
Notes
Article history
The research reported in this issue of the journal was commissioned and funded by the HTA programme on behalf of NICE as project number 10/70/01. The protocol was agreed in December 2010. The assessment report began editorial review in April 2011 and was accepted for publication in November 2011.The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the referees for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
During the past 5 years William Simpson has received unrestricted educational grants from Schering Plough, honoraria for lectures and advisory boards from AstraZeneca, Menarini, MSD, Randox and Schering Plough, and sponsorship for attendance at scientific meetings from Genzyme and Siemens Diagnostics. During the past 5 years Zosia Miedzybrodzka has received sponsorship for attendance at an educational seminar from AstraZeneca and has attended Scottish Lipid Forum educational meetings sponsored by Schering Plough and MSD. The other authors have no competing interests.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2012. This work was produced by Sharma et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This journal is a member of and subscribes to the principles of the Committee on Publication Ethics (COPE) (http://www.publicationethics.org/). This journal may be freely reproduced for the purposes of private research and study and may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NETSCC, Health Technology Assessment, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2012 Queen’s Printer and Controller of HMSO
Chapter 1 Background and definition of the decision problem
Description of health problem
Introduction
Familial hypercholesterolaemia (FH) is a genetic condition in which people inherit an abnormal (mutant) gene that affects the rate at which cholesterol is cleared from the blood, giving rise to a high level of cholesterol in the bloodstream. An individual can inherit a mutant gene either from one parent (a condition known as heterozygous FH) or from both parents (a condition termed as homozygous FH or compound heterozygous FH). Homozygous FH occurs if a person inherits two copies of exactly the same gene alteration from each parent. Compound heterozygous FH occurs when a person inherits two different types of gene alterations, one from each parent. A person with homozygous FH or compound heterozygous FH usually has a much more severe form of the disease than someone with heterozygous FH. Almost all people with FH have heterozygous FH.
Affected individuals have raised cholesterol concentrations from birth, and this leads to early development of atherosclerosis and coronary heart disease (CHD), and high risk of premature death. FH is generally characterised by the presence of physical symptoms such as tendon xanthomata (cholesterol deposits) and arcus cornealis (cholesterol deposits in eyes) and clinical symptoms (high cholesterol levels).
However, treatment from late childhood with statin therapy, combined with lifestyle changes such as stopping smoking, healthy eating and exercising, can restore normal life expectancy. A recent National Institute for Health and Clinical Excellence (NICE) guideline on the identification and management of FH reviewed strategies for case ascertainment and effective treatment. 1 A key element was the recommendation that cascade testing of first-, second- and if possible third-degree relatives of affected individuals should be offered. Such cascade testing should be carried out either by offering DNA-based testing to consenting individuals or by biochemical measurement of cholesterol levels. 1
Aetiology, pathology and prognosis
The major aetiological determinant of FH is the presence of a highly penetrant mutation (penetrance refers to the proportion of individuals with the mutation who exhibit clinical symptoms) in a gene important in cholesterol metabolism. FH is mainly caused by a mutation in the low-density lipoprotein receptor (LDLR) gene, which is found on the short form of chromosome 19 and is responsible for primary hepatic low-density lipoprotein cholesterol (LDL-C) uptake, processing up to 70% of circulating LDL-C. LDL-C is bound to the receptor (a structural protein molecule on the cell surface that binds to a specific factor, such as a drug or other molecules) and then transported into the cell, where it is metabolised. High-affinity LDLRs are found in the endothelium, smooth muscle cells and liver. In FH, there are four groups of mutations leading to a high level of total cholesterol (TC) and LDL-C:
-
those resulting in impaired receptor synthesis
-
those resulting in impaired transport of receptors to the cell surface
-
those resulting in failure of LDL-C to bind the LDLR properly
-
those resulting in failure to transport bound LDL-C into the cell.
Mutations associated with FH have also been found in the apolipoprotein B (APOB) and protein convertase subtilisin/kexin 9 (PCSK9) genes but with fewer variants than in the LDLR gene. The APOB gene makes a protein that helps hold cholesterol-carrying lipoproteins together in the blood. If there is an alteration in this gene, the LDL does not bind well to the LDLRs on the surface of the liver and it is removed only slowly from the blood. If there is an alteration to the PCSK9 gene, more LDLRs are broken down in the liver, resulting in fewer to remove LDL from the blood. The result in both cases is that the level of LDL-C in the blood remains high. The overall effect of these gene alterations is that the liver is less able to take up excess cholesterol from the blood, meaning that less is excreted into the intestines, from where it can be removed from the body. 2
As the gene is inherited in an autosomal dominant manner, the probability of inheriting the condition is 50% in first-degree biological relatives (parents, siblings, children), 25% in second-degree relatives (aunts, uncles, grandparents, nieces, nephews) and 12.5% in third-degree relatives (first cousins and siblings of grandparents). 3
High cholesterol levels in the blood have complex causes, with genetic and environmental causes operating simultaneously4. As with all genetic conditions, there are several other genes and metabolic and environmental factors contributing to the clinical course of the condition:4
-
examples of genetic causes: specific mutations leading to the FH phenotype, genetic factors that influence lipoprotein metabolism, genetic factors that influence CHD
-
examples of metabolic causes: hormonal, diet/body weight, lipoproteins and enzymes and apolipoproteins modulating their metabolism, factors involved in inflammation, clotting and thrombosis
-
examples of environmental causes: prevalence of CHD in the community, drugs affecting lipoprotein metabolism used without identifying FH.
There is strong evidence that smoking greatly increases the risk of CHD in FH and modest evidence that diet is an important contributory factor.
Familial hypercholesterolaemia is latent (presymptomatic period) from birth to the second decade of life and if diagnosed by then can be successfully treated. FH is usually evident (by blood cholesterol levels) in the first year of life and physical signs such as xanthomata are seen in the second decade of life. Tendon xanthomata are frequent but not always present. Symptomatic CHD usually appears by the fourth decade of life. People with heterozygous FH usually have LDL-C levels that are double the normal level (with TC often between 7.5 and 10 mmol/l), and receptor activity that is about half the normal level. 5 People with homozygous FH typically present with very severe hypercholesterolaemia, with LDL-C levels six times the normal level (i.e. LDL-C levels 15–20 mmol/l) and early onset of disease in childhood. 5
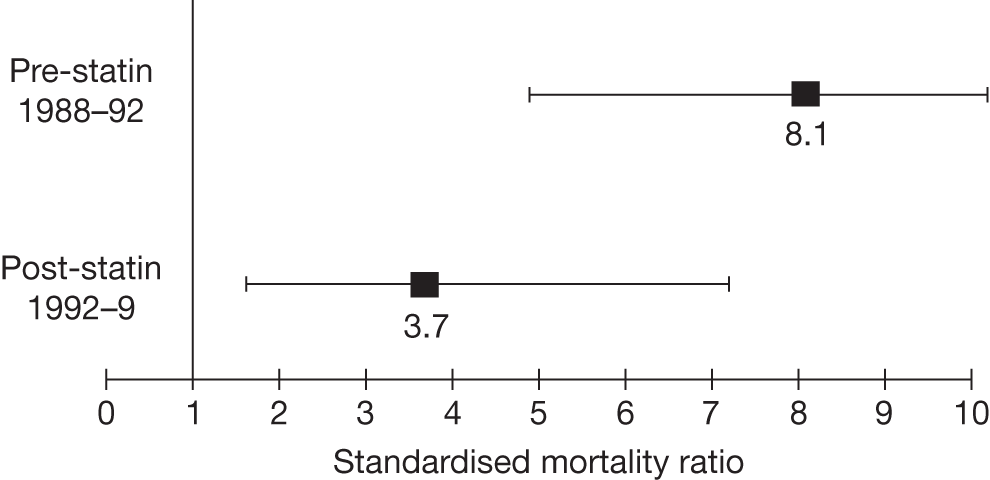
If untreated, approximately 50% of men and 30% of women with FH will develop CHD by age 60 years6 and around 50% of men will die before the age of 60 years. 7 People with homozygous FH have a significantly poorer prognosis than those with heterozygous FH and most will die before the age of 30 years. However, the risk of CHD can be greatly reduced if FH is diagnosed before the onset of the condition, by treatment with lipid-modifying drug therapy (statins) in combination with lifestyle changes. 1 Statins have been shown to be effective in lowering the risk of mortality from CHD in patients with clinical FH (see Figures 1 and 2). 8,9
FIGURE 1.
Kaplan–Meier curve showing the cumulative event-free survival in patients with and without statin treatment. Source: Versmissen et al. 8
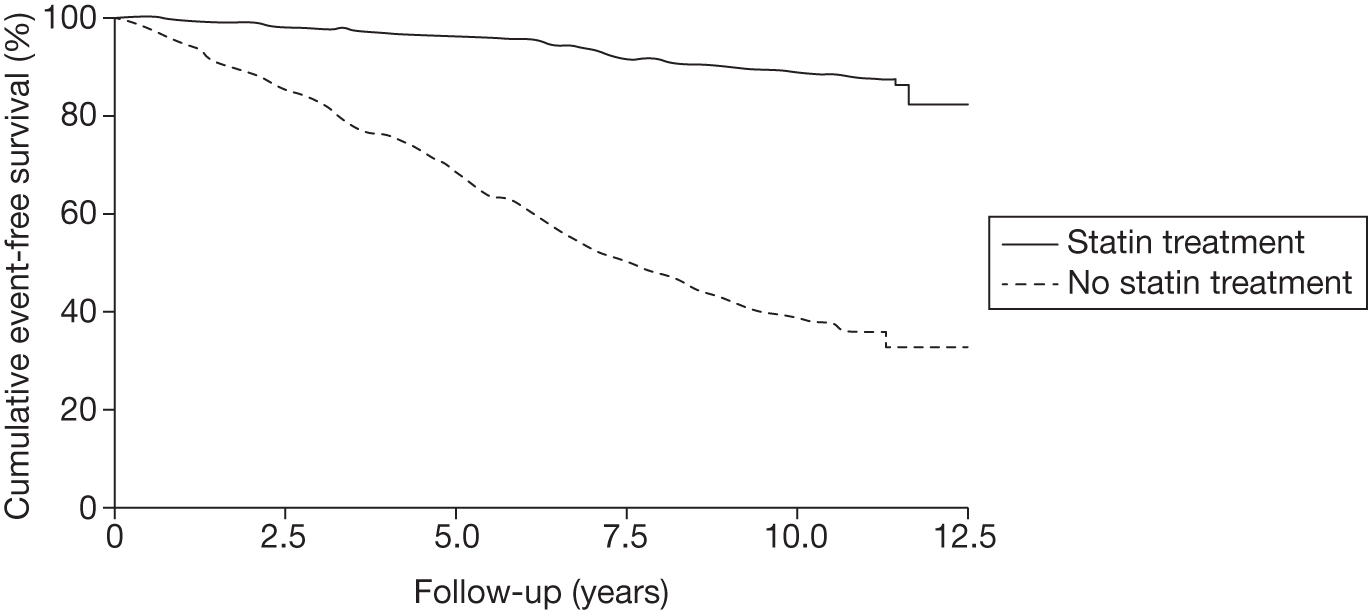
FIGURE 2.
Pre- and post-statin death rates in FH patients (20–59 years). Source: Department of Health Familial Hypercholesterolemia Cascade Testing Audit Project. 10
Epidemiology, incidence and prevalence
It has been estimated that worldwide around 10 million people have FH, of whom around 200,000 die each year from CHD. 11 The prevalence of heterozygous FH varies in different populations. In the UK, prevalence is estimated at 1 in 500, affecting around 100,000 people in England, around 6000 in Wales and approximately 10,000 in Scotland. Homozygous FH and compound heterozygous FH are much rarer, with a prevalence of 1 in 1 million. 12 The frequency of FH-causing mutations can vary by country and within countries by ethnicity. The Centre for Cardiovascular Genetics (University College London)13 keeps an up-to-date database of genetic mutations associated with FH.
The LDLR, APOB and PCSK9 genes are most frequently implicated, but other genes remain to be discovered. Therefore, it is possible that in some people other, yet undiscovered, mutations will not be detected using current genetic strategies. Approximately 1400 unique mutations have been identified worldwide so far, of which over 200 have been reported in the UK population. 14 Approximately 93% of genetic mutations associated with FH occur in the LDLR gene, whereas mutations in the APOB and PCSK9 genes account for approximately 5% and 2% of cases respectively. 15
Impact of the health problem
People with FH have consistently been shown to be at high risk of cardiovascular-associated morbidity and mortality. 9,16 Adults with FH aged 20–39 years have a 100-fold increased risk of dying from CHD. 17 FH is an underdiagnosed condition. It has been estimated that > 85% (around 102,000) of the 120,000 people in the UK thought to be affected with FH are undiagnosed,18 putting them at increased risk of CHD. Often a diagnosis is made too late for an individual to benefit from treatment. 1 A definitive diagnosis through DNA screening of suspected FH patients and then testing of their relatives has been identified as the best possible approach to improve diagnosis of FH. 19
Measurement of disease
Clinical diagnosis
Different sets of clinical criteria have been developed for the diagnosis of FH. These criteria primarily include a combination of high cholesterol, presence of tendon xanthomata in the patient or first-degree relative and a family history of premature CHD or high cholesterol.
The most widely utilised and validated sets of clinical criteria are:
-
the UK Simon Broome Register criteria
-
the US MedPed (make early diagnosis, prevent early death) criteria
-
the Dutch Lipid Clinic Screening Network criteria.
Simon Broome criteria
The Simon Broome criteria include a combination of family history of CHD, physical signs such as tendon xanthomata, cholesterol concentration and DNA testing for the diagnosis of FH (Table 1). 5,20 This approach categorises FH as ‘definite’ or ‘possible’. The major distinction between definite and possible FH is the presence of tendon xanthomata in the definite FH cases. DNA-based evidence was subsequently introduced into the criteria for provision of an unequivocal diagnosis of FH. However, around 10% of people with FH do not meet the Simon Broome criteria.
| Criteria required for clinical diagnosis of FH | Definite FH | Possible FH |
|---|---|---|
|
Cholesterol concentration Child/young person: TC > 6.7 mmol/l, LDL-C > 4 mmol/l; adult: TC > 7.5 mmol/l, LDL-C > 4.9 mmol/l |
Yes | Yes |
|
Clinical symptoms Tendon xanthomata or evidence of these signs in first- or second-degree relative |
Yes | No |
|
Family history of MI in second-degree relative aged < 50 years or in first-degree relative aged < 60 years or Raised TC (> 7.5 mmol/l in adult first- or second-degree relative or > 6.7 mmol/l in child or sibling < 16 years) |
No | Yes (at least one of these criteria) |
The Simon Broome Register was set up, utilising an endowment donated by his wife Katherine, after his premature death from cardiovascular disease, when he was found to have FH. 21
MedPed criteria
The US MedPed criteria take account of the prior probability of a LDLR mutation, which is different for first-, second- and third-degree relatives and the general population. For each of these groups and for four age groups, different cholesterol level cut off points were then designated (Table 2). 5 FH is diagnosed if TC levels exceed the cut off point.
| Age (years) | LDL-C (mmol/l) | |||
|---|---|---|---|---|
| First-degree relatives with FH | Second-degree relatives with FH | Third-degree relatives with FH) | General population | |
| < 18 | 5.7 | 5.9 | 6.2 | 7.0 |
| 20 | 6.2 | 6.5 | 6.7 | 7.5 |
| 30 | 7.0 | 7.2 | 7.5 | 8.8 |
| 40 | 7.5 | 7.8 | 8.0 | 9.3 |
Dutch Lipid Clinic Screening Network criteria
The Dutch criteria5 are similar to the Simon Broome criteria except that a scoring system is used to distinguish between definite, possible or probable FH (Table 3). A diagnosis of FH is definite if the score is > 8 points, probable if the score is 6–8 points and possible if the score is 3–5 points. A score of < 3 points is considered non-FH. The only difference between the Dutch criteria and the Simon Broome criteria is the requirement of tendon xanthomata in the Simon Broome criteria for a diagnosis of definite FH (if a mutation has not been identified).
| Criteria | Point |
|---|---|
| Family history | |
|
First-degree relative with known premature (< 55 years men, < 60 years women) coronary and vascular disease or First-degree relative with known LDL-C > 95th percentile and/or First-degree relative with tendon xanthomata and/or arcus cornealis or |
1 |
| Children < 18 years with LDL-C > 95th percentile | 2 |
| Clinical history | |
| Patient has premature (< 55 years men, <60 years women) coronary artery disease | 2 |
| Patient has premature (< 55 years men, <60 years women) cerebral or peripheral vascular disease | 1 |
| Physical examination | |
| Tendon xanthomata | 6 |
| Arcus cornealis < 45 years | 4 |
| Cholesterol (mmol/l) | |
| LDL-C ≥ 8.5 | 8 |
| LDL-C ≥ 6.5–8.4 | 5 |
| LDL-C ≥ 5.0–6.4 | 3 |
| LDL-C ≥ 4.0–4.9 | 1 |
| DNA analysis | |
| Functional mutation in the LDLR present | 8 |
However, identification of patients by elevated cholesterol levels is not fully reliable. An overlap in blood cholesterol levels between people with FH and those with non-genetic polygenic hypercholesterolaemia has been reported. 22,23 In some FH cases, LDL-C levels are not elevated, resulting in a false-negative diagnosis. 20,24
Genetic diagnosis
DNA-based mutation screening methods provide a definitive diagnosis of FH by identifying a causative mutation and confirming the clinical diagnosis. 25 DNA testing adds clinical certainty to a diagnosis among relatives. Mutations associated with FH have been mostly found in the LDLR gene and rarely in the APOB and PCSK9 genes. 26 The LDLR gene is divided into 18 exons (coding regions in a gene) and 17 introns (non-coding regions in a gene). 27 There are different types of mutations. Large rearrangements or deletions in the LDLR gene have been reported in 5% of FH patients in the UK. 5 Different genetic screening systems are used to screen the entire coding region for the LDLR gene, such as single-strand conformation polymorphism (SSCP) analysis, denaturing gradient gel electrophoresis (DGGE), DNA sequencing and RNA analysis. 27 None of these techniques has been reported to be 100% accurate, with detection rates of 75–85%. Techniques such as Southern blot analysis27 or multiplex ligation-dependent probe amplification (MLPA) are used to identify larger rearrangements and deletions. MLPA analysis, being a simple and rapid method for detecting large rearrangements, has been recommended to be included in the comprehensive genetic analysis (CGA) testing strategy for FH. 28
Current service provision
Diagnosis and management
The NICE clinical guideline1 on the identification and management of FH recommends that diagnosis should be based upon the Simon Broome criteria. Health-care professionals should inform people with a diagnosis of FH based on the Simon Broome criteria that they have a clinical diagnosis of FH. To confirm a diagnosis of FH, health-care professionals should undertake two measurements of LDL-C concentration because biological and analytical variability occurs.
The NICE guideline1 recommends that health-care professionals should inform all people who have an identified mutation diagnostic of FH that they have an unequivocal diagnosis of FH even if their LDL-C concentration does not meet the diagnostic criteria. Health-care professionals should offer all people with FH a referral to a specialist with expertise in FH for confirmation of diagnosis and initiation of cascade testing in relatives.
Cascade testing using a combination of DNA testing and LDL-C concentration measurement is recommended to identify affected relatives of those index cases with a clinical diagnosis of FH. This should include at least the first- and second- and, when possible, third-degree biological relatives. In families in which a mutation has been identified, the mutation and not LDL-C concentration should be used to identify affected relatives. In the absence of a DNA diagnosis, cascade testing using LDL-C concentration measurements should be undertaken to identify people with FH. To diagnose FH in relatives of an index case, age- and gender-specific criteria for LDL-C concentration should be used, as using the Simon Broome LDL-C criteria for index cases would result in underdiagnosis. The age- and gender-specific LDL-C levels are split into three zones: green (relatives unlikely to have FH), red (relatives are likely to have a clinical diagnosis of FH) and grey (uncertain)1 (see Appendices 1 and 2).
For the management of adults, the NICE guideline1 recommends that a high-intensity statin should be prescribed to achieve a recommended reduction in LDL-C concentration of > 50% from baseline. Health-care professionals should offer all children and young people diagnosed with, or being investigated for, FH a referral to a specialist with expertise in FH in children and young people. This should be in an appropriate child/young person-focused setting that meets the standards within the National Service Framework for Children, Young People and Maternity Services. 29
Current service cost
Currently, the majority of cascade testing is conducted using LDL-C. This is relatively inexpensive compared with DNA testing; however, it is associated with test inaccuracies. Costs are estimated to occur over a 5- to 10-year period, after which time the number of cascade tests would be expected to fall. The estimated cost implications for implementing the current NICE guidance in the NHS are shown in Table 4.
| Recurrent costs | Year 1 (£M) | Year 2 (£M) | Year 3 (£M) | Year 4 (£M) | Year 5 (£M) | Year 6 (£M) | Year 7 (£M) | Year 8 (£M) | Year 9 (£M) | Year 10 (£M) |
|---|---|---|---|---|---|---|---|---|---|---|
| Cascade testing | 4.73 | 4.73 | 4.73 | 4.73 | 4.73 | 4.73 | 4.73 | 4.73 | 4.73 | 4.73 |
| Drug therapy for people diagnosed with FH | 2.55 | 5.11 | 7.66 | 10.21 | 12.76 | 15.31 | 17.86 | 20.41 | 22.96 | 25.51 |
| Specialist referrals for people diagnosed with FH through cascade testing | 0.69 | 0.69 | 0.69 | 0.69 | 0.69 | 0.69 | 0.69 | 0.69 | 0.69 | 0.69 |
| Annual review meetings | 0 | 0.60 | 1.19 | 1.79 | 2.39 | 2.99 | 3.59 | 4.19 | 4.79 | 5.39 |
| Coronary events avoided | –0.45 | –0.91 | –1.36 | –1.8 | –2.25 | –2.7 | –3.15 | –3.6 | –4.05 | –4.5 |
| Net resource impact of guideline | 7.52 | 10.22 | 12.91 | 15.62 | 18.32 | 21.02 | 23.72 | 26.42 | 29.12 | 31.82 |
Cost implications associated with cascade testing will probably be most relevant to secondary care. Savings from reductions in coronary events are likely to apply to both primary and secondary care. It is estimated that the cascade testing process will take approximately 5–10 years. Therefore, costs in year 3 would be expected to be extrapolated in a similar pattern out to 10 years, after which overall cost implications would start to fall as fewer people would require testing and savings from coronary events avoided would continue to increase. The costing report referenced in Table 4 did not extrapolate over a 10-year time horizon and these numbers are based on strong assumptions about how these costs might change over time. For example, treatment costs are likely to be less than shown owing to the reduction in prices associated with next-generation gene sequencing and the forthcoming reduction in the cost of atorvastatin as it comes off patent. ‘Next generation’ refers to the emergence in recent years of new (non-Sanger-based) DNA sequencing techniques. This allows higher throughput in genetics laboratories to test for more mutations, more quickly, and hence reduce costs. Early estimates suggest that the emergence of next-generation sequencing may reduce the sequencing costs in the testing of FH by approximately 40%. Therefore, the results presented are a guideline only and should be interpreted with caution. They are not an estimate of the resource use implications from implementing the recommendations of this report. Further details of how the above were derived are available from the NICE website. 1
Variation in services and/or uncertainty about best practice
A 2004 census of clinics providing specialist lipid services in the UK30 reported that, of the 165 clinics on Heart UK’s database, 144 provided specialist lipid services; however, the service provision was reported to be patchy, with < 10% of the estimated FH patients in the UK recorded on the computerised system. In such a scenario, the implementation of fully effective national cascade testing would be impeded. 30 Furthermore, it was reported that 64% of these clinics employed only one doctor and > 20% did not employ a nurse, with only 22% providing two or more sessions per week (see also Current usage in the NHS).
Relevant national guidelines and related documents
These include:
-
Identification and Management of Familial Hypercholesterolaemia, NICE clinical guideline 71, 20081
-
The National Audit of the Management of Familial Hypercholesterolaemia 2010, Royal College of Physicians18
-
Primary Care Service Framework: Familial Hypercholesterolaemia, Primary Care Commissioning, 201031
-
Model of Care: Familial Hypercholesterolaemia, Western Australia Program Committee, 20086
-
Familial Hypercholesterolemia: Screening, Diagnosis and Management of Paediatric and Adult Patients, National Lipid Association Expert Panel on Familial Hypercholesterolemia, 200832
-
Screening for Lipid Disorders in Children, US Preventive Services Task Force, 2007. 33
Description of technologies under assessment
Elucigene FH20™ (Gen-Probe Life Sciences, UK) and LIPOchip® (Progenika Biopharma, Spain) have been designed to reduce the need for CGA for the detection of genetic mutations associated with FH. These kits detect fewer genetic mutations than CGA.
Summary of Elucigene FH20
The Elucigene FH20 kit detects 20 genetic mutations associated with FH commonly found in the UK population. These mutations, with a frequency ranging from 1.3% to 11.4%, were identified from a cohort study in the UK involving 400 patients with FH. 26 Of these 20 mutations, 18 are found in the LDLR gene and one each in the APOB and PCSK9 genes (Table 5).
| Gene | Mutation |
|---|---|
| LDLR | P664L, L458P, R329X, E207X, D200G, E80K, IVS3+1G>A, D461H, ∆G197, fs206, Q363X, W66G, V408M, D206E, C656R, K290RfsX20, C163Y, D461N |
| APOB | R3500Q |
| PCSK9 | D374Y |
The kit uses ARMS™ (AstraZenera, UK) allele-specific amplification technology, which detects point mutations, insertions or deletions in the LDLR, APOB and PCSK9 genes in human whole blood. The principle of ARMS technology is that oligonucleotides with a 3′ mismatched residue will not function as polymerase chain reaction (PCR) primers under specified conditions. Selection of appropriate oligonucleotides allows specific mutant or normal DNA sequences to be amplified and detected by fluorescent analysis using capillary electrophoresis (a technique for separating substances from a fluid substrate). Elucigene FH20 can also be processed using gel-based analysis. The gel-based version is currently the only version available in the UK.
Mutations detected in the Elucigene FH20 assay are believed to be pathogenic; in other words, if the individual tests positive on the Elucigene FH20 kit, they have a confirmed diagnosis of FH.
A limitation of the kit is that it tests for only 20 genetic mutations associated with FH commonly found in the UK population. Hence, less frequently occurring FH-causing mutations will not be detected. Worldwide, approximately 1400 FH-causing mutations have been identified,14 of which over 200 have been reported in the UK population. Therefore, in terms of the number of different FH-causing mutations found in the UK population, Elucigene FH20 would detect only around 10% of them.
Summary of LIPOchip
LIPOchip is a genetic test that uses DNA array technology as part of a tiered system (LIPOchip platform). The current version (version 10) of the chip tests for 189 mutations in the three principal genes causing FH, i.e. LDLR, APOB and PCSK9, known to occur in the UK population. The chip is designed to detect both point mutations and copy number changes of the LDLR gene that are associated with FH. The LIPOchip platform involves the following steps:
-
Samples are analysed using the DNA array system, which is designed to detect targeted mutations in the LDLR, APOB and PCSK9 genes as well as copy number variations.
-
If these mutations are not detected the samples are fully sequenced for the mutations in the LDLR gene.
To process the chip, a thermal cycler, hybridisation station 4800™ (Tecan, Switzerland) and a glass-slide scanner are required. The data are analysed by the LIPOchip software, which generates a report containing information on the pathogenicity of detected mutations based on either scientific publications or bioinformatics analysis.
The manufacturer of LIPOchip also offers a sample testing service in its laboratory in Spain. The laboratory has achieved ISO 9001:2008 certification. Two processing options are available. The first is to run the LIPOchip test only (as described in step 1 above). The second runs the LIPOchip test and, in addition, for samples that are negative for a mutation after the LIPOchip test, carries out automated sequencing of the 18 exons of the LDLR gene (as described in steps 1 and 2 above). If step 2 fails to detect any mutations then the sample is confirmed as FH negative by the manufacturer.
Comparators
Low-density lipoprotein cholesterol concentration measurement (Simon Broome criteria)
Low-density lipoprotein cholesterol is most commonly assessed using an estimated figure calculated from the TC and high-density lipoprotein (HDL) cholesterol values and the triglyceride level, the combination commonly being referred to as ‘lipids’. Because triglyceride measurements vary with fasting status, assessments are usually performed after an overnight fast. LDL-C by itself is neither fully sensitive nor specific for the diagnosis of FH, with considerable overlap between FH and non-FH individuals. LDL-C assessment would be recommended whether or not a genetic test is being undertaken, as other hyperlipidaemias (and the small proportion, perhaps around 5%, of patients with gene-negative FH) would have to be managed on the basis of lipid analysis, and the response to treatment would also be gauged by measuring lipids.
Comprehensive genetic analysis
Comprehensive genetic analysis is defined as the most complete genetic analysis generally available for FH within a diagnostic setting and is expected to detect almost all known FH-causing mutations. This analysis includes DNA sequence analysis of the promoter, all exons and the exon/intron boundaries and into the 3′ untranslated region of the LDLR gene, which will detect the majority (around 88%) of detectable FH mutations, MLPA for each exon and the promoter region of the LDLR gene to detect deletions and duplications (around 5% of detectable FH mutations) plus analysis for the common APOB p.Arg3527Gln gene mutation (around 5% of FH mutations) and the PCSK9 p.Asp374Tyr gene mutation (around 2% of FH mutations).
Targeted gene sequencing
Targeted gene sequencing is used to describe the genetic test for sequencing a specific part of the gene where the family mutation is found. Targeted gene sequencing may be used for cascade testing to identify FH in the biological relatives of index cases.
Identification of important subgroups
There are few data on mutation frequencies in different ethnic groupings across the UK. Extrapolation from genetic studies of a range of other diseases would suggest that it is likely that mutation frequencies could vary markedly between different ethnic groups.
Current usage in the NHS
At present, because of current NHS commissioning arrangements for genetic tests and in common with much specialist genetic testing across the UK, only a small number of laboratories offer genetic testing for FH. As a result, the main test currently used to diagnose FH is measurement of LDL-C concentration. Those laboratories that do offer genetic testing for FH include hospitals in Aberdeen, Belfast, Birmingham, Bristol, Cardiff, Great Ormond Street and Salisbury. Most laboratories proceed straight to CGA rather than using a pre-screen, and most perform MLPA in addition to DNA sequencing.
UK national audit of the management of familial hypercholesterolaemia
Following the publication of the NICE guideline for FH in 2008,1 a national clinical audit investigating the care received by individual patients with FH was undertaken by the Royal College of Physicians, with the results published in 2010. 18 A 2008 survey had shown that around 15,000 adults and 500 children were being managed in UK lipid clinics and the audit examined around 15% (n = 2324) of the adults and 30% (n = 147) of the children. 34
The results, key findings and recommendations of the audit in relation to cascade and DNA testing are detailed below.
Results
-
A total of 42% of sites reported having no database for FH patients.
-
Only 12% of sites had a commissioned cascade testing service.
-
Only 15% of sites received NHS funding for DNA testing.
-
In individuals in whom DNA testing was carried out, a mutation was detected in 62% of adults and 65% of children.
-
When the family mutation was known the child had been offered a DNA test in 94% of cases.
Key findings
-
Current resources were inadequate to cope with the identification of the predicted FH relatives of affected cases UK-wide. This included access to trained staff (86% of sites had no lipid specialist nurses), IT provision and pedigree drawing.
-
There was a major lack of family ‘cascade’ testing, whether carried out on the basis of lipid levels or, more effectively, of a DNA diagnosis.
-
Although there was good access to DNA diagnosis and funding for DNA testing in Scotland, Northern Ireland and Wales, access in England was poor.
Key recommendations
-
Additional resources would be needed to cope with the care of new FH patients identified by cascade testing. Training to address the shortage of staff with key skills would be required.
-
Systems needed to be developed and implemented to carry out comprehensive ‘cascade’ testing. This would require trained health professionals with the appropriate skills to follow up the families of index patients, improved IT resources, including a FH patient database, and pedigree drawing.
-
Resources were needed for DNA diagnosis and clinical genetics input.
-
Based on published data, cascade testing alone would find < 50% of the predicted 100,000 unidentified FH patients in the UK, and other methods for finding FH index cases would need to be explored.
-
Given that FH families were geographically dispersed, cascade testing might be facilitated by a specifically funded UK FH Register to which all FH cases would be notified.
Anticipated costs associated with the intervention(s)
Diagnostic technologies
With regards to genetic tests, two novel screening techniques have emerged (Elucigene FH20 and LIPOchip). Some reports suggest that DNA testing for FH costs approximately £400, whereas other work estimates that the process could cost between £500 and £1000 per test. The main reasons for the large variation in reported costs are (1) the definition of DNA testing has varied in previous reports with differences in the genes sequenced and whether or not genes were screened for deletions or duplications and (2) the cost of DNA sequencing has reduced over time as laboratories build up economies of scale and improve equipment allowing for faster processing and reporting times; as a result, previous cost estimates for testing for FH have varied greatly across reports and studies.
The Elucigene FH20 kit is available at a cost of £15 per test and LIPOchip is available at a cost of €250 or approximately £198. However, these costs do not account for staff time to process samples, consumables or overheads. Therefore, the costs of Elucigene FH20 and LIPOchip will be much greater in practice than just their unit test cost.
A standard NHS tariff does not exist per se for genetic tests; however, a recently developed system is now increasingly used by genetics laboratories across the UK to apportion costs to genetic testing services. This ‘MOLU’ (MOLecular Units) pricing system is the most commonly used costing mechanism for genetic testing of FH in laboratories across the UK. Genetic testing strategies vary in complexity depending on the type and volume of analysis required for different reports (genetic tests). The PCR amplicon or equivalent was chosen as a measure of complexity, which is transparent and easily counted. Reports are grouped into a total of six ‘bands’ (A–F). Bands are assigned and given a weighting according to the number of amplicons analysed to produce a report in that band. The number of reports multiplied by the appropriate band weight produces a final number of MOLUs. The total number of MOLUs derived from the exercise can be divided into the total laboratory budget to give an approximate monetary value to MOLUs. This in turn produces an indicative cost for the various testing strategies. Laboratories that can keep their budget constant or can reduce it but increase the number of MOLUs produced will have lower unit costs. It is estimated that the average cost per MOLU is between £30 and £35. Costs of all genetic tests including targeted gene sequencing for relatives can be estimated in this way.
Although the MOLU costing approach has been decided upon as the most appropriate and generally accepted method to cost these test strategies, it is far from ideal. The approach does not necessarily account for full economic costing or indeed opportunity costs of resources. The MOLU approach is basically a price banding agreed upon in collaboration between the laboratories from a UK Genetic Testing Network (UKGTN) group and the Clinical Molecular Genetics Society (CMGS). This has limitations in terms of the accuracy of the costs produced; however, in the absence of any more robust costing methods for these genetic tests, the MOLU classification system has been deemed the most appropriate method with which to compare these testing strategies.
Costs of LDL-C measurement will need to take into account the costs of resource use to retrieve samples and the costs of testing the samples by a laboratory. LDL-C testing is relatively inexpensive compared with genetic testing. These assays are performed routinely in most laboratories using current fully automated equipment (e.g. the laboratory in Aberdeen Royal Infirmary performs > 100,000 per annum) and the reagent cost is minimal (pence), so the overall cost of the procedure consists almost entirely of the general costs associated with processing any sample (around £3–10).
Ancillary costs
The genetic equipment required to process the tests is assumed to be readily available in UK laboratories. However, should this not be the case as standard, the costs of one-off purchases of this equipment will be included in the laboratory budget and thus indirectly accounted for using the MOLU system identified above.
Treatments
As per recommendations from NICE clinical guideline CG71,1 the recommended treatment for patients with FH is high-intensity statin therapy (usually atorvastatin 80 mg). For patients at risk of CHD based on high lipid levels but who do not have FH, the recommended treatment is low-intensity statin therapy (e.g. simvastatin 40 mg). The cost of atorvastatin is due to decrease during the course of this assessment and is likely to be equivalent to that of generic simvastatin. The implications of this are explored in the cost-effectiveness analysis. Costs of a number of other statin-based therapies such as rosuvastatin, pravastatin, etc. are considered. Other treatments include ezetimibe (evidence of efficacy uncertain) and bile acid sequestrants (costly).
Other costs
Other cost considerations include the cost of health-care professionals to identify family pedigree and the costs of initiating contact with relatives for cascade testing. Costs of annual follow-ups for patients diagnosed with FH are also considered in the analysis.
Care pathways
The care pathway for this evaluation is determined by NICE clinical guideline CG711 on the identification and management of FH. The key elements from the care pathway are as follows:
-
A diagnosis of FH should be made on the basis of a combination of the Simon Broome criteria for a clinical diagnosis and a DNA test to confirm this diagnosis unequivocally. This confirmation should include two measures of LDL-C because of biological and analytical variability of the tests.
-
The children of adults identified with FH should be offered a DNA test if the family mutation is known; alternatively, if the mutation is unknown, LDL-C testing should be carried out and repeated after puberty.
-
Cascade testing of at-risk relatives is recommended using a combination of DNA testing and LDL-C concentration measurement in first-, second- and possibly third-degree biological relatives. If the family mutation is known then DNA testing and not LDL-C should be used to identify relatives.
-
Prescription of a high-intensity statin should be considered to achieve a recommended reduction in LDL-C concentration of > 50% for patients with FH. Lipid-modifying treatment in children with FH should be considered by age 10 years and initial treatment should be statin therapy.
It is important to note that, in practice, the guideline is not very well implemented across the UK because of a lack of funding for the genetic testing of patients with FH and cascade genetic testing of identified relatives. In many cases, LDL-C is the most commonly administered test to identify FH but is subject to poor accuracy and reliability.
Definition of the decision problem
Purpose of the decision to be made
The purpose of this assessment is to address the following questions:
-
What are the most effective and cost-effective strategies for confirming a diagnosis of FH in index cases and for cascade testing of relatives?
-
In cascade testing of relatives for mutations identified in index cases by Elucigene FH20 or LIPOchip, would it be more cost-effective to use those tests rather than targeted gene sequencing?
Definition of the intervention
The interventions are described in Description of technologies under assessment.
Populations and relevant subgroups
Populations and relevant subgroups are described in Chapter 2, Inclusion and exclusion criteria.
Place of the interventions in the treatment pathway(s)
The care pathway for this assessment is based on NICE clinical guideline CG711 on the identification and management of FH.
Index cases
The assessment investigates the use of diagnostic strategies including Elucigene FH20 and/or LIPOchip for providing an unequivocal diagnosis of FH for those with a clinical diagnosis based on the Simon Broome criteria.
Cascade testing of relatives
The assessment investigates the use of diagnostic strategies including Elucigene FH20 and LIPOchip for cascade testing to identify FH in the relatives of index cases. The use of Elucigene FH20 or LIPOchip for cascade testing depends on the mutation detected in the index case and the cost of targeted gene sequencing. (In index cases with an identified genetic mutation, targeted gene sequencing is also considered for cascade testing of relatives. In index cases without an identified genetic mutation, cascade testing using LDL-C concentration measurement is considered.)
A scenario encompassing a single test strategy (Elucigene FH20 or LIPOchip) that does not end in CGA for test-negatives may not detect all cases of FH. In such a scenario there may be implications for test-negative patients in terms of how their condition is managed.
Relevant comparators
Relevant comparators are described in Description of technologies under assessment.
Overall aim and objectives of the assessment
The overall aim of the assessment is to assess the diagnostic accuracy, effect on patient outcomes and cost-effectiveness of Elucigene FH20, LIPOchip and comparators for the diagnosis of FH.
The objectives of the assessment are to:
-
systematically review the evidence on the test performance and clinical effectiveness of Elucigene FH20, LIPOchip and comparators in confirming a diagnosis of FH in patients with a clinical diagnosis of FH
-
systematically review the evidence on the test performance and clinical effectiveness of Elucigene FH20, LIPOchip and comparators in cascade testing of relatives of index cases with a confirmed diagnosis of FH
-
review the evidence on the cost-effectiveness of Elucigene FH20 and LIPOchip for the identification of index cases and cascade testing of relatives
-
estimate the costs of different diagnostic strategies for detecting FH in index cases and for cascade testing of relatives of index cases with a diagnosis of FH
-
develop a comprehensive health economic model to link test accuracy of various diagnostic testing strategies to lifelong cost and treatment outcomes using a linked evidence approach to the modelling process
-
determine the most cost-effective testing strategy relative to current practice (LDL-C) and also to investigate which strategies may be cost-effective compared with current NICE guideline recommendations (i.e. DNA testing), akin to CGA in the context of this assessment.
Chapter 2 Assessment design and results: test performance
Methods for reviewing test performance
Identification of studies
Studies were identified by searching electronic databases and relevant websites, contact with experts in the field and the scrutiny of bibliographies of retrieved papers. Highly sensitive electronic searches were conducted to identify reports of published and ongoing studies on the diagnostic accuracy and clinical effectiveness of tests for FH in index cases and for cascade testing of relatives. The search strategy excluded studies published before 2000.
The databases searched were MEDLINE (1948 to Week 1 2011), MEDLINE In-Process & Other Non-Indexed Citations (10 January 2011), EMBASE (1980 to 2011 Week 1), BIOSIS (1956 to 10 January 2011), Science Citation Index (1970 to 10 January 2011), Conference Proceedings Citation Index – Science (1990 to 10 January 2011) and Cochrane Controlled Trials Register (The Cochrane Library, Issue 1, 2011), as well as current research registers: Current Controlled Trials (January 2011), Clinical Trials (January 2011) and the World Health Organization International Clinical Trials Registry (January 2011). Additional databases searched for systematic reviews and other background information included the Cochrane Database of Systematic Reviews (The Cochrane Library, Issue 1, 2011), Database of Abstracts of Reviews of Effects (January 2011) and Health Technology Assessment database (January 2011). Recent conference proceedings were also searched. Full details of the search strategies used and websites consulted are documented in Appendix 3. In addition, reference lists of all included studies were scanned to identify additional potentially relevant studies.
Inclusion and exclusion criteria
Population
The population considered was adults and children with a clinical diagnosis of FH (the index cases/probands) based on the Simon Broome, Dutch or MedPed criteria and, for cascade testing, the first-, second- and third-degree biological relatives of the index case. (In the protocol for the review we stated that we would consider those with a clinical diagnosis based on the Simon Broome criteria as recommended for clinical diagnosis of FH in the UK. However, we also identified a few studies based on the Dutch and MedPed criteria and in consultation with our clinical advisers we relaxed our inclusion criteria to also include studies in which participants had received a clinical diagnosis of FH based on these criteria, as clinical advice suggested that these criteria were sufficiently similar to the Simon Broome criteria and if consistently applied would also provide potentially useful evidence.)
Given sufficient evidence, subgroup analysis was to be undertaken on the performance of Elucigene FH20 and LIPOchip in ethnic populations.
Setting
The settings considered were secondary or tertiary care.
Interventions and comparators
The interventions considered were Elucigene FH20 and LIPOchip for index cases and cascade testing of relatives. The comparators considered for testing in index cases were (1) CGA and (2) LDL-C concentration measurement (Simon Broome, Dutch or MedPed criteria). The comparators considered for cascade testing of relatives were (1) targeted gene sequencing and (2) LDL-C concentration measurement (age- and gender-specific criteria as recommended in NICE clinical guideline CG711).
Reference standard
The reference standard was CGA in combination with the Simon Broome, Dutch or MedPed criteria. CGA was defined as the ‘most complete genetic analysis’ generally available for FH within a diagnostic setting and is expected to detect almost all known FH-causing mutations. This analysis includes DNA sequence analysis of the promoter, all exons and the exon/intron boundaries and into the 3′ untranslated region of the LDLR gene, which will detect the majority (∼88%) of detectable FH mutations, MLPA for each exon and the promoter region of the LDLR gene to detect deletions and duplications (∼5% detectable FH mutations) plus analysis for the common APOB p.Arg3527Gln gene mutation (∼5% of FH mutations) and the PCSK9 p.Asp374Tyr gene mutation (∼2% of FH mutations).
During the screening process it was ascertained that some studies reporting genetic analysis did not fulfil all of the above criteria for CGA, for example:
-
LDLR, APOB and PCSK9 gene analysis but testing for deletion/duplication was carried out using a process other than MLPA such as Southern blot analysis or quantitative multiplex PCR methodology (QMFSP)
-
LDLR and APOB gene analysis, but no PCSK9 analysis.
Therefore, we took a pragmatic decision to still include studies reporting such an ‘incomplete CGA’ and to assess the quality of such a reference standard in terms of comprehensiveness and variations in test accuracy.
Studies reporting the following single genetic analyses were excluded:
-
APOB gene analysis only
-
PCSK9 gene analysis only
-
test for deletion/duplication only.
In the event of a sequential mutational detection strategy used for the diagnosis of FH, for example Elucigene FH20 followed by gene sequencing for those negative on Elucigene FH20 and then followed by MLPA tests for those negative on gene sequencing, the combination of these sequences could be considered to be CGA.
Low-density lipoprotein cholesterol measurement as part of the clinical diagnosis was one of the comparators. Estimates of the accuracy of LDL-C using the reference standard of CGA plus a clinical diagnosis that includes LDL-C measurement are likely to be inflated compared with the estimates of accuracy of other index tests being evaluated. Therefore, for inclusion of studies reporting the diagnostic accuracy of LDL-C (which is a part of the clinical diagnosis), we considered the estimates of accuracy of LDL-C against a reference standard of CGA (either most complete or incomplete) only.
Outcomes
The following outcomes were considered:
-
test accuracy: sensitivity, specificity, positive likelihood ratio and negative likelihood ratio.
In any studies reporting the above outcomes the following outcomes were also considered:
-
proportion of cases with an unequivocal diagnosis identified by Elucigene FH20 and LIPOchip
-
proportion requiring CGA after Elucigene FH20 and LIPOchip
-
proportion of FH identified from cascade testing
-
acceptability of the tests
-
interpretability of the tests.
Test accuracy data on the absolute numbers of true-positives, false-positives, false-negatives and true-negatives were extracted or calculated from the information provided in the studies. We also considered studies in which derivation of a complete 2 × 2 diagnostic table was not possible but which reported data to allow derivation of one of the test accuracy measures, for example sensitivity but not specificity.
Study design
The following types of studies were considered:
-
direct (head-to-head) studies in which the index test, comparator test and reference standard test were carried out independently in the same group of people
-
randomised controlled trials (RCTs) in which people were randomised to the index and comparator test(s) and all received the reference standard test.
In case of insufficient evidence from direct and randomised studies, indirect (between-study) comparisons in the following types of study were also considered:
-
diagnostic cross-sectional studies comparing the index test or comparator test against a reference standard test
-
case–control studies in which two groups were created, one known to have the target disease and one known not to have the target disease, in which it was reasonable for all included to go through the tests.
Exclusion criteria
The following types of reports were excluded:
-
preclinical and biological studies
-
reviews, editorials and opinions
-
case reports
-
reports investigating technical aspects of a test.
Non-English-language reports were excluded.
Data extraction strategy
Two reviewers (PS and GM) independently screened the titles and abstracts of all reports identified by the search strategy. Full-text copies of all studies deemed to be potentially relevant were obtained and two reviewers (PS and GM) independently assessed them for inclusion. Disagreements were resolved by consensus or arbitration by a third party (ZM and WS).
A data extraction form was developed and piloted (see Appendix 4). One reviewer (PS) extracted the details of study design, participants, index, comparator, reference standard tests and outcome data. A second reviewer (GM) checked the data extraction. Any disagreements were resolved by consensus or arbitration by a third party (ZM and WS). Any study data requested and received from the manufacturers that met the inclusion criteria were to be extracted and quality assessed in accordance with the procedures outlined in the protocol for the assessment.
Quality assessment strategy
The methodological quality of the included diagnostic studies was assessed using QUADAS,35 a quality assessment tool developed for use in systematic reviews of diagnostic studies. QUADAS was developed through a formal consensus method and was based on empirical evidence. The checklist was adapted for the purposes of this review (it is designed to be adapted to make it more applicable to a specific review topic) (see Appendix 5 for the modified QUADAS checklist). The original QUADAS checklist contained 14 questions. Questions 1, 3, 5–7 and 10–14 of the original QUADAS tool were retained (questions 1–10 in the modified version). Three questions in the original QUADAS tool that related to the quality of reporting rather than methodological quality were omitted from the modified version (questions 2, 8 and 9). These questions related to the description of (1) the selection criteria, (2) the execution of the index test and (3) the execution of the reference standard test. A fourth question relating to whether or not the time period between the reference standard and index test was short enough to be reasonably sure that the target condition did not change between the two tests was also omitted. This question was not considered to be relevant as a person will either have or not have FH.
Three questions were added to the modified checklist on (1) whether or not cut-off values were established before the study was started, (2) whether or not the technology of the index test was unchanged since the study was carried out and (3) whether or not the study provided a clear definition of what was considered to be a ‘positive’ result. Three questions in the modified checklist were considered to be relevant to studies reporting LDL-C but not applicable to studies reporting Elucigene FH20 or LIPOchip owing to the nature of these tests: question 8, ‘Were the same clinical data available when test results were interpreted as would be available when the test is used in practice?’; question 11, ‘Were cut-off values established before the study was started?’; and question 13, ‘Did the study provide a clear definition of what was considered to be a “positive” result?’.
Two reviewers independently assessed the quality of all included full-text diagnostic studies using the modified version of QUADAS. Each question was checked as ‘yes’, ‘no’ or ‘unclear’, or, for questions 8, 11 and 13, ‘not applicable’ for reports of Elucigene FH20 or LIPOchip. Any disagreements were resolved by consensus or arbitration by a third party. Studies were not included or excluded on the basis of their methodological quality. Conference abstracts were not quality assessed on the basis that they were not considered to contain sufficient information to allow for an adequate assessment of their methodological quality.
Data analysis
Analysis focused on the ability of Elucigene FH20, LIPOchip and its comparators to confirm FH in index cases of FH diagnosed clinically. Two-by-two tables were extracted from each of the included studies in which information was provided on the numbers of true- and false-positives and -negatives for the index and/or comparator test compared with the reference standard for detecting those mutations that the index and/or comparator test are designed to identify. For each study, where there was sufficient information, sensitivity, specificity, positive and negative likelihood ratios and their confidence intervals (CIs) were calculated.
Where appropriate and given sufficient information, we had planned to use summary receiver operating characteristic (SROC) curves for the meta-analysis of data from studies reporting estimates of true- and false-positives and -negatives. Where appropriate, it was planned to fit models using the hierarchical summary receiver operating characteristic (HSROC) framework, which takes proper account of the diseased and non-diseased sample sizes in each study, and allows estimation of random effects for the threshold and accuracy effects, and testing of the impact of potential sources of heterogeneity. However, there was insufficient information to enable pooling of results or to provide SROC curves as planned and so forest plots of sensitivity and specificity were used to visualise the heterogeneity amongst the included studies. No formal meta-analysis was therefore carried out.
Diagnostic accuracy metrics
For the purpose of this assessment, we define test-positive and test-negative as follows:
-
Elucigene FH20/LIPOchip tests: those with a FH-causing mutation detected by Elucigene FH20 or LIPOchip were defined as ‘test-positive’ and those with no mutations detected were defined as ‘test-negative’.
-
LDL-C tests (as a part of the Simon Broome criteria): we assumed that people with positive clinical criteria would have positive cut-offs of LDL-C as suggested in the definition of the criteria. A minimum LDL-C level of 4 mmol/l is required to diagnose index cases.
-
Age- and gender-specific LDL-C test (as recommended in NICE guideline): those with LDL-C levels greater than the cut-offs were defined as ‘test-positive’ and those with LDL-C levels lower than the cut-offs were defined as ‘test-negative’.
-
True-positives: people with clinical FH who are positive on tests (Elucigene FH20 or LIPOchip or LDL-C as part of the Simon Broome criteria or age- and gender-specific LDL-C) and positive on CGA.
-
False-negatives: people with clinical FH who are negative on tests, but positive on CGA.
-
False-positives: people with clinical FH who are positive on tests, but negative on CGA.
-
True-negatives: people with clinical FH who are negative on tests and negative on CGA.
-
Sensitivity = true-positive/(true-positive + false-negative) × 100.
-
Specificity = true-negative/(true-negative + false-positive) × 100.
Results of test performance
Quantity of research available
Quantity of studies identified
The searches identified 1529 records for the review of test performance. Following screening of titles and abstracts, 1296 articles were excluded and full-text reports of the remaining 233 articles were obtained for further assessment. Figure 3 shows a flow diagram outlining the screening process.
FIGURE 3.
Flow diagram outlining the screening process.
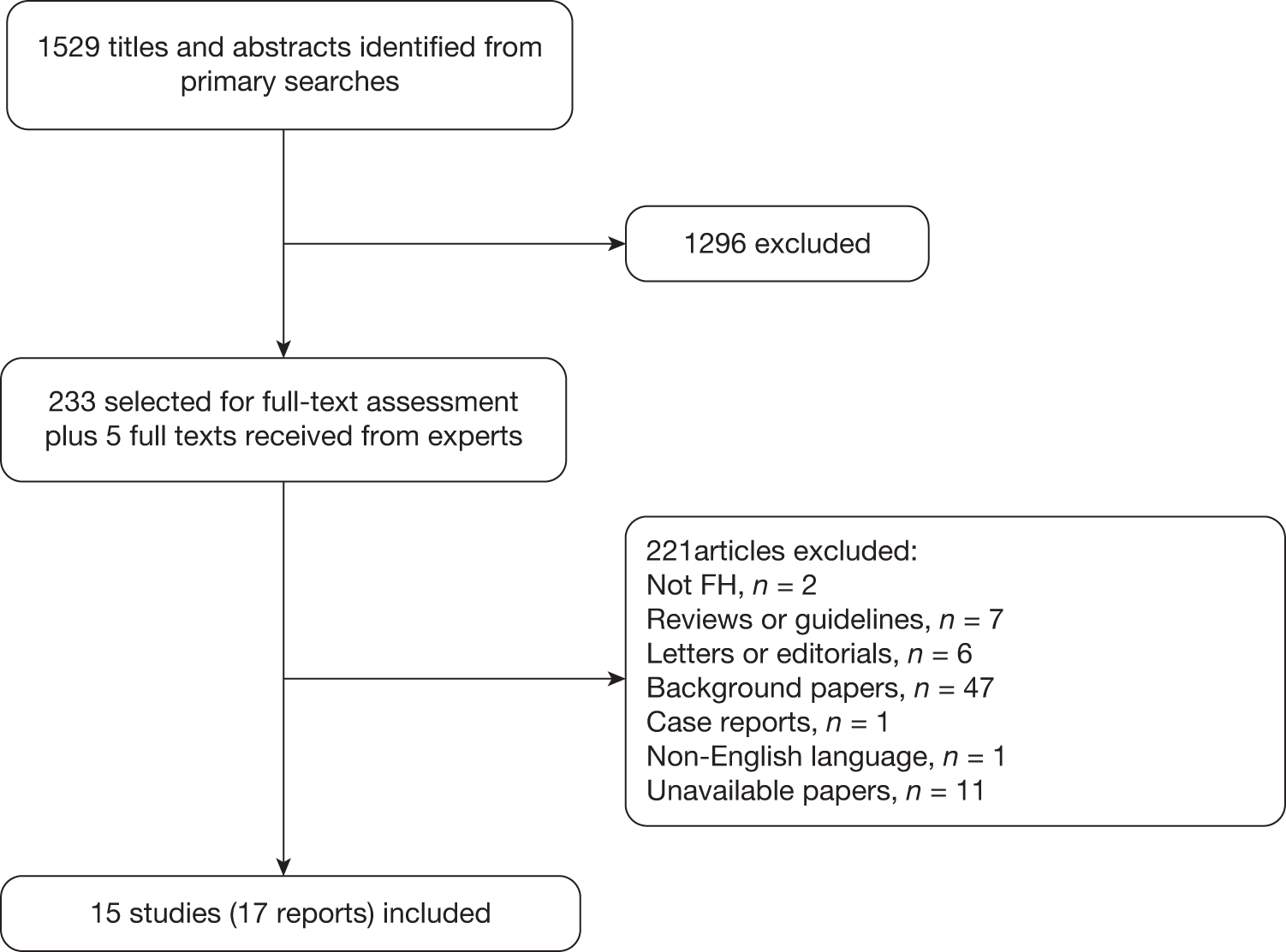
Appendix 6 lists the 15 studies (17 reports) that were included in the review of test performance (Table 6 lists the studies, tests evaluated, publication status and other linked reports). Of the 15 studies, three (four reports) reported Elucigene FH20,36–38 five (six reports) evaluated LIPOchip,39–43 four reported LDL-C compared with genetic analysis44–47 and three reported age- and gender-specific LDL-C for cascade testing of relatives. 48–50 We did not identify any studies reporting a combination of the index tests, that is Elucigene FH20 and LIPOchip.
| Main studya | Test(s) evaluated | Publication status | Other reports linked to the study (not included in the review) |
|---|---|---|---|
| Alonso 200939 | LIPOchip | Full text | |
| Callaway 201040 | LIPOchip | Presentation plus information from author | |
| Civeira 200844 | LDL-C | Full text plus information from author | |
| Damgaard 200545 | LDL-C, targeted sequencing | Full text | |
| Hooper 200936 | Elucigene FH20 | Abstract | |
| Lee 201048 | LDL-C age and gender specific (NICE criteria) | Abstract and information from author | |
| Mabuchi 200546 | LDL-C | Full text | Yu 200251 |
| Palacios 201041 [Stef 201052] | LIPOchip | Abstract and poster plus manufacturer data | |
| Starr 2008,49 Damgaard 200545 | LDL-C age and gender specific (NICE criteria), targeted sequencing | Full text | Leren 2004,53 Umans-Eckenhausen 200119 |
| Stef 200942 | LIPOchip | Abstract | |
| Taylor 201037 [Taylor 200754] | Elucigene FH20, targeted sequencing | Full text plus information from author | Taylor 2009,28 Tabrah 200555 |
| Tejedor 200543 | LIPOchip | Full text | Tejedor 2006,56 Oliva 200957 |
| Widhalm 200747 | LDL-C | Full text | |
| Wiegman 200350 | LDL-C age specific, targeted sequencing | Full text | Fouchier 200158 |
| Yarram 201038 | Elucigene FH20, cascade test | Presentation |
Number and type of studies excluded
A list of the 221 potentially relevant studies identified by the search strategy for which full-text papers were obtained but which subsequently failed to meet the inclusion criteria is given in Appendix 7.
Characteristics of the included studies
Appendix 8 shows the characteristics of the individual included studies.
Study design
All of the studies were diagnostic cross-sectional studies evaluating the performance of Elucigene FH20,36–38 LIPOchip39–43 or LDL-C44–50 against a reference standard of genetic analysis (either incomplete or complete in terms of the definition of CGA as stated in Inclusion and exclusion criteria) in which all participants received a clinical diagnosis using Simon Broome, Dutch or MedPed criteria. No RCTs were identified that randomised participants to any of the tests of interest with all receiving a reference standard test.
Country and setting
Of the eight studies evaluating Elucigene FH20 or LIPOchip, four were conducted in the UK37,38,40,41 (two37,38 reporting Elucigene FH20 and two40,41 reporting LIPOchip) and one was conducted in Australia36 (evaluating Elucigene FH20), with the remaining three taking place in Spain39,42,43 (all of which reported LIPOchip). Of the seven studies reporting the performance of LDL-C (in index cases or for cascade testing of relatives), one each was conducted in the UK,48 Spain,44 Denmark,45 Austria,47 Japan46 and the Netherlands. 50 The study by Starr and colleagues49 included participants from the Netherlands, Denmark and Norway. When reported (seven studies), the clinical diagnosis was performed in lipid clinics.
Clinical diagnosis
The clinical diagnostic criteria used tended to differ according to the country where the study was carried out. The studies by Palacios and colleagues,41 Callaway and colleagues,40 Taylor and colleagues,37 Yarram38 and Lee and colleagues48 were conducted in the UK and their participants had a clinical diagnosis based on the Simon Broome criteria. Of three studies set in Spain, those by Stef and colleagues42 and Alonso and colleagues39 used the Dutch criteria, whereas the study by Tejedor and colleagues43 employed the MedPed criteria. The study by Hooper and colleagues,36 set in Australia, used the Dutch criteria whereas the study by Widhalm and colleagues,47 set in Austria, used the MedPed criteria.
In the studies by Civeira and colleagues44 and Damgaard and colleagues45 patients were given a clinical diagnosis followed by a genetic diagnosis and were then retrospectively classified by the Simon Broome, Dutch and MedPed criteria. Civeira and colleagues44 used an initial clinical diagnosis based on the MedPed criteria, whereas Damgaard and colleagues45 included participants who fulfilled two of the following three criteria: (1) LDL-C > 6 mmol/l, TC > 8 mmol/l and triglycerides < 2.5 mmol/l; (2) tendon xanthomata; and (3) a history of coronary artery disease before the age of 60 years in the patient and/or in a first-degree relative and/or hypercholesterolaemia in a first-degree relative.
In the studies by Starr and colleagues49 and Mabuchi and colleagues46 a genetically tested cohort of relatives was recruited to study the test performance of age- and gender-specific LDL-C cut-offs and a cut-off of 4 mmol/l, which is the minimum cut-off required by Simon Broome criteria respectively. In the study by Starr and colleagues, clinically diagnosed index cases based on the Dutch criteria (the Netherlands) and a combination of lipid levels, clinical characteristics and family history (Norway and Denmark) were included, whereas the study by Mabuchi and colleagues46 included clinically diagnosed index cases based on TC (≥ 5.9 mmol/l and < 12.9 mmol/l) with tendon xanthomata or primary hypercholesterolaemia with/without tendon xanthomata in a family with FH patients among first-degree relatives. The study by Wiegman and colleagues50 recruited relatives of index cases with a clinical diagnosis of FH based on the MedPed criteria or from a genetic diagnosis.
Participants
In the studies by Taylor and colleagues,37 Damgaard and colleagues,45 Mabuchi and colleagues,46 and Tejedor and colleagues43 the participants were all adults. In the study by Wiegman and colleagues50 the participants were children. In the studies by Starr and colleagues49 and Widhalm and colleagues47 the participants were a mixture of adults, adolescents and children, whereas in the study by Civeira and colleagues44 they were adults and adolescents. The remaining seven studies36,38–42,48 (six abstracts and one full text) did not specify whether the participants (index patients or relatives) were adults, children or adolescents.
Eight studies reported diagnostic accuracy in index cases only,36,39–44,46 whereas four37,38,45,47 reported this information both for index cases and for cascade testing of relatives, with the remaining three studies48–50 reporting test performance for cascade testing of relatives only. In studies reporting cascade testing of relatives these were all first-degree relatives apart from the two studies by Damgaard and colleagues45 and Lee and colleagues,48 in which this information was not specified.
For studies evaluating the test performance of Elucigene FH20 and LIPOchip, the sample size ranged from 22 patients40 to 2462 patients. 42 In studies reporting test performance of LDL-C, the sample size ranged from 26347 to 3294. 49 In studies reporting cascade tests through targeted sequencing the sample size of relatives ranged from 27 relatives (from 104 index cases)38 to 1034 relatives (from 591 index cases). 50
In the study by Lee and colleagues48 all included relatives were heterozygous FH (coming from homozygous FH index cases), whereas in the study by Mabuchi and colleagues46 none was homozygous. The rest of the studies did not report on the status of FH patients. Three studies43–45 reported the number of participants at baseline with coronary artery disease, which ranged from 15% to 20%, and xanthomata, which ranged from 16% to 56%. The mean LDL-C concentration of participants as reported in two studies ranged from 4.3 mmol/l to 5.7 mmol/l. 47,49
Only one study reported the proportion of participants by ethnic group. 37 In this study most of the patients were white British (85.4%), 5.8% were of European origin and very few were from ethnic minorities, including 1.7% of Middle Eastern origin, 4.5% of Indian-Asian origin, 1.3% of African or Afro-Caribbean origin and 0.8% from the Far East.
Characteristics of the tests reported by the included studies
Table 7 summarises the characteristics of the studies reporting Elucigene FH20 or LIPOchip, whereas Table 8 summarises the characteristics of the studies reporting LDL-C.
| Study, country | Study design, total sample (n) | Study population | Methods (number evaluated for each test) | Genes tested | Setting (prevalence of FHa) |
|---|---|---|---|---|---|
| Elucigene FH20 | |||||
|
Taylor 201037 UK |
Cross-sectional evaluation (consecutive), 635 |
Clinically diagnosed definite FH, possible FH or unclassified FH based on Simon Broome criteria First-degree relatives of index cases |
Elucigene FH20 (635), SSCP/dHPLC/sequencing (533), MLPA (414) Targeted sequencing of relatives (296) |
LDLR, APOB, PCSK9 | Six lipid clinics, laboratory in UK, one genetics laboratory (36.5%) |
|
Hooper 201036 Australia |
Cross-sectional evaluation, 63 | Clinically diagnosed patients with definite FH based on Dutch criteria | Elucigene FH20 (63), MLPA (not reported), sequencing | LDLR, APOB, PCSK9 | Not reported (77.8%) |
|
UK |
Cross-sectional evaluation, 104 | Clinically diagnosed definite FH, possible FH, unclassified FH or criteria unmet based on Simon Broome criteria |
Elucigene FH20 (104), sequencing, MLPA (not reported) Cascade (27) |
LDLR, APOB, PCSK9 | One genetics laboratory in Bristol, UK (48%) |
| LIPOchip | |||||
|
Alonso 200939 Spain |
Cross-sectional comparative, 808 | Clinically diagnosed patients with definite FH or probable FH based on Dutch criteria | LIPOchip platform (195 mutations): DNA array (808), QMFSP (389), sequencing (312) | LDLR, APOB, | Eleven lipid clinics, one genetics laboratory in Spain (66.5%) |
|
Callaway 201040 UK |
Cross-sectional comparative (validation study), 22 | Clinically diagnosed definite FH based on Simon Broome criteria or cholesterol > 8 mmol/l plus family history of high cholesterol or cardiovascular disease (these were negatives on Elucigene FH20) |
LIPOchip platform (251 mutations) (22) dHPLC/sequencing/MLPA (22) (All received Elucigene FH20, LIPOchip and sequencing/MLPA) |
LDLR, APOB, PCSK9 | One genetics laboratory in, Wessex, UK (not calculable) |
|
Palacios 201041 UK |
Cross-sectional comparative, 126 | Clinically diagnosed patients with Simon Broome criteria and tested with Elucigene FH20 + SSCP/dHPLC/direct sequencing |
LIPOchip platform (251 mutations): LIPOchip (126), sequencing (not reported) LIPOchip version 10 (UK) (126), data from manufacturer |
LDLR, APOB, PCSK9 | Two centres, one genetics laboratory in Spain (51.6%) |
|
Stef 200942 Spain |
Cross-sectional evaluation, 2462 | Clinically diagnosed patients with Dutch–MedPed criteria | LIPOchip platform (247 mutations): LIPOchip (2462), sequencing (not reported) | LDLR, APOB, PCSK9 | Not reported (49.0%) |
|
Tejedor 200543 Spain |
Cross-sectional comparative, 407 phenotyped, 1180 genotyped | Clinically diagnosed definite FH or probable FH based on Dutch–MedPed criteria (genotyped FH identified by SSCP/sequencing used to test performance of chip) |
LIPOchip (118 mutations): DNA array (407), sequencing (123 with DFH) SSCP/sequencing (1180) |
LDLR, APOB | Seventy lipid clinics, genetics laboratory (45.9%) |
| Study, country | Study design, total sample (n) | Study population | Methods (number evaluated for each test) | Genes tested | Setting, (prevalence of FHa) |
|---|---|---|---|---|---|
|
Civeira 200844 Spain |
Cross-sectional comparative (consecutive), 825 (index cases) |
Clinically diagnosed patients (≥ 14 years) who underwent genetic testing were retrospectively categorised based on Simon Broome, Dutch or MedPed criteria (definite, possible or probable FH) Adults and adolescents |
Test 1 (825): LIPOchip platform (203 mutations) (DNA array/QMFSP/sequencing) Test 2 (825): LDL-C test as part of Simon Broome, Dutch, MedPed criteria |
LDLR, APOB, PCSK9 | Three lipid clinics, one genetics laboratory in Spain (55.6%) |
|
Damgaard 200545 Denmark |
Cross-sectional comparative, 408 (index cases), 385 (relatives) |
Clinically diagnosed patients categorised based on Simon Broome, Dutch or MedPed criteria before genetic analysis (definite, possible or probable FH) Adults |
Test 1 (408): LDL-C test as part of Simon Broome, Dutch, MedPed criteria Test 2 (408): CGA (screening of three common mutations in Danish population/SSCP/sequencing/APOB analysis/MLPA) Test 3 (385): targeted sequencing of relatives |
LDLR, APOB | One lipid clinic, genetics laboratory in Denmark (33.1%) |
|
Lee 201048 UK |
Cross-sectional comparative, 30 (index cases, all homozygous), 90 (relatives, all heterozygotes) | Clinically diagnosed index cases based on Simon Broome criteria and genetic test and their relatives |
Test 1 (90): CGA (Elucigene FH20/dHPLC/MLPA or LIPOchip/sequencing or iPLEX/sequencing/MLPA) Test 2 (90): age- and gender-specific LDL-C cut-offs (NICE guideline1) |
LDLR, APOB, PCSK9 | Three (two UK, one Spain) genetics laboratories (not calculable) |
|
Mabuchi 200546 Japan |
Cross-sectional comparative, 281 (index cases) |
Clinically diagnosed index cases based on TC ≥ 5.9 mmol/l and < 12.9 mmol/l) with tendon xanthomata or primary hypercholesterolaemia with/without tendon xanthomata in a family with FH patients among first-degree relatives (Yu 200251) and genetic test (LDLR gene mutation) and unaffected first- and second-degree relatives Adults |
Test 1 (281): CGA (PCR/DGGE/direct sequencing of LDLR gene/Southern blot analysis) Test 2 (281): LDL-C cut-offs > 4.0 mmol/l |
LDLR | (Probably) Japan (64.4%) |
|
Starr 200849 UK |
Cross-sectional comparative, all relatives, the Netherlands = 3294, Denmark = 321, Norway = 1116 |
Clinically diagnosed index cases based on Dutch criteria (the Netherlands), a combination of lipid levels, clinical characteristics and family history (Norway and Denmark) and genetically tested cohort of first-degree relatives from three European countries Adults, adolescents and children |
Test 1: CGA (DGGE/direct sequencing/PCR or screening of three common mutations in Danish population/SSCP/sequencing/MLPA or sequencing/MLPA) Test 2: age- and gender-specific LDL-C cut-offs (NICE guideline1) Test 3: MedPed age-specific LDL-C cut-offs (the Netherlands = 3294, Denmark = 321, Norway = 1116; all received tests 1, 2 and 3) |
LDLR, APOB | Laboratories in the Netherlands, Denmark and Norway (the Netherlands = 25.1%, Denmark = 9.8%, Norway = 34.0%) |
|
Wiegman 200350 The Netherlands |
Cross-sectional comparative, 591 (index cases), 1034 (first-degree relatives – children) | Children of index parents with definite FH (LDLR gene mutation or clinical diagnosis) |
Test 1 (1034): CGA (PCR/DGGE/sequencing/Southern blot) Test 2 (282): age- and gender-specific LDL-C (≥ 3.50 mmol/l) cut-offs for those whose genetic test not yet established |
LDLR | Lipid clinics in the Netherlands (76.6%) |
|
Widhalm 200747 Austria |
Cross-sectional comparative, 263 (index cases – adults = 147, children = 116) |
Clinically diagnosed based on MedPed (definite or possible FH) Adults and children |
Test 1 (119): LDL-C > 5.1 mmol/l in index cases, LDL-C > 4.0 mmol/l in relatives Test 2 (263): CGA (PCR/DGGE/sequencing) |
LDLR, APOB | Tests were performed in Vienna (45.3%; adults 42.2%, children 49.1%) |
Elucigene FH20
Three studies, by Taylor and colleagues,37 Hooper and colleagues36 and Yarram,38 reported Elucigene FH20 (Gen-Probe, UK) as a pre-screen genetic tool for the diagnosis of FH. In all three studies, the genetic screening of clinically diagnosed patients took place in three stages of tests: (1) Elucigene FH20 to screen for 20 common genetic mutations found in the UK (18 LDLR, one PCSK9, one APOB); (2) MLPA to screen for deletions and duplications in the LDLR gene for those negative on Elucigene FH20; and (3) sequencing of the entire LDLR gene for those negative on MLPA. In the study by Taylor and colleagues37 sequencing was performed using SSCP, denaturing high-performance liquid chromatography (dHPLC) and direct sequencing (promoter, all exons, the exon intron boundaries, 3′ untranslated region). Hooper and colleagues36 reported exon-by-exon sequencing of the LDLR gene, whereas Yarram38 reported sequencing of all 18 LDLR exons and the promoter region.
Taylor and colleagues37 and Yarram38 included unrelated patients who were clinically diagnosed as having definite FH or possible FH based on the Simon Broome criteria. These studies also reported on clinical cases who could not be classified as having definite or possible FH because of insufficient information provided from the lipid clinics (usually because of missing untreated cholesterol data), grouped as unclassified FH. Additionally, Yarram38 included 18% (19/104) of patients who did not met Simon Broome criteria in the analysis. Hooper and colleagues,36 on the other hand, included patients with a diagnosis of definite FH based on the Dutch criteria who were enrolled in the FH Western Australia (FHWA) pilot programme.
A paper54 relating to the study by Taylor and colleagues (for which Taylor and colleagues37 is considered the primary reference) reported results on an earlier version of the Elucigene FH20 kit (Elucigene FH013 B1), which screened for 13 common genetic mutations found in the UK population (11 LDLR, one PCSK9 and one APOB). Detection rate data from this test were included with those from Elucigene FH20 in the results reported by Taylor and colleagues37 but were treated as if all samples had been tested with Elucigene FH20. Results from the Taylor and colleagues 2007 report54 were included because all study participants received both Elucigene FH20 and also a reference standard of sequencing of the LDLR gene, unlike the Taylor and colleagues 2010 report,37 in which only test-negatives on Elucigene FH20 went on to receive CGA.
Both of the studies by Taylor and colleagues37 and Hooper and colleagues36 reported, for index cases with a clinical diagnosis of FH, the detection rate using Elucigene FH20 as a pre-screening test alone and when used in combination with sequencing and MLPA. Yarram38 reported the sensitivity of Elucigene FH20 against CGA. Taylor and colleagues37 additionally reported detection rates for FH by ethnicity. The studies by Taylor and colleagues37 and Yarram38 also reported results for cascade testing of relatives, in which the index cases had been identified using Elucigene FH20 initially followed by genetic screening in the form of sequencing and then MLPA.
LIPOchip
Five studies evaluated various versions of the LIPOchip platform (Progenika Biopharma, Spain). 39–43 In two studies, by Palacios and colleagues41 and Stef and colleagues,42 the LIPOchip platform comprised detection of point mutations in the LDLR, APOB and PCSK9 genes by the LIPOchip DNA array and copy number changes in the LDLR gene followed by sequencing of the LDLR gene for test-negatives on the chip. In the study by Alonso and colleagues,39 LIPOchip detected mutations in the LDLR and APOB genes and also large rearrangements, followed by sequencing of the LDLR gene for test-negatives. Callaway and colleagues40 reported the performance of LIPOchip against dHPLC/sequencing and MLPA in one of the genetic laboratories in the UK, in which all samples (negative on Elucigene FH20) received LIPOchip and also sequencing and MLPA, analysing all three genes. The study by Tejedor and colleagues43 reported only the performance of the DNA array in detecting point mutations in the LDLR and APOB genes.
The studies by Palacios and colleagues41 and Callaway and colleagues40 reported detection of FH mutations in a UK population using version 8 of LIPOchip, which included 251 of the most prevalent mutations in Spain, the Netherlands, Italy and the UK. Information on version 10 of LIPOchip, which was developed by analysing 1000 patients from several cohorts, was obtained from the manufacturer based on the former study. 41 This version of the chip detects 189 of the most frequent FH mutations known to occur in the UK population and can also detect copy number changes in the LDLR gene. Palacios and colleagues41 analysed samples from Newcastle and from Wales using version 8, and version 8 or version 9 of LIPOchip, respectively; however, the Welsh samples did not have information on clinical diagnosis (response by manufacturer to queries) and therefore did not meet the review’s inclusion criteria.
Stef and colleagues42 reported on a Spanish version of LIPOchip containing 247 of the most frequent Spanish FH mutations (238 LDLR, three APOB and six PCSK9) designed to detect point mutations in the LDLR, APOB and PCSK9 genes and copy number changes in the LDLR gene. Alonso and colleagues39 also evaluated the performance of a Spanish version of the LIPOchip platform containing a DNA array designed to detect 191 different point mutations in the LDLR gene and four different mutations in the APOB genes and adapted QMFSP for the analysis of large deletions or insertions in the LDLR gene. Tejedor and colleagues43 reported the earliest version of LIPOchip comprising a DNA array including 118 mutations (117 LDLR and one APOB) as identified from SSCP/sequencing/restriction polymorphism analysis, with more than half of these mutations having been reported in Holland, France, Germany, Italy, Greece, the UK and the USA.
In all of these studies (except in the study by Callaway and colleagues40), the analysis was performed in the manufacturer’s laboratory in Spain.
The study by Palacios and colleagues41 was the only study that used DNA samples from patients with a clinical diagnosis based on the Simon Broome criteria. All of the samples had previously undergone genetic testing comprising Elucigene FH20 followed by, for test-negatives, SSCP/dHPLC/direct sequencing of all exons and finally MLPA for test-negatives on the previous test. In the study by Callaway and colleagues40 selection criteria included one or more of the following: clinical diagnosis of Simon Broome ‘definite FH’ or high cholesterol (> 8 mmol/l) with family history of high cholesterol or cardiovascular disease. Alonso and colleagues39 included unrelated cases with a clinical diagnosis of definite or probable FH based on the Dutch criteria (all participants had a score of ≥ 6 points). The studies by Stef and colleagues42 and Tejedor and colleagues43 included participants with a clinical diagnosis based on Dutch–MedPed criteria. Tejedor and colleagues43 included patients with definite FH (score ≥ 8 points) and probable or possible FH (score 4–8 points).
Four studies reported the detection rate of FH by LIPOchip but only three39,41,42 provided true-positive test data for each stage of testing and overall, to allow calculation of the sensitivity of LIPOchip in the diagnosis of FH against CGA. One study reported true-positive, true-negative, false-positive and false-negative data along with sensitivity and specificity. 40 The studies by Alonso and colleagues39 and Tejedor and colleagues43 reported the diagnostic accuracy (sensitivity and specificity) of the LIPOchip DNA array at the mutational level. The sensitivity of the array was determined by the number of mutations detected by sequencing LDLR in the samples in which the DNA array failed to detect mutations, whereas specificity was determined by random verification of DNA array-positive samples by automatic sequencing.
The studies by Palacios and colleagues41 and Alonso and colleagues39 provided information on the time taken to obtain LIPOchip platform results.
None of the LIPOchip studies reported results for cascade testing of relatives.
Low-density lipoprotein cholesterol as part of the Simon Broome criteria
The studies by Civeira and colleagues,44 Damgaard and colleagues,45 Mabuchi and colleagues46 and Widhalm and colleagues47 reported the diagnostic accuracy of LDL-C using the Simon Broome criteria cut-offs against a reference standard of genetic analysis. However, only the study by Civeira and colleagues44 reported the analysis of all three genes (LDLR, APOB, PCSK9), using the LIPOchip platform designed to detect 203 mutations. Two studies analysed LDLR and APOB genes by using screening of three common mutations in a Danish population/SSCP/sequencing/APOB analysis/MLPA45 or PCR/DGGE/sequencing,47 whereas the study by Mabuchi and colleagues46 reported an analysis of the LDLR gene only, by using PCR/DGGE/direct sequencing/Southern blot analysis.
The study by Damgaard and colleagues45 also reported cascade testing by targeted sequencing of relatives.
Age-and gender-specific low-density lipoprotein cholesterol cut-offs according to the NICE clinical guideline
The studies by Lee and colleagues,48 Starr and colleagues49 and Wiegman and colleagues50 reported the test performance of age- and gender-specific LDL-C cut-offs according to the NICE clinical guideline CG711 against a reference standard of CGA, in cascade testing of relatives for the diagnosis of FH.
Lee and colleagues48 evaluated the validity of these cut-offs in a Welsh population and compared them with genetic testing. This study included index cases with a definite diagnosis of homozygous FH based on the Simon Broome criteria and genetic testing from an ongoing national cascade testing project in Wales, and genetically tested relatives of genotyped FH index cases. Genetic tests were performed in three different laboratories (two in the UK and one in Spain), which included screening the LDLR, APOB and PCSK9 genes using Elucigene FH20/dHPLC/MLPA or LIPOchip/sequencing or multiple MassARRAY spectrometry (iPLEX) (50 mutations)/sequencing/MLPA.
In the study by Starr and colleagues,49 age- and gender-specific LDL-C cut-offs were derived from a genetically tested large Dutch cohort of relatives with known mutational status and validated against genetically tested cohorts from Denmark and Norway in which the participants were first-degree relatives of index cases with a definite genotyped diagnosis of FH, and also compared with the MedPed age-specific LDL-C cut-offs. Genetic testing of cohorts was performed in three different countries (the Netherlands, Norway and Denmark) and included analysis of the LDLR and APOB genes using (1) screening for three common mutations in the Danish cohort using SSCP/sequencing/MLPA or (2) sequencing of all exons/MLPA in the Norwegian cohort or (3) PCR/DGGE/direct sequencing in the Dutch cohort.
In the study by Wiegman and colleagues,50 age-specific LDL-C cut-offs ≥ 3.50 mmol/l were derived from children who had been genetically tested (PCR/DGGE/sequencing/Southern blot of the LDLR and APOB genes) and who came from families with a definite diagnosis of FH based on either (1) a documented LDL mutation or (2) plasma LDL-C levels above the 95th percentile for age and gender in a family with a history of premature cardiovascular disease (PCVD) along with (3) tendon xanthomata. The LDL-C cut-offs used in this study represented the red zone of the age- and gender-specific criteria as recommended by the NICE guideline, in which children are likely to have a clinical diagnosis of FH.
Quality of the included studies
Figures 4, 5 and 6 summarise the results of the quality assessment of the full-text studies reporting Elucigene FH20 (one study), LIPOchip (two studies) and LDL-C (six studies) respectively. Quality assessment results for the individual studies (nine full text) are summarised in Appendix 9. For the purposes of the quality assessment, Elucigene FH20, LIPOchip and LDL-C were considered to be index tests and CGA the reference standard. Three questions were considered to be not applicable to studies reporting Elucigene FH20 or LIPOchip:
-
Q8: ‘Were the same clinical data available when test results were interpreted as would be available when the test is used in practice?’ This question was considered not applicable because this information would have no effect on the results of the tests.
-
Q11: ‘Were cut-off values established before the study was started?’ This question was considered not applicable as there is no range of cut-off values applied, but rather a mutation is either detected or not.
-
Q13: ‘Did the study provide a clear definition of what was considered to be a “positive” result?’ This question was considered to be not applicable as, similar to above, a mutation is either detected or not.
FIGURE 4.
Summary of quality assessment of Elucigene FH20 studies (n = 1).
FIGURE 5.
Summary of quality assessment of LIPOchip studies (n = 2).
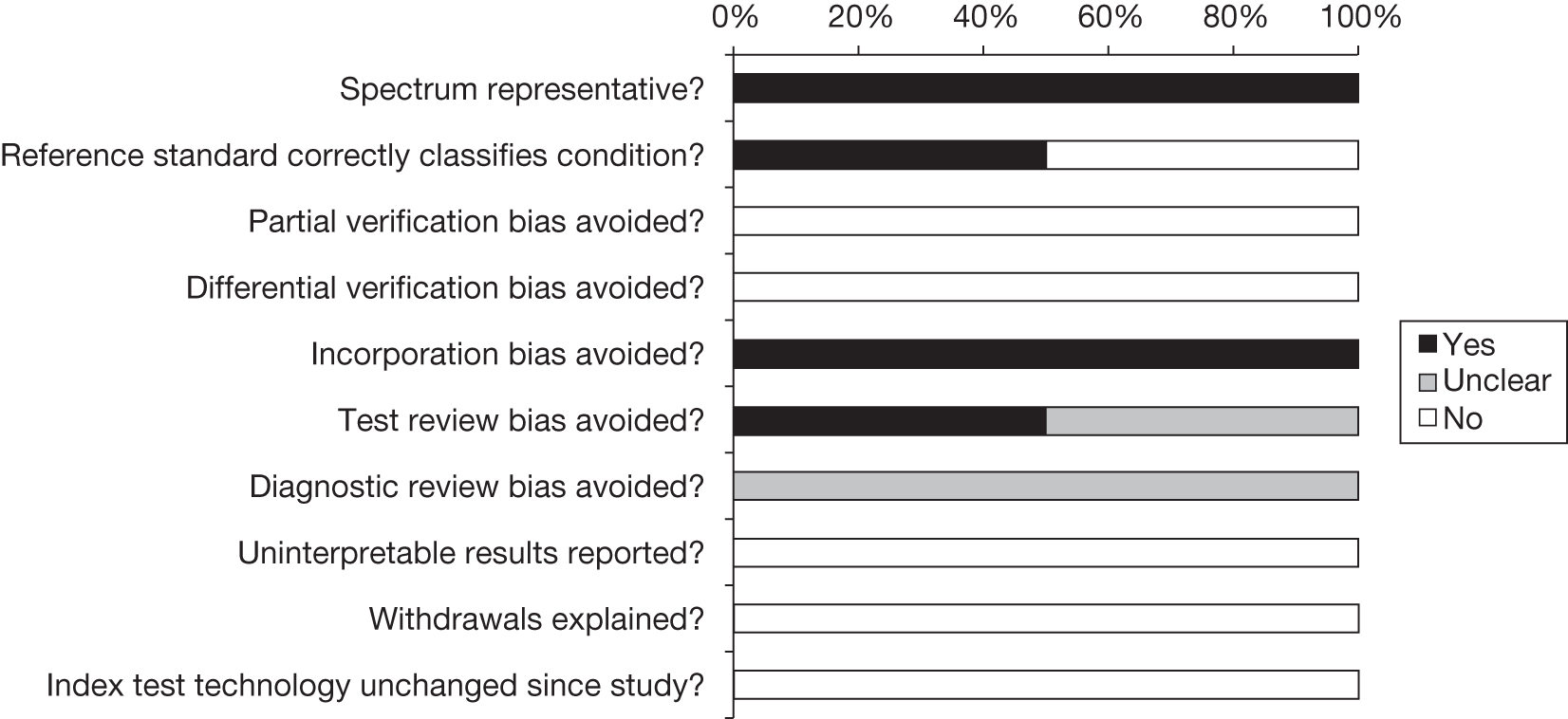
FIGURE 6.
Summary of quality assessment of LDL-C studies (n = 6).
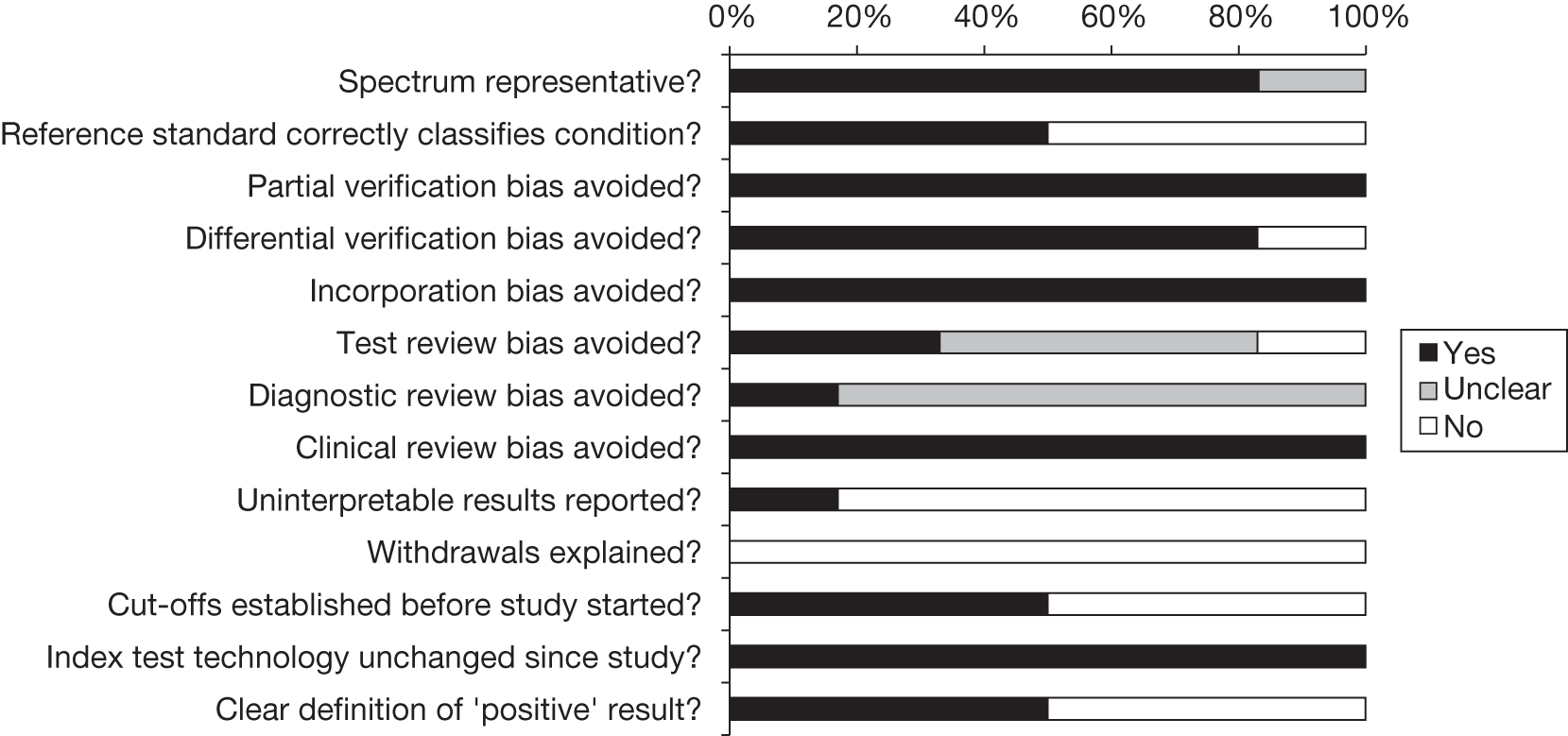
In the study by Taylor and colleagues37 reporting Elucigene FH20, both studies reporting LIPOchip and 83% (n = 5) of the studies reporting LDL-C, the spectrum of patients was considered to be representative of those who would receive the test in practice. For this question patients were considered to be representative if they had received a clinical diagnosis of FH (index cases) or were relatives of index cases with confirmed FH. In the Elucigene FH20 study, one of the two LIPOchip studies and 50% (n = 3) of the LDL-C studies the reference standard used was considered likely to correctly classify FH. Given that the FH-causing PCSK9 gene is rare and was discovered only fairly recently, for this question those studies that included DNA sequence analysis of the promoter, all exons, the exon/intron boundaries and into the 3′ untranslated region of the LDLR gene; MLPA for each exon and the promoter region of the LDLR gene to detect deletions and duplications; and APOB p.Arg3527Gln gene mutation analysis but without assessing the PCSK9 p.Asp374Tyr gene mutation were still considered to be comprehensive and were considered to correctly classify FH in this assessment. The studies that were considered not to classify FH correctly either were missing a test for deletions and duplications in the LDLR gene (one LIPOchip study,43 one LDL-C study47) or did not undertake APOB p.Arg3527Gln gene mutation analysis (two LDL-C studies46,50).
Partial verification bias was avoided in all LDL-C studies in that all patients who underwent LDL-C also received a reference standard, which was not the case with Elucigene FH20 or LIPOchip, for which only test-negatives went on to receive further genetic tests. In practical terms this meant that it was not possible to calculate the specificity of these studies, other than making an assumption of no false-positives and therefore 100% specificity. Differential verification bias was avoided (patients received the same reference standard test regardless of the index test results) in 83% (n = 5) of the LDL-C studies but none of the Elucigene FH20 or LIPOchip studies. In all nine studies incorporation bias was avoided in that the index test was considered to be independent of the reference standard test, even though, for Elucigene FH20 and LIPOchip, these tests formed part of a sequence of tests.
Test review bias was avoided (the results of the index test were interpreted without knowledge of the results of the reference standard) in the study reporting Elucigene FH20, one of the two studies reporting LIPOchip but only 33% (n = 2) of the LDL-C studies. It was unclear in the Elucigene FH20 study, both LIPOchip studies and 83% (n = 5) of the LDL-C studies whether or not diagnostic review bias had been avoided (the results of the reference standard being interpreted without knowledge of the results of the index test).
Clinical review bias was avoided (the same clinical data were available when the index test result were interpreted as would be available when the test was used in practice) in all of the LDL-C studies. In the Elucigene FH20 study, both LIPOchip studies and 83% (n = 5) of the LDL-C studies either un-interpretable test results were not reported or there were none, whereas in all nine studies either an explanation was not given for any withdrawals from the study or there were none. In three LDL-C studies (50%) cut-off values were established before the start of the study. The technology of the index test remained unchanged for Elucigene FH20 as this study reported the FH20 kit; however, the two LIPOchip studies reported earlier versions of this technology. Finally, in 50% (n = 3) of the LDL-C studies a clear definition of a positive result was given.
Assessment of test performance
Overview
This section reports the performance of the index tests Elucigene FH20 and LIPOchip and comparator test LDL-C against a reference standard in the diagnosis of FH in index cases and for cascade testing of relatives. Elucigene FH20 and LIPOchip are designed to detect mutations that are most frequent in the European Caucasian population and which have already been identified using sequencing techniques, a ‘gold standard’ of genetic tests, in this population. Therefore, the mutational analysis of these techniques against the gold standard is most likely to give 100% sensitivity and 100% specificity in this population. Therefore, the results focus mainly on patient-level analysis at trial level. Studies evaluating Elucigene FH20 or LIPOchip comprised a sequence of genetic tests in which the participants received the index test as a pre-screen and test-negatives would then receive further genetic tests such as gene sequencing and MLPA. None of the studies directly compared Elucigene FH20 or LIPOchip with LDL-C, and none reported the acceptability or interpretability of the test or the clinical effectiveness outcomes resulting from use of the test. The results for each of the different tests are reported under the broad headings of Diagnosis of index cases and Cascade testing of relatives. This is followed by sections on other outcomes and subgroup analysis, including by ethnicity, region and type of gene, followed by a brief summary of the chapter. Individual study results are given in Appendix 10.
Diagnosis of index cases
Elucigene FH20
Table 9 shows the test performance results for the three studies that reported Elucigene FH20, by Taylor and colleagues,37 Hooper and colleagues36 and Yarram,38 involving 802 participants. Taylor and colleagues37 and Yarram38 reported the performance of Elucigene FH20 in detecting FH-causing mutations in patients with a clinical diagnosis of definite FH and possible FH and overall for both (Simon Broome criteria) against CGA, whereas Hooper and colleagues36 reported the performance of Elucigene FH20 for those with a clinical diagnosis of definite FH (Dutch criteria) against CGA. Sensitivity of Elucigene FH20 in detecting FH-causing mutations in overall clinical diagnosis ranged from 44.0% in the study by Taylor and colleagues37 to 52.0% in the study by Yarram. 38 Data were not pooled because there was no information on true- and false-positives and negatives in the study by Yarram38 to compute a CI.
| Study, country | Diagnosis | Criteria | n | Sensitivity (%) |
|---|---|---|---|---|
|
Hooper 200936 Australia |
DFH | Dutch | 63 | 28.6 |
|
England |
DFH | Simon Broome | 190 | 48.6 |
|
England |
PFH | Simon Broome | 394 | 40.2 |
|
England |
UFH | Simon Broome | 51 | 38.5 |
|
England |
DFH/PFH/UFH | Simon Broome | 635 | 44.0 |
|
Yarram 201038 England |
DFH/PFH/UFH | Simon Broome | 104 | 52.0 |
Taylor and colleagues37 reported sensitivities of 48.6% and 40.2% of Elucigene FH20 in detecting FH in patients with a clinical diagnosis of ‘definite’ and ‘possible’ FH respectively. Hooper and colleagues36 reported a lower sensitivity of Elucigene FH20 of 28.6% in detecting FH in those with a clinical diagnosis of definite FH. The difference may be explained at least in part by the fact that the two studies used different clinical diagnostic criteria and that the Elucigene FH20 kit was used in two different populations (UK and Australia), given that the kit is designed to screen for the most common mutations in the UK population. For the same reason, a pooled estimate was not calculated but the estimated sensitivities of Elucigene FH20 in confirming FH-causing mutations of definite FH are represented graphically to visualise the heterogeneity (Figure 7). The specificity of Elucigene FH20 in these studies could not be calculated as test-positives did not go on to receive a reference standard test.
FIGURE 7.
Forest plot of Elucigene FH20 sensitivity for patients with a clinical diagnosis of definite FH.
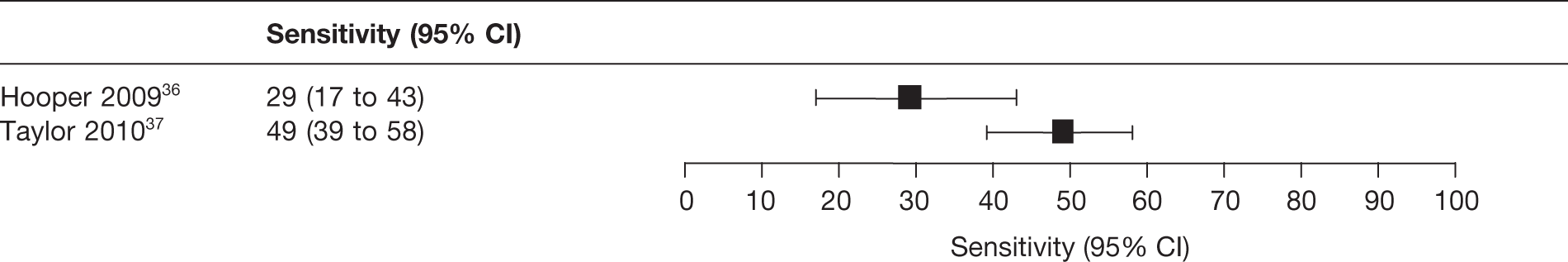
A previous report54 to Taylor and colleagues37 provided information on an earlier version of Elucigene (FH13). In this report the FH13 kit was validated against a reference standard of sequencing the LDLR gene in a patient population in which all patients were clinically diagnosed with definite or possible FH based on the Simon Broome criteria and all received testing with the kit and sequencing of the LDLR gene. The sensitivity of the kit was found to be 30% for patients with a clinical diagnosis of possible FH, 52% for patients with definite FH and 38% for those with a clinical diagnosis of definite or possible FH (Table 10).
| Study | Diagnosis | Test(s) evaluated | n | Sensitivity (%) |
|---|---|---|---|---|
| Taylor 200754 (linked to Taylor 201037) | DFH | Elucigene FH13 | 400 | 52 |
| PFH | Elucigene FH13 | 400 | 30 | |
| DFH/PFH | Elucigene FH13 | 400 | 38 | |
| DFH/PFH | SSCP/dHPLC (LDLR only) | 400 | 62 |
The previous report54 to the study by Taylor and colleagues37 reported that there were no false-positive and no false-negative results from the Elucigene FH13 kit for detection of FH-causing mutations in patients.
In the study by Taylor and colleagues,37 99 mutations plus eight different deletions and duplications were identified in total. Of the 20 mutations present in the Elucigene FH20 kit, three were not identified in any of the participants. Taylor and colleagues37 also reported the prevalence of each mutation that was present in the Elucigene FH20 kit and was identified in the study. The most frequently identified mutations were the mutation in the APOB gene, with a prevalence of 12%, and the three LDLR mutations, p.Gly218del, the intron 3 splice variant c.313+1G>A and the p.Pro685Leu exon 14 variant, with a prevalence of 5% of the total mutations detected.
LIPOchip
Table 11 shows the test performance results for the four studies that reported LIPOchip, by Alonso and colleagues,39 Callaway and colleagues,40 Palacios and colleagues41 and Stef and colleagues,42 and involving 3418 participants. The studies used different versions of LIPOchip. Palacios and colleagues41 and Callaway and colleagues40 reported results for LIPOchip version 8 (Spanish version but study conducted in the UK). Based on the sample reported in Palacios and colleagues,41 the manufacturer of LIPOchip provided further information on version 10, which contains mutations specific to the UK population. Alonso and colleagues39 and Stef and colleagues42 used Spanish versions but did not provide information on the version number. Although the Spanish versions are not specific to the UK, they cover the mutations that are more frequent in Western Europe including the UK.
| Study, country | LIPOchip version | Diagnosis | Criteria | n | Sensitivity (%) | Specificity (%) |
|---|---|---|---|---|---|---|
|
Palacios 201041 and data received from the manufacturer UK |
Version 10, UK mutations | NR | Simon Broome | 126 | 78.5 | NC |
|
Callaway 201040 UK |
Version 8 (251 mutations) | DFH or possible FH | Simon Broome | 22 | 33.3 | 93.8 |
|
Palacios 201041 UK |
Version 8 (251 mutations) | NR | Simon Broome | 120 | 56.9 | NC |
|
Stef 200942 Spain |
247 mutations | NR | Dutch–MedPed | 2462 | 94.5 | NC |
|
Alonso 200939 Spain |
195 mutations | DFH or probable FH | Dutch criteria | 808 | 78.0 | NC |
The sensitivity reported by the studies ranged from 33.3%40 (LIPOchip version 8, Simon Broome criteria) to 94.5%42 (Spanish version designed to detect 247 mutations, Dutch–MedPed criteria). Palacios and colleagues41 reported sensitivity of 56.9% for LIPOchip version 8, which was based on 126 samples (120 analysed). Based on the above sample, the sensitivity of LIPOchip UK version 10 would be 78.5% (51/65) (Progenika, 2011, personal communication) There was heterogeneity across the studies, particularly in relation to Palacios 201041 (version 8 LIPOchip) and Stef 2009,42 and therefore a pooled estimate was not calculated. The estimated sensitivities (with 95% CIs) of LIPOchip in confirming FH-causing mutations are graphically represented to visualise the heterogeneity (Figure 8). The heterogeneity may be explained at least in part by the fact that different versions of LIPOchip and different clinical diagnostic criteria were used. In addition, the difference may also be explained by the fact that the LIPOchip kit was used in two different populations (UK and Spain), given that the prevalence of the FH-causing mutations varies according to the country of origin. The specificity of FH detection by LIPOchip in three studies could not be calculated as test-positives did not go on to receive a reference standard test. 39,41,42 In the study by Callaway and colleagues40 the specificity of LIPOchip version 8 was reported to be 93.8% with one false-positive diagnosis.
FIGURE 8.
Forest plot of LIPOchip sensitivity for patients with a clinical diagnosis of FH.
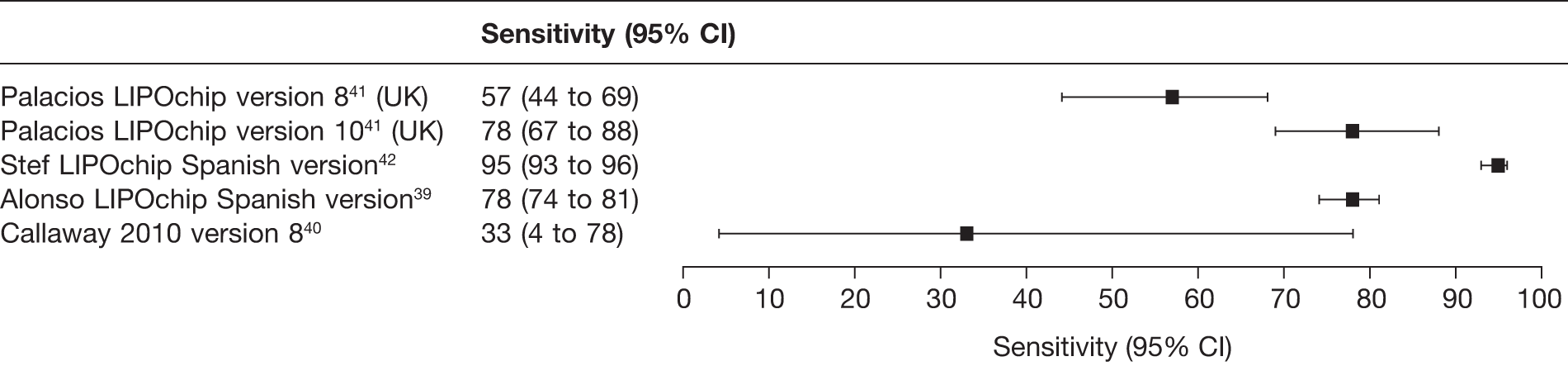
None of the included LIPOchip studies reported accuracy data according to definite or possible FH.
In a mutational-level analysis, the studies by Alonso and colleagues,39 Palacios and colleagues41 and Tejedor and colleagues43 reported sensitivity and specificity of LIPOchip of around 100%. Results on validation with mutation-negative samples and LIPOchip-positive samples by sequencing and QMFSP against MLPA were reported in these studies. See Appendix 10 for the tabulated results.
In the study by Tejedor and colleagues,43 59/118 mutations were detected using this earliest version of LIPOchip. Palacios and colleagues41 reported that in 37 patients 17/251 mutations were picked up by LIPOchip and in 28 patients 25/251 mutations were picked up by sequencing. Overall, the mutation detection rate (by the LIPOchip platform) was 42/251 mutations in 65 patients. 41
Low-density lipoprotein cholesterol
Table 12 shows the test performance results for the four studies that reported LDL-C, by Civeira and colleagues,44 Damgaard and colleagues,45 Mabuchi and colleagues,46 and Widhalm and colleagues,47 involving 1777 participants. The sensitivity of LDL-C as part of the Simon Broome criteria for those with a clinical diagnosis of possible or definite FH was 90%45 and 93%,44 although specificity was much lower at 29% and 28% respectively. The sensitivity of LDL-C as part of the Dutch criteria was 88%44 and 99%,45 although specificity again was also much lower at 18% and 6% respectively. For LDL-C as part of the MedPed criteria the sensitivity reported was 54%45 and 91%,44 although specificity was 83% and 53% respectively. Widhalm and colleagues47 reported sensitivity of LDL-C as part of the MedPed criteria separately for adults and children (66% and 81% respectively), whereas Mabuchi and colleagues46 in a study from Japan reported sensitivity of 98% for LDL-C at a cut-off of > 4 mmol/l. We could not reproduce the specificity of 98.5%, as was reported in this study, from the given values.
| Study | Criteria | Diagnosis | n | Sensitivity (%) | Specificity (%) | LR+ | LR– | Reference standard: CGA |
|---|---|---|---|---|---|---|---|---|
| Damgaard 200545 | Simon Broome | Overall | 408 | 90 | 29 | 1.3 | 0.3 | Screening of three common mutation in Danish population/SSCP/sequencing/MLPA (LDLR, APOB) |
| Dutch | 408 | 99 | 6 | 1.1 | 0.1 | |||
| MedPed | 408 | 54 | 83 | 3.1 | 0.6 | |||
| Civeira 200844 | Simon Broome | Overall | 825 | 93 | 28 | 1.3 | 0.3 | DNA array/QMFSP/sequencing (LDLR, APOB, PCSK9) |
| DFH | 59 | 93 | 8.4 | 0.4 | ||||
| PFH | 90 | 27 | 1.2 | 0.4 | ||||
| Dutch | Overall | 825 | 88 | 18 | 1.1 | 0.7 | ||
| DFH | 72 | 83 | 4.2 | 0.3 | ||||
| MedPed | Overall | 825 | 91 | 53 | 1.9 | 0.2 | ||
| Widhalm 200347 | MedPed | Adults | 147 | 66 | NC | 0.7 | NC | PCR/DGGE/sequencing (LDLR, APOB); no test for deletion and duplication |
| Children | 116 | 81 | NC | 0.8 | NC | |||
| Mabuchi 200546 | LDLC > 4 mmol/la | 281 | 98 | NCb | NC | 0.0 | PCR/DGGE/sequencing/Southern blot analysis (LDLR) |
Cascade testing of relatives
Age- and gender-specific low-density lipoprotein cholesterol cut-offs
Table 13 shows the results for the three studies that used LDL-C age- and gender-specific cut-offs as recommended in NICE clinical guideline CG711 for cascade testing of relatives of index cases with FH.
| Study | Country | Participants | n | Sensitivity (%) | Specificity (%) | Reference standard: CGA |
|---|---|---|---|---|---|---|
| Lee 201048 | UK | Relatives | 90 | 91.5 | 93.0 | Elucigene/dHPLC/MLPA or LIPOchip/sequencing or iPLEX/sequencing/MLPA (LDLR, APOB, PCSK9) |
| 45–54 years | 80.0 | 70.0 | ||||
| Starr 200849 | The Netherlands | First-degree relatives | 3294 | 68.0 | 85.2 | DGGE/sequencing/PCR (LDLR, APOB) |
| Denmark | First-degree relatives | 321 | 79.4 | 85.1 | Screening of three common mutations in Danish population/SSCP/sequencing/MLPA (LDLR, APOB) | |
| Norway | First-degree relatives | 1116 | 83.7 | 83.8 | Sequencing/MLPA (LDLR) | |
| Wiegman 200350 | The Netherlands | Children of definite FH parents | 611 | 96.0 | NC | PCR/DGGE/sequencing/Southern blot (LDLR) |
Lee and colleagues48 reported sensitivity and specificity of 91.5% and 93%, respectively, in the UK cohort.
Wiegman and colleagues50 reported 96% sensitivity for those with LDL-C cut-offs ≥ 3.50 mmol/l (age adjusted), which represents the LDL-C cut-off value in children as stated in NICE clinical guideline CG71. 1 Because of the lack of information on false-positive diagnosis, specificity could not be calculated for this LDL-C cut off. All of the parents of these children had a definite diagnosis of FH. Wiegman and colleagues50 further reported that, out of 228 children of genetically or clinically diagnosed FH parents, 131 (57%) had LDLC ≥ 3.50 mmol/l.
Starr and colleagues,49 for the first-degree relatives, reported sensitivity of 68.0% (the Netherlands), 79.4% (Denmark) and 83.7% (Norway), with specificity of around 85% for all three groups. Starr and colleagues49 also reported test performance by age band including 0–14 years, 15–24 years, 35–44 years, 45–54 years and ≥ 55 years (Table 14). In the Netherlands (n = 3294) and Norway (n = 1116) cohorts, the test performance of LDL-C decreased as the age increased, with a sensitivity ranging from 84.7% (specificity 93.4%) in the < 15 years group to 38.2% (specificity 85.6%) in the 55+ years group (the Netherlands cohort) and sensitivity of 92.5% (specificity 93.5%) in the < 15 years group to 66.7% (specificity 79%) in the 55+ years group (Norway cohort). In the Danish cohort (n = 321) the sensitivity increased as age increased with 95.5% sensitivity in the older group (55+ years) and 76.2% sensitivity in the younger group (15–24 years). Test specificity in this cohort varied across groups, at 72.4% in the 45–54 years group to 94.4% in the 25–34 years group. Starr and colleagues also reported the performance of MedPed LDL-C cut-offs in these cohorts, reporting low sensitivity but consistently higher specificity compared with the age- and gender-specific LDL-C cut-offs.
| n | +ve/–ve mutation | Sensitivity (95% CI) (%) | Specificity (95% CI) (%) | False-negative (95% CI) (%) | False-positive (95% CI) (%) | |
|---|---|---|---|---|---|---|
| The Netherlands | ||||||
| 0–14 years | 183/243 | 84.7 (78.7 to 89.6) | 93.4 (89.5 to 96.2) | 15.3 (10.4 to 21.3) | 6.6 (3.8 to 10.5) | |
| 15–24 years | 187/276 | 71.1 (64.1 to 77.5) | 85.1 (80.4 to 89.1) | 28.9 (22.5 to 35.9) | 14.9 (10.9 to 19.6) | |
| 25–34 years | 138/293 | 64.5 (55.9 to 72.4) | 82.6 (77.8 to 86.8) | 35.5 (27.6 to 44.1) | 17.4 (13.2 to 22.2) | |
| 35–44 years | 136/471 | 71.3 (62.9 to 78.7) | 83.4 (79.8 to 86.7) | 28.7 (21.3 to 37.1) | 16.6 (13.3 to 20.2) | |
| 45–54 years | 92/449 | 57.6 (46.9 to 67.9) | 83.7 (80.0 to 87.0) | 42.4 (32.1 to 53.1) | 16.3 (13.0 to 20.0) | |
| 55+ years | 89/737 | 38.2 (28.1 to 49.1) | 85.6 (82.9 to 88.1) | 61.8 (50.9 to 71.9) | 14.4 (11.9 to 17.1) | |
| Overall | 3294 | 825/2469 | 68.0 (64.7 to 71.2) | 85.2 (83.8 to 86.6) | 32 (28.3 to 35.3) | 14.8 (13.4 to 16.2) |
| Denmarka | ||||||
| 15–24 years | 42/23 | 76.2 (60.5 to 87.9) | 91.3 (72.0 to 98.9) | 23.8 (12.1 to 39.5) | 8.7 (1.1 to 28) | |
| 25–34 years | 34/36 | 58.8 (40.7 to 75.4) | 94.4 (81.3 to 99.3) | 41.2 (24.6 to 59.3) | 5.6 (0.7 to 18.7) | |
| 35–44 years | 39/27 | 89.7 (75.8 to 97.1) | 81.5 (61.9 to 93.7) | 10.3 (2.9 to 24.2) | 18.5 (6.3 to 38.1) | |
| 45–54 years | 18/29 | 88.9 (65.3 to 98.6) | 72.4 (52.8 to 87.3) | 11.1 (1.4 to 34.7) | 27.6 (12.7 to 47.2) | |
| 55+ years | 22/40 | 95.5 (77.2 to 99.9) | 90.0 (76.3 to 97.2) | 4.6 (0.1 to 22.8) | 10.0 (2.8 to 23.7) | |
| Overall | 321 | 160 /161 | 79.4 (72.3 to 85.4) | 85.1 (78.6 to 90.2) | 20.6 (14.6 to 27.7) | 14.9 (9.8 to 21.4) |
| Norway | ||||||
| 0–14 years | 106/107 | 92.5 (85.7 to 96.7) | 93.5 (87.0 to 97.3) | 7.6 (3.3 to 14.3) | 6.5 (2.7 to 13.0) | |
| 15–24 years | 82/103 | 86.6 (77.3 to 93.1) | 91.3 (84.1 to 95.9) | 13.4 (6.9 to 22.7) | 8.7 (4.1 to 15.9) | |
| 25–34 years | 69/124 | 87.0 (76.7 to 93.9) | 85.5 (78.0 to 91.2) | 13.0 (6.1 to 23.3) | 14.5 (8.8 to 22.0) | |
| 35–44 years | 51/145 | 78.4 (64.7 to 88.7) | 82.8 (75.6 to 88.5) | 21.6 (11.3 to 35.3) | 17.2 (11.5 to 24.4) | |
| 45–54 years | 39/120 | 66.7 (49.8 to 80.9) | 74.2 (65.4 to 81.7) | 33.3 (19.1 to 50.2) | 25.8 (18.3 to 34.6) | |
| 55+ years | 27/143 | 66.7 (46.0 to 83.5) | 79.0 (71.4 to 85.4) | 33.3 (16.5 to 54.0) | 21.0 (14.6 to 28.6) | |
| Overall | 1116 | 374/742 | 83.7 (79.5 to 87.3) | 83.8 (81.0 to 86.4) | 16.3 (12.7 to 20.5) | 16.2 (13.6 to 19.0) |
| MedPed age-specific LDL-C cut-offs | ||||||
| The Netherlandsb | 3294 | 42.3 | 97.8 | 57.7 | 2.2 | |
| Denmark | 321 | 68.8 | 89.4 | 31.3 | 10.6 | |
| Norwayb | 1116 | 74.9 | 92.7 | 25.1 | 7.3 | |
Targeted gene sequencing for a mutation found in a family member
Table 15 shows the results of the four studies that investigated cascade testing of relatives. Three studies reported cascade testing of relatives using targeted gene sequencing for a mutation in a family member. 37,45,50 One study published as a presentation did not specify whether or not cascade testing was carried out by targeted sequencing. 38 Three of these studies reported that 53–56% of relatives were positive for FH,37,38,45 which was more or less consistent with the expected 50% probability of diagnosis in relatives.
| Study | Country | Study participants | Test | Number of index cases/families | Number of relatives tested | Number identified with FH (%) |
|---|---|---|---|---|---|---|
| Damgaard 200545 | Denmark | Relatives of index cases in whom a mutation was identified |
Index cases: screening of initial three common mutations + SSCP + sequencing + APOB analysis + MLPA (LDLR/APOB) Relatives: targeted sequencing in relatives |
408 | 385 | 205 (53) |
| Taylor 201037 | UK | First-degree relatives of index cases in whom a mutation was identified |
Index cases: Elucigene FH20 + SSCP/dHPLC/sequencing for test-negatives with Elucigene FH20 + MLPA for test-negatives with sequencing (LDLR/APOB/PCSK9) Relatives: targeted sequencing in relatives |
100 families | 296 | 166 (56) |
| DFH = 47 | 138 | 75 (54) | ||||
| PFH = 47 | 146 | 84 (58) | ||||
| UFH = 6 | 12 | 7 (58) | ||||
| Wiegman 200350a | The Netherlands | Children of families with a documented LDLR mutation |
Index parents: PCR/DGGE/sequencing/Southern blot of LDLR Targeted sequencing in children |
591 families | 806 | 617 (77) |
| LDLC ≥ 3.50 mmol/l for children (age and sex adjusted) without confirmed FH | 228 | 131 (57) | ||||
| Targeted sequencing + LDLC ≥ 3.50 mmol/l for children (age and sex adjusted) without known FH | 1034 | 748 (72) | ||||
| Yarram 201038 | UK | Relatives |
Index cases: Elucigene FH20 + SSCP/dHPLC/sequencing for test-negatives with Elucigene FH20 + MLPA for test-negatives with sequencing (LDLR/APOB/PCSK9) Cascade testing of relatives |
104 | 27 | 15 (56) |
Taylor and colleagues37 reported results of cascade testing of relatives of index cases with a documented mutation who had received an initial clinical diagnosis based on the Simon Broome criteria. This study used a sequence of tests for detecting mutations in index cases that included Elucigene FH20 as a pre-screen test and then sequencing for test-negatives on Elucigene FH20, which in turn was followed by MLPA for test-negatives on sequencing. Relatives of the index cases received targeted gene sequencing for the specific mutation found in the family member. A total of 296 first-degree relatives from 100 families were recruited and a FH-causing mutation was identified in 56%. The detection rate was similar (around 55%) in relatives from families with an initial diagnosis of definite FH or an initial diagnosis of possible FH. Yarram38 used a similar approach as in the study by Taylor and colleagues37 to diagnose index cases and reported that 27 relatives from 104 index cases were identified through cascade testing and 56% had a FH-causing mutation.
The fourth study, by Wiegman and colleagues,50 used conventional sequencing of the LDLR gene in children of heterozygous (a documented LDLR mutation) or clinically diagnosed (plasma LDL-C levels above the 95th percentile for age and gender in a family with a history of PCVD in conjunction with tendon xanthomata) parents (n = 591) and reported 77% to have a FH-causing mutation. The authors suggested that the high proportion diagnosed might be due to the fact that siblings with very low levels of LDL-C were not referred to the paediatric clinic. Moreover, the paediatric hyperlipidaemic are less likely to have polygenic cases of hyperlipidaemia and are therefore more likely to have a higher mutation detection rate.
There were no studies using LIPOchip in relatives.
Other outcomes
Table 16 gives the proportions with an unequivocal diagnosis by Elucigene FH20 and LIPOchip and the proportions that would subsequently require sequencing as reported in six studies. 36,37,39,41–43 Elucigene FH20 identified a mutation in only 16% of cases in a cohort of 635 clinically diagnosed FH cases, with > 80% still requiring sequencing for confirmation of diseases. 37 In Spanish studies, 46%42,43 to 52%39 were confirmed to have a mutation using LIPOchip. LIPOchip version 8 confirmed 29% of cases in a UK setting but in the same population 40% would be identified with a FH-causing mutation by LIPOchip version 10, with 60% still requiring sequencing. 41 Elucigene FH20 or LIPOchip detected, almost twice as many people with FH-causing mutations who were definite FH than those classed as possible FH (27% vs 11% in the study by Taylor and colleagues;37 51% vs 37% in the study by Tejedor and colleagues43).
| Study | Country | Test | FH diagnosis | Total tested | Number with unequivocal diagnosis (%) | Number requiring sequencing (%) |
|---|---|---|---|---|---|---|
| Hooper 200936 | Australia | Elucigene FH20 | DFH | 63 | 14 (22) | 49 (78) |
| Taylor 201037 | UK | Elucigene FH20 | DFH | 190 | 52 (27) | 138 (73) |
| PFH | 394 | 45 (11) | 349 (89) | |||
| UFH | 51 | 5 (10) | 46 (90) | |||
| Total | 635 | 102 (16) | 533 (84) | |||
| Alonso 200939 | Spain | LIPOchip (195 Spanish mutations) | DFH or probable FH | 808 | 419 (52) | 389 (48) |
| Palacios 201041 | UK | LIPOchip version 8 | NR | 126 | 37 (29) | 89 (71) |
| LIPOchip version 10 | NR | 126 | 51 (40) | 75 (60) | ||
| Stef 200942 | Spain | LIPOchip (247 mutations) | NR | 2462 | 1140 (46) | 1322 (54) |
| Tejedor 200543 | Spain | DNA array (118 mutations) | DFH | 252 | 129 (51) | 123 (49) |
| Possible/probable FH | 155 | 58 (37) | 97 (63) | |||
| Total | 407 | 187 (46) | 220 (54) |
Two studies on LIPOchip reported the time taken to obtain test results. 39,41 The time taken to obtain positive test results with LIPOchip (including data extraction and analysis) ranged from 10 days41 to an average of 15 days. 39 Additionally, the time taken to detect rearrangements was 7 days and then 3041–4539 days for sequencing. Palacios and colleagues41 reported that 2 months was required to obtain results by sequencing in conjunction with MLPA. An average of 68 days (range 10–93 days) was reported for obtaining complete results with the LIPOchip platform, with the majority of mutations being detected within 15–22 days after the start of the analysis. 39
Subgroup analysis
The mutation detection rate may vary in ethnic groups as FH-causing mutations may be more frequent in one group of people than in another. The Elucigene FH20 and LIPOchip genetic tests are designed to detect mutations that are more frequent in European Caucasian populations.
Only the study reporting Elucigene FH20 by Taylor and colleagues37 reported the detection rate of FH for ethnic groups. By using a sequence of tests in which Elucigene FH20 was used as a pre-screen followed by sequencing for test-negatives on Elucigene FH20 and then MLPA for test-negatives on sequencing, the mutation detection rate in a population of Indian Asian origin was 32.3% (n = 31) and in a population of African origin was 25% (n = 8). The study suggested that detection rates were lower for these groups than for white British groups, but the difference was not statistically significant (p = 0.63). The study population also comprised those of Middle East, Far East and non-British European origin, although detection rates for these groups were not reported.
In total, 10 out of 20 FH-causing mutations were identified in 31 patients of Indian Asian origin and only 1 out of 20 FH-causing mutations was identified in four patients of African origin. Only 3 out of the 10 mutations detected in the Indian Asian group were detected by Elucigene FH20.
Taylor and colleagues37 also reported the overall detection rate by CGA across six different centres in the UK. The detection of FH ranged from 8.3% to 73.6% among definite FH (p = 0.001) and from 21.7% to 39.5% for those with possible FH (p = 0.13). The authors further reported that when a centre with the smallest sample size was removed from the analysis the difference was no longer significant for the definite FH category (p = 0.07).
Five studies reported FH detection according to the type of gene (Table 17). In patients with a genetic diagnosis of FH, most mutations detected by Elucigene FH20 or LIPOchip were in the LDLR gene (range 69–97%), followed by the APOB gene (range 3–27%) and the PCSK9 gene [4% and 6% (two studies)].
| Study | Test | Total analysed | Total detected | Detected with LDLR gene, n (%) | Detected with APOB gene, n (%) | Detected with PCSK9 gene, n (%) |
|---|---|---|---|---|---|---|
| Taylor 201037 | Elucigene FH20 | 635 | 102 | 70 (69) | 28 (27) | 4 (4) |
|
Palacios 201041 Newcastle sample |
LIPOchip version 8 + sequencing | 120 | 65 | 52 + 1 CNC (80 + 2) | 8 (12) | 4 (6) |
| Stef 200942 | LIPOchip Spanish version (247 mutations) | 2462 | NR | 94% + 6% CNC | 0 | 0 |
| Tejedor 200543 | LIPOchip earlier version (118 mutations) | 407 | 187 | 181 (97) | 6 (3) | NAn |
| Alonso 200939 | LIPOchip Spanish version (191 mutations) | 808 | 537 | 521 (97) | 16 (3) | NAn |
| DNA array | 808 | 419 | 403 (96) | 16 (4) | NAn |
Summary
In total, 15 studies (17 reports) were included. Three studies (four reports) evaluated Elucigene FH20, five studies (six reports) evaluated various versions of LIPOchip, four studies reported data on the performance of LDL-C as part of the Simon Broome criteria or LDL-C cut-offs of > 4 mmol/l and three studies reported age-and gender-specific LDL-C cut-offs for cascade testing of relatives. Five studies conducted in the UK recruited participants who had received a clinical diagnosis of FH based on the Simon Broome criteria, reporting Elucigene FH20, LIPOchip and age- and gender-specific LDL-C cut-offs. Three studies reported targeted gene sequencing for a mutation found in a family member.
Only studies reported as full-text papers (n = 9) were quality assessed. In the studies reporting Elucigene FH20 (n = 1) and LIPOchip (n = 2) and five of the six studies reporting LDL-C, the participants were representative of those who would receive the tests in practice. As Elucigene FH20 and LIPOchip were used as a pre-screen with only test-negatives going on to receive further genetic tests, these studies suffered from partial verification bias, whereas in all of the LDL-C studies all of the participants who received the index test (LDL-C) also received a reference standard test. Patients received the same reference standard regardless of the index test result in 83% (n = 5) of the LDL-C studies but none of the Elucigene FH20 or LIPOchip studies (differential verification bias).
For Elucigene FH20, two studies, one by Taylor and colleagues37 involving 635 participants and another by Yarram38 involving 104 participants, reported 44% and 52% sensitivity, respectively, in detecting FH-causing mutations in patients with a Simon Broome clinical diagnosis of possible or definite FH. The kit had higher sensitivity in those with a clinical diagnosis of definite FH (49%) than in those with possible FH (40%). 37 Hooper and colleagues,36 in a study set in Australia, reported a lower sensitivity of 29% for Elucigene FH20 in detecting FH-causing mutations in patients with a clinical diagnosis of definite FH based on the Dutch criteria.
Four studies reported the sensitivity of different versions of LIPOchip. LIPOchip version 10, containing mutations frequent in the UK population, showed sensitivity of 78.5% (n = 126) (based on hypothetical data received from the manufacturer) in detecting FH-causing mutations in those with a Simon Broome clinical diagnosis, whereas the LIPOchip version designed to detect 251 mutations that were not specific to the UK showed from 33.3% (n = 22) to 56.9% (n = 120) sensitivity. The sensitivity of two Spanish versions of LIPOchip containing 195 mutations and 247 mutations was reported as 78% (n = 808) and 95% (n = 2462), respectively, with the clinical diagnosis being made according to Dutch criteria or Dutch–MedPed criteria respectively.
One study reporting the performance of LIPOchip version 8 against CGA reported one false-positive, with specificity of 93.8%. In all other studies evaluating the performance of Elucigene FH20 and LIPOchip in detecting patients with FH-causing mutations, specificity was not calculable because none of the test-positives went on to receive CGA and therefore it was not known whether or not there were any false-positive results.
In two studies, the LDL-C test as part of the Simon Broome criteria had high sensitivity (90% and 93%) in detecting FH compared with a reference standard of CGA; however, both studies also reported high rates of false-positives, resulting in low specificity (28% and 29%). In these studies, LDL-C as part of the Dutch clinical diagnostic criteria was also shown to be highly sensitive (88% and 99%) in detecting FH but with very low specificity (18% and 6%) compared with CGA. The reported sensitivity of LDL-C as part of the MedPed diagnostic criteria varied. Widhalm and colleagues47 reported that the sensitivity of LDL-C cut-offs as part of the MedPed criteria was higher in children (81%) than in adults (66%) in detecting FH. Mabuchi and colleagues46 reported higher accuracy of LDL-C cut-offs of 4.1 mmol/l (sensitivity 98.5%, specificity 98.5%) among genetically diagnosed FH patients and unaffected relatives.
Three studies reported data for age- and gender-specific LDL-C cut-offs for cascade testing compared with a reference standard of CGA. Lee and colleagues48 reported sensitivity of 91% and specificity of 93% for cascade testing in a cohort from the UK. Starr and colleagues49 reported sensitivities of 68%, 79% and 84% and specificities of 85%, 85% and 84% in cohorts of first-degree relatives from the Netherlands, Denmark and Norway respectively. Wiegman and colleagues50 reported high sensitivity of 96% in children. Using an approach of targeted gene sequencing for a mutation found in a family member, 53–77% of relatives with FH were identified in different study populations. There were no studies using LIPOchip in relatives.
Chapter 3 Assessment design and results: cost-effectiveness
Review of cost-effectiveness studies
Search strategy
Two separate searches were conducted for studies considering the cost-effectiveness of any of the intervention tests (Elucigene FH20 or LIPOchip) for proband testing or for the cascade testing of relatives. Studies were sourced from searching a range of electronic databases and websites. This was supplemented with a quality-of-life search. Contact with experts in the field and the scrutiny of bibliographies of retrieved papers were also used to identify any additional studies. Highly sensitive electronic searches were conducted to identify reports of published studies on the cost-effectiveness of tests for FH in index cases and for cascade testing of relatives. The search focused on identifying RCTs and comparative studies and the results were restricted to articles written in English. The search strategy included searches of all relevant journals since inception.
The databases searched were MEDLINE (1948 to Week 1 2011), MEDLINE In-Process & Other Non-Indexed Citations (10 January 2011), EMBASE (1980 to 2011 Week 1), BIOSIS (1956 to 10 January 2011), Science Citation Index (1970 to 10 January 2011), Conference Proceedings Citation Index – Science (1990 to 10 January 2011), Centre for Reviews and Dissemination databases including Database of Abstracts of Reviews of Effects, NHS Economic Evaluation Database (NHS EED) and Health Technology Assessment database. Searches were also carried out of the Cost-Effectiveness Analysis Registry. A supplementary quality-of-life search was also undertaken, including MEDLINE (1948 to Week 1 2011), MEDLINE In-Process & Other Non-Indexed Citations (10 January 2011), EMBASE (1980 to 2011 Week 1) and IDEAS Economics and Finance Research (February 2011). Full details of the search strategies used and websites consulted are documented in Appendix 3. In addition, reference lists of all included studies were scanned to identify additional potentially relevant studies
Methods (inclusion and exclusion criteria)
Studies were deemed to be relevant for the cost-effectiveness review if they included a measure of cost-effectiveness of the intervention tests (Elucigene FH20 – or alternative earlier versions or LIPOchip version 8–version 10) relative to any of the included clinical diagnostic criteria (Simon Broome, MedPed or Dutch criteria). The population and setting for the studies retrieved for further investigation were as described in Chapter 2. In terms of outcomes, the preferred type of analysis was cost-effectiveness measured as cost–utility analysis [cost per quality-adjusted life-year (QALY) gained]. However, because of a lack of data, we also considered other measures of cost-effectiveness, including cost per case detected or cost per diagnostic accuracy measurement. Study type inclusion and exclusion criteria were limited as we did not want to exclude any potentially relevant studies at this stage, the principal requirement being that studies were for a population of index cases or relatives of index cases with a clinical diagnosis of FH. Titles and abstracts of all reports identified by the search strategy were screened. Full-text copies of all studies deemed to be potentially relevant were obtained and assessed for inclusion. Disagreements were resolved by consensus or arbitration by a local clinical advisor. A data extraction form was developed, with data extracted by one health economist. A second health economist checked the data extraction and any disagreements were resolved by consensus among the review team. Additional further studies that did not meet our specific inclusion criteria but were none the less informative for development and population of the economic model were also retained. As these additional included studies did not form a vital part of the assessment, they have not been systematically critically appraised in depth but are included and narratively described in the following sections.
Results of the cost-effectiveness searches
A total of 258 papers were initially identified through the database searches, with a further 11 potentially relevant titles identified through the diagnostic accuracy search. However, on reading the titles and abstracts, only nine were judged potentially relevant to the cost-effectiveness review, with the remaining 260 not meeting the inclusion criteria of health economic analysis (cost-effectiveness or cost–utility) of a genetic test. We requested full-text articles of these nine papers that reported the cost-effectiveness of genetic testing and cascade testing techniques. These papers were further assessed by reading the full text of each retrieved paper and reapplying the inclusion and exclusion criteria. At this stage, only one study reported the cost-effectiveness of any of the comparators for this assessment. Of the remaining eight papers, three did not include any measure of cost-effectiveness and only briefly referred to cost implications, thus leaving a total of five relevant studies. Four of the five studies retrieved have been summarised in the previous systematic review undertaken as part of NICE clinical guidance CG71. 1 Data are extracted and published in appendix D of the clinical guidance document. The remaining study, which was not previously summarised as part of CG71,1 is discussed below. In relation to additional searches for utility of diagnostic information, effect of mutation type on treatment choice and the efficacy of statins in children, potentially relevant full-text papers were retrieved and read in full, and have been considered in the economic modelling process and/or discussion where appropriate.
Discussion of included studies evaluating the cost-effectiveness of Elucigene FH20 and/or LIPOchip
One study57 was identified that met our inclusion criteria and evaluated the cost-effectiveness of one of the intervention tests. This study assessed the cost-effectiveness of LIPOchip in identifying and testing first-degree relatives of index cases identified with FH in a Spanish population. The analysis also included subsequent treatment with statins of test-positive individuals. Screening and treatment management was compared with a strategy of no screening and the perspective of the analysis was that of the national health system (payer). Cost-effectiveness outcome was measured as incremental cost per life-year gained. Clinical diagnosis of at-risk individuals was based on a uniform protocol for clinical diagnosis and genetic testing of index cases was carried out using the LIPOchip platform, which included the following diagnostic steps:
-
LIPOchip DNA array
-
multiplex quantitative PCR used to identify significant gene rearrangements (applied if DNA array was negative)
-
complete sequencing of the LDLR gene (applied if the previous two steps were negative).
Among confirmed cases, the DNA array had a specificity and sensitivity of 99.7% and 99.9%, respectively, for all 118 mutations tested. Once index patients were identified, first-degree relatives were tested using steps 1 and 2 above only. Effectiveness among relatives was based on relative risks adjusted for age and sex59 and applied to national mortality rates. Once identified and treated, it was assumed that mortality risk reduced relative to untreated patients. The total cost of detecting a positive case was €1447 based on the assumption that, to detect one positive case, 3.4 relatives would need to be tested. This was combined with treatment costs based on simvastatin 40 mg and costs of acute myocardial infarctions (MIs) avoided based on risks calculated from Wonderling and colleagues. 59 The cost-effectiveness was thus estimated based on cost per life-year gained as €3243 in the base-case analysis. Sensitivity analyses conducted varied the incremental cost-effectiveness ratio (ICER) from €1073 to €7235 per life-year gained. Probabilistic analyses indicated a 95% probability of cost-effectiveness at a societal willingness to pay for a life-year gained of > €7400 and a probability of 45% at a willingness to pay of €3450. The results suggest that genetic screening of first-degree relatives with LIPOchip in Spain is a cost-effective use of resources. The main limitation to this study in terms of this assessment is that there is no active comparator – it is assumed that no screening would take place in routine care. However, the study is useful and informative regarding the potential of LIPOchip. No studies were available reporting on cost-effectiveness for any of the other intervention tests.
Discussion of supplementary cost-effectiveness evidence
The remaining supplementary papers detailing cost-effectiveness of cascade testing among relatives using targeted cascade testing and other methods are briefly summarised and discussed below. Full data extraction pertaining to these reports is available from the NICE website as appendix D to the NICE clinical guideline document CG71. 1 None of these studies evaluated the cost-effectiveness of any of the tests specifically in index patients; however, all indicated cost-effectiveness of cascade testing for FH among relatives of known FH index patients. The five included studies are discussed briefly below.
Marang-van de Mheen and colleagues60 compared five screening options in the Dutch population compared with no screening: treating (1) all patients with cholesterol level > the 95th percentile for the general Dutch population; (2) individuals fulfilling treatment criteria based on Dutch Institute of Health Care Improvement guidelines on hypercholesterolaemia; (3) (1) above but only those untreated at screening; (4) (2) above but only those untreated at screening; and (5) all FH-positive patients. The Framingham equation61 was used to estimate risk, survival and costs and the economic outcome measure is cost per life-year gained. This is explicitly not recommended as part of CG711 for calculating risk in the Simon Broome population. The most cost-effective option is option (2) with an associated ICER of €24,376 per life-year gained. Discounting was not conducted and there are questions relating to generalisability to a NHS perspective.
Marks and colleagues62 completed a cost-effectiveness analysis of screening for FH patients aged 16–24 years from the perspective of the NHS. Strategies evaluated were universal screening, opportunistic screening (unrelated reasons), opportunistic screening (patients with premature MI) and full screening of all first-degree relatives diagnosed with FH. The main comparison for the analysis was no screening. The primary outcome measure was cost per life-year gained and the study showed that tracing family members (first-degree relatives) systematically was the most cost-effective strategy with an ICER of £3097 per life-year gained.
Marks and colleagues63 conducted additional work over a 10-year period estimating the cost-effectiveness of (1) family tracing of index cases and (2) systematically screening all 16-year-olds. Primary economic outcomes were cost per case detected and cost per death averted. The main comparison for the analysis was no screening and no incremental analyses were conducted between groups. Costs per case identified were £3505 (family tracing) and £13,141 (universal screening). Costs per death averted were £3187 and £1.6M for the family tracing and universal options respectively. Therefore, the authors conclude that a more targeted screening programme identifying relatives of index cases is more cost-effective.
Wonderling59 used data from the Dutch screening programme from year 2000 in a sample of 18- to 60-year-olds to estimate the cost-effectiveness of screening compared with no screening. Treatment was administered using statins and it was estimated that screening would prevent 26 MIs per 100 patients receiving statin therapy. Primary outcome measures for the economic analysis were cost per case detected and cost per life-year gained, which were $7500 and $8800 respectively. Results were sensitive to the price of statins and a worst-case scenario estimated that the ICER could increase to $38,300 per life-year gained.
The additional included study was an older version of the currently included Marks study. 64 Therefore, the up-to-date data have been reported. Other studies, including those by Leren,65 Humphries and colleagues66 and Hadfield and colleagues67 all suggest that genetic screening is a cost-effective use of NHS resources and should be implemented across the UK.
The main background for the economic modelling of these candidate tests comes from NICE clinical guideline CG71,1 in which an economic model was developed to compare DNA testing with LDL-C testing. The results showed that DNA testing was cost-effective with an associated ICER of £2676 per QALY gained. This model has been updated and integrated to account for the testing of Elucigene FH20 and LIPOchip and other plausible scenarios for the identification of FH and is described in more detail in the following sections.
We did not identify any other health economic models for the identification of FH that would be informative to the development of this assessment.
Summary
NICE clinical guidance (CG71)1 concluded that genetic testing of relatives of index cases with FH is cost-effective. There was, however, no available evidence detailing the cost-effectiveness of genetic testing of index patients specifically using any of the candidate tests in this review (i.e. Elucigene FH20 or LIPOchip). One study evaluated the cost-effectiveness of an intervention test for cascade testing of relatives. 57 This is cascade testing based on LIPOchip; however, there are less costly methods of cascade testing of relatives (targeted sequencing) and so this analysis may be of limited use for informing the economic evaluation for this appraisal. A number of supplementary studies discussed provide strong evidence that cascade testing of relatives of index cases with FH is cost-effective. Based on this evidence together with the results of CG71,1 we have developed an economic model to assess the cost-effectiveness of Elucigene FH20, LIPOchip and comparators (including CGA and LDL-C) for the identification and treatment of index cases with FH and the identification of relatives by cascade testing.
Methods for economic analysis
The care pathway for this economic evaluation has been defined by NICE clinical guidance (CG71)1 and is as summarised in Chapter 1 (see Care pathways). In brief, the key points set out in this guideline that have implications for the economic evaluation recommend:
-
DNA testing to confirm clinical diagnosis of FH based on Simon Broome criteria in index (proband) patients suspected of having FH. A clinical diagnosis will include two LDL-C concentration measurements.
-
DNA testing for identified mutations in first-, second- and possibly third-degree family relatives.
-
Patients identified with FH should be offered a high-intensity statin therapy option.
A number of diagnostic pathways were specified as part of the NICE scope and review group protocol for analysis and are used to develop the economic modelling for this assessment; they are presented in Table 18.
| Strategya | Stage 1 | Stage 2 | Stage 3 |
|---|---|---|---|
| 1 | Clinical diagnosis of FH (LDL-C test required) | Elucigene FH20 | Treatment decision for index case and initiation of cascade testing for test-positive first-, second- and possibly third-degree biological relatives of the index case |
| 2 | LIPOchip | ||
| 3 | CGA | ||
| 4 | Elucigene FH20 then LIPOchip for negatives | ||
| 5 | Elucigene FH20 then CGA for negatives | ||
| 6 | LIPOchip then CGA for negatives | ||
| 7 | Elucigene FH20 then LIPOchip for negatives then CGA for negatives | ||
| 8 | Elucigene FH20 then MLPA for negatives | ||
| 9 | LIPOchip then MLPA for negatives | ||
| 10 | Elucigene FH20 then LIPOchip for negatives then MLPA for negatives | ||
| 11 | LDL-C test |
Using these care pathways we developed an economic model to estimate the cost-effectiveness of several diagnostic strategies for the confirmation of clinical diagnosis of FH among index cases and the subsequent identification and treatment of FH-positive first-, second- and third-degree biological relatives of the index case.
Model structure
The model structure was developed based on clinical advice in line with the NICE scoping document and assessment group protocol. As diagnostic strategies in themselves do not lead to quality-of-life implications directly, the model follows a linked evidence approach in which intermediate outcomes (diagnostic accuracy) are linked to treatment outcomes and hence QALY gains. By a linked evidence approach we mean that, based on diagnostic test result, a patient will be either positive or negative. Positive-testing patients receive a high-intensity treatment and negative-testing index cases receive a low-intensity treatment as they will still be at risk of cardiovascular events based on high LDL-C levels. The treatment received by each group (true-positive, true-negative, false-positive, false-negative) will determine their cardiovascular events avoided and hence their QALYs gained from that treatment decision. The outcomes on the index diagnostic test also determine whether or not the relatives will receive targeted sequencing in combination with LDL-C or LDL-C alone as the cascade test of choice. Therefore, we can say that the diagnostic test outcome of the index case is ‘linked’ to treatment choice and overall health outcomes over a lifetime horizon.
A decision tree model has been developed to identify the most cost-effective method of identification of index cases and subsequent testing and identification of at-risk relatives. Diagnostic accuracy outcomes are linked to treatment outcomes and hence QALY gains using a previously developed economic Markov model used for clinical guidance (NICE CG711).
One of the most important advantages of genetic testing is the identification of family members for cascade testing. The test used to cascade test relatives of index cases will depend on the test used to identify the index case. Three tests (targeted gene sequencing, LIPOchip and Elucigene FH20) are substantially cheaper than CGA and may be used for cascade testing. For the majority of genetically confirmed index cases, targeted sequencing for the culprit mutation is the most commonly applied genetic cascade testing method (Dr Zosia Miedzybrodzka, University of Aberdeen, 2011, personal communication). For relatives of index cases identified using the Elucigene FH20 or LIPOchip tests, the cheaper test designed to detect the identified mutation (LIPOchip, Elucigene FH20 or targeted sequencing) may be used to cascade test relatives. LIPOchip or Elucigene FH20 may also be used to cascade known mutations picked up on other tests that would also be detected by the candidate tests. 68 This scenario would apply only if LIPOchip or Elucigene FH20 were cheaper than targeted sequencing. For index cases identified based on Simon Broome criteria and not a genetic test, then LDL-C concentration measurement is the most common method used to cascade test relatives. The model structure for relatives assumes that, once a patient has a confirmed diagnosis, his/her close relatives will be identified and cascade testing will begin, first testing all first-degree relatives. For the base-case analysis it is assumed that each index case will have on average five first-degree relatives and each first-degree relative will have on average a further two first-degree relatives (second-degree relatives of each index case) who will require testing. For the purposes of this assessment we assume that once a first-degree relative tests positive, the process moves on to second-degree relatives and similarly on to third-degree relatives if appropriate. If a first-degree relative tests negative for FH, then the cascade testing process stops irrespective of the test used for cascading.
A copy of the model decision tree is illustrated in Figure 9, detailing the identification strategies for index cases in the model. Each circle represents a chance node at which probabilities of positive and negative test results are assigned. Index cases receive cost and QALY payoffs at each terminal node (triangle), at which point relatives are identified for cascade testing as described above.
FIGURE 9.
Economic decision tree model for index cases. DFH, definite familial hypercholesterolaemia; PFH, possible familial hypercholesterolaemia.
Identification of probabilities for the decision model
The probabilities used to populate this model were estimated using standard conventions of Bayes’ theorem. Basically, once we know the sensitivity and specificity of a test as well as the a priori probability of disease in the target population, we can calculate positive, negative, true-positive, true-negative and thus false-positive and false-negative values for the model. The formulae used for the calculation of each branch of the tree for single test strategies (e.g. CGA alone) are described in Table 19.
| Test results for decision tree | Calculation |
|---|---|
| Positive | Sensitivity × prevalence + (1 – specificity) × (1 – prevalence) |
| Negative | 1 – positive |
| False-positive | 1 – PPV |
| False-negative | 1 – NPV |
When tests are connected in series as add-ons to each other (i.e. the second test detects the same mutations as the first test plus additional FH-causing mutations), the theory is essentially the same but will be represented by the associated values of the second test. Taking Elucigene FH20 followed by CGA as an example, the positive rate will be [(proportion testing positive on Elucigene FH20 + proportion testing positive on CGA) – proportion testing positive on Elucigene FH20]. The proportions testing positive on Elucigene FH20 cancel each other out as they are incorporated in CGA and CGA detects all the mutations detected by Elucigene FH20 and more; therefore, the proportion testing positive on this example strategy is simply the value of the most comprehensive test in the strategy (i.e. CGA). A similar argument applies to Elucigene FH20 followed by LIPOchip.
For strategies in which MLPA is used as an add-on test to Elucigene FH20 or LIPOchip, the calculations are slightly different. As MLPA detects additional cases not detected using Elucigene FH20 or LIPOchip (we assume here that the detection of deletions and duplications on LIPOchip is inadequate and MLPA will still be needed to give a more robust estimate), the effect of the two tests in series is not as before. Therefore, for the calculation of true-negatives on Elucigene FH20 followed by MLPA, the effect will be multiplicative and can be calculated as [(1 – prevalence) × (specificity of Elucigene FH20) × (specificity of MLPA)]. The MLPA test has not been considered separately from CGA because, by definition, CGA will already include MLPA as part of the process.
Sensitivity and specificity values used in the calculations of the model are presented in Table 20 for information. More detailed information on sensitivity and specificity for all included studies is presented in Chapter 2. Studies chosen to inform the economic modelling fulfilled two main criteria: (1) they were based on patients with a Simon Broome definite FH or possible FH clinical diagnosis of FH (preferably in a UK population) where possible and (2) when tests were conducted in a number of different countries (outwith the UK) in a study, we have chosen the cohort with the largest sample size (unless some explicit reason existed why this would not be appropriate). These were assumed to offer the most robust estimates in the absence of UK data. When studies did not report clinical diagnosis based on the Simon Broome criteria or when evidence was of poor quality and limited usability, we obtained parameter values from Dutch and MedPed criteria instead. For reasons discussed in the statistical analysis, it has not been possible to pool estimates of sensitivity and specificity for a combination of definite FH and possible FH diagnoses across studies in a robust way because of study heterogeneity (see Chapter 2, Assessment of test performance). Therefore, single studies have been chosen based on the best available evidence and the most recent version of each test analysed. The impact of these choices on our base-case conclusions will be explored through the use of lowest and highest estimates available from all of the included studies, based on all clinical criteria (MedPed and Dutch criteria included), in sensitivity analysis.
| Test | Sensitivitya | Specificitya | Source used for economic modelling | Justification for choice of source |
|---|---|---|---|---|
| Elucigene FH20 | 0.44 | 1 | Taylor 201037 | This is the most up-to-date test for Elucigene FH20 |
| LIPOchip | 0.79 | 1 | Palacios 201041 | Only available data based on UK version 10 of LIPOchip |
| CGA | 1 | 1 | Assumption | Based on clinical expert opinion, this will correctly detect all known mutations causing FH; it is assumed, therefore, that if a patient tests negative he/she will not have FH |
| LDL-C (Index)b | 0.90 | 0.29 | Damgaard 200545 | This was the best available data based on Simon Broome criteria |
| LDL-C (Relatives) | 0.68 | 0.85 | Starr 200849 the Netherlands group | The Netherlands group chosen as it represented the greatest number of patients being tested (sensitivity analysis explores high and low estimates of both sensitivity and specificity based on all studies) |
| MLPAc | 0.12 | 1 | Calculation | Relates to a stand-alone detection rate of approximately 5%, confirmed through clinical expert opinion (Dr Gail Norbury, Guy’s Hospital, London, 2011 and Dr Zosia Miedzybrodzka, University of Aberdeen, Aberdeen, 2010) |
It is important to note that there is likely to be some correlation between those patients detected on MLPA and those detected using LIPOchip. Clinical expert opinion (Dr Zosia Miedzybrodzka, University of Aberdeen, personal communication) suggests that the LIPOchip test may be inadequate to detect deletions and duplications and in practice MLPA may be required to give a more accurate diagnosis.
LIPOchip can be used within the model in two separate ways. First, the strategy ‘LIPOchip’ refers to the test purchased by a laboratory in the UK from the manufacturer and processed at the UK laboratory. Additionally, the manufacturer offers a service whereby blood samples can be sent to the manufacturer’s plant in Spain for analysis using a two-stage process, first testing with LIPOchip and then sequencing of the LDLR gene for those testing negative. This is referred to as LIPOchip platform (Spain). Because of its second stage, at an additional cost of €100, this test has a higher sensitivity. It is, however, not CGA as the process does not include MLPA. Therefore, the sensitivity is less than that of CGA. Clinical expert opinion in the UK suggests that, to be able to fully detect all deletions and duplications of the gene, the MLPA test would be required as LIPOchip’s own method of detecting these cases may be inadequate. Additional data presented at the spring meeting of the CMGS70 suggest that (using data from Bristol’s NHS Hospital Genetics Laboratory) LIPOchip version 10 may be inadequate to detect copy number changes compared with MLPA, with only two cases out of a sample of seven correctly identified using LIPOchip.
In addition, there is much debate about the true prevalence of detectable FH-causing mutations among patients testing positive (definite FH or possible FH) based on the Simon Broome criteria. There is also great variation in this number between laboratories and this is likely to be because of issues of ethnicity as some tests will have different detection rates based on different ethnic groups (see Chapter 2, Assessment of test performance for additional information). For the purposes of our base-case analysis, we have assumed that 36.5% of clinically diagnosed patients (Simon Broome definite FH or possible FH) will have an identifiable mutation. 37 Data from four regional Scottish genetics services (Aberdeen, Dundee, Edinburgh and Glasgow; Dr Zosia Miedzybrodzka, University of Aberdeen, 2010, personal communication) suggest that, between 2007 and 2010, this value was approximately 35% for the whole of Scotland based on data classifiable as definite FH or possible FH. This has been confirmed in personal communication with Dr Zosia Miedzybrodzka, who estimates that, for every three patients tested in Aberdeen using CGA, on average only one will have a detectable FH-causing mutation. NICE CG711 estimates, using data extracted from the UK FH Cascade Audit Project (FHCAP),70 that 80% of patients clinically diagnosed with definite FH will have a detectable FH-causing mutation and 30% of those diagnosed as possible FH will have a detectable mutation. Given that the FH audit 201018 identifies 36% as definite FH and 58% as possible FH (the remainder being homozygous or not stated), this would suggest that 46.2% of patients clinically diagnosed as definite FH or possible FH would have an identifiable genetic mutation using CGA. Other studies quote varying estimates of these values and so maximum and minimum values will be explored in the sensitivity analysis. It is estimated that 50% of first-degree relatives of an index case will have an inherited mutation. This evidence for first-degree relatives has been applied to second- and third-degree relatives in the model. The reason for this is that the process of cascade testing is an iterative approach. Second-degree relatives will not be tested using targeted sequencing unless a first-degree relative has an identified mutation. Therefore, it is assumed that the second-degree relative is in fact the first-degree relative of an individual with an identified FH-causing mutation and so will also have a 50% probability of having inherited that mutation.
Markov model
The Markov model for this assessment has been adapted from the model used for the estimation of treatment effect used to inform NICE CG71. 1 The model was developed by the Royal College of Physicians Guideline Development Group and is updated in this assessment. This model calculated the lifelong treatment costs and outcomes of high-intensity statin therapy for the management of FH and low-intensity statin therapy for the management of others at risk of CHD because of elevated lipid levels. In addition to those who were classed as well, a total of eight further health states were modelled [unstable angina, MI, peripheral arterial disease (PAD), stroke, heart failure, revascularisation, cardiovascular death and other death]. Baseline risks were sourced from NICE technology appraisal 9471 and relative risks were sourced from the Simon Broome register. Utility weights were sourced from the literature and validated by the health economist working on this assessment. Utility of the general population was taken from the Health Survey for England 1996,72 which is the most up-to-date data source for the UK general population, and was adjusted for age and sex differentials. Beneficial health outcomes were used to estimate QALYs based on reduced risks of cardiovascular incidents. These treatment effects were sourced from a meta-analysis of two RCTs, the Incremental Decrease in Clinical Endpoints Through Aggressive Lipid Lowering (IDEAL) and the Treating to New Targets (TNT) trials conducted as part of the NICE CG71 assessment. 1 Data from Versmissen and colleagues8 were checked against and found to be consistent with the assumptions and data used for CG71,1 in so far as they show the efficacy of statins in improving the clinical causes of cardiovascular disease and by extension the reduction in serious cardiovascular events such as MI. However, they do not describe the exact causal relationship between the improved clinical outcome and reduced events. The data from Versmissen and colleagues8 are consistent with those of the CG711 assessment in that they suggest efficacy of statins and by extension the reduction in serious cardiovascular events such as MI. Costs and outcome data have been updated to current values using the latest available literature in the field or inflated to current prices (2010/2011) if no updated literature was available. Further details of the model structure are available from the NICE website (appendix E to the clinical guideline document1). The perspective of this economic evaluation is that of the UK NHS and all costs and resource use are applied in accordance with NICE guidelines on the methods of technology appraisal. NICE recommends that, where possible, the desired economic outcome is cost per QALY gained. Treatment costs and QALYs gained are extrapolated to the patient’s lifetime horizon and discounted at a rate of 3.5% per annum in line with standard NICE methods. It was not deemed necessary to discount diagnostic costs for each individual as the time taken for diagnosis is < 1 year. Sensitivity analyses explore the impact of varying the discount rate for both costs and QALYs between 0% and 6%. All other follow-up clinical costs that are expected to occur annually once a diagnosis of FH has been made are discounted as described.
Relevant patient populations
The relevant patient population for the base-case analyses is adults with heterozygous FH, focusing on index patients with a clinical diagnosis of FH based on the Simon Broome criteria (either definite or possible FH). Sensitivity and specificity of the tests included for the economic modelling both implicitly account for patients with either definite or possible FH. Data showing separate sensitivity and specificity rates for definite FH and possible FH were not available for all tests under consideration, thus making accurate subgroup analysis difficult. The data that were available are detailed in Tables 9–14 (index cases) and Table 15 (testing of relatives). Children with a clinical diagnosis are considered as a separate age subgroup in line with current CG71 recommendations. 1 Patients with an identified mutation causing FH are informed of their diagnosis and first-, second- and third-degree biological relatives are identified. Sensitivity analysis explores a situation in which only first- and second-degree biological relatives are cascade tested.
Treatment options to be evaluated
Treatment options to be evaluated are based on NICE CG71,1 which recommends that a patient with FH should be offered a high-intensity statin therapy for the aggressive lowering of lipid levels by a recommended 50%. Index cases who have elevated lipids on the basis of the Simon Broome criteria (i.e. the majority of patients) will benefit from statin therapy as they are at a ≥ 20% 10-year risk of cardiovascular disease events. 71 We assume that 10% of relatives testing negative on targeted sequencing will also require some cholesterol-lowering therapy. This is an author assumption based on clinical expert opinion and previous NICE guidance and is varied between 0% and 50% in the sensitivity analyses. This refers to the estimated percentage of relatives without an identified mutation who will require treatment on the basis of high cholesterol levels. Such cases receive a low-intensity treatment in the model. As relatives are not clinically diagnosed with FH based on the Simon Broome criteria, it would be inappropriate to treat all patients, as only a percentage will have elevated lipids. The impact of varying this assumption is explored in sensitivity analysis. There is, however, much debate among clinicians over how to treat FH and patients at an increased risk of cardiovascular disease as a result of elevated lipids, with some choosing a ‘start low’ treatment option (starting all patients on a low-intensity statin such as simvastatin 40 mg) and others giving everyone a high-intensity statin (e.g. atorvastatin 80 mg or rosuvastatin). For the base-case analysis, we have assumed a multitreatment regimen for FH patients based on and adapted from the FH clinical audit 2010. 18 Patients with a Simon Broome-positive diagnosis but who have no genetic confirmation of FH will receive low-intensity statin therapy to reduce their elevated lipid levels. Such cases (especially those relatives who are false-positive) may also respond adequately to exercise and diet therapy, the effects on quality of life of which are beyond the scope of this assessment. Cole and colleagues73 have conducted a detailed systematic review of the literature to explore the evidence in relation to the effects of dietary and lifestyle interventions in chronic heart disease risk reduction. Also, NICE guidance on dietary interventions in CHD provides additional information in the UK. Personal communications from Dr Anthony Wierzbicki (2011, Guy’s and St Thomas’ Hospitals NHS Trust) and Dr William Simpson (2011, NHS Grampian) are used in sensitivity analyses to explore the sensitivity of the model to treatment choice in practice.
Resource use estimation
Clinical resource use
For the purposes of this evaluation, we have assumed that all index cases will have received a clinical diagnosis of FH based on the Simon Broome criteria. Resource use and costs associated with this diagnosis are common to all tests being evaluated and so are not included. This is standard economic evaluation practice to include only resource-use estimations which differ between tests under consideration. However, the resource use associated with tests after the initial diagnosis is important and has been considered in the analysis. It is assumed that, once the proband has a genetic test or LDL-C confirmation of FH, he or she will attend a lipid clinic to discuss treatment and lifestyle management of the condition. It is at this point that family pedigree will be identified and contact with relatives will be initiated. It is assumed that initially only first-degree relatives will be contacted as there would be no point in contacting second-degree relatives until a diagnosis was confirmed in first-degree relatives using a genetic screen. Table 21 details resource use and cost estimation for this process based on clinical expert opinion and Hadfield and colleagues. 70
| Health-care professional | Unit cost/hour (£) | Time (hours) index case | Cost index case (£) | Time (hours) relatives | Cost per relative positive (£) | Cost per relative negative (£) | Source for unit costs |
|---|---|---|---|---|---|---|---|
| Consultation with lipid specialist | 222.00 | 222.00 | 222.00 | 0 | PBR, cardiologist, first attendance74 | ||
| Clinical nurse specialist, grade 7, to confirm family pedigree and discuss | 57.00 | 1.86 | 106.02 | 1.20 | 68.40 | 68.40 | PSSRU 2010,75 cost per hour of client contact, Hadfield 200870 |
| Clerk time to initiate contact with relativesa | 26.00 | 0.25 | 48.75 | 0.25 | 39.00 | 0 | PSSRU 2010,75 band 5 administrator |
| Cost of consumables to initiate contact with relatives | 0.78 | 5.85 | 4.68 | 0 | Cost per letter, NICE CG711 | ||
| Two lipid profile tests to confirm diagnosis | 8.00 | 16.00 | 16.00 | 16.00 | Personal communication, Dr William Simpson, 2011, NHS Grampian | ||
| Cost of processing the lipid tests | 1.60 | 3.20 | 3.20 | 3.20 | PBR national tariff for clinical biochemistry74 | ||
| Cost of GP consultation to take second cholesterol measure for confirmation | 36.00 | 36.00 | 36.00 | 36.00 | 11.7 minutes, surgery consultation, PSSRU 201075 | ||
| Total | 438.00 | 389.00 | 124.00 |
Index cases or relatives diagnosed with FH are offered an annual follow-up appointment with a lipids specialist at an outpatient clinic. In the absence of a specific unit cost tariff for a lipids specialist, this service is assumed similar to a cardiologist appointment (Dr William Simpson, University of Aberdeen, 2011, personal communication) and is costed at £222 per outpatient consultation.
Diagnostic resource use
A new national activity unit has been developed for molecular genetics and cytogenetic tests in the UK. This is based on a weighted report and uses for molecular genetics an amplicon as the base unit. All molecular genetic tests are then assigned a relative number of units that slot into bands with some efficiency built in as the number of amplicons increases. This new methodology for measuring activity for molecular genetic tests was developed by collaboration between the CMGS and the UKGTN. The objective was to devise a transparent and consensus system for measuring molecular test activity that could be implemented by all laboratories. Tests are weighted by complexity so that, for example, simply booking in a sample has the lowest weight and sequencing a gene of over 100 exons, for example RYR2, the highest. All realisable costs of each laboratory are collated and a total cost of the service is then calculated including salaries, consumables, overheads, etc. Each laboratory can derive its own unit cost, based on dividing budget by activity, and thus in effect derive a cost per test. For example, a £1.2M service producing 30,000 MOLUs will have a unit cost of £40.00. This system of pricing has been modelled by most of the laboratories in the UK and has been accepted by the professional bodies and UKGTN as a suitable approach to establishing a national tariff for genetic tests. Details of the national MOLU bands are included in Appendix 11 for information. The MOLU system is not a perfect system of estimating costs, however, and the limitations are outlined in Chapters 1 and 5.
For the base-case analysis, transportation costs of samples (preferably blood samples) for DNA testing and blood samples for LDL-C testing are included. Based on clinical expert advice (Dr Gail Norbury, Guy’s Hospital, London, 2011, personal communication), an increasing number of genetics samples are tested by processing saliva samples. Saliva-based samples are less costly to transport as they are more stable and require only first-class postage; however, the kits to extract the DNA are substantially more expensive. These resource use differences, however, will be included in the MOLU consumables mentioned above based on 1 MOLU for DNA extraction. The majority of tests are carried out in the UK; however, LIPOchip may be processed by the manufacturer on site in Spain. The additional resource and transportation costs associated with sending a blood sample overseas via air are considered for the LIPOchip platform processed in Spain. This was assumed to take a cost of 1 MOLU, commonly applied in genetic testing to cost transferring samples to laboratories overseas. Therefore, a cost of £30 has been applied in the base case. Additionally, there may be extra costs associated with resampling an estimated 3% of samples (Progenika, 2011, personal communication). These costs are also incorporated.
Unit cost estimation
Clinical costs
Costs of clinician time for treating patients, identifying a family pedigree, counselling relatives on the importance of their condition and contacting relatives themselves are estimated using Payment by Results (PBR) national tariffs where available (e.g. for a first appointment with a lipid specialist). For all other resource use, including clinical nurse specialist (to identify pedigree and counsel patients), GP time to confirm second LDL-C test and administrator time to contact relatives, costs are estimated using Personal Social Services Research Unit (PSSRU) unit costs of health and social care. 75 Costs are based on the median of the appropriate agenda for change pay scale and include overheads, training costs, insurance, annual leave, etc.
Diagnostic costs
Costs of genetic testing strategies vary greatly among laboratories, especially based on their area of expertise and also in relation to their size – the greater the laboratory size, the greater the throughput of samples tested and thus the lower the costs based on economies of scale through mass genetic testing. Laboratories that can keep their budget constant or can reduce it but increase the number of MOLUs produced will have lower unit costs. The incentive then is to reduce the total budget while maintaining or increasing output. This system is simplistic and transparent and is the method adopted by most laboratories in the UK in setting their genetic testing tariffs (Dr Zosia Miedzybrodzka, University of Aberdeen, and Dr Gail Norbury, Guy’s Hospital, London, 2011, personal communication). For the purposes of the base-case analysis, it is assumed that the MOLU cost is £30 per MOLU (Dr Kevin Kelly, University of Aberdeen, 2011, personal communication). The cost of each MOLU will be varied in sensitivity analysis provided by Dr Gail Norbury (£33 per MOLU). Unit cost estimation is adjusted within the model for strategies that have more than one test in order to account for the cost differentials associated with earlier positive test identification. The cost of DNA extraction is also incorporated into the analysis and receives a unit of 1 MOLU. Details of MOLU units applied and the associated costs for each test strategy are presented in Table 22. The cost of testing a hypothetical cohort of 1000 index cases with combination strategies is dependent on the numbers testing positive on the first test in that strategy. For example, in a strategy such as Elucigene FH20 followed by CGA for negatives, an index case who tests positive on Elucigene FH20 will not receive the second more comprehensive test.
| Testing strategy | MOLUs test 1 (including extraction) | Number positive test 1a | Number receiving test 2 | MOLUs test 2 | Number positive test 2b | Number receiving test 3 | MOLUs test 3 | Total MOLUs for a cohort of 1000 index cases testedc | Total cost per MOLU | Total cost (£) |
|---|---|---|---|---|---|---|---|---|---|---|
| Elucigene FH20 | 5 | 161 | 5000 | 30 | 150,000 | |||||
| LIPOchip | 11 | 287 | 11,000 | 30 | 330,000 | |||||
| CGA (including MLPA) | 16 | 365 | 16,000 | 30 | 480,000 | |||||
| Elucigene FH20 followed by LIPOchip for negatives | 5 | 161 | 839 | 10 | 126 | 13,390 | 30 | 401,700 | ||
| Elucigene FH20 followed by CGA for negatives | 5 | 161 | 839 | 15 | 205 | 17,585 | 30 | 527,550 | ||
| LIPOchip followed by CGA for negatives | 11 | 287 | 713 | 15 | 79 | 21,695 | 30 | 650,850 | ||
| Elucigene FH20 followed by LIPOchip followed by CGA for negatives | 5 | 161 | 839 | 10 | 126 | 713 | 15 | 24,085 | 30 | 722,550 |
| Elucigene FH20 followed by MLPA for negatives | 5 | 161 | 839 | 2 | 45 | 6678 | 30 | 200,340 | ||
| LIPOchip followed by MLPA for negatives | 11 | 287 | 713 | 2 | 45 | 12,426 | 30 | 372,780 | ||
| Elucigene FH20 followed by LIPOchip followed by MLPA for negatives | 5 | 161 | 839 | 10 | 126 | 713 | 2 | 14,816 | 30 | 444,480 |
In addition to the tests outlined above, the LIPOchip platform (Spain) as a genetic testing platform is a potential alternative to CGA. The test, which involves using the LIPOchip followed by sequencing of test-negative cases, is offered by the manufacturer (Progenika) at a cost of €250 for a LIPOchip test and €350 for the whole process. The associated costs are incorporated into the analysis using an exchange rate of €1 = £0.89. The LIPOchip platform processed in Spain is explained in Chapter 1. Briefly, this is a two-stage process whereby, if the sample is positive on LIPOchip, no further testing takes place. If the sample is negative on LIPOchip then the sample is sequenced for an additional €100. Therefore, assuming that the sensitivity of LIPOchip is the same regardless of where it is processed and using similar methodology to that in Table 22, we estimate the total cost of the strategy (before transportation of samples costs) as (1000 × 250 × 0.89) + (713 × 100 × 0.89) = £285,957.
The cost of targeted sequencing may also be estimated using the MOLU system. Targeted sequencing (including DNA extraction) is allocated a MOLU of 3. At a cost of £30 per MOLU, this would amount to £90 per targeted sequencing test. Based on the MOLU system, targeted sequencing is cheaper than Elucigene FH20 and is therefore the strategy of choice for cascading relatives.
Low-density lipoprotein cholesterol concentration measurements will be taken for all members of the study, regardless of testing strategy. Additional measures will, however, be carried out to confirm the diagnosis. Therefore, an additional two LDL-C tests will be required (at least one of which will be a fasting blood sample) to confirm the Simon Broome diagnosis if this is the method of diagnosis being adopted. It is assumed that, in order to get an extra blood test taken for the additional LDL-C measurement, an additional visit to a GP will be required. It is not expected that transportation costs of samples sent to laboratories for analysis will differ significantly between LDL-C and genetic tests as both require the transportation of potentially hazardous blood specimens.
Treatment costs
As discussed in Treatment options to be evaluated and as recommended by CG71,1 treatment will be of either high or low intensity, predominantly with statins. Should a patient be intolerant to statins, treatment may be administered using ezetimibe as per the NICE CG711 guideline. There is, however, some debate as to the relative effectiveness of ezetimibe monotherapy; therefore, only a small proportion of patients are likely to receive this treatment in practice (Dr William Simpson, NHS Grampian, personal communication). Also based on personal communication (Dr Anthony Wierzbicki), ezetimibe as monotherapy is ineffective and patients who have an inadequate response to statins may need to be treated with ezetimibe plus bile acid sequestrants. A number of FH patients will receive polypharmacy incorporating treatment with statins and ezetimibe. Table 23 details the unit costs per year of treatment with all of the potential drugs included in the modelling process with costs sourced from the British National Formulary (BNF). 76 To reflect differential treatment practice among clinicians, various combinations of these drugs (based on clinical expert opinions) are explored in the model. The most common combination therapies are included in Table 23.
| Treatment strategy | Number of tablets per pack | Cost per pack (£) | Cost per year (£) | Source |
|---|---|---|---|---|
| Atorvastatin monotherapy 40 mga | 28 | 24.64 | 321.20 | BNF 201176 |
| Atorvastatin monotherapy 80 mga | 28 | 28.21 | 367.74 | BNF 201176 |
| Rosuvastatin monotherapy 10 mg | 28 | 18.03 | 235.03 | BNF 201176 |
| Rosuvastatin monotherapy 20 mg | 28 | 26.02 | 339.19 | BNF 201176 |
| Rosuvastatin monotherapy 40 mg | 28 | 29.69 | 387.03 | BNF 201176 |
| Simvastatin monotherapy 20 mg | 28 | 1.01 | 13.17 | BNF 201176 |
| Simvastatin monotherapy 40 mg | 28 | 1.32 | 17.21 | BNF 201176 |
| Simvastatin monotherapy 80 mg | 28 | 2.29 | 29.85 | BNF 201176 |
| Ezetimibe monotherapy | 28 | 26.31 | 342.97 | BNF 201176 |
| Rosuvastatin 20 mg + ezetimibe | 28 | 52.33 | 682.16 | BNF 201176 |
| Simvastatin 40 mg + ezetimibe | 28 | 27.63 | 360.18 | BNF 201176 |
| Atorvastatin 40 mg + ezetimibea | 28 | 50.95 | 664.17 | BNF 201176 |
| Simvastatin 40 mg + ezetimibe | 28 | 38.98 | 508.13 | BNF 201176 |
For the base-case analysis, we used data from the FH audit 2010,18 the most up-to-date data source on FH treatment in practice. We also use data from clinical experts (Dr Anthony Wierzbicki, Guy’s and St Thomas’ Hospitals NHS Trust, 2011, personal communication, and Dr William Simpson, NHS Grampian, personal communication) to conduct sensitivity analysis surrounding the proportions of patients on each treatment as part of either a high- or a low-intensity statin therapy. The cost impact of atorvastatin, which is due to come off patent during the course of this assessment, will have implications for treatment costs in the model. This will be explored in sensitivity analyses.
Costs of cardiovascular events avoided as a result of treatment
Table 24 details the costs of cardiovascular events avoided. For the base-case analysis, these costs have been calculated using weighted averages of all Health Resources Group (HRG) codes pertaining to each cardiovascular event avoided. Elective and non-elective tariffs from PBR data for 2010–1174 are used and weighted for the numbers of elective and non-elective cases sourced from the Hospital Episodes Statistics online database (www.hesonline.nhs.uk/Ease/servlet/ContentServer?siteID=1937%26categoryID=192).
| Event | Cost (£) | Source |
|---|---|---|
| No event | 74 | NICE 20081 |
| MI (first year) | 3780 | Department of Health 201174 |
| MI (subsequent) | 500 | NICE 20081 |
| Stroke (first year) | 4335 | Department of Health 201174 |
| Stroke (subsequent) | 2336 | Department of Health 201174 |
| PAD (first year) | 2212 | Department of Health 201174 |
| PAD (subsequent) | 285 | NICE 20081 |
| Heart failure | 4379 | Department of Health 201174 |
| Heart failure (subsequent) | 500 | Assumption |
| Revascularisation | 8610 | Department of Health 201174 |
| Revascularisation (subsequent) | 500 | As MI (subsequent) |
| Unstable angina (first year) | 2074 | Department of Health 201174 |
| Unstable angina (subsequent) | 500 | As MI subsequent |
| Cardiovascular death | 0 | NICE 20081 |
| Death, other | 0 | NICE 20081 |
Data sourced from current NICE guidelines1 such as for subsequent MI are not available as part of PBR nor do any national tariff prices exist for these events. Therefore, values have been sourced from CG711 and inflated to current price levels for use in the model. Costs of cardiovascular death or other deaths have been assumed to be equal to £0 as it is not envisaged that this would have cost implications for the NHS. However, such deaths avoided would have great impact on the results from a societal perspective.
List of assumptions
A number of assumptions have been made throughout the modelling exercise and for the base-case model; the impact of each will be explored in relevant sensitivity analyses. Table 25 summarises the main assumptions made throughout the health economic modelling process.
| Assumption | Justification for assumption | Additional comments |
|---|---|---|
| Cascade testing is of first-, second- and third-degree relatives of the index proband case | This is the widest spectrum of relatives recommended by NICE clinical guideline CG711 and is recommended if possible | Sensitivity analysis will explore cascade testing of first- and second-degree relatives only |
| The percentage of probands providing family history and agreeing for the initiation of contact with relatives is 60% and the proportion of relatives agreeing to be tested is 65% | Assumption based on NICE clinical guideline CG711 | Assumption will be adapted and varied in sensitivity analyses based on data from Hadfield and colleagues67 |
| Cost of atorvastatin is based on BNF values | BNF | Cost of atorvastatin based on reduced pricing as a result of coming off patent will be explored in sensitivity analysis |
| 10% of negative relatives receive low-intensity statin therapy | Relatives who are negative for FH are test-negative and are unlikely to require treatment (author assumption) | In sensitivity analysis a proportion of negatives will receive lipid-lowering therapy based on low-intensity statins (this will not assume costs of annual follow-up in secondary care). A range of 0–50% will be explored |
| No QALY decrements for patients testing false-positive for FH | Author assumption | Patients who test false-positive may incur a QALY decrement due to stress and anxiety associated with having a condition; however, if they have high LDL-C levels it is likely that this will be offset by the knowledge that they are being treated for their high cholesterol and will be at reduced risk of cardiovascular disease |
| Prevalence, sensitivities and specificities for cascade testing using LDL-C are assumed to be the same for cascade testing from index test-negatives and index test-positives | Author assumption | All index cases have a clinical diagnosis of FH regardless of whether or not they have a detectable mutation. Sensitivity analysis varies all estimates of test sensitivity and specificity in the model |
| All index cases will require treatment of some kind | As patients will be positive for FH, they will have elevated cholesterol levels by definition and will be at increased risk of cardiovascular events | Sensitivity analyses will assume a fraction of these patients are treated (i.e. only those with a genetically confirmed mutation) |
Data analysis
Base-case analysis
For the base-case analysis, we analyse an index patient of age 50 years, with an assumed average first-degree relative age of 50 years. The decision model is run on the basis of a hypothetical cohort of 1000 patients with a clinical diagnosis of FH based on the Simon Broome criteria (including both definite FH and possible FH). Cost and QALY values are estimated as described in the preceding sections and applied to the number of people passing through each branch of the decision tree illustrated in Figure 9. On the basis of test accuracy, a proportion of all 1000 index patients are positive (true-positive or false-positive) or negative (true-negative or false-negative). These patients are assigned the relevant cost and QALY values as described and total costs and QALYs are generated for the full cohort.
Test strategies are ranked in ascending order of cost. Those strategies that are more costly and less effective are excluded on the basis of simple dominance. Additional tests that are dominated by a combination or two or more alternative strategies are excluded by extended dominance. ICERs are calculated as incremental costs divided by incremental QALYs between non-dominated strategies. This is the most common method of presenting ICERs and relates the options sequentially ranked by costs. For the purposes of this assessment, the most relevant comparators are:
-
CGA, recommended indirectly by NICE guidance CG71. 1
-
LDL-C concentration measurement only. The reason for this is that, in practice, LDL-C is the main method of identification presently adopted in the UK (although genetic testing is more common in Scotland, Wales and Northern Ireland than in England).
Therefore, ICERs are presented as cost per QALY compared with the two suggested reference standards for this evaluation (LDL-C and CGA).
This process is applied to two distinct research questions. First, we investigate the cost-effectiveness of each of the 12 strategies for index cases alone. However, of greater importance and thus the primary focus of the analysis is to present cost-effectiveness estimates for the complete process of index case confirmation of clinical diagnosis but also for the identification and testing of relatives (i.e. the whole cascade testing process).
Subgroup and additional scenario analysis
The cost and QALY results for different age groups are explored in this section for the full cascading project only (i.e. index and relative cases). Results for index cases alone are presented in Appendix 12.
These subgroup analyses include a range of age profiles and also include the incorporation of any available evidence relating to the efficacy of statins in the treatment of children. To this end, we have completed a structured search of the literature, which has identified four systematic reviews of the efficacy of statins in children, the most recent of which is a Cochrane review of high quality that is used to inform the discussion and the model. 77 The data suggest that statins are efficacious in children in reducing cholesterol and have non-significantly different adverse events to placebo. Therefore, statins are likely to be safe in children with FH although long-term follow-up of this patient group is required. As data relating directly to CHD are lacking, treatment effect relative to CHD is assumed to be similar to that of a young adult (equivalent to a 30-year old index case in the economic model).
A number of age-specific subgroups were considered (probands aged 15, 30, 50, 65, 75 and 85 years). These age subgroups are similar to those used in previous economic modelling for FH1 and represent a good distribution of the ages of the population who may present for testing. Table 26 details the calculated number of relatives for each index case and their average age used in the model.
| Age of index case years | Number of first-degree relatives | Number of second-degree relatives | Number of third-degree relatives | Average age of all relatives years |
|---|---|---|---|---|
| 15 | 3 | 6 | 8 | 50 |
| 30 | 5 | 4 | 4 | 30 |
| 50 | 5 | 4 | 4 | 50 |
| 65 | 3 | 6 | 8 | 30 |
| 75 | 3 | 6 | 8 | 50 |
| 85 | 2 | 6 | 4 | 30 |
As discussed in Model structure, there may be alternative estimates of cost-effectiveness based on whether the index case is identified as definite FH or possible FH as their clinical diagnosis. It should be noted, however, that because of a lack of sensitivity data for each test separated into definite FH and possible FH subgroups, it was not possible to conduct robust analyses of FH cases split by clinical diagnosis subgroup. We have, however, conducted threshold analyses which show the combination of mutation prevalence and test sensitivity that would be required for the candidate test to be considered cost-effective as a pre-screen to CGA. The probabilistic sensitivity analysis accounts for the combined variation in all of the input parameters.
Sensitivity analyses
As many assumptions are made throughout the modelling process and selective data are chosen to inform the parameters, it is possible that the results generated will be sensitive to some of the judgement calls, assumptions and decisions made in the analysis. Therefore, we carry out a range of sensitivity analyses to determine the sensitivity of the base-case results to changes in our assumptions. A range of univariant deterministic analyses are presented in Appendix 14, the main results of which are reported and discussed in Analysis of uncertainty, including probabilistic sensitivity analysis. In addition, a probabilistic sensitivity analysis is also presented to explore uncertainty in the model.
Areas of uncertainty that are explored include:
-
Prevalence rates of FH-causing mutations among clinically diagnosed index cases and at-risk relatives.
-
Treatment differences for those with genetically confirmed FH and those without a genetic confirmation. The implication of forthcoming price reductions of atorvastatin is also explored.
-
Uncertainty surrounding the proportion of probands and relatives with a given test result receiving treatment (e.g. the proportion of those with a false-negative or true-negative test result receiving statin therapy).
-
The costs of diagnostic strategies, especially issues of uncertainty surrounding the MOLU pricing system and the likely cost of a 1-unit MOLU output.
-
Key assumptions relating to the model structure, including cascade testing only of first- and second-degree relatives, discount rates applied to costs and effects, the impact of not cascade testing negative index cases and the proportion of index and relative cases agreeing to participate in the identification and testing process.
-
Uncertainty associated with assumptions listed in Table 25 including structural assumptions regarding management of negative-testing index and relative cases.
Probabilistic sensitivity analysis
Deterministic one-way sensitivity analyses and point estimates of ICERs do not adequately provide information on the true impact of uncertainty surrounding the model parameters. Because of imperfect information on both the resource use and effectiveness of each treatment strategy, costs and QALYs are highly likely to be subject to at least some degree of uncertainty. Therefore, we conducted additional probabilistic sensitivity analysis using Monte Carlo simulation (5000 repetitions). Distributions were fitted to each of the parameters based on published studies (where available), CG71 data1 and a number of assumptions where no data were available. For example, where insufficient data existed in published sources to fit distributions to parameters, standard errors were assumed in order to calculate alpha and beta values. This may slightly under- or overestimate the variation in some of the parameters; however, it is not likely to impact greatly on resultant cost-effectiveness acceptability curves (CEACs). For sensitivity of test strategies (Elucigene FH20 and LIPOchip) the analysis was bounded by the highest and lowest reported mean values in all of the studies identified from the systematic review of the literature. Full details of probabilistic sensitivity analysis parameters are presented in Appendix 16.
The net benefit framework was used to estimate net monetary benefits for each simulation as described in Briggs. 78 The defining characteristic of this approach is that all strategies add to a probability of cost-effectiveness equal to 1. This uncertainty is illustrated in the form of CEACs for each of the non-dominated strategies of testing. CEACs for the base-case analysis are presented in the text, with supplementary analyses following the same approach for each age subgroup in the model presented in Appendix 15 for completeness. The analysis is presented for non-dominated test strategies only. The comparison for the calculation of incremental costs and QALYs for this analysis is LDL-C as this is current practice in the NHS. As the remit of this report is primarily to assess the cost-effectiveness for index cases and relatives, we have not conducted probabilistic sensitivity analysis for index cases alone. In addition, CEACs are presented for 5%, 10%, 20% and 50% mutation prevalence rates in order to reflect the uncertainty surrounding mutation detection rates in various subgroups of the population, primarily varying based on ethnic background.
Results of economic analysis
Results presented for the base-case analysis are subject to the assumptions listed in Table 25.
Summary of test results for a hypothetical cohort of 1000 familial hypercholesterolaemia patients
Table 27 details the flow of a hypothetical cohort of 1000 patients through the model based on those testing false-positive, true-positive, false-negative and true-negative. The values for sensitivity and specificity are combined values for all definite FH or possible FH patients based on the Simon Broome criteria. We have used estimates of sensitivity and specificity derived from the clinical effectiveness review and applied these to the model as discussed in Methods for economic analysis.
| Diagnostic test | Index cases | Relatives of positive index cases (tested using targeted sequencing)b | Relatives of negative index cases (tested using LDL-C)b | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TP | FP | TN | FN | Total | TP | FP | TN | FN | Total | TP | FP | TN | FN | Total | |
| Elucigene FH20 | 161 | 0 | 635 | 205 | 1000 | 374 | 0 | 374 | 0 | 748 | 900 | 255 | 1307 | 662 | 3124 |
| Elucigene FH20_LIPOchip | 287 | 0 | 635 | 79 | 1000 | 667 | 0 | 667 | 0 | 1335 | 765 | 216 | 1111 | 563 | 2655 |
| Elucigene FH20_CGA | 365 | 0 | 635 | 0 | 1000 | 851 | 0 | 851 | 0 | 1701 | 680 | 193 | 989 | 501 | 2362 |
| Elucigene FH20_LIPOchip_CGA | 365 | 0 | 635 | 0 | 1000 | 851 | 0 | 851 | 0 | 1701 | 680 | 193 | 989 | 501 | 2362 |
| Elucigene FH20_MLPA | 205 | 0 | 635 | 160 | 1000 | 478 | 0 | 478 | 0 | 956 | 852 | 241 | 1238 | 627 | 2958 |
| Elucigene FH20_LIPOchip_MLPA | 331 | 0 | 635 | 34 | 1000 | 772 | 0 | 772 | 0 | 1543 | 717 | 203 | 1042 | 528 | 2489 |
| LIPOchip | 287 | 0 | 635 | 79 | 1000 | 667 | 0 | 667 | 0 | 1335 | 765 | 216 | 1111 | 563 | 2655 |
| LIPOchip platform (Spain) | 321 | 0 | 635 | 45 | 1000 | 747 | 0 | 747 | 0 | 1493 | 728 | 206 | 1058 | 536 | 2529 |
| LIPOchip_CGA | 365 | 0 | 635 | 0 | 1000 | 851 | 0 | 851 | 0 | 1701 | 680 | 193 | 989 | 501 | 2362 |
| LIPOchip_MLPA | 331 | 0 | 635 | 34 | 1000 | 772 | 0 | 772 | 0 | 1543 | 717 | 203 | 1042 | 528 | 2489 |
| CGA | 365 | 0 | 635 | 0 | 1000 | 851 | 0 | 851 | 0 | 1701 | 680 | 193 | 989 | 501 | 2362 |
| LDL-C | 329 | 451 | 184 | 37 | 1000 | 835 | 236 | 1214 | 615 | 2901 | 236 | 67 | 344 | 174 | 821 |
Assumptions relating to the incidence of FH among the tested population are discussed in Model structure; however, for the base-case model we assume a mutation detection rate of 36.5%37 among people who are either possible or definite FH using the Simon Broome clinical diagnosis (i.e. approximately one in three reporting for testing will test positive based on CGA). Sensitivity analysis explores the variation in this estimate.
As we have assumed that each genetic test is associated with specificity equal to 1, there are no false-positives for the base-case analysis. However, data suggest that LDL-C among index cases has a specificity of 0.29,44 indicating that a substantial number of positive test results will in fact be false-positives.
Mean cost and mean treatment effects associated with each diagnostic strategy
Index (proband) familial hypercholesterolaemia patients
Table 28 presents total costs and total QALYs for each treatment strategy for index cases alone ranked according to cost with dominance or otherwise indicated. Patients without FH will have a slightly longer survival prognosis and will thus receive slightly greater QALY gains than those with FH. Such patients are clinically diagnosed as having FH, have high lipid levels and are at an increased risk of CHD and so will have a positive response to cholesterol-lowering therapy.
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 14,192,370 | 13,005 | |||
| Elucigene FH20_MLPA | 14,462,441 | 13,016 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 14,991,529 | 13,037 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 15,063,229 | 13,037 | Dominated | Dominated | Dominated |
| LIPOchip platform (Spain) | 15,154,374 | 13,045 | 962,004 | 40 | 24,025 |
| LIPOchip_MLPA | 15,254,040 | 13,048 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 15,325,740 | 13,048 | Dominated | Dominated | Dominated |
| CGA | 15,528,212 | 13,056 | 373,838 | 11 | 33,402 |
| Elucigene FH20_CGA | 15,575,762 | 13,056 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 15,699,062 | 13,056 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 15,770,762 | 13,056 | Dominated | Dominated | Dominated |
| LDL-C | 17,678,183 | 13,079 | 2,149,970 | 23 | 93,518 |
The Elucigene FH20 diagnostic strategy alone generates the lowest costs for identifying index patients for two reasons: first, it is the cheapest genetic diagnostic test available and, second, it detects the lowest number of true-positive index cases. Therefore, it confirms the clinical diagnosis in the fewest index cases with FH and for that reason is associated with the lowest QALYs of all of the tests included. LDL-C identifies the largest proportion of positives (not necessarily true-positives for FH – although all index cases are technically true-positives based on their clinical diagnosis) and has the highest QALY gain as it detects the greatest number of patients at increased risk of CHD. Of the non-dominated sequences, LIPOchip platform (Spain) and CGA are both associated with ICERs between £20,000 and £35,000 per QALY gained.
Index cases and relatives
However, where genetic testing has the greatest advantage over LDL-C is in the identification of relatives for cascade testing. Cascade testing using LDL-C alone is less likely to be cost-effective in this population because of the large number of false-positives (including relatives incorrectly identified as having FH) who may be treated using high-intensity statin therapy when low-intensity therapy would have sufficed to reduce their cholesterol levels. Assumptions regarding false-positive relatives are detailed in Table 25 and can be tested in sensitivity analysis within the model. Table 29 presents total costs and QALYs for index and relative cases combined (i.e. a whole integrated strategy for the identification and management of index cases and relatives with FH). Cascade testing of relatives of an index case with an identified mutation is by targeted sequencing. This is because targeted sequencing is less costly than both of the other candidate tests. Therefore, as all tests would detect the identified mutation they are supposed to in the relatives, targeted sequencing is the most cost-effective way to do this in relatives of a mutation-positive index case.
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 43,371,985 | 36,653 | |||
| LDL-C | 43,880,789 | 34,744 | Dominated | Dominated | Dominated |
| Elucigene FH20_MLPA | 44,470,770 | 37,216 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 46,506,304 | 38,240 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 46,578,004 | 38,240 | Dominated | Dominated | Dominated |
| LIPOchip platform (Spain) | 47,298,810 | 38,668 | 3,926,825 | 2015 | 1949 |
| LIPOchip_MLPA | 47,597,529 | 38,803 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 47,669,229 | 38,803 | Dominated | Dominated | Dominated |
| CGA | 48,501,362 | 39,231 | 1,202,552 | 563 | 2135 |
| Elucigene FH20_CGA | 48,548,912 | 39,231 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 48,672,212 | 39,231 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 48,743,912 | 39,231 | Dominated | Dominated | Dominated |
In the analysis presented in Table 29, LDL-C is the least effective of all tests. Elucigene FH20 is the least costly genetic testing strategy and is also the most cost-effective of all non-dominated genetic testing strategies relative to LDL-C, being less costly, more effective and thus dominant. CGA is the most effective non-dominated strategy in terms of QALYs gained, with an associated ICER of £2135 per QALY gained relative to the next most effective non-dominated strategy (LIPOchip platform, Spain). Combination genetic tests are dominated by single genetic test strategies. For example, Elucigene FH20 followed by LIPOchip is dominated by LIPOchip alone-meaning that the extra cost of pretesting with Elucigene FH20 does not add any additional QALYs over and above LIPOchip. The reason for this is that LIPOchip will detect the same mutations and cost more when added to Elucigene FH20. A similar argument can be made for tests used for pre-screening prior to CGA. The case for test strategies including MLPA as a component is slightly different in that MLPA detects deletions and duplications of the gene and so detects approximately an extra 5% of mutations that would not otherwise be detected by Elucigene FH20. MLPA is incorporated and included in the CGA process and has not been considered separately here. In relation to LIPOchip, there is some uncertainty in relation to the detection of deletions and duplications of the gene. Therefore, we have taken a pragmatic approach and included LIPOchip alone and LIPOchip followed by MLPA. This will allow the reader to decide, based on further investigation of LIPOchip, whether or not MLPA would be required to obtain a definitive diagnosis among positive test results (i.e. a specificity of 1) as assumed in the model.
Incremental analysis for reference case and other scenarios
Index cases
Tables 30 and 31 evaluate the non-dominated sequences compared with the relevant comparators for index cases. The scope and protocol for this assessment define two important comparators: (1) the comparator recommended as part of the NICE clinical guidelines – full genetic DNA testing (or CGA as defined in our protocol) and (2) LDL-C, which is currently the most commonly used method as part of the Simon Broome criteria to identify FH in practice in the UK. Currently, DNA testing is available only in 15% of UK primary care trusts (UK FH audit project 201018) and therefore LDL-C is deemed an appropriate comparator based on current clinical practice in the UK (NICE diagnostic advisory group, 2011, personal communication).
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 14,192,370 | 13,005 | –3,485,812 | –74 |
| LIPOchip platform (Spain) | 15,154,374 | 13,045 | –2,523,808 | –34 |
| CGA | 15,528,212 | 13,056 | –2,149,970 | –23 |
| LDL-C | 17,678,183 | 13,079 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 14,192,370 | 13,005 | –1,335,842 | –51 | |
| LIPOchip platform (Spain) | 15,154,374 | 13,045 | –373,838 | –11 | |
| CGA | 15,528,212 | 13,056 | |||
| LDL-C | 17,678,183 | 13,079 | 2,149,970 | 23 | 93,518 |
Of the non-dominated sequences, LDL-C is the most costly and most effective test overall (see Table 30). Elucigene FH20 is the least costly but also the least effective test in terms of QALYs. In fact, all of the non-dominated testing strategies are cheaper overall and generate fewer QALYs than LDL-C. Although diagnosis costs for LDL-C are lower than the alternatives presented, treatment costs are much higher. This is because as all index patients will technically have FH based on their clinical diagnosis on the Simon Broome criteria, they will benefit from statin therapy. Additionally, even if they were not true FH, they would still be at an increased risk of coronary artery disease based on their cholesterol levels and so would benefit from treatment. LDL-C is therefore also associated with the greatest number of QALYs gained for index cases. This is because, should a negative diagnosis be based on a genetic test, patients who test false-negative may be inappropriately treated and would thus gain fewer QALYs than if they were prescribed high-intensity treatment for their FH based on LDL-C levels.
Table 31 presents similar information for index cases alone when the comparator of interest is CGA.
When the comparison of interest for index cases alone is CGA, all other non-dominated genetic tests are less costly and less effective than CGA. The question for a decision-maker in this scenario would thus be whether or not the cost savings are worth the associated QALY loss. ICERs are not reported in informing such a question as there is lack of evidence regarding how much society is willing to accept in compensation (in the form of cost savings) for a QALY loss.
Figure 10 presents the cost-effectiveness plane comparing all tests for index cases. This confirms the results alluded to in the tables of results above.
FIGURE 10.
Cost-effectiveness plane (index cases).
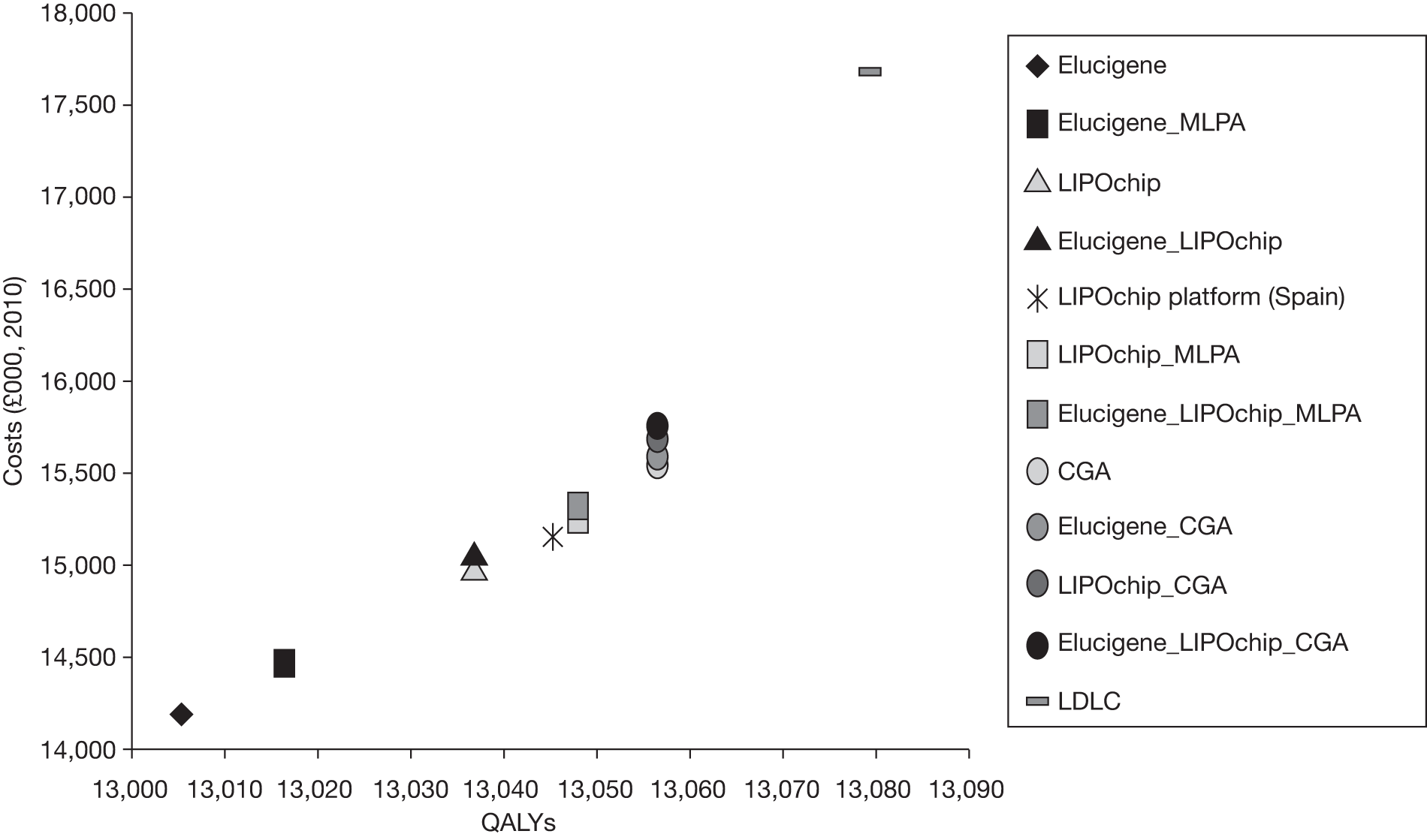
There are two important things to note from this illustration. First, LDL-C is the most costly strategy (driven by high-intensity statin treatment costs). As all patients are at risk of cardiovascular disease, however, QALY gain is highest driven by the extra-intensive treatment based on false-positive diagnoses of FH by LDL-C. These patients benefit from the increased statin therapy as they are at increased risk of cardiovascular disease based on their cholesterol levels. Second, the graph illustrates the dominance of single test strategies over similar strategies preceded by less-sensitive screening tests such as Elucigene FH20. As all strategies ending in CGA generate the same QALY gains, dominance is due to greater costs amongst multiple test strategies.
Confirmation of clinical diagnosis in index cases and cascade testing of relatives
Tables 32 and 33 evaluate the non-dominated sequences compared with the relevant comparators for the full process of index case confirmation of the clinical diagnosis and cascade testing of relatives. The comparators are LDL-C and CGA as in the preceding section.
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 43,371,985 | 36,653 | –508,805 | 1909 | Dominant |
| LDL-C | 43,880,789 | 34,744 | |||
| LIPOchip platform (Spain) | 47,298,810 | 38,668 | 3,418,020 | 3924 | 871 |
| CGA | 48,501,362 | 39,231 | 4,620,573 | 4487 | 1030 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 43,371,985 | 36,653 | –5,129,377 | –2578 |
| LIPOchip platform (Spain) | 47,298,810 | 38,668 | –1,202,552 | –563 |
| CGA | 48,501,362 | 39,231 |
Multiple testing strategies are dominated by single testing strategies generating the same sensitivity and test-positive rate overall. All non-dominated genetic tests are highly cost-effective compared with LDL-C in the identification of index cases with FH and cascade testing of relatives (assuming that society’s willingness to pay for a QALY gain is £20,000). The Elucigene FH20 single test strategy is the most cost-effective, being less costly and more effective and thus dominant over LDL-C (see Table 32). However, should a decision-maker wish to have a DNA test with a definitive genetic diagnosis, (i.e. CGA) then this is more expensive but generates the most QALYs gained compared with LDL-C. Relative to LDL-C (current practice), CGA could be considered a cost-effective testing strategy with an associated ICER of only £1030 per QALY gained. This is also well below a willingness-to-pay value of £20,000 per QALY gained.
When cost and QALY pairs are compared with CGA (current NICE recommendations) for the whole process of identification of index cases and cascade testing of relatives, all non-dominated tests are less costly and less effective than CGA. Table 33 presents this comparison.
Again, as discussed previously, the reporting of ICERs for this scenario does not inform the question of what reduction in QALYs a decision-maker is willing to accept in order to achieve a predefined cost saving. All non-dominated testing strategies are less costly and also less effective than CGA.
Figure 11 presents the cost-effectiveness plane comparing all tests for index cases and cascade testing of first-, second- and third-degree biological relatives. This confirms the results alluded to in the tables of results above.
FIGURE 11.
Cost-effectiveness plane for index cases and cascade testing of relatives.
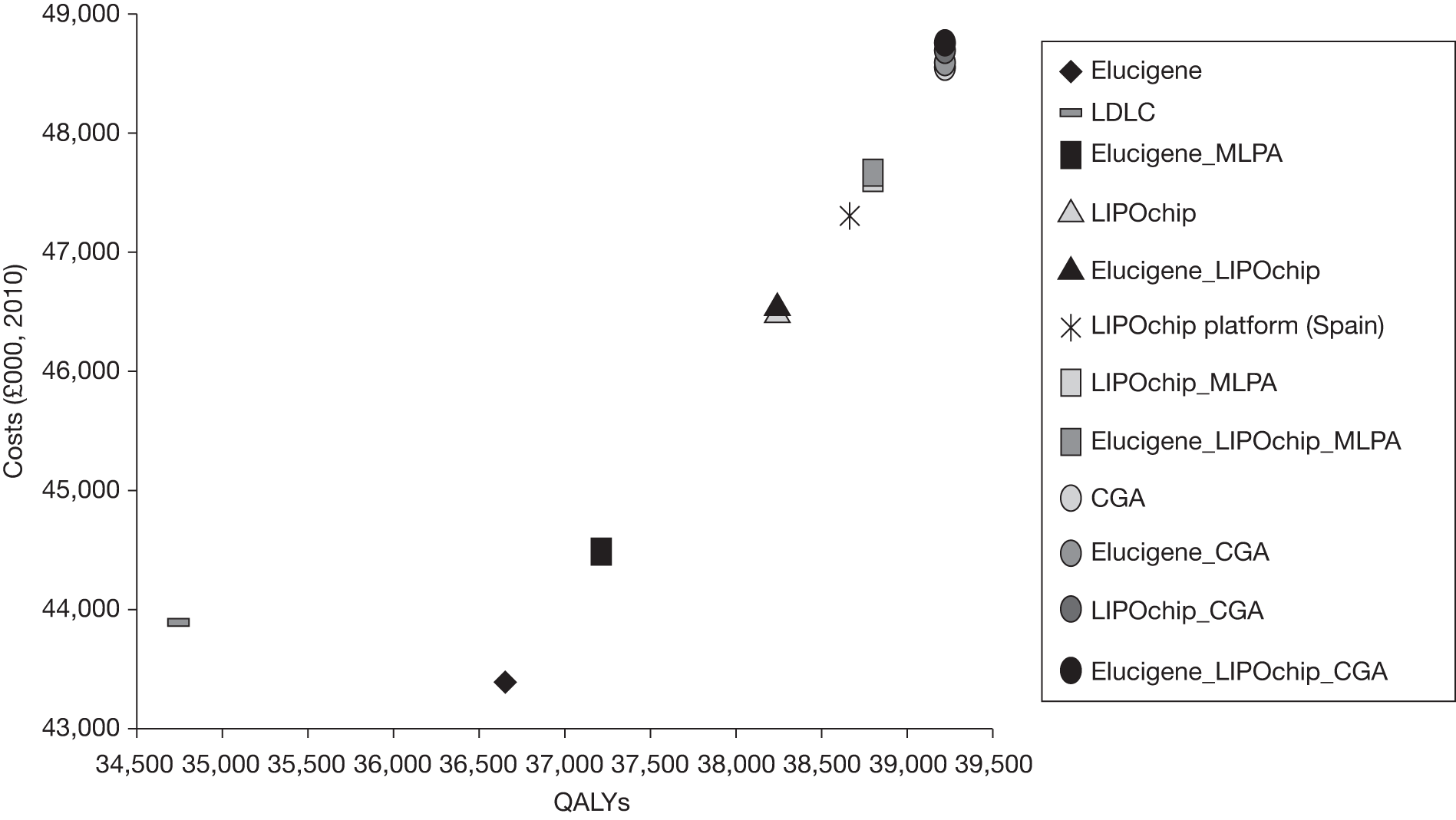
In this scenario (including cascade testing of relatives in the analysis), LDL-C used as a method of identification of relatives is less costly than all other tests (with the exception of Elucigene FH20) but does not generate the same QALY gains as any of the genetics-based tests. LDL-C is an inexpensive test to carry out (relative to other more costly genetic options); however, LDL-C alone will falsely diagnose many index cases as having FH and hence many relatives will be cascade tested unnecessarily for fewer QALYs gained. LDL-C is thus dominated by the lower-cost Elucigene FH20 test.
Differential results for subgroups
The impact of varying the age of the index case and associated average age of relatives is explored in this section. As with the base-case analysis, results are presented sequentially and also incrementally relative to LDL-C and CGA. Analyses for index cases only are presented in Appendix 12.
For index cases alone, the results are quite difficult to interpret and there appears to be much variability in the ICERs depending on age (see results tables in Appendix 12). As in all other analyses, all pre-screen tests are dominated by more effective tests that generate cost savings due to treatment effects. For all index case ages, non-dominated test strategies appear to be less costly and less effective than LDL-C, with the exception of an 85-year-old index case, for which genetic tests are dominant over LDL-C. These results should, however, be interpreted with caution. The wide variability in the presented ICERs is due to small or indeed negligible QALY differences between strategies. This is because, for index cases alone, most if not all patients will be at risk of cardiovascular events and all will have a clinical diagnosis of FH.
Genetic testing has the advantage in the identification and treatment of relatives through the cascade testing process for all age subgroups and this is evident from associated tables (for index cases and relatives combined) reported in Appendix 13 and discussed in the following paragraph.
The results presented suggest that, as in the base case, all pre-screening strategies are dominated by single test strategies detecting the same number of people, regardless of age. The reason for this is that costs associated with savings on test-positive cases are offset by submitting a whole cohort of negative patients through two or maybe three tests. As only a proportion will have a genetic mutation, these additional costs outweigh cost savings from those tested positive on pre-screens such as Elucigene FH20 or LIPOchip. This confirms the base-case results presented in Tables 29, 32 and 33. As reported for the base case in Table 29, relative to LDL-C, Elucigene FH20 is the most cost-effective option for all age groups analysed, with all ICERs under £1400 per QALY gained. This probably represents a highly cost-effective use of NHS resources. The next most cost-effective testing options after Elucigene FH20 are LIPOchip (platform processed in Spain), for which the costs per QALY gained are between £714 and £2513 irrespective of age group analysed, and CGA, with ICERs only slightly higher than those of LIPOchip (platform processed in Spain). Therefore, as in the base case, there are a number of tests that could be deemed cost-effective, all with very low ICERs relative to LDL-C. As discussed, should we wish to achieve a definitive diagnosis and generate the greatest QALY gain then CGA is a cost-effective means to achieve such an objective.
Summarising these results together, all of the age group analyses are consistent with the conclusions of the base-case analysis for an index case aged 50 years. Therefore, one may conclude that the conclusions of the model for index and relative cases are not sensitive to the age of the index case or associated relatives. CEACs based on probabilistic sensitivity analysis of the age subgroup results show some uncertainty at low threshold values of willingness to pay but, at threshold values > £5000 per QALY gained, CGA is the most likely cost-effective testing strategy, increasing to almost 100% as the threshold value increases towards a threshold ceiling ratio of £20,000 per QALY gained. CEACs reporting these results are presented for illustration in Appendix 15.
Because of a lack of good-quality data differentiating the sensitivities of the tests for definite and possible FH, we have conducted a threshold analysis indicating the prevalence and sensitivity that would be required for the candidate tests to be cost-effective as a pre-screen for CGA. Additional sensitivity analysis around the maximum and minimum values of all reported studies is presented in the following section. At the current estimate of sensitivity of Elucigene FH20, there would need to be an underlying prevalence of mutations of 61% at current prices of CGA. Should the price of CGA drop in the future as a result of next-generation sequencing, the required prevalence of underlying mutations would need to be 93%. This is based on an assumed price reduction of 40% in the cost of DNA sequencing in the future. The results for LIPOchip are less favourable at current levels of sensitivity as the lower cost of Elucigene FH20 followed by CGA would dominate LIPOchip followed by CGA at high prevalence rates, irrespective of whether or not we apply a cost reduction of 40% to DNA sequencing as part of CGA.
From an alternative perspective, one may be interested in the sensitivity of Elucigene FH20 and/or LIPOchip that would be required to generate cost savings as a pre-screen to CGA at current levels of mutation prevalence. Elucigene FH20 would be required to have a sensitivity of at least 73% to be a cost-saving pre-screen to CGA for a mutation prevalence rate of 36.5% as used in the base-case economic model. LIPOchip would not be cost-effective as a pre-screen to CGA for any plausible sensitivity values at this mutation prevalence level. Plausible values are defined as those sensitivities below the sensitivity of CGA. The reason for this is that, because of the relatively low prevalence of mutations, even at a sensitivity of 90%, only 33% of cases would be positive, with the remaining 67% requiring CGA to confirm the presence or otherwise of a FH-causing genetic mutation.
Therefore, if the goal is to gain an unequivocal diagnosis, for low mutation prevalence rates, pre-screening with Elucigene FH20 or LIPOchip prior to CGA is not cost-effective. At high prevalence rates, > 61%, Elucigene FH20 may offer a cost-effective option; however, this is less likely once the costs of next-generation sequencing fall.
Analysis of uncertainty, including probabilistic sensitivity analysis
One-way deterministic sensitivity analyses
A range of one-way deterministic analyses are presented to investigate the sensitivity of the model to uncertainty in some of the key parameters and in relation to model structural assumptions as outlined in Table 25. All deterministic sensitivity analyses were carried out on the base case of an age 50 years index case.
A full range of sensitivity analyses in relation to treatment effect have been carried out previously in the NICE clinical guidance CG71. 1 The model was found to be insensitive to a range of sensitivity analyses including assumptions surrounding nurse and consultant time with patients, costs of cholesterol testing, costs of letters to relatives for cascading, cascading from alternative numbers of relatives (first and second degree), relative risks of non-cardiovascular disease deaths and treatment effect used in the model. As data from the CG711 assessment have been updated and used for the purposes of this report, it is highly unlikely that these sensitivity analyses will have any impact on the sequences of ICERs for this analysis. We have additionally explored the impact of including a cost of £80 (standard A&E tariff) for those patients who die in the model. This is to reflect any additional costs that may be involved over the £0 assumed in the base-case analysis. The results are not sensitive to this assumed value.
Therefore, the focus of sensitivity analyses for this assessment centres on parameters and assumptions that we hypothesise may have an impact on the sequence of ICERs or on the overall cost-effectiveness conclusions. Many parameters alter the cost-effectiveness of identifying index cases alone; however, as the remit for this report is primarily the detection and treatment of relatives with FH, we focus mainly on analyses that affect the overall outcome (i.e. the confirmation of index cases and the cascade testing of at-risk relatives). Full analyses for both groups are included in Appendix 14 for information. In the appendix, results for the index case analysis are presented first, followed by results for index cases and relatives together. The order of tables follows the sequence of results presented below.
The following discussion refers to index cases and relatives together.
Prevalence of familial hypercholesterolaemia-causing mutations among index cases and relatives
Prevalence of FH-causing mutations among index cases is varied between 28%79 and 52%. 41 For both low and high estimates of mutation prevalence, the order of the ICERs remains unchanged compared with the base case. Elucigene FH20 remains the most cost-effective strategy relative to LDL-C (associated ICERs = dominant and £395 per QALY gained for low and high estimates respectively). The next most cost-effective options after Elucigene FH20 are LIPOchip (platform processed in Spain) and CGA for both low and high mutation prevalence rates with all ICERs < £1300 per QALY gained. See Differential results for subgroups for a threshold analysis estimating the prevalence required for Elucigene FH20 or LIPOchip to be deemed a cost-effective pre-screen to CGA.
Prevalence of FH-causing mutations among relatives of index cases is an uncertain parameter that is generally held to be approximately 50%, based on the logic that one out of every two offspring will inherit a genetic mutation. Sensitivity analysis varied this assumption by ±20% to between 40% and 60% of first-degree relatives inheriting the culprit gene (author assumption). A low estimate suggests that Elucigene FH20 is dominant, being less costly and generating more QALYs than LDL-C. After that, as in the base-case analysis, LIPOchip platform (processed in Spain) and CGA remain the next most cost-effective testing strategies. A higher estimate of mutation prevalence among relatives of 60% suggests the same three non-dominated test strategies as in the base case, all with ICERs of < £1200 per QALY gained. Therefore, as similar tests are recommended as being cost-effective for all prevalence values considered, the base-case conclusions remain insensitive to any assumptions surrounding prevalence rates in either index cases or relatives with all ICERs for non-dominated strategies < £1300 per QALY gained relative to LDL-C.
Familial hypercholesterolaemia treatment
Analyses reducing the cost of atorvastatin did not change the base-case conclusions, with no difference in the sequence of the presented ICERs. Elucigene FH20 remains the most cost-effective option relative to LDL-C; LIPOchip (platform processed in Spain) is the next most cost-effective option followed by CGA, as was reported in the base-case analysis. ICERs for all three non-dominated tests are insensitive to changes in the cost of treatment used in the model (all reported ICERs are < £1100 per QALY gained relative to LDL-C).
Data in relation to the base-case model sourced treatment proportions for FH from the FH audit 201018 and assumed generic simvastatin treatment for those without confirmed FH. This assumption was tested using treatment proportions provided by Dr Anthony Wierzbicki (personal communication, Guy’s and St Thomas’ Hospitals NHS Trust, 2011). This assumed that both genetically confirmed FH and genetically non-confirmed FH patients would receive a range of treatments. This included polypharmacy for some patients including treatment with ezetimibe as well as statins. The order and magnitude of the ICERs relative to LDL-C remain similar to that in the base-case analysis.
The conclusions drawn are therefore not sensitive to changes in treatment pattern or to costs of treatment administered to patients.
The impact of the decision to treat negative-testing relatives or index cases
The base-case analysis assumes that 10% of negative-testing relatives will require treatment. However, it may be that 0% or at least no more than in the general population will require treatment. Therefore, sensitivity analysis investigates a scenario in which none of these relatives would receive statin therapy. In this scenario, the magnitude and order of the ICERs are very similar to those in the base-case analysis, with Elucigene FH20 remaining the most cost-effective strategy, dominating LDL-C. LIPOchip (platform processed in Spain) and CGA are the next most cost-effective options (ICERs of £902 and £1062 per QALY gained, respectively, relative to LDL-C). Hypothetically increasing this proportion to 50% does not lead to any significant change in the order or magnitude of the ICERs presented.
In an unlikely situation that negative index cases do not receive treatment or clinical follow-up, Elucigene FH20 is the only non-dominated genetic testing strategy and is less costly but less effective than LDL-C, the reason being that index cases testing negative for a FH-causing genetic mutation are still at significant risk of cardiovascular events and so not treating based on genetic mutation alone would lead to large numbers of at-risk individuals being missed, hence the reason LDL-C would be the most cost-effective strategy. It is important to note, however, that the above-mentioned analysis is for illustration only and is not necessarily a reflection of the true care pathway.
Costs of diagnostic strategies
Increasing or decreasing the MOLU costs associated with each test by ±£10 (varying cost per MOLU from £20 to £40) does not impact on the overall test order, with only minimal changes in the relevant ICERs. This is because the model is determined primarily around lifelong costs and health outcomes associated with treatment for FH or otherwise.
Sensitivity of key assumptions (model structure)
The assumption that cascade testing takes place of first-, second- and third-degree biological relatives of the index case is tested by assuming that the process stops after the second-degree relative regardless of test result. All genetic tests are even more cost-effective in this scenario. Elucigene FH20 is less costly and generates greater QALYs than LDL-C and is thus dominant. LIPOchip (Spain) and CGA are both associated with ICERs of < £800 per QALY gained.
The base-case analysis assumes that all index patients with a clinical diagnosis will have their family pedigree investigated, with cascade testing using targeted sequencing for relatives of genetically confirmed index cases. However, those that do not receive a genetic test or are test-negative will still be cascade tested using LDL-C. This is because, although a genetic mutation may not be detected, it is possible that such individuals have mutations or genes that have not yet been identified as causing FH. However, as a sensitivity analysis, we have explored the impact on the results of not cascade testing from such genetically test-negative index cases. In this scenario, all non-dominated genetic tests are actually less costly and less effective than LDL-C testing. Although the results are sensitive to this aspect of the model, clinical advice suggests that this would be highly unlikely in practice as cascade testing from negative index cases is a very important part of the cascade process. The results are not sensitive to assumptions regarding the proportion of index and/or relatives agreeing to have their family history investigated or agreeing for cascade testing to take place.
We varied the discount rate between 0% and 6% for costs and benefits as is standard practice in economic modelling to test our model to assumptions regarding uncertainty surrounding the value of future costs and health gains accrued over a lifetime horizon. For a discount of both 0% and 6% the order of the ICERs relative to LDL-C remained the same as in the base-case analysis. The magnitude of these ICERs showed no significant changes either. The results for the base-case analysis present estimates of cost-effectiveness based on current costs of CGA. However, the cost of genetic DNA sequencing will fall in the coming months and years with the development of next-generation (non-Sanger based) sequencing techniques. Therefore, we have explored the impact on the results of reducing the cost of sequencing by an estimated 40% (Dr Gail Norbury, Guy’s Hospital, London, 2011, personal communication). In this scenario, LIPOchip (platform processed in Spain) becomes extendedly dominated. Elucigene FH20 is dominant and CGA is associated with an ICER of £995 per QALY gained relative to LDL-C.
Sensitivity relating to diagnostic test accuracy
For each of the main tests we have investigated the cost-effectiveness based on studies reporting the highest and lowest sensitivity values for Elucigene FH20 and for LIPOchip. This gives a greater picture of the uncertainty across studies and the impact on associated cost-effectiveness results. It also reflects the sensitivity of our analyses to different population groups, some of whom may have greater sensitivity on Elucigene FH20, with others doing better with LIPOchip.
In relation to Elucigene FH20, the upper limit of the sensitivity analysis (0.5238) increases the ICER associated with Elucigene FH20 relative to LDL-C. This suggests higher proportionate increases in costs relative to proportionate increases in QALYs, thereby increasing the ICER between the two tests. Lower estimates (0.28636) work in the opposite direction and lead to Elucigene FH20 being dominant over LDL-C. Such findings are somewhat counterintuitive, with there usually being a positive relationship between higher test sensitivity and improvements in cost-effectiveness. The situation here, however, is more complex because of the clinical benefit (and QALY gain) of LDL-C at minimal cost as well as the addition of cascade testing. Higher sensitivity tests lead to a greater number of positive relatives being given a targeted sequencing test (which is more expensive). Although this test detects more true FH cases and generates greater QALY gain, this is offset somewhat by the advantages of LDL-C (individuals will gain improvements in QALYs regardless of whether or not they have FH, through statin-based therapy for their high cholesterol) that form part of the comparator testing. A similar situation arises with LIPOchip strategies relative to LDL-C. However, in all of these analyses, the rank ordering of Elucigene FH20, LIPOchip and CGA in terms of effectiveness and cost-effectiveness remains the same. As the sensitivity of Elucigene FH20 and LIPOchip increases, their associated ICERs approach that of CGA.
The sensitivity and specificity of LDL-C among relatives are taken from Starr and colleagues49 and varied according to the upper and lower bounds of the 95% CIs. In both scenarios, Elucigene is the most cost-effective option relative to LDL-C. ICERs tend to be slightly lower for genetic tests using the higher bound of the CI for sensitivity and slightly higher for the lower bound. These differences are, however, small in magnitude and the counterintuitive effect of test sensitivity in relation to the ICER can be explained as discussed above. Similar analysis of the specificity of LDL-C among relatives does not alter the sequences of the ICERs or the conclusions drawn from the relevant comparisons. Again, all non-dominated sequences are highly cost-effective relative to LDL-C.
In conclusion, based on the above analyses, the results show some sensitivity to changes in some parameters and structure for the confirmation of index cases alone, but are more robust to variations in key parameters when index cases and relatives are analysed together. In all scenarios presented, Elucigene FH20, LIPOchip (Spain) and CGA are cost-effective uses of NHS resources relative to LDL-C. There is some uncertainty surrounding the direction of movement of the ICER as a result of changes in the sensitivity of the tests that may seem counterintuitive. The results of the one-way sensitivity analyses should therefore be interpreted with caution and, for a more accurate measure of overall model uncertainty, probabilistic sensitivity analysis is likely to offer a better estimate.
Probabilistic sensitivity analysis
Probabilistic sensitivity analysis is carried out for the base case as described in the methods section. Figure 12 illustrates the results in the form of a CEAC.
FIGURE 12.
Cost-effectiveness acceptability curves for the base-case analysis.
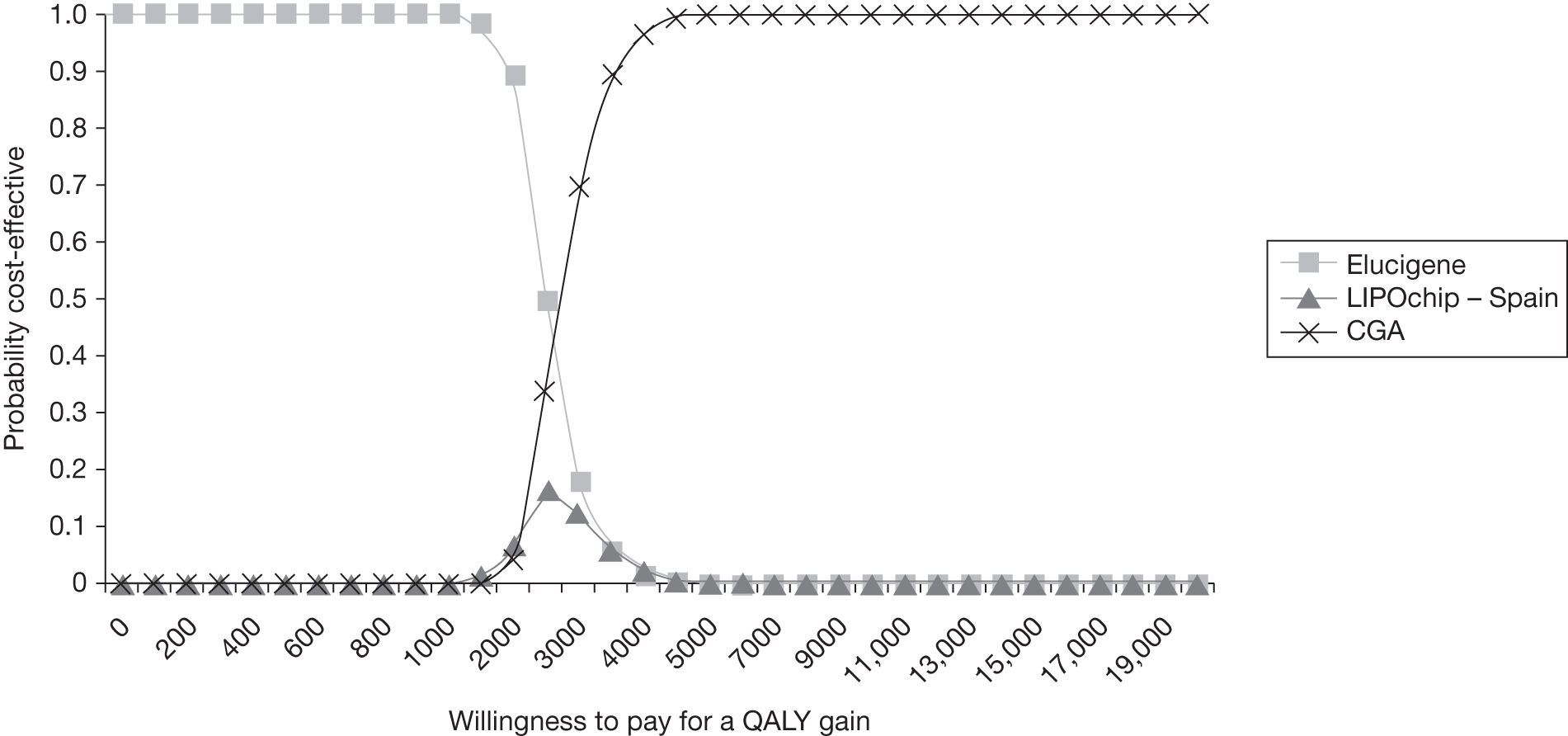
This figure shows that, at low threshold values of willingness to pay for a QALY gain relative to LDL-C (< £2500), Elucigene FH20 has the highest probability of being cost-effective, but this reduces as the willingness to pay for an additional QALY increases. CGA is the most cost-effective option at threshold values > £2500 and is associated with a > 90% probability of being cost-effective at all threshold values > £3500 per QALY gained (Figure 12 is scaled down to aid discussion of low threshold values). The other non-dominated strategy, LIPOchip platform processed in Spain, is never associated with a probability of cost-effectiveness > 20%. Probabilistic analysis also generates similar results and conclusions for each age subgroup in the analysis (see Appendix 15). At threshold values of willingness to pay for a QALY gain approaching £20,000, CGA is always the most cost-effective option. This is an important point and confirms the generalisability of the base-case probabilistic results to other age groups.
In addition to deterministic analysis surrounding the mutation detection rate among clinically diagnosed FH patients, we considered some extra analysis based on input from Dr Anthony Wierzbicki (Guy’s and St Thomas’ Hospitals NHS Trust, 2011, personal communication), who states that, among his patient group, the majority of patients are possible FH and he estimates that the proportion likely to be detected by CGA is approximately 20–25% or may even fall to 5% in some population groups. With this in mind, we have conducted probabilistic sensitivity analysis for a range of potential mutation detection rates for CGA. The relevant CEACs are presented in Appendix 17 and show that the results are somewhat sensitive to this value in the model, especially at low threshold values and for lower rates of prevalence. For the lowest prevalence rate considered (5%), there is quite a bit of uncertainty at threshold values < £6000 per QALY gained. At very low values of willingness to pay (< £3000 per QALY), Elucigene FH20 is the strategy most likely to be cost-effective. LIPOchip platform processed in Spain is less likely to be cost effective except at very specific values of willingness to pay between £3000 and £4000 per QALY gained and at a low prevalence rate of 5%. However, this test is never associated with a probability of cost-effectiveness of > 50% regardless of prevalence rate or willingness-to-pay threshold. For higher estimates of prevalence (i.e. 10–50%), the results mirror those of the base-case analysis. However, of greater importance is that, for all prevalence rates of FH considered, CGA is the most cost-effective strategy at threshold values of > £5000 per QALY gained, increasing to 70% at the conventional value of willingness to pay of £20,000 per QALY gained for a prevalence of 5%. This probability increases to almost 100% for all other prevalence rates considered (i.e. 10%, 20% and 50%). Therefore, although there is some uncertainty surrounding the results based on varying mutation detection rates in clinically diagnosed index cases, probabilistic analysis shows CGA to be the most likely cost-effective use of NHS resources. The conclusion of the cost-effectiveness of CGA confirms the results of the previous NICE guidance in that the most comprehensive test for FH is cost-effective. NICE CG711 estimated that CGA was cost-effective with an associated ICER of £2676 per QALY gained versus LDL-C. Our results generate similar conclusions with a lower estimate of the ICER of £1030 per QALY gained relative to LDL-C. This is likely to be because of the cost reductions in CGA and in treatment over time.
Summary
Base-case results from deterministic analyses show that Elucigene FH20 is the most cost-effective diagnostic test, being less costly and more effective and thus dominant over LDL-C. However, this test strategy is less effective than recommended alternatives such as CGA (the most comprehensive diagnostic test for FH). Other non-dominated test strategies, LIPOchip platform processed in Spain and CGA, are also highly cost-effective. The latter strategy generates the greatest QALY gain but at additional cost. The sequences of the ICERs remain robust to the majority of deterministic sensitivity analyses; however, some plausible variations change the magnitude of these ICERs slightly. It is likely that CGA will become more cost-effective going forward because of the emergence of next-generation sequencing techniques, reducing the time and cost required to conduct large gene sequencing operations. More important, though, is the fact that all three non-dominated test strategies are cost-effective at all conceivable threshold values of willingness to pay for a QALY gain. In all cases it is more cost-effective to cascade test relatives using targeted sequencing instead of either Elucigene FH20 or LIPOchip. This is because of the relative diagnostic cost savings for the same high level of accuracy in a targeted group of relatives.
Probabilistic sensitivity analysis more clearly shows the relative cost-effectiveness of the three test strategies mentioned above. At usual threshold values of willingness to pay for a QALY gain of £20,000, CGA is the most cost-effective test strategy. Although the probabilistic sensitivity analysis shows some uncertainties surrounding alternative mutation detection rates among clinically diagnosed index cases, CGA still remains the most likely option to be cost-effective. The probabilistic results are not sensitive to the age of the index case or associated average age of relatives.
Amongst the test strategies identified as being cost-effective, there are other factors that may need further consideration before arriving at a judgement on which strategy to recommend. For example, there may be practical and resource issues associated with full-scale implementation of CGA if this is recommended as a test strategy for all. If so, then judgement is required on whether it is ethical to implement cascading based on an index test result that is not as accurate as alternative more effective and cost-effective strategies such as CGA. In addition, cost-effectiveness will also depend on how clinicians view the outcome of tests such as Elucigene FH20, which detect only approximately 44% of cases with a FH-causing mutation; for example, there is the potential for missing cases, especially at-risk relatives who may not show high LDL-C levels when tested but who may have a FH-causing mutation. These patients may forgo potentially life-saving treatment if index cases are managed only on the basis of their clinical diagnosis as opposed to their genetic test. This issue does not arise for CGA for which an unequivocal diagnosis is reported in so far as this method detects all known FH-causing mutations.
Chapter 4 Assessment of factors relevant to the NHS and other parties
Factors relevant to the NHS
Funding of the DNA testing
The current NICE clinical guideline (CG71)1 identifies DNA testing as the recommended method for confirming a clinical diagnosis of FH among Simon Broome definite FH and possible FH probands and also (and perhaps most importantly) the identification of first-, second- and possibly third-degree relatives of the index case for testing using a targeted sequencing test. However, findings from the 2010 audit of FH services18 suggest that the current NICE guideline is not being widely implemented, primarily because of shortages in funding at a local level. The 2010 audit found that, although 97% of sites have access to an accredited laboratory for lipid measurement, only15% had access to funded DNA testing.
Our results confirm that CGA is the most sensitive testing strategy for identifying at-risk relatives and, based on the results from probabilistic sensitivity analysis, is likely to be the most cost-effective testing strategy. This is in line with recommendations from CG71. 1 CGA is, however, the most costly diagnostic test (although the cost implications can be partially offset against cost savings emanating from reduced cardiovascular events treated and more appropriate targeted treatments for these people). With concerns about access to funding for DNA testing being raised in the FH audit18 there may be perceived barriers to the widespread adoption of CGA as the strategy of choice.
The adoption of less costly approaches than CGA is possible. Other non-dominated strategies also appear cost-effective at points below conventional willingness to pay for a QALY values. However, strategies such as Elucigene FH20 and LIPOchip are imperfect methods for detecting gene deletions and duplications, such that a MLPA test would be required with Elucigene FH20 and may well be pragmatically required in addition to LIPOchip to confirm the diagnosis for these cases. On the plus side, however, such strategies may be simpler and cheaper than CGA.
It is probable, however, that the cost of implementing testing with CGA will reduce in future years. It is estimated from previous guidance that the cost of cascade testing of all at-risk individuals would be approximately £12.913M per year80 over 5–10 years, after which time costs would fall further as more and more of the current 100,000 or so patients with FH would be detected. After this 5- to 10-year period, cascade testing would be on a case-by-case basis of those who had not previously been tested. With reductions in costs associated with next-generation gene sequencing, these cost estimates have fallen over recent years and are likely to fall further in coming years. Additionally, atorvastatin therapy is coming off patent in 2011, which will also ease the financial burden of implementing the guidance. It may therefore be a more efficient use of NHS resources to adopt a comprehensive testing programme now to avoid the additional costs of delaying and retesting patients currently cascaded using LDL-C with genetic tests in the future. There may therefore be some savings to the NHS that have not as yet been identified. It is difficult to quantify such potential savings as this would depend on future NICE guidance and whether or not primary care trusts implement the current guidelines as per CG711 to use at least some form of DNA testing strategy.
Financial burden to NHS of (as yet) undiagnosed patients
The NHS needs to be aware of the financial burden of the significant number of individuals (estimated to be around 100,000) who have FH and are as yet undiagnosed (but who would subsequently be diagnosed through cascade testing). The management and treatment of these cases, once identified, will generate a significant resource burden to already tight NHS budgets. Also, it is unclear whether or not the capacity currently exists in lipid clinics to identify cases, trace family history and refer all those requiring testing. Clinical expert opinion suggests that capacity is available within the genetics laboratories in the UK to conduct all relevant tests.
Factors relevant to other parties
Benefits to individuals of a definitive diagnosis
Should the widespread implementation of cascade testing be achieved in the UK, there are a number of benefits that individuals identified with FH through that process can expect to incur. For example, appropriate treatment can be started quickly, cholesterol levels can be monitored and managed, the risk of getting CHD and having a heart attack is reduced and close family members can be screened and treatment started if necessary. It is also known that, if treatment can be started early, before CHD is established, this reduces the risk of dying prematurely. 2
Possible adverse sequelae of a definitive diagnosis
Despite the benefits that a definitive diagnosis can bring, it is also well known that psychological sequelae can arise for individuals and their family following the formal diagnosis of a clinical condition. There are issues of anxiety associated with being diagnosed with a genetic disease; however, equally there may be a sense of closure for the patient, who will be able to proceed with an action plan to manage his or her FH using appropriate treatment methods. Although no evidence exists linking psychological impact to QALY gain for FH patients, as FH is very treatable once identified, it is likely that the psychological impact of the genetic testing would be positive for the patient. Individuals, especially parents, may also gain positive views from the knowledge that a relative, especially their children, will be treated correctly should they be diagnosed with FH.
Insurance for those diagnosed with familial hypercholesterolaemia
If you are being treated for a medical condition you usually have to declare it to your insurance company, otherwise it could invalidate your insurance. Having a diagnosis of FH may affect how a person is treated when they apply for life assurance or travel insurance and could also have an impact on mortgage applications. Some insurance companies may decide that a person with FH has a higher risk of getting CHD and may charge higher premiums. Also some insurance companies may not differentiate between high cholesterol as a result of poor diet and other lifestyle factors and high cholesterol caused by an inherited condition such as FH. 2
Other issues
The use of strategies involving Elucigene FH20 and/or LIPOchip could provide advantages to patients in terms of early detection of disease and provision of an unequivocal diagnosis, allowing cascade testing for the early identification and treatment of relatives. Although the benefits of such tests in achieving a definitive diagnosis are clearly evident, there are some ethical and equity issues arising from the recommendation of a less than fully sensitive and specific genetic test. As reported in Chapter 3 (compared with LDL-C), although strategies such as Elucigene FH20 or LIPOchip (platform processed in Spain) appear to offer a cost-effective use of NHS resources at less than usual threshold values of willingness to pay, their recommendation as a single test may raise ethical concerns. Elucigene FH20 and LIPOchip both detect a limited set of FH-causing mutations. Owing to concerns over LIPOchip’s ability to detect copy number changes as accurately as MLPA, some relatives of index cases with less commonly occurring mutations may go undetected. Such individuals would be disadvantaged owing to the documented inadequacies of the use of LDL-C to give a definitive diagnosis of FH.
Chapter 5 Discussion
The first section of this chapter includes discussion of diagnostic accuracy test performance (see Chapter 2); this is followed by discussion of the results of the cost-effectiveness analysis (see Chapter 3).
Discussion of test performance results
Statement of principal findings
Fifteen studies were included in this assessment. Three studies (four reports) evaluated Elucigene FH20, five studies (six reports) evaluated various versions of LIPOchip, four studies reported data on the performance of LDL-C as a part of the Simon Broome criteria or LDL-C cut-offs of > 4 mmol/l and three studies reported age-and gender-specific LDL-C cut-offs for cascade testing of relatives.
Elucigene FH20 and LIPOchip studies
The included studies on Elucigene FH20 and LIPOchip reported a sequential genotyping test in which (1) the participants received a clinical diagnosis of FH followed by the index test (as a pre-screen) and then (2) those who tested negative received further genetic investigations such as gene sequencing and MLPA. CGA was the reference standard considered in the review. Assessment of the methodological quality of the included studies showed that overall the participants were representative of those who would receive the tests in practice.
Based on the data from the included studies we were able to deduce true-positive, true-negative and false-negative rates for each test. False-positive results could not be derived for any of the studies evaluating Elucigene FH20 or LIPOchip as only those who initially tested negative went on to receive the reference standard. Therefore, only sensitivity, and not specificity, of Elucigene FH20 or LIPOchip could be deduced and reported (sensitivity represented the percentage of cases with mutations found by CGA that are also detected by the candidate test).
Because of the sparse data on overall clinical diagnosis and variability in the LIPOchip versions used, sensitivity data could not be pooled. Therefore, we have provided a narrative overview and graphical presentation of the diagnostic performance of Elucigene FH20 and LIPOchip.
Low-density lipoprotein cholesterol studies
In general, the included studies on LDL-C (as part of the Simon Broome criteria for the diagnosis of probands or age-and gender-specific LDL-C cut-offs for the diagnosis of relatives as recommended by NICE guideline CG711) provided data on true- and false-positives and -negatives, allowing the calculation of both sensitivity and specificity. Again, because of the variability in both the clinical diagnosis and the comprehensiveness of the genetic tests used, sensitivities and specificities could not be pooled to provide a single estimate.
Diagnostic accuracy
The sensitivity of Elucigene FH20 and LIPOchip in detecting FH varied. Amongst UK populations with a clinical diagnosis based on the Simon Broome criteria, Elucigene FH20 was reported to detect 44% and 52% of those with FH-causing mutations that were detected by CGA. The UK has a population with a wide mutational spectrum and, as Elucigene FH20 is designed to detect a limited number of mutations, the sensitivity of the kit is largely dependent upon the prevalence of these specific FH-causing mutations in the population, hence resulting in variations. For example, predicted sensitivities of 32% in Wales48 and approximately 33% in Aberdeen, Scotland (prevalence with CGA 28%),79 were reported for Elucigene FH20 (by tallying the mutations that are covered in the Elucigene kit against the mutations that were picked up by CGA in those setting), which is lower than sensitivities reported by included studies. Moreover, it has been suggested that interpretation of the Simon Broome diagnostic criteria is not uniform throughout the lipid clinics in the UK and this may also lead to variation in the detection of FH. 70
Variation was observed across countries in the sensitivity of Elucigene FH20 in detecting FH in those with a clinical diagnosis of definite FH. Elucigene FH20 showed sensitivity of 49% in confirming FH in those with a clinical diagnosis of definite FH in a UK population (the prevalence of FH in the study was 28%), whereas sensitivity of only 29% was observed in an Australian population in which the prevalence of definite FH in the study was very high (78%). These differences could possibly be explained at least in part by the following two factors: (1) mutations included in the Elucigene FH20 kit were selected based on their frequencies in a sample of around 400 patients who were diagnosed as definite FH based on a Simon Broome diagnosis26 and (2) differences in the definitions of definite FH used in the two study populations (in the Dutch criteria a clinical diagnosis of definite FH does not require the presence of xanthomata, unlike the Simon Broome criteria).
The sensitivity of LIPOchip ranged from 33.3% (UK population) to 94.5% (Spanish population) using various versions. Using LIPOchip version 8, which contains 251 of the mutations most prevalent in a European population, the sensitivity observed in a UK population with a clinical diagnosis of FH based on the Simon Broome criteria ranged from 33.3%40 to 56.9%,41 while specificity was reported as 93.8%. 40 It should be noted that LIPOchip version 8 does not detect five mutations that are detected by Elucigene FH20 and which are common in the UK population; therefore, the sensitivity of the UK version of LIPOchip is likely to be higher. In the version of LIPOchip including frequent UK mutations (version 10), sensitivity would be improved to 78.5%41 (Progenika, personal communication) in detecting FH in a UK population. However, this was based on a very small sample size (n = 120) and only those with a confirmed genetic diagnosis were included and therefore the results should be interpreted with caution. None of the included LIPOchip studies reported accuracy data separated by definite FH or possible FH.
The sensitivity of LDL-C as part of the Simon Broome criteria compared with CGA was high (90–93%); however, specificity was low (28–29%) with a large number of false-positives observed. Nevertheless, only four of the LDL-C studies (three full text and one abstract) used the most complete CGA as defined in the assessment. The implications of this high false-positive rate are that potentially unnecessary additional tests or treatments may be given to those who do not have (genetically diagnosed) FH.
A risk of 10–20% of either incorrect diagnosis or misdiagnosis of FH has been reported. 81,82 In the UK, an overlap in LDL-C distributions amongst those with and without FH has been reported. It has been suggested that, because of the overlap in LDL-C levels, no cut-offs are 100% accurate. 22 Mabuchi and colleagues46 reported higher accuracy of LDL-C using a cut-off of 4.1 mmol/l (sensitivity and specificity of > 98%) among genetically diagnosed FH patients and unaffected relatives in Japan. The mean LDL-C levels amongst those with and without FH may differ from country to country, with some studies reporting an overlap in LDL-C distributions while others do not. 49 In the study by Mabuchi and colleagues46 the mean LDL-C level was 6.7 (SD 1.52) mmol/l in those with FH compared with 2.97 (SD 0.65) mmol/l in those without FH, with almost no overlap in LDL-C distributions. This could partly explain why using a LDL-C cut-off of 4.1 mmol/l was found to be highly sensitive in this population.
Cascade testing
In a family with FH, 50% of first-degree relatives are likely to have the condition. One of the advantages of genetic testing is that, if a mutation is identified in probands, targeted gene sequencing can be used in cascade testing of relatives to detect the culprit mutation and provide an unequivocal diagnosis of FH. Using targeted gene sequencing, the observed detection rate of FH in relatives ranged from 53% to 56% in two studies,37,45 which is broadly consistent with rates reported by others (37–56%; see Appendix 18). 11,19,67,83–85 A study by Wiegman and colleagues50 reported that a high detection rate (77%) in children from families in whom a mutation was identified in probands was observed through targeted sequencing. However, the authors of the study suggested that one possible reason for the high detection rate was that siblings with very low LDL-C levels were not taken to the clinic to undergo targeted sequencing. Moreover, children are present with monogenic causes of hypercholesterolaemia and are likely to have a higher detection rate of FH-causing mutations.
High sensitivity and specificity of age- and gender-specific LDL-C cut-offs compared with CGA were reported in cascade testing of relatives, suggesting the clinical utility of this approach in the absence of genetic diagnosis. In the study by Lee and colleagues,48 91% sensitivity and 93% specificity of cascade testing of relatives were reported using age- and gender-specific LDL-C cut-offs, although one explanation for the high values reported is that the included index participants were all homozygous for FH. In a subgroup analysis, Wiegman and colleagues50 reported sensitivity of 96% using LDL-C cut-offs of ≥ 3.5 mmol/l in children of parents with a clinical diagnosis of definite FH. 48,50 The authors of the study suggested that this sensitivity would apply to those children in a family with definite diagnosis of FH only.
Strengths and limitations of the assessment
In terms of strengths of the assessment, screening of articles and quality assessment of full-text papers were performed independently by two reviewers. Conference abstracts were included. To avoid missing potentially relevant studies reporting Elucigene FH20 or LIPOchip, in addition to studies reporting a clinical diagnosis of FH based on the Simon Broome criteria, those reporting a clinical diagnosis of FH based on the Dutch or MedPed criteria were also included. We also contacted study authors to obtain clarification on aspects of their reports or in an attempt to obtain missing data.
In terms of limitations of the assessment, non-English-language studies were excluded. A limitation of the literature was that, because the tests evaluated are still new and evolving, a limited amount of evidence was identified reporting Elucigene FH20 and LIPOchip in a UK population, with sample sizes as low as 22 patients40 and not all published as peer-reviewed full reports. One possible mechanism we could have adopted to indirectly infer additional information on the tests would have been to back-calculate data from those studies that had undertaken CGA (by tallying the mutations that are covered in the Elucigene FH20 kit or LIPOchip kit against the mutations that were picked up by CGA) and thence calculate the predicted sensitivity of the tests. However, because of time constrains we were unable to do so. Also, we would have had to acknowledge the inferred nature of those calculations had they been undertaken.
The available evidence varied in terms of the diagnostic criteria used to provide a clinical diagnosis of probands, the versions of LIPOchip used, the comprehensiveness of the genetic analysis (specifically for studies reporting LDL-C compared with CGA) and the threshold of LDL-C cut-offs used to define a positive test result. Because of this heterogeneity it was not considered appropriate to calculate pooled estimates.
Methodological quality of the included studies
We did not find any studies that directly compared Elucigene FH20 and/or LIPOchip with LDL-C (either as part of the Simon Broome criteria for the diagnosis of probands or age- and gender-specific LDL-C for the diagnosis of relatives) against a reference standard of CGA. A RCT86 (and its secondary report84) was identified, conducted in the UK, in which all participants received a clinical diagnosis based on the Simon Broome criteria and one group received a genetic test while the other received a LDL-C test using Simon Broome cut-offs. However, there were insufficient data to calculate the sensitivity and specificity of these tests in index cases, and also Simon Broome LDL-C cut-offs were used in testing relatives instead of age- and gender-specific LDL-C cut-offs; hence, this study was excluded from the assessment.
All of the included studies were cross-sectional in nature, with only two studies recruiting consecutive patients. 37,44 Abstracts were not quality assessed as they were not considered to contain sufficient information to allow for an adequate assessment of study methodology. In all but one study (LDL-C test46), patients were representative of the spectrum of those who would receive the test in practice. These were patients with a clinical diagnosis of FH based on the Simon Broome, Dutch or MedPed criteria or, for cascade testing, the relatives of those index cases with a confirmed clinical diagnosis of FH. The results from studies in which participants are clinically diagnosed based on the Simon Broome criteria would be of specific interest to UK practice.
An incomplete genetic testing strategy may result in mutations not being detected because of the limitations of the testing strategy. Only one study reporting Elucigene FH20,37 one study reporting LIPOchip39 and 50% (three out of six) of the LDL-C studies44,45,49 used genetic analysis that comprised DNA sequence analysis of the LDLR and APOB genes in conjunction with MLPA. Given that the FH-causing PCSK9 gene is rare and was discovered only fairly recently, those studies that otherwise met the definition of CGA but without assessing the PCSK9 gene were judged to include an acceptable reference standard in terms of classifying the target condition in this assessment. However, only two of the above studies that employed CGA did not perform PCSK9 analysis. 45,49 With respect to the reference standard used, all six abstracts (two reporting Elucigene FH20, three reporting LIPOchip and one reporting LDL-C) used adequately defined CGA, which comprised DNA sequence analysis of the LDLR and APOB genes (plus PCSK9 gene) in conjunction with MLPA that was likely to classify the target condition.
In the Elucigene FH20 and LIPOchip studies, only those who tested negative went on to receive further genetic investigations; thus, none of the test-positives received the reference standard (differential verification bias), giving rise to the possibility of overestimation of test performance. Because of the sequential nature of the tests used, these studies were also at risk of partial verification bias as neither the whole sample nor a random sample received verification with a reference standard. All of the LDL-C studies were free of partial verification bias and one was free of differential verification bias. 44 Test review bias (the results of the index test are interpreted with knowledge of the results of the reference standard test) was avoided in the Elucigene FH20 study, one of the LIPOchip studies and two of the LDL-C studies. It has been suggested that both test review bias and diagnostic review bias (in which the results of the reference standard test are interpreted with knowledge of the index test) may lead to higher values being reported for sensitivity. 87 However, these biases are of more importance in tests in which the results are based on subjective interpretation rather than automatically generated.
Uncertainties
The spectrum of patients considered in this assessment was those with a clinical diagnosis of either definite or possible FH (including relatives of confirmed FH cases). Therefore, any evidence from this review is not generalisable to the wider, asymptomatic general population. The inclusion of population-based screening studies was beyond the scope of this review.
Assessment of a new technique – iPLEX – was also beyond the scope of this review as this was not CE marked at the time that the review was conducted. iPLEX is a rapid genetic testing kit developed to cover 56 mutations (54 in the LDLR gene, one in the APOB gene and one in the PCSK9 gene) most commonly found in the UK population. It has been reported that this kit has an average detection rate of 75% (n = 150 patients) with a false-positive rate in a ‘no mutational control group’ of 0.015%,88 and that the kit can produce a test result within 1 hour.
Analysis of the sensitivity of Elucigene FH20 and LIPOchip in relation to homozygous or compound heterozygous FH was similarly beyond the scope of this assessment. People with compound heterozygous FH carry more than one mutation and pre-screening with Elucigene FH20 or LIPOchip may miss the second mutation if it is not covered by the genetic testing kit and genetic testing stops after the first mutation is identified. In such circumstances relatives of the diagnosed proband may carry a different mutation to the one identified by the pre-screen and could be misdiagnosed as non-FH if only the mutation identified on the pre-screen is checked in cascade testing of relatives. A case study by Taylor and colleagues89 reported that compound heterozygous FH gave rise to a severe phenotype and suggested that the presence of additional mutations in families should be considered when relatives have varying phenotypes. Although such FH cases are very rare in the UK, with a prevalence of around 1 in 1 million, recognition of the issue is important.
A wide range of approximately 3027–95%90 of patients with a clinical diagnosis has been reported to have a mutation confirmed by genetic diagnosis. In some people with FH the results of CGA might still be negative because full sequencing of the APOB and PCSK9 genes is not routinely undertaken, and there may be other genes as yet unrecognised that give rise to the FH phenotype. Moreover, a number of other high-penetrance genes may harbour quite rare mutations (as is emerging in schizophrenia) or alternatively familial clustering of low-penetrance alleles may cause a FH phenotype, as reported in familial breast cancer. The recent report found that approximately 95% of children meeting the Dutch criteria for FH had a genetic mutation,90 whereas only approximately 3027–50%37 of patients meeting the Simon Broome criteria had a mutation confirmed by genetic diagnosis.
None of the included studies reporting Elucigene FH20 or the LIPOchip UK version provided information on clinical effectiveness outcomes. Other studies have shown clinical improvements in patients in whom the diagnosis of FH has been confirmed after cascade screening. 11,19 In a large genetic screening study with 1 year of follow-up, a very high proportion of patients (93%) identified with FH started on lipid-lowering medication, showing the effectiveness of the genetic testing. 19 Significant reductions in TC and LDL-C were observed in those with identified FH 6 months after genetic screening. 11
Other relevant factors
Psychological impact and acceptability of genetic testing
Evidence has suggested that genetic diagnosis of FH has no clinically relevant adverse psychological effects. 11,91 A RCT conducted in the UK that included probands with FH and their relatives found that there was no significant effect of genetic diagnosis on perceptions of control over FH, fatalism of FH, control over cholesterol or control over heart disease and adherence to risk-reducing behaviour; however, those with a confirmed genetic diagnosis had a strong belief in the efficacy of cholesterol-lowering drugs and a less strong belief in the efficacy of diet. 86 Another prospective comparative study conducted in the Netherlands among participants in a family-based genetic screening programme, however, found that those with an identified mutation perceived that they were at greater risk of heart disease than those with no mutation. The result was influenced by age, education, cholesterol level and cardiovascular disease in the family. 92
Cascade screening studies have reported high participation rates of 7311–90%. 19 In a UK cascade screening study in Oxfordshire, 97% of parents asked for their children to be screened. 93 In terms of approaches used, directly contacting relatives from clinics has reported higher participation rates19 than contacting relatives through probands. 93 Using both approaches resulted in a higher participation rate (73%) than contact through probands but was not higher than directly contacting relatives. However, there may also be concerns about the possible consequences of receiving a positive genetic test result. In a large genetic screening study in the Netherlands, 10% of individuals declined genetic testing because of the fear of negative effects on employment or insurance. 19
Although genetic diagnosis may be generally acceptable to patients, for clinicians making a diagnosis of FH still remains a challenge and dependent upon their judgement in circumstances in which patients have raised cholesterol levels but no identified FH-causing mutation. 94
Risk associated with different types of mutation
Based on the lipoprotein levels, mutations have been categorised as either ‘severe’ (functional null alleles and missense in exon 3/4; or functional null alleles plus splice variants) or ‘mild’ (missense outside exons 3/4 and splice; or any missense mutation). 84 A null mutation has been identified as one of the important risk factors associated with PCVD in FH patients. 43,95 A significantly higher risk of PCVD, recurrence of cardiovascular events and family history of PCVD was reported in patients carrying null mutations compared with patients with defective mutations. 95 The relative risk of PCVD in patients with a null mutation was 3.1 times higher than that in patients with a missense mutation. 43 The mean PCVD-free survival time in those with null mutations was 51–53 years, in those with missense mutations was 58 years and in those carrying defective mutations was 53 years (p < 0.01). 43,95
Taylor and colleagues37 reported a similar prevalence of severe mutations across study participants with a clinical diagnosis of definite FH, possible FH and also unclassified FH.
Discussion of cost-effectiveness results
Statement of principal findings
Index cases only
The base-case analysis refers to an index case aged 50 years and a mutation detection rate for FH equal to 36.5% of cases with a Simon Broome possible or definite FH diagnosis. With regards to the identification of index cases alone, Elucigene FH20 was the least costly test but also generated the least QALY gain because of the high number of false-negatives associated with this test. Accounting for the inclusion of both diagnostic and treatment costs (including clinical management), LDL-C was the most costly option for index cases alone but also generated the greatest number of QALYs gained. The reason for this is that all patients who meet the Simon Broome clinical diagnosis of FH will have elevated cholesterol levels as part of that diagnosis. The diagnosis is not definitive but patients with false-positive test results on LDL-C for FH will still gain from statin therapy on the basis of them having high cholesterol; the difficulty, however, with this strategy arises when cascade testing incorrectly takes place from false-positive index cases.
Index cases and relatives
Of greater relevance, however, to the decision problem is the identification of at-risk relatives of index cases in whom cascade testing should be carried out. Each 50-year-old index case will have on average five first-degree relatives, four second-degree relatives and four third-degree relatives still alive and eligible for contact for cascade testing. These numbers refer to the base-case scenario and will vary depending on the average age of the index case. For example, older people may have more second-degree relatives eligible for testing as they are likely to have living grandchildren. Similarly, younger people will have more grandparents alive than an index case aged 50 years. This variation is incorporated in the model results.
In the analysis of index cases and relatives, LDL-C as a stand-alone test is the least costly testing option. CGA dominates pre-screen tests also including CGA as part of the strategy of testing. This is because of the assumption that QALY gains for FH are not time sensitive and the extra time taken to deliver a tiered-strategy diagnostic test will have no implications for treatment or QALY impact. This suggests that, although pre-screen tests such as Elucigene FH20 and LIPOchip (Spain) are less costly in their own right, they do not offer overall cost savings as a pretest to CGA, suggesting that the extra costs associated with running negative samples through all tests in a sequence outweigh the cost savings of those detected as positive on either Elucigene FH20 or LIPOchip. Only at very specific prevalence and sensitivity combinations would Elucigene FH20 be a cost-effective pre-screen to CGA. As the cost of gene sequencing falls in the future, it is less likely that targeted tests (at current prices) will offer a cost-effective pre-screen strategy for the majority of the population in whom testing would be carried out.
Of greater interest, however, is the comparison of the non-dominated tests with the relevant comparators for this assessment. CG711 recommended that DNA testing in combination with LDL-C testing was the most cost-effective strategy to test index cases and identify relatives for cascade testing. However, in practice, uptake of DNA testing for FH has been very slow, especially in England, where the 2010 FH audit18 suggests that only 12% of trusts have access to a formal system of cascade testing, with a further 14% stating that such a system is in development. One issue for this may be a lack of funding in the area, and clinical advice suggests that, in reality, LDL-C testing in combination with the Simon Broome criteria is a more realistic reference standard for this assessment. With this in mind, the main comparison of non-dominated sequences is with LDL-C. However, we also report sequential results ordered by cost and a comparison of cost and QALY differences against CGA for completeness.
When compared with LDL-C, CGA is a highly cost-effective diagnostic test and is the only option that gives an unequivocal diagnosis of FH. This strategy is estimated to cost £1030 per QALY gained relative to LDL-C and thus at usual thresholds would appear to be a highly cost-effective use of NHS resources, and this is further confirmed through probabilistic sensitivity analysis. This finding is in agreement with similar findings from NICE clinical guideline CG71,1 which also found that DNA testing was highly cost-effective in the identification of relatives of index cases with FH. As reported in Chapter 2, there is always the potential that there exist undiscovered genetic mutations and culprit genes that may lead to FH. For this reason, cascade testing would be carried out from mutation-negative index cases using LDL-C testing as they are still technically FH positive based on their clinical diagnosis.
The LIPOchip manufacturer, Progenika, offers a service for testing samples in Spain. This platform offers LIPOchip as a pre-screen and a follow-up screen of all negative samples using sequencing of the LDLR gene. As LIPOchip tests for duplications of and deletions in the gene, this may be described by the manufacturer as CGA; however, there is much clinical uncertainty in relation to the accuracy of the LIPOchip method of detecting deletions and large rearrangements of the gene. Additional evidence suggests that LIPOchip will correctly detect only two out of seven exon copy number changes compared with MLPA. 69 The authors acknowledge that these data are based on small sample numbers; however, they raise further questions with regard to the accuracy of LIPOchip compared with MLPA. Therefore, MLPA would also be required to obtain a completely unequivocal diagnosis. The LIPOchip platform, processed in Spain, does not offer MLPA as part of the process and so it is assumed that the diagnosis obtained from this strategy would be inferior to that of CGA as described in this analysis. As this platform is slightly less sensitive, it is likely that there will be more uncertainty in the test result in about 5% of patients in whom the MLPA test would be required for additional confirmation. The LIPOchip platform processed in Spain is also found to be a cost-effective strategy, with an ICER of £871. Information provided by the manufacturer of LIPOchip suggests that 80.5% of index cases would be detected using the LIPOchip test and the remaining 19.5% would need gene sequencing to confirm the diagnosis (Progenika, 2011, personal communication to NICE). These figures refer to a definite FH sample being tested. However, it is estimated that of all the samples presenting for genetic testing, only 30–50% will have an identifiable mutation. Therefore, the estimates of cost provided by the manufacturer may have underestimated the number of samples testing negative on the LIPOchip test that would also require gene sequencing as the second stage of analysis. Equally, it may be that these costs would be reasonable because of manufacturer economies of scale. Should this strategy be recommended, it is imperative that the issue of price be confirmed before any decision is made.
The results do, however, suggest that if the recommendation to undertake CGA for all was considered impractical or too expensive (e.g. sufficiently large increases in CGA testing might lead to a requirement for extra laboratory space and associated infrastructure, not captured by the existing unit cost assumptions) and an alternative test (Elucigene FH20 or LIPOchip platform) is deemed appropriate, then either or both could be recommended. The test chosen for a particular population would pragmatically reflect the sensitivity of each of these tests within a particular clinic catchment area as it is found that tests may perform differently on groups of different ethnic origin. It may also reflect local resource conditions and clinical judgement, as there is a trade-off between costs and effects. The difficulty, however, in adopting such a strategy is that not all tests detect all individuals. Therefore, should any of these less-than-perfect testing strategies be accepted, they may miss out a number of potentially important cases. This may raise ethical and equity concerns as only a proportion of people will have the culprit gene identified and have their relatives followed up for genetic cascade testing.
The economic model results are more sensitive to changes in parameters for index cases alone than for index and relative cases together. As the latter is the main focus of the analysis of this project, these results are given the most weight in the discussion and reporting of results.
Relative to the comparator of LDL-C, probabilistic sensitivity analysis reveals that CGA is the most cost-effective diagnostic test strategy for the identification and testing of relatives with FH, with a probability of cost-effectiveness of > 90% for all age groups at all conventional values of willingness to pay for a QALY gain. Some variation is identified in the probabilistic analysis based on mutation prevalence rates, which may vary greatly in practice between different geographical areas. Although there is some variability in the results at low threshold values and at very low mutation prevalence rates, CGA does, however, remain the most likely strategy to be cost-effective relative to LDL-C (never dropping to < 50% probability at threshold values of societal willingness to pay of > £5000 per QALY gained).
Strengths and limitations
The economic analysis has a number of strengths and limitations for the confirmation of FH among index cases and the identification of first-, second- and possibly third-degree biological relatives for cascade testing. As the modelling processes used to generate this economic model are adapted from a previously developed Markov model for these patient groups, many of the relevant strengths and limitations of this economic evaluation have already been reported in appendix E to the NICE clinical guideline CG71. 1 In addition to those already reported and published, the following discussion relates explicitly to additional issues relative to the model that may not have been reported elsewhere.
Strengths
This work is important and builds on the health economics evidence generated as part of the NICE CG71 development process. 1 This economic modelling exercise identifies a range of potential diagnostic strategies that were not available for the CG711 assessment (namely LIPOchip and Elucigene FH20) and investigates their cost-effectiveness either as stand-alone tests or as pre-screens to reduce costs associated with CGA. However, one issue of importance is that the cost of CGA, and gene sequencing in particular, has fallen since the CG711 assessment and is likely to fall still further with the evolution of next-generation sequencing. This favours the CGA approach and means that, in the future, going forward, the costs of gene sequencing may well fall further with the evolution of new methods and new technologies. However, even at current prices (approximately £480 per test), CGA is still a cost-effective method of cascade testing. It is the most cost-effective strategy in terms of attaining an unequivocal genetic diagnosis of FH among index cases and for cascade testing those relatives at greatest risk of having the disease. The analysis considered all currently available and approved diagnostic tests and linked test accuracy with final treatment outcomes measured in terms of QALYs. As additional tests become available, it will be possible to incorporate these into the model and re-run the analysis incorporating newly available evidence and tests.
The model structure is a key strength of the analysis in that it presents a linked evidence approach linking intermediate outcomes (i.e. diagnostic accuracy of each testing strategy) to the associated lifelong costs and health outcomes associated with each test result and whether tests were true-positive, false-positive, true-negative or false-negative. This was explored for a total of 12 alternative testing strategies through decision tree and Markov model analysis.
A structured literature search was carried out to identify existing cost-effectiveness evaluations of these tests and of the cascade testing of relatives. No studies directly compared the interventions under consideration, and only one study detailed the cost-effectiveness of any of the new interventions relative with no testing. This referred to the LIPOchip test but was used in relatives. As targeted sequencing is a much lower cost method of testing identified relatives, this LIPOchip study was not relevant to this analysis. Our results, however, do confirm the findings from a number of other studies evaluating the cost-effectiveness of the cascade testing process more generally and also the findings of the previous NICE guideline. 1
Methods used to identify and obtain parameter estimates for the economic modelling sought to identify and utilise published sources and best available information; however, this was not possible in all cases. In such scenarios, for example the proportion of patients receiving various statin therapies and how treatment changes based on diagnosis, we have relied on clinical expert opinion from two or more clinical experts. Estimates of parameters are also sourced from the CG711 analysis where available and tested in sensitivity analyses.
Limitations
The model structure focused on the identification and treatment of index cases and relatives of index cases with FH as clinically diagnosed using the Simon Broome criteria. The costs and benefits of identifying other causes of similar symptoms have not been modelled in detail with the exception of the prescription of statin therapy for all index cases with high cholesterol levels. The impact of additional therapies such as diet, exercise, smoking cessation, etc. has not been included and is beyond the scope of this assessment. The effect of not including such detail in the model is uncertain; however, it is generally widely acknowledged that all patients with a clinical diagnosis of FH will require some form of active treatment, generally statin therapy.
Another challenge related to the analysis of subgroups of the population, especially in relation to FH in children. Relative risks associated with children aged 15 years are assumed to be similar to those of adults aged 30 years. There is little evidence linking the efficacy of statins directly to cardiovascular events avoided, and for this reason we have assumed risks similar to those of a 30-year-old in the model. This probably creates some uncertainty in the estimation of quality of life in this subgroup of the population. Although data do exist relating to clinical outcomes in children (i.e. TC level), these are not linked directly to cases avoided. As Avis and colleagues96 show, statins are efficacious in reducing cholesterol in children with FH and are not associated with significantly different adverse events to placebo; it is therefore assumed that health effects are similar to those of the next youngest age group in the model (aged 20–39 years). It is unlikely that this assumption greatly impacts on the cost-effectiveness results and associated conclusions drawn.
In terms of the estimation of costs for each testing strategy in the economic model, we have used the MOLU classification system to assign tests to bands and apply MOLUs to each band. This is an agreed costing mechanism devised by the UKGTN and CMGS with some built-in flexibility in pricing for each laboratory’s individual circumstances. This is not necessarily an accurate reflection of true economic costs or indeed opportunity costs associated with testing for FH. However, in the absence of price data for all combinations of tests considered, robust costing methods for genetic testing for FH or a NHS tariff for the tests as well as uncertainty surrounding variability from laboratory to laboratory, it was impossible to cost all testing strategies fairly using any other universally acceptable approach. Using the cost of the test alone would be insufficient as it would not account for staff time and consumables required. Therefore, although acknowledging its limitations, we have on the basis of expert opinion relied on the MOLU system for the estimation of diagnostic testing costs in the economic model. Studies retrieved from the cost-effectiveness searches showed great variability in the costs of testing for FH. The reason for this is that testing for FH and genetic testing more generally is a rapidly evolving discipline. Many studies presented alternative definitions of DNA testing and the costs of completing the tests have fallen almost yearly in recent years. Therefore, older estimates would be an overestimate of the true costs. This is another reason why the MOLU system was used for this assessment.
A number of other assumptions were made in relation to the clinical management of patients with and without disease. Much uncertainty exists among clinicians in the treatment of FH, with many advocating a start low approach followed by an increase in treatment intensity if a satisfactory response is not achieved. Others believe that, as statin therapy generates very few adverse events, it would be appropriate to treat everyone with a high-intensity statin (e.g. atorvastatin). The impact of this uncertainty is explored in sensitivity analyses and is found not to alter our base-case results and conclusions, with only very small differences in ICERs.
Further, in relation to the utility of diagnostic information, there is no published evidence that links the outcome from the results of a genetic test for FH (e.g. increased anxiety) to quality-of-life outcomes and hence QALYs gained. A number of plausible scenarios are possible, including reduction in QALYs emanating from the shock of knowing that one has a genetic disorder. Equally, however, people diagnosed with FH could gain some reassurance from the fact that they know what is causing their illness and can aim to develop a plan of action in dealing with this. Additionally, parents may place a positive value on knowing the source of a child’s illness. It is, however, likely that these factors would cancel each other out or favour genetic testing and would be much smaller in comparison with the QALY gains associated with being treated for a life-threatening condition such as FH.
An additional limitation of our analysis is that it is on an average patient level. We are aware that some mutations cause greater harm and are associated with greater risk of cardiovascular events. For example, mutations of the PCSK9 gene are associated with greater clinical risk. Also, there are differences between missense and nonsense genetic mutations. Although these are likely to have implications for the prognosis of individual patients, this has not been modelled as there is insufficient evidence available to estimate how this would impact on quality of life. Further, as cost and QALY differentials are based primarily around treatment decisions and the general insensitivity of the model to small changes, it is unlikely that analysing the data on a mutation level for the purposes of cost-effectiveness would generate great differences in results or conclusions. Further, such data would be available for only few if any mutations and the inclusion of these only would generate further ‘noise’ into the analysis.
There are limited data available for the sensitivity of the LIPOchip test, with wide variation in all of the reported studies for various versions of the test. This generates a lot of uncertainty surrounding the true sensitivity of LIPOchip as used to populate the economic model. As many of the tests were analysed at the manufacturer’s own laboratory, there is a lack of academic peer-reviewed information on this input for the economic model. To deal with the associated uncertainty we have conducted wide variation in the probabilistic sensitivity analysis, which should counter any biases that may have arisen as a result of a lack of critique of the estimate of sensitivity used for LIPOchip version 10 in the model.
A further limitation of the analysis refers to the accuracy of test sensitivity and specificity differentials between those relatives of genetically negative index cases and those relatives of genetically confirmed index cases. For the purposes of the economic modelling, we have assumed, because of a lack of relevant usable data, that sensitivity and specificity of LDL-C testing in both groups are similar. Although this may overestimate the sensitivity and specificity of LDL-C as a test for relatives of genetically negative index cases, it is justifiable on the basis that all index cases are clinically diagnosed with FH and so may well have genetic mutations that as yet may not have been discovered or have not been detected using any of the tests for this assessment. In terms of the cost-effectiveness results, this could represent a bias of uncertain magnitude in favour of CGA, although in the context of insensitivity to variations in this parameter in probabilistic analysis it is unlikely to alter overall results and conclusions.
Uncertainties
Although the cost-effectiveness analysis was performed using the best available data, there was nonetheless some uncertainty surrounding some of the parameters used in the model. Current NICE guidelines recommend that cascade testing is undertaken for all relatives of index cases (using targeted sequencing for those genetically identified and LDL-C for those with no genetic test or those with a negative genetic test). As discussed, because of our assumption that similar proportions of relatives test positive on LDL-C regardless of the index case’s genetic result, this represents some uncertainty. Other issues of uncertainty reflect the parameters and assumptions varied in the deterministic and probabilistic analysis. Results, however, indicate that, in general, the model is insensitive to a range of plausible changes in structure and parameters.
Chapter 6 Conclusions
Implications for service provision
Based on the available, albeit limited, evidence, Elucigene FH20 and LIPOchip version 10 (designed to detect 189 UK-specific mutations) will detect approximately 44–52% and 78.5%, respectively, of FH-causing mutations that are also detected by CGA amongst people with a clinical diagnosis of FH based on Simon Broome criteria. As targeted tests are designed to detect a limited number of genetic mutations, Elucigene FH20 and LIPOchip cannot detect all cases of FH; therefore, further genetic screening using MLPA and sequencing is still required to give an unequivocal diagnosis of FH. This implies that using these targeted tests alone for diagnosis of probands would miss up to approximately 50% (using Elucigene FH20) and 20% (using LIPOchip version 10) of patients with FH-causing mutations who are at risk of developing CHD. As such, these individuals may not receive appropriate treatment and other members of their extended families will also be missed (as they will not be identified for cascade testing).
Using the LDL-C test (high sensitivity and low specificity) as part of the Simon Broome criteria means that a large number of people will receive a clinical diagnosis of FH who will not have a detectable FH-causing mutation. Hence, using LDL-C alone for the diagnosis of FH may lead to inappropriate treatment. In a small UK cohort, age- and gender-specific LDL-C was shown to perform well in the relatives of homozygous FH probands, suggesting the utility of this test for cascade testing (in the absence of genetic tests) among those with a strong phenotype.
As the Elucigene FH20 and LIPOchip kits are designed to detect targeted gene mutations, the sensitivities of both of the kits are largely dependent upon the prevalence of these specific FH-causing mutations in the population. Sensitivities observed in this assessment may not, therefore, be generalisable to other populations or ethnic groups.
At conventional values of willingness to pay for a QALY, CGA is the most cost-effective method of confirmation of clinical diagnosis of FH among Simon Broome possible or definite FH index cases and for the associated cascade testing of first-, second- and third-degree biological relatives. The associated ICER (relative to current practice – LDL-C) is £1030 per QALY gained. The LIPOchip platform and Elucigene FH20 have an even lower reported point estimate of the ICER but are associated with fewer QALY gains. However, there may be practical and resource issues associated with full-scale implementation of CGA if it is recommended as a test strategy for all. If so, then a judgement is required on whether or not it is ethical to implement cascade testing based on an index test result that is not as accurate as alternative more accurate cost-effective options. In addition, a decision-maker needs to be aware that clinicians may or may not base treatment decisions on the outcome of tests such as Elucigene FH20, which detect only around 44% of cases with a FH-causing mutation; for example, there is the potential for missing cases (especially at-risk relatives who may not show high LDL-C levels when tested but may have a FH-causing mutation). These patients may forgo potentially life-saving treatment if index cases are identified only on the basis of their clinical diagnosis as opposed to their genetic test. This issue does not arise in CGA, for which an unequivocal diagnosis is reported. Should a decision-maker deem CGA too expensive given current NHS budgets, Elucigene FH20 and/or LIPOchip platform (processed in Spain) could be recommended as cost-effective strategies. Probabilistic sensitivity analysis shows CGA to be associated with a probability of cost-effectiveness that is > 90% at threshold values of willingness to pay of > £5000 per QALY gained. Although some variation exists depending on mutation prevalence rates among varying populations, CGA remains the most likely cost-effective testing strategy when the ultimate goal is the identification and treatment of relatives with FH.
It is likely that there would be significant resource use implications associated with implementing the findings of this assessment. As there are approximately 100,000 people with FH as yet undiagnosed, this will provide a substantial resource burden to already tight NHS budgets; however, costs associated with genetic testing are reducing and will continue to do so with the emergence of next-generation sequencing techniques. Similarly, costs of treatment are also likely to reduce going forward as atorvastatin is due to come off patent in 2011 with an expected retail cost similar to that of generic simvastatin.
Currently, CGA is used as the method of cascade testing of choice in only a small number of centres in the UK. The use of CGA is observed to be funded less often by primary care trusts in England than in other parts of the UK where adoption of the technology as part of current practice is much higher. In England, currently 97% of audited sites have access to dedicated lipid measurement services; however, only 12% have access to a dedicated genetic testing service for FH. As the initial cost of CGA is quite high, less costly tests may appear more attractive; however, a judgement call would be required as to what QALY loss would be acceptable to a decision-maker in order to generate cost savings.
Suggested research priorities
There are a number of potential areas in which further research and data would be useful.
-
The test performance results of the UK version of LIPOchip were hypothetical and were derived based on a small sample size from one centre where subjects had all been tested genetically. There was no evidence on the performance of LIPOchip version 10 across different regions of the UK or in different ethnic groups. Limited evidence was identified on the sensitivity of Elucigene FH20 across different regions of the UK and in different ethnic groups. As the UK has a population with a wide mutational spectrum, the sensitivity observed with these tests in different populations may vary. Therefore, a prospective multicentre study comparing the performance of Elucigene FH20 and LIPOchip with the LDL-C test in patients with a clinical diagnosis of FH based on the Simon Broome criteria, in which both test-positives and test-negatives are verified against a reference standard of CGA, would be informative. Such a study should also include subgroup analysis of the performance of the tests in different ethnic groups, if possible have a period of follow-up to allow provision of relevant longer-term clinical effectiveness outcomes and incorporate an economic evaluation. The economic evaluation should aim to include a measure of utility of diagnostic information, (especially in relation to the impact of false-negative or false-positive test results on quality-of-life estimates). Such information could be used to assess the impact on QALYs of future modelling exercises.
-
There is little evidence linking efficacy of statins in children to the onset of CHD. Although systematic reviews show that statins are efficacious in lowering cholesterol, we have assumed that this leads to similar reductions in cardiovascular events as in the young adult population group. There is a need to assess the relative risks of onset of disease in this group of patients.
-
There are many mutations that may have a varying impact in terms of risk of CHD. Evidence on the effect of these mutations is lacking and is an ongoing area of research. There is a need for a systematic review of all of the FH-causing mutations currently detectable in the UK population as a whole and in specific ethnic groups and their associated impact on risk of CHD.
-
There is a requirement for continuing research into finding new, as yet unknown, FH-causing genetic mutations. As only approximately 30–50% of patients with a clinical diagnosis have a mutation confirmed by genetic diagnosis, it is possible that there are many genetic causes of FH as yet undiscovered. This is an area that is progressing and further research is required to inform and update the positive detection rate of CGA based on ongoing clinical research.
-
It was outwith the scope of the review to consider tests such as iPLEX, which may also be used for detecting FH but are not as yet CE marked for this purpose. Therefore, further research into the diagnostic accuracy and cost-effectiveness of this test would be informative.
Acknowledgements
We thank the study authors we contacted who provided additional details of their studies, the Royal College of Physicians for supplying access to the economic model used to inform previous NICE guidance, the NICE assessment subgroup specialist members for their responses to our queries and Lara Kemp for secretarial support. We would also like to thank Dr Kevin Kelly for providing guidance and advice on the development of the MOLU costing methodology used in this project. Cynthia Fraser supervised the work involved in developing and running the search strategies. Graeme MacLennan provided oversight to the statistical analysis. The Health Services Research Unit and Health Economics Research Unit, Institute of Applied Health Sciences, University of Aberdeen, are both core funded by the Chief Scientist Office of the Scottish Government Health Directorates.
Contribution of authors
Pawana Sharma (Research Fellow) and Graham Mowatt (Senior Research Fellow) screened the search results, assessed full-text studies for inclusion and undertook data extraction and quality assessment. Pawana Sharma drafted the chapter reporting the results of the diagnostic accuracy studies. Dwayne Boyers (Research Fellow) undertook the economic modelling and drafted the chapter on cost-effectiveness, along with Mary Kilonzo (Research Fellow) and Paul McNamee (Reader in Health Economics). Zosia Miedzybrodzka (Reader in Medical Genetics) and William Simpson (Consultant Chemical Pathologist) drafted the background chapter and provided expert advice on clinical aspects of the review. Charles Boachie (Statistician) conducted the statistical analysis. Fiona Stewart (Information Specialist) developed and ran the search strategies, obtained papers and formatted the references. All authors assisted in preparing the manuscript and commenting on drafts.
Disclaimers
The views expressed in this publication are those of the authors and not necessarily those of the HTA programme or the Department of Health
References
- National Institute for Health and Clinical Excellence . Identification and Management of Familial Hypercholesterolaemia 2008. www.nice.org.uk/nicemedia/live/12048/41700/41700.pdf (accessed April 2011).
- Main L. Inherited Heart Conditions: Familial Hypercholesterolaemia 2011. www.heartuk.org.uk/index.php?/healthy_living/inherited_heart_conditions_-_familial_hypercholesterolaemia/ (accessed April 2011).
- Bhatnagar D. Familial hypercholesterolaemia: the case for finding the patients and finding their genes. Preston: Cardiac and Stroke Networks Prevention and Detection Conference; 2009.
- Bhatnagar D. Diagnosis and screening for familial hypercholesterolaemia: finding the patients, finding the genes. Ann Clin Biochem 2006;43:441-56.
- Marks D, Thorogood M, Neil HAW, Humphries SE. A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemia. Atherosclerosis 2003;168:1-14.
- Western Australian Program Committee . Model of Care: Familial Hypercholesterolaemia 2008. www.healthnetworks.health.wa.gov.au/modelsofcare/docs/Familial%20Hypercholesterolaemia%20Model%20of%20Care.pdf (accessed April 2011).
- Connor M, Ferguson-Smith M. Essential medical genetics. Oxford: Blackwell; 1997.
- Versmissen J, Oosterveer DM, Yazdanpanah M, Defesche JC, Basart DCG, Liem AH, et al. Efficacy of statins in familial hypercholesterolaemia: a long term cohort study. BMJ 2008;337.
- Mortality in treated heterozygous familial hypercholesterolaemia: implications for clinical management. Scientific Steering Committee on behalf of the Simon Broome Register Group. Atherosclerosis 1999;142:105-12.
- Department of Health (DoH) . Familial Hypercholesterolemia Cascade Testing Project. Recommendations to the DoH from the Steering Group of the DH FH Cascade Testing Audit Project 2007. www.heartuk.org.uk/HealthProfessionals/index.php/fh_cascade_testing_project/ (accessed April 2011).
- Leren TP, Finborud TH, Manshaus TE, Ose L, Berge KE. Diagnosis of familial hypercholesterolemia in general practice using clinical diagnostic criteria or genetic testing as part of cascade genetic screening. Community Genet 2008;11:26-35.
- Austin MA, Hutter CM, Zimmern RL, Humphries SE. Genetic causes of monogenic heterozygous familial hypercholesterolemia: a HuGE prevalence review. Am J Epidemiol 2004;160:407-20.
- Centre for Cardiovascular Genetics, University College London . LDLR (low Density Lipoprotein Receptor) Database @ LOVD (Leiden Open Variation Database) 2011. www.ucl.ac.uk/ldlr (accessed April 2011).
- Leigh SE, Foster AH, Whittall RA, Hubbart CS, Humphries SE. Update and analysis of the University College London low density lipoprotein receptor familial hypercholesterolemia database. Ann Hum Genet 2008;72:4-98.
- Varret M, Abifadel M, Rabes J-P, Boileau C. Genetic heterogeneity of autosomal dominant hypercholesterolemia. Clin Genet 2008;73:1-13.
- Sijbrands EJ, Westendorp RG, Defesche JC, de Meier PH, Smelt AH, Kastelein JJ. Mortality over two centuries in large pedigree with familial hypercholesterolaemia: family tree mortality study. BMJ 2001;322:1019-23.
- Warner M. Child–parent screening for familial hypercholesterolaemia: screening strategy based on a meta-analysis. Ann Clin Biochem 2008;45.
- Royal College of Physicians . The National Audit of the Management of Familial Hypercholesterolaemia 2010 2010. www.rcplondon.ac.uk/sites/default/files/fh-full-audit-report-2011.pdf (accessed April 2011).
- Umans-Eckenhausen MA, Defesche JC, Sijbrands EJ, Scheerder RL, Kastelein JJ. Review of first 5 years of screening for familial hypercholesterolaemia in the Netherlands. Lancet 2001;357:165-8.
- Risk of fatal coronary heart disease in familial hypercholesterolaemia. Scientific Steering Committee on behalf of the Simon Broome Register Group. BMJ 1991;303:893-6.
- Department of Primary Care Health Sciences, University of Oxford . Simon Broome FH Register 2011. www.primarycare.ox.ac.uk/research/vascular/Research/simon_broome/ (accessed October 2011).
- Kwiterovich PO, Fredrickson DS, Levy RI. Familial hypercholesterolemia (one form of familial type II hyperlipoproteinemia). A study of its biochemical, genetic and clinical presentation in childhood. J Clin Invest 1974;53:1237-49.
- Leonard JV, Whitelaw AG, Wolff OH. Diagnosing familial hypercholesterolaemia in childhood by measuring serum cholesterol. Br Med J 1977;1:1566-8.
- Marks D, Wonderling D, Thorogood M, Lambert H, Humphries SE, Neil HA. Screening for hypercholesterolaemia versus case finding for familial hypercholesterolaemia: a systematic review and cost-effectiveness analysis. Health Technol Assess 2000;4.
- Humphries SE, Galton D, Nicholls P. Genetic testing for familial hypercholesterolaemia: practical and ethical issues. QJM 1997;90:169-81.
- Humphries SE, Whittall RA, Hubbart CS, Maplebeck S, Cooper JA, Soutar AK, et al. Genetic causes of familial hypercholesterolaemia in patients in the UK: relation to plasma lipid levels and coronary heart disease risk. J Med Genet 2006;43:943-9.
- Heath KE, Humphries SE, Middleton-Price H, Boxer M. A molecular genetic service for diagnosing individuals with familial hypercholesterolaemia (FH) in the United Kingdom. Eur J Hum Genet 2001;9:244-52.
- Taylor A, Martin B, Wang D, Patel K, Humphries SE, Norbury G. Multiplex ligation-dependent probe amplification analysis to screen for deletions and duplications of the LDLR gene in patients with familial hypercholesterolaemia. Clin Genet 2009;76:69-75.
- Department of Health . National Service Framework for Children, Young People and Maternity Services: Core Standard 2007. www.dh.gov.uk/en/Publicationsandstatistics/Publications/PublicationsPolicyAndGuidance/Browsable/DH_4094329 (accessed April 2011).
- Marks D, Thorogood M, Farrer JM, Humphries SE. Census of clinics providing specialist lipid services in the United Kingdom. J Public Health 2004;26:353-4.
- Department of Health . Primary Care Service Framework: Familial Hypercholesterolaemia 2010. www.pcc.nhs.uk/uploads/commissioning/2010/02/primary_care_service_framework_familial_hypercholesterolaemia.pdf (accessed April 2011).
- Goldberg AC, Hopkins PN, Toth PP, Ballantyne C, Rader DJ, Robinson JG, et al. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol 2011;5:133-40.
- US Preventive Services Task Force . Screening for Lipid Disorders in Children 2007. www.uspreventiveservicestaskforce.org/uspstf/uspschlip.htm (accessed April 2011).
- Heart UK. Looking forward in lipids. Maidenhead: Heart UK; 2008.
- Whiting P, Rutjes AW, Reitsma JB, Bossuyt PM, Kleijnen J. The development of QUADAS: a tool for the quality assessment of studies of diagnostic accuracy included in systematic reviews. BMC Med Res Methodol 2003;3:1-13.
- Hooper AJ, Nguyen LT, Burnett JR, Van Bockxmeer FM. Molecular screening approach for identification of mutations causing familial hypercholesterolaemia in Western Australia. Twin Res Hum Genet 2009;12.
- Taylor A, Wang D, Patel K, Whittall R, Wood G, Farrer M, et al. Mutation detection rate and spectrum in familial hypercholesterolaemia patients in the UK pilot cascade project. Clin Genet 2010;77:572-80.
- Yarram L. Familial Hypercholesterolaemia: LIPOchip Experience n.d. www.cmgs.org/Restricted%20access%20area/CMGS%20members/CMGS%202010/SP1_5/SP01%20FH%20Lipochip%20experience%20CMGS.ppt (accessed April 2011).
- Alonso R, Defesche JC, Tejedor D, Castillo S, Stef M, Mata N, et al. Genetic diagnosis of familial hypercholesterolemia using a DNA-array based platform. Clin Biochem 2009;42:899-903.
- Callaway J, Wood O, Cross E, Skinner AC, Harvey JF. Validation of a novel mutation screening strategy for familial hypercholesterolaemia LIPOchip, a DNA-array based system. J Med Genet 2010;47.
- Palacios L, Stef M, Taylor A, Humphries SE, Cuevas N, McAnulty C, et al. Rapid and accurate genetic diagnosis by Lipochip (R) in UK FH patients. Atheroscler Suppl 2010;11.
- Stef M, Palacios L, Tejedor D, Martinez A. The LIPOchip experience in Spain. Atheroscler Suppl 2009;10.
- Tejedor D, Castillo S, Mozas P, Jimenez E, Lopez M, Tejedor MT, et al. Reliable low-density DNA array based on allele-specific probes for detection of 118 mutations causing familial hypercholesterolemia. Clin Chem 2005;51:1137-44.
- Civeira F, Ros E, Jarauta E, Plana N, Zambon D, Puzo J, et al. Comparison of genetic versus clinical diagnosis in familial hypercholesterolemia. Am J Cardiol 2008;102:1187-93.
- Damgaard D, Larsen ML, Nissen PH, Jensen JM, Jensen HK, Soerensen VR, et al. The relationship of molecular genetic to clinical diagnosis of familial hypercholesterolemia in a Danish population. Atherosclerosis 2005;180:155-60.
- Mabuchi H, Higashikata T, Nohara A, Lu H, Yu WX, Nozue T, et al. Cutoff point separating affected and unaffected familial hypercholesterolemic patients validated by LDL-receptor gene mutants. J Atheroscler Thromb 2005;12:35-40.
- Widhalm K, Dirisamer A, Lindemayr A, Kostner G. Diagnosis of families with familial hypercholesterolaemia and/or Apo B-100 defect by means of DNA analysis of LDL-receptor gene mutations. J Inherit Metab Dis 2007;30:239-47.
- Lee WP, Ong BB, Haralambos K, Townsend D, Rees JAE, Williams EJ, et al. Familial hypercholesterolaemia screening – application of genetic testing and diagnostic LDL-C cut-off values for relatives of FH patients in a Welsh population. Eur Heart J Suppl 2010;12:F20-21.
- Starr B, Hadfield SG, Hutten BA, Lansberg PJ, Leren TP, Damgaard D, et al. Development of sensitive and specific age- and gender-specific low-density lipoprotein cholesterol cutoffs for diagnosis of first-degree relatives with familial hypercholesterolaemia in cascade testing. Clin Chem Lab Med 2008;46:791-803.
- Wiegman A, Rodenburg J, de Jongh S, Defesche JC, Bakker HD, Kastelein JJP, et al. Family history and cardiovascular risk in familial hypercholesterolemia: data in more than 1000 children. Circulation 2003;107:1473-8.
- Yu W, Nohara A, Higashikata T, Lu H, Inazu A, Mabuchi H. Molecular genetic analysis of familial hypercholesterolemia: spectrum and regional difference of LDL receptor gene mutations in Japanese population. Atherosclerosis 2002;165:335-42.
- Stef M. Rapid diagnosis of familial hypercholesterolemia in British patients. New and developing technologies for genetic diagnostics. Salisbury; 2010.
- Leren TP, Manshaus T, Skovholt U, Skodje T, Nossen IE, Teie C, et al. Application of molecular genetics for diagnosing familial hypercholesterolemia in Norway: results from a family-based screening program. Semin Vasc Med 2004;4:75-8.
- Taylor A, Tabrah S, Wang D, Sozen M, Duxbury N, Whittall R, et al. Multiplex ARMS analysis to detect 13 common mutations in familial hypercholesterolaemia. Clin Genet 2007;71:561-8.
- Tabrah S, Tabrah S, Taylor A, Humphries SE, Norbury G. A strategy for molecular genetic mutation screening for familial hypercholesterolaemia. J Med Genet 2005;42.
- Tejedor D, Castillo S, Mozas P, Jimenez E, Lopez M, Tejedor MT, et al. Comparison of DNA array platform vs DNA sequencing as genetic diagnosis tools for familial hypercholesterolemia. Clin Chem 2006;52:1971-2.
- Oliva J, Lopez-Bastida J, Moreno SG, Mata P, Alonso R. Cost-effectiveness analysis of a genetic screening program in the close relatives of Spanish patients with familial hypercholesterolemia. Rev Esp Cardiol 2009;62:57-65.
- Fouchier SW, Defesche JC, Umans-Eckenhausen MAW, Kastelein JJP. The molecular basis of familial hypercholesterolemia in the Netherlands. Hum Genet 2001;109:602-15.
- Wonderling D, Umans-Eckenhausen MAW, Marks D, Defesche JC, Kastelein JJP, Thorogood M. Cost-effectiveness analysis of the genetic screening program for familial hypercholesterolemia in the Netherlands. Semin Vasc Med 2004;4:97-104.
- Marang-van de Mheen PJ, Ten Asbroek AHA, Bonneux L, Bonsel GJ, Klazinga NS. Cost-effectiveness of a family and DNA based screening programme on familial hypercholesterolaemia in The Netherlands. Eur Heart J 2002;23:1922-30.
- D’Agostino RB, Vasan RS, Pencina MJ, Wolf PA, Cobain M, Massaro JM, et al. General cardiovascular risk profile for use in primary care: the Framingham Heart Study. Circulation 2008;117:743-53.
- Marks D, Wonderling D, Thorogood M, Lambert H, Humphries SE, Neil HAW. Cost effectiveness analysis of different approaches of screening for familial hypercholesterolaemia. BMJ 2002;324:1303-6.
- Marks D, Thorogood M, Neil AW, Wonderling D, Humphries SE. Comparing costs and benefits over a 10 year period of strategies for familial hypercholesterolaemia screening. J Public Health Med 2003;25:47-52.
- Marks D, Wonderling D, Thorogood M, Lambert H, Humphries SE, Neil HAW. Screening for hypercholesterolaemia versus case finding for familial hypercholesterolaemia: a systematic review and cost effectiveness analysis. analysis. Health Technol Assess 2000;4.
- Leren TP. Cascade genetic screening for familial hypercholesterolemia. Clin Genet 2004;66:483-7.
- Humphries SE, Norbury G, Leigh S, Hadfield SG, Nair D. What is the clinical utility of DNA testing in patients with familial hypercholesterolaemia?. Curr Opin Lipidol 2008;19:362-8.
- Hadfield SG, Horara S, Starr BJ, Yazdgerdi S, Marks D, Bhatnagar D, et al. Family tracing to identify patients with familial hypercholesterolaemia: the second audit of the Department of Health Familial Hypercholesterolaemia Cascade Testing Project. Ann Clin Biochem 2009;46:24-32.
- NICE . Elucigene FH20 and LIPOchip for the Diagnosis of Familial Hypercholesterolaemia: Final Scope 2010. http://guidance.nice.org.uk/DT/2/Scope/pdf/English (accessed April 2011).
- Stockdale C, Yarram L, Williams M. Application of LIPOchip Technology to Genetic Screening in Autosomal Dominant Hypercholesterolaemia Referrals n.d. www.springmeeting.cytogenetics.org.uk/posters%5CM7.chris.stockdale.pdf (accessed April 2011).
- Hadfield SG, Horara S, Starr BJ, Yazdgerdi S, Bhatnagar D, Cramb R, et al. Are patients with familial hypercholesterolaemia well managed in lipid clinics? An audit of eleven clinics from the Department of Health Familial Hypercholesterolaemia Cascade Testing project. Ann Clin Biochem 2008;45:199-205.
- NICE . Statins for the Prevention of Cardiovascular Events. TA94 2006. www.nice.org.uk/nicemedia/live/11564/33151/33151.pdf (accessed April 2011).
- Department of Health . Health Survey for England 1996 1998. www.archive.official-documents.co.uk/document/doh/survey96/ehtitle.htm (accessed April 2011).
- Cole JA, Smith SM, Hart N, Cupples ME. Systematic review of the effect of diet and exercise lifestyle interventions in the secondary prevention of coronary heart disease. Cardiol Res Pract 2011;1. https://doi.org/10.4061/2011/232351.
- Department of Health . NHS Payment by Results 2010–11 National Tariff Information 2011. www.dh.gov.uk/prod_consum_dh/groups/dh_digitalassets/@dh/@en/@ps/documents/digitalasset/dh_113900.xls (accessed April 2011).
- Curtis L. Unit costs of health and social care. Canterbury, Kent: Personal Social Services Research Unit, University of Kent; 2010.
- British Medical Association and Royal Pharmaceutical Society of Great Britain . British National Formulary 2011. www.bnf.org/ (accessed April 2011).
- Vuorio A, Kuoppala J, Kovanen PT, Humphries SE, Strandberg T, Tonstad S, et al. Statins for children with familial hypercholesterolemia. Cochrane Database Syst Rev 2010.
- Briggs A. A Bayesian approach to stochastic cost-effectiveness analysis. Health Econ 1999;8:257-61.
- Tennant S, Bell C, Brown C, Kelly K, Hailey H, Gregory H, et al. Familial Hypercholesterolaemia Testing in Scotland [poster Presentation] n.d.
- National Institute for Health and Clinical Excellence . Identification and Management of Familial Hypercholesterolaemia. Implementing NICE Guidance: Costing Report 2009. http://guidance.nice.org.uk/CG71/CostingReport/pdf/English (accessed April 2011).
- Koivisto PV, Koivisto UM, Miettinen TA, Kontula K. Diagnosis of heterozygous familial hypercholesterolemia. DNA analysis complements clinical examination and analysis of serum lipid levels. Arterioscler Thromb 1992;12:584-92.
- Ward AJ, O’Kane M, Nicholls DP, Young IS, Nevin NC, Graham CA. A novel single base deletion in the LDLR gene (211delG): effect on serum lipid profiles and the influence of other genetic polymorphisms in the ACE, APOE and APOB genes. Atherosclerosis 1996;120:83-91.
- Vergotine J, Thiart R, Kotze MJ. Clinical versus molecular diagnosis of heterozygous familial hypercholesterolaemia in the diverse South African population. S Afr Med J 2001;91:1053-9.
- Humphries SE, Cranston T, Allen M, Middleton-Price H, Fernandez MC, Senior V, et al. Mutational analysis in UK patients with a clinical diagnosis of familial hypercholesterolaemia: relationship with plasma lipid traits, heart disease risk and utility in relative tracing. J Mol Med 2006;84:203-14.
- Bourbon M, Alves AC, Medeiros AM, Silva S, Soutar AK. Familial hypercholesterolaemia in Portugal. Atherosclerosis 2008;196:633-42.
- Marteau T, Senior V, Humphries SE, Bobrow M, Cranston T, Crook MA, et al. Psychological impact of genetic testing for familial hyperecholesterolemia within a previously aware population: a randomized controlled trial. Am J Med Genet 2004;128A:285-93.
- Whiting P, Rutjes AW, Reitsma JB, Glas AS, Bossuyt PM, Kleijnen J. Sources of variation and bias in studies of diagnostic accuracy: a systematic review. Ann Intern Med 2004;140:189-202.
- Wright WT, Heggarty SV, Young IS, Nicholls DP, Whittall R, Humphries SE, et al. Multiplex MassARRAY spectrometry (iPLEX) produces a fast and economical test for 56 familial hypercholesterolaemia-causing mutations. Clin Genet 2008;74:463-8.
- Taylor A, Bayly G, Patel K, Yarram L, Williams M, Hamilton-Shield J, et al. A double heterozygote for familial hypercholesterolaemia and familial defective apolipoprotein B-100. Ann Clin Biochem 2010;47:5-90.
- Van Der Graaf A, Avis HJ, Kusters DM, Vissers MN, Hutten BA, Defesche JC, et al. Molecular basis of autosomal dominant hypercholesterolemia: assessment in a large cohort of hypercholesterolemic children. Circulation 2011;123:1167-73. 10.1161/CIRCULATIONAHA.110.979450.
- Van Maarle MC, Stouthard MEA, Mheen PJM, Klazinga NS, Bonsel GJ. How disturbing is it to be approached for a genetic cascade screening programme for familial hypercholesterolaemia?: psychological impact and screenees’ views. Community Genet 2001;4:244-52.
- Van Maarle MC, Stouthard MEA, Bonsel GJ. Risk perception of participants in a family-based genetic screening program on familial hypercholesterolemia. Am J Med Genet 2003;116A:136-43.
- Marks D, Thorogood M, Neil SM, Humphries SE, Neil HAW. Cascade screening for familial hypercholesterolaemia: implications of a pilot study for national screening programmes. J Med Screen 2006;13:156-9.
- Will CM, Armstrong D, Marteau TM. Genetic unexceptionalism: clinician accounts of genetic testing for familial hypercholesterolaemia. Soc Sci Med 2010;71:910-17.
- Alonso R, Mata N, Castillo S, Fuentes F, Saenz P, Muniz O, et al. Cardiovascular disease in familial hypercholesterolaemia: influence of low-density lipoprotein receptor mutation type and classic risk factors. Atherosclerosis 2008;200:315-21.
- Avis HJ, Huijgen R, Hutten BA, Vissers MN, Janssen TH, Kusters DM, et al. Follow-up of children after positive genetic testing in the Dutch national screening program for familial hypercholesterolemia. Atheroscler Suppl 2010;11.
- Poke S, Watts G, Maxwell S, Brameld K, O’Leary P. Familial hypercholesterolaemia (FH) pilot cascade screening project. Twin Res Hum Genet 2009;12.
- Koeijvoets KCMC, Wiegman A, Rodenburg J, Defesche JC, Kastelein JJP, Sijbrands EJG. Effect of low-density lipoprotein receptor mutation on lipoproteins and cardiovascular disease risk: a parent–offspring study. Atherosclerosis 2005;180:93-9.
Appendix 1 Age-specific low-density lipoprotein cholesterol cut-offs for females
| LDL-C cut-off (mmol/l) | |||||
|---|---|---|---|---|---|
| Age (years) | |||||
| 0–14 | 15–24 | 25–34 | 35–44 | 45–54 | 55+ |
| 5.3 | 5.3 | 5.3 | 5.3 | 5.3 | 5.3 |
| 5.2 | 5.2 | 5.2 | 5.2 | 5.2 | 5.2 |
| 5.1 | 5.1 | 5.1 | 5.1 | 5.1 | 5.1 |
| 5.0 | 5.0 | 5.0 | 5.0 | 5.0 | 5.0 |
| 4.9 | 4.9 | 4.9 | 4.9 | 4.9 | 4.9 |
| 4.8 | 4.8 | 4.8 | 4.8 | 4.8 | 4.8 |
| 4.7 | 4.7 | 4.7 | 4.7 | 4.7 | 4.7 |
| 4.6 | 4.6 | 4.6 | 4.6 | 4.6 | 4.6 |
| 4.5 | 4.5 | 4.5 | 4.5 | 4.5 | 4.5 |
| 4.4 | 4.4 | 4.4 | 4.4 | 4.4 | 4.4 |
| 4.3 | 4.3 | 4.3 | 4.3 | 4.3 | 4.3 |
| 4.2 | 4.2 | 4.2 | 4.2 | 4.2 | 4.2 |
| 4.1 | 4.1 | 4.1 | 4.1 | 4.1 | 4.1 |
| 4.0 | 4.0 | 4.0 | 4.0 | 4.0 | 4.0 |
| 3.9 | 3.9 | 3.9 | 3.9 | 3.9 | 3.9 |
| 3.8 | 3.8 | 3.8 | 3.8 | 3.8 | 3.8 |
| 3.7 | 3.7 | 3.7 | 3.7 | 3.7 | 3.7 |
| 3.6 | 3.6 | 3.6 | 3.6 | 3.6 | 3.6 |
| 3.5 | 3.5 | 3.5 | 3.5 | 3.5 | 3.5 |
| 3.4 | 3.4 | 3.4 | 3.4 | 3.4 | 3.4 |
| 3.3 | 3.3 | 3.3 | 3.3 | 3.3 | 3.3 |
| 3.2 | 3.2 | 3.2 | 3.2 | 3.2 | 3.2 |
Appendix 2 Age-specific low-density lipoprotein cholesterol cut-offs for males
| LDL-C cut-off (mmol/l) | |||||
|---|---|---|---|---|---|
| Age (years) | |||||
| 0–14 | 15–24 | 25–34 | 35–44 | 45–54 | 55+ |
| 5.3 | 5.3 | 5.3 | 5.3 | 5.3 | 5.3 |
| 5.2 | 5.2 | 5.2 | 5.2 | 5.2 | 5.2 |
| 5.1 | 5.1 | 5.1 | 5.1 | 5.1 | 5.1 |
| 5.0 | 5.0 | 5.0 | 5.0 | 5.0 | 5.0 |
| 4.9 | 4.9 | 4.9 | 4.9 | 4.9 | 4.9 |
| 4.8 | 4.8 | 4.8 | 4.8 | 4.8 | 4.8 |
| 4.7 | 4.7 | 4.7 | 4.7 | 4.7 | 4.7 |
| 4.6 | 4.6 | 4.6 | 4.6 | 4.6 | 4.6 |
| 4.5 | 4.5 | 4.5 | 4.5 | 4.5 | 4.5 |
| 4.4 | 4.4 | 4.4 | 4.4 | 4.4 | 4.4 |
| 4.3 | 4.3 | 4.3 | 4.3 | 4.3 | 4.3 |
| 4.2 | 4.2 | 4.2 | 4.2 | 4.2 | 4.2 |
| 4.1 | 4.1 | 4.1 | 4.1 | 4.1 | 4.1 |
| 4.0 | 4.0 | 4.0 | 4.0 | 4.0 | 4.0 |
| 3.9 | 3.9 | 3.9 | 3.9 | 3.9 | 3.9 |
| 3.8 | 3.8 | 3.8 | 3.8 | 3.8 | 3.8 |
| 3.7 | 3.7 | 3.7 | 3.7 | 3.7 | 3.7 |
| 3.6 | 3.6 | 3.6 | 3.6 | 3.6 | 3.6 |
| 3.5 | 3.5 | 3.5 | 3.5 | 3.5 | 3.5 |
| 3.4 | 3.4 | 3.4 | 3.4 | 3.4 | 3.4 |
| 3.3 | 3.3 | 3.3 | 3.3 | 3.3 | 3.3 |
| 3.2 | 3.2 | 3.2 | 3.2 | 3.2 | 3.2 |
| 3.1 | 3.1 | 3.1 | 3.1 | 3.1 | 3.1 |
| 3.0 | 3.0 | 3.0 | 3.0 | 3.0 | 3.0 |
Appendix 3 Search strategy
Diagnostic accuracy and clinical effectiveness
MEDLINE (1948 to Week 1 2011), MEDLINE In-Process & Other Non-Indexed Citations (10 January 2011), EMBASE (1980 to 2011 Week 1)
Ovid multifile search
-
Hyperlipoproteinemia Type II/di
-
familial hypercholesterolemia/di
-
lipochip.tw.
-
elucigene.tw.
-
or/1-4
-
Hyperlipoproteinemia Type II/ use prmz
-
familial hypercholesterolemia/ use emez
-
(autosomal dominant adj5 hypercholesterol?emia).tw.
-
familial hypercholesterol?emia.tw.
-
hyperlipoprotein?emia.tw.
-
(familial adj5 apolipoprotein$).tw.
-
or/6-11
-
exp Genetic Predisposition to Disease/ use prmz
-
Genetic Testing/
-
Gene Amplification/ use prmz
-
exp Gene Amplification/ use emez
-
exp Nucleic Acid Amplification Techniques/ use prmz
-
dna microarray/ use emez
-
sequence analysis/ use emez
-
exp polymerase chain reaction/
-
exp sequence analysis/ use prmz
-
base sequence/ use prmz
-
(dna adj3 test$).tw.
-
gene sequencing.tw.
-
comprehensive genetic analysis.tw.
-
mutation screen$.tw.
-
direct sequencing.tw.
-
fragment analysis.tw.
-
(sanger adj3 (method or sequenc$)).tw.
-
(target$ adj3 gene$ sequenc$).tw.
-
(sequenc$ adj3 analysis).tw.
-
(cascade adj3 (test$ or screen$)).tw.
-
(genetic adj3 (test$ or screen$)).tw.
-
(arms or amplification refractory mutation system).tw.
-
(PCR or polymerase chain reaction).tw.
-
Polymorphism, Single-Stranded Conformational/
-
(sscp or single-stranded conformation polymorphism).tw.
-
(mlpa or Multiplex ligation-dependent probe amplification).tw.
-
(hrm or high resolution melt analysis).tw.
-
(DGGE or denaturing gradient gel electrophoresis).tw.
-
(dhplc or denaturing high performance liquid chromatography).tw.
-
Cholesterol, LDL/ use prmz
-
low density lipoprotein cholesterol/ use emez
-
ldl-c.tw.
-
simon broome.tw.
-
or/13-45
-
12 and 46
-
“sensitivity and specificity”/
-
roc curve/
-
receiver operating characteristic/ use emez
-
predictive value of tests/
-
diagnostic errors/ use emez
-
false positive reactions/ use prmz
-
false negative reactions/ use prmz
-
diagnostic accuracy/ use emez
-
diagnostic value/ use emez
-
du.fs. use prmz
-
sensitivity.tw.
-
distinguish$.tw.
-
differentiat$.tw.
-
identif$.tw.
-
detect$.tw.
-
diagnos$.tw.
-
(predictive adj4 value$).tw.
-
accura$.tw.
-
comparison.tw.
-
or/48-66
-
47 and 67
-
exp clinical trial/ use emez
-
randomized controlled trial.pt.
-
controlled clinical trial.pt.
-
randomization/ use emez
-
randomi?ed.ab.
-
randomly.ab.
-
trial.ab.
-
groups.ab.
-
or/69-76
-
exp animals/ not humans/
-
77 not 78
-
79 and 47
-
comparative study/ use prmz
-
major clinical study/ use emez
-
controlled study/ use emez
-
clinical trial/ use emez
-
(compare$ or compara$).tw.
-
or/81-85
-
86 and 47
-
5 or 68 or 80 or 87
-
remove duplicates from 88
-
limit 89 to yr=“2000-current”
-
limit 90 to english language
Science Citation Index (1970 to 10 January 2011), Conference Proceedings Citation Index – Science (1990 to 10 January 2011)
URL: www.isiknowledge.com
-
#1 TS=hyperlipoprotein*emia
-
#2 TS=(familial SAME hyperlipid*emia)
-
#3 TS=familial hypercholesterol*emia
-
#4 TS=((autosomal dominant) SAME hypercholesterol*emia)
-
#5 TS=(familial SAME apolipoprotein*)
-
#6 #5 OR #4 OR #3 OR #2 OR #1
-
#7 TS=low-density lipoprotein cholesterol
-
#8 TS=ldl-c
-
#9 TS=ldl cholesterol
-
#10 TS=simon broome
-
#11 TS=gene amplification
-
#12 TS= (DNA same “sequence analysis”)
-
#13 TS=(genetic SAME (test* or screen*))
-
#14 TS=(cascade SAME (test* or screen*))
-
#15 TS=mutation screen*
-
#16 TS=(#6 AND genetic analysis)
-
#17 TS=(#6 AND gene sequenc*)
-
#18 #17 OR #16 OR #15 OR #14 OR #13 OR #12 OR #11 OR #10 OR #9 OR #8 OR #7
-
#19 TS=(elucigene or lipochip)
-
#20 #18 and #6
-
#21 TS=(#6 SAME (diagnos* or test* or screen* or identif* or detect* or accura* or false positive or false negative))
-
#22 TS=(#6 SAME (trial* or random* or comparison or compare or comparative))
-
#23 #22 OR #21
-
#24 #23 AND #20
-
#25 #24 OR #19
-
#26 #24 OR #19 Refined by: Languages=(ENGLISH)
-
#27 #24 OR #19 Refined by: Languages=(ENGLISH) AND [excluding] Publication Years=(1999)
BIOSIS (1956 to 10 January 2011)
URL: www.isiknowledge.com
-
#1 TS=hyperlipoprotein*emia
-
#2 TS=(familial SAME hyperlipid*emia)
-
#3 TS=familial hypercholesterol*emia
-
#4 TS=((autosomal dominant) SAME hypercholesterol*emia)
-
#5 TS=(familial SAME apolipoprotein*)
-
#6 #5 OR #4 OR #3 OR #2 OR #1
-
#7 TS=ldl-c
-
#8 TS=ldl cholesterol
-
#9 TS=simon broome
-
#10 TS=gene amplification
-
#11 TS= (DNA same “sequence analysis”)
-
#12 TS=(genetic SAME (test* or screen*))
-
#13 TS=(cascade SAME (test* or screen*))
-
#14 TS=(#6 AND genetic analysis)
-
#15 TS=(#6 AND gene sequenc*)
-
#16 TS=(#6 AND low-density lipoprotein cholesterol)
-
#17 TS=(#6 AND mutation screen*)
-
#18 #17 OR #16 OR #15 OR #14 OR #13 OR #12 OR #11 OR #10 OR #9 OR #8 OR #7
-
#19 TS=(elucigene or lipochip)
-
#20 #18 and #6
-
#21 TS=(#6 SAME (diagnos* or test* or screen* or identif* or detect* or accura* or false positive or false negative))
-
#22 TS=(#6 SAME (trial* or random* or comparison or compare or comparative))
-
#23 #22 OR #21
-
#24 #23 AND #20
-
#25 #24 OR #19
-
#26 #24 OR #19 Refined by: [excluding] Publication Years=(1999 OR 1998) AND Languages=(ENGLISH)
Cochrane Controlled Trials Register, Cochrane Database of Systematic Reviews (The Cochrane Library, Issue 1, 2011)
URL: www.thecochranelibrary.com
-
#1 MeSH descriptor Hyperlipoproteinemia Type II, this term only with qualifier: DI
-
#2 Lipochip
-
#3 Elucigene
-
#4 #1 or #2 or #3
-
#5 MeSH descriptor Hyperlipoproteinemia Type II, this term only
-
#6 MeSH descriptor Hyperlipidemia, Familial Combined, this term only
-
#7 familial hyperlipid*emia
-
#8 familial hypercholesterol*emia
-
#9 #5 or #6 or #7 or #8
-
#10 MeSH descriptor Genetic Predisposition to Disease explode tree 1
-
#11 MeSH descriptor Genetic Testing, this term only
-
#12 MeSH descriptor Gene Amplification, this term only
-
#13 MeSH descriptor Nucleic Acid Amplification Techniques explode all trees
-
#14 MeSH descriptor Oligonucleotide Array Sequence Analysis explode tree 4
-
#15 MeSH descriptor Sequence Analysis, DNA explode all trees
-
#16 dna near/3 test*
-
#17 gene sequencing
-
#18 comprehensive genetic analysis
-
#19 target* near/3 gene* sequenc*
-
#20 sequenc* near/3 analysis
-
#21 cascade near/3 (test* or screen*)
-
#22 genetic near/3 (test$ or screen*)
-
#23 arms or “amplification refractory mutation system”
-
#24 PCR or “polymerase chain reaction”
-
#25 MeSH descriptor Polymorphism, Single-Stranded Conformational, this term only
-
#26 sscp or “single-stranded conformation polymorphism”
-
#27 mlpa or “Multiplex ligation-dependent probe amplification”
-
#28 MeSH descriptor Cholesterol, LDL, this term only
-
#29 ldl-c
-
#30 #10 or #11 or #12 or #13 or #14 or#15 or #16 or #17 or #18 or #19 or #20 or #21 or #22 or #23 or #24 or #25 or #26 or #27 or #28 or #29
-
#31 (#9 AND #30)
-
#32 (#4 OR #31)
-
#33 (#32), from 2000 to 2010
Database of Abstracts of Reviews of Effects, Health Technology Assessment database (January 2011), Centre for Reviews and Dissemination
-
#1 MeSH Hyperlipoproteinemia Type II QUALIFIERS DI
-
#2 elucigene OR lipochip
-
#3 #1 or #2
-
#4 MeSH Hyperlipoproteinemia Type II
-
#5 familial AND hypercholesterolemia
-
#6 familial AND hypercholesterolaemia
-
#7 hyperlipoproteinemia
-
#8 hyperlipoproteinaemia
-
#9 familial AND hyperlipidemia
-
#10 familial AND hyperlipidaemia
-
#11 #3 or #4 or #5 or #6 or #7 or #8 or #9 or #10
-
#12 #3 or #11 RESTRICT YR 2000 2011
ClinicalTrials.gov (December 2010)
familial hypercholesterolemia OR familial hypercholesterolaemia OR hyperlipoproteinemia type II OR familial combined hyperlipidemia
Controlled Trials (December 2010)
URL: www.controlledtrials.com/mrct
familial hypercholesterolaemia OR familial hypercholesterolemia OR hyperlipoproteinemia type II OR hyperlipoproteinaemia type II OR familial combined hyperlipidemia OR familial combined hyperlipidaemia
International Clinical Trials Registry (December 2010)
URL: http://apps.who.int/trialsearch/
familial hypercholesterolaemia OR familial hypercholesterolemia OR hyperlipoproteinemia type II OR familial combined hyperlipidemia
Websites consulted
American Association for Clinical Chemistry (December 2010)
URL: www.aacc.org
2010 annual meeting
2009 annual meeting
Atherosclerosis Supplements (December 2010)
URL: www.sciencedirect.com/science/journal/15675688
78th European Atherosclerosis Society Congress, Atherosclerosis Supplements 2010;11(2)
XII Brazilian Congress of Atherosclerosis, Brazilian Society of Cardiology, Atherosclerosis Supplements 2009;10(3)
XV International Symposium on Atherosclerosis, Atherosclerosis Supplements 2009;10(2)
European Society of Human Genetics (December 2010)
URL: www.eshg.org
European Human Genetics Conference 2010, European Journal of Human Genetics 2010;18(Suppl. 1)
European Human Genetics Conference 2009
Fonazione Giovanni Lorenzini (December 2010)
URL: www.lorenzinifoundation.org
4th International Conference of Biomarkers in Chronic Diseases (Diabetes, Obesity and Cardiovascular Diseases) 2010
National Genetics Reference Laboratory (December 2010)
URL: www.ngrl.org.uk/Wessex/tech_meeting10.html
New and Developing Technologies for Genetic Diagnostics ‘10
Cost-effectiveness
MEDLINE (1948 to Week 4 2011), MEDLINE In-Process & Other Non-Indexed Citations (2 February 2011), EMBASE (1980 to 2011 Week 4)
Ovid multifile search
-
Hyperlipoproteinemia Type II/di [Diagnosis]
-
familial hypercholesterolemia/di
-
lipochip.tw.
-
elucigene.tw.
-
or/1-4
-
Hyperlipoproteinemia Type II/ use prmz
-
familial hypercholesterolemia/ use emez
-
(autosomal dominant adj5 hypercholesterol?emia).tw.
-
familial hypercholesterol?emia.tw.
-
hyperlipoprotein?emia.tw.
-
(familial adj5 apolipoprotein$).tw.
-
or/6-11
-
exp Genetic Predisposition to Disease/ use prmz
-
Genetic Testing/
-
Gene Amplification/ use prmz
-
exp Gene Amplification/ use emez
-
exp Nucleic Acid Amplification Techniques/ use prmz
-
dna microarray/ use emez
-
sequence analysis/ use emez
-
exp polymerase chain reaction/
-
exp sequence analysis/ use prmz
-
base sequence/ use prmz
-
(dna adj3 test$).tw.
-
gene sequencing.tw.
-
comprehensive genetic analysis.tw.
-
mutation screen$.tw.
-
direct sequencing.tw.
-
fragment analysis.tw.
-
(sanger adj3 (method or sequenc$)).tw.
-
(target$ adj3 gene$ sequenc$).tw.
-
(sequenc$ adj3 analysis).tw.
-
(cascade adj3 (test$ or screen$)).tw.
-
(genetic adj3 (test$ or screen$)).tw.
-
(arms or amplification refractory mutation system).tw.
-
(PCR or polymerase chain reaction).tw.
-
Polymorphism, Single-Stranded Conformational/
-
(sscp or single-stranded conformation polymorphism).tw.
-
(mlpa or Multiplex ligation-dependent probe amplification).tw.
-
(hrm or high resolution melt analysis).tw.
-
(DGGE or denaturing gradient gel electrophoresis).tw.
-
(dhplc or denaturing high performance liquid chromatography).tw.
-
Cholesterol, LDL/ use prmz
-
low density lipoprotein cholesterol/ use emez
-
ldl-c.tw.
-
simon broome.tw.
-
or/13-45
-
12 and 5 and 46
-
exp “costs and cost analysis”/ use prmz
-
economics/
-
exp economic evaluation/ use emez
-
exp models, economic/
-
exp decision theory/
-
ec.fs.
-
monte carlo method/
-
markov chains/
-
exp health status indicators/
-
cost$.ti.
-
(cost$ adj2 (effective$ or utilit$ or benefit$ or minimis$)).ab.
-
economic$ model$.tw.
-
(price$ or pricing).tw.
-
(financial or finance or finances or financed).tw.
-
markov$.tw.
-
monte carlo.tw.
-
(decision$ adj2 (tree? or analy$ or model$)).tw.
-
(standard adj1 gamble).tw.
-
trade off.tw.
-
or/48-66
-
47 and 67
-
limit 68 to yr=“2000 -Current”
-
limit 69 to english language
-
remove duplicates from 70
Science Citation Index (1970 to 2 February 2011), Conference Proceedings Citation Index – Science (1990 to 2 February 2011)
URL: www.isiknowledge.com
-
#1 TS=hyperlipoprotein*emia
-
#2 TS=(familial SAME hyperlipid*emia)
-
#3 TS=familial hypercholesterol*emia
-
#4 TS=((autosomal dominant) SAME hypercholesterol*emia)
-
#5 TS=(familial SAME apolipoprotein*)
-
#6 #5 OR #4 OR #3 OR #2 OR #1
-
#7 TS=low-density lipoprotein cholesterol
-
#8 TS=ldl-c
-
#9 TS=ldl cholesterol
-
#10 TS=simon broome
-
#11 TS=gene amplification
-
#12 TS= (DNA same “sequence analysis”)
-
#13 TS=(genetic SAME (test* or screen*))
-
#14 TS=(cascade SAME (test* or screen*))
-
#15 TS=mutation screen*
-
#16 TS=(#6 AND genetic analysis)
-
#17 TS=(#6 AND gene sequenc*)
-
#18 #17 OR #16 OR #15 OR #14 OR #13 OR #12 OR #11 OR #10 OR #9 OR #8 OR #7
-
#19 TS=(elucigene or lipochip)
-
#20 #18 and #6
-
#21 TS=(#20 AND economic*)
-
#22 TS=(#20 AND cost*)
-
#23 TS=(#20 AND price*)
-
#24 TS=(#20 AND pricing*)
-
#25 TS=(#20 AND financ*)
-
#26 TS=(#20 AND markov*)
-
#27 TS=(#20 AND monte carlo)
-
#28 TS=(decision SAME (tree* OR analy* OR model*))
-
#29 #28 OR #27 OR #26 OR #25 OR #24 OR #23 OR #22 OR #21
-
#30 #29 AND #20
-
#31 #30 OR #19
-
#32 #30 OR #19 Refined by: Languages=( ENGLISH ) AND [excluding] Publication Years=(1999)
NHS Economic Evaluation Database (February 2011), Centre for Reviews and Dissemination
-
#1 MeSH Hyperlipoproteinemia Type II QUALIFIERS DI
-
#2 elucigene OR lipochip
-
#3 #1 or #2
-
#4 MeSH Hyperlipoproteinemia Type II
-
#5 familial AND hypercholesterolemia
-
#6 familial AND hypercholesterolaemia
-
#7 hyperlipoproteinemia
-
#8 hyperlipoproteinaemia
-
#9 familial AND hyperlipidemia
-
#10 familial AND hyperlipidaemia
-
#11 #3 or #4 or #5 or #6 or #7 or #8 or #9 or #10
-
#12 #3 or #11 RESTRICT YR 2000 2011
Cost-effectiveness Analysis Registry (February 2011)
URL: https://research.tufts-nemc.org/cear4/default.aspx
Search terms: familial hypercholesterolaemia OR familial hypercholesterolemia
Quality-of-life and cost data for model
MEDLINE (1948 to Week 4 2011), MEDLINE In-Process & Other Non-Indexed Citations (2 February 2011), EMBASE (1980 to 2011 Week 4)
Ovid multifile search
-
Hyperlipoproteinemia Type II/di [Diagnosis]
-
familial hypercholesterolemia/di
-
lipochip.tw.
-
elucigene.tw.
-
or/1-4
-
Hyperlipoproteinemia Type II/ use prmz
-
familial hypercholesterolemia/ use emez
-
(autosomal dominant adj5 hypercholesterol?emia).tw.
-
familial hypercholesterol?emia.tw.
-
hyperlipoprotein?emia.tw.
-
(familial adj5 apolipoprotein$).tw.
-
or/6-11
-
exp Genetic Predisposition to Disease/ use prmz
-
Genetic Testing/
-
Gene Amplification/ use prmz
-
exp Gene Amplification/ use emez
-
exp Nucleic Acid Amplification Techniques/ use prmz
-
dna microarray/ use emez
-
sequence analysis/ use emez
-
exp polymerase chain reaction/
-
exp sequence analysis/ use prmz
-
base sequence/ use prmz
-
(dna adj3 test$).tw.
-
gene sequencing.tw.
-
comprehensive genetic analysis.tw.
-
mutation screen$.tw.
-
direct sequencing.tw.
-
fragment analysis.tw.
-
(sanger adj3 (method or sequenc$)).tw.
-
(target$ adj3 gene$ sequenc$).tw.
-
(sequenc$ adj3 analysis).tw.
-
(cascade adj3 (test$ or screen$)).tw.
-
(genetic adj3 (test$ or screen$)).tw.
-
(arms or amplification refractory mutation system).tw.
-
(PCR or polymerase chain reaction).tw.
-
Polymorphism, Single-Stranded Conformational/
-
(sscp or single-stranded conformation polymorphism).tw.
-
(mlpa or Multiplex ligation-dependent probe amplification).tw.
-
(hrm or high resolution melt analysis).tw.
-
(DGGE or denaturing gradient gel electrophoresis).tw.
-
(dhplc or denaturing high performance liquid chromatography).tw.
-
Cholesterol, LDL/ use prmz
-
low density lipoprotein cholesterol/ use emez
-
ldl-c.tw.
-
simon broome.tw.
-
or/13-45
-
5 and 12 and 46
-
quality of life/
-
quality adjusted life year/
-
“Value of Life”/ use prmz
-
health status indicators/ use prmz
-
health status/ use emez
-
sickness impact profile/ use prmz
-
disability evaluation/ use prmz
-
disability/ use emez
-
activities of daily living/ use prmz
-
exp daily life activity/ use emez
-
cost utility analysis/ use emez
-
rating scale/
-
questionnaires/
-
(quality adj1 life).tw.
-
quality adjusted life.tw.
-
disability adjusted life.tw.
-
(qaly? or qald? or qale? or qtime? or daly?).tw.
-
(euroqol or euro qol or eq5d or eq 5d).tw.
-
(hql or hqol or h qol or hrqol or hr qol).tw.
-
(hye or hyes).tw.
-
health$ year$ equivalent$.tw.
-
(hui or hui1 or hui2 or hui3).tw.
-
(health adj3 (utilit$ or disutili$)).tw.
-
(health adj3 (state or status)).tw.
-
(sf36 or sf 36 or short form 36 or shortform 36).tw.
-
(sf6 or sf 6 or short form 6 or shortform 6).tw.
-
(sf12 or sf 12 or short form 12 or shortform 12).tw.
-
(sf16 or sf 16 or short form 16 or shortform 16).tw.
-
(sf20 or sf 20 or short form 20 or shortform 20).tw.
-
willingness to pay.tw.
-
standard gamble.tw.
-
trade off.tw.
-
conjoint analys?s.tw.
-
discrete choice.tw.
-
or/48-81
-
47 and 82
-
limit 83 to yr=“2000-current”
-
limit 84 to english language
IDEAS (February 2011)
Efficacy of statins
MEDLINE (1948 to Week 9 2011), MEDLINE In-Process & Other Non-Indexed Citations (9 March 2011), EMBASE (1980 to 2011 Week 9)
Ovid multifile search
-
Hyperlipoproteinemia Type II/ use prmz
-
familial hypercholesterolemia/ use emez
-
(autosomal dominant adj5 hypercholesterol?emia).tw.
-
familial hypercholesterol?emia.tw.
-
hyperlipoprotein?emia.tw.
-
(familial adj5 apolipoprotein$).tw.
-
or/1-6
-
exp Hydroxymethylglutaryl-CoA Reductase Inhibitors/ use prmz
-
hydroxymethylglutaryl coenzyme A reductase inhibitor/ use emez
-
Simvastatin/
-
Pravastatin/
-
rosuvastatin/ use emez
-
fluindostatin/ use emez
-
atorvastatin/ use emez
-
(simvastatin or pravastatin or rosuvastatin or fluvastatin or atorvastatin or statin$).tw.
-
hmg-coa.tw.
-
or/8-12
-
7 and 17
-
exp animals/ not humans/
-
18 not 19
-
limit 20 to yr=“2008-current”
-
2008$.ed.
-
2008$.em.
-
22 or 23
-
20 and 24
-
21 or 25
-
(letter or comment or editorial).pt.
-
26 not 27
-
remove duplicates from 28
-
limit 29 to english language
-
limit 28 to yr=“2000-current”
Database of Abstracts of Reviews of Effects, NHS Economic Evaluation Database, Health Technology Assessment database (March 2011), Centre for Reviews and Dissemination
-
#1 MeSH Hyperlipoproteinemia Type II
-
#2 familial AND hypercholesterolemia
-
#3 familial AND hypercholesterolaemia
-
#4 hyperlipoproteinemia
-
#5 hyperlipoproteinaemia
-
#6 familial AND hyperlipidemia
-
#7 familial AND hyperlipidaemia
-
#8 #1 or #2 or #3 or #4 or #5 or #6 or #7
-
#9 MeSH Hydroxymethylglutaryl-CoA Reductase Inhibitors
-
#10 MeSH Simvastatin
-
#11 MeSH Pravastatin
-
#12 simvastatin OR pravastatin OR rosuvastatin OR fluvastatin OR atorvastatin OR statin*
-
#13 hmg-coa
-
#14 #9 or #10 or #11 or #12 or #13
-
#15 #8 and #14 RESTRICT YR 2000 2011
Cochrane Database of Systematic Reviews (The Cochrane Library, Issue 3, 2011)
URL: www.thecochranelibrary.com
-
#1 MeSH descriptor Hyperlipoproteinemia Type II, this term only
-
#2 MeSH descriptor Hyperlipidemia, Familial Combined, this term only
-
#3 familial hyperlipid*emia
-
#4 familial hypercholesterol*emia
-
#5 (#1 OR #2 OR #3 OR #4)
-
#6 MeSH descriptor Hydroxymethylglutaryl-CoA Reductase Inhibitors explode tree 1
-
#7 MeSH descriptor Simvastatin, this term only
-
#8 MeSH descriptor Pravastatin, this term only
-
#9 simvastatin OR pravastatin OR rosuvastatin OR fluvastatin OR atorvastatin OR statin*
-
#10 hmg-coa
-
#11 (#6 OR #7 OR #8 OR #9 OR #10)
-
#12 (#5 AND #11), from 2007 to 2011
Effect of mutation type on treatment choice
MEDLINE (1948 to March Week 1 2011), MEDLINE In-Process & Other Non-Indexed Citations (14 March 2011), EMBASE (1980 to 2011 Week 10)
Ovid multifile search
-
Hyperlipoproteinemia Type II/ use prmz
-
familial hypercholesterolemia/ use emez
-
(autosomal dominant adj5 hypercholesterol?emia).tw.
-
familial hypercholesterol?emia.tw.
-
hyperlipoprotein?emia.tw.
-
(familial adj5 apolipoprotein$).tw.
-
or/1-6
-
Hyperlipoproteinemia Type II/dt [Drug Therapy]
-
familial hypercholesterolemia/dt [Drug Therapy]
-
or/8-9
-
exp Mutation/
-
(mutation$ adj2 variation$).tw.
-
(mutation$ adj2 type$).tw.
-
or/11-13
-
Niacin/ use prmz
-
nicotinic acid/ use emez
-
(niacin or nicotonic acid).tw.
-
exp Fibric Acids/ use prmz
-
exp fibric acid derivative/ use emez
-
fibrate$.tw.
-
exp Fish Oils/ use prmz
-
fish oil/ use emez
-
omega 3 fatty acid/ use emez
-
fish oil$.tw.
-
omega 3.tw.
-
exp Blood Component Removal/ use prmz
-
exp apheresis/ use emez
-
(aphersis or plasmapheresis).tw.
-
resin/ use emez
-
resin$.tw.
-
ezetimibe/ use emez
-
ezetimibe.tw.
-
or/15-32
-
10 and 14
-
7 and 14 and 33
-
34 or 35
-
(comment or letter or editorial).pt.
-
36 not 37
-
limit 36 to english language
-
limit 39 to yr=“2000 -Current”
-
remove duplicates from 40
Cochrane Database of Systematic Reviews (The Cochrane Library, Issue 3, 2011)
URL: www.thecochranelibrary.com
-
#1 MeSH descriptor Hyperlipoproteinemia Type II, this term only
-
#2 MeSH descriptor Hyperlipidemia, Familial Combined, this term only
-
#3 familial hyperlipid*emia
-
#4 familial hypercholesterol*emia
-
#5 (#1 OR #2 OR #3 OR #4)
-
#6 MeSH descriptor Hyperlipoproteinemia Type II, this term only with qualifier: DT
-
#7 MeSH descriptor Mutation explode all trees
-
#8 mutation* variation*
-
#9 mutation* type*
-
#10 (#7 OR #8 OR #9)
-
#11 (#5 AND #10)
-
#12 (#6 AND #10)
-
#13 (#11 OR #12)
Database of Abstracts of Reviews of Effects, NHS Economic Evaluation Database, Health Technology Assessment database (March 2011), Centre for Reviews and Dissemination
-
#1 MeSH Hyperlipoproteinemia Type II
-
#2 familial AND hypercholesterolemia
-
#3 familial AND hypercholesterolaemia
-
#4 familial AND hyperlipidemia
-
#5 familial AND hyperlipidaemia
-
#6 #1 #2 or #3 or #4 or #5
-
#7 MeSH Hyperlipoproteinemia Type II QUALIFIERS DT
-
#8 MeSH Mutation EXPLODE 1
-
#9 mutation* AND type*
-
#10 mutation* AND variation*
-
#11 #8 or #9 or #10
-
#12 #6 and #11
-
#13 #7 or #12
Utility of diagnostic information
MEDLINE (1996 to February Week 4 2011)
Ovid multifile search
-
*genetic testing/
-
*quality of life/px
-
*psychology/
-
*patient satisfaction/
-
*patient acceptance of health care/
-
*attitude to health/
-
*rating scale/
-
*questionnaires/
-
(quality adj1 life).tw
-
(patient? adj1 (preferenc$ or experienc$ or perception$ or satisfaction$)).ti.
-
quality of life/
-
quality adjusted life year/
-
“Value of Life”/
-
health status indicators/
-
sickness impact profile/
-
quality adjusted life.tw.
-
disability adjusted life.tw.
-
(qaly? or qald? or qale? or qtime? or daly?).tw.
-
(euroqol or euro qol or eq5d or eq 5d).tw
-
(hql or hqol or h qol or hrqol or hr qol).tw.
-
(hye or hyes).tw
-
health$ year$ equivalent$.tw
-
(hui or hui1 or hui2 or hui3).tw.
-
(health adj3 (utilit$ or disutili$)).tw.
-
(health adj3 (state or status)).tw.
-
(sf36 or sf 36 or short form 36 or shortform 36).tw.
-
(sf6 or sf 6 or short form 6 or shortform 6).tw.
-
(sf12 or sf 12 or short form 12 or shortform 12).tw.
-
(sf16 or sf 16 or short form 16 or shortform 16).tw.
-
(sf20 or sf 20 or short form 20 or shortform 20).tw.
-
willingness to pay.tw.
-
standard gamble.tw.
-
trade off.tw.
-
conjoint analys?s.tw.
-
discrete choice.tw.
-
or/2-35
-
1 and 36
-
limit 37 to (english language and yr=“2000 -Current”)
Database of Abstracts of Reviews of Effects, NHS Economic Evaluation Database, Health Technology Assessment database (March 2011), Centre for Reviews and Dissemination
-
#1 MeSH Genetic Screening EXPLODE 1 2 3 4 5 6 7 RESTRICT YR 2000 2011
-
#2 cost:ty
-
#3 #1 NOT #2
Appendix 4 Data extraction form
Appendix 5 Modified QUADAS checklist
| Item | Yes | No | Unclear | |
|---|---|---|---|---|
| 1 | Was the spectrum of patients representative of the patients who will receive the test in practice? | |||
| 2 | Is the reference standard likely to correctly classify the target condition?a | |||
| 3 | Did the whole sample or a random selection of the sample receive verification using a reference standard of diagnosis? | |||
| 4 | Did patients receive the same reference standard regardless of the index test result? | |||
| 5 | Was the reference standard independent of the index test (i.e. the index test did not form part of the reference standard)? | |||
| 6 | Were the index test results interpreted without knowledge of the results of the reference standard? | |||
| 7 | Were the reference standard results interpreted without knowledge of the results of the index test? | |||
| 8 | Were the same clinical data available when test results were interpreted as would be available when the test is used in practice?b | |||
| 9 | Were uninterpretable/intermediate/test results reported? | |||
| 10 | Were withdrawals from the study explained? | |||
| 11 | Were cut-off values established before the study was started? | |||
| 12 | Is the technology of the index test unchanged since the study was carried out? | |||
| 13 | Did the study provide a clear definition of what was considered to be a ‘positive’ result?c | |||
Please also note if the paper reported details of any of the following issues:
-
MLPA: the location of probes in the intron; single nucleotide polymorphism (SNP) at probe binding site
-
Elucigene FH20: inadequate electrophoretic separation and misidentification of the FH20 mutations
-
LIPOchip: assessment of batch capacity; assessment of training requirements; assessment of instrumentation required, maintenance, etc.
Appendix 6 List of included studies
Alonso 2009
Alonso R, Defesche JC, Tejedor D, Castillo S, Stef M, Mata N, et al. Genetic diagnosis of familial hypercholesterolemia using a DNA-array based platform. Clin Biochem 2009;42:899–903.
Callaway 2010
Callaway J, Wood O, Cross E, Skinner AC, Harvey JF. Validation of a novel mutation screening strategy for familial hypercholesterolaemia LIPOchip, a DNA-array based system. J Med Genet 2010;47:S62.
Civeira 2008
Civeira F, Ros E, Jarauta E, Plana N, Zambon D, Puzo J, et al. Comparison of genetic versus clinical diagnosis in familial hypercholesterolemia. Am J Cardiol 2008;102:1187–93.
Damgaard 2005
Damgaard D, Larsen ML, Nissen PH, Jensen JM, Jensen HK, Soerensen VR, et al. The relationship of molecular genetic to clinical diagnosis of familial hypercholesterolemia in a Danish population. Atherosclerosis 2005;180:155–60.
Hooper 2009
Hooper AJ, Nguyen LT, Burnett JR, Van Bockxmeer FM. Molecular screening approach for identification of mutations causing familial hypercholesterolaemia in Western Australia. Twin Res Hum Genet 2009;12:218.
Lee 2010
Lee WP, Ong BB, Haralambos K, Townsend D, Rees JAE, Williams EJ, et al. Familial hypercholesterolaemia screening – application of genetic testing and diagnostic LDL-C cut-off values for relatives of FH patients in a Welsh population. Eur Heart J Suppl 2010;12:F20–1.
Mabuchi 2005
Mabuchi H, Higashikata T, Nohara A, Lu H, Yu WX, Nozue T, et al. Cutoff point separating affected and unaffected familial hypercholesterolemic patients validated by LDL-receptor gene mutants. J Atheroscler Thromb 2005;12:35–40.
Palacios 2010
Primary study
Palacios L, Stef M, Taylor A, Humphries SE, Cuevas N, McAnulty C, et al. Rapid and accurate genetic diagnosis by LIPOchip (R) in UK FH patients. Atheroscler Suppl 2010;11:31.
Secondary study
Stef M. Rapid diagnosis of familial hypercholesterolemia in British patients. New and Developing Technologies for Genetic Diagnostics, Salisbury, 5–6 July 2010. URL: www.ngrl.org.uk/Wessex/downloads/tm10/TM10-S2-2%20Marianne%20Stef.pdf (accessed March 2011).
Starr 2008
Starr B, Hadfield SG, Hutten BA, Lansberg PJ, Leren TP, Damgaard D, et al. Development of sensitive and specific age- and gender-specific low-density lipoprotein cholesterol cutoffs for diagnosis of first-degree relatives with familial hypercholesterolaemia in cascade testing. Clin Chem Lab Med 2008;46:791–803.
Stef 2009
Stef M, Palacios L, Tejedor D, Martinez A. The LIPOchip experience in Spain. Atheroscler Suppl 2009;10:e1001.
Taylor 2010
Primary study
Taylor A, Wang D, Patel K, Whittall R, Wood G, Farrer M, et al. Mutation detection rate and spectrum in familial hypercholesterolaemia patients in the UK pilot cascade project. Clin Genet 2010;77:572–80.
Secondary study
Taylor A, Tabrah S, Wang D, Sozen M, Duxbury N, Whittall R, et al. Multiplex ARMS analysis to detect 13 common mutations in familial hypercholesterolaemia. Clin Genet 2007;71:561–8.
Tejedor 2005
Tejedor D, Castillo S, Mozas P, Jimenez E, Lopez M, Tejedor MT, et al. Reliable low-density DNA array based on allele-specific probes for detection of 118 mutations causing familial hypercholesterolemia. Clin Chem 2005;51:1137–44.
Widham 2007
Widhalm K, Dirisamer A, Lindemayr A, Kostner G. Diagnosis of families with familial hypercholesterolaemia and/or Apo B-100 defect by means of DNA analysis of LDL-receptor gene mutations. J Inherit Metab Dis 2007;30:239–47.
Wiegman 2003
Wiegman A, Rodenburg J, De Jongh S, Defesche JC, Bakker HD, Kastelein JJP, et al. Family history and cardiovascular risk in familial hypercholesterolemia: data in more than 1000 children. Circulation 2003;107:1473–8.
Yarram 2010
Yarram L. Familial hypercholesterolaemia: LIPOchip experience. Clinical Molecular Genetics Society meeting, St Catherine’s College, Oxford, April 2010. URL: www.cmgs.org/Restricted%20access%20area/CMGS%20members/CMGS%202010/SP1_5/SP01%20FH%20Lipochip%20experience%20CMGS.ppt (accessed April 2011).
Appendix 7 List of excluded studies
Not a required reference standard (APOB or PCSK9 or deletion/duplication only)
Cantafora A, Blotta I, Pino E, Pisciotta L, Calandra S, Bertolini S. Quantitative polymerase chain reaction and microchip electrophoresis to detect major rearrangements of the low-density lipoprotein receptor gene causing familial hypercholesterolemia. Electrophoresis 2004;25:3882–9.
Garcia-Garcia AB, Blesa S, Martinez-Hervas S, Mansego ML, Gonzalez-Albert V, Ascaso JF, et al. Semiquantitative multiplex PCR: a useful tool for large rearrangement screening and characterization. Hum Mutat 2006;27:822–8.
Garcia-Otin AL, Strunk M, Pueyo M, Solanas M, Fiddyment S, Aceves M, et al. Screening for PCSK9 mutations in Spanish patients with autosomal dominant hypercholesterolemia unrelated to LDLR or APOB. Atheroscler Suppl 2009;10:e1228.
Goldmann R, Tichy L, Freiberger T, Zapletalova P, Letocha O, Soska V, et al. Genomic characterization of large rearrangements of the LDLR gene in Czech patients with familial hypercholesterolemia. BMC Med Genet 2010;11:115.
Heath KE, Day INM, Humphries SE. Universal primer quantitative fluorescent multiplex (UPQFM) PCR: a method to detect major and minor rearrangements of the low density lipoprotein receptor gene. J Med Genet 2000;37:272–80.
Holla OL, Teie C, Berge KE, Leren TP. Identification of deletions and duplications in the low density lipoprotein receptor gene by MLPA. Clin Chim Acta 2005;356:164–71.
Kalina A, Csaszar A, Czeizel AE, Romics L, Szaboki F, Szalai C, et al. Frequency of the R3500Q mutation of the apolipoprotein B-100 gene in a sample screened clinically for familial hypercholesterolemia in Hungary. Atherosclerosis 2001;154:247–51.
Liyanage KE, Hooper AJ, Defesche JC, Burnett JR, Van Bockxmeer FM. High-resolution melting analysis for detection of familial ligand-defective apolipoprotein B-100 mutations. Ann Clin Biochem 2008;45:170–6.
Merino-Ibarra E, Castillo S, Mozas P, Cenarro A, Martorell E, Diaz JL, et al. Screening of APOB gene mutations in subjects with clinical diagnosis of familial hypercholesterolemia. Hum Biol 2005;77:663–73.
Meshkov A, Stambolsky D, Malyshev P, Kotkina T, Boitsov S, Kukharchuk V. The prevalence of apolipoprotein B-100 gene mutations in Russian familial hypercholesterolemia patients. Atheroscler Suppl 2009;10:e990.
Taylor A, Martin B, Wang D, Patel K, Humphries SE, Norbury G. Multiplex ligation-dependent probe amplification analysis to screen for deletions and duplications of the LDLR gene in patients with familial hypercholesterolaemia. Clin Genet 2009;76:69–75.
Case report
Taylor A, Bayly G, Patel K, Yarram L, Williams M, Hamilton-Shield J, et al. A double heterozygote for familial hypercholesterolaemia and familial defective apolipoprotein B-100. Ann Clin Biochem 2010;47:5–90.
Single test (comprehensive genetic analysis) or insufficient/not usable data to allow calculation of test performance
Alonso R, Mata N, Castillo S, Fuentes F, Saenz P, Muniz O, et al. Cardiovascular disease in familial hypercholesterolaemia: influence of low-density lipoprotein receptor mutation type and classic risk factors. Atherosclerosis 2008;200:315–21.
Alves A, Medeiros A, Francisco V, Bourbon M. Familial hypercholesterolaemia: a perspective of 10 years of study in Portugal. Atherosclerosis 2009;10:e1219.
Alves AC, Medeiros AM, Francisco V, Gaspar IM, Rato Q, Bourbon M. Molecular diagnosis of familial hypercholesterolemia: an important tool for cardiovascular risk stratification. Rev Port Cardiol 2010;29:907–21.
Arraiz N, Bermudez V, Rondon N, Reyes F, Borjas L, Solis E, et al. Novel mutations identification in exon 4 of LDLR gene in patients with moderate hypercholesterolemia in a Venezuelan population. Am J Ther 2010;17:325–9.
Bertolini S, Cantafora A, Averna M, Cortese C, Motti C, Martini S, et al. Clinical expression of familial hypercholesterolemia in clusters of mutations of the LDL receptor gene that cause a receptor-defective or receptor-negative phenotype. Arterioscler Thromb Vasc Biol 2000;20:E41–52.
Bhatnagar D, Morgan J, Siddiq S, Mackness MI, Miller JP, Durrington PN. Outcome of case finding among relatives of patients with known heterozygous familial hypercholesterolaemia. BMJ 2000;321:1497–500.
Blesa S, Garcia-Garcia AB, Martinez-Hervas S, Mansego ML, Gonzalez-Albert V, Ascaso JF, et al. Analysis of sequence variations in the LDL receptor gene in Spain: general gene screening or search for specific alterations? Clin Chem 2006;52:1021–5.
Bourbon M, Rato Q. Portuguese familial hypercholesterolemia study: presentation of the study and preliminary results. Rev Port Cardiol 2006;25:999–1013.
Bourbon M, Alves AC, Medeiros AM, Silva S, Soutar AK. Familial hypercholesterolaemia in Portugal. Atherosclerosis 2008;196:633–42.
Briffaut D, Tounian P, Benlian P, Girardet J. Molecular-based diagnostic criteria of familial hypercholesterolemia in children with dominantly-inherited hypercholesterolemia. J Pediatr Gastroenterol Nutr 2000;31:S209.
Brusgaard K, Jordan P, Hansen H, Hansen AB, Horder M. Molecular genetic analysis of 1053 Danish individuals with clinical signs of familial hypercholesterolemia. Clin Genet 2006;69:277–83.
Bunn CF, Lintott CJ, Scott RS, George PM. Comparison of SSCP and DHPLC for the detection of LDLR mutations in a New Zealand cohort. Hum Mutat 2002;19:311.
Campagna F, Martino F, Bifolco M, Montali A, Martino E, Morrone F, et al. Detection of familial hypercholesterolemia in a cohort of children with hypercholesterolemia: results of a family and DNA-based screening. Atherosclerosis 2008;196:356–64.
Cefalu AB, Emmanuele G, Marino G, Fiore B, Caldarella R, Vivona N, et al. Effectiveness of screening for known mutations in Sicilian patients with ‘probable’ familial hypercholesterolemia. Nutr Metab Cardiovasc Dis 2001;11:394–400.
Charng MJ, Chiou KR, Chang HM, Cheng HM, Ye ZX, Lin SJ. Identification and characterization of novel low-density lipoprotein receptor mutations of familial hypercholesterolaemia patients in Taiwan. Eur J Clin Invest 2006;36:866–74.
Chaves FJ, Real JT, Garcia-Garcia AB, Civera M, Armengod ME, Ascaso JF, et al. Genetic diagnosis of familial hypercholesterolemia in a South European outbreed population: influence of low-density lipoprotein (LDL) receptor gene mutations on treatment response to simvastatin in total, LDL, and high-density lipoprotein cholesterol. J Clin Endocrinol Metab 2001;86:4926–32.
Chiou K-R, Charng M-J. Detection of mutations and large rearrangements of the low-density lipoprotein receptor gene in Taiwanese patients with familial hypercholesterolemia. Am J Cardiol 2010;105:1752–8.
Chmara M, Wasag B, Zuk M, Kubalska J, Wegrzyn A, Bednarska-Makaruk M, et al. Molecular characterization of Polish patients with familial hypercholesterolemia: novel and recurrent LDLR mutations. J Appl Genet 2010;51:95–106.
Cohen H, Harats D, Wael N, Anikster Y, Mazor-Aronovitch K, Pinhas-Hamiel O. LDL receptor mutation in a druze kindred – clinical, biochemical and genetic characteristics. Atheroscler Suppl 2009;10:e1222.
Damgaard D, Nissen PH, Jensen LG, Nielsen GG, Stenderup A, Larsen ML, et al. Detection of large deletions in the LDL receptor gene with quantitative PCR methods. BMC Med Genet 2005;6:15.
De Castro-Ors I, Palacios L, Pampn S, Plana N, Masana L, Stef M, et al. Functional analysis of LDLR promoter mutations associated with familial hypercholesterolemia. Atheroscler Suppl 2010;11:112–3.
Dedoussis GVZ, Skoumas J, Pitsavos C, Choumerianou DM, Genschel J, Schmidt H, et al. FH clinical phenotype in Greek patients with LDL-R defective vs. negative mutations. Eur J Clin Invest 2004;34:402–9.
Defesche JC. Is molecular genetic testing for familial hypercholesterolemia cost effective and clinically useful? Atheroscler Suppl 2006;7:19.
Diakou M, Miltiadous G, Xenophontos S, Cariolou M, Elisaf M. Mutational analysis in northwestern Greece patients with clinical diagnosis of familial hypercholesterolemia. Atheroscler 2009;10(Suppl.):e1224.
Diakou M, Miltiadous G, Xenophontos S, Cariolou M, Heta N, Korita I, et al. Characterization of low density lipoprotein receptor (LDLR) gene mutations in Albania. Arch Med Sci 2010;6:198–200.
Ejarque I, Real JT, Martinez-Hervas S, Chaves FJ, Blesa S, Garcia-Garcia AB, et al. Evaluation of clinical diagnosis criteria of familial ligand defective apoB 100 and lipoprotein phenotype comparison between LDL receptor gene mutations affecting ligand-binding domain and the R3500Q mutation of the apoB gene in patients from a South European population. Transl Res 2008;151:162–7.
El MM, Ait CK, Chater R, Vallve JC, Bennis F, Hafidi A, et al. Familial hypercholesterolemia in Morocco: first report of mutations in the LDL receptor gene. J Hum Genet 2003;48:199–203.
Emi M, Hirayama T, Tsuji M, Hata A. Novel mutations of the LDL receptor gene in familial hypercholesterolemia pedigrees in Hokkaido. In Kita T, Yokode M, editors. Lipoprotein metabolism and atherogenesis. Springer Verlag; 2000. pp. 48–50.
Fouchier SW, Defesche JC, Umans-Eckenhausen MAW, Kastelein JJP. The molecular basis of familial hypercholesterolemia in the Netherlands. Hum Genet 2001;109:602–15.
Fouchier SW, Kastelein JJP, Defesche JC. Update of the molecular basis of familial hypercholesterolemia in The Netherlands. Hum Mutat 2005;26:550–6.
Garcia-Garcia AB, Real JT, Puig O, Cebolla E, Marin-Garcia P, Martinez Ferrandis JI, et al. Molecular genetics of familial hypercholesterolemia in Spain: ten novel LDLR mutations and population analysis. Hum Mutat 2001;18:458–9.
Graham CA, McIlhatton BP, Kirk CW, Beattie ED, Lyttle K, Hart P, et al. Genetic screening protocol for familial hypercholesterolemia which includes splicing defects gives an improved mutation detection rate. Atherosclerosis 2005;182:331–40.
Guardamagna O, Restagno G, Rolfo E, Pederiva C, Martini S, Abello F, et al. The type of LDLR gene mutation predicts cardiovascular risk in children with familial hypercholesterolemia. J Pediatr 2009;155:199–204.
Hadfield SG, Horara S, Starr BJ, Yazdgerdi S, Bhatnagar D, Cramb R, et al. Are patients with familial hypercholesterolaemia well managed in lipid clinics? An audit of eleven clinics from the Department of Health Familial Hypercholesterolaemia Cascade Testing project. Ann Clin Biochem 2008;45:199–205.
Hadfield SG, Horara S, Starr BJ, Yazdgerdi S, Marks D, Bhatnagar D, et al. Family tracing to identify patients with familial hypercholesterolaemia: the second audit of the Department of Health Familial hypercholesterolaemia Cascade Testing project. Ann Clin Biochem 2009;46:24–32.
Heath KE, Humphries SE, Middleton-Price H, Boxer M. A molecular genetic service for diagnosing individuals with familial hypercholesterolaemia (FH) in the United Kingdom. Eur J Hum Genet 2001;9:244–52.
Higgins S, Stay C, Duxbury N, Smit C. Validation of Elucigene QST*R, a QF-PCR Assay. J Med Genet 2006;43:S102.
Huijgen R, Kindt I, Fouchier S, Defesche J, Hutten B, Kastelein J, et al. Non-functionality of three LDL-R and APOB gene mutations that were assumed to cause familial hypercholesterolemia. Atheroscler Suppl 2009;10:e1231.
Huijgen R, Versmissen J, Oosterveer DM, Kindt I, Sijbrands EJG, Kastelein JJP. Efficacy of 15 years of genetic cascade screening for familial hypercholesterolemia in the Netherlands in prevention of coronary artery disease. Atheroscler Suppl 2010;11:L6.
Humphries SE, Cranston T, Allen M, Middleton-Price H, Fernandez MC, Senior V, et al. Mutational analysis in UK patients with a clinical diagnosis of familial hypercholesterolaemia: relationship with plasma lipid traits, heart disease risk and utility in relative tracing. J Mol Med 2006;84:203–14.
Humphries SE, Whittall RA, Hubbart CS, Maplebeck S, Cooper JA, Soutar AK, et al. Genetic causes of familial hypercholesterolaemia in patients in the UK: relation to plasma lipid levels and coronary heart disease risk. J Med Genet 2006;43:943–9.
Jelassi A, Slimani A, Jguirim I, Najah M, Abid A, Boughamoura L, et al. Moderate phenotypic expression of familial hypercholesterolemia in Tunisia. Clin Chim Acta 2010;411:735–8.
Junyent M, Mateo R, Gilabert R, Cofan M, Nunez I, Pocovi M, et al. Impact of LDL receptor mutational class on carotid atherosclerosis in familial hypercholesterolemia (FH). Atheroscler Suppl 2009;10:e1232.
Junyent M, Gilabert R, Jarauta E, Nunez I, Cofan M, Civeira F, et al. Impact of low-density lipoprotein receptor mutational class on carotid atherosclerosis in patients with familial hypercholesterolemia. Atherosclerosis 2010;208:437–41.
Khoo KL, van Acker P, Defesche JC, Tan H, van De Kerkhof L, Heijnen-van Eijk SJ, et al. Low-density lipoprotein receptor gene mutations in a Southeast Asian population with familial hypercholesterolemia. Clin Genet 2000;58:98–105.
Koeijvoets KCMC, Wiegman A, Rodenburg J, Defesche JC, Kastelein JJP, Sijbrands EJG. Effect of low-density lipoprotein receptor mutation on lipoproteins and cardiovascular disease risk: a parent–offspring study. Atherosclerosis 2005;180:93–9.
Kuhrova V, Francova H, Zapletalova P, Freiberger T, Fajkusova L, Hrabincova E, et al. Spectrum of low density lipoprotein receptor mutations in Czech hypercholesterolemic patients. Hum Mutat 2001;18:253.
Kuhrova V, Francova H, Zapletalova P, Freiberger T, Fajkusova L, Hrabincova E, et al. Spectrum of low density lipoprotein receptor mutations in Czech hypercholesterolemic patients. Hum Mutat 2002;19:80.
Lagerstedt SA, Train LJ, Jordan K, Baudhuin LM. Full gene sequencing of the low density lipoprotein receptor (LDLR) gene for the clinical diagnosis of familial hypercholesterolemia. Clin Chem 2007;53:C-151.
Laurie AD, Scott RS, George PM. Genetic screening of patients with familial hypercholesterolaemia (FH): a New Zealand perspective. Atheroscler Suppl 2004;5:13–15.
Leren TP, Manshaus TE, Berge KE. Application of genetic testing in cascade genetic screening for familial hypercholesterolemia. Atheroscler Suppl 2005;6:178.
Leren TP, Manshaus TE, Ose L, Berge KE. Application of genetic testing in cascade genetic screening for familial hypercholesterolemia. Atheroscler Suppl 2006;7:36–7.
Lind S, Rystedt E, Eriksson M, Wiklund O, Angelin B, Eggertsen G. Genetic characterization of Swedish patients with familial hypercholesterolemia: a heterogeneous pattern of mutations in the LDL receptor gene. Atherosclerosis 2002;163:399–407.
Lindemayr AC, Dirisamer A, Schmidt H, Widhalm K. Familial hypercholesterolemia: diagnosis in childhood by means of DNA-analysis. Pediatr Res 2000;47:206A.
Liyanage K, Burnett J, Van BF. Detection of familial hypercholesterolaemia in a cohort of coronary heart disease patients in Western Australia. Atheroscler Suppl 2009;10:e989.
Lombardi MP, Redeker EJW, Defesche JC, Kamerling SW, Trip MD, Mannens MMAM, et al. Molecular genetic testing for familial hypercholesterolemia: spectrum of LDL receptor gene mutations in the Netherlands. Clin Genet 2000;57:116–24.
Lombardi MP, Redeker EJW, Van Gent DHM, Smeele KL, Weerdesteijn R, Mannens MMAM. Molecular genetic testing for familial hypercholesterolemia in the Netherlands: a stepwise screening strategy enhances the mutation detection rate. Genet Test 2006;10:77–84.
Mabuchi H, Nohara A, Noguchi T, Kobayashi J, Kawashiri M-A, Tada H, et al. Usefulness of DNA analysis for the diagnosis of familial hypercholesterolemia (FH) and extraordinarily high frequency of FH in Japan. Atheroscler Suppl 2010;11:115.
Marduel M, Carrie A, Sassolas A, Devillers M, Carreau V, Di Filippo M, et al. Molecular spectrum of autosomal dominant hypercholesterolemia in France. Hum Mutat 2010;31:E1811–24.
Marteau T, Senior V, Humphries SE, Bobrow M, Cranston T, Crook MA, et al. Psychological impact of genetic testing for familial hyperecholesterolemia within a previously aware population: a randomized controlled trial. Am J Med Genet 2004;128A:285–93.
Mata P, Alonso R, Castillo S, Pocovi M. MEDPED and the Spanish familial hypercholesterolemia foundation. Atheroscler Suppl 2002;2:9–11.
Medeiros AM, Alves AC, Francisco V, Bourbon M. Update of the Portuguese Familial Hypercholesterolaemia Study. Atherosclerosis 2010;212:553–8.
Miltiadous G, Cariolou MA, Elisaf M. Familial hypercholesterolaemia: taking advantage of a founder effect for early diagnosis and treatment. Community Genet 2001;4:123–4.
Miltiadous G, Xenophontos S, Demetriou N, Bairaktari E, Seferiadis K, Cariolou M, et al. Familial hypercholesterolaemia in north-western Greece. Hellenic J Cardiol 2004;45:299–304.
Mozas P, Cenarro A, Civeira F, Castillo S, Ros E, Pocovi M. Mutation analysis in 36 unrelated Spanish subjects with familial hypercholesterolemia: identification of 3 novel mutations in the LDL receptor gene. Hum Mutat 2000;15:483–4.
Mozas P, Castillo S, Tejedor D, Reyes G, Alonso R, Franco M, et al. Molecular characterization of familial hypercholesterolemia in Spain: identification of 39 novel and 77 recurrent mutations in LDLR. Hum Mutat 2004;24:187.
Nauck MS, Koster W, Dorfer K, Eckes J, Scharnagl H, Gierens H, et al. Identification of recurrent and novel mutations in the LDL receptor gene in German patients with familial hypercholesterolemia. Hum Mutat 2001;18:165–6.
Nicholls DP, Cather M, Byrne C, Graham CA, Young IS. Diagnosis of heterozygous familial hypercholesterolaemia in children. Int J Clin Pract 2008;62:990–4.
Plewa R, Luczak M, Bolewski A, Wierzchowiecki J, Siminiak T. Detection of the low-density lipoprotein receptor and apolipoprotein B gene mutation of patients with familial hypercholesterolaemia using real time polymerase chain reaction and SYBER Green chemistry. Eur Heart J 2003;24:459.
Plewa R, Luczak M, Burchardt P, Bolewski A, Wierzchowiecki J, Siminiak T. Monogenic hypercholesterolaemias – an evaluation of apolipoprotein B100 and LDL receptor gene polymorphisms. Kardiol Pol 2006;64:127–33.
Pocsai Z, Paragh G, Adany R. Multiplex PCR assay for screening deletions in the low density lipoprotein receptor gene. Clin Chim Acta 2001;309:7–12.
Punzalan FE, Sy RG, Santos RS, Cutiongco EM, Gosiengfiao S, Fadriguilan E, et al. Low density lipoprotein-receptor (LDL-R) gene mutations among Filipinos with familial hypercholesterolemia. J Atheroscler Thromb 2005;12:276–83.
Romano M, Di Taranto MD, D’Agostino MN, Marotta G, Gentile M, Abate G, et al. Identification and functional characterization of LDLR mutations in familial hypercholesterolemia patients from Southern Italy. Atherosclerosis 2010;210:493–6.
Salazar LA, Hirata MH, Hirata RDC. Increasing the sensitivity of single-strand conformation polymorphism analysis of the LDLR gene mutations in Brazilian patients with familial hypercholesterolemia. Clin Chem Lab Med 2002;40:441–5.
Shin JA, Kim SH, Kim UK, Chae JJ, Choe SJ, Namkoong Y, et al. Identification of four novel mutations of the low-density lipoprotein receptor gene in Korean patients with familial hypercholesterolemia. Clin Genet 2000;57:225–9.
Sozen M, Whittall R, Humphries SE. Mutation detection in patients with familial hypercholesterolaemia using heteroduplex and single strand conformation polymorphism analysis by capillary electrophoresis. Atheroscler Suppl 2004;5:7–11.
Sozen MM, Whittall R, Oner C, Tokatli A, Kalkanoglu HS, Dursun A, et al. The molecular basis of familial hypercholesterolaemia in Turkish patients. Atherosclerosis 2005;180:63–71.
Sozen M, Whittall R, Webb MBT, Norbury G, Tabrah S, Taylor A, et al. An arms-based diagnostic kit for genetic testing for familial hypercholesterolemia in the UK. Atheroscler Suppl 2006;7:129.
Stef M, Palacios L, Viegas M, Tejedor D, Martinez A, Pocovi M, et al. A unique tool to detect LDLr, APOB and PCSK9 point mutations as well as copy number change in the LDLr gene. Atheroscler Suppl 2009;10:e273.
Stoll M, Esperon P, Raggio V, Lorenzo M. Molecular basis of familial hypercholesterolemia in Uruguay. Circulation 2008;118:E398.
Tabrah S, Tabrah S, Taylor A, Humphries SE, Norbury G. A strategy for molecular genetic mutation screening for familial hypercholesterolaemia. J Med Genet 2005;42:S110.
Takada D, Emi M, Ezura Y, Nobe Y, Kawamura K, Iino Y, et al. Interaction between the LDL-receptor gene bearing a novel mutation and a variant in the apolipoprotein A-II promoter: molecular study in a 1135-member familial hypercholesterolemia kindred. J Hum Genet 2002;47:656–64.
Tarantino E, Mabille S, Bruniaux M, Descamps O. Molecular study of familial hypercholesterolemia in Belgium. Atheroscler Suppl 2010;11:117.
Ten Asbroek AHA, Marang-van de Mheen P, Defesche JC, Kastelein JJP, Gunning-Schepers LJ. Results from a family and DNA based active identification programme for familial hypercholesterolaemia. J Epidemiol Community Health 2001;55:500–2.
Tennant S, Bell C, Brown C, Kelly K, Hailey H, Gregory H, et al. Familial hypercholesterolaemia testing in Scotland. British Society for Human Genetics, Warwick University, September 2010.
Thiart R, Scholtz CL, Vergotine J, Hoogendijk CF, de Villiers JN, Nissen H et al. Predominance of a 6 bp deletion in exon 2 of the LDL receptor gene in Africans with familial hypercholesterolaemia. J Med Genet 2000;37:514-9.
Thiart R, Varret M, Lintott CJ, Scott RS, Loubser O, Du PL, et al. Mutation analysis in a small cohort of New Zealand patients originating from the United Kingdom demonstrates genetic heterogeneity in familial hypercholesterolemia. Mol Cell Probes 2000;14:299–304.
Tosi I, Toledo-Leiva P, Neuwirth C, Naoumova RP, Soutar AK. Genetic defects causing familial hypercholesterolaemia: identification of deletions and duplications in the LDL-receptor gene and summary of all mutations found in patients attending the Hammersmith Hospital Lipid Clinic. Atherosclerosis 2007;194:102–11.
Tybjaerg-Hansen A, Jensen HK, Benn M, Steffensen R, Jensen G, Nordestgaard B. Phenotype of heterozygotes for low-density lipoprotein receptor mutations identified in different background populations. Arterioscler Thromb Vasc Biol 2005;25:211–15.
Umans-Eckenhausen MA, Defesche JC, Sijbrands EJ, Scheerder RL, Kastelein JJ. A nation-wide screening program for familial hypercholesterolemia in the Netherlands: five years experience. Circulation 2000;102:II-873.
Umans-Eckenhausen MAW, Sijbrands EJG, Kastelein JJP, Defesche JC. Low-density lipoprotein receptor gene mutations and cardiovascular risk in a large genetic cascade screening population. Circulation 2002;106:3031–6.
Vaca G, Vzquez A, Magaa MT, Ramrez ML, Dvalos IP, Marn B, et al. Study on the molecular basis of familial hypercholesterolemia in Mexico. Advances of project. Atheroscler Suppl 2010;11:124.
Van Aalst-Cohen ES, Jansen ACM, Tanck MWT, Defesche JC, Trip MD, Lansberg PJ, et al. Diagnosing familial hypercholesterolaemia: the relevance of genetic testing. Eur Heart J 2006;27:2240–6.
Van der Graaf A, Avis H, Vissers M, Hutten B, Defesche J, Wiegman A, et al. Phenotype and genotype of familial hypercholesterolemia in over 2000 children. Atheroscler Suppl 2009;10:e202.
Van der Graaf A, Avis HJ, Kusters DM, Vissers MN, Hutten BA, Defesche JC, et al. Molecular basis of autosomal dominant hypercholesterolemia: assessment in a large cohort of hypercholesterolemic children. Circulation 2011;123:1167–73.
Vergotine J, Thiart R, Kotze MJ. Clinical versus molecular diagnosis of heterozygous familial hypercholesterolaemia in the diverse South African population. S Afr Med J 2001;91:1053–9.
Vergotine J, Thiart R, Langenhoven E, Hillermann R, De Jong G, Kotze MJ. Prenatal diagnosis of familial hypercholesterolemia: importance of DNA analysis in the high-risk South African population. Genet Counsel 2001;12:121–7.
Vladimirova-Kitova LG, Deneva TI, Angelova E, Bichev AN, Nikolov FP. Role of some biomarkers of atherogenic risk in the screening for molecular defects in the low density lipoprotein receptor in severe hypercholesterolemia. Folia Med (Plovdiv) 2008;50:14–22.
Voevoda MI, Kulikov IV, Shakhtshneider EV, Maksimov VN, Pilipenko IV, Tereschenko IP, et al. The spectrum of mutations in the low-density lipoprotein receptor gene in the Russian population. Russ J Genet 2008;44:1191–4.
Vohnout B, Raslova K, Gasparovic J, Franekova J, Fabryova L, Belosovicova M, et al. Lipid levels and their genetic regulation in patients with familial hypercholesterolemia and familial defective apolipoprotein B-100: the MEDPED Slovakia Project. Atheroscler Suppl 2003;4:3–5.
Wang J, Ban MR, Hegele RA. Multiplex ligation-dependent probe amplification of LDLR enhances molecular diagnosis of familial hypercholesterolemia. J Lipid Res 2005;46:366–72.
Weiss N, Binder G, Keller C. Mutations in the low-density-lipoprotein receptor gene in German patients with familial hypercholesterolaemia. J Inherit Metab Dis 2000;23:778–90.
Yamwong P, Pongrapeeporn KU, Thepsuriyanont P, Amornrattana A, Somkasettrin A, Sribhen K. Mutation analysis of exon 9 of the LDL receptor gene in Thai subjects with primary hypercholesterolemia. J Med Assoc Thai 2000;83:S81–8.
Yu W, Nohara A, Higashikata T, Lu H, Inazu A, Mabuchi H. Molecular genetic analysis of familial hypercholesterolemia: spectrum and regional difference of LDL receptor gene mutations in Japanese population. Atherosclerosis 2002;165:335–42.
Zakharova FM, Damgaard D, Mandelshtam MY, Golubkov VI, Nissen PH, Nilsen GG, et al. Familial hypercholesterolemia in St.-Petersburg: the known and novel mutations found in the low density lipoprotein receptor gene in Russia. BMC Med Genet 2005;6:6.
Zrinski TR, Ferencak G, Sucic M, Grskovic B, Stavljenic Rukavina A. Detection of LDL-receptor gene mutations causing familial hypercholesterolemia in Croatia. Clin Chem Lab Med 2001;39:S318.
Zrinski TR, Gornik O, FloGel M, Sertic J, Stavljenic RA. Low density lipoprotein receptor gene mutations in Croatian hypercholesterolemic patients. Period Biol 2004;106:295–9.
Low-density lipoprotein cholesterol cut-off values or clinical diagnosis not reported
Descamps OS, Gilbeau JP, Luwaert R, Heller FR. Impact of genetic defects on coronary atherosclerosis in patients suspected of having familial hypercholesterolaemia. Eur J Clin Invest 2003;33:1–9.
Garcia-Otin AL, Cofan M, Junyent M, Recalde D, Cenarro A, Pocovi M, et al. Increased intestinal cholesterol absorption in autosomal dominant hypercholesterolemia and no mutations in the low-density lipoprotein receptor or apolipoprotein B genes. J Clin Endocrinol Metab 2007;92:3667–73.
Real JT, Chaves FJ, Ejarque I, Garia-Garcia AB, Valldecabres C, Ascaso JF, et al. Influence of LDL receptor gene mutations and the R3500Q mutation of the apoB gene on lipoprotein phenotype of familial hypercholesterolemic patients from a South European population. Eur J Hum Genet 2003;11:959–65.
Robles-Osorio L, Ordonez M, Aguilar-Salinas CA, Auron-Gomez M, Tusie-Luna M, Gomez-Perez FJ, et al. Familial hypercholesterolemia due to ligand-defective apolipoprotein B100. First case report in a Mexican family. Arch Med Res 2003;34:70–5.
Salazar LA, Hirata MH, Cavalli SA, Nakandakare ER, Forti N, Diament J, et al. Molecular basis of familial hypercholesterolemia in Brazil: identification of seven novel LDLR gene mutations. Hum Mutat 2002;19:462–3.
Thorsson B, Sigurdsson G, Gudnason V. Systematic family screening for familial hypercholesterolemia in Iceland. Arterioscler Thromb Vasc Biol 2003;23:335–8.
Tounian P, Briffaut D, Benlian P, Girardet J. Molecular-based diagnostic criteria of familial hypercholesterolaemia in children with dominantly-inherited hypercholesterolaemia. Clin Nutr (Edinburgh) 2000;19:48–9.
Yang K-C, Su Y-N, Shew J-Y, Yang K-Y, Tseng W-K, Wu C-C, et al. LDLR and ApoB are major genetic causes of autosomal dominant hypercholesterolemia in a Taiwanese population. J Formos Med Assoc 2007;106:799–807.
Letter/editorial
Graesdal A, Ostli L, Arnesen K-E. Familial hypercholesterolemia in Norway – substantial variation in frequency of genetic testing and degree of follow up in different geographical regions. Atheroscler Suppl 2009;10:e1230.
Hopcroft KA. Child–parent screening may have adverse psychological effects. BMJ 2007;335:683.
Kotze MJ. High specificity makes DNA screening the method of choice for diagnosis of familial hypercholesterolaemia. S Afr Med J 2001;91:1042.
Kwiterovich J. Clinical implications of the molecular basis of familial hypercholesterolemia and other inherited dyslipidemias. Circulation 2011;123:1153–5.
Taylor A, Patel K, Tsedeke J, Humphries SE, Norbury G. Mutation screening in patients for familial hypercholesterolaemia (ADH). Clin Genet 2010;77:97–9.
Tejedor D, Castillo S, Mozas P, Jimenez E, Lopez M, Tejedor MT, et al. Comparison of DNA array platform vs DNA sequencing as genetic diagnosis tools for familial hypercholesterolemia. Clin Chem 2006;52:1971–2.
No relevant test of interest
Azian M, Hapizah MN, Khalid BA, Khalid Y, Rosli A, Jamal R. Use of the denaturing gradient gel electrophoresis (DGGE) method for mutational screening of patients with familial hypercholesterolaemia (FH) and familial defective apolipoprotein B100 (FDB). Malays J Pathol 2006;28:7–15.
Bodamer OA, Bercovich D, Schlabach M, Ballantyne C, Zoch D, Beaudet AL. Use of denaturing HPLC to provide efficient detection of mutations causing familial hypercholesterolemia. Clin Chem 2002;48:1913–18.
Fard-Esfahani P, Khatami S, Zeinali C, Taghikhani M, Allahyari M. A modified conformation sensitive gel electrophoresis (CSGE) method for rapid and accurate detection of low density lipoprotein (LDL) receptor gene mutations in familial hypercholesterolemia. Clin Biochem 2005;38:579–83.
Fouchier SW, Defesche JC, Kastelein JJP, Sijbrands EJG. Familial defective apolipoprotein B versus familial hypercholesterolemia: an assessment of risk. Semin Vasc Med 2004;4:259–64.
Graham C, Wright W, Heggarty SV, Byrne C, Young I, Nicholls D. iPLEX mass array, MLPA and high throughput sequencing provide a comprehensive mutation detection service for familial hypercholesterolaemia. J Med Genet 2007;44:S88.
Hovland A, Hardersen R, Nielsen EW, Mollnes TE, Lappegard KT. Hematologic and hemostatic changes induced by different columns during LDL apheresis. J Clin Apheresis 2010;25:294–300.
Laurie AD, George PM. Evaluation of high-resolution melting analysis for screening the LDL receptor gene. Clin Biochem 2009;42:528–35.
Liguori R, Bianco AM, Argiriou A, Pauciullo P, Giannino A, Rubba P, et al. LDL receptor cDNA sequence analysis in familial hypercholesterolemia patients: 5 novel mutations with high prevalence in families originating from southern Italy. Hum Mutat 2001;17:433.
Liguori R, Argiriou A, Simone VD. A rapid method for detecting mutations of the human LDL receptor gene by complete cDNA sequencing. Mol Cell Probes 2003;17:15–20.
Whittall RA, Scartezini M, Li K, Hubbart C, Reiner Z, Abraha A, et al. Development of a high-resolution melting method for mutation detection in familial hypercholesterolaemia patients. Ann Clin Biochem 2010;47:44–55.
Wiesbauer F, Blessberger H, Azar D, Goliasch G, Wagner O, Gerhold L, et al. Familial-combined hyperlipidaemia in very young myocardial infarction survivors (< or = 40 years of age). Eur Heart J 2009;30:1073–9.
Non-English-language
Andalusian Agency for Health Technology Assessment. Molecular genetics diagnosis for familial hypercholesterolemia using LIPOCHIP – review (brief record). Sevilla: Andalusian Agency for Health Technology Assessment; 2004. URL: www.juntadeandalucia.es/salud/servicios/contenidos/nuevaaetsa/up/AETSA_P_2004_10_Lipochip.pdf (accessed January 2011).
Not familial hypercholesterolaemia
Civeira F, Jarauta E, Cenarro A, Garcia-Otin AL, Tejedor D, Zambon D, et al. Frequency of low-density lipoprotein receptor gene mutations in patients with a clinical diagnosis of familial combined hyperlipidemia in a clinical setting. J Am Coll Cardiol 2008;52:1546–53.
Glynou K, Laios E, Drogari E, Tsaoussis V. Development of a universal chemiluminometric genotyping method for high-throughput detection of 7 LDLR gene mutations in Greek population. Clin Biochem 2008;41:335–42.
Reviews/guidelines
Adelaide Health Technology Assessment. Horizon scanning technology prioritising summary: LIPOchip DNA microarray for the detection of familial hypercholesterolaemia. Adelaide: Australia and New Zealand Horizon Scanning Network; 2009. URL: www.horizonscanning.gov.au (accessed 12 January 2011).
Datta BN, McDowell IFW, Rees A. Integrating provision of specialist lipid services with cascade testing for familial hypercholesterolaemia. Curr Opin Lipidol 2010;21:366–71.
Goldberg AC, Hopkins PN, Toth PP, Ballantyne C, Rader DJ, Robinson JG, et al. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol 2011;5:133–40.
Hegele RA. Genetic susceptibility to heart disease in Canada: lessons from patients with familial hypercholesterolemia. Genome 2006;49:1343–50.
Lebenthal Y, Horvath A, Dziechciarz P, Szajewska H, Shamir R. Are treatment targets for hypercholesterolemia evidence based? Systematic review and meta-analysis of randomised controlled trials. Arch Dis Child 2010;95:673–80.
Leren TP, Berge KE. Comparison of clinical and molecular genetic criteria for diagnosing familial hypercholesterolemia. Clin Lipidol 2009;4:303–10.
Wald DS, Bestwick JP, Wald NJ. Child–parent screening for familial hypercholesterolaemia: screening strategy based on a meta-analysis. BMJ 2007;335:599–603.
Test unclear, no response from author
Alonso R, Villar J, Fuentes F, Zambon D, Mata P. Genetic diagnosis of familial hypercholesterolemia (FH) and LDL-cholesterol goal achievement. Atheroscler Suppl 2009;10:e272.
Farrer JM, Nair D, Norbury G, Taylor A, Humphries SE. Does DNA information complement lipid measures in cascade testing in familial hypercholesterolaemia patients? Atherosclerosis 2005;182:S7.
Poke S, Watts G, Maxwell S, Brameld K, O’Leary P. Familial hypercholesterolaemia (FH) pilot cascade screening project. Twin Res Hum Genet 2009;12:229.
Total cholesterol not low-density lipoprotein cholesterol used as a test
Benlian P, Turquet A, Carrat F, Amsellem S, Sanchez L, Briffaut D, et al. Diagnosis scoring for clinical identification of children with heterozygous familial hypercholesterolemia. J Pediatr Gastroenterol Nutr 2009;48:456–63.
Chater R, Ait CK, Rabes JP, Varret M, Chabraoui L, El JY, et al. Mutational heterogeneity in low-density lipoprotein receptor gene related to familial hypercholesterolemia in Morocco. Clin Chim Acta 2006;373:62–9.
Freiberger T, Ravcukova B, Plotena M, Fajkusov L. Sensitivity and specificity of cholesterol measurements in familial hypercholesterolemia diagnosis in Czech population. Atheroscler Suppl 2010;11:194.
Leren TP, Finborud TH, Manshaus TE, Ose L, Berge KE. Diagnosis of familial hypercholesterolemia in general practice using clinical diagnostic criteria or genetic testing as part of cascade genetic screening. Community Genet 2008;11:26–35.
Marks D, Thorogood M, Neil SM, Humphries SE, Neil HAW. Cascade screening for familial hypercholesterolaemia: implications of a pilot study for national screening programmes. J Med Screen 2006;13:156–9.
Umans-Eckenhausen MA, Defesche JC, Sijbrands EJ, Scheerder RL, Kastelein JJ. Review of first 5 years of screening for familial hypercholesterolaemia in the Netherlands. Lancet 2001;357:165–8.
Unavailable papers
Defesche JC, Lansberg PJ, Umans-Eckenhausen MAW, Kastelein JJP. Advanced method for the identification of patients with inherited hypercholesterolemia. Semin Vasc Med 2004;4:59–65.
Haralambos K, Townsend D, Williams B, Rees A, Williams J, Diaz J, et al. Development of register and website resources to support a familial hypercholesterolaemia (FH) family cascade testing programme. J Med Genet 2008;45:S109.
Leren TP, Manshaus T, Skovholt U, Skodje T, Nossen IE, Teie C, et al. Application of molecular genetics for diagnosing familial hypercholesterolemia in Norway: results from a family-based screening program. Semin Vasc Med 2004;4:75–85.
Marteau TM, Senior V, Humphries S. Impact on perceived control and risk-reducing behaviour of genetic testing for familial hypercholesterolaemia (FH): a randomised controlled trial. Eur J Hum Genet 2002;10:306–7.
Martin B, Wang D, Taylor A, Humphries S, Norbury G. MLPA analysis to detect large deletions and duplications in familial hypercholesterolaemia. J Med Genet 2007;44:S81.
Miller J. Familial hypercholesterolaemia cascade testing: a sociological study. J Med Genet 2010;47:S101.
Muir LA, George PM, Laurie AD, Reid N, Whitehead L. Preventing cardiovascular disease: a review of the effectiveness of identifying the people with familial hypercholesterolaemia in New Zealand. N Z Med J 2010;123:97–102.
Ose L. Diagnostic, clinical, and therapeutic aspects of familial hypercholesterolemia in children. Semin Vasc Med 2004;4:51–7.
Pocovi M, Civeira F, Alonso R, Mata P. Familial hypercholesterolemia in Spain: case-finding program, clinical and genetic aspects. Semin Vasc Med 2004;4:67–74.
van Maarle M, Stouthard M, Klazinga N, Bonsel G. Risk perception of participants in a family based genetic screening programme on familial hypercholesterolemia. Eur J Hum Genet 2001;9:1058.
Vergopoulos A, Knoblauch H, Schuster H. DNA testing for familial hypercholesterolemia: improving disease recognition and patient care. Am J Pharmacogenomics 2002;2:253–62.
Background
Adelaide Health Technology Assessment. Horizon scanning technology – horizon scanning report: genetic screening for familial hypercholesterolaemia. Adelaide: Australia and New Zealand Horizon Scanning Network; 2007. URL: www.horizonscanning.gov.au (accessed 12 January 2011).
Ara R, Tumur I, Pandor A, Duenas A, Williams R, Wilkinson A, et al. Ezetimibe for the treatment of hypercholesterolaemia: a systematic review and economic evaluation. Health Technol Assess 2008;12(21).
Arambepola C, Farmer A, Perera R, Neil HAW. Statin treatment for children and adolescents with heterozygous familial hypercholesterolaemia: a systematic review and meta-analysis. Atherosclerosis 2007;195:339–47.
Austin MA, Hutter CM, Zimmern RL, Humphries SE. Genetic causes of monogenic heterozygous familial hypercholesterolemia: a HuGE prevalence review. Am J Epidemiol 2004;160:407–20.
Avis HJ, Vissers MN, Stein EA, Wijburg FA, Trip MD, Kastelein JJP, et al. A systematic review and meta-analysis of statin therapy in children with familial hypercholesterolemia. Arterioscler Thromb Vasc Biol 2007;27:1803–10.
Bhatnagar D. Diagnosis and screening for familial hypercholesterolaemia: finding the patients, finding the genes. Ann Clin Biochem 2006;43:441–56.
Catapano A, Brady WE, King TR, Palmisano J. Lipid altering-efficacy of ezetimibe co-administered with simvastatin compared with rosuvastatin: a meta-analysis of pooled data from 14 clinical trials. Curr Med Res Opin 2005;21:1123–30.
Civeira F, Pocovi M, Alegria E, Alonso R, Carmena R, Casasnovas JA, et al. Guidelines for the diagnosis and management of heterozygous familial hypercholesterolemia. Atherosclerosis 2004;173:55–68.
Civeira F, Castillo S, Alonso R, Merino-Ibarra E, Cenarro A, Artied M, et al. Tendon xanthomas in familial hypercholesterolemia are associated with cardiovascular risk independently of the low-density lipoprotein receptor gene mutation. Arterioscler Thromb Vasc Biol 2005;25:1960–5.
Finnie RM. Cascade screening for familial hypercholesterolaemia in Scotland. Br J Diabet Vasc Dis 2010;10:123–5.
Gray J, Jaiyeola A, Whiting M, Modell M, Wierzbicki AS. Identifying patients with familial hypercholesterolaemia in primary care: an informatics-based approach in one primary care centre. Heart 2008;94:754–8.
Hadfield SG, Humphries SE. Implementation of cascade testing for the detection of familial hypercholesterolaemia. Curr Opin Lipidol 2005;16:428–33.
Herman K, Van HC, Wile D. Cascade screening for familial hypercholesterolaemia and its effectiveness in the prevention of vascular disease. Br J Diabet Vasc Dis 2009;9:171–4.
Homsma SJM, Huijgen R, Middeldorp S, Sijbrands EJG, Kastelein JJP. Molecular screening for familial hypercholesterolaemia: consequences for life and disability insurance. Eur J Hum Genet 2008;16:14–17.
Hopcroft KA. Familial hypercholesterolaemia: child–parent screening may have adverse psychological effects. BMJ 2007;335:683.
Humphries SE, Norbury G, Leigh S, Hadfield SG, Nair D. What is the clinical utility of DNA testing in patients with familial hypercholesterolaemia? Curr Opin Lipidol 2008;19:362–8.
Humphries SE, Neil HAW. Developing and applying clinically useful approaches to identify individuals with familial hypercholesterolemia in the UK. Clin Lipidol 2010;5:497–507.
Huxley RR, Hawkins MH, Humphries SE, Karpe F, Neil HAW. Risk of fatal stroke in patients with treated familial hypercholesterolemia: a prospective registry study. Stroke 2003;34:22–5.
Jansen ACM, Van Aalst-Cohen ES, Tanck MW, Trip MD, Lansberg PJ, Liem AH, et al. The contribution of classical risk factors to cardiovascular disease in familial hypercholesterolaemia: data in 2400 patients. J Intern Med 2004;256:482–90.
Jensen HK. The molecular genetic basis and diagnosis of familial hypercholesterolemia in Denmark. Dan Med Bull 2002;49:318–45.
Leren TP. Cascade genetic screening for familial hypercholesterolemia. Clin Genet 2004;66:483–7.
Marang-van de Mheen PJ, Van Maarle MC, Stouthard MEA. Getting insurance after genetic screening on familial hypercholesterolaemia; the need to educate both insurers and the public to increase adherence to national guidelines in the Netherlands. J Epidemiol Community Health 2002;56:145–7.
Marks D, Wonderling D, Thorogood M, Lambert H, Humphries SE, Neil HA. Screening for hypercholesterolaemia versus case finding for familial hypercholesterolaemia: a systematic review and cost-effectiveness analysis. Health Technol Assess 2000;4(29).
Marks D, Thorogood M, Neil AW, Wonderling D, Humphries SE. Comparing costs and benefits over a 10 year period of strategies for familial hypercholesterolaemia screening. J Public Health Med 2003;25:47–52.
Marks D, Thorogood M, Neil HAW, Humphries SE. A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemia. Atherosclerosis 2003;168:1–14.
Marks D, Thorogood M, Farrer JM, Humphries SE. Census of clinics providing specialist lipid services in the United Kingdom. J Public Health 2004;26:353–4.
Maxwell S, Molster C, Poke S, O’Leary P. Community views on the communication of FH genetic information within families. Twin Res Hum Genet 2009;12:223.
Maxwell SJ, Molster CM, Poke SJ, O’Leary P. Communicating familial hypercholesterolemia genetic information within families. Genet Test Mol Biomarkers 2009;13:301–6.
McGowan M. NLA Symposium screening and treatment of familial hypercholesterolemia: how can we do better? Opening and introductions. J Clin Lipidol 2010;4:335–7.
Morrell J. Familial hypercholesterolaemia: recognising the unrecognised. Br J Cardiol 2008;15:79–81.
Moruisi KG, Oosthuizen W, Opperman AM. Phytosterols/stanols lower cholesterol concentrations in familial hypercholesterolemic subjects: a systematic review with meta-analysis. J Am Coll Nutr 2006;25:41–8.
Muller PY, Miserez AR. Large heterogeneity of mutations in the gene encoding the low-density lipoprotein receptor in subjects with familial hypercholesterolaemia. Atheroscler Suppl 2004;5:1–5.
Neil HA, Huxley RR, Hawkins MM, Durrington PN, Betteridge DJ, Humphries SE, et al. Comparison of the risk of fatal coronary heart disease in treated xanthomatous and non-xanthomatous heterozygous familial hypercholesterolaemia: a prospective registry study. Atherosclerosis 2003;170:73–8.
Newson AJ, Humphries SE. Cascade testing in familial hypercholesterolaemia: how should family members be contacted? Eur J Hum Genet 2005;13:401–8.
O’Gorman CS, Higgins MF, O’Neill MB. Systematic review and metaanalysis of statins for heterozygous familial hypercholesterolemia in children: evaluation of cholesterol changes and side effects. Pediatr Cardiol 2009;30:482–9.
Oosterveer DM, Versmissen J, Yazdanpanah M, Hamza TH, Sijbrands EJG. Differences in characteristics and risk of cardiovascular disease in familial hypercholesterolemia patients with and without tendon xanthomas: a systematic review and meta-analysis. Atherosclerosis 2009;207:311–17.
Shafiq N, Bhasin B, Pattanaik S, Pandhi P, Venkateshan SP, Singh M, et al. A meta-analysis to evaluate the efficacy of statins in children with familial hypercholesterolemia.[Erratum appears in Int J Clin Pharmacol Ther 2008;46:41.] Int J Clin Pharmacol Ther 2007;45:548–55.
Shafiq N, Singh M, Kaur S, Khosla P, Malhotra S. Dietary treatment for familial hypercholesterolaemia. Cochrane Database Syst Rev 2010; Issue 1, Art No. CD001918. DOI: 10.1002/14651858.CD001918.pub2.
van den Nieuwenhoff HW, Mesters I, Nellissen JJ, Stalenhoef AF, de Vries NK. The importance of written information packages in support of case-finding within families at risk for inherited high cholesterol. J Genet Couns 2006;15:29–40.
Van Maarle MC, Stouthard MEA, Mheen PJM, Klazinga NS, Bonsel GJ. How disturbing is it to be approached for a genetic cascade screening programme for familial hypercholesterolaemia?: psychological impact and screenees’ views. Community Genet 2001;4:244–52.
Van Maarle MC, Stouthard MEA, Bonsel GJ. Risk perception of participants in a family-based genetic screening program on familial hypercholesterolemia. Am J Med Genet 2003;116A:136–43.
Vuorio A, Kuoppala J, Kovanen P. Statins for children with familial hypercholesterolemia. Cochrane Database Syst Rev 2010; Issue 7, Art No. CD006401. DOI: 10.1002/14651858.CD006401.pub2.
Warner M. Child–parent screening for familial hypercholesterolaemia: screening strategy based on a meta-analysis. Ann Clin Biochem 2008;45:114.
Watts GF, Van Bockxmeer FM, Bates T, Burnett JR, Juniper A, O’Leary P. A new model of care for familial hypercholesterolaemia from Western Australia: closing a major gap in preventive cardiology. Heart Lung Circ 2010;19:419–22.
Weiner K, Durrington PN. Patients’ understandings and experiences of familial hypercholesterolemia. Community Genet 2008;11:273–82.
Will CM, Armstrong D, Marteau TM. Genetic unexceptionalism: clinician accounts of genetic testing for familial hypercholesterolaemia. Soc Sci Med 2010;71:910–17.
Wright WT, Heggarty SV, Young IS, Nicholls DP, Whittall R, Humphries SE, et al. Multiplex MassARRAY spectrometry (iPLEX) produces a fast and economical test for 56 familial hypercholesterolaemia-causing mutations. Clin Genet 2008;74:463–8.
Appendix 8 Characteristics of the included studies
| Study details | Participants | Test characteristics | Outcomes reported | Comments | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Alonso 200939 Study design: cross-sectional comparative Publication type: full text Other related reports: none Number of study centres: 11 Spanish regions Setting: NR Country: Spain Recruitment date: screening started in 2004 Patients recruited consecutively, Y/N: NR Source of funding: NR |
Inclusion criteria: unrelated cases with clinical diagnosis of FH after the affiliation of the Dutch Lipid Clinic Network criteria with a score of ≥ 6 points Exclusion criteria: NR Clinical diagnosis of index cases: Dutch criteria Participants: all index cases FH diagnosis: definite FH or probable FH Index casesEnrolled, n808Analysed, n808 |
Index cases | Enrolled, n | 808 | Analysed, n | 808 |
Test 1: LIPOchip platform, which comprises: Stage 1 (index test): DNA array that can detect 191 different point mutations in LDLR and four different mutations in APOB (no PCSK9 analysis) Stage 2 (part of CGA): adapted QMFSP for the analysis of large deletions or insertions Stage 3 (part of CGA): sequencing of LDLR gene using MegaBace 1000 apparatus Stage 1Stage 2Stage 3Received test, n808389312 Reproducibility evaluation: Blind analysis of FH samples from Spain and the Netherlands was performed. Dutch FH samples (LDLR point mutations) analysed with DNA array = 53; Spanish FH samples (positive for large deletions and insertions) analysed with MLPA in Dutch laboratory = 43 (not all received CGA) |
Stage 1 | Stage 2 | Stage 3 | Received test, n | 808 | 389 | 312 |
Diagnostic accuracy (sensitivity and specificity) of the DNA array Reproducibility Number diagnosed with positive test for each stage of test and total test Time taken to complete the LIPOchip platform and to obtain each test result |
Sensitivity of the DNA array: determined by the number of mutations detected by sequencing LDLR in the samples in which the DNA array failed to detect mutations Specificity of the DNA array: 125 different DNA array-positive samples were randomly selected and sequenced automatically |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Index cases | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Enrolled, n | 808 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Analysed, n | 808 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Stage 1 | Stage 2 | Stage 3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Received test, n | 808 | 389 | 312 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Callaway 201040 Study design: cross-sectional comparative Publication type: presentation plus information from author Other related reports: none Number of study centres: 1 Setting: NR Country: UK Recruitment date: NR Patients recruited consecutively, Y/N: NR Source of funding: NR |
Inclusion criteria: samples selected for validation were 10 normal control, 6 Elucigene FH20 positive control, 22 Elucigene FH20 negative control subjects Exclusion criteria: NR Clinical diagnosis of index cases: definite FH on referral card; or high cholesterol level (> 8 mmol/l) plus either (1) family history of high cholesterol or (2) family history of cardiovascular disease Participants: NR FH diagnosis: definite FH or probable FH PatientsEnrolled, n22 |
Patients | Enrolled, n | 22 |
Test 1: LIPOchip platform consisting of two stages: Stage 1 (index test): LIPOchip version 8 detects point mutations in the LDLR, APOB and PCSK9 genes and copy number changes in the LDLR gene. Includes 251 mutations most prevalent in Spain, the Netherlands, Norway, Italy and the UK Stage 2 (component of CGA): sequencing of LDLR if no mutation was detected by LIPOchip Test 2: Elucigene FH20 (included APOB and PCSK9)/dHPLC/ sequencing of LDLR gene/MLPA Test 1Test 2Received test, n2222 |
Test 1 | Test 2 | Received test, n | 22 | 22 | Diagnostic accuracy (sensitivity and specificity) of LIPOchip | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Patients | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Enrolled, n | 22 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Test 1 | Test 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Received test, n | 22 | 22 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Civeira 200844 Study design: cross-sectional comparative, retrospective Publication type: full text Other related reports: Tejedor 2005,43 Tejedor 200656 (for LIPOchip test) Number of study centres: 3 Setting: lipid clinics Country: Spain Study dates: May 2004–November 2007 Patients recruited consecutively, Y/N: Y Source of funding: NR |
Inclusion criteria: patients aged ≥ 14 years, evaluated at three large clinics for clinical diagnosis and underwent the FH genetic diagnostic procedure using LIPOchip. Patients were retrospectively categorised into Simon Broome criteria, Dutch criteria and MedPed criteria Exclusion criteria: NR Participants: adults and adolescents, index cases Clinical diagnosis: MedPed criteria for initial diagnosis (by attending physicians), which includes very high TC or LDL-C levels, with or without tendon xanthomata, with or without familial or personal histories of premature CAD FH diagnosis: definite FH or possible FH Index casesEnrolled, n825Analysed, n825Age, yearsSex, M/F438/387CAD, n17Personal history of CAD, n105Xanthomata, n195 |
Index cases | Enrolled, n | 825 | Analysed, n | 825 | Age, years | Sex, M/F | 438/387 | CAD, n | 17 | Personal history of CAD, n | 105 | Xanthomata, n | 195 |
Test 1: LIPOchip platform (203 mutations in LDLR and 4 mutations in APOB) as follows: (1) DNA array (2) Quantitative fluorescence-based multiplex polymerase chain reaction for analysis of large gene rearrangements in samples with negative results from DNA array (3) sequencing of the promoter, all exons and the exon/intron boundaries of the LDLR gene for samples with negative results from above test Test was carried out in Spain Test 2: lipid measurements were determined using standard enzymatic methods. LDL-C concentration was estimated from a fasting blood sample using the Friedwald equation when serum triglyceride levels were < 400 mg/dl Test 1Test 2Enrolled, n825825Analysed, n825825 |
Test 1 | Test 2 | Enrolled, n | 825 | 825 | Analysed, n | 825 | 825 |
Diagnostic accuracy of Simon Broome, MedPed and Dutch criteria Detection rate of FH by LIPOchip platform |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Index cases | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Enrolled, n | 825 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Analysed, n | 825 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Age, years | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Sex, M/F | 438/387 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CAD, n | 17 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Personal history of CAD, n | 105 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xanthomata, n | 195 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Test 1 | Test 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Enrolled, n | 825 | 825 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Analysed, n | 825 | 825 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Damgaard 200545 Study design: cross-sectional comparative, retrospective Publication type: full text Other related reports: Starr 200849 Number of study centres: 1 Setting: lipid clinics Country: Denmark Study dates: January 1995–December 2003 Patients recruited consecutively, Y/N: Y Sources of funding: Danish Heart Foundation and Institute of Experimental Clinical Research at the University of Aarhus |
Inclusion criteria: patient referred for FH to the lipid clinic. Patient with any two of the following criteria: LDL-C cut-offs > 6 mmol/l, TC > 8 mmol/l, tendon xanthomata and history of CAD before the age of 60 years in the patient and/or in a first-degree relative and/or hypercholesterolaemia in a first-degree relative Exclusion criteria: secondary hypercholesterolaemia, renal failure, nephritic syndrome, liver disease, hypothyroidism and diabetes Participants: adults; index cases and relatives of index cases in whom a mutation was identified Clinical diagnosis: index patients were categorised retrospectively based on lipid measurements and clinical examination before genetic analysis into Simon Broome, Dutch and MedPed criteria FH diagnosis: definite FH, possible FH or probable FH FHNot FHEnrolled, nIndex cases408Relatives385Analysed, nIndex casesa133273Relatives203180Age, years, meanLDLR = 42.6 APOB = 45.552Sex, M, n140266CAD, n3461Tendon xanthomata, n4125a |
FH | Not FH | Enrolled, n | Index cases | 408 | Relatives | 385 | Analysed, n | Index casesa | 133 | 273 | Relatives | 203 | 180 | Age, years, mean | LDLR = 42.6 APOB = 45.5 | 52 | Sex, M, n | 140 | 266 | CAD, n | 34 | 61 | Tendon xanthomata, n | 41 | 25 | a |
Test 1: (comparator test): LDL-C concentration estimated from a fasting blood sample using Friedwald equation LDL-C as part of Simon Broome DNA sequence analysis Genes tested: LDLR (including deletions) and APOB Test 2: (reference standard): genetic test performed in following stages: (1) routine screening for three common mutations in the LDLR gene in Danish population (all) (2) SSCP for those with negative mutation (3) sequencing for those with negative mutation (69 patients) (4) analysis of APOB gene (all) (5) MLPA for those with negative mutation Test 3: targeted sequencing of relatives for the mutation found in the index patient of the family: Tests were performed in Denmark Test 1Test 2Test 3Analysed, nIndex cases408408Relatives385 |
Test 1 | Test 2 | Test 3 | Analysed, n | Index cases | 408 | 408 | Relatives | 385 | Diagnostic accuracy of LDL-C as part of Simon Broome and Dutch criteria and LDL-C age- and gender-specific MedPed criteria to diagnose FH | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FH | Not FH | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Enrolled, n | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Index cases | 408 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Relatives | 385 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Analysed, n | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Index casesa | 133 | 273 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Relatives | 203 | 180 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Age, years, mean | LDLR = 42.6 APOB = 45.5 | 52 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Sex, M, n | 140 | 266 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CAD, n | 34 | 61 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Tendon xanthomata, n | 41 | 25 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Test 1 | Test 2 | Test 3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Analysed, n | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Index cases | 408 | 408 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Relatives | 385 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Hooper 200936 Study design: cross-sectional Publication type: conference abstract Other related reports: Poke 200997 Number of study centres: NR Setting: NR Country: Australia Recruitment date: NR Patients recruited consecutively, Y/N: NR Source of funding: NR |
Inclusion criteria: patients with a diagnosis of definite FH based on Dutch Lipid Clinic criteria enrolled in the FH Western Australia (FHWA) pilot programme Exclusion criteria: NR Clinical diagnosis: Dutch criteria FH diagnosis: all definite FH Index casesEnrolled, n63Analysed, n63 |
Index cases | Enrolled, n | 63 | Analysed, n | 63 |
Test 1: genetic test consisting of three stages: Stage 1 (index test): Elucigene FH20 for the detection of 20 common mutations Stage 2 (part of CGA): MLPA for the detection of deletions/duplications Stage 3 (part of CGA): Exon-by-exon sequencing of the LDLR gene Stage 1Stage 2Stage 3Received test, n634943 Not all received sequencing and MLPA |
Stage 1 | Stage 2 | Stage 3 | Received test, n | 63 | 49 | 43 | Number of patients with mutation detected by each stage of test and combination of tests | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Index cases | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Enrolled, n | 63 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Analysed, n | 63 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Stage 1 | Stage 2 | Stage 3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Received test, n | 63 | 49 | 43 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Lee 201048 Study design: cross-sectional, retrospective Publication type: abstract (additional information from author) Other related reports: none Number of study centres: NR Setting: NR Country: UK Study dates: 2005–10 Patients recruited consecutively, Y/N: NR Source of funding: NR |
Inclusion criteria: index patients with a definite diagnosis of homozygous FH based on Simon Broome criteria and genetic test, recruited from an ongoing national cascade testing project, and their relatives Exclusion criteria: NR Participants: relatives of genotyped FH index cases Clinical diagnosis: Simon Broome and then Dutch scoring FH diagnosis: definite FH or possible FH Index casesRelativesEnrolled, n3090Analysed, n3090Homozygous FH, n30NR |
Index cases | Relatives | Enrolled, n | 30 | 90 | Analysed, n | 30 | 90 | Homozygous FH, n | 30 | NR |
Test 1: genetic test as follows in three separate laboratories: Laboratory 1 (biochemistry, Wales): Elucigene FH20/dHPLC/MLPA Laboratory 2 (Progenika): LIPOchip/sequencing Laboratory 3 (Belfast): iPLEX (50 mutations)/sequencing/MLPA Genes tested: LDLR, APOB, PCSK9 Test 2: Sex- and age-adjusted LDL-C values based on Dutch scoring criteria to determine eligibility for genotyping of relatives Test 1Test 2Analysed, n9090 |
Test 1 | Test 2 | Analysed, n | 90 | 90 |
Detection rate of Elucigene FH20 of index cases as of 2010 (34/104 index patients = 32%) Diagnostic accuracy of test 2 on relatives |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Index cases | Relatives | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Enrolled, n | 30 | 90 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Analysed, n | 30 | 90 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Homozygous FH, n | 30 | NR | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Test 1 | Test 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Analysed, n | 90 | 90 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Mabuchi 200546 Study design: case–control Publication type: full text Other reports this study may link with: Yu 200251 Number of study centres: NR Setting: NR Country: Japan Recruitment date: NR Patients recruited consecutively, Y/N: NR Source of funding: NR |
Inclusion criteria: patients with definite FH (LDLR gene mutation) and unaffected first- and second-degree relatives (without LDLR gene mutation) Exclusion criteria: those who had a disease affecting serum lipid concentrations Participants: adults; index cases and first- and second-degree unaffected relatives Clinical diagnosis: NR FH diagnosis: NRFHNo FHEnrolled, n181100Analysed, n18110Age, years, mean (SD)41.9 (16.7)35.1 (17.7)Sex, M/F, n91/9051/49Homozygous FH, n0N/A |
FH | No FH | Enrolled, n | 181 | 100 | Analysed, n | 181 | 10 | Age, years, mean (SD) | 41.9 (16.7) | 35.1 (17.7) | Sex, M/F, n | 91/90 | 51/49 | Homozygous FH, n | 0 | N/A |
Test 1: genetic test as follows: (1) PCR/DGGE/direct sequencing of LDLR gene (2) Southern blot analysis to detect large rearrangements Test 2: LDL-C estimated from a fasting blood sample using Friedwald equation. LDL-C cut-offs > 4.0 mmol/l and TC > 5.8 mmol/l established through bimodal distributions of TC and LDL-C Test 1Test 2Received test, n281281FH, n181No FH, n100 |
Test 1 | Test 2 | Received test, n | 281 | 281 | FH, n | 181 | No FH, n | 100 | Diagnostic test accuracy of LDL-C concentration | Studies with genetically identified patients in whom clinical cut-offs were retrospectively applied | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FH | No FH | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Enrolled, n | 181 | 100 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Analysed, n | 181 | 10 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Age, years, mean (SD) | 41.9 (16.7) | 35.1 (17.7) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Sex, M/F, n | 91/90 | 51/49 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Homozygous FH, n | 0 | N/A | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Test 1 | Test 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Received test, n | 281 | 281 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FH, n | 181 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| No FH, n | 100 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Study design: cross-sectional, comparative Recruitment data: NR Patients recruited consecutively, Y/N: NR Publication type: abstract Other related reports: Stef 201052 (presentation from Progenika Biopharma), plus some additional data from Progenika Biopharma Number of study centres: 2 Setting: NR Country: UK Recruitment date: Patients recruited consecutively, Y/N: Source of funding: Progenika Biopharma Stef 2010:52 samples from two centres were included (Newcastle and Wales); however, the Welsh samples do not have information on clinical diagnosis and also do not have previous genetic diagnosis (according to response to queries from the manufacturer), hence study was not included for the review purpose |
Inclusion criteria: DNA samples from patients with clinical diagnosis based on Simon Broome criteria and previously tested with genetic test (ARMS + SSCP/dHPLC/direct sequencing + MLPA) Exclusion criteria: NR Clinical diagnosis: Simon Broome criteria FH diagnosis: NR Index casesEnrolled, n126Analysed, n120 |
Index cases | Enrolled, n | 126 | Analysed, n | 120 |
Test 1: LIPOchip platform consisting of two stages: Stage 1 (index test): LIPOchip version 8 detects point mutations in the LDLR, APOB and PCSK9 genes and copy number changes in the LDLR gene. Includes 251 mutations most prevalent in Spain, the Netherlands, Norway, Italy and the UK Stage 2 (component of CGA): sequencing of LDLR if no mutation was detected by LIPOchip Test 2: genetic screening in three stages (CGA): Stage 1: Elucigene (APOB, PCSK9 included) Stage 2: SSCP/dHPLC/direct sequencing of all exons if no mutation was detected Stage 3: MLPA if no mutation was detected Test 1Test 2Stage 1Stage 2Enrolled, n126126 mutation positiveReceived test, n12089 |
Test 1 | Test 2 | Stage 1 | Stage 2 | Enrolled, n | 126 | 126 mutation positive | Received test, n | 120 | 89 |
Patients with mutation detected by each test Diagnostic accuracy Time taken to obtain positive results with LIPOchip platform (test was performed by Progenika in Spain) |
LIPOchip UK version 10 was developed by analysing 1000 patients from several cohorts. The chip contains the most frequent mutations found in the UK as well as having the ability to detect copy number changes in the LDLR gene | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Index cases | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Enrolled, n | 126 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Analysed, n | 120 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Test 1 | Test 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Stage 1 | Stage 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Enrolled, n | 126 | 126 mutation positive | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Received test, n | 120 | 89 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Starr 200849 Study design: cross-sectional comparative, retrospective Publication type: full text Other reports this paper may link with: Leren 200453 (Norway), Umans-Eckenhausen 200119 (the Netherlands), Damgaard 200545 (Denmark); samples come from these studies. Number of study centres: 3 Setting: lipid clinics Country: UK Recruitment date: NR Patients recruited consecutively, Y/N: NR Sources of funding: Department of Health, British Heart Foundation. One author supported by the Department of Trade and Industry for the IDEAS Genetics Knowledge Park |
Inclusion criteria: a data set of a cohort of relatives of known mutation status (previously genetically tested) from a European country (the Netherlands, Norway, Denmark) Exclusion criteria: those on lipid-lowering therapy, non-fasting samples and those with triglyceride levels of > 2 mmol/l Participants: first-degree relatives with known mutational status of FH index cases in whom a mutation had been found Clinical diagnosis: based on Dutch criteria (the Netherlands); a combination of lipid levels, clinical characteristics and family history (Norway and Denmark) FH diagnosis: definite FH or possible FH The NetherlandsMut+Mut–Enrolled, n8252469Analysed, n8252469Age, years, mean (SD)30.7 (18.1)42.8 (19.3)Sex, M %5044LDL-C, mmol/l, mean (SD)4.36 (1.39)3.02 (0.94)TC, mmol/l, mean (SD)6.23 (1.43)4.99 (1.05)DenmarkMut+Mut–Analysed, n160161Age, years, mean (SD)35.5 (15.0)41.2 (16.9)Sex, M %4443LDL-C, mmol/l, mean (SD)5.72 (1.86)3.56 (1.14)TC, mmol/l, mean (SD)7.74 (1.88)5.64 (1.28) |
The Netherlands | Mut+ | Mut– | Enrolled, n | 825 | 2469 | Analysed, n | 825 | 2469 | Age, years, mean (SD) | 30.7 (18.1) | 42.8 (19.3) | Sex, M % | 50 | 44 | LDL-C, mmol/l, mean (SD) | 4.36 (1.39) | 3.02 (0.94) | TC, mmol/l, mean (SD) | 6.23 (1.43) | 4.99 (1.05) | Denmark | Mut+ | Mut– | Analysed, n | 160 | 161 | Age, years, mean (SD) | 35.5 (15.0) | 41.2 (16.9) | Sex, M % | 44 | 43 | LDL-C, mmol/l, mean (SD) | 5.72 (1.86) | 3.56 (1.14) | TC, mmol/l, mean (SD) | 7.74 (1.88) | 5.64 (1.28) |
Test 1: CGA (DGGE/direct sequencing/PCR or screening of three common mutations in Danish population/SSCP/sequencing/MLPA or sequencing/MLPA) Test 2: lipid tests The Netherlands: blood sample measured using cholesterol analysis equipment (calibrated). Subjects refrained from eating for 2 hours before test Norway: cholesterol was measured using Roche modular Pinstrument (method standardised). Results from non-fasting samples excluded Denmark: validated with reference standard LDL-C was calculated according to the Friedwald formula Age- and gender-specific LDL-C cut-offs according to NICE guideline used Test 3: MedPed age-specific LDL-C cut-offs for relatives For all tests: Received test, nMut+Mut–The Netherlands82524690–14 years18324315–24 years18727625–34 years13829335–44 years13647145–54 years9244955+ years89737 |
Received test, n | Mut+ | Mut– | The Netherlands | 825 | 2469 | 0–14 years | 183 | 243 | 15–24 years | 187 | 276 | 25–34 years | 138 | 293 | 35–44 years | 136 | 471 | 45–54 years | 92 | 449 | 55+ years | 89 | 737 | Diagnostic accuracy of age- and gender-specific LDL-C vs genetic tests | |||||||||||||||||||||||||||||||||||||||||||||||||||
| The Netherlands | Mut+ | Mut– | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Enrolled, n | 825 | 2469 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Analysed, n | 825 | 2469 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Age, years, mean (SD) | 30.7 (18.1) | 42.8 (19.3) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Sex, M % | 50 | 44 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LDL-C, mmol/l, mean (SD) | 4.36 (1.39) | 3.02 (0.94) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TC, mmol/l, mean (SD) | 6.23 (1.43) | 4.99 (1.05) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Denmark | Mut+ | Mut– | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Analysed, n | 160 | 161 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Age, years, mean (SD) | 35.5 (15.0) | 41.2 (16.9) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Sex, M % | 44 | 43 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LDL-C, mmol/l, mean (SD) | 5.72 (1.86) | 3.56 (1.14) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TC, mmol/l, mean (SD) | 7.74 (1.88) | 5.64 (1.28) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Received test, n | Mut+ | Mut– | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| The Netherlands | 825 | 2469 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 0–14 years | 183 | 243 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 15–24 years | 187 | 276 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 25–34 years | 138 | 293 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 35–44 years | 136 | 471 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 45–54 years | 92 | 449 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 55+ years | 89 | 737 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| NorwayMut+Mut-Analysed, n374742Age, years, mean (SD)27.3 (17.2)37.0 (18.5)Sex, M/F %4646LDL-C, mmol/l, mean (SD)5.71 (1.69)3.4 (1.03)TC, mmol/l, mean (SD)7.74 (1.88)5.64 (1.28) | Norway | Mut+ | Mut- | Analysed, n | 374 | 742 | Age, years, mean (SD) | 27.3 (17.2) | 37.0 (18.5) | Sex, M/F % | 46 | 46 | LDL-C, mmol/l, mean (SD) | 5.71 (1.69) | 3.4 (1.03) | TC, mmol/l, mean (SD) | 7.74 (1.88) | 5.64 (1.28) | Received test, nMut+Mut–Denmarka1601610–14 years15–24 years422325–34 years343635–44 years392745–54 years182955+ years2250Norway3747420–14 years10610715–24 years8210325–34 years6912435–44 years5114545–54 years3912055+ years27143aPlease note that all the figures presented in this table were sourced from Starr and colleagues,49 where an error was observed in the total number reported for the Denmark group (the age subgroups do not add to the total reported). Authors were unable to get the correct values from the original source | Received test, n | Mut+ | Mut– | Denmarka | 160 | 161 | 0–14 years | 15–24 years | 42 | 23 | 25–34 years | 34 | 36 | 35–44 years | 39 | 27 | 45–54 years | 18 | 29 | 55+ years | 22 | 50 | Norway | 374 | 742 | 0–14 years | 106 | 107 | 15–24 years | 82 | 103 | 25–34 years | 69 | 124 | 35–44 years | 51 | 145 | 45–54 years | 39 | 120 | 55+ years | 27 | 143 | aPlease note that all the figures presented in this table were sourced from Starr and colleagues,49 where an error was observed in the total number reported for the Denmark group (the age subgroups do not add to the total reported). Authors were unable to get the correct values from the original source | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Norway | Mut+ | Mut- | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Analysed, n | 374 | 742 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Age, years, mean (SD) | 27.3 (17.2) | 37.0 (18.5) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Sex, M/F % | 46 | 46 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LDL-C, mmol/l, mean (SD) | 5.71 (1.69) | 3.4 (1.03) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TC, mmol/l, mean (SD) | 7.74 (1.88) | 5.64 (1.28) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Received test, n | Mut+ | Mut– | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Denmarka | 160 | 161 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 0–14 years | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 15–24 years | 42 | 23 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 25–34 years | 34 | 36 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 35–44 years | 39 | 27 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 45–54 years | 18 | 29 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 55+ years | 22 | 50 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Norway | 374 | 742 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 0–14 years | 106 | 107 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 15–24 years | 82 | 103 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 25–34 years | 69 | 124 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 35–44 years | 51 | 145 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 45–54 years | 39 | 120 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 55+ years | 27 | 143 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| aPlease note that all the figures presented in this table were sourced from Starr and colleagues,49 where an error was observed in the total number reported for the Denmark group (the age subgroups do not add to the total reported). Authors were unable to get the correct values from the original source | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Stef 200942 Study design: cross-sectional Publication type: conference abstract Other reports: none Number of study centres: NR Setting: NR Country: Spain Recruitment date: NR Patients recruited consecutively, Y/N: NR Source of funding: Progenika Biopharma |
Inclusion criteria: NR Exclusion criteria: NR Clinical diagnosis: Dutch–MedPed criteria FH diagnosis: NR Index casesEnrolled, n2462Analysed, n2462 |
Index cases | Enrolled, n | 2462 | Analysed, n | 2462 |
Test 1: LIPOchip platform consisting of two stages: Stage 1 (index test): LIPOchip Spanish version containing 247 most frequent Spanish mutations (238 LDLR, three APOB and six PCSK9). LIPOchip detects point mutations in the LDLR, APOB and PCSK9 genes and copy number changes in the LDLR gene Stage 2 (part of CGA): sequencing of the entire LDLR gene for those negative on LIPOchip Number receiving test for each stage of tests not reported Not all received sequencing |
Percentage of patients with mutation detected by LIPOchip and combination of tests | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Index cases | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Enrolled, n | 2462 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Analysed, n | 2462 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Tejedor 200543 Study design: cross-sectional comparative Publication type: full text Other reports: Oliva 2009,57 Tejedor 200656 Number of study centres: 70 Setting: lipid clinics Country: Spain Recruitment date: NR Patients recruited consecutively, Y/N: NR Source of funding: NR |
Inclusion criteria: genotyped FH identified by SSCP/sequencing/restricted polymorphism and non-genotyped FH based on Dutch–MedPed criteria from Spanish national register Exclusion criteria: NR Clinical diagnosis: Dutch–MedPed criteria FH diagnosis: definite FH based on clinical score of ≥ 8 points and probable or possible FH with score of 4–8 Phenotyped (n = 407)DFHPFHEnrolled, n252155AdultsChildrenReceived test, n252155Analysed, n252155Age, years, mean (SD)47.6 (14.2)46.0 (18.1)Sex, M/F, n126/12662/93PCVD18.7%6.8%Tendon xanthomata33.1%23.7% |
Phenotyped (n = 407) | DFH | PFH | Enrolled, n | 252 | 155 | Adults | Children | Received test, n | 252 | 155 | Analysed, n | 252 | 155 | Age, years, mean (SD) | 47.6 (14.2) | 46.0 (18.1) | Sex, M/F, n | 126/126 | 62/93 | PCVD | 18.7% | 6.8% | Tendon xanthomata | 33.1% | 23.7% |
Test 1: LIPOchip, earliest Spanish version Stage 1 (index test): DNA array including 118 mutations (117 LDLR and 1 APOB) as identified from SSCP/sequencing/restriction polymorphism analysis (more than half of these mutations have been reported in Western Europe (Holland, France, Germany, Italy, Greece and the UK) and the USA Stage 2 (part of CGA): sequencing of LDLR gene for mutation-negative samples on DNA array Stage 1Stage 2Patients analysed, n407123 (DFH) Not all received sequencing Genotyped patients (1180) were used to test specificity and sensitivity of DNA array |
Stage 1 | Stage 2 | Patients analysed, n | 407 | 123 (DFH) |
Number of patients with mutation detected by each stage of test PCVD-free survival time depending on the type of mutation based on the Kaplan–Meier curves with age limits of 55 years in men and 65 years in women for DNA array Sensitivity and specificity of DNA array to detect mutations that are included in the DNA array (tests were performed in Spain) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phenotyped (n = 407) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| DFH | PFH | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Enrolled, n | 252 | 155 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Adults | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Children | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Received test, n | 252 | 155 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Analysed, n | 252 | 155 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Age, years, mean (SD) | 47.6 (14.2) | 46.0 (18.1) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Sex, M/F, n | 126/126 | 62/93 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PCVD | 18.7% | 6.8% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Tendon xanthomata | 33.1% | 23.7% | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Stage 1 | Stage 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Patients analysed, n | 407 | 123 (DFH) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Study design: cross-sectional Publication type: full text Other related reports: Tabrah 2005,55 Taylor 2009,28 Taylor 200754 Number of study centres: 6 (5 for cascade testing) Setting: lipid clinics Country: UK Study date: 3 years Patients recruited consecutively, Y/N: Y Sources of funding: Departments of Health and Trade Industry for the IDEAS Genetics Knowledge Park and British Heart Foundation |
Inclusion criteria: sample of unrelated patients attending one of six clinics in the UK and all received DNA analysis Exclusion criteria: NR Participants: adults; index cases and first-degree relatives Clinical diagnosis: Simon Broome criteria FH diagnosis: definite FH, possible FH or unclassified FH (unclassifiable because of the information provided, usually missing untreated cholesterol data) DFHPFHUFHTotalEnrolled, nIndex cases19039451635Relatives13814612296Analysed, nIndex cases19039451635Relatives13814612296Ethnicity, nWhite Britisha397Europe37Middle East11Indian Asian31Afro-Caribbean8Far Eastb6NR145ab |
DFH | PFH | UFH | Total | Enrolled, n | Index cases | 190 | 394 | 51 | 635 | Relatives | 138 | 146 | 12 | 296 | Analysed, n | Index cases | 190 | 394 | 51 | 635 | Relatives | 138 | 146 | 12 | 296 | Ethnicity, n | White Britisha | 397 | Europe | 37 | Middle East | 11 | Indian Asian | 31 | Afro-Caribbean | 8 | Far Eastb | 6 | NR | 145 | ab |
Genes tested: LDLR, APOB, PCSK9 Test 1: genetic tests performed in following sequence: Stage 1 (index test): Elucigene FH20 Stage 2 (part of reference standard): DNA sequence analysis of promoter, all exons, the exon/intron boundaries, 3ʹ untranslated region of the LDLR gene using SSCP/dHPLC/direct sequencing for those with negative mutation on Elucigene FH20 Stage 3 (part of reference standard): MLPA for the detection of deletions/duplications for those with negative mutation on sequencing Test 2: targeted sequencing of relatives of index cases with identified mutation Tests performed in the UK Test 1Test 2Stage 1Stage 2Stage 3Analysed, nDFH19013887138PFH394349287146UFH51464012Total635533414296 |
Test 1 | Test 2 | Stage 1 | Stage 2 | Stage 3 | Analysed, n | DFH | 190 | 138 | 87 | 138 | PFH | 394 | 349 | 287 | 146 | UFH | 51 | 46 | 40 | 12 | Total | 635 | 533 | 414 | 296 |
Detection rate of FH Detection rate of FH by ethnicity Detection rate of cascade testing Mutation detection rate of Elucigene FH20 |
Elucigene FH013 B1 kit (11 LDLR + 1 APOB + 1 PCSK9) was used for initial testing. Detection rate data from these tests were combined as if samples were tested using FH20 | |||||||||||||||||||||||||||||||||||||||||||||
| DFH | PFH | UFH | Total | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Enrolled, n | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Index cases | 190 | 394 | 51 | 635 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Relatives | 138 | 146 | 12 | 296 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Analysed, n | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Index cases | 190 | 394 | 51 | 635 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Relatives | 138 | 146 | 12 | 296 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ethnicity, n | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| White Britisha | 397 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Europe | 37 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Middle East | 11 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Indian Asian | 31 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Afro-Caribbean | 8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Far Eastb | 6 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| NR | 145 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ab | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Test 1 | Test 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Stage 1 | Stage 2 | Stage 3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Analysed, n | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| DFH | 190 | 138 | 87 | 138 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PFH | 394 | 349 | 287 | 146 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| UFH | 51 | 46 | 40 | 12 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Total | 635 | 533 | 414 | 296 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Wiegman 200350 Study design: cross-sectional comparative Publication type: full text Other reports: Fouchier 2001,58 Koeijvoets 200598 Number of study centres: NR Setting: lipid clinics Country: the Netherlands Study dates: July 1989–July 2001 Patients recruited consecutively, Y/N: NR Source of funding: NR |
Inclusion criteria: children from parents with a diagnosis of heterozygous FH referred to a lipid clinic Exclusion criteria: NR Participants: children of FH parents Clinical diagnosis: parents’ diagnosis of heterozygous FH was based on a documented LDLR mutation or plasma LDL-C levels > 95th percentile for age and gender in a family with a history of PCVD in conjunction with tendon xanthomata. Children’s diagnosis was based on age- and gender-specific LDL-C levels > 3.50 mmol/l cut-offs for those whose genetic test not yet calculated FH diagnosis: definite FH or possible FHFH genetic diagnosisFH not yet confirmed with genetic testTotalEnrolled, nIndex cases591Children8062281034Analysed8062281034 |
FH genetic diagnosis | FH not yet confirmed with genetic test | Total | Enrolled, n | Index cases | 591 | Children | 806 | 228 | 1034 | Analysed | 806 | 228 | 1034 |
Test 1 (reference standard): genetic testing includes DNA sequence analysis of the coding region of the LDLR gene Test 2: clinical test that includes age- and gender-specific LDL-C levels > 3.50 mmol/l cut-offs Plasma TC, high-density lipoprotein cholesterol and triglycerides measured using commercial kits. LDL-C calculated by Friedwald equation Test 1Test 2Enrolled, n1034282Analysed, n806228 |
Test 1 | Test 2 | Enrolled, n | 1034 | 282 | Analysed, n | 806 | 228 |
FH detection rates of children by both tests Test accuracy data |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FH genetic diagnosis | FH not yet confirmed with genetic test | Total | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Enrolled, n | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Index cases | 591 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Children | 806 | 228 | 1034 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Analysed | 806 | 228 | 1034 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Test 1 | Test 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Enrolled, n | 1034 | 282 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Analysed, n | 806 | 228 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Widhalm 200747 Study design: cross-sectional comparative Publication type: full text Other related reports: none Number of study centres: NR Setting: lipid clinics Country: Austria Recruitment date:1997 Patients recruited consecutively, Y/N: NR Source of funding: NR |
Inclusion criteria: children and adolescents (< 18 years) and their families with definite FH or possible FH Exclusion criteria: NR Participants: index cases < 18 years; relatives adult (all presented as index cases in the study) Clinical diagnosis: MedPed criteria FH diagnosis: definite FH (family had at least one member with confirmed FH) and possible FH (whose family had no member with proven FH) Index casesEnrolled, nTotal263Adults147Children116Sex, M/FAdults83/64Children59/57Current treatment for high cholesterol14 on statins; DFH received dietary treatmentLDL-C, mg/dl, mean (SD)Adults, M/F203 (70)/187 (52)Children, M/F170(60)/166 (68)TC, mmol/l, mean (SD)Adults, M/F292 (100)/268 (59)Children, M/F250 (74)/240 (67) |
Index cases | Enrolled, n | Total | 263 | Adults | 147 | Children | 116 | Sex, M/F | Adults | 83/64 | Children | 59/57 | Current treatment for high cholesterol | 14 on statins; DFH received dietary treatment | LDL-C, mg/dl, mean (SD) | Adults, M/F | 203 (70)/187 (52) | Children, M/F | 170(60)/166 (68) | TC, mmol/l, mean (SD) | Adults, M/F | 292 (100)/268 (59) | Children, M/F | 250 (74)/240 (67) |
Test 1: clinical test Index cases = LDL-C levels > 5.1 mmol/l, relatives = LDL-C levels > 4 mmol/l, age specific (all treated as index cases) Lipid measurements using commercial kit. LDL-C concentration measurement estimated from fasting blood sample using the Friedwald equation. LDL-C measured twice Test was performed in the Lipoprotein Research Laboratory, Vienna Test 2: genetic test as follows: (1) PCR/DGGE/sequencing of the LDLR gene (2) APOB analysis Test was performed in the Institute of Medical Biochemistry, AustriaTest 1Test 2Received test, n119a263Adults, n62147Children, n57116Analysed, n119263a |
Test 1 | Test 2 | Received test, n | 119a | 263 | Adults, n | 62 | 147 | Children, n | 57 | 116 | Analysed, n | 119 | 263 | a | FH detection rates by clinical and genetic tests | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Index cases | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Enrolled, n | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Total | 263 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Adults | 147 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Children | 116 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Sex, M/F | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Adults | 83/64 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Children | 59/57 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Current treatment for high cholesterol | 14 on statins; DFH received dietary treatment | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LDL-C, mg/dl, mean (SD) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Adults, M/F | 203 (70)/187 (52) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Children, M/F | 170(60)/166 (68) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| TC, mmol/l, mean (SD) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Adults, M/F | 292 (100)/268 (59) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Children, M/F | 250 (74)/240 (67) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Test 1 | Test 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Received test, n | 119a | 263 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Adults, n | 62 | 147 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Children, n | 57 | 116 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Analysed, n | 119 | 263 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| a | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Yarram 201038 Study design: cross-sectional Publication type: presentation Other related reports: none Number of study centres: 1 Setting: lipid clinics Country: UK Study date: NR Patients recruited consecutively, Y/N: NR Source of funding: NR |
Inclusion criteria: NR Exclusion criteria; NR Participants: index cases and relatives Clinical diagnosis: Simon Broome criteria with patients diagnosed as definite FH, possible FH, unclassified FH and not meeting criteria DFHPFHUFHNo FHEnrolled, n131Index cases, n15531719Relatives, n27 |
DFH | PFH | UFH | No FH | Enrolled, n | 131 | Index cases, n | 15 | 53 | 17 | 19 | Relatives, n | 27 |
Test 1: genetic tests performed in following sequence: Stage 1 (index test): Elucigene FH20 Stage 2 (part of reference standard): DNA sequence analysis of promoter and all exons of the LDLR gene using sequencing for those with negative mutation on Elucigene FH20 Those with positive mutation on Elucigene FH20 were confirmed with sequencing Stage 3 (part of reference standard): MLPA for the detection of deletions/duplications for those with negative mutation on sequencing Test 2: cascade test Test 1Test 2Enrolled, n10427 |
Test 1 | Test 2 | Enrolled, n | 104 | 27 |
Sensitivity of Elucigene FH20 Sensitivity of LIPOchip (data not included in the report as were not cleared; author was contacted but no response so far) Number testing positive Proportion identified from cascade testing |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| DFH | PFH | UFH | No FH | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Enrolled, n | 131 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Index cases, n | 15 | 53 | 17 | 19 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Relatives, n | 27 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Test 1 | Test 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Enrolled, n | 104 | 27 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Appendix 9 Quality assessment results for the individual studies (full text)
| Q1 | Q2 | Q3 | Q4 | Q5 | Q6 | Q7 | Q8a | Q9 | Q10 | Q11a | Q12 | Q13a | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Taylor 201037 | + | + | – | – | + | + | ? | N/A | – | – | N/A | + | N/A |
| Alonso 200939 | + | + | – | – | + | + | ? | N/A | – | – | N/A | – | N/A |
| Tejedor 200543 | + | – | – | – | + | ? | ? | N/A | – | – | N/A | – | N/A |
| Civeira 200844 | + | + | + | – | + | + | ? | + | – | – | + | + | + |
| Damgaard 200545 | + | + | + | + | + | ? | ? | + | + | – | + | + | + |
| Mabuchi 200546 | ? | – | + | + | + | ? | ? | + | – | – | – | + | – |
| bStarr 200849 | + | + | + | + | + | – | + | + | – | – | – | + | – |
| Widhalm 200747 | + | – | + | + | + | + | ? | + | – | – | + | + | + |
| Wiegman 200350 | + | – | + | + | + | ? | ? | + | – | – | – | + | – |
Appendix 10 Individual study results for Elucigene FH20/LIPOchip
| Study | Diagnosis | Criteria | Genes | Tests | Methods | Enrolled | Tested | Positive | Negative | TP | FP | FN | TN | Sensitivity | Specificity |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hooper 200936 | DFH | Dutch | LDLR/APOB/PCSK9 | Reference standard (1 + 2 + 3) | Elucigene FH20 + MLPA for test-negative with Elucigene FH20 + sequencing for test-negative with MLPA | 63 | 49 | 14 | |||||||
| DFH | LDLR/APOB/PCSK9 | 1 | Elucigene FH20 | 63 | 14 | 49 | 14 | 35 | 14 | 0.285714 | NC | ||||
| DFH | LDLR | 2 | MLPA for test-negative with Elucigene FH20 | 49 | 6 | 43 | |||||||||
| DFH | LDLR | 3 | Sequencing for test-negative with MLPA | 43 | 29 | 14 | |||||||||
| Taylor 200754 | DFH | Simon Broome | LDLR/APOB/PCSK9 | Reference standard (1 + 2) | Elucigene FH13 + SSCP/dHPLC | 400 | 400 | 54 | 54 | 87 | 259 | NC | |||
| PFH | LDLR/APOB/PCSK9 | Reference standard (1 + 2) | As above | 400 | 400 | 87 | 87 | 54 | 259 | NC | |||||
| Total | LDLR/APOB/PCSK9 | Reference standard (1 + 2) | As above | 400 | 400 | 141 | 259 | ||||||||
| DFH | LDLR/APOB/PCSK9 | 1 | Elucigene FH13 | 400 | 400 | 28 | 28 | 113 | 259 | 0.518518 | NC | ||||
| PFH | LDLR/APOB/PCSK9 | 1 | As above | 400 | 400 | 26 | 26 | 115 | 259 | 0.298850 | NC | ||||
| Total | LDLR/APOB/PCSK9 | 1 | As above | 400 | 400 | 54 | 346 | 54 | 87 | 0.382979 | |||||
| Total | LDLR | 2 | SSCP/dHPLC | 400 | 400 | 87 | 313 | 87 | 54 | 259 | 0.617021 | NC | |||
| Taylor 201037 | DFH | Simon Broome | LDLR/APOB/PCSK9 | Reference standard (1 + 2 + 3) | Elucigene FH20 + SSCP/dHPLC/sequencing for test-negative with Elucigene FH20 + MLPA for test-negative with sequencing | 190 | 107 | 83 | |||||||
| PFH | LDLR/APOB/PCSK9 | Reference standard (1 + 2 + 3) | As above | 394 | 112 | 282 | |||||||||
| UFH | LDLR/APOB/PCSK9 | Reference standard (1 + 2 + 3) | As above | 51 | 13 | 38 | |||||||||
| Total | LDLR/APOB/PCSK9 | Reference standard (1 + 2 + 3) | As above | 635 | 232 | 403 | |||||||||
| DFH | LDLR/APOB/PCSK9 | 1 | Elucigene FH20 | 190 | 52 | 138 | 52 | 55 | 83 | 0.485981 | NC | ||||
| PFH | LDLR/APOB/PCSK9 | 1 | As above | 394 | 45 | 349 | 45 | 67 | 282 | 0.401786 | NC | ||||
| UFH | LDLR/APOB/PCSK9 | 1 | As above | 51 | 5 | 46 | 5 | 8 | 38 | 0.384615 | NC | ||||
| Total | LDLR/APOB/PCSK9 | 1 | As above | 635 | 102 | 533 | 102 | 130 | 403 | 0.439655 | NC | ||||
| DFH | LDLR | 2 | SSCP/dHPLC/sequencing for test-negative with Elucigene FH20 | 138 | 51 | 87 | |||||||||
| PFH | LDLR | 2 | As above | 349 | 62 | 287 | |||||||||
| UFH | LDLR | 2 | As above | 46 | 6 | 40 | |||||||||
| Total | LDLR | 2 | As above | 533 | 119 | 414 | |||||||||
| DFH | LDLR | 3 | MLPA for test-negative with sequencing | 87 | 4 | 83 | |||||||||
| PFH | LDLR | 3 | As above | 287 | 5 | 282 | |||||||||
| UFH | LDLR | 3 | As above | 40 | 2 | 38 | |||||||||
| Total | LDLR | 3 | As above | 414 | 11 | 403 | |||||||||
|
Palacios 201041 Newcastle samples |
Total | Simon Broome | LDLR/APOB/PCSK9 | Reference standard (1 + 2) | LIPOchip version 8 + sequencing for test negative with chip | 126 | 120 | 65 | 55 | ||||||
| Total | LDLR/APOB/PCSK9 | 1 | LIPOchip version 8 (UK version) | 126 | 120 | 37 | 83 | 37 | 0 | 28 | 55 | 0.569231 | NC | ||
| 2 | Sequencing for test-negative with chip | 83 | 28 | 55 | |||||||||||
| Total | LDLR/APOB/PCSK9 | 3 | Elucigene FH20 + SSCP/dHPLC/direct sequencing + MLPA | 126 | 126 | 62 | 64 | 62 | 0 | 3 | 61 | 0.953846 | NC | ||
| Total | LDLR/APOB/PCSK9 | 4 | LIPOchip version 10 (UK) | 126 | 51 | 51 | 0 | 14 | 55 | 0.784615 | NC | ||||
| Stef 200942 | Total | Dutch–MedPed | LDLR/APOB/PCSK9 | Reference standard | LIPOchip platform (LIPOchip + sequencing for test-negative with chip) | 2462 | 1206 | 1265 | |||||||
| Total | LDLR/APOB/PCSK9 | 1 | LIPOchip Spanish version | 2462 | 1140 | 1322 | 1140 | 0 | 66 | 1265 | 0.945274 | NC | |||
| Alonso 200939 | DFH/probable | Dutch | LDLR/APOB | Reference standard (1+2+3) | LIPOchip platform (DNA array + QMFSP + sequencing) Spanish version | 808 | 808 | 537 | 271 | ||||||
| Total | LDLR/APOB | 1 | DNA array for 195 mutations | 808 | 419 | 389 | 419 | 0 | 118 | 271 | 0.780261 | NC | |||
| Total | Re-arrangements in LDLR | 2 | QMFSP for test-negative with DNA array | 389 | 77 | 312 | |||||||||
| Total | LDLR | 3 | Sequencing for test-negative with QMFSP | 312 | 41 | 271 | |||||||||
| Tejedor 200543 | DFH/PFH | Dutch–MedPed | LDLR/APOB | Reference standard | LIPOchip (DNA array only + sequencing) Spanish version | NR | |||||||||
| DFH | LDLR/APOB | 1 | DNA array for 118 mutations | 252 | 252 | 129 | |||||||||
| PFH | LDLR/APOB | 1 | As above | 155 | 155 | 58 | |||||||||
| Total | LDLR/APOB | 1 | As above | 407 | 407 | 187 | 220 | NC | NC | ||||||
| DFH | LDLR/APOB | 2 | Sequencing for test-negative with DNA array | 123 | 123 | 43 | 80 | ||||||||
| Manufacturer data for version 10 LIPOchip | NR | NR | LDLR/APOB/PCSK9 | LIPOchip platform (LIPOchip + sequencing) + MLPA | NR | ||||||||||
| LIPOchip version 10 | 138 samples | 138 | 198 | 130 | NC | NC |
| Study | Criteria | Total, n | Mutation samples | TP | FP | TN | FN | Sensitivity (%) | Specificity (%) |
|---|---|---|---|---|---|---|---|---|---|
| Alonso 200939 | Dutch criteria – DFH or probable FH | 808 phenotyped cases | 178 real positive calls; 442 real negative calls | 177 | 1 | 441 | 1 | 99.8 | 99.5 |
| Tejedor 200543 | Dutch–MedPed criteria | 1180 genotyped; 407 blind phenotyped samples | 118 of the LDLR mutations tested with 1180 previously sequenced DNA samples and 10 control DNA samples | NR | NR | NR | NR | 99.9 | 99.7 |
| Manufacturer data for version 10 LIPOchip | Against the mutation already present in LIPOchip version 9 | NR | NR | NR | NR | 100 | 100 |
| Study | LIPOchip version | Samples used | Total samples | Test used to verify | Number correctly detected | Comments |
|---|---|---|---|---|---|---|
| Alonso 200939 | Validation of LIPOchip (194 mutations) | Dutch samples (positive LDLR point mutations) | 53 | DNA array | 51 | One false-positive and one false-negative result (false-negative identified by sequencing) – based on mutational-level analysis |
| Spanish samples (positive deletions in LDLR by QMFSP) | 43 | MLPA | 42 | One sample – detected deletions between exons varied | ||
| Spanish random sample (positive on DNA array) | 125 | Re-sequencing | 125 | |||
| Tejedor 200543 | To identify unidentified mutation that could be introduced into DNA array | Samples with negative mutations on DNA array | 123 | Sequencing | 43 | 28 new mutations identified not detected previously in Spanish population |
| Manufacturer of LIPOchip | LIPOchip version 10 – a technical validation to evaluate its reproducibility | NR | NR | NR | For point mutations and CNV, the reproducibility obtained was 99.49% and 98.33% respectively | |
| Internal validation of LIPOchip version 9 | Samples negative on LIPOchip | 130 | Sequencing | All the mutations revealed by sequencing were not present on the chip (12 new mutations on sequencing, two discrepancies with MLPA) | ||
| Samples positive for point mutation/negative for CNV on LIPOchip | 30 | MLPA | 29 | One discrepancy | ||
| Point mutation and CNV positives on LIPOchip | 5 | Sequencing | 4 | |||
| Point mutation and CNV positives on LIPOchip | 5 | MLPA | 4 | |||
| Samples positive for CNV/ negative for point mutation on LIPOchip | 30 | Sequencing | 30 | |||
| Samples positive for CNV/ negative for point mutation on LIPOchip | 30 | MLPA | 29 | One discrepancy |
| Study | FH diagnosis | Genes | Methods | Enrolled | Tested | Positive | Negative | TP | FN | FP | TN | Sensitivity | Specificity | LR+ | LR– |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Damgaard 200545 | Total | Clinical tests | 408 | 408 | 317 | 91 | |||||||||
| Total | LDLR, APOB | Genetic test: screening of initial three common mutations + SSCP + sequencing + APOB analysis + MLPA | 408 | 408 | 135 | 273 | |||||||||
| Simon Broome criteria | |||||||||||||||
| DFH | 75 | ||||||||||||||
| PFH | 242 | ||||||||||||||
| Total | 408 | 408 | 317 | 91 | |||||||||||
| Genetic test of those with FH from Simon Broome criteria | |||||||||||||||
| DFH | 75 | 46 | 29 | 46 | 29 | ||||||||||
| PFH | 242 | 76 | 166 | 76 | 166 | ||||||||||
| Total | 317 | 122 | 195 | 122 | 195 | ||||||||||
| Total | Genetic test of those without FH from Simon Broome criteria | 91 | 13 | 78 | 13 | 78 | |||||||||
| Total Simon Broome criteria against reference standard | 122 | 13 | 195 | 78 | 0.90 | 0.29 | 1.265185 | 0.337037 | |||||||
| Dutch criteria | |||||||||||||||
| DFH | 89 | ||||||||||||||
| PFH | 204 | ||||||||||||||
| UFH | 98 | ||||||||||||||
| Total | 408 | 391 | 17 | ||||||||||||
| Genetic test of those with FH from the Dutch criteria | |||||||||||||||
| DFH | 89 | 56 | 33 | ||||||||||||
| PFH | 204 | 44 | 160 | ||||||||||||
| UFH | 98 | 34 | 64 | ||||||||||||
| Total | 391 | 134 | 257 | 134 | 257 | ||||||||||
| Total | Genetic test of those without FH from the Dutch criteria | 17 | 1 | 16 | 1 | 16 | |||||||||
| Total Dutch criteria against reference standard | 134 | 1 | 257 | 16 | 0.99 | 0.06 | 1.054388 | 0.126389 | |||||||
| MedPed criteria | 408 | 177 | 231 | ||||||||||||
| Total | Genetic test of those with FH from the MedPed criteria | 135 | 95 | 40 | 95 | 40 | |||||||||
| Total | Genetic test of those without FH from the MedPed criteria | 273 | 82 | 191 | 82 | 191 | |||||||||
| Total | Total MedPed against reference standard | 95 | 82 | 40 | 191 | 0.54 | 0.83 | 3.099576 | 0.560298 | ||||||
| Civeira 200844 | LDLR/APOB/PCSK9 | Genetic tests: LIPOchip (207 mutations in Spain) + PCR for negative results + sequencing for further negative results | 825 | 459 | 366 | ||||||||||
| DFH | Simon Broome criteria | 0.59 | 0.93 | 8.428571 | 0.44086 | ||||||||||
| PFH | 0.90 | 0.27 | 1.232877 | 0.37037 | |||||||||||
| Total | 0.93 | 0.28 | 1.291667 | 0.25 | |||||||||||
| DFH | Dutch criteria | 0.72 | 0.83 | 4.235294 | 0.337349 | ||||||||||
| Total | 0.88 | 0.18 | 1.073171 | 0.666667 | |||||||||||
| Total | MedPed criteria | 0.91 | 0.53 | 1.93617 | 0.169811 | ||||||||||
| Widhalm 200747 | PCR/DGGE/sequencing – total | 263 | 119 | 144 | |||||||||||
| Adults | 147 | 62 | 85 | ||||||||||||
| Children | 116 | 57 | 59 | ||||||||||||
| Adults with FH | 62 | 41 | 21 | 41 | 21 | 0.66 | 0.66129 | ||||||||
| Children with FH | 57 | 46 | 11 | 46 | 11 | 0.81 | 0.807018 | ||||||||
| Mabuchi 200546 | DFHa | LDLR | Genetic tests: PCR/sequencing/ Southern blot analysis | 281 | 181 | 100 | |||||||||
| LDL-C cut-offs ≥ 4.0 mmol/l (with FH-causing mutation) | 181 | 178 | 3 | 178 | 3 | 0 | 100 | 0.98 | 1.00 | 0.016575 | |||||
| LDL-C cut-offs ≥ 4.0 mmol/l (without FH-causing mutation) | 100 | 0 | 100 |
Appendix 11 MOLU classification of genetic tests
Clinical Molecular Genetics Society, MOLU workload units guide version 2.2, March 2010. Available from http://www.cmgs.org/GeneralDownloads/MOLUsystemv2.2.pdf (accessed March 2011).
| Band | MOLU score | General examples | Specific examples |
|---|---|---|---|
| A | 1 |
|
|
| B | 2 |
|
|
| C | 4 |
|
|
| D | 10 |
|
|
| E | 15 |
|
|
| F | 25 |
|
|
| G | 40 |
|
|
Appendix 12 Age-specific analysis for index cases only
In each of the following analyses, costs and incremental costs are rounded to the nearest whole pounds sterling. QALYs and incremental QALYs are rounded to the nearest whole QALY. ICERs are also rounded to the nearest £/QALY gained from the economic model. Therefore, the ratio of reported costs divided by reported QALYs may not always reflect an exact recalculation of the numbers from the tables. This is caused due to rounding; however, the ICERs reported are the true and exact ICERs as generated by the economic model and so minimise the impact of rounding errors on our results.
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 21,356,696 | 17,842 | |||
| Elucigene FH20_MLPA | 21,615,298 | 17,844 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 22,123,539 | 17,847 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 22,195,239 | 17,847 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 22,277,670 | 17,848 | 920,975 | 7 | 137,963 |
| LIPOchip_MLPA | 22,374,581 | 17,849 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 22,446,281 | 17,849 | Dominated | Dominated | Dominated |
| CGA | 22,640,040 | 17,850 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_CGA | 22,687,590 | 17,850 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 22,810,890 | 17,850 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 22,882,590 | 17,850 | Dominated | Dominated | Dominated |
| LDL-C | 25,788,322 | 17,873 | 3,510,651 | 25 | 142,303 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 21,356,696 | 17,842 | –4,431,626 | –31 |
| LIPOchip platform – Spain | 22,277,670 | 17,848 | –3,510,651 | –25 |
| LDL-C | 25,788,322 | 17,873 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 21,356,696 | 17,842 | –1,283,344 | –9 | |
| LIPOchip platform – Spain | 22,277,670 | 17,848 | –362,370 | –2 | |
| CGA | 22,640,040 | 17,850 | |||
| LDL-C | 25,788,322 | 17,873 | 3,148,282 | 23 | 138,056 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 19,797,956 | 15,873 | |||
| Elucigene FH20_MLPA | 20,047,551 | 15,875 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 20,539,419 | 15,877 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 20,611,119 | 15,877 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 20,686,707 | 15,878 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip_MLPA | 20,781,455 | 15,878 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 20,853,155 | 15,878 | Dominated | Dominated | Dominated |
| CGA | 21,040,070 | 15,879 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_CGA | 21,087,620 | 15,879 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 21,210,920 | 15,879 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 21,282,620 | 15,879 | Dominated | Dominated | Dominated |
| LDL-C | 23,864,524 | 15,925 | 4,066,569 | 51 | 79,053 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 19,797,956 | 15,873 | –4,066,569 | –51 |
| LDL-C | 23,864,524 | 15,925 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 19,797,956 | 15,873 | –1,242,114 | –5 | |
| CGA | 21,040,070 | 15,879 | |||
| LDL-C | 23,864,524 | 15,925 | 2,824,455 | 46 | 61,363 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 10,501,988 | 9395 | |||
| Elucigene FH20_MLPA | 10,737,632 | 9401 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 11,204,139 | 9411 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 11,275,839 | 9411 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 11,340,826 | 9415 | 838,838 | 21 | 40,607 |
| LIPOchip_MLPA | 11,432,222 | 9417 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 11,503,922 | 9417 | Dominated | Dominated | Dominated |
| CGA | 11,680,237 | 9421 | 339,411 | 6 | 58,782 |
| Elucigene FH20_CGA | 11,727,787 | 9421 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 11,851,087 | 9421 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 11,922,787 | 9421 | Dominated | Dominated | Dominated |
| LDL-C | 13,140,258 | 9434 | 1,460,021 | 13 | 109,771 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 10,501,988 | 9395 | –2,638,270 | –40 |
| LIPOchip platform – Spain | 11,340,826 | 9415 | –1,799,432 | –19 |
| CGA | 11,680,237 | 9421 | –1,460,021 | –13 |
| LDL-C | 13,140,258 | 9434 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 10,501,988 | 9395 | –1,178,249 | –26 | |
| LIPOchip platform – Spain | 11,340,826 | 9415 | –339,411 | –6 | |
| CGA | 11,680,237 | 9421 | |||
| LDL-C | 13,140,258 | 9434 | 1,460,021 | 13 | 109,771 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 7,645,809 | 6624 | |||
| Elucigene FH20_MLPA | 7,832,406 | 6628 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 8,209,757 | 6634 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 8,281,457 | 6634 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 8,309,178 | 6637 | 663,369 | 12 | 53,738 |
| LIPOchip_MLPA | 8,388,794 | 6637 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 8,460,494 | 6637 | Dominated | Dominated | Dominated |
| CGA | 8,599,543 | 6640 | 290,365 | 3 | 84,152 |
| Elucigene FH20_CGA | 8,647,093 | 6640 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 8,770,393 | 6640 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 8,842,093 | 6640 | Dominated | Dominated | Dominated |
| LDL-C | 9,545,838 | 6641 | 946,294 | 1 | 1,183,172 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALY |
|---|---|---|---|---|
| Elucigene FH20 | 7,645,809 | 6624 | –1,900,029 | –17 |
| LIPOchip platform – Spain | 8,309,178 | 6637 | –1,236,659 | –4 |
| CGA | 8,599,543 | 6640 | –946,294 | –1 |
| LDL-C | 9,545,838 | 6641 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALY | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 7,645,809 | 6624 | –953,734 | –16 | |
| LIPOchip platform – Spain | 8,309,178 | 6637 | –290,365 | –3 | |
| CGA | 8,599,543 | 6640 | |||
| LDL-C | 9,545,838 | 6641 | 946,294 | 1 | 1,183,172 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 5,006,181 | 4036 | |||
| Elucigene FH20_MLPA | 5,139,959 | 4038 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 5,421,293 | 4041 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip platform – Spain | 5,480,582 | 4042 | 474,401 | 6 | 78,151 |
| Elucigene FH20_LIPOchip | 5,492,993 | 4041 | Dominated | Dominated | Dominated |
| LIPOchip_MLPA | 5,547,511 | 4043 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 5,619,211 | 4043 | Dominated | Dominated | Dominated |
| CGA | 5,718,127 | 4044 | 237,545 | 2 | 139,999 |
| Elucigene FH20_CGA | 5,765,677 | 4044 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 5,888,977 | 4044 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 5,960,677 | 4044 | Dominated | Dominated | Dominated |
| LDL-C | 6,159,018 | 4032 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 5,006,181 | 4036 | –1,152,837 | 4 | Dominant |
| LIPOchip platform – Spain | 5,480,582 | 4042 | –678,436 | 10 | Dominant |
| CGA | 5,718,127 | 4044 | –440,891 | 12 | Dominant |
| LDL-C | 6,159,018 | 4032 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 5,006,181 | 4036 | –711,946 | –8 |
| LIPOchip platform – Spain | 5,480,582 | 4042 | –237,545 | –2 |
| CGA | 5,718,127 | 4044 |
Appendix 13 Age-specific analysis for index cases and relatives
In each of the following analyses, costs and incremental costs are rounded to the nearest whole pounds sterling. QALYs and incremental QALYs are rounded to the nearest whole QALY. ICERs are also rounded to the nearest £/QALY gained from the economic model. Therefore, the ratio of reported costs divided by reported QALYs may not always reflect an exact recalculation of the numbers from the tables. This is caused due to rounding; however, the ICERs reported are the true and exact ICERs as generated by the economic model and so minimise the impact of rounding errors on our results.
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 48,382,902 | 39,682 | |||
| LDL-C | 49,384,423 | 37,360 | Dominated | Dominated | Dominated |
| Elucigene FH20_MLPA | 49,596,348 | 40,338 | Ext | Ext Dom | Ext Dom |
| LIPOchip | 51,840,314 | 41,532 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 51,912,014 | 41,532 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 52,719,939 | 42,031 | 4,337,038 | 2350 | 1846 |
| LIPOchip_MLPA | 53,046,200 | 42,189 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 53,117,900 | 42,189 | Dominated | Dominated | Dominated |
| CGA | 54,037,153 | 42,688 | 1,317,213 | 657 | 2005 |
| Elucigene FH20_CGA | 54,084,703 | 42,688 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 54,208,003 | 42,688 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 54,279,703 | 42,688 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 48,382,902 | 39,682 | –1,001,521 | 2321 | Dominant |
| LDL-C | 49,384,423 | 37,360 | |||
| LIPOchip platform – Spain | 52,719,939 | 42,031 | 3,335,516 | 4671 | 714 |
| CGA | 54,037,153 | 42,688 | 4,652,730 | 5328 | 873 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 72,338,134 | 44,143 | |||
| LDL-C | 72,671,492 | 43,013 | Dominated | Dominated | Dominated |
| Elucigene FH20_MLPA | 73,626,950 | 44,473 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 76,007,923 | 45,073 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 76,079,623 | 45,073 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 76,944,814 | 45,324 | 4,606,680 | 1181 | 3901 |
| LIPOchip_MLPA | 77,289,179 | 45,403 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 77,360,879 | 45,403 | Dominated | Dominated | Dominated |
| CGA | 78,337,397 | 45,654 | 1,392,583 | 330 | 4219 |
| Elucigene FH20_CGA | 78,384,947 | 45,654 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 78,508,247 | 45,654 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 78,579,947 | 45,654 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 72,338,134 | 44,143 | –333,358 | 1130 | Dominant |
| LDL-C | 72,671,492 | 43,013 | |||
| LIPOchip platform – Spain | 76,944,814 | 45,324 | 4,273,322 | 2311 | 1849 |
| CGA | 78,337,397 | 45,654 | 5,665,905 | 2641 | 2145 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LDL-C | 58,704,114 | 34,704 | |||
| Elucigene FH20 | 60,370,422 | 36,158 | 1,666,308 | 1454 | 1146 |
| Elucigene FH20_MLPA | 61,804,338 | 36,580 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 64,449,077 | 37,346 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 64,520,777 | 37,346 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 65,496,216 | 37,667 | 5,125,794 | 1508 | 3398 |
| LIPOchip_MLPA | 65,875,433 | 37,768 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 65,947,133 | 37,768 | Dominated | Dominated | Dominated |
| CGA | 67,033,899 | 38,088 | 1,537,683 | 422 | 3647 |
| Elucigene FH20_CGA | 67,081,449 | 38,088 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 67,204,749 | 38,088 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 67,276,449 | 38,088 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LDL-C | 58,704,114 | 34,704 | |||
| Elucigene FH20 | 60,370,422 | 36,158 | 1,666,308 | 1454 | 1146 |
| LIPOchip platform – Spain | 65,496,216 | 37,667 | 6,792,102 | 2962 | 2293 |
| CGA | 67,033,899 | 38,088 | 8,329,786 | 3384 | 2462 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LDL-C | 33,141,939 | 26,128 | |||
| Elucigene FH20 | 34,672,015 | 28,464 | 1,530,076 | 2336 | 655 |
| Elucigene FH20_MLPA | 35,813,456 | 29,123 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 37,926,532 | 30,319 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 37,998,232 | 30,319 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 38,751,448 | 30,820 | 4,079,432 | 2355 | 1732 |
| LIPOchip_MLPA | 39,060,413 | 30,978 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 39,132,113 | 30,978 | Dominated | Dominated | Dominated |
| CGA | 39,996,656 | 31,478 | 1,245,209 | 658 | 1891 |
| Elucigene FH20_CGA | 40,044,206 | 31,478 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 40,167,506 | 31,478 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 40,239,206 | 31,478 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LDL-C | 33,141,939 | 26,128 | |||
| Elucigene FH20 | 34,672,015 | 28,464 | 1,530,076 | 2336 | 655 |
| LIPOchip platform – Spain | 38,751,448 | 30,820 | 5,609,509 | 4692 | 1196 |
| CGA | 39,996,656 | 31,478 | 6,854,717 | 5350 | 1281 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LDL-C | 30,692,288 | 17,638 | |||
| Elucigene FH20 | 31,577,622 | 18,313 | 885,334 | 675 | 1312 |
| Elucigene FH20_MLPA | 32,278,769 | 18,501 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 33,591,472 | 18,844 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 33,663,172 | 18,844 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 34,081,850 | 18,987 | 2,504,228 | 674 | 3715 |
| LIPOchip_MLPA | 34,285,059 | 19,032 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 34,356,759 | 19,032 | Dominated | Dominated | Dominated |
| CGA | 34,886,764 | 19,175 | 804,914 | 188 | 4272 |
| Elucigene FH20_CGA | 34,934,314 | 19,175 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 35,057,614 | 19,175 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 35,129,314 | 19,175 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LDL-C | 30,692,288 | 17,638 | |||
| Elucigene FH20 | 31,577,622 | 18,313 | 885,334 | 675 | 1312 |
| LIPOchip platform – Spain | 34,081,850 | 18,987 | 3,389,562 | 1349 | 2513 |
| CGA | 34,886,764 | 19,175 | 4,194,476 | 1537 | 2728 |
Appendix 14 One-way sensitivity analyses carried out on the base-case model
In each of the following analyses, costs and incremental costs are rounded to the nearest whole pounds sterling. QALYs and incremental QALYs are rounded to the nearest whole QALY. ICERs are also rounded to the nearest £/QALY gained from the economic model. Therefore, the ratio of reported costs divided by reported QALYs may not always reflect an exact recalculation of the numbers from the tables. This is caused due to rounding; however, the ICERs reported are the true and exact ICERs as generated by the economic model and so minimise the impact of rounding errors on our results.
The analysis presented in Tables 47 and 48 refers to the variation in prevalence of FH in the UK. Base-case analysis uses data from Taylor and colleagues,37 supported by the expert clinical opinion of Dr Zosia Miedzybrodzka (University of Aberdeen). The following analysis uses values ranging from 28%79 to 52%. 41
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 13,834,026 | 13,126 | |||
| Elucigene FH20_MLPA | 14,055,008 | 13,135 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 14,488,607 | 13,150 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 14,571,407 | 13,150 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 14,618,431 | 13,157 | 784,405 | 31 | 25,558 |
| LIPOchip_MLPA | 14,703,829 | 13,159 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 14,786,569 | 13,159 | Dominated | Dominated | Dominated |
| CGA | 14,934,997 | 13,166 | 316,566 | 9 | 36,901 |
| Elucigene FH20_CGA | 14,999,197 | 13,166 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 15,135,997 | 13,166 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 15,218,347 | 13,166 | Dominated | Dominated | Dominated |
| LDL-C | 17,501,754 | 13,195 | 2,566,757 | 29 | 87,175 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 13,834,026 | 13,126 | –3,667,728 | –69 |
| LIPOchip platform – Spain | 14,618,431 | 13,157 | –2,883,323 | –38 |
| CGA | 14,934,997 | 13,166 | –2,566,757 | –29 |
| LDL-C | 17,501,754 | 13,195 | – | – |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 13,834,026 | 13,126 | –1,100,971 | –39 | |
| LIPOchip platform – Spain | 14,618,431 | 13,157 | –316,566 | –9 | |
| CGA | 14,934,997 | 13,166 | – | – | |
| LDL-C | 17,501,754 | 13,195 | 2,566,757 | 29 | 87,175 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 42,318,489 | 36,311 | |||
| Elucigene FH20_MLPA | 43,174,675 | 36,743 | Ext Dom | Ext Dom | Ext Dom |
| LDL-C | 43,704,361 | 34,860 | Dominated | Dominated | Dominated |
| LIPOchip | 44,762,955 | 37,527 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 44,845,755 | 37,527 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 45,375,409 | 37,855 | 3,056,920 | 1544 | 1979 |
| LIPOchip_MLPA | 45,613,381 | 37,959 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 45,696,121 | 37,959 | Dominated | Dominated | Dominated |
| CGA | 46,327,179 | 38,287 | 951,770 | 432 | 2205 |
| Elucigene FH20_CGA | 46,391,379 | 38,287 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 46,528,179 | 38,287 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 46,610,529 | 38,287 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 42,318,489 | 36,311 | –1,385,872 | 1451 | Dominant |
| LDL-C | 43,704,361 | 34,860 | |||
| LIPOchip platform – Spain | 45,375,409 | 37,855 | 1,671,048 | 2996 | 558 |
| CGA | 46,327,179 | 38,287 | 2,622,819 | 3427 | 765 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 42,318,489 | 36,311 | –4,008,691 | –1976 |
| LIPOchip platform – Spain | 45,375,409 | 37,855 | –951,770 | –432 |
| CGA | 46,327,179 | 38,287 | – | – |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 14,842,263 | 12,786 | |||
| Elucigene FH20_MLPA | 15,201,307 | 12,802 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 15,903,629 | 12,830 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 15,954,929 | 12,830 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 16,126,407 | 12,843 | 1,284,143 | 57 | 22,530 |
| LIPOchip_MLPA | 16,251,932 | 12,846 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 16,303,172 | 12,846 | Dominated | Dominated | Dominated |
| CGA | 16,604,067 | 12,859 | 477,660 | 16 | 29,981 |
| Elucigene FH20_CGA | 16,621,017 | 12,859 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 16,720,467 | 12,859 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 16,771,317 | 12,859 | Dominated | Dominated | Dominated |
| LDL-C | 17,998,153 | 12,870 | 1,394,087 | 11 | 123,530 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 14,842,263 | 12,786 | –3,155,890 | –84 |
| LIPOchip platform – Spain | 16,126,407 | 12,843 | –1,871,747 | –27 |
| CGA | 16,604,067 | 12,859 | –1,394,087 | –11 |
| LDL-C | 17,998,153 | 12,870 | – | – |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 14,842,263 | 12,786 | –1,761,804 | –73 | |
| LIPOchip platform – Spain | 16,126,407 | 12,843 | –477,660 | –16 | |
| CGA | 16,604,067 | 12,859 | |||
| LDL-C | 17,998,153 | 12,870 | 1,394,087 | 11 | 123,530 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LDL-C | 44,200,760 | 34,535 | |||
| Elucigene FH20 | 45,282,603 | 37,273 | 1,081,843 | 2739 | 395 |
| Elucigene FH20_MLPA | 46,821,312 | 38,075 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 49,668,040 | 39,532 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 49,719,340 | 39,532 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 50,787,132 | 40,141 | 5,504,529 | 2868 | 1919 |
| LIPOchip_MLPA | 51,196,009 | 40,334 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 51,247,249 | 40,334 | Dominated | Dominated | Dominated |
| CGA | 52,444,457 | 40,943 | 1,657,325 | 802 | 2067 |
| Elucigene FH20_CGA | 52,461,407 | 40,943 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 52,560,857 | 40,943 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 52,611,707 | 40,943 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LDL-C | 44,200,760 | 34,535 | |||
| Elucigene FH20 | 45,282,603 | 37,273 | 1,081,843 | 2739 | 395 |
| LIPOchip platform – Spain | 50,787,132 | 40,141 | 6,586,372 | 5607 | 1175 |
| CGA | 52,444,457 | 40,943 | 8,243,697 | 6409 | 1286 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| LDL-C | 44,200,760 | 34,535 | –8,243,697 | –6409 |
| Elucigene FH20 | 45,282,603 | 37,273 | –7,161,854 | –3670 |
| LIPOchip platform – Spain | 50,787,132 | 40,141 | –1,657,325 | –802 |
| CGA | 52,444,457 | 40,943 | – | – |
The base-case assumption in our model is that, on average, 50% of first-degree relatives of a diagnosed index case will possess the culprit genetic mutation causing FH. This, however, is an assumption based on expert opinion; therefore, in our model we need to assume some variance around this estimate. In Tables 49 and 50 the prevalence of FH among relatives is varied by ±20%, that is, between 40% and 60%. As these results refer only to relatives, there is no impact on index cases.
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 37,215,919 | 32,419 | |||
| Elucigene FH20_MLPA | 37,891,155 | 32,677 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 39,156,757 | 33,146 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 39,228,457 | 33,146 | Dominated | Dominated | Dominated |
| LDL-C | 39,246,248 | 31,607 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 39,627,448 | 33,342 | 2,411,529 | 923 | 2613 |
| LIPOchip_MLPA | 39,824,433 | 33,404 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 39,896,133 | 33,404 | Dominated | Dominated | Dominated |
| CGA | 40,406,451 | 33,600 | 779,003 | 258 | 3020 |
| Elucigene FH20_CGA | 40,454,001 | 33,600 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 40,577,301 | 33,600 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 40,649,001 | 33,600 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 37,215,919 | 32,419 | –2,030,329 | 812 | Dominant |
| LDL-C | 39,246,248 | 31,607 | |||
| LIPOchip platform – Spain | 39,627,448 | 33,342 | 381,200 | 1735 | 220 |
| CGA | 40,406,451 | 33,600 | 1,160,203 | 1993 | 582 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 37,215,919 | 32,419 | –3,190,532 | –1181 |
| LIPOchip platform – Spain | 39,627,448 | 33,342 | –779,003 | –258 |
| CGA | 40,406,451 | 33,600 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LDL-C | 49,065,143 | 38,227 | |||
| Elucigene FH20 | 50,587,294 | 41,583 | 1,522,151 | 3356 | 454 |
| Elucigene FH20_MLPA | 52,251,440 | 42,549 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 55,314,692 | 44,305 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 55,386,392 | 44,305 | Dominant | Dominant | Dominant |
| LIPOchip platform – Spain | 56,536,761 | 45,039 | 5,949,467 | 3456 | 1721 |
| LIPOchip_MLPA | 56,971,278 | 45,271 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 57,042,978 | 45,271 | Dominant | Dominant | Dominant |
| CGA | 58,304,674 | 46,006 | 1,767,913 | 966 | 1830 |
| Elucigene FH20_CGA | 58,352,224 | 46,006 | Dominant | Dominant | Dominant |
| LIPOchip_CGA | 58,475,524 | 46,006 | Dominant | Dominant | Dominant |
| Elucigene FH20_LIPOchip_CGA | 58,547,224 | 46,006 | Dominant | Dominant | Dominant |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LDL-C | 49,065,143 | 38,227 | |||
| Elucigene FH20 | 50,587,294 | 41,583 | 1,522,151 | 3356 | 454 |
| LIPOchip platform – Spain | 56,536,761 | 45,039 | 7,471,618 | 6812 | 1097 |
| CGA | 58,304,674 | 46,006 | 9,239,531 | 7778 | 1188 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| LDL-C | 49,065,143 | 38,227 | –9,239,531 | –7778 |
| Elucigene FH20 | 50,587,294 | 41,583 | –7,717,380 | –4422 |
| LIPOchip platform – Spain | 56,536,761 | 45,039 | –1,767,913 | –966 |
| CGA | 58,304,674 | 46,006 | – | – |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 13,878,892 | 13,005 | |||
| Elucigene FH20_MLPA | 14,061,700 | 13,016 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 14,432,160 | 13,037 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 14,503,860 | 13,037 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 14,528,702 | 13,045 | 649,809 | 40 | 16,229 |
| LIPOchip_MLPA | 14,607,407 | 13,048 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 14,697,107 | 13,048 | Dominated | Dominated | Dominated |
| CGA | 14,815,276 | 13,056 | 286,575 | 11 | 25,605 |
| Elucigene FH20_CGA | 14,862,826 | 13,056 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 14,986,126 | 13,056 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 15,057,826 | 13,056 | Dominated | Dominated | Dominated |
| LDL-C | 16,109,186 | 13,079 | 1,293,910 | 23 | 56,282 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 13,878,892 | 13,005 | –2,230,294 | –74 |
| LIPOchip platform – Spain | 14,528,702 | 13,045 | –1,580,485 | –34 |
| CGA | 14,815,276 | 13,056 | –1,293,910 | –23 |
| LDL-C | 16,109,186 | 13,079 | – | – |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 13,878,892 | 13,005 | –936,384 | –51 | |
| LIPOchip platform – Spain | 14,528,702 | 13,045 | –286,575 | –11 | |
| CGA | 14,815,276 | 13,056 | – | – | |
| LDL-C | 16,109,186 | 13,079 | 1,293,910 | 23 | 56,282 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LDL-C | 39,531,598 | 34,744 | |||
| Elucigene FH20 | 39,976,768 | 36,653 | 445,170 | 1909 | 233 |
| Elucigene FH20_MLPA | 40,904,348 | 37,216 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 42,628,666 | 38,240 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 42,700,366 | 38,240 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 43,291,089 | 38,668 | 3,314,321 | 2015 | 1645 |
| LIPOchip_MLPA | 43,548,686 | 38,803 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 43,620,386 | 38,803 | Dominated | Dominated | Dominated |
| CGA | 44,322,437 | 39,231 | 1,031,348 | 563 | 1831 |
| Elucigene FH20_CGA | 44,369,987 | 39,231 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 44,493,287 | 39,231 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 44,564,987 | 39,231 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LDL-C | 39,531,598 | 34,744 | |||
| Elucigene FH20 | 39,976,768 | 36,653 | 445,170 | 1909 | 233 |
| LIPOchip platform – Spain | 43,291,089 | 38,668 | 3,759,492 | 3924 | 958 |
| CGA | 44,322,437 | 39,231 | 4,790,839 | 4487 | 1068 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| LDL-C | 39,531,598 | 34,744 | –4,790,839 | –4487 |
| Elucigene FH20 | 39,976,768 | 36,653 | –4,345,669 | –2578 |
| LIPOchip platform – Spain | 43,291,089 | 38,668 | –1,031,348 | –563 |
| CGA | 44,322,437 | 39,231 | – | – |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 19,077,991 | 13,005 | |||
| Elucigene FH20_MLPA | 19,123,772 | 13,016 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip platform – Spain | 19,237,573 | 13,045 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 19,245,144 | 13,037 | Dominated | Dominated | Dominated |
| LIPOchip_MLPA | 19,283,365 | 13,048 | Ext Dom | Ext Dom | Ext Dom |
| LDL-C | 19,311,875 | 13,079 | 233,884 | 74 | 3161 |
| Elucigene FH20_LIPOchip | 19,316,844 | 13,037 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_MLPA | 19,355,065 | 13,048 | Dominated | Dominated | Dominated |
| CGA | 19,387,121 | 13,056 | Dominated | Dominated | Dominated |
| Elucigene FH20_CGA | 19,434,671 | 13,056 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 19,557,971 | 13,056 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 19,629,671 | 13,056 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 19,077,991 | 13,005 | –233,884 | –74 |
| LDL–C | 19,311,875 | 13,079 | – | – |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 19,077,991 | 13,005 | –309,130 | –51 | |
| LDL-C | 19,311,875 | 13,079 | –75,246 | 23 | Dominant |
| CGA | 19,387,121 | 13,056 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LDL-C | 47,524,644 | 34,744 | |||
| Elucigene FH20 | 50,339,916 | 36,653 | 2,815,272 | 1909 | 1475 |
| Elucigene FH20_MLPA | 51,234,495 | 37,216 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 52,898,822 | 38,240 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 52,970,522 | 38,240 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 53,536,172 | 38,668 | 3,196,256 | 2015 | 1586 |
| LIPOchip_MLPA | 53,785,842 | 38,803 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 53,857,542 | 38,803 | Dominated | Dominated | Dominated |
| CGA | 54,534,518 | 39,231 | 998,347 | 563 | 1773 |
| Elucigene FH20_CGA | 54,582,068 | 39,231 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 54,705,368 | 39,231 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 54,777,068 | 39,231 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LDL-C | 47,524,644 | 34,744 | |||
| Elucigene FH20 | 50,339,916 | 36,653 | 2,815,272 | 1909 | 1475 |
| LIPOchip platform – Spain | 53,536,172 | 38,668 | 6,011,528 | 3924 | 1532 |
| CGA | 54,534,518 | 39,231 | 7,009,875 | 4487 | 1562 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| LDL-C | 47,524,644 | 34,744 | –7,009,875 | –4487 |
| Elucigene FH20 | 50,339,916 | 36,653 | –4,194,603 | –2578 |
| LIPOchip platform – Spain | 53,536,172 | 38,668 | –998,347 | –563 |
| CGA | 54,534,518 | 39,231 | – | – |
The analyses presented in Table 53 relate to the potential treatment of negative-testing relatives. This value is assumed to be 10% in the model. As this is purely an assumption based on author opinion, this value is varied between 0% and 50% in sensitivity analysis. As this has no effect on the results for index cases alone, results are presented only for index cases and relatives combined. It is assumed that if negative relatives are treated then their treatment of choice will be low-intensity statin therapy as defined for the base-case model.
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 42,251,638 | 33,421 | |||
| LDL-C | 42,709,251 | 31,523 | Dominated | Dominated | Dominated |
| Elucigene FH20_MLPA | 43,364,673 | 33,981 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 45,426,111 | 34,999 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 45,497,811 | 34,999 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 46,229,444 | 35,425 | 3,977,806 | 2004 | 1985 |
| LIPOchip_MLPA | 46,531,587 | 35,559 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 46,603,287 | 35,559 | Dominated | Dominated | Dominated |
| CGA | 47,446,247 | 35,985 | 1,216,803 | 560 | 2172 |
| Elucigene FH20_CGA | 47,493,797 | 35,985 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 47,617,097 | 35,985 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 47,688,797 | 35,985 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental effects | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 42,251,638 | 33,421 | –457,614 | 1898 | Dominant |
| LDL-C | 42,709,251 | 31,523 | |||
| LIPOchip platform – Spain | 46,229,444 | 35,425 | 3,520,193 | 3902 | 902 |
| CGA | 47,446,247 | 35,985 | 4,736,995 | 4462 | 1062 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 42,251,638 | 33,421 | –5,194,609 | –2564 |
| LIPOchip platform – Spain | 46,229,444 | 35,425 | –1,216,803 | –560 |
| CGA | 47,446,247 | 35,985 | – | – |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 47,853,372 | 49,583 | |||
| LDL-C | 48,566,941 | 47,630 | Dominated | Dominated | Dominated |
| Elucigene FH20_MLPA | 48,895,157 | 50,158 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 50,827,075 | 51,204 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 50,898,775 | 51,204 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 51,576,271 | 51,641 | 3,722,899 | 2059 | 1808 |
| LIPOchip_MLPA | 51,861,300 | 51,780 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 51,933,000 | 51,780 | Dominated | Dominated | Dominated |
| CGA | 52,721,823 | 52,217 | 1,145,552 | 575 | 1991 |
| Elucigene FH20_CGA | 52,769,373 | 52,217 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 52,892,673 | 52,217 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 52,964,373 | 52,217 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 47,853,372 | 49,583 | –713,569 | 1953 | Dominant |
| LDL-C | 48,566,941 | 47,630 | |||
| LIPOchip platform – Spain | 51,576,271 | 51,641 | 3,009,331 | 4011 | 750 |
| CGA | 52,721,823 | 52,217 | 4,154,883 | 4587 | 906 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 47,853,372 | 49,583 | –4,868,451 | –2634 |
| LIPOchip in Spain | 51,576,271 | 51,641 | –1,145,552 | –575 |
| CGA | 52,721,823 | 52,217 |
Base-case analysis assumes that all test-negative index cases will require some treatment. The justification for this assumption is that all these patients will have elevated lipids and will thus be at increased risk of CHD. However, it is possible that these patients could be managed effectively using diet and exercise interventions, the evaluation of which is beyond the scope of this report. Therefore, to assess the impact of this assumption on our results, Table 54 presents the analysis assuming an extreme case scenario in which none of the test-negative index cases will receive treatment. Index cases are therefore not followed up clinically in this scenario.
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 8,205,446 | 1967 | |||
| Elucigene FH20_MLPA | 8,863,412 | 2514 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 10,097,620 | 3509 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 10,169,320 | 3509 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 10,555,189 | 3925 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip_MLPA | 10,748,025 | 4057 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 10,819,725 | 4057 | Dominated | Dominated | Dominated |
| CGA | 11,316,921 | 4473 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_CGA | 11,364,471 | 4473 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 11,487,771 | 4473 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 11,559,471 | 4473 | Dominated | Dominated | Dominated |
| LDL-C | 16,140,001 | 10,152 | 7,934,555 | 8185 | 969 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 8,205,446 | 1967 | –7,934,555 | –8185 |
| LDL-C | 16,140,001 | 10,152 | – | – |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 8,205,446 | 1967 | –3,111,475 | –2506 | |
| CGA | 11,316,921 | 4473 | – | – | |
| LDL-C | 16,140,001 | 10,152 | 4,823,079 | 5679 | 849 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 37,385,060 | 25,614 | |||
| Elucigene FH20_MLPA | 38,871,740 | 26,714 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 41,612,394 | 28,713 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip | 41,684,094 | 28,713 | Dominated | Dominated | Dominated |
| LDL-C | 42,342,607 | 31,817 | 4,957,547 | 6202 | 799 |
| LIPOchip platform – Spain | 42,699,624 | 29,548 | Dominated | Dominated | Dominated |
| LIPOchip_MLPA | 43,091,514 | 29,812 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_MLPA | 43,163,214 | 29,812 | Dominated | Dominated | Dominated |
| CGA | 44,290,071 | 30,648 | Dominated | Dominated | Dominated |
| Elucigene FH20_CGA | 44,337,621 | 30,648 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 44,460,921 | 30,648 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 44,532,621 | 30,648 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 37,385,060 | 25,614 | –4,957,547 | –6202 | |
| LDL-C | 42,342,607 | 31,817 | – | – | – |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 37,385,060 | 25,614 | –6,905,011 | –5033 | |
| LDL-C | 42,342,607 | 31,817 | –1,947,464 | 1169 | Dominant |
| CGA | 44,290,071 | 30,648 | – | – |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 14,242,370 | 13,005 | |||
| Elucigene FH20_MLPA | 14,529,221 | 13,016 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 15,101,529 | 13,037 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip platform – Spain | 15,154,374 | 13,045 | 912,004 | 40 | 22,777 |
| Elucigene FH20_LIPOchip | 15,197,129 | 13,037 | Dominated | Dominated | Dominated |
| LIPOchip_MLPA | 15,378,300 | 13,048 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 15,473,900 | 13,048 | Dominated | Dominated | Dominated |
| CGA | 15,688,212 | 13,056 | 533,838 | 11 | 47,698 |
| Elucigene FH20_CGA | 15,751,612 | 13,056 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 15,916,012 | 13,056 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 16,011,612 | 13,056 | Dominated | Dominated | Dominated |
| LDL-C | 17,678,183 | 13,079 | 1,989,970 | 23 | 86,558 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 14,242,370 | 13,005 | –3,435,812 | –74 |
| LIPOchip platform – Spain | 15,154,374 | 13,045 | –2,523,808 | –34 |
| CGA | 15,688,212 | 13,056 | –1,989,970 | –23 |
| LDL–C | 17,678,183 | 13,079 | – | – |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 14,242,370 | 13,005 | –1,445,842 | –51 | |
| LIPOchip platform – Spain | 15,154,374 | 13,045 | –533,838 | –11 | |
| CGA | 15,688,212 | 13,056 | |||
| LDL-C | 17,678,183 | 13,079 | 1,989,970 | 23 | 86,558 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 43,444,427 | 36,653 | |||
| LDL-C | 43,880,789 | 34,744 | Dominated | Dominated | Dominated |
| Elucigene FH20_MLPA | 44,566,239 | 37,216 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 46,656,350 | 38,240 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 46,751,950 | 38,240 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 47,343,602 | 38,668 | 3,899,175 | 2015 | 1935 |
| LIPOchip_MLPA | 47,768,083 | 38,803 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 47,863,683 | 38,803 | Dominated | Dominated | Dominated |
| CGA | 48,712,402 | 39,231 | 1,368,800 | 563 | 2430 |
| Elucigene FH20_CGA | 48,775,802 | 39,231 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 48,940,202 | 39,231 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 49,035,802 | 39,231 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 43,444,427 | 36,653 | –436,362 | 1909 | Dominant |
| LDL-C | 43,880,789 | 34,744 | |||
| LIPOchip platform – Spain | 47,343,602 | 38,668 | 3,462,813 | 3924 | 883 |
| CGA | 48,712,402 | 39,231 | 4,831,613 | 4487 | 1077 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 43,444,427 | 36,653 | –5,267,975 | –2578 |
| LIPOchip platform – Spain | 47,343,602 | 38,668 | –1,368,800 | –563 |
| CGA | 48,712,402 | 39,231 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 14,142,370 | 13,005 | |||
| Elucigene FH20_MLPA | 14,395,661 | 13,016 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 14,881,529 | 13,037 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 14,929,329 | 13,037 | Dominated | Dominated | Dominated |
| LIPOchip_MLPA | 15,129,780 | 13,048 | 987,410 | 43 | 23,108 |
| LIPOchip platform – Spain | 15,154,374 | 13,045 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_MLPA | 15,177,580 | 13,048 | Dominated | Dominated | Dominated |
| CGA | 15,368,212 | 13,056 | 238,432 | 9 | 28,038 |
| Elucigene FH20_CGA | 15,399,912 | 13,056 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 15,482,112 | 13,056 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 15,529,912 | 13,056 | Dominated | Dominated | Dominated |
| LDL-C | 17,678,183 | 13,079 | 2,309,970 | 23 | 100,474 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 14,142,370 | 13,005 | –3,535,812 | –74 |
| LIPOchip_MLPA | 15,129,780 | 13,048 | –2,548,403 | –31 |
| CGA | 15,368,212 | 13,056 | –2,309,970 | –23 |
| LDL-C | 17,678,183 | 13,079 | – | – |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 14,142,370 | 13,005 | –1,225,842 | –51 | |
| LIPOchip_MLPA | 15,129,780 | 13,048 | –238,432 | –9 | |
| CGA | 15,368,212 | 13,056 | – | ||
| LDL-C | 17,678,183 | 13,079 | 2,309,970 | 23 | 100,477 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 43,299,542 | 36,653 | |||
| LDL-C | 43,880,789 | 34,744 | Dominated | Dominated | Dominated |
| Elucigene FH20_MLPA | 44,375,300 | 37,216 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 46,356,258 | 38,240 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 46,404,058 | 38,240 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 47,254,017 | 38,668 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip_MLPA | 47,426,976 | 38,803 | 4,127,434 | 2150 | 1920 |
| Elucigene FH20_LIPOchip_MLPA | 47,474,776 | 38,803 | Dominated | Dominated | Dominated |
| CGA | 48,290,322 | 39,231 | 863,346 | 428 | 2017 |
| Elucigene FH20_CGA | 48,322,022 | 39,231 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 48,404,222 | 39,231 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 48,452,022 | 39,231 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 43,299,542 | 36,653 | –581,247 | 1909 | Dominant |
| LDL-C | 43,880,789 | 34,744 | |||
| LIPOchip_MLPA | 47,426,976 | 38,803 | 3,546,187 | 4059 | 874 |
| CGA | 48,290,322 | 39,231 | 4,409,532 | 4487 | 983 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 43,299,542 | 36,653 | –4,990,780 | –2578 |
| LIPOchip_MLPA | 47,426,976 | 38,803 | –863,346 | –428 |
| CGA | 48,290,322 | 39,231 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 37,712,894 | 32,096 | |||
| Elucigene FH20_MLPA | 38,506,088 | 32,440 | Ext Dom | Ext Dom | Ext Dom |
| LDL-C | 39,319,481 | 30,973 | Dominated | Dominated | Dominated |
| LIPOchip | 39,986,115 | 33,066 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 40,057,815 | 33,066 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 40,546,431 | 33,328 | 2,833,537 | 1232 | 2299 |
| LIPOchip_MLPA | 40,771,748 | 33,411 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 40,843,448 | 33,411 | Dominated | Dominated | Dominated |
| CGA | 41,443,392 | 33,673 | 896,961 | 344 | 2604 |
| Elucigene FH20_CGA | 41,490,942 | 33,673 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 41,614,242 | 33,673 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 41,685,942 | 33,673 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 37,712,894 | 32,096 | –1,606,587 | 1123 | Dominant |
| LDL-C | 39,319,481 | 30,973 | |||
| LIPOchip platform – Spain | 40,546,431 | 33,328 | 1,226,950 | 2355 | 521 |
| CGA | 41,443,392 | 33,673 | 2,123,911 | 2700 | 787 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 37,712,894 | 32,096 | –3,730,498 | –1577 |
| LIPOchip platform – Spain | 40,546,431 | 33,328 | –896,961 | –344 |
| CGA | 41,443,392 | 33,673 |
The sensitivity analyses in Table 58 refers to not cascade testing from index cases in whom a genetic mutation has not been identified.
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 14,192,370 | 13,005 | |||
| Elucigene FH20_MLPA | 14,462,441 | 13,016 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 14,991,529 | 13,037 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 15,063,229 | 13,037 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 15,154,374 | 13,045 | 962,004 | 40 | 24,025 |
| LIPOchip_MLPA | 15,254,040 | 13,048 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 15,325,740 | 13,048 | Dominated | Dominated | Dominated |
| CGA | 15,528,212 | 13,056 | 373,838 | 11 | 33,402 |
| Elucigene FH20_CGA | 15,575,762 | 13,056 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 15,699,062 | 13,056 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 15,770,762 | 13,056 | Dominated | Dominated | Dominated |
| LDL-C | 17,678,183 | 13,079 | 2,149,970 | 23 | 93,518 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 14,192,370 | 13,005 | –3,485,812 | –74 |
| LIPOchip platform – Spain | 15,154,374 | 13,045 | –2,523,808 | –34 |
| CGA | 15,528,212 | 13,056 | –2,149,970 | –23 |
| LDL-C | 17,678,183 | 13,079 | – | – |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 14,192,370 | 13,005 | –1,335,842 | –51 | |
| LIPOchip platform – Spain | 15,154,374 | 13,045 | –373,838 | –11 | |
| CGA | 15,528,212 | 13,056 | |||
| LDL-C | 17,678,183 | 13,079 | 2,149,970 | 23 | 93,518 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 21,378,104 | 18,468 | |||
| Elucigene FH20_MLPA | 23,648,479 | 20,000 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 27,813,741 | 22,785 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 27,885,441 | 22,785 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 29,496,425 | 23,949 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip_MLPA | 30,076,556 | 24,317 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 30,148,256 | 24,317 | Dominated | Dominated | Dominated |
| CGA | 31,870,568 | 25,481 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_CGA | 31,918,118 | 25,481 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 32,041,418 | 25,481 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 32,113,118 | 25,481 | Dominated | Dominated | Dominated |
| LDL-C | 38,100,678 | 29,965 | 16,722,574 | 11,497 | 1455 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 21,378,104 | 18,468 | –16,722,574 | –11,497 |
| LDL-C | 38,100,678 | 29,965 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 21,378,104 | 18,468 | –10,492,464 | –7012 | |
| CGA | 31,870,568 | 25,481 | |||
| LDL-C | 38,100,678 | 29,965 | 6,230,110 | 4484 | 1389 |
Hadfeld and colleagues66 estimate that 69% of index cases and also 69% of qualifiable relatives will agree to genetic testing being carried out. There will not be any implications here for index cases alone and therefore the results in Table 59 are for index and relative cases combined only.
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LDL-C | 54,528,198 | 43,548 | |||
| Elucigene FH20 | 55,623,568 | 46,562 | 1,095,370 | 3015 | 363 |
| Elucigene FH20_MLPA | 57,168,909 | 47,433 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 60,016,199 | 49,016 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 60,087,899 | 49,016 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 61,148,000 | 49,678 | 5,524,433 | 3116 | 1773 |
| LIPOchip_MLPA | 61,553,981 | 49,887 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 61,625,681 | 49,887 | Dominated | Dominated | Dominated |
| CGA | 62,797,109 | 50,549 | 1,649,109 | 871 | 1893 |
| Elucigene FH20_CGA | 62,844,659 | 50,549 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 62,967,959 | 50,549 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 63,039,659 | 50,549 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LDL-C | 54,528,198 | 43,548 | |||
| Elucigene FH20 | 55,623,568 | 46,562 | 1,095,370 | 3015 | 363 |
| LIPOchip platform – Spain | 61,148,000 | 49,678 | 6,619,802 | 6131 | 1080 |
| CGA | 62,797,109 | 50,549 | 8,268,912 | 7002 | 1181 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| LDL-C | 54,528,198 | 43,548 | –8,268,912 | –7002 |
| Elucigene FH20 | 55,623,568 | 46,562 | –7,173,542 | –3987 |
| LIPOchip platform – Spain | 61,148,000 | 49,678 | –1,649,109 | –871 |
| CGA | 62,797,109 | 50,549 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 27,204,825 | 20,571 | |||
| Elucigene FH20_MLPA | 27,611,712 | 20,594 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 28,389,507 | 20,636 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 28,461,207 | 20,636 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 28,656,305 | 20,654 | 1,451,480 | 84 | 17,377 |
| LIPOchip_MLPA | 28,788,833 | 20,660 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 28,860,533 | 20,660 | Dominated | Dominated | Dominated |
| CGA | 29,166,959 | 20,678 | 510,654 | 23 | 21,872 |
| Elucigene FH20_CGA | 29,214,509 | 20,678 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 29,337,809 | 20,678 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 29,409,509 | 20,678 | Dominated | Dominated | Dominated |
| LDL_C | 32,978,473 | 20,728 | 3,811,514 | 50 | 75,678 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 27,204,825 | 20,571 | –5,773,648 | –157 |
| LIPOchip platform – Spain | 28,656,305 | 20,654 | –4,322,169 | –74 |
| CGA | 29,166,959 | 20,678 | –3,811,514 | –50 |
| LDL-C | 32,978,473 | 20,728 | – | – |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 27,204,825 | 20,571 | –1,962,134 | –107 | |
| LIPOchip platform – Spain | 28,656,305 | 20,654 | –510,654 | –23 | |
| CGA | 29,166,959 | 20,678 | – | – | |
| LDL-C | 32,978,473 | 20,728 | 3,811,514 | 50 | 75,678 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 80,370,221 | 58,348 | |||
| LDL-C | 80,740,629 | 55,359 | Dominated | Dominated | Dominated |
| Elucigene FH20_MLPA | 82,281,317 | 59,248 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 85,793,478 | 60,883 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 85,865,178 | 60,883 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 87,203,180 | 61,566 | 6,832,959 | 3218 | 2123 |
| LIPOchip_MLPA | 87,697,014 | 61,782 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 87,768,714 | 61,782 | Dominated | Dominated | Dominated |
| CGA | 89,218,043 | 62,466 | 2,014,863 | 899 | 2240 |
| Elucigene FH20_CGA | 89,265,593 | 62,466 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 89,388,893 | 62,466 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 89,460,593 | 62,466 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 80,370,221 | 58,348 | –370,048 | 2990 | Dominant |
| LDL-C | 80,740,629 | 55,359 | |||
| LIPOchip platform – Spain | 87,203,180 | 61,566 | 6,462,911 | 6208 | 1041 |
| CGA | 89,218,043 | 62,466 | 8,477,774 | 7107 | 1193 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 80,370,221 | 58,348 | –8,814,822 | –4117 |
| LIPOchip platform – Spain | 87,203,180 | 61,566 | –2,014,863 | –899 |
| CGA | 89,218,043 | 62,466 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 10,029,010 | 10,117 | |||
| Elucigene FH20_MLPA | 10,248,696 | 10,125 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 10,686,196 | 10,138 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 10,757,896 | 10,138 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 10,810,758 | 10,143 | 781,748 | 26 | 30,023 |
| LIPOchip_MLPA | 10,898,322 | 10,145 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 10,970,022 | 10,145 | Dominated | Dominated | Dominated |
| CGA | 11,134,212 | 10,151 | 323,454 | 7 | 44,442 |
| Elucigene FH20_CGA | 11,181,762 | 10,151 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 11,305,062 | 10,151 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 11,376,762 | 10,151 | Dominated | Dominated | Dominated |
| LDL-C | 12,668,695 | 10,165 | 1,534,483 | 14 | 109,720 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 10,029,010 | 10,117 | –2,639,685 | –47 |
| LIPOchip platform – Spain | 10,810,758 | 10,143 | –1,857,937 | –21 |
| CGA | 11,134,212 | 10,151 | –1,534,483 | –14 |
| LDL-C | 12,668,695 | 10,165 | – | – |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 10,029,010 | 10,117 | –1,105,202 | –33 | |
| LIPOchip platform – Spain | 10,810,758 | 10,143 | –323,454 | –7 | |
| CGA | 11,134,212 | 10,151 | – | – | |
| LDL-C | 12,668,695 | 10,165 | 1,534,483 | 14 | 109,720 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 31,278,303 | 28,428 | |||
| LDL-C | 31,745,080 | 26,934 | Dominated | Dominated | Dominated |
| Elucigene FH20_MLPA | 32,102,865 | 28,865 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 33,639,915 | 29,658 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 33,711,615 | 29,658 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 34,224,064 | 29,989 | 2,945,761 | 1561 | 1887 |
| LIPOchip_MLPA | 34,456,917 | 30,094 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 34,528,617 | 30,094 | Dominated | Dominated | Dominated |
| CGA | 35,152,394 | 30,426 | 928,329 | 436 | 2127 |
| Elucigene FH20_CGA | 35,199,944 | 30,426 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 35,323,244 | 30,426 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 35,394,944 | 30,426 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 31,278,303 | 28,428 | –466,777 | 1494 | Dominant |
| LDL-C | 31,745,080 | 26,934 | |||
| LIPOchip platform – Spain | 34,224,064 | 29,989 | 2,478,984 | 3055 | 811 |
| CGA | 35,152,394 | 30,426 | 3,407,313 | 3492 | 976 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 31,278,303 | 28,428 | –3,874,090 | –1998 |
| LIPOchip platform – Spain | 34,224,064 | 29,989 | –928,329 | –436 |
| CGA | 35,152,394 | 30,426 |
The base-case analysis assumes that there will be no reduction in the cost of next-generation sequencing. Clinical advice is that there will be a reduction in cost; however, we are unsure as to how much that reduction will be in practice. Table 62 presents the results of the sensitivity analysis assuming that next-generation sequencing costs will reduce by 40% into the future.
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 14,192,370 | 13,005 | |||
| Elucigene FH20_MLPA | 14,462,441 | 13,016 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 14,991,529 | 13,037 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 15,063,229 | 13,037 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 15,154,374 | 13,045 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip_MLPA | 15,254,040 | 13,048 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 15,325,740 | 13,048 | Dominated | Dominated | Dominated |
| CGA | 15,372,212 | 13,056 | 1,179,842 | 51 | 23,029 |
| Elucigene FH20_CGA | 15,444,878 | 13,056 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 15,587,834 | 13,056 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 15,659,534 | 13,056 | Dominated | Dominated | Dominated |
| LDL-C | 17,678,183 | 13,079 | 2,305,970 | 23 | 100,303 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 14,192,370 | 13,005 | –3,485,812 | –74 |
| CGA | 15,372,212 | 13,056 | –2,305,970 | –23 |
| LDL-C | 17,678,183 | 13,079 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 14,192,370 | 13,005 | –1,179,842 | –51 | |
| CGA | 15,372,212 | 13,056 | |||
| LDL-C | 17,678,183 | 13,079 | 2,305,970 | 23 | 100,303 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 43,371,985 | 36,653 | |||
| LDL-C | 43,880,789 | 34,744 | Dominated | Dominated | Dominated |
| Elucigene FH20_MLPA | 44,470,770 | 37,216 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 46,506,304 | 38,240 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 46,578,004 | 38,240 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 47,298,810 | 38,668 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip_MLPA | 47,597,529 | 38,803 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 47,669,229 | 38,803 | Dominated | Dominated | Dominated |
| CGA | 48,345,362 | 39,231 | 4,973,377 | 2578 | 1929 |
| Elucigene FH20_CGA | 48,418,028 | 39,231 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 48,560,984 | 39,231 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 48,632,684 | 39,231 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 43,371,985 | 36,653 | –508,805 | 1909 | Dominant |
| LDL-C | 43,880,789 | 34,744 | |||
| CGA | 48,345,362 | 39,231 | 4,464,573 | 4487 | 995 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 43,371,985 | 36,653 | –4,973,377 | –2578 |
| CGA | 48,345,362 | 39,231 |
Table 63 details the effect of a high value for the sensitivity of Elucigene FH20. This is the highest estimate from the clinical effectiveness review and is 52%. 38
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 14,336,524 | 13,013 | |||
| Elucigene FH20_MLPA | 14,604,854 | 13,024 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 14,991,529 | 13,037 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 15,054,529 | 13,037 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 15,154,374 | 13,045 | 817,851 | 33 | 25,012 |
| LIPOchip_MLPA | 15,254,040 | 13,048 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 15,317,040 | 13,048 | Dominated | Dominated | Dominated |
| CGA | 15,528,212 | 13,056 | 373,838 | 11 | 33,402 |
| Elucigene FH20_CGA | 15,562,712 | 13,056 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 15,699,062 | 13,056 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 15,762,062 | 13,056 | Dominated | Dominated | Dominated |
| LDL-C | 17,678,183 | 13,079 | 2,149,970 | 23 | 93,518 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 14,336,524 | 13,013 | –3,341,659 | –67 |
| LIPOchip platform – Spain | 15,154,374 | 13,045 | –2,523,808 | –34 |
| CGA | 15,528,212 | 13,056 | –2,149,970 | –23 |
| LDL-C | 17,678,183 | 13,079 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 14,336,524 | 13,013 | –1,191,689 | –44 | |
| LIPOchip platform – Spain | 15,154,374 | 13,045 | –373,838 | –11 | |
| CGA | 15,528,212 | 13,056 | |||
| LDL-C | 17,678,183 | 13,079 | 2,149,970 | 23 | 93,518 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LDL-C | 43,880,789 | 34,744 | |||
| Elucigene FH20 | 44,059,813 | 37,022 | 179,023 | 2278 | 79 |
| Elucigene FH20_MLPA | 45,156,858 | 37,586 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 46,506,304 | 38,240 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 46,569,304 | 38,240 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 47,298,810 | 38,668 | 3,238,997 | 1645 | 1968 |
| LIPOchip_MLPA | 47,597,529 | 38,803 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 47,660,529 | 38,803 | Dominated | Dominated | Dominated |
| CGA | 48,501,362 | 39,231 | 1,202,552 | 563 | 2135 |
| Elucigene FH20_CGA | 48,535,862 | 39,231 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 48,672,212 | 39,231 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 48,735,212 | 39,231 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LDL-C | 43,880,789 | 34,744 | |||
| Elucigene FH20 | 44,059,813 | 37,022 | 179,023 | 2278 | 79 |
| LIPOchip platform – Spain | 47,298,810 | 38,668 | 3,418,020 | 3924 | 871 |
| CGA | 48,501,362 | 39,231 | 4,620,573 | 4487 | 1030 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| LDL-C | 43,880,789 | 34,744 | –4,620,573 | –4487 |
| Elucigene FH20 | 44,059,813 | 37,022 | –4,441,549 | –2209 |
| LIPOchip platform – Spain | 47,298,810 | 38,668 | –1,202,552 | –563 |
| CGA | 48,501,362 | 39,231 |
Table 64 presents the results of sensitivity analysis in which the lower limit of the sensitivity of Elucigene FH20 is used (i.e. a sensitivity of 0.286, taken from Hooper and colleagues36).
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 13,916,450 | 12,991 | |||
| Elucigene FH20_MLPA | 14,189,881 | 13,002 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 14,991,529 | 13,037 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 15,080,029 | 13,037 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 15,154,374 | 13,045 | 1,237,924 | 54 | 22,884 |
| LIPOchip_MLPA | 15,254,040 | 13,048 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 15,342,480 | 13,048 | Dominated | Dominated | Dominated |
| CGA | 15,528,212 | 13,056 | 373,838 | 11 | 33,402 |
| Elucigene FH20_CGA | 15,600,962 | 13,056 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 15,699,062 | 13,056 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 15,787,112 | 13,056 | Dominated | Dominated | Dominated |
| LDL-C | 17,678,183 | 13,079 | 2,149,970 | 23 | 93,518 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 13,916,450 | 12,991 | –3,761,732 | –88 |
| LIPOchip platform – Spain | 15,154,374 | 13,045 | –2,523,808 | –34 |
| CGA | 15,528,212 | 13,056 | –2,149,970 | –23 |
| LDL-C | 17,678,183 | 13,079 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 13,916,450 | 12,991 | –1,611,762 | –65 | |
| LIPOchip platform – Spain | 15,154,374 | 13,045 | –373,838 | –11 | |
| CGA | 15,528,212 | 13,056 | |||
| LDL-C | 17,678,183 | 13,079 | 2,149,970 | 23 | 93,518 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 42,055,432 | 35,946 | |||
| Elucigene FH20_MLPA | 43,157,577 | 36,509 | Ext Dom | Ext Dom | Ext Dom |
| LDL-C | 43,880,789 | 34,744 | Dominated | Dominated | Dominated |
| LIPOchip | 46,506,304 | 38,240 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 46,594,804 | 38,240 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 47,298,810 | 38,668 | 5,243,377 | 2722 | 1926 |
| LIPOchip_MLPA | 47,597,529 | 38,803 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 47,685,969 | 38,803 | Dominated | Dominated | Dominated |
| CGA | 48,501,362 | 39,231 | 1,202,552 | 563 | 2135 |
| Elucigene FH20_CGA | 48,574,112 | 39,231 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 48,672,212 | 39,231 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 48,760,262 | 39,231 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 42,055,432 | 35,946 | –1,825,357 | 1202 | Dominant |
| LDL-C | 43,880,789 | 34,744 | |||
| LIPOchip platform – Spain | 47,298,810 | 38,668 | 3,418,020 | 3924 | 871 |
| CGA | 48,501,362 | 39,231 | 4,620,573 | 4487 | 1030 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 42,055,432 | 35,946 | –6,445,390 | –3285 |
| LIPOchip platform – Spain | 47,298,810 | 38,668 | –1,202,552 | –563 |
| CGA | 48,501,362 | 39,231 |
Tables 65 and 66 report deterministic sensitivity analyses for LIPOchip sensitivity. The high value is taken from Stef and colleagues42 and the low value is taken from Callaway and colleagues. 40
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 14,192,370 | 13,005 | |||
| Elucigene FH20_MLPA | 14,462,411 | 13,016 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip platform – Spain | 15,149,123 | 13,045 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 15,279,477 | 13,051 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 15,351,177 | 13,051 | Dominated | Dominated | Dominated |
| CGA | 15,528,212 | 13,056 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip_MLPAa | 15,538,448 | 13,063 | 1,346,078 | 57 | 23,452 |
| Elucigene FH20_CGA | 15,575,762 | 13,056 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_MLPA | 15,610,148 | 13,063 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 15,672,512 | 13,056 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 15,744,212 | 13,056 | Dominated | Dominated | Dominated |
| LDL-C | 17,678,183 | 13,079 | 2,139,735 | 17 | 127,161 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 14,192,370 | 13,005 | –3,485,812 | –74 |
| LIPOchip_MLPAa | 15,538,448 | 13,063 | –2,139,735 | –17 |
| LDL-C | 17,678,183 | 13,079 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 14,192,370 | 13,005 | –1,335,842 | –51 | |
| CGA | 15,528,212 | 13,056 | |||
| LIPOchip_MLPAa | 15,538,448 | 13,063 | 10,235 | 6 | 1661 |
| LDL-C | 17,678,183 | 13,079 | 2,149,970 | 23 | 93,518 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 43,371,985 | 36,653 | |||
| LDL-C | 43,880,789 | 34,744 | Dominated | Dominated | Dominated |
| Elucigene FH20_MLPA | 44,470,770 | 37,216 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip platform – Spain | 47,293,559 | 38,668 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 47,880,247 | 38,978 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 47,951,947 | 38,978 | Dominated | Dominated | Dominated |
| CGA | 48,501,362 | 39,231 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_CGA | 48,548,912 | 39,231 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 48,645,662 | 39,231 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 48,717,362 | 39,231 | Dominated | Dominated | Dominated |
| LIPOchip_MLPAa | 48,967,932 | 39,541 | 5,595,947 | 2888 | 1937 |
| Elucigene FH20_LIPOchip_MLPA | 49,039,632 | 39,541 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 43,371,985 | 36,653 | –508,805 | 1909 | Dominant |
| LDL-C | 43,880,789 | 34,744 | |||
| LIPOchip_MLPAa | 48,967,932 | 39,541 | 5,087,143 | 4797 | 1060 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 43,371,985 | 36,653 | –5,129,377 | –2578 | |
| CGA | 48,501,362 | 39,231 | – | – | |
| LIPOchip_MLPAa | 48,967,932 | 39,541 | 466,570 | 310 | 1504 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LIPOchip | 14,175,438 | 12,995 | |||
| Elucigene FH20 | 14,192,370 | 13,005 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 14,247,138 | 12,995 | Dominated | Dominated | Dominated |
| LIPOchip_MLPA | 14,447,909 | 13,006 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_MLPA | 14,462,441 | 13,016 | 287,002 | 21 | 13,523 |
| Elucigene FH20_LIPOchip_MLPA | 14,519,669 | 13,006 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 15,169,148 | 13,045 | 706,707 | 29 | 24,497 |
| CGA | 15,528,212 | 13,056 | 359,064 | 11 | 32,082 |
| Elucigene FH20_CGA | 15,575,762 | 13,056 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 15,773,762 | 13,056 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 15,845,912 | 13,056 | Dominated | Dominated | Dominated |
| LDL-C | 17,678,183 | 13,079 | 2,149,970 | 23 | 93,518 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| LIPOchip | 14,175,438 | 12,995 | –3,502,744 | –84 |
| Elucigene FH20_MLPA | 14,462,441 | 13,016 | –3,215,742 | –63 |
| LIPOchip platform – Spain | 15,169,148 | 13,045 | –2,509,034 | –34 |
| CGA | 15,528,212 | 13,056 | –2,149,970 | –23 |
| LDL-C | 17,678,183 | 13,079 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LIPOchip | 14,175,438 | 12,995 | –1,352,774 | –61 | |
| Elucigene FH20_MLPA | 14,462,441 | 13,016 | –1,065,771 | –40 | |
| LIPOchip platform – Spain | 15,169,148 | 13,045 | –359,064 | –11 | |
| CGA | 15,528,212 | 13,056 | |||
| LDL-C | 17,678,183 | 13,079 | 2,149,970 | 23 | 93,518 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LIPOchip | 42,612,324 | 36,148 | |||
| Elucigene FH20_LIPOchip | 42,684,024 | 36,148 | Dominated | Dominated | Dominated |
| Elucigene FH20 | 43,371,985 | 36,653 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip_MLPA | 43,713,509 | 36,711 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 43,785,269 | 36,711 | Dominated | Dominated | Dominated |
| LDL-C | 43,880,789 | 34,744 | Dominated | Dominated | Dominated |
| Elucigene FH20_MLPA | 44,470,770 | 37,216 | 1,858,445 | 1068 | 1740 |
| LIPOchip platform – Spain | 47,313,584 | 38,668 | 2,842,814 | 1452 | 1958 |
| CGA | 48,501,362 | 39,231 | 1,187,778 | 563 | 2109 |
| Elucigene FH20_CGA | 48,548,912 | 39,231 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 48,746,912 | 39,231 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 48,819,062 | 39,231 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LIPOchip | 42,612,324 | 36,148 | –1,268,465 | 1404 | Dominant |
| LDL-C | 43,880,789 | 34,744 | |||
| Elucigene FH20_MLPA | 44,470,770 | 37,216 | 589,980 | 2472 | 239 |
| LIPOchip platform – Spain | 47,313,584 | 38,668 | 3,432,794 | 3924 | 875 |
| CGA | 48,501,362 | 39,231 | 4,620,573 | 4487 | 1030 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| LIPOchip | 42,612,324 | 36,148 | –5,889,038 | –3083 |
| Elucigene FH20_MLPA | 44,470,770 | 37,216 | –4,030,592 | –2015 |
| LIPOchip platform – Spain | 47,313,584 | 38,668 | –1,187,778 | –563 |
| CGA | 48,501,362 | 39,231 |
Table 67 details the results of sensitivity analysis using the upper bound of the CI for the sensitivity of LDL-C among relatives from Starr and colleagues. 49 This applies only to relatives and therefore results for index cases alone will not change in this analysis.
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 48,545,084 | 40,457 | |||
| Elucigene FH20_MLPA | 49,368,304 | 40,817 | Ext Dom | Ext Dom | Ext Dom |
| LDL-C | 50,043,807 | 39,276 | Dominated | Dominated | Dominated |
| LIPOchip | 50,902,913 | 41,473 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 50,974,613 | 41,473 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 51,486,042 | 41,747 | 2,940,959 | 1290 | 2280 |
| LIPOchip_MLPA | 51,718,572 | 41,833 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 51,790,272 | 41,833 | Dominated | Dominated | Dominated |
| CGA | 52,413,029 | 42,107 | 926,987 | 361 | 2571 |
| Elucigene FH20_CGA | 52,460,579 | 42,107 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 52,583,879 | 42,107 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 52,655,579 | 42,107 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 48,545,084 | 40,457 | –1,498,723 | 1181 | Dominant |
| LDL-C | 50,043,807 | 39,276 | |||
| LIPOchip platform – Spain | 51,486,042 | 41,747 | 1,442,235 | 2471 | 584 |
| CGA | 52,413,029 | 42,107 | 2,369,222 | 2832 | 837 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 48,545,084 | 40,457 | –3,867,946 | –1651 |
| LIPOchip platform – Spain | 51,486,042 | 41,747 | –926,987 | –361 |
| CGA | 52,413,029 | 42,107 |
Table 68 details the results of sensitivity analysis using the lower bound of the CI for the sensitivity of LDL-C among relatives from Starr and colleagues. 49 This applies only to relatives and therefore results for index cases alone will not change in this analysis.
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LDL-C | 38,314,283 | 30,629 | |||
| Elucigene FH20 | 38,699,584 | 33,199 | 385,301 | 2570 | 150 |
| Elucigene FH20_MLPA | 40,047,263 | 33,946 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 42,535,238 | 35,304 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 42,606,938 | 35,304 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 43,516,854 | 35,872 | 4,817,270 | 2673 | 1802 |
| LIPOchip_MLPA | 43,875,357 | 36,051 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 43,947,057 | 36,051 | Dominated | Dominated | Dominated |
| CGA | 44,968,300 | 36,619 | 1,451,446 | 747 | 1942 |
| Elucigene FH20_CGA | 45,015,850 | 36,619 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 45,139,150 | 36,619 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 45,210,850 | 36,619 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LDL-C | 38,314,283 | 30,629 | |||
| Elucigene FH20 | 38,699,584 | 33,199 | 385,301 | 2570 | 150 |
| LIPOchip platform – Spain | 43,516,854 | 35,872 | 5,202,571 | 5243 | 992 |
| CGA | 44,968,300 | 36,619 | 6,654,018 | 5990 | 1111 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| LDL-C | 38,314,283 | 30,629 | –6,654,018 | –5990 |
| Elucigene FH20 | 38,699,584 | 33,199 | –6,268,716 | –3420 |
| LIPOchip platform – Spain | 43,516,854 | 35,872 | –1,451,446 | –747 |
| CGA | 44,968,300 | 36,619 |
Table 69 presents analysis for the high estimate of the sensitivity of LDL-C in index cases (high value = 1), assuming that if the LDL-C test result is negative then the index case is a true negative; however, this is not always the case in reality.
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 14,192,370 | 13,005 | |||
| Elucigene FH20_MLPA | 14,462,441 | 13,016 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 14,991,529 | 13,037 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 15,063,229 | 13,037 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 15,154,374 | 13,045 | 962,004 | 40 | 24,025 |
| LIPOchip_MLPA | 15,254,040 | 13,048 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 15,325,740 | 13,048 | Dominated | Dominated | Dominated |
| CGA | 15,528,212 | 13,056 | 373,838 | 11 | 33,402 |
| Elucigene FH20_CGA | 15,575,762 | 13,056 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 15,699,062 | 13,056 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 15,770,762 | 13,056 | Dominated | Dominated | Dominated |
| LDL-C | 17,857,701 | 13,089 | 2,329,489 | 32 | 72,493 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 14,192,370 | 13,005 | –3,665,331 | –83 |
| LIPOchip platform – Spain | 15,154,374 | 13,045 | –2,703,327 | –43 |
| CGA | 15,528,212 | 13,056 | –2,329,489 | –32 |
| LDL-C | 17,857,701 | 13,089 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 14,192,370 | 13,005 | –1,335,842 | –51 |
| LIPOchip platform – Spain | 15,154,374 | 13,045 | –373,838 | –11 |
| CGA | 15,528,212 | 13,056 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 43,371,985 | 36,653 | |||
| LDL-C | 44,060,308 | 34,753 | Dominated | Dominated | Dominated |
| Elucigene FH20_MLPA | 44,470,770 | 37,216 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 46,506,304 | 38,240 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 46,578,004 | 38,240 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 47,298,810 | 38,668 | 3,926,825 | 2015 | 1949 |
| LIPOchip_MLPA | 47,597,529 | 38,803 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 47,669,229 | 38,803 | Dominated | Dominated | Dominated |
| CGA | 48,501,362 | 39,231 | 1,202,552 | 563 | 2135 |
| Elucigene FH20_CGA | 48,548,912 | 39,231 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 48,672,212 | 39,231 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 48,743,912 | 39,231 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 43,371,985 | 36,653 | –688,323 | 1900 | Dominant |
| LDL-C | 44,060,308 | 34,753 | |||
| LIPOchip platform – Spain | 47,298,810 | 38,668 | 3,238,502 | 3915 | 827 |
| CGA | 48,501,362 | 39,231 | 4,441,054 | 4478 | 992 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 43,371,985 | 36,653 | –5,129,377 | –2578 |
| LIPOchip platform – Spain | 47,298,810 | 38,668 | –1,202,552 | –563 |
| CGA | 48,501,362 | 39,231 |
Table 70 presents analysis for the low estimate of sensitivity of LDL-C among index cases (low value = 0.54,45 MedPed criteria).
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 14,192,370 | 13,005 | |||
| Elucigene FH20_MLPA | 14,462,441 | 13,016 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 14,991,529 | 13,037 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 15,063,229 | 13,037 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 15,154,374 | 13,045 | 962,004 | 40 | 24,025 |
| LIPOchip_MLPA | 15,254,040 | 13,048 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 15,325,740 | 13,048 | Dominated | Dominated | Dominated |
| CGA | 15,528,212 | 13,056 | 373,838 | 11 | 33,402 |
| Elucigene FH20_CGA | 15,575,762 | 13,056 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 15,699,062 | 13,056 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 15,770,762 | 13,056 | Dominated | Dominated | Dominated |
| LDL-C | 17,031,916 | 13,047 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 14,192,370 | 13,005 | –2,839,546 | –41 | |
| LIPOchip platform – Spain | 15,154,374 | 13,045 | –1,877,542 | –1 | |
| CGA | 15,528,212 | 13,056 | –1,503,704 | 10 | Dominant |
| LDL-C | 17,031,916 | 13,047 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 14,192,370 | 13,005 | –1,335,842 | –51 |
| LIPOchip platform – Spain | 15,154,374 | 13,045 | –373,838 | –11 |
| CGA | 15,528,212 | 13,056 |
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LDL-C | 43,234,523 | 34,711 | |||
| Elucigene FH20 | 43,371,985 | 36,653 | 137,462 | 1942 | 71 |
| Elucigene FH20_MLPA | 44,470,770 | 37,216 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 46,506,304 | 38,240 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 46,578,004 | 38,240 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 47,298,810 | 38,668 | 3,926,825 | 2015 | 1949 |
| LIPOchip_MLPA | 47,597,529 | 38,803 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 47,669,229 | 38,803 | Dominated | Dominated | Dominated |
| CGA | 48,501,362 | 39,231 | 1,202,552 | 563 | 2135 |
| Elucigene FH20_CGA | 48,548,912 | 39,231 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 48,672,212 | 39,231 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 48,743,912 | 39,231 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| LDL-C | 43,234,523 | 34,711 | |||
| Elucigene FH20 | 43,371,985 | 36,653 | 137,462 | 1942 | 71 |
| LIPOchip platform – Spain | 47,298,810 | 38,668 | 4,064,287 | 3957 | 1027 |
| CGA | 48,501,362 | 39,231 | 5,266,839 | 4520 | 1165 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| LDL-C | 43,234,523 | 34,711 | –5,266,839 | –4520 |
| Elucigene FH20 | 43,371,985 | 36,653 | –5,129,377 | –2578 |
| LIPOchip platform – Spain | 47,298,810 | 38,668 | –1,202,552 | –563 |
| CGA | 48,501,362 | 39,231 |
Table 71 details the results of sensitivity analysis using the upper bound of the CI for the specificity of LDL-C among relatives (values are taken from Starr and colleagues49 and apply only to index cases and relatives together). Results refer to relatives of a 50-year-old index case as in the base-case model.
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 41,972,619 | 35,492 | |||
| LDL-C | 42,213,642 | 33,361 | Dominated | Dominated | Dominated |
| Elucigene FH20_MLPA | 43,145,947 | 36,117 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 45,316,985 | 37,253 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 45,388,685 | 37,253 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 46,166,129 | 37,728 | 4,193,510 | 2236 | 1875 |
| LIPOchip_MLPA | 46,482,753 | 37,878 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 46,554,453 | 37,878 | Dominated | Dominated | Dominated |
| CGA | 47,443,224 | 38,353 | 1,277,095 | 625 | 2043 |
| Elucigene FH20_CGA | 47,490,774 | 38,353 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 47,614,074 | 38,353 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 47,685,774 | 38,353 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 41,972,619 | 35,492 | –241,023 | 2131 | Dominant |
| LDL-C | 42,213,642 | 33,361 | |||
| LIPOchip platform – Spain | 46,166,129 | 37,728 | 3,952,486 | 4367 | 905 |
| CGA | 47,443,224 | 38,353 | 5,229,581 | 4992 | 1048 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 41,972,619 | 35,492 | –5,470,605 | –2861 |
| LIPOchip platform – Spain | 46,166,129 | 37,728 | –1,277,095 | –625 |
| CGA | 47,443,224 | 38,353 |
Table 72 details the results of sensitivity analysis using the lower bound of the CI for the specificity of LDL-C among relatives (values are taken from Starr and colleagues49 and apply only to index cases and relatives together). Results refer to relatives of a 50-year-old index case as in the base-case model.
| Test strategy | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 45,013,836 | 38,015 | |||
| LDL-C | 45,836,823 | 36,367 | Dominated | Dominated | Dominated |
| Elucigene FH20_MLPA | 46,025,161 | 38,506 | Ext Dom | Ext Dom | Ext Dom |
| LIPOchip | 47,901,711 | 39,398 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip | 48,973,411 | 39,398 | Dominated | Dominated | Dominated |
| LIPOchip platform – Spain | 48,627,764 | 39,771 | 3,613,928 | 1755 | 2059 |
| LIPOchip_MLPA | 48,905,476 | 39,888 | Ext Dom | Ext Dom | Ext Dom |
| Elucigene FH20_LIPOchip_MLPA | 48,977,176 | 39,888 | Dominated | Dominated | Dominated |
| CGA | 49,742,857 | 40,261 | 1,115,093 | 491 | 2273 |
| Elucigene FH20_CGA | 49,790,407 | 40,261 | Dominated | Dominated | Dominated |
| LIPOchip_CGA | 49,913,707 | 40,261 | Dominated | Dominated | Dominated |
| Elucigene FH20_LIPOchip_CGA | 49,985,407 | 40,261 | Dominated | Dominated | Dominated |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICER (£/QALY) |
|---|---|---|---|---|---|
| Elucigene FH20 | 45,013,836 | 38,015 | –822,988 | 1,648 | Dominant |
| LDL-C | 45,836,823 | 36,367 | |||
| LIPOchip platform – Spain | 48,627,764 | 39,771 | 2,790,941 | 3403 | 820 |
| CGA | 49,742,857 | 40,261 | 3,906,033 | 3894 | 1003 |
| Test | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs |
|---|---|---|---|---|
| Elucigene FH20 | 45,013,836 | 38,015 | –4,729,021 | –2246 |
| LIPOchip platform – Spain | 48,627,764 | 39,771 | –1,115,093 | –491 |
| CGA | 49,742,857 | 40,261 |
Appendix 15 Cost-effectiveness acceptability curves for each age subgroup
FIGURE 13.
Cost-effectiveness acceptability curve: age 15 years, index case.
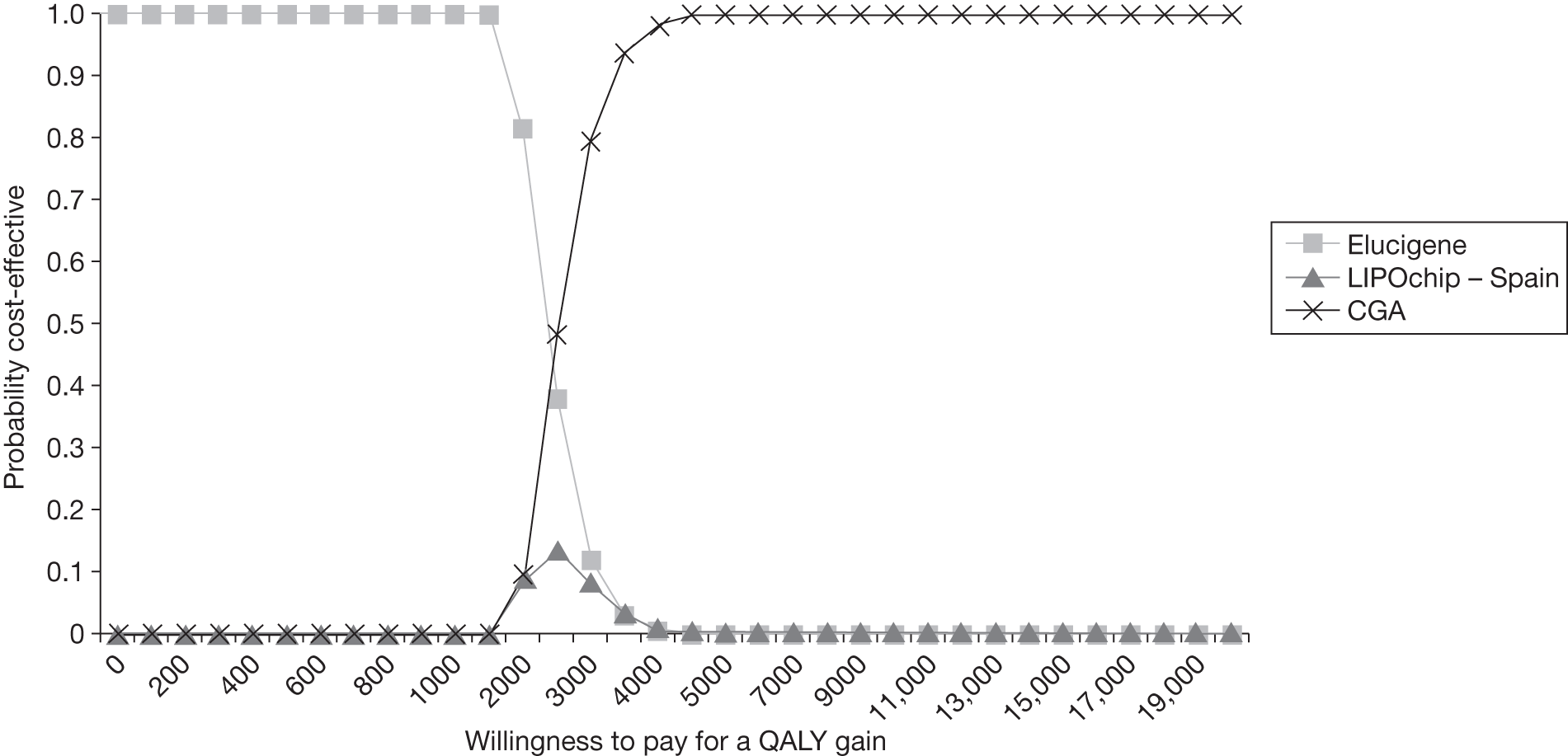
FIGURE 14.
Cost-effectiveness acceptability curve: age 30 years, index case.
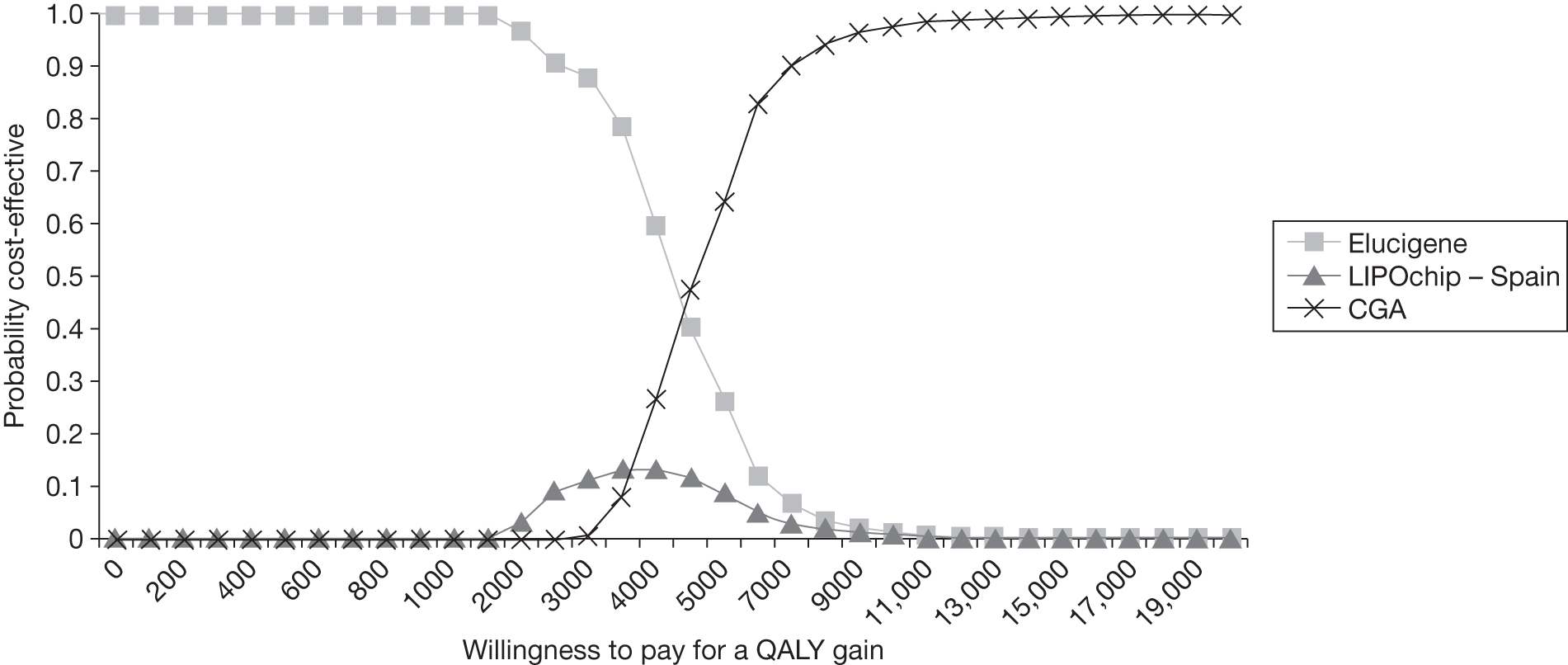
FIGURE 15.
Cost-effectiveness acceptability curve: age 65 years, index case.
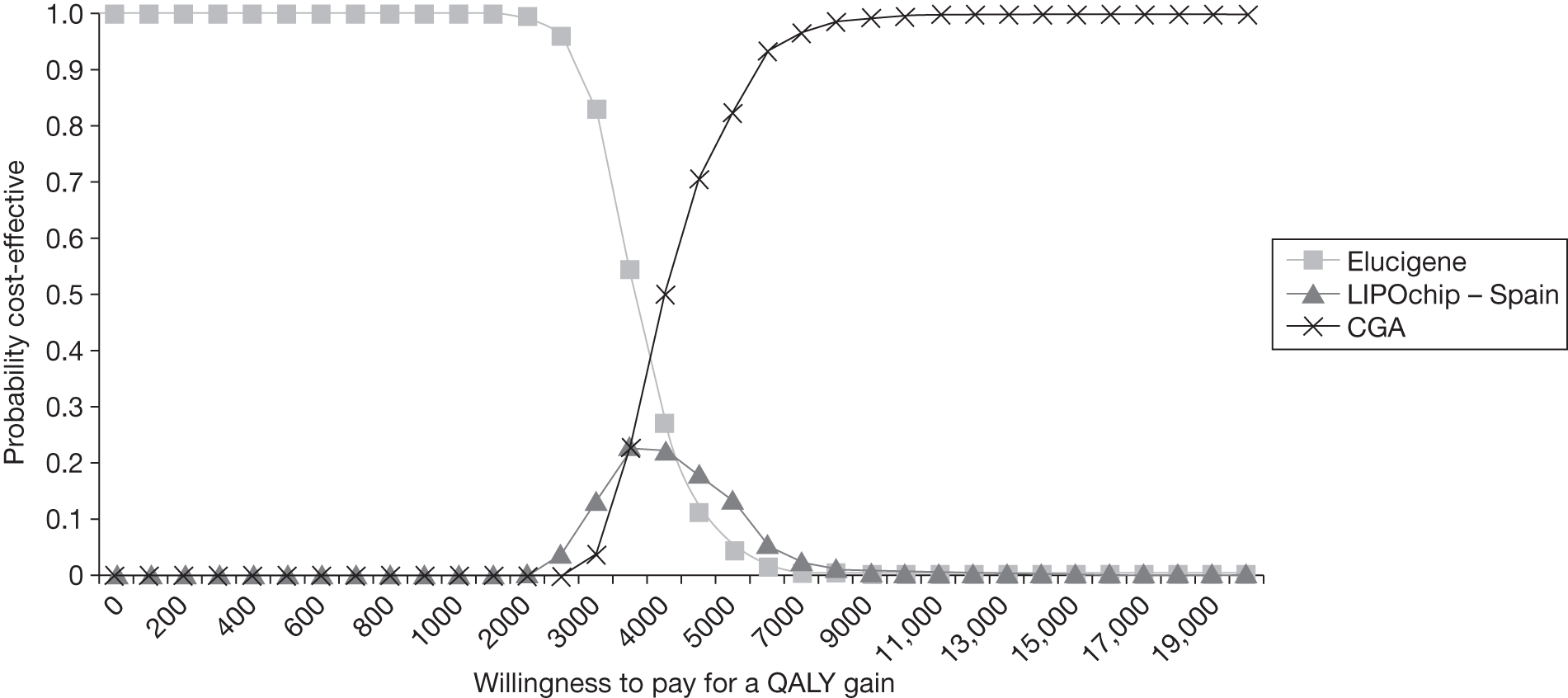
FIGURE 16.
Cost-effectiveness acceptability curve: age 75 years, index case.
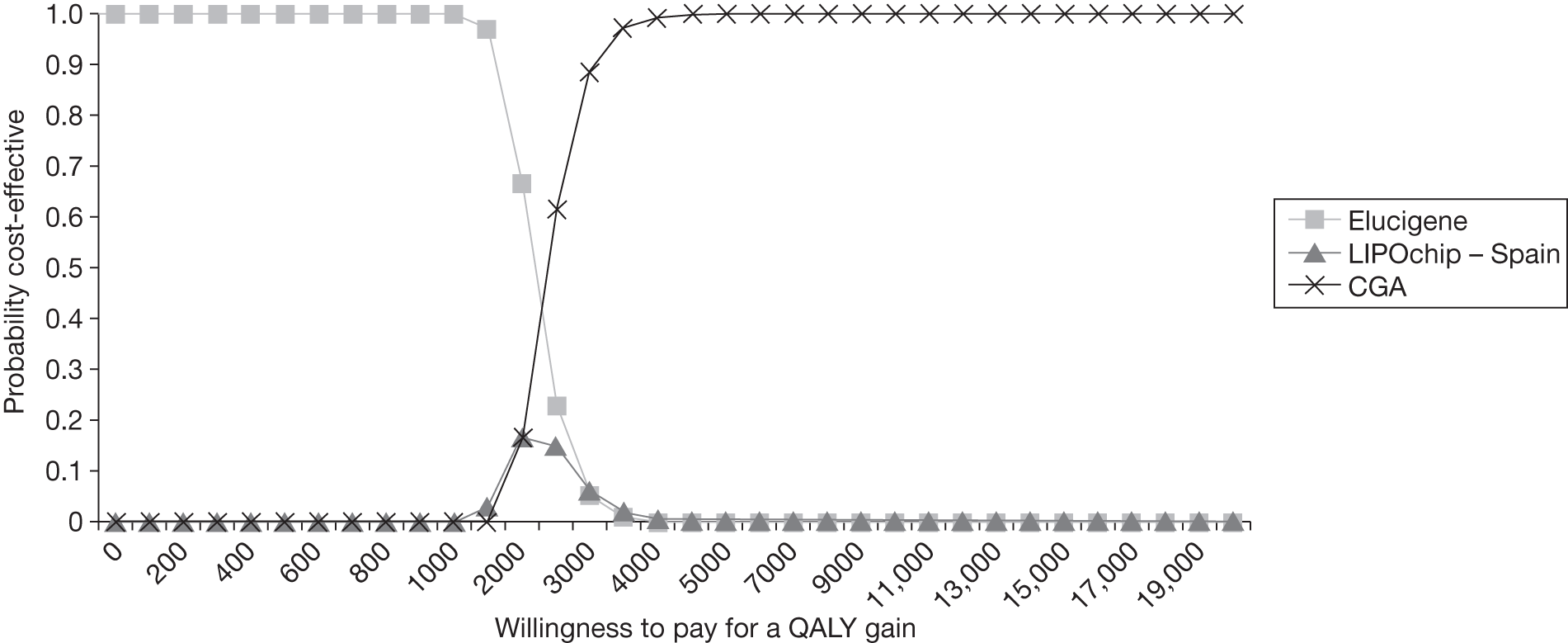
FIGURE 17.
Cost-effectiveness acceptability curve: age 85 years, index case.
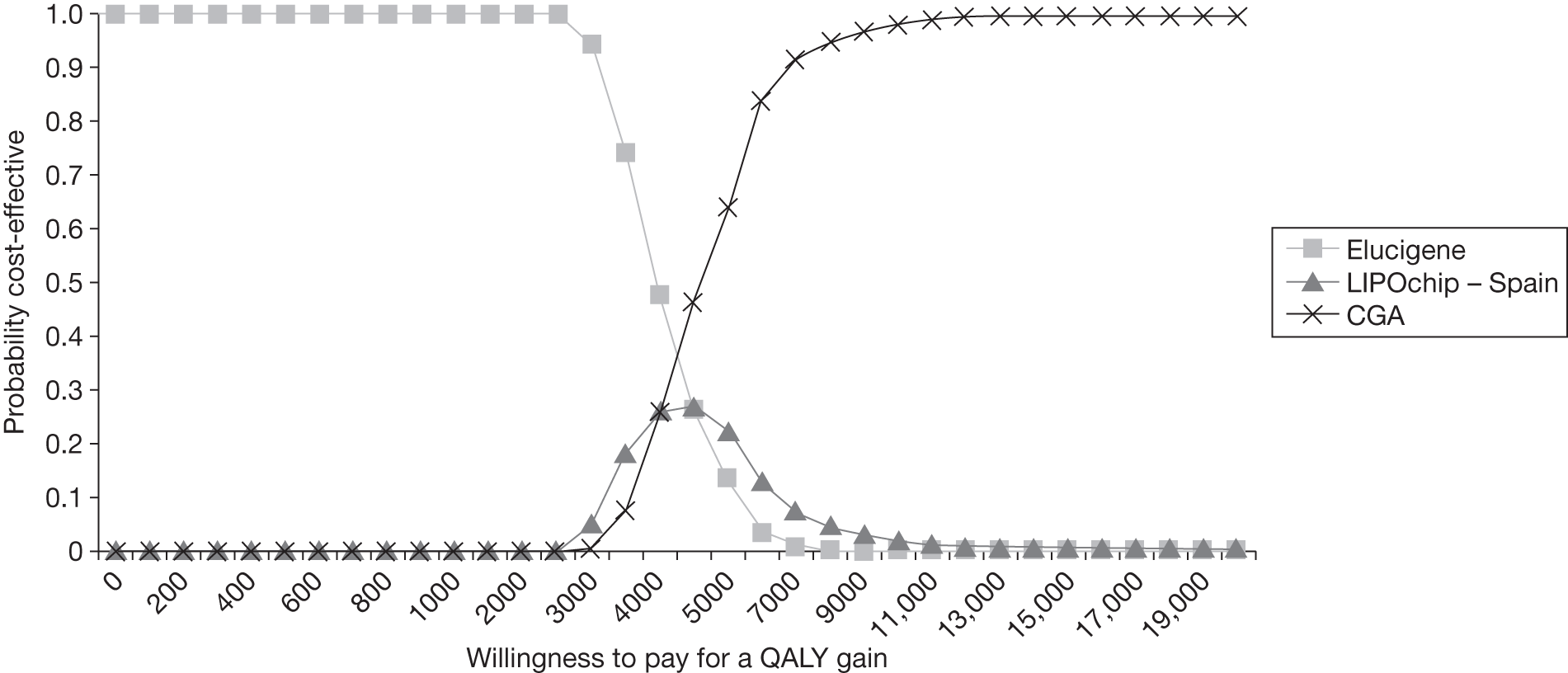
Appendix 16 Parameters for estimation of the distributions for the probabilistic model (base case)
| Parameter | Value | Low | High | Source | Distribution | Alpha | Beta |
|---|---|---|---|---|---|---|---|
| Costs of cardiovascular events (£) | |||||||
| No event | 74 | NICE 20081 | Gamma | 25 | 2.96 | ||
| MI (first year) | 3780 | NICE 20081 | Gamma | 25 | 151.1952 | ||
| MI (subsequent) | 500 | NICE 20081 | Gamma | 25 | 20 | ||
| Stroke (first year) | 4335 | NICE 20081 | Gamma | 25 | 173.4137 | ||
| Stroke (subsequent) | 2336 | NICE 20081 | Gamma | 25 | 93.44554 | ||
| PAD (first year) | 2212 | NICE 20081 | Gamma | 25 | 88.4974 | ||
| PAD (subsequent) | 285 | NICE 20081 | Gamma | 25 | 11.4053 | ||
| Heart failure (first year) | 4379 | NICE 20081 | Gamma | 25 | 175.1699 | ||
| Heart failure (subsequent) | 500 | NICE 20081 | Gamma | 25 | 20 | ||
| Revascularisation (first year) | 8610 | NICE 20081 | Gamma | 25 | 344.3940 | ||
| Revascularisation (subsequent) | 500 | NICE 20081 | Gamma | 25 | 20 | ||
| Unstable angina (first year) | 2074 | NICE 20081 | Gamma | 25 | 82.9677 | ||
| Unstable angina (subsequent) | 500 | NICE 20081 | Gamma | 25 | 20 | ||
| Additional cost parameters | |||||||
| Cost per MOLU | 30 | 20 | 40 | Personal communication | Beta | 1 | 1 |
| Cost LDL-C | 19.97 | 15.97 | 23.96 | Assumed high and low | Beta | 1 | 1 |
| Cost low-intensity statins | 17.21 | 13.77 | 20.65 | Assumed high and low | Beta | 1 | 1 |
| Cost high-intensity statins | 377 | 302 | 453 | Assumed standard error | Gamma | 25 | 15.0993 |
| Test sensitivity and specificity | |||||||
| Elucigene sensitivity | 0.4397 | 0.286a | 0.52a | Taylor 201037 | Beta | 102 | 130 |
| LIPOchip sensitivity | 0.7846 | 0.33a | 0.945a | Palacios 201041 | Beta | 57 | 8 |
| LIPOchip platform – Spain, sensitivity | 0.8776 | 0.805 | 1 | Assumption | Beta | 57 | 8 |
| LDL-C index cases, sensitivity | 0.9 | 0.72 | 1 | Damgaard, 200545 | Beta | 1 | 1 |
| LDL-C (relatives) sensitivity | 0.576 | 0.469 | 0.679 | Starr, 200849 | Beta | 50.36 | 37.07 |
| LDL-C (relatives) specificity | 0.837 | 0.8 | 0.87 | Starr, 200849 | Beta | 401.99 | 78.29 |
| Health-state multipliers | |||||||
| MI | 0.76 | 0.56 | 0.96 | NICE 20081 | Beta | 427.09 | 134.8711 |
| Post MI | 0.88 | 0.78 | 1.00 | NICE 20081 | Beta | 285.93 | 38.9911 |
| Stroke | 0.629 | 0.43 | 0.83 | NICE 20081 | Beta | 91.1103 | 53.7391 |
| Post stroke | 0.629 | 0.43 | 0.83 | NICE 20081 | Beta | 91.1103 | 53.7391 |
| PAD | 0.9 | 0.86 | 0.98 | NICE 20081 | Beta | 201.6 | 22.4 |
| Post PAD | 0.9 | 0.86 | 0.98 | NICE 20081 | Beta | 201.6 | 22.4 |
| Heart failure | 0.683 | 0.48 | 0.88 | NICE 20081 | Beta | 369.0095 | 171.268 |
| Post heart failure | 0.683 | 0.48 | 0.88 | NICE 20081 | Beta | 369.0095 | 171.268 |
| Revascularisation | 0.93 | 0.74 | 1.00 | NICE 20081 | Beta | 31.3118 | 2.3568 |
| Post revascularisation | 0.93 | 0.74 | 1.00 | NICE 20081 | Beta | 40.9973 | 3.0858 |
| Unstable angina | 0.77 | 0.57 | 0.97 | NICE 20081 | Beta | 420.1158 | 125.4891 |
| Post unstable angina | 0.88 | 0.78 | 1.00 | NICE 20081 | Beta | 285.9348 | 38.9911 |
| General population quality of life | |||||||
| < 25 years | 0.94 | 0.705 | 1 | NICE 20081 | Beta | 1 | 1 |
| 25–34 years | 0.93 | 0.636 | 1 | NICE 20081 | Beta | 1 | 1 |
| 35–44 years | 0.91 | 0.596 | 1 | NICE 20081 | Beta | 1 | 1 |
| 45–54 years | 0.85 | 0.36 | 1 | NICE 20081 | Beta | 1 | 1 |
| 55–64 years | 0.8 | 0.29 | 1 | NICE 20081 | Beta | 1 | 1 |
| 65–74 years | 0.78 | 0.27 | 1 | NICE 20081 | Beta | 1 | 1 |
| 75+ years | 0.73 | 0.20 | 1 | NICE 20081 | Beta | 1 | 1 |
| Treatment effect for each health state in the model | |||||||
| MI | 0.81 | 0.72 | 0.91 | Assumed standard error | Beta | 47.079 | 11.0432 |
| Stroke | 0.82 | 0.70 | 0.96 | Assumed standard error | Beta | 22.902 | 5.0273 |
| TIA | 0.79 | 0.65 | 0.94 | Assumed standard error | Beta | 21.587 | 5.7383 |
| PAD | 0.87 | 0.69 | 1.00 | Assumed standard error | Beta | 21.497 | 3.2122 |
| Heart failure | 0.77 | 0.65 | 0.92 | Assumed standard error | Beta | 22.513 | 6.7247 |
| Revascularisation | 0.78 | 0.69 | 1.00 | Assumed standard error | Beta | 9.8438 | 2.7764 |
| Unstable angina | 0.84 | 0.71 | 0.86 | Assumed standard error | Beta | 1083.4132 | 206.3644 |
| CVD death | 0.92 | 0.72 | 1.00 | Assumed standard error | Beta | 39.7241 | 3.4543 |
| Death other | 1.00 | 0.80 | 1.00 | Assumed standard error | Beta | 1 | 1 |
Appendix 17 Cost-effectiveness acceptability curves for alternative mutation test-positive rates on comprehensive genetic analysis
FIGURE 18.
Cost-effectiveness acceptability curve: incidence of genetic mutation = 5%.
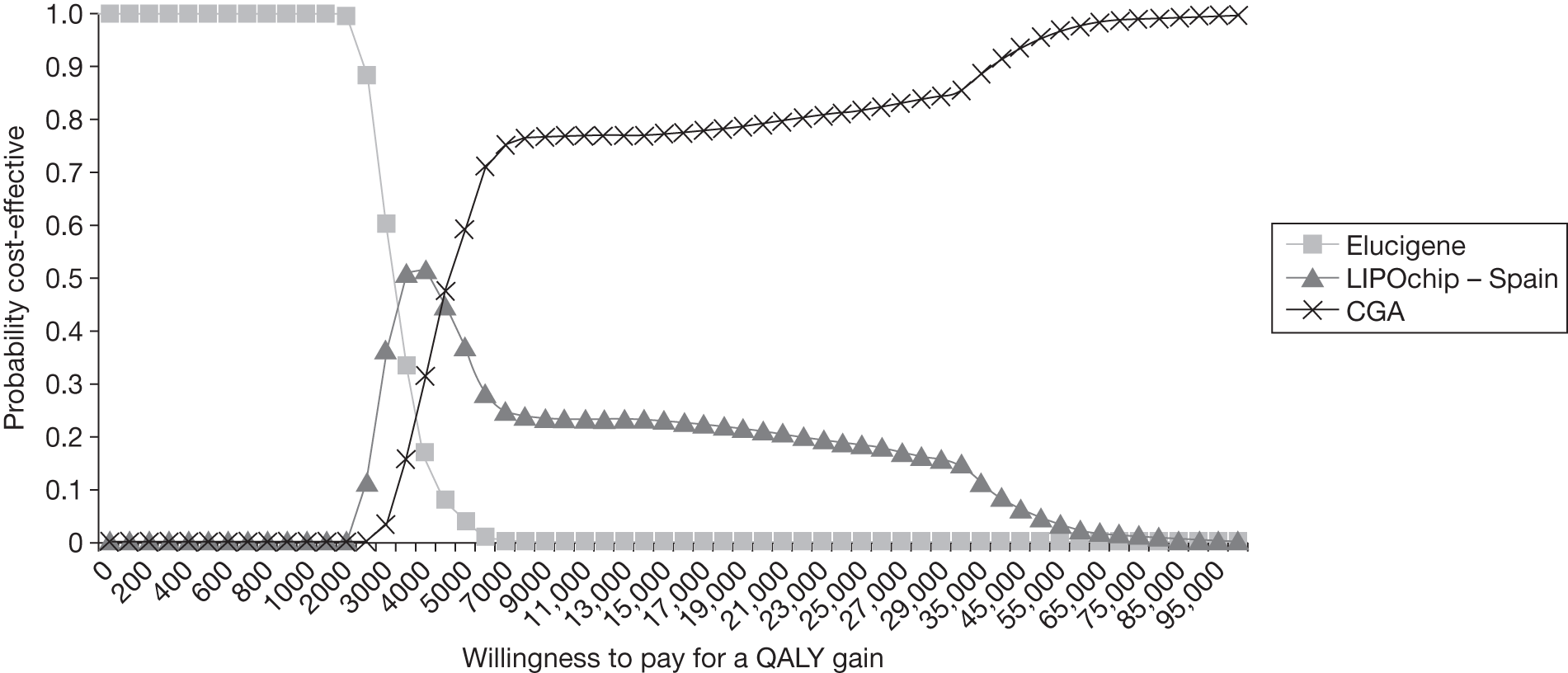
FIGURE 19.
Cost-effectiveness acceptability curve: incidence of genetic mutation = 10%.
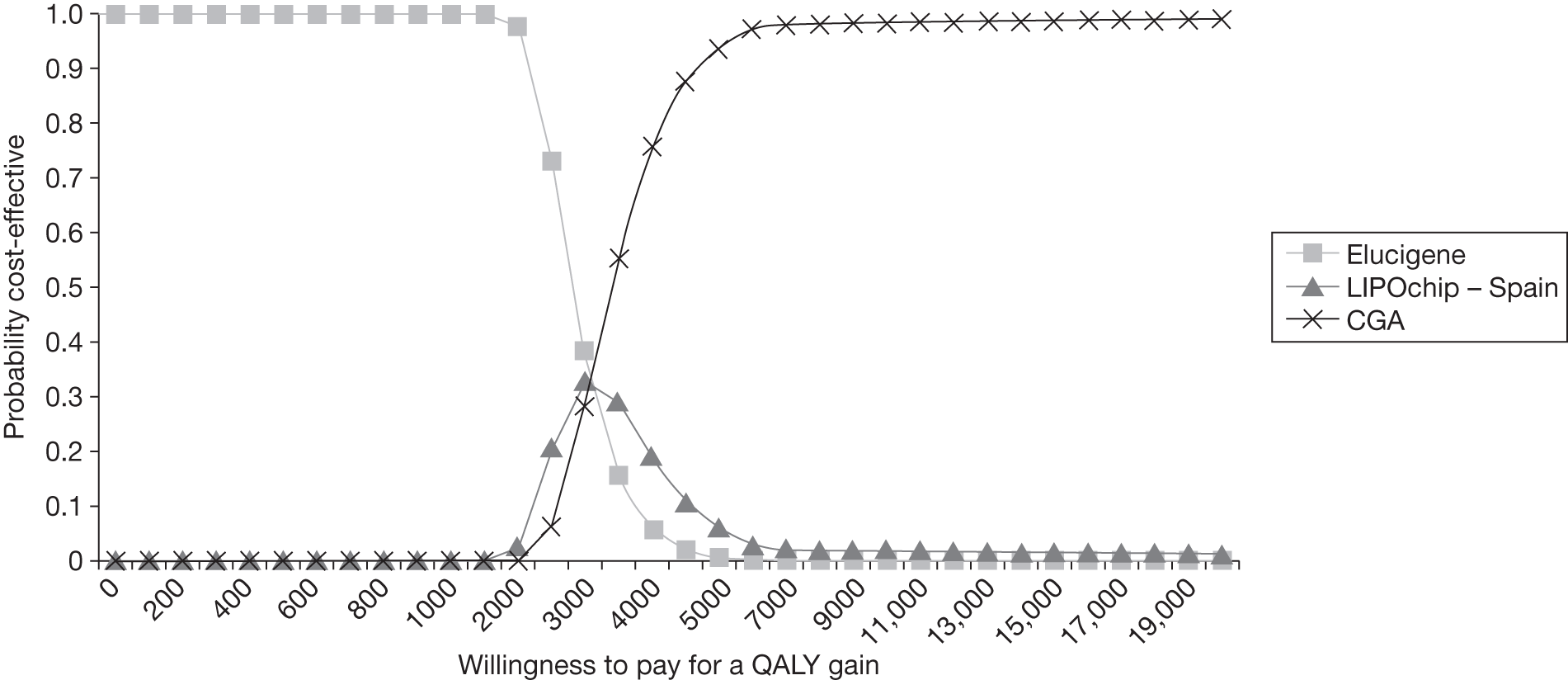
FIGURE 20.
Cost-effectiveness acceptability curve: incidence of genetic mutation = 20%.
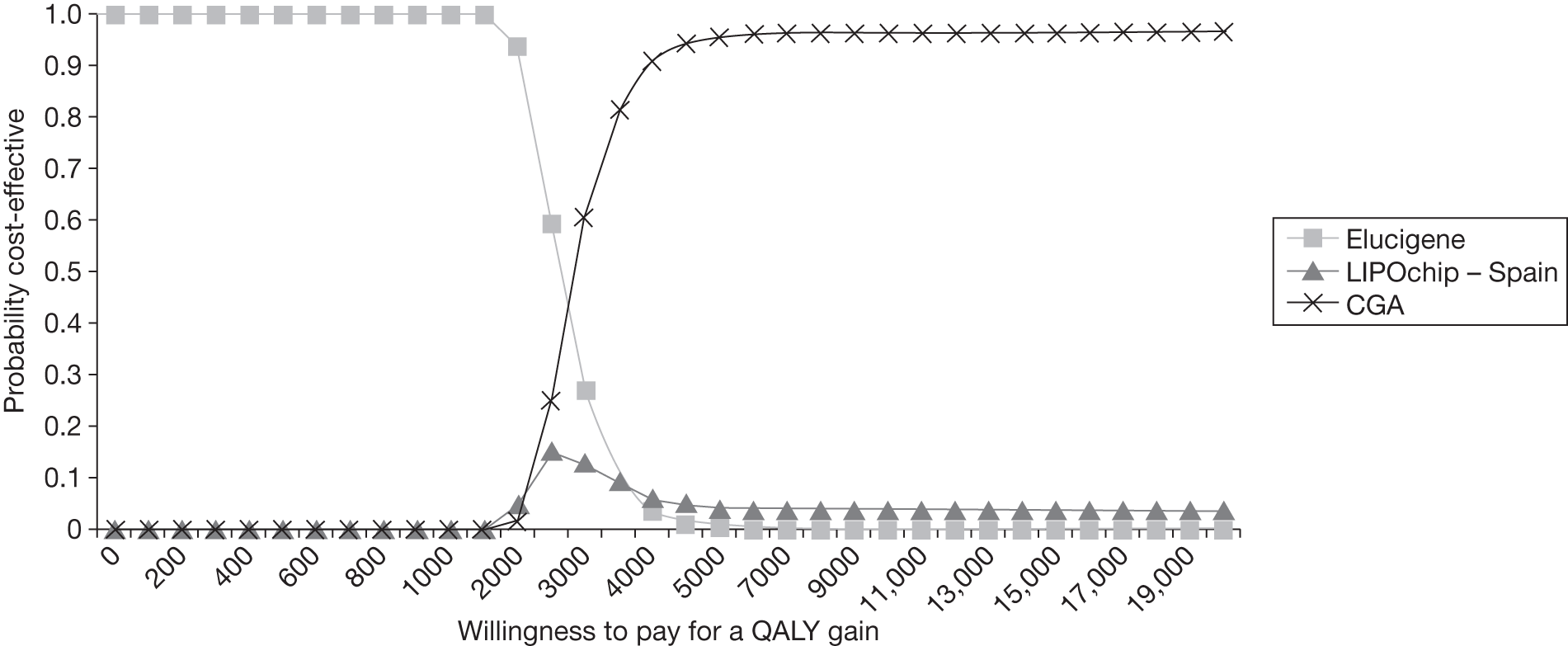
FIGURE 21.
Cost-effectiveness acceptability curve: incidence of genetic mutation = 50%.
Appendix 18 Proportion identified by cascade testing using comprehensive genetic analysis (targeted sequencing) or age- and gender- specific low-density lipoprotein cholesterol cut-offs
| Study | Country | Clinical diagnosis of index cases | Study participants | Number of participants | Test for cascading | Proportion identified from cascade testing |
|---|---|---|---|---|---|---|
| Bourbon 200885 | Portugal | Simon Broome criteria | Relatives of index cases |
Index cases = 88; relatives = 206 Families = 165; relatives = 226 |
Targeted sequencing (LDLR/APOB/PCSK9). Index cases tested with dHPLC/sequencing/MLPA | 56% (116/206) (as of 2008); 51% (226/443) (as of 2010) |
| Hadfield 200966 | UK | Definite or possible Simon Broome criteria | First-degree relatives of index cases | Index cases = 931; relatives = 591 | LDL-C age- and gender-specific cut-offs according to NICE guideline. Living in catchment area | Likely plus uncertain = 42% (250/591); likely = 28% (168/591); uncertain = 14% (82/591) |
| Relatives = 178 | LDL-C age- and gender-specific cut-offs according to NICE guideline. Living in non-catchment area | Likely plus uncertain = 40% (72/178); likely = 29% (51/178); uncertain = 12% (21/178) | ||||
| Humphries 200684 | UK | Definite or possible Simon Broome criteria | First-degree relatives of index cases | Index cases = 69; relatives = 54 | Targeted sequencing (LDLR/APOB). Index cases tested with SSCP/sequencing/UPQFM-PCR | 50% (27/54) |
| Leren 200811 | Norway | Not reported | First-degree relatives of index cases | Index cases = 440; relatives = 1805 | Targeted sequencing (LDLR/APOB). Index cases tested with sequencing/MLPA/PCR for APOB | 45% (808/1805) |
| Umans-Eckenhausen 200119 | The Netherlands | Dutch criteria | First- and second-degree relatives of index cases | 237 families; relatives = 2039 | Targeted sequencing (LDLR/APOB). Index cases tested with DGGE/sequencing/restriction digest analysis | 37% (2039/5442) |
| Vergotine 200183 | South Africa | Families of index cases | Index cases = 379; relatives = 790 | Targeted sequencing (LDLR) | 43% (338/790) |
Appendix 19 Protocol
Final protocol, 16th December 2010
1. Title of the project
The clinical and cost-effectiveness of Elucigene FH20 and LIPOchip for the diagnosis of familial hypercholesterolaemia: systematic review and economic evaluation.
2. Name of External Assessment Group (EAG) and project lead
Aberdeen Technology Assessment Group
Pawana Sharma
Research Fellow
Health Services Research Unit
3rd Floor
University of Aberdeen
Health Sciences Building
Foresterhill
Aberdeen
AB25 2ZD
Tel: 01224 559055
Email: p.sharma@abdn.ac.uk
Reserve contact:
Graham Mowatt
Senior Research Fellow
Health Services Research Unit
3rd Floor
University of Aberdeen
Health Sciences Building
Foresterhill
Aberdeen
AB25 2ZD
Tel: 01224 552494
Email: g.mowatt@abdn.ac.uk
3. Plain English Summary
Familial hypercholesterolaemia (FH) is an inherited (genetic) condition resulting in raised levels of cholesterol in the blood. A person can either inherit the genetic defect from one parent (heterozygous FH) or from both parents (homozygous FH). In the UK heterozygous FH has a frequency of 1 in 500, affecting around 100,000 people in England, while homozygous FH is much rarer, with a frequency of 1 in one million. 1 The condition is transmitted from generation to generation, so that the siblings or children of a person with FH have a 50% risk of inheriting the genetic defect.
The raised levels of cholesterol in the blood that characterise heterozygous FH lead to a greater than 50% risk of coronary heart disease (CHD) by the age of 50 in men and at least 30% risk in women by the age of 60. 2 If untreated, around 50% of men will die before the age of 60. 3 People with homozygous FH have a significantly poorer prognosis than those with heterozygous FH.
FH is generally characterised by the presence of increased levels of cholesterol concentration and clinical symptoms such as tendon xanthomata (yellowish skin lesions on the tendons of the hands and feet) and a family history of CHD. However there are variations in the time at which clinical signs and CHD appear. 4 Tendon xanthomata, which are frequent but not always present, may be seen in the second decade of life, while CHD is usually present by the fourth decade. Diagnosis of FH by cholesterol concentration is not entirely reliable3 with a 10% risk of misdiagnosis. 5 FH is an underdiagnosed condition, with at least 75% of people in the UK with heterozygous FH remaining undiagnosed. 6
The NICE clinical guideline on identification and management of familial hypercholesterolaemia recommends that a diagnosis of FH should be made using the Simon Broome criteria, which include a combination of family history, clinical signs, cholesterol concentration and DNA testing, to improve diagnosis and early identification of FH. 7 Cascade testing (a mechanism for identifying people at risk of FH by a process of family tracing) using a combination of DNA testing and low density lipoprotein cholesterol (LDL-C) concentration measurement is recommended to identify affected relatives of those individuals with a clinical diagnosis of FH. 7 The aim of early identification is to reduce the risk of vascular diseases by starting treatment with cholesterol-lowering drugs such as statins and by allowing management by lifestyle changes and diet modification. 8 The use of statins, even in lower doses than recommended, can reduce the risk of CHD in patients with FH. 9 The standard method of DNA testing is comprehensive genetic analysis, which is the most complete genetic analysis generally available for FH within a diagnostic setting; however the process is slow and expensive (estimated at around £500 to £1000 per patient in the UK setting). 10,11 Elucigene FH2012 and LIPOchip13 are recently developed rapid genetic testing kits that are designed to detect a more limited number of genetic mutations associated with FH that are commonly found in the UK population.
This systematic review will assess the clinical and cost-effectiveness of the Elucigene FH20 kit, LIPOchip, and comparators, for the diagnosis and cascade testing of FH.
4. Decision problem
4.1 Purpose of the decision to be made
The purpose of this appraisal is to address the following questions:
-
What are the most effective and cost-effective strategies for confirming a diagnosis of FH in index individuals and for cascade testing of relatives?
-
In cascade testing of relatives for mutations identified in index individuals by Elucigene FH20 or LIPOchip, would it be more cost-effective to use those tests rather than targeted gene sequencing?
4.2 Clear definition of the intervention
Elucigene FH20
The Elucigene FH20 kit (Gen-Probe Life Sciences, UK), using the principle of an amplification refractory mutation system (ARMS), is designed to detect 20 genetic mutations associated with FH that are commonly found in the UK population (see Table 1). 14 These mutations, with a frequency ranging from 1.3% to 11.4%, were identified from a cohort study involving 400 patients in the UK with FH. 15 Of the 20 mutations, 18 are found in the Low-density lipoprotein receptor (LDLR) gene, one in the Apolipoprotein B (APOB) gene and one in the Protein convertase subtilisin/kexin 9 (PCSK) gene (Table 1). 14
| Gene | Mutation |
|---|---|
| Low-density lipoprotein receptor (LDLR) | P664L, L458P, R329X, E207X, D200G, E80K, IVS3+1G>A, D461H, ∆G197, fs206, Q363X, W66G, V408M, D206E, C656R, K290RfsX20, C163Y and D461N |
| Apolipoprotein B (APOB) | R3500Q |
| Protein convertase subtilisin/kexin 9 (PCSK9) | D374Y |
By using ARMS, the Elucigene FH20 kit combines the amplification step and diagnostic steps,16 making the process faster. A limitation of the kit is that it only tests for 20 FH mutations. Worldwide approximately 1200 FH-causing mutations have been identified,17 of which over 200 have been reported in the UK population.
LIPOchip
LIPOchip (Progenika Biopharma, Spain) is an alternative genetic test designed to diagnose FH. 13 LIPOchip is a tiered system that uses DNA array technology. The chip can detect point mutations, copy number changes and variation of number of copies of the LDLR gene. The current version (version 10) tests for 189 mutations in the LDLR, APOB and PCSK genes that are known to occur in the UK population.
The LIPOchip platform involves the following steps:
-
Firstly, samples are analysed using the DNA array which is designed to detect 189 mutations in the LDLR and APOB genes.
-
If the samples fail to detect these mutations they are analysed for large gene re-arrangements.
-
If the first two steps fail to detect mutations then samples are analysed by automated sequencing of the LDLR.
-
If all three of the above steps fail to detect mutations then the sample is confirmed as FH negative.
-
Finally, the LIPOchip software generates a report containing information on the pathogenicity of detected mutations.
The manufacturer also offers a LIPOchip test processing service from its laboratory in Spain.
4.3 Populations and relevant subgroups
The populations considered are adults and children with a clinical diagnosis of FH (the index individuals/probands) based on the Simon Broome criteria, and, for cascade testing, first-, second- and third-degree biological relatives.
4.4 Place of the interventions in the treatment pathway(s)
The care pathway for this assessment is based on the NICE clinical guideline on the identification and management of FH. 7
Index individuals
The assessment will investigate the effect of diagnostic strategies including Elucigene FH20 and/or LIPOchip for providing an unequivocal diagnosis of FH for those with a clinical diagnosis based on the Simon Broome criteria.
Cascade testing of relatives
The assessment will investigate the effect of diagnostic strategies including Elucigene FH20 for cascade testing to identify FH in the relatives of index individuals. The use of Elucigene FH20 for cascade testing will depend on the mutation detected in the index individual and the cost of targeted gene sequencing. (In index individuals with an identified genetic mutation, depending on the test used to detect the mutation, targeted gene sequencing will also be considered for cascade testing of relatives. In index individuals without an identified genetic mutation, cascade testing using LDL-C concentration measurement will be considered.)
A scenario encompassing a single test strategy (Elucigene FH20 or LIPOchip) that does not end in comprehensive genetic analysis for test negatives may not detect all cases of FH. In such a scenario there may be implications for test negative patients in terms of how their condition is managed.
4.5 Relevant comparators
Comprehensive genetic analysis
Comprehensive genetic analysis is defined as the most complete genetic analysis generally available for FH within a diagnostic setting and is expected to detect almost all known FH causing mutations. This analysis will include DNA sequence analysis of the promoter, all exons, the exon/intron boundaries and into 3′ untranslated region of the LDLR gene that will detect the majority (∼88%) of detectable FH mutations, multiplex ligation-dependent probe amplification (MLPA)18 for each exon and the promoter region of the LDLR gene to detect deletions and duplications (∼5% detectable FH mutations) plus analysis for the common APOB p.Arg3527Gin gene mutation (∼5% FH mutations) and the PCSK9 p.Asp374Tyr gene mutation (∼2% FH mutations).
Multiplex ligation-dependent probe amplification (MLPA) (MRC-Holland) is a commercial kit that enhances the molecular diagnosis of FH with an ability to detect large deletions and or duplications for each of the LDLR 18 exons. 18 Comprehensive genetic analysis including DNA sequencing with MLPA is considered to be the ‘gold standard’ of genetic testing.
Targeted gene sequencing
Targeted gene sequencing (the genetic test for sequencing a specific part of the gene where a family mutation is found) may be used for cascade testing to identify FH in the relatives of index individuals. The use of targeted sequencing for cascade testing will depend on the test used to detect a genetic mutation in the index individual.
LDL-C concentration as part of the Simon Broome criteria
In UK a clinical diagnosis of FH should be made based on the Simon Broome criteria,7 which include a combination of family history of CHD, clinical signs such as tendon xanthomata, cholesterol concentration and DNA testing11,19 (Table 2). This approach categorises FH as ‘definite’ or ‘possible’. DNA based evidence was subsequently introduced into the criteria for provision of an unequivocal diagnosis of FH. However, around 10% of people with FH do not meet the Simon Broome criteria.
| Criteria required for clinical diagnosis of FH | Definite FH | Possible FH |
|---|---|---|
|
Cholesterol concentration: Child/young person: Total cholesterol (TC) > 6.7 mmol/L, Low density lipoprotein cholesterol (LDL-C) > 4 mmol/L; Adult: TC > 7.5 mmol/L, LDL-C: > 4.9 mmol/L |
Yes | Yes |
|
Clinical symptoms: Tendon xanthomata, or evidence of these signs in first- or second-degree relative |
Yes | No |
Family history of:
|
No | Yes (at least one of these criteria) |
LDL-C concentration is usually estimated from a fasting blood sample using the Friedwald equation. Due to NHS commissioning arrangements of genetic tests, LDL-C concentration measurement is the main test currently used to diagnosis FH in index cases and for cascade testing of relatives. 20 However, it has some limitations in terms of diagnostic accuracy, including:
-
There is an overlap in LDL-C levels between affected and unaffected individuals, and the cut-offs used can result in diagnostic ambiguity in an estimated 15% of children (aged 5–15 years) and in nearly 50% of adults (aged 45–55 years). 21,22
-
In children who are at risk of FH, cholesterol levels may appear normal initially with the levels rising only later in life. 23
-
Girls generally have lower cholesterol concentration than boys at an early age but may go on to develop CHD in later years. 22
Age adjusted LDL-C measurement has been found to give better clinical diagnosis of FH, with a sensitivity of 72% and specificity of 71%. 24 The gender- and age-specific LDL-C criteria rather than the Simon Broome LDL-C criteria are the recommended criteria for cascade testing of relatives of index individuals. 7
4.6 Key factors to be addressed
This systematic review will aim to:
-
Assess the diagnostic accuracy and clinical effectiveness of Elucigene FH20, LIPOchip and comparators in confirming a diagnosis of FH in patients with a clinical diagnosis of FH.
-
Assess the diagnostic accuracy and clinical effectiveness of Elucigene FH20 and comparators in cascade testing of relatives of index individuals with a confirmed diagnosis of FH.
-
Estimate the costs of different diagnostic strategies for detecting FH in index individuals and for cascade testing of relatives of index individuals with a confirmed diagnosis of FH.
5. Report methods for assessing the outcomes arising from the use of the interventions
A systematic review of the evidence on Elucigene FH20 and LIPOchip for the diagnosis of familial hypercholesterolaemia will be undertaken following the general principles of the Centre for Reviews and Dissemination (CRD) guidance for conducting reviews in health care25 and NICE Diagnostics Assessment Programme interim methods statement. 26
5.1 Inclusion and Exclusion criteria
Population
The populations considered are adults and children with a clinical diagnosis of FH (the index cases/probands) based on the Simon Broome criteria, and, for cascade testing, first-, second- and third-degree biological relatives of the index individual.
If the evidence allows, subgroup analysis will be undertaken on the performance of Elucigene FH20 and LIPOchip in ethnic populations.
Setting
The setting considered is secondary or tertiary care.
Interventions
The interventions considered are Elucigene FH20 and LIPOchip for index cases and Elucigene FH20 for cascade testing.
Comparators
The comparators for testing in index individuals are (i) comprehensive genetic analysis and (ii) LDL-C concentration measurement (Simon Broome criteria). The comparators for cascade testing of relatives are (i) targeted gene sequencing and (ii) LDL-C concentration measurement (gender- and age-specific criteria as recommended in NICE CG71).
Reference standard
The reference standard is comprehensive genetic analysis in combination with the Simon Broome Criteria.
Outcomes
The following outcomes will be considered:
-
Test accuracy;
-
Mutation detection rate – proportion of cases with an unequivocal diagnosis identified by Elucigene and LIPOchip;
-
Proportion requiring comprehensive genetic analysis after Elucigene and LIPOchip; and
-
Proportion of FH identified from cascade testing;
In any studies reporting the above outcomes the following outcomes will also be considered if reported:
-
Acceptability of the tests; and
-
Interpretability of the tests.
Studies reporting test accuracy must report the absolute numbers of true positives, false positives, false negatives and true negatives, or provide information allowing their calculation.
Study design
The following types of studies will be included:
-
Direct (head-to-head) studies in which the index test, comparator test and reference standard test are done independently in the same group of people.
-
Randomised controlled trials (RCTs) in which people are randomised to the index and comparator test(s) and all receive the reference standard test.
In case of insufficient evidence from direct and randomised studies, we will consider indirect (between-study) comparisons of the following types of study:
-
Diagnostic cross-sectional studies comparing the index test or comparator test against a reference standard test.
-
Case–control studies in which two groups are created, one known to have the target disease and one known not to have the target disease, where it is reasonable for all included to go through the tests.
Exclusion criteria
We will exclude the following types of report:
-
Preclinical and biological studies
-
Reviews, editorials and opinions
-
Case reports
-
Reports investigating technical aspects of a test
Non-English language reports may be excluded if the evidence base containing English-language reports is sufficiently large.
5.2 Search strategy
Extensive electronic searches will be conducted to identify reports of published and ongoing studies on Elucigene FH20 and LIPOchip for the detection and cascade testing of FH. The search strategies will be designed to retrieve all studies that assess the diagnostic accuracy and clinical effectiveness of the index, comparator and reference standard tests. Searches will be restricted to publications from 2000 onwards. Both full-text papers and recent conference abstracts will be sought. Potentially relevant non-English-language studies will be excluded and listed in an appendix to the review, unless the English-language evidence base is deemed to be insufficient in which case they will be included. Databases to be searched will include: MEDLINE, EMBASE, Science Citation Index, Biosis and the Cochrane Controlled Trials Register. A preliminary MEDLINE search strategy is shown in Appendix A and will be adapted for use in other databases.
A search for systematic reviews and other background publications will also be undertaken. Sources will include the Cochrane Database of Systematic Reviews, HTA Database and DARE.
Current research registers, including Current Controlled Trials, Clinical Trials and WHO International Clinical Trials Registry will be searched. Recent conference proceedings of key organisations will also be screened and will include the European Society of Human Genetics, American Association for Clinical Chemistry, International Atherosclerosis Society and Heart UK.
In addition, an internet search using Copernic Agent will be undertaken and will also include key professional organisations.
5.3 Data extraction strategy
Two reviewers will independently screen the titles (and abstracts if available) of all reports identified by the search strategy. Full-text copies of all studies deemed to be potentially relevant will be obtained, and two reviewers will independently assess them for inclusion. Any disagreements will be resolved by consensus or arbitration by a third party.
A data extraction form will be developed and piloted. One reviewer will extract details of study design, participants, index, comparator, reference standard tests and outcome data. A second reviewer will check the data extraction. Any disagreements will be resolved by consensus or arbitration by a third party.
Study data requested and received from the manufacturers that meet the inclusion criteria, and are received in time to be incorporated into the review, will be extracted and quality assessed in accordance with the procedures outlined in this protocol.
5.4 Quality assessment strategy
Two reviewers will independently assess the methodological quality of the included diagnostic studies. Any disagreements will be resolved by consensus or arbitration by a third party. Studies will not be included or excluded on the basis of methodological quality.
Various quality assessment tools will be used depending upon the type of studies included. For instance, included diagnostic studies will be quality assessed using QUADAS, a quality assessment tool developed for use in systematic reviews of diagnostic studies. 27 The quality assessment tool will be adapted to make it more applicable to assess the quality of studies of tests for detecting FH.
5.5 Methods of analysis/synthesis
Analysis will focus on the ability of Elucigene FH20, LIPOchip and relevant comparators to detect FH. Where appropriate two by two tables will be extracted from each included study where information is provided on the numbers of true and false-positives and negatives for the index and/or comparator test compared with the reference standard for detecting those mutations that the index and/or comparator test are designed to identify. For each study we will attempt to calculate sensitivity, specificity, positive and negative likelihood ratios and diagnostic odds ratios and their confidence intervals.
Where appropriate and given sufficient information, we will use summary receiver operating characteristic (SROC) curves for the meta-analysis of data from studies reporting estimates of true and false-positives and negatives. This approach characterises the relationship between sensitivity and 1–specificity across studies and takes into account variation in the threshold for test positivity between studies. ROC curves will be generated, where possible, for each testing procedure. Where data are available, potential sources of heterogeneity will be investigated by extending the SROC regression models to include study level covariates. These potential sources of heterogeneity include characteristics of the population such as age, race, family history and whether the test is cascade testing.
Where appropriate, models will be fitted using the hierarchical summary receiver operating characteristic (HSROC) framework, which takes proper account of the diseased and non-diseased sample sizes in each study, and allows estimation of random effects for the threshold and accuracy effects, and testing of the impact of potential sources of heterogeneity. Estimates and their CI’s for the average operating points, expressed as sensitivity, specificity and likelihood ratios will be obtained by combining these estimates. 28
Average and ranges of feasible operating points will be identified on the fitted ROC points to convert ROC curve values into estimates of true positive and false positive rates which will serve as parameters within the economic model.
5.6 Methods for estimating quality of life – relevance to the decision analysis
Quality of life estimates used in the economic model will be informed by the current NICE guideline on the identification and management of familial hypercholesterolaemia7 and relevant literature searches together with clinical expert opinion as appropriate. As FH is a chronic disease requiring long-term care, we will extrapolate cost and QALY values over a life-time horizon and discount both cost and QALYs at a rate of 3.5% as recommended by NICE. This will use a linked evidence approach linking diagnostic accuracy of the various strategies with any potential changes in clinical management and thus life-time final health outcomes. The economic model informing current NICE guideline CG71 for treatment of FH will be validated and used to estimate the final treatment outcomes.
6. Report methods for synthesising evidence of cost-effectiveness
A systematic search for existing cost-effectiveness literature will be undertaken for diagnostic assessment strategies for the detection of genetic mutations causing familial hypercholesterolaemia.
6.1 Identifying and systematically searching published cost-effectiveness studies.
Studies will be sought, reporting both costs and outcomes for diagnostic assessment strategies, from a systematic review of the literature. No language restrictions or limitations to searches will be imposed.
Databases to be searched will include MEDLINE, EMBASE, Science Citation Index, NHS EED, HTA Database, Health Management Information Consortium and the CEA Registry. In addition, reference lists of all included studies will be scanned to identify additional potentially relevant studies. A draft MEDLINE search strategy is appended and will be adapted for use in the other databases.
6.2 Evaluation of costs and cost-effectiveness
The evidence on costs and cost-effectiveness will be evaluated using the NICE Diagnostics Assessment Programme interim methods. 26 An economic model will be developed to estimate the cost-effectiveness of each care pathway and link this to final treatment outcomes. Current NICE guideline CG71 will be used to inform the development of this approach.
6.3 Development of a health economic model
An economic evaluation of the cost-effectiveness of Elucigene, LIPOchip and identified comparators will be conducted. An economic model will be developed to determine which diagnostic and treatment strategy is the most cost-effective use of scarce NHS resources for genetic testing for FH among proband cases (identified using the Simon Broome criteria) and cascade testing of relatives.
The primary economic model output will be incremental cost per quality adjusted life year (QALY) gained associated with the use of a variety of genetic testing strategies for the detection of FH. A life-time horizon will be used in the model and costs and benefits will be discounted at a rate of 3.5% as recommended by NICE. 29 The development of this economic model will be an iterative approach and it will be developed in a way that is adaptable to the analysis of new and emerging technologies. A possible scenario for the modelling is presented in Appendix B for the index cases and Appendix C for the cascade testing of their relatives (Appendix B, Appendix C). A range of diagnostic strategies will be explored initially for index patients with a clinical diagnosis based on the Simon Broome criteria. The model will further estimate the most cost-effective method of cascade testing for FH in first-, second-, and possibly third-degree relatives of the index patient. This too will be presented as incremental cost per QALY gained. We note that the diagnostic test used to detect the family mutation may not be the same as that used to detect the mutation in the index individual. This is due to the potential for cost savings among alternative cheaper tests for cascade testing (e.g. Elucigene) once the FH-causing family mutation has been identified. Our analysis will be from the perspective of the NHS as well as a personal social services perspective as appropriate. Any assumptions made in the modelling approach and parameter development will be taken primarily from the literature and supplemented by clinical expert opinion as appropriate/required.
Health related quality of life and QALY data for lifelong health outcomes have already been modelled in terms of management of FH in cascade testing and treatment strategy. These data will be validated, updated as necessary and used to help populate the economic model being developed. Any evidence on detection rates and diagnostic accuracy of the comparators will be sourced from the literature. As it is unlikely that a large evidence base exists in the literature, data will be supplemented by clinical expert opinion as required. A key challenge in terms of diagnostic accuracy of the genetic testing kits will be to generalise detection rates to the general UK population. It is likely that detection rates will vary depending on ethnicity and so this will need to be fully understood and uncertainties explored through sensitivity analyses. Data from the genetic bank held in London, together with manufacturer and clinical expert supplied input will be used to estimate detection rates of the different strategies.
Resource use and costs for detection are likely to be the major driver of the cost-effectiveness results. It will be important to fully incorporate all economic costs associated with testing and processing diagnostic samples for each treatment strategy and the range of scenarios required by the model. A combination of national resources such as NHS reference costs, the Personal Social Services Research Unit (PSSRU) and the British National Formulary (BNF) will be used as appropriate together with any other relevant sources of data identified. Costs of diagnostic kits will be sourced from the manufacturers and costs of processing samples sourced from a combination of manufacturer and clinical expert data. As obtaining test results is not time sensitive due to the clinical nature of FH, the base case analysis will assume genetic laboratories will batch test to gain maximum efficiency (i.e. minimum cost). The impact of operating testing procedures below maximum efficiency will be considered in model sensitivity analyses. A key challenge will be to generalise the cost of comprehensive genetic analysis across the UK, where various laboratories report different unit workload costs. The effect of alternative costing strategies will be explored through model sensitivity analyses.
The development of this economic model will be an iterative approach. As the evidence base changes and new evidence arises, the economic model structure and parameters will evolve to reflect this. We further suspect that the evidence base will be lacking for some of the model parameters. With this in mind, uncertainty in model parameters will be explored in terms of their outputs through a range of one-way and multi-way sensitivity analyses deemed appropriate as the modelling progresses. As we anticipate a lack of evidence to inform the model, we will explore parameter uncertainty through probabilistic sensitivity analyses, with the generation of cost-effectiveness acceptability curves illustrating this uncertainty graphically.
7. Handling information from the companies
Following a request for information, any ‘commercial in confidence’ data provided by a manufacturer and specified as such will be highlighted in blue and underlined in the assessment report (followed by an indication of the relevant company name e.g. in brackets).
8. Competing interests of authors
None
9. Timetable/milestones
| Milestones | Date to be completed |
|---|---|
| Draft protocol | 24/11/10 |
| Final protocol | 14/12/10 |
| Progress report | w/c 18/02/11 |
| Draft version of report | 01/04/11 |
| Final version of report | 28/04/11 |
10. References
- Austin MA, Hutter CM, Zimmern RL, Humphries SE. Genetic causes of monogenic heterozygous familial hypercholesterolemia: A HuGE prevalence review. Am J Epidemiol 2004;160:407-20.
- Model of care: familial hypercholesterolaemia [document on the Internet]. Perth: Office of Population Health Genomics, Department of Health, Government of Western Australia; 2008.
- Connor M, Ferguson-Smith M. Essential medical genetics. Oxford: Blackwell; 2010.
- Heath KE, Humphries SE, Middleton-Price H, Boxer M. A molecular genetic service for diagnosing individuals with familial hypercholesterolaemia (FH) in the United Kingdom. Eur J Hum Genet 2001;9:244-52.
- Koivisto PVI, Koivisto UM, Miettinen TA, Kontula K. Diagnosis of heterozygous familial hypercholesterolemia: DNA analysis complements clinical examination and analysis of serum lipid levels. Arterioscler Thromb 1992;12:584-92.
- Marks D, Thorogood M, Farrer JM, Humphries SE. Census of clinics providing specialist lipid services in the United Kingdom. J Public Health 2004;26:353-4.
- CG71 Identification and management of familiar hypercholesterolaemia [document on the Internet]. London: National Institute for Health and Clinical Excellence; 2008.
- Shafiq N, Singh M, Kaur S, Khosla P, Malhotra S. Dietary treatment for familial hypercholesterolaemia. Cochrane Database of System Rev 2010. URL: 10.1002./14.
- Versmissen J, Oosterveer DM, Yazdanpanah M, Defesche JC, Basart DCG, Liem AH, et al. Efficacy of statins in familial hypercholesterolaemia: A long term cohort study. BMJ 2009;338:223-6.
- Centre for Evidence-based Purchasing . CEP 10034 Evidence Review: Rapid Genetic Testing for Familial Hypercholesteraemia 2010.
- Marks D, Thorogood M, Neil HAW, Humphries SE. A review on the diagnosis, natural history, and treatment of familial hypercholesterolaemia. Atherosclerosis 2003;168:1-14.
- Taylor A, Patel K, Tsedeke J, Humphries SE, Norbury G. Mutation screening in patients for familial hypercholesterolaemia (ADH). Clin Genet 2010;77:97-9.
- Alonso R, Defesche JC, Tejedor D, Castillo S, Stef M, Mata N, et al. Genetic diagnosis of familial hypercholesterolemia using a DNA-array based platform. Clin Biochem 2009;42:899-903.
- Taylor A, Martin B, Wang D, Patel K, Humphries SE, Norbury G. Multiplex ligation-dependent probe amplification analysis to screen for deletions and duplications of the LDLR gene in patients with familial hypercholesterolaemia. Clin Genet 2009;76:69-75.
- Humphries SE, Whittall RA, Hubbart CS, Maplebeck S, Cooper JA, Soutar AK, et al. Genetic causes of familial hypercholesterolaemia in patients in the UK: Relation to plasma lipid levels and coronary heart disease risk. J Med Genet 2006;43:943-9.
- Lo YMD, LovYMD. The amplification refractory mutation system. Totawa, NJ: Humana; 1998.
- Leigh SEA, Foster AH, Whittall RA, Hubbart CS, Humphries SE. Update and analysis of the university college London low density lipoprotein receptor familial hypercholesterolemia database. Ann Hum Genet 2008;72:485-98.
- Wang J, Ban MR, Hegele RA. Multiplex ligation-dependent probe amplification of LDLR enhances molecular diagnosis of familial hypercholesterolemia. J Lipid Res 2005;46:366-72.
- Betteridge DJ, Broome K, Durrington PN, Mann JI, Miller JP, Neil HAW, et al. Risk of fatal coronary heart disease in familial hypercholesterolaemia. Br Med J 1991;303:893-6.
- National Clinical Audit of the Management of Familial Hypercholesterolaemia 2009: Pilot [document on the Internet] 2009. URL: http://www.rcplondon.ac.uk/clinical-standards/ceeu/Current-work/Documents/FH%20Pilot%20Audit%20Report%20v8%20Full%20Report.pdf.
- Kwiterovich PO, Fredrickson DS, Levy RI. Familial hypercholesterolemia (one form of familial type II hyperlipoproteinemia). A study of its biochemical, genetic and clinical presentation in childhood. J Clin Invest 1974;53:1237-49.
- Leonard JV, Whitelaw AGL, Wolff OH. Diagnosing familial hypercholesterolaemia in childhood by measuring serum cholesterol. Br Med J 1977;1:1566-8.
- Kessling AM, Seed M, Taylor R, Wynn V, Humphries SE. Rising cholesterol levels in children with familial hypercholesterolaemia. Biomed Pharmacother 1990;44:373-9.
- Civeira F, Ros E, Jarauta E, Plana N, Zambon D, Puzo J, et al. Comparison of Genetic Versus Clinical Diagnosis in Familial Hypercholesterolemia. Am J Cardiol 2008;102:1187-93.
- Centre of Reviews and Dissemination . Systematic Reviews: CRD’s Guidance for Undertaking Reviews in Health Care [Internet] 2009. URL: http://www.york.ac.uk/inst/crd/systematic_reviews_book.htm.
- Diagnostics Assessment programme – interim methods statement (programme) [document on the Internet]. London: National Institute for Health and Clinical Excellence; 2010.
- Whiting P, Rutjes AW, Reitsma JB, Bossuyt PM, Kleijnen J. The development of QUADAS: a tool for the quality assessment of studies of diagnostic accuracy included in systematic reviews. BMC Med Res Methodol 2003;3.
- Rutter CM, Gatsonis CA. A hierarchical regression approach to meta-analysis of diagnostic test accuracy evaluations. Stat Med 2001;20:2865-84.
- Updated Guide to the Methods of Technology Appraisal [document on the Internet] 2008. URL: http://www.nice.org.uk/media/B52/A7/TAMethodsGuideUpdatedJune2008.pdf.
11. Appendices
Appendix A
Preliminary MEDLINE strategy
Diagnostic Accuracy and Clinical Effectiveness of Elucigene FH20, LIPOchip and Comparators
-
Hyperlipoproteinemia Type II/di [Diagnosis]
-
lipochip.tw.
-
elucigene.tw.
-
Hyperlipoproteinemia Type II/
-
hyperlipidemia, familial combined/
-
familial hypercholesterol?emia.tw.
-
hyperlipoprotein?emia.tw.
-
familial hyperlipid?emia.tw.
-
or/4-8
-
exp Genetic Predisposition to Disease/
-
Genetic Testing/
-
Gene Amplification/
-
exp Nucleic Acid Amplification Techniques/
-
exp oligonucleotide array sequence analysis/ or exp sequence analysis, dna/
-
(dna adj3 test$).tw.
-
gene sequencing.tw.
-
(sequenc$ adj3 analysis).tw.
-
(cascade adj3 (test$ or screen$)).tw.
-
(genetic adj3 (test$ or screen$)).tw.
-
(arms or amplification refractory mutation system).tw.
-
(PCR or polymerase chain reaction).tw.
-
Polymorphism, Single-Stranded Conformational/
-
(sscp or single-stranded conformation polymorphism).tw.
-
(mlpa or Multiplex ligation-dependent probe amplification).tw.
-
Cholesterol, LDL/
-
ldl-c.tw.
-
or/10-26
-
9 and 27
-
“sensitivity and specificity”/
-
roc curve/
-
predictive value of tests/
-
false positive reactions/
-
false negative reactions/
-
du.fs.
-
sensitivity.tw.
-
distinguish$.tw.
-
differentiat$.tw.
-
identif$.tw.
-
detect$.tw.
-
diagnos$.tw.
-
(predictive adj4 value$).tw.
-
accura$.tw.
-
comparison.tw.
-
or/29-43
-
28 and 44
-
1 or 2 or 3 or 45
-
limit 46 to yr=”2000 -Current»
-
randomized controlled trial.pt.
-
controlled clinical trial.pt.
-
randomi?ed.ab.
-
placebo.ab.
-
drug therapy.fs.
-
randomly.ab.
-
trial.ab.
-
groups.ab.
-
or/48-55
-
exp animals/ not humans/
-
56 not 57
-
28 and 58
-
limit 59 to yr=”2000 -Current»
-
46 or 60
Preliminary MEDLINE strategy
Economic evaluations of Elucigene FH20, LIPOchip and Comparators
-
Hyperlipoproteinemia Type II/di
-
elucigene.tw
-
lipochip.tw
-
Hyperlipoproteinemia Type II/
-
hyperlipidemia, familial combined/
-
familial hypercholesterol?emia.tw.
-
hyperlipoprotein?emia.tw.
-
familial hyperlipid?emia.tw.
-
or/4-8
-
genetic predisposition to disease/
-
genetic testing/
-
(genetic adj3 (test$ or screen$)).tw.
-
(cascade adj3 (test$ or screen$)).tw.
-
(dna adj3 test$).tw
-
gene amplification/
-
exp Nucleic Acid Amplification Techniques/
-
exp sequence analysis,dna/
-
exp oligonucleotide array sequence analysis/
-
(arms or amplification refractory mutation system).tw.
-
(PCR or polymerase chain reaction).tw
-
(sscp or single-stranded conformation polymorphism).tw
-
(mlpa or Multiplex ligation-dependent probe amplification).tw.
-
gene sequencing.tw.
-
sequence analys?s.tw.
-
ldl-c.tw.
-
or/10-25
-
9 and 26
-
or/1-3,27
-
exp “costs and cost analysis»/
-
economics/
-
exp economics,medical/
-
economics,pharmaceutical/
-
exp budgets/
-
exp models, economic/
-
exp decision theory/
-
monte carlo method/
-
markov chains/
-
exp technology assessment, biomedical/
-
cost$.ti.
-
(cost$ adj2 (effective$ or utilit$ or benefit$ or minimis$)).ab.
-
economics model$.tw.
-
economic$ .tw.
-
(price or prices or pricing).tw.
-
(value adj1 money).tw.
-
markov$.tw.
-
monte carlo.tw.
-
(decision$ adj2 (tree? or analy$ or model$)).tw.
-
or/29-47
-
28 and 48
Appendix B
Patient care pathways (Index cases with a clinical diagnosis of FH using the Simon Broome criteria – including a LDL-c test)*
*The above is a guideline to the main strategies, there may be exceptions to these strategies which will be explored as the analysis progresses.
| 1. | Elucigene | → | Treatment decision | ||||
| 2. | Elucigene | → | Lipochip for negatives | → | Treatment decision | ||
| 3. | Elucigene | → | MLPA for negatives | → | Treatment decision | ||
| 4. | Elucigene | → | CGA for negatives | → | Treatment decision | ||
| 5. | Elucigene | → | Lipochip for negatives | → | GA for negatives | → | Treatment decision |
| 6. | Elucigene | → | Lipochip for negatives | → | MLPA for negatives | → | Treatment decision |
| 7. | Lipochip | → | Treatment decision | ||||
| 8. | Lipochip | → | CGA for negatives | → | Treatment decision | ||
| 9. | Lipochip | → | MLPA for negatives | → | Treatment decision | ||
| 10. | CGA | → | Treatment decision | ||||
| 11. | LDL-c | → | Treatment decision (current practice) | ||||
Appendix C
Patient care pathways (Cascade testing of relatives of FH identified index patients)**
**Once a relative is found to be negative for the mutation being tested for, cascade testing stops and further cascade testing is not conducted
| Index case identified by | Cascade testing of relatives | Clinical management | |
|---|---|---|---|
| Elucigene | Elucigene | → | Treatment decision |
| Elucigene | Targeted Sequencing | → | Treatment decision |
| Lipochip | Elucigene | → | Treatment decision |
| Lipochip | Targeted Sequencing | → | Treatment decision |
| CGA | Elucigene | → | Treatment decision |
| CGA | Targeted Sequencing | → | Treatment decision |
Glossary
Technical terms and abbreviations are used throughout this report. The meaning is usually clear from the context, but a glossary is provided for the non-specialist reader.
- Autosomal dominant pattern of inheritance
- An affected individual has one copy of a mutant gene and one normal gene on a pair of autosomal (i.e. non-sex) chromosomes. Therefore, one copy of the mutant gene is sufficient to express the phenotype. Individuals with autosomal dominant diseases have a 50 : 50 chance of passing the mutant gene, and therefore the disorder, on to each of their children.
- Cascade testing
- A mechanism for identifying people at risk of a genetic condition by a process of family tracing. Relatives of the individual diagnosed with familial hypercholesterolaemia are tested for the condition, as are their relatives; ideally, cascade testing should be undertaken in first-, second- and third-degree relatives. For familial hypercholesterolaemia the test employed is measurement of (low-density lipoprotein) cholesterol in the blood and/or a DNA test if a disease-causing mutation has been identified in the proband/index.
- Coronary heart disease
- An abnormal condition characterised by narrowing of the small blood vessels that supply blood and oxygen to the heart (coronary heart disease is synonymous with coronary artery disease).
- First-degree relatives
- A person’s biological parents, brothers and sisters and children.
- Heterozygous familial hypercholesterolaemia
- High low-density lipoprotein cholesterol concentration in the blood caused by an inherited mutation from one parent only.
- Homozygous familial hypercholesterolaemia
- Very high low-density lipoprotein cholesterol level in the blood caused by an inherited mutation from both parents. When a person inherits exactly the same affected gene from both parents this is called truly ‘homozygous’ familial hypercholesterolaemia. When the mutations in the low-density lipoprotein receptor gene (or equivalent) are different, this state is called ‘compound heterozygous’.
- Mutation
- An identified change in the DNA sequence of a gene that is predicted to damage the normal function of the gene and so cause disease.
- p-value
- The probability that an observed difference could have occurred by chance if the null hypothesis is true. A p-value of < 0.05 is conventionally considered to be statistically significant.
- Proband
- The affected (index) individual through whom a family with a genetic disorder is ascertained. The terms ‘index case’, ‘index individual’, ‘index patient’ and ‘proband’ are synonymous with one another in this report.
- Second-degree relatives
- A person’s biological grandparent, uncle, aunt, niece, nephew, half-sister or half-brother.
- Tendon xanthoma/xanthomata
- A clinically detectable nodularity and/or thickening of the tendons caused by infiltration with lipid-laden histiocytes (macrophages in connective tissue). A distinctive feature of familial hypercholesterolaemia that most frequently affects the Achilles tendons but can also involve tendons on the back of the hands, elbows and knees.
- Third-degree relatives
- A person’s biological great-grandparent, great-grandchild, great-aunt, great-uncle, first cousin, grand-nephew or grand-niece.
List of abbreviations
- APOB
- apolipoprotein B
- ARMS
- amplification refractory mutation system
- BNF
- British National Formulary
- CEAC
- cost-effectiveness acceptability curve
- CG71
- clinical guideline number 71
- CGA
- comprehensive genetic analysis
- CHD
- coronary heart disease
- CI
- confidence interval
- CMGS
- Clinical Molecular Genetics Society
- DFH
- definite familial hypercholesterolaemia
- DGGE
- denaturing gradient gel electrophoresis
- dHPLC
- denaturing high-performance liquid chromatography
- Ext Dom
- Extendedly dominated
- FH
- familial hypercholesterolaemia
- ICER
- incremental cost-effectiveness ratio
- iPLEX
- multiple MassARRAY spectrometry
- LDL-C
- low-density lipoprotein cholesterol
- LDLR
- low-density lipoprotein receptor
- MedPed
- make early diagnosis, prevent early death
- MI
- myocardial infarction
- MLPA
- multiplex ligation-dependent probe amplification
- MOLU
- MOLecular Unit
- N/A
- not applicable
- NA
- not available
- NC
- not calculable
- NICE
- National Institute for Health and Clinical Excellence
- NR
- not reported
- PAD
- peripheral arterial disease
- PBR
- Payment by Results
- PCR
- polymerase chain reaction
- PCSK
- protein convertase subtilisin/kexin
- PCVD
- premature cardiovascular disease
- PFH
- possible familial hypercholesterolaemia
- PSSRU
- Personal Social Services Research Unit
- QALY
- quality-adjusted life-year
- QMFSP
- quantitative multiplex PCR methodology
- RCT
- randomised controlled trial
- SROC
- summary receiver operating characteristic
- SSCP
- single-strand conformation polymorphism
- TC
- total cholesterol
- UFH
- unclassified familial hypercholesterolaemia
- UKGTN
- United Kingdom Genetic Testing Network
All abbreviations that have been used in this report are listed here unless the abbreviation is well known (e.g. NHS), or it has been used only once, or it is a non-standard abbreviation used only in figures/tables/appendices, in which case the abbreviation is defined in the figure legend or in the notes at the end of the table.
Notes
Health Technology Assessment programme
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
Prioritisation Group
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Imti Choonara, Professor in Child Health, Academic Division of Child Health, University of Nottingham
Chair – Pharmaceuticals Panel
-
Dr Bob Coates, Consultant Advisor – Disease Prevention Panel
-
Dr Andrew Cook, Consultant Advisor – Intervention Procedures Panel
-
Dr Peter Davidson, Director of NETSCC, Health Technology Assessment
-
Dr Nick Hicks, Consultant Adviser – Diagnostic Technologies and Screening Panel, Consultant Advisor–Psychological and Community Therapies Panel
-
Ms Susan Hird, Consultant Advisor, External Devices and Physical Therapies Panel
-
Professor Sallie Lamb, Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick
Chair – HTA Clinical Evaluation and Trials Board
-
Professor Jonathan Michaels, Professor of Vascular Surgery, Sheffield Vascular Institute, University of Sheffield
Chair – Interventional Procedures Panel
-
Professor Ruairidh Milne, Director – External Relations
-
Dr John Pounsford, Consultant Physician, Directorate of Medical Services, North Bristol NHS Trust
Chair – External Devices and Physical Therapies Panel
-
Dr Vaughan Thomas, Consultant Advisor – Pharmaceuticals Panel, Clinical
Lead – Clinical Evaluation Trials Prioritisation Group
-
Professor Margaret Thorogood, Professor of Epidemiology, Health Sciences Research Institute, University of Warwick
Chair – Disease Prevention Panel
-
Professor Lindsay Turnbull, Professor of Radiology, Centre for the MR Investigations, University of Hull
Chair – Diagnostic Technologies and Screening Panel
-
Professor Scott Weich, Professor of Psychiatry, Health Sciences Research Institute, University of Warwick
Chair – Psychological and Community Therapies Panel
-
Professor Hywel Williams, Director of Nottingham Clinical Trials Unit, Centre of Evidence-Based Dermatology, University of Nottingham
Chair – HTA Commissioning Board
Deputy HTA Programme Director
HTA Commissioning Board
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
-
Department of Public Health and Epidemiology, University of Birmingham
-
Professor of Clinical Pharmacology, Director, NIHR HTA programme, University of Liverpool
-
Professor Ann Ashburn, Professor of Rehabilitation and Head of Research, Southampton General Hospital
-
Professor Judith Bliss, Director of ICR-Clinical Trials and Statistics Unit, The Institute of Cancer Research
-
Professor Peter Brocklehurst, Professor of Women’s Health, Institute for Women’s Health, University College London
-
Professor David Fitzmaurice, Professor of Primary Care Research, Department of Primary Care Clinical Sciences, University of Birmingham
-
Professor John W Gregory, Professor in Paediatric Endocrinology, Department of Child Health, Wales School of Medicine, Cardiff University
-
Professor Steve Halligan, Professor of Gastrointestinal Radiology, University College Hospital, London
-
Professor Angela Harden, Professor of Community and Family Health, Institute for Health and Human Development, University of East London
-
Dr Martin J Landray, Reader in Epidemiology, Honorary Consultant Physician, Clinical Trial Service Unit, University of Oxford
-
Dr Joanne Lord, Reader, Health Economics Research Group, Brunel University
-
Professor Stephen Morris, Professor of Health Economics, University College London, Research Department of Epidemiology and Public Health, University College London
-
Professor Dion Morton, Professor of Surgery, Academic Department of Surgery, University of Birmingham
-
Professor Gail Mountain, Professor of Health Services Research, Rehabilitation and Assistive Technologies Group, University of Sheffield
-
Professor Irwin Nazareth, Professor of Primary Care and Head of Department, Department of Primary Care and Population Sciences, University College London
-
Professor E Andrea Nelson, Professor of Wound Healing and Director of Research, School of Healthcare, University of Leeds
-
Professor John David Norrie, Chair in Clinical Trials and Biostatistics, Robertson Centre for Biostatistics, University of Glasgow
-
Dr Rafael Perera, Lecturer in Medical Statisitics, Department of Primary Health Care, University of Oxford
-
Professor Barney Reeves, Professorial Research Fellow in Health Services Research, Department of Clinical Science, University of Bristol
-
Professor Peter Tyrer, Professor of Community Psychiatry, Centre for Mental Health, Imperial College London
-
Professor Martin Underwood, Professor of Primary Care Research, Warwick Medical School, University of Warwick
-
Professor Caroline Watkins, Professor of Stroke and Older People’s Care, Chair of UK Forum for Stroke Training, Stroke Practice Research Unit, University of Central Lancashire
-
Dr Duncan Young, Senior Clinical Lecturer and Consultant, Nuffield Department of Anaesthetics, University of Oxford
-
Dr Tom Foulks, Medical Research Council
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
HTA Clinical Evaluation and Trials Board
-
Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick and Professor of Rehabilitation, Nuffield Department of Orthopaedic, Rheumatology and Musculoskeletal Sciences, University of Oxford
-
Professor of the Psychology of Health Care, Leeds Institute of Health Sciences, University of Leeds
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Keith Abrams, Professor of Medical Statistics, Department of Health Sciences, University of Leicester
-
Professor Martin Bland, Professor of Health Statistics, Department of Health Sciences, University of York
-
Professor Jane Blazeby, Professor of Surgery and Consultant Upper GI Surgeon, Department of Social Medicine, University of Bristol
-
Professor Julia M Brown, Director, Clinical Trials Research Unit, University of Leeds
-
Professor Alistair Burns, Professor of Old Age Psychiatry, Psychiatry Research Group, School of Community-Based Medicine, The University of Manchester & National Clinical Director for Dementia, Department of Health
-
Dr Jennifer Burr, Director, Centre for Healthcare Randomised trials (CHART), University of Aberdeen
-
Professor Linda Davies, Professor of Health Economics, Health Sciences Research Group, University of Manchester
-
Professor Simon Gilbody, Prof of Psych Medicine and Health Services Research, Department of Health Sciences, University of York
-
Professor Steven Goodacre, Professor and Consultant in Emergency Medicine, School of Health and Related Research, University of Sheffield
-
Professor Dyfrig Hughes, Professor of Pharmacoeconomics, Centre for Economics and Policy in Health, Institute of Medical and Social Care Research, Bangor University
-
Professor Paul Jones, Professor of Respiratory Medicine, Department of Cardiac and Vascular Science, St George‘s Hospital Medical School, University of London
-
Professor Khalid Khan, Professor of Women’s Health and Clinical Epidemiology, Barts and the London School of Medicine, Queen Mary, University of London
-
Professor Richard J McManus, Professor of Primary Care Cardiovascular Research, Primary Care Clinical Sciences Building, University of Birmingham
-
Professor Helen Rodgers, Professor of Stroke Care, Institute for Ageing and Health, Newcastle University
-
Professor Ken Stein, Professor of Public Health, Peninsula Technology Assessment Group, Peninsula College of Medicine and Dentistry, Universities of Exeter and Plymouth
-
Professor Jonathan Sterne, Professor of Medical Statistics and Epidemiology, Department of Social Medicine, University of Bristol
-
Mr Andy Vail, Senior Lecturer, Health Sciences Research Group, University of Manchester
-
Professor Clare Wilkinson, Professor of General Practice and Director of Research North Wales Clinical School, Department of Primary Care and Public Health, Cardiff University
-
Dr Ian B Wilkinson, Senior Lecturer and Honorary Consultant, Clinical Pharmacology Unit, Department of Medicine, University of Cambridge
-
Ms Kate Law, Director of Clinical Trials, Cancer Research UK
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
Diagnostic Technologies and Screening Panel
-
Scientific Director of the Centre for Magnetic Resonance Investigations and YCR Professor of Radiology, Hull Royal Infirmary
-
Professor Judith E Adams, Consultant Radiologist, Manchester Royal Infirmary, Central Manchester & Manchester Children’s University Hospitals NHS Trust, and Professor of Diagnostic Radiology, University of Manchester
-
Mr Angus S Arunkalaivanan, Honorary Senior Lecturer, University of Birmingham and Consultant Urogynaecologist and Obstetrician, City Hospital, Birmingham
-
Dr Diana Baralle, Consultant and Senior Lecturer in Clinical Genetics, University of Southampton
-
Dr Stephanie Dancer, Consultant Microbiologist, Hairmyres Hospital, East Kilbride
-
Dr Diane Eccles, Professor of Cancer Genetics, Wessex Clinical Genetics Service, Princess Anne Hospital
-
Dr Trevor Friedman, Consultant Liason Psychiatrist, Brandon Unit, Leicester General Hospital
-
Dr Ron Gray, Consultant, National Perinatal Epidemiology Unit, Institute of Health Sciences, University of Oxford
-
Professor Paul D Griffiths, Professor of Radiology, Academic Unit of Radiology, University of Sheffield
-
Mr Martin Hooper, Public contributor
-
Professor Anthony Robert Kendrick, Associate Dean for Clinical Research and Professor of Primary Medical Care, University of Southampton
-
Dr Nicola Lennard, Senior Medical Officer, MHRA
-
Dr Anne Mackie, Director of Programmes, UK National Screening Committee, London
-
Mr David Mathew, Public contributor
-
Dr Michael Millar, Consultant Senior Lecturer in Microbiology, Department of Pathology & Microbiology, Barts and The London NHS Trust, Royal London Hospital
-
Mrs Una Rennard, Public contributor
-
Dr Stuart Smellie, Consultant in Clinical Pathology, Bishop Auckland General Hospital
-
Ms Jane Smith, Consultant Ultrasound Practitioner, Leeds Teaching Hospital NHS Trust, Leeds
-
Dr Allison Streetly, Programme Director, NHS Sickle Cell and Thalassaemia Screening Programme, King’s College School of Medicine
-
Dr Matthew Thompson, Senior Clinical Scientist and GP, Department of Primary Health Care, University of Oxford
-
Dr Alan J Williams, Consultant Physician, General and Respiratory Medicine, The Royal Bournemouth Hospital
-
Dr Tim Elliott, Team Leader, Cancer Screening, Department of Health
-
Dr Joanna Jenkinson, Board Secretary, Neurosciences and Mental Health Board (NMHB), Medical Research Council
-
Professor Julietta Patrick, Director, NHS Cancer Screening Programme, Sheffield
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Disease Prevention Panel
-
Professor of Epidemiology, University of Warwick Medical School, Coventry
-
Dr Robert Cook, Clinical Programmes Director, Bazian Ltd, London
-
Dr Colin Greaves, Senior Research Fellow, Peninsula Medical School (Primary Care)
-
Mr Michael Head, Public contributor
-
Professor Cathy Jackson, Professor of Primary Care Medicine, Bute Medical School, University of St Andrews
-
Dr Russell Jago, Senior Lecturer in Exercise, Nutrition and Health, Centre for Sport, Exercise and Health, University of Bristol
-
Dr Julie Mytton, Consultant in Child Public Health, NHS Bristol
-
Professor Irwin Nazareth, Professor of Primary Care and Director, Department of Primary Care and Population Sciences, University College London
-
Dr Richard Richards, Assistant Director of Public Health, Derbyshire County Primary Care Trust
-
Professor Ian Roberts, Professor of Epidemiology and Public Health, London School of Hygiene & Tropical Medicine
-
Dr Kenneth Robertson, Consultant Paediatrician, Royal Hospital for Sick Children, Glasgow
-
Dr Catherine Swann, Associate Director, Centre for Public Health Excellence, NICE
-
Mrs Jean Thurston, Public contributor
-
Professor David Weller, Head, School of Clinical Science and Community Health, University of Edinburgh
-
Ms Christine McGuire, Research & Development, Department of Health
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
External Devices and Physical Therapies Panel
-
Consultant Physician North Bristol NHS Trust
-
Reader in Wound Healing and Director of Research, University of Leeds
-
Professor Bipin Bhakta, Charterhouse Professor in Rehabilitation Medicine, University of Leeds
-
Mrs Penny Calder, Public contributor
-
Dr Dawn Carnes, Senior Research Fellow, Barts and the London School of Medicine and Dentistry
-
Dr Emma Clark, Clinician Scientist Fellow & Cons. Rheumatologist, University of Bristol
-
Mrs Anthea De Barton-Watson, Public contributor
-
Professor Nadine Foster, Professor of Musculoskeletal Health in Primary Care Arthritis Research, Keele University
-
Dr Shaheen Hamdy, Clinical Senior Lecturer and Consultant Physician, University of Manchester
-
Professor Christine Norton, Professor of Clinical Nursing Innovation, Bucks New University and Imperial College Healthcare NHS Trust
-
Dr Lorraine Pinnigton, Associate Professor in Rehabilitation, University of Nottingham
-
Dr Kate Radford, Senior Lecturer (Research), University of Central Lancashire
-
Mr Jim Reece, Public contributor
-
Professor Maria Stokes, Professor of Neuromusculoskeletal Rehabilitation, University of Southampton
-
Dr Pippa Tyrrell, Senior Lecturer/Consultant, Salford Royal Foundation Hospitals’ Trust and University of Manchester
-
Dr Nefyn Williams, Clinical Senior Lecturer, Cardiff University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Interventional Procedures Panel
-
Professor of Vascular Surgery, University of Sheffield
-
Consultant Colorectal Surgeon, Bristol Royal Infirmary
-
Mrs Isabel Boyer, Public contributor
-
Mr Sankaran Chandra Sekharan, Consultant Surgeon, Breast Surgery, Colchester Hospital University NHS Foundation Trust
-
Professor Nicholas Clarke, Consultant Orthopaedic Surgeon, Southampton University Hospitals NHS Trust
-
Ms Leonie Cooke, Public contributor
-
Mr Seumas Eckford, Consultant in Obstetrics & Gynaecology, North Devon District Hospital
-
Professor Sam Eljamel, Consultant Neurosurgeon, Ninewells Hospital and Medical School, Dundee
-
Dr Adele Fielding, Senior Lecturer and Honorary Consultant in Haematology, University College London Medical School
-
Dr Matthew Hatton, Consultant in Clinical Oncology, Sheffield Teaching Hospital Foundation Trust
-
Dr John Holden, General Practitioner, Garswood Surgery, Wigan
-
Dr Fiona Lecky, Senior Lecturer/Honorary Consultant in Emergency Medicine, University of Manchester/Salford Royal Hospitals NHS Foundation Trust
-
Dr Nadim Malik, Consultant Cardiologist/Honorary Lecturer, University of Manchester
-
Mr Hisham Mehanna, Consultant & Honorary Associate Professor, University Hospitals Coventry & Warwickshire NHS Trust
-
Dr Jane Montgomery, Consultant in Anaesthetics and Critical Care, South Devon Healthcare NHS Foundation Trust
-
Professor Jon Moss, Consultant Interventional Radiologist, North Glasgow Hospitals University NHS Trust
-
Dr Simon Padley, Consultant Radiologist, Chelsea & Westminster Hospital
-
Dr Ashish Paul, Medical Director, Bedfordshire PCT
-
Dr Sarah Purdy, Consultant Senior Lecturer, University of Bristol
-
Dr Matthew Wilson, Consultant Anaesthetist, Sheffield Teaching Hospitals NHS Foundation Trust
-
Professor Yit Chiun Yang, Consultant Ophthalmologist, Royal Wolverhampton Hospitals NHS Trust
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Pharmaceuticals Panel
-
Professor in Child Health, University of Nottingham
-
Senior Lecturer in Clinical Pharmacology, University of East Anglia
-
Dr Martin Ashton-Key, Medical Advisor, National Commissioning Group, NHS London
-
Dr Peter Elton, Director of Public Health, Bury Primary Care Trust
-
Dr Ben Goldacre, Research Fellow, Division of Psychological Medicine and Psychiatry, King’s College London
-
Dr James Gray, Consultant Microbiologist, Department of Microbiology, Birmingham Children’s Hospital NHS Foundation Trust
-
Dr Jurjees Hasan, Consultant in Medical Oncology, The Christie, Manchester
-
Dr Carl Heneghan, Deputy Director Centre for Evidence-Based Medicine and Clinical Lecturer, Department of Primary Health Care, University of Oxford
-
Dr Dyfrig Hughes, Reader in Pharmacoeconomics and Deputy Director, Centre for Economics and Policy in Health, IMSCaR, Bangor University
-
Dr Maria Kouimtzi, Pharmacy and Informatics Director, Global Clinical Solutions, Wiley-Blackwell
-
Professor Femi Oyebode, Consultant Psychiatrist and Head of Department, University of Birmingham
-
Dr Andrew Prentice, Senior Lecturer and Consultant Obstetrician and Gynaecologist, The Rosie Hospital, University of Cambridge
-
Ms Amanda Roberts, Public contributor
-
Dr Gillian Shepherd, Director, Health and Clinical Excellence, Merck Serono Ltd
-
Mrs Katrina Simister, Assistant Director New Medicines, National Prescribing Centre, Liverpool
-
Professor Donald Singer, Professor of Clinical Pharmacology and Therapeutics, Clinical Sciences Research Institute, CSB, University of Warwick Medical School
-
Mr David Symes, Public contributor
-
Dr Arnold Zermansky, General Practitioner, Senior Research Fellow, Pharmacy Practice and Medicines Management Group, Leeds University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Mr Simon Reeve, Head of Clinical and Cost-Effectiveness, Medicines, Pharmacy and Industry Group, Department of Health
-
Dr Heike Weber, Programme Manager, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Psychological and Community Therapies Panel
-
Professor of Psychiatry, University of Warwick, Coventry
-
Consultant & University Lecturer in Psychiatry, University of Cambridge
-
Professor Jane Barlow, Professor of Public Health in the Early Years, Health Sciences Research Institute, Warwick Medical School
-
Dr Sabyasachi Bhaumik, Consultant Psychiatrist, Leicestershire Partnership NHS Trust
-
Mrs Val Carlill, Public contributor
-
Dr Steve Cunningham, Consultant Respiratory Paediatrician, Lothian Health Board
-
Dr Anne Hesketh, Senior Clinical Lecturer in Speech and Language Therapy, University of Manchester
-
Dr Peter Langdon, Senior Clinical Lecturer, School of Medicine, Health Policy and Practice, University of East Anglia
-
Dr Yann Lefeuvre, GP Partner, Burrage Road Surgery, London
-
Dr Jeremy J Murphy, Consultant Physician and Cardiologist, County Durham and Darlington Foundation Trust
-
Dr Richard Neal, Clinical Senior Lecturer in General Practice, Cardiff University
-
Mr John Needham, Public contributor
-
Ms Mary Nettle, Mental Health User Consultant
-
Professor John Potter, Professor of Ageing and Stroke Medicine, University of East Anglia
-
Dr Greta Rait, Senior Clinical Lecturer and General Practitioner, University College London
-
Dr Paul Ramchandani, Senior Research Fellow/Cons. Child Psychiatrist, University of Oxford
-
Dr Karen Roberts, Nurse/Consultant, Dunston Hill Hospital, Tyne and Wear
-
Dr Karim Saad, Consultant in Old Age Psychiatry, Coventry and Warwickshire Partnership Trust
-
Dr Lesley Stockton, Lecturer, School of Health Sciences, University of Liverpool
-
Dr Simon Wright, GP Partner, Walkden Medical Centre, Manchester
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health