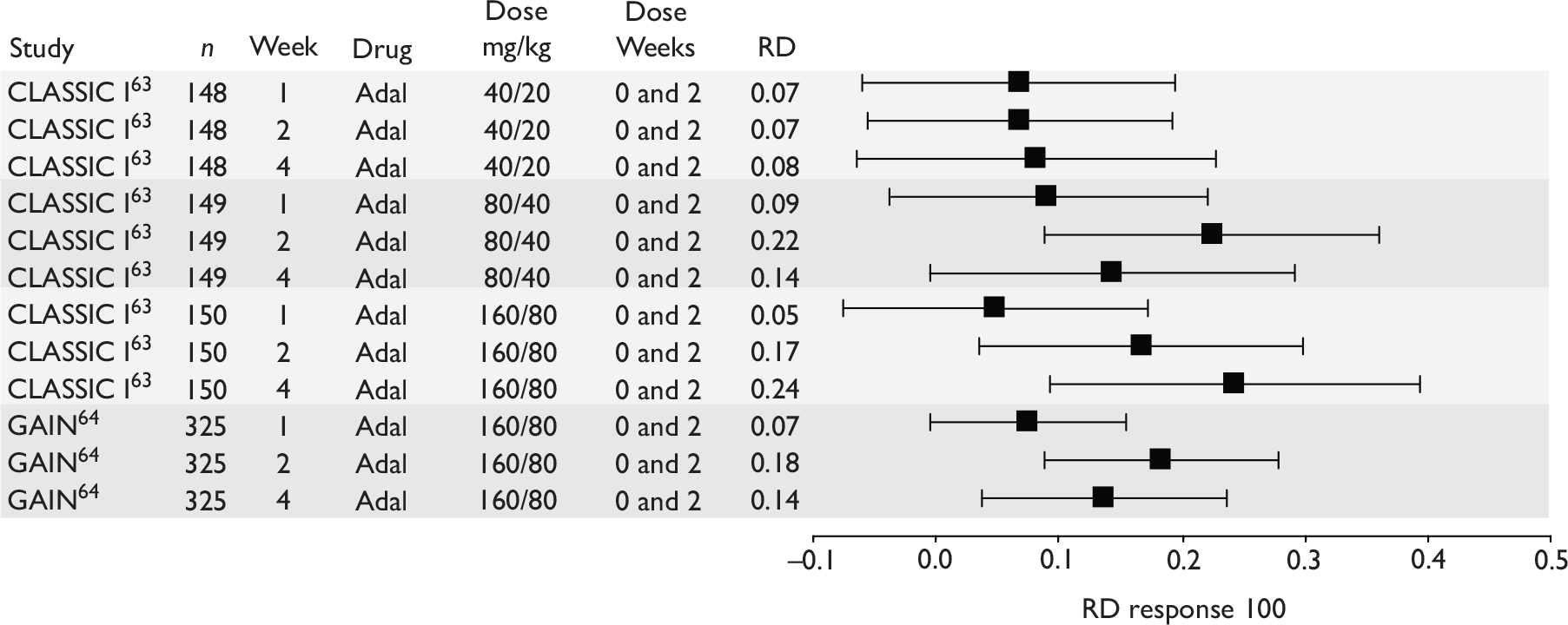
FIGURE 50.
Induction trials – risk ratio response 70. Ada, adalimumab; Inflix, infliximab; RR, risk ratio.
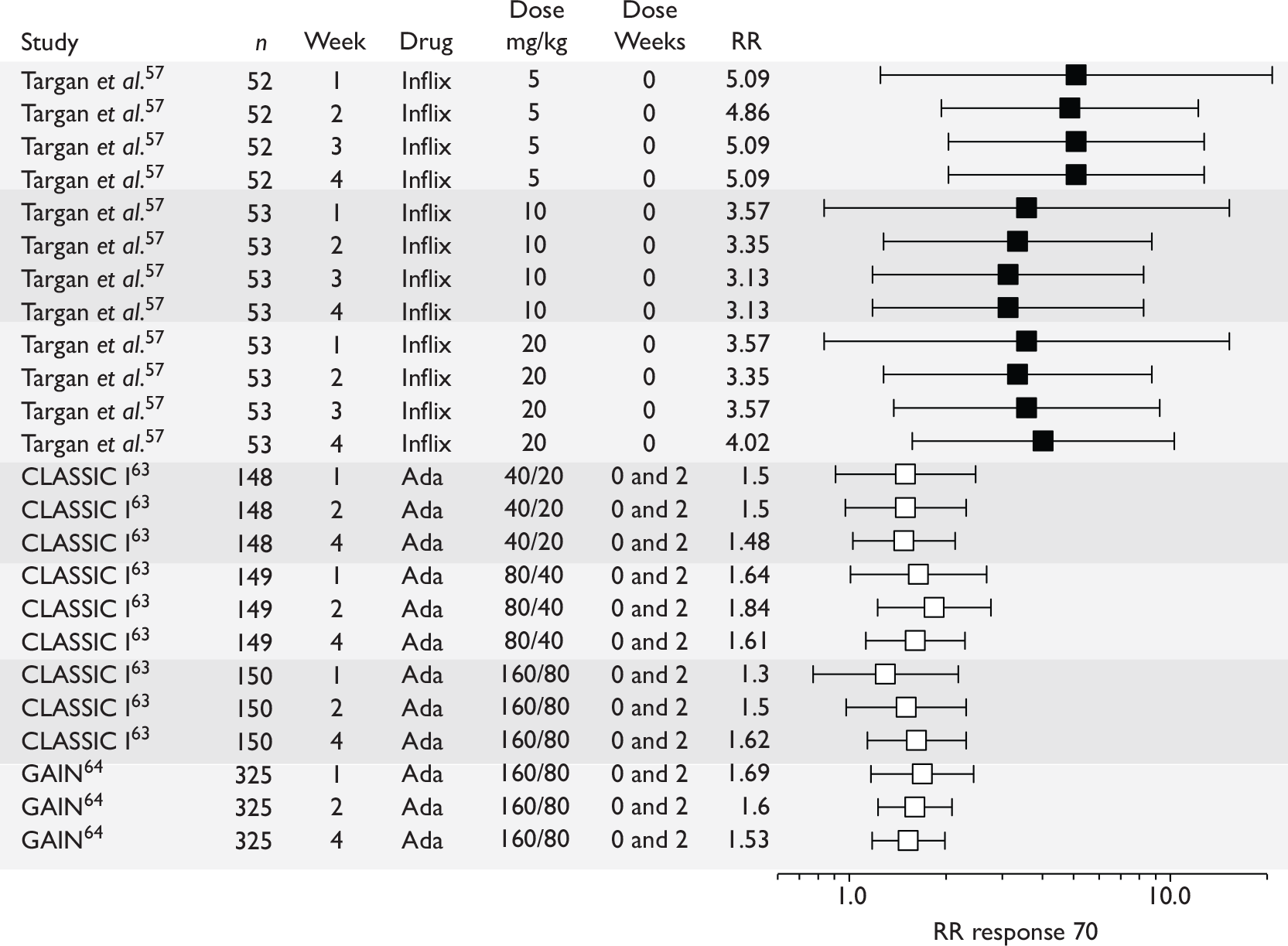
FIGURE 51.
Induction trials – risk difference response 70. Ada, adalimumab; Infl, infliximab; RD, risk difference.
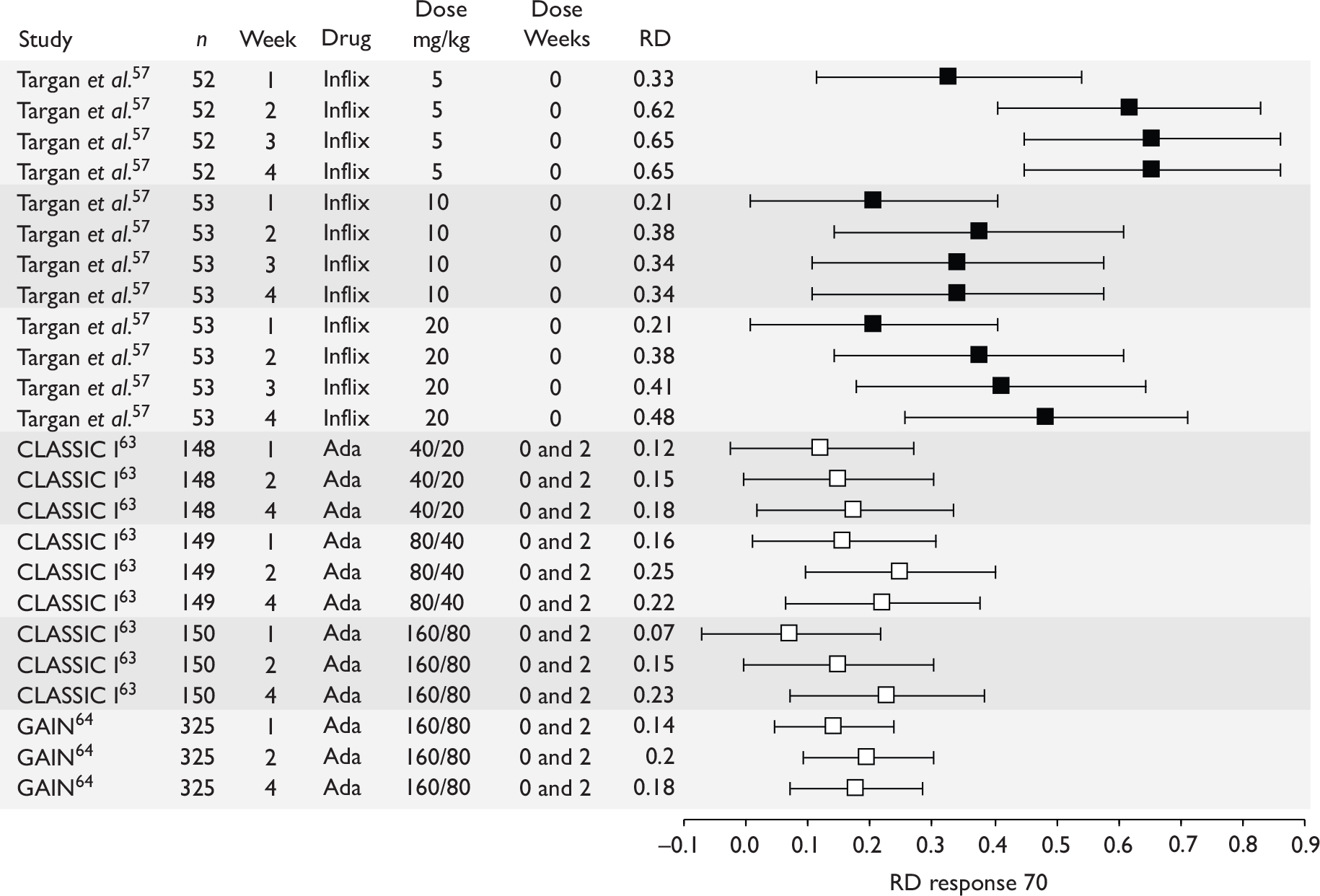
FIGURE 52.
Induction trials – CDAI scores (mean scores). Ada, adalimumab; Inflix, infliximab.
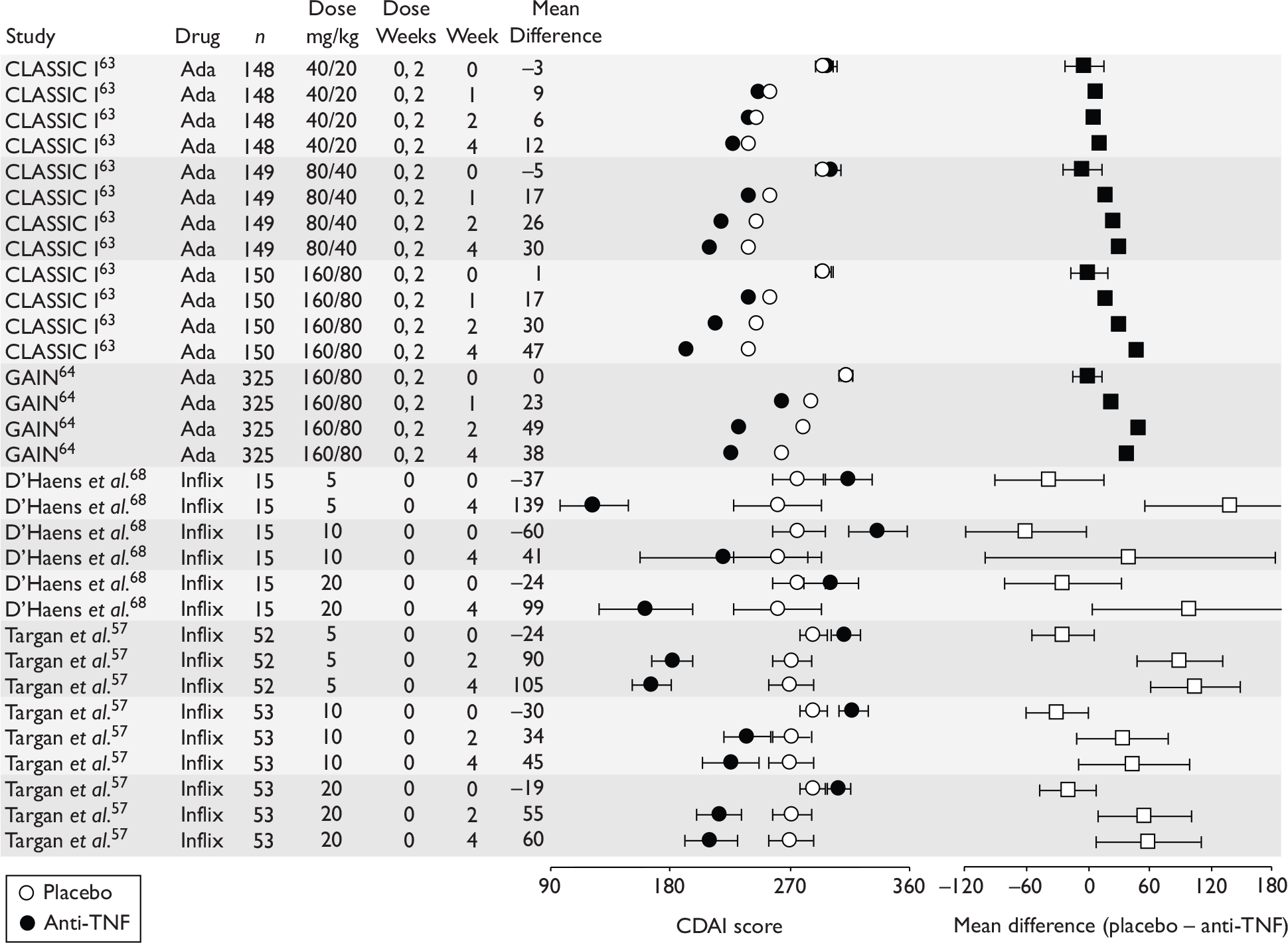
FIGURE 53.
Induction trials – IBDQ scores. Ada, adalimumab; Inflix, infliximab.
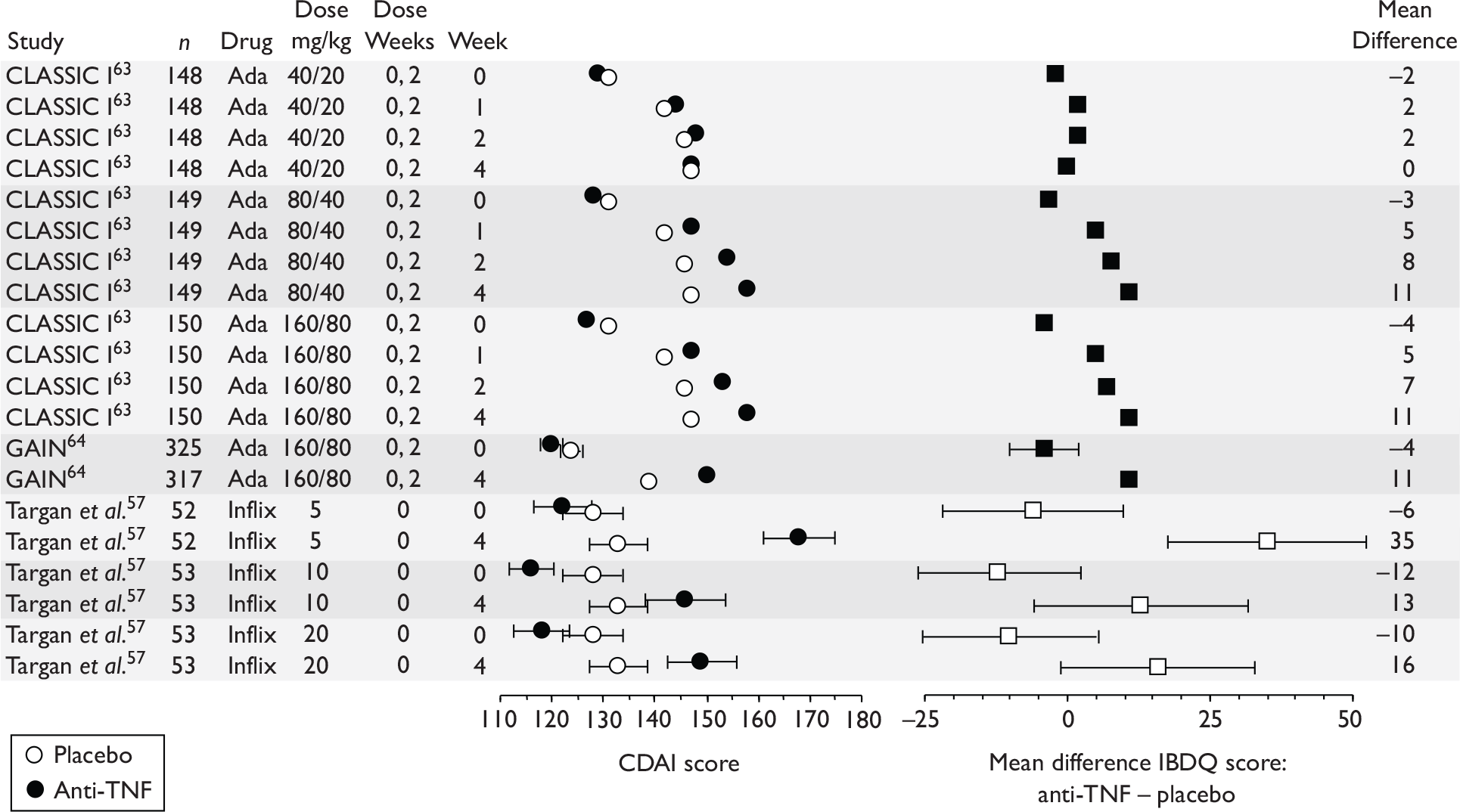
Maintenance trials (unless stated ‘dose weeks’ refers to post-randomisation doses)
FIGURE 54.
Maintenance trials – risk ratio remission (responders). ?, the dosing schedule was not clear from the published papers; Ada, adalimumab; Inflix, infliximab; RR, risk ratio.
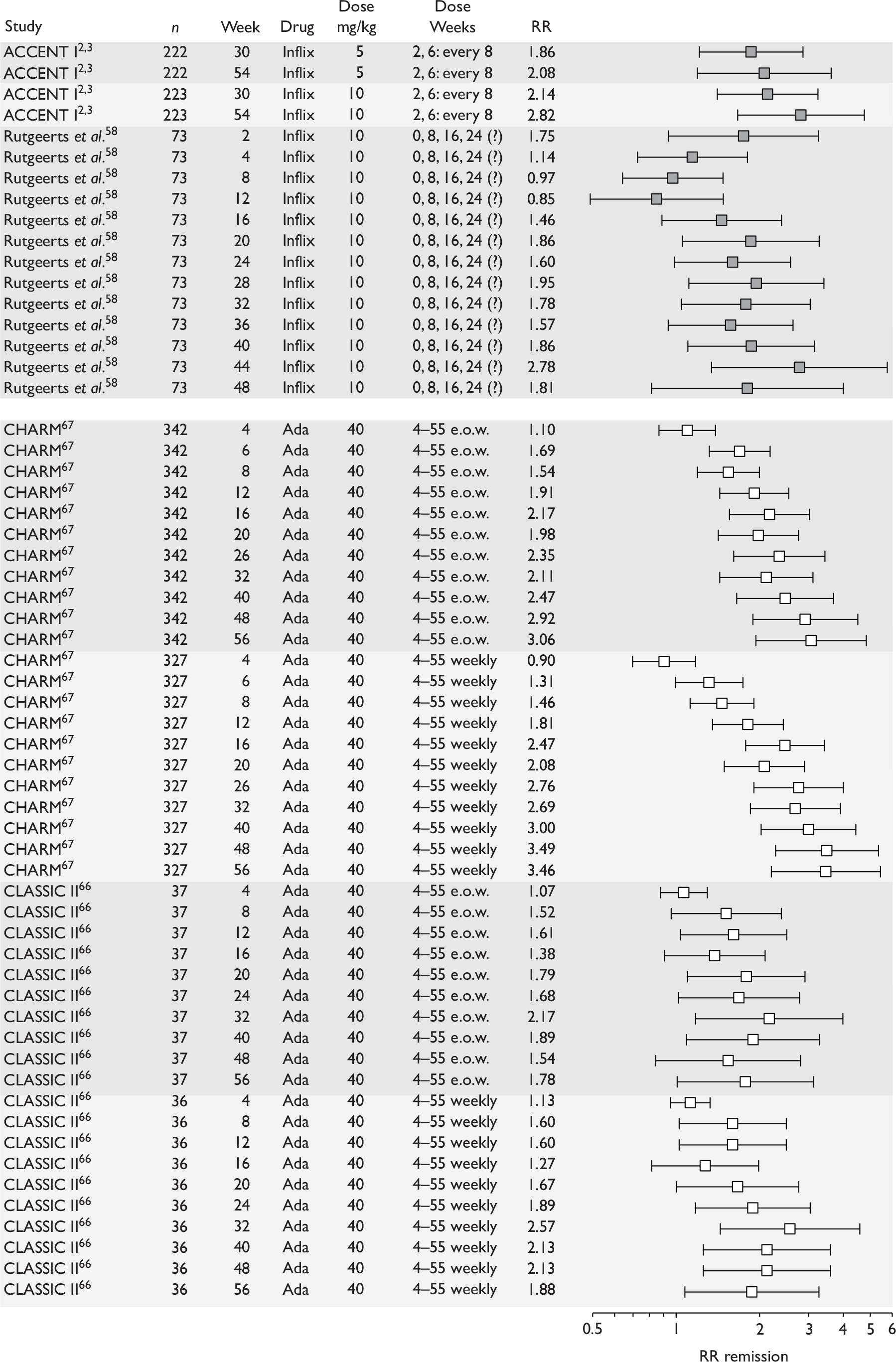
FIGURE 55.
Maintenance trials – risk difference remission (responders). ?, the dosing schedule was not clear from the published papers; Ada, adalimumab; Inflix, infliximab; RD, risk difference.
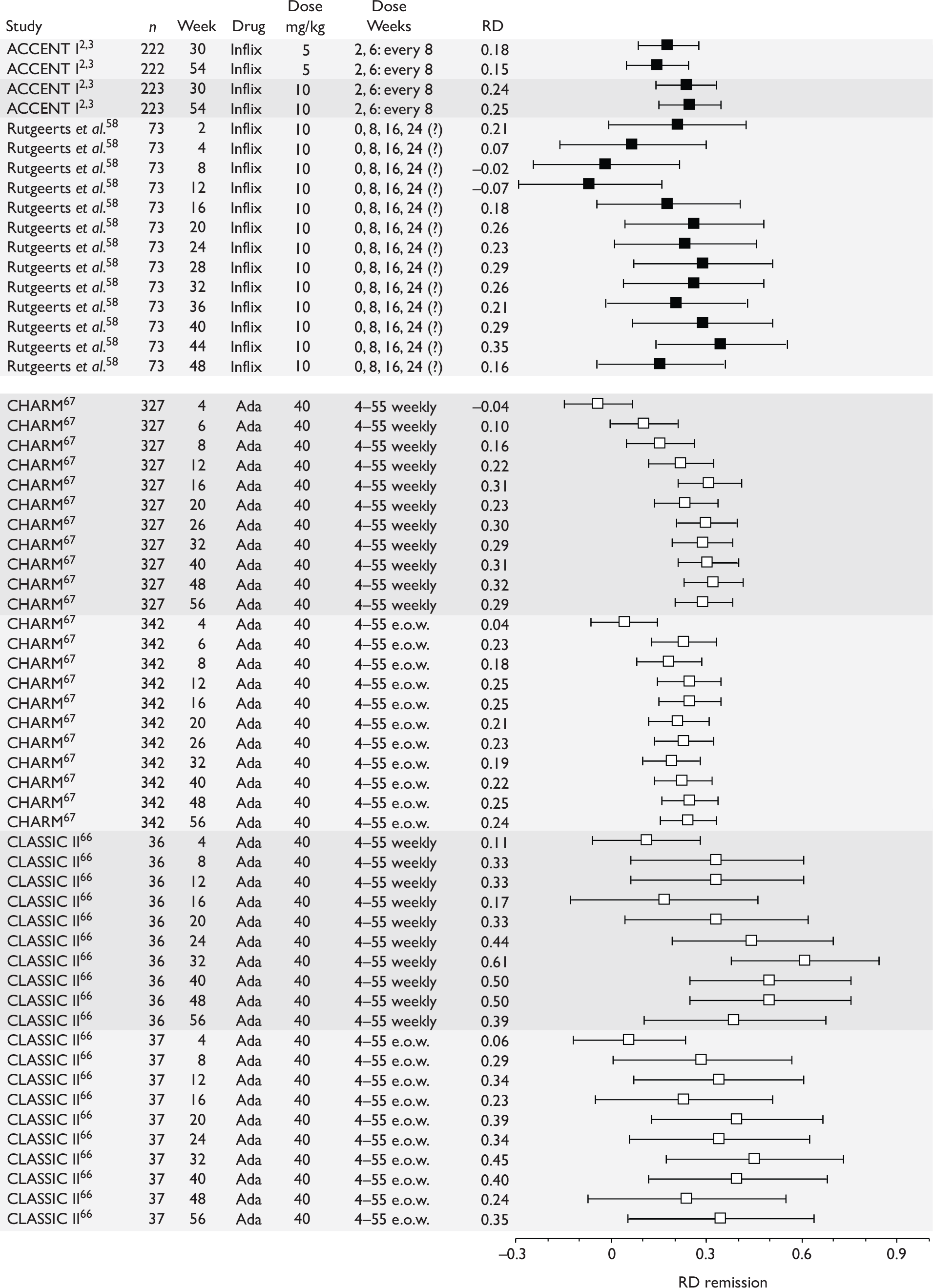
FIGURE 56.
Maintenance trials – risk ratio remission, all patients. Inflix, infliximab; RR, risk ratio.
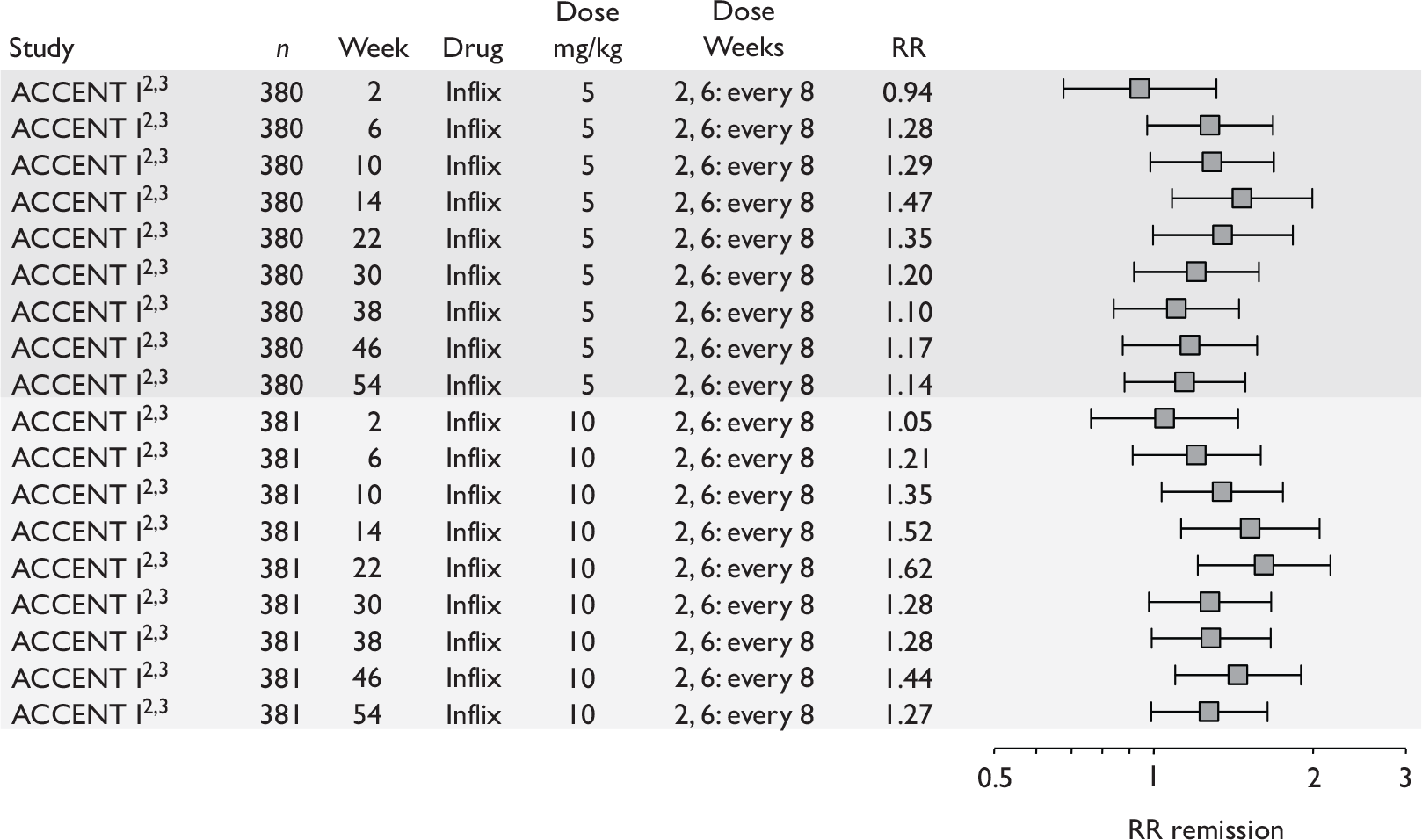
FIGURE 57.
Maintenance trials – risk difference remission, all patients. Inflix, infliximab; RD, risk difference.
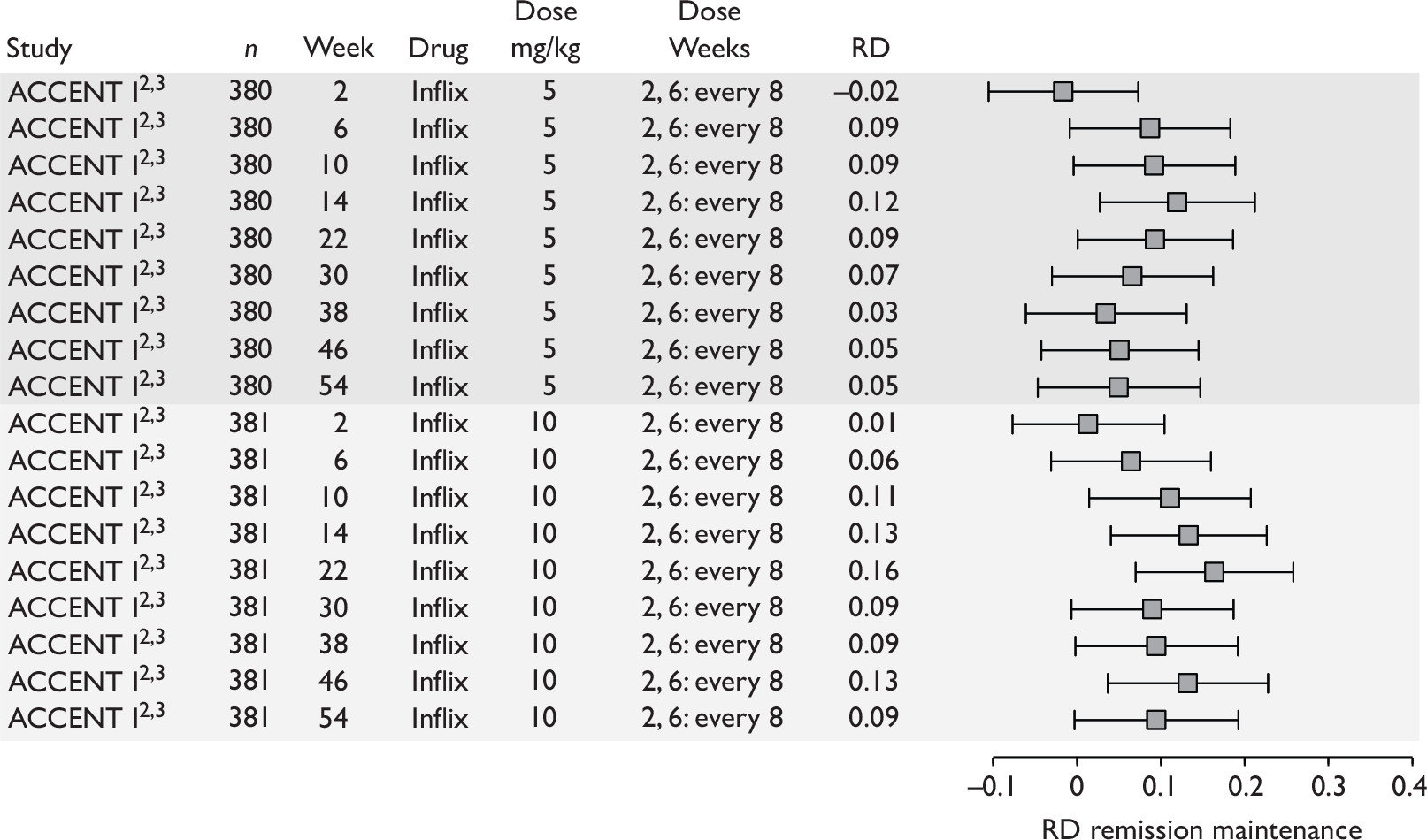
FIGURE 58.
Maintenance trials – risk ratio response 100. Ada, adalimumab; RR, risk ratio.
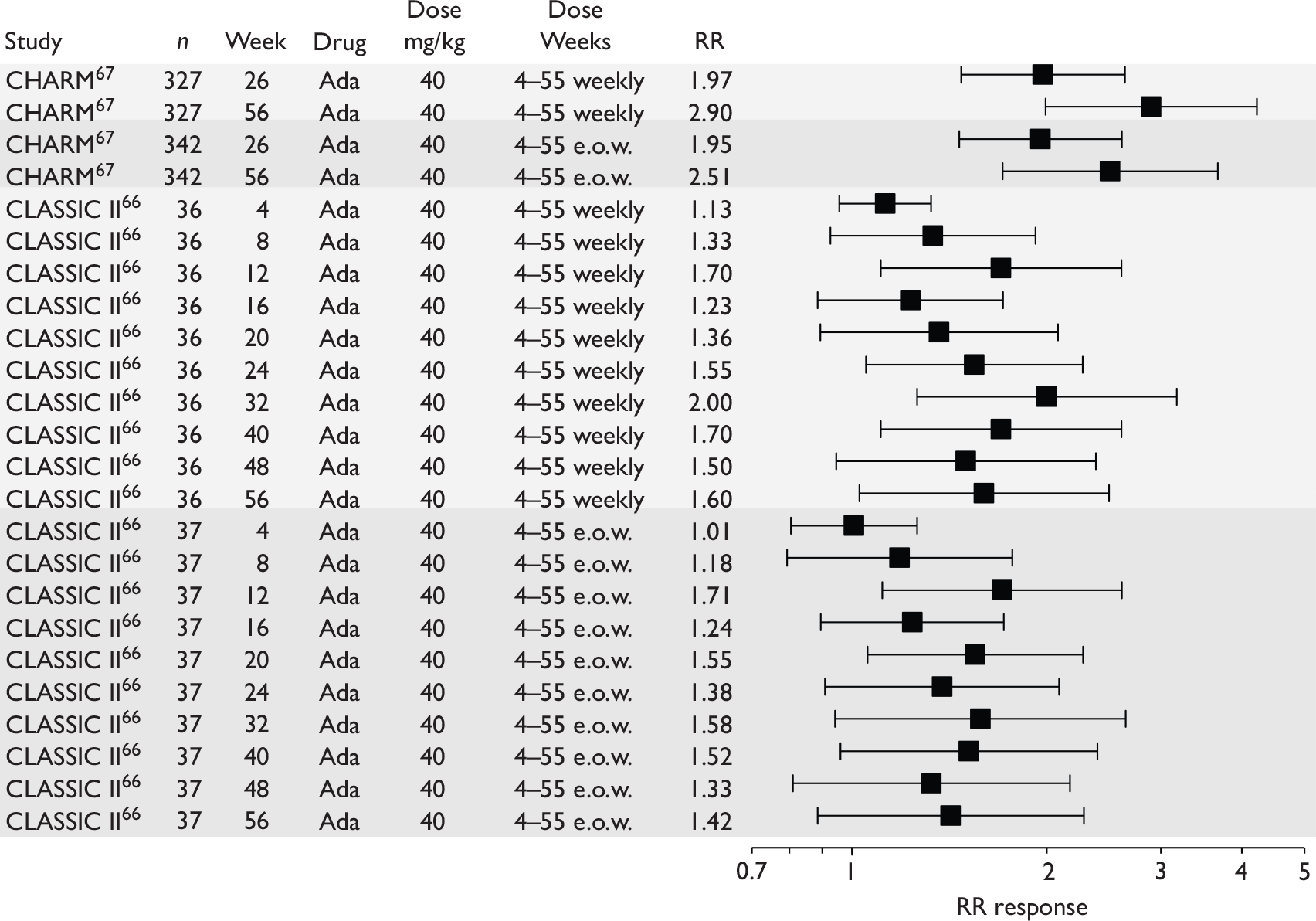
FIGURE 59.
Maintenance trials – risk difference response 100. Ada, adalimumab; RD, risk difference.
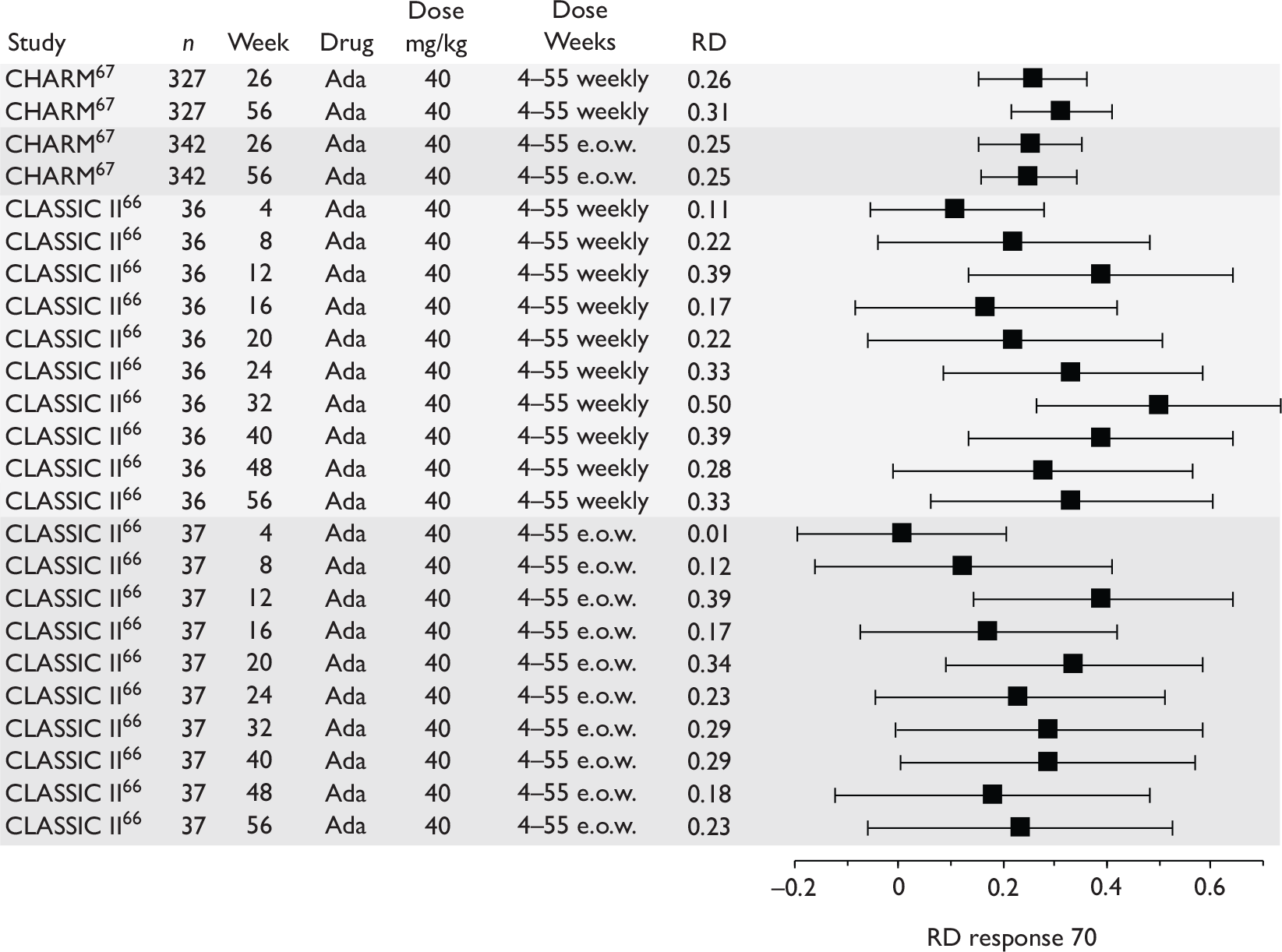
FIGURE 60.
Maintenance trials – risk ratio response 70 (responders). (‘Dose weeks’ for Rutgeerts et al. 58 refers to all scheduled dose weeks including those prior to randomisation.) ?, the dosing schedule was not clear from the published papers; Ada, adalimumab; Inflix, infliximab; RR, risk ratio.
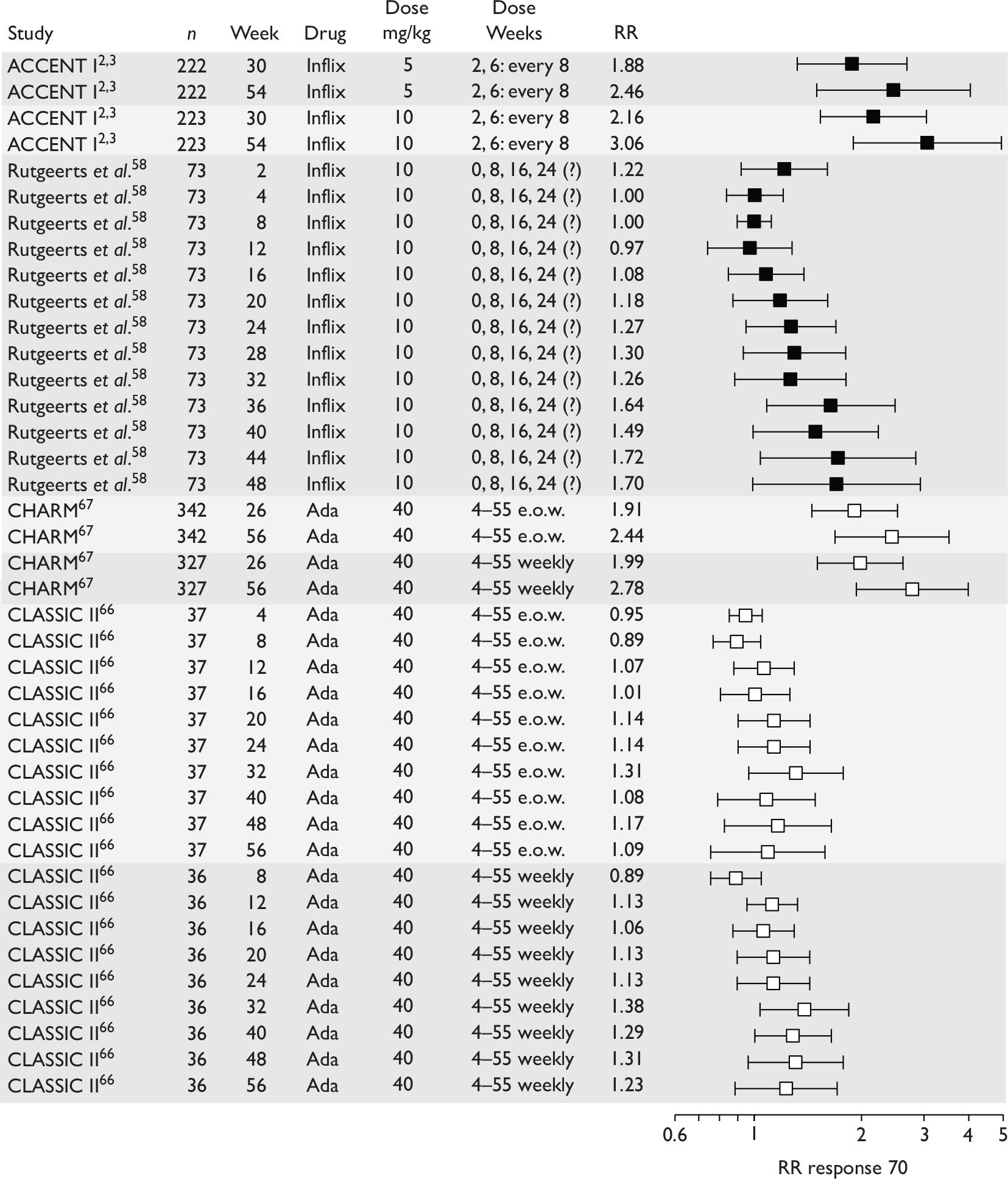
FIGURE 61.
Maintenance trials – risk difference response 70 (responders). (‘Dose weeks’ for Rutgeerts et al. 58 refers to all scheduled dose weeks including those prior to randomisation.) ?, the dosing schedule was not clear from the published papers; Ada, adalimumab; Inflix, infliximab; RD, risk difference.
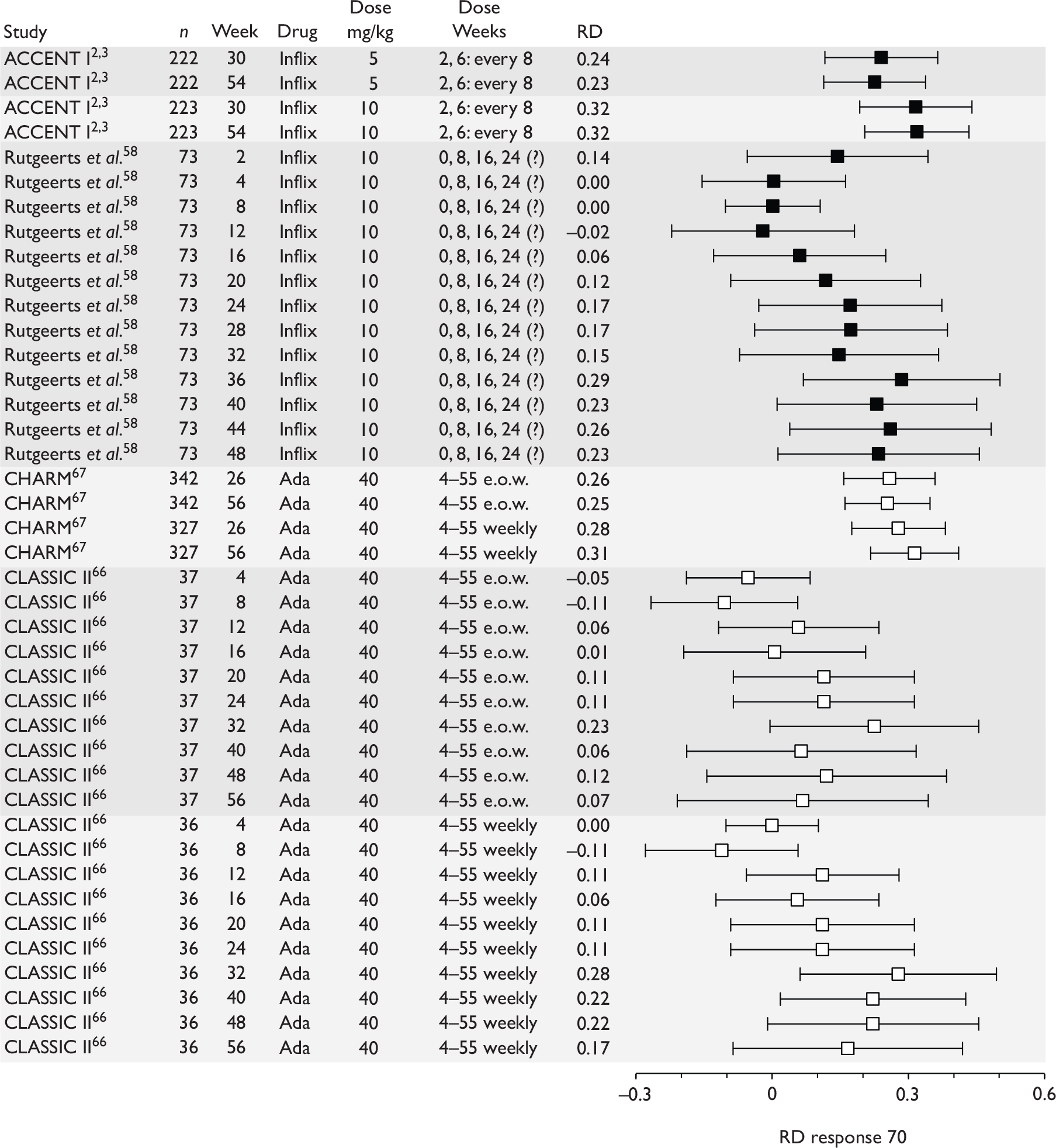
FIGURE 62.
Maintenance trials – risk ratio response 70 all patients. Inflix, infliximab; RR, risk ratio.
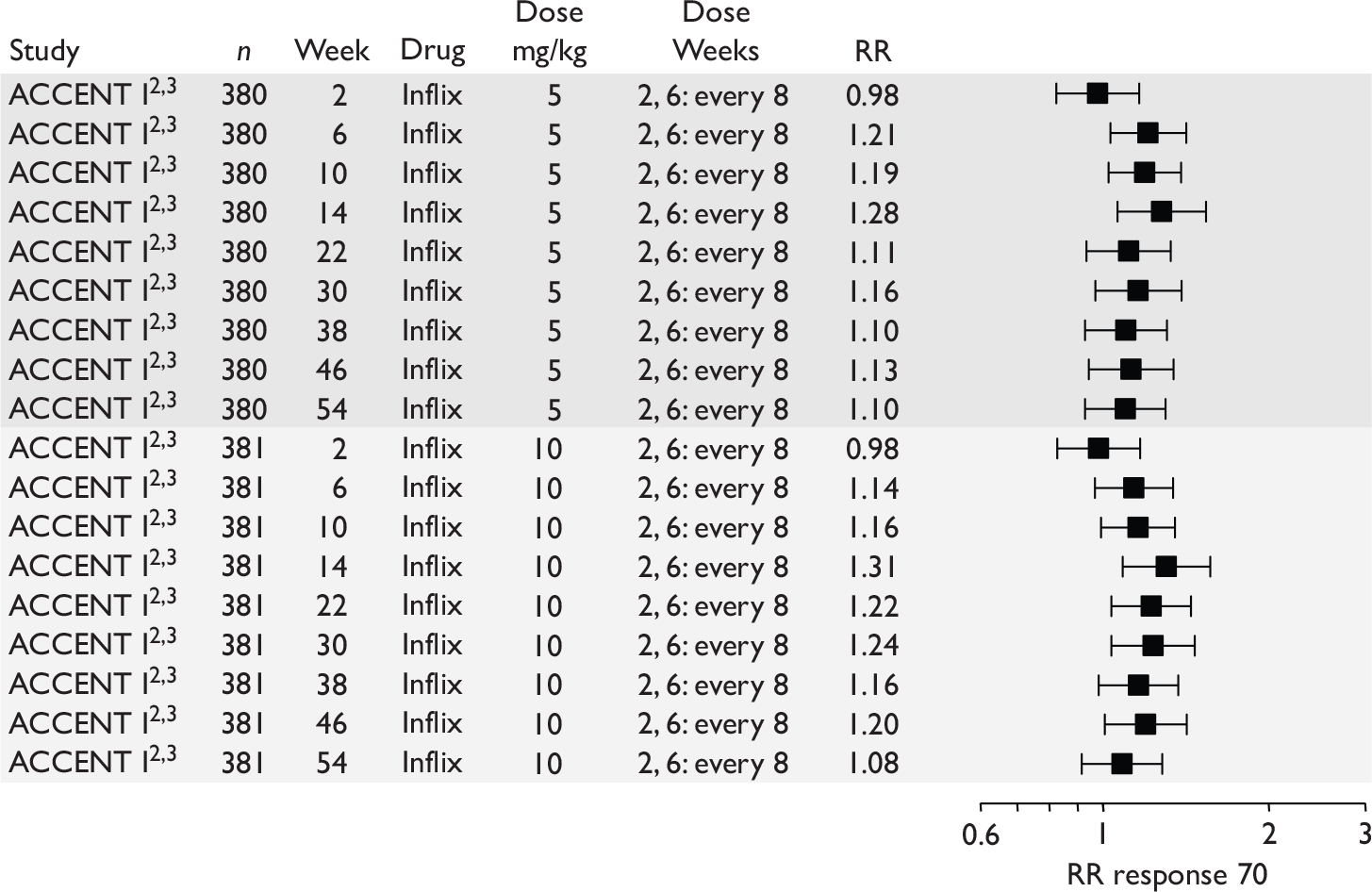
FIGURE 63.
Maintenance trials – risk difference response 70 all patients. Inflix, infliximab; RD, risk difference.
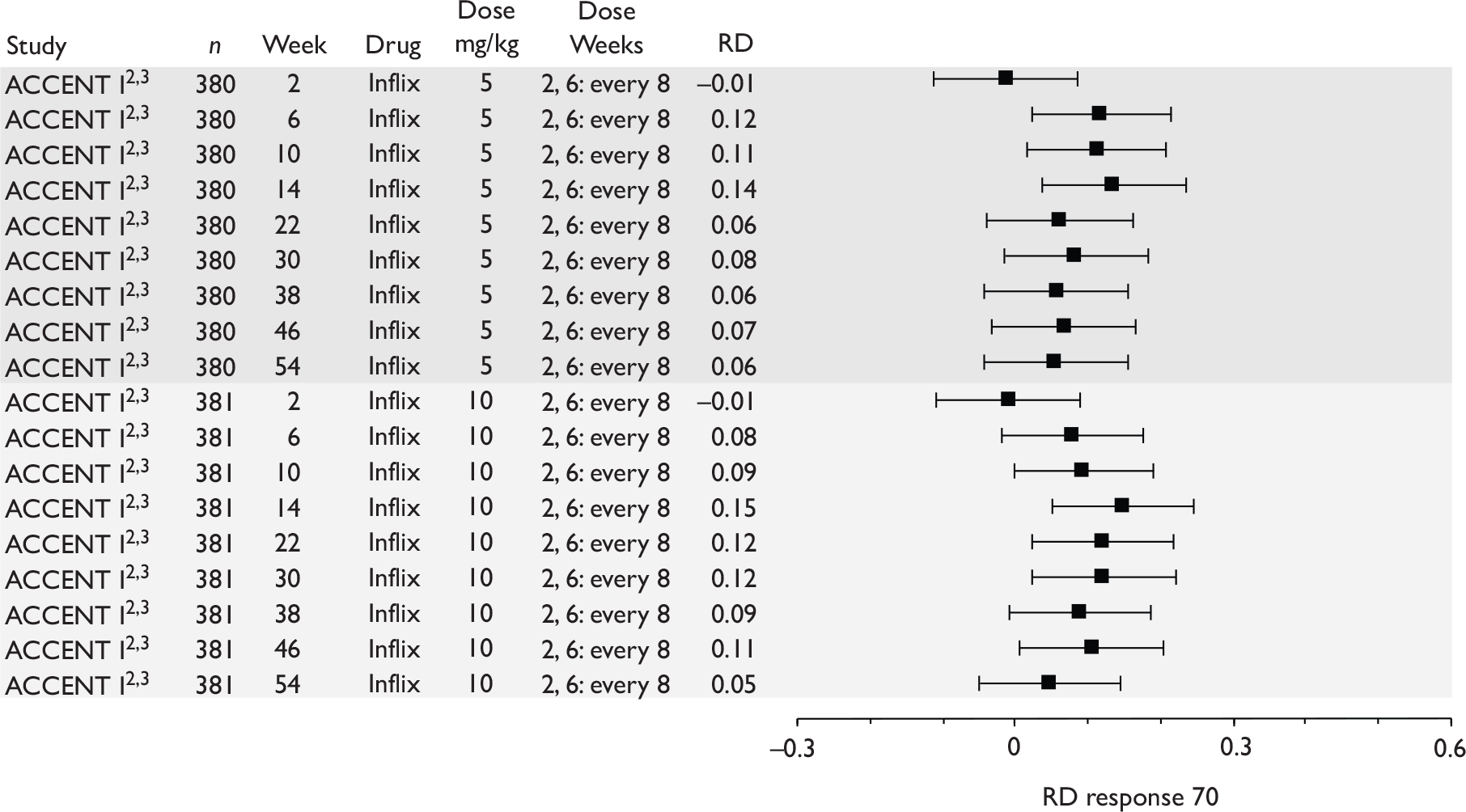
FIGURE 64.
Maintenance trials – CDAI scores for trials reporting median scores (results for responders). ?, the dosing schedule was not clear from the published papers; Inflix, infliximab.
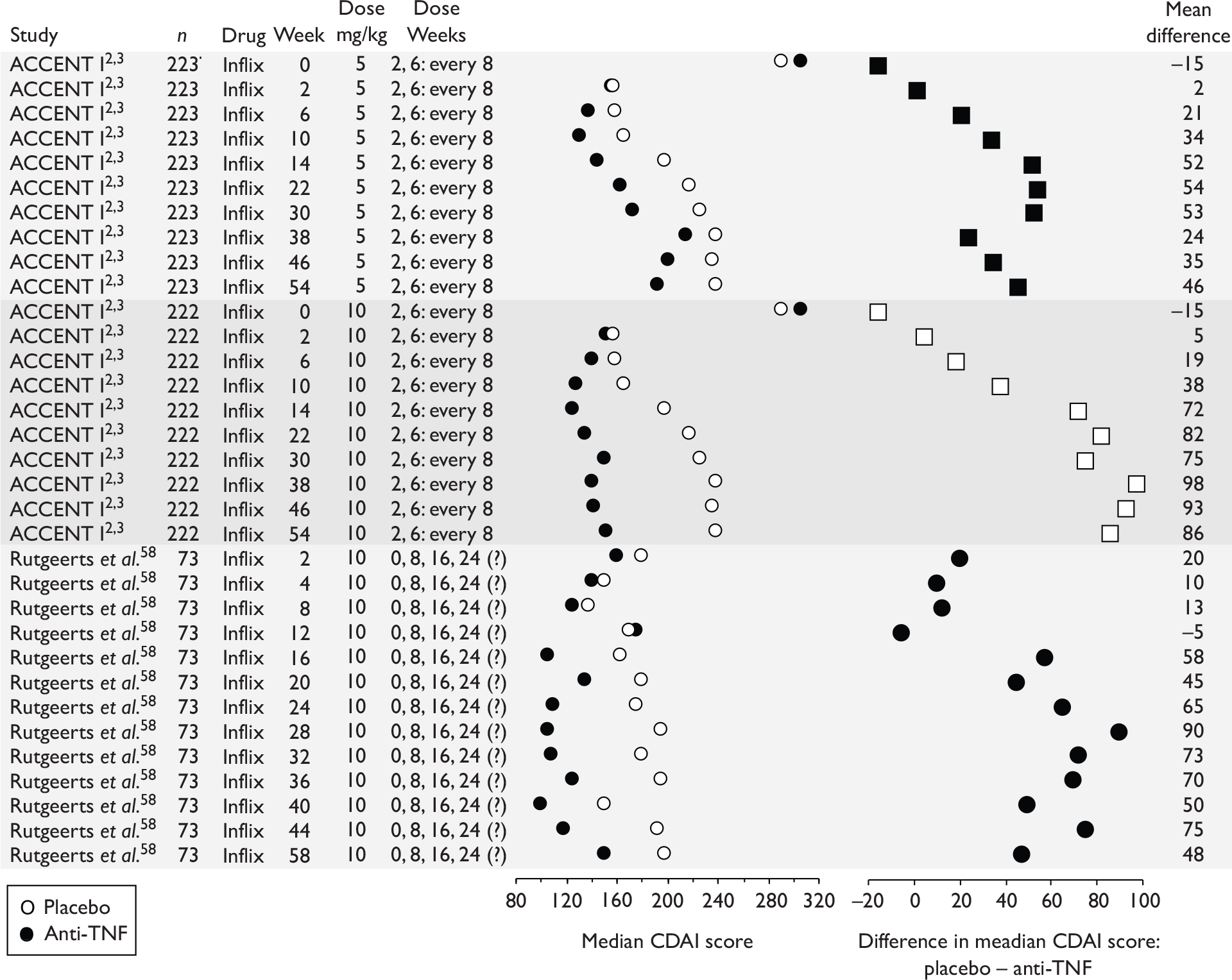
FIGURE 65.
Maintenance trials – CDAI scores for trials reporting median scores (results for all patients). Inflix, infliximab.
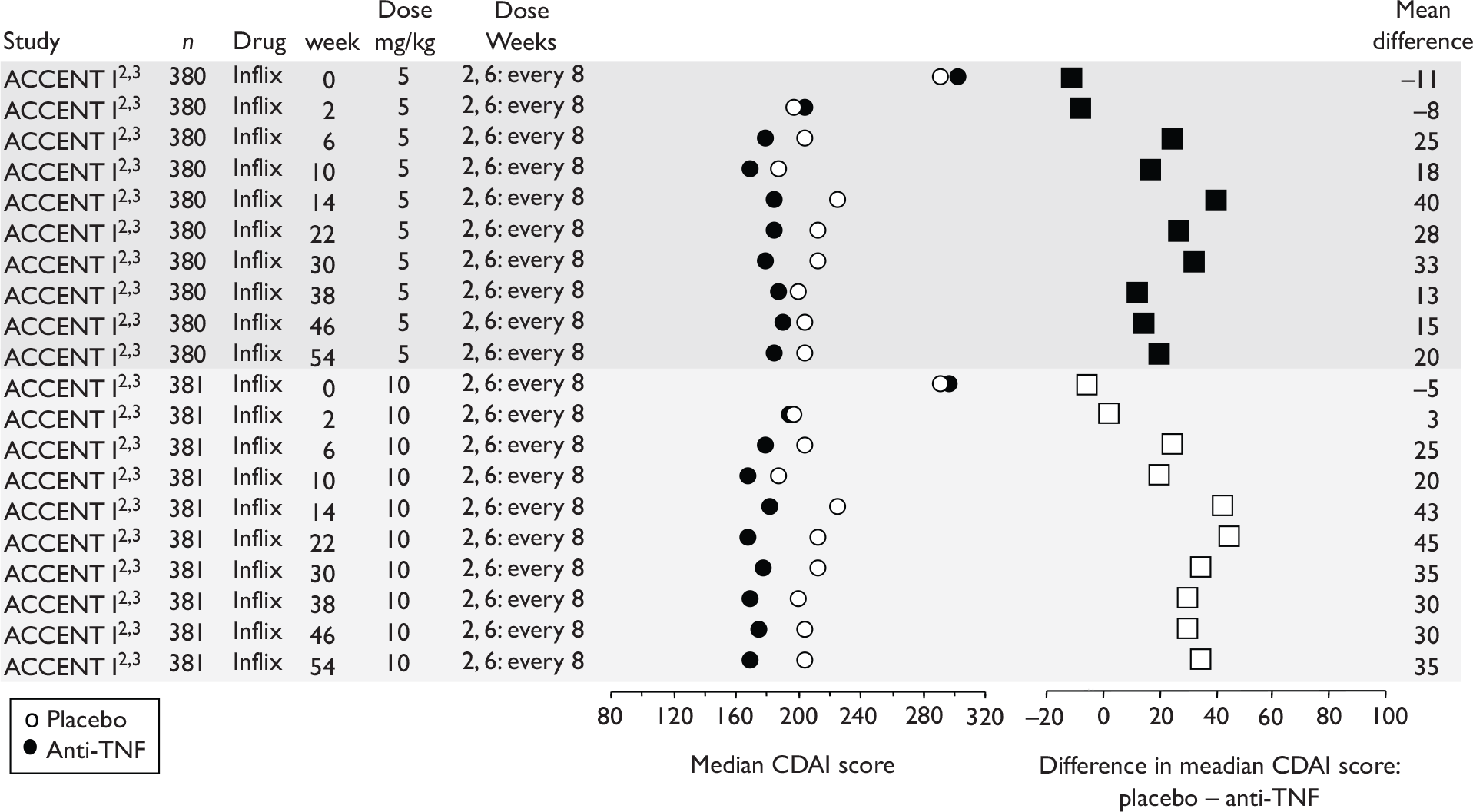
FIGURE 66.
Maintenance trials – CDAI scores for trials reporting mean scores.
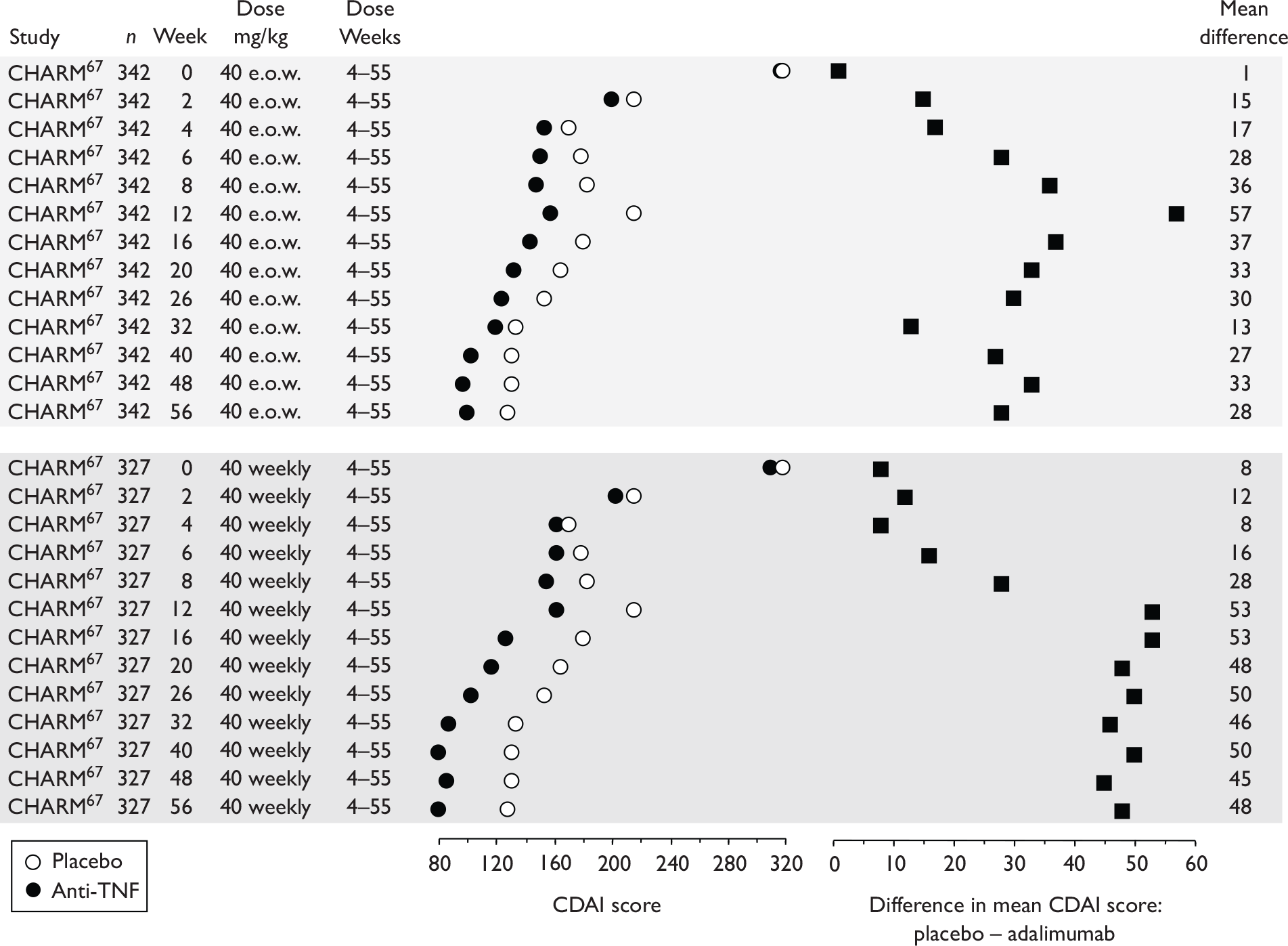
FIGURE 67.
Maintenance trials – IBDQ scores for trials reporting mean scores. Ada, adalimumab.
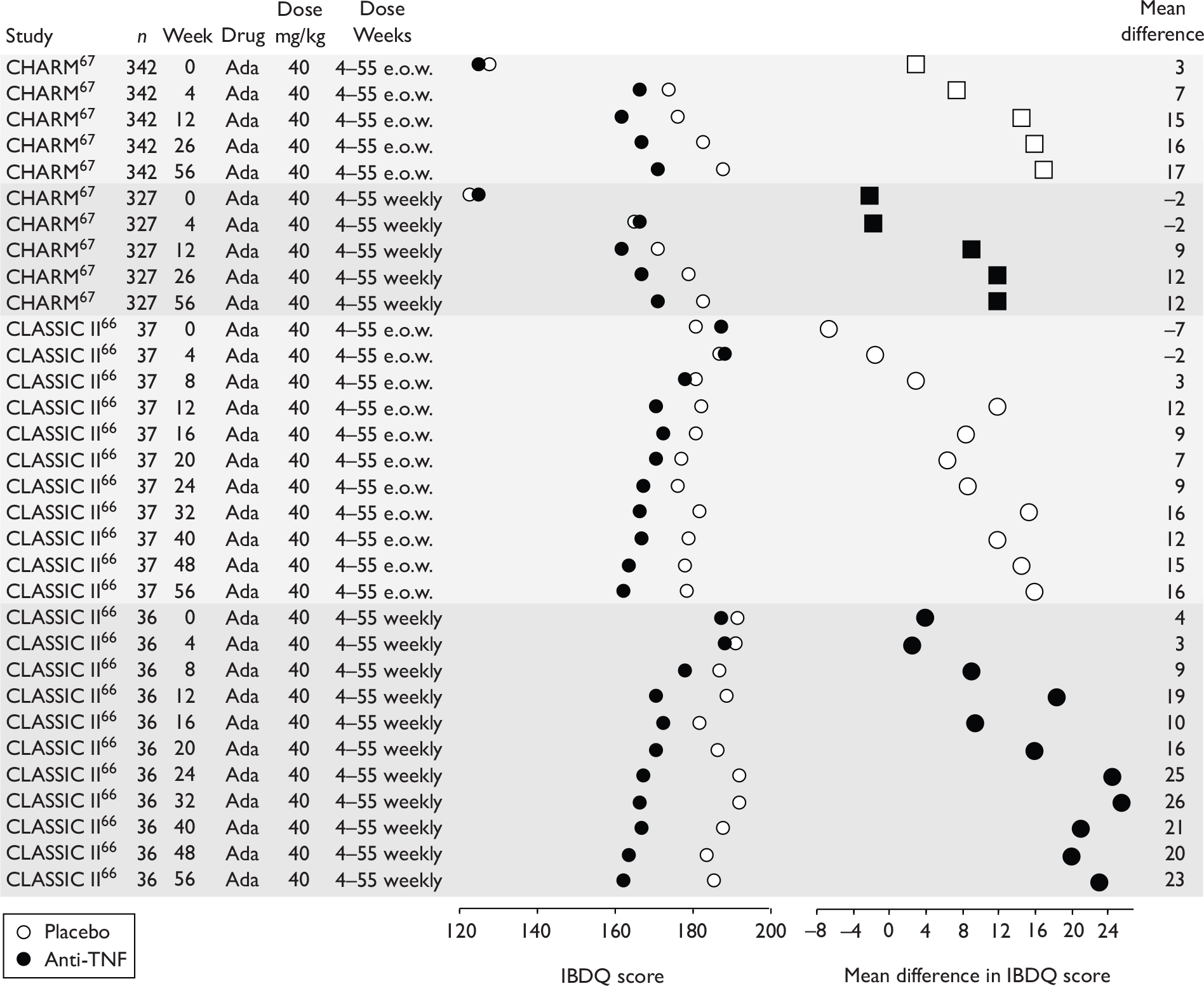
FIGURE 68.
Maintenance trials – IBDQ scores for trials reporting median scores. (‘Dose weeks’ refers to all scheduled doses including those prior to randomisation.) ?, the dosing schedule was not clear from the published papers; Inflix, infliximab.
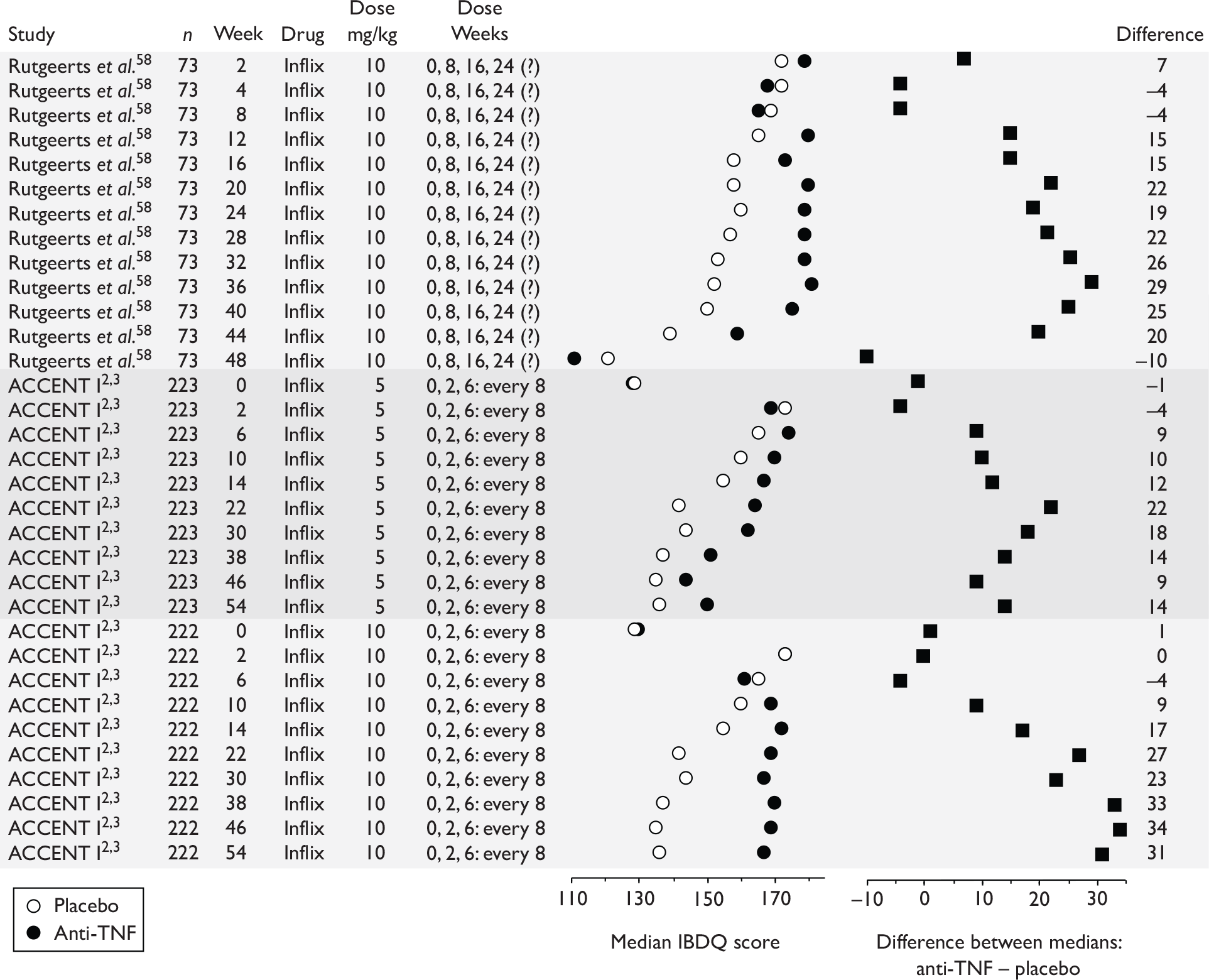
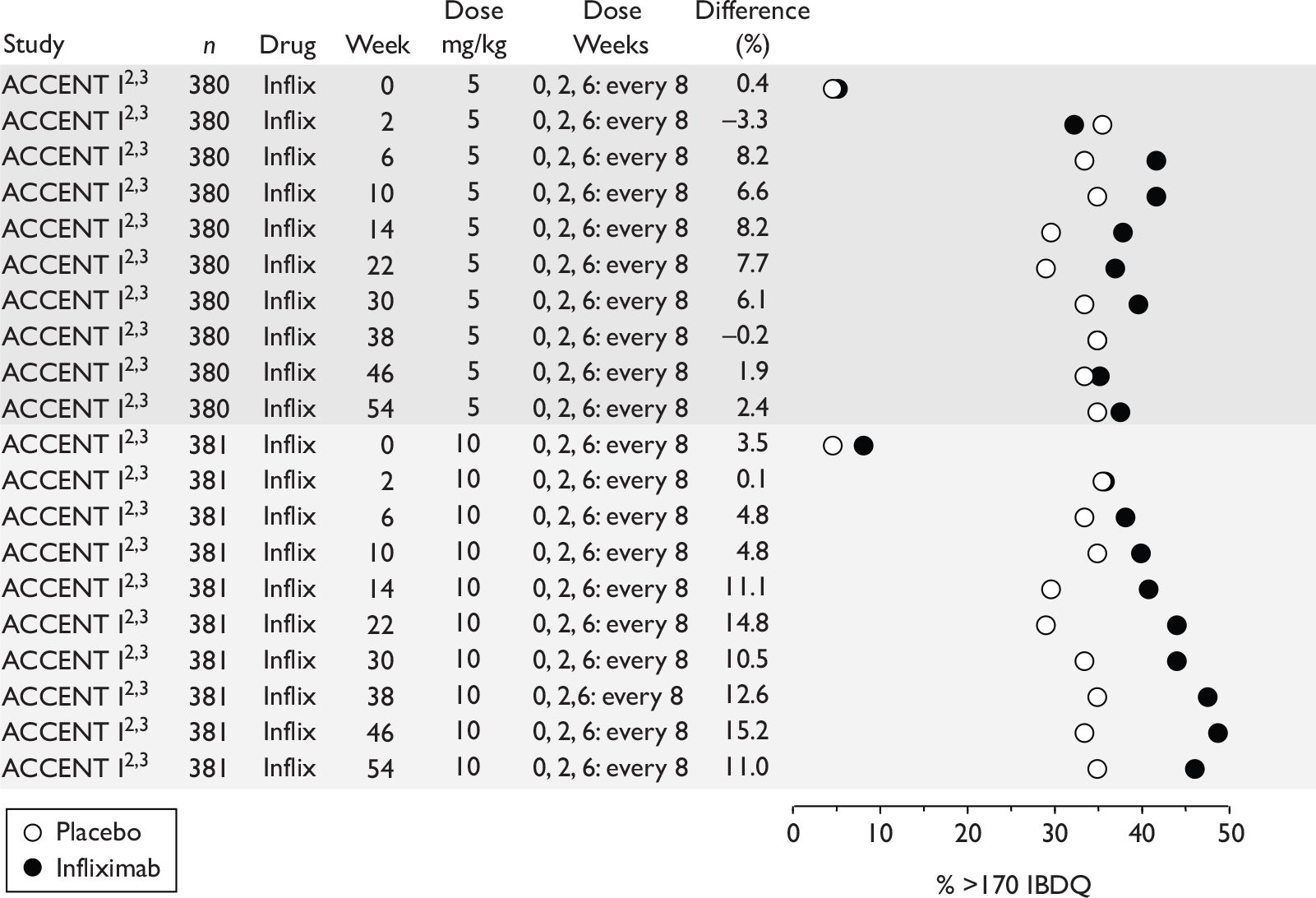
Appendix 11 Response rates among non-responders in maintenance trials
In this appendix, results for non-responders in the ACCENT I2,3 trial (infliximab) are presented, followed by results for non-responders and for all patients in the CHARM67 trial (adalimumab).
ACCENT I trial CDAI scores
Examination of median CDAI scores in the two separate publications2,3 allowed an approximation of the response to treatment in ‘non-responders’ at least to week 14, after which crossover to increased infliximab dosage was allowed for relapsing patients. The pertinent results for median CDAI scores are summarised in Figure 70.
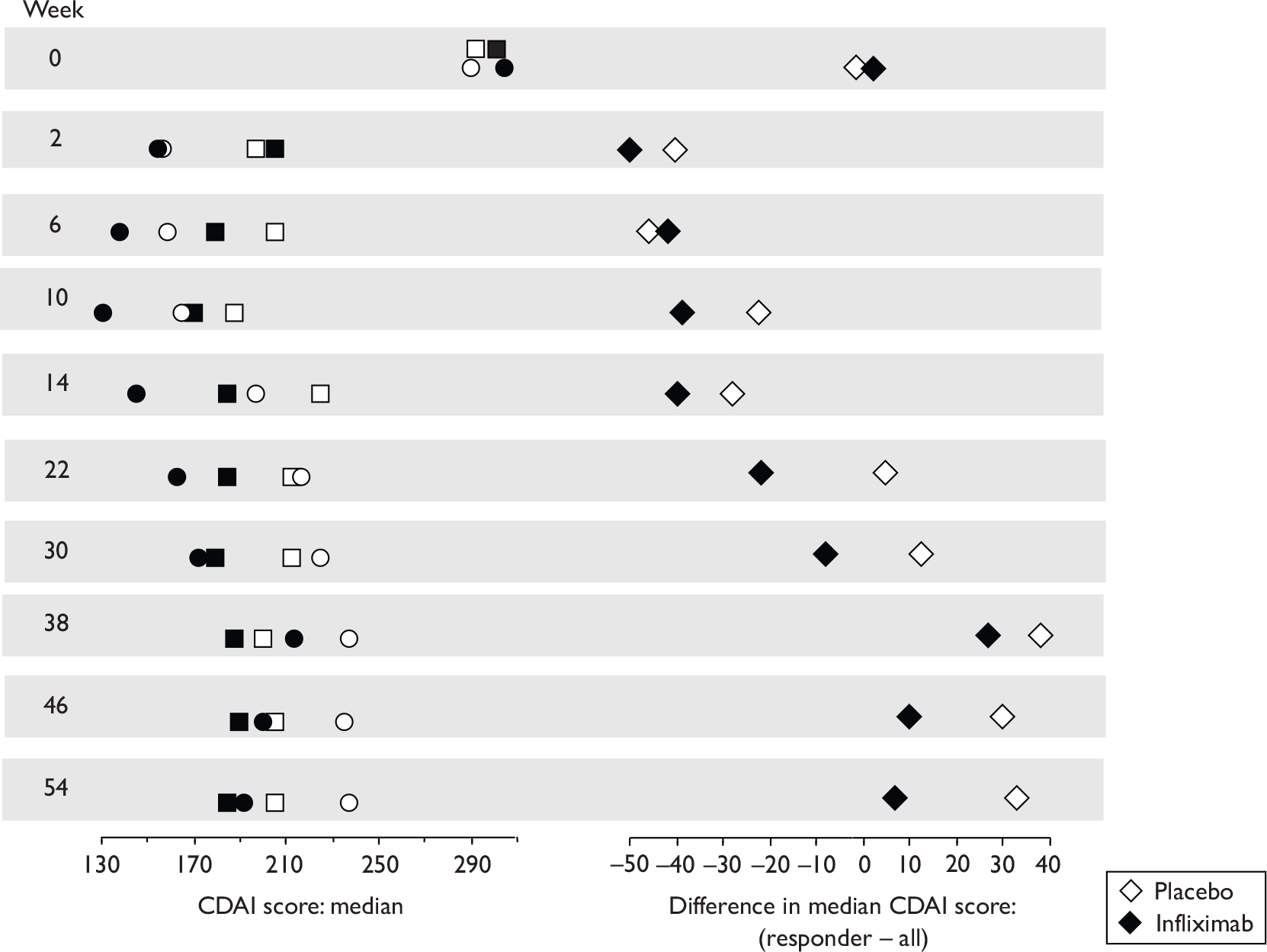
At randomisation (week 2), the difference in CDAI score of ‘responders’ minus ‘all’ was at its maximum, 50 points for infliximab and 40 for placebo, and was determined by patient selection. After randomisation up to week 14, the difference for the infliximab-treated patients (responders – all) remained fairly stable (at approximately 40 points), implying that during this phase of the trial, ‘responders’ and ‘non-responders’ fare about equally well with respect to their CDAI score at randomisation. After the introduction of permitted crossover for the ‘all-patient’ analysis at week 14 both infliximab- and placebo-treated groups exhibited striking increases in the score difference ‘responders’ – ‘all’. Given that increased CDAI implies a worse disease state, this trend implies non-responders were able to respond to treatment better than responders during this phase of the trial.
ACCENT I remission and responder 70 rates
Figure 71 shows the placebo and intervention response 70 rates for responders and calculated for non-responders. At week 2 a very large difference was evident as would be expected from the act of dichotomising patients into subgroups. Thereafter to week 14, response rates in the intervention and placebo arms gradually approached more closely. For non-responders there was only weak evidence that intervention was better than placebo. At week 14 both placebo groups and the non-responder intervention arm had a response rate of about 50%. The major difference between the responder and non-responder subgroups appeared to be the much larger proportion of placebo responders in the non-responder group; or, conversely among the responder group, there was a greater proportion of patients who required early doses of infliximab to achieve response. Unfortunately, information beyond 14 weeks was not available except for responders.
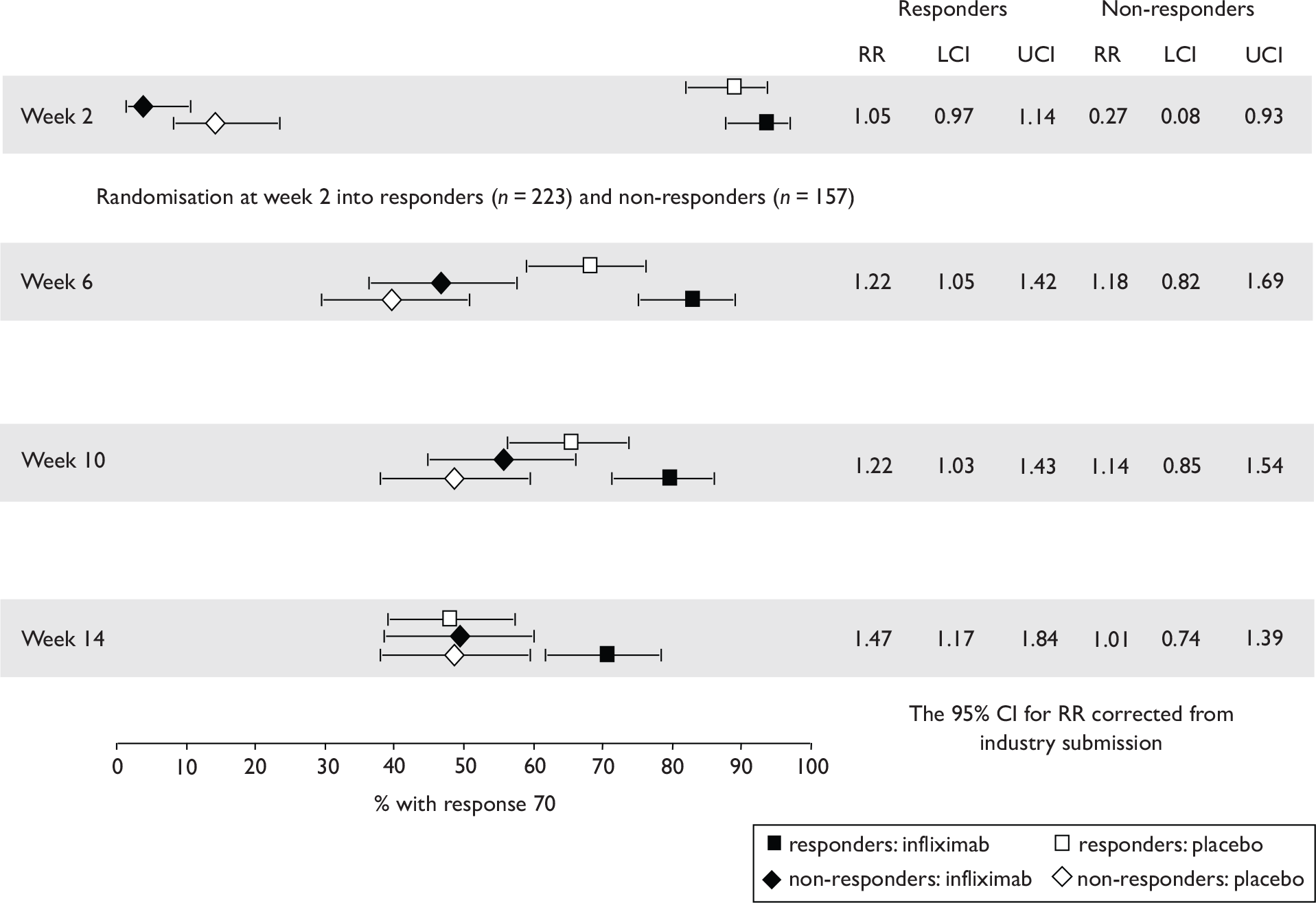
Similar results were seen for remission (Figure 72). At week 14, 20% of non-responders had attained remission irrespective of treatment. At week 14, the responder intervention arm exhibited 20% more patients in remission than the responder placebo group; thereafter this difference diminished. Unfortunately, no information for non-responders was available beyond 14 weeks.
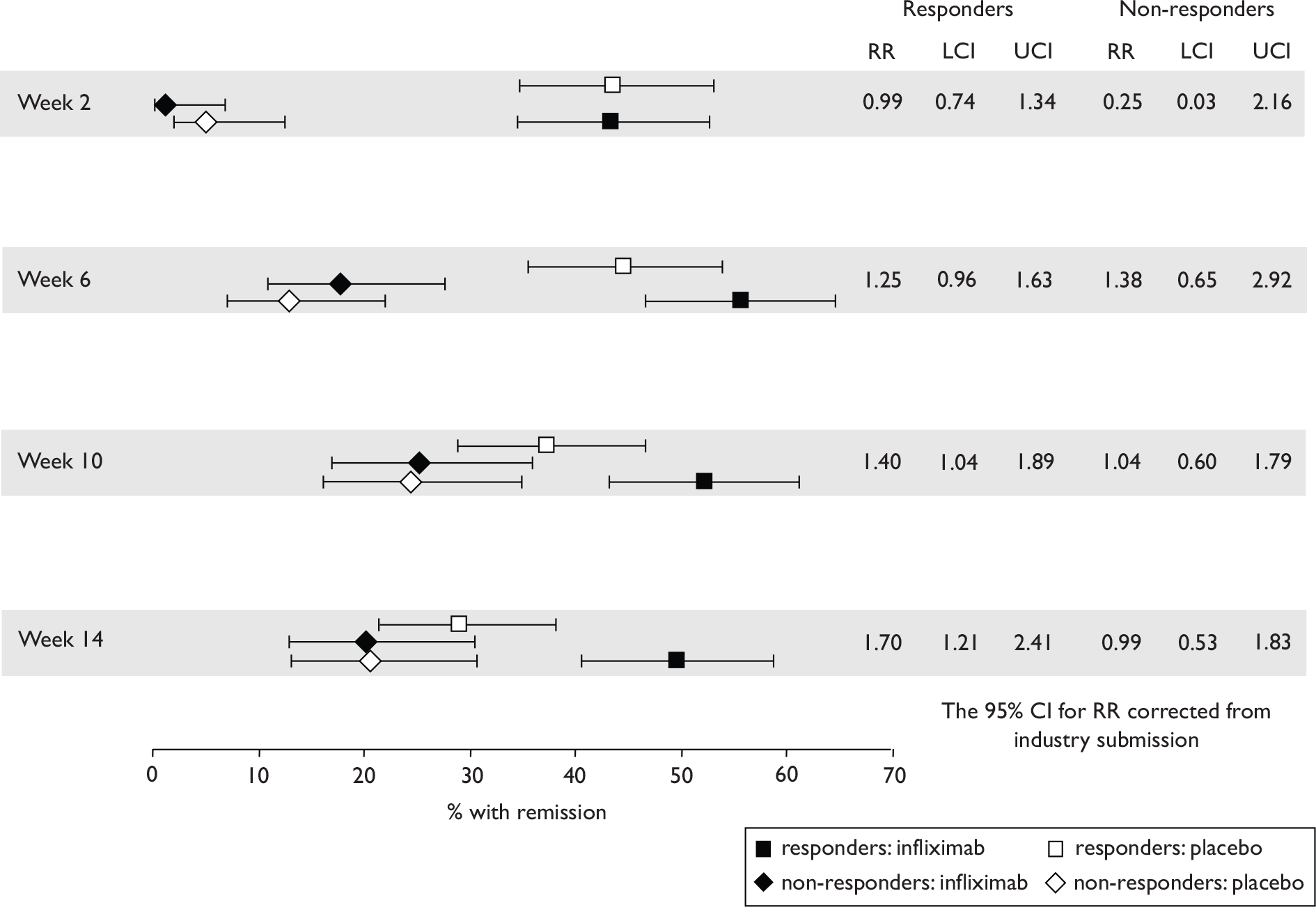
At week 14 the yield in percentage of patients with response 70 per dose, and in percentage of patients with remission per dose for strategies in which all patients received one dose, all patients received three doses or responders received three doses and non-responders a single dose was 49%, 21% and 25% for response 70 respectively, and 25%, 13% and 17% for remission respectively.
CHARM trial remission and response 70 and 100 rates
(CiC information has been removed.)
FIGURE 73.
(CiC information has been removed.)
FIGURE 74.
(CiC information has been removed.)
Appendix 12 Quality assessment of trials
| Study | Trial design | Blinding | Handling of missing data (binary and continuous); ITT | % of withdrawals and/or crossovers and loss to follow-up Other comments |
|---|---|---|---|---|
| Induction | ||||
|
Hanauer et al. , 200663 CLASSIC I (Adalimumab) |
4-week multicentre, randomised, double-blind, placebo-controlled trial; 299 patients randomised to placebo, or 40/20 mg (weeks 0 and 2), 80/40 mg or 160/80 mg adalimumab |
|
All 299 patients included in efficacy analyses. Those with missing data at week 4 classified as remission failures (assume that also counted as response failures but not explicitly stated) Also states that ‘all analyses were as observed with the exception of the IBDQ data that assessed the last observation carried forward’ (unclear what ‘as observed’ means) |
284/299 (95%) patients completed the trial; remaining patients withdrew. No loss to follow-up. Unclear how many patients contributing to each analysis |
|
Sandborn et al. , 200764 GAIN (Adalimumab) |
4-week multicentre, randomised, double-blind, placebo-controlled trial; 325 patients randomised to placebo or 160/80 mg adalimumab |
|
For clinical remission and response measures, all patients included: considered patients with missing data to be non-responders For continuous variables included only those patients with complete data |
No loss to follow-up; 14/325 (4%) discontinued intervention or placebo; unclear how many patients were counted as non-responders owing to missing data or how many did not contribute to continuous outcome data |
|
D’Haens et al. , 199968 (Infliximab) |
4-week multicentre, randomised, double-blind, placebo-controlled trial; 30 patients randomised to placebo, 5, 10 or 20 mg/kg infliximab |
|
Unclear if missing data or how missing data were handled; states that second colonoscopy could not be performed in two patients; states that ‘only biopsy specimens from patients who underwent two endoscopic procedures and biopsy sampling were used for the final analysis (n = 9)’. Unclear which analysis this refers to | % of withdrawal/loss to follow-up unclear |
|
Targan et al. , 199757 (Infliximab) |
4-week multicentre, randomised, double-blind, placebo-controlled trial; 108 patients randomised to placebo, 5, 10 or 20 mg/kg infliximab Patients without a response at week 4 were enrolled in a parallel, open-label study and were followed for 12 additional weeks |
(Refers to first 4 weeks) |
Unclear how missing data were handled States that the original study protocol did not specify the use of ITT analysis, but that patients were analysed according to assignment (except two patients who did not receive treatment and were excluded from the analysis). For assessing the response and remission rates in all evaluation periods after the initial blinded infusion, patients who received an open-label infusion or those with a change in concomitantly administered medication were considered non-responders. It is unclear if patients who did not contribute data during the blinded period were also counted as non-responders Not clear how missing continuous data were handled |
Based on data in Figure 1 it appears that at most 7/108 (6%) patients were not evaluated for results (at week 2) 100% of patients completed 4 weeks of double-blind therapy |
| Maintenance | ||||
|
Hanauer et al. , 20023 and Rutgeerts et al. , 20042 ACCENT I (Infliximab) |
54-week, multicentre, randomised, double-blind placebo-controlled trial; all patients initially received 5 mg/kg (week 0), then randomised to placebo (‘episodic treatment’) or 5 mg/kg (5 mg/kg weeks 2 and 6, then every 8 weeks) or 10 mg/kg infliximab (5 mg/kg weeks 2 and 6, then 10 mg/kg every 8 weeks) All patients included in analysis – responders and non-responders (in Rutgeerts et al. 2) Responders only analysis in Hanauer et al. 3 Note: at week 14 or later patients who had responded at any time to infliximab therapy but then worsened were eligible to cross over to ‘active episodic’ treatment as needed with infliximab 5, 10 or 15 mg/kg for patients originally assigned to episodic, 5 mg/kg or 10 mg/kg respectively |
|
Rutgeerts et al. 2 – responder and non-responder analysis: Hanauer et al. 3 responder analysis: |
124/573 (22%) patients had withdrawn by week 54; 201/573 (35%) had crossed over to active episodic treatment by week 54 [92/188 (49%) of patients crossed over from placebo to episodic treatment] No loss to follow-up Results include any patients with a response or in remission at different time points, not just patients maintaining a response (also includes non-responders) |
|
Rutgeerts et al. , 199958 (Infliximab) |
36-week, multicentre, randomised, double-blind placebo-controlled trial; patients randomised to placebo or 10 mg/kg infliximab; eligible patients had previously shown a response in the RCT by Targan et al. 57 (see induction trials) or, if initial non-response, a response in an 8 week open-label extension of Targan et al. ;57 unclear if this included any patients who had shown a response to placebo, or if all had received infliximab Responders only randomised |
No explicit statement regarding blinding of other parties |
Treatment was considered a failure in patients who underwent surgery or were treated with medication regimens excluded from the study regardless of CDAI Last measure carried forward for continuous measures (CDAI, IBDQ) in patients who discontinued follow-up, had a CD-related surgical procedure or had non-permitted medication change |
Results include any patients with a response at different time points, not just patients maintaining a response; unclear why data do not start with 100% of patients with a response as only responders included 24/73 (33%) patients withdrawn by end of study. No details regarding potential crossovers |
|
Colombel et al. , 200767 CHARM (Adalimumab) |
56-week, multicentre, randomised, double-blind placebo-controlled trial; all patients received adalimumab 80 mg subcutaneously at week 0, followed by 40-mg dose at week 2; randomisation at week 4, stratified by responder status to placebo or 40-mg adalimumab weekly or 40-mg adalimumab e.o.w.; only responders included in efficacy analysis States that secondary efficacy analyses include non-responders also, but present results for responders only in this publication; only fistula results include non-responders Note: those patients who experienced a disease flare or sustained non-response at or after week 12 were switched to open-label treatment (40 mg e.o.w., which could be escalated to 40 mg weekly) |
|
Patients who switched to open-label therapy or withdrew from the study were counted as remission failures. Patients without CDAI assessments at weeks 26 or 56 were classified as remission failures (remission = primary end point) Unclear if patients also counted as treatment failures for secondary outcomes (response) No details on how continuous data were handled |
Results include any patients in remission at different time points, not just patients maintaining remission 505/778 (65%) of patients completed study (note: paper states 59% unclear); 50% of these remained on double-blind therapy, 50% completed the study on open-label treatment Of 499 patients (responders) included in efficacy analysis, 29% withdrew and 38% were still on double-blind therapy. Assume 33% therefore on open-label treatment, though not clearly stated No details on loss-to follow-up |
|
Sandborn et al. , 200766 CLASSIC II (Adalimumab) |
56-week, multicentre, randomised, double-blind placebo-controlled trial; all patients from CLASSIC I63 trial eligible if they demonstrated remission at weeks 0 and 4 (unclear if this includes patients from placebo group in remission); randomisation to placebo, 40-mg adalimumab weekly or 40-mg adalimumab eow (unclear if placebo weekly or e.o.w.) Randomised patients in remission only Note: randomised patients experiencing a flare or with continued non-response could switch to open-label adalimumab 40 mg e.o.w.; patients on open-label adalimumab e.o.w. could switch to adalimumab 40 mg weekly |
|
Efficacy analysis included all randomised patients; patients who switched to open label or with missing data were classified in a ‘no maintenance of remission’ category Secondary analyses used ‘last observation carried forward’ |
10/55 (18%) withdrew (of these, one lost to follow-up) 32/55 (58%) patients completed 56 weeks of double-blind therapy (6/18, 33% of patients in placebo group completed 56 weeks of double-blind therapy); remainder completed study on open-label therapy Results include any patients in remission at different time points, not just patients maintaining remission |
| Fistulising | ||||
|
Present et al. , 199962 (Infliximab) |
18-week multicentre, randomised, double-blind placebo-controlled trial; patients randomised to placebo, 5 mg/kg infliximab or 10 mg/kg infliximab |
No details on blinding (other than to state that this was a double-blind trial) |
Treatment considered to have failed in patients who had changes in medication that were not permitted, who underwent surgery related to CD or who did not return for follow-up visits For continuous variables, measurements from the last evaluation were carried forward |
88/94 (94%) completed trial; no loss to follow-up Appear to be no crossovers |
|
Sands et al. , 200465 ACCENT II (Infliximab) |
54-week multicentre, randomised, double-blind placebo-controlled trial; all patients received 5 mg/kg infliximab at weeks 0, 2 and 6; responders at week 14 randomised to placebo or 5 mg/kg infliximab every 8 weeks Responders only included in primary analysis Non-responders also randomised for secondary analysis From week 22, patients could crossover from placebo to 5 mg/kg or from 5 mg/kg to 10 mg/kg infliximab |
|
All patients included in analysis. Data for patients who crossed over from placebo to infliximab were censored before crossover occurred. Not stated for patients who crossed over from lower to higher infliximab dose For continuous variables (CDAI, IBDQ), measurements from the last evaluation were carried forward |
95/282 (34%) crossed over (total randomised population; 2223 from placebo group to treatment) 78/195 (40%) crossed over (responder only) by week 54 (28% from placebo group to treatment) No details on withdrawals or loss to follow-up post randomisation |
| Paediatric | ||||
|
Baldassano et al. , 200346 (Infliximab) |
12-week multicentre, randomised (no placebo control); 21 patients randomised to 1 mg/kg, 5 mg/kg or 10 mg/kg infliximab |
|
No details. Numbers included in different analyses vary at different time points | 19/21 (90%) of patients completed trial. No further details |
|
Hyams et al. , 200745 REACH (Infliximab) |
54-week multicentre, randomised, open-label (no placebo control); 112 patients received induction therapy (5 mg/kg infliximab) for 10 weeks; only patients with response (n = 103) randomised at week 10 to 5 mg/kg infliximab every 8 weeks or 5 mg/kg infliximab every 12 weeks; patients losing clinical response eligible to cross over one time to receive treatment more frequently or at higher dose (10 mg/kg every 8 weeks) | No blinding: open-label study |
All analyses based on ITT principle. Patients who lost response and crossed over were considered non-responders (treatment failures) for the remainder of the study Last non-missing score used for continuous data where patients discontinued study or had insufficient data |
59/103 (57%) patients in treatment arms as randomised at study end. 35 patients (34%) crossed over in total and nine (9%) withdrew No loss to follow-up |
Appendix 13 Rates of response and remission in placebo arms of induction trials for anti-TNF interventions
FIGURE 75.
Placebo rates for response 100 (upper panel) and response 70 (lower panel) in induction trials. Ada, adalimumab; certo, certolizumab; inflix, infliximab.
FIGURE 76.
Placebo rates for remission in induction trials. Ada, adalimumab; certo, certolizumab; inflix, infliximab.
Appendix 14 Search strategy for economic evaluation
Note: certolizumab pegol and natalizumab were originally part of this appraisal; they were subsequently excluded after searching had been completed
Source – MEDLINE (Ovid), 1950 to week 4 May 2007
-
(adalimumab or humira).mp. (540)
-
(certolizumab or cimzia).mp. (19)
-
(infliximab or remicade).mp. (3096)
-
(natalizumab or tysabri).mp. (208)
-
or/1-4 (3473)
-
Crohn Disease/ (21,624)
-
crohn$.mp. (25,626)
-
or/6-7 (25,626)
-
5 and 8 (1046)
-
economics/ (24,885)
-
exp “costs and cost analysis”/ (129,414)
-
cost of illness/ (9149)
-
exp health care costs/ (28,541)
-
economic value of life/ (4847)
-
exp economics medical/ (11,355)
-
exp economics hospital/ (14,731)
-
economics pharmaceutical/ (1764)
-
exp “fees and charges”/ (22,970)
-
(econom$or cost or costs or costly or costing or price or pricing or pharmacoeconomic$).tw. (244,897)
-
(expenditure$not energy).tw. (10,410)
-
(value adj1 money).tw. (10)
-
budget$.tw. (10,892)
-
or/10-22 (358,461)
-
9 and 23 (51)
-
limit 24 to yr=“2000 - 2007” (48)
Source – MEDLINE (Ovid), 1950 to week 3 June 2007*
-
ca2.mp. (105,908)
-
d2e7.mp. (23)
-
cdp870.mp. (26)
-
pha-738144.mp. (0)
-
pha 738144.mp. (0)
-
(anti adj2 4 integrin).mp. (45)
-
anti alpha4 integrin.mp. (49)
-
anti alpha 4 integrin.mp. (32)
-
or/1-8 (106,047)
-
crohn disease/ (21,699)
-
crohn$.mp. (25,732)
-
or/10-11 (25,732)
-
9 and 12 (66)
-
economics/ (24,922)
-
exp “costs and cost analysis”/ (130,028)
-
cost of illness/ (9244)
-
exp health care costs/ (28,753)
-
economic value of life/ (4854)
-
exp economics medical/ (11,385)
-
exp economics hospital/ (14,773)
-
economics pharmaceutical/ (1786)
-
exp “fees and charges”/ (23,036)
-
(econom$or cost or costs or costly or costing or price or pricing or pharmacoeconomic$).tw. (246,746)
-
(expenditure$not energy).tw. (10,484)
-
(value adj1 money).tw. (10)
-
budget$.tw. (10,945)
-
or/14-26 (360,657)
-
13 and 27 (1)
*Additional search to account for alternative terminology used for the drugs.
Source – EMBASE (Ovid), 1980 to 2007 week 22
-
(adalimumab or humira).mp. (2036)
-
(certolizumab or cimzia).mp. (230)
-
(infliximab or remicade).mp. (7811)
-
(natalizumab or tysabri).mp. (843)
-
or/1-4 (8685)
-
Crohn Disease/ (20,817)
-
crohn$.mp. (23,756)
-
or/6-7 (23,756)
-
5 and 8 (2554)
-
cost benefit analysis/ (26,197)
-
cost effectiveness analysis/ (48,867)
-
cost minimization analysis/ (1140)
-
cost utility analysis/ (1927)
-
economic evaluation/ (3621)
-
(cost or costs or costed or costly or costing).tw. (146,502)
-
(economic$or pharmacoeconomic$or price$or pricing).tw. (70,477)
-
(technology adj assessment$).tw. (1366)
-
or/10-17 (223,990)
-
9 and 18 (151)
-
limit 19 to yr=“2000 - 2007” (149)
Source – EMBASE (Ovid), 1980 to 2007 week 25*
-
ca2.mp. (115,879)
-
d2e7.mp. (65)
-
cdp870.mp. (16)
-
pha-738144.mp. (1)
-
pha 738144.mp. (1)
-
(anti adj2 4 integrin).mp. (9)
-
anti alpha4 integrin.mp. (37)
-
anti alpha 4 integrin.mp. (2)
-
or/1-8 (116,001)
-
crohn disease/ (20,928)
-
crohn$.mp. (23,876)
-
or/10-11 (23,876)
-
9 and 12 (72)
-
cost benefit analysis/ (26,342)
-
cost effectiveness analysis/ (49,154)
-
cost minimization analysis/ (1156)
-
cost utility analysis/ (1947)
-
economic evaluation/ (3637)
-
(cost or costs or costed or costly or costing).tw. (147,257)
-
(economic$or pharmacoeconomic$or price$or pricing).tw. (70,847)
-
(technology adj assessment$).tw. (1367)
-
or/14-21 (225,192)
-
13 and 22 (3)
*Additional search to account for alternative terminology used for the drugs.
Quality of life
Source – MEDLINE (Ovid), 1950 to week 4 May 2007
-
(adalimumab or humira).mp.
-
(certolizumab or cimzia).mp.
-
(infliximab or remicade).mp.
-
(natalizumab or tysabri).mp.
-
or/1-4 (3473)
-
Crohn Disease/ (21,624)
-
crohn$.mp. (25,626)
-
or/6-7 (25,626)
-
5 and 8 (1046)
-
quality of life/ (59,486)
-
life style/ (25,902)
-
health status/ (33,125)
-
health status indicators/ (10,984)
-
value of life/ (4847)
-
quality adjusted life.mp. (3912)
-
or/10-15 (124,425)
-
8 and 16 (427)
-
limit 17 to yr=“2000 - 2007” (246)
-
from 18 keep 1-246 (246)
Source – EMBASE (Ovid), 1980 to 2007 week 22
-
(adalimumab or humira).mp. (2036)
-
(certolizumab or cimzia).mp. (230)
-
(infliximab or remicade).mp. (7811)
-
(natalizumab or tysabri).mp. (843)
-
or/1-4 (8685)
-
Crohn Disease/ (20,817)
-
crohn$.mp. (23,756)
-
or/6-7 (23,756)
-
5 and 8 (2554)
-
quality of life/ (75,452)
-
quality adjusted life year/ (3013)
-
health status/ (31,455)
-
health status indicator$.mp. (129)
-
or/10-13 (104,174)
-
8 and 14 (624)
-
limit 15 to yr=“2000 - 2007” (481)
Source – HEED, June 2007
Search terms: (adalimumab OR humira OR certolizumab OR cimzia OR infliximab OR remicade OR natalizumab OR tysabri OR ca2 OR d2e7 OR cdp870 OR pha-738144 OR pha 738144 OR anti 4 integrin OR anti alpha4 integrin OR anti alpha 4 integrin) AND (crohn OR crohns)
Cohort studies of infliximab and Crohn’s disease
Source – MEDLINE (Ovid), 1950 to week 4 May 2007
-
(infliximab or remicade).mp. (3096)
-
crohn$.mp. (25,626)
-
crohn disease/ (21,624)
-
or/2-3 (25,626)
-
1 and 4 (992)
-
cohort studies/ (73,136)
-
Risk/ (74,606)
-
cohort$.mp. (132,834)
-
risk$.mp. (848,754)
-
or/6-9 (922,221)
-
5 and 10 (186)
Clinical guidelines
Source – MEDLINE (Ovid), 1950 to week 5 May 2007
-
Crohn Disease/ (21,659)
-
crohn$.mp. (25,672)
-
or/1-2 (25,672)
-
exp “guideline [publication type]”/ (15,854)
-
exp “consensus development conference [publication type]”/ (5531)
-
guideline$.mp. (137,797)
-
recommend$.mp. (215,591)
-
consensus.mp. (63,448)
-
or/4-8 (381,390)
-
3 and 9 (631)
-
or/4-5 (20,617)
-
3 and 11 (51)
Appendix 15 Details of studies included in cost-effectiveness review
| Study | Type of evaluation and synthesis | Interventions | Study population | Country | Duration of study |
|---|---|---|---|---|---|
| Jaisson-Hot et al.7 (2004; non-fistulising) | CUA |
|
Adult patients with non-responsive, non-fistulising CD, (CDAI between 220 and 440) 38 years old at baseline | France | Lifetime |
| Clark et al.5 (2003; non-fistulising) | CUA |
|
Adult patients (70 kg) with non-responsive, non-fistulising CD, 37 years at baseline | UK | Unclear, probably 1 year |
| Marshall et al.6 (2002; non-fistulising) | CUA |
|
Adult patients (70 kg) with CD resistant to conventional medical therapy | Canada | 1 year |
| Clark et al.5 (2003; fistulising) | CUA |
|
Adult patients with fistulising CD | UK | 1 year |
| Arseneau et al.90 (2001; fistulising) | CUA |
|
Adult patients (70 kg) with symptomatic perianal fistulas | USA | 1 year |
| Study | Type of model | Perspective | Model assumptions | |
|---|---|---|---|---|
| Outcomes | Costs and resource use | |||
| Jaisson-Hot et al.7 (2004; non-fistulising) | Markov model, cycle length of 2 months | Third-party payer perspective | Lifetime model but no stated mortality assumptions |
Infliximab dose at 5 mg/kg per infusion Maintenance treatment every 8 weeks |
| Clark et al.5 (2003; non-fistulising) |
Modified industry submission Markov model, cycle length of 2 months |
Unclear | Benefits related to the numbers in remitted health state (CDAI < 150). Report also gives outcomes under industry assumption (benefit = reduction of 70 CDAI points) | Unclear |
| Marshall et al.6 (2002; non-fistulising) | Markov model: initial cycle length of 12 weeks, with subsequent cycles at 8 weeks | Third-party (Canadian provision ministry of health) perspective |
US data (Olmstead County) used to estimate transition probabilities in usual care No transitions between remission and drug-responsive states (owing to data limitations) Retreatment strategy assumed to have equivalent effectiveness to initial dosage. All infliximab dosages (5 mg/kg, 10 mg/kg, 20 mg/kg) treated as equally effective |
20% of patients in drug-refractory state would be admitted to hospital, with the remaining 80% receiving outpatient care Only 5 mg/kg infliximab dosages used Acute infusion reactions are mild, and have no effect on treatment efficacy or cost Methotrexate and ciclosporin not used by the model cohort No medication given in the period following surgery as post-operative prophylaxis |
| Clark et al.5 (2003; fistulising) | Unclear | Unclear | Time spent with fistulas closure | Infliximab dose (unclear) offset by possible savings in surgery |
| Arseneau et al.90 (2001; fistulising) | Markov model, cycle length of 1 month | Third-party payer perspective |
Episodic remission figures assumed to equal remission from initial infusion Benefits from initial infusion assumed to occur within the first month following infusion The chance of fistula recurrence increases by 3% per month after 4 months Pancreatitis state includes 1 week with acute pancreatitis, and 3 weeks of fistula/improved fistula |
Initial infliximab infusions at 5 mg/kg (weeks 0, 2 and 6), according to FDA-approved protocol |
| Study | Cost items | Cost data sources | Resource use | Resource data source | Currency and currency year | Discount rate |
|---|---|---|---|---|---|---|
| Jaisson-Hot et al.7 (2004; non-fistulising) | Hospitalisations Outpatient care (physicians’ visits, nursing care, laboratory), medications, and patient transportation | Some unit costs based on diagnosis-related group estimates and negotiated prices | Based on expert opinion. No details given | Not given | Not given | 5% |
| Clark et al.5 (2003; non-fistulising) |
Drug and administration costs Other items unclear |
Not given | Not given | Not given | Not given | Not discounted (probably 1 year) |
| Marshall et al.6 (2002; non-fistulising) | Infliximab infusion; CD-related outpatient prescriptions; outpatient physician visits; medical hospital admissions for CD; surgical hospital admissions for CD | Unit costs based on: 2001 Drug Benefits Formulary, McMaster University Medical Centre outpatient pharmacy | Appears in appendices to CCOHTA report |
Three-member expert panel of gastroenterologists based on text description Surgical costs from patient-level database |
Canadian dollars, 2001 | Not discounted (1 year) |
| Clark et al.5 (2003; fistulising) |
Drug costs; surgery Other items unclear |
Not given | Not given | Not given | Not given | Not discounted (1 year) |
| Arseneau et al.90 (2001; fistulising) | Diagnostic, physician, medication. Surgical costs in abscess state only |
Administrative database of hospital and physician billing data Cost data calculated according to hospital cost–charge ratios |
Usage split by state and treatment | Not given | US dollars, 1999 | 3% |
| Study | Efficacy data | Efficacy data sources | Health outcomes/utility | Health outcome data sources | Discount rate |
|---|---|---|---|---|---|
| Jaisson-Hot et al.7 (2004; non-fistulising) | Derived from published data and expert opinion |
Unclear Some figures from Targan et al. 57 |
QALY QoL figures unclear |
Gregor et al.92 | 5% |
| Clark et al.5 (2003; non-fistulising) |
Response from treatment continued for 80 days (median) in both initial and subsequent treatment. 100% success of initial responders in retreatment. Large numbers of data removed because of confidentiality Scenario 1: uses company’s effectiveness estimates. 19.4% more patients achieve remission (CDAI < 150) over infliximab arms Scenario 2: uses estimates on remission at different dosages to infer the proportion of those achieving mild disease at a 5-mg dosage. 28.7% more patients achieve remission (CDAI < 150) under 5-mg infliximab |
Olmstead County data (usual care) |
Interpolation used to SG utilities Mild disease: 0.86 Drug-refractory disease: 0.74 |
Gregor et al.92 plus Olmstead County data | Not discounted (probably 1 year) |
| Marshall et al.6 (2002; non-fistulising) |
8-week transitions: usual care Drug-refractory from remission: 0.2150 Remission from drug-refractory: 0.0524 Remission from drug-dependent: 0.0540 8-week transitions: infliximab Probability of remaining in clinical response (remission or drug-responsive) over 8 weeks = 0.796 (single dose) 0.937 (maintenance) Remission at CDAI < 150 |
Olmstead County data (usual care) Targan et al. 57 trial (infliximab, initial values 46) Rutgeerts et al. 58 trial (infliximab, after 12 weeks) |
Mild (0.82) used for remission states and mild disease Moderate (0.73) used for drug-responsive/dependent states Severe (0.54) used for drug-refractory and surgery states |
Gregor et al.92 provides standard gamble utility values for three states (mild, moderate and severe) | Not discounted (1 year) |
| Clark et al.5 (2003; fistulising) | Based on Present et al.62 study into time spent with close fistulas in first 12 months after treatment | Present et al.62 study | Based on CDAI and PDAI scores using an unpublished algorithm provided in industry submission | Unpublished data | Not discounted (1 year) |
| Arseneau et al.90 (2001; fistulising) |
Fistula recurrence based on clinical data in first 4 months (18% per month), then 3% in subsequent months Monthly transitions: |
Various studies (named) |
QALY QoL figures from patients: Infliximab:6MP/met: |
Standard gamble utilities from 32 CD patients (17 fistulising, 15 non-fistulising) Descriptions of valued states not given |
3% |
| Study | Cost of anti-TNF-α therapy | Total costs | Total incremental costs | Total outcome | Total incremental outcomes | Cost-effectiveness ratios |
|---|---|---|---|---|---|---|
| Jaisson-Hot et al.7 (2004; non-fistulising) | Not given |
|
B vs A: €48,505.16 C vs A: infliximab (maintenance) €615,790.52 |
Not given | Not given |
B vs A: infliximab (episodic) €63,700.82/QALY C vs A: infliximab (maintenance) vs usual care €784,057.49/QALY |
| Clark et al.5 (2003; non-fistulising) | £1457 per dose | Not given |
vs placebo Single treatment £1457 per patient Episodic treatment (vs placebo) £3861 |
Not given |
QALY vs placebo Single treatment Scenario 1: 0.006 Scenario 2: 0.009 Episodic treatment Scenario 1: 0.043 Scenario 2: 0.067 |
vs placebo Single treatment Scenario 1: £244,756 per QALY Scenario 2: £165,445 per QALY Episodic treatment Scenario 1: £72,261 per QALY Scenario 2: £62,016 per QALY |
| Marshall et al.6 (2002; non-fistulising) | Single dose cost C$5064.11 |
|
B vs A: C$2762 C vs B: C$1037 D vs C: C$7858 |
0.6281 0.6433 0.6455 0.6568 |
B vs A: 0.0152 C vs B: 0.0022 D vs C: 0.00132762 |
B vs A: C$181,201/QALY C vs B: C$480,111/QALY D vs C: C$696,078/QALY |
| Clark et al.5 (2003; fistulising) | Unclear | Not given | Not given | Not given | Not given | Initial treatment vs placebo is £102,000–123,000 per QALY depending on cost offsets |
| Arseneau et al.90 (2001; fistulising) | Single dose cost $2030 for 5 mg/kg dose, 70-kg person |
|
All vs comparator: B vs A: $7109 C vs A: $7218 D vs A: $3770 |
0.76 0.78 0.78 0.77 |
Not given |
All vs comparator: B vs A: $355,450 C vs A: $360,900 D vs A: $377,000 |
| Study | Sensitivity analysis methods | Sensitivity analysis results |
|---|---|---|
| Jaisson-Hot et al.7 (2004; non-fistulising) |
‘Influential’ variables considered, but choice of variables not justified. Tornado diagram used to identify utility weights for ‘post-surgery remission’ and ‘remission not following surgery’ as important Only one-way sensitivity analyses reported |
Surgery and non-infliximab treatment become dominant where post-surgery remission receives utility value 0.92. No dominance found when varying the value for non-surgical remission utility |
| Clark et al.5 (2003; non-fistulising) | One-way sensitivity analyses for utility (to 0.20 from 0.12), duration of response (120 days from 80 days), averted surgery (50% averted surgeries) | None of the one-way sensitivity analyses reduced the ICERs below £40,000 per QALY |
| Marshall et al.6 (2002; non-fistulising) | Probabilistic sensitivity analysis conducted in addition to one-way sensitivity analysis: use of medical/surgical treatment in drug-refractory state (varying 0%–100% from 20% baseline). Surgical admissions varied (0%–100%, 13% baseline). Infliximab cost (0%–100% of baseline cost) |
Rate of surgical admission for drug-refractory CD found to have little effect on ICER Proportion of patients with drug-refractory disease treated medically fell to C$39,000/QALY at 60% for B vs A At 75% of baseline cost, ICERs are: (B vs A) C$98,186; (C vs B) C$329,204; (D vs C) C$522,511/QALY. Usual care dominated by Strategy B (one single dose) where prices reduced to 25% of baseline cost. Usual care favoured for maximum willingness to pay per QALY (l) < C$180,000 One single dose of infliximab (B) favoured for C$180,000 < l < C$430,000 |
| Clark et al.5 (2003; fistulising) | Success rate for retreatment and reclosure of fistulas varied, alongside the level of costs offset due to averted surgery | Even at the most favourable assumptions, the ICER remains above £80,000 per QALY |
| Arseneau et al.90 (2001; fistulising) |
One-way sensitivity analyses for all cost, probability and utility estimates in the model Cost estimates varied by 25%, probability and utility estimates over 95% CI One-way sensitivity analyses as assumptions varying fistula recurrence > 4 months after infliximab usage (0% or 18% recurrence) One-way sensitivity analysis on the effectiveness of infliximab as first- and second- line therapy (0%–100%) Tornado diagram used to identify influential variables (not given) Threshold analysis on the cost of a single dose of infliximab Utility estimates from healthy volunteers |
All ICERs remain above $100,000 per QALY, except where comparator treatment dominates (equal or more effective, lower cost) ICER above $100,000 per QALY even with 100% chance of improvement following either first-line, second-line, or reinfused infliximab Assuming 18% recurrence rate of fistulas after infliximab following month 4 increases ICERs to: $736,400, $409,500 and $412,700 per QALY (Interventions I, II and III vs comparator respectively) Assuming 0% recurrence rates of fistulas after infliximab following month 4 decreases ICERs to: $339,450, $218,133 and $361,200 per QALY (Interventions I, II and III vs comparator respectively) Intervention II) Infliximab (infusions + episodic infliximab reinfusions for treatment failures/relapse) falls beneath $100,000 per QALY where infliximab dose is reduced in price by 75% (to $508/dose) |
| Study | Author conclusions | Industry author affiliation |
|---|---|---|
| Jaisson-Hot et al.7 (2004; non-fistulising) | Infliximab treatment (episodic) could be cost-effective but infliximab treatment (maintenance) may not justify increased cost | None declared |
| Clark et al.5 (2003; non-fistulising) | Re-estimation of the cost-effectiveness using company estimates for the proportion of patients gave a cost/QALY for episodic treatment of £72,000 when using efficacy data from all infliximab arms, and £62,000 when using 5 mg/kg dosing. These findings were relatively insensitive to major changes in key assumptions. The key issue appears to be the duration of benefit from treatment | None declared. Study funded by UK HTA |
| Marshall et al.6 (2002; non-fistulising) | For cost-effectiveness thresholds < C$180,000, usual care was more likely to maximise net benefit than infliximab treatment strategies | None declared. Study funded by CCOHTA |
| Clark et al.5 (2003; fistulising) | The cost per QALY estimates from the industry model were high, at £82,000 even in the most favourable re-treatment assumptions on closure rates | None declared. Study funded by UK HTA |
| Arseneau et al.90 (2001; fistulising) | The ICER for infliximab is > $350,000 per QALY, driven by both the high cost of infliximab and the similar effectiveness of infliximab and 6MP/met treatment strategies | None declared |
| Jaisson-Hot et al. (2004)7 | Marshall et al. (2002)6 | Arseneau et al. (2001)90 | |
|---|---|---|---|
| (1) The research question is stated | Yes | Yes | Yes |
| (2) The economic importance of the research question is stated | Yes | Yes | Yes |
| (3) The viewpoint(s) of the analysis are clearly stated and justified | Yes | Yes | Yes |
| (4) The rationale for choosing the alternative programmes or interventions compared is stated | Yes | Yes | Unclear |
| (5) The alternatives being compared are clearly described | Yes | Yes | Yes |
| (6) The form of economic evaluation used is stated | Yes | Yes | Yes |
| (7) The choice of form of economic evaluation is justified in relation to the questions addressed | Yes | Yes | Yes |
| (8) The source(s) of effectiveness estimates used are stated | Unclear | Yes | Yes |
| (9) Details of the design and results of effectiveness study are given (if based on a single study) | No | NA | NA |
| (10) Details of the method of synthesis or meta-analysis of estimates are given (if based on an overview of a number of effectiveness studies | NA | Yes | Yes |
| (11) The primary outcome measure(s) for the economic evaluation are clearly stated | Yes | Yes | Unclear |
| (12) Methods to value health states and other benefits are stated | Yes | Yes | Yes |
| (13) Details of the subjects from whom valuations were obtained are given | Yes | Yes | Yes |
| (14) Productivity changes (if included) are reported separately | NA | NA | NA |
| (15) The relevance of productivity changes to the study question is discussed | Yes | Yes | Yes |
| (16) Quantities of resources are reported separately from their unit costs | No | Yes | Yes |
| (17) Methods for the estimation of quantities and unit costs are described | No | Yes | Yes |
| (18) Currency and price data are recorded | No | Yes | Yes |
| (19) Details of currency of price adjustments for inflation or currency conversion are given | No | Yes | NA |
| (20) Details of any model used are given | Unclear | Yes | Yes |
| (21) The choice of model used and the key parameters on which it is based are justified | No | Yes | Yes |
| (22) Time horizon of costs and benefits is stated | Yes | Yes | Yes |
| (23) The discount rate(s) are stated | Yes | 1 year | Yes |
| (24) The choice of rate(s) are justified | No | 1 year | No |
| (25) An explanation is given if costs or benefits are not discounted | NA | 1 year | NA |
| (26) Details of statistical tests and confidence intervals are given for stochastic data | No | Yes | Partial |
| (27) The approach to sensitivity analysis is given | Yes | Yes | Yes |
| (28) The choice of variables for sensitivity analysis is justified | Yes | Yes | Yes |
| (29) The ranges over which the variables are varied are stated | No | Yes | Yes |
| (30) Relevant alternatives are compared | Yes | Yes | Yes |
| (31) Incremental analysis is reported | No | Yes | No |
| (32) Major outcomes are presented in a disaggregated as well as aggregated form | No | Yes | No |
| (33) The answer to the study question is given | Yes | Yes | Yes |
| (34) Conclusions follow from the data reported | Yes | Yes | Yes |
| (35) Conclusions are accompanied by the appropriate caveats | Yes | Yes | Yes |
| 18/35 | 34/35 | 26/35 |
| Paper | Included/Excluded | Reason |
|---|---|---|
| Arseneau et al. (2001)90 | Included | |
| Clark et al. (2003)5 | Included | |
| Jaisson-Hot et al. (2004)7 | Included | |
| Marshall et al. (2002)6 | Included | |
| Marshall et al. (2002)91 | Excluded | See Marshall et al. (2002)6 |
| Dubinsky et al. (2005) | Excluded | Comparators not relevant |
| Williams et al. (2000) | Excluded | Comparators not relevant |
| Condino et al. (2005) | Excluded | Comparators not relevant |
| Harrison and Rubensteini (2003) | Excluded | Abstract only |
| Wong (1999) | Excluded | Abstract only |
| Andersson et al. (2003) | Excluded | Not ee |
| Arnott et al. (2001) | Excluded | Not ee |
| Balfour Sartor (2004) | Excluded | Not ee |
| Barkun (2002) | Excluded | Not ee |
| Bassi et al. (2004) | Excluded | Not ee |
| Bernklev et al. (2005) | Excluded | Not ee |
| Bernklev et al. (2006) | Excluded | Not ee |
| Bodger (2002) | Excluded | Not ee |
| Bodger (2005) | Excluded | Not ee |
| Broering et al. (2001a) | Excluded | Not ee |
| Broering et al. (2001b) | Excluded | Not ee |
| Buller (2001) | Excluded | Not ee |
| Cadahia et al. (2004) | Excluded | Not ee |
| Caprilli et al. (2006) | Excluded | Not ee |
| Casellas (2000) | Excluded | Not ee |
| Casellas et al. (2003) | Excluded | Not ee |
| Casellas et al. (2005a) | Excluded | Not ee |
| Casellas et al. (2005b) | Excluded | Not ee |
| Cohen (2002a) | Excluded | Not ee |
| Cohen (2002b) | Excluded | Not ee |
| Cohen (2003) | Excluded | Not ee |
| Cohen (2006) | Excluded | Not ee |
| Cohen et al. (2002) | Excluded | Not ee |
| Colombel et al. (2007) | Excluded | Not ee |
| D’Haens (2002) | Excluded | Not ee |
| Etienney et al. (2004) | Excluded | Not ee |
| Feagan (2001) | Excluded | Not ee |
| Feagan et al. (2003) | Excluded | Not ee |
| Feagan et al. (2005) | Excluded | Not ee |
| Fleurence and Spackman (2006) | Excluded | Not ee |
| Garnett and Yunker (2001) | Excluded | Not ee |
| Ghosh (2003) | Excluded | Not ee |
| Goldfarb et al. (2004) | Excluded | Not ee |
| Gregor et al. (1997) | Excluded | Not ee |
| Hanauer (2005) | Excluded | Not ee |
| Hanauer (2007) | Excluded | Not ee |
| Hilsden (2002) | Excluded | Not ee |
| Hyams (2003) | Excluded | Not ee |
| Inadomi and Terdiman (2006) | Excluded | Not ee |
| Jewel et al. (2005) | Excluded | Not ee |
| Kam (2000) | Excluded | Not ee |
| Kay (2003) | Excluded | Not ee |
| Kennedy et al. (2000) | Excluded | Not ee |
| Kennedy et al. (2004) | Excluded | Not ee |
| Koelewijn et al. (2006) | Excluded | Not ee |
| Leshno (2001) | Excluded | Not ee |
| Lichtenstein (2004) | Excluded | Not ee |
| Lichtenstein (2005) | Excluded | Not ee |
| Lichtenstein et al. (2004) | Excluded | Not ee |
| Lichtenstein et al. (2006) | Excluded | Not ee |
| Luces and Bodger (2006) | Excluded | Not ee |
| Marshall (2002a) | Excluded | Not ee |
| Mealy and Bayes (2005) | Excluded | Not ee |
| Mitton (2002) | Excluded | Not ee |
| Nahar et al. (2003) | Excluded | Not ee |
| Nash and Florin (2005) | Excluded | Not ee |
| Odes et al. (2006) | Excluded | Not ee |
| Ollendorf and Lidsky (2006) | Excluded | Not ee |
| Rubenstein et al. (2002) | Excluded | Not ee |
| Rutgeerts et al. (2004) | Excluded | Not ee |
| Sartor (2004) | Excluded | Not ee |
| Siegel et al. (2006) | Excluded | Not ee |
| Silverstein et al. (1999) | Excluded | Not ee |
| Strong (2001) | Excluded | Not ee |
| Thaler et al. (2005) | Excluded | Not ee |
| van Balkom et al. (2002) | Excluded | Not ee |
| Wicks (2002) | Excluded | Not ee |
| Williams and Meyers (2002) | Excluded | Not ee |
Glossary
There is some difficulty with using the term ‘episodic treatment’ because it has several possible definitions, depending on where it is being used. Possible definitions include the following:
-
Giving treatment when patients experience a disease relapse (if signs and symptoms recur) [see previous National Institute for Health and Clinical Excellence (NICE) guidance1]. The relapse could occur once in several years or much more frequently, such as every 11 weeks [see Rutgeerts et al. ’s2 report of ACCENT (A Crohn’s disease Clinical trial Evaluating infliximab in a New long-term Treatment regimen) I – median time interval between episodic infusions].
-
Treatment given to the comparator arm (i.e. placebo arm) of the ACCENT I2,3 trial (see diagram for the treatment given in Appendix 9). This includes patients who were given placebo and patients who were given infliximab, i.e. crossovers. It also does not distinguish between responders and non-responders.
-
Treatment ‘as needed with infliximab’ (see Rutgeerts et al. ’s2 report of ACCENT I).
-
‘Intermittent therapy’ or ‘induction only/reinitiation therapy’ (see Abbott’s industry submission response to the West Midlands Health Technology Assessment Collaboration (WMHTAC) Technology Assessment Report (TAR), top of page 2). 4
-
Three retreatments for those who initially respond but subsequently relapse (see economic model in previous TAR, p. 34). 5
-
Retreatment with a single dose of infliximab (see Marshall et al. 6 model).
-
Retreatment when patients relapse or do not respond (see Jaisson-Hot et al. 7 model).
There is also some difficulty with the term ‘maintenance treatment’. Generally this is thought to mean keeping patients who have initially responded to treatment in continuing response or remission. However, the following definitions have also been used:
-
Any scheduled maintenance treatment [see most randomised controlled trial (RCT) reports and Jaisson-Hot et al. 7 cost-effectiveness analysis].
-
Any continuing treatment (to distinguish between induction and maintenance therapy) – this treatment can be episodic or scheduled maintenance (see ACCENT I2,3 trial).
-
Any treatment that includes an induction and a maintenance phase (see Schering-Plough response to WMHTAC TAR, p. 3).
In this report, the term episodic treatment has been used in different places in the clinical effectiveness section, particularly with reference to the ACCENT I trial, but does not specify what was meant by the term. In the critical appraisal of the infliximab industry submission, the term ‘infliximab clinical discretion’ has been used for clarity because the precise definition of episodic treatment that was being used in the model could not be determined.
List of abbreviations
- 6MP / met
- 6-mercaptopurine / metronidazole
- ACCENT
- A Crohn’s disease Clinical trial Evaluating infliximab in a New long-term Treatment regimen
- AE
- adverse event
- CD
- Crohn’s disease
- CDAI
- Crohn’s Disease Activity Index
- CDEIS
- Crohn’s Disease Endoscopic Index of Severity
- CEAC
- cost-effectiveness acceptability curve
- CENTRAL
- Cochrane Central Register of Controlled Trials
- CHARM
- Crohn’s Trial of the Fully Human Antibody Adalimumab for Remission Maintenance
- CI
- confidence interval
- CiC
- commercial-in-confidence
- CLASSIC
- CLinical assessment of Adalimumab Safety and efficacy Studied as Induction therapy in Crohn’s disease
- CRP
- C-reactive protein
- DNA
- deoxyribonucleic acid
- EMEA
- European Medicines Agency
- e.o.w.
- every other week
- EQ-5D
- EuroQoL-5 Dimensions
- FDA
- US Food and Drug Administration
- GAIN
- Gauging Adalimumab efficacy in Infliximab Nonresponders
- HBI
- Harvey–Bradshaw Index
- HRQoL
- health-related quality of life
- HTA
- Health Technology Assessment
- IBD
- inflammatory bowel disease
- IBDQ
- Inflammatory Bowel Disease Questionnaire
- ICD
- infliximab clinical discretion
- ICER
- incremental cost-effectiveness ratio
- IMT
- infliximab maintenance treatment
- IND
- induction (in Chapter 4 only when referring to the Markov model)
- IQR
- interquartile range
- ITT
- intention to treat
- i.v.
- intravenous
- LVCF
- last value carried forward
- MNT
- maintenance (in Chapter 4 only when referring to the Markov model)
- NACC
- National Association for Colitis and Crohn’s Disease
- NICE
- National Institute for Health and Clinical Excellence
- PCDAI
- Paediatric Crohn’s Disease Activity Index
- PDAI
- Perianal Disease Activity Index
- PSS
- public social service
- PSSRU
- Personal Social Services Research Unit
- QALY
- quality-adjusted life-year
- QoL
- quality of life
- RCT
- randomised controlled trial
- REACH
- A randomized, multicenter, open-label study to evaluate the safety and efficacy of anti-TNFα chimeric monoclonal antibody (infliximab, Remicade®) in pediatric subjects with moderate-to-severe Crohn’s disease
- Rx
- active treatment (anti-TNF) group
- SC
- standard care
- SD
- standard deviation
- SF-36
- Short Form (36) Health Survey questionnaire
- SPCs
- Summary of Product Characteristics
- TAR
- Technology Assessment Report
- TNF
- tumour necrosis factor
- WMHTAC
- West Midlands Health Technology Assessment Collaboration
All abbreviations that have been used in this report are listed here unless the abbreviation is well known (e.g. NHS), or it has been used only once, or it is a non-standard abbreviation used only in figures/tables/appendices, in which case the abbreviation is defined in the figure legend or in the notes at the end of the table.
NoteThis monograph is based on the Technology Assessment Report produced for NICE. The full report contained a considerable amount of data that were deemed commercial-in-confidence. The full report was used by the Appraisal Committee at NICE in their deliberations. The full report with each piece of commercial-in-confidence data removed and replaced by the statement ‘commercial-in-confidence information removed’ is available on the NICE website: www.nice.org.uk.
The present monograph presents as full a version of the report as is possible while retaining readability, but some sections, sentences, tables and figures have been removed. Readers should bear in mind that the discussion, conclusions and implications for practice and research are based on all the data considered in the original full NICE report.
Notes
Health Technology Assessment programme
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
Prioritisation Group
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Imti Choonara, Professor in Child Health, Academic Division of Child Health, University of Nottingham
Chair – Pharmaceuticals Panel
-
Dr Bob Coates, Consultant Advisor – Disease Prevention Panel
-
Dr Andrew Cook, Consultant Advisor – Intervention Procedures Panel
-
Dr Peter Davidson, Director of NETSCC, Health Technology Assessment
-
Dr Nick Hicks, Consultant Adviser – Diagnostic Technologies and Screening Panel, Consultant Advisor–Psychological and Community Therapies Panel
-
Ms Susan Hird, Consultant Advisor, External Devices and Physical Therapies Panel
-
Professor Sallie Lamb, Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick
Chair – HTA Clinical Evaluation and Trials Board
-
Professor Jonathan Michaels, Professor of Vascular Surgery, Sheffield Vascular Institute, University of Sheffield
Chair – Interventional Procedures Panel
-
Professor Ruairidh Milne, Director – External Relations
-
Dr John Pounsford, Consultant Physician, Directorate of Medical Services, North Bristol NHS Trust
Chair – External Devices and Physical Therapies Panel
-
Dr Vaughan Thomas, Consultant Advisor – Pharmaceuticals Panel, Clinical
Lead – Clinical Evaluation Trials Prioritisation Group
-
Professor Margaret Thorogood, Professor of Epidemiology, Health Sciences Research Institute, University of Warwick
Chair – Disease Prevention Panel
-
Professor Lindsay Turnbull, Professor of Radiology, Centre for the MR Investigations, University of Hull
Chair – Diagnostic Technologies and Screening Panel
-
Professor Scott Weich, Professor of Psychiatry, Health Sciences Research Institute, University of Warwick
Chair – Psychological and Community Therapies Panel
-
Professor Hywel Williams, Director of Nottingham Clinical Trials Unit, Centre of Evidence-Based Dermatology, University of Nottingham
Chair – HTA Commissioning Board
Deputy HTA Programme Director
HTA Commissioning Board
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
-
Professor of General Practice, Department of Primary Health Care, University of Oxford Programme Director,
-
Professor of Clinical Pharmacology, Director, NIHR HTA programme, University of Liverpool
-
Professor Ann Ashburn, Professor of Rehabilitation and Head of Research, Southampton General Hospital
-
Professor Deborah Ashby, Professor of Medical Statistics and Clinical Trials, Queen Mary, Department of Epidemiology and Public Health, Imperial College London
-
Professor Peter Brocklehurst, Director, National Perinatal Epidemiology Unit, University of Oxford
-
Professor John Cairns, Professor of Health Economics, London School of Hygiene and Tropical Medicine
-
Professor Peter Croft, Director of Primary Care Sciences Research Centre, Keele University
-
Professor Jenny Donovan, Professor of Social Medicine, University of Bristol
-
Professor Jonathan Green, Professor and Acting Head of Department, Child and Adolescent Psychiatry, University of Manchester Medical School
-
Professor John W Gregory, Professor in Paediatric Endocrinology, Department of Child Health, Wales School of Medicine, Cardiff University
-
Professor Steve Halligan, Professor of Gastrointestinal Radiology, University College Hospital, London
-
Professor Freddie Hamdy, Professor of Urology, Head of Nuffield Department of Surgery, University of Oxford
-
Professor Allan House, Professor of Liaison Psychiatry, University of Leeds
-
Dr Martin J Landray, Reader in Epidemiology, Honorary Consultant Physician, Clinical Trial Service Unit, University of Oxford
-
Professor Stephen Morris, Professor of Health Economics, University College London, Research Department of Epidemiology and Public Health, University College London
-
Professor E Andrea Nelson, Professor of Wound Healing and Director of Research, School of Healthcare, University of Leeds
-
Professor John David Norris, Chair in Clinical Trials and Biostatistics, Robertson Centre for Biostatistics, University of Glasgow
-
Dr Rafael Perera, Lecturer in Medical Statisitics, Department of Primary Health Care, University of Oxford
-
Professor James Raftery, Chair of NETSCC and Director of the Wessex Institute, University of Southampton
-
Professor Barney Reeves, Professorial Research Fellow in Health Services Research, Department of Clinical Science, University of Bristol
-
Professor Martin Underwood, Warwick Medical School, University of Warwick
-
Professor Marion Walker, Professor in Stroke Rehabilitation, Associate Director UK Stroke Research Network, University of Nottingham
-
Dr Duncan Young, Senior Clinical Lecturer and Consultant, Nuffield Department of Anaesthetics, University of Oxford
-
Professor Stephen Morris, Professor of Health Economics, University College London, Research Department of Epidemiology and Public Health, University College London
-
Professor E Andrea Nelson, Professor of Wound Healing and Director of Research, School of Healthcare, University of Leeds
-
Professor John David Norris Chair in Clinical Trials and Biostatistics, Robertson Centre for Biostatistics, University of Glasgow
-
Dr Rafael Perera, Lecturer in Medical Statisitics, Department of Primary Health Care, University of Oxford
-
Professor James Raftery, Chair of NETSCC and Director of the Wessex Institute, University of Southampton
-
Professor Barney Reeves, Professorial Research Fellow in Health Services Research, Department of Clinical Science, University of Bristol
-
Professor Martin Underwood, Warwick Medical School, University of Warwick
-
Professor Marion Walker, Professor in Stroke Rehabilitation, Associate Director UK Stroke Research Network, University of Nottingham
-
Dr Duncan Young, Senior Clinical Lecturer and Consultant, Nuffield Department of Anaesthetics, University of Oxford
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
HTA Clinical Evaluation and Trials Board
-
Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick and Professor of Rehabilitation, Nuffield Department of Orthopaedic, Rheumatology and Musculoskeletal Sciences, University of Oxford
-
Professor of the Psychology of Health Care, Leeds Institute of Health Sciences, University of Leeds
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Keith Abrams, Professor of Medical Statistics, Department of Health Sciences, University of Leicester
-
Professor Martin Bland, Professor of Health Statistics, Department of Health Sciences, University of York
-
Professor Jane Blazeby, Professor of Surgery and Consultant Upper GI Surgeon, Department of Social Medicine, University of Bristol
-
Professor Julia M Brown, Director, Clinical Trials Research Unit, University of Leeds
-
Professor Alistair Burns, Professor of Old Age Psychiatry, Psychiatry Research Group, School of Community-Based Medicine, The University of Manchester & National Clinical Director for Dementia, Department of Health
-
Dr Jennifer Burr, Director, Centre for Healthcare Randomised trials (CHART), University of Aberdeen
-
Professor Linda Davies, Professor of Health Economics, Health Sciences Research Group, University of Manchester
-
Professor Simon Gilbody, Prof of Psych Medicine and Health Services Research, Department of Health Sciences, University of York
-
Professor Steven Goodacre, Professor and Consultant in Emergency Medicine, School of Health and Related Research, University of Sheffield
-
Professor Dyfrig Hughes, Professor of Pharmacoeconomics, Centre for Economics and Policy in Health, Institute of Medical and Social Care Research, Bangor University
-
Professor Paul Jones, Professor of Respiratory Medicine, Department of Cardiac and Vascular Science, St George‘s Hospital Medical School, University of London
-
Professor Khalid Khan, Professor of Women’s Health and Clinical Epidemiology, Barts and the London School of Medicine, Queen Mary, University of London
-
Professor Richard J McManus, Professor of Primary Care Cardiovascular Research, Primary Care Clinical Sciences Building, University of Birmingham
-
Professor Helen Rodgers, Professor of Stroke Care, Institute for Ageing and Health, Newcastle University
-
Professor Ken Stein, Professor of Public Health, Peninsula Technology Assessment Group, Peninsula College of Medicine and Dentistry, Universities of Exeter and Plymouth
-
Professor Jonathan Sterne, Professor of Medical Statistics and Epidemiology, Department of Social Medicine, University of Bristol
-
Mr Andy Vail, Senior Lecturer, Health Sciences Research Group, University of Manchester
-
Professor Clare Wilkinson, Professor of General Practice and Director of Research North Wales Clinical School, Department of Primary Care and Public Health, Cardiff University
-
Dr Ian B Wilkinson, Senior Lecturer and Honorary Consultant, Clinical Pharmacology Unit, Department of Medicine, University of Cambridge
-
Ms Kate Law, Director of Clinical Trials, Cancer Research UK
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
Diagnostic Technologies and Screening Panel
-
Scientific Director of the Centre for Magnetic Resonance Investigations and YCR Professor of Radiology, Hull Royal Infirmary
-
Professor Judith E Adams, Consultant Radiologist, Manchester Royal Infirmary, Central Manchester & Manchester Children’s University Hospitals NHS Trust, and Professor of Diagnostic Radiology, University of Manchester
-
Mr Angus S Arunkalaivanan, Honorary Senior Lecturer, University of Birmingham and Consultant Urogynaecologist and Obstetrician, City Hospital, Birmingham
-
Dr Stephanie Dancer, Consultant Microbiologist, Hairmyres Hospital, East Kilbride
-
Dr Diane Eccles, Professor of Cancer Genetics, Wessex Clinical Genetics Service, Princess Anne Hospital
-
Dr Trevor Friedman, Consultant Liason Psychiatrist, Brandon Unit, Leicester General Hospital
-
Dr Ron Gray, Consultant, National Perinatal Epidemiology Unit, Institute of Health Sciences, University of Oxford
-
Professor Paul D Griffiths, Professor of Radiology, Academic Unit of Radiology, University of Sheffield
-
Mr Martin Hooper, Public contributor
-
Professor Anthony Robert Kendrick, Associate Dean for Clinical Research and Professor of Primary Medical Care, University of Southampton
-
Dr Anne Mackie, Director of Programmes, UK National Screening Committee, London
-
Mr David Mathew, Public contributor
-
Dr Michael Millar, Consultant Senior Lecturer in Microbiology, Department of Pathology & Microbiology, Barts and The London NHS Trust, Royal London Hospital
-
Mrs Una Rennard, Public contributor
-
Dr Stuart Smellie, Consultant in Clinical Pathology, Bishop Auckland General Hospital
-
Ms Jane Smith, Consultant Ultrasound Practitioner, Leeds Teaching Hospital NHS Trust, Leeds
-
Dr Allison Streetly, Programme Director, NHS Sickle Cell and Thalassaemia Screening Programme, King’s College School of Medicine
-
Dr Alan J Williams, Consultant Physician, General and Respiratory Medicine, The Royal Bournemouth Hospital
-
Dr Tim Elliott, Team Leader, Cancer Screening, Department of Health
-
Dr Catherine Moody, Programme Manager, Medical Research Council
-
Professor Julietta Patrick, Director, NHS Cancer Screening Programme, Sheffield
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Disease Prevention Panel
-
Professor of Epidemiology, University of Warwick Medical School, Coventry
-
Dr Robert Cook, Clinical Programmes Director, Bazian Ltd, London
-
Dr Colin Greaves, Senior Research Fellow, Peninsula Medical School (Primary Care)
-
Mr Michael Head, Public contributor
-
Professor Cathy Jackson, Professor of Primary Care Medicine, Bute Medical School, University of St Andrews
-
Dr Russell Jago, Senior Lecturer in Exercise, Nutrition and Health, Centre for Sport, Exercise and Health, University of Bristol
-
Dr Julie Mytton, Consultant in Child Public Health, NHS Bristol
-
Professor Irwin Nazareth, Professor of Primary Care and Director, Department of Primary Care and Population Sciences, University College London
-
Dr Richard Richards, Assistant Director of Public Health, Derbyshire Country Primary Care Trust
-
Professor Ian Roberts, Professor of Epidemiology and Public Health, London School of Hygiene & Tropical Medicine
-
Dr Kenneth Robertson, Consultant Paediatrician, Royal Hospital for Sick Children, Glasgow
-
Dr Catherine Swann, Associate Director, Centre for Public Health Excellence, NICE
-
Professor Carol Tannahill, Glasgow Centre for Population Health
-
Mrs Jean Thurston, Public contributor
-
Professor David Weller, Head, School of Clinical Science and Community Health, University of Edinburgh
-
Ms Christine McGuire, Research & Development, Department of Health
-
Dr Kay Pattison Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
External Devices and Physical Therapies Panel
-
Consultant Physician North Bristol NHS Trust
-
Reader in Wound Healing and Director of Research, University of Leeds
-
Professor Bipin Bhakta, Charterhouse Professor in Rehabilitation Medicine, University of Leeds
-
Mrs Penny Calder, Public contributor
-
Dr Dawn Carnes, Senior Research Fellow, Barts and the London School of Medicine and Dentistry
-
Dr Emma Clark, Clinician Scientist Fellow & Cons. Rheumatologist, University of Bristol
-
Mrs Anthea De Barton-Watson, Public contributor
-
Professor Nadine Foster, Professor of Musculoskeletal Health in Primary Care Arthritis Research, Keele University
-
Dr Shaheen Hamdy, Clinical Senior Lecturer and Consultant Physician, University of Manchester
-
Professor Christine Norton, Professor of Clinical Nursing Innovation, Bucks New University and Imperial College Healthcare NHS Trust
-
Dr Lorraine Pinnigton, Associate Professor in Rehabilitation, University of Nottingham
-
Dr Kate Radford, Senior Lecturer (Research), University of Central Lancashire
-
Mr Jim Reece, Public contributor
-
Professor Maria Stokes, Professor of Neuromusculoskeletal Rehabilitation, University of Southampton
-
Dr Pippa Tyrrell, Senior Lecturer/Consultant, Salford Royal Foundation Hospitals’ Trust and University of Manchester
-
Dr Sarah Tyson, Senior Research Fellow & Associate Head of School, University of Salford
-
Dr Nefyn Williams, Clinical Senior Lecturer, Cardiff University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Interventional Procedures Panel
-
Professor of Vascular Surgery, University of Sheffield
-
Consultant Colorectal Surgeon, Bristol Royal Infirmary
-
Mrs Isabel Boyer, Public contributor
-
Mr David P Britt, Public contributor
-
Mr Sankaran ChandraSekharan, Consultant Surgeon, Breast Surgery, Colchester Hospital University NHS Foundation Trust
-
Professor Nicholas Clarke, Consultant Orthopaedic Surgeon, Southampton University Hospitals NHS Trust
-
Ms Leonie Cooke, Public contributor
-
Mr Seamus Eckford, Consultant in Obstetrics & Gynaecology, North Devon District Hospital
-
Professor David Taggart, Consultant Cardiothoracic Surgeon, John Radcliffe Hospital
-
Professor Sam Eljamel, Consultant Neurosurgeon, Ninewells Hospital and Medical School, Dundee
-
Dr Adele Fielding, Senior Lecturer and Honorary Consultant in Haematology, University College London Medical School
-
Dr Matthew Hatton, Consultant in Clinical Oncology, Sheffield Teaching Hospital Foundation Trust
-
Dr John Holden, General Practitioner, Garswood Surgery, Wigan
-
Professor Nicholas James, Professor of Clinical Oncology, School of Cancer Sciences, University of Birmingham
-
Dr Fiona Lecky, Senior Lecturer/Honorary Consultant in Emergency Medicine, University of Manchester/Salford Royal Hospitals NHS Foundation Trust
-
Dr Nadim Malik, Consultant Cardiologist/ Honorary Lecturer, University of Manchester
-
Mr Hisham Mehanna, Consultant & Honorary Associate Professor, University Hospitals Coventry & Warwickshire NHS Trust
-
Dr Jane Montgomery, Consultant in Anaesthetics and Critical Care, South Devon Healthcare NHS Foundation Trust
-
Professor Jon Moss, Consultant Interventional Radiologist, North Glasgow Hospitals University NHS Trust
-
Dr Simon Padley, Consultant Radiologist, Chelsea & Westminster Hospital
-
Dr Ashish Paul, Medical Director, Bedfordshire PCT
-
Dr Sarah Purdy, Consultant Senior Lecturer, University of Bristol
-
Professor Yit Chiun Yang, Consultant Ophthalmologist, Royal Wolverhampton Hospitals NHS Trust
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Pharmaceuticals Panel
-
Professor in Child Health, University of Nottingham
-
Senior Lecturer in Clinical Pharmacology, University of East Anglia
-
Dr Martin Ashton-Key, Medical Advisor, National Commissioning Group, NHS London
-
Mr John Chapman, Public contributor
-
Dr Peter Elton, Director of Public Health, Bury Primary Care Trust
-
Dr Peter Elton, Director of Public Health, Bury Primary Care Trust
-
Dr Ben Goldacre, Research Fellow, Division of Psychological Medicine and Psychiatry, King’s College London
-
Dr James Gray, Consultant Microbiologist, Department of Microbiology, Birmingham Children’s Hospital NHS Foundation Trust
-
Ms Kylie Gyertson, Oncology and Haematology Clinical Trials Manager, Guy’s and St Thomas’ NHS Foundation Trust London
-
Dr Jurjees Hasan, Consultant in Medical Oncology, The Christie, Manchester
-
Dr Carl Heneghan Deputy Director Centre for Evidence-Based Medicine and Clinical Lecturer, Department of Primary Health Care, University of Oxford
-
Dr Dyfrig Hughes, Reader in Pharmacoeconomics and Deputy Director, Centre for Economics and Policy in Health, IMSCaR, Bangor University
-
Dr Maria Kouimtzi, Pharmacy and Informatics Director, Global Clinical Solutions, Wiley-Blackwell
-
Professor Femi Oyebode, Consultant Psychiatrist and Head of Department, University of Birmingham
-
Dr Andrew Prentice, Senior Lecturer and Consultant Obstetrician and Gynaecologist, The Rosie Hospital, University of Cambridge
-
Ms Amanda Roberts, Public contributor
-
Dr Martin Shelly, General Practitioner, Silver Lane Surgery, Leeds
-
Dr Gillian Shepherd, Director, Health and Clinical Excellence, Merck Serono Ltd
-
Mrs Katrina Simister, Assistant Director New Medicines, National Prescribing Centre, Liverpool
-
Professor Donald Singer Professor of Clinical Pharmacology and Therapeutics, Clinical Sciences Research Institute, CSB, University of Warwick Medical School
-
Mr David Symes, Public contributor
-
Dr Arnold Zermansky, General Practitioner, Senior Research Fellow, Pharmacy Practice and Medicines Management Group, Leeds University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Mr Simon Reeve, Head of Clinical and Cost-Effectiveness, Medicines, Pharmacy and Industry Group, Department of Health
-
Dr Heike Weber, Programme Manager, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Psychological and Community Therapies Panel
-
Professor of Psychiatry, University of Warwick, Coventry
-
Consultant & University Lecturer in Psychiatry, University of Cambridge
-
Professor Jane Barlow, Professor of Public Health in the Early Years, Health Sciences Research Institute, Warwick Medical School
-
Dr Sabyasachi Bhaumik, Consultant Psychiatrist, Leicestershire Partnership NHS Trust
-
Mrs Val Carlill, Public contributor
-
Dr Steve Cunningham, Consultant Respiratory Paediatrician, Lothian Health Board
-
Dr Anne Hesketh, Senior Clinical Lecturer in Speech and Language Therapy, University of Manchester
-
Dr Peter Langdon, Senior Clinical Lecturer, School of Medicine, Health Policy and Practice, University of East Anglia
-
Dr Yann Lefeuvre, GP Partner, Burrage Road Surgery, London
-
Dr Jeremy J Murphy, Consultant Physician and Cardiologist, County Durham and Darlington Foundation Trust
-
Dr Richard Neal, Clinical Senior Lecturer in General Practice, Cardiff University
-
Mr John Needham, Public contributor
-
Ms Mary Nettle, Mental Health User Consultant
-
Professor John Potter, Professor of Ageing and Stroke Medicine, University of East Anglia
-
Dr Greta Rait, Senior Clinical Lecturer and General Practitioner, University College London
-
Dr Paul Ramchandani, Senior Research Fellow/Cons. Child Psychiatrist, University of Oxford
-
Dr Karen Roberts, Nurse/Consultant, Dunston Hill Hospital, Tyne and Wear
-
Dr Karim Saad, Consultant in Old Age Psychiatry, Coventry and Warwickshire Partnership Trust
-
Dr Lesley Stockton, Lecturer, School of Health Sciences, University of Liverpool
-
Dr Simon Wright, GP Partner, Walkden Medical Centre, Manchester
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Expert Advisory Network
-
Professor Douglas Altman, Professor of Statistics in Medicine, Centre for Statistics in Medicine, University of Oxford
-
Professor John Bond, Professor of Social Gerontology & Health Services Research, University of Newcastle upon Tyne
-
Professor Andrew Bradbury, Professor of Vascular Surgery, Solihull Hospital, Birmingham
-
Mr Shaun Brogan, Chief Executive, Ridgeway Primary Care Group, Aylesbury
-
Mrs Stella Burnside OBE, Chief Executive, Regulation and Improvement Authority, Belfast
-
Ms Tracy Bury, Project Manager, World Confederation of Physical Therapy, London
-
Professor Iain T Cameron, Professor of Obstetrics and Gynaecology and Head of the School of Medicine, University of Southampton
-
Professor Bruce Campbell, Consultant Vascular & General Surgeon, Royal Devon & Exeter Hospital, Wonford
-
Dr Christine Clark, Medical Writer and Consultant Pharmacist, Rossendale
-
Professor Collette Clifford, Professor of Nursing and Head of Research, The Medical School, University of Birmingham
-
Professor Barry Cookson, Director, Laboratory of Hospital Infection, Public Health Laboratory Service, London
-
Dr Carl Counsell, Clinical Senior Lecturer in Neurology, University of Aberdeen
-
Professor Howard Cuckle, Professor of Reproductive Epidemiology, Department of Paediatrics, Obstetrics & Gynaecology, University of Leeds
-
Professor Carol Dezateux, Professor of Paediatric Epidemiology, Institute of Child Health, London
-
Mr John Dunning, Consultant Cardiothoracic Surgeon, Papworth Hospital NHS Trust, Cambridge
-
Mr Jonothan Earnshaw, Consultant Vascular Surgeon, Gloucestershire Royal Hospital, Gloucester
-
Professor Martin Eccles, Professor of Clinical Effectiveness, Centre for Health Services Research, University of Newcastle upon Tyne
-
Professor Pam Enderby, Dean of Faculty of Medicine, Institute of General Practice and Primary Care, University of Sheffield
-
Professor Gene Feder, Professor of Primary Care Research & Development, Centre for Health Sciences, Barts and The London School of Medicine and Dentistry
-
Mr Leonard R Fenwick, Chief Executive, Freeman Hospital, Newcastle upon Tyne
-
Mrs Gillian Fletcher, Antenatal Teacher and Tutor and President, National Childbirth Trust, Henfield
-
Professor Jayne Franklyn, Professor of Medicine, University of Birmingham
-
Mr Tam Fry, Honorary Chairman, Child Growth Foundation, London
-
Professor Fiona Gilbert, Consultant Radiologist and NCRN Member, University of Aberdeen
-
Professor Paul Gregg, Professor of Orthopaedic Surgical Science, South Tees Hospital NHS Trust
-
Bec Hanley, Co-director, TwoCan Associates, West Sussex
-
Dr Maryann L Hardy, Senior Lecturer, University of Bradford
-
Mrs Sharon Hart, Healthcare Management Consultant, Reading
-
Professor Robert E Hawkins, CRC Professor and Director of Medical Oncology, Christie CRC Research Centre, Christie Hospital NHS Trust, Manchester
-
Professor Richard Hobbs, Head of Department of Primary Care & General Practice, University of Birmingham
-
Professor Alan Horwich, Dean and Section Chairman, The Institute of Cancer Research, London
-
Professor Allen Hutchinson, Director of Public Health and Deputy Dean of ScHARR, University of Sheffield
-
Professor Peter Jones, Professor of Psychiatry, University of Cambridge, Cambridge
-
Professor Stan Kaye, Cancer Research UK Professor of Medical Oncology, Royal Marsden Hospital and Institute of Cancer Research, Surrey
-
Dr Duncan Keeley, General Practitioner (Dr Burch & Ptnrs), The Health Centre, Thame
-
Dr Donna Lamping, Research Degrees Programme Director and Reader in Psychology, Health Services Research Unit, London School of Hygiene and Tropical Medicine, London
-
Professor James Lindesay, Professor of Psychiatry for the Elderly, University of Leicester
-
Professor Julian Little, Professor of Human Genome Epidemiology, University of Ottawa
-
Professor Alistaire McGuire, Professor of Health Economics, London School of Economics
-
Professor Neill McIntosh, Edward Clark Professor of Child Life and Health, University of Edinburgh
-
Professor Rajan Madhok, Consultant in Public Health, South Manchester Primary Care Trust
-
Professor Sir Alexander Markham, Director, Molecular Medicine Unit, St James’s University Hospital, Leeds
-
Dr Peter Moore, Freelance Science Writer, Ashtead
-
Dr Andrew Mortimore, Public Health Director, Southampton City Primary Care Trust
-
Dr Sue Moss, Associate Director, Cancer Screening Evaluation Unit, Institute of Cancer Research, Sutton
-
Professor Miranda Mugford, Professor of Health Economics and Group Co-ordinator, University of East Anglia
-
Professor Jim Neilson, Head of School of Reproductive & Developmental Medicine and Professor of Obstetrics and Gynaecology, University of Liverpool
-
Mrs Julietta Patnick, Director, NHS Cancer Screening Programmes, Sheffield
-
Professor Robert Peveler, Professor of Liaison Psychiatry, Royal South Hants Hospital, Southampton
-
Professor Chris Price, Director of Clinical Research, Bayer Diagnostics Europe, Stoke Poges
-
Professor William Rosenberg, Professor of Hepatology and Consultant Physician, University of Southampton
-
Professor Peter Sandercock, Professor of Medical Neurology, Department of Clinical Neurosciences, University of Edinburgh
-
Dr Philip Shackley, Senior Lecturer in Health Economics, Sheffield Vascular Institute, University of Sheffield
-
Dr Eamonn Sheridan, Consultant in Clinical Genetics, St James’s University Hospital, Leeds
-
Dr Margaret Somerville, Director of Public Health Learning, Peninsula Medical School, University of Plymouth
-
Professor Sarah Stewart-Brown, Professor of Public Health, Division of Health in the Community, University of Warwick, Coventry
-
Dr Nick Summerton, GP Appraiser and Codirector, Research Network, Yorkshire Clinical Consultant, Primary Care and Public Health, University of Oxford
-
Professor Ala Szczepura, Professor of Health Service Research, Centre for Health Services Studies, University of Warwick, Coventry
-
Dr Ross Taylor, Senior Lecturer, University of Aberdeen
-
Dr Richard Tiner, Medical Director, Medical Department, Association of the British Pharmaceutical Industry
-
Mrs Joan Webster, Consumer Member, Southern Derbyshire Community Health Council
-
Professor Martin Whittle, Clinical Co-director, National Co-ordinating Centre for Women’s and Children’s Health, Lymington