
Notes
Article history
The research reported in this issue of the journal was commissioned and funded by the HTA programme on behalf of NICE as project number 08/98/01. The protocol was agreed in July 2009. The assessment report began editorial review in December 2009 and was accepted for publication in July 2010. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the referees for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
none
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2011. This work was produced by Hartwell et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This journal is a member of and subscribes to the principles of the Committee on Publication Ethics (COPE) (http://www.publicationethics.org/). This journal may be freely reproduced for the purposes of private research and study and may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NETSCC, Health Technology Assessment, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2011 Queen’s Printer and Controller of HMSO
Chapter 1 Background
Description of underlying health problem
Hepatitis C is a slowly progressing infectious disease of the liver arising from the blood-borne hepatitis C virus (HCV). First identified in 1989, HCV belongs to the Flaviviridae family of viruses. It is a ribonucleic acid (RNA) virus, of which there are six genetic variations, known as genotypes (e.g. 1, 2, 3, etc.), the prevalence of which varies considerably between countries. 1,2 In England and Wales, the most prevalent genotypes are 1 and 3, representing more than 90% of all diagnosed infections. 3 Genotype 3a remains the most common, with a prevalence of 39%, followed by genotype 1a (22%). 3 Response to treatment is strongly influenced by HCV genotype (see Current service provision and Description of technology under assessment).
There are two main phases of infection: acute and chronic. Acute HCV refers to the period immediately after HCV infection, whereas chronic HCV (the focus of this report) is defined as infection persisting for > 6 months. Of those exposed to HCV, approximately 20% will clear the virus spontaneously, although the remaining 80% will go on to develop chronic infection. Chronic HCV is categorised as mild, moderate or severe according to the extent of liver damage, based on both the level of fibrosis (scarring) that has occurred in the liver as well as the degree of necroinflammation (inflammation and destruction of liver tissue) (see Disease progression and prognosis). Symptoms in people with chronic HCV are typically mild and non-specific, and include fatigue, flu-like symptoms, anorexia, depression, sleep disturbance, cognitive impairment, right upper quadrant pain, itching and nausea. 4,5 Although the symptoms are mild in some people, in others they can cause a significant decrease in quality of life (QoL) irrespective of the degree of liver damage. 6 Symptoms and signs of chronic HCV-related liver damage may occur later in the disease when scarring of the liver has progressed.
Aetiology
Hepatitis C virus is transmitted parenterally (i.e. via routes other than the digestive tract) and is acquired primarily through exposure to contaminated blood. The most common source of HCV transmission in the UK is through the sharing of injecting paraphernalia during illicit intravenous drug use, accounting for around 90% of cases. 3 Other less common sources of infection include mother–baby transmission, occupational exposure (e.g. via needlestick injury), tattooing and body piercing. Before the introduction of blood screening in 1991, it was also spread through the use of contaminated blood products or organ transplantation. In some resource-poor countries it is thought that infections may occur through the use of unsterilised needles in health-care settings. The risk of sexual transmission has been thought, traditionally, to be low. For example, the Health Protection Agency (HPA) estimates that only 1.4% of infections identified through laboratory reports between 1996 and 2007 were attributed to sexual exposure. 3 However, increasing numbers of acute infections in human immunodeficiency virus (HIV)-positive men who have sex with men (MSM) suggests potential for transmission associated with high-risk sexual practices probably involving blood (see below). 7
Epidemiology
Prevalence
The estimated global prevalence of chronic HCV is around 2%–3%, corresponding to about 130–170 million people. 1,8 In England and Wales, the HPA3 estimates that, based on statistical model data for the year 2003, around 191,000 [95% credible interval (CrI) 124.00 to 311.00] people aged 15–59 years are HCV antibody positive, with 142,000 people chronically infected, a prevalence of 0.44% (95% CrI 0.29% to 0.72%) in this age group.
Prevalence estimates vary geographically in England and Wales, with highest numbers of laboratory reports (from public health and UK NHS laboratories in England and Wales under a voluntary surveillance scheme) returned in the north-west, followed by London and the south-east of England. 3
The prevalence of chronic HCV also varies according to different population groups. For example, HCV is more common in men and in the 25–44 years age group. Estimates of the number of current injecting drug users (IDUs) in England vary between 100,000 and 217,000, and it is estimated that around 40% of IDUs are infected with chronic HCV, based on the Unlinked Anonymous Prevalence Monitoring Programme’s Survey of Injecting Drug Users in 2006. 9 There are limited data on prevalence in minority ethnic populations. However, it is thought that the prevalence of HCV is higher in migrants who will have acquired the infection while overseas, notably Pakistan. 3
Evidence suggests varying rates of HCV in people with HIV infection. For example, Mohsen and colleagues10 reviewed the international literature on the epidemiology of HCV/HIV co-infected patients. They included 12 HCV seroprevalence studies carried out in people infected with HIV-1 in Europe and the USA. HCV prevalence ranged from 7% to 57%, largely influenced by risk factors in the study populations. Prevalence was highest in people with a history of injecting drug use (> 80%). It has been suggested that up to 10% of all HCV-infected people are co-infected with HIV. 11
Prevalence is difficult to estimate because symptoms of HCV are frequently absent or non-specific and thus people can remain undiagnosed for many years. Between 1992 and 2007 there were 62,000 laboratory-confirmed diagnoses of HCV in England, and 3688 in Wales (from 1996). 3 It is thought that a proportion of those who are undiagnosed are ex-IDUs who used drugs transiently in the past. Sentinel surveillance by the HPA suggests that the number of people diagnosed with HCV in all settings is increasing, which may, in part, reflect awareness-raising campaigns to encourage uptake of testing. 3
Incidence
The incidence of chronic HCV is likely to be driven by two main sources – newly acquired infections in current UK residents (largely IDUs) and inward migration of chronically infected individuals from other countries. Up-to-date estimates of overall incidence are not yet available but recent studies in IDUs suggest that 3%–42% of susceptible injectors become infected each year. 3 The HPA reports that the number of laboratory-confirmed diagnoses of HCV in England and Wales in 2007 was 7540, representing a 12% increase from 2006. 3 This does not, however, necessarily represent an increase in rates of incidence but may be attributed to testing rates.
Recent rises in HCV infection in HIV-positive MSM have generated increased interest in the role of sexual transmission of HCV. HCV RNA can be detected in the semen of HCV-infected men, with higher levels in HIV-positive men, suggesting the possibility of transmission during certain sexual practices. Increases in cases of acute co-infection in HIV-positive MSM in urban centres in Europe and the USA have been reported in recent years. 12 A study of genitourinary medicine (GUM) clinics in London and the south-east of England found a 20% average annual increase in the number of HIV-positive MSM diagnosed with HCV between January 2002 and June 2006. 7 The prevalence of HCV in HIV-positive MSM is estimated to be between 4% and 11.5%. 12
Disease progression and prognosis
Chronic HCV infection is associated with progression to liver failure in some, but not all, people. Progressive liver disease is characterised by inflammation of the liver, which leads to gradual fibrosis, which, in its severe form, produces cirrhosis. Cirrhosis can progress from a compensated state (where the liver is still functioning despite the fibrosis) to a decompensated state (where the functioning of the liver is seriously impaired). Decompensation is characterised by complications such as ascites (large accumulations of fluid in the abdominal cavity), variceal bleeding (enlarged and bleeding veins around the oesophagus) and hepatic encephalopathy (neuropsychiatric abnormalities, such as cognitive impairment associated with liver dysfunction). There are a number of commonly used systems for classifying the severity of HCV-related liver disease from biopsy samples. Some share common characteristics and are derived from the same systems. 13 Three commonly cited systems are the Knodell histological activity index (HAI),14 the Ishak revised HAI15 and the METAVIR system. 16 The Ishak system,15 for example, classifies mild HCV as a fibrosis score of ≤ 2 and a necro-inflammation score of between 1 and 8, moderate HCV as a fibrosis score of 3–5 and a necro-inflammation score of 0–18 (moderate/severe), and severe HCV as a fibrosis score of 6 (cirrhosis). If the fibrosis score reaches ‘6’ the patient is classified as having severe HCV-related liver damage, irrespective of the necro-inflammation score (see our previous technology assessment report17 on antiviral treatment for mild HCV for further detail on liver biopsy classification systems).
Cirrhosis can develop rapidly, within 1–2 years of exposure (although this is rare), but more usually develops slowly over two to three decades. A recent Markov modelling study18 of three different observational cohorts in the UK estimated that between 6% and 23% of people will progress to cirrhosis after 20 years of infection. The estimates were highly sensitive to the type of cohort used, with lower estimates from the HCV National Register lookback cohort, comprising individuals identified from blood screening and donor surveillance schemes, and highest estimates from a London-based tertiary referral centre in which patients underwent a biopsy. Estimates of progression to cirrhosis from retrospective studies are higher, with between 17% and 55% of patients progressing between 10 and 30 years following infection. 19 It is estimated that 6%–10% of cirrhotic patients will progress to decompensated cirrhosis. 19
A recent modelling study estimated that in England the number of HCV-infected people living with compensated cirrhosis will rise from 3705 (95% CrI 2820 to 4975) in 2005 to 7550 (95% CrI 5120 to 116,400) in 2015. 20
Patients with HCV-related cirrhosis are at risk of developing hepatocellular carcinoma (HCC) with an annual incidence of 1%–4%. 21 Some patients with decompensated cirrhosis or HCC may require liver transplantation. In 2007, 482 liver transplants were conducted in England, of which 13% (n = 64) were classified as first liver transplants with post-HCV cirrhosis at registration/HCV positive at registration or transplant. 3 However, demand for liver donors remains high and not all patients will be considered for transplantation. The number of people with decompensated cirrhosis and/or HCC is also estimated to rise from 1150 (95% CrI 1055 to 1250) in 2005 to 2540 (95% CrI 2035 to 3310) in 2015. 20
Risk factors associated with rapid disease progression include male gender, excessive alcohol consumption and age at infection. 19 For example, Poynard and colleagues22 studied a cohort of 2313 untreated patients and reported that increasing age at infection was independently associated with disease progression. Two per cent of those infected before the age of 20 years developed cirrhosis over a 20-year period compared with 6% of those infected between the ages of 31 and 40 years, 37% infected between 41 and 50 years, and 63% infected after the age of 50 years. HCV genotypes and HCV RNA viral load, although important in governing the effectiveness of treatment regimens (see Shortening the course of treatment), are not thought to influence the natural course of infection. 19
Co-infection with HIV is also associated with rapid HCV-related disease progression. 23–25 Since the introduction of highly active antiretroviral therapy (HAART) in the mid to late 1990s, patients with HIV infection are living longer and therefore those who are co-infected are becoming at risk of long-term chronic HCV-related liver disease. Mohsen and colleagues26 reported a study of 153 HCV-infected and 55 HCV/HIV co-infected patients (72% of whom were receiving antiretroviral therapy at time of liver biopsy) from two London hospitals. The estimated median fibrosis progression rate was 0.17 units/year in HCV/HIV co-infected patients and 0.13 in HCV mono-infected patients (p = 0.01). This equates to estimated times from HCV infection to cirrhosis of 23 and 32 years, respectively. HIV positivity and a low CD4 cell count were among a number of factors that were independently related to fibrosis progression. A retrospective analysis by Poynard and colleagues27 of 4852 patients with chronic liver disease of a variety of causes found that HCV/HIV co-infection was associated with the fastest fibrosis progression compared with causes such as genetic haemochromatosis, primary biliary cirrhosis and alcoholic liver disease. Despite the findings of these studies it has been suggested that the effective immune restoration observed with HAART can, indirectly, reduce the rate of liver fibrosis to a value that is comparable with the rate of HCV mono-infected people,12 although a systematic review of natural history studies in co-infected patients concluded that this was not necessarily the case. 28
Given the slowing of HIV-related disease progression and extended survival associated with HAART29 it could be assumed that HCV is now one of the major causes of mortality in people with HIV. 11 However, although there has been an increase in liver disease-related deaths in co-infected patients, it is not clear whether this is associated with HAART-related toxicity or HCV-related liver disease, as studies have shown mixed findings. 30,31
Diagnosis
Presence of HCV infection may be detected through the identification of antibodies using enzyme-linked immunosorbent assays (ELISAs) and then confirmed through the identification of HCV RNA in serum. 32 The latter can be carried out using sensitive molecular assays, such as polymerase chain reaction (PCR). A detectable HCV viral load of 50 international units (IU)/ml or above is generally considered indicative of infection, although newer assays have a lower threshold of detectability of 12–20 IU/ml. As part of the diagnostic process, patients receive testing to determine their genotype, as this is associated with the efficacy of treatment and will govern the duration of therapy (see Shortening the course of treatment). Alanine aminotransferase (ALT) biochemical tests are also used to indicate potential HCV-related liver damage, but are not necessarily used to determine eligibility for treatment.
Traditionally, a liver biopsy has been used to gauge the extent of HCV-related liver damage in order to guide treatment decisions. If the biopsy sample showed significant fibrosis or cirrhosis, clinicians would probably commence antiviral treatment. However, there has been a shift away from using biopsy in recent years for a number of reasons, including the risk of complications (e.g. a small risk of hepatic bleeding), the pain and discomfort to the patient, the lack of interobserver reliability between pathologists, and the suggestion that it may discourage some patients from presenting for assessment. Furthermore, guidance from organisations such as the National Institute for Health and Clinical Excellence (NICE)33 to extend the provision of treatment to those with mild HCV means that it is no longer necessary to use biopsy to gauge disease severity in order to determine when to begin treatment.
Nevertheless, some clinicians find liver biopsy a useful tool to detect the presence or absence of steatosis (fatty liver) and other potential confounding liver diseases. This is reflected by NICE’s guidance, which states that clinicians may conduct a biopsy, if required, for other reasons,33 and also by Scottish guidelines on the management of HCV, which state that liver biopsy should be considered if there is concern about additional causes of liver disease. 32 Patients may also seek a biopsy to determine the extent of any fibrosis to help them decide whether or not to commence treatment.
The development of non-invasive serum markers and other technologies (e.g. ultrasound) as an alternative to biopsy has generated interest in recent years, although their clinical effectiveness and cost-effectiveness have not yet been appraised at policy level in England and Wales.
Current service provision
Antiviral treatment
The majority of people with chronic HCV will not clear the virus spontaneously and will need to be assessed for possible antiviral treatment. Patients with chronic HCV are generally managed in specialist hepatology centres. They may also be managed by gastroenterologists and specialists in infectious diseases. Specialist hepatology nurses are also involved, particularly in the administration of antiviral treatment.
The primary aim of treatment is to clear the virus from the blood, and success is usually taken to be a sustained virological response (SVR), defined as a drop in serum HCV RNA to undetectable levels (e.g. below 50 IU/ml) 6 months after the end of treatment. An SVR is generally considered to indicate permanent resolution of infection, although relapse may occur in around 5% of cases after 5 years. 34 Studies (mostly observational) have reported that people who achieve an SVR have a lower probability of developing HCC35 and liver-related death36 than those that do not. However, the validity of SVR as a surrogate for long-term clinical outcomes – such as decompensated liver disease, HCC and death – has been questioned. 37 It is suggested that this is because of an absence of randomised controlled trials (RCTs) in which the effects of antiviral treatment, in terms of SVR, have been correlated with long-term clinical outcomes. The exception is for cirrhotic patients in whom some evidence of a correlation between SVR and HCC has been identified (based on studies of treatment with interferon alfa monotherapy). 37 It is recommended that several RCTs of antiviral treatment, with long-term follow-up over a number of years, are required to determine the validity of a surrogate outcome. 37 Given that this is unlikely to be practical, and the general acceptance of SVR as being the most reliable measure of HCV infection resolution, it is pragmatic to assume that an SVR, in most people, will reduce the likelihood of morbidity and mortality.
Interferon alfa, originally as monotherapy and then as combination therapy with ribavirin, was the mainstay of treatment until the pegylated forms of interferon (peginterferon alfa or α) were introduced in 2002. The peginterferons are cytokines, the mechanism of which is to assist the immune response by inhibiting viral replication. Two forms are available: peginterferon alfa-2a (Pegasys®, Roche Products) and peginterferon alfa-2b (ViraferonPeg®, Schering-Plough). Ribavirin is a synthetic nucleoside analogue that is available in three forms: Copegus® (Roche Products), Rebetol® (Schering-Plough) and Ribavirin Teva (Teva UK). Copegus is licensed only for combination therapy with peginterferon alfa-2a, whereas Rebetol and Ribavirin Teva are licensed only for combination therapy with peginterferon alfa-2b.
The current NICE guidance [Technology Appraisal (TA) 106,33 an extension of TA 7538] recommends combination therapy with ribavirin and either peginterferon alfa-2a or peginterferon alfa-2b for adult patients with chronic HCV, regardless of disease severity. Monotherapy with peginterferon alfa-2a or peginterferon alfa-2b is recommended for patients who are unable to tolerate ribavirin or for whom ribavirin is contraindicated. For those with mild HCV, the decision whether to treat immediately or adopt an approach of ‘watchful waiting’ is made by the patient and clinician on an individual basis. The standard duration of treatment is 24 or 48 weeks, depending on a combination of factors including the genotype, initial viral load, and rapid and early virological response (EVR) to treatment. Treatment is currently restricted to patients who:
-
are treatment naive
-
have previously been treated with non-peginterferon alfa combination therapy or monotherapy
-
have previously been treated with peginterferon alfa monotherapy but did not respond or subsequently relapsed.
It is not thought that there are substantial variations in practice across the country in terms of antiviral treatment, although clinical management of chronic HCV may vary according to the availability of hepatologists and specialist clinics.
There are a number of specific areas in which the clinical management of HCV infection is evolving, including prescribing shorter treatment courses, re-treating patients who have not responded or relapsed to a previous course, and treating patients who are co-infected with HCV/HIV. These areas are discussed in the following subsections.
Shortening the course of treatment
In recent years, one of the key aims of the management of HCV has been to maximise the likelihood of an SVR while minimising potential adverse effects of treatment. The adverse effects associated with interferon-based antiviral treatment (e.g. flu-like symptoms, nausea, vomiting, depression) and ribavirin (e.g. anaemia) can be significant, and some patients describe it as a very unpleasant experience, disrupting their social and family life, and, in some cases, impairing their ability to work. Sparing them the potential adverse effects through shorter but effective treatment courses will make therapy more tolerable, and may have the additional advantage of encouraging more people with suspected HCV to present for diagnosis, assessment and treatment.
To demonstrate the efficacy of shortened courses of treatment, clinical trials have measured viral response at interim time points after commencement of therapy to determine the likelihood of an SVR. An EVR is measured after 12 weeks of therapy and is generally defined as either a negative HCV RNA (complete EVR) or a minimum two log10 drop in quantitative HCV RNA levels (partial EVR). 39 EVR tends to be measured in genotype 1 patients to determine whether to stop treatment at 12 weeks in non-responders (patients who do not achieve an EVR generally do not go on to achieve an SVR with continued treatment) or to continue for 48 weeks in those who have responded.
Recently, there has been a focus on identifying responders earlier than 12 weeks. A rapid virological response (RVR) is measured at week 4 of therapy and is generally defined as a negative qualitative HCV RNA. Thresholds for negativity vary according to the assay, with some assays using a lower limit of detectability of 50 IU/ml, and others using thresholds as low as 12 IU/ml. RVR tends to be measured in genotype 2 or 3 patients in order to determine whether treatment can be shortened from 24 to 16 weeks, and in genotype 1 or 4 patients to determine whether treatment can be shortened from 48 to 24 weeks.
Decisions regarding the most appropriate length of treatment may also take into account baseline viral load in addition to genotype. Low viral loads (LVLs) have generally been associated with increased likelihood of an SVR in some clinical trials. 40,41 There does not appear to be a consensus regarding what constitutes a low or high viral load. However, the manufacturers of peginterferon alfa-2a and peginterferon alfa-2b consider LVL as being HCV RNA ≤ 800,000 and < 600,000 IU/ml, respectively. 42,43
Re-treatment of non-responders and relapsers
Given the fact that, on average, SVRs are achieved by between only 50% and 60% of patients receiving antiviral therapy17,44 (with variations according to factors such as genotype, baseline viral load and treatment regimen), it is important to establish the efficacy of re-treatment with a subsequent course for those who did not respond or who relapsed. A non-responder is a patient who has detectable HCV RNA throughout a course of antiviral treatment. A relapser is defined as a patient who achieves loss of detectable HCV RNA during treatment, but in whom HCV RNA reappears either while still on therapy or once therapy is stopped.
Current NICE guidance recommends the re-treatment of patients who have failed previous treatment with non-peginterferon alfa and ribavirin combination therapy or non-peginterferon alfa monotherapy, or peginterferon alfa monotherapy, providing they achieve an EVR (as defined above in Shortening the course of treatment). 33 However, the guidance does not currently make provision for patients who have not responded to, or failed, a previous course of, peginterferon alfa and ribavirin combination therapy.
If re-treatment with peginterferon alfa (with or without ribavirin, depending on contraindication) does not achieve an SVR, then it is unlikely that maintenance treatment to reduce progressive liver damage will be considered. At the present time there are no other licensed drugs that could be used as second-line treatment in patients with HCV.
Treatment of HCV/HIV co-infected patients
Effective clinical management of people co-infected with HCV and HIV is important, given the increased rate of HCV-related disease progression in this group (as discussed in Disease progression and prognosis). For example, treatment decisions need to take into account any possible drug interactions between HCV antiviral medication and HAART [e.g. didanosine (Videx®, Bristol–Myers Squibb), which is contraindicated in co-infected patients taking antiviral treatment for HCV]. 45 There is potential for significant HAART-associated hepatotoxicity in co-infected patients, which in serious cases may necessitate the withdrawal of HAART, with subsequent potential for the development of resistance to HIV medication. 11 The adverse effects of HCV antiviral medication may be more pronounced in co-infected patients, notably depression.
Given the complexity of managing both infections, clinical guidelines on the management of HCV/HIV co-infected people recommend that treatment be led by specialists in both HIV and HCV. 46 Treatment with peginterferon alfa and ribavirin in combination is recommended unless contraindicated. 45,46 Although HCV/HIV co-infected people were not the focus of NICE’s previous technology appraisals, the guidance does recommend antiviral treatment for this group, in common with that for HCV mono-infected people. 33,38
Description of technology under assessment
The intervention under assessment in this report is peginterferon alfa-2a and alfa-2b in combination with ribavirin (or as monotherapy if ribavirin is contraindicated). Peginterferon alfa-2a was licensed in June 2002, with extensions to the licence granted in June 2007. The recommended dose is 180 µg once per week, administered subcutaneously, for 16, 24 or 48 weeks, dependent on genotype, baseline viral load and treatment response. Peginterferon alfa-2b was licensed in February 2002, with extensions to the licence granted in May 2005. The recommended dose is 1.5 µg/kg body weight once per week, administered subcutaneously for 24 or 48 weeks, dependent on genotype, baseline viral load and treatment response.
The three forms of ribavirin (Rebetol, Copegus and Ribavirin Teva) were licensed in May 1999, November 2002 and March 2009, respectively. The recommended dose of ribavirin ranges from 800 mg to 1400 mg taken orally each day in two divided doses (200-mg capsules), with the dose depending on the patient’s body weight. The dose of Copegus also varies according to genotype [800 mg per day for genotype 2/3 and 1000–1200 mg per day (depending on body weight: 1000 mg for weight < 75 kg, 1200 mg for weight ≥ 75 kg) for genotype 1].
For both forms of peginterferon alfa, the therapeutic indication is the treatment of adult patients with chronic HCV who are positive for serum HCV RNA, including those with clinically stable HIV co-infection. The preferred indication is in combination with ribavirin, but monotherapy is indicated in cases of intolerance or contraindication to ribavirin. Patients may be treatment naive or may have failed previous monotherapy or combination treatment.
For peginterferon alfa-2a, genotype 1 patients with detectable HCV RNA at 4 weeks (i.e. no RVR) should receive 48 weeks’ treatment. Those with genotype 2/3 and detectable HCV RNA at 4 weeks should receive 24 weeks’ treatment. The licence extensions allow genotype 1 patients with LVL, an RVR and undetectable HCV RNA at week 24 to complete treatment at week 24 rather than receive the standard 48 weeks’ treatment. It also allows genotype 2/3 patients with LVL (≤ 800,000 IU/ml), an RVR and undetectable HCV RNA at week 16 to finish treatment at week 16 rather than receive the standard 24 weeks’ treatment. Those with genotype 4 may be treated as genotype 1, without the requirement for LVL. It is recommended that patients receiving peginterferon monotherapy be treated for 48 weeks.
For peginterferon alfa-2b, genotype 1 patients with an EVR (at week 12) should receive 48 weeks’ treatment. Those without an EVR are considered unlikely to achieve an SVR and consideration should be given to withdrawal of treatment. Genotype 2/3 patients should be treated for 24 weeks. Licence extensions permit genotype 1 patients with LVL (< 600,000 IU/ml) and an RVR and undetectable HCV RNA at week 24 to receive 24 weeks’ treatment rather than 48. The licence does not permit, however, shorter courses of treatment in genotype 2, 3 or 4 patients. Patients receiving peginterferon monotherapy who achieve an EVR should continue treatment for another 3 months. Extension of treatment to 1 year should be based on prognostic factors such as age and genotype.
For both peginterferon alfa-2a and alfa-2b, patients co-infected with HIV should be treated for 48 weeks, regardless of genotype. Full details of the indications, dosages and duration of treatment are given in the summaries of product characteristics (SPCs). 42,43
In terms of costs, a 180-µg prefilled syringe of peginterferon alfa-2a (the recommended weekly dose) costs £126.91. A 168 × 200 mg-tab pack of ribavirin (Copegus) costs £444.43. The weekly cost of Copegus would be £111 for genotype 1 (based on 1200 mg per day for an average body weight of 79 kg) and £74 for genotype 2/3 (based on 800 mg per day for an average body weight of 79 kg). A 120-µg prefilled injection pen of peginterferon alfa-2b costs £162.60. This would be the weekly cost for an average patient weighing 79 kg (1.5 µg per kg). A 168 × 200 mg-tab pack of ribavirin (Rebetol) costs £327. The weekly cost of for Rebetol would be £68, based on 1000 mg per day for an average body weight of 79 kg. All costs are from the British National Formulary (BNF), No. 58, September 2009. 47 [See Chapter 5 (Cost data) for full details of the drug costs estimated in our independent economic evaluation.]
Chapter 2 Definition of the decision problem
Interferon alfa (pegylated and non-pegylated) and ribavirin for the treatment of moderate to severe HCV was appraised by NICE in 2004 (TA75),38 and an appraisal specifically for mild HCV was carried out in 2006 (TA106). 33 Both appraisals were based on our independent assessment reports. 17,44 Since NICE’s clinical guidance was published, there have been extensions to the licences for peginterferon alfa-2a and alfa-2b. This health technology assessment (HTA) is a part-review of the current NICE guidance and is restricted to the patient subgroups that are affected by the licence extensions, as below.
Decision problem
The decision problem is based on the scope of the appraisal as set by NICE. The relevant intervention is peginterferon alfa (2a and 2b) in combination with ribavirin, or peginterferon alfa monotherapy where ribavirin is contraindicated. The population of interest is adult patients with chronic HCV infection in one or more of the following patient groups – those who (1) meet the licensed criteria for receiving shortened courses of combination therapy; (2) have been previously treated with peginterferon alfa and ribavirin in combination and who either did not respond or who responded but relapsed; and (3) are co-infected with HIV.
The relevant comparator for studies evaluating the efficacy of shortened treatment courses is standard treatment duration (e.g. 48 weeks for genotype 1 patients, 24 weeks for genotype 2/3 patients). For the other two patient groups the comparator is best supportive care (BSC). Relevant outcomes include virological response (e.g. during treatment, 6 months post treatment), biochemical response (e.g. ALT levels), histological improvement (fibrosis and inflammation), survival, adverse effects of treatment, and health-related quality of life (HRQoL).
Overall aims and objectives of assessment
The aim of this HTA is to assess the clinical effectiveness and cost-effectiveness of peginterferon alfa and ribavirin for the treatment of chronic HCV in three specific patient groups: those eligible for shortened treatment courses; those eligible for re-treatment following previous non-response or relapse; and those who are co-infected with HIV.
Chapter 3 Methods
The a priori methods for systematically reviewing the evidence of clinical effectiveness and cost-effectiveness were described in a research protocol (see Appendix 1), which was sent to experts for comment. Minor amendments were made as appropriate but no comments that identified specific problems with the methods of the review were received. The methods of the Southampton Health Technology Assessments Centre (SHTAC) economic evaluation can be seen in Chapter 5 (Methods for SHTAC independent economic analysis).
Identification of studies
A sensitive search strategy was developed and refined by an experienced information scientist and was based upon that used in previous technology assessment reports. 17,44 Separate searches were conducted to identify studies of clinical effectiveness, cost-effectiveness, QoL, resource use/costs and epidemiology. The different search strategies are provided in Appendix 2.
Searches for clinical effectiveness and cost-effectiveness literature were undertaken from April 2007 (the date the most recent search was conducted48) to October 2009. References identified in the previous hepatitis C technology assessment reports17,44 in which literature searching extended back to the year 2000 were incorporated into the searches. Search filters were run, where possible, to locate RCTs and searches were restricted to the English language. The strategies were applied to the following databases:
-
Cochrane Database of Systematic Reviews (CDSR)
-
Cochrane Central Register of Controlled Trials (CENTRAL)
-
Centre for Reviews and Dissemination (CRD) (University of York) databases: Database of Abstracts of Reviews of Effects (DARE), NHS Economic Evaluation Database (NHS EED) and the HTA database
-
MEDLINE (Ovid)
-
EMBASE (Ovid)
-
PREMEDLINE In-Process & Other Non-Indexed Citations (Ovid)
-
Web of Science with Conference Proceedings: Science Citation Index Expanded (SCIE) and Conference Proceedings Citation Index – Science (CPCI) (ISI Web of Knowledge)
-
Biosis Previews (ISI Web of Knowledge)
-
National Institute for Health Research (NIHR) Clinical Research Network Portfolio
-
ClinicalTrials.gov
-
Current Controlled Trials.
Bibliographies of retrieved papers were screened for relevant studies, and the manufacturers’ submissions (MSs) to NICE were assessed for any additional studies [see Appendix 3 for a critique of the clinical effectiveness section of the MS, and Chapter 5 (Review of manufacturers’ submissions) for further discussion of the cost-effectiveness section]. Experts who were contacted for advice and peer review were also asked to identify additional published and unpublished references. All search results were downloaded into a reference manager (Thomson Reuters, New York, NY, USA) database.
Key hepatitis C websites and symposia were also searched for completed or ongoing studies and background resources. These included:
-
European Association for the Study of the Liver (EASL)
-
British Association for the Study of the Liver (BASL)
-
American Association for the Study of Liver Diseases (AASLD)
-
British Viral Hepatitis Group (BVHG)
-
British Liver Trust
-
British Society of Gastroenterology (BSG)
-
International HIV and Hepatitis Co-infection workshop
-
Health Protection Agency
-
Hepatitis C Trust.
Inclusion process
Titles and abstracts identified by the search strategy for the clinical effectiveness section of the review were assessed for possible eligibility by one reviewer using an inclusion worksheet (see Appendix 4) based on the inclusion/exclusion criteria detailed below. The full texts of relevant papers were then obtained and inclusion criteria were applied independently by two reviewers. Any disagreements over eligibility were resolved by consensus. References identified from our previous searches were rescreened according to the inclusion criteria for the current review.
Titles and abstracts identified by the search strategy for the cost-effectiveness section of the review were assessed for potential eligibility by two reviewers independently. Economic evaluations were considered for inclusion if they reported both health service costs and effectiveness, or presented a systematic review of such evaluations. Full papers were formally assessed for inclusion by two reviewers independently. Data extraction was undertaken by one reviewer and checked by a second.
Inclusion criteria
Study design
Randomised controlled trials were included for the clinical effectiveness review. Trials published as abstracts or conference presentations from 2007 onwards were included only if sufficient details were presented to allow an appraisal of the methodology and the assessment of results to be undertaken. Systematic reviews were used only as a source of references. For the systematic review of cost-effectiveness, studies were eligible for inclusion if they reported the results of full economic evaluations [cost-effectiveness analyses (reporting cost per life-year gained), cost–utility analyses or cost–benefit analyses]. For studies reporting QoL and epidemiology/natural history, a range of study designs were eligible (e.g. cohort studies, cross-sectional surveys).
Interventions
-
Combination therapy comprising ribavirin and either peginterferon alfa-2a or peginterferon alfa-2b.
-
Peginterferon alfa-2a or peginterferon alfa-2b monotherapy (for patients who are unable to tolerate or are contraindicated to ribavirin).
Comparators
For patients who have been previously treated with combination therapy, and for HCV/HIV co-infected patients:
-
BSC (e.g. symptomatic treatment, monitoring, treatment without any form of interferon therapy).
For patients who meet the criteria for receiving shortened courses of combination therapy:
-
standard-duration courses of peginterferon alfa and ribavirin combination therapy (up to 24 or 48 weeks, as appropriate).
Population
Adults with chronic HCV, restricted to people who:
-
have been previously treated with peginterferon alfa and ribavirin in combination but who relapsed/did not respond
-
have HCV/HIV co-infection
-
meet the criteria within the marketing authorisation for receiving shortened courses of peginterferon alfa and ribavirin in combination, namely patients with:
-
– genotype 2 or 3 with LVL* at the start of treatment and an RVR (defined as HCV RNA undetectable by week 4) – shortened course of 16 weeks†
-
– genotype 1 with LVL* and an RVR (defined as HCV RNA undetectable by week 4 and at week 24) – shortened course of 24 weeks
-
– genotype 4 with an RVR (defined as HCV RNA undetectable by week 4 and at week 24) – shortened course of 24 weeks.†
-
(*For peginterferon alfa-2a, LVL is defined as ≤ 800,000 IU/ml;42 for peginterferon alfa-2b, LVL is defined as ≤ 600,000 IU/ml. 43 †Applies only to peginterferon alfa-2a.)
Outcomes
Studies had to report SVR (defined as undetectable HCV RNA at least 6 months after treatment cessation). The following outcomes were also included:
-
virological response (e.g. during treatment)
-
biochemical response (e.g. ALT levels)
-
histological improvement (fibrosis and inflammation)
-
survival
-
adverse effects of treatment
-
HRQoL
-
cost-effectiveness (incremental cost per life-year gained) or cost–utility [incremental cost per quality-adjusted life-year (QALY) gained].
Data extraction and critical appraisal strategy
Data from included studies were extracted by one reviewer using a standardised data extraction form and checked by a second reviewer. The quality of included RCTs was assessed using criteria recommended by CRD49 (see Appendix 5). Quality criteria were applied by one reviewer and checked by a second reviewer. At each stage, any differences in opinion were resolved through discussion.
Methods of data analysis/synthesis
Data were synthesised through a narrative review with tabulation of results of all included studies. Full data extraction forms are presented in Appendix 6. It was not considered appropriate to combine the RCTs in a meta-analysis owing to differences in the drug regimens and also because the population of interest (i.e. patients with LVL and RVR) were often subgroups of the main treatment arms. Any meta-analyses would therefore compromise intention-to-treat (ITT) principles and the data may be biased and not valid.
Consideration was given to performing a pairwise indirect comparison of peginterferon alfa with or without ribavirin with a trial featuring no active treatment (analogous to BSC). For this to be possible, an RCT featuring an arm in which patients were treated with peginterferon alfa would be required, in addition to an RCT featuring a ‘no active treatment’ (e.g. placebo) in patients with HCV/HIV co-infection or previous non-responders or relapsers. A comparator arm common to both RCTs would be necessary, such as non-peginterferon alfa. However, as will be discussed in the following chapter, we did not identify any such studies from our database of RCTs of both peginterferon and non-peginterferon alfa (which we have amassed from our previous technology assessment reports on antiviral treatment for hepatitis C for NICE since 2000). Furthermore, none of the systematic reviews of HCV/HIV co-infected patients identified in our search identified any trials in which a non-active treatment arm was included. 50,51
As antiviral treatment for HCV has been available for some time – first with interferon alfa monotherapy, followed by the addition of ribavirin as combination therapy, and latterly with the introduction of peginterferon alfa and ribavirin – it is unlikely that any studies, whether randomised or not, will have included a non-active treatment arm, as withholding treatment would not be considered ethical.
Chapter 4 Clinical effectiveness
Results
Quantity and quality of research available
Literature searches identified 1317 references, after the removal of duplicates. A further 1389 references identified from searches conducted for our previous hepatitis C technology assessment reports17,44 were screened according to the inclusion criteria for the present review. After further de-duplication, the total number of records screened was 2400. Following initial screening of titles and abstracts, 2310 references were excluded because they did not meet the inclusion criteria and full copies of 90 articles were retrieved. Of these, 82 were excluded on further inspection, leaving eight included studies. The total number of published papers included at each stage of the systematic review is shown in the flow chart in Figure 1; the list of excluded studies can be seen in Appendix 7.
FIGURE 1.
Flow chart of identification of studies for inclusion in the review. *Includes total number of studies identified in updated searches and searches from previous hepatitis C assessment reports (n = 2706); further de-duplication left n = 2400 for screening.
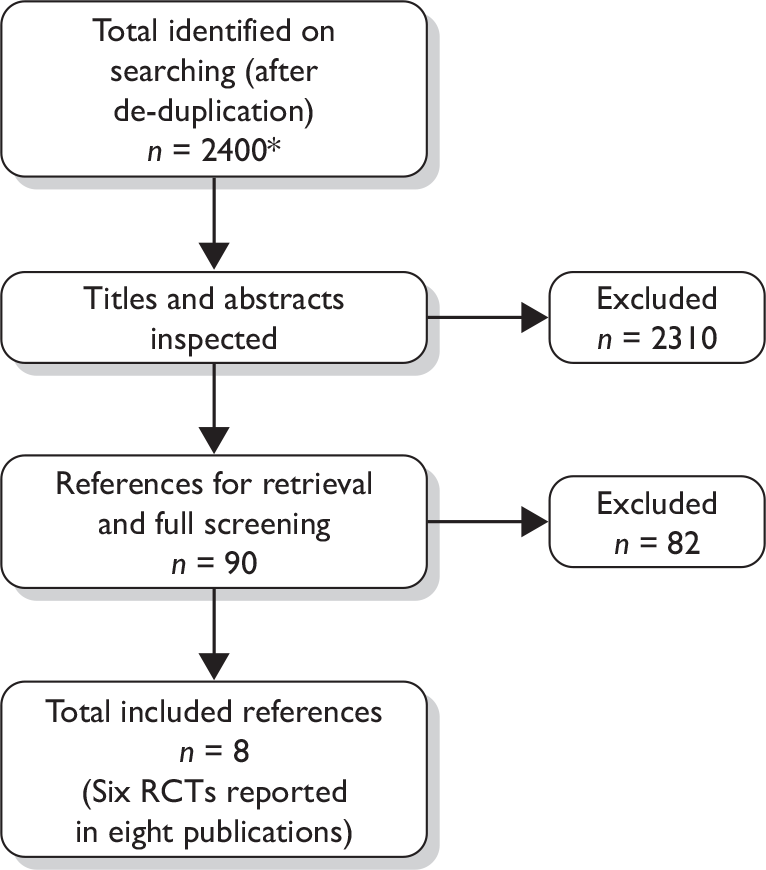
Eight publications describing six RCTs met the inclusion criteria of the review. 52–59 Two of the articles were abstracts57,58 linked to full publications. 53,60 All of the included studies report peginterferon and ribavirin combination therapy in patients eligible for shortened treatment duration (i.e. those with specific genotypes as described in Chapter 3, Inclusion crtieria). No RCTs comparing peginterferon alfa with or without ribavirin compared with BSC for the other two population groups specified in the NICE scope (i.e. re-treatment following previous non-response or relapse, and HCV/HIV co-infection) were identified through our searches. A number of RCTs comparing peginterferon alfa with or without ribavirin to active treatment comparators were identified (e.g. peginterferon alfa and ribavirin vs non-peginterferon alfa and ribavirin) but these did not meet the inclusion criteria for the review, which was based on the scope of the appraisal issued by NICE. 61
The remainder of this chapter describes the six trials in patients who were eligible for shortened courses of treatment.
Description of the included trials
The key characteristics of the RCTs are shown in Table 1. Four of the included studies evaluated peginterferon alfa-2a in combination with ribavirin,53–56 one trial (Berg and colleagues59) evaluated peginterferon alfa-2b and ribavirin, and one trial evaluated peginterferon alfa-2a or peginterferon alfa-2b in combination with ribavirin (Mangia and colleagues52). The comparator in all the studies was the same intervention for a shorter duration. The dose of peginterferon alfa-2a was the same in all the trials (180 µg/week, subcutaneously), as was the dose of peginterferon alfa-2b (1.5 µg/kg/week). Ribavirin was administered orally, according to body weight, at a dose of 1000 mg/day for patients weighing ≤ 75 kg and 1200 mg/day for patients weighing > 75 kg in four studies,52–55 or 800 mg/day for patients weighing ≤ 65 kg, 1000 mg/day for patients weighing 65–85 kg, and 1200 mg/day for patients weighing > 85 kg in one study. 56 Berg and colleagues59 reported only that patients received 800–1400 mg/day ribavirin and it is assumed that the dose was administered according to body weight. It should be noted that in two trials,55,56 the doses of ribavirin used are higher than those stipulated in the current licence for peginterferon alfa-2a and ribavirin combination treatment (800 mg/day for genotype 2/342,62) owing to changes in the licence since these studies were carried out.
| Study | Methods | Key inclusion criteria | Key patient characteristics | Outcomes |
|---|---|---|---|---|
| Berg and colleagues 2009 59 |
Design: open-label, multicentre RCT No. of centres: 19 Country: Germany Sponsor: Essex Pharma (subsidiary of Schering-Plough), Bayer Diagnostics, German Competence Network for viral hepatitis Interventions: PEG α-2b + RBV for 48 weeks vs PEG α-2b + RBV for 18, 24, 30, 36, 42 or 48 weeks Follow-up: 24 weeks after treatment cessation No. of participants: n = 433 |
Inclusion criteria: Treatment-naive adults with compensated chronic HCV, genotype 1 Anti-HCV positive HCV RNA > 1000 IU/ml by quantitative reverse transcription PCR Increased ALT levels at screening Liver biopsy consistent with chronic HCV within preceding 24 months Neutrophils ≥ 1500 µl Platelets ≥ 80,000 µl Hb ≥ 12 g/dl for women, ≥ 13 g/dl for men Creatinine < 1.5 mg/dl |
Mean viral load (log10 IU/ml): 5.7 Group 1, 5.7 Group 2 Mean serum ALT × ULN, IU/l: 2.6 Group 1, 2.6 Group 2 Fibrosis score 0–2: 87% Group 1, 85% Group 2 Genotype 1: 100% Mean age: 42 years Gender: 55% male Mode of infection: NR Ethnicity: NR |
Primary outcome: SVR Secondary outcomes: Biochemical responsea On-treatment virological response (RVR, EOT) Relapse rate Adverse events |
| Mangia and colleagues 2008 52 |
Design: multicentre RCT No. of centres: 11 Country: Italy Sponsor: NR Interventions: PEG α-2a or PEG α-2b + RBV for 48 weeks vs PEG α-2a or PEG α-2b + RBV for 24, 48 or 72 weeks Follow-up: 24 weeks after treatment cessation No. of participants: n = 696 |
Inclusion criteria: Treatment-naive adults with compensated chronic HCV, genotype 1 HCV RNA positive Anti-HCV positive Neutrophils ≥ 1500 µl Platelets ≥ 90,000 µl Hb ≥ 12 g/dl for women, ≥ 13 g/dl for men Creatinine < 1.5 mg/dl |
Serum HCV RNA < 400,000 IU/ml: 26% Group 1, 22% Group 2 Serum ALT ≥ 3 ULN: 19% Group 1, 16% Group 2 Fibrosis score 0–2: 62% Group 1, 65% Group 2 Genotype 1a: 9%, 1b: 91% Mean age: 52 years Gender: 56% male Mode of infection: blood transfusion 21%, drug abuse 7%, unknown 72% Ethnicity: NR Treatment: PEG α-2a 46% Group 1, 49% Group 2; PEG α-2b 53% Group 1, 51% Group 2 |
Primary outcome: SVR Secondary outcomes: RVR EOT virological response SVR according to virological response at weeks 4, 8 and 12 Relapse rate Adverse events |
| Liu and colleagues 2008; 53 2008 abstract 57 |
Design: multicentre RCT No. of centres: 5 Country: Taiwan Sponsor: National Taiwan University Hospital, National Science Council & Department of Health, Executive Yuan, Taiwan Interventions: PEG α-2a + RBV for 24 weeks vs PEG α-2a + RBV for 48 weeks Follow-up: 24 weeks after treatment cessation No. of participants: n = 308 |
Inclusion criteria: Treatment-naive adults with chronic HCV, genotype 1 Liver biopsy consistent with chronic HCV within previous 3 months Detectable HCV RNA for > 6 months Presence of anti-HCV antibody Serum ALT > ULN |
Mean viral load (log10 IU/ml): 5.7 Group 1, 5.8 Group 2 Mean serum ALN × ULN: 3.2 Group 1, 3.0 Group 2 Fibrosis score ≥ 3: 77% Genotype 1a: 2%, 1b: 94%, 1a and 1b: 4% Mean age: 54 years Gender: 57% male Mode of infection: NR Ethnicity: 100% Asian |
Primary outcome: SVR Secondary outcomes: RVR EVR EOT virological response Relapse rate Biochemical response Histological response Adverse events |
| Yu and colleagues 2008; 54 2007 abstract 58 |
Design: open-label, multicentre RCT No. of centres: 4 Country: Taiwan Sponsor: Taiwan Liver Research Foundation Interventions: PEG α-2a + RBV for 24 weeks vs PEG α-2a + RBV for 48 weeks Follow-up: 24 weeks after treatment cessation No. of participants: n = 200 |
Treatment-naive adults with chronic HCV, genotype 1 Liver biopsy consistent with chronic HCV within ≤ 1 year of study entry HCV RNA positive Positive for HCV antibodies Elevated serum ALT ≥ 2 measurements within ≤ 6 months of study entry Neutrophils ≥ 1500 mm–3 Platelets ≥ 90,000 µl Hb > 12 g/dl for women, > 11 g/dl for men Creatinine < 1.5 mg/dl |
Mean viral load (log10 IU/ml): 5.43 Group 1, 5.66 Group 2 Serum HCV RNA < 400,000 IU/ml: 55% Serum ALT IU/l: 156 Group 1, 137 Group 2 Fibrosis score 0–2: 75% Group 1, 81% Group 2 Genotype 1: 100% Mean age: 49 years Gender: 57% male Mode of infection: NR Ethnicity: NR |
Primary outcome: SVR Secondary outcomes: RVR EVR EOT virological response Relapse rate Adverse events |
| Yu and colleagues 2007 55 |
Design: open-label, multicentre RCT No. of centres: 4 Country: Taiwan Sponsor: Taiwan Liver Research Foundation Interventions: PEG α-2a + RBV for 24 weeks vs PEG α-2a + RBV for 16 weeks Follow-up: 24 weeks after treatment cessation No. of participants: n = 150 |
Treatment-naive adults with chronic HCV, genotype 2 Liver biopsy consistent with chronic HCV within ≤ 1 year of study entry Seropositive for HCV RNA Seropositive for HCV antibodies Increased serum ALT ≥ 1.5 × ULN for ≤ 2 measurements within 6 months before study entry Neutrophils > 1500 mm–3 Platelets > 9 × 104 mm–3 Hb > 12 g/dl for women, > 11 g/dl for men Creatinine < 1.5 mg/dl |
Mean viral load (log10 IU/ml): 4.88 Group 1, 4.98 Group 2 Serum ALT IU/l: 108.9 Group 1, 107 Group 2 Fibrosis score 0–2: 80% Group 1, 78% Group 2 Genotype 2: 100% Mean age: 50 years Gender: 60% male Mode of infection: NR Ethnicity: 100% Asian (Taiwanese) |
Primary outcome: SVR Secondary outcomes: RVR EOT virological response Relapse rate Adverse events |
| von Wagner and colleagues 2005 56 |
Design: multicentre, Phase IIIb RCT No. of centres: 6 Country: Germany Sponsor: Hoffmann-La Roche and German Hepatitis Network of Competence (Hep-Net) Interventions: PEG α-2a + RBV for 16 weeks vs PEG α-2a + RBV for 24 weeks (RVR) vs PEG α-2a + RBV for 24 weeks (no RVR) Follow-up: 24 weeks after treatment cessation No. of participants: n = 142 |
Treatment-naive adults with compensated chronic HCV, genotype 2 or 3 Liver biopsy consistent with chronic HCV within ≤ 18 months before study entry HCV RNA positive (> 600 IU/ml) Positive for anti-HCV antibodies Elevated serum ALT at screening or study entry Neutrophils > 1500/µl Platelets > 90,000/µl Hb ≥ 12 g/dl for women, ≥ 13 g/dl for men |
Mean viral load (log10 IU/ml): 5.8 Group 1, 5.8 Group 2 Serum ALT × ULN IU/l: 2.8 Group 1, 2.8 Group 2 Mean fibrosis score: 1.6 Group 1, 1.6 Group 2 Genotype 2: 27%, genotype 3: 73% Mean age: 38 years Gender: 65% male Mode of infection: NR Ethnicity: NR |
Primary outcome: SVR Secondary outcomes: RVR EOT virological response Biochemical response Adverse events |
Four trials evaluated treatment in patients with genotype 1,52–54,59 with two of these53,54 comparing the standard 48 weeks’ treatment duration with a shorter 24 weeks’ treatment duration. The other two genotype 1 studies52,59 randomised patients to the standard 48 weeks’ treatment duration or to a variable treatment duration based on the time when HCV RNA first became undetectable. In the Mangia and colleagues trial,52 patients who were first HCV RNA negative at weeks 4, 8 and 12 were treated for 24, 48 and 72 weeks, respectively; in the Berg and colleagues trial,59 time to first HCV RNA negativity was multiplied by a factor of 6, such that patients who were first HCV RNA negative at weeks 3, 4, 5, 6, 7 or 8 were treated for 18, 24, 30, 36, 42 or 48 weeks, respectively. One trial by Yu and colleagues55 assessed treatment in patients with genotype 2, comparing the standard 24 weeks’ treatment duration with a shorter 16 weeks’ treatment duration. The sixth trial by von Wagner and colleagues56 evaluated treatment in patients with genotypes 2 and 3 and had three treatment arms. All patients were treated with combination therapy for an initial period of 8 weeks, and those with an RVR at week 4 were randomised (at week 8) to receive either a further 8 or 16 weeks’ treatment (giving a total treatment duration of 16 vs 24 weeks, respectively). Patients without an RVR at week 4 were allocated (at week 8) to receive a further 16 weeks’ treatment (giving a total treatment duration of 24 weeks).
In five of the RCTs,53–56,59 patients had LVL at baseline (based on the mean viral load) ranging from 4.98 log10 HCV RNA (95,500 IU/ml) to 5.8 log10 HCV RNA (631,000 IU/ml). In the trial by Mangia and colleagues,52 only 24% of patients were reported to have LVL (HCV RNA < 400,000 IU/ml) at baseline. However, the study was included because results were reported for the subgroup of patients with LVL and RVR. The two trials of genotype 2/3 patients55,56 used a cut-off HCV RNA level of ≤ 800,000 IU/ml to differentiate LVL and high viral load. The Berg and colleagues trial59 in genotype 1 patients also used a cut-off of < 800,000 IU/ml, although it should be noted that this threshold for LVL is higher than the threshold of < 600,000 IU/ml specified in the SPC for peginterferon alfa-2b. 43 Two of the trials in genotype 1 patients52,54 used a cut-off of < 400,000 IU/ml. The sixth genotype 1 trial (Liu and colleagues53) presented results for viral load of between 400,000 and 1,000,000 IU/ml, at 200,000 IU/ml intervals, but in the published paper the authors appear to use a cut-off of < 800,000 IU/ml to define LVL. The trials varied in their lower limits of detection of serum HCV RNA. For RVR, a lower limit of < 50 IU/ml was used in three trials,52,54,55 < 25 IU/ml was used in one trial,53 < 600 IU/ml in one trial56 and < 615 IU/ml in the sixth trial. 59 For SVR, most of the trials had a threshold of < 50 IU/ml,52,54–56 whereas Liu and colleagues53 used a lower limit of < 25 IU/ml. In the Berg and colleagues trial,59 HCV RNA negativity was verified using a highly sensitive transcription-mediated amplification (TMA) assay with a detection limit of < 5.3 IU/ml.
All of the included studies were multicentre trials (ranging from 4 to 19 centres), recruiting patients from medical centres, hospitals and/or tertiary referral centres in Taiwan,53–55 Italy52 and Germany. 56,59 The trial by Mangia and colleagues52 was the largest, recruiting 696 patients, followed by Berg and colleagues (n = 433)59 and Liu and colleagues (n = 308). 53 The numbers of participants in the three smaller trials ranged from 142 to 200. Two of the studies received partial funding from the drug manufacturers: von Wagner and colleagues56 were partially sponsored by Hoffmann-La Roche, and Berg and colleagues59 were partially sponsored by Essex Pharma (a subsidiary of Schering-Plough).
All of the trials were based on middle-aged (mean age range 39–53 years) adult patients, with the proportion of male participants ranging from 55% to 73%. Patients were treatment naive in all studies. Two of the studies53,55 reported that 100% of patients were of Asian ethnicity, and it can be assumed that this was also the case for the third Taiwanese study. 54 The ethnicity groups of the three European studies52,56,59 were not reported. Only one trial52 reported the source of infection, although for nearly three-quarters of patients this was unknown: approximately 20% were infected by blood transfusion and 7% via intravenous drug use. The proportion of patients with a fibrosis score of 0–2 was similar in four trials52,54,55,59 (range 62%–87%), with one-fifth56 reporting a mean fibrosis score of 1.6. In contrast, more than three-quarters of patients in the study by Liu and colleagues53 had a fibrosis score of ≥ 3, indicating a greater degree of liver damage.
In general, all six trials had similar inclusion criteria, with patients required to have chronic HCV (as determined by liver biopsy in five trials53–56,59), be positive for anti-HCV antibodies, be HCV RNA positive and have elevated serum ALT levels. 53–56,59 The other primary inclusion criterion was a specific HCV genotype, with patients required to have HCV genotype 1,52–54,59 genotype 255 or genotype 2 or 3. 56
Exclusion criteria were similar across the included trials. All six trials excluded patients with significant comorbidities, such as chronic hepatitis B or HIV infection, autoimmune liver disease or other causes of liver disease, as well as organ transplant, excessive alcohol intake or pregnancy. All except one study52 excluded patients with psychiatric conditions, and four studies52,53,56,59 excluded patients with drug abuse. Further details on exclusion criteria can be found in the data extraction forms in Appendix 6.
All of the trials stipulated certain laboratory readings in their inclusion/exclusion criteria, most of which are related to conditions that are consistent with decompensated liver cirrhosis, such as thrombocytopenia, anaemia and neutropenia. Patients were required to have a neutrophil count of > 1500 cells/mm3, a platelet count ranging from at least 70,000 cells/mm3 to at least 90,000 cells/mm3, haemoglobin (Hb) levels of ≥ 11–12 g/dl for women and ≥ 12–13 g/dl for men, and creatinine level of < 1.5 mg/dl. 53–55,59,63
All six RCTs reported SVR as the primary outcome measure. In terms of secondary outcomes, RVR and end-of-treatment (EOT) virological response were reported by all six trials, with some trials also reporting EVR at week 12 of therapy53,54 and relapse rate. 53–55,59 Biochemical response (ALT levels) was reported by two trials53,56 and histological response by one trial. 53 Five RCTs52–54,56,59 presented SVR rates according to RVR and viral load. All six trials reported adverse events in some way but none reported HRQoL.
Characteristics for the third treatment arm in the von Wagner and colleagues trial56 are not discussed here, as this group did not achieve an RVR and thus is not relevant to this review. It is not possible to report baseline characteristics for the 24-week subset of the variable treatment duration groups in the trials by Mangia and colleagues52 and Berg and colleagues,59 as these were not reported separately by the authors.
Quality assessment of included studies
The methodological quality of reporting in the included studies was assessed using criteria set by CRD at the University of York,49 and is shown in Table 2. On the whole, the methodological quality of the trials was good, particularly for the two studies by Yu and colleagues. 54,55 Four trials explicitly reported a computer-generated randomisation procedure that assured true random assignment to treatment groups, while in two studies56,59 details were not reported. The use of a central randomisation procedure assured adequate concealment of allocation in only two trials. 54,55
| Quality criteria | Berg 200959 | Mangia 200852 | Liu 200853 | Yu 200854 | Yu 200755 | von Wagner 200556 |
|---|---|---|---|---|---|---|
| Adequate randomisation | Unclear | Yes | Yes | Yes | Yes | Unclear |
| Adequate allocation concealment | Unclear | Unclear | Unclear | Yes | Yes | Unclear |
| Similarity of baseline prognostic factors | Yesa | Yesa | Yes | Yes | Yes | Yes |
| Blinding of outcome assessors | Unclear | Unclear | Unclear | Unclear | Unclear | Unclear |
| Blinding of care provider | No | No | No | No | No | No |
| Blinding of patient | No | No | No | No | No | No |
| Unexpected imbalances in dropouts | No | No | No | No | No | No |
| More outcomes measured than reported | Yes | No | No | No | No | No |
| ITT analysis included: | Yes | Yes | Yes | Yes | Yes | Yes |
| Appropriate | Unclear | Yes | Yes | Yes | Yes | Yes |
| Missing data accounted for | Unclear | Unclear | Unclear | Yes | Yes | Yes |
The groups appeared similar at baseline on demographic, biochemical and virological characteristics, with most presenting supporting statistical comparisons. However, in the studies by Berg and colleagues59 and Mangia and colleagues,52 the comparability of the standard-treatment-duration group (48 weeks) versus the 24 weeks’ subset of the variable-treatment-duration group is unknown, as characteristics for this subset were not presented. Neither patients nor caregivers were blinded to treatment in any of the trials, but this would not be possible given the treatment regimens. Although the blinding of outcome assessors was unclear in all trials, the possibility of detection bias would be minimal, given the objective hard end point of virological response.
There were no unexpected imbalances in dropouts between groups in any of the studies, nor was there any evidence to suggest that the authors measured more outcomes than they reported, with the exception of the Berg and colleagues study,59 where sustained biochemical response was reported by the authors as a secondary outcome but no results were presented in the publication. All six RCTs undertook an appropriate ITT data analysis for the primary efficacy outcome, although appropriate methods were used to account for missing data in only three trials. 54–56 All of the trials were statistically powered (at 80%) for the primary outcome of SVR between treatment groups as a whole. However, none performed a power calculation for patient subgroups (such as those with RVR and LVL), and therefore these results in the following sections should be interpreted with caution.
Assessment of clinical effectiveness
The results in the following sections relate to the included trials of patients eligible for shortened courses of treatment with the focus on the subgroup of patients with an RVR and LVL, where reported. Results presented in the tables are ordered by genotype.
Sustained virological response
Sustained virological response was defined as undetectable serum HCV RNA (< 50 IU/ml,52,56 25 IU/ml,53 < 5.3 IU/ml59) at the end of 24 weeks’ follow-up in four trials, and as HCV RNA negative (< 50 IU/ml) at the end of treatment and end of follow-up in two trials. 54,55
Sustained virological response was the primary outcome in all six included RCTs. Four of the trials52–54,56 separately reported SVR in the subgroup of patients who achieved an RVR and had LVL at baseline, which is the patient subgroup meeting the licensed criteria for receiving shortened courses of combination therapy (Table 3). Yu and colleagues55 reported SVR for patients who achieved an RVR, but did not further stratify this subset by baseline viral load. However, it can be assumed that rates would be similar to SVR by RVR rates, as the mean baseline viral load was low for both treatment arms, and approximately 83% of the study population had LVL at baseline (< 800,000 IU/ml). Although the trial by Berg and colleagues59 reported SVR in the subgroup of patients who achieved an RVR and had LVL at baseline, the threshold used was either ≤ 800,000 IU/ml or > 800,000 IU/ml, which differs from the threshold of < 600,000 IU/ml specified in the SPC for the study drug peginterferon alfa-2b. 43 For this reason we do not present the results for this subgroup, but instead present the SVRs for the subgroup that achieved an RVR irrespective of the baseline viral load. As the mean viral load for the study sample, as a whole, was log10 5.7 IU/ml (calculated to be around 500,000 IU/ml), these SVRs can be considered, overall, to reflect LVL in accordance with the SPC.
| Study details | Group 1 | Group 2 | p-value | |
|---|---|---|---|---|
| Genotype 1 | ||||
| Berg and colleagues 2009 59 | PEG α-2b + RBV a | PEG α-2b + RBV | ||
| 48 weeks, n = 225 | 24 weeks, n = 28 b | |||
| SVR by RVR, % (n/N ) | 42 (8/19) | 57 (16/28) | NR | |
| Mangia and colleagues 2008 52 | PEG α-2a or α-2b + RBV | PEG α-2a or α-2b + RBV | ||
| 48 weeks, n = 237 | 24 weeks, n = 123 c | |||
| SVR by RVR and baseline viral load,% (n/N ) | < 400,000 IU/ml | 83.3 (20/24) | 84.4 (38/45) | 0.83 |
| ≥ 400,000 IU/ml | 86.8 (33/38) | 73.1 (57/78) | 0.14 | |
| Liu and colleagues 2008 53 | PEG α-2a + RBV | PEG α-2a + RBV | ||
| 48 weeks, n = 154 | 24 weeks, n = 154 | |||
| SVR by RVR and baseline viral load,% (n) | < 400,000 IU/ml | 100 (42) | 94 (49) | 0.25 |
| < 600,000 IU/ml | 100 (50) | 93 (61) | 0.13 | |
| < 800,000 IU/ml | 100 (57) | 94 (69) | 0.13 | |
| < 1,000,000 IU/ml | 100 (61) | 92 (71) | 0.03 | |
| Yu and colleagues 2008 54 | PEG α-2a + RBV | PEG α-2a + RBV | ||
| 48 weeks, n = 100 | 24 weeks, n = 100 | |||
| SVR by RVR and baseline viral load, % (n/N ) | < 400,000 IU/ml (n = 52) | 100 (24/24) | 96.4 (27/28) | 1.000d |
| ≥ 400,000 IU/ml (n = 35) | 100 (18/18) | 76.5 (13/17) | 0.045 | |
| Genotype 2/3 | ||||
| Yu and colleagues 2007 55 | PEG α-2a + RBV | PEG α-2a + RBV | ||
| 24 weeks, n = 100 | 16 weeks, n = 50 | |||
| SVR by RVR, % (n/N ) | RVR | 98 (85/87) | 100 (43/43) | 1 |
| No RVR | 77 (10/13) | 57 (4/7) | 0.610 | |
| von Wagner and colleagues 2005 56 | PEG α-2a + RBV | PEG α-2a + RBV | ||
| 24 weeks, RVR n = 71 e | 16 weeks, RVR n = 71 e | |||
| SVR by RVR and baseline viral load, % (n/N ) | ≤ 800,000 IU/ml (n = 66) | 87 (27/31) | 94 (33/35) | NR |
| > 800,000 IU/ml (n = 75) | 75 (30/40) | 69 (24/35) | NR | |
Results for SVR for treatment groups as a whole, SVR by RVR, and SVR by viral load can be seen in the data extraction forms in Appendix 6.
In patients with LVL (≤ 800,000 IU/ml) who attained an RVR, SVR rates were comparable between groups who received the standard duration of treatment and those who received shortened courses, for both genotype 1 and genotypes 2 and 3. Rates were similar in five trials, ranging from 83% to 100% for standard treatment duration compared with 84%–96% for shortened treatment duration, with no statistically significant differences between treatment arms. In addition, SVRs were broadly similar regardless of genotype with the exception of the trial by Berg and colleagues,59 in which SVRs were lower than in the other studies. This may be due to the fact that these rates are only for those who first became HCV RNA negative at week 4 and do not include those who became HCV RNA negative during weeks 1–3 (as a consequence of the study design), whereas in all of the other trials the rates reflect all patients who became negative up to week 4. It should also be noted that patient numbers in these subgroups were small, and none of the trials was powered for this subgroup analysis. In the trial by Mangia and colleagues,52 in particular, only 10% of patients had an RVR and LVL.
For those with high baseline viral load, lower SVR rates were observed in patients who were treated for a shorter duration, although this was reported to be statistically significant in only two trials (100% vs 92%, p = 0.03, at < 1,000,000 IU/ml;53 100% vs 76.5% , p = 0.045, at ≥ 400,000 IU/ml54 for standard vs shortened treatment, respectively).
Virological response during treatment
The included trials varied in their lower limits of detection, with RVR defined as undetectable serum HCV RNA (< 25 IU/ml),53 serum HCV RNA negative (< 50 IU/ml),52,54,55 serum HCV RNA < 600 IU/ml56 or < 615 IU/ml,59 all at week 4 of therapy.
Table 4 presents RVR rates for each of the six included RCTs. There were no statistically significant differences between treatment groups who received the standard duration of treatment compared with those who received shortened courses, for both genotype 1 and genotypes 2 and 3.
| Study details | Group 1 | Group 2 | p-value | ||
|---|---|---|---|---|---|
| Genotype 1 | |||||
| Berg and colleagues 2009 59 | PEG α-2b + RBV | PEG α-2b + RBV | |||
| 48 weeks, n = 225 | 24 weeks, n = 28 a | ||||
| Percentage with response (n/N ):b RVR | 8.4 (19/225)c | 13.5 (28/208)c | NR | ||
| 35 (78/225)d | 37 (76/208)d | ||||
| Mangia and colleagues 2008 52 | PEG α-2a or α-2b + RBV | PEG α-2a or α-2b + RBV | |||
| 48 weeks, n = 237 | 24 weeks, n = 123 e | ||||
| Percentage with response (n/N ): RVR | 26.2 (62/237) | 26.8 (123/459)f | 0.90 | ||
| 100 (123/123)g | |||||
| Liu and colleagues 2008 53 | PEG α-2a + RBV | PEG α-2a + RBV | |||
| 48 weeks, n = 154 | 24 weeks, n = 154 | ||||
| Percentage with response (n): RVR | 63 (97) | 68 (104) | 0.47 | ||
| Yu and colleagues 2008 54 | PEG α-2a + RBV | PEG α-2a + RBV | |||
| 48 weeks, n = 100 | 24 weeks, n = 100 | ||||
| Percentage with response (n): RVR | 42 | 45 | NR | ||
| Genotype 2/3 | |||||
| Yu and colleagues 2007 55 | PEG α-2a + RBV | PEG α-2a + RBV | |||
| 24 weeks, n = 100 | 16 weeks, n = 50 | ||||
| Percentage with response (n/N ): RVR | 87 (87/100) | 86 (43/50) | NR | ||
| von Wagner and colleagues 2005 56 | PEG α-2a + RBV | PEG α-2a + RBV | PEG α-2a + RBV | ||
| 24 weeks, RVR n = 71 h | 16 weeks, RVR n = 71 h | 24 weeks, no RVR, n = 11 h | |||
| Percentage with response: RVR | 100 | 100 | 0 | NR | |
There was a large range in reported RVR between the studies, with rates in genotype 1 patients generally being lower than in genotype 2/3 patients. In the four genotype 1 trials,52–54,59 26%–68% of patients achieved an RVR, although in the subset of patients treated for 24 weeks in the Mangia and colleagues trial52 all of the patients achieved an RVR as per the study design (see Description of the included trials). The rates in this trial were lower than in the other five trials, and this may be due to the smaller proportion of patients (24%) having LVL at baseline. In the trial of genotype 2 patients by Yu and colleagues,55 rates were much higher at 86%. In the study of genotype 2/3 patients,56 two of the three treatment arms had RVR rates of 100% owing to the nature of the study design, whereby patients who achieved an RVR at week 4 were randomised (at week 8) to a total of 16 or 24 weeks’ treatment. In the trial by Mangia and colleagues,52 it is also reported that RVR rates were not significantly different between those treated with peginterferon alfa-2a compared with peginterferon alfa-2b (24% vs 29%, respectively, p = 0.14) (see Appendix 6), although results were not reported for the different treatment arms for the two peginterferons.
Early virological response rates and EOT response rates were similar for patients receiving shortened and standard duration treatment, with no statistically significant differences (where significance values were reported). As these results were presented for all patients rather than the subgroup of patients with RVR and LVL of interest to this systematic review, we have not presented these data here. However, for information they can be found in Appendix 6.
Relapse rate
Relapse was defined as the re-appearance of serum HCV RNA during the 24-week follow-up period in patients who achieved an EOT response. The RCT by Yu and colleagues54 was the only included trial to report the relapse rate in the subgroup of patients with an RVR and LVL (Table 5). In this subgroup, rates of relapse were low and were not statistically significantly different between treatment arms [3.6% vs 0% for 24 vs 48 weeks, respectively, difference 3.6%, 95% confidence interval (CI) –7.2 to 6.6, p = 1.000]. In those with an RVR and high viral load, shortening the duration of therapy resulted in higher rates of relapse, reaching statistical significance (23.5% vs 0 for 24 weeks vs 48 weeks, respectively, p = 0.045).
| Study details | Group 1 (PEG α-2a + RBV 48 weeks, n = 100) | Group 2 (PEG α-2a + RBV 24 weeks, n = 100) | p-value | |
|---|---|---|---|---|
| Relapse rate by RVR and baseline viral load, % (n/N ) | < 400,000 IU/ml (n = 52) | 0 (0/24) | 3.6 (1/28) | 1.000a |
| ≥ 400,000 IU/ml (n = 35) | 0 (0/18) | 23.5 (4/17) | 0.045 | |
Relapse rates for the other included RCTs can be found in Appendix 6. These have not been presented here because they were reported for the study groups as a whole rather than the subgroup of patients of relevance to this systematic review (i.e. those with both LVL and an RVR).
Biochemical response
Two RCTs reported biochemical response rate (normalisation of ALT levels) (Table 6). 53,56 In one trial of genotype 1 patients (Liu and colleagues53), data were analysed for 248 patients with available paired ALT levels (baseline and end of follow-up). Treatment for 24 weeks resulted in a lower ALT normalisation rate compared with 48 weeks of treatment, with the difference being statistically significant (51% vs 72%, respectively, p < 0.001). However, the study did not report the response rate for the subgroup of patients with an RVR or RVR and LVL. In the trial of genotype 2/3 patients (von Wagner and colleagues56) there was no statistically significant difference in sustained biochemical response rates between groups who achieved an RVR.
| Study details | Group 1 | Group 2 | p-value |
|---|---|---|---|
| Genotype 1 | |||
| Liu and colleagues 2008 53 | PEG α-2a + RBV | PEG α-2a + RBV | |
| 48 weeks | 24 weeks | ||
| Percentage with response (n) | 72 (107) | 51 (75) | < 0.001 |
| Genotype 2/3 | |||
| von Wagner and colleagues 2005 56 | PEG α-2a + RBV | PEG α-2a + RBV | |
| 24 weeks, RVR n = 71 | 16 weeks, RVR n = 71 | ||
| Percentage with response | 87 | 89 | NR |
Histological response
Histological response was reported by one trial in patients with genotype 1 HCV (Liu and colleagues53), and was analysed for 295 patients with available paired liver biopsy specimens (baseline and end of follow-up). However, the numbers in each treatment arm were not reported by the authors. Patients who received the shortened treatment regimen had a significantly lower histological response than those treated for the standard duration of 48 weeks (59% vs 78%, respectively, p = 0.001). Again, the study did not report the response rate specifically for the subgroup of patients with an RVR or RVR and LVL (Table 7).
| Study details | Group 1 (PEG α-2a + RBV 48 weeks) | Group 2 (PEG α-2a + RBV 24 weeks) | p-value |
|---|---|---|---|
| Percentage with response (n) | 78 (97) | 59 (71) | 0.001 |
Adverse events
Adverse events for the included studies are presented in Table 8. All of the trials presented adverse events for treatment groups as a whole, not for the subgroup of patients achieving an RVR and with LVL.
| Reported adverse events: % (n) of patients affected | Genotype 1 | Genotype 2 | Genotype 2/3 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Berg and colleagues 200959 (PEG α-2b + RBV) | Mangia and colleagues 200852 (PEG α-2a or α-2b + RBV) | Liu and colleagues 200853 (PEG α-2a + RBV) | Yu and colleagues 200854 (PEG α-2a + RBV) | Yu and colleagues 200755 (PEG α-2a + RBV) | von Wagner and colleagues 200556 (PEG α-2a + RBV) | ||||||||
| 48 weeks (n = 225) | Variablea (n = 208) | 48 weeks (n = 237) | Variablea (n = 459) | 48 weeks (n = 154) | 24 weeks (n = 154) | 48 weeks (n = 100) | 24 weeks (n = 100) | 24 weeks (n = 100) | 16 weeks (n = 50) | 24 weeks, RVR (n = 71)b | 16 weeks, RVR (n = 71)b | ||
| Dose discontinuation: | 10 (24) | 13 (59) | 10 (10) | 3 (3)c | 1 (1) | 0 | 8 (6) | 1 (1) | |||||
| Adverse event | 3 (7) | 2 (4) | 7 (16) | 7 (30) | 9 (14) | 4 (6) | 8 (8) | 3 (3) | 1 (1) | 0 | 1 (1) | 1 (1) | |
| Other reason | 3 (8) | 6 (29) | 2 (2) | 0 | 0 | 0 | 7 (5) | 0 | |||||
| Dose modificationd for adverse events/lab abnormalities | PEG α-2a | NR | NR | NR | NR | NR | NR | 24 (24) | 22 (22) | 9 (9) | 8 (4) | 19 (13) | 7 (5) |
| RBV | NR | NR | NR | NR | NR | NR | 60 (60) | 49 (49) | 51 (51) | 46 (23) | 11 (8) | 8 (6) | |
| PEG α-2a or RBV | NR | NR | NR | NR | NR | NR | 65 (65) | 54 (54) | 54 (54) | 52 (26) | NR | NR | |
| PEG α-2b or RBV | 16 | 15 | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | |
| Dose reduction for any adverse event | NR | NR | 14 (32) | 10 (47) | 53 (82) | 45 (69) | NR | NR | NR | NR | NR | NR | |
| Serious adverse events | 6.6 | 2.6 | NR | NR | 3 (4) | 7 (11) | 1 (1) | 1 (1) | 0 | 0 | 5 (7)e | ||
| Deaths, n | NR | NR | NR | NR | 1 | 0 | NR | NR | NR | NR | NR | NR | |
The incidence of dose discontinuations as a result of adverse events was reported by all six RCTs and was low across treatment groups, ranging from 0% to 9%. For three trials (all genotype 153,54,59) there appeared to be fewer discontinuations due to adverse events in those patients treated for a shortened duration, although this was not significantly different in one trial (p = 0.10)53 and not statistically tested in the other two. 54,59 However, Yu and colleagues54 found a statistically significant difference in the total incidence of treatment discontinuations (for adverse events and other reasons combined) in favour of the shortened treatment regimen (10% vs 3%, p = 0.045). In the trial by von Wagner and colleagues56 in genotype 2/3 patients, the total incidence of treatment discontinuations also appeared to favour the shortened-treatment-duration group (1% vs 8% for 16 weeks vs 24 weeks, respectively), although this was not statistically tested.
For four trials,54–56,59 the incidence of drug dose modifications for adverse events/laboratory abnormalities [classified by the studies as either peginterferon alfa-2a, RBV (ribavirin), peginterferon alfa-2a or RBV, or peginterferon alfa-2b or RBV] was observed to be lower in patients treated for a shortened duration, as might be expected, although the differences were not statistically significant54,55 or not tested. 56,59 The same trend was observed in the two trials that presented the incidence of drug dose reductions for any adverse event, but differences between treatment arms were not statistically tested. 52,53
The incidence of serious adverse events was low (range 0%–7%) as reported by five trials. 53–56,59 Frequencies were not different between treatment arms although statistical tests were generally not reported. In two trials54,56 it is not clear whether the events were related to treatment or not, although in one trial53 12 of the 15 events were considered to be treatment related (3 out of 4 vs 9 out of 11 in 48 weeks vs 24 weeks, respectively, p = 0.11). von Wagner and colleagues56 did not differentiate between the three treatment groups when reporting this outcome, so the proportion in each group is unknown. Only one death was reported,53 which was due to reactivation of pulmonary tuberculosis in a patient with a history of pulmonary tuberculosis and prolonged fever, dyspnoea and weight loss.
All of the trials reported the frequency of specific adverse events (see full data extractions in Appendix 6 for more details) with the exception of Berg and colleagues,59 who did not present the data. Most adverse events reported were typical of those commonly associated with peginterferon-based treatment. The most frequently occurring adverse events were similar across trials and included influenza-like symptoms, such as headache, fatigue and fever, insomnia, anorexia, dermatological symptoms such as skin rash/dry skin and alopecia. On the whole, the frequency of adverse events was not statistically different between treatment arms, although in three studies53,54,56 there was a trend for a lower incidence of events in patients treated for a shorter duration. Two trials54,55 reported statistical tests for comparison between groups for all the reported adverse events, two trials reported statistical comparisons for some adverse events,52,53 while two trials56,59 presented no statistical comparison between treatment groups. Liu and colleagues53 found that body weight loss (weight reduction of > 10% from baseline weight) was encountered less frequently in those receiving treatment for 24 weeks than in those receiving treatment for 48 weeks (19% vs 30%, respectively, p = 0.03). In the trial by Yu and colleagues,55 the incidence of alopecia was significantly lower in the 16-week group than in the 24-week group (20% vs 49%, respectively, p = 0.001).
Clinical effectiveness: summary
-
All six included RCTs were in patients who were eligible for shortened treatment duration. No RCTs comparing peginterferon alfa with or without ribavirin with BSC were identified for the HCV/HIV co-infection or re-treatment patient groups.
-
In the subgroup of patients who achieved an RVR and had LVL at baseline, SVR rates were comparable (i.e. no statistically significant differences) between groups who received the standard duration of treatment and those who received shortened courses, for both genotype 1 and genotypes 2 and 3. This implies that this patient group can receive shortened courses of peginterferon combination therapy without compromising SVR rates.
-
For both genotype 1 and genotype 2 and 3 patients, there were no statistically significant differences in rates of RVR between treatment groups who received the standard duration of treatment and those who received shortened courses. Rates of RVR in genotype 2/3 patients were observed to be generally higher than in genotype 1 patients.
-
Relapse rates in the subgroup of patients with LVL and RVR (one trial) were low and not significantly different between those treated for 24 versus 48 weeks.
-
Treatment for 24 weeks resulted in a significantly lower biochemical response rate (reduction of ALT to normal levels) and histological response rate than 48 weeks of treatment in one trial of genotype 1 patients. Shortening the treatment duration had no effect on biochemical response in one trial of genotype 2/3 patients. Rates of biochemical and histological response should be treated with caution, as the results relate only to those patients with available data and rates were not reported in the subgroup of patients with LVL and RVR.
-
Adverse events were presented for treatment groups as a whole and the reporting of statistical tests varied. However, the most frequently occurring adverse events were similar across all the trials and included flu-like symptoms, insomnia, anorexia, dermatological symptoms and alopecia.
-
There was a trend for a lower incidence of adverse events in patients who were treated for a shorter duration (three trials), although statistically they were comparable between treatment arms. The incidence of dose discontinuations was significantly lower in those receiving a shortened treatment regimen in one trial.
-
None of the studies was powered for subgroup analysis and therefore the results should be interpreted with caution.
Ongoing studies
The following study was identified in searches and is currently ongoing:
-
NCT 00532701. Peginterferon alfa-2a and ribavirin in patients with genotype 2 chronic hepatitis C: a randomised study of treatment duration and ribavirin dose stratified by rapid virological response. Study type: Phase IV, open-label, parallel RCT. Sample size: 700. Start date: June 2006. Estimated study completion date: June 2009. Status: currently recruiting participants. Funding: National Taiwan University Hospital. Funding amount: not stated.
Chapter 5 Economic analysis
The aim of this section is to assess the cost-effectiveness of peginterferon alfa and ribavirin in patients with chronic HCV who are:
-
eligible for a shortened course of treatment compared with standard length of treatment
-
eligible for re-treatment following previous non-response or relapse to treatment, compared with BSC
-
co-infected with HIV, compared with BSC.
The economic analysis comprises:
-
a systematic review of the literature on the cost-effectiveness of peginterferon and ribavirin treatment
-
a review of studies of the HRQoL of patients with chronic HCV from the above patient groups
-
a review of the drug manufacturers’ submissions to NICE
-
our independent economic model and cost-effectiveness evaluation (the SHTAC model).
Systematic review of existing cost-effectiveness evidence
A systematic review was undertaken to identify economic evaluations of peginterferon alfa and ribavirin in patients with chronic HCV in the subgroups outlined above (see Chapter 3 for methods). The details of the search strategy are documented in Appendix 2.
Quantity and quality of the research available
A total of 142 references were identified by the search, of which one full paper and one conference abstract were retrieved for further inspection. The full paper was included, and the conference abstract was excluded. A second full paper was identified on searching the references of the included study, and this study met the inclusion criteria. Therefore, two full economic evaluations64,65 met the inclusion criteria for the review. The study characteristics are presented in Table 9.
| Kuehne and colleagues 200264 | Campos and colleagues 200765 | |
|---|---|---|
| Publication year | 2002 | 2007 |
| Country | USA | USA |
| Study type | CUA model | CEA model |
| Study population | A cohort of HCV/HIV co-infected individuals | A treatment-eligible urban cohort, co-infected with HCV/HIV |
| Interventions |
|
|
| Treatment effect modelled |
Patients were assumed to have: |
SVR (in combination PEG α-2a and RBV) of 40%, based on one trial66 |
| Currency base | US$ | 2004, US$ |
The two included studies evaluated treatment of HCV/HIV co-infected cohorts. No economic evaluations were identified in re-treated cohorts, or in patients who were eligible for shortened courses of treatment. Both included studies were conducted in the USA, and each of the studies compared peginterferon alfa and ribavirin with peginterferon alfa monotherapy, combined non-peginterferon alfa and ribavirin, and no treatment. An additional interferon alfa monotherapy arm was included in the Kuehne and colleagues study. 64 Kuehne and colleagues64 present a cost–utility analysis, while in the more recent Campos and colleagues paper65 a cost-effectiveness analysis is reported.
The included studies were assessed based on a checklist suggested for the critical appraisal of cost-effectiveness analysis by Drummond and colleagues,67 the requirements of NICE for submissions on cost-effectiveness (reference case)68 and a suggested guideline for good practice in decision modelling by Philips and colleagues. 69
Judgements of the methodological quality of the included studies are shown in Table 10. Overall, the methodological quality of the two papers was judged to be variable.
| Kuehne and colleagues 200264 | Campos and colleagues 200765 | |
|---|---|---|
| Is there a clear statement of the decision problem? | Yes | Yes |
| Is the perspective of the model clearly stated? | Unclear | Yes |
| Is the model structure appropriate and does it fit with the clinical theory of the disease process? | Yes | Yes |
| Are assumptions reasonable and appropriate? | Yes | Yes |
| Is the comparator routinely used in the UK NHS? | Yes | Yes |
| Is the study type and modelling methodology reasonable? | Yes | Yes |
| Is the patient group in the study similar to those of interest in the UK NHS? | Yes | Yes |
| Is the health-care system or setting comparable to the UK? | No | No |
| Have the costs and outcomes been discounted? | NR | Yes |
| Are the health states and parameters used in the model described clearly? | No | Unclear |
| Is the effectiveness of the intervention established based on a systematic review? | No | No |
| Are health benefits measured in QALYs using a standardised and validated generic instrument? | Unclear | No |
| Are the resource costs reasonable? | Unclear | Unclear |
| Has uncertainty been assessed? | Yes | Yes |
| Has the model been validated? | Yes | Yes |
Neither of the included studies derived the treatment effectiveness measure used in the evaluation from a systematic review. Kuehne and colleagues64 cite several sources for the treatment efficacy measure. The study by Kuehne and colleagues64 was conducted prior to the publication of trials of antiviral treatment in co-infected patients. The treatment efficacy measure therefore comes from studies of the treatment of mono-infected patients, and should therefore be viewed with caution. No details are reported on how, or if, these results have been statistically pooled. Campos and colleagues65 used an effectiveness measure from a large RCT of co-infected patients: APRICOT (AIDS Pegasys Ribavirin International Co-infection Trial). 66 The use of the efficacy measure from this trial has not been justified within the paper.
Both of the included studies provide a clear statement of the decision problem, which is to assess the cost-effectiveness of the various interventions for HCV in a co-infected cohort, and adopt an appropriate model structure in order to address this.
Both models have been validated by comparing the predicted rate of future cirrhosis progression with those found in published cohort studies. Kuehne and colleagues64 found a comparable rate of cirrhosis progression when comparing the model’s predictions with a published cross-sectional study70 (16.9% vs 14.9%, respectively). Campos and colleagues’ model65 was compared with a study of co-infected former injection drug-using patients by Di Martino and colleagues. 71 The rates were similar: 17.5% in the Di Martino and colleagues study71 versus 16% in Campos and colleagues,65 over the same follow-up period.
Kuehne and colleagues64 stated that a societal perspective had been adopted, but with no indication of patient-borne costs. Costs and outcomes are discounted in the Campos and colleagues study65 at 3%; no discount rate is reported in the Kuehne and colleagues paper. 64 Campos and colleagues65 also clearly describe the perspective of the model as societal, with patient time costs being excluded.
The initial assumptions in both studies appear reasonable and appropriate, although several of the assumptions listed by Kuehne and colleagues64 did not have any sources attached. While the assumptions adopted in both papers are broadly similar, the fibrosis rate in the absence of effective treatment was conditional on age and sex, and patients with decompensated cirrhosis were eligible for liver transplantation in the Campos and colleagues paper. 65 Kuehne and colleagues64 assumed that minor adverse effects of treatment resulted in additional costs and a temporary decrease in QoL, and that major toxicity would result in discontinuation of treatment. This disutility is not defined in the paper, although the authors state that data from several studies are used to derive a ‘plausible range’ for the risk.
The health states used in the Campos and colleagues model65 are described clearly, and appear relevant to the UK. The cost parameters and disease progression transition probabilities are reported, but how these are derived is unclear. The authors stated that they have been modified from previously published data, but did not elaborate further on the methods used, with the exception of the assumption that the rate of progression to decompensated cirrhosis was comparable between co-infected and mono-infected patients.
It is unclear whether the disease progression rates in the study by Kuehne and colleagues64 have been derived from the literature or are empirically calibrated to the observed data. The relative risks (RRs) used in the base-case analysis for progression of cirrhosis in co-infected patients compared with mono-infected patients are not justified in the paper, although these are tested in the sensitivity analysis. The liver disease utility values are sourced from several references, but, again, the methods used in pooling these results are not reported, and there is no explanation of how rates for co-infected patients have been derived from those of mono-infected patients.
The probabilities of SVR in the groups according to genotype and treatment in the two included studies are presented in Table 11.
| Treatment strategy | Base-case probabilities (%) | |
|---|---|---|
| Kuehne and colleagues 200264 | Campos and colleagues 200765 | |
| Interferon alfa (48 weeks) | ||
| Genotype 1 | 6 (2–8) | |
| Genotype non-1 | 27 (15–28) | Not applicable |
| Interferon alfa + ribavirin (24 weeks) | ||
| Genotype 1 | 16 (14–28) | Not applicable |
| Genotype non-1 | 69 (62–73) | |
| Interferon alfa + ribavirin (48 weeks) | ||
| Genotype 1 | 33 (28–40) | 7 |
| Genotype non-1 | 75 (61–85) | 18 |
| Peginterferon alfa (48 weeks) | ||
| Genotype 1 | 14 (12–31) | 14 |
| Genotype non-1 | 46 (40–67) | 31 |
| Peginterferon alfa + ribavirin (48 weeks) | ||
| Genotype 1 | 42 (34–45) | 29 |
| Genotype non-1 | 79 (76–88) | 58 |
Kuehne and colleagues’ annual SVR probabilities64 are considerably higher than those reported by Campos and colleagues65 for interferon alfa and ribavirin for 48 weeks’ duration [33% (28%–40%) vs 7%, respectively, in genotype 1]. This gap is more pronounced in the same treatment strategy for patients with genotype non-1: Kuehne and colleagues64 reported 75% (61%–85%) compared with Campos and colleagues65 reporting 18% for this group. The SVR probabilities are similar between the studies for peginterferon monotherapy and genotype 1: both studies reported 14% for genotype 1 and 46% by Kuehne and colleagues64 versus 31% by Campos and colleagues65 for genotype non-1. In patients receiving peginterferon combined with ribavirin these probabilities were again higher in the Kuehne study:64 in genotype 1, Kuehne and colleagues64 reported 42% (34%–45%) versus 29% in the Campos study,65 and in genotype non-1 they were 79% versus 58% respectively. In all treatment strategies and genotypes, with the exception of genotype 1 and peginterferon alfa monotherapy, Kuehne and colleagues64 employed higher probabilities of SVR, with the difference in the case of interferon alfa and ribavirin for 48 weeks being substantial. As mentioned earlier, these SVRs were based on mono-infected patients.
Health benefits in the Kuehne and colleagues study64 are measured in years of life saved (YLS), in quality-adjusted life-months and QALYs. The authors stated that the quality weights for HCV-specific health states were derived from published studies using the visual analogue scale, and that HIV health states were derived from studies based upon the HIV Cost and Services Utilization Study. 72–75 It is not reported what instrument was used in this study. Campos and colleagues65 measured health benefits in YLS.
Whether the selected resource costs are reasonable is judged to be unclear in both of the included studies. In both cases the costs are relevant to the US health-care system. Both studies used cost-of-care estimates from a study published in 1997,76 which, in turn, modelled the cost-effectiveness of interferon alfa-2b, and in which the resources were based on estimates by a panel of hepatologists. A base year for costs is not given, but the authors state that all costs were converted to constant dollars. Hepatitis C costs were previously published costs, again based upon estimates from an expert panel. The costs of HIV care were based upon previously published studies; the authors stated that the estimates derived were similar to those given in other sources of costs incurred by HIV/AIDS.
Uncertainty is assessed in both of the included studies through sensitivity analyses. Univariate and multivariate sensitivity analyses were carried out in both studies to compare the effect of alternative assumptions compared with those in the base case. Selected results are reported. Neither study has reported a probabilistic sensitivity analysis (PSA) or cost-effectiveness acceptability curve (CEAC).
Relevance of the studies to the UK
The patient group in the model is similar to one of those currently of interest in this appraisal – patients co-infected with HIV. However, the Kuehne and colleagues study64 focuses on patients with moderate HCV liver-related disease, whereas the current NICE guidance covers patients with moderate to severe38 and mild chronic HCV. 33 The US health system, in which both of the studies are based, is not comparable to the UK NHS, and this will therefore extend to the costs incurred within it.
Assessment of cost-effectiveness
The base-case results reported by Kuehne and colleagues64 are presented in Tables 12 and 13, below, with a summary of those reported by Campos and colleagues65 presented in Table 14. It is difficult to directly compare the results of the two studies, as they are reported very differently. Campos and colleagues65 have reported their results by sex and genotype, whereas Kuehne and colleagues64 have reported results by CD4 cell count (350 and 200 cells/µl), by mild or moderate disease, and by genotype 1 or genotype non-1. Both studies report the incremental cost by YLS, and Kuehne and colleagues64 additionally present incremental costs per QALY for each subgroup.
| Patient group | Treatment type | US$/YLS | US$/QALY |
|---|---|---|---|
| Co-infected patients with CD4 cell counts of 350 cells/µl and mild chronic HCV | No treatment | – | – |
| IFN 48 weeks | Dominateda | Dominateda | |
| IFN + RBV 24 weeks | Dominateda | Dominateda | |
| IFN + RBV 48 weeks | Dominatedb | Dominatedb | |
| PEG 48 weeks | 107,900 | 35,900 | |
| PEG + RBV 48 weeks | 349,900 | 113,100 | |
| Co-infected patients with CD4 cell counts of 200 cells/µl and mild chronic HCV | No treatment | – | – |
| IFN 48 weeks | Dominateda | Dominatedb | |
| IFN + RBV 24 weeks | Dominateda | Dominateda | |
| IFN + RBV 48 weeks | Dominatedb | Dominatedb | |
| PEG 48 weeks | 1,401,200 | 340,600 | |
| PEG + RBV 48 weeks | 4,293,900 | 937,200 | |
| Co-infected patients with CD4 cell counts of 350 cells/µl and moderate chronic HCV | No treatment | – | – |
| IFN 48 weeks | Dominateda | Dominateda | |
| IFN + RBV 24 weeks | Dominateda | Dominateda | |
| IFN + RBV 48 weeks | 18,500 | 11,600 | |
| PEG 48 weeks | Dominatedb | Dominatedb | |
| PEG + RBV 48 weeks | 65,100 | 40,000 | |
| Co-infected patients with CD4 cell counts of 200 cells/µl and moderate chronic HCV | No treatment | – | – |
| IFN 48 weeks | Dominateda | Dominateda | |
| IFN + RBV 24 weeks | Dominateda | Dominateda | |
| IFN + RBV 48 weeks | Dominatedb | Dominatedb | |
| PEG 48 weeks | 184,200 | 85,900 | |
| PEG + RBV 48 weeks | 594,800 | 267,200 |
| Patient group | Treatment type | US$/YLS | US$/QALY |
|---|---|---|---|
| Co-infected patients with CD4 cell counts of 350 cells/µl and mild chronic HCV | No treatment | – | – |
| IFN 48 weeks | Dominateda | Dominateda | |
| IFN + RBV 24 weeks | 37,400 | 11,900 | |
| IFN + RBV 48 weeks | 347,000 | 112,100 | |
| PEG 48 weeks | Dominatedb | Dominatedb | |
| PEG + RBV 48 weeks | 894,000 | 300,800 | |
| Co-infected patients with CD4 cell counts of 200 cells/µl and mild chronic HCV | No treatment | – | – |
| IFN 48 weeks | Dominateda | Dominateda | |
| IFN + RBV 24 weeks | 541,300 | 104,400 | |
| IFN + RBV 48 weeks | 3,865,600 | 1,088,500 | |
| PEG 48 weeks | Dominatedb | Dominatedb | |
| PEG + RBV 48 weeks | 11,827,300 | 4,000,000 | |
| Co-infected patients with CD4 cell counts of 350 cells/µl and moderate chronic HCV | No treatment | – | – |
| IFN 48 weeks | 4700 | 2900 | |
| IFN + RBV 24 weeks | Dominatedb | Dominatedb | |
| IFN + RBV 48 weeks | 63,500 | 38,800 | |
| PEG 48 weeks | Dominatedb | Dominatedb | |
| PEG + RBV 48 weeks | 169,700 | 105,300 | |
| Co-infected patients with CD4 cell counts of 200 cells/µl and moderate chronic HCV | No treatment | – | – |
| IFN 48 weeks | Dominateda | Dominateda | |
| IFN + RBV 24 weeks | 67,900 | 29,800 | |
| IFN + RBV 48 weeks | 561,200 | 265,100 | |
| PEG 48 weeks | Dominatedb | Dominatedb | |
| PEG + RBV 48 weeks | 1,558,800 | 771,200 |
| Patient group | Treatment strategy | Incremental cost per YLS (US$) |
|---|---|---|
| Men | ||
| Genotype 1 | PEG + RBV | 73,000 |
| Genotype non-1 | PEG + RBV | 39,700 |
| Women | ||
| Genotype 1 | PEG + RBV | 70,000 |
| Genotype non-1 | PEG + RBV | 39,300 |
In the Kuehne and colleagues study,64 both peginterferon alfa monotherapy and peginterferon alfa plus ribavirin in combination dominated the other strategies in genotype 1 patients, with CD4 cell counts of 350 cells/µl and 200 cells/µl and mild chronic HCV, and in patients with a CD4 cell count of 200 cells/µl and moderate HCV. Peginterferon alfa monotherapy was the more cost-effective in each case. In patients with CD4 cell counts of 350 cells/µl and moderate HCV, peginterferon plus ribavirin and interferon plus ribavirin dominated, whereas the latter was the most cost-effective at US$11,600 versus US$40,000 for peginterferon in combination per QALY gained.
In the base-case analysis for genotype non-1 patients, again peginterferon alfa plus ribavirin in combination was not the most cost-effective of the treatment strategies tested. In patients in this group with mild disease, the monotherapies were dominated in each case. In patients with CD4 cell counts of 350 and 200 cells/µl, the lowest cost per QALY gained came from the 24-week course of interferon plus ribavirin at US$11,900 and US$104,400, respectively. In both cases, peginterferon and ribavirin (48 weeks) was the least cost-effective of the dominating strategies at US$300,800 in patients with 350 cells/µl, and US$4,000,000 in patients with 200 cells/l.
In patients with genotype non-1 moderate HCV and a CD4 cell count of 350/µl, interferon monotherapy for 48 weeks was most cost-effective at US$2900 per QALY. Interferon in combination with ribavirin for 24 weeks was the most cost-effective strategy in patients with CD4 cell counts of 200 cells/µl and with moderate HCV.
Peginterferon alfa in combination with ribavirin dominated all other strategies (all assumed to have been received for 48 weeks) in each patient subgroup reported in the model by Campos and colleagues. 65 The authors’ results suggested that the incremental cost per YLS of peginterferon with ribavirin in patients with genotype non-1 is approximately one-half of that of the incremental cost in patients with genotype 1. This is the case for both men and women.
In the Campos and colleagues study,65 incremental costs per YLS saved were comparable between men and women with the same genotype of HCV virus: US$73,000 (men) versus US$70,000 (women) in genotype 1, and US$39,700 (men) versus US$39,300 (women) in genotype non-1. The incremental costs per YLS for each of the other strategies were not reported in detail, as they were dominated by peginterferon and ribavirin. 65
Sensitivity analyses
The authors of both studies report that the results are sensitive to the discount rate. In Kuehne and colleagues,64 this is a variable to which the results appear most sensitive; however, the discount rate applied in the base-case analysis was not reported. Campos and colleagues65 describe their results as sensitive to the discount rate: a 0% rate resulted in an ICER 60% lower than the base case, whereas a 5% discount rate resulted in an ICER of 140% higher than the base case.
The results in both the included studies are sensitive to the fibrosis progression rates. In a two-way sensitivity analysis with the effectiveness of combination peginterferon alfa plus ribavirin and disease progression, Campos and colleagues65 reported that cost-effectiveness ratios were < US$50,000 per YLS, regardless of fibrosis, when treatment efficacy exceeded 50%. This is higher than the base-case treatment efficacy of 40%. Where treatment efficacy was < 25%, cost-effectiveness ratios were < US$100,000 across the range of RRs, although these are described as having had ‘slightly more influence’ (p. 277). 65 No further details of how, or the degree to which, the RR is influential are reported. Kuehne and colleagues64 reported that in mild HCV and peginterferon plus ribavirin the difference in their ICER from the base case was largest when the RR was between 1 and 2, with less sensitivity to RRs > 3. Changes in the RR of progression had a greater effect on the ICER in patients with mild HCV than in those with moderate HCV.
The order of the strategies described in the study by Campos and colleagues65 remained the same when it was assumed that treatment was discontinued in the absence of an EVR – US$59,300 per YLS for men in genotype 1 versus US$33,100 per YLS for men in genotype non-1; the results in women again reflected this. The results were reported by the authors as being most sensitive to variation in the annual excess death rate owing to HIV, fibrosis progression rates and treatment efficacies in non-cirrhotic patients, and as being ‘moderately’ sensitive to drug costs. None of these was reported in detail across the patient subgroups or intervention strategies. Where no discount rate was applied this resulted in an ICER that was 60% lower than the base-case analysis; a 5% discount rate saw the ICER increase to 140% higher. The cost of the peginterferon alfa and ribavirin strategy was varied from 50% to 150% of the base-case value, which resulted in ICERs of between US$56,300 and US$88,000 per YLS, respectively. The variation in death rate owing to HIV was illustrated by an example of the excess mortality being reduced by 97%, reducing the ICER to US$41,000 per YLS. No justification for this reduction is described. However, where this was increased 11-fold to reflect death rates in patients with a history of severe opportunistic infections, treatment is dominated by non-treatment. No results are reported for the fibrosis progression rates or treatment efficacy one-way analyses.
Campos and colleagues65 further describe a two-way sensitivity analysis whereby the effectiveness of the combination therapy of peginterferon alfa plus ribavirin and the RR of fibrosis progression due to co-infection were varied. Where efficacy was increased by 50%, the cost-effectiveness ratios decreased to < US$50,000 per YLS, and this was not sensitive to the variation in fibrosis progression. Where efficacy was decreased by 25% the cost-effectiveness ratios decreased to < US$100,000 per YLS. The authors state that this was ‘slightly more’ sensitive to fibrosis progression.
Kuehne and colleagues64 performed a number of one-way sensitivity analyses in patients who were receiving interferon alfa plus ribavirin and peginterferon plus ribavirin. The ICERs were found to be most sensitive to the RR of progression to cirrhosis compared with mono-infected patients. In the figure in the study publication, the ICER appears most sensitive to the discount rate, HAART efficacy, relapse after a sustained response and cost of ribavirin in patients receiving interferon alfa combination therapy for 48 weeks compared with 24 weeks, as well as discount rate, relapse rate and HAART efficacy in peginterferon alfa plus ribavirin compared with interferon alfa plus ribavirin for 48 weeks. Minor adverse events are reported as having little impact on the ICERs, while major toxicity in 20% of patients receiving 48 weeks of peginterferon combination therapy increased this ICER from US$40,000 to US$69,000. Decreasing utility estimates by 10% during 48 weeks of therapy led to this strategy dominating the non-peginterferon-based treatments.
The authors further reported that the ICER was minimally sensitive to minor toxic effects, with no further details. Major toxicity could affect the effectiveness of HAART in this group, and a sensitivity analysis was undertaken: if the effectiveness of HAART was reduced by 50% in 20% of the patients receiving peginterferon and ribavirin, the ICER increased from US$40,600 to US$69,000 per QALY.
Summary
-
Two economic evaluations64,65 of treatment strategies in patients co-infected with HCV/HIV were included in the review. No studies assessing the cost-effectiveness of shortened courses of treatment or re-treating patients who had not-responded to, or failed, previous therapy were identified.
-
The papers were found to be of mixed methodological quality overall. The authors presented a clear decision problem in co-infected patients, using an appropriate study design and model structure. These were state-transition models with SVR as the main measure of treatment effectiveness. It is not clear how the effectiveness measures have been derived.
-
The studies are both based in the USA and therefore both the setting and costs are unlikely to be generalisable to the UK NHS.
-
There are notable differences in the SVR probabilities used by the two included studies. This is likely to be owing to SVRs in the study by Kuehne and colleagues64 being derived from studies of mono-infected patients.
-
The costs in both papers appear to have been taken from a previous published study in which resource use was estimated by expert opinion. The ICERs in the Campos and colleagues study65 are sensitive to these costs.
-
Sensitivity analyses have been reported in both studies, but a PSA has not been conducted in the Campos and colleagues paper,65 where this would now be considered standard practice.
-
Kuehne and colleagues64 reported that their results in HCV/HIV co-infected patients were most sensitive to the RR of progression to cirrhosis compared with HCV mono-infected patients. It is difficult to ascertain from the paper how these RRs were derived.
-
In the Kuehne and colleagues study,64 a clear pattern does not emerge over the reported subgroups. While peginterferon plus ribavirin is a dominant strategy in each subgroup, it is not the most cost-effective strategy in any of these groups.
-
Campos and colleagues65 concluded that peginterferon alfa plus ribavirin is the dominating strategy in all patient subgroups reported. In contrast, Kuehne and colleagues64 reported varied results across their subgroups, but in each, peginterferon and ribavirin combination therapy was the least cost-effective of the dominating strategies.
-
These results should be viewed with caution owing to the mixed methodological quality of the included studies.
Review of manufacturers’ submissions
Roche submission to NICE: cost-effectiveness analysis
Overview
The Roche submission to NICE in support of peginterferon alfa-2a consists of a 226-page written document (containing submitted evidence on the clinical effectiveness and a cost-effectiveness analysis) and a fully executable, electronic copy of the manufacturer’s economic model. The MS reports cost-effectiveness results for the three populations covered by the scope for this NICE appraisal:
-
Patients who have previously been treated with peginterferon alfa, including both those who did not respond to previous treatment (by viral genotype) and those who relapsed on previous treatment. Costs and outcomes for these patients are compared with supportive care, in line with the scope for this appraisal.
-
Patients with LVL and RVR who receive shortened courses of treatment with peginterferon alfa (by viral genotype). Costs and outcomes for these patients are compared with treatment for the same group of patients receiving the standard duration of treatment, in line with the scope for this appraisal.
-
Patients co-infected with HCV/HIV. Costs and outcomes for these patients are compared with treatment for the same group of patients receiving non-peginterferon alfa therapy, which is not consistent with the scope for this appraisal.
The perspective of the analysis is not stated, but appears to be consistent with the NICE reference case68 of the NHS and Personal Social Services (PSS), capturing direct costs and benefits only. The submission reports lifetime costs and outcomes (reported as life expectancy and QALYs) for each treatment arm and the incremental costs and outcomes for peginterferon alfa-2a combined with ribavirin compared with usual care (which varies between patient populations, as stated above).
Below we outline the approach taken by the manufacturer and provide an outline review based on a checklist suggested for the critical appraisal of cost-effectiveness analysis by Drummond and colleagues,67 the requirements of NICE for submissions on cost-effectiveness (reference case),68 and a suggested guideline for good practice in decision modelling by Philips and colleagues. 69
Modelling approach
The cost-effectiveness analysis model adopted for the MS is a state-transition model that is structurally similar to published models previously used in the population of patients with chronic HCV,76–81 including our previous assessment report17 for NICE (TA106). The model has a lifetime horizon (in the base-case analysis the cohort simulation is truncated at patient age of 99 years), with a cycle length of 1 year, and is used to estimate the morbidity and cost resulting from progressive liver disease and treatment costs (up to a maximum duration of treatment with peginterferon alfa-2a of 72 weeks). The model has five health states indicating progressive liver disease (HCV, compensated cirrhosis, decompensated cirrhosis, HCC and liver transplantation), one state representing a treatment response (SVR) and one absorbing state (death), although this last state is broken down to differentiate deaths from progressive liver disease and deaths from all other causes. Unlike the model adopted in our previous assessment,17 the model developed for the MS does not distinguish the stage of liver disease in non-cirrhotic patients with chronic HCV (i.e. there is no distinction between mild and moderate HCV). The impact of this structural assumption is not discussed in the MS.
The main treatment effect applied in the model is the SVR for treated patients, with the proportion of patients in each of the modelled populations achieving an SVR based on data from clinical trials conducted in the relevant patient populations, reported in the MS (discussed in Data inputs; see also Appendix 3). Patients who achieve an SVR are assumed in the model to be ‘cured’ and do not face any risk of reactivation of disease or any excess risk of progressive liver disease (above that of a general population). Age-specific mortality risks for the general population, weighted for the proportion of men in the baseline cohort, are applied to patients achieving an SVR. Patients who do not achieve an SVR are at risk of progressive liver disease and are assumed to face the same risks of disease progression as untreated patients. Risks of disease progression and, where relevant, excess mortality risks associated with advanced liver disease states in the model have been drawn from natural history studies.
The base-case population in each analysis is the same, with all patients entering the model being non-cirrhotic, with chronic HCV. The simulated patient cohort has a mean age of 45 years, with 70% being male. These assumptions have no impact on response to treatment (i.e. SVRs in the model are not broken down by age or sex), but affect the all-cause mortality rates applied in the model. Patient weight is assumed to be greater than 75 kg – again this has no impact on the patient response to treatment, but has an impact on the cost of treatment, as ribavirin dosage is weight related. The MS discusses these assumptions in relation to the characteristics of patients recruited to the clinical trials used to estimate the SVRs applied in the model. However, there is no discussion of the relevance of these characteristics to the population of UK patients with chronic HCV or in the modelled populations.
Health-state utilities applied to the chronic HCV and progressive liver disease states in the model were taken from the UK Mild Hepatitis C Trial. 82 Age-specific utility values [reported for a general population survey using the European Quality of Life-5 Dimensions (EQ-5D83) and valued using a UK general population tariff84] were applied for only the SVR state. The MS does not discuss the possible implications of using age-specific utility values for one state and not for others (discussed in Data inputs). The model does not include treatment-related adverse events, other than to reduce utility in the year of treatment by 0.11 (from 0.66 to 0.55).
The costs applied in the submission were made up of two components. Treatment-related costs (which for peginterferon alfa-2a combination therapy consist of drug acquisition costs, monitoring of patients on treatment and surveillance of patients once treatment has stopped) were estimated separately from health-state costs. The latter relate to service use associated with management of progressive liver disease, associated with chronic HCV infection in patients who do not respond to treatment and for patients whose disease progresses despite demonstrating a response to treatment.
Drug usage for peginterferon alfa-2a was based on a dosage of 180 µg/week, supplied in a prefilled syringe and self-administered by patients, at a cost of £126.91. The dose of ribavirin used in combination with peginterferon alfa-2a varies by patient group and by weight, in the case of genotype 1 patients (Table 15). Expected duration of treatment with the combination of peginterferon alfa-2a plus ribavirin also varies by patient group (see Table 15 for a summary).
| Patient group included in model | RBV dose per day (mg) | RBV cost per week (£) | Treatment duration (weeks) |
|---|---|---|---|
| Re-treatment of non-responding patients | |||
| Genotype 1 | 1000/1200a | 84.15/100.98 | 72 |
| Genotype non-1 | 800 | 67.32 | 48 |
| Re-treatment of relapsed patients | |||
| Genotype 1 | 1000/1200a | 84.15/100.98 | 48 |
| Genotype non-1 | 800 | 67.32 | 48 |
| Shortened duration of treatment | |||
| Genotype 1 | 1000/1200a | 84.15/100.98 | 48/24b |
| Genotype 2/3 | 800 | 67.32 | 24/16b |
| HCV/HIV co-infected | |||
| All genotypes | 800 | 67.32 | 48 |
Resource use for patient monitoring associated with peginterferon alfa-2a and ribavirin combination therapy and surveillance of patients following treatment cessation was estimated using management protocols, which were developed using expert opinion for our previous report17 for NICE (TA106). The original costing protocols were slightly modified (to include quantitative, rather than qualitative, HCV viral load at key assessment stages) and were inflated to 2007–8 prices using the Hospital and Community Health Services (HCHS) Pay and Prices Index85 (Table 16).
| Duration of treatment | Cost (£) |
|---|---|
| On-treatment monitoring (weeks) | |
| 12 weeks | 568 |
| 16 weeks | 600 |
| 24 weeks | 795 |
| 48 weeks | 1473 |
| 72 weeks | 1711 |
| Post-treatment surveillance | |
| Non-responders | 102 |
| Responders (SVR) | 167 |
Health-state costs in the model are based on values adopted in our previous assessment,17 inflated from 2003–4 to 2007–8 prices using the HCHS Pay and Prices Index85 (Table 17).
| Health state | Health-state cost (£) |
|---|---|
| Moderate chronic hepatitis C | 843 |
| CC | 1338 |
| DC | 10,725 |
| HCC | 9557 |
| Liver transplantation, first year | 43,263 |
| Liver transplantation, subsequent years | 1628 |
Model/cost-effectiveness results
The MS reports total costs (broken down as treatment-related costs and future costs of medical care for HCV) and outcomes (life expectancy and QALYs) for peginterferon alfa-2a combination therapy and each comparator modelled separately, as well as an incremental analysis (these are summarised in Table 18). Scatter plots showing the cost-effectiveness plane (incremental cost and incremental QALYs for peginterferon alfa-2a combination therapy) from PSA are also reported for each patient population, as well as CEACs for re-treatment of patients who failed to respond to previous treatment with peginterferon.
| Patient group | Genotype | Treatment | Cost (£) | QALYs | ICER (£ per QALY gained) |
|---|---|---|---|---|---|
| Non-responders | 1 | No treatment | 27,114 | 11.06 | 3334 |
| PEG α-2a + RBVa | 29,224 | 11.69 | |||
| Non-1 | No treatment | 27,114 | 11.06 | 809 | |
| PEG α-2a + RBVb | 27,942 | 12.08 | |||
| Relapsed on previous treatment | All | No treatment | 27,114 | 11.06 | Dominant |
| PEG α-2a + RBV | 21,199 | 13.74 | |||
| Shortened treatment duration for patients with LVL and RVR | 1 + 4 | PEG α-2a + RBV 48 weeks | 13,387 | 15.78 | 15,472 |
| PEG α-2a + RBV 24 weeks | 8866 | 15.49 | |||
| 2 + 3 | PEG α-2a + RBV 24 weeks | 8053 | 15.64 | 2719 | |
| PEG α-2a + RBV 16 weeks | 7391 | 15.39 | |||
| HCV/HIV co-infected patients | All | IFN α-2a + RBV | 32,431 | 11.62 | Dominant |
| PEG α-2a + RBV | 28,786 | 12.99 |
The MS states that peginterferon alfa-2a in combination with ribavirin is cost-effective in all modelled comparisons for all populations (below a threshold of £15,000), emphasising that treatment dominates the ‘current standard of care’ for relapsed patients and for those with HCV/HIV co-infection. These conclusions are reflected in the manufacturer’s PSA where:
-
the probability of peginterferon alfa-2a combination being cost-effective (at a threshold of £20,000) was 100% for re-treating patients who failed to respond to previous peginterferon treatment (both for genotype 1 and genotype non-1 patient subgroups)
-
treatment for patients who relapsed on previous peginterferon alfa treatment and for HCV/HIV co-infected patients was dominated in the majority (99%) of simulations. However, as stated earlier, the comparator included in the model for HCV/HIV co-infected patients was non-peginterferon alfa combination therapy – not supportive care as specified in the scope.
The interpretation of the results of the model for patients receiving shortened duration of treatment is complicated by the fact that, although shortened treatment duration is associated with significant savings in treatment costs, it incurs a penalty in terms of a reduced SVR compared with standard durations (from 97% to 91% for genotypes 1 and 4 and from 94% to 89% for genotypes 2/3). As a result, the incremental cost and incremental QALYs associated with shortened treatment duration are negative (for genotypes 1 and 4 the total cost is reduced by £4500 and total QALYs are 0.29 lower, whereas for genotypes 2 and 3 the total cost is reduced by £660 and total QALYs are 0.25 lower), yielding a positive ICER. However, this cannot be interpreted using the commonly assumed decision rule – is the ICER below a given (arbitrary) threshold – as the manufacturer’s have done in their conclusions (section 7.5, p. 182 of the MS), selecting a threshold of £15,000 per QALY gained. In this situation the logic is reversed whereby ICERs below the threshold are rejected. 86 This can perhaps be better understood by considering the analysis using the net benefits framework where we would accept an intervention with positive incremental net benefit, i.e. where the value of incremental benefits exceeds the incremental costs (see Appendix 8 for more details). This requires costs and benefits to be valued on the same scale – commonly achieved by multiplying the incremental effect (incremental QALYs) by a given threshold value (willingness to pay per QALY gained), as below:
where ΔE is incremental QALYs, ΔC is incremental cost and λ is the threshold.
Applying this framework to the analysis of patients receiving shortened duration of treatment presented by the manufacturer, for a range of threshold values (λ) from £0 to £30,000 per QALY gained (Table 19), the incremental net monetary benefit for shortened duration of treatment is positive for genotype 2/3 at only comparatively low threshold values (below the ICER value of £2719). For genotype 1/4 patients the incremental net monetary benefit is positive over a wider range of willingness-to-pay values (below the ICER value of £15,472).
| ΔC | ΔE | 0 | £10,000 | £20,000 | £30,000 | |
|---|---|---|---|---|---|---|
| Genotypes 1 + 4 | –£4521 | –0.29 | 4521 | 1599 | –1323 | –4245 |
| Genotypes 2 + 3 | –£662 | –0.24 | 662 | –1773 | –4208 | –6643 |
Outline appraisal of the cost-effectiveness analysis undertaken
The NICE reference case requirements (Roche) are shown in Table 20.
| NICE reference case requirements68 | Included in submission |
|---|---|
| Decision problem: as per the scope developed by NICE | ✗a |
| Comparator: alternative therapies routinely used in the UK NHS | ✓ |
| Perspective on costs: NHS and PSS | ✓ |
| Perspective on outcomes: all health effects on individuals | ✓ |
| Type of economic evaluation: cost-effectiveness analysis | ✓ |
| Synthesis of evidence on outcomes: based on a systematic review | ×b |
| Measure of health benefits: QALYs | ✓ |
| Description of health states for QALY calculations: use of a standardised and validated generic instrument | ✓ |
| Method of preference elicitation for health-state values: choice-based method (e.g. TTO, SG, not rating scale) | ✓ |
| Source of preference data: representative sample of the public | ✓c |
| Discount rate: 3.5% pa for costs and health effects | ✓ |
Outline review of modelling approach
Model structure/structural assumptions
The MS reports that update searches of MEDLINE and EMBASE (based on the search strategies from our previous assessment17) were conducted to identify economic evaluations published since the searches reported in our previous assessment. 17 This search is not discussed in the main body of the submission, but is included in an appendix. The appendix to the MS states that the purpose of this review was to identify more recent sources (for transition probabilities, costs and utilities) to populate the economic model. The MS does not report full details on any of the economic evaluations identified by this search, nor whether any of these were conducted for patient populations covered by this review. The MS does not present a review of published economic evaluations or discuss alternative approaches to modelling the cost-effectiveness of antiviral treatment for chronic HCV infection.
The manufacturer’s model is structurally similar to published models previously used in the population of patients with chronic HCV,76–81 including our previous assessment. 17 The states representing more advanced liver disease in the model (compensated cirrhosis, decompensated cirrhosis, HCC and liver transplantation) are commonly accepted as distinct stages of progressive liver disease, which can be distinguished by their impact on QoL, resource use or excess mortality risk. However, this model does not distinguish the stage of disease in non-cirrhotic patients with chronic HCV. In terms of the health-state utility value (0.66) and the transition probability for progressing to compensated cirrhosis (0.037), this health state has the characteristics of moderate HCV. There is no discussion in the MS of the rationale for adopting this structure nor of the possible implications, for the cost-effectiveness analysis, of assuming that all patients enter the model with moderate HCV (as opposed to mild or severe HCV). The MS does not report any evidence of approaches to establish the internal consistency of the model, or any evidence of external validation (by expert clinical opinion or by comparison with other published economic evaluations).
The effect of treatment is to induce an SVR in a proportion of patients, which is assumed to be a permanent cure. This approach is in accordance with previously published models in this patient population and would agree with long-term follow-up studies of patients achieving SVR on treatment. However, recent publications have highlighted a risk of liver cancer in patients who have undergone SVR – particularly in patients with compensated cirrhosis at baseline – which, while lower than for non-responding patients, is not completely eradicated. A retrospective study of 920 Italian patients with cirrhosis treated with interferon reported HCC incidence rates of 0.66 per 100 person-years of follow-up (95% CI 0.27 to 1.37) for those who achieved an SVR, and 2.10 per 100 person-years of follow-up (95% CI 1.75 to 2.51) for non-SVR patients. 87 The hazard ratio for HCC in non-SVR patients compared with those who achieved an SVR was 2.59 (95% CI 1.13 to 5.97). The manufacturer’s model assumes a zero risk of HCC for patients in the SVR state: this may be reasonable given that all patients were assumed to enter the model in the chronic HCV state. However, under current guidance, patients with compensated cirrhosis may undergo treatment with peginterferon alfa, and a model that allowed patients to enter in all treatment-eligible states would be likely to produce more generalisable results.
Treatment-related adverse events are not included in the model, other than to reduce utility in the year of treatment by 0.11 (from 0.66 to 0.55). The exclusion of the costs of adverse events from the model is justified in the MS on the basis that the most commonly occurring treatment-related adverse events are unlikely to be associated with substantial treatment costs, and that no specific subgroup of adverse events accounts for more than 2% of the populations in any of their included clinical trials. The exclusion of treatment costs for adverse events is in line with the approach adopted in previously published economic evaluations of antiviral treatment for chronic HCV.
Data inputs
The main treatment effect applied in the model is the SVR for treated patients. For patients who failed to respond or relapsed on previous peginterferon therapy, the SVRs for treated patients were taken from clinical trials (Jensen and colleagues88 for non-responders and Berg and colleagues89 for patients who relapsed – see Appendix 3). The SVR for the no treatment group was assumed to be zero, as none of the trials was placebo controlled or contained BSC arms. As acknowledged in the MS, the SVR reported by Berg and colleagues89 may be higher than would be expected in more generalisable populations of relapsed patients. Genotype 1 patients, who were subsequently enrolled in the study by Berg and colleagues,89 had relapsed following initial treatment that was less intensive than would be regarded as the current standard of care for this patient group [they received 24 weeks, rather than 48 weeks, of peginterferon combination treatment with a lower dosage of ribavirin (800 mg) than is recommended].
For patients receiving shortened durations of treatment, the SVRs for both groups in the model were taken from unpublished subgroup analyses of clinical trial subjects. For genotype 2 and 3 patients the subgroups were taken from the trial reported by Shiffman and colleagues,90 and for genotype 1 and 4 subgroups appear to have been taken from the trial reported by Hadziyannis and colleagues91 (referred to as trial NV15942 in the submission; these data are reported in table 8 of the SPC for peginterferon alfa-2a42). For patients with HCV/HIV co-infection, the SVRs were taken from a clinical trial comparing non-peginterferon alfa-2a with peginterferon alfa-2a66 – as stated earlier, this is not consistent with the scope for this appraisal.
The EVRs applied in the model for re-treated patients were taken from the same trials (Jensen and colleagues88 for non-responders and Berg and colleagues89 for patients who relapsed). These have the effect of reducing the cost of treatment by ceasing drug treatment in patients who do not show an EVR (in line with the SPC for peginterferon alfa-2a; see p. 4 of SPC 42).
As discussed above, update searches for economic evaluations published since our previous assessment17 are reported in an appendix to the MS, which states that the purpose of this review was to identify more recent sources (for transition probabilities, costs and utilities) to populate the economic model. It further states that ‘13 full publications were considered for further informing the economic evaluation in this submission’ (p. 216), but gives only brief details (lead author and date of publication) for six publications (one of which is our previous assessment and a further three are referenced in our previous assessment). The transition probabilities for the natural history model appear to have been taken from our previous assessment. 17
The MS reports that an update search (based on the search strategy from our previous assessment17) of MEDLINE, EMBASE, PREMEDLINE and EMBASE Alert was conducted to find newer health-state utility values than those used in our previous assessment. 17 This search is not discussed in the main body of the submission but is included in an appendix, which concludes that no new relevant utility data have been published. The manufacturer does not appear to have attempted targeted searches for QoL or utility data in the specific populations of patients included in its submission. As a result, the model uses utility values from the UK Mild Hepatitis C Trial,82 for chronic HCV and advanced liver disease states. However, age-specific utilities values from a general population were applied for the SVR state, which may lead to an overestimation of the utility gain, for two reasons:
-
The age-specific utility values are substantially higher than the utility values for the SVR state reported from the UK Mild Hepatitis C Trial. 82 The age-specific utility for a 45-year-old patient achieving an SVR in the manufacturer’s model is 0.85, declining to 0.73 once the patient is aged > 75 years. In contrast, the utility for patients achieving an SVR from moderate HCV, using the UK Mild Hepatitis C Trial82 valuations, is 0.72. The equivalent value for patients achieving an SVR from mild HCV is 0.82.
-
All patients enter the model with moderate HCV, which has a utility value of 0.66, whereas the value for mild HCV is 0.77.
The MS reports that no new sources for health-state costs were identified by their updated searches and that they have used the values from our previous assessment,17 inflated from 2003–4 to 2007–8 prices using the HCHS Pay and Prices Index. 85
Assessment of uncertainty
Uncertainty is addressed using deterministic sensitivity analysis (DSA) and PSA. The DSAs were limited in scope and focused on characteristics not included in the PSA, and might more accurately be termed ‘scenario analyses’, as they deal with alternative assumptions rather than variability in input parameters. These analyses address issues of methodological uncertainty (varying discount rates), parameter uncertainty (using alternative assumptions for baseline characteristics including patients’ mean age and weight as well as the proportion of women in the baseline cohort) and structural uncertainty (duration of surveillance for patients following cessation of treatment). The MS reports the incremental cost and effect, as well as the ICER, for each of the sensitivity analyses to facilitate interpretation of changes in the ICER in relation to alternative assumptions. The ICERs were largely insensitive to changes assessed in the DSA and none of these analyses would lead to a change in conclusion from the base-case analysis. The greatest variation was associated with differences in the starting age for the cohort (where incremental cost tended to reduce and incremental effect tended to increase with younger starting ages) and discounting practice (where re-treatment of non-responding patients became dominant for discount rates of 0% for both costs and effects). Additional DSAs were conducted for the HCV/HIV co-infected cohort to consider alternative assumptions regarding the excess death rate for co-infected patients. In the analysis presented in the MS peginterferon alfa-2a, treatment was dominant for all scenarios; however, this was for the comparison with non-peginterferon alfa, rather than with BSC.
Parameter uncertainty is also addressed in a PSA. The majority of parameters in the model are included in the PSA, including transition probabilities in the natural history model, health-state utilities, health-state costs, on-treatment monitoring and post-treatment surveillance costs, as well as SVR and (where appropriate) EVR probabilities. The choice of distribution applied to model parameters appears appropriate, beta distributions for utilities and probabilities and log-normal distributions for costs. However, the parameterisation for many of the distributions does not make best use of the available data. The SVR and EVR probabilities have been parameterised using the point estimate from the base-case analysis as the mean of the distribution, as would be expected, with the standard error (SE) assumed to be 0.02 (the implications of this assumption are discussed below). The rationale for this assumption is not discussed in the MS. The MS presents (for each trial used to derive the base-case SVR and EVR for each modelled population) the total number of patients in each arm, and the number achieving SVR and, where relevant, EVR, but it is not clear why these observed values were not used to parameterise the distributions. Similarly, the SEs for health-state costs have been assumed at 20% of the mean value, without any justification for this assumption. Standard deviations and the number of observations for the health-state costs are reported by the UK Mild Hepatitis C Trial82 and could have been used to parameterise the distributions. Scatter plots of incremental cost and incremental QALYs are presented for all comparisons, while CEACs are presented only for re-treatment of patients who failed to respond to previous peginterferon treatment. There is no discussion of this in the MS and the presentation of the PSA is generally inadequate in the context of current NICE methodological guidance. 68
The key source of heterogeneity in the modelled populations, in terms of response to treatment, has been taken into account through the presentation of separate analyses for viral genotype – either characterised as genotype 1 and genotype non-1 in the case of re-treatment of patients who did not respond to prior peginterferon treatment, or as genotype 1/4 and genotype 2/3 for shortened treatment duration. The remaining analyses (re-treatment of patients who did not relapse following prior peginterferon treatment and HCV/HIV co-infected patients) were not stratified by genotype. The MS does not discuss how representative the overall SVR from included clinical trials (which will reflect the genotype distribution of patients in the trial population) is of the overall SVR expected in a UK population of patients with chronic HCV, which may have a different genotype distribution. The MS has not considered another important source of heterogeneity, in terms of response to treatment, which is the stage of disease at treatment. Where trials have analysed SVR by stage of disease they tend to indicate that response is lower in patients with cirrhosis.
Summary of general concerns
-
The manufacturer’s model appears likely to overestimate the QALY gain from achieving SVR by:
-
– applying age-specific utilities to the SVR state and not applying age-specific utilities to other health states
-
– collapsing the HCV state into one, rather than differentiating mild and moderate HCV (which appear to have different health-state values).
-
-
The model assumes that all patients start treatment in the moderate HCV state. It is likely that some patients will present at other stages of liver disease, including compensated cirrhosis. The base-case results, applying to patients with moderate liver disease, may not apply to this group.
-
The manufacturer’s model does not include the cost of the health state patients are in when they start treatment.
-
The cost applied for surveillance of patients who achieve an SVR is low compared with that estimated in the UK Mild Hepatitis C Trial. This cost is applied only for the year following transition to the SVR state.
-
The manufacturer’s model appears to be applying an incorrect cost for ribavirin (for genotype 2/3 patients and for the HCV/HIV co-infected group).
-
The parameterisation of some distributions in the PSA is based on assumed values and could be improved on. Additionally, some logically related parameters appear to be sampled independently in the PSA, which is likely to give misleading results.
Additional analyses undertaken by SHTAC
The assessment group undertook additional analyses using the manufacturer’s model to address some of the concerns raised in the previous section. Table 21 reports the results of the additional analyses undertaken for the population of patients eligible for shortened duration of treatment. All of the changes made to the manufacturer’s model have the effect of increasing the value of the ICER. However, it needs to be borne in mind when interpreting these results that the incremental costs and outcome when comparing shortened with standard treatment duration are negative. The majority of the changes in assumptions in the model reduce the incremental QALYs between standard treatment and shortened duration – the exception is the change in the distribution of patients across stages of disease (to assume 32% of the cohort have cirrhosis prior to starting treatment).
| Genotypes 1 + 4 | Genotypes 2 + 3 | ||||
|---|---|---|---|---|---|
| Cost (£) | Outcome | Cost (£) | Outcome | ||
| Original | Standard | 13,387 | 15.78 | 8053 | 15.63 |
| Shortened | 8866 | 15.49 | 7391 | 15.39 | |
| ICER | 15,472 | 2719 | |||
| Do not use age-specific utility | Standard | 13,387 | 14.16 | 8053 | 14.07 |
| Shortened | 8866 | 13.97 | 7391 | 13.91 | |
| ICER | 23,541 | 4137 | |||
| Stage distribution (50 : 50, mild/moderate) | Standard | 13,125 | 15.83 | 7529 | 15.73 |
| Shortened | 8081 | 15.64 | 6431 | 15.57 | |
| ICER | 26,146 | 6830 | |||
| Stage distribution (33 : 35 : 32, mild/moderate/CC) | Standard | 13,358 | 15.78 | 7995 | 15.62 |
| Shortened | 8780 | 15.47 | 7285 | 15.37 | |
| ICER | 15,071 | 2805 | |||
| Add cost of original health state to year 1 SVR | Standard | 13,796 | 15.78 | 8449 | 15.63 |
| Shortened | 8866 | 15.49 | 7391 | 15.39 | |
| ICER | 16,872 | 4347 | |||
| All togethera | Standard | 13,735 | 14.16 | 8360 | 14.05 |
| Shortened | 8780 | 13.95 | 7285 | 13.88 | |
| ICER | 24,334 | 6336 | |||
Table 22 reports the results of the additional analyses undertaken for the population of non-responding or relapsing patients undergoing re-treatment. For non-responding patients the ICER increases in value for each of the scenarios examined, with the results for both genotype groupings being most sensitive to changes in the distribution of patients across stages of disease at baseline. However, although these analyses suggest that the ICER for re-treating patients with peginterferon alfa-2a combination therapy may be higher than the manufacturer’s base case, they do not substantially alter the conclusions from the analysis. In all of the alternative scenarios, re-treatment of relapsing patients remains dominant.
| Non-responders | Relapsers | ||||||
|---|---|---|---|---|---|---|---|
| Genotype 1 | Genotype non-1 | All genotypes | |||||
| Cost (£) | Outcome | Cost (£) | Outcome | Cost (£) | Outcome | ||
| Original | BSC | 27,114 | 11.06 | 27,114 | 11.06 | 27,114 | 11.06 |
| PEG α-2a | 29,225 | 11.69 | 27,942 | 12.08 | 21,199 | 13.74 | |
| ICER | 3334 | 809 | PEG dominates | ||||
| Do not use age-specific utility | BSC | 27,114 | 11.06 | 27,114 | 11.06 | 27,114 | 11.06 |
| PEG α-2a | 29,225 | 11.47 | 27,942 | 11.73 | 21,199 | 12.82 | |
| ICER | 5073 | 1232 | PEG dominates | ||||
| Stage distribution (50 : 50 mild and moderate) | BSC | 18,392 | 12.71 | 18,392 | 12.71 | 18,392 | 12.71 |
| PEG α-2a | 21,637 | 13.13 | 21,052 | 13.39 | 17,274 | 14.48 | |
| ICER | 7763 | 3939 | PEG dominates | ||||
| Stage distribution (33 : 35 : 32 mild/moderate/CC) | BSC | 26,153 | 10.86 | 26,153 | 10.86 | 26,153 | 10.86 |
| PEG α-2a | 28,389 | 11.52 | 27,183 | 11.93 | 20,766 | 13.65 | |
| ICER | 3397 | 968 | PEG dominates | ||||
| Add cost of original health state to year 1 SVR | BSC | 27,114 | 11.06 | 27,114 | 11.06 | 27,114 | 11.06 |
| PEG α-2a | 29,280 | 11.69 | 28,030 | 12.08 | 21,431 | 13.74 | |
| ICER | 3421 | 896 | PEG dominates | ||||
| All togethera | BSC | 26,153 | 10.86 | 26,153 | 10.86 | 26,153 | 10.86 |
| PEG α-2a | 28,440 | 11.31 | 27,265 | 11.58 | 20,980 | 12.73 | |
| ICER | 5182 | 1559 | PEG dominates | ||||
Table 23 reports the results of the additional analyses undertaken for the population of HCV/HIV co-infected patients. The results are similar to those for other patient groups – the ICER increases in value for each of the scenarios examined, with the results for both genotype groupings being most sensitive to changes in the distribution of patients across stages of disease at baseline. As before, while these analyses suggest that the ICER for treating HCV/HIV co-infected patients with peginterferon alfa-2a combination therapy may be higher than the manufacturer’s base case, they do not substantially alter the conclusions from the analysis.
| Cost (£) | Outcome | ||
|---|---|---|---|
| Original | BSC | 27,022 | 11.03 |
| PEG α-2a | 28,786 | 12.99 | |
| IFN α-2a | 32,431 | 11.62 | |
| ICER | 903 | ||
| Do not use age-specific utility | BSC | 27,022 | 11.03 |
| PEG α-2a | 28,786 | 12.32 | |
| IFN α-2a | 32,431 | 11.42 | |
| ICER | 1372 | ||
| Stage distribution (50 : 50, mild/moderate) | BSC | 18,320 | 12.68 |
| PEG α-2a | 23,565 | 13.97 | |
| IFN α-2a | 24,773 | 13.07 | |
| ICER | 4050 | ||
| Stage distribution (33 : 35 : 32, mild/moderate/CC) | BSC | 26,080 | 10.84 |
| PEG α-2a | 28,221 | 12.87 | |
| IFN α-2a | 31,602 | 11.45 | |
| ICER | 1054 | ||
| Add cost of original health state to year 1 SVR | BSC | 27,022 | 11.03 |
| PEG α-2a | 28,955 | 12.99 | |
| IFN α-2a | 32,431 | 11.62 | |
| ICER | 989 | ||
| All together | BSC | 26,080 | 10.84 |
| PEG α-2a | 28,377 | 12.20 | |
| IFN α-2a | 31,602 | 11.25 | |
| ICER | 1684 | ||
Schering-Plough submission to NICE: cost-effectiveness analysis
Overview
The Schering-Plough submission92 to NICE consists of a 69-page written document (containing submitted evidence on the clinical effectiveness and a cost-effectiveness analysis) and a fully executable, electronic copy of the manufacturer’s economic model. The MS reports cost-effectiveness results for two populations covered by the scope of the NICE appraisal:
-
Patients who have been treated previously with peginterferon, and who did not respond to previous treatment or who relapsed on previous treatment. This analysis is reported for all patients (a cohort including patients of all viral genotypes) and broken down by broad genotype categories (genotypes 1 and 4 combined or genotypes 2 and 3 combined). Costs and outcomes for these patients is compared with BSC, in line with the scope for this appraisal.
-
Patients co-infected with HCV/HIV. This analysis is reported for all patients (a cohort including patients of all viral genotypes) and broken down by broad genotype categories (genotypes 1 and 4 combined or genotypes 2 and 3 combined). Costs and outcomes for these patients are compared with BSC, in line with the scope for this appraisal.
No assessment is presented on the cost-effectiveness of shortened versus standard treatment duration. The reason for this omission is not discussed by the manufacturer, though it may be due to peginterferon alfa-2b being licensed only for shorter treatment durations in genotype 1 (as opposed to genotypes 2, 3 and 4).
The perspective of the analysis is stated as being that of the NHS and PSS, consistent with the NICE reference case. 68 The submission reports lifetime costs and outcomes (reported as QALYs) for each treatment arm and the incremental costs and outcomes for peginterferon alfa-2b combined with ribavirin compared with BSC.
The MS does not report whether a systematic search was undertaken for economic evaluations of peginterferon alfa-2b or other treatments for chronic HCV in the patient populations covered by the scope, nor does it report any detail on the development and validation (including any details of clinical validation) of the model adopted for the MS.
Below we describe the approach taken for the model and provide an outline review based on a checklist suggested for the critical appraisal of cost-effectiveness analysis by Drummond and colleagues,67 the requirements of NICE for submissions on cost-effectiveness (reference case)68 and a suggested guideline for good practice in decision modelling by Philips and colleagues. 69
Modelling approach
The model consists of an initial decision tree covering the first year in the model, where patients are eligible to receive treatment. The decision tree incorporates two chance nodes: the first of these applies a probability of patients achieving an EVR, the second applies a probability of patients who achieved an EVR (and therefore remained on treatment) achieving an SVR. A state-transition model is then used to model patients’ costs and outcomes, depending on the state in which they emerge from the decision – with an SVR, remaining with HCV/compensated cirrhosis, or dead from all causes. The state-transition model is structurally similar to published models previously used in the population of patients with HCV, including the previous assessment report for NICE. 17 The model has six health states (mild HCV, moderate HCV, compensated cirrhosis, decompensated cirrhosis, HCC and liver transplantation) indicating progressive liver disease, one state representing a treatment response (SVR) and one absorbing state (death), although this last state is broken down to differentiate deaths from progressive liver disease and deaths from all other causes.
The model does not differentiate the SVR state according to patients’ stage of disease prior to SVR. However, QoL data reported by the UK Mild Hepatitis C Trial82 would suggest that there are differences in health-state utility for patients who enter the SVR state from both mild and moderate chronic HCV, and it may be more appropriate to structure the model to identify prior stage of disease (given that patients with compensated cirrhosis are eligible to receive treatment, as well as those with mild or moderate chronic HCV).
The main treatment effect applied in the model is the SVR for treated patients, with the proportion of patients in each of the modelled populations achieving an SVR being based on data from clinical trials conducted in the relevant patient populations, reported in the MS. Patients who achieve an SVR are assumed in the model to be ‘cured’ and do not face any risk of reactivation of disease or any excess risk of progressive liver disease (above that of a general population). Age-specific mortality risks for the general population, weighted for the proportion of men in the baseline cohort, are applied to patients achieving an SVR. Patients who do not achieve an SVR are at risk of progressive liver disease and are assumed to face the same risks of disease progression as untreated patients. Risks of disease progression and, where relevant, excess mortality risks associated with advanced liver disease states in the model have been drawn from natural history studies.
The base-case population characteristics (in terms of age at entry to the model, weight and the proportion that are male) differ between the patient subgroups modelled and are based on the baseline populations in the relevant clinical trials. These assumptions have no impact on the patient response to treatment (i.e. SVRs in the model are not broken down by age or sex), but age and the proportion of men affect the all-cause mortality rates applied in the model, while patient weight has an impact on cost of treatment, as peginterferon alfa-2b and ribavirin dosage is weight related.
The health-state utilities have been derived from the UK Mild Hepatitis C Trial,82 and a study of the cost-effectiveness of liver transplantation. 93 There is no systematic search for these values reported in the submission. The EQ-5D was completed by 130 patients within the Mild Hepatitis C Trial,82 at 24 or 48 weeks post treatment or control. A linked observational study used cases recruited to the costing study in order to estimate the HRQoL for patients with moderate disease, compensated cirrhosis or decompensated cirrhosis. HRQoL for liver transplant patients and post-liver transplant health states was taken from a cost-effectiveness study of liver transplantation. This latter study also utilised the EQ-5D to estimate HRQoL in liver transplant patients; however, its applicability may be limited, as the included patients did not have HCV.
The model applies a disutility of 0.13 (owing to treatment-related adverse effects) to the utility score for treatment-eligible health states for the year in which patients undergo treatment. In their example, moderate HCV was assigned a baseline utility of 0.66, and this was reduced to 0.53 during treatment. This was based on the overall mean difference in EQ-5D utility score for treated and control patients at 12 or 24 weeks following randomisation in the UK Mild Hepatitis C Trial. 82 The disutility associated with treatment was adjusted for duration of treatment, so that a lower utility decrement would apply for patients (who fail to demonstrate an EVR) stopping treatment at 12 weeks.
The costs applied in the submission were made up of two components. Treatment-related costs (which consist of drug acquisition costs and monitoring of patients on treatment) were estimated separately from health-state costs. Health-state costs include resource use associated with the management of progressive liver disease.
Drug usage for peginterferon alfa-2b was based on a dosage of 1.5 µg per kg per week and was assumed to be supplied in prefilled pens. As dosage of both peginterferon alfa-2b and ribavirin is weight based (Table 24) the MS needed to assume an average weight for each of the modelled patient populations. A mean weight of 80 kg was applied in the base-case analysis for the re-treated group, and of 63 kg in the HCV/HIV co-infected group. The values for the weight of the HCV/HIV co-infected group were taken from the Laguno study;94 however, the default value in the MS is reported as 68.29 kg.
| Body weight (kg) | PEG α-2b | Ribavirin | ||
|---|---|---|---|---|
| Vial/pen strength (µg/ml) | Administer once weekly (ml) | Total daily dose (mg) | No. of capsules (200 mg) | |
| < 40 | 50 | 0.5 | 800 | 4 |
| 40–50 | 80 | 0.5 | 800 | 4 |
| 51–64 | 80 | 0.5 | 800 | 4 |
| 65–75 | 100 | 0.5 | 1000 | 5 |
| 76–85 | 120 | 0.5 | 1000 | 5 |
| 86–105 | 150 | 0.5 | 1200 | 6 |
| > 105 | 150 | 0.5 | 1400 | 7 |
The drug acquisition costs for peginterferon alfa-2b and ribavirin adopted in the Schering-Plough submission92 (taken from the BNF, No. 57, March 2009) are presented in Tables 25 and 26 below. With an assumed body weight of 80 kg for re-treated patients, and assuming a preference for prefilled pens for peginterferon alfa-2b and tablets for ribavirin, the weekly treatment costs are £165.73 for peginterferon alfa-2b and £114.85 for ribavirin. The equivalent cost for the HCV/HIV co-infected patients is £118 for peginterferon alfa-2b and £91.88 for ribavirin.
| PEG α-2b (µg/bottle) | Pack costs (powder for reconstitution) (£) | Pack cost (prefilled pens) (£) |
|---|---|---|
| 50 | 62.78 | 69.05 |
| 80 | 100.44 | 118.00 |
| 100 | 125.55 | 138.11 |
| 120 | 150.66 | 165.73 |
| 150 | 188.33 | 207.16 |
| Tablet size (mg) | Caps/pack | Pack cost (£) |
|---|---|---|
| 200 | 84 | 275.65 |
| 200 | 140 | 459.42 |
| 200 | 168 | 551.30 |
The costs of initial investigations and monitoring included liver biopsy, an overnight stay in hospital for this procedure, and regular outpatient consultations and investigations. These costs were all taken from the Mild Hepatitis C Trial. 82 The initial investigations were calculated to cost £822.27 per patient assessed. As a result of interviews with clinicians, suggesting that between one and five patients would be assessed for each patient treated, this was then tripled to account for that (range 1–5). The monitoring costs of patients being treated was £489, which was inflated to £587.85 per patient treated (2007–8 values).
Costs for each disease state were again taken from the UK Mild Hepatitis C Trial,82 or in the case of moderate and more severe disease from the observational costing study conducted within that trial.
The baseline population differs between the two patient groups modelled, and, for the majority of these characteristics, is based on the baseline population of the relevant clinical trials. The simulated cohort of re-treated patients has a mean age of 49 years, with 71% being male. Patient weight is assumed to be greater than 81 kg and it is assumed that 85% of re-treated patients have genotype 1 or 4 (the remainder with viral genotypes 2 and 3). This last assumption has an impact on outcome for the overall cohort of patients, as patients with viral genotypes 1 and 4 have a lower probability of SVR than those with viral genotype 2 or 3. For patients with HCV/HIV co-infection, the base-case characteristics are based on the RCT by Laguno and colleagues,95 with a mean age of 40 years, and 68% being male. Patient weight is substantially lower at 63 kg, although as mentioned above this only affects the drug costs. While these characteristics have been drawn from clinical trials conducted in relevant subgroups, the MS does not discuss how relevant these characteristics may be to the population of UK patients with chronic HCV, in general, or how relevant they may be to the UK population of patients to be re-treated following non-response to, or relapse following, prior peginterferon treatment or those with HCV/HIV co-infection.
In both analyses, patients enter the model in one of three states – with mild HCV (33%), moderate HCV (33%) or compensated cirrhosis (34%). Each of these is a treatment-eligible health state and the probability of EVR or SVR is assumed to be equal for each possible starting state.
Model/cost-effectiveness results
The MS reports total costs and QALYs for a cohort of 100 re-treated patients and 100 HCV/HIV co-infected patients. Both cohorts include genotype 1, 2, 3 and 4 patients. To estimate costs and outcomes for these cohorts of mixed genotypes, treatment efficacy estimates (SVR and, where relevant, EVR) for genotype subgroups were used to estimate response for each subgroup. The overall results for the cohort were then calculated as weighted totals (based on the proportion of the total cohort in each subgroup). The model results are also presented as an average cost and average QALYs per patient for the cohort including all genotypes and for subgroups of genotypes 1 and 4 and of genotypes 2 and 3. The distribution of patients across viral genotypes is based on the populations recruited to the clinical trials used to derive the efficacy data for the model. The MS does not discuss how generalisable these proportions may be to UK populations of UK patients with chronic HCV infection.
Table 27 reports the base-case results, including the ICER, from the Schering-Plough model92 for re-treatment of patients who did not respond or relapsed following previous interferon therapy and for HCV/HIV co-infected patients. Scatter plots showing the cost-effectiveness plane (incremental cost and incremental QALYs for peginterferon alfa-2b combination therapy) and CEACs are presented in a separate section of the MS reporting the results of the PSA.
| Patient group | Genotypes | Treatment | Cost (£) | QALYs | ICER (£ per QALY gained) |
|---|---|---|---|---|---|
| Non-responders/relapsers | 1 + 4 | No treatment | 22,130 | 9.97 | 7177 |
| PEG α-2b + RBV | 27,125 | 10.67 | |||
| 2 + 3 | No treatment | 22,130 | 9.97 | 783 | |
| PEG α-2b + RBV | 24,301 | 12.75 | |||
| All | No treatment | 22,130 | 9.97 | 4387 | |
| PEG α-2b + RBV | 26,666 | 11.01 | |||
| HCV/HIV co-infection | 1 + 4 | No treatment | 24,494 | 10.90 | 1637 |
| PEG α-2b + RBV | 27,790 | 12.91 | |||
| 2 + 3 | No treatment | 24,494 | 10.90 | 403 | |
| PEG α-2b + RBV | 25,645 | 13.75 | |||
| All | No treatment | 24,494 | 10.90 | 1077 | |
| PEG α-2b + RBV | 26,997 | 13.22 |
The MS presents a further analysis for the cohort of re-treated patients, reporting separate analyses for previous relapsers, previous non-responders and previous treatment failures (although the definition of previous treatment failure is not very clear, and is described in the MS as referring to ‘patients who could not be classified as relapsers or non-responders due to missing data or other reasons’). The results for these subgroups are presented in Table 28 and show that previous non-responders have a lower QALY gain (and higher incremental cost) than previous treatment failures and relapsing patients.
| Patient group | Cost (£) | QALYs | ICER (£ per QALY gained) | |
|---|---|---|---|---|
| Previous relapsers | No treatment | 22,130 | 9.97 | 2048 |
| PEG α-2b + RBV | 25,996 | 11.86 | ||
| Previous non-responders | No treatment | 22,130 | 9.97 | 7581 |
| PEG α-2b + RBV | 27,009 | 10.62 | ||
| Prior treatment failures | No treatment | 22,130 | 9.97 | 3013 |
| PEG α-2b + RBV | 26,157 | 11.31 | ||
The MS states that peginterferon alfa-2b in combination with ribavirin is cost-effective for adults with HCV/HIV co-infection and for patients whose previous treatment was unsuccessful. These conclusions draw on evidence from the base-case analyses presented above, from DSAs (where ICERs remained below £20,000 per QALY gained in the scenarios tested) and from PSAs where the probability for peginterferon alfa-2b being cost-effective, compared with no active treatment, for re-treating patients who did not respond or relapsed following previous interferon therapy was estimated at 95% at a willingness-to-pay threshold of £20,000 per QALY and the probability for peginterferon alfa-2b being cost-effective, compared with no active treatment, for HCV/HIV co-infected patients, was estimated at 98% at a willingness-to-pay threshold of £20,000 per QALY.
Outline appraisal of the cost-effectiveness analysis undertaken
The NICE reference case requirements (Schering-Plough) are shown in Table 29.
| NICE reference case requirements68 | Included in submission |
|---|---|
| Decision problem: as per the scope developed by NICE | ✓ |
| Comparator: alternative therapies routinely used in the UK NHS | ✓ |
| Perspective on costs: NHS and PSS | ✓ |
| Perspective on outcomes: all health effects on individuals | ✓ |
| Type of economic evaluation: cost-effectiveness analysis | ✓ |
| Synthesis of evidence on outcomes: based on a systematic review | ?a |
| Measure of health benefits: QALYs | ✓ |
| Description of health states for QALY calculations: use of a standardised and validated generic instrument | ✓ |
| Method of preference elicitation for health-state values: choice-based method (e.g. TTO, SG, not rating scale) | ✓ |
| Source of preference data: representative sample of the public | ✓ |
| Discount rate: 3.5% pa for costs and health effects | ✓ |
Outline review of modelling approach
Model structure/structural assumptions
No review of previous models has been reported, although the authors state that their model has adopted a similar structure to those used in previous assessment reports for NICE and for the economic evaluation alongside the Mild Hepatitis C trial. 82 The states representing more advanced liver disease in the model (compensated cirrhosis, decompensated cirrhosis, HCC and liver transplantation) are commonly accepted as distinct stages of progressive liver disease. These can be distinguished by their impact on QoL, resource use or excess mortality risk.
The effect of treatment is to induce an SVR in a proportion of patients, which is assumed to be a permanent cure. This agrees with previously published models in this patient population and is supported by long-term follow-up studies of patients achieving SVR on treatment. However, recent publications have highlighted a risk of liver cancer in patients in patients who have undergone SVR – particularly in patients with compensated cirrhosis at baseline, which, although lower than for non-responding patients, is not completely eradicated. As patients can enter the model in the compensated cirrhosis state (and receive treatment), excluding a transition from the SVR state (for patients who had developed cirrhosis at baseline) may overestimate the benefits from an SVR.
The model does not differentiate the SVR state according to patients’ stage of disease prior to SVR. However, QoL data reported by the UK Mild Hepatitis C Trial82 would suggest that there are differences in health-state utility for patients who enter the SVR state from mild and from moderate chronic HCV, and it may be more appropriate to structure the model to identify prior stage of disease (given that patients with compensated cirrhosis are eligible to receive treatment, as well as those with mild or moderate chronic HCV). Whether including only one SVR state is likely to over- or underestimate QoL or health-state utility will depend on the value assigned to the state.
Treatment-related adverse events are not included in the model, other than through the use of a decrement to utility while patients are on treatment. The exclusion of the costs of managing adverse events in the model is not discussed in the MS. However, the exclusion of treatment costs for adverse events is in line with the approach adopted in previously published economic evaluations of antiviral treatment for chronic HCV.
The MS does not report any evidence of approaches to establish the internal consistency of the model or any evidence of external validation (by expert clinical opinion or by comparison with other published economic evaluations).
Data inputs
The main treatment effect applied in the model is the SVR for treated patients. For patients who failed to respond to or relapsed following previous interferon therapy the SVRs were taken from the EPIC3 study,96 which is an open-label, single-arm study. The SVR for BSC was assumed to be zero for patients with moderate chronic HCV or compensated cirrhosis, but a low spontaneous SVR probability was applied for patients with mild chronic HCV. The spontaneous SVR probability is applied to both the treatment and the BSC cohorts. The spontaneous clearance of HCV is not discussed in the MS and the value (and derivation) of the transition probability is not included in table 35 of the MS, which lists the transition probabilities in the model. The MS does not discuss the relevance of data from the EPIC3 study,96 with inclusion criteria that patients had prior failure (either non-response or relapse) on previous combination therapy with ribavirin and (non-pegylated or pegylated) interferon. The study does not appear strictly to meet the scope for the appraisal, which identifies the population considered for retreatment to be those previously treated with peginterferon alfa and ribavirin.
For patients with HCV/HIV co-infection, data on response to treatment were taken from an RCT reported by Laguno and colleagues,95 which recruited treatment-naive (naive to combination therapy) patients with histologically verified liver disease who were HIV positive with controlled disease. In the trial, patients were randomised either to non-peginterferon combination therapy or peginterferon combination therapy. In the absence of a placebo or no active treatment control, the SVR for BSC was assumed to be zero for moderate chronic HCV, but with a low spontaneous SVR probability for patients with mild chronic HCV, as discussed earlier.
The EVR applied for re-treated patients was also derived from the EPIC3 study. 96
The MS does not report any systematic or targeted searches to identify new data or to update parameter inputs derived from the model developed for our previous assessment17 or that developed for the UK Mild Hepatitis C Trial,82 nor does it report undertaking targeted searches for parameter inputs specific to the patient groups within the scope of this assessment.
The utility scores used for each disease state in the model were based on the values reported in the Mild Hepatitis C Trial,82 which evaluated non-peginterferon alfa and ribavirin. These were generated in this trial using the standard EQ-5D time trade-off (TTO) tariff. The mean values are higher than those that have been used in the base-case analysis. All patients were assumed to experience a 0.13 reduction in utility due to treatment adverse effects. The disutility was applied to all patients receiving all treatment strategies, as there are no published data for patients’ QoL while receiving peginterferon alfa. The drug costs were taken from the SPC. The Mild Hepatitis C Trial82 was used to inform further costs: those taken into account included liver biopsy to assess eligibility for treatment and regular outpatient appointments and investigations for those who would not be eligible for further treatment. These costs were inflated to 2007–8 values. The discontinuation rates due to adverse events were taken from EPIC396 and Laguno and colleagues. 95 The transitions between health states came again from the Mild Hepatitis C Trial,82 from the UK study of patients undergoing transplantation, and on a range of additional studies, referenced within the submission. It is unclear how these have been derived, and from which studies.
In this submission patients are distributed across the treatment-eligible states, but SVR/EVR are not adjusted according to the stage of disease in the base case or sensitivity analysis. This is despite evidence that SVR/EVR do vary according to disease severity, which is alluded to in the manufacturers’ own submission. For example, on p. 16 the authors stated that key predictors of SVR included fibrosis level, and on p. 17 they stated that two significant predictors of SVR were identified, namely genotype and fibrosis score.
Assessment of uncertainty
Schering-Plough have tested response to therapy (where the EVR and SVR are varied) and drug dosing requirements (varying patient weight and drug administration method) in their DSA. In addition, the disutility and distribution of disease severity and distribution of genotype at baseline were varied. Two scenario analyses have also been reported: one in which the re-treatment group is presented as non-responders and relapsed patients and a further scenario in which discounting is removed.
The ICERs for both the re-treated group and the HCV/HIV co-infection group were both sensitive to variation in the EVR and SVR, and to changes in patient weight. The proportion of patients achieving EVR and SVR in the re-treatment group were varied between the upper and lower CIs from the included studies. The ICER in this group then changed from £4387 in the base-case analysis to £4842, where EVR proportion was ‘low’, and to £4003, where this was ‘high’. The SVR was similarly varied, and the ICER changed to £5227 in the ‘low-value’ group and to £3615’ in the ‘high-value’ group.
The peginterferon alfa-2b arm of the Scotto and colleagues97 study was also used to calculate ICERs for re-treated patients. This resulted in a greatly increased ICER of £19,004 per QALY in the genotype 1 and 4 group, a decreased ICER of £2520 in the genotype 2 + 3 group, and £10,742 for all patients. The manufacturers state that this is owing to the re-treated patients in this study being previous non-responders.
Incremental cost-effectiveness ratios in the HCV/HIV co-infection group were sensitive to the SVR rate (the authors state that the EVR rate was unavailable in this group). Again, values for the sensitivity analysis were taken from the upper and lower CIs reported in the included study. The base-case ICER was £1077. In the ‘low-value’ group this increased to £4065 per QALY, and in the ‘high-value’ group, peginterferon alfa-2b was dominant. When the EVR and SVR values from the more recent Laguno and colleagues’ RCT94 were applied, the manufacturers report ICERs of £6140 per QALY in genotypes 1 and 4, £422 per QALY for genotypes 2 and 3, and £2311 for all genotypes. It is not clear if both EVR and SVR have been adjusted here.
Changes in the distribution of patients with different liver disease severity produced smaller variation in the ICERs in the re-treatment group. In the case of mild disease the percentage of patients decreases from 33% to 27%, for moderate disease this proportion decreased from 33% to 31%, and for compensated cirrhosis this proportion increased from 33% to 42%. This variation is quite small, and had the effect of decreasing the ICER to £3596 per QALY from £4387 per QALY.
A sensitivity analysis was performed on distribution of genotype at baseline. The treatment response of genotypes 1 and 4, and then 2 and 3, are applied to all patients. The treatment response of genotypes 1 and 4 applied to the entire cohort resulted in an ICER of £7176 per QALY, and that of genotypes 2 and 3 resulted in an ICER of £782, in the re-treatment group. In the HCV/HIV co-infected group, the ICERs became £1637 and £403. The ICERs are the same as those presented in the base-case analysis, and it is unclear what has been added to this analysis by the reporting of this scenario.
The first scenario analysis presented ICERs for the re-treatment ‘subgroups’: previous relapsers and non-responders to treatment. The base-case ICER for this group was £4387. For the ‘previous relapser’ group alone it was £2048 and for ‘previous non-responders’ alone in this scenario it was £7581, with the higher ICER thought by the authors to reflect the lower expected level of success in this group.
The second scenario analysis examined the effects of not discounting costs and outcomes. Where discounting is removed, the ICER is reduced to £1265 per QALY in the re-treatment group, and the intervention becomes dominant (more effective and less costly) in the HCV/HIV co-infection group.
Parameter uncertainty is also addressed in a PSA. The majority of parameters in the model are included in the PSA, including transition probabilities in the natural history model, health-state utilities, health-state costs, probability of discontinuing treatment as well as SVR and (where relevant) EVR. The choice of distribution applied to the parameters appears appropriate, using beta distributions for probabilities and utilities, and gamma distributions for costs. The electronic model appears to use an implementation of the Dirichlet distribution for sampling transition probabilities in the model that are competing risks (e.g. patients with compensated cirrhosis may remain in that state, may progress or may develop HCC), although this is not discussed in the submission. The written submission contains an appendix that lists the parameters included in the PSA, their mean value, SE and the choice of distribution, but not the parameterisation of the distribution.
The MS reports three PSAs for each patient group (re-treated and HCV/HIV co-infected patients), each based on 10,000 simulations. The first analysis applies to the overall cohort, followed by separate analysis for genotype subgroups (genotypes 1 and 4 and genotypes 2 and 3). Cost-effectiveness scatter plots are presented along with CEACs for each of these analyses. The MS also reports the probability of the intervention of interest being cost-effective at willingness-to-pay thresholds of £20,000 per QALY gained and at £30,000 per QALY gained. The presentation of the PSA appears generally to be in accordance with NICE methodological guidance68 but does not report mean costs and outcome for the PSAs.
The key source of heterogeneity in the modelled populations, in terms of response to treatment, has been taken into account through the presentation of separate analyses for viral genotype. The MS has not considered another important source of heterogeneity, in terms of response to treatment, which is the stage of disease at treatment. Where trials have analysed SVR by stage of disease they tend to indicate that response is lower in patients with cirrhosis.
Summary of general concerns
-
The Schering-Plough model appears to underestimate the SVR in each analysis, as a result of applying an unnecessary adjustment for treatment discontinuation, but appears to overestimate the utility gain through treatment by not applying an adjustment for treatment discontinuation:
-
– The observed SVR for a given patient population (e.g. 38% for HCV/HIV co-infected patients with genotype 1 or 4) is applied to the proportion of patients expected to be in that population (63% of HCV/HIV co-infected patients are assumed to be genotype 1 or 4 in a cohort of 100 patients, i.e. 63 people) – therefore the expected number of SVRs is 24. This value is then multiplied by the probability of not discontinuing treatment (probability of discontinuing is 0.1731, therefore probability of not discontinuing = 1 – 0.1731), which gives a value of 20 (the number of SVRs adjusted for discontinuation), resulting in an SVR rate of 31.42% (20/63). As the original SVR rate of 38% was based on the observed data reported in the RCT by Laguno and colleagues,95 adjusting by the discontinuation probability seems unnecessary.
-
-
There is an implicit assumption that patients achieve an SVR immediately after treatment is initiated and therefore accrue health benefits on entering the model. It might be more reasonable to assume that transitions occur mid-cycle (essentially applying half-cycle adjustment). This would mean adjusting cycle lengths (currently annual) to cope with treatments that are significantly less than 52 weeks, or calculating a weighted combination of the utility for the initial state and the utility for the appropriate SVR state (weighted according to what proportion of the cycle is spent in the initial health state and what proportion in the SVR state).
-
The model collapses the SVR state into one and therefore does not track whether patients have achieved SVR from mild HCV, moderate HCV or compensated cirrhosis. It applies the same health-state utility to patients achieving an SVR, irrespective of their stage of liver disease when treatment was initiated. This does not accord with utility data from the UK Mild Hepatitis C trial, which reported a lower mean utility for patients achieving SVR from moderate liver disease than those achieving SVR from mild liver disease.
-
The model assumes that the SVR health-state cost is applied for all cycles the patient remains in the SVR state. This differs from the assumption applied in our previous assessment report,17 where it was assumed that the SVR cost applied only for the year following treatment response.
-
The model appears to have underestimated the cost of ribavirin (tables 31 and 32 of the MS report weekly cost of ribavirin as £16.41 for re-treated patients and £13.13 for HCV/HIV co-infected patients). These are derived using an estimated average cost per 200-mg tablet of ribavirin of approximately £3.28. However, the figures used in the MS are the daily, not weekly, costs.
Additional analyses undertaken by SHTAC
The assessment group undertook additional analyses using the manufacturer’s model to address some of the concerns raised in the previous section. Table 30 reports the results of the additional analyses undertaken for HCV/HIV co-infected patients. Removing treatment discontinuation from the calculation of the SVR probability and applying only the SVR health-state cost in year after SVR occurs reduces the ICER – making treatment for genotype 2 + 3 patients dominant. In contrast, correcting the calculation of ribavirin costs and splitting the SVR state to apply utility values that take account of disease stage prior to SVR increase the ICER.
| Genotypes 1 + 4 | Genotypes 2 + 3 | All genotypes | |||||
|---|---|---|---|---|---|---|---|
| Cost | Outcome | Cost | Outcome | Cost | Outcome | ||
| Original | BSC | 24,494 | 10.90 | 24,494 | 10.90 | 24,494 | 10.90 |
| PEG | 27,790 | 12.91 | 25,645 | 13.75 | 26,997 | 13.22 | |
| ICER | 1637 | 403 | 1077 | ||||
| Use observed SVR (remove discontinuation) | BSC | 24,494 | 10.90 | 24,494 | 10.90 | 24,494 | 10.90 |
| PEG | 26,653 | 13.36 | 24,058 | 14.37 | 25,693 | 13.73 | |
| ICER | 878 | –126 | 423 | ||||
| Allow for different utility for SVR states | BSC | 24,494 | 10.90 | 24,494 | 10.90 | 24,494 | 10.90 |
| PEG | 27,790 | 12.21 | 25,645 | 12.77 | 26,997 | 12.42 | |
| ICER | 2511 | 613 | 1645 | ||||
| Correct RBV cost | BSC | 24,494 | 10.90 | 24,494 | 10.90 | 24,494 | 10.90 |
| PEG | 31,407 | 12.91 | 29,262 | 13.75 | 30,613 | 13.22 | |
| ICER | 3434 | 1671 | 2633 | ||||
| Only apply SVR cost for year following SVR | BSC | 24,446 | 10.90 | 24,446 | 10.90 | 24,446 | 10.90 |
| PEG | 25,747 | 12.91 | 22,814 | 13.75 | 24,661 | 13.22 | |
| ICER | 646 | –572 | 93 | ||||
| Adjust SVR for disease stage | BSC | 24,494 | 10.90 | 24,494 | 10.90 | 24,494 | 10.90 |
| PEG | 28,192 | 12.75 | 26,205 | 13.53 | 27,457 | 13.04 | |
| ICER | 1992 | 649 | 1382 | ||||
| All togethera | BSC | 24,446 | 10.90 | 24,446 | 10.90 | 24,446 | 10.90 |
| PEG | 28,296 | 12.41 | 24,942 | 13.06 | 27,055 | 12.65 | |
| ICER | 2541 | 230 | 1488 | ||||
The same SVR was applied to all treated patients in the manufacturer’s model, regardless of stage of fibrosis. However, analyses of response to treatment, by stage of disease, typically suggest that treatment response is lower in patients with fibrosis. Ratios of the relative effectiveness of treatment for patients with fibrosis stages F2, F3 and F4 (derived using data reported in the MS for the EPIC3 study95) were used to examine the effect, on the cost-effectiveness results, of reducing the SVR for cirrhotic patients. This is labelled in Table 30 as ‘Adjust SVR for disease stage’.
Table 31 reports the results of the additional analyses undertaken for re-treated patients. Removing treatment discontinuation from the calculation of the SVR probability is less influential than in the analysis for HCV/HIV co-infected patients. Applying the SVR health-state cost in the year after SVR occurs reduces the ICER, while correcting the calculation of ribavirin costs and splitting the SVR state to apply utility values that take account of disease stage prior to SVR increase the ICER. Adjusting the SVR for disease stage has relatively little impact on the cost-effectiveness results. Overall, while these analyses suggest that the ICER for treating HCV/HIV co-infected patients with peginterferon alfa-2b combination therapy may be higher than in the manufacturer’s base case, they do not substantially alter the conclusions from the analysis.
| Genotypes 1 + 4 | Genotypes 2 + 3 | All genotypes | |||||
|---|---|---|---|---|---|---|---|
| Cost | Outcome | Cost | Outcome | Cost | Outcome | ||
| Original | BSC | 22,130 | 9.97 | 22,130 | 9.97 | 22,130 | 9.97 |
| PEG | 27,125 | 10.67 | 24,301 | 12.75 | 26,666 | 11.01 | |
| ICER | 7177 | 783 | 4387 | ||||
| Use observed SVR (remove discontinuation) | BSC | 22,130 | 9.97 | 22,130 | 9.97 | 22,130 | 9.97 |
| PEG | 26,974 | 10.72 | 23,723 | 12.95 | 26,445 | 11.09 | |
| ICER | 6463 | 535 | 3881 | ||||
| Allow for different utility for SVR states | BSC | 22,130 | 9.97 | 22,130 | 9.97 | 22,130 | 9.97 |
| PEG | 27,125 | 10.40 | 24,301 | 11.74 | 26,666 | 10.62 | |
| ICER | 11,586 | 1232 | 7006 | ||||
| Correct Rebetol cost | BSC | 22,130 | 9.97 | 22,130 | 9.97 | 22,130 | 9.97 |
| PEG | 29,324 | 10.67 | 28,221 | 12.75 | 29,145 | 11.01 | |
| ICER | 10,336 | 2195 | 6785 | ||||
| Only apply SVR cost for year following SVR | BSC | 22,093 | 9.97 | 22,093 | 9.97 | 22,093 | 9.97 |
| PEG | 26,337 | 10.67 | 21,396 | 12.75 | 25,534 | 11.01 | |
| ICER | 6099 | –251 | 3329 | ||||
| Adjust SVR for disease stage | BSC | 22,130 | 9.97 | 22,130 | 9.97 | 22,130 | 9.97 |
| PEG | 27,301 | 10.61 | 24,975 | 12.53 | 26,923 | 10.92 | |
| ICER | 8102 | 1114 | 5047 | ||||
| All together | BSC | 22,093 | 9.97 | 22,093 | 9.97 | 22,093 | 9.97 |
| PEG | 28,521 | 10.41 | 25,258 | 11.75 | 27,991 | 10.63 | |
| ICER | 14,773 | 1781 | 9027 | ||||
Summary of manufacturers’ models, compared with SHTAC model from previous assessment report
Table 32 summarises the transition probabilities used in the manufacturers’ models and in our previous assessment of peginterferon alfa combination treatment for chronic HCV infection. 17 It is clear from the table that identical values have been used for the majority of transition probabilities modelling the natural history of progressive liver disease. These are primarily drawn from studies reported by Sweeting and colleagues,18 Wright and colleagues82 and Fattovich and colleagues. 98 The principal differences between the three models are that:
-
Patients enter the Roche model with moderate HCV, so that the transition probability from mild-to-moderate disease is not relevant.
-
The Schering-Plough model includes a small risk for non-cirrhotic patients (with moderate disease) developing HCC, based on a previously published economic evaluation by Bennett and colleagues. 76
-
The Schering-Plough model uses a higher excess mortality risk for HCC than is applied in the other models, based on a previously published economic evaluation99 and cancer mortality statistics. 100
-
The Schering-Plough model uses a slightly higher probability for developing HCC for patients with cirrhosis. The value in Table 32 is used in the electronic model, while a lower value of 0.014 (identical to that used by Roche and in our previous assessment) is reported in tables included in the main submission document. This discrepancy is not explained in the submission or the electronic model.
| Health state | Roche | Schering-Plough | SHTAC17 | ||||
|---|---|---|---|---|---|---|---|
| From | To | ||||||
| SVR | SVR | a | a | a | |||
| Mortality | Liver disease | 0 | 0 | 0 | |||
| All cause | Age-/sex-specific | Age-/sex-specific | Age-/sex-specific | ||||
| HCV | SVR | See Table 33 | See Table 33 | ||||
| HCV | a | a | 0 | a | 0 | ||
| 0.025 | a | 0.025 | a | ||||
| CC | 0.037 | 0 | 0.037 | 0 | 0.037 | ||
| HCC | 0 | 0 | 0.001 | 0 | 0 | ||
| Mortality | Liver disease | 0 | 0 | 0 | |||
| All cause | Age-/sex-specific | Age-/sex-specific | Age-/sex-specific | ||||
| CC | SVR | 0 | See Table 33 | ||||
| CC | a | a | a | ||||
| DC | 0.039 | 0.039 | 0.039 | ||||
| HCC | 0.014 | 0.01441 | 0.014 | ||||
| Mortality | Liver disease | 0 | 0.02 | 0 | |||
| All cause | Age-/sex-specific | Age-/sex-specific | Age-/sex-specific | ||||
| DC | DC | a | a | 0.039 | |||
| HCC | 0.014 | 0.01441 | 0.014 | ||||
| LT | 0.02 | 0.022 | 0.02 | ||||
| Mortality | Liver disease | 0.129 | 0.130 | 0.130 | |||
| All cause | 0 | Age-/sex-specific | Age-/sex-specific | ||||
| HCC | HCC | a | a | a | |||
| LT | 0 | 0.02 | 0 | ||||
| Mortality | Liver disease | 0.427 | 0.560 | 0.43 | |||
| All cause | 0 | Age-/sex-specific | Age-/sex-specific | ||||
| LT | LT | a | a | a | |||
| Mortality | Liver disease | 0.210 | 0.150 | 0.150 | |||
| All cause | 0 | Age-/sex-specific | Age-/sex-specific | ||||
| Post LT | Post LT | a | a | a | |||
| Mortality | Liver disease | 0.057 | 0.057 | 0.057 | |||
| All cause | 0 | Age-/sex-specific | Age-/sex-specific | ||||
Sustained virological responses used in manufacturers’ models are shown below in Table 33.
| Patient group | Roche | Schering-Plough | ||
|---|---|---|---|---|
| Genotype 1 | Genotype non-1 | Genotypes 1 + 4 | Genotypes 1 + 3 | |
| Non-responders (%) | 13 | 21 | 48.65 | 69.95 |
| Relapsed (%) | 55 | 55 | 48.65 | 69.95 |
| Shortened duration (%) | 91a | 89b | NA | NA |
| HIV co-infected (%) | 40 | 40 | 38 | 53 |
Tables 34 and 35 report the health-state utility values applied in the three models and the impact on utility applied while patients are on treatment. The impact of structural assumptions in the models (particularly the inclusion of a single SVR state, which does not distinguish the stage of disease prior to SVR) and the selection of utility values applied to the SVR state have been discussed in the previous sections, appraising each of the manufacturer’s models separately. Table 34 shows that, in all but one case, identical utility values have been applied to the health states relating to more advanced liver disease, while there is considerable difference in the utility values applied to patients achieving an SVR [and, to a lesser extent, the HCV health state(s)].
| Health state | Roche | Schering-Plough | SHTAC17 |
|---|---|---|---|
| SVR |
0.91 (< 45) 0.85 (45–54) 0.80 (55–64) 0.78 (65–74) 0.73 (≥ 75) |
0.82 |
0.82 (from mild) 0.72 (from moderate) 0.60 (from CC) |
| HCV | 0.66 | 0.77 (mild) | 0.77 (mild) |
| 0.66 (moderate) | 0.66 (moderate) | ||
| CC | 0.55 | 0.55 | 0.55 |
| DC | 0.45 | 0.45 | 0.45 |
| HCC | 0.45 | 0.45 | 0.45 |
| LT | 0.45 | 0.45 | 0.45 |
| Post LT | 0.45 | 0.67 | 0.67 |
| Health state | Roche | aSchering-Plough | bSHTAC17 |
|---|---|---|---|
| Treatment-year utility | 0.55 | 0.64 (mild) | 0.66 (mild) |
| 0.53 (moderate) | 0.55 (moderate) | ||
| 0.42 (CC) | 0.44 (CC) |
Table 35 indicates that, although there are sizable differences in the utility values applied to the HCV and SVR health states, there is more agreement on the on-treatment utility reduction associated with peginterferon alfa and ribavirin. All three models have based their valuations on data from the UK Mild Hepatitis C Trial,82 which reported health-state valuations for treated and untreated patients by stage of disease (adopted by Roche and in our previous assessment) and an overall mean difference in EQ-5D utility score, for treated and control patients, at 12 weeks or 24 weeks following randomisation (adopted by Schering-Plough).
Table 36 summarises the health-state costs applied in the three models. The main differences between the three models relate to:
-
Structural assumptions in the models (patients enter the Roche model with moderate HCV, so cost of mild HCV is not relevant, while both manufacturers collapse the SVR state and do not track the stage of disease prior to SVR).
-
Sources of costs that have been inflated to current prices. Health-state costs in all three models are based on those reported for the UK Mild Hepatitis C Trial. 82 Roche have inflated health-state costs reported in our previous assessment (which had been inflated from 2002–3 to 2003–4 prices), whereas Schering-Plough inflated the original health-state costs reported for the trial. The discrepancies between the two sets of costs arises from slight adjustments that have been made to the HCHS Pay and Prices Index85 over time.
| Health state | Roche (2007–8) | Schering-Plough (year not stated)a | SHTAC17 (2003–4 prices) | SHTAC (2007–8 prices)b |
|---|---|---|---|---|
| SVR | 0 | 311 | 267 (mild)c | 311 |
| 267 (moderate) | 311 | |||
| 585.50 (CC) | 684 | |||
| HCV | 843.38 | 166 (mild) | 142 (mild) | 166 |
| 862 (moderate) | 738 (moderate) | 862 | ||
| CC | 1338.21 | 1368 | 1171 | 1368 |
| DC | 10,725.12 | 10,965 | 9385 | 10,964 |
| HCC | 9557.18 | 9770 | 8363 | 9770 |
| LT | 43,262.74 | 44,953 | 37,857 | 44,225 |
| Post LT | 1628.48 | 1665 | 1425 | 1665 |
Methods for SHTAC independent economic analysis
Statement of the decision problem and perspective for the cost-effectiveness analysis
We adapted our previously published economic model17 to estimate the cost-effectiveness of peginterferon alfa-2a and peginterferon alfa-2b for the treatment of chronic HCV, compared with current practice, in subgroups of adults who:
-
were eligible for a shortened duration of treatment with peginterferon alfa-2a
-
had failed to show a SVR on previous treatment with peginterferon alfa-2a or peginterferon alfa-2b
-
were co-infected with HCV/HIV.
The perspective of the cost-effectiveness analysis is that of the NHS and PSS.
Strategies/comparators
The scope for the appraisal, as issued by NICE, states that the interventions to be considered are:
-
combination therapy with peginterferon alfa and ribavirin
-
peginterferon alfa monotherapy (for those who cannot tolerate ribavirin).
The comparators for these interventions are BSC (for people who have been previously treated with peginterferon alfa and ribavirin in combination, and for people with HCV/HIV co-infection), or standard duration courses of combination therapy (for people who meet the criteria for receiving shortened courses of combination therapy with peginterferon alfa and ribavirin).
Model type and rationale for the model structure
The principal outcome of interest in the clinical trials systematically reviewed in Chapter 4 is the SVR, defined as undetectable HCV RNA in the serum for at least 6 months after treatment cessation. To estimate the impact of this intermediate effect on final outcomes for patients we required an appropriate model of the natural history of chronic HCV. We adapted our previously published model,17 which was used in NICE TA106. 33
The state-transition diagram describing the health states within the model and the allowable transitions between these states is shown Figure 2. The diagram shows seven non-absorbing health states. For clarity, mortality (the absorbing state) has not been included. In this diagram, ellipses indicate health states and arrows indicate allowable transitions between health states. The shaded ellipses indicate health states with excess mortality risks attributable to chronic liver disease.
FIGURE 2.
State-transition diagram for SHTAC economic model.
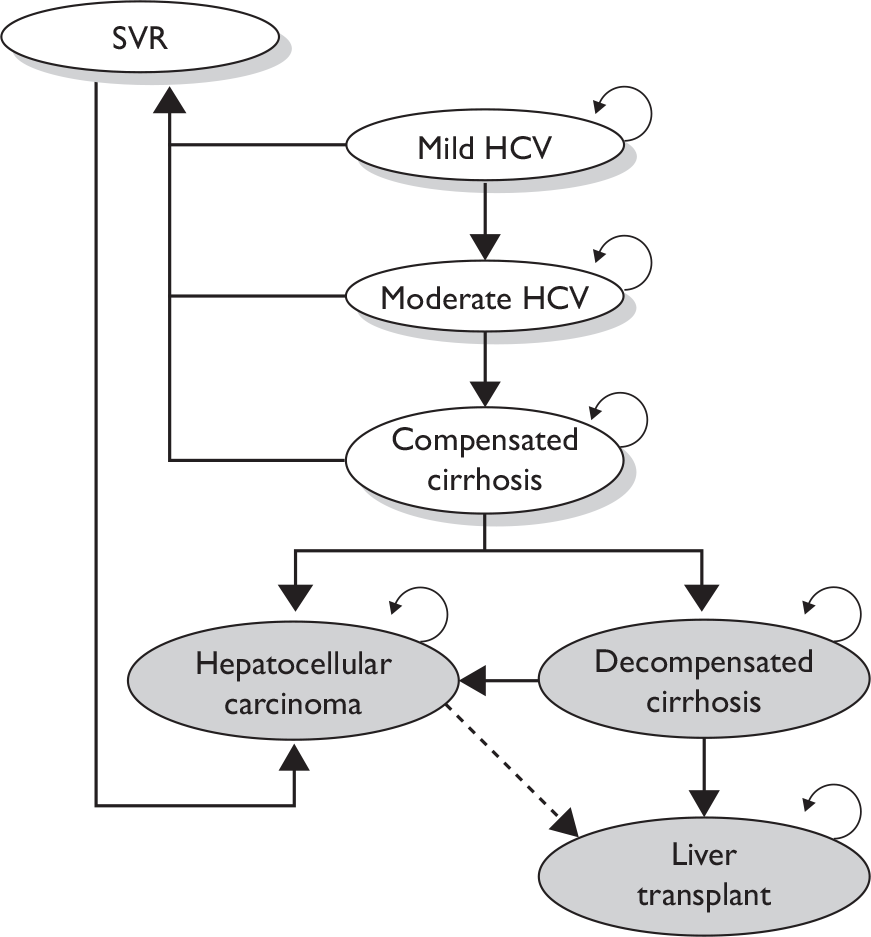
The diagram indicates that, in the absence of successful treatment, patients with chronic HCV or compensated cirrhosis may remain in their current health state or progress to more severe stages of liver disease. In our model the health state labelled SVR is divided into three, to differentiate the stage of disease (mild chronic HCV, moderate chronic HCV or compensated cirrhosis) prior to successful treatment. This is to take account of differences in risk for patients entering the SVR state from different stages of chronic liver disease (patients who achieve an SVR from mild or moderate chronic HCV are assumed to have the same risk of developing HCC as the general population, whereas those who had progressed to cirrhosis are assumed to have an excess risk of HCC). The SVR state is assumed to be a permanent condition, with no spontaneous reactivation of HCV infection, although individuals are not immune from re-infection (note: our analysis does not consider the impact of onwards transmission of, or re-infection with, HCV). Individuals in this health state are assumed to face the same mortality risks as the general population and face no greater risk of liver cancer than the general population.
Patients with mild or moderate chronic HCV, as well as those with compensated cirrhosis, face the same mortality risk as the general population. However, patients with decompensated liver disease, HCC and those who undergo liver transplantation face higher mortality rates related to their stage of liver disease than the general population. A dotted line has been drawn between HCC and liver transplantation to indicate that this transition is often not included in treatment models used for economic evaluations in chronic HCV, and has been excluded from this analysis.
The model has a lifetime horizon and a cycle length of 1 year, with a half-cycle correction applied. To take account of adverse effects of antiviral treatment on HRQoL, health-state utilities are reduced during the year in which treatment occurs.
Baseline cohort of adult chronic HCV patients
Baseline characteristics of the modelled populations were taken from a range of studies reporting relevant characteristics for UK populations of people with chronic HCV infection. 101,102 Patients eligible for shortened treatment durations and those with HCV/HIV co-infection have a mean age at entry to the model of 40 years, while re-treated patients have a mean age of 45 years. Seventy per cent of the cohort is male. The distribution of patients across stages of liver disease is taken from data reported from a clinical audit of patients attending for treatment at a liver unit at a London teaching hospital. 102 While this paper pre-dates current NICE guidance on the treatment of patients with chronic HCV infection, no other studies reporting the distribution of UK patients across stages of disease were identified in our searches. Table 37 reports the distribution across disease stages for existing patients (taken to represent population of patients previously treated with peginterferon alfa and ribavirin) and new patients (taken to represent population of patients with HCV/HIV co-infection and those eligible to receive shortened courses of combination therapy).
| Mild, % (n) | Moderate, % (n) | Cirrhosis, % (n) | |
|---|---|---|---|
| Existing patients | 33 (38) | 35 (40) | 32 (35) |
| New patients | 46 (21) | 44 (20) | 10 (5) |
Data sources
Effectiveness data
Table 38 reports the transition probabilities adopted in the natural history model for this economic evaluation. They represent the complete set of transition probabilities for the BSC comparator and are taken from our previous assessment report. 17
| Health state | Transition probability (SE) | Source | |
|---|---|---|---|
| From | To | ||
| Mild disease | Mild disease | a | |
| Moderate disease | 0.025 (0.004) | Wright and colleagues,82 Grieve and colleagues81 | |
| Moderate disease | Moderate disease | a | |
| CC | 0.037 (0.007) | Wright and colleagues,82 Grieve and colleagues81 | |
| CC | CC | a | |
| DC | 0.039 (0.010) | Fattovich and colleagues98 | |
| HCC | 0.014 (0.010) | Fattovich and colleagues98 | |
| DC | DC | a | |
| HCC | 0.014 (0.010) | Fattovich and colleagues98 | |
| LT | 0.020 | Grieve and colleagues,81 Siebert and colleagues103 | |
| Death | 0.130 (0.010) | Fattovich and colleagues98 | |
| HCC | HCC | a | |
| Death | 0.430 (0.030) | Fattovich and colleagues98 | |
| LT | a | ||
| LT | Death | Year 1 = 0.150, year 2 = 0.057 | Grieve and colleagues,81 Bennett and colleagues76 |
The transition probabilities from mild-to-moderate disease, and from moderate disease to compensated cirrhosis, were derived for the economic evaluation undertaken alongside the UK Mild Hepatitis C Trial82 and were based on a re-analysis of data from UK cross-sectional and longitudinal data sets. The remaining transition probabilities were taken from the literature on natural history and previous economic evaluations. 76,81,98,103 Targeted searches, undertaken as part of this assessment, did not identify new natural history evidence relating to progression or management of chronic HCV to update the model parameters.
Table 39 reports the treatment effects (proportion of patients achieving SVR) that have been applied, in the model, to estimate the effectiveness of peginterferon alfa and ribavirin combination therapy in the treatment strategies and patient subgroups being considered. The studies used to estimate the effectiveness of treatment have typically reported SVRs for all patients in the relevant subgroup and have not indicated the effect of stage of liver disease on response to treatment. For the base-case analyses we have assumed that the same SVR applies for patients with mild or moderate HCV, and for those patients with compensated cirrhosis. We examine the effect of cirrhosis, on reducing the response to treatment, in sensitivity analyses.
| Patient group/treatment strategy | Intervention | Genotype (s) | SVR | EVR | Withdrawal | Source | ||
|---|---|---|---|---|---|---|---|---|
| Standard duration (n, %) | Shortened duration (n, %) | Standard duration (n, %) | Shortened duration (n, %) | |||||
| Shortened treatment duration | PEG α-2a + RBV | 1 | 57/57 (100) | 69/73 (94.5) | NA | 14/154 (9.1)a | 6/154 (3.1)a | Liu and colleagues53 Yu and colleagues, 200754 |
| 24/24 (100) | 27/28 (96.4) | 8/100 (8.0)a | 3/100 (3.0)a | |||||
| 2 | 85/87 (97.7) | 43/43 (100) | NA | 1/100 (1.0)a | 0/50 (0.0)a | Yu and colleagues, 200855 von Wagner and colleagues56 | ||
| 2/3 | 27/31 (87.1) | 33/35 (94.3) | 1/71 (1.4)a | 0/71 (0.0)a | ||||
| PEG α-2b + RBV | 1 | 8/19, (42.1) | 16/28, (57.1) | NA | 7/255 (2.7)a | 4/208 (1.9)a | Berg and colleagues59 | |
| SVR (n, %) | EVR (n, %) | Withdrawal (n, %) | ||||||
| Re-treated | PEG α-2a + RBV | 1 | 18/142 (12.7) | 21/142 (14.8) | 20/316 (6.3) | Jensen and colleagues,88 Roche104 | ||
| Non-1 | 6/29 (20.7) | 10/29 (34.5) | ||||||
| PEG α-2b + RBV | 1 + 4 | 162/1121 (14.5) | 333/1121 (29.7) | 89/1341 (6.6) | Schering-Plough92 | |||
| 2 + 3 | 117/206 (56.8) | 162/206 (78.6) | ||||||
| HCV/HIV co-infected | PEG α-2a + RBV | 1 + 4 | 64/245 (26.1) | NA | 91/606 (15.0),b 99/606 (15.3)c | Kim and colleagues,51 Zhao and colleagues50 | ||
| 2 + 3 | 59/95 (62.1) | |||||||
| PEG α-2b + RBV | 1 + 4 | 55/233 (23.6) | ||||||
| 2 + 3 | 71/152 (46.7) | |||||||
Sustained virological response estimates for patients receiving shortened courses of treatment are based on those used in our systematic review of clinical effectiveness (see Chapter 4, Assessment of clinical effectiveness). SVR estimates for patients co-infected with HCV/HIV were based on those reported from two recent systematic reviews of antiviral treatment in this patient group (further details can be found in Appendix 9). SVR estimates for patients re-treated following non-response to, or relapse from, a previous course of peginterferon alfa-2a were taken from the trial by Jensen and colleagues,88 supplemented with information from the Roche submission to NICE (see Appendix 9). SVRs for re-treatment with peginterferon alfa-2b were from the EPIC3 study,96 as summarised in the Schering-Plough submission to NICE (see Appendix 9).
Health-state values/utilities
A systematic search of the literature (see Appendix 2 for search strategy) and targeted searches did not find new utility data to update our model. In particular, the searches did not identify utility data that were specific to the patient populations within the scope of this assessment. As a result we have adopted the same utility values as for our previous assessment (Table 40). 17 These data are appropriate to the NICE reference case68 for measuring and valuing health benefits, in that the QoL measurements were undertaken using the EQ-5D in patients with chronic HCV recruited to the UK Mild HCV Trial,82 an observational study of patients with more severe liver disease conducted alongside the trial and a UK study of costs and outcomes following liver transplantation. 93 The QoL measurements were valued using a tariff derived in a general population. 84 While the use of these data has the advantage of consistency with those applied for the previous assessment,17 they are not specific to the patient populations in the scope of this assessment. It may be argued that values derived from HCV mono-infected patients may overestimate the heath–state utility for HCV/HIV co-infected patients or that values from treatment-naive patients (as in the UK Mild HCV Trial82) may not be representative of utilities for patients who have been previously treated.
| Health state | Utility |
|---|---|
| SVR (from mild disease) | 0.82 |
| SVR (from moderate disease) | 0.72 |
| Mild HCV | 0.77 |
| Treatment for mild HCV | 0.66 |
| Moderate HCV | 0.66 |
| Treatment for moderate HCV | 0.55 |
| Cirrhosis | 0.55 |
| DC | 0.45 |
| HCC | 0.45 |
| LT | 0.45 |
| Post LT | 0.67 |
Cost data
Intervention costs
Protocols describing the frequency and intensity of monitoring of patients being treated with peginterferon were developed for the previous assessment, based on clinical guidelines and discussion with hepatologists/specialist nurses at Southampton University Hospitals Trust, and are described in full in the previous assessment report. 17 Additional costs for patient management (including the initial evaluation of a new patient with chronic HCV, further investigations required to assess suitability for treatment, costs of clinical decision-making regarding choice of treatment and final tests prior to commencing treatment) were also identified. These costs have been uprated to 2007–8 values (from 2003–4 prices) using the HCHS Pay and Prices Index85 and are reported in Table 41.
| On-treatment monitoring (weeks) | Cost (£) |
|---|---|
| 12 | 649 |
| 16 | 782 |
| 24 | 792 |
| 48 | 1051 |
| 72 | 1039 |
In addition to the excess costs of health service contacts for patients undergoing treatment, drug costs also need to be estimated. Drug unit costs were taken from the BNF, No. 58, September 2009. 47
Drug costs for peginterferon alfa-2a (Pegasys) were calculated for a dosage of 180 µg/0.5 ml, self-administered by patients once per week, corresponding to a weekly cost of £126.91. The total drug cost for a 24-week course of treatment for genotype 2/3 patients is £3046 for monotherapy, and for 48 weeks is £6092. Drug costs for ribavirin (Copegus) for dual therapy with peginterferon alfa-2a were calculated for a dosage of 800 mg per day for genotype 2/3 and 1000–1200 mg per day (depending on body weight: 1000 mg for weight < 75 kg and 1200 mg for weight ≥ 75 kg) for genotype 1. Patients co-infected with HCV and HIV also receive 800 mg of ribavirin per day, irrespective of genotype. A 168-tab packet of 200-mg tablets costs £444.43. This corresponds to a weekly cost of £111 for genotype 1 (based on an average body weight of 79 kg) and £74 for genotype 2/3. The total drug costs estimated for 24 weeks of dual therapy are £4824, and are £11,425 for 48 weeks of dual therapy (or £9647 for HCV/HIV co-infected patients having 48 weeks of dual therapy).
Drug costs for peginterferon alfa-2b (ViraferonPeg) were calculated for a patient weighing 79 kg (at a dosage of 1.5 µg/kg for dual therapy). Weekly costs were estimated as the unit cost for the appropriate dosage using a pre-filled pen (£162.60 for dual therapy). The total drug cost for a 24-week course of treatment is £3902 and for 48 weeks is £7805. Dosage of ribavirin (Rebetol), used in dual therapy with peginterferon alfa-2b, is also weight based (Table 42). Drug costs for ribavirin, in combination with peginterferon alfa-2b, were calculated for a dosage of 1000 mg per day, based on an average body weight of 79 kg. A 168-tab packet of 200-mg tablets costs £327.60, which corresponds to a weekly cost of £68. Combined with the costs estimated above this gives a total drug cost for combination therapy (peginterferon alfa-2b plus ribavirin) of £5540 for 24 weeks of treatment for genotype 2/3 patients, and £11,081 for 48 weeks of treatment for genotype 1 patients.
| Body weight (kg) | Total daily dose of Rebetol (mg) |
|---|---|
| < 65 | 800 |
| 65–85 | 1000 |
| 86–105 | 1200 |
| > 105 | 1400 |
Health-state costs
Health-state costs for SVR, chronic HCV, compensated cirrhosis, decompensated cirrhosis and HCC have been taken from the observational study conducted during the UK Mild HCV Trial (Table 43). 82 Costs for liver transplantation and post liver transplantation were taken from a Department of Health-funded study of the costs of liver transplantation. 105 Costs for 2002–3 have been updated to 2007–8 costs using the HCHS Pay and Prices Index. 85
| Health state | Cost (£/year), 2007–8 prices |
|---|---|
| SVR | 311 |
| Mild chronic HCV | 142 |
| Moderate chronic HCV | 862 |
| CC | 1368 |
| DC | 10,964 |
| HCC | 9770 |
| LT | 44,225 |
| Post LT | 1665 |
Discounting of future costs and benefits
A discount rate of 3.5% was applied to future costs and benefits, in line with current methodological guidance from NICE. 68 Discount rates of 0% (for both costs and benefits), 6% (for costs) and 1.5% (for benefits) were applied in the sensitivity analyses.
Presentation of results
We report findings on the cost-effectiveness of interventions based on analysis of a cohort of patients with age, sex and genotype characteristics as reported above (see Baseline cohort of adult chronic HCV patients). For HCV/HIV co-infected patients and re-treatment of patients who failed to respond to, or relapse from, prior peginterferon alfa therapy, the interventions assessed in this report are compared with BSC (i.e. without any form of interferon alfa therapy) as specified in the NICE scope. For patients who are eligible for shortened courses of peginterferon alfa, results are presented in comparison with the usual duration of treatment.
We report the results of these comparisons in terms of the incremental gain in QALYs and the incremental costs determined in the cohort analysis.
Assessment of uncertainty in the SHTAC analysis (sensitivity analysis)
Parameter uncertainty is addressed using PSA. Probability distributions are assigned to the point estimates used in the base-case analysis. The point estimates for state transitions and treatment effects are reported in Tables 38 and 39, for health-state utilities in Table 40 and for health-state costs in Table 43. Appendix 10 reports the variables included in the PSA, the form of distribution used for sampling and the parameters of the distribution.
Univariate sensitivity analysis is used to address particular areas of uncertainty in the model related to:
-
model structure
-
methodological assumptions
-
transition probabilities, around which there is considerable uncertainty or which may be expected, a priori, to have disproportionate impact on study results.
The purpose of this analysis is to identify clearly the impact of this uncertainty and to test the robustness of the cost-effectiveness results to variation in structural assumptions and parameter inputs.
Results of SHTAC independent economic analysis
Shortened treatment duration
Peginterferon alfa-2a
Costs and outcomes modelled for patients who were eligible for shortened duration of treatment with peginterferon alfa-2a and ribavirin combination therapy are presented in Table 44 for genotype 1 patients and in Table 45 for patients with genotype 2 or 3. As it was not considered appropriate to conduct a meta-analysis of RCTs, we present separate results for each trial included in our systematic review of clinical effectiveness (with the exception of the RCT by Mangia and colleagues,52 which used both peginterferon alfa-2a and alfa-2b within the same trial; as the two drugs are considered pharmacologically different, we present cost-effectiveness estimates for peginterferon alfa-2a based on RCTs of alfa-2a, and peginterferon alfa-2b based on RCTs of alfa-2b). The comparator in each of the analyses is the standard duration of peginterferon alfa-2a combination therapy (48 weeks for genotype 1 patients and 24 weeks for patients with genotype 2 or 3). The tables report total costs (antiviral treatment and supportive care), health outcomes (in terms of life-years and QALYs) and the incremental cost-per-QALY ratios. Costs and health outcomes are discounted at 3.5%.
| RCT | Treatment duration | Cost (£) | Outcome (life-years) | Outcome (QALYs) | ICER (£/QALY gained) |
|---|---|---|---|---|---|
| Liu and colleagues 200853 | Standard (48 weeks) | 14,206 | 20.86 | 15.68 | |
| Shortened (24 weeks) | 9399 | 20.76 | 15.54 | ||
| Incremental | –4807 | –0.11 | –0.14 | 34,510 | |
| Yu and colleagues 200854 | Standard (48 weeks) | 14,206 | 20.86 | 15.68 | |
| Shortened (24 weeks) | 8994 | 20.80 | 15.60 | ||
| Incremental | –5212 | –0.07 | –0.08 | 64,880 |
| RCT | Treatment duration | Cost (£) | Outcome (life-years) | Outcome (QALYs) | ICER (£/QALY gained) |
|---|---|---|---|---|---|
| Yu and colleagues 200755 | Standard (24 weeks) | 7834 | 20.82 | 15.64 | |
| Shortened (16 weeks) | 5728 | 20.86 | 15.72 | ||
| Incremental | –2107 | 0.04 | 0.08 | Shortened duration dominates | |
| von Wagner and colleagues 200556 | Standard (24 weeks) | 10,089 | 20.61 | 15.31 | |
| Shortened (16 weeks) | 6943 | 20.75 | 15.54 | ||
| Incremental | –3146 | 0.14 | 0.23 | Shortened duration dominates |
In both of the included trials of shortened treatment duration for genotype 1 patients, standard 48-week treatment was associated with an SVR of 100%. However, this high SVR is only applicable to the subgroup of patients who had baseline LVL (< 800,000 IU/ml in the trial by Liu and colleagues53 and < 400,000 IU/ml in the trial by Yu and colleagues, 200854 – the SPC for peginterferon alfa-2a42 defines LVL as ≤ 800,000 IU/ml at baseline) and who also demonstrate an RVR. Shortened treatment duration was associated with a slight reduction in SVR (to 94% in the trial by Liu and colleagues53 and 96% in the trial by Yu and colleagues54). Shorter duration of 24 weeks of treatment is associated with a reduction in total costs between £4800 and £5200. This is primarily due to the reduction in drug acquisition costs, although there is some additional reduction in cost of on-treatment monitoring. While the small reduction in SVR means that there are some additional costs associated with disease progression for the cohort of patients receiving shorter duration of treatment, these are not sufficient to offset the cost reduction associated with the shorter duration of treatment.
Given that the SVR is lower, and therefore there is a greater risk of progressive liver disease, there is a reduction in total QALYs between 0.08 and 0.14 (depending on trial) associated with shorter duration of treatment. This is not offset by the QoL impact of treatment-related adverse events estimated for the cohort of patients receiving shorter duration of treatment. As total costs and total QALYs are reduced in the cohort of patients receiving shorter duration of treatment, the ICER is positive but is located in the south-west (cost- and outcome-reducing) quadrant of the cost-effectiveness map rather than the more familiar north-east (cost-increasing and outcome-gaining) quadrant. This has implications for the interpretation of the results from the base-case (deterministic) analysis and from the PSA, including the interpretation of the CEACs.
In both of the included trials for genotype 2 or 3 patients, shortened treatment duration was associated with a higher SVR than was the case for standard treatment. Shorter duration of treatment is associated with a reduction in total costs between £2100 and £3150. This is primarily due to the reduction in drug acquisition costs and a reduction in the cost of on-treatment monitoring. Given that the SVR is higher for the shorter duration of treatment, there are small reductions in total cost associated with a reduced risk of disease progression for the cohort of patients receiving shorter duration of treatment. The higher SVR for the shorter duration of treatment also results in improvements in modelled outcomes associated with shorter duration of treatment so that the strategy of shortened treatment duration for this group of patients dominates standard duration treatment.
Deterministic sensitivity analysis
Table 46 reports the results of a DSA for genotype 1 patients who were eligible for shortened treatment duration, and Table 47 reports the results for genotype 2/3 patients. These are predominantly univariate sensitivity analyses – that is, varying one parameter at a time, from its base-case value, leaving all other variables unchanged. The table is divided to distinguish between analyses undertaken owing to structural uncertainties in the model, uncertainties over the composition of the baseline cohort and uncertainty over parameter values.
| Genotype 1 | ||||||
|---|---|---|---|---|---|---|
| Liu and colleagues53 | Yu and colleagues 200854 | |||||
| Incremental cost (£) | Incremental QALY | ICER | Incremental cost (£) | Incremental QALY | ICER | |
| Base case | –4807 | –0.14 | 34,510 | –5212 | –0.08 | 64,880 |
| Structural uncertainty | ||||||
| Spontaneous SVR from mild (0.002) | –4851 | –0.13 | 37,420 | –5241 | –0.07 | 70,779 |
| Spontaneous SVR from mild (0.010) | –4831 | –0.13 | 36,033 | –5228 | –0.08 | 67,953 |
| Discount cost and outcome at 0% | –3605 | –0.38 | 9543 | –4429 | –0.24 | 18,785 |
| Discount cost at 6%, outcome at 1.5% | –5187 | –0.24 | 21,447 | –5460 | –0.15 | 37,096 |
| Baseline cohort characteristics | ||||||
| Cohort 80% male | –4813 | –0.14 | 34,917 | –5216 | –0.08 | 65,702 |
| Cohort 40% male | –4788 | –0.14 | 33,281 | –5200 | –0.08 | 62,412 |
| Change average age of cohort at start of simulation (base case 40 years old) | ||||||
| –10 years | –4674 | –0.18 | 26,429 | –5126 | –0.10 | 48,901 |
| +5 years | –4892 | –0.12 | 41,051 | –5268 | –0.07 | 78,362 |
| +10 years | –4989 | –0.10 | 50,551 | –5331 | –0.05 | 98,940 |
| +15 years | –5098 | –0.08 | 65,021 | –5402 | –0.04 | 132,866 |
| Change distribution of cohort across disease stages at start of simulation | ||||||
| Cohort 100% mild chronic HCV | –5396 | –0.09 | 57,661 | –5597 | –0.05 | 110,708 |
| Cohort 100% moderate HCV | –4415 | –0.17 | 26,641 | –4957 | –0.10 | 50,807 |
| Cohort 100% CC | –3817 | –0.23 | 16,371 | –4567 | –0.14 | 32,270 |
| Parameter uncertainty | ||||||
| Assume SVR is 25% lower in patients with CC | –4860 | –0.13 | 36,627 | –5247 | –0.08 | 69,001 |
| Assume SVR is 50% lower in patients with CC | –4914 | –0.13 | 38,964 | –5282 | –0.07 | 73,614 |
| Cohort 100% CC, assume SVR is 25% lower in patients with CC | –4356 | –0.17 | 26,023 | –4918 | –0.10 | 49,855 |
| Cohort 100% CC, assume SVR is 50% lower in patients with CC | –4894 | –0.10 | 48,177 | –5269 | –0.06 | 94,485 |
| Transition probability from mild-to-moderate disease = 0.04 | –4733 | –0.15 | 31,168 | –5164 | –0.09 | 58,333 |
| Transition probability from moderate disease to CC = 0.073 | –4650 | –0.18 | 26,516 | –5110 | –0.10 | 49,205 |
| Cost of SVR state = £0 | –4790 | –0.14 | 34,392 | –5201 | –0.08 | 64,747 |
| Reduce cost of PEG α-2a by 20% | –4197 | –0.14 | 30,136 | –4603 | –0.08 | 57,298 |
| Reduce cost of PEG α-2a by 30% | –3893 | –0.14 | 27,949 | –4298 | –0.08 | 53,506 |
| Reduce cost of RBV by 20% | –4273 | –0.14 | 30,681 | –4679 | –0.08 | 58,242 |
| Reduce cost of RBV by 20% | –4007 | –0.14 | 28,766 | –4412 | –0.08 | 54,922 |
| Genotype 2 | Genotype 2/3 | |||||
|---|---|---|---|---|---|---|
| Yu and colleagues 200755 | von Wagner and colleagues 200556 | |||||
| Incremental cost (£) | Incremental QALY | ICER | Incremental cost (£) | Incremental QALY | ICER | |
| Base case | –2107 | 0.08 | –26,000 | –3146 | 0.23 | –13,555 |
| Structural uncertainty | ||||||
| Spontaneous SVR from mild (0.002) | –2088 | 0.08 | –27,124 | –3088 | 0.22 | –14,071 |
| Spontaneous SVR from mild (0.010) | –2096 | 0.08 | –26,595 | –3115 | 0.23 | –13,827 |
| Discount cost and outcome at 0% | –2610 | 0.18 | –14,416 | –4722 | 0.55 | –8664 |
| Discount cost at 6%, outcome at 1.5% | –1947 | 0.12 | –15,695 | –2647 | 0.37 | –7220 |
| Baseline cohort characteristics | ||||||
| Cohort 80% male | –2104 | 0.08 | –26,165 | –3138 | 0.23 | –13,632 |
| Cohort 40% male | –2115 | 0.08 | –25,495 | –3171 | 0.24 | –13,319 |
| Change average age of cohort at start of simulation (base case 40 years old) | ||||||
| –10 years | –2162 | 0.10 | –22,343 | –3320 | 0.28 | –11,801 |
| +5 years | –2071 | 0.07 | –28,532 | –3034 | 0.21 | –14,752 |
| +10 years | –2030 | 0.06 | –31,720 | –2906 | 0.18 | –16,250 |
| +15 years | –1984 | 0.06 | –35,763 | –2763 | 0.15 | –18,153 |
| Change distribution of cohort across disease stages at start of simulation | ||||||
| Cohort 100% mild chronic HCV | –1859 | 0.06 | –30,058 | –2372 | 0.17 | –13,780 |
| Cohort 100% moderate HCV | –2271 | 0.09 | –24,655 | –3660 | 0.27 | –13,721 |
| Cohort 100% CC | –2522 | 0.12 | –20,940 | –4444 | 0.36 | –12,507 |
| Parameter uncertainty | ||||||
| Assume SVR is 25% lower in patients with CC | –2084 | 0.08 | –26,629 | –3075 | 0.22 | –13,763 |
| Assume SVR is 50% lower in patients with CC | –2061 | 0.08 | –27,303 | –3005 | 0.21 | –13,987 |
| Cohort 100% CC, assume SVR is 25% lower in patients with CC | –2296 | 0.09 | –24,734 | –3737 | 0.27 | –13,895 |
| Cohort 100% CC, assume SVR is 50% lower in patients with CC | –2070 | 0.07 | –31,740 | –3031 | 0.18 | –16,594 |
| Transition probability from mild-to-moderate disease = 0.04 | –2137 | 0.09 | –24,769 | –3243 | 0.25 | –13,045 |
| Transition probability from moderate disease to CC = 0.073 | –2172 | 0.10 | –22,591 | –3352 | 0.28 | –11,994 |
| Cost of SVR state = £0 | –2113 | 0.08 | –26,085 | –3168 | 0.23 | –13,648 |
| Reduce cost of PEG α-2a by 20% | –1903 | 0.08 | –23,494 | –2943 | 0.23 | –12,680 |
| Reduce cost of PEG α-2a by 30% | –1802 | 0.08 | –22,241 | –2842 | 0.23 | –12,243 |
| Reduce cost of RBV by 20% | –1988 | 0.08 | –24,537 | –3028 | 0.23 | –13,045 |
| Reduce cost of RBV by 20% | –1929 | 0.08 | –23,806 | –2968 | 0.23 | –12,789 |
The DSA suggests that the results are robust to a change in structural assumptions (allowing spontaneous SVR from the mild chronic HCV state), the proportion of the baseline cohort that is male and the cost associated with the SVR health state. Reducing drug acquisition costs has the effect of reducing the cost-effectiveness of shortened treatment duration, as it reduces the cost saving between standard and shortened treatment duration while the outcome difference is unchanged.
The greatest variability in ICERs is associated with changes in two assumptions regarding baseline characteristics of the cohort of treatment-eligible patients. Increasing the mean age of patients at the start of the simulation up to 15 years leads to an approximate doubling of the ICER. This occurs because the QALY difference between the standard and shortened treatment duration reduces rapidly [from –0.14 at a starting age of 40 years to –0.08 at a starting age of 55 years (a reduction of 43%) using efficacy data from the RCT by Liu and colleagues53]. The difference in costs between standard and shortened treatment duration is less responsive to changes in starting age [from –£4807 at a starting age of 40 to –£5098 at a starting age of 55 (a reduction of 6%) using the same efficacy data]. Alternative assumptions regarding the stage of liver disease in patients starting treatment also has a large impact on the ICER, with shortened treatment duration being more cost-effective in patients with less severe disease than in those with cirrhosis. This arises because, all other things being equal, the higher the proportion of a cohort starting the simulation with cirrhosis, the greater the proportion that will progress to advanced liver disease. As a result, the penalty (in terms of a reduction in total QALYs) associated with a lower SVR for shortened treatment duration will be greater in cohorts that contain a higher proportion of patients with cirrhosis.
The pattern of results for genotype 2/3 patients, reported in Table 47, is similar to those for genotype 1 patients. The results are largely insensitive to changes in input parameters, other than baseline assumptions relating to age and stage of disease at start of treatment. Shortened treatment duration remains dominant in all the scenarios tested in Table 47, using efficacy data from either of the included trials.
As the included trials give contradictory results (with shortened duration less effective than standard duration for genotype 1 patients, but more effective for genotype 2/3 patients) and, in the case of genotype 2/3 patients, potentially counterintuitive results, we conducted an additional scenario analysis on the impact of the difference in SVR on the cost-effectiveness results, assuming that the SVR for shortened treatment duration is less than or equal to that for standard treatment duration (Table 48).
| SVR difference (%) | Incremental cost (£) | Incremental QALY | ICER |
|---|---|---|---|
| Genotype 1 | |||
| 0 | –5971 | 0.03 | –199,047 |
| 1 | –5759 | 0.00a | 6,442,219 |
| 3 | –5334 | –0.06 | 85,091 |
| 5 | –4908 | –0.12 | 39,435 |
| Genotype 2/3 | |||
| 0 | –1618 | 0.01 | –161,785 |
| 1 | –1405 | –0.02 | 67,257 |
| 3 | –980 | –0.08 | 11,854 |
| 5 | –555 | –0.14 | 3841 |
This suggests that shortened treatment duration may be a highly cost-effective option, where there is no difference (or a very small difference) in SVR between shortened and standard treatment duration, but as the SVR difference increases the cost reduction decreases and the QALY loss increases rapidly – particularly in the case of genotype 2/3 patients.
Probabilistic sensitivity analysis
In a PSA, where the probabilities of achieving SVR, health-state costs, health-state utility values and transition probabilities for the natural history parameters were sampled probabilistically, shortened duration of treatment with peginterferon alfa-2a and ribavirin combination therapy was generally associated with reduced QALYs. For genotype 1 patients incremental QALYs associated with shortened duration of treatment were negative for the majority of simulations – 95% of simulations using efficacy data (proportion of patients with SVR) from Liu and colleagues,53 and 99.5% of simulations using efficacy data from Yu and colleagues. 54 The opposite is true in the analysis of genotype 2 or 3 patients. Approximately 2% of simulations were associated with negative incremental QALYs, for genotype 2 patients, using efficacy data from Yu and colleagues,55 whereas none of the simulations resulted in negative incremental QALYs using efficacy data from von Wagner and colleagues. 56 Incremental costs associated with shortened duration of treatment were negative in all simulations – ranging from –£2500 to –£6000 for genotype 1 patients and from –£550 to –£5200 for genotype 2/3 patients. Table 49 reports summary information for the PSAs and Figures 3–6 show the scatter plots for each analysis, including 95% confidence ellipses.
| RCT | Treatment duration | Lifetime costs (£) | QALYs |
|---|---|---|---|
| Genotype 1 | |||
| Liu and colleagues 200853 | Standard duration | 14,566 (14,020 to 15,708) | 15.60 (14.39 to 16.77) |
| Shortened duration | 9,815 (8546 to 12,040) | 15.45 (14.32 to 16.56) | |
| Incremental | –4752 (–5582 to –3658) | –0.14 (–0.32 to –0.02) | |
| Yu and colleagues 200854 | Standard duration | 15,062 (14,067 to 17,639) | 15.55 (14.38 to 16.71) |
| Shortened duration | 9701 (8309 to 12,538) | 15.50 (14.36 to 16.66) | |
| Incremental | –5361 (–5810 to –4922) | –0.06 (–0.13 to 0.01) | |
| Genotype 2 | |||
| Yu and colleagues 200755 | Standard duration | 8056 (7360 to 9300) | 15.61 (14.48 to 16.78) |
| Shortened duration | 6201 (5577 to 7659) | 15.65 (14.49 to 16.84) | |
| Incremental | –1855 (–2019 to –1,576) | 0.04 (0.00 to 0.07) | |
| Genotypes 2 + 3 | |||
| von Wagner and colleagues 200556 | Standard duration | 10,072 (8048 to 13,552) | 15.33 (14.25 to 16.42) |
| Shortened duration | 6931 (5794 to 9160) | 15.56 (14.41 to 16.66) | |
| Incremental | –3141 (–4287 to –2242) | 0.23 (0.08 to 0.40) | |
FIGURE 3.
Cost-effectiveness plane for genotype 1 patients – incremental cost and incremental QALYs for shortened treatment duration using peginterferon alfa-2a and ribavirin combination therapy (24 vs 48 weeks of treatment) – efficacy from Liu and colleagues. 53
FIGURE 4.
Cost-effectiveness plane for genotype 1 patients – incremental cost and incremental QALYs for shortened treatment duration using peginterferon alfa-2a and ribavirin combination therapy (24 vs 48 weeks of treatment) – efficacy from Yu and colleagues. 54
FIGURE 5.
Cost-effectiveness plane for genotype 2 patients – incremental cost and incremental QALYs for shortened treatment duration using peginterferon alfa-2a and ribavirin combination therapy (16 vs 24 weeks of treatment) – efficacy from Yu and colleagues. 55
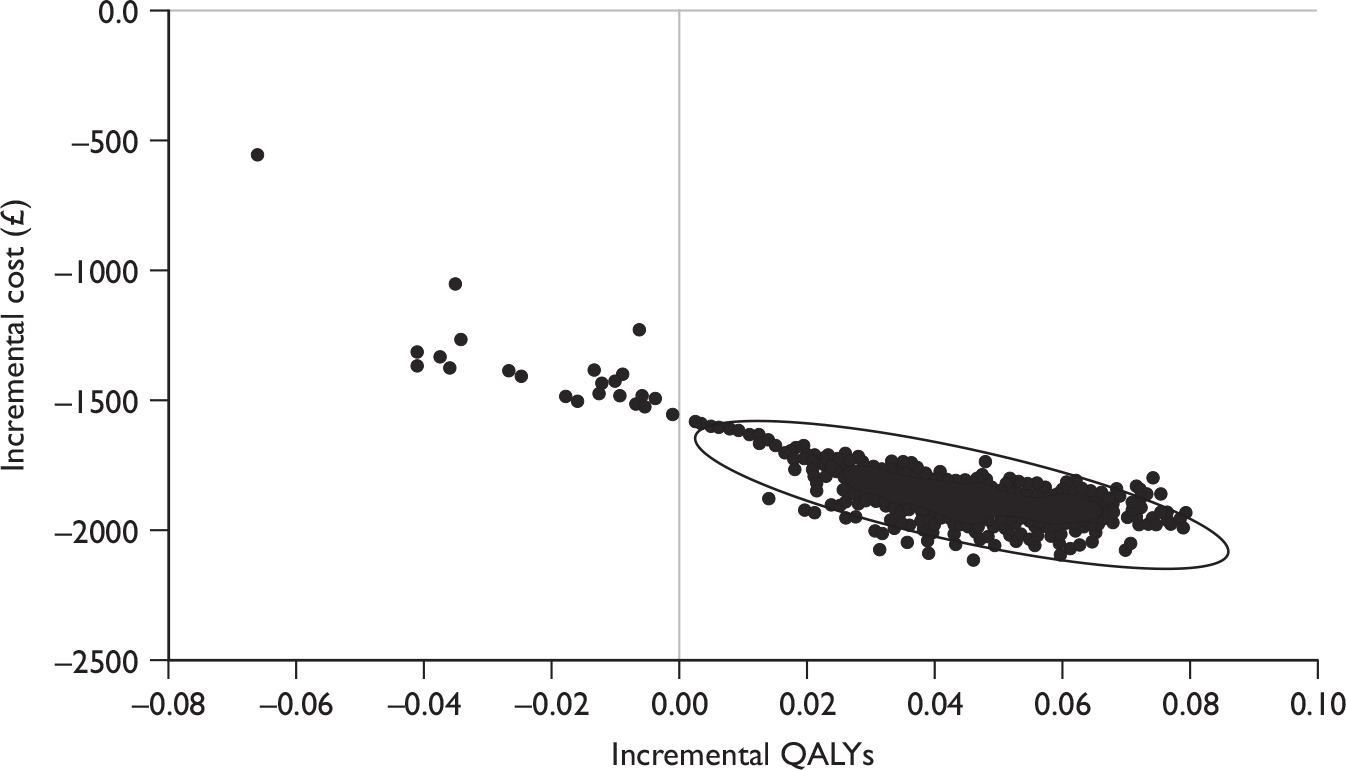
FIGURE 6.
Cost-effectiveness plane for genotypes 2/3 patients – incremental cost and incremental QALYs for shortened treatment duration using peginterferon alfa-2a and ribavirin combination therapy (16 vs 24 weeks of treatment) – efficacy from von Wagner and colleagues. 56
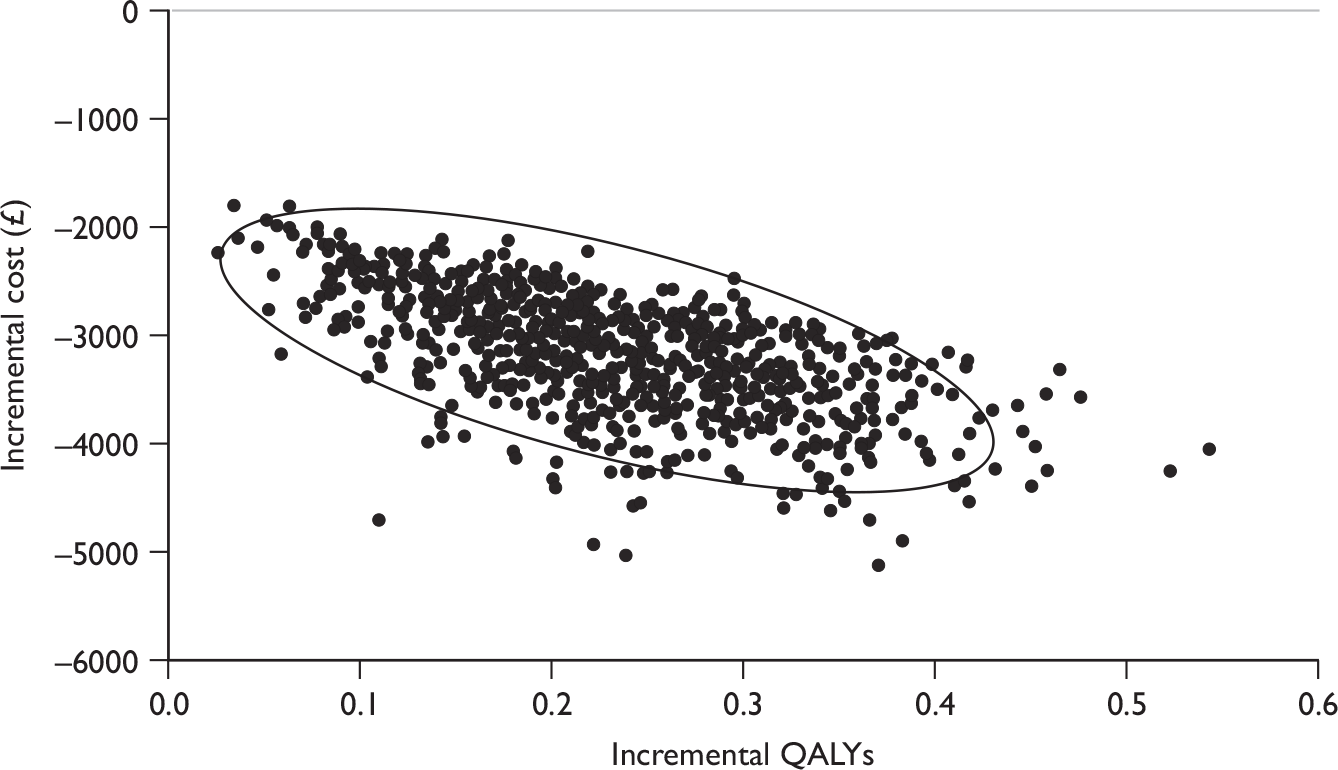
In this analysis, shortened duration of treatment using peginterferon alfa-2a and ribavirin combination therapy for genotype 1 patients had a probability of being cost-effective [compared with the standard duration (48 weeks) of treatment] of 83% at a willingness-to-pay threshold of £20,000 per QALY and 59% at a willingness-to-pay threshold of £30,000, using efficacy data from the trial reported by Liu and colleagues53 (Figure 7). The equivalent values using efficacy data from the trial reported by Yu and colleagues 200854 are 100% at willingness-to-pay thresholds of £20,000 and £30,000 per QALY.
FIGURE 7.
Cost-effectiveness acceptability curves for shortened treatment duration with peginterferon alfa-2a and ribavirin combination therapy (24 vs 48 weeks of treatment) for genotype 1 patients.
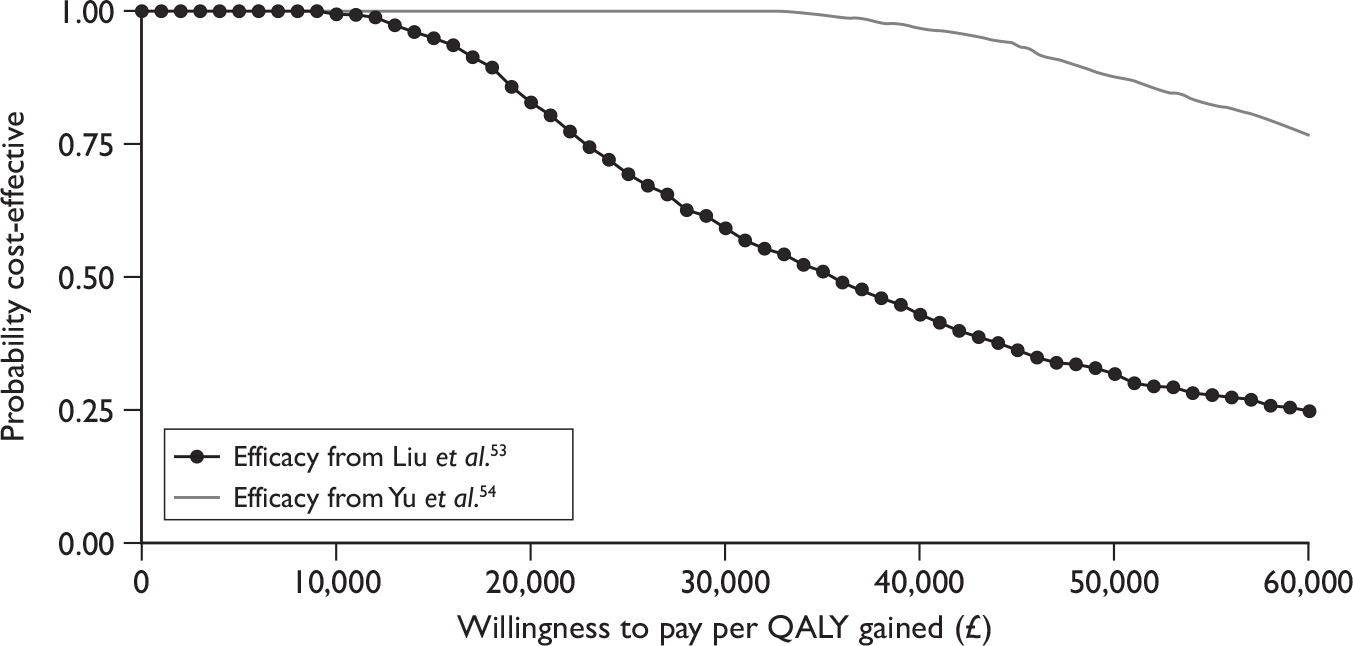
For patients with genotypes 2 and 3, the probability of being cost-effective [compared with the standard duration (24 weeks) of treatment] was 100% at willingness-to-pay thresholds of £20,000 and £30,000 per QALY, using efficacy data from either the trial reported by Yu and colleagues, 200755 or the trial by von Wagner and colleagues56 (Figure 8). This reflects the proportion of simulations located in the south-east quadrant of the cost-effectiveness map (where the intervention dominates the comparator) – see Figures 5 and 6.
FIGURE 8.
Cost-effectiveness acceptability curves for shortened treatment duration with peginterferon alfa-2a and ribavirin combination therapy (16 vs 24 weeks of treatment) for genotypes 2/3.
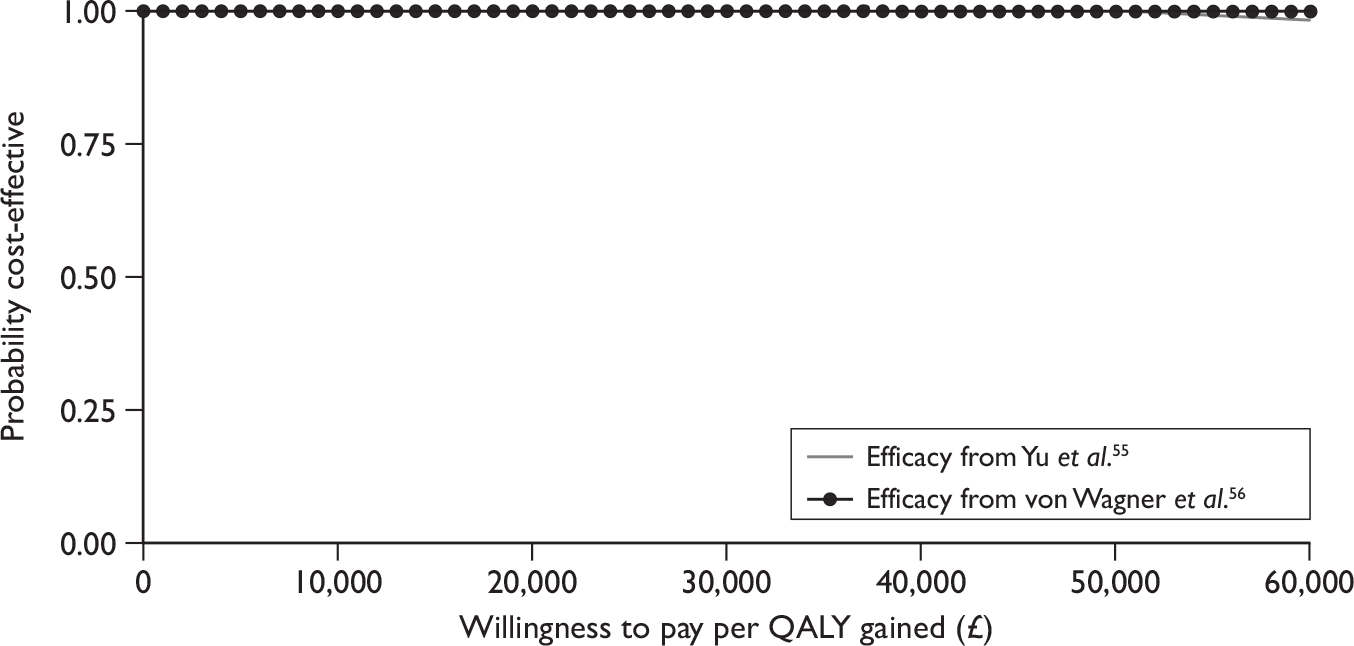
Peginterferon alfa-2b
Costs and outcomes modelled for genotype 1 patients eligible for shortened duration of treatment on peginterferon alfa-2b and ribavirin combination therapy are presented in Table 50.
| RCT | Treatment duration | Cost (£) | Outcome (life-years) | Outcome (QALYs) | ICER (£/QALY gained) |
|---|---|---|---|---|---|
| Berg and colleagues59 | Standard (48 weeks) | 26,169 | 19.74 | 13.89 | |
| Shortened (24 weeks) | 17,173 | 20.03 | 14.38 | ||
| Incremental | –8996 | 0.29 | 0.49 | Shortened duration dominates |
In the trial reported by Berg and colleagues,59 shortened treatment duration was associated with a higher SVR than was the case for standard treatment. Shorter duration of treatment is associated with a reduction in total costs of approximately £9000. This is primarily due to the reduction in drug acquisition costs and a reduction in cost of on-treatment monitoring. Given that the SVR is higher for the shorter duration of treatment, there are also small reductions in total cost associated with a reduced risk of disease progression for the cohort of patients receiving shorter duration of treatment. The higher SVR for the shorter duration of treatment also results in improvements in modelled outcomes associated with shorter duration of treatment so that the strategy of shortened treatment duration for this group of patients dominates standard treatment.
Deterministic sensitivity analysis
Table 51 reports the results of a DSA for genotype 1 patients who were eligible for shortened treatment duration with peginterferon alfa-2b and ribavirin combination therapy. The DSAs suggest that the results are generally insensitive to changes in structural assumptions and input parameter values. The greatest variability in ICERs is associated with changes in the mean age of patients at the start of the simulation and the initial distribution of patients across stages of liver disease.
| Genotype 1 | |||
|---|---|---|---|
| Incremental cost (£) | Incremental QALY | ICER | |
| Base case | –8996 | 0.49 | Shortened duration dominates |
| Structural uncertainty | |||
| Spontaneous SVR from mild (0.002) | –8874 | 0.47 | Shortened duration dominates |
| Spontaneous SVR from mild (0.010) | –8930 | 0.48 | |
| Discount cost and outcome at 0% | –12,292 | 1.15 | |
| Discount cost at 6%, outcome at 1.5% | –7952 | 0.78 | |
| Baseline cohort characteristics | |||
| Cohort 80% male | –8979 | 0.49 | Shortened duration dominates |
| Cohort 40% male | –9048 | 0.51 | |
| Change average age of cohort at start of simulation (base case 40 years old) | |||
| –10 years | –9361 | 0.60 | Shortened duration dominates |
| +5 years | –8762 | 0.44 | |
| +10 years | –8495 | 0.38 | |
| +15 years | –8196 | 0.33 | |
| Change distribution of cohort across disease stages at start of simulation | |||
| Cohort 100% mild chronic HCV | –7377 | 0.37 | Shortened duration dominates |
| Cohort 100% moderate HCV | –10,072 | 0.57 | |
| Cohort 100% CC | –11,711 | 0.75 | |
| Parameter uncertainty | |||
| Assume SVR is 25% lower in patients with CC | –8848 | 0.48 | Shortened duration dominates |
| Assume SVR is 50% lower in patients with CC | –8701 | 0.46 | |
| Cohort 100% CC, assume SVR is 25% lower in patients with CC | –10,233 | 0.57 | |
| Cohort 100% CC, assume SVR is 50% lower in patients with CC | –8755 | 0.39 | |
| Transition probability from mild-to-moderate disease | –9198 | 0.53 | |
| Transition probability from moderate disease to CC | –9426 | 0.59 | |
| Cost of SVR state = £0 | –9041 | 0.49 | |
| Reduce cost of PEG α-2b by 20% | –8216 | 0.49 | |
| Reduce cost of PEG α-2b by 30% | –7825 | 0.49 | |
| Reduce cost of RBV by 20% | –8669 | 0.49 | |
| Reduce cost of RBV by 20% | –8505 | 0.49 | |
As the included trial by Berg and colleagues59 gives a potentially counterintuitive result (with shortened treatment duration being more effective than standard duration), we conducted an additional scenario analysis on the impact of the difference in SVR on the cost-effectiveness results, assuming that the SVR for shortened treatment duration is less than or equal to that for standard treatment duration (Table 52).
| SVR difference (%) | Incremental cost (£) | Incremental QALY | ICER |
|---|---|---|---|
| 0 | –5799 | 0.03 | Shortened duration dominates |
| 1 | –5587 | 0.00a | 6,249,786 |
| 3 | –5162 | –0.06 | 82,347 |
| 5 | –4736 | –0.12 | 38,053 |
This analysis suggests that shortened treatment duration may be a highly cost-effective option, where there is no difference (or a very small difference) in SVR between shortened and standard treatment duration. Where there is no difference in SVR, shortened duration of treatment dominates standard duration by reducing the utility loss associated with treatment.
Probabilistic sensitivity analysis
In a PSA, where the probabilities of achieving SVR, health-state costs, health-state utility values, and transition probabilities for the natural history parameters were sampled probabilistically, shortened duration of treatment with peginterferon alfa-2b and ribavirin combination therapy, for genotype 1 patients with baseline LVL and who achieve an RVR, is associated with reduced costs and increased QALYs in all simulations (using efficacy data from Berg and colleagues59). Table 53 reports summary information for the PSAs and Figure 9 shows the cost-effectiveness plane, including 95% confidence ellipses.
| RCT | Treatment duration | Lifetime costs (£) | QALYs |
|---|---|---|---|
| Berg and colleagues 200959 | Standard duration | 26,256 (20,507 to 33,463) | 13.90 (12.96 to 14.85) |
| Shortened duration | 17,247 (12,786 to 22,987) | 14.38 (13.43 to 15.34) | |
| Incremental | –9009 (–10,506 to –7717) | 0.49 (0.25 to 0.75) |
FIGURE 9.
Cost-effectiveness plane for genotype 1 patients – incremental cost and incremental quality-adjusted life-years for shortened treatment duration using peginterferon alfa-2b and ribavirin combination therapy (24 vs 48 weeks of treatment) – efficacy from Berg and colleagues. 59
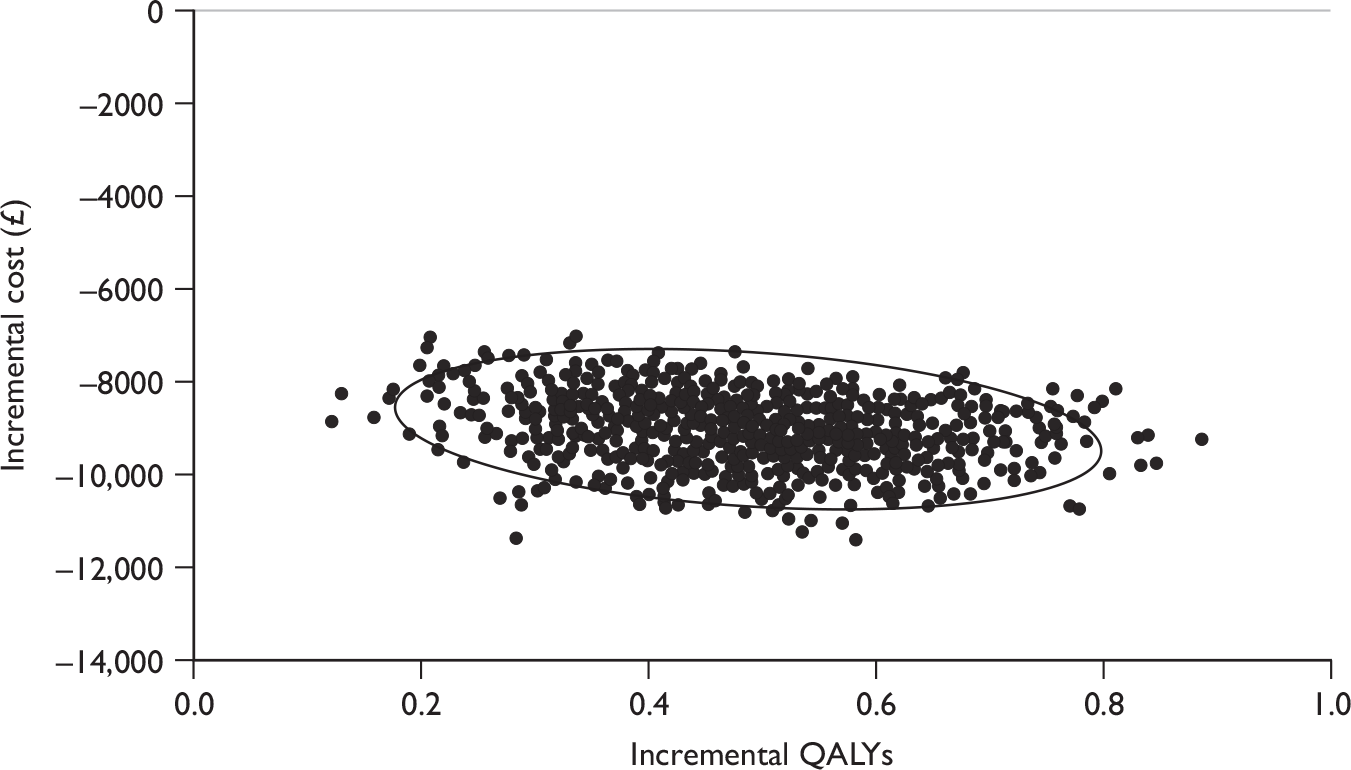
In this analysis of shortened duration of treatment using peginterferon alfa-2b and ribavirin combination therapy for genotype 1 patients, all simulations were in the south-east quadrant of the cost-effectiveness map, where the comparator [in this case standard duration (48 weeks) of treatment] is dominated.
Re-treated patients
Baseline characteristics (starting age and distribution of patients across stages of chronic liver disease) for re-treated patients in the model are based on those reported for existing patients in the clinical audit at St Mary’s Hospital, London,102 as this group of patients is expected to be older and will probably have more advanced liver disease than would be the case for treatment-naive groups.
Peginterferon alfa-2a
Sustained virological responses for this patient population are taken from the RCT reported by Jensen and colleagues,88 which compared re-treatment with varying doses and duration of peginterferon alfa-2a in patients who had previously failed to respond to, or relapsed on, peginterferon or non-peginterferon alfa plus ribavirin. This trial was not included in our systematic review of clinical effectiveness, which specified, in line with the scope issued by NICE, that the comparator in trials of re-treated patients should be BSC (i.e. excluding active treatment with interferon alfa). For this analysis, in the absence of any relevant trial data, we assumed that the SVR for the cohort of re-treated patients receiving BSC would be zero. The assumed treatment duration for genotype 1 patients is 72 weeks, based on the SPC for peginterferon alfa-2a. 42 For genotype non-1 patients the treatment duration is 48 weeks (see Appendix 8).
Costs and outcomes modelled for re-treatment in patients previously treated with peginterferon alfa-2a and ribavirin combination therapy are presented in Table 54. This table reports total costs (antiviral treatment and supportive care), health outcomes (in terms of life-years and QALYs) and the incremental cost-per-QALYs ratios.
| Re-treatment | Cost (£) | Outcome (life-years) | Outcome (QALYs) | ICER (£/QALY gained) |
|---|---|---|---|---|
| Genotype 1 | ||||
| BSC | 26,221 | 16.75 | 10.74 | |
| PEG α-2a | 42,350 | 17.07 | 11.05 | |
| Incremental | 16,130 | 0.33 | 0.31 | 52,587 |
| Genotype non-1 | ||||
| BSC | 26,221 | 16.75 | 10.74 | |
| PEG α-2a | 32,640 | 17.28 | 11.33 | |
| Incremental | 6419 | 0.54 | 0.59 | 10,926 |
The impact of re-treating this group of patients is to improve the predicted outcome (by 0.31 and 0.59 QALYs for genotype 1 and genotype non-1, respectively) and to increase lifetime costs (by £16,130 and £6419 QALYs for genotype 1 and genotype non-1, respectively). The reduction in supportive care costs associated with disease progression in both groups of patients (genotype 1 and genotype non-1) is insufficient to fully offset the additional costs of antiviral treatment.
The cost-effectiveness results in Table 54 do not take account of patients withdrawing from treatment owing to adverse events, nor do they consider the impact of treatment stopping rules (e.g. ceasing treatment at 12 weeks in patients who do not demonstrate an EVR). Table 55 reports cost-effectiveness results for re-treated patients, allowing for patient withdrawals owing to adverse effects of treatment with peginterferon alfa and ribavirin combination therapy. This has a marginal impact on the cost-effectiveness results, with the ICER for patients with genotype 1 remaining high.
| Re-treatment | Cost (£) | Outcome (life-years) | Outcome (QALYs) | ICER (£/QALY gained) |
|---|---|---|---|---|
| Genotype 1 | ||||
| BSC | 26,221 | 16.75 | 10.74 | |
| PEG α-2a | 41,900 | 17.07 | 11.05 | |
| Incremental | 15,680 | 0.33 | 0.31 | 50,730 |
| Genotype non-1 | ||||
| BSC | 26,221 | 16.75 | 10.74 | |
| PEG α-2a | 32,488 | 17.28 | 11.33 | |
| Incremental | 6267 | 0.54 | 0.59 | 10,650 |
Table 56 reports cost-effectiveness results for re-treated patients, allowing for the adoption of early stopping rules whereby patients who do not demonstrate an EVR stop treatment at 12 weeks. This has a substantial impact on the cost-effectiveness results, reducing the increase in total costs for patients treated with peginterferon alfa and ribavirin combination therapy to between £1415 and £3398, depending on genotype grouping, while also increasing the QALY gain by approximately 0.06 QALYs. As a result the ICER for patients with genotype 1 falls to £9169.
The EVRs used in the analysis reported in Table 56 are taken from the Roche submission to NICE,104 as Jensen and colleagues88 do not report EVR separately for the genotype groupings used in this analysis. The interpretation of the data available in the MS is difficult, as the number of patients achieving SVR is not reported according to whether patients demonstrated an EVR, for each treatment arm. Rather, the submission reports only predictive values for patients achieving full viral suppression at week 12. The analysis in Table 56 assumes that all patients who achieve an SVR demonstrated an EVR.
| Re-treatment | Cost (£) | Outcome (life-years) | Outcome (QALYs) | ICER (£/QALY gained) |
|---|---|---|---|---|
| Genotype 1 | ||||
| BSC | 26,221 | 16.75 | 10.74 | |
| PEG α-2a | 29,619 | 17.07 | 11.11 | |
| Incremental | 3398 | 0.33 | 0.37 | 9169 |
| Genotype non-1 | ||||
| BSC | 26,221 | 16.75 | 10.74 | |
| PEG α-2a | 27,636 | 17.28 | 11.36 | |
| Incremental | 1415 | 0.54 | 0.62 | 2294 |
Deterministic sensitivity analysis
Table 57 reports the results of a DSA for re-treatment using peginterferon alfa-2a and ribavirin combination therapy in previously treated patients. These are predominantly univariate sensitivity analyses, varying one parameter at a time from its base-case value, leaving all other variables unchanged.
| Genotype 1 | Genotype non-1 | |||||
|---|---|---|---|---|---|---|
| Incremental cost (£) | Incremental QALY | ICER | Incremental cost (£) | Incremental QALY | ICER | |
| Base case | 3398 | 0.37 | 9169 | 1415 | 0.62 | 2294 |
| Structural uncertainty | ||||||
| Spontaneous SVR from mild (0.002) | 3460 | 0.36 | 9685 | 1516 | 0.60 | 2547 |
| Spontaneous SVR from mild (0.010) | 3431 | 0.36 | 9442 | 1469 | 0.61 | 2428 |
| Discount cost and outcome at 0% | 945 | 0.84 | 1121 | –2588 | 1.39 | –1864 |
| Discount cost at 6%, outcome at 1.5% | 4270 | 0.58 | 7355 | 2838 | 0.96 | 2957 |
| Baseline cohort characteristics | ||||||
| Cohort 80% male | 3414 | 0.37 | 9306 | 1441 | 0.61 | 2360 |
| Cohort 40% male | 3348 | 0.38 | 8754 | 1334 | 0.64 | 2097 |
| Change average age of cohort at start of simulation (base case 40 years old) | ||||||
| –10 years | 3052 | 0.47 | 6500 | 851 | 0.78 | 1093 |
| +5 years | 3624 | 0.32 | 11,401 | 1784 | 0.53 | 3361 |
| +10 years | 3887 | 0.26 | 14,685 | 2213 | 0.44 | 4983 |
| +15 years | 4188 | 0.21 | 19,740 | 2705 | 0.36 | 7547 |
| Change distribution of cohort across disease stages at start of simulation | ||||||
| Cohort 100% mild chronic HCV | 5325 | 0.23 | 23,560 | 4560 | 0.38 | 11,970 |
| Cohort 100% moderate HCV | 3152 | 0.37 | 8508 | 1014 | 0.62 | 1644 |
| Cohort 100% CC | 1680 | 0.52 | 3232 | –1389 | 0.86 | –1614 |
| Parameter uncertainty | ||||||
| Assume SVR is 25% lower in patients with CC | 3784 | 0.33 | 11,573 | 2045 | 0.55 | 3747 |
| Assume SVR is 50% lower in patients with CC | 4170 | 0.28 | 14,720 | 2675 | 0.47 | 5638 |
| Cohort 100% CC, assume SVR is 25% lower in patients with CC | 2886 | 0.38 | 7526 | 579 | 0.64 | 907 |
| Cohort 100% CC, assume SVR is 50% lower in patients with CC | 4091 | 0.25 | 16,570 | 2546 | 0.42 | 6135 |
| Transition probability from mild-to-moderate disease = 0.04 | 3288 | 0.39 | 8462 | 1236 | 0.65 | 1912 |
| Transition probability from moderate disease to CC = 0.073 | 3126 | 0.43 | 7313 | 971 | 0.71 | 1368 |
| Cost of SVR state = £0 | 3360 | 0.37 | 9066 | 1353 | 0.62 | 2193 |
| Reduce cost of PEG α-2a by 20% | 2868 | 0.37 | 7739 | 796 | 0.62 | 1290 |
| Reduce cost of PEG α-2a by 30% | 2603 | 0.37 | 7024 | 486 | 0.62 | 787 |
| Reduce cost of RBV by 20% | 2316 | 0.37 | 6248 | 1054 | 0.62 | 1708 |
| Reduce cost of RBV by 20% | 2161 | 0.37 | 5831 | 873 | 0.62 | 1415 |
The DSA suggests that the results are robust to a change in structural assumptions (allowing spontaneous SVR from the mild chronic HCV state), the proportion of the baseline cohort that is male and variation in early disease transition probabilities. Reducing drug acquisition costs has the effect of improving the cost-effectiveness of re-treatment, as would be expected, by reducing incremental costs while leaving incremental outcome unchanged.
The results are highly sensitive to two assumptions regarding baseline cohort characteristics. Increasing age at entry to the model is associated with a substantial increase in the ICER – the ICER value approximately doubles if age at entry is increased by 15 years from the base case. This arises as the QALY gain from re-treatment is reduced by approximately 43%, while incremental cost increases by 23%, for genotype 1 patients. The results also appear to be sensitive to the distribution of patients across liver disease stages, at entry to the model. Higher QALY gains are associated with more advanced disease stage, with lower incremental costs; however, in this analysis we have assumed the same SVR in patients with and without cirrhosis. Subsequent analyses suggest that the ICER is also sensitive to variation in the SVR applied for patients with cirrhosis at baseline.
Probabilistic sensitivity analysis
In a PSA where the probabilities of achieving EVR and SVR, health-state costs, health-state utility values and transition probabilities for the natural history parameters were sampled probabilistically, re-treatment using peginterferon alfa-2a and ribavirin combination therapy is associated with increased QALYs (with a range from 0.05 to 0.59 QALYs for genotype 1 patients, and from 0.05 to 2.94 QALYs for genotype non-1 patients), but for genotype 1 patients is typically also associated with increased costs when compared with BSC. Table 58 provides summary information and Figures 10 and 11 scatter plots that also show the 95% confidence ellipses. The incremental cost was negative in approximately 25% of simulations for genotype non-1 patients.
| Re-treatment | Lifetime costs (£) | QALYs |
|---|---|---|
| Genotype 1 | ||
| BSC | 26,183 (17,678 to 35,971) | 10.79 (9.89 to 11.73) |
| PEG α-2a | 29,552 (22,032 to 38,284) | 11.15 (10.27 to 12.02) |
| Incremental | 3369 (1573 to 4509) | 0.37 (0.13 to 0.67) |
| Genotype non-1 | ||
| BSC | 26,005 (17,302 to 36,253) | 10.81 (9.91 to 11.74) |
| PEG α-2a | 27,186 (19,864 to 36,507) | 11.44 (10.41 to 12.51) |
| Incremental | 1,181 (–4127 to 4030) | 0.63 (0.14 to 1.49) |
FIGURE 10.
Cost-effectiveness plane for genotype 1 – incremental cost and incremental QALYs for re-treatment using peginterferon alfa-2a and ribavirin combination therapy (applying early stopping rule based on early virological response).
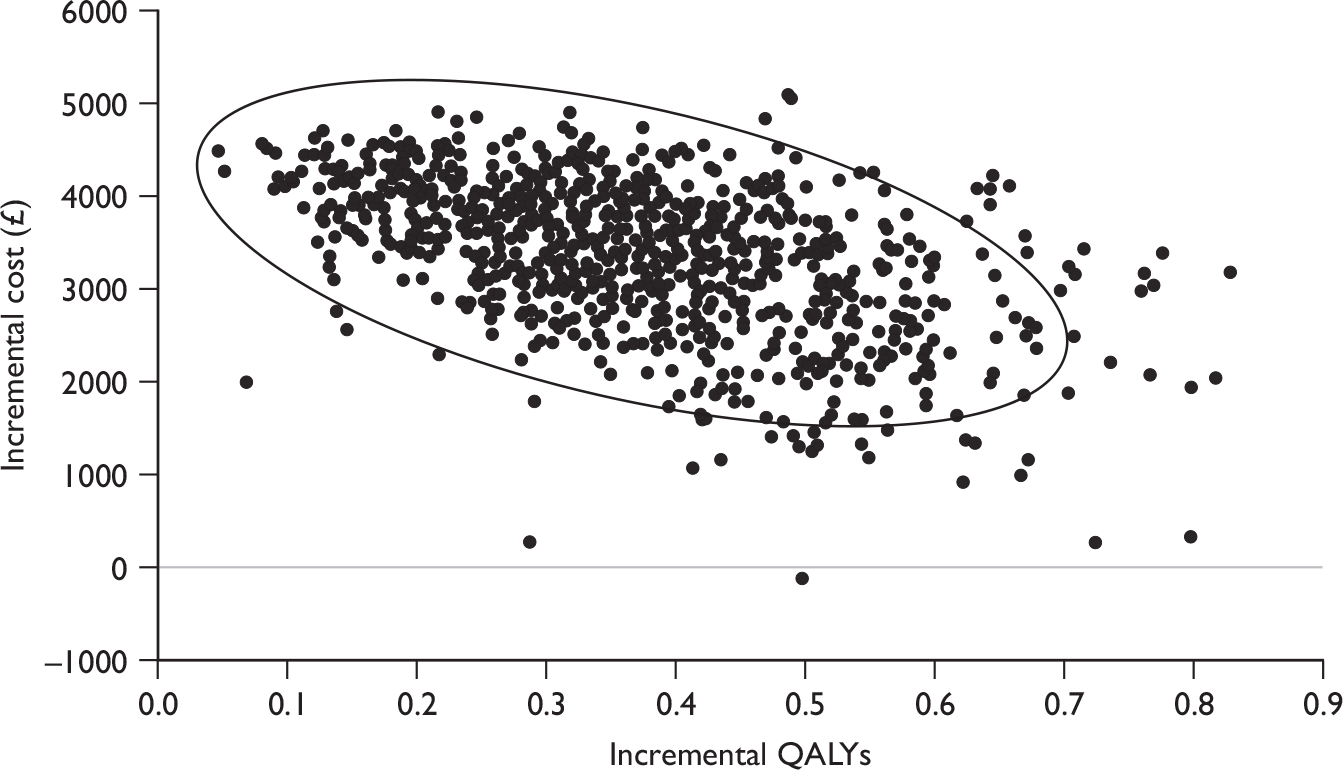
FIGURE 11.
Cost-effectiveness plane for genotype non-1 – incremental cost and incremental QALYs for re-treatment using peginterferon alfa-2a and ribavirin combination therapy (applying early stopping rule based on early virological response).
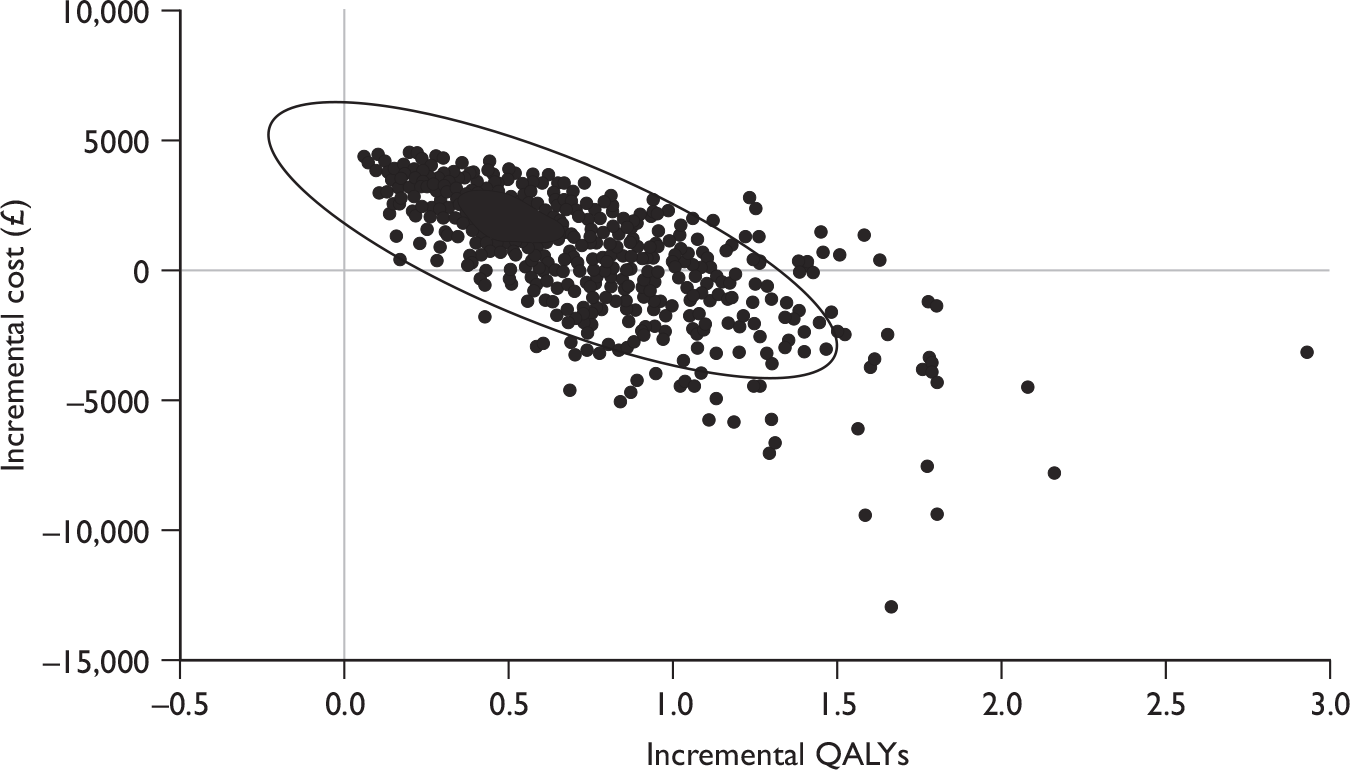
In this analysis, re-treatment using peginterferon alfa and ribavirin combination therapy for genotype 1 patients has a probability of being cost-effective (compared with BSC) of 90% at a willingness-to-pay threshold of £20,000 per QALY and 98% at a willingness-to-pay threshold of £30,000 if a stopping rule based on EVR is adopted. If patients are treated for the full 72 weeks, regardless of EVR, the equivalent figures are 2% and 11% (Figure 12). For genotype non-1 patients, the probability of re-treatment using peginterferon alfa plus ribavirin being cost-effective (compared with BSC) was 96% at a willingness-to-pay threshold of £20,000 per QALY and 98% at a willingness-to-pay threshold of £30,000, when adopting the stopping rule based on EVR (Figure 13).
FIGURE 12.
Cost-effectiveness acceptability curves for re-treatment of genotype 1 patients with peginterferon alfa-2a, with and without stopping rules based on EVR.
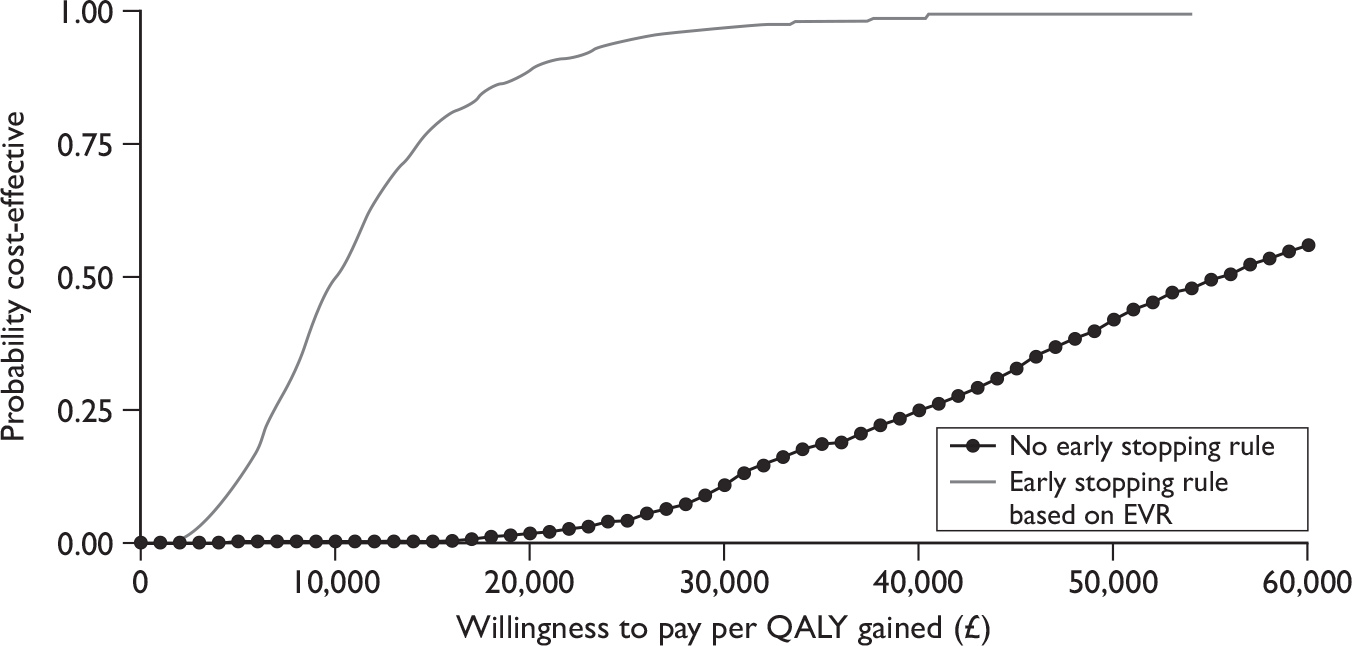
FIGURE 13.
Cost-effectiveness acceptability curves for re-treatment of genotype non-1 patients with peginterferon alfa-2a, with and without stopping rules based on EVR.
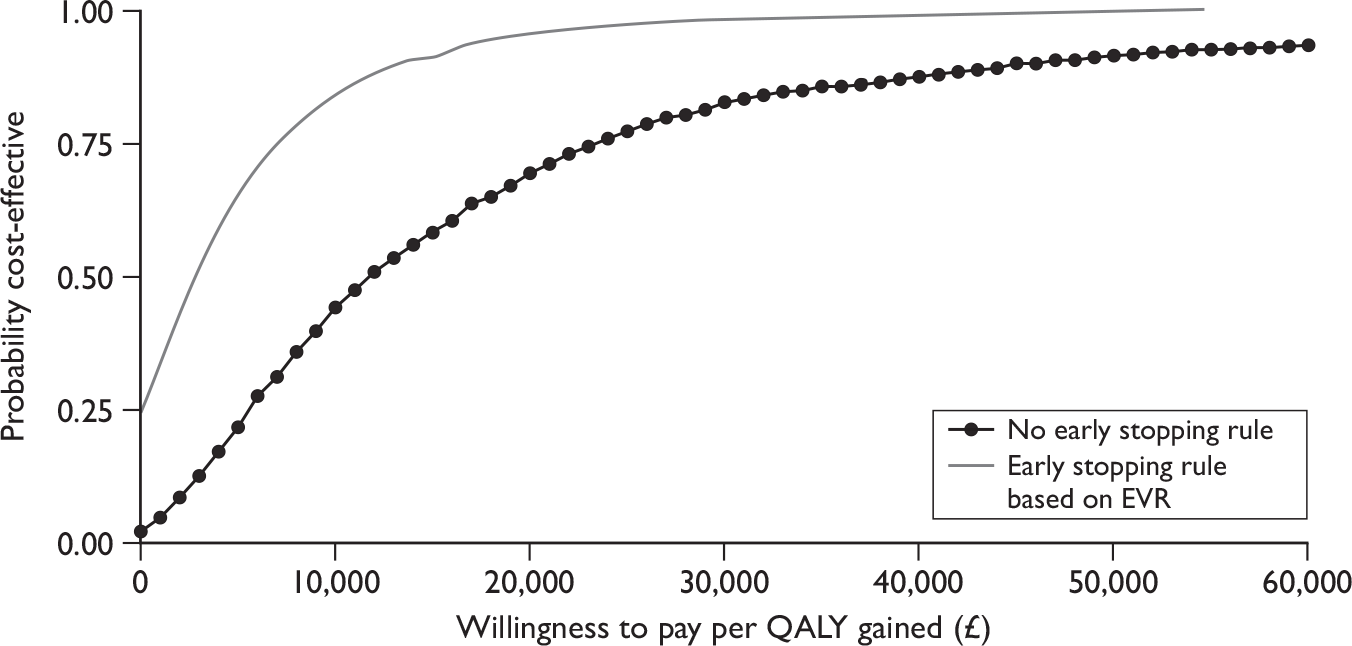
Peginterferon alfa-2b
Sustained virological responses for this patient population are taken from the MS by Schering-Plough, which reported treatment outcomes for the EPIC3 study96 (a multicentre, non-randomised open-label uncontrolled study). This study did not meet the inclusion criteria for our systematic review of clinical effectiveness (see Appendix 8 for an explanation of the choice of clinical evidence in this patient group). The assumed treatment duration for all patients is 48 weeks, and the SVR for the cohort of patients receiving BSC is assumed to be zero.
Costs and outcomes modelled for re-treatment in patients previously treated with peginterferon alfa-2b and ribavirin combination therapy are presented in Table 59. This table reports total costs (antiviral treatment and supportive care), health outcomes (in terms of life-years and QALYs) and the incremental cost-per-QALY ratios.
| Re-treatment | Cost (£) | Outcome (life-years) | Outcome (QALYs) | ICER (£/QALY gained) |
|---|---|---|---|---|
| Genotypes 1 + 4 | ||||
| BSC | 26,221 | 16.75 | 10.74 | |
| PEG α-2b | 35,601 | 17.12 | 11.14 | |
| Incremental | 9380 | 0.37 | 0.39 | 23,912 |
| Genotypes 2 + 3 | ||||
| BSC | 26,221 | 16.75 | 10.74 | |
| PEG α-2b | 25,232 | 18.21 | 12.46 | |
| Incremental | –989 | 1.47 | 1.72 | PEG α-2b dominates |
The impact of re-treating this group of patients is to improve the predicted outcome (by 0.39 and 1.72 QALYs for genotypes 1 and 4 and genotypes 2 and 3, respectively) and to increase lifetime costs (by £9380 for genotypes 1 and 4). The reduction in supportive care costs associated with disease progression in genotype 2 + 3 patients, associated with re-treatment with peginterferon alfa-2b and ribavirin combination therapy, is sufficient to fully offset the additional costs of antiviral treatment. This is due to the high SVR reported for genotype 2 + 3 patients (58.3% overall and 56.8% in those demonstrating an EVR) reported for the EPIC3 study,96 in the MS.
The cost-effectiveness results in Table 59 do not take account of patients withdrawing from treatment owing to adverse events or consider the impact of treatment stopping rules (e.g. ceasing treatment at 12 weeks in patients who do not demonstrate an EVR). Table 60 reports cost-effectiveness results for re-treated patients, allowing for patient withdrawals owing to adverse effects of treatment with peginterferon alfa-2b and ribavirin combination therapy; this has a marginal impact on the cost-effectiveness results.
| Re-treatment | Cost (£) | Outcome (life-years) | Outcome (QALYs) | ICER (£/QALY gained) |
|---|---|---|---|---|
| Genotypes 1 + 4 | ||||
| BSC | 26,221 | 16.75 | 10.74 | |
| PEG α-2b | 35,417 | 17.12 | 11.14 | |
| Incremental | 9197 | 0.37 | 0.39 | 23,384 |
| Genotypes 2 + 3 | ||||
| BSC | 26,221 | 16.75 | 10.74 | |
| PEG α-2b | 25,048 | 18.21 | 12.46 | |
| Incremental | –1173 | 1.47 | 1.72 | PEG α-2b dominates |
Table 61 reports cost-effectiveness results for re-treated patients, allowing for the adoption of early stopping rules whereby patients who do not demonstrate an EVR stop treatment at 12 weeks. This has a substantial impact on the cost-effectiveness results, reducing the increase in total costs for genotype 1 and 4 patients treated with peginterferon alfa-2b and ribavirin combination therapy to £3256. As a result the ICER for patients with genotype 1 falls to £7681.
| Re-treatment | Cost (£) | Outcome (life-years) | Outcome (QALYs) | ICER (£/QALY gained) |
|---|---|---|---|---|
| Genotypes 1 + 4 | ||||
| BSC | 26,221 | 16.75 | 10.74 | |
| PEG α-2b | 29,476 | 17.12 | 11.17 | |
| Incremental | 3256 | 0.37 | 0.42 | 7681 |
| Genotypes 2 + 3 | ||||
| BSC | 26,221 | 16.75 | 10.74 | |
| PEG α-2b | 23,371 | 18.21 | 12.47 | |
| Incremental | –2850 | 1.47 | 1.73 | PEG α-2b dominates |
Deterministic sensitivity analysis
Table 62 reports the results of a DSA for re-treatment using peginterferon alfa-2b and ribavirin combination therapy in previously treated patients. These are predominantly univariate sensitivity analyses – that is, varying one parameter at a time, from its base-case value, leaving all of the other variables unchanged. The DSA suggests that the results are robust to a change in structural assumptions (allowing spontaneous SVR from the mild chronic HCV state), the proportion of the baseline cohort that is male and transition probabilities for early disease states. Reducing drug acquisition costs has the effect of reducing the ICER, as might be expected, as it reduces the drug costs while the outcome difference is unchanged.
| Genotypes 1 + 4 | Genotypes 2 + 3 | |||||
|---|---|---|---|---|---|---|
| Incremental cost (£) | Incremental QALY | ICER | Incremental cost (£) | Incremental QALY | ICER | |
| Base case | 3256 | 0.42 | 7681 | –2850 | 1.73 | –1650 |
| Structural uncertainty | ||||||
| Spontaneous SVR from mild (0.002) | 3326 | 0.41 | 8,139 | –2575 | 1.67 | –1545 |
| Spontaneous SVR from mild (0.010) | 3294 | 0.42 | 7923 | –2702 | 1.69 | –1594 |
| Discount cost and outcome at 0% | 460 | 0.96 | 477 | –13,840 | 3.84 | –3600 |
| Discount cost at 6%, outcome at 1.5% | 4250 | 0.66 | 6408 | 1055 | 2.67 | 396 |
| Baseline cohort characteristics | ||||||
| Cohort 80% male | 3274 | 0.42 | 7802 | –2778 | 1.71 | –1624 |
| Cohort 40% male | 3199 | 0.44 | 7313 | –3073 | 1.78 | –1726 |
| Change average age of cohort at start of simulation (base case 40 years old) | ||||||
| –10 years | 2862 | 0.54 | 5331 | –4399 | 2.17 | –2027 |
| +5 years | 3514 | 0.36 | 9658 | –1837 | 1.49 | –1232 |
| +10 years | 3813 | 0.30 | 12,579 | –660 | 1.25 | –527 |
| +15 years | 4156 | 0.24 | 17,087 | 690 | 1.02 | 678 |
| Change distribution of cohort across disease stages at start of simulation | ||||||
| Cohort 100% mild chronic HCV | 5453 | 0.26 | 21,048 | 5783 | 1.08 | 5359 |
| Cohort 100% moderate HCV | 2976 | 0.42 | 7022 | –3951 | 1.73 | –2289 |
| Cohort 100% CC | 1297 | 0.59 | 2184 | –10,548 | 2.40 | –4402 |
| Parameter uncertainty | ||||||
| Assume SVR is 25% lower in patients with CC | 3696 | 0.37 | 9878 | –1121 | 1.53 | –732 |
| Assume SVR is 50% lower in patients with CC | 4136 | 0.32 | 12,751 | 607 | 1.34 | 455 |
| Cohort 100% CC, assume SVR is 25% lower in patients with CC | 2672 | 0.44 | 6093 | –5146 | 1.78 | –2884 |
| Cohort 100% CC, assume SVR is 50% lower in patients with CC | 4046 | 0.28 | 14,304 | 255 | 1.17 | 218 |
| Transition probability from mild-to-moderate disease = 0.04 | 3131 | 0.44 | 7044 | –3342 | 1.81 | –1849 |
| Transition probability from moderate disease to CC = 0.073 | 2946 | 0.49 | 6028 | –4069 | 1.98 | –2053 |
| Cost of SVR state = £0 | 3213 | 0.42 | 7578 | –3020 | 1.73 | –1749 |
| Reduce cost of PEG α-2b by 20% | 2518 | 0.42 | 5940 | –4161 | 1.73 | –2409 |
| Reduce cost of PEG α-2b by 30% | 2149 | 0.42 | 5069 | –4816 | 1.73 | –2789 |
| Reduce cost of RBV by 20% | 2946 | 0.42 | 6950 | –3400 | 1.73 | –1969 |
| Reduce cost of RBV by 20% | 2791 | 0.42 | 6584 | –3675 | 1.73 | –2128 |
The greatest variability in ICERs is associated with changes in the age at which patients enter the model, the distribution of patients across disease stages and (to a lesser extent) response to treatment (SVR) for patients with cirrhosis. Increasing the mean age of patients at the start of the simulation up to 15 years leads to an approximate doubling of the ICER for genotype 1 and 4 patients and results in a positive, though low-value, ICER for genotype 2 and 3 patients. In both cases, the QALY gain with treatment is approximately halved. Similarly, alternative assumptions regarding the stage of liver disease in which patients enter the model has a large impact on the ICER, with less favourable results associated with patients being in the earlier (lower fibrosis) stages of disease. For genotype 2 and 3 patients the ICER becomes positive if all patients in the modelled cohorts have mild chronic HCV (rather than moderate chronic HCV or compensated cirrhosis).
Probabilistic sensitivity analysis
In a PSA, where the probabilities of achieving EVR and SVR, health-state costs, health-state utility values, and transition probabilities for the natural history parameters were sampled probabilistically, re-treatment using peginterferon alfa and ribavirin combination therapy is associated with increased QALYs (with a range from 0.08 to 0.80 QALYs for genotypes 1 and 4 and from 0.28 to 3.06 QALYs for patients with genotypes 2 and 3), but for genotype 1 and 4 patients is typically also associated with increased costs when compared with BSC (Table 63 provides summary information and Figures 14 and 15 scatter plots, which also show the 95% confidence ellipses). The incremental cost was negative in approximately 84% of simulations for genotype 2 and 3 patients.
| Re-treatment | Lifetime costs (£) | QALYs |
|---|---|---|
| Genotypes 1 + 4 | ||
| BSC | 25,820 (17,909 to 35,424) | 10.78 (9.86 to 11.72) |
| PEG α-2b | 29,118 (22,213 to 37,485) | 11.20 (10.38 to 12.01) |
| Incremental | 3298 (1785 to 4480) | 0.42 (0.22 to 0.66) |
| Genotypes 2 + 3 | ||
| BSC | 25,914 (17,928 to 35,721) | 10.78 (9.89 to 11.69) |
| PEG α-2b | 23,250 (19,240 to 28,246) | 12.48 (11.65 to 13.32) |
| Incremental | –2664 (–8971 to 1846) | 1.69 (0.88 to 2.48) |
FIGURE 14.
Cost-effectiveness plane for genotypes 1 and 4 – incremental cost and incremental QALYs for re-treatment using peginterferon alfa-2b and ribavirin combination therapy (applying early stopping rule based on EVR).
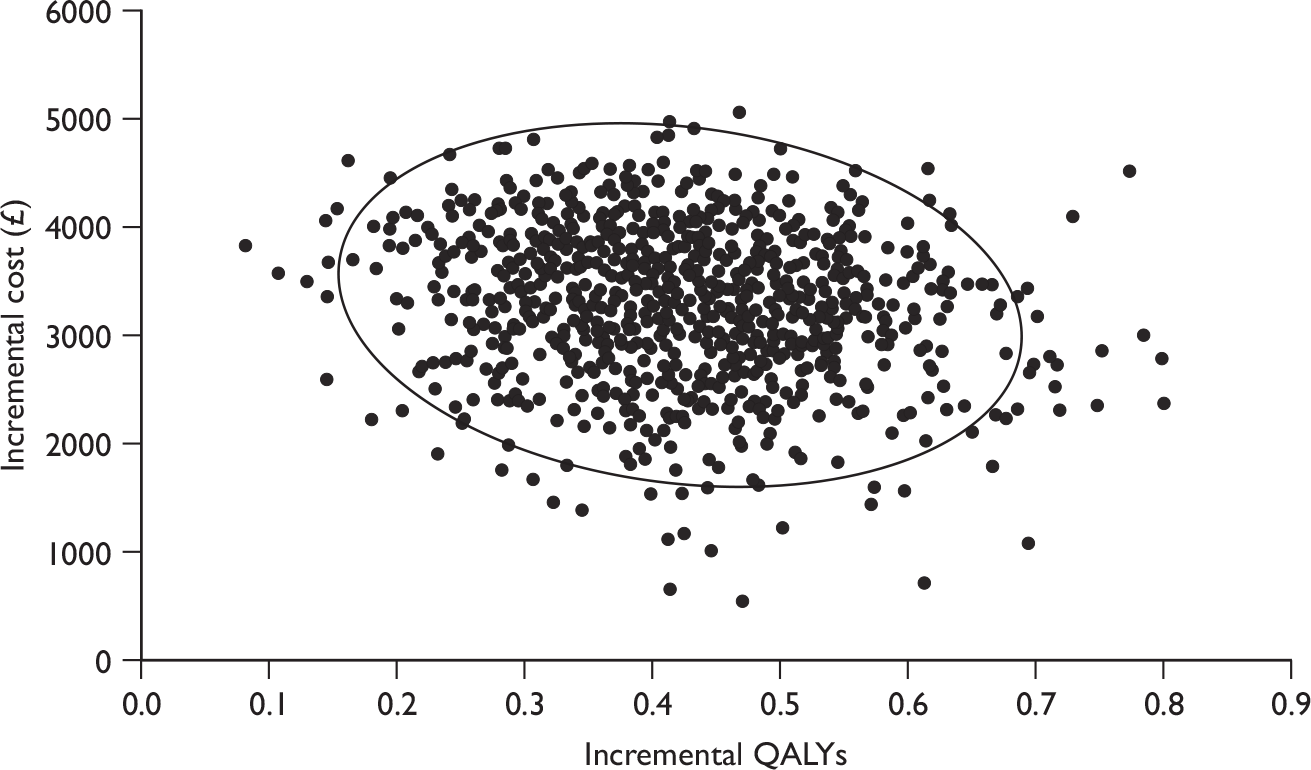
FIGURE 15.
Cost-effectiveness plane for genotypes 2 and 3 – incremental cost and incremental QALYs for re-treatment using peginterferon alfa-2b and ribavirin combination therapy (applying early stopping rule based on EVR).
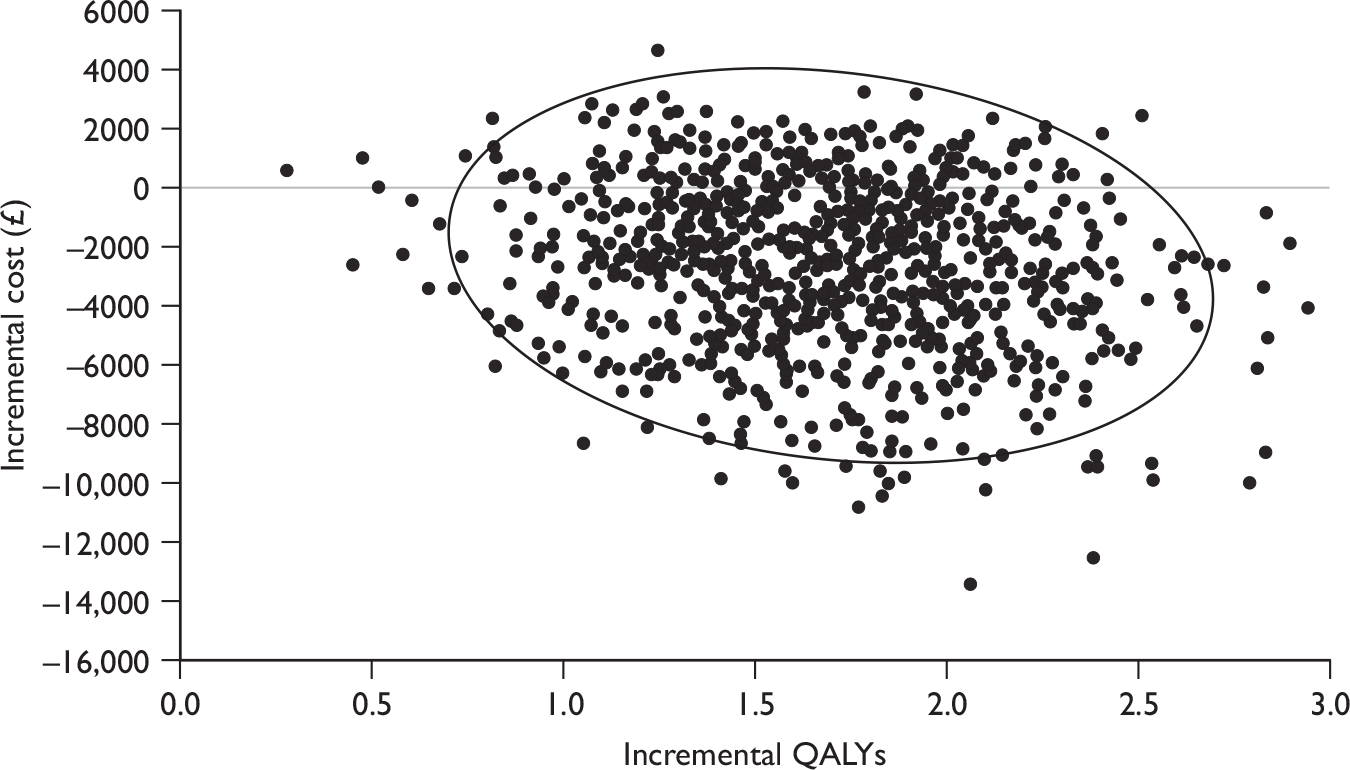
In this analysis, re-treatment using peginterferon alfa-2b and ribavirin combination therapy for genotype 1 and 4 patients had a probability of being cost-effective (compared with BSC) of 99% at a willingness-to-pay threshold of £20,000 per QALY, and 100% at a willingness-to-pay threshold of £30,000 if a stopping rule based on EVR is adopted. If patients are treated for the full 48 weeks, regardless of EVR, the equivalent figures are 24% and 74% (Figure 16). For genotype 2 and 3 patients the probability of re-treatment using peginterferon alfa-2b and ribavirin being cost-effective (compared with BSC) was 100% at a willingness-to-pay threshold of £20,000 per QALY, and at a willingness-to-pay threshold of £30,000 when adopting the stopping rule based on EVR (Figure 17).
FIGURE 16.
Cost-effectiveness acceptability curves for re-treatment of genotype 1 and 4 patients with peginterferon alfa-2b, with and without stopping rules based on EVR.
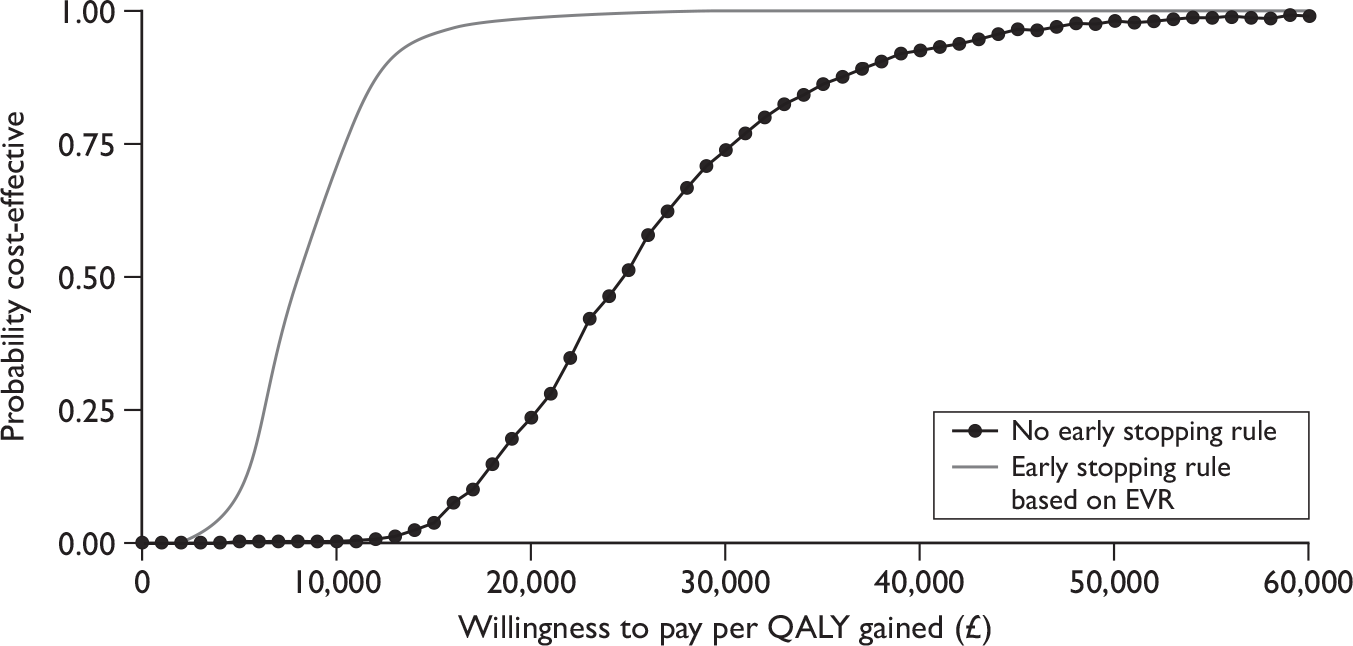
FIGURE 17.
Cost-effectiveness acceptability curves for re-treatment of genotype 2 and 3 patients with peginterferon alfa-2b, with and without stopping rules based on EVR.

HCV/HIV co-infected patients
No data reporting the distribution of treatment-eligible HCV/HIV co-infected patients across liver disease stages were identified in our searches. The distribution of HCV/HIV co-infected patients across stages of chronic liver disease, at entry to the model, is based on that reported for new mono-infected patients in the clinical audit at St Mary’s Hospital. 102 SVRs for this patient population are based on those reported in two recent systematic reviews of antiviral treatment with peginterferon alfa in HCV/HIV co-infected patients, which included trials with active treatment comparators50,51 (see Appendix 9). The systematic review of clinical effectiveness in this report (see Chapter 4) specified, in line with the scope issued by NICE, that the comparator in trials of HCV/HIV co-infected patients should be BSC (excluding active treatment with interferon alfa). For this analysis, in the absence of any relevant trial data, we assumed that the SVR for the cohort of re-treated patients receiving BSC would be zero.
The tables in this section report lifetime costs (antiviral treatment and BSC), health outcomes (in terms of life-years and QALYs) and the incremental cost-per-QALY ratios. The assumed treatment duration for all patients in the base case is 48 weeks, regardless of genotype. This is in accordance with the SPC for peginterferon alfa-2a42 and for peginterferon alfa-2b. 42
Peginterferon alfa-2a
Costs and outcomes modelled for patients co-infected with HCV/HIV receiving combination therapy with peginterferon alfa-2a plus ribavirin are presented in Table 64.
| Genotype | Treatment | Cost (£) | Outcome (life-years) | Outcome (QALYs) | ICER (£/QALY gained) |
|---|---|---|---|---|---|
| Genotypes 1 + 4 | BSC | 22,201 | 18.93 | 12.65 | |
| PEG α-2a | 28,133 | 19.43 | 13.40 | ||
| Incremental | 5932 | 0.51 | 0.75 | 7941 | |
| Genotypes 2 + 3 | BSC | 22,201 | 18.93 | 12.65 | |
| PEG α-2a | 20,484 | 20.13 | 14.51 | ||
| Incremental | –1717 | 1.20 | 1.86 | PEG α-2a dominates |
The impact of treating this group is to improve the predicted outcome (by 0.75 and 1.86 QALYs for genotypes 1 and 4 and genotypes 2 and 3, respectively) and to increase lifetime costs for patients with genotypes 1 and 4 (by £5932). However, in patients with genotypes 2 and 3 the modelled reduction in supportive care costs (in the peginterferon-treated cohort) offsets the additional costs of antiviral treatment; in this situation the strategy of providing antiviral treatment dominates.
The cost-effectiveness results in Table 64 do not take account of uncertainties regarding the potential impact of HIV co-infection on the natural history of HCV infection, overall mortality, utility gains from successful treatment or additional costs of on-treatment monitoring.
A published meta-analysis25 suggests that a RR for cirrhosis of 2.07 (95% CI 1.4 to 3.07) and a RR for decompensation of 6.14 (95% CI 2.86 to 13.20) in HCV/HIV co-infected patients compared with that in HCV mono-infected patients. Table 65 reports the cost-effectiveness results from the model when these RRs for liver disease progression are applied to the baseline risks in the natural history model. This suggests that treatment using peginterferon alfa-2a and ribavirin combination therapy will be more cost-effective in HCV/HIV co-infected patients, if the risks of fibrosis progression are greater than for mono-infected patients.
| Genotype | Treatment | Cost (£) | Outcome (life-years) | Outcome (QALYs) | ICER (£/QALY gained) |
|---|---|---|---|---|---|
| Genotypes 1 + 4 | BSC | 31,839 | 16.91 | 10.90 | |
| PEG α-2a | 35,254 | 17.94 | 12.10 | ||
| Incremental | 3415 | 1.03 | 1.21 | 2833 | |
| Genotypes 2 + 3 | BSC | 31,839 | 16.91 | 10.90 | |
| PEG α-2a | 24,137 | 19.37 | 13.84 | ||
| Incremental | –7703 | 2.46 | 2.95 | PEG α-2a dominates |
Table 66 reports the cost-effectiveness results from the model when the age-specific mortality risks are doubled for HCV/HIV co-infected patients. This would result in an age-specific life expectancy of 33.2 years at the age of 40 years for an HIV infected person (in the absence of chronic liver disease) compared with 39.8 years if the age-specific mortality risks for the general population are applied (as in the base-case analysis). This reduces lifetime costs and QALYs both for peginterferon treated and BSC cohorts. This suggests that treatment using peginterferon alfa-2a and ribavirin combination therapy will be less cost-effective in HCV/HIV co-infected patients, if mortality risk is greater than for mono-infected patients. However, while the incremental cost for peginterferon treatment increases and the QALY gain is reduced, with higher mortality risk for HCV/HIV co-infected patients, treatment with peginterferon still dominates BSC for genotype 2 and 3 patients.
| Genotype | Treatment | Cost (£) | Outcome (life-years) | Outcome (QALYs) | ICER (£/QALY gained) |
|---|---|---|---|---|---|
| Genotypes 1 + 4 | BSC | 19,865 | 17.46 | 11.70 | |
| PEG α-2a | 26,398 | 17.84 | 12.31 | ||
| Incremental | 6534 | 0.38 | 0.61 | 10,704 | |
| Genotypes 2 + 3 | BSC | 19,865 | 17.46 | 11.70 | |
| PEG α-2a | 19,578 | 18.36 | 13.23 | ||
| Incremental | –287 | 0.91 | 1.53 | PEG α-2a dominates |
Tables 67 and 68 report cost-effectiveness results from alternative assumptions on the utility gain for HCV/HIV co-infected patients who achieve an SVR. In the first case, the utility gain is assumed to be one-half of that reported for HCV mono-infected patients, and in the second case the utility gain is assumed to be zero. In both cases the QALY gain from treatment with peginterferon is reduced, indicating that treatment using peginterferon alfa-2a and ribavirin combination therapy will be less cost-effective in HCV/HIV co-infected patients, if utility gain from SVR is lower in HCV/HIV co-infected patients than in mono-infected patients.
| Genotype | Treatment | Cost (£) | Outcome (life-years) | Outcome (QALYs) | ICER (£/QALY gained) |
|---|---|---|---|---|---|
| Genotypes 1 + 4 | BSC | 22,201 | 18.93 | 12.65 | |
| PEG α-2a | 28,133 | 19.43 | 13.25 | ||
| Incremental | 5932 | 0.51 | 0.60 | 9889 | |
| Genotypes 2 + 3 | BSC | 22,201 | 18.93 | 12.65 | |
| PEG α-2a | 20,484 | 20.13 | 14.16 | ||
| Incremental | –1717 | 1.20 | 1.51 | PEG α-2a dominates |
| Genotype | Treatment | Cost (£) | Outcome (life-years) | Outcome (QALYs) | ICER (£/QALY gained) |
|---|---|---|---|---|---|
| Genotypes 1 + 4 | BSC | 22,201 | 18.93 | 12.65 | |
| PEG α-2a | 28,133 | 19.43 | 13.10 | ||
| Incremental | 5932 | 0.51 | 0.45 | 13,103 | |
| Genotypes 2 + 3 | BSC | 22,201 | 18.93 | 12.65 | |
| PEG α-2a | 20,484 | 20.13 | 13.81 | ||
| Incremental | –1717 | 1.20 | 1.16 | PEG α-2a dominates |
A final scenario analysis was performed to consider the impact of on-treatment monitoring costs on the cost-effectiveness of antiviral treatment for HCV/HIV co-infected patients. Table 69 reports the cost-effectiveness results from the model if on-treatment costs for HCV/HIV co-infected patients are assumed to be double those for HCV mono-infected patients. As with the previous analyses, this assumption suggests that treatment using peginterferon alfa-2a and ribavirin combination therapy is less cost-effective than in the base-case analysis. However, treatment with peginterferon still dominates BSC for genotype 2 and 3 patients.
| Genotype | Treatment | Cost (£) | Outcome (life-years) | Outcome (QALYs) | ICER (£/QALY gained) |
|---|---|---|---|---|---|
| Genotypes 1 + 4 | BSC | 22,201 | 18.93 | 12.65 | |
| PEG α-2a | 29,184 | 19.43 | 13.40 | ||
| Incremental | 6983 | 0.51 | 0.75 | 9348 | |
| Genotypes 2 + 3 | BSC | 22,201 | 18.93 | 12.65 | |
| PEG α-2a | 21,535 | 20.13 | 14.51 | ||
| Incremental | –666 | 1.20 | 1.86 | PEG α-2a dominates |
Deterministic sensitivity analysis
Table 70 reports the results of a DSA for treatment of HCV/HIV co-infected patients using peginterferon alfa-2a and ribavirin combination therapy. These suggest that the results are robust to a change in structural assumptions (allowing spontaneous SVR from the mild chronic HCV state), the proportion of the baseline cohort that is male, transition probabilities for early disease states and cost of the SVR health state. Reducing drug acquisition costs has the effect of reducing the ICER, as might be expected, as it reduces the drug costs while the outcome difference is unchanged.
| Genotype 1 | Genotypes 2 + 3 | |||||
|---|---|---|---|---|---|---|
| Incremental cost (£) | Incremental QALY | ICER | Incremental cost (£) | Incremental QALY | ICER | |
| Base case | 5932 | 0.75 | 7941 | –1717 | 1.86 | –924 |
| Structural uncertainty | ||||||
| Spontaneous SVR from mild (0.002) | 6144 | 0.70 | 8765 | –1213 | 1.75 | –693 |
| Spontaneous SVR from mild (0.010) | 6047 | 0.72 | 8374 | –1445 | 1.80 | –803 |
| Discount cost and outcome at 0% | 206 | 1.88 | 109 | –15,331 | 4.56 | –3360 |
| Discount cost at 6%, outcome at 1.5% | 7745 | 1.24 | 6266 | 2593 | 3.02 | 858 |
| Baseline cohort characteristics | ||||||
| Cohort 80% male | 5961 | 0.74 | 8055 | –1648 | 1.84 | –895 |
| Cohort 40% male | 5842 | 0.77 | 7598 | –1933 | 1.91 | –1012 |
| Change average age of cohort at start of simulation (base case 40 years old) | ||||||
| –10 years | 5299 | 0.93 | 5722 | –3223 | 2.28 | –1411 |
| +5 years | 6338 | 0.65 | 9734 | –752 | 1.63 | –461 |
| +10 years | 6804 | 0.55 | 12,291 | 354 | 1.40 | 253 |
| +15 years | 7323 | 0.46 | 16,029 | 1588 | 1.17 | 1359 |
| Change distribution of cohort across disease stages at start of simulation | ||||||
| Cohort 100% mild chronic HCV | 8744 | 0.53 | 16,524 | 4969 | 1.34 | 3706 |
| Cohort 100% moderate HCV | 4064 | 0.87 | 4655 | –6159 | 2.16 | –2854 |
| Cohort 100% CC | 1217 | 1.19 | 1018 | –12,928 | 2.92 | –4423 |
| Parameter uncertainty | ||||||
| Assume SVR is 25% lower in patients with CC | 6189 | 0.72 | 8648 | –1107 | 1.78 | –620 |
| Assume SVR is 50% lower in patients with CC | 6446 | 0.68 | 9420 | –496 | 1.71 | –290 |
| Cohort 100% CC, assume SVR is 25% lower in patients with CC | 3784 | 0.88 | 4295 | –6825 | 2.18 | –3135 |
| Cohort 100% CC, assume SVR is 50% lower in patients with CC | 6351 | 0.57 | 11,194 | –721 | 1.43 | –504 |
| Transition probability from mild-to-moderate disease = 0.04 | 5581 | 0.81 | 6916 | –2552 | 2.00 | –1275 |
| Transition probability from moderate disease to CC = 0.073 | 5186 | 0.92 | 5642 | –3492 | 2.27 | –1540 |
| Cost of SVR state = £0 | 5854 | 0.75 | 7836 | –1904 | 1.86 | –1024 |
| Reduce cost of PEG α-2a by 20% | 4714 | 0.75 | 6310 | –2935 | 1.86 | –1579 |
| Reduce cost of PEG α-2a by 30% | 4105 | 0.75 | 5495 | –3545 | 1.86 | –1907 |
| Reduce cost of RBV by 20% | 5221 | 0.75 | 6989 | –2428 | 1.86 | –1306 |
| Reduce cost of RBV by 20% | 4866 | 0.75 | 6513 | –2784 | 1.86 | –1498 |
The greatest variability in ICERs is associated with changes in the age at which patients enter the model, the distribution of patients across disease stages (to a lesser extent) and response to treatment (SVR) for patients with cirrhosis. For genotype 2 and 3 patients the ICER becomes positive (positive incremental cost and positive incremental QALYs) for the scenarios where age at entry is increased by 10 years and where all treated patients have mild chronic HCV. These are the only scenarios (other than a change in discounting practice where costs are discounted at 6% and outcomes at 1.5%), where treatment for genotype 2 and 3 patients with HCV/HIV co-infection is not dominant.
Increasing the age at which patients enter the model by 15 years leads to an approximate doubling of the ICER for genotype 1 and 4 patients – the QALY gain with treatment is reduced by around one-third. Similarly, alternative assumptions regarding the stage of liver disease in which patients enter the model has a large impact on the ICER, with less favourable results associated with patients being in the earlier (lower fibrosis) stages of disease. Reducing response to treatment for patients with cirrhosis at baseline also leads to less favourable cost-effectiveness estimates.
Probabilistic sensitivity analysis
In a PSA, where the probabilities of achieving SVR, health-state costs, health-state utility values, and transition probabilities for the natural history parameters are sampled probabilistically, treatment of co-infected patients with genotypes 1 and 4 is associated with increased QALYs (with a range from 0.09 to 1.49 QALYs), but typically also increased costs (ranging from –£447 to £9022) when compared with BSC (Table 71 and Figure 18). While treatment for patients with genotypes 2 and 3 is also associated with increased QALYs (from 0.09 to 3.63 QALYs gained), in approximately 70% of simulations the incremental cost was negative (Figure 19).
| Genotype | Treatment | Lifetime costs (£) | QALYs |
|---|---|---|---|
| Genotypes 1 + 4 | BSC | 22,049 (15,040 to 30,554) | 12.68 (11.71 to 13.48) |
| PEG α-2a | 28,035 (22,764 to 34,429) | 13.42 (12.63 to 14.11) | |
| Incremental | 5986 (3332 to 7993) | 0.74 (0.33 to 1.15) | |
| Genotypes 2 + 3 | BSC | 22,031 (15,443 to 30,254) | 12.69 (11.81 to 13.53) |
| PEG α-2a | 20,456 (17,298 to 24,327) | 14.51 (13.62 to 15.41) | |
| Incremental | –1575 (–7275 to 2673) | 1.82 (0.91 to 2.81) |
FIGURE 18.
Cost-effectiveness plane for genotypes 1 and 4 – incremental cost and incremental QALYs for treatment of HCV/HIV co-infected patients with peginterferon alfa-2a and ribavirin combination therapy.
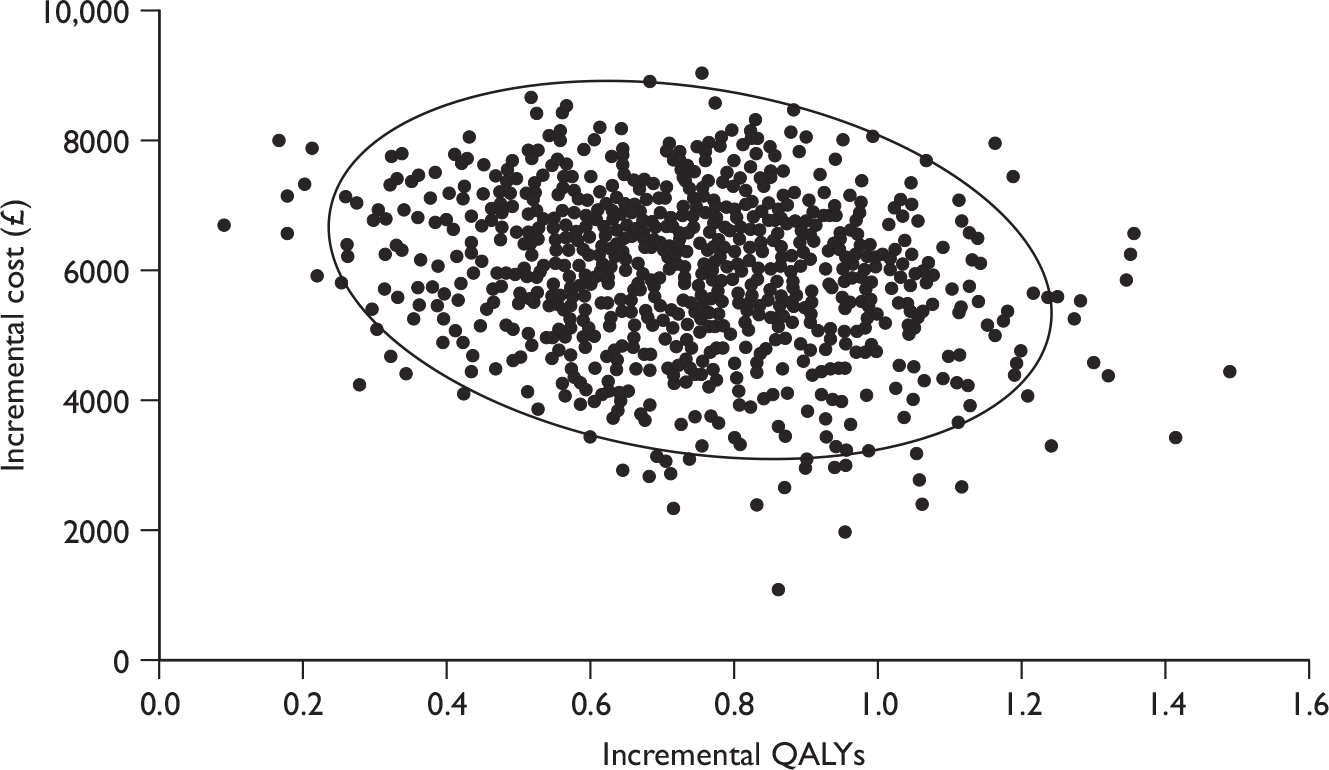
FIGURE 19.
Cost-effectiveness plane for genotypes 2 and 3 – incremental cost and incremental QALYs for treatment of HCV/HIV co-infected patients with peginterferon alfa-2a and ribavirin combination therapy.
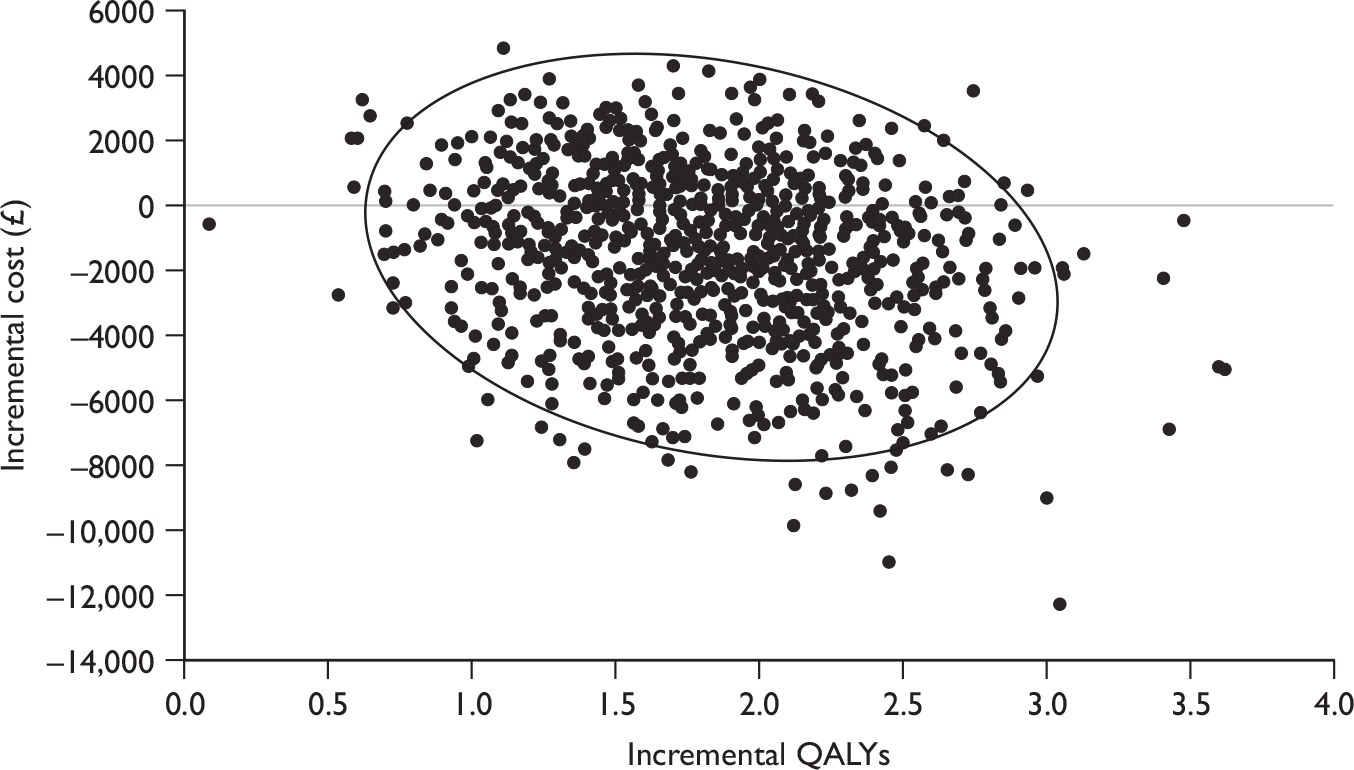
In this analysis, treatment using peginterferon alfa-2a and ribavirin combination therapy, for HCV/HIV co-infected patients with genotypes 1 and 4, had a probability of being cost-effective (compared with BSC) was 98% at a willingness-to-pay threshold of £20,000 per QALY, and 99% at a willingness-to-pay threshold of £30,000 per QALY (Figure 20). For patients with genotypes 2 and 3, treatment using peginterferon alfa-2a and ribavirin combination therapy had a probability of being cost-effective (compared with BSC) of 100% at a willingness-to-pay threshold of £20,000 per QALY.
FIGURE 20.
Cost-effectiveness acceptability curves for treatment of HCV/HIV co-infected patients with peginterferon alfa-2a and ribavirin combination therapy.
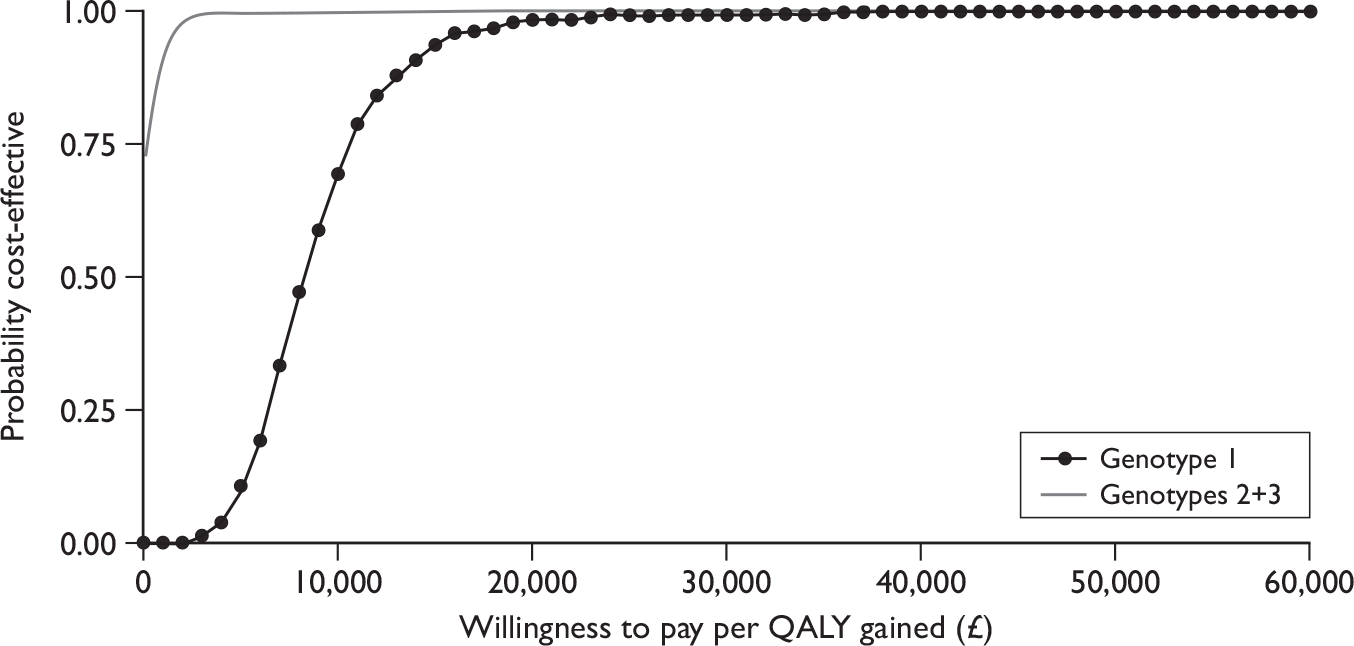
Peginterferon alfa-2b
Costs and outcomes modelled for patients co-infected with HCV/HIV receiving combination therapy with peginterferon alfa-2b and ribavirin are presented in Table 72.
| Genotype | Treatment | Cost (£) | Outcome (life-years) | Outcome (QALYs) | ICER (£/QALY gained) |
|---|---|---|---|---|---|
| Genotypes 1 + 4 | BSC | 22,201 | 18.93 | 12.65 | |
| PEG α-2b | 30,102 | 19.38 | 13.32 | ||
| Incremental | 7901 | 0.46 | 0.67 | 11,806 | |
| Genotypes 2 + 3 | BSC | 22,201 | 18.93 | 12.65 | |
| PEG α-2b | 25,190 | 19.83 | 14.03 | ||
| Incremental | 2989 | 0.91 | 1.38 | 2161 |
The impact of treating this group of patients is to improve the predicted outcome (by 0.67 and 1.38 QALYs for genotypes 1 and 4 and genotypes 2 and 3, respectively) and to increase lifetime costs (by £7901 and £2989 QALYs for genotypes 1 and 4 and genotypes 2 and 3, respectively). The reduction in supportive care costs associated with disease progression in both groups of patients (genotypes 1 and 4 and genotypes 2 and 3) is insufficient fully to offset the additional costs of antiviral treatment.
As described above, the cost-effectiveness results in Table 72 do not take account of uncertainties regarding the potential impact of HIV co-infection on the natural history of HCV infection, overall mortality, utility gains from successful treatment or additional costs of on-treatment monitoring. Table 73 reports the cost-effectiveness results from the model after applying the RRs for disease progression25 to the baseline risks in the natural history model. This suggests that treatment using peginterferon alfa-2b and ribavirin combination therapy will be more cost-effective in HCV/HIV co-infected patients, if the risks of fibrosis progression are greater than for mono-infected patients, as the incremental cost associated with providing treatment is lower and incremental QALY gain is greater than in the base case. In this analysis peginterferon alfa-2b is dominant (produces improved outcomes at lower cost) compared with supportive care for patients with genotypes 2 and 3.
| Genotype | Treatment | Cost (£) | Outcome (life-years) | Outcome (QALYs) | ICER (£/QALY gained) |
|---|---|---|---|---|---|
| Genotypes 1 + 4 | BSC | 31,839 | 16.91 | 10.90 | |
| PEG α-2b | 37,465 | 17.84 | 11.98 | ||
| Incremental | 5626 | 0.93 | 1.08 | 5193 | |
| Genotypes 2 + 3 | BSC | 31,839 | 16.91 | 10.90 | |
| PEG α-2b | 30,327 | 18.76 | 13.10 | ||
| Incremental | –1513 | 1.85 | 2.20 | PEG α-2b dominates |
Table 74 reports the cost-effectiveness results from the model when the age-specific mortality risks are doubled, for HCV/HIV co-infected patients. This reduces lifetime costs and QALYs for both peginterferon-treated and BSC cohorts, and would suggest that treatment using peginterferon alfa-2b and ribavirin combination therapy will be less cost-effective in HCV/HIV co-infected patients, if mortality risk is greater than for mono-infected patients.
| Genotype | Treatment | Cost (£) | Outcome (life-years) | Outcome (QALYs) | ICER (£/QALY gained) |
|---|---|---|---|---|---|
| Genotypes 1 + 4 | BSC | 19,865 | 17.46 | 11.70 | |
| PEG α-2b | 28,309 | 17.80 | 12.25 | ||
| Incremental | 8445 | 0.35 | 0.55 | 15,472 | |
| Genotypes 2 + 3 | BSC | 19,865 | 17.46 | 11.70 | |
| PEG α-2b | 23,929 | 18.14 | 12.84 | ||
| Incremental | 4065 | 0.68 | 1.14 | 3570 |
Tables 75 and 76 report cost-effectiveness results from alternative assumptions on the utility gain for HCV/HIV co-infected patients who achieve an SVR – in the first case the utility gain is assumed to be half that reported for HCV mono-infected patients and in the second case the utility gain is assumed to be zero. In both cases the QALY gain from treatment with peginterferon is reduced, indicating that treatment using peginterferon alfa-2b and ribavirin combination therapy will be less cost-effective in HCV/HIV co-infected patients, if utility gain from SVR is lower in HCV/HIV co-infected patients than for mono-infected patients.
| Genotype | Treatment | Cost (£) | Outcome (life-years) | Outcome (QALYs) | ICER (£/QALY gained) |
|---|---|---|---|---|---|
| Genotypes 1 + 4 | BSC | 22,201 | 18.93 | 12.65 | |
| PEG α-2b | 30,102 | 19.38 | 13.19 | ||
| Incremental | 7901 | 0.46 | 0.54 | 14,733 | |
| Genotypes 2 + 3 | BSC | 22,201 | 18.93 | 12.65 | |
| PEG α-2b | 25,190 | 19.83 | 13.77 | ||
| Incremental | 2989 | 0.91 | 1.12 | 2669 |
| Genotype | Treatment | Cost (£) | Outcome (life-years) | Outcome (QALYs) | ICER (£/QALY gained) |
|---|---|---|---|---|---|
| Genotypes 1 + 4 | BSC | 22,201 | 18.93 | 12.65 | |
| PEG α-2b | 30,102 | 19.38 | 13.05 | ||
| Incremental | 7901 | 0.46 | 0.40 | 19,590 | |
| Genotypes 2 + 3 | BSC | 22,201 | 18.93 | 12.65 | |
| PEG α-2b | 25,190 | 19.83 | 13.51 | ||
| Incremental | 2989 | 0.91 | 0.86 | 3489 |
A final scenario analysis was performed to consider the impact of on-treatment monitoring costs on the cost-effectiveness of antiviral treatment for HCV/HIV co-infected patients. Table 77 reports the cost-effectiveness results from the model if on-treatment costs for HCV/HIV co-infected patients are assumed to be double those for HCV mono-infected patients. As with the previous analyses this assumption suggests that treatment using peginterferon alfa-2b and ribavirin combination therapy is less cost-effective than in the base case.
| Genotype | Treatment | Cost (£) | Outcome (life-years) | Outcome (QALYs) | ICER (£/QALY gained) |
|---|---|---|---|---|---|
| Genotypes 1 + 4 | BSC | 22,201 | 18.93 | 12.65 | |
| PEG α-2b | 31,153 | 19.38 | 13.32 | ||
| Incremental | 8952 | 0.46 | 0.67 | 13,376 | |
| Genotypes 2 + 3 | BSC | 22,201 | 18.93 | 12.65 | |
| PEG α-2b | 26,241 | 19.83 | 14.03 | ||
| Incremental | 4040 | 0.91 | 1.38 | 2921 |
Deterministic sensitivity analysis
Table 78 reports the results of a DSA for treatment of HCV/HIV co-infected patients using peginterferon alfa-2b and ribavirin combination therapy. These suggest that the results are robust to a change in structural assumptions (allowing spontaneous SVR from the mild chronic HCV state), the proportion of the baseline cohort that is male and cost of the SVR health state. Reducing drug acquisition costs has the effect of reducing the ICER, as might be expected as it reduces the drug costs while the outcome difference is unchanged.
| Genotype 1 | Genotypes 2 + 3 | |||||
|---|---|---|---|---|---|---|
| Incremental cost (£) | Incremental QALY | ICER | Incremental cost (£) | Incremental QALY | ICER | |
| Base case | 7901 | 0.67 | 11,806 | 2989 | 1.38 | 2161 |
| Structural uncertainty | ||||||
| Spontaneous SVR from mild (0.002) | 8093 | 0.63 | 12,893 | 3369 | 1.30 | 2590 |
| Spontaneous SVR from mild (0.010) | 8005 | 0.65 | 12,376 | 3194 | 1.34 | 2386 |
| Discount cost and outcome at 0% | 2727 | 1.70 | 1607 | –7250 | 3.42 | –2122 |
| Discount cost at 6%, outcome at 1.5% | 9539 | 1.11 | 8586 | 6231 | 2.26 | 2760 |
| Baseline cohort characteristics | ||||||
| Cohort 80% male | 7927 | 0.66 | 11,957 | 3,041 | 1.37 | 2219 |
| Cohort 40% male | 7819 | 0.69 | 11,350 | 2,827 | 1.42 | 1988 |
| Change average age of cohort at start of simulation (base case 40 years old) | ||||||
| –10 years | 7329 | 0.83 | 8819 | 1857 | 1.70 | 1090 |
| +5 years | 8268 | 0.58 | 14,192 | 3715 | 1.21 | 3066 |
| +10 years | 8688 | 0.49 | 17,573 | 4547 | 1.04 | 4384 |
| +15 years | 9157 | 0.41 | 22,499 | 5475 | 0.86 | 6336 |
| Change distribution of cohort across disease stages at start of simulation | ||||||
| Cohort 100% mild chronic HCV | 10,442 | 0.47 | 22,104 | 8018 | 0.99 | 8070 |
| Cohort 100% moderate HCV | 6213 | 0.78 | 7934 | –351 | 1.61 | –218 |
| Cohort 100% CC | 3640 | 1.07 | 3390 | –5443 | 2.18 | –2493 |
| Parameter uncertainty | ||||||
| Assume SVR is 25% lower in patients with CC | 8133 | 0.64 | 12,690 | 3448 | 1.33 | 2599 |
| Assume SVR is 50% lower in patients with CC | 8365 | 0.61 | 13,656 | 3907 | 1.27 | 3075 |
| Cohort 100% CC, assume SVR is 25% lower in patients with CC | 5960 | 0.79 | 7541 | –852 | 1.62 | –525 |
| Cohort 100% CC, assume SVR is 50% lower in patients with CC | 8280 | 0.51 | 16,334 | 3738 | 1.06 | 3521 |
| Transition probability from mild-to-moderate disease = 0.04 | 7584 | 0.72 | 10,483 | 2361 | 1.49 | 1585 |
| Transition probability from moderate disease to CC = 0.073 | 7226 | 0.82 | 8762 | 1654 | 1.69 | 978 |
| Cost of SVR state = £0 | 7830 | 0.67 | 11,700 | 2849 | 1.38 | 2060 |
| Reduce cost of PEG α-2b by 20% | 6340 | 0.67 | 9473 | 1428 | 1.38 | 1033 |
| Reduce cost of PEG α-2b by 30% | 5560 | 0.67 | 8307 | 648 | 1.38 | 468 |
| Reduce cost of RBV by 20% | 7246 | 0.67 | 10,827 | 2334 | 1.38 | 1688 |
| Reduce cost of RBV by 30% | 6918 | 0.67 | 10,337 | 2006 | 1.38 | 1451 |
The greatest variability in ICERs is associated with changes in the age at which patients enter the model, the distribution of patients across disease stages (to a lesser extent) and response to treatment (SVR) for patients with cirrhosis. Increasing the age at which patients enter the model by 15 years leads to an approximate doubling of the ICER for genotype 1 and 4 patients – the QALY gain with treatment is reduced by around one-half for both genotype 1 and 4 patients and genotype 2 and 3 patients. Alternative assumptions regarding the stage of liver disease in which patients enter the model has a large impact on the ICER, with less favourable results associated with patients being in the earlier (lower fibrosis) stages of disease. Reducing response to treatment for patients with cirrhosis at baseline also leads to less favourable cost-effectiveness estimates, while increasing the probability of fibrosis progression for early disease states leads to more favourable cost-effectiveness results.
Probabilistic sensitivity analysis
In a PSA, where the probabilities of achieving SVR, health-state costs, health-state utility values and transition probabilities for the natural history parameters were sampled probabilistically, treatment of co-infected patients with genotypes 1 and 4 is associated with increased QALYs (with a range from 0.1 to 1.41 QALYs), but also increased costs (ranging from £4260 to £10,560) in all simulations when compared with BSC (Table 79 and Figure 21). Treatment for patients with genotypes 2 and 3 is also associated with increased QALYs (from 0.22 to 2.72 QALYs gained) and generally with increased costs. In approximately 7% of simulations the incremental cost was negative (Figure 22).
| Genotype | Treatment | Lifetime costs (£) | QALYs |
|---|---|---|---|
| Genotypes 1 + 4 | BSC | 22,175 (15,557 to 30,351) | 12.70 (11.89 to 13.51) |
| PEG α-2b | 30,086 (24,839 to 36,244) | 13.37 (12.66 to 14.07) | |
| Incremental | 7910 (5593 to 9673) | 0.66 (0.32 to 1.06) | |
| Genotypes 2 + 3 | BSC | 22,010 (15,706 to 30,199) | 12.70 (11.85 to 13.56) |
| PEG α-2b | 25,105 (21,202 to 30,212) | 14.06 (13.26 to 14.85) | |
| Incremental | 3095 (–1241 to 6340) | 1.36 (0.69 to 2.01) |
FIGURE 21.
Cost-effectiveness plane for genotypes 1 and 4 – incremental cost and incremental QALYs for treatment of HCV/HIV co-infected patients with peginterferon alfa-2b and ribavirin combination therapy.
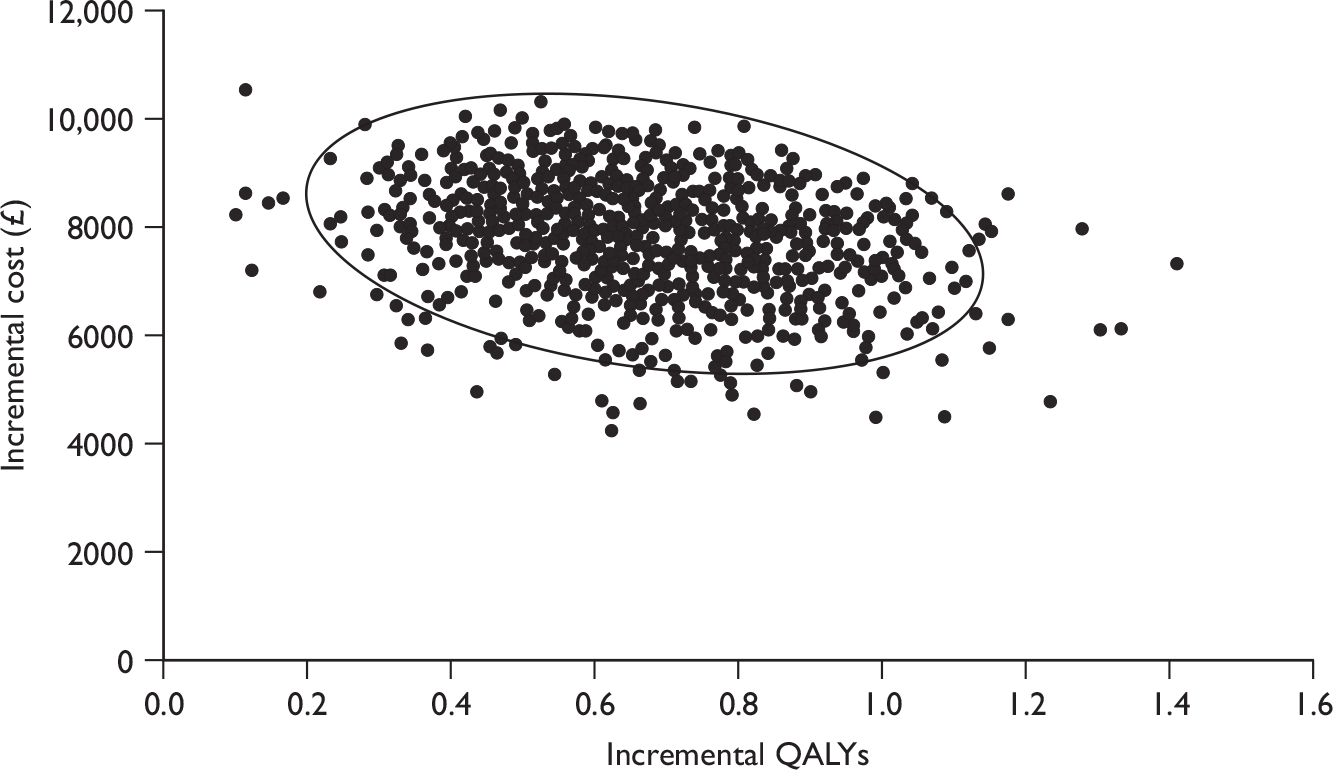
FIGURE 22.
Cost-effectiveness plane for genotypes 2 and 3 – incremental cost and incremental QALYs for treatment of HCV/HIV co-infected patients with peginterferon alfa-2b and ribavirin combination therapy.
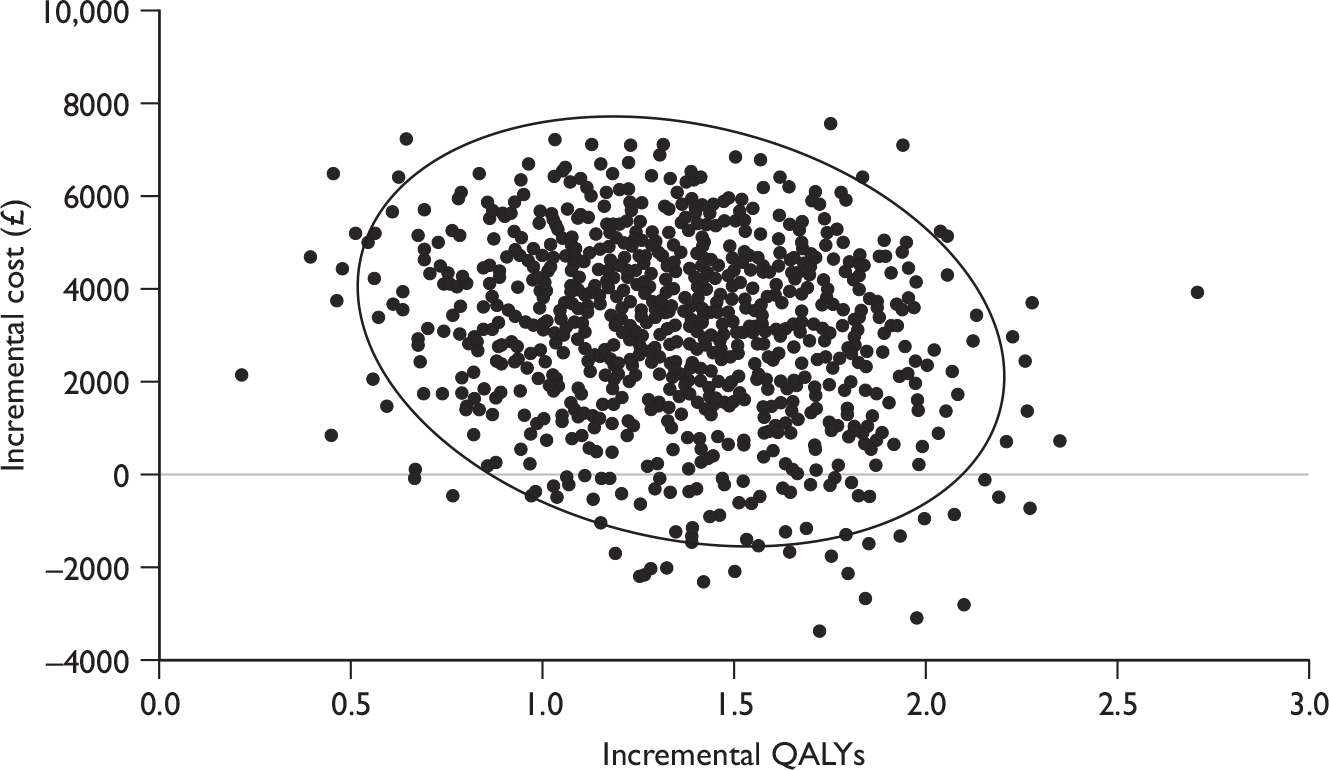
In this analysis, treatment using peginterferon alfa-2b and ribavirin combination therapy for patients with genotypes 2 and 3 had a probability of being cost-effective (compared with BSC) of 100% at a willingness-to-pay threshold of £20,000 per QALY – see Figure 23. For patients with genotypes 1 and 4 the probability of being cost-effective (compared with BSC) was 90% at a willingness-to-pay threshold of £20,000 per QALY and 99% at a willingness-to-pay threshold of £30,000 per QALY.
FIGURE 23.
Cost-effectiveness acceptability curves for treatment of HCV/HIV co-infected patients with peginterferon alfa-2b and ribavirin combination therapy.
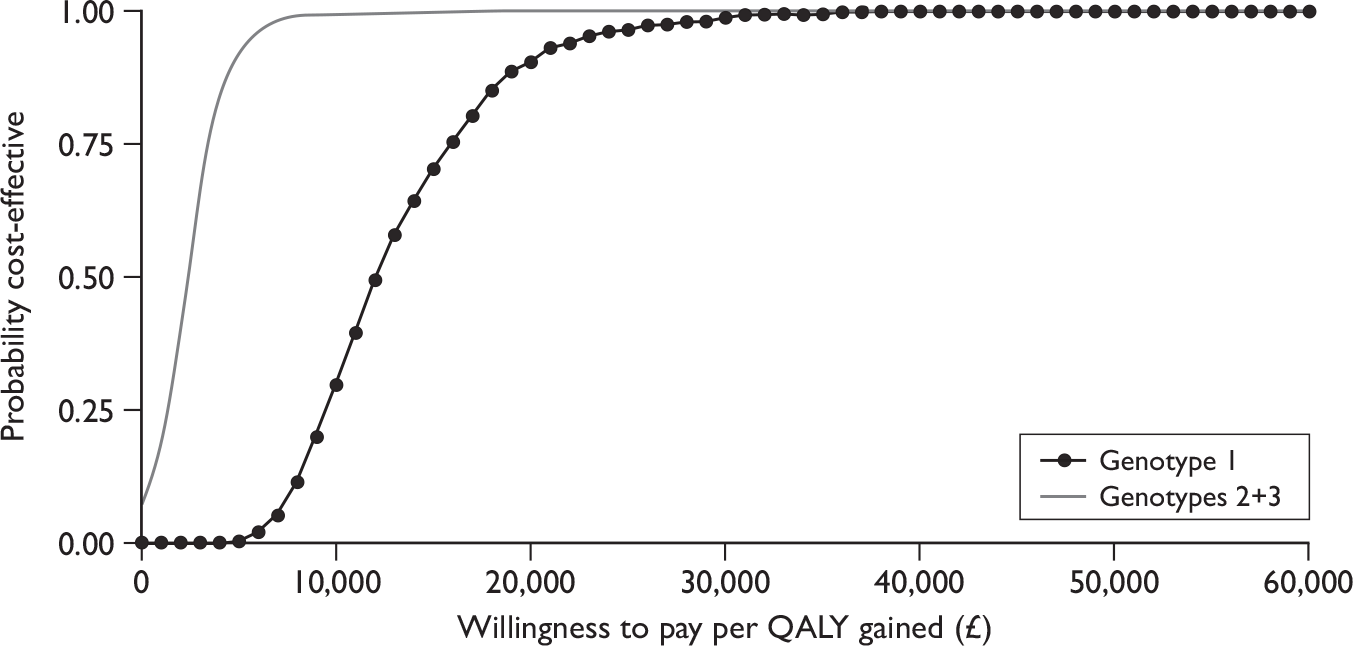
Summary of key results
Systematic review of published cost-effectiveness and QoL evidence
-
A systematic search of the literature found two fully published economic evaluations that were relevant to the scope of this assessment. Both economic evaluations used Markov models to extrapolate from SVRs, reported in clinical trials, to life expectancy and (in one case) quality-adjusted life expectancy gains associated with antiviral treatment strategies for patients who were co-infected with HCV and HIV. One of the evaluations64 based its analysis on data from trials that included only patients mono-infected with HCV, while the other65 used data from trials including co-infected patients. Both evaluations indicated that HCV antiviral treatment was associated with gains in life expectancy for HCV/HIV co-infected patients. Both evaluations were conducted in the context of the US health-care system.
-
A systematic search for published studies of HRQoL found no relevant studies.
Roche submission to NICE
-
Roche submitted a dossier104 in support of peginterferon alfa-2a combined with ribavirin in three subgroups of patients:
-
– shortened duration of treatment for patients with LVL who exhibit an RVR
-
– re-treatment in patients who did not respond or relapsed on previous treatment with peginterferon
-
– treatment of patients with HCV/HIV co-infection.
-
-
The submission included model-based economic evaluations using clinical effectiveness data from published RCTs, although effectiveness evidence for shortened treatment duration was derived from subgroup analyses. A number of the clinical effectiveness studies included by the manufacturer do not make the comparisons specified by NICE (patients who did not respond88 or relapsed,89 and patients with HCV/HIV co-infection66). Most commonly, these trials had an active comparator, rather than supportive care. In the majority of situations the comparison with supportive care assumed that the spontaneous SVR rate will be zero, which generally accords with clinical opinion.
-
Roche’s model is structurally similar to that used in our previous assessment report17 for NICE TA106. The natural history parameters in the model are also similar to our previous assessment report,17 as are the health-state utilities – except for the SVR state which in the manufacturer’s model are age-specific values derived in a general population. The differences in structural assumptions and utility values appear likely to produce higher estimates of utility gain associated with SVR.
-
The economic evaluation section of the MS does not indicate clearly where the clinical effectiveness parameters (EVR and SVR) are presented and critically appraised in the clinical effectiveness section of the MS. As a result there is no discussion or critical analysis of the reliability or generalisability of the clinical effectiveness evidence used to populate the model.
-
Shortening the duration of treatment results in a QALY loss compared with standard treatment duration, as a result of a slight reduction in SVR, as well as a reduction in costs. As both costs and outcomes are lower with shortened treatment duration, the ICERs are positive (in the south-west quadrant of the cost-effectiveness plane) – £15,472 for genotype 1 and 4 patients and £2719 for genotype 2 and 3 patients. The MS does not discuss the appropriate approach or decision rules to interpret ICERs for cost-saving and QALY-reducing interventions.
-
Two separate populations of re-treated patients were modelled: patients who relapsed following treatment with peginterferon and patients who did not respond to initial treatment with peginterferon. For relapsing patients the model estimates a QALY gain and a reduction in total costs, compared with BSC, suggesting that re-treatment with peginterferon is dominant. This is based on data from an RCT that may not be generalisable to all relapsed patients. Re-treatment of non-responding patients results in QALY gains compared with BSC, but also increased costs – the estimated reduction in costs of managing progressive liver disease in the cohort of patients receiving antiviral treatment does not fully offset treatment costs – resulting in positive ICERs (in the north-east quadrant of the cost-effectiveness plane).
-
For patients with HCV/HIV co-infection the MS reports a comparison with non-peginterferon, using effectiveness data from APRICOT,66 suggesting that peginterferon dominates non-peginterferon. This does not meet the scope issued by NICE, which specifies that peginterferon be compared with BSC. We extended the analysis conducted by the manufacturer (applying the same assumption as that adopted for non-responding or relapsing patients – that the SVR rate for untreated patients would be zero), estimating a QALY gain (using the manufacturer’s model) of 1.95 and incremental cost of £1765, resulting in an ICER of £903 per QALY gained.
-
Deterministic sensitivity analyses reported in the MS suggested that the results are generally robust to variation in a limited number of parameters that were not included in the PSAs. These included longer duration of surveillance following SVR, average patient weight, start age and proportion of women in the modelled cohort.
-
We undertook further analyses of the manufacturer’s model examining the robustness of the results in the MS to changes in assumptions regarding the:
-
– utility value for patients achieving an SVR
-
– distribution of patients across stages of progressive liver disease
-
– inclusion of chronic disease management costs alongside treatment costs.
-
-
These additional analyses generally resulted in less favourable ICERs but did not substantially alter the conclusions from the MS.
Schering-Plough submission to NICE
-
Schering-Plough submitted a dossier92 in support of peginterferon alfa-2b combined with ribavirin in two of the three subgroups of patients within the scope of the NICE appraisal:
-
– re-treatment in patients who did not respond or relapsed on previous treatment with peginterferon
-
– treatment of patients with HCV/HIV co-infection.
-
-
The submission included model-based economic evaluations based on clinical data from a multicentre, non-randomised, open-label uncontrolled study (for re-treatment in non-responding or relapsing patients) and a Phase III open-label trial95 (for patients with HCV/HIV co-infection). As the included studies do not make the comparisons specified by NICE (antiviral treatment compared with BSC), the manufacturer has assumed that the spontaneous SVR rate for moderate chronic HCV and compensated cirrhosis (applied to BSC patients) will be zero; this would generally accord with clinical opinion. The model includes a low spontaneous SVR probability for patients with mild chronic HCV; this is applied to patients in the BSC and active treatment cohorts.
-
The manufacturer’s model is structurally similar to that used in our previous assessment report for NICE. 17 However, it does not distinguish between patients achieving an SVR from any of the treatment-eligible states (mild or moderate HCV and compensated cirrhosis). Utility estimates published from the UK Mild Hepatitis C Trial82 would suggest that these states should be separate. The natural history parameters in the model are similar to those adopted for our previous assessment report for NICE,17 as are the health-state utilities and health-state costs (inflated from 2003–4 to 2007–8 costs using the HCHS Pay and Prices Index85).
-
No systematic searches for health-state utilities or costs are reported. The manufacturers did not report a critical appraisal of the EPIC3,96 Scotto and colleagues97 or Laguno and colleagues95 trials, which provided the clinical effectiveness data for the model and sensitivity analyses. It is therefore difficult to judge the reliability or generalisability of the data used to populate the model. Costs and health-state utilities were primarily derived from the Mild Hepatitis C Trial. 82
-
Two groups of patients were modelled in the Schering-Plough submission;92 the first of these was re-treated and relapsed patients, each based upon data from the EPIC396 clinical study report. In the group of ‘non-responders’ overall in the Schering-Plough submission,92 peginterferon and ribavirin combination therapy cost £26,666, with a QALY gain of 1.04 over no treatment, resulting in an ICER of £4387 per QALY gained. For genotypes 1 and 4, these results were £27,125, with a 0.7 QALY gain and an ICER of £7177 per QALY gained. For genotypes 2 and 3, costs of £24,301 and a QALY gain of 2.78 resulted in an ICER of £783 per QALY gained.
-
The second group included in this submission was patients co-infected with HCV/HIV, and modelled using effectiveness data from the Laguno and colleagues trial. 95 In this group overall, peginterferon and ribavirin combination therapy cost £26,997, with a QALY gain of 2.32, which resulted in an ICER of £1077. For genotypes 1 and 4 in this group, peginterferon-plus-ribavirin combination therapy cost £27,790, with a QALY gain of 2.01, giving an ICER of £1637; in genotypes 2 and 3, a cost of £25,645 and QALY gain of 2.85 resulted in an ICER of £403 per QALY gained.
-
The DSA showed that the ICERs in both the re-treated and co-infected cohorts were sensitive to variation in the EVR and SVR, and to changes in patient weight. In the re-treatment group, ICERs showed a small increase in response to changes in disease severity distribution within the patient group. The ICERs in this group appeared very sensitive to (and increased substantially with) the substitution of data from the Scotto and colleagues study97 with the EPIC3 trial. 96 Where discounting was removed, the ICER reduced to £1265 per QALY gained in the re-treatment group. In the HCV/HIV co-infection group, peginterferon and ribavirin combination therapy was dominant where discounting was removed.
-
Probabilistic sensitivity analyses were conducted including the majority of parameters in the model. Appropriate distributions appear to have been used. Three PSAs are presented for each patient group (re-treated and HCV/HIV co-infected), including the overall cohort, and then separate analyses for genotype subgroups. The PSA reports high probabilities (over 90%) of treatment with peginterferon alfa-2b being cost-effective at a willingness-to-pay threshold of £20,000 and £30,000.
SHTAC independent economic analysis
-
We adapted a previously published model to undertake an independent economic assessment of shortened treatment duration with peginterferon alfa, using clinical effectiveness data included in this review. Our economic model was structurally similar to those developed by the manufacturers, using similar input parameters to model disease progression, health-state costs and utility. The model consists of nine non-absorbing health states representing stages of chronic liver disease and one absorbing state representing death.
-
The economic model contains three health states (SVR) representing cure of chronic HCV, which are differentiated by the patient’s stage of disease (mild HCV, moderate HCV and compensated cirrhosis) prior to treatment, as these are expected to have an impact on subsequent risk of progressive liver disease, post-treatment surveillance and also HRQoL. The remaining six, non-absorbing states (mild HCV, moderate HCV, compensated and decompensated cirrhosis, HCC and liver transplant) represent stages of progressive liver disease. Patients not exhibiting an SVR are expected to face the same risk of disease progression as untreated patients. These assumptions are all consistent with our previous assessments, and other published economic evaluations of antiviral treatment for chronic HCV. The model has a cycle length of 1 year and incorporates a half-cycle adjustment.
-
Baseline populations in the model were based on a clinical audit undertaken at a London teaching hospital. These differentiated between new and existing patients in terms of average age and the distribution of patients across stages of chronic liver disease (mild HCV, moderate HCV and compensated cirrhosis). The proportion of men in the baseline cohort was based on our previous assessment. The majority of these assumptions do not affect response to treatment, but relate to patients’ risk of all-cause mortality. The influence of stage of chronic liver disease on response to treatment (and the effect on cost-effectiveness of intervention) was assessed in a sensitivity analysis.
-
Sustained virological responses extracted from clinical trials included in the clinical effectiveness review are used in the model to estimate the probability of treatment-eligible patients transitioning to a relevant SVR state. Where applicable, EVRs are used to estimate the average duration of treatment and total drug acquisition costs for each antiviral treatment strategy. Early stopping of treatment in patients unlikely to achieve an SVR can have a significant impact on the cost-effectiveness of treatment with peginterferon alfa.
-
Our clinical effectiveness systematic review included five trials of shortened treatment duration used in our economic evaluation (three for genotype 1 patients, one for genotype 2 only and one for genotypes 2 and 3 combined):
-
– Shorter duration of treatment (from 48 to 24 weeks) with peginterferon alfa-2a for the subgroup of genotype 1 patients with baseline LVL and who achieve an RVR reduced total costs by approximately one-third, but was also associated with slightly poorer outcome. The ICERs were positive (as both incremental cost and incremental QALYs are negative) and ranged from around £34,000 per incremental QALY to £65,000 per incremental QALY. As these ICERs are derived as the ratio of two negative numbers, the commonly assumed decision rule – Is the ICER below a given (arbitrary) threshold? – does not hold. In this situation the logic is reversed and ICERs below the threshold are rejected. This can be better interpreted using the net benefits framework.
-
– Shorter duration of treatment (from 24 to 16 weeks) with peginterferon alfa-2a for genotype 2 and 3 patients reduced total costs by approximately one-quarter, and was associated with better outcome in the included trials. In these scenarios, there was shortened treatment duration for the subgroup of genotype 2 or 3 patients with low baseline viral load and who achieved an RVR-dominated standard duration.
-
– Shorter duration of treatment (from 48 to 24 weeks) with peginterferon alfa-2b for the subgroup of genotype 1 patients with baseline LVL and who achieve an RVR was associated with a reduction in costs of approximately £9000. Combined with a QALY gain increase of 0.49, this resulted in peginterferon alfa-2b dominating the standard 48-week duration of treatment.
-
-
None of the RCTs identified by our searches, which examined re-treatment of patients previously treated with peginterferon or that assessed peginterferon treatment in patients with HCV/HIV co-infection, met the inclusion criteria. The analyses of these patient subgroups have used data that have not been formally quality assessed in the same way as for the review of shortened treatment duration.
-
Re-treatment, with peginterferon alfa-2a, of patients who did not respond to previous peginterferon therapy increased costs (by approximately 62% in patients with genotype 1, and approximately 25% in genotype non-1 patients). The QALY gain from treatment was 0.31 for genotype 1 patients and 0.59 for genotype non-1 patients. This resulted in positive ICERs for both groups: in genotype 1 patients this was £52,587 per QALY gained and in genotype non-1 patients this was £10,926 per QALY gained.
-
Where an ‘early stopping rule’ at 12 weeks for patients not demonstrating an EVR was applied to re-treated patients the incremental cost increase was substantially reduced by approximately 12% (£3398) in genotype 1 patients, and by approximately 5% (£1415) in genotype non-1 patients. The QALY gain increased slightly in both groups (to 0.37 in genotype 1 and 0.62 in genotype non-1). Accordingly the ICERs for each group, while remaining positive, reduced to £9169 per QALY gained in genotype 1 and £2294 per QALY gained in genotype non-1.
-
Sustained virological responses for the re-treated patients receiving peginterferon alfa-2b were taken from the Schering-Plough MS. The impact of re-treating patients with genotypes 1 and 4, was an increase in costs of £9380, and in QALYs of 0.39, resulting in an ICER of £23,912. For genotypes 2 and 3 these costs were reduced by £989 and QALYs increased by 1.72, resulting in peginterferon alfa-2b dominating BSC.
-
Where an early stopping rule is applied for patients not demonstrating an EVR in genotypes 1 and 4, the incremental costs reduce to £3256 and the QALY gain increases to 0.42, resulting in an ICER of £7681. In genotypes 2 and 3 the incremental costs are reduced further, to –£2850, and the QALY gain increased slightly.
-
For patients that are co-infected with HCV/HIV, treatment with peginterferon alfa-2a resulted in a QALY gain of 0.75 for genotypes 1 and 4, and 1.86 for genotypes 2 and 3. Costs also increased by approximately 27% (£5932) in genotypes 1 and 4, which resulted in a positive ICER of £7941 per QALY gained in this group. Costs decreased overall as a result of treating genotypes 2 and 3 by approximately 8%, a reduction of £1717. This resulted in peginterferon alfa-2a dominating BSC in this group of patients.
-
For patients that are co-infected with HCV/HIV, treatment with peginterferon alfa-2b resulted in increased costs for both genotypes 1 and 4 and genotypes 2 and 3 (of £7901 and £2989, respectively); the QALY gain also increased by 0.67 and 1.38, respectively. ICERs for both groups were positive: in genotypes 1 and 4 this was £11,806 per QALY gained, and in genotypes 2 and 3 this was £2161 per QALY gained.
Strengths, limitations and generalisability
-
The majority of the clinical trials used to model response to treatment (SVR and, where relevant, EVR) were not included in our systematic review, and have not been fully critically appraised. Only clinical trials relating to shortened treatment duration were included. In the case of re-treated patients and those with HCV/HIV co-infection, no trials were found that met the scope for this appraisal (of having placebo or supportive care control arms). As a result, the model uses clinical trial data that have not been assessed for risk of bias. The effectiveness data for patients with HCV/HIV co-infection have been extracted from published systematic reviews/meta-analyses (see Appendix 9) and, although these were quality assessed during the process of the published reviews, they have not been quality assessed or critically assessed in our current review.
-
Some of the effectiveness data included in the model have been taken from comparatively small trials (20–40 patients per arm) that were not adequately powered to detect differences in SVR, or were derived from subgroups of patients in larger trials. In some cases the reporting of outcomes has not been consistent – for example, von Wagner and colleagues56 report SVR for patients with RVR and LVL, whereas Yu and colleagues55 report SVR for patients with RVR but do not stratify this result by viral load.
-
The proportion of patients with different genotypes in multinational clinical trials is unlikely to be reflective of the genotype distribution in the UK. Hence, the overall SVR is unlikely to provide a good indication of response. As a result, where possible, patient genotypes have been modelled separately adopting commonly used groupings of ‘difficult-to-treat’ genotypes (genotype 1 and, occasionally, genotype 4) and more responsive genotypes (2 and 3).
-
Baseline populations applied in the economic model were based on data for new and existing patients from a clinical audit in a liver unit at a London teaching hospital. 102 Clinical advisors to this project confirmed that the distribution of patients across disease stages agreed with their clinical experience. However, it is not clear how closely these distributions, or the assumed mean age of patients at the start of the model, relate to the characteristics of patients in the subgroups of patients covered by this review. The clinical audit data pre-date NICE guidance on the use of peginterferons in patients with chronic HCV (TA7538 and TA10633) and it is not clear how the distribution of patients across disease stages may have changed, particularly given recent guidance on treating patients with mild disease (TA10633). However, there is generally very little information on the age and stage of disease for treated patients – the latter becoming less relevant to decisions to initiate treatment but remaining relevant to modelling response to treatment where patients with cirrhosis appear less likely to achieve SVR.
-
Disease progression parameters included in the model were derived from large cohort studies in relevant (European) populations. The parameters have been used in previous economic evaluations17,81 and ensure consistency between appraisals. Input parameters for fibrosis progression (from mild-to-moderate and from moderate-to-compensated cirrhosis) were taken from a recent analysis using biopsy data from a UK cohort study. 18,82 Where evidence suggests that differential progression rates should be applied for the subgroups covered by this assessment (e.g. fibrosis progression in HCV/HIV co-infected patients), this has been addressed in additional analyses in this report.
-
Quality of life/health-state utility weights in the model were taken from reports on a multicentre trial and observational study,81,82 conducted using the EQ-5D and valued using the UK general population tariff. 84 The population of patients recruited to the UK trial were treatment-naive patients with mild HCV, and this was supplemented by an observational study recruiting patients with compensated and decompensated cirrhosis. It is not clear how applicable these QoL weights are to some of the subgroups of patients in the current assessment – re-treated patients are likely to be older, whereas QoL assessments for mono-infected patients may not be directly applicable to those with HCV/HIV co-infection.
-
Health-state costs included in the model, taken from the UK Mild Hepatitis C Trial,81,82 were developed in an observational study alongside the trial. Intervention costs were based on treatment protocols developed as part of our previous assessment17 in collaboration with UK clinical experts and valued using reference costs from an NHS hospital trust. All costs were inflated to current costs using the HCHS Pay and Prices Index. 85 It is not clear how adequately the treatment protocols may capture the complexity of managing patients with HCV/HIV co-infection – the sensitivity of the cost-effectiveness results to the costs of managing antiviral treatment in this group of patients was addressed in a sensitivity analysis.
Chapter 6 Assessment of factors relevant to the NHS and other parties
It should be acknowledged that the lower limits of detection for HCV RNA in terms of RVR and SVR differed slightly between the RCTs included in our systematic review of clinical effectiveness, according to the different assays used. For example, RVR was defined as HCV RNA < 50 IU/ml in three of the trials, < 25 IU/ml in one trial, < 600 IU/ml in one trial and < 615 IU/ml in another. Although a detectable HCV viral load of 50 IU/ml or above is generally considered indicative of infection, thresholds of detectability are becoming lower as more sophisticated assays are being produced. It is therefore important to achieve standardisation in definitions of virological response, particularly given the increased emphasis on using RVR to determine optimum treatment duration. Similarly, there is a lack of clarity regarding thresholds for LVL and high viral load. The SPC for the two peginterferons vary in terms of what they consider to be LVL (varying between < 600,000 IU/ml and ≤ 800,000 IU/ml). Again, clarity is needed regarding viral load thresholds to ensure consistent clinical management of patients.
If patients with specific genotypes meeting the licence criteria received shortened courses of antiviral treatment, then they would benefit in terms of reduced exposure to adverse effects, which can be very unpleasant and have a profound impact on a person’s day-to-day life, as well as that of family and carers. Consequently, it may also mean that less time is lost from work, thereby having an impact on economic circumstances.
Initiatives to encourage people who may have put themselves at risk of HCV infection, such as the Department of Health’s ‘FaCe It’ campaign, need to be maintained to reduce the substantial pool of undiagnosed infection. As well as the government, the voluntary sector also plays a key role in public awareness raising. Efforts to identify HCV infections need to be augmented by appropriate methods of referral to specialist care for further investigation and, if appropriate, antiviral treatment. Referral mechanisms need to be effective to ensure that as many eligible patients progress through the care pathway to be successfully treated. Strategies are also needed to motivate patients to attend assessment appointments and to complete the full course of therapy. This may be more problematic for patients with co-infection with HIV, who may not perceive their infection to be serious enough to undergo further assessment and treatment, particularly given the unpleasant adverse effects associated with interferon. Motivation is also particularly important for people who use drugs and alcohol, whose lifestyles are often unpredictable, making concordance with treatment regimes difficult. Such responsibilities may fall to specialist hepatology nurses, as well as general practitioners and other services. However, these may be time and resource intensive, and will be subject to budget constraints.
In terms of implementation issues, there do not appear to be any significant barriers to diffusion of the appraised treatments into routine practice. Peginterferon alfa has been the standard of care for some time. Specialist hepatology nurses will already be familiar with the administration of these drugs in the treatment of HCV. However, management protocols will need to be updated, where necessary, to ensure efficient testing for RVR and viral load to identify which patients are likely to be successfully treated with shorter courses.
Chapter 7 Discussion
Statement of principal findings
Clinical effectiveness
The results of six RCTs were included in this systematic review, all in patients who were eligible for shortened treatment duration. Treatment in patients with genotype 1 was evaluated in four trials,52–54,59 genotype 2 in one trial55 and genotypes 2 and 3 in one trial. 56 All studies compared standard treatment duration (48 weeks for genotype 1, 24 weeks for genotypes 2 and 3) to a shorter duration (24 or 16 weeks, respectively). In five of the RCTs the patients had LVL at baseline (based on mean viral load), while in one RCT52 less than one-quarter of patients had LVL (defined as HCV RNA < 400,000 IU/ml) at baseline. However, it was included in our systematic review because SVRs were presented for the subgroup of those with LVL who attained an RVR (i.e. the patient subgroup meeting the licensed criteria for receiving shortened courses of therapy, and thus within the scope of the NICE appraisal). Note, however, that this subgroup constituted only 10% of the total study population. In only one trial56 did all randomised patients consist of those with LVL and who achieved an RVR. In addition, none of the studies was powered for this subgroup and results should therefore be interpreted with caution.
In terms of demographic characteristics, three of the studies53–55 were carried out in Asian (Taiwanese) populations and may therefore not be generalisable to the likely eligible population in a UK setting. In addition, the mode of HCV infection was not reported in most of the studies, which may have implications for the relevance to the UK HCV population. The methodological reporting and study quality varied between the included trials but was generally good, although there was a risk of selection bias in two studies,56,59 where the randomisation procedure was unclear.
All of the trials reported SVR as the primary outcome measure. The evidence showed that in the subgroup of patients who achieved an RVR and had LVL at baseline, there were no statistically significant differences in SVR rates between groups who received the standard duration of treatment and those who received shortened courses, for both genotype 1 and genotypes 2 and 3. The SVR rates in genotype 1 patients are much higher than would normally be expected for this genotype, probably due to the fact that it is a highly select group of patients with favourable factors that increase the chance of response (e.g. LVL and RVR, generally mild-to-moderate HCV-related liver damage, absence of significant comorbidities or co-infections, absence of drug or alcohol abuse).
The evidence does suggest that patients in this subgroup can receive shorter courses of combination therapy without compromising SVR rates. However, only two of the trials52,59 were designed to establish non-inferiority (one of which became a superiority trial when a significant difference in overall SVR rates was observed),59 so it cannot necessarily be assumed that shortened and standard duration treatment are comparable. It should also be remembered that SVRs according to baseline LVL and RVR are based on subgroups (of varying sizes) of the randomised patients and are likely to be underpowered. The results of the trials in these subgroups should therefore be regarded as speculative.
Other outcome measures included virological response during treatment, relapse rate, biochemical response, histological response and adverse effects of treatment. The proportion of patients achieving an RVR was not statistically significantly different between treatment groups who received the standard duration of treatment compared with those who received shortened courses, regardless of genotype. Rates of RVR in genotype 2/3 patients were generally higher than in genotype 1 patients. In the one trial54 reporting relapse rates in the subgroup of patients with LVL and RVR, rates were low and not significantly different between those treated for 24 versus 48 weeks. Rates of adverse events were reported only for treatment groups as a whole (rather than subgroups based on LVL and RVR). There was a trend for a lower incidence of adverse events in patients treated for a shorter duration in three trials,53,54,56 although, on the whole, there were no statistically significant differences between treatment arms (where reported).
As stated in Chapter 4 (Quantity and quality of research available), no RCTs in patients co-infected with HCV/HIV comparing peginterferon alfa with BSC met our inclusion criteria. There were also no RCTs of the re-treatment of patients who had failed to respond to, or relapsed from, peginterferon alfa with a subsequent course of peginterferon alfa, comparing against BSC. However, it should be acknowledged that there is a wider evidence base in these patient groups, notably for co-infected people, in whom peginterferon alfa is compared with non-peginterferon alfa. For example, Kim and colleagues51 and Zhao and colleagues50 both included the same six RCTs in their systematic review of the effectiveness of peginterferon alfa in the treatment of HCV/HIV co-infection (see Appendix 8). All but one of the six RCTs in these two systematic reviews compared peginterferon alfa (2a or 2b) with non-peginterferon alfa. Furthermore, studies evaluating shortened treatment courses were eligible for inclusion in this review only if they reported SVR in patients with RVR and LVL. There are likely to be other studies evaluating shortened treatment courses but which were not restricted to patients with LVL. It should also be acknowledged that there were no RCTs of peginterferon alfa monotherapy that met the inclusion criteria for our systematic review, for any of the patient groups considered in this NICE appraisal, thus limiting what can be recommended for this patient group.
Cost-effectiveness
Systematic review of existing cost-effectiveness evidence
A systematic search of the literature for published economic evaluations that were relevant to the scope of this assessment identified two studies – both in HCV/HIV co-infected patients. Both studies included non-peginterferon (in combination with ribavirin or monotherapy) as well as peginterferon (in combination with ribavirin or monotherapy) and no treatment (supportive care), and used Markov models to extrapolate from SVRs, reported in clinical trials, to life expectancy and to QALYs (in one of the studies). Only one of the evaluations65 based its analysis on data from clinical trials including HCV/HIV co-infected patients. Both evaluations were conducted in the context of the US health-care system and were considered to be of limited relevance to the current assessment.
Cost-effectiveness evidence submitted by manufacturers
Two manufacturers submitted evidence to NICE, with respect to this assessment.
Roche submitted a dossier104 in support of peginterferon alfa-2a combined with ribavirin in three subgroups of patients:
-
shortened duration of treatment for patients with LVL who exhibit an RVR
-
re-treatment in patients who did not respond or relapsed on previous treatment with peginterferon; relapsing and non-responding patients were treated as separate subgroups, using data from different clinical trials
-
treatment of patients with HCV/HIV co-infection.
Schering-Plough submitted a dossier92 in support of peginterferon alfa-2b combined with ribavirin in two subgroups of patients:
-
re-treatment in patients who did not respond or relapsed on previous treatment with peginterferon
-
treatment of patients with HCV/HIV co-infection.
In some cases the studies used by the manufacturers to estimate response to treatment with peginterferon do not make the comparisons specified by NICE (patients who did not respond or relapsed and also patients with HCV/HIV co-infection, where the specified comparator is supportive care). In the majority of situations the manufacturer has conducted the comparison with supportive care by assuming that the spontaneous SVR rate will be zero – this would generally accord with clinical opinion.
The manufacturers’ economic models were structurally similar, but not identical, to that adopted for the previous assessment report for NICE17 and generally adopted similar natural history parameters, health-state utilities and health-state costs. The structural differences, and the differences in parameter inputs between the manufacturers’ models and that adopted for our previous assessment,17 were considered to be likely to overestimate the utility gain from treatment. The assessment group undertook additional analyses to quantify the impact of these differences on the QALY gains from treatment and on the resulting ICER.
Roche submission104
Shorter treatment duration resulted in substantial reductions in antiviral treatment costs (49% lower for genotype 1 and 4 patients and 31% lower for genotype 2 and 3 patients) and lower total costs (including costs of managing progressive liver disease associated with chronic HCV infection). However, there was also a reduction in total QALYs for shorter treatment duration compared with standard treatment duration, as a result of a reduction in SVR. As both costs and outcomes are lower with shortened treatment duration, the ICERs are positive (in the south-west quadrant of the cost-effectiveness plane) – £15,472 for genotype 1 and 4 patients and £2719 for genotype 2 and 3 patients. The submission did not discuss the complications of interpreting ICERs for cost- and outcome-reducing strategies.
Re-treating patients who relapsed following previous peginterferon treatment was reported as dominating supportive care – yielding a gain of 2.7 QALYs while reducing total costs by approximately £6000. This arises from a high SVR observed in one trial, which may not be generalisable to other populations of relapsed patients. The majority of patients in the study were genotype 1 patients who had received a shorter duration of treatment than the current standard of care (24 rather than 48 weeks). The SVRs applied in the model for re-treatment of patients who did not respond to previous peginterferon treatment were lower than for relapsed patients. While treatment resulted in QALY gains compared with BSC, the estimated reduction in costs of managing progressive liver disease did not fully offset treatment costs, resulting in positive ICERs (in the north-east quadrant of the cost-effectiveness plane) – £3334 for genotype 1 patients and £809 for genotype non-1 patients. The majority of patients recruited to the trial of non-responders to previous peginterferon treatment were genotype 1. There were only 29 genotype non-1 patients (9% of the arm used to estimate effectiveness of treatment in the model), the majority (66%) of whom were genotype 4.
For patients with HCV/HIV co-infection, treatment with peginterferon was estimated to dominate non-peginterferon, using direct effectiveness evidence from the APRICOT study. 66 However, this is not the comparison specified in the scope issued by NICE. The assessment group extended the analysis – assuming that the SVR rate for untreated patients would be zero – estimating a QALY gain (using the manufacturer’s model) of 1.95 and incremental cost of £1765, for peginterferon compared with BSC, resulting in an ICER of £903 per QALY gained.
The cost-effectiveness results were generally robust to variation in a limited number of parameters included in a DSA reported in the MS. PSAs were conducted, including the majority of parameters in the model. While appropriate distributions appear to have been used for the PSA, the parameterisation of the distributions for some inputs does not appear to make best use of data reported in the submission. Moreover, there seems to have been a lack of consideration regarding logical relationships and potential correlation between model inputs. Rather than report the probability of cost-effectiveness at certain willingness-to-pay thresholds, the submission identified a maximum threshold of £15,000 for all analyses. Further analyses of the manufacturer’s model undertaken by the assessment group generally resulted in less favourable ICERs, but did not substantially alter the conclusions from the MS.
Schering-Plough submission92
Re-treating patients who did not respond or relapsed following previous interferon treatment was estimated to result in a QALY gain of 1.03, compared with supportive care, at an incremental cost of £4536, resulting in an ICER of £4387. These results were reported for a combined cohort of genotype 1 and 4 (84% of total) patients and genotype 2 and 3 patients. Separate results are also reported for the two genotype subgroups: the ICERs were £7177 per QALY gained for genotype 1 and 4 patients and £783 per QALY gained for genotype 2 and 3 patients. The submission also reports subgroup analyses (not stratified by genotype) for non-responding and relapsed patients separately – suggesting that the QALY gain is higher for relapsed than for non-responding patients. Effectiveness data for this group of patients were taken from the unpublished EPIC3 study,96 which recruited patients who had been previously treated with non-peginterferon as well as peginterferon. The effectiveness data in the model appear not strictly to meet the scope issued by NICE, as they appear to be based on all patients in the EPIC3 study, not just those who were previously treated with peginterferon.
For a cohort of patients (of all genotypes) co-infected with HCV/HIV, treatment with peginterferon was estimated to result in a gain of 2.32 QALYs compared with no treatment, at an incremental cost of £2502, resulting in an ICER of £1077. For patients with genotypes 1 and 4 the ICER was estimated at £1637 per QALY gained, while for patients with genotypes 2 and 3 the ICER was £403 per QALY gained.
The DSA showed that the ICERs in both the re-treated and the co-infected cohorts were sensitive to variation in the EVR and SVR, and to changes in patient weight, as dosing of both peginterferon alfa-2b and ribavirin are weight based. In the re-treatment group ICERs showed a small increase in response to changes in disease severity distribution within the patient group.
Probabilistic sensitivity analyses were conducted including the majority of parameters in the model. The choice of distribution applied to parameters appears to have been appropriate. Three PSAs are reported for each patient group (re-treated and HCV/HIV co-infected patients) – the first is for the overall cohort of patients followed by separate analyses for genotype subgroups. The PSA reports high probability (> 90%) of treatment with peginterferon alfa-2b being cost-effective for all analyses, at willingness-to-pay thresholds of £20,000 and £30,000.
Independent economic assessment
We adapted a previously published model17 to undertake an independent economic assessment of shortened treatment duration with peginterferon alfa, based on SVRs extracted from clinical trials included in our clinical effectiveness review. Our economic model was structurally similar to those developed by the manufacturers, using similar input parameters to model disease progression, health-state costs and utility. The model consists of nine non-absorbing health states representing stages of chronic liver disease and one absorbing state representing death. The model has a cycle length of 1 year and incorporates a half-cycle adjustment.
Baseline populations in the model were based on a clinical audit undertaken at a London teaching hospital, differentiating between new and existing patients in terms of average age and the distribution of patients across stages of chronic liver disease. 101 The proportion of men in the baseline cohort was based on our previous assessment.
For the subgroup of genotype 1 patients with baseline LVL and who achieve an RVR, shorter duration of treatment with peginterferon alfa-2a (from 48 to 24 weeks) reduced total costs by approximately one-third (approximately £5000) but was also associated with slightly poorer outcome (4%–6% lower SVR, resulting in a reduction in total QALYs of 0.08 to 0.14). The ICERs were positive (as both incremental cost and incremental QALYs are negative) and range from around £35,000 per incremental QALY to £65,000 per incremental QALY. As these ICERs are derived as a ratio of two negative values, the commonly assumed decision rule – Is the ICER below a given threshold? – does not hold. In this situation the logic is reversed and ICERs below the threshold are rejected. This can be better understood using the net benefits framework.
Shorter duration of treatment with peginterferon alfa-2a (from 24 to 16 weeks) for genotype 2 and 3 patients reduced total costs by approximately one-quarter (between £2000 and £3000) and was associated with better outcome in the included trials (2%–7% higher SVR, resulting in an increase in total QALYs of 0.08 to 0.23). In these scenarios, shortened treatment duration dominated standard care for the subgroup of genotype 2 or 3 patients with baseline LVL and who achieve an RVR.
For genotype 1 patients with baseline LVL and who achieve an RVR, shorter duration of treatment with peginterferon alfa-2b (from 48 to 24 weeks) reduced total costs by approximately one-third (approximately £9000), and was associated with better outcome in the included trial [15% higher SVR (8/19 vs 16/28), resulting in an increase in total QALYs of 0.49]. This results in shortened treatment with peginterferon alfa-2b dominating standard duration of treatment for this patient group.
No RCTs of re-treatment of patients previously treated with peginterferon, or of treatment in patients with HCV/HIV co-infection, met the inclusion criteria for our review of clinical effectiveness. The analyses of these patient subgroups have used data that have not been formally quality assessed in the same way as for the review of shortened treatment duration.
For peginterferon alfa-2a, the analysis of re-treatment of patients who did not respond to previous peginterferon therapy, and was based on data included in the submission by Roche,104 provided further detail on the trial reported by Jensen and colleagues. 88 In this analysis, re-treatment using peginterferon alfa-2a resulted in increased costs and increased QALYs. The ICER for genotype 1 patients was £52,587 per QALY gained, and for genotype non-1 patients was £10,926 per QALY gained. The ICERs changed marginally when accounting for patients withdrawing from treatment owing to adverse events. Adopting an early stopping rule based on EVR led to a substantial reduction in incremental costs for treated patients. The ICERs for each group reduced to £9169 per QALY gained in genotype 1 and £2294 per QALY gained in genotype non-1.
For peginterferon alfa-2b, the analysis of re-treatment of patients who did not respond to previous peginterferon therapy was based on data included in the submission by Schering-Plough92 reporting evidence from the EPIC3 study. 96 In this analysis, re-treating patients with genotypes 1 and 4 increased costs by £9380, and increased QALYs by 0.39, resulting in an ICER of £23,912. For genotypes 2 and 3, these costs were reduced by £989 and QALYs increased by 1.72, resulting in peginterferon alfa-2b dominating BSC. Adopting an early stopping rule based on EVR led to a substantial reduction in incremental costs for treated patients. The ICERs for the group including genotypes 1 and 4 patients reduced to £7681 per QALY gained.
For patients who were co-infected with HCV/HIV, treatment with peginterferon alfa-2a resulted in a QALY gain of 0.75 for genotypes 1 and 4, and 1.86 for genotypes 2 and 3. Costs also increased by approximately 27% (£5932) in genotypes 1 and 4, which resulted in a positive ICER of £7941 per QALY gained in this group. Costs decreased overall as a result of treating genotypes 2 and 3 by approximately 8%, a reduction of £1717. This resulted in peginterferon alfa-2a dominating BSC in this group of patients.
For patients who are co-infected with HCV/HIV, treatment with peginterferon alfa-2b resulted in increased costs for both genotypes 1 and 4 and genotypes 2 and 3 (£7901 and £2989, respectively). The QALY gain also increased by 0.67 and 1.38, respectively. ICERs for both groups were positive: in genotypes 1 and 4 this was £11,806 per QALY gained, and in genotypes 2 and 3 it was £2161 per QALY gained.
Summary of cost-effectiveness evidence
All three models used in this assessment (the two manufacturers’ models and the independent model adopted by the assessment group) were structurally similar and used similar parameter inputs for the chronic HCV natural history model. However, there were key differences in structural assumptions regarding the SVR state, health-state utility and characteristics of the baseline cohorts entering the models that led to differences in cost-effectiveness results.
For patients co-infected with HCV/HIV, and in patients who did not respond to previous peginterferon alfa combination therapy, the cost-effectiveness results are broadly consistent, but with less favourable ICERs in the independent economic assessment. For the subgroup of patients who were eligible for shortened duration of treatment, the results of the manufacturer’s analysis and the independent economic assessment are inconsistent. In the manufacturer’s analysis, shortened duration of treatment would not be a cost-effective treatment option at conventionally accepted threshold values, whereas in the independent economic assessment shortened treatment duration would be a cost-effective option. The difference in the results of these analyses arises from the differences in structural assumptions, health-state utility and baseline cohort characteristics, as well as differences in effectiveness data used in the two models. The interpretation of the ICER results is complicated by the fact that shortened duration of treatment would most typically be associated with a QALY loss (compared with standard duration) owing to a reduction in effectiveness (lower SVR). Interpretation of such cost-saving and QALY-reducing options can be aided by adopting the net benefits approach, rather than relying on ICERs.
Strengths and limitations of the assessment
-
In terms of strengths, this technology assessment report has been undertaken following standard principles for conducting a systematic review. 49 The methods were set out in a research protocol that defined the research question, inclusion criteria, quality criteria, data extraction process and methods to be used at different stages of the review (see Appendix 1). An advisory group has informed the review from its initiation. The research protocol was informed by comments received from the advisory group, and the advisory group has reviewed and commented on the final report.
-
The report brings together the evidence for the clinical effectiveness and cost-effectiveness of peginterferon alfa and ribavirin for chronic HCV in three specific patient groups. This evidence has been critically appraised and presented in a consistent and transparent manner.
-
An economic model has been developed following recognised guidelines, and systematic searches have been conducted to identify data for the economic model. The main results have been summarised and presented. The report is also independent of any vested interest.
-
In terms of limitations it should be acknowledged that outcome data, in terms of SVR according to RVR and LVL, in the studies evaluating shortened courses of treatment, were based on patient subgroups as opposed to all randomised patients. It is unlikely that the RCTs were statistically powered with respect to these subgroups so caution is advised in the interpretation of data.
Two of the RCTs of peginterferon alfa-2a included in our systematic review of clinical effectiveness used doses of ribavirin according to body weight, which is no longer within the licence indication. Both of these trials restricted inclusion to genotype 255 or genotype 2/3 patients. 56 The product licence for peginterferon alfa-2a specifies that ribavirin should be given in a fixed dose of 800 mg to patients with genotypes 2 and 3. Both trials appear to have been designed and executed before the licence variation. Exclusion of these RCTs solely on this basis would have further reduced the evidence base in our systematic review such that there would be no evidence of the impact of shortened treatment durations in patients with genotypes 2 or 3.
The majority of studies used to derive estimates of response to treatment with peginterferon alfa did not make the comparisons specified by NICE. For re-treatment of patients who did not respond or relapsed following previous treatment and also patients with HCV/HIV co-infection the specified comparator was supportive care, while the clinical trials have active comparators. We were unable to construct evidence networks that included placebo (or supportive care) controlled trials. As a result, in common with the manufacturers, we have conducted the comparison with supportive care by assuming that the spontaneous SVR rate will be zero. While this is generally supported by clinical opinion, it remains an assumption and is not supported by robust evidence.
Parameters in the model (disease progression, utility and health-state cost) have not been derived for the specific patient subgroups in this assessment. Targeted searches undertaken for this review did not identify suitable data, for the relevant patient groups, for the majority of parameters in the model. It is not clear how applicable health-state utility values for HCV mono-infected patients are to those with HCV/HIV co-infection. Similarly, treatment costs based on protocols for mono-infected patients may underestimate the resource use required for on-treatment management of HCV/HIV co-infected patients. We have attempted to address this through sensitivity analyses.
In common with our previous technology assessment reports,17,44 we have presented the results of this report separately for peginterferon alfa-2a and alfa-2b, as these agents are generally considered to be pharmacologically distinct from each other. It should be acknowledged that one of the RCTs included in the systematic review of clinical effectiveness, Mangia and colleagues,52 treated patients with either peginterferon alfa-2a or -2b in both of its arms, as opposed to the other RCTs, each of which evaluated either alfa-2a or alfa-2b but not both in the same trial.
Uncertainties
Across the included trials, RVR and LVL were not consistently defined, with the lower limits of detection of the virus being different between studies. RVR was defined as undetectable serum HCV RNA but the lower threshold for detection varied from < 25 to < 615 IU/ml. Similarly, the threshold for LVL differed between trials with a cut-off HCV RNA level of < 400,000 or < 800,000 IU/ml being used to differentiate between low and high viral load. This variability in cut-off limits has implications for the number of patients rightly classified as having LVL or achieving an RVR. In clinical practice, an HCV RNA < 30 IU/ml at week 4 of treatment is generally regarded as an RVR.
Sustained virological response has not been reported according to stage of liver disease in the included studies. However, peginterferon alfa treatment is indicated for patients with compensated liver disease and is therefore likely to be provided to patients with compensated cirrhosis. Fibrosis stage (particularly cirrhosis) has been shown consistently (in other populations of patients with chronic HCV) to be associated with poorer outcome in terms of SVR. We have attempted to address this by including sensitivity analyses adopting a lower probability of SVR in patients with cirrhosis.
Quality of life/health-state utility weights in the model were taken from reports on a multicentre trial, which recruited treatment-naive patients with mild HCV and this was supplemented by an observational study recruiting patients with compensated and decompensated cirrhosis. It is not clear how applicable these QoL weights are to some of the subgroups of patients in the current assessment – re-treated patients are likely to be older while QoL assessments for mono-infected patients may not be directly applicable to those with HCV/HIV co-infection. Similarly, the health-state costs included in the model were developed in an observational study conducted alongside the UK Mild Hepatitis C Trial,81,82 whereas intervention costs were based on treatment protocols developed as part of our previous assessment17 in collaboration with UK clinical experts, and valued using reference costs from an NHS Hospital Trust. It is not clear how adequately the treatment protocols may capture the complexity of managing patients with HCV/HIV co-infection – the sensitivity of the cost-effectiveness results to the costs of managing antiviral treatment in this group of patients was addressed in a sensitivity analysis.
There is very limited information on the baseline characteristics of patients undergoing treatment for chronic HCV. We found no information on characteristics for patients in the relevant subgroups and have used baseline characteristics from our previous assessment and a small audit undertaken in a London teaching hospital. Clinical experts for this review regarded these assumptions as reasonable, but this remains an assumption and is not supported by robust evidence.
Chapter 8 Conclusions
Implications for service provision
A recommendation to extend antiviral treatment to patients who did not respond to, or who relapsed from, a previous course of peginterferon alfa and ribavirin combination therapy may increase the number of eligible patients in some areas, with resultant budget implications for primary care trusts and increased use of hepatology services.
For patients co-infected with HCV/HIV, there would be implications for the availability of resources and a need for HIV specialists to work closely with hepatitis specialists. The complexity of this joint management is probably achievable in many tertiary centres but may pose some difficulties for isolated centres. Furthermore, the reality of provision of joint clinics, and other aspects of joint management, could pose significant logistical challenges for service managers, particularly if the dominance of one disease specialist in a patient’s care is to be avoided and a more holistic approach adopted.
Suggested research priorities
Further RCTs are required to assess the clinical effectiveness of re-treating people who have not responded to, or have relapsed following, a previous course of peginterferon alfa. Trials of new pharmacological agents should be conducted, particularly for patients in whom re-treatment with a subsequent course of peginterferon alfa is not successful in terms of achieving undetectable levels of virus. It is important to increase the number of treatment options for this group, as currently there are no other licensed agents available. Phase II and III trials are currently in progress, evaluating the safety and efficacy of protease inhibitors for chronic HCV, which can be used in combination with peginterferon alfa in both treatment-naive and treatment-experienced patients, such as telaprevir and boceprevir. In Phase III development is the nucleoside analogue taribavirin (a prodrug of ribavirin), which is being evaluated for use in combination with peginterferon alfa. Also being trialled is albinterferon alfa-2b, a genetic fusion of human albumin and interferon, which can be administered via injection every 2 weeks, in contrast to peginterferon alfa, which is given once a week. New agents such as these, once licensed, may be eligible for appraisal by NICE so that guidance can be issued to the NHS on their use.
Acknowledgements
We would like to thank members of our advisory group panel who provided expert advice and comments on the protocol and a draft of this report:
-
Dr Steve Ryder, Consultant Hepatologist, Nottingham University Hospitals NHS Trust, Nottingham.
-
Professor Will Irving, Professor of Virology, Queen’s Medical Centre, Nottingham.
-
Dr Kathryn Nash, Consultant Hepatologist, Southampton General Hospital, Southampton.
-
Dr Sanjay Bhagani, Consultant in Infectious Diseases and HIV Medicine, Royal Free Hospital, London. [Dr Bhagani has received speaker’s fees and conference attendance support and has participated on advisory boards for Roche and Schering-Plough.]
-
Dr Kirsten Major, Chief Economist, NHS North West, Manchester.
-
Mr Richard Denny, Patient Advocate, Hepatitis C Trust, London. [The Hepatitis C Trust has received funding from Schering-Plough and Roche.]
We are also grateful to Karen Welch, Information Scientist, SHTAC, University of Southampton, for generating and running the literature searches and Emma Loveman, Senior Research Fellow, SHTAC, University of Southampton, for reviewing a draft of this report.
Contribution of authors
D Hartwell (Research Fellow) developed the research protocol, contributed to the background section, assisted in the development of the search strategy, assessed studies for inclusion, extracted data from and quality assessed included studies, synthesised evidence, drafted and edited the final report, and project managed the study.
J Jones (Principal Research Fellow) developed the research protocol, assessed studies for inclusion, extracted data from and quality assessed included studies, synthesised evidence, developed the economic evaluation and drafted the report.
L Baxter (Research Fellow) assessed studies for inclusion, extracted data from and quality assessed included studies, synthesised evidence, assisted in developing the economic evaluation and drafted the report.
J Shepherd (Principal Research Fellow) contributed to developing the research protocol, drafted the background section, assessed studies for inclusion, extracted data from and quality assessed included studies, synthesised evidence and drafted the report.
Disclaimers
The views expressed in this publication are those of the authors and not necessarily those of the HTA programme or the Department of Health.
References
- Thomson BJ, Finch RG. Hepatitis C virus infection. Clin Microbiol Infect 2005;11:86-94.
- Flamm SL. Chronic hepatitis C virus infection. JAMA 2003;289:2413-17.
- Health Protection Agency (HPA) . Hepatitis C in the UK: 2008 Report 2008.
- Booth J, O’Grady J, Neuberger J. on behalf of the Royal College of Physicians of London and the British Society of Gastroenterology . Clinical guidelines on the management of hepatitis C. Gut 2001;49:1-21.
- Hoofnagle JH. Hepatitis C: the clinical spectrum of disease. Hepatology 1997;26:S15-20.
- Foster GR, Goldin RD, Thomas HC. Chronic hepatitis C virus infection causes a significant reduction in quality of life in the absence of cirrhosis. Hepatology 1998;27:209-12.
- Giraudon I, Ruf M, Maguire H, Charlett A, Ncube F, Turner J, et al. Increase in diagnosed newly acquired hepatitis C in HIV-positive men who have sex with men across London and Brighton, 2002–2006: is this an outbreak?. Sex Transm Infect 2008;84:111-15.
- Global Burden Of Hepatitis C Working Group . Global burden of disease (GBD) for hepatitis C. J Clin Pharmacol 2004;44:20-9.
- Health Protection Agency Centre for Infections, Health Protection Scotland, National Public Health Service for Wales, Communicable Disease Surveillance Centre, Northern Ireland, Centre for Research on Drugs & Health Behaviour, London School of Hygiene & Tropical Medicine . Shooting Up: Infections Among Injecting Drug Users in the United Kingdom 2006 2007.
- Mohsen A, Easterbrook P, Taylor C, Norris S. Hepatitis C and HIV-1 coinfection. Gut 2002;51:601-8.
- Pol S, Soriano V. Management of chronic hepatitis C virus infection in HIV-infected patients. Clin Infect Dis 2008;47:94-101.
- Thomson EC, Main J. Epidemiology of hepatitis C virus infection in HIV-infected individuals. J Viral Hepat 2008;15:773-81.
- Desmet VJ. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. J Hepatol 2003;38:382-6.
- Knodell RG, Ishak KG, Black WC, Chen TS, Craig R, Kaplowitz N, et al. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. Hepatology 1981;1:431-5.
- Ishak K, Baptista A, Bianchi L, Callea F, De Groote J, Gudat F, et al. Histological grading and staging of chronic hepatitis. J Hepatol 1995;22:696-9.
- Bedossa P, Poynard T. An algorithm for the grading of activity in chronic hepatitis C. The METAVIR Cooperative Study Group. Hepatology 1996;24:289-93.
- Shepherd J, Jones J, Hartwell D, Davidson P, Price A, Waugh N. Interferon alfa (pegylated and non-pegylated) and ribavirin for the treatment of mild chronic hepatitis C: a systematic review and economic evaluation. Health Technol Assess 2007;11.
- Sweeting MJ, De Angelis D, Neal KR, Ramsay ME, Irving WL, Wright M, et al. Estimated progression rates in three United Kingdom hepatitis C cohorts differed according to method of recruitment. J Clin Epidemiol 2006;59:144-52.
- Alberti A, Vario A, Ferrari A, Pistis R. Review article: chronic hepatitis C – natural history and co-factors. Aliment Pharmacol Ther 2005;22:74-8.
- Sweeting MJ, De Angelis D, Brant LJ, Harris HE, Mann AG, Ramsay ME. The burden of hepatitis C in England. J Viral Hepat 2007;14:570-6.
- Di Bisceglie AM. Hepatitis C. Lancet 1998;351:351-5.
- Poynard T, Ratziu V, Charlotte F, Goodman Z, McHutchison J, Albrecht J. Rates and risk factors of liver fibrosis progression in patients with chronic hepatitis C. J Hepatol 2001;34:730-9.
- Martinez-Sierra C, Arizcorreta A, Diaz F, Roldan R, Martin-Herrera L, Perez-Guzman E, et al. Progression of chronic hepatitis C to liver fibrosis and cirrhosis in patients coinfected with hepatitis C virus and human immunodeficiency virus. Clin Infect Dis 2003;36:491-8.
- Martin-Carbonero L, Benhamou Y, Puoti M, Berenguer J, Mallolas J, Quereda C, et al. Incidence and predictors of severe liver fibrosis in human immunodeficiency virus-infected patients with chronic hepatitis C: a European collaborative study. Clin Infect Dis 2004;38:128-33.
- Graham CS, Baden LR, Yu E, Mrus JM, Carnie J, Heeren T, et al. Influence of human immunodeficiency virus infection on the course of hepatitis C virus infection: a meta-analysis. Clin Infect Dis 2001;33:562-9.
- Mohsen AH, Easterbrook PJ, Taylor C, Portmann B, Kulasegaram R, Murad S, et al. Impact of human immunodeficiency virus (HIV) infection on the progression of liver fibrosis in hepatitis C virus infected patients. Gut 2003;52:1035-40.
- Poynard T, Mathurin P, Lai CL, Guyader D, Poupon R, Tainturier MH, et al. A comparison of fibrosis progression in chronic liver diseases. J Hepatol 2003;38:257-65.
- Thein HH, Yi Q, Dore GJ, Krahn MD. Natural history of hepatitis C virus infection in HIV-infected individuals and the impact of HIV in the era of highly active antiretroviral therapy: a meta-analysis. AIDS 2008;22:1979-91.
- The Antiretroviral Therapy Cohort Collaboration . Life expectancy of individuals on combination antiretroviral therapy in high-income countries: a collaborative analysis of 14 cohort studies. Lancet 2008;372:293-9.
- Macias J, Melguizo I, Fernandez-Rivera FJ, Garcia-Garcia A, Mira JA, Ramos AJ, et al. Mortality due to liver failure and impact on survival of hepatitis virus infections in HIV-infected patients receiving potent antiretroviral therapy. Eur J Clin Microbiol Infect Dis 2002;21:775-81.
- Mocroft A, Soriano V, Rockstroh J, Reiss P, Kirk O, De WS, et al. Is there evidence for an increase in the death rate from liver-related disease in patients with HIV?. AIDS 2005;19:2117-25.
- Scottish Intercollegiate Guidelines Network (SIGN) . Management of Hepatitis C 2006.
- National Institute for Health and Clinical Excellence (NICE) . Peginterferon Alfa and Ribavirin for the Treatment of Mild Chronic Hepatitis C 2006.
- Veldt BJ, Saracco G, Boyer N, Camma C, Bellobuono A, Hopf U, et al. Long term clinical outcome of chronic hepatitis C patients with sustained virological response to interferon monotherapy. Gut 2004;53:1504-8.
- Chander G, Sulkowski MS, Jenckes MW, Torbenson MS, Herlong HF, Bass EB, et al. Treatment of chronic hepatitis C: a systematic review. Hepatology 2002;36:S135-44.
- Yoshida H, Arakawa Y, Sata M, Nishiguchi S, Yano M, Fujiyama S, et al. Interferon therapy prolonged life expectancy among chronic hepatitis C patients. Gastroenterol 2002;123:483-91.
- Gluud C, Brok J, Gong Y, Koretz RL. Hepatology may have problems with putative surrogate outcome measures. J Hepatol 2007;46:734-42.
- National Institute for Clinical Excellence (NICE) . Interferon Alfa (pegylated and Non-Pegylated) and Ribavirin for the Treatment of Chronic Hepatitis C 2004.
- Davis GL. Monitoring of viral levels during therapy of hepatitis C. Hepatology 2002;36:S145-51.
- Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 2001;358:958-65.
- Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Goncales FL, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med 2002;347:975-82.
- Roche UK . Summary of Product Characteristics – Peginterferon Alfa-2a (Pegasys) 2009. http://emc.medicines.org.uk/medicine/10081/SPC/Pegasys±135mcg±and±180mcg±±solution±for±injection±in±Pre-filled±Syringe/ (accessed 18 May 2009).
- Schering-Plough . Summary of Product Characteristics – Peginterferon Alfa-2b (ViraferonPeg) 2009. http://emc.medicines.org.uk/medicine/10321/SPC/ViraferonPeg±Pen±50%2c±80%2c±100%2c±120±or±150±micrograms±±powder±and±solvent±for±solution±for±injection±in±pre-filled±pen/ (accessed 18 May 2009).
- Shepherd J, Brodin H, Cave C, Waugh N, Price A, Gabbay J. Pegylated interferon α-2a and -2b in combination with ribavirin in the treatment of chronic hepatitis C: a systematic review and economic evaluation. Health Technol Assess 2004;8.
- Rockstroh JK, Bhagani S, Benhamou Y, Bruno R, Mauss S, Peters L, et al. European AIDS Clinical Society (EACS) guidelines for the clinical management and treatment of chronic hepatitis B and C coinfection in HIV-infected adults. HIV Med 2008;9:82-8.
- Nelson M, Matthews G, Brook MG, Main J, Bateman P, Gilson R, et al. BHIVA guidelines on HIV and chronic hepatitis: coinfection with HIV and hepatitis C virus infection (2005). HIV Med 2005;6:96-106.
- British Medical Association and Royal Pharmaceutical Society of Great Britain . British National Formulary. 2009.
- Hartwell D, Shepherd J. Pegylated and non-pegylated interferon-alfa and ribavirin for the treatment of mild chronic hepatitis C: a systematic review and meta-analysis. Int J Technol Assess Health Care 2009;25:56-62.
- Centre for Reviews and Dissemination (CRD) . Systematic Reviews: CRD’s Guidance for Undertaking Reviews in Health Care 2009.
- Zhao S, Cheng D, Liu E, Yu H, Yang H, Xue X, et al. Peginterferon vs. interferon in the treatment of different HCV genotype infections in HIV patients. Eur J Clin Microbiol Infect Dis 2008;27:1183-92.
- Kim AI, Dorn A, Bouajram R, Saab S. The treatment of chronic hepatitis C in HIV-infected patients: a meta-analysis. HIV Med 2007;8:312-21.
- Mangia A, Minerva N, Bacca D, Cozzolongo R, Ricci GL, Carretta V, et al. Individualized treatment duration for hepatitis C genotype 1 patients: a randomized controlled trial. Hepatology 2008;47:43-50.
- Liu C-H, Liu C-J, Lin C, Liang C-C, Hsu S-J, Yang S-S, et al. Pegylated interferon-alpha-2a plus ribavirin for treatment-naive Asian patients with hepatitis C virus genotype 1 infection: a multicenter, randomized controlled trial. Clin Infect Dis 2008;47:1260-9.
- Yu ML, Dai CY, Huang JF, Chiu CF, Yang YH, Hou NJ, et al. Rapid virological response and treatment duration for chronic hepatitis C genotype 1 patients: a randomized trial. Hepatology 2008;47:1884-93.
- Yu ML, Dai CY, Huang JF, Hou NJ, Lee LP, Hsieh MY, et al. A randomised study of peginterferon and ribavirin for 16 versus 24 weeks in patients with genotype 2 chronic hepatitis C. Gut 2007;56:553-9.
- von Wagner M, Huber M, Berg T, Hinrichsen H, Rasenack J, Heintges T, et al. Peginterferon-alpha-2a (40KD) and ribavirin for 16 or 24 weeks in patients with genotype 2 or 3 chronic hepatitis C. Gastroenterology 2005;129:522-7.
- Liu CH, Liu CJ, Lin CL, Liang CC, Hsu SJ, Yang SS, et al. A randomized trial of 24- versus 48-week peginterferon alfa-2A plus ribavirin for treatment-naive Taiwanese patients with hepatitis C virus genotype 1 infection. Hepatology 2008;48.
- Yu M, Dai C, Huang J, Lee L, Hsieh M, Chiu C, et al. High chance of cure in HCV genotype 1 patients with a low viral load achieving an RVR treated for 24 wks with peginterferon alfa-2a (PEGASYS®) plus ribavirin (COPEGUS®): prospective, randomized, controlled study comparing 24 and 48 weeks of treatment. Hepatology 2007;46:822-3A.
- Berg T, Weich V, Teuber G, Klinker H, Moller B, Rasenack J, et al. Individualized treatment strategy according to early viral kinetics in hepatitis C virus type 1-infected patients. Hepatology 2009;50:369-77.
- Younossi ZM, Nader FH, Bai C, Sjogren R, Ong JP, Collantes R, et al. A phase II dose finding study of darbepoetin alpha and filgrastim for the management of anaemia and neutropenia in chronic hepatitis C treatment. J Viral Hepat 2008;15:370-8.
- National Institute for Health and Clinical Excellence (NICE) . Peginterferon Alfa and Ribavirin for the Treatment of Chronic Hepatitis C (part-Review of TA75 and TA106): Final Scope n.d. www.nice.org.uk/guidance/index.jsp?action=download%26o=44230 (accessed May 2009).
- Roche UK . Summary of Product Characteristics – Ribavirin (Copegus) 2009. http://emc.medicines.org.uk/medicine/11755/SPC/Copegus±200mg/ (accessed 18 May 2009).
- Nguyen-Khac E, Capron D, Castelain S, Francois C, Braillon A. Personalized therapy for chronic viral hepatitis C in the naive patient: how can we optimize treatment duration as a function of viral genotype?. Eur J Inter Med 2007;18:510-15.
- Kuehne FC, Bethe U, Freedberg K, Goldie SJ. Treatment for hepatitis C virus in human immunodeficiency virus-infected patients: clinical benefits and cost-effectiveness. Arch Intern Med 2002;162:2545-56.
- Campos NG, Salomon JA, Servoss JC, Nunes DP, Samet JH, Freedberg KA, et al. Cost-effectiveness of treatment for hepatitis C in an urban cohort co-infected with HIV. Am J Med 2007;120:272-9.
- Torriani FJ, Rodriguez-Torres M, Rockstroh JK, Lissen E, Gonzalez-Garcia J, Lazzarin A, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection in HIV-infected patients. N Engl J Med 2004;351:438-50.
- Drummond MF, Sculpher MJ, O’Brien BJ, Stoddart GL, Torrance GW. Methods for the economic evaluation of health care programmes. Oxford: Oxford University Press; 2005.
- National Institute for Health and Clinical Excellence (NICE) . Guide to the Methods of Technology Appraisal 2008. www.nice.org.uk/media/B52/A7/TAMethodsGuideUpdatedJune2008.pdf (accessed August 2009).
- Philips Z, Ginnelly L, Sculpher M, Claxton K, Golder S, Riemsma R, et al. Review of guidelines for good practice in decision-analytic modelling in health technology assessment. Health Technol Assess 2004;8.
- Soto B, Sanchez-Quijano A, Rodrigo L. Human immunodeficiency virus infection modifies the natural history of chronic parenterally acquired hepatitis C with an unusually rapid progression to cirrhosis. J Hepatol 1997;26:1-5.
- Di Martino V, Rufat P, Boyer N. The influence of human immunodeficiency virus coinfection on chronic hepatitis C in injection drug users: a long-term retrospective cohort study. Hepatology 2001;34:1193-9.
- Schackman BR, Goldie SJ, Freedberg KA, Losina E, Brazier J, Weinstein MC. Comparison of health state utilities using community and patient preference weights derived from a survey of patients with HIV/AIDS. Med Decis Making 2002;22:27-38.
- Bozzette, S, Berry, SH, Duan N, Frankel MR, Leibowitz AA, Lefkowitz D, et al. for the HIV Cost and Services Utilization Study Consortium. The care of HIV-infected adults in the United States. N Engl J Med 1998;339:1897-904.
- Multiattribute utility function for a comprehensive health status classification system: Health Utilities Index Mark 2. Med Care 1996;34:702-22.
- Brazier J, Usherwood T, Harper R, Thomas K. Deriving a preference-based single index from the UK SF-36 Health Survey. J Clin Epidemiol 1998;51:1115-28.
- Bennett WG, Inoue Y, Beck JR, Wong JB, Pauker SG, Davis GL. Estimates of the cost-effectiveness of a single course of interferon-alpha 2b in patients with histologically mild chronic hepatitis C. Ann Intern Med 1997;127:855-65.
- Davis GL, Beck JR, Farrell G, Poynard T. Prolonged treatment with interferon in patients with histologically mild chronic hepatitis C: a decision analysis. J Viral Hepat 1998;5:313-21.
- Wong JB, Koff RS. Watchful waiting with periodic liver biopsy versus immediate empirical therapy for histologically mild chronic hepatitis C: a cost-effectiveness analysis. Ann Intern Med 2000;133:665-75.
- Salomon JA, Weinstein MC, Hammitt JK, Goldie SJ. Cost-effectiveness of treatment for chronic hepatitis C infection in an evolving patient population. JAMA 2003;290:228-37.
- Grieve R, Roberts J. Economic evaluation for hepatitis C. Acta Gastroenterol Belg 2002;65:104-9.
- Grieve R, Roberts J, Wright M, Sweeting M, Deagelis D, Rosenberg W, et al. Cost-effectiveness of interferon alpha or peginterferon alpha with ribavirin for histologically mild chronic hepatitis C. Gut 2006;55:1332-8.
- Wright M, Grieve R, Roberts J, Main J, Thomas HC. UK Mild Hepatitis C Trial Investigators . Health benefits of antiviral therapy for mild chronic hepatitis C: randomised controlled trial and economic evaluation. Health Technol Assess 2006;10.
- Kind P, Hardman G, Macran S. UK Population norms for EQ-5D. York: Centre for Health Economics, University of York; 1999.
- Dolan P, Gudex C, Kind P, Williams A. A social tariff for EuroQol: results from a UK general population survey. York: Centre for Health Economics, University of York; 1995.
- Curtis L. Unit costs of health and social care. Canterbury: Personal Social Services Research Unit, University of Kent; 2008.
- Stinnett AA, Mullahy J. Net health benefits: a new framework for the analysis of uncertainty in cost-effectiveness analysis. Med Dec Making 2009;18:S68-80.
- Bruno S, Stroffolini T, Colombo M, Bollani S, Benvegnù L, Mazzella G, et al. Sustained virological response to interferon-alpha is associated with improved outcome in HCV-related cirrhosis: a retrospective study. Hepatology 2007;45:579-87.
- Jensen DM, Marcellin P, Freilich B, Andreone P, Di BA, Brandao-Mello CE, et al. Re-treatment of patients with chronic hepatitis C who do not respond to peginterferon-alpha2b: a randomized trial. Ann Intern Med 2009;150:528-40.
- Berg C, Goncales FL, Bernstein DE, Sette H, Rasenack J, Diago M, et al. Re-treatment of chronic hepatitis C patients after relapse: efficacy of peginterferon-alpha-2a (40 kDa) and ribavirin. J Viral Hepat 2006;13:435-40.
- Shiffman ML, Suter F, Bacon BR, Nelson D, Harley H, Sola R, et al. Peginterferon alfa-2a and ribavirin for 16 or 24 weeks in HCV genotype 2 or 3. N Engl J Med 2007;357:124-34.
- Hadziyannis SJ, Sette H, Morgan TR, Balan V, Diago M, Marcellin P, et al. Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: a randomized study of treatment duration and ribavirin dose. Ann Intern Med 2004;140:346-55.
- Schering-Plough Ltd . Pegylated Interferon Alfa-2b and Ribavirin for Hepatitis C: Re-Treatment of Patients Previously Failing Therapy and Treatment of HIV HCV Co-Infected Patients 2009.
- Ratcliffe J, Longworth L, Young T, Bryan S, Burroughs A, Buxton M, et al. Assessing health-related quality of life pre- and post-liver transplantation: a prospective multicenter study. Liver Transpl 2002;8:263-70.
- Laguno M, Cifuentes C, Murillas J, Veloso S, Larrousse M, Payeras A, et al. Randomized trial comparing pegylated interferon alpha-2b versus pegylated interferon alpha-2a, both plus ribavirin, to treat chronic hepatitis C in human immunodeficiency virus patients. Hepatology 2009;49:22-31.
- Laguno M, Murillas J, Blanco JL, Martinez E, Miquel R, Sanchez-Tapias JM, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for treatment of HIV/HCV co-infected patients. AIDS 2004;18:F27-36.
- Poynard T, Schiff E, Terg R, Moreno Otero R, Flamm S, Schmidt W, et al. Sustained Viral Response (SVR) Is Dependent on Baseline Characteristics in the Re-Treatment of Previous Alfa Interferon Ribavirin (I R) Nonresponders (NR): Final Results from the EPIC3 Program n.d.
- Scotto G, Fazio V, Fornabaio C, Tartaglia A, Di TR, Saracino A, et al. Peg-interferon alpha-2a versus Peg-interferon alpha-2b in nonresponders with HCV active chronic hepatitis: a pilot study. J Interferon Cytokine Res 2008;28:623-9.
- Fattovich G, Giustina G, Degos F, Tremolada F, Diodati G, Almasio P, et al. Morbidity and mortality in compensated cirrhosis type C: a retrospective follow-up study of 384 patients. Gastroenterol 1997;112:463-72.
- Sonnenberg FA, Gregory P, Yomtovian R, Russell LB, Tierney W, Kosmin M, et al. The cost-effectiveness of autologous transfusion revisited: implications of an increased risk of bacterial infection with allogeneic transfusion. Transfusion 1999;39:808-7.
- Boring CC, Squires TS, Tong T. Cancer statistics, 1993. CA Cancer J Clin 1993;43:7-26.
- Mohsen AH. Trent HCV Study Group . The epidemiology of hepatitis C in a UK health regional population of 5.12 million. Gut 2001;48:707-13.
- Foster GR, Goldin RD, Main J, Murray-Lyon I, Hargreaves S, Thomas HC. Management of chronic hepatitis C: clinical audit of biopsy based management algorithm. BMJ 1997;315:453-8.
- Siebert U, Sroczynski G, Rossol S, Wasem J, Ravens-Sieberer U, Kurth BM, et al. Cost-effectiveness of peginterferon alpha-2b plus ribavirin versus interferon alpha-2b plus ribavirin for initial treatment of chronic hepatitis C. Gut 2003;52:425-32.
- Roche . Pegasys (peginterferon Alfa-2a) and Ribavirin for the Treatment of Chronic Hepatitis C (part-Review of TA75 and TA106) 2009.
- Longworth L, Young T, Ratcliffe J, Bryan S, Buxton M. Economic evaluation of the transplantation programme in England and Wales: an assessment of the costs of liver transplantation. Unpublished report to the Department of Health; 2001.
- NHS Centre for Reviews and Dissemination (CRD) . NHS Economic Evaluation Database Handbook n.d. www.york.ac.uk/inst/crd/pdf/nhseed-handb07.pdf (accessed 20 August 2008).
- Shiffman ML, Di Bisceglie AM, Lindsay KL, Morishima C, Wright EC, Everson GT, et al. Peginterferon alfa-2a and ribavirin in patients with chronic hepatitis C who have failed prior treatment. Gastroenterology 2004;126:1015-23.
- Everson GT, Hoefs JC, Seeff LB, Bonkovsky HL, Naishadham D, Shiffman ML, et al. Impact of disease severity on outcome of antiviral therapy for chronic hepatitis C: Lessons from the HALT-C trial. Hepatology 2006;44:1675-84.
- Shiffman ML, Mihas AA, Millwala F, Sterling RK, Luketic VA, Stravitz RT, et al. Treatment of chronic hepatitis C virus in African Americans with genotypes 2 and 3. Am J Gastroenterol 2007;102:761-6.
- Yoshida EM, Sherman M, Bain VG, Cooper CL, Deschenes M, Marotta PJ, et al. Retreatment with pegylated interferon alpha-2a and ribavirin in patients with chronic hepatitis C who have relapsed or not responded to a first course of pegylated interferon-based therapy. Can J Gastroenterol 2009;23:180-4.
- Parise ER, de Oliveira AC, Conceicao RDO, Amaral AC, Leite K. Response to treatment with interferon-alpha and ribavirin in patients with chronic hepatitis C virus genotypes 2 and 3 depends on the degree of hepatic fibrosis. Braz J Infect Dis 2006;10:78-81.
- Zeuzem S, Pawlotsky JM, Lukasiewicz E, von WM, Goulis I, Lurie Y, et al. International, multicenter, randomized, controlled study comparing dynamically individualized versus standard treatment in patients with chronic hepatitis C. J Hepatol 2005;43:250-7.
- Jensen DM, Morgan TR, Marcellin P, Pockros PJ, Reddy KR, Hadziyannis SJ, et al. Early identification of HCV genotype 1 patients responding to 24 weeks peginterferon alpha-2a (40 kd)/ribavirin therapy. Hepatology 2006;43:954-60.
- Ferenci P, Fried MW, Shiffman ML, Smith CI, Marinos G, Goncales FL, et al. Predicting sustained virological responses in chronic hepatitis C patients treated with peginterferon alfa-2a (40 KD)/ribavirin. J Hepatol 2005;43:425-33.
- Ferenci P, Laferl H, Scherzer TM, Gschwantler M, Maieron A, Brunner H, et al. Peginterferon alfa-2a and ribavirin for 24 weeks in hepatitis C type 1 and 4 patients with rapid virological response. Gastroenterology 2008;135:451-8.
- Carrat F, Bani-Sadr F, Pol S, Rosenthal E, Lunel-Fabiani F, Benzekri A, et al. Pegylated interferon alfa-2b vs standard interferon alfa-2b, plus ribavirin, for chronic hepatitis C in HIV-infected patients: a randomized controlled trial. JAMA 2004;292:2839-48.
- Pol S, Carrat F, Bani-Sadr F, Rosenthal E, Lunel F, Morand P, et al. Final results of ANRS HC02-ribavic: A randomized controlled trial of pegylated-interferon alpha-2b plus ribavirin vs interferon alpha-2b plus ribavirin for naive HCV-HIV co- infected patients. J Hepatol 2005;42:10-1.
- Chung RT, Andersen J, Volberding P, Robbins GK, Liu T, Sherman KE, et al. Peginterferon Alfa-2a plus ribavirin versus interferon alfa-2a plus ribavirin for chronic hepatitis C in HIV-coinfected persons. N Engl J Med 2004;351:451-9.
- Crespo M, Sauleda S, Esteban JI, Juarez A, Ribera E, Andreu AL, et al. Peginterferon alpha-2b plus ribavirin vs interferon alpha-2b plus ribavirin for chronic hepatitis C in HIV-coinfected patients. J Viral Hepat 2007;14:228-38.
- Cargnel A, Angeli E, Mainini A, Gubertini G, Giorgi R, Schiavini M, et al. Open, randomized, multicentre Italian trial on PEG-IFN plus ribavirin versus PEG-IFN monotherapy for chronic hepatitis C in HIV-coinfected patients on HAART. Antivir Ther 2005;10:309-17.
- Briggs A, Claxton K, Sculpher M. Decision modelling for health economic evaluation. Handbooks in Health Economic Evaluation. Oxford: Oxford University Press; 2006.
Appendix 1 Methods from research protocol
Title of the project
Peginterferon alfa and ribavirin for the treatment of chronic hepatitis C (part review of TA75 and TA106).
Report methods for synthesis of evidence of clinical effectiveness
A review of the evidence for clinical effectiveness and cost-effectiveness will be undertaken systematically following the general principles outlined in the Centre for Reviews and Dissemination (CRD) guidance for undertaking reviews in health care. 49
Search strategy
A search strategy will be developed and tested by an experienced information scientist. The strategy will be designed to identify: (1) clinical effectiveness studies reporting on comparisons between peginterferon and ribavirin combination therapy (or peginterferon monotherapy for those who cannot tolerate ribavirin) and best supportive care (BSC) or standard-duration courses of peginterferon/ribavirin (as described in Inclusion and exclusion criteria); and (2) studies reporting on the cost-effectiveness of peginterferon and ribavirin, and the relative comparisons. The search strategy will also identify studies reporting resource use and costs, epidemiology and natural history.
The following electronic databases will be searched: the Cochrane Library, including the Cochrane Database of Systematic Reviews (CDSR) and the Cochrane Central Register of Controlled Trials (CENTRAL); Centre for Reviews and Dissemination (CRD, University of York); Database of Abstracts of Reviews of Effects (DARE); the NHS Economic Evaluation Database (NHS EED); the Health Technology Assessment (HTA) database; MEDLINE (Ovid); EMBASE (Ovid); PREMEDLINE In-Process & Other Non-Indexed Citations (Ovid); Web of Science with Conference Proceedings: Science Citation Index Expanded (SCIE) and Conference Proceedings Citation Index – Science (CPCI-S) (ISI Web of Knowledge); Biosis Previews (ISI Web of Knowledge); NIHR-Clinical Research Network Portfolio; ClinicalTrials.gov; and Current Controlled Trials. Relevant hepatitis C symposia will also be searched. The draft search strategy for MEDLINE will be adapted for other databases.
Bibliographies of related papers will be assessed for relevant studies where possible. The manufacturers’ submissions to the National Institute for Health and Clinical Excellence (NICE) will be assessed for any additional studies that meet the inclusion criteria. Experts will be contacted to identify additional published and unpublished evidence.
Literature searches will be carried out from April 2007 (the date the most recent search was conducted48 to the present and will be limited to randomised controlled trials (RCTs) and the English language (note: the search will incorporate the references identified in our previous technology assessment reports17,44 in which literature searching extended back to the year 2000; these references will be rescreened according to the inclusion criteria for the current assessment). For the cost-effectiveness assessment, searches for other evidence to inform cost-effectiveness modelling will be conducted as required and may include a wider range of study types (including non-randomised studies). All searches will be updated when the draft report is under review, prior to submission of the final report.
Inclusion and exclusion criteria
The following criteria are those stipulated in the final scope issued by NICE. 61
Population
Adults with chronic hepatitis C virus (HCV) infection, restricted to:
-
people who have been previously treated with peginterferon alfa and ribavirin in combination but who relapsed/did not respond
-
people who meet the criteria within the marketing authorisation for receiving shortened courses of peginterferon alfa and ribavirin in combination, namely:
-
– patients with genotype 2 or 3 with a low viral load (LVL) at the start of treatment and a rapid viral response (defined as HCV RNA undetectable by week 4)*
-
– patients with genotype 1 with a LVL and a rapid viral response (defined as HCV RNA undetectable by week 4 and at week 24)
-
– patients with genotype 4 and a rapid viral response
-
-
people with HCV/HIV co-infection.
The subgroups are not mutually exclusive.
(*Applies only to peginterferon alfa-2a.)
Intervention
-
Combination therapy comprising ribavirin and either peginterferon alfa-2a or peginterferon alfa-2b.
-
Peginterferon alfa-2a or peginterferon alfa-2b monotherapy (for patients who are unable to tolerate or are contraindicated to ribavirin).
Comparators
For patients who have been previously treated with combination therapy, and for HCV/HIV co-infected patients:
-
Best supportive care (e.g. symptomatic treatment, monitoring, treatment without any form of interferon therapy).
For patients who meet the criteria for receiving shortened courses of combination therapy:
-
Standard duration courses of peginterferon alfa and ribavirin combination therapy (up to 24 or 48 weeks as appropriate).
Outcomes
Studies must report sustained virological response (SVR) (defined as undetectable HCV RNA at least 6 months after treatment cessation). Studies may also include one or more of the following outcomes:
-
virological response (e.g. during treatment, end of treatment)
-
biochemical response [e.g. alanine aminotransferase (ALT) levels]
-
histological improvement (fibrosis and inflammation)
-
survival
-
adverse effects of treatment
-
health-related quality of life (QoL)
-
cost-effectiveness [incremental cost per quality-adjusted life-year (QALY)].
Types of studies
-
Fully published RCTs will be included.
-
Studies published as abstracts or conference presentations from 2007 onwards will be included only if sufficient details are presented to allow an appraisal of the methodology and the assessment of results to be undertaken.
-
For the systematic review of cost-effectiveness, studies will only be included if they report the results of full economic evaluations [cost-effectiveness analyses (reporting cost per life-year gained), cost–utility analyses or cost–benefit analyses].
-
Systematic reviews will only be used as a source of references.
-
Case series, case studies, narrative reviews, editorials and opinions will not be included.
-
Non-English language studies will be excluded.
Screening and data extraction process
Reference screening
The titles and abstracts of studies identified by the search strategy will be assessed for potential eligibility using the inclusion/exclusion criteria detailed above. This will be performed by one reviewer. Full papers of studies that appear potentially relevant will be requested for further assessment, and these will be screened by one reviewer and checked by a second. Any disagreements will be resolved by discussion, with involvement of a third reviewer where necessary.
Data extraction
Data will be extracted by one reviewer using a standardised data extraction form. Extracted data will be checked by a second reviewer. Discrepancies will be resolved by discussion, with recourse to a third reviewer when necessary.
Quality assessment strategy
The quality of the clinical effectiveness studies will be assessed according to criteria based on that used by the CRD (University of York). 49 Economic evaluations will be assessed using criteria recommended by Drummond and colleagues67 and/or the format recommended and applied in the CRD NHS Economic Evaluation Database (NHS EED) (using principles outlined in the NHS EED Handbook106). For any studies based on decision models we will also make use of the checklist for assessing good practice in decision analytic modelling (Philips and colleagues69). Published studies carried out from the UK NHS and Personal and Social Services (PSS) perspective will be examined in more detail.
The quality of the individual studies will be assessed by one reviewer, and independently checked for agreement by a second reviewer. Any disagreements will be resolved by consensus, and if necessary a third reviewer will be consulted.
Methods of data analysis/synthesis of clinical effectiveness data
Clinical effectiveness data will be synthesised through a narrative review with tabulation of the results of included studies. Where data are of sufficient quality and homogeneity, a meta-analysis of the clinical effectiveness studies will be performed to estimate a summary measure of effect on relevant outcomes. If a meta-analysis is appropriate, it will be performed using Cochrane Review Manager ([sc]revman[/sc] 5) software. Where data allow, clinical effectiveness and cost-effectiveness will be assessed according to patient subgroups (e.g. by genotype, baseline viral load).
Methods for synthesising evidence of cost-effectiveness
Published and submitted economic evaluations
Narrative synthesis, supported by the data extraction tables, will be used to summarise the evidence base from published economic evaluations. Any economic evaluation included in sponsor submissions to NICE will be assessed using the same quality criteria as for published economic evaluations, but will be reported separately.
Economic modelling
Where appropriate, an economic model will be constructed by adapting an existing model or developing a new one using best available evidence. The Markov model developed by the Southampton Health Technology Assessments Centre (SHTAC) for a previous NICE assessment of treatment for mild chronic hepatitis C17 will be reviewed to assess its applicability to the patient subgroups within the scope of the current review. If the model structure is considered appropriate, the model will be further reviewed to determine whether updated parameter estimates for disease progression, health-state utility or resource use/cost are required. All updated parameter estimates will be derived from the best available published literature, NHS sources (including Finance Department at Southampton University Hospitals Trust) and industry submissions, where applicable.
The perspective for the analysis will be that of the NHS and PSS. The incremental cost-effectiveness of the interventions will be estimated in terms of cost per QALY gained, as well as the cost per life-year gained if data permit. Both cost and outcomes will be discounted at 3.5%.
Parameter values for the model will be obtained from relevant research literature, including our own systematic review of clinical effectiveness. Where required parameters are not available from good-quality published studies in the relevant patient group, we may use data from sponsor submissions to NICE or experts’ clinical opinion. Searches for additional information regarding model parameters, patient preferences and other topics will be conducted as required. Sources for parameters will be stated clearly.
Resource use will be specified and valued from the perspective of the NHS and PSS. Cost data will be derived from local sources, extracted from published sources or from sponsor submissions to NICE, as appropriate.
The simulated population will be defined on the basis of the published evidence about the characteristics of UK chronic HCV patients, within the scope of the current review, and the populations for which good-quality clinical effectiveness is available. The base-case results will be presented separately for the subgroups of patients:
-
who have been previously treated with peginterferon alfa and ribavirin in combination and did not respond or responded but relapsed
-
who meet the licensed criteria for receiving shortened courses of combination therapy
-
with HCV/HIV co-infection.
The time horizon for our analysis will initially be governed by the outcomes reported, and the follow-up data available from included clinical trials – we will investigate the feasibility of extrapolating treatment effects beyond the clinical trials.
Methods for estimating QoL
Where presented, QoL information as well as incidence of adverse events and side effects of treatment will be extracted from included RCTs. Adverse effects of treatment that are likely to have a substantial impact on patients’ QoL, will be included in estimates of health-state utility while on treatment. Where QoL data are insufficient to calculate utility estimates, data will be derived from the broader literature or estimated from other sources. Ideally, utility values will be taken from studies that have been based on ‘public’ (as opposed to patient or clinician) preferences elicited using a choice-based method (in accordance with NICE methodological guidance). 68
Analysis of uncertainty
Analysis of uncertainty will focus on cost–utility, assuming the cost per QALY can be estimated. Uncertainty will be explored through one-way sensitivity analysis and, if the data and modelling approach permit, probabilistic sensitivity analysis (PSA). The outputs of PSA will be presented both using plots on the cost-effectiveness plane and cost-effectiveness acceptability curves (CEACs).
Handling the company submission(s)
All data submitted by the manufacturers will be considered if received by the technolgoy assessment report (TAR) team no later than 27 August 2009. Data arriving after this date will not be considered. If the data meet the inclusion criteria for the review, they will be extracted and quality assessed in accordance with the procedures outlined in this protocol. Any economic evaluations included in the company submission, provided it complies with NICE’s guidance on presentation,68 will be assessed for clinical validity, reasonableness of assumptions and appropriateness of the data used in the economic model.
Methods adopted, and incremental cost-effectiveness ratios (ICERs) estimated from models supporting the company submission will be compared with published economic evaluations of peginterferon and ribavirin included in the assessment report and with the results from the Assessment Group’s analysis. Reasons for large discrepancies in estimated ICERs will be explored and, where possible, explained.
Any ‘academic-in-confidence’ data or ‘commercial-in-confidence’ data taken from a company submission will be underlined and highlighted in the assessment report.
Appendix 2 Search strategies
The following strategies were used to search MEDLINE (Ovid) and EMBASE (Ovid) 2007–9 (searches from the previous assessment reports17,44 covered the period 2000–7). The strategies were translated to search the other databases listed in Chapter 3 (Identification of studies).
Clinical effectiveness searches
MEDLINE (Ovid)
-
(hepatitis c or HCV).mp. (35,528)
-
exp Hepatitis C/ (26,263)
-
Hepatitis C, Chronic/ (9982)
-
Hepacivirus/ (12,474)
-
or/1-4 (35,757)
-
Ribavirin/ (4279)
-
(ribavirin or copegus or rebetol).ti,ab,nm. (5452)
-
(peginterferon$or peg-ifn or peg-interferon$or (pegylat$adj3 interferon$) or peg$or (polyethylene glycol adj3 interferon$) or ViraferonPeg or pegintron or Pegasys).mp. (15,918)
-
Interferon Alfa-2a/ (2560)
-
Interferon Alfa-2b/ (3487)
-
Polyethylene Glycols/ (13,117)
-
11 and (9 or 10) (1364)
-
6 or 7 or 8 or 12 (19,230)
-
5 and 13 (4722)
-
limit 14 to (english language and humans and yr=“2007 - 2009”) (1144)
-
(systematic$adj2 review$).mp. (18,692)
-
(systematic$adj2 overview$).mp. (354)
-
meta-analysis/ (18,478)
-
(meta analysis or metaanalysis).ab,pt,ti. (25398)
-
randomized controlled trial.pt. (172,423)
-
Randomized Controlled Trial/ (172,423)
-
random allocation/ (29,335)
-
random*.ti,ab. (310,605)
-
controlled clinical trial.pt. (33,195)
-
Controlled Clinical Trial/ (33,195)
-
randomized controlled trials/ (51,594)
-
Single-Blind Method/ (10,146)
-
Double-Blind Method/ (55,762)
-
((singl$or doubl$or tripl$or trebl$) adj5 (blind$or mask$)).tw. (53,879)
-
exp placebos/ (9753)
-
placebo*.ti,ab. (69,682)
-
exp research design/ (154,706)
-
or/16-32 (520,197)
-
15 and 33 (233)
-
(letter or comment or editorial).pt. (551,934)
-
34 not 35 (225)
-
from 36 keep 1-222 (222)
(222 in search on 20 May 2009; re-ran for strategy on 2 June 2009 – extra three results)
Added in interferon terms on 2 June 2009:
-
38 interferon alpha/ (8733)
-
(interferon alpha or interferon alfa or roferon or intron or viraferon).ti,ab. (27,829)
-
5 and (38 or 39) (4209)
-
limit 40 to (english language and humans and yr=“2007 - 2009”) (520)
-
41 not 36 (438)
-
33 and 42 (6)
-
exp interferon alpha/ (13,975)
-
5 and (39 or 44) (5932)
-
limit 45 to (english language and humans) (5023)
-
33 and 46 (999)
-
limit 47 to yr=“2007 - 2009” (207)
-
(letter or comment or editorial).pt. (551,934)
-
48 not 49 (198)
-
50 not 36 (9)
-
from 36 keep 1-3 (3)
-
51 or 52 (12)
-
from 53 keep 1-12 (12)
EMBASE (Ovid)
-
(hepatitis C or hcv).mp. (40,260)
-
exp Hepatitis C/or exp Hepatitis C virus/ (37,333)
-
1 or 2 (40,260)
-
(peginterferon$or peg-ifn or peg-interferon$or (peg$adj3 interferon$) or (polyethylene glycol adj3 interferon$) or Pegasys or pegintron or viraferonpeg).mp. (5786)
-
peginterferon/or peginterferon alpha2a/or peginterferon alpha2b/ (5285)
-
(interferon alpha or interferon alfa or roferon or intron or viraferon).ti,ab. (25,587)
-
exp Alpha Interferon/ (21,113)
-
Recombinant Alpha2a Interferon/ (1749)
-
Recombinant Alpha2b Interferon/ (2660)
-
interferon/or alpha2a interferon/or alpha2b interferon/or alpha interferon/ (36,974)
-
or/4-10 (58,971)
-
3 and 11 (12,123)
-
limit 12 to (human and english language and yr=“2007 - 2009”) (2516)
-
(systematic$adj2 review$).mp. (35,802)
-
(systematic$adj2 overview$).mp. (341)
-
(meta analy$or metaanaly$).ti,ab,pt. (21,234)
-
exp meta analysis/ (31,882)
-
randomized controlled trial/ (139,490)
-
controlled clinical trial/ (61,251)
-
exp randomization/ (24,841)
-
exp double blind procedure/ (53,393)
-
exp single blind procedure/ (7234)
-
placebo*.tw. (70,462)
-
random*.tw. (295,710)
-
((singl$or doubl$or tripl$or trebl$) adj5 (blind$or mask$)).tw. (55,235)
-
((hand or manual or computer or electronic or database) adj2 search*).ti,ab. (8649)
-
or/14-26 (410,504)
-
13 and 27 (337)
-
(comment or editiorial or letter).pt. (305,933)
-
28 not 29 (334)
-
from 30 keep 1-334 (334)
Cost-effectiveness searches
MEDLINE (Ovid)
-
“hepatitis C” or HCV).mp. (35,682)
-
exp hepatitis C/or Hepatitis C, Chronic/or exp Hepacivirus/ (29,926)
-
or/1-2 (35,914)
-
exp “Costs and Cost Analysis”/ (82,109)
-
exp Cost–benefit Analysis/ (30,108)
-
exp health care costs/ (26,090)
-
Economics, Medical/or Economics, Pharmaceutical/ (2298)
-
(pharmacoeconomic* or pharma economic*).tw. (1724)
-
(cost$adj2 (benefit* or utilit* or minim*)).tw. (9235)
-
(decision adj1 (tree* or analys* or model*)).tw. (3934)
-
Markov Chains/ (4817)
-
Monte Carlo Method/ (10,228)
-
or/4-12 (102,814)
-
3 and 13 (486)
-
limit 14 to (english language and humans and yr=“2007 - 2009”) (76)
-
Ribavirin/ (4308)
-
(ribavirin or copegus or rebetol).ti,ab,nm. (5488)
-
(peginterferon$or peg-ifn or peg-interferon$or (pegylat$adj3 interferon$) or peg$or (polyethylene glycol adj3 interferon$) or ViraferonPeg or pegintron or Pegasys).mp. (16,012)
-
Interferon Alfa-2a/ (2577)
-
Interferon Alfa-2b/ (3506)
-
Polyethylene Glycols/ (13,172)
-
21 and (19 or 20) (1379)
-
((interferon adj1 alpha) or (interferon adj1 alfa)).ti,ab. (11,238)
-
(roferon or intron or viraferon).ti,ab. (18,563)
-
hepatitis c/dt (2820)
-
hepatitis c chronic/dt (4565)
-
or/16-18,22-26 (49,445)
-
15 and 27 (29)
EMBASE (Ovid)
-
(hepatitis C or hcv).mp. (40,384)
-
exp Hepatitis C/or exp Hepatitis C virus/ (37,440)
-
1 or 2 (40,384)
-
(peginterferon$or peg-ifn or peg-interferon$or (peg$adj3 interferon$) or (polyethylene glycol adj3 interferon$) or Pegasys or pegintron or viraferonpeg).mp. (5811)
-
peginterferon/or peginterferon alpha2a/or peginterferon alpha2b/ (5299)
-
(interferon alpha or interferon alfa or roferon or intron or viraferon).ti,ab. (25,641)
-
exp Alpha Interferon/ (21,178)
-
Recombinant Alpha2a Interferon/ (1751)
-
Recombinant Alpha2b Interferon/ (2663)
-
interferon/or alpha2a interferon/or alpha2b interferon/or alpha interferon/ (37,087)
-
or/4-10 (59,136)
-
3 and 11 (12,158)
-
*Economics/ (449)
-
monte carlo method/ (7621)
-
markov.ti,ab. (4291)
-
cost minimization analysis/ (1493)
-
cost of illness/ (5027)
-
cost utility analysis/ (2561)
-
drug cost/ (30,500)
-
economic evaluation/ (4615)
-
pharmacoeconomics/ (870)
-
budget/ (6833)
-
“resource use”.ti,ab. (2058)
-
(cost or economic*).ti. (27,489)
-
*health economics/ (2099)
-
*health care cost/ (7402)
-
or/13-26 (81,064)
-
12 and 27 (326)
-
(cost and effective* and “hepatitis C”).ti. (101)
-
(cost and effective* and “hepatitis C”).ab. (312)
-
11 and (29 or 30) (188)
-
28 or 31 (391)
-
limit 32 to (human and english language and yr=“2007 - 2009”) (66)
-
(letter or editorial).pt. (489,386)
-
33 not 34 (63)
Quality of life searches
MEDLINE (Ovid)
-
value of life/ (1918)
-
quality adjusted life year/ (3675)
-
quality adjusted life.ti,ab. (2613)
-
(qaly$or qald$or qale$or qtime$).ti,ab. (2126)
-
disability adjusted life.ti,ab. (515)
-
daly$.ti,ab. (520)
-
health status indicators/ (10,595)
-
(sf36 or sf 36 or short form 36 or shortform 36 or sf thirtysix or sf thirty six or shortform thirstysix or shortform thirty six or short form thirty six or short form thirtysix or short form thirty six).ti,ab. (8391)
-
(sf6 or sf 6 or short form 6 or shortform 6 or sf six or sfsix or shortform six or short form six).ti,ab. (400)
-
(sf12 or sf 12 or short form 12 or shortform 12 or sf twelve of sftwelve or shortform twelve or short form twelve).ti,ab. (1125)
-
(sf16 or sf 16 or short form 16 or shortform 16 or sf sixteen or sfsixteen or shortform sixteen or short form sixteen).ti,ab. (5)
-
(sf20 or sf 20 or short form 20 or shortform 20 or sf twenty of sftwenty or shortform twenty of short form twenty).ti,ab. (164)
-
(euroqol or euro qol or eq5d or eq 5d).ti,ab. (1471)
-
(hql or hqol or h qol or hrqol or hr qol).ti,ab. (3294)
-
(hye or hyes).ti,ab. (20)
-
health$year$equivalent$.ti,ab. (14)
-
health utilit$.ab. (502)
-
(hui or hui1 or hui2 or hui3).ti,ab. (417)
-
disutil$.ti,ab. (86)
-
rosser.ti,ab. (35)
-
quality of well being.ti,ab. (169)
-
quality of wellbeing.ti,ab. (1)
-
qwb.ti,ab. (99)
-
willingness to pay.ti,ab. (973)
-
standard gamble$.ti,ab. (448)
-
time trade off.ti,ab. (378)
-
time tradeoff.ti,ab. (150)
-
tto.ti,ab. (282)
-
(index adj2 well being).mp. (261)
-
(quality adj2 well being).mp. (468)
-
(health adj3 utilit$ind$).mp. (381)
-
((multiattribute$or multi attribute$) adj3 (health ind$or theor$or health state$or utilit$or analys$)).mp. (109)
-
quality adjusted life year$.mp. (4680)
-
(15D or 15 dimension$).mp. (705)
-
(12D or 12 dimension$).mp. (152)
-
rating scale$.mp. (37,389)
-
linear scal$.mp. (292)
-
linear analog$.mp. (349)
-
visual analog$.mp. (14,997)
-
(categor$adj2 scal$).mp. (595)
-
or/1-40 (81,641)
-
(letter or editorial or comment).pt. (556,056)
-
41 not 42 (79,235)
-
(hepatitis C or hcv).mp. (35,792)
-
exp Hepatitis C/or Hepatitis C, Chronic/or exp Hepacivirus/ (30,013)
-
43 and (44 or 45) (311)
-
limit 46 to (english language and humans and yr=“2007 - 2009”) (72)
-
“quality of life”.ti. (19,254)
-
(“hepatitis C” or HCV or “hepacivurs”).ti. (22,969)
-
48 and 49 (100)
-
limit 50 to (english language and humans and yr=“2007 - 2009”) (26)
-
47 or 51 (80)
EMBASE (Ovid)
-
quality adjusted life year/ (4254)
-
quality adjusted life.ti,ab. (2661)
-
(qaly$or qald$or qale$or qtime$).ti,ab. (2184)
-
disability adjusted life.ti,ab. (472)
-
daly*.ti,ab. (482)
-
(sf36 or sf 36 or short form 36 or shortform 36 or sf thirtysix or sf thirty six or shortform thirtysix or shortform thirty six or short form thirty six or short form thirtysix or short form thirty six).ti,ab. (8281)
-
(sf6 or sf 6 or short form 6 or shortform 6 or sf six or sfsix or shortform six or short form six).ti,ab. (498)
-
(sf12 or sf 12 or short form 12 or shortform 12 or sf twelve or sftwelve or shortform twelve or short form twelve).ti,ab. (1055)
-
(sf16 or sf 16 or short form 16 or shortform 16 or sf sixteen or sfsixteen or shortform sixteen or short form sixteen).ti,ab. (3)
-
(sf20 or sf 20 or short form 20 or shortform 20 or sf twenty or sftwenty or shortform twenty or short form twenty).ti,ab. (145)
-
(euroqol or “euro qol” or “eq5d” or “eq 5d”).ti,ab. (1485)
-
(hql or hqol or “h qol” or hrqol or “hr qol”).ti,ab. (3251)
-
(“hye” or “hyes”).ti,ab. (16)
-
health* year* equivalent*.ti,ab. (16)
-
health utilit*.ti,ab. (525)
-
(hui or hui1 or hui2 or hui3).ti,ab. (378)
-
disutil*.ti,ab. (82)
-
rosser.ti,ab. (31)
-
quality of well being.ti,ab. (161)
-
quality of wellbeing.ti,ab. (5)
-
qwb.ti,ab. (98)
-
willingness to pay.ti,ab. (970)
-
standard gamble*.ti,ab. (430)
-
time trade off.ti,ab. (387)
-
time tradeoff.ti,ab. (140)
-
tto.ti,ab. (299)
-
(index adj2 well being).mp. (258)
-
(quality adj2 well being).mp. (453)
-
(health adj3 util* adj ind*).mp. (389)
-
((multiattribute* or multi attribute*) adj3 (health ind* or theor* or health state* or util* or analys*)).mp. (112)
-
quality adjusted life year*.mp. (4976)
-
health status indicator*.ti,ab. (95)
-
(15D or 15 dimension*).mp. (737)
-
(12D or 12 dimension*).mp. (160)
-
“health related quality of living”.ti,ab. (2)
-
“health related quality of life”.ti,ab. (9742)
-
rating scale*.mp. (58,665)
-
visual analog*.mp. (19,195)
-
(categor* adj scale*).mp. (255)
-
linear scal*.mp. (214)
-
linear analog*.mp. (345)
-
or/1-41 (97,590)
-
(editorial or letter or comment).pt. (491,976)
-
42 not 43 (94,514)
-
exp hepatitis C/or exp hepacivirus/ (37,643)
-
(“Hepatitis C” or HCV).mp. (40,598)
-
44 and (45 or 46) (434)
-
limit 47 to (human and english language and yr=“2007 -Current”) (111)
-
(“quality of life” and (HCV or Hepatitis C or hepacivirus)).ti. (102)
-
limit 49 to (human and english language and yr=“2007 -Current”) (27)
-
48 or 50 (115)
-
from 51 keep 1-115 (115)
Epidemiology searches
MEDLINE (Ovid)
-
*Hepatitis C, Chronic/ep
-
(“hepatitis C” adj4 (incidence or prevalence or epidemiolog* or “natural history”)).ti,ab.
-
((natural* or disease*) adj4 (progres* or course* or histor*)).ti,ab.
-
hepatitis C chronic/
-
3 and 4
-
2 and chronic.ti,ab.
-
1 or 5 or 6
-
limit 7 to (english language and humans and yr=“2007 - 2009”)
EMBASE (Ovid)
-
(“hepatitis C” and (epidemiolog* or incidence or prevalence or statistic*)).ti.
-
limit 1 to (human and english language and yr=“2007 - 2009”)
EMBASE (Ovid) – strategy specifically relating to HCV/HIV co-infection
-
coinfection.tw.
-
co?infection*.tw.
-
(hiv and (hepatitis C or HCV)).ti,ab.
-
3 and (1 or 2)
-
(incidence or prevalence or epidemiol* or “natural history” or rate*).tw.
-
4 and 5
-
limit 6 to (english language and humans and yr=“2005 - 2006”)
-
(mortality or morbidity).tw.
-
4 and 8
-
7 or 9
-
limit 10 to (human and english language and yr=“2005 - 2006”)
-
(co?infection* adj5 (incidence or prevalence or epidemiol* or “natural history” or mortality or morbidy)).tw.
-
3 and 12
-
limit 13 to (human and english language and yr=“2005 - 2006”)
-
(“hepatitis C” adj5 (incidence or prevalence or epidemiol* or “natural history” or mortality or morbidity or survival)).tw.
-
(HCV adj5 (incidence or prevalence or epidemiol* or “natural history” or mortality or morbidity or survival)).tw.
-
15 or 16
-
limit 17 to (human and english language and yr=“2005 - 2006”)
-
(“hepatitis C” or HCV).ti.
-
18 and 19
-
chronic.ti,ab.
-
20 and 21
-
*hepatitis C/ep [Epidemiology]
-
(chronic adj2 “hepatitis C”).ti,ab.
-
23 and 24
-
(“chronic hepatitis C” or “chronic HCV”).ti.
-
(incidence or prevalence or epidemiol* or “natural history” or mortality or morbidity or survival).ti.
-
26 and 27
-
limit 28 to (human and english language and yr=“2005 - 2006”)
-
14 or 29
-
(“chronic hepatitis C” or “chronic HCV”).ab.
-
27 and 31
-
limit 32 to (human and english language and yr=“2005 - 2006”)
-
30 or 33
-
risk factor*.ti,ab.
-
26 and 35
-
limit 36 to (human and english language and yr=“2005 - 2006”)
-
34 or 37
Additional searching
All references of the five included trials were checked to ensure that no eligible studies had been missed.
Appendix 3 SHTAC peer review of clinical effectiveness in the manufacturers’ submissions of peginterferon and ribavirin for chronic hepatitis C
Roche hepatitis C submission to NICE 2009
Summary
-
Manufacturer’s submission (MS) does not present itself as a systematic review.
-
Manufacturer reports a simple EMBASE search using what appear to be free-text terms. No search results presented (in terms of number of hits screened, etc.).
-
No explicit inclusion criteria are used except ‘When possible predominantly data from prospective, randomised, active control studies with good statistical power and similar to UK patient population were considered’ (p. 28). There is no evidence of any systematic process for applying this rule.
-
With the exception of some uncontrolled studies, all of the trials included had active comparators and for the re-treatment and HCV/HIV co-infection patient groups this contravenes the scope of the NICE appraisal.
-
There is no mention of the possibility of conducting an indirect comparison with no active treatment for the re-treatment and HCV/HIV co-infection patient groups.
-
A number of retrospective subgroup analyses are included, some of which appear to have been funded by Roche (and published), and some which are ‘data on file’.
-
Of the six RCTs currently included in the SHTAC systematic review of clinical effectiveness, only two have been included by Roche.
Re-treatment studies
| Study included in MS | Eligible for inclusion in the current SHTAC systematic review of clinical effectiveness? | Study details | Comments | |
|---|---|---|---|---|
| MV17150 REPEAT study, Jensen and colleagues (2009)88 | No |
In total, 942 patients treated, all non-responders to prior PEG Four-arm trial: PEG α-2a, 360 µg/week, for 12 weeks then 180 µg/week to complete 72 weeks (group A) or 48 weeks (group B); or PEG α-2a, 180 µg/week for 72 weeks (group C) or 48 weeks (group D) |
Active comparator study (different induction doses/lengths of PEG) In the economic model, SVRs are used from a subgroup of non-responders from this study (data on file) |
|
| HALT-C (lead-in phase) | Shiffman and colleagues (2004)107 | No | Reports first 604 patients entering lead-in phase of HALT-C | The majority of patients in HALT-C were non-responders to IFN monotherapy (24%) or IFN–RBV combination therapy (66%); this contravenes the scope of the NICE appraisal |
| Everson and colleagues (2006)108 |
Described as an updated publication data set used as part of the European Medicines Agency filing in February 2008 Reports 1046 patients who had RNA assessments at weeks 20 and 72; analyses results in four subgroups of patients subdivided by increasing liver disease severity |
|||
| Shiffman and colleagues (2007)109 | Subgroup of 936 gastrointestinal patients with RNA assessments at weeks 20 and 72 (a subgroup of the 1046 in Everson and colleagues108) | |||
| WV16143, Berg and colleagues (2006)89 | No |
Described as a ‘supporting study’ Uncontrolled trial in 64 patients. Patients had originally been in the NV15942 trial by Hadziyannis and colleagues (2004),91 but had relapsed |
Uncontrolled | |
| Yoshida and colleagues (2009)110 | No |
Described as a ‘supporting study’ Post hoc analysis of a Canadian, multicentre, open-label study 87 non-responders/relapsers |
||
| Parise and colleagues (2006)111 | No |
Described as a ‘supporting study’ 134 Brazilian relapsers/non-responders to non-PEG/RBV |
||
Shorter courses studies: genotypes 2 and 3
| Study included in MS | Eligible for inclusion in the current SHTAC systematic review of clinical effectiveness? | Study details | Comments | |
|---|---|---|---|---|
| ACCELERATE NV17317 | Shiffman and colleagues (2007)90 | No |
1469 patients 16 vs 24 weeks of PEG + RBV 31% of patients had LVL at baseline (≤ 800,000 IU/ml) |
Paper was excluded from TAR because SVRs were not presented for patients with LVL and RVR SVRs from this trial are used in manufacturer’s economic model |
|
Retrospective analysis Zeuzem and colleagues (2005)112 |
No |
Mentions a retrospective research report 1026369, which reports results for patients with a RVR and LVL 216 patients with RVR and LVL in 16-week arm, 200 patients with RVR and LVL in 24-week arm |
Attributes this retrospective analysis to Zeuzem et al. 112 Manufacturer uses SVRs from this study in their economic model (89% for 16-week group vs 94% for 24-week group) These SVRs are similar to those used in the von Wagner and colleagues56 and Yu and colleagues55 studies below |
|
| von Wagner and colleagues (2005)56 | Yes | Both studies described as ‘supportive’ evidence in the submission. Mentions that both used unlicensed weight-based RBV doses for genotype 2/3 (hence why not included in their main analysis) | von Wagner and colleagues56 RBV dose = 800/1000/1200 mg | |
| Yu and colleagues (2007)55 | Yes | Yu and colleagues55 RBV dose = 1000/1200 mg | ||
Shorter courses studies: genotype 1
| Study included in MS | Eligible for inclusion in the current SHTAC systematic review of clinical effectiveness? | Details | Comments |
|---|---|---|---|
| Jensen and colleagues (2006)113 | No |
Retrospective analysis based on the one-third of genotype 1 patients who achieved an SVR after 24 weeks’ treatment in the Hadziyannis trial91 (incorrectly referred to as 2006 on p. 97 of submission) Purpose was to assess factors associated with RVR and an SVR in genotype 1 patients treated for 24 weeks |
SVRs from this study are used in manufacturer’s economic model |
| Ferenci and colleagues (2005)114 | No | Retrospective analysis of data from an RCT of 48 weeks’ PEG + RBV treatment in 1121 patients (compared with IFN + RBV) |
Shorter courses studies: genotype 4
| Study included in MS | Eligible for inclusion in the current SHTAC systematic review of clinical effectiveness? | Details | Comments |
|---|---|---|---|
| Ferenci and colleagues (2008)115 | No |
Retrospective analysis of NV15801 (Jensen and colleagues 2006) and NV 15942 (Hadziyannis trial91) Note: On p. 106 they refer to this as being a Research Report 1023045 data on file, and refer to the clinical trials as NV15801 (Fried and colleagues41) and NV 15942 (Yu and colleagues54). There is some confusion here regarding the identity of the trials |
Study was excluded from our review because it is not a randomised comparison of 24 vs 48 weeks. Patients with RVR were treated for 24 weeks, those without were then randomised at weeks 12–48 or 72 weeks. The journal paper presents SVRs for only the 24-week group anyway (ongoing trial) |
HCV/HIV co-infection
| Study included in MS | Eligible for inclusion in the current SHTAC systematic review of clinical effectiveness? | Details | Comments |
|---|---|---|---|
| APRICOT Torriani and colleagues (2004)66 | No | 868 treatment-naive co-infected patients randomised to receive: PEG α-2a (180 µg/week) plus RBV (800 mg per day); PEG α-2a plus placebo, or IFN α-2a (3 million IU three times/week) plus RBV | |
| Laguno and colleagues (2009)94 | No | Prospective multicentre RCT in Spain; compares PEG α-2a with PEG α-2b | Described in the MS as a ‘supporting study’ |
Schering-Plough hepatitis C submission to NICE 2009
-
Manufacturer’s submission only covers the re-treatment and HCV/HIV co-infection patient groups of the appraisal, not the shortened courses patient group (no explanation given for this).
-
Submission describes itself as a ‘systematic review’ conducted for the company’s own use as well as for NICE, so therefore it considers evidence beyond the scope of the appraisal including trials with active comparators (though for the purposes of the appraisal it does not use all of the trial arms). Although it provides details of its search strategy it does not describe the methods for screening and data extracting studies. Not clear on what basis they selected studies other than they were ones that were ‘pivotal’ in their licence extension application.
Re-treatment studies
| Study included in MS | Eligible for inclusion in the current SHTAC systematic review of clinical effectiveness? | Details | Comments |
|---|---|---|---|
| EPIC3 study (clinical study report on file), P02370, P02569, P02570 | No | Short-term non-randomised uncontrolled efficacy phase (P02370) followed by long-term maintenance stage (PEG mono vs no treatment) to prevent disease progression (P02569 and P02570) | Similar trial to HALT-C, but uses PEG α-2b |
| Submission presents short-term results of first efficacy cohort | |||
| Patients re-treated after failing previous IFN + RBV or PEG + RBV | |||
| Data from this trial are used in their economic evaluation: genotypes 1 and 4 EVR/SVR = 29.76%/48.65%; genotypes 2 and 3 = 79.13% and 69.95%, respectively | |||
| Scotto and colleagues (2008)97 | No | RCT PEG α-2a + RBV vs PEG α-2b + RBV for 48 weeks in previous IFN + RBV non-responders | Does not meet scope of the appraisal as patients are not re-treated following PEG |
HCV/HIV co-infection
| Study included in MS | Eligible for inclusion in the current SHTAC systematic review of clinical effectiveness? | Details | Comments |
|---|---|---|---|
| P01017 (Carrat and colleagues 2004,116 Pol and colleagues 2005117) | No | RCT PEG α-2b + RBV vs IFN + RBV | Active comparator studies, not within scope of appraisal |
| P02080 (Laguno and colleagues 200495) | No |
RCT PEG α-2b + RBV vs IFN + RBV Efficacy estimates from this trial used in their economic evaluation: genotypes 1 and 4 = 38%; genotypes 2 and 3 = 53% |
|
| Laguno and colleagues (2009)94 | No |
RCT PEG α-2a + RBV vs PEG α-2b + RBV Efficacy estimates from this trial used in sensitivity analysis |
Appendix 4 Inclusion criteria worksheet for systematic review of clinical effectiveness
| Trial name or number | ||||
|---|---|---|---|---|
|
Design: RCT or systematic review Exclude any conference abstracts from 2006 or earlier |
Yes ↓ Next question |
Unclear ↓ Next question |
No → EXCLUDE |
EXCLUDE1 (E1) (not the appropriate study design) |
|
Population: Adult patients with chronic hepatitis C, restricted to one or more of the following groups: (Note: Can be mild/moderate or severe hepatitis C) |
Yes ↓ Next question |
Unclear ↓ Next question |
No → EXCLUDE |
EXCLUDE2 (E2) (not the appropriate patient group) |
|
Intervention: Patients re-treated following previous relapse or non-response to PEG and RBV (or PEG monotherapy, and/or HIV/HCV co-infected) PEG + RBV PEG monotherapy Compared with BSC/placebo Intervention: (Patients eligible for shortened course of treatment): Compared with ‘standard’ duration courses of PEG/RBV (up to 24 or 48 weeks as appropriate) |
Yes ↓ Next question |
Unclear ↓ Next question |
No → EXCLUDE |
EXCLUDE3 (E3) (not the appropriate intervention) |
| Outcomes: SVR, defined as undetectable HCV RNA for at least 6 months after treatment cessation |
Yes ↓ Next question |
Unclear ↓ Next question |
No → EXCLUDE |
EXCLUDE4 (E4) (not the appropriate outcome measures) |
| Final decision | INCLUDE | UNCLEAR (Discuss) | EXCLUDE | Results of discussion: |
Appendix 5 Quality assessment criteria
CRD criteria for assessment of risk of bias in RCTs49
-
Was the method used to generate random allocations adequate?
-
Was the allocation adequately concealed?
-
Were the groups similar at the outset of the study in terms of prognostic factors, for example severity of disease?
-
Were the care providers, participants and outcome assessors blind to treatment allocation? If any of these people were not blinded, what might be the likely impact on the risk of bias (for each outcome)?
-
Were there any unexpected imbalances in dropouts between groups? If so, were they explained or adjusted for?
-
Is there any evidence to suggest that the authors measured more outcomes than they reported?
-
Did the analysis include an ITT analysis? If so, was this appropriate and were appropriate methods used to account for missing data?
Appendix 6 Data extraction forms and critical appraisal
Berg and colleagues59
| Reviewer 1: JS 16 November 2009, reviewer 2: DH 16 November 2009 | |||
|---|---|---|---|
| Reference and design | Intervention | Participants | Outcome measures |
|
Author: Berg and colleagues59 Year: 2009 Study design: Open-label, multicentre RCT No. of centres: 19 Country: Germany Sponsor: Essex Pharma (subsidiary of Schering-Plough), Bayer diagnostics, German Competence Network for Viral Hepatitis (German Ministry of Education and Research) |
Group 1: standard treatment duration n = 225 Drug 1: PEG α-2b Dose: 1.5 µg/kg/week Duration: 48 weeks Drug 2: RBV Dose: 800–1400 mg/day Duration: 48 weeks Group 2: variable treatment duration n = 208 Drug 1: PEG α-2b Dose: 1.5 µg/kg/week Duration: 18, 24, 30, 36, 42 or 48 weeksa Drug 2: RBV Dose: 800–1400 mg/day Duration: 18, 24, 30, 36, 42 or 48 weeksa |
Total numbers involved: 438 patients screened, 433 randomised Treatment naive/non-responders/relapsers: Treatment naive Previous treatment: NA HCV/HIV co-infection: No Recruitment: December 2001 and July 2003 Inclusion criteria: 18–70 years, compensated chronic HCV genotype 1, previously untreated with any type of IFN alfa and/or RBV, anti-HCV positive, HCV RNA > 1000 IU/ml by quantitative reverse-transcription PCR, increased serum ALT levels at screening, liver biopsy within preceding 24 months confirming chronic hepatitis, neutrophil count ≥ 1500/l and platelet count ≥ 80,000 l, Hb ≥ 12 g/dl for females and ≥ 13 g/dl for males, creatinine levels < 1.5 mg/dl Exclusion criteria: Patients with HCV type other than type 1, decompensated liver disease, hepatitis B or HIV co-infection or other causes of liver disease, autoimmune disorders, concomitant immunosuppressive medication, clinically significant bleeding disorders, clinically significant cardiac or cardiovascular abnormalities, organ grafts, systemic infections, pre-existing severe psychiatric conditions, evidence of malignant neoplastic diseases, excessive daily intake of alcohol (≥ 40 g/day in women and ≥ 60 g/day in men), drug abuse within past year, or unwillingness to practice contraception Baseline measurements: Viral load log (IU/ml), mean ± SD Group 1: 5.7 ± 0.49, range 2.79–7.8 Group 2: 5.7 ± 0.45, range 3–7.6 Serum ALT × ULN (IU/l), mean ± SD Group 1: 2.6 ± 0.2, range 0.5–28.8 Group 2: 2.6 ± 0.4, range 0.4–1.6 Histology: Fibrosis stage 0–2, n (%)b: Group 1: 177 (87%) Group 2: 161 (85.1%) Fibrosis stage 3–4, n (%)b: Group 1: 34 (13%) Group 2: 31 (14.9%) Necroinflammatory score, mean (± SD): NR Genotypes, n (%): 1 (100) Gender male, n (%): Group 1: 128 (57) Group 2: 113 (54.3) Age (years), mean ± SD, range: Group 1: 42.8 ± 0.8, 18–73 Group 2: 42.7 ± 11.69, 19 –66 Ethnic groups, n (%): NR Mode of infection, n (%): NR Losses to follow-up, n (%): Group 1: Therapy and follow-up completed 150 (67) Therapy completed 154 (68) Follow-up completed 189 (84) Group 2: Therapy and follow-up completed 135 (65) Therapy completed 145 (70) Follow-up completed 174 (84) Compliance, n: Therapy discontinuations (n = 71) Group 1: Therapy failure 39 Adverse events 7 Lost to follow-up 24 Other reason 1 Therapy discontinuations ( n = 63) Group 2: Therapy failure 42 Adverse events 4 Lost to follow-up 15 Other reason 2 |
Primary outcomes: SVR Secondary outcomes: Sustained biochemical response (ALT normalisation at end of follow-up) On-treatment virological response rates (RVR and EOT) Relapse rate Adverse events Length of follow-up: 24 weeks after cessation of treatment Methods of assessing outcomes: HCV RNA levels were quantified at baseline and weekly until week 8 as well as at weeks 12, 24 and 48 by bDNA assay (detection limit 615 IU/ml) SVR HCV RNA negativity verified using highly sensitive qualitative TMA assay (detection limit < 5.3 IU/ml). This assay was reserved only for those patients who had HCV RNA levels of < 1000 IU/ml by the bDNA test. The cut-off of 1000 IU/ml, instead of 615 IU/ml, was chosen to improve the specificity of the bDNA assay. Patients with HCV RNA levels of between 615 and 1000 IU/ml but being HCV RNA negative on TMA were considered bDNA undetectable after confirmation by re-testing HCV genotyping performed by reverse hybridisation; histological results classified using standard criteria (Desmet 1994 cited) |
|
HVL, high viral load (> 800,000 IU/ml); LVL, low viral load (≤ 800,000 IU/ml); PEG α, peginterferon alfa; RBV, ribavirin; RVR, rapid virological response (defined as HCV RNA negativity < 615 IU/ml at week 4); SVR, sustained virological response [defined as negative qualitative HCV RNA (< 5.3 IU/ml by sensitive TMA assay) 24 weeks after the end of treatment]. a Individualised duration based on time to first HCV RNA negativity by bDNA assay multiplied by a factor of 6. First negative at week 3, 4, 5, 6, 7 or 8 corresponded to a treatment duration of 18, 24, 30, 36, 42 or 48 weeks, respectively (n = 28 appear to have been treated for 24 weeks). b It is not clear from the trial publication what the denominators were for these percentages. The percentages given are not for the total randomised in each study group. It therefore does not appear that all patients randomised underwent liver biopsy at baseline. |
|||
|
Treatment failures: Breakthrough (reappearance of HCV viraemia during antiviral treatment); relapse (reappearance of HCV RNA during follow-up after stopping therapy in patients with an EOT virological response) or non-response (patients testing HCV RNA positive at any time point during the study). |
|||
| Outcome | Group 1 standard (n = 225), (48 weeks) | Group 2 variable (n = 208), (18–48 weeks) | p-value |
| Viral response, % (n/N), 95% CI: | |||
| 4-week (RVR)c | 8.4d (19/225) | 13.5d (28/208) | NR |
| RVR (weeks 1–3 + week 4)e | 35d (78/225) | 37d (76/208) | NR |
| 12-week (EVR) | – | – | |
| End of treatment | 65 (146/225) 58.3 to 71.1 | 64 (133/208) 57 to 70.5 | NR |
| End of follow-up (SVR) | 48 (108/225) 41.3 to 54.7 | 35 (72/208) 28.2 to 41.5 | 0.005 |
| SVR by RVR, % (n/N )f | 42 (8/19) | 57 (16/28) | NR |
| SVR by baseline viral load, % (n/N ) | – | – | – |
| SVR by baseline viral load and RVR, % (n/N)g | |||
| ≤ 800,000 IU/ml (low) | 75 (3/4) | 69 (11/16) | NR |
| > 800,000 IU/ml (high) | 33 (5/15) | 42 (5/12) | NR |
| Non-response % (n/N), 95% CI | 18 (41/225) 13.4 to 23.9 | 20 (41/208) 14.5 to 25.8 | NR |
| Virological relapse % (n/N), 95% CI | 14 (32/225) 9.9 to 19.5 | 33 (68/208) 26.4 to 39.5 | < 0.0005 |
| Breakthrough % (n/N), 95% CI | 5 (11/225) 2.5 to 8.6 | 3 (7/208) 1.4 to 6.8 | NR |
|
c Time to first HCV RNA < 615 IU/ml at week 4 by bDNA assay (not including those who became first negative between weeks 1 and 3). d Percentage calculated by reviewer from numbers presented in trial publication. e Total number of patients first becoming HCV RNA negative between weeks 1 and 3 (n = 59 in Group 1, n = 48 in Group 2) and those becoming first negative at week 4 (n = 19 in Group 1, n = 28 in Group 2) combined to give total number of patients becoming HCV RNA negative by week 4. f Numerator calculated by reviewer from figures presented in trial publication. g Study defines LVL as ≤ 800,000 IU/ml and HVL as > 800,000 IU/ml – this threshold for LVL is higher than the threshold of < 600,000 IU/ml specified in the SPC for PEG α-2b. |
|||
| Biochemical response, % (n/N ): | NR | ||
| End of treatment | |||
| End of follow-up | |||
| Histology (proportion with improvement) | NR | ||
| Adverse events: | |||
| Dose discontinuation for any adverse event | 3% (7/225) | 2% (4/208) | |
| Dose reduction for any adverse event or lab abnormality | 16% | 15% | |
| Serious adverse events | 6.6%: Anaemia n =1, appendectomy n = 1, sinusitis n = 1, pneumonia n = 2, psychiatric disorder n = 7, subileus n = 1, wound infection n = 1 | 2.6%: Ankle fracture n = 1, retina ablation n = 1, pneumonia n = 2, psychiatric disorder n = 1 | 0.243 |
| Additional results/comments | |||
|
Authors report that percentage of patients reporting adverse events was similar in the two treatment groups. Both the type and severity of treatment side effects (typical of IFN-based treatment) were not statistically different between the two groups (data not shown) Most commonly observed causes of dose modifications of PEG α-2a and RBV were neutropenia and anaemia, respectively Results (in terms of SVR by RVR, and SVR by RVR and baseline viral load) are also presented according to time to HCV RNA negativity as measured by the TMA assay (< 5.3 IU/ml). The purpose was to explore differences in treatment effect between the two assays. However, the SVRs according to TMA negativity at week 4 (i.e. RVR) in Group 2 are based on some patients who only received 18 weeks’ treatment rather than 24 weeks’ treatment. Treatment for less than 24 weeks in patients with genotype 1 (as a comparator to 48 weeks’ treatment) is not within the scope of this systematic review and therefore the results have not been extracted here |
|||
| Methodological comments | |||
|
Allocation to treatment groups: Randomised by stratification for baseline viraemia (≤ 800,000 vs > 800,000 IU/ml). No further detail given on randomisation procedure Allocation concealment: No details given Blinding: No details given, but due to the differences in regimens it is unlikely that patient or investigator blinding would be possible. No mention is made about whether outcome assessors (e.g. liver biopsy pathologists) were blinded to treatment allocation Analysis by ITT: States that an ITT analysis was conducted, although does not provide a definition of what ITT considered to be. Patients were classified as unknown with respect to treatment response in the case of missing relevant data for exact and reliable categorisation Comparability of treatment groups at baseline: Authors report that treatment groups were well matched and differed only slightly with respect to relevant variables by univariate between-group analyses (although statistics not presented) Method of data analysis: Descriptive statistics used for all relevant dependent variables including absolute and relative frequencies for categorical data and means, standard deviations and ranges for continuous scaled data. Statistical comparisons between the two treatment groups were made using the chi-squared test. Multiple logistic regression was used to analyse the influence of independent predictive factors on the occurrence of an SVR Sample size/power analysis: Study was originally designed to be a non-inferiority trial. An SVR of approximately 45% was estimated for the standard fixed duration of 48 weeks. A difference in SVR rates of up to 12.5% across both study arms was considered as still being equivalent. Under this assumption 436 patients were required if a level of significance of α = 0.05, a minimal power of 80% and a drop out rate of 10% are assumed. Because there was a significantly higher SVR rate in the standard treatment arm (Group 1) the trial was switched to a superiority trial in accordance with guidance from the European Medicines Agency Attrition/dropout: Rates of therapy and follow-up completion were 150 (67%) in Group 1 and 135 (65%) in Group 2 |
|||
| General comments | |||
|
Generalisability: Results applicable to treatment-naive European genotype 1 patients with mild-to-moderate HCV-related fibrosis. Mean baseline viral load was low (log10 5.7 = 501,187 IU/ml) Intercentre variability: NR Conflict of interests: NR |
|||
Quality criteria for assessment (updated CRD guidance)a
| 1. | Was the method used to generate random allocations adequate? | Unclear |
| 2. | Was the allocation adequately concealed? | Unclear |
| 3. | Were the groups similar at the outset of the study in terms of prognostic factors, for example severity of disease? | Yes |
| 4. | Were outcome assessors blinded to the treatment allocation? | Unclear |
| 5. | Was the care provider blinded? | No |
| 6. | Was the patient blinded? | No |
| 7. | Were there any unexpected imbalances in dropouts between groups? If so, were they explained or adjusted for? | No |
| 8. | Is there any evidence to suggest that the authors measured more outcomes than they reported? | Yes |
| 9. | Did the analysis include an ITT analysis? | Yes |
| If so, was this appropriate? | Unclear | |
| If so, were appropriate methods used to account for missing data? | Unclear |
Mangia and colleagues52
| Reference and design | Intervention | Participants | Outcome measures |
|---|---|---|---|
|
Author: Mangia and colleagues52 Year: 2008 Study design: Multicentre RCT No. of centres: 11 Country: Italy Sponsor: NR (but states no support was received from pharmaceutical companies) |
Intervention 1: standard group (48 weeks) n = 237 Drug 1: PEG α-2a or α-2b Dose: α-2a 180 µg/week; α-2b 1.5 µg/kg/week Duration: 48 weeks Drug 2: RBV Dose: 1000 mg/day for patients ≤ 75 kg, 1200 mg/day for patients > 75 kg Duration: 48 weeks Intervention 2: variable group (24, 48 or 72 weeks a ) n = 459 Drug 1: PEG α-2a or α-2b Dose: α-2a 180 µg/week; α-2b 1.5 µg/kg/week Duration: 24, 48 or 72 weeksa Drug 2: RBV Dose: 1000 mg/day for patients ≤ 75 kg, 1200 mg/day for patients > 75 kg Duration: 24, 48 or 72 weeksa |
Total numbers involved: 711 enrolled, 696 randomised; n = 237 Group 1, n = 459 Group 2 Treatment naive/non-responders/relapsers: Treatment naive Previous treatment: NA HCV/HIV co-infection: No Recruitment: 11 centres in southern Italy between June 2004 and December 2005 Inclusion criteria: Previously untreated adults (18–70 years) with compensated chronic HCV genotype 1, anti-HCV positive, HCV RNA positive, neutrophil count ≥ 1500 µl, platelet count ≥ 90,000 µl, Hb ≥ 12 g/dl for women and ≥ 13g/dl for men, creatinine < 1.5 mg/dl Exclusion criteria: Other causes of liver disease, hepatitis B, HIV, autoimmune disorders, clinically significant cardiac or cardiovascular abnormalities, systemic infection, organ graft, clinically significant bleeding disorders, evidence of malignant diseases, concomitant immunosuppressive medication, excessive alcohol intake or concomitant drug abuse, pregnancy, lactation or male partners of pregnant women Baseline measurements: Serum HCV RNA, n (%): < 400,000 IU/ml: 62 (26%) Group 1, 103 (22%) Group 2, p = 0.30 ≥ 400,000 IU/ml: 175 (74%) Group 1, 356 (78%) Group 2 Serum ALT, n (%): < 3 ULN: 193 (81%) Group 1, 385 (84%) Group 2, p = 0.39 ≥ 3 ULN: 44 (19%) Group 1, 74 (16%) Group 2 Histology: Fibrosis stage, n (%):b 0–2: 140 (62%) Group 1, 258 (65%) Group 2, p = 0.33 3–4: 87 (38%) Group 1, 134 (34%) Group 2 Grade of activity, n (%):c 0–2: 167 (76%) Group 1, 306 (78%) Group 2, p = 0.42 3: 54 (24%) Group 1, 89 (22%) Group 2 Steatosis:d Yes: 70 (31%) Group 1, 103 (26%) Group 2, p = 0.07 No: 151 (68%) Group 1, 295 (74%) Group 2 Genotypes, n (%): 1a: 15 (6%) Group 1, 49 (11%) Group 2, p = 0.08 1b: 222 (94%) Group 1, 410 (89%) Group 2 Gender, n (%): Female: 105 (44%) Group 1, 201 (44%) Group 2, p = 0.93 Male: 132 (56%) Group 1, 258 (56%) Group 2 Age (years), mean (± SD): 52.6 (± 11.8) Group 1, 51.1 (± 12.1) Group 2, p = 0.12 Ethnic groups, n (%): NR Mode of infection, n (%): Blood transfusion: 50 (21%) Group 1, 93 (20%) Group 2 Drug abuse: 17 (7%) Group 1, 37 (8%) Group 2, p = 0.81 Unknown: 170 (72%) Group 1, 329 (72%) Group 2 Treatment, n (%): PEG α-2b: 127 (53%) Group 1, 235 (51%) Group 2, p = 0.52 PEG α-2a: 110 (46%) Group 1, 224 (49%) Group 2 Losses to follow-up: n = 6 (Group 2) Compliance: n = 83 (12%) discontinued treatment (24 Group 1, 59 Group 2) due to adverse events (16 Group 1, 30 Group 2) or no compliance (8 Group 1, 29 Group 2) |
Primary outcomes: SVR Secondary outcomes: SVR according to virological response at weeks 4, 8 and 12 RVR EOT Adverse events Length of follow-up: 24 weeks after cessation of treatment Methods of assessing outcomes: HCV-RNA levels quantified at baseline (lower limit of detection 600 IU/ml) and qualitatively analysed by PCR assay (lower limit of detection 50 IU/ml) during and off therapy; HCV RNA of 400,000 IU/ml chosen as cut-off for LVL or high viral load. HCV genotyping performed by reverse hybridisation, histological results classified using standard criteria (Desmet cited); platelet counts < 140,000/mm3 were taken as evidence of advanced fibrosis in patients without biopsy as per cited literature |
|
EOT, end-of-treatment virological response; PEG α, peginterferon alfa; RBV, ribavirin; RVR, rapid virological response (defined as HCV RNA negative at week 4); SVR, sustained virological response (defined as undetectable serum HCV RNA at the end of 24 weeks’ follow-up); ULN, upper limit of normal. ‘Non-responders’ defined as patients who were viraemic at week 24 and also patients with a < 2-log decline at week 12. ‘Treatment failures’ defined as relapse (reappearance of HCV RNA during follow-up period after an EOT response), non-response or discontinuation. a Treatment duration was based on time when HCV RNA first became undetectable; patients who were first HCV RNA negative at: week 4 treated for 24 weeks, week 8 treated for 48 weeks, week 12 treated for 72 weeks. b Data unavailable from 67 patients (10 Group 1, 57 Group 2). c Data unavailable from 78 patients (14 Group 1, 64 Group 2). Data missing from 67 patients (6 Group 1, 61 Group 2). |
|||
| Outcome | Group 1 standard (n = 237), 48 weeks | Group 2 variable (n = 123), 24 weeks | p-value |
| Note: Data have been extracted only for Group 1 vs 24-week subset of Group 2, as results for Group 2 as a whole (n = 459) are not relevant to this review. | |||
| Viral response, % (n/N, 95% CI): | |||
| EOT by RVR | 96.7 (60/62, 92.3 to 100) | 95.1 (117/123, 92.3 to 99.4) | 0.42 |
| SVR by RVR | 87.1 (54/62, 78.7 to 95.4) | 77.2 (95/123, 69.8 to 84.6) | 0.12; difference –9.9 (10.5 to 9.2) |
| SVR by RVR and baseline viral load, (n/N ): | |||
| ≥ 400,000 IU/ml | 86.8 (33/38) | 73.1 (57/78) | 0.14 |
| < 400,000 IU/ml | 83.3 (20/24) | 84.4 (38/45) | 0.83 |
| Other viral response outcomes: | |||
| Relapse rate (whole group), % (n/N ) | 19.1 (25/131) | 19.4 (54/278) | 1.0 |
| Relapse rate by RVR, % (n/N ) | 10% (6/62) | 18.8 (22/123) (Reviewer: should be 17.9.) | 0.13 |
| Biochemical response, % (n/N ) | NR | NR | |
| Histology (proportion with improvement) | NR | NR | |
| Adverse events (for Group 1 vs Group 2, not 24-week subset of Group 2) | Group 1, 48 weeks, n = 237, n (%) | Group 2, 24, 48 or 72 weeks, n = 459, n (%) | |
| Dose discontinuation: | 24 (10.1) | 59 (12.9) | 0.19 |
| For any adverse event | 16 (6.7) | 30 (6.5) | |
| For no compliance | 8 (3.4) | 29 (6.3) | 0.49 |
| Dose reduction | 32 (13.5) | 47 (10.2) | |
| Specific adverse events, n (%): | |||
| Asthenia | 101 (42.6) | 183 (39.8) | |
| Flu-like symptoms | 34 (14.3) | 87 (18.9) | |
| Dermatological symptoms | 29 (12.2) | 60 (13.0) | |
| Psychiatric symptoms | 4 (1.7) | 7 (1.5) | |
| Anaemia | 20 (8.4) | 33 (7.1) | |
| Leucopenia and thrombocytopenia | 58 (24.4) | 35 (7.6) | |
| Thyroid diseases | 7 (2.9) | 11 (2.3) | |
| Decrease in Hb to < 9.5 g/dl | 20 (8.4) | 33 (7.1) | 0.66 |
| Neutrophil count < 1000/mm3 (requiring PEG dose reduction) | 12 (5.1) | 19 (4.1) | 0.69 |
| Additional results/comments (e.g. early response factors, QoL) | |||
| Virological response | |||
|
Results for EOT, SVR and predictive factors were reported for Group 1 vs Group 2, as well as Group 1 vs the 48-week and 72-week subsets of Group 2, but these have not been extracted In the entire population (n = 696), 185 (26.6%) had undetectable HCV RNA at week 4 (i.e. RVR), comprising 62 (26.2%) Group 1 and 123 (26.8%) Group 2 (whole Group), p = 0.90. An EOT response was achieved by 55.3% (131/237) and 60.6% (278/459) of the standard and variable treatment groups, respectively RVR was achieved in 29% (105/362) patients treated with PEG α-2b and 24% (80/334) patients treated with PEG α-2a (p = 0.14) In univariate analysis (in entire population), factors associated with RVR were young age (p = 0.004), low viraemia levels (p = 0.0001) and fibrosis stage ≤ 2 (p = 0.0001). In multivariate analysis (entire population), independent predictors of RVR were serum HCV RNA levels < 400,000 IU/ml [odds ratio (OR) 2.27, 95% CI 1.49 to 3.41] and absence of advanced fibrosis (OR 1.40, 95% CI 1.15 to 1.64) The only independent predictor of SVR in RVR patients was a mild-to-moderate degree of fibrosis (OR 2.60, 95% CI 1.09 to 6.17). Off therapy, 24.4% of patients with high viraemia, and 8.9% of patients with low viraemia, relapsed (p = 0.05) |
|||
| Methodological comments | |||
|
Allocation to treatment groups: Patients were allocated 1 : 2 in blocks of five, using a computer-generated randomisation list that was sent to each participating centre. PEG α-2a or α-2b was prescribed on a 1 : 1 basis Allocation concealment: No details reported Blinding: Blinding of participants and care providers not possible and blinding of outcome assessors not reported Analysis by ITT: ITT analysis – all randomised patients who received at least one dose of study medication were used for analysis of primary and secondary outcomes Comparability of treatment groups at baseline: Participant baseline demographic, biological and virological characteristics were well matched between Group 1 and Group 2, with no statistically significant differences (p-values reported). Also reports that baseline characteristics did not differ between patients treated with PEG α-2a and PEG α-2b (but data not presented). However, comparability of Group 1 (48 weeks) vs 24 weeks subset is unknown Method of data analysis: The descriptive analysis included absolute and relative frequencies for grouped data and means ± SD for continuous scaled data. Statistical comparison between patients with and without SVR used the chi-squared test and the t-test (continuous data). Level of significance was 0.05 (two-sided) for all statistical tests; all CIs provided are at 95%. spss was used for statistical analysis Sample size/power analysis: Study designed as a non-inferiority analysis comparing standard and variable treatment duration. An SVR rate of 45% was expected on the basis of data from previous cited international studies. Sample size of 212 patients per treatment group was estimated to show that the variable treatment duration is no more than 5% different than the standard duration, with one-sided 95% CI and 80% power. With a dropout rate of 10%, 237 patients per group were required. Given that the secondary aim of investigating SVR rates according to on-treatment virological response, double this number (474) of patients were assumed to be recruited into the variable group for meaningful subgroup comparisons. [Important note: Only 69 (9.9%) patients (24 Group 1, 45 Group 2 24-week subset) had LVL (< 400,000 IU/ml) and RVR and thus the study was likely not powered for this subgroup.] Attrition/dropout: Numbers and reasons provided for those discontinuing treatment; numbers provided for those lost to follow-up. Numbers reported for those completing treatment are not consistent between figure 1 and table 3. Figure 1 reports 144 completed, 24 discontinued, 69 HCV RNA positive at week 24 (Group 1); 297 completed, 59 discontinued, 103 HCV RNA positive at week 24 (Group 2). Table 3 reports 122 completed treatment, 24 discontinued, 91 no response at week 24 (Group 1); 237 completed treatment, 59 discontinued, 163 no response at week 24 (Group 2) |
|||
| General comments | |||
|
Generalisability: Treatment-naive, Italian patients with genotype 1 HCV. Only 24% had LVL at baseline and only 10% had LVL and RVR Intercentre variability: Reports that HCV RNA testing carried out at individual centres provided that all centres used the same assay. However, no intercentre variability reported. For better comparisons between different histopathologists, individual fibrosis stage was documented as significant (cirrhosis/transition to cirrhosis) or not significant (no cirrhosis) Conflict of interests: None reported Other: This is not a standard 48-week vs 24-week study in genotype 1 patients as Group 2 included patients treated for 24, 48 and 72 weeks, although some results were reported separately. Also, as noted above, only 10% of patients fulfilled the inclusion criteria of having LVL and RVR – the study was included because SVR rates were reported separately for this subgroup, but results should be treated with caution |
|||
Quality criteria for assessment (updated CRD guidance)a
| 1. | Was the method used to generate random allocations adequate? | Yes |
| 2. | Was the allocation adequately concealed? | Unclear |
| 3. | Were the groups similar at the outset of the study in terms of prognostic factors, for example severity of disease? | Yes (Group 1 vs Group 2); unclear for Group 1 vs 24-week subset |
| 4. | Were outcome assessors blinded to the treatment allocation? | Unclear |
| 5. | Was the care provider blinded? | No |
| 6. | Was the patient blinded? | No |
| 7. | Were there any unexpected imbalances in dropouts between groups? If so, were they explained or adjusted for? | No |
| 8. | Is there any evidence to suggest that the authors measured more outcomes than they reported? | No |
| 9. | Did the analysis include an ITT analysis? | Yes |
| If so, was this appropriate? | Yes | |
| If so, were appropriate methods used to account for missing data? | Unclear |
Liu and colleagues53
| Reference and design | Intervention | Participants | Outcome measures |
|---|---|---|---|
|
Author: Liu and colleagues, Liu and colleagues (abstract)53,57 Year: 2008, 2008 abstract Study design: Open-label, multicentre RCT No. of centres: 5 Country: Taiwan Sponsor: National Taiwan University Hospital, National Science Council, and Department of Health, Executive Yuan, Taiwan |
Group 1: 24 weeks n =154 Drug 1: PEG α-2a Dose: 180 g/week s.c. Duration: 24 weeks Drug 2: RBV Dose: 1000 mg/day for body weight < 75 kg and 1200 mg/day for body weight ≥ 75kg Duration: 24 weeks Group 2: 48 weeks n = 154 Drug 1: PEG α-2a Dose: 180 µg/week s.c. aDuration: 48 weeks Drug 2: RBV Dose: 1000 mg/day for body weight < 75 kg and 1200 mg/day for body weight ≥ 75kg Duration: 48 weeks |
Total numbers involved: 308 patients n = 154 Group 1, n = 154 Group 2 Treatment naive/non-responders/relapsers: Treatment naive Previous treatment: NA HCV/HIV co-infection: No Recruitment: Five academic centres (in Taiwan hospitals) between June 2006 and March 2008 Inclusion criteria: Patients with genotype 1 aged > 18 years, presence of anti-HCV antibody and detectable serum HCV RNA level for > 6 months, serum ALT level > ULN, liver histological characteristics consistent with chronic viral hepatitis within the last 3 months Exclusion criteria: Anaemia: < 13 g/dl for men; < 12 g/dl for women, neutropenia (neutrophil count < 1500 cells/mm3), thrombocytopenia (platelet count < 70,000 cells/mm3), mixed infection with HCV-1 and another genotype of HCV, co-infection with hepatitis B virus or HIV, chronic alcohol abuse (daily alcohol consumption > 20 g/day), DC (Child–Pugh class B or C), serum creatinine level > 1.5 times the ULN, autoimmune liver disease, neoplastic disease, organ transplantation or immunosuppressive therapy, evidence of drug abuse, pregnancy, poorly controlled autoimmune disease, cardiopulmonary disease, neuropsychiatric disorders, diabetes mellitus with retinopathy, unwillingness to receive contraception during the study period Baseline measurements: Viral load (IU/ml), mean log 10 (± SD): 5.7 ± 0.7 (Group 1), 5.8 ± 0.7 (Group 2), p = 0.83 Serum ALT: mean value × ULN ± SD: 3.2 ± 2.6 (Group 1), 3.0 ± 2.1 (Group 2), p = 0.91 Histology: Fibrosis score n (%): ≥ 3: 121 (78.6) (Group 1), 117 (76.0) (Group 2), p = 0.68 6: 35 (22.7) (Group 1), 31 (20.1) (Group 2), p = 0.68 (≥ 3 = significant fibrosis, 6 = cirrhosis) Mean total modified HAI score, (±SD): 12.7 ± 3.3 (Group 1), 12.3 ± 3.7 (Group 2), p = 0.43 Genotypes, n (%): 1a: 4 (2.6) Group 1, 3 (1.9) Group 2 1b: 143 (92.9) Group 1, 145 (94.2) Group 2 1a and 1b: 7 (4.5) Group 1, 6 (3.9) Group 2 Gender male, n (%): 88 (57.1) (Group 1), 87 (56.5) (Group 2), p > 0.99 Age (years), mean (range): 54 ± 10 (Group 1), 53 ± 11 (Group 2), p = 0.41 Ethnic groups, n (%): Asian (no further details reported) Mode of infection, n (%): NR Losses to follow up: Group 1: 7 discontinued prior to treatment completion, 0 after treatment completion Group 2: 4 discontinued prior to treatment completion, 15 after treatment completion Compliance: NR |
Primary outcomes: SVR rate Secondary outcomes: RVR EVR EOT virological response Relapse rate ALT normalisation Histological response Length of follow-up: Additional 24 weeks of follow-up after end of therapy Methods of assessing outcomes: Patients received outpatients visits to assess the efficacy and safety at weeks 1, 2, 4, 6 and 8 of the study and then monthly until the end of the follow-up period Serum HCV RNA levels quantitatively assessed at baseline, weeks 4, 12, end of treatment and 24 weeks after end of treatment (lower limit of detection 25 IU/ml). Patients in 48-week group had an additional HCV RNA test at week 24 of treatment Liver biopsies were performed at baseline and at the end of the follow-up period and assessed in accordance with Brunt’s classification, and the modified HAI |
|
Complete EVR, an undetectable serum HCV RNA level at week 12 of therapy in patients who did not achieve RVR, and partial EVR was defined as at least a 2-log reduction in serum HCV RNA level from baseline to week 12 of therapy in those who did not achieve RVR at week 4 and did not achieve an undetectable serum HCV RNA level at week 12 of therapy; EVR, early virological response, defined as at least a 2 log reduction in serum HCV RNA level from baseline to week 12 of therapy. ALT, alanine aminotransferase; DC, decompensated cirrhosis; EOT, virological response defined as an undetectable serum HCV RNA at the end of treatment; HAI, histological activity index; PEG α, peginterferon alfa; RBV, ribavirin; RVR, rapid virological response defined as an undetectable serum HCV RNA level (< 25 IU/ml) at week 4 of therapy; s.c., subcutaneously; SVR, an undetectable serum HCV RNA at the end of the follow-up period; histological response rate defined as at least two-point reduction in the modified histological activity index from baseline to follow up; relapse included patients with an undetectable HCV RNA level at the end of treatment but with a detectable level at the end of follow-up; ULN, upper limit of normal. a Treatment was prematurely discontinued in patients who were randomised to 48 weeks of treatment but who continued to have HCV viraemia at week 24 of therapy, because they had minimal chance of achieving SVR with continued therapy. |
|||
| Outcome | Group 1 (24 weeks’ treatment) | Group 2 (48 weeks’ treatment) | p-value |
| Viral response, n (%): | |||
| 4-week (RVR) | 104 (68) | 97 (63) | 0.47 |
| 12-week (EVR) | 142 (94) | 148 (97) | 0.17 |
| End of treatment | 136 (91) | 142 (97) | 0.06 |
| End of follow-up (SVR) | 87 (56) | 117 (76) | < 0.001 |
| Relapse rate | 46 (34) | 24 (17) | 0.001 |
| Percentages reported by paper (above) are incorrect if denominator is 154. For RVR, EVR, EOT, SVR and relapse rate, the percentages would be 67%, 92%, 88%, 56% and 29% for 24-week group, and 62%, 96%, 92%, 75% and 15% for 48-week group respectively. | |||
| Group 1 (24 weeks’ treatment) | Group 2 (48 weeks’ treatment) | p-value | |
| Predictability of SVR during treatment with RVR stratified by baseline viral load | |||
| SVR by RVR, n (%): | |||
| RVR | 104 (76) | 97 (98) | < 0.001 |
| No RVR | 49 (16) | 56 (39) | 0.01 |
| < 400, 000 IU/ml | 49 (94) | 42 (100) | 0.25 |
| < 600, 000 IU/ml | 61 (93) | 50 (100) | 0.13 |
| < 800, 000 IU/ml | 69 (94) | 57 (100) | 0.13 |
| < 1,000,000 IU/ml | 71 (92) | 61 (100) | 0.03 |
| ALT normalisation, n (%) | 75 (51) | 107 (72) | < 0.001 |
| Histological response, n (%) | 71 (59) | 97 (78) | 0.001 |
| RVR | SVR | ||||||
|---|---|---|---|---|---|---|---|
| Univariate analysis (p-value) | Multivariate analysis | Univariate analysis (p-value) | Multivariate analysis | ||||
| OR (95% CI) | p-value | OR (95% CI) | p-value | ||||
| HCV RNA level (< 800, 000 vs ≥ 800,000 IU/ml) | < 0.001 | 3.33 (1.96 to 5.64) | < 0.001 | < 0.001 | 10.51 (5.47 to 20.21) | < 0.001 | |
| Group 1 (24 weeks) | Group 2 (48 weeks) | p-value | |||||
| Adverse events: | |||||||
| Dose discontinuation for any adverse event n (%) | 6 (4) | 14 (9) | 0.10 | ||||
| Dose reduction for: | |||||||
| Any adverse event | 69 (45) | 82(53) | |||||
| Anaemia | 60 (39) | 68 (44) | |||||
| Neutropenia | 34 (22) | 42 (27) | |||||
| Serious adverse events, %: | 3 | 7 | 0.11 | ||||
| Death, n | 0 | 1 | |||||
| All, n | 4 | 11 | |||||
| Treatment related, n | 3 | 9 | |||||
| Specific adverse events, n (%): | |||||||
| Fever | 35 (23) | 33 (21) | |||||
| Rigour | 19 (12) | 13 (8) | |||||
| Fatigue | 88 (57) | 100 (65) | |||||
| Headache | 28 (18) | 35 (23) | |||||
| Myalgia | 40 (26) | 36 (23) | |||||
| Arthralgia | 8 (5) | 13 (8) | |||||
| Insomnia | 61 (40) | 69 (45) | |||||
| Irritability | 19 (12) | 22 (14) | |||||
| Depression | 36 (23) | 26 (17) | |||||
| Anorexia | 63 (41) | 80 (52) | |||||
| Constipation | 10 (6) | 15 (10) | |||||
| Diarrhoea | 14 (9) | 18 (12) | |||||
| Body weight lossa | 19 (19) | 46 (30) | 0.03 | ||||
| Hair loss/alopecia | 24 (16) | 36 (23) | |||||
| Aphthous ulcer | 22 (14) | 34 (22) | |||||
| Cough | 28 (18) | 32 (21) | |||||
| Nasal congestion | 13 (8) | 17 (11) | |||||
| Tinnitus | 13 (8) | 20 (13) | |||||
| Dermatitis | 44 (29) | 48 (31) | |||||
| Injection reaction | 22 (14) | 29 (19) | |||||
| Anaemia | 60 (39) | 68 (44) | |||||
| Neutropenia | 34 (22) | 42 (27) | |||||
| Thrombocytopenia | 25 (16) | 23 (15) | |||||
| a Weight reduction of > 10% from the baseline weight. | |||||||
| Adverse events | |||||||
|
In the 24-week group, severe adverse events included retinal ischaemia, hepatic decompensation, major depression and HCC (the first three events were considered to be treatment related). In the 48-week group, severe adverse events included hepatic decompensation in three patients and major depression, renal abscess, interstitial pneumonitis, diabetes mellitus, empyema, pulmonary tuberculosis, HCC, and acute pancreatitis in one patient each (the first nine events were considered to be treatment related) Fifteen patients experienced serious adverse events during the study period; 12 (80%) were considered to be treatment related Four patients developed hepatic decompensation, with ascites and hepatic encephalopathy, requiring cessation of PEG. Three of these had cirrhosis and one of them had advanced fibrosis One death due to reactivation of pulmonary tuberculosis at week 36 of therapy was reported in the 48-week group |
|||||||
| Methodological comments | |||||||
|
Allocation to treatment groups: Eligible patients were assigned 1 : 1. Randomisation was performed with the use of block sizes of 4 or 6 by computer-generated assignment Allocation concealment: NR Blinding: Open-label trial. Biopsy pathologist was blind to clinical status of study participants. Not stated whether other outcome assessors were blinded Analysis by ITT: Authors state analysis was by ITT for the primary efficacy end point. The secondary efficacy end points were analysed only for patients who had undergone paired biopsies or for patients with available baseline and follow up ALT levels. Treatment was prematurely discontinued in patients who were randomised to 48 weeks of treatment but continued to have HCV viraemia at week 24 of therapy, because they had minimal chance of achieving SVR with continued therapy. A total of 88% completed 48 weeks of therapy Comparability of treatment groups at baseline: Groups appear comparable at baseline Method of data analysis: The baseline characteristics of treatment groups were compared using the chi-squared test, Fisher’s exact test, or Student’s t-test. Treatment responses, including efficacy and safety, were compared using Fisher’s exact test. A p-value < 0.05 was considered to be statistically significant, all statistical tests were two tailed Sample size/power analysis: The sample size was estimated to be 152 patients in each group on the basis of a type I error rate of α = 0.05 and a type 2 error rate of β = 0.20 for a primary two-sided test with the assumption of a 15% difference in SVR rates (60% and 75% for 24 and 48 weeks of treatment, respectively) Attrition/dropout: Participants were considered withdrawn from the study if the investigator was concerned about treatment safety or if the patient missed four consecutive weeks of therapy. In Group 1, seven patients discontinued treatment, six due to adverse events or laboratory abnormalities, one declined treatment. In Group 2, four discontinued treatment due to adverse events or laboratory abnormalities. Fifteen patients in Group 2 discontinued after treatment completion: 10 were due to adverse events or laboratory abnormalities, two patients had a positive HCV RNA at week 24, one declined treatment and two were lost to follow-up |
|||||||
| General comments | |||||||
|
Generalisability: The study appears generalisable to Asian patients with genotype 1 only. Mean baseline viral load [log10 5.7 = 501,000 IU/ml (Group 1) and log10 5.8 = 630,957 IU/ml (Group 2)] was low and approximately 65% had RVR at week 4 Intercentre variability: NR Conflict of interests: One author has been a consultant for Novartis and Roche, one author has been a consultant for Novartis and GSK. Another has been a consultant for Bristol–Myers Squibb (BMS), GSK, Novartis, Omrix, Roche and Schering-Plough and served on the speakers’ bureau for Roche, BMS and GSK Other: The percentages reported by the paper for RVR, EVR, EOT, SVR, relapse rate and SVR according to baseline viral load for both treatment groups are incorrect if the number of patients are calculated as a proportion of the whole group (n = 154). It is unclear what the denominator is |
|||||||
Quality criteria for assessment (updated CRD guidance)a
| 1. | Was the method used to generate random allocations adequate? | Yes |
| 2. | Was the allocation adequately concealed? | Unclear |
| 3. | Were the groups similar at the outset of the study in terms of prognostic factors, for example severity of disease? | Yes |
| 4. | Were outcome assessors blinded to the treatment allocation? | Unclear |
| 5. | Was the care provider blinded? | No |
| 6. | Was the patient blinded? | No |
| 7. | Were there any unexpected imbalances in dropouts between groups? If so, were they explained or adjusted for? | No |
| 8. | Is there any evidence to suggest that the authors measured more outcomes than they reported? | No |
| 9. | Did the analysis include an ITT analysis? | Yes |
| If so, was this appropriate? | Yes | |
| If so, were appropriate methods used to account for missing data? | Unclear |
Yu and colleagues54
| Reference and design | Intervention | Participants | Outcome measures | |
|---|---|---|---|---|
|
Author: Yu and colleagues54,58 Year: 2008 (2007) abstract Study design: Open-label, multicentre RCT No. of centres: Four Country: Taiwan Sponsor: Taiwan Liver Research Foundation |
Group 1: 24 weeks n = 100 Drug 1: PEG α-2a Dose: 180 µg/week s.c. Duration: 24 weeks Drug 2: RBV Dose: 1000 mg/day for body weight ≤ 75 kg and 1200 mg/day for body weight > 75 kg, oral, two divided doses Duration: 24 weeks Group 2: 48 weeks n = 100 Drug 1: PEG α-2a Dose: 180 µg/week s.c. Duration: 48 weeks Drug 2: RBV Dose: 1000 mg/day for body weight ≤ 75 kg and 1200 mg/day for body weight > 75 kg, oral, two divided doses Duration: 48 weeks |
Total numbers involved: 200 Intervention 1: 100 Intervention 2: 100 Treatment naive/non-responders/relapsers: Treatment naive Previous treatment: NA HCV/HIV co-infection: No Recruitment: One medical centre and three regional hospitals in Taiwan from April 2005 to May 2007 Inclusion criteria: Previously untreated Taiwanese patients with HCV aged 18–65 years. Seropositive for HCV antibodies and HCV RNA, had undergone liver biopsy that was consistent with HCV within 1 year before entry, elevated serum ALT for ≥ 2 measurements within 6 months before trial entry, genotype 1 infection, neutrophil count > 1500 mm–3, platelet count > 9 × 104 mm–3, Hb level > 12 g/dl men and > 11 g/dl for women, serum creatinine level < 1.5 mg/dl, no pregnancy/lactation, and use of reliable method of contraception Exclusion criteria: HCV genotype infections other than HCV-1, hepatitis B surface antigen, HIV infection, autoimmune hepatitis, primary biliary cirrhosis, sclerosing cholangitis, Wilson disease, alfa1-antitryspin deficiency, DC, overt hepatic failure, a current or history of alcohol abuse (≥ 20 g daily), psychiatric conditions, previous LT, or with evidence of HCC Baseline measurements: Viral load (log IU/ml), mean (± SD): Group 1: 5.43 ± 1.00 Group 2: 5.66 ± 0.95 p = 0.104 Lower viral load, < 400,000 IU/ml, n (%): Group 1: 55 (55%) Group 2: 56 (56%), p = not reported Serum ALT (IU/l) mean (± SD): Group 1: 156 ± 84 Group 2: 137 ± 92, p-value = 0.145 Histology: Fibrosis score, n (%): overall p = 0.306 F 0–2: Group 1: 75 (75), Group 2: 81 (81) F 3–4: Group 1: 25 (25), Group 2: 19 (19) Necroinflammatory score, mean (± SD): Group 1: 4.82 ± 2.55 Group 2: 4.41 ± 2.29 p = 0.241 Genotypes, n (%): 1 : 200 (100) 1a: 199 1b: 1 Gender male, n (%): Group 1: 57 (57%) Group 2: 58 (58%), p-value =0.886 Age (years), mean (± SD): Group 1: 49.7 ± 11.6 Group 2: 49.1 ± 12, p-value = 0.729 Ethnic groups, n (%): NR Mode of infection, n (%): NR Losses to follow-up: Group 1: Treatment terminated early n = 3 (adverse events n = 3), lost to follow up n = 0 Group 2: Treatment terminated early n = 10 (Adverse events n = 8, laboratory abnormalities n = 1, insufficient response n = 1, lost to follow up n = 1) Compliance: NR |
Primary outcomes: SVR Secondary outcomes: RVR EVR EOT virological response Relapse rate Adverse events Length of follow-up: 24 weeks (following treatment end) Methods of assessing outcomes: Bi-weekly outpatient visits in the first month then monthly visits during remaining treatment period and follow-up At each visit patients underwent physical examination and adverse events were recorded HCV genotypes determined by Okamoto. Serum HCV RNA at baseline, weeks 4 and 12, end of treatment and 24 weeks after treatment determined by qualitative PCR. Serum HCV RNA at baseline measured by qualitative PCR (limit 615 IU/ml). Liver histology according to Knodell and Scheuer |
|
|
PEG α, peginterferon alfa; RBV, ribavirin; s.c., subcutaneously. EVR was defined as PCR-negative or at least a 2-log10 decline from baseline of serum HCV RNA at 12 weeks of treatment; EOT virological response was defined as PCR-negative serum HCV-RNA (< 50 IU/ml) at the end of treatment; ‘relapse’ was defined as HCV RNA re-appearance during the follow-up period in patients who achieved an EOT virological response. RVR was defined by PCR-negative serum HCV RNA (< 50 IU/ml) at 4 weeks of therapy; ‘serum HCV RNA at baseline’, weeks 4, 12 end of treatment and 24 weeks after therapy, were determined by qualitative PCR – levels at baseline and week 12 of treatment were measured using the branched DNA assay (Versant HCV RNA 3.0, Bayer, Tarrytown, NJ; quantification limit: 615 IU/ml); SVR was defined as HCV RNA PCR-seronegative by the end of treatment and throughout the follow-up period. |
||||
| Outcome | Group 1, 24-week treatment (n = 100) | Group 2, 48-week treatment (n = 100) | p-value | |
| Viral response, % (95% CI) | ||||
| 4-week (RVR) | 45 (35 to 55) | 42 (32 to 52) | ||
| EVR | 95.9 (92 to 100) | 93 (88 to 98) | ||
| End of treatment | 93 (88 to 98) | 90 (84 to 96) | ||
| Relapse | 36.6 (27 to 47) | 12.2 (5 to 19) | < 0.0001 | |
| End of follow-up (SVR) | 59 (49 to 69) | 79 (71 to 87) | 0.002 | |
| SVR by RVR | ||||
| RVR, % (n/N, 95% CI) | 88.9 (40/45, 0.8 to 0.98) | 100 (42/42) | 0.056a | |
| No RVR, % (n/E, 95% CI) | 34.5 (19/55, 0.22 to 0.47) | 63.8 (37/58, 0.51 to 0.76) | 0.002b | |
|
a Difference 11.1%, (95% CI –22.6% to 4.2%). b Difference 29.2% (95% CI –48% to –13.4%). |
||||
| Group 1: 24 weeks | Group 2: 48 weeks | p-value | ||
| SVR by viral load and RVR, % (n/N, 95% CI): | ||||
| RVR and LVL (n =52) | 96.4% (27/28, 89 to 103) | 100% (24/24) | Difference –3.6% (–14.3% to –0.6%), p = 1.000 | |
| RVR and HVL (n = 35) | 76.5 (13/17, 56 to 97) | 100 (18/18) | 0.045 | |
| Relapse rate, % (n/N, 95% CI) | 36.6 (34/93, 27 to 47) | 12.2 (11/90, 5 to 19) | < 0.0001 | |
| Relapse rate by RVR: | ||||
| RVR | 11.1 (5/45, 0.02 to 0.2) | 0 (0/42) | Difference 11.1 (–0.4 to 18) | |
| No RVR | 60.4 (29/48, 0.46 to 0.74) | 22.9 (11/48, 0.11 to 0.35) | Difference 37.5 (17.2 to 53.7) | |
| Relapse rate by viral load and RVR, % (n/N, 95% CI): | ||||
| RVR and LVL (n = 52) | 3.6 (1/28, –3 to 11) | 0 (0/24) | Difference 3.6 (–7.2 to 6.6), p = 1.000 | |
| RVR and HVL (n = 35) | 23.5 (4/17, 3 to 44) | 0 (0/18) | 0.045 | |
| Adverse events n (%): | ||||
| Serious adverse events | 1 (1) | 1 (1) | ||
| Discontinuation | 3 (3) | 10c (10) | 0.045 | |
| Dose modification or transient interruption for adverse events or laboratory abnormalities: | ||||
| PEG α-2a | 22 (22) | 24 (24) | 0.737 | |
| RBV | 49 (49) | 60 (60) | 0.118 | |
| PEG α-2a or RBV | 54 (54) | 65 (65) | 0.113 | |
| Influenza-like symptoms including fever, chills, headache | 76 (76) | 74 (74) | 0.744 | |
| Gastrointestinal symptoms: | ||||
| Anorexia or nausea | 50 (50) | 53 (53) | 0.671 | |
| Diarrhoea | 18 (18) | 26 (26) | 0.172 | |
| Psychiatric symptoms: | ||||
| Anxiety | 31 (32) | 36 (36) | 0.454 | |
| Depression | 24 (24) | 34 (34) | 0.119 | |
| Insomnia | 59 (59) | 65 (65) | 0.382 | |
| Dermatological symptoms | ||||
| Hair loss | 66 (66) | 72 (72) | 0.359 | |
| Skin rash | 54 (55) | 66 (66) | 0.083 | |
| Haematological abnormality: | ||||
| Leucopenia (white cell count < 1500/mm3) | 5 (5) | 8 (8) | 0.39 | |
| Anaemia (Hb < 10 g/dl) | 39 (39) | 48 (48) | 0.199 | |
| Thrombocytopenia (< 50/mm3) | 2 (2) | 6(6) | 0.279 | |
| Abnormal thyroid tests | 13 (13) | 15 (15) | 0.684 | |
|
c Eight of these were owing to adverse events, one to insufficient serum creatinine level and one because of insufficient response. Serious adverse event: one patient with cirrhosis experienced variceal bleeding at EOT, one patient experienced severe myalgias over the lower back, resulting in disability of gait during treatment. |
||||
| Group 1: 24 weeks | Group 2: 48 weeks | |||
| SVR(–) | SVR(+) | SVR(–) | SVR(+) | |
| Additional results/comments | ||||
| Baseline HCV RNA level, log IU/ml | 5.92 ± 0.60 | 5.09 ± 1.08d | 5.93 ± 0.86 | 5.58 ± 0.96e |
| < 400,000 IU/ml, n (%) | 11 (26) | 34 (57.6)f | 8 (38.1) | 36 (45.6)g |
| ≥ 400,000 IU/ml, n (%) | 30 (73.2) | 25 (42.4)h | 13 (61.9) | 43 (54.4)h |
|
d p < 0.0001 between those with and without SVR in 24-week group (Group 1). e p = 0.132 between those with and without SVR in 48-week group (Group 2). f p = 0.002 between those with and without SVR in 24-week group (Group 1). g p = 0.540 between those with and without SVR in 48-week group (Group 2). h p-value not reported. |
||||
|
Adverse events were graded as mild, moderate, severe or potentially life threatening Significantly more patients with a lower baseline viral load (< 400, 000 IU/ml) achieved an RVR [RVR(+) 59.8% vs RVR(–) 32.7% p < 0.0001] Lower baseline viral load (< 400,000 IU/ml) was the only significant factor associated with RVR with an odds ratio of 3.052 (95% CI 1.706 to 5.458) The influence of other factors associated with the RVR (baseline demographical characteristics, ALT, liver histopathology, fibrosis and mean dose of RBV) were reported in the publication but none was significant and are not presented here In the 24-week group, RVR (p < 0.0001), lower viraemia (< 400,000 IU/ml) (p = 0.002), younger age (p = 0.055) and 80/80/80 adherence (p = 0.056) were predictive factors associated with a higher SVR rate (reviewer note: last two factors are borderline significance). Other factors predictive of SVR (baseline demographical characteristics, ALT, liver histopathology and fibrosis) were reported in the publication but these were not significant and are not reported here Independent predictors of SVR in the 24-week group were RVR and lower viraemia with odds ratios (CI) of 10.84 (3.189 to 36.82) and 3.087 (1.031 to 9.239), respectively. In the 48-week group, RVR was the only independent predictor of SVR, with an odds ratio of ‘infinity’ Independent predictors for SVR for all 200 patients (by logistic regression analysis) were RVR, followed by treatment duration, RBV dose and baseline viral load For 148 patients with either high viraemia or without an RVR the relapse rate was significantly higher in the 24-week group (50.8%, 95% CI 39 to 63) than in the 48-week group (16.7%, 95% CI 8 to 26) p < 0.0001). The SVR rate was significantly lower in the 24-week group (44.4%, 95% CI 33 to 56) than in the 48-week group (71.4%, 95% CI 62 to 82, p = 0.001) |
||||
| Methodological comments | ||||
|
Allocation to treatment groups: Randomly by computer coding, 1 : 1 randomisation ratio. The randomisation sequence was centrally accessed through telephone or direct office visit Allocation concealment: The details of the series were contained in a set of sealed envelopes and unknown to the investigators who enrolled subjects Blinding: Open label, therefore no blinding of participants or care providers. Liver histology graded and staged by single pathologist blinded to treatment, no further details of blinding of outcome assessors Analysis by ITT: ITT All patients receiving one dose of either study drug were analysed Comparability of treatment groups at baseline: Groups were comparable at baseline, there were no statistically significant differences (p-values were reported) Method of data analysis: Frequency was compared between groups using the chi-squared test with the Yates correction, or Fisher’s exact test. Groups means, presented as mean ± SD, were compared using analysis of variance and Student t-test, or Mann–Whitney test when appropriate. Serum HCV RNA levels were expressed after logarithmic transformation of original values. Analysis on spss. All statistical analyses were based on two-sided hypothesis test with a significance level of p < 0.05 Sample size/power analysis: The study was designed to detect a difference of 12% with 80% power or more, anticipating a 10% dropout rate.Attrition/dropout: 199/200 patients completed the study. One patient in the 48-week group was lost to follow-up 2 months after cessation of treatment and was classified as a non-responder for final analysis |
||||
| General comments | ||||
|
Generalisability: The study appears generalisable to treatment-naive, Asian patients with genotype 1 HCV. Mean baseline viral load (log 5.43 = 269,153 IU/ml and log 5.66 = 457,088 IU/ml for 24 weeks and 48 weeks, respectively) was low and approximately 55% had LVL (< 400,000 IU/ml). Approximately 43% had RVR at week 4 Inter-centre variability: NR Conflict of interests: It is stated that the sponsor did not participate in the study design, patient collection, analysis or interpretation |
||||
Quality criteria for assessment (updated CRD guidance)a
| 1. | Was the method used to generate random allocations adequate? | Yes |
| 2. | Was the allocation adequately concealed? | Yes |
| 3. | Were the groups similar at the outset of the study in terms of prognostic factors, for example severity of disease? | Yes |
| 4. | Were outcome assessors blinded to the treatment allocation? | Unclear |
| 5. | Was the care provider blinded? | No |
| 6. | Was the patient blinded? | No |
| 7. | Were there any unexpected imbalances in dropouts between groups? If so, were they explained or adjusted for? | No |
| 8. | Is there any evidence to suggest that the authors measured more outcomes than they reported? | No |
| 9. | Did the analysis include an ITT analysis? | Yes |
| If so, was this appropriate? | Yes | |
| If so, were appropriate methods used to account for missing data? | Yes |
Yu and colleagues55
| Reference and design | Intervention | Participants | Outcome measures |
|---|---|---|---|
|
Author: Yu and colleagues55 Year: 2007 Study design: Open-label, multicentre RCT No. of centres: 4 Country: Taiwan Sponsor: Taiwan Liver Research Foundation |
Intervention 1: 24 weeks n = 100 Drug 1: PEG α-2a Dose: 180 µg once/week, s.c. Duration: 24 weeks Drug 2: RBV Dose: 1000 mg/day for patients ≤ 75 kg, 1200 mg/day for patients > 75 kg (oral, two divided doses) Duration: 24 weeks Intervention 2: 16 weeks n = 50 Drug 1: PEG α-2a Dose: 180 µg once/week, s.c. Duration: 16 weeks Drug 2: RBV Dose: 1000 mg/day for patients ≤ 75 kg, 1200 mg/day for patients > 75 kg (oral, two divided doses) Duration: 16 weeks |
Total numbers involved: 326 screened, 150 eligible and randomised. n = 100 Group 1, n = 50 Group 2 Treatment naive/non-responders/relapsers: Treatment naive Previous treatment: NA HCV/HIV co-infection: No Recruitment: A medical centre and three regional core hospitals in Taiwan between September 2003 and December 2005 Inclusion criteria: Previously untreated adults (18–65 years) with HCV genotype 2, seropositive for HCV antibodies and for HCV RNA PCR, undergone liver biopsy within 1 year before entry (with result of chronic hepatitis C), increased serum ALT defined as ≥ 1.5 times the ULN for two or more measurements within 6 months preceding trial entry, neutrophil count > 1500/mm3, platelet count > 9 × 104/mm3, Hb > 12 g/dl for men and 11 g/dl for women, serum creatinine < 1.5 mg/dl, no pregnancy or lactation and using reliable contraception for women Exclusion criteria: HCV genotype other than type 2, hepatitis B surface antigen, HIV infection, autoimmune hepatitis, primary biliary cirrhosis, sclerosing cholangitis, Wilson’s disease α-antitrypsin deficiency, DC (Child–Pugh class B or C), overt hepatic failure, current or history of alcohol misuse (≥ 20 g/day) psychiatric condition, previous liver transplant, evidence of HCC Baseline measurements: Viral load (log IU/ml), mean (± SD): 4.88 (1.07) Group 1, 4.98 (1.08) Group 2, p = 0.62 Serum ALT (IU/l), mean (± SD): 108.9 (68.75) Group 1, 107 (64.6) Group 2, p = 0.857 Histology: Fibrosis, n (%): p = 0.832 F 0–2: 80 (80) Group 1, 39 (78) Group 2 F 3–4: 20 (20) Group 1, 11 (22) Group 2 Necroinflammatory score, mean (± SD): 4.84 (2.34) Group 1, 5.48 (3.32) Group 2, p = 0.226 Steatosis, n (%): p = 1 None (0): 67 (67) Group 1, 34 (68) Group 2 Mild (1): 28 (28) Group 1, 13 (26) Group 2 Moderate – severe (2–3): 5 (5) Group 1, 3 (6) Group 2 Genotypes, n (%): 100% genotype 2 Gender male, n (%): 58 (58%) Group 1, 32 (64%) Group 2 Age (years), mean (± SD): 49.9 (10.69) Group 1, 50.8 (9.74) Group 2, p = 0.621 Ethnic groups, n (%): 100% Asian (Taiwanese) Mode of infection, n (%): NR Losses to follow-up: 0 Compliance: 80/80/80 adherence, n (%): 73 (73) Group 1, 43 (86) Group 2 |
Primary outcomes: SVR Secondary outcomes: RVR ETVR (EOT virological response) Relapse rate Adverse events Length of follow-up: 24 weeks after cessation of treatment Methods of assessing outcomes: Patients had bi-monthly out-patient visits during the first month and monthly visits thereafter where they underwent a physical examination and adverse events were recorded. A citation was given (ref. 18) for the method by which HCV genotypes 1a, 1b, 2a, 2b and 3a were determined. Serum HCV RNA levels at baseline and during treatment week 4 were measured using the branched DNA assay, quantification limit 615 IU/ml. Serum HCV RNA at baseline, during treatment weeks 4, 12, EOT and at follow-up was determined by standardised automated qualitative PCR, detection limit 50 IU/ml. Scheuer and Knodell scoring system used for liver histology |
| ALT, alanine transaminase level; ETVR, EOT virological response (defined as PCR-negative serum HCV RNA at end of treatment); HCC, hepatocellular carcinoma; PCR, polymerase chain reaction assay; 80/80/80 adherence, patients who had received > 80% of expected PegIFN and RBV doses and completed at least 80% of expected duration; PEG α, peginterferon alfa; RBV, ribavirin; RVR, defined as PCR-negative serum HCV RNA at 4 weeks of treatment); s.c., subcutaneously; SVR, defined as PCR-negative serum HCV RNA by end of treatment and end of follow-up); ‘non-response’ defined as not achieving SVR; ‘relapse’ defined as re-appearance of HCV RNA during follow-up period in patients who achieved an ETVR; ULN, upper limit of normal. | |||
| Outcome | Intervention 1 (24 weeks) | Intervention 2 (16 weeks) | p-value |
| Viral response, % (n/N, 95% CI): | |||
| 4-week (RVR) | 87 (87/100, 80 to 94) | 86 (43/50, 76 to 96) | |
| 12-week (EVR) | – | – | |
| End of treatment (ETVR) | 98 (98/100, 95 to 100) | 100 | |
| End of follow-up (SVR) | 95% (95/100, 91 to 99) | 94 (47/50, 87 to 100) | Difference –1%, 95% CI 9 to 7 |
| SVR by RVR, % (n/N ): | |||
| RVR | 98 (85/87) | 100 (43/43) | 1 |
| No RVR | 77 (10/13) | 57 (4/7) | 0.610 |
| SVR by baseline HCV RNA, % (n/N ): | |||
| < 800,000 IU/ml | 95 (81/85) | 95 (39/41) | 1 |
| > 800,000 IU/ml | 93 (14/15) | 89 (8/9) | 1 |
| SVR by viral load & RVR, % (n/N ) | NR | NR | |
| Other viral response outcomes: | |||
| Relapse rate, % (n/N, 95% CI) | 3.1 (3/98, –1 to 13) | 6 (3/50, 0 to 7) | Difference (not reported) 95% CI –10.4 to 4.5 |
| Relapse rate by baseline HCV RNA, % (n/N ): | |||
| < 800,000 IU/ml | 3.6 (3/84) | 4.9 (2/41) | 1.000 |
| > 800,000 IU/ml | 0 (0/14) | 11.1 (1/9) | 0.391 |
| Relapse rate by RVR,% (n/N ): | |||
| RVR | 2.3 (2/87) | 0 (0/43) | 0.554 |
| No RVR | 9.1 (1/11) | 42.9 (3/7) | 0.245 |
| Biochemical response, % (n/N ): | NR | NR | 1 |
| End of treatment | |||
| End of follow-up | |||
| Histology (proportion with improvement): | NR | NR | 1 |
| Inflammation, mean change | |||
| Fibrosis, mean change | |||
| Adverse events, n (%) | |||
| Dose discontinuation for any adverse event | 1 (1%) | 0 | 1 |
| Dose modification for adverse events or lab abnormalities: | |||
| PEG | 9 (9) | 4 (8) | 1 |
| RBV | 51 (51) | 23 (46) | 0.564 |
| PEG or RBV | 54 (54) | 26 (52) | 0.817 |
| Specific adverse events | |||
| Flu-like symptoms: | |||
| Fever | 55 (55) | 29 (58) | 0.727 |
| Chills | 28 (28) | 12 (24) | 0.602 |
| Headache | 39 (39) | 21 (42) | 0.724 |
| Gastrointestinal symptoms: | |||
| Anorexia | 46 (46) | 20 (40) | 0.601 |
| Nausea | 15 (15) | 3 (6) | 0.181 |
| Diarrhoea | 9 (9) | 5 (10) | 1 |
| Psychiatric symptoms: | |||
| Anxiety | 7 (7) | 4 (8) | 1 |
| Depression | 10 (10) | 3 (6) | 0.545 |
| Insomnia | 57 (57) | 23 (46) | 0.227 |
| Dermatological symptoms: | |||
| Hair loss | 49 (49) | 10 (20) | 0.001 |
| Skin rash | 54 (54) | 22 (44) | 0.248 |
| Haematological abnormality: | |||
| Leucopenia (white cell count < 1500/mm3) | 2 (2) | 1 (2) | 1 |
| Anaemia (Hb < 10 g/dl) | 53 (53) | 27 (54) | 1 |
| Thrombocytopenia (< 50,000/mm3) | 1 (1) | 0 | 1 |
| Abnormal thyroid function tests | 13 (13) | 4 (8) | 0.362 |
| Additional results/comments (e.g. early response factors, QoL) | |||
| Virological response | |||
|
Within treatment groups, mean (± SD) baseline HCV RNA level was not significantly different in patients who achieved an SVR compared to those who did not for both the 24-week Group (4.86 ± 1.08 vs 5.33 ± 0.55, p = 0.342) and the 16-week Group (4.93 ± 1.1 vs 5.63 ± 0.35, p = 0.283) Within treatment groups, significantly more patients who achieved an SVR had an RVR at 4 weeks compared with those who did not achieve an SVR in both the 24-week Group [90% (85/95) vs 40% (2/5), p = 0.015] and the 16-week Group [92% (43/47) vs 0% (0/3), p = 0.002]. No other baseline factors were significantly associated with an SVR Factors significantly associated with SVR were RVR at week 4 (OR 40.76, 95% CI 5.964 to 278.6) and patient’s age (OR 0.834, 95% CI 0.721 to 0.965). Treatment duration was not associated with SVR (OR 1.241, 95% CI 0.186 to 8.279) For patients without an RVR, the relapse rate was higher in the 16-week Group (42.9%, 95% CI –7% to 92%) than in the 24-week Group (9.1%, 95% CI –11% to 29%), and the SVR rate was lower in the 16-week Group (57%, 95% CI 20 to 94) than in the 24-week Group (77%, 95% CI 54 to 99), but neither were statistically significant The influence of a number of other prognostic factors (baseline demographical characteristics, liver histopathology, 80/80/80 adherence and received doses of PEG and RBV) on the SVR rate were reported in the publication, but none was significant and they are not presented here. Similarly, between group differences in relapse rate and SVR rate were reported by age, sex, body mass index, fibrosis, steatosis, 80/80/80 adherence, received RBV doses and dose modifications, but none was significant Results were reported for mean RBV dose throughout the treatment period stratified by RVR, SVR and treatment duration (but these are not presented here) |
|||
| Safety | |||
|
Treatment was discontinued by 1 patient (24-week Group) due to anaemia and leucopenia at week 23 PEG dose reductions were due to adverse events (n = 5), leucopenia (n = 3), anaemia (n = 4) and thrombocytopenia (n = 1) Adverse events were typical of those previously reported for PEG and RBV combination treatment No serious adverse event was reported |
|||
| Methodological comments | |||
|
Allocation to treatment groups: Patients were assigned randomly by computer coding in a 1 : 2 randomisation ratio Allocation concealment: The computer-generated code was generated by a contract research organisation independent of the study and was centrally accessed through telephone or direct office visit. Details of the series were contained in sealed envelopes and were unknown to any of the investigators who enrolled patients for the study Blinding: No blinding of participants and care providers (open label) and blinding of outcome assessors not reported, except for biopsy pathologists Analysis by ITT: States that evaluation of efficacy was based on ITT analysis and that all patients receiving a treatment dose of PEG or RBV were analysed. SVR was reported for all 150 randomised patients Comparability of treatment groups at baseline: Participant baseline demographics were well matched between arms with no statistically significant differences (p-values reported). Patients in 16-week Group had slightly higher 80/80/80 adherence than the 24-week Group (86% vs 73%, p = 0.073), but the difference was not significant Method of data analysis: Frequency was compared between groups using the chi-squared test, with the Yates correction, or Fisher’s exact test. Group means were compared using analysis of variance and Student’s t-test or non-parametric Mann–Whitney U-test when appropriate. Serum HCV RNA levels were expressed after log transformation of original values. Stepwise logistical regression was used to analyse which variables had a better predictive value for SVR (using spss version12.0). All statistical analyses were based on two-sided hypothesis tests with a significance level of p < 0.05 Sample size/power analysis: Assuming an SVR rate of 82% for 24 weeks’ treatment and no SVR if untreated, the study was powered to detect a difference of ≥ 24.6% with 80% power, anticipating a 10% dropout rate. It is reported that this margin is equivalent to other published data (reference cited) Attrition/dropout: Reasons for the one dropout were provided |
|||
| General comments | |||
|
Generalisability: Treatment-naive, Taiwanese Asian patients with genotype 2 HCV. Mean baseline viral load (log 4.88 = 75,860 IU/ml and log 4.98 = 95,500 IU/ml for 24 weeks and 16 weeks, respectively) was low, and when SVR was measured (at 24-week follow-up) approximately 83% had baseline LVL (< 800,000 IU/ml). The majority (86%) had RVR at week 4 Inter-centre variability: NR Conflict of interests: None |
|||
Quality criteria for assessment (updated CRD guidance)a
| 1. | Was the method used to generate random allocations adequate? | Yes |
| 2. | Was the allocation adequately concealed? | Yes |
| 3. | Were the groups similar at the outset of the study in terms of prognostic factors, for example severity of disease? | Yes |
| 4. | Were outcome assessors blinded to the treatment allocation? | Unclear |
| 5. | Was the care provider blinded? | No |
| 6. | Was the patient blinded? | No |
| 7. | Were there any unexpected imbalances in dropouts between groups? If so, were they explained or adjusted for? | No |
| 8. | Is there any evidence to suggest that the authors measured more outcomes than they reported? | No |
| 9. | Did the analysis include an ITT analysis? | Yes |
| If so, was this appropriate? | Yes | |
| If so, were appropriate methods used to account for missing data? | Yes |
von Wagner and colleagues56
| Reference and design | Intervention | Participants | Outcome measures |
|---|---|---|---|
|
Author: von Wagner and colleagues56 Year: 2005 Study design: Multicentre, Phase IIIb RCT No. of centres: 6 Country: Germany Sponsor: Hoffmann-La Roche and the German Hepatitis Network of Competence (Hep-Net) |
n = 153 PEG -2a Dose: 180 µg/week, s.c. Duration: 8 weeks RBV Dose: 800 mg/day for patients ≤ 65kg, 1000 mg/day for patients 65–85 kg, 1200 mg/day for patients > 85 kg, oral Duration: 8 weeks Those with RVR at week 4 randomised at week 8 to: Intervention 1: 16 weeks, RVR (Group A) n = 71 Drug 1: PEG α-2a Dose: 180 µg/week, s.c. Duration: 8 weeks Drug 2: RBV Dose: 800 mg/day for patients ≤ 65 kg, 1000 mg/day for patients 65–85 kg, 1200 mg/day for patients > 85 kg; oral Duration: 8 weeks (total duration 16 weeks) Intervention 2: 24 weeks, RVR (Group B) n = 71 Drug 1: PEG α-2a Dose: 180 µg/week, s.c. Duration: 16 weeks Drug 2: RBV Dose: 800 mg/day for patients ≤ 65kg, 1000 mg/day for patients 65–85 kg, 1200 mg/day for patients > 85 kg; oral Duration: 16 weeks (total duration 24 weeks) Patients without an RVR at week 4 allocated at week 8 to: Intervention 3: 24 weeks, no RVR (Group C) a n = 11 Drug 1: PEG α-2a Dose: 180 µg/week, s.c. Duration: 16 weeks Drug 2: RBV Dose: 800 mg/day for patients ≤ 65kg, 1000 mg/day for patients 65–85 kg, 1200 mg/day for patients > 85 kg; oral Duration: 16 weeks (total duration 24 weeks) |
Total numbers involved: 153 enrolled; 142 randomised at week 8 (Groups A and B) Treatment naive/non-responders/relapsers: Treatment naive Previous treatment: NA HCV/HIV co-infection: No Recruitment: Six tertiary referral centres in Germany between January 2002 and March 2004 Inclusion criteria: Adults (> 18 years), not previously treated with IFN and/or RBV, with compensated chronic HCV genotype 2 or 3, positive for anti-HCV antibody and HCV RNA > 600 IU/ml, liver biopsy within 18 months prior to screening, ≥ 1 serum ALT level elevated at screening or study entry, neutrophil count ≥ 1500/l, platelet count ≥ 90,000/l, Hb ≥ 13g/dl for men and ≥ 12g/dl for women Exclusion criteria: Any other cause of liver disease or other relevant disorders including HIV or hepatitis B co-infection, clinically significant haematological, hepatic, metabolic, renal, rheumatological, neurological or psychiatric disease, clinically significant cardiac or cardiovascular abnormalities, organ grafts, systemic infection, clinically significant bleeding disorders, evidence of malignant neoplastic disease, concomitant immunosuppressive medication, excessive daily intake of alcohol or drug abuse within past year, pregnancy, lactation, male partners of pregnant women Baseline measurements: Viral load (log IU/ml), mean (± SD): 5.8 (±0.7) Group A 5.8 (±0.8) Group B 5.7 (±0.5) Group C Serum ALT × ULN (IU/l), mean (± SD): 2.8 (± 2.9) Group A 2.8 (± 2.0) Group B 2.4 (± 0.9) Group C Histology: Classification system used: Ishak Fibrosis score, mean (± SD): 1.6 (±1.4) Group A 1.6 (±1.1) Group B 2.4 (±2.3) Group C Necroinflammatory score (total inflammation), mean (± SD): 4.3 (±2.4) Group A 4.6 (±2.4) Group B 5.0 (±4.0) Group C Genotypes, n (%): Genotype 2, genotype 3: 19/71 (27%) Group A,a 51/71 (72%) Group A 19/71 (27%) Group B, 52/71 (73%) Group B 1/11 (9%) Group C, 10/11 (91%) Group C (aG2 or 3 could not be differentiated in one patient) Gender male, n (%): 52 (73%) Group A 41 (58%) Group B 4 (36%) Group C Age (years), mean (± SD): 38 (± 9) Group A 39 (± 11) Group B 42 (± 10) Group C Ethnic groups, n (%): NR Mode of infection, n (%): NR Losses to follow-up: 144/153 (94%) completed treatment; n = 9 (3 Group A, 6 Group B) lost to follow-up. However, those who withdrew prematurely from treatment were encouraged to return for follow-up. 142/153 (93%) completed follow-up (68 Group A, 65 Group B and 9 Group C) Compliance: n = 9 discontinued treatment (1 Group A, 6 Group B, 2 Group C), n = 8 prematurely withdrew for non-safety reasons (1 Group A, 5 Group B, 2 Group C) |
Primary outcomes: SVR Secondary outcomes: RVR EOT virological response Sustained biochemical response Virological response according to genotype and baseline viraemia Adverse events Length of follow-up: 24 weeks after end of treatment Methods of assessing outcomes: Evaluated at weeks 2, 4, 8, 12, 16, 20 and 24 (Groups B and C) during treatment and at weeks 4, 12 and 24 following end of treatment. During treatment, HCV RNA quantified by PCR assay, end of treatment and SVR assessed by qualitative PCR assay (lower detection limit 50 IU/ml). HCV genotyping performed by reverse hybridisation; histology classified according to Ishak |
|
ALT, alanine transaminase level; PCR, polymerase chain reaction assay; PEG α, peginterferon alfa; RBV, ribavirin; RVR, defined as serum HCV RNA < 600 IU/ml at 4 weeks of treatment; SVR, defined as undetectable serum HCV RNA 24 weeks after end of treatment; s.c., subcutaneously; ULN, upper limit of normal. a Not randomised. |
|||
| Outcome | Group A (n = 71), 16 weeks, RVR | Group B (n = 71), 24 weeks, RVR | Group C (n = 11), 24 weeks, no RVR |
| Viral response, % (n/N ): | |||
| 4-week (RVR) | 100 | 100 | 0 |
| 12-week (EVR) | – | – | – |
| End of treatment | 94 (67/71) | 85 (60/71) | 72c |
| End of follow-up (SVR) | 82 (58/71)b | 80 (57/71) | 36d |
| SVR by genotype and baseline viral load, % (n/N ): | |||
| Genotype HCV-2 (n = 38): | |||
| ≤ 800,000 IU/ml | 100 (6/6) | 100 (6/6) | – |
| > 800,000 IU/ml | 93 (12/13) | 93 (12/13) | – |
| Genotype HCV-3 (n = 103): | |||
| ≤ 800,000 IU/ml | 93 (27/29) | 84 (21/25) | – |
| > 800,000 IU/ml | 54 (12/22) | 67 (18/27)e | – |
| SVR by baseline viral load and RVR, % (n/N ): | |||
| ≤ 800,000 IU/ml (n = 66) | 94 (33/35) | 87 (27/31) | – |
| > 800,000 IU/ml (n = 75) | 69 (24/35) | 75 (30/40) | – |
| Biochemical response, % (n/N ): | |||
| End of treatment | – | – | – |
| End of follow–up | 89 | 87 | 67 |
| Histology: | |||
| Inflammation | NR | NR | NR |
| Fibrosis | NR | NR | NR |
| Discontinuation: | |||
| For adverse events | 0 | 1 (1.4%)f | 0 |
| For other reason | 1 (1.4%) | 5 (7.0%) | 2 (18.2%) |
| Dose modification for adverse events/laboratory abnormalities: | |||
| PEG | 5 (7.0%) | 13 (18.8%) | 4 (36.4%) |
| RBV | 6 (8.5%) | 8 (11.3%) | 3 (27.3%) |
| Specific adverse events:g | |||
| Flu-like symptoms | 37 (52.1%) | 33 (46.5%) | 2 (18.2%) |
| Fatigue | 26 (36.6%) | 30 (42.3%) | 8 (72.7%) |
| Pruritus | 19 (26.8%) | 24 (33.8%) | 3 (27.3%) |
| Headache | 18 (25.4%) | 22 (31.0%) | 6 (54.5%) |
| Anorexia | 16 (22.5%) | 19 (26.8%) | 3 (27.3%) |
| Alopecia | 15 (21.1%) | 18 (25.4%) | 2 (18.2%) |
| Asthenia | 12 (16.9%) | 18 (25.4%) | 2 (18.2%) |
| Pain | 9 (12.7%) | 16 (22.5%) | 5 (45.5%) |
| Dyspnoea | 10 (14.1%) | 16 (22.5%) | 3 (27.3%) |
| Sleeping disturbance | 9 (12.7%) | 16 (22.5%) | 4 (36.4%) |
| Pyrexia | 10 (14.1%) | 13 (18.3%) | 3 (27.3%) |
| Dry skin | 13 (18.3%) | 9 (12.7%) | 0 |
| Aggressivity | 8 (11.3%) | 12 (16.9%) | 0 |
| Depression | 8 (11.3%) | 10 (14.1%) | 2 (18.2%) |
| Chills | 10 (14.1%) | 8 (11.3%) | 1 (9.1%) |
| Nausea | 5 (7.0%) | 11 (15.5%) | 3 (27.3%) |
| Dry mouth | 4 (5.6%) | 8 (11.3%) | 4 (36.4%) |
|
b Difference of at most 11.5% (97.5% one-sided CI) for Group A vs B. c p = NS for Group B vs C. d p = 0.005 for Group B vs C. e p > 0.2 for Group A vs B. f Intravenous drug abuse. g Related to treatment, as judged by investigators, that occurred in at least 10% of patients who received at least one dose of study medication. |
|||
| Additional results/comments (e.g. early response factors, QoL) | |||
| Virological response | |||
|
After first 4 weeks of treatment, RVR (HCV RNA < 600 IU/ml) was achieved by 142/152 (93%) patients, made up of 37/38 (97%) genotype 2 and 103/112 (92%) genotype 3 (p > 0.2). These patients and one patient who was negative at week 2 with a missing HCV RNA result at week 4 were randomised to groups A (n = 71) and B (n = 71) An overall ITT EOT response was achieved in 135/153 patients (88%), and an SVR in 119/153 patients (78%) |
|||
| SVR according to genotype and pre-treatment viraemia | |||
|
SVR in genotype HCV-2 patients were higher than in HCV-3 patients (92% vs 73%, respectively) (no p-value reported), and were not affected by pretreatment viraemia. However, HCV-3 patients with a baseline viraemia > 800,000 IU/ml achieved a significantly lower SVR than patients with baseline viraemia ≤ 800,000 IU/ml (59% vs 85%, respectively, p = 0.003) There were no significant differences between groups A and B for SVR rates for patients with either HCV-2 or HCV-3 |
|||
| Predictors of SVR | |||
| From multivariate logistic regression analysis of all patients, genotype HCV-2, LVL and low γ-glutamyltransferase (GGT) value were independent factors of SVR. Based on patients with HCV-3 only, baseline viral load (p = 0.01) and GGT value (p = 0.02) remained as independent negative predictors for SVR. Fibrosis score and GGT were slightly higher in patients without a RVR (Group C) compared with patients with RVR (Groups A and B); however, differences did not reach statistical significance | |||
| Biochemical response | |||
| Sustained biochemical response was observed in 110/115 sustained virological responders (96%), whereas five sustained virological responders did not show a biochemical response with ALT levels ranging up to 2.95 times the upper limit of normal. Each of the five subjects was infected with genotype HCV-2 | |||
| Adverse events | |||
|
Seven serious adverse events were reported (bacterial infection, carcinoma, diverticulitis, paranoid reaction, pneumonia, pregnancy of partner, tuberculosis) Adverse events were similar to those previously reported for PEG + RBV. In general, the frequency of adverse events was lower in Group A than in Groups B and C. (Reviewer note: No statistical comparison reported.) Neutropenia (3%) and anaemia (6%) were the most common adverse events leading to dose modification |
|||
| Methodological comments | |||
|
Allocation to treatment groups: No details about the randomisation method were reported. Patients with a RVR at week 4 of therapy were randomised 1 : 1 at week 8. Patients were stratified according to baseline viraemia (≤ 800,000 IU/ml vs > 800,000 IU/ml) and treatment centre. Allocation concealment: NR Blinding: Patients randomised at week 8 were informed about their treatment group assignment at the next clinical visit and were therefore not blinded. No details reported regarding blinding of outcome assessors Analysis by ITT: ITT analysis for efficacy and safety variables (n = 153). One patient with a negative HCV RNA result at week 2 and missing data at week 4 was allocated to the RVR group (not reported whether Group A or B). One patient with missing data at weeks 2 and 4 was allocated to Group C Comparability of treatment groups at baseline: Generally baseline demographic and disease characteristics were comparable across treatment groups. However, mean fibrosis score was higher in Group C vs Groups A and B (2.4 vs 1.6 and 1.6, respectively); also the proportion of genotype 3 patients was higher in Group C vs Groups A and B (91% vs 72% and 73%, respectively). Characteristics for Group A vs B were comparable. No statistical comparison was presented Method of data analysis: The primary statistical analysis was the determination of a one-sided 97.5% CI for the difference in SVR rates between treatment groups A and B. Fisher’s exact test and chi-squared tests were applied to compare different rates. Multivariate logistic regression was performed to identify independent predictors of RVR and SVR. Unless stated otherwise, p-values of < 0.05 were considered significant Sample size/power analysis: The study was powered to detect a difference of 25% or more with a power of at least 80% Attrition/dropout: Numbers reported but reasons not fully reported Other: SVR rates reported for Group B in text are not consistent. Top right paragraph on p. 524 reports an SVR of 81% and EOT response of 84% for Group B, but in the previous paragraph reported 80% and 85%, respectively. Differences are possibly due to rounding of figures |
|||
| General comments | |||
|
Generalisability: Treatment-naive patients with genotype 2 and 3 HCV. Mean baseline viral load (log10 5.8 = 631,000 IU/ml, log10 5.8 = 631,000 and log10 5.7 = 501,200 IU/ml for Group A, Group B and Group C, respectively) was low and all patients in Groups A and B had RVR at week 4 Intercentre variability: NR Conflict of interests: The study was partly supported by the drug manufacturer (Roche) |
|||
Quality criteria for assessment (updated CRD guidance)a
| 1. | Was the method used to generate random allocations adequate? | Unclear |
| 2. | Was the allocation adequately concealed? | Unclear |
| 3. | Were the groups similar at the outset of the study in terms of prognostic factors, for example severity of disease? | Yes |
| 4. | Were outcome assessors blinded to the treatment allocation? | Unclear |
| 5. | Was the care provider blinded? | No |
| 6. | Was the patient blinded? | No |
| 7. | Were there any unexpected imbalances in dropouts between groups? If so, were they explained or adjusted for? | No |
| 8. | Is there any evidence to suggest that the authors measured more outcomes than they reported? | No |
| 9. | Did the analysis include an ITT analysis? | Yes |
| If so, was this appropriate? | Yes | |
| If so, were appropriate methods used to account for missing data? | Yes |
Appendix 7 List of excluded studies
The reasons for study exclusion were applied in the order given in the inclusion criteria worksheet (see Appendix 4). Studies may have been excluded on more than one criterion but only the primary reason is given.
Reason for exclusion: study design
Aguilera V, Rubin A, Benlloch S, Zamora PA, Ortiz C, Prieto M, et al. Systematic review of the treatment of established recurrent hepatitis C with pegylated interferon in combination with ribavirin. Liver Transpl 2008;14:S178.
Alberti A, Zehnter E, Lee S, Hadziyannis S, Zeuzem S, Rizzetto M, et al. Sustained virological response rates with peginterferon alpha-2a (40 kd) (PEGASYS®) plus ribavirin (COPEGUS®) in randomised controlled clinical trials are replicated in the clinical practice setting. J Hepatol 2007;46(Suppl. 1).
Andriulli A, Mangia A, Iacobellis A, Ippolito A, Leandro G, Zeuzem S. Meta-analysis: the outcome of anti-viral therapy in HCV genotype 2 and genotype 3 infected patients with chronic hepatitis. Aliment Pharmacol Ther 2008;28:397–404.
Berg C, Goncales FL Jr, Bernstein DE, Sette H Jr, Rasenack J, Diago M, et al. Re-treatment of chronic hepatitis C patients after relapse: efficacy of peginterferon-alpha-2a (40 kDa) and ribavirin. J Viral Hepat 2006;13:435–40.
Cervoni J, Richou C, Thevenot T, Di Martino V. Shortened course of therapy for chronic hepatitis C genotype 1 (G1) patients developing rapid virological response (RVR): meta-analysis of randomized controlled trials (RCTs). J Hepatol 2009;50(Suppl. 1):220–1.
Condat B. Peginterferon alpha-2b plus ribavirine compared with interferon alpha-2 and ribavirine for the treatment of chronic hepatitis C: a randomized trial. Hepatogastoenterology 2002;9:141–2.
Derbala M, Amer A, Bener A, Lopez AC, Omar M, El GM. Pegylated interferon-alpha 2b-ribavirin combination in Egyptian patients with genotype 4 chronic hepatitis. J Viral Hepat 2005;12:380–5.
Di Martino V, Richou C, Thevenot T, Sanchez-Tapias JM, Ferenci P. Modulations of peg-interferon plus ribavirin duration according to HCV-genotype and virologic response at w4 and w12: meta-analyses of RCTs with individual data. Hepatology 2008;48:404A.
Grewal AS, Choudhary A, Bechtold ML, Puli SR, Othman MO, Roy PK. Peginterferon and ribavirin for treatment of hepatitis C and HIV co-infection: a meta-analysis of randomized controlled trials. Gastroenterology 2008;134(Suppl. 1).
Mohsen A, Norris S. Hepatitis C (chronic). Clin Evid Handbook 2007:262–4.
Moreno L, Quereda C, Moreno A, Perez-Elias MJ, Antela A, Casado JL, et al. Pegylated interferon alpha 2b plus ribavirin for the treatment of chronic hepatitis C in HIV-infected patients. AIDS 2004;18:67–73.
Nunez M, Marino A, Miralles C, Berdun MA, Sola J, Hernandez-Burruezo JJ, et al. Baseline serum hepatitis C virus (HCV) RNA level and response at week 4 are the best predictors of relapse after treatment with pegylated interferon plus ribavirin in HIV/HCV co-infected patients. J Acquir Immune Defic Syndr 2007;45:439–44.
Nunez M, Miralles C, Berdun MA, Losada E, Aguirrebengoa K, Ocampo A, et al. Role of weight-based ribavirin dosing and extended duration of therapy in chronic hepatitis C in HIV-infected patients: The PRESCO trial. AIDS Res Hum Retroviruses 2007;23:972–82.
Opravil M, Sasadeusz J, Cooper DA, Rockstroh JK, Clumeck N, Clotet B, et al. Effect of baseline CD4 cell count on the efficacy and safety of peginterferon alfa-2a (40kd) plus ribavirin in patients with HIV/hepatitis C virus co-infection. J Acquir Immune Defic Syndr 2008;47:36–49.
Perronne C, Carrat F, Banisadr F, Morand P, Lunel F, Rosenthal E, et al. ANRS HC02-Ribavic: a randomized controlled trial of pegylated interferon alpha-2b plus ribavirin vs interferon alpha-2b plus ribavirin as primary treatment of chronic hepatitis C in HIV co-infected patients. Hepatology 2002;36:283A.
Poynard T, Schiff E, Terg R, Moreno Otero R, Flamm S, Schmidt W, et al. Sustained viral response (SVR) is dependent on baseline characteristics in the re-treatment of previous alfa interferon/ribavirin (I/R) nonresponders (NR): final results from the EPIC3 program. 43rd Annual Meeting of the European Association for the Study of the Liver (EASL), 23–27 April 2008, Milan, Italy.
Rodriguez-Torres M, Rodriguez-Orengo JF, Rios-Bedoya CF, Fernandez-Carbia A, Gonzalez-Lassalle E, Salgado-Mercado R, et al. Efficacy and safety of peg-IFN alfa-2a with ribavirin for the treatment of HCV/HIV co-infected patients who failed previous IFN based therapy. J Clin Virol 2007;38:32–8.
Sanchez-Tapias JM, Diago M, Escartin P, Enriquez J, Romero-Gomez M, Barcena R, et al. Peginterferon-alfa2a plus ribavirin for 48 versus 72 weeks in patients with detectable hepatitis C virus RNA at week 4 of treatment. Gastroenterology 2006;131:451–60.
Sarrazin C, Schwendy S, Moller B, Dikopoulos N, Buggisch P, Encke J, et al. Individualized treatment duration with peginterferon-alfa-2B and ribavirin for 24, 30 or 36 weeks in HCV genotype 1-infected patients with undetectable HCV-RNA early during therapy (INDIV-2 STUDY). J Hepatol 2009;50(Suppl. 1):236.
Schiff E, Poordad F, Jacobson I, Flamm S, Bacon B, Lawitz E, et al. Boceprevir (B) combination therapy in null responders (NR): response dependent on interferon responsiveness. J Hepatol 2008;48(Suppl. 2):46.
Shiffman ML, Mansbach H, Hammond J, O’Neill M. The effect of complete and partial response at week 12 on sustained virologic response: results from controlled trials in naive HCV genotype 1 patients treated with pegylated interferon and ribavirin. Hepatology 2007;46(Suppl.):824A–5A.
Shire NJ, Welge JA, Sherman KE. Response rates to pegylated interferon and ribavirin in HCV/HIV co-infection: a research synthesis (Cochrane provisional abstract). J Viral Hepat 2007;14:239–48.
Slavenburg S, Weggelaar I, van Oijen MGH and Drenth JPH. Optimal length of antiviral therapy in patients with hepatitis C virus genotype 2 and 3: a meta-analysis. EASL 44th Annual Meeting, 22–26 April 2009, Copenhagen, Denmark.
Yoshida EM, Sherman M, Bain VG, Cooper CL, Deschenes M, Marotta PJ, et al. Retreatment with pegylated interferon alpha-2a and ribavirin in patients with chronic hepatitis C who have relapsed or not responded to a first course of pegylated interferon-based therapy. Can J Gastroenterol 2009;23:180–4.
Yu ML, Dai CY, Huang JF, Hou NJ, Lee LP, Hsieh MY, et al. A randomized, controlled, open-label study of peginterferon alfa-2A (40KD) (PEGASYS®) plus ribavirin (COPEGUS®) for 16 vs. 24 weeks in patients with genotype 2 hepatitis C infection. Hepatology 2006;44:267A.
Zoulim F. Treatment of patients with chronic hepatitis C who relapsed or did not respond to a previous treatment. Gastroenterol Clin Biol 2002;26:225–230.
Reason for exclusion: population
Berak H, Kolakowska-Rzadzka A, Wasilewski M, Kowalska J, Stanczak JJ, Bardadin K, et al. Randomized, open label trial comparing efficacy and safety of pegylated interferon alfa 2b vs alfa 2b treatment of patients with chronic hepatitis C infected with non 2/3 genotypes: final analysis. J Hepatol 2007;46 (Suppl. 1):217–18.
Brady DE, An JW, Lawitz EJ, Harrison S. Does induction pegylated interferon alfa-2b in combination with ribavirin enhance the sustained response rates in patients with genotype 1 and 4 chronic hepatitis C? Results from a prospective, randomized, multi-center, open-label treatment study. Hepatology 2008;48:402A.
Brandao C, Barone A, Carrilho F, Silva A, Patelli M, Caramori C, et al. The results of a randomized trial looking at 24 weeks vs 48 weeks of treatment with peginterferon alpha-2a (40 kDa) and ribavirin combination therapy in patients with chronic hepatitis C genotype 1. J Viral Hepat 2006;13:552–9.
Dalgard O, Bjoro K, Ring-Larsen H, Verbaan H. Peginterferon alpha-2b and ribavirin for 14 or 24 weeks in patients with HCV genotype 2 or 3 and rapid virological response. The North-C trial. J Hepatol 2007;46(Suppl. 1):S57.
Dalgard O, Bjoro K, Ring-Larsen H, Bjornsson E, Holberg-Petersen M, Skovlund E, et al. Pegylated interferon alfa and ribavirin for 14 versus 24 weeks in patients with hepatitis C virus genotype 2 or 3 and rapid virological response. Hepatology 2008;47:35–42.
Diago M, Crespo J, Olveira A, Perez R, Barcena R, Sanchez-Tapias JM, et al. Clinical trial: pharmacodynamics and pharmacokinetics of re-treatment with fixed-dose induction of peginterferon alpha-2a in hepatitis C virus genotype 1 true non-responder patients. Aliment Pharmacol Ther 2007;26:1131–8.
Everson GT, Hoefs JC, Seeff LB, Bonkovsky HL, Naishadham D, Shiffman ML, et al. Impact of disease severity on outcome of antiviral therapy for chronic hepatitis C: lessons from the HALT-C trial. Hepatology 2006;44:1675–84.
Ferenci P, Laferl H, Scherzer TM, Maieron A, Gschwantler M, Brunner H, et al. Customizing treatment with peginterferon alfa-2a (40 kd) (PEGASYS®) plus ribavirin (COPEGUS®) in patients with HCV genotype 1 or 4 infection. Interim results of a prospective randomized trial. Hepatology 2006;44:336A.
Jacobson IM, Brown RS Jr, Freilich B, Afdhal N, Kwo PY, Santoro J, et al. Peginterferon alfa-2b and weight-based or flat-dose ribavirin in chronic hepatitis C patients: a randomized trial. Hepatology 2007;46:971–81.
Kamal SM, El Tawil AA, Nakano T, He Q, Rasenack J, Hakam SA, et al. Peginterferon alpha-2b and ribavirin therapy in chronic hepatitis C genotype 4: impact of treatment duration and viral kinetics on sustained virological response. Gut 2005;54:858–66.
Kamal SM, El Kamary SS, Shardell MD, Hashem M, Ahmed IN, Muhammadi M, et al. Pegylated interferon alpha-2b plus ribavirin in patients with genotype 4 chronic hepatitis C: The role of rapid and early virologic response. Hepatology 2007;46:1732–40.
Lagging M, Pedersen C, Rauning BM, Färkkilä M, Langeland N, Mørch K, et al. Comparison of peginterferon alpha-2a and ribavirin for 12 or 24 weeks in patients with HCV genotype 2/3: the Nordynamic trial. J Hepatol 2007;46(Suppl. 1):229.
Lagging M, Pedersen C, Rauning BM, Färkkilä M, Langeland N, Mørch K, et al. Peginterferon alfa-2A and ribavirin for 12 or 24 weeks in patients with HCV genotype 2/3: the Nordynamic trial. Hepatology 2007;46(Suppl. 1):815A–16A.
Lagging M, Langeland N, Pedersen C, Färkkilä M, Buhl MR, Mørch K, et al. Randomized comparison of 12 or 24 weeks of peginterferon alpha-2a and ribavirin in chronic hepatitis C virus genotype 2/3 infection. Hepatology 2008;47:1837–45.
Mangia A, Santoro R, Minerva N, Ricci GL, Carretta V, Persico M, et al. Peginterferon alfa-2b and ribavirin for 12 vs. 24 weeks in HCV genotype 2 or 3. N Engl J Med 2005;352:2609–17.
Mangia A, Minerva N, Carretta V, Bacca D, Ricci GL, Vinelli F, et al. Predictors of rapid virologic response (RVR) in HCV genotype 1 chronic infected patients: results of a randomized controlled trial on individualized treatment. Hepatology 2006;44(Suppl. 1):606A–7A.
Manns M, Zeuzem S, Sood A, Lurie Y, Cornberg M, Klinker H, et al. Reduced dose and duration of peginterferon alfa-2B + weight-based ribavirin in European and Asian genotype 2 and 3 chronic hepatitis C patients (REDD 2/3 trial). J Hepatol 2009;50(Suppl. 1): 59.
Mathew A, Peiffer LP, Rhoades K, McGarrity T. Sustained viral response to pegylated interferon alpha-2b and ribavirin in chronic hepatitis C refractory to prior treatment. Dig Dis Sci 2006;51:1956–61.
Mecenate F, Barbaro G, Pellicelli A, Barlattani A, Mazzoni E, Bonaventura M, et al. Comparison of peginterferon alfa-2a and ribavirin for 12 or 24 weeks in patients with HCV genotype 2 or 3: the CLEO trial. Hepatology 2007;46(Suppl. 1):828A.
Rumi M, Aghemo A, Prati GM, D’Ambrosio R, Donato MF, Russo A, et al. Randomized study comparing peginterferon-alfa2a plus ribavirin and peginterferon-alfa2b plus ribavirin in naive patients with chronic hepatitis c: final results of the Milan safety tolerability (MIST) study. Hepatology 2008;48:404A.
Sakai T, Iino S, Omata M, Kiyosawa K, Kumada H. Peginterferon alfa-2a (40KD) (PEGASYS®) plus ribavirin (COPEGUS®) in treatment-naive Japanese chronic hepatitis C patients: efficacy and safety of a randomised, double-blind, multicentre, phase III trial. J Gastroenterol Hepatol 2006;21:A25.
Shiffman ML, Nelson DR, Hooper G, Messinger D, Zeuzem S. HCV Patients with genotype 2 or 3 who do not achieve a rapid virologic response (RVR) with peginterferon alfa-2a (40KD)(PEGASYS®) and ribavirin (COPEGUS®) are not easy to treat: an analysis of non-RVR patients from the ACCELERATE study. Hepatology 2007;46(Suppl. 1):309A–10A.
Shiffman ML, Suter F, Bacon BR, Nelson D, Harley H, Sola R, et al. Peginterferon alfa-2a and ribavirin for 16 or 24 weeks in HCV genotype 2 or 3. N Engl J Med 2007;357:124–34.
Shiha G, Abdel KE, Abbas B, Elshennawy H, Zalata KH. Sustained virological response of peginterferon alpha-2a plus ribavirin for chronic hepatitis C virus genotype 4. J Hepatol 2007;46(Suppl. 1):215.
Siebert U, Sroczynski G, Aidelsburger P, Rossol S, Wasem J, Manns M, et al. Clinical effectiveness and cost-effectiveness of tailoring chronic hepatitis C treatment with peginterferon alpha-2b plus ribavirin to HCV genotype and early viral response: a decision analysis based on German guidelines. Pharmacoeconomics 2009;27:341–54.
Tang KH, Herrmann E, Pachiadakis I, Paulon E, Tatman N, Zeuzem S, et al. Clinical trial: individualized treatment duration for hepatitis C virus genotype 1 with peginterferon-alpha 2a plus ribavirin. Aliment Pharmacol Ther 2008;27:810–19.
Toyoda H, Kumada T, Kiriyama S, Sone Y, Tanikawa M, Hisanaga Y, et al. Eight-week regimen of antiviral combination therapy with peginterferon and ribavirin for patients with chronic hepatitis C with hepatitis C virus genotype 2 and a rapid virological response. Liver Int 2009;29:120–5.
Tsubota A, Satoh K, Aizawa M, Takamatsu S, Namiki Y, Ohkusa T, et al. Four-week pegylated interferon alpha-2a monotherapy for chronic hepatitis C with genotype 2 and low viral load: a pilot, randomized study. World J Gastroenterol 2008;14:7220–4.
Reason for exclusion: intervention
Behler CM, Vittinghoff E, Lin F, Chung RT, Peters MG, Robbins GK, et al. Hematologic toxicity associated with interferon-based hepatitis C therapy in HIV type 1-coinfected subjects. Clin Infect Dis 2007;44:1375–83.
Cargnel A, Angeli E, Mainini A, Gubertini G, Giorgi R, Schiavini M, et al. Open, randomized, multicentre Italian trial on PEG-IFN plus ribavirin versus PEG-IFN monotherapy for chronic hepatitis C in HIV-coinfected patients on HAART. Antivir Ther 2005;10:309–17.
Carrat F, Bani-Sadr F, Pol S, Rosenthal E, Lunel-Fabiani F, Benzekri A, et al. Pegylated interferon alfa-2b vs standard interferon alfa-2b, plus ribavirin, for chronic hepatitis C in HIV-infected patients: a randomized controlled trial. JAMA 2004;292:2839–48.
Chung RT, Andersen J, Volberding P, Robbins GK, Liu T, Sherman KE, et al. Peginterferon alfa-2a plus ribavirin versus interferon alfa-2a plus ribavirin for chronic hepatitis C in HIV-co-infected persons. N Engl J Med 2004;351:451–9.
Crespo M, Sauleda S, Esteban JI, Juarez A, Ribera E, Andreu AL, et al. Peginterferon alpha-2b plus ribavirin vs interferon alpha-2b plus ribavirin for chronic hepatitis C in HIV-co-infected patients. J Viral Hepat 2007;14:228–38.
Dalgard O, Bjoro K, Ring-Larsen H, Verbaan H, North C. Is sustained virological response to HCV treatment associated with a clinical important improvement in vitality? J Hepatol 2008;48(Suppl. 2):272.
De Compadri P, Koleva D, Mangia A, Motterlini N, Garattini L. Cost minimisation analysis of 12 or 24 weeks of peginterferon alfa-2b + ribavirin for hepatitis C virus. J Med Economics 2008;11:151–63.
Di Bisceglie AM, Shiffman ML, Everson GT, Lindsay KL, Everhart JE, Wright EC, et al. Prolonged therapy of advanced chronic hepatitis C with low-dose peginterferon. N Engl J Med 2008;359:2429–41.
Fuster D, Planas R, Gonzalez J, Force L, Cervantes M, Vilaro J, et al. Results of a study of prolonging treatment with pegylated interferon-alpha2a plus ribavirin in HIV/HCV co-infected patients with no early virological response. Antivir Ther 2006;11:473–82.
Graham CS, Wells A, Liu T, Sherman KE, Peters M, Chung RT, et al. Relationships between cellular immune responses and treatment outcomes with interferon and ribavirin in HIV/hepatitis C virus co-infection. AIDS 2006;20:345–51.
Haque M, Yoshida EM. Hepatitis C antiviral long-term treatment against cirrhosis (HALT-C) trial. Ann Hepatol 2009;8:78–9.
Hezode C, Foucher J, Bronowicki JP, Leroy V, Tran A, Larrey DG, et al. Efficacy and safety of an intensified regimen of pegylated interferon alfa-2A plus ribavirin in HCV genotype 1-infected patients who did not respond to a prior standard regimen of pegylated interferon alfa-2A plus ribavirin: an interim analysis of the multicentric, randomized, controlled SYREN trial. Hepatology 2008;48(Suppl.):1148A–9A.
Jensen D, Craxi A, Brandao-Mello C, Di Bisceglie A, Andreone P, Freilich B, et al. Identifying patients with a high likelihood of achieving an SVR following re-treatment with peginterferon alfa-2a (40 Kd) plus ribavirin: use of positive prognostic factors (PPFs). J Hepatol 2009;50(Suppl. 1):228.
Jensen DM, Marcellin P, Freilich B, Andreone P, DiBisceglie A, Brandao-Mello CE, et al. Re-treatment of patients with chronic hepatitis C who do not respond to peginterferon-alpha 2b: a randomized trial. Ann Intern Med 2009;150:528–40.
Kallinowski B, Stein K, Boecher W, Goeser T, Spengler U, Link R, et al. Comparison of a shortened ribavirin treatment vs a 24 week standard combination therapy in HCV infected patients with genotype 2/3. J Hepatol 2007;46(Suppl. 1):228–9.
Khalili M, Bernstein D, Lentz E, Barylski C, Hoffman-Terry M. Pegylated interferon alpha-2a with or without ribavirin in HCV/HIV co-infection: partially blinded, randomized multicenter trial. Dig Dis Sci 2005;50:1148–55.
Kim AI, Dorn A, Bouajram R, Saab S. The treatment of chronic hepatitis C in HIV-infected patients: a meta-analysis. HIV Med 2007;8:312–21.
Laguno M, Murillas J, Blanco JL, Martinez E, Miquel R, Sanchez-Tapias JM, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for treatment of HIV/HCV co-infected patients. AIDS 2004;18:F27–36.
Laguno M, Larrousse M, Murillas J, Blanco JL, Leon A, Milinkovic A, et al. Predictive value of early virologic response in HIV/hepatitis C virus-coinfected patients treated with an interferon-based regimen plus ribavirin. J Acquir Immune Defic Syndr 2007;44:174–8.
Laguno M, Cifuentes C, Murillas J, Veloso S, Larrousse M, Payeras A, et al. Randomized trial comparing pegylated interferon alpha-2b versus pegylated interferon alpha-2a, both plus ribavirin, to treat chronic hepatitis C in human immunodeficiency virus patients. Hepatology 2009;49:22–31.
Lissen E, Clumeck N, Sola R, Mendes-Correa M, Montaner J, Nelson M, et al. Histological response to pegIFNalpha-2a (40KD) plus ribavirin in HIV-hepatitis C virus co-infection. AIDS 2006;20:2175–81.
Marcellin P, Freilich B, Andreone P, DiBisceglie A, Brandao CE, Reddy KR, et al. HCV-RNA status at week 12 of treatment with peginterferon alfa2a/RBV predicts SVR in patients with prior non-response to pegylated interferon alfa-2b/RBV: results from repeat study. J Hepatol 2008;48(Suppl. 2):301.
Marcellin P, Freilich B, Andreone P, DiBisceglie A, Brandao CE, Reddy KR, et al. Type of response to prior pegylated interferon alfa-2B (12KD)/RBV predicts subsequent response to retreatment with peginterferon alfa-2A (40 Kd)/RBV. J Hepatol 2008;48(Suppl. 2):306.
Puoti M, Zanini B, De Luca A, Quinzan GP, Allegri R, Bruno R, et al. Results of a randomized controlled trial on the impact of prolonged combination anti-HCV treatment in HIV/HCV co-infected patients. J Hepatol 2008;48(Suppl. 2):309.
Rodriguez-Torres M, Torriani F, Rockstroh J, DePamphilis J, Carosi G, Dieterich DT. Patients co-infected with HCV and HIV who achieve an RVR (HCV RNA < 50 IU/ml at week 4) or cEVR (HCV RNA < 50 IU/ml at week 12) have similar rates of SVR to monoinfected patients treated with peginterferon alfa-2a (40KD) (PEGASYS®) and ribavirin (COPEGUS®). Hepatology 2007;46(Suppl. 1):814A.
Torriani FJ, Rodriguez-Torres M, Rockstroh JK, Lissen E, Gonzalez-Garcia J, Lazzarin A, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection in HIV-infected patients. N Engl J Med 2004;351:438–50.
Zhao S, Cheng D, Liu E, Yu H, Yang H, Xue X, et al. Peginterferon vs. interferon in the treatment of different HCV genotype infections in HIV patients. Eur J Clin Microbiol Infect Dis 2008;27:1183–92.
Reason for exclusion: outcomes
Shiffman ML, Morishima C, Lindsay KL, Hoefs JC, Dienstag JL, Szabo G, et al. Suppression of serum HCV RNA levels during maintenance peginterferon (PEGIFN) alfa-2a therapy and clinical outcomes in the HALT-C trial. J Hepatol 2008;48(Suppl. 2):62.
Appendix 8 Net benefit framework
Cost-effectiveness decision rules and the incremental cost-effectiveness ratio
Standard decision rules for considering the cost-effectiveness of an intervention (I), compared with a given comparator (C), focus on the difference in effect (ΔE = EI – EC also referred to as the incremental effect) and the difference in cost (ΔC = CI – CC also referred to as the incremental cost). The decision rules are outlined using the cost-effectiveness plane as shown in Figure 24, below.
FIGURE 24.
Cost-effectiveness plane for intervention (I) compared with comparator (C).
If the incremental cost is negative and the incremental effect is positive (SE quadrant), the intervention is unequivocally cost-effective (it is dominant, achieving better outcomes at lower cost).
If the incremental cost is positive and the incremental effect is negative (NW quadrant), the intervention is unequivocally not cost-effective (it is dominated, achieving poorer outcomes at higher cost).
If both the incremental cost and the incremental effect are negative (SW quadrant) or both the incremental cost and the incremental effect are positive (NE quadrant) no such unequivocal statements can be made. Determining whether the intervention is cost-effective depends on a threshold value (λ), defined as the maximum amount society is willing to pay for an incremental health gain or, equivalently, as the minimum amount society is willing to accept for foregoing an incremental health gain. The intervention would be regarded as cost-effective if its incremental cost-effectiveness ratio is lower than the threshold (ΔC/ΔE < λ) for ICERs in the NE quadrant or higher than the threshold (ΔC/ΔE > λ) for ICERs in the SW quadrant.
Cost-effectiveness decision rules and incremental net benefit
The inequalities (ΔC/ΔE < λ for ICERs in the NE quadrant and ΔC/ΔE > λ for ICERs in the SW quadrant) can be re-arranged to give equivalent inequalities on the cost scale (incremental net monetary benefit) or on the effect scale (incremental net health benefit) (see Briggs and colleagues 2006):
One of the drawbacks of the incremental cost-effectiveness ratio is that the location of negative ICERs [whether they are in the SE (dominant) or NW (dominated) quadrant] cannot be determined without reference to other contextual information (the incremental cost and incremental effectiveness underlying the ratio or the quadrant of the cost-effectiveness plane). Similarly, positive ICERs cannot be interpreted (given that the decision rules depend on whether the ICER lies in the NE or the SW quadrant) without such additional information. In contrast, the incremental net benefit (regardless of the scale) provides an unambiguous decision rule, although this implies knowledge of the threshold value (λ), which has been a subject of considerable debate in the context of NICE decision making (see Appleby and colleagues 2007, McCabe and colleagues 2008, Raftery 2009 and Towse 2009). Current NICE methodological guidance suggests presenting expected net monetary health benefits using values of £20,000 and £30,000 per QALY for λ.
Example This report presents cost-effectiveness results for shortened treatment duration using peginterferon alfa-2a combination therapy in genotype 1 patients (see Table 44), peginterferon alfa-2a combination therapy in genotype 2 or 3 patients (see Table 45) and peginterferon alfa-2b combination therapy in genotype 1 patients (see Table 50). The table below presents these results along with the incremental net benefits.
For genotype 1 patients treated with peginterferon alfa-2a, the ICER is positive and ranges from approximately £35,000 to £65,000 per QALY gained. Without reference to the incremental cost and incremental QALY estimates we cannot determine which quadrant (NE or SW) the ICER is located in – therefore we do not know which cost-effectiveness decision rule to apply. However, the incremental net monetary benefits (or equivalently the incremental net health benefits) are positive at both suggested threshold values, suggesting that reduced duration of treatment is a cost-effective option for genotype 1 patients treated with peginterferon alfa-2a.
For genotype 1 patients treated with peginterferon alfa-2b and genotype 2 or 3 patients treated with peginterferon alfa-2a the ICER is negative. Again, without reference to the incremental cost and incremental QALY estimates we cannot determine which quadrant (NW or SE) the ICER is located in – therefore we do not know whether shortened treatment duration dominates or is dominated by standard duration. The incremental net benefits are positive at both suggested threshold values, suggesting that reduced duration of treatment is a cost-effective option in these patient groups.
Shortened treatment duration with peginterferon in different patient groups
The table below provides details of the incremental cost and outcome, ICERs and incremental net monetary (health) benefits for shortened treatment duration with peginterferon in different patient groups.
| Treatment: patient group | RCT | Incremental | ICER (£ per QALY gained) | Incremental net benefit | ||||
|---|---|---|---|---|---|---|---|---|
| Monetary | Health | |||||||
| Cost (£) | Outcome (QALYs) | λ = 20,000 | λ = 30,000 | λ = 20,000 | λ = 30,000 | |||
| PEG α-2a: genotype 1 | Liu and colleagues53 | –4807 | –0.14 | 34,510 | 2007 | 607 | 0.10 | 0.02 |
| Yu and colleagues 200854 | –5212 | –0.08 | 64,880 | 4526 | 4722 | 0.18 | 0.09 | |
| PEG α-2b: genotype 1 | Berg and colleagues59 | –8996 | 0.49 | –18,359 | 18,796 | 23,696 | 0.94 | 0.79 |
| PEG α-2a: genotype 2 or 3 | Yu and colleagues55 | –2107 | 0.08 | –26,338 | 3707 | 4507 | 0.19 | 0.15 |
| von Wagner and colleagues 200854 | –3146 | 0.23 | –13,678 | 4288 | 4176 | 0.39 | 0.33 | |
References
- Appleby J, Devlin N, Parkin D. NICE’s cost-effectiveness threshold. How high should it be?. BMJ 2007;335:358-9.
- Briggs A, Claxton K, Sculpher M. Decision modelling for health economic evaluation. Oxford: Oxford University Press; 2006.
- McCabe C, Claxton K, Culyer AJ. The NICE cost-effectiveness threshold: what it is and what that means. Pharmacoeconomics 2008;26:733-44.
- Raftery J. Should NICE’s threshold range for cost per QALY be raised? No. BMJ 2009;338.
- Towse A. Should NICE’s threshold range for cost per QALY be raised? Yes. BMJ 2009;338.
Appendix 9 SVR estimates for re-treated, and for HCV/HIV co-infected, patients used in the SHTAC economic model
Re-treated patients
As explained in Chapter 3 (Methods of data analysis/synthesis) of this report, no RCTs of re-treatment with peginterferon alfa and ribavirin following non-response to, or relapse from, a previous course of peginterferon alfa and ribavirin met the inclusion criteria for the systematic review of clinical effectiveness. This was because no RCTs used BSC as a comparator, and it was not possible to conduct an adjusted indirect comparison. Our search did identify one RCT (evaluating peginterferon alfa-2a) that met all of the criteria, with the exception that it had an active comparator (different regimens of peginterferon alfa-2a plus ribavirin). 88 For the purposes of economic modelling we have used the SVR reported for Group C of the trial (peginterferon alfa-2a 180 µg/week, plus ribavirin for 72 weeks) for genotype 1 patients (the SPC recommends 72 weeks re-treatment for genotype 1 patients). 42 For genotype non-1 patients SVRs were taken from Group D of the trial (peginterferon alfa-2a 180 µg/week, plus ribavirin for 48 weeks) (the SPC recommends 48 weeks re-treatment for genotype non-1 patients).
We did not identify any published RCTs of re-treatment with peginterferon alfa-2b plus ribavirin, irrespective of whether an active or inactive comparator was used. However, in order to model the cost-effectiveness of this drug we used SVRs from the currently unpublished EPIC3 study,96 which was also used by Schering-Plough in their submission to NICE. EPIC3 is an uncontrolled study that evaluates peginterferon alfa-2b and ribavirin for 48 weeks in over 2000 patients who had failed to respond to, or relapsed on, previous treatment (around two-thirds had received non-peginterferon alfa).
For both peginterferon alfa-2a and 2b we assumed that no patients receiving only BSC would achieve an SVR. Caution is therefore necessary in the interpretation of the ICERs, given that they are not based on an adjusted indirect comparison.
HCV/HIV co-infected patients
Given that no RCTs of antiviral treatment in HCV/HIV co-infected patients met the inclusion criteria for our systematic review of clinical effectiveness, we have taken SVR estimates for patients treated with peginterferon alfa and ribavirin from two recent published systematic reviews in co-infected patients. 50,51 These reviews were identified from the search conducted for our clinical effectiveness systematic review. Both reviews comprise the same six RCTs in which peginterferon alfa plus ribavirin was compared to either peginterferon alfa monotherapy or to non-peginterferon alfa plus ribavirin. We have extracted and tabulated the SVRs for the individual RCTs presented in the systematic reviews according to type of peginterferon alfa (2a or 2b) and genotype (see tables below). As it has not been possible to perform an adjusted indirect comparison between peginterferon alfa and ribavirin and BSC [as explained in Chapter 3 (Methods of data analysis/synthesis], we have assumed that no patients receiving only BSC will achieve an SVR. Again, caution is therefore necessary in the interpretation of the ICERs given that they are based upon an unadjusted indirect comparison.
Overall SVRs for HCV/HIV co-infected patients
| Study | No. with SVR (%) | Total no. of patients |
|---|---|---|
| Peginterferon α-2a | ||
| Chung and colleagues (2004)118 | 18 (27) | 66 |
| Torriani and colleagues (2004)66 | 116 (40) | 289 |
| Combined | 134 (38) | 355 |
| Peginterferon α-2b | ||
| Carrat and colleagues (2004)116 | 56 (27) | 205 |
| Laguno and colleagues (2004)95 | 23 (44) | 52 |
| Crespo and colleagues (2007)119 | 33 (55) | 60 |
| Cargnel and colleagues (2005)120 | 15 (22) | 69 |
| Combined | 127 (33) | 386 |
Genotypes 1 and 4
| Study | No. with SVR (%) | Total no. of patients |
|---|---|---|
| Peginterferon α-2a | ||
| Chung and colleagues (2004)118 | 7 (14) | 51 |
| Torriani and colleagues (2004)66 | 57 (30) | 194 |
| Combined | 64 (26) | 245 |
| Peginterferon α-2b | ||
| Carrat and colleagues (2004)116 | 21 (17) | 125 |
| Laguno and colleagues (2004)95 | 12 (38) | 32 |
| Crespo and colleagues (2007)119 | 18 (46) | 39 |
| Cargnel and colleagues (2005)120 | 4 (11) | 37 |
| Combined | 55 (24) | 233 |
Genotypes 2 and 3
| Study | No. with SVR (%) | Total no. patients |
|---|---|---|
| Peginterferon α-2a | ||
| Chung and colleagues (2004)118 | NA | NA |
| Torriani and colleagues (2004)66 | 59 (62) | 95 |
| Combined | 59 (62) | 95 |
| Peginterferon α-2b | ||
| Carrat and colleagues (2004)116 | 35 (44) | 80 |
| Laguno and colleagues (2004)95 | 10 (57) | 19 |
| Crespo and colleagues (2007)119 | 15 (71) | 21 |
| Cargnel and colleagues (2005)120 | 11 (34) | 32 |
| Combined | 71 (47) | 152 |
Data for the Cargnel and colleagues study120 have been added in, but these were not used in the respective meta-analyses of peginterferon alfa and ribavirin compared with non-peginterferon alfa by Kim and colleagues51 and Zhao and colleagues,50 as the study compared peginterferon alfa and ribavirin with peginterferon alfa monotherapy. Also data for the Chung and colleagues study118 for genotype 1 and 4 patients was not used in the meta-analysis by Kim and colleagues,51 but have been added in here. Therefore, the combined SVR results presented below are not strictly comparable with those in the published meta-analyses.
Appendix 10 Variables and probability distributions used in the probabilistic model
In the PSA, transition probabilities and utilities are sampled from beta distributions and costs are sampled from gamma distributions. 121 Parameters for sampling distributions were derived from point estimate and SEs for each variable, using the ‘method of moments’. 121
| Name | Distribution | Alfa | Beta |
|---|---|---|---|
| Transition probabilities | |||
| Mild-to-moderate chronic HCV | Beta | 38.08594 | 1485.35156 |
| Moderate chronic HCV to CC | 26.90504 | 700.25822 | |
| Compensated to DC | 14.61681 | 360.17319 | |
| CC to HCC | 1.93256 | 136.10744 | |
| DC to HCC | 1.93256 | 136.10744 | |
| DC excess mortality | 147.03000 | 983.97000 | |
| HCC excess mortality | 117.10333 | 155.23000 | |
| Utilities | |||
| Utility of SVR (from mild chronic HCV) | Beta | 65.86776 | 14.45878 |
| Utility of SVR (from moderate chronic HCV) | 58.06080 | 22.57920 | |
| Utility of SVR (from CC) – by assumption | 58.04760 | 37.11240 | |
| Utility of mild chronic HCV | 521.23750 | 155.69432 | |
| Utility of moderate chronic HCV | 168.24614 | 86.67226 | |
| Utility of CC | 47.10208 | 38.53806 | |
| Utility of DC | 123.75000 | 151.25000 | |
| Utility of HCC | 123.75000 | 151.25000 | |
| Utility of liver transplant | 123.75000 | 151.25000 | |
| Utility of post liver transplant | 59.25480 | 29.18520 | |
| Health-state costs | |||
| Cost of SVR state | Gamma | 28.81409 | 8.98866 |
| Cost of mild chronic HCV | 25.69952 | 5.36975 | |
| Cost of moderate chronic HCV | 88.85025 | 8.06976 | |
| Cost of CC | 24.23423 | 46.95836 | |
| Cost of DC | 36.03281 | 253.13041 | |
| Cost of HCC | 18.10811 | 448.80449 | |
| Cost of liver transplant | 89.75357 | 304.50042 | |
| Cost of care in year of liver transplant | 13.77880 | 686.41683 | |
| Cost of care in years after liver transplant | 15.21890 | 91.00529 | |
In the PSA treatment effects (probability of SVR and, where relevant, EVR) are sampled from beta distributions. The parameters of the sampling distributions are the number of events of interest (EVR or SVR) in the relevant population. 121
| Treatment effects | Distribution | Events | Population |
|---|---|---|---|
| Shortened treatment duration peginterferon α-2a | |||
| Liu and colleagues53 | |||
| SVR – standard duration | Beta | 57 | 58 |
| SVR – shortened duration | 69 | 74 | |
| Yu and colleagues 200755 | |||
| SVR – standard duration | Beta | 24 | 25 |
| SVR – shortened duration | 27 | 29 | |
| Yu and colleagues 200854 | |||
| SVR – standard duration | Beta | 85 | 88 |
| SVR – shortened duration | 43 | 44 | |
| Yu and colleagues 200755 | |||
| SVR – standard duration | Beta | 27 | 31 |
| SVR – shortened duration | 33 | 35 | |
| Shortened treatment duration peginterferon α-2b | |||
| SVR – standard duration | Beta | 8 | 19 |
| SVR – shortened duration | 16 | 28 | |
| Re-treatment with peginterferon α-2a | |||
| EVR – genotype 1 | Beta | 21 | 142 |
| SVR – genotype 1 | 18 | 21 | |
| EVR – genotype non-1 | 10 | 29 | |
| SVR – genotype non-1 | 6 | 10 | |
| Re-treatment with peginterferon α-2b | |||
| EVR – genotype 1 | Beta | 333 | 1121 |
| SVR – genotype 1 | 162 | 333 | |
| EVR – genotype non-1 | 162 | 206 | |
| SVR – genotype non-1 | 117 | 162 | |
| HCV/HIV co-infected treated with peginterferon α-2a | |||
| SVR – genotypes 1 and 4 | Beta | 64 | 245 |
| SVR – genotypes 2 and 3 | 59 | 95 | |
| HCV/HIV co-infected treated with peginterferon α-2b | |||
| SVR – genotypes 1 and 4 | Beta | 55 | 233 |
| SVR – genotypes 2 and 3 | 71 | 152 | |
List of abbreviations
- ALT
- alanine aminotransferase
- BNF
- British National Formulary
- BSC
- best supportive care
- CC
- compensated cirrhosis
- CEA
- cost-effectiveness analysis
- CEAC
- cost-effectiveness acceptability curve
- CI
- confidence interval
- CrI
- credible interval
- CUA
- cost–utility analysis
- DC
- decompensated cirrhosis
- DSA
- deterministic sensitivity analysis
- EOT
- end of treatment (virological response)
- EQ-5D
- European Quality of Life-5 Dimensions
- EVR
- early virological response
- GGT
- γ-glutamyltransferase
- Gp
- group
- HAART
- highly active antiretroviral therapy
- HAI
- histological activity index
- Hb
- haemoglobin
- HCC
- hepatocellular carcinoma
- HCHS
- hospital and community health services
- HCV
- hepatitis C virus
- HIV
- human immunodeficiency virus
- HPA
- Health Protection Agency
- HRQoL
- health-related quality of life
- HTA
- health technology assessment
- ICER
- incremental cost-effectiveness ratio
- IDU
- injecting drug user
- ITT
- intention to treat
- IU
- international unit
- LT
- liver transplantation
- LVL
- low viral load
- MS
- manufacturer’s submission
- MSM
- men who have sex with men
- NA
- not available
- NICE
- National Institute for Health and Clinical Excellence
- NR
- not reported
- pa
- per annum
- PCR
- polymerase chain reaction
- Post LT
- post liver transplantation
- PSA
- probabilistic sensitivity analysis
- PSS
- Personal and Social Services
- QALY
- quality-adjusted life-year
- QoL
- quality of life
- RCT
- randomised controlled trial
- RNA
- ribonucleic acid
- RR
- relative risk/risk ratio
- RVR
- rapid virological response
- SE
- standard error
- SG
- standard gamble
- SHTAC
- Southampton Health Technology Assessments Centre
- SPC
- summary of product characteristics
- SVR
- sustained virological response
- TA
- Technology Appraisal
- TAR
- technology assessment report
- TMA
- transcription-mediated amplification
- TTO
- time trade-off
- ULN
- upper limit of normal
- YLS
- years of life saved
All abbreviations that have been used in this report are listed here unless the abbreviation is well known (e.g. NHS), or it has been used only once, or it is a non-standard abbreviation used only in figures/tables/appendices, in which case the abbreviation is defined in the figure legend or in the notes at the end of the table.
Notes
Health Technology Assessment programme
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
Prioritisation Group
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Imti Choonara, Professor in Child Health, Academic Division of Child Health, University of Nottingham
Chair – Pharmaceuticals Panel
-
Dr Bob Coates, Consultant Advisor – Disease Prevention Panel
-
Dr Andrew Cook, Consultant Advisor – Intervention Procedures Panel
-
Dr Peter Davidson, Director of NETSCC, Health Technology Assessment
-
Dr Nick Hicks, Consultant Adviser – Diagnostic Technologies and Screening Panel, Consultant Advisor–Psychological and Community Therapies Panel
-
Ms Susan Hird, Consultant Advisor, External Devices and Physical Therapies Panel
-
Professor Sallie Lamb, Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick
Chair – HTA Clinical Evaluation and Trials Board
-
Professor Jonathan Michaels, Professor of Vascular Surgery, Sheffield Vascular Institute, University of Sheffield
Chair – Interventional Procedures Panel
-
Professor Ruairidh Milne, Director – External Relations
-
Dr John Pounsford, Consultant Physician, Directorate of Medical Services, North Bristol NHS Trust
Chair – External Devices and Physical Therapies Panel
-
Dr Vaughan Thomas, Consultant Advisor – Pharmaceuticals Panel, Clinical
Lead – Clinical Evaluation Trials Prioritisation Group
-
Professor Margaret Thorogood, Professor of Epidemiology, Health Sciences Research Institute, University of Warwick
Chair – Disease Prevention Panel
-
Professor Lindsay Turnbull, Professor of Radiology, Centre for the MR Investigations, University of Hull
Chair – Diagnostic Technologies and Screening Panel
-
Professor Scott Weich, Professor of Psychiatry, Health Sciences Research Institute, University of Warwick
Chair – Psychological and Community Therapies Panel
-
Professor Hywel Williams, Director of Nottingham Clinical Trials Unit, Centre of Evidence-Based Dermatology, University of Nottingham
Chair – HTA Commissioning Board
Deputy HTA Programme Director
HTA Commissioning Board
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
-
Professor of General Practice, Department of Primary Health Care, University of Oxford Programme Director,
-
Professor of Clinical Pharmacology, Director, NIHR HTA programme, University of Liverpool
-
Professor Ann Ashburn, Professor of Rehabilitation and Head of Research, Southampton General Hospital
-
Professor Deborah Ashby, Professor of Medical Statistics and Clinical Trials, Queen Mary, Department of Epidemiology and Public Health, Imperial College London
-
Professor Peter Brocklehurst, Director, National Perinatal Epidemiology Unit, University of Oxford
-
Professor John Cairns, Professor of Health Economics, London School of Hygiene and Tropical Medicine
-
Professor Peter Croft, Director of Primary Care Sciences Research Centre, Keele University
-
Professor Jenny Donovan, Professor of Social Medicine, University of Bristol
-
Professor Jonathan Green, Professor and Acting Head of Department, Child and Adolescent Psychiatry, University of Manchester Medical School
-
Professor John W Gregory, Professor in Paediatric Endocrinology, Department of Child Health, Wales School of Medicine, Cardiff University
-
Professor Steve Halligan, Professor of Gastrointestinal Radiology, University College Hospital, London
-
Professor Freddie Hamdy, Professor of Urology, Head of Nuffield Department of Surgery, University of Oxford
-
Professor Allan House, Professor of Liaison Psychiatry, University of Leeds
-
Dr Martin J Landray, Reader in Epidemiology, Honorary Consultant Physician, Clinical Trial Service Unit, University of Oxford
-
Professor Stephen Morris, Professor of Health Economics, University College London, Research Department of Epidemiology and Public Health, University College London
-
Professor E Andrea Nelson, Professor of Wound Healing and Director of Research, School of Healthcare, University of Leeds
-
Professor John David Norris, Chair in Clinical Trials and Biostatistics, Robertson Centre for Biostatistics, University of Glasgow
-
Dr Rafael Perera, Lecturer in Medical Statisitics, Department of Primary Health Care, University of Oxford
-
Professor James Raftery, Chair of NETSCC and Director of the Wessex Institute, University of Southampton
-
Professor Barney Reeves, Professorial Research Fellow in Health Services Research, Department of Clinical Science, University of Bristol
-
Professor Martin Underwood, Warwick Medical School, University of Warwick
-
Professor Marion Walker, Professor in Stroke Rehabilitation, Associate Director UK Stroke Research Network, University of Nottingham
-
Dr Duncan Young, Senior Clinical Lecturer and Consultant, Nuffield Department of Anaesthetics, University of Oxford
-
Professor Stephen Morris, Professor of Health Economics, University College London, Research Department of Epidemiology and Public Health, University College London
-
Professor E Andrea Nelson, Professor of Wound Healing and Director of Research, School of Healthcare, University of Leeds
-
Professor John David Norris Chair in Clinical Trials and Biostatistics, Robertson Centre for Biostatistics, University of Glasgow
-
Dr Rafael Perera, Lecturer in Medical Statisitics, Department of Primary Health Care, University of Oxford
-
Professor James Raftery, Chair of NETSCC and Director of the Wessex Institute, University of Southampton
-
Professor Barney Reeves, Professorial Research Fellow in Health Services Research, Department of Clinical Science, University of Bristol
-
Professor Martin Underwood, Warwick Medical School, University of Warwick
-
Professor Marion Walker, Professor in Stroke Rehabilitation, Associate Director UK Stroke Research Network, University of Nottingham
-
Dr Duncan Young, Senior Clinical Lecturer and Consultant, Nuffield Department of Anaesthetics, University of Oxford
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
HTA Clinical Evaluation and Trials Board
-
Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick and Professor of Rehabilitation, Nuffield Department of Orthopaedic, Rheumatology and Musculoskeletal Sciences, University of Oxford
-
Professor of the Psychology of Health Care, Leeds Institute of Health Sciences, University of Leeds
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Keith Abrams, Professor of Medical Statistics, Department of Health Sciences, University of Leicester
-
Professor Martin Bland, Professor of Health Statistics, Department of Health Sciences, University of York
-
Professor Jane Blazeby, Professor of Surgery and Consultant Upper GI Surgeon, Department of Social Medicine, University of Bristol
-
Professor Julia M Brown, Director, Clinical Trials Research Unit, University of Leeds
-
Professor Alistair Burns, Professor of Old Age Psychiatry, Psychiatry Research Group, School of Community-Based Medicine, The University of Manchester & National Clinical Director for Dementia, Department of Health
-
Dr Jennifer Burr, Director, Centre for Healthcare Randomised trials (CHART), University of Aberdeen
-
Professor Linda Davies, Professor of Health Economics, Health Sciences Research Group, University of Manchester
-
Professor Simon Gilbody, Prof of Psych Medicine and Health Services Research, Department of Health Sciences, University of York
-
Professor Steven Goodacre, Professor and Consultant in Emergency Medicine, School of Health and Related Research, University of Sheffield
-
Professor Dyfrig Hughes, Professor of Pharmacoeconomics, Centre for Economics and Policy in Health, Institute of Medical and Social Care Research, Bangor University
-
Professor Paul Jones, Professor of Respiratory Medicine, Department of Cardiac and Vascular Science, St George‘s Hospital Medical School, University of London
-
Professor Khalid Khan, Professor of Women’s Health and Clinical Epidemiology, Barts and the London School of Medicine, Queen Mary, University of London
-
Professor Richard J McManus, Professor of Primary Care Cardiovascular Research, Primary Care Clinical Sciences Building, University of Birmingham
-
Professor Helen Rodgers, Professor of Stroke Care, Institute for Ageing and Health, Newcastle University
-
Professor Ken Stein, Professor of Public Health, Peninsula Technology Assessment Group, Peninsula College of Medicine and Dentistry, Universities of Exeter and Plymouth
-
Professor Jonathan Sterne, Professor of Medical Statistics and Epidemiology, Department of Social Medicine, University of Bristol
-
Mr Andy Vail, Senior Lecturer, Health Sciences Research Group, University of Manchester
-
Professor Clare Wilkinson, Professor of General Practice and Director of Research North Wales Clinical School, Department of Primary Care and Public Health, Cardiff University
-
Dr Ian B Wilkinson, Senior Lecturer and Honorary Consultant, Clinical Pharmacology Unit, Department of Medicine, University of Cambridge
-
Ms Kate Law, Director of Clinical Trials, Cancer Research UK
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
Diagnostic Technologies and Screening Panel
-
Scientific Director of the Centre for Magnetic Resonance Investigations and YCR Professor of Radiology, Hull Royal Infirmary
-
Professor Judith E Adams, Consultant Radiologist, Manchester Royal Infirmary, Central Manchester & Manchester Children’s University Hospitals NHS Trust, and Professor of Diagnostic Radiology, University of Manchester
-
Mr Angus S Arunkalaivanan, Honorary Senior Lecturer, University of Birmingham and Consultant Urogynaecologist and Obstetrician, City Hospital, Birmingham
-
Dr Stephanie Dancer, Consultant Microbiologist, Hairmyres Hospital, East Kilbride
-
Dr Diane Eccles, Professor of Cancer Genetics, Wessex Clinical Genetics Service, Princess Anne Hospital
-
Dr Trevor Friedman, Consultant Liason Psychiatrist, Brandon Unit, Leicester General Hospital
-
Dr Ron Gray, Consultant, National Perinatal Epidemiology Unit, Institute of Health Sciences, University of Oxford
-
Professor Paul D Griffiths, Professor of Radiology, Academic Unit of Radiology, University of Sheffield
-
Mr Martin Hooper, Public contributor
-
Professor Anthony Robert Kendrick, Associate Dean for Clinical Research and Professor of Primary Medical Care, University of Southampton
-
Dr Anne Mackie, Director of Programmes, UK National Screening Committee, London
-
Mr David Mathew, Public contributor
-
Dr Michael Millar, Consultant Senior Lecturer in Microbiology, Department of Pathology & Microbiology, Barts and The London NHS Trust, Royal London Hospital
-
Mrs Una Rennard, Public contributor
-
Dr Stuart Smellie, Consultant in Clinical Pathology, Bishop Auckland General Hospital
-
Ms Jane Smith, Consultant Ultrasound Practitioner, Leeds Teaching Hospital NHS Trust, Leeds
-
Dr Allison Streetly, Programme Director, NHS Sickle Cell and Thalassaemia Screening Programme, King’s College School of Medicine
-
Dr Alan J Williams, Consultant Physician, General and Respiratory Medicine, The Royal Bournemouth Hospital
-
Dr Tim Elliott, Team Leader, Cancer Screening, Department of Health
-
Dr Catherine Moody, Programme Manager, Medical Research Council
-
Professor Julietta Patrick, Director, NHS Cancer Screening Programme, Sheffield
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Disease Prevention Panel
-
Professor of Epidemiology, University of Warwick Medical School, Coventry
-
Dr Robert Cook, Clinical Programmes Director, Bazian Ltd, London
-
Dr Colin Greaves, Senior Research Fellow, Peninsula Medical School (Primary Care)
-
Mr Michael Head, Public contributor
-
Professor Cathy Jackson, Professor of Primary Care Medicine, Bute Medical School, University of St Andrews
-
Dr Russell Jago, Senior Lecturer in Exercise, Nutrition and Health, Centre for Sport, Exercise and Health, University of Bristol
-
Dr Julie Mytton, Consultant in Child Public Health, NHS Bristol
-
Professor Irwin Nazareth, Professor of Primary Care and Director, Department of Primary Care and Population Sciences, University College London
-
Dr Richard Richards, Assistant Director of Public Health, Derbyshire Country Primary Care Trust
-
Professor Ian Roberts, Professor of Epidemiology and Public Health, London School of Hygiene & Tropical Medicine
-
Dr Kenneth Robertson, Consultant Paediatrician, Royal Hospital for Sick Children, Glasgow
-
Dr Catherine Swann, Associate Director, Centre for Public Health Excellence, NICE
-
Professor Carol Tannahill, Glasgow Centre for Population Health
-
Mrs Jean Thurston, Public contributor
-
Professor David Weller, Head, School of Clinical Science and Community Health, University of Edinburgh
-
Ms Christine McGuire, Research & Development, Department of Health
-
Dr Kay Pattison Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
External Devices and Physical Therapies Panel
-
Consultant Physician North Bristol NHS Trust
-
Reader in Wound Healing and Director of Research, University of Leeds
-
Professor Bipin Bhakta, Charterhouse Professor in Rehabilitation Medicine, University of Leeds
-
Mrs Penny Calder, Public contributor
-
Dr Dawn Carnes, Senior Research Fellow, Barts and the London School of Medicine and Dentistry
-
Dr Emma Clark, Clinician Scientist Fellow & Cons. Rheumatologist, University of Bristol
-
Mrs Anthea De Barton-Watson, Public contributor
-
Professor Nadine Foster, Professor of Musculoskeletal Health in Primary Care Arthritis Research, Keele University
-
Dr Shaheen Hamdy, Clinical Senior Lecturer and Consultant Physician, University of Manchester
-
Professor Christine Norton, Professor of Clinical Nursing Innovation, Bucks New University and Imperial College Healthcare NHS Trust
-
Dr Lorraine Pinnigton, Associate Professor in Rehabilitation, University of Nottingham
-
Dr Kate Radford, Senior Lecturer (Research), University of Central Lancashire
-
Mr Jim Reece, Public contributor
-
Professor Maria Stokes, Professor of Neuromusculoskeletal Rehabilitation, University of Southampton
-
Dr Pippa Tyrrell, Senior Lecturer/Consultant, Salford Royal Foundation Hospitals’ Trust and University of Manchester
-
Dr Sarah Tyson, Senior Research Fellow & Associate Head of School, University of Salford
-
Dr Nefyn Williams, Clinical Senior Lecturer, Cardiff University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Interventional Procedures Panel
-
Professor of Vascular Surgery, University of Sheffield
-
Consultant Colorectal Surgeon, Bristol Royal Infirmary
-
Mrs Isabel Boyer, Public contributor
-
Mr David P Britt, Public contributor
-
Mr Sankaran ChandraSekharan, Consultant Surgeon, Breast Surgery, Colchester Hospital University NHS Foundation Trust
-
Professor Nicholas Clarke, Consultant Orthopaedic Surgeon, Southampton University Hospitals NHS Trust
-
Ms Leonie Cooke, Public contributor
-
Mr Seamus Eckford, Consultant in Obstetrics & Gynaecology, North Devon District Hospital
-
Professor David Taggart, Consultant Cardiothoracic Surgeon, John Radcliffe Hospital
-
Professor Sam Eljamel, Consultant Neurosurgeon, Ninewells Hospital and Medical School, Dundee
-
Dr Adele Fielding, Senior Lecturer and Honorary Consultant in Haematology, University College London Medical School
-
Dr Matthew Hatton, Consultant in Clinical Oncology, Sheffield Teaching Hospital Foundation Trust
-
Dr John Holden, General Practitioner, Garswood Surgery, Wigan
-
Professor Nicholas James, Professor of Clinical Oncology, School of Cancer Sciences, University of Birmingham
-
Dr Fiona Lecky, Senior Lecturer/Honorary Consultant in Emergency Medicine, University of Manchester/Salford Royal Hospitals NHS Foundation Trust
-
Dr Nadim Malik, Consultant Cardiologist/ Honorary Lecturer, University of Manchester
-
Mr Hisham Mehanna, Consultant & Honorary Associate Professor, University Hospitals Coventry & Warwickshire NHS Trust
-
Dr Jane Montgomery, Consultant in Anaesthetics and Critical Care, South Devon Healthcare NHS Foundation Trust
-
Professor Jon Moss, Consultant Interventional Radiologist, North Glasgow Hospitals University NHS Trust
-
Dr Simon Padley, Consultant Radiologist, Chelsea & Westminster Hospital
-
Dr Ashish Paul, Medical Director, Bedfordshire PCT
-
Dr Sarah Purdy, Consultant Senior Lecturer, University of Bristol
-
Professor Yit Chiun Yang, Consultant Ophthalmologist, Royal Wolverhampton Hospitals NHS Trust
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Pharmaceuticals Panel
-
Professor in Child Health, University of Nottingham
-
Senior Lecturer in Clinical Pharmacology, University of East Anglia
-
Dr Martin Ashton-Key, Medical Advisor, National Commissioning Group, NHS London
-
Mr John Chapman, Public contributor
-
Dr Peter Elton, Director of Public Health, Bury Primary Care Trust
-
Dr Peter Elton, Director of Public Health, Bury Primary Care Trust
-
Dr Ben Goldacre, Research Fellow, Division of Psychological Medicine and Psychiatry, King’s College London
-
Dr James Gray, Consultant Microbiologist, Department of Microbiology, Birmingham Children’s Hospital NHS Foundation Trust
-
Ms Kylie Gyertson, Oncology and Haematology Clinical Trials Manager, Guy’s and St Thomas’ NHS Foundation Trust London
-
Dr Jurjees Hasan, Consultant in Medical Oncology, The Christie, Manchester
-
Dr Carl Heneghan Deputy Director Centre for Evidence-Based Medicine and Clinical Lecturer, Department of Primary Health Care, University of Oxford
-
Dr Dyfrig Hughes, Reader in Pharmacoeconomics and Deputy Director, Centre for Economics and Policy in Health, IMSCaR, Bangor University
-
Dr Maria Kouimtzi, Pharmacy and Informatics Director, Global Clinical Solutions, Wiley-Blackwell
-
Professor Femi Oyebode, Consultant Psychiatrist and Head of Department, University of Birmingham
-
Dr Andrew Prentice, Senior Lecturer and Consultant Obstetrician and Gynaecologist, The Rosie Hospital, University of Cambridge
-
Ms Amanda Roberts, Public contributor
-
Dr Martin Shelly, General Practitioner, Silver Lane Surgery, Leeds
-
Dr Gillian Shepherd, Director, Health and Clinical Excellence, Merck Serono Ltd
-
Mrs Katrina Simister, Assistant Director New Medicines, National Prescribing Centre, Liverpool
-
Professor Donald Singer Professor of Clinical Pharmacology and Therapeutics, Clinical Sciences Research Institute, CSB, University of Warwick Medical School
-
Mr David Symes, Public contributor
-
Dr Arnold Zermansky, General Practitioner, Senior Research Fellow, Pharmacy Practice and Medicines Management Group, Leeds University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Mr Simon Reeve, Head of Clinical and Cost-Effectiveness, Medicines, Pharmacy and Industry Group, Department of Health
-
Dr Heike Weber, Programme Manager, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Psychological and Community Therapies Panel
-
Professor of Psychiatry, University of Warwick, Coventry
-
Consultant & University Lecturer in Psychiatry, University of Cambridge
-
Professor Jane Barlow, Professor of Public Health in the Early Years, Health Sciences Research Institute, Warwick Medical School
-
Dr Sabyasachi Bhaumik, Consultant Psychiatrist, Leicestershire Partnership NHS Trust
-
Mrs Val Carlill, Public contributor
-
Dr Steve Cunningham, Consultant Respiratory Paediatrician, Lothian Health Board
-
Dr Anne Hesketh, Senior Clinical Lecturer in Speech and Language Therapy, University of Manchester
-
Dr Peter Langdon, Senior Clinical Lecturer, School of Medicine, Health Policy and Practice, University of East Anglia
-
Dr Yann Lefeuvre, GP Partner, Burrage Road Surgery, London
-
Dr Jeremy J Murphy, Consultant Physician and Cardiologist, County Durham and Darlington Foundation Trust
-
Dr Richard Neal, Clinical Senior Lecturer in General Practice, Cardiff University
-
Mr John Needham, Public contributor
-
Ms Mary Nettle, Mental Health User Consultant
-
Professor John Potter, Professor of Ageing and Stroke Medicine, University of East Anglia
-
Dr Greta Rait, Senior Clinical Lecturer and General Practitioner, University College London
-
Dr Paul Ramchandani, Senior Research Fellow/Cons. Child Psychiatrist, University of Oxford
-
Dr Karen Roberts, Nurse/Consultant, Dunston Hill Hospital, Tyne and Wear
-
Dr Karim Saad, Consultant in Old Age Psychiatry, Coventry and Warwickshire Partnership Trust
-
Dr Lesley Stockton, Lecturer, School of Health Sciences, University of Liverpool
-
Dr Simon Wright, GP Partner, Walkden Medical Centre, Manchester
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Expert Advisory Network
-
Professor Douglas Altman, Professor of Statistics in Medicine, Centre for Statistics in Medicine, University of Oxford
-
Professor John Bond, Professor of Social Gerontology & Health Services Research, University of Newcastle upon Tyne
-
Professor Andrew Bradbury, Professor of Vascular Surgery, Solihull Hospital, Birmingham
-
Mr Shaun Brogan, Chief Executive, Ridgeway Primary Care Group, Aylesbury
-
Mrs Stella Burnside OBE, Chief Executive, Regulation and Improvement Authority, Belfast
-
Ms Tracy Bury, Project Manager, World Confederation of Physical Therapy, London
-
Professor Iain T Cameron, Professor of Obstetrics and Gynaecology and Head of the School of Medicine, University of Southampton
-
Professor Bruce Campbell, Consultant Vascular & General Surgeon, Royal Devon & Exeter Hospital, Wonford
-
Dr Christine Clark, Medical Writer and Consultant Pharmacist, Rossendale
-
Professor Collette Clifford, Professor of Nursing and Head of Research, The Medical School, University of Birmingham
-
Professor Barry Cookson, Director, Laboratory of Hospital Infection, Public Health Laboratory Service, London
-
Dr Carl Counsell, Clinical Senior Lecturer in Neurology, University of Aberdeen
-
Professor Howard Cuckle, Professor of Reproductive Epidemiology, Department of Paediatrics, Obstetrics & Gynaecology, University of Leeds
-
Professor Carol Dezateux, Professor of Paediatric Epidemiology, Institute of Child Health, London
-
Mr John Dunning, Consultant Cardiothoracic Surgeon, Papworth Hospital NHS Trust, Cambridge
-
Mr Jonothan Earnshaw, Consultant Vascular Surgeon, Gloucestershire Royal Hospital, Gloucester
-
Professor Martin Eccles, Professor of Clinical Effectiveness, Centre for Health Services Research, University of Newcastle upon Tyne
-
Professor Pam Enderby, Dean of Faculty of Medicine, Institute of General Practice and Primary Care, University of Sheffield
-
Professor Gene Feder, Professor of Primary Care Research & Development, Centre for Health Sciences, Barts and The London School of Medicine and Dentistry
-
Mr Leonard R Fenwick, Chief Executive, Freeman Hospital, Newcastle upon Tyne
-
Mrs Gillian Fletcher, Antenatal Teacher and Tutor and President, National Childbirth Trust, Henfield
-
Professor Jayne Franklyn, Professor of Medicine, University of Birmingham
-
Mr Tam Fry, Honorary Chairman, Child Growth Foundation, London
-
Professor Fiona Gilbert, Consultant Radiologist and NCRN Member, University of Aberdeen
-
Professor Paul Gregg, Professor of Orthopaedic Surgical Science, South Tees Hospital NHS Trust
-
Bec Hanley, Co-director, TwoCan Associates, West Sussex
-
Dr Maryann L Hardy, Senior Lecturer, University of Bradford
-
Mrs Sharon Hart, Healthcare Management Consultant, Reading
-
Professor Robert E Hawkins, CRC Professor and Director of Medical Oncology, Christie CRC Research Centre, Christie Hospital NHS Trust, Manchester
-
Professor Richard Hobbs, Head of Department of Primary Care & General Practice, University of Birmingham
-
Professor Alan Horwich, Dean and Section Chairman, The Institute of Cancer Research, London
-
Professor Allen Hutchinson, Director of Public Health and Deputy Dean of ScHARR, University of Sheffield
-
Professor Peter Jones, Professor of Psychiatry, University of Cambridge, Cambridge
-
Professor Stan Kaye, Cancer Research UK Professor of Medical Oncology, Royal Marsden Hospital and Institute of Cancer Research, Surrey
-
Dr Duncan Keeley, General Practitioner (Dr Burch & Ptnrs), The Health Centre, Thame
-
Dr Donna Lamping, Research Degrees Programme Director and Reader in Psychology, Health Services Research Unit, London School of Hygiene and Tropical Medicine, London
-
Professor James Lindesay, Professor of Psychiatry for the Elderly, University of Leicester
-
Professor Julian Little, Professor of Human Genome Epidemiology, University of Ottawa
-
Professor Alistaire McGuire, Professor of Health Economics, London School of Economics
-
Professor Neill McIntosh, Edward Clark Professor of Child Life and Health, University of Edinburgh
-
Professor Rajan Madhok, Consultant in Public Health, South Manchester Primary Care Trust
-
Professor Sir Alexander Markham, Director, Molecular Medicine Unit, St James’s University Hospital, Leeds
-
Dr Peter Moore, Freelance Science Writer, Ashtead
-
Dr Andrew Mortimore, Public Health Director, Southampton City Primary Care Trust
-
Dr Sue Moss, Associate Director, Cancer Screening Evaluation Unit, Institute of Cancer Research, Sutton
-
Professor Miranda Mugford, Professor of Health Economics and Group Co-ordinator, University of East Anglia
-
Professor Jim Neilson, Head of School of Reproductive & Developmental Medicine and Professor of Obstetrics and Gynaecology, University of Liverpool
-
Mrs Julietta Patnick, Director, NHS Cancer Screening Programmes, Sheffield
-
Professor Robert Peveler, Professor of Liaison Psychiatry, Royal South Hants Hospital, Southampton
-
Professor Chris Price, Director of Clinical Research, Bayer Diagnostics Europe, Stoke Poges
-
Professor William Rosenberg, Professor of Hepatology and Consultant Physician, University of Southampton
-
Professor Peter Sandercock, Professor of Medical Neurology, Department of Clinical Neurosciences, University of Edinburgh
-
Dr Philip Shackley, Senior Lecturer in Health Economics, Sheffield Vascular Institute, University of Sheffield
-
Dr Eamonn Sheridan, Consultant in Clinical Genetics, St James’s University Hospital, Leeds
-
Dr Margaret Somerville, Director of Public Health Learning, Peninsula Medical School, University of Plymouth
-
Professor Sarah Stewart-Brown, Professor of Public Health, Division of Health in the Community, University of Warwick, Coventry
-
Dr Nick Summerton, GP Appraiser and Codirector, Research Network, Yorkshire Clinical Consultant, Primary Care and Public Health, University of Oxford
-
Professor Ala Szczepura, Professor of Health Service Research, Centre for Health Services Studies, University of Warwick, Coventry
-
Dr Ross Taylor, Senior Lecturer, University of Aberdeen
-
Dr Richard Tiner, Medical Director, Medical Department, Association of the British Pharmaceutical Industry
-
Mrs Joan Webster, Consumer Member, Southern Derbyshire Community Health Council
-
Professor Martin Whittle, Clinical Co-director, National Co-ordinating Centre for Women’s and Children’s Health, Lymington