
Notes
Article history
The research reported in this issue of the journal was commissioned and funded by the HTA programme on behalf of NICE as project number 09/21/01. The protocol was agreed in October 2009. The assessment report began editorial review in March 2010 and was accepted for publication in October 2010. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the referees for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
none
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2011. This work was produced by Hislop et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This journal is a member of and subscribes to the principles of the Committee on Publication Ethics (COPE) (http://www.publicationethics.org/). This journal may be freely reproduced for the purposes of private research and study and may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NETSCC, Health Technology Assessment, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2011 Queen’s Printer and Controller of HMSO
Chapter 1 Background
Description of health problem
Introduction
Gastrointestinal stromal tumours (GISTs) are tumours of mesenchymal origin that arise in the gastrointestinal tract (GI tract). Historically, and based upon morphological appearance alone, GISTs were considered to be of smooth muscle origin and regarded as leiomyomas or leiomyosarcomas. Subsequently, electron microscopic and molecular analysis has demonstrated that GISTs are a distinct tumour type arising from the interstitial cells of Cajal, and characterised by the expression of receptor tyrosine kinase KIT (CD117) protein demonstrated by immunohistochemistry. 1 CD117/KIT immunoreactivity now provides the diagnostic criteria for GISTs, although there is recognition that a small proportion of GISTs (4%) are KIT immunoreactive negative. 2,3
Aetiology, pathology and prognosis
Recent investigation has provided clinically significant insights into the molecular pathogenesis of GISTs. This has allowed the rational development of systemic therapies (including imatinib and sunitinib), provided robust diagnostic criteria for GISTs, and demonstrated the ability of certain pathogenic gene mutations to predict clinical behaviour and response to therapy in GISTs, therefore having potential application as predictive biomarkers.
Activating mutations in the KIT proto-oncogene are an early and key event in the pathogenesis of GISTs, and present in up to 95% of cases. 4–10 The protein product is a member of the receptor tyrosine kinase family and a transmembrane receptor for stem cell factor (SCF). 11 Extracellular binding of SCF to the receptor results in dimerisation of KIT and subsequent activation of the intracellular KIT kinase domain,9 leading to activation of intracellular signalling cascades controlling cell proliferation, adhesion and differentiation. KIT mutation is necessary but not sufficient for the pathogenesis of GISTs; other mutations are essential, and KIT mutation is absent in a minority of cases (< 5%). 12,13 In the majority of KIT mutation-negative cases, mutational activation of the closely related tyrosine kinase platelet-derived growth factor receptor alpha (PDGFRA) is the pathogenic event, and KIT and PDGFRA activation have similar biological effects. 12,13
It has been demonstrated that KIT and PDGFRA gene mutations are mutually exclusive7,8,10,14 and GISTs with no KIT mutations have either PDGFRA-activating mutations or no identified kinase mutations. 13 GISTs that lack KIT mutations may still have high KIT kinase activity and so may have KIT mutations that are not detected by conventional screening methods. Alternatively, KIT kinase activation may be due to non-mutational mechanisms. 6
Diagnosis of GIST is made when morphological and clinical features of the tumour are consistent and the tumour has positive KIT/CD117 protein expression. 15 However, as noted above, approximately 4% of GISTs have clinical and morphological features of GIST but have negative KIT immunoreactivity. 2 These KIT-negative GISTs are more likely to contain PDGFRA mutations. 2 It is important in these cases, when KIT/CD117 staining is negative, that other markers are investigated to confirm GIST diagnosis. Recent studies have shown that a novel protein DOG1 is highly expressed in both KIT and PDGFRA mutant GISTs,16,17 and immunostaining for DOG1 can be used in conjunction with CD117 staining, and diagnosis of GIST made on the basis of KIT and/or DOG1 immunoreactivity. 15 PDGRFA immunohistochemistry should also be performed and positivity can assist with diagnosis. Mutational analysis also plays a role in the diagnosis of KIT/CD117-negative suspected GISTs, as with consistent morphological and clinical features, positive mutation analysis for either KIT or PDGFRA is diagnostic. 15
Without treatment GISTs are progressive and will eventually metastasise to distant organs and so are invariably fatal without any intervention. GISTs are resistant to ‘conventional’ oncology treatments of cytotoxic chemotherapy and radiotherapy. Prognosis is highly dependent on the resectability of the tumour; however, only 50% of GIST patients have resectable disease at first presentation. 18,19 Ten-year survival for resectable/non-metastatic tumours is 30–50%, and at least 50% will relapse within 5 years of surgery, but for unresectable tumours prognosis is very poor, with survival generally < 2 years without further treatment. 18,19
Epidemiology and incidence
While GISTs are the most common mesenchymal tumour of the GI tract, overall they are a rare cancer, accounting for less than 1% of all cancers of the GI tract. 20 GISTs can occur anywhere in the GI tract from the oesophagus to the rectum, but most arise in the stomach or small intestine. 21 They are rare before the age of 40 years and very rare in children, with a median age at diagnosis of 50–60 years. 22,23 Some data show a slight male predominance but this is not a consistent finding. 22,24,25
Retrospective studies carried out using KIT immunoreactivity as a diagnostic criterion have shown that GISTs have been underdiagnosed in the past. 26,27 These retrospective population-based reclassification studies provide the most reliable and accurate current estimate of an annual incidence of 15 cases per million, which would equate to 900 cases in the UK. 15
Impact of health problem
The symptoms of GISTs depend on the size and location of the primary tumour and any metastatic deposits. While one-third of cases are asymptomatic and discovered incidentally during investigations or surgical procedures for unrelated disease, severe and debilitating symptoms occur in many patients and are invariable in those patients who have (or develop) metastatic disease. 28
Gastrointestinal stromal tumours of < 2 cm in size with no metastatic disease are usually asymptomatic. Larger primary tumours and those of patients with metastatic disease are usually symptomatic and the most common symptom is GI tract bleeding, which occurs in 50% of patients, 25% of these patients presenting as emergencies with acute GI haemorrhage, either into the intestine or peritoneum. 29 Abdominal discomfort is a feature of larger tumours. 30 Oesophageal GISTs typically present with dysphagia, which represents the main symptomatic problem in these cases, and colorectal GISTs may cause bowel obstruction. In metastatic disease, debilitating systemic symptoms, such as fever, night sweats and weight loss, are common.
Current service provision
Management of disease
There is wide consensus that the management of GISTs should be undertaken in the context of discussion of individual cases by a multidisciplinary team. 15,31
Management of resectable disease
Surgical resection is the primary treatment for GISTs and offers the only possibility of cure. Surgical resection is undertaken with the aim of achieving a complete microscopic resection (R0 resection). Evaluation of the suitability and possibility of a complete microscopic resection of a GIST is made after appropriate preoperative assessment to determine stage and also the fitness of the patient for the procedure required. Preoperative assessment for staging includes (as a minimum) a computerised tomography (CT) scan of the chest, abdomen and pelvis, and, in specific circumstances, there is a role for endoscopic ultrasound, laparoscopy and angiography.
After resection patients are followed up with protocols involving clinical examination and/or surveillance imaging, based upon relapse risk stratification by means of histopathological criteria of the resected tumour. 15,32 Preliminary results from one randomised, placebo-controlled Phase III trial suggest that adjuvant therapy with imatinib (400 mg/day for 1 year) increases recurrence-free survival following resection, and it is therefore suggested that adjuvant imatinib may have an important role to play in the prevention of recurrence of GISTs after resection. 33 The results of other similar adjuvant trials are awaited. 15 At present imatinib is licensed for adjuvant treatment of patients who are at a significant risk of relapse,34 but although Scottish guidelines recommend adjuvant imatinib (400 mg/day) in patients considered to be of moderate or high risk of relapse, according to histopathological criteria,15 a National Institute for Health and Clinical Excellence (NICE) Technology Appraisal for this indication is still ongoing,35 and it is acknowledged that, until more data are available from ongoing adjuvant studies, there is still uncertainty regarding the optimal duration of treatment, and also the subgroups of patients who may or may not benefit from adjuvant therapy. The use of imatinib as an adjuvant therapy may have implications, for example with regard to the development of drug resistance, for the subsequent systemic treatment of GISTs upon recurrence. 36
Studies are ongoing to determine the role of imatinib as preoperative therapy in resectable tumours. 37 Nevertheless, the use of imatinib preoperatively to downstage tumours from unresectable to resectable is considered safe and clinically worthwhile. 15 Similarly, preoperative imatinib has also been recommended to limit the extent and (accordingly) morbidity of resection in specific circumstances, for example to facilitate sphincter-sparing resection in rectal GISTs.
Management of unresectable and metastatic disease
Conventional cytotoxic chemotherapy and radiotherapy are ineffective in the treatment of advanced GISTs. Similarly, initial debulking surgery is not recommended unless there is an immediate clinical need, such as to remove an obstructing tumour.
Imatinib (Glivec®, Novartis Pharmaceuticals UK) is a rationally designed small molecule inhibitor of several tyrosine kinases, including KIT and PDGFRA, and has provided the first clinically effective systemic therapy for GISTs. The European licence for imatinib was based on a Phase II study of 147 patients who were randomised to receive imatinib at either 400 or 600 mg orally taken once daily. 38 The treatment was well tolerated, objective response rate was the primary efficacy outcome and an overall partial response (PR) rate of 67% was demonstrated with no difference between treatment arms. Long-term results revealed median survival of 57 months for all patients. 39 A concurrent study investigated dose escalation and established 800 mg daily as the maximum tolerated dose. 40 Phase III trials performed both in Europe and Australasia [European Organisation for Research and Treatment of Cancer (EORTC) 62005 study], and in North America (S0033 Intergroup study), confirmed the efficacy of imatinib in a larger patient population, and established the starting dose of 400 mg orally per day. 41,42
Primary resistance to imatinib is uncommon, but acquired resistance is highly likely, and manifest clinically by the observation of disease progression. 41–45 Guidelines suggest that patients should have a CT scan every 3 months while on therapy. 15 Measurement of response by conventional criteria, such as Response Evaluation Criteria in Solid Tumors (RECIST), based on objectively measured changes in tumour size, may not occur, or may happen only after many months of treatment. This means that definitive evidence of patient response, and therefore clinical benefit, can be difficult to ascertain (at least initially). This has been addressed by the development of alternate methods of GIST response assessment, such as the ‘Choi criteria’ based upon tumour density as well as tumour size. 46,47 Similarly, fluorodeoxyglucose-positron emission tomography (FDG-PET) has demonstrated some efficacy in predicting early response to imatinib therapy,48 although it should be noted that PET scanning is not widely available in the UK as very few NHS centres have access to this technology.
In addition, the assessment of progression of GISTs may be problematic if based on RECIST-based tumour size criteria, as tumour liquefaction (cystic degeneration) can occur, which may give the appearance of progressive disease (PD) although the tumour is actually responding. 47 Accordingly, it is recognised that experienced radiologists should assess CT scans before confirming progression.
It has been demonstrated that interruption of treatment results in rapid disease progression in many patients with advanced GISTs. 45 This includes patients with disease progression in whom a symptomatic worsening or ‘flare’ has been described. 49 Therefore, continuation of imatinib in these patients has been common practice despite progression, as part of best supportive care (BSC).
Several studies have reported further disease control after progression on an initial imatinib dose of 400 mg orally per day with dose escalation of imatinib to 800 mg orally per day, and this has also become common practice. 39,44 However, it should be noted that current NICE guidelines for imatinib do not actually recommend dose escalation for patients with unresectable and/or metastatic GISTs who progress on an initial dose of 400 mg/day. 50
Recently, sunitinib (Sutent®, Pfizer UK), another molecular-based treatment for GIST, became available, and has been approved by NICE for patients with unresectable and/or metastatic GIST who have progressed on treatment with imatinib. 51 The NICE advice follows a randomised, double-blind, placebo-controlled, multicentre Phase II trial in which 312 patients, who were resistant or intolerant to imatinib, received either sunitinib (50-mg starting dose in 6-week cycles; 4 weeks on and 2 weeks off treatment) or placebo;52 the trial was unblinded early when interim analysis showed a significantly longer time to tumour progression (the primary end point) with sunitinib.
To date, no randomised trial has been conducted comparing imatinib and sunitinib. One had been planned but was stopped owing to poor recruitment. 53 As new options for management of patients with unresectable and/or metastatic GIST have developed since the initial 2004 publication of NICE guidance for GIST treatment with imatinib, a review of the evidence available on treatments currently used in clinical practice is required.
Current service cost and anticipated costs associated with the intervention
As GIST affects mostly the middle-aged and older age population, the loss of productivity from the middle-aged population suffering from GIST is of concern. The median age of the GIST patients was found to be between 50 and 60 years,22,23 and incidence of GIST was found to increase with increase in age. 54 The cost of different treatment strategies needs thorough investigation in a robust economic evaluation.
Treatment with imatinib per patient within an NHS setting has been estimated at £18,896 and £24,368 annually for patients on 400 and 600 mg/day, respectively. 55 Other associated annual costs of treatment (including the treatment of adverse events) were estimated at £2730 (price year not stated). Estimates from previous disease models suggest that in 2 years it would cost the NHS approximately £31,160 to treat a patient with imatinib, and for 10 years this figure would be £56,146 (2002 price year). 54,55 Costs would differ when patients who fail to respond to imatinib are provided with higher doses or alternative treatments (e.g. sunitinib). 50
The costs of treating patients with unresectable and/or metastatic GIST using imatinib were estimated at between £1557 and £3115 per month per patient, resulting in a cost to the NHS (England and Wales) of between approximately £5.6M and £11.2M per year (2002 price year). 55 Another study estimates that the total costs over 10 years for managing GIST patients with molecularly targeted treatment would be between £47,521 and £56,146 per patient compared with a cost of between £4047 and £4230 per patient when managed with BSC (price year not stated). 54
Variation in service and uncertainty about best practice
The treatment of GISTs after progression on imatinib is generally decided on a case-by-case basis by multidisciplinary teams, and the alternatives are dose escalation of imatinib, sunitinib at 50 mg/day (4 weeks out of 6 weeks) or, alternatively, BSC only (although due to the ‘symptomatic flare’ already mentioned this may include continuation of imatinib at 400 mg/day). Many clinicians advocate initial dose escalation of imatinib and then consider sunitinib on subsequent progression, but there will be variation in clinical practice depending on the specific needs of individual patients.
Description of technology under assessment
Summary of intervention
Imatinib
Imatinib (Glivec) is a rationally designed small molecule inhibitor of several oncogenic tyrosine kinases: c-Abl, PDGFRA and the KIT tyrosine kinases. Its therapeutic activity in GISTs relates to inhibition of KIT, although in cases with no KIT mutation the inhibition of PDGFRA is likely to be of therapeutic importance. 2 Imatinib is a derivative of 2-phenylaminopyrimidine, and a competitive antagonist of adenosine triphospate (ATP) binding, which blocks the ability of KIT to transfer phosphate groups from ATP to tyrosine residues on substrate proteins. This interrupts KIT-mediated signal transduction, which is the key pathogenic driver for many GISTs. The inhibitory activity of imatinib on KIT is highly selective, and minimal inhibition of other kinases that are important in normal cell function occurs, thereby affording a good toxicity and safety profile.
Imatinib is licensed and approved for use in the UK NHS in KIT-immunoreactive positive advanced/unresectable GISTs. 50,57
Sunitinib
Sunitinib malate (Sutent), is a tyrosine kinase inhibitor targeting KIT, PDGFRA, all three isoforms of vascular endothelial growth factor receptor (VEGFR), FMS-like tyrosine kinase 3 (FLT3) colony-stimulating factor 1 receptor (CSF-1R) and glial cell line-derived neurotrophic factor receptor. 58 Sunitinib activity in GISTs may predominantly relate to inhibition of KIT and/or PDGFRA, and ex vivo investigation has shown that sunitinib can inhibit the kinase activity of KIT molecules harbouring secondary mutations conferring imatinib resistance. 59 However, the potent antiangiogenic activity of sunitinib as a consequence of strong VEGFR inhibition may also be important for clinical activity in GISTs.
Best supportive care
Best supportive care is not well defined or standardised, and can also be referred to as ‘supportive care’ or ‘active symptom control’. 55 It usually involves interventions to manage pain and treat fever, anaemia (due to GI haemorrhage) and GI obstruction,50 and can include palliative measures. 60 A Cochrane review of supportive care for patients with GI cancer defined supportive care as ‘the multi-professional attention to the individual’s overall physical, psychosocial, spiritual and cultural needs’. 61 It was argued that this type of care should ethically be made available to all treatment groups, meaning that treatment with imatinib or sunitinib could not be provided without concomitant supportive care as well in clinical practice for patients with GIST, although it is possible that treatment with BSC could be provided without additional drug treatment with either imatinib or sunitinib. It should be noted that the amount of care required as part of BSC is likely to increase as the disease progresses and symptoms become worse.
Identification of important subgroups
The differential benefit from imatinib and sunitinib in subgroups of patients with GIST, whose tumours have different primary and secondary KIT mutations, has suggested possible benefits in personalising first- and second-line therapy.
Primary KIT mutations are those that are pathogenic and present before any systemic treatment, while secondary mutations are those that have been identified after imatinib treatment and confer resistance to imatinib. Identification of secondary mutations requires rebiopsy of tumours, and studies have suggested that the emergence of secondary (or acquired) imatinib resistance is polyclonal, so patients with GIST may acquire more than one secondary KIT mutation. 62
A meta-analysis of 1640 patients revealed that patients with KIT exon 9 primary mutations have a better outcome if treated at the escalated dose of 800 mg daily. 63 Similarly, objective response rates to imatinib 400 mg/day are higher in patients with exon 11 primary mutations than in those with exon 9 mutations, or those with no detectable KIT or PDGFR mutation. 14,41 Therefore, advanced GIST patients with exon 9 mutations may benefit from immediate dose escalation of imatinib, and the benefit of dose escalation on progression may be more significant in this subgroup of patients and thereby have implications for therapeutic alternatives and choices on progression in different groups of patients defined by KIT mutations. Recent studies have indicated that plasma monitoring in GIST patients could assist clinicians’ decision-making with regard to whether or not dose escalation of imatinib is required for particular patients, including those with mutations in KIT. 64–66
Secondary mutations in KIT exons 13, 14, 17 and 18 are associated with acquired resistance to imatinib. 43 Sunitinib activity after progression on imatinib has been demonstrated in GIST patients with imatinib resistance conferring secondary KIT mutations. 62 However, both the primary KIT mutation genotype and secondary KIT mutations may influence the clinical benefit effect of sunitinib in GIST patients who have progressed on imatinib. 62 Interestingly, in contrast to imatinib, greater benefit from sunitinib (after imatinib failure) is seen in patients with primary exon 9 mutations or wild-type KIT as opposed to primary exon 11 mutations. 62 However, it is not clear how dose-escalated imatinib (800 mg/day) compares with sunitinib in patients with primary exon 9 KIT mutations. While the polyclonal emergence of resistance is an investigational and clinical challenge, it appears that GIST patients with secondary KIT mutations associated with acquired imatinib resistance in exons 13 or 14 (which involve the KIT–ATP binding pocket) appear to gain greater clinical benefit from sunitinib after imatinib failure than those patients with exon 17 or 18 imatinib resistance secondary mutations (which involve the KIT activation loop). 62
Changes in FDG (fluorodeoxyglucose) avidity of GISTs measured by FDG-PET occur earlier than anatomical changes in GISTs and so may also have a role as a predictive biomarker for imatinib response, and also for detecting early disease progression49 in the future as the technology becomes more widely available in NHS settings.
Current usage in the NHS
Current practice is to commence patients on imatinib 400 mg/day, and on confirmed disease progression the options are dose escalation of imatinib up to 800 mg/day or sunitinib, or BSC only. Practice is variable, and decided on a case-by-case basis. Some clinicians proceed with dose escalation of imatinib initially and then, on further progression, use sunitinib. Some guidelines and clinicians advocate returning to imatinib for symptomatic benefit, when there are no other therapeutic options, and the cessation of imatinib in the absence of alternative treatment options is not recommended owing to the tumour flare phenomenon, with rapid deterioration in symptoms observed in some patients.
Chapter 2 Definition of the decision problem
Decision problem
Specific information on the population, interventions, comparators and relevant outcomes considered for this review are discussed in detail in Chapter 4 (see Identification of studies).
Until the licensing of imatinib, the prognosis for people with unresectable and/or metastatic GISTs was poor. 19 Since 2002, the clinical effectiveness of treatment for GIST with imatinib at a dose of 400 mg/day has been well documented. 50,55 There is also clinical trial evidence showing that patients with unresectable and/or metastatic GIST can also respond to higher doses of imatinib, up to a maximum tolerated dose of 800 mg/day,40 and that patients with different exon mutations in the KIT gene may differ in their response to imatinib at both standard and escalated doses. 14
Guidance from NICE does not currently recommend the prescription of escalated doses of imatinib upon progression on the standard 400 mg/day dose,50 although it is common in clinical practice. 15,32 Most of the evidence relating to dose-escalated imatinib comes from randomised trials where participants were randomised to doses greater that 400 mg/day, as opposed to receiving these higher doses upon disease progression on the 400 mg/day dose. However, evidence suggests that tolerability of higher doses may depend on the extent of prior exposure to the drug,66 and if in clinical practice escalated doses are prescribed only upon progression, these trial data may not provide reliable estimates of response, progression-free survival (PFS) and overall survival (OS), quality-of-life effects or the extent of adverse event occurrence. In addition, if patients with unresectable and/or metastatic GIST are likely to attain different levels of clinical benefit from different imatinib doses then clinicians’ decision-making on appropriate dosages for individual patients should be informed by the best available evidence.
The development of imatinib has represented a paradigm shift in the treatment of unresectable and/or metastatic GIST, as, prior to its introduction onto the market, the only available treatment remaining for this population group was BSC, which, given the severity of this disease, represents essentially palliative intervention. Since the introduction of imatinib, other new treatments for unresectable and/or metastatic GIST have become available, including sunitinib, which has been recommended by NICE as the second-line treatment for the population of interest, after failure on treatment with imatinib. 51 As there are now various options available for treating unresectable and/or metastatic GIST, it is therefore necessary to review the available evidence on imatinib at escalated doses, when compared with sunitinib, for patients with unresectable and/or metastatic GIST, whose disease has progressed on the standard imatinib dose of 400 mg/day.
Overall aims and objectives
The aim of this review was to assess the clinical effectiveness and cost-effectiveness of imatinib at escalated doses (i.e. 600 or 800 mg/day) within its licensed indication,67 for the treatment of patients with unresectable and/or metastatic GISTs, who have progressed on imatinib at a dose of 400 mg/day.
The objectives of this review will help facilitate decision-making on the most appropriate treatment(s) for patients with unresectable and/or metastatic GIST who have progressed on imatinib at a dose of 400 mg/day, by:
-
conducting a systematic review of the evidence available on the clinical effectiveness of imatinib at dosages of 600 or 800 mg/day compared with sunitinib and/or BSC
-
conducting a systematic review of the cost-effectiveness of imatinib at dosages of 600 or 800 mg/day compared with sunitinib and/or BSC
-
analysing available outcome data for particular subgroups of interest (e.g. patients with different KIT mutations) in order to establish any differences in clinical effectiveness for specific groups
-
developing an economic model to compare the cost-effectiveness and cost–utility of imatinib at a dose of 600 or 800 mg/day with those of sunitinib (within its recommended dose range) or BSC only.
This report contains reference to confidential information provided as part of the NICE appraisal process. This information has been removed from the report and the results, discussions and conclusions of the report do not include the confidential information. These sections are clearly marked in the report.
Chapter 3 Critique of the manufacturer submission
The manufacturer of imatinib (Novartis) did not provide an economic analysis in their submission, stating that, owing to the limited amount of data available from the key clinical studies and the dearth of data comparing imatinib dose escalation with sunitinib and BSC, they were unable to submit a sufficiently robust economic analysis that met the scope for the appraisal. However, they did provide a summary of clinical evidence and implications for the economic analysis. With the exception of the Executive Summary section, and most of the References section, a large proportion of the submission document was highlighted as commercial in confidence (CiC). Electronic copies of all the papers cited in the References section, including two labelled as CiC by the manufacturer, were provided. Apart from both of the CiC documents, these studies had already been retrieved by our searching process and are discussed in Chapter 4.
Of the two CiC reports provided, one (CiC information has been removed) was a report on the randomised, Phase II, B2222 trial comparing imatinib at doses of 400 and 600 mg/day. Patient data from this trial that are relevant to this review have since been published by Blanke et al. 39 in the Journal of Clinical Oncology. The remaining CiC report (CiC information has been removed) provided a meta-analysis of data from the randomised, Phase III, intergroup S0033 trial comparing imatinib at doses of 400 and 800 mg/day, and the randomised, Phase III, EORTC-ISG (Italian Sarcoma Group)-AGITG (Australasian Gastro-Intestinal Trials Group) trial, also comparing imatinib at these doses. Crossover data from the S0033 trial have been published separately,41,68 as have crossover data from the EORTC-ISG-AGITG trial. 44 (CiC information has been removed.)
(CiC information has been removed.) All relevant results pertaining to the population of interest for this review have been provided in Chapter 4(Assessment of clinical effectiveness). (CiC information has been removed) but as more recent results for the study population of interest have been published, only study characteristics information was used in Chapter 4 of this review.
The key points made in the manufacturer submission were as follows:
-
The limited number of data available from the key clinical studies and the paucity of data comparing imatinib dose escalation with sunitinib and BSC prevent, in the opinion of the manufacturer, the submission of a sufficiently robust economic analysis which meets the scope of the appraisal.
-
There are currently no head-to-head trial data comparing imatinib with sunitinib.
-
Sunitinib represents a third-line treatment, rather than second line as per the scope of the evaluation, making it difficult, if not impossible, to conduct a robust and plausible indirect comparison of the two technologies. UK National GIST Guidelines56 recommend that changing treatment to sunitinib should be considered only after patients have shown progression on imatinib dose escalation.
-
Since the publication of TA86 clinical practice has evolved to consider dose escalation to a daily dose of 600 or 800 mg, when patients progress on the standard daily dose of 400 mg, and this change in clinical practice is reflected within UK National GIST Guidelines. 56
-
(CiC information has been removed.)
-
(CiC information has been removed.)
-
(CiC information has been removed.)
Chapter 4 Assessment of clinical effectiveness
Methods for reviewing effectiveness
Identification of studies
Extensive sensitive electronic searches were conducted to identify reports of published and ongoing studies on the clinical effectiveness of imatinib. The searches were also designed to retrieve clinical effectiveness studies of the comparator treatments (sunitinib and BSC). In addition, reference lists of retrieved papers and submissions from industry and other consultees were scrutinised to identify additional potentially relevant studies.
The databases searched were MEDLINE (1966 – September, week 3, 2009), MEDLINE In-Process (25 September 2009), EMBASE (1980 – week 39, 2009), Cumulative Index to Nursing and Allied Health Literature (CINAHL) (September 2009), Science Citation Index (SCI) (2000 – 26 September 2009), BIOSIS (2000 – 24 September 2009), Health Management Information Consortium (September 2009), and the Cochrane Controlled Trials Register for primary research and the Database of Abstracts of Reviews of Effects (DARE) (October 2009), the Cochrane Database of Systematic Reviews (CDSR) (Issue 3, 2009) and the Health Technology Assessment (HTA) database (October 2009).
Ongoing and recently completed trials were searched in the following databases: current research registers, including Clinical Trials, Current Controlled Trials (CCT), National Institute of Health Research (NIHR) Portfolio, World Health Organization (WHO) International Clinical Trials Registry Platform, International Federation of Pharmaceutical Manufacturers & Associations (IFPMA) Clinical Trials and the Association of the British Pharmaceutical Industry (ABPI) database. Recent conference proceedings of key oncology and GI organisations, including the American Society for Clinical Oncology (ASCO), European Society for Medical Oncology (ESMO) and the European Cancer Organisation, were screened. Websites of the GIST Support International, and the drug manufacturers Pfizer and Novartis were also scrutinised.
Full details of the search strategies used are reproduced in Appendix 1.
Inclusion and exclusion criteria
Types of studies
An initial scoping search suggested that there would be few studies looking specifically at either of the named interventions (imatinib 600 or 800 mg/day). Therefore, we considered all of the following types of studies for the assessment of clinical effectiveness:
-
randomised controlled trials (RCTs)
-
non-randomised comparative studies, and
-
case series.
If the number of studies meeting our inclusion criteria was sufficiently large, consideration was to be given to limiting them by type of study design, and also possibly other factors (e.g. sample size). Additionally, we planned to exclude non-English language papers, and/or reports published as meeting abstracts, if the evidence base of English language and/or full-text reports was sufficiently large.
Types of participants
Participants considered were people with KIT (CD117)-positive unresectable and/or metastatic malignant GISTs, whose disease had progressed on treatment with imatinib at a dose of 400 mg/day. If sufficient evidence was available, subgroup analysis was to be undertaken for those patients with different mutations of CD117, as there is some evidence to suggest this may affect their response to escalated doses of imatinib14,41,63 (see Chapter 1, Identification of important subgroups). In addition, subgroup analysis was also to be undertaken on methods used to identify response or resistance (e.g. FDG-PET or CT scanning) and the use of imatinib in a neoadjuvant or adjuvant setting for patients with previously resectable GIST, where sufficient data were available.
Types of intervention and comparators
The interventions considered were imatinib at escalated doses of 600 and 800 mg/day, respectively, being prescribed with BSC. The comparators considered were sunitinib, prescribed within its recommended dose range of 27–75 mg and provided with BSC, and BSC only. As previously stated, BSC is defined as ‘the multi-professional attention to the individual’s overall physical, psychosocial, spiritual and cultural needs’. 61
Types of outcomes
For the assessment of clinical effectiveness, the following outcomes were considered:
-
overall response
-
overall survival
-
disease-free survival
-
progression-free survival
-
time to treatment failure
-
health-related quality of life (HRQoL) [e.g. European Quality of Life-5 Dimensions (EQ-5D) scores]
-
adverse effects of treatment (e.g. number of discontinuations due to adverse events).
Exclusion criteria
We excluded studies of animal models, preclinical and biological studies, reviews, editorials, opinions, case reports and reports investigating technical aspects of the interventions.
Data extraction strategy
The titles and abstracts (where available) of all records identified by the search strategy were screened by two reviewers independently. Full-text copies of all potentially relevant reports were retrieved. The full-text reports were assessed against the inclusion and exclusion criteria by two reviewers independently. Full-text papers and conference abstracts were assessed using a screening form that was developed and piloted for this purpose. Any disagreements were resolved by consensus or arbitration by a third party. A copy of the screening form used can be found in Appendix 2.
A data extraction form was developed and piloted (Appendix 3). One reviewer extracted details of the study design, participants, intervention, comparator and outcomes, and a second reviewer checked the data extraction for accuracy. Any disagreements were resolved by consensus or arbitration by a third party.
Quality assessment strategy
Two reviewers independently assessed the methodological quality of the included full-text studies. Non-randomised comparative studies were assessed using an 18-question checklist, with the same checklist minus four questions used to assess the methodological quality of case series. This checklist for non-randomised studies and case series was adapted from several sources, including the Centre for Reviews and Dissemination’s guidance for those carrying out or commissioning reviews,69 Verhagen et al. ,70 Downs and Black,71 and the Generic Appraisal Tool for Epidemiology (GATE). It assesses bias and generalisability, sample definition and selection, description of the intervention, outcome assessment, adequacy of follow-up, and performance of the analysis. The checklist was developed through the Review Body for Interventional Procedures (ReBIP). ReBIP is a joint venture between Health Services Research at Sheffield University and the Health Services Research Unit at the University of Aberdeen, and works under the auspices of the NICE Interventional Procedures Programme.
We planned to assess the quality of RCTs using the Cochrane Collaboration’s tool for assessing risk of bias. 72 The tool addresses six specific domains: sequence generation, allocation concealment, blinding, incomplete outcome data, selective outcome reporting and other issues. Each quality assessment item had three possible responses: ‘yes’, ‘no’ or ‘unclear’, with space for additional comments. Disagreements between reviewers over study quality were to be resolved by consensus and, if necessary, arbitration by a third party. Abstracts were not quality assessed because they were considered unlikely to provide sufficient methodological information to enable an accurate assessment of study quality. Methodological quality did not form part of the criteria for the inclusion or exclusion of studies. A copy of the quality assessment tool can be found in Appendix 4.
Data analysis
The type of data analysis considered was dependent on the number of studies meeting the specified inclusion criteria, and study design. Where a quantitative synthesis was considered inappropriate or not feasible, it was planned that a narrative synthesis of results would be provided instead.
For relevant outcomes from randomised comparisons, it was decided that meta-analysis (where appropriate) would be used to estimate a summary measure of effect. Dichotomous outcome data for the overall response outcome would be combined using the Mantel–Haenszel relative risk (RR) method, and continuous outcomes by using the inverse variance weighted mean difference (WMD) method. For both of these estimates, 95% confidence intervals (CIs) and p-values would also be calculated. Chi-squared tests and I2-statistics were to be used to explore statistical heterogeneity across studies, with possible reasons for heterogeneity explored using sensitivity analysis. Where no obvious reason for heterogeneity was found, the implications would be explored using random effects methods.
The pooled weighted ratio of median survival would be derived for OS, disease-free survival and PFS. The hazard ratio (HR) is the most appropriate statistic for time-to-event outcomes (i.e. for time to treatment failure). If available, the HR would be extracted directly from the trial publications, but if not reported it would be extracted if possible from other available summary statistics or from data extracted from published Kaplan–Meier curves using methods described by Parmar et al. 73 A pooled HR from available RCTs could then be obtained by combining the observed (O) minus expected (E) number of events and the variance obtained for each trial using a fixed effects model. 74 A weighted average of survival duration across studies was to be calculated. The chi-squared test for heterogeneity was to be used to test for statistical heterogeneity between studies.
Where no RCT data were available, but non-randomised studies had reported relevant data for survival outcomes, assessment of the risk of bias and heterogeneity was to be undertaken using meta-regression analysis.
It was expected that few studies, if any, would report direct comparisons of the intervention and comparators, so (depending on feasibility and appropriateness) it was decided that, where non-randomised evidence was available, meta-analysis models would be used to model survival rates for interventions and comparators. A ‘cross-design’ approach was to be adopted to allow non-randomised evidence to be included, while avoiding the strong assumption of the equivalence of studies. Evidence suggests that this approach would allow data from RCTs, non-randomised comparative studies and case series to be included. 75 Differences between treatments for survival outcomes were to be assessed via the corresponding odds ratio and 95% credible intervals. These results are ‘unadjusted odds ratios’, but meta-analysis models adjusting for study type were also to be used. The results from these models produce ‘adjusted’ odds ratios. 76 Winbugs software (MRC Biostatics Unit, Cambridge, UK) was to be used for the analysis.
Any reported data on adverse effects of treatment and quality of life (QoL) that were collected were to be combined, using standardised mean difference, where appropriate.
In addition, and taking into account the type of evidence, the feasibility of using a mixed treatment comparison model for indirect comparisons was to be considered.
Results
Number of studies identified
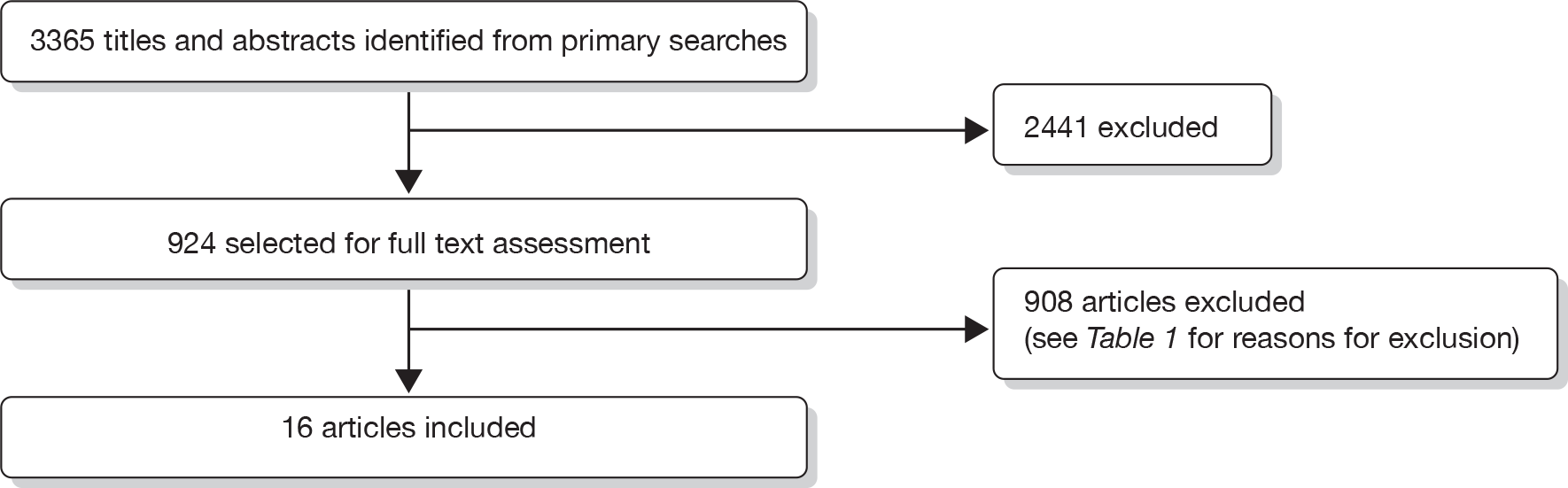
We identified 3365 records from the primary searches for the review of clinical effectiveness. After title and abstract screening, 2441 articles were considered not to be relevant for this review and were excluded. The full-text papers of 924 records were obtained and screened. One hundred and twenty-three of these full-text papers were non-English language publications. In total, six full-text papers and 10 abstracts reporting four separate clinical trials and one additional retrospective cohort met our inclusion criteria. An additional 49 papers were retained for background information. The reasons for exclusion of assessed full-text papers are given in Table 1. A flow diagram of the screening process is outlined in Figure 1. Information on the reasons for excluding individual studies is provided in Appendix 5.
| Reason for exclusion | No. of studies excluded |
|---|---|
| Patient had resectable GIST | 24 |
| Outcomes not reported separately for patients with GIST | 10 |
| < 10 patients in relevant study population | 46 |
| Imatinib dose is 400 mg/day | 13 |
| No/insufficient data reported for escalated dose patients | 65 |
| No imatinib dose reported | 84 |
| No relevant interventions | 15 |
| Treatment not evaluated | 11 |
| No outcomes of relevance | 10 |
| Other reason | 61 |
| 339 | |
| Retained for background information | 49 |
| Review articles | 169 |
| Letter/editorial/correspondence/symposium articles/meeting reports/expert views/comments | 117 |
| Case study/case series < 10 patients | 64 |
| Non-English language exclusions | 123 |
| Not obtained | 47 |
| Total | 908 |
FIGURE 1.
Flow diagram outlining the screening process for the review of clinical effectiveness.
Included studies
See Appendix 6 for a list of studies that were included in the review of clinical effectiveness. We did not identify any RCTs, or non-randomised comparative studies, comparing the effectiveness of escalated doses of imatinib (600 or 800 mg/day) with sunitinib or BSC that met our inclusion criteria. One ongoing trial was identified comparing imatinib and sunitinib. However, this study was stopped owing to poor recruitment. 53 We identified five full-text reports of three randomised trials of imatinib that contained relevant data for this review. 14,38,39,41,44 The studies by Zalcberg et al. ,44 Blanke et al. (S0033)41 and Blanke et al. (B2222)39 were designated as the primary reports for the EORTC-ISG-AGITG (62005) trial, the S0033 trial and the B2222 trial, respectively. The study by Debiec-Rychter et al. 14 met our inclusion criteria and provided additional information from the EORTC-ISG-AGITG (62005) study on response following crossover, while the study by Demetri et al. 52 met our inclusion criteria and provided interim data from the B2222 trial on response following crossover.
An additional three abstracts were identified, with two68,77 reporting interim data for the S0033 trial, and one reporting interim data for the EORTC-ISG-AGITG 62005 trial. 78
All of these included studies contained a treatment arm of 400 mg/day, and reported data separately for participants who received an escalated dose of imatinib upon progression at this randomised dose. One additional full-text paper detailing the results of a non-randomised retrospective study by Park et al. 79 was also included. This study met our inclusion criteria as it also provided separate outcome data for patients with metastatic or unresectable GIST, who received escalated doses of imatinib on progression at an initial dose of 400 mg/day.
For the comparator treatment of sunitinib, we identified seven abstract reports meeting our inclusion criteria. All were interim results of an ongoing, open-label sunitinib trial reporting information on participants recruited to the trial following failure at different doses of imatinib, including doses of ≤ 400 mg/day. 80–86 We designated the abstract by Seddon et al. 86 to be the primary report for this trial, as it was thought to contain its most recent results.
For the comparator treatment of BSC, no randomised, non-randomised or case series studies were identified that compared either of the interventions (imatinib at a dose of 600 mg/day or imatinib at 800 mg/day) with BSC, or provided data on relevant outcomes for the population of interest for BSC only. It should be noted that studies published on the clinical effectiveness of BSC prior to the licensing of imatinib18,19 were not eligible for this review as our population of interest was those who had failed on imatinib at 400 mg/day; therefore all studies published prior to the availability of imatinib automatically failed to meet our inclusion criteria because BSC at that time could not possibly have been provided following failure of treatment with imatinib at a dose of 400 mg/day.
Corresponding authors for each of the included trials were contacted in order to determine whether any additional data could be provided specifically for the population of interest (i.e. those participants failing on an imatinib dose of 400 mg/day and receiving either an escalated dose of imatinib 600 or 800 mg/day or, alternatively, sunitinib). For the ongoing, open-label sunitinib study, the corresponding author replied that no further information could be provided as the study was an official, ongoing trial by the manufacturer (Pfizer). For the imatinib trials, in the case of both studies by Blanke et al. 39,41 our requests for information were forwarded to the statistics team involved in the trials. The requested data for the S0033 trial were provided on 17 February 2010. For the study by Zalcberg et al. ,44 a response to our request was received, explaining that an official data request form must be completed. This was submitted, and a further response was received on 9 April 2010 explaining that the data could not be provided until September 2010 (and then only if the request were approved). It was decided not to pursue the request for data further, given the timelines for this project.
Two additional reports (CiC information has been removed) to the ones identified through our search strategy were provided for this review by the manufacturer and have been discussed in Chapter 3, and are also discussed below. Both of these reports were marked as CiC.
Excluded studies
A list of 340 studies, originally identified as potentially relevant but subsequently failing to meet our inclusion criteria, is provided in Appendix 5. The studies were excluded because they failed to meet one or more of the inclusion criteria in terms of the type of study, participants, intervention, comparator or outcomes reported. It should be noted that all full-text screened studies on plasma monitoring, as well as those on the use of FDG-PET technology for evaluating PD, did not meet our inclusion criteria. In addition, the types of participants were limited to an adult population, therefore studies involving children with GIST were excluded. However, it should be noted that the age range provided in the baseline data for the included study by Seddon et al. 86 indicates that at least one child was recruited on to this trial, but, as the median age reported indicates that the majority of patients in this trial were adults, the study was not excluded.
Studies with a relevant population of fewer than 10 patients were also excluded. Changes to our original protocol were reported to NIHR in a progress report submitted on 9 December 2009.
In addition to the included studies identified above, nine studies (reported in 14 papers) reported sufficient information with regard to our inclusion criteria to be considered for potential inclusion in this review, subject to clarification from the study authors regarding specific aspects of the study. Corresponding authors for each of the nine studies were therefore contacted. Responses were received from four corresponding authors (GD Demetri, Ludwig Center at Dana-Farber/Harvard Cancer Center and Sarcoma Center, Boston, MA, USA, 2010; YK Kang, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea, 2010; P Rutkowski, Sklodowska-Curie Memorial Cancer Center and Institute of Oncology Department of Soft Tissue/Bone Sarcoma and Melanoma, Warsaw, Poland, 2010; P Wolter, UZ Leuven, Leuven, Belgium, 2010; personal communication). In the cases of two responses, this resulted in the exclusion of the studies (five papers in total) from the review (P Rutkowski, P Wolter, personal communication) In the remaining two studies (four papers), the responses did not result in clarification, as the authors requested that we wait for a further response from them or their colleagues (GD Demetri, YK Kang, personal communication). In the case of correspondence with YK Kang, it was decided that the study by Park et al. 79 could be included in the review without further clarification from the corresponding author.
Of the correspondences that did not result in responses, one e-mail could not be sent successfully87 and the remaining four authors did not respond. 88–91
Characteristics of the included studies
Study characteristics data were available for the four full-text included imatinib studies39,41,44,79 and the primary report of the included sunitinib trial. 86 However, of these studies, only the studies by Zalcberg et al. 44 and Park et al. 79 gave specific baseline information for the crossover subgroup of interest. Therefore, Table 2 provides details of all characteristics information provided for each crossover group, while Table 3 provides details of the same characteristics for all patients in the treatment arms of interest (initial randomisation to a dose of 400 mg/day). In the case of the EORTC-ISG-AGITG trial reported by Zalcberg et al. ,44 relevant study characteristic data for participants initially randomised to the 400 mg/day dose were not available. However, these data were reported in a paper by Verweij et al. 42 for the same trial. The paper by Verweij et al. 42 failed to meet the inclusion criteria for this review as it did not provide any outcome data for patients receiving an escalated dose of 800 mg/day imatinib upon progression at a 400 mg/day dose, but as it provides information on the characteristics of all randomised patients (of whom a proportion went on to receive an escalated dose of 800 mg/day and formed the study population of the included study by Zalcberg et al. 44), it was felt that the baseline data from this excluded study could still be used.
| Zalcberg 200544 | Blanke S003341 | Blanke B222239 | Park 200979 | Seddon 200886 | |
|---|---|---|---|---|---|
| Drug assessed | Imatinib | Imatinib | Imatinib | Imatinib | Sunitinib |
| Doses given | 400 mg/day, 800 mg/day | 400 mg/day, 800 mg/day | 400 mg/day, 600 mg/day | 600 mg/day, 800 mg/day | Cycle of 50 mg/day for 4 weeks, then 0 mg/day for 2 weeks |
| Start date | (CiC information has been removed) | December 2000 | July 2000 | June 2001 | Unspecified |
| End date | April 2004 | (CiC information has been removed) | May 2006 | June 2006 | December 2007 |
| Study countries | Australia, Belgium, Denmark, France, Germany, Italy, the Netherlands, New Zealand, Poland, Singapore, Spain, Switzerland, UK | Canada, USA | Finland, USA | Seoul, South Korea | Unspecified but ‘worldwide’ and ‘multicentre’ |
| No. of institutions involved (no. of countries involved) | (CiC information has been removed) | 148 (2) | 4 (2) | 1 (1) | 96 (33) |
| Length of follow-up at time of analysis | Median of 25 months (maximum of 35 months) | Median of 4.5 years | Median of 63 months (maximum of 71 months) | Median of 8 months (range 1.4–22.3) | Median of 51 weeks (range 0.1–159) |
| Number receiving escalated dose of imatinib after failure of imatinib at 400 mg/day, out of all of those randomised to receive 400 mg/day | 133/473 (28.1%) | 118/345 (34.2%) | 43/73 (58.9%) | 24/24 (100.0%) | NA |
| Number receiving sunitinib after failure of imatinib at ≤ 400 mg/day, out of all of those receiving sunitinib | NA | NA | NA | NA | 351/1117 (31.4%) |
| Included in this analysis | aEORTC-ISG-AGITG42 | Blanke S003341 | Blanke B222239 (CiC information has been removed) | Park 200979 | Seddon 200886 | |
|---|---|---|---|---|---|---|
| All those randomised to 400 mg/day | All those who received escalated doses of imatinib on progression at a dose of 400 mg/dayb | All those receiving sunitinib | ||||
| Number included | 473 | 345 | 73 | 24 | 1117 | |
| Age in years: median (range) | 59 (49–67) | 61.9 (18–87) | (CiC information has been removed) | 52 (31–73) | 59 (10–92) | |
| Sex: M/F | 283/190 | 187/158 | (CiC information has been removed) | 18/6 | 665/451 | |
| ECOG/WHO Performance Status Score: | 0 | 217 | (CiC information has been removed) | 4 | 420 | |
| 1 | 191 | (CiC information has been removed) | 18 | 515 | ||
| 2 | 48 | (CiC information has been removed) | 2 | 134 | ||
| ≤ 2 | (456) | 332 | (CiC information has been removed) | (1069) | ||
| > 2 | 17 | 13 | (CiC information has been removed) | 38 | ||
| Missing | 10 | |||||
| Race/ethnicity (n) | NR | NR | NR | |||
| White | 273 | CiC information has been removed) | ||||
| Black | 37 | (CiC information has been removed) | ||||
| Asian | 25 | (CiC information has been removed) | ||||
| Other/unknown | 10 | (CiC information has been removed) | ||||
| Number having previous chemotherapy | 156 (32.9%) | NR | (CiC information has been removed) | 3 (12.5%) | 225 (26.8%) | |
| Number having previous radiotherapy | 26 (5.5%) | NR | (CiC information has been removed) | NR | 78 (7.9%) | |
| Number having prior surgery | 410 (86.7%) | (CiC information has been removed) | 20 (83.3%) | NR | ||
Four of the included trials reported data for imatinib,39,41,44,79 while the remaining trial reported data for sunitinib. 86 Two of the imatinib trials randomised patients to imatinib doses of either 400 or 800 mg/day,41,44 one randomised patients to imatinib doses of either 400 or 600 mg/day,39 and the other was a retrospective study looking only at patients with GIST who had received escalated doses of imatinib at either 600 or 800 mg/day on progression at a dose of 400 mg/day. 79 The sunitinib trial is an ongoing, non-randomised, open-label study and participants are provided with a 6-week cycle of sunitinib, at a dose of 50 mg/day for 4 weeks followed by 2 weeks without the drug. 86
The study start date was reported for three out of the four included imatinib trials39,41,79 and was made available for the study by Zalcberg et al. 44 by the manufacturer (CiC information has been removed). From this it can be seen that the earliest study start date is that of the study (CiC information has been removed)39 (CiC information has been removed). The included sunitinib abstract did not report a start date.
Three out of the four included imatinib studies reported an end date,39,44,79 and in the case of the sunitinib study by Seddon et al. 86 a date was reported for the most recent analysis. The manufacturer also made this information available for the study by Blanke et al. 41 (CiC information has been removed). The ongoing sunitinib trial has the most recent update, while the study by Zalcberg et al. was completed first, in April 2004. 44
With the exception of the study by Park et al. ,79 which involved one centre in one country, all trials were international and multicentre,39,41,44,86 with the sunitinib trial involving the most countries85 and the S0033 trial involving the most institutions. 41 The B2222 trial involved the fewest countries and fewest institutions. 39
The longest length of follow-up occurred in the B2222 trial reported by Blanke et al. ,39 in which patients were followed up for a median of 63 months, while the shortest length of follow-up was found in the study by Park et al. ,79 which gave a median follow-up for the study population of 8 months.
Among the imatinib trials, 133/473 (28.1%), 118/345 (34.2%) and 43/73 (58.9%) of those initially randomised to imatinib at 400 mg/day progressed and were given an escalated dose. 39,41,44 In the imatinib study by Park et al. ,79 the study population comprised only those who were given escalated doses of imatinib so 24/24 (100%) received an escalated dose. In the sunitinib study by Seddon et al. ,86 351/1117 (31.4%) of those who failed on imatinib and were entered into the trial had failed on a dose of 400 mg/day or less. Therefore, the study with the largest relevant population was the sunitinib trial,86 while the study by Park et al. 79 had the smallest study population.
The Park study79 had the youngest population, whereas the S0033 trial41 had the oldest study population. In (CiC information has been removed) studies, the number of male patients was higher than the number of female patients, which concurs with the epidemiological trends in gender associated with this disease (see Chapter 1, Epidemiology and incidence).
(CiC information has been removed) studies reported data on the performance status score of participants, although the study by Blanke et al. for the S0033 trial41 had combined the Eastern Cooperative Oncology Group (ECOG) performance status categories 0–2. Doing the same for the remaining studies shows that the vast majority of participants, 456/473 (96.4%), 332/345 (96.2%), (CiC information has been removed), 24/24 (100%) and 1069/1107 (96.6%) in the EORTC-ISG-AGITG trial,42 S0033 trial,41 B2222 trial,39 (CiC information has been removed) Park study79 and the sunitinib trial,86 respectively, had a performance status score of ≤ 2.
(CiC information has been removed.)
In terms of prior treatment, (CiC information has been removed) two reported the number having previous radiotherapy,42,86 (CiC information has been removed). For the imatinib studies, 3/24 (12.5%), 156/473 (32.9%) and (CiC information has been removed) of participants had undergone previous chemotherapy in the study by Park et al. ,79 the EORTC-ISG-AGITG trial42 and the B2222 trial39 (CiC information has been removed), respectively, while 26.8% (225/1117) of patients had received prior chemotherapy in the study by Seddon et al. 86 With regard to radiotherapy, 26/473 (5.5%) of patients in the EORTC-ISG-AGITG trial42 and 78/1117 (7.9%) of patients in the sunitinib trial86 had received prior radiotherapy. (CiC information has been removed) of participants involved in the B2222 trial reportedly had received prior surgery, (CiC information has been removed) while this figure was 86.7% (410/473) for participants in the EORTC-ISG-AGITG trial,42 and 83.3% (20/24) in the study by Park et al. 79
Quality of the included studies
Results of the quality assessment for all four included full-text papers are summarised in Figure 2. No third party arbitration for quality assessment was required. The results of the quality assessment for each individual study are provided in Appendix 9. Three full-text studies assessed for quality assessment were included in the review because they provided crossover data on a subset of patients who were originally randomised to a dose of 400 mg/day, but progressed and received an escalated dose of either 600 mg/day39 or 800 mg/day. 41,44 The fourth study79 was assessed for quality because it included a retrospective analysis of a subgroup of a cohort of patients given treatment with imatinib at 400 mg/day. The subgroup were patients who received escalated doses of 600 mg/day and/or 800 mg/day after progression on the 400 mg/day dose.
FIGURE 2.
Quality assessment results summary.
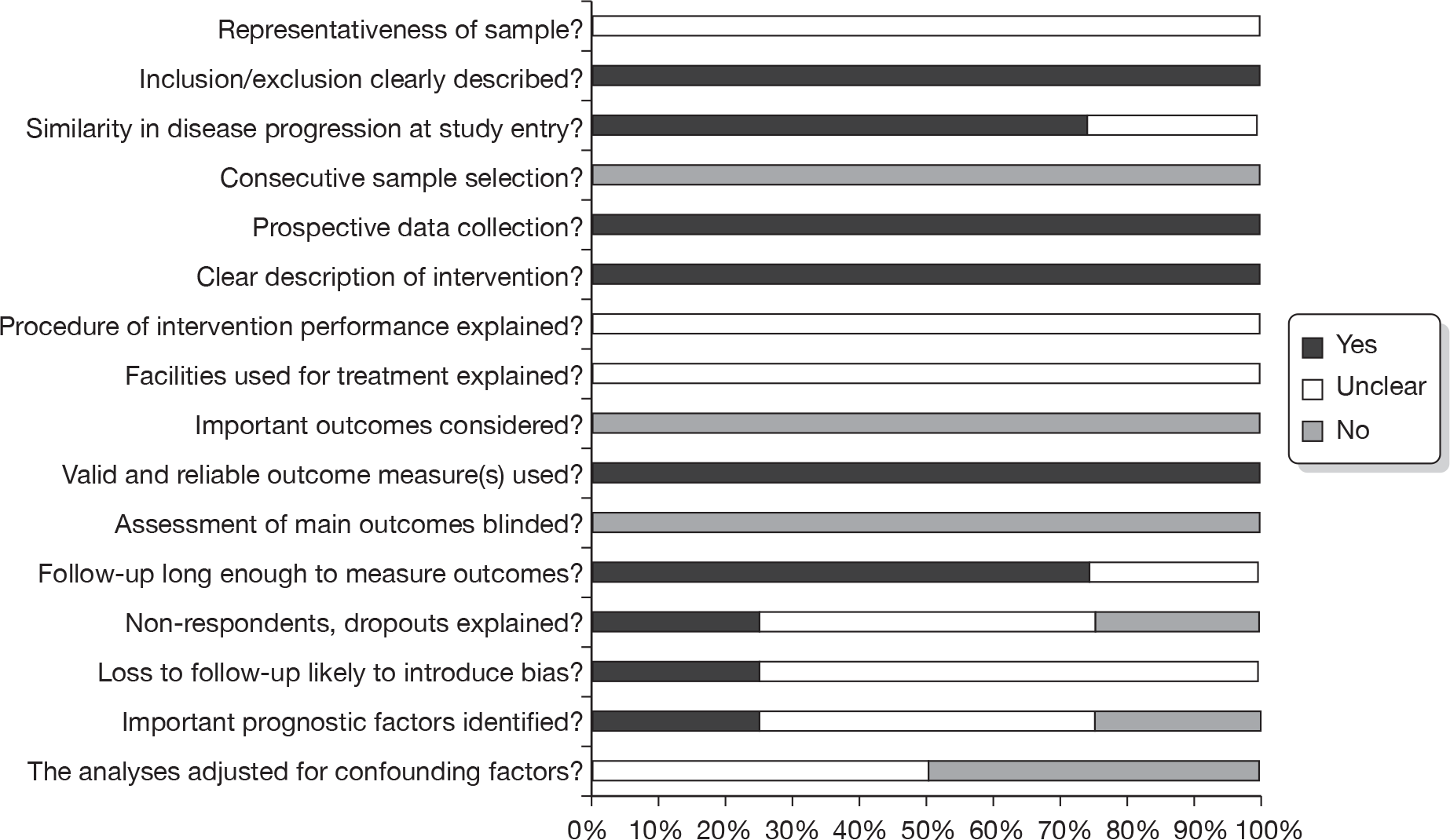
As the study populations of interest were not the original randomised populations, but the crossover subgroup in three studies,39,41,44 and a subgroup of consecutively treated patients in the remaining study,79 quality was assessed using the checklist for non-randomised studies (detailed in Methods, above). Questions within this checklist that were specific to non-randomised comparative groups (i.e. Q6 and Q16) were not considered applicable to the crossover subset population included in our review, and were therefore not summarised (see Appendix 4).
However, two specific domains were assessed using the Cochrane Collaboration’s tool for assessing risk of bias, namely sequence generation and allocation concealment, as these would check for selection bias at trial level.
Sample definition and selection
In three studies39,41,44 the included subgroups of participants were randomised at trial level, but crossover patients were not randomly selected, and so it is unclear the extent to which this group can be considered representative of the relevant patient population (Q1). The other study provided inadequate information to allow judgement of the representativeness of the sample. 79 With regard to the randomisation process at trial level, the studies by Blanke et al. 41 and Zalcberg et al. 44 used methods that adequately generated the allocation sequence to avoid influence of confounding factors while Blanke et al. 39 did not report sufficient data on the randomisation process. In the study by Zalcberg et al. ,44 allocation to treatment was not concealed. Both the B222239 and S003341 studies by Blanke et al. reported inadequate information on allocation concealment. All four studies adequately described inclusion and exclusion criteria (Q2). To consider whether participants entered the study at a similar point in their disease progression, we looked at data on their performance status. Three of the studies39,41,79 involved participants whose performance status at the time of study entry was similar, while the study by Zalcberg et al. 44 included participants with different performance status at study entry (Q3), although most of the participants in all populations had a performance status of < 2, meaning they were ambulatory and awake for at least 50% of their waking hours. None of the studies undertook consecutive selection of patients (Q4). Data were collected prospectively in all of the four studies (Q5).
Description of the intervention
The intervention was adequately defined by all studies (Q7). However, no study provided sufficient data describing supervision of the intervention (Q8) and no information was provided describing the types of staff involved, or the facilities used (Q9).
Outcome assessment
The quality of all four studies was similar in terms of outcome assessment (Q10). None of the studies had considered all of the outcomes of interest, but all reported the objective response of escalated imatinib dosing in patients with GIST, while one41 reported OS and two41,44 measured PFS. The study by Park et al. 79 reported time to progression, and the study by Zalcberg et al. 44 was the only study that also reported adverse events for those on an escalated dose of imatinib. No study reported outcomes related to QoL.
All four studies used valid and reliable outcome measures (Q11), such as RECIST to assess objective response or Kaplan–Meier methods to estimate survival curves, minimising detection bias. Assessment of main outcomes was not blinded in any of the studies (Q12).
Follow-up and attrition bias
Follow-up was considered long enough to detect important effects on outcomes of interest in all but one study where follow-up information was not provided and so this was unclear79 (Q13). Information on those lost to follow-up was either not provided39 (and thereby likely to introduce bias) or not provided at a sufficient level of detail41,44,79 to judge whether those lost to follow-up would be likely to introduce bias (Q14 and Q15).
Performance of the analysis
For both studies by Blanke et al. ,39,41 important prognostic factors such as sex, performance status, neutrophils counts, etc., were investigated and multivariate analyses were performed at trial level but this was not carried out for the subset of patients who crossed over. Similarly, Park et al. 79 identified possible prognostic factors (but did not adjust for confounding factors during analysis). The study by Zalcberg et al. 44 also did not identify any prognostic factors or their effect on analyses, or adjust for confounding factors (Q17 and Q18). Hence we considered the quality of reporting ambiguous in terms of the performance of the analyses.
Assessment of effectiveness
Response
For imatinib at an escalated dose of 600 mg/day following progression at a dose of 400 mg/day, response is reported in the B2222 study by Blanke et al. 39 and the study by Park et al. 79 In the study by Blanke et al. ,39 the median follow-up at this time was 63 months (maximum 71 months), and, at that time, 43 patients had crossed over from 400 to 600 mg/day. Of these 43 patients, 11 (25.6%) showed either PR or stable disease (SD). However, it should be noted that one patient showed response only after further escalation from 600 to 800 mg/day. Some of the 43 patients who crossed over would have had an initial response to 400 mg/day before progression, as only 11 patients in the 400 mg/day arm showed a best response of PD. 39 Interim data for this study population are provided in the study by Demetri et al. ,38 where, after a median follow-up of 288 days (maximum 9 months), nine patients had crossed over, with one showing PR at that point, and two with SD. 38
In the study by Park et al. ,79 median follow-up was eight months (range 1.4–22.3 months) and, of the 12 patients who received an escalated dose of 600 mg/day of imatinib, five (41.7%) showed either PR or SD.
With regard to response data provided by the manufacturer, (CiC information has been removed). As a result, these data from the manufacturer’s submission were not used in our review.
For imatinib at a dose of 800 mg/day following progression at a dose of 400 mg/day, response data are available from the S0033 study by Blanke et al. ,41 the EORTC-ITG-AGITG trial by Zalcberg et al. 44 and the study by Park et al. 79 Of the crossover populations in the S003341 and EORTC trials44 (117 and 133 patients, respectively), three patients in each trial (i.e. six in total) had a PR, while 33 patients in the S0033 trial41 and 36 patients in the EORTC-ISG-AGITG trial44 had SD as a best response. This means that out of a total of 250 patients, 75 (30%) had a response after escalation from 400 mg to 800 mg/day.
Response information from the study by Park et al. 79 did not provide separate data for those with SD and those achieving PR. However, it did state that four out of the 12 patients (33.3%) receiving an escalated imatinib dose of 800 mg/day upon progression at the 400 mg/day dose achieved either PR or SD. 79
Some of the patients receiving dose-escalated imatinib to 800 mg/day would have had an initial response to the 400 mg/day dose, because only 42/345 patients (12.2%) in the S0033 trial 400-mg arm had a best/only response of PD (or ‘early death’),41 and in the study by Zalcberg et al. 44 this figure was 61/473 (12.9%). 42
Interim data for the EORTC-ISG-AGITG trial were provided for a data cut-off point of 7 December 2003, at which point there were 2/97 (2.1%) patients showing a PR, 30/97 (30.9%) patients with SD, and 65/97 (67.0%) patients with PD. 78 Interim data for the S0033 trial, also from December 2003, showed that there were 5/68 (7.4%) patients with PR, and 20/68 (29.4%) patients with SD, during crossover treatment with 800 mg/day of imatinib, following failure of treatment at 400 mg/day. 68
In addition, secondary analysis for the EORTC-ISG-AGITG trial in the study by Debiec-Rychter et al. 14 indicated, without stating the number of patients involved, that response following crossover was significantly more likely to occur in patients with wild-type GIST than with KIT exon 11 mutation (p = 0.0012), and response following crossover was also significantly more likely to occur in patients with KIT exon 9 mutation compared with exon 11 mutation (p = 0.0017). 14
No response data were provided for treatment with sunitinib at a dose of 50 mg/day (as part of a 4 weeks-on-treatment/2 weeks-off-treatment 6-week cycle), following progression on an imatinib dose of 400 mg/day.
Overall survival
For imatinib at an escalated dose of 600 mg/day following progression at a dose of 400 mg/day, OS data were not reported by Blanke et al. 39 (CiC information has been removed) for the B2222 trial.
For imatinib at a dose of 800 mg/day following progression at a dose of 400 mg/day, the EORTC-ISG-AGITG trial by Zalcberg et al. 44 did not report OS outcomes. However, the S0033 trial by Blanke et al. 41 reported relevant outcome data, and at the time of the analysis (median follow-up of 4.5 years) noted that 76/118 (64.4%) of patients had died. 41 Median OS was 19 months (95% CI 13 to 23 months) starting from the commencement of crossover. Interim data for the S0033 trial were also provided in the study by Rankin et al. ,68 which stated that median OS at December 2003 was 19 months. 68
(CiC information has been removed.)
(CiC information has been removed.)
(CiC information has been removed.)
(CiC information has been removed.)
For sunitinib, OS data were available for those on 50 mg/day of sunitinib who failed on a prior imatinib dose of ≤ 400 mg/day from two abstracts of the same trial, taken at different follow-up periods. 82,86 The data from the study by Reichardt et al. 82 were analysed after a median of four cycles. Median survival at this point was 93 weeks (95% CI 72 to 100 weeks) and 231/339 (68.1%) of patients were still alive. 82 The data from the report by Seddon et al. 86 were analysed after a median of 51 weeks (range 0.1–159 weeks). Median survival at that time was 90 weeks (95% CI 73 to 106 weeks) and 193/351 (55%) were still alive. 86 It should also be noted that further interim OS data were provided in another study by Seddon et al. ,85 but although the date of analysis is the same month as that reported by the studies by Reichardt et al. 82 and Rutkowski et al. ,83 the median OS reported differed, at 80.4 weeks (95% CI 60.3 to NA weeks), while the population who had failed on doses of imatinib of ≤ 400 mg/day was also less (307 patients). 85
It was possible to compare OS with an escalated dose of 800 mg/day, from the S0033 trial reported by Blanke et al. ,41 with that with sunitinib at a dose of 50 mg/day (provided in 4 weeks-on/2 weeks-off cycles of 6 weeks), for patients who had progressed on imatinib at a dose of 400 mg/day. Quarterly OS estimates for the sunitinib participants reported in a Kaplan–Meier chart by Seddon et al. 86 were obtained using the method proposed by Parmar et al. 73 and compared with OS estimates for the S0033 trial provided by the authors. The results are provided in Figure 3.
FIGURE 3.
Comparison of OS estimates for imatinib at 800 mg/day and sunitinib at 50 mg/day.
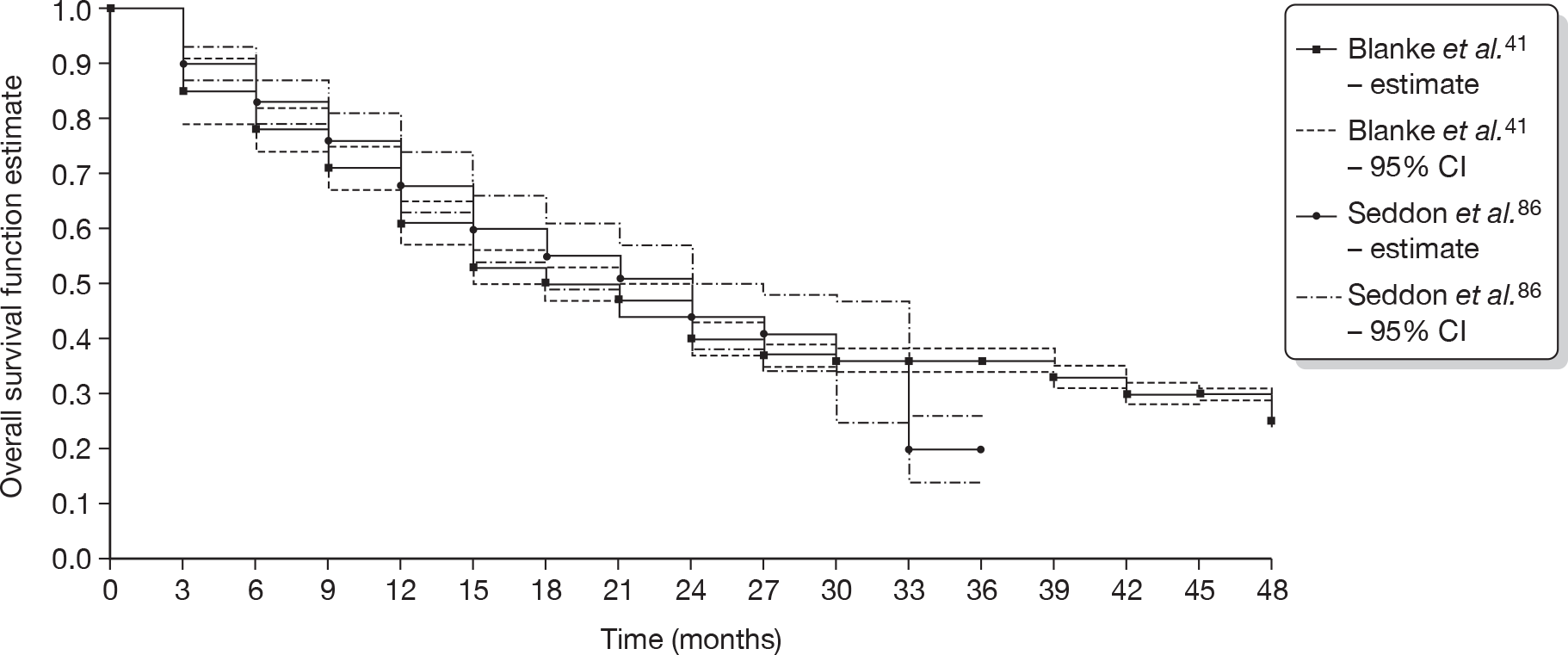
The study by Zalcberg et al. did not report information on OS and was therefore not included in the comparison in Figure 3. However, data are available from the (CiC information has been removed), and data from the study by Seddon et al. 86 on treatment with sunitinib are provided in Table 6.
| No. years elapsed | Seddon 200886 (n = 351) | (CiC information has been removed) | |||
|---|---|---|---|---|---|
| Survival estimate | 95% CI | (CiC information has been removed) | (CiC information has been removed) | ||
| 1 | 0.684 | 0.626 to 0.741 | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) |
| 2 | 0.441 | 0.379 to 0.503 | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) |
| 3 | 0.200 | 0.140 to 0.261 | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) |
| 4 | NR | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | |
Disease-free survival
No data were reported for this outcome on account of no patient in any of the included studies having a complete response.
Progression-free survival
For imatinib at an escalated dose of 600 mg/day following progression at a dose of 400 mg/day, PFS data were not reported by Blanke et al. 39 (CiC information has been removed) for the B2222 trial.
For imatinib at an escalated dose of 800 mg/day following progression at a dose of 400 mg/day, data were reported for the S0033 trial by Blanke et al. ,41 and for the EORTC-ISG-AGITG trial by Zalcberg et al. 44
For the S0033 trial, at the time of the analysis, median follow-up of 4.5 years (54 months), 99/118 (83.9%) of the crossover cohort for whom data were available had progressed. 41 Median PFS was estimated to be 5 months (95% CI 2 to 10 months). Of the 99 patients who had PD or had died at the time of the analysis, 23/99 (23.2%) had progressed but were still alive. Interim data from this trial, at a data cut-off point of December 2003, gave median PFS to be 4 months following crossover, for 68 patients. 68
For the EORTC-ISG-AGITG trial, median follow-up was 25 months (maximum follow-up was 35 months), and, at that time, 108/133 (81.2%) of the crossover cohort with data available had progressed. Median PFS was 81 days. Sixty-seven patients (50.4%) had progressed or died within 3 months (Kaplan–Meier survival estimate 0.467). At 1 year, the Kaplan–Meier survival estimate was 0.181. 44
(CiC information has been removed.)
The estimates of PFS provided at 3-month intervals by the authors of the S0033 study,41 and available as a Kaplan–Meier chart in the published paper of this study by Blanke et al. ,41 were compared with PFS estimates at 3-month intervals that were measured from an enlarged copy of the plot of the Kaplan–Meier survival function estimate given in the paper by Zalcberg et al. 44 The number of events in each time period was then calculated using the method proposed by Parmar et al. ,73 corrected to ensure that the total number of patients censored was consistent with the number reported in the published paper. 44 For both trials the standard error of the survival function estimates was estimated from the quarterly numbers for events and patients at risk using Greenwood’s formula. Figure 4 shows the survival functions from each trial, together with 95% CIs for each.
FIGURE 4.
Kaplan–Meier plot for PFS with 800 mg/day imatinib.
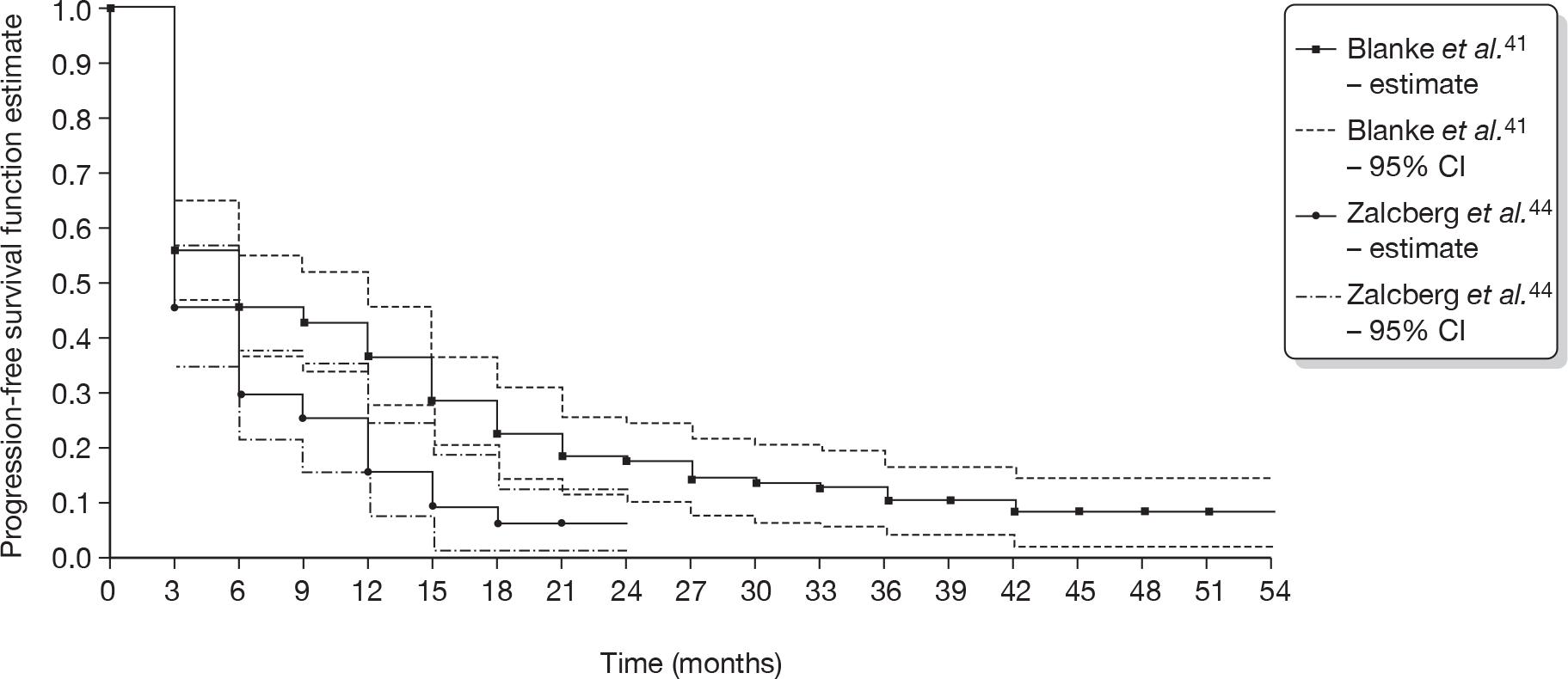
A meta-analysis of these two survival curves was attempted, using the methods described in Arends et al. 92 However, no valid results could be achieved owing to the lack of data.
For sunitinib at a dose of 50 mg/day for a 6-week cycle, no progression data were available specifically for trial participants who had failed on a prior dose of imatinib at ≤ 400 mg/day.
Time to treatment failure
Data on the duration of response/time to treatment failure were available from the study by Park et al. ,79 which showed that, of the 12 patients who had their imatinib dose escalated to 600 mg/day following progression at the 400 mg/day dose, one patient died of a cause unrelated to both their disease and imatinib treatment, while the remaining 11 patients eventually progressed on imatinib treatment at the escalated dose after a median of 1.7 months (range 0.7–24.9 months).
For those receiving an escalated dose of 800 mg/day of imatinib following progression at an initial dose of 400 mg/day, data were available from the EORTC-ISG-AGITG trial showing that, of those who achieved PR or SD after crossover, the median duration of ‘stabilisation’ (i.e. PR or SD after crossover) was 153 days (range 37–574 days). 44 Interim data from this trial (7 December 2003 data cut-off) gave a median time to progression of 78 days. 78
For the sunitinib trial, the specific median treatment duration for those given sunitinib after failure on imatinib at a dose of ≤ 400 mg/day was not provided, but interim median treatment duration for the whole cohort was reported at 126 days (range 1–618), and at that time point (median follow-up not stated) it was noted that median treatment duration ‘did not significantly differ based on the dose of prior imatinib therapy (≤ 400 vs > 400 mg/day). 80
Health-related quality of life
No data were reported for this outcome by any of the included studies.
Adverse events
Data on adverse events were not reported for participants receiving an escalated dose of 600 mg/day of imatinib following progression at an initial dose of 400 mg/day.
For those receiving an escalated dose of 800 mg/day of imatinib following progression at an initial dose of 400 mg/day, data were available from the EORTC-ISG-AGITG trial reported by Zalcberg et al. ,44 and there was some information on dose reductions in the S0033 trial report by Dileo et al. 77
The number of discontinuations due to adverse events was not explicitly stated for the EORTC-ISG-AGITG trial44 reported in the study by Zalcberg et al. , but they did report that the vast majority of discontinuations (88.4%, i.e. approximately 86/97 withdrawals) were due to disease progression, suggesting that the maximum possible adverse event withdrawals possible would be 11.6% of all 97 withdrawals, i.e. 11 patients. Interim data for this trial at a December 2003 data cut-off point showed that there were two toxicity withdrawals at that time. 78
Data from this trial on specific adverse events following crossover are shown in Table 7 for those patients with 60 days’ follow-up data.
| Adverse event | No. with adverse event | Less severe after crossover (n, %) | More severe after crossover (n, %) | No. achieving new grade 3- to grade 4-level adverse event |
|---|---|---|---|---|
| Oedema | 99 | 25/99 (25.3) | 33/99 (33.3) | 7 |
| Skin rash | 45 | 23/45 (51.1) | 19/45 (42.2) | 2 |
| Fatigue | 102 | 21/102 (20.6) | 47/102 (46.1) | 10 (p < 0.001) |
| Dyspnoea | 30 | 8/30 (26.7) | 14/30 (46.7) | 1 |
| Infection | 20 | 9/20 (45.0) | 9/20 (45.0) | 1 |
| Nausea | 82 | 38/82 (46.3) | 26/82 (31.7) | 3 |
| Leucopenia | 56 | 25/56 (44.6) | 16/56(28.6) | 0 |
| Neutropenia | 49 | 30/49 (61.2) | 13/49 (26.5) | 0 (p = 0.002) |
| Thrombocytopenia | 7 | 4/7 (57.1) | 2/7 (28.6) | 0 |
| Anaemia | 119 | 15/119 (12.6) | 51/119 (42.9) | 17 (p = 0.015) |
A higher proportion of those with skin rash, nausea, leucopenia, neutropenia and thrombocytopenia had reduced severity from these effects following crossover to the 800 mg/day dose of imatinib, compared with the proportion who had increased severity from these effects following crossover (though, with the exception of neutropenia, these differences were not significant at the 0.05 level). The same proportion of people with infection had increased and decreased severity from this following crossover. For all other adverse events, a higher proportion of sufferers had increased severity from these effects than improvement, and in the case of anaemia and fatigue the increase in severity following crossover was significant at the 0.05 level. 44
Interim data reported by Zalcberg et al. 78 for this trial showed that 31% of patients (exact number not calculable) required a dose reduction (note: stated as ‘cumulative incidence’). No information was provided on the dose given following dose reduction.
Interim data for the S0033 trial reported by Dileo et al. 77 showed that, of the 77 patients who had crossed over from an imatinib dose of 400 to 800 mg/day at that time, 18 (23.3%) had at least one dose delay, and 12 (15.6%) had at least one dose reduction, due to oedema and rash. No information was provided on the dose given following dose reduction.
(CiC information has been removed.)
(CiC information has been removed.)
For sunitinib at a dose of 50 mg/day for a 6-week cycle, no progression data were available specifically for trial participants who had failed on a prior dose of imatinib at ≤ 400 mg/day.
A summary of the results for all outcomes with the exception of adverse events is provided in Table 10.
| Drug/dose | Median follow-up (range): months | n (%) with PR or SD | Duration of response/time to treatment failure | Median OS (95% CI) | n (%) still alive | Median progression-free survival (95% CI) | n (%) progression free | Reference source |
|---|---|---|---|---|---|---|---|---|
| Sunitinib at 50 mg/day | 4.5 (0–22.1) | Median treatment duration did not differ based on prior imatinib dose | Kang 200780 | |||||
| < 6 ? | 20.1 months (15.1 to N/A months) | ?/307 | Seddon 200785 | |||||
| 6 | 23.3 months (18–25 months) | 231/339 (68.1) | Reichardt 200882 | |||||
| Imatinib at 600 mg/day | 8 | 5/12 (41.6) | 1.7 months (range 0.7–24.9 months) | Park 200979 | ||||
| Imatinib at 800 mg/day | 8 | 4/12 (33.3) | Park 200979 | |||||
| Imatinib at 600 mg/day | 9.5 (?–9) | 3/9 (33.3) | Demetri 200238 | |||||
| Sunitinib at 50 mg/day | 12 (0–39.8) | 22.5 months (18.3–26.5 months) | 193/351 (55) | Seddon 200886 | ||||
| Imatinib at 800 mg/day | < 25 (< ? to < 35) | 32/65 (49.2) | 2.8 months | Zalcberg 200478 | ||||
| 25 (?–35) | 39/133 (29.3) | 5.5 months (range 1.3–20.5 months) | 2.9 months | 25/133 (18.8) | Zalcberg 200544 | |||
| < 54 | 25/68 (36.8) | 19 months (not stated) | Rankin 200468 | |||||
| 54 | 36/117 (30.8) | 19 months (13–23 months) | 42/118 (35.6) | 5 months (2–10 months) | 19/118 (16.1) | Blanke S003341 | ||
| Imatinib at 600 mg/day | 63 (?–71) | 11/43 (25.6) | Blanke B222239 | |||||
| Imatinib at 800 mg/day | Significantly more likely to occur in patients with wild-type and exon 9 mutations than exon 11 mutations | Debiec-Rychter 200614 |
Chapter 5 Assessment of cost-effectiveness
The aim of this chapter is to assess the cost-effectiveness of alternative treatment strategies for people with KIT (CD117)-positive unresectable and/or metastatic GISTs, whose disease has progressed on treatment with imatinib at a dose of 400 mg/day.
The specific objectives are:
-
To determine, by undertaking a systematic review of the literature, the cost-effectiveness of using imatinib at an escalated dose of 600 or 800 mg/day to treat patients with unresectable and/or metastatic GISTs (whose disease has progressed with imatinib at a dose of 400 mg/day), compared with treatment with sunitinib (within its recommended dose range) or BSC.
-
To develop an economic model to compare the cost-effectiveness and cost–utility of imatinib at a dose of 600 or 800 mg/day; the use of sunitinib (within its recommended dose range); or BSC only, for people with KIT (CD117)-positive unresectable and/or metastatic GISTs whose disease has progressed on treatment with imatinib at a dose of 400 mg/day or those whose treatment with imatinib has failed owing to intolerance.
Systematic review of existing cost-effectiveness evidence
The purpose of the review of economic evaluation studies was to identify published studies and assess their quality and usefulness for comparisons of treatments of GISTs; inform the methodology of the proposed economic model; and identify data on the parameters of the proposed economic model (e.g. utilities for different health states, costs and epidemiological data).
Methods
Search strategy for identification of published reports
A comprehensive search was undertaken to identify studies that assessed the cost or cost-effectiveness of the alternative treatments used for GISTs. Databases searched included: MEDLINE, MEDLINE In-Process, EMBASE, SCI, Health Management Information Consortium, NHS Economic Evaluation Database (NHS EED), the HTA database, Cost-effectiveness Analysis (CEA) Registry and the Research Papers in Economics (RePEc). There were no language restrictions in the search strategy and all databases were searched from 2000 onwards.
The search strategy used is provided in Appendix 10. The abstracts of the International Society for Pharmacoeconomics and Outcomes Research (ISPOR) conferences from 2006 were also searched and, in addition, websites of key professional organisations, GIST Support International and the drug manufacturers Pfizer and Novartis were scrutinised.
The reference lists of all identified studies and evidence syntheses, as well as submissions from industry and other consultees, were also checked for additional potentially relevant references. The methods for how the industry submissions were to be handled are described below, although, as noted in Chapter 3, no industry submission was reviewed for this technology assessment review (TAR). The full texts of potentially relevant reports were obtained and assessed in terms of their relevance to the economic evaluation or cost analysis.
Quality assessment
Included studies were assessed using the guidelines of the Centre for Reviews and Dissemination. 69 Modelling studies were assessed against the Philips checklist. 93
Inclusion and exclusion criteria
To be included, studies had to include a cost analysis or a cost-effectiveness analysis (CEA) of alternative treatments for GISTs. Non-English language studies were excluded.
Data extraction
Information and relevant data were extracted by an economist according to the guidelines produced by the Centre for Reviews and Dissemination for the critical appraisal of economic evaluations. Where an economic evaluation was based on a modelling exercise, additional data extraction criteria developed by Philips et al. were applied. 93,94
Handling industry submissions
Information from the manufacturer was to be considered if it was submitted in accordance with the 3 December 2009 deadline set by NICE. Any economic evaluations included in the company submission, provided they complied with NICE’s guidance on presentation, would be assessed for clinical validity, reasonableness of assumptions and appropriateness of the data used in the economic model, using the methods outlined above. The strengths and weaknesses in terms of the methodology adopted, and reporting of results and conclusions, would be described. The conclusions derived from the company submissions were then to be compared with those provided by the review of the other existing evidence and the model reported in Economic modelling (below), highlighting any differences in results. Any ‘CiC’ data taken from a company submission were to be reported in accordance with NICE guidelines. 94
Synthesising evidence
Data from the included studies on economic analysis and economic evaluation were summarised in order to identify common results, and to summarise the variations and differences between studies. The studies that used economic modelling were critically reviewed with regard to, for example, model structure use, and how these models dealt with uncertainties while predicting results.
Results
Results of literature search
In total there were 250 papers identified from the initial search (Table 11). Of these, 18 were selected as potentially relevant abstracts, and 13 were included for further screening. From these papers, nine were selected for the review. Appendix 11 summarises the included studies.
| Database | No. retrieved |
|---|---|
| MEDLINE (2000 – October, week 4, 2009), EMBASE (2000 – week 44, 2009) | 227 |
| MEDLINE In-Process (3 November 2009) (after de-duplication in Ovid) | 0 |
| SCIa (2000 to 3 November 2009) | 16 |
| Health Management Information Consortiuma (September 2009) | 0 |
| NHS EEDa (October 2009) | 0 |
| HTA database (October 2009) | 0 |
| ISPOR conference abstracts 2006–9 | 7 |
| Total | 250 |
As already noted no submission was received from industry reporting relevant evidence.
Characteristics of included studies
Out of the nine studies, seven55,95–100 reported a full economic evaluation that assessed both the costs and cost-effectiveness of the alternatives compared. Of the remaining two studies, the study by Reddy54 is a review reporting information related to costs and health outcomes reported in other studies and did not undertake an economic evaluation. The other study,101 which is also a review of the management of GIST with sunitinib, reports on, amongst other things, the cost of treatment with sunitinib.
Five studies55,95,96,99,100 conducted a modelling exercise rather than incorporating data from actual patient follow-up. Two studies96,98 used non-randomised or non-trial patient data (from retrospective cohorts) to inform their economic evaluations.
One study55 reported an economic evaluation in a UK context, which was based on an industry submission to NICE for a previous TAR. Two studies95,98 reported a Canadian context, and one study was from a US context. 97 The remaining three studies were conducted in the context of Mexico,96 Spain99 and Brazil,100 respectively. Table 12 summarises the main features of the included studies.
| Study | Country, currency, price year | Perspectives | Comparisons | Patient failed on imatinib? | Outcomes reported | Modelling | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Imatinib 400 mg/day | Imatinib 600 mg/day | Imatinib 800 mg/day | BSC | Sunitinib | OS | Median OS | Survival rate | PFS | Progression-freelife-years | Time toprogression | PFM | Life-years gained | QALY | Cost-effectiveness ratio | ICER | |||||
| Chabot 200895 | Canada, Canadian $, 2005 | Provincial health authority | ✓ | ✓ | Yes | ✓ | ✓ | ✓ | ✓ | ✓ | Markov model | |||||||||
| Contreras-Hernandez 200896 | Mexico, US$, 2006a | Health insurance system | ✓ | ✓ | ✓ | Yes | ✓ | ✓ | ✓ | Markov model | ||||||||||
| Mabasa 200898 | Canada, Canadian $, 2006 | BCCA | ✓ | ✓ | No | ✓ | ✓ | ✓ | ✓ | ✓ | CEA using cost-effectiveness ratios and ICERs | |||||||||
| Paz-Ares 200899 | Spain, €, 2007 | Health-care system | ✓ | ✓ | Yes | ✓ | ✓ | ✓ | ✓ | ✓ | Markov model | |||||||||
| Huse 200797 | USA, US$, 2005 | Societal perspective (payers for health care) | ✓ | ✓ | NA | ✓ | ✓ | CEA | ||||||||||||
| Teich 2009100 | Brazil, Brazilian $ (R$), 2005b | Health-care system | ✓ | ✓ | ✓ | Yes | ✓ | ✓ | ✓ | Markov model | ||||||||||
| Wilson 200555 | UK, GB£, (2004?) | Health-care system | ✓ | ✓ | ✓ | Yes | ✓ | ✓ | ✓ | Markov model | ||||||||||
Comparative studies
Imatinib and best supportive care
Three studies55,97,98 compared imatinib with BSC. The study by Wilson et al. 55 used the manufacturer submissions (Novartis model) and compared imatinib and BSC, but in the imatinib group allowed for escalation of doses from 400 to 600 mg/day for those who failed to respond or were intolerant to imatinib at the 400 mg/day dose. The study by Mabasa et al. 98 noted that patients included from retrospective cohorts in their analysis were given imatinib 400 mg/day until disease progression, and later were allowed escalated doses of between 600 and 800 mg/day. Six out of 56 patients in the imatinib groups of patients considered in this economic evaluation were then allowed to switch to sunitinib therapy. The economic evaluation by Huse et al. 97 considered imatinib at 400 mg/day (see Table 12).
Imatinib, sunitinib and best supportive care
Two studies96,100 compared sunitinib, escalated doses of imatinib, and BSC or palliative care as comparators for their economic evaluations. The Contreras-Hernandez et al. 96 study compared treatment with imatinib, sunitinib and palliative care. Both treatments (sunitinib and imatinib) were compared with BSC in a model-based analysis. The doses for both the treatments were clearly specified (imatinib at 800 mg/day and sunitinib at 50 mg/day) as the study was based on primary data collected from hospital records. The study did not include dose escalation with imatinib at a 600 mg/day dose. Teich et al. 100 compared sunitinib, imatinib at 800 mg/day and BSC (see Table 12).
Sunitinib and best supportive care
The studies by Chabot et al. 95 and Paz-Ares et al. 99 compared treatment with sunitinib and BSC for patients with GIST who were imatinib resistant or intolerant. Chabot et al. 95 did not specify the dose of sunitinib used, or mention whether patients who were imatinib resistant or intolerant were initially treated with 400 mg/day and then with escalated imatinib doses (e.g. 600 or 800 mg/day). Paz-Ares et al. 99 specified a dose of 50 mg/day for the patients in the sunitinib group. The patients in the sunitinib group were provided with BSC. Therefore, this study compared sunitinib plus BSC with BSC alone. BSC in this study included diagnostic tests and routine palliative treatment. 99
The definition of BSC in the economic evaluation studies was not the same across the studies. Chabot et al. 95 did not clearly define what BSC included, while Contreras-Hernandez et al. 96 defined clearly that BSC included treatment with imatinib. Paz-Ares et al. 99 defined BSC as essentially consisting of diagnostic tests and routine palliative care. In the other three studies,55,97,98 the control group of patients, who are considered as effectively being treated with BSC, were not provided with treatment with imatinib. As a full-text paper of the study by Teich et al. 100 was not available, information on how this study defined BSC was not available.
All treatments
We did not find any studies that conducted an economic evaluation of all of the alternative treatments (e.g. escalated doses of imatinib 600 mg/day or imatinib 800 mg/day, sunitinib and BSC) for patients who failed on imatinib at a dose of 400 mg/day.
Study design
Among the seven studies that conducted a full economic evaluation, five used Markov modelling. 55,95,96,99,100 Huse et al. 97 used a very simple modelling framework and Mabasa et al. 98 also used patient-level data and had 46 and 47 patients in their imatinib and BSC (historical group) groups, respectively. Contreras-Hernandez et al. 96 also used patient-level data (for 21 patients) collected at the Hospital de Oncología to estimate the costs of care associated with imatinib, BSC and other procedures, and used these costs in their model.
Perspective
Three studies55,99,100 adopted the perspective of a national health-care system. The study by Contreras-Hernandez et al. 96 was from Mexico’s health insurance system’s perspective. The study by Huse et al. 97 did not specifically mention whether it was from a health insurance system perspective; however, it mentioned that it had been conducted from a US societal perspective. The studies by Chabot et al. 95 and Mabasa et al. 98 considered a provincial health authority and a specialised agency (British Columbia Cancer Agency) perspective, respectively, for their economic evaluations. None of the seven studies55,95–100 that conducted full economic evaluations reported indirect non-medical resource use, or indirect costs to society in terms of productivity loss, costs to carers, and other indirect costs associated with GIST.
Health outcome measures
The major outcome measures used in the seven studies reporting full economic evaluations were PFS,95,96,98–100 OS,95,98 life-years gained95,96,98–100 and quality-adjusted life-years (QALYs). 55,95,97,99 Four studies55,95,97,99 reported the incremental cost per QALY gained. The remaining three studies96,98,100 used incremental cost per life-year gained, and incremental cost per progression-free life-year gained.
Data sources
Most of the studies,95,96,99 which were based on modelling exercises, used effectiveness or health outcome data from major trials38,52,102–104 and adapted them for their specific contexts. The sources of cost data were mainly from relevant patients’ records, and health-care cost databases. Wilson et al. 55 used data from an industry submission (Novartis trial). Table 13 summarises the data sources used for the studies. A full paper of the study by Teich et al. 100 was not available and so information on the data sources used was unknown.
| Study | Unit costs | Resource use for treatment | Effective/health outcomes |
|---|---|---|---|
| Chabot 200895 | Published literature and Canadian government benefit schedule and medical oncologist | Published literature and Canadian government benefit schedule and medical oncologist | Phase III trial NCT0007521852 |
| Contreras-Hernandez 200896 | Hospital records (Hospital de Oncología) for 21 patients in Mexico, IMSS pricing and reimbursement procedure, and cost of sunitinib from Pfizer Laboratories | Patients’ medical charts, associated information from IMSS (Mexican insurance system) | Phase III trial52,104 |
| Mabasa 200898 | BCCA | BCCA registry | Patients’ data in two arms (imatinib groups and 46 non-imatinib group) were compared with Demetri 200238 and Verweij 2003102 |
| Paz-Ares 200899 | Health costs database eSalud (for administration, radiotherapy, nephrectomy and monitoring costs). General Council of Pharmacists Official Colleges for drug costs. Ojeda et al. (2003)105 for unit costs of adverse events | Data reported by expert panel on number of visits to oncology clinic, laboratory tests, CT scans, nurse visits and visits to palliative units, and analgesic drugs | Demetri 2006,52 adverse events105 |
| Huse 200797 | Drug acquisition costs: published average wholesale price (Red Book: Pharmacy’s Fundamental Reference. Montvale, NJ: Thomson Health Care; 2005, and Physicians’ Desk Reference 2005. Montvale, NJ: Thomson PDR; 2005) | Based on the resources used by patients with pancreatic cancer (not advanced in US context) to determine the resources used for medical management in the absence of data on resource use by GIST patients | Demetri 2002,38 Phase II and Blanke 2006103 |
| Wilson 200555 | Industry submission: Novartis model – Novartis submission to NICE 2003 | Novartis model – Novartis submission to NICE 2003 | QoL based on ECOG data from B2222 trial,39 and Goss study (data AiC) |
Time horizon
The studies that used models in their economic evaluations used different time horizons and treatment cycle lengths for the Markov model. The two studies95,99 that had sunitinib and BSC as comparator treatments used a time horizon of 6 years and a treatment cycle length of 6 weeks in the modelling exercise. Of the other studies, the study by Contreras-Hernandez et al. ,96 which had sunitinib as a comparator along with imatinib and BSC, used a lifetime time horizon and also a 6-week cycle of treatment (to be consistent with the sunitinib treatment cycle of 6 weeks). Huse et al. 97 used a 10-year time horizon for the analysis, while Teich et al. 100 used a 6-year time horizon, and a 6-week treatment cycle.
Discount rate
A 5% discount rate for costs and health outcomes was used in two studies. 95,96 Wilson et al. 55 in their model discounted costs by 6% and QALYs by 1.5%, as per NICE methods guidance at the time the work was conducted. Paz-Ares et al. 99 and Huse et al. 97 used 3% and 3.5%, respectively. Mabasa et al. 98 used 3% for discounting costs and outcomes. The abstract by Teich et al. 100 did not report the discount rate used in their modelling exercise.
Findings on costs and cost-effectiveness
The cost of treatment and cost per different health outcome under different alternatives are presented in Table 14. As regards cost in relation to the health outcomes, the incremental cost-effectiveness ratios (ICERs) from the studies are noted in the table with respect to the main outcomes, i.e. life-year saved (LYS), PFS and QALYs. Although the Contreras-Hernandez et al. study96 considered three alternative treatments (sunitinib, imatinib and BSC), it did not report an ICER for imatinib versus BSC.
| Study | Comparator | Mean cost of treatment per patient | ICER1 | ICER2 |
|---|---|---|---|---|
| Chabot 200895 | Sunitinib | C$46,125 | SUN vs BSC | SUN vs BSC |
| Costs in Canadian $ at 2005 prices | C$49,826 per life-year saved | C$79,884 per QALY | ||
| BSC | C$11,632 | |||
|
Contreras-Hernandez 200896 Costs in US$ at 2006 prices |
Sunitinib |
US$17,806 Standard deviation US$695 95% CI US$15,377 to 19,816 |
SUN vs BSC $15,734 per patient treated with sunitinib and $56,612 per year of PFS, and $46,108 per life-year gained |
|
| Imatinib |
US$35,057 SD US$1253 95% CI US$31,381 to 38,705 |
|||
| BSC |
US$2071 Standard deviation US$473 95% CI US$1543 to 2869 |
|||
| Mabasa 200898 | Imatinib | C$79,839 | Imatinib vs BSC (control) | |
| Costs in Canadian $ at 2006 prices | BSC | C$1743 | C$15,882 per life-year saved | |
| Paz-Ares 200899 | Sunitinib | €23,259 | SUN vs BSC | SUN vs BSC |
| Costs in € at 2007 prices | €30,242 per life-year saved |
€4090 per PFM €49,090 per QALY gained |
||
| BSC | €1622 | |||
| Huse 200797 | Imatinib | US$416,255 | ||
| Cost in US$ at 2005 price | BSC | US$341,886 | ||
|
Wilson 200555 Cost in £ at 2004 prices |
Imatinib |
£18,896 (400 mg/day) £24,368 (600 mg/day) Other cost of treatment £1136 |
Cost per QALY: £70,206 (year 2), £51,514 (year 3), £36,479 (year 5) and £25,859 (year 10) | |
| BSC | £562 |
Higher doses of imatinib versus BSC
The Contreras-Hernandez et al. 96 study suggested that a higher dose of imatinib (800 mg/day) might be cost-effective compared with BSC (where BSC included treatment with imatinib at a lower dose). Wilson et al. ,55 using the modified Novartis model in a UK context and from an NHS perspective, estimated the incremental cost per QALY gained at £51,515–98,889 at 2 years, and £27,331–44,236 at 5 years compared with BSC.
Sunitinib versus higher dose of imatinib and/or BSC
Sunitinib treatment was associated with an estimated gain of 0.7 years and 0.4 QALYs compared with BSC. 95 Sunitinib treatment also resulted in a higher number of progression-free months (PFMs) than both the imatinib and BSC therapies. The mean number of PFMs was found to be 5.64 for sunitinib, while it was 5.28 and 2.58, respectively, for imatinib and BSC. The incremental effectiveness of sunitinib therapy compared with BSC was 3.1 PFMs and compared with a high dose of imatinib was 0.3 PFMs. Over the 5-year treatment horizon, Contreras-Hernandez et al. 96 found that patients with sunitinib had a mean life-year gain (LYG) of 1.4 compared with 1.31 and 1.08 for imatinib and BSC, respectively. The study also suggested that patients taking imatinib at a dose of 800 mg/day had the highest mean costs of treatment. Teich et al. 100 reported that sunitinib was cost-effective compared with imatinib at a dose of 800 mg/day for a 6-year time horizon. Their study suggested that sunitinib increased life-years and progression-free life-years by 0.3 and 0.26, respectively, with an incremental cost of R$86,756 (Brazilian dollars) [US$61,968 purchasing power parity (PPP) 2005] in comparison with BSC. They found that sunitinib was both more effective showing a gain in life-years of 0.02 and progression-free life-years of 0.47, and less costly than imatinib over 6 years.
Assessment of uncertainty
All six full-text studies55,95–99 used some form of sensitivity analysis. Chabot et al. 95 varied the most influential model parameters, i.e. utility of progression and no progression, OS (HR), PFS, positron emission tomography (PET) at initiation of sunitinib treatment, the cost of palliative care and the cost of PET. The model assumed the acquisition cost of sunitinib was certain and did not vary this in the sensitivity analysis. The sensitivity analysis suggested that the results of the economic evaluation were most sensitive to the health-state utility value and rate of OS and PFS. The sensitivity analysis also suggested that the results were robust. Contreras-Hernandez et al. 96 conducted probabilistic sensitivity analysis with data obtained from the Markov model. An acceptability curve was derived and reported the cost-effectiveness ratios for sunitinib in comparison with palliative care. In the absence of any threshold for cancer therapy in Mexico, they used three hypothetical re-imbursement cut-points equivalent to US$27,723, US$36,364 and US$45,455 to derive acceptability curves. These hypothetical values were based on taking 5%, 14% and 40% of the upper threshold that NICE reimburses for imatinib as first-line treatment. Mabasa et al. 98 varied the median OS rate, the rate of PFS and years of life expectancy, and conducted univariate sensitivity analysis. They found that the model used for the analysis remained robust. The ICER for each median life-year gained was found to be within the range of C$0–550 (Canadian dollars), and for each median progression-free year it ranged from C$0 to C$75,505. Paz-Ares et al. 99 also conducted univariate sensitivity analysis. Their model results were calculated in a probabilistic analysis considering the impact of uncertainty on the values of each variable included in the model, by assuming different distributions of these variables. The study conducted sensitivity analysis of the results by adding the cost of imatinib to the BSC group, by assuming all patients in the palliative care group would be given imatinib 400 mg/day. The most sensitive variables affecting the results were efficacy of treatment, and the unit cost of sunitinib. The study by Huse et al. 97 also used univariate sensitivity analysis and examined the impact of considering the upper and lower values of the cost of the drugs, the cost of treatment, the utilities of successful treatment and PD, the time horizon and the annual rate of discount in their analysis. They used imatinib at a 600 mg/day dose to examine the impact of results variation as an alternative scenario for the sensitivity analysis. The study by Wilson et al. 55 fitted a Weibull curve to estimate progression and death due to GIST in their sensitivity analysis and found that the ICER, based on a Weibull curve, was £26,427, and with an exponential fitting was £21,707.
Summary of the review
We found that most of the economic evaluation studies reviewed used a modelling exercise. However, only two studies96,100 compared both imatinib and sunitinib with BSC for patients who had failed or become resistant to imatinib 400 mg/day. The full paper for only one of these96 was available. Among the five studies55,95,96,99,100 that used modelling exercises, Contreras-Hernandez et al. 96 and Teich et al. 100 did not use QALYs as health outcome measures. Although Contreras-Hernandez et al. 96 used patient-level data as the basis of their cost estimates, they used survival and PFS as effectiveness measures in their model, which was based on the studies by Motzer et al. 104 and Demetri et al. 52
The two studies95,99 that used modelling exercises to compare the cost-effectiveness of sunitinib only with BSC used the same trial data (A6181004). 52 Their utility data were based on responses to the EQ-5D instrument provided by participants in this trial.
In our review we did not identify any published economic evaluation studies in a UK context comparing all the relevant interventions. The study that included an economic evaluation of higher dose imatinib in a UK context55 did not actually have as a comparator those who failed with imatinib 400 mg/day; rather the model allowed patients who failed on 400 mg/day to cross over to a higher dose of imatinib 600 mg/day rather than 800 mg/day.
The definition of BSC in the economic evaluation studies reviewed was not the same across the studies and cost-effectiveness of treatments compared with that of BSC cannot be easily compared. In addition, the pattern of resources used including the drugs for treatment was reported in different ways in different studies.
For a comprehensive economic evaluation of the alternative treatments for GIST patients who fail on or become resistant to imatinib 400 mg/day, further evidence is needed to fill in gaps in the evidence base. The challenge is to obtain appropriate and sufficient information on survival rates and responses to treatments with escalated doses of imatinib, and sunitinib. The economic evaluations which were identified based on modelling exercises have limitations. For example, all extrapolated clinical trial data from a short time horizon were used to predict cost-effectiveness results for a longer period. There is a need for empirical patient-level data for future economic evaluations. The outcome measures for disease severity can be considered as important surrogate end points. In cases where the patients in placebo groups or in BSC arms of trials are allowed to cross over to an experimental group (either escalated doses of imatinib or sunitinib) it could be argued that an intention to treat analysis would result in an underestimation of the survival benefit of patients randomised in the treatment groups, and the cost of the treatment for these patients who were assigned to placebo/BSC groups is often not accounted for in economic evaluations.
There has been no consideration of the patients’ and society’s costs/resource use in the studies reviewed. A wider perspective might be informative. However, NICE’s guidance94 suggests that costs and resources falling to the NHS and Personal Social Services (PSS) should be used. This approach by NICE is not universally accepted and costs and benefits falling on other groups may be relevant, for example in helping to illustrate additional choices and trade-offs that a decision-maker may wish to consider.
Economic modelling
Model structure
The structure of the model was informed by the modelling studies identified as part of the systematic review of economic evaluations, the review of clinical effectiveness, and other existing evidence including previous NICE TARs. We have also drawn upon advice from health-care professionals within the research team in this regard.
The model was developed to compare the alternative treatment strategies for people with KIT (CD117)-positive unresectable and/or metastatic GISTs whose disease has progressed on treatment with imatinib at a dose of 400 mg/day or those whose treatment with imatinib has failed owing to intolerance. According to the scope for the review the treatment strategies to be compared in the models were:
-
treatment with an escalated dose of 600 mg/day, regulating symptoms with BSC
-
treatment with an escalated dose of 800 mg/day, regulating symptoms with BSC
-
treatment with sunitinib (within its recommended dose range), regulating symptoms with BSC
-
regulating symptoms with BSC only.
The assumed pathway of the model
We considered a range of different alternative pathways for patients who progressed on imatinib at a dose of 400 mg/day, which led to the creation of nine alternative pathways, and, following advice from our clinical advisers, we determined seven clinically plausible pathways (Figure 5). The model is based on these seven clinically plausible care pathways. Circles represent health states that individuals may return to, rectangles represent health states during which treatment is administered, and the arrows show the possible directions in which individuals could move at the end of each cycle, depending on the transition probabilities. The states considered in the model were those thought to reflect care pathways for people with GIST. Patients entering the pathways are those who failed on imatinib 400 mg/day. The alternative treatments considered were dose T1 = imatinib 600 mg/day, T2 = imatinib 800 mg/day, T3 = sunitinib (with recommended dose 50 mg/day) and BSC.
FIGURE 5.
Markov model for GIST patients who have failed with imatinib 400 mg/day.
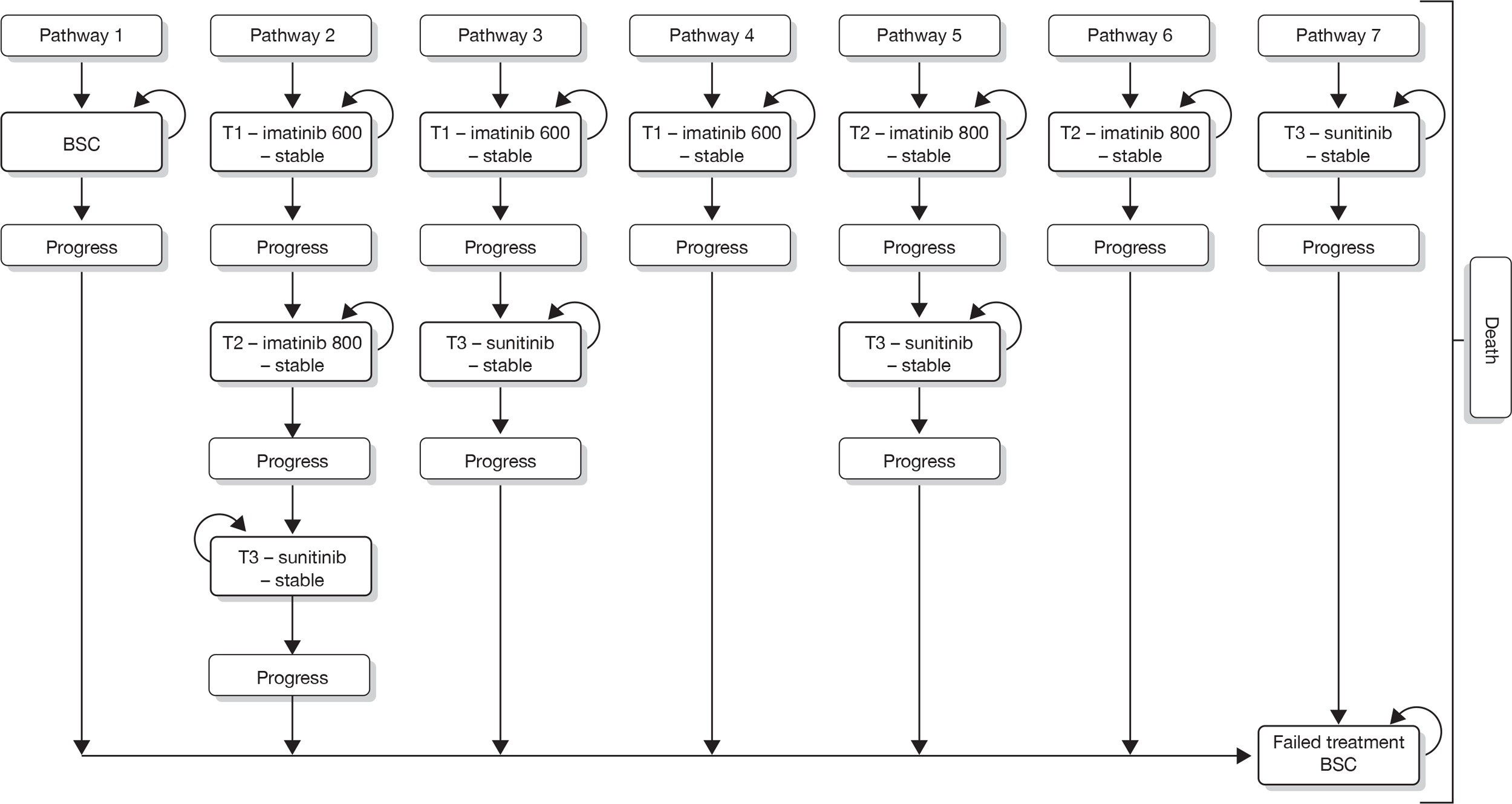
A Markov model was developed to model these care pathways using Treeage Pro 2009 (TreeAge Software Inc., Williamstown, MA, USA). In this model, patients whose disease has progressed on treatment with imatinib at a dose of 400 mg/day or those whose treatment with imatinib has failed owing to intolerance enter one of the seven care pathways. Figure 6 is an illustrative example of the model structure for Path-4, where patients are treated with imatinib 600 mg/day, and if the disease progresses on this treatment the patients are treated with BSC. Appendix 12 illustrates the model for all seven pathways of alternative treatments.
FIGURE 6.
Example of model structure for care pathway Path-4 (imatinib 600 mg/day – BSC).
Path-1 shows the patients with BSC treatment. It is assumed that the patients with BSC are still treated with imatinib and palliative care. Path-2 represents treatment options where escalated doses of imatinib (600 and 800 mg/day) and treatment with sunitinib are provided to the cohort of patients. All patients start the treatment with imatinib 600 mg/day. If they survive and respond to imatinib 600 mg/day then they will continue with the dose until they move to a state of stable condition with complete response or PR (CR/Stable IM 600). From this point, a proportion of patients will survive and continue to respond to treatment. Those who stop responding to imatinib 600 mg/day move to a state where they receive imatinib 800 mg/day (PD at IM 600). A proportion of patients will remain with the escalated dose of imatinib 800 mg daily (CR/Stable IM 800). If patients fail to respond on imatinib 800 mg daily, they are switched to treatment with sunitinib (PD with sunitinib). If they respond to sunitinib then they will continue with the treatment and move to a state of stable condition with complete response or PR (CR/Stable with sunitinib). From this point, a proportion of patients may continue to respond to the treatment and remain stable, or they may stop responding to sunitinib and receive BSC for the remainder of their life.
Path-3 represents treatment options through which an escalated dose of imatinib (imatinib 600 mg/day only) and treatment with sunitinib are provided. In this pathway, all patients also start the treatment with imatinib 600 mg/day (PD initial treatment IM 600). If they respond to imatinib 600 mg/day then they will continue with the dose and move to a state of stable condition with complete response or PR (CR/Stable IM 600). If a patient treated with imatinib 600 mg/day fails to respond, or ceases to respond, then instead of trying further dose escalation with imatinib they are switched to treatment with sunitinib (PD with sunitinib). If they respond to sunitinib they will continue with the treatment and move to a state of stable condition with complete response or PR (CR/Stable with sunitinib). Should they fail to respond to sunitinib or if at some point they cease to respond they continue with BSC for the remainder of their life.
In Path-4, all patients start the treatment with imatinib 600 mg/day and no switching to other treatments is considered. If they respond to imatinib 600 mg/day then they continue with this treatment until the GIST progresses or they die (CR/Stable IM 600). If at any point they do not respond to imatinib 600 mg/day they continue with BSC for the remainder of their life. This option has been considered as a treatment option in the model, although actual clinical practice may favour further escalation to 800 mg/day. Nevertheless, for this model, care pathways were developed with clinical advice on plausible pathways of care, some of which may be more typical of current practice than others.
The remaining care pathways are variants of earlier pathways. Path-5 is similar to Path-3 with respect to the combination of escalated dose of imatinib and sunitinib, the main difference in this case is that the escalated dose is imatinib 800 mg/day. Apart from this difference the pathways are identical. Path-6 is similar to Path-4. However, in this pathway the escalated dose is imatinib 800 mg/day instead of imatinib 600 mg/day. Path-7 is similar to Path-4. In this pathway, however, instead of being treated with imatinib 600 mg/day, patients receive sunitinib. Apart from this change, the care pathways are identical (see Appendix 12).
The care pathways chosen for our model are not exhaustive and do not include every single possible clinical intervention available to oncologists treating GIST after failure at 400 mg/day of imatinib, but the care pathways chosen reflect the scope of this research as agreed with NICE. Other possible treatments (e.g. surgery for those whose tumours become resectable following treatment with escalated doses of imatinib) were not considered.
Key assumptions of the modelling exercise
The key assumptions of the model are:
-
The time horizon of the model is 10 years, over which time all patients are expected to die, and the cycle length is 1 month.
-
The model assumes that patients entering a pathway will remain in a health state and on the treatment for one cycle only. If they respond and remain stable they continue on the treatment in the next cycle. If they do not respond but survive in the treatment arm they are considered to move to an escalated dose, move to another alternative (if allowed by a treatment pathway) or continue with BSC for the remaining time horizon of the model.
-
The model assumes that the probabilities of progressing and dying do not change over time. This assumption was made because of the limited data available.
-
The utilities of the health outcome from treatment with imatinib 600 mg/day, imatinib 800 mg/day and sunitinib are assumed to be the same. This assumption was made because of the limited data available.
-
All patients failing or not responding to the treatment in any of the treatment arms of the model continue with BSC for the remainder of the model time horizon or until they die, and are assumed to derive the same utility as from the health state of progression. Owing to lack of data, time-dependent changes in transition rates of response were not built into the model.
Data requirements and model inputs
For our model, data on the clinical effectiveness of interventions were based upon the systematic review of clinical effectiveness described earlier. These data were combined within the model with health-state utilities data to provide estimates of QALYs for the alternative treatment strategies for patients with GIST.
With respect to clinical effectiveness, data were required for the model on the probability of death per cycle and the probability of not responding to treatment per cycle.
Probability of death
As described in the systematic review of effectiveness few data were available for any of the treatments, few of which were based on direct comparisons. Therefore, the data available are imprecise and potentially biased. The direction and magnitude of any bias is unknown. As a consequence the data used to derive probabilities of death for each therapy under consideration should be treated cautiously.
Probability of death for BSC
The data for BSC were taken from two studies106,107 and a pooled weighted estimate suggested that 87.9% (51/58) died during the observation period of 60 months. A monthly rate was derived using an exponential function which assumes the probability of death per month is constant over time. The same value was used in circumstances where patients moved on to BSC after previously being treated with imatinib at an escalated dose or with sunitinib. The data from the review of clinical effectiveness were not appropriate to populate the BSC states in the model, as no BSC studies met the inclusion criteria. The second best source of data would have been information on follow-up of non-crossover patients following failure at 400 mg/day, but these patients were not followed up in any of the included studies looking at dose escalation of imatinib. The only other data available on BSC come from the pre-imatinib era (where it would not have been possible for people to have failed on 400 mg/day of imatinib). The two sources chosen from the studies identified (see Appendix 13) were chosen because they had larger sample sizes and longer median follow-up times. However, one of the sources relates to a study conducted before there was awareness of GIST as being a distinct tumour. Alternative data for this parameter are outlined in Appendix 13; however, it is likely that these data would provide similar, imprecise and potentially biased estimates for this probability.
Probability of death for imatinib at 600 and 800 mg/day
The data on mortality for the imatinib 600 mg/day treatment groups were taken from the available trial data39 and 55% (6/11) of those who crossed over to imatinib 600 mg/day died over the trial period of 60 months. Although the sample size is very small, in the absence of any better alternative it has been used in the model. The data on mortality for imatinib 800 mg/day were taken from Blanke et al. 41 [where the data suggest that 64.41% (76/118) died in the imatinib 800 mg/day group]. Again the monthly mortality rate was derived as an exponential rate. It should be noted that the study was not designed to assess dose escalation, and the use of data from the crossover groups from the studies used are not ideal estimates for probability of death for these patients. In the absence of other suitable data we have used these data for our model.
Probability of death for sunitinib
The mortality data for those treated with sunitinib came from Reichardt et al. 81 In this study 231/331 patients receiving sunitinib survived. The monthly mortality rate was derived assuming an exponential rate. In the analysis it was also assumed that the mortality rate for those receiving sunitinib was the same regardless of any possible differences in prior treatment. It should also be noted that the survival estimate from this trial was based on those who failed on imatinib at doses of ≤ 400 mg/day, but it is not clear whether the patients failed on the 400 mg/day dose or at lower doses.
Response rates for the treatments
For our model, response to treatment was also taken to include PR, complete response and those reported to be in a stable condition.
The response rate for imatinib 600 mg/day was based upon data from the B2222 trial. 39 This study reported that 25.5% (11/43) of patients had responded and remained stable during a median follow-up of 63 months. The sample size of this study was small, but these were the only data available for the specific population of interest. It should be noted that the B2222 trial39 was not designed to assess dose escalation and there was no randomisation of patients at the point of disease progression.
The S003341 and EORTC44 trials data were used to provide evidence of the response rate for imatinib 800 mg/day. These studies reported that 30% (75/250) of patients responded to treatment with imatinib 800 mg/day and showed PR or stable condition after a median follow-up of 54 months.
For sunitinib none of the studies meeting the inclusion criteria for the review of effectiveness reported data on response rate. Therefore, this parameter was estimated from the weighted average response rate from two studies reporting this outcome. 38,108 In these two studies in total 266/382 patients responded, and a simple weighted mean was used to derive the pooled response rate. This response rate was assumed to be unaffected by prior treatment received. It should be noted that the patient groups in these two studies may not be the same. The Prior et al. study108 does not report the previous imatinib dose for participants, whereas in Demetri et al. 38 most of the population failed on 800 mg/day imatinib. As there was no statistically significant difference in the response rates, we took these two studies as a second-best source. The non-response data for each treatment were converted into monthly transition probabilities by assuming an exponential function.
Cost data
Resources used by the selected treatment strategies were identified from relevant sources [e.g. NHS reference costs, the British National Formulary (BNF), etc.] and the review of economic evaluations. Costs have been considered from a NHS perspective only. An identification of the potential direct and indirect resource costs for the NHS and PSS that would be expected from the introduction of the technology is presented.
We included the costs of drugs, i.e. costs of imatinib 400 mg/day, 600 mg/day, 800 mg/day and sunitinib 50 mg/day. As the sunitinib treatment process involved taking medication for 4 weeks and then no medication for the following 2 weeks, we estimated the yearly medication costs of this drug and then equally proportioned this cost to each month within that year. Data on cost of drugs were obtained from the BNF no. 58. 109 It has been assumed that patients on BSC still receive medications and it has been assumed that the cost of these is equivalent to the cost of imatinib at 400 mg/day.
Resource use by the treatments was based on the study by Wilson et al. ,55 which suggested that there are GP visits (£40 per year), outpatient visits including tests (£440 per year) and CT scans (£656 per year) and cost of management of adverse events (£159 per year), at 2004 prices. These cost estimates were used for our model after adjusting for inflation with the Hospital and Community Health Services (HCHS) Index for pay and prices inflation for the year 2008–9. 110 Based on these estimates, the total monthly cost of management with imatinib treatment is £128.16. These other treatment costs represent approximately 5% of the total cost of the drug itself. In the absence of better data these costs have been used for imatinib at both 600 and 800 mg/day.
For the sunitinib group we have used the resources based on the Pfizer single technology assessment submission60 for patient monitoring, outpatient and GP visits (£799.73 per year), CT imaging (£336 for 7.3 months) and management of adverse events (£159 per year). These costs are at 2008 prices and were adjusted to 2009 prices using the same methods as described above. Based on these data the estimated total monthly cost of this care used within the model is £185.
For BSC, data from the Pfizer submission were again used:60 the suggested costs in 2008 prices for patient monitoring, outpatient and GP visits were £249 per year, and £105 per year for CT imaging. These costs were inflated to 2009 prices using the same methods described above.
The different estimates for the costs of CT scanning between the two drugs can be accounted for by the fact that different sources were used to derive the costs of CT scanning. When inflated to 2008–9 prices, this gave the monthly cost of CT scans as £15.01 for BSC groups, £64.92 for the imatinib groups and £48.04 for the sunitinib groups. 51,55
The monthly cost of adverse events in the model is £13.25 for the imatinib groups (600 and 800 mg/day) and £21.78 for sunitinib, which is about 10% of all other costs for imatinib and 12% of all other costs for sunitinib. There were insufficient data on disutility to incorporate this as a parameter within the model, despite evidence to suggest differences in the adverse event profiles of imatinib and sunitinib that could influence disutility. 111
Utility data
There were few data relating to health-state utilities. Our model has used data in which the health-state valuations are derived from the EQ-5D, and the values used were taken from Wilson et al. 55 and Chabot et al. 95 The utility associated with PFS for those responding to imatinib (regardless of dose) was 0.935. 55 The utility for those receiving BSC was taken from Chabot et al. 95 and was taken to be 0.577. In the absence of alternative data it has been assumed that the utility for those who have not progressed on sunitinib is the same as that assumed for imatinib, i.e. 0.935.
Table 15 describes the parameter inputs used within the model. It also describes the sources of data, alternative valuations and data used to inform the probabilistic sensitivity analysis (described in more detail below).
| Parameters | Description | Value | For sensitivity analysis | Distribution | Values | Data source and assumptions | |
|---|---|---|---|---|---|---|---|
| Low | High | ||||||
| Cost parameters (£) | |||||||
| cImat600 | Cost of drugs: imatinib 600 | 2406 | 2005 | 2406 | BNF 58:109 low value is average of imatinib 400 and 600 mg | ||
| cImat800 |
Cost of drugs: imatinib 800 |
3208 | 2807 | 3208 | BNF 58:109 low value is average of imatinib 600 and 800 mg | ||
| CNott |
Cost of BSC |
1604 | 1283 | 1604 | Includes cost of imatinib 400 mg (BNF 58109) | ||
| CSunb | Cost of drugs: sunitinib | 3138.8 | 2092.5 | 3138.8 | BNF 58:109 low value is average of reduced dose of sunitinib | ||
| OthCostBSC | Other costs and management of treatment in BSC arm | 50.61 | Resource use in the treatment was based on the study by Wilson 200555 | ||||
| OthCostIm | Other costs and management of treatment in imatinib treatment arm | 128.16 | Resource use in the treatment was based on the study by Wilson 2005.55 Assumed to be same for imatinib 600 and imatinib 800 | ||||
| OthCostSun | Other costs and management of treatment in sunitinib treatment arm | 185.11 | Resource use in the treatment was based on the study by Wilson 200555 and single technology appraisal of Pfizer60 | ||||
| Mortality and response to treatment | |||||||
| deathBSC | Probability of death in the BSC treatment arm | 0.014627 | Beta |
α = 0.8448898 β = 57.775 |
Pooled weighted rate106,107 | ||
| dth600 | Probability of death in imatinib 600 treatment arm | 0.007472 | Beta |
α = 0.08162 β = 10.91838 |
B2222 study39 | ||
| dth800 | Probability of death in imatinib 800 treatment arm | 0.011857 | Beta |
α = 1.39948 β = 116.600 |
S0033 study41 | ||
| Dthsun | Death due to GIST: sunitinib | 0.026706 | Beta |
α = 9.3284 β = 341.62 |
Reichardt 200881 | ||
| nonrespIm600 | Transition probability of non-response to imatinib 600 | 0.011743 | Beta |
α = 0.504949 β = 42.495051 |
B2222 study39 | ||
| nonrespIm800 | Transition probability of non-response to imatinib 800 | 0.012879 | Beta |
α = 3.21875 β = 246.780 |
S0033 study41 and Zalcberg 200544 | ||
| nonrespSun | Transition probability of non-response to sunitinib | 0.080959 | Beta |
α = 12.30 β = 139.6945 |
Weighted average response rate52,108 | ||
| uImat600 | Utility with imatinib 600 | 0.935 | 0.712 | 0.939 | Wilson 200555 | ||
| uImat800 | Utility with imatinib 800 | 0.935 | 0.712 | 0.939 | Wilson 200555 | ||
| uProg | Utility for PD | 0.577 | 0.52 | 0.712 | Chabot 200895 and Wilson 200555 for lower level values for sensitivity analysis | ||
| uSun | Utility with sunitinib treatment | 0.935 | 0.712 | 0.939 | Chabot 200895 | ||
| Structural and methodological parameters | |||||||
| Cycle length | Time period that utilities, costs and probabilities relate to | 1 month | Assumption | ||||
| Length of run | No. of cycles model is run for | 120 (10 years) | 72 (6 years) | 144 (12 years) | Assumption | ||
| DR | Discount rate | 0.002917 | 0 | 0.005 | NICE guideline94 | ||
In a sensitivity analysis, the high value of the costs of drugs (imatinib and sunitinib) has been assumed to be similar to the value based on the BNF price,109 which we used in our model for the base-case analysis. For the lower value, we have taken an average of the price of the higher and lower doses assuming that there may be a need to lower the dose in the treatment pathways assumed in our model. For sunitinib, during the sensitivity analysis the price of the lower dose is assumed.
Time horizon for the model
The model looked at the costs and consequences directly attributable to GIST. As reported earlier the typical survival of such patients is relatively short and hence the time horizon of the model was limited to 10 years. The cycle length was 1 month to reflect the natural history of the condition.
Analysis methods
The results of the model are presented in terms of the incremental cost per QALY. The costs and outcomes were discounted at 3.5% in accordance with NICE guidelines. As described below, both deterministic and probabilistic sensitivity analyses were conducted with a net benefit framework being used to compare the different treatment strategies.
Sensitivity analysis
Probabilistic sensitivity analyses
Probabilistic sensitivity analyses of the base-case scenario were conducted by assuming a beta distribution of the probability of death and non-response to treatment in the different treatment strategies. The values used to define these distributions are reported in Table 15 and are derived from the data reported in Data requirements and model inputs, above.
The beta distribution as defined above might arguably be considered to be too precise and to not truly reflect the degree of uncertainty that exists. To examine the uncertainties around the distribution assumed for the base-case scenario, sensitivity analysis was conducted by assigning a uniform distribution to these parameters, where the low and high value of probability of death and non-response rate were assumed to be 90% less than and 90% more than the mean value used in our model, respectively. The justification for this distribution was that comparisons of interventions that are based on non-randomised and non-comparative data are potentially biased and that both the magnitude and direction of bias are uncertain.
Deterministic sensitivity analyses
Sensitivity analysis was conducted with respect to methodological and structural assumptions. First, the discount rate for costs and effectiveness was changed to 0% and 6% in the sensitivity analysis. The time horizon was also varied between 6 and 12 years (data are presented in the results for 6- and 12-year time horizons).
Sensitivity analysis was also conducted to examine the uncertainties around the values used for the cost of drugs (which are major components of the cost of treatment for different treatment strategies) and the utility values for the different health states of the model. The values used in the sensitivity analysis are reported in Table 15.
A further area of uncertainty relates to the very limited data available for imatinib 600 mg/day. In the base-case analysis the effectiveness (in terms of survival and response rates) is better for imatinib 600 mg/day than with imatinib at 800 mg/day. As this was based on non-randomised, non-comparative data the relative difference is potentially biased. Therefore, in this sensitivity analysis a more conservative assumption was taken that the survival rate and the response rate for treatment with imatinib 800 mg/day also applied to imatinib 600 mg/day.
Results
Base-case analysis
Table 16 shows the mean estimates of cost and effectiveness of the seven alternative treatment strategies modelled. As this table shows, effectiveness has been reported in two ways: life-years and QALYs. Path-4, imatinib 600 mg/day has an incremental cost per QALY that is < £30,000 compared with Path-1, BSC. The only other non-dominated or non-extendedly dominated strategy is Path-2, imatinib 600 to imatinib 800 mg/day to sunitinib. However, in this case the incremental cost per QALY (compared with the next most costly option of Path-4, imatinib 600 mg/day) is in excess of £40,000.
| Strategies | Cost (£) | Incremental cost (£) | Life-years | Incremental life-years | QALYs | Incremental QALYs | Incremental cost per QALY (£) |
|---|---|---|---|---|---|---|---|
| Path-1 BSC | 92,811 | 4.154 | 2.397 | ||||
| Path-7 Sunitinib | 96,688 | 3877 | 3.716 | (Dominated) | 2.411 | 0.014 | 272,365 |
| Path-4 Imatinib 600 mg | 147,060 | 50,372 | 5.211 | 1.057 | 4.256 | 1.845 | 27,304 |
| Path-3 Imatinib 600 mg to sunitinib | 149,200 | 2139 | 5.032 | Dominated | 4.286 | 0.030 | 71,723 |
| Path 6 Imatinib 800 mg | 153,901 | 4702 | 4.506 | Dominated | 3.635 | –0.651 | Dominated |
| Path-5 Imatinib 800 mg to sunitinib | 155,828 | 6628 | 4.336 | Dominated | 3.659 | –0.627 | Dominated |
| Path-2 Imatinib 600 mg to 800 mg to sunitinib | 172,152 | 22,953 | 5.278 | 0.067 | 4.803 | 0.517 | 44,359 |
| With dominated and extendedly dominated options removed | |||||||
| Path-1 BSC | 92,811 | 4.154 | 2.397 | ||||
| Path-4 Imatinib 600 mg | 147,060 | 54,249 | 5.211 | 1.057 | 4.256 | 1.859 | 29,181 |
| Path-2 Imatinib 600 mg to 800 mg to sunitinib | 172,152 | 25,092 | 5.278 | 0.067 | 4.803 | 0.547 | 45,850 |
Of note is that in the base-case analysis treatment with sunitinib for those who failed with imatinib 400 mg/day (Path-7) was estimated to have a lower life expectancy than BSC but greater QALYs. The reason for this was that the estimates of survival for sunitinib were based upon limited non-randomised and non-comparative data (as was the case for all the other comparators). Hence, any comparison should be treated cautiously.
The finding that sunitinib was dominated by BSC when effectiveness was measured in life-years but not dominated when effectiveness was measured in QALYs illustrates the importance of the utility estimates used within the model. Again, such data were sparse and, particularly for sunitinib, do not reflect the potentially worse side effect profile. Other things remaining unchanged the inclusion of side effects would have reduced the QALYs obtained from pathways containing sunitinib and potentially led to Path-7 being dominated by BSC (at the very least the incremental cost per QALY would have increased from the £272,365 reported in Table 16).
The results reported in Table 16 are surrounded by considerable imprecision. One of the main sources of the imprecision in the analysis surrounds the clinical effectiveness data. Therefore, a partial probabilistic sensitivity analysis was conducted, with the imprecision surrounding response rates and mortality rates being characterised by beta distributions. Figure 7 shows the cost-effectiveness acceptability curve and illustrates that the pathway with the highest likelihood of being considered cost-effective when society’s willingness to pay for a QALY is less than approximately £25,000 is Path-1, BSC. When society’s willingness to pay for a QALY is between approximately £25,000 and £45,000 then Path-4, imatinib 600 mg/day, is most likely to be considered cost-effective. Beyond a threshold of approximately £45,000, Path-2, imatinib 600 mg/day to imatinib 800 mg/day to sunitinib, is most likely to be cost-effective.
FIGURE 7.
Cost-effectiveness acceptability curve for alternative treatments over the 10-year time horizon. Pathways with a low probability of being cost-effective over the range of willingness to pay for a QALY values considered have not been shown.
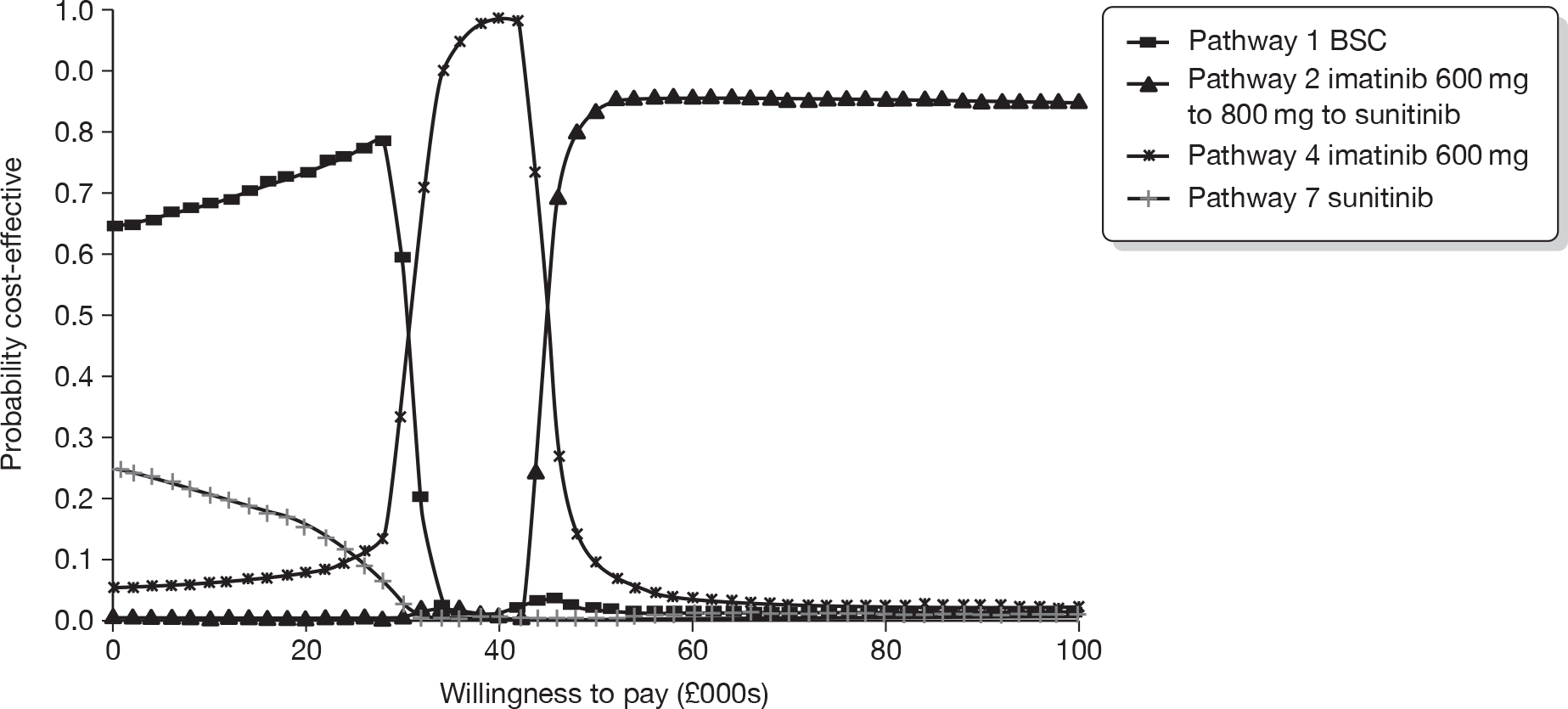
Sensitivity analysis
Uncertainty around the distributions used for mortality and response rates
The beta distributions used to generate Figure 7 potentially do not fully characterise the extent of the uncertainty surrounding the estimates of mortality and response used within the model. As noted in the previous section (see Probabilistic sensitivity analyses), this is because the data are used essentially as if they came from non-randomised, non-comparative sources, and hence any comparisons drawn may be highly biased. For this reason, in this sensitivity analysis uniform distributions were substituted for the beta distributions (Figure 8). It should be noted that these uniform distributions were assumed to be symmetrical around the point estimates used in the base-case analysis.
FIGURE 8.
Cost-effectiveness acceptability curve for alternative treatments over the 10-year time horizon assuming uniform distributions for mortality and response rates. Pathways with a low probability of being cost-effective over the range of willingness to pay for a QALY values considered have not been shown.
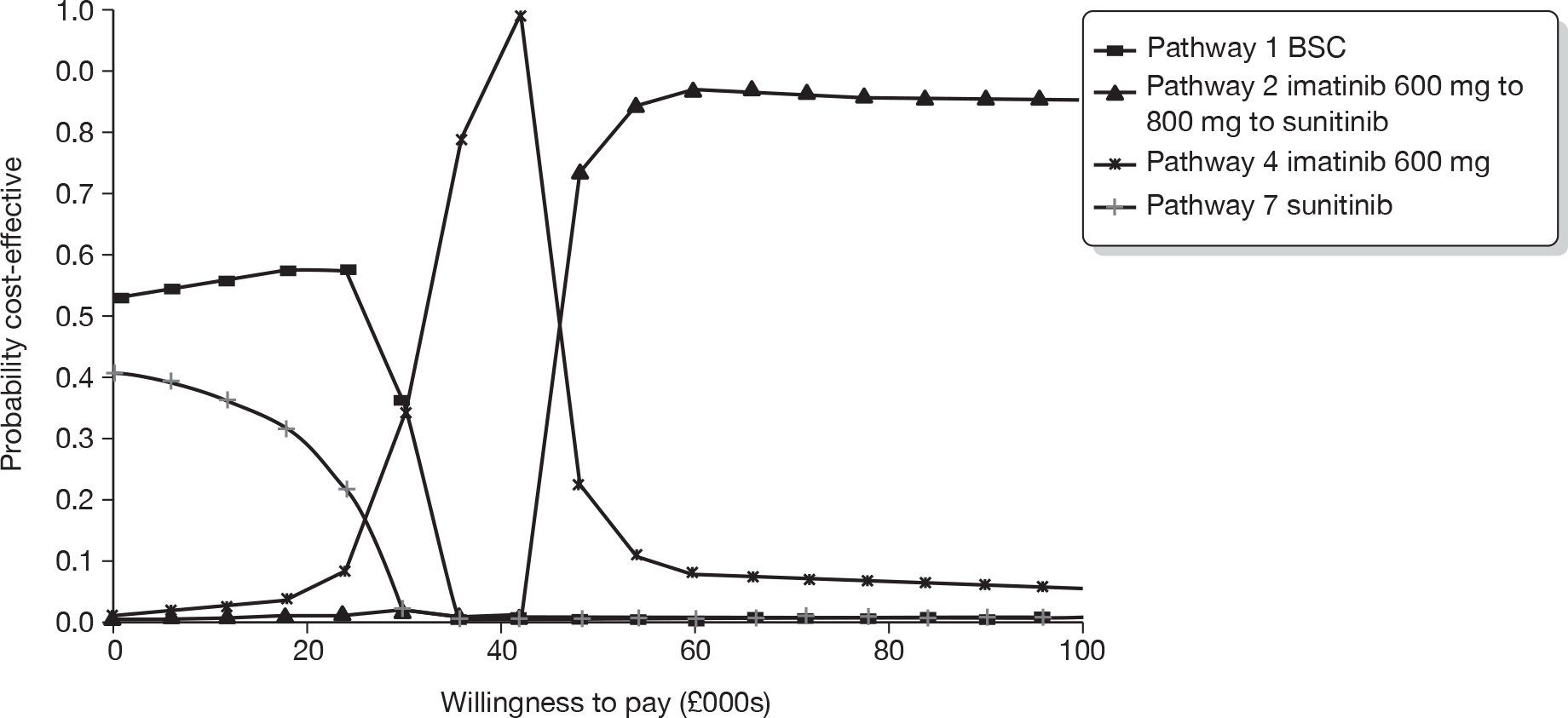
As Figure 8 illustrates, the basic pattern of the cost-effectiveness acceptability curve is the same as that depicted in Figure 7. At low threshold values for the willingness to pay for a QALY, Path-1, BSC, is still the most likely to be considered cost-effective. However, Path-7, sunitinib, is more likely to cost-effective at low thresholds. It should be noted that even though the distributions surrounding mortality weights are very wide in this analysis sunitinib is still associated with a trend towards a slightly higher mortality rate than BSC. As previously noted this trend is based upon sparse and potentially unreliable data on the performance of sunitinib. At a threshold value of approximately £36,000 Path-3, imatinib 600 mg daily to sunitinib, has a similar probability of being considered cost-effective as Path-1, BSC, and Path-4, imatinib 600 mg/day. Between a threshold of £36,000 and £48,000, Path-4, imatinib 600 mg/day, is most likely to be cost-effective, and beyond that threshold value Path-2, imatinib 600 mg/day to imatinib 800 mg/day to sunitinib, is most likely to be cost-effective.
Uncertainty surrounding structure and methodological assumptions around distribution
Two different discount rates have been applied to costs and benefits to examine the sensitivity of the results to plausible changes in the discount rate (Table 17). At a 0% discount rate there is no change in the options that are dominated or extendedly dominated, and the incremental cost per QALY for Path-4, imatinib 600 mg/day, compared with Path-1, BSC, increases to £31,183. The incremental cost per QALY for Path-2, imatinib 600 mg/day to 800 mg/day to sunitinib, compared with Path-4, imatinib 600 mg/day, increases to £54,715.
| Strategy | Cost (£) | QALYs | Incremental cost per QALY (£) | |
|---|---|---|---|---|
| Base case, i.e. discount rates = 3.5% on cost and benefit; time horizon = 10 years | Path-1 BSC | 92,811 | 2.397 | |
| Path-7 Sunitinib | 96,688 | 2.411 | 272,365 | |
| Path-4 Imatinib 600 mg | 147,060 | 4.256 | 27,304 | |
| Path-3 Imatinib 600 mg to sunitinib | 149,200 | 4.286 | 71,723 | |
| Path-6 Imatinib 800 mg | 153,901 | 3.635 | Dominated | |
| Path-5 Imatinib 800 mg to sunitinib | 155,828 | 3.659 | Dominated | |
| Path-2 Imatinib 600 to 800 mg to sunitinib | 172,152 | 4.803 | 44,359 | |
| Sensitivity analysis 1, i.e. discount rates = 0% on cost and benefit; time horizon = 10 years | Path-1 BSC | 93,137 | 2.706 | |
| Path-7 Sunitinib | 97,719 | 2.672 | Dominated | |
| Path-4 Imatinib 600 mg | 159,462 | 4.833 | 31,183 | |
| Path-3 Imatinib 600 mg to sunitinib | 163,601 | 4.859 | Extendedly dominated | |
| Path 6 Imatinib 800 mg | 165,641 | 4.087 | Dominated | |
| Path-5 Imatinib 800 mg to sunitinib | 169,210 | 4.105 | Dominated | |
| Path-2 Imatinib 600 to 800 mg to sunitinib | 195,193 | 5.486 | 54,715 | |
| Sensitivity analysis 2, i.e. discount rates = 6%; time horizon = 10 years | Path-1 BSC | 92,614 | 2.209 | |
| Path- 7 Sunitinib | 96,007 | 2.254 | Extendedly dominated | |
| Path-4 Imatinib 600 mg | 139,473 | 3.908 | 27,593 | |
| Path-3 Imatinib 600 mg to sunitinib | 140,394 | 3.940 | 28,801 | |
| Path 6 Imatinib 800 mg | 146,627 | 3.360 | Dominated | |
| Path-5 Imatinib 800 mg to sunitinib | 147,542 | 3.387 | Dominated | |
| Path-2 Imatinib 600 to 800 mg to sunitinib | 158,271 | 4.392 | 39,480 |
When the discount rate is changed to 6%, the incremental cost per QALYs for the non-dominated strategies falls compared with the base-case analysis. The key change is that Path-3, imatinib 600 mg/day to sunitinib, is no longer extendedly dominated by Path 4, imatinib 600 mg/day. Furthermore, the incremental cost per QALY for this comparison is < £30,000. Overall, the sensitivity analysis around discount rates illustrates that the results are sensitive to the choice of discount rate.
Table 18 reports the results of the sensitivity analysis around the time horizon of the model. When the time horizon is reduced to 6 years (base case = 10 years) the incremental cost per QALYs associated with the non-dominated options increases slightly. When the time horizon increases, the incremental cost per QALY for Path-4, imatinib 600 mg/day, compared with Path-1, BSC, increases slightly. The incremental cost per QALY for Path-2, imatinib 600 mg/day to imatinib 800 mg/day to sunitinib, compared with Path-4, imatinib 600 mg/day, is virtually unchanged.
| Strategy | Cost (£) | QALYs | Incremental cost per QALY (£) | |
|---|---|---|---|---|
| Base case, i.e. discount rates = 3.5% on cost and benefit; time horizon = 10 years | Path-1 BSC | 92,811 | 2.397 | |
| Path-7 Sunitinib | 96,688 | 2.411 | 272,365 | |
| Path-4 Imatinib 600 mg | 147,060 | 4.256 | 27,304 | |
| Path-3 Imatinib 600 mg to sunitinib | 149,200 | 4.286 | 71,723 | |
| Path-6 Imatinib 800 mg | 153,901 | 3.635 | Dominated | |
| Path-5 Imatinib 800 mg to sunitinib | 155,828 | 3.659 | Dominated | |
| Path-2 Imatinib 600 to 800 mg to sunitinib | 172,152 | 4.803 | 44,359 | |
| Sensitivity analysis 3, i.e. discount rates = 3.5%; time horizon = 6 years | Path-1 BSC | 73,246 | 1.960 | |
| Path-7 Sunitinib | 79,720 | 2.032 | Extendedly dominated | |
| Path-4 Imatinib 600 mg | 114,433 | 3.402 | 28,560 | |
| Path-3 Imatinib 600 mg to sunitinib | 117,729 | 3.455 | Extendedly dominated | |
| Path-6 Imatinib 800 mg | 126,750 | 3.017 | Dominated | |
| Path-5 Imatinib 800 mg to sunitinib | 129,873 | 3.066 | Dominated | |
| Path-2 Imatinib 600 to 800 mg to sunitinib | 131,848 | 3.758 | 48,969 | |
| Sensitivity analysis 4, i.e. discount rates = 3.5%; time horizon = 12 years | Path-1 BSC | 98,464 | 2.510 | |
| Path-7 Sunitinib | 101,589 | 2.509 | Dominated | |
| Path-4 Imatinib 600 mg | 156,943 | 4.489 | 29,553 | |
| Path-3 Imatinib 600 mg to sunitinib | 158,421 | 4.507 | Extendedly dominated | |
| Path-6 Imatinib 800 mg | 161,295 | 3.790 | Dominated | |
| Path-5 Imatinib 800 mg to sunitinib | 162,637 | 3.803 | Dominated | |
| Path-2 Imatinib 600 to 800 mg to sunitinib | 183,961 | 5.093 | 44,736 |
Uncertainty surrounding transition probabilities of survival and response to treatment with imatinib 600 mg/day
The data available for imatinib given at a dose of 600 mg/day were sparse and what few data there were suggested a superior effectiveness compared with imatinib 800 mg/day. These data are (1) potentially unreliable because they are based upon non-randomised and non-comparative data and (2) potentially counterintuitive (in a direct comparison would we expect imatinib 800 mg/day to perform worse than imatinib 600 mg/day?). Therefore, in this sensitivity analysis it was assumed that the mortality and response to treatment with imatinib 600 mg/day was the same as that with imatinib 800 mg/day.
As Table 19 shows the incremental cost per QALY for Path-4, imatinib 600 mg/day, compared with Path-1, BSC, falls. This is because there is a reduction in the cost of medications as the probabilities that patients die or make the transition to BSC increases, which more than compensates for the fall in QALYs. The QALYs associated with Path-3, imatinib 600 mg/day to imatinib 800 mg/day to sunitinib, fall but the incremental cost per QALY compared with Path 4, imatinib 600 mg/day, is virtually unchanged.
| Strategy | Cost (£) | QALYs | Incremental cost per QALY (£) | |
|---|---|---|---|---|
| Base case | Path-1 BSC | 92,811 | 2.397 | |
| Path-7 Sunitinib | 96,688 | 2.411 | 272,365 | |
| Path-4 Imatinib 600 mg | 147,060 | 4.256 | 27,304 | |
| Path-3 Imatinib 600 mg to sunitinib | 149,200 | 4.286 | 71,723 | |
| Path-6 Imatinib 800 mg | 153,901 | 3.635 | Dominated | |
| Path-5 Imatinib 800 mg to sunitinib | 155,828 | 3.659 | Dominated | |
| Path-2 Imatinib 600 to 800 mg to sunitinib | 172,152 | 4.803 | 44,359 | |
| Sensitivity analysis 5: survival rate and response rate to imatinib 600 mg treatment same as that with imatinib 800 mg | Path-1 BSC | 92,811 | 2.397 | |
| Path-7 Sunitinib | 96,688 | 2.411 | 272,365 | |
| Path-4 Imatinib 600 mg | 126,074 | 3.635 | 24,019 | |
| Path-3 Imatinib 600 mg to sunitinib | 128,001 | 3.659 | 80,476 | |
| Path-2 Imatinib 600 to 800 mg to sunitinib | 149,703 | 4.145 | 44,603 | |
| Path-6 Imatinib 800 mg | 153,901 | 3.635 | Dominated | |
| Path-5 Imatinib 800 to sunitinib | 155,828 | 3.659 | Dominated | |
| Sensitivity analysis 6: survival rate for BSC = 0.02349 | Path-1 BSC | 65,412 | 1.729 | |
| Path-7 Sunitinib | 77,669 | 1.954 | Dominated | |
| Path-4 Imatinib 600 mg | 137,060 | 4.022 | 31,239 | |
| Path-3 Imatinib 600 mg to sunitinib | 142,643 | 4.134 | Extendedly dominated | |
| Path-2 Imatinib 600 to 800 mg to sunitinib | 144,349 | 3.411 | Dominated | |
| Path-6 Imatinib 800 mg | 149,517 | 3.512 | Dominated | |
| Path-5 Imatinib 800 to sunitinib | 170,340 | 4.762 | 44,603 |
As noted in Data requirements and model inputs (above), the two sources106,107 of mortality data that we have used for BSC were chosen because these studies had larger sample sizes and longer median follow-up times (Appendix 13). We conducted sensitivity analysis using different sources of mortality data for BSC, i.e. using a pooled mortality estimate of 19.8% that is based on survival estimates from Pierie et al. ,107 Dougherty et al. 112 and Artyan et al. 113 The monthly mortality rate would be higher (0.02349) than what we have used in the base case (0.014627). As mortality for BSC increases, the cost and QALYs for the pathways fall because BSC is part of each pathway. As a consequence the incremental cost per QALYs do not change greatly although all slightly increase compared with less costly but less effective pathways because the increase in mortality for BSC has proportionately a greater effect on costs than on QALYs.
Uncertainty surrounding utility values
The sensitivity of a lower and higher value of utility for the health status of disease progression was examined. In this analysis the lower value was 0.52 and a higher utility value for those patients who progressed with GIST of 0.712 was assumed instead of 0.577, as was used in the base case (Table 20). Reducing the utility value increased the QALYs for treatments that had higher probabilities of response. The incremental cost per QALY for Path-4, imatinib 600 mg/day, compared with Path-1, BSC, slightly falls and the incremental cost per QALY for Path-2, imatinib 600 mg/day to imatinib 800 mg/day to sunitinib, compared with Path-4, imatinib 600 mg/day, falls to approximately £40,000.
| Strategy | Cost (£) | QALYs | Incremental cost per QALY (£) | |
|---|---|---|---|---|
| Base case: utility of progressive state = 0.577 | Path-1 BSC | 92,811 | 2.397 | |
| Path-7 Sunitinib | 96,688 | 2.411 | 272,365 | |
| Path-4 Imatinib 600 mg | 147,060 | 4.256 | 27,304 | |
| Path-3 Imatinib 600 mg to sunitinib | 149,200 | 4.286 | 71,723 | |
| Path 6 Imatinib 800 mg | 153,901 | 3.635 | Dominated | |
| Path-5 Imatinib 800 mg to sunitinib | 155,828 | 3.659 | Dominated | |
| Path-2 Imatinib 600 to 800 mg to sunitinib | 172,152 | 4.803 | 44,359 | |
| Sensitivity analysis 6: utility of progressive state = 0.52 | Path-1 BSC | 92,811 | 2.160 | |
| Path-7 Sunitinib | 96,688 | 2.242 | Extendedly dominated | |
| Path-4 Imatinib 600 mg | 147,060 | 4.158 | 27,156 | |
| Path-3 Imatinib 600 mg to sunitinib | 149,200 | 4.219 | 34,911 | |
| Path 6 Imatinib 800 mg | 153,901 | 3.543 | Dominated | |
| Path-5 Imatinib 800 mg to sunitinib | 155,828 | 3.596 | Dominated | |
| Path-2 Imatinib 600 to 800 mg to sunitinib | 172,152 | 4.782 | 40,759 | |
| Sensitivity analysis 7: utility of progressive state = 0.712 | Path-1 BSC | 92,811 | 2.958 | |
| Path-7 Sunitinib | 96,688 | 2.812 | Dominated | |
| Path-4 Imatinib 600 mg | 147,060 | 4.488 | 35,440 | |
| Path-3 Imatinib 600 mg to sunitinib | 149,200 | 4.444 | Dominated | |
| Path 6 Imatinib 800 mg | 153,901 | 3.853 | Dominated | |
| Path-5 Imatinib 800 mg to sunitinib | 155,828 | 3.808 | Dominated | |
| Path-2 Imatinib 600 to 800 mg to sunitinib | 172,152 | 4.853 | 68,837 |
Conversely, increasing the utility associated with PD reduced the opportunity for pathways that are clinically more effective to generate additional QALYs. As a consequence, in this sensitivity analysis the incremental cost per QALYs for the non-dominated pathways increase.
Uncertainty surrounding the cost of imatinib and sunitinib
In this set of sensitivity analyses reductions in the cost of imatinib 600 mg/day, imatinib 800 mg/day and sunitinib are explored (Table 21). Over most of these sensitivity analyses the pathways that are dominated or are extendedly dominated do not change. As would be expected reducing the costs of each medication individually reduces the cost of pathways involving that medication. Over all these sensitivity analyses there are only relatively modest changes in the ICERs reported. One of the more substantive changes is that when the cost of sunitinib is reduced, Path-7, sunitinib, becomes the least costly option. This is primarily because this pathway uses potentially unreliable data on mortality for sunitinib which means that patients on this pathway do not survive long enough to incur higher costs.
| Strategy | Cost (£) | QALYs | Incremental cost per QALY (£) | |
|---|---|---|---|---|
|
Base case: Imatinib 600 mg £2406 Imatinib 800 mg $3208 Sunitinib £3138.8 |
Path-1 BSC | 92,811 | 2.397 | |
| Path-7 Sunitinib | 96,688 | 2.411 | 272,365 | |
| Path-4 Imatinib 600 mg | 147,060 | 4.256 | 27,304 | |
| Path-3 Imatinib 600 mg to sunitinib | 149,200 | 4.286 | 71,723 | |
| Path-6 Imatinib 800 mg | 153,901 | 3.635 | Dominated | |
| Path-5 Imatinib 800 mg to sunitinib | 155,828 | 3.659 | Dominated | |
| Path-2 Imatinib 600 to 800 mg to sunitinib | 172,152 | 4.803 | 44,359 | |
|
Sensitivity analysis 8 (change in imatinib 600 mg price): Imatinib 600 mg £2005 Imatinib 800 mg $3208.16 Sunitinib £3138.8 |
Path-1 BSC | 92,811 | 2.397 | |
| Path-7 Sunitinib | 96,688 | 2.411 | Extendedly dominated | |
| Path-4 Imatinib 600 mg | 130,272 | 4.256 | 20,150 | |
| Path-3 Imatinib 600 mg to sunitinib | 132,412 | 4.286 | Extendedly dominated | |
| Path-6 Imatinib 800 mg | 153,901 | 3.635 | Dominated | |
| Path-2 Imatinib 600 to 800 mg to sunitinib | 155,364 | 4.803 | 45,850 | |
| Path-5 Imatinib 800 mg to sunitinib | 155,828 | 3.659 | Dominated | |
|
Sensitivity analysis 9 (change in imatinib 800 mg price): Imatinib 600 mg £2406 Imatinib 800mg $2807 Sunitinib £3138.8 |
Path-1 BSC | 92,811 | 2.397 | |
| Path-7 Sunitinib | 96,688 | 2.411 | Extendedly dominated | |
| Path-6 Imatinib 800 mg | 139,988 | 3.635 | Extendedly dominated | |
| Path-5 Imatinib 800 mg to sunitinib | 141,915 | 3.659 | Extendedly dominated | |
| Path-4 Imatinib 600 mg | 147,060 | 4.256 | 29,181 | |
| Path-3 Imatinib 600 mg to sunitinib | 149,200 | 4.286 | Extendedly dominated | |
| Path-2 Imatinib 600 to 800 mg to sunitinib | 166,000 | 4.803 | 34,609 | |
|
Sensitivity analysis 10 (change in sunitinib price): Imatinib 600 mg £2406 Imatinib 800mg $3208.16 Sunitinib £2092 |
Path-7 Sunitinib | 87,533 | 2.411 | |
| Path-1 BSC | 92,811 | 2.397 | Dominated | |
| Path-3 Imatinib 600 mg to sunitinib | 144,524 | 4.286 | 30,400 | |
| Path-4 Imatinib 600 mg | 147,060 | 4.256 | Dominated | |
| Path-5 Imatinib 800 mg to sunitinib | 151,560 | 3.659 | Dominated | |
| Path-6 Imatinib 800 mg | 153,901 | 3.635 | Dominated | |
| Path-2 Imatinib 600 to 800 mg to sunitinib | 170,364 | 4.803 | 49,940 |
Summary
The systematic review of economic evaluations reported in this chapter was not especially informative. This was anticipated at the outset and hence an economic modelling exercise was planned. The modelling exercise compared alternative treatment pathways for patients with unresectable GIST who failed to respond to imatinib 400 mg/day. Over almost all the sensitivity analyses Path-1, BSC, is the least costly and least effective intervention. Similarly, Path-4, imatinib 600 mg/day, typically has an incremental cost per QALY that is less than £30,000 compared with Path-1, BSC. Path-2 (imatinib 600 mg/day to imatinib 800 mg/day to sunitinib) is the only other pathway which is not dominated or extendedly dominated over most of the analyses conducted. However, in this case the incremental cost per QALY (compared with the next most costly option – Path-4: imatinib 600 mg/day) tends to be in excess of £40,000.
When society’s willingness to pay for a QALY is less than approximately £25,000 Path-1, BSC, is the most cost-effective. When society’s willingness to pay for a QALY is between approximately £25,000 and £45,000 then Path-4, imatinib 600 mg/day, is most likely to be considered cost-effective. Beyond a threshold of approximately £45,000, Path-2, imatinib 600 mg/day to imatinib 800 mg/day to sunitinib, is most likely to be cost-effective.
The results of the economic analysis are based upon sparse data that are potentially biased and are surrounded by considerable imprecision. In particular, data for sunitinib and for imatinib 600 mg/day are the most suspect. The analysis has also not considered two main areas of uncertainty due to lack of data:
-
The considerable uncertainty around the extrapolation from the sparse data on death and response rate and the impact of alternative assumptions about how probabilities of death and response change over time.
-
Reductions in utility associated with adverse effects of treatment.
By assuming constant probabilities over time, death may be overestimated at earlier time points and underestimated at later stages of the time horizon of the model. The probability of death may increase over time as the disease progresses. Similarly the probability of non-response may increase over time and patients may have an increasing need for an escalated dose of imatinib, or sunitinib. The assumption of constant probability over time generally delays the transition to patients’ deaths. This means that our analysis has a bigger impact on the most effective treatments. These treatments will also incur high treatment costs over a longer period. The net impact of these two changes on cost-effectiveness is unclear.
The net impact of adjusting scores for adverse effects is also uncertain but it might be expected that it will reduce the QALYs associated with each medication and, although there are limited data available from the systematic review of clinical effectiveness, this reduction may be greater for pathways involving sunitinib because its adverse effect profile is believed to be worse than that of imatinib. 111
Owing to sparse data for this analysis, few data were available on the utility values for defined disease states in the model. Furthermore, the disease states selected in the model may not be complete and exhaustive as data on alternative plausible disease states were not available. Sensitivity analysis explored uncertainty in key parameter estimates but clearly this does not investigate the influence of structural assumptions such as the limited number of disease states chosen for the modelling. A more sophisticated model would have allowed further sensitivity analysis but without at least some data to guide assumptions we would have needed to identify threshold values for many individual parameters and combinations of parameters. This would have resulted in a substantially expanded economics chapter reporting extensive speculative results that would have been very difficult to interpret.
A further factor not considered by the economic model was the cost-effectiveness of treatment for those with specific gene mutations. Again this was not addressed owing to lack of data, and as there were also no data available to assess the impact of plasma monitoring on the study population of interest, this was also not considered by the economic model.
Finally, the economic evaluation has assumed that patients who move on to BSC remain on treatment to prevent tumour flare. This has the impact of increasing the cost of BSC. It is further assumed that there is no impact on effectiveness (the implicit assumption is that discontinuing the medication would reduce life expectancy). Within the analysis it has been assumed that all patients on BSC or moving on to BSC after failing to respond on a medication would receive imatinib 400 mg/day. This assumption appears reasonable for Path-1, BSC, but may not be appropriate for the other pathways where patients would move on to BSC after failing to respond on an escalated dose of imatinib or on sunitinib. Should these patients continue with the last active medication that they received then costs, and incremental costs per QALY, would increase.
Chapter 6 Assessment of factors relevant to the NHS and other parties
Gastrointestinal stromal tumours are a rare cancer, accounting for < 1% of all cancers of the GI tract. The incidence and subsequent overall burden on the NHS is not large, and only a small proportion of patients with GIST will have unresectable and/or metastatic disease that progresses on imatinib at a dose of 400 mg/day. NICE guidance on imatinib for the treatment of unresectable and/or metastatic GIST does not recommend an increase in the dose of imatinib for people receiving imatinib who develop PD after initially responding at the 400 mg/day dose. 50 Some guidelines, however, do advocate dose escalation for such patients, particularly those with KIT exon 9 mutations, indicating that escalated doses may help this group of GIST patients and offer them the opportunity to continue with a normal life for a longer period of time. 15,114,115
Since the availability of sunitinib, guidance on the treatment of patients with unresectable and/or metastatic GIST has been adapted to take account of this drug as a possible second-line treatment15 in circumstances where patients either are intolerant to imatinib, or have progressed on treatment with imatinib at a 400 mg/day dose. NICE guidance recommends sunitinib as a treatment option for people with unresectable and/or metastatic malignant GIST if imatinib treatment has failed because of resistance or intolerance, and the drug cost of sunitinib for the first treatment cycle is met by the manufacturer.
In clinical practice the treatment of patients with unresectable and/or metastatic GIST is generally decided on a case-by-case basis by multidisciplinary teams. Many clinicians advocate initial dose escalation of imatinib and then consider sunitinib on subsequent progression, although practice will vary depending on the specific needs of individual patients.
Chapter 7 Discussion
Statement of principal findings
Review of clinical effectiveness
This review is a part update of a previous review on imatinib for the treatment of patients with unresectable and/or metastatic GISTs. 55 We focused on patients with KIT (CD117)-positive, unresectable and/or metastatic GISTs whose disease had progressed on treatment with imatinib at a dose of 400 mg/day. Five studies involving 669 patients from within the relevant treatment arms met the inclusion criteria. Of these studies, four involving 318 patients reported imatinib outcomes and one involving 351 patients, who had received a prior imatinib dose of ≤ 400 mg/day, reported sunitinib outcomes. No studies reporting BSC were identified that met our inclusion criteria.
Although the study designs for most of the included trials were RCTs (plus one retrospective cohort study) none of these trials had, as their primary objective, the assessment of the effects of dose escalation following progression on 400 mg/day imatinib. Only a proportion of the overall patient populations received an escalated dose, and these patients were not randomised at the point of dose escalation to receive either an escalated dose of imatinib or remain on 400 mg/day. Therefore, the nature of the evidence base for patients who progress on 400 mg/day imatinib and receive escalated doses of 600 or 800 mg/day is observational and open to bias.
The sample sizes of the studies from which the 669 patients were drawn from ranged from 2479 to 111786 participants. Each study had more male than female participants. The vast majority of participants in each study had an ECOG performance status of ≤ 2, meaning that they were ambulatory and confined to bed for less than 50% of their waking hours. 116 Of the studies that reported the proportion of the study population receiving prior surgery,39,44,79 most patients had undergone prior surgery for treatment of their disease. Information on the characteristics of all the 669 patients relevant to this review was not provided separately.
From the data on imatinib it can be seen that approximately one-third of patients progressing on 400 mg/day of imatinib will respond to escalated doses. With 600 mg/day, between 25.6% (11/43)39 and 41.7% (5/12)79 of patients with unresectable and/or metastatic GIST, who had previously progressed on a dose of 400 mg/day of imatinib, either developed a PR or maintained SD. With 800 mg/day, the proportions achieving PR or SD ranged between 29.3%44 and 33.3%. 79 These data were used to inform transition probabilities of non-response to imatinib at escalated doses of 600 and 800 mg/day, respectively. However, response data were not available for patients receiving sunitinib following treatment with imatinib at a dose of ≤ 400 mg/day. As an alternative to excluding sunitinib entirely, which could arguably have been appropriate given the lack of data, the economic model used data that did not meet the inclusion criteria for the review of clinical effectiveness because it failed to report response data separately for those progressing on a 400 mg/day dose. A further assumption made in the economic model was that response was unaffected by prior treatment received. This assumption was made because of a lack of data on how response might change over time and be affected by prior treatments other than imatinib at 400 mg/day.
Median OS data were not reported for those receiving an escalated imatinib dose of 600 mg/day upon progression at a 400 mg/day dose. Therefore, the economic model calculated the probability of death from the available trial data on median OS according to best response, and the proportion of patients receiving escalated doses who will have had a response to imatinib at the initial 400 mg/day dose prior to eventual progression and dose escalation.
For those receiving an escalated imatinib dose of 800 mg/day upon progression, median OS was reported to be 19 months (95% CI 13 to 23 months) in the S0033 trial. 41 Median OS was not reported for the EORTC-ISG-AGITG study44 for the population of interest, (CiC information has been removed). For those receiving sunitinib after a prior imatinib dose of ≤ 400 mg/day, median OS was reported as 22.5 months (95% CI 18.3 to 26.5 months). 86
Figure 3 provided a visual comparison of the median OS times for imatinib at an escalated dose of 800 mg/day and sunitinib, showing overlapping CIs until 33 months from commencement of treatment, at which point the estimated proportion of sunitinib patients surviving appeared to be less than the proportion surviving on the 800 mg/day imatinib dose. (CiC information has been removed.) It is difficult to draw any conclusions with regard to possible differences in OS between imatinib at an escalated dose of 800 mg/day and sunitinib at 50 mg/day (with a 4-weeks-on/2-weeks-off cycle), owing to the lack of data, but as the 95% CIs for median OS overlap, there does not appear to be any significant difference in median OS with dose escalation, compared with sunitinib.
The median time to progression and PFS was reported for imatinib 600 mg/day as 1.7 months (range 0.7–24.9 months),79 and for imatinib 800 mg/day it ranged between 2.9 months (reported without CIs as ‘81 days’)44 and 5 months (95% CI 2 to 10 months). 41 A visual representation of these data for imatinib 800 mg/day in Figure 4 gives 95% CIs that do not overlap, for all time points between 12 and 21 months, indicating that PFS was significantly shorter in the EORTC-ISG-AGITG study reported by Zalcberg et al. 44 than in the S0033 trial reported by Blanke et al. 41
In addition, those studies looking at an 800 mg/day dose of imatinib reported that between 16.1% (19/118) and 18.8% (25/133) of patients were progression free at the time of the analysis. This represented a proportion of between 52.8% (19/36) and 64.1% (25/39) of all of those achieving PR and SD on the 800 mg/day dose. This suggests that a small proportion (i.e. < 20%) of those receiving an escalated dose of 800 mg/day of imatinib on progression may maintain their response/SD for a median time period of at least 25 months (i.e. the shorter of the median follow-up times reported by these trials), and those who achieve a response or maintain SD on the escalated dose may have a greater than 50% likelihood of maintaining this in the longer term.
For those receiving an escalated dose of 800 mg/day, the study by Zalcberg et al. 44 reported a median duration of ‘stabilisation’ among those showing response or SD with treatment of 153 days (range 37–574 days). For sunitinib, the treatment duration for all patients receiving sunitinib (i.e. regardless of the dose of prior imatinib therapy) was 126 days (range 1–618 days). 86
Data on adverse events were not available from any of the studies where the population of interest received imatinib at 600 mg/day or sunitinib following progression at 400 mg/day. For the trials reporting outcomes following dose escalation from 400 to 800 mg/day after progression at the lower dose, it was reported that the vast majority (88.4%) of study discontinuations were due to disease progression and not study drug toxicity. 44 (CiC information has been removed.)
Nevertheless, it was also reported that between 15.6%77 and 31%78 of patients receiving an escalated imatinib dose of 800 mg/day required a dose reduction. It was also reported that 23.3% (18/77) of patients required at least one dose delay. 77 However, it was not possible to take possible dose reductions into account with regard to any of the outcomes. This was because information on the dose provided following reduction, the median duration of any dose delay or dose reduction, and any other factors, besides toxicity, contributing to any of the dose delays or reductions were not reported.
These data on discontinuations and dose modifications indicate that, although disease progression is far more likely than adverse events to contribute to the decision to stop escalated imatinib treatment at the 800 mg/day dose, approximately one-third of patients will require dose modifications (i.e. dose reduction or interruption) during treatment at this escalated dose.
With regard to specific adverse events, data were reported by Zalcberg et al. 44 showing that a higher proportion of patients with skin rash, nausea, leucopenia, neutropenia and thrombocytopenia reported a reduction in the severity of these events following dose escalation compared with the proportion of patients reporting an increase in these events. This reduction was significant in the case of neutropenia (p = 0.002). However, the proportion of patients with oedema, fatigue, dyspnoea and anaemia who reported an increase in severity of these events following dose escalation was greater than the proportion of patients who reported a reduction in these events. This increase in severity was significant in the case of fatigue (p < 0.001) and anaemia (p = 0.015). 44 (CiC information has been removed.) It is difficult to draw any conclusions about specific adverse events from these data, aside from noting that fatigue and anaemia may significantly increase upon dose escalation from 400 mg/day imatinib to 800 mg/day.
The only data available for any of the prespecified subgroups of interest were reported by Debiec-Rychter et al. 14 for the EORTC-ISG-AGITG trial, which looked at imatinib dose escalation from 400 to 800 mg/day following progression at the lower dose. They noted that patients with wild type, and those with exon 9 mutations, were significantly more likely to have a response to dose escalation than those with exon 11 mutations, but no numerical data were reported for the population of interest. (CiC information has been removed.) Furthermore, it has been argued that subgroups with certain exon mutations might have improved response and/or survival outcomes if they initially receive an escalated imatinib dose, rather than receiving dose escalation only if there is progression at the 400 mg/day dose. 114
It was outwith the remit of this review to consider outcomes for patients receiving escalated dosing other than following progression on the initial 400 mg/day dose. The lack of data available meant it was not possible to assess for specific mutational population subgroups the effects of escalation to an imatinib dose of 800 mg/day following progression at the initial 400 mg/day dose.
Review of cost-effectiveness
The economic component of this study included both a review of the existing economic evaluations and an economic modelling exercise. The evidence from the review of economic evaluations was sparse and there was no published economic evaluation conducted for a UK context that compared all of the interventions for the patient group of interest.
The modelling exercise compared alternative treatment pathways for patients with unresectable GIST who failed to respond to imatinib 400 mg/day. Over almost all the sensitivity analyses, Path-1, BSC, is the least costly and least effective intervention. Similarly, Path-4, imatinib 600 mg/day, typically has an incremental cost per QALY that is less than £30,000 compared with Path-1, BSC. Path-2 (imatinib 600 mg/day to 800 mg/day to sunitinib) is the only other pathway that is not dominated or extendedly dominated over most of the analyses conducted. However, in this case the incremental cost per QALY (compared with the next most costly option: Path-4, imatinib 600 mg/day) tends to be > £40,000.
When society’s willingness to pay for a QALY is < ∼£25,000, Path-1, BSC, is the most cost-effective intervention. When society’s willingness to pay for a QALY is between approximately £25,000 and £45,000, Path-4, imatinib 600 mg/day, is most likely to be considered cost-effective. Beyond a threshold of approximately £45,000, Path-2, imatinib 600 mg/day to imatinib 800 mg/day to sunitinib, is most likely to be cost-effective.
As discussed below, these data should be treated cautiously, as the data used are observational and non-comparative. Furthermore, the data on sunitinib and imatinib 600 mg/day are particularly sparse and potentially unreliable. For example, data on treatment with sunitinib show a lower life expectancy than those on treatment with BSC (although sunitinib has greater QALYs). This means that when the cost of sunitinib is reduced it becomes more cost-effective than BSC, as the potentially unreliable source data for life expectancy on sunitinib mean that patients on sunitinib will not survive long enough to incur higher costs of treatment. Although sufficient evidence on the effectiveness of sunitinib compared with BSC following treatment on imatinib at a 400 mg/day dose was not available, evidence on the effectiveness of sunitinib compared with BSC regardless of prior imatinib dose suggests that life expectancy with sunitinib is superior.
In addition, the data available for imatinib at a dose of 600 mg/day suggested superior effectiveness compared with the 800 mg/day dose. This is because the evidence on imatinib at the 600 mg/day dose was based on a smaller sample size (43 patients), making the model results for this pathway potentially counterintuitive if we expect higher drug doses to have greater effectiveness than lower doses.
Strengths and limitations of the assessment
In terms of strengths, the review of the evidence base was detailed and thorough. It was unclear from the information provided in a substantial number of abstracts whether the studies met the inclusion criteria and full-text papers for all of these reports were obtained and assessed. Non-English language studies were not excluded. Authors were contacted in an attempt to obtain additional information concerning their studies. For the review of economic evaluations, a rigorous systematic approach was adopted. The economic model considered a large number of plausible alternative treatments and also incorporated both probabilistic and deterministic estimates of cost effectiveness. The former was limited to clinical effectiveness parameters but this limitation was chosen specifically to draw attention to the uncertainties surrounding these data.
In terms of limitations, there was a dearth of evidence available on the specific population of interest, despite the overall large evidence base on the treatment of GISTs with imatinib or sunitinib. The quality of reporting of dose information in reports of imatinib or sunitinib for GISTs was poor and the data on the population of interest for the studies that were included were non-randomised, non-comparative and therefore observational. Therefore, lack of quality data, as well as lack of data itself, severely limited both assessments of clinical effectiveness and cost-effectiveness.
There was also a lack of evidence on QoL outcomes, which may be of fundamental importance to patients given the potentially palliative nature of treatment following progression, and there was also a lack of evidence on BSC. This is important as since the development of imatinib and sunitinib, it no longer represents the only treatment option for those with unresectable/metastatic disease. There was little evidence on response to escalated doses of imatinib based on mutational status, specifically for those who had already failed on an initial imatinib dose of 400 mg/day. It was also not possible to account for the effects of required dose interruptions and reductions, or the effects of sunitinib on those intolerant to imatinib, owing to the lack of available data. This lack of data also prevented comparative analysis of adverse events between the intervention and comparator treatments.
For sunitinib, it was also necessary to assume that the vast majority of those receiving sunitinib after imatinib treatment at ≤ 400 mg/day had actually received imatinib at 400 mg/day, which may not be a valid assumption. However, it was not possible to confirm the validity of the assumption despite contacting the study authors (P Reichardt, HELIOS Klnikum Bad Saarow, Germany, 2010, personal correspondence). In addition, much of the evidence base for sunitinib generally relates to its use following the failure of escalated doses of imatinib rather than failure on 400 mg/day, suggesting that the role of sunitinib is seen more as a third-line treatment rather than a potential comparator to 600 mg/day or 800 mg/day imatinib treatment. This was highlighted by the manufacturer of imatinib in their submission for this technology appraisal, and is noted in Chapter 3 of this report.
For the economic model, sufficient sound comparative data for the different plausible treatments were not available, despite conducting an extensive review of relevant studies. This necessitated a number of simplifying assumptions being made with respect to the model and also the use of data that were potentially unreliable. The model assumes that patients entering a pathway will remain in that treatment for one cycle only if they do not respond and survive in the treatment arm. In these cases they are considered to move to the escalated doses, move to another alternative (if allowed by a treatment pathway) or continue with BSC for the remainder of the model time horizon or until they die. The care pathways considered in the economic model are not an exhaustive list of all possible treatment options available but represent plausible treatment scenarios. Some are likely to be more representative of clinical practice than others. Whilst additional clinical advice during the development of the care pathways might have increased the extent to which the chosen scenarios reflect true clinical practice, it may also have increased the level of complexity required within the model. Given the lack of robust data it was felt that a more sophisticated model would be difficult to populate.
Within the model, several simplifying assumptions had to be made for individual parameters. For example, it was necessary to consider the costs and utilities associated with BSC as consistent across all care pathways despite the fact that in clinical practice the costs of BSC may increase as an individual’s health deteriorates. Unfortunately, there were no data available to model how costs of BSC might increase and QoL might fall over time.
A further simplifying assumption was not to model the complications and side effects of therapy. This latter assumption was made owing to the very limited evidence available. This is coupled with the assumption made that the utility associated with stable response or progression did not vary between treatments. One impact of this assumption is that no utility decrement has been assumed for the arguably worse side effect profile of sunitinib. This means that pathways involving sunitinib may overestimate QALYs.
Perhaps a more important limitation is caused by the limited evidence base available. With respect to the clinical effectiveness data used to derive transition probabilities these data, as already noted, were based upon non-randomised, non-comparative data. Such data are potentially biased as well as being imprecise. In particular, it is worth noting that point estimates of death and response used within the model may be misleading, for example the point estimates used suggest that sunitinib has a higher mortality rate than BSC.
Uncertainties
For the assessment of clinical effectiveness:
-
The diagnosis of GIST as stated in the final scope document was based on a positive KIT (CD117) test. However, this is not a perfect test and in a small (< 5%) number of cases a patient can have a GIST despite having a negative KIT (CD117) test. 4,7,25 More recent tests (e.g. PDGRFA and DOG1) may clarify diagnosis. However, the WHO classification of GI tumours recommends that a diagnosis of GIST should only apply to those patients testing positive for the KIT (CD117) protein.
-
It was not possible to conduct any subgroup analysis for patients with particular mutations, or consider the methods used to identify response (e.g. FDG-PET or CT scanning), or possible factors related to the provision of dose-escalated imatinib in an adjuvant or neoadjuvant setting.
-
It was not possible within the time frame of this review for sufficient information to be provided that would have enabled meta-analysis of outcomes for the 800 mg/day dose of imatinib. This evidence may have enabled more robust estimates of survival following dose escalation to 800 mg/day. However, the data would still be prone to bias (being taken from data from a non-randomised patient population) and uncertainty surrounding other parameters (e.g. BSC, sunitinib and imatinib at 600 mg/day) would still be likely to make the model difficult to interpret.
-
Following progression, the proportion of patients subsequently progressing on escalated doses, who are kept on the study drug on the basis that progression of disease might be slower than if the patient were to be taken off the drug, is not known. It is also not clear whether there is a standard dose used for this purpose. Within the economic model it has been assumed that this would be the case (400 mg/day).
-
This review only considered drug treatments that were licensed for patients with GISTs and did not consider other drugs that may be being used in the treatment of GISTs, or licensed drugs that are being used ‘off licence’ to treat GIST (e.g. imatinib at doses exceeding 800 mg/day, or sunitinib provided in a continuous daily dosing regime).
-
Surgical interventions were also not considered even though surgery is an important treatment option for GIST patients, and even though those with unresectable disease may be eligible for surgery if their tumours become resectable following treatment with an escalated dose of imatinib. The role of emergency surgery as part of BSC was also not considered.
The economic model has also not considered three main areas of uncertainty due to lack of data:
-
alternative assumptions about how probabilities of death and response change over time
-
reductions in utility associated with side effects of treatment
-
impact on cost-effectiveness for people with different gene mutations.
The impact of making alternative assumptions about how probabilities for death and response change is unknown but it is anticipated that the assumption of constant probabilities over time will exaggerate estimated life expectancy (and hence QALYs and cost) for all pathways. The net impact on relative cost-effectiveness is unclear as it depends upon the magnitude of any changes in both costs and QALYs that might occur.
A further uncertainty is the probability of death for BSC. No studies for this comparator met the inclusion criteria for the review. The only sources available for this parameter were from studies published in the pre-imatinib era where the population could not have been exposed to a prior 400 mg/day dose of imatinib, and the proportion of the study populations with KIT-positive GIST was not known. With regard to the impact of this uncertainty on the economic model, it is reasonable to assume that if, for example, there was an increase in mortality for BSC, the costs and QALYs associated with each of the pathways would fall because BSC is included within each pathway.
The net impact of adjusting utility scores for side effects is also uncertain but it might be expected that it will reduce the QALYs associated with each medication and, although there are limited data available from the systematic review of effectiveness, this reduction may be greater for pathways involving sunitinib because its side effect profile is believed to be worse than that of imatinib.
A further factor not considered by the economic model was the cost-effectiveness of treating patients with specific gene mutations. Again this was not addressed owing to lack of data.
No studies looking at plasma monitoring met our inclusion criteria, but its potential, along with that of mutation testing, as an early predictor of the need for escalated imatinib dosing may have implications for both the costs and effects of escalated doses, because it may allow the identification of those people who are expected to respond better to escalated doses quickly and hence they may be given escalated doses immediately rather than waiting for progression to occur at the 400 mg/day dose. If either of these practices become widely adopted within the NHS then the evidence on the effect of imatinib dose escalation following progression at the standard 400 mg/day dose will become less relevant to clinical practice. Should mutation testing and plasma monitoring allow the tailoring of dose escalation then we might expect the benefits to those who receive therapy to be increased, particularly at earlier stages of treatment (although this also means that there may be fewer remaining treatment options following failure at the escalated dose). Costs would also increase owing to both the cost of mutation testing or plasma monitoring, and also the costs of escalated doses that are incurred earlier. The net impact of this on cost-effectiveness is unclear.
Finally, the economic evaluation has assumed that patients who move on to BSC still receive medication to prevent tumour flare. This has the impact of increasing the cost of BSC. It is further assumed that there is no impact on effectiveness (the implicit assumption is that discontinuing the medication would reduce life expectancy). Within the analysis it has been assumed that all patients on BSC or moving on to BSC after failing to respond to a medication would receive imatinib 400 mg/day. This assumption appears reasonable for Path-1, BSC only, but may not be appropriate for the other pathways where patients would move on to BSC after failing to respond to an escalated dose of imatinib, or on sunitinib. Should these patients continue with the last active medication that they received then costs, and incremental costs per QALY, would increase.
Chapter 8 Conclusions
Implications for service provision
-
There was very limited evidence available from very few studies on the effects of escalated doses of imatinib 600 mg/day and 800 mg/day or treatment with sunitinib for people with unresectable and/or metastatic GIST, whose disease had progressed on the 400 mg/day dose. The evidence that was available was essentially observational in nature and subject to the biases associated with such data, consisting mostly of reporting of subgroups of patients in RCTs that were not designed to assess the effects of dose escalation.
-
The limited evidence base suggests that around one-third of patients with unresectable and/or metastatic GIST who have failed on a dose of 400 mg/day may show response or SD with escalated doses of imatinib, and those who do respond may have a reasonable chance of maintaining this response over a longer period of time than would otherwise have been the case.
-
For all patients receiving either dose-escalated imatinib or sunitinib, the median OS, where reported, was < 2 years.
-
There is a need to interpret all results from the economic model with caution owing to the limitations of the evidence base. The results themselves indicate that should society’s threshold for willingness to pay be less than £25,000 per QALY a pathway of BSC only has the highest probability of being cost-effective. Between a threshold of £25,000 and £45,000 provision of an escalated dose of imatinib would be most likely to be cost-effective. Above a threshold of £45,000 a pathway of escalated doses of imatinib followed by sunitinib, if necessary, would be most likely to be cost-effective.
-
In terms of policy-making, the results of this review and economic model show that the current evidence available on the effectiveness of imatinib dose escalation for GIST patients following progression on the standard 400 mg/day dose is characterised by such a high degree of uncertainty that, in the authors’ opinion, it would be inappropriate to conclude that dose escalation of imatinib would be a cost-effective strategy for the NHS.
Recommendations for research
Further evidence is needed in order to provide a comprehensive assessment of the effectiveness and cost-effectiveness of the alternative treatments for patients with GIST who fail on or become resistant to imatinib 400 mg/day. Ideally, such data would come from RCTs involving patients who progress on 400 mg/day of imatinib, where patients are randomised to 600 mg/day imatinib, 800 mg/day imatinib or sunitinib, or to remain on 400 mg/day imatinib. However, such a study may be difficult to organise, as neither patients nor practitioners may be in equipoise. Dose escalation appears to be used within the NHS already and hence health-care professionals may not find it acceptable that their patients could be randomised to ‘BSC’. Therefore, alternative quasi-experimental or observational designs should be considered but with sufficient focus on understanding and controlling for selection biases.
The pathways most likely to be cost-effective at thresholds society might be willing to pay and hence potentially the most useful to assess would be dose escalation with imatinib and dose escalation with imatinib followed by sunitinib if necessary. Such studies should as a matter of course include an economic evaluation and measurement of health-state utilities (where there is currently a dearth of evidence for each of the relevant health states for GIST patients), and would need to measure outcomes over a sufficiently long time period to capture the main impact on costs and outcomes. Where possible further studies should also report outcomes for subgroups of patients with specific KIT mutations.
With respect to costs, should further comparative studies be conducted, estimates of the usage of health services might usefully be collected. A wider perspective on the consideration of costs might also be informative, for example costs that fall on PSS (which would be relevant for NICE to consider) and costs for patients and their families (which goes beyond NICE’s reference case).
Acknowledgements
We thank the study authors we contacted who provided additional details of their studies, Lara Kemp (Health Services Research Unit) for secretarial support, and Cathryn Rankin (Southwest Oncology Group) for providing additional data from the S0033 trial. The Health Services Research Unit, and the Health Economics Research Unit, Institute of Applied Health Sciences, University of Aberdeen, are both core funded by the Chief Scientist Office of the Scottish Government Health Directorates. The views expressed are those of the authors and not necessarily those of the funding bodies. Any errors are the responsibility of the authors.
Contributions of authors
Jenni Hislop (Research Fellow) and Pawana Sharma (Research Fellow) screened the search results for clinical effectiveness, assessed full-text studies for inclusion, and undertook data extraction and quality assessment. Jenni Hislop drafted the chapters of the report other than the background and cost-effectiveness chapters, and coordinated the review. Pawana Sharma contributed to the chapter on clinical effectiveness and the appendices. Graham Mowatt (Senior Research Fellow) and Luke Vale (Professor of Health Technology Assessment) commented on drafts. Zahidul Quayyum (Research Fellow) screened the search results on cost-effectiveness, undertook data extraction and quality assessment, drafted the chapter on cost-effectiveness and developed the economic model, supervised by Luke Vale. Russell Petty (Clinical Senior Lecturer in Medical Oncology) drafted the background chapter, and provided expert advice on clinical aspects of the review. David Jenkinson and Andrew Elders (Statisticians) contributed to the data analysis section of the assessment of clinical effectiveness and conducted the statistical analysis. Cynthia Fraser (Information Specialist) developed and ran the search strategies, obtained papers and formatted the references. All authors assisted in preparing the manuscript and commenting on drafts. Graham Mowatt is guarantor.
Disclaimers
The views expressed in this publication are those of the authors and not necessarily those of the HTA programme or the Department of Health.
References
- Kindblom LG, Remotti HE, Aldenborg F, Meis-Kindblom JM. Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol 1998;152:1259-69.
- Medeiros F, Corless CL, Duensing A, Hornick JL, Oliveira AM, Heinrich MC, et al. KIT-negative gastrointestinal stromal tumors: proof of concept and therapeutic implications. Am J Surg Pathol 2004;28:889-94.
- Miettinen M, Majidi M, Lasota J. Pathology and diagnostic criteria of gastrointestinal stromal tumors (GISTs): a review. Eur J Cancer 2002;38:S39-51.
- Corless CL, McGreevey L, Haley A, Town A, Heinrich MC. KIT mutations are common in incidental gastrointestinal stromal tumors one centimeter or less in size. Am J Pathol 2002;160:1567-72.
- Duensing A, Medeiros F, McConarty B, Joseph NE, Panigrahy D, Singer S, et al. Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumors (GISTs). Oncogene 2004;23:3999-4006.
- Heinrich MC, Rubin BP, Longley BJ, Fletcher JA. Biology and genetic aspects of gastrointestinal stromal tumors: KIT activation and cytogenetic alterations. Hum Pathol 2002;33:484-95.
- Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 2003;21:4342-9.
- Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998;279:577-80.
- Rubin BP, Fletcher JA, Fletcher CDM. Molecular insights into the histogenesis and pathogenesis of gastrointestinal stromal tumors. Int J Surg Pathol 2000;8:5-10.
- Rubin BP, Singer S, Tsao C, Duensing A, Lux ML, Ruiz R, et al. KIT activation is a ubiquitous feature of gastrointestinal stromal tumors. Cancer Res 2001;61:8118-21.
- Zsebo KM, Williams DA, Geissler EN, Broudy VC, Martin FH, Atkins HL, et al. Stem cell factor is encoded at the Sl locus of the mouse and is the ligand for the c-kit tyrosine kinase receptor. Cell 1990;63:213-24.
- Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003;299:708-10.
- Hirota S, Ohashi A, Nishida T, Isozaki K, Kinoshita K, Shinomura Y, et al. Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology 2003;125:660-7.
- Debiec-Rychter M, Sciot R, Le Cesne A, Schlemmer M, Hohenberger P, Van Oosterom AT, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer 2006;42:1093-103.
- Scottish Sarcoma Network . Guidelines for the Management of Gastrointestinal Stromal Tumours (GISTs) in Scotland 2009. www.ssn.scot.nhs.uk/protocolsandguidelines/Scottish%20GIST%20guidelines_NOV545_no% (accessed February 2010).
- Miettinen M, Wang ZF, Lasota J. DOG1 antibody in the differential diagnosis of gastrointestinal stromal tumors: a study of 1840 cases. Am J Surg Pathol 2009;33:1401-8.
- West RB, Corless CL, Chen X, Rubin BP, Subramanian S, Montgomery K, et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutation status. Am J Pathol 2004;165:107-13.
- Conlon KC, Casper ES, Brennan MF. Primary gastrointestinal sarcomas: analysis of prognostic variables. Ann Surg Oncol 1995;2:26-31.
- Dematteo RP, Lewis JJ, Leung D, Mudan SS, Woodruff JM, Brennan MF. Two hundred gastrointestinal stromal tumors: recurrence patterns and prognostic factors for survival. Ann Surg 2000;231:51-8.
- Graadt van Roggen JF, van Velthuysen ML, Hogendoorn PC. The histopathological differential diagnosis of gastrointestinal stromal tumours. J Clin Pathol 2001;54:96-102.
- Fletcher CD, Berman JJ, Corless C, Gorstein F, Lasota J, Longley BJ, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Hum Pathol 2002;33:459-65.
- Miettinen M, Sarlomo-Rikala M, Lasota J. Gastrointestinal stromal tumors: recent advances in understanding of their biology. Hum Pathol 1999;30:1213-20.
- Miettinen M, Lasota J. Gastrointestinal stromal tumors: definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Arch 2001;438:1-12.
- Dematteo RP. The GIST of targeted cancer therapy: a tumor (gastrointestinal stromal tumor), a mutated gene (c-kit), and a molecular inhibitor (STI571). Ann Surg Oncol 2002;9:831-9.
- Miettinen M, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol 2005;29:52-68.
- Nilsson B, Bumming P, Meis-Kindblom JM, Oden A, Dortok A, Gustavsson B, et al. Gastrointestinal stromal tumors: the incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era: a population-based study in western Sweden. Cancer 2005;103:821-9.
- Tryggvason G, Gislason HG, Magnusson MK, Jonasson JG. Gastrointestinal stromal tumors in Iceland, 1990–2003: the Icelandic GIST study, a population-based incidence and pathologic risk stratification study. Int J Cancer 2005;117:289-93.
- Reichardt P. Practical aspects of managing gastrointestinal stromal tumours. Monogr Gastronintest Stromal Tumours 2003;1:3-8.
- Lehnert T. Gastrointestinal sarcoma (GIST): a review of surgical management. Ann Chir Gynaecol 1998;87:297-305.
- Dematteo RP, Heinrich MC, El Rifai WM, Demetri G. Clinical management of gastrointestinal stromal tumors: before and after STI-571. Hum Pathol 2002;33:466-77.
- Judson I. A guideline for the management of gastrointestinal stromal tumour (GIST). Sarcoma 2002;6:83-7.
- Blay JY, Bonvalot S, Casali P, Choi H, Debiec-Richter M, Dei Tos AP, et al. Consensus meeting for the management of gastrointestinal stromal tumors. Report of the GIST Consensus Conference of 20–21 March 2004, under the auspices of ESMO. Ann Oncol 2005;16:566-78.
- Dematteo RP, Ballman KV, Antonescu CR, Maki RG, Pisters PW, Demetri GD, et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet 2009;373:1097-104.
- Committee for Medicinal Products for Human Use . Monthly Report. CHMP 174243 2009 2009. www.ema.europa.eu/pressoffice/chmp.htm (accessed June 2010).
- National Institute for Health and Clinical Excellence (NICE) . Imatinib for the Adjuvant Treatment of Gastrointestinal Stromal Tumours. Final Appraisal Determination 2010. http://guidance.nice.org.uk/TA/Wave20/79/FAD (accessed June 2010).
- Van den Abbeele AD, Badawi RD, Manola J, Morgan JA, Desai J, Kazanovicz A, et al. Effects of cessation of imatinib mesylate (IM) therapy in patients (pts) with IM-refractory gastrointestinal stromal tumors (GIST) as visualized by FDG-PET scanning. J Clin Oncol 2004;22.
- Eisenberg BL, Harris J, Blanke CD, Demetri GD, Heinrich MC, Watson JC, et al. Phase II trial of neoadjuvant/adjuvant imatinib mesylate (IM) for advanced primary and metastatic/recurrent operable gastrointestinal stromal tumor (GIST): early results of RTOG 0132/ACRIN 6665. J Surg Oncol 2009;99:42-7.
- Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 2002;347:472-80.
- Blanke CD, Demetri GD, von Mehren M, Heinrich MC, Eisenberg B, Fletcher JA, et al. Long-term results from a randomized phase II trial of standard versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol 2008;26:620-5.
- Van Oosterom AT, Judson I, Verweij J, Stroobants S, Donato dP, Dimitrijevic S, et al. Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: a phase I study. Lancet 2001;358:1421-3.
- Blanke CD, Rankin C, Demetri GD, Ryan CW, von Mehren M, Benjamin RS, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol 2008;26:626-32.
- Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet 2004;364:1127-34.
- Gramza AW, Corless CL, Heinrich MC. Resistance to tyrosine kinase inhibitors in gastrointestinal stromal tumors. Clin Cancer Res 2009;15:7510-18.
- Zalcberg JR, Verweij J, Casali PG, Le Cesne A, Reichardt P, Blay JY, et al. Outcome of patients with advanced gastro-intestinal stromal tumours crossing over to a daily imatinib dose of 800 mg after progression on 400 mg. Eur J Cancer 2005;41:1751-7.
- Blay JY, Le Cesne A, Ray-Coquard I, Bui B, Duffaud F, Delbaldo C, et al. Prospective multicentric randomized phase III study of imatinib in patients with advanced gastrointestinal stromal tumors comparing interruption versus continuation of treatment beyond 1 year: the French Sarcoma Group. J Clin Oncol 2007;25:1107-13.
- Choi H, Charnsangavej C, Faria SC, Macapinlac HA, Burgess MA, Patel SR, et al. Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteria. J Clin Oncol 2007;25:1753-9.
- Choi H. Response evaluation of gastrointestinal stromal tumors. Oncologist 2008;13:4-7.
- Gayed I, Vu T, Iyer R, Johnson M, Macapinlac H, Swanston N, et al. The role of 18F-FDG PET in staging and early prediction of response to therapy of recurrent gastrointestinal stromal tumors. J Nucl Med 2004;45:17-21.
- Van den Abbeele AD. The lessons of GIST: PET and PET/CT: a new paradigm for imaging. Oncologist 2008;13:8-13.
- National Institute for Health and Clinical Excellence (NICE) . NICE Guidance TA86 [document on the Internet]. Gastro-Intestinal Stromal Tumours (GIST) – Imatinib 2004. http://guidance.nice.org.uk/TA86/Guidance/pdf/English (accessed August 2009).
- National Institute for Health and Clinical Excellence (NICE) . Sunitinib for the Treatment of Gastrointestinal Stromal Tumours 2009. www.nice.org.uk/nicemedia/pdf/TA179Guidance.pdf (accessed February 2010).
- Demetri GD, Van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 2006;368:1329-38.
- Pfizer . Safety and Effectiveness of Daily Dosing With Sunitinib or Imatinib in Patients With Gastrointestinal Stromal Tumors 2006. http://clinicaltrials.gov/ (accessed February 2010).
- Reddy P. The epidemiologic, health-related quality of life, and economic burden of gastrointestinal stromal tumours. J Clin Pharm Ther 2007;32:557-65.
- Wilson J, Connock M, Song F, Yao G, Fry-Smith A, Raftery J, et al. Imatinib for the treatment of patients with unresectable and/or metastatic gastrointestinal stromal tumours: systematic review and economic evaluation. Health Technol Assess 2005;9.
- Association of Upper Gastrointestinal Surgeons of Great Britain and Ireland (AUGIS) . Guidelines for the Management of Gastrointestinal Stromal Tumours (GISTs) 2005. www.augis.org/news_guidelines/management_guidelines.htm (accessed February 2010).
- Scottish Medicines Consortium . Imatinib for GIST (Glivec®) 2002. www.scottishmedicines.org.uk/files/GlivecGIST_v2.pdf (accessed February 2010).
- O’Farrell AM, Abrams TJ, Yuen HA, Ngai TJ, Louie SG, Yee KW, et al. SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood 2003;101:3597-605.
- Prenen H, Cools J, Mentens N, Folens C, Sciot R, Schoffski P, et al. Efficacy of the kinase inhibitor SU11248 against gastrointestinal stromal tumor mutants refractory to imatinib mesylate. Clin Cancer Res 2006;12:2622-7.
- Pfizer . Single Technology Appraisal of Sunitinib for the Treatment of Gastrointestinal Stromal Tumours 2008. www.nice.org.uk/guidance/index.jsp?action=download%26o=43440 (accessed August 2009).
- Ahmed N, Ahmedzai S, Vora V, Hillam S, Paz S. Supportive care for patients with gastrointestinal cancer. Cochrane Database Syst Rev 2004;1. 10.1002/14651858.CD003445.pub2.
- Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol 2008;26:5352-9.
- Van Glabbeke M, Owzar K, Rankin C, Simes J, Crowley J. GIST Meta-analysis Group (MetaGIST) . Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumours (GIST): a meta-analysis based on 1640 patients. J Clin Oncol 2007;25.
- Demetri G, Wang Y, Wehre E, Blanke C, Joensuu H, von Mehren M. Correlation of Imatinib Plasma Levels With Clinical Benefit in Patients (Pts) With Unresectable Metastatic Gastrointestinal Stromal Tumors (GIST) n.d.
- Egorin MJ, Mauro MJ, Trent JC. Drug plasma monitoring in CML and GIST: a case-based discussion. Clin Adv Hematol Oncol 2009;7:S1-1.
- Widmer N, Decosterd LA, Leyvraz S, Duchosal MA, Rosselet A, Debiec-Rychter M, et al. Relationship of imatinib-free plasma levels and target genotype with efficacy and tolerability. Br J Cancer 2008;98:1633-40.
- Novartis . Glivec: Summary of Product Characteristics 2009. http://emc.medicines.org.uk/history/15014/SPC/GLIVEC+Tablets#03/06/2009%20to%20Current (accessed February 2010).
- Rankin C, von Mehren M, Blanke C, Benjamin R, Fletcher CDM, Bramwell V, et al. Dose effect of imatinib (IM) in patients (pts) with metastatic GIST: phase III Sarcoma Group Study S0033. J Clin Oncol 2004;22.
- Centre for Reviews and Dissemination (CRD) . Systematic Reviews: CRD’s Guidance for Undertaking Systematic Reviews in Health Care 2009. www.york.ac.uk/inst/crd/SysRev/!SSL!/WebHelp/SysRev3.htm (accessed January 2010).
- Verhagen AP, de Vet HC, de Bie RA, Kessels AG, Boers M, Bouter LM, et al. The Delphi list: a criteria list for quality assessment of randomized clinical trials for conducting systematic reviews developed by Delphi consensus. J Clin Epidemiol 1998;51:1235-41.
- Downs SH, Black N. The feasibility of creating a checklist for the assessment of the methodological quality both of randomised and non-randomised studies of health care interventions. J Epidemiol Community Health 1998;52:377-84.
- Higgins JP, Green S. Cochrane handbook for systematic reviews of interventions version 5.0.2. The Cochrane Collaboration; 2009.
- Parmar MK, Torri V, Stewary L. Extracting summary statistics to perform meta-analyses of the published literature for survival endpoints. Stat Med 1998;17:2815-34.
- Yusuf S, Peto R, Lewis J, Colins R, Sleight P. Beta blockade during and after myocardial infarction. An overview of randomized trials. Prog Cardiovasc Dis 1985;27:335-71.
- Droitcour J, Silberman G, Chelimsky E. Cross-design synthesis: a new form of meta-analysis for combining results from randomized clinical trials and medical-practice databases. Int J Technol Assess Health 1993;9:440-9.
- Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease. J Natl Cancer Inst 1959;22:719-48.
- Dileo P, Rankin CJ, Benjamin RS, von Mehren M, Blanke C, Bramwell V, et al. Incidence and reasons for dose modification of standard-dose vs. high-dose imatinib mesylate (IM) in the Phase III Intergroup Study S0033 of patients (pts) with unresectable or metastatic gastrointestinal stromal tumor (GIST). J Clin Oncol 2005;23.
- Zalcberg JR, Verweij J, Casali PG, Le Cesne A, Reichardt P, Blay JY, et al. Outcome of patients with advanced gastro-intestinal stromal tumors (GIST) crossing over to a daily imatinib dose of 800mg (HD) after progression on 400mg (LD): an international, intergroup study of the EORTC, ISG and AGITG. J Clin Oncol 2004;22.
- Park I, Ryu MH, Sym SJ, Lee SS, Jang G, Kim TW, et al. Dose escalation of imatinib after failure of standard dose in Korean patients with metastatic or unresectable gastrointestinal stromal tumor. Jpn J Clin Oncol 2009;39:105-10.
- Kang Y, Reichardt P, Ruka W, Seddon B, Baum C, Demetri G. Efficacy and safety of sunitinib in a worldwide treatment-use trial of GIST patients following imatinib failure. Ann Oncol 2007;18.
- Reichardt P, Kang Y, Ruka W, Seddon B, Nieto A, Breazna A, et al. Sunitinib (Su) in a worldwide treatment-use trial of patients with GIST: updated efficacy and safety analysis. Ann Oncol 2008;19.
- Reichardt P, Kang Y, Ruka W, Seddon B, Guerriero A, Breazna A, et al. Detailed analysis of survival and safety with sunitinib (SU) in a worldwide treatment-use trial of patients with advanced GIST. J Clin Oncol 2008;26.
- Rutkowski P, Reichardt P, Kang Y, Ruka W, Seddon B, Guerriero A, et al. Sunitinib in a worldwide treatment-use trial of patients with imatinib-resistant/intolerant gastrointestinal stromal tumor: detailed analysis of survival and safety. Ann Oncol 2008;19.
- Schutte J, Reichardt P, Schlemmer M, Wendtner CM, Demetri GD. Efficacy and safety of sunitinib in patients with gastrointestinal stromal tumour resistant or intolerant of prior imatinib therapy: results from a worldwide treatment-use study. Onkologie 2008;31.
- Seddon B, Reichardt P, Ruka W, Kang YK, Baum CM, Demetri GD. Safety and efficacy results of sunitinib from a worldwide treatment use trial of gastrointestinal stromal tumour (GIST) patients (pts) with resistance or intolerance to prior imatinib therapy. Eur J Cancer 2007;5.
- Seddon B, Reichardt P, Kang YK, Ruka W, Nieto A, Breazna A, et al. Detailed Anlaysis of Survival and Safety With Sunitinib in a Worldwide Treatment-Use Trial of Patients With Advanced Imatinib-Resistant Intolerant GIST n.d.
- An JY, Choi MG, Noh JH, Sohn TS, Kang WK, Park CK, et al. Gastric GIST: a single institutional retrospective experience with surgical treatment for primary disease. Eur J Surg Oncol 2007;33:1030-5.
- Chu TF, Rupnick MA, Kerkela R, Dallabrida SM, Zurakowski D, Nguyen L, et al. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet 2007;370:2011-19.
- Goerres GW, Stupp R, Barghouth G, Hany TF, Pestalozzi B, Dizendorf E, et al. The value of PET, CT and in-line PET/CT in patients with gastrointestinal stromal tumours: long-term outcome of treatment with imatinib mesylate. Eur J Nucl Med Molecul Imag 2005;32:153-62.
- Nishida T, Shirao K, Sawaki A, Koseki M, Okamura T, Ohtsu A, et al. Efficacy and safety profile of imatinib mesylate (ST1571) in Japanese patients with advanced gastrointestinal stromal tumors: a phase II study (STI571B1202). Int J Clin Oncol 2008;13:244-51.
- Phongkitkarun S, Phaisanphrukkun C, Jatchavala J, Sirachainan E. Assessment of gastrointestinal stromal tumors with computed tomography following treatment with imatinib mesylate. World J Gastroenterol 2008;14:892-8.
- Arends LR, Hunink MG, Stijnen T. Meta-analysis of summary survival curve data. Stat Med 2008;27:4381-96.
- Philips Z, Ginnelly L, Sculpher M, Claxton K, Golder S, Riemsma R, et al. Review of guidelines for good practice in decision-analytic modelling in health technology assessment. Health Technol Assess 2004;8.
- National Institute for Health and Clinical Excellence (NICE) . Guide to the Methods of Technology Appraisal 2008. www.nice.org.uk/media/B52/A7/TAMethodsGuideUpdatedJune2008.pdf (accessed February 2010).
- Chabot I, LeLorier J, Blackstein ME. The challenge of conducting pharmacoeconomic evaluations in oncology using crossover trials: the example of sunitinib for gastrointestinal stromal tumour. Eur J Cancer 2008;44:972-7.
- Contreras-Hernandez I, Mould-Quevedo JF, Silva A, Salinas-Escudero G, Villasis-Keever MA, Granados-Garcia V, et al. A pharmaco-economic analysis of second-line treatment with imatinib or sunitinib in patients with advanced gastrointestinal stromal tumours. Br J Cancer 2008;98:1762-8.
- Huse DM, von Mehren M, Lenhart G, Joensuu H, Blanke C, Feng W, et al. Cost effectiveness of imatinib mesylate in the treatment of advanced gastrointestinal stromal tumours. Clin Drug Invest 2007;27:85-93.
- Mabasa VH, Taylor SCM, Chu CCY, Moravan V, Johnston K, Peacock S, et al. Verification of imatinib cost-effectiveness in advanced gastrointestinal stromal tumor in British Columbia (VINCE-BC study). J Oncol Pharm Pract 2008;14:105-12.
- Paz-Ares L, Garcia del Muro X, Grande E, Gonzalez P, Brosa M, Diaz S. Cost-effectiveness analysis of sunitinib in patients with metastatic and/or unresectable gastrointestinal stroma tumours (GIST) after progression or intolerance with imatinib. Clin Trans Oncol 2008;10:831-9.
- Teich N, Hashizume C, Follador W. Economic evaluation of sunitinib vs. imatinib in second line for gastrointestinal tumor (GIST) in Brazil. Value Health 2009;12.
- Hopkins TG, Marples M, Stark D. Sunitinib in the management of gastrointestinal stromal tumours (GISTs). Eur J Surg Oncol 2008;34:844-50.
- Verweij J, Casali PG, Zalcberg J, Le Cesne A, Reichard P, Blay J, et al. Early Efficacy Comparison of Two Doses of Imatinib for the Treatment of Advanced Gastro-Intestinal Stromal Tumors (GIST): Interim Results of a Randomized Phase III Trial from the EORTC-STBSG, ISG and AGITG Abstract n.d.
- Blanke CD, Demetri GD, von Mehren M, Heinrich MC, Eisenberg BL, Fletcher J, et al. Long-term follow-up of a phase II randomized trial in advanced gastrointestinal stromal tumor (GIST) patients (pts) treated with imatinib mesylate. J Clin Oncol 2006;24.
- Motzer RJ, Hoosen S, Bello CL, Christensen JG. Sunitinib malate for the treatment of solid tumours: a review of current clinical data. Expert Opin Investig Drugs 2006;15:553-61.
- Ojeda B, de Sande LM, Casado A, Merino P, Casado MA. Cost-minimisation analysis of pegylated liposomal doxorubicin hydrochloride versus topotecan in the treatment of patients with recurrent epithelial ovarian cancer in Spain. Br J Cancer 2003;89:1002-7.
- McGrath PC, Neifeld JP, Lawrence W, Kay S, Horsley JS, Parker GA. Gastrointestinal sarcomas. Analysis of prognostic factors. Ann Surg 1987;206:706-10.
- Pierie JP, Choudry U, Muzikansky A, Yeap BY, Souba WW, Ott MJ. The effect of surgery and grade on outcome of gastrointestinal stromal tumors. Arch Surg 2001;136:383-9.
- Prior JO, Montemurro M, Orcurto MV, Michielin O, Luthi F, Benhattar J, et al. Early prediction of response to sunitinib after imatinib failure by 18F-fluorodeoxyglucose positron emission tomography in patients with gastrointestinal stromal tumor. J Clin Oncol 2009;27:439-45.
- British Medical Association and Royal Pharmaceutical Society of Great Britain . British National Formulary 2009.
- Curtis L. Unit costs of health and social care 2009. Canterbury: Personal Social Services Research Unit; 2009.
- Nilsson B, Nilsson O, Ahlman H. Treatment of gastrointestinal stromal tumours: imatinib, sunitinib: and then?. Expert Opin Investig Drugs 2009;18:457-68.
- Dougherty MJ, Compton C, Talbert M, Wood WC. Sarcomas of the gastrointestinal tract. Separation into favorable and unfavorable prognostic groups by mitotic count. Ann Surg 1991;214:569-74.
- Artinyan A, Kim J, Soriano P, Chow W, Bhatia S, Ellenhorn JD. Metastatic gastrointestinal stromal tumors in the era of imatinib: improved survival and elimination of socioeconomic survival disparities. Cancer Epidemiol Biomarkers Prev 2008;17:2194-201.
- Casali PG. Gastrointestinal stromal tumours: ESMO Clinical Recommendations for diagnosis, treatment and follow-up. Ann Oncol 2009;20:iv64-7.
- Demetri GD, Benjamin RS, Blanke CD, Blay JY, Casali P, Choi H, et al. NCCN Task Force report: management of patients with gastrointestinal stromal tumor (GIST): update of the NCCN clinical practice guidelines. J Natl Compr Cancer Netw 2007;5:S1-29.
- Oken MM, Creech RH, Tormey DC, Horton J, Davis TE, McFadden ET, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol 1982;5:649-55.
- Blanke CD, Joensuu H, Demetri GD, Heinrich MC, Eisenberg B, Fletcher J, et al. Outcome of advanced gastrointestinal stromal tumor (GIST) patients treated with imatinib mesylate: four year follow-up of a phase II randomized trial [abstract]. J Clin Oncol 2006;24.
- Maki R, Fletcher JA, Heinrich M, Morgan JA, George S, Desai J, et al. Results from a continuation trial of SU11248 in patients with imatinib (IM)-resistant gastrointestinal stromal tumor (GIST). J Clin Oncol 2005;23.
- George S, Blay J, Casali P, Le Cesne A, Morgan J, Pokela J, et al. Continuous daily dosing (CDD) of sunitinib malate (SU) compares favorably with intermittent dosing in patients with advanced GIST. J Clin Oncol 2007;25.
- Scottish Medicines Consortium . Sunitinib 50 mg Capsule (Sutent) No.275 06 2006. www.scottishmedicines.org.uk/SMC_Advice/Advice/Sunitinib__Sutent_/sunitinib_50mg_capsule__Sutent_.
- de Mestier P, des Guetz G. Treatment of gastrointestinal stromal tumors with imatinib mesylate: a major breakthrough in the understanding of tumor-specific molecular characteristics. World J Surg 2005;29:357-61.
- von Mehren M. Imatinib-refractory gastrointestinal stromal tumors: the clinical problem and therapeutic strategies. Curr Oncol Rep 2006;8:192-7.
- Plaat BE, Hollema H, Molenaar WM, Torn Broers GH, Pijpe J, Mastik MF, et al. Soft tissue leiomyosarcomas and malignant gastrointestinal stromal tumors: differences in clinical outcome and expression of multidrug resistance proteins. J Clin Oncol 2000;18:3211-20.
- Pidhorecky I, Cheney RT, Kraybill WG, Gibbs JF. Gastrointestinal stromal tumors: current diagnosis, biologic behavior, and management. Ann Surg Oncol 2000;7:705-12.
- Comandone A, Boglione A. Biology, diagnosis and therapeutic options in gastrointestinal stromal tumours. Minerva Chir 2005;60:197-203.
- Duffaud F, Blay JY. Gastrointestinal stromal tumors: biology and treatment. Oncology 2003;65:187-97.
- Cohen MH, Dagher R, Griebel DJ, Ibrahim A, Martin A, Scher NS, et al. US Food and Drug Administration drug approval summaries: imatinib mesylate, mesna tablets, and zoledronic acid. Oncologist 2002;7:393-400.
- Totman MB. STEER: Succinct and Timely Evaluated Evidence Reviews. Southampton: Wessex Institute for Health Research & Development, University of Southampton; 2002.
- Katz SC. Gastrointestinal stromal tumors and leiomyosarcomas. J Surg Oncol 2008;97:350-9.
- Trent JC, Beach J, Burgess MA, Papadopolous N, Chen LL, Benjamin RS, et al. A two-arm phase II study of temozolomide in patients with advanced gastrointestinal stromal tumors and other soft tissue sarcomas. Cancer 2003;98:2693-9.
- Le Cesne A, Van Glabbeke M, Verweij J, Casali PG, Findlay M, Reichardt P, et al. Absence of progression as assessed by response evaluation criteria in solid tumors predicts survival in advanced GI stromal tumors treated with imatinib mesylate: the intergroup EORTC-ISG-AGITG phase III trial. J Clin Oncol 2009;27:3969-74.
Appendix 1 Search strategies
MEDLINE (2000 – September, week 3, 2009), EMBASE (2000–9, week 39), MEDLINE In-Process (25 September 2009)
Ovid Multifile Search URL: https://shibboleth.ovid.com/
-
Gastrointestinal Stromal Tumors/use mesz
-
Gastrointestinal Stromal Tumor/use emez
-
gastrointestinal neoplasms/use mesz
-
exp digestive system tumor/use emez
-
gist.tw
-
((gastro$or gastric) adj3 stromal).tw.
-
(3 or 4) and (kit or cd117 or cd 117).tw.
-
(3 or 4) and (stromal or connective or mesenchymal).tw.
-
or/1-2,5-8
-
imatinib.tw,rn.
-
gleevec.tw,rn.
-
glivec.tw,rn.
-
(sti571 or sti 571).tw,rn.
-
or/10-13
-
sunitinib.tw,rn.
-
sutent.tw,rn.
-
(su11248 or su 11248).tw,rn
-
or/15-17
-
dt.fs
-
9 and 19
-
20 not (14 or 18)
-
Palliative Care/
-
((palliative or support$) adj3 (care or treatment)).tw.
-
(symptom$adj3 control$).tw.
-
or/21-24
-
9 and 14
-
9 and 18
-
9 and 25
-
or/26-28
-
exp clinical trial/
-
randomized controlled trial.pt.
-
controlled clinical trial.pt.
-
randomization/use emez
-
randomi?ed.ab.
-
placebo.ab.
-
drug therapy.fs.
-
randomly.ab.
-
trial.ab
-
groups.ab.
-
or/30-39
-
comparative study/use mesz
-
follow-up studies/use mesz
-
time factors/use mesz
-
Treatment outcome/use emez
-
major clinical study/use emez
-
controlled study/use emez
-
clinical trial/use emez
-
(preoperat$or pre operat$).mp. use mesz
-
(chang$or evaluat$or reviewed or baseline).tw
-
(prospective$or retrospective$).tw. use mesz
-
(cohort$or case series).tw. use mesz
-
(compare$or compara$).tw. use emez
-
or/41-52
-
29 and (40 or 53)
-
animals/not (humans/and animals/)
-
nonhuman/not (human/and nonhuman)
-
54 not (55 or 56)
-
remove duplicates from 57
-
limit 58 to yr=“2000 -Current”
SCI (2000 – 26 September 2009), BIOSIS (2000 – 24 September 2009), ISI Proceedings (2000 – 26 September 2009)
Web of Knowledge URL: http://wok.mimas.ac.uk/
-
#1 ts=gist
-
#2 ts=((gastric or gastro*) SAME stromal)
-
#3 ts=((gastric or gastro*) AND (KIT or cd117 or cd 117))
-
#4 ts=((gastic or gastro*) and mesenchymal)
-
#5 #1 OR #2 OR #3 OR #4
-
#6 ts=(imatinib or gleevec or glivec or sti571 or sti 571)
-
#7 #5 AND #6
-
#8 ts=(sunitinib or sutent or su11248 or su 11248)
-
#9 #5 AND #8
-
#10 ts=(palliative same (care or treatment))
-
#11 #5 AND #10
-
#12 ts=(support* SAME (care or treatment))
-
#13 #5 AND #12
-
#14 ts=(symptom* SAME control*)
-
#15 #5 AND #14
-
#16 #15 OR #13 OR #11 OR #9 OR #7
-
#17 #16 CPCI-S Timespan=2000–2009
CINAHL (September 2009)
EBSCOhost URL: http://web.ebscohost.com/
-
S1 (MH “Gastrointestinal Neoplasms+”)
-
S2 TX gastric or gastro*
-
S3 S1 OR S2
-
S4 TX (stromal or connective or mesenchymal)
-
S5 S3 and S4
-
S6 TX kit or cd117 or cd 117
-
S7 S3 and S6
-
S8 S5 or S7
-
S9 TX gist
-
S10 (S8 or S9)
-
S11 TX (imatinib or gleevec or glivec or sti571 or sti 571)
-
S12 S10 and S11
-
S13 TX (sunitinib or sutent or su11248 or su 11248)
-
S14 S10 and S13
-
S15 (MH “Palliative Care”)
-
S16 (MH “Hospice and Palliative Nursing
-
S17 TX (palliative N3 care) OR (palliative N3 treatment)
-
S18 TX (support* N3 care) OR (support* N3 treatment)
-
S19 TX (symptom* N3 control*)
-
S20 (S15 or S16 or S17 or S18 or S19)
-
S21 S10 and S20
-
S22 S12 OR S14 OR S21
Cochrane Library Issue 3, 2009 [Cochrane Central Register of Controlled Trials (CENTRAL) and CDSR]
URL: www3.interscience.wiley.com/cgi-bin/mrwhome/106568753/HOME
-
#1 MeSH descriptor Gastrointestinal Stromal Tumors, this term only
-
#2 (gist)
-
#3 (gastric or gastro*) NEAR/3 stromal
-
#4 MeSH descriptor Gastrointestinal Neoplasms explode all trees
-
#5 (kit or cd117 or cd 117) or (stromal or connective or mesenchymal)
-
#6 (#4 AND #5)
-
#7 (#1 OR #2 OR #3 OR #6)
-
#8 (imatinib or gleevec or glivec or sti571 or sti 571) or (sunitinib or sutent or su11248 or su 11248)
-
#9 (#7 AND #8)
-
#10 Any MeSH descriptor with qualifier: DT
-
#11 (#7 AND #10)
-
#12 MeSH descriptor Palliative Care, this term only
-
#13 (symptom* NEAR/3 control*) or (palliative NEAR/3 (care or treatment)) or (support* NEAR/3 (care or treatment))
-
#14 (#7 AND (#12 OR #13))
-
#15 (#9 OR #11 OR #14)
DARE and HTA Databases (October 2009)
NHS Centre for Reviews and Dissemination URL: http://nhscrd.york.ac.uk/welcome.htm
-
# 1 MeSH Gastrointestinal Stromal Tumors EXPLODE 1 2 3
-
# 2 gist
-
# 3 (gastric OR gastro*) AND (kit OR cd117 OR cd AND 117)
-
# 4 (gastric OR gastro*) AND (stromal OR connective OR mesenchymal)
-
# 5 #1 or #2 or #3 or #4
-
# 6 (imatinib OR gleevec OR glivec OR sti571 OR sti AND 571)
-
# 7 #5 and #6
-
# 8 (sunitinib OR sutent OR su11248 OR su AND 11248)
-
# 9 #5 and #8
-
# 10 MeSH Palliative Care EXPLODE 1 2
-
# 11 palliative
-
# 12 #5 and (#10 or #11)
-
# 13 #7 or #9 or #12
Health Management Information Consortium (September 2009)
Ovid Multifile Search URL: http://gateway.ovid.com/athens
-
gist.tw.
-
((gastro$or gastric$) adj3 stromal).tw.
-
gastrointestinal cancer/94
-
3 and (kit or CD117 or cd 117).tw.
-
3 and (stromal or connective or mesenchymal).tw.
-
or/1–2,4–5
Clinical Trials (September 2009)
URL: http://clinicaltrials.gov/ct/gui/c/r
“GIST”:Topic
CCT (September 2009)
URL: www.controlled-trials.com/
Gastro% stromal OR GIST
WHO International Clinical Trials Registry Platform (ICTRP) (September 2009)
URL: www.who.int/ictrp/en/
Gastro% stromal OR GIST
Clinical Study Results Database (September 2009)
Association of the British Pharmaceutical Industry (ABPI) (September 2009)
URL: www.cmrinteract.com/clintrial
Sutent or gleevec or glivec
International Federation of Pharmaceutical Manufacturers & Associations (IFPMA) (September 2009)
URL: http://clinicaltrials.ifpma.org
Sutent or gleevec or glivec
Conference proceedings
American Society of Clinical Oncology
Annual Meeting, Chicago, 1–5 June 2007.
Annual Meeting, Chicago, 30 May to 3 June 2008.
Annual Meeting, Orlando, 29 May to 2 June 2009.
European Society for Medical Oncology
9th World Congress on Gastrointestinal Cancer, Barcelona, 28 June to 1 July 2007.
10th World Congress on Gastrointestinal Cancer, Barcelona, 25–28 June 2008.
33rd Congress, Stockholm, 12–16 September 2008.
European Cancer Organisation
ECCO 14: European Cancer Conference, Barcelona, 22–27 September 2007.
ECCO 15: European Cancer Conference, Berlin, 24–29 September 2009.
Appendix 2 Full-paper screening tool
Appendix 3 Data extraction form
Appendix 4 Quality assessment tool
| Criteria | Yes | No | Unclear | Comments |
|---|---|---|---|---|
| Participants: sample definition and selection | ||||
| 1. Were participants a representative sample selected from a relevant patient population? | ||||
| 2. Were the inclusion/exclusion criteria of participants clearly described? | ||||
| 3. Were participants entering the study at a similar point in their disease progression? | ||||
| 4. Was selection of patients consecutive? | ||||
| 5. Was data collection undertaken prospectively? | ||||
| 6. Were the groups comparable on demographic characteristics and clinical features? | ||||
| Intervention | ||||
| 7. Was the intervention (and comparison) clearly defined? | ||||
| 8. Was the intervention undertaken by someone experienced at performing the procedure? | ||||
| 9. Were the staff, place and facilities where the patients were treated appropriate for performing the procedure? (e.g. access to back-up facilities) | ||||
| Outcome measures | ||||
| 10. Were all the important outcomes considered? | ||||
| 11. Were objective (valid and reliable) outcome measure/s used? | ||||
| 12. Was the assessment of main outcomes blind? | ||||
| Follow-up | ||||
| 13. Was follow-up long enough to detect important effects on outcomes of interest? | ||||
| 14. Was information provided on non-respondents, dropouts? | ||||
| 15. Were participants lost to follow-up likely to introduce bias? (e.g. high drop-out rate; differential dropout; no description of those lost) | ||||
| 16. Was length of follow-up similar between comparison groups? | ||||
| Analysis | ||||
| 17. Were important prognostic factors identified? | ||||
| 18. Were the analyses adjusted for confounding factors? | ||||
| Quality criteria | Yes | No | Unclear | Comments |
|---|---|---|---|---|
1. Was the allocation sequence adequately generated? (RevMan5, selection bias)
|
||||
2 Was allocation adequately concealed? (quality of random allocation concealment)
|
Appendix 5 Information on the reasons for exclusion
Resectable GIST (n = 24)
Agaram NP, Besmer P, Wong GC, Guo T, Socci ND, Maki RG, et al. Pathologic and molecular heterogeneity in imatinib-stable or imatinib-responsive gastrointestinal stromal tumors. Clin Cancer Res 2007;13:170–81.
Antonescu CR, Besmer P, Guo T, Arkun K, Hom G, Koryotowski B, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res 2005;11:4182–90.
Aparicio T, Boige V, Sabourin JC, Crenn P, Ducreux M, Le Cesne A, et al. Prognostic factors after surgery of primary resectable gastrointestinal stromal tumours. Eur J Surg Oncol 2004;30:1098–103.
Besana-Ciani I. Outcome and long term results of surgical resection for gastrointestinal stromal tumors (GIST). Scand J Surg 2003;92:195–9.
Bolukbasi H, Nazli O, Tansug T, Bozdag AD, Isgiider AS, Yaman I, et al. Gastrointestinal stromal tumors (GISTs): analysis of 20 cases. Hepatogastroenterology 2006;53:385–8.
Bucher P, Villiger P, Egger JF, Buhler L, Morel P. Results of primary surgical treatment of gastrointestinal stromal tumors. Gastroenterology 2004;126(Suppl. 2):58.
Date RS, Stylianides NA, Pursnani KG, Ward JB, Mughal MM. Management of gastrointestinal stromal tumours in the imatinib era: a surgeon’s perspective. World J Surg Oncol 2008;6:77–81.
Goh B, Chow P, Chuah K, Yap W, Foo K, Wong W. Pathological, radiological and PET scan response of gastrointestinal stromal tumors after neoadjuvant treatment with imatinib mesylate: a review of 37 cases. Ann Oncol 2006;17(Suppl. 6):101–2.
Gupta M, Sheppard BC, Corless CL, MacDonell KR, Blanke CD, Billingsley KG. Outcome following surgical therapy for gastrointestinal stromal tumors. J Gastrointest Surg 2006;10:1099–105.
Hou YY, Grabellus F, Weber F, Zhou Y, Tan YS, Li J, et al. Impact of KIT and PDGFRA gene mutations on prognosis of patients with gastrointestinal stromal tumors after complete primary tumor resection. J Gastrointest Surg 2009;13:1583–92.
Jindal S, Singhal D, Kakodkar R, Gupta S, Soin A, Chaudhary A, et al. Management of gastrointestinal stromal tumors: a single center experience in the pre-imatinib era. J Gastroenterol Hepatol 2008;23:A162–3.
McAuliffe JC, Hunt KK, Lazar AJ, Choi H, Qiao W, Thall P, et al. A randomized, phase II study of preoperative plus postoperative imatinib in GIST: evidence of rapid radiographic response and temporal induction of tumor cell apoptosis. Ann Surg Oncol 2009;16:910–19.
Machado-Aranda D, Malamet M, Chang YJ, Jacobs MJ, Ferguson L, Silapaswan S, et al. Prevalence and management of gastrointestinal stromal tumors. Am Surgeon 2009;75:55–60.
Mudan SS, Conlon KC, Woodruff JM, Lewis JJ, Brennan MF. Salvage surgery for patients with recurrent gastrointestinal sarcoma: prognostic factors to guide patient selection. Cancer 2000;88:66–74.
Nieto EA. Gastrointestinal stromal tumors: experience in 49 patients. Clin Translat Oncol 2006;8:594–8.
Nikfarjam M, Kimchi E, Shereef S, Gusani NJ, Jiang Y, Liang J, et al. Surgical outcomes of patients with gastrointestinal stromal tumors in the era of targeted drug therapy. J Gastrointest Surg 2008;12:2023–31.
Nilsson B, Bumming P, Meis-Kindblom JM, Oden A, Dortok A, Gustavsson B, et al. Gastrointestinal stromal tumors: the incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era: a population-based study in western Sweden. Cancer 2005;103:821–9.
Nishida T. Phase II trial of adjuvant imatinib mesylate after resection of localized primary high risk GIST. Eur J Cancer 2009;7:S594.
Nunobe S, Sano T, Shimada K, Sakamoto Y, Kosuge T. Surgery including liver resection for metastatic gastrointestinal stromal tumors or gastrointestinal leiomyosarcomas. Jpn J Clin Oncol 2005;35:338–41.
Perez EA, Gutierrez JC, Jin X, Lee DJ, Rocha-Lima C, Livingstone AS, et al. Surgical outcomes of gastrointestinal sarcoma including gastrointestinal stromal tumors: a population-based examination. J Gastrointest Surg 2007;11:114–25.
Rutkowski P, Nowecki Z, Nyckowski P, Dziewirski W, Grzesiakowska U, Nasierowska-Guttmejer A, et al. Surgical treatment of patients with initially inoperable and/or metastatic gastrointestinal stromal tumors (GIST) during therapy with imatinib mesylate. J Surg Oncol 2006;93:304–11.
Rutkowski P, Nowecki ZI, Michej W, Debiec-Rychter M, Wozniak A, Limon J, et al. Risk criteria and prognostic factors for predicting recurrences after resection of primary gastrointestinal stromal tumor. Ann Surg Oncol 2007;14:2018–27.
Sujendran V, Fearnhead N, De Pennington N, Warren BF, Maynard ND. Proposals for the management of gastrointestinal stromal tumours of the stomach. Surgeon 2007;5:149–53.
Zhan WH. Efficacy and safety of adjuvant post-surgical therapy with imatinib. J Clin Oncol 2007;25(18S):Abstract 10045.
Outcomes not reported separately for patients with GIST (n = 10)
Berman E, Nicolaides M, Maki RG, Fleisher M, Chanel S, Scheu K, et al. Altered bone and mineral metabolism in patients receiving imatinib mesylate. N Engl J Med 2006;354:2006–13.
Billemont B. Scrotal cutaneous side effects of sunitinib. N Engl J Med 2008;359:975–6.
Chugh R, Wathen JK, Maki RG, Benjamin RS, Patel SR, Myers PA, et al. Phase II multicenter trial of imatinib in 10 histologic subtypes of sarcoma using a Bayesian hierarchical statistical model. J Clin Oncol 2009;27:3148–53.
Fraunfelder FW, Solomon J, Druker BJ, Esmaeli B, Kuyl J. Ocular side-effects associated with imatinib mesylate [Gleevec (R)]. J Ocul Pharmacol Ther 2003;19:371–5.
Heinrich MC, Joensuu H, Demetri GD, Corless CL, Apperley J, Fletcher JA, et al. Phase II, open-label study evaluating the activity of imatinib in treating life-threatening malignancies known to be associated with imatinib-sensitive tyrosine kinases. Clin Cancer Res 2008;14:2717–25.
Kaneko T, Goto S, Kushima Y, Miyamoto Y, Eriguchi M, Nieda M, et al. A report of three patients treated with immunocell therapy with imatinib mesylate. Anticancer Res 2004;24:3303–9.
Oudard S, Negrier S, Ferrero JM, Gravis G, Blay JY, Le Cesne A, et al. Safety profile of single-agent sunitinib malate from the French Temporary Authorization for Use program (Cohort ATU) in metastatic renal cell carcinoma (MRCC) after failure of treatment with cytokines and gastrointestinal stromal tumor (GIST) patients after failure of imatinib mesylate treatment. Eur J Cancer 2007;5:S4526.
Polyzos A. Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma and various other solid tumors. J Steroid Biochem Mol Biol 2008;108:261–6.
Radaelli F, Vener C, Ripamonti F, Iurlo A, Colombi M, Artoni A, et al. Conjunctival hemorrhagic events associated with imatinib mesylate. Int J Hematol 2007;86:390–3.
Wong E, Rosen LS, Mulay M, Vanvugt A, Dinolfo M, Tomoda C, et al. Sunitinib induces hypothyroidism in advanced cancer patients and may inhibit thyroid peroxidase activity. Thyroid 2007;17:351–5.
Fewer than 10 patients in study population (n = 46)
Armbrust T. Does imatinib turn recurrent and/or metastasized gastrointestinal stromal tumors into a chronic disease? – Single center experience. Eur J Gastroenterol Hepatol 2009;21:819–23.
Banzo I, Quirce R, Martinez-Rodriguez I, Jimenez-Bonilla J, Sainz-Esteban A, Barragan J, et al. 18F FDG PET/CT in patients with gastrointestinal stromal tumors treated with imatinib mesylate. Eur J Nucl Med Mol Imaging 2007;34(Suppl. 2):298.
Bertolini V, Chiaravalli AM, Klersy C, Placidi C, Marchet S, Boni L, et al. Gastrointestinal stromal tumors – frequency, malignancy, and new prognostic factors: the experience of a single institution. Path Res Pract 2008;204:219–33.
Britten CD, Kabbinavar F, Hecht JR, Bello CL, Li J, Baum C, et al. A phase I and pharmacokinetic study of sunitinib administered daily for 2 weeks, followed by a 1-week off period. Cancer Chemother Pharmacol 2008;61:515–24.
Bui BN. Trough imatinib plasma levels in patients treated for advanced gastrintestinal stromal tumors evidence of large interpatient variations under treatment with standard doses. J Clin Oncol 2008;26:Abstract 10564.
Bulusu VR, Basu BJ, Hatcher H, Parkinson C, Sherbourne K, Earl HM, et al. Imatinib mesylate (IM) in locally advanced and metastatic gastrointestinal stromal tumours (GISTs): Cambridge GIST study group experience. Ann Oncol 2005;16(Suppl. 2):311.
Bulusu V, Fawcett S, Cook N, Hatcher H, Moyle P, Carroll N, et al. Size does not matter! Patterns of response and progression in patients (PTS) with metastatic gastrointestinal stromal tumours (GISTS) on imatinib mesylate (IM). Ann Oncol 2006;17(Suppl. 6):100.
Casali P, Fumagalli E, Bello A, George S. Safety and tolerability of sunitinib (SU) initiated 24 h after the last dose of imatinib (IM) in advanced GIST. J Clin Oncol 2008;26(Suppl. 5):Abstract 10557.
Chen C-W. Surgical management and clinical outcome of gastro-intestinal stromal tumor of the colon and rectum. Z Gastroenterol 2008;46:760–5.
Chen MY, Bechtold RE, Savage PD. Cystic changes in hepatic metastases from gastrointestinal stromal tumors (GISTs) treated with Gleevec (imatinib mesylate). Am J Roentgenol 2002;179:1059–62.
Chen TW, Liu HD, Shyu RY, Yu JC, Shih ML, Chang TM, et al. Giant malignant gastrointestinal stromal tumors: recurrence and effects of treatment with STI-571. World J Gastroenterol 2005;11:260–3.
Cipolla C, Fulfaro F, Sandonato L, Fricano S, Pantuso G, Grassi N, et al. Clinical presentation and treatment of gastrointestinal stromal tumors. Tumori 2006;92:279–84.
De Giorgi U, Aliberti C, Benea G, Conti M, Marangolo M. Effect of angiosonography to monitor response during imatinib treatment in patients with metastatic gastrointestinal stromal tumors. Clin Cancer Res 2005;11:6171–6.
Gibbons J, Egorin MJ, Ramanathan RK, Fu PF, Mulkerin DL, Shibata S, et al. Phase I and pharmacokinetic study of imatinib mesylate in patients with advanced malignancies and varying degrees of renal dysfunction: a study by the National Cancer Institute Organ Dysfunction Working Group. J Clin Oncol 2008;26:570–6.
Gronchi A, Fiore M, Miselli F, Lagonigro MS, Coco P, Messina A, et al. Surgery of residual disease following molecular-targeted therapy with imatinib mesylate in advanced/metastatic GIST. Ann Surg 2007;245:341–6.
Harney J, Wong WI, Short S. A preliminary evaluation of the potential role of FDG-PET to assess response to Glivec (TM) in gastrointestinal stromal tumours (GISTs). Br J Cancer 2003;88(Suppl. 1):S41.
Hasegawa J, Kanda T, Hirota S, Fukuda M, Nishitani A, Takahashi T, et al. Surgical interventions for focal progression of advanced gastrointestinal stromal tumors during imatinib therapy. Int J Clin Oncol 2007;12:212–17.
Hassan I, You YN, Dozois EJ, Shayyan R, Smyrk TC, Okuno SH, et al. Clinical, pathologic, and immunohistochemical characteristics of gastrointestinal stromal tumors of the colon and rectum: implications for surgical management and adjuvant therapies. Dis Colon Rectum 2006;49:609–15.
Hsiao HH, Liu YC, Tsai HJ, Chen LT, Lee CP, Chuan CH, et al. Imatinib mesylate therapy in advanced gastrointestinal stromal tumors: experience from a single institute. Kaohsiung J Med Sci 2006;22:599–603.
Kasper B, Kallinowski B, Herrmann T, Lehnert T, Mechtersheimer G, Geer T, et al. Treatment of gastrointestinal stromal tumor with imatinib mesylate: a retrospective single-center experience in Heidelberg. Dig Dis 2006;24:207–11.
Lasota J, vel Dobosz AJ, Wasag B, Wozniak A, Kraszewska E, Michej W, et al. Presence of homozygous KIT exon 11 mutations is strongly associated with malignant clinical behavior in gastrointestinal stromal tumors. Lab Invest 2007;87:1029–41.
Maehara N, Chijiiwa K, Eto T, Funagayama M, Uchiyama S, Nakashima S, et al. Surgical treatment for gastric GIST with special reference to liver metastases. Hepatogastroenterology 2008;55:512–16.
Mearadji A, den Bakker MA, van Geel AN, Eggermont AM, Sleijfer S, Verweij J, et al. Decrease of CD117 expression as possible prognostic marker for recurrence in the resected specimen after imatinib treatment in patients with initially unresectable gastrointestinal stromal tumors: a clinicopathological analysis. Anticancer Drugs 2008;19:607–12.
Mussi C, Schildhaus HU, Gronchi A, Wardelmann E, Hohenberger P. Therapeutic consequences from molecular biology for gastrointestinal stromal tumor patients affected by neurofibromatosis type 1. Clin Cancer Res 2008;14:4550–5.
Neagu S, Costea R, Sajin M, Ardelean C, Zarnescu NO. Gastric stromal tumors: histopathological, clinical and therapeutical aspects. Proceedings of the 8th International Gastric Cancer Congress, Krakow, Poland, 10–13 June 2009, pp. 85–7.
Nishida T, Kanda T, Nishitani A, Takahashi T, Nakajima K, Ishikawa T, et al. Secondary mutations in the kinase domain of the KIT gene are predominant in imatinib-resistant gastrointestinal stromal tumor. Cancer Sci 2008;99:799–804.
Nishida T, Takahashi T, Nishitani A, Doi T, Shirao K, Komatsu Y, et al. Sunitinib-resistant gastrointestinal stromal tumors harbor cis-mutations in the activation loop of the KIT gene. Int J Clin Oncol 2009;14:143–9.
Ohnishi K, Sakai F, Kudoh S, Ohno R. Twenty-seven cases of drug-induced interstitial lung disease associated with imatinib mesylate. Leukemia 2006;20:1162–4.
Petricevic B, Vrbanec D, Belev B, Plestina S, Herceg D, Dedic-Plavetic N. Imatinib mesylate in treatment of advanced gastrointestinal stromal tumours. Ann Oncol 2005;16(Suppl. 2):313.
Prakash S, Sarran L, Socci N, Dematteo RP, Eisenstat J, Greco AM, et al. Gastrointestinal stromal tumors in children and young adults: a clinicopathologic, molecular, and genomic study of 15 cases and review of the literature. J Pediatr Hematol Oncol 2005;27:179–87.
Raut C, Van den Abbeele A, Ramaiya N, Morgan J, George S, Quigley M, et al. PET imaging demonstrates two patterns of response in GIST patients benefiting from long-term sunitinib therapy. Ann Oncol 2006;17(Suppl. 6):103.
Richter KK, Schmid C, Thompson-Fawcett M, Settmacher U, Altendorf-Hofmann A. Long-term follow-up in 54 surgically treated patients with gastrointestinal stromal tumours. Langenbecks Arch Surg 2008;393:949–55.
Rutkowski P, Nyckowski P, Grzesiakowska U, Nowecki ZI, Nasierowska-Guttmejer A, Pienkowski A, et al. The clinical characteristics and the role of surgery and imatinib treatment in patients with liver metastases from c-Kit positive gastrointestinal stromal tumors (GIST). Neoplasma 2003;50:438–42.
Salazar LIF, Gago TA, Rubiales AS, Jimenez BV, de la Fuente RA, Hernandez JMG. Gastrointestinal stromal tumors (GIST): clinical aspects. Rev Esp Enferm Dig 2007;99:19–24.
Scaife CL, Hunt KK, Patel SR, Benjamin RS, Burgess MA, Chen LL, et al. Is there a role for surgery in patients with ‘unresectable’ cKIT+ gastrointestinal stromal tumors treated with imatinib mesylate? Am J Surg 2003;186:665–9.
Schindler CG, Armbrust T, Gunawan B, Langer C, Fuzesi L, Ramadori G. Gastrointestinal stromal tumor (GIST): single center experience of prolonged treatment with imatinib. Z Gastroenterol 2005;43:267–73.
Sym SJ, Ryu MH, Lee JL, Chang HM, Kim TW, Kim HC, et al. Surgical intervention following imatinib treatment in patients with advanced gastrointestinal stromal tumors (GISTs). J Surg Oncol 2008;98:27–33.
Tham C-K. Gastrointestinal stromal tumour in the elderly. Crit Rev Oncol Hematol 2009;70:256–61.
Tsukuda K, Hirai R, Miyake T, Takagi S, Ikeda E, Kunitomo T, et al. The outcome of gastrointestinal stromal tumors (GISTs) after a surgical resection in our institute. Surg Today 2007;37:953–7.
Van Oosterom AT, Judson I, Verweij J, Stroobants S, Donato dP, Dimitrijevic S, et al. Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: a phase I study. Lancet 2001;358:1421–3.
Wang CM, Fu H, Zhao GF, Zhou XY, Du CY, Dong RZ, et al. Secondary resistance to imatinib in patients with gastrointestinal stromal tumors through an acquired KIT exon 17 mutation. Mol Med Report 2009;2:455–60.
Warakaulle DR, Gleeson F. MDCT appearance of gastrointestinal stromal tumors after therapy with imatinib mesylate. Am J Roentgenol 2006;186:510–15.
Wolter P, Stefan C, Decallonne B, Dumez H, Bex M, Carmeliet P, et al. The clinical implications of sunitinib-induced hypothyroidism: a prospective evaluation. Br J Cancer 2008;99:448–54.
Wong DW, Lupton SC, Bhatt L, Gross L, Taniere P, Peake DR, et al. Use of imatinib mesylate in gastrointestinal stromal tumours: Pan-Birmingham Cancer Network experience. Clin Oncol 2008;20:517–22.
Wu A-W. Gastrointestinal stromal tumors of the anorectum – a special entity: GISTs of the anorectum. Chinese J Cancer Res 2006;18:38–44.
Zhu J, Wang Y, Hou M, Li HY, Zhang J. Imatinib mesylate treatment for advanced gastrointestinal stromal tumor: a pilot study focusing on patients experiencing sole liver metastasis after a prior radical resection. Oncology 2007;73:324–7.
400 mg/day imatinib dose only (n = 13)
Blay JY, Le Cesne A, Ray-Coquard I, Bui B, Duffaud F, Delbaldo C, et al. Prospective multicentric randomized phase III study of imatinib in patients with advanced gastrointestinal stromal tumors comparing interruption versus continuation of treatment beyond 1 year: the French Sarcoma Group. J Clin Oncol 2007;25:1107–13.
Bumming P, Andersson J, Meis-Kindblom JM, Klingenstierna H, Engstrom K, Stierner U, et al. Neoadjuvant, adjuvant and palliative treatment of gastrointestinal stromal tumours (GIST) with imatinib: a centre-based study of 17 patients. Br J Cancer 2003;89:460–4.
Choi H, Charnsangavej C, de Castro FS, Tamm EP, Benjamin RS, Johnson MM, et al. CT evaluation of the response of gastrointestinal stromal tumors after imatinib mesylate treatment: a quantitative analysis correlated with FDG PET finding. Am J Roentgenol 2004;183:1619–28.
Delbaldo C, Chatelut E, Re M, Deroussent A, Jambu A, Mackrodt A, et al. Inflammatory response affects the pharmacokinetics (PK) and pharmacodynamics (PD) of imatinib and CGP 74588 in patients with advanced gastrointestinal sarcoma (GIST). J Clin Oncol 2004;22:2070.
Dematteo RP, Antonescu CR, Chadaram V, You YN, McCall L, Maki R, et al. Adjuvant imatinib mesylate in patients with primary high risk gastrointestinal stromal tumor (GIST) following complete resection: safety results from the US Intergroup Phase II trial ACOSOG Z9000. J Clin Oncol 2005;23:S818.
Kim TW, Ryu MH, Lee H, Sym SJ, Lee JL, Chang HM, et al. Kinase mutations and efficacy of imatinib in Korean patients with advanced gastrointestinal stromal tumors. Oncologist 2009;14:540–7.
Le Cesne A, Rav-Coquard I, Bul B, Duffaud F, Deligny N, Cupissol D, et al. Interruption of imatinib (IM) in responding patients after one year treatment does not influence overall survival of patients with advanced GIST: updated results of the French Sarcoma Group randomized phase III BFR14 trial. Eur J Cancer 2005;3:S202.
Le Cesne A, Perol D, Ray-Coquard I, Bui B, Duffaud F, Rios M, et al. Interruption of imatinib (IM) in GIST patients with advanced disease: updated results of the prospective French Sarcoma Group randomized phase III trial on survival and quality of life. J Clin Oncol 2005;23:823S.
Steinert DM, Oyarzo M, Wang X, Choi H, Thall PF, Medeiros LJ, et al. Expression of Bcl-2 in gastrointestinal stromal tumors: correlation with progression-free survival in 81 patients treated with imatinib mesylate. Cancer 2006;106:1617–23.
Van den Abbeele AD, Badawi RD, Tetrault RJ, Cliche JP, Manola J, Spangler T, et al. FDG-PET as a surrogate marker for response to Gleevec (TM) (imatinib mesylate) in patients with advanced gastrointestinal stromal tumors (GIST). J Nucl Med 2003;44:77.
Widmer N, Decosterd LA, Leyvraz S, Duchosal MA, Rosselet A, Debiec-Rychter M, et al. Relationship of imatinib-free plasma levels and target genotype with efficacy and tolerability. Br J Cancer 2008;98:1633–40.
Yeh CN, Chen TW, Liu FY, Jan YY, Chen MF. Genetic changes in advanced gastrointestinal stromal tumor (GIST) patients during imatinib mesylate treatment. Langenbecks Arch Surg 2006;391:615–21.
Yeh CN, Chen TW, Wu TJ, Hsueh S, Jan YY. Treatment of patients with advanced gastrointestinal stromal tumor of small bowel: implications of imatinib mesylate. World J Gastroenterol 2006;12:3760–5.
No/insufficient data for escalated dose patients (n = 65)
Aliberti S, Grignani G, Allione P, Vormola R, Bucci AS, Porrino G, et al. Erythrocyte macrocytosis is a rather common, apparently uneventful yet unexplained finding in GIST imatinib (I) chronic therapy. J Clin Oncol 2005;23:S827.
Allione P, Grignani G, Aliberti S, Scalabrini DR, Schianca FC, Capaldi A, et al. Increase of creatine kinase value (CK) correlates with muscoloskeletal complaints (MSC) in GIST patients during imatinib therapy. J Clin Oncol 2006;24:9509.
An JY, Choi MG, Noh JH, Sohn TS, Kang WK, Park CK, et al. Gastric GIST: a single institutional retrospective experience with surgical treatment for primary disease. Eur J Surg Oncol 2007;33:1030–5.
Barman B, Gupta P, Sen AN, Deb S, Sarkar S, Mukhopadhyay S, et al. Imatinib mesylate as first-line therapy in patients with gastro intestinal stomach tumour (GIST): an experience of 24 cases from India. Ann Oncol 2005;16(Suppl. 2):311.
Benjamin RS, Rankin C, Fletcher C, Blanke C, von Mehren M, Maki R, et al. Phase III dose-randomized study of imatinib mesylate (STI571) for GIST: intergroup S0033 early results abstract. 39th Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, 31 May to 3 June 2003, abstract no. 3271.
Bhattacharjee C, Mukhopadhyay S, Dey S, Basak J, Dutta S, Mukhopadhyay A. Tolerance of imatinib mesylate inpatients with gastro intestinal stromal tumour (GIST): an Indian experience. Ann Oncol 2008;19(Suppl. 5):167.
Blanke CD, von Mehren M, Joensuu H, Roberts PJ, Eisenberg B, Heinrich M, et al. Evaluation of the safety and efficacy of an oral molecularly-targeted therapy, STI571, in patients (pts) with unresectable or metastatic gastrointestinal stromal tumors (GISTS) expressing C-KIT (CD117). 37th Annual Meeting of the American Society of Clinical Oncology, San Francisco, CA, 12–15 May 2001, abstract no. 1.
Blanke CD, Demetri GD, von Mehren M, Heinrich MC, Eisenberg BL, Fletcher J, et al. Long-term follow-up of a phase II randomized trial in advanced gastrointestinal stromal tumor (GIST) patients (pts) treated with imatinib mesylate. J Clin Oncol 2006;24:9528.
Casali PG, Verwell J, Zalcberg J, LeCesne A, Reichardt P, Ray Coquard I, et al. Imatinib (Glivec) 400 vs 800 mg daily in patients with gastrointestinal stromal tumours (GIST): a randomized phase III trial from the EORTC Soft Tissue & Bone Sarcoma Group, the Italian Sarcoma Group (ISG) and the Australasian Gastro-Intestinal Trials Group (AGITG). A toxicity report abstract. 38th Annual Meeting of the American Society of Clinical Oncology, Orlando, FL, 18–21 May 2002, abstract no. 413.
Casali PG, Verweij J, Kotasek D, LeCesne A, Reichardt P, Blay JY, et al. Imatinib mesylate in advanced gastrointestinal stromal tumors (GIST): survival analysis of the intergroup EORTC/ISG/AGITG randomized trial in 946 patients. Eur J Cancer 2005;3:S201–2.
Choi D, Yoo EY, Kim KM, Sohn TS, Lee WJ, Lee JY, et al. Residual and recurrent gastrointestinal stromal tumors with KIT mutations: findings at first follow-up CT after imatinib treatment. Am J Roentgenol 2009;193:W100–5.
Debiec-Rychter M, Dumez H, Judson I, Wasag B, Verweij J, Brown M, et al. Use of c-KIT/PDGFRA mutational analysis to predict the clinical response to imatinib in patients with advanced gastrointestinal stromal tumours entered on phase I and II studies of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer 2004;40:689–95.
DeMatteo R. Adjuvant imatinib mesylate increases recurrence free survival in patients with completely resected localized primary gastrointestinal stromal tumor. J Clin Oncol 2007;25(Suppl. 18):Abstract 10079.
Demetri GD, Rankin C, Fletcher C, Benjamin RS, Blanke C, Von Mehren N, et al. Phase III dose-randomized study of imatinib mesylate (Gleevec, STI571) for GIST: intergroup S0033 early results abstract. 38th Annual Meeting of the American Society of Clinical Oncology, Orlando, FL, 18–21 May 2002, abstract no. 1651.
Demetri GD, Wang Y, Wehrle E, Racine A, Nikolova Z, Blanke CD, et al. Imatinib plasma levels are correlated with clinical benefit in patients with unresectable/metastatic gastrointestinal stromal tumors. J Clin Oncol 2009;27:3141–7.
Dincer S, Akboru H, Teke F, Eren B, Askaroglu B, Demir C, et al. Evaluation of gastrointestinal stromal tumor patients and imatinib therapy. Ann Oncol 2008;19(Suppl. 8):270–1.
Doi T, Nishida T, Hirota S, Sugiyama T, Yamao K, Koseki M, et al. Phase II clinical study of STI571 in Japanese (Jpn) patients (pts) with malignant gastrointestinal stromal tumors (GIST): results of the B 1201 study abstract. 40th Annual Meeting of the American Society of Clinical Oncology, New Orleans, LA, 5–8 June 2004, abstract no. 4078.
Duffaud F, Le Cesne A. Imatinib in the treatment of solid tumours. Target Oncol 2009;4:45–56.
Goerres GW, Stupp R, Barghouth G, Hany TF, Pestalozzi B, Dizendorf E, et al. The value of PET, CT and in-line PET/CT in patients with gastrointestinal stromal tumours: long-term outcome of treatment with imatinib mesylate. Eur J Nucl Med Mol Imaging 2005;32:153–62.
Hartmann JT, Heidel F, Stoehlmacher J, Duex M, Izbicki JR, Elsaid A, et al. Pattern of progression and its impact on outcome in patients with gastrointestinal stromal tumors after initial response to imatinib mesylate: a retrospective multicenter long-term follow-up study. J Clin Oncol 2006;24:9541.
Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol 2003;21:4342–9.
Heinrich MC, Shoemaker JS, Corless CL, Hollis D, Demetri GD, Bertagnolli MM, et al. Correlation of target kinase genotype with clinical activity of imatinib mesylate (IM) in patients with metastatic GI stromal tumors (GISTs) expressing KIT (KIT+). J Clin Oncol 2005;23(Suppl. 16):7.
Heinrich MC, Owzar K, Corless CL, Hollis D, Borden EC, Fletcher CD, et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup phase III trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol 2008;26:5360–7.
Hohenberger P. Neoadjuvant imatinib and organ preservation in locally advanced gastrointestinal stromal tumors (GIST). J Clin Oncol 2009;27(Suppl. 15):Abstract 10550.
Joensuu H, De Braud F, Coco P, De Pas T, Spreafico C, Bono P, et al. A phase II, open-label study of PTK787/ZK222584 in the treatment of metastatic gastrointestinal stromal tumors (GISTs) resistant to imatinib mesylate. J Clin Oncol 2006;24:9531.
Joensuu H, Demetri GD, Heinrich MC, Eisenberg BL, Fletcher JA, Corless CL, et al. Up to 6 years’ follow-up of patients receiving imatinib mesylate (Glivec) to treat unresectable or metastatic gastrointestinal stromal tumors (GISTs). Eur J Cancer 2007;5:S7506.
Judson IR, Verweij J, Van Oosterom A, Blay JY, Rodenhuis S, van der Graaf W, et al. Imatinib (Gleevec) an active agent for gastrointestinal stromal tumours (GIST), but not for other soft tissue sarcoma (STS) subtypes not characterized for KIT and PGDF-R expression: results of EORTC phase II studies. Proc Am Soc Clin Oncol 2002;21:Abstract 1609.
Kang B. A phase II study of imatinib mesylate as adjuvant treatment for curatively resected high-risk localized GIST. J Clin Oncol 2009;27(Suppl.):Abstract e21515.
Le Cesne A. Continuous versus interruption of imatinib in responding patients. J Clin Oncol 2007;25(Suppl. 18):Abstract 10005.
Le Cesne A, Van Glabbeke M, Verweij J, Casali P, Zalcberg J, Reichardt P, et al. Is a stable disease according to RECIST criteria a real stable disease in GIST patients treated with imatinib mesylate (IM) included in the intergroup EORTC/ISG/AGITG trial? J Clin Oncol 2006;24:9510.
Le Cesne A, Van Glabbeke M, Verweij J, Casali PG, Findlay M, Reichardt P, et al. Absence of progression as assessed by response evaluation criteria in solid tumors predicts survival in advanced GI stromal tumors treated with imatinib mesylate: the intergroup EORTC-ISG-AGITG phase III trial. J Clin Oncol 2009;27:3969–74.
Lee JL, Ryu MH, Chang HM, Kim TW, Kang HJ, Sohn HJ, et al. Clinical outcome in gastrointestinal stromal tumor patients who interrupted imatinib after achieving stable disease or better response. Jpn J Clin Oncol 2006;36:704–11.
Li J. Adjuvant therapy with imatinib in gastrointestinal stromal tumor (GIST) patients with intermediate or high risk. J Clin Oncol 2009;27(15S):Abstract 10556.
Menard C, Blay JY, Borg C, Michiels S, Ghiringhelli F, Robert C, et al. Natural killer cell IFN-gamma levels predict long-term survival with imatinib mesylate therapy in gastrointestinal stromal tumor-bearing patients. Cancer Res 2009;69:3563–9.
Nishida T. Preliminary results of the phase II clinical trial of imatinib mesylate for advanced gastrointestinal stromal tumors in Japan. Gann Mono Cancer Res 2004;53:151–7.
Nishida T, Shirao K, Sawaki A, Koseki M, Okamura T, Ohtsu A, et al. Efficacy and safety profile of imatinib mesylate (ST1571) in Japanese patients with advanced gastrointestinal stromal tumors: a phase II study (STI571B1202). Int J Clin Oncol 2008;13:244–51.
Perol D. Does interruption of imatinib in responding GIST patients after one year of treatment influence the secondary resistance to IM after its reintroduction? J Clin Oncol 2008;26(Suppl. 5):Abstract 10556.
Phongkitkarun S, Phaisanphrukkun C, Jatchavala J, Sirachainan E. Assessment of gastrointestinal stromal tumors with computed tomography following treatment with imatinib mesylate. World J Gastroenterol 2008;14:892–8.
Rios M. Interruption of imatinib in GIST patients with advanced disease after one year of treatment. J Clin Oncol 2007;25(Suppl. 18):Abstract 10016.
Romeo S, Debiec-Rychter M, Van Glabbeke M, Van Paassen H, Comite P, Van Eijk R, et al. Cell cycle/apoptosis molecule expression correlates with imatinib response in patients with advanced gastrointestinal stromal tumors. Clin Cancer Res 2009;15:4191–8.
Ruka W. The outcomes of patients with metastatic/inoperable gastrointestinal stromal tumors (GIST) treated with imatinib: an interim multicenter analysis of Polish Clinical GIST Registry. Nowotwory 2005;55:195–9.
Rutkowski P, Debiec-Rychter M, Nowecki ZI, Wozniak A, Michej W, Limon J, et al. Different factors are responsible for predicting relapses after primary tumors resection and for imatinib treatment outcomes in gastrointestinal stromal tumors. Med Sci Monitor 2007;13:CR515–22.
Rutkowski P, Nowecki ZI, Debiec-Rychter M, Grzesiakowska U, Michej W, Wozniak A, et al. Predictive factors for long-term effects of imatinib therapy in patients with inoperable/metastatic CD117(+) gastrointestinal stromal tumors (GISTs). J Cancer Res Clin Oncol 2007;133:589–97.
Rutkowski P, Nowecki ZI, Grzesiakowska U, Michej W, Debiec-Rychter M, Wozniak A, et al. Long-term effects of imatinib therapy and impact of surgery on patients (pts) with CD117(+) gastrointestinal stromal tumours (GIST) without early progression on imatinib. Society of Surgical Oncology 60th Annual Cancer Symposium March 2007, Washington DC, USA, poster P280.
Ryu M, Lee J, Chang H, Kim T, Kang H, Sohn H, et al. Patterns of progression in gastrointestinal stromal tumor treated with imatinib mesylate. Eur J Cancer 2005;3:S23.
Ryu MH, Lee JL, Chang HM, Kim TW, Kang HJ, Sohn HJ, et al. Patterns of progression in gastrointestinal stromal tumor treated with imatinib mesylate. Jpn J Clin Oncol 2006;36:17–24.
Ryu MH, Kang WK, Bang YJ, Lee KH, Shin DB, Ryoo BY, et al. A prospective, multicenter, phase 2 study of imatinib mesylate in Korean patients with metastatic or unresectable gastrointestinal stromal tumor. Oncology 2009;76:326–32.
Sawaki A. Recurrence after imatinib treatment for patients with gastrointestinal stromal tumor. J Gastroenterol Hepatol 2006;21(Suppl. 6):A499.
Sciot R, Debiec-Rychter M, Daugaard S, Fisher C, Collin F, Van Glabbeke M, et al. Distribution and prognostic value of histopathologic data and immunohistochemical markers in gastrointestinal stromal tumours (GISTs): an analysis of the EORTC phase III trial of treatment of metastatic GISTs with imatinib mesylate. Eur J Cancer 2008;44:1855–60.
Stroobants S, Goeminne J, Seegers M, Dimitrijevic S, Dupont P, Nuyts J, et al. 18FDG-Positron emission tomography for the early prediction of response in advanced soft tissue sarcoma treated with imatinib mesylate (Glivec). Eur J Cancer 2003;39:2012–20.
Tamas KR, Papai Z, Vegh EA, Petranyi AE, Szucs Z, Rohanszky M, et al. One institutional experience with imatinib mesylate in the treatment of gastrointestinal stromal tumors. Ann Oncol 2006;17(Suppl. 9):164.
van Erp N, Gelderblom H, Van Glabbeke M, Van Oosterom A, Verweij J, Guchelaar HJ, et al. Effect of cigarette smoking on imatinib in patients in the soft tissue and bone sarcoma group of the EORTC. Clin Cancer Res 2008;14:8308–13.
Van Glabbeke MM, Verweij J, Casali PG, Zalcberg J, Le Cesne A, Reichard P, et al. Prognostic factors of toxicity and efficacy in patients with gastro-intestinal stromal tumors (GIST) treated with imatinib: a study of the EORTC-STBSG, ISG and AGITG. Proc Am Soc Clin Oncol 2003;818:Abstract 3288.
Van Glabbeke M, Verweij J, Casali PG, Le Cesne A, Hohenberger P, Ray-Coquard I, et al. Initial and late resistance to imatinib in advanced gastrointestinal stromal tumors are predicted by different prognostic factors: a European Organisation for Research and Treatment of Cancer-Italian Sarcoma Group-Australasian Gastrointestinal Trials Group study. J Clin Oncol 2005;23:5795–804.
Van Glabbeke M, Verweij J, Casali PG, Simes J, Le Cesne A, Reichardt P, et al. Predicting toxicities for patients with advanced gastrointestinal stromal tumours treated with imatinib: a study of the European Organisation for Research and Treatment of Cancer, the Italian Sarcoma Group, and the Australasian Gastro-Intestinal Trials Group (EORTC-ISG-AGITG). Eur J Cancer 2006;42:2277–85.
Van Glabbeke M, Owzar K, Rankin C, Simes J, Crowley J, GIST Meta-analysis Group (MetaGIST). Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumours (GIST): a meta-analysis based on 1640 patients. J Clin Oncol 2007;25(Suppl. 18):Abstract 10004.
Van Oosterom AT, Judson IR, Verweij J, Stroobants S, Dumez H, Donato dP, et al. Update of phase I study of imatinib (STI571) in advanced soft tissue sarcomas and gastrointestinal stromal tumors: a report of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer 2002;38(Suppl. 5):S83–7.
Verweij J, Casali PG, Zalcberg J, Le Cesne A, Reichard P, Blay J, et al. Early efficacy comparison of two doses of imatinib for the treatment of advanced gastro-intestinal stromal tumors (GIST): interim results of a randomized phase III trial from the EORTC-STBSG, ISG and AGITG. Proc Am Soc Clin Oncol 2003;22:Abstract 3272.
Verweij J, Van Oosterom A, Blay JY, Judson I, Rodenhuis S, van der GW, et al. Imatinib mesylate (STI-571 Glivec, Gleevec) is an active agent for gastrointestinal stromal tumours, but does not yield responses in other soft-tissue sarcomas that are unselected for a molecular target. Results from an EORTC Soft Tissue and Bone Sarcoma Group phase II study. Eur J Cancer 2003;39:2006–11.
Verweij J, Casali PG, Kotasek D, Le Cesne A, Reichard P, Judson IR, et al. Imatinib does not induce cardiac left ventricular failure in gastrointestinal stromal tumours patients: analysis of EORTC-ISG-AGITG study 62005. Eur J Cancer 2007;43:974–8.
Villalobos R. Clinical response to imatinib mesylate in recurrent and metastaic gastrointestinal stromal tumors. J Clin Oncol 2007;25(Suppl. 18):Abstract 20528.
von Mehren M. Imatinib response linked to variations in drug exposure. Oncology Report 2008;fall:31.
Yeh CN, Chen TW, Lee HL, Liu YY, Chao TC, Hwang TL, et al. Kinase mutations and imatinib mesylate response for 64 Taiwanese with advanced GIST: preliminary experience from Chang Gung Memorial Hospital. Ann Surg Oncol 2007;14:1123–8.
Yildiz I, Mandel NM, Demir G, Ozguroglu M, Toptas T, Buyukunal E, et al. Imatinib mesylate response in gastrointestinal stromal tumors: experience of Cerrahpasa Medical Faculty. Ann Oncol 2006;17(Suppl. 9):164.
Zekri JM, Robinson MH. Relative hypocalcaemia and muscle cramps in patients receiving imatinib for gastrointestinal stromal tumour. Sarcoma 2006;2006:Article ID 48948.
Imatinib dose not reported (n = 84)
Al Batran SE, Hartmann JT, Heidel F, Stoehlmacher J, Wardelmann E, Dechow C, et al. Focal progression in patients with gastrointestinal stromal tumors after initial response to imatinib mesylate: a three-center-based study of 38 patients. Gastric Cancer 2007;10:145–52.
Artinyan A, Kim J, Soriano P, Ellenhorn J. Survival from metastatic gastrointestinal stromal tumors in the era of imatinib. Ann Surg Oncol 2008;15(Suppl. 2):92.
Bachet J, Hostein I, Le Cesne A, Beauchet A, Brahimi S, Tabone-Eglinger S, et al. GIST with deletion of TYR568 and TYR570 of kit have similar prognosis and response to imatinib as those with DELWK557–558, but are mainly extra-gastric. Ann Oncol 2008;19(Suppl. 6):9.
Bachet JB, Hostein I, Le Cesne A, Brahimi S, Beauchet A, Tabone-Eglinger S, et al. Prognosis and predictive value of KIT exon 11 deletion in GISTs. Br J Cancer 2009;101:7–11.
Bao X, Kamo N, Hao H, Sakurama K, Noma K, Shirakawa Y, et al. The upregulation of FAK contributes to tumor progression and a resistance to imatinib in gastrointestinal stromal tumor. Proceedings of the American Association for Cancer Research Annual Meeting 2009, p. 50.
Basu S, Balaji S, Bennett DH, Davies N. Gastrointestinal stromal tumors (GIST) and laparoscopic resection. Surg Endosc 2007;21:1685–9.
Bearzi I, Mandolesi A, Arduini F, Costagliola A, Ranaldi R. Gastrointestinal stromal tumor. A study of 158 cases: clinicopathological features and prognostic factors. Anal Quant Cytol Histol 2006;28:137–47.
Benjamin RS, Choi H, Macapinlac HA, Burgess MA, Patel SR, Chen LL, et al. Response of gastrointestinal stromal tumors (GISTs) to imatinib by Choi criteria and response evaluation criteria in solid tumors (RECIST) as surrogates for survival and time to progression. J Clin Oncol 2006;24:9506.
Benjamin RS, Choi H, Macapinlac HA, Burgess MA, Patel SR, Chen LL, et al. We should desist using RECIST, at least in GIST. J Clin Oncol 2007;25:1760–4.
Blackstein M, Huang X, Demetri G, Casali P, Garrett C, Schoffski P, et al. Investigation of soluble KIT as a potential surrogate marker for TTP in sunitinib-treated patients with GIST. Ann Oncol 2007;18(Suppl. B):VII16.
Boulos BM, Jajeh A, Nawaz U, Osafo D, Tamkus D, Ogundipe O, et al. Retrospective analysis of the use of imatinib mesylate in the treatment of chronic myeloid leukemia (CML) and gastrointestinal stromal tumor (GIST). FASEB J 2007;21:A1189.
Bui BN, Le Cesne A, Ray-Coquard I, Duffaud F, Rios M, Adenis A, et al. Do patients with initially resected metastatic GIST benefit from ‘adjuvant’ imatinib (IM) treatment? Results of the prospective BFR14 French Sarcoma Group randomized phase III trial. J Clin Oncol 2006;24:9501.
Casali P, Garrett C, Blackstein M, Shah M, Verweij J, McArthur G, et al. A phase III trial of sunitinib in GIST patients following failure of imatinib mesylate: updated trial results. Ann Oncol 2006;17(Suppl. 6):21–2.
Casali PG, Fumagalli E, Messina A, Spreafico C, Comandini D, Camadone A, et al. Tumor response to imatinib mesylate in advanced GIST. J Clin Oncol 2004;22:9028.
Casali PG, Garrett CR, Blackstein ME, Shah M, Verweij J, McArthur G, et al. Updated results from a phase III trial of sunitinib in GIST patients (pts) for whom imatinib (IM) therapy has failed due to resistance or intolerance. J Clin Oncol 2006;24:9513.
Cassier P, Fayette J, Ray-Coquard I, Bui B, Duffaud F, Adonis A, et al. Outcome of patients with locally advanced GIST receiving imatinib mesylate in a prospective trial. Ann Oncol 2007;18(Suppl. 7):VII105–6.
Chevreau C. Outcome of patients with advanced GIST achieving a complete remission. J Clin Oncol 2009;27(Suppl. 15):Abstract 10549.
Choi H, Tamm EP, Macapinlac HA, Faria S, Podoloff DA, Broemeling LD, et al. The role of CT density measurement to monitor the gastrointestinal stromal tumors after treatment with STI-571: a quantitative analysis. Clin Cancer Res 2001;7:233.
Choi H, Faria SC, Benjamin RS, Podoloff DA, Macapinlac HA, Charnsangavej C. Monitoring treatment effects of STI-571 on gastrointestinal stromal tumors (GIST) with CT and PET: a quantitative analysis. Radiology 2002;225(Suppl. S):583.
Choi H, Charnsangavej C, Faria SC, Macapinlac HA, Burgess MA, Patel SR, et al. Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteria. J Clin Oncol 2007;25:1753–9.
Chu TF, Rupnick MA, Kerkela R, Dallabrida SM, Zurakowski D, Nguyen L, et al. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet 2007;370:2011–19.
Cioffi A. Long term progression-free survval correlates with KIT/PDGFR. J Clin Oncol 2007;25(Suppl. 18):Abstract 10053.
Cioffi A. Outcomes for patients with advanced GIST. J Clin Oncol 2008;26(Suppl. 5):Abstract 10550.
Davis D, Heymach J, McConkey D, Desai J, George S, Jackson J, et al. Receptor tyrosine kinase activity and apoptosis in gastrointestinal stromal tumours: a pharmacodynamic analysis of response to sunitinib malate (SU11248) therapy. Clin Cancer Res 2005;11:S9027.
De Giorgi U, Aliberti C, Benea G, Kopf B, Marangolo M. Effect of angio-echography with a second-generation contrast agent to assess tumor response to imatinib treatment in patients with advanced gastrointestinal stromal tumor (GIST): comparison with computerized tomography (CT). J Clin Oncol 2004;22:4216.
Demetri G, Huang X, Garrett C, Schoffski P, Blackstein M, Shah MH, et al. Novel statistical analysis of long-term survival to account for crossover in a phase III trial of sunitinib (SU) vs placebo (PL) in advanced GIST after imatinib failure. J Clin Oncol 2009;26(Suppl. 5):Abstract 10524.
Demetri GD, Desai J, Fletcher JA, Morgan JA, Fletcher CDM, Kazanovicz A, et al. SU11248, a multi-targeted tyrosine kinase inhibitor, can overcome imatinib (IM) resistance caused by diverse genomic mechanisms in patients (pts) with metastatic gastrointestinal stromal tumor (GIST). J Clin Oncol 2004;22:3001.
Demetri GD, Van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 2006;368:1329–38.
Demetri GD, Heinrich MC, Fletcher JA, Fletcher CD, Van den Abbeele AD, Corless CL, et al. Molecular target modulation, imaging, and clinical evaluation of gastrointestinal stromal tumor patients treated with sunitinib malate after imatinib failure. Clin Cancer Res 2009;15:5902–9.
Deprimo SE, Huang X, Blackstein ME, Garrett CR, Harmon CS, Schoffski P, et al. Circulating levels of soluble KIT serve as a biomarker for clinical outcome in gastrointestinal stromal tumor patients receiving sunitinib following imatinib failure. Clin Cancer Res 2009;15:5869–77.
Desai J, Dileo P, Morgan JA, Larsen PR, Chen MH, George S, et al. Hypothyroidism may accompany SU11248 therapy in a subset of patients (pts) with metastatic (met) gastrointestinal stromal tumors (GIST) and is manageable with replacement therapy. J Clin Oncol 2005;23:S201.
Desai J, Yassa L, Marqusee E, George S, Frates MC, Chen MH, et al. Hypothyroidism after sunitinib treatment for patients with gastrointestinal stromal tumors. Ann Intern Med 2006;145:660–4.
Dileo P, Morgan JA, Garrett CR, Schutte HJ, Hurwitz H, Rosen LS, et al. Updated results from a ‘treatment-use’ trial of sunitinib in advanced gastrointestinal stromal tumor (GIST). Ann Oncol 2006;17(Suppl. 9):162–3.
Doi V, Shirao K, Yamada Y, Muro K, Fuse N, Euda E, et al. Sunitinib in Japanese patients with GIST after prior treatment with imatinib mesylate: a phase I/II dose-escalation study. Ann Oncol 2006;17(Suppl. 6):101.
Efthirniou E, Mudan S, Hayes A, Judson I, Thomas M. Pre-operative imatinib for recurrent, metastatic and locally advanced gastrointestinal tumours (GIST). Ann Oncol 2008;19(Suppl. 6):72.
Ferreira TC, Vieira MR, Salazar M, Nobre-Leitao C. Importance of F-18 fluorodeoxyglucose (FDG) positron emission tomography (PET) scan in the evaluation of patients (pts) with gastrointestinal stromal tumour (GIST) under imatinib mesylate (IMT): one year of experience. Eur J Nucl Med Mol Imaging 2004;31(Suppl. 2):335.
Fiore M, Palassini E, Fumagalli E, Pilotti S, Tamborini E, Stacchiotti S, et al. Preoperative imatinib mesylate for unresectable or locally advanced primary gastrointestinal stromal tumors (GIST). Eur J Surg Oncol 2009;35:739–45.
Garrett C, Huang X, Casali P, Schoffski P, Blackstein M, Shah M, et al. Long-term survival in a phase III trial of sunitinib in imatinib-resistant/intolerant gastrointestinal stromal tumor with novel statistical analysis to account for crossover. Ann Oncol 2008;19(Suppl. 6):12.
Gomez D, Al Mukthar A, Menon KV, Toogood GJ, Lodge JP, Prasad KR. Aggressive surgical resection for the management of hepatic metastases from gastrointestinal stromal tumours: a single centre experience. HPB 2007;9:64–70.
Gong JS, Zuo M, Yang P, Zang D, Zhang Y, Xia L, et al. Value of CT in the diagnosis and follow-up of gastrointestinal stromal tumors. Clin Imaging 2008;32:172–7.
Heinrich MC. Does tumor mutational status correlate with clinical response to imatinib? Commentary. Nature Clin Pract Oncol 2006;3:600–1.
Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol 2008;26:5352–9.
Hohenberger P, Langer C, Pistorius S, Iesalnieks I, Wardelmann E, Reichardt P. Indication and results of surgery following imatinib treatment of locally advanced or metastatic GI stromal tumors (GIST). J Clin Oncol 2006;24:9500.
Holdsworth CH, Manola J, Badawi RD, Israel DA, Blanke C, von Mehren M, et al. Use of computerized tomography (CT) as an early prognostic indicator of response to imatinib mesylate (IM) in patients with gastrointestinal stromal tumors (GIST). J Clin Oncol 2004;22:3011.
Hong X, Choi H, Loyer EM, Benjamin RS, Trent JC, Charnsangavej C. Gastrointestinal stromal tumor: role of CT in diagnosis and in response evaluation and surveillance after treatment with imatinib. Radiographics 2006;26:481–95.
Janicek MJ, Potter A, Merriam P, Demetri GD. Does pattern of metastases and early measurable changes after treatment predict therapy effect in gastrointestinal stromal tumor (GIST) on STI571 (imatinib)? Radiology 2002;225(Suppl. S):583.
Jin J, Robinson A, Willis J, Hardacre J, Kim J. Does imatinib mesylate (IM) affect longterm outcome in patients with gastrointestinal stromal tumors (GISTs)? J Am Coll Surg 2007;205:S14.
Judson IR, Casali PG, Garrett CR, Blackstein ME, Shah M, Verweij J, et al. Updated results from a phase III trial of sunitinib in advanced gastrointestinal stromal tumor (GIST). Ann Oncol 2006;17(Suppl. 9):162.
Kaneta T, Takahashi S, Fukuda H, Arisaka Y, Oriuchi N, Hayashi T, et al. Clinical significance of performing F-18-FDG PET on patients with gastrointestinal stromal tumors: a summary of a Japanese multicenter study. Ann Nucl Med 2009;23:459–64.
Kim TW, Ryu MH, Lee H, Lee SS, Sym SJ, Kim MK, et al. No relationship between C-kit mutation and response to imatinib in Korean patients with metastatic gastrointestinal stromal tumor. Ann Oncol 2006;17(Suppl. 9):162.
Kleinbaum EP, Lazar AJ, Tamborini E, McAuliffe JC, Sylvestre PB, Sunnenberg TD, et al. Clinical, histopathologic, molecular and therapeutic findings in a large kindred with gastrointestinal stromal tumor. Int J Cancer 2008;122:711–18.
Kuhlmann KFD, Kerst JM, Cats A, van Coevorden F. Is there a role for post-imatinib surgery in advanced gastrointestinal stromal tumours? Eur J Gastroenterol Hepatol 2009;21:A3.
Lamuraglia M, Le Cesne A, Chami L, Bonvalot S, Terrier P, Tursz T, et al. Dynamic contrast-enhanced Doppler ultrasound (DCE-US) is a useful radiological assessment to early predict the outcome of patients with gastrointestinal stromal tumors (GIST) treated with imatinib (IM). J Clin Oncol 2006;24:9539.
Lassau N, Lamuraglia M, Leclere J, Bonvalot S, Vanel D, Robert C, et al. Doppler-ultrasonography with perfusion software and contrast medium injection as an early evaluation tool of gastro intestinal stromal tumor (GIST) treated by imatinib: results of a prospective study. J Clin Oncol 2004;22:9048.
Lassau N, Lamuraglia M, Chami L, Le Cesne A, Roche A, Leclere L, et al. Interest of Doppler-ultrasonography with perfusion software and contrast injection to early evaluation and detection of secondary resistence of gastrointestinal stromal tumors treated with imatinib. Eur J Cancer 2005;3:S320.
Lee J, Ryu M, Chang H, Kim T, Kang H, Sohn H, et al. Clinical outcome in gastrointestinal stromal tumor patients who interrupted imatinib after achieving stable disease or better response. Eur J Cancer 2005;3(Suppl.):64.
Lichinitser M. Molecular biomarkers and imatinib efficacy in metastatic gastrointestinal stromal tumors. J Clin Oncol 2008;26(Suppl. 5):Abstract 21500.
Maki R, Fletcher JA, Heinrich M, Morgan JA, George S, Desai J, et al. Results from a continuation trial of SU11248 in patients with imatinib (IM)-resistant gastrointestinal stromal tumor (GIST). J Clin Oncol 2005;23(Suppl.):Abstract 9011.
Mannavola D, Coco P, Vannucchi G, Bertuelli R, Carletto M, Casali PG, et al. A novel tyrosine-kinase selective inhibitor, sunitinib, induces transient hypothyroidism by blocking iodine uptake. J Clin Endocrinol Metabol 2007;92:3531–4.
Morgan JA, Demetri GD, Fletcher JA, George S, Desai J, Maki RG, et al. Patients with imatinib mesylate-resistant GIST exhibit durable responses to sunitinib malate (SU11248). Eur J Cancer 2005;3:S421.
Morgan JA, Garrett CR, Schutte HJ, Hurwitz H, Rosen LS, Ruka W, et al. Sunitinib for patients (pts) with advanced imatinib (IM)-refractory GIST: early results from a ‘treatment-use’ trial. J Clin Oncol 2006;24:9540.
Nishida T, Doi T, Komatsu Y, Ueda E, Baum C, Shirao K. Sunitinib treatment of GIST in Japanese patients after failure of prior imatinib treatment: a phase II trial. Ann Oncol 2007;18(Suppl. 7):VII110.
Park SS, Ryu JS, Oh SY, Kim WB, Lee JH, Chae YS, et al. Surgical outcomes and immunohistochemical features for gastrointestinal stromal tumors (GISTS) of the stomach: with special reference to prognostic factors. Hepatogastroenterology 2007;54:1454–7.
Pawlik TM, Vauthey JN, Abdalla EK, Pollock RE, Ellis LM, Curley SA. Results of a single-center experience with resection and ablation for sarcoma metastatic to the liver. Arch Surg 2006;141:537–43.
Perez EA, Livingstone AS, Franceschi D, Rocha-Lima C, Lee DJ, Hodgson N, et al. Current incidence and outcomes of gastrointestinal mesenchymal tumors including gastrointestinal stromal tumors. J Am Coll Surg 2006;202:623–9.
Prior JO, Montemurro M, Orcurto MV, Michielin O, Luthi F, Benhattar J, et al. Early prediction of response to sunitinib after imatinib failure by 18F-fluorodeoxyglucose positron emission tomography in patients with gastrointestinal stromal tumor. J Clin Oncol 2009;27:439–45.
Raut C. Clinical experience with periopertaive sunitinib and extensive resection of imatinib resistant metastatic GIST. Ann Oncol 2007;18(Suppl. 7):123.
Raut C. Perioperative sunitinib dosing around extensive resections of imatinib-resistant metastatic gastrointestinal stromal tumors. J Clin Oncol 2007;25(Suppl. 18):Abstract 10044.
Reichardt P, Kang Y, Ruka W, Seddon B, Baum C, Demetri G. Subpopulation analysis in a world wide treatment-use trial of sunitinib (SU) in GIST patients with resistance or intolerance to prior imatinib (IM) therapy. J Clin Oncol 2007;25(Suppl. 18):Abstract 10022.
Rutkowski P, Nowecki Z, Nyckowski P, Dziewirski W, Nasierowska-Guttmejer A, Grzesiakowska U, et al. Surgical treatment of patients (pts) with gastrointestinal stromal tumors (GIST) after imatinib mesylate (IM) therapy. J Clin Oncol 2005;23:S825.
Rutkowski P, Nowecki ZI, Michej W, Debiec-Rychter M, Limon J, Siedlecki JA, et al. The criteria of aggressiveness and other prognostic factors for predicting relapses of primary tumors and imatinib (IM) treatment outcomes in advanced KIT immunopositive gastrointestinal stromal tumors (GIST): a report of the Polish Clinical GIST Registry (PCGR). J Clin Oncol 2006;24(Suppl. 18):Abstract 9544.
Ryu MH, Lee JL, Chang HM, Kim TW, Kim HC, Yook JH, et al. Surgical resection after imatinib treatment in patients with metastatic or unresectable gastrointestinal stromal tumors. Ann Oncol 2006;17(Suppl. 9):164.
Schoffski P, Huang X, Casali PG, Garrett CR, Blackstein ME, Shah MH, et al. Phase III trial of sunitinib (Su) in imatinib (Im)-resistant/intolerant GIST with novel statistical analysis of long-term survival to account for crossover. Ann Oncol 2008;19(Suppl. 8):266.
Schurr P, Kohrs D, Reichelt U, Kaifi J, Vashist Y, Bachmann K, et al. Repeated surgery improves survival in recurrent gastrointestinal stromal tumors: a retrospective analysis of 144 patients. Dig Surg 2009;26:229–35.
Shankar S, Stay RM, vanSonnenberg E, Dipiro PJ, Janicek MJ, Demetri GD. CT features of gastrointestinal stromal tumors following treatment with STI-571. Radiology 2002;225(Suppl.):583–4.
Shankar S, vanSonnenberg E, Desai J, Dipiro PJ, Van den AA, Demetri GD. Gastrointestinal stromal tumor: new nodule-within-a-mass pattern of recurrence after partial response to imatinib mesylate. Radiology 2005;235:892–8.
Steigen SE, Eide TJ, Wasag B, Lasota J, Miettinen M. Mutations in gastrointestinal stromal tumors: a population-based study from Northern Norway. APMIS 2007;115:289–98.
Steinert DM, Blakely LJ, Patel SR, Burgess MA, Chen LL, Trent JC, et al. Outcomes of gastrointestinal stromal tumors (GIST) and other intra-abdominal sarcomas (IAS) in the era of imatinib therapy. J Clin Oncol 2004;22:9047.
Tryggvason G. Clinical study on gastrointestinal stromal tumors (GIST) in Iceland, 1990–2003. Dig Dis Sci 2007;52:2249–53.
Tzen CY. Spectrum and prognostication of KIT and PDGFRA mutation in gastrointestinal stromal tumors. Eur J Surg Oncol 2008;34:563–8.
Van den Abbeele AD, Badawi RD, Cliche J, Israel DA, Dimitrijevic S, Demetri GD. Response to imatinib mesylate (GleevecTM) therapy in patients with advanced gastrointestinal stromal tumors (GIST) is demonstrated by F-18-FDG-PET prior to anatomic imaging with CT. Radiology 2002;225(Suppl.):424.
Van den Abbeele AD, Badawi RD, Manola J, Morgan JA, Desai J, Kazanovicz A, et al. Effects of cessation of imatinib mesylate (IM) therapy in patients (pts) with IM-refractory gastrointestinal stromal tumors (GIST) as visualized by FDG-PET scanning. J Clin Oncol 2004;22:3012.
Vanel D, Albiter M, Shapeero L, Le Cesne A, Bonvalot S, Le Pechoux C, et al. Role of computed tomography in the follow-up of hepatic and peritoneal metastases of GIST under imatinib mesylate treatment: a prospective study of 54 patients. Eur J Radiol 2005;54:118–23.
Zalinski S, Palavecino M, Abdalla EK. Hepatic resection for gastrointestinal stromal tumor liver metastases. Hematol Oncol Clin N Am 2009;23:115.
Intervention not relevant (n = 15)
Awasthi R, Forrest J, Toy E, Sarsfield P. A review of gastro-intestinal stromal turnours (GISTs) presenting at a single hospital in a twenty year period. J Pathol 2006;208(Suppl. S):54.
Bauer S, Hubert C, Heinrich MC, Cohen P, Bertagnolli M, Demetri GD, et al. KIT hyperactivation in imatinib-resistant GIST: implications for salvage therapies. J Clin Oncol 2005;23:S824.
Bilimoria KY. Small bowel cancer in the United States: changes in epidemiology, treatment, and survival over the last 20 years. Ann Surg 2009;249:63–71.
Chirieac LR, Trent JC, Steinert DM, Choi H, Yang Y, Zhang J, et al. Correlation of immunophenotype with progression-free survival in patients with gastrointestinal stromal tumors treated with imatinib mesylate. Cancer 2006;107:2237–44.
Egberts J-H. Small bowel cancer: single-centre results over a period of 12 years. Hepatogastroenterology 2007;54:129–34.
Gupta M, Sheppard BC, Corless CL, Blanke CD, Billingsley KG. Outcome following aggressive surgical therapy for gastrointestinal stromal tumor. Gastroenterology 2005;128(Suppl. 2):63.
Joensuu H, De Braud F, Coco P, De Pas T, Putzu C, Spreafico C, et al. Phase II, open-label study of PTK787/ZK222584 for the treatment of metastatic gastrointestinal stromal tumors resistant to imatinib mesylate. Ann Oncol 2008;19:173–7.
Lau S, Lui CY, Yeung YP, Lam HS, Mak KL. Gastrointestinal stromal tumor of rectum: a report of 2 cases. J Comput Assist Tomogr 2003;27:609–15.
Leahy M, Ray-Coquard I, Verweij J, Le Cesne A, Duffaud F, Hogendoorn PC, et al. Brostallicin, an agent with potential activity in metastatic soft tissue sarcoma: a phase II study from the European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. Eur J Cancer 2007;43:308–15.
Pauls K, Hohenberger P, Merkelbach-Bruse S, Pietsch T, Heinicke T, Buettner R, et al. Correlation of the c-kit mutational status of gastrointestinal stromal tumors with the clinical response to the tyrosine kinase inhibitor imatinib. Path Res Pract 2004;200:316–17.
Unalp HR, Derici H, Kamer E, Bozdag AD, Tarcan E, Onal MA. Gastrointestinal stromal tumours: outcomes of surgical management and analysis of prognostic variables. Can J Surg 2009;52:31–8.
Wagner AJ, Yazji S, Morgan JA, Choy E, George S, Hohos M, et al. A phase II study of the KIT inhibitor XL820 in patients with advanced gastrointestinal stromal tumors (GIST) resistant to or intolerant of imatinib and/or sunitinib. Eur J Cancer 2008;6(Suppl.):67.
Wise SC, Smith BD, Booth R, Kaufman MD, Lu WP, Petillo PA, et al. Small molecule modulators of KIT kinase for treatment of gastrointestinal stromal tumors (GIST). Inhibitors of juxtamembrane domain, D816V, T670I and V654A mutant forms. Proceedings of the American Association for Cancer Research Annual Meeting 2009, Denver, CO, 18–22 April, p. 50.
Wozniak A, Rutkowski P, Sciot R, De IW, Ruka W, Schoffski P, et al. Analysis of genomic imbalances in imatinib-resistant, progressive gastrointestinal stromal tumors by array comparative genomic hybridization (aCGH). Cell Oncol 2008;30:280–1.
You YQ, Hassan I, Shyyan R, Que FG, Smyrk TC, Donohue JH. Surgical pathology and outcome of gastrointestinal stromal tumors of the stomach. Gastroenterology 2004;126(Suppl. 2):61.
Treatment not evaluated (n = 11)
Bumming P, Ahlman H, Andersson J, Meis-Kindblom JM, Kindblom LG, Nilsson B. Population-based study of the diagnosis and treatment of gastrointestinal stromal tumours. Br J Surg 2006;93:836–43.
Changchien CR, Wu MC, Tasi WS, Tang R, Chiang JM, Chen JS, et al. Evaluation of prognosis for malignant rectal gastrointestinal stromal tumor by clinical parameters and immunohistochemical staining. Dis Colon Rectum 2004;47:1922–9.
Dupart J, Trent JC, Cohen P, Zhang W. Imatinib mesylate activates IGFBP3 expression in gastrointestinal stromal tumors. Clin Cancer Res 2005;11:S8987.
Dupart JJ, Trent JC, Lee HY, Chiao PJ, Godwin AK, Zhang W. IGFBP3 induces apoptosis in gastrointestinal stromal tumors but does not mediate response to imatinib mesylate. Proceedings of the American Association for Cancer Research Annual Meeting 2009, Denver, CO, 18–22 April, p. 50.
Fernandez A, Sanguino A, Peng Z, Ozturk E, Chen J, Crespo A, et al. An anticancer C-Kit kinase inhibitor is reengineered to make it more active and less cardiotoxic. J Clin Invest 2007;117:4044–54.
Fletcher JA, Corless CL, Liegl B, Fletcher CD, Raut CP, Donsky R, et al. KIT mutations and sunitinib resistance in gastrointestinal stromal tumors (GISTs). Eur J Cancer 2007;5:S7503.
Koay MHE. Gastrointestinal stromal tumours (GISTs): a clinicopathological and molecular study of 66 cases. Pathology 2005;37:22–31.
Kurtzman D, Malladevan D. Small molecule therapy: overcoming imatinib resistance in gastrointestinal stromal tumors. J Investig Med 2008;56:80.
Liegl B, Kepten I, Donsky R, Fletcher CDM, Corless CL, Fletcher JA. Morphologic and molecular heterogeneity in imatinib-resistant GIST. Mod Pathol 2008;21(Suppl. 1):51.
Miettinen M, Makhlouf H, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the jejunum and ileum: a clinicopathologic, immunohistochemical, and molecular genetic study of 906 cases before imatinib with long-term follow-up. Am J Surg Pathol 2006;30:477–89.
Yokoi K, Tanaka N, Shoji K, Ishikawa N, Seya T, Horiba K, et al. A study of histopathological assessment criteria for assessing malignancy of gastrointestinal stromal tumor, from a clinical standpoint. J Gastroenterol 2005;40:467–73.
No relevant outcomes (n = 10)
Abecasis MM, Lage P, Salazar M, Freire J, Moreira A, Jorge M, et al. Unlike CML patients, patients with gastrointestinal stromal tumors (GIST) treated with imatinib do not develop hematopoietic clonal cytogenetic abnormalities. Blood 2004;104:4702.
Antonescu CR, Guo T, Arkun K, Dematteo RP, Besmer P. Acquired resistance to imatinib in gastrointestinal stromal tumor (GIST) occurs through secondary gene mutation. Mod Pathol 2005;18(Suppl. 1):33.
Antonescu CR, Guo T, Arkun K, Dematteo RP, Besmer P. Acquired resistance to imatinib in gastrointestinal stromal tumor (GIST) occurs through secondary gene mutation. Lab Invest 2005;85(Suppl. 1):33.
Pricl S, Ferrone M, Fermeglia M, Tamborini E, Delia D, Pierotti MA, et al. C-kit mutants in GISTs and their interaction with STI 571: insights from computer simulations and clinical trials. Abs Pap Am Chem Soc 2003;225(1–2):24.
Raut C, Morgan J, Quigley M, George S, Wagner A, Demetri G, et al. Clinical experience with perioperative sunitinib and extensive resection of imatinib-resistant metastatic GIST. Ann Oncol 2007;18(Suppl. 7):VII123.
Rink L. Correlation of gastrointestinal stromal tumor (GIST) gene expression signatures and response to imatinib mesylate in the RTOG phase II clinical trial S-0132. J Clin Oncol 2009;27(15S):Abstract 10533.
Schoffski P, Huang X, Demetri GD, Casali R, Garrett CR, Blackstein M, et al. Assessment of plasma levels of soluble KIT (sKIT) as a potential surrogate marker for TTP in patients (pts) with advanced gastrointestinal stromal tumor (GIST) treated with sunitinib. Eur J Cancer 2007;5(Suppl.):7502.
Van den Abbeele AD, Melenevsky Y, de Vries D, Manola J, Dileo P, Tetrault R, et al. FDG-PET imaging demonstrates kinase target inhibition by sunitinib malate (SU11248) in GIST patients resistant to or intolerant of imatinib mesylate. Eur J Cancer 2005;3:S202–3.
Van den Abbeele A, Melenevsky Y, de Vries D, Manola J, Dileo P, Tetrault R, et al. Imaging kinase target inhibition with SU11248 by FDG-PET in patients (pts) with imatinib-resistant gastrointestinal stromal tumors (I-R GIST). J Clin Oncol 2005;23:S817.
Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet 2004;364:1127–34.
Other reasons (n = 61)
Andtbacka RH, Ng CS, Scaife CL, Cormier JN, Hunt KK, Pisters PW, et al. Surgical resection of gastrointestinal stromal tumors after treatment with imatinib. Ann Surg Oncol 2007;14:14–24.
Artinyan A, Kim J, Soriano P, Chow W, Bhatia S, Ellenhorn JD. Metastatic gastrointestinal stromal tumors in the era of imatinib: improved survival and elimination of socioeconomic survival disparities. Cancer Epidemiol Biomark Prevent 2008;17:2194–201.
Bakshi CA, Jain RA, Sastry PS, Sainani AR, Advani SH. Imatinib in gastrointestinal stromal tumors. J Assoc Physicians India 2004;52:403–9.
Basu S, Mohandas KM, Peshwe H, Asopa R, Vyawahare M. FDG-PET and PET/CT in the clinical management of gastrointestinal stromal tumor. Nucl Med Commun 2008;29:1026–39.
Bauer S, Hartmann JT, Lang H, Antoch G, Dirsch O, Ebeling P, et al. Imatinib may enable complete resection in previously unresectable or metastatic GIST. J Clin Oncol 2004;22:9023.
Bauer S, Hartmann JT, de Wit M, Lang H, Grabellus F, Antoch G, et al. Resection of residual disease in patients with metastatic gastrointestinal stromal tumors responding to treatment with imatinib. Int J Cancer 2005;117:316–25.
Berman J, O’Leary TJ. Gastrointestinal stromal tumor workshop. Hum Pathol 2001;32:578–82.
Blay JY, Le Cesne A. Gastrointestinal stromal tumors: ESMO Clinical Recommendations for diagnosis, treatment and follow-up. Ann Oncol 2007;18:27–9.
Blay J, George S, Casali P, Le Cesne A, Morgan J, Pokela J, et al. Clinical activity and tolerability of continuous daily dosing of sunitinib in patients with advanced GIST. Ann Oncol 2007;18(Suppl. 7):VII24.
Blay J, George S, Casali PG, Le Cesne A, Ray-Coquard I, Harmon CS, et al. Efficacy, safety, pharmacokinetic and pharmacodynamic analysis of sunitinib (Su) adminstered on a continuous daily dosing (CDD) schedule in patients with advanced GIST. Ann Oncol 2008;19(Suppl. 8):267–8.
Blay JY, Berthaud P, Perol D, Ray-Coquard I, Bui B, Duffaud F, et al. Continuous vs intermittent imatinib treatment in advanced GIST after one year: a prospective randomized phase III trial of the French Sarcoma Group. J Clin Oncol 2004;22:9006.
Blay JY, George S, Casali PG, Le Cesne A, Morgan JA, Tyler A, et al. Clinical benefit of continuous daily dosing of sunitinib in patients (pts) with advanced gastrointestinal stromal tumor (GIST). Ann Oncol 2006;17(Suppl. 9):163.
Blay JY, George S, Casali R, Le Cesne A, Morgan JA, Pokela J, et al. Continuous daily dosing (CDD) study of sunitinib malate (SU) in patients (pts) with advanced GIST compares favorably with intermittent dosing. Eur J Cancer 2007;5:S7501.
Boukovinas IP. Advancing treatment of gastrointestinal stromal tumors (GIST). Ann Gastroenterol 2007;20:75–7.
Casali P, Fumagalli E, Bello A, George S. Initiation of sunitinib treatment 24 hours following a final dose of imatinib in patients with advanced gastrointestinal stromal tumor: safety and tolerability. Ann Oncol 2008;19(Suppl. 8):74.
Das S, Mukhopadhyay S, Garai J, Biswas S, Sarkar R, Ghosh P, et al. Tolerance of imatinib mesylate in chronic myeloid leukemia and gastro intestinal stromal tumours patients: an experience from India. Ann Oncol 2006;17(Suppl. 3):51.
Davis DW, McConkey DJ, Heymach JV, Desai J, George S, Deprimo SE, et al. Correlation of receptor tyrosine kinase (RTK) activity and apoptosis with response to sunitinib treatment in patients with gastrointestinal stromal tumor (GIST). Eur J Cancer 2006;4:S57.
Demetri GD, Van Oosterom AT, Blackstein M, Garrett C, Shah M, Heinrich M, et al. Phase 3, multicenter, randomized, double-blind, placebo-controlled trial of SU11248 in patients (pts) following failure of imatinib for metastatic GIST. J Clin Oncol 2005;23:S308.
Desai J, Shankar S, Heinrich MC, Fletcher JA, Fletcher CD, Manola J, et al. Clonal evolution of resistance to imatinib in patients with metastatic gastrointestinal stromal tumors. Clin Cancer Res 2007;13:5398–405.
Duffaud F, LeCesne A, Ray-Coquard I, Bompass E, Assi K, Berthaud P, et al. Erythropoietin for anemia treatment of patients with GIST receiving imatinib. J Clin Oncol 2004;22:9046.
Eilber FC, Rosen G, Forscher C, Nelson SD, Dorey F, Eilber FR. Recurrent gastrointestinal stromal sarcomas. Surg Oncol 2000;9:71–5.
Eisenberg BL, Harris J, Blanke CD, Demetri GD, Heinrich MC, Watson JC, et al. Phase II trial of neoadjuvant/adjuvant imatinib mesylate (IM) for advanced primary and metastatic/recurrent operable gastrointestinal stromal tumor (GIST): early results of RTOG 0132/ACRIN 6665. J Surg Oncol 2009;99:42–7.
Faivre S, Delbaldo C, Vera K, Robert C, Lozahic S, Lassau N, et al. Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J Clin Oncol 2006;24:25–35.
George S, Casali P, Blay J, Le Cesne A, Tyler A, Quigley M, et al. Continuous daily dosing of sunitinib in patients with advanced GIST: initial results of a phase II study. Ann Oncol 2006;17(Suppl. 6):28.
George S, Casali PG, Blay J, Le Cesne A, Tyler AR, Quigley MT, et al. Phase II study of sunitinib administered in a continuous daily dosing regimen in patients (pts) with advanced GIST. J Clin Oncol 2006;24:9532.
George S, Blay J, Casali P, Le Cesne A, Morgan J, Pokela J, et al. Continuous daily dosing (CDD) of sunitinib malate (SU) compares favorably with intermittent dosing in pts with advanced GIST. J Clin Oncol 2007;25(Suppl. 18):Abstract 10015.
George S, Blay JY, Casali P, Le Cesne A, Deprimo S, Harmon CS, et al. Continuous daily dosing of sunitinib in patients with advanced GIST: updated efficacy safety PK and pharmacodynamic analysis. J Clin Oncol 2008;26(Suppl. 5): Abstract 10554.
George S, Blay JY, Casali PG, Le Cesne A, Stephenson P, Deprimo SE, et al. Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumour after imatinib failure. Eur J Cancer 2009;45:1959–68.
Gold JS. Outcome of metastatic GIST in the era before tyrosine kinase inhibitors. Ann Surg Oncol 2007;14:134–42.
Gold JS, van der Zwan SM, Gonen M, Maki RG, Singer S, Brennan MF, et al. Outcome of metastatic GIST in the era before imatinib mesylate. Ann Surg Oncol 2006;13:29.
Gronchi A, Fiore M, Bertulli R, Colecchia M, Tamborini E, Pilotti S, et al. Surgery of residual disease following imatinib mesylate in advanced gastrointestinal stromal tumors (GIST). J Clin Oncol 2005;23:825S.
Holdsworth CH, Badawi RD, Manola JB, Kijewski MF, Israel DA, Demetri GD, et al. CT and PET: early prognostic indicators of response to imatinib mesylate in patients with gastrointestinal stromal tumor. Am J Roentgenol 2007;189:W324–30.
Houk BE, Bello C, Garrett M, Poland B, Wagg J, Wada R, et al. Population pharmacokinetic (PK)-pharmacodynamic (PD) meta-analysis of sunitinib malate (SU11248) efficacy and tolerability endpoints in gastrointestinal stromal tumor (GIST), metastatic renal cell carcinoma (MRCC) and solid tumor patients. Clin Cancer Res 2005;11:S9075-6.
Houk B, Bello C, Cohen D, Casali P, Shirao K, Demetri G. An exposure-response-based meta-analysis of the efficacy of sunitinib in patients with gastrointestinal stromal tumor. Ann Oncol 2008;19(Suppl. 8):11–12.
Houk BE, Bello C, Cohen D, Kuwabara-Wagg J, Poland B. A sunitinib exposure-effect based meta-analysis of treatment-related adverse events (TRAEs) in patients with metastatic renal cell carcinoma (RCC) and gastrointestinal stromal tumor (GIST). Clin Pharmacol Ther 2008;83(Suppl. 1):53.
Houk BE, Bello CL, Demetri GD, Michaelson M, Casali PG, Bukowski RM, et al. Comparative efficacy of sunitinib administered on an intermittent or a continuous daily dosing schedule in metastatic renal cell carcinoma (mRCC) and gastrointestinal stromal tumor (GIST) patients predicted using population PK approaches. Proceedings of the American Association for Cancer Research Annual Meeting, San Diego, CA, 12–16 April 2008: 49.
Houk B, Kang D, Bello C, Cohen D, Poland B. Impact of demographic and clinical factors on the pharmacokinetics (PK) of sunitinib in patients and healthy volunteers. Clin Pharmacol Ther 2008;83(Suppl. 1):53–4.
Houk BE, Bello CL, Kang D, Amantea M. A population pharmacokinetic meta-analysis of sunitinib malate (SU11248) and its primary metabolite (SU12662) in healthy volunteers and oncology patients. Clin Cancer Res 2009;15:2497–506.
Hsieh CB, Shih ML, Yu JC, Lee HS, Chen DW, Chao TY, et al. Giant malignant gastrointestinal stromal tumors (> 10cm): recurrent factors and effect of STI 571 treatment. XXXIII World Congress of the International College of Surgeons 2002;119–23.
Huang CC, Yang CY, Lai IR, Chen CN, Lee PH, Lin MT. Gastrointestinal stromal tumor of the small intestine: a clinicopathologic study of 70 cases in the postimatinib era. World J Surg 2009;33:828–34.
Judson I. Imatinib in the treatment of gastrointestinal stromal tumour. Anal Cell Pathol 2003;25:23.
Kang Y, Kang BW, Ryu M, Im S, Park SR, Kang WK, et al. A phase II study of imatinib mesylate as adjuvant treatment for curatively resected high-risk localized gastrointestinal stromal tumors with C-Kit exon 11 mutation. Ann Oncol 2008;19(Suppl. 8):169.
Keun PC, Lee EJ, Kim M, Lim HY, Choi DI, Noh JH, et al. Prognostic stratification of high-risk gastrointestinal stromal tumors in the era of targeted therapy. Ann Surg 2008;247:1011–18.
Mabille M, Vanel D, Albiter M, Le Cesne A, Bonvalot S, Le Pechoux C, et al. Follow-up of hepatic and peritoneal metastases of gastrointestinal tumors (GIST) under imatinib therapy requires different criteria of radiological evaluation (size is not everything!!!). Eur J Radiol 2009;69:204–8.
McAuliffe JC, Lazar AJ, Yang D, Steinert DM, Qiao W, Thall PF, et al. Association of intratumoral vascular endothelial growth factor expression and clinical outcome for patients with gastrointestinal stromal tumors treated with imatinib mesylate. Clin Cancer Res 2007;13:6727–34.
Miettinen M, Makhlouf H, Sobin LH, Lasota J. Gastrointestinal stromal tumors (GISTs) of the jejunum and ileum: a clinicopathologic, immunohistochemical and molecular genetic study of 906 cases prior to imatinib with long-term follow-up. Lab Invest 2006;86:56.
Miettinen M, Makhlouf H, Sobin L, Lasota J. Gastrointestinal stromal tumors (GISTs) of the jejunum and ileum: a clinicopathologic, immunohistochemical and molecular genetic study of 906 cases prior to imatinib with long-term follow-up. Mod Pathol 2006;19(Suppl. 1):15A.
Mindikoglu AL, Regev A, Bejarano PA, Martinez EJ, Jeffers LJ, Schiff ER. Imatinib mesylate (Gleevec) hepatotoxicity. Dig Dis Sci 2007;52:598–601.
Mukhopadhyay A, Barman B, Sen A, Sarkar S, Gupta P, Deb A, et al. Tolerance of imatinib mesylate in chronic myeloid leukemia and gastrointestinal stromal tumours patients: an experience from India. Ann Oncol 2005;16(Suppl. 2):304.
Paul C, Mukhoapdhyay S, Mondal AK, Chitalkar PG, Mukhoapdhyay A. Imatinib mesylate as first line therapy in patients with gastrointestinal stromal tumour (GIST). An experience from India. Ann Oncol 2007;18(Suppl. 4):40.
Perik PJ, Rikhof B, de Jong FA, Verweij J, Gietema JA, Van der Graaf WT. Results of plasma N-terminal pro B-type natriuretic peptide and cardiac troponin monitoring in GIST patients do not support the existence of imatinib-induced cardiotoxicity. Ann Oncol 2008;19:359–61.
Raut C, Morgan JA, Quigley MT, George S, Wagner AJ, Demetri G, et al. Postoperative experience in patients with metastatic GIST are similar in patients while on sunitinib or imatinib. Eur J Cancer 2007;5:S7522.
Raut CP, Posner M, Desai J, Morgan JA, George S, Zahrieh D, et al. Surgical management of advanced gastrointestinal stromal tumors after treatment with targeted systemic therapy using kinase inhibitors. J Clin Oncol 2006;24:2325–31.
Reichardt P, Pink D, Lindner T, Heinrich MC, Cohen PS, Wang Y, et al. A phase I/II trial of the oral PKC-inhibitor PKC412 (PKC) in combination with imatinib mesylate (IM) in patients (pts) with gastrointestinal stromal tumor (GIST) refractory to IM. J Clin Oncol 2005;23:S196.
Rock EP, Goodman V, Jiang JX, Mahjoob K, Verbois SL, Morse D, et al. Food and Drug Administration drug approval summary: sunitinib malate for the treatment of gastrointestinal stromal tumor and advanced renal cell carcinoma. Oncologist 2007;12:107–13.
Shen L, Li J, Li J, Gong J, Wu A. Adjuvant post-surgery therapy with imatinib in intermediate or high-risk gastrointestinal stromal tumour (GIST) patients: interim analysis from a single centre comparison study. Ann Oncol 2008;19(Suppl. 8):267.
Siehl J, Thiel E. C-kit, GIST, and imatinib. Recent Results Cancer Res 2007;176:145–51.
Trent JC, Choi H, Hunt K, Macapinlac H, McConkey D, Charnsangravej C, et al. Apoptotic and anti-vascular activity of imatinib in GIST patients. J Clin Oncol 2005;23:S816.
Ullrich A. Targeted cancer therapies: herceptin and SUTENT. FEBS J 2008;275(Suppl. 1):11.
Usman M. Hematological and nonhematological toxicities of imatinib mesylate in patients with chronic myeloid leukemia and gastrointestinal stromal tumor. Ind J Pharmacol 2007;39:192–5.
Wu PC, Langerman A, Ryan CW, Hart J, Swiger S, Posner MC. Surgical treatment of gastrointestinal stromal tumors in the imatinib (STI-571) era. Surgery 2003;134:656–65.
Not obtained/received too late (n = 47)
ASCO 2007 updates. Sunitinib in the treatment of metastatic renal carcinoma and the gastrointestinal stromal tumors. Tumori 2007;93(Suppl. 5):1–16.
Don’t interrupt imatinib in GIST patients. Oncol News Int 2007;16:1.
Focus on hematology, Gleevec postsurgery sharply reduces GIST recurrences. Oncol News Int 2007;16:42.
Focus on hematology. Long-term imatinib recommended for metastatic GIST even after complete resection, French study shows. Oncol News Int 2006;15:30.
Imatinib for the treatment of life-threatening gastrointestinal cancer. Dtsch Apoth Ztg 2002;142:40–2.
Aliberti S, Grignani G, Bordonaro R. TK-inhibitors in the clinical management of GIST. What’s up, what’s next? Supp Palliat Cancer Care 2006;2:143–6.
Anton CR, Balan G. [Gastrointestinal stromal tumors. Actual diagnosis and treatment.] Rev Med Chir Soc MedNatIasi 2005;109:223–9.
Baldo S, Rivoire M, Sobrero A, Comandini D, Civalleri D, Stella M, et al. [Surgical resection of gastrointestinal stromal tumor after treatment with imatinib: clinical case.] Suppl Tumori 2005;4:S97.
Beheshti M. The potential value of F-18 FDG PET in comparison to CT in early prediction of response to imatinib (STI571) therapy in patients with gastrointestinal stromal tumors. Iran J Nucl Med 2007;15:34–42.
Beijnen JH. Recent developments in pharmacotherapy of cancer. Pharm Weekbl 2007;142:2–7.
Belev B, Vrbanec D, Kralik M, Plestina S, Petricevic B, Sirotkovic-Skerlev M. Gastrointestinal stromal tumors (GIST): a paradigm of successful targeted therapy of solid tumors. Acta Med Croatica 2006;60:471–5.
Buchner-Steudel P, Fleig WE. [Diagnosis and conservative treatment of gastrointestinal stromal tumors (GIST).] Deutsche Medizinische Wochenschrift 2004;129:1808–10.
Cabebe E, Wakelee H. Sunitinib: a newly approved small-molecule inhibitor of angiogenesis. Drugs Today 2006;42:387–98.
Casali PG. Selective tyrosine kinase inhibitors: imatinib in GIST. Tumori 2003;89:S42–3.
Delmonte L. Good results continue for Gleevec for GIST… Metastatic gastrointestinal stromal tumors. Oncology Times 2002;13–14.
Eisenberg B. GIST adjuvant therapy – some answers and more questions. Nature Rev Clin Oncol 2009;6:441–2.
Fath R. Effectiveness of imatinib in GIST continues for the long-term. Z Gastroenterol 2006;44:9.
Feldman B. Getting the gist of GIST… gastrointestinal stromal tumor. J GENCA 2007;17:13–17.
Fuerst ML. GIST: molecular tests predict response to imatinib. Oncology Times 2005;18.
Gale S. 27th Annual JPMorgan Healthcare Conference – Biogen Idec. IDrugs 2009;12:140–1.
Gluszek S, Rylski R, Kot M, Stanislavek J, Karoz W, Urbaniak A, et al. GIST: risk of recurrence and dissemination. PRZ Gastroenterol 2008;3:176–84.
Gordon JK, Magid SK, Berman E. Elevations in creatine kinase occur frequently in patients treated with imatinib mesylate (Gleevec). Arthritis Rheum 2008;58:25.
Helwick C. Focus on hematology. Good survival with adjuvant imatinib for high-risk GIST. Oncol News Int 2008;17:28.
Houk BE, Bello C, Cohen DP, Demetri GD, Casali PG, Shirao K. Efficacy of sunitinib in patients with gastrointestinal stromal tumor (GIST): an exposure-response based meta-analysis. Mol Cancer Ther 2007;6:S3589.
Jimeno J. Gastrointestinal stromal tumors: surgical treatment. Gastroenterologia y Hepatologia Continuada 2009;8:82–6.
Joensuu H. Treatment of inoperable gastrointestinal stromal tumor (GIST) with imatinib (Glivec, Gleevec). Med Klin 2002;97(Suppl.):128–30.
Jost D, Stroszczynski C, Chmelik P, Gaffke G, Schlecht I, Pink D, et al. [Morphology of gastrointestinal stromal tumors in advanced stages of the disease: baseline findings before chemotherapy with imatinib]. Rofo 2003;175:791–8.
Kale SS. A case and literature review of complicated gastrointestinal stromal tumors. Gastroenterol and Hepatol 2008;4:650–7.
Khadija B. Gastrointestinal stromal tumor: epidemiology and outcome study in 40 cases. Tunis Med 2006;84:26–9.
Kitamuru Y, Hirota S, Nishida T. Gastrointestinal stromal tumors (GIST): a model for molecule-based diagnosis and treatment of solid tumors. Gann Monograph on Cancer Research no. 53: gastrointestinal stromal tumor (GIST): from pathology to molecular target therapy. Karger; 2004. pp. 27–40.
Le Cesne A. C-KIT and GIST: rational use of Glivec in gastrointestinal stromal tumors. Ann Pathol 2002;22:S1–4.
McIntyre JA. Sunitinib malate: oncolytic drug multitargeted tyrosine kinase inhibitor. Drugs Future 2005;30:785–92.
Mansueto G, Longo F. [News on ASCO 2007. Sunitinib in the treatment of metastatic renal carcinoma and gastrointestinal stromal tumors.] Tumori 2007;93(Suppl.):16.
Masche UP. Sunitinib. Pharma-Kritik 2006;28:39.
Mosnier J-F. Gastrointestinal stromal tumors: from gene to treatment. Hepatogastroenterology 2002;9:403–6.
Motzer RJ. Sunitinib. Drugs 2006;66:2267.
Prenen H, Dumez H, Stefan C, Hoeben A, Wouters C, Van Lierde MA, et al. Imatinib for the treatment of patients with unresectable or metastatic malignant KIT-positive gastrointestinal stromal tumours: an open-label Belgian trial. Acta Gastroenterol Belg 2006;69:367–71.
Raccah E, Merimsky O, Kuten A, Apter S, Catane R. [Imatinib mesylate (glivec) as a treatment for gastrointestinal stromal tumor (GIST): long term follow-up.] Harefuah 2007;146:329–34.
Raut CP, Hornick JL, Bertagnolli MM. Gastrointestinal stromal tumors of gastric origin. In Wang TC, Fox JG, Giraud AS, editors. The biology of gastric cancers. New York, NY: Springer; 2009. pp. 135–63.
Raut CP. Advanced gastrointestinal stromal tumor: potential benefits of aggressive surgery combined with targeted tyrosine kinase inhibitor therapy. Am J Oncol Rev 2006;5:707–12.
Rutkowski P. Gastrointestinal stromal tumors: clinical and morphological features. Pol Przegl Chir 2003;75:374–84.
Sanchez MRG. Gastrointestinal stromal tumors: medical treatment. Gastroenterologia y Hepatologia Continuada 2009;8:87–90.
Schutte J. Gastrointestinal stromal tumors (GIST): systemic treatment with imatinib and treatment monitoring. Tumor Diagnostik und Therapie 2004;25:174–6.
Sebastian M. Aggressive surgery combined with targeted tyrosine kinase inhibitor therapy in advanced gastrointestinal stromal tumor: a commentary. Am J Oncol Rev 2006;5:715–16.
Szawlowski AW. Observation results of patients with gastrointestinal stromal tumors (GIST) subjected to surgical treatment: commentary. Pol Przegl Chir 2006;78:581–2.
Van den Abbeele AD. FDG-PET to measure response to targeted therapy: the example of gastrointestinal stromal tumor and imatinib mesylate (Gleevec). PET Clinics 2008;3:77–87.
Yokoyama A, Dairaku N, Kusano M, Koshita S, Shimada N, Yamagiwa T, et al. [Two cases of primary unresectable and/or recurrent gastrointestinal stromal tumors of small intestine presenting hemoperitoneum caused by administration of imatinib mesylate.] Nippon Shokakibyo Gakkai Zasshi – Japn J Gastroenterol 2008;105:1619–26.
Appendix 6 List of included studies
Blanke B2222 study
Primary reference
Blanke CD, Demetri GD, von Mehren M, Heinrich MC, Eisenberg B, Fletcher JA, et al. Long-term results from a randomized phase II trial of standard versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol 2008;26:620–5.
Secondary reference
Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med 2002;347:472–80.
Blanke S0033 study
Primary reference
Blanke CD, Rankin C, Demetri GD, Ryan CW, von Mehren M, Benjamin RS, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol 2008;26:626–32.
Secondary references
Dileo P, Rankin CJ, Benjamin RS, von Mehren M, Blanke C, Bramwell V, et al. Incidence and reasons for dose modification of standard-dose vs. high-dose imatinib mesylate (IM) in the Phase III Intergroup Study S0033 of patients (pts) with unresectable or metastatic gastrointestinal stromal tumor (GIST). J Clin Oncol 2005;23(Suppl. 16):824S.
Rankin C, von Mehren M, Blanke C, Benjamin R, Fletcher CDM, Bramwell V, et al. Dose effect of imatinib (IM) in patients (pts) with metastatic GIST – phase III Sarcoma Group Study S0033. J Clin Oncol 2004;22(Suppl. 14):Abstract 9005.
EORTC-ISG-AGITG (62005) study
Primary reference
Zalcberg JR, Verweij J, Casali PG, Le Cesne A, Reichardt P, Blay JY, et al. Outcome of patients with advanced gastro-intestinal stromal tumours crossing over to a daily imatinib dose of 800 mg after progression on 400 mg. Eur J Cancer 2005;41:1751–7.
Secondary references
Debiec-Rychter M, Sciot R, Le Cesne A, Schlemmer M, Hohenberger P, Van Oosterom AT, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer 2006;42:1093–103.
Zalcberg JR, Verweij J, Casali PG, Le Cesne A, Reichardt P, Blay JY, et al. Outcome of patients with advanced gastro-intestinal stromal tumors (GIST) crossing over to a daily imatinib dose of 800mg (HD) after progression on 400mg (LD): an international, intergroup study of the EORTC, ISG and AGITG. J Clin Oncol 2004;22(Suppl. 14):9004.
Park 2009
Primary reference
Park I, Ryu MH, Sym SJ, Lee SS, Jang G, Kim TW, et al. Dose escalation of imatinib after failure of standard dose in Korean patients with metastatic or unresectable gastrointestinal stromal tumor. Jpn J Clin Oncol 2009;39:105–10.
Seddon 2008
Primary reference
Seddon B, Reichardt P, Kang YK, Ruka W, Nieto A, Breazna A, et al. Detailed anlaysis of survival and safety with sunitinib in a worldwide treatment-use trial of patients with advanced imatinib-resistant/intolerant GIST. Connective Tissue Oncology Society, 14th Annual Meeting, London, November 2008, abstract no. 34980.
Secondary references
Kang Y, Reichardt P, Ruka W, Seddon B, Baum C, Demetri G. Efficacy and safety of sunitinib in a worldwide treatment-use trial of GIST patients following imatinib failure. Ann Oncol 2007;18(Suppl. 7):vii16-Abstract O-0017.
Reichardt P, Kang Y, Ruka W, Seddon B, Guerriero A, Breazna A, et al. Detailed analysis of survival and safety with sunitinib (SU) in a worldwide treatment-use trial of patients with advanced GIST. J Clin Oncol 2008;26(Suppl. 15):Abstract 10548.
Reichardt P, Kang Y, Ruka W, Seddon B, Nieto A, Breazna A, et al. Sunitinib (Su) in a worldwide treatment-use trial of patients with GIST: updated efficacy and safety analysis. Ann Oncol 2008;19(Suppl. 8):viii267-Abstract 869PD.
Rutkowski P, Reichardt P, Kang Y, Ruka W, Seddon B, Guerriero A, et al. Sunitinib in a worldwide treatment-use trial of patients with imatinib-resistant/intolerant gastrointestinal stromal tumor: detailed analysis of survival and safety. Ann Oncol 2008;19(Suppl. 6):vii12-Abstract O-013.
Schutte J, Reichardt P, Schlemmer M, Wendtner CM, Demetri GD. Efficacy and safety of sunitinib in patients with gastrointestinal stromal tumour resistant or intolerant of prior imatinib therapy: results from a worldwide treatment-use study. Onkologie 2008;31(Suppl. 1):77-Abstract OP130.
Seddon B, Reichardt P, Ruka W, Kang YK, Baum CM, Demetri GD. Safety and efficacy results of sunitinib from a worldwide treatment use trial of gastrointestinal stromal tumour (GIST) patients (pts) with resistance or intolerance to prior imatinib therapy. Eur J Cancer Supplements 2007;5:405-Abstract 7511.
Appendix 7 Protocol (4 September 2009, HTA 09/21/01)
Title of the project
-
Imatinib at escalated doses of 600 mg/day or 800 mg/day for the treatment of people with unresectable and/or metastatic GISTs whose disease has progressed on treatment with imatinib at a dose of 400 mg/day: systematic review and economic evaluation
Name of technology assessment review (TAR) team and ‘lead’
Aberdeen HTA Group
Jenni Hislop
Research Fellow, Systematic Reviewer
Health Services Research Unit (HSRU)
3rd Floor
University of Aberdeen
Health Sciences Building
Foresterhill
Aberdeen
AB25 2ZD
Reserve contact
Graham Mowatt
Senior Research Fellow
HSRU
University of Aberdeen
3rd Floor
Health Sciences Building
Foresterhill
Aberdeen
AB25 2ZD
Plain English summary
Gastrointestinal stromal tumours (GISTs) are a rare type of cancerous tumours that most commonly arise in the stomach or small intestine. People will be diagnosed with this type of cancer only if a biopsy of their tumours tests positive for a particular protein (called ‘KIT’ or ‘CD117’). In around half of all cases it is possible to remove the tumour surgically; however, overall at least 50% of those operated on will develop recurrent disease within 5 years. In these patients with recurrence, and other patients with inoperable disease at diagnosis, survival beyond a period of 2 years is uncommon without further treatment. The usual treatment for patients with inoperable GISTs is the drug imatinib, prescribed at a dose of 400 mg per day. Over 75% of patients will show either response or stable disease (SD) with the standard dose of imatinib, which typically provides control of the GISTs for a period of 2–3 years. Approximately 50% of patients will survive 5 years or more with this treatment. However, in all patients, resistance of the GISTs to imatinib will eventually occur and the disease will then progress. Genetic differences, for example whether certain mutations in the c-KIT or CD117 gene are present in patients or not, may help clinicians’ understanding of who is more likely to be able to tolerate the drug and/or have least resistance to it. Fluorodeoxyglucose-positron emission tomography (FDG-PET) scans may also be useful to detect early response or resistance to imatinib and these measures may allow more individualised treatment approaches. At present, increasing the dose of imatinib, when 400 mg per day ceases to improve a patient’s condition, is not officially recommended (although in practice it is usually tried). An alternative drug (sunitinib) is recommended to be prescribed in cases where imatinib has failed. The only other alternative to these treatments for patients with inoperable GISTs is to provide BSC through management of the patient’s pain and other symptoms, and attend to their needs and general well-being, without providing treatment to actively fight the cancer itself. However, in reality it is likely that all patients (including those receiving active treatment) will receive supportive care as part of this treatment.
This review will look at two alternative doses of imatinib (600 and 800 mg per day) and compare these with the current recommended treatment alternatives (i.e. sunitinib and/or BSC) for those patients with inoperable GISTs whose disease progresses while on imatinib at a dose of 400 mg per day.
Decision problem
Gastrointestinal stromal tumours are tumours of the connective tissue of the gastrointestinal (GI) tract arising in the interstitial cells of Cajal. They are rare cancers and estimated to account for 1% of all tumours arising in the GI tract. 1 It is estimated that the vast majority (between 60% and 70%) will arise in the stomach, though they can also occur in the small bowel (25–35%), colon and rectum (5%), and, to a lesser extent, the oesophagus. 2 Estimates of the number of people affected by GIST vary, but it is thought that the annual incidence is unlikely to exceed 240. 3 However, previous estimates have suggested that it could be as high as 2000 cases per year. 3 The median age at time of first presentation is approximately 60 years. 4 Prognosis for patients with GISTs is highly dependent on the resectability of the tumour and approximately half of patients with GISTs will have resectable disease at first presentation. GISTs are resistant to the ‘conventional’ oncology treatments of cytotoxic chemotherapy and radiotherapy. For resectable/non-metastatic tumours, prognosis gives a 10-year survival rate of 30–50% of patients, and at least 50% will relapse within 5 years,5 but for unresectable tumours prognosis is poor, with survival generally < 2 years without further treatment. 6
For a GIST to be diagnosed, it is widely accepted that a positive test result (at protein level) for the marker KIT (CD117) is required. KIT (CD117) is a tyrosine kinase receptor that provides a major pathogenic drive for the majority of GISTs by promoting tumour growth and inhibiting tumour cell death. There has been some debate on the definition of a GIST, as it has been noted that in extremely rare cases (< 5%) a patient can have a GIST despite testing negative for c-KIT protein expression and in most of these cases a mutation of the platelet-derived growth factor receptor alpha (PDGFRA) gene has been detected. 7–9 However, the World Health Organization (WHO) classification of GI tumours recommends that a diagnosis of GIST should only apply to those patients testing positive for the KIT (CD117) protein. 10
Imatinib is manufactured by Novartis under the names Glivec® (in Europe) and Gleevec® (in the USA). Having originally been licensed as a treatment for chronic myeloid leukaemia, it was first licensed for treatment of GIST in 2002 and is now the standard first-line treatment for ‘locally advanced, inoperable patients and metastatic patients’ with GIST. 11 The 2004 National Institute for Health and Clinical Excellence (NICE) Technology Appraisal no. 86 on the use of imatinib for the treatment of unresectable and/or metastatic GISTs recommends 400 mg per day as first-line management. At present the NICE guidance does not recommend dose escalation of imatinib for those whose disease progresses after initially responding at the 400 mg per day dose, although dose escalation has been noted to be the standard approach to disease progression where patient non-adherence or intolerance to imatinib are not factors in disease progression. 11
The alternative treatments available for unresectable and/or metastatic GISTs are sunitinib (manufactured by Pfizer) and best supportive care (BSC). Sunitinib is recommended for patients with unresectable and/or metastatic GISTs if treatment with imatinib has failed because of resistance or intolerance, and the drug cost for the first treatment cycle will be met by the manufacturer. 12 BSC is less well defined or standardised in different clinical trials or treatment protocols, and has also been referred to as ‘active symptom control’. 2 It has been said to involve interventions to manage pain, treat fever, anaemia (due to GI haemorrhage) and GI obstruction,1 and can include palliative measures. 13 In a Cochrane review of supportive care for patients with GI cancer, supportive care was defined as ‘the multi-professional attention to the individual’s overall physical, psychosocial, spiritual and cultural needs’. 14 It was argued that this type of care should ethically be made available to all treatment groups, meaning that in practice for patients with GISTs, treatment with imatinib or sunitinib would not be provided without supportive care as well, though it is possible that treatment with BSC could be provided without additional drug treatment with either imatinib or sunitinib.
The survival of patients with GISTs is largely dependent on whether or not the tumour is resectable. For patients with unresectable and/or metastatic disease, the treatment options are imatinib, sunitinib or BSC. Guidance is available on the effectiveness of imatinib at the 400 mg per day dose. 1 However, assessment is required of the clinical effectiveness of imatinib at higher dosages (i.e. 600 and 800 mg per day) in patients whose disease has progressed on treatment with the 400 mg dose, given that an estimated 16% of patients will experience primary resistance to imatinib, and all will develop resistance and progressive disease (PD) at a later stage. 15 In evaluating the effectiveness of escalated doses of imatinib or other alternate treatments it is also necessary to consider subgroups of patients with specific KIT mutations who may respond differently to treatment, and also note how rapidly, and by what method (e.g. FDG-PET scans), these patients were identified.
This review will assess the clinical effectiveness and cost-effectiveness of imatinib at escalated doses of 600 mg per day, and 800 mg per day, compared with treatment using sunitinib, or BSC, in patients with KIT (CD117)-positive unresectable and/or metastatic malignant GISTs, whose disease has progressed on treatment with imatinib at a dose of 400 mg per day.
Report methods for synthesis of evidence of clinical effectiveness
A systematic review of the evidence of the clinical effectiveness of imatinib at escalated doses of 600 or 800 mg per day will be undertaken following the gen eral principles of the guidance of the Centre for Reviews and Dissemination (CRD) for undertaking reviews in health care16 and reported in accordance with the PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) statement. 17
Inclusion and exclusion criteria
Types of studies
The types of studies considered will be randomised controlled trials (RCTs), non-randomised comparative studies and case series. If the number of studies meeting our inclusion criteria is sufficiently large, we may consider limiting them by type of study design and taking into account the importance of other factors, such as sample size.
Scoping searches have already been conducted and fewer than 40 potentially relevant studies were found looking specifically at either of the named interventions (i.e. imatinib at 600 or 800 mg per day).
Population
The population considered will be people with KIT (CD117)-positive unresectable and/or metastatic malignant GISTs, whose disease has progressed on treatment with imatinib at a dose of 400 mg per day.
If there is sufficient evidence, subgroup analysis will be undertaken for those patients with different mutations of CD117 that are likely to affect their response to escalated doses of imatinib. Data will also be recorded on the methods used to identify response or resistance (e.g. FDG-PET or CT scanning), and whether or not imatinib had been prescribed in a neoadjuvant or adjuvant setting for patients with previously resectable GIST.
Intervention
The intervention considered will be imatinib at escalated doses of 600 and 800 mg per day, being prescribed in addition to BSC.
Comparators
The comparators considered will be sunitinib, prescribed within its recommended dose range of 27–75 mg, and provided with BSC, and BSC only. BSC has been defined above (see Decision problem).
Outcomes
The following outcomes will be considered:
-
overall response
-
overall survival (OS)
-
disease-free survival
-
progression-free survival (PFS)
-
time to treatment failure
-
health-related quality of life (HRQoL)
-
adverse effects of treatment.
Exclusion criteria
We will exclude the following types of studies:
-
animal models
-
preclinical and biological studies
-
reviews, editorials, opinions
-
case reports
-
reports investigating technical aspects of the intervention.
In addition, we may consider excluding non-English language papers, and/or reports published as meeting abstracts, if the evidence base containing English language and/or full-text reports is sufficiently large.
Search strategy
Extensive sensitive electronic searches will be conducted to identify reports of published and ongoing studies on the clinical effectiveness of imatinib. The searches will also be designed to retrieve clinical effectiveness studies of the comparator treatments. Databases to be searched will include: MEDLINE, MEDLINE In-Process, EMBASE, Cumulative Index to Nursing and Allied Health Literature (CINAHL), Science Citation Index (SCI), BIOSIS, Health Management Information Consortium, and the Cochrane Controlled Trials Register for primary research and the Database of Abstracts of Reviews of Effects (DARE), the Cochrane Database of Systematic Reviews (CDSR) and the HTA database for relevant evidence synthesis.
A preliminary MEDLINE search strategy is provided in the appendix** and will be adapted for use in the other databases. Current research registers, including Clinical Trials, Current Controlled Trials, UK Clinical Research Network Study Portfolio, WHO International Clinical Trials Registry Platform, International Federation of Pharmaceutical Manufacturers & Associations Clinical Trials and the Association of the British Pharmaceutical Industry (ABPI) database will be searched to identify ongoing and recently completed trials. Recent conference proceedings of key oncology and GI organisations will also be screened and will include the American Society for Clinical Oncology, the International Society of Gastrointestinal Oncology, and the National Cancer Research Institute.
Protocol appendices were not provided but are available from the authors on request.
In addition, an Internet search using copernic agent will be undertaken, and will include the websites of key professional organisations, GIST Support International, and the drug manufacturers Pfizer and Novartis.
There will be no language restriction and all databases will be searched from 2000 onwards.
The reference lists of all identified studies and evidence syntheses, as well as submissions from industry and other consultees, will be checked for additional references.
Data extraction strategy
One reviewer will screen the titles (and abstracts if available) of all reports identified by the search strategy. Full-text copies of all studies deemed to be potentially relevant will be obtained, and two reviewers will independently assess them for inclusion. Any disagreements will be resolved by consensus or arbitration by a third party.
A data extraction form will be developed and piloted. One reviewer will extract details of study design, participants, intervention, comparator and outcomes. A second reviewer will check the data extraction. Any disagreements will be resolved by consensus or arbitration by a third party.
Quality assessment strategy
Two reviewers will independently assess the methodological quality of the included studies. Any disagreements will be resolved by consensus or arbitration by a third party. Studies will not be included or excluded on the basis of methodological quality.
Randomised controlled trials will be assessed using the Cochrane Collaboration’s tool for assessing risk of bias. 18 The tool addresses six specific domains: sequence generation, allocation concealment, blinding, incomplete outcome data, selective outcome reporting and ‘other issues’. Non-randomised comparative studies will be assessed using an 18-question checklist, with the same checklist minus four questions used to assess the quality of case series. The checklist for non-randomised studies and case series was adapted from several sources, including the CRD’s guidance for those carrying out or commissioning reviews,16 Verhagen et al. ,19 Downs and Black20 and the Generic Appraisal Tool for Epidemiology (GATE), which assesses bias and generalisability, sample definition and selection, description of the intervention, outcome assessment, adequacy of follow-up and performance of the analysis. The checklist was developed through the Review Body for Interventional Procedures (ReBIP). ReBIP is a joint venture between Health Services Research at Sheffield University and the Health Services Research Unit at the University of Aberdeen, and works under the auspices of the NICE Interventional Procedures Programme (IPP).
Methods of analysis/synthesis
For relevant outcomes from randomised studies, where appropriate, meta-analysis will be used to estimate a summary measure of effect. Dichotomous outcome data for the overall response outcome will be combined using the Mantel–Haenszel relative risk (RR) method and continuous outcomes will be combined using the inverse-variance weighted mean difference (WMD) method. For the estimates of RR and WMD 95% CIs and p-values will be calculated. Chi-squared tests and I2-statistics will be used to explore statistical heterogeneity across studies. Possible reasons for heterogeneity will be explored using sensitivity analysis. Where there is no obvious reason for heterogeneity, the implications will be explored using random effects methods.
Pooled weighted ratio of median survival will be derived for OS, disease-free survival and PFS. The hazard ratio (HR) is the most appropriate statistic for time-to-event outcomes (i.e. for time to treatment failure). If available, the HR will be extracted directly from the trial publications. If not reported the HR will be extracted from other available summary statistics or from data extracted from published Kaplan–Meier curves using methods described by Parmar et al. 21 A pooled HR from available RCTs will be obtained by combining the observed (O) minus expected (E) number of events and the variance obtained for each trial using a fixed effects model. 22 A weighted average of survival duration across studies will then be calculated. The chi-squared test for heterogeneity will be used to test for statistical heterogeneity between studies. If no RCT data are available, but non-randomised studies have reported relevant data for this outcome, then assessment of the risk of bias and heterogeneity will be undertaken using meta-regression analysis.
Data on adverse effects of treatment and quality of life (QoL) will be collected and combined, ideally using standardised mean difference to compare QoL, where there are available data to do so.
It is expected that studies with direct comparisons of the intervention and comparators are likely to be limited. If feasible, and appropriate where we have non-randomised evidence, meta-analysis models will be used to model survival rates for interventions and comparators. A ‘cross-design’ approach will be adopted to allow non-randomised evidence to be included, while avoiding the strong assumption of the equivalence of studies. This approach will enable evidence from RCTs, non-randomised comparative studies and case series to be included. 23 Differences between treatments for survival outcomes will be assessed by the corresponding odds ratio and 95% credible intervals. These results will be ‘unadjusted odds ratios’, but meta-analysis models adjusting for study type will also be used. The results from these models will produce ‘adjusted’ odds ratios using winbugs software. 24
If appropriate, and where there are sufficient data to do so, we will consider using a mixed-treatment comparison model for indirect comparisons.
Where a quantitative synthesis is considered to be inappropriate or not feasible, a narrative synthesis of results will be provided.
Report methods for synthesising evidence of cost-effectiveness
Economic evaluation
The economic impact of GISTs for the UK NHS is associated with its incidence rate, and the proportion of patients who may have unresectable disease (and the consequent resource use by the health systems), and burden in terms of patient outcome. Information from the work on an economic model for the UK, mainly from an industry submission, is based on the assumption that the incidence rate is 15 per million population, and 10–30% of all patients with GISTs are likely to have resectable disease. If these patients (between 80 and 240 people) are treated with imatinib, the annual drug costs per patient to the NHS have been estimated at £18,896 and £24,368 for patients on 400 and 600 mg per day, respectively. Other associated yearly costs with the treatment (including the treatment of adverse events) were estimated at £2730. The model estimates suggest that in 2 years it would cost the NHS £31,160 to treat a patient with imatinib, and in 10 years it would cost the NHS £56,146. 2,25 An estimate suggests that the total yearly cost to the NHS (England and Wales) for treating with imatinib would be between £5.6M and £11.2M. The cost to the NHS would differ when patients who fail to progress with imatinib are provided with higher doses, or other alternative treatments (e.g. treatment with sunitinib). NICE estimates suggest the number of new cases of unresectable and/or metastatic GISTs to be around 240 people per year. 3 The economic impact of different treatment strategies needs thorough investigation for a robust economic evaluation.
Objectives
The aim is to assess the clinical effectiveness and cost-effectiveness of alternative treatment strategies for people with KIT (CD117)-positive unresectable and/or metastatic gastrointestinal tumours (GISTs), whose disease has progressed on treatment with imatinib at a dose of 400 mg per day.
The specific objectives are:
-
To determine, by undertaking a systematic review of the literature, the clinical effectiveness and cost-effectiveness of using imatinib at an escalated dose of 600 or 800 mg per day to treat patients with GISTs (whose disease has progressed with imatinib at a dose of 400 mg per day), compared with treating them with sunitinib and BSC.
-
To develop an economic model to compare the cost-effectiveness and cost–utility of use of imatinib at a dose of 600 or 800 mg per day, or use of sunitinib, or BSC only, for treating people with KIT (CD117)-positive unresectable and/or metastatic gastrointestinal tumours (GISTs) whose disease has progressed on treatment with imatinib at a dose of 400 mg per day.
The economic assessment will be a comparison of alternative treatments for people with GISTs whose disease has progressed despite treatment with imatinib at a dose of 400 mg per day, or those whose treatment with imatinib has failed owing to resistance or intolerance. The alternative treatments that will be considered are (1) treating with escalated doses of 600 or 800 mg per day; (2) treating with sunitinib (within its recommended dosage); and (3) providing BSC to manage symptoms. It should be noted here that BSC is often not provided exclusively. For treatment with imatinib, and treatment with sunitinib, it will be assumed that BSC would be provided alongside these treatments.
The economic assessment will be based on two components: (1) a systematic review of existing economic evaluations of the above alternative treatments and (2) an economic evaluation modelling exercise. More specifically, the economic assessment will consider alternative treatment strategies used for treatment of GISTs (particularly for patients whose disease has progressed with imatinib at a dose of 400 mg per day).
The purpose of the review of studies on economic analysis, or economic evaluation, will be to identify published studies and assess their quality and usefulness for comparisons of alternative treatment of GISTs; inform the methodology of the proposed economic model; and identify data on the parameters of the proposed economic model (e.g. utilities for different health states, costs and epidemiological data).
Data sought
With respect to costs, data will be sought to gather information on costs to the health services (NHS) in treating patients with GISTs and on costs to patients, in order to estimate overall mean costs. Specific information will also be collected on (1) the cost of treating the different clinical outcomes (e.g. cost of achieving total survival for patients with GISTs whose disease has progressed on treatment with imatinib at a dose of 400 mg per day – the base case); (2) the costs of maintaining patients with GISTs at a disease progression-free state for a specific period of time under alternative treatment strategies; and (3) the cost per life-year gained under alternative treatment strategies. Data will be sought on the costs associated with each alternative. For costs to the health services this will include, for example, the mean number of visits to the oncologist, number of laboratory tests and examinations, radiology examinations, the number of inpatient-days and the costs of drugs. Costs associated with the treatment of adverse effects will be included within the costs of treatment under different strategies (most of the adverse effects noted in the literature include fatigue and fever, hypertension, GI illnesses, dermatological, haemorrhagic events, etc.), and data will be sought accordingly. Data on costs to patients in seeking care and for BSC under different strategies will also be collected.
With respect to effectiveness, data will be sought on the same outcomes (OS, disease-free survival or PFS, adverse effects of the treatments, time to treatment failure or time to tumour progression, and overall response rate) as noted in the review of effectiveness of different strategies (see Inclusion and exclusion criteria, above). This will aid comparison of the results of individual economic evaluations with pooled estimates of effectiveness. In addition to this, we will also seek information on the quality-adjusted life-years (QALYs) associated with each treatment strategy, and for different relevant health states noted.
More specifically, we will seek to identify any data on the QALY loss caused by GI cancer or GISTs, tumour progression, and adverse effects of the different treatment strategies.
Types of studies
Economic evaluations and cost analyses comparing the above mentioned alternative treatment strategies will be included. Non-UK studies will also be included provided that they report interventions or involve populations relevant to the scope of the study.
Search strategy for identification of published reports
A comprehensive search will be undertaken to identify studies that assess the cost or cost-effectiveness of the alternative treatments used for GISTs. Databases to be searched will include MEDLINE, MEDLINE In-Process, EMBASE, SCI, Health Management Information Consortium, NHS EED, the HTA database, the Cost-effectiveness Analysis (CEA) Registry and the Research Papers in Economics (RePEc). There will be no language restriction and all databases will be searched from 2000 onwards.
A preliminary MEDLINE search strategy is provided in the Appendix and will be adapted for use in the other databases. In addition, an Internet search using copernic agent will be undertaken and will include the websites of key professional organisations, GIST Support International and the drug manufacturers Pfizer and Novartis.
The references lists of all identified studies and evidence syntheses, as well as submissions from industry and other consultees, will be checked for additional potentially relevant references.
The description of how the industry submissions will be handled is described below [see Handling the company submission(s)].
Quality assessment
All included studies will be assessed using the guidelines of the CRD. 16 Modelling studies will also be quality assessed against the Philips checklist. 26
Report methods for synthesising evidence of cost-effectiveness
The titles and abstracts of all published reports, literature and industry submissions identified by the search strategy will be examined to select relevant studies. The full texts of potentially relevant reports, publications and industry submissions will be obtained and assessed in terms of their relevance to the economic evaluation or cost analysis. Data will be extracted by an economist according to the guidelines produced by the CRD for the critical appraisal of economic evaluations. Where the economic evaluation has been based on a modelling exercise, additional data extraction criteria developed by Philips et al. will apply. 26,27
Data from the included studies on economic analysis and economic evaluation will be summarised in order to identify common results, and to summarise the variations and weaknesses between studies. The studies that use economic modelling will be critically reviewed with regard to, for example, model structure use, parameterisation and how these models have dealt with uncertainty. This critical review will assist us in developing methods that can be used to structure our model.
Economic modelling
Model structure
The structure of the model will be informed by the modelling studies identified as part of the systematic review of economic evaluations, the review of clinical effectiveness and other existing evidence including previous NICE TARs. We will also draw upon advice from health-care professional members of our research team. However, the scope of the study suggests that treatment strategies to be compared in the model are:
-
Treatment of GIST patients (whose disease has progressed on treatment with imatinib at a dose of 400 mg per day) with an escalated dose of 600 mg per day, regulating symptoms with BSC.
-
Treatment of GIST patients (whose disease has progressed on treatment with imatinib at a dose of 400 mg per day) with an escalated dose of 800 mg per day, regulating symptoms with BSC.
-
Treatment with sunitinib (within its recommended dose range), regulating symptoms with BSC.
-
Regulating symptoms with BSC only.
The model will consider the above treatment strategies as different types of intervention, and will consider the costs and consequences of patients following these different pathways of care. When building the model we will also consider whether the use of FDG-PET to predict non-response should be built into the model. The inclusion of this imaging technology may alter estimates of cost-effectiveness because (1) it is costly and (2) it may provide an early indication of non-responders who may benefit from the early introduction of an alternative therapy.
Consideration will be given to estimating relative differences between treatments based on non-directly comparative data, if direct evidence is not identified within the literature.
The model used will be a Markov model, where the following health states will be considered (all are associated with clinical effectiveness): OS, treatment failure, time to tumour progression, and PFS. In an earlier HTA of imatinib at a dose of 400 mg per day,2 and other studies,28 the health states within the economic model were (1) ‘imatinib treatment’ with different doses or ‘sunitinib treatment’ that stops disease progression, or at least leads to a partial response (PR); (2) PD; and (3) death. It is likely that the health states used in our model will be similar to these analyses, although the final choice will depend upon advice and also the literature as described in the section Economic evaluation. Where evidence is available, subgroup analysis will be undertaken on patients with different gene mutation types that may affect their response to escalated doses of imatinib.
Data requirements
For our model, data on the relative effectiveness of interventions will be based upon the systematic review. Resource use of the selected treatment strategies, and for baseline (patients whose disease has progressed on treatment with imatinib at a dose of 400 mg/day), will be identified from relevant sources (NHS cost data, NHS tariff), the review of economic evaluations and advice from experts. Data on resource use can generally be classified into different groups: for example, resource use in the treatment strategy of the escalated doses of imatinib, secondary care resource use related to secondary level of care or services other than the interventions, for example side effect management and other associated treatments, laboratory and other examinations, and resource use for other health care. Data/information on unit costs will be obtained from NHS national reference costs and from studies that will be identified as described in the section entitled Economic evaluation. Additional focused searching for relevant cost data will also be conducted.
A cost–utility analysis will be conducted, with outcomes estimated in terms of QALYs for patients, where the European Quality of Life-5 Dimensions (EQ-5D) health-state profile can be used from the information expected to be available from the review of economic evaluation studies on such treatments. Each health state of the state transition model will require a utility estimated using the best available data [EQ-5D, Eastern Cooperative Oncology Group (ECOG) category mapped to QALY]. These data will be identified from the systematic review, additional focused searches and routine data sources. Where necessary we may need to make assumptions in order to use utility values derived from different patient populations.
Time horizon for the model
The model will look at the costs and consequences directly attributable to the events occurring for patients with GIST (whose disease progression takes place despite treatment with imatinib at 400 mg per day) and treating them with alternative strategies up to the end of the patient’s lifetime. Although the time horizon used will be the patient’s lifetime, it is expected that this is unlikely to exceed 6 years (the maximum number of years patients are expected to live after they are diagnosed with unresectable and/or metastatic GISTs).
Analysis methods
The results of the model will be presented in terms of a cost–consequence analysis and cost–utility analysis. The cost–consequence analysis will examine the costs and effects on natural and clinical measures. The likely consequences that are expected to be included in the analysis would include OS and PFS. In the cost–utility analysis, results will be presented in terms of an incremental cost per QALY, incremental cost per OS (life-years gained) and incremental cost per months/year of PFS.
Where appropriate, costs and outcomes will be discounted at 3.5% for both the cost–consequence and cost–utility analyses. 27 The economic evaluation will consider the different subgroups noted earlier.
Both deterministic and probabilistic sensitivity analysis will be conducted for the uncertainty surrounding parameters, and a net benefit framework will be used to compare the different treatment strategies.
Handling the company submission(s)
Information from the manufacturer will be considered if submitted in accordance with the 3 December 2009 deadline set by NICE. Following receipt of the submission, members of the Aberdeen TAR team will critically appraise sections of the report according to each member’s own area of expertise. Studies reported in the manufacturer’s submission that meet the inclusion criteria for the review will be data extracted and quality assessed in accordance with the procedures outlined in this protocol, and included in the data analysis.
Any economic evaluations included in the company submission, provided they comply with NICE’s guidance on presentation, will be assessed for clinical validity, reasonableness of assumptions and appropriateness of the data used in the economic model, again using the methods outlined in this protocol. Strengths and weaknesses in terms of methodology adopted, reporting of results and conclusions will be described. The default position of the TAR team is that further modelling work will be necessary and if the TAR team judge that the existing economic evidence is not robust then further work will be undertaken, either by adapting what already exists or developing de novo modelling (as described in Economic modelling, above). The conclusions derived from the company submission may then be compared with those provided by the review of the other existing evidence and any model we develop so that differences in results can be highlighted. If the model we may develop differs substantively from that submitted by any company, we shall justify any assumptions made.
Any ‘CiC’ data taken from a company submission will be reported in accordance with NICE guidelines.
Competing interests of authors
None.
Reference list
- National Institute for Health and Clinical Excellence (NICE) . Gastro-Intestinal Stromal Tumours (GIST) – Imatinib: Guidance TA86 2004. URL: http://guidance.nice.org.uk/TA86/Guidance/pdf/English (accessed August 2009).
- Wilson J, Connock M, Song F, Yao G, Fry-Smith A, Raftery J, et al. Imatinib for the treatment of patients with unresectable and/or metastatic gastrointestinal stromal tumours: systematic review and economic evaluation. Health Technol Assess 2005;9.
- National Institute for Health and Clinical Excellence (NICE) . TA86 Gastro-Intestinal Stromal Tumours (GIST) – Imatinib: Quick Reference Guide 2004. URL: http://guidance.nice.org.uk/TA86/QuickRefGuide/pdf/English (accessed August 2009).
- King DM. The radiology of gastrointestinal stromal tumours (GIST). Cancer Imaging 2005;5:150-6.
- Judson I, Leahy M, Whelan J, Lorigan P, Verrill M, Grimer R. A guideline for the management of gastrointestinal stromal tumour (GIST). Sarcoma 2002;6:83-7.
- DeMatteo RP, Lewis JJ, Leung D, Mudan SS, Woodruff JM, Brennan MF. Two hundred gastrointestinal stromal tumors: recurrence patterns and prognostic factors for survival. Ann Surg 2000;231:51-8.
- Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol 2004;22:3813-25.
- Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen C-J, Joseph N, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003;299:708-10.
- Miettinen M, Lasota J. Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med 2006;130:1466-78.
- Hamilton SR. World Health Organization classification of tumours: pathology and genetics of tumours of the digestive system. Geneva: IARC Press; 2000.
- Casali PG, Jost L, Reichardt P, Schlemmer M, Blay JY. ESMO Guidelines Working Group . Gastrointestinal stromal tumours: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol 1920;20:64-7.
- National Institute for Health and Clinical Excellence (NICE) . Gastrointestinal Stromal Tumours – Sunitinib: Final Appraisal Determination 2009. URL: www.nice.org.uk/guidance/index.jsp?action=download%26o=45125 (accessed August 2009).
- National Institute for Health and Clinical Excellence (NICE) . Single Technology Appraisal of Sunitinib for the Treatment of Gastrointestinal Stromal Tumours 2008. URL: www.nice.org.uk/guidance/index.jsp?action=download%26o=43440 (accessed August 2009).
- Ahmed N, Ahmedzai S, Vora V, Hillam S, Paz S. Supportive care for patients with gastrointestinal cancer. Cochrane Database Syst Rev 2004. URL: 10.1002/14651858.CD003445.pub2.
- Blanke CD, Demetri GD, Von Mehren M, Heinrich MC, Eisenberg B, Fletcher JA, et al. Long-term results from a randomized phase II trial of standard versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol 2008;26:620-5.
- Centre for Reviews and Dissemination (CRD) . Systematic Reviews: CRD’s Guidance for Undertaking Systematic Reviews in Health Care 2009. URL: www.york.ac.uk/inst/crd/SysRev/!SSL!/WebHelp/SysRev3.htm (accessed August 2009).
- Moher D, Liberati A, Tetzlaff J, Altman DG. PRISMA Group . Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ 2009;339:332-9.
- The Cochrane Collaboration . The Cochrane Collaboration’s tool for assessing risk of bias. The Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.1 2008. URL: www.cochrane-handbook.org/ (accessed August 2009).
- Verhagen AP, de Vet HC, de Bie RA, Kessels AG, Boers M, Bouter LM, et al. The Delphi list: a criteria list for quality assessment of randomized clinical trials for conducting systematic reviews developed by Delphi consensus. J Clin Epidemiol 1998;51:1235-41.
- Downs SH, Black N. The feasibility of creating a checklist for the assessment of the methodological quality both of randomised and non-randomised studies of health care interventions. J Epidemiol Community Health 1998;52:377-84.
- Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta-analyses of the published literature for survival endpoints. Stat Med 1998;17:2815-34.
- Yusuf S, Peto R, Lewis J, Collins R, Sleight P. Beta blockade during and after myocardial infarction: an overview of the randomized trials. Prog Cardiovasc Dis 1985;27:335-71.
- Sutton AJ, Abrams KR. Bayesian methods in meta-analysis and evidence synthesis. Stat Methods Med Res 2001;10:277-303.
- Lunn DG, Thomas A, Best N, Spiegelhalter D. Winbugs: a Bayesian modelling framework: concepts, structure, and extensibility. Stat Comput 2000;10:325-37.
- Reddy P, Boci K, Charbonneau C. The epidemiologic, health-related quality of life, and economic burden of gastrointestinal stromal tumours. J Clin Pharm Ther 2007;32:557-65.
- Philips Z, Ginnelly L, Sculpher M, Claxton K, Golder S, Riemsma R, et al. Review of guidelines for good practice in decision-analytic modelling in health technology assessment. Health Technol Assess 2004;8.
- National Institute for Health and Clinical Excellence (NICE) . NICE Guide to the Methods of Technology Appraisal 2008. URL: www.nice.org.uk/media/B52/A7/TAMethodsGuideUpdatedJune2008.pdf (accessed September 2008).
- Contreras-Hernandez I, Mould-Quevedo JF, Silva A, Salinas-Escudero G, Villasis-Keever MA, Granados-Garcia V, et al. A pharmaco-economic analysis of second-line treatment with imatinib or sunitinib in patients with advanced gastrointestinal stromal tumours. Br J Cancer 2008;98:1762-8.
Appendix 8 Characteristics of included studies
| Study ID | Participants | Intervention(s) and comparators | Outcomes summary |
|---|---|---|---|
|
Time period: July 2000 to May 2006 Countries involved: 2 (Finland, USA) No. of institutions involved: 4 |
n receiving intervention(s): 43 n receiving comparator(s): 0 Baseline characteristics: not stated |
Escalated dose intervention(s): imatinib at 600 mg/day Comparator(s): NA |
n (%) showing response or SD: 11/43 (25.6%) |
|
Time period: December 2000 to (CiC information has been removed) Countries involved: 2 (Canada, USA) No. of institutions involved: 148 |
n receiving intervention(s): 118 n receiving comparator(s): 0 Baseline characteristics: not stated |
Escalated dose intervention(s): imatinib at 800 mg/day Comparator(s): NA |
n (%) showing response or SD: 36/117 (30.8%) Median OS: 19 months (95% CI 13 to 23 months) n (%) still alive at data cut-off point: 42/118 (35.6%) Median PFS: 5 months (2–10 months) n (%) still progression free at data cut-off point: 19/118 (16.1%) |
|
Park 2009 79 Time period: June 2001 to June 2006 Countries involved: 1 (Republic of Korea) No. of institutions involved: 1 |
n receiving intervention: 24 n receiving comparator(s): 0 Baseline characteristics: Age: Median, years (range): 52 (31–73) Sex: n (%) male: 18 (75.0%) n (%) female: 6 (25.0%) ECOG performance status: 0: 4 (16.7%) 1: 18 (75.0%) 2: 2 (8.3%) Primary tumour site: Stomach: 5 (20.8%) Small bowel: 15 (62.5%) Colon or rectum: 3 (12.5%) Omentum: 1 (4.2%) n receiving previous treatment of: Surgery: 20 (83.3%) Conventional chemotherapy: 3 (12.5%) Radiofrequency ablation: 1 (4.2%) Transarterial chemoembolization: 1 (4.2%) Site(s) of metastases at time of dose escalation: Liver: 20 (83.3%) Peritoneum: 15 (62.5%) Retroperitoneum: 5 (20.8%) n (%) with prior response to standard-dose imatinib of: PR: 9 (37.5%) SD: 8 (33.3%) PD: 7 (29.2%) n (%) whose time to progression (TTP) with standard-dose imatinib was: ≤ 6 months: 8 (33.3%) > 6 months: 16 (66.7%) n (%) given initial escalated dose of imatinib at: 600 mg/day: 12 (50.0%) 800 mg/day: 12 (50.0%) |
Escalated dose intervention(s): imatinib at 600 mg/day; imatinib at 800 mg/day Comparator(s): NA |
n (%) showing response or SD: at 600 mg/day – 5/12 (41.6%); at 800 mg/day – 4/12 (33.3%) Median time to progression: at 600 mg/day – 1.7 months (range 0.7–24.9 months). |
|
Time period: not stated to December 2007 Countries involved: 33 (not stated) No. of institutions involved: 96 |
n receiving intervention: 0 n receiving comparator(s): 351 Baseline characteristics: not stated |
Escalated dose intervention(s): NA Comparator(s): sunitinib at 50 mg/day in a 6-week cycle of 4 weeks on treatment/2 weeks off treatment |
Median OS: 90 weeks (95% CI 73 to 106 weeks) n (%) still alive at data cut-off point: 193/351 (55.0%) |
|
Zalcberg 2005 44 Time period: (CiC information has been removed) to April 2004 Countries involved: 13: (Australia, Belgium, Denmark, France, Germany, Italy, the Netherlands, New Zealand, Poland, Singapore, Spain, Switzerland, UK) No. of institutions involved: 56 |
n receiving intervention: 133 n receiving comparator(s): 0 Baseline characteristics: Age: Median, years (range): 59 (20–85) Sex: n (%) male: 87 (65%) n (%) female: 46 (36%) ECOG performance status: 0: 63 (47%) 1: 49 (37%) 2: 12 (9%) 3: 9 (7%) n (%) whose primary tumour site was: GI: 109 (82%) Gastric: 34 (26%) Small bowel: 35 (26%) Duodenum: 20 (15%) Other GI: 20 (15%) Other abdominal 20 (15%) Retroperitoneal: 4 (3%) n (%) with time since primary diagnosis of: < 12 months: 70 (53%) 12–24 months: 29 (22%) > 24 months: 34 (26%) n (%) with site(s) of active disease at study entry in: Site of primary tumour: 50 (38%) Liver: 96 (72%) Lung: 16 (12%) Ascites: 12 (9%) Pleura: 4 (3%) Bone: 3 (2%) Skin: 3 (2%) n (%) receiving previous treatment of: Surgery: 116 (87%) Radiotherapy: 6 (5%) Chemotherapy: 51 (38%) |
Escalated dose intervention(s): imatinib at 800 mg/day Comparator(s): NA |
n (%) showing response or SD: 39/133 (29.3%) ‘Response to cross-over … occurred significantly more often in wild-type cases (83%) compared with KIT exon 11 mutants (7%) (p = 0.0012, Fisher’s exact test), and in KIT exon 9 mutants (57%) compared to KIT exon 11 mutants (p = 0.0017, Fisher’s exact test)’ Median PFS: 81 days n (%) still progression free at data cut-off point: 24/133 (18.8%) Median duration of response: 153 days (range 37–574 days) n (%) of patients requiring at least one dose reduction: 12/77 (15.6%) n (%) of patients requiring at least one dose delay: 18/77 (23.4%) n (%) with adverse events: Oedema: 99/124 (79.8%) Skin rash: 45/124 (36.3%) Fatigue: 102/124 (82.3%) Dyspnoea: 30/124 (24.2%) Infection: 20/124 (16.1%) Nausea: 82/124 (66.1%) Leucopenia: 56/121 (46.3%) Neutropenia: 49/121 (40.5%) Thrombocytopenia: 7/121 (5.8%) Anaemia: 119/121 (98.3%) n (%) with adverse event reporting decreased severity after crossover: Oedema: 25/99 (25.3%) Skin rash: 23/45 (51.1%) Fatigue: 21/102 (20.6%) Dyspnoea: 8/30 (26.7%) Infection: 9/20 (45.0%) Nausea: 38/82 (46.3%) Leucopenia: 25/56 (44.6%) Neutropenia: 30/49 (61.2%) Thrombocytopenia: 4/7 (57.1%) Anaemia: 15/119 (12.6%) n (%) with adverse event reporting increased severity after crossover: Oedema: 33/99 (33.3%) Skin rash: 19/45 (42.2%) Fatigue: 47/102 (46.1%) Dyspnoea: 14/30 (46.7%) Infection: 9/20 (45.0%) Nausea: 26/82 (31.7%) Leucopenia: 16.56 (28.6%) Neutropenia: 13/49 (26.5%) Thrombocytopenia: 2/7 (28.6%) Anaemia: 51/119 (42.9%) n (%) with adverse event achieving increased severity to grade 3- to grade-4 level: Oedema: 7/99 (7.1%) Skin rash: 2/45 (4.4%) Fatigue: 10/102 (9.8%) Dyspnoea: 1/30 (3.3%) Infection: 1/20 (5.0%) Nausea: 3/82 (3.7%) Leucopenia: 0/56 (0.0%) Neutropenia: 0/49 (0.0%) Thrombocytopenia: 0/7 (0.0%) Anaemia: 17/119 (14.3%) |
Appendix 9 Quality assessment of the individual full-text studies
| Quality criteria | Study ID | |||
|---|---|---|---|---|
| Blanke 200839 (B2222) | Blanke 200841 (S0033) | Park 200979 | Zalcberg 200544 | |
| Q1: Were participants a representative sample selected from a relevant patient population? | ? | ? | ? | ? |
| Q2: Were the inclusion/exclusion criteria of participants clearly described? | + | + | + | + |
| Q3: Were participants entering the study at a similar point in their disease progression? | + | + | + | ? |
| Q4: Was selection of patients consecutive? | – | – | – | – |
| Q5: Was data collection undertaken prospectively? | + | + | + | + |
| Q6: Were the groups comparable on demographic characteristics and clinical features? | N/A | N/A | N/A | N/A |
| Q7: Was the intervention (and comparison) clearly defined? | + | + | + | + |
| Q8: Was the intervention undertaken by someone experienced at performing the procedure? | ? | ? | ? | ? |
| Q9: Were the staff, place and facilities where the patients were treated appropriate for performing the procedure? (e.g. access to back-up facilities) | ? | ? | ? | ? |
| Q10: Were all the important outcomes considered? | – | – | – | – |
| Q11: Were objective (valid and reliable) outcome measure(s) used? | + | + | + | + |
| Q12: Was the assessment of main outcomes blind? | – | – | – | – |
| Q13: Was follow-up long enough to detect important effects on outcomes of interest? | + | + | ? | + |
| Q14: Was information provided on non-respondents, dropouts? | – | + | ? | ? |
| Q15: Were participants lost to follow-up likely to introduce bias? (e.g. high dropout rate; differential dropout; no description of those lost) | + | ? | ? | ? |
| Q16: Was length of follow-up similar between comparison groups? | N/A | N/A | N/A | N/A |
| Q17: Were important prognostic factors identified? | ? | ? | + | – |
| Q18: Were the analyses adjusted for confounding factors? | ? | ? | – | – |
Appendix 10 Search strategies for review of economic analysis studies, cost-effectiveness analysis
MEDLINE (2000 – October, week 4 2009), EMBASE (2000–9, week 44), MEDLINE In-Process (3 November 2009)
Ovid Multifile Search URL: https://shibboleth.ovid.com/
-
Gastrointestinal Stromal Tumors/use mesz
-
Gastrointestinal Stromal Tumor/use emez
-
gastrointestinal neoplasms/use mesz
-
exp digestive system tumor/use emez
-
gist.tw.
-
((gastro$or gastric) adj3 stromal).tw.
-
(3 or 4) and (kit or cd117 or cd 117).tw.
-
(3 or 4) and (stromal or connective or mesenchymal).tw.
-
or/1-2,5-8
-
exp “costs and cost analysis”/
-
exp economic evaluation/use emez
-
economics/
-
exp economics,hospital/
-
exp economics,medical/
-
economics,pharmaceutical/
-
exp budgets/
-
exp models, economic/
-
exp decision theory/
-
ec.fs. use mesz
-
monte carlo method/
-
markov chains/
-
exp technology assessment, biomedical/
-
cost$.ti.
-
(cost$adj2 (effective$or utilit$or benefit$or minimis$))
-
economics model$.tw.
-
(economics$or pharmacoeconomic$or pharmo-economic$).ti.
-
(price$or pricing$).tw.
-
(financial or finance or finances or financed).tw.
-
(value adj2 (money or monetary)).tw.
-
markov$.tw.
-
monte carlo.tw.
-
(decision$adj2 (tree? or analy$or model$)).tw.
-
or/10-32
-
9 and 33
-
limit 34 to yr=“2000 -Current”
-
quality of life/
-
quality adjusted life year/
-
“Value of Life”/use mesz
-
health status indicators/use mesz
-
health status/use emez
-
sickness impact profile/use mesz
-
disability evaluation/use mesz
-
disability/use emez
-
activities of daily living/use mesz
-
exp daily life activity/use emez
-
cost utility analysis/use emez
-
rating scale/
-
questionnaires/
-
(quality adj1 life).tw.
-
quality adjusted life.tw.
-
disability adjusted life.tw.
-
(qaly? or qald? or qale? or qtime? or daly?).tw.
-
(euroqol or euro qol or eq5d or eq 5d).tw.
-
(hql or hqol or h qol or hrqol or hr qol).tw.
-
(hye or hyes).tw.
-
health$year$equivalent$.tw.
-
(hui or hui1 or hui2 or hui3).tw.
-
(health adj3 (utilit$or disutili$)).tw.
-
(health adj3 (state or status)).tw.
-
(sf36 or sf 36 or short form 36 or shortform 36).tw.
-
(sf6 or sf 6 or short form 6 or shortform 6).tw.
-
(sf12 or sf 12 or short form 12 or shortform 12).tw.
-
(sf16 or sf 16 or short form 16 or shortform 16).tw.
-
(sf20 or sf 20 or short form 20 or shortform 20).tw.
-
willingness to pay.tw.
-
standard gamble.tw.
-
trade off.tw.
-
conjoint analys?s.tw.
-
discrete choice.tw.
-
or/36-69
-
9 and 70
-
limit 71 to yr=“2000 -Current”
-
35 or 72
Science Citation Index (2000, 3 November 2009)
Web of Knowledge URL: http://wok.mimas.ac.uk/
-
# 1 TS=gist
-
# 2 TS=((gastric or gastro*) SAME stromal)
-
# 3 TS=((gastric or gastro*) SAME (kit or cd117 or cd 117))
-
# 4 TS=((gastric or gastro*) SAME mesenchymal)
-
# 5 #1 or #2 or #3 or #4
-
# 6 #5 and TS=economic*
-
# 7 #5 and TS=cost*
-
# 8 #5 and TS=(price* or pricing)
-
# 9 #5 and TS=(financial or finance*)
-
# 10 #5 and TS=(decision* SAME (tree* OR analy* or model*))
-
# 11 #5 and TS=markov*
-
# 12 #5 and TS=monte carlo
-
# 13 #5 and TS=conjoint analys*
-
# 14 #5 and TS=discrete choice*
-
# 15 #5 and TS=standard gamble
-
# 16 #5 and TS=trade off
-
# 17 #5 and TS=willingness to pay
-
# 18 #5 and TS=(health SAME (indicator* or status or utilit*))
-
# 19 #5 and TS=quality of life
-
# 20 #5 and TS=quality adjusted life
-
# 21 #5 and TS=disability adjusted life
-
# 22 #5 and TS=(qaly* or qald* or qale* or qtime* or daly*)
-
# 23 #5 and TS=(euroqol* or euro qol* or eq5d or eq 5d)
-
# 24 #5 and TS=(hql or hqol or h qol or hrqol or hr qol)
-
# 25 #5 and TS=(hye or hyes)
-
# 26 #25 OR #24 OR #23 OR #22 OR #21 OR #20 OR #19 OR #18 OR #17 OR #16 OR #15 OR #14 OR #13 OR #12 OR #11 OR #10 OR #9 OR #8 OR #7 OR #6
-
#27 #26 CPCI-S Timespan=2000–2009
Health Management Information Consortium (September 2009)
Ovid Multifile Search URL: http://gateway.ovid.com/athens
-
gist.tw.
-
((gastro$or gastric$) adj3 stromal).tw.
-
gastrointestinal cancer/94
-
3 and (kit or CD117 or cd 117).tw.
-
3 and (stromal or connective or mesenchymal).tw.
-
or/1-2,4-5
NHS Economic Evaluation Database (October 2009), HTA Database (October 2009)
NHS Centre for Reviews and Dissemination URL: http://nhscrd.york.ac.uk/welcome.htm
-
# 1 MeSH Gastrointestinal Stromal Tumors EXPLODE 1 2 3
-
# 2 gist
-
# 3 (gastric OR gastro*) AND (kit OR cd117 OR cd AND 117)
-
# 4 (gastric OR gastro*) AND (stromal OR connective OR mesenchymal)
-
# 5 #1 or #2 or #3 or #4
IDEAS (October 2009)
RePEC URL: http://ideas.repec.org/
Gist or gastrointestinal stromal
Conference proceedings
International Society for Pharamoeconomics and Outcomes Research
9th Annual European Congress, Copenhagen, October 2006
10th Annual European Congress, Dublin, October 2007
11th Annual European Congress, Athens, November 2008
12th Annual European Congress, Paris, October 2009
11th Annual International Meeting, Philadelphia, May 2006
12th Annual International Meeting, Arlington, May 2007
13th Annual International Meeting, Toronto, May 2008
14th Annual International Meeting, Orlando, May 2009
Websites consulted (accessed October 2009)
Association of Upper Gastrointestinal Surgeons of Great Britain and Ireland
URL: www.augis.org/
Department of Health
GIST Support International
URL: www.gistsupport.org/
Glivec
Medicines and Healthcare products Regulatory Agency (MHRA)
URL: www.mhra.gov.uk/
National Cancer Institute
URL: www.cancer.gov/
National Comprehensive Cancer Network
National Institute for Health and Clinical Excellence
NHS Evidence
NHS Knowledge Network Scotland
Novaritis UK
URL: www.novartis.co.uk/
Pfizer UK
Scottish Sarcoma Network
URL: www.ssn.scot.nhs.uk/
Appendix 11 Summary of the included economic analysis and economic evaluation studies
| Study identification | Author and year | Chabot 200895 |
| Intervention studied/comparators | BSC vs sunitinib for imatinib-resistant or -intolerant patients | |
| Hypothesis/question | Examine the challenges to undertake cost-effectiveness study in oncology using crossover trial, and presented the submission to the CDR of a cost-effectiveness evaluation of sunitinib vs BSC for treatment of GIST in patients who are imatinib resistant or intolerant | |
| Key features of the study | Type of study | Descriptive, and a full economic evaluation (cost-effectiveness analysis) |
| Target population/sample population | Patients who failed or are intolerant to imatinib | |
| Context/settings | Canada, hypothetical population at provincial level | |
| Date to which the data of the study relate | 2005 | |
| Source of effectiveness data |
Clinical effectiveness from Phase III clinical trials (NCT00075218)52 Health outcome – QALY-based utility measured by EQ-5D questionnaire administered on clinical trial patients |
|
| Modelling | Markov modelling | |
| Link between effectiveness and costs data | Costs in the model include costs of sunitinib acquisition, and health-care resource use for BSC, cost of routine follow-up for patients receiving sunitinib, cost of adverse events, and end-life costs. Information on health-care resource use and corresponding unit costs were derived from published literature, medical oncologist and Canadian Government Schedule | |
| Information on the clinical evidence and effectiveness – main outcome of the study | Sample patients/study sample/patient groups | Cohort population in the model |
| Study design | Modelling for cost–utility analysis | |
| Effectiveness analysis |
The following trial end points were used for the valuation of the outcomes (effectiveness): (a) PFS, defined as the time from randomisation to the point when the tumour progressed or death was due to GIST (b) OS (c) utility, measured by the EQ-5D (d) treatment-related adverse events |
|
| Effectiveness measures and results/outcome measures | Sunitinib compared with BSC for the patients who failed or did not respond to imatinib and found sunitinib more effective than BSC – in terms of OS, PFS, LYG, LYS and QALY | |
| Primary end points/outcome and secondary end points/outcome |
Mean survival sunitinib group,1.6 years; mean progression-free health state, 0.5 years; and 1.1 years with PD Patients in BSC group spent on average 0.2 years in the progression-free health state and 0.7 years with PD; and had mean survival of 0.9 years Sunitinib treatment resulted in 0.7 LYG, and 0.4 QALYs compared with BSC |
|
| Statistical precision of these outcomes |
Utilities associated with sunitinib: No progression during 4 weeks’ sunitinib: 0.712 ± 0.2 Next 2 weeks’ utility improvement: 0.081 ± 0.02 No progression BSC: 0.781 ± 0.2 Progression: 0.577 + 0.3 |
|
| Clinical recommendations and conclusion | The initial CDR recommendation based on the economic evaluation was ‘not to reimburse’ sunitinib in Canada. This was reversed owing to the fact that patients who are resistant to imatinib have no other treatment options. Based on review of the quality, safety and efficacy data, Health Canada concluded that sunitinib had favourable risk–benefit profile for the treatment of GIST after failure or intolerance of imatinib treatment | |
| Economic analysis | Measures of health outcome/benefits used in the economic analysis | QALY based on EQ-5D from UK study55 |
| Direct costs and its components | Cost per 6-week cycle | |
|
Prospective or retrospective (depend on study design) Whether values were imputed in for certain cases How hospital stay was defined, and whether any classifications were used or not Costing of complications or side effects Estimations of unit costs and source/methods |
Sunitinib treatment standard dose: C$6947.99 Sunitinib treatment reduced dose for adverse event management: C$5210.99 Sunitinib treatment medical follow-up: cycle 1 C$2275.13, cycle 2 726.47, cycle 3+ 1072.11 Terminal phase – end-of-life cost C$3752. Cost of serious adverse event with sunitinib $42.84 |
|
| Indirect costs and its components | ||
| Cost of productivity, cost of volunteer care and support for the patient | Not considered | |
| Currency, year prices | C$, at 2005 prices | |
| Statistical analysis/cost | Mean and standard deviation of the progression and progression-free time | |
| Sensitivity analysis | Univariate sensitivity analysis was conducted by varying the most influential model parameters, namely utility of progression and no progression, OS (HR), PFS, PET at initiation of sunitinib treatment, the cost of palliative care and the cost of PET. The model assumed the cost of acquisition of sunitinib is certain and did not vary this in sensitivity analysis. The sensitivity analysis suggests that results of the economic evaluation were most sensitive to health-state utility value and rate of OS and PFS | |
| Results/major findings | Benefits results from the economic evaluation |
Mean QALYs: Sunitinib 0.97 BSC 0.54 ICER ($/LYS) 49,826 ICUR ($/QALYs) 79,884 These (ICER, ICUR lies between an estimated thresholds boundary of $26,433–132,166) |
| Costs results used in the economic evaluation | Mean costs in C$ | |
|
Cost of treatment, costs to health sector (cost to NHS) Major determinants of costs, the principle costs drivers |
Sunitinib $46,125 BSC $11,632 |
|
| Synthesis of costs and benefits | Cost-effectiveness of sunitinib vs BSC | |
| Any attempt to consider the uncertainty surrounding estimates of effects |
ICER ($/LYS) 49,826 ICUR ($/QALYs) 79,884 Sensitivity analysis – sensitivity uncertainty in the OS advantage for sunitinib? As patients were allowed to cross over |
|
| Author conclusion/recommendations |
Sunitinib cost-effective The decision of approval for sunitinib from Health Canada was based on the recognition of sunitinib’s clinical benefits for the imatinib-intolerant group. The paper suggests reliance on cost-effectiveness methodology is unsatisfactory Guidance is needed on how better to reconcile the best available clinical trial data with the cost-effectiveness requirements and the objectives of prompt access to oncology medicine |
| Study identification | Author and year | Contreras-Hermandez 200896 |
| Intervention studied/comparators | Sunitinib 50 mg/day, imatinib 800 mg/day and BSC | |
| Hypothesis/question | Examine the cost-effectiveness to compare the alternatives (imatinib 800 mg/day, sunitinib 50 mg/day) as second line of treatment for those who failed or became intolerant with imatinib 400 mg/day. The study examined whether it is worth it for the Mexican insurance system to reimburse for sunitinib or higher dose of imatinib | |
| Key features of the study | Type of study | Model-based (Markov) full economic evaluation (cost-effectiveness analysis) |
| Target population/sample population | Twenty-one advanced GIST patients who were treated at Hospital de Oncología IMSS, Mexico. Treatment examined over 5 years | |
| Context/settings | Mexico, 21 advanced GIST patients who were treated at Hospital de Oncología IMSS | |
| Dates to which the data of the study relate | January 2005 to 31 December 2007 | |
| Source of effectiveness data |
Clinical trial and published literature Motzer et al. 2006104 – sunitinib Phase III study and study by Demetri et al. 200652 mainly from survival data and 21 advanced GIST patients who were treated at Hospital de Oncología IMSS |
|
| Modelling | Markov model. Model utilised the effectiveness data from Motzer et al. 2006104 (review of sunitinib treatment) – sunitinib Phase III study and study by Demetri et al. 200652 | |
| Link between effectiveness and costs data | All costs used in the model (except for the cost of sunitinib) were based on the information from IMSS pricing and reimbursement procedures. For cost of sunitinib, as it was not available in the Mexican market at the time of the analysis, the cost information was provided by Pfizer Laboratories. Costs included cost of mean number of visits to the oncologist, laboratory examinations, and radiology procedures, and cost of mean length of stay | |
| Information on the clinical evidence and effectiveness, main outcome of the study | Sample patients/study sample/patient groups | Twenty-one advanced GIST patients who were treated at Hospital de Oncología IMSS and hypothetical cohort of 1000 patients for modelling exercise |
| Study design | Observation study based on 21 patients and Markov modelling with a follow-up period of 5 years | |
| Effectiveness analysis | PFMs, PFS, LYG | |
| Effectiveness measures and results/outcome measures | ||
|
Primary end points/outcome and secondary end points/outcome Statistical precision of these outcomes |
PFMs 5.64 and 1.4 LYG (95% CI 1.3 to 1.6) for sunitinib Imatinib – PFM = 5.28 and 1.31 LYG (95% CI 1.1 to 1.4) BSC – PFM = 2.52 and 1.08 LYG (95% CI 1.0 to 1.3) |
|
| Clinical recommendations and conclusion | Sunitinib as second line of treatment for those who failed with 400 mg | |
| Economic analysis | Measures of health outcome/benefits used in the economic analysis |
PFMs LYGs |
| Direct costs and its components |
Direct costs estimated from treatment follow-up, health systems perspective Imatinib higher dose: expected costs per patient US$35,225 (SD US$1253) Sunitinib: expected costs per patient US$17,805 (SD US$694.83) BSC: expected cost per patient US$2071.86 (SD US$472.88) Using IMSS data, the estimated annual cost per patient for medical consultation, hospitalisation, laboratory examination and radiology procedures was $2424.32, $2657.57, $566.99 and $2392.67, respectively |
|
| Indirect costs and its components | ||
| Cost of productivity, cost of volunteer care and support for the patient | Not taken into consideration | |
| Currency, year prices | US$, at 2006 prices | |
| Statistical analysis/cost (whether parametric or non-parametric bootstrap used to generate the CIs around each difference in costs and differences in total costs | Standard deviation of the mean costs, and mean life-years saved, and CI of the mean life-years saved | |
| Sensitivity analysis: one way or two way |
Monte Carlo second order sensitivity analysis, probabilistic sensitivity analysis conducted Results from the sensitivity analysis were used to develop the acceptability curve |
|
| Results/major findings | Benefits results from the economic evaluation |
Sunitinib resulted in mean PFMs of 5.64, and 1.4 LYG For imatinib, PFM = 5.28, and 1.31 LYG For BSC, PFM = 2.52, and 1.08 LYG Incrementally, sunitinib yielded 0.32 LYG when compared with BSC ICER: sunitinib vs BSC $15,734.23 per patient treated with sunitinib and $56,612.55 per year of PFS and $46,108.89 per LYG |
| Costs results used in the economic evaluation |
Imatinib higher dose: expected cost per patient US$35,225 (SD US$1253) Sunitinib: expected cost per patient US$17,805 (SD US$694.83) BSC: expected cost per patient – US$2071.86 (SD US$472.88) Using IMSS data, the estimated annual cost per patient for medical consultation, hospitalisation, laboratory examination and radiology procedures was $2424.32, $2657.57, $566.99 and $2392.67, respectively |
|
| Author conclusion/recommendations | Reimbursing sunitinib over high dose of imatinib would deliver cost savings to the IMSS and greater survival benefits |
| Study identification | Author and year | Mabasa 200898 |
| Intervention studied/comparators | Imatinib vs no imatinib (BSC) in GISTs | |
| Hypothesis/question | Examine the cost-effectiveness of imatinib | |
| Key features of the study | Type of study | Full economic evaluation (cost-effectiveness analysis) |
| Target population/sample population | Patients in British Columbia, BCCA patients with advanced GIST who received imatinib or historical treatment | |
| Context/settings | BCCA-registered patients with advanced GIST, British Columbia, Canada | |
| Dates to which the data of the study relate |
1996–2001 for non-imatinib cases 2002–5 imatinib cases. Follow-up periods: 60 months and 44 months, respectively |
|
| Source of effectiveness data | Data derived from medical records of the patients | |
| Modelling | No modelling, patient-level data used for CEA | |
| Link between effectiveness and costs data |
All costs used were based on the information on the BCCA patients followed and included on an intention-to-treat basis. The mean and median duration of follow-up for the imatinib group were significantly longer than for the historical group Costs of treatment include cost of drugs, cost per cycle of 1 month, cost of labour and supply (not clearly specified what it includes) and cost of counselling Costing was based on BCCA registry: |
|
| Information on the clinical evidence and effectiveness – main outcome of the study | Sample patients/study sample/patient groups |
46 imatinib group 47 no imatinib (historical) group |
| Study design | Retrospective follow up case–control study based on medical records | |
| Effectiveness analysis | Kaplan–Meier estimates of OS and imatinib and historical groups | |
| Effectiveness measures and results/outcome measures | ||
|
Primary end points/outcome and secondary end points/outcome Statistical precision of these outcomes |
Median OS (months) Imatinib 66.7 No imatinib 7.7 Median PFS (months) Imatinib 45.3 No imatinib 5.6 OS at 1 year Imatinib 95.4% No imatinib 32.6% PFS at 1 year Imatinib 81.4% No imatinib 17.4% |
|
| Clinical recommendations and conclusion | Patient receiving imatinib had significantly longer median OS and median PFS, and higher 1-year OS and 1-year PFS than the historical group | |
| Economic analysis | Measures of health outcome/benefits used in the economic analysis | OS, PFS and life-year gained |
| Direct costs and its components | Details provided in methods section on actual cost of drugs, labour and supply, but no results given | |
|
Prospective or retrospective (depend on study design) Whether values were imputed in for certain cases How hospital stay was defined, and whether any classifications were used or not |
Mean costs per patient: $79,829 imatinib; $1743 no imatinib Costs of surgery or radiotherapy not included (though similar in both arms) |
|
|
Costing of complications or side effects Estimations of unit costs and source/methods |
Did not include the cost of side effects, cost of health-care visits, or supportive care Cost of drugs presumably include cost of side effects treatment |
|
| Indirect costs and its components | ||
| Cost of productivity, cost of volunteer care and support for the patient | Not included | |
| Currency, year prices | C$, 2006 prices | |
| Sensitivity analysis | Conducted univariate sensitivity analysis to examine the impact of upper and lower values of the cost of the drugs, the cost of treatment, the utilities of successful treatment and PD, the time horizon, and the annual rate of discount. They used imatinib at a 600 mg/day dose to examine the impact of results variation as an alternative scenario for the sensitivity analysis | |
| Results/major findings | Benefits results from the economic evaluation |
Mean OS from imatinib 66.7 months, and historical control group 7.7 months Mean PFS – 45.3 months vs 5.6 months |
| Costs results used in the economic evaluation |
Cost of treatment, costs to health sector (cost to NHS) Major determinants of costs, the principle costs drivers |
|
| Synthesis of costs and benefits | Conducted the sensitivity analysis | |
| Author conclusion/recommendations | Imatinib cost-effective in treatment of GIST with an ICER of $15,882 |
| Study identification | Author and year | Paz-Ares 200899 |
| Intervention studied/comparators | Sunitinib (50 mg/day) with BSC and BSC alone | |
| Hypothesis/question | Assess cost-effectiveness of sunitinib vs BSC as second line of treatment | |
| Key features of the study | Type of study | Full economic evaluation (cost-effectiveness analysis) |
| Target population/sample population | Hypothetical cohort of Spanish population with GIST after progression with imatinib. Perspective – Spanish national health system | |
| Context/settings | Patients with advanced unresectable GIST, intolerant to or with diseases progressing during treatment with imatinib | |
| Dates to which the data of the study relate | Used Demetri et al. 2006 study52 | |
| Source of effectiveness data |
Used Demetri et al. 2006 study52 Expert panel, three pathology experts, three health economists |
|
| Modelling | Markov model | |
| Link between effectiveness and costs data | Data reported by expert panel on number of visits to oncology clinic, laboratory tests, CT scans, nurse visits, visits to palliative units and analgesic drugs. QoL obtained from EQ-5D scores of A6181004 (Demetri study population) | |
| Information on the clinical evidence and effectiveness – main outcome of the study | Sample patients/study sample/patient groups | Hypothetical cohort of patients with advanced unresectable GIST, intolerant to or with disease progressing during treatment with imatinib (same as Demetri study??) |
| Study design | Decision model analysis, based on the trial52 | |
| Effectiveness analysis |
LYG, QALY Progression-free life-years Total mean cost per patient Cost per QALY gained ICER |
|
| Effectiveness measures and results/outcome measures | ||
|
Primary end points/outcome and secondary end points/outcome Statistical precision of these outcomes |
OS, LYG PFS Incidence and treatment of adverse effects |
|
| Clinical recommendations and conclusion | According to oncology thresholds for oncology patients, sunitinib is considered better | |
| Economic analysis | Measures of health outcome/benefits used in the economic analysis | QoL obtained from EQ-5D scores |
| Direct costs and its components | Total mean costs/patient | |
| €23,259 in sunitinib group (including costs of adverse events) as against €1622 for BSC | ||
| Indirect costs and its components | ||
| Cost of productivity, cost of volunteer care and support for the patient | Not included | |
| Currency, year prices | €, 2007 prices | |
| Statistical analysis/cost (whether parametric or non-parametric boot strap used to generate the CIs around each difference in costs and differences in total costs | Deterministic | |
| Sensitivity analysis | Univariate sensitivity analysis | |
| Results/major findings | Benefits results from the economic evaluation |
Patients benefits in LYG: 1.59 (for sunitinib + BSC) vs 0.88 (BSC) Progression-free life-years: 0.50 (sunitinib) vs 0.24 (BSC) QALY 1 vs 0.55 |
| Costs results used in the economic evaluation |
Total mean costs/patients: €23,259 vs €1622 |
|
| Synthesis of cost and benefits |
Treatment with sunitinib vs BSC resulted in patients’ benefits of 0.26 progression-free life-years, 0.71 LYG and 0.45 QALYs gained with the cost difference of €21,637/per patient between both treatments ICER of sunitinib vs BSC:Univariate sensitivity analysis The most important variables: OS HR Cost of sunitinib Utility value during active treatment and after progression |
|
| Any attempt to consider the uncertainty surrounding estimates of effects |
Yes, considered the uncertainty surrounding estimates of effects Considering ± 25% variation on the OS, the parameter most influencing the model results, the ICER/QALY gained would oscillate between €39,201 and €62,806 |
|
| Author conclusion/recommendations |
Sunitinib can be considered cost-effective vs BSC with acceptable cost per LYG and QALY gained Notes the limitation in using an extrapolated survival curve |
| Study identification | Author and year | Huse 200797 |
| Intervention studied/comparators | Imatinib in the treatment of advanced GIST | |
| Hypothesis/question | Estimated the cost-effectiveness of imatinib mesylate in treatment of unresectable GIST using trials data elsewhere and using them in US context | |
| Key features of the study | Type of study | Cost-effectiveness modelling for decision analysis |
| Target population/sample population | Advanced GIST patients | |
| Context/settings | USA, imatinib mesylate treatment vs no treatment of advanced hypothetical GIST population in USA | |
| Dates to which the data of the study relates to | Mostly trial data used: Demetri et al. 200238 trial data and Blanke trial39,103,117 data and Phase II clinical trial data | |
| Source of effectiveness data | Demetri et al. 200238 trial data and Blanke trial39,103,117 data | |
| Modelling | Decision modelling | |
| Link between effectiveness and costs data |
Imatinib cost: Pharmacy’s Fundamental Reference. Montvale, NJ: Thomson Health Care; 2005, and Physicians’ Desk Reference 2005. Montvale, NJ: Thomson PDR; 2005 Cost of medical management for pancreatic cancer was used in absence of data for GIST management Cost data for diseases specific For palliative care – as GIST-specific palliative care data not available, information on palliative care for pancreatic cancer was used |
|
| Information on the clinical evidence and effectiveness – main outcome of the study | Sample patients/study sample/patient groups | Hypothetical cohort population with advanced GIST |
| Study design | Decision model | |
| Effectiveness analysis | QALY | |
| Effectiveness measures and results/outcome measures | Used from UK study (Wilson et al.55) | |
|
Primary end points/outcome and secondary end points/outcome Statistical precision of these outcomes |
Utilities 0.875 for PD (lower bound 0.75 to 1.00 upper) 0.935 for successful treatment (0.4 to 1.00) |
|
| Clinical recommendations and conclusion | Imatinib is cost-effective in advanced GIST patients | |
| Economic analysis | Measures of health outcome/benefits used in the economic analysis | QALY, OS, cost, cost per LYG and cost per QALY gained |
| Indirect costs and its components | ||
| Cost of productivity, cost of volunteer care and support for the patient | Not included | |
| Currency, year prices | US$, 2005 prices | |
| Sensitivity analysis | One-way sensitivity analysis | |
| Results/major findings | Benefits results from the economic evaluation |
Effectiveness QALYs – 4.15 for imatinib, 2.23 for untreated Difference (treated – untreated) 1.92 The net discounted cost of achieving the survival benefit of 2.2 QALY (PV of 1.9 QALY) is US$74,369 per imatinib-treated patient CER – US$38,723 |
|
Costs results used in the economic evaluation Cost of treatment, costs to health sector (cost to NHS) Major determinants of costs, the principle costs drivers |
Imatinib treatment US$416,255 Untreated US$341,886 Weekly cost of imatinib: $US685 (685 to 1028) Weekly costs of care successfully treated patients: US$359 (226 to 492) Weekly cost of care for PD: US$2575 (1700 to 3450) Utilities of successful treatment and PD: 0.935, 0.875, respectively Time horizon (years): 10, 20 in sensitivity analysis Major cost drivers – cost of drugs |
|
|
Synthesis of cost and benefits Any attempt to consider the uncertainty surrounding estimates of effects |
The cost-effectiveness ratio was most sensitive to variation in the cost estimates and time horizon for the analysis CER ratios were estimated for the upper and lower bound of the parameters |
|
| Author conclusion/recommendations | Over 10 years’ time horizon, imatinib treatment increases mean quality-adjusted survival from 2.4 to 4.6 QALYs, this gain of 2.2 QALYs (undiscounted) with PV of 1.92 QALYs. Net undiscounted cost of achieving this survival benefit is US$74,369 per imatinib-treated patient, yielding a cost-effectiveness ratio of US$38,723 per QALY |
| Study identification | Author and year | Teich 2009100 |
| Intervention studied/comparators | Sunitinib vs imatinib 800 mg/day, and BSC for those who failed with imatinib 400 mg/day | |
| Hypothesis/question | What is the cost-effectiveness of sunitinib vs imatinib in second-line treatment for GIST in Brazil | |
| Key features of the study | Type of study | Model analysis |
| Target population/sample population | Cohort population failed with imatinib 400 mg/day | |
| Dates to which the data of the study relate | Not specified, 2005 prices used | |
| Modelling | Markov model | |
| Link between effectiveness and costs data |
Cost per LYGs, cost per progression-free life-years ICER |
|
| Information on the clinical evidence and effectiveness – main outcome of the study | Sample patients/study sample/patient groups | Cohort population number 1000 |
| Study design | Modelling | |
| Effectiveness analysis |
In comparison with BSC sunitinib increases life-years and progression-free life-years by 0.3 and 0.26 years, respectively With incremental costs of R$86,756 (US$61,968, PPP 2005) In comparison with imatinib, sunitinib was more effective and cost-effective with increased life-year of 0.02 and progression-free LYG of 0.47, and less costly over 6 years |
|
| Results/major findings | Author conclusion/recommendations | Sunitinib is cost-effective when compared with imatinib 800 mg/day and BSC |
| Study identification | Author and year | Wilson 200555 |
| Intervention studied/comparators | Cost-effectiveness of imatinib in the treatment of unresectable and/or metastatic KIT-positive GIST relative to current standard practice | |
| Hypothesis/question | Assess the clinical effectiveness and cost-effectiveness of imatinib in the treatment of unresectable and/or metastatic KIT-positive GIST relative to current standard practice | |
| Key features of the study | Type of study | Systematic review of clinical effectiveness and economic evaluation |
| Target population/sample population | Hypothetical cohort population with unresectable GIST in UK | |
| Context/settings | UK NHS perspective | |
| Dates to which the data of the study relates to | 2004? | |
| Source of effectiveness data |
Trials Novartis model from clinical trial |
|
| Modelling |
Markov modelling Reporting results from two modelling works |
|
| Link between effectiveness and costs data | ICER, cost per QALY | |
| Information on the clinical evidence and effectiveness – main outcome of the study | Sample patients/study sample/patient groups |
Trial patients – 147 patients with malignant unresectable and/or metastatic GISTs with median follow-up 25 months Modelled for 10 years |
| Study design | Open-label multicentre trial compared two imatinib doses: 400 or 600 mg/day | |
| Effectiveness analysis | The survival rate was 88% after 1 year and 78% after 2 years | |
| Clinical recommendations and conclusion | The survival rate was 88% after 1 year and 78% after 2 years | |
| Economic analysis | Measures of health outcome/benefits used in the economic analysis | QALYs from ECOG performance of the trial patients |
| Direct costs and its components | ||
| Prospective or retrospective (depend on study design) | Prospective as trial data | |
|
Whether values were imputed in for certain cases How hospital stay was defined, and whether any classifications were used or not |
Values were not imputed as patients’ data were used from trials | |
| Costing of complications or side effects | Costs of side effects were available from patients’ data | |
| Estimations of unit costs and source/methods |
From Novartis model Drug cost of imatinib £20,000 Costs of outpatient visits £440 per year Cost of CT scan £656 for imatinib patients and £82 for patients with PD Cost of GP visits £40 per year Cost of management of adverse events £159 per year (range £127.20–190.80) Costs discounted at 6% (sensitivity – 3% and 6%) QALY discounted at 1.5% (sensitivity – 1.5–3%) Birmingham model developed for this report 4 weeks Cost of adverse event £12.23 Cost of imatinib 400 mg £1453.54 Cost of imatinib 600 mg £1874.49 Costs of no treatment (BSC) £43.23 Cost of terminal disease (death) £2730 Discounted rate for cost 0.0046154 Discounted rate for QALY 0.0011538 Other costs for imatinib-treated patients £87.38 Utility for imatinib 0.935 Utility for progressive state 0.875 Using incidence rate used by Novartis (15 per million population) and assuming 10–30% of all GIST patients expected to have metastatic and/or unresectable disease, the number of patients treated with metastatic and/or unresectable disease would be between 80 and 240, and the budgetary impact on the NHS is estimated at between £2.4M and £11.8M per year. The costs to the NHS per patient at £20,400 per year |
|
| Indirect costs and its components | Not included | |
| Currency, year prices | £, 2004 prices | |
| Results/major findings | Benefits results from the economic evaluation |
The cost per QALY ranged from £51,515 to £98,889 after 2 years and from £27,331 to £44,236 after 5 years and from £21,404 to £33,976 after 10 years Results from Birmingham model ICER changes depending whether Weibull or exponential distribution is used Weibull ICER – £26,427 Exponential ICER £21,707 |
|
Costs results used in the economic evaluation Cost of treatment, costs to health sector (cost to NHS) Major determinants of costs, the principle costs drivers |
From Novartis model Drug cost of imatinib £20,000 Costs of outpatient visits £440 per year Cost of CT scan £656 for imatinib patients and £82 for patients with PD Cost of GP visits £40 per year Cost of management of adverse events £159 per year (range £127.20–190.80) Weekly cost of imatinib (pooled trial data) £420.38 (£420.38–370.38; 400 mg per day start dose) Other costs per imatinib-treated patients £1136 (£1786–570) Others costs per PD patients £562 (£1498–233) Utilities: Imatinib treated 0.935 (0.900–0.935) Progressive 0.875 (0.875) Birmingham model developed for this report 4 weeks Cost of adverse event £12.23 Cost of imatinib 400 mg £1453.54 Cost of imatinib 600 mg £1874.49 Costs of no treatment (BSC) £43.23 Cost of terminal disease (death) £2730 Discounted rate for cost 0.0046154 Discounted rate for QALY 0.0011538 Other costs for imatinib-treated patients £87.38 Utility for imatinib 0.935 Utility for progressive state 0.875 |
|
|
Synthesis of cost and benefits Any attempt to consider the uncertainty surrounding estimates of effects |
Yes costs, discount rate, cost for acquisition of drugs | |
| Author conclusion/recommendations |
The Novartis model suggested that the costs per QALY gained ranged from £51,515 to £98,889 after 2 years, from £27,331 to £44,236 after 5 years and from £21,404 to £33,976 after 10 years. This range of estimates may still not reflect the uncertainty, as the estimates after 2 years are mainly based on mathematical extrapolation beyond observed data. The results from the Birmingham model confirm the findings of the Novartis model Because there were no directly controlled trials the results for the model cannot be very conclusive owing to the uncertainties |
| Study identification | Author and year | Reddy 200754 |
| Intervention studied/comparators | NA | |
| Hypothesis/question | NA | |
| Key features of the study | Type of study |
Systematic review to identify, summarise and evaluate published studies and abstracts describing the epidemiological, HRQoL and economic impact of GIST 2000–6 34 publications 29 provided data on epidemiology One provided cost data Three reported HRQoL One reported cost and HRQoL |
| Target population/sample population | NA | |
| Context/settings | NA | |
| Economic analysis | Measures of health outcome/benefits used in the economic analysis | Performance stated was assessed using ECOG scale performance take from Demetri et al. study52 |
| Results/major findings |
Costs results used in the economic evaluation Cost of treatment, costs to health sector (cost to NHS) Major determinants of costs, the principle costs drivers |
The acquisition costs of imatinib were estimated at $18 per 100-mg tablet in the USA and €23 in France Annual cost $32,850 in the USA and €41,975 in France (assuming 50% of patients each received 400 or 600 mg/day) UK study Annual drug cost £20,000 Outpatient visits including laboratory tests £440 GP visits £40 per year CT scans £656 for imatinib patients and £82 for patients with PD Management of adverse events: £159 (range £127–191) Another study (model base Wilson et al. 55) Annual costs of imatinib were £18,896 and £24,368 for patients on 400 and 600 mg daily, respectively |
|
Synthesis of cost and benefits Any attempt to consider the uncertainty surrounding estimates of effects |
Total costs with imatinib over 2 years £30,295 and for 10 years £47,521 BSC – £1949 at 2 years and £4047 at 10 years Cost QALY gained £85,224 after 2 years and £29,789 after 10 years Total costs were £31,160 at 2 years compared with £56,146 at 10 years with imatinib vs £1998 and £4230 at 2 and 10 years, respectively, with BSC The cost per QALY gain varied from £45,533 to £70,206 at 2 years and from £21,708 to £25,859 at 10 years |
| Study identification | Author and year | Hopkins 2008101 |
| Intervention studied/comparators | Sunitinib and imatinib, and placebo (different studies reviewed) | |
| Hypothesis/question | Review the new developments in therapeutic cancer drugs | |
| Key features of the study | Type of study | Review |
| Target population/sample population |
GIST patients, patients with diseases resistant to imatinib 800 mg/day or intolerant of imatinib Sample not applicable |
|
| Context/settings |
Settings of the clinical trials for sunitinib Three trials Phase III, 56 sites, Europe, America, Asia and Australia |
|
| Dates to which the data of the study relate | 2003, 2004, 2005 and 2009 | |
| Source of effectiveness data | Reviewed from all the studies mentioned | |
| Modelling | Not applicable | |
| Link between effectiveness and costs data | Not relevant | |
| Information on the clinical evidence and effectiveness – main outcome of the study | Sample patients/study sample/patient groups |
Maki118 2005 – 97 Demetri 200652 – 207 and 105 (placebo) George119 2007 – 60 |
| Clinical recommendations and conclusion |
Initial results for use of sunitinib are promising; however, too early to draw conclusion Important to consider the secondary resistance in GIST Mutational status should be determined before treatment in order to decide the initial dosage of kinase inhibitor |
|
| Economic analysis | Measures of health outcome/benefits used in the economic analysis | Referred to SMC study120 |
| Direct costs and its components | ||
|
Prospective or retrospective (depend on study design) Whether values were imputed in for certain cases How hospital stay was defined, and whether any classifications were used or not Costing of complications or side effects Estimations of unit costs and source/methods |
Not relevant – did not use or refer to studies with costing of the intervention Refer to SMC study120 Drug costs for one 6-week cycle of sunitinib 50 mg – £3304 for the 4–2 regimen – 4-cycle costing over £1300 |
|
| Indirect costs and its components | ||
| Cost of productivity, cost of volunteer care and support for the patient | Not considered | |
| Currency, year prices | Drug costs at 2006 prices | |
| Statistical analysis/cost | ||
| Sensitivity analysis | ||
| Results/major findings | Benefits results from the economic evaluation | |
| Costs results used in the economic evaluation | Drugs costs – UK NHS | |
|
Cost of treatment, costs to health sector (cost to NHS) Major determinants of costs, the principle costs drivers |
The total costs were not reported for the study reviewed. The costs are not from study reviewed | |
| Synthesis of cost and benefits: |
There was not a complete economic evaluation either referred or modelled in this study So synthesising not relevant |
|
| Any attempt to consider the uncertainty surrounding estimates of effects | No | |
| Author conclusion/recommendations | No recommendation from economic evaluation |
Appendix 12 Model structure
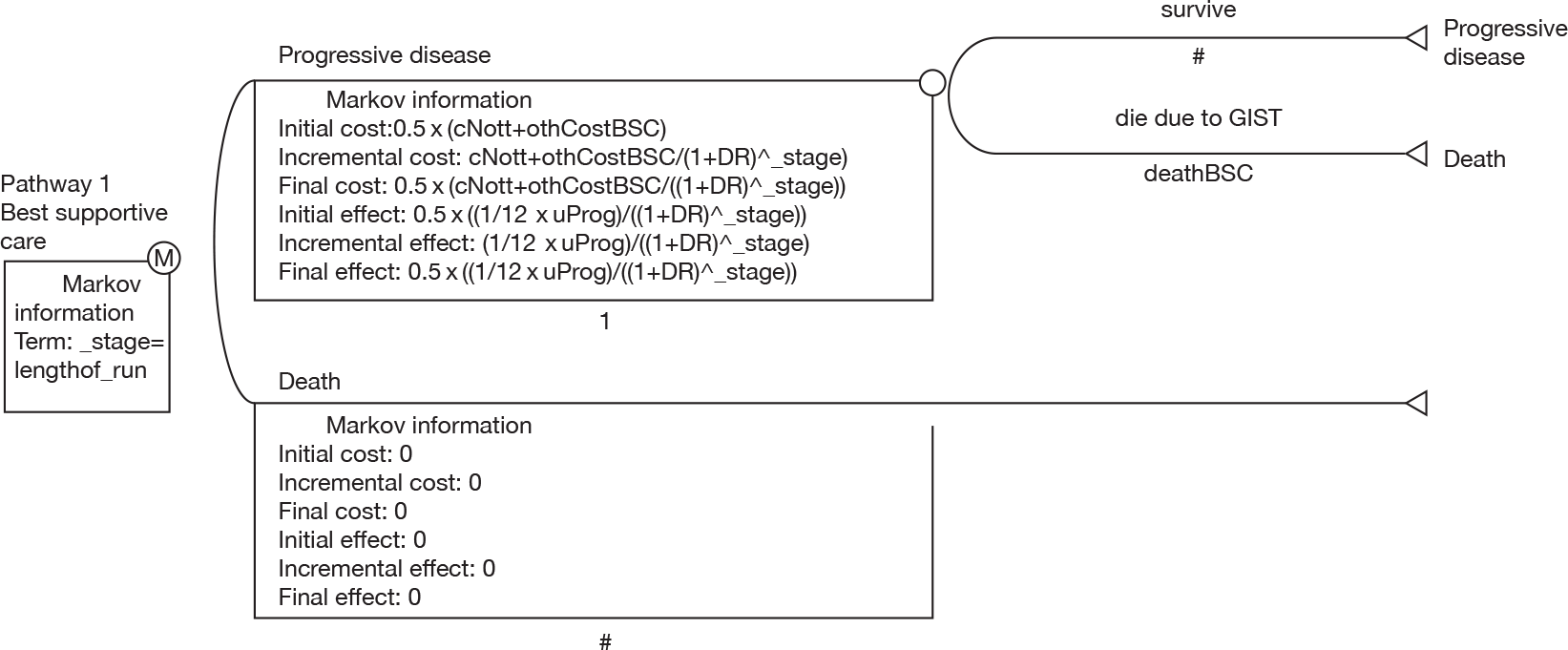
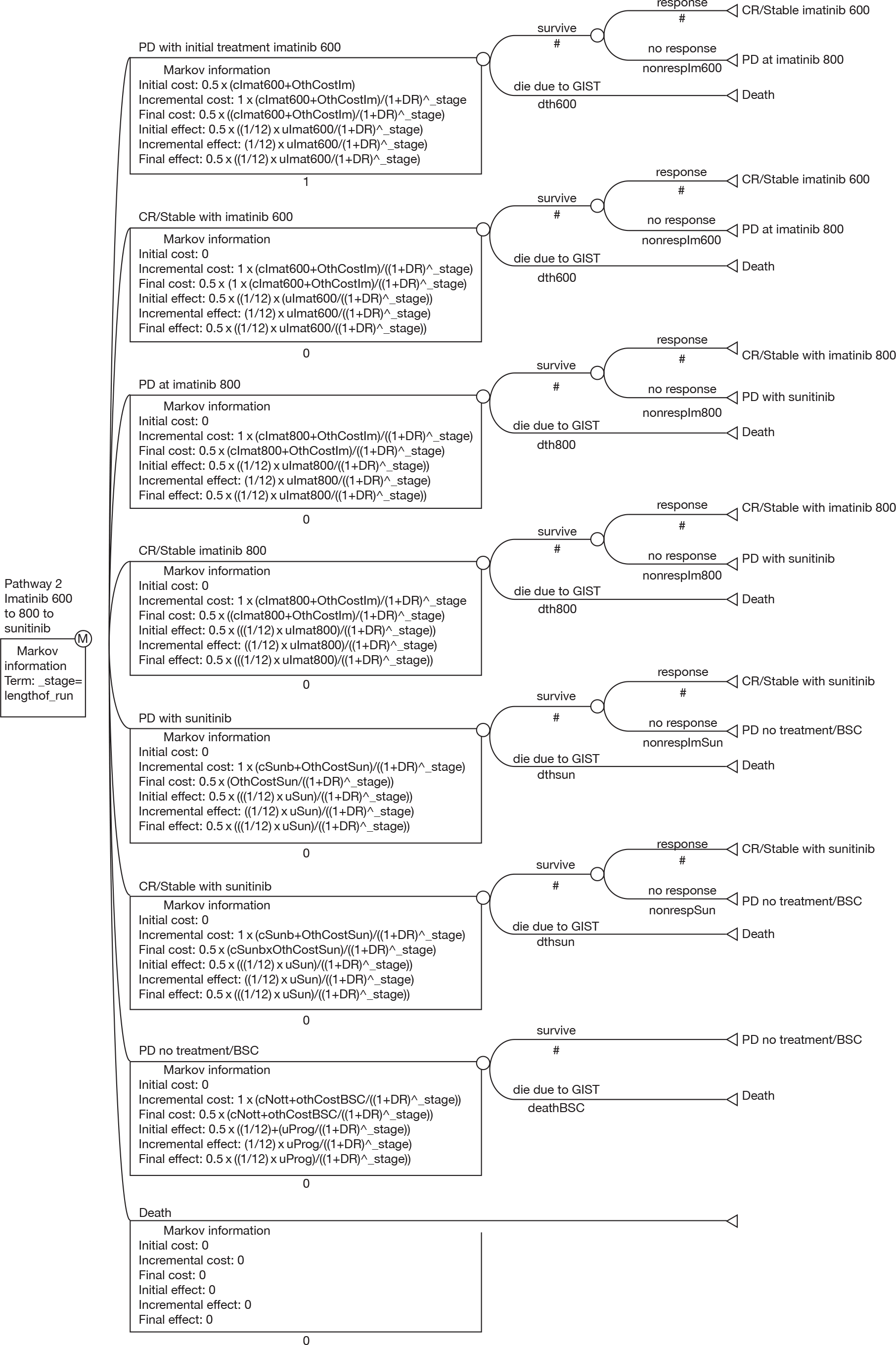
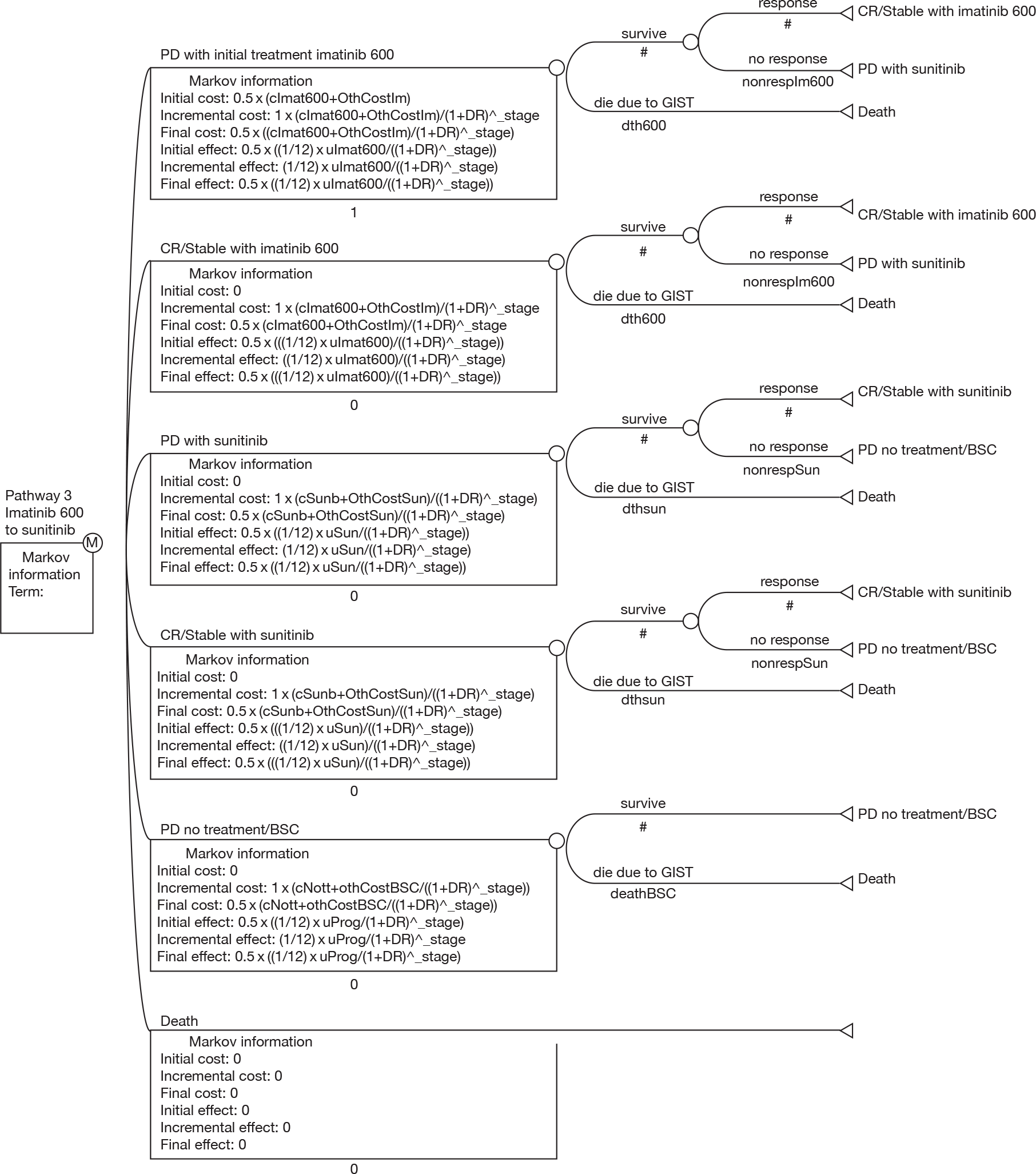
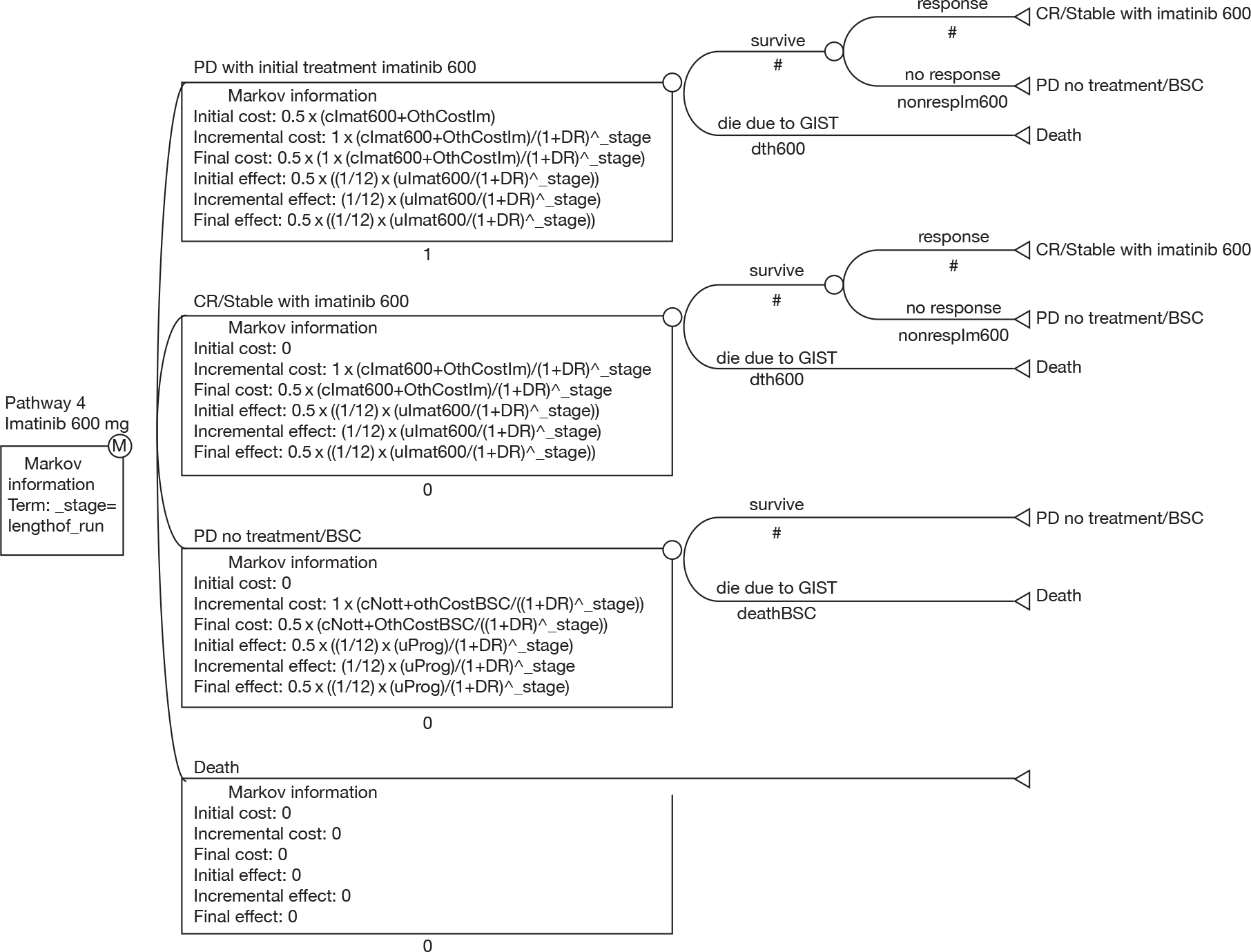
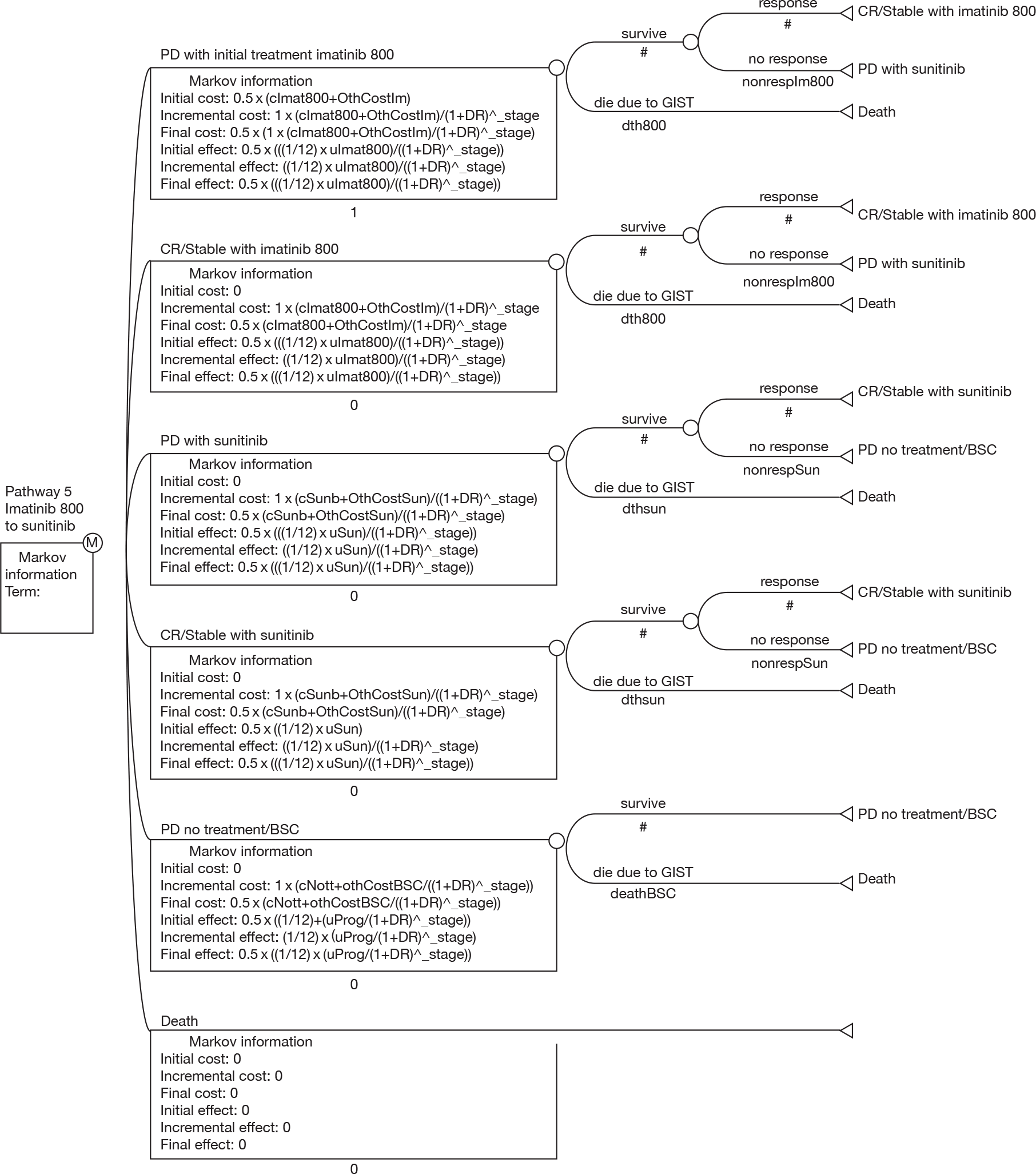
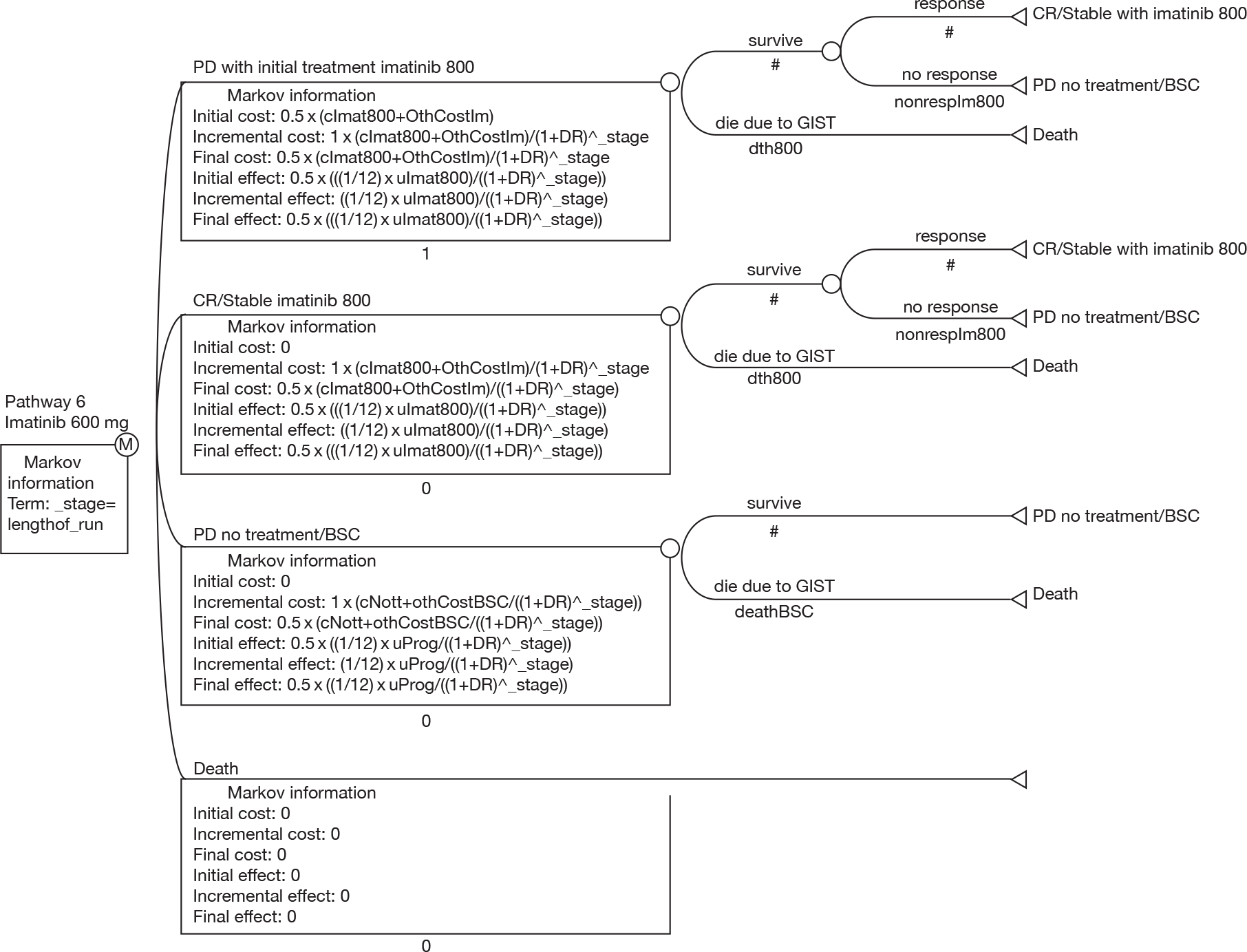
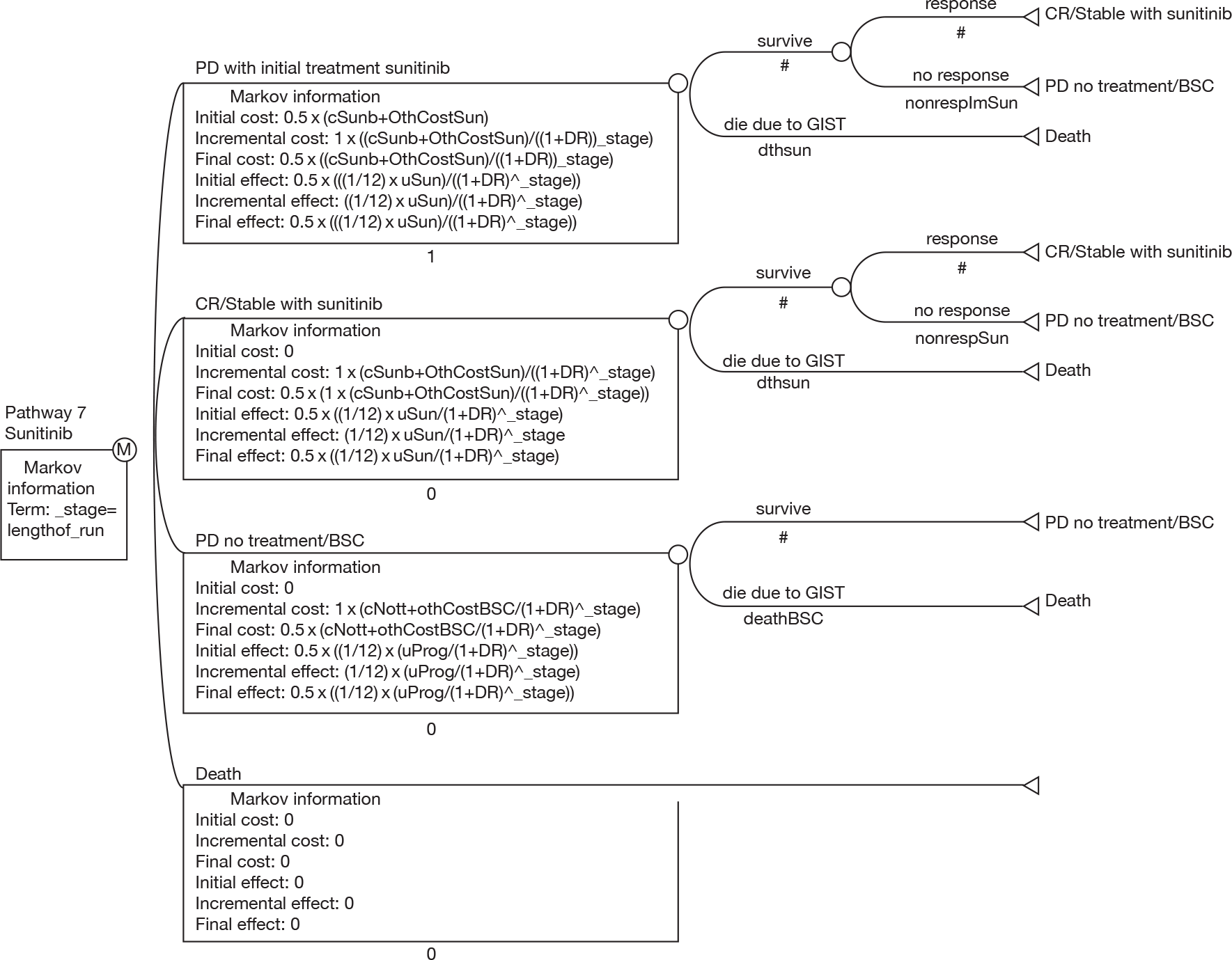
Appendix 13 Alternative best supportive care survival estimates
| Source | Year | Definition of population for which survival outcome is given | No. in sample | Follow-up time | Median OS | Percentage surviving |
|---|---|---|---|---|---|---|
| Conlon18 | 1995 | Those not having a complete resection | 38 | 5 years | 0 | |
| Dematteo19 | 2000 | Metastatic (including 28/94 who had complete resection) | 94 | 14 months | 19 months | |
| de Mestier121/Dematteo19 | 2005/2000 | Those not having a complete resection | 86 | 14 months | 12 months | < 30 at 1 year |
| Demetri52 | 2006 | Receiving placebo after median prior imatinib dose of 800 mg | 105 | 7.2 months | 62.5 | |
| Nilsson26 | 2005 | Those with overtly malignant GISTs | 29 | 1.4 years | 5/29 | |
| Von Mehren122 | 2006 | Those who had metastic GIST or recurrence after primary resection | 6–18 months | |||
| Plaat123 | 2000 | Those with malignant GIST (18/26 had metastatic disease) | 26 | 28 months | ||
| 2 years | 58.5 | |||||
| 5 years | 13.0 | |||||
| Pidhorecky124 | 2000 | Those undergoing palliative surgical procedure/biopsy | 11 | 5 years | 15 months | 10 |
| Those with unresectable metastatic GIST | 40 months | |||||
| Comandone125 | 2005 | Metastatic GIST | 6 months | |||
| Pierie107 | 2001 | Those with incomplete resection (41% had metastatic disease) | 69 | 3 years | 13 | |
| 5 years | 9 | |||||
| Duffaud126 | 2003 | Those with unresectable disease | 10–20 months | |||
| Cohen127 | 2002 | Those with metastatic or recurrent disease | 12–19 months | |||
| Totman128/Van%%Oosterom40 | 2001 | Those with unresectable or metastatic sarcoma (including GIST) | 53 weeks | |||
| Katz129 | 2008 | Those who could not undergo complete resection | 9–12 months | |||
| Trent130 | 2003 | Those with advanced/metastatic GIST treated with temozolomide, of which none responded | 17 | 2 years | 26.4 months | 62a |
| Le Cesne131/Verweij42 | 2009/2004 | Those presenting with incurable advanced disease | 2 years | 10.25 monthsa | 25 | |
| McGrath106 | 1987 | Those with partial resection | 21 | 5 years | 9 months | 10 |
| Those with distant metastases | 28 | 5 years | 10 months | 0 | ||
| Dougherty112 | 1991 | Presenting with unresectable disease | 15 | 2.167 years | 12 months | 3/15 |
| 2.75 years | 2/15 | |||||
| 4.167 years | 1/15 | |||||
| Artinyan113 | 2008 | Those with metastatic GIST | 140 | 3 years | 12 months | 24 |
| 3 years 11 months | 21 |
List of abbreviations
- AGITG
- Australasian Gastro-Intestinal Trials Group
- AiC
- academic in confidence
- ATP
- adenosine triphosphate
- BNF
- British National Formulary
- BSC
- best supportive care
- c-KIT
- cytokine-tyrosine kinase receptor
- CEA
- cost-effectiveness analysis
- CI
- confidence interval
- CiC
- commercial in confidence
- CT
- computerised tomography
- ECOG
- Eastern Cooperative Oncology Group
- EORTC
- European Organisation for Research and Treatment of Cancer
- EQ-5D
- European Quality of Life-5 Dimensions
- ESMO
- European Society for Medical Oncology
- FDG-PET
- fluorodeoxyglucose-positron emission tomography
- GI
- gastrointestinal
- GIST
- gastrointestinal stromal tumour
- HR
- hazard ratio
- HRQoL
- health-related quality of life
- ICER
- incremental cost-effectiveness ratio
- IM
- imatinib
- ISG
- Italian Sarcoma Group
- ISPOR
- International Society for Pharmacoeconomics and Outcomes Research
- KIT
- tyrosine kinase
- LYG
- life-year gain
- LYS
- life-year saved
- N/A
- not applicable
- NA
- not available
- NICE
- National Institute for Health and Clinical Excellence
- NIHR
- National Institute for Health Research
- NR
- not reported
- OS
- overall survival
- PD
- progressive disease
- PDGFRA
- platelet-derived growth factor receptor alpha
- PFM
- progression-free month
- PFS
- progression-free survival
- PR
- partial response
- QALY
- quality-adjusted life-year
- QoL
- quality of life
- RCT
- randomised controlled trial
- ReBIP
- Review Body for Interventional Procedures
- RECIST
- Response Evaluation Criteria in Solid Tumors
- RR
- relative risk
- SCF
- stem cell factor
- SD
- stable disease
- SMC
- Scottish Medicine Consortium
- TAR
- technology assessment review
- VEGFR
- vascular endothelial growth factor receptor
- WHO
- World Health Organization
- WMD
- weighted mean difference
All abbreviations that have been used in this report are listed here unless the abbreviation is well known (e.g. NHS), or it has been used only once, or it is a non-standard abbreviation used only in figures/tables/appendices, in which case the abbreviation is defined in the figure legend or in the notes at the end of the table.
NoteThis monograph is based on the Technology Assessment Report produced for NICE. The full report contained a considerable amount of data that was deemed commercial-in-confidence. The full report was used by the Appraisal Committee at NICE in their deliberations. The full report with each piece of commercial-in-confidence data removed and replaced by the statement ‘commercial-in-confidence information (or data) removed’ is available on the NICE website: www.nice.org.uk.
The present monograph presents as full a version of the report as is possible while retaining readability, but some sections, sentences, tables and figures have been removed. Readers should bear in mind that the discussion, conclusions and implications for practice and research are based on all the data considered in the original full NICE report.
Notes
Health Technology Assessment programme
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
Prioritisation Group
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Imti Choonara, Professor in Child Health, Academic Division of Child Health, University of Nottingham
Chair – Pharmaceuticals Panel
-
Dr Bob Coates, Consultant Advisor – Disease Prevention Panel
-
Dr Andrew Cook, Consultant Advisor – Intervention Procedures Panel
-
Dr Peter Davidson, Director of NETSCC, Health Technology Assessment
-
Dr Nick Hicks, Consultant Adviser – Diagnostic Technologies and Screening Panel, Consultant Advisor–Psychological and Community Therapies Panel
-
Ms Susan Hird, Consultant Advisor, External Devices and Physical Therapies Panel
-
Professor Sallie Lamb, Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick
Chair – HTA Clinical Evaluation and Trials Board
-
Professor Jonathan Michaels, Professor of Vascular Surgery, Sheffield Vascular Institute, University of Sheffield
Chair – Interventional Procedures Panel
-
Professor Ruairidh Milne, Director – External Relations
-
Dr John Pounsford, Consultant Physician, Directorate of Medical Services, North Bristol NHS Trust
Chair – External Devices and Physical Therapies Panel
-
Dr Vaughan Thomas, Consultant Advisor – Pharmaceuticals Panel, Clinical
Lead – Clinical Evaluation Trials Prioritisation Group
-
Professor Margaret Thorogood, Professor of Epidemiology, Health Sciences Research Institute, University of Warwick
Chair – Disease Prevention Panel
-
Professor Lindsay Turnbull, Professor of Radiology, Centre for the MR Investigations, University of Hull
Chair – Diagnostic Technologies and Screening Panel
-
Professor Scott Weich, Professor of Psychiatry, Health Sciences Research Institute, University of Warwick
Chair – Psychological and Community Therapies Panel
-
Professor Hywel Williams, Director of Nottingham Clinical Trials Unit, Centre of Evidence-Based Dermatology, University of Nottingham
Chair – HTA Commissioning Board
Deputy HTA Programme Director
HTA Commissioning Board
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
-
Department of Public Health and Epidemiology, University of Birmingham
-
Professor of Clinical Pharmacology, Director, NIHR HTA programme, University of Liverpool
-
Professor Ann Ashburn, Professor of Rehabilitation and Head of Research, Southampton General Hospital
-
Professor Peter Brocklehurst, Professor of Women’s Health, Institute for Women’s Health, University College London
-
Professor Jenny Donovan, Professor of Social Medicine, University of Bristol
-
Professor Jonathan Green, Professor and Acting Head of Department, Child and Adolescent Psychiatry, University of Manchester Medical School
-
Professor John W Gregory, Professor in Paediatric Endocrinology, Department of Child Health, Wales School of Medicine, Cardiff University
-
Professor Steve Halligan, Professor of Gastrointestinal Radiology, University College Hospital, London
-
Professor Freddie Hamdy, Professor of Urology, Head of Nuffield Department of Surgery, University of Oxford
-
Professor Allan House, Professor of Liaison Psychiatry, University of Leeds
-
Dr Martin J Landray, Reader in Epidemiology, Honorary Consultant Physician, Clinical Trial Service Unit, University of Oxford
-
Professor Stephen Morris, Professor of Health Economics, University College London, Research Department of Epidemiology and Public Health, University College London
-
Professor Irwin Nazareth, Professor of Primary Care and Head of Department, Department of Primary Care and Population Sciences, University College London
-
Professor E Andrea Nelson, Professor of Wound Healing and Director of Research, School of Healthcare, University of Leeds
-
Professor John David Norrie, Chair in Clinical Trials and Biostatistics, Robertson Centre for Biostatistics, University of Glasgow
-
Dr Rafael Perera, Lecturer in Medical Statisitics, Department of Primary Health Care, University of Oxford
-
Professor Barney Reeves, Professorial Research Fellow in Health Services Research, Department of Clinical Science, University of Bristol
-
Professor Martin Underwood, Professor of Primary Care Research, Warwick Medical School, University of Warwick
-
Professor Marion Walker, Professor in Stroke Rehabilitation, Associate Director UK Stroke Research Network, University of Nottingham
-
Dr Duncan Young, Senior Clinical Lecturer and Consultant, Nuffield Department of Anaesthetics, University of Oxford
-
Dr Tom Foulks, Medical Research Council
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
HTA Clinical Evaluation and Trials Board
-
Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick and Professor of Rehabilitation, Nuffield Department of Orthopaedic, Rheumatology and Musculoskeletal Sciences, University of Oxford
-
Professor of the Psychology of Health Care, Leeds Institute of Health Sciences, University of Leeds
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Keith Abrams, Professor of Medical Statistics, Department of Health Sciences, University of Leicester
-
Professor Martin Bland, Professor of Health Statistics, Department of Health Sciences, University of York
-
Professor Jane Blazeby, Professor of Surgery and Consultant Upper GI Surgeon, Department of Social Medicine, University of Bristol
-
Professor Julia M Brown, Director, Clinical Trials Research Unit, University of Leeds
-
Professor Alistair Burns, Professor of Old Age Psychiatry, Psychiatry Research Group, School of Community-Based Medicine, The University of Manchester & National Clinical Director for Dementia, Department of Health
-
Dr Jennifer Burr, Director, Centre for Healthcare Randomised trials (CHART), University of Aberdeen
-
Professor Linda Davies, Professor of Health Economics, Health Sciences Research Group, University of Manchester
-
Professor Simon Gilbody, Prof of Psych Medicine and Health Services Research, Department of Health Sciences, University of York
-
Professor Steven Goodacre, Professor and Consultant in Emergency Medicine, School of Health and Related Research, University of Sheffield
-
Professor Dyfrig Hughes, Professor of Pharmacoeconomics, Centre for Economics and Policy in Health, Institute of Medical and Social Care Research, Bangor University
-
Professor Paul Jones, Professor of Respiratory Medicine, Department of Cardiac and Vascular Science, St George‘s Hospital Medical School, University of London
-
Professor Khalid Khan, Professor of Women’s Health and Clinical Epidemiology, Barts and the London School of Medicine, Queen Mary, University of London
-
Professor Richard J McManus, Professor of Primary Care Cardiovascular Research, Primary Care Clinical Sciences Building, University of Birmingham
-
Professor Helen Rodgers, Professor of Stroke Care, Institute for Ageing and Health, Newcastle University
-
Professor Ken Stein, Professor of Public Health, Peninsula Technology Assessment Group, Peninsula College of Medicine and Dentistry, Universities of Exeter and Plymouth
-
Professor Jonathan Sterne, Professor of Medical Statistics and Epidemiology, Department of Social Medicine, University of Bristol
-
Mr Andy Vail, Senior Lecturer, Health Sciences Research Group, University of Manchester
-
Professor Clare Wilkinson, Professor of General Practice and Director of Research North Wales Clinical School, Department of Primary Care and Public Health, Cardiff University
-
Dr Ian B Wilkinson, Senior Lecturer and Honorary Consultant, Clinical Pharmacology Unit, Department of Medicine, University of Cambridge
-
Ms Kate Law, Director of Clinical Trials, Cancer Research UK
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
Diagnostic Technologies and Screening Panel
-
Scientific Director of the Centre for Magnetic Resonance Investigations and YCR Professor of Radiology, Hull Royal Infirmary
-
Professor Judith E Adams, Consultant Radiologist, Manchester Royal Infirmary, Central Manchester & Manchester Children’s University Hospitals NHS Trust, and Professor of Diagnostic Radiology, University of Manchester
-
Mr Angus S Arunkalaivanan, Honorary Senior Lecturer, University of Birmingham and Consultant Urogynaecologist and Obstetrician, City Hospital, Birmingham
-
Dr Diana Baralle, Consultant and Senior Lecturer in Clinical Genetics, University of Southampton
-
Dr Stephanie Dancer, Consultant Microbiologist, Hairmyres Hospital, East Kilbride
-
Dr Diane Eccles, Professor of Cancer Genetics, Wessex Clinical Genetics Service, Princess Anne Hospital
-
Dr Trevor Friedman, Consultant Liason Psychiatrist, Brandon Unit, Leicester General Hospital
-
Dr Ron Gray, Consultant, National Perinatal Epidemiology Unit, Institute of Health Sciences, University of Oxford
-
Professor Paul D Griffiths, Professor of Radiology, Academic Unit of Radiology, University of Sheffield
-
Mr Martin Hooper, Public contributor
-
Professor Anthony Robert Kendrick, Associate Dean for Clinical Research and Professor of Primary Medical Care, University of Southampton
-
Dr Nicola Lennard, Senior Medical Officer, MHRA
-
Dr Anne Mackie, Director of Programmes, UK National Screening Committee, London
-
Mr David Mathew, Public contributor
-
Dr Michael Millar, Consultant Senior Lecturer in Microbiology, Department of Pathology & Microbiology, Barts and The London NHS Trust, Royal London Hospital
-
Mrs Una Rennard, Public contributor
-
Dr Stuart Smellie, Consultant in Clinical Pathology, Bishop Auckland General Hospital
-
Ms Jane Smith, Consultant Ultrasound Practitioner, Leeds Teaching Hospital NHS Trust, Leeds
-
Dr Allison Streetly, Programme Director, NHS Sickle Cell and Thalassaemia Screening Programme, King’s College School of Medicine
-
Dr Matthew Thompson, Senior Clinical Scientist and GP, Department of Primary Health Care, University of Oxford
-
Dr Alan J Williams, Consultant Physician, General and Respiratory Medicine, The Royal Bournemouth Hospital
-
Dr Tim Elliott, Team Leader, Cancer Screening, Department of Health
-
Dr Joanna Jenkinson, Board Secretary, Neurosciences and Mental Health Board (NMHB), Medical Research Council
-
Professor Julietta Patrick, Director, NHS Cancer Screening Programme, Sheffield
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Disease Prevention Panel
-
Professor of Epidemiology, University of Warwick Medical School, Coventry
-
Dr Robert Cook, Clinical Programmes Director, Bazian Ltd, London
-
Dr Colin Greaves, Senior Research Fellow, Peninsula Medical School (Primary Care)
-
Mr Michael Head, Public contributor
-
Professor Cathy Jackson, Professor of Primary Care Medicine, Bute Medical School, University of St Andrews
-
Dr Russell Jago, Senior Lecturer in Exercise, Nutrition and Health, Centre for Sport, Exercise and Health, University of Bristol
-
Dr Julie Mytton, Consultant in Child Public Health, NHS Bristol
-
Professor Irwin Nazareth, Professor of Primary Care and Director, Department of Primary Care and Population Sciences, University College London
-
Dr Richard Richards, Assistant Director of Public Health, Derbyshire County Primary Care Trust
-
Professor Ian Roberts, Professor of Epidemiology and Public Health, London School of Hygiene & Tropical Medicine
-
Dr Kenneth Robertson, Consultant Paediatrician, Royal Hospital for Sick Children, Glasgow
-
Dr Catherine Swann, Associate Director, Centre for Public Health Excellence, NICE
-
Mrs Jean Thurston, Public contributor
-
Professor David Weller, Head, School of Clinical Science and Community Health, University of Edinburgh
-
Ms Christine McGuire, Research & Development, Department of Health
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
External Devices and Physical Therapies Panel
-
Consultant Physician North Bristol NHS Trust
-
Reader in Wound Healing and Director of Research, University of Leeds
-
Professor Bipin Bhakta, Charterhouse Professor in Rehabilitation Medicine, University of Leeds
-
Mrs Penny Calder, Public contributor
-
Dr Dawn Carnes, Senior Research Fellow, Barts and the London School of Medicine and Dentistry
-
Dr Emma Clark, Clinician Scientist Fellow & Cons. Rheumatologist, University of Bristol
-
Mrs Anthea De Barton-Watson, Public contributor
-
Professor Nadine Foster, Professor of Musculoskeletal Health in Primary Care Arthritis Research, Keele University
-
Dr Shaheen Hamdy, Clinical Senior Lecturer and Consultant Physician, University of Manchester
-
Professor Christine Norton, Professor of Clinical Nursing Innovation, Bucks New University and Imperial College Healthcare NHS Trust
-
Dr Lorraine Pinnigton, Associate Professor in Rehabilitation, University of Nottingham
-
Dr Kate Radford, Senior Lecturer (Research), University of Central Lancashire
-
Mr Jim Reece, Public contributor
-
Professor Maria Stokes, Professor of Neuromusculoskeletal Rehabilitation, University of Southampton
-
Dr Pippa Tyrrell, Senior Lecturer/Consultant, Salford Royal Foundation Hospitals’ Trust and University of Manchester
-
Dr Nefyn Williams, Clinical Senior Lecturer, Cardiff University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Interventional Procedures Panel
-
Professor of Vascular Surgery, University of Sheffield
-
Consultant Colorectal Surgeon, Bristol Royal Infirmary
-
Mrs Isabel Boyer, Public contributor
-
Mr Sankaran Chandra Sekharan, Consultant Surgeon, Breast Surgery, Colchester Hospital University NHS Foundation Trust
-
Professor Nicholas Clarke, Consultant Orthopaedic Surgeon, Southampton University Hospitals NHS Trust
-
Ms Leonie Cooke, Public contributor
-
Mr Seumas Eckford, Consultant in Obstetrics & Gynaecology, North Devon District Hospital
-
Professor Sam Eljamel, Consultant Neurosurgeon, Ninewells Hospital and Medical School, Dundee
-
Dr Adele Fielding, Senior Lecturer and Honorary Consultant in Haematology, University College London Medical School
-
Dr Matthew Hatton, Consultant in Clinical Oncology, Sheffield Teaching Hospital Foundation Trust
-
Dr John Holden, General Practitioner, Garswood Surgery, Wigan
-
Dr Fiona Lecky, Senior Lecturer/Honorary Consultant in Emergency Medicine, University of Manchester/Salford Royal Hospitals NHS Foundation Trust
-
Dr Nadim Malik, Consultant Cardiologist/Honorary Lecturer, University of Manchester
-
Mr Hisham Mehanna, Consultant & Honorary Associate Professor, University Hospitals Coventry & Warwickshire NHS Trust
-
Dr Jane Montgomery, Consultant in Anaesthetics and Critical Care, South Devon Healthcare NHS Foundation Trust
-
Professor Jon Moss, Consultant Interventional Radiologist, North Glasgow Hospitals University NHS Trust
-
Dr Simon Padley, Consultant Radiologist, Chelsea & Westminster Hospital
-
Dr Ashish Paul, Medical Director, Bedfordshire PCT
-
Dr Sarah Purdy, Consultant Senior Lecturer, University of Bristol
-
Dr Matthew Wilson, Consultant Anaesthetist, Sheffield Teaching Hospitals NHS Foundation Trust
-
Professor Yit Chiun Yang, Consultant Ophthalmologist, Royal Wolverhampton Hospitals NHS Trust
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Pharmaceuticals Panel
-
Professor in Child Health, University of Nottingham
-
Senior Lecturer in Clinical Pharmacology, University of East Anglia
-
Dr Martin Ashton-Key, Medical Advisor, National Commissioning Group, NHS London
-
Dr Peter Elton, Director of Public Health, Bury Primary Care Trust
-
Dr Ben Goldacre, Research Fellow, Division of Psychological Medicine and Psychiatry, King’s College London
-
Dr James Gray, Consultant Microbiologist, Department of Microbiology, Birmingham Children’s Hospital NHS Foundation Trust
-
Dr Jurjees Hasan, Consultant in Medical Oncology, The Christie, Manchester
-
Dr Carl Heneghan, Deputy Director Centre for Evidence-Based Medicine and Clinical Lecturer, Department of Primary Health Care, University of Oxford
-
Dr Dyfrig Hughes, Reader in Pharmacoeconomics and Deputy Director, Centre for Economics and Policy in Health, IMSCaR, Bangor University
-
Dr Maria Kouimtzi, Pharmacy and Informatics Director, Global Clinical Solutions, Wiley-Blackwell
-
Professor Femi Oyebode, Consultant Psychiatrist and Head of Department, University of Birmingham
-
Dr Andrew Prentice, Senior Lecturer and Consultant Obstetrician and Gynaecologist, The Rosie Hospital, University of Cambridge
-
Ms Amanda Roberts, Public contributor
-
Dr Gillian Shepherd, Director, Health and Clinical Excellence, Merck Serono Ltd
-
Mrs Katrina Simister, Assistant Director New Medicines, National Prescribing Centre, Liverpool
-
Professor Donald Singer, Professor of Clinical Pharmacology and Therapeutics, Clinical Sciences Research Institute, CSB, University of Warwick Medical School
-
Mr David Symes, Public contributor
-
Dr Arnold Zermansky, General Practitioner, Senior Research Fellow, Pharmacy Practice and Medicines Management Group, Leeds University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Mr Simon Reeve, Head of Clinical and Cost-Effectiveness, Medicines, Pharmacy and Industry Group, Department of Health
-
Dr Heike Weber, Programme Manager, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Psychological and Community Therapies Panel
-
Professor of Psychiatry, University of Warwick, Coventry
-
Consultant & University Lecturer in Psychiatry, University of Cambridge
-
Professor Jane Barlow, Professor of Public Health in the Early Years, Health Sciences Research Institute, Warwick Medical School
-
Dr Sabyasachi Bhaumik, Consultant Psychiatrist, Leicestershire Partnership NHS Trust
-
Mrs Val Carlill, Public contributor
-
Dr Steve Cunningham, Consultant Respiratory Paediatrician, Lothian Health Board
-
Dr Anne Hesketh, Senior Clinical Lecturer in Speech and Language Therapy, University of Manchester
-
Dr Peter Langdon, Senior Clinical Lecturer, School of Medicine, Health Policy and Practice, University of East Anglia
-
Dr Yann Lefeuvre, GP Partner, Burrage Road Surgery, London
-
Dr Jeremy J Murphy, Consultant Physician and Cardiologist, County Durham and Darlington Foundation Trust
-
Dr Richard Neal, Clinical Senior Lecturer in General Practice, Cardiff University
-
Mr John Needham, Public contributor
-
Ms Mary Nettle, Mental Health User Consultant
-
Professor John Potter, Professor of Ageing and Stroke Medicine, University of East Anglia
-
Dr Greta Rait, Senior Clinical Lecturer and General Practitioner, University College London
-
Dr Paul Ramchandani, Senior Research Fellow/Cons. Child Psychiatrist, University of Oxford
-
Dr Karen Roberts, Nurse/Consultant, Dunston Hill Hospital, Tyne and Wear
-
Dr Karim Saad, Consultant in Old Age Psychiatry, Coventry and Warwickshire Partnership Trust
-
Dr Lesley Stockton, Lecturer, School of Health Sciences, University of Liverpool
-
Dr Simon Wright, GP Partner, Walkden Medical Centre, Manchester
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Expert Advisory Network
-
Professor Douglas Altman, Professor of Statistics in Medicine, Centre for Statistics in Medicine, University of Oxford
-
Professor John Bond, Professor of Social Gerontology & Health Services Research, University of Newcastle upon Tyne
-
Professor Andrew Bradbury, Professor of Vascular Surgery, Solihull Hospital, Birmingham
-
Mr Shaun Brogan, Chief Executive, Ridgeway Primary Care Group, Aylesbury
-
Mrs Stella Burnside OBE, Chief Executive, Regulation and Improvement Authority, Belfast
-
Ms Tracy Bury, Project Manager, World Confederation of Physical Therapy, London
-
Professor Iain T Cameron, Professor of Obstetrics and Gynaecology and Head of the School of Medicine, University of Southampton
-
Professor Bruce Campbell, Consultant Vascular & General Surgeon, Royal Devon & Exeter Hospital, Wonford
-
Dr Christine Clark, Medical Writer and Consultant Pharmacist, Rossendale
-
Professor Collette Clifford, Professor of Nursing and Head of Research, The Medical School, University of Birmingham
-
Professor Barry Cookson, Director, Laboratory of Hospital Infection, Public Health Laboratory Service, London
-
Dr Carl Counsell, Clinical Senior Lecturer in Neurology, University of Aberdeen
-
Professor Howard Cuckle, Professor of Reproductive Epidemiology, Department of Paediatrics, Obstetrics & Gynaecology, University of Leeds
-
Professor Carol Dezateux, Professor of Paediatric Epidemiology, Institute of Child Health, London
-
Mr John Dunning, Consultant Cardiothoracic Surgeon, Papworth Hospital NHS Trust, Cambridge
-
Mr Jonothan Earnshaw, Consultant Vascular Surgeon, Gloucestershire Royal Hospital, Gloucester
-
Professor Martin Eccles, Professor of Clinical Effectiveness, Centre for Health Services Research, University of Newcastle upon Tyne
-
Professor Pam Enderby, Dean of Faculty of Medicine, Institute of General Practice and Primary Care, University of Sheffield
-
Professor Gene Feder, Professor of Primary Care Research & Development, Centre for Health Sciences, Barts and The London School of Medicine and Dentistry
-
Mr Leonard R Fenwick, Chief Executive, Freeman Hospital, Newcastle upon Tyne
-
Mrs Gillian Fletcher, Antenatal Teacher and Tutor and President, National Childbirth Trust, Henfield
-
Professor Jayne Franklyn, Professor of Medicine, University of Birmingham
-
Mr Tam Fry, Honorary Chairman, Child Growth Foundation, London
-
Professor Fiona Gilbert, Consultant Radiologist and NCRN Member, University of Aberdeen
-
Professor Paul Gregg, Professor of Orthopaedic Surgical Science, South Tees Hospital NHS Trust
-
Bec Hanley, Co-director, TwoCan Associates, West Sussex
-
Dr Maryann L Hardy, Senior Lecturer, University of Bradford
-
Mrs Sharon Hart, Healthcare Management Consultant, Reading
-
Professor Robert E Hawkins, CRC Professor and Director of Medical Oncology, Christie CRC Research Centre, Christie Hospital NHS Trust, Manchester
-
Professor Richard Hobbs, Head of Department of Primary Care & General Practice, University of Birmingham
-
Professor Alan Horwich, Dean and Section Chairman, The Institute of Cancer Research, London
-
Professor Allen Hutchinson, Director of Public Health and Deputy Dean of ScHARR, University of Sheffield
-
Professor Peter Jones, Professor of Psychiatry, University of Cambridge, Cambridge
-
Professor Stan Kaye, Cancer Research UK Professor of Medical Oncology, Royal Marsden Hospital and Institute of Cancer Research, Surrey
-
Dr Duncan Keeley, General Practitioner (Dr Burch & Ptnrs), The Health Centre, Thame
-
Dr Donna Lamping, Research Degrees Programme Director and Reader in Psychology, Health Services Research Unit, London School of Hygiene and Tropical Medicine, London
-
Professor James Lindesay, Professor of Psychiatry for the Elderly, University of Leicester
-
Professor Julian Little, Professor of Human Genome Epidemiology, University of Ottawa
-
Professor Alistaire McGuire, Professor of Health Economics, London School of Economics
-
Professor Neill McIntosh, Edward Clark Professor of Child Life and Health, University of Edinburgh
-
Professor Rajan Madhok, Consultant in Public Health, South Manchester Primary Care Trust
-
Professor Sir Alexander Markham, Director, Molecular Medicine Unit, St James’s University Hospital, Leeds
-
Dr Peter Moore, Freelance Science Writer, Ashtead
-
Dr Andrew Mortimore, Public Health Director, Southampton City Primary Care Trust
-
Dr Sue Moss, Associate Director, Cancer Screening Evaluation Unit, Institute of Cancer Research, Sutton
-
Professor Miranda Mugford, Professor of Health Economics and Group Co-ordinator, University of East Anglia
-
Professor Jim Neilson, Head of School of Reproductive & Developmental Medicine and Professor of Obstetrics and Gynaecology, University of Liverpool
-
Mrs Julietta Patnick, Director, NHS Cancer Screening Programmes, Sheffield
-
Professor Robert Peveler, Professor of Liaison Psychiatry, Royal South Hants Hospital, Southampton
-
Professor Chris Price, Director of Clinical Research, Bayer Diagnostics Europe, Stoke Poges
-
Professor William Rosenberg, Professor of Hepatology and Consultant Physician, University of Southampton
-
Professor Peter Sandercock, Professor of Medical Neurology, Department of Clinical Neurosciences, University of Edinburgh
-
Dr Philip Shackley, Senior Lecturer in Health Economics, Sheffield Vascular Institute, University of Sheffield
-
Dr Eamonn Sheridan, Consultant in Clinical Genetics, St James’s University Hospital, Leeds
-
Dr Margaret Somerville, Director of Public Health Learning, Peninsula Medical School, University of Plymouth
-
Professor Sarah Stewart-Brown, Professor of Public Health, Division of Health in the Community, University of Warwick, Coventry
-
Dr Nick Summerton, GP Appraiser and Codirector, Research Network, Yorkshire Clinical Consultant, Primary Care and Public Health, University of Oxford
-
Professor Ala Szczepura, Professor of Health Service Research, Centre for Health Services Studies, University of Warwick, Coventry
-
Dr Ross Taylor, Senior Lecturer, University of Aberdeen
-
Dr Richard Tiner, Medical Director, Medical Department, Association of the British Pharmaceutical Industry
-
Mrs Joan Webster, Consumer Member, Southern Derbyshire Community Health Council
-
Professor Martin Whittle, Clinical Co-director, National Co-ordinating Centre for Women’s and Children’s Health, Lymington