
Notes
Article history
The research reported in this issue of the journal was commissioned and funded by the HTA programme on behalf of NICE as project number 09/87/01. The protocol was agreed in January 2010. The assessment report began editorial review in January 2011 and was accepted for publication in January 2011. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the referees for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
none
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2012. This work was produced by Bond et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This journal is a member of and subscribes to the principles of the Committee on Publication Ethics (COPE) (http://www.publicationethics.org/). This journal may be freely reproduced for the purposes of private research and study and may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NETSCC, Health Technology Assessment, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2012 Queen’s Printer and Controller of HMSO
Chapter 1 Background
Aim of the review
The aim of this assessment is to review and update, as necessary, the National Institute for Health and Clinical Excellence (NICE) guidance to the NHS in England and Wales (issued November 2006, amended September 2007 and August 20091) on the clinical effectiveness and cost-effectiveness of donepezil, galantamine and rivastigmine for mild-to-moderate Alzheimer’s disease (AD), and memantine for moderate-to-severe AD. 1
This previous guidance was primarily based on evidence presented to NICE in the assessment report by Loveman and colleagues2 in 2004. We will summarise the evidence presented in this previous report, and review and report new evidence from 2004 to the present.
Description of health problem
Pathology
Definitions
Dementia is usually a disease of later life and has been defined as:
A syndrome consisting of progressive impairment in memory and at least one other cognitive deficit (aphasia, apraxia, agnosia, or disturbance in executive function) in the absence of another explanatory central nervous system disorder, depression, or delirium.
Diagnostic and Statistical Manual of Mental Disorders-Fourth Edition (DSM-IV)3
People with dementia may also show other symptoms, such as depression, psychosis, wandering and aggression.
Alzheimer’s disease is the most common form of dementia, and is additionally characterised by the presence of neurofibrillary tangles and amyloid plaques in the cerebral cortex, observed at post-mortem.
Diagnosis
In the distant past, diagnosis of AD before death had been on the basis of excluding other causes. However, there are now agreed criteria that accurately predict up to 90% of AD cases (Box 1). Alternatively, diagnosis can be made from ICD-10 (International Classification of Diseases, 10th Revision4) and DSM-IV(Diagnostic and Statistical Manual of Mental Disorders-Fourth Edition3).
Dementia established by clinical examination, documented by the MMSE or similar and confirmed by neuropsychological tests
Decline in memory and at least one non-memory intellectual function
Decline from previous level and continuing decline
Onset between 40 and 90 years of age
No disturbance in consciousnessAbsence of systemic disorders or other brain diseases that in, and of, themselves could account for the progressive deficits in memory and cognition
Definite AD
Clinical criteria of probable ADHistopathological evidence of AD at post-mortem or biopsy
Possible ADPatient has dementia syndrome with no other cause, but clinical variation from typical AD
Patient had second disorder that is sufficient to produce dementia, but not considered the cause of the dementia
Single gradually progressive cognitive deficit in absence of other causes
The diagnosis of dementia may happen many months after onset, as the development of symptoms is usually insidious. It may take some time for the individual to realise that significant memory, mood or ability changes are taking place. Other possible diagnoses, such as depression, delirium, vitamin B12 deficiency and hypothyroidism, have to be excluded first. Further testing is necessary to determine the particular cause of dementia. 5
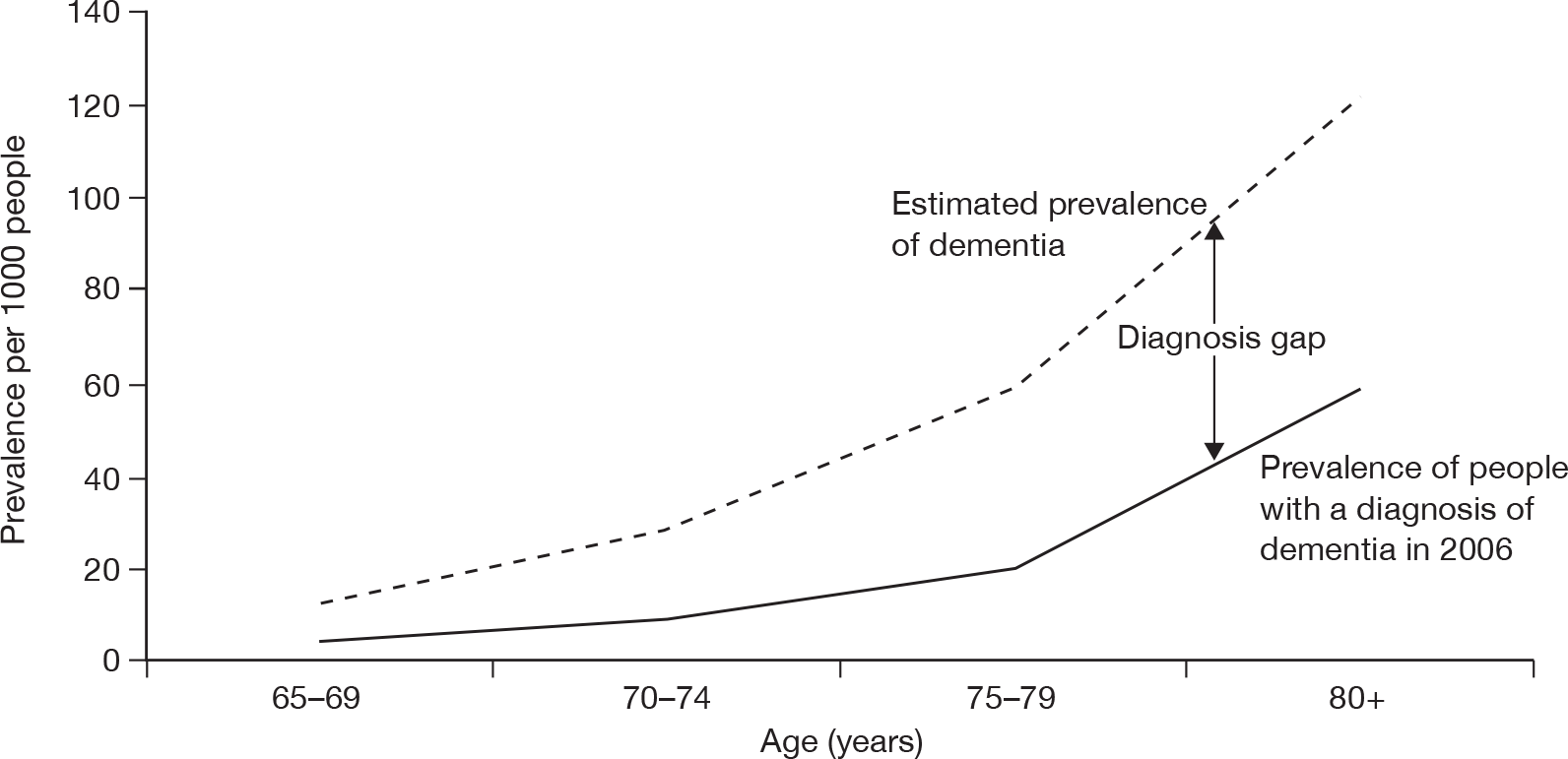
Examination by the National Audit Office (NAO) of data from primary care trusts (PCTs), the Dementia UK report8 and the Office for National Statistics (ONS) has indicated that, in England, more than 50% of people with dementia never receive a correct diagnosis. 9 See Figure 1 for 2006 estimates of the diagnosis gap.
FIGURE 1.
The gap between prevalence and diagnosis of dementia in England. Source: Knapp et al. 8 Dementia UK report to the Alzheimer’s Society, King’s College London and London School of Economics and Political Science (estimated actual average prevalence) and General Practice Research Database report to the NAO (reported prevalence based on diagnoses). The graph shows reported prevalence of dementia, based on levels of diagnosis within PCTs, for ages 65 years and upwards in 2006. The estimated actual average prevalence has been calculated using data from the 2007 Dementia UK report8 in conjunction with population estimates from the ONS. The latter does not take into consideration those aged 85 years and above, owing to restrictions on the data available, and as such forms a very prudent estimate of dementia in the > 65-year population.
Epidemiology
Alzheimer’s disease is predominantly a disease of later life, with some 5% of the UK population over 65 years affected. Early-onset AD can be found in younger people; this is a rare condition, accounting for only an estimated 2.2% of those with dementia. 8 Currently, there are estimated to be about 820,000 people in the UK with dementia (1.3% of the population); of these approximately 520,000 (62%) will have AD, of whom approximately 423,000 (83%) live in England and 26,000 (5%) live in Wales. 8,10 AD is more commonly found in women than men in the UK, with 67% of women with dementia having AD, but only 55% of men. 8 However, the association with gender is completely explained by the shorter life expectancy of men. 11
The incidence of dementia, therefore, increases with age. In England and Wales, among people aged 65–69 years, the incidence is estimated to be 7.4 per 1000 person-years [95% confidence interval (CI) 3.6 to16.1 per 1000 person-years]; this rises to 84.9 per 1000 person-years (95% CI 63.0 to 107.8 per 1000 person-years) at 85 years old and above. 12 These rates predict 180,000 new cases of dementia per year, and if 62% of these have AD (see above) then there are approximately 111,600 new cases of AD in England and Wales per year. The Medical Research Council’s Cognitive Function and Ageing Study (2006) found that in England and Wales increasing age was the greatest risk factor for dementia, with gender weakly associated. Having Parkinson’s disease increased the risk of dementia by three times [odds ratio 3.5 (95% CI 1.3 to 9.3)], but rating your own health as poor was a greater risk factor [odds ratio 3.9 (95% CI 2.2 to 6.9)]. Better education was a marginally protective factors [odds ratio 0.7 (95% CI 0.5 to 1.0)]. 11
Table 1 shows the combined numbers of diagnosed and undiagnosed cases of dementia in the UK in 2006 estimated by the Dementia 2010 study. 10
| Age group (years) | Gender | Total | |
|---|---|---|---|
| Male | Female | ||
| 30–59 | 19,840 | 11,381 | 31,221 |
| 60–64 | 25,034 | 7782 | 32,816 |
| 65–69 | 28,056 | 15,378 | 43,434 |
| 70–74 | 50,085 | 48,319 | 98,404 |
| 75–79 | 42,805 | 74,037 | 116,842 |
| 80–84 | 68,343 | 120,482 | 188,825 |
| 85–89 | 50,439 | 124,465 | 174,904 |
| 90–94 | 28,399 | 78,606 | 107,005 |
| 95–99 | 5008 | 23,424 | 28,432 |
| Total | 318,009 | 503,874 | 821,883 |
Aetiology
The cause of AD is uncertain. However, it is generally believed that the condition develops from multiple factors, with increasing age bringing the greatest risk. Up to 5% of cases are linked to genetic causes; medical history and lifestyle are also contributing factors. 12 At least three genes have been identified that are associated with the rare condition of early-onset AD. 14–16 A genetic link is also likely for those with a family history of late-onset AD, although a particular gene for this has not yet been identified. 5
There is evidence that it may be possible to prevent some incidence of AD; it is thought that due to the cerebrovascular contribution to brain pathology, managing cardiovascular risk factors (high cholesterol, high blood pressure, type 2 diabetes and being overweight) may delay or prevent the onset of AD. Other possibly preventative factors include regular exercise, a low-fat diet and a good social network. 17–19
Prognosis
There is currently no cure for AD. There is variation in the time it takes from diagnosis to death. The estimated median survival for AD from onset has been calculated as 7.1 years (95% CI 6.7 to 7.5 years) in the USA by Fitzpatrick and colleagues20 and is reported in Warrell and colleagues7 as about 10 years in the UK. Although, survival figures are varied and depend on whether or not they are from the time of reported onset or the time of actual diagnosis; in general, a diagnosis of AD halves life expectancy.
The contribution of AD to these survival figures is difficult to know, as people with AD frequently have comorbidities that will influence their longevity. The proportion of deaths estimated to be due to AD increases with age and varies with gender. At 65 years old, 1% of women and 2% of men are likely to die from dementia; at 85–89 years old, this rises to 23% of women and 18% of men. 8
Impact of health problem
Significance for patients
It can take several years of slow deterioration for the full effects of AD to be felt. 21 In the early stages there can be severe memory loss for recent events with associated repetitive questioning and loss of the ability to learn. 21,22 There may be a general deterioration in the ability to socialise, which can be difficult for both sufferer and carer to cope with. 23,24 As mild AD takes hold, normal activities of daily living, such as shopping or managing finances, become increasingly difficult as cognitive function deteriorates. 25 Communication also becomes a problem as vocabulary shrinks and fluency falters. 25,26 At this stage the sufferer may still be aware of their failing abilities, and the experience and known outcome of AD can frequently lead to associated depression.
Disease progression to moderate AD leads to further loss of cognitive abilities, including the ability to remember and/or understand words. Activities of daily living become increasingly affected as the ability to perform purposeful movements decreases, for example getting dressed or cooking. Commonly there are also neuropsychiatric symptoms such as anxiety, wandering, irritability, disinhibition and apathy. Visual and auditory hallucinations occur in about 30–59% of sufferers. 7 Managing these symptoms can be a very difficult burden for carers, who may well be elderly themselves. Indeed, the main predictors of full-time institutional care are caregiver exhaustion,27 the degree of patient dependence28 and the rate of disease progression. 29
In developed countries, sufferers of AD usually end their days in institutional care, as the last stages of AD bring complete dependence. This final stage is characterised by limitations such as inability to walk, manage personal care, mutism, inability to recognise familiar people and objects, and incontinence. There may also be seizures and involuntary twitching.
Significance for carers
Being the main carer for a person with AD can have an enormous impact on physical, psychological and social well-being. 30 From the early frustrations, prior to diagnosis, of living with others’ impaired cognitive function, through the devastating diagnosis, to the knowledge that the relation/friend is going to get progressively worse and die, the outlook for carers is bleak. Many carers are elderly spouses, perhaps with health concerns of their own or grown-up children who now have their own families to care for as well. 31 Carers may cope reasonably well with the early stages of the disease, but as the behavioural and psychological symptoms of dementia (BPSD) become more severe, full-time institutional care becomes increasingly likely. 5 For some carers this brings feelings of guilt and depression,32,33 possibly leading to the cognitive decline of the carers themselves. 34 Behavioural and psychological symptoms are common in AD and may be difficult to manage, causing distress to carers and patients alike. They have been shown to be better predictors of institutionalisation35 and carer distress36 than cognitive symptoms.
As AD progresses, increasing grief and feelings of loss may be experienced by carers. 37 Findings from the EUROCARE European study of co-resident spouse carers of dementia sufferers showed that co-resident carers carried a heavy burden and that mental distress was high. They concluded that issues of behavioural disturbance, negative social reactions, financial worries and younger spouse carers predicted greater distress. 38 However, there is evidence that enhanced counselling and support can relieve symptoms of depression in caregivers and delay admission to institutional care of people with AD. 39
Significance for the NHS and social services
With an increasingly elderly population, the burden of AD upon the NHS and social services is considerable. Of the estimated 520,000 people in the UK with AD,10 It is estimated that approximately 63.5% live at home and 36.5% are in residential care. 8 Unsurprisingly, the risk of moving into residential care increases with age and disease severity. The proportion of people with severe dementia increases from 6.3% among those between 65 and 69 years old to 23.5% among those aged 95 years or older. 8 Consequently, the proportion of people in the UK with dementia who live in residential care rises from 26.6% of those aged 65–74 years to 27.8% of those aged 75–84 years, 40.9% of those aged 85–89 years and 60.8% of those aged 90 years or older. 8
As the disease progresses, the balance of burden of care shifts from predominantly falling on the informal carers to the NHS and social services as patients are sustained with medication and support at home, until finally financial costs fall mostly on social services as patients move into institutional care, although a proportion of this cost may be borne by the carer or their family. Another proportion of people will qualify for the NHS Continuing Care programme, which will meet their care needs either in the community or in an institution. A longitudinal cohort study by Banerjee and colleagues40 has found that when a person with dementia lives with their main carer they are 20 times less likely, over the course of a year, to move into residential care than those who do not [odds ratio 0.05 (95% CI 0.01 to 0.42)]. They also found that the carer’s psychological quality of life (QoL) and the severity of behavioural problems shown by the patient were predictors of institutionalisation [odds ratios 1.10 (95% CI 1.02 to 1.19) and 1.08 (95% CI 1.01 to 1.15), respectively]. 40 However, in a similar study, de Vugt and colleagues41 found that it was the carer’s response to the behavioural symptoms (rather than the symptoms themselves) that predicted institutionalisation.
Measurement of disease
Details of individual measures used in the included trials can be found in Appendix 1.
A review of outcome measures used in clinical trials of drugs for AD by Wolfson and colleagues42 revealed a number of shortcomings in these measures. In particular, they found that several of the scales had weak psychometric properties, for example lack of responsiveness to change. Some studies had small sample sizes and others used inappropriate statistical analyses. 43
The progress and symptoms of AD can be measured through cognitive tests, behavioural measures, measures of functional ability/QoL and global rating scales.
A thorough assessment of cognitive ability would include measures of attention, processing speed, visuospatial function, praxis, language, executive function and abstraction. The most commonly used scales for this domain are the Mini Mental State Examination44 (MMSE) and the Alzheimer’s Disease Assessment Scale – Cognitive Subscale45 (ADAS-cog). Whereas the MMSE’s validity and reliability as a screening tool for AD have been established,44 it has problems with identifying change over time and scores are affected by people’s level of education. 46,47 Similarly, the ADAS-cog has been criticised for its insensitivity to change in cognitive ability at either end of the severity continuum. 48 It is concerning that the most commonly used instruments to measure change in cognitive function in drug trials for AD should be insensitive to change.
The measurement of behaviour change is important as it is these symptoms that many caregivers find most difficult to cope with, precipitating the transition into institutional care. 35,36 The most frequently used measure of behavioural change in AD trials is the Neuropsychiatric Inventory (NPI). 49 This is a proxy-rated scale, usually completed by the main carer; its validity and reliability have been demonstrated by Cummings and colleagues. 49
There are two kinds of global rating scales for AD: those that measure the severity of illness at a point in time, for example the Clinical Dementia Rating (CDR) Scale50 and can, if used repeatedly, plot mental deterioration over time; the other sort of global instruments are change scales, such as the Clinician’s Interview-based Impression of Change – plus Caregiver Input (CIBIC-plus). 51 These measure broad changes in AD. However, their use may be biased towards cognitive abilities, as Claus and colleagues52 have found that clinicians may have a bias towards this aspect of AD, whereas carers place more emphasis on behavioural and psychological symptoms and functional ability. However, the use of CIBIC-plus may help to overcome this.
Measures of functional status in clinical trials are most commonly taken using the Activities of Daily Living (ADL) Scale53 or the Instrumental Activities of Daily Living (IADL). 54 Their reliability and validity has been described by McDowell and Newell. 55 However, this is not in the specific context of dementia.
Although the DEMQOL has been validated as a measure of health-related quality of life (HRQoL) in people with dementia,56 in clinical trials the most frequently used measure is the patient-rated QoL scale. This is a seven-item patient-rated scale that measures feelings of well-being in the domains of relationships, eating, sleeping and social and leisure activities, on a 0–50 analogue scale. 57
Care for people with Alzheimer’s disease
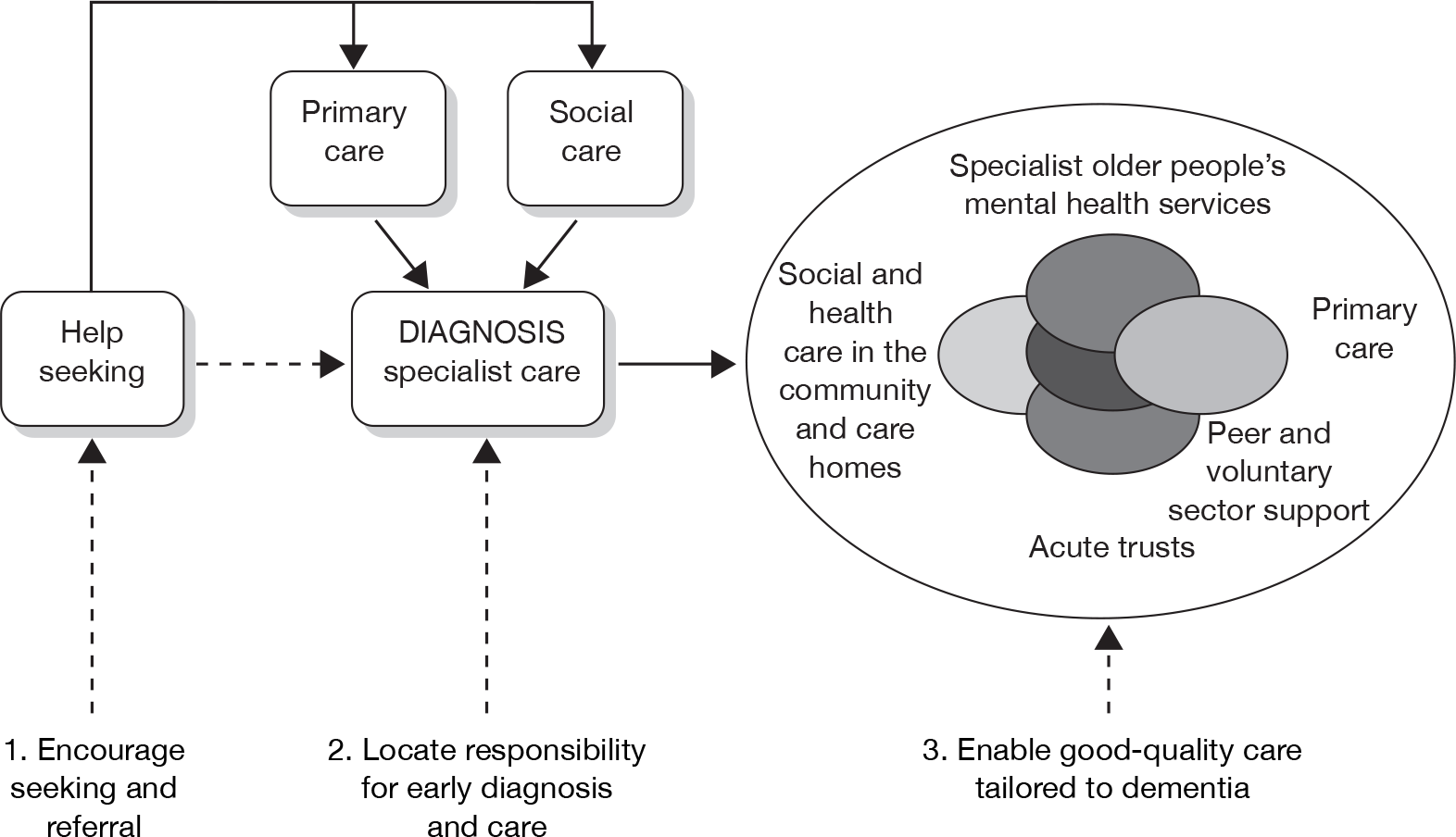
The National Dementia Strategy for England58 says that everyone with suspected dementia should have access to ‘A rapid and competent specialist assessment; an accurate diagnosis sensitively communicated to the person with dementia and their carers; and treatment, care and support provided as needed following diagnosis’. The system needs to have the capacity to see all new cases of dementia in the area.
In order to achieve this goal the Department of Health has set out the following care pathway,58 as shown in Figure 2. A separate Dementia Action Plan has been developed for Wales.
FIGURE 2.
Care pathway summarising the three themes of the National Dementia Strategy and the commissioning challenges. Source: Department of Health 2009. 58
The provision of care for people with AD is complex, as it is shared between informal voluntary care, private care, social services and the NHS.
Informal care
Carers
An analysis of the General Household Survey (1998–9) data estimated that 53% of people over 65 years of age who could not live completely independently were supported by unpaid carers. 59 This estimate translates to approximately 4 million carers in England, most of working age. 60 Changing demographic patterns, with children living a considerable distance from their parents and more single people, may mean that this caring resource is reduced in the future. 61 Reports in the last decade have promoted support for carers: Support for Carers of Older People;60 Caring about Carers: A Strategy for Carers in Wales (Implementation Plan)62 and The NHS Plan. 63 However, many carers feel unsupported and isolated. 60 The burden of caring can affect the health and well-being of carers,64 possibly with high levels of depression,65 although another study found that over a 2-year period carers’ psychological well-being did not deteriorate. 66 Another effect of caring is the reduction of the capacity of the carer to earn a living. 67 The Medical Research Council’s Cognitive Function and Ageing Study (MRC CFAS) also found that 9% of carers of people with dementia had reduced their hours of work and one-fifth of carers who were younger than the statutory retirement age had given up work completely. 66
Formal care
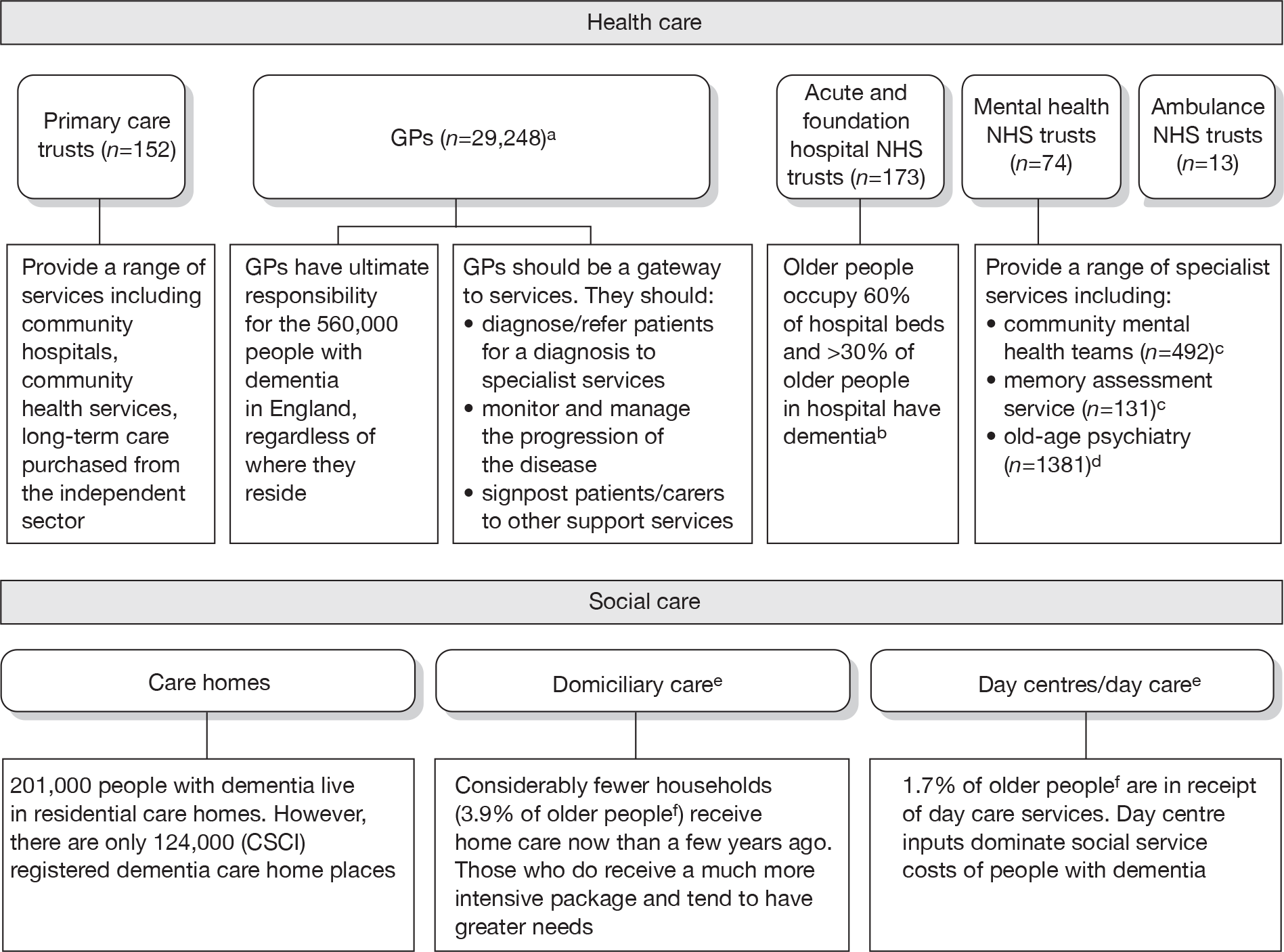
The formal care of people with AD falls mainly to the NHS and social services, although private and voluntary sector agencies are also involved. The NAO has produced a diagram to show the types of providers that are currently involved in dementia care (Figure 3).
FIGURE 3.
Key public providers involved in the formal care of people with dementia. Source: NAO. 9 a, Royal College of General Practitioners;68 b, Royal College of Psychiatrists. 69 c, Barnes and Lombardo. 70 d, Membership Office. 71 e, Patients receiving domiciliary care may also be in receipt of day care and vice versa. f, Knapp et al. 8 Dementia UK report to the Alzheimer’s Society, King’s College London and London School of Economics and Political Science (older people are aged 65 years and over). The 2005 census data and Dementia UK both report that there are over 8 million older people in England.
In addition to informal care provided by unpaid carers, at any one time people with dementia will have, or need to, access to a range of health and social care services. For example, a person with dementia may receive services from the district nurse or community matron, see their GP, attend day care and be in receipt of domiciliary care. As the disease progresses, the needs of most people with dementia for domiciliary and other care and support will increase. Ultimately, a care home may become appropriate or the person with dementia may stay in their own home supported by various aspects of health and social care.
NHS services
The medical needs of people with AD span a wide range of specialties; as people with AD are usually elderly, comorbidities are common and their treatment is frequently complicated by the dementia. However, the main specialty involved is old age psychiatry, although, in the UK, a geriatrician may be responsible for diagnosis and treatment. The first contact a person with symptoms has is usually with primary care; a correct and early provisional diagnosis here is vital as this is the way into specialist care. 8
There are no nationally agreed criteria for referral; therefore, the burden of care falls differentially on primary and secondary care depending on location. The kinds of care provided can be grouped as either those for ‘serious mental illness’ or ‘early intervention’, depending on the severity of symptoms. The initial assessment of someone who may have AD is ideally conducted in their home, although many people with early stages of the disease are now seen in memory clinics. The home is a preferable setting to an outpatient department because it enables the assessor to see how the person functions in everyday situations. It also enables risk assessment of potential dangers in the home and is more likely to take place, as the possibly confused and forgetful person may lack understanding of their need to attend an assessment appointment.
Social services
Apart from cognitive and psychological decline, people with AD face a gradual loss of their ability to live independently. Initial support with everyday activities frequently comes from family and friends. However, where this is not available, and when the disease progresses, such support predominantly comes from social services, although private agencies may be involved.
There are no statistics about the total number of people with AD who are supported at home either by social services or the private sector. However, in England, there has been an increase in recent years, in the volume of home care bought by local authorities, a decrease in the numbers of people supported and an increase of support from private providers. 8 This means that fewer people are receiving help at home from social services than in the recent past, but those who do generally have greater needs and are receiving more comprehensive support. A consequence of this is that people are entering full-time residential care at later stages of AD. In Wales the picture is different, with a decrease in the amount spent on home care by local authorities. 8
In recent years the supply of residential care homes has been in decline in England and the balance of ownership has changed, with more homes now being in private hands. 8 Also the average size of a care home has increased to 34 beds and the quality of care provided continues to be variable; areas of concern include unstimulating environments and a low paid, poorly trained work force that has a high turnover, which undermines the building of relationships between staff and residents. 60 Standards have begun to rise as a result of regulation, according to the Audit Commission, but there is still a long way to go.
The cost of care: overview
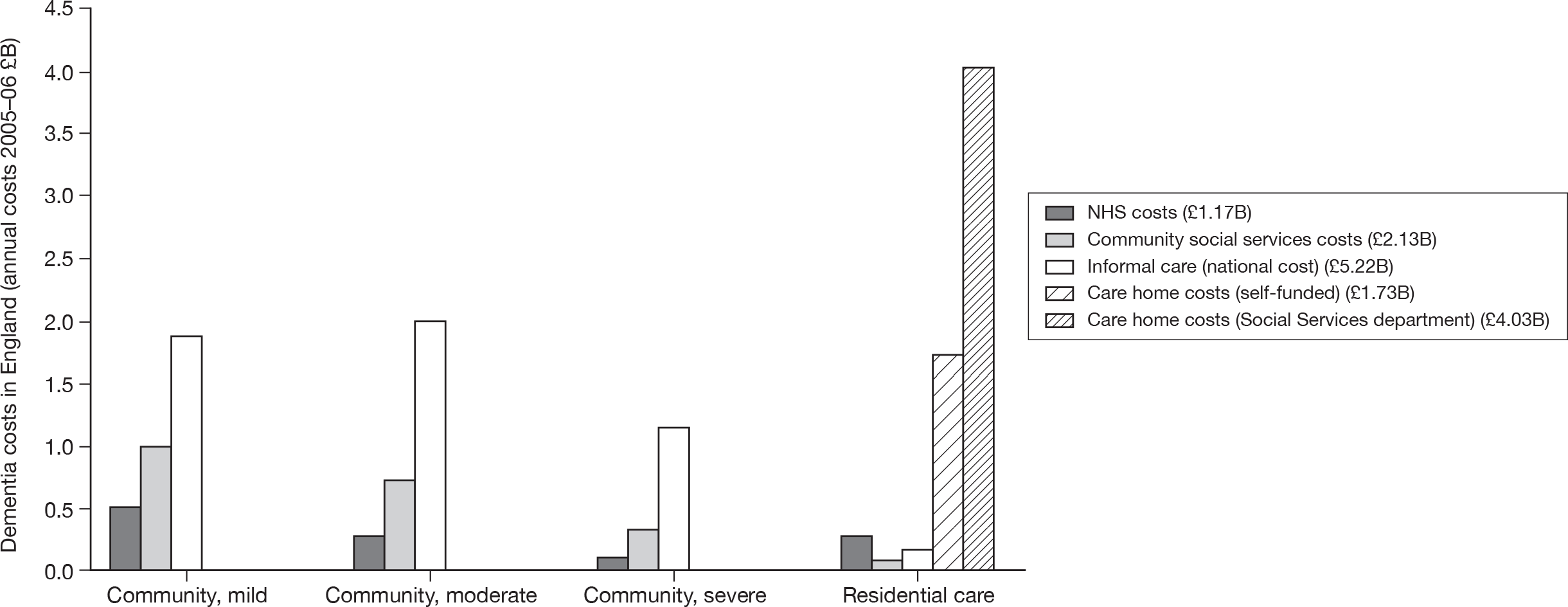
Around two-thirds of the care for people with AD comes informally from the community and it is the family who bares the greatest burden of cost. 9 The following diagram from the NAO report (Figure 4) shows how the cost of dementia was spread in England in 2005–6. [The categories of mild, moderate and severe dementia are based on the schedule named the Cambridge Mental Disorders of the Elderly Examination (CAMDEX). 72]
FIGURE 4.
Dementia costs in England 2005–6 by severity and place of residence. Source: NAO. 9 Dementia UK report to the Alzheimer’s Society, King’s College London, and London School of Economics and Political Science. Informal care is costed using (1) the hourly cost of a home care worker for specific tasks and (2) the minimum wage for time spent on general tasks and supervisory activities. The estimated service cost of late-onset dementia is over £14B per annum. Most of this is accounted for by supported accommodation (i.e. care home) costs and informal care (i.e. notional) costs. Some £6.87B is borne by people with dementia and their families.
It has been estimated in the recent Dementia 2010 report that these costs have risen to an annual cost to the UK economy of £23B per year (2007–8) (this compares with £12B per year for cancer and £8B per year for heart disease). The majority of this £12.4B cost fell on unpaid carers (55%); social services funded 40% (£9B) of the cost and the NHS funded 5% (£1.2B). 10
Presuming that 62% of people with dementia have AD (see Epidemiology), this translates to an annual cost to the UK economy of AD of over £14B per year. For each person with AD this gives an estimated annual cost of £27,647 – more than cancer (£6000), stroke (£5000) and heart disease patients (£3500) put together. 10 A full description of the costs of care for people with AD can be found in Chapter 7 (see Cost of health and social care received by Alzheimer’s disease patients).
Variation in services
The Dementia UK report8 indicated that there is a wide variation in the prescribing of anti-dementia medication in England and Wales. 8 Information specific to AD was not available, but it may be reasonable to suggest that the picture would be similar. The data were collected by IMS, a medical information consultancy, between October 2005 and September 2006 from 50% of the pharmacies in England and Wales, and represent 90% of all UK prescribing. This information is a reflection of national commissioning practice and shows, by PCT, the likelihood of being prescribed medication for dementia. The number of prescriptions per person with dementia in primary care varied from 12.0 prescriptions per year in Knowsley to 0.4 in West Berkshire. Most PCTs (75%) prescribed between 1.0 and 4.0 prescriptions per year. 8 The reason for this variation in provision is unclear.
National guidelines, guidance and reports
The following national guidelines, guidance and reports are related to this technology appraisal (TA).
-
Dementia 2010: The economic burden of dementia and associated research funding in the United Kingdom. 10
-
Living well with dementia: a national dementia strategy (2009). 58
-
NICE TA No. 111 entitled Donepezil, galantamine, rivastigmine (review) and memantine for the treatment of AD (amended August 2009). 1
-
Dementia UK: the full report (2007). 8
-
Dementia: The NICE-SCIE Guideline on Supporting People with Dementia and their Carers in Health and Social Care (2007). 5
-
Improving services and support for people with dementia (2007). 9
-
Everybody’s Business – Integrated mental health services for older adults: a service development guide (2005). 73
-
Forget-me-not 2002: mental health services for older people – the Audit Commission. 74
-
National Service Framework for Older People (2001). 75
-
Forget-me-not: mental health services for older people – the Audit Commission (2002). 76
Description of technology under assessment
Summary of interventions
Three licensed acetylcholinesterase inhibitors
This technology assessment report (TAR) will consider four pharmaceutical interventions. Three have marketing authorisations in the UK for the treatment of adults with mild-to-moderately severe AD (measured by the MMSE score 26–10). These are donepezil (Aricept®, manufactured by Eisai Ltd), rivastigmine (Exelon®, manufactured by Novartis) and galantamine (Reminyl®, manufactured by Shire Pharma). They are acetylcholinesterase inhibitors (AChEIs), which work by restricting the cholinesterase enzyme from breaking down acetylcholine, thus increasing the concentration and duration of acetylcholine at sites of neurotransmission.
Donepezil hydrochloride
Donepezil hydrochloride (Aricept®) is manufactured by Eisai Ltd and co-marketed with Pfizer Ltd. It was the first drug to be licensed in the UK specifically for AD. Donepezil is a reversible, specific AChEI. Donepezil is easily absorbed by the body and can be taken once a day, initially at 5 mg and then, after 4 weeks’ use, titrated up to 10 mg per day if necessary. Possible side effects associated with donepezil include, bradycardia (particularly in people with sick sinus syndrome or other supraventricular cardiac conduction conditions), seizures, nausea, vomiting, diarrhoea, muscle cramp, urinary incontinence, fatigue, insomnia and dizziness.
Rivastigmine tartrate
Rivastigmine tartrate (Exelon®) made by Novartis pharmaceuticals, is a selective inhibitor of acetylcholinesterase and also butrylcholinesterase, another enzyme. Owing to its short half-life (1.5 hours) it has to be taken twice a day. Doses start at 3 mg per day and increase gradually to between 6 and 12 mg per day. It can be taken orally or by a transdermal patch, with doses of either 4.6 or 9.5 mg/24 hours. Care should be used with people with renal disease, mild or moderate liver disease, sick sinus syndrome, conduction abnormalities, gastric or duodenal ulcers and a history of asthma or obstructive pulmonary disease. The main possible side effects found are nausea and vomiting, usually in the dose escalation phase.
Galantamine
Galantamine (Reminyl®) is manufactured by the Shire Pharmaceuticals Group. Galantamine was originally made from snowdrop and narcissus bulbs, but is now synthetically produced. It is a reversible inhibitor of acetylcholinesterase, with a half-life of about 7 hours, indicating that it should be taken twice a day at the recommended dose of 16–24 mg each time. An alternative version (Reminyl XL) is taken once a day at doses of 8, 16 or 24 mg. The side effects from galantamine are similar to those of the other AChEIs and are mainly gastrointestinal – abdominal pain, diarrhoea, nausea and vomiting – although bradycardia and dizziness have been reported.
Memantine hydrochloride
The fourth drug, memantine hydrochloride (Ebixa®), manufactured by Lundbeck, has a UK marketing authorisation for the treatment of people with moderate-to-severe AD (measured by the MMSE, score of ≤ 20). It is a voltage-dependent, moderate-affinity, uncompetitive N-methyl-d-aspartate (NMDA) receptor antagonist that blocks the effects of pathologically elevated tonic levels of glutamate which may lead to neuronal dysfunction. Memantine is taken orally twice a day. The starting dose is 10 mg/day and this can be increased to a maximum daily dose of 20 mg/day. Caution should be used when prescribing memantine for people with renal failure or epilepsy; it is also contraindicated for people with severe renal impairment. Side effects may include dizziness, confusion, headache and incontinence.
Chapter 2 Definition of the decision problem
Decision problem
The inclusion criteria for this assessment are as follows.
Population
The population for this assessment is adults with AD. However, as in the assessment that informed TA No. 111 (TA111),1 where trials have included participants with mixed dementias, these trials will be included where the dominant dementia is AD. Papers will be considered on a case-by-case basis.
Intervention
The intervention to be included is dependent on the severity of AD, measured by the MMSE criteria:
-
Mild AD (MMSE 21–26) – donepezil, galantamine and rivastigmine.
-
Moderate AD (MMSE 10–20) – donepezil, galantamine, rivastigmine and memantine.
-
Severe AD (MMSE < 10) – memantine.
Comparators
The comparators are again dependent on the severity of the AD:
-
Mild AD (MMSE 21–26) – placebo or best supportive care (BSC). (Best supportive care includes social support and assistance with day-to-day activities. These include information and education; carer support groups; community dementia teams; home nursing and personal care; community services, such as Meals on Wheels; befriending services; day centres, respite and care homes.)
-
Moderate AD (MMSE 10–20) – donepezil, galantamine, rivastigmine, memantine, placebo or BSC.
-
Severe AD (MMSE < 10) – placebo or BSC.
Outcomes
The outcomes of interest include measures of:
-
severity of disease and response to treatment
-
behavioural symptoms
-
mortality
-
ability to remain independent
-
likelihood of admission to residential/nursing care
-
HRQoL of patients and carers (where data permit, analysis will be carried out separately for patients alone, and for patients and carers combined)
-
adverse effects of treatment
-
cost-effectiveness and costs (review of economic studies).
Key issues
All medicines will be considered according only to their UK marketing authorisation.
Overall aims and objectives of assessment
The purpose of this assessment was to review and update as necessary guidance to the NHS in England and Wales on the clinical effectiveness and cost-effectiveness of donepezil, galantamine, rivastigmine and memantine, within their UK licensed indications, for the treatment of AD, which was issued in November 2006, and amended in September 2001 and August 2009.
Chapter 3 Assessment of clinical effectiveness
The purpose of the systematic review of clinical effectiveness is to record the studies found by Loveman and colleagues2 in 2004 and to update their findings with the results of subsequent trials.
This chapter has been arranged as follows:
-
methods for reviewing effectiveness
-
results of the systematic review
-
manufacturers’ reviews of clinical effectiveness
-
results – pair-wise comparisons
-
donepezil versus placebo
-
galantamine versus placebo
-
rivastigmine versus placebo
-
memantine versus placebo
-
-
head-to-head comparisons
-
combination therapy
-
results – multiple treatment comparisons
-
cognitive
-
functional
-
behavioural
-
global
-
-
summary of clinical effectiveness.
Methods for reviewing effectiveness
The clinical effectiveness of donepezil, galantamine, rivastigmine and memantine for AD was assessed by a systematic review of research evidence. The review was undertaken following the principles published by the NHS Centre for Reviews and Dissemination (CRD). 77 The study protocol can be viewed on the NICE website: www.nice.org.uk.
Identification of studies
Electronic databases were searched for systematic reviews and/or meta-analyses, randomised controlled trials (RCTs) and ongoing research in November 2009 and updated in March 2010; this updated search revealed no includable new studies. Appendix 2 shows the databases searched and the strategies in full. These included The Cochrane Library (2009 Issue 4, Cochrane Database of Systematic Reviews and Cochrane Central Register of Controlled Trials), MEDLINE, MEDLINE In-Process & Other Non-Indexed Citations, EMBASE, PsycINFO, EconLit, ISI Web of Science Databases – Science Citation Index, Conference Proceedings Citation Index, and BIOSIS, and the CRD databases – NHS Economic Evaluation Database (NHS EED), Health Technology Assessment, and Database of Abstracts of Reviews of Effects databases. Where possible, a controlled trials and human filter was added. As this is an update of a previous review the searches were run in the time frame 2004 to current. The meta-register of controlled trials and ‘clinicaltrials.gov’ were searched for ongoing trials. Bibliographies of included studies were searched for further relevant studies. The reference lists of the industry submissions were also scrutinised for additional studies. Owing to resource limitations the search was restricted to English-language papers only. All references were managed using Reference Manager (Professional Edition, version 11; Thomson ISI ResearchSoft, Thomson Reuters, New York, NY, USA) and Microsoft Access 2003 software (Microsoft Corporation, Redmond, WA, USA).
Relevant studies were identified in two stages. Titles and abstracts returned by the search strategy were examined independently by two researchers (GR and MB) and screened for possible inclusion. Disagreements were resolved by discussion. Full texts of the identified studies were obtained. Two researchers (GR and MB) examined these independently for inclusion or exclusion, and disagreements were again resolved by discussion. A third reviewer was available to resolve disagreements, but this was not necessary.
Inclusion and exclusion criteria
Study design
Inclusion criteria
For the review of clinical effectiveness, only systematic reviews of RCTs and RCTs were considered. The review protocol made provision for broadening the search criteria to include some observational evidence if insufficient systematic reviews or RCTs were identified; however, this proved unnecessary in view of the reasonable yield of evidence of a preferred design (see below).
Systematic reviews were used as a source for finding further RCTs and to compare with our systematic review. For the purpose of this review, a systematic review77–79 was defined as a review that has:
-
a focused research question
-
explicit search criteria that are available to review, either in the document or on application
-
explicit inclusion/exclusion criteria, defining the population(s), intervention(s), comparator(s) and outcome(s) of interest
-
a critical appraisal of included studies, including consideration of internal and external validity of the research
-
a synthesis of the included evidence, whether narrative or quantitative.
Exclusion criteria
Studies were excluded if they did not match the inclusion criteria, and in particular:
-
non-randomised studies [except for adverse events (AEs)]
-
animal models
-
preclinical and biological studies
-
narrative reviews, editorials, opinions
-
non-English-language papers
-
reports published as meeting abstracts only, where insufficient methodological details were reported to allow critical appraisal of study quality.
Population
Studies were included if they reported a population comprising adults with AD. Following the 2004 review, trials that included participants with mixed dementia were included if the predominant dementia was AD.
Participants in included trials were required to meet the definitions of disease severity specified in the technologies’ UK marketing authorisations (MMSE 26–10 for donepezil, galantamine and rivastigmine; MMSE 20–0 for memantine).
The exact inclusion criterion adopted for MMSE scores was defined as an approximation of the principle that at least 80% of a study’s participants should be within the specified range. This approach relied on the assumption that reported baseline MMSE scores were normally distributed. On this basis, studies were included if the predefined thresholds were not exceeded by the reported mean baseline MMSE score ± 0.8416 standard deviation (SD), where 0.8416 is the inverse of the standard normal distribution corresponding to a probability of 0.8.
Interventions and comparators
Studies were included if the technologies they assessed fulfilled the following criteria:
-
Interventions The four technologies under review were considered within their UK marketing authorisations:
-
– mild-to-moderately severe AD (measured by the MMSE 26–10) – donepezil, galantamine and rivastigmine
-
– moderate-to-severe AD (measured by the MMSE 20–0) – memantine.
-
-
Comparators For people with mild AD the comparators of interest were placebo and/or BSC (i.e. treatment without AChEIs and without memantine). For people with moderate AD the comparators were donepezil, galantamine, rivastigmine, memantine and placebo and/or BSC (i.e. treatment without AChEIs). For people with severe AD the comparator was treatment without memantine.
Outcomes
Studies were included if they reported data on one or more of the following outcomes:
-
measures of severity and response to treatment
-
behavioural symptoms
-
mortality
-
ability to remain independent
-
likelihood of admission to residential/nursing care
-
HRQoL of patients and carers
-
adverse events of treatment.
Data extraction strategy
Data were extracted by GR into forms in bespoke software and checked by MB. Disagreements were resolved by discussion. The items extracted can be found in the data extraction forms of included studies which are available in Appendix 3.
Critical appraisal strategy
Assessments of study quality were performed according to the instrument developed for the 2004 review (which was based on criteria recommended by the NHS CRD77). The instrument is summarised below; for full details, see Appendix 5 of the 2004 review. 2 Results were tabulated and the relevant aspects described in the data extraction forms.
Internal validity
The instrument sought to assess the following considerations:
-
Was the assignment to the treatment groups really random?
-
Was the treatment allocation concealed?
-
Were the groups similar at baseline in terms of prognostic factors?
-
Were the eligibility criteria specified?
-
Were outcome assessors blinded to the treatment allocation?
-
Was the care provider blinded?
-
Was the patient blinded?
-
Were point estimates and a measure of variability presented for the primary outcome measure?
-
Did the analyses include an intention-to-treat (ITT) analysis?
-
Were withdrawals and dropouts completely described?
In addition, methodological notes were made for each included study, including the reviewer’s observations on sample size and power calculations, participant attrition, methods of data analysis, and conflicts of interest.
External validity
External validity was judged according to the ability of a reader to consider the applicability of findings to a patient group and service setting. Study findings can only be generalisable if they describe a cohort that is representative of the affected population at large. Studies that appeared representative of the UK AD population with regard to these considerations were judged to be externally valid.
Methods of quantitative synthesis
Where data permitted, the results of individual trials were pooled using the methods described in the following section.
Pair-wise meta-analysis
We used random-effects meta-analyses (DerSimonian and Laird model80) only, regardless of any statistical evidence of interstudy homogeneity. Heterogeneity was explored by visualisation of results and, in statistical terms, both Cochran’s Q-test (compared with a chi-squared distribution)81 and the I2-statistic. 82,83 Small-study effects (including publication bias) were visualised using funnel plots and quantified using Egger’s test84 (see Appendix 4). Analyses were conducted using bespoke software, written in Microsoft Visual basic for Applications (version 6) and applied in both Microsoft Access 2003 and Microsoft Excel 2003 (Microsoft Corporation, Redmond, WA, USA). Stata 10.1 (StataCorp LP, College Station, TX, USA) was used to generate forest plots (‘metan’ command) and to assess small-study effects (‘metabias’ command).
Where more than one arm of a contributing trial was relevant to any analysis, data were pooled to form a single meta-arm as the unit of analysis, as recommended in the Cochrane Handbook for Systematic Reviews of Interventions (section 16.5.4). 85 For the continuous outcome measures reported in this review, the mean for the combined arm is estimated as the weighted mean from the multiple separate arms (where the numbers in each arm provide the weights), and the SD for the combined arm is calculated according to the usual formula:
where i indexes a total of k arms being combined, ni is the number of participants in each arm, and si is the SD for that arm.
All meta-analyses were stratified according to the measurement population. Where multiple-measurement populations were reported in an individual study, we used the highest ranking according to a prespecified hierarchy:
-
true ITT
-
last observation carried forward (LOCF)
-
observed cases (OCs) only.
The issue of how to deal with missing data point from dropouts in ITT analysis of dementia patients is a contentious one. Owing to the natural course of this degenerative disease, the assumption of LOCF that disease progression stops at the last data point clearly does not reflect reality. Similarly, to use OCs only (i.e. not estimating any data points after dropout) may give misleading results. A better solution may be to apply the rate of decline found in the control group to all dropouts. 86
We performed separate analyses for different periods of follow-up. The two lengths of follow-up for which data were generally available were approximately 3 months (12–16 weeks of treatment) and approximately 6 months (21–28 weeks) (figures showing these results are in the body of the text).
Where different dosages of drugs were found in various studies, we meta-analysed comparable groups separately (figures for commonly used doses in the UK are in the body of the text). We also performed a single analysis in which all dosages were combined (figures from these analyses are in Appendix 5). For continuous outcomes measured over a longitudinal period of follow-up, it is possible for investigators to report outcomes in two ways: the mean of each participant’s observed change from a measured baseline score (mean change from baseline) or absolute measurements at the relevant juncture (absolute value). If randomisation is adequate, the difference between these values should be the same (i.e. the mean of the differences will be the same as the difference in the means). However, the dispersion of each measure may vary. It is stated in the Cochrane Handbook for Systematic Reviews of Interventions (section 9.4.5.2) that ‘[t]here is no statistical reason why studies with change-from-baseline outcomes should not be combined in a meta-analysis with studies with final measurement outcomes’. 85 However, exploratory analyses showed that the inclusion of both types of data led to large differences in the results of meta-analyses, although this may be because the studies that only report final measurement data tend to be of a lower methodological standard (and, therefore, may also be more susceptible to biases that would distort reported treatment effect). As a result, we were not prepared to pool the two types of measurement, and all of our meta-analyses rely on studies reporting mean change from baseline only.
Pooling of multiple outcome measures
In addition to pair-wise meta-analyses of treatment effect pooled on each outcome’s natural scale [weighted mean difference (WMD)], we combined outcomes in a series of broad domains – cognitive, functional, behavioural and global – to investigate the overall characteristics of reported effectiveness evidence in each area (see figures in the body of the text and data sets used in the meta-analysis of pooled multiple outcome measures in Appendix 6).
In order to combine studies using different outcome measures within each domain, effect sizes were expressed as a standardised mean difference (SMD). The SMD expresses the size of the treatment effect in each trial relative to the variability observed in that trial. Accordingly, for a given trial i,
where m1i and m2i represent the reported means in active treatment and control cohorts, respectively, and si is the pooled SD across both groups, estimated as
where n1i, n2i and Ni represent the sample sizes of treated, control and combined cohorts, respectively, and the reported SDs of measurements in treated and control groups are SD1i and SD2i. In order to pool SMDs, it is necessary to derive the standard error (SE), which is estimated as follows:
The method assumes that the differences in SDs between studies reflect differences in measurement scales and not real differences in variability among study populations.
Where studies reported more than one outcome contributing to the same domain, a weighted average of all SMDs was calculated, using the precision of the estimates as the weighting factor (this could be seen as a submeta-analysis, adopting a fixed-effects model with inverse variance weighting). So that such studies were not given spurious weight, the sample size for each outcome measure was divided by the total number of outcomes.
This approach has the advantage of enabling a broader evidence base to be combined, but it has the disadvantage of requiring estimates to be pooled on a scale that has no direct clinical meaning. Accordingly, we used these analyses solely to explore the characteristics of the evidence base, and not to draw direct conclusions about the magnitude of relative effectiveness of the comparators. In particular, we used the analyses as a basis for metaregression (see below), and for assessing small-study effects.
Metaregression
Where there was sufficient evidence (at least five individual data points in a meta-analysis), study-level regression (‘metaregression’) was used to explore the statistical heterogeneity across studies. Three prespecified covariates were explored: population gender, population gender, and baseline disease severity (as measured by MMSE). Because of inconsistencies in the evidence base, it was not possible to undertake multivariate analyses, so regressions were conducted solely on a univariate basis. Metaregression was undertaken in Stata 10.1 (‘metareg’ command), using the restricted maximum likelihood estimator, as recommended. 87,88 These figures are in Appendix 7.
Mixed-treatment comparison: indirect comparison
In addition to pair-wise meta-analyses, where sufficient data were available, we synthesised information on all technologies and their comparators simultaneously, in a mixed-treatment comparison (MTC) using Bayesian Markov Chain Monte Carlo (MCMC) sampling. 89–92 The analyses were performed using WinBUGS 1.4.1 (MRC Biostatistics Unit, Cambridge UK) (model code is reproduced in Appendix 8).
Vague prior distributions were used in the analyses (Normal[0, 0.000001] for mean difference between treatments; Uniform[0,2] for SD of random-effects distribution). Point estimates and 95% credible intervals were calculated from 100,000 simulated draws from the posterior distribution after a burn-in of 10,000 iterations.
Outputs are presented in terms of treatment effect compared with a common baseline. In each case in the presented analyses, the available evidence networks included at least one placebo arm; therefore, the baseline treatment is always placebo. This is helpful, as it enables all MTC outputs to be interpreted on a common level. In addition to treatment effect relative to placebo, the posterior probability that each treatment is most effective is presented, simply calculated as the proportion of MCMC trials in which the given treatment had the highest (or lowest, for negative scales) estimated treatment of all comparators.
This approach assumes ‘exchangeability’ of treatment effect across all included trials, such that the observed treatment effect for any comparison could have been expected to arise if it had been measured in the populations reported in all other included trials. Exchangeability was judged through examination of the trial populations and comparability of outcomes in the common treatment group facilitating the comparison. Figures representing these analyses are in the body of the text.
As for pair-wise syntheses, we generated separate MTCs for different periods of follow-up (12–16 weeks and 21–28 weeks). We also generated separate analyses according to measurement population: LOCF only; ITT plus LOCF; OC only; and all measurement populations combined (see Appendix 9). Where multiple measurement populations were reported in an individual study and more than one was pertinent to one of these analyses, we used the highest ranking according to the hierarchy given above. Multiple relevant arms within a single study were pooled according to the methods detailed above (see Pair-wise meta-analysis), before being entered into the MTC.
Graphical representation of summary trial information
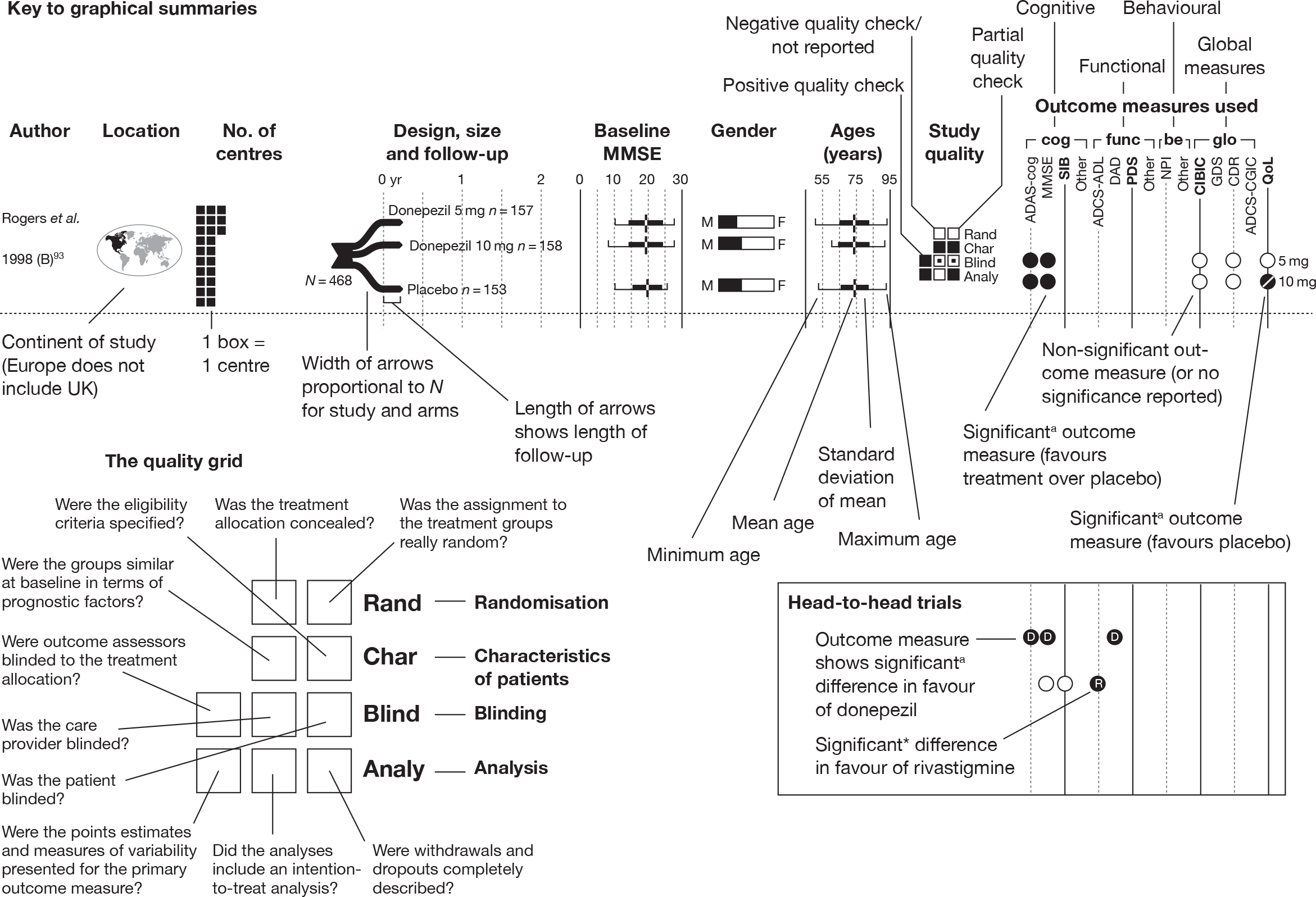
We present a novel approach to summarising the complex information relating to each trial at the end of each comparison section. These figures graphically represent the location, size, MMSE score at baseline, gender, age, study quality and results in a format that allows quick comparison between trials. A key to understanding the graphics is presented below in Figure 5.
FIGURE 5.
Key to graphical summaries. aSignificance displayed if mean change scores reported show significance of p < 0.05. In the case of multiple analyses, ITT is favoured over LOCF, which is, in turn, favoured over OC analyses. ADCS-CGIC, Alzheimer’s Disease Cooperative Study–Clinical Global Impression of Change; be, behavioural; cog, cognitive; DAD, Disability Assessment for Dementia Scale; GDS, Global Deterioration Scale; F, female; func, functional; glo, global; M, male; PDS, Progressive Deterioration Scale.
Results of the systematic review: identification of evidence
From screening the titles and abstracts of the 1843 references identified by our searches and additional sources, we retrieved 191 papers for detailed consideration, of which 21 were judged to meet the inclusion criteria for the review. The process is illustrated in detail in Figure 6. In assessing the titles and abstracts, agreement between the two reviewers was moderately good (κ = 0.642). At the full-text stage, agreement was moderate (κ = 0.538). At both stages, initial disagreements were easily resolved by consensus.
FIGURE 6.
Identification of published evidence for review. IPD, individual patient data; SR, systematic review.

The submissions from Eisai/Pfizer and Lundbeck contained a number of published and unpublished items that we have excluded from our review because they did not meet our inclusion criteria. A list of these items, with reasons for their exclusion, can be found in Appendix 10. A list of ongoing trials can be found in Appendix 11.
Results: systematic reviews
Our searches found four systematic reviews that met our inclusion criteria. These were critically appraised using the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement checklist, which describes 27 items that a report of a systematic review or meta-analysis should contain. 78 A summary table of whether or not these quality indicators were present in these systematic reviews can be found in Appendix 12. The references of each systematic review were checked to see if they held any includable additional trials, no further includable studies were found. A brief summary of each systematic review can be seen below.
Summary of included systematic reviews and meta-analyses
Donepezil
No systematic reviews of donepezil were found that matched our inclusion criteria.
Galantamine
No systematic reviews of galantamine were found that matched our inclusion criteria.
Rivastigmine
Birks and colleagues94 conducted a Cochrane review of rivastigmine compared with placebo for people with mild-to-moderate AD. They found nine trials with a total of 4775 participants. The review found that the use of rivastigmine (6–12 mg daily) was associated with a two-point improvement on the ADAS-cog compared with placebo [ITT WMD –1.99 (95% CI –2.49 to –1.50)] and a 2.2-point improvement on the Progressive Deterioration Scale (PDS) for ADL [ITT WMD –2.15 (95% CI –3.16 to –1.13)] at 26 weeks. The authors concluded that rivastigmine gave benefit to people with mild-to-moderate AD when compared with placebo. The review also considered delivery of the drug by transdermal patch. It found that the lower dose patch (9.6 mg/day) was associated with fewer side effects than the capsules or the higher dose patch (17.4 mg/day), and produced similar efficacy. The main AEs were gastrointestinal (nausea and vomiting), usually occurring during the titration phase.
All cholinesterase inhibitors
The German Institute for Quality and Efficiency in Health Care (IQWiG) conducted a systematic review and meta-analysis of all of the cholinesterase inhibitors included in this report for people with mild-to-moderate AD. 95 They included RCTs up to June 2006 in their systematic review, and found 27 studies with a total of 9883 participants. Only four of these trials met our inclusion criteria. 96–99 The IQWiG concluded that all the AChEIs provided benefit in improving or maintaining cognitive function and ADLs, and that galantamine alleviated psychological symptoms. However, none of the studies provided evidence of improvement in QoL. A summary table of these results can be found in Appendix 13.
Memantine
A systematic review of memantine for dementia was carried out by Raina and colleagues. 100 Their inclusion criteria were broader than ours and included all major types of dementia, patients with mild-to-moderate disease severity and all drugs for treating dementia. Of the 59 studies they included, only two met our inclusion criteria for trials. 96,101 The data syntheses from this systematic review are not relevant to this TAR and will not be discussed.
All included drugs
Hansen and colleagues102 conducted a systematic review and meta-analysis of functional outcomes from the use of donepezil, galantamine, rivastigmine and memantine for people with mild-to-moderate AD. They included 13 RCTs, 12 of which were included in the previous TAR (No. 111). The new study, which is included in this review, is Brodaty and colleagues. 93 Overall, they found a small effect size (d = 0.1–0.4) favouring drug treatment. A metaregression showed that this effect was not affected by disease severity, age, gender and drug dose. AEs were most commonly gastrointestinal.
Manufacturers’ reviews of clinical effectiveness
Three reviews were presented summarising evidence on the effectiveness of donepezil, galantamine and memantine by the manufacturers of each of the drugs. Although not part of the Peninsula Technology Assessment Group (PenTAG)’s systematic review they are presented here for convenience and because their findings are compared with our own review. Each submission is briefly discussed in the sections below.
Donepezil
Eisai and Pfizer submitted a systematic review as part of their joint submission on donepezil. It included both RCTs and targeted non-RCT/observational studies. Concerning the effect of donepezil relative to placebo the reported results of effect on cognition, function, behaviour and global impact were consistent with the results of the PenTAG review. There was, however, limited information on any summary estimates of effect in the manufacturer submission. Challenges to the validity of the AD2000 trial103 were re-emphasised.
Published meta-analyses were used to explore whether or not the effect of donepezil varied depending on the severity of AD, particularly the effectiveness in patients with mild AD. These suggested that a beneficial effect of donepezil relative to placebo on cognition, global impact and behaviour was present for patients with mild AD. The summary estimates quoted for mild AD were broadly similar to the overall summary estimates calculated in the PenTAG systematic review.
Results from non-RCT and observational data were presented to support the following additional aspects of the effectiveness of donepezil:
-
duration of effectiveness extending beyond 6 months up to at least 3 years
-
worsening of symptoms following withdrawal of treatment
-
emergence of benefit after initial absence of changes suggesting response
-
impact on carers, particularly caregiver stress and carer time
-
trends towards reductions in antipsychotic medication use
-
reductions in mortality.
Galantamine
Shire Pharmaceuticals presented a summary of all available RCTs (not just those from 2004 onwards) comparing galantamine with placebo, but did not indicate how the review had been conducted. They emphasised the importance of newer dosing regimens and highlighted deficiencies in the previous systematic review by the Southampton Health Technology Assessment Centre (SHTAC), particularly concerning failure to include a study directly comparing galantamine with donepezil.
The pooled summary estimates presented for the effect of galantamine on cognition, behaviour and function were consistent with the summary estimates in the PenTAG systematic review. The Shire Pharmaceuticals submission provided additional analyses, indicating an increase in effect with increasing severity of disease. Similar analyses could not be done in the PenTAG systematic review because of the requirement for individual patient data (IPD).
Memantine
Lundbeck presented a meta-analysis of pivotal trials as part of its submission. Although some details on the methods of analysis were provided, there was no information on how the pivotal trials were ascertained. The inclusion criteria were given and in essence the included studies were double-blind RCTs comparing memantine with placebo measuring cognition, disability, global health state and behaviour at 3 or 6 months. The need for IPD was further stipulated to allow subgroup analysis. There were six included studies in the main analysis covering all periods, not just 2004 onwards. The reasons why some studies were included in the Lundbeck analysis but not included in the PenTAG meta-analysis are documented in Appendix 10. Briefly, these were that Lundbeck’s pooling methods relied on the availability of IPD to which PenTAG did not have access, and Lundbeck were prepared to pool data from trials of memantine plus AChEIs versus AChEIs alone with data from trials of memantine monotherapy versus placebo to produce a single estimate of memantine effect. PenTAG were not comfortable with the assumptions necessary to justify such a single analysis. Notwithstanding this, the direction and size of effect of memantine relative to placebo on cognition, disability, global health state and behaviour are consistent between the Lundbeck and PenTAG analyses. In this, account needs to be taken of the fact that the results in the Lundbeck submission are presented as SMDs, whereas those in the PenTAG analysis were WMDs. Approximate interconversion is achieved by multiplying or dividing by the pooled SD. The 95% CIs are narrower in the Lundbeck analysis because of the greater number of included studies. The submission identified no evidence that the effectiveness varied by severity of AD, by past use of AChEIs or by concurrent use AChEIs. These analyses could not be repeated in the PenTAG systematic review because they depend on IPD.
Lundbeck also examined whether or not there was evidence of a difference in effectiveness depending on the presence of agitation/aggression and/or psychotic symptoms (APS), defined by the baseline NPI score being ≥ 3 (as opposed to the definition of > 0 used in the last the submission for the last NICE guidance). The results suggested that there is greater effectiveness in patients with APS but, again, these analyses could not be repeated in the PenTAG systematic review because they depend on IPD.
Unavailable evidence
Subgroup analyses
The study protocol specified that if evidence allowed subgroups based on disease severity, response to treatment, behavioural disturbance and comorbidities should be considered. However, none of the included trials reported any of these subgroup analyses. Therefore, we are unable to comment on them.
Outcomes
None of the included trials reported mortality or institutionalisation outcomes, or reported on outcomes beyond 28 weeks.
Results: pair-wise comparisons
Donepezil versus placebo
Identified evidence
The 2004 review2 identified 14 RCTs investigating the effectiveness of donepezil compared with placebo. 93,103–116
Notes:
-
The Nunez and colleagues trial111,112 was reviewed in poster form in 2003; a full publication, authored by Johannsen and colleagues,112 is now available, from which we have extracted data; however, for consistency with the 2004 review, we continue to refer to this RCT as ‘Nunez and colleagues’.
-
The Seltzer and colleagues trial115 was reviewed on a commercial-in-confidence (CiC) basis using information supplied by the manufacturer in 2004 – a full publication, authored by Seltzer and colleagues. 115
-
Winblad and colleagues,116 with additional information contained in the trial of Wimo and colleagues. 117
Our searches identified an additional five RCTs. 118–122 A summary of their design characteristics can be found in Table 2 and the interventions, comparators and baseline characteristics of the participants in Table 3. Critical appraisal of these small studies showed that none was of good quality; neither study was reporting adequate randomisation nor allocation concealment. A summary of the markers of internal validity is presented in Table 4.
| Study details | Inclusion criteria | Exclusion criteria | Methodological notes | Other |
|---|---|---|---|---|
|
Mazza et al. (2006) 118 Design: Parallel double-blind RCT Country: Italy? No. of centres: 1 No. randomised: 76 Maximum follow-up: 24 MMSE range included: 13–25 Funding: Not reported |
AD (DSM-IV criteria) Brief Cognitive Rating scale mean score 3–5 Hachinski Ischaemic Score < 4 Adequate level of premorbid intelligence (IG > 80, global assessment) |
Dementia of other aetiology Severe organic diseases (tumours, severe infectious diseases, brain trauma, epilepsy, cerebrovascular malformations, alcohol or drug abuse) Pseudodementia or a history of schizophrenic or affective psychoses (Geriatric Depression Scale, 15-item version, total score < 9) Vasoactive drugs, nootropics and long-term treatment with other drugs were proscribed during the study, with the exception of low doses of benzodiazepines and neuroleptic drugs in the treatment of behavioural disturbances |
Sample attrition/dropout: 60 of 76 randomised patients completed the study (a further 41 were excluded during the run-in period). Reasons for dropout were not reported Randomisation and allocation: Randomisation computer generated (whether or not unreadable before allocation is not stated). Appearance of pills and placebo not reported Power calculation: Not reported |
Therapy common to all participants: Single-blind placebo 4-week run-in period (in order to exclude placebo responders) Study funding: Not reported Other conflicts: Not reported |
|
dos Santos Moraes et al. (2006) 119 Design: Parallel double-blind RCT Country: Brazil No. of centres: 1 No. randomised: 35 Maximum follow-up: 26 MMSE range included: Not reported Funding: FAPESP, AFIP |
Probable AD (ADRDA criteria) CDR (Brazilian version) 1–2 (mild to moderate) |
Other causes of dementia Other current severe medical or psychiatric disease Evidence of moderate to severe sleep disorders, based on medical, sleep, and psychiatric interviews Apnoea–hypoapnoea index > 10/hour and periodic leg movement index > 5/hour at baseline polysomnographic recording Psychoactive drugs in the month prior to entering the study |
Sample attrition/dropout: Eight patients left the study owing to technical difficulties in polysomnography recordings Randomisation and allocation: Randomisation process not reported. Individual responsible for the random allocation of patients to the trial arms was blind to the treatment code (how blinding was attained is not reported). Appearance of donepezil and placebo tablets is not described Power calculation: Data from 10 patients were initially analysed for sample size estimation (procedure not reported). Based on this analysis, a sample size of 15 subjects in each group was calculated to set out a difference of eight percentage points in REM sleep percentage (significance level of 1% and power of 95%). To assess the interaction term in the ANOVA model, 27 people were required in each group (sample size not attained) – power of 80% was possible with the sample size analysed |
Therapy common to all participants: Two nights of polysomnographic recording (for purposes of habituation) Study funding: FAPESP, AFIP Other conflicts: Authors state no financial conflicts of interest No financial support from industry for study |
|
Moraes et al. (2008) 120 Design: Parallel double-blind RCT Country: Brazil No. of centres: 1 No. randomised: 23 Maximum follow-up: 12 MMSE range included: 6–27 Funding: FAPESP, AFIP |
AD (ADRDA criteria) Rating of 1–2 (mild to moderate) on Brazilian version of CDR |
Rating of ≥ 3 on Brazilian version of CDR Other causes of dementia Other current severe medical or psychiatric disease Psychoactive drugs in the month prior to entering the study |
Sample attrition/dropout: Not reported Randomisation and allocation: Randomisation performed using computer-generated random number list (0–1) with uniform distribution, with patients consecutively allocated to the two treatment groups (≤ 0.5 to group A, > 0.5 to group B). Donepezil and placebo pills were ‘packed in the same fashion’, but precise appearance of pills not reported Power calculation: Not reported |
Therapy common to all participants: Two nights of polysomnographic recording (for purposes of habituation) Study funding: FAPESP, AFIP Other conflicts: Authors state no conflicts of interest to disclose |
|
Peng et al. (2005) 121 Design: Parallel double-blind RCT Country: China No. of centres: 15 hospitals in Beijing, Shanghai, and Guangzhou No. randomised: 90 Maximum follow-up: 12 MMSE range included: 10–24 Funding: Not reported |
AD (NINCDS-ADRDA and DSM-IVR criteria) ≥ 55 years old In female patients, menopause ≥ 2 years Sufficient vision and hearing to complete assessments |
Other disease that may lead to dementia Severe heart or kidney dysfunction, active peptic ulcer or active epilepsy Allergy to cholinergic drugs |
Sample attrition/dropout: 89 of 90 completed the study. n = 1 dropped out due to AE (dizziness) Randomisation and allocation: Randomisation procedure not described. Placebo described as having the same colour, shape, flavour and size as donepezil Power calculation: Not reported |
Therapy common to all participants: None Study funding: Not reported Other conflicts: Not reported |
|
Winstein et al. (2007) 122 Design: Parallel double-blind RCT Country: USA No. of centres: 1 No. randomised: 10 Maximum follow-up: 4 MMSE range included: 11–26 |
Probable AD diagnosis (criteria not reported) Independent in ambulation Alert Able to follow simple instructions |
Delirium Familial tremor Parkinson’s disease Stroke Peripheral neuropathy Dementia due to other than probable AD Use of any concurrent pharmaceutical treatment for cognitive dysfunction |
Sample attrition/dropout: 10 of 10 completed study Randomisation and allocation: Randomisation procedure not described. Placebo described as identical in appearance to donepezil Power calculation: Not reported |
Therapy common to all participants: None Study funding: University of Southern California Alzheimer’s Disease Research Centre, Alzheimer’s Disease Research Centres of California, and Pfizer, Inc. Other conflicts: None reported |
| Study | Arm | Dose (mg/day) | Dosage details | N | Age, years (SD) | Gender, n male (%) | Race, n white (%) | Weight, kg (SD) | Education, years (SD) | Duration of dementia, months (SD) | ADAS-cog, score (SD) | MMSE, score (SD) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| dos Santos Moraes et al. (2006)119 | Donepezil | 5–10 | Starting daily dose of 5 mg for the first month, increased to 10 mg/day in the second month | 17 | 77.4 (6.60) | 4 (23.5) | 4.40 (3.60) | 35.6 (13.7) | ||||
| Placebo | – | Single daily dose | 18 | 74.5 (9.80) | 7 (38.9) | 6.00 (5.20) | 39.0 (18.5) | |||||
| Mazza et al. (2006)118 | Donepezil | 5 | 5 mg daily | 25 | 64.5 (6.00) | 13 (52.0) | 18.6 (3.47) | |||||
| Placebo | – | Not reported | 26 | 69.8 (3.00) | 10 (38.5) | 18.8 (3.63) | ||||||
| Moraes et al. (2008)120 | Donepezil | 5–10 | Single dose of 5 mg (administered at bedtime) in the first month, increased to single dose of 10 mg in second month | 11 | 76.8 (6.20) | 3 (27.3) | 34.5 (15.8) | 19.0 (3.60) | ||||
| Placebo | – | Single dose administered at bedtime | 12 | 72.6 (11.0) | 5 (41.7) | 29.3 (17.3) | 17.2 (7.80) | |||||
| Peng et al. (2005)121 | Donepezil | 5 | Same dose administered throughout duration of study | 46 | 72.6 (6.80) | 21 (45.7) | 17.8 (2.30) | |||||
| Placebo | – | – | 43 | 71.8 (8.20) | 19 (44.2) | 18.2 (2.70) | ||||||
| Winstein et al. (2007)122 | Donepezil | 5 | One tablet taken nightly | 5 | 84.2 (8.67) | 2 (40.0) | 24.0 (3.08) | 19.2 (3.35) | ||||
| Placebo | 5 | 88.0 (7.62) | 1 (20.0) | 26.0 (11.6) | 20.2 (4.09) |
| Study | Was the assignment to the treatment groups really random? | Was the treatment allocation concealed? | Were the groups similar at baseline in terms of prognostic factors? | Were the eligibility criteria specified? | Were outcome assessors blinded to the treatment allocation? | Was the care provider blinded? | Was the patient blinded? | Were the point estimates and measure of variability presented for the primary outcome measure? | Did the analyses include an ITT analysis? | Were withdrawals and dropouts completely described? |
|---|---|---|---|---|---|---|---|---|---|---|
| Mazza et al. (2006)118 | Partial | Inadequate | Reported – yes | Inadequate | Partial | Partial | Partial | Adequate | Partial | Partial |
| dos Santos Moraes et al. (2006)119 | Unknown | Inadequate | Reported – yes | Inadequate | Partial | Partial | Partial | Adequate | Unknown | Partial |
| Moraes et al. (2008)120 | Inadequate | Inadequate | Reported – yes | Unknown | Partial | Adequate | Adequate | Adequate | Unknown | Inadequate |
| Peng et al. (2005)121 | Unknown | Unknown | Reported – yes | Unknown | Unknown | Adequate | Adequate | Adequate | Inadequate | Adequate |
| Winstein et al. (2007)122 | Unknown | Unknown | Reported – yes | Inadequate | Unknown | Adequate | Adequate | Inadequate | Partial | Adequate |
Evidence of clinical effectiveness
Cognition
In 2004, Loveman and colleagues2 summarised the evidence they found for donepezil versus placebo for cognitive outcomes as follows:
Six RCTs showed that donepezil appears to confer a statistically significant benefit to participants on the ADAS-cog scale when compared to placebo. The benefit varies according to the dose of donepezil with higher doses of donepezil tending to show increasing benefit. Because the mean change scores varied quite considerably between the included studies, this dose-related trend can particularly be seen within individual trials, although no direct statistical comparisons were made in any of these. The mean change scores were however varied between the included studies. Eight RCTs showed trends towards better MMSE score in the donepezil treated groups when compared to the placebo groups, although this was not always demonstrated to be statistically significant. These trends were mirrored in one unpublished trial of people with mild Alzheimer’s disease.
In the studies we found published since 2004, four showed significant cognitive benefit for donepezil versus placebo. 118–121 However, only two of these trials118,122 estimated the missing values from dropouts using ITT analysis. The others made estimates using OCs only, thus potentially magnifying any benefit from donepezil and biasing their results in favour of the intervention. A summary of the results from cognitive measures can be seen in Table 5.
| Study | Subgroup | Outcome | Type | Donepezil | Placebo | p-value | ||
|---|---|---|---|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | |||||
| Moraes et al. (2008)120 | OC population | ADAS-cog – 13 weeks | A | 11 | 29.7 (15.7) | 12 | 31.8 (18.5) | < 0.05a |
| Winstein et al. (2007)122 | ITT population | ADAS-cog – 4 weeks | MC | 5 | –5 (2) | 5 | 0 (4.85) | 0.066b |
| Serial Reaction Time Task – 4 weeks | MC | 5 | 3.325 (8.39) | 5 | 1.65 (10.1) | 0.782b | ||
| dos Santos Moraes et al. (2006)119 | OC population | ADAS-cog – 13 weeks | A | 17 | 30.7 (13.9) | 18 | 40.9 (19.4) | 0.085b |
| ADAS-cog – 26 weeks | A | 17 | 28.3 (12.3) | 18 | 42.8 (18.7) | < 0.01c | ||
| Mazza et al. (2006)118 | ITT population | MMSE – 24 weeks | A | 25 | 19.8 (3.16) | 26 | 18.6 (3.66) | NSd |
| MC | 25 | 1.2 (12.2) | 26 | –0.25 (5)e | 0.06d | |||
| SKT – 24 weeks | A | 25 | 11.8 (2.9) | 26 | 16.9 (3.9) | 0.01d | ||
| MC | 25 | –3.3 (–2.55) | 26 | 0.9 (1.3) | < 0.001d | |||
| CGI-item 2 (cognitive) – 24 weeks | A | 25 | 3.6 (0.94) | 26 | 5.2 (0.95) | 0.01d | ||
| MC | 25 | –0.9 (1.02) | 26 | 0.15 (0.338) | < 0.001d | |||
| Peng et al. (2005)121 | OC population | MMSE – 12 weeks | A | 46 | 22.1 (2) | 43 | 18.7 (2.4) | < 0.01f |
The data from the new trials were synthesised with those from Loveman and colleagues report2 by random-effects meta-analysis. This was conducted considering ADAS-cog and then MMSE as the outcomes, measuring differences between donepezil (all doses) and placebo at 12 and 24 weeks post randomisation. The results can be seen in Figures 7 and 8. We also meta-analysed the data by 5 mg/day and all doses combined; these results can be found in Appendix 5. We then went on to explore the effect of pooling the entire cognitive outcome measures at 24–26 weeks; the results of this can be seen below (see Figure 11).
FIGURE 7.
Random-effects meta-analysis: ADAS-cog at 12 weeks (mean change from baseline): donepezil (10 mg/day) vsplacebo.
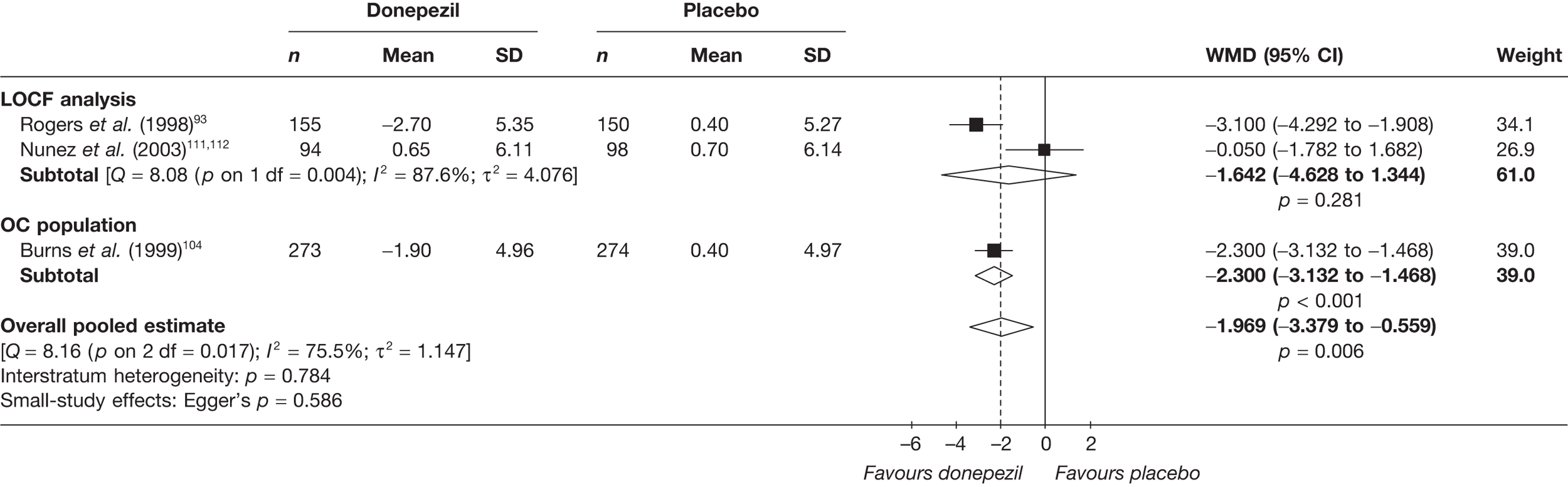
FIGURE 8.
Random-effects meta-analysis: ADAS-cog at 24 weeks (mean change from baseline): donepezil (10 mg/day) vsplacebo.
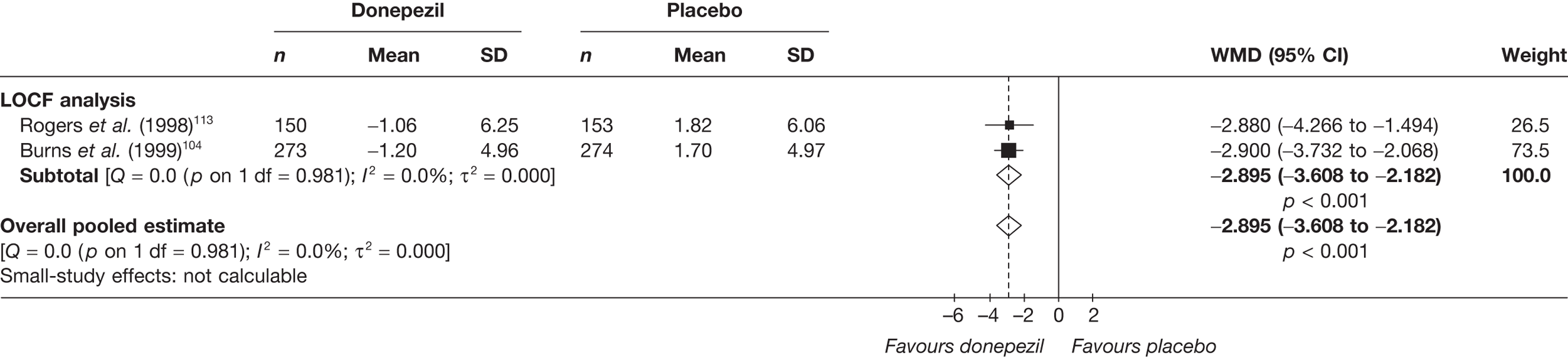
We found no new studies reporting the ADAS-cog at 12 or 24 weeks. The meta-analyses presented below are of studies included in the previous assessment report. The overall pooled estimates shows a benefit from donepezil compared with placebo, which increases over time: 12 weeks’ WMD = –1.97 (95% CI –3.38 to –0.56), p = 0.006; 24 weeks’ WMD = –2.90 (95% CI –3.61 to –2.18), p < 0.001 (Figures 7 and 8).
Similarly, no new evidence was found for the outcome measure MMSE at 12 weeks post randomisation, but one new study was found with measures at 24 weeks’ follow-up. The meta-analyses below show an overall benefit from donepezil versus placebo when measured on the MMSE: 12 weeks’ (10 mg/day) WMD = –1.17 (95% CI 0.88 to 1.45), p < 0.001; 24 weeks’ (5 and 10 mg/day) WMD = 1.21 (95% CI 0.84 to 1.57), p < 0.001 (Figures 9 and 10).
FIGURE 9.
Random-effects meta-analysis: MMSE at 12 weeks (mean change from baseline): donepezil (10 mg/day) vsplacebo.
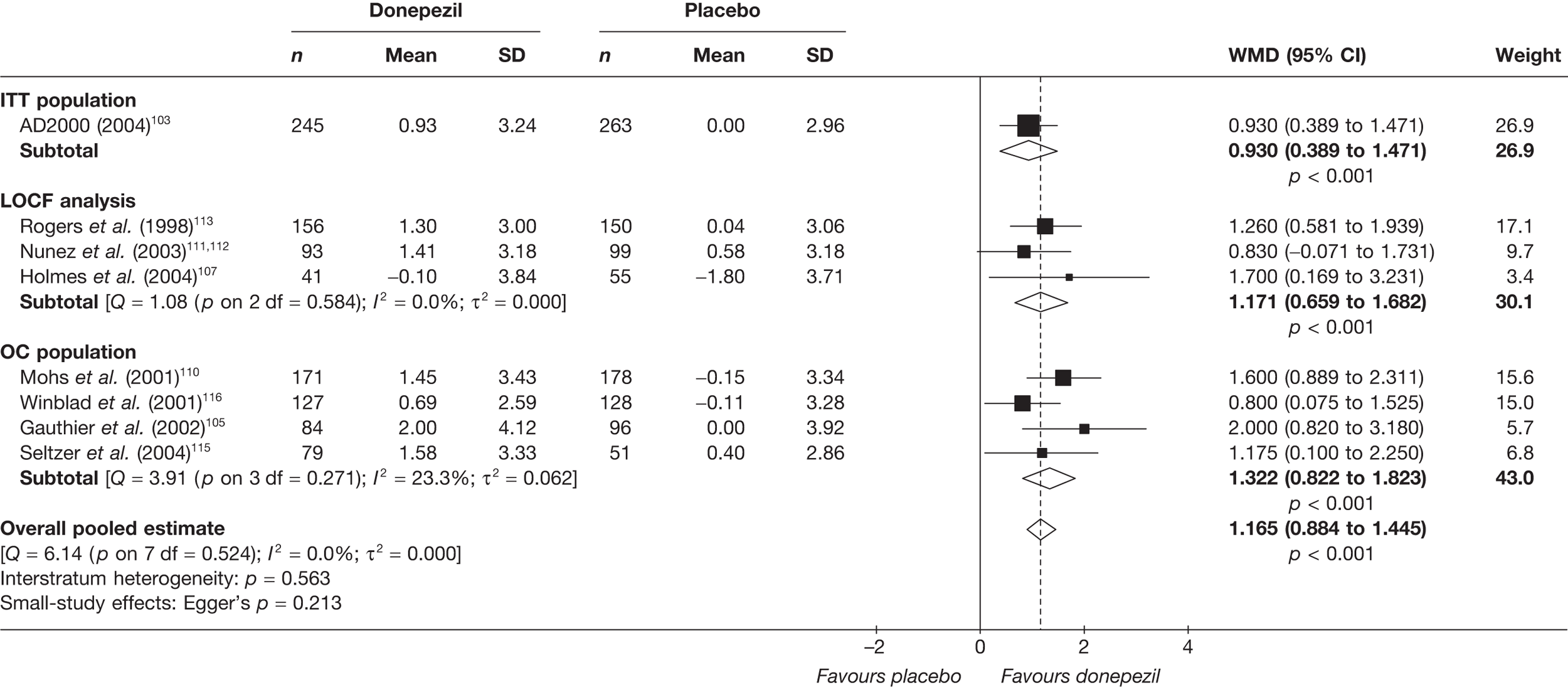
FIGURE 10.
Random-effects meta-analysis: MMSE at 24 weeks (mean change from baseline): donepezil (all dosages) vsplacebo. a, WMD and error bars provided in publication; SE estimated on assumption that error bars represent 95% CIs; b, Pooled 5 and 10 mg/day arms.
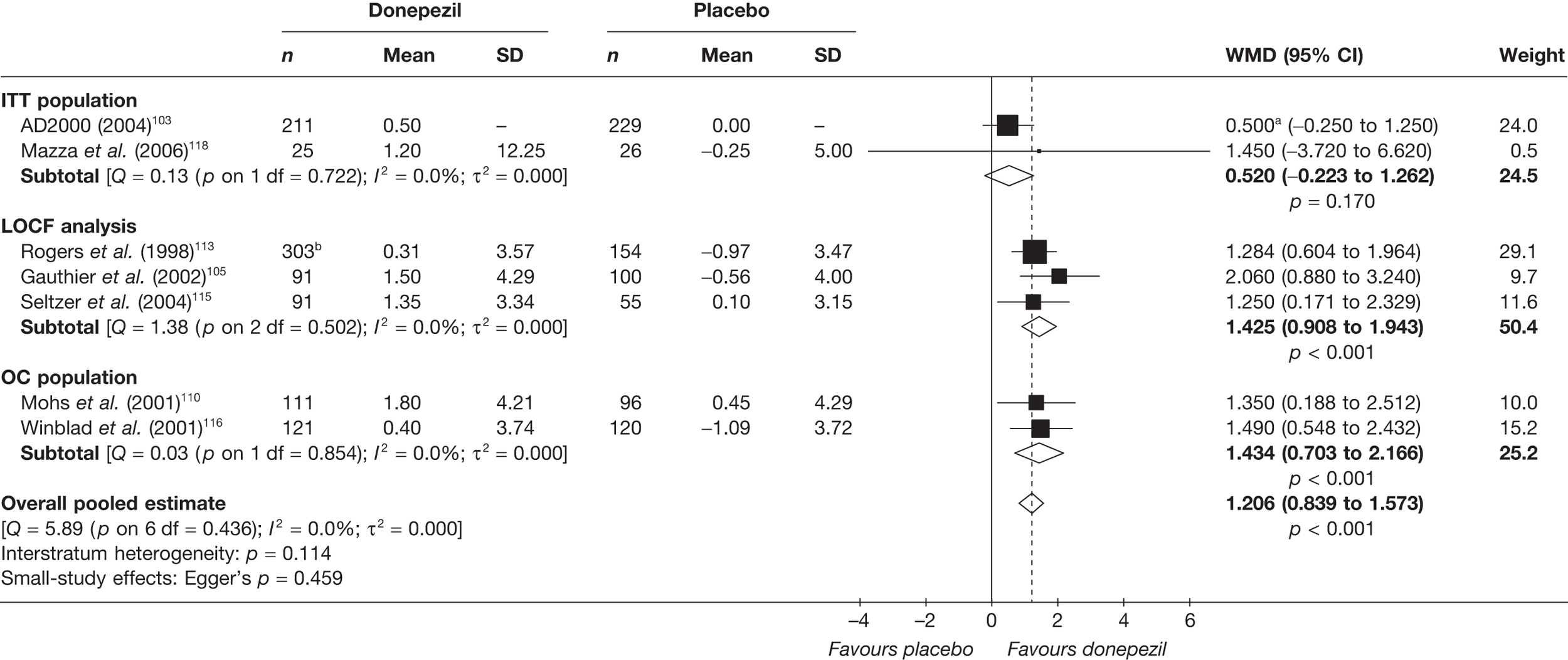
Two new studies were found to add to this combined meta-analysis of cognitive outcome measures at 24–26 weeks. The overall pooled estimate showed a significant cognitive benefit from donepezil compared with placebo: SMD = 0.40 (95% CI 0.29 to 0.50), p < 0.001 (Figure 11). The data set used in this meta-analysis can be found in Appendix 6.
FIGURE 11.
Random-effects meta-analysis: cognitive outcomes (SMD) at 24–26 weeks: donepezil (all dosages) vs placebo.
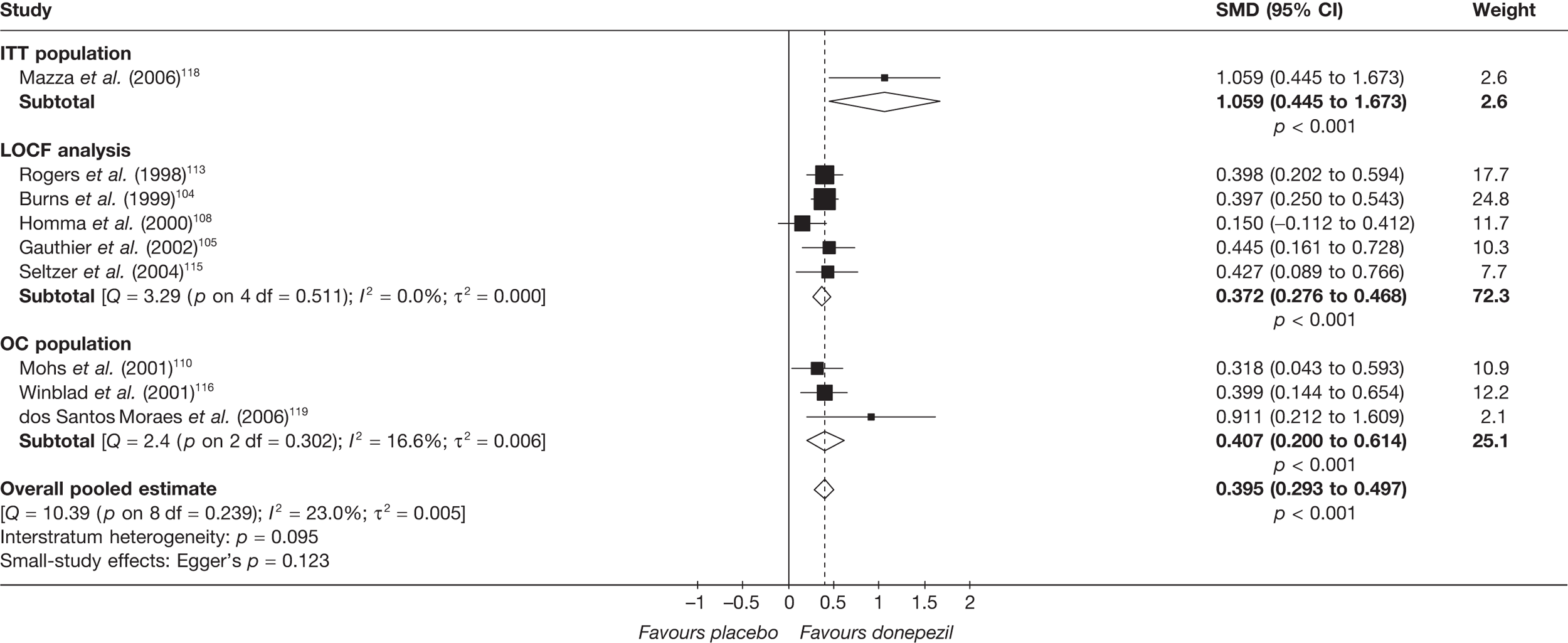
Functional
The 2004 assessment report2 found that:
A variety of functional measures were used in eight RCTs. Donepezil had some effect in improving or limiting further deterioration on ADLs when compared with placebo, but this was not always statistically significant, particularly over longer durations of follow-up. One trial reported time to loss of ADL and/or time to institutional care and found that donepezil conferred no advantage to placebo.
We found only one new RCT measuring functional outcomes for this comparison. This small, poorly reported trial showed a significant benefit from donepezil (5 mg/day) for ADLs in an OC-measured population at 12 weeks’ follow-up – mean difference: I = 40.5 (SD 7.6), C = 49.5 (SD 6.3), p < 0.01 (Table 6).
When the 2004 and post-2004 evidence bases were collected together, there was an extremely heterogeneous collection of outcome measures for this domain. As a result, we have not been able to perform any quantitative synthesis of individual outcome measures on a natural scale.
There were no new studies that measured functional outcomes at 24 weeks; therefore, we pooled the functional outcome data from the studies in the previous assessment. This showed a significant benefit for donepezil at all doses compared with placebo: SMD = 0.30 (95% CI 0.14 to 0.45), p < 0.001 (Figure 12). The data set used for this meta-analysis can be found in Appendix 6.
FIGURE 12.
Random-effects meta-analysis: functional outcomes (SMD) at 24 weeks: donepezil (all dosages) vs placebo.
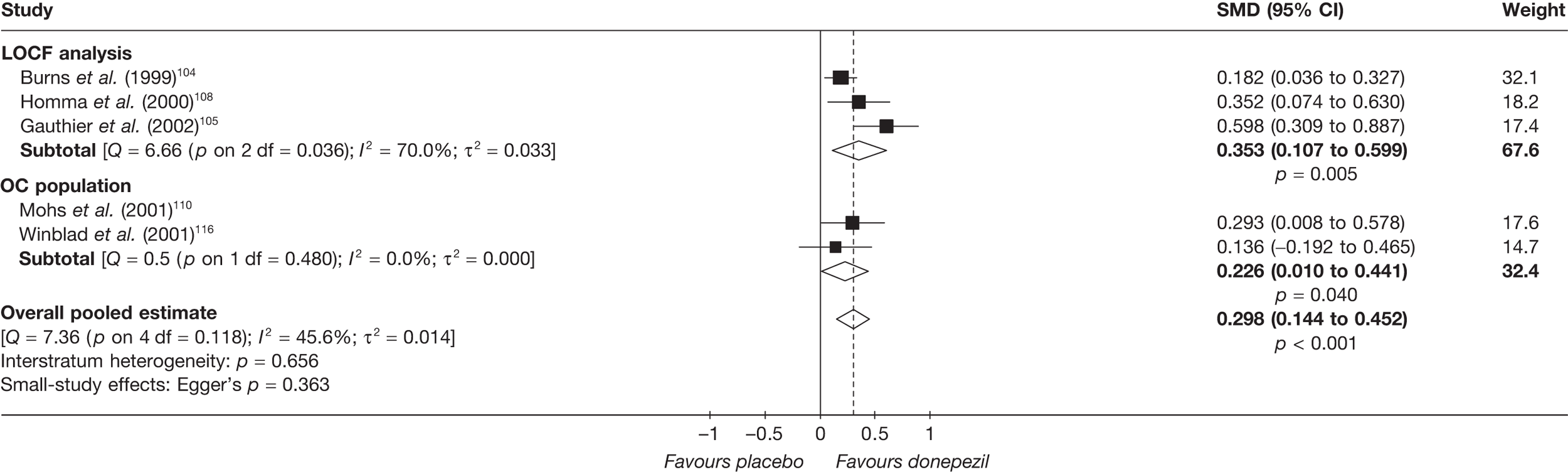
Behavioural and mood
In 2004 the assessment group2 reported that:
The NPI was used as a measure of mood and behaviour in four RCTs. Data were varied but suggested that donepezil may have some effect in improving or limiting further deterioration on the NPI scale compared to placebo, at least over shorter durations of follow-up.
None of the newly identified studies provided any additional data on the effect of donepezil as indicated by measures of behavioural function. Therefore, we conducted random-effects meta-analysis of the studies included in 2004 for the NPI at 12 and 24 weeks, which showed no significant benefit from donepezil measured by the NPI (Figures 13 and 14).
FIGURE 13.
Random-effects meta-analysis: NPI at 12 weeks (mean change from baseline): donepezil [all dosages (all are 10 mg/day)] vsplacebo. a, Score inverted from published figure to reflect usual direction of NPI; b, WMD and error bars provided in publication – SE estimated on assumption that error bars represent 95% CIs.
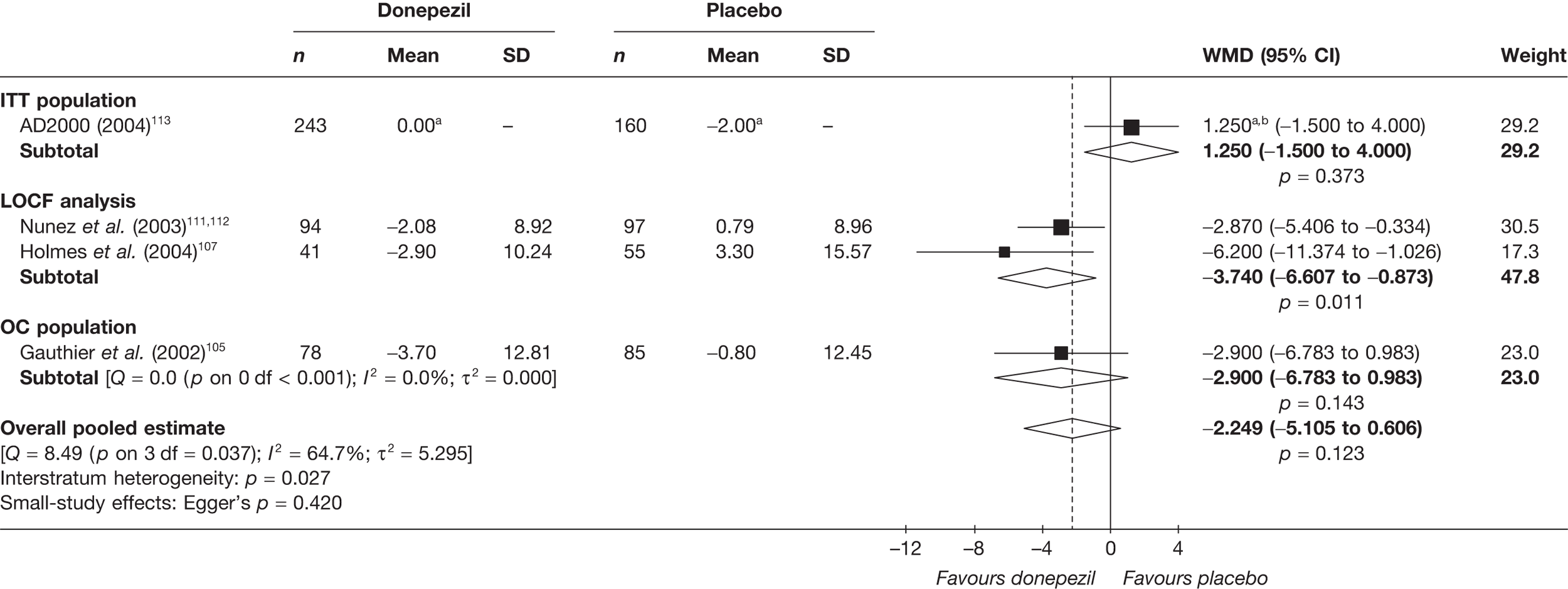
FIGURE 14.
Random-effects meta-analysis: NPI at 24 weeks (mean change from baseline): donepezil [all dosages (all are 10 mg/day)] vs placebo. a, Score inverted from published figure to reflect usual of direction NPI; b, WMD and error bars provided in publication – SE estimated on assumption that error bars represent 95% CIs.
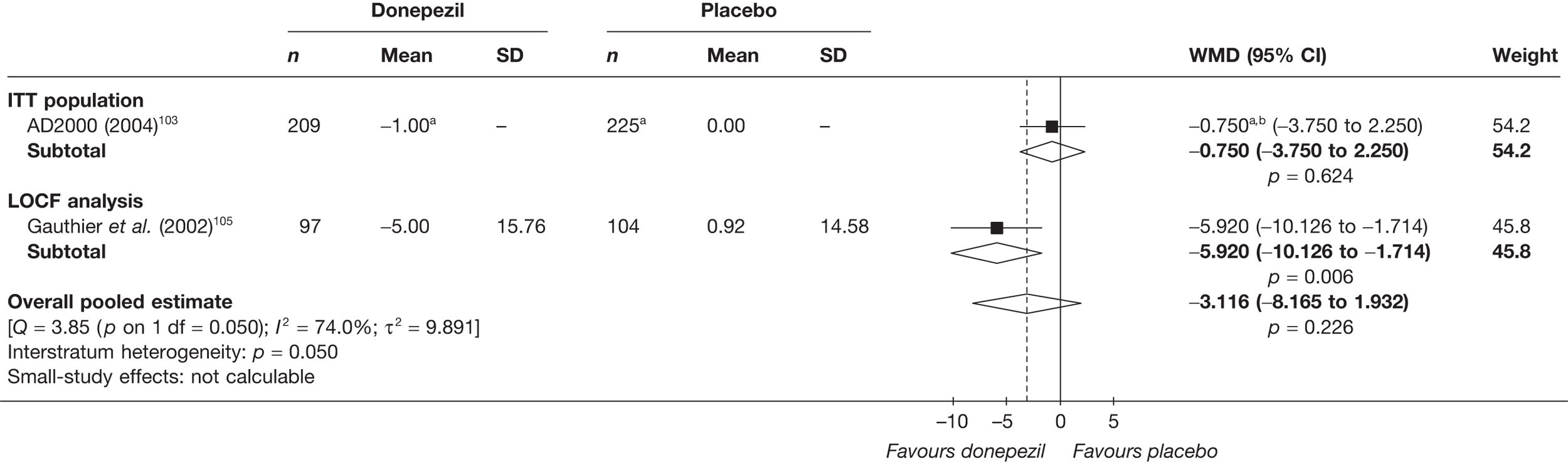
Because NPI is the only outcome measure used in this domain of the evidence base, it was not necessary to pool outcomes on a standardised level.
Global effect
Loveman and colleagues2 summarised their findings on global outcomes comparing donepezil and placebo as:
Seven RCTs assessed the effect of donepezil compared with placebo on the Clinical Global Impression of Change (CGIC) or CIBIC plus, showing overall that CGIC/CIBIC-plus scores were statistically significantly better with donepezil. The range of scores varied between the included studies. Higher proportions of participants receiving donepezil were considered as responders to treatment, although this was not compared statistically in many cases. On the CDR scale trends were also demonstrated towards improved global function in the donepezil-treated groups compared with the placebo groups in five trials but statistical significance was not demonstrated. In one unpublished trial with participants with mild Alzheimer’s disease, no benefit on the CDR was noted in the donepezil treated group.
Only one of the new studies measured global outcomes. 121 They also found significant benefit on the CDR: I = 1.2 (SD 0.2), C = 2.0 (SD 0.2), p < 0.01 (Table 7).
Only the previously included studies had data for meta-analysis of the CIBIC-plus. We pooled studies at 12 and 24 weeks and found that at both time points there was a significant overall pooled estimate of benefit from donepezil at 10 mg/day compared with placebo [12 weeks’ WMD = –0.38 (95% CI –0.49 to –0.26), p < 0.001; 24 weeks’ WMD = –0.43 (95% CI –0.55 to –0.31), p < 0.001] (Figures 15 and 16). Meta-analyses of CIBIC-plus results for 5 mg/day and all doses combined can be found in Appendix 5.
FIGURE 15.
Random-effects meta-analysis: CIBIC-plus at 12 weeks (mean change from baseline): donepezil (10 mg/day) vs placebo.
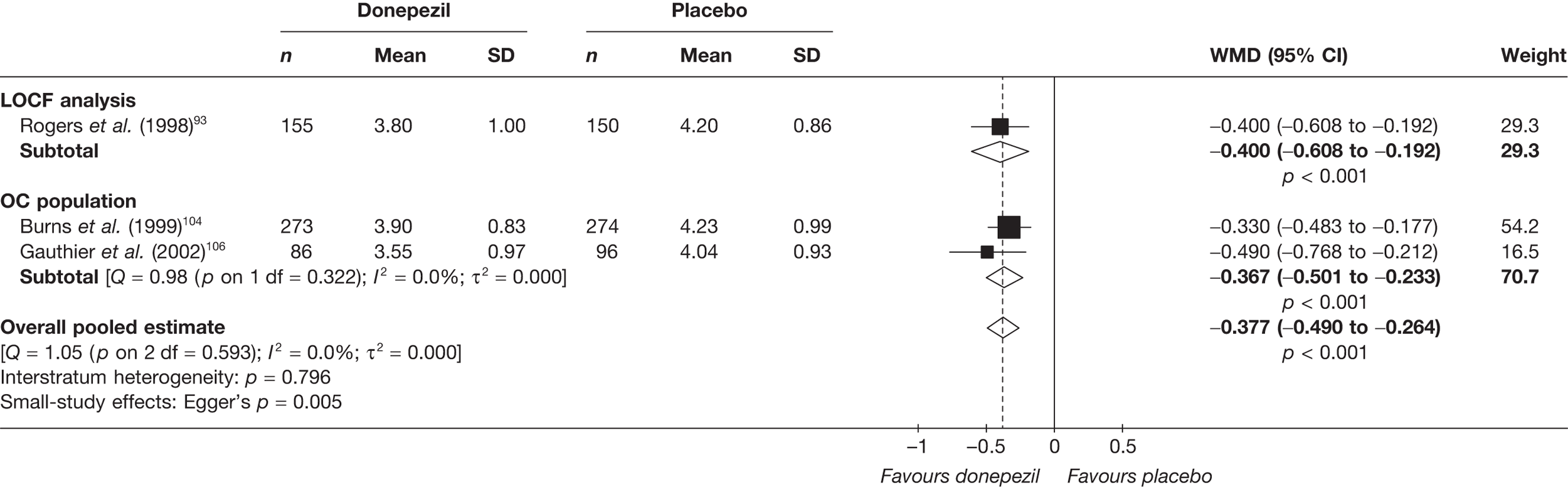
FIGURE 16.
Random-effects meta-analysis: CIBIC-plus at 24 weeks (mean change from baseline): donepezil (10 mg/day) vs placebo.
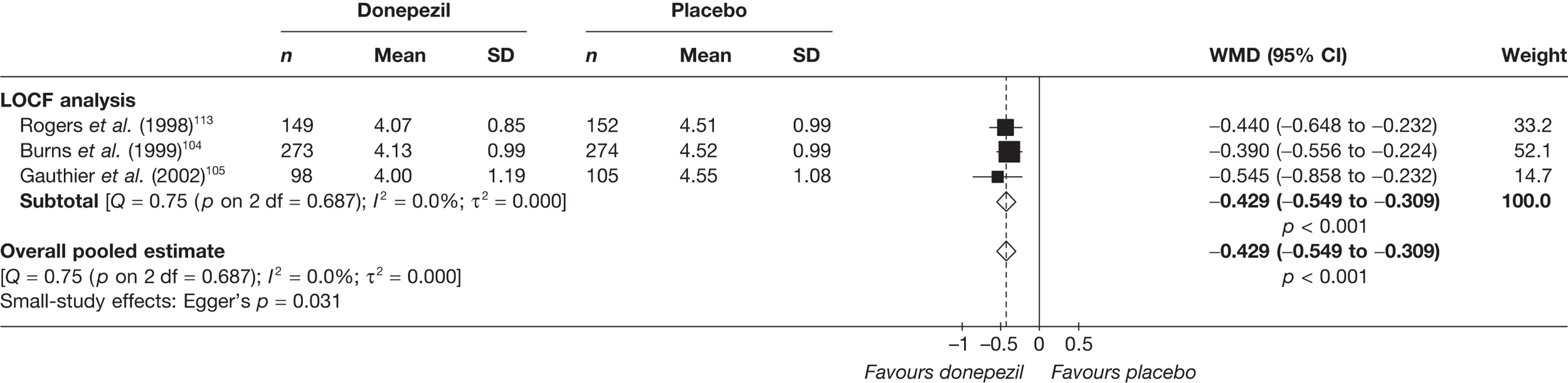
The pooled results on the CDR scale showed a significant advantage from taking donepezil at 12 and 24 weeks’ follow-up: 12 weeks’ WMD = –0.26 (95% CI –0.44 to –0.09), p < 0.003 and 24 weeks’ WMD = –0.57 (95% CI –0.85 to –0.29), p < 0.001 (Figures 17 and 18).
FIGURE 17.
Random-effects meta-analysis: CDR at 12 weeks (mean change from baseline): donepezil (all dosages) vs placebo. a, Pooled 5- and 10 mg/day arms.
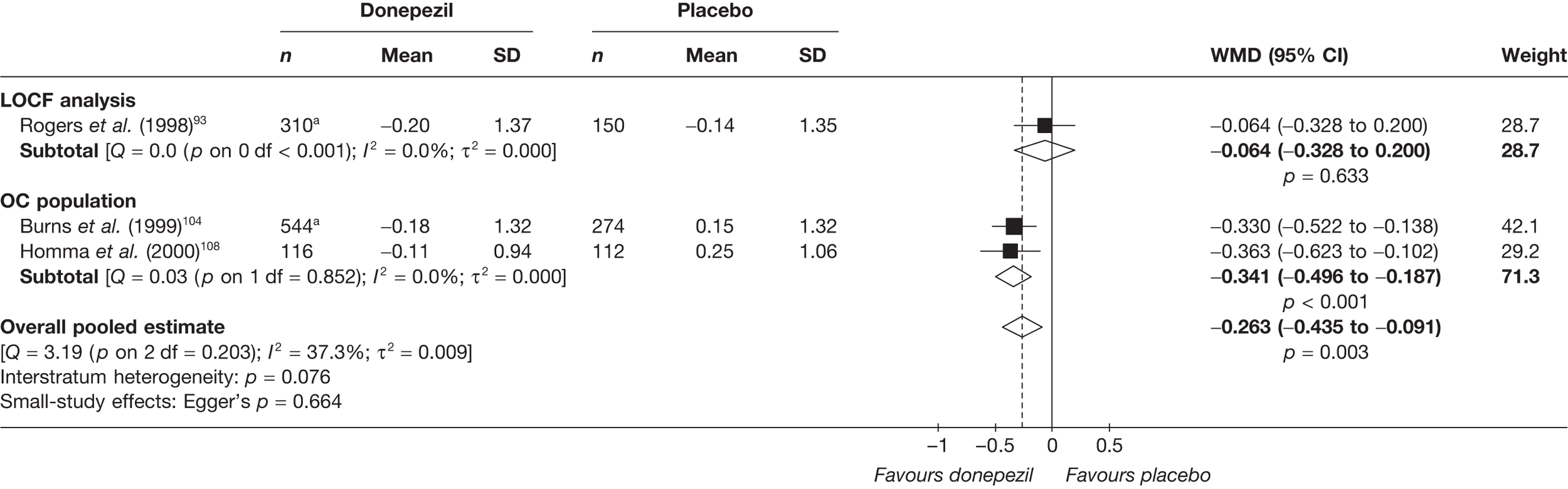
FIGURE 18.
Random-effects meta-analysis: CDR at 24 weeks (mean change from baseline): donepezil (all dosages) vs placebo. a, 5 and 10 mg/day arms pooled.
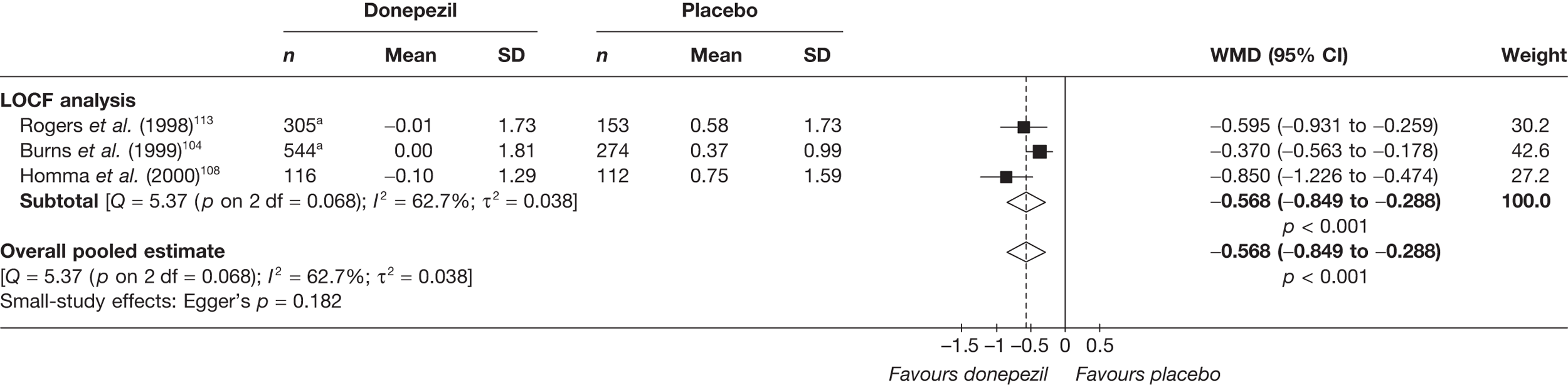
We did not find any new studies that measured global outcomes at 24–26 weeks; therefore, we pooled the global outcome data from the studies in the previous assessment. This showed a significant benefit for donepezil at all doses compared with placebo: SMD = 0.38 (95% CI 0.27 to 0.48), p < 0.001 (Figure 19). The data set used in this meta-analysis can be found in Appendix 6.
FIGURE 19.
Random-effects meta-analysis: global outcomes (SMD) at 24–26 weeks: donepezil (all dosages) vs placebo.
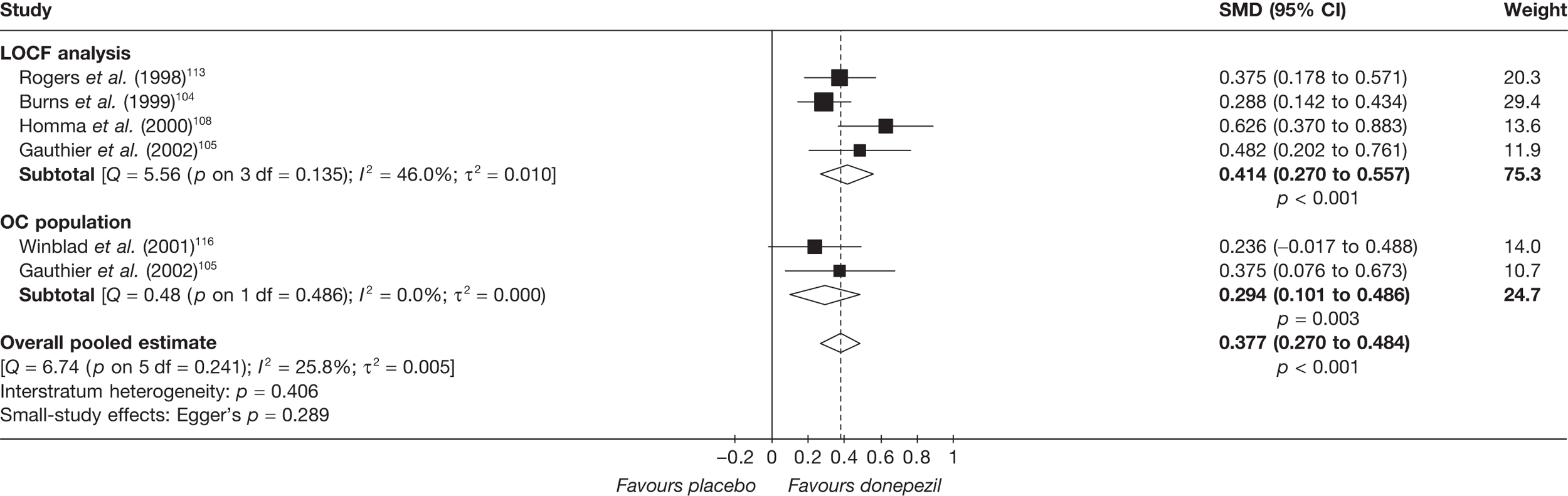
We also conducted metaregression analysis to explore the statistical heterogeneity across studies, looking at age, age and baseline MMSE (as a proxy for disease severity). Only one graph showed a significant relationship; this was between baseline MMSE and functional outcomes at 24 weeks for all doses of donepezil giving a metaregression estimate of α = 1.456, β = –0.065, p = 0.009. However, owing to the small number of studies in each analysis and the fact that the data were assessed at a population level (which may not reflect the individual level) we felt that these results may be ambiguous and so have placed them in Appendix 7 in case they are of interest.
Quality of life
None of the included studies provided any additional data on QoL with donepezil compared with placebo. Accordingly, the data presented in the 2004 review2 (p. 32) represent a complete and current summary of the randomised evidence on this subject.
Safety
None of the five newly identified studies118–122 provide data on AEs observed under randomised conditions.
Peng and colleagues121 present limited safety data conflating their randomised study with data from a parallel observational study. Among the total of 145 individuals who took donepezil, seven (4.8%) experienced an AE (it appears that one each experienced dizziness, nausea, inappetence, mild diarrhoea, constipation, fatigue and agitation). Four of these seven patients stopped taking medicine. Among patients in the placebo group of the randomised trial, two (4.7%) experienced dizziness and stopped medication for this reason.
Summary: donepezil versus placebo
We found an additional five RCTs118–122 to add to the 14 previously found by Loveman and colleagues;2 none of the new studies was of good quality or had a follow-up of longer than 6 months.
Pooled cognitive outcomes showed a significant benefit from donepezil measured by the ADAS-cog and MMSE with greater benefit shown at 24 weeks [ADAS-cog: WMD = –2.90 (95% CI –3.61 to –2.18), p < 0.001; MMSE: WMD = 1.21 (95% CI 0.84 to 1.57), p < 0.001]. The pooled estimates of all cognitive outcomes likewise showed a benefit from donepezil at 24–26 weeks’ follow-up.
Only one new study looked at functional outcomes for this comparison; at 12 weeks this showed a significant gain for those taking donepezil [mean difference: I = 40.5 (SD 7.6), C = 49.5 (SD 6.3), p < 0.01)]. At 24 weeks there were data from only the 2004 assessment trials, and the results from all the studies reporting functional outcomes were pooled; this analysis again showed a significant benefit from taking donepezil.
None of the new studies measured behavioural outcomes; the pooled estimates from the previous assessment, using the NPI, failed to show a significant gain on behavioural outcomes at either 12 or 24 weeks.
Just one new study121 looked at global outcomes; this showed a benefit from taking donepezil on the CDR [I = 1.2 (SD 0.2), C = 2.0 (SD 0.2), p < 0.01)]. The pooled results for the CIBIC-plus scale were only from the previous TAR and they showed a significant advantage from donepezil at 12 and 24 weeks’ follow-up [24 weeks: WMD = –0.43 (95% CI –0.55 to –0.31), p < 0.001]. When both the global outcome measures were pooled at 24–26 weeks, the results again showed a significant benefit from donepezil.
None of the new studies provided additional data on QoL or safety under randomised conditions.
The new studies found have added to the body of evidence showing a benefit from donepezil compared with placebo for cognitive, functional and global outcomes. However, there is no new or pooled evidence to show a behavioural benefit from donepezil versus placebo in people with mild-to-moderate AD. All but two of the studies included in these meta-analyses calculated their missing data points using LOCF or OC methods, thereby potentially biasing their results in favour of donepezil.
Graphical summary of donepezil versus placebo
The first thing that is noticeable when comparing Figures 20–22 is that no new large studies have been undertaken since 2004 and that only one study was multicentre. Closer examination shows that the quality of trials has not improved and, with the exception of Peng and colleagues, studies measured only cognitive abilities. As in the previous review, these outcomes showed that patients benefited from taking donepezil. Although the new trials add to the precision of our knowledge of the effects of donepezil on cognitive measures in mixed mild/moderate AD populations, none of the new studies was in the mild AD population. This means that we are still dependent on one moderately sized RCT from the 2004 review that looked at cognitive outcomes in the mild AD population, which showed a cognitive benefit for this group from taking 10 mg/day donepezil.
FIGURE 20.
Summary of studies included in the 2004 review: donepezil vs placebo. ADCS-ADL, Alzheimer’s Disease Cooperative Study – Activities of Daily Living Inventory; ADCS-CGIC, Alzheimer’s Disease Cooperative Study – Clinical Global Impression of Change; Analy, analysis; be, behavioural; Blind, blinding; Char, characteristics of patients; cog, cognitive; DAD, Disability Assessment for Dementia Scale; F, female; func, functional; GDS, Global Deterioration Scale; glo, global; M, male; Rand, randomisation; SIB, Severe Impairment Battery.
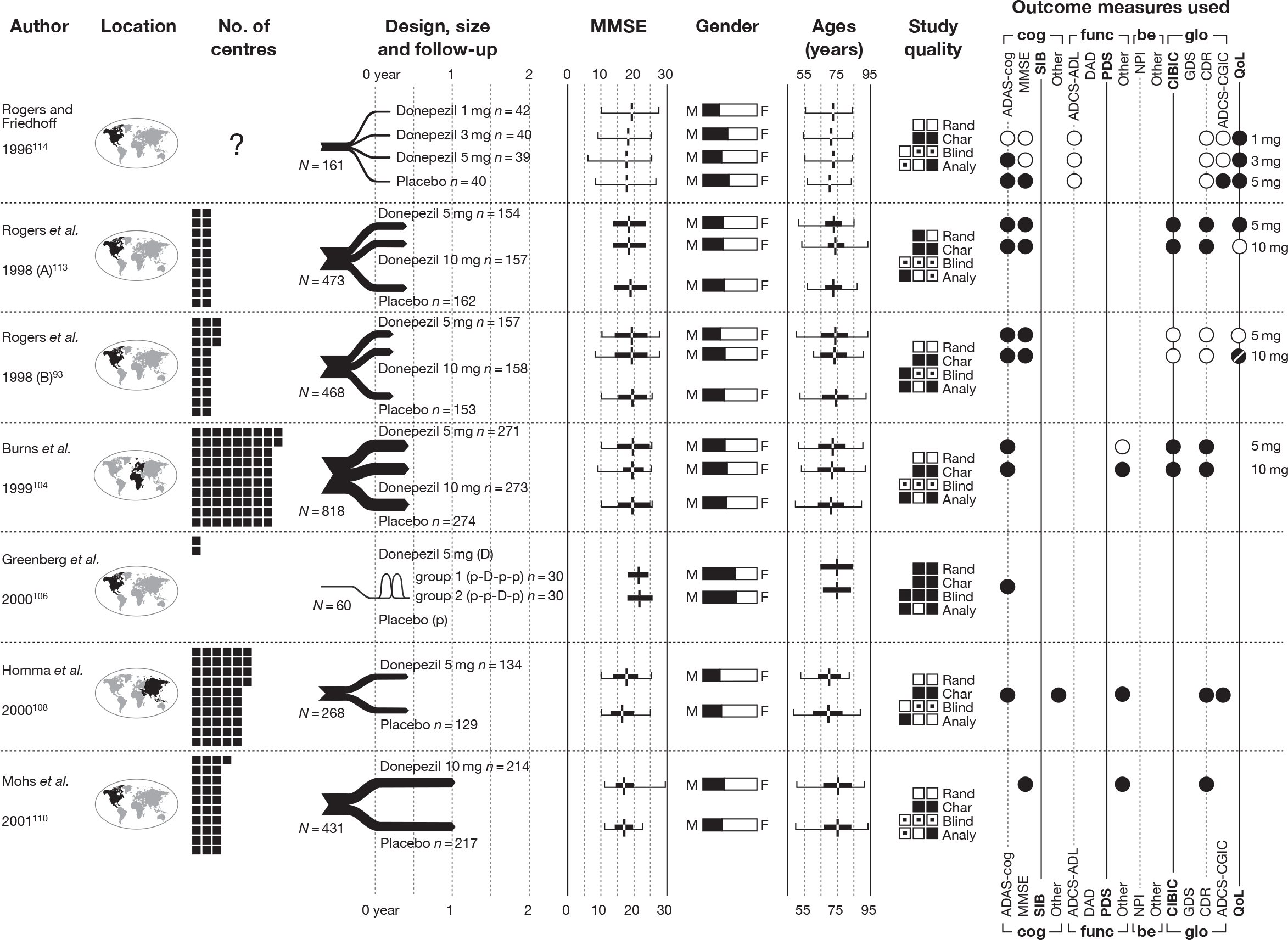
FIGURE 21.
Summary of studies included in the 2004 review: donepezil vs placebo (cont.). ADCS-ADL, Alzheimer’s Disease Cooperative Study – Activities of Daily Living Inventory; ADCS-CGIC, Alzheimer’s Disease Cooperative Study – Clinical Global Impression of Change; Analy, analysis; be, behavioural; Blind, blinding; Char, characteristics of patients; cog, cognitive; DAD, Disability Assessment for Dementia Scale; F, female; func, functional; GDS, Global Deterioration Scale; glo, global; M, male; Rand, randomisation; SIB, Severe Impairment Battery. *Median value reported instead of mean.
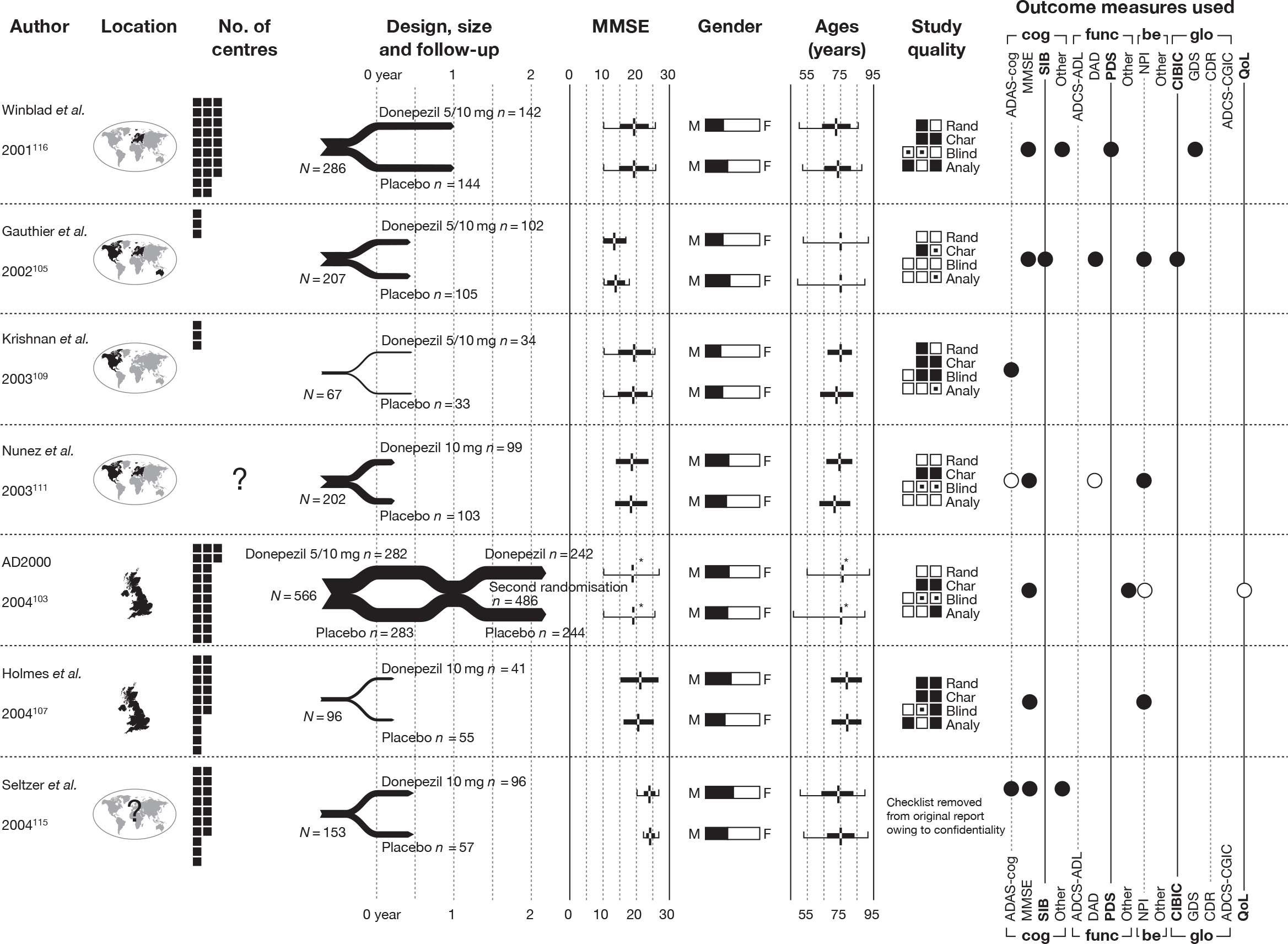
FIGURE 22.
Summary of new studies included in the 2010 review: donepezil vs placebo. ADCS-ADL, Alzheimer’s Disease Cooperative Study – Activities of Daily Living Inventory; ADCS-CGIC, Alzheimer’s Disease Cooperative Study – Clinical Global Impression of Change; Analy, analysis; be, behavioural; Blind, blinding; Char, characteristics of patients; cog, cognitive; DAD, Disability Assessment for Dementia Scale; F, female; func, functional; GDS, Global Deterioration Scale; glo, global; M, male; Rand, randomisation; SIB, Severe Impairment Battery.
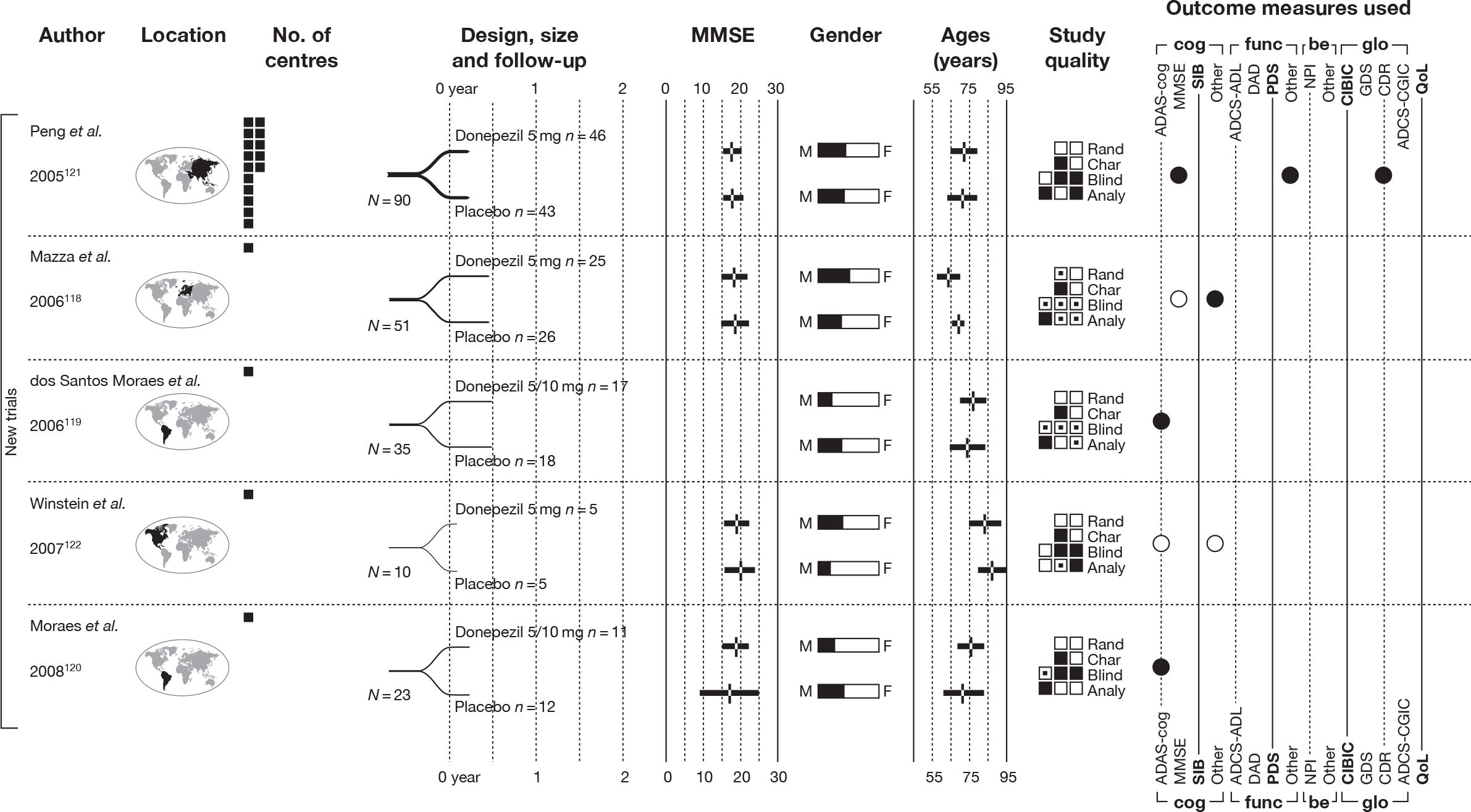
Galantamine versus placebo
Identified evidence
For the pair-wise comparison between galantamine and placebo, the 2004 review included six RCTs. 123–129 However, it is apparent that two of these publications124,129 report the same population. Accordingly, we have entered only Rockwood and colleagues’ primary publication124 in syntheses, below.
We identified an additional three moderately good-to-poor-quality RCTs, meeting few of the quality criteria indicated above (see Critical appraisal strategy).
The primary publication of the GAL-INT-6 study, written by Erkinjuntti and colleagues in 2002,130 was correctly excluded from the 2004 review because it conflated participants with multiple forms of dementia. However, we were able to include a subsequent paper – Bullock and colleagues 2004101 – reporting the AD-specific subgroup of this trial. A summary of their design characteristics can be found in Table 8 and the interventions, comparators and baseline characteristics of the participants in Table 9. A summary of the markers of internal validity is presented in Table 10.
| Study details | Inclusion criteria | Exclusion criteria | Methodological notes | Other |
|---|---|---|---|---|
|
Brodaty et al. (2005) 96 Design: Parallel double-blind RCT Countries: USA, Australia, Canada, South Africa and New Zealand No. of centres: 93 No. randomised: 971 Maximum follow-up: 26 MMSE range included: 10–24 Funding: None reported |
Mild-to-moderate probable AD (NINCDS-ADRDA) ADAS-cog/11 ≥ 18 History of cognitive decline that was gradual in onset and progressive over a period of ≥ 6 months Living with, or regular daily visits from, a responsible caregiver (≥ 5 days/week) |
Other neurodegenerative disorders or cognitive impairment due to acute cerebral trauma, hypoxic cerebral damage, vitamin deficiency states, infection, primary or metastatic cerebral neoplasia, significant endocrine or metabolic disease or mental retardation Vascular dementia or evidence of clinically active cerebrovascular disease History of epilepsy or convulsions; current clinically significant psychiatric disease; active peptic ulcer; clinically significant hepatic, renal, pulmonary, metabolic, or endocrine disturbances; clinically significant urinary outflow obstruction; clinically significant cardiovascular disease Use of any agent for the treatment of dementia (approved, experimental, or over the counter) including, but not limited to, nootropic agents, cholinomimetic agents, oestrogens taken without medical need, chronic non-steroidal anti-inflammatory agents or cyclo-oxygenase-2 inhibitors (> 30 consecutive days, regardless of indication) and vitamin E (unless a stable dose had been taken for ≥ 6 months prior to trial initiation) |
Sample attrition/dropout: 768 of 971 completed study. 203 withdrew after allocation: did not receive treatment (n = 6); AE (n = 67); withdrew consent (n = 62); non-compliance (n = 29); lost to follow-up (n = 10); insufficent response (n = 10); death (n = 5); other reasons (n = 3). No differences between groups Randomisation and allocation: Randomisation to treatment was determined by calling an interactive voice-response system. The subject number and treatment code (which corresponded to a specific medication kit) was randomly generated after the caller at the site provided the requested subject details. All treatments were supplied in opaque, size 0 gelatin capsules that were identical in appearance, taste and smell. All subjects received one capsule twice a day Power calculation: Powered at > 95% to detect a 2.5-point (SD 6.2) difference in ADAS-cog/11 score and at 90% to detect a 15% difference between active and placebo groups in their CIBIC-plus responder rates, assuming a 55% placebo responder rate (no change/improved CIBIC-plus score). Required sample size not explicitly reported |
Therapy common to all participants: 1-month placebo run-in prior to treatment allocation Study funding: None reported Other conflicts: Lead author declares consultancy fees, a grant and sponsored speaking engagements from Janssen |
|
Bullock et al. (2004) 101 Design: Parallel double-blind RCT Countries: ‘Including’ Canada, Denmark, Finland, France, Germany, Israel, the Netherlands, Poland and the UK No. of centres: 62 No. randomised: 285 Maximum follow-up: 26 MMSE range included: 10–25 Funding: None reported |
Probable vascular dementia (NINDS-AIREN definition) or AD + CVD (NINCDS-ADRDA definition) (with CVD evidenced by CT or MRI) Score ≥ 12 on 11-item subscale of of AD Assessment Scale Presence of focal neurological signs Disease onset at between 40 and 90 years of age |
Neurogenerative disorders Cognitive impairment resulting from other cerebral trauma Cerebral neoplasia Mental retardation Vitamin deficiency Significant endocrine or metabolic disease Clinically significant co-exitsng medical conditions Significant CVD that would likely limit the patient’s ability to complete the study Current use of agents for the treatment of dementia Recent history (within 30 days) of treatment with other investigational agents History of alcohol or drug abuse |
Sample attrition/dropout: 230 of 285 completed study Randomisation and allocation: Randomisation was conducted using a ‘computer-generated code’ (no further details provided) No details provided about appearance, taste, or smell of placebo Power calculation: Not reported |
Therapy common to all participants: 1-month’s single-blind placebo run-in prior to treatment allocation Study funding: None reported Other conflicts: None reported Notes: Follow-up also at 32 and 52 weeks during the open-label phase of the trial Unable to calculate attrition n, as using percentages quoted in the text gives non-whole numbers This paper deals with the subgroup of people with AD + CVD |
|
Rockwood et al. (2006) 131 Design: Parallel double-blind RCT Country: Canada No. of centres: 10 No. randomised: 130 Maximum follow-up: 16 MMSE range included: 10–25 Funding: Janssen-Ortho Canada (80%) and the Canadian Institutes of Health Research (20%) (grant no. DCT-49981). The sponsor provided all medications and matching placebos, conducted on-site monitoring and gathered and electronically coded the case report forms. All data are held by the principal investigator (Kenneth Rockwood), who initiated and supervised all analyses. Janssen-Ortho received the paper 45 days before submission to verify protocol details. At the authors’ request, Janssen-Ortho statisticians answered questions about the use of the mixed-effects model, but had no other input in the analyses |
Probable AD (NINCDS-ADRDA criteria) ADAS-cog score ≥ 18 Daily contact with a responsible caregiver |
Resident in nursing home Disabling communication difficulties (problems in language, speech, vision or hearing) Other active medical issues or competing causes of dementia Patients who had taken anti-dementia medications within 30 days before screening for study enrolment Hypersensitivity to cholinomimetic agents or bromide Participation in other galantamine trials |
Sample attrition/dropout: 109 of 130 completed study. 21 withdrew after allocation: AE n = 7; non-compliance n = 6; insufficient response n = 4; lost to follow-up n = 1; withdrew consent n = 2; died n = 1. More patients in the galantamine group (n = 5) withdrew due to AEs than in the placebo group (n = 2), otherwise no difference between groups Randomisation and allocation: Randomisation was determined immediately before medication was administered by research nurse telephoning into a contracted, interactive voice-response system for an assignment number. Nurse was blind to the number’s meaning in terms of treatment assignment. Randomisation was in blocks of two, by site, to decrease the chance of incomplete blocks (the GAS instrument was new to investigators at the study sites and some sites might have had to withdraw if investigators did not know how to complete it) Power calculation: Authors state that on the basis that the GAS instrument can be more responsive than standard measures because it is personalised, this attribute had not been tested in a controlled trial in dementia. For the exploratory analysis, the sample size was estimated from the authors’ limited experience with GAS in anti-dementia drug trials. Assuming a moderate effect size of about 0.524 and a 15% dropout at 4 months, it was determined that 152 subjects would be required to detect differences at the 5% significance level (two-tailed) with 80% power. Authors recognised that this might not result in statistically significant results for the secondary outcomes, which were used to compare with the primary outcomes and with results from other studies |
Therapy common to all participants: None reported Other conflicts: Lead author has undertaken consultancies and received honoraria from Janssen-Ortho, the study’s co-sponsor, and from Pfizer, Novartis and Merck, and was also lead author of an earlier galantamine study. Lead author owns no stock in pharmaceutical companies. Lead author is part owner of Dementia Guide, which is developing a website to aid in goal setting for people with dementia Co-authors: CM has received research grants from Janssen-Ortho, Pfizer, Lundbeck and Novartis, but has received no personal payments; MG has received honoraria and travel grants from Janssen-Ortho, Pfizer and Merck; SF and XS have no conflicts of interest to declare Notes: Five patients (two in galantamine group, three in placebo group) had MMSE scores that were outside the 10–25 range stipulated in the inclusion criteria; one had a MMSE score of < 10, the other 4 had MMSE scores > 25 Seven patients (four in galantamine group, three in placebo group) had ADAS-cog scores that were outside the > 17 range stipulated in the inclusion criteria; in each case the score was below the lower limit, which indicated milder impairment |
| Study | Arm | Dose (mg/day) | Dosage details | N | Age, years (SD) | Gender, n male (%) | Race, n white (%) | Weight, kg (SD) | Education (years) | Duration of dementia, months (SD) | ADAS-cog, score (SD) | MMSE, score (SD) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Brodaty et al. (2005)96 | Galantamine PRCa | 8–24 |
Titrated from an initial dosage of 8 mg/day for the first 4 weeks up to a maximum of 24 mg/day in increments of 8 mg/day every 4 weeks after the placebo run-in Whole dose given in single capsule am; placebo given pm |
319 | 76.6 (7.64) | 114 (35.7) | 297 (93.1) | 68.6 (14.2)a | 18.0 (3.97) | |||
| – | 8–24 |
Titrated from an initial dosage of 8 mg/day for the first 4 weeks up to a maximum of 24 mg/day in increments of 8 mg/day every 4 weeks after the placebo run-in Single capsules am and pm |
326 | 76.5 (7.77) | 118 (36.2) | 293 (89.9) | 68.3 (15.9) | 17.8 (4.14) | ||||
| Placebo | – | Single placebo dose am and pm | 320 | 76.3 (8.03) | 115 (35.9) | 289 (90.3) | 67.8 (14.6)b | 18.1 (4.08) | ||||
| Bullock et al. (2004)101 | Galantamine | 4–24 | Titrated upwards in weekly 4-mg increments over a period of 6 weeks, and then continued at this maintenance dose (24 mg/day) for an additional 4.5 months | 152 | 75.8 (6.78) | 73 (48.0) | 69.9 (12.9) | 22.7 (9.37)c | 20.5 (3.95) | |||
| Placebo | – | Single placebo dose am and pm | 86 | 77.6 (6.12) | 42 (48.8) | 67.0 (13.0) | 23.9 (9.92)d | 20.2 (3.52) | ||||
| Rockwood et al. (2006)131 | Galantamine | 8–24 | Initial dose of 8 mg/day (4 mg twice daily) for 4 weeks, followed by 16 mg/day for another 4 weeks. At the end of week 8, dose could be increased to 24 mg/day depending on tolerability. At week 12, patients were re-evaluated; the dose could then be reduced to 16 mg/day if necessary, after which time it could not be changed | 64 | 77.0 (8.00) | 23 (35.9) | 11.0 (3.00) | 24.2 (6.40) | 20.8 (3.30) | |||
| Placebo | – | Sham titration schedule | 66 | 78.0 (8.00) | 25 (37.9) | 11.0 (3.00) | 27.9 (8.40) | 19.9 (4.20) |
| Study | Was the assignment to the treatment groups really random? | Was the treatment allocation concealed? | Were the groups similar at baseline in terms of prognostic factors? | Were the eligibility criteria specified? | Were outcome assessors blinded to the treatment allocation? | Was the care provider blinded? | Was the patient blinded? | Were the point estimates and measure of variability presented for the primary outcome measure? | Did the analyses include an ITT analysis? | Were withdrawals and dropouts completely described? |
|---|---|---|---|---|---|---|---|---|---|---|
| Brodaty et al. (2005)96 | Adequate | Adequate | Reported – yes | Adequate | Adequatea | Adequate | Adequate | Partialb | Partialc | Adequate |
| Bullock et al. (2004)101 | Partiald | Unknown | Reported – yes | Unknown | Unknown | Unknown | Partial | Adequate | Partiale | Adequate |
| Rockwood et al. (2006)131 | Adequate | Adequate | Reported – nof | Inadequate | Partial | Partial | Partial | Adequate | Adequate | Adequate |
Evidence of clinical effectiveness
Cognition
The previous review2 summarised the evidence from cognition outcomes thus:
Six published RCTs showed that galantamine appears to confer a statistically significant benefit to participants on the ADAS-cog scale when compared to placebo, whether reducing the deterioration or leading to some improvement in their condition. The benefit varies depending upon the dose of galantamine. The galantamine–placebo differences in ADAS-cog for 8 mg/day was 1.3 points, 16 mg/day 3.1 points, 18 mg/day 1.7 points, 16 or 24 mg/day 2.5 to 2.8 points; 24 to 32 mg/day 1.7 to 3.4 points and 36 mg/day 2.3 points. The one unpublished RCT mirrored these positive effects of galantamine versus placebo. In addition, some 14–17% more of galantamine participants were classified as responders (improving by 4 or more points on the ADAS-cog) than those on placebo.
The results from the three new studies96,101,131 show that overall those treated with galantamine had improved scores on the ADAS-cog, whereas those in the control group remained stable or declined. However, missing data were accounted for using LOCF and OC methods, leading to a potential overestimate of the treatment effect. 86 Table 11 summarises the cognitive results from the new studies.
| Study | Subgroup | Outcome | Type | Arm | Galantamine | Placebo | p-value | ||
|---|---|---|---|---|---|---|---|---|---|
| N | Mean, n (SD) | N | Mean, n (SD) | ||||||
| Bullock et al. (2004)101 | OC population | ADAS-cog 6 weeks | 148 | –0.5 (4.62)a | 85 | 0.15 (6.26)a | 0.366b | ||
| ADAS-cog 13 weeks | MC | 148 | –1.48 (4.32)a | 85 | 0 (6.03)a | 0.031b | |||
| ADAS-cog 26 weeks | MC | 147 | –1.1 (5.79) | 83 | 2 (5.56) | < 0.001b | |||
| A | 147 | 21.5 (10.5) | 83 | 25.7 (12) | 0.006b | ||||
| Brodaty et al. (2005)96 | LOCF analysis | ADAS-cog 8 weeks | MC | o.d.c | 287 | –1.5 (5.08) | 293 | 0 (5.14) | – |
| b.i.d.d | 294 | –1.7 (4.97) | – | ||||||
| ADAS-cog 12 weeks | MC | o.d.c | 290 | –2f (5.28) | 296 | 0.2 (5.33) | – | ||
| b.i.d.d | 296 | –2.5 (5.16) | – | ||||||
| ADAS-cog 26 weeks | MC | o.d.c | 291 | –1.3 (5.29) | 296 | 1.2 (5.68) | < 0.001f | ||
| b.i.d.d | 296 | –1.6 (6.19) | < 0.001f | ||||||
| OC population | ADAS-cog 8 weeks | MC | o.d.c | 284 | –1.5 (5.06) | 289 | 0 (5.1) | – | |
| b.i.d.d | 286 | –1.7 (5.07) | – | ||||||
| ADAS-cog 12 weeks | MC | o.d.c | 269 | –2.2 (5.25) | 275 | 0 (5.14) | – | ||
| b.i.d.d | 268 | –2.6 (5.07) | – | ||||||
| ADAS-cog 26 weeks | MC | o.d.c | 240 | –1.4 (5.27) | 248 | 1.3 (5.67) | < 0.001f | ||
| b.i.d.d | 227 | –1.8 (6.33) | < 0.01f | ||||||
| Rockwood et al. (2006)131 | LOCF analysis | ADAS-cog 8 weeks | MC | 62 | –1.85 (4.18) | 65 | –0.25 (4.97) | – | |
| ADAS-cog 16 weeks | MC | 62 | –1.6 (5.38) | 65 | 0.325 (5.49) | – | |||
We pooled data from the new studies96,101,131 with those of the 2004 assessment2 by random-effects meta-analysis using the ADAS-cog as the outcome measure for ≤ 24 mg/day at 12–16 weeks and 21–26 weeks post randomisation. No studies reported the MMSE. We also meta-analysed the data by > 12 mg/day and all doses combined; these results can be found in Appendix 5.
Two new studies were included in the meta-analysis. The overall pooled estimates showed a benefit from galantamine compared with placebo at 12–16 and 21–26 weeks: 12–16 weeks WMD = –2.39 (95% CI –2.80 to –1.97), p < 0.001; 21–26 weeks WMD = –2.96 (95% CI –3.41 to –2.51), p < 0.001 (Figures 23 and 24).
FIGURE 23.
Random-effects meta-analysis: ADAS-cog at 12–16 weeks (mean change from baseline) – galantamine (maximum dose ≤ 24 mg/day) vsplacebo. a, 18 and 24 mg/day arms pooled; b, once daily prolonged-release formulation and twice daily standard formulation pooled; c, 8, 16 and 24 mg/day arms pooled.
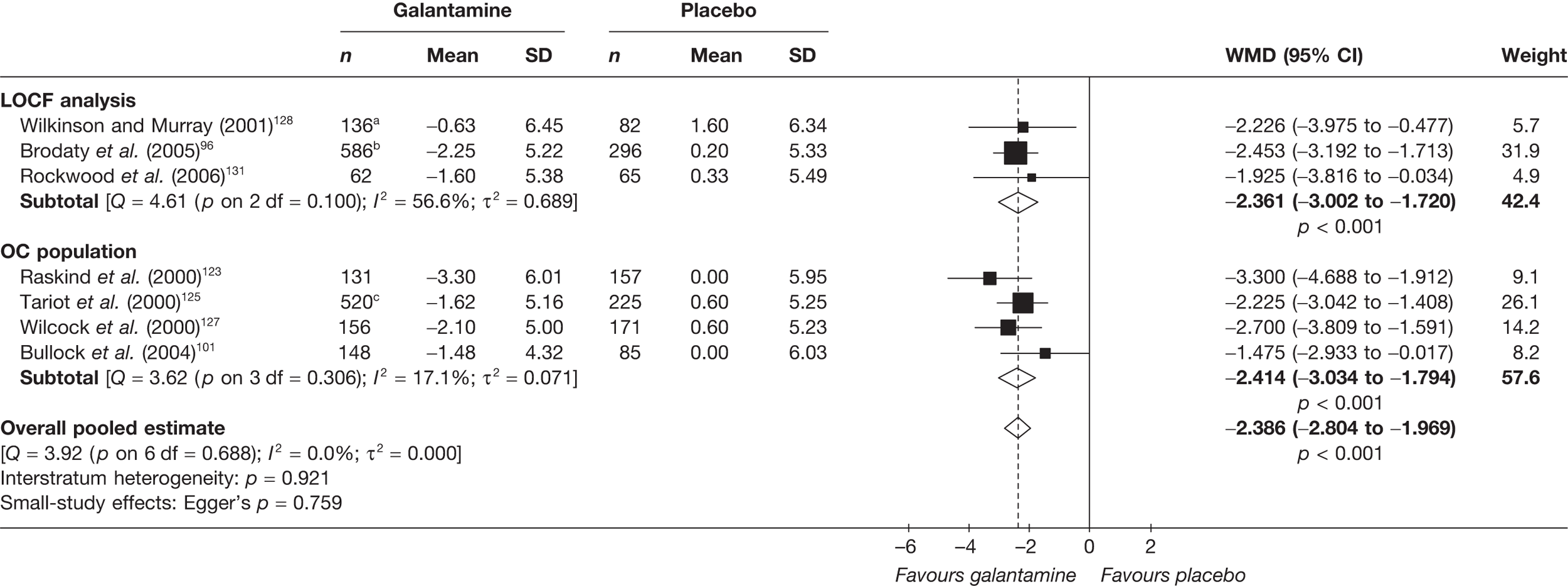
FIGURE 24.
Random-effects meta-analysis: ADAS-cog at 21–26 weeks (mean change from baseline) – galantamine (maximum dose ≤ 24 mg/day) vs placebo. a, 8, 16 and 24 mg/day arms pooled; b, once daily prolonged-release formulation and twice daily standard formulation pooled.
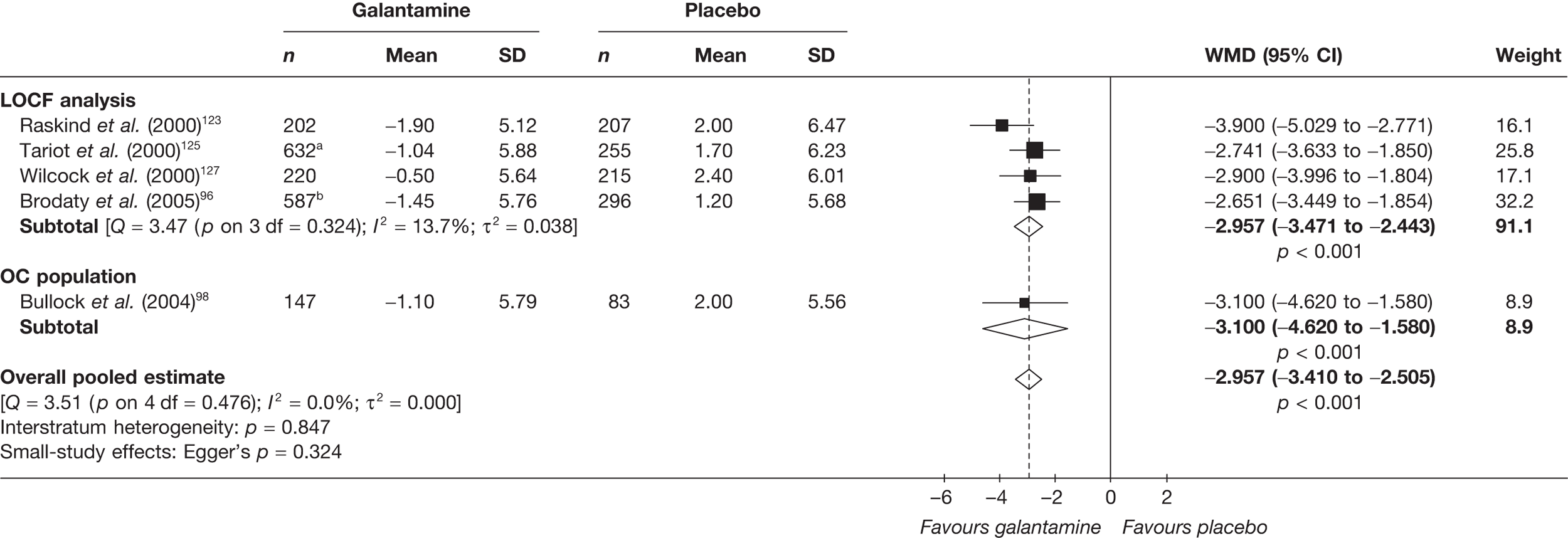
Because ADAS-cog is the only outcome measure used to assess cognitive effect in the placebo-controlled trials of galantamine, it was not necessary to pool outcomes on a standardised level for this domain.
Functional
In 20042 the assessment group reported for functional outcomes that:
Three RCTs assessed mean changes from baseline on the DAD scale, all reporting statistically significantly slower deterioration for those receiving galantamine 24–32 mg/day compared to placebo. Two RCTs found that participants receiving 16 mg/day and/or 24 mg/day galantamine experienced a statistically significantly smaller deterioration on the ADCS/ADL compared to placebo.
All three new RCTs measured functional outcomes. 96,101,131 They found that functional abilities had improved significantly more in the treatment group. Table 12 summarises the results.
| Study | Subgroup | Outcome | Type | Arm | Galantamine | Placebo | p-value | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| N | Mean, n (SD%) | N | Mean, n (SD %) | ||||||||
| Bullock et al. (2004)101 | LOCF analysis | DAD | 26 weeks | MC | 188 | –1 (15.8)a | 97 | –6 (14.5)a | < 0.01b | ||
| Brodaty et al. (2005)96 | LOCF analysis | ADCS-ADL | 26 weeks | MC | o.d.c | 245d | 0 (7.51) | 258d | –2.7 (0.899) | < 0.001e | |
| MC | b.i.d.f | 242d | –1 (0.778) | 0.018e | |||||||
| OC population | ADCS-ADL | 8 weeks | MC | o.d.c | 280 | 0.8 (6.86) | 294 | –0.7 (7.72) | – | ||
| MC | b.i.d.f | 292 | 0.9 (7.18) | – | |||||||
| 12 weeks | MC | o.d.c | 276 | 0.4 (6.65) | 281 | –0.3 (7.71) | – | ||||
| MC | b.i.d.f | 279 | 1.1 (7.85) | – | |||||||
| 26 weeks | MC | o.d.c | 245 | 0 (8.61) | 258 | –2.4 (9.64) | 0.003e | ||||
| MC | b.i.d.f | 242 | –1 (8.87)g | 0.088e | |||||||
| Rockwood et al. (2006)131 | LOCF analysis | GAS (CR)h | 8 weeks | A | 61 | 52.5 (9.12) | 66 | 52.2 (6.97) | – | ||
| 16 weeks | A | 61 | 54.8 (9.36) | 66 | 50.9 (9.74) | 0.02i | |||||
| GAS (PCR)j | 8 weeks | A | 61 | 54.6 (7.97) | 66 | 52.5 (8.57) | – | ||||
| 16 weeks | A | 61 | 54.2 (10.8) | 66 | 52.3 (9.12) | 0.27i | |||||
| OC populationk | GAS-VR | Improved | 16 weeks | D | 20 | 14 (70.0) | 30 | 8 (26.7) | < 0.01l | ||
| Worsened | 16 weeks | D | 20 | 2 (10.0) | 30 | 10 (33.3) | – | ||||
| No change | 16 weeks | D | 20 | 4 (20.0) | 30 | 12 (40.0) | – | ||||
The data from the new trials96,101,131 were pooled, by random-effects meta-analysis, with those of the studies found in 2004. The outcome measures considered were the Alzheimer’s Disease Cooperative Study – Activities of Daily Living (ADCS-ADL) Inventory and the Disability Assessment of Dementia (DAD) Scale.
Results from the ADCS-ADL were pooled at 12–13 weeks’ follow-up and at 21–26 weeks’ follow-up. The overall pooled estimates showed functional benefit from galantamine compared with placebo: 12–13 weeks’ WMD = 1.39 (95% CI 0.59 to 2.20), p < 0.001; 21–26 weeks WMD = 2.23 (95% CI 1.33 to 3.14), p < 0.001 (Figures 25 and 26).
FIGURE 25.
Random-effects meta-analysis: ADCS-ADL at 12–13 weeks (mean change from baseline): galantamine (≤ 24 mg/day]) vs placebo. a, 8, 16 and 24 mg/day arms pooled; b, once daily prolonged-release formulation and twice daily standard formulation pooled.
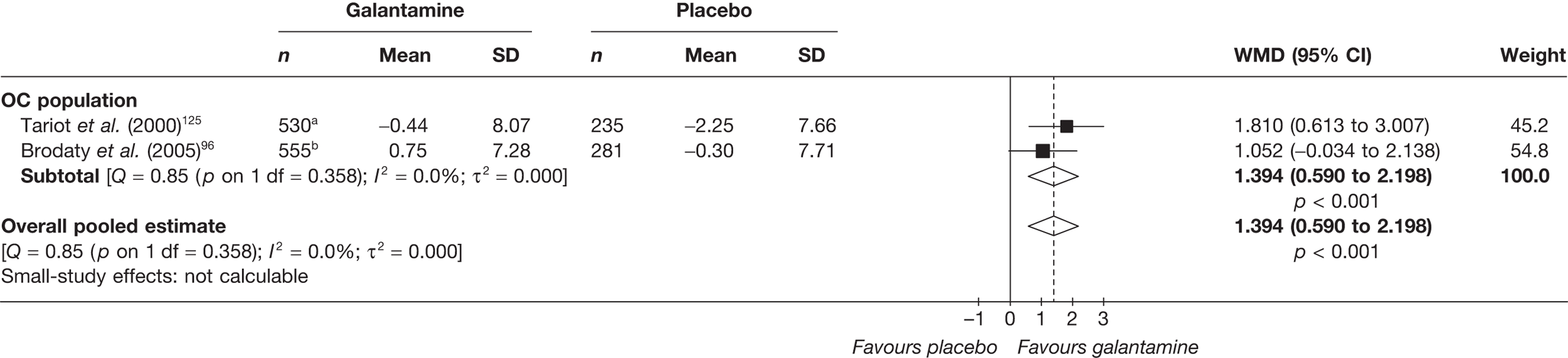
FIGURE 26.
Random-effects meta-analysis: ADCS-ADL at 21–26 weeks (mean change from baseline): galantamine (≤ 24 mg/day]) vs placebo. a, 8, 16 and 24 mg/day arms pooled; b, once daily prolonged-release formulation and twice daily standard formulation pooled.
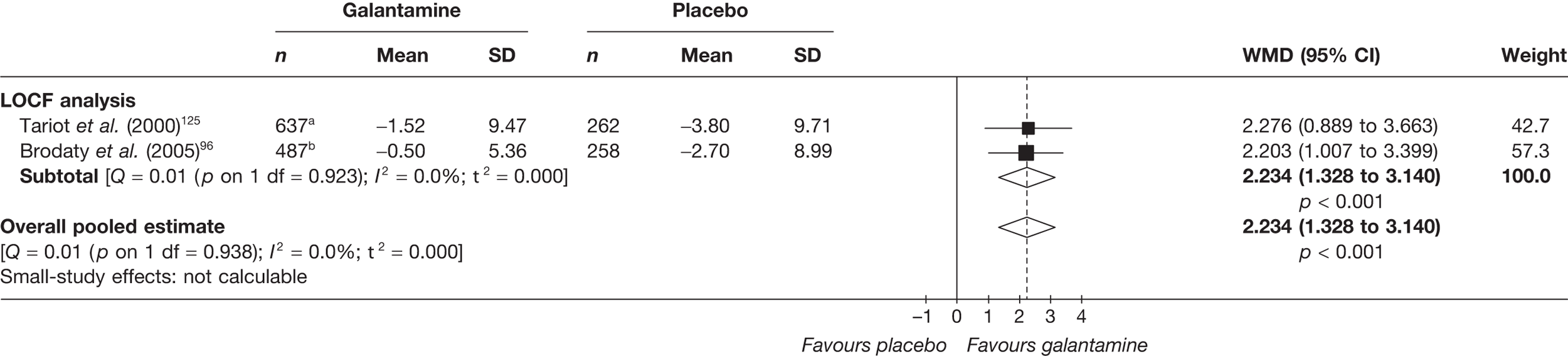
The results from the DAD were pooled at 21–26 weeks’ follow-up. They again showed a significant benefit from galantamine compared with placebo: WMD = 3.76 (95% CI 1.66 to 5.86), p < 0.001 (Figure 27).
FIGURE 27.
Random-effects meta-analysis: DAD at 21–26 weeks (mean change from baseline): galantamine (≤ 4 mg/day) vs placebo. a, 24 and 32 mg/day arms pooled.
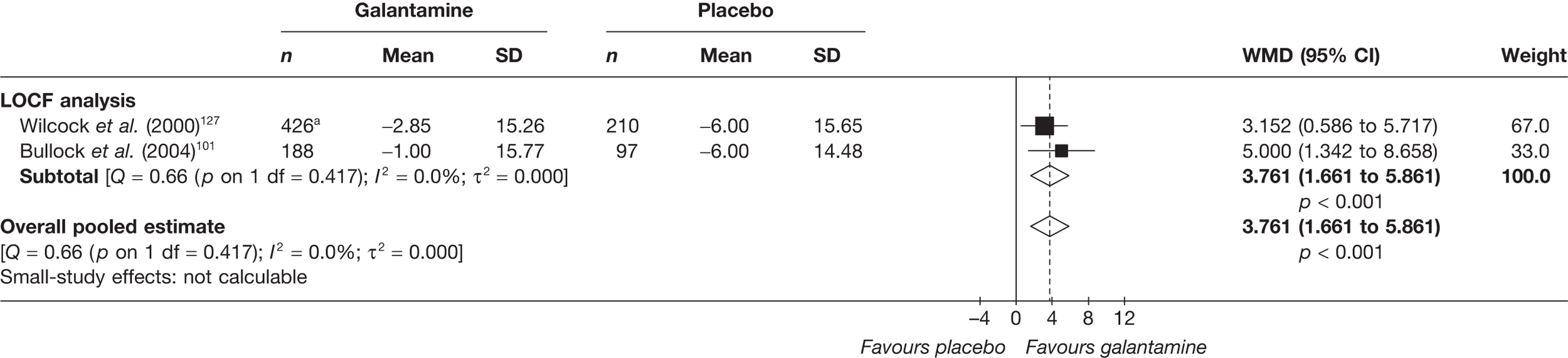
Two new studies were added to the meta-analysis of combined functional outcome measures at 21–26 weeks. The overall pooled estimate showed a significant functional benefit from galantamine compared with placebo: SMD = 0.27 (95% CI 0.18 to 0.34), p < 0.001 (Figure 28). The data set used in this meta-analysis can be found in Appendix 6.
FIGURE 28.
Random-effects meta-analysis: functional outcomes (SMD) at 21–26 weeks: galantamine (all dosages) vs placebo.
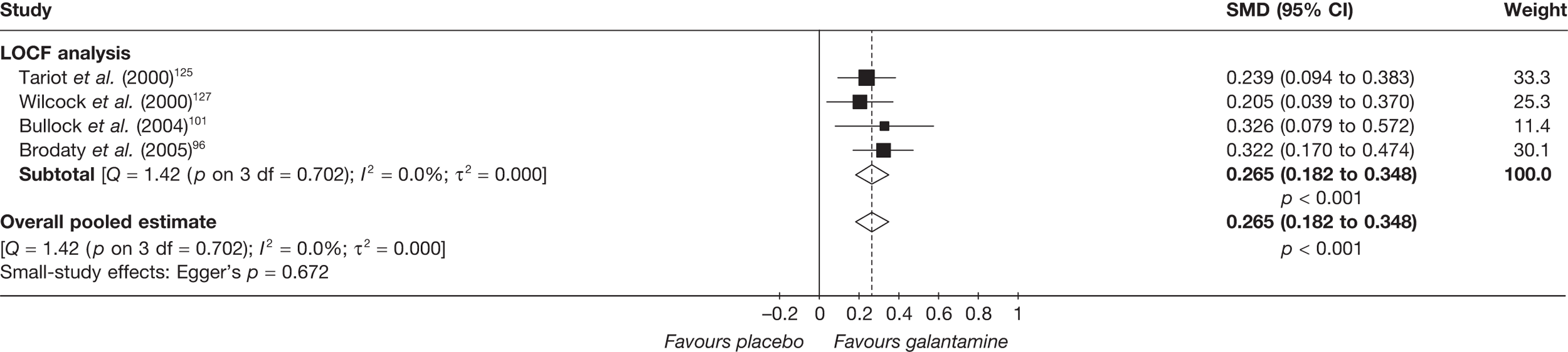
With only four data points in this evidence base, it would not be informative to perform metaregression.
Behavioural and mood
In 2004, Loveman and colleagues2 summarised the behavioural results from this comparison as:
Two published and one unpublished RCTs found that galantamine had some effect in improving or limiting further deterioration on the NPI scale compared to placebo. Differences in the mean change from baseline were statistically significant for doses of 16 mg/day or over in one of the three studies.
Only one included study96 provided additional data on the effectiveness of galantamine in relieving behavioural symptoms of AD when compared with placebo. However, this failed to show any statistically significant benefit. Data are shown in Table 13.
| Study | Subgroup | Outcome | Type | Arm | Galantamine | Placebo | p-value | ||
|---|---|---|---|---|---|---|---|---|---|
| N | Mean, n (%) | N | Mean, n (%) | ||||||
| Brodaty et al. (2005)96 | LOCF analysis | NPI – 26 weeks | MC | o.d.a | 245b | –0.6 (10.3) | 258b | 0.6 (9.96) | 0.941c |
| b.i.d.d | 242b | –0.9 (11.4) | 0.102c | ||||||
| OC population | NPI – 26 weeks | MC | o.d.a | 245 | –0.6 (10.8) | 258 | 0.1 (13.2) | 0.451c | |
| b.i.d.d | 242 | –1.2 (12.9) | 0.203c | ||||||
Only one new study added evidence to this meta-analysis. We looked for estimates of effectiveness at 13 and 21–26 weeks; at 13 weeks no significant benefit was found. However, at 21–26 weeks the overall pooled estimate favoured galantamine: WMD = –1.46 (95% CI –2.59 to –0.34), p = 0.012 (Figures 29 and 30).
FIGURE 29.
Random-effects meta-analysis: NPI at 13 weeks (mean change from baseline) – galantamine (all dosages) vs placebo. a, 8, 16 and 24 mg/day arms pooled.
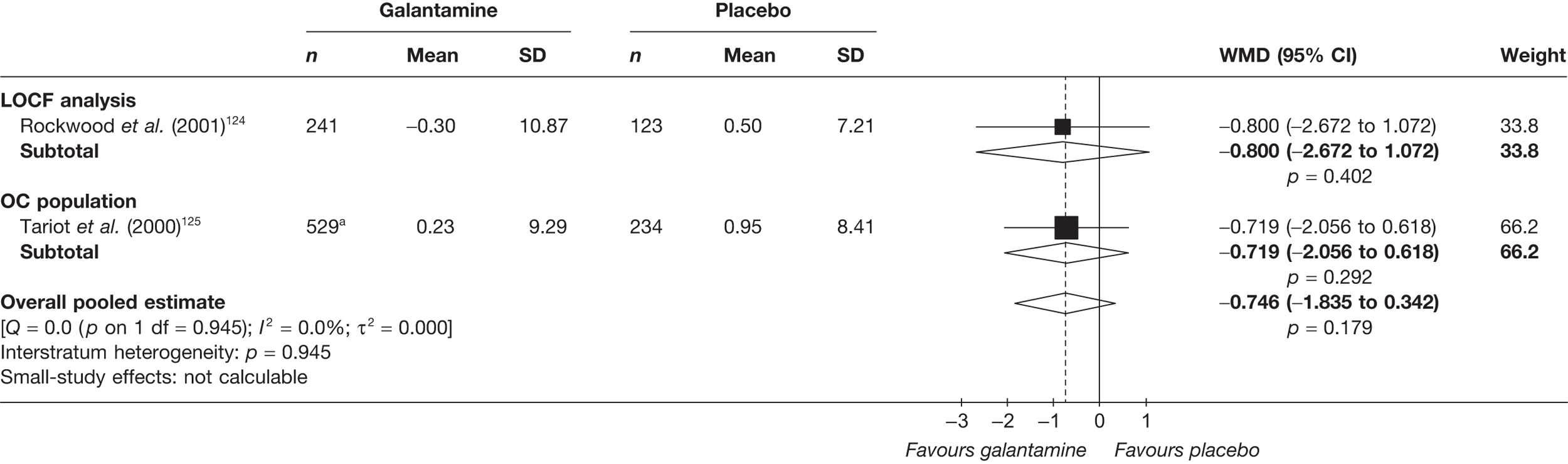
FIGURE 30.
Random-effects meta-analysis: NPI at 21–26 weeks (mean change from baseline) – galantamine (all dosages) vs placebo. a, 8, 16 and 24 mg/day arms pooled; b, once daily prolonged-release formulation and twice daily standard formulation pooled.
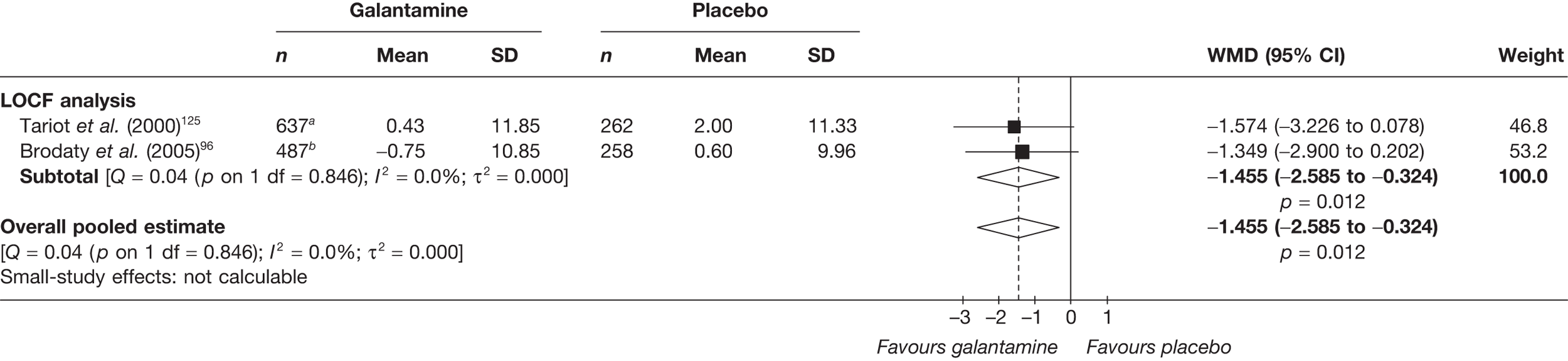
Because the NPI is the only outcome measure used in this domain of the evidence base, it was not necessary to pool outcomes on a standardised level.
Global effect
The previous assessment in 20042 summarised the effectiveness of galantamine on global outcomes:
Five published and one unpublished RCT assessed the effect of galantamine compared to placebo on the CIBIC plus, individually showing that higher proportions of participants receiving galantamine experience improvement in their condition compared to those on placebo (0% to 6.5% more participants). In contrast, a higher proportion of placebo participants tend to deteriorate (4% to 18% more participants). Also, a higher proportion of galantamine compared to placebo participants were considered to be responders to treatment with differences of between 4% (8 mg/day) and 17% (24 mg/day). When studies are pooled there are no statistically significant effects demonstrated.
Two new studies were found that measured global outcomes. 96,131 Rockwood and colleagues131 found a benefit from galantamine measured by the CIBIC-plus compared with placebo at 13–16 weeks (Table 14).
| Study | Subgroup | Outcome | Type | Arm | Galantamine | Placebo | p-value | ||
|---|---|---|---|---|---|---|---|---|---|
| N | Mean, n (%) | N | Mean, n (%) | ||||||
| Brodaty et al. (2005)96 | LOCF analysis | CIBIC-plus score – 26 weeks | A | o.d.a | 291 | 4.21 (SD 1.1) | 301 | 4.35 (SD 1.14) | NSb |
| b.i.d.c | 302 | 4.21 (SD 1.07) | NSb | ||||||
| CIBIC-plus: markedly improved – 26 weeks | D | o.d.a | 291 | 3 (1.0) | 301 | 3 (1.0) | 0.712d | ||
| b.i.d.c | 302 | 3 (1.0) | 0.685d | ||||||
| CIBIC-plus: moderately improved – 26 weeks | D | o.d.a | 291 | 14 (4.8) | 301 | 11 (3.7) | 0.621d | ||
| b.i.d.c | 302 | 15 (5.0) | 0.553d | ||||||
| CIBIC-plus: minimally improved – 26 weeks | D | o.d.a | 291 | 49 (16.8) | 301 | 48 (15.9) | 0.856d | ||
| b.i.d.c | 302 | 46 (15.2) | 0.897d | ||||||
| CIBIC-plus: no change – 26 weeks | D | o.d.a | 291 | 114 (39.2) | 301 | 111 (36.9) | 0.623d | ||
| b.i.d.c | 302 | 127 (42.1) | 0.224d | ||||||
| CIBIC-plus: minimally worse – 26 weeks | D | o.d.a | 291 | 81 (27.8) | 301 | 80 (26.6) | 0.802d | ||
| b.i.d.c | 302 | 78 (25.8) | 0.907d | ||||||
| CIBIC-plus: moderately worse – 26 weeks | D | o.d.a | 291 | 24 (8.2) | 301 | 41 (13.6) | 0.050d | ||
| b.i.d.c | 302 | 30 (9.9) | 0.201d | ||||||
| CIBIC-plus: markedly worse – 26 weeks | D | o.d.a | 291 | 6 (2.1) | 301 | 7 (2.3) | 0.951d | ||
| b.i.d.c | 302 | 3 (1.0) | 0.336d | ||||||
| OC population | CIBIC-plus score – 26 weeks | A | o.d.a | 246 | 4.19 (SD 1.13) | 259 | 4.36 (SD 1.15) | NSb | |
| b.i.d.c | 240 | 4.21 (SD 1.11) | NSb | ||||||
| CIBIC-plus: markedly improved – 26 weeks | D | o.d.a | 246 | 3 (1.2) | 259 | 3 (1.2) | 0.728d | ||
| b.i.d.c | 240 | 3 (1.3) | 0.751d | ||||||
| CIBIC-plus: moderately improved – 26 weeks | D | o.d.a | 246 | 14 (5.7) | 259 | 9 (3.5) | 0.327d | ||
| b.i.d.c | 240 | 24 (5.8) | 0.298d | ||||||
| CIBIC-plus: minimally improved – 26 weeks | D | o.d.a | 246 | 43 (17.5) | 259 | 41 (15.8) | 0.705d | ||
| b.i.d.c | 240 | 36 (15.0) | 0.895d | ||||||
| CIBIC-plus: no change – 26 weeks | D | o.d.a | 246 | 90 (36.6) | 259 | 94 (36.3) | 0.981d | ||
| b.i.d.c | 240 | 93 (38.8) | 0.636d | ||||||
| CIBIC-plus: minimally worse – 26 weeks | D | o.d.a | 246 | 69 (28.0) | 259 | 70 (27.0) | 0.875d | ||
| b.i.d.c | 240 | 67 (27.9) | 0.903d | ||||||
| CIBIC-plus: moderately worse – 26 weeks | D | o.d.a | 246 | 23 (9.3) | 259 | 36 (13.9) | 0.146d | ||
| b.i.d.c | 240 | 25 (10.4) | 0.294d | ||||||
| CIBIC-plus: markedly worse – 26 weeks | D | o.d.a | 246 | 4 (1.6) | 259 | 6 (2.3) | 0.812d | ||
| b.i.d.c | 240 | 2 (0.8) | 0.336d | ||||||
| Rockwood et al. (2006)131 | LOCF analysis | CIBIC-plus score – 8 weeks | A | 61 | 3.64 (SD 0.797) | 65 | 4.17 (SD 0.905) | – | |
| CIBIC-plus score – 16 weeks | A | 61 | 3.67 (SD 0.996) | 65 | 4.12 (SD 0.987) | 0.03e | |||
When the new studies’ data were pooled with the existing evidence base, the overall pooled estimates of the CIBIC-plus at 26 weeks showed a benefit from galantamine compared with placebo: WMD = –0.20 (95% CI –0.30 to –0.09), p < 0.001 (Figure 31).
FIGURE 31.
Random-effects meta-analysis: CIBIC-plus at 26 weeks – galantamine (maximum dose ≥ 24 mg/day) vs placebo. a, once daily prolonged-release formulation and twice daily standard formulation pooled.
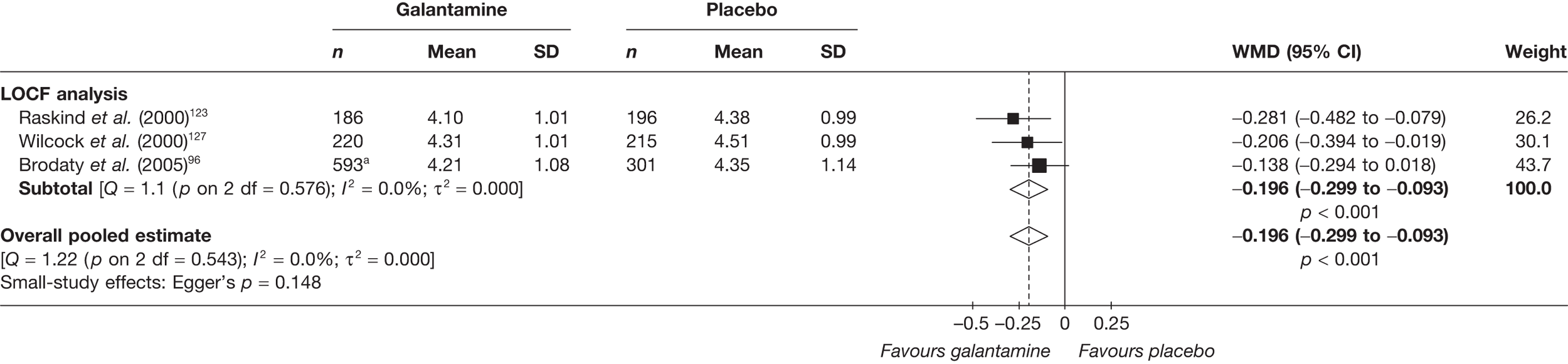
Because CIBIC-plus is the only outcome measure used to assess global effect in the placebo-controlled trials of galantamine, there was no reason to pool outcomes on a standardised level for this domain.
Quality of life
None of the included studies provided any randomised evidence on QoL with galantamine compared with placebo and no such data were identified in the 2004 review. 2
Safety
Overall, there was a high percentage of any AE in both studies in treatment and control groups (any AE: Brodaty,96 once a day treatment = 79%, placebo = 70%; Rockwood,131 treatment = 84%, placebo = 62%). The main AEs were gastrointestinal. A summary of all the AEs reported can be found in Table 15.
| AEs | Galantamine | Placebo | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| o.d. | b.i.d. | ||||||||||||
| Brodaty et al. (2005)96 | Brodaty et al. (2005)96 | Rockwood et al. (2006)131 | Brodaty et al. (2005)96 | Rockwood et al. (2006)131 | |||||||||
| N | n (%) | p-valuea | N | n (%) | p-valuea | N | n (%) | p-valuea | N | n (%) | N | n (%) | |
| Any AE | 319 | 253 (79.3) | 0.009 | 326 | 235 (72.1) | 0.619 | 64 | 54 (84.4) | 0.008 | 320 | 224 (70.0) | 66 | 41 (62.1) |
| Any gastrointestinal | 319 | 111 (34.8) | 0.009 | 326 | 114 (35.0) | 0.007 | 320 | 80 (25.0) | |||||
| Any psychiatric | 319 | 73 (22.9) | 0.551 | 326 | 58 (17.8) | 0.415 | 320 | 66 (20.6) | |||||
| Any general | 319 | 76 (23.8) | 0.141 | 326 | 62 (19.0) | 0.989 | 320 | 60 (18.8) | |||||
| Any central/peripheral nervous system | 319 | 77 (24.1) | 0.017 | 326 | 69 (21.2) | 0.134 | 320 | 52 (16.3) | |||||
| Any respiratory | 319 | 45 (14.1) | 0.896 | 326 | 41 (12.6) | 0.835 | 320 | 43 (13.4) | |||||
| Any metabolic/nutritional | 319 | 42 (13.2) | 0.536 | 326 | 43 (13.2) | 0.527 | 320 | 36 (11.3) | |||||
| Any urinary | 319 | 40 (12.5) | 0.892 | 326 | 39 (12.0) | 0.931 | 320 | 38 (11.9) | |||||
| Any secondary term | 319 | 28 (8.8) | 0.201 | 326 | 30 (9.2) | 0.271 | 320 | 39 (12.2) | |||||
| Anorexia | 319 | 19 (6.0) | 0.048 | 326 | 22 (6.7) | 0.017 | 64 | 7 (10.9) | 0.061 | 320 | 8 (2.5) | 66 | 1 (1.5) |
| Nausea | 319 | 54 (16.9) | 0.000 | 326 | 45 (13.8) | 0.000 | 64 | 15 (23.4) | 0.011 | 320 | 16 (5.0) | 66 | 4 (6.1) |
| Diarrhoea | 319 | 15 (4.7) | 0.314 | 326 | 22 (6.7) | 0.926 | 320 | 22 (6.9) | |||||
| Vomiting | 319 | 21 (6.6) | 0.012 | 326 | 28 (8.6) | 0.001 | 64 | 11 (17.2) | 0.017 | 320 | 7 (2.2) | 66 | 2 (3.0) |
| Agitation | 319 | 22 (6.9) | 0.992 | 326 | 20 (6.1) | 0.951 | 320 | 21 (6.6) | |||||
| Depression | 319 | 18 (5.6) | 0.070 | 326 | 16 (4.9) | 0.159 | 320 | 8 (2.5) | |||||
| Injury | 319 | 24 (7.5) | 0.419 | 326 | 12 (3.7) | 0.324 | 320 | 18 (5.6) | |||||
| Dizziness | 319 | 33 (10.3) | 0.006 | 326 | 24 (7.4) | 0.148 | 320 | 14 (4.4) | |||||
| Headache | 319 | 29 (9.1) | 0.127 | 326 | 18 (5.5) | 0.909 | 320 | 18 (5.6) | |||||
| Upper respiratory tract infection | 319 | 15 (4.7) | 0.993 | 326 | 12 (3.7) | 0.529 | 64 | 8 (12.5) | 0.090 | 320 | 16 (5.0) | 66 | 2 (3.0) |
| Weight decrease | 319 | 14 (4.4) | 0.031 | 326 | 17 (5.2) | 0.009 | 320 | 4 (1.3) | |||||
| Urinary tract infection | 319 | 22 (6.9) | 0.661 | 326 | 22 (6.7) | 0.605 | 320 | 26 (8.1) | |||||
| Fall | 319 | 20 (6.3) | 0.992 | 326 | 20 (6.1) | 0.952 | 320 | 19 (5.9) | |||||
Summary: galantamine versus placebo
We found an additional three RCTs96,101,131 to add to the five reported in the 2004 review. 2
Overall cognitive results from the new studies using ADAS-cog showed improvements for those taking galantamine. When these studies were pooled with the existing evidence the benefit remained and increased with time, with greater benefit seen at 21–26 weeks [WMD = –2.96 (95% CI –3.41 to –2.51), p < 0.001] than 12–16 weeks [WMD = –2.39 (95% CI –2.80 to –1.97), p < 0.001].
All of the new studies reported functional outcomes. Those measured by the DAD and ADCS-ADL scales generally showed significant improvement; those measured by the Goal Attainment Scale (GAS) were rather more ambiguous. Pooled results of the ADCS-ADL and the DAD at 21–26 weeks continued to show benefit from galantamine compared with placebo [WMD = 2.23 (95% CI 1.33 to 3.14), p < 0.001; WMD = 3.76 (95% CI 1.66 to 5.86), p < 0.001, respectively]. When data from both these outcome measures were pooled, results still favoured galantamine.
Behavioural outcomes from one new study, measured by the NPI, failed to show a benefit from galantamine. This lack of benefit was also seen from the pooled results at 13 weeks from follow-up. However, when the new data were pooled with those of the previous assessment2 a significant difference favouring galantamine was found at 21–26 weeks [WMD = –1.46 (95% CI –2.59 to –0.34), p = 0.012].
Two of the new studies measured global outcomes; one found that it produced a significant benefit on the CIBIC-plus. When these data were pooled with the data from the previous review, significant benefit was found on the CIBIC-plus at 26 weeks’ follow-up: WMD = –0.20 (95% CI –0.30 to –0.09), p < 0.001, with doses of ≤ 24 mg/day.
No QoL data were reported in either the new or the old studies for this comparison. The main AEs found were gastrointestinal.
The evidence from the new studies confirmed the benefits from galantamine for cognitive outcomes and, although there was some ambiguity with functional measures, the overall pooled estimates were favourable. Although none of the new studies showed significant benefit for behavioural outcomes, when these data were pooled with existing evidence, gains were shown at later follow-up times. Again, although the new trial data varied in its significance, pooling suggested that there were gains to be made on global outcomes. However, it should be noted that in all these studies estimates for missing data were calculated using LOCF or OC methods and may therefore have inflated the benefits from galantamine.
Graphical summary of galantamine versus placebo
In contrast with the donepezil research, one large study has been conducted since 2004 (comparing two different methods of delivery versus placebo), and the quality of the studies has improved overall (Figures 32 and 33). As in the previous review, the larger studies are more likely to show a benefit from galantamine on cognitive outcomes. The evidence for an effect on functional outcomes continues to be mixed: previously there appeared to be a relationship to the size of the dose – in the new study by Brodaty and colleagues96 effectiveness at 24 mg/day was linked to mode of delivery, with the prolonged-release capsule (PRC) showing a significant gain over placebo, which the regular capsule did not. Similar to the 2004 review,2 the results on global outcomes were mixed; thus it remains unclear what effect galantamine has on these outcomes.
FIGURE 32.
Summary of studies included in the 2004 review:2 galantamine vs placebo. ADCS-CGIC, Alzheimer’s Disease Cooperative Study – Clinical Global Impression of Change; Analy, analysis; be, behavioural; Blind, blinding; Char, characteristics of patients; cog, cognitive; F, female; func, functional; GDS, Global Deterioration Scale; glo, global ; M, male; Rand, randomisation, SIB, Severe Impairment Battery.
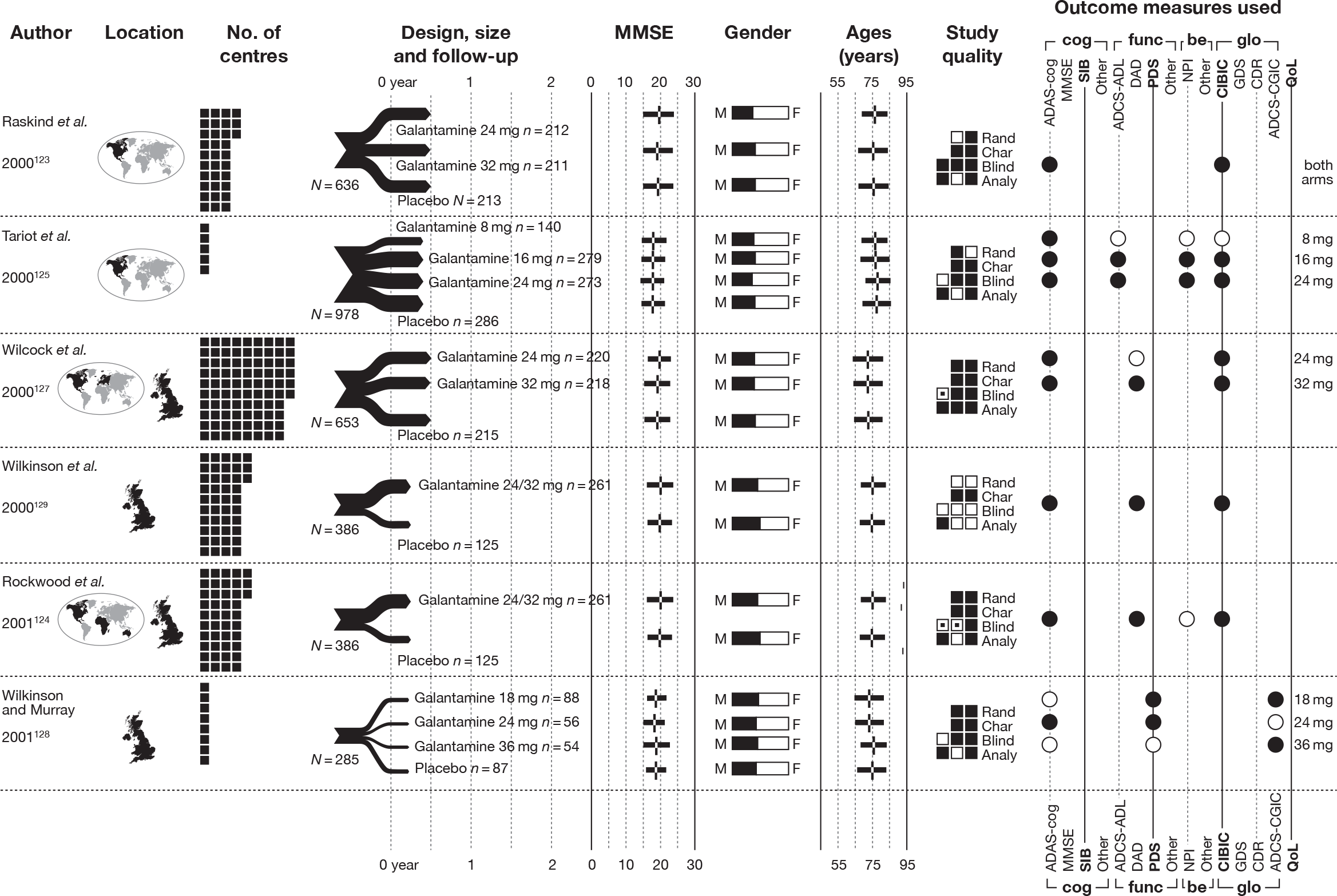
FIGURE 33.
Summary of new studies included in the 2010 review: galantamine vs placebo. a. Significant only for ’clinician-rated’ outcome measure, not for ’patient/caregiver rated’. ADCS-CGIC, Alzheimer’s Disease Cooperative Study – Clinical Global Impression of Change; Analy, analysis; be, behavioural; Blind, blinding; Char, characteristics of patients; cog, cognitive; F, female; func, functional; GDS, Global Deterioration Scale; glo, global; M, male; Rand, randomisation; SIB, Severe Impairment Battery.
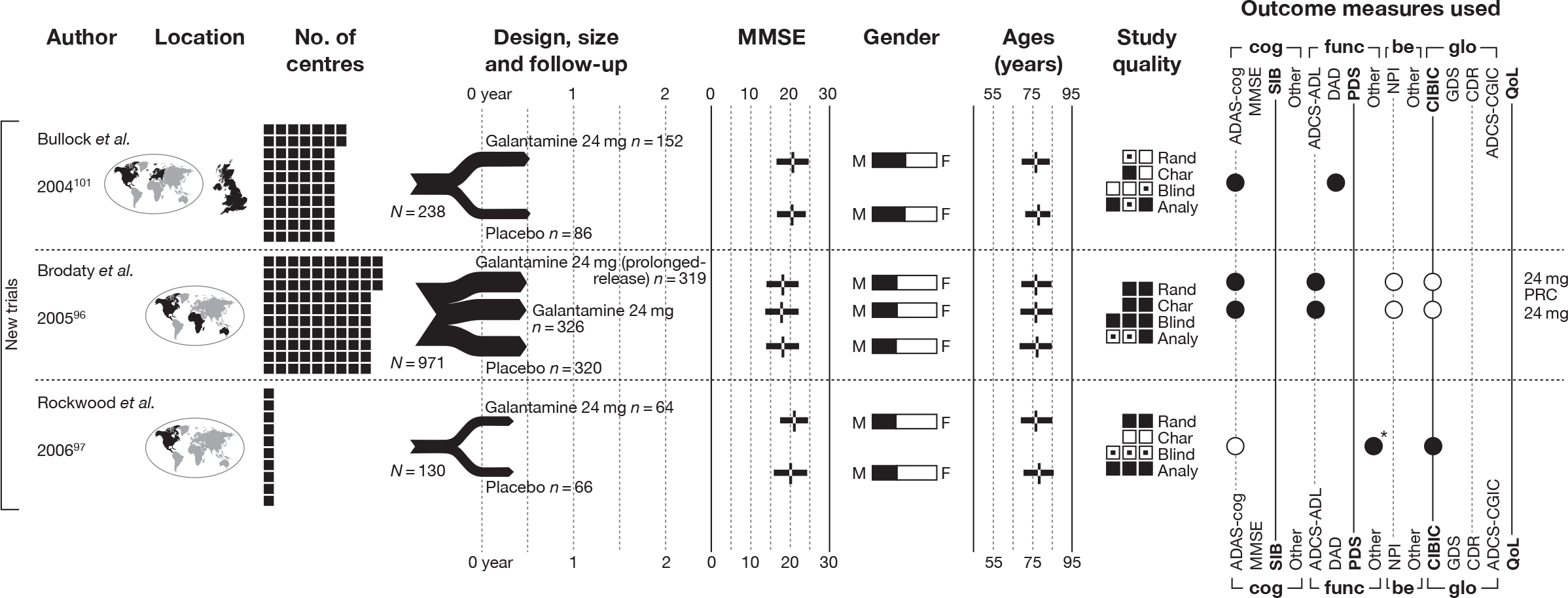
Rivastigmine versus placebo
Identified evidence
The 20042 review included four RCTs. 134–137
Our searches identified three additional relevant trials,138–140 details of which are tabulated in Table 16. Table 17 contains information about studies’ interventions, comparators and baseline characteristics and Table 18 gives key markers of internal validity for the included studies, which were moderately good to poor quality.
| Study details | Inclusion criteria | Exclusion criteria | Methodological notes | Other |
|---|---|---|---|---|
|
Feldman and Lane (2007) 138 Design: Parallel double-blind RCT Countries: Australia, Canada, Ireland, Italy, South Africa and the UK No. of centres: 37 No. randomised: 678 Maximum follow-up: 26 MMSE range included: 10–26 Funding: Commissioned by Novartis Pharma AG (Switzerland) |
AD (DSM-IV criteria) and probable AD (NINCDS-ADRDA) Responsible caregiver |
Severe and unstable cardiac disease Severe and obstructuive pulmonary disease Other life-threatening conditions Use of anticholoinergic drugs, health food supplements containing ACh precursors, putative memory enhancers or insulin Use of psychotropic drugs, with the exception of chloral hydrate, short-acting benzodiazepines and haloperidol ≤ 3 days in succession and not < 72 hours before any efficacy assessment) |
Sample attrition/dropout: 553 of 678 completed study. In total, 125 withdrew after allocation: AEs (n = 83); ECG abnormalities (n = 4); laboratory abnormalities (n = 1); withdrawn consent (n = 14); protocol violation (n = 8); treatment failure (n = 2); failure to attend (n = 7); other reasons (n = 6). Differences between groups was only on AEs (rivastigmine t.i.d. 11%; rivastigmine b.i.d. 17%; placebo 9%) Randomisation and allocation: Randomisation procedure not described. Rivastigmine and placebo tablets were identical and the number taken was the same at each dose in all groups Power calculation: The study sample size was determined on the basis of an estimated three-point difference between rivastigmine administered b.i.d. and placebo on the ADAS-cog, an estimated 0.4-point difference between b.i.d. and placebo on the CIBIC-plus and an increased proportion of responders with CIBIC-plus ratings of > 4 of 20% within the b.i.d. rivastigmine group (35% rivastigmine vs 15% placebo). Sample sizes of 192 per group were required. For practical reasons the sample size was chosen as 200 [(ITT) population]. An individual power of 90% guaranteed protection of the global power in view of the requirement that both ADAS-cog and CIBIC-plus analyses should be significant at the 0.0499 level |
Therapy common to all participants: None Study funding: Commissioned by Novartis Pharma AG (Switzerland) Other conflicts: HF has received honoraria for consulting, advisory boards and for participation in CME programmes sponsored by Novartis. HF has also received grant-in-aid funding for research from Novartis. RL is an employee of Novartis. The study was commissioned by Novartis Pharma AG in Switzerland |
|
Mowla et al. (2007) 139 Design: Parallel double-blind RCT Country: Not reported. Lead author based in the Islamic Republic of Iran No. of centres: Not reported No. randomised: 122 Maximum follow-up: 12 MMSE range included: 10–24 Funding: Shiraz University of Medical Sciences |
AD (DSM-IV criteria) Brief Cognitive Rating Score mean 3–5 Hachinski Ischaemic Score < 4 Adequate level of premorbid intelligence (IG > 80, global assessment) |
Dementia of other aetiology Severe organic disease (tumours, severe infectious disease, brain trauma, epilepsy, cerebrovascular malformations, alcohol or drug abuse) Other psychiatric disorders (Hamilton Depression Scale, 17-item version, total score < 10) |
Sample attrition/dropout: 98 of 122 completed study. Dropouts: rivastigmine arm n = 7; fluoxetine plus rivastigmine n = 9; placebo n = 8. Major cause of withdrawal in fluoxetine plus rivastigmine arm was AEs; in placebo arm it was loss of efficacy Randomisation and allocation: Computer-generated (on-site) randomisation – whether researchers were able to view randomisation sequence prior to allocation is not reported. Same number of pills for all trial arms, but appearance of these pills not reported (simply described as ‘similar’) Power calculation: Not reported |
Therapy common to all participants: Single-blind placebo 6-week run-in period to exclude placebo responders Study funding: Shiraz University of Medical Sciences Other conflicts: Not reported Notes: 12-week mean MMSE/WMS/ADL/HAM scores in the fluoxetine plus rivastigmine arm were much lower than in the other arms – potential error? |
|
Winblad et al. (2007) 140 Design: Parallel double-blind RCT Countries: Chile, Czech Republic, Denmark, Finland, Germany, Guatemala, Israel, Italy, Republic of Korea, Mexico, Norway, Peru, Poland, Portugal, Russian Federation, Slovak Republic, Sweden, Taiwan (Province of China), the USA, Uruguay and Venezuela No. of centres: 100 No. randomised: 1195 Maximum follow-up: 24 MMSE range included: 10–20 Funding: Novartis Pharma AG, Basel, Switzerland |
AD (DSM-IV criteria) and probable AD (NINCDS-ADRDA criteria) [brain scan (MRI or CT) used for establishing these criteria must have been done within 1-year prior to randomisation] Age 50–85 years Living with someone in the community or, if living alone, in daily contact with a responsible caregiver |
Advanced, severe, progressive, or unstable disease of any type that could interfere with study assessments or put the patient at special risk Any condition other than AD that could explain the dementia Use of any investigational drugs, new psychotropic or dopaminergic agents, cholinesterase inhibitors or anticholinergic agents during the 4 weeks prior to randomisation |
Sample attrition/dropout: 970 of 1195 patients completed study. Reasons for dropout: AEs, withdrawn consent, lost to follow-up, death and unsatisfactory therapeutic effect. No difference between groups Randomisation and allocation: Automated random assignment of treatment using an interactive voice-response system. Blocking was done on a study centre basis. All personnel directly involved in the conduct of the study remained unaware of the active treatment groups until all data had been retrieved and finalised for analysis Appearance of tablets, patches and placebo not reported Power calculation: In previous placebo-controlled trials of the rivastigmine capsule in patients with AD, a treatment difference to placebo in the ADAS-cog change from baseline of approximately 2.5 points was observed in the ITT analysis. In the current trial, a non-inferiority margin was predefined as 1.25 points on the ADAS-cog to preserve 50% of this effect, which was considered the smallest value that could represent a clinically meaningful difference. To determine the power of this study, the assumptions on delta (difference in means) and SD for the change in ADAS-cog and ADCS-CGIC from baseline were based on 24-week data from the rivastigmine capsule studies that used the ADAS-cog and CIBIC-plus. The ADCS-CGIC scale is comparable to the CIBIC-plus, which was used in previous rivastigmine capsule studies. To ensure that the study had adequate power, 1040 evaluable patients were needed. In order to reach an overall power of 80% for all of the first three hypotheses (which is defined as the product of the individual powers), the sample size was 260 patients per treatment group |
Therapy common to all participants: None reported Study funding: Novartis Pharma AG, Basel, Switzerland Other conflicts: Three co-authors (SZ, JN, RL) are employees of Novartis. Remaining authors were investigators (BW, NA, GG, MO, CS) and/or Study Publication Committee members (BW, JC, NA, GG, MO, SZ, JN, RL). BW, JC, NA, GG, MO and CS have provided consultation services to many pharmaceutical companies that develop dementia drugs, including Novartis. A writing committee prepared an initial draft of the manuscript, based on a report provided by Novartis, and all authors contributed to its finalisation through interactive review Data were collected by investigators and co-investigators, entered into a central database using electronic data-capture software and analysed by Novartis Pharma AG, which vouches for the data and the analysis |
| Study | Arm | Dose (mg/day) | Dosage details | N | Age, years (SD) | Gender, n male (%) | Race, n white (%) | Weight, kg (SD) | Education (years) | Duration of dementia, months (SD) | ADAS-cog, score (SD) | MMSE, score (SD) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Feldman and Lane (2007)138 | Rivastigmine t.i.d. | 2–12 | Dose administered three times a day. Titrated from an initial dose of 2 mg/day for the first week up to a maximum of 12 mg in 1 mg/day steps at weekly intervals. Patients unable to tolerate 2 mg/day by day 10 were withdrawn from the study. Tolerability could be optimised by maintaining a dose level for periods of up to 2 weeks | 227 | 71.4 (7.90) | 91 (40.1)a | 65.9 (12.9) | 38.4 (24.5) | 28.1 (12.5) | 18.3 (4.50) | ||
| Rivastigmine b.i.d. | 2–12 | Dose administered two times a day (plus one placebo tablet). Titrated from an initial dose of 2 mg/day for the first week up to a maximum of 12 mg in 1 mg/day steps at weekly intervals. Patients unable to tolerate 2 mg/day by day 10 were withdrawn from the study. Tolerability could be optimised by maintaining a dose level for periods of up to 2 weeks | 229 | 71.0 (8.20) | 98 (42.8)a | 66.7 (12.2) | 40.6 (31.2) | 27.7 (12.3)b | 18.8 (4.60) | |||
| Placebo | – | – | 222 | 71.7 (8.70) | 89 (40.1)a | 65.9 (12.3) | 39.7 (28.2) | 28.5 (12.3)c | 18.7 (4.60) | |||
| Mowla et al. (2007)139 | Rivastigmine | 3–12 |
Titrated from initial dose of 1.5 mg twice a day, doubled every 2 weeks until maximum dose of 6 mg twice a day reached (or dose which patient could tolerate) No details of placebo fluoxetine administration |
41 | 69.2d | 65 (53.3)e,f | 16.3 (4.10) | |||||
| Rivastigmine plus fluoxetine | 3–12 |
Titrated from initial dose of 1.5 mg twice a day, doubled every 2 weeks until maximum dose of 6 mg twice a day reached (or dose which patient could tolerate) Fluoxetine 20 mg/day |
41 | 69.2d | 15.6 (0.730) | |||||||
| Placebo | – | – | 40 | 69.2d | 16.5 (3.60) | |||||||
| Winblad et al. (2007)140 | Rivastigmine patch (9.5 mg/day) | 4.75–9.5 |
Titrated from initial 5-cm2 dose up to 9.5 mg/day patch in 5-cm2 step at 4-week intervals, followed by an 8-week maintenance phase Dose adjustments permitted to address perceived safety or tolerability issues Placebo capsules administered according to regimen for active capsule arm |
291 | 73.6 (7.90) | 93 (32.0) | 220 (75.6) | 9.90 (4.30) | 13.2 (16.8) | 27.0 (10.3)g | 16.6 (3.10) | |
| Rivastigmine patch (17.4 mg/day) | 4.75–17.4 |
Titrated from initial 5-cm2 dose up to 17.4 mg/day patch in 5-cm2 steps at 4-week intervals, followed by an 8-week maintenance phase Dose adjustments permitted to address perceived safety or tolerability issues Placebo capsules administered according to regimen for active capsule arm |
303 | 74.2 (7.70) | 103 (34.1)f | 227 (74.9) | 9.90 (4.40) | 13.2 (16.8) | 27.4 (9.70)h | 16.6 (2.90) | ||
| Rivastigmine capsules | 3–12 |
Initial dosage of 3 mg/day titrated upwards in steps of 3 mg/day up to a maximum of 12 mg/day Dose adjustments permitted to address perceived safety or tolerability issues Placebo patch |
294 | 72.8 (8.20) | 101 (34.4) | 219 (74.5) | 9.90 (4.40) | 13.2 (16.8) | 27.9 (9.40)i | 16.4 (3.10) | ||
| Placebo | – | Placebo capsules plus placebo patch | 302 | 73.9 (7.30) | 101 (33.4) | 227 (75.2) | 9.90 (4.30) | 13.2 (16.8) | 28.6 (9.90)j | 16.4 (3.00) |
| Study | Was the assignment to the treatment groups really random? | Was the treatment allocation concealed? | Were the groups similar at baseline in terms of prognostic factors? | Were the eligibility criteria specified? | Were outcome assessors blinded to the treatment allocation? | Was the care provider blinded? | Was the patient blinded? | Were the point estimates and measure of variability presented for the primary outcome measure? | Did the analyses include an ITT analysis? | Were withdrawals and dropouts completely described? |
|---|---|---|---|---|---|---|---|---|---|---|
| Feldman and Lane (2007)138 | Unknown | Unknown | Reported – yes | Unknown | Unknown | Adequate | Adequate | Adequate | Adequate | Adequate |
| Mowla et al. (2007)139 | Partial | Adequate | Reported – yes | Unknown | Partial | Partial | Partial | Adequate | Inadequate | Adequate |
| Winblad et al. (2007)140 | Adequate | Adequate | Reported – yes | Adequate | Adequate | Partial | Partial | Adequate | Adequate | Adequate |
Evidence of clinical effectiveness
Cognition
The 2004 assessment report by Loveman and colleagues2 summarised the evidence they found on cognitive outcomes as:
Statistically significant differences between the 6–12 mg/day treatment groups and placebo were reported by two of three published trials which reported ADAS-cog and MMSE. No statistically significant effects were seen in the low-dose treatment groups in these studies. However, sample sizes were very low (< 30 participants in each group) and this study presented no information on power calculations.
The unpublished studies both found statistically better mean changes from ADAS-cog baseline scores in participants taking rivastigmine compared with placebo groups, a statistically significantly higher percentage of participants receiving t.i.d. (× 3 daily). Rivastigmine showed an improvement of at least four points compared with the placebo group, but there was no statistically significant difference between the b.i.d. (× 2 daily) group and placebo. One of the studies also reported on ADAS-cogA, and found statistically significant differences in both mean change from baseline and percentage of improvers for both b.i.d. and t.i.d. treatment groups compared with placebo.
Both unpublished studies reported MMSE as an outcome measure and found a statistically significant improvement in participants receiving rivastigmine compared with those receiving a placebo, with the exception of the 9 mg/day rivastigmine group.
In the three studies138–140 we found that had been published since 2004, comparing rivastigmine with placebo for mild-to-moderate AD, all studies reported benefits for the treatment group on cognitive outcome measures. These results appear to be dose dependent (as in the previous report), with doses ≥ 12 mg/day showing a greater effect (see Appendix 5). However, only one study measured missing outcomes with an ITT population. 138 Table 19 shows the summary results for cognition in the included studies. It should be noted that in the study by Winblad and colleagues140 only the 10-cm patch is currently licensed in the UK.
| Study | Subgroup | Outcome | Type | Arm | Rivastigmine | Placebo | p-value | ||
|---|---|---|---|---|---|---|---|---|---|
| n 1 | Mean 1 (SD) | n 2 | Mean 2 (SD) | ||||||
| Feldman and Lane (2007)138 | ITT population | ADAS-cog – 12 weeks | MC | b.i.d. | 227 | –1.9 (6.66)a | 220 | 0.9 (5.93)a | < 0.001b |
| t.i.d. | 228 | –0.8 (6.04)a | < 0.05b | ||||||
| ADAS-cog – 18 weeks | MC | b.i.d. | 227 | –1.6 (6.66)a | 220 | 1.8 (6.67)a | < 0.001c | ||
| t.i.d. | 228 | –0.1 (6.79)a | < 0.001c | ||||||
| ADAS-cog – 26 weeks | MC | b.i.d. | 227 | –0.2 (7.3) | 220 | 2.8 (7.2) | ≤ 0.001c | ||
| t.i.d. | 228 | 1.2 (7.2) | < 0.05c | ||||||
| ADAS-cog: any improvement – 12 weeks | D | b.i.d. | 227 | 68 (30.0%)a | 220 | 36 (16.4%)a | ≤ 0.001d | ||
| t.i.d. | 228 | 52 (22.8%)a | < 0.05d | ||||||
| ADAS-cog: any improvement – 18 weeks | D | b.i.d. | 227 | 75 (33.0%)a | 220 | 28 (12.7%)a | ≤ 0.001d | ||
| t.i.d. | 228 | 57 (25.0%)a | ≤ 0.001d | ||||||
| ADAS-cog: any improvement – 26 weeks | D | b.i.d. | 227 | 52 (22.9%)a | 220 | 28 (12.7%)a | NSd | ||
| t.i.d. | 228 | 41 (18.0%)a | < 0.05d | ||||||
| ADAS-cogA – 26 weeks | MC | b.i.d. | 227 | –0.1 (7.9) | 220 | 3.2 (7.8) | ≤ 0.001c | ||
| t.i.d. | 228 | 1.5 (7.8) | < 0.05c | ||||||
| MMSE – 26 weeks | MC | b.i.d. | 227 | 0.3 (3.6) | 220 | –1.4 (3.6) | ≤ 0.001b | ||
| t.i.d. | 227 | –0.6 (3.6) | < 0.05b | ||||||
| LOCF analysis | ADAS-cog – 26 weeks | MC | b.i.d. | 209 | –0.7 (6.9) | 208 | 2.7 (6.8) | ≤ 0.001c | |
| t.i.d. | 199 | 0.8 (6.9) | < 0.05c | ||||||
| ADAS-cogA – 26 weeks | MC | b.i.d. | 209 | –0.6 (7.5) | 208 | 3.1 (7.4) | ≤ 0.001c | ||
| t.i.d. | 199 | 1 (7.5) | < 0.05c | ||||||
| MMSE – 26 weeks | MC | b.i.d. | 193 | 0.4 (3.4) | 198 | –1.4 (3.5) | ≤ 0.001b | ||
| t.i.d. | 186 | –0.4 (3.5) | < 0.05b | ||||||
| OC population | ADAS-cog – 26 weeks | MC | b.i.d. | 180 | –0.9 (6.8) | 183 | 2.1 (6.8) | ≤ 0.001c | |
| t.i.d. | 173 | 0.9 (7) | NSc | ||||||
| Mowla et al. (2007)139 | OC populationf | MMSE – 12 weeks | A | 34 | 17.4 (3.7) | 32 | 16 (3.7) | 0.129f | |
| MC | 34 | 1.1 (1.4) | 32 | –0.5 (0.5) | < 0.001g | ||||
| WMS-III – 12 weeks | A | 34 | 8.7 (2.2) | 32 | 7.5 (1.4) | 0.011f | |||
| MC | 34 | 0.97 (1.7) | 32 | –0.66 (1.1) | < 0.001g | ||||
| CGIC-item 2 (cognitive) – 12 weeks | A | 34 | 3.1 (0.96) | 32 | 3.7 (0.67) | 0.005f | |||
| Winblad et al. (2007)140 | LOCF analysis | ADAS-cog – 16 weeks | MC | 10h | 248 | –0.825 (6.3)a | 281 | 0 (6.71)a | 0.09i |
| 20j | 262 | –1.39 (6.47)a | < 0.05i | ||||||
| Oral | 253 | –0.5 (6.36)a | NSi | ||||||
| ADAS-cog – 24 weeks | MC | 10k | 248 | –0.6 (6.4) | 281 | 1 (6.8) | 0.005i | ||
| 20j | 262 | –1.6 (6.5) | < 0.001i | ||||||
| Oral | 253 | –0.6 (6.2) | 0.003i | ||||||
| MMSE – 24 weeks | MC | 10i | 250 | 1.1 (3.3) | 281 | 0 (3.5) | 0.002k | ||
| 20j | 262 | 0.9 (3.4) | < 0.001k | ||||||
| Oral | 256 | 0.8 (3.2) | 0.002k | ||||||
| Ten-point clock-drawing test – 24 weeks | MC | 10i | 245 | 0.3 (3.4) | 269 | –0.1 (3.2) | 0.08k | ||
| 20j | 251 | 0.1 (3.1) | 0.08k | ||||||
| Oral | 246 | 0.2 (2.9) | 0.15k | ||||||
| TMT – 24 weeks | MC | 10i | 241 | –12.3 (55.1) | 258 | 7.7 (56.6) | < 0.001i | ||
| 20j | 238 | –6.5 (55.9) | 0.005i | ||||||
| Oral | 240 | –9.8 (66.1) | < 0.001i | ||||||
The data from the new trials were synthesised with those of the previous assessment using random-effects meta-analysis. The measures of ADAS-cog and MMSE were first considered separately and then combined in a pooled multiple outcome measure analysis at ≥ 12 mg/day. We also meta-analysed the data by ≤ 10 mg/day, 4 mg/day and combined doses; results can be found in Appendix 5.
The meta-analyses of ADAS-cog scores at 24–26 weeks showed a significant benefit from rivastigmine (≥ 12 mg/day) compared with placebo: WMD = –2.46 (95% CI –3.37 to –1.56), p < 0.001 (Figure 34).
FIGURE 34.
Random-effects meta-analysis: ADAS-cog at 24–26 weeks (mean change from baseline) – rivastigmine (≥ 12 mg/day) vs placebo. a, b.i.d. (twice a day) and t.i.d. (three times a day) arms pooled; b, the 17.4 mg/day patch and 12 mg/day capsules arms pooled.
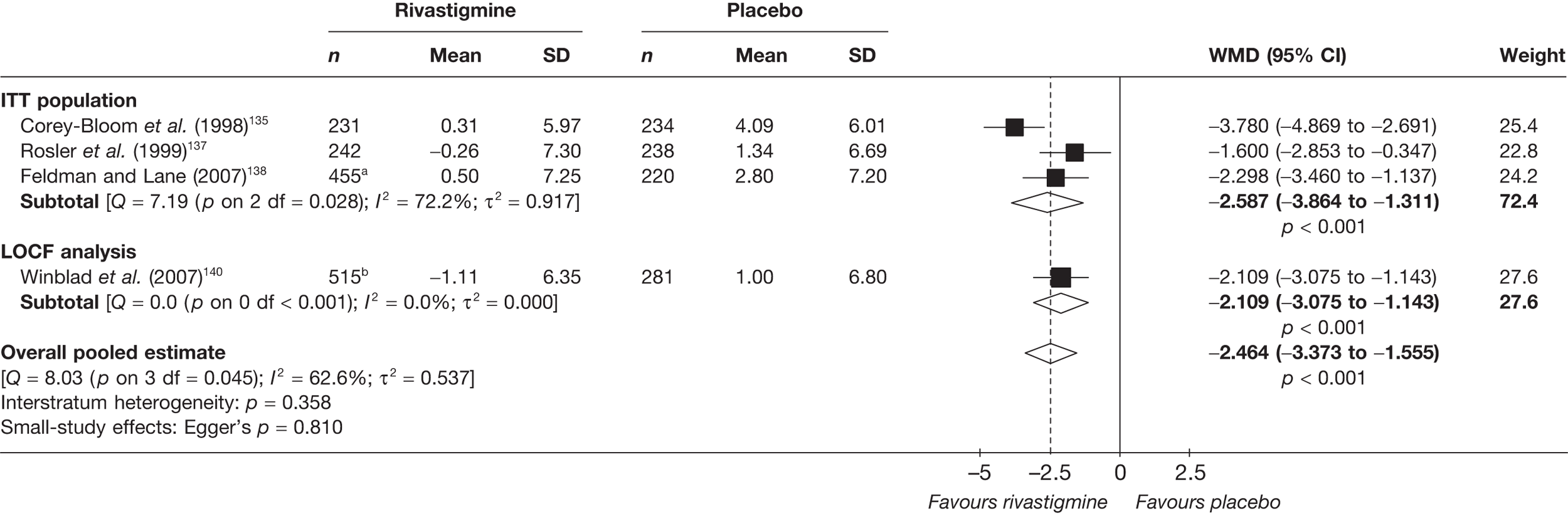
At 24–26 weeks’ follow-up the pooled estimate of effect showed a benefit from rivastigmine: WMD = 1.02 (95% CI 0.63 to 1.41), p < 0.001 (Figure 35).
FIGURE 35.
Random-effects meta-analysis: MMSE at 24–26 weeks (mean change from baseline) – rivastigmine (≥ 12 mg/day) vs placebo. a, b.i.d. (twice a day) and t.i.d. (three times a day) arms pooled; b, the 20-cm2 patch and 12 mg/day capsule arms pooled.
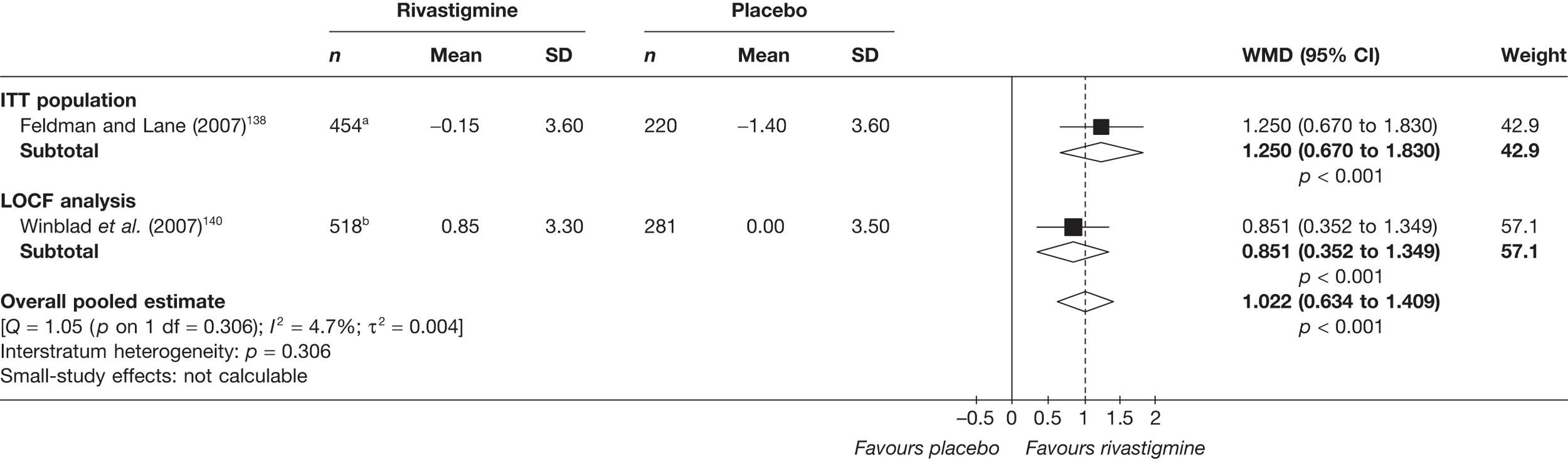
When we pooled all the results for the cognitive outcomes from the new and existing studies, we found that the overall pooled estimate showed a significant benefit from rivastigmine compared with placebo: SMD = 0.28 (95% CI 0.14 to 0.42), p < 0.001 (Figure 36). The data set used in this meta-analysis can be found in Appendix 6.
FIGURE 36.
Random-effects meta-analysis – cognitive outcomes (SMD) at 24–26 weeks: rivastigmine (all dosages) vs placebo.
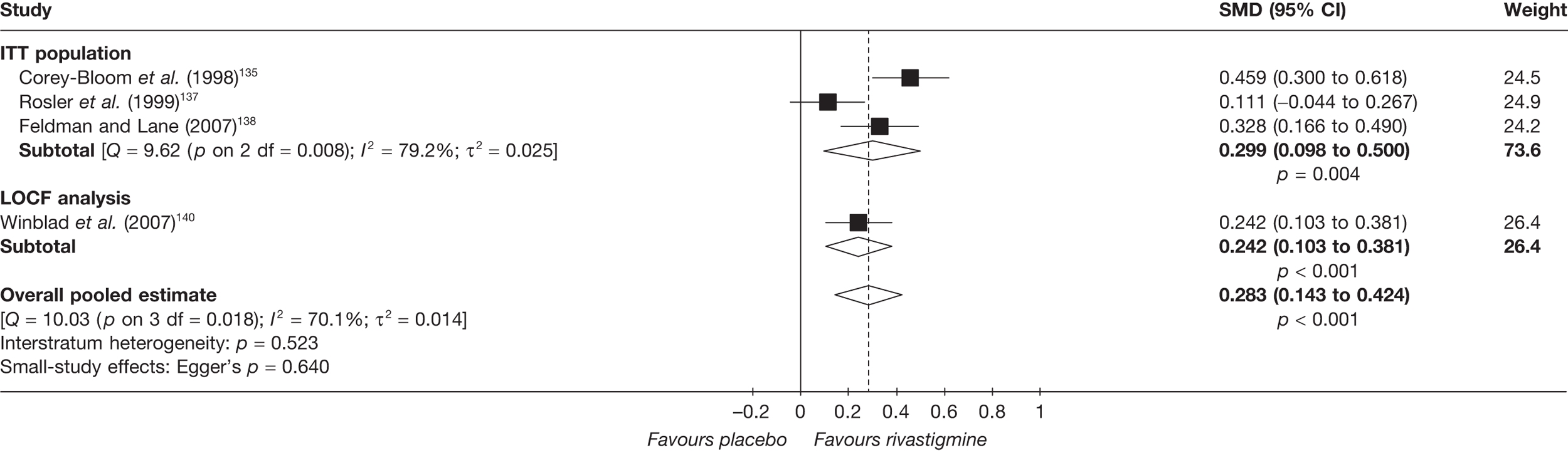
With only four data points in this evidence base, it would not be informative to perform metaregression.
Functional
In 2004 Loveman and colleagues2 reported that:
Two published studies reported the PDS as a functional outcome measure. One of these found a statistically significant improvement in participants treated with 6–12 mg/day rivastigmine compared with placebo, and the other reported that a statistically significantly higher percentage of these high dose participants than placebo participants showed an improvement of at least 10%.
Two of the three new studies found since 2004 reported significant functional benefit from rivastigmine compared with placebo. These used PDS and ADCS-ADL as their outcome measures. A summary table of results can be found below in Table 20.
| Study | Subgroup | Outcome | Type | Arm | Rivastigmine | Placebo | p-value | ||
|---|---|---|---|---|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | ||||||
| Feldman and Lane (2007)138 | ITT population | PDS – 26 weeks | MC | b.i.d. | 227 | –2.6 (11.1) | 221 | –4.9 (11.2) | ≤ 0.001a |
| t.i.d. | 225 | –1.5 (11.2) | 221 | –4.9 (11.2) | < 0.05a | ||||
| LOCF analysis | PDS – 26 weeks | MC | b.i.d. | 207 | –1 (11.4) | 209 | –4.7 (11.3) | ≤ 0.001a | |
| t.i.d. | 195 | –2.3 (11.5) | 209 | –4.7 (11.3) | < 0.05a | ||||
| Mowla et al. (2007)139 | OC population | ADL – 12 weeks | A | 34 | 25.3 (6.6) | 32 | 27.1 (6.9) | 0.283b | |
| MC | 34 | 1.2 (2.6) | 32 | –0.68 (1.3) | 0.58c | ||||
| Winblad et al. (2007)140 | LOCF analysis | ADCS-ADL – 16 weeks | MC | 10d | 247 | –0.6 (9.43)e | 281 | –1.6 (7.96)f | NSg |
| 20g | 263 | 0.4 (9.73)e | 281 | –1.6 (7.96)d | < 0.05e | ||||
| Oral | 254 | –0.4 (7.97)e | 281 | –1.6 (7.96)e | NSf | ||||
| ADCS-ADL – 24 weeks | MC | 10e | 247 | –0.1 (9.1) | 281 | –2.3 (9.4) | 0.01g | ||
| 20h | 263 | 0 (11.6) | 281 | –2.3 (9.4) | 0.02g | ||||
| Oral | 254 | –0.5 (9.5) | 281 | –2.3 (9.4) | 0.04g | ||||
Data from the existing evidence were synthesised with the new data in a meta-analysis of the PDS scores.
The overall pooled estimate at 24–26 weeks showed a significant benefit from rivastigmine: WMD = 3.10 (95% CI 1.81 to 4.40), p = 0.001 (Figure 37).
FIGURE 37.
Random-effects meta-analysis – PDS at 24–26 weeks (mean change from baseline): rivastigmine (12 mg/day) vs placebo. a,b.i.d. (twice a day) and t.i.d. (three times a day) arms pooled.
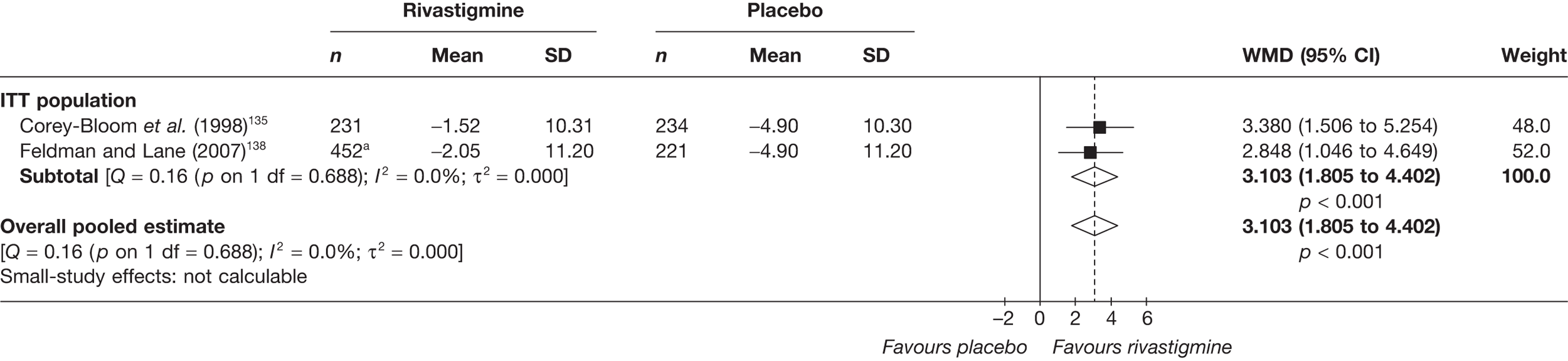
Two new studies were found to add to this combined meta-analysis of functional outcomes at 24–26 weeks. Again, the overall pooled estimate showed a benefit from rivastigmine compared with placebo: SMD = 0.21 (95% CI 0.12 to 0.29), p < 0.001 (Figure 38). The data set used in this meta-analysis can be found in Appendix 6.
FIGURE 38.
Random-effects meta-analysis: functional outcomes (SMD) at 24–26 weeks: rivastigmine (all dosages) vs placebo.
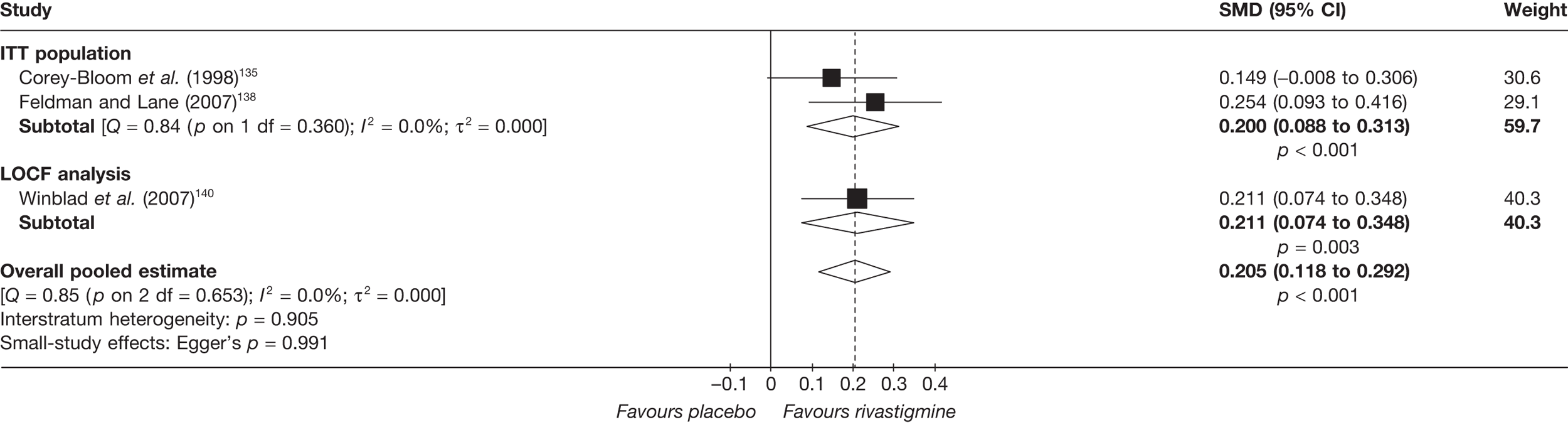
With only three data points in this evidence base, it would not be informative to perform metaregression.
Behavioural and mood
The 2004 systematic review2 summarised the behavioural results as:
On measures of behaviour and mood no statistically significant benefit was demonstrated in the rivastigmine treated groups compared to the placebo groups.
Two new studies139,140 were found that measured behavioural outcomes. One small study by Mowla and colleagues139 found a significant benefit from rivastigmine; the other much larger study did not. 140 Table 21 below shows the summary outcome data.
| Study | Subgroup | Outcome | Type | Arm | Rivastigmine | Placebo | p-value | ||
|---|---|---|---|---|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | ||||||
| Mowla et al. (2007)139 | OC population | Hamilton Depression Scale – 12 weeks | A | 34 | 6.26 (2.9) | 32 | 8.33 (1.12) | < 0.001a | |
| Winblad et al. (2007)140 | LOCF analysis | NPI – 24 weeks | MC | 10b | 248 | –1.7 (11.5) | 281 | –1.7 (13.8) | 0.74c |
| NPI – 24 weeks | MC | 20d | 263 | –2.3 (13.3) | 281 | –1.7 (13.8) | 0.69c | ||
| NPI – 24 weeks | MC | Oral | 253 | –2.2 (11.9) | 281 | –1.7 (13.8) | 0.51c | ||
| NPI – caregiver distress – 24 weeks | MC | 10b | 248 | –1 (5.5) | 281 | –1.1 (6.3) | 0.37c | ||
| NPI – caregiver distress – 24 weeks | MC | 20d | 263 | –1.1 (6.4) | 281 | –1.1 (6.3) | 0.98c | ||
| NPI – caregiver distress – 24 weeks | MC | Oral | 253 | –1.1 (6.6) | 281 | –1.1 (6.3) | 0.12c | ||
Synthesis with existing evidence base
The data identified by this review and the 2004 review2 are sparse and too heterogeneous to permit meaningful quantitative synthesis.
Global effect
The evidence from the 2004 assessment2 was summarised thus:
Both of the published studies which included CIBIC-plus as a global outcome measure reported a statistically significant improvement in high dose participants (6–12 mg/day) compared with placebo participants. One study also reported a statistically significantly greater proportion of ‘responders’ among participants treated with rivastigmine compared against placebo participants. Another study reported that a greater proportion of high dose rivastigmine participants than placebo participants had a ‘successful’ CIGIC assessment, i.e. scoring one or two on the scale. Two trials found a statistically significant improvement on the Global Deterioration Scale (GDS) measure in participants treated with 6–12 mg/day of rivastigmine compared with placebo participants.
The two new studies138,140 in this comparison that reported global outcomes had conflicting results. Feldman and Lane138 found mostly significantly favourable results with the CIBIC-plus and the Global Deterioration Scale (GDS), whereas Winblad and colleagues’ results were mostly non-significant140 (Table 22).
| Study | Subgroup | Outcome | Type | Arm | Rivastigmine | Placebo | p-value | ||
|---|---|---|---|---|---|---|---|---|---|
| N | Mean, n (%) | N | Mean, n (%) | ||||||
| Feldman and Lane (2007)138 | ITT population | CIBIC-plus score – 18 weeks | A | b.i.d. | 215 | 4.1 (SD 1.03)a | 213 | 4.5 (SD 1.02)a | ≤ 0.001b |
| t.i.d. | 220 | 3.9 (SD 1.04)a | ≤ 0.001b | ||||||
| CIBIC-plus score – 26 weeks | A | b.i.d. | 222 | 4.1 (SD 1.3) | 216 | 4.5 (SD 1.3) | ≤ 0.001c | ||
| t.i.d. | 222 | 3.9 (SD 1.3) | < 0.05c | ||||||
| CIBIC-plus: any improvement – 12 weeks | D | b.i.d. | 220 | 66 (30.0)a | 213 | 34 (16.0)a | ≤ 0.001d | ||
| t.i.d. | 215 | 62 (28.8)a | < 0.05d | ||||||
| CIBIC-plus: any improvement – 18 weeks | D | b.i.d. | 220 | 68 (30.9)a | 213 | 40 (18.8)a | ≤ 0.001d | ||
| t.i.d. | 215 | 47 (21.9)a | NSd | ||||||
| CIBIC-plus: any improvement – 26 weeks | D | b.i.d. | 220 | 68 (30.9)a | 213 | 40 (18.8)a | < 0.05d | ||
| t.i.d. | 215 | 49 (22.8)a | NSd | ||||||
| GDS – 26 weeks | MC | b.i.d. | 227 | 0 (SD 0.7) | 222 | –0.3 (SD 0.7) | < 0.05b | ||
| t.i.d. | 229 | –0.2 (SD 0.7) | NSb | ||||||
| LOCF analysis | CIBIC-plus score – 26 weeks | A | b.i.d. | 206 | 3.9 (SD 1.2) | 205 | 4.5 (SD 1.2) | ≤ 0.001c | |
| t.i.d. | 198 | 4.1 (SD 1.2) | < 0.05c | ||||||
| GDS – 26 weeks | MC | b.i.d. | 195 | 0 (SD 0.7) | 202 | –0.3 (SD 0.7) | < 0.05b | ||
| t.i.d. | 188 | –0.1 (SD 0.7) | NSb | ||||||
| OC population | CIBIC-plus score – 26 weeks | A | b.i.d. | 177 | 3.9 (SD 1.2) | 179 | 4.4 (SD 1.2) | ≤ 0.001c | |
| t.i.d. | 167 | 4.1 (SD 1.2) | < 0.05c | ||||||
| Winblad et al. (2007)140 | LOCF analysis | ADCS-CGIC: score – 16 weeks | A | 10e | 248 | 3.9 (SD 1.14)f | 278 | 4.35 (SD 1.25)f | NSg |
| 20h | 260 | 3.93 (SD 1.17)f | NSg | ||||||
| Oral | 253 | 4.25 (SD 1.11)f | NSg | ||||||
| ADCS-CGIC: score – 24 weeks | A | 10e | 248 | 3.9 (SD 1.2) | 278 | 4.2 (SD 1.3) | 0.01g | ||
| 20h | 260 | 4 (SD 1.3) | 0.054g | ||||||
| Oral | 253 | 3.9 (SD 1.3) | 0.009g | ||||||
| ADCS-CGIC: markedly improved – 24 weeks | D | 10e | 248 | 5 (2.0) | 278 | 2 (0.7) | 0.361i | ||
| 20h | 260 | 5 (1.9) | 0.395i | ||||||
| Oral | 253 | 3 (1.2) | 0.916i | ||||||
| ADCS-CGIC: moderately improved – 24 weeks | D | 10e | 248 | 29 (11.7) | 278 | 26 (9.4) | 0.463i | ||
| 20h | 260 | 32 (12.3) | 0.334i | ||||||
| Oral | 253 | 29 (11.5) | 0.513i | ||||||
| ADCS-CGIC: minimally improved – 24 weeks | D | 10e | 248 | 43 (17.3) | 278 | 50 (18.0) | 0.937i | ||
| 20h | 260 | 48 (18.5) | 0.975i | ||||||
| Oral | 253 | 60 (23.7) | 0.129i | ||||||
| ADCS-CGIC: unchanged – 24 weeks | D | 10e | 248 | 105 (42.3) | 278 | 91 (32.7) | 0.029i | ||
| 20h | 260 | 94 (36.2) | 0.457i | ||||||
| Oral | 253 | 96 (37.9) | 0.244i | ||||||
| ADCS-CGIC: minimally worse – 24 weeks | D | 10e | 248 | 41 (16.5) | 278 | 65 (23.4) | 0.065i | ||
| 20h | 260 | 50 (19.2) | 0.285i | ||||||
| Oral | 253 | 30 (11.9) | < 0.001i | ||||||
| ADCS-CGIC: moderately worse – 24 weeks | D | 10e | 248 | 22 (8.9) | 278 | 36 (12.9) | 0.177i | ||
| 20h | 260 | 27 (10.4) | 0.429i | ||||||
| Oral | 253 | 30 (11.9) | 0.803i | ||||||
| ADCS-CGIC: markedly worse – 24 weeks | D | 10e | 248 | 3 (1.2) | 278 | 8 (2.9) | 0.303i | ||
| 20h | 260 | 4 (1.5) | 0.448i | ||||||
| Oral | 253 | 5 (2.0) | 0.696i | ||||||
Data from the new studies were pooled with the existing evidence in random-effects meta-analyses using the CIBIC-plus at 26 weeks and the GDS at 26 weeks. The results can be seen in Figures 39 and 40.
FIGURE 39.
Random-effects meta-analysis – CIBIC-plus at 26 weeks: rivastigmine (12 mg/day) vs placebo. a, b.i.d. (twice a day) and t.i.d. (three times a day) arms pooled.
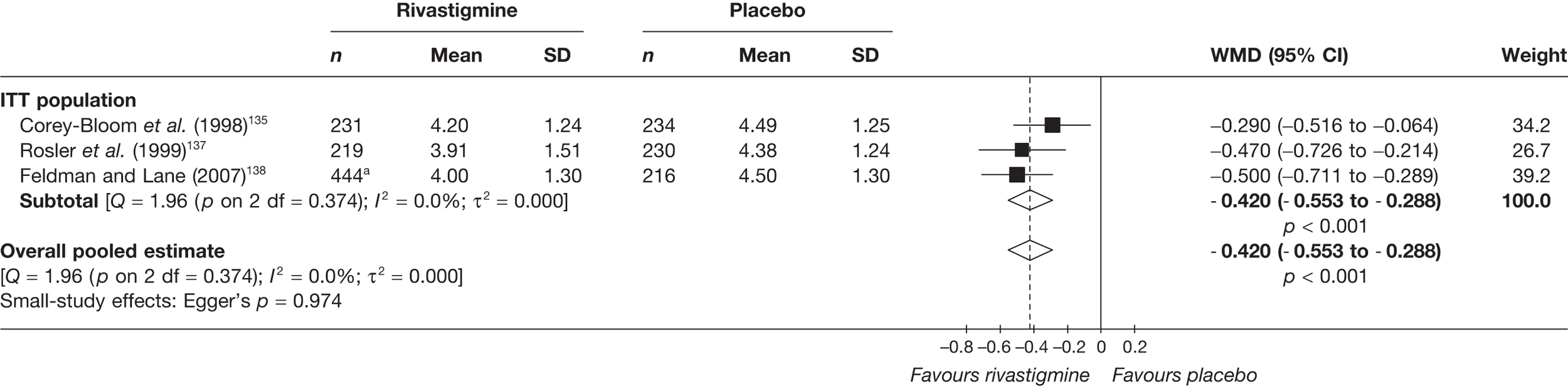
FIGURE 40.
Random-effects meta-analysis – GDS at 26 weeks (mean change from baseline): rivastigmine (12 mg/day) vs placebo. a, b.i.d. (twice a day) and t.i.d. (three times a day) arms pooled.
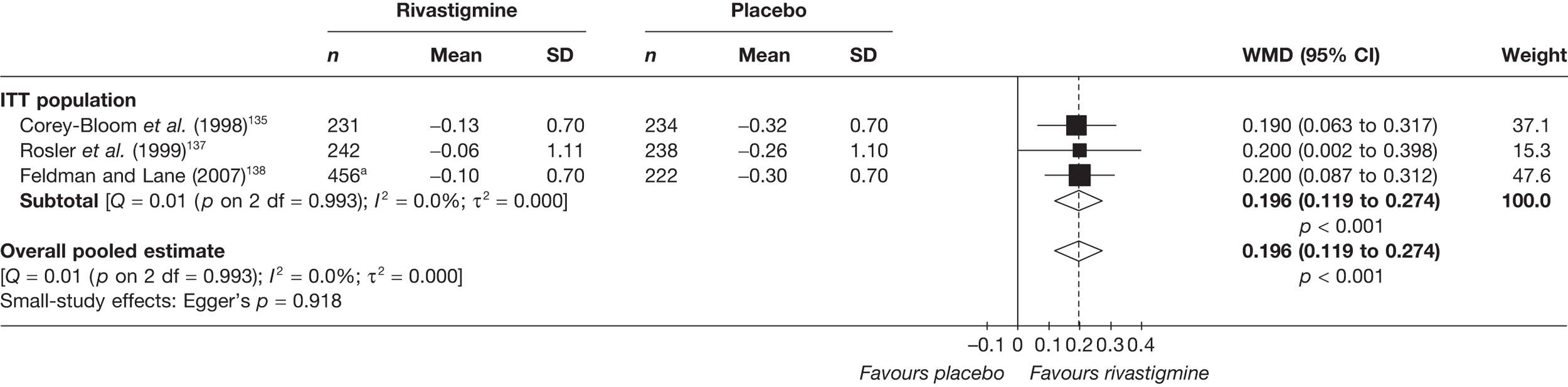
The meta-analysis showed a significant benefit from rivastigmine at 26 weeks: WMD = –0.42 (95% CI –0.55 to –0.29), p < 0.001.
This meta-analysis also showed a significant benefit from rivastigmine at 26 weeks: WMD = 0.20 (95% CI 0.12 to 0.27), p < 0.001.
We then pooled the results from both outcomes; the results from this can be seen in Figure 41 and showed an overall pooled estimate of SMD = 0.23 (95% CI 0.16 to 0.31), p < 0.001. The data set that was used in this meta-analysis can be found in Appendix 6.
FIGURE 41.
Random-effects meta-analysis – global outcomes (SMD) at 24–26 weeks: rivastigmine (all dosages) vs placebo.
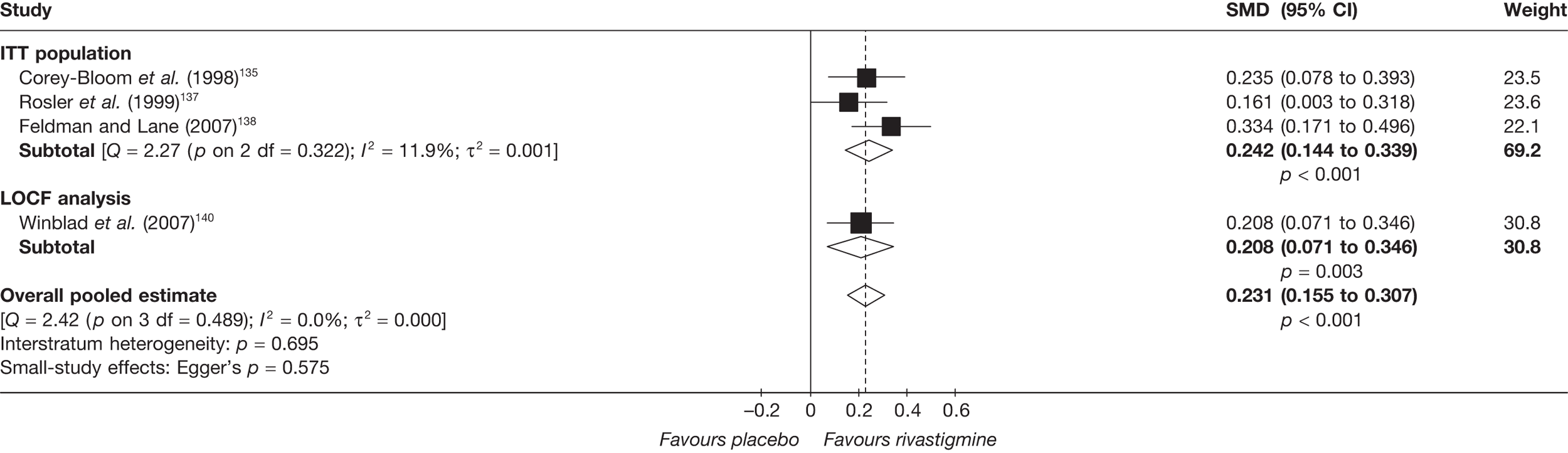
With only four data points in this evidence base, it would not be informative to perform metaregression.
Quality of life
None of the included studies provided any randomised evidence on QoL with rivastigmine compared with placebo, and no such data were identified in the 2004 review. 2
Safety
Overall, there was a high percentage of any AEs, ranging from 51% to 91% in the treatment groups and from 46% to 76% in control groups. The main AEs were gastrointestinal: the lower dose (9.5 mg/day) transdermal patch produced fewer side effects than the capsule (12 mg/day). A summary of all the AEs reported can be found in Table 23.
| AE | Rivastigmine | Placebo | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| t.i.d. (≤ 12 mg/day) | b.i.d. (≤ 12 mg/day) | Patch | |||||||||||||||||
| 9.5 mg/day | 17.4 mg/day | ||||||||||||||||||
| Feldman and Lane (2007)138 | Feldman and Lane (2007)138 | Winblad et al. (2007)140 | Winblad et al. (2007)140 | Winblad et al. (2007)140 | Feldman and Lane (2007)138 | Winblad et al. (2007)140 | |||||||||||||
| N | n (%) | p-valuea | N | n (%) | p-value | N | n (%) | p-value | N | n (%) | p-value | N | n (%) | p-value | N | n (%) | N | n (%) | |
| Any AE | 227 | 208 (91.6) | < 0.05b | 228 | 208 (91.2) | < 0.05b | 294 | 186 (63.3) | ≤ 0.001c | 291 | 147 (50.5) | NSc | 303 | 200 (66.0) | ≤ 0.001c | 222 | 169 (76.1) | 302 | 139 (46.0) |
| Any serious AE | 227 | 40 (17.6) | NSb | 228 | 40 (17.5) | NSb | 222 | 33 (14.9) | |||||||||||
| Anorexia | 227 | 42 (18.5) | < 0.05b | 228 | 47 (20.6) | < 0.05b | 222 | 6 (2.7) | |||||||||||
| Nausea | 227 | 109 (48.0) | < 0.05b | 228 | 123 (53.9) | < 0.05b | 294 | 68 (23.1) | ≤ 0.001c | 291 | 21 (7.2) | NSc | 303 | 64 (21.1) | ≤ 0.001c | 222 | 31 (14.0) | 302 | 15 (5.0) |
| Diarrhoea | 227 | 38 (16.7) | < 0.05b | 228 | 40 (17.5) | < 0.05b | 294 | 16 (5.4) | NSc | 291 | 18 (6.2) | NSc | 303 | 31 (10.2) | ≤ 0.001c | 222 | 20 (9.0) | 302 | 10 (3.3) |
| Vomiting | 227 | 68 (30.0) | < 0.05b | 228 | 88 (38.6) | < 0.05b | 294 | 50 (17.0) | ≤ 0.001c | 291 | 18 (6.2) | NSc | 303 | 57 (18.8) | ≤ 0.001c | 222 | 14 (6.3) | 302 | 10 (3.3) |
| Abdominal pain | 227 | 26 (11.5) | < 0.05b | 228 | 34 (14.9) | < 0.05b | 222 | 12 (5.4) | |||||||||||
| Agitation | 227 | 14 (6.2) | < 0.05b | 228 | 21 (9.2) | NSb | 222 | 26 (11.7) | |||||||||||
| Anxiety | 227 | 8 (3.5) | NSb | 228 | 13 (5.7) | < 0.05b | 222 | 3 (1.4) | |||||||||||
| Dizziness | 227 | 39 (17.2) | < 0.05b | 228 | 42 (18.4) | < 0.05b | 294 | 22 (7.5) | ≤ 0.01c | 291 | 7 (2.4) | NSc | 303 | 21 (6.9) | ≤ 0.05c | 222 | 16 (7.2) | 302 | 7 (2.3) |
| Headache | 227 | 36 (15.9) | NSb | 228 | 40 (17.5) | < 0.05b | 294 | 18 (6.1) | ≤ 0.01c | 291 | 10 (3.4) | NSc | 303 | 13 (4.3) | NSc | 222 | 23 (10.4) | 302 | 5 (1.7) |
| Flatulence | 227 | 15 (6.6) | < 0.05b | 228 | 11 (4.8) | NSb | 222 | 4 (1.8) | |||||||||||
| Haemorrhoids | 227 | 2 (0.9) | NSb | 228 | 0 (0.0) | < 0.05b | 222 | 6 (2.7) | |||||||||||
| Weight loss | 294 | 16 (5.4) | ≤ 0.01c | 291 | 8 (2.7) | NSc | 303 | 23 (7.6) | ≤ 0.001c | 302 | 4 (1.3) | ||||||||
| Decreased appetite | 294 | 12 (4.1) | ≤ 0.05c | 291 | 2 (0.7) | NSc | 303 | 15 (5.0) | ≤ 0.01c | 302 | 3 (1.0) | ||||||||
| Asthenia | 294 | 17 (5.8) | ≤ 0.001c | 291 | 5 (1.7) | NSc | 303 | 9 (3.0) | NSc | 302 | 3 (1.0) | ||||||||
Summary: rivastigmine versus placebo
Our update searches identified three new RCTs138–140 to add to the four134–137 included in the previous review. 2
All three studies138–140 showed benefits from rivastigmine on the ADAS-cog and MMSE, although these benefits were dependent on dose, with greater benefits seen at 12 mg/day than at 6 mg/day. When these data were pooled with the existing evidence, significant differences favouring rivastigmine continued to be seen on the ADAS-cog at 24–26 weeks (≥ 12 mg/day), WMD = –2.46 (95% CI –3.37 to –1.56), p < 0.001. However, the benefits from rivastigmine were not apparent on MMSE scores until 24–26 weeks’ follow-up [WMD = 1.02 (95% CI 0.63 to 1.41), p < 0.001]; this may be due to the MMSE’s difficulties with detecting change. When the outcomes from both cognitive measures were combined they continued to show an advantage from taking rivastigmine on cognitive outcomes.
Two of the three new studies reporting functional outcomes showed significant gains for these measures. When these new data were synthesised with existing evidence using the PDS, significant gains were shown at 24–26 weeks [WMD = 3.10 (95% CI 1.81 to 4.40), p = 0.001].
The data on behavioural outcomes from the new studies were unclear, with the smaller study139 showing a benefit from rivastigmine that the larger one140 did not. The existing evidence was too heterogeneous for meta-analysis, so the overall effectiveness of rivastigmine for behavioural outcomes is unknown.
The results for global outcomes were also mixed. Results from the CIBIC-plus were almost universally significant, whereas those measured by the Alzheimer’s Disease Cooperative Study–Clinical Global Impression of Change (ADCS-CGIC) were almost universally not; those using the GDS showed no significant gain from rivastigmine. However, when these data were pooled with the existing evidence, the overall estimates favoured rivastigmine on the CIBIC-plus [WMD = –0.42 (95% CI –0.55 to –0.29), p < 0.001] and the GDS [WMD = 0.20 (95% CI 0.12 to 0.27), p < 0.001]. When results from both these outcome measures were combined, the result continued to show significant benefit from rivastigmine, these results are based on a robust ITT population.
When rivastigmine patches were compared with capsules, the results showed that the 9.5 mg/day patch was similarly effective as the 12.5 mg/day capsule, but with fewer side effects.
None of the included studies in either the updated or the original review reported QoL outcomes.
As in the other AChEIs, the main AEs were gastrointestinal.
Overall, pooled estimates of cognitive benefits from rivastigmine were favourable, but were shown to be dose dependent as in the previous review. The results from functional and global outcomes also showed significant gains. However, the results from individual trials of behavioural outcomes were mixed (pooling was not possible owing to heterogeneity). The lower dose transdermal patch (9.5 mg/day) was shown to be as effective as the capsule (12 mg/day), but with fewer side effects.
Graphical summary of rivastigmine versus placebo
The graphical summary in Figure 42 shows that two large studies138,140 and one small study139 have been added to the evidence for rivastigmine since 2004. The figure illustrates how the new studies have added to the precision of our knowledge of the effects of rivastigmine in AD. Previously, the results for cognitive outcomes were ambiguous; however, the results from the new trials all show cognitive benefits. The smaller new study139 showed a gain on behavioural outcomes that had not been seen in the previous studies, but the results for functional and global outcomes continued to be mixed.
FIGURE 42.
Summary of all included studies in the 2004 and 2010 reviews: rivastigmine vs placebo. *Significant only for ’TMT’, not ’ten-point clock-drawing test’. Analy, analysis; be, behavioural; b.i.d., twice a day; Blind, blinding; Char, characteristics of patients; cog, cognitive; F, female; func, functional; glo, global; M, male; Rand, randomisation; SIB, Severe Impairment Battery; t.i.d., three times a day; TMT, trail-making test.
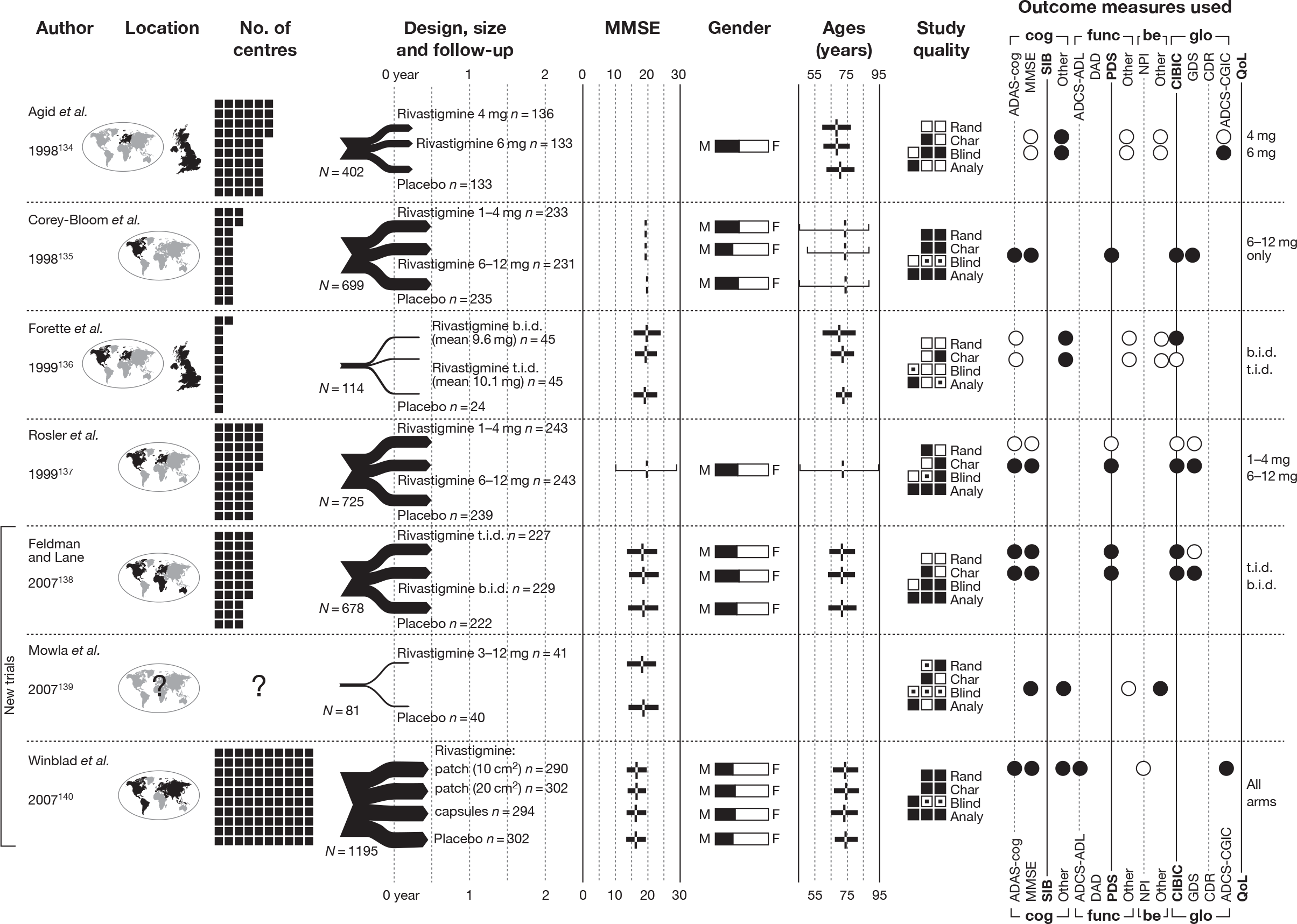
Memantine versus placebo
Differing approaches to pooling data
A key difference to our memantine findings and Lundbeck’s lies in our differing approaches to pooling data. In particular, Lundbeck have pooled together memantine plus AChEI versus placebo plus AChEI trials with memantine monotherapy versus placebo studies; we were not comfortable with this approach owing to the heterogeneity of the data. Nevertheless, we have followed this approach for completeness and present the results in Appendix 14. The effect of pooling data in this way is to show a more favourable response to memantine.
Identified evidence
The 2004 review2 lists two RCTs141,142 as investigations into the effectiveness of memantine in AD. However, one of those studies141 addressed the effectiveness of memantine in combination with donepezil; accordingly, this study is considered as part of our assessment of combination therapy (see Summary: head-to-head comparisons).
We identified one additional RCT143 of relevance to this comparison, details of which are presented in Table 24. This study’s interventions, comparators and baseline characteristics can be seen in Table 25 and the markers of internal validity in Table 26.
| Study details | Inclusion criteria | Exclusion criteria | Methodological notes | Other |
|---|---|---|---|---|
|
Van Dyck et al. (2007) 143 Design: Parallel double-blind RCT Country: USA No. of centres: 35 No. randomised: 350 Maximum follow-up: 24 MMSE range included: 5–14 Funding: Forest Laboratories Inc., provided all financial and material support for the study, as well as statistical and editorial support for the manuscript |
Probable AD (NINCDS-ADRDA criteria) Age ≥ 50 years Brain imaging evaluation (CT or MRI performed within 12 months before study entry) consistent with probable AD A knowledgable and reliable caregiver to accompany the participant to all study visits and supervise administration of the study drug Ability to ambulate Sufficient vision and hearing to comply with assessments Medical stability Stable doses of the following medications were allowed: antihypertensives, anti-inflammatories, diuretics, laxatives, antidepressants, atypical antipsychotics, tocopherol |
Significant and active pulmonary, gastrointestinal, renal, hepatic, endocrine, or cardiovascular disease Clinically significant vitamin B12 or folate deficiency Evidence of any psychiatric or neurological disorder other than AD Hachinski Ischaemic Score > 4 Delusions or delirium (DSM-IV criteria) Active malignancy History of substance abuse within 10 years Likelihood of nursing home placement within 6 months Previous memantine treatment Treatment with an investigational drug within 30 days (or five drug half-lives, whichever was longer) of screening Postmenopausal > 2 years or surgically sterile (female participants) |
Sample attrition/dropout: 260 of 350 completed study. 90 withdrew after allocation: AEs (n = 45), consent withdrawn (n = 26), protocol violation (n = 8), insufficient therapeutic response (n = 3), other (n = 8). No differences between groups Randomisation and allocation: Randomisation procedure not reported Power calculation: Assuming an effect size of 0.35, at least 340 participants were needed to provide 90% power at an alpha level of 0.05 (two sided) on the basis of a two-sample t-test for change from baseline to week 24 in SIB and ADCS-ADL scores |
Therapy common to all participants: 1–2 weeks single-blind placebo lead-in phase to assess compliance and minimise treatment response at baseline Study funding: Forest Laboratories Inc., provided all financial and material support for the study, as well as statistical and editorial support for the manuscript Other conflicts: Lead author (CD) and two co-authors (PT, BM) have received grant support and honoraria from Forest Laboratories Inc. One co-author (PT) has given expert testimony related to memantine. One author (EM) is an employee of Forest Laboratories Inc. |
| Study | Arm | Dose (mg/day) | Dosage details | N | Age, years (SD) | Gender, n male (%) | Race, n white (%) | Weight, kg (SD) | Education (years) | Duration of dementia, months (SD) | ADAS-cog, score (SD) | MMSE, score (SD) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Van Dyck et al. (2007)143 | Memantine | 5–20 |
Initial dosage of 5 mg/day with titration in 5-mg weekly increments to a final dosage of 20 mg/day (administered as two 5-mg tablets twice a day). Dose adjustments were permitted between weeks 3 and 8 for participants with AEs. Participants unable to tolerate 20 mg/day by the end of week 8 were discontinued from the study Compliance monitored by inventory of returned individual blister packs, and protocol adherence by routine assessment of concomitant medication use |
178 | 78.1 (8.20) | 49 (27.5) | 142 (79.8) | 64.4 (13.5)a | 10.0 (2.80) | |||
| Placebo | – | – | 172 | 78.3 (7.60) | 51 (29.7) | 141 (82.0) | 65.8 (12.8) | 10.3 (3.10) |
| Study | Was the assignment to the treatment groups really random? | Was the treatment allocation concealed? | Were the groups similar at baseline in terms of prognostic factors? | Were the eligibility criteria specified? | Were outcome assessors blinded to the treatment allocation? | Was the care provider blinded? | Was the patient blinded? | Were the point estimates and measure of variability presented for the primary outcome measure? | Did the analyses include an ITT analysis? | Were withdrawals and dropouts completely described? |
|---|---|---|---|---|---|---|---|---|---|---|
| Van Dyck et al. (2007)143 | Unknown | Unknown | Reported – yes | Yes | Unknown | Partial | Partial | Adequate | Adequate | Adequate |
Evidence of clinical effectiveness
Cognition
The 2004 report by Loveman and colleagues2 found two studies of memantine; they summarised their results as follows:
Both studies used the SIB as a measure of cognitive outcome. Statistically significant differences in favour of the use of memantine over placebo were apparent in the two studies. MMSE scores deteriorated in both the memantine group and the placebo group and the degree of deterioration was not statistically significantly different between the two groups.
The data from the new trial143 showed a significant effect only from memantine on one of six analyses. However, this was in an OCs-only analysis that may have biased the results; Table 27 summarises the results.
| Study | Subgroup | Outcome | Type | Memantine | Placebo | p-value | ||
|---|---|---|---|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | |||||
| Van Dyck et al. (2007)143 | LOCF analysis | SIB – 24 weeks | MC | 170 | –2 (13) | 165 | –2.5 (12.8) | 0.616a |
| OC population | SIB – 4 weeks | MC | 167 | 0.875 (7.43)b | 164 | –0.3 (6.4)b | 0.146a | |
| SIB – 8 weeks | 158 | 2.08 (7.86)b | 155 | 0.375 (7.16)b | 0.064a | |||
| SIB – 12 weeks | 146 | 1.65 (9.06)b | 150 | –0.825 (8.27)b | 0.008a | |||
| SIB – 18 weeks | 140 | 0 (8.28)b | 139 | –2.12 (9.14)b | 0.065a | |||
| SIB – 24 weeks | 131 | –1.8 (12.6) | 126 | –2.4 (13.5) | 0.617a | |||
The data from the new trial were pooled with that of the existing studies in random-effects meta-analyses of the Severe Impairment Battery (SIB) at 12 weeks and 24–28 weeks. The results showed a significant effect at 12 weeks [WMD = 4.15 (95% CI 0.52 to 7.78), p = 0.025], but not at 24–28 weeks (Figures 43 and 44).
FIGURE 43.
Random-effects meta-analysis – SIB at 12 weeks (mean change from baseline): memantine vs placebo.
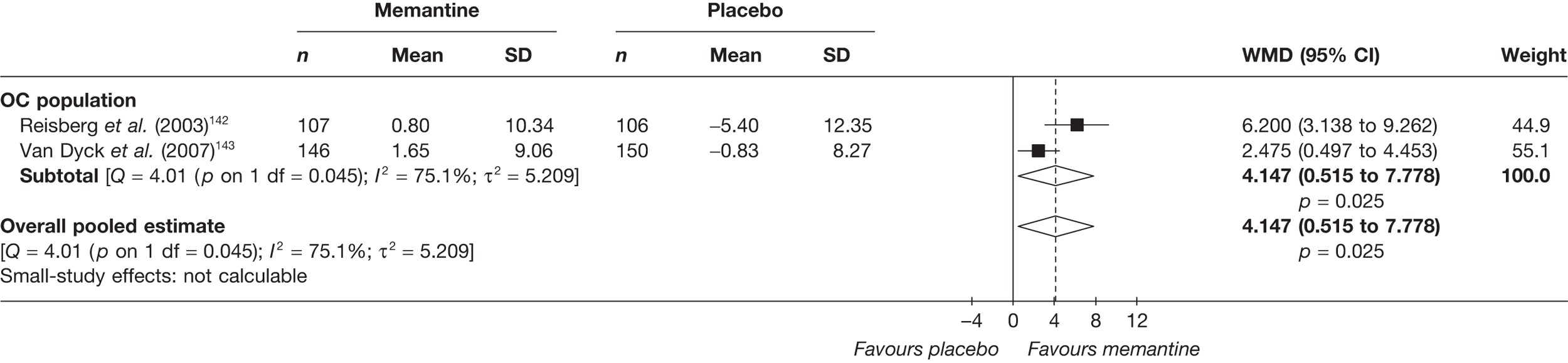
FIGURE 44.
Random-effects meta-analysis – SIB at 24–28 weeks (mean change from baseline): memantine vs placebo.
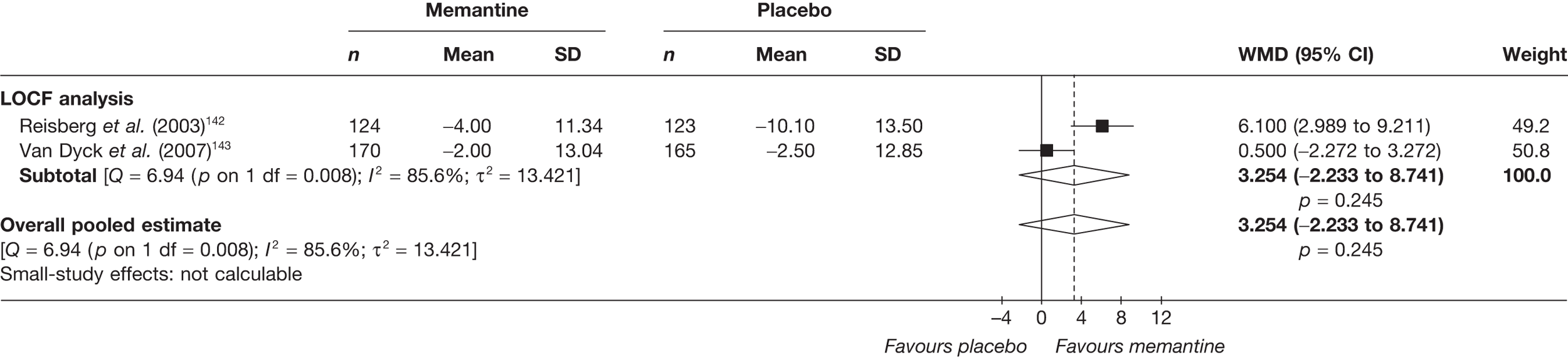
Functional
The previous assessment report2 summarised the findings about the effects of memantine on functional outcomes as:
Both studies demonstrated that memantine appears to show a statistically significant benefit to participants on the ADCS-ADL when compared to placebo, with a reduction in the level of deterioration.
The results from the new study showed no significant benefit on functional outcomes for memantine compared with placebo (Table 28).
| Study | Subgroup | Outcome | Type | Memantine | Placebo | p-value | ||
|---|---|---|---|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | |||||
| Van Dyck et al. (2007)143 | LOCF analysis | ADCS-ADL – 24 weeks | MC | 171 | –2 (7.85) | 165 | –2.7 (7.71) | 0.282a |
| FAST – 24 weeks | 151 | 0.3 (1.23) | 141 | 0.6 (1.19) | 0.093a | |||
| OC population | ADCS-ADL – 4 weeks | MC | 168 | 0.312 (4.37)b | 164 | 0.512 (4)b | 0.801a | |
| ADCS-ADL – 8 weeks | 159 | –0.0875 (5.2)b | 156 | –0.188 (4.84)b | 0.665a | |||
| ADCS-ADL – 12 weeks | 147 | 0 (5.46)b | 150 | –0.488 (5.05)b | 0.155a | |||
| ADCS-ADL – 18 weeks | 142 | –0.688 (7.3)b | 140 | –1.38 (5.62)b | 0.357a | |||
| ADCS-ADL – 24 weeks | 133 | –1.3 (6.92) | 127 | –2.3 (6.76) | 0.188a | |||
| FAST – 24 weeks | 133 | 0.3 (1.15) | 127 | 0.6 (1.13) | 0.074a | |||
The data from the new studies were synthesised with the existing evidence in a random-effects meta-analysis.
Two studies provide data on functional effect as measured by ADCS-ADL; both report the modified ADCS-ADL19 version of the instrument, consisting of 19 items that have been individually validated in cases of more severe dementia. The data were meta-analysed at 12 weeks’ and 24–28 weeks’ follow-up. The results were not significant at 12 weeks and barely significant at 24–28 weeks, especially considering that the population analysed were LOCF: WMD = 1.41 (95% CI 0.04 to 2.78), p = 0.044 (Figures 45 and 46).
FIGURE 45.
Random-effects meta-analysis – ADCS-ADL19 at 12 weeks (mean change from baseline): memantine vs placebo.
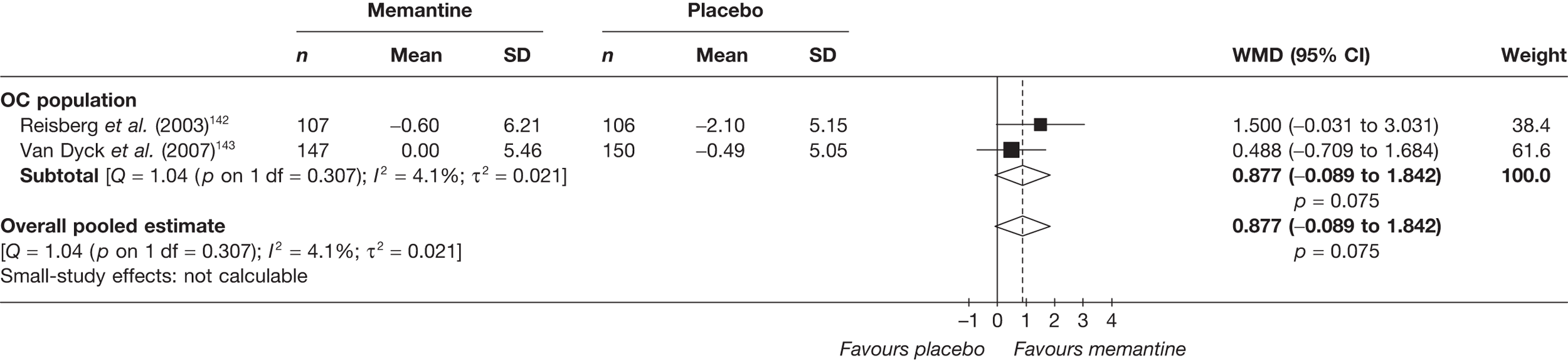
FIGURE 46.
Random-effects meta-analysis – ADCS-ADL19 at 24–28 weeks (mean change from baseline): memantine vs placebo.
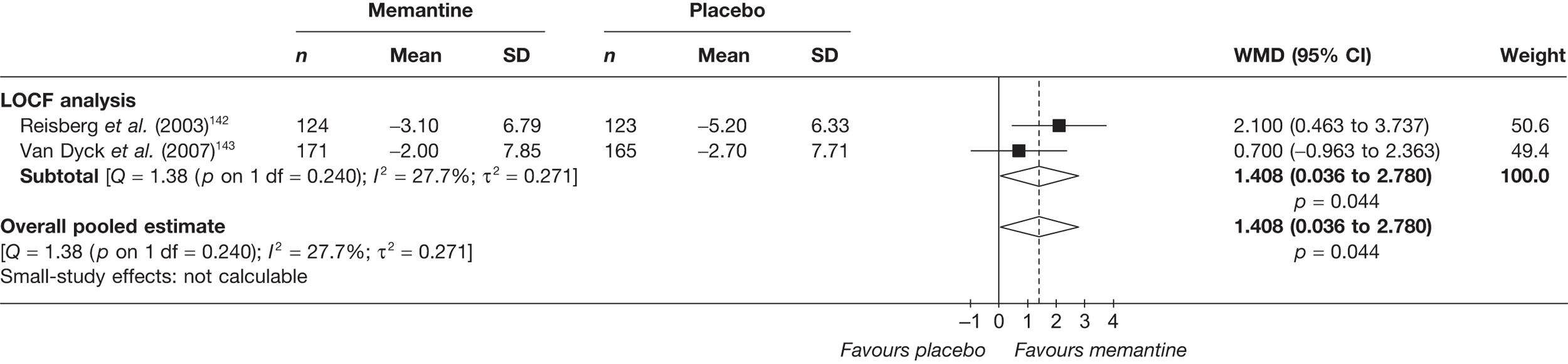
Another meta-analysis was conducted with data from existing and new studies using the Functional Assessment Staging Tool (FAST) at 24–28 weeks’ follow-up. The overall pooled estimate showed a significant benefit from memantine compared with placebo, WMD = –0.34 (95% CI –0.55 to –0.13), p = 0.002 (Figure 47).
FIGURE 47.
Random-effects meta-analysis – FAST at 24–28 weeks (mean change from baseline): memantine vs placebo.
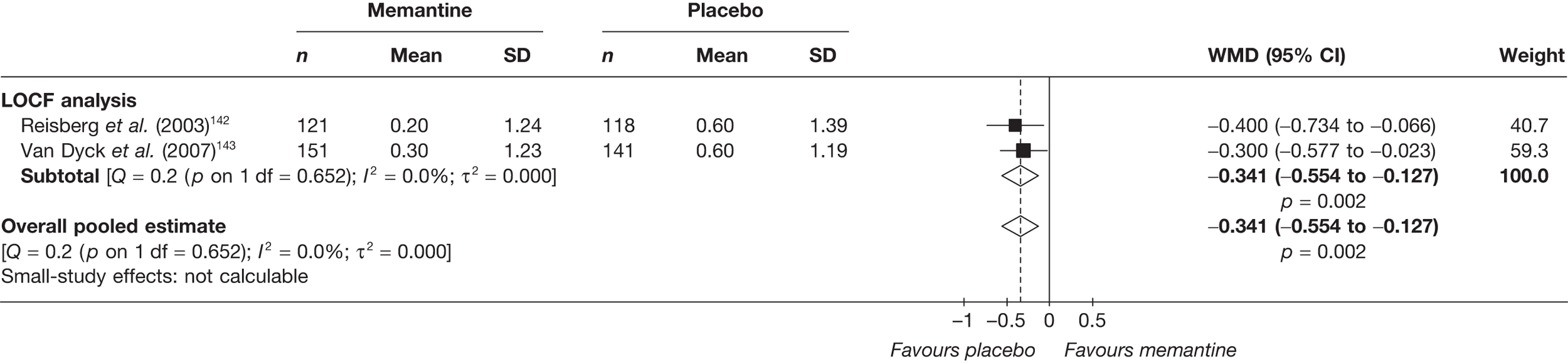
Behavioural and mood
The 2004 assessment report2 summarised the finding for behavioural outcomes comparing memantine with placebo as:
It appears that participants receiving memantine and already receiving a steady dose of donepezil have a statistically significantly lower NPI score than placebo. Those on memantine only however, showed no statistically significant difference compared to placebo.
The study143 that was published after 2004 measured behavioural outcomes using the NPI and the Behavioural Rating Scale for Geriatric Patients (BGP). Neither measure showed a significant benefit from memantine (Table 29).
| Study | Subgroup | Outcome | Type | Memantine | Placebo | p-value | ||
|---|---|---|---|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | |||||
| Van Dyck et al. (2007)143 | LOCF analysis | NPI – 24 weeks | MC | 161 | 1 (16.5) | 154 | 1.1 (17.4) | 0.963a |
| BGP: total – 24 weeks | 151 | 0.6 (6.14) | 141 | 1.5 (7.12) | 0.197a | |||
| BGP: care dependency – 24 weeks | 151 | 0.5 (4.92) | 141 | 1.4 (4.75) | 0.076a | |||
| OC population | NPI – 24 weeks | 133 | 0.5 (15) | 127 | 1 (15.8) | 0.782a | ||
| BGP: total – 24 weeks | 133 | 0.4 (6.92) | 127 | 1.1 (6.76) | 0.312a | |||
| BGP: care dependency – 24 weeks | 133 | 0.4 (4.61) | 127 | 1.2 (5.63) | 0.138a | |||
The NPI data from van Dyck and colleagues143 were pooled with the existing data at 24–28 weeks’ follow-up in a random-effects meta-analysis. This analysis also failed to show a significant gain from memantine compared with placebo (Figure 48).
FIGURE 48.
Random-effects meta-analysis – NPI at 24–28 weeks (mean change from baseline): memantine vs placebo.
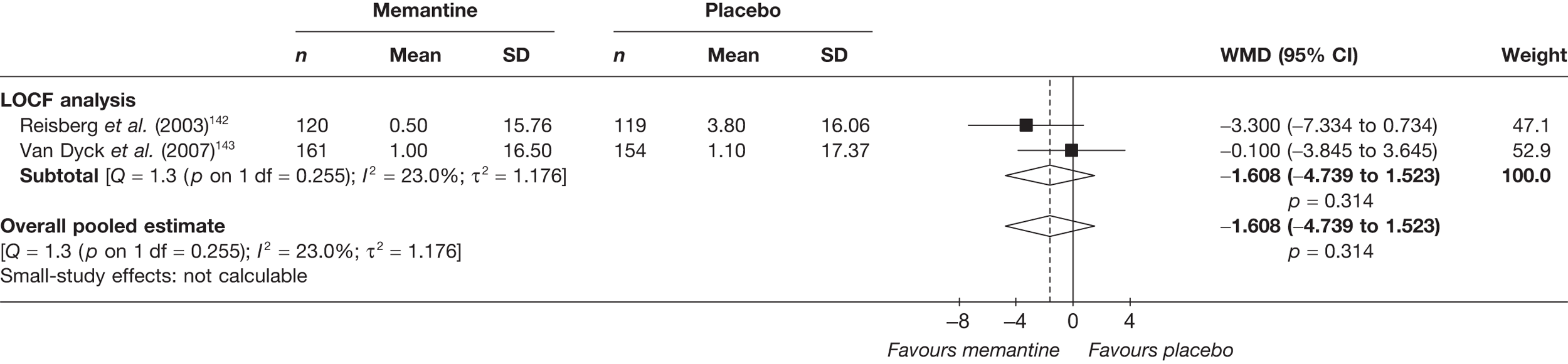
Global effect
In 2004, Loveman and colleagues2 summarised the results for global measures comparing memantine with placebo as:
Both studies used the CIBIC-plus as a measure of global outcome, and, in both cases, memantine appeared to be effective.
Van Dyck and colleagues143 also measured global outcomes with the CIBIC-plus; however, the differences they found were not significant (Table 30).
| Study | Subgroup | Outcome | Type | Memantine | Placebo | p-value | ||
|---|---|---|---|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | |||||
| Van Dyck et al. (2007)143 | LOCF analysis | CIBIC-plus score – 24 weeks | C | 171 | 4.3 (13.1) | 163 | 4.6 (12.8) | 0.182a |
| OC population | CIBIC-plus score – 24 weeks | C | 134 | 4.3 (12.7) | 127 | 4.6 (11.3) | 0.089a | |
When the new data were pooled with the existing studies in a random-effects meta-analysis the overall pooled estimate showed a significant beneficial effect from memantine compared with placebo: WMD = –0.30 (95% CI –0.47 to –0.13), p < 0.001 (Figure 49).
FIGURE 49.
Random-effects meta-analysis – CIBIC-plus at 24–28 weeks: memantine vs placebo.
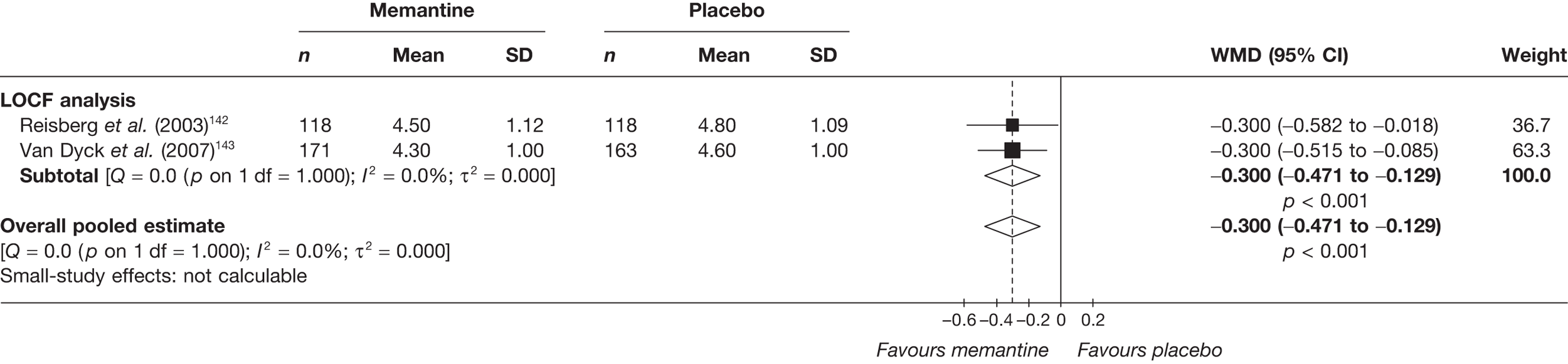
Quality of life
None of the included studies provided any randomised evidence on QoL with memantine compared with placebo and no such data were identified in the 2004 review.
Safety
The proportion of any AEs were similar in the treatment and control groups (T = 74%, C = 73%). The main AEs were agitation and hypertension in the memantine group, and agitation and falls in the control group (Table 31).
| AE | Van Dyck et al. (2007)143 | ||||
|---|---|---|---|---|---|
| Memantine | Placebo | p-value | |||
| N | n (%) | N | n (%) | ||
| Any AE | 178 | 131 (73.6) | 172 | 125 (72.7) | 0.941a |
| Any serious AE | 178 | 26 (14.6) | 172 | 29 (16.9) | 0.666a |
| Diarrhoea | 178 | 10 (5.6) | 172 | 8 (4.7) | 0.867a |
| Agitation | 178 | 16 (9.0) | 172 | 24 (14.0) | 0.197a |
| Anxiety | 178 | 10 (5.6) | 172 | 6 (3.5) | 0.485a |
| Depression | 178 | 9 (5.1) | 172 | 5 (2.9) | 0.451a |
| Injury | 178 | 10 (5.6) | 172 | 13 (7.6) | 0.605a |
| Dizziness | 178 | 12 (6.7) | 172 | 11 (6.4) | 0.932a |
| Headache | 178 | 3 (1.7) | 172 | 11 (6.4) | 0.048a |
| Urinary tract infection | 178 | 9 (5.1) | 172 | 9 (5.2) | 0.867a |
| Fall | 178 | 10 (5.6) | 172 | 17 (9.9) | 0.195a |
| Influenza-like symptoms | 178 | 10 (5.6) | 172 | 8 (4.7) | 0.867a |
| Confusion | 178 | 9 (5.1) | 172 | 8 (4.7) | 0.942a |
| Hypertension | 178 | 14 (7.9) | 172 | 4 (2.3) | 0.035a |
| Peripheral oedema | 178 | 12 (6.7) | 172 | 8 (4.7) | 0.541a |
| Constipation | 178 | 11 (6.2) | 172 | 8 (4.7) | 0.693a |
| Insomnia | 178 | 4 (2.2) | 172 | 9 (5.2) | 0.233a |
Summary: memantine versus placebo
One new moderate-to-poor-quality study was found to add to the existing evidence for memantine versus placebo.
The pooled results for cognitive abilities measured by the SIB showed a significant benefit from memantine at 12 weeks’ follow-up [WMD = 4.15 (95% CI 0.52 to 7.78), p = 0.025]. However, at 24 weeks the data pooled with that of the previous review showed no significant benefit.
Similar to the previous review,2 the new study143 found a significant benefit from memantine from the FAST functional outcome, although not with the ADCS-ADL at 12 weeks. When the FAST data from new and existing studies were pooled, a significant relationship was found between memantine and an improvement in scores [WMD = –0.34 (95% CI –0.55 to –0.13), p = 0.002]. A marginally significant benefit was seen from memantine when pooled ADCS-ADL data were measured at 24–28 weeks [WMD = 1.41 (95% CI 0.04 to 2.78) p = 0.044].
The results from behavioural outcomes in the new study,141 similar to the previous review,2 failed to show a significant benefit from memantine, either singly or when the data were pooled.
Although the results of the CIBIC-plus failed to show a significant gain from memantine, when these data were pooled with that from the previous review a significant effect was found [WMD = –3.00 (95% CI –0.471 to –0.129), p < 0.001].
No studies reported QoL outcomes. The main AEs were agitation and hypertension.
The meta-analysis of memantine versus placebo studies showed benefit from memantine at 12 weeks’ follow-up on the SIB. However, treatment gain, measured by functional outcome, depended on the type of instrument used, and no benefit was seen from behavioural outcomes. Nevertheless, pooled estimates of global outcomes showed a benefit from taking memantine at 24–28 weeks. Overall, the pooled results from these moderate-to-poor-quality studies showed inconclusive results for cognitive and behavioural outcomes. The results for functional outcomes were dependent on the measure used, but the pooled results of the new and existing evidence for global outcomes showed significant benefit from using memantine.
Graphical summary of memantine versus placebo
Figure 50, below, illustrates how little the evidence has changed for memantine versus placebo. The cognitive benefits found in 2004 failed to be replicated; indeed the new study143 favoured memantine on only one outcome measure. However, the quality of the new143 and existing study142 was not high; thus, these results cannot be considered conclusive.
FIGURE 50.
Summary of all included studies in the 2004 and current reviews: memantine vs placebo. Analy, analysis; be, behavioural; Blind, blinding; Char, characteristics of patients; cog, cognitive; F, female; func, functional; glo, global; M, male; Rand, randomisation.
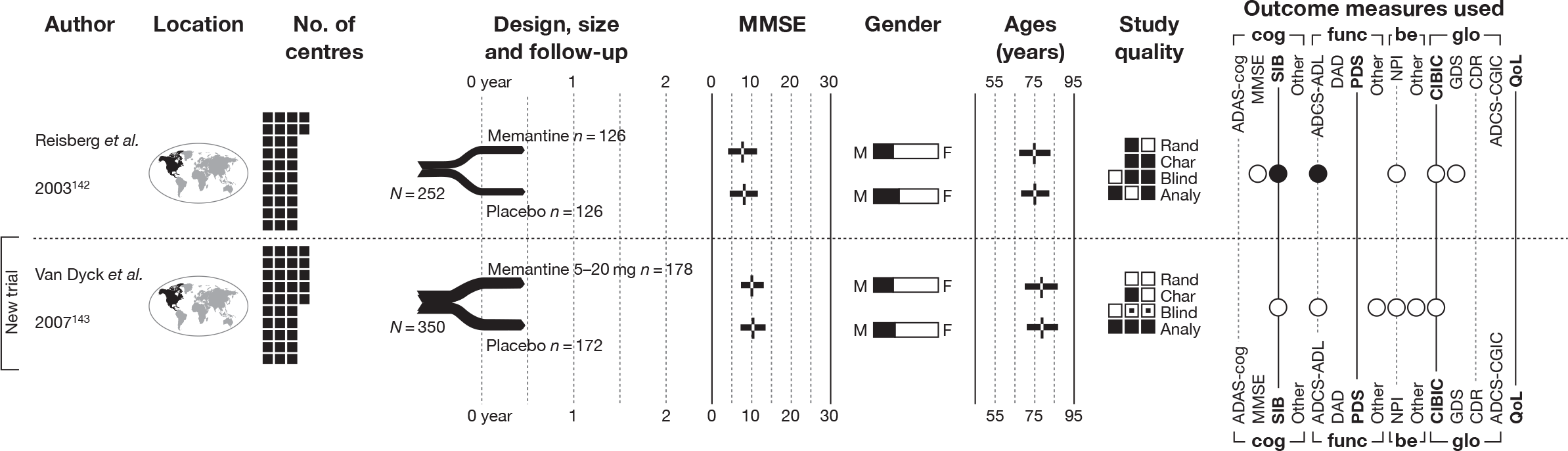
Head-to-head comparisons
Identified evidence
Alongside placebo-controlled trials, a certain amount of randomised evidence provides direct, head-to-head comparisons of two or more of the technologies under review. Three such RCTs were included in the 2004 review:2 donepezil versus rivastigmine,144 donepezil versus rivastigmine,145 and donepezil versus galantamine. 146
Our searches identified a further four RCTs of this type. 99,147–149 Details of the design of these trials are tabulated in Table 32, and a summary of treatments and baseline characteristics of participants can be found in Table 33. Two of the new RCTs99,147 compared all three AChEIs (although Nordberg and colleagues’ trial147 is of relevance to the current review only for its safety data). One trial investigated donepezil versus rivastigmine148 and the last was concerned with donepezil versus galantamine. 149
| Study details | Inclusion criteria | Exclusion criteria | Methodological notes | Other |
|---|---|---|---|---|
|
Ancoli-Israel et al. (2005) 149 Design: Parallel double-blind RCT Country: Not reported. All study authors based in the USA No. of centres: Not reported No. randomised: 63 Maximum follow-up: 8 MMSE range included: 10–24 Funding: Janssen Medical Affairs |
Mild-to-moderate AD (criteria not reported) Age ≥ 60 years Resident with a responsible caregiver who agreed to participate and monitor sleep and answer questionnaires |
Other neurodegenerative disease contributing to dementia (including multi-infarct dementia or clinically active cerebrovascular disease) Other medical condittions causing cognitive impairment Clinically significant co-existing medical conditions Use of a muscarinic-1 agonist or AChEI within 30 days prior to involvement |
Sample attrition/dropout: 54 of 63 completed study; discontinued due to AE (n = 3 in galantamine arm; n = 4 in donepezil arm); discontinued due to severe AE possibly related to trial drug (hepatic failure, n = 1 in donepezil arm); death (judged to be unrelated to trial drug, n = 1) Randomisation and allocation: Randomisation procedure not described Power calculation: None |
Therapy common to all participants: 2-week, single-blind, placebo run-in Study funding: Janssen Medical Affairs Other conflicts: Lead author declares no financial disclosure; co-authors are employees of funder (Janssen Medical Affairs) |
|
Bullock et al. (2005) 148 Design: Parallel double-blind RCT Countries: Australia, Canada, France, Germany, Italy, Spain and the UK No. of centres: 94 No. randomised: 998 Maximum follow-up: 104 MMSE range included: 10–20 Funding: Study supported by Novartis Pharma AG |
Male or female outpatients aged 50–85 years AD (DSM-IV criteria) or probable AD (NINCDS-ADRDA criteria) Contact with a responsible caregiver at least once a day Patients with AD who also had symptoms suggestive of concomitant Lewy body disease (McKeith et al. criteria) were also permitted to enter the study |
Current diagnosis of any primary neurodegenerative disorder other than AD (including Parkinson’s disease) Any advance, severe, progressive or unstable disease or disability A major depressive episode Active, uncontrolled seizure disorder or peptic ulceration Acute, severe or unstable asthmatic conditions Severe or unstable cardiovascular disease History or diagnosis of cerebrovascular disease Known hyperensitivity to drugs similar to rivastigmine or donepezil in structure or pharmacological action Use of any cholinesterase inhibitor or other approved treatment for AD in the 6 weeks prior to randomisation Use of any investigational drug, any drug or treatment known to cause major organ system toxicity, or any new psychotropic medication during the 4 weeks prior to randomisation Anticholinergic drugs at randomisation |
Sample attrition/dropout: 578 of 994 (58.1%) completed study [rivastigmine 261 of 495 (52.7%), donepezil 317 of 499 (63.5%)] (998 were randomised, four withdrew before receiving treatment) Reasons for non-completion: Rivastigmine – AEs (n = 129); abnormal laboratory values (n = 1); unsatisfactory therapeutic effect (n = 19); protocol violation (n = 12); withdrawn consent (n = 34); lost to follow-up (n = 10); administrative problems (n = 4); and death (n = 26) Donezepil – AEs (n = 80); abnormal laboratory values (n = 1); unsatisfactory therapeutic effect (n = 17); protocol violation (n = 9); withdrawn consent (n = 22); lost to follow-up (n = 13); administrative problems (n = 6); and death (n = 34) Randomisation and allocation: Performed using an interactive voice-response system that automated the random assignment of treatment groups to randomisation numbers. Randomisation was stratified with respect to severity, i.e. was done separately with MMSE scores of 10–14 and 15–20 All treatments were supplied as capsules that were identical in size, shape and colour, and all patients received the same number of capsules per day Power calculation: Powered at 85% to detect a statistically significant (significance level 5%, two-sided) difference in SIB of four points between the two groups (assuming a SD of 20 on change from baseline in mean SIB scores, as observed in previous trials), sample size of 450 patients per treatment group was required |
Therapy common to all participants: None Study funding: Study supported by Novartis Pharma AG Four of the study authors (YH, JN, GR, RL) are employees of Novartis The remaining four authors (RB, JT, HB, GG) did not receive remuneration for taking part in the study or writing the manuscript Other conflicts: Study supported by Novartis Pharma AG Four of the study authors (YH, JN, GR, RL) are employees of Novartis The remaining four authors (RB, JT, HB, GG) did not receive remuneration for taking part in the study or writing the manuscript |
|
Cumbo (2005) 99 Design: – Country: Funded by an Italian health agency, but not stated whether or not study conducted in Italy or elsewhere No. of centres: Not stated. Small sample size suggests single centre. No. randomised: 101 Maximum follow-up: 78 MMSE range included: 10–27 Funding: Supported by Department of Neuroscience (NHS District of Caltanissetta) |
Probable AD (NINCDS-ARDRA) ≥ 3-year duration of disease No behavioural symptoms Carer who could ensure compliance to treatment and attendance and provide the information required for psychometric and behavioural assessments |
History of primary neurological or psychiatric disease other than AD Drug or alcohol abuse Clinically significant medical or surgical disorders independently of stability Previous therapy for dementia Concomitant treatment with cholinomimetic or anticholinergic drugs, investigational drugs, tricyclic antidepressants or neuroleptics Refusal to give informed consent in writing |
Sample attrition/dropout: None Randomisation and allocation: No details of randomisation procedure reported Open-label trial Power calculation: None reported |
Therapy common to all participants: None Study funding: Supported by Department of Neuroscience (NHS District of Caltanissetta) Novartis Farma SpA supported the English editing of the manuscript Other conflicts: Supported by Department of Neuroscience (NHS District of Caltanissetta) Novartis Farma SpA supported the English editing of the manuscript |
|
Nordberg et al. (2009) 147 Design: – Country: Not reported No. of centres: Not reported No. randomised: 63 Maximum follow-up: 13 MMSE range included: 10–20 Funding: Novartis Pharmaceuticals; Swedish Research Council; KI foundations, L-H Osterman and Stohne’s Foundations supported two co-authors (AN, TDS). Alpha-Plus provided editorial assistance with the production of the manuscript |
AD (DSM-IV criteria) and probable or possible AD (NINCDS-ADRDA criteria) Age 50–85 years Provided the dose had been stabilised for the past month, treatment with psychotropics was permitted |
Prior exposure to rivastigmine, donepezil or galantamine Advance, severe or unstable disease of any type that might interfere with study evaluation or put the patient at special risk Imaging findings consistent with a condition other than AD that would explain the patient’s dementia Current treatment with coumarin derivatives Blood clotting abnormalities or inadequate platelet function |
Sample attrition/dropout: 53 of 63 completed study. 10 withdrew after allocation; AEs (n = 8), withdrew consent (n = 1), lost to follow-up (n = 1) Randomisation and allocation: Randomisation procedure not described. Open-label trial (although laboratory personnel who processed CSF samples were blinded) Power calculation: Assuming a mean treatment difference of 0.3 U/l (primary outcome variable), SD 0.28 and two-sided significance level of 0.025, z-test showed approximately 20 patients per treatment group were required to achieve a power of 0.85 for detecting a significant pair-wise treatment difference |
Therapy common to all participants: None Other conflicts: Three co-authors (AN, TD-S, MM) were responsible for the enzyme analysis and received research sponsorship from Novartis. One co-author’s (HS) institute received research sponsorship from Novartis for this study. Two co-authors (GE, RL) are full-time employees of Novartis |
| Study | Arm | Dose (mg/day) | Dosage details | N | Age, years (SD) | Gender, n male (%) | Race, n white (%) | Weight, kg (SD) | Education (years) | Duration of dementia, months (SD) | ADAS-cog, score (SD) | MMSE, score (SD) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Bullock et al. 2005148 | Rivastigmine | 3–12 | Titrated from 3 mg/day for the first 4 weeks, up to a maximum of 12 mg/day in increments of 3 mg/day every 4 weeks | 495 | 75.9 (6.60) | 154 (31.1) | 33.6 (22.2) | 15.1 (3.00) | ||||
| Donepezil | 5–10 |
Titrated from 5 mg/day for weeks 1–8 up to 10 mg/day in weeks 9–16 For patients who did not achieve the maximum dose during the titration period, investigators were asked to make at least one attempt during the maintenance period to increase the dose to the next highest dose level |
499 | 75.8 (6.80) | 157 (31.5) | 34.2 (26.5) | 15.1 (2.90) | |||||
| Cumbo (2005)99 | Rivastigmine | 9a | Dosage/titration scheme not reported | 101b | 76.4 (rng 66–83)b | 43 (42.6)b | 5.00 (rng 3–12)b | 61.1 (rng 36–108)b | 16.6b | |||
| Galantamine | 16a | Dosage/titration scheme not reported | ||||||||||
| Donepezil | 10a | Dosage/titration scheme not reported | ||||||||||
| Ancoli-Israel et al. (2005)149 | Donepezil | 5–10 | Dose titrated from 5 mg once a day at night for the first 4 weeks up to 10 mg once a day at night for the remainder of study | 32 | 77.8 (6.20) | 14 (43.8) | 26 (81.3) | c | 19.4 (rng 13–24) | |||
| Galantamine | 8–16 | Dose titrated from 4 mg twice a day for the first 4 weeks up to 8 mg twice a day for remainder of study | 31 | 76.5 (7.70) | 10 (32.3) | 25 (80.6) | d | 19.3 (rng 11–24) | ||||
| Nordberg et al. 2009147 | Donepezil | 5–10 | Starting dose 5 mg q.d.; after ≥ 4 weeks, if tolerated, up-titrated to 10 mg q.d.; no subsequent up-titrations | 20 | 74.0 (8.00) | 9 (45.0) | 20 (100.0) | 65.2 (8.00) | 32.4 (19.2) | 20.0 (3.50) | ||
| Galantamine | 8–24 | Starting dose 4 mg b.i.d.; after ≥ 4 weeks, if tolerated, up-titrated to 8 mg b.i.d.; subsequent up-titrations could be made after ≥ 4 weeks at each dose, based upon the patient’s well-being and tolerability to a maximum of 12 mg b.i.d. | 21 | 73.7 (6.50) | 5 (23.8) | 21 (100.0) | 65.7 (11.5) | 39.6 (25.2) | 19.2 (3.10) | |||
| Rivastigmine | 3–12 | Starting dose 1.5 mg b.i.d.; after ≥ 4 weeks, if tolerated, up-titrated to 3 mg b.i.d.; subsequent up-titrations could be made after ≥ 4 weeks at each dose, based upon the patient’s well-being and tolerability to a maximum of 6 mg b.i.d. | 22 | 76.8 (8.90) | 5 (22.7) | 21 (95.5) | 65.1 (9.70) | 34.8 (25.2) | 18.8 (3.80) |
The quality of the newly identified RCTs in this category (Table 34) tended to be low. Cumbo’s three-way examination of donepezil, galantamine and rivastigmine99 is of especially dubious validity, with no description of randomisation or allocation, and an open-label treatment period. Moreover, most of the outcomes reported by this trial – concentrating on distribution of symptoms among participants experiencing behavioural disturbance – are of little help for our purpose of establishing the relative effectiveness of the technologies.
| Study | Was the assignment to the treatment groups really random? | Was the treatment allocation concealed? | Were the groups similar at baseline in terms of prognostic factors? | Were the eligibility criteria specified? | Were outcome assessors blinded to the treatment allocation? | Was the care provider blinded? | Was the patient blinded? | Were the point estimates and measure of variability presented for the primary outcome measure? | Did the analyses include an ITT analysis? | Were withdrawals and dropouts completely described? |
|---|---|---|---|---|---|---|---|---|---|---|
| Bullock et al. (2005)147 | Adequate | Adequate | Reported – yes | Inadequate | Partial | Adequate | Adequate | Adequate | Adequate | Adequate |
| Cumbo (2005)99 | Unknown | Unknown | Unknowna | Unknown | Unknown | Unknownb | Unknownb | Adequate | Adequatec | Adequated |
| Ancoli-Israel et al. (2005)149 | Unknown | Unknown | Reported – yes | Unknown | Partial | Partial | Partial | Adequate | Partial | Adequate |
| Nordberg et al. (2009)147 | Unknown | Unknown | Reported – yese | Unknown | Inadequatef,g | Inadequate | Inadequatef | Adequate | Inadequate | Adequate |
Of the RCTs we identified, Bullock and colleagues’148 2-year, double-blind comparison of donepezil and rivastigmine was judged to be much the least susceptible to bias. Robust randomisation, allocation and assessment methods are reported, and the study was of a good size, with each treatment arm comprising almost 500 individuals.
Evidence of clinical effectiveness
Cognition
Only one148 of the newly identified RCTs reports outcome measures assessing the cognitive function of participants. Bullock and colleagues148 report that, following 2 years of double-blind treatment, a similar cognitive decline was seen in individuals who had been randomised to donepezil or rivastigmine (Table 35).
| Study | Subgroup | Outcome | Type | Rivastigmine | Donepezil | p-value | ||
|---|---|---|---|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | |||||
| Bullock et al. (2005)148 | LOCF analysis | MMSE – 104 weeks | MC | 471 | –2.35 (6.51) | 484 | –2.85 (6.6) | 0.089,a 0.106b |
| SIB – 104 weeks | MC | 471 | –9.3 (23.9) | 483 | –9.91 (24.2) | 0.609,a 0.738b | ||
It was not possible to amalgamate the new and existing evidence in quantitative synthesis, because the Bullock and colleagues trial148 featured much more extensive follow-up than the 12- to 30-week donepezil versus rivastigmine RCTs identified in 2004. 144,145 Unfortunately, Bullock and colleagues148 do not report findings on cognitive measures over the course of their trial (one figure showing SIB decline is provided, but does not give any indication of dispersion at each juncture), so their findings cannot be combined at earlier follow-up either.
Functional
Again, Bullock and colleagues’ RCT148 provides the only new evidence on the relative effectiveness of the technologies under review in the functional domain. In the primary – ITT LOCF – analysis, a significant advantage for rivastigmine over donepezil after 2 years’ treatment was detected. Individuals who had been randomised to receive rivastigmine declined by around two fewer points on the ADCS-ADL instrument (Table 36). It should be noted, however, that this finding was not replicated in the secondary analyses, which relied on evaluable cases (all participants who were treated for at least 16 weeks, with LOCF imputation for subsequent missing values) and OCs.
| Study | Subgroup | Outcome | Type | Rivastigmine | Donepezil | p-value | ||
|---|---|---|---|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | |||||
| Bullock et al. (2005)148 | LOCF analysis | ADCS-ADL – 104 weeks | MC | 454 | –12.8 (19.2) | 475 | –14.9 (19.6) | 0.047,a 0.007b |
Behavioural and mood
Bullock and colleagues148 found no significant difference between donepezil and rivastigmine on the NPI scale, with participants in both groups declining by an average of between two and three points over 2 years’ treatment (Table 37). Cumbo’s trial99 is explicitly focused on behavioural disturbance in individuals taking AChEIs. However, the paper mostly concentrates on the profile of individuals who were adjudged to experience behavioural and psychological symptoms, rather than the incidence of such events in the whole population. It was found that most categories of behavioural event happened with lower frequency among those taking rivastigmine; however, no tests of the magnitude of such differences are presented. We have found that, in most cases, any discrepancy would be insufficient to fulfil conventional definitions of statistical significance (i.e. p < 0.05 by chi-squared test), with the exceptions of the night-time behaviour subdomain of the NPI and the diurnal cycle disturbances item of the Behavioral Pathology in Alzheimer’s disease (BEHAVE-AD) scale (see Table 37). The high probability of type 1 error in the presence of multiple comparisons must clearly be borne in mind here.
| Study | Subgroup | Outcome | Type | Rivastigmine | Galantamine | Donepezil | p-value | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | Mean, n (%) | N | Mean, n (%) | N | Mean, n (%) | Rivastigmine vs galantamine | Rivastigmine vs donepezil | Galantamine vs donepezil | ||||
| Bullock et al. (2005)148 | LOCF analysis | NPI – 104 weeks | MC | 471 | 2.4 (17.4) | – | – | 484 | 2.94 (17.6) | – | 0.554,a 0.505b | – |
| Cumbo (2005)99 | ITT population | Probability of being BPSD-free at 78 weeks | TTE | 37 | 0.622 (SEM 0.080) | 33 | 0.546 (SEM 0.087) | 31 | 0.484 (SEM 0.090) | 0.235c | 0.055c | 0.365c |
| NPI – delusions – 78 weeks | D | 37 | 1 (2.7) | 33 | 4 (12.1) | 31 | 5 (16.1) | 0.288d | 0.130d | 0.919d | ||
| NPI – hallucinations – 78 weeks | D | 37 | 0 (0.0) | 33 | 0 (0.0) | 31 | 3 (9.7) | 0.341d | 0.226d | 0.274d | ||
| NPI – agitation/aggression – 78 weeks | D | 37 | 4 (10.8) | 33 | 9 (27.3) | 31 | 7 (22.6) | 0.144d | 0.326d | 0.885d | ||
| NPI – depression/dysphoria – 78 weeks | D | 37 | 13 (35.1) | 33 | 10 (30.3) | 31 | 13 (41.9) | 0.861d | 0.746d | 0.479d | ||
| NPI – anxiety – 78 weeks | D | 37 | 14 (37.8) | 33 | 15 (45.5) | 31 | 14 (45.2) | 0.687d | 0.716d | 0.820d | ||
| NPI – elation/euphoria – 78 weeks | D | 37 | 0 (0.0) | 33 | 0 (0.0) | 31 | 1 (3.2) | 0.341d | 0.902d | 0.965d | ||
| NPI – apathy/indifference – 78 weeks | D | 37 | 7 (18.9) | 33 | 7 (21.2) | 31 | 8 (25.8) | 0.952d | 0.698d | 0.890d | ||
| NPI – disinhibition – 78 weeks | D | 37 | 0 (0.0) | 33 | 3 (9.1) | 31 | 1 (3.2) | 0.252d | 0.902d | 0.651d | ||
| NPI – irritability/lability – 78 weeks | D | 37 | 12 (32.4) | 33 | 14 (42.4) | 31 | 15 (48.4) | 0.538d | 0.276d | 0.820d | ||
| NPI – aberrant motor behaviour – 78 weeks | D | 37 | 0(0.0) | 33 | 0 (0.0) | 31 | 0 (0.0) | 0.341d | 0.355d | 0.328d | ||
| NPI – night-time behaviour – 78 weeks | D | 37 | 1( 2.7) | 33 | 9 (27.3) | 31 | 0 (0.0) | 0.010d | 0.902d | 0.008d | ||
| NPI – appetite/eating change – 78 weeks | D | 37 | 0 (0.0) | 33 | 1 (3.0) | 31 | 1 (3.2) | 0.936d | 0.902d | 0.500d | ||
| Developing BPSD – 78 weeks | D | 37 | 14 (37.8) | 33 | 15 (45.5) | 31 | 16 (51.6) | 0.687d | 0.371d | 0.808d | ||
| BEHAVE-AD – delusional and paranoid ideation – 78 weeks | D | 37 | 1 (2.7) | 33 | 4 (12.1) | 31 | 5 (16.1) | 0.288d | 0.130d | 0.919d | ||
| BEHAVE-AD – hallucinations – 78 weeks | D | 37 | 0 (0.0) | 33 | 0 (0.0) | 31 | 3 (9.7) | 0.341d | 0.226d | 0.274d | ||
| BEHAVE-AD – activity disturbances – 78 weeks | D | 37 | 0 (0.0) | 33 | 0 (0.0) | 31 | 0 (0.0) | 0.341d | 0.355d | 0.328d | ||
| BEHAVE-AD – aggression – 78 weeks | D | 37 | 4 (10.8) | 33 | 9 (27.3) | 31 | 7 (22.6) | 0.144d | 0.326d | 0.885d | ||
| BEHAVE-AD – diurnal cycle disturbances – 78 weeks | D | 37 | 1 (2.7) | 33 | 9 (27.3) | 31 | 10 (32.3) | 0.010d | 0.003d | 0.871d | ||
| BEHAVE-AD – affective disturbances – 78 weeks | D | 37 | 13 (35.1) | 33 | 10 (30.3) | 31 | 13 (41.9) | 0.861d | 0.746d | 0.479d | ||
| BEHAVE-AD – anxiety and phobias – 78 weeks | D | 37 | 14 (37.8) | 33 | 15 (45.5) | 31 | 15 (48.4) | 0.687d | 0.529d | 0.988d | ||
Individuals taking rivastigmine were also reported to have a higher probability of remaining free of behavioural symptoms at 18 months than those taking donepezil in Cumbo’s RCT,99 although the methods adopted in the time-to-event (TTE) analysis are unclear.
Once more, heterogeneity of measures reported and follow-up times at which data are available makes it impossible to perform meaningful synthesis within or between the newly identified evidence base and that reported in 2004.
Global effect
Bullock and colleagues148 used the GDS to measure overall effect. They found that, over the 2-year trial, individuals who had been randomised to donepezil deteriorated by around 0.1 points more than those taking rivastigmine. As with the difference found on their chosen functional measure, this discrepancy appeared significant in the ITT LOCF analysis (p < 0.05 by Wilcoxon’s rank-sum test), but this finding was not repeated in secondary analyses based on evaluable and OCs.
In the Ancoli-Israel and colleagues RCT,149 none of the individuals taking galantamine experienced a global decline, according to the CIBIC-plus, over the 8 weeks of treatment, whereas 13% of those taking donepezil deteriorated on the same measure, although this difference does not appear to be a significant one (Table 38).
| Study | Subgroup | Outcome | Type | Rivastigmine | Donepezil | Galantamine | p-value | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| N | Mean, n (%) | N | Mean, n (%) | N | Mean, n (%) | Rivastigmine vs donepezil | Donepezil vs galantamine | ||||
| Bullock et al. (2005)148 | LOCF analysis | GDS – 104 weeks | MC | 471 | 0.58 (0.9) | 483 | 0.69 (0.9) | – | – | 0.049a | – |
| Ancoli-Israel et al. (2005)149 | OC population | CIBIC-plus score – 8 weeks | A | – | – | 29 | 3.97 (1.02) | 27 | 3.59 (0.64) | – | 0.106b |
| CIBIC-plus: markedly improved – 8 weeks | D | – | – | 29 | 0 (0.0) | 27 | 0 (0.0) | – | 0.330c | ||
| CIBIC-plus: moderately improved – 8 weeks | – | – | 29 | 3 (10.3) | 27 | 2 (7.4) | – | 0.933c | |||
| CIBIC-plus: minimally improved – 8 weeks | – | – | 29 | 4 (13.8) | 27 | 7 (25.9) | – | 0.421c | |||
| CIBIC-plus: no change – 8 weeks | – | – | 29 | 18 (62.1) | 27 | 18 (66.7) | – | 0.936c | |||
| CIBIC-plus: minimally worse – 8 weeks | – | – | 29 | 3 (10.3) | 27 | 0 (0.0) | – | 0.334c | |||
| CIBIC-plus: moderately worse – 8 weeks | – | – | 29 | 3 (10.3) | 27 | 0 (0.0) | – | 0.334c | |||
| CIBIC-plus: markedly worse – 8 weeks | – | – | 29 | 0 (0.0) | 27 | 0 (0.0) | – | 0.330c | |||
Quantitative synthesis combining the newly identified evidence and/or that reported in 2004 was not possible, owing to heterogeneity of measures reported and follow-up times at which data are available.
Quality of life
None of the newly identified,99,147–149 head-to-head, randomised studies investigated QoL with the technologies under assessment, and no such data were identified in the 2004 review. 2
Safety
A variety of AEs were reported in the included studies; the most common were nausea, diarrhoea, vomiting and headache (Tables 39–41).
| AE | Donepezil | Galantamine | Donepezil vs galantamine | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ancoli-Israel et al. (2005)149 | Cumbo (2005)99 | Nordberg et al. (2009)147 | Ancoli-Israel et al. (2005)149 | Cumbo (2005)99 | Nordberg et al. (2009)147 | Ancoli-Israel et al. (2005)149 | Cumbo (2005)99 | Nordberg et al. (2009)147 | |||||||
| N | n (%) | N | n (%) | N | n (%) | N | n (%) | N | n (%) | N | n (%) | p-valuea | |||
| Abdominal pain | 20 | 2 (10.0) | 21 | 0 (0.0) | 0.447 | ||||||||||
| Anorexia | 31 | 0 (0.0) | 33 | 1 (3.0) | 0.975 | ||||||||||
| Bronchitis | 32 | 0 (0.0) | 31 | 3 (9.7) | 0.226 | ||||||||||
| Constipation | 32 | 3 (9.4) | 31 | 0 (0.0) | 0.248 | ||||||||||
| Diarrhoea | 32 | 5 (15.6) | 20 | 0 (0.0) | 31 | 1 (3.2) | 21 | 6 (28.6) | 0.212 | 0.032 | |||||
| Dizziness | 20 | 1 (5.0) | 21 | 3 (14.3) | 0.635 | ||||||||||
| Headache | 32 | 3 (9.4) | 31 | 2 (6.5) | 20 | 2 (10.0) | 31 | 2 (6.5) | 33 | 0 (0.0) | 21 | 2 (9.5) | 0.970 | 0.445 | 0.635 |
| Influenza | 20 | 0 (0.0) | 21 | 2 (9.5) | 0.490 | ||||||||||
| Injury | 32 | 2 (6.3) | 31 | 2 (6.5) | 0.628 | ||||||||||
| Insomnia | 20 | 2 (10.0) | 21 | 2 (9.5) | 0.635 | ||||||||||
| Muscle spasms | 20 | 3 (15.0) | 21 | 1 (4.8) | 0.563 | ||||||||||
| Nausea | 32 | 1 (3.1) | 31 | 2 (6.5) | 20 | 2 (10.0) | 31 | 3 (9.7) | 33 | 2 (6.1) | 21 | 6 (28.6) | 0.583 | 0.651 | 0.269 |
| Painb | 32 | 3 (9.4) | 31 | 2 (6.5) | 0.970 | ||||||||||
| Upper respiratory tract infection | 20 | 1 (5.0) | 21 | 0 (0.0) | 0.980 | ||||||||||
| Vomiting | 31 | 0 (0.0) | 20 | 0 (0.0) | 33 | 1 (3.0) | 21 | 3 (14.3) | 0.975 | 0.248 | |||||
| Weight decrease | 31 | 0 (0.0) | 33 | 1 (3.0) | 0.975 | ||||||||||
| Weight loss | 20 | 1 (5.0) | 21 | 1 (4.8) | 0.490 | ||||||||||
| AE | Donepezil | Rivastigmine | Donepezil vs rivastigmine | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Bullock et al. (2005)148 | Cumbo (2005)99 | Nordberg et al. (2009)147 | Bullock et al. (2005)148 | Cumbo (2005)99 | Nordberg et al. (2009)147 | Bullock et al. (2005)148 | Cumbo (2005)99 | Nordberg et al. (2009)147 | |||||||
| N | n (%) | N | n (%) | N | n (%) | N | n (%) | N | n (%) | N | n (%) | p-valuea | |||
| Any AE | 453 | 349 (77.0) | 404 | 318 (78.7) | 0.613 | ||||||||||
| Any serious AE | 499 | 162 (32.5) | 495 | 157 (31.7) | 0.854 | ||||||||||
| Abdominal pain | 20 | 2 (10.0) | 22 | 0 (0.0) | 0.427 | ||||||||||
| Aggression | 453 | 25 (5.5) | 404 | 19 (4.7) | 0.700 | ||||||||||
| Agitation | 453 | 47 (10.4) | 404 | 34 (8.4) | 0.389 | ||||||||||
| Anorexia | 453 | 14 (3.1) | 31 | 0 (0.0) | 404 | 26 (6.4) | 37 | 1 (2.7) | 0.031 | 0.929 | |||||
| Depression | 453 | 16 (3.5) | 404 | 21 (5.2) | 0.303 | ||||||||||
| Diarrhoea | 453 | 30 (6.6) | 20 | 0 (0.0) | 404 | 26 (6.4) | 22 | 2 (9.1) | 0.978 | 0.512 | |||||
| Dizziness | 20 | 1 (5.0) | 22 | 3 (13.6) | 0.67 | ||||||||||
| Fall | 453 | 44 (9.7) | 404 | 33 (8.2) | 0.503 | ||||||||||
| Headache | 453 | 12 (2.6) | 31 | 2 (6.5) | 20 | 2 (10.0) | 404 | 13 (3.2) | 37 | 1 (2.7) | 22 | 3 (13.6) | 0.771 | 0.875 | 0.91 |
| Hypertension | 453 | 18 (4.0) | 404 | 21 (5.2) | 0.487 | ||||||||||
| Influenza | 20 | 0 (0.0) | 22 | 1 (4.5) | 0.962 | ||||||||||
| Injury | |||||||||||||||
| Insomnia | 20 | 2 (10.0) | 22 | 1 (4.5) | 0.932 | ||||||||||
| Muscle spasms | 20 | 3 (15.0) | 22 | 0 (0.0) | 0.199 | ||||||||||
| Nausea | 453 | 24 (5.3) | 31 | 2 (6.5) | 20 | 2 (10.0) | 404 | 52 (12.9) | 37 | 3 (8.1) | 22 | 10 (45.5) | < 0.001 | 0.837 | 0.028 |
| Upper respiratory tract infection | 20 | 1 (5.0) | 22 | 2 (9.1) | 0.932 | ||||||||||
| Urinary tract infection | 453 | 26 (5.7) | 404 | 18 (4.5) | 0.487 | ||||||||||
| Vomiting | 453 | 20 (4.4) | 31 | 0 (0.0) | 20 | 0 (0.0) | 404 | 62 (15.3) | 37 | 1 (2.7) | 22 | 4 (18.2) | < 0.001 | 0.929 | 0.139 |
| Weight decrease | 453 | 43 (9.5) | 31 | 0 (0.0) | 404 | 36 (8.9) | 37 | 0 (0.0) | 0.861 | ||||||
| Weight loss | 20 | 1 (5.0) | 22 | 2 (9.1) | 0.932 | ||||||||||
| AE | Galantamine | Rivastigmine | Galantamine vs rivstigmine | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cumbo (2005)99 | Nordberg et al. (2009)147 | Cumbo (2005)99 | Nordberg et al. (2009)147 | Cumbo (2005)99 | Nordberg et al. (2009)147 | |||||
| N | n (%) | N | n (%) | N | n (%) | N | n (%) | p-valuea | ||
| Abdominal pain | 21 | 0 (0.0) | 22 | 0 (0.0) | – | |||||
| Anorexia | 33 | 1 (3.0) | 37 | 1 (2.7) | 0.524 | |||||
| Diarrhoea | 21 | 6 (28.6) | 22 | 2 (9.1) | 0.212 | |||||
| Dizziness | 21 | 3 (14.3) | 22 | 3 (13.6) | 0.705 | |||||
| Headache | 33 | 0 (0.0) | 21 | 2 (9.5) | 37 | 1 (2.7) | 22 | 3 (13.6) | 0.954 | 0.956 |
| Influenza | 21 | 2 (9.5) | 22 | 1 (4.5) | 0.967 | |||||
| Insomnia | 21 | 2 (9.5) | 22 | 1 (4.5) | 0.967 | |||||
| Muscle spasms | 21 | 1 (4.8) | 22 | 0 (0.0) | 0.981 | |||||
| Nausea | 33 | 2 (6.1) | 21 | 6 (28.6) | 37 | 3 (8.1) | 22 | 10 (45.5) | 0.894 | 0.407 |
| Upper respiratory tract infection | 21 | 0 (0.0) | 22 | 2 (9.1) | 0.490 | |||||
| Vomiting | 33 | 1 (3.0) | 21 | 3 (14.3) | 37 | 1 (2.7) | 22 | 4 (18.2) | 0.524 | 0.946 |
| Weight decrease | 33 | 1 (3.0) | 37 | 0 (0.0) | 0.954 | |||||
| Weight loss | 21 | 1 (4.8) | 22 | 2 (9.1) | 0.967 | |||||
Summary: head-to-head comparisons
Four new head to head RCTs were found;99,147–149 two compared all included AChEIs,99,147 one compared donepezil to rivastigmine148 and one compared donepezil to galantamine. 149 Pooling of data from head-to-head trials was not possible owing to the heterogeneity of the data. The quality of the evidence they provide is limited because of the poor quality of most of the trials. The exception to this was Bullock and colleagues,148 whose good-quality study found no significant difference between donepezil and rivastigmine for cognitive or behavioural outcomes. However, when they looked at functional and global outcomes, patients taking rivastigmine fared significantly better than those taking donepezil in the primary analysis.
Graphical summary of head-to-head comparisons
Figure 51 clearly shows that this group of studies is dominated by the new comparison of rivastigmine with donepezil by Bullock and colleagues. 148 The small studies from the previous review indicate that there is no difference between donepezil and galantamine on cognitive outcomes. These earlier results support those of the much larger study by Bullock and colleagues,148 which also shows that rivastigmine is significantly better than donepezil on functional and global outcomes. Previously, when donepezil and galantamine were compared in a small, poor-quality trial, the results favoured donepezil on cognitive and functional outcomes; no new evidence for this comparison for these outcomes was found. There was no good or even moderate evidence comparing all three AChEIs.
FIGURE 51.
Summary of all included head-to-head studies in the 2004 and 2010 reviews. Analy, analysis; be, behavioural; Blind, blinding; Char, characteristics of patients; cog, cognitive; F, female; glo, global effect; GvD, galantamine vs donepezil; M, male; Rand, randomisation; RvD, rivastigmine vs donepezil; RvG, rivastigmine vs galantamine. a, Significant difference in 1 of 12 categories only (night-time behaviour). b, Significant difference in one out of seven categories only (diurnal cycle disturbances).
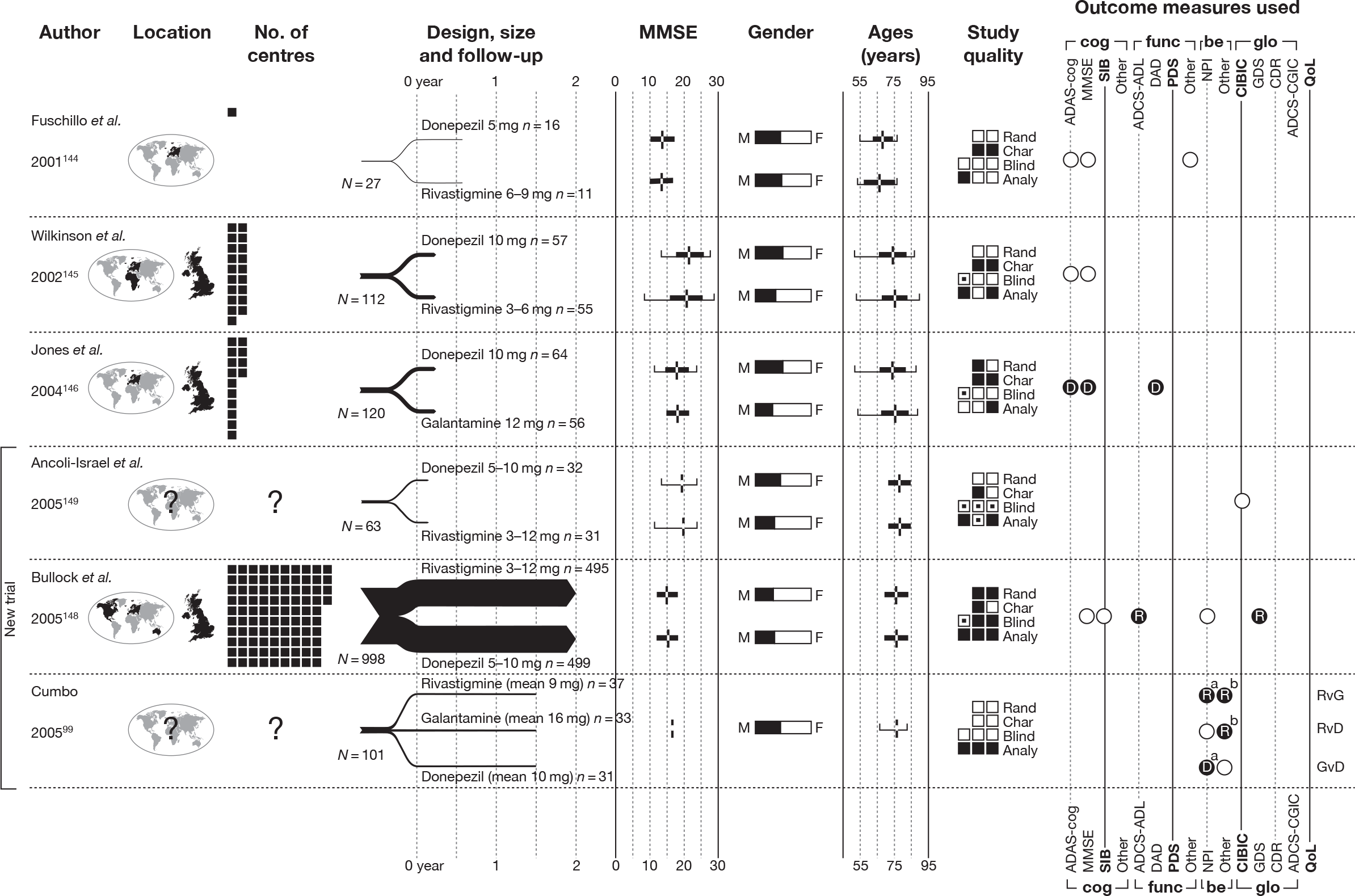
Combination therapy
Identified evidence
Our searches identified a single new randomised trial addressing the effectiveness of combination therapy consisting of two of the technologies under review. 150 Details of the design and characteristics are presented in Tables 42 and 43, and an assessment of study quality in Table 44.
| Study details | Inclusion criteria | Exclusion criteria | Methodological notes | Other |
|---|---|---|---|---|
|
Porsteinsson et al. (2008) 150 Design: Parallel double-blind RCT Country: USA No. of centres: 38 No. randomised: 433 Maximum follow-up: 24 MMSE range included: 10–22 Funding: Forest Laboratories Inc. (New York, NY, USA) provided all financial and material support for research and analyses – and assisted the Memantine Study Group in the development of the trial design, implementation, data collection, post hoc analyses and manuscript development |
Probable AD (NINCDS-ADRDA criteria) Age ≥ 50 years MRI or CT scan results consistent with AD diagnosis and acquired within 1 year of study Treatment with cholinesterase inhibitors for ≥ 6 months, and a stable-dosing regimen for ≥ 3 months (donepezil 5 or 10 mg/day; rivastigmine 6, 9 or 12 mg/day; galantamine 16 or 24 mg/day) A knowledgable and reliable caregiver to acompany the participant to all study visits and supervise administraton of study drug Ability to ambulate Vision and hearing sufficient to permit compliance with assessments Montgomery–Åsberg Depression Rating Scale (MADRS) score < 22 Medically stable Postmenopausal for ≥ 2 years, or surgically sterile (female participants) |
Clinically significant and active pulmonary, gastrointestinal, renal, hepatic, endocrine or cardiovascular disease Clinically significant vitamin B12 or folate deficiency Evidence (including CT/MRI) of other psychiatric or neurological disorders Dementia complicated by organic disease or AD with delusions or delirium Undergoing treatment for an oncology diagnosis or completion of treatment within 6 months of screening Modified Hachinski Ischaemic Score > 4 Poorly controlled hypertension Substance abuse Participation in an investigational drug study or use of an investigational drug within 30 days (or five half-lives, whichever is longer) of screening Depot neuroleptic use within 6 months of screening Positive urine drug test Likely institutionalisation during trial Previous memantine treatment or participation in an investgational study of memantine Likely cessation of cholinesterase inhibitors during the trial |
Sample attrition/dropout: 385 of 433 completed study. Dropouts in memantine arm: AEs n = 13, withdrew consent n = 4, protocol violation n = 5, insufficient therapeutic response n = 1; dropouts in placebo arm: AEs n = 17, withdrew consent n = 4, protocol violation n = 1, insufficient therapeutic response n = 1 and other n = 2. No differences between groups Randomisation and allocation: Randomised in permuted blocks of four in accordance with randomisation list generated and retained by Forest Research Institute, Department of Statistical Programming. Participants were sequentially assigned randomisation numbers at the baseline visit. No individual participant randomisation code was revealed during the trial. Memantine and placebo tablets described as being identical in appearance Power calculation: Assuming an effect size (defined as difference of mean scores between treatment groups on ADAS-cog at end point (LOCF), relative to pooled SD, of 0.325, at least 400 participants were needed to provide 90% power at an alpha level of 0.05 (two-sided), based on a two-sided t-test. The total patient population, consisting of all participants randomised into the study (n = 433) was identical to the safety population, which consisted of randomised participants who received at least one dose of double-blind study medication. The ITT population (n = 427) comprised participants in the safety population who completed at least one postbaseline ADAS-cog or CIBIC-plus assessment |
Therapy common to all participants: All participants continued to take cholinesterase inhibitor (donepezil, galantamine or rivastigmine) 1- to 2-week single-blind placebo lead-in phase completed before randomisation to assess compliance Other conflicts: One co-author’s (JO) affilliation is Novartis Inc. |
| Study | Arm | Dose (mg/day) | Dosage details | N | Age, years (SD) | Gender, n male (%) | Race, n white (%) | Weight, kg (SD) | Education (years) | Duration of dementia, months (SD) | ADAS-cog, score (SD) | MMSE, score (SD) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Porsteinsson et al. (2008)150 | Memantine | 5–20 |
Titrated from an initial dosage of 5 mg/day in 5-mg weekly increments to a maximum dose of 20 mg/day (administered as four 5 mg tablets q.d. at bedtime) AChEIs administered separately as part of participants’ ongoing maintenance therapy |
217 | 74.9 (7.64) | 100 (46.1) | 70.0 (14.9) | 16.7 (3.67) | ||||
| Placebo | – |
Placebo had identical appearance to memantine AChEIs administered separately as part of participants’ ongoing maintenance therapy |
216 | 76.0 (8.43) | 107 (49.5) | 72.2 (14.7) | 17.0 (3.64) |
| Study | Was the assignment to the treatment groups really random? | Was the treatment allocation concealed? | Were the groups similar at baseline in terms of prognostic factors? | Were the eligibility criteria specified? | Were outcome assessors blinded to the treatment allocation? | Was the care provider blinded? | Was the patient blinded? | Were the point estimates and measure of variability presented for the primary outcome measure? | Did the analyses include an ITT analysis? | Were withdrawals and dropouts completely described? |
|---|---|---|---|---|---|---|---|---|---|---|
| Porsteinsson et al. (2008)150 | Adequate | Adequate | Reported – yes | Inadequate | Unknown | Adequate | Adequate | Adequate | Adequate | Adequate |
One included study in the 2004 review141 addressed the effectiveness of donepezil plus memantine versus donepezil plus placebo. In the 2004 review,2 this is considered among the evidence of effectiveness of memantine. We have not followed this approach, as we prefer to assess monotherapy and combination regimens separately, because the effect of multiple agents may or may not be straightforwardly additive.
Evidence of clinical effectiveness
Cognition
The new study by Porsteinsson and colleagues150 failed to show any benefit on cognitive outcomes from combining memantine with an AChEI (Table 45). One reason for this may be an underlying pharmacological interaction between galantamine and memantine that could neutralise their effects.
| Study | Subgroup | Outcome | Type | AChEI + memantine | AChEI + placebo | p-value | ||
|---|---|---|---|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | |||||
| Porsteinsson et al. (2008)150 | LOCF analysis | ADAS-cog – 24 weeks | C | 214 | 28.5 (12.8) | 213 | 28 (11.9) | 0.184a |
| MMSE – 24 weeks | C | 210 | 16.5 (5.38) | 198 | 16.4 (5.08) | 0.123a | ||
| OC population | ADAS-cog – 24 weeks | C | 192 | 28.2 (12.8) | 188 | 27.6 (11.7) | 0.186a | |
| MMSE – 24 weeks | C | 193 | 16.6 (5.41) | 188 | 16.4 (5.08) | 0.190a | ||
Because the previously identified study141 relies on SIB to estimate the effect of combination therapy on cognition, whereas Porsteinsson and colleagues150 report MMSE and ADAS-cog, it was not possible to combine the two studies in a WMD meta-analysis, nor would it have been informative to combine the two RCTs on a standardised scale.
Functional
Similarly, functional outcomes failed to show a significant difference between combination therapy and an AChEI plus placebo (Table 46).
| Study | Subgroup | Outcome | Type | AChEI + memantine | AChEI + placebo | p-value | ||
|---|---|---|---|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | |||||
| Porsteinsson et al. (2008)150 | LOCF analysis | ADCS-ADL – 24 weeks | C | 214 | 51.8 (15.9) | 213 | 52 (15.7) | 0.816a |
| OC population | 193 | 51.8 (16) | 189 | 53.6 (14.6) | 0.741a | |||
Although both relevant studies use the ADCS-ADL to measure the functional effectiveness of combination therapy, different versions of the instrument are adopted: Porsteinsson and colleagues150 rely on the 23-item scale, whereas Tariot and colleagues141 use the 19-item version. Accordingly, it is not valid to synthesise these data on their original scales.
Behavioural and mood
Porsteinsson and colleagues150 also failed to show any benefit from combination therapy when behavioural outcomes were measured with the NPI (Table 47).
| Study | Subgroup | Outcome | Type | AChEI + memantine | AChEI + placebo | p-value | ||
|---|---|---|---|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | |||||
| Porsteinsson et al. (2008)150 | LOCF analysis | NPI – 24 weeks | MC | 212 | 0.70 (12.01)a | 209 | 0.40 (12.29)a | NSb |
| C | 212 | 12.9 (14.5) | 209 | 12.6 (14.6) | 0.743b | |||
| OC population | NPI – 12 weeks | MC | 193 | 0.80 (10.77)a | 189 | 0.30 (10.65)a | NSb | |
| NPI – 24 weeks | MC | 193 | 0.00 (11.81)a | 189 | 0.00 (11.69)a | NSb | ||
| NPI – 24 weeks | C | 193 | 12.3 (13.7) | 189 | 11.9 (13.5) | 0.985b | ||
When the data from Porsteinsson and colleagues150 were pooled with the existing data from the NPI at 12 and 24 weeks, the overall pooled estimates showed no significant gain from combination therapy (Figures 52 and 53).
FIGURE 52.
Random-effects meta-analysis – NPI at 12 weeks (mean change from baseline): AChEI + memantine vs AChEI + placebo.
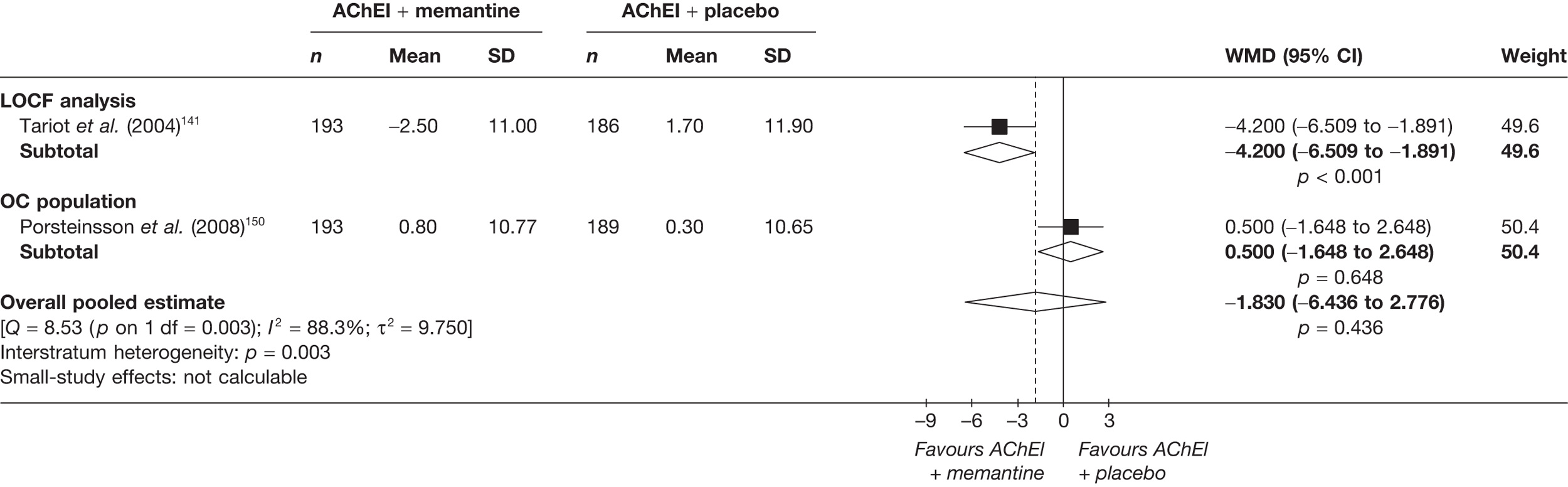
FIGURE 53.
Random-effects meta-analysis – NPI at 24 weeks (mean change from baseline): AChEI + memantine vs AChEI + placebo.
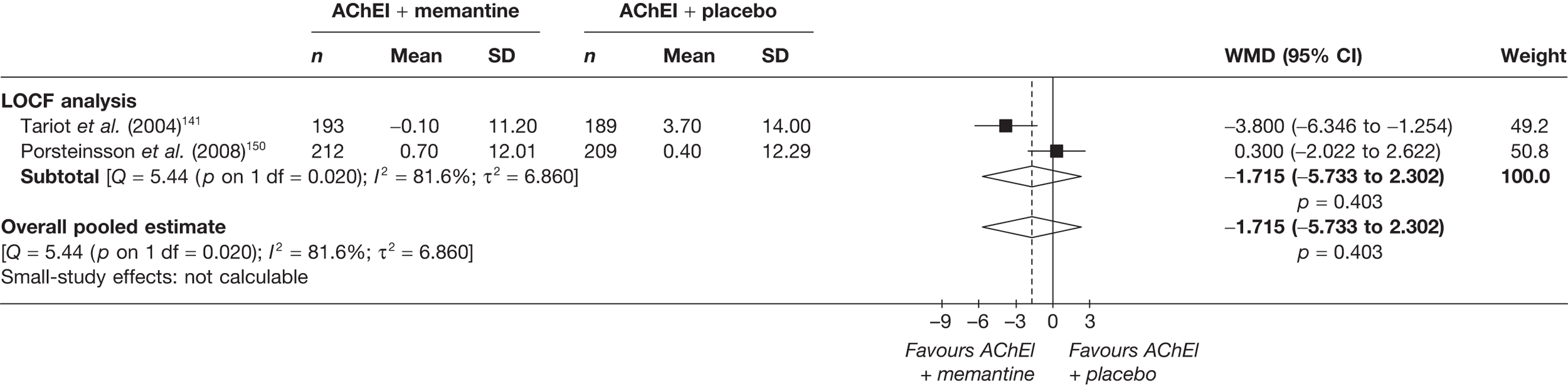
Global effect
Again, with global outcomes, no additional benefit was found from combination therapy (Table 48).
| Study | Subgroup | Outcome | Type | AChEI + memantine | AChEI + placebo | p-value | ||
|---|---|---|---|---|---|---|---|---|
| n | Mean (SD) | n | Mean (SD) | |||||
| Porsteinsson et al. (2008)150 | LOCF analysis | CIBIC-plus score – 24 weeks | C | 214 | 4.38 (1) | 213 | 4.42 (0.96) | 0.843a |
| OC population | 192 | 4.36 (1.01) | 189 | 4.4 (0.96) | 0.650a | |||
A synthesis of new and existing evidence for global outcomes showed no overall benefit from combination therapy (see Table 45).
FIGURE 54.
Random-effects meta-analysis – CIBIC-plus at 24 weeks: AChEI + memantine vs AChEI + placebo.
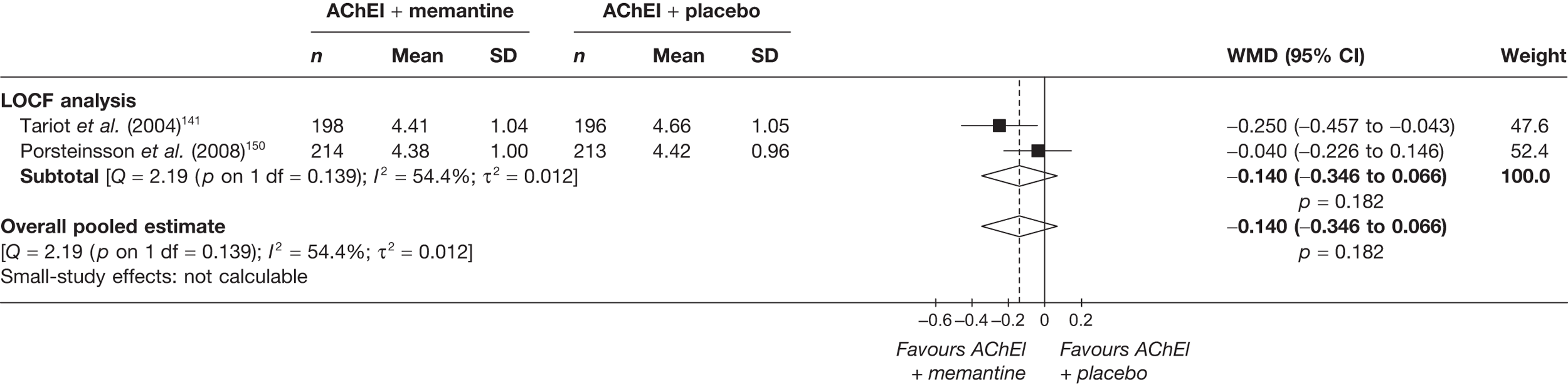
Quality of life
The new included study did not provide any randomised evidence on QoL with combination therapy and no such data were identified in the 2004 review. 2
Safety
The proportion of AEs did not significantly vary between groups. The most common AEs were falls and injury (Table 49).
| AE | Porsteinsson et al. (2008)148 | ||||
|---|---|---|---|---|---|
| AChEI + memantine | AChEI + placebo | p-valuea | |||
| N | n (%) | N | n (%) | ||
| Any serious AE | 217 | 27 (12.4) | 216 | 30 (13.9) | 0.762 |
| Diarrhoea | 217 | 12 (5.5) | 216 | 14 (6.5) | 0.830 |
| Agitation | 217 | 17 (7.8) | 216 | 17 (7.9) | 0.869 |
| Depression | 217 | 14 (6.5) | 216 | 15 (6.9) | 0.990 |
| Injury | 217 | 20 (9.2) | 216 | 16 (7.4) | 0.612 |
| Dizziness | 217 | 16 (7.4) | 216 | 16 (7.4) | 0.865 |
| Upper respiratory tract infection | 217 | 12 (5.5) | 216 | 6 (2.8) | 0.233 |
| Fall | 217 | 22 (10.1) | 216 | 15 (6.9) | 0.309 |
| Influenza-like symptoms | 217 | 15 (6.9) | 216 | 12 (5.6) | 0.700 |
| Abnormal gait | 217 | 14 (6.5) | 216 | 9 (4.2) | 0.398 |
| Confusion | 217 | 12 (5.5) | 216 | 9 (4.2) | 0.662 |
| Fatigue | 217 | 11 (5.1) | 216 | 7 (3.2) | 0.476 |
| Hypertension | 217 | 11 (5.1) | 216 | 6 (2.8) | 0.327 |
Summary: combination therapy
Our searches found one new trial that compared memantine plus an AChEI with an AChEI. 148 This failed to show any benefit from combining memantine with an AChEI on cognitive, functional, behavioural or global outcomes. Pooling these data with previous trails also failed to show any additional benefit from combination therapy.
Graphical summary of combination therapy
Figure 55 clearly illustrates the similarities between the new and existing evidence for combination therapy; what is striking is the difference in results. Some of the variation may be explained by the use of different outcome measures or versions of outcome measures. However, it is unclear why the behavioural and global outcome results are different. The designs of these studies differed in that Porsteinsson and colleagues150 combined memantine with any of the three included AChEIs, whereas Tariot and colleagues141 only combined memantine with donepezil. The other notable difference is that the 2004141 authors analysed a modified ITT population, whereas the 2008150 study authors analysed a full ITT population. Whether or not these differences are sufficient to account for these differences in apparently similar populations is unknown.
FIGURE 55.
Summary of all studies included in the 2004 and 2010: combination therapy. Analy, analysis; be, behavioural; Blind, blinding; Char, characteristics of patients; cog, cognitive; F, female; glo, global effect; M, male; Rand, randomisation.
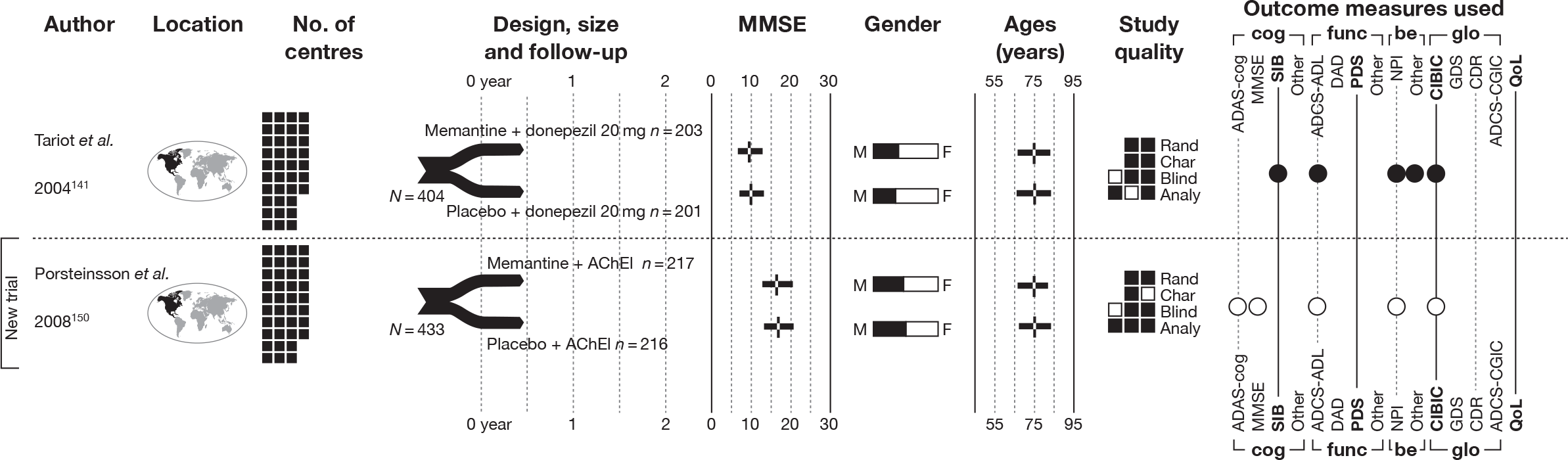
Mixed-treatment comparisons: indirect comparisons
Where there were sufficient data we pooled information on all technologies and their comparators simultaneously, in a MTC using Bayesian MCMC sampling. 89–92 The results are shown in terms of treatment effect compared with a common baseline. The evidence network shows the comparisons that were available and the quantity of those comparisons (by the thickness of the connecting lines). More details can be found in Methods of quantitative analysis/Mixed-treatment comparisons: indirect comparisons. MTCs of the technologies performed in specified measurement populations can be found in Appendix 9.
Cognitive
Alzheimer’s Disease Assessment Scale – Cognitive Subscale
Table 50 shows the studies pooled in this MTC at 12–16 weeks’ follow-up, with their evidence network and effectiveness estimates. The results in Table 51 give the relative effectiveness of each technology compared with placebo, indicating that donepezil and galantamine are certainly more effective than placebo and that donepezil is probably the most effective of these (0.48).
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Rogers et al. (1998)93 | –2.799 | –3.831 to –1.767 |
| Burns et al. (1999)104 | –2.151 | –2.871 to –1.430 | ||
| Homma et al. (2000)108 | –2.175 | –3.527 to –0.823 | ||
| Nunez et al. (2003)111,112 | –0.050 | –1.782 to 1.682 | ||
| Galantamine vs placebo | Raskind et al. (2000)123 | –3.158 | –4.371 to –1.946 | |
| Tariot et al. (2000)125 | –2.225 | –3.042 to –1.408 | ||
| Wilcock et al. (2000)127 | –2.848 | –3.829 to –1.867 | ||
| Rockwood et al. (2001)124 | –1.600 | –2.704 to –0.496 | ||
| Wilkinson and Murray (2001)128 | –2.246 | –3.872 to –0.620 | ||
| Bullock et al. (2004)101 | –1.475 | –2.933 to –0.017 | ||
| Brodaty et al. (2005)96 | –2.453 | –3.192 to –1.713 | ||
| Rockwood et al. (2006)131 | –1.925 | –3.816 to –0.034 | ||
| Rivastigmine vs placebo | Feldman and Lane (2007)138 | –2.249 | –3.226 to –1.271 | |
| Winblad et al. (2007)140 | –0.911 | –1.817 to –0.006 | ||
| Donepezil vs rivastigmine | Wilkinson et al. (2002)145 | 0.150 | –1.561 to 1.861 | |
| Donepezil vs galantamine | Jones et al. (2004)146 | –2.225 | –4.131 to –0.319 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.000 |
| Donepezil | –2.209 | –2.951 to –1.452 | 1.000 | 0.475 |
| Galantamine | –2.176 | –2.725 to –1.540 | 1.000 | 0.421 |
| Rivastigmine | –1.700 | –2.728 to –0.751 | 0.999 | 0.104 |
| Memantine | – | – | – | – |
At 21–26 weeks’ follow-up the MTC showed that all the treatments were more effective than placebo, with galantamine probably the most effective (0.89) (Tables 52 and 53).
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Rogers et al. (1998)113 | –2.684 | –3.876 to –1.491 |
| Burns et al. (1999)104 | –2.203 | –2.968 to –1.438 | ||
| Homma et al. (2000)108 | –2.540 | –3.427 to –1.653 | ||
| Galantamine vs placebo | Raskind et al. (2000)123 | –3.653 | –4.696 to –2.611 | |
| Tariot et al. (2000)125 | –2.741 | –3.633 to –1.850 | ||
| Wilcock et al. (2000)127 | –3.049 | –4.030 to –2.068 | ||
| Bullock et al. (2004)101 | –3.100 | –4.620 to –1.580 | ||
| Brodaty et al. (2005)96 | –2.651 | –3.449 to –1.854 | ||
| Rivastigmine vs placebo | Corey-Bloom et al. (1998)135 | –2.751 | –3.694 to –1.808 | |
| Rosler et al. (1999)137 | –0.785 | –1.851 to 0.281 | ||
| Feldman and Lane (2007)138 | –2.298 | –3.460 to –1.137 | ||
| Winblad et al. (2007)140 | –1.943 | –2.858 to –1.029 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.000 |
| Donepezil | –2.431 | –3.174 to –1.709 | 1.000 | 0.107 |
| Galantamine | –2.986 | –3.591 to –2.405 | 1.000 | 0.885 |
| Rivastigmine | –1.978 | –2.630 to –1.303 | 1.000 | 0.009 |
| Memantine | – | – | – | – |
Mini Mental State Examination
The data used for the 12- to 13-week MTC for MMSE can be seen in Table 54. The results in Table 55 show that at this early follow-up donepezil is the only treatment that is certainly more effective than placebo and consequently probably the most effective treatment overall (0.54).
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
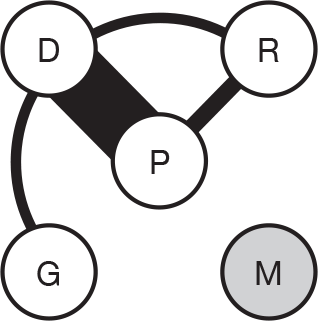
|
Donepezil vs placebo | Rogers et al. (1998)93 | 1.110 | 0.514 to 1.706 |
| Mohs et al. (2001)110 | 1.600 | 0.889 to 2.311 | ||
| Winblad et al. (2001)116 | 0.800 | 0.075 to 1.525 | ||
| Gauthier et al. (2002)105 | 2.000 | 0.820 to 3.180 | ||
| Nunez et al. (2003)111,112 | 0.830 | –0.071 to 1.731 | ||
| AD2000 (2004)103 | 0.930 | 0.389 to 1.471 | ||
| Holmes et al. (2004)107 | 1.700 | 0.169 to 3.231 | ||
| Seltzer et al. (2004)115 | 1.175 | 0.100 to 2.250 | ||
| Rivastigmine vs placebo | Agid et al. (1998)134 | 0.144 | –0.493 to 0.782 | |
| Mowla et al. (2007)139 | 1.600 | 1.099 to 2.101 | ||
| Donepezil vs rivastigmine | Wilkinson et al. (2002)145 | –0.490 | –1.825 to 0.845 | |
| Donepezil vs galantamine | Jones et al. (2004)146 | 0.888 | 0.004 to 1.771 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.000 |
| Donepezil | 1.145 | 0.677 to 1.637 | 1.000 | 0.537 |
| Galantamine | 0.259 | –1.214 to 1.761 | 0.646 | 0.075 |
| Rivastigmine | 1.057 | 0.283 to 1.852 | 0.993 | 0.389 |
| Memantine | – | – | – | – |
At 24–26 weeks from baseline there is no evidence from galantamine, and donepezil continues to show that it is probably the most effective treatment (0.67) (Tables 56 and 57).
| Evidence Network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
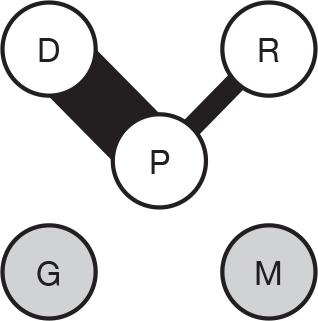
|
Donepezil vs placebo | Rogers et al. (1998)113 | 1.284 | 0.604 to 1.964 |
| Mohs et al. (2001)110 | 1.350 | 0.188 to 2.512 | ||
| Winblad et al. (2001)116 | 1.490 | 0.548 to 2.432 | ||
| Gauthier et al. (2002)105 | 2.060 | 0.880 to 3.240 | ||
| AD2000 (2004)103 | 0.500 | –0.250 to 1.250 | ||
| Seltzer et al. (2004)115 | 1.250 | 0.171 to 2.329 | ||
| Mazza et al. (2006)118 | 1.450 | –3.720 to 6.620 | ||
| Rivastigmine vs placebo | Feldman and Lane (2007)138 | 1.250 | 0.670 to 1.830 | |
| Winblad et al. (2007)140 | 0.932 | 0.461 to 1.403 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.000 |
| Donepezil | 1.235 | 0.747 to 1.778 | 1.000 | 0.670 |
| Galantamine | – | – | – | – |
| Rivastigmine | 1.073 | 0.358 to 1.809 | 0.993 | 0.330 |
| Memantine | – | – | – | – |
Functional
Alzheimer’s Disease Cooperative Study – Activities of Daily Living Index
Mixed-treatment comparisons were conducted for the ADCS-ADL. At 12–16 weeks, galantamine and rivastigmine were shown to be almost equally effective compared with placebo, with rivastigmine possibly being the most effective (0.50) (Tables 58 and 59).
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
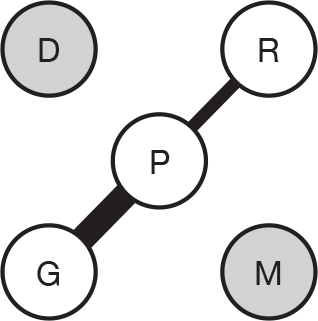
|
Galantamine vs placebo | Tariot et al. (2000)125 | 1.810 | 0.613 to 3.007 |
| Brodaty et al. (2005)96 | 1.052 | –0.034 to 2.138 | ||
| Rivastigmine vs placebo | Winblad et al. (2007)140 | 1.411 | 0.279 to 2.543 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.008 |
| Donepezil | – | – | – | – |
| Galantamine | 1.410 | –0.316 to 3.148 | 0.956 | 0.494 |
| Rivastigmine | 1.410 | –1.033 to 3.842 | 0.907 | 0.498 |
| Memantine | – | – | – | – |
At 21–26 weeks’ follow-up, the situation had changed, with galantamine showing a slightly greater probability of being the most effective technology (0.55) (Tables 60 and 61).
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
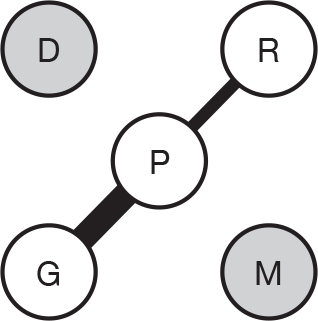
|
Galantamine vs placebo | Tariot et al. (2000)125 | 2.276 | 0.889 to 3.663 |
| Brodaty et al. (2005)96 | 2.203 | 1.007 to 3.399 | ||
| Rivastigmine vs placebo | Winblad et al. (2007)140 | 2.101 | 0.788 to 3.415 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.001 |
| Donepezil | – | – | – | – |
| Galantamine | 2.238 | 0.528 to 3.943 | 0.990 | 0.547 |
| Rivastigmine | 2.091 | –0.322 to 4.519 | 0.962 | 0.451 |
| Memantine | – | – | – | – |
Behavioural
Neuropsychiatric Inventory
The MTC for behavioural outcomes were measured using the NPI. At 12–13 weeks from baseline, donepezil was probably more effective than galantamine at controlling behavioural symptoms (0.78) (Tables 62 and 63).
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Gauthier et al. (2002)105 | –2.900 | –6.783 to 0.983 |
| Nunez et al. (2003)111,112 | –2.870 | –5.406 to –0.334 | ||
| AD2000 (2004)103 | 1.250 | 1.500 to 4.000 | ||
| Holmes et al. (2004)107 | –6.200 | –11.374 to –1.026 | ||
| Galantamine vs placebo | Tariot et al. (2000)125 | –0.719 | –2.056 to 0.618 | |
| Rockwood et al. (2001)124 | –0.900 | –2.688 to 0.888 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.006 |
| Donepezil | –1.960 | –4.095 to 0.033 | 0.973 | 0.799 |
| Galantamine | –0.788 | –2.872 to 1.267 | 0.810 | 0.195 |
| Rivastigmine | – | – | – | – |
| Memantine | – | – | – | – |
At 21–28 weeks’ follow-up there were also data on rivastigmine and memantine to put into the MTC. However, donepezil was still probably the most effective treatment (0.57) (Tables 64 and 65).
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Gauthier et al. (2002)105 | –5.920 | –10.126 to –1.714 |
| AD2000 (2004)103 | –0.750 | –3.750 to 2.250 | ||
| Galantamine vs placebo | Tariot et al. (2000)125 | –1.574 | –3.226 to 0.078 | |
| Brodaty et al. (2005)96 | –1.349 | –2.900 to 0.202 | ||
| Rivastigmine vs placebo | Winblad et al. (2007)140 | –0.372 | –2.205 to 1.461 | |
| Memantine vs placebo | Reisberg et al. (2003)142 | –3.300 | –7.334 to 0.734 | |
| Van Dyck et al. (2007)143 | –0.100 | –3.845 to 3.645 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.000 |
| Donepezil | –2.683 | –5.673 to 0.207 | 0.966 | 0.576 |
| Galantamine | –1.462 | –3.438 to 0.526 | 0.940 | 0.129 |
| Rivastigmine | –0.366 | –3.308 to 2.554 | 0.612 | 0.052 |
| Memantine | –1.600 | –4.762 to 1.540 | 0.845 | 0.243 |
Global
For global outcomes MTC was carried out using the CIBIC-plus and the GDS.
Clinician’s Interview-based Impression of Change plus Caregiver Input
At 12–16 weeks post baseline, a MTC of all the treatments showed that galantamine was probably the most effective treatment (0.54) (Tables 66 and 67).
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Rogers et al. (1998)93 | –0.350 | –0.527 to –0.174 |
| Burns et al. (1999)104 | –0.265 | –0.406 to –0.125 | ||
| Gauthier et al. (2002)106 | –0.490 | –0.768 to –0.212 | ||
| Galantamine vs placebo | Rockwood et al. (2001)124 | –0.335 | –0.524 to –0.146 | |
| Rockwood et al. (2006)131 | –0.450 | –0.797 to –0.103 | ||
| Rivastigmine vs placebo | Rosler et al. (1999)137 | –0.007 | –0.186 to 0.172 | |
| Memantine vs placebo | Reisberg et al. (2003)142 | –0.070 | –0.347 to 0.207 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.001 |
| Donepezil | –0.338 | –0.647 to –0.079 | 0.985 | 0.373 |
| Galantamine | –0.370 | –0.746 to –0.025 | 0.978 | 0.541 |
| Rivastigmine | –0.007 | –0.492 to 0.477 | 0.520 | 0.027 |
| Memantine | –0.071 | –0.591 to 0.448 | 0.647 | 0.058 |
However, at 24–28 weeks’ follow-up, donepezil was probably the most effective (0.55) (Tables 68 and 69).
| Evidence Network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Rogers et al. (1998)113 | –0.400 | –0.593 to –0.207 |
| Burns et al. (1999)104 | –0.340 | –0.484 to –0.196 | ||
| Gauthier et al. (2002)105 | –0.545 | –0.858 to –0.232 | ||
| Galantamine vs placebo | Raskind et al. (2000)123 | –0.248 | –0.419 to –0.077 | |
| Wilcock et al. (2000)127 | –0.288 | –0.450 to –0.127 | ||
| Brodaty et al. (2005)96 | –0.138 | –0.294 to 0.018 | ||
| Rivastigmine vs placebo | Corey-Bloom et al. (1998)135 | –0.275 | –0.471 to –0.079 | |
| Rosler et al. (1999)137 | –0.300 | –0.519 to –0.081 | ||
| Feldman and Lane (2007)138 | –0.500 | –0.711 to –0.289 | ||
| Memantine vs placebo | Reisberg et al. (2003)147 | –0.300 | –0.582 to –0.018 | |
| Van Dyck et al. (2007)143 | –0.300 | –0.515 to –0.085 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.000 |
| Donepezil | –0.392 | –0.549 to –0.251 | 1.000 | 0.546 |
| Galantamine | –0.222 | –0.356 to –0.091 | 0.997 | 0.010 |
| Rivastigmine | –0.354 | –0.508 to –0.203 | 1.000 | 0.285 |
| Memantine | –0.300 | –0.507 to –0.100 | 0.996 | 0.159 |
Global Deterioration Scale
There were only data from the GDS at 24–28 weeks from baseline. This indicated that rivastigmine was probably more effective than donepezil or memantine for global outcomes (0.49) (Tables 70 and 71).
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Winblad et al. (2001)116 | 0.160 | –0.006 to 0.326 |
| Rivastigmine vs placebo | Corey-Bloom et al. (1998)135 | 0.175 | 0.065 to 0.285 | |
| Rosler et al. (1999)137 | 0.120 | –0.042 to 0.282 | ||
| Feldman and Lane (2007)138 | 0.200 | 0.087 to 0.312 | ||
| Memantine vs placebo | Reisberg et al. (2003)142 | –0.100 | –0.220 to 0.020 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.012 |
| Donepezil | 0.161 | –0.402 to 0.720 | 0.866 | 0.453 |
| Galantamine | – | – | – | – |
| Rivastigmine | 0.171 | –0.159 to 0.486 | 0.941 | 0.491 |
| Memantine | –0.099 | –0.662 to 0.450 | 0.189 | 0.043 |
Summary: mixed-treatment comparisons
The MTC results for cognitive outcomes varied with follow-up time and the measure used. Donepezil was shown to be probably the most effective treatment at short-term follow-up on the ADAS-cog and MMSE, and this remained the case for the MMSE at 24–26 weeks; however, the ADAS-cog favoured galantamine at this later follow-up time. Functional outcomes measured with the ADCS-ADL showed equal effectiveness from galantamine and rivastigmine at 12–16 weeks, but by 21–26 weeks galantamine was probably the most effective treatment. For behavioural outcomes donepezil came out most favourably. For global outcomes the results were less clear, with galantamine probably the best treatment at 12–16 weeks when measured by the CIBIC-plus, but donepezil taking over by 24–28 weeks. However, when global outcomes were measured with the GDS, rivastigmine came out as the most effective.
Summary of clinical effectiveness evidence
-
From 1843 titles and abstracts screened, four systematic reviews and 17 RCTs were found that matched our inclusion criteria, which had been published since 2004.
-
Overall, the quality of the trials was disappointing, and there was insufficient evidence to suggest that one treatment is better than another. We, therefore, suggest that the AChEIs are taken as a class of drugs.
-
When combined with data from the previous review in 2004,2 donepezil was shown to provide gains on cognitive, functional and global outcomes when compared with placebo.
-
Similar pooling of data from galantamine studies was conducted, showing clear benefits from cognitive, functional and global outcomes. Additionally, results favouring treatment were seen for behavioural outcomes at later (6-month) follow-up.
-
Pooled estimates of cognitive benefits from rivastigmine were favourable, but were shown to be dose dependent as in the previous review in 2004. 2 The results from functional and global outcomes also showed significant gains. However, results from individual trials of behavioural outcomes were mixed (pooling was not possible owing to heterogeneity). The lower dose transdermal patch (9.5 mg/day) was shown to be as effective as the capsule (12 mg/day), but with fewer side effects.
-
The meta-analysis of memantine versus placebo showed benefit from memantine at 12 weeks’ follow-up on the SIB. However, treatment gain, measured by functional outcome, depended on the type of instrument used, and no benefit on behavioural outcomes was seen. Nevertheless, pooled estimates of global outcomes showed a benefit from taking memantine.
-
Pooling of data from head-to-head trials was not possible owing to the heterogeneity of the data. Results from the one reasonably good-quality trial showed no significant difference between donepezil and rivastigmine for cognitive or behavioural outcomes. However, when looking at functional and global outcomes, patients taking rivastigmine fared significantly better than those taking donepezil in the primary analysis.
-
Pooling data from trials combining memantine plus an AChEI versus an AChEI failed to show any additional benefit from combination therapy.
-
The MTC results for cognitive outcomes varied. Donepezil and galantamine were both probably the most effective for cognitive outcomes depending on the measure used and the length of follow-up. Similarly, depending on the follow-up time, galantamine and rivastigmine were either equally effective or galantamine was more effective on functional outcomes. For longer-term outcomes donepezil or rivastigmine were probably the most effective treatments, depending on the measure used.
As found in the previous review,2 the main AEs for the AChEIs were gastrointestinal, and agitation and hypertension for memantine. However, the source of this evidence was limited to the included RCTs. The trial populations and their experience of AEs may not reflect those of people with AD who were not in trials.
Table 72, summarising the change in the evidence of effectiveness, as measured by statistical significance at p < 0.05, should be interpreted with caution. The fewer the studies contributing to a category of outcome measure, for example cognitive, the more likely it is to have a positive or negative result, and the more studies there are in a category, the more likely it is that their results will go in different directions. Thus, there appears to be a possibly false sense of certainty about the memantine results when this may simply be an artefact of the number of studies.
| Outcome | Data | Donepezil (no. of studies) | Galantamine (no. of studies) | Rivastigmine (no. of studies) | Memantine (no. of studies) |
|---|---|---|---|---|---|
| Cognitive | New | ∼ (5) | ✓ (3) | ✓ (3) | ✗ (1) |
| Existing151 | ∼ (6) | ✓ (6) | ∼ (3) | ∼ (1) | |
| Pooled | ✓ | ✓ | ✓ | ∼a | |
| Functional | New | ✓ (1) | ∼ (3) | ✓ (3) | ✗ (1) |
| Existing | ∼ (8) | ✓ (3) | ∼ (2) | ✓ (1) | |
| Pooled | ✓ | ✓ | ✓ | ∼b | |
| Behavioural | New | – | ✗ (1) | ✗ (2) | ✗ (1) |
| Existing | ∼ (4) | ∼ (2) | ✗ (2) | ✗ (1) | |
| Pooled | ✗c | ∼d | – | ✗ | |
| Global | New | ✓ (1) | ∼ (2) | ✓ (2) | ✗ (1) |
| Existing | ∼ (7) | ✓ (5) | ✓ (3) | ✓ (1) | |
| Pooled | ✓ | ✓ | ✓ | ✓ | |
| Change in direction of evidence | All | ↑ | ↓ | ↑ | ↔ |
| Change in amount of evidence | All | ↑ | ↑ | ↑ | ↑ |
| Increased precision | All | ↑ | ↑ | ↑ | ↔e |
Chapter 4 Assessment of cost-effectiveness
Introduction
The aim of this chapter is to assess the cost-effectiveness of donepezil, rivastigmine, galantamine and memantine for AD, updating the last guidance, NICE TA111,1 which considered evidence up to July 2004. The economic analysis comprises a systematic review of the literature on cost-effectiveness, a review of the manufacturer’s submissions to NICE on cost-effectiveness and this Technology Assessment Group’s independent economic model.
The focus of the economic analysis is on the evidence and analyses that have been produced since 2004. We do not review work that would already have been considered in previous technology assessments. Duplicate publications after 2004 of economic analyses or models originally published before 2004 (and included in the original economic analysis) would be clear examples of this.
Cost-effectiveness evidence that supported existing guidance
The starting point is thus the economic findings in the last guidance, which we summarise as follows. These have been taken from the text of TA1111 (amended September 2007, August 2009) – donepezil, galantamine, rivastigmine (review) and memantine for the treatment of AD (amended) – recognising that this represents a summary of a highly complex series of deliberations over a number of years, starting with the TAR group report and industry submissions, incorporating additional analyses from NICE, considering responses from consultees, taking into account feedback from a judicial review and then responses to this from the Decision Support Unit (DSU).
Concerning the acetylcholinesterase inhibitors (donepezil, galantamine and rivastigmine)
Main guidance
The three acetylcholinesterase inhibitors donepezil, galantamine and rivastigmine are recommended as options in the management of patients with Alzheimer’s disease of moderate severity only (that is, subject to section 1.2 below, those with a Mini Mental State Examination [MMSE] score of between 10 and 20 points), and under specified conditions [as stated in 1.1].
Key economic considerations
Different incremental cost-effectiveness ratios (ICERs) for moderate and mild AD were recognised. 4.3.10.8 states:
… It (the appraisal committee) therefore considered whether it might be possible to define, prospectively, subgroups of people with Alzheimer’s disease who might benefit more than average, and for whom AChEIs might be a relatively cost-effective treatment … In accepting the subgroup analyses using severity of cognitive impairment, the Committee reviewed the estimates of cost-effectiveness. It noted that for people with moderate Alzheimer’s disease these estimates ranged from £23,000 to £35,000 depending on the choice of AChE inhibitor and by including carer benefits in the augmented base-case. Conversely, the Committee noted that for the subgroup of people with mild Alzheimer’s disease estimates of cost-effectiveness ranged from £56,000 to £72,000 depending on the choice of AChE inhibitor and by including carer benefits in the augmented base-case …
The specific ICERs given for moderate AD were:
For moderate disease treated with donepezil, the augmented base-case ICER was £31,550 per QALY gained [from DSU augmented base-case as stated in 4.2.8.6].
The impact of additional sensitivity analyses were also explored in the DSU analysis, but this did not appear to have a major effect on the ICERs for moderate Alzheimer’s disease [as stated in 4.2.8.8 to 4.2.11 inclusively].
Further detail on the ICERs for mild AD included:
The Committee concluded that the cumulative impact of the changes it considered appropriate reduced the base-case ICER for mild Alzheimer’s disease to approximately £55,000 to £58,000 per QALY gained (for galantamine and donepezil, respectively) which is further reduced by approximately £1500 when using the appropriate starting age of the full-time care (FTC) index. The Committee noted the sensitivity analyses on estimates of health-related utility performed by the DSU but did not consider that the results of these were appropriate to consider as base-case estimates of the ICERs for the AChE inhibitors. It accepted that the ICERs could be lower than the base-case but concluded that the amendments had not reduced the ICERs for the subgroup of people with mild Alzheimer’s disease to within the range normally accepted as a cost-effective use of NHS resources [as stated in 4.3.37].
Concerning memantine
Main guidance
Memantine is not recommended as a treatment option for patients with moderately severe to severe Alzheimer’s disease except as part of well-designed clinical studies [as stated in 1.4].
Key economic considerations
The main evidence on cost-effectiveness was derived from the model submitted by the manufacturer.
In the probabilistic model submitted by the manufacturer, disease states were described by severity, level of dependency (dependent or independent), whether people were in institutional care or not and death. The people in the model made transitions between the states. The time horizon was 2 years. The transition probabilities between health states (defined as categories of MMSE score) were derived from a single RCT of memantine monotherapy. The odds ratio associated with institutionalisation was also derived from this single RCT and was not adjusted for differences in disease severity. The manufacturer calculated from this model that memantine dominated placebo for the total population as well as the subgroups except the subgroup of severe and dependent people with Alzheimer’s disease for which an estimate of approximately £4000 was reported for the CQG. [as stated in 4.2.9.2].
The influential estimates of cost-effectiveness were, however, generated by the use of more plausible parameters in this model.
The Assessment Group re-ran the model using a set of assumptions similar to those used in its own model for AChE inhibitors, and the CQG estimates were between £37,000 and £53,000. Further changes to transition probabilities in relation to the available trial evidence for, and costs of care associated with, memantine raised the estimated CQG in the manufacturer’s model substantially above £53,000 [as stated in 4.2.9.3].
This in turn led to the Committee concluding:
… on the basis of current evidence on clinical effectiveness memantine could not reasonably be considered a cost-effective therapy for moderately severe to severe Alzheimer’s disease [as stated in 4.3.49].
The economic analysis for this report thus specifically considers whether or not new evidence would alter any of these conclusions and so lead the Appraisal Committee to reconsider the guidance. In this respect it should also be noted that the scope for memantine has changed slightly. In the previous guidance the licensed indication was in moderately severe-to-severe AD, whereas for this report the licensed indication is moderate-to-severe AD.
Systematic review of existing economic evaluations
Method
General
This followed the process set out in the protocol from which there were no major departures.
This systematic review aimed to update the systematic review of cost-effectiveness studies,2 which was conducted in 2004 as part of the review of evidence to inform the NICE’s earlier guidance on these drugs (TA111). 1
The review aimed to summarise the main results of the included studies, and identify any key economic costs and trade-offs relevant to the decision problem. It also indicated the strengths and weaknesses of different modelling approaches in this treatment area.
It fully extracted study data and assessed study quality only for those economic evaluations or costing studies published since 2004, which were of relevance to the current decision problem. Further, these were not to have duplicated work or analyses considered in the original guidance [last sentence added for clarification and did not appear in the original protocol].
Search strategy
The range of sources searched included those for clinical effectiveness and in addition NHS EED and EconLit. Full details of the search strategies are provided in Appendix 2.
Study selection criteria and procedures
The inclusion and exclusion criteria for the systematic review of economic evaluations were identical to those for the systematic review of clinical effectiveness, except:
-
Non-randomised studies were included (e.g. decision model-based analyses or analyses of patient-level cost and effectiveness data alongside observational studies).
-
Full cost-effectiveness analyses, cost–utility analyses, cost–benefit analyses and cost–consequence analyses were included. (Economic evaluations which only report average cost–effectiveness ratios will only be included if the incremental ratios can be easily calculated from the published data.)
-
Stand-alone cost analyses based in the UK NHS were also sought and appraised.
-
Based on the above inclusion/exclusion criteria, study selection was made by one reviewer (CH).
-
Study quality assessment – the methodological quality of the economic evaluations were assessed according to internationally accepted criteria such as the City Health Economics Centre (CHEC) list questions developed by Evers and colleagues. 152 Any studies based on decision models were assessed against the International Society for Pharmacoeconomic and Outcomes Research (ISPOR) guidelines for good practice in decision analytic modelling. 153
-
Data extraction strategy – for those studies which were of relevance to the current decision problem, data were extracted by one researcher (CH) into two summary tables: one to describe the study design of each economic evaluation and the other to describe the main results. (These have been merged for this report.)
In the study design table the components were author and year; model type or trial-based; study design [e.g. cost-effectiveness analysis (CEA), cost–utility analysis (CUA) or cost-analysis]; service setting/country, study population, comparators, research question; perspective, time horizon, and discounting; main costs included; main outcomes included; sensitivity analyses conducted; and other notable design features.
For the modelling-based economic evaluations, a supplementary study design table recorded further descriptions of model structure (and note its consistency with the study perspective, and knowledge of disease/treatment processes), sources of transition and chance node probabilities, sources of utility values, sources of resource use and unit costs, handling of heterogeneity in populations and evidence of validation (e.g. debugging, calibration against external data, comparison with other models).
In the results table the components for each comparator were incremental cost, incremental effectiveness/utility and ICERs. Excluded comparators on the basis of dominance or extended dominance will also be noted. The original authors’ conclusions were noted and also any issues raised concerning the generalisability of results. Finally, the reviewers’ comments on study quality and generalisability (in relation to the TAR scope) of their results were recorded.
Synthesis of extracted evidence
Narrative synthesis, supported by the data extraction tables, was used to summarise the evidence base.
Results
The flow of papers is summarised in Figure 56. In brief, over 1400 citations were identified from the searches, 71 of which were ordered in full; two of these could not be retrieved, but from the information in the tile seemed to offer a low probability of representing additional included studies. Of the 69 that were retrieved, 42 were excluded. The most common reasons for exclusion were the paper was not an economic evaluation or the paper had been considered in the previous guidance. Further details and references for these excluded papers are available on request.
FIGURE 56.
Flow diagram for search, retrieval and inclusion of articles in the systematic review of evidence on the economic evaluations of AChEIs and memantine for treatment of AD.

A total of 27 papers, describing 23 studies, were included, and are detailed in the following sections. No additional potentially includable studies were identified from checking of reference lists of included systematic reviews or manufacturer submissions.
Results
General papers not specific to one particular drug
There were five studies in this category. 154–158
There were two systematic reviews of economic evaluations. 154,157 Green154 offered a slightly extended search period to the systematic review of cost-effectiveness studies presented in the last TAR report,2 searching to the end of 2005. Two additional studies were identified in the period 2004–5, both of which are included in this review. Oremus157 searched to the end of 2007. He identified 11 studies published in the period from 2004 to the end of his review. Four of these were included in the original TAR report and the remaining seven are included in this review. Both systematic reviews concluded that further research on the cost-effectiveness of AD was the priority. Green154 suggested the need for improved model structures and model parameters and Oremus157 the need for more economic evaluation alongside RCTs. A further systematic review154 addressed cost studies, subdividing these by country in which the estimate was derived. All of the UK-based studies had already been captured and discussed in detail in the last TAR report. The review reinforced the highly variable nature of costs from country to country.
Two further papers provided information on cost-effectiveness of AD medication. 155,158 Although these were not considered of sufficiently direct relevance to be appraised in detail for this guidance, they are noted because of the potential relevance to future guidance. The first paper, by Sheehan et al. ,155 is a randomised trial protocol for ‘DOMINO-AD’ [donepezil and memantine in moderate-to-severe AD; International Standard Randomised Controlled Trial Number (ISRCTN) 49545035]. It addresses the question of what treatment course should be pursued in the face of advancing AD in patients who are already receiving an AChEI such as donepezil. It will test the clinical effectiveness and cost-effectiveness of alternative approaches, such as continuing or stopping donepezil and/or starting memantine. Economic evaluation will be conducted alongside the trial. The second paper158 is a Monte Carlo model assessing the cost–benefit of screening for, and then applying, unspecified anti-AD interventions including drug treatments. A further abstract on the use of donepezil in this context was also found and is mentioned in the relevant subsection below. 159
Donepezil
There were nine included studies addressing the cost-effectiveness of donepezil. 159–168
Three further included papers were interim reports of169,170 or correspondence on171 the main study by Feldman and colleagues. 160 One further included paper172 was a conference abstract of the study by Teipel and colleagues. 168 This contained very little information and was thus not considered further.
Finally, as already mentioned in the general section, a further included study reports an ongoing economic evaluation alongside a trial investigating the cost-effectiveness of stopping donepezil, continuing donepezil, continuing donepezil with the addition of memantine, or stopping donepezil and starting memantine in AD patients who have stopped responding to donepezil. 155
The general features of the nine main included cost-effectiveness studies159–168 for donepezil are set out in Table 73.
| Item | Feldman et al. (2004)160,169–171 | Fuh et al. (2008)161 | Getsios et al. (2009)162,163 | Getsios et al. (2009)159 | Lopez-Bastida et al. (2009)164 | Lu et al. (2005)165 | Mesterton et al. (2009)166 | Teipel et al. (2007)168,172 |
|---|---|---|---|---|---|---|---|---|
| Publication type | Full paper | Full paper | Abstract | Abstract | Full paper | Full paper | Abstract | Full paper |
| Study purpose | Investigate costs to society of AD | Apply existing model to Taiwan (Province of China) | Enhance method of modelling cost-effectiveness | Assess cost-effectiveness of screening + treatment | Assess cost-effectiveness in Spain | Estimate impact on health-care costs | Update estimates of cost-effectiveness using recently collected data | Apply existing model to Germany and extend time frame |
| Country setting | Canada, France and Australia | Taiwan (Province of China) | UK | UK | Spain | USA | Sweden | Germany |
| Base-year prices | 1998 CDN$ | 2006 US$ | 2007 GB£ | Not stated | 2006 € | 1999–2002 US$ | Not stated; SEK | 2004 € |
| Intervention/comparator | Donepezil 5–10 mg for 24 weeks vs placebo | Donepezil vs no pharmacological treatment | Donepezil 10 mg vs standard care | Screen and treat with donepezil 10 mg vs treat alone | Donepezil 5–10 mg vs no drug treatment | Donepezil vs no donepezil | Donepezil for 1 year vs placebo | Donepezil 10 mg for 1 year vs placebo |
| Study type | Economic evaluation alongside RCT | Markov model | Discrete-event simulation model | Discrete-event simulation model | Markov model | Case–control study | Markov model | Markov model |
| Model duration/cycle length | N/A | 5 years; 12 months | 10 years; N/A | 10 years; N/A | 0.5, 1, 1.5, 2 and 2.5 years; 1 month | N/A | 5 years; 6 months | 5 and 10 years; 12 months |
| No. of states | N/A | 4 | N/A | N/A | 4 | N/A | 12 | 5 |
| Study group – AD | Moderate-to-severe AD; MMSE 5–17 | Mild and moderate AD | Mild-to-moderately severe AD; MMSE 10–26 | Patients without AD, > 65 years and having memory complaints are screened | Mild or moderate AD | ICD-9-CM diagnosis of dementia | Mild-to-moderate AD | Mild-to-moderate AD |
| Perspective | Societal | Societal | Health-care system and societal | Health-care system and societal | Health service and societal | Health-care payer | Not stated; implied societal | Societal |
| Discount rate p.a. (costs/benefits) | N/A | 3%; 3% | 3.5%; applied to ‘all outcomes’ (i.e. both benefits and costs) | Not stated | 3%; 3% | N/A | Not stated | 5%; unclear whether or not applied to both benefits and costs |
| Industry role | Funded by manufacturers | Funded by manufacturer | Company employee listed as author | Company employee listed as author | Funded by Ministry of Health | Funded by manufacturer | Company employee listed as authors | German Centre of Gerontology. Statement of ‘no competing interests’ |
| Study base-case ‘headline’ findings | Donepezil cost saving | Donepezil cost-saving from societal perspective | Donepezil highly cost-effective | Screening and treating is cost-effective | Donepezil cost-saving from societal perspective and cost-effective from health service perspective for mild AD. Not cost-effective for moderate AD | Reduced health-care costs associated with donepezil use in the Medicare-managed plan studied | Donepezil cost saving | Donepezil may be cost-effective, but considerable uncertainties remain |
The review of cost-effectiveness studies reveals some potentially valuable new evidence since the last guidance. The studies fall into four categories:
-
Primary economic evaluations There were two studies in this category. 160,165 Of the two, the study by Feldman and colleagues160 is the more robust, representing a bottom-up costing alongside a RCT173 in Canada, Australia and France in which 144 patients with moderate-to-severe AD (MMSE 5–17) were randomised to donepezil for 24 weeks and 146 to placebo. The societal cost per patient was CDN$9904 in the donepezil group and CDN$10,236 in the placebo group, representing a net saving of CDN$332. When caregiver costs were excluded, the cost was CDN$4355 in the donepezil group and CDN$4321 in placebo, representing a net increase of CDN$34 with donepezil. Conference abstracts169,170 present results for costs in the 145 patients with severe AD (MMSE 5–12) in which there was a net cost saving associated with donepezil of CDN$467. Thus, in turn this suggests that there would also be a societal cost-saving in the moderate group of AD (MMSE 12–17) of approximately CDN $200. The study by Lu and colleagues165 was an observational study, and, hence, much more open to bias and confounding, but, nonetheless, also suggested that prescription of donepezil was associated with lower costs to a large Medicare-managed health-care plan. The difference in costs was US$2500 (95% CI US$330 to US$4671) and was adjusted for differences in a patient characteristics between cases and controls. Both studies were funded by the manufacturer of donepezil.
-
Application of existing models to different settings Three studies of donepezil161,164,168 essentially apply existing model structures to new settings, defined in terms of the health-care systems in different countries. Parameters, where country-specific estimates exist, were substituted for the parameters and assumptions in the parent models. The conclusions are consistent with the parent models that were reported in the last guidance, indicating that donepezil is cost saving, particularly when a societal perspective is considered. The study based in Germany168 was perhaps more cautious in its conclusions than past models, acknowledging the enormous impact of uncertainty on its cost-effectiveness estimates and also suggesting that implementation might not be justified in the context of the German reimbursement system. The Spanish study164 was interesting in that it suggested that cost-effectiveness might be better in mild AD, but not in moderate AD. This is the opposite conclusion to that reached by NICE in its last guidance. In terms of industry involvement, only the model by Fuh and colleagues161 was supported by the manufacturers. The models by Lopez-Bastida/Teipel and colleagues164,168 represent two of the few economic evaluations apparently performed independently of manufacturer influence.
-
Newly developed or updated models Three conference abstracts representing two models donepezil159,162,166 appear to represent novel approaches to modelling. The analysis of donepezil from the UK perspective has also been published as a full paper in early 2010. 163 In the model by Getsios and colleagues,159,162,163 a discrete-event simulation approach has been developed to deal with limitations of the previous models. There is very limited information in the abstracts about the details of the model, but it seems clear from the full-paper version163 that the approach adopted is very similar, or even identical, to the manufacturer’s submission for donepezil for this NICE guidance. For this reason we did not explore it further at this stage, relying instead on the working model supplied by the manufacturer. The study by Mesterton and colleagues166 also provided very limited details to support the view that it genuinely provides an updated approach using new data on costs and utilities. Concerning results, both models in this category suggest that donepezil produces health benefits and is cost-saving, and so dominates the no-drug treatment alternative. In the second abstract and the full paper using the Getsios model,159 the new model is applied to the question of whether or not screening for AD, followed by donepezil treatment is cost-effective relative to donepezil treatment in those presenting with AD. The screening approach is claimed to be cost-effective, although this is not an issue of direct interest in this appraisal. Both models in this group of studies have been developed with the support of the manufacturer.
-
Other There was one poorly described model,167 which claimed to have assessed the cost-effectiveness of donepezil, high-dose rivastigmine and low-dose rivastigmine relative to no-drug treatment in a Thai private hospital. The details were so scant, however, that it is debatable whether or not the conclusions can be given any credibility.
Results: rivastigmine
There were only two included studies claiming to provide new evidence on the cost-effectiveness of rivastigmine. 167,174 Their details are summarised in Table 74.
| Item | Brennan et al. 2007174 | Pattanaprateep et al. 2005167 |
|---|---|---|
| Publication type | Abstract | Abstract |
| Study purpose | Assess cost-effectiveness of Exelon patch | Assess cost-effectiveness in Thailand |
| Country setting | UK | Thailand |
| Base-year prices | Not stated; GB£ | Not stated; bhat and US$ |
| Intervention/comparator | Exelon patch vs BSC |
Donepezil 10 mg vs high or low dose Rivastigmine vs no drug treatment |
| Study type | Model (type not stated) | Decision tree analysis |
| Model duration/cycle length | Not stated | Not stated; N/A |
| No. of states | Not stated | Not stated |
| Study group – AD | Moderate AD | Mild-to-moderate AD |
| Perspective | UK NHS | Health service |
| Discount rate p.a. (costs/benefits) | Not stated | Not stated |
| Industry role | Company employee listed as author | Not stated |
| Study base-case ‘headline’ findings | Exelon patch cost-effective | Cost-effectiveness of high-dose rivastigmine greater than donepezil, greater in turn than low-dose rivastigmine |
The first study174 was a model that claimed to assess the cost-effectiveness of a rivastigmine patch. Unfortunately, the scant methodological details undermine the credibility of its findings that the Exelon patch was cost-effective with a cost per quality-adjusted life-year (QALY) of about £13,000 from a UK NHS perspective. The study had support from the manufacturer.
The second study167 attempted to compare rivastigmine at high and low doses with donepezil, and has already been described in the donepezil section. As already indicated, the details of the modelling process are so scant that the credibility of the conclusion that high-dose rivastigmine is more cost-effective than donepezil, which, in turn, is more cost-effective than low-dose rivastigmine must be questioned.
Results: galantamine
There were again only two included studies claiming to provide new evidence on the cost-effectiveness of galantamine. 175,176 Their details are summarised in Table 75.
| Item | Suh et al. 2008175 | Suh 2009176 |
|---|---|---|
| Publication type | Full paper | Abstract |
| Study purpose | Assess clinical and economic benefits of galantamine | Apply existing model to Korean setting |
| Country setting | Republic of Korea | Republic of Korea |
| Base-year prices | 2002; KRW and US$ | 2007; US$ |
| Intervention/comparator | Galantamine 8–24 mg/day vs community control over 1 year | Galantamine vs usual care |
| Study type | Economic evaluation alongside controlled trial (non-randomised) | Markov model |
| Model duration/cycle length | N/A | 5 years; not stated |
| No. of states | N/A | 3 |
| Study group – AD | Mild-to-moderate AD (MMSE 10–22) | Mild-to-moderately severe AD |
| Perspective | Societal | Third-party payer |
| Discount rate p.a. (costs/benefits) | N/A | 6%; 1.5% |
| Industry role | Sponsored by manufacturer | No financial support. Statement of no conflicts of interest |
| Study base-case ‘headline’ findings | Galantamine is cost-saving | Galantamine is cost-effective relative to usual care |
Both studies were by Suh and colleagues175,176 The first175 was an industry-sponsored economic evaluation alongside a controlled trial in which the costs of galantamine administered in the context of a RCT comparing different galantamine doses were compared with the costs in a community-derived untreated control group. The duration of the study was 1 year and showed a cost saving of US$5372.
The second study by Suh176 is an economic model, in which an existing framework is applied to the Korean setting. The results suggest that, from the perspective of a third-party payer over 5 years, galantamine is cost-effective relative to usual care (cost per QALY US$4939). The author claims that there are no conflicts to declare, but this is somewhat inconsistent with the manufacturer sponsorship of the previously mentioned economic evaluation alongside the RCT175 in which the same author is the lead.
Results: memantine
There were six main included studies addressing the cost-effectiveness of memantine. 177–182
In addition, as already mentioned in the general section, a further included study reports an ongoing economic evaluation alongside a trial investigating the cost-effectiveness of starting memantine, with or without donepezil, in AD patients who have stopped responding to donepezil. 155
The general features of the main included studies for memantine are recorded in Table 76.
| Item | Antonanzas et al. (2006)177 | Gagnon et al. (2007)178 | Guilhaume et al. (2005)179 | Jonsson (2006)180 | Toumi et al. (2009)181 | Weycker et al. (2007)182 |
|---|---|---|---|---|---|---|
| Publication type | Full paper | Full paper | Abstract | Full paper | Abstract | Full paper |
| Study purpose | Apply existing model to Spanish setting | Apply existing model to Canadian health-care setting | Model validation | Apply existing model to Sweden | Model using updated predictive equations for time to FTC | Consider cost-effectiveness of memantine added to donepezil |
| Country setting | Spain | Canada | UK | Sweden | Norway | USA |
| Base-year prices | 2005 € | 2005 CDN$ | N/A | 2004 SEK | 2008 € and NOK | 2005 US$ |
| Intervention/comparator | Memantine vs standard care | Memantine vs standard care | Memantine vs standard care | Memantine vs no pharmacological treatment | Memantine vs standard care | Memantine + donepezil vs donepezil |
| Study type | Markov model | Markov model | Modelled outputs compared with actual outputs | Markov model | Markov model | Microsimulation model |
| Model duration/cycle length | 2 years; 6 months | 2 years; 6 months | Unclear | 5 years; 6 months | 5 years; not stated | Lifetime; not stated |
| No. of states | 7 | 5 | Not stated | 13 | 3 | N/A |
| Study group – AD | Moderately severe and severe AD | Moderate-to-severe AD; MMSE < 19 | MMSE < 14 | Moderately severe and severe AD | Moderate-to-severe AD | Moderate-to-severe AD; MMSE 5–14 |
| Perspective | Societal | Societal | Not stated | Swedish public health-care payer | Societal | Societal |
| Discount rate p.a. (costs/benefits) | 6%; 6% | 5% (unclear whether or not applied to both costs and benefits) | Not stated | 3%; unclear whether or not also applied to benefits | 3%; 3% | 3%; 3% |
| Industry role | Company employees listed as authors | Company employees listed as authors | Company employees listed as authors | Supported by unrestricted grant from company | Company employees listed as authors | Unclear |
| Study base-case ‘headline’ findings | Memantine cost-saving | Health benefits with no additional costs | Modelled and actual disease course similar over 18 months | Health benefits achieved with cost saving | Higher benefits with no additional costs | Improved clinical outcomes with reduced costs of health care |
Half of the papers177,178,180 focused on the application of the analytical approach used in the previous guidance to different settings, and were thus thought unlikely to provide estimates of cost-effectiveness that responded to the criticisms raised in the last guidance. This was compounded by the likelihood that the analyses were not independent. All papers repeated the conclusion put forward by the manufacturer at the time of the last guidance that benefits were achieved at reduced cost.
The study by Guilhaume and colleagues179 was of interest in providing reassurance that extrapolation of natural history by the Markov model corresponded with actually observed states, but was limited by the small amount of information available in the abstract. The study by Tuomi and colleagues181 appeared to represent a new approach to estimating the cost-effectiveness of memantine relative to standard care, but was again limited by the small amount of information available in the abstract. Normally, we would have pursued additional information, but did not do so in this case because the modelling approach appeared similar to that adopted in the industry submission. This has been appraised in detail in Chapter 7. The study by Weycker and colleagues182 also appeared to offer an updated approach relative to those encountered in the last guidance and did not have an obvious connection with the manufacturer. It did, however, address a question not directly relevant to the decision question of interest.
Summary
The systematic review of cost-effectiveness studies published since the last guidance raises the following key points:
-
There have been further publications on cost-effectiveness of pharmacological interventions for AD in the general medical literature. These are generally supportive of the cost-effectiveness of the acetylcholinesterase inhibitors (donepezil in particular) and memantine in the treatment of AD at all stages of disease. Most work is supported by the manufacturers as it was in the last appraisal. There are, however, a few more examples of independent assessments,164,168 which, although more cautious, also support the cost-effectiveness of drug treatments for AD.
-
Many studies apply existing models to new settings and, as such, appear to add little further general understanding concerning the cost-effectiveness of AD drug treatments outside the new setting considered.
-
There are some new economic evaluations alongside trials and other studies, which appear to offer new evidence. 160,165,175 They support the cost-effectiveness of donepezil and memantine, in contrast with the AD2000 study102 in the last guidance, but are all manufacturer supported.
-
There also appear to be a small number of novel approaches to modelling, attempting to overcome problems observed with previous models. The most obvious of these is the discrete-event simulation model of the cost-effectiveness of donepezil. 162 This will be considered in closer detail as part of the assessment of the manufacturer’s submission.
Chapter 5 Assessment of industry submissions to NICE
Introduction
Four manufacturer submissions were potentially available for this multiple TA (MTA). However, Novartis did not submit an economic evaluation and Shire provided only a critique of aspects of the previous SHTAC model that they felt remained unaddressed. The remaining two manufacturers both submitted economic evaluations based on decision models, and they are both critiqued in this chapter. A critique of their clinical effectiveness evidence reviews can be found in Chapter 3 (see Manufacturers’ reviews of clinical effectiveness).
Decision Support Unit involvement
The DSU was asked to examine the technical accuracy of the Eisai/Pfizer economic evaluation for donepezil, as it was produced using software (Arena; Rockwell Automation, Wexford, PA, USA) with which the Technology Assessment Group (TAG) were unfamiliar. According to section 3.2.10 of the Guide to the MTA process, models should be submitted in standard software, and if manufacturers plan to submit models in non-standard software then prior agreement should by sought. Pfizer requested that its model be submitted in Arena software, which NICE accepted on the basis that training would be provided to the Assessment Group. Although some training was provided, further expertise in Arena software was required, thus the DSU were asked to help complete this task. The DSU report has been fully integrated into this chapter. The DSU did not examine Lundbeck’s economic model.
Lundbeck (memantine): critique of economic submission
The decision problem
The manufacturer of memantine submitted a model-based economic evaluation comparing it with standard care, defined as ‘no pharmacological treatment or any background Alzheimer’s disease therapy’. The model is based on a Markov approach and health outcomes were expressed as QALYs. A NHS and Personal Social Services (PSS) cost perspective was used and future health effects and costs were both discounted at 3.5% per annum. The patient cohort consists of individuals with moderate-to-severe AD as measured using a number of functional and behavioural instruments, but not MMSE. The model was run for two patient populations, with the starting characteristics (shown in Table 79) for (1) a ‘general moderate-to-severe AD’ group and (2) a group considered to have baseline symptoms of agitation, aggression or psychosis as defined as a score of ≥ 3 on the NPI (referred to as the APS subgroup). The APS subgroup was included because the manufacturer believes there is evidence that treatments are particularly effective in this group. A similar argument was put forward in Lundbeck’s submission in the previous appraisal, although the Appraisal Committee was critical of the ‘overly broad’ way the subgroup had been defined, an issue that was also raised at the appeal hearing. This point is acknowledged in Lundbeck’s current submission.
An overview of how the model works
Three health states are defined: pre-FTC, FTC and death. The model cycles monthly over 5 years. This structure is in line with the Assessment of Health Economics in Alzheimer’s disease (AHEAD) model, used in the previous appraisal, although memantine was not evaluated using it and the definition of FTC varies. All individuals enter the model in the health state and are assumed to have moderate AD. Patients who receive memantine do so at the beginning of the model and remain on it all times unless they enter the FTC health state or die. The baseline (no-treatment arm), probability of moving between the pre-FTC and FTC health states was assessed using a risk equation, derived from a non-controlled longitudinal UK-based prevalent cohort study (the LASER-AD study183) of people with AD. The probability of death was also derived from this source, estimated using a Weibull function: the same equation is applied to both pre-FTC and FTC health states, meaning that the probability of death does not change with increasingly progressive disease. WMDs derived from a meta-analysis of six RCTs are applied to the risk equation as a method of incorporating memantine’s treatment effect. Utilities were estimated using a mapping exercise and data from the LASER-AD study (n = 98) relating to people with moderate-to-severe AD.
The model is run probabilistically, although not all of the appropriate variables are specified as distributions, for example the coefficients in the predictive equations. Memantine was predicted to be less costly and more effective than standard treatment in the base case for both patient groups (Table 77). The base-case cost-effectiveness acceptability curves are not shown in the submission, but generated directly from the model programming and taken at face value, suggesting that the probability of memantine being cost-effective is > 90% for both subgroups at all willingness-to-pay (WTP) thresholds for an additional QALY.
| Cost (£) (2009) | QALYs | ICERa | |
|---|---|---|---|
| General group | |||
| Memantine | 92,971 | 1.534 | Dominant |
| Standard care | 94,687 | 1.503 | |
| APS subgroup | |||
| Memantine | 93,663 | 1.566 | Dominant |
| Standard care | 98,639 | 1.496 | |
Comparator treatment options
As previously stated, the model compares standard care, defined as no pharmacological treatment or any background AD therapy. Implicitly this means that the use of memantine monotherapy, combination therapy and a range of comparators has been considered, and a mix (if not comprehensive) of RCTs evaluating different treatment regimens problems has been pooled together. This approach, i.e. the mixing or blending of different decision problems (with respect to the technologies at hand and the patient populations) within the framework of a single model, is not considered to be appropriate, as it potentially masks differences in incremental costs and effects of mutually exclusive treatment options. Moreover, it means that interpretation of the model results is difficult.
The risk equation: estimating the monthly probability of entering full-time care
One element(s), if not the key element, to the model is the risk equation used to estimate the monthly probability of moving to the FTC health state. The risk equation was derived using a subsection of patients from the LASER-AD study. The LASER-AD study included a total of 224 individuals at various stages of disease. This particular analysis was restricted to 117 (52%) of individuals, as the remaining 107 were already considered to require FTC at the time of enrolment. A statistical model was developed using the corresponding data set to estimate time-dependent probabilities of moving between the pre-FTC and FTC health states, based on a number of patient characteristics and time. The final baseline equation is shown in Table 78 and the baseline starting characteristics of the two populations are show in Table 79.
| Variable | Coefficient | SE | p > |z| |
|---|---|---|---|
| Ln (time in months) | 3.3195 | 0.4965 | < 0.001 |
| Baseline ADAS cog total score | 0.0330 | 0.0147 | 0.0247 |
| Baseline ADCS-ADL total score | –0.0877 | 0.0164 | < 0.001 |
| Baseline NPI total score | 0.0377 | 0.0154 | 0.0140 |
| ADAS-cog total score (slope) | 0.8122 | 0.2798 | 0.0037 |
| ADCS-ADL total score (slope) | –2.4072 | –0.3995 | < 0.001 |
| Intercept | –11.1343 | 1.8284 | < 0.001 |
| Parameter | Mean | SDa |
|---|---|---|
| General population | ||
| ADAS-cog baseline | 36.30 | 1.70 |
| ADCS-ADL baseline | 45.00 | 1.87 |
| NPI baseline | 18.54 | 1.86 |
| ADAS-cog slope | 0.6116 | 0.0809 |
| ADCS-ADL slope | –0.7503 | 0.0876 |
| APS subpopulation | ||
| ADAS-cog baseline | 40.30 | 2.66 |
| ADCS-ADL baseline | 45.60 | 2.31 |
| NPI baseline | 22.45 | 2.21 |
| ADAS-cog slope | 0.6179 | 0.1216 |
| ADCS-ADL slope | –0.7775 | 0.1157 |
More details of how the equation was derived are supplied in appendix O of the manufacturer’s submission, marked ‘academic-in-confidence’. In terms of the data collection exercise, the model was said to be based on data collected at months 6, 18, 30, 42 and 54 after baseline. The methods also state that exact dates when FTC was required were unknown; only changes in FTC requirements at the above corresponding time points are given, meaning that the data were analysed using discrete-grouped data methods rather than a continuous time model.
The TAG has the following concerns with the derivation and use of this risk equation, although they are not listed in any particular order of importance.
-
It is unclear how representative the patient sample is with respects to the general moderate-to-severe AD population in the UK.
-
Approximately two-thirds of the LASER-AD patients were receiving AChEIs; any related treatment effect does not seem to have been taken into account when constructing the equation.
-
Full-time care was defined as either entering an ‘institution’ or when individuals were considered to be ‘dependent’ in terms of requiring FTC from others. Although the latter assessment was said to be based on domains on the ADCS-ADL (basic activities, domestic activities and communication), the details of this categorisation process are unclear, for example the threshold value for requiring dependence. This is important, as one-third of patients over the 54 months were classified as becoming ‘dependent’.
-
No sensitivity analysis was undertaken to test the robustness of the final risk model to alternative assumptions regarding the definition of dependence.
-
Fifty-nine per cent of patients whose details were used in this specific analysis were said to have mild AD at baseline, thus the sample used to derive the risk equation is not representative of the baseline decision problem (treatment for moderate-to-severe AD). Whereas it is possible to hypothesise that exclusion of mild AD patients may lead to an increased time to institutionalisation, and therefore greater relative treatments effects, it is not clear this is the case as the probability of entering FTC and different stages of disease and disease progression might not be constant or linear. As something of an indicator of this potential issue, it is worth noting that although 58% of patients in the risk equation study were classified as requiring FTC over the 54-month period, examination of the Markov trace for the general AD population showed that approximately 58% of patients in the general AD population model had moved to the FTC health state in the standard treatment arm by month 25. One-way sensitivity analysis undertaken by the TAG showed that if the probability of death from both health states was set to 0 (to isolate the independent effects of the risk equation), 58% of patients moved to the FTC health state by approximately month 23, and by month 54, 99% of patients had entered FTC. Thus, it is not clear that the risk equation, when used in the model, accurately predicts the probability of requiring FTC in terms of being consistent with the source data.
-
The predictive equation has not been validated against an external data source, therefore the degree to which the results are generalisable is unclear.
-
The programming of the statistical model is poorly described, meaning there is concern that it may not have been used appropriately. Specifically, in addition to the baseline ADAS-cog total score, baseline ADCS-ADL total score and NPI baseline score, the rate of change of ADAS-cog and ADCS-ADL were also significant predictors of time to FTC (the submission refers to these variables as slope parameters – Table 78). These values were then multiplied by what is also referred to as mean ASDS-cog and ADCS-ADL slope scores (see Table 79). This second set of variables were also said to have been derived from the LASER-AD study, but there is no explanation of the methods used to derive these values or what indeed these values represent. Examination of the basic risk equation described on p. 268 of the full manufacturer’s submission suggests that they are likely to/could represent the natural progression of the variables over time. For example, the value of –0.7503 might represent the change in ADSC-ADL per time interval. However, the equation on p. 268 also suggests that these variables should change over time, as they are specified to the jth time interval, but the programming in the model does not allow for these values to change. A more standard approach to applying risk equations in economic models is to multiply relevant coefficients by the current values on an outcome to predict the probability of a future event, and then to recalculate this probability every time the value of the underlying outcome changes. However, this basic approach does not appear to have been undertaken. An alternative approach to this would be to multiply the rate of change (i.e. the slope) by time to assess over all change, as indeed the manufacturer has done in the pre-FTC utility function.
Estimating relative treatment effects
Treatment effects were added to the underlying equation (Table 80) using results from a meta-analysis of six RCTS (MRZ 9001–9605/1, MEM-MD-01, MEM-MD-02, 99679, MEM-MD-10 and MEM-MD-12). Specifically, changes on the ADAS-cog baseline, ADCS-ADL baseline and NPI baseline scores were meta-analysed and added to the related baseline variables in the risk equation.
| Parameter | Treatment effect | SD |
|---|---|---|
| General population group | ||
| Baseline ADAS cog total score | –1.54 | 0.31 |
| Baseline ADCS-ADL total score | 1.53 | 0.62 |
| NPI baseline | –1.34a | 0.93 |
| APS subgroup | ||
| Baseline ADAS cog total score | –2.08 | 0.59 |
| Baseline ADCS-ADL total score | 3.59 | 0.85 |
| NPI baseline | –2.49a | 1.65 |
As with the derivation of the baseline risk equation, a number of criticisms can be levied at this meta-analysis. In three of the studies, patients were said to have mild-to-moderate AD. 150,184,185 Although the submission acknowledges this and states that these individuals were removed from the analysis, it is unclear how this was done.
Only two142,143 of the six compared studies are strictly in accordance with the stated decision problem: memantine monotherapy compared with placebo alone. Concerns with respect to pooling the data for all six RCTs have already been raised in the clinical evidence section of this report.
A related issue is that ADAS-cog is not measured in either the Reisberg and colleagues study142 or the van Dyck and colleagues study. 143 Instead, it is stated that SIB scores from the two studies were transformed into ADAS-cog scores using a linear regression model computed on data from the LASER-AD study. No useful details of this transformation process are provided. However, one-way sensitivity analysis performed by the TAG showed that setting the mean ADAS-cog coefficient to 0 instead of –1.54 (therefore removing any treatment effect on this variable) did little to change the results.
The Reisberg and van Dyck studies142,143 measured functional status using the ADCS-ADL19 (scores ranging between 0 and 54), not the ADCS-ADL23 (scores ranging between 0 and 78), which is the version used in the evidence synthesis. The manufacturer states that scores from the shorter version were ‘rescaled’ into scores for the longer version. However, there is no discussion of the methods used to do this or the possible errors this might introduce. One-way sensitivity analysis conducted by the TAG suggests that the results are particularly sensitive to the ADCS-ADL23 component of the risk equation. For example, replacing the coefficient of 1.53 (see Table 80) in the general AD population base case with 0 increased the ICER to about £33,000 per QALY from being dominant. There are two further points to note on this issue. First, visual examination of the forest plots provided by Lundbeck suggests that smaller mean effects are likely to have resulted if the Reisberg and colleagues’ study142 was excluded from the meta-analysis. Second, the meta-analysis on ADCS-ADL19 results conducted by the TAG using LOCF analysis on week 24–28 data showed marginally statistically significant results (WMD 1.408, p = 0.044), meaning that it is not at all clear that memantine monotherapy is associated with improvements in functioning.
Lastly, the results from the baseline risk equation analysis showed that the NPI hallucination score was a significant predictor of time to FTC, not the NPI total score. It is unclear, however, which of these variables was estimated in the meta-analysis, but it is most likely to be the latter. If this is true, there is a disjoint between the treatment effects estimated by the evidence synthesis and the underlying risk equation, as the NPI total score was not found to independently predict outcome. It should also be noted that results from the TAG’s own meta-analysis, when restricted to RCTs that included individuals with moderate-to-severe AD who either received memantine monotherapy or placebo showed a non-statistically significant difference in NPI total score in favour of memantine (WMD –1.6; 95% CI –4.739 to 1.523). However, despite all this, basic one-way sensitivity analysis suggests that the base case was not sensitive to different parameter values (setting the effect of memantine to 0 on the corresponding risk coefficient) had a negligible impact on the results.
The probability of death
The base-case probability of death was estimated using a sample of the LASER-AD population, and specified using a Weibull function, where the hazard is a function of increasing time, but no other independent variables. Specifically, patients who were not institutionalised or dependent at baseline were said to be included in the analysis. The same Weibull function was applied to both pre-FTC and FTC health states, therefore the probability of death within the model was not considered to change with increasing severity of disease per se. The base-case general AD population analysis showed that 50% of people died in the no-treatment arm at approximately month 60.
There were a number of specific concerns with this part of the model. One-third of patients in the LASER-AD study had mild AD, meaning that the function might overestimate survival in people with moderate-to-severe AD. However, a crude one-way sensitivity analysis undertaken by the TAG suggests that the results were not sensitive to the probability of death each month if the probability is assumed to be independent of AD severity.
Costs
Memantine treatment costs were said to be £2.16 per day in the manufacturer’s submission, regardless of dosage or pack size, but it is not clear that this is the case. The March 2010 Monthly Index of Medical Specialities states that a 28-tablet 10-mg pack costs £34.50. Thus, 20 mg per day is equal to (£34.50/28) × 2 = £2.46. Although one-way sensitivity analysis by the TAG suggests that this increased cost had little bearing on the base-case cost-effectiveness results, clearly its importance will be magnified if other changes are simultaneously made to the model, such as lessening the effect of memantine. The manufacturer also included the cost of a psychiatrist at the start of memantine treatment (£126) and a GP monitoring cost (£35) every 6 months.
The costs associated with pre-FTC and FTC were estimated using resource data from the LASER-AD study, and by combining this information with unit costs from the most recent Personal Social Services Research Unit (PSSRU) publication. 186 Resource-use data were said to have been collected using the Client Service Receipt Inventory (CSRI), by interviewing patients/their carers every 3 months. In general, the resource-use study is poorly described. For example, little is said about how many people provided resource-use data and how missing data were handled. Thus, it is difficult to assess the validity of the results.
The monthly pre-FTC and FTC were calculated to be £724 and £3267 per month, respectively (or £8688 and £39,204 per year). The value of £3367 is a weighted average of people who were considered to have received FTC in the community (£852 × n = 29/98) and people who were considered to be institutionalised (£4282 × n = 69/98). The annual values used in the previous Assessment Group’s economic model were £3397 and £11,247, respectively. Thus, even without allowing for inflation in the latter, these estimates appear to be very different. One reason for the large discrepancy is that the industry submission appears to include the costs that are borne by individuals, rather than the state – an issue in the previous appraisal – but the percentage is not explicit.
Examination of the manufacturers costing exercise shows that the main difference in costs between the pre-FTC and FTC health states is the time individuals spent in ‘day hospitalisation’ and nursing homes. Specifically, an average of 0.63 days in pre-FTC and 0.87 (if individuals were community based) and 8.97 days (if they were in an institution) for people in FTC, using a cost of £281 per bed-day. For individuals who were considered to be in institutionalised FTC, an extra cost of £1760 per month was added to this amount. The table referring to the references for the unit costs of £281 per hospital bed per day and £573 per week in an institution refer to other pages in the submission. However, referring to the other pages revealed no further details.
Utilities
Health benefits to individuals with AD were measured and valued within the analysis, but potential benefits to carers were not included. Patient utilities were estimated using results from individual items on three different instruments mapped on to the European Quality of Life-5 Dimensions (EQ-5D) five-domain classification system (as direct EQ-5D scores were said to be absent). However, it should be noted that the EQ-5D has previously been directly used to estimate mean utility values for people with AD.
The mapping methods were considered by the TAG to be particularly poorly described, thus the values should be treated with some caution. For example, it is said that data relating a sample from the LASER-AD cohort were used, but the basic sample demographics are not reported. Indeed, many other important methodological issues are not discussed including: why the (unspecified) mapping approach was chosen, who did the mapping, why these particular instruments were chosen in the first instance or how different model specifications could lead to different results.
From the mapping exercise, a mean utility value for the FTC health state of 0.336 was derived (the equivalent value in the previous SHTAC base case was 0.34). Values for patients in the pre-FTC health state were not set at a static amount – rather they were adjusted according to ADCS-ADL total score each cycle, using the results from a regression analysis. However, although the LASER-AD study was said to be the data source, few other details are provided. For example, basic sample demographics are not provided, the ADCS-ADL total score was said to be ‘the strongest’ predictor of utility, but it would be useful to understand the relationship between utility and other explanatory variables. Moreover, no assessment of goodness of fit is provided or whether or not alternative models would have better fitted the data.
Pre-FTC utility = 0.202 + 0.008 (baseline ADCS-ADL total scores + ADCS-ADL change × time in months), where the ADCS-ADL total score relates to the specific treatment strategy. Because in the base case the ADCS-ADL total score was assumed to be higher for memantine, this, in effect, means that memantine patients accumulate more QALYs per time period whereas in the pre-FTC health state than when in the standard-care arm.
On investigation it was discovered that this specification leads to some logical problems. For example, when time is 0, the pre-FTC utility score is 0.562, but when time is > 40 months, the predicted value is lower than the (mean of) 0.33 associated with FTC. Moreover, no justification is given for having utility levels based on a function of declining ADS-ADL total score for one health state and a mean (fixed) value in the other.
Extra sensitivity analysis on the general population base case
A number of additional deterministic sensitivity analyses were undertaken by the TAG, some of which have already been reported in the text. In this section, a number of further analyses have been undertaken.
-
Using previous SHTAC costs and inflating to 2009 prices, and using indices provided by Lundbeck, produces an ICER of about £20,000 per additional QALY.
-
Simultaneously making the above change and changing the utility values so that the utility equation for pre-FTC also extends to include patients in the FTC health state, produces an ICER of approximately £30,600 per additional QALY.
-
Changing to both the previous SHTAC inflated costs and original SHTAC utility values produces an ICER of about £23,000 per additional QALY.
-
Extending the time horizon to consider long periods of time had negligible difference on the results.
Summary of memantine model comments
The submitted economic evaluation of memantine was based on a three-state Markov model, with many of the inputs relating to a UK-based (LASER-UK) study. The base-case submitted analysis suggested that memantine generated more QALYs at lower cost than standard treatment for both a general population of individuals with severe to moderate AD and for individuals in an APS subgroup. The results were particularly sensitive to treatment effects as measured using the ADCS-ADL, as both the monthly probability of entering FTC and utility values were conditional upon it. However, the TAGs general view is that the base-case results should be treated with some caution – broadly speaking for the following main reasons:
-
The model is poorly described in many places. Particularly with respect to the derivation and implementation of the underlying risk equation, the methods used to derive the utility functions and to transform some outcome scores from one scale (from the RCTs) to other scales (which were specified in the risk equation). Many of the model inputs were derived from the LASER-AD study, but it is unclear how representative it is of the general AD population and whether or not appropriate subgroups have been used for the various substudies. The results from the TAG’s own systematic review of the memantine monotherapy RCTs compared with placebo shows almost no statistically significant advantage of using memantine, only on the CIBC-plus, which is not included in this model. Thus, at a face value, it is difficult to believe that there is at least a 90% probability that memantine is cost-effective at all WTP thresholds as the results from this model suggest. No attempt has been made to compare the cost-effectiveness of memantine with the AChEIs in individuals with moderate AD.
-
Lastly, and perhaps most importantly, the decision problem and meta-analysis relate to the use of memantine in a number of possible scenarios, yet this consideration is contained within a single model. For example, the comparator in the model is said to be ‘standard care’ comprising any treatment or background therapy other than memantine. Moreover, the model (implicitly) assumes that memantine can be used as an adjunct to other treatments (including an AChEI). Thus, the decision problem is confused, and even if technically correct, is its unclear what the decision problem is that the model is addressing. At the very least, this is not considered to be an appropriate method of evaluation – different models should be used to address discrete decision problems or one model should be used to evaluate all different and distinct treatment options.
Eisai/Pfizer (donepezil): critique of economic submission
The manufacturer of donepezil submitted a model-based economic evaluation, which was built in Arena software and incorporated a Microsoft Excel data input sheet. Numerous other text files were also included that are created from the Excel input sheet by a Visual Basic for Applications macro. This model was critiqued by the DSU at the University of Sheffield.
The decision problem
The model evaluated the use of 10 mg daily of donepezil compared with ‘no AChEI’ treatment. All individuals were assumed to stop treatment at a MMSE score of 10 if they had not already done so. No attempt was made to compare the relative effects of the three different AChEIs using mixed-treatment methods or against memantine in individuals with moderate AD. The patient cohort consists of individuals with mild-to-moderate AD, as measured using MMSE (mild MMSE 20–26, moderate MMSE 10–19). The model runs over a lifetime horizon (set in the base case to 25 years). In the base case, the model suggests that treatment is less costly and more effective than no treatment, for individuals with mild or moderate AD.
All health outcomes were expressed in terms of QALYs, where total expected QALYs are a summation of associated patient and caregiver values. A NHS and PSS cost perspective was said to have been used in the base case, although this is later acknowledged in the submission as not strictly true. Future health effects and costs were both discounted at 3.5% per annum over a lifetime horizon.
Rationale for choice of modelling framework
The model is based on a discrete-event simulation (DES) approach. In a Markov-type analysis, individuals move between a set of predefined mutually exclusive health states over a fixed unit of time according to a set of transition probabilities, thus they are often referred to as discrete-time models. This is in contrast with DESs, where a set of possible events is defined (along with associated costs and health outcomes), but the time between each event is variable in a first-order sense (i.e. representing individual variability rather than parameter uncertainty). Thus, DESs estimate times between events, with the sum of these intervals typically representing total life expectancy.
Both discrete-time and -event models are useful when treatment costs and benefits are likely to accrue over relatively long periods of times. However, the limitation with Markov models is that the probability of moving from one health state to another is typically not based on an individual’s prior experiences. A further limitation with Markov-type models is that they become inefficient and demanding in a programming and data-requirement sense if multiple health outcomes are possible, as increasingly more complex sets of health states are needed (e.g. changes on different AD scales that are considered to be important predictors of costs and health outcomes). DESs potentially overcome both these problems, thus it is considered to be an appropriate modelling approach in this AD context (note: later comments in this chapter suggest, however, that this model is not a DES in the truest sense).
An overview of how the model works
The Arena model submitted by the manufacturer is a generic model, which has a variety of other modules/logic that are not relevant for the current decision problem. For example, it includes a screening module, and an option for people to restart treatment as well as having the provision to estimate costs/utilities of two additional drugs along with donepezil and no treatment. Notwithstanding the model’s capability to perform different analyses, this critique focuses on the issues in the model that are directly related to the CEA of donepezil against no treatment.
How people are selected
The model utilises a weighted sampling approach to sample the people in the model from the trial population. The trial population consists of 826 trial participants and there is a provision to select the people based on different characteristics such as age, gender, MMSE, etc. The two main subgroups utilised are a mild patient group (221 people with 20 ≤ MMSE ≤ 26) and a moderate patient group (542 people with 10 ≤ MMSE < 20). The model utilises 1000 people and these are sampled from the corresponding subgroup using a weighted approach, i.e. if using a mild population, 1000 people are sampled from the 221 people with mild AD and are assigned the characteristics of the corresponding trial patient. These characteristics include age, gender, race, MMSE, NPI, ADL, IADL, previous MMSE and the change in MMSE in the previous year, as well as other information, such as whether or not they are on psychiatric medications, whether or not they are living with their primary caregiver and, if so, the caregiver’s age and gender. These characteristics are specific to the individual people and are assigned to people as attributes.
As there are fewer people in the trial population than in the sampled model population, it is likely that the same trial patient will be included more than once in the modelled population. As the sampling is weighted to achieve an age and gender distribution that is consistent with the UK AD population, this may mean that some people whose characteristics are rare in the trial data set, but common in the UK AD population, may be sampled multiple times and their individual characteristics may have a disproportionate influence on the overall results.
The people are then cloned, i.e. each patient is separated into two identical people with the exact same characteristics. One of the hypothetical people is then allocated to the donepezil arm of the model and the other is allocated to the no-treatment arm. The deterministic results are based on the average costs and QALYs after 20 runs of 1000 people, although the manufacturer’s submission notes that the results are stable after five runs of 1000 people. The manufacturer’s probabilistic sensitivity analysis (PSA) results are based on 350 parameter samples, each run for 5000 people.
Model updates/disease progression
Disease progression is measured using a variety of outcome measures (referred to as attributes). The attributes of each patient are updated at different time intervals in order to replicate the progression of the disease and are then used by the model to perform CEA. The model keeps track of four disease measures: MMSE, NPI, ADL and IADL (Figure 57). It should be noted that the MMSE equation is parameterised in terms of an annual rate of change. The change in MMSE since the last update is estimated by calculating the annual rate of change and scaling this to account for the time since the last update. The other three equations use time as a continuous variable to estimate the new values.
FIGURE 57.
Simplified representation of the AD model taken directly from the Eisai/Pfizer submission.
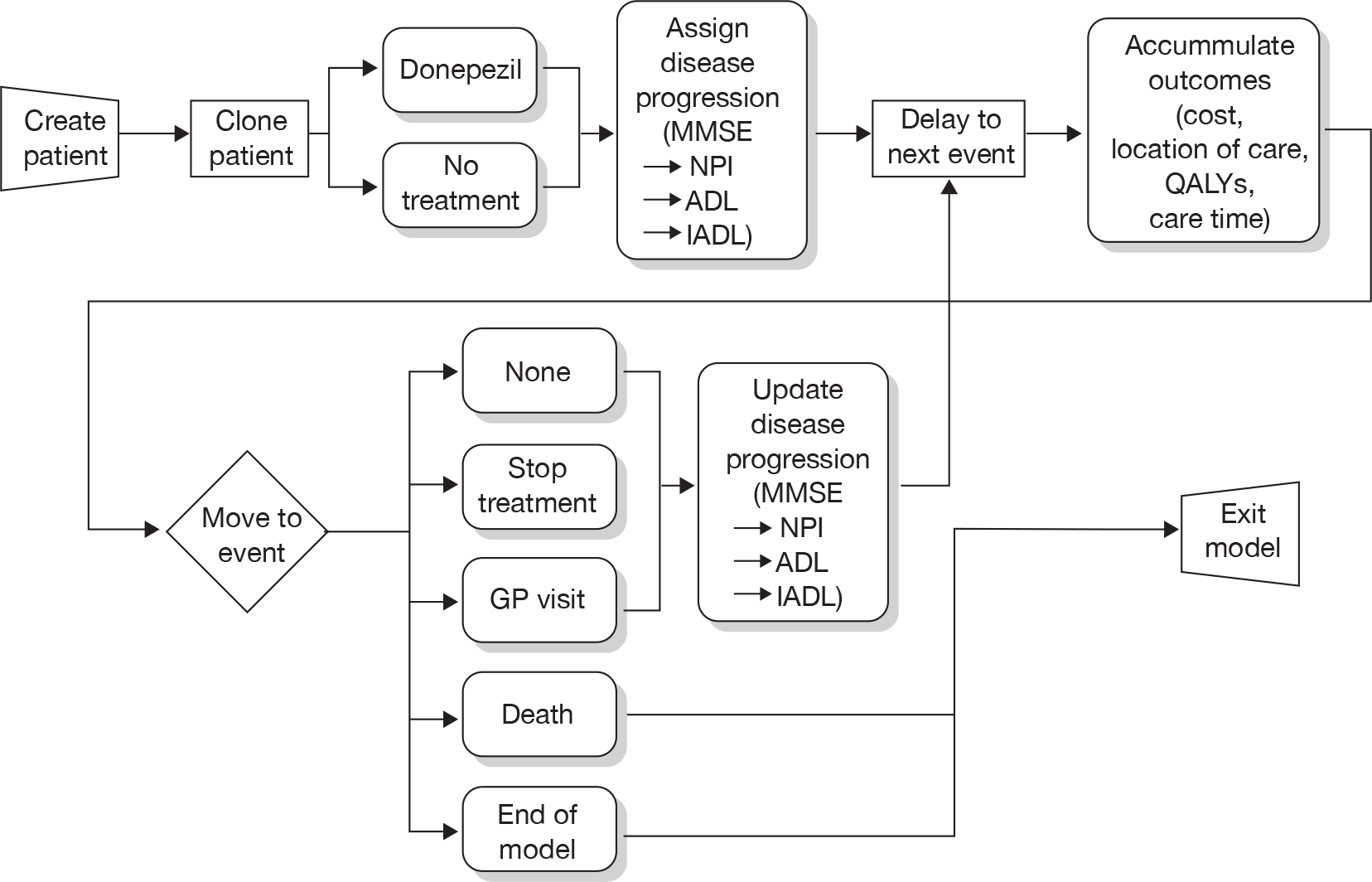
Annual changes in MMSE are first calculated, changes on the remaining three measures then follow, predicted by the change in MMSE as each of the other three equations includes current MMSE values as an individual term. Note that mortality is not dependent on choice of treatment.
The underlying progression equations for MMSE are defined as follows.
For the untreated cohort:
For the treated cohort:
where T_eff is 6.1583 if time is < 20 weeks, and 2.4671 otherwise. The treatment effects last for only 1 year, after which it is assumed to be zero.
The underlying NPI equation in the model is defined as:
where Psymed is a dummy variable indicating whether or not individuals were receiving psychiatric medications, black/white are indicators of ethnic background and 1.44 is a scaling factor to convert the normalised scale of 0–100 scores to 0–144 (note that the reasoning/appropriateness of this transformation is not described in detail).
The underlying ADL equation in the model is defined as:
The underlying IADL equation in the model is defined as:
The term norm(0,x) appearing in each of the disease progression equations is a random intercept parameter that is included to introduce patient-level variation to the disease progression. This random variation is in addition to the variation provided by each patient having unique characteristics.
The people are assigned a severity level based on their MMSE scores after every update. The severity categories and their MMSE ranges are shown in Table 81. The time spent in different severity levels are accumulated for all the people in the donepezil arm as well as the no-treatment arm. The proportion of people in institutional care is dependent on the severity level is shown in Table 81.
| MMSE score | Severity scale | Home (%) | Institutional care (%) |
|---|---|---|---|
| 25–30 | Mild | 87.1 | 12.9 |
| 20–24 | Mild to moderate | 74.4 | 25.6 |
| 15–19 | Moderate | 61.7 | 38.3 |
| 10–14 | Moderate to severe | 49.0 | 51.0 |
| 0–9 | Severe | 30.0 | 70.0 |
Even although the model utilises an individual patient approach, the patient and caregiver utilities are estimated using average values. For example, in the patient utility equation the ‘Institutionalised’ covariate is, strictly speaking, a factor or dummy variable that takes a value of ‘1’ if the patient is institutionalised and ‘0’ if not. The cost-effectiveness model does not classify individual people as institutionalised or not; rather they are assigned a probability of being institutionalised based on their MMSE score.
Quality-adjusted life-years are estimated for both individuals with AD and a caregiver. The utility functions are specified as follows:
Where Psymed is a dummy variable indicating whether or not people with AD were receiving antipsychotic medications.
The costs for both people treated and untreated with donepezil are estimated by accumulating the treatment costs (for people under treatment) and the patient-care costs for home or institutional care. These monthly patient-care costs are based on severity level, as seen in Table 82. Again, although the model is based on an individual patient approach, patient-care costs are estimated using a weighted average of the costs for institutionalised and non-institutionalised patients, according to the probability of institutionalisation for the individuals’ severity state.
| Severity | Monthly medical costs (£) | |
|---|---|---|
| Home | Institutional | |
| Mild | 687 | 2801 |
| Mild to moderate | 742 | 2801 |
| Moderate | 798 | 2801 |
| Moderate to severe | 878 | 2801 |
| Severe | 957 | 2801 |
Drug treatment costs are accrued according to the number of days on treatment. In addition to the drug treatment costs, people also incur the cost of a medical consultation every 6 months while on treatment.
The caregiver times are estimated by the model, but the caregiver costs are not taken into account. Hence, the total costs are calculated by adding the treatment costs to the patient-care costs and the model estimates both discounted and undiscounted values of total costs. The discounted and undiscounted costs accumulated in different severity levels are also calculated.
Possible events
Patient characteristics are updated and the costs along with QALYs are calculated every time the patient undergoes an event. The events that occur in the life of a patient and the times when they occur are presented in Table 83.
| Event | Time |
|---|---|
| Start treatment | 0.01 days |
| Checks for discontinuationa | 0.02, 91.3, 182.6 and 365.25 days |
| Regular updates | Every 3 months |
| Doctor visit | Every 6 months while in treatment |
| Stop treatment | Patient specific |
| Death | Patient specific |
| Last update/model end | 9131.25 days |
General concerns with the model and estimation of model inputs
The annual change in Mini Mental State Examination regression equation
The MMSE regression equation has been derived from a sample of people with AD (721/1094) from a US patient registry [the CERAD (Consortium to establish a registry for AD) study; http://cerad.mc.duke.edu/] note that manufacturer’s submission states that the registry includes only individuals who have never received treatment for their AD. Although the principle of estimating a risk equation from a cohort study (and applying a treatment effect derived from RCT evidence) is considered to be sound, there are a number of concerns with the way the manufacturer has undertaken this analysis, meaning it is difficult to critique.
There is an overall lack of detail as to how the equation was constructed; appendix J of the submission contains few additional details to the main submission. Specifically, the participants in the US CERAD study are not described in any detail, thus it is unclear how representative they are of UK individuals with mild-to-moderate AD. For example, it is stated on p. 89 of the main submission that the CERAD database does not include ‘treated’ people. Little further discussion of this point is provided, but it suggests that individuals included in the study might not necessarily be representative of a typical mild-to-moderate AD population. Additionally, corresponding model statistics, such as goodness of fit, are not provided and there has been no to attempt to validate the MMSE risk equation against external data sources, a point noted by the authors of the original economic model (Getsios and colleagues161).
In appendix H of the submission, the manufacturer notes that the annual rate of change in MMSE was notably different when RCT data were used instead of individuals from the CERAD study; this point is illustrated, to some extent, in Figure 58 (figure 4 in appendix H of the submission). Specifically, the submission states that using the alternative source of data led to ‘no change or a small annual change in MMSE scores < 20 and potentially large declines for those with values above 20’. A reason for this possible discrepancy is suggested – shorter measurement intervals in controlled studies – but it is uncertain that this in itself is sufficient justification for choosing one source over another, or whether or not it indeed suggests more reason to use it as the primary source. Furthermore, the patterns of change observed in CERAD were broadly more in line with what has been previously reported, as the trial data indicated an improvement in MMSE in some untreated people (see Figure 58).
FIGURE 58.
Relationship between annual rate of change in MMSE and source data (taken from the Eisai/Pfizer submission).
Changes in Neuropsychiatric Inventory, Activities of Daily Living and Instrumental Activities of Daily Living scores
As already noted, three regression equations similar to the MMSE equation are used to predict the progression of NPI, ADL and IADL over time. For the NPI scale, data from four RCTs were used (not CERAD). The submission is not specific about the source of information used to estimate changes on the ADL and IADL scales. Indeed, although few methodological are details are provided, one point of concern is that the ADL and IADL scales appear to be a composite of a number of different instruments, although there is no discussion of how these transformations were undertaken, employed in terms of adding in treatment effects or the errors this process might introduce. The following sentence has been taken from the MS: ‘clinical trials measuring ADL and IADL used a variety of scales so “standardised scales” were constructed using items from the various measures in order to link trial results to the utility function’.
Estimating treatment benefits
The effects of donepezil are included in the model through terms in the MMSE, NPI, ADL and IADL regression equations. All four equations include a direct donepezil treatment effect. The NPI, ADL and IADL equations also contained a MMSE term, meaning that changes in MMSE caused changes in the decline of these scores. More specifically, better maintenance of MMSE scores are predictive of a slower decline on the other disease measures. The IADL predictive equation also included an interaction term between donepezil treatment and duration of treatment, indicating that donepezil’s effect increases over time.
The treatment effects on the NPI, ADL and IADL scales appear to have been estimated at the same time that baseline disease progression was estimated, as RCT was evidence used. However, the manufacturer’s submission states that the terms used to estimate treatment effects of donepezil on MMSE were estimated using a ‘similar’ model to that derived for the baseline, as CERAD did not include treated people. Appendix H of the submission suggests that results from eight RCTs were included in this analysis, but few other details are useful details of this ‘similar’ model are provided.
There is some concern that effects of donepezil have been double counted. Although treatment affects both MMSE, NPI, ADL and IADL directly as covariates in the four regression equations, there is also an additional link between the measures as MMSE is also a covariate in the NPI, ADL and IADL regression equations.
Treatment effects were assumed to be different after week 20, compared with weeks 21–52. However, the rationale for this cut-off point is broadly stated to be ‘after careful consideration of the data, and an attempt to maximise goodness of fit given insufficient data to consider to alternative functional forms’ (p. 89 of the main submission). Few other details are provided.
The MMSE treatment effect is modelled as an ‘absolute’ benefit rather than using relative risk methodology, i.e. for each period on treatment there is a fixed, absolute change in MMSE. This assumes that all people receive the same benefit, with a mean value derived from trial populations irrespective of their characteristics, such as severity, age and gender. This absolute benefit is then applied to the untreated progression, which is based on the CERAD data.
It is often assumed when building models that the relative risks from a trial are independent of the baseline risks and can therefore be applied to baseline risks estimated from cohorts that may be more representative of the population being treated. However, there is some concern about the approach taken in this instance as it is questionable that the reduction in progression achieved by treatment and estimated from the trial data are independent of the underlying rate of progression. MMSE treatment effect is one of the key drivers in this model, meaning that if the absolute treatment effect is not transferable from the trial participants to the CERAD cohort participants, the ICERs could be substantially different from those reported. However, in the absence of a sensitivity analysis examining this assumption, it is difficult to assess the size and direction of any potential bias.
Patient utilities
Utilities are assigned within the model using an algorithm published in a Swedish study187 consisting of 208 from 272 people with AD and their carers, who were surveyed over a 12-month period. Utility values were measured using the EQ-5D and valued using a normative UK tariff. A number of statistical models are presented, but the one used in the evaluation relates to data at all follow-up points but is based on carer responses to the health status classification part of the EQ-5D. Note that age was not shown to independently predict utility values and the publication does not present statistical models based on patient responses.
The EQ-5D and associated valuation method were considered to be the appropriate methods of assigning utility scores. However, concerns with the use of these algorithms included the following. MMSE score is used as an independent determinant of the change in baseline NPI score. However, both MMSE and NPI scores are used in the patient-utility function, meaning that there is some concern that the effects of MMSE score have been double counted.
The published utility algorithm makes reference to the brief NPI. However, as most of the trials used the longer version of this instrument, the manufacturer converted the NPI coefficient to an alternative score (–0.018 became –0.004) – the transformation is poorly described and it is unclear whether or not it was appropriately undertaken.
The above patient-utility function is based on proxy responses from AD carers, the publication does not show equivalent models based on AD patient responses. However, it is clear from within the publication that responses from individuals with AD and their carers were markedly different. Indeed, non-adjusted results presented by MMSE strata suggest that the choice of data set is likely to be an important determinant of utility (Table 84), an issue acknowledged by the original authors. For example, using the patient-rated data set, there are few differences in utility scores between people with mild and moderate disease. The differences are much more pronounced in the caregiver-related utility data set.
Caregiver utilities
Caregiver utilities were derived using Short Form questionnaire-36 items (SF-36) scores and the Brazier algorithm, using data from three clinical trials. The base-case results suggest that caregiver QALYs contributed a much smaller amount to total QALYs than to patient-related values (about one-tenth, depending on the exact scenario). However, the TAG has some concerns with the final statistical model. For example, finer details of how the final utility equation was derived are not provided, such as how the independent variables were chosen in the first instance or the overall goodness of fit. Moreover, the patient-utility function suggests that entering an ‘institution’ significantly reduces people with AD utility values. However, the caregiver-utility function does not include this term, when it is plausible to believe that such an event could increase carer’s utility levels. It is unclear whether or not this was excluded because the relationship was not examined or because no such relationship was found. It is also worth noting that although ADL and IADL scales were shown to independently predict carer utility levels they are likely to incorporate broadly the same domains meaning there some reason to believe patient utilities are being doubled counted. The patient age coefficient is also positive, suggesting that caregiver utilities increase with increasing patient age. Altogether, there is some concern over the robustness and underlying logic of the caregiver-utility function.
The probability of entering institutionalised full-time care and associated costs
A daily treatment cost of £3 was included for 10 mg of donepezil, along with 6-monthly costs of a ‘doctor’ visit (£62.29 per visit, purported based on NHS reference costs for a geriatrician appointment; the assessment group could not confirm this unit cost from the source cited, and most consultant-led outpatient appointments cost from £100 to £170 in the national schedule of reference costs). However, the expected costs of care are by far the larger and more important costs components. Specifically, the model includes the possibility individuals enter institutionalised FTC (IFTC). As previously noted, the time to entering IFTC in this model is not an explicit event in a DES sense – rather it is modelled purely as a function of MMSE score. More specifically, individuals in one of the five MMSE strata have a probability of either being cared for in the community or in IFTC, with increasing MMSE scores associated with a higher probability of being in IFTC (see Table 81). The latter is also associated with higher costs (see Table 82). Note that the costs of IFTC were not assumed to vary according to the severity of the disease.
The cost estimates in the industry submission are all taken from a report commissioned by the Alzheimer’s Society (Dementia UK report8) in 2007, inflated to current prices. They include both health-care and PSS costs, but, unlike the original SHTAC model, no adjustments are made for the proportion of these costs for which the AD patient or their family is liable. Note that the costs of IFTC inputted into this model are approximately three times higher than those inputted into the original SHTAC model. Specific criticisms that can be levied at the accuracy and use of these cost estimates include the following. The costs are estimated on retrospectively collected resource-use data for 114 individuals between January 1997 and June 1999. Thus, not only is the sample size arguably small, they may not represent contemporary standards of care. Similarly, unit costs have effectively been inflated from 1998 until the present year and are liable to similar criticisms. The authors of the report themselves state that care arrangements are likely to have changed during this time.
The disease severity in the report was classified as mild, moderate and severe disease using the CDR Scale. However, the model is divided into five severity groupings, dependent on MMSE score – no details are provided as to how the costs were divided up, but further investigation of these costs in the Dementia UK report8 indicate that Eisai have assumed that mild on the CDR scale is equivalent to MMSE > 25, moderate on the CDR scale is equivalent to 15 < MMSE < 20, and severe on the CDR scale is equivalent to MMSE < 10. The two further categories of 19 < MMSE < 26 and < 9 MMSE < 16 are calculated as the means of the costs in their adjacent severity groups, i.e. the cost for 19 < MMSE < 26 is the mean of the cost for 15 < MMSE < 20 and MMSE > 25. Additionally, although perhaps not the largest concern, the resource-use study was based on people with dementia rather than individuals with AD.
Information on the proportion of individuals living in the community and IFTC by severity of disease was estimated using a published report in 2007 of 445 individuals in a UK nursing homes (described in the study as being care homes for the elderly mentally infirm, but excluding ‘specialist’ residences). 188 However, this (important) component of the model is considered by the TAG to be particularly poorly described, as the original report does not include specific statistics relating to the proportion of individuals who are living in the community or in institutionalised care. This issue is acknowledged in the manufacturer’s submission; however, the assumptions and calculations used to generate the proportions in Table 85 are lacking in any detail. This said, an obvious criticism of the use of this evidence is that the study was completed in individuals who were already in nursing homes – it did not include people who had not been admitted to care. Thus, it is difficult to understand how these proportions could be accurately derived from this data set in the first instance. Also note that one-quarter of the study participants was estimated not to have dementia. The importance of this evidence as a driver for cost-effectiveness is discussed below.
| Risk factor | Months | |||
|---|---|---|---|---|
| 0–3 | 3–6 | 6–12 | After 12 | |
| Baseline MMSE | 18.8 | 18.8 | 18.8 | 1 |
| Current MMSE | 19.3 | 18.8 | 17.8 | 1 |
| Annualised change in MMSE | 4.31 | –2.15 | –2.69 | 1 |
General technical concerns with the model
Patient population
The modelled population is sampled from individual-level data from three RCTs, but it is weighting by age and gender to match the distribution of these variables in the UK AD population. The weighted sampling is done from the patient population after it has been filtered to include only people with mild or only moderate AD. Therefore, it should produce age and gender distributions that are similar in each severity category. However, the simulated moderate population has a better mean survival than the simulated mild population (4.603 vs 4.110 years). The manufacturer states (in section 3.4.14 of its submission) that this is because the simulated moderate population is younger and has a higher proportion of women. This may produce misleading results if people with mild disease are actually more likely to be younger than people with moderate disease in the UK AD population. It also suggests that the method used to weight the sampling to match the age and gender distribution in the UK is not functioning effectively. This could be because there are insufficient people in the data set from which the population is sampled, as previously discussed.
There is also some concern that other characteristics of the sampled population may not match the UK AD population, such as the likelihood of living with a carer, use of psychiatric medications, ethnicity, etc. For example, everyone lives with the carer in the sampled patient population, which is unlikely to be representative of people with AD in the UK. It has not been possible to investigate the sensitivity of the model to changes in the patient characteristics owing to the way the model samples its patient population from the trial data.
Model structure
The DES approach has been used to track multiple patient characteristics, but these are updated at fixed intervals (e.g. 3 months). In a Markov model, a half-cycle correction would be applied to estimate the costs and QALYs based on the distribution of people across the health states at the mid-point of each time-cycle. In this DES model, there is effectively a 3-month time cycle, but no equivalent ‘half-cycle type’ correction is applied. Therefore, if the time since the last update is 3 months then the costs and utilities applied during those 3 months are based on patient variables at the end of the 3 months.
Even although it is claimed that this is a DES approach, the model calculates two of the most important parameters in determining costs and effects (patient-care costs and utilities) using weighted averages in the same manner as a cohort model. Location of care (home or institutionalised care) is not modelled on an individual level, but is based on the mean rate for people according to severity. The model is not a pure DES-type model, but incorporates elements of individual sampling and cohort modelling approaches.
Time to discontinuation of treatment
Different discontinuation rates are applied for different time periods within the model. The rates are presented in table 8 of the manufacturer’s submission as fixed probabilities over discrete time periods. In the model, it is assumed that the hazard is constant over each of these discrete time periods, allowing the hazard to be calculated from an exponential survival distribution. The hazard is then adjusted for three continuous risk factors that increase the risk of discontinuation. The individual’s time to discontinuation, Td, is then sampled using:
This time to discontinuation is resampled at the start of each discrete time period (0, 3, 6 and 12 months). Each time a new sample is taken from the uniform distribution, meaning that an individual who is sampled to have a higher than average risk of discontinuation in the first time interval (0–3 months) can then be sampled to have a lower than average risk of discontinuation in the next interval (3–6 months), even before the discontinuation risk has been adjusted to account for their individual risk factors. Using the same sample from the uniform distribution for each time interval would allow the risk of discontinuation to be estimated more consistently for the individual over the course of their lifetime, but still allow the hazard to be updated according to changes in their risk factor profile during the first year.
Error suppressions in calculations
There is an extensive use of various functions such as MIN, MAX, etc. to suppress any implausible values that arise during calculations. For example, in utility and MMSE calculations (MX(0, utility)), MN(30,MMSE) and other similar expression are used to suppress any negative utilities values or any MMSE values > 30. It is the TAG/DSU view that any implausible values predicted by the model to have been recorded as errors and investigated rather than being suppressed in the calculations.
Redundant programming syntax
The model submitted by the manufacturer is a generic model, which has a variety of other modules/logic which are not relevant for the present TA. This redundancy is present throughout the model, which has hampered the review process. For example, although the utility equation in the model is correctly implemented, it is defined as a combination of five different equations. This general lack of transparency means it is almost impossible to be certain that all the issues in the model have been identified.
Specific technical errors in the model
Life expectancy
The manufacturer’s submission states that expected survival was calculated by fitting functions of the form to the MRC CFAS data:
The median survival estimates from the MRC CFAS data are given in table 10 of the manufacturer’s submission, and the A and B parameters for men and women according to their age group are given in table 11 of appendix H to its submission. The model samples the life expectancy of the people as follows:
where A and B are selected from table 11 of appendix H for the appropriate age and gender of the patient. The following mistakes were made in estimating the life expectancy of the people in the model.
Male survival estimates are applied to women in one age category
For women aged 70–79 years, the expression (eTimeEvDeath), which is being used to select the appropriate A and B, is referring to the data for men rather than women. Therefore, the model is underestimating survival in this group, as median survival is greater for women in this age category. This error affects both treated and untreated people.
No survival estimate for age 90 years
The expression (eTimeEvDeath), which is used in the model to select the appropriate A and B values according to age and gender, defines the oldest age category as age > 90 years rather than age ≥ 90 years. Therefore, it does not generate an expected survival for people aged 90 years. This effectively set the expected survival to zero for people who start the model with age = 90 years. This error will essentially remove some people from the model before they incur any costs or accrue any QALYs and again, it affects both treated and untreated people. There are four people aged 90 years in the set of 826 trial participants from which the modelled population is sampled, but it is unclear how many times these people are included within the sampled population.
Mini Mental State Examination scaling
The PrevMMSEChange term used in estimating the updated MMSE (equation) is the annual rate of change and, therefore, the change since the last update has to be scaled to give an annual rate. This is calculated in the model as:
However, it is our belief that it should be calculated as:
Given that updates usually occur at 3-monthly intervals, the PrevMMSEChange scores is being underestimated by factor of 16.
Application of hazard ratios for discontinuation
The hazard for discontinuation of treatment is adjusted for three risk factors that increase the risk of discontinuation. These risk factors are baseline MMSE, current MMSE and annualised change in MMSE. The hazard ratios (HRs) for these risk factors are specified for different time periods during the first year. The hazard ratios for these risk factors are only applied during the first year of treatment and are then set to unity. In appendix H, of the manufacturer’s submission, it is stated that a Cox regression model was used to estimate the HRs. In a Cox regression model, the natural logarithm of the hazard is assumed to be a linear function of the form:
Given that the risk factors included within the analysis are all continuous variables, it would be usual to present either the regression coefficient (beta) or the HRs for an increase in one unit along the scale of the continuous variable [HR = exp(beta)]. It can be seen from the equation above that the HR for a decrease in one unit is the reciprocal of the HR for an increase in one unit. Likewise, the HR for an increase in two units is the square of the HR for an increase in one unit. More generally, if HR1 is the HR for an increase in one unit from the reference range then the HR for y units difference from the reference range is defined as follows:
In the model, the expressions aHRb, aHRc and aHRr are used to calculate the HR for the patient’s baseline MMSE, current MMSE and annualised change in MMSE compared with the reference range for each of these variables. However, these are not being calculated in a manner which is consistent with a Cox proportional hazards model. Instead, the following is being calculated:
Therefore, in the model MMSE scores that are lower and higher than the reference range both increase the risk of discontinuation rather than lower ones decreasing the risk and higher ones increasing the risk. The reference ranges used in the model are given in Table 85 for information, as these are not reported in the manufacturer’s submission.
Discrepancies between the model and the submission
Five instances where the data in the manufacturer’s submission does not match that being used in the model have been identified. The differences found were as follows:
-
The constant in calculating the annual rate of decline in MMSE is –5.4663 in the model calculations instead of 5.4663, as mentioned on p. 89 of the submission.
-
In the NPI equation, the coefficient for the interaction term, baseNPI × weeks, in the model is –0.0012 instead of –0.59, as reported in NICE submission (p. 90 of the submission). The same coefficient is reported as 0.0012 in appendix H, table 5.
-
The coefficient for the interaction term, baseIADL × weeks, in the IADL equation is –0.002 in the model instead of 0.002 as mentioned in table 7 of appendix H.
-
The caregiver utility equation uses 0.013 as coefficient for PsyMed instead of –0.01 in the report (p. 93 of the submission) and in table 15 of appendix H.
-
The caregiver utility equation has a patient age term with a coefficient of 0.0014 in the model. Also, it has no term for patient gender as reported in p. 93 of the submission.
The first four discrepancies listed above were confirmed by the manufacturer to be typographical errors in the report and, therefore, do not alter the reported results. The fifth discrepancy affects utilities of treated and untreated patients equally and, therefore, does not affect the incremental cost-effectiveness.
Probabilistic sensitivity analysis
The PSA was replicated as detailed in the report, i.e. 350 runs with 5000 people each run for people with mild and moderate AD separately. Jack-knifing189 has been performed on the results to identify the CIs and they are reported in Table 86.
| AD | Deterministic | Stochastic | ||||
|---|---|---|---|---|---|---|
| Cost (£) | QALYs | ICER (£) | Cost (£) | QALYs | ICER (£) (95% CI) | |
| Mild | –3386 | 0.147 | –22,975 | –1786 | 0.130 | –13,764 (–18,873 to –8768) |
| Moderate | –1883 | 0.109 | –17,310 | –1316 | 0.105 | –12,585 (–17,727 to –7553) |
It can be observed that the deterministic mean is quite different to the stochastic mean, even although all of the ICERs indicate that donepezil dominates standard care. In fact, the mean cost savings and QALY gains are smaller in the PSA analysis for people with both mild and moderate AD which means that the base-case results presented in the submission are quite optimistic relative to the probabilistic mean. This would suggest that the deterministic ICERs cannot be used as a good estimate of the expected cost-effectiveness and that the PSA analysis is the most appropriate to use. This said, there are concerns with the implementation of the PSA analysis in the model. In the health-utility equations, all of the terms in the equation are varied within the PSA, but each term is allowed to vary independently of the others, removing any correlation between the terms. For the disease progression equations, only the intercept term and the treatment effects are varied within the PSA analysis. Again this removes any correlation between the intercept term and the other terms that are fixed. There are also specific concerns regarding the beta distributions used to describe the probability of institutional care, as described below. The results of the PSA analysis should, therefore, be interpreted with caution.
Beta distributions for institutional care
The model uses beta distributions to describe the uncertainty in the proportion of people receiving institutional care for each severity state. The alpha and beta parameters used to define the beta distribution are < 1 for all severity states and are similar, but not exactly equivalent, to the average proportions in home and institutional care used in the deterministic analysis. When the alpha and beta parameters are both < 1, this produces a U-shaped beta distribution with asymptotes at 0 and 1, which does not seem to be a realistic distribution for this parameter. No details are provided on how the alpha and beta parameters, which are given in Table 87, have been derived.
| MMSE | Living in the community (%) | Institutionalised (%) | Distribution used in manufacturer model |
|---|---|---|---|
| Mild | 87.1 | 12.9 | Beta(0.86229,0.12771) |
| Mild to moderate | 74.4 | 25.6 | Beta(0.73656, 0.25344) |
| Moderate | 61.7 | 38.3 | Beta(0.61083,0.37917) |
| Moderate to severe | 49.0 | 51.0 | Beta(0.4851,0.5049) |
| Severe | 30.0 | 70.0 | Beta(0.297,0.693) |
Amendments made to the base case
Given the above concerns with the model, a number of corrections were attempted and additional sensitivity analyses were performed to examine the robustness of the results to alternative assumptions.
Mini Mental State Examination scaling
The PrevMMSEChange term used in estimating the updated MMSE (equation) is estimated using a corrected scaling factor. The costs and QALYs estimated using the updated equation are presented against the base-case model in Table 88, based on a deterministic ICER after 20 runs with 1000 people.
| AD | Base-case model | Base case with corrected MMSE scaling | ||||
|---|---|---|---|---|---|---|
| Cost (£)a | QALYsa | ICER (£) | New cost (£)a | New QALYsa | New ICER (£) | |
| Mild | –3386 | 0.147 | –22,975 | –2953 | 0.137 | –21,554 |
| Moderate | –1883 | 0.109 | –17,310 | –1612 | 0.102 | –15,813 |
Life expectancy
The expression eTimeEvDeath was changed to include people aged 90 years in the fourth age category and to select the appropriate estimates for A and B for women aged 70–79 years. The impact on results from these two combined changes is seen in Table 89 based on a deterministic ICER after 20 runs with 1000 people. The increase in cost-effectiveness of donepezil can be attributed to using the correct life expectancy for women, which was underestimated in the base case.
Hazard calculations
Revised costs and QALYs were calculated by amending the expressions aHRb, aHRc and aHRr to use the correct method for calculating the HRs as previously detailed. The effect on results from these changes is seen in Table 90.
New (deterministic) base-case results
The new base-case model is obtained by correcting the three errors identified in the manufacturer’s model simultaneously. The model is 20 runs with 1000 people (for both mild and moderate categories) and the ICERs are presented in Table 91. Note that the combined corrections have made little difference to the results.
New probabilistic sensitivity results
More appropriate beta distributions relating to the probability of being institutionalised were entered into the model. However, as appropriate measures of variance were not available, the following was undertaken, as outlined in appendix H of the submission:
Where a standard error was not available, we used ±25% of the parameter mean to assign a 95% CI and calculate the corresponding standard error estimate.
Using this method, we have calculated the 95% CI for the proportion receiving institutional care and used these to derive alpha and beta parameters for the proportion receiving care at home as shown in Table 92.
| MMSE | Living in the community (%) | Institutionalised (95% CI)a | Distribution used in DSU analysis for proportion living in community |
|---|---|---|---|
| Mild | 87.1 | 12.9% (9.7% to 16.1%) | Beta(360.6,53.4) |
| Mild to moderate | 74.4 | 25.6% (19.2% to 32.0%) | Beta(132.2,45.5) |
| Moderate | 61.7 | 38.3% (28.7% to 47.9%) | Beta(60.5,37.5) |
| Moderate to severe | 49.0 | 51.0% (38.3% to 63.8%) | Beta(28.4,29.6) |
| Severe | 30.0 | 70.0% (52.5% to 87.5%) | Beta(7.6,17.7) |
The model was then run 350 times for 5000 people and jack-knifing was performed to calculate the CIs. These results incorporate the revised beta functions in addition to the corrections made to the base case to produce the deterministic results. It can be observed from Table 93 that the CI is smaller for the new base case and this can be attributed to the fact that it uses the updated beta functions. The deterministic value is still towards the lower end of the interval obtained through the PSA analysis. These results incorporate the revised beta functions in addition to the corrections made to the base case to produce the deterministic results.
| AD | Deterministic | Stochastic | ||||
|---|---|---|---|---|---|---|
| Cost (£) | QALYs | ICER (£) | Cost (£) | QALYs | ICER (£) (95% CI) | |
| Mild | ||||||
| Base-case model | –3386 | 0.147 | –22,975 | –1786 | 0.130 | –13,764 (–18,873 to –8768) |
| New base-case model | –3563 | 0.164 | –21,713 | –3166 | 0.156 | –20,282 (–22,837 to –17,730) |
| Moderate | ||||||
| Base-case model | –1883 | 0.109 | –17,310 | –1316 | 0.105 | –12,585 (–17,728 to –7553) |
| New base-case model | –1763 | 0.111 | –15,824 | –1380 | 0.109 | –12,678 (–15,309 to –10,057) |
The model was also run 1000 times with 5000 people to gather more accurate results and these are presented in Table 94. It can be observed that the CI is smaller when using 1000 PSA samples rather than 350 PSA samples. Also, the stochastic mean is closer to the deterministic mean. These results suggest that it is necessary to run more than 350 samples to obtain an unbiased estimate using the PSA analysis.
| AD | Cost (£) | QALYs | ICER (£) (95% CI) |
|---|---|---|---|
| Mild | |||
| Deterministic | –3563 | 0.164 | –21,725 |
| PSA with 350 samples | –3166 | 0.156 | –20,282 (–22,837 to –17,730) |
| PSA with 1000 samples | –3415 | 0.159 | –21,433 (–22,354 to –20,515) |
| Moderate | |||
| Deterministic | –1763 | 0.111 | –15,882 |
| PSA with 350 samples | –1380 | 0.109 | –12,678 (–15,309 to –10,057) |
| PSA with 1000 samples | –1703 | 0.111 | –15,285 (–16,686 to –13,888) |
Exploratory analyses on the new base case
In addition to the above technical corrections, a number of exploratory sensitivity analyses were also run to examine the robustness of the results to alternative assumptions.
Proportion institutionalised
The proportion of people institutionalised is dependent on the severity level, as shown in Table 81. The people are assigned a severity level based on their MMSE scores alone. As there was concern regarding the evidence used to estimate these proportions, further analyses were undertaken. Specifically, the assumption was made that disease severity levels (as measured using MMSE) has no effect on the probability of institutionalisation. This was implemented in the model by having the same proportion of people institutionalised (36.5%) at all severity levels. The value of 36.5% is reported by the manufacturer as the overall percentage of institutionalised people with AD in the UK. Although it is unlikely that there is no relationship between MMSE and probability of institutionalisation, this analysis demonstrates how important this relationship is as a driver of cost-effectiveness. The costs and QALYs are calculated and presented in Table 95.
| AD | New base case | New base case with proportion of institutionalised patients set to 36.5% across all severity categories | ||||
|---|---|---|---|---|---|---|
| Cost (£)a | QALYsa | ICER (£) | New cost (£)a | New QALYsa | New ICER (£) | |
| Mild | –3563 | 0.164 | –21,713 | 2186 | 0.113 | 19,389 |
| Moderate | –1763 | 0.111 | –15,824 | 1826 | 0.077 | 23,676 |
Assuming MMSE has no effect on institutionalisation, the ICERs for the mild and moderate populations have become £19,339 per QALY and £23,676 per additional QALY, respectively.
Impact of institutionalisation on caregiver utility
As previously mentioned, caregiver utility is calculated using an equation which includes caregiver age and gender, the four main patient disease measures (MMSE, NPI, ADL, IADL) and use of psychiatric medicine. It does not contain any terms that relate to whether or not the carer is living with the patient and providing care in the home or whether or not the patient is living in an institution. The manufacturer’s submission states that the caregiver utility equation has been derived using data from the Nordic 324 and 312 trials,110,190 which are the same trials used to provide the patient data set from which the modelled population is sampled. Looking at this data set it would appear that all of the people with AD have the variable ‘living with patient’ set to ‘1’, suggesting that all people had a caregiver living with them at the start of the study. They also state that information was not available on the impact of institutionalisation on caregiver utility. Therefore, caregiver utility is estimated in the model to be the same regardless of whether or not the caregiver is living with the patient and regardless of whether or not the patient is receiving home care or institutional care.
Treatment reduces progression to more severe disease states, which are associated with a higher risk of institutional care in the model. If institutional care is associated with an increase in carer utility owing to a lower burden of care being placed on the primary caregiver then reducing disease progression and lowering the average time spent in institutional care will reduce expected QALYs for the caregivers. The sensitivity of the model to alternative assumptions regarding caregiver utility was investigated by removing the utility decrement associated with NPI, ADL and IADL for people receiving institutional care. This improved caregiver utility in both arms of the model, but the incremental effect of treatment on caregiver utility became negative as treatment delays institutionalisation, which is associated with gains in caregiver utility. The incremental costs and QALYs are calculated and presented in Table 96. Although this sensitivity analysis may overestimate the utility gains associated with reduced carer burden after institutionalisation, it does demonstrate that carer utility after institutionalisation is not a significant driver of cost-effectiveness.
| AD | New base case | New base case with improved carer utility after institutionalisation | |||||
|---|---|---|---|---|---|---|---|
| Cost (£)a | Carer QALYsa | Total QALYsa | Cost (£)a | Carer QALYsa | Total QALYsa | ICER (£) | |
| Mild | –3563 | 0.016 | 0.164 | –3563 | –0.010 | 0.138 | –25,844 |
| Moderate | –1763 | 0.011 | 0.111 | –1763 | –0.010 | 0.091 | –19,399 |
Potential overestimation of treatment effect
As noted before, the NPI, ADL and IADL expressions have a MMSE term, as well as having a treatment benefit term. Therefore, it is possible that the effect of treatment is being overestimated. The importance of this structural assumption was investigated by using untreated MMSE values in the NPI, ADL and IADL progression equations for treated patients. The incremental costs and QALYs presented in Table 97 show the same costs, but with a reduction in QALYs as expected.
| AD | New base case | New base case without MMSE treatment effects carrying over into NPI, ADL and IADL | ||||
|---|---|---|---|---|---|---|
| Cost (£)a | QALYsa | ICER (£) | New cost (£)a | New QALYsa | New ICER (£) | |
| Mild | –3563 | 0.164 | –21,713 | –3563 | 0.136 | –26,130 |
| Moderate | –1763 | 0.111 | –15,824 | –1763 | 0.093 | –19,001 |
Combined effect of the exploratory studies so far
This section presents the results of the new base-case model after making several changes to the assumptions to explore the combined effect. These were (1) fixing the proportion of people institutionalised across the severity levels; (2) including the impact of institutionalisation on caregiver utility and (3) removing the MMSE treatment effect from the NPI, ADL and IADL progression equations (Table 98).
Regular update interval
Patient’s disease status is updated regularly, every 3 months, and at these time points the costs and QALYs accrued since the last update are calculated. Updates are also made when other events occur, such as stopping treatment or death, but the timing of these events is unique to each patient. The new base-case model was run for different update intervals and the results are presented in Table 99. There seems to be a clear pattern, as the update period increases the cost savings and QALY benefits decrease and vice versa. However, we cannot be sure why the costs and QALYs vary in a systematic way in relation to the time period between updates. One possibility is that it may be due to the fact that the patient’s attributes at the end of the period are applied to the whole period, as the last update without any type of half-cycle correction being used to reflect the fact that their attributes have been changing over that time period. If the patient’s utility is falling over time, this would systematically underestimate the QALYs accrued by the patient. If the patient’s utility is falling faster in the untreated arm than in the treated arm, one would expect this error to overestimate the QALYs gained by treatment more for less frequent updates, although here we see that the QALY gains are greater for more frequent updates. The cause of this behaviour has not been identified during our examination of the model and, therefore, we cannot exclude the possibility that it may be because of an error in the model logic.
| Update period | Population | |||
|---|---|---|---|---|
| Mild | Moderate | |||
| Cost (£) | QALYs | Cost (£) | QALYs | |
| 30 days | –4247 | 0.184 | –2172 | 0.126 |
| 60 days | –3600 | 0.166 | –1784 | 0.113 |
| New base case (90 days) | –3563 | 0.164 | –1763 | 0.111 |
| 120 days | –2942 | 0.149 | –1481 | 0.102 |
Distribution of sampled life expectancy estimates
Samples of 5000 people were generated using the distributions that are applied in the model (using Excel 2007) and the summary parameters for these were compared with the MRC CFAS data. The median and interquartile ranges (IQRs) for each are presented in Table 100. The median survival estimates appear to match closely at older ages, but there are differences of up to 0.8 years in some age categories between the trial data and the sampled population which is being used to represent the distribution observed in the trial.
| Age (years) | MRC CFAS study (table 10, appendix H of manufacturer’s submission) | 5000 people sampled from the distribution used in the model | ||
|---|---|---|---|---|
| Women | Men | Women | Men | |
| 65–69 | 7.5 (4.8 to NA) | NA (9.1 to NA) | 8.1 (5.5 to 10.0) | 11.8 (9.1 to 11.3) |
| 70–79 | 5.8 (3.6 to 8.3) | 4.6 (3.0 to 8.6) | 6.0 (3.6 to 8.1) | 5.4 (2.9 to 7.8) |
| 80–89 | 4.4 (2.8 to 7.0) | 3.7 (2.5 to 6.3) | 4.8 (2.7 to 6.6) | 4.2 (2.4 to 5.8) |
| ≥ 90 | 3.9 (2.4 to 5.2) | 3.4 (1.5 to 5.5) | 3.9 (2.4 to 5.2) | 3.4 (1.5 to 5.5) |
The sensitivity of the model to differences in the survival estimates was investigated by running the model with the survival times fixed at the median and interquartile values taken from the MRC CFAS study (Table 101).
| Item | Cost (£) | QALYs | ICER (£) |
|---|---|---|---|
| Mild | |||
| New base case | –3563 | 0.164 | –21,713 |
| New base case with survival fixed at median survival | –3857 | 0.180 | –21,395 |
| New base case with survival fixed at lower IQR | –2669 | 0.129 | –20,631 |
| New base case with survival fixed at upper IQR | –4721 | 0.214 | –22,102 |
| Moderate | |||
| New base case | –1763 | 0.111 | –15,824 |
| New base case with survival fixed at median survival | –2085 | 0.127 | –16,475 |
| New base case with survival fixed at lower IQR | –1580 | 0.105 | –15,056 |
| New base case with survival fixed at upper IQR | –2239 | 0.133 | –16,880 |
For males aged < 70 years, no median or upper IQR are provided, so it was assumed that the width of the IQR from males aged 70–80 years could be applied to estimate the median and upper IQRs as 10.7 years and 14.7 years, respectively. Additionally, for women aged < 70 years, no upper IQR is provided, so it was again assumed that the width of the IQR from women aged 70–80 years could be applied to estimate the upper IQR as 10 years. The results show that although the cost-effectiveness estimate is sensitive to changes in the survival inputs, treatment still dominates no treatment even when applying the lower IQR for survival from the MRC CFAS study.
Summary of donepezil model comments
The version of the economic model submitted in the manufacturer’s submission suggests that treatment with donepezil is less costly and more effective compared with no treatment for individuals with mild or moderate AD. However, inspection of the manuscript and programming syntax suggests a number of important issues that should be considered alongside this claim.
Disease progression is modelled using four regression equations. First, changes in MMSE are predicted conditional on a number of independent variables (including treatment), followed by changes on NPI, ADL and IADL scales, also dependent on a number of variables (including treatment and current MMSE). However, there are a number of concerns with the appropriateness of the CERAD study used to estimate these equations and the possibility of double counting of treatment effects, as MMSE was included as an independent term in the NPI, ADL and IADL scales. Moreover, the manufacturer’s submission refers to the ADL/IADL scales as composite measures without fully explaining how they were derived or how estimates of treatment effect measured using specific ADL/IADL scales in the various RCTs were linked to this equation.
The patient-utility function includes a utility decrement if individuals enter institutionalised care. However, no such consideration is given to the possibility that a caregiver’s utility could increase at this time.
The model includes a probability that individuals at various MMSE strata require institutionalised care. However, on inspection the source data used to estimate these proportions includes only individuals who are already said to be in nursing homes. Thus, it is unclear how these data have been used to estimate these proportions. Sensitivity analysis suggests that the base-case ICERs are sensitive to these proportions (see Table 95).
Closer inspection of the model also suggests that is not a pure DES approach, but actually incorporates elements of individual sampling alongside some cohort modelling methods. In particular, it uses a cohort approach to estimate the costs of care and patient utilities, based on the probability of institutionalisation rather than sampling the location of care for each patient.
The deterministic estimates of the ICER overestimated the cost-effectiveness of donepezil compared with the expected ICER obtained from the PSA analysis. This suggests that a robust PSA analysis is needed to determine the cost-effectiveness of donepezil. However, we also had significant concerns regarding the implementation of the PSA analysis and, therefore, the PSA results should be treated with caution.
A number of exploratory sensitivity analyses were undertaken to establish what the cost-effectiveness results would be if changes were made to some of the more important model assumptions, specifically where there was concern with respects to the quality of the inputted data. They included the relationship between MMSE and institutionalisation, the impact of institutionalisation on caregiver utility and the potential overestimation of treatment effects that may be caused by the inclusion of the MMSE treatment effect within the NPI, ADL and IADL progression equations. Exploratory sensitivity analyses showed that the ICER for donepezil compared with no treatment could be as high as £26,000 per QALY in mild AD and £31,000 per QALY in moderate AD, if alternative assumptions are made for each of these key model assumptions. This exploratory analysis demonstrates the range of ICERs that are possible, given the areas of uncertainty identified in the model inputs and assumptions, rather than indicating the most likely ICER.
Lastly, a number of technical errors within the Arena program were detected. Although the corresponding corrections did not significantly alter the cost-effectiveness estimates or the implied decision, concerns remain that there may be further errors within the model, as behaviour was identified that could not be explained when examining the use of an alternative update frequency. There were also concerns regarding the age distribution in the modelled population, which may have resulted from the way in which the model samples its population from a limited set of trial patients.
Chapter 6 The Peninsula Technology Assessment Group cost–utility assessment
Defining the decision problem(s)
Interventions and comparators
The aim of this assessment is to review, and update as necessary, NICE guidance1 to the NHS in England and Wales on the clinical effectiveness and cost-effectiveness of donepezil, galantamine, rivastigmine, for mild-to-moderate AD, and memantine, for moderate-to-severe AD, which was issued in November 2006, and amended in September 2007 and August 2009.
Given the different licensed indications of the four drugs in the UK, for people with different levels of severity of AD, this means that there are:
-
four alternative possible treatments/comparators for people with mild AD (the three AChEIs plus BSC)
-
five alternative possible treatments/comparators for people with moderate AD (i.e. the three AChEIs plus memantine plus BSC)
-
two alternative possible treatments/comparators for people with severe AD (i.e. memantine plus BSC).
Assuming that:
-
the three AChEIs should initially be treated as separate technologies (i.e. with different effectiveness estimates and different intervention costs)
-
there may be subgroup evidence of their differential effectiveness for people with mild, moderate or severe AD (as defined by MMSE).
Then, there are, in theory, 4 × 5 × 2 = 40 alternative technology adoption policies that might need to be modelled (i.e. accounting for all the possible sequences of treatments across the three levels of disease severity). This is clearly an impractical initial range of policy options to model, not least because the evidence of the effectiveness of the four drugs is unlikely to be available for all severity subgroups. Furthermore, evidence of the effectiveness for people switching treatments, for example the effectiveness of switching from one AChEI to another when moving from mild-to-moderate disease, is also highly unlikely to exist in published trials. Ultimately, we found no published clinical effectiveness research that would support either of these potential modelling analyses.
Another new issue since the 2004 technology assessment’s economic modelling is that the range of disease severity that is treatable within the licences of the three AChEIs now overlaps with the severity range treatable with memantine – people with moderate AD (MMSE 10–20). This means that, in theory, people with mild AD who progress to moderate AD could now switch to or start on memantine instead of continuing with their present treatment. Again, whether or not existing published trials would allow reliable estimates of the relative effectiveness of these treatment alternatives is doubtful (e.g. such estimates should ideally come from a RCT that had recruited only people either diagnosed with moderate AD or progressing to it, and allocates them to either memantine or one of the three AChEIs).
Therefore, we have necessarily simplified our initial decision problem and expanded it when only relevant research evidence was found, which justified a more complex specification of the problem. This was necessary, for example, when the considerable difference in cost between patches and capsules for achieving the same daily dose of rivastigmine became apparent and needed to be reflected (see next section).
The decision problems to be modelled
Table 102 shows the main alternatives that will be modelled in terms of the patient populations starting in the model and the treatment comparators. We have taken it as a given that the costs and outcomes (QALYs) to be estimated are those specified in the scope for this TA.
| Decision problem | Simulated population | Starting comparators | Treatment continuation or switching |
|---|---|---|---|
|
Decision problem 1a (treating mild and moderate AD) |
Existing (i.e. prevalent case) AD patients whose disease meets the eligibility criteria for receiving one of the three AChEIs (i.e. MMSE score-based mild or moderate AD (MMSE = 26–10) |
BSC (BSC) Donepezil Rivastigmine (× 2)a Galantamine |
Those who start on BSC stay on it Drug treatment is continued until either (1) clinical decision to stop treatment (e.g. no longer responding) or (2) patient progresses to severe AD |
|
Decision problem 1b (treating mild AD) |
Existing (i.e. prevalent case) AD patients with mild AD (MMSE = 26–21) |
BSC (BSC) Donepezil Rivastigmine (× 2)a Galantamine |
As for decision problem 1a |
|
Decision problem 1c (treating moderate AD) |
Existing (i.e. prevalent case) AD patients with moderate AD (MMSE = 20–10) |
BSC (BSC) Donepezil Rivastigmine (× 2)a Galantamine |
As for decision problem 1a |
|
Decision problem 2a (treating people with moderate and severe AD) |
Existing AD patients with moderate-to-severe AD (MMSE = 20–0) |
BSC Memantine |
Those who start on BSC stay on it Drug treatment is continued until the clinical decision to stop treatment (e.g. no longer responding) |
|
Decision problem 2b (treating people with severe AD) |
Existing AD patients with severe AD (MMSE < 10) |
BSC Memantine |
Those who start on BSC stay on it Drug treatment is continued until the clinical decision to stop treatment (e.g. no longer responding) |
|
Decision problem 3 (treating people with moderate AD) |
Existing AD patients with moderate AD (MMSE = 20–10) |
BSC Donepezil Rivastigmine (× 2)a Galantamine Memantine (if trial data for moderate only) |
Those who start on BSC stay on it Drug treatment is continued until either (1) clinical decision to stop treatment (e.g. no longer responding) or, for AChEIs, (2) patient progresses to severe AD |
Note that in decision problem 1 there is no option of switching to memantine when people progress from mild-to-moderate disease, even although this is a possibility under the current licensed indications for memantine. The cost-effectiveness of each drug is assessed using the doses reported in the RCTs.
Overview of decision model development
The process of developing a decision model for this technology assessment had five main stages. These were:
-
Preparation and familiarisation:
-
familiarisation (by JP and RA) with past economic modelling studies in AD and, in particular, the perceived strengths and weaknesses of the modelling approaches used by the manufacturers and the TAG in the 2004
-
rapid reviews to re-assess what factors drive (or what are associated with) changes in care costs or changes in HRQoL during the progression of AD
-
contact with experts in the field.
-
-
Choosing between discrete-event simulation or Markov (discrete state) modelling.
-
Exploring the possible development of a ‘two-dimensional’ Markov model of AD progression – i.e. a natural history disease model that simulated change through stages of both cognitive status and either functional status or behavioural symptoms. Given the typically univariate reporting of trial outcomes, this approach would probably require access to IPD.
-
Taking the 2004 SHTAC-AHEAD model from the previous technology assessment – which was based around a multivariate model for predicting time to FTC – and both updating the model parameters and adapting the model to try and address some of the more substantial criticisms made of it.
-
Developing a new Markov model that is structurally quite similar to the SHTAC-AHEAD model, but has been based on a time-to-institutionalisation equation based on a cohort of UK Alzheimer’s patients.
Stages 1–4 are described more fully in the TAR for NICE. 191 Stage 5, which led directly to the final model design use, is described below. 187
Building a time-to-institutionalisation model based on UK data
Following correspondence with the authors and principal investigators, in March 2010 we were kindly sent the full data set of the London-based LASER-AD study (principal investigator, Professor Gill Livingston, UCL, London, UK), and on the 9 March 2010 we were also kindly sent the full data set of the Oxfordshire AD data (health economist, Dr Jane Wolstenholme, University of Oxford, Oxford, UK).
The availability of both these UK data sets of IPD about patients with AD and their care and outcomes opened up a number of possibilities for our modelling. In particular, it provided the possibility of using UK data to develop a multivariate regression model of time to institutionalisation (or time to FTC) to replace the US (AHEAD) study-based equations in the SHTAC-AHEAD model. Importantly, also, it allowed us to explore for ourselves possible relationships between time to institutionalisation and MMSE, and care costs, with a view to further informing model assumptions about gradually increasing care costs, and gradually decreasing HRQoL in the time before patients become institutionalised. The following sections describe the methods, and then results, of the final modelling approach that we developed. Ultimately, for various reasons, we made more use of the Oxfordshire AD data set than the LASER-AD study192 data, and we explain how and why in the relevant sections.
Methods
Model structure
The above development stages, therefore, led to the development of a decision model based broadly on the structure of the three-state Markov model described in the previous TAR. 2 An exploration of the SHTAC model in light of the various criticisms and issues raised during and since the 2004 review process, has led to the development of a model based upon time to institutionalisation, parameterised with updated estimates of effectiveness, costs and utilities. A review of all documentation (from manufacturers, interest groups, NICE and the published literature) relating to the decision model described in the previous TAR was undertaken. From this review a list of the various criticisms and issues associated with the SHTAC model was created and is shown in Table 103. Using this list, a number of changes to the SHTAC model structure and the parameter values used in the three-state Markov model were explored. These alterations are described in the appropriate sections below. The model was developed in Microsoft Excel 2007 with additional analyses undertaken in the statistical software package R (The R Foundation for Statistical Computing, Vienna, Austria).
| Criticism of SHTAC model | Addressed in PenTAG model | Method used to try and address the criticism | Relevant section of report | |
|---|---|---|---|---|
| AD progression | ||||
| 1 | Generalisability of risk equations | Yes | Used a UK-based data set55 to model progression in AD | Health-state occupancy |
| 2 | Implicit assumption in SHTAC model that FTC = severe AD | Yes | This assumption has been justified using the IPD from Wolstenholme et al.,55 which suggests MMSE of 9 reached at 0.04 years prior to institutionalisation | Model assumptions |
| 3 | Baseline characteristics – change cohort characteristics | Yes | Base-case baseline characteristics are taken from the Wolstenholme et al.’s IPD; baseline characteristics from LASER-AD study were used in sensitivity analyses | Modelled population |
| Cost data | ||||
| 4 | Query the costs used: inaccurate, out of date, not UK based | No | The only sources of evidence for resource use and costs are from many years ago. Cost data have been inflated to 2009 prices | Cost of health and social care received by Alzheimer’s disease patients |
| 5 | Pre-FTC too heterogeneous a state for a single cost value | Yes | The relationship between costs and time to pre-institutionalisation has been modelled allowing costs in the pre-institutionalised state to be dependent on time to institutionalisation | Cost of health and social care received by Alzheimer’s disease patients |
| 6 | Query the proportion of people in FTC that are institutionalised | No longer relevant | This is no longer relevant as the UK data use time to institutionalisation, rather than FTC | |
| 7 | Query the exclusion of costs for those in institutionalised care who pay privately | Not completely | Based on the Dementia UK report,8 a number of assumptions have been made and assessed | Cost of health and social care received by Alzheimer’s disease patients |
| 8 | No inclusion of carer’s costs | No | No data on the NHS/PSS costs for carer’s of people with AD could be identified | Cost estimates |
| QoL data | ||||
| 9 | No daily health benefit associated with treatment | Yes | The relationship between MMSE and time to institutionalisation has been modelled allowing health benefit to accrue in the pre-institutionalised state | Quality of life of the individual with Alzheimer’s disease |
| 10 | No benefit for those going straight from pre-FTC to death (related to above point) | Yes | As above | Quality of life of the individual with Alzheimer’s disease |
| 11 | Pre-FTC too heterogeneous a state for a single-utility value | Yes | As above | Quality of life of the individual with Alzheimer’s disease |
| 12 | Query the values used | Yes | Utility values by MMSE assessed to be reasonably similar across different studies and the different utility values by MMSE will be investigated in sensitivity analyses | Quality of life of the individual with Alzheimer’s disease |
| 13 | No inclusion of carer’s QoL | Yes | Incorporated carer’s utility as a sensitivity analysis; evidence from one study only | Quality of life of the carer |
| Treatment and effectiveness | ||||
| 14 | Assume treatment stops once enter FTC | Yes | Analysis of the Wolstenholme et al.’s55 IPD suggests that institutionalisation is a good proxy for severe AD (see point 2 above) | Model assumptions |
| 15 | No consideration of treatment dropout, non-responders, AEs | Yes | The PenTAG model allows for a proportion of the total cohort to discontinue treatment each month from the start of treatment; this assumption is constant across all drugs | Treatment discontinuation |
| 16 | No treatment effect observed in psychiatric symptoms | No | Baseline characteristics for the prediction of institutionalisation from the UK data do not include variables for psychiatric symptoms, therefore no treatment effects on psychiatric symptoms are assumed. However, the PenTAG model does incorporate a treatment on functional symptoms in addition to cognitive symptoms | Clinical effectiveness |
| 17 | No treatment benefit beyond 6 months | To an extent | For consistency across drugs, trial data with 6 months’ follow-up have been used; sensitivity analyses for donepezil have incorporated longer-term follow-up | Clinical effectiveness |
| 18 | Placebo effect observed in trials | No | ||
| 19 | Responder analyses not included | No | No data identified from the RCTs | |
| Modelling | ||||
| 20 | Time horizon longer than 5 years | Yes | Time horizon is 20 years, where it is estimated that < 5% of the cohort are still alive | Time horizon |
| 21 | Constant mortality assumed | Yes | Mortality in the PenTAG model is based on age, starting MMSE and ADL, and is the same for treated and untreated patients in the base-case analysis | Health-state |
| 22 | Overestimated mortality | Not addressed directly, but see 21 above | ||
| 23 | Lots of queries regarding the PSA | Yes | Only parameters with uncertainty have associated distributions in the PSA | Results |
| 24 | Inclusion of multiway sensitivity analyses | Not undertaken formally | Some multiway sensitivity analyses were undertaken for comparison with the SHTAC, Eisai/Pfizer and Lundbeck models | SHTAC, Eisai/Pfizer and Lundbeck comparisons |
| 25 | Individual vs population characteristics | Not addressed directly | Cohorts are split by age groups | Model assumptions |
| 26 | No monitoring of MMSE/ADL, etc. – cannot model current NICE guidance | Yes | Inclusion of time to pre-institutionalisation by MMSE allows assessment of disease progression over time by MMSE | Quality of life: utility estimates |
| 27 | Accounted costs during initial treatment period, but not any health benefits | Yes | Both costs and health benefits in the initial treatment period are accounted for (i.e. during the 6 months up to the point of estimation of the treatment effect) | Model assumptions |
Model states
The Markov model consists of three states: pre-institutionalisation, institutionalisation and death (Figure 59). Note that this model differs from that of the SHTAC model, as progression is based on time to institutionalisation in the PenTAG model, not time to FTC (defined by ‘equivalent institutional care’,193 including day and night ‘supervision of personal care, safety or medical care’194) as it was in the SHTAC model. Institutionalisation is defined in the IPD from Wolstenholme and colleagues,195 and thus in the PenTAG model, as ‘Living in a residential home or a nursing home (not as short respite care) or in hospital on a long-term or permanent basis’ (Jane Wolstenholme, personal communication). A particular criticism of the SHTAC model was whether or not the risk equations used to predict FTC from the US study by Stern and colleagues193 could be generalised to England and Wales. Furthermore, the definition of FTC was queried regarding its relationship to institutionalisation, the major step change in costs associated with AD as identified by a review of costs in AD mentioned above. In an attempt to address these concerns, the PenTAG model is based upon UK data predicting time to institutionalisation. Depending on severity, at the beginning of the model individuals in the cohort start in either the pre-institutionalisation state or the institutionalisation state, for example for the base-case analysis for mild-to-moderate severity (decision problem 1a – see Table 102) 90% of the cohort are assumed to start in the pre-institutionalisation state. Transition to death from either of the alive states can occur at any point in time. It is assumed that once an individual becomes institutionalised they do not return to the pre-institutionalised state, thus there are no backward transitions in this model. Note that because of treatment discontinuations some individuals may be on treatment while in the pre-institutionalised state, whereas others may not be on treatment (this is further explained below – see Treatment discontinuation). The three-state Markov model was applied to a cohort of 1000 individuals with mild-to-moderate AD to model the cost–utility of the AChEIs (decision problem 1a in Table 102) and moderate-to-severe AD to model the cost–utility of treatment with memantine (decision problem 2a in Table 102). Information on the characteristics of the modelled population is given in the section below.
FIGURE 59.
Diagram of the three-state Markov model.
Modelled population
For the three cholinesterase inhibitors – donepezil, rivastigmine (capsules and patches) and galantamine – the base-case analysis modelled a cohort of people with mild-to-moderate AD (MMSE 26–10). For memantine, the base-case analysis concerned people with moderate-to-severe AD (MMSE 10–20). In exploratory sensitivity analyses, the cost-effectiveness of treatment with donepezil, rivastigmine (capsules and patches) and galantamine was investigated for a cohort of people with mild AD. Further exploratory sensitivity analyses investigated the cost–utility of donepezil, rivastigmine (capsules and patches), galantamine and memantine for people with moderate-only AD (MMSE 10–20) and the cost-effectiveness of memantine in the treatment of people with severe-only AD (MMSE < 10).
A prevalent cohort is assumed for this decision model, as the data informing disease progression for untreated patients are from a prevalent cohort. Thus, the decision problem only considers the costs and QALYs of a treatment change for the prevalent cohort of individuals with AD. It does not consider the costs and QALYs of individuals diagnosed in the future with AD, which will typically differ from patients in the prevalent cohort. 195 Therefore, it is necessary to state from the outset that this model is based on an assumption that individuals have had a diagnosis of AD for a mean of 4.9 years (IPD from Wolstenholme and colleagues196). Data regarding the characteristics of people with AD were primarily based on IPD from the study by Wolstenholme and colleagues. 196 This study is used to inform much of the PenTAG decision model (including disease progression and cost estimates; see Quality of life – utility estimates and Cost estimates, below). It was chosen as it contains data on untreated people with AD in England and was made available to us by Wolstenholme and colleagues. 196 A UK-based epidemiological cohort study, such as that by Wolstenholme and colleagues,196 was preferred over clinical trial data to avoid any biases of assuming disease progression based on RCT populations, which are subject to a number of inclusion and exclusion criteria not representative of our target population: people with AD in England and Wales. Furthermore, longer follow-up data were available from the Wolstenholme data set188 than those available from clinical trial data. A second UK-based epidemiological data set was available from the LASER-AD study. 192 This study was not used to predict disease progression, as many participants were taking cholinesterase inhibitors and/or memantine during the study period. However, data from the LASER-AD study192 were used to justify and/or corroborate a number of assumptions in the model.
The 1997–8 UK-based study by Wolstenholme and colleagues195 provided estimates of the NHS and PSS costs associated with AD. This was a retrospective cohort analysis of people diagnosed with AD or vascular dementia. Having access to the IPD from this data set made it possible to restrict all analyses to only those people with AD (excluding 8 out of 100 individuals who had vascular dementia). The study participants were recruited through GPs, community psychiatric nurses and consultant geriatricians in the Oxfordshire area during 1988–9. Up to 11 years’ follow-up data are available from this cohort. These data represent a prevalent cohort of 92 patients with AD. At the time of study entry, patients were diagnosed with AD at a median of 4.0 years and a mean of 4.9 years ago.
For each patient, the time from study entry to institutionalisation and death was recorded: 82 of the 92 patients died before the end of the study; 16 patients died before becoming institutionalised and 72 of the 92 patients were institutionalised. At the time of study entry, among a number of outcome measures, the MMSE, Barthel ADL Index and age of the patient were recorded.
The population characteristics from an analysis of the IPD from Wolstenholme and colleagues196 are shown in Table 104. These values were used in the PenTAG model to inform various parameter values for the base-case analyses. In exploratory sensitivity analyses, the cost-effectiveness of treatment with donepezil, galantamine and rivastigmine (capsules and patches) in a cohort of people with characteristics of mild AD was assessed (decision problem 1b in Table 102), as was the cost-effectiveness of memantine compared with BSC for a cohort of people with severe AD (decision problem 2b in Table 102) and the cost-effectiveness of donepezil, galantamine, rivastigmine (capsules and patches) and memantine in a cohort of individuals with the characteristics of moderate AD (decision problem 3 in Table 102). The parameter values for the population characteristics from the Wolstenholme IPD196 used in the base-case and exploratory sensitivity analyses are shown in Table 104.
| Severity of AD | |||||
|---|---|---|---|---|---|
| Mild to moderate (MMSE 26–10) | Moderate to severe (MMSE 20–0) | Mild (MMSE 26–21) | Moderate (MMSE 20–10) | Severe (MMSE 9–0) | |
| n | 71 | 70 | 22 | 49 | 21 |
| Mean age (years) | 77.7 | 78.57 | 76.55 | 78.22 | 79.38 |
| Mean MMSE | 17 | 11.73 | 23.04 | 14.43 | 5.43 |
| Mean Barthel ADL Index | 17.52 | 16.34 | 18.88 | 16.94 | 14.92 |
Note that the data informing disease progression are those from a prevalent cohort of patients living in the community,196 and are therefore not fully representative of the target population of patients in England and Wales living in the community and in institutionalised care. It was, therefore, felt that the model should account for the fact that some individuals in the prevalent cohort are likely to be in institutional care. Data indicating the proportion of people with AD who are institutionalised was available from the LASER-AD study. 192 Livingston and colleagues197 reported that 5.6% of individuals with MMSE > 19, 27.1% of individuals with MMSE 15–19 and 59% of individuals with MMSE < 15 were in institutional care at baseline. This translates to 13% for MMSE > 14 (slightly different to the usual definition of MMSE > 9 for mild-to-moderate disease) and 46% for MMSE < 20 (the usual definition for moderate-to-severe disease). However, it is unclear from the LASER-AD study192 whether or not a prevalent or incident cohort are described and analysed, therefore there are questions as to how the baseline characteristics of the LASER-AD192 population compare with the baseline characteristics of the Wolstenholme study. 196 In addition to this, recent evidence indicates that the number of individuals in institutional care is falling (see Knapp and colleagues,6 chapter 4, p. 50). Therefore, in one-way sensitivity analyses, the LASER-AD study192 results are used as a guide to assume that 10% of the mild-to-moderate cohort and 40% of the moderate-to-severe cohort are institutionalised at the start of the model. In the PenTAG model, both time to institutionalisation and death are significantly dependent on age (see Health-state occupancy), and so the cohort model allows three subgroups defined by age to be included, allowing some degree of heterogeneity to be modelled within the cohort. Table 105 shows the baseline population characteristic parameter values for mild-to-moderate and moderate-to-severe cohorts. Note that, as would be expected, the more severe cohort has a slightly older profile. The parameter values are assessed for their impact on the cost–utility findings in one-way sensitivity analyses.
| Severity of AD | Source | ||
|---|---|---|---|
| Mild to moderate | Moderate to severe | ||
| Mean age (years) | |||
| Age group 1 | 69 | 69 | Wolstenholme et al. IPD194 |
| Age group 2 | 77 | 78 | |
| Age group 3 | 86 | 87 | |
| Proportion in age group | |||
| Age group 1 | 0.25 | 0.25 | Wolstenholme et al. IPD194 |
| Age group 2 | 0.50 | 0.50 | |
| Age group 3 | 0.25 | 0.25 | |
| Mean MMSE | 17 | 11.73 | |
| Mean Barthel ADL Index | 17.52 | 16.34 | |
| Proportion starting in institutionalisation | 0.1 | 0.4 | Informed by data from the LASER-AD study189 |
Model population parameters and assumptions used in sensitivity analyses
In one-way sensitivity analyses, the characteristics of the modelled cohort for mild-to-moderate and moderate-to-severe analyses were changed to represent a different cohort using baseline data from the LASER-AD study. 197 The alternative mean parameter values for the age, MMSE and ADCS-ADL of the cohort are shown in Table 106. Participants in the LASER-AD study197 are older than those from the Wolstenholme et al. study,196 but the mild-to-moderate cohort from the LASER-AD study197 has slightly less cognitive impairment than that from the Wolstenholme et al. study196 (MMSE 19.21 from LASER-AD study197 vs 17.52 from Wolstenholme et al. study196), whereas the moderate-to-severe cohort are slightly more cognitively compared in the LASER-AD study197 compared with the Wolstenholme et al. study196 (MMSE 10.91 from LASER-AD study197 vs 11.73 from Wolstenholme study196).
| Severity of AD | ||
|---|---|---|
| Mild to moderate (MMSE 26–10) | Moderate to severe (MMSE 20–0) | |
| Mean age (years) | ||
| Age group 1; 25% cohort | 71 | 72 |
| Age group 2: 50% cohort | 81 | 82 |
| Age group 3: 25% cohort | 90 | 91 |
| Mean MMSE | 19.21 | 10.91 |
| Mean ADCS-ADL | 44.52 | 27.59 |
As noted above, further sensitivity analyses on the modelled cohort were undertaken to explore decision problems 1b, 1c, 2b and 3, as defined in Table 102. The parameter values for the cohort of mild, moderate and severe AD patients are shown in the final three columns of Table 104. However, caution is needed when interpreting the results from these exploratory analyses, as the populations from which the estimates of effectiveness were obtained were not restricted to the mild, moderate or severe populations. For instance, only one RCT reports effectiveness data within a mild population,115 and this is for only one drug (donepezil) and for only one outcome (MMSE). As the estimate for an effect on MMSE reported in Seltzer and colleagues115 is similar to the pooled estimate for the mild-to-moderate cohort, but with greater uncertainty [compare 1.25 (95% CI 0.17 to 2.33) from Seltzer and colleagues115 with 1.24 (95% CI 0.81 to 1.66) in Table 107], an exploration of the cost–utility of donepezil, galantamine and rivastigmine in a mild population was undertaken using the same effectiveness estimates as that for the mild to moderate population – only the characteristics of the population are altered. Therefore, the sensitivity analysis results for a mild cohort should be interpreted with caution. Similarly, the comparisons between drugs in a moderate cohort should be interpreted with caution, as the populations in the RCTs informing the effectiveness are not entirely comparable, those reporting effect estimates for the AChEIs are based on mild-to-moderate populations, whereas that for memantine is from a single study with a moderate population.
| Treatment | Outcome measure | WMD (95% CI) | Analysis type | Source |
|---|---|---|---|---|
| Donepezil (10 mg) | MMSE | 1.24 (0.81 to 1.66) | M-A result | AD2000 (2004),103 Rogers et al. (1998),113 Gauthier et al. (2002),105 Seltzer et al. (2004),115 Mohs et al. (2001),110 Winblad et al. (2001)116 (appendix 5, figure 15) |
| ADCS-ADLa | 2.02 (1.06 to 3.28) | Average of estimate from galantamine (24 mg) and rivastigmine (≤ 12 mg) | ||
| ADAS-cog | –2.90 (–3.61 to –2.18) | M-A results | ||
| Galantamine (16–24 mg) | MMSE | 1.13 (0.72 to 1.54) | Average of donepezil (10 mg) and rivastigmine (≤ 12 mg) | |
| ADCS-ADLa | 2.23 (1.33 to 3.14) | M-A result | Tariot et al. (2000),141 Brodaty et al. (2005)96 (figure 26) | |
| ADAS-cog | –3.05 (–3.52 to –2.57) | M-A result | ||
| Rivastigmine capsules (9–12 mg) | MMSE | 1.02 (0.63 to 1.41) | M-A result | Feldman and Lane (2007),138 Winblad et al. (2007)140 (figure 35) |
| ADCS-ADLa | 1.80 (0.20 to 3.40) | Single study | Winblad et al. (2007)140 | |
| ADAS-cog | –2.34 (–3.38 to –1.30) | M-A result | ||
| Rivastigmine patches (9.5 mg/day) | MMSE | 1.10 (0.52 to 1.68) | Single study | Winblad et al. (2007)140 |
| ADCS-ADLa | 2.20 (0.62 to 3.78) | Single study | Winblad et al. (2007)140 | |
| ADAS-cog | –1.60 (–2.73 to –0.47) | Single study | Winblad et al. (2007)140 | |
| Memantine (15–20 mg) | MMSE | 0.70 (0.02 to 1.38) | Single study | Reisberg et al. (2003).142 Note: only data from memantine vs placebo RCTs |
| ADCS-ADLb | 1.41 (0.04 to 2.78) | M-A result | Reisberg et al. (2003),142 Van Dyck et al. (2007)143 (figure 46) Note: only data from memantine vs placebo RCTs |
Model assumptions
The model starts when treatment begins for the treated cohorts (point A in Figure 60). For people in the BSC cohort, mean time to institutionalisation (point B in Figure 60) and mean time to death are predicted using the mean baseline characteristics of the cohort: age, MMSE and Barthel ADL Index. The effect of treatment with AChEIs or memantine is assumed to delay institutionalisation. In sensitivity analyses, treatment with AChEIs and memantine is also assumed to increase survival.
FIGURE 60.
Timeline of model for typical individual with AD. A, model starts; B, individual becomes institutionalised.

Figure 61 shows a simple disease progression trajectory based on MMSE scores for two individuals, both starting with a MMSE score of 26 (point a). The thick line represents the individual receiving BSC and the thin line represents the individual receiving treatment, and we assume that both individuals will become institutionalised when their MMSE score reaches 10. If we assume from 6-month RCT data that we know that treated individuals have a difference in MMSE of + x compared with untreated individuals, we can then calculate point b (given that the treated individual started at point a and is + x MMSE points compared with the untreated individual at 6 months). In the absence of effectiveness data beyond our assumed 6-month time period, we assume that the rate of decline for treated individuals from 6 months is the same as the rate of decline of untreated individuals. Thus, the two trajectories are parallel from point b onwards. This then leads to a delay to institutionalisation of z months for treated individuals compared with untreated individuals (see Figure 61). Calculation of this delay to institutionalisation for the PenTAG model is described below (see State occupancy for treated cohorts). Note that both MMSE and ADL scores are used to predict the delay to institutionalisation due to treatment.
FIGURE 61.
Simple disease progression trajectory for an untreated individual and a treated individual.
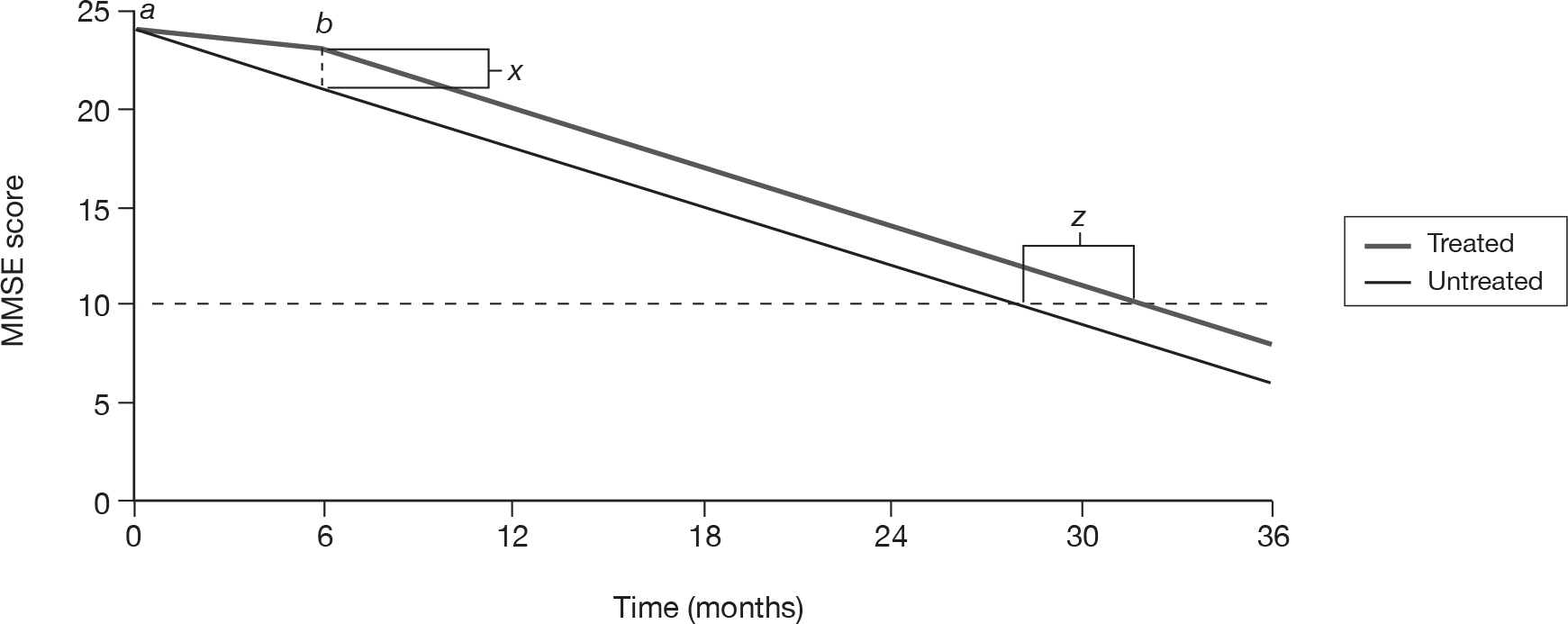
Implicit in our method of modelling the treatment effect on time to institutionalisation and time to death is an important assumption. As shown in Figure 61, distance x represents the treatment effect on the MMSE scale (reported in the RCTs), and the time difference z represents the treatment effect in terms of delay in time to institutionalisation or death, as calculated in our revised methodology. Our critical assumption is that the rate of decline in treated individuals equals the rate of decline in untreated individuals after 6 months. However, at this time point some individuals in the treated cohort will still be receiving treatment, whereas others will have stopped. It could be argued that there could be a ‘bounce-back’ effect for times after point b, that is, when individuals stop drug treatment, their MMSE score declines at a greater rate than individuals on BSC. This scenario is not modelled in this report, but if this were the case, all drugs would be found to be less cost-effective compared with BSC than those estimates in Results suggest. We are not aware of any data to help further investigate this assumption, i.e. to quantify the difference in MMSE at later times.
In the 2004 SHTAC model, only costs during the initial treatment period were accrued, not utilities (see no. 27 in Table 103). In the PenTAG model, both treatment costs and utilities are accrued during the initial treatment period.
As time to death is predicted by age, MMSE and ADL, and given that the treatments affect MMSE and ADL scores, it is possible to assume that the treatments delay death as well as delaying institutionalisation. There is, however, no evidence from the RCTs that treatment increases survival. Neither is there any epidemiological evidence to suggest a treatment effect on survival. Therefore, for the base-case analysis, it is assumed that treatment with donepezil, rivastigmine (capsules and patches), galantamine or memantine delays time to institutionalisation, but has no impact on survival. In the sensitivity analyses, a treatment effect on survival is assumed and the results are presented in similar detail to the base-case analyses.
A criticism of the SHTAC model from the previous MTA (see no. 9 in Table 103) was that there was no daily benefit accrued by treated individuals prior to the point of needing FTC. In the SHTAC model, any individuals dying while in the pre-FTC state or remaining in the pre-FTC state at the end of the model did not contribute any health benefit, yet accrued treatment costs. To overcome this criticism, IPD from the Oxfordshire study by Wolstenholme and colleagues196 was used to refine the pre-institutionalised state of the PenTAG model to allow for gradual increases in costs and gradual reductions in HRQoL during the pre-institutionalised state. Therefore, rather than a single utility or cost value being assigned to the pre-institutionalisation state (as in the SHTAC model), pre-institutionalised utility and cost are dependent upon time to institutionalisation. This allows individuals predicted to be close to being institutionalised to have higher costs and lower utility than those individuals who are predicted to be years away from being institutionalised (see Quality of life: utility estimates and Cost estimates, for further details).
The PenTAG model allows for treatment discontinuations (see Treatment discontinuation), and assumes that for the three cholinesterase inhibitors treatment stops once they enter institutionalisation. Thus, the model implicitly assumes that institutionalisation is equivalent to severe AD (MMSE < 10). Therefore, once in an institution, patients’ QoL and utility are assumed to be that of people with severe AD (MMSE < 10). At MMSE < 10 both the marketing licence and current guidance recommends that patients be taken off the cholinesterase inhibitors. This equivalent assumption was made in the SHTAC model for patients entering FTC and criticised (see no. 2 in Table 103). However, analysis of the IPD from Wolstenholme and colleagues196 suggests that entering institutionalisation is a good proxy for severe AD (as measured by the MMSE): the mean time at which participants reached a MMSE score of 9 is 0.04 years prior to institutionalisation. No such assumption is required to model memantine, as the drug is licensed for moderate-to-severe AD, therefore unless treatment is discontinued (see Treatment discontinuation), memantine is assumed to be taken by individuals until they die.
Time horizon
A monthly time cycle was used in the model and the time horizon was 20 years. By this time it was estimated that < 5% of the cohort would be alive.
Discount rates
In the base-case analyses, discount rates of 3.5% were applied to both costs and health benefits. In the sensitivity analyses, differential discount rates were explored such that health benefits were discounted at 1.5% and costs at 3.5%.
Sources of effectiveness data
Clinical effectiveness
For estimates of clinical effectiveness the highest quality evidence was required. Therefore, only estimates from those RCTs identified in Chapter 4 contributed to the parameterisation of the model. The estimates of clinical effectiveness sought were those from head-to-head trials (as per NICE methods guide198), reporting on:
-
cognition – MMSE in particular
-
functional ability – ADCS-ADL in particular.
The longest follow-up consistent across the different drugs and outcomes was 6 months. Therefore, treatment effect estimates at this time point were used in the base-case analysis, and so the time between points A and B in Figure 60 is 6 months. Longer follow-up data for donepezil were available and were assessed in sensitivity analyses when compared directly with BSC. The longer follow-up data were not used to compare across other AChEIs, as the effectiveness data would not be comparable.
Estimates of effectiveness as measured on the MMSE and ADCS-ADL scales, reported in Chapter 3 were used in the cost-effectiveness modelling. For treated cohorts, the mean difference in MMSE and ADCS-ADL from RCTs was applied to the baseline estimate of MMSE and ADCS-ADL used in the BSC cohort. Thus, the mean baseline MMSE score for treated cohorts was expected to be greater than that for the BSC cohort, as a larger MMSE score indicates better cognitive function than a smaller MMSE score. Similarly, the mean difference in ADCS-ADL from the RCT evidence was added to the mean ADCS-ADL score in the BSC cohort, with ADCS-ADL scores expected to be higher in the treated cohorts. The effectiveness estimates used in the decision model are given in Table 107. Note that the only RCT providing effectiveness evidence on ADAS-cog for memantine did not restrict participants to use of memantine only; participants also received AChEIs (see Combination therapy), therefore these data are not included Table 107.
The sources of effectiveness were also decided upon by comparison with dose levels reported in RCTs and those available in the British National Formulary (BNF). For instance, there was effectiveness evidence for 32 mg/day of galantamine; however, this was not available as a dose regime in the BNF. Therefore, effectiveness data regarding galantamine at these dose levels (32 and 36 mg/day) were excluded from being considered as inputs for clinical effectiveness in the cost–utility model.
To help obtain informative findings for the different drugs, it was felt that an assessment of the cost–utility would be more appropriate for defined dose levels rather than considering a large mix of doses. Dose levels used in the RCTs providing MMSE and ADCS-ADL outcomes for each drug were noted.
-
Donepezil Daily doses were reported to be 5 or 10 mg in the RCTs described in Table 107; however, the majority of RCTs reported treatment effects for participants receiving 10 mg. As donepezil at a daily dose of 10 mg is included in the BNF, 10 mg was taken to be the dose of donepezil to be considered in the CUA. The limited effectiveness evidence for a 5-mg dose is assessed in the sensitivity analyses.
-
Galantamine Two RCTs reported on the clinical effectiveness of galantamine as measured on the ADCS-ADL. 96,125 Both of these RCTs contained individuals taking 8–24 mg/day. In Brodaty and colleagues,96 treated participants received 8 mg/day for the first 4 weeks, with treatment then titrated to a maximum of 24 mg/day. The average daily dose received by participants and reported in Brodaty and colleagues96 was approximately 17 mg. As the BNF indicates that 16 mg/day is the lower recommended dose, the assessment of galantamine is assumed to be for 16–24 mg/day doses and the drug costs are calculated based on an average of the two doses (for further details on drug costs, see Drug costs for Alzheimer’s disease).
-
Rivastigmine Two RCTs reported clinical effectiveness on MMSE or ADCS-ADL. 138,140 Feldman and Lane138 reported only on the use of capsules (2–12 mg/day), whereas Winblad and colleagues140 reported on the effectiveness of both capsules (3–12 mg) and patches (9.5 mg/day and 17.4 mg/day). For the capsules, participants were titrated from a dose of 2 mg/day and 3 mg/day, respectively, to 12 mg/day in the studies by Feldman and Lane138 and Winblad and colleagues. 140 The mean daily doses received by participants were reported to be approximately 9 mg in the Feldman and Lane study138 and 9.7 mg in Winblad and colleagues. 140 Therefore, the drug costs are based on a combination of 9 and 12 mg/day doses. Assessment of the mean differences between the 9.5 and 17.4 mg/day patches for MMSE and ADCS-ADL outcomes at 6 months suggested little difference between the effectiveness of the patches compared with placebo (Table 108). Furthermore, the 17.4 mg/day patch is not a dose regime in the BNF. Therefore, all assessments of the rivastigmine patch are based on the 9.5 mg/day patch. Comparison of the effectiveness and the costs of rivastigmine capsules and patches indicate that these are different technologies and so both are considered in the cost–utility analyses.
-
Memantine The two RCTs contributing to the effectiveness data for memantine both compared memantine to placebo without the additional use of cholinesterase inhibitors. 142,143 The dose used in the RCT of Reisberg and colleagues142 is reported to be 20 mg, whereas in the RCT of Van Dyck and colleagues143 an initial dose of 5 mg is assumed with subsequent incremental doses leading to the target dose of 20 mg. Therefore, the cost–utility of memantine reported here is based on an average of the 15 and 20 mg/day costs.
| Rivastigmine | MMSEa | ADCS-ADLa |
|---|---|---|
| 9.5 mg/day patch | 1.1 (0.52 to 1.68) | 2.2 (0.612 to 3.78) |
| 17.4 mg/day patch | 0.9 (0.32 to 1.48) | 2.3 (0.53 to 4.07) |
A consequence of using the UK data set from Wolstenholme and colleagues196 is that functional capacity is measured on the Barthel ADL Index, an index that is not used or reported in any of the included RCTs. To incorporate this information, the effectiveness evidence from the ADCS-ADL scale used in the RCTs had to be translated on to the Barthel ADL Index. The Barthel ADL Index includes the following ADLs: toileting, bathing, grooming, dressing, feeding, transferring from a sitting to a standing position, mobility and use of stairs. We are not aware of a mapping from the ADCS-ADL Index to the Barthel ADL Index in the literature. It is tempting to assume a direct proportionality between scores on the ADCS-ADL and Barthel ADL Indices, with the constant of proportionality equal to the ratio of the maximum score on the Barthel ADL Index (20) and the maximum score on the ADCS-ADL Index (78). However, this would be a strong assumption that would not be evidence based. Instead, we estimated a quadratic mapping from the ADCS-ADL Index to the Barthel ADL Index, using data from Galasko and colleagues,199 which gives the mean scores over 145 patients in the USA at each of three time points (t = 0, 6 and 12 months) for each of the 19 questions of the ADCS-ADL-Severe Index. From these data, we first estimated the corresponding scores at each of the three time points on the Barthel ADL Index. Next, we estimated the corresponding scores at each of the three time points on the ADCS-ADL Index. Together this gave us three data points for the mapping from the ADCS-ADL to the Barthel ADL Index.
All data were taken from table 1 in Galasko and colleagues. 199 First, the three Barthel scores, corresponding to times 0, 6 and 12 months were derived as follows. Each question of the Barthel ADL Index (bowels, bladder, grooming, toilet, feeding, transfer, mobility, dressing, stairs and bathing) was taken in turn. For each question, one question of the ADCS-ADL-Severe Index was identified which most closely correlated with the question on the Barthel ADL Index. In many cases, there is an exactly analogous question on the ADCS-ADL-Severe Index, for example bathing. Next, a simple relationship was derived between the score on the question on the ADCS-ADL-Severe Index and the score of the question on the Barthel ADL Index. This was usually, but not always, a simple direct proportional relationship, with the constant of proportionality equal to the ratio of the maximum score on the Barthel ADL Index and the maximum score on the ADCS-ADL-Severe Index. For example, the ratio for bathing was set to 1 : 3, given a maximum score of 1 on the Barthel ADL Index and 3 on the ADCS-ADL-Severe Index.
Next, the score for each time point for a given question on the Barthel ADL Index was calculated as the score for the correlating question on the ADCS-ADL-Severe Index multiplied by the relationship between the scores on the Barthel and ADCS-ADL-Severe Indices described in the previous paragraph.
When no single question on the ADCS-ADL-Severe Index could be identified which closely correlated with a given question on the Barthel ADL Index, we set the score for that question on the Barthel ADL Index equal to the maximum score for that question on the Barthel ADL Index multiplied by the ratio of the total score over all questions on the ADCS-ADL-Severe Index and the maximum possible total score on the ADCS-ADL-Severe Index (equal to 54). In this way, the scores on each of the ADCS-ADL-Severe Index questions influenced the score for the single question on the Barthel ADL Index. This procedure yielded the following values on the Barthel ADL Index, corresponding to patient times 0, 6 and 12 months, respectively: 13.16, 10.33 and 7.74.
Next, the three ADCS-ADL scores, corresponding to times 0, 6 and 12 months, were derived in the way described above for the Barthel scores, except when no single question on the ADCS-ADL-Severe Index could be identified which closely correlated with a given question on the ADCS-ADL Index, we set the score for that question on the ADCS-ADL Index exactly as described above [i.e. equal to the maximum score for that question on the ADCS-ADL Index multiplied by the ratio of the total score over all questions on the ADCS-ADL-Severe Index and the maximum possible total score on the ADCS-ADL-Severe Index (equal to 54)], then multiplied by 50%. It was necessary to multiply by 50% because only 50% of patients in the study of Galasko and colleagues199 even attempted to answer the question. These questions on the ADCS-ADL Index, for which there is no correlating question on the ADCS-ADL-Severe Index (e.g. making a meal, shopping, reading, writing), are activities that are very rarely attempted by people with moderate-to-severe AD. Indeed, these questions are omitted from the ADCS-ADL-Severe Index because the ADCS-ADL-Severe Index is designed for moderate-to-severe AD. We acknowledge that the 50% factor is an approximation, but this is our best estimate given the lack of further data.
This procedure yielded the following values on the ADCS-ADL Index, corresponding to times 0, 6 and 12 months, respectively: 29.60, 23.09 and 17.48. In addition, we know that when the maximum score of 78 is achieved on the ADCS-ADL Index, the maximum score of 20 must be achieved on the Barthel ADL Index.
We then fitted a statistical model in R software to these four data points with the Barthel score as the response variable and a quadratic in the ADCS-ADL score as the explanatory variables, where the intercept was constrained to equal zero (as the function must pass through the origin). The deterministic mapping is given as follows (Figure 62); Barthel score = 0.534 × (ADCS-ADL score) – 0.0036 × (ADCS-ADL score). 2
FIGURE 62.
Statistical relationship between the ADCS-ADL Index and the Barthel ADL Index. The thick curved line shows the relationship used in our base case calculated from Galasko and colleagues. 199
For the PSA, it is necessary to model the uncertainty in the covariate coefficients. This was achieved as follows. First, the Cholesky matrix C, corresponding to the variance/covariance matrix of the parameter coefficients was calculated as:
The rows and columns of C correspond to the linear and quadratic terms in the ADCS-ADL score, respectively. Probabilistic covariate coefficients were then simulated as y + Cz, where y is the vector of coefficient means (given in the deterministic equations above) and z is a vector of independent standard normal variables. 200
The ADL effectiveness data for memantine from both RCTs142,143 used ADCS-ADL-Severe Index. Thus, the above procedure was repeated to obtain a quadratic mapping from the ADCS-ADL-Severe scale to the Barthel scale (Figure 63). The quadratic equation is given by: Barthel = 0.5835 (ADCS-ADL-severe) – 0.0039 (ADCS-ADL-severe). 2
FIGURE 63.
Statistical relationship between ADCS-ADL-Severe Index and Barthel ADL Index.
For the PSA, the Cholesky matrix C, corresponding to the variance/covariance matrix of the parameter coefficients, was calculated as
As above, the rows and columns of C correspond to the linear and quadratic terms in the ADCS-ADL-Severe score, respectively. Probabilistic covariate coefficients were then simulated as y + Cz, where y is the vector of coefficient means (given in the deterministic equations above) and z is a vector of independent standard normal variables. 200
There were two instances of missing data across the five treatments and two outcomes:
-
an estimate of effect on ADCS-ADL at 6 months for donepezil (10 mg)
-
an estimate of effect on MMSE at 6 months for galantamine (16–24 mg).
It was assumed that this was a lack of evidence for an effect rather than a lack of effect. The average treatment effect from the same class of drugs was used for these two instances of missing data. In other words, the effectiveness of donepezil based on ADCS-ADL score was taken as an average of the effectiveness of galantamine and rivastigmine capsules. Similarly, the effectiveness of galantamine based on MMSE score was assumed to be an average of the MMSE effectiveness estimate from rivastigmine (capsules) and donepezil.
A treatment effect on cognition as measured by MMSE was used in the base case as it was the scale used in the study by Wolstenholme and colleagues,196 and therefore used in the PenTAG model to predict time to institutionalisation and time to death. In the sensitivity analyses, a treatment effect on cognition as measured by the ADAS-cog score was assumed for all AChEIs and translated onto the MMSE scale using an equation published by Doraiswamy and colleagues. 201 Further sensitivity analyses explore the impact of increasing and decreasing the ADCS-ADL and MMSE effectiveness estimates for all technologies.
Treatment discontinuation
For all effectiveness estimates it is assumed that an ITT analysis has been undertaken, so that estimates relate to all participants and not only those continuing to take treatment. Given that many RCTs did not report an ITT analysis, this assumption is likely to overestimate any treatment effects in the decision model.
Data on the proportions of individuals discontinuing treatment were available from the RCTs included in the systematic review. There was a great deal of information across different dose levels and follow-up; however, each RCT reported only discontinuations at the last follow-up within each study. The available data are given in Figure 64 for each drug. As can be seen, the data are not entirely consistent across studies, with higher discontinuations observed at shorter follow-up than at longer follow-up (e.g. galantamine 16–24 mg). In the base-case analysis a constant rate of treatment discontinuation was, therefore, assumed for all drugs at all doses. The basis for this value was a mixture of the evidence from the RCTs (indicating that if discontinuations carried on as reported at 6 months, by about 2 years most patients would have discontinued treatment) and clinical opinion on the length of time patients would generally spend on treatment. It is possible that the pattern of treatment discontinuations is not linear as assumed in the PenTAG model and that it may be more likely that many more patients discontinue treatment at the earlier stages of treatment. However, as noted, the only data available are those at a single time point, and a more complex relationship between discontinuations and time (other than a linear relationship) would require further, currently untestable, assumptions to be made. It is, therefore, assumed that 4% of the total cohort discontinue treatment each month, so that after 2 years of treatment almost all individuals are no longer receiving treatment (Figure 65). In the PenTAG model, the proportion of participants discontinuing treatment was applied to the treatment and monitoring cost estimates. The impact of different discontinuation rates was assessed in sensitivity analyses. These were based on the minimum (2.3%) and maximum (5.7%) slopes across all technologies in Figure 65, and informed the distribution placed on this value in the PSAs.
FIGURE 64.
Proportion of participants discontinuing treatment by time from start of treatment (weeks) from the RCT data. Each solid line represents a single RCT. The dotted line in each plot is the estimate used in the base-case analyses.
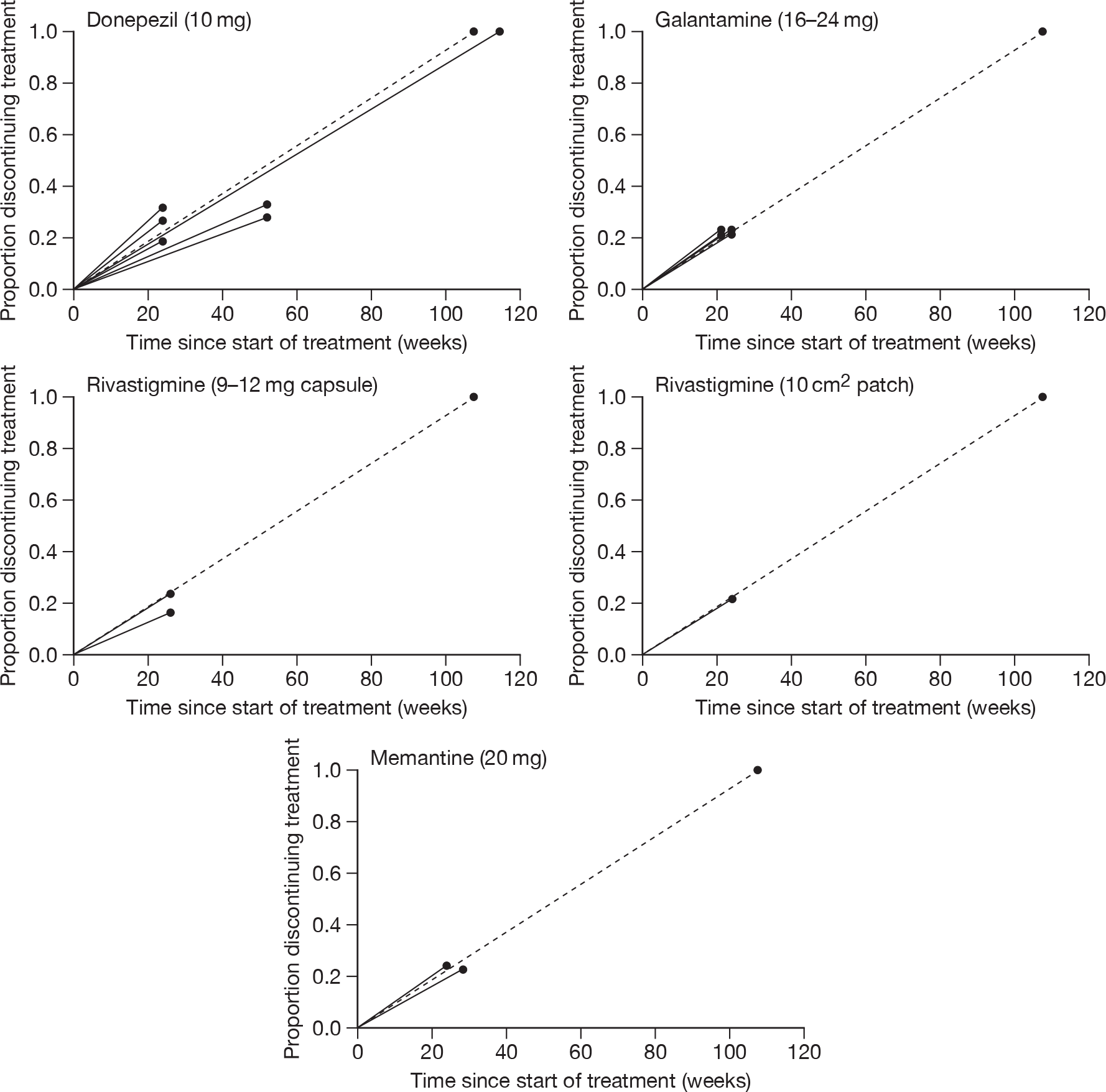
FIGURE 65.
Assumed pattern of treatment discontinuation for all drugs (base-case analysis shown by bold line and sensitivity analyses shown by dotted lines).
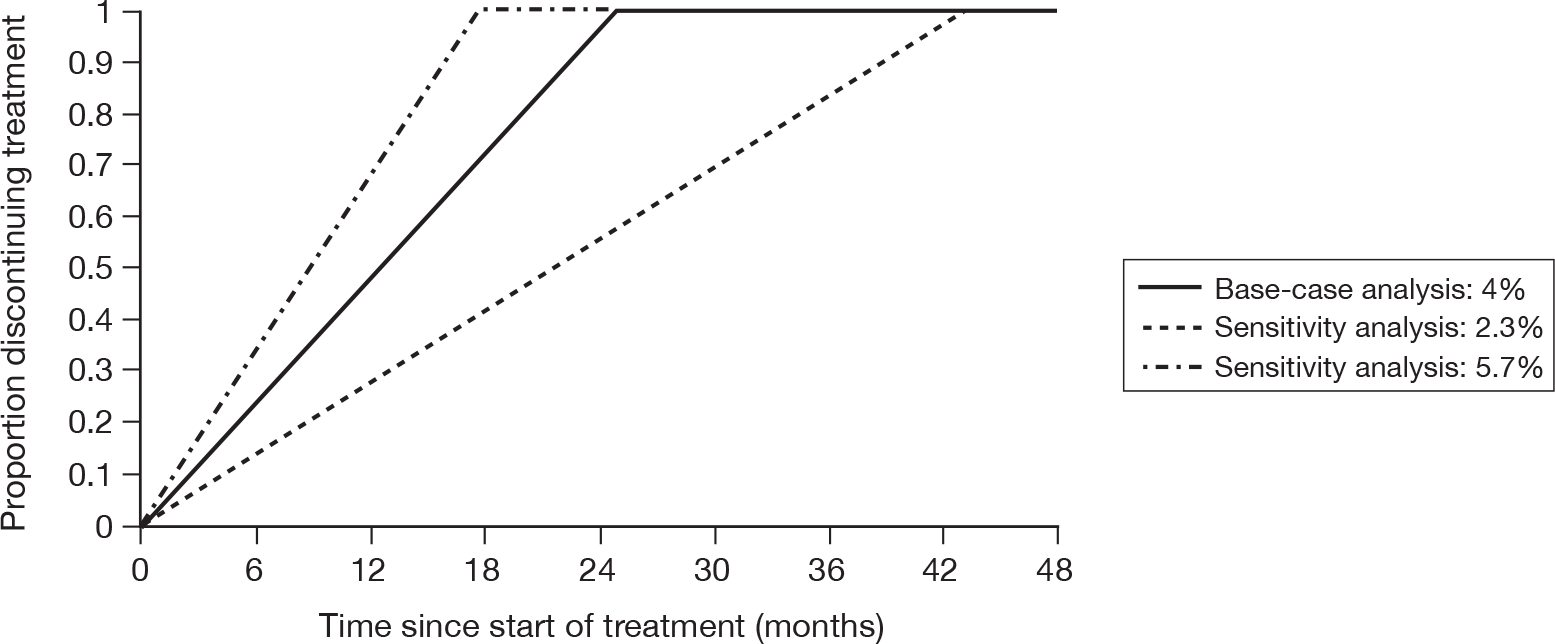
Health-state occupancy
State occupancy for best supportive care cohorts
Individual personal development data from the study by Wolstenholme and colleagues196 was used to estimate the proportion of the total cohort in each of the following three health states at each cycle of the model (1 month): pre-institutionalised, institutionalised and dead. A summary of the data from Wolstenholme and colleagues196 was given above (see Modelled population). For all 92 participants in the IPD, the median and mean time to end of pre-institutionalisation was 1.8 and 2.4 years, respectively (see Figure 66). The median and mean overall survival was 2.7 and 3.3 years, respectively (see Figure 67). To calculate an equation representing time to end of pre-institutionalisation, an exponential survival regression model (‘survreg’ routine from the ‘survival’ R package) was fitted, with time to end of pre-institutionalisation as the response variable, and MMSE, Barthel ADL Index and age at the start of study as covariates. Note that the phrase ‘time to end of pre-institutionalisation’ is not quite the same definition as time to institutionalisation, as some individuals died before entering institutionalisation. For simplicity, the exponential distribution was chosen, rather than more complex two-parameter functions. Age was found to be a highly statistically significant predictor of time until end of pre-institutionalisation. Although MMSE and Barthel ADL Index at the start of the study were not identified as statistically significant variables in explaining the variance of time to end of pre-institutionalisation, both were retained in the model so that a treatment effect could be incorporated into the decision model. In the deterministic case, the time to pre-institutionalisation was described by an exponential distribution with the following rate parameter:
FIGURE 66.
Proportion of cohort pre-institutionalised over time with model fit by MMSE (a), Barthel ADL Index (b) and age (c) all at the start of the study. (a) Lower curve MMSE = 10, middle curve MMSE = 14.5 (mean), upper curve MMSE = 20; (b) lower curve Barthel ADL Index = 10, middle curve Barthel ADL Index = 17 (mean), upper curve Barthel ADL Index = 20; (c) lower curve age = 85 years, middle curve age = 78 years (mean), upper curve age = 70 years.
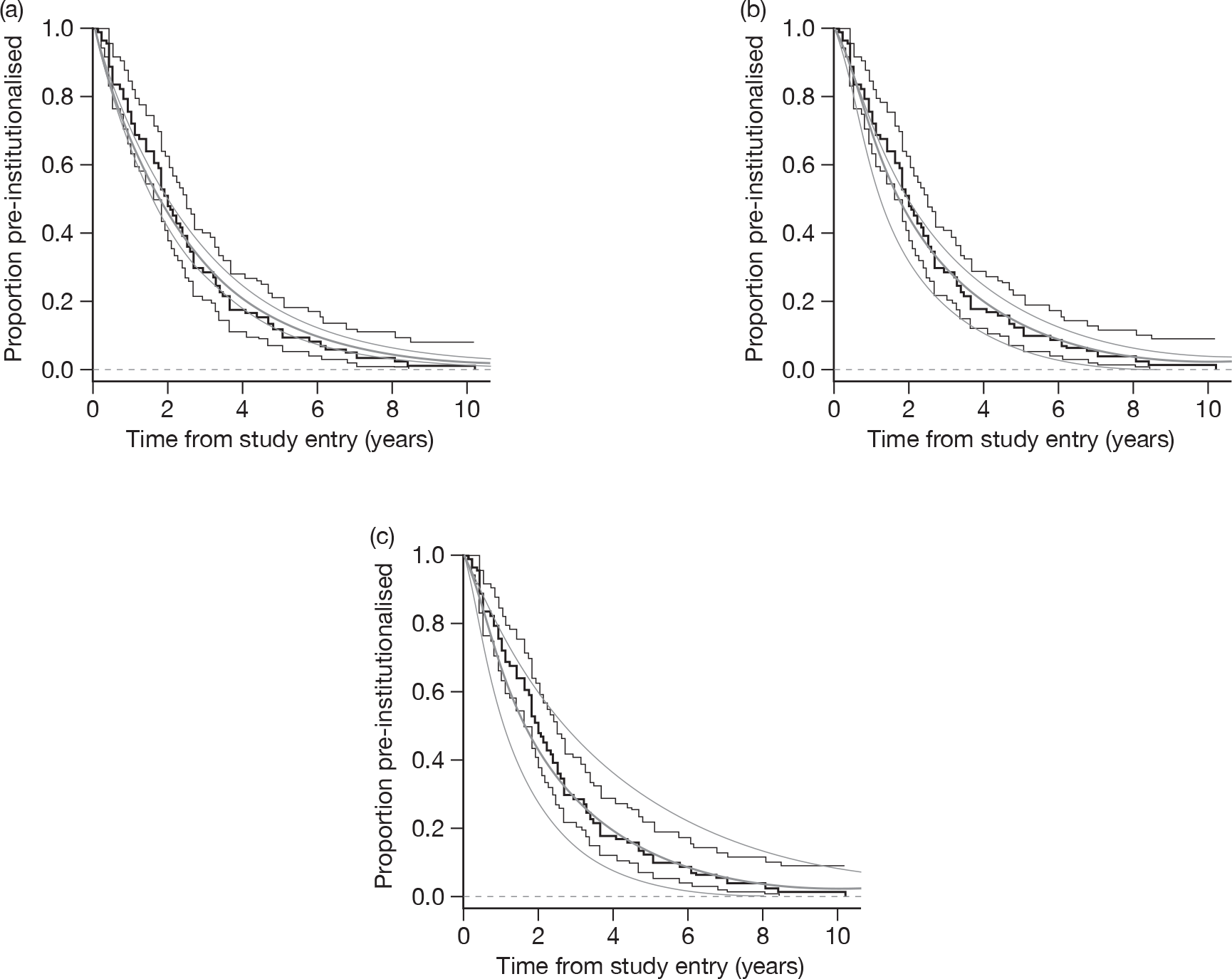
FIGURE 67.
Proportion of cohort alive over time with model fit by MMSE (a), Barthel ADL Index (b) and age all at the start of the study (c). (a) lower curve MMSE = 10, middle curve MMSE = 14.5 (mean), upper curve MMSE = 20; (b) lower curve Barthel = 10, middle curve Barthel = 17 (mean), upper curve Barthel = 20; (c) lower curve age = 85 years, middle curve age = 78 years (mean), upper curve age = 70 years.
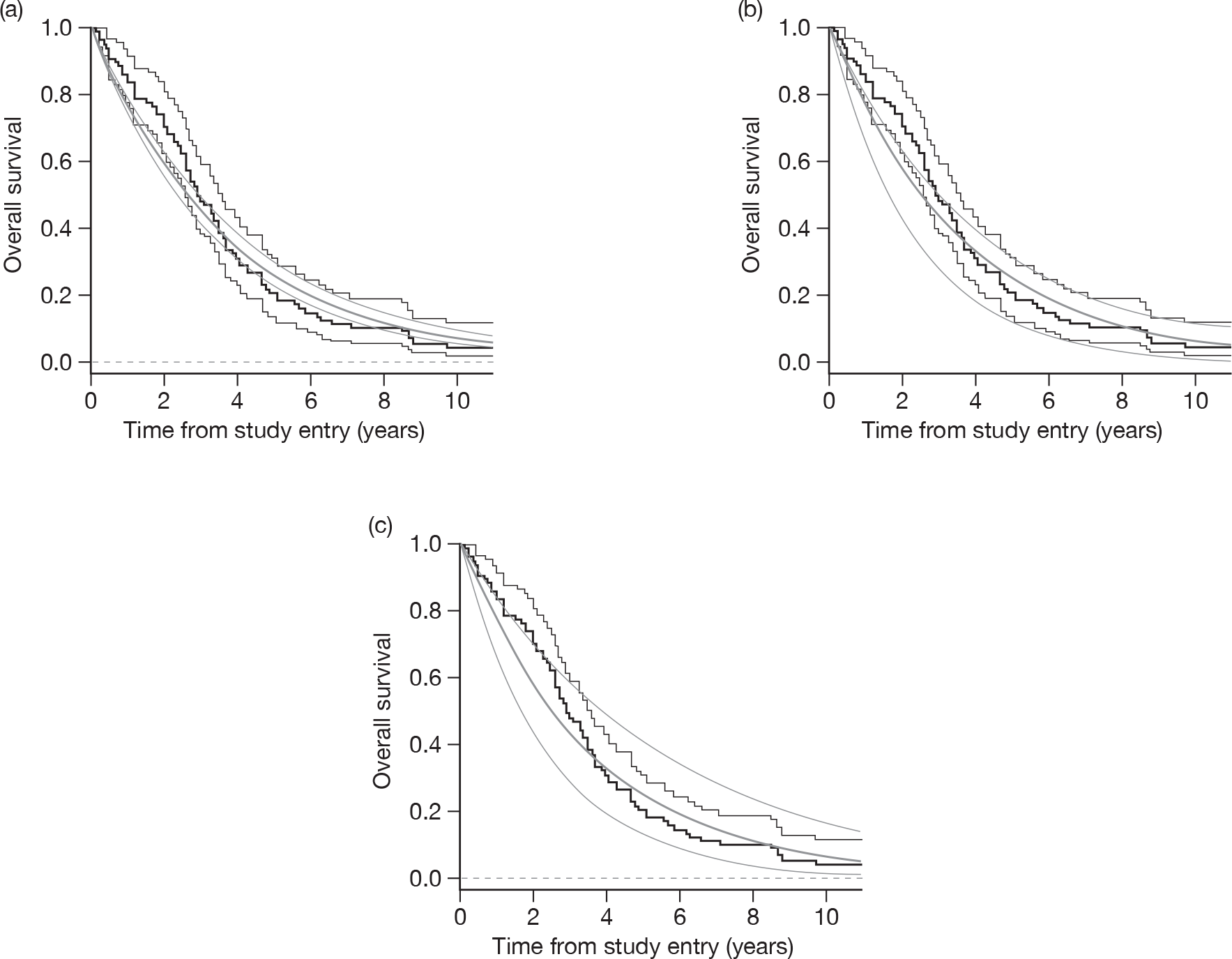
As expected, the greater the MMSE score, the greater the Barthel ADL Index score, and the lower the age of the individual at study entry, the longer that individual remained pre-institutionalised (Figure 66). For the PSA, it is necessary to model the uncertainty in the covariate coefficients. This was achieved as follows. First, the Cholesky matrix C, corresponding to the variance/covariance matrix of the parameter coefficients was calculated as:
where the rows and columns of C correspond to the intercept, MMSE score at start of study, Barthel ADL Index score at start of study, and age at start of study, respectively. Probabilistic covariate coefficients were then simulated as y + Cz, where y is the vector of coefficient means (given in the equation for λpre-inst above), and z is a vector of independent standard normal variables. 200
To model overall survival, an exponential distribution with the same covariates was fitted to the data with overall survival as the response variable. Overall survival was described by an exponential distribution with the following rate parameter:
Again, as expected, the greater the MMSE score, the greater the Barthel ADL Index score, and the lower the age of an individual at study entry, the longer that individual survived (Figure 67). For the PSA, the Cholesky matrix C was calculated as:
Probabilistic covariate coefficients were then simulated as before, using the vector of coefficient means given in the equation for λOS above. For the PSA, we assumed no correlation between the covariate coefficients for pre-institutionalisation and overall survival.
Thus, assuming exponential distributions with the above two rate parameters, the proportion of the cohort in any of the three states at any time period could be obtained. For the BSC cohort, the baseline age, MMSE and Barthel ADL Index scores given in Table 104 were inputted into these rate parameters.
State occupancy for treated cohorts
Using the Wolstenholme and colleagues IPD,196 a linear mixed-effects model (from the ‘nlme’ R package) was fitted with time to end of pre-institutionalisation (or overall survival) as the response variable, MMSE and Barthel as explanatory variables, and patient as a random effect. For each patient there were typically several observations. Variations in the intercept and slopes of the effects of MMSE and Barthel across patients were modelled as random variables, as normal distributions. In addition, a covariate, MMSE at the start of the study was included in the model. The following equations were obtained:
The uncertainty in these equations was modelled for the PSA by recording the Cholesky matrices from the statistical analyses. These are given below.
For time to institutionalisation:
For time to death:
For mild-to-moderate patients, with a mean baseline MMSE of 17 (refer back to Table 105), the equations give:
And for moderate-to-severe patients, with a mean baseline MMSE of 11.7 (refer back to Table 105);
The impact of (time-varying) MMSE on time to institutionalisation can be seen from Appendix 18, Figure 165. From the equations above, the mean increase in the time (years) to institutionalisation for a given drug is calculated for mild-to-moderate patients as 0.1032(ΔMMSE) + 0.0781(ΔBarthel), and for moderate-to-severe patients as 0.0910(ΔMMSE) + 0.1159(ΔBarthel), where ΔMMSE and ΔBarthel are the treatment effects on the MMSE and Barthel scales.
Also, the mean increase in the time to death for a given drug is calculated for mild-to-moderate patients as 0.1270(ΔMMSE) + 0.1021(ΔBarthel), and for moderate-to-severe patients as 0.1138(ΔMMSE) + 0.1595(ΔBarthel). Note that we do not assume that drugs affect overall survival in the base case. Instead, this is addressed in a sensitivity analysis.
The time to institutionalisation and the time to death for each drug were assumed to follow exponential distributions in the treated cohorts, as for the BSC cohorts. The mean time to institutionalisation for a given drug is calculated as the mean time to institutionalisation for BSC plus the mean increase in the time to institutionalisation for the drug (as calculated above). Similarly, for the sensitivity analysis, the mean time to death for a given drug is calculated as the mean time to death for BSC plus the mean increase in the time to death for the drug.
Transitions to institutionalisation and death are both based on data from Wolstenholme and colleagues. 196 It is likely that the individuals described in this study are not as ill as those in the general population for two important reasons. First, inclusion criteria stated that individuals were not living in institutional care, and, second, individuals have had a diagnosis of AD for a mean of 4.9 years.
Quality of life: utility estimates
In the base-case analyses only utilities of people with AD are considered. In the sensitivity analyses, data concerning carer utility are also included. Evidence on the QoL of people with AD is reviewed and discussed in the next section, with the limited evidence available for carer’s QoL reviewed and discussed below (see Quality of life of the carer).
Quality of life of the individual with Alzheimer’s disease
Since the previous review of the four drug treatments for AD,2 a number of papers reporting utility values for people with AD have been published. This literature is reviewed below (and summarised in Table 109), grouped by whether or not just one or multiple utilities are reported. The actual utility values from each of these studies are given in Appendix 17. For completeness and comparison, the utilities from the studies reviewed in the previous health technology assessment (HTA) are also provided in Appendix 17.
| Source | Sample | Scale | Categories | Comments |
|---|---|---|---|---|
| Kerner et al.211 |
AD 159 AD spousal carer-proxy USA |
QWB |
AD patients Control subject |
|
| Miller et al.202 |
AD Up to 359 carer-proxy USA |
HUI-3 |
Baseline 3 months 6 months 9 months |
60% of patients on cholinesterase inhibitors |
| Naglie et al.203 |
AD 60 self and carer-proxy Toronto |
EQ-5D QWB HUI-3 |
Only patients able to complete two interviews are included | |
| Jonsson et al.187 |
AD 208 self and carer-proxy Sweden, Denmark, Finland, Norway |
EQ-5D | MMSE: 26–30, 21–25, 15–20, 10–15, 0–9 | |
| Sano et al.212 |
AD AD experts and students USA |
TTO and VAS | CDR: 1 and 3 | |
| Ekman et al.204 |
Dementia General public (45–84 years) Sweden |
TTO | CDR: 0.5, 1, 2, 3 | Response rate of only 30% |
| Neumann et al.213 |
AD 679 carer-proxy USA |
HUI-2 | CDR: 0.5, 1, 2, 3, 4, 5 | |
| Wlodarczyk et al.205 |
AD 100 residents self and carer-proxy Australia |
AQoL |
MMSE: 0–10, 10–15, 15–20, 20–25, 25+ IADL: 0–2, 3–5, 6–8 |
Part of global donepezil trial |
| Andersen et al.206 |
Dementia 244 combined carer and patient answers Denmark |
EQ-5D |
MMSE: 21+, 10–20, 0–9 Dependency: independent/dependent Residential status: community/institution |
Mapped from health status questionnaire and ADL answers |
| Karlawish et al.207 |
AD Self and carer-proxy USA |
EQ-5D HUI-2 |
MMSE: 24–29, 20–23, 11–19 IADL: 8–10, 11–14, 15–27 BADLS: 6, 7–14 |
Utilities also reported by other measures: 3MS, QoL-AD, GDS and SF-12 |
One-state health utility: having Alzheimer’s disease
In an assessment of clinical and demographic correlates with utility scores, Miller and colleagues202 report (carer proxy) Health Utilities Index version 3 (HUI-3) utilities for ‘having Alzheimer’s’ at baseline, 3, 6 and 9 months for up to 359 patients from the USA. Patients had a mean MMSE score of 15 (SD 5.8; range 4–29) at baseline. Miller and colleagues202 note that the utilities they report (0.184, range 0.29–1) are lower than those reported elsewhere using HUI-3 or other scales and suggest this may be because of a higher proportion of patients in their study having serious psychiatric symptoms compared with other studies. Miller and colleagues202 also point out that HUI-3 utility estimates are often found to be lower than those from alternative scales [i.e. Health Utilities Index version 2 (HUI-2), Quality of Well-being (QWB) and EQ-5D], as there is a ‘greater emphasis on cognition’ with HUI-3 than with the other scales. Approximately 60% of patients were on treatment with a cholinesterase inhibitor at baseline.
Naglie and colleagues203 report self- and carer-proxy-rated health-state values for ‘having Alzheimer’s’ comparing the EQ-5D, QWB and HUI-3 in 60 people with AD in Toronto, Canada. Participants had a mean MMSE score of 18.9 (SD 4.5). Naglie and colleagues203 point out that their findings (ranging from a utility of 0.86 from patients to 0.42 from carer-proxies) may not be generalisable to all patients with AD, as only those able to complete two facilitated interviews were included in this study.
Multiple-state health utility: by cognition
Jonsson and colleagues187 provide EQ-5D, EQ-5D VAS (visual analogue scale) and QoL-AD (Quality of Life in Alzheimer’s Disease Scale) health-state utilities by MMSE ranges, based on self-ratings and carer-proxy ratings for 208 people with AD in Sweden, Denmark, Finland and Norway. The utilities are reported where both patient and carer ratings are available or where only patient or only carer ratings are available. These utilities, ranging from 0.21 to 1, are from a prospective observational study, with 71% of patients receiving cholinesterase inhibitors at baseline.
Community-based time trade-off (TTO) health-state values were elicited for dementia (not specifically AD) by Ekman and colleagues204 from members of the Swedish public aged 45–84 years old. Ekman and colleagues204 report the TTO values by age group (45–54, 55–64, 65–74 and 75–84 years) and gender of the members of public contributing to the utility values. The TTO values are reported by severity based on the CDR. Ekman and colleagues204 found that age, gender and self-assessed health status were associated with the utilities elicited; however, there was no consistent pattern across severity, for example age was a significant factor only for utilities elicited for patients with mild cognitive impairment and mild dementia. The questionnaire from which the TTO values were obtained was validated by colleague and participant comments. Four vignettes based on the CDR scale were given. The response rate for the TTO section of the study was just 30%, with participants stating that ‘it was impossible to imagine living with dementia’. This study was funded by Novartis.
Multiple-state health utility: by cognition and dependency
In an examination of the relationship between MMSE, IADL and QoL in 100 people with AD from elderly care centres in Australia, Wlodarczyk and colleagues205 report mean utility from the self-reported Assessment of Quality of Life (AQoL) by MMSE and IADL for self- and carer-proxy ratings. This sample is a subset of patients from a global donepezil trial where patients received open-label treatment daily for 24 weeks. The ‘weights from the AQoL are determined using TTO from a weighted sample of Victorian population, designed to ensure representativeness of the Australia population’.
Multiple-state health utility: by cognition, dependency and residential status
Andersen and colleagues206 mapped answers from health status and ADL questions from a cross-sectional survey of 244 demented patients (67.2% of which had AD) in Denmark to EQ-5D. This study was reviewed in the previous MTA, although was then unpublished. Issues with the mapping of these answers to EQ-5D have been highlighted elsewhere,151 including, as pointed out by the authors themselves, that ‘questions in the study included an aspect of time that the EQ-5D does not’. Health-state utilities are reported by severity (MMSE), dependency and residential setting. The interviews were undertaken by a nurse in the participant’s home where a family or professional carer either helped answer the questions or later verified them.
Karlawish and colleagues207 have assessed both self-ratings and carer-proxy ratings207 for health-status values using EQ-5D and HUI-2 for people with AD. The patient-rated utility values were based on 93 respondents with a mean MMSE score of 21.3 (SD 4.3). Only patients with a MMSE score > 11 who were not in a nursing home and had an identifiable carer were included in the study, which was based in the USA. Utility values are reported by MMSE, modified MMSE, QoL-AD, IADL, BADLS (Bristol Activities of Daily Living Scale), GDS and SF-12 (Short Form questionnaire-12 items) from the EQ-5D and HUI-2. Results for the carer-proxy ratings of health-state utilities are based on responses from 100 carers, again reported by MMSE, modified MMSE, QOL-AD, IADL, BADLS and SF-12. Only utilities derived from patient self-ratings and carer-proxy ratings by MMSE, IADL and BADLS are reported in Table 109.
From the above review it is clear that a decision between the use of patient self-rated QoL and carer-proxy-rated QoL is needed. Differences in utilities derived from patient’s own or carer-proxy-based QoL ratings have been found in a number of studies of people with AD. 182,203,204,207 Such differences have been noted in other disease areas. For dementia and AD authors have noted that these differences may be explained by carer depression and/or burden (Karlawish and colleagues208 and Sands and colleagues,209 as cited in Vogel and colleagues210) or lack of insight on the part of the patient. 210 There is no evidence that for people with moderate-to-severe AD, reliable and consistent self-ratings of utility can be obtained. For example, Jonsson and colleagues187 report self-rated utilities of 1 and 0.94 in patients with MMSE scores of 10–15 and MMSE scores < 10, respectively. Although some may caution the use of carer-proxy ratings,214 given the inconsistent QoL observed with self-ratings of people with AD, the base-case analysis uses carer-proxy ratings. In the sensitivity analyses, results using patient self-reports are provided.
None of the studies reporting utilities have been carried out in the UK and only four have used the EQ-5D tool preferred in economic evaluations by NICE:198 Andersen and colleagues,206 Karlawish and colleagues207 and Jonsson and colleagues. 187 Note that Andersen and colleagues206 mapped responses to general questionnaires to EQ-5D, which involves some uncertainty as to the appropriateness of the mapping and the meaningfulness of the subsequent EQ-5D values.
With regard to utility, a particular criticism concerning the SHTAC model was that pre-FTC was too heterogeneous a state for a single utility (see no. 11 in Table 103). Use of the IPD from Wolstenholme and colleagues196 provided a relationship between MMSE and time to end of pre-institutionalisation. As a number of sources have reported utility values by MMSE it is possible to map time prior to the end of pre-institutionalisation to utility.
Within the IPD from Wolstenholme and colleagues,196 MMSE is recorded for each patient at multiple follow-ups from study entry, and so is a repeated measures data set. A linear mixed-effects model (from the ‘nlme’ R package) was fitted with MMSE as the response variable, time to end of pre-institutionalisation as a fixed effect, and patient as a random effect. Variation in the intercept and slope across patients were modelled as normal distributions. A fixed-effect variable indicating whether a patient had mild-to-moderate AD at the start of the study, or moderate-to-severe AD, was included in the model. Assuming t = years before the end of pre-institutionalisation, as above, for patients with mild-to-moderate AD, the following equation was obtained: MMSE = 8.34 + 4.17t. The corresponding equation for patients with moderate-to-severe AD was MMSE = 5.18 + 3.55t (Figure 68). Note that higher order terms for t were not modelled as they explained little of the variance in MMSE.
FIGURE 68.
Mini Mental State Examination score as a function of time until end of pre-institutionalisation with model fit for (a) mild-to-moderate AD and (b) moderate-to-severe AD.
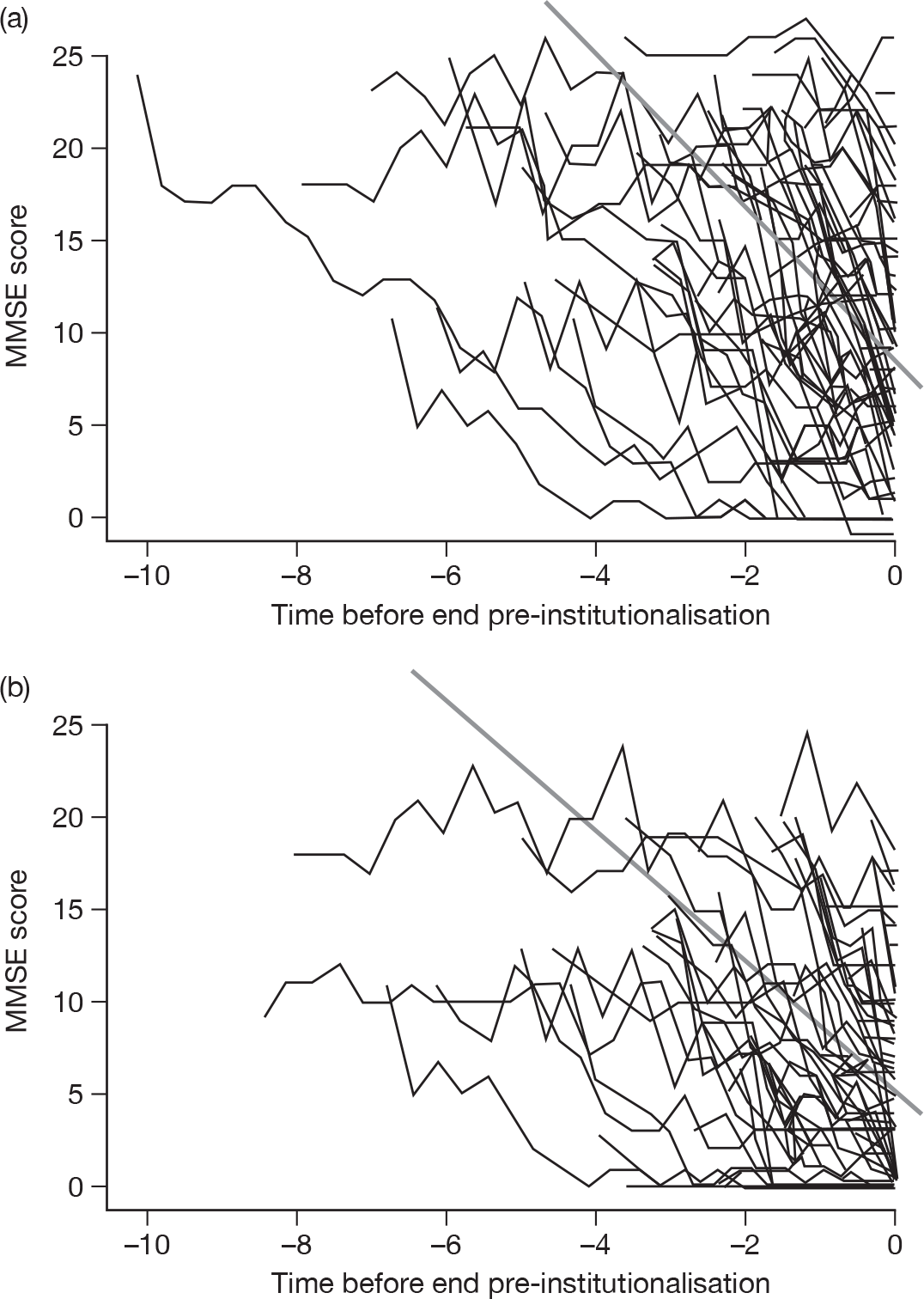
For the PSA, it was necessary to model the uncertainty in the covariate coefficients. The Cholesky matrix C, corresponding to the variance/covariance matrix of the parameter coefficients was calculated as:
where C1 corresponds to mild-to-moderate AD and C2 corresponds to moderate-to-severe AD. The rows and columns of C1 and C2 correspond to the intercept, time to institutionalisation, indicator for mild-to-moderate or moderate-to-severe AD to add to the intercept, and indicator for mild-to-moderate or moderate-to-severe AD to add to the gradient respectively. Probabilistic covariate coefficients were then simulated as y + Cz, where y is the vector of coefficient means (given in the deterministic equations above), and z is a vector of independent standard normal variables. 200
Utilities corresponding to these MMSE scores were required. Utility data by MMSE was available from five published studies187,205–207 two of which considered the same population of people with AD, but reported patient self-ratings207 and carer-proxy ratings207 separately. The utility weights based on the carer-proxy ratings from these four studies are shown in Figure 69.
FIGURE 69.
Carer-proxy utility weights for people with AD by MMSE score.
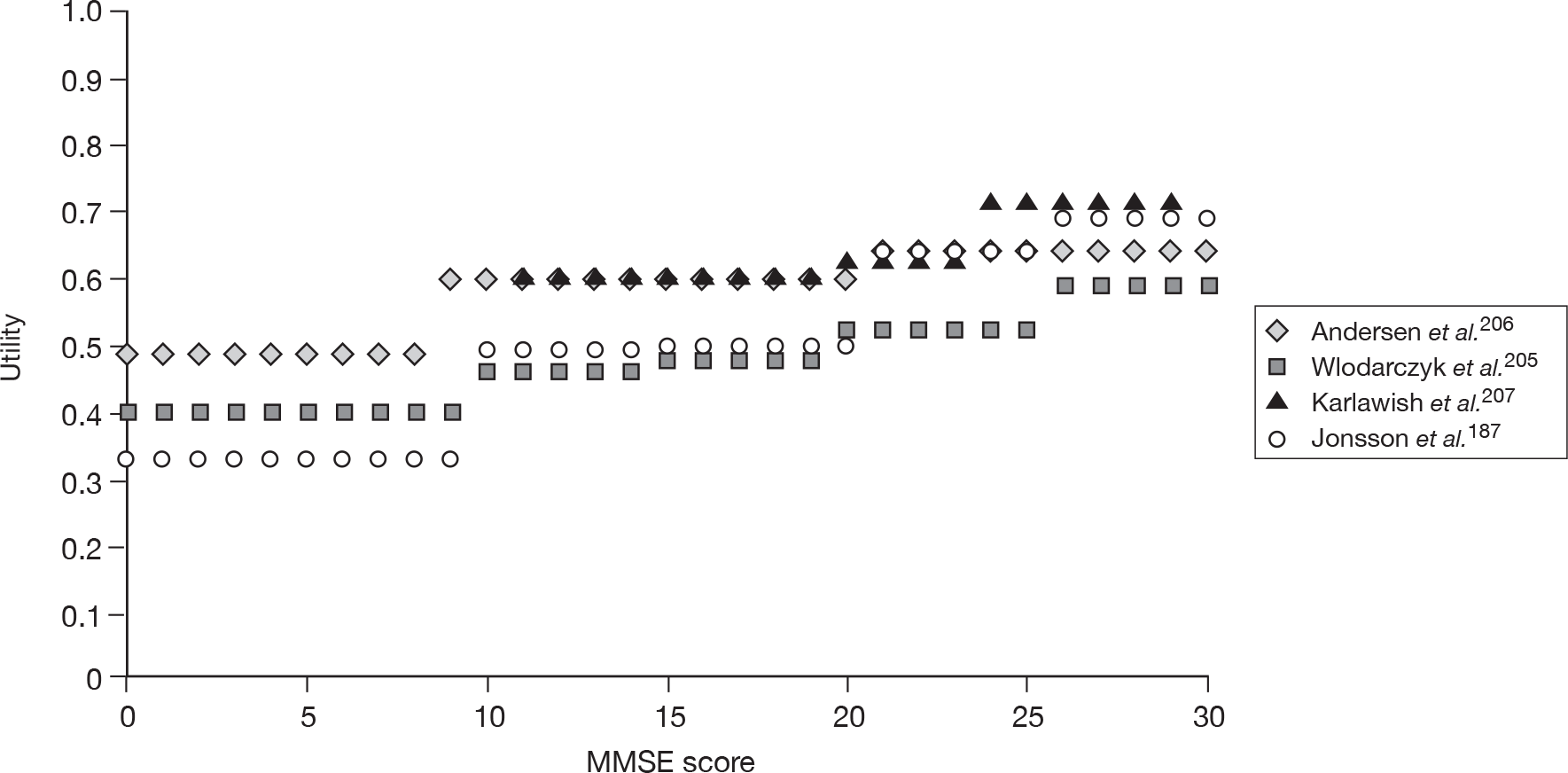
Jonsson and colleagues187 report EQ-5D valuations of utility across all MMSE scores. The average carer-proxy-rated utilities (from EQ-5D, EQ-5D VAS and QoL-AD) from Jonsson and colleagues187 range from 0.69 for MMSE 30–26 to 0.33 for severe AD. The evidence suggests that the utility weights from Jonsson and colleagues187 are not particularly different to those from the rest of the literature. Interestingly, the utility weight for MMSE < 10 from Jonsson and colleagues187 is very similar to the utility weight for people with AD who are defined as dependent from the study by Andersen and colleagues:206 0.33 from Jonsson and colleagues187 and 0.343 from Andersen and colleagues. 206 Similarly, when compared with the EQ-5D ratings when both carer-proxy and patient self-ratings are available (the second column of utility values), the utilities are very similar for all MMSE scales except for severe AD (0.33 from the average of scales and 0.4 from where both carer-proxy and patient self-ratings were available). When patient self-ratings are not available (the third column of utility values), the carer-proxy utilities are inconsistent across MMSE. Sensitivity analyses using the EQ-5D values where both carer-proxy and patient self-rating are available, and, where just carer-proxy ratings are available, are undertaken in the sensitivity analyses (Table 110).
| MMSE score | Average carer-proxy EQ-5D, EQ-5D VAS and QoL-AD utilitiesa | EQ-5D where both carer and patient ratings were available | EQ-5D where just carer ratings were available |
|---|---|---|---|
| 26–30 | 0.69 | 0.7 | 0.5 |
| 21–25 | 0.64 | 0.65 | 0.19 |
| 15–20 | 0.5 | 0.52 | 0.21 |
| 10–14 | 0.49 | 0.51 | 0.39 |
| 0–9 | 0.33 | 0.4 | 0.22 |
Mapping utility weights from Jonsson and colleagues187 on to time until the end of pre-institutionalisation from the equations above shows that, as expected, the shorter the time until the end of pre-institutionalisation, the lower the utility weight (Figure 70). The utility values used in the base-case analysis are shown in Table 111. Note that utility in the institutionalised state is the same as for MMSE < 10 because, as noted above, individuals have, on average, a MMSE < 10 before being institutionalised.
FIGURE 70.
Plot of utility from Jonsson and colleagues187 by time to end of pre-institutionalisation used in the base-case analysis.
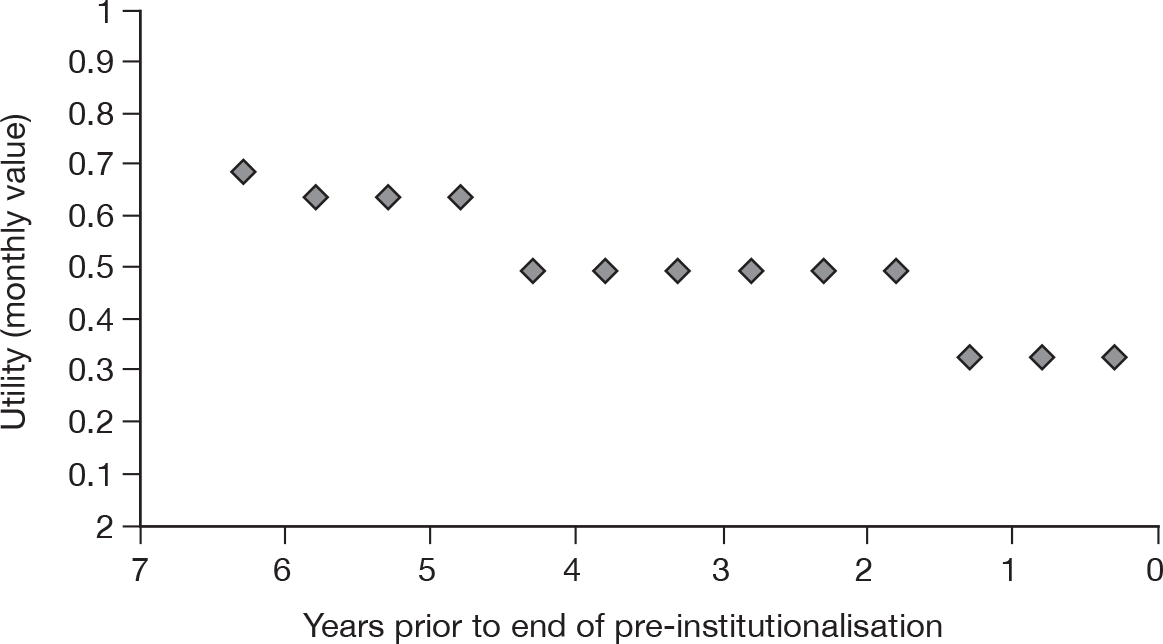
| Health state | Value | n | PenTAG estimates of SD | Patient-rated QoL |
|---|---|---|---|---|
| Pre-institutionalisation by MMSE score | ||||
| 0–9 | 0.33 | 44 | 0.151 | 0.78 |
| 10–14 | 0.49 | 88 | 0.107 | 0.73 |
| 15–20 | 0.5 | 83 | 0.110 | 0.83 |
| 21–25 | 0.49 | 25 | 0.200 | 0.85 |
| 26–30 | 0.69 | 22 | 0.213 | 0.84 |
| Institutionalisation (MMSE 0–9) | 0.33 | 44 | 0.151 | 0.78 |
| Dead | 0 | |||
In sensitivity analyses the patient’s self-rated utility from Jonsson and colleagues187 were assessed for their impact on the cost–utility findings. Similarly, utility estimates from the AQoL utilities as reported by Wlodarczyk and colleagues205 were assessed in one-way sensitivity analyses.
For the PSA, estimates of the uncertainty of the utility values from Jonsson and colleagues187 were obtained by assuming that the SD of these values was equivalent to 1/√N. Although this is not ideal, it produced figures of the same magnitude as the SDs reported by Andersen and colleagues206 for their utility estimates.
Quality of life of the carer
Very few data on the utility of the carers of people with AD were identified from the literature. A study by Neumann and colleagues,213 which was reviewed in the previous MTA, contained some evidence regarding carer utility (measured on the HUI-2 scale) by patient progression (measured on the CDR scale). This evidence suggests that the carer’s utility scores remain fairly stable until the patient’s disease progresses to a score of 3 on the CDR (indicating severe AD) when the carer’s QoL starts to improve (Figure 71). This is most likely owing to the patient being placed in institutionalised care. However, note that this was a cross-sectional study that did not follow patients and their carer’s over time, so may not be fully representative of carer’s utility as the patient progresses.
FIGURE 71.
Health Utilities Index version 2 utility scores of patients (carers providing proxy scores) and carers by CDR from a cross-sectional study in the USA.
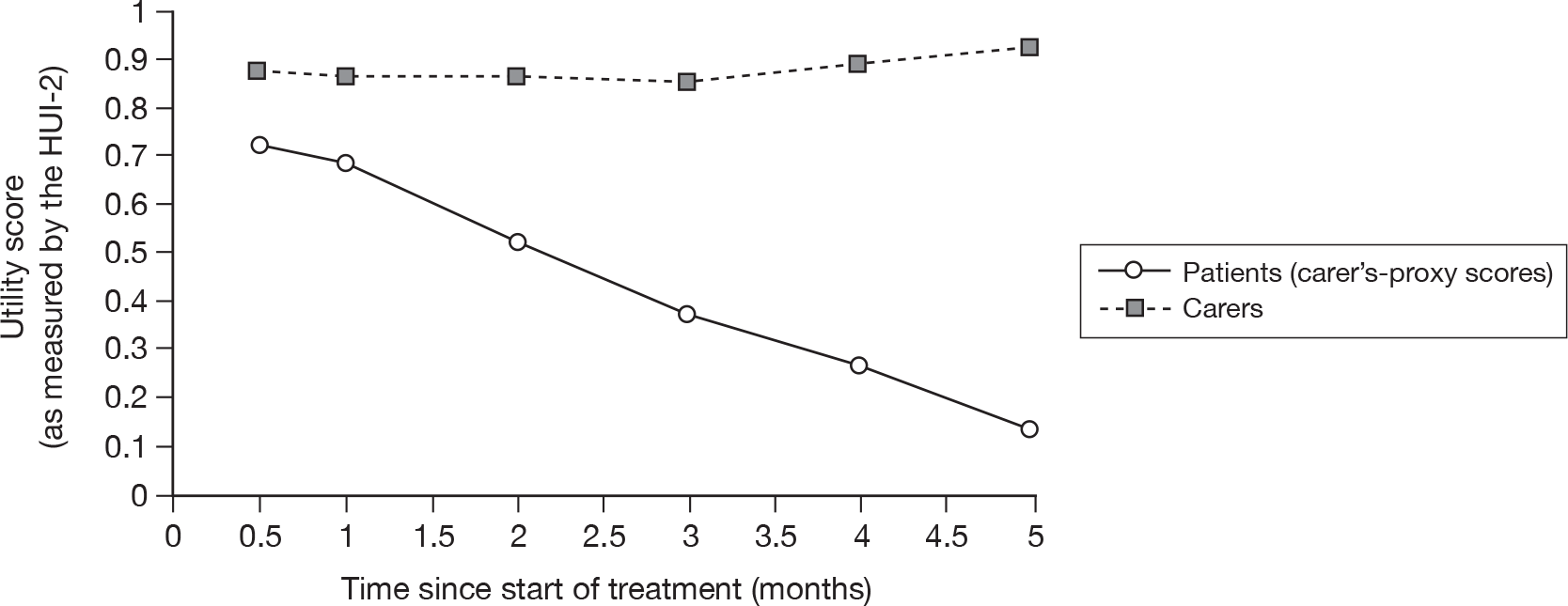
To include data on carer’s utility in the PenTAG cost–utility model, the CDR scale was mapped on to the MMSE scale using the findings of a study by Perneczky and colleagues. 215 Perneczky and colleagues215 demonstrated that the CDR scale can be used to map on to MMSE ranges for people with dementia. Their analysis indicated substantial agreement (as measured by Cohen’s kappa, κ) between the MMSE ranges and CDR stages for the mild, moderate and severe stages of dementia (κ > 0.6). These results are reproduced below in Table 112.
| CDR stage | MMSE range | Cohen’s kappa, κ | HUI-2 utility |
|---|---|---|---|
| 0 – no dementia | 30 | 0.44 | – |
| 0.5 – questionable dementia | 26–29 | 0.28 | 0.88 |
| 1 – mild dementia | 21–25 | 0.62 | 0.87 |
| 2 – moderate dementia | 11–20 | 0.69 | 0.87 |
| 3 – severe dementia | 0–10 | 0.76 | 0.86 |
Assuming these mappings in Table 112, the carer’s utility by the patient’s MMSE score was obtained and then, as in Quality of life of the individual with Alzheimer’s disease, above, mapped on to time prior to institutionalisation for the person with AD.
Cost estimates
The costs considered in the decision model are those that fall on the NHS and PSS. They are the drug costs, monthly costs of care (pre-institutionalised and institutionalised) and the costs of a 6-monthly monitoring outpatient visit for those treated with donepezil, galantamine, rivastigmine or memantine. It is assumed that any AEs are mild, do not require further treatment, and so do not induce further costs. All costs are for 2009 to avoid incorporating further uncertainty for the inflation to 2010 costs, where such costs are not yet published [e.g. Unit Costs of Health and Social Care (2009)216 and NHS Reference Costs 2008–9217]. Note that the relevant drug costs do not differ between BNF 58218 (fourth quarter 2009) and BNF 59219 (first quarter 2010). A review of evidence on the costs associated with AD did not identify data on the NHS and PSS costs of carers of people with AD. Therefore, no such carers’ costs were included in the PenTAG decision model.
Drug costs for Alzheimer’s disease
Monthly drug costs were calculated from costs reported in the BNF 58 for the specific doses of interest. For galantamine, rivastigmine capsules and memantine, a mix of doses were assumed, which is reflected in the monthly costs presented in Table 113. It was also assumed that treated individuals would have a 6-monthly outpatient monitoring visit. The cost of such a visit was obtained from the NHS Reference Costs 2008–9. 217
| Cost component | £, 2009 | Source |
|---|---|---|
| Drugs (monthly cost) | ||
| Donepezil (10 mg/day) | 97 | Calculated from the daily drug cost of £3.18 (BNF 58218) |
| Galantamine (16–24 mg/day) | 83 | Calculated as an average of daily costs for 16 mg (£2.44) and 24 mg (£3.66), leading to a daily cost of £2.72 (BNF 58218) |
| Rivastigmine capsules (9–12 mg/day) | 72 | Calculated as a weighted average of daily costs for 9 mg (0.7 × £2.38) and 12 mg (0.3 × £2.37), leading to daily drug cost of £2.38 |
| Rivastigmine patches (9.5 mg/day) | 79 | Calculated from the daily cost of £2.60 |
| Memantine (15–20 mg/day) | 71 | Calculated as a weighted average of daily costs for 15 mg (0.2 × £1.85) and 20 mg (0.8 × £2.46), leading to daily drug cost of £2.34 |
| Outpatient visit | ||
| Six-monthly visit | 158 | NHS Reference Costs 2008–9.217 NHS Trusts consultant led; Follow-up attendance non-admitted face to face |
| Monthly cost | 26 | |
| NHS and PSS | ||
| Pre-institutionalised | See equations 39 and 40 | IPD from Wolstenholme et al.196 |
| Institutionalised | 2941 | |
All of the costs used in the base-case analysis are shown in Table 113. As noted in Sources of effectiveness data, the drug costs and associated 6-monthly outpatient costs are adjusted in the model to reflect treatment discontinuation by a proportion of the treated cohort.
Cost of health and social care received by Alzheimer’s disease patients
A review of published research evidence failed to identify up-to-date estimates of the NHS and PSS costs associated with AD. For the PenTAG model it was therefore necessary to use available data from a number of years ago and inflate the relevant costs as appropriate. Data from the Oxfordshire cohort study by Wolstenholme and colleagues196 was used to provide NHS and PSS costs for the PenTAG model. Information on the following resource use was recorded by Wolstenholme and colleagues196 at baseline and each subsequent follow-up interview:
-
number and duration of acute hospitalisations and respite care
-
number of outpatient visits
-
day care and home attendances by district nurses, community psychiatric nurses, home helps or other care assistants
-
number of visits by or to the GP or practice nurse.
All of these items of service use were recorded, whether or not they were related to AD or other health problems. Data on the use of wheelchairs, bath or bed hoists and incontinence pads and sheets were recorded. The individual’s current place of residence was also noted so that accommodation costs could be calculated. Wolstenholme and colleagues196 report that unit costs were taken mainly from the Unit Costs for Health and Social Care 1998220 and 1999,221 supplemented by hospital trust financial returns and the Chartered Institute of Public Finance and Accounting in addition to data from surveys of local hospitals, and from specific residential and nursing homes, carried out by Wolstenholme and colleagues196 (see table 1 of Wolstenholme and colleagues196).
To be used in the PenTAG model, the monthly costs calculated by Wolstenholme and colleagues196 (implicitly 1998 costs) were inflated to 2009 costs using an inflation factor of 1.54 (= 267.0/173.5) from the inflation indices for hospital and community health services in Unit Costs of Health and Social Care 2009 and 2004). 216,222 Pre-institutionalisation costs were all assumed to fall on the NHS or PSS budget, but post institutionalisation a proportion of the accommodation costs were assumed to be self-funded (i.e. paid by patients or their families). On the basis of data in the Dementia UK report (as cited by the 2007 NAO report8,9), we assume that being in residential care6 is equivalent to being institutionalised in our model, and 94% of the non-informal care costs of being institutionalised are accommodation (or ‘care home costs’). Of these, 30% were reported in 2007 to be self-funded (i.e. not NHS or social services department). So, in our base-case analysis we assume that 28% of post-institutionalisation costs (excluding informal care costs) are self funded, and 72% are NHS and PSS funded.
A criticism of the SHTAC model that was explored in the PenTAG model was that the pre-FTC health state was too heterogeneous to represent a single NHS and PSS cost (see no. 5 in Table 103). The IPD from Wolstenholme and colleagues196 allowed an exploration of this criticism by developing a relationship between the inflated cost per month and the time before the end of pre-institutionalisation. See Appendix 18.
A linear mixed-effects model with the inflated cost per month as the response variable, a cubic equation for the time to end of pre-institutionalisation as a fixed effect (higher order terms were non-significant), and patient as a random effect, where variation in the intercept across patients was modelled as a normal distribution. An indicator explanatory variable was included for whether a patient had mild-to-moderate AD at the start of the study, or moderate-to-severe AD.
For patients with mild-to-moderate AD, the following equation was obtained [Figure 72, part (a)]:
where t = years before the end of pre-institutionalisation. The corresponding equation for patients with moderate-to-severe AD was [see Figure 72, part (b)]:
FIGURE 72.
Monthly inflated cost as a function of time until pre-institutionalisation showing model fit for mild-to-moderate AD (a) and moderate-to-severe AD (b).
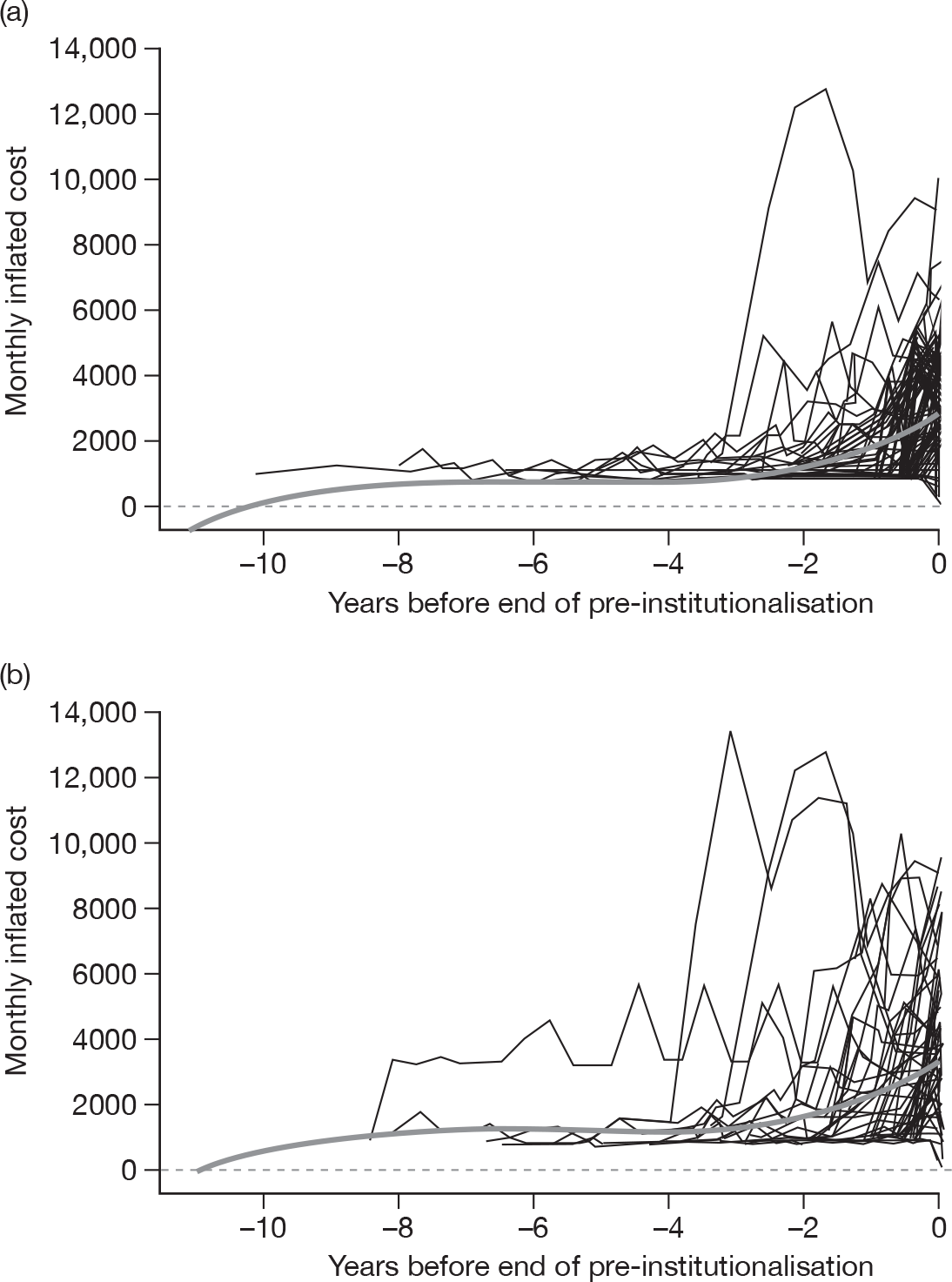
For the PSA, it is necessary to model the uncertainty in the covariate coefficients, and this was done by calculating the Cholesky matrix C, corresponding to the variance/covariance matrix of the parameter coefficients, as:
where C1 corresponds to mild-to-moderate AD and C2 corresponds to moderate-to-severe AD. The rows and columns of C1 and C2 correspond to the intercept, time to institutionalisation, square of time to institutionalisation, cube of time to institutionalisation, and indicator for mild-to-moderate or moderate-to-severe AD to add to the intercept. Probabilistic covariate coefficients were then simulated as y + Cz, where y is the vector of coefficient means (given in the deterministic equations above), and z is a vector of independent standard normal variables. 198
Thus, at 1 year until the end of pre-institutionalisation, the mean NHS and PSS cost per participant for mild-to-moderate participants was £1938 per month, while for moderate-to-severe participants the cost was £2427 per month. In the cost–utility model, at each cycle, the proportion of the cohort within 6-monthly time periods of leaving the pre-institutionalised state was calculated. The time periods were 0–6 months, 7–12 months, 13–18 months, and so on, until 72 months. The mid-points of these 6-monthly time-periods were used to calculate MMSE and costs prior to institutionalisation. Any individuals > 72 months prior to institutionalisation were assumed to be 75 months prior to institutionalisation. A plot of the monthly pre-institutionalised costs by time to institutionalisation used in the PenTAG model is given in Figure 73.
FIGURE 73.
Monthly costs (£, 2009) by time to institutionalisation used in the base-case analyses.
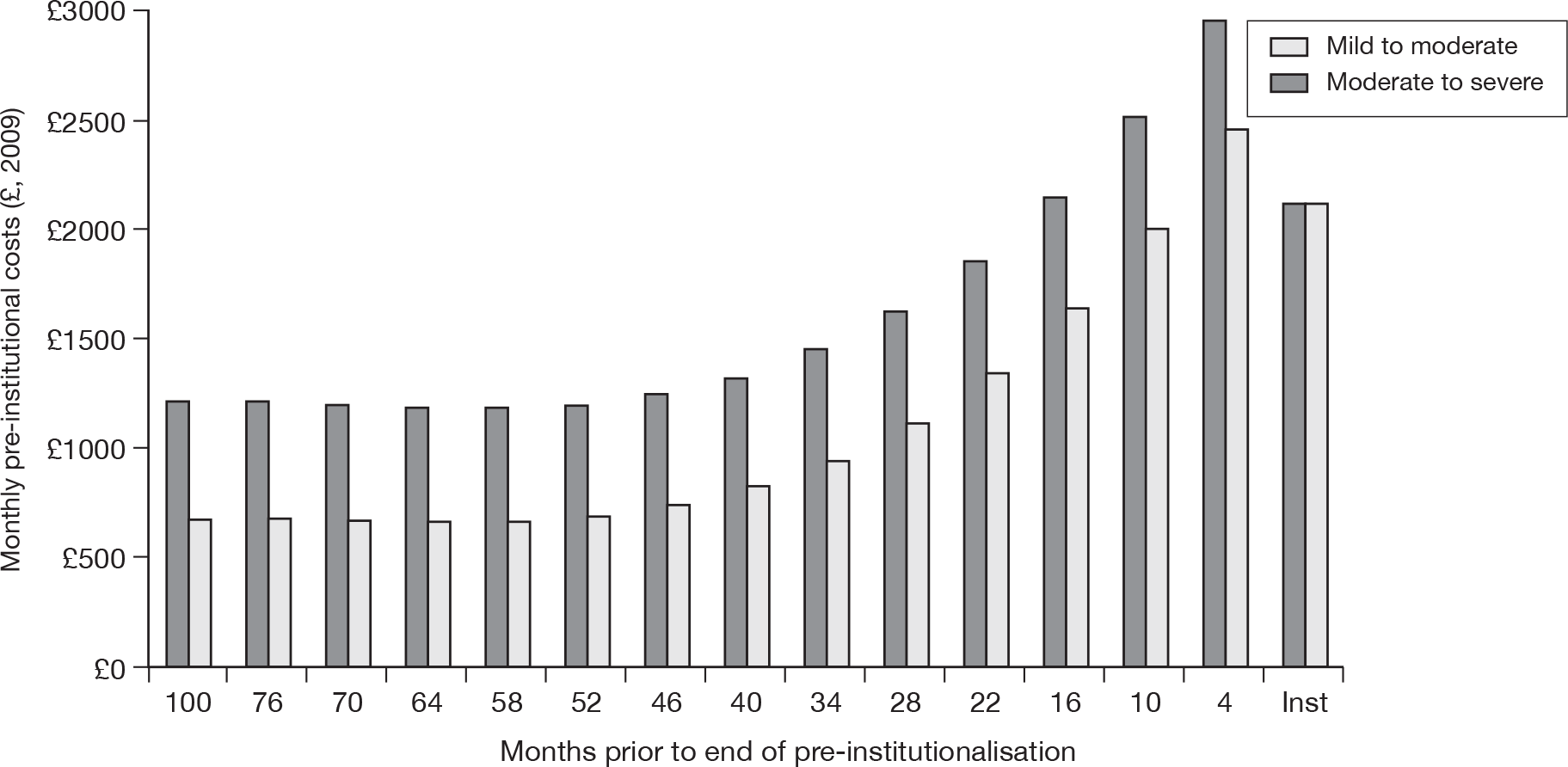
To calculate a mean monthly cost while in institutional care, a linear mixed-effects model was fitted, with the inflated cost per month as the response variable, fitting just the intercept term, with patient ID as a random effect. The overall cost was assumed independent of disease severity, and independent of time since institutionalised. Visual inspection suggested that the second point was approximately true. The overall mean monthly cost was £2941. For the PSA, the monthly cost was assumed normally distributed with SE of £180. Only 72% of this monthly cost for institutional care is included in the PenTAG model to account for the 28% of institutionalised costs being privately funded as described above. Thus, the monthly cost of institutional care is £2117.
Note that when costs for institutional care are compared with the pre-institutionalised costs (final data point in Figure 73), it can be seen that institutional care costs are lower than the pre-institutionalised costs for mild-to-moderate individuals within 4 months of being institutionalised, and moderate-to-severe individuals within 16 months of being institutionalised. The total institutional care costs in the PenTAG model have been subjected to a 28% reduction to account for the fact that not all of the institutional care costs are NHS/PSS funded. The pre-institutionalised costs have not been subjected to this reduction. The pattern of care costs shown in Figure 73 reflects increases in care costs to the NHS/PSS as individual progress to requiring institutionalisation. Once in institutional care, fewer NHS/PSS costs are incurred.
Cost estimates used in sensitivity analyses
The drug costs reported by Lundbeck and Eisai in their industry submission are used in one-way sensitivity analyses, as are the industry estimates of the monthly cost of institutionalisation and the 6-monthly outpatient follow-up visit cost. The impact of assuming that 72% of the monthly £2941 cost of institutionalisation is NHS/PSS funded is also assessed. Where in the base case it is assumed that 30% of accommodation costs were self funded, in sensitivity analyses it is assumed that 50% or only 10% of accommodation costs are self funded, leading to assumptions that 53% or 91% of institutionalised costs are NHS/PSS costs, respectively. Note that these percentages do not refer to the industry cited costs as these are assumed to have already accounted for the fact that not all institutionalisation costs are funded by the NHS/PSS.
Key assumptions of the Peninsula Technology Assessment Group’s model
To summarise, the key assumptions in the base-case analysis are that there was no treatment effect on survival, ADCS-ADL was transformed onto the Barthel ADL Index, time to institutionalisation and overall survival in clinical practice are similar to that experienced in the Oxfordshire study by Wolstenholme and colleagues196 and carer-proxy utility values were used for patient QoL. The pre-institutionalised state allowed for a relationship between (1) utility and time prior to end of pre-institutionalisation and (2) cost and time prior to pre-institutionalisation. Costs and utilities were assumed to be constant within the institutionalised state. All parameter values used in the base-case analyses for mild and moderate AD and moderate-to-severe AD are presented in Tables 114 and 115, respectively. PSA was undertaken for both of the base-case analyses: mild-to-moderate and moderate-to-severe AD. In both PSAs, distributions were placed on uncertain parameters. The distributions used are also presented in Tables 114s, 115, and are shown graphically in Appendix 19. For each PSA, 10,000 simulations of the cost–utility model were undertaken.
| Parameter | Value | SE | Source | Justification | Distribution for PSAa |
|---|---|---|---|---|---|
| Cohort characteristics | |||||
| Mean age | |||||
| Group 1: 25% cohort | 69 | NA | IPD from Wolstenholme et al.196 | Based on data from a UK epidemiological study on which AD progression is modelled for mild-to-moderate severity of AD | NA |
| Group 2: 50% cohort | 77 | ||||
| Group 3: 25% cohort | 86 | ||||
| Mean MMSE | 17 | NA | IPD from Wolstenholme et al.196 | Based on data from the UK epidemiological study on which AD progression is modelled for mild-to-moderate severity of AD | NA |
| Mean Barthel ADL Index | 17.52 | NA | IPD from Wolstenholme et al.196 | Based on data from the UK epidemiological study on which AD progression is modelled for mild-to-moderate severity of AD | NA |
| Proportion starting model in institutionalised state | 0.1 | LASER-AD study197 | Based on data from a UK epidemiological study, and evidence of a reduction in the number of individuals institutionalised for mild-to-moderate severity of AD | Beta(1,9) | |
| Time horizon | 20 years | Estimated that < 5% of cohort are still alive | NA | ||
| Discounting costs | 3.5% | NICE methods guide198 | As stated in NICE methods guide | NA | |
| Discounting benefits | 3.5% | NICE methods guide198 | As stated in NICE methods guide | NA | |
| Clinical effectiveness | |||||
| Donepezil (10 mg) | |||||
| MMSE | 1.24 | 0.216 | Meta-analysis result from Chapter 5 | Based on data from a systematic review of the evidence |
Normal(1.24, 0.216) From 95% CI of pooled estimate |
| ADCS-ADL | 2.02 | 0.470 | Average of value from galantamine and rivastigmine capsules | Since no evidence were identified, this is based on an assumption of a class effect |
Normal(2.17, 0.470) From 95% CI of pooled estimate |
| Galantamine (16–24 mg) | |||||
| MMSE | 1.13 | 0.156 | Average of value from donepezil and rivastigmine capsules | As no evidence were identified, this is based on an assumption of a class effect |
Normal(1.13, 0.156) From 95% CI of pooled estimate |
| ADCS-ADL | 2.23 | 0.462 | Meta-analysis result from Chapter 5 | Based on data from a systematic review of the evidence |
Normal(2.23, 0.462) From 95% CI of pooled estimate |
| Rivastigmine capsules (9−12 mg) | |||||
| MMSE | 1.02 | 0.225 | Meta-analysis result from Chapter 5 | Based on data from a systematic review of the evidence |
Normal(1.02, 0.225) From 95% CI of pooled estimate |
| ADCS-ADL | 1.80 | 0.818 | Single study | Based on only RCT reporting outcome |
Normal(1.8, 0.818) From 95% CI of pooled estimate |
| Rivastigmine patches (9.5 mg/day) | |||||
| MMSE | 1.10 | 0.296 | Single study | Based on only RCT reporting outcome |
Normal(1.1, 0.296) From 95% CI of estimate |
| ADCS-ADL | 2.20 | 0.808 | Single study | Based on only RCT reporting outcome |
Normal(2.2, 0.808) From 95% CI of estimate |
| Percentage of total cohort discontinuing treatment each month | 4% | Based on mixture of evidence from RCTs and clinical opinion | Assumes most participants have discontinued treatment by 2 years (similar to AD2000100 results) | Beta(12,290) | |
| Health-state utilities | |||||
| Pre-institutionalised (by MMSE) | Linear equation with time | MMSE by time prior to pre-institutionalised calculated from IPD of Wolstenholme et al.196 | Cholesky matrix C shown in Chapter 6 (see Quality of life of the individual with Alzheimer’s disease) | ||
| MMSE: 0–9 | 0.33 | 0.044 | Jonsson et al.187 | Utilities reported in Jonsson et al.181 are similar to those reported by MMSE in other studies | Beta(36.59, 74.28) |
| MMSE: 10–14 | 0.49 | 0.039 | Beta(78.04, 81.22) | ||
| MMSE: 15–20 | 0.5 | 0.012 | Beta(856.27, 856.27) | ||
| MMSE: 21–25 | 0.64 | 0.011 | Beta(1137.19, 639.67) | ||
| MMSE: 26–30 | 0.69 | 0.023 | Beta(282.51, 126.92) | ||
| Institutionalised (MMSE 0–9) | 0.33 | 0.044 | Jonsson et al.187 | Analysis of IPD from Wolstenholme et al.196 suggests that participants had an average MMSE score of 9 when 0.04 years prior to institutionalisation, therefore institutionalisation used as proxy for severe AD | Beta(36.59, 74.28) |
| Dead | 0 | NA | |||
| Monthly drug costs | |||||
| Donepezil (10 mg) | £97 | BNF 58218 | NA | ||
|
Galantamine (16–24 mg) % 16-mg costs % 24-mg costs |
£83 50 50 |
BNF 58218 | Author judgement on % RCT participants having mean dose of 16 mg or 24 mg |
NA % 16 mg = Normal(0.5,0.1) % 24 mg = 1 –% 16 mg |
|
|
Rivastigmine (≤ 12 mg) % 9-mg costs %12-mg costs |
£72 70 30 |
BNF 58218 | Author judgement on % RCT participants having mean dose of 9 mg or 12 mg |
NA % 9 mg = Normal(0.3, 0.1) %12 mg = 1 –% 9 mg |
|
| Rivastigmine patch | £79 | BNF 58218 | NA | ||
| 6-monthly monitoring outpatient visit cost | £158 | NHS Reference Costs 2008–9217 | NHS Trusts consultant led; follow-up attendance non-admitted face to face | Gamma(4.94, 32) | |
| Monthly monitoring outpatient visit cost | £26 | Calculated from 6-monthly cost of £158 for a single visit | |||
| Pre-institutionalised NHS/PSS costs | Cubic equation with time | Relationship between NHS and PSS costs and time prior to institutionalisation calculated from IPD of Wolstenholme et al.196 | Cholesky matrix C shown in Cost of health and social care received by Alzheimer’s disease patients | ||
| Institutionalised | £2941 | £108 | Calculated from IPD of Wolstenholme et al.194 | Monthly cost of institutional care. Only 72% of these costs are assumed to be NHS/PSS | Normal(2941, 108) |
| Percentage institutionalised costs funded by NHS/PSS | 0.72 | Dementia UK report (as cited by the 2007 NAO report8,9) | Assumes that 28% of institutional care costs are not funded by the NHS/PSS | Beta(15, 5.83) | |
| Parameter | Value | SE | Source | Justification | Distribution for PSAa |
|---|---|---|---|---|---|
| Cohort characteristics | |||||
| Mean age (years) | |||||
| Group 1: 25% cohort | 69 | IPD from Wolstenholme et al.196 | Based on data from a UK epidemiological study on which AD progression is modelled for moderate-to-severe AD | NA | |
| Group 2: 50% cohort | 78 | ||||
| Group 3: 25% cohort | 87 | ||||
| Mean MMSE | 11.73 | IPD from Wolstenholme et al.196 | Based on data from a UK epidemiological study on which AD progression is modelled for moderate-to-severe AD | NA | |
| Mean Barthel ADL Index | 16.34 | IPD from Wolstenholme et al.196 | Based on data from a UK epidemiological study on which AD progression is modelled for moderate-to-severe AD | NA | |
| Proportion starting model in institutionalised state | 0.4 | LASER-AD study197 | Based on data from a UK epidemiological study, and evidence of a reduction in the number of individuals institutionalised for moderate-to-severe AD | Beta(4,6) | |
| Time horizon | 20 years | Estimated that < 5% of cohort are still alive | NA | ||
| Discounting costs | 3.5% | NICE methods guide198 | As stated in NICE methods | NA | |
| Discounting benefits | 3.5% | NICE methods guide198 | As stated in NICE methods | NA | |
| Clinical effectiveness | |||||
| Memantine (20 mg) | |||||
| MMSE | 0.70 | 0.346 | Meta-analysis result from Chapter 5 | Based on data from systematic review of evidence | Normal(0.70, 0.346) |
| ADCS-ADL | 1.41 | 0.70 | Meta-analysis result from Chapter 5 | Based on data from systematic review of evidence | Normal(1.41, 0.70) |
| Percentage of total cohort discontinuing treatment each month | 4% | Based on a mixture of evidence from RCTs and clinical opinion | Assumes most participants have discontinued treatment by 2 years (similar to AD2000103 results) | Beta(12,290) | |
| Health-state utilities | |||||
| Pre-institutionalised (by MMSE) | Linear equation with time | MMSE by time prior to pre-institutionalised calculated from IPD of Wolstenholme et al.196 | Cholesky matrix C shown in Quality of life of the individual with Alzheimer’s disease | ||
| MMSE: 0–9 | 0.33 | 0.044 | Jonsson et al.187 | Utilities reported in Jonsson et al. are similar to those reported by MMSE in other studies | Beta(36.59, 74.28) |
| MMSE: 10–14 | 0.49 | 0.039 | Beta(78.04, 81.22) | ||
| MMSE: 15–20 | 0.5 | 0.012 | Beta(856.27, 856.27) | ||
| MMSE: 21–25 | 0.64 | 0.011 | Beta(1137.19, 639.67) | ||
| MMSE: 26–30 | 0.69 | 0.023 | Beta(282.51, 126.92) | ||
| Institutionalised (MMSE 0–9) | 0.33 | 0.044 | Jonsson et al.187 | Analysis of IPD from Wolstenholme et al. suggests participants had an average MMSE score of 9 when 0.04 years prior to institutionalisation, therefore institutionalisation used as proxy for severe AD | Beta(36.59, 74.28) |
| Dead | 0 | ||||
| Monthly drug costs | |||||
|
Memantine (20 mg) %15 mg %20 mg |
£75 | BNF 58218 | Author judgement on% RCT participants having mean dose of 15 mg or 20 mg |
%15 mg = Normal(0.2,0.05) %20 mg = 1 –%15 mg |
|
| 6-monthly monitoring outpatient visit cost | £158 | NHS Reference Costs 2008–9217 | NHS Trusts consultant led; follow-up attendance non-admitted face to face | Gamma(4.94, 32) | |
| Monthly monitoring outpatient visit cost | £26 | Calculated from 6-monthly cost of £158 for single visit | |||
| Pre-institutionalised NHS/PSS costs | Cubic equation with time | Relationship between NHS and PSS costs and time prior to institutionalisation calculated from IPD of Wolstenholme et al.196 | Cholesky matrix C shown in Cost of Health and social care received by Alzheimer’s disease patients | ||
| Institutionalised | £2941 | £108 | Calculated from IPD of Wolstenholme et al.196 | Monthly cost of institutional care. Only 72% of these costs are assumed to be NHS/PSS | Normal(2941, 108) |
| Percentage institutionalised costs funded by NHS/PSS | 0.72 | Dementia UK report (as cited by the 2007 NAO report8,9) | Assumes that 28% of institutional care costs are not funded by the NHS/PSS | Beta(15, 5.83) | |
Results
The full results for the CUA of AChEIs are presented first, followed by the full results for the cost–utility of memantine. Owing to the many assumptions associated with the parameter estimates in the PenTAG model, it is important to be fully aware of the uncertainty in the model. Because of this, the first set of analyses presented in this section are those from the PSAs of the base-case parameter values. These results are followed by the deterministic base-case results, which are compared with the corresponding mean estimates from the PSA. Note that there is a great deal of structural uncertainty in the PenTAG model that cannot be accounted for in the PSA. Deterministic sensitivity analyses have been undertaken to explore some of the structural and further parameter uncertainty.
Mild-to-moderate Alzheimer’s disease: cholinesterase inhibitors (decision problem 1a)
Probabilistic sensitivity analysis
The cost-effectiveness results of 10,000 simulations for the base-case analysis of the cost–utility of AChEIs in people with mild-to-moderate AD are presented in Figure 74, showing a great deal of uncertainty. However, the cost-effectiveness acceptability curve shown in Figure 75 demonstrates that there is a very low probability that BSC is the most cost-effective technology, regardless of the threshold WTP per QALY gained. At a WTP of £30,000 per QALY gained there is a 0.3% probability that BSC is the best treatment option, thus indicating that there is > 99% probability that it is not the most cost-effective treatment option. At WTPs of £30,000 and £20,000 per QALY gained, rivastigmine patches have the highest probability of being cost-effective at 32%. Donepezil has a probability of 28% of being the most cost-effective treatment option at a WTP of £30,000 and 27% at a WTP of £20,000.
FIGURE 74.
Base-case cost-effectiveness plane for treatment with AChEIs in people with mild-to-moderate AD.
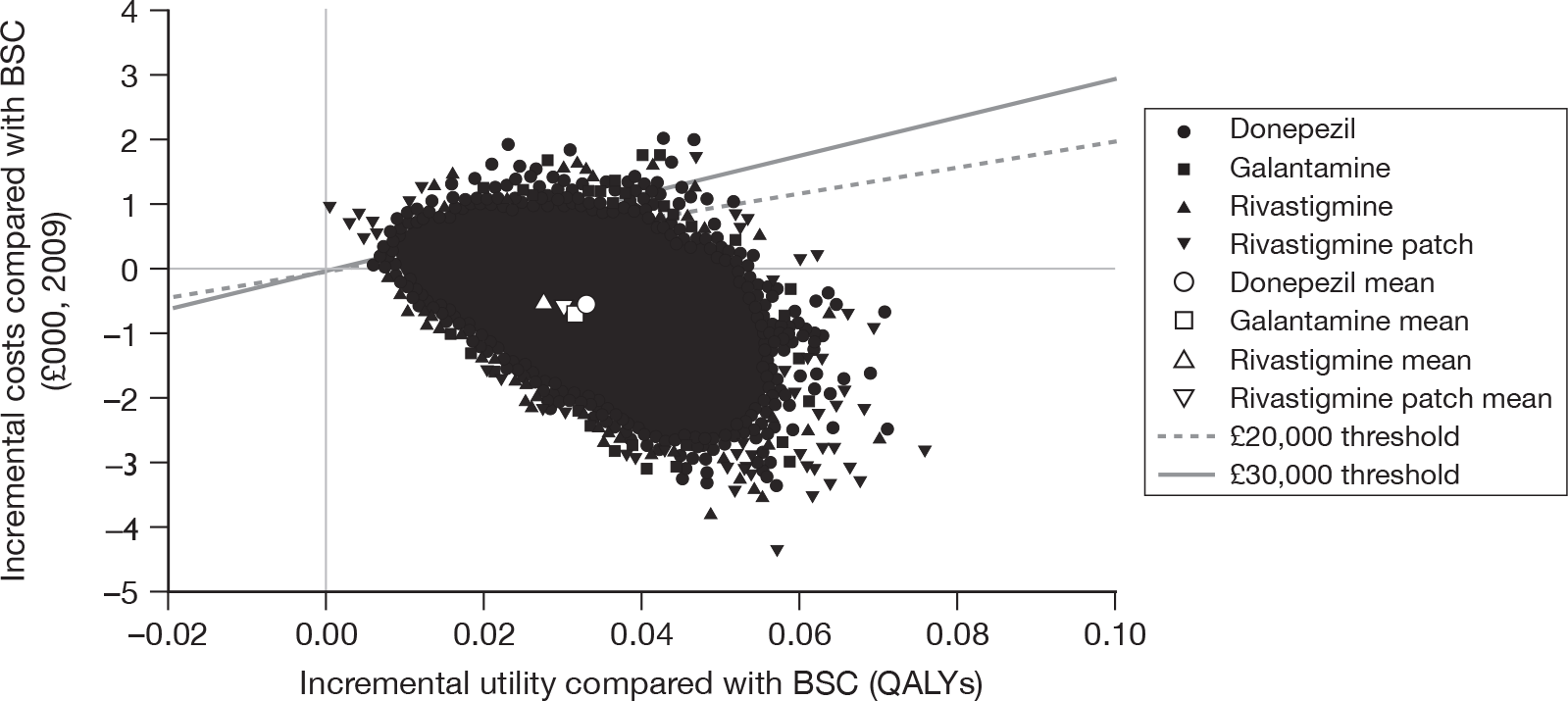
FIGURE 75.
Base-case cost-effectiveness acceptability curve for AChEIs in people with mild-to-moderate AD.
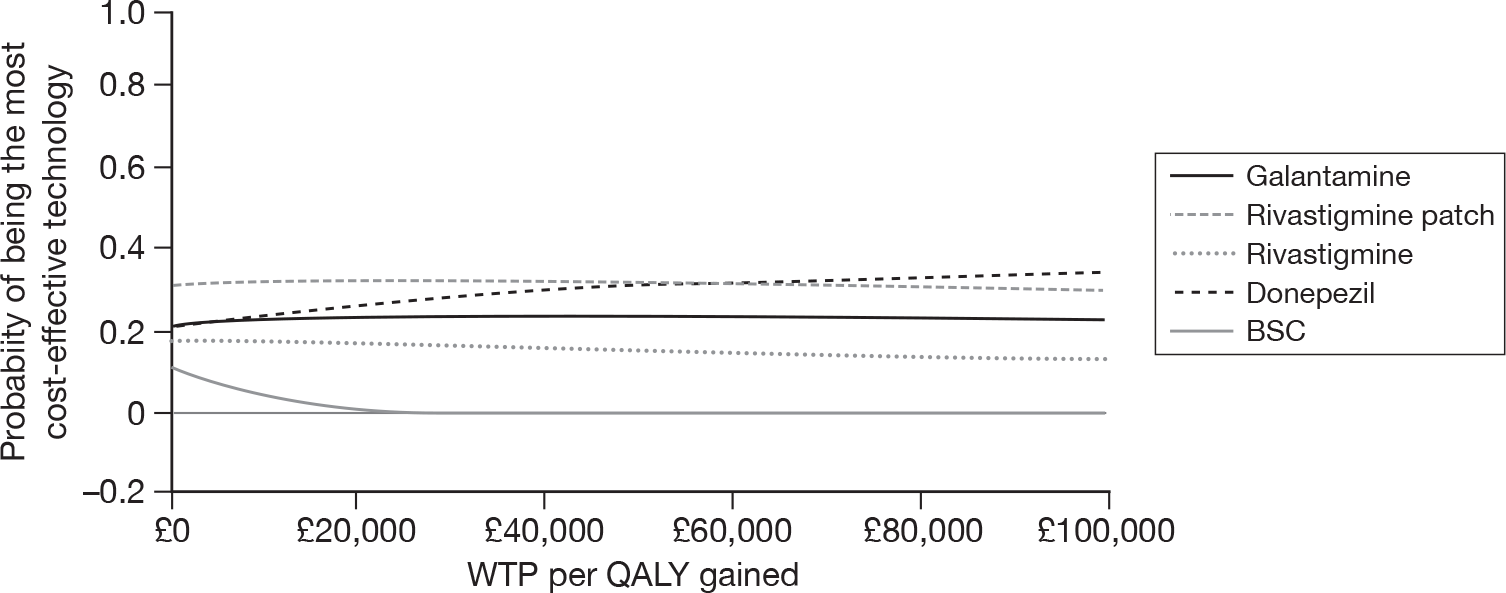
The findings from the PSA indicate that, on average, BSC, rivastigmine and rivastigmine patches are dominated as they are more expensive and less effective than donepezil and/or galantamine. Galantamine is estimated to be the cheapest option, but with donepezil providing the greatest QALY gains at an ICER of £23,453 per QALY compared with galantamine.
Deterministic analysis
A graph of the progression of individuals with mild-to-moderate AD from the BSC cohort through the three-state Markov model is shown in Figure 76 for the middle age group, having a mean starting age of 77 years (representing 50% of the cohort). Ten per cent of the cohort start the model in the institutionalised state. Across all three age groups, the mean overall survival for the total prevalent cohort is 3.84 years. This is regardless of the treatment received as, in the base-case analysis, it is assumed that there is no treatment effect on survival.
FIGURE 76.
Progression of the BSC cohort for the base-case analysis (mild-to-moderate AD, age group 2).
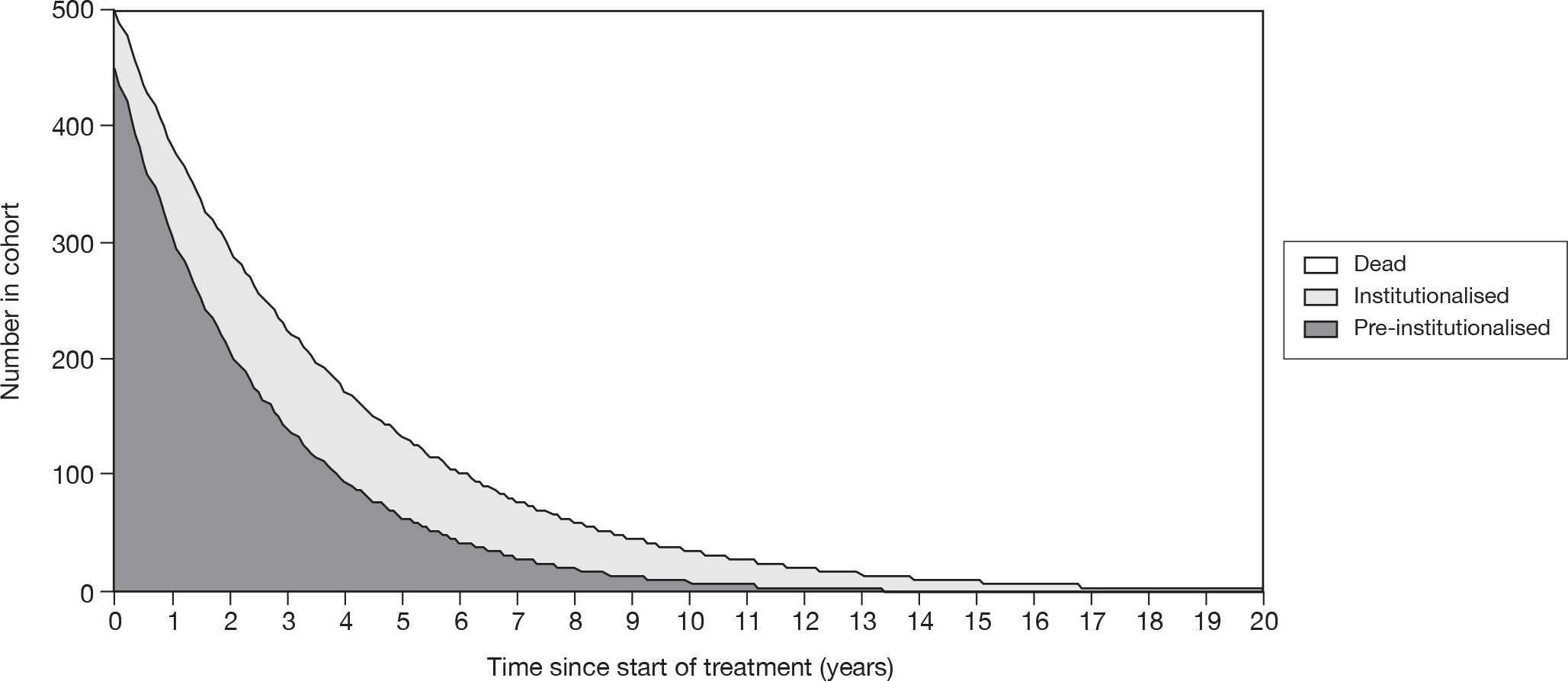
The mean time until the end of pre-institutionalisation for the treated cohorts is given in Table 116, alongside the total cost and QALY estimates from the deterministic analysis. There is very little difference between the three cholinesterase inhibitors (as further demonstrated in Figure 77, where BSC is the lower line and the AChEIs are indistinguishable from each other, appearing as the solid upper line). This is expected given the similar magnitude of effectiveness for MMSE and ADCS-ADL (refer back to Table 107), with treatment leading to a mean of 1.4–1.7 months (42–51 days) delay in becoming institutionalised.
| Treatment | Mean months to institutional carec | Months’ delay to institutional care compared with BSC | Total costs (£) | Total QALYs | Incremental costs (£) | Incremental QALYs | ICERa,b (£) |
|---|---|---|---|---|---|---|---|
| Galantamine (16–24 mg) | 30.4 | 1.6 | 69,592 | 1.617 | |||
| Rivastigmine patch (9.5 mg/day) | 30.3 | 1.5 | 69,598 | 1.616 | Dominated | ||
| Donepezil (10 mg) | 30.5 | 1.7 | 69,624 | 1.619 | 32 | 0.002 | 17,900 |
| Rivastigmine capsules (9–12 mg) | 30.2 | 1.4 | 69,678 | 1.613 | Dominated | ||
| BSC | 28.8 | NA | 70,212 | 1.584 | Dominated |
FIGURE 77.
Time to institutionalisation for a mild-to-moderate cohort.
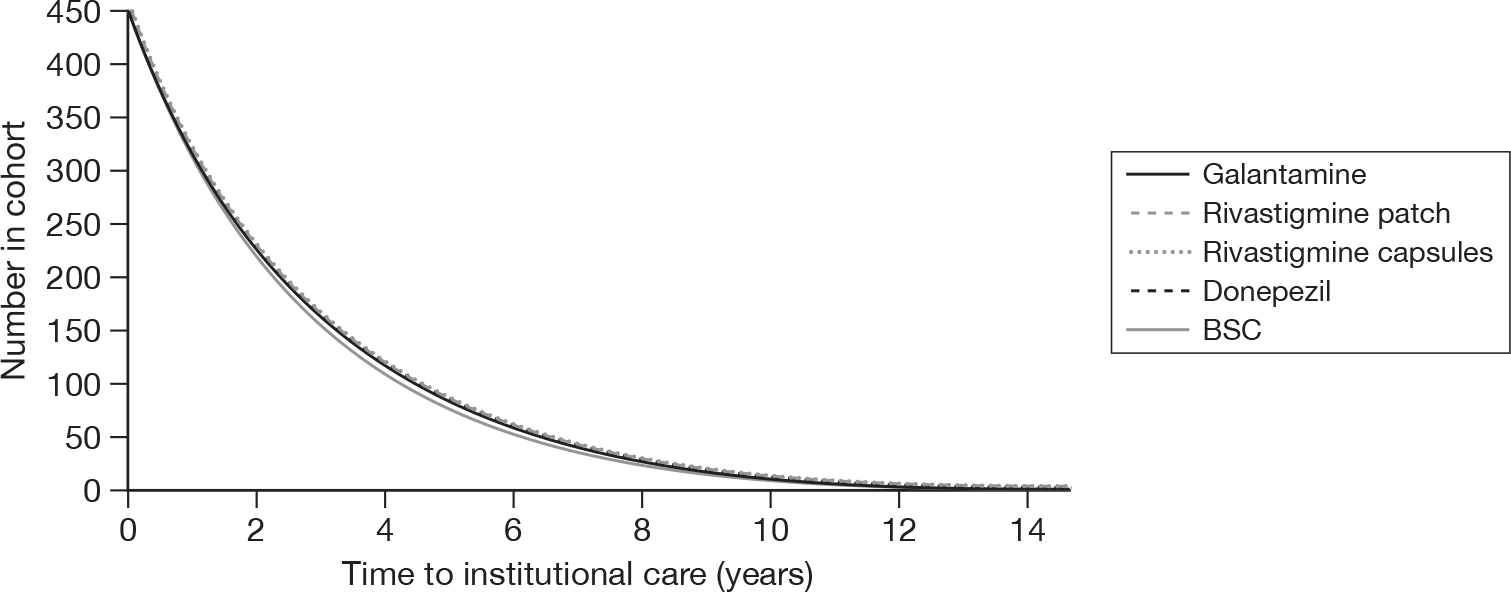
The base-case results for the incremental cost–utility of the AChEIs compared with the next cheapest, non-dominated technology are given in Table 116. The cost-effectiveness frontier is shown in Figure 78. It is estimated that over a person’s lifetime the options of treating with BSC, rivastigmine and rivastigmine patches are dominated by donepezil and galantamine. BSC and rivastigmine are associated with greater costs and fewer QALYs than donepezil, whereas rivastigmine patches are associated with greater costs and fewer QALYs compared with galantamine. Treatment with galantamine is estimated as the cheapest option, with total costs of £69,598 and total QALYs of 1.617. However, treatment with donepezil is estimated to have the most QALY gains over someone’s lifetime (1.619 QALYs) with a total cost of £69,624. Thus the ICER for donepezil compared with galantamine is £17,900 per QALY. This ICER is smaller than that from the PSA (£23,500/QALY) owing to non-linearities in the PSA. Nevertheless, all reference to the base-case analysis will refer to the deterministic ICERs and not the PSA ICERs.
FIGURE 78.
Base-case cost-effectiveness plane for the CUA for mild-to-moderate AD. C-E frontier, cost-effectiveness frontier.
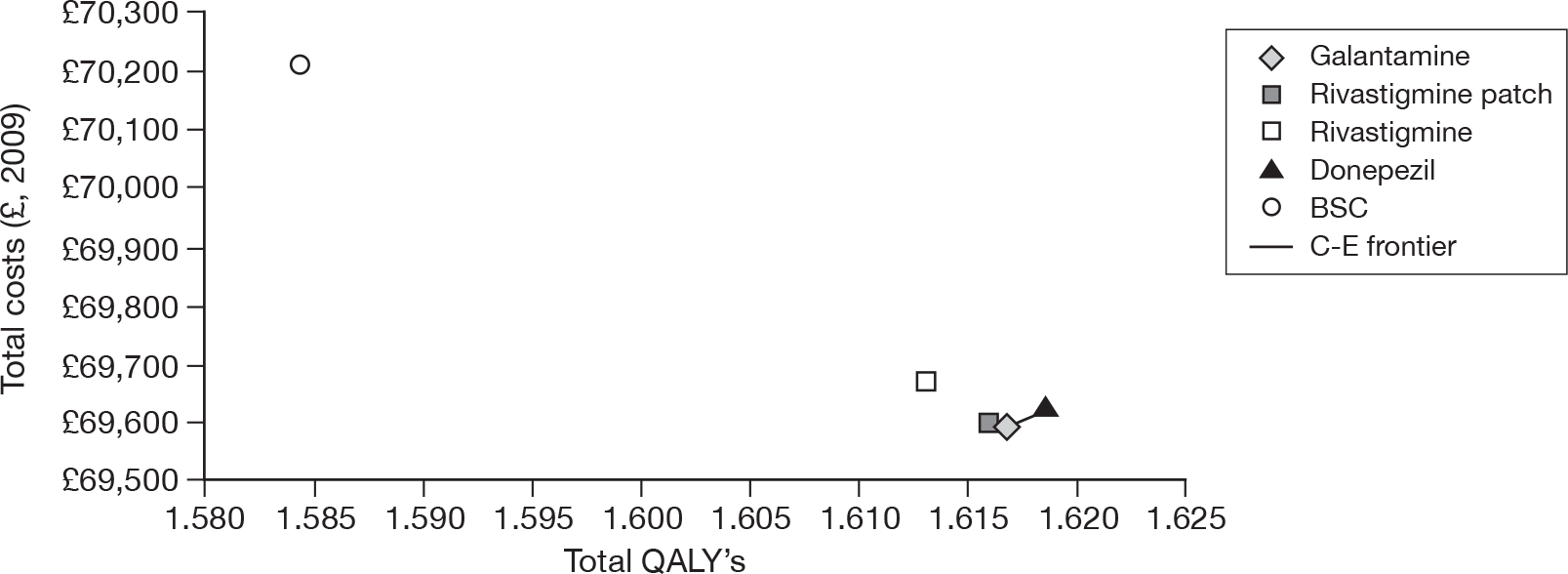
The differences in the component costs between the four AChEIs when each is compared with BSC are shown in Figure 79. For all four technologies, the largest saving is for the costs associated with being in institutional care. This is as expected, as the technologies are estimated to delay institutionalisation for 1.4–1.7 months. As overall survival is not assumed to be affected by the AChEIs, an individual’s total time spent in institutional care is reduced by receiving treatment. The delay to institutionalisation is also reflected in the higher costs incurred for the pre-institutionalised state when compared with BSC (Figure 79). The costs saved from delaying institutionalisation are greater than the combined costs of the drugs, monitoring and increased costs of care in the pre-institution state when compared with BSC. Thus, treatment with any of the AChEIs leads to cost-savings compared with BSC. Figure 79 highlights a slight difference in total drug costs per patient between the AChEIs, with rivastigmine capsules being the cheapest and donepezil the most expensive. Note also that the larger cost saved in institutional care for donepezil compared with the other AChEIs is owing to the slightly greater delay to institutionalisation assumed with donepezil than with other AChEIs (1.7 months: refer back to Table 116).
FIGURE 79.
Base-casecost components for the cholinesterase inhibitors compared with BSC for mild-to-moderate AD.
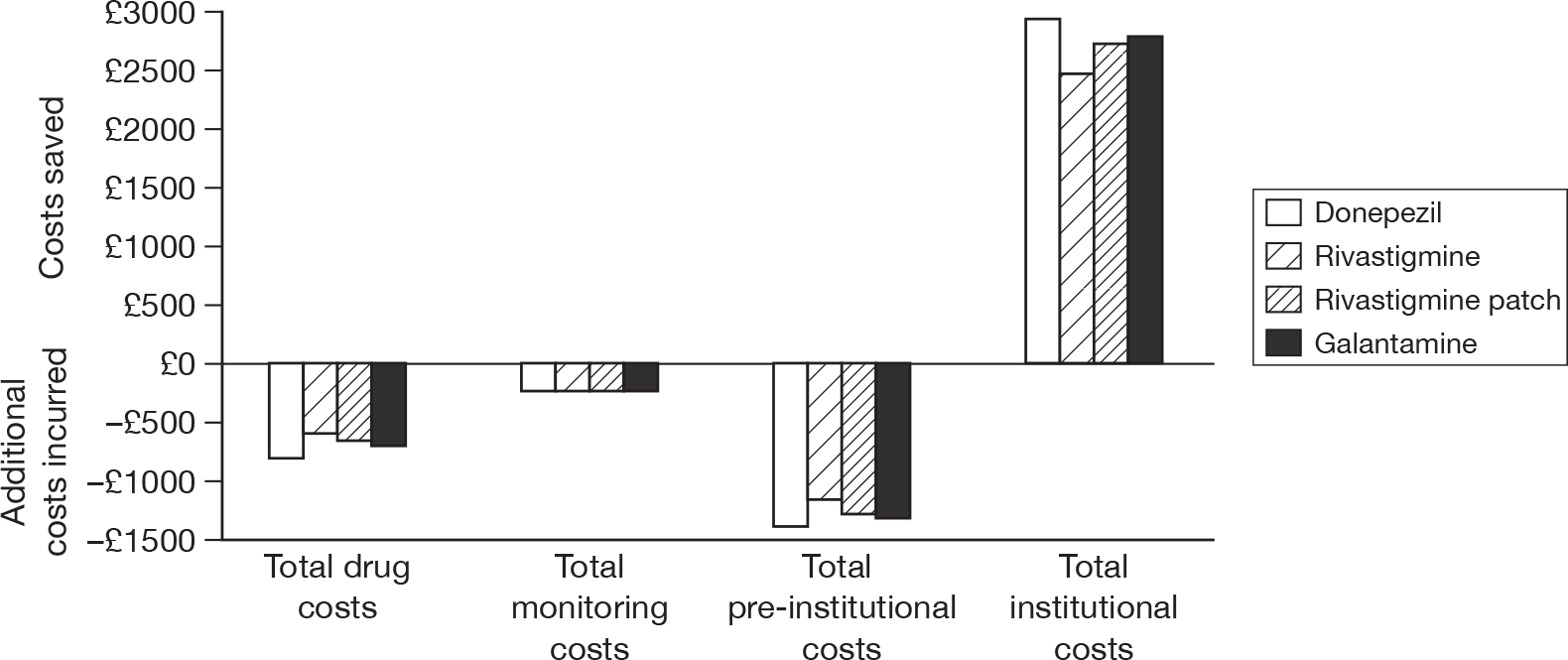
The additional QALY gains over BSC for the four technologies are all in the pre-institutionalised state (Figure 80). The QALYs lost in the institutionalised state with treatment with the AChEIs compared with BSC reflect the reduced time spent in institutionalisation for those on treatment (because the base-case assumptions include no treatment effect on overall survival). The QALY gains before institutionalisation are greater than the QALY losses when in the institutionalised state because the utilities before institutionalisation are greater than the utility while institutionalised.
FIGURE 80.
Base-case QALY components for the cholinesterase inhibitors compared with BSC in mild-to-moderate AD.
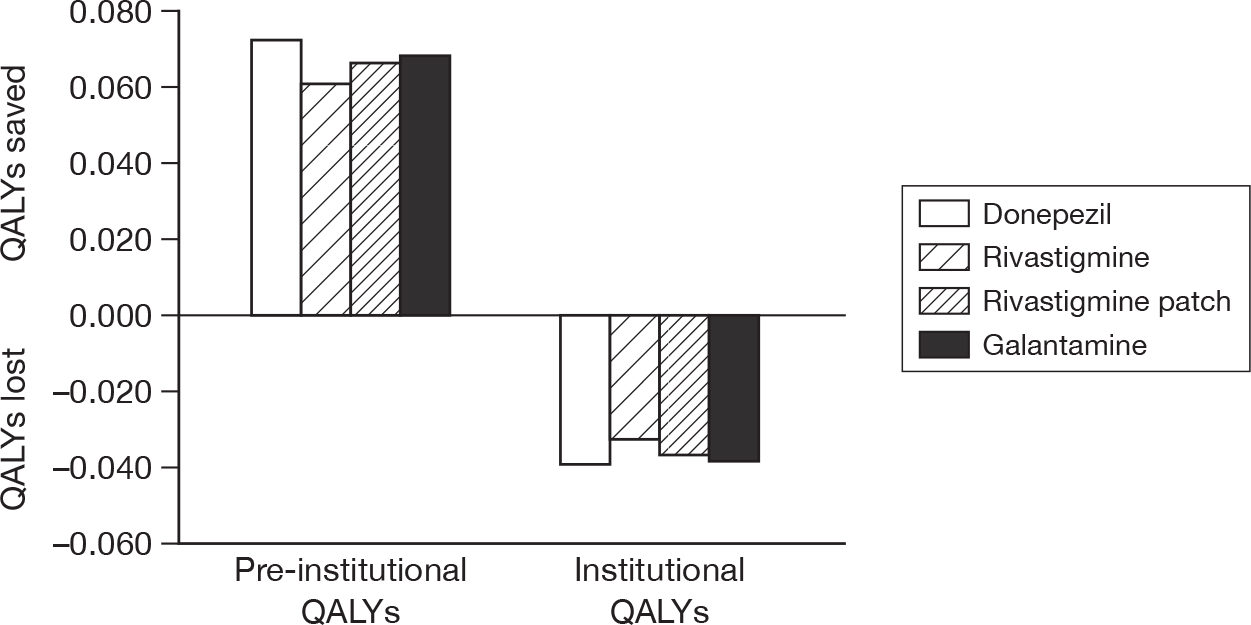
Summary of the deterministic and probabilistic analyses
The PSA and the deterministic base-case analyses indicate that all AChEIs dominate BSC. Galantamine is associated with the least costs, but donepezil is associated with the greatest QALY gains. Note that the incremental costs and QALYs between the AChEIs are very small. Furthermore, the PSA results do not indicate a particular AChEI as having a much greater probability of being cost-effective compared with any of the other AChEIs. This is the case across a range of WTP values.
In Table 117, the deterministic and probabilistic ICERs from the PenTAG model are presented.
| Treatment | Deterministicb (£) | Probabilisticb (£) | Deterministic vs BSC |
|---|---|---|---|
| Galantamine (16–24 mg) | More effective and less costly | ||
| Rivastigmine patches (9.5 mg/day) | Dominated | Dominated | |
| Donepezil (10 mg) | 17,900 | 23,500 | |
| Rivastigmine capsules (9–12 mg) | Dominated | Dominated | |
| BSC | NA |
One-way deterministic sensitivity analyses
Treatment effect on mortality
In the base-case analysis, it was assumed that there was no treatment effect on overall survival. However, analysis of the IPD from Wolstenholme and colleagues196 for predicting time to death in the BSC cohort used baseline MMSE, Barthel ADL Index and age as independent variables, and the effectiveness data indicate that AChEI treatment affects MMSE and ADL. Thus, as a sensitivity analysis it is assumed that treatment effect measured by MMSE and the Barthel ADL Index does affect overall survival. The mean times to institutionalisation do not change from the base-case analysis (see Table 116), but the mean time to death is extended and given in Table 118 for each treatment cohort. All treatments delay death by 1.9 to 2.2 months compared with BSC and this difference can be seen in Figure 81, where BSC is the lower line and the AChEIs are indistinguishable from each other, appearing as the solid upper line.
| Treatment | Mean time (months) to deatha | Extended life (months) compared with BSC | Costs (£) | QALYs | Incremental costs (£) | Incremental QALYs | ICERb,c |
|---|---|---|---|---|---|---|---|
| BSC | 46.0 | 70,212 | 1.584 | ||||
| Rivastigmine (9–12 mg) | 47.9 | 1.9 | 72,807 | 1.654 | Extended dominated | ||
| Rivastigmine patch (9.5 mg/day) | 48.1 | 2.1 | 73,052 | 1.661 | 2840 | 0.077 | 37,100 |
| Galantamine (16–24 mg) | 48.1 | 2.1 | 73,129 | 1.663 | 77 | 0.002 | 41,800 |
| Donepezil (10 mg) | 48.2 | 2.2 | 73,346 | 1.667 | 217 | 0.004 | 51,800 |
FIGURE 81.
Overall survival for a mild-to-moderate cohort.
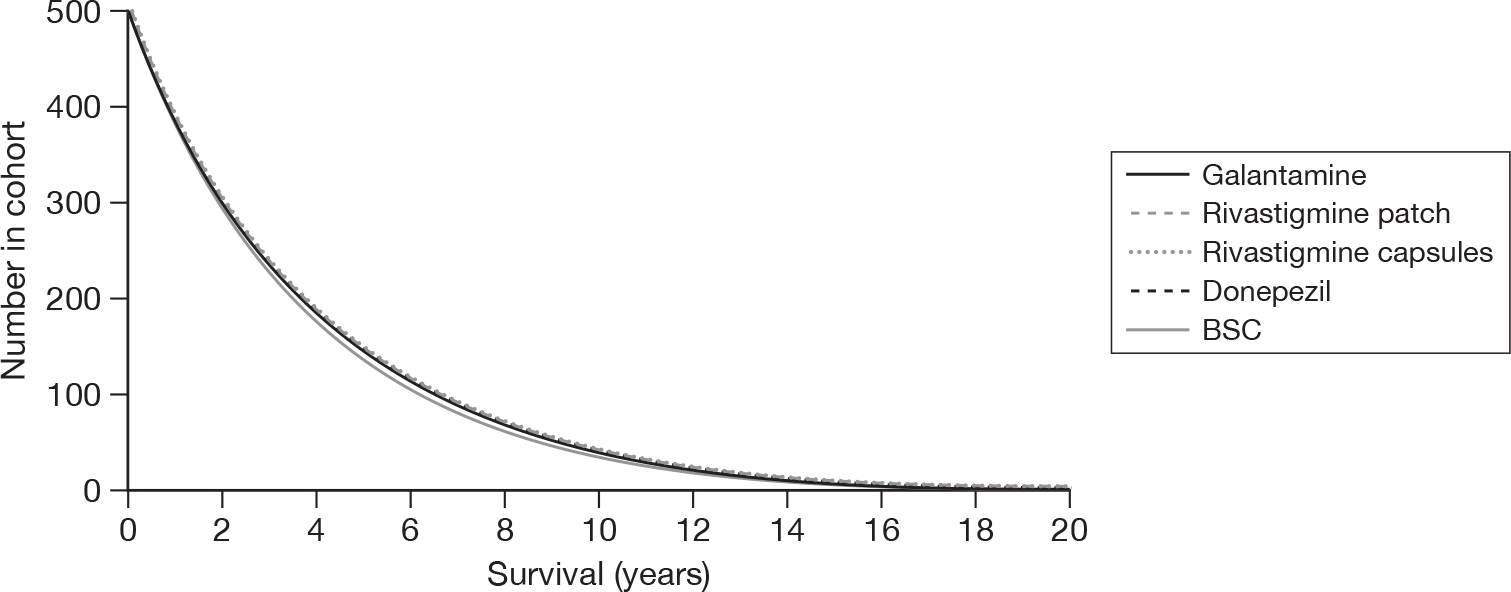
The CUA results assuming a treatment effect on survival are shown in Table 118 for ICERs compared with the next cheapest non-dominated technology. ICERs are presented also in Table 119 for comparison with BSC, demonstrating very little difference between the AChEIs.
| Treatment | Costs (£) | QALYs | Incremental costs (£) | Incremental QALYs | ICERa,b |
|---|---|---|---|---|---|
| BSC | 70,212 | 1.584 | |||
| Rivastigmine (9–12 mg) | 72,807 | 1.654 | 2595 | 0.069 | 37,400 |
| Rivastigmine patch (9.5 mg/day) | 73,052 | 1.661 | 2840 | 0.077 | 37,100 |
| Galantamine (16–24 mg) | 73,129 | 1.663 | 2917 | 0.078 | 37,200 |
| Donepezil (10 mg) | 73,346 | 1.667 | 3134 | 0.083 | 37,900 |
Under the assumption of a treatment effect on mortality, greater costs and QALYs are associated with the AChEIs, but BSC is no longer dominated. It is estimated that treatment with rivastigmine patches provides an additional 0.077 QALYs per patient over BSC, with additional costs of £2840, leading to an ICER of £37,100/QALY. Treatment with galantamine or donepezil provides additional QALYs over rivastigmine patches, but at additional costs leading to ICERs of £41,800/QALY for galantamine compared with rivastigmine patches, and £51,800/QALY for donepezil compared with galantamine. Rivastigmine capsules are extended dominated by rivastigmine patches and BSC.
In comparison to the base-case analysis, more QALYs are gained when a treatment effect on survival is assumed, owing to additional life, but this gain is spent in a more expensive state – institutional care (Figures 82 and 83). Indeed, given that institutional care is a highly cost-ineffective state, when we allow the drugs to increase overall survival, all drugs become far less cost-effective against BSC (see Table 119).
FIGURE 82.
Cost components for the cholinesterase inhibitors compared with BSC, when a treatment effect on survival is assumed.
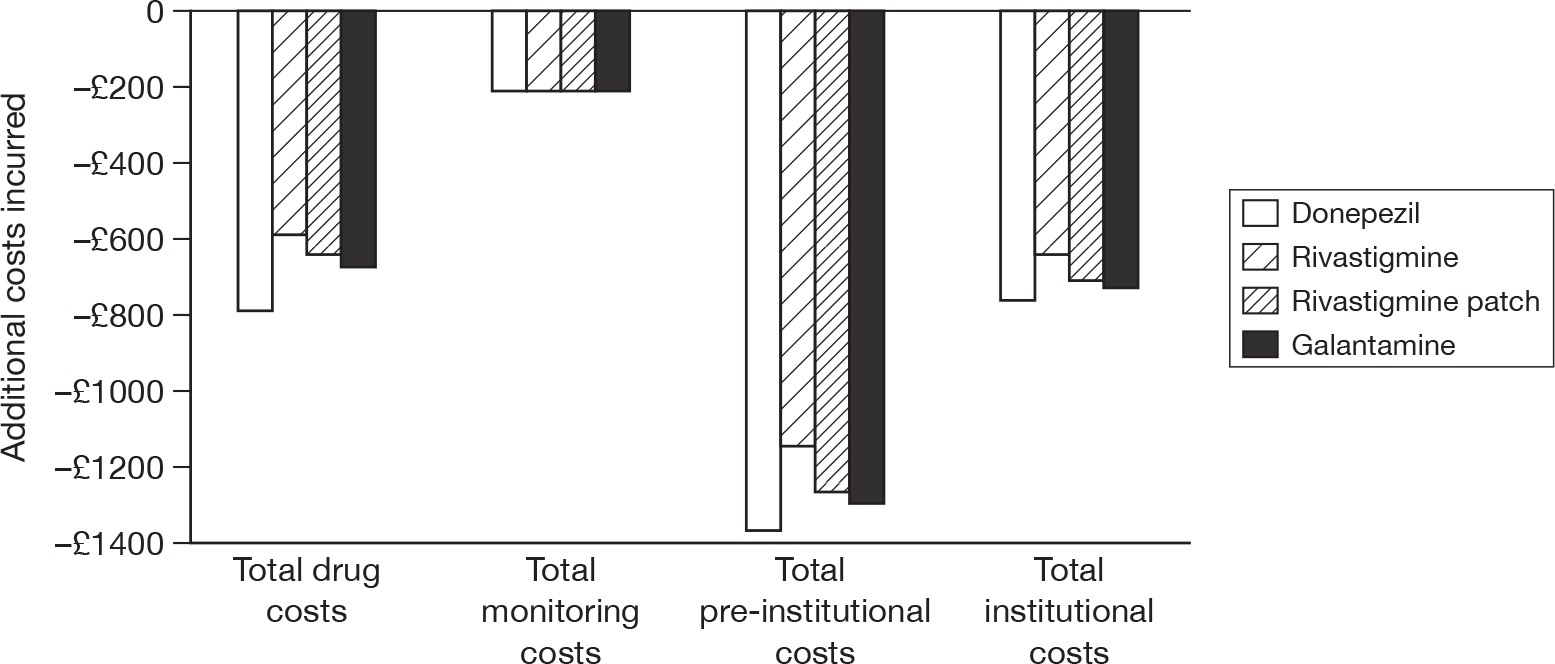
FIGURE 83.
Quality-adjusted life-year components for the cholinesterase inhibitors compared with BSC, assuming a treatment effect on survival.
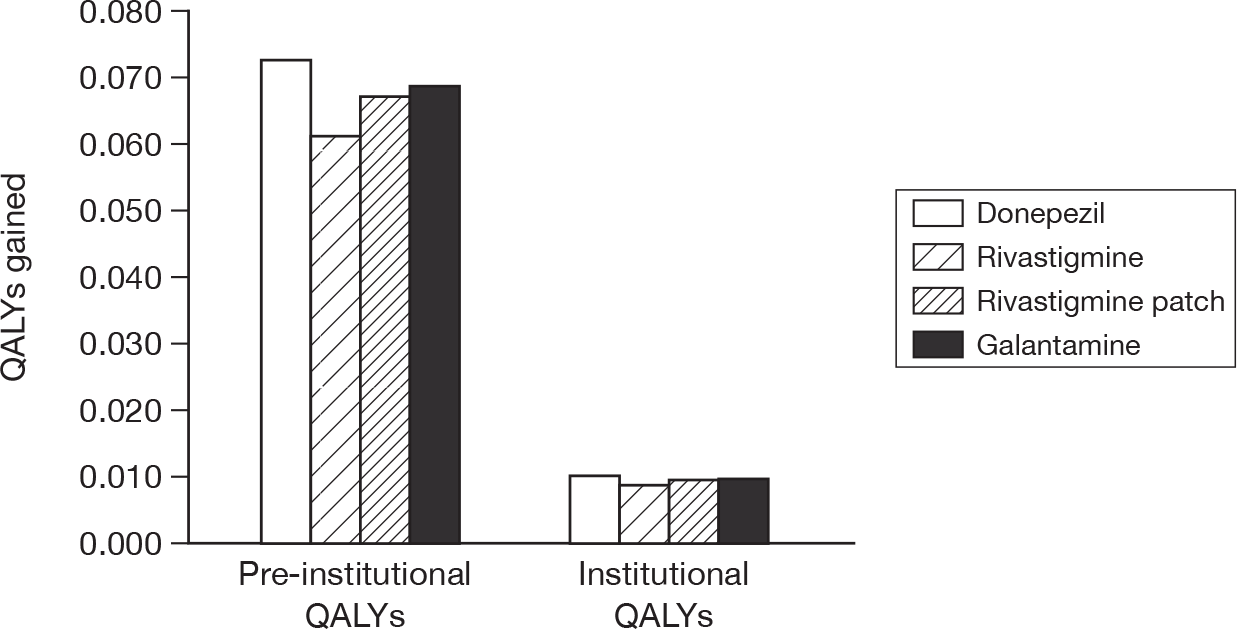
Importance of the effectiveness on the Mini Mental State Examination
There has been debate regarding the appropriateness of the methods used to map ADCS-ADL to the Barthel ADL Index. To assess the impact of a treatment effect on ADL in the PenTAG model for AChEIs, it was assumed that treatment affected only the MMSE. In other words, the treatment effect for ADL for all drugs was set to zero. In these sensitivity analyses, the overall findings from the base case do not change: all AChEIs dominate BSC (Table 120). This identifies the treatment effect on MMSE as an important driver of the PenTAG model.
| Treatment | Costs (£) | QALYs | Incremental costs (£) | Incremental QALYs | ICERa,b (£) |
|---|---|---|---|---|---|
| Galantamine (24 mg) | 69,914 | 1.610 | |||
| Rivastigmine patch (9.5 mg/day) | 69,915 | 1.609 | Dominated | ||
| Donepezil (10 mg) | 69,916 | 1.612 | 2 | 0.002 | 718 |
| Rivastigmine (≤ 12 mg) | 69,939 | 1.607 | Dominated | ||
| BSC | 70,212 | 1.584 | Dominated |
Alternatively, when it is assumed that a treatment effect only occurs on ADL (i.e. treatment effect for MMSE is set to zero), none of the AChEIs dominates BSC (Table 121). In fact, donepezil is dominated by galantamine, and rivastigmine capsules are dominated by rivastigmine patches and BSC. Rivastigmine patches give additional gains of 0.007 QALYs over BSC, but at a cost of £532 leading to an ICER of £78,024/QALY. An additional QALY gain of < 0.001 is provided by galantamine compared with rivastigmine patches, but at a cost that leads to an ICER of £247,800/QALY for galantamine compared with rivastigmine patches.
| Treatment | Costs (£) | QALYs | Incremental costs (£) | Incremental QALYs | ICERa,b |
|---|---|---|---|---|---|
| BSC | 70,212 | 1.584 | |||
| Rivastigmine (≤ 12 mg) | 70,743 | 1.590 | Dominated | ||
| Rivastigmine patch (9.5 mg/day) | 70,745 | 1.591 | 532 | 0.007 | 78,000 |
| Galantamine (24 mg) | 70,770 | 1.591 | 25 | < 0.001 | 247,800 |
| Donepezil (10 mg) | 70,912 | 1.591 | Dominated |
Further one-way sensitivity analyses
The parameter values and assumptions explored in the following one-way sensitivity analyses are shown in Table 122. Analyses are presented as incremental net monetary benefits at a WTP of £30,000 per QALY for donepezil compared with galantamine (Figure 84), the next cheapest non-dominated technology (as in the above analyses) and for donepezil compared with BSC (Figure 85). In the majority of one-way sensitivity analyses, rivastigmine patches and capsules and BSC were dominated by donepezil and/or galantamine, therefore no tornado plots are shown for the incremental net monetary benefits of these treatment options over the next cheapest non-dominated technology. Tornado plots for galantamine, rivastigmine patches and capsules compared with BSC are provided in Appendix 20 and follow the same general trend as donepezil compared with BSC (see Figure 85).
| Parameter/assumption | Base case | Deterministic sensitivity analysis | Reference in tornado plots |
|---|---|---|---|
| Survival effect | Independent of treatment | Depends on treatment | Survival effect |
| Drug costs | See Table 113 | Industry cost for donepezil; 9-mg cost for rivastigmine capsules; 16-mg cost for galantamine; 24-mg cost for galantamine | Drug cost |
| Cost in institutional care | £2941 per month estimated from Wolstenholme et al.196 IPD | £3267 from Lundbeck submission; £2801 from Eisai submission | Inst cost £ |
| Percentage institutional costs NHS/PSS | 0.72 | 0.53 and 0.906 | % NHS/PSS cost |
| Treatment discontinuations | 4% of the total cohort per month | The maximum (5.7%) and minimum (2.34%) from the RCTs discontinue each month, as well as 0% discontinuing | % discontinuations |
| Cost in pre-institution state | Based on relationship from Wolstenholme et al.196 IPD | Transformed industry pre-institutionalisation costs by MMSE to time to institutionalisation | Industry pre-inst costs by MMSE |
| Population characteristics | Based on Wolstenholme et al.196 IPD | Based on LASER-AD IPD | LASER-AD cohort |
| Severity of cohort | Mild to moderate | Mild or moderate | Cohort severity |
| Monitoring costs | From NHS Reference Costs 2008–9,217 £158 per visit | £185, upper value of IQR from NHS Reference Costs 2008–9;217 £62.29, Lundbeck estimate of subsequent outpatient visit | Monitoring cost (£) |
| Patient utility weights | Average of EQ-5D, VAS and QoL-AD carer-proxy utilities | Patient self-rated EQ-5D utility and of carer-proxy EQ-5D utility | Patient self-rated utility; Carer-proxy utility (patient and proxy available; only proxy available) |
| Carer utility weights | Not included | HUI-2 | Carer utility |
| Effectiveness estimates | Treatment effects MMSE and ADL | Treatment only affects MMSE or only ADL | MMSE effect only; ADL effect only |
| Discounting | 3.5% for costs and benefits | 3.5% for costs and 1.5% for benefits | Discounting |
FIGURE 84.
One-way sensitivity analyses for the incremental net monetary benefit of donepezil compared with galantamine for mild-to-moderate AD. See Table 122 for a description of the individual sensitivity analyses undertaken.
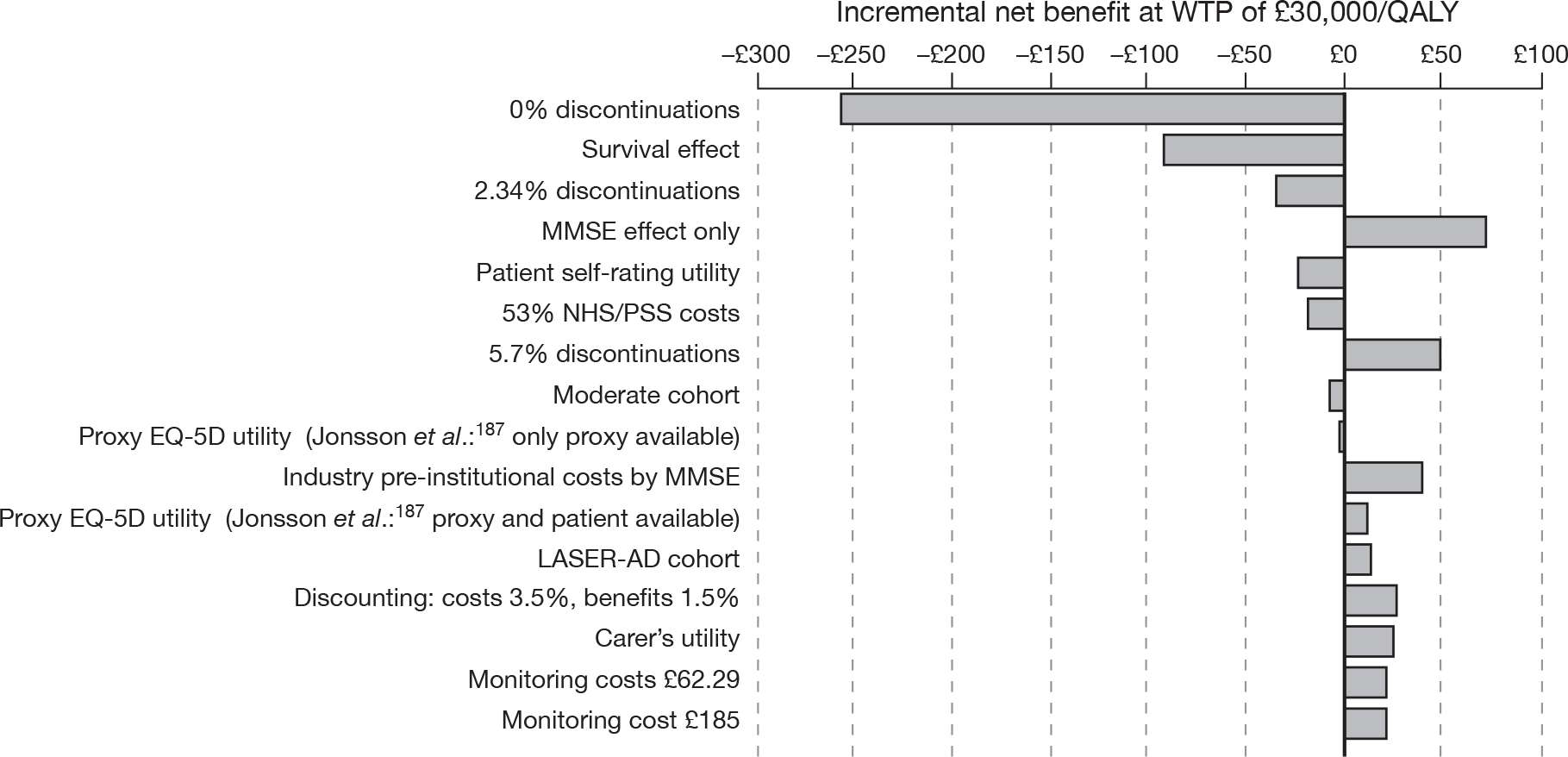
FIGURE 85.
One-way sensitivity analyses for the incremental net monetary benefit of donepezil compared with BSC. See Table 122 for explanation of description of individual sensitivity analyses undertaken.
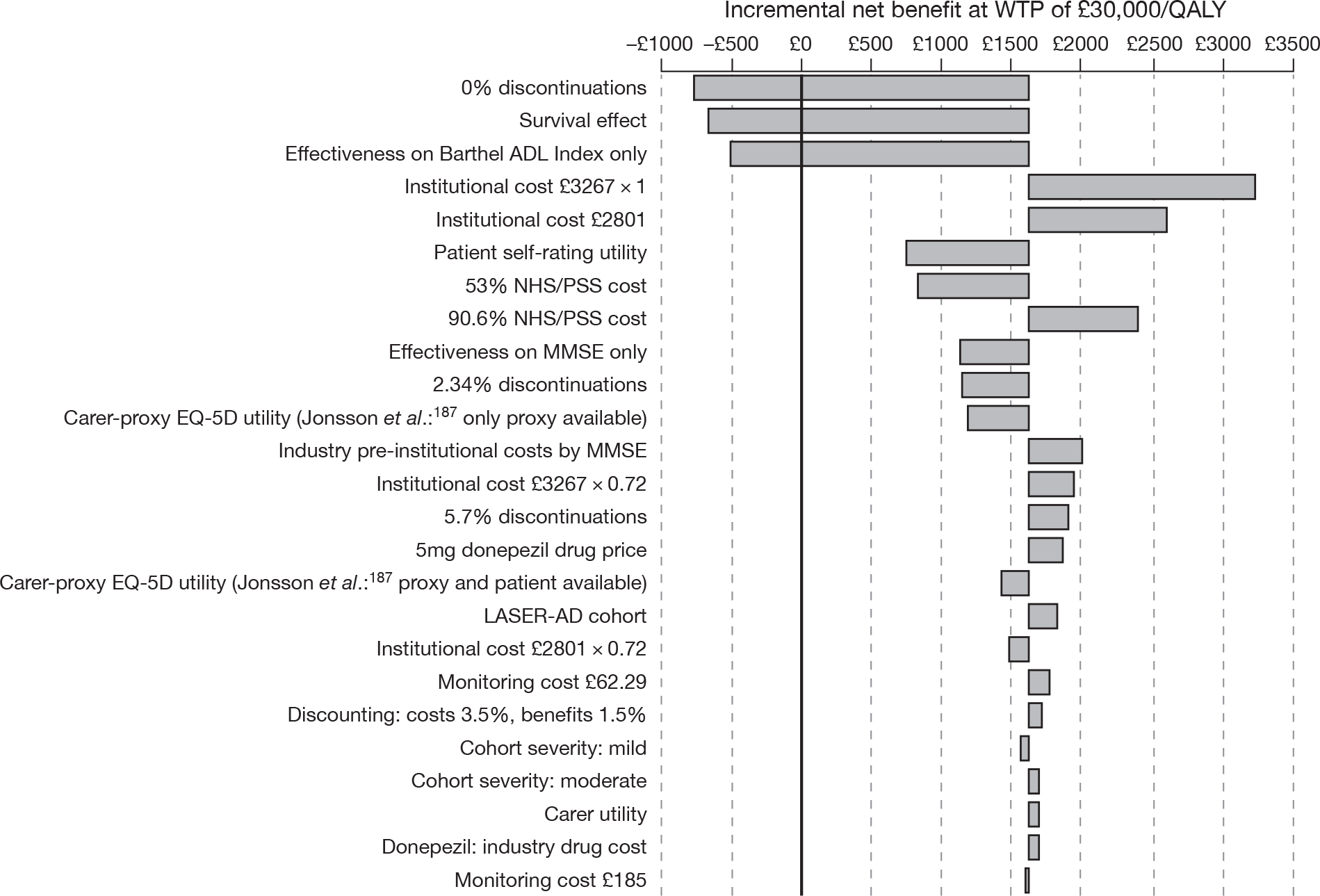
In the base-case analysis, at a WTP of £30,000 per QALY gained, donepezil has an incremental net benefit of £22 compared with galantamine, whereas galantamine dominates BSC, rivastigmine capsules and patches. At a WTP of £30,000 per QALY, donepezil has an incremental net benefit of £1616 compared with BSC. Note that there is no comparison for a number of scenarios (the mild cohort, where 90.6% of institutional care costs are assumed to be NHS/PSS, where industry costs of institutional care are assumed or where the 5-mg cost of donepezil is assumed), as donepezil dominates galantamine in all of these scenarios.
The assumption having the largest impact on the net benefit of donepezil compared with galantamine (see Figure 84) and for donepezil compared with BSC (see Figure 85) is the assumption that all patients continue treatment until they enter institutional care (0% discontinuations assumed). As pointed out above (see Treatment discontinuation), we assume that a discontinuation rate for treatment only affects the costs associated with treatment, not the effectiveness, as it is assumed that the effect estimates are based on an ITT analysis. Therefore, lower estimates of this percentage lead to greater treatment and monitoring costs, resulting in a negative net benefit for the AChEIs. Higher estimates (such as 5.7% as shown in Figures 84 and 85) led to fewer costs and greater net benefit associated with the AChEIs.
As discussed above, the assumption of a treatment effect on survival leads to the AChEIs having a larger cost per QALY gained than in the base-case analysis and no longer dominating BSC.
Assumptions on the costs of care in the institutionalised state have a large impact on the results as would be expected. As it is assumed that the AChEIs delay and, therefore, in the base-case analysis reduce time spent in institutionalised care, this cost is important. Assuming a lower cost for institutional care compared with pre-institutional care leads to fewer costs saved by the treatments. This is demonstrated in Figure 84, where lower costs in institutionalised care (either by assuming a lower total cost or by decreasing the percentage of institutionalised costs funded by NHS/PSS to 53%) led to a smaller net benefit at a WTP of £30,000 per QALY gained.
There is also some uncertainty about the utility estimates used. Alternative estimates of carer-proxy utility led to lower estimates of net benefit, as these estimates provide less of a change in utilities as the disease progresses. This is especially the case for the EQ-5D utilities from Jonsson and colleagues,187 where only proxy utilities are available. Therefore, by delaying disease progression, a greater utility gain is obtained when there is a larger difference between utility for mild disease compared with severe disease. The estimates used in the base-case analysis span a large range of utility weights across severity, from 0.69 for MMSE > 25 to 0.33 for MMSE < 10. These utility estimates are, therefore, more favourable to the AChEIs in the PenTAG model, as a delay to more severe stages of AD leads to a bigger gain in utility than would be obtained using alternative care-proxy estimates having a narrower range of values across severity. Use of patient’s self-rated QoL leads to lower estimates of net benefit for the AChEIs, as even for the most severe state, a utility of 0.78 was reported compared with 0.84 for MMSE of 26–30. Thus, fewer QALY gains by delaying entry into institutional care are obtained when assuming patient-rated QoL estimates. This is not a surprising result, as the most severe state was estimated to have greater utility by patients than the adjacent less severe state (See Quality of life: utility estimates).
Inclusion of carer’s own QoL estimates led to a very small increase in the net benefit of the AChEIs. This is as expected given that these estimates are based on data indicating that there is very little change in the carer’s QoL as the disease progresses.
Summary of one-way sensitivity analyses
In Table 123 the degree of uncertainty in the decision model and the impact of these parameters on the cost-effectiveness of the AChEIs is presented for people with mild-to-moderate AD. The most important items are those discussed above, the main one being whether or not a treatment effect on survival is assumed and the rate at which patients discontinue treatment.
| Issue | Evidence source | Level of uncertainty in data | Impact of uncertainty in model | Overall rating of importance in cost-effectiveness results |
|---|---|---|---|---|
| Assuming a treatment effect on survival | No published RCT or epidemiological evidence. Survival prediction allows treatment survival effect | High | High | Very important |
| Treatment discontinuations | Final time point data from RCTs | High | High | Very important |
| Costs in institutional care | Inflated 20-year-old estimates from 92 individuals | High | Moderate | Important |
| Effectiveness evidence | Mix of different quality RCTs | Moderate | Low | Moderate |
| Patient’s health-state utility | Proxy respondents or self-rated from published literature | Moderate | Moderate | Important |
| Carer’s health-state utility | Poor published evidence | High | Low | Moderate |
| Percentage of costs in institutional care funded by NHS/PSS | Poor published evidence plus expert opinion | High | Moderate | Moderate |
| Costs in pre-institutional state | Inflated 11- to 20-year-old estimates from 92 individuals | High | Moderate | Moderate |
| Cost of treatment-monitoring visit | NHS Reference Costs 2008–9 217 | Low | Low | Low |
| Percentage starting model in institutional care | Published epidemiological study and author assumption | High | Low | Not important |
| Baseline characteristics | Statistical analysis of 92 individuals | Low | Low | Not important |
| Cost of drugs | BNF compared with some poor reporting of doses used in RCTs | Moderate | Moderate | Moderate |
Moderate-to-severe Alzheimer’s disease: memantine (decision problem 2a)
Probabilistic sensitivity analysis
There is a great deal of uncertainty associated with the estimation of costs and QALYs of treatment with memantine compared with BSC for people with moderate-to-severe AD (Figure 86). The cost-effectiveness acceptability curve (Figure 87) indicates that memantine would be the most cost-effective option when compared with BSC, for a WTP per QALY gained greater than £44,000. There is a 38% probability that memantine is the most cost-effective treatment at a WTP threshold of £30,000 per QALY compared with BSC. At a WTP of £20,000 per QALY, memantine has a probability of 28% of being the most cost-effective treatment option. The ICER from the PSA for memantine compared with BSC is £36,700/QALY.
FIGURE 86.
Base-case cost-effectiveness plane for memantine in people with moderate-to-severe AD.
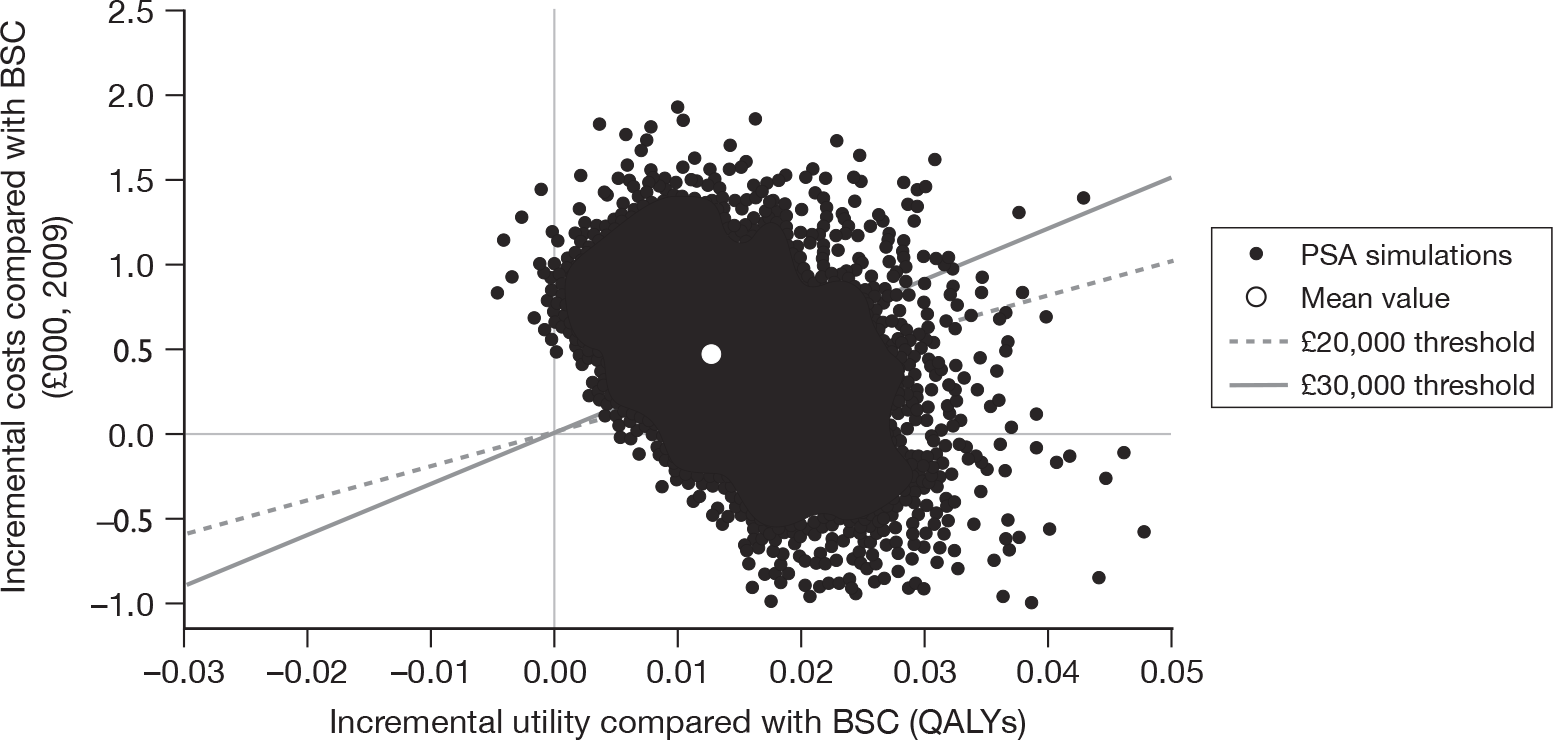
FIGURE 87.
Base-case cost-effectiveness acceptability curve for memantine in people with moderate-to-severe AD.
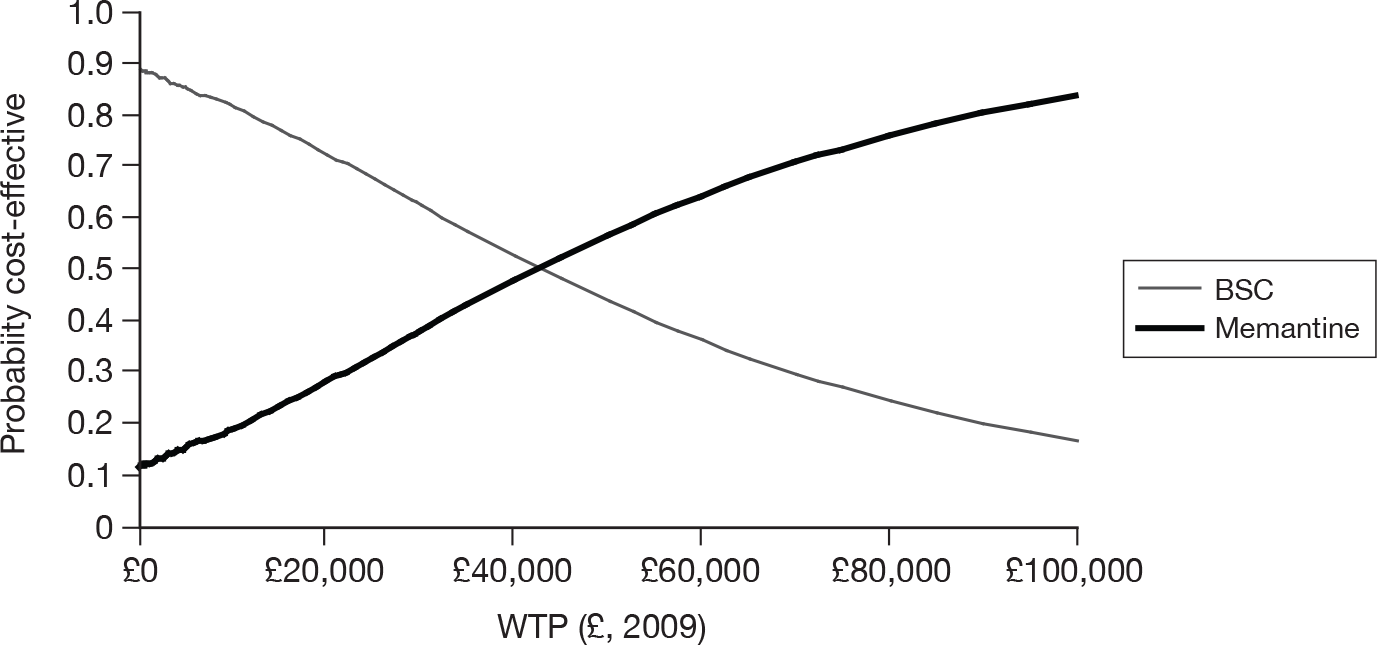
Deterministic analysis
The progression of a proportion of the moderate-to-severe cohort on BSC through the model is represented graphically in Figure 88 as an example of the time spent within each state of the model. Figure 88 is based on data for individuals with a mean starting age of 78 years (representing 50% of the cohort). Forty per cent of the cohort are assumed to be in institutional care at the start of the model. The mean overall survival across all three age cohorts for moderate-to-severe AD is 42.1 months. The mean time to institutionalisation for the BSC cohort is 17.7 months, whereas for the memantine cohort this is 18.5 months, leading to a delay to institutionalisation of 0.8 months (about 23 days).
FIGURE 88.
Progression of the BSC cohort in the base case (moderate-to-severe AD, age group 2).
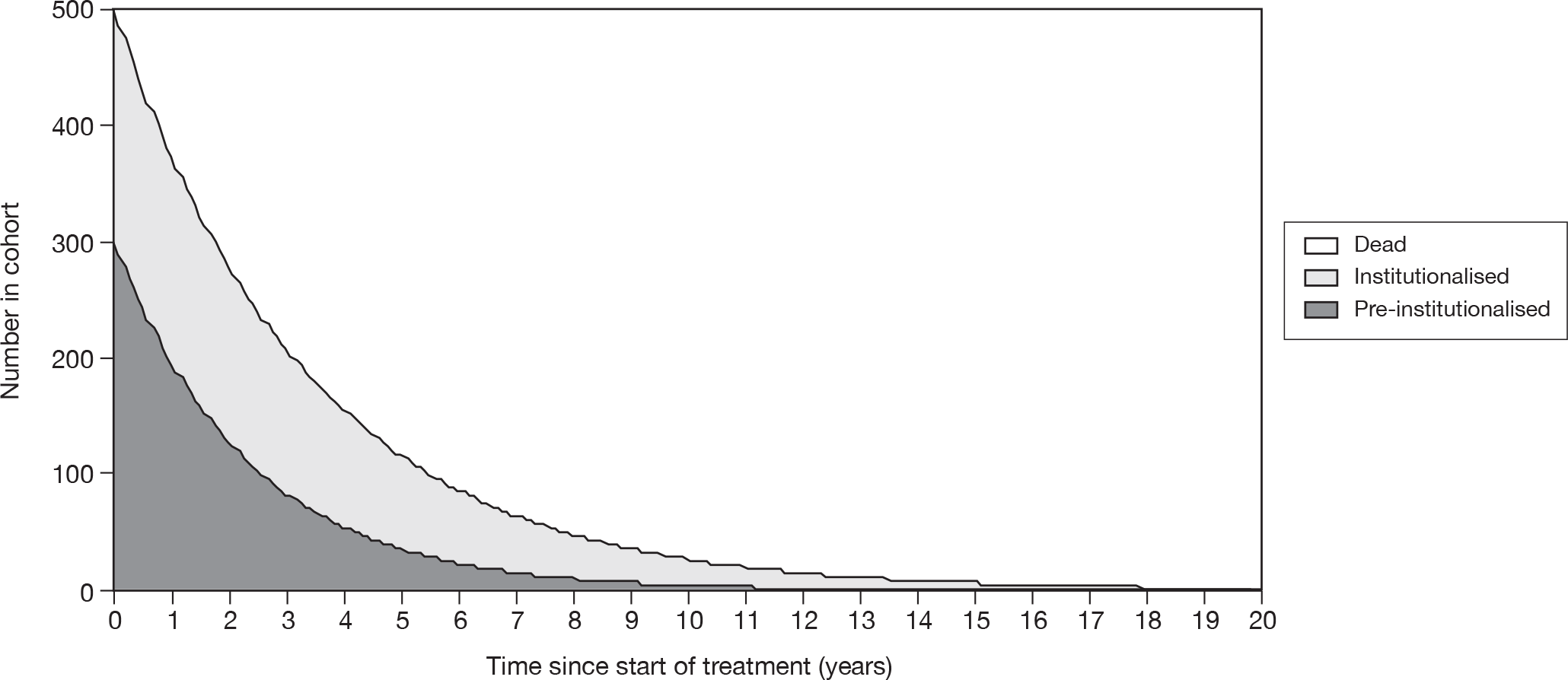
The base-case CUA result for memantine compared with BSC for people with moderate-to-severe AD (MMSE 20–0) is given in Table 124. For a gain of 0.013 QALYs over a patient’s lifetime when treated with memantine compared with BSC, the extra cost is £405, leading to an estimated cost per QALY of £32,100 from the deterministic base-case analysis. The cost components detailed in Figure 89 demonstrate that, as with the AChEIs, the cost savings of treatment with memantine occur when the individual is in institutionalised care. However, the drug, monitoring and incremental pre-institutionalised costs combined are greater than the incremental institutionalisation costs leading to memantine being more costly than BSC. The gains in QALYs with memantine over BSC (Figure 90) are seen in the pre-institutionalised state, as longer time is spent in this state for memantine-treated individuals.
| Treatment | Costs (£) | QALYs | Incremental costs (£) | Incremental QALYs | ICERa (£) |
|---|---|---|---|---|---|
| BSC | 78,123 | 1.215 | |||
| Memantine (20 mg) | 78,528 | 1.227 | 405 | 0.013 | 32,100 |
FIGURE 89.
Base-case cost components for memantine compared with BSC for moderate-to-severe AD.
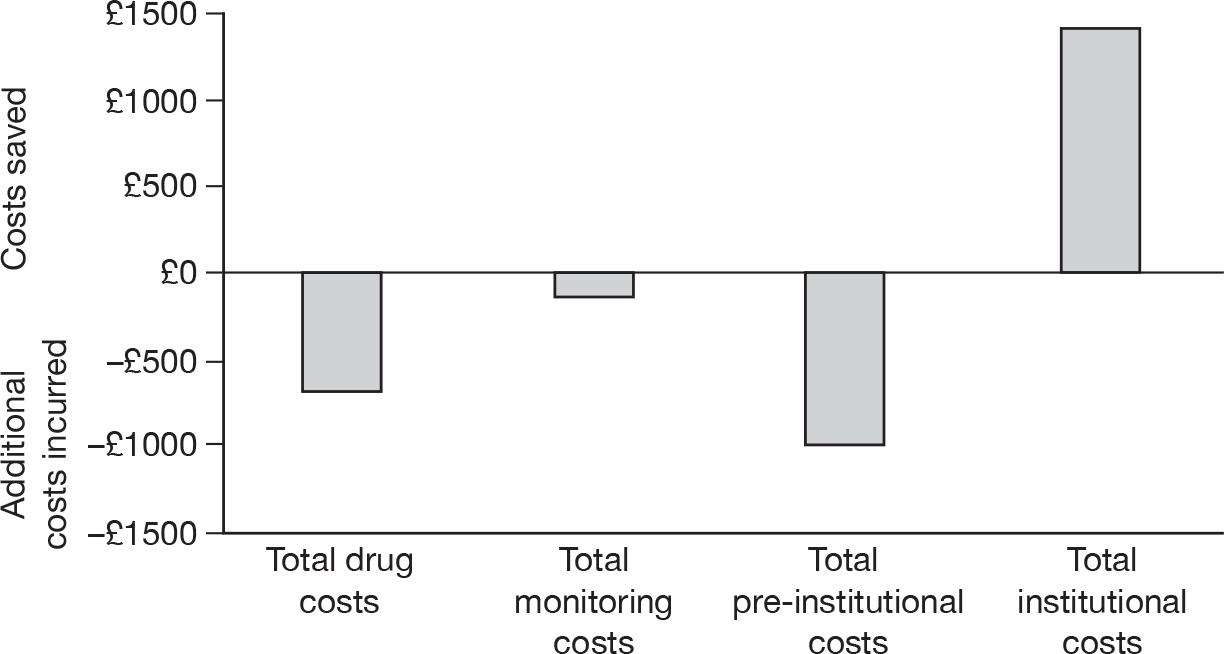
FIGURE 90.
Base-case QALY components of memantine compared with BSC for moderate-to-severe AD.
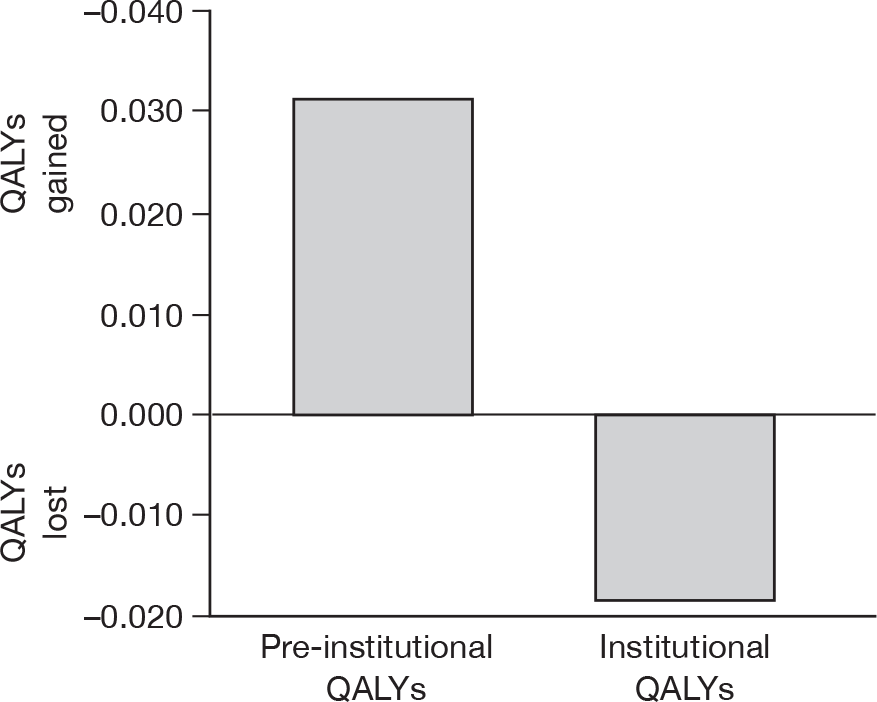
Summary of probabilistic sensitivity analysis and deterministic analysis
As with the AChEIs, there is a great deal of parameter uncertainty in the cost–utility of memantine compared with BSC. However, at a WTP of £30,000 per QALY gained, memantine has a 38% probability of being cost-effective. Only for WTP thresholds > £44,000 per QALY is there a > 50% probability that memantine is the most cost-effective treatment option.
One-way sensitivity analysis
Treatment effect on mortality
Assuming a treatment effect on survival leads to a mean estimate of overall survival of 42.1 months for BSC and 43.7 months for treatment with memantine: an additional 1.7 months of life. It is estimated that treatment with memantine provides an additional 0.049 QALYS compared with BSC over a patient’s lifetime when a treatment effect on survival is assumed. However, these QALY gains cost an additional £3235, leading to a cost per QALY of £65,600 for memantine compared with BSC (Table 125).
| Treatment | Costs (£) | QALYs | Incremental costs (£) | Incremental QALYs | ICERa (£) |
|---|---|---|---|---|---|
| BSC | 78,123 | 1.215 | |||
| Memantine (20 mg) | 81,358 | 1.264 | 3235 | 0.049 | 65,619 |
The assumption of a treatment effect on survival leads to a larger cost per QALY than that estimated in the base-case analysis (ICER of £32,100/QALY). Examination of the cost components in Figure 91 reveals that there are no cost savings associated with memantine over BSC. However, Figure 92 demonstrates that there are QALY gains in both states – pre-institutionalised and institutionalised. This is in contrast with the base-case analysis where there are QALY losses in the institutionalised state (refer back to Figure 90), as longer time is spent in the institutionalised state when a treatment effect on survival is assumed.
FIGURE 91.
Cost components for memantine compared with BSC assuming a treatment effect on survival.
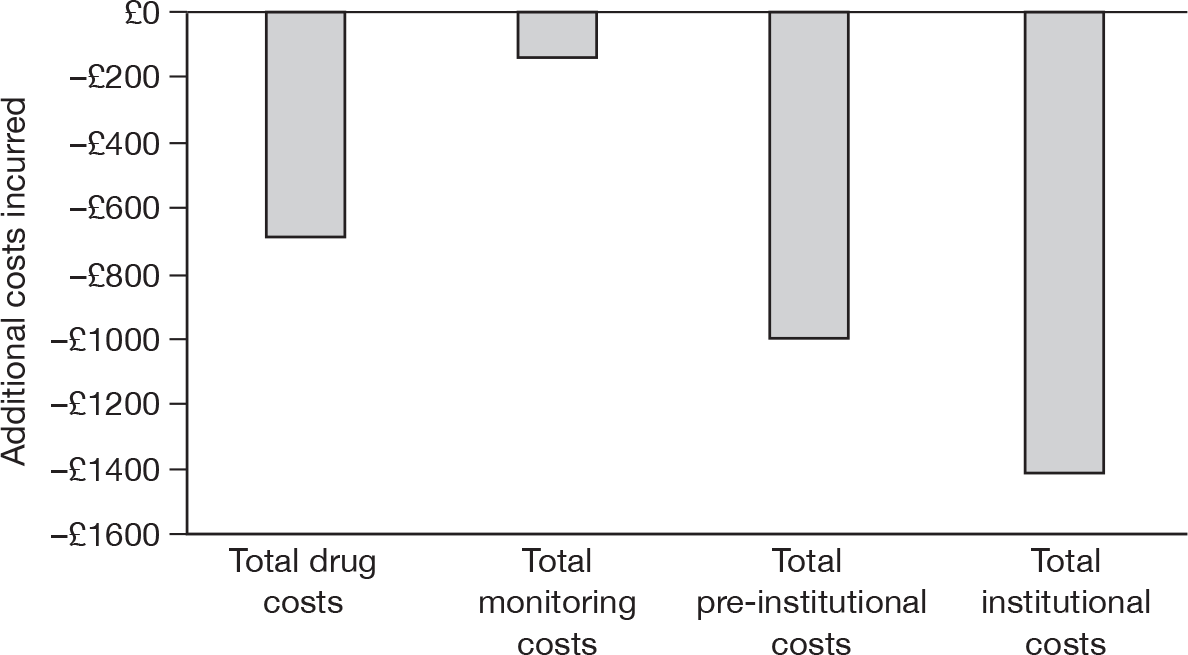
FIGURE 92.
Quality-adjusted life-year components for memantine compared with BSC, assuming a treatment effect on survival.
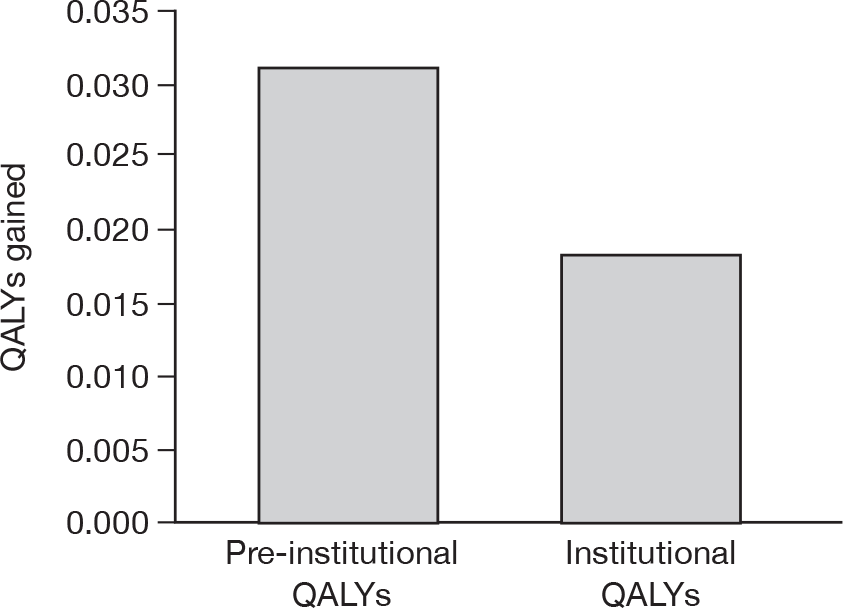
Importance of Mini Mental State Examination effectiveness
As with the AChEIs, an assumption that treatment with memantine impacts only upon MMSE was made (Table 126). This led to an ICER of £79,600 per QALY for memantine compared with BSC. Thus, the effectiveness for MMSE is especially important in the cost-effectiveness of memantine, as with the AChEIs.
| Treatment | Costs (£) | QALYs | Incremental costs (£) | Incremental QALYs | ICERa (£) |
|---|---|---|---|---|---|
| BSC | 78,123 | 1.215 | |||
| Memantine (20 mg) | 78,703 | 1.222 | 579 | 0.007 | 79,600 |
Assuming no treatment effect on MMSE, only an effect on the Barthel ADL Index, gives a much larger ICER for memantine – £122,200/QALY. An additional issue to bear in mind is that the estimate of effectiveness used in this decision model for MMSE in patients treated with memantine is based on just one study, Reisberg and colleagues,142 as the other available study reporting an effect on MMSE included patients who had been treated with AChEIs as well as memantine. 150 However, inclusion of the data from this study reduces the overall effectiveness of memantine on MMSE from 0.7 to 0.5 (see Appendix 7, Figure 148) and increases the ICER to £45,000 (Table 127).
| Treatment | Costs (£) | QALYs | Incremental costs (£) | Incremental QALYs | ICERa (£) |
|---|---|---|---|---|---|
| BSC | 78,123 | 1.215 | |||
| Memantine (20 mg) | 78,597 | 1.225 | 474 | 0.011 | 45,000 |
Further one-way sensitivity analyses
As with the AChELs, a number of one-way sensitivity analyses have been undertaken to assess important assumptions and parameters in the model. The assumptions outlined in Table 122 are applied to the memantine model, except for the assumption of different drug costs and the proportion of the cohort starting the model, in institutional care (Table 128). A tornado plot showing the impact on the cost-effectiveness of changing individual parameters and assumptions is given in Figure 93. Assessment of the cost-effectiveness of memantine for different severity cohorts is described and discussed below (see Exploratory subgroup cost–utility analyses).
| Parameter/assumption | Base case | Deterministic sensitivity analysis | Reference in tornado plots |
|---|---|---|---|
| Drug costs | See Table 113 | Industry cost for memantine; 20-mg cost for memantine | Drug cost |
| Percentage start in institutional care | 40 | 20 | % start inst |
The assumption of a survival effect has a very large impact on the net benefit from the base-case analysis of memantine in moderate-to-severe AD, along with the differing assumptions for treatment discontinuations. Using proxy EQ-5D utilities or patient EQ-5D utilities from Jonsson and colleagues,187 rather than the average EQ-5D, VAS and QoL-AD utilities used in the base case, leads to larger negative net benefits associated with memantine treatment. The pattern of the importance of different assumptions is similar to that for the AChEIs and so the reader is referred to Table 123, which summarises the importance of these assumptions.
Summary of one-way sensitivity analysis
There are many uncertainties in the PenTAG model for treatment with memantine in people with AD. Note that although many of the one-way sensitivity analyses shown in Figure 93 lead to positive net benefits for memantine compared with BSC, a number do lead to negative net benefits. These are:
-
the utility values used (average vs EQ-5D)
-
the estimate of effectiveness on MMSE (0.5 vs 0.7)
-
the assumption of the rate of discontinuations (especially assuming 0%)
-
a possible survival effect from treatment.
FIGURE 93.
One-way sensitivity analyses for the incremental net benefit of memantine compared with BSC.
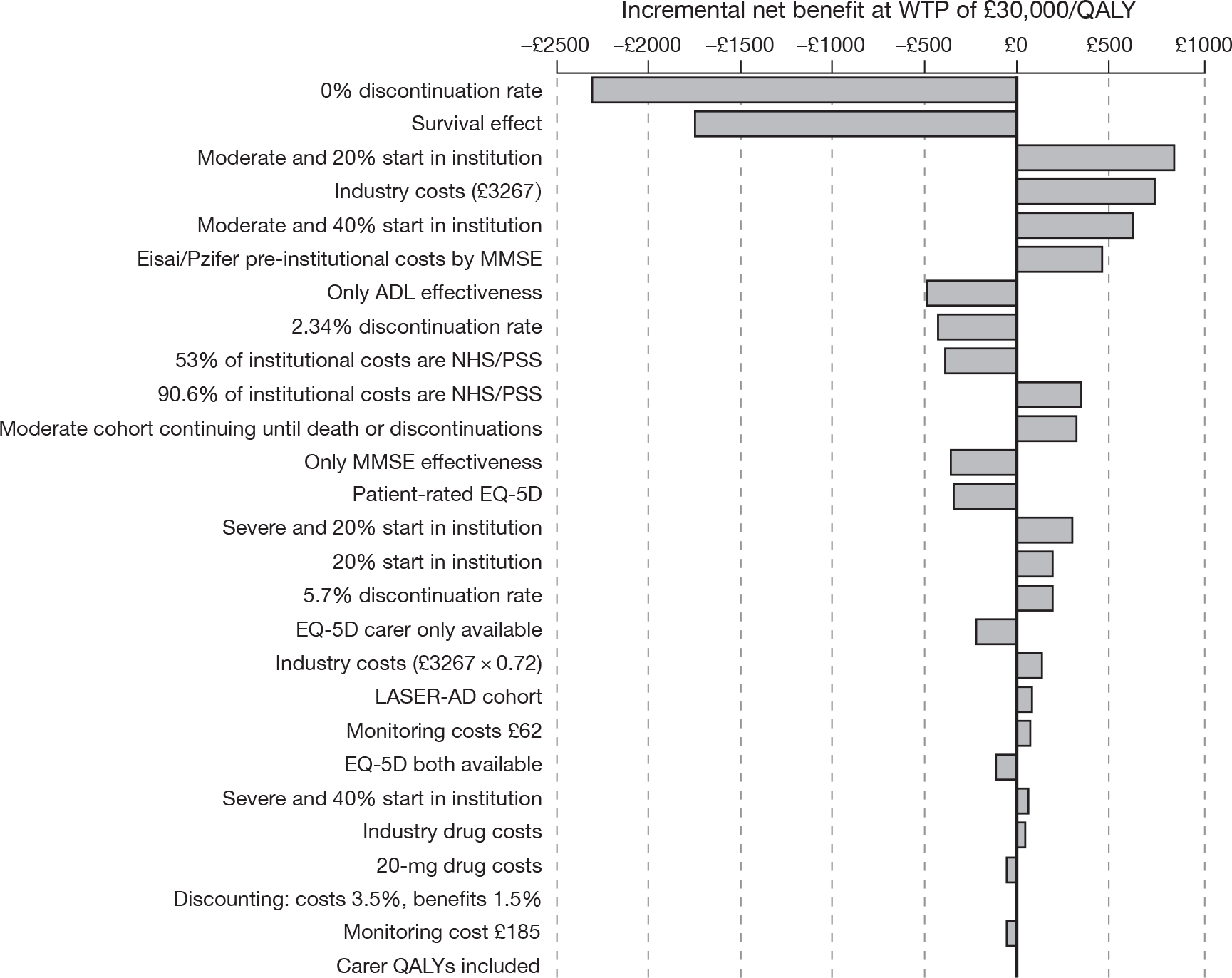
The assumption of a survival effect with treatment has one of the largest impacts on the cost-effectiveness findings. As noted above, there is no direct evidence from RCTs that memantine extends survival; however, memantine does influence the covariates explaining some of the variation in overall survival.
Exploratory subgroup cost–utility analyses
Exploratory subgroup analyses were undertaken to assess:
-
decision problem 1b in Table 102: treatment of mild AD with AChEIs
-
decision problem 1c in Table 102: treatment of moderate AD with AChEIs
-
decision problem 2a in Table 102: treatment of moderate AD with memantine
-
decision problem 3 in Table 102: treatment of moderate AD with AChEIs or memantine.
Caution should be used in the interpretation of these results, as the effectiveness estimates used are not restricted to the severities assessed. That is, they have not been derived from trials that have recruited patients of that disease severity or from trial subgroup analyses. Therefore, the main differences between these analyses and the base-case analyses are the baseline population characteristics. Furthermore, the methods mapping Barthel ADL Index to ADCS-ADL are dependent upon baseline Barthel, therefore differences in Barthel ADL Index effectiveness can be seen for different severity cohorts, given the same treatment effect on ADCS-ADL (see Clinical effectiveness). This explains the finding that treatment with memantine in the moderate-to-severe cohort has a larger ICER than treatment of a severe cohort. Additionally, the methods used to incorporate the treatment effects into the decision model induce an effect by severity. As severity increases, the MMSE coefficient for delaying time to institutionalisation decreases, whereas the Barthel coefficient increases (see State occupancy for treated cohorts).
Treatment of mild Alzheimer’s disease (decision problem 1b)
The results of an exploratory CUA of AChEIs for a cohort of people starting the model with mild AD are presented in Table 129. In this analysis a cohort with mild AD starts the model and continues treatment until they enter institutional care or discontinue for other reasons. The results indicate that donepezil is estimated to dominate all other treatment options, including BSC. Furthermore, all drugs dominate BSC.
| Treatment | Costs (£) | QALYs | Incremental costs (£) | Incremental QALYs | ICER |
|---|---|---|---|---|---|
| Donepezil (10 mg) | 74,919 | 1.784 | |||
| Galantamine (16–24 mg) | 74,922 | 1.781 | Dominated | ||
| Rivastigmine patch (9.5 mg/day) | 74,928 | 1.780 | Dominated | ||
| Rivastigmine capsules (9–12 mg) | 74,979 | 1.778 | Dominated | ||
| BSC | 75,470 | 1.750 | Dominated |
Treatment of moderate Alzheimer’s disease (decision problems 1c and 3)
The results of an exploratory CUA of AChEIs or memantine for a cohort of people starting the model with moderate AD are presented in Table 130. In this analysis it is assumed that 10% of the cohort start in institutional care and that treatment with all drugs stops once patients enter institutional care or discontinue for other reasons. Memantine is dominated and so the results presented in Table 130 address both decision problems 1c and 3. The results indicate that all treatment options are dominated by donepezil. Total costs and QALYs are smaller for the moderate than the mild group (compare with Table 129), as survival is lower and disease severity is greater for the moderate cohort. Also note that all drugs dominate BSC.
| Treatment | Costs (£) | QALYs | Incremental costs (£) | Incremental QALYs | ICERa,b (£) |
|---|---|---|---|---|---|
| Galantamine (16–24 mg) | 66,847 | 1.533 | |||
| Rivastigmine patch (9.5 mg/day) | 66,853 | 1.533 | Dominated | ||
| Donepezil (10 mg) | 66,896 | 1.535 | 49 | 0.001 | 35,300 |
| Rivastigmine | 66,948 | 1.529 | Dominated | ||
| Memantine (15–20 mg) | 67,249 | 1.523 | Dominated | ||
| BSC | 67,517 | 1.500 | Dominated |
Treatment of severe Alzheimer’s disease (decision problem 2b)
The results of an exploratory CUA of memantine for a cohort of people starting the model with severe AD are presented in Table 131. In this analysis it is assumed that individuals continue treatment until they die or discontinue for other reasons. The resultant ICER of £26,500 per QALY is slightly lower than that for the cohort of people with moderate-to-severe AD (base-case analysis – see Table 124). This is because there are lower incremental costs associated with treatment with memantine in the severe cohort than in the moderate-to-severe cohort, but the QALYs gained are the same (0.013). The data informing the effectiveness of memantine in this severe cohort is from a trial where the participant population ranged from moderate-to-severe AD. Therefore, these results should be treated with caution, as with the results presented in Tables 129 and 130.
| Treatment | Costs (£) | QALYs | Incremental costs (£) | Incremental QALYs | ICERa (£) |
|---|---|---|---|---|---|
| BSC | 67,988 | 1.012 | |||
| Memantine (15–20 mg) | 68,342 | 1.025 | 354 | 0.013 | 26,500 |
Summary of cost-effectiveness findings
The cost–utility results for AChEIs in people with mild-to-moderate AD [see Mild-to-moderate Alzheimer’s disease: cholinesterase inhibitors (decision problem 1a)] and memantine in people with moderate-to-severe AD [see Moderate-to-severe Alzheimer’s disease: memantine (decision problem 2a)] indicate a great deal of uncertainty, only some of which is expressed in the PSA. Nevertheless, when considering the AChEIs, there is > 99% probability that BSC is not the most cost-effective treatment option at a WTP of £30,000 per QALY for the base-case analysis. However, this analysis does not account for uncertainty as to whether or not the treatment impacts upon the survival of AD patients. If this is assumed, the AChEIs no longer dominate BSC, and ICERs for the AChEIs are approximately £37,000.
The probability that memantine is cost-effective in a moderate-to-severe cohort compared with BSC (see Moderate to severe Alzheimer’s disease: memantine (decision problem 2a)] at a WTP of £30,000 per QALY is 38% (and 28% at a WTP of £20,000 per QALY). Above a WTP of around £44,000/QALY, the probability of memantine being more cost-effective than BSC is > 50% and this increases as the WTP threshold increases.
Base-case deterministic and probabilistic ICERs for treating mild-to-moderate and moderate-to-severe AD are presented in Table 132. Note that the incremental costs and QALYs estimated in the PenTAG model are very small compared with those often reported in MTAs and single technology assessments (STAs) (Figure 94).
| Treatment | Severity of AD | |||
|---|---|---|---|---|
| Mild to moderate | Moderate to severe | |||
| Deterministic | Probabilistic | Deterministic | Probabilistic | |
| Galantamine (16–24 mg) | Dominated | Dominated | N/A | N/A |
| Rivastigmine patches (9.5 mg/day) | N/A | N/A | ||
| Donepezil (10 mg) | £17,900a | £23,500a | N/A | N/A |
| Rivastigmine capsules (9–12 mg) | Dominated | Dominated | N/A | N/A |
| BSC | Dominated | Dominated | ||
| Memantine (15–20 mg) | NA | NA | £32,100b | £36,700b |
FIGURE 94.
Cost-effectiveness plane for the base-case estimate for rivastigmine patches in the current CUA and base-case estimates from recent MTAs and STAs. Source: www.nice.org.uk (cited June 11 2010).
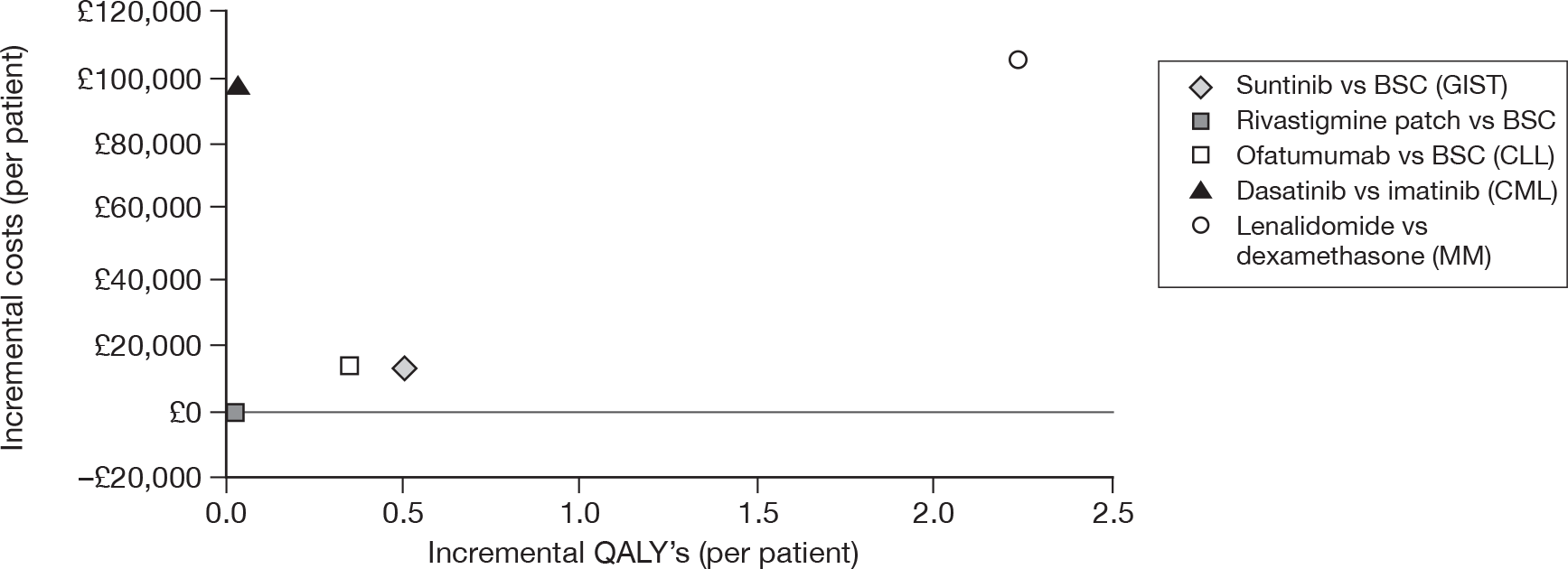
Comparison of the Peninsula Technology Assessment Group’s model with the Southampton Health Technology Assessment Centre’s model
In Table 133 the cost–utility estimates of the AChEIs provided by the PenTAG model (first column of results) and those published in the Health Technology Assessment journal151 by SHTAC in the previous assessment (second column of results). As can be seen, these estimates are very different to each other.
| Treatment | PenTAG model | SHTAC model | ||
|---|---|---|---|---|
| Deterministic base-case results | As reported in the Health Technology Assessment journal149 (£) | 3.5% discount rates; probabilities as probabilities 20-year time horizon (£) | As previous column plus discontinuations and PenTAG effectiveness and cost estimates (£) | |
| Donepezil (10 mg) | Dominates BSC | 80,900 | 66,500 | 33,400 |
| Galantamine (16–24 mg) | 68,000 | 55,000 | 27,600 | |
| Rivastigmine capsules (9–12 mg) | 58,000 | 46,100 | 50,400 | |
| Rivastigmine patches (9.5 mg/day)b | ||||
We have gone some way to obtain estimates from the SHTAC model to account for current discount rates (3.5% for both costs and benefits), a longer time horizon (20 years as in the PenTAG model) and have amended the SHTAC model so that all probabilities are represented as probabilities (and not hazards as done in the previous review). These updated estimates are also shown in Table 133 (third column of results), alongside further updated estimates from the SHTAC model (last column) for the inclusion of treatment discontinuations and updated effectiveness and cost parameters. Table 134 provides a comparison of effectiveness and cost parameter inputs between the SHTAC and PenTAG models. The results from the SHTAC model using current cost and effectiveness estimates (final column) are lower than those reported in the previous HTA, but still greater than those estimated in the PenTAG model. To help explain these differences a comparison of outputs for the cost-effectiveness of donepezil from the PenTAG model and the updated SHTAC model are given in Table 135.
| Parameter | SHTAC 2004 value | PenTAG 2010 value |
|---|---|---|
| ADAS-cog effectiveness | ||
| Donepezil | 3.01 | 2.90 |
| Rivastigmine | 3.08 | 2.34 |
| Galantamine | 3.28 | 3.05 |
| Monthly drug costs (£) | ||
| Donepezil | 97 | 97 |
| Rivastigmine | 74 | 98 |
| Galantamine | 91 | 83 |
| 6-monthly monitoring visit cost | 108 | 158 |
| Monthly pre-FTC/inst cost | 328 | 2051 |
| Monthly FTC/inst cost | 937 | 2117 (£2941 × 72%) |
| Output | Treatment | Model outputs | Incremental values | ||
|---|---|---|---|---|---|
| Updated SHTACb | PenTAG | Updated SHTACb | PenTAG | ||
| ICER | 33,359 | Donepezil dominates | |||
| Total costs (£) | Donepezil | 140,456 | 69,624 | ||
| No treatment | 139,095 | 70,212 | 1361 | –588 | |
| Total QALYs | Donepezil | 2.513 | 1.619 | ||
| No treatment | 2.472 | 1.584 | 0.041 | 0.035 | |
| Undiscounted total life-years | 6.500 | 3.84 | |||
| Undiscounted life-years in community | Donepezil | 2.581 | 2.539 | ||
| No treatment | 2.403 | 2.401 | 0.178 | 0.138 | |
| Undiscounted years in institutional care | Donepezil | 3.916 | 1.297 | ||
| No treatment | 4.094 | 1.436 | –0.178 | –0.138 | |
| Total drug costs (£) | 1169 | 790 | |||
| Total monitoring costs (£) | Donepezil | 318 | 215 | ||
| No treatment | 0 | 0 | 318 | 215 | |
| Total pre-inst costs (£) | Donepezil | 59,432 | 41,298 | ||
| No treatment | 55,568 | 39,360 | 3864 | 1938 | |
| Total inst costs (£) | Donepezil | 79,538 | 27,321 | ||
| No treatment | 83,527 | 30,282 | –3989 | –2961 | |
The updated SHTAC model estimates greater total costs and greater total QALYs than the PenTAG model. This is largely because of overall survival being almost twice as large in the updated SHTAC model compared with the PenTAG model (6.5 vs 3.8 years). A slightly longer delay to FTC/institutionalisation is estimated by the updated SHTAC compared with the PenTAG model (0.18 vs 0.14 years), and this is reflected in the greater incremental QALYs associated with the updated SHTAC model than with the PenTAG model (0.041 vs 0.035). However, it is the differences in the incremental costs that lead to the updated SHTAC model having an ICER of £33,400 per QALY compared with the PenTAG model, which estimates that donepezil dominates BSC.
To help explain these differences it is important to consider two further structural differences between the updated SHTAC and the PenTAG models. First, the updated SHTAC model assumes that all individuals start in the pre-FTC state, whereas the PenTAG model assumes that 10% of the cohort start the model in the institutionalised state. Thus, greater total treatment and monitoring costs are estimated in the SHTAC model compared with the PenTAG model. Second, the PenTAG model allows pre-institutionalised costs to depend upon severity, whereas the cost of pre-FTC is fixed regardless of severity in the SHTAC model. This leads to quite different average pre-institutionalised costs per person estimated by the models: £23,000 (£59,432/2.581) for donepezil-treated patients in the updated SHAC model compared with £16,300 (£41,298/2.539) for donepezil-treated patients in the PenTAG model. As the cost of institutional/FTC is the same for both models, the PenTAG model will lead to more savings than the SHTAC model, as delaying institutionalisation leads to greater institutional costs saved in the PenTAG model.
The SHTAC and PenTAG models use data from two different studies to predict institutionalisation/FTC and differences between these studies are noted in Table 136. Even though the estimated difference in delay to institutionalisation/FTC is not large between the models, it is interesting to note that the SHTAC model incorporated an effect on only cognition, through the ADAS-cog, whereas the PenTAG model incorporated an effect on cognition (MMSE) and function (Barthel ADL Index). Neither model incorporated a treatment effect on behaviour. For comparison, details on the LASER-AD study192 used by Lundbeck in their submission are also shown in Table 136.
| Study characteristic | Stern et al.193 data set | Wolstenholme et al.196 data set | Livingston et al.192 data set |
|---|---|---|---|
| Geographical setting | USA: New York, Baltimore and Boston | UK: Oxfordshire | UK: North London and Essex |
| Event definition | Requiring FTC; ‘equivalent institutional care’ | Institutionalisation | Entering 24-hour care |
| AD sample size | 236 | 92 | 224 |
| Available care-cost estimates | No | Yes | Yes |
| Start data collection | Not reported | 1988/9 | Not reported |
| Length of follow-up (years) | Up to 7 | Up to 11 | Up to 4.5 |
| Predictors of time to event (and stat significance) | Modified MMSE (p < 0.1); psychosis (p < 0.1) ; age at onset (p < 0.1); extrapyramidal symptoms (p < 0.1); duration of illness (p < 0.1) |
MMSE (ns) Barthel ADL Index (ns) Age (p = 0.0009) |
MMSE (p = 0.001); hours spend caring (p = –0.03); level of education (p = 0.004); relationship to carer (partner vs family p = 0.04; partner vs paid carer, p = 0.001; family vs paid carer, p = 0.001) |
| Mean age at study entry (years) | 73.1 (SD 8.9) | 78.1 (SD 6.9) | 81 (SD 7.4) |
| Average severity at study entry | Mild at study entry (MMSE > 15) | MMSE = 14.4 (SD 6.7) |
30% MMSE < 15 40% 14 < MMSE < 20 30% MMSE > 19 |
| Time since onset/diagnosis (years) | Average time since onset (not diagnosis): 3.9 | Average time since diagnosis: 4.9 | Unclear |
Comparison of the Peninsula Technology Assessment Group’s model with industry models
Eisai/Pfizer versus Peninsula Technology Assessment Group models: donepezil
The Eisai/Pfizer model structure and simulation method is very different to the structure of the PenTAG model. To allow comparison between the base-case results of the two models, outputs from these models for a moderate cohort are presented in Table 137, and those for a mild cohort are presented in Table 138. Note that in the Eisai/Pfizer base-case analysis, carer’s utility is included alongside patient utility. Only patient utility is included in the PenTAG base-case model. Also note that the Eisai/Pfizer model predicts a shorter survival for the mild cohort than for the moderate cohort. This inconsistency was noted by the DSU in Chapter 5 [see Eisai/Pfizer (donepezil): critique of economic submission]. The results from the models of Eisai/Pfizer and PenTAG for the cost-effectiveness of donepezil in a mild cohort and a moderate cohort estimate that donepezil is less costly and more effective than BSC.
| Output | Treatment | Model outputs | Incremental values | ||
|---|---|---|---|---|---|
| Eisai/Pfizer | PenTAG | Eisai/Pfizer | PenTAG | ||
| ICER | Donepezil dominates | Donepezil dominates | |||
| Total costs (£) | Donepezil | 102,086 | 66,896 | ||
| No treatment | 103,969 | 67,517 | –1883 | –621 | |
| Total QALYs | Donepezil | 4.353 (patient + carer)b | 1.535 | ||
| No treatment | 4.245 (patient + carer)b | 1.500 | 0.108 | 0.035 | |
| Undiscounted total life-years | 4.603 | 3.633 | |||
| Undiscounted life-years in community | Donepezil | 1.852 | 2.418 | ||
| No treatment | 1.685 | 2.276 | 0.167 | 0.142 | |
| Undiscounted years in institutional care | Donepezil | 2.751 | 1.215 | ||
| No treatment | 2.918 | 1.357 | –0.167 | –0.142 | |
| Mean treatment duration (years) | 1.89 | 0.67 | |||
| Total drug costs (£) | 1973 | 780 | |||
| Total monitoring costs (£) | Donepezil | 208 | 212 | ||
| No treatment | 0 | 0 | 208 | 212 | |
| Total pre-inst costs (£) | Donepezil | 39,201 | 40,135 | ||
| No treatment | 37,413 | 38,690 | 1788 | 1445 | |
| Total inst costs (£) | Donepezil | 60,705 | 25,769 | ||
| No treatment | 66,556 | 28,827 | 5851 | –3058 | |
For a moderate cohort (see Table 137), total costs are greater in the Eisai/Pfizer model compared with the PenTAG model. This is because of the greater total survival estimated in the Eisai/Pfizer model than in the PenTAG model (4.6 vs 3.6 years). In particular, the higher total costs from the Eisai/Pfizer model are a result of the greater time spent in institutional care and the greater costs associated with institutional care assumed by Eisai/Pfizer.
Total QALYs are also estimated to be greater from the Eisai/Pfizer model compared with the PenTAG model; however, Eisai/Pfizer include both patient and carer QALYs in their base case (whereas the PenTAG base-case model includes only patient QALYs). Comparison of total patient QALYs only indicates that the average utility for patients is higher in the PenTAG model than in the Eisai/Pfizer model. This is because greater total QALYs are estimated in the PenTAG model, yet survival is lower in the PenTAG model [the estimated average utility for patients treated with donepezil is 0.29 (1.332/4.603) from the Eisai/Pfizer model and 0.42 (1.535/3.633) from the PenTAG model]. It is not possible to disaggregate these utilities to those attributed to pre-institutional or institutional in the Eisai/Pfizer model.
Although both models suggest that donepezil costs less and is more effective in the base-case analyses, the cost savings and QALY gains estimated from the Eisai/Pfizer model are about three times greater than those estimated from the PenTAG model. The greater delay to institutionalisation in the Eisai/Pfizer model compared with the PenTAG model (0.167 vs 0.142 years) explains some of these differences in the incremental costs and QALYs. However, there is a greater than expected difference between the institutional costs of donepezil-treated patients and BSC patients in the Eisai/Pfizer model, which cannot be explained.
Other differences between the two models are the cost inputs, particularly the NHS/PSS care costs in the community and in institutional care. The Eisai/Pfizer pre-institutionalised care costs are reported by MMSE, whereas the PenTAG pre-institutionalised care costs are calculated by time to institutionalisation. Using the equation described in Chapter 6 (see Quality of life of the individual with Alzheimer’s disease) to relate MMSE to time to institutionalisation, it is possible to compare the community-living costs from each model defined by MMSE. As can be seen clearly from Figure 95, the community costs assumed in the PenTAG model are much larger than the assumed Eisai/Pfizer costs and allow for more change in costs as individuals progress over time. However, there is some concern that the community costs used by Eisai/Pfizer have not been appropriately translated from the CDR scale to the MMSE scale.
FIGURE 95.
Monthly NHS/PSS costs by MMSE for individuals living in the community from the Eisai/Pfizer and the PenTAG models.
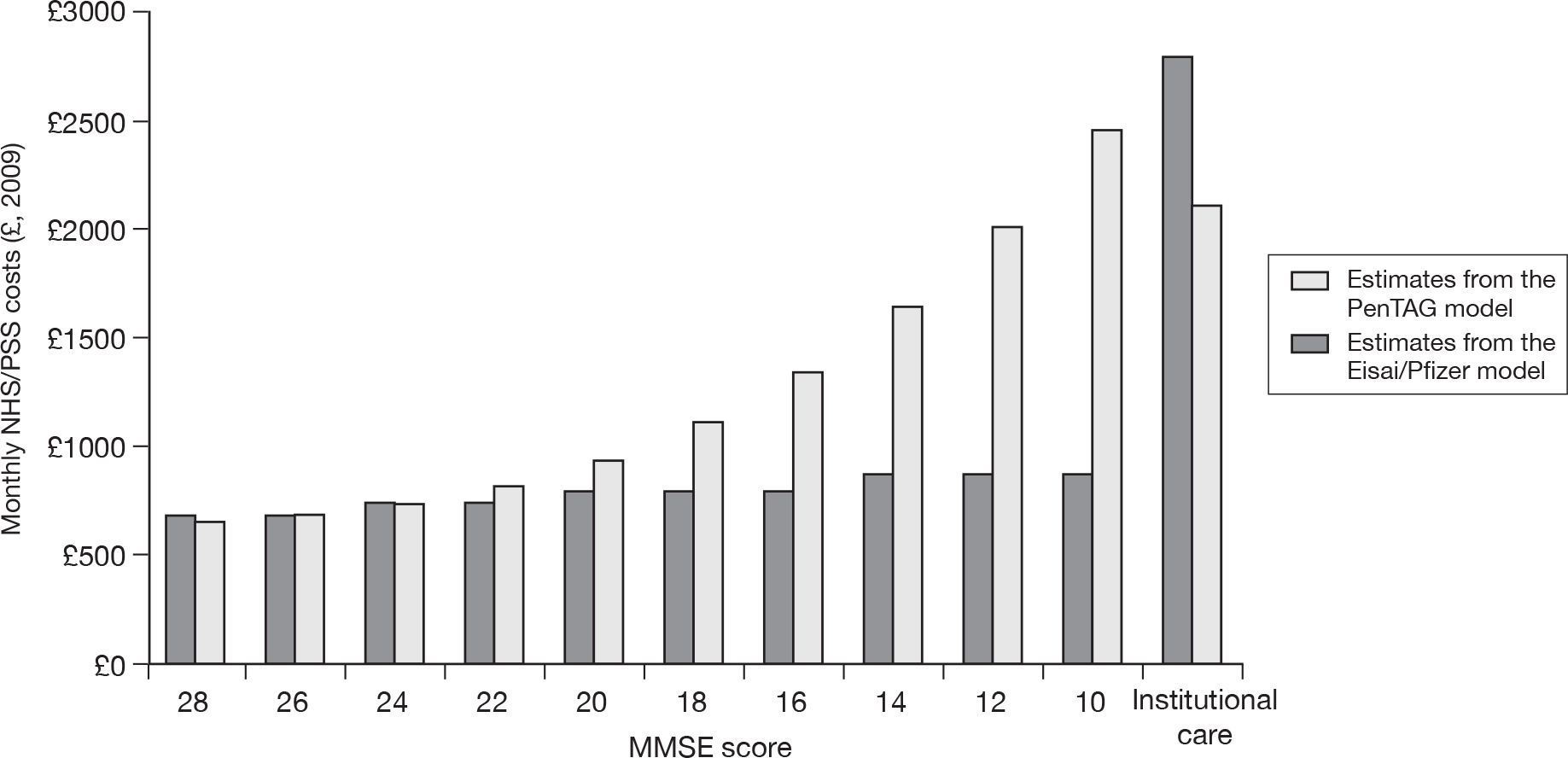
In their industry submission, Eisai/Pfizer cite Knapp and colleagues,8 who report the annual cost of community care by CDR. To obtain their cost estimates Eisai/Pfizer have assumed that mild on the CDR scale is equivalent to MMSE > 25, moderate on the CDR scale is equivalent to MMSE > 15 and MMSE < 20, and severe on the CDR scale is equivalent to MMSE < 10. To calculate community costs of care for the remaining severities defined by Eisai/Pfizer, 19 < MMSE < 26 and < 9 MMSE < 16, they have interpolated the mean cost values from the adjacent severities. Perneczky and colleagues215 indicate that the severities defined by CDR mild, moderate and severe are a good approximation to the MMSE severities of mild (MMSE 25–21), moderate (MMSE 20–11) and severe (MMSE 10–0). These approximations do not relate as expected to the Eisai/Pfizer cost estimates and there is no indication of any other published evidence used by Eisai/Pfizer to approximate CDR and MMSE scores.
The monthly costs of institutional care with the Eisai/Pfizer model assume costs of £2801 per month, whereas the PenTAG model assumes costs of £2117 per month (accounting for the 28% of institutional costs assumed to be privately funded). Again, there is concern that the monthly costs for institutional care in the Eisai/Pfizer model are not solely costs funded by the NHS/PSS. Adjusting the cost of institutional care in the Eisai/Pfizer model to that used in the PenTAG leads to reduced cost savings associated with donepezil (£588 compared with £1883 in the base case), thus identifying this cost as an important factor between the models.
In both models a 6-monthly outpatient monitoring visit is accounted for. Eisai/Pfizer describe this as a geriatrician visit, although it is described as a consultant-led outpatient visit for the PenTAG model. The cost for this monitoring visit is very different between the Eisai/Pfizer and PenTAG models: £62 from Eisai/Pfizer versus £158 from PenTAG. Both costs are cited from the National Schedule of Reference Costs (service code 430 Geriatric Medicine), with the Eisai/Pfizer cost cited as 2007–8 and the PenTAG cost as 2008–9. There are also slight differences in the daily drug costs for 10 mg donepezil between the two models. The Eisai/Pfizer submission reports a daily cost of £3 from the NHS drug tariff, compared with £3.18 used in the PenTAG model from the BNF 58. However, given the magnitude of the pre-institutional and institutional care costs, these differences in monitoring and drug costs are unlikely to explain further differences between the results of the Eisai/Pfizer and PenTAG models.
A similar pattern in the cost-effectiveness of donepezil between the Eisai/Pfizer and PenTAG models is seen for the mild cohort (Table 138). However, the delay to institutionalisation due to treatment with donepezil estimated in the Eisai/Pfizer model is almost twice as large as that estimated by the PenTAG model. Nevertheless, the unexplained greater than expected incremental institutional costs estimated in the Eisai/Pfizer model are also a feature of the model for the mild cohort.
| Output | Treatment | Model outputs | Incremental values | ||
|---|---|---|---|---|---|
| Eisai/Pfizer | PenTAG | Eisai/Pfizer | PenTAG | ||
| ICER | Donepezil dominates | Donepezil dominates | |||
| Total costs (£) | Donepezil | 79,023 | 74,919 | ||
| No treatment | 82,409 | 75,470 | –3386 | –552 | |
| Total QALYs | Donepezil | 4.267 (patient + carer)b | 1.784 | ||
| No treatment | 4.120 (patient + carer)b | 1.750 | 0.147 | 0.034 | |
| Undiscounted total life-years | 4.110 | 4.243 | |||
| Undiscounted life-years in community | Donepezil | 2.161 | 2.777 | ||
| No treatment | 1.926 | 2.642 | 0.235 | 0.135 | |
| Undiscounted years in institutional care | Donepezil | 1.949 | 1.466 | ||
| No treatment | 2.184 | 1.600 | –0.235 | –0.135 | |
| Mean treatment duration (years) | 2.23 | 0.69 | |||
| Total drug costs (£) | 2281 | 807 | |||
| Total monitoring costs (£) | Donepezil | 240 | 220 | ||
| No treatment | 0 | 0 | 240 | 220 | |
| Total pre-inst costs (£) | Donepezil | 37,938 | 43,427 | ||
| No treatment | 37,128 | 42,160 | 810 | 1267 | |
| Total inst costs (£) | Donepezil | 38,564 | 30,465 | ||
| No treatment | 45,282 | 33,310 | –6718 | –2845 | |
Lundbeck versus Peninsula Technology Assessment Group models: memantine
Although the structures of the Lundbeck and PenTAG models are similar, different conclusions are reached in the base-case analyses: the Lundbeck model estimates memantine to be less costly and more effective than the comparator, whereas the PenTAG model estimates an ICER of £32,100 per QALY gained (Table 139). Outputs from both models are shown in Table 140 for comparison. Greater total costs and QALYs are estimated by the Lundbeck model, which also estimates slightly longer overall survival (3.7 vs 3.5 years). However, time spent in institutional care is almost identical between the two models (1.97 years for memantine-treated individuals; for non-memantine-treated individuals it is 2.05 years from the Lundbeck model and 2.03 years from the PenTAG model).The Lundbeck model estimates a greater delay to institutionalisation for those treated with memantine than in the PenTAG model (0.08 vs 0.065 years); hence, larger incremental costs and QALYs are estimated in the Lundbeck model.
| Item | Lundbeck model | PenTAG model |
|---|---|---|
| Base-case estimate | Memantine dominates | ICER of £32,100 per QALY |
| Modelling assumption | ||
| Definition of alive states | Pre-FTC/FTCa | Pre-institutionalisation/institutionalisationb |
| Data describing AD progress | LASER-AD study192 | IPD from Wolstenholme et al.196 |
| Severity at start of model | Moderate to severe | Moderate to severe |
| Treatment stopping rules | Stop when in FTC | Continue until death or treatment discontinuations |
| Parameter value | ||
| Monthly pre-FTC/institutionalised care costs (£) | 724 | Dependent on severity, > 724 |
| Monthly FTC/institutional care costs (£) | 3201 | 2117 |
| Monthly memantine drug costs (£) | 64.80 | 71.28 |
| Output | Treatment | Model outputs | Incremental values | ||
|---|---|---|---|---|---|
| Lundbeck | PenTAG | Lundbeck | PenTAG | ||
| ICER (£) | Memantine dominates | 32,084 | |||
| Total costs (£) | Memantine | 93,076 | 78,528 | ||
| No treatment | 94,787 | 78,123 | –1711 | £405 | |
| Total QALYs | Memantine | 1.533 | 1.227 | ||
| No treatment | 1.502 | 1.215 | 0.031 | 0.013 | |
| Total pre-inst/FTC QALYs | Memantine | 0.870 | 0.665 | ||
| No treatment | 0.813 | 0.634 | 0.057 | 0.031 | |
| Total inst/FTC QALYs | Memantine | 0.661 | 0.562 | ||
| No treatment | 0.690 | 0.581 | –0.029 | –0.018 | |
| Expected overall survival (years) | 3.7 | 3.5 | |||
| Expected time to FTC/institutional care (years) | Memantine | 1.73 | 1.538 | ||
| No treatment | 1.65 | 1.473 | 0.08 | 0.065 | |
| Time in FTC/institutional care | Memantine | 1.97 | 1.966 | ||
| No treatment | 2.05 | 2.032 | –0.08 | –0.065 | |
| Mean treatment duration | 1.73 | 0.79 | |||
| Total drug costs (£) | 1348 | 678 | |||
| Total monitoring costs (£) | Memantine | 106 | 140 | ||
| No treatment | 0 | 0 | 106 | 140 | |
| Total pre-inst costs (£) | Memantine | 16,642 | 34,413 | ||
| No treatment | 14,324 | 33,414 | 2318 | 999 | |
| Total inst costs (£) | Memantine | 77,133 | 43,298 | ||
| No treatment | 80,464 | 44,710 | –3331 | –1412 | |
A further difference between the models is that the average cost of care pre-institutionalised, as estimated by the models, is much lower in the Lundbeck model than in the PenTAG model, yet the institutionalised cost of care in the Lundbeck model is greater than in the PenTAG model (see parameter values in Table 139). Thus, greater costs are saved by delaying institutionalisation in the Lundbeck model than in the PenTAG model. This is seen in the larger incremental total costs between memantine-treated patients and those not treated with memantine in the Lundbeck model compared with the PenTAG model.
A further one-way sensitivity analysis of the Lundbeck model, assuming 28% of the FTC cost of £3201 from the Lundbeck model, led to a reduction in the total costs saved with memantine treatment to £521, yet memantine remained dominant. Assuming £2117 for FTC costs in the Lundbeck model (as assumed in the PenTAG model) also led to a reduction in the total costs saved with treatment, just £210, but, again, memantine remained dominant.
Chapter 7 Other factors relevant to the NHS
The care and treatment of people with AD is complex and goes beyond the patient themselves to include carers to a degree not seen in many other conditions. The extra burden to the NHS, social care services and the economy posed by the ill health of carers because of suboptimal service provision for Alzheimer’s patients is unknown, but must be considerable and growing. Unfortunately, none of the trials included in the clinical effectiveness systematic review measured the effects of AD on carers.
With respect to the economic evaluation, many of the factors that would often be mentioned as ‘other factors’ in this section, such as impact on carers, have already been highlighted in previous appraisals and directly considered in the modelling exercises. Such themes have been further pursued in the analysis in this report and are thus not mentioned here. Taking a wider societal perspective in the economic analyses is an issue that has been raised previously in relation to this topic. The reasons why such a broad perspective is not appropriate for the decisions made by NICE have already been clearly expressed and tested. To be consistent with this the main focus of analyses in this report has been from a NHS and PSS perspective. This is not to deny the value of taking a wider perspective in the context of other decisions outside NICE.
Chapter 8 Discussion
Statement of principal findings
Aim
The remit for this report has been to update the evidence used to inform the last NICE guidance on donepezil, galantamine, rivastigmine and memantine for the treatment of AD, particularly as laid out in the report by SHTAC. In general, they considered evidence up to 2004, and this is the start date that we have used for this report.
In this section we will not re-state the previous evidence, but assume that it will be read in the context of the previous evidence summaries and the decisions that flowed from them. Similarly, the conclusions will focus on whether or not the new evidence on effectiveness and cost-effectiveness is likely to change the current guidance. A complete re-examination of all the available evidence from scratch was beyond the scope of this report.
Effectiveness review
In the previous assessment report in 2004, there was evidence for the effectiveness of donepezil, galantamine and rivastigmine on improving cognition, function, behaviour and global impact over the short term and evidence on the effectiveness of memantine was much more uncertain. Important gaps in the evidence were identified concerning long-term outcomes, impact on QoL, carers and time to institutionalisation.
Overall, we found that although more evidence has accumulated over the last 6 years, its impact on conclusions about effectiveness appears small. An enduring problem is that of trying to predict what will happen to people over the course of 5 years or more on the basis of 6 months or less information. The quality of many of the recent trials is a contributing factor to this; some good-quality trials have been conducted, but most of the new studies were of moderate-to-poor quality. A particular criticism is the use of LOCF and OC methods to account for missing data; these methods are inappropriate in a condition that naturally declines to death and may lead to an overestimation of the treatment effect. Methods of randomisation and allocation concealment were frequently not reported.
In total, 17 new RCTs were included in the clinical effectiveness systematic review: there were 12 pair-wise comparisons with placebo (donepezil 5, n = 234; galantamine 3, n = 1386; rivastigmine 3, n = 1995 and memantine 1, n = 350); four head-to-head studies and one combination therapy study (memantine added to AChEIs) were also found. The amount of evidence for these treatments has thus increased and has particularly consolidated the evidence on effectiveness of galantamine and rivastigmine relative to placebo. Evidence on the effectiveness of memantine does not appear to have been greatly strengthened. None of the gaps in evidence noted previously has been closed by new RCTs and no new evidence has emerged on differential effectiveness by subgroup, particularly disease severity and there is no evidence that these treatments increase longevity. Concerning comparative research, although there is one good-quality new head-to-head trial, comparing donepezil and rivastigmine, showing results for rivastigmine on functional and global outcomes were significantly better than those for donepezil, more generally the case for one AChEI being more effective than another remains unconvincing. Our view, overall, is that these drugs should be treated as a class. The evidence about memantine hinged on two trials (one new and one from the previous review). The new study did not find any significant gain from memantine on any outcome. Although, pooling of these data with the previous review showed some inconsistent, partly positive, evidence on cognitive, functional and global outcomes.
In 2004 the assessment group found that donepezil improved cognitive and global outcomes, with increased benefit from higher doses; in some cases this benefit was maintained over a year. There was weaker evidence for a significant effect with functional and behavioural outcomes. The current systematic review found five small poor-quality studies that have added to the evidence base. They had a maximum of 6 months’ follow-up. All studies measured cognitive outcomes. A dose-related beneficial effect was found at 10 mg/day. One study measured functional and global outcomes, but it was of such poor quality that the positive findings lack credibility.
We found an additional three variable-quality RCTs of galantamine versus placebo to add to the evidence base of six studies included in 2004. The previous review found a dose–response relationship for cognitive, functional and global outcomes. In the two trials reporting behavioural outcomes, one found a significant gain, the other did not. The studies included in our review all found significant benefit on cognitive outcomes; the results for functional and global outcomes were inconclusive, and no significantly positive gain was found for behavioural outcomes. However, when the results from these studies were pooled, significant gains for people taking galantamine were found for cognitive, functional and global outcomes.
The evidence for the effectiveness of rivastigmine in the previous review was varied; there was some evidence of benefit at 6–12 mg/day with cognitive, functional and global outcomes, but no gain was reported on behavioural measures. Our update review found three more studies; one of these was of reasonable size and quality. Positive benefits from rivastigmine were found on cognitive, functional and global outcomes, but, as before, not on behavioural ones. The lower-dose transdermal patch (9.5 mg/day) was shown to be as effective as the capsule (12 mg/day), but with fewer side effects.
There was some evidence, from a single study, in the previous review that memantine was more effective on cognitive and functional outcomes than placebo; although, as this study’s results were not analysed by ITT, they may be unreliable. However, the new, poorer-quality study, failed to show any benefit from memantine on any outcome measure. When the data were pooled, a significant benefit from memantine was found from global outcomes. It should be noted that these results are based on two moderate-to-poor-quality trials and may be untrustworthy.
Three new head-to-head comparisons were found in addition to the three in the previous review. Only one of the new studies was large and of reasonable quality, this compared donepezil to rivastigmine. It measured cognitive, functional, behavioural and global outcomes, but only found statistically significant differences on functional and global outcomes, both favouring rivastigmine. This is in contrast with the much smaller and poorer-quality studies found in the previous review, which showed no significant differences between the treatments. One new study and one previous study compared donepezil with galantamine; neither was of good quality. The trial from the previous review found that donepezil had greater effects on cognitive and functional outcomes. The new study only looked at global outcomes and found no difference between the treatments. One very poor-quality study, looking at behavioural outcomes, compared all three AChEIs; it found that rivastigmine was significantly better than donepezil or galantamine.
We also found one new, reasonably good study comparing combined memantine with an AChEI against AChEI and placebo. This showed no significant advantage to combining these treatments. This contrasts with the results from the previous review, which found significant benefits from combination therapy on cognitive, functional, behavioural and global outcomes. The reason for this difference in outcomes may be as a result of an underlying pharmacological interaction between galantamine and memantine – which neutralises their respective effects – in the new trial, which used all three AChEIs, whereas the existing trial combined only memantine and donepezil. The other difference between these studies is the lack of ITT analysis in the former one, which may have led to more favourable results for combination therapy.
Mixed-treatment comparison results varied depending on the outcome measure used. There was evidence for both donepezil and galantamine being probably the most effective treatment on cognitive outcomes. A similarly unclear picture for functional measures emerged with galantamine or rivastigmine possibly being equally effective. The amount of uncertainty in these results means it is impossible to say whether or not one AChEI is better than another at treating AD.
Comparison with other systematic reviews
The findings of our systematic review comparing rivastigmine with placebo were similar to those of Birks and colleagues;94 that rivastigmine confers benefit for those with mild-to-moderate AD and that the benefit increases with increasing dose up to 12 mg/day, if the side effects can be tolerated. The transdermal patch, which confers similar clinical benefit to the capsule, but with fewer adverse effects, may be a solution for some people who find that they cannot endure the capsules. Our findings differed slightly from those of the IQWiG (the German Federal Agency for assessing health technologies),95 in that we did not find new evidence to support the assertion that galantamine could relieve psychological symptoms. This difference can be explained by their broader study design inclusion criteria. Otherwise, we agreed that the AChEIs provided some help with the cognitive and functional symptoms from AD. Hansen and colleagues,102 who conducted a systematic review of functional outcomes from all the Alzheimer’s drugs included in this review, found an overall benefit from treatment. This broadly agrees with our findings, although, the evidence for benefit from galantamine was inconsistent.
Three effectiveness reviews were submitted as part of manufacturer submissions in support of donepezil, galantamine and memantine. Full details on the review method were provided only for the systematic review on donepezil. All reviews focused on RCTs, although some non-RCT literature was also included. The comprehensiveness of the identification of this non-RCT literature was unclear. The new studies identified in the manufacturer submissions were consistent with those included in the PenTAG systematic review. The direction and size of effect relative to placebo on cognition, function, behaviour and global impact were consistent between the manufacturer submissions and the PenTAG systematic review. In the case of memantine, the summary estimates of effect were more precise in the manufacturer’s submission. Subgroup analyses for galantamine indicated that there was generally greater effectiveness for more severe AD. These analyses could not be done in the PenTAG systematic review. Evidence for the equal effectiveness of donepezil in mild AD relative to other severity groups argued against the presence of a subgroup effect. Subgroup analyses for memantine also did not show any difference in effectiveness by severity of AD, but did show a difference depending on the presence of APS. Again, these analyses could not be undertaken in the PenTAG systematic review. Additional effects supported by non-RCT and observational data on duration of effectiveness, effects on carers, anti-psychotic drug use, institutionalisation and mortality were also claimed.
Economic evaluations
Initial estimates
The starting point for estimates of cost-effectiveness of donepezil, galantamine, rivastigmine and memantine is complicated by the fact that the ICERs presented in the 2004 SHTAC report were considerably modified by discussions, debate and further work undertaken as part of the NICE appraisal process. The directly quoted cost per QALY gained in the NICE guidance document that underpinned the final decisions were:
-
AChEIs for AD of moderate severity – £31,550 per QALY (the cost per QALY quoted was specifically for donepezil)
-
AChEIs for AD of mild severity – £55,000–58,000 per QALY, but with note that the true value was probably less than this, but not within the range normally considered cost-effective
-
memantine for severe AD – ‘above £53,000 per QALY’.
Published economic evaluations
A systematic review of economic evaluations was conducted, which identified 23 included studies published since 2004, over one-third of which were published only as abstracts and could not be considered in depth. Of the remainder, most addressed the costs and cost-effectiveness of either donepezil or memantine. Of these, the majority reapplied modelling approaches considered as part of the last guidance to the circumstances applying in other countries and were thus felt to add little to this update reconsidering cost-effectiveness in England and Wales. Enhanced modelling approaches were presented for both donepezil and memantine, but in both cases the publications closely mirrored the economic models submitted as part of the industry submissions, which we discuss in detail in the next section.
The included economic evaluations also provide some additional evidence on the impact on resource use and cost alongside trials. They provide support for the conclusion that use of donepezil or galantamine can be cost saving in the short term (6 months to 1 year). They conflict with the conclusion of the AD2000 study,103 the main economic evaluation alongside a trial included in the SHTAC report, which concluded that introduction of donepezil would increase costs over 2 years.
Industry submissions
Two companies offered models of cost-effectiveness: Eisai Ltd and Pfizer Ltd for donepezil and Lundbeck for memantine. Shire for galantamine made a submission focusing on effectiveness and emphasising issues concerning cost-effectiveness raised in the last appraisal; there was no submission for rivastigmine.
The model for donepezil has been described as a discrete-event simulation model. This is a modelling approach that could overcome, theoretically, a number of challenges facing the assessment of the cost-effectiveness of drug treatments for AD, particularly dealing with multiple interdependent outcomes. However, the model does not use a pure discrete-event simulation approach and actually incorporates elements of individual sampling alongside some cohort modelling methods. The manufacturer’s conclusion is that donepezil provides benefits at reduced costs relative to BSC, and is thus dominant, in both mild and moderately severe AD, a conclusion that is robust to the sensitivity analyses conducted by the manufacturer. However, the review of the submitted model identified several areas where there was concern with respect to the quality of the inputted data or the validity of the model assumptions. Exploratory sensitivity analyses suggest that the ICER could be at the margins of what would normally be considered cost-effective by NICE when applying more pessimistic assumptions in the areas where there was considerable uncertainty regarding the validity of the model inputs or assumptions.
The model for memantine used a more traditional Markov approach with three states: pre-FTC, FTC and death. It concludes that memantine provides benefits at reduced costs relative to BSC, and is thus dominant, in moderate and severe AD. Detailed appraisal, again, suggests that considerable caution is required in accepting this result with simple sensitivity analyses conducted by the report authors indicating ICERs that would not normally be considered cost-effective by NICE.
The Peninsula Technology Assessment Group’s cost–utility model
Notwithstanding the uncertainty of our findings, in contrast with the previous TAR, we have found, in the base case, that the AChEIs are probably cost-saving at a WTP threshold of £30,000 per QALY for people with mild-to-moderate AD. For this class of drugs, there is a > 99% probability that the AChEIs are more cost-effective than BSC. These analyses assume that the AChEIs have no effect on survival. If a survival effect is assumed, the AChEIs no longer dominate BSC and ICERs for the AChEIs are approximately £37,000. However, as we have not been able to find any relevant studies that measure survival, these ICERs are purely speculative.
For the AChEIs, in people with mild-to-moderate AD, the PSAs suggested that donepezil is the most cost-effective of the AChEIs, but only with a probability of 28% of being the most cost-effective option at a WTP of £30,000 per QALY (27% at a WTP of £20,000 per QALY). At a WTP of £30,000 per QALY gained, there is only a 0.3% probability that BSC is the best treatment option. In the deterministic results, donepezil dominates the other drugs and BSC, which, along with rivastigmine patches are associated with greater costs and fewer QALYs. Thus, although galantamine has a slightly cheaper total cost than donepezil (£69,592 vs £69,624), the very slightly greater QALY gains from donepezil (1.616 vs 1.617) are enough for donepezil to dominate galantamine.
The probability that memantine is cost-effective in a moderate-to-severe cohort compared with BSC (see Moderate-to-severe Alzheimer’s disease: memantine (decision problem 2a) at a WTP of £30,000 per QALY is 38% (and 28% at a WTP of £20,000 per QALY). The deterministic ICER for memantine is £32,100 per/QALY and the probabilistic £36,700 per/QALY. Sensitivity analyses, assuming that memantine gave an additional 1.7 months of life, changed the ICER to £65,619 per QALY, owing to the modest utility gains and greater additional cost.
In considering the strengths and weaknesses of the PenTAG model-based analyses, compared with the manufacturer and other models (see below), there should be no initial presumption that the model from the independent review group is somehow more valid or reliable than the others. Rather, in this complex disease area, the diversity of models is partly a reflection of evident structural uncertainty regarding how to simulate this disease and its consequences, as well as differences in the rationales and context for developing each model.
Strengths and limitations of the systematic review of studies of effectiveness
The strengths of this systematic review are that is was conducted by an independent research team using the latest evidence.
There are a number of limitations:
-
The length of follow-up of the trials was a maximum of 6 months, which makes it very difficult to reliably extrapolate findings for years ahead.
-
There has been a lack of evidence from the trials on key outcomes such as mortality, institutionalisation, the impact on carer’s time and the prescription of anti-psychotic drugs.
-
None of the trials conducted subgroup analyses based on disease severity, making us unable to comment on the effectiveness of treatments for mild, moderate or severe AD separately.
-
Overall the quality of the trials was moderate to poor, with lack of reporting of key measures of trial quality, thus adding to the uncertainty of the results.
-
The use of LOCF and OC methods for accounting for missing data may have overestimated the treatment benefit from the drugs.
-
Some of the measures used in the trials are insensitive to change in AD (ADAS-cog, MMSE). Therefore, the effects of treatment may have been underestimated in some cases.
-
The searches were limited to the English language due to resource limitations, which may have led us to exclude important studies.
Strengths and limitations of the economic modelling by the Peninsula Technology Assessment Group
Although we believe we have made a number of improvements on the previous SHTAC-AHEAD model, and attempted to address some of the specific criticisms of the previous model (as detailed in Appendix 12), there still remain limitations. In particular these are as follows.
-
The underlying disease model captures just the two dimensions of cognitive status and functional status/ADL. Behavioural and psychological symptoms are not incorporated into the model and, therefore, any treatment effects and QoL impacts related to these symptoms will not be captured.
-
The expression of treatment effectiveness, although based on a multivariate formula of, patient age, ADL status and cognitive status, is mainly based on predicting delays in time to institutionalisation. Although there is good evidence that this event/transition marks a key change in care costs, the evidence that it is also a key marker of decline in QoL is uncertain.
-
Although the model now incorporates more graduated declines in patient utility, and more graduated increases in NHS and PSS costs prior to institutionalisation, assuming that all of these time-related cost and utility changes will be delayed by the same amount of time that institutionalisation is delayed is a key assumption in the model (especially bearing in mind that many of the health-care costs will not be related to AD).
-
The main database of IPD from the UK that the time-to-institutionalisation model and key cost parameters are largely based upon is relatively old (1988–99), small (n = 92 with AD) and from a limited part of the UK (Oxfordshire). Its generalisability to the whole of England and Wales in 2010, therefore, has to be considered (see below).
-
Unlike the 2004 SHTAC analysis, utility benefits pre-institutionalisation have been accounted for, as utilities are based upon MMSE, and both costs and MMSE prior to institutionalisation are conditional on the time until institutionalisation. However, as with the previous model, basing the structure of the model around living in the community (i.e. at home), or living in a nursing or residential home (or long-term hospitalisation), means estimating the benefits of drug treatments for those already in residential care is problematic. This is a more considerable weakness of this modelling approach for evaluating the cost-effectiveness of memantine.
-
In attempting to overcome a criticism of the SHTAC model where AD progression was based on US data, AD progression in the PenTAG model is based on UK IPD. However, the generalisability of these data should be questioned for a number of reasons: (1) the data are from just 92 individuals; (2) it is collected from the Oxfordshire area only; and (3) these data were collected between 1988–9 and 1999. Not only are these data used to inform AD progression, but also they are used as a basis for the NHS/PSS costs of care (in the community and in institutions). This has an advantage in one respect, as there is no need to incorporate an additional source of evidence, with its own uncertainties, into the model. However, if the data from Wolstenholme and colleagues196 cannot be generalised to the whole of England and Wales in 2010, it is likely that the model will not be generalisable either, even although few options were available as the basis for predicting disease progression. In addition to considering the US data used in the SHTAC model, RCT data were considered, but felt not to be ideal because of the restricted populations from inclusion/exclusion criteria. The available UK epidemiological evidence was either from Wolstenholme and colleagues196 or a longitudinal cohort study where many participants were receiving AChEI and/or memantine treatment and had a shorter follow-up time (i.e. the LASER-AD study192).
-
Note also that we have assumed that 100% of the pre-institutional care costs are funded by the NHS/PSS. This is unlikely to be the case in reality, with some costs attributed to personal care and domestic help.
-
An important assumption of our model is that once individuals stop treatment they decline at the same rate as an individual who did not receive treatment. We were unable to identify evidence to inform what happened after treatment discontinues, but note that if a ‘bounce-back’ effect occurred, all drugs would be associated with larger costs and fewer QALYs, possibly leading to the AChEIs no longer dominating BSC.
-
The treatment effects incorporated into the PenTAG model are absolute effects. There has been no accounting for differential effects for baseline severity, but there was some, albeit exploratory, evidence of an association between baseline MMSE and functional outcomes identified in Chapter 3 (see Appendix 7).
-
A further limitation relates to effectiveness data availability. No relevant ADL data for donepezil and no relevant MMSE for galantamine at 21–26 weeks were identified from the clinical effectiveness review. Thus, it was assumed that this was a lack of evidence for an effect, rather than lack of effect and a class effect was assumed (i.e. the effectiveness was assumed to be the same as the other AChEIs).
Strengths and limitations of the economic modelling in the Eisai/Pfizer submission
A strength of the Eisai/Pfizer model is that it is able to track changes on cognitive status, functional status (using both ADL and IADL), and behavioural and psychological symptoms. However, there were a number of concerns with the appropriateness of the data used to predict progression on each of these scales and the possibility of double-counting treatment effects, as changes on one scale were used as an independent term to predict progression on the other scales. The electronic version of the model contained many features that were not used in the submission and the presence of these redundant features reduced the transparency of the model, making it very difficult to review. Although some errors were identified, we cannot be entirely confident that no other errors remain unidentified. The review, as a whole, cannot be considered as an endorsement of the validity of the model. We were unable to explain some features of the behaviour of the model and, therefore, retain a degree of caution about its functioning.
The most significant weakness with the model is that the data used in the model to relate cognitive function (MMSE) to the probability of institutionalisation appear to have been derived from a study that only included institutionalised patients and insufficient details are provided to explain how the data used in the model could have been derived from this study. This is a significant weakness, as it is a major driver of cost-effectiveness.
Uncertainties
There continue to be many uncertainties, indeed it is likely that the nature and extent of these uncertainties is similar to those operating when the last TAR was compiled. The most influential of these are:
-
effect of anti-AD drugs in the longer term on any outcome, especially beyond 1 year.
-
effect of anti-AD drugs on outcomes beyond cognition, function, behaviour and global impact, particularly QoL, impact on carers, effect on admission to FTC and impact on resource use
-
whether or not the effects vary substantially by subgroup, particularly severity of AD
-
whether or not the future cost of the anti-AD drugs will be affected by the entry of generic formulations. 223
Chapter 9 Conclusions
The additional clinical effectiveness evidence identified in this updated systematic review continues to suggest that there is clinical benefit from the AChEIs in alleviating AD symptoms, although there is considerable debate about the magnitude of the effect. There is also some evidence that they have an impact on controlling disease progression. However, there is only randomised evidence for this up to 6 months’ follow-up and the quality of these news studies remains mostly moderate at best.
Although there is also new evidence on the effectiveness of memantine, it remains less supportive of this drug’s use than the evidence for AChEIs.
The conclusions concerning cost-effectiveness are quite different from the previous TAR assessment. This is because both the changes in effectiveness and costs, between drug use and non-drug use, underlying the ICERs are very small. This leads to highly uncertain results that are very sensitive to change and about all model parameters.
Implications for service provision
These are not clear and will ultimately rest on the interpretation of the new evidence from a variety of sources, including this report, in the forthcoming NICE appraisal on this topic.
Suggested research priorities
New research in the following areas could reduce the uncertainty noted:
-
Good-quality longer-term RCTs (following CONSORT; consolidated standards of reporting trials) to include mortality, time to institutionalisation and HRQoL as outcomes and sufficiently powered for subgroup analysis by disease severity, response to treatment, behavioural disturbance and comorbidities. We have identified that a limited number of major RCTs addressing relevant issues, such as management when patients fail to respond to AChEIs, are already in progress (DOMINO-AD).
-
Such good-quality trials should aim to use the same standardised measures of cognitive status, functional status/ADL, and behavioural/psychiatric symptoms.
-
Systematic reviews of non-RCT evidence on the impact of anti-AD treatments on resource use, institutionalisation and mortality.
-
Further independent comparison of different methodological approaches to the modelling of cost-effectiveness of anti-AD treatments.
-
Research into cognitive measures that are sensitive to change in dementia.
-
Studies should measure HRQoL with measures validated for people with dementia, for example DEMQOL. Work is needed to derive utility values from such validated measures.
In addition, this report highlights some wider methodological issues that would benefit from further investigation: research into more valid ways of accounting for missing data than LOCF and OC, particularly in degenerative diseases such as AD.
Acknowledgements
We would like to thank Dr Jane Wolstenholme (Health Economics Research Centre, Oxford) and the principal investigators of the Oxfordshire AD cohort study for sharing their study’s data set, Professor Gill Livingston (UCL) for sharing the LASER-AD study data set with us, Professor Douglas Galasko (University of California San Diego, developer of the ADCS-ADL and the ADCS-ADL-sev functional status instruments), Researchers at the Personal Social Services Research Unit, University of Kent (in relation to clarifying aspects of the costs in the Unit Costs of Health and Social Care 2009 report), and Professor Douglas Galasko and Dr Steven Stokes from the University of California, San Diego, CA, for help with the use of the ADCS-ADL questionnaires.
We would also like to acknowledge the help of Martin Pitt for model checking, Mark Pearson and Harriet Hunt for supporting the systematic reviews, Louise Crathorne for proof-reading, Will Stahl-Timmins for summary graphics, Colin Green for general support in helping us to understand this complex disease and modelling area, and Sue Whiffin and Jenny Lowe for their administrative support, all from the Peninsula Medical School, University of Exeter.
About the Peninsula Technology Assessment Group
The Peninsula Technology Assessment Group is part of the Institute of Health Service Research at the Peninsula College of Medicine & Dentistry. PenTAG was established in 2000 and currently has four major work streams: independent HTAs for NICE and the National Institute for Health Research HTA programme, systematic reviews and economic analyses for the NICE Centre for Public Health Excellence, systematic reviews as part of the Cochrane Collaboration Heart Group, and evidence synthesis work in relation to the needs of the SW Peninsula Collaboration for Applied Health Research and Care (PenCLAHRC), as well as for other local and national decision-makers.
The group is multidisciplinary and draws on individuals’ backgrounds in public health, health services research, computing and decision analysis, systematic reviewing, statistics and health economics. The Peninsula College of Medicine & Dentistry is a school within the Universities of Plymouth and Exeter. The Institute of Health Research is made up of discrete, but methodologically related research groups, among which HTA is a strong and recurring theme. Recent projects include the following.
Health technology assessment
-
Dasatinib and nilotinib for imatinib resistant or intolerant chronic myeloid leukaemia. 224
-
Bevacizumab, sorafenib tosylate, sunitinib and temsirolimus for renal cell carcinoma: a systematic review and economic evaluation. 225
-
Systematic review of the effectiveness and cost-effectiveness of weight management schemes for the under fives. 226
-
The effectiveness and cost-effectiveness of cochlear implants for severe to profound deafness in children and adults: a systematic review and economic model. 227
-
The effectiveness and cost-effectiveness of methods of storing donated kidneys from deceased donors: a systematic review and economic model. 228
-
The harmful health effects of recreational ecstasy: a systematic review of observational evidence. 229
-
The use of surrogate outcomes in model-based cost-effectiveness analyses: a survey of UK health TARs. 230
-
Systematic review and economic analysis of the comparative effectiveness of different inhaled corticosteroids and their usage with long-acting beta2 agonists for the treatment of chronic asthma in children under the age of 12 years. 231
-
The clinical effectiveness and cost-effectiveness of cardiac resynchronisation (biventricular pacing) for Heart Failure: a systematic review and economic model. 232
Synthesising public health evidence
-
Prevention of unintentional injuries to children in outdoor play and leisure environments. 233
Expert Advisory Group
We would particularly like to thank Professor Peter Passmore who acted as our Expert Advisor for his help throughout the project.
Competing interests Professor Peter Passmore has received fees for consultancy, honoraria and assistance with study leave from Pfizer, Eisai, Shire, Jansen and Jansen, Novartis and Lundbeck, and has given expert testimony in relation to the last judicial review as an expert for Eisai.
Contributions of authors
Mary Bond provided overall project management, wrote the protocol, assessed abstracts and titles for inclusion, contributed to the clinical effectiveness systematic review, contributed to the design of the model and the writing and editing of the report.
Gabriel Rogers assessed abstracts and titles for inclusion, led the systematic review of clinical effectiveness, contributed to the design and execution of the economic model, and contributed to the writing and editing of the report.
Jaime Peters led the design, development and execution of the economic model and contributed to the writing and editing of the report.
Rob Anderson oversaw the cost-effectiveness aspects of the analysis and report, advised on obtaining costs and utilities for the model, contributed to the design of the model and contributed to the editing of the report.
Martin Hoyle contributed to the design, parameterisation of the model and model checking, and contributed to the editing of the report.
Alec Miners critically appraised industry submissions and contributed to the editing of the report.
Tiffany Moxham wrote and ran all the search strategies.
Sarah Davis provided critical appraisal of the cost-effectiveness model submitted by Eisai and contributed to the writing of the final report.
Praveen Thokala provided critical appraisal of the cost-effectiveness model submitted by Eisai and contributed to the writing of the final report.
Allan Wailoo provided critical appraisal of the cost-effectiveness model submitted by Eisai and contributed to the writing of the final report.
Mike Jeffreys provided clinical input into the design of the model, advised on clinical matters and contributed to the editing of the report.
Chris Hyde led the systematic review of economic evaluations, contributed to the design of the model, contributed to the writing and editing of the report, and was overall director of the project and guarantor of the report.
Disclaimers
The views expressed in this publication are those of the authors and not necessarily those of the HTA programme or the Department of Health.
References
- National Institute of Health and Clinical Excellence (NICE) . Donepezil, Galantamine, Rivastigmine (review) and Memantine for the Treatment of Alzheimer’s Disease (amended) 2009.
- Loveman E, Green C, Kirby J, Takeda A, Picot J, Payne E, et al. The clinical and cost-effectiveness of donepezil, rivastigmine, galantamine and memantine for Alzheimer’s disease. Southampton: University of Southampton; 2004.
- American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorder 2000.
- World Health Organization (WHO) . The ICD 10 Classification of Mental and Behavioural Disorders: Diagnostic Criteria for Research 1993.
- National Collaborating Centre for Mental Health . Dementia: A NICE-SCIE Guideline on Supporting People With Dementia and Their Carers in Health and Social Care 2007.
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984;34:939-44.
- Warrell DA, Cox TM, Firth JD. Oxford textbook of medicine. Oxford: Oxford University Press; 2003.
- Knapp M, Prince M, Albanese E, Banerjee S, Dhanasiri S, Fernandez J, et al. Dementia UK: The Full Report 2007.
- National Audit Office . Improving Services and Support for People With Dementia 2007.
- Luengo-Fernandez R, Leal J, Gray A. Dementia 2010: The economic burden of dementia and associated research funding in the United Kingdom. Cambridge: Alzheimer’s Research Trust; 2010.
- Yip AG, Brayne C, Matthews FE. Risk factors for incident dementia in England and Wales: The Medical Research Council Cognitive Function and Ageing Study. A population-based nested case–control study. Age Ageing 2006;35:154-60.
- Matthews F, Brayne C. The incidence of dementia in England and Wales: findings from the five identical sites of the MRC CFA Study. PLoS Med 2005;2.
- Alzheimer’s Association . Alzheimer’s disease facts and figures. Alzheimers Dement 2009;5:234-70.
- Hardy J. New insights into the genetics of Alzheimer’s disease. Ann Med 1996;28:255-8.
- Cruts M, van Duijn CM, Backhovens H, Van den BM, Wehnert A, Serneels S, et al. Estimation of the genetic contribution of presenilin-1 and -2 mutations in a population-based study of presenile Alzheimer disease. Hum Mol Genet 1998;7:43-51.
- Schellenberg G, Anderson L, O’dahl S. APP717, APP693 and PRIP gene mutations are rare in Alzheimer’s disease. Am J Hum Genet 1991;49:511-17.
- Takashi ASAD. Prevention of Alzheimer’s disease: putative nutritive factors. Psychogeriatrics 2007;7:125-31.
- Rolland YM, Pillard FM, Klapouszczak AM, Reynish EM, Thomas DM, Andrieu SM, et al. Exercise program for nursing home residents with Alzheimer’s disease: a 1-year randomized, controlled trial. J Am Geriatr Soc 2007;55:158-65.
- Fratiglioni L, Paillard-Borg S, Winblad B. An active and socially integrated lifestyle in late life might protect against dementia. Lancet Neurol 2004;3:343-53.
- Fitzpatrick AL, Kuller LH, Lopez OL, Kawas CH, Jagust W. Survival following dementia onset: Alzheimer’s disease and vascular dementia. J Neurol Sci 2005;229:43-9.
- Backman L, Jones S, Berger AK, Laukka EJ, Small BJ. Multiple cognitive deficits during the transition to Alzheimer’s disease. J Intern Med 2004;256:195-204.
- Arnaiz E, Almkvist O. Neuropsychological features of mild cognitive impairment and preclinical Alzheimer’s disease. Acta Neurol Scand 2003:34-41.
- Ritchie K, Lovestone S. The dementias. Lancet 2002;360:1759-66.
- Brodaty H, Hadzi-Pavlovic D. Psychosocial effects on carers of living with persons with dementia. Aust N Z J Psychiatry 1990;24:351-61.
- Carlesimo GA, Oscar-Berman M. Memory deficits in Alzheimer’s patients: a comprehensive review. Neuropsychol Rev 1992;3:119-69.
- Frank EM. Effect of Alzheimer’s disease on communication function. J S C Med Assoc 1994;90:417-23.
- Wimo A, Gustafsson L, Mattson B. Predictive validity of factors influencing the institutionalization of elderly people with psycho-geriatric disorders. Scand J Prim Health Care 1992;10:185-91.
- Drachman DA, O’Donnell BF, Lew RA, Swearer JM. The prognosis in Alzheimer’s disease. ‘How far’ rather than ‘how fast’ best predicts the course. Arch Neurol 1990;47:851-6.
- Brodaty H, McGilchrist C, Harris L, Peters KE. Time until institutionalization and death in patients with dementia: role of caregiver training and risk factors. Arch Neurol 1993;50:643-50.
- Thompson C, Spilsbury K, Hall J, Birks Y, Barnes C, Adamson J. Systematic review of information and support interventions for caregivers of people with dementia. BMC Geriatrics 2007;7.
- Aguglia E, Onor. Stress in the caregivers of Alzheimer’s patients: An experimental investigation in Italy. Am J Alzheimers Dis Other Demen 2004;19:248-52.
- Holland JM, Currier JM, Gallagher-Thompson D. Outcomes From the Resources for Enhancing Alzheimer’s Caregiver Health (REACH) Program for Bereaved Caregivers. Psychol Aging 2009;24:190-202.
- Garity JE. Caring for a Family Member with Alzheimer’s Disease: Coping with Caregiver Burden Post-Nursing Home Placement. J Gerontol Nurs 2006;32:39-48.
- Vitaliano PP, Echeverria D, Yi J, Phillips PEM, Young H, Siegler IC. Psychophysiological Mediators of Caregiver Stress and Differential Cognitive Decline. Psychol Aging 2005;20:402-11.
- Bianchetti A, Scuratti A, Zanetti O, Binetti G, Frisoni GB, Magni E, et al. Predictors of mortality and institutionalization in Alzheimer disease patients 1 year after discharge from an Alzheimer dementia unit. Dementia 1995;6:108-12.
- Donaldson C, Tarrier N, Burns A. The impact of the symptoms of dementia on caregivers. Br J Psychiatry 1997;170:62-8.
- Adams KB, Sanders SA. Alzheimer’s caregiver differences in experience of loss, grief reactions and depressive symptoms across stage of disease: a mixed-method analysis. Dementia 2004;3:195-210.
- Schneider J, Murray J, Banerjee S, Mann A. EUROCARE: a cross-national study of co-resident spouse carers for people with Alzheimer’s disease: I--Factors associated with carer burden. Int J Geriatr Psychiatry 1999;14:651-61.
- Mittelman MS, Haley WE, Clay OJM, Roth DLP. Improving caregiver well-being delays nursing home placement of patients with Alzheimer disease. Neurology 2006;67:1592-9.
- Banerjee S, Murray J, Foley B, Atkins L, Schneider J, Mann A. Predictors of institutionalisation in people with dementia. J Neurol Neurosurg Psychiatry 2003;74:1315-16.
- de Vugt ME, Stevens F, Aalten P, Lousberg R, Jaspers N, Verhey FRJ. A prospective study of the effects of behavioral symptoms on the institutionalization of patients with dementia. Int Psychogeriatr 2005;17:577-89.
- Wolfson C, Moride Y, Perrault A. Drug treatments for Alzheimer’s disease. II: a review of outcome measures in clinical trials. Ottawa, ON: Canadian Coordinating Office for Health Technology Assessment; 2000.
- Wolfson C, Oremus M, Shukla V, Momoli F, Demers L, Perrault A, et al. Donepezil and rivastigmine in the treatment of Alzheimer’s disease: a best-evidence synthesis of the published data on their efficacy and cost effectiveness. Clin Ther 2002;24:862-86.
- Folstein MF, Folstein SE, McHugh PR. ‘Mini-mental state’: a practical method for grading the cognitive state of patients for the clinician. J Psychiatry Res 1975;12:189-98.
- Mohs RC, Rosen WG, Davis KL. The Alzheimer’s disease assessment scale: an instrument for assessing treatment efficacy. Psychopharmacol Bull 1983;19:448-50.
- Roselli F, Tartaglione B, Federico F, Lepore V, Defazio G, Livrea P, et al. Rate of MMSE score change in Alzheimer’s disease: influence of education and vascular risk factors. Clin Neurol Neurosurg 2009;111:327-30.
- Tombaugh TN. Test-retest reliable coefficients and 5-year change scores for the MMSE and 3MS. Arch Clin Neuropsychol 2005;20:485-503.
- Irizarry MC, Webb DJ, Bains C, Barrett SJ, Lai RY, Laroche JP, et al. Predictors of placebo group decline in the Alzheimer’s disease Assessment Scale-cognitive subscale (ADAS-Cog) in 24 week clinical trials of Alzheimer’s disease. J Alzheimers Dis 2008;14:301-11.
- Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA, Gornbein J. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology 1994;44:2308-14.
- Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A new clinical scale for the staging of dementia. Br J Psychiatry 1982;140:566-72.
- Boothby H, Mann AH, Barker A. Factors determining inter-rater agreement with rating global change in dementia: CIBIC-plus. Int J Geriatr Psychiatry 1995;10:1037-45.
- Claus JJ, Teunisse S, Walstra GJM, van Gool WA. Determinants of Global Clinical Change Assessment in Patients with Early Alzheimer’s Disease. Dement Geriatr Cogn Discord 1998;9:157-63.
- Gauthier S, Bodick N, Erzigkeit E, Feldman H, Geldmacher DS, Huff J, et al. Activities of daily living as an outcome measure in clinical trials of dementia drugs. Position paper from the International Working Group on Harmonization of Dementia Drug Guidelines. Alzheimer Dis Assoc Disord 1997;11:6-7.
- Nygard L, Wimo A, Jonnson B, Karlsson G, Winblad B. Health economics of dementia. New York, NY: John Wiley and Sons; 1998.
- McDowell I, Newell C. Measuring health: a guide to rating scales and questionnaires. New York, NY: Oxford University Press; 1996.
- Smith SC, Lampling DL, Banerjee S, Harwood R, Foley B, Smith P, et al. Measurement of health-related quality of life for people with dementia: development of a new instrument (DEMQOL) and an evaluation of current methodology. Health Technol Assess 2005;9.
- Blau TH. Quality of life, social indicators and criteria of change. Prof Psychol 1977;8:464-73.
- Department of Health . Living Well With Dementia: A National Dementia Strategy 2009.
- Pickard L, Wittenburg R, Commas-Herrera A, Davis B, Darton R. Community care for frail older people: analysis using the 1998/99 General Household Survey. Quality in later life: rights, rhetoric and reality. Stirling: University of Stirling; 2001.
- Audit Commission . Support for Carers of Older People 2004.
- Comas-Herrera A, Wittenberg R, Pickard L, Knapp M. Cognitive impairment in older people: future demand for long-term care services and the associated costs. Int J Geriatr Psychiat 2007;22:1037-45.
- National Assembly for Wales . Caring about Carers: A Strategy for Carers in Wales. Implementation Plan 2000.
- Secretary of State for Health . The NHS Plan. Cm 4818 2000.
- Morris RG, Morris LW, Britton PG. Factors affecting the emotional wellbeing of the caregivers of dementia sufferers. Br J Psychiatry 1988;153:147-56.
- Livingston G, Manela M, Katona C. Depression and other psychiatric morbidity in carers of elderly people living at home. BMJ 1996;312:153-6.
- Medical Research Council . Cognitive function and dementia in six areas of England and Wales: the distribution of MMSE and prevalence of GMS organicity level in the MRC CFA Study. Psychol Med 1998;28:319-35.
- Evandrou M, Phillips J. Working carers. Aldershot: Avebury; 1995.
- Royal College of General Practitioners . Information Sheet. Profile of General Practitioners 2006.
- Royal College of Psychiatrists . Who Cares Wins. Improving the Outcome for Older People Admitted to the General Hospital: Guidelines for the Development of Liaison Mental Health Services for Older People 2005.
- Barnes D, Lombardo C. A Profile of Older people’s Mental Health Services: Report of Service Mapping 2006 2006.
- Membership Office . Royal College of Psychiatrists 2007.
- Roth M, Tym E, Mountjoy CQ, Huppert FA, Hendrie H, Verma S, et al. CAMDEX . A standardised instrument for the diagnosis of mental disorder in the elderly with special reference to the early detection of dementia. Br J Psychiatry 1986;149:698-709.
- Department of Health, Care Services Improvement Partnership . Everybody’s Business. Integrated Mental Health Service for Older Adults: A Service Development Guide 2005.
- Audit Commission . Forget-Me-Not 2002: Mental Health Services for Older People 2002.
- Department of Health . National Service Framework for Older People 2001.
- Audit Commission . Forget-Me-Not: Mental Health Services for Older People 2000.
- NHS Centre for Reviews and Dissemination . Systematic Reviews: CRD’s Guidance for Undertaking Reviews in Healthcare 2009.
- Moher D, Liberati A, Tetzlaff J, Altman DG. for the PRISMA Group . Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ 2009;339.
- Moher D, Cook DJ, Eastwood S, Olkin I, Rennie D, Stroup DF. Improving the quality of reports of meta-analyses of randomised controlled trials: the QUOROM statement. Lancet 1999;354:1896-900.
- DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials 1986;7:177-88.
- Cochran WG. The Combination of Estimates from Different Experiments. Biometrics 1954;10:101-29.
- Higgins JPT, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med 2002;21:1539-58.
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ 2003;327:557-60.
- Egger M, Smith GD, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. BMJ 1997;315:629-34.
- The Cochrane Collaboration . Cochrane Handbook for Systematic Reviews of Interventions 2009.
- Molnar FJ, Hutton B, Fergusson D. Does analysis using ‘last observation carried forward’ introduce bias in dementia research?. Can Med Assoc J 2008;179:751-3.
- Thompson SG, Sharp SJ. Explaining heterogeneity in meta-analysis: a comparison of methods. Stat Med 1999;18:2693-708.
- Egger M, Smith GD, Altman DG. Systematic reviews in health care: meta-analysis in context. London: BMJ Books; 2001.
- Ades AE. A chain of evidence with mixed comparisons: models for multi-parameter synthesis and consistency of evidence. Stat Med 2003;22:2995-3016.
- Caldwell DM, Ades AE, Higgins JP. Simultaneous comparison of multiple treatments: combining direct and indirect evidence. BMJ 2005;331:897-900.
- Lu G, Ades AE. Combination of direct and indirect evidence in mixed treatment comparisons. Stat Med 2004;23:3105-24.
- Higgins JP, Whitehead A. Borrowing strength from external trials in a meta-analysis. Stat Med 1996;15:2733-49.
- Rogers SL, Doody RS, Mohs RC, Friedhoff LT. Donepezil improves cognition and global function in Alzheimer disease: a 15-week, double-blind, placebo-controlled study. Arch Int Med 1998;158.
- Birks J, Evans JG, Iakovidou V, Tsolaki M. Rivastigmine for Alzheimer’s disease. Cochrane Database Syst Rev 2009;2.
- Institute for Quality and Efficiency in Health Care (IQWiG) . Cholinesterase Inhibitors in Alzheimer’s Disease. Cologne: Institut für Qualität Und Wirtschaftlichkeit Im Gesundheitswesen (IQWiG) 2007.
- Brodaty H, Corey-Bloom J, Potocnik FCV, Truyen L, Gold M, Damaraju CRV. Galantamine prolonged-release formulation in the treatment of mild to moderate Alzheimer’s disease. Dement Geriatr Cogn Disord 2005;20:120-32.
- Rockwood K, Fay S, Song X, MacKnight C, Gorman M, . Video-Imaging Synthesis of Treating Alzheimer’s Disease (VISTA) Investigators . Attainment of treatment goals by people with Alzheimer’s disease receiving galantamine: a randomized controlled trial. CMAJ 2006;174:1099-105.
- Bullock R. Treatment of behavioural and psychiatric symptoms in dementia: implications of recent safety warnings. Curr Med Res Opin 2005;21:1-10.
- Cumbo E. Differential effects or rivastigmine, galantamine and donepezil on behavioral and psychological symptoms in patients with Alzheimer’s disease: 18-month, randomized, open-label trial. Prim Care Community Psychiatr 2005;10:95-102.
- Raina P, Santaguida P, Ismaila A, Patterson C, Cowan D, Levine M, et al. Effectiveness of cholinesterase inhibitors and memantine for treating dementia: Evidence review for a clinical practice guideline. Ann Int Med 2008;148:379-97.
- Bullock R, Erkinjuntti T, Lilienfeld S. Management of patients with Alzheimer’s disease plus cerebrovascular disease: 12-Month treatment with galantamine. Dement Geriatr Cogn Disord 2004;17:29-34.
- Hansen RA, Gartlehner G, Lohr KN, Kaufer DI, Hansen RA, Gartlehner G, et al. Functional outcomes of drug treatment in Alzheimer’s disease: a systematic review and meta-analysis. Drugs Aging 2007;24:155-67.
- Bentham P, Gray R, Raftery J, Hills R, Sellwood E, Courtney C, et al. Long-term donepezil treatment in 565 patients with Alzheimer’s disease (AD2000): randomised double-blind trial. Lancet 2004;363:2105-15.
- Burns A, Rossor M, Hecker J, Gauthier S, Petit H, Moeller HJ, et al. The effects of donepezil in Alzheimer’s disease: results from a multinational trial. Dement Geriatr Cogn Disord 1999;10:237-44.
- Gauthier S, Feldman H, Hecker J, Vellas B, Emir B, Subbiah P, et al. Functional, cognitive and behavioral effects of donepezil in patients with moderate Alzheimer’s disease. Curr Med Res Opin 2002;18.
- Greenberg SM, Tennis MK, Brown LB, Gomez-Isla T, Hayden DL, Schoenfeld DA, et al. Donepezil therapy in clinical practice: a randomized crossover study. Arch Neurol 2000;57:94-9.
- Holmes C, Wilkinson D, Dean C, Vethanayagam S, Olivieri S, Langley A, et al. The efficacy of donepezil in the treatment of neuropsychiatric symptoms in Alzheimer disease. Neurology 2004;63:214-19.
- Homma A, Takeda M, Imai Y, Udaka F, Hasegawa K, Kameyama M, et al. Clinical efficacy and safety of donepezil on cognitive and global function in patients with Alzheimer’s disease. A 24-week, multicenter, double-blind, placebo-controlled study in Japan. E2020 Study Group. Dement Geriatr Cogn Disord 2000;11.
- Krishnan K, Charles HC, Doraiswamy PM, Mintzer J, Weisler R, Yu X, et al. Randomized, placebo-controlled trial of the effects of donepezil on neuronal markers and hippocampal volumes in Alzheimer’s disease. Am J Psychiatry 2003;160:2003-11.
- Mohs RC, Doody RS, Morris JC, Ieni JR, Rogers SL, Perdomo CA, et al. A 1-year, placebo-controlled preservation of function survival study of donepezil in AD patients. Neurology 2001;57.
- Johannsen P, Barcikowska M, Dautzenberg P, Hampel H, Hasselbalch S, Sakka P, et al. Donepezil-Treated Alzheimer’s Disease Patients With Apparent Initial Cognitive Decline Demonstrate Significant Benefits When Therapy Is Continued: Results from a Randomised, Placebo-Controlled Trial n.d.
- Johannsen P, Salmon E, Hampel H, Xu Y, Richardson S, Qvitzau S, et al. Assessing therapeutic efficacy in a progressive disease: a study of donepezil in Alzheimer’s disease. CNS Drugs 2006;20:311-25.
- Rogers SL, Farlow MR, Doody RS, Mohs R, Friedhoff LT. A 24-week, double-blind, placebo-controlled trial of donepezil in patients with Alzheimer’s disease. Neurology 1998;50.
- Rogers SL, Friedhoff LT. The efficacy and safety of donepezil in patients with Alzheimer’s disease: results of a US multicentre, randomized, double-blind, placebo-controlled trial. Dementia 1996;7:293-30.
- Seltzer B, Zolnouni P, Nunez M, Goldman R, Kumar D, Ieni J, et al. Efficacy of donepezil in early-stage Alzheimer disease: a randomized placebo-controlled trial. Arch Neurol 2004;61:1852-6.
- Winblad B, Engedal K, Soininen H, Verhey F, Waldemar G, Wimo A, et al. A 1-year, randomized, placebo-controlled study of donepezil in patients with mild to moderate AD. Neurology 2001;57.
- Wimo A, Winblad B, Engedal K, Soininen H, Verhey F, Waldemar G, et al. An economic evaluation of donepezil in mild to moderate Alzheimer’s disease: results of a 1-year, double-blind, randomized trial. Dement Geriatr Cogn Disord 2003;15.
- Mazza M, Capuano A, Bria P, Mazza S. Ginkgo biloba and donepezil: a comparison in the treatment of Alzheimer’s dementia in a randomized placebo-controlled double-blind study. Eur J Neurol 2006;13:981-5.
- dos Santos Moraes WA, Poyares DR, Guilleminault C, Ramos LR, Ferreira Bertolucci PH, Tufik S. The effect of donepezil on sleep and REM sleep EEG in patients with Alzheimer disease: a double-blind placebo-controlled study. Sleep 2006;29:199-205.
- Moraes W, Poyares D, Sukys-Claudino L, Guilleminault C, Tufik S. Donepezil improves obstructive sleep apnea in Alzheimer disease: a double-blind, placebo-controlled study. Chest 2008;133:677-83.
- Peng DT, Xu XH, Wang LN. Efficiency and safety assessment of donepezil for treating mild and moderate Alzheimer disease. Chin J Clin Rehabil 2005;9:170-2.
- Winstein CJ, Bentzen KR, Boyd L, Schneider LS. Does the cholinesterase inhibitor, donepezil, benefit both declarative and non-declarative processes in mild to moderate Alzheimer’s disease?. Curr Alzheimer Res 2007;4:273-6.
- Raskind MA, Peskind ER, Wessel T, Yuan W. Galantamine in AD: a 6-month randomized, placebo-controlled trial with a 6-month extension. Neurology 2000;54.
- Rockwood K, Mintzer J, Truyen L, Wessel T, Wilkinson D. Effects of a flexible galantamine dose in Alzheimer’s disease: a randomised, controlled trial. BMJ 2001;71.
- Tariot PN, Solomon PR, Morris JC, Kershaw P, Lilienfeld S, Ding C. A 5-month, randomized, placebo-controlled trial of galantamine in AD. The Galantamine USA-10 Study Group. Neurology 2000;54.
- Cummings JL, Schneider L, Tariot PN, Kershaw PR, Yuan W. Reduction of behavioral disturbances and caregiver distress by galantamine in patients with Alzheimer’s disease. Am J Psychiatry 2004;161:532-8.
- Wilcock GK, Lilienfeld S, Gaens E. Efficacy and safety of galantamine in patients with mild to moderate Alzheimer’s disease: multicentre randomised controlled trial. BMJ 2000;321.
- Wilkinson D, Murray J. Galantamine: a randomized, double-blind, dose comparison in patients with Alzheimer’s disease. Int J Geriatr Psychiatry 2001;16.
- Wilkinson D, Lilienfeld S, Truyen L. Galantamine improves activities of daily living in patients with Alzheimer’s disease: a 3-month placebo-controlled study 2000.
- Erkinjuntti T, Kurz A, Gauthier S, Bullock R, Lilienfeld S, Damaraju CV. Efficacy of galantamine in probable vascular dementia and Alzheimer’s disease combined with cerebrovascular disease: a randomised trial. Lancet 2002;359:1283-90.
- Rockwood K, Fay S, Song X, MacKnight C, Gorman M. Attainment of treatment goals by people with Alzheimer’s disease receiving galantamine: A randomized controlled trial. CMAJ 2006;174:1099-105.
- Feldman HH, Van Baelen B, Kavanagh S. Effects of galantamine on activities of daily living in Alzheimer’s disease: evidence from six randomised double-blind placebo-controlled trials. Res Pract Alzheimers Dis 2005;10:234-8.
- Rockwood K, Fay S, Jarrett P, Asp E. Effect of galantamine on verbal repetition in AD: a secondary analysis of the VISTA trial. Neurology 2007;68:1116-21.
- Agid Y, Dubois B, Anand R, Gharabawi G. Efficacy and tolerability of rivastigmine in patients with dementia of the Alzheimer type. Curr Ther Res 1998;59:837-45.
- Corey-Bloom J, Anand R, Veach J. A randomized trial evaluating the efficacy and safety of ENA 713(rivastigmine tartrate), a new acetylcholinesterase inhibitor, in patients with mild to moderately severe Alzheimer’s disease. Int J Geriatr Psychopharmacol 1998;1:55-6.
- Forette F, Anand R, Gharabawi G. A phase II study in patients with Alzheimer’s disease to assess the preliminary efficacy and maximum tolerated dose of rivastigmine (Exelon®). Eur J Neurol 1999;6:423-9.
- Rosler M, Anand R, Cicin-Sain A, Gauthier S, Agid Y, Dal-Bianco P, et al. Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: international randomised controlled trial – Commentary: Another piece of the Alzheimer’s jigsaw. BMJ 1999;318.
- Feldman HH, Lane R. Rivastigmine: a placebo controlled trial of twice daily and three times daily regimens in patients with Alzheimer’s disease. J Neurol Neurosurg Psychiatry 2007;78:1056-63.
- Mowla A, Mosavinasab M, Haghshenas H, Haghighi AB. Does serotonin augmentation have any effect on cognition and activities of daily living in Alzheimer’s dementia? A double-blind, placebo-controlled clinical trial. J Clin Psychopharmacol 2007;27:484-7.
- Winblad B, Cummings J, Andreasen N, Grossberg G, Onofrj M, Sadowsky C, et al. A six-month double-blind, randomized, placebo-controlled study of a transdermal patch in Alzheimer’s disease: rivastigmine patch versus capsule. Int J Geriatr Psychiatry 2007;22:456-67.
- Tariot PN, Farlow MR, Grossberg GT, Graham SM, McDonald S, Gergel I. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA 2004;291:317-24.
- Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ. Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med 2003;348.
- Van Dyck CH, Tariot PN, Meyers B, Malca Resnick E. A 24-week randomized, controlled trial of memantine in patients with moderate-to-severe Alzheimer disease. Alzheimer Dis Assoc Disord 2007;21:136-43.
- Fuschillo C, La PS, Campana F, Pinto A, De SL. Cognitive deficits in Alzheimer’s disease: treatment with acetylcholinesterase inhibitor agents. Arch Gerontol Geriatr 2001;33:151-8.
- Wilkinson DG, Passmore AP, Bullock R, Hopker SW, Smith R, Potocnik FCV, et al. A multinational, randomised, 12-week, comparative study of donepezil and rivastigmine in patients with mild to moderate Alzheimer’s disease. Int J Clin Pract 2002;56:441-6.
- Jones RW, Soininen H, Hager K, Aarsland D, Passmore P, Murthy A, et al. A multinational, randomised, 12-week study comparing the effects of donepezil and galantamine in patients with mild to moderate Alzheimer’s disease. Int J Geriatr Psychiatry 2004;19:58-67.
- Nordberg A, Darreh-Shori T, Peskind E, Soininen H, Mousavi M, Eagle G, et al. Different cholinesterase inhibitor effects on CSF cholinesterases in Alzheimer patients. Curr Alzheimer Res 2009;6:4-14.
- Bullock R, Touchon J, Bergman H, Gambina G, He Y, Rapatz G, et al. Rivastigmine and donepezil treatment in moderate to moderately severe Alzheimer’s disease over a 2-year period. Curr Med Res Opin 2005;21:1317-27.
- Ancoli-Israel S, Amatniek J, Ascher S, Sadik K, Ramaswamy K. Effects of galantamine versus donepezil on sleep in patients with mild to moderate Alzheimer disease and their caregivers: a double-blind, head-to-head, randomized pilot study. Alzheimer Dis Assoc Disord 2005;19:240-5.
- Porsteinsson AP, Grossberg GT, Mintzer J, Olin JT. Memantine treatment in patients with mild to moderate Alzheimer’s disease already receiving a cholinesterase inhibitor: a randomized, double-blind, placebo-controlled trial. Curr Alzheimer Res 2008;5:83-9.
- Loveman E, Green C, Kirby J, Takeda A, Picot J, Payne E, et al. The clinical and cost-effectiveness of donepezil, rivastigmine, galantamine and memantine for Alzheimer’s disease. Health Technol Assess 2006;10.
- Evers S, Goossens M, de Vet H, van Tulder M, Ament A. Criteria list for assessment of methodological quality of economic evaluations: Consensus on Health Economic Criteria. Int J Technol Assess Health Care 2005;21:240-5.
- Weinstein MC, O’Brien B, Hornberger J, Jackson J, Johannesson M, McCabe C, et al. Principles of good practice for decision analytic modeling in health-care evaluation: report of the ISPOR Task Force on Good Research Practice: modeling studies. Value Health 2003;6:9-17.
- Green C. Modelling disease progression in Alzheimer’s disease: a review of modelling methods used for cost-effectiveness analysis. Pharmacoeconomics 2007;25:735-50.
- Sheehan B, Phillips P, Juszczak E, Adams J, Baldwin A, Ballard C, et al. DOMINO-AD protocol: donepezil and memantine in moderate to severe Alzheimer’s disease – a multicentre RCT. Trials 2009;10.
- Jonsson L, Wimo A. The cost of dementia in Europe. A review of the evidence, and methodological considerations. Pharmacoeconomics 2009;27:391-403.
- Oremus M. Systematic review of economic evaluations of Alzheimer’s disease medications. Expert Rev Pharmacoecon Outcomes Res 2008;8:273-89.
- Weimer DL, Sager MA. Early identification and treatment of Alzheimer’s disease: Social and fiscal outcomes. Alzheimers Dement 2009;5:215-26.
- Getsios D, Blume S, Ishak KJ, MacLaine G. Cost-effectiveness of screening and treatment of Alzheimer’s disease with donepezil in the United Kingdom. Value Health 2009;12.
- Feldman H, Gauthier S, Hecker J, Vellas B, Hux M, Xu Y, et al. Economic evaluation of donepezil in moderate to severe Alzheimer disease. Neurology 2004;63:644-50.
- Fuh JL, Wang SJ, Fuh JL, Wang SJ. Cost-effectiveness analysis of donepezil for mild to moderate Alzheimer’s disease in Taiwan. Int J Geriatr Psychiatry 2008;23:73-8.
- Getsios D, Blume S, Ishak KJ, MacLaine G. Cost-effectiveness study in patients with mild to moderately severe Alzheimer’s disease: projected benefits of donepezil in the United Kingdom. Value Health 2009;12.
- Getsios D, Blume S, Ishak KJ, MacLaine G. Cost-effectiveness of donepezil in the treatment of mild to moderate Alzheimer’s disease: a UK evaluation using discrete-event simulation. Pharmacoeconomics 2010;28:411-27.
- Lopez-Bastida J, Hart W, Garcia-Perez L, Linertova R. Cost-effectiveness of donepezil in the treatment of mild or moderate Alzheimer’s disease. J Alzheimers Dis 2009;16:399-407.
- Lu S, Hill J, Fillit H. Impact of donepezil use in routine clinical practice on health care costs in patients with Alzheimer’s disease and related dementias enrolled in a large medicare managed care plan: a case–control study. Am J Geriatr Pharmacother 2005;3:92-102.
- Mesterton J, By A, Sandelin R, Jonsson L. Cost-effectiveness of donepezil in Alzheimer’s disease in Sweden. Value Health 2009;12.
- Pattanaprateep O, Phongchareonsuk P, Chaikledkaew U. The cost-effectiveness of donepezil and rivastigmine in the treatment of Alzheimer’s disease in Thailand private hospital. Value Health 2005;8:314-15.
- Teipel SJ, Ewers M, Reisig V, Schweikert B, Hampel H, Happich M. Long-term cost-effectiveness of donepezil for the treatment of Alzheimer’s disease. Eur Arch Psychiatry Clin Neurosci 2007;257:330-6.
- Feldman H, Gauthier S, Hecker J, Vellas B, Hux M, Xu Y, et al. Pharmacoeconomic benefits of donepezil treatment in severe Alzheimer’s disease. J Am Geriatr Soc 2004;52.
- Shah SN, Feldman H, Gauthier S, Hecker J, Vellas B, Hux M, et al. Pharmacoeconomic benefits of donepezil treatment in severe Alzheimer’s disease. Neurobiol Aging 2004;25.
- Feldman H, Hux M, Schwam EM. Economic evaluation of donepezil in moderate to severe Alzheimer disease – reply. Neurology 2005;64.
- Faltraco F, Happich M, Reisig V, Teipel SJ, Moeller HJ, Hampel H. Treatment of Alzheimer’s disease with cholinesterase inhibitors in Germany: a cost-effectiveness study. Neurobiol Aging 2004;25.
- Gauthier S, Feldman H, Hecker J, Vellas B, Ames D, Subbiah P, et al. Efficacy of donepezil on behavioral symptoms in patients with moderate to severe Alzheimer’s disease. Int Psychogeriatr 2002;14:389-404.
- Brennan A, Nagy B, Brandtmuller A, Thomas SK, Sullivan SD, Akehurst R. The cost-utility of exelon patch in the management of patients with moderate Alzheimer’s disease in the United Kingdom. Value Health 2007;10.
- Suh GH, Jung HY, Lee CU, Choi S, Suh GH, . Korean Galantamine Study Group . Economic and clinical benefits of galantamine in the treatment of mild to moderate Alzheimer’s disease in a Korean population: a 52-week prospective study. J Korean Med Sci 2008;23:10-7.
- Suh GH. Modeling the cost-effectiveness of galantamine for mild to moderately severe Alzheimer’s disease in Korea. Value Health 2009;12:49-54.
- Antonanzas F, Rive B, Badenas JM, Gomez-Lus S, Guilhaume C. Cost-effectiveness of memantine in community-based Alzheimer’s disease patients: An adaptation in Spain. Eur J Health Econ 2006;7:137-44.
- Gagnon M, Rive B, Hux M, Guilhaume C. Cost-effectiveness of memantine compared with standard care in moderate-to-severe Alzheimer disease in Canada. Can J Psychiatry 2007;52:519-26.
- Guilhaume C, Rive B, Francois C, Livingston G, Katona C. External validation of the probabilistic Markov model estimating the cost effectiveness of memantine versus standard care in Alzheimer disease from a UK perspective. Value Health 2005;8.
- Jonsson L. Cost-effectiveness of memantine for moderate to severe Alzheimer’s disease in Sweden. Am J Geriatr Pharmacother 2005;3:77-86.
- Toumi M, Lamure M, Grishchenko M, Cochran J, Rive B. Cost-effectiveness of memantine in the treatment of moderate to severe Alzheimer’s disease in Norway. Value Health 2009;12.
- Weycker D, Taneja C, Edelsberg J, Erder MH, Schmitt FA, Setyawan J, et al. Cost-effectiveness of memantine in moderate-to-severe Alzheimer’s disease patients receiving donepezil. Curr Med Res Opin 2007;23:1187-97.
- Paradise M, Walker Z, Cooper C, Blizard R, Regan C, Katona C, et al. Prediction of survival in Alzheimer’s disease – the LASER-AD longitudinal study. Int J Geriatr Psychiatry 2009;24:739-47.
- Bakchine S, Loft H. Memantine treatment in patients with mild to moderate Alzheimer’s disease: results from a randomised, double-blind, placebo-controlled 6-month study. J Alzheimers Dis 2008;13:97-107.
- Peskind ER, Potkin SG, Pomara N, Ott BR, Graham SM, Olin JT, et al. Memantine treatment in mild to moderate Alzheimer disease: a 24-week randomized, controlled trial. Am J Geriatr Psychiatry 2006;14:704-15.
- Curtis L. Unit Costs of Health Social Care 2010. Canterbury: PSSRU, University of Kent; 2010.
- Jonsson L, Andreasen N, Kilander L, Soininen H, Waldemar G, Nygaard H, et al. Patient- and proxy-reported utility in Alzheimer disease using the EuroQoL. Alzheimer Dis Assoc Disord 2006;20:49-55.
- Macdonald A, Cooper B. Long-term care dementia services: an impending crisis. Age Ageing 2007;36:16-22.
- Iglehart D. Simulating Stable Stochastic Systems, V: comparison of ratio estimators. Naval Res Logist Quart 1975;22:553-65.
- Feldman H, Gauthier S, Hecker J, Vellas B, Emir B, Mastey V, et al. Donepezil MSAD Study Investigators Group. Efficacy of donepezil on maintenance of activities of daily living in patients with moderate to severe Alzheimer’s disease and the effect on caregiver burden. J Am Geriatr Soc 2003;51:737-44.
- Bond M, Rogers G, Peters J, Anderson R, Hoyle M, Miners A, et al. The Effectiveness and Cost-Effectiveness of Donepezil, Galantamine, Rivastigmine and Memantine for the Treatment of Alzheimer’s Disease (review of TA111): A Systematic Review and Economic Model n.d. www.nice.org.uk/guidance/index.jsp?action=folder%26o=51051 (accessed 6 December 2010).
- Livingston G, Katona C, Roch B, Guilhaume C, Rive B. A dependency model for patients with Alzheimer’s disease: its validation and relationship to the costs of care – the LASER-AD study. Curr Med Res Opin 2004;20:1007-16.
- Stern Y, Tang MX, Albert MS, Brandt J, Jacobs DM, Bell K, et al. Predicting time to nursing home care and death in individuals with Alzheimer disease. JAMA 1997;277:806-12.
- Stern Y, Albert SM, Sano M, Richards M, Miller L, Folstein M, et al. Assessing patient dependence in Alzheimer’s disease. J Gerontol 1994;49:216-22.
- Hoyle M, Anderson R. Whose costs and benefits? Why economic evaluations should simulate both prevalent and all future incident patient cohorts. Med Decis Making 2010;30:426-37.
- Wolstenholme J, Fenn P, Gray A, Keene J, Jacoby R, Hope T. Estimating the relationship between disease progression and cost of care in dementia. Br J Psychiatry 2002;181:36-42.
- Livingston G, Katona C, Francois C, Guilhaume C, Cochran J, Sapin C. Characteristics and health status change over 6 months in people with moderately severe to severe Alzheimer’s disease in the UK. Int Psychogeriatr 2006;18:527-38.
- National Institute for Health and Clinical Excellence (NICE) . Guide to the Methods of Technology Appraisal 2008.
- Galasko D, Schmitt F, Thomas R, Jin S, Bennett D, Ferris S. Detailed assessment of activities of daily living in moderate to severe Alzheimer’s disease. J Int Neuropsychol Soc 2005;11:446-53.
- Briggs A, Claxton K, Sculpher M. Decision modelling for health economic evaluation. Oxford: Oxford University Press; 2008.
- Doraiswamy PM, Bieber F, Kaiser L, Krishnan KR, Reuning-Scherer J, Gulanski B. The Alzheimer’s Disease Assessment Scale: patterns and predictors of baseline cognitive performance in multicenter Alzheimer’s disease trials. Neurology 1997;48:1511-17.
- Miller EA, Schneider LS, Zbrozek A, Rosenheck RA. Sociodemographic and Clinical Correlates of Utility Scores in Alzheimer’s Disease. Value Health 2008;11:1120-30.
- Naglie G, Tomlinson G, Tansey C, Irvine J, Ritvo P, Black SE, et al. Utility-based quality of life measures in Alzheimer’s disease. Qual Life Res 2006;15:631-43.
- Ekman M, Berg J, Wimo A, Jonsson L, McBurney C. Health utilities in mild cognitive impairment and dementia: a population study in Sweden. Int J Geriatr Psychiatry 2007;22:649-55.
- Wlodarczyk JH, Brodaty H, Hawthorne G, Wlodarczyk JH, Brodaty H, Hawthorne G. The relationship between quality of life, Mini-Mental State Examination, and the Instrumental Activities of Daily Living in patients with Alzheimer’s disease. Arch Gerontol Geriatr 2004;39:25-33.
- Andersen CK, Wittrup-Jensen KU, Lolk A, Andersen K, Kragh S, Andersen CK, et al. Ability to perform activities of daily living is the main factor affecting quality of life in patients with dementia. Health Qual Life Outcomes 2004;2.
- Karlawish JH, Zbrozek A, Kinosian B, Gregory A, Ferguson A, Low DV, et al. Caregivers’ assessments of preference-based quality of life in Alzheimer’s disease. Alzheimers Dement 2008;4:203-11.
- Karlawish JH, Casarett D, Klocinski J, Clark CM. The relationship between caregivers’ global ratings of Alzheimer’s disease patients’ quality of life, disease severity, and the caregiving experience. J Am Geriatr Soc 2001;49:1066-70.
- Sands LP, Ferreira P, Stewart AL, Brod M, Yaffe K. What explains differences between dementia patients’ and their caregivers’ ratings of patients’ quality of life?. Am J Geriatr Psychiatry 2004;12:272-80.
- Vogel A, Mortensen EL, Hasselbalch SG, Andersen BB, Waldemar G. Patient versus informant reported quality of life in the earliest phases of Alzheimer’s disease. Int J Geriatr Psychiatry 2006;21:1132-8.
- Kerner DN, Patterson TL, Grant I, Kaplan RM. Validity of the Quality of Well-Being Scale for Patients with Alzheimer’s disease. J Aging Health 1998;10:44-61.
- Sano M, Albert SM, Tractenberg RE, Schittini M. Developing utilities: quantifying quality of life for stages of Alzheimer’s disease as measured by the clinical dementia rating. J Mental Health Aging 1999;5:59-68.
- Neumann PJ, Kuntz KM, Leon J, Araki SS, Hermann RC, Hsu MA, et al. Health utilities in Alzheimer’s disease: a cross-sectional study of patients and caregivers. Med Care 1999;37:27-32.
- Huang HL, Chang MY, Tang JSH, Chiu YC, Weng LC. Determinants of the discrepancy in patient- and caregiver-rated quality of life for persons with dementia. J Clin Nurs 2008;18:3107-17.
- Perneczky R, Wagenpfeil S, Komossa K, Grimmer T, Diehl J, Kurz A, et al. Mapping scores onto stages: mini-mental state examination and clinical dementia rating. Am J Geriatr Psychiatry 2006;14:139-44.
- Curtis L. Unit Costs of Health Social Care 2009. Canterbury: PSSRU, University of Kent; 2009.
- Department of Health . NHS Reference Costs 2008–9 2010.
- British Medical Association and Royal Pharmaceutical Society of Great Britain . British National Formulary 2009.
- British Medical Association and Royal Pharmaceutical Society of Great Britain . British National Formulary 2010.
- Curtis L. Unit Costs of Health Social Care 1998. Canterbury: PSSRU, University of Kent; 1998.
- Curtis L. Unit Costs of Health Social Care 1999. Canterbury: PSSRU, University of Kent; 1999.
- Curtis L, Netten A. Unit Costs of Health Social Care 2004. PSSRU, University of Kent; 2004.
- Hoyle M. Future drug prices and cost-effectiveness analyses. Pharmacoeconomics 2008;26:589-602.
- Rogers G, Hoyle M, Thompson-Coon J, Pitt M, Moxham T, Liu Z, et al. Dasatinib and nilotinib for imatinib-resistant or intolerant chronic myeloid leukaemia: a systematic review and economic evaluation. Health Technol Assess 2012.
- Thompson-Coon J, Hoyle M, Green C, Liu Z, Welch K, Moxham T, et al. Bevacizumab, sorafenib tosylate, sunitinib and temsirolimus for renal cell carcinoma: a systematic review and economic evaluation. Health Technol Assess 2012.
- Bond M, Wyatt K, Lloyd J, Welch K, Taylor RS. Systematic review of the effectiveness and cost-effectiveness of weight management schemes for the under fives. Health Technol Assess 2009;13.
- Bond M, Mealing S, Anderson R, Elston J, Weiner G, Taylor RS, et al. The effectiveness and cost-effectiveness of cochlear implants for severe to profound deafness in children and adults: a systematic review and economic model. Health Technol Assess 2009;13.
- Bond M, Pitt M, Akoh J, Moxham T, Hoyle M, Anderson R. The effectiveness and cost-effectiveness of methods of storing donated kidneys from deceased donors: a systematic review and economic model. Health Technol Assess 2009;13.
- Rogers G, Elston J, Garside R, Roome C, Taylor RS, Younger P, et al. The harmful health effects of recreational ecstasy: a systematic review of observational evidence. Health Technol Assess 2009;13.
- Taylor RS, Elston J. The use of surrogate outcomes in model-based cost-effectiveness analyses: a survey of UK health technology assessment reports. Health Technol Assess 2009;13.
- Main C, Shepherd J, Anderson R, Rogers G, Thompson-Coon J, Liu Z, et al. Systematic review and economic analysis of the comparative effectiveness of different inhaled corticosteroids and their usage with long-acting beta2 agonists for the treatment of chronic asthma in children under the age of 12 years. Health Technol Assess 2008;12.
- Fox M, Mealing S, Anderson R, Dean J, Stein K, Price A, et al. The Clinical Effectiveness and Cost-effectiveness of Cardiac Resynchronisation (Biventricular Pacing) for Heart Failure: a systematic review and economic model. Health Technol Assess 2007;11.
- Pearson M, Garside R, Moxham T, Anderson R. Preventing unintentional injuries to children in the home: a systematic review of the effectiveness of programmes supplying and/or installing home safety equipment. Health Prom Int 2011;26:376-92.
- Mecocci P, Bladstrom A, Stender K. Effects of memantine on cognition in patients with moderate to severe Alzheimer’s disease: post-hoc analyses of ADAS-cog and SIB total and single-item scores from six randomized, double-blind, placebo-controlled studies. Int J Geriatr Psychiatry 2009;24:532-8.
- Gauthier S, Loft H, Cummings J. Improvement in behavioural symptoms in patients with moderate to severe Alzheimer’s disease by memantine: a pooled data analysis. Int J Geriatr Psychiatry 2008;23:537-45.
- Wilcock GK, Ballard CG, Cooper JA, Loft H. Memantine for agitation/aggression and psychosis in moderately severe to severe Alzheimer’s disease: A pooled analysis of 3 studies. J Clin Psychiatry 2008;69:341-8.
- Farlow MR, Graham SM, Alva G. Memantine for the treatment of Alzheimer’s disease. Drug Safety 2008;31:577-85.
- Quentin W, Riedel-Heller S, Luppa M, Rudolph A, König H-H. Cost-of illness studies of dementia: a systematic review focusing on stage dependency of costs. Acta Psychiatr Scand 2010;121:243-59.
- Lowin A, Knapp M, McCrone P. Alzheimer’s disease in the UK: comparative evidence on cost of illness and volume of health services research funding. Int J Geriatr Psychiatry 2001;16:1143-8.
- Souêtre E, Thwaites R, Yeardley H. Economic impact of Alzheimer’s disease in the United Kingdom: Cost of care and disease deverity for non-institutionalised patients with Alzheimer’s disease. Br J Psychiatry 1999;174:51-5.
- Parliamentary Office of Science and Technology (POST) . Alzheimer’s & Dementia 2007.
- Kavanagh S, Knapp M. Costs and cognitive disability: modelling the underlying associations. Br J Psychiatry 2002;180:120-5.
- Martin J, Meltzer H, Elliot D. The Prevalence of Disability Among Adults: OPCS Surveys of Disability in Great Britain Report I 1988.
- Mendiondo MS, Ashford JW, Kryscio RJ, Schmitt FA. Modelling mini mental state examination changes in Alzheimer’s disease. Stat Med 2000;19:1607-16.
Appendix 1 Outcome measures
These tables of outcome measures have been copied from the previous TAR (TA111, appendix 6). 151
Global outcome measures
| Type | Construct measure and scoring | Critical appraisal |
|---|---|---|
| CDR and CDR-SB |
Cognitive impairment in memory, orientation, judgement/problem-solving, community affairs, home/hobbies, and personal care 0 = none, 0.5 = questionable, 1 = mild, 2 = moderate, 3 = severe CDR-SB is a modified form that sums the ratings in the six performance categories to give a global dementia ranking |
Provides physicians with a global rating that encompasses a broad range of patient characteristics and can be used by neurologists, psychiatrists and psychologists, and focuses on cognition, not on items that may be related to other medical, emotional or social conditions Good inter-rater reliability and fair-to-good concurrent validity. Although no work has been done on test–retest reliability, nothing so far suggests that researchers should avoid this scale when trying to stage AD. The CDR can be used as an eligibility criterion for trial participation or as an outcome measure |
| GDS |
Progressive stages of cognitive impairment 1 (no cognitive decline) to 7 (very severe cognitive decline) |
Most frequently used, but ratings can mis-state a patient’s severity. Problems might arise when the GDS is used as an inclusion criterion for participation in a RCT. The ability to enrol desired patients could be threatened if the GDS misidentifies the stages of dementia The GDS should not be used to stage dementia in AD drug trials |
| CGIC scale and the global improvement index with interviewing of patients CIBIC and with caregiver input (CIBIC-M or –Plus) |
Overall improvement in patient health status assessed by clinician (with caregiver) 1 (very much improved) to 7 (very much worse) A number of different variations are available Scale is non-parametric and of a non-interval nature |
Fair-to-good test–retest and inter-rater reliability and concurrent validity. Results may arise from fact that groups providing global assessments do not base their ratings on the same domains. Physicians take clinical psychopathology as the basis of determining global improvement, nurses believe the amount of work needed to care for patients was important. This instrument also includes a caregiver opinion, results may differ depending on whether or not the rater first interviews the patient or caregiver. The number of different variations may have reduced the validity |
| GBS |
Motor function, intellectual function, emotional function and symptoms common to demented patients 0 (normal function or absence of symptoms) to 6 (maximal disturbance or presence of symptoms) |
Psychometric properties range from fair to good. Scale is useful mean of quantifying dementia in drug trials. GBS should not be used as a diagnostic tool |
| MENFIS | A modification of the GBS prepared by the study authors for a previous study. Scores range from 0 to 78, with a higher score indicating a greater degree of deficit | Unable to source data on reliability and validity |
| PGA | Seven-point Likert scale ranges from 1 (very much improved) through 4 (no change) to 7 (very much worse) | Unable to source data on reliability and validity |
Cognitive outcome measurement scales
| Type | Construct measure and scoring | Critical appraisal |
|---|---|---|
| ADAS-cog |
Orientation, memory, language and praxis 0–70, with higher scores indicating greater impairment |
Limited in its ability to detect change at one end or the other of the severity continuum. For many subtests, detection of improvement appears only possible for a restricted range of severity levels Limitations should be considered when used as a drug efficacy measure. The rate of decline of AD using ADAS-cog suggests that the decline is non-linear and not a constant, but is dependent on the stage of the disease. Content and ecological validity are lacking |
| BVRT | Assesses visual perception, visual memory and visuoconstructive abilities. The test has three alternate forms, each consisting of 10 designs. In addition, there are four possible modes of administration. Scoring is based on an assessment of the number and types of errors made compared with the expected scores found in the norm tables. The wider the discrepancy in favour of the expected score, the more probable it is that the participant has suffered neurological impairment |
The interscorer agreement for total error score is high and for major categories of errors reliability is moderate to high. A correlation of 0.42 was found between the Benton and the Digit Span Wechsler Adult Intelligence Scale subtest. This low correlation indicates discriminate validity, as the Benton was created to supplement the Digit Span test Educational level may influence a participant’s score on the test. Participants with higher educational levels tend to use a more exhaustive exploration strategy during the recognition phase of the test, allowing them to perform better than participants with lower educational levels. The executive working memory component is more efficient in participants with higher educational levels |
| Computerised Memory Battery (CMBT) | A computerised version of the Memory Assessment Clinical (MAC) Battery designed to simulate critical cognitive tasks: name–face association (delayed recall and total acquisition); first and last names (total acquisition), facial recognition (first miss and total correct); telephone number recall (seven-digit and 10-digit number correct); house and object placement task (total acquisition and first trial) | The MAC-Q questionnaire demonstrates internal consistency and test–retest reliability |
| Clinical Global Impression-item 2 (CGI-2) | This rating instrument expresses the global change in observable cognitive functioning directly on a transitional scale ranging from 1 (very much improved) to 7 (very much deteriorated) as rated by a clinician | This is a subtest of the CGI; it is easy and quick to administer, and is widely used in clinical and trial settings |
| Digit symbol substitution subtest (DSST) of the Wechsler Adult Intelligence Scale-Revised | Participants fill in a grid of 100 blank squares, each paired with a randomly assigned number from 1 to 9, using a key that pairs each number with a different symbol. The score is the number of correct answers after 90 seconds | Performance on this test is affected by many different components, so the test lacks specificity. Participants with impaired vision or visuomotor coordination, pronounced motor slowing or low education levels are at a disadvantage |
| Fuld object–memory evaluation (FOME) | Ten-item assessment with 10 common objects in a bag are presented ‘to determine whether the patient can identify objects by touch’ (stereognosis). The test was developed while testing large samples of aged adults, nursing home residents and community active people, for whom norms are provided | Unable to source data on reliability and validity |
| MMSE |
Eleven questions on orientation, memory, concentration, language and praxis Scale ranges from 0 to 30. Higher score indicates less impairment. There is no range of scores that can be rigidly and universally applied to indicate dementia severity, i.e. as a marker of mild, moderate and severe dementia. In clinical trials often a score of 21–26 is associated with mild AD, moderate AD is associated with a MMSE score of 10–20 and severe AD is usually associated with a MMSE score of < 10. This may be less suitable within routine daily practice |
Good reliability and validity for its original purpose of screening for dementia; short screening scales are not designed to measure more subtle aspects of cognition. Short scales such as the MMSE may indicate little or no change over time in subjects who would otherwise be shown to have declined substantially if another scale had been used to measure change in status. Not an ideal outcome measure for AD drug trials, especially if the expected benefits are not large. It has dependence on intact language ability and there are no available validated versions in languages suitable for use with ethnic minorities. It cannot be used effectively in people with low IQs or learning disabilities |
| SIB | A measure of cognition that was developed to assess a range of cognitive functioning in individuals who are too impaired to complete standard neuropsychological tests and takes into account specific behavioural and cognitive deficits associated with severe dementia. It is composed of 40 simple one-step commands that are scored on a three-point scale and are presented in conjunction with gestural cues. The SIB also allows for non-verbal and partially correct responses. The six major subscales are attention, orientation, language, memory, visuospatial ability and construction. Overall scores range from 0 to 100, with positive scores indicating clinical improvement | The SIB has been shown to be psychometrically reliable and clinical norms are available. No further details of reliability and validity have been sourced |
| SKT | A psychometric test battery for the assessment of memory and attention. The SKT consists of nine 1-minute subtests that are partly speed oriented and partly span orientated: scaled subtest scores are aggregated to a SKT total status score ranging from 1 (very good) to 27 (very poor) | This test has shown good test–retest reliability. Correlations with other cognitive measures support its validity as a cognitive outcome measure for AD |
| Ten-point clock-drawing test | This is a screening test for dementia in particular for assessing visuospatial and executive functions. Patients have to drawn in the numbers of digits placed in a predrawn circle | This test has been shown to be both reliable and valid and is simple and easy to administer with good sensitivity and specificity |
| TMT | Assesses speed of visual search, attention, mental flexibility and motor function. The test has two parts (A) drawing a line linking numbers in sequence and (B) drawing a line linking letters in sequence. The reviewer calls any mistakes to the attention of the participant and these must be corrected before progressing. The score is the time taken to successfully complete a test | Reliability is reported to be higher for part A than for part B, which requires more information-processing ability and is more sensitive to brain damage. Reliability is restricted owing to the use of time scores rather than both error counts and time scores, as error correction may take longer in some participants than others. Scores are strongly affected by the participant’s education level |
| Wechsler logical memory test | This test is one of 13 subtests of the Wechsler Memory Scale-Revised (WMS-R). The first subtest is for screening purposes, and the other 12 are grouped into five separate memory areas. The test manual provides guidelines for scoring and weighting, and provides norms for individuals aged 16–74 years, with information about significant differences between any two scores | Test–retest reliability and concurrent validity with a verbal learning test are adequate for the whole WMS-R test. Level of education affects a participant’s score. Normative data for those aged 75 years and over is lacking. The score is more heavily influenced by verbal memory performance than by other memory components |
Functional and quality of life outcome measurement scales
| Type | Construct measure and scoring | Critical appraisal |
|---|---|---|
| ADCS-ADL | This rating scale is a 23-item assessment of ADLs that is scored from 0 (greatest impairment) to 78. It evaluates activities of daily living | The ADCS-ADL is a structured questionnaire originally created to assess functional capacity over a broad range of severity of dementia. The ADAS-ADL19 is a subset of the original inventory and focuses on items appropriate for the assessment of later stages of dementia. The sensitivity and reliability of this modification has been established |
| ADFACS | Scale consists of 10 items for instrumental ADL: ability to use the telephone, performing household tasks, using household appliances, handling money, shopping, preparing food, ability to get around both inside and outside the home, pursuing hobbies and leisure activities, handling personal mail, grasping situations or explanations. Scale has a range of 0 to 54, where lower scores correspond to better function. Test takes approximately 20 minutes to complete |
Full assessment of psychometric properties not yet published. Has face validity for those with mild to moderate AD The ADL items chosen for this scale have been demonstrated to be sensitive to change over 12 months, correlate well with MMSE scores, and have good test–retest reliability (although several questions have been modified in the scale) |
| BGP | Consists of 35 items (scored 0, 1 or 2) assessing observable aspects of cognition, function and behaviour. A high score indicates worse function | Unable to source data on reliability and validity |
| BADLS | Caregiver assessment of 20 ADLs. Categories included are food, eating, drinks, drinking, dressing, hygiene, teeth, bath, toilet, transferring, mobility, orientation to time and space, communication, telephone, housework/gardening, shopping, finances, hobbies and transport. Scores range from 0 to 60, with higher scores indicating better function | Designed specifically for use with patients with dementia. Face validity was measured by asking carers whether or not items were important, and construct validity was confirmed by principal components analysis. Concurrent validity was assessed by observed performance, the test has good content validity, and there is good test–retest reliability. The test is shown to correlate well with performance ADLs and tests of cognitive function |
| CMCS | A modified CGRS. This a seven-item scale using a Likert-type scoring method. Questions include comprehension to time and place, carrying out conversation, cooperation, restlessness, dressing, social activities and leisure. Negative change relates to clinical improvement | Reliability demonstrated. Unable to source data on validity |
| DAD | This rating scale is a 46-item structured interview or questionnaire for the caregiver that is scored from 0 to 100 (least impairment). It evaluates ADLs and takes approximately 20 minutes to complete. It is based on a recognised conceptual definition of disability from the WHO | The DAD scale demonstrates a high degree of internal consistency and excellent inter-rater and test–retest reliability. Full details of concurrent and construct validity not yet published |
| FAST | Assesses the magnitude of progressive functional deterioration in patients with dementia by identifying characteristic progressive disabilities. Seven major stages range from normal (stage 1) to severe dementia (stage 7) | FAST has been shown to be a reliable and valid assessment technique for evaluating functional deterioration in AD patients throughout the entire course of the illness. Because the elements of functional capacity incorporated in FAST are relatively universal and readily ascertainable, as well as characteristic of the course of AD, FAST can serve as a strong diagnostic and differential diagnostic aid for clinicians |
| GHQ-30 | The GHQ is a self-report psychiatric screening test, and items include questions on depression and unhappiness, anxiety and felt psychological disturbance, social impairment and hypochondriasis. Participants rate themselves on a four-point severity scale, according to how they have recently experienced each GHQ item: better than usual, same as usual, worse than usual, or much worse than usual. Normally each item is scored either 0 or 1, depending on which severity choice is selected. Individual items are summed to give the total score | GHQ-30 is based on Medical Outcomes Study SF-36, which is extensively validated |
| IADL |
For women, the set of behaviour assessed include telephoning, shopping, food preparation, housekeeping, laundering, use of transport, use of medicine and ability to handle money. For men, the areas of food preparation, housekeeping and laundering are excluded. Each of the behavioural areas is given a score of 0 or 1, leading to an overall score that ranges from 0 to 8 for women and from 0 to 5 for men |
The IADL is a very frequently used and often cited instrument for assessing the instrumental competence of elderly patients. The scale is well anchored from a theoretical point of view and the behaviours that are included are likely to be affected in the first stages of dementia |
| IDDD |
The IDDD measures functional disability in self-care (16 items such as washing, dressing and eating) and complex activities (17 items such as shopping, writing and answering the telephone) Severity of impairment is rated on a seven-point scale, where 1–2 = no or slight impairment, 3–4 = mild impairment, 5–6 = moderate impairment, 7 = severe impairment, giving a total range score of 22–231 |
This scale appears to be appropriate to assess community-living patients with mild and moderate levels of dementia. It assesses a substantial proportion of complex activities likely to be affected during the first stages of the AD. The number of non-redundant items in the scale is viewed positively, as it may increase the sensitivity of the tool. Empirical information on the testing of the IDDD and its measurement properties is seriously lacking |
| PSMS | Measured through competence of six behaviours: toileting, feeding, dressing, grooming, locomotion and bathing. It can be completed by untrained staff based on information from subjects, caregivers, friends, etc. Each behavioural area is given a score of 1 or 0, with over score ranging from 0 to 6. Using Guttman scaling, each scale point has five descriptive scale points | Brief assessment of activities of daily living. Theoretically well grounded, it has been proven useful for evaluation of institutionalised elderly, but has a ceiling effect for those living in the community. Testing of psychometric properties is incomplete |
| PDS |
PDS examines activities of daily living and instrumental activities of daily living. Examples are extent to which a patient can leave the immediate neighbourhood, use of familiar household implements, involvement in family finances and budgeting Each question is scored by measuring the distance along the line on a scale from 0 to 100, with higher scores reflecting better functionality. A composite score is derived from averaging across the items for a maximal score of 100 The scale is sometimes classified as a measure of QoL |
This scale has been shown to be sensitive to three severity stages of dementia, although there is some debate on whether or not the content is adequate to assess those with moderately severe AD. The scale was systematically developed and tested on a fairly large sample of AD patients (although the mean age of the final test group was only 69.5 years) Test–retest reliability was determined in 123 patients, giving stage correlations (rs) of 0.889 for early AD (14 participants), 0.775 for 44 middle-stage participants and 0.775 for 65 late-stage participants. A moderate degree of correlation has been demonstrated between PDS and ADAS-cog scores (rp = –0.57 to –0.64) There is considerable reduplication within the scale – four questions relate to handling finances, but there are no items pertaining to basic activities such as washing, dressing and toileting. The scale is, therefore, not thought to have adequate content to assess people with moderately severe AD as it does not assess the wide range of daily living skills affected at different stages of the disease. There are high levels of between and within patient variability (in the order of 12 points), which may make it less suited to detect differences over short time periods |
| QoL (patient and caregiver scales) | This assessment was a seven-item patient-rated scale evaluating the patients perceptions of their well-being in terms of relationships, eating and sleeping, and social and leisure activities. The tests is conducted by interview. Scored on an analogue scale between 0 (worst quality) to 50 (best quality) | This instrument has not been validated in patients with AD, but was selected because no QOL instrument has been validated in this population |
| Unified ADL |
All self-care and mobility variables commonly used to assess patient’s functional status A 20-item scale was produced. The need for assistance is scored for every item, on a 10-point scale |
The psychometric properties of this scale, resulting from the combination of existing evaluations, have not been published |
Behaviour and mood outcome measurement scales
| Type | Construct measure and scoring | Critical appraisal |
|---|---|---|
| BEHAVE-AD | A measure of the severity of behavioural symptoms in AD. It consists of 25 symptoms group on to seven categories. Each symptom is scored on the basis of severity on a four-point scale | The BEHAVE-AD has been shown to be reliable and valid |
| BGP | A 35-item rating scale that is more commonly used in European trials | No information about the reliability or validity of this scale was found |
| NOSGER | Contains 30 items of behaviour, each rated on a five-point scale according to frequency of occurrence. Item scores are summarised into six dimension scores (memory, instrumental activities of daily life, self-care, mood, social behaviour and disturbing behaviour) | This scale has been validated, and has high inter-rater and test–retest reliability. The test correlates well with clinician’s global rating of change |
| NPI | Currently evaluates 12 items: delusions, hallucinations, dysphoria, anxiety, agitation, euphoria, apathy, irritability, disinhibition, aberrant motor behaviour, night-time behaviour and changes in appetite/eating behaviour. Psychometric properties were established on first 10 items. Total score for each domain is calculated by multiplying frequency rating by severity rating, adding domain scores to get a total score. Higher scores represent more problems. Maximum scores is 12 per domain, with either 10 or 12 domains assessed | Content validity has been established, reliability and validity are satisfactory. Limitations included: poor description of appraisal period for behavioural symptoms; no justification for scoring system; and, inter-rater reliability was poorly deserved |
Appendix 2 Literature search strategies
Clinical effectiveness search strategy
The MEDLINE search strategy below was translated and run in:
| Database | Search date |
|---|---|
| MEDLINE (Ovid) and MEDLINE In-Process & Other Non-Indexed Citations: 1950 to present | 16 November 2009 |
| EMBASE (Ovid): 1980 to 2009 week 46 | |
| PsycINFO (Ovid): 2002 to November week 2 2009 | |
| CCTR: 2009 Issue 4 | 13 November 2009 |
| CDSR: 2009 Issue 4 | |
| CRD databases: NHS EED, HTA, DARE | 16 November 2009 |
| ISI Web of Science: SCI | |
| ISI Web of Science: CPCI | |
| BIOSIS – via ISI Web of Science |
All searches were then rerun on 31 March 2010.
MEDLINE Ovid 1950 to present
Search date: 16 November 2009; rerun search date 31 March 2010.
-
Alzheimer Disease/
-
alzheimer*.tw.
-
1 or 2
-
Memantine/
-
Memantine.mp.
-
ebixa.mp.
-
axura.mp.
-
namenda*.mp.
-
or/4-8
-
Galantamine/
-
galantamin*.mp.
-
galanthamine.mp.
-
Epigalanthamin.mp.
-
Jilkon*.mp.
-
Lycoremin*.mp.
-
Nivalin*.mp.
-
Razadyne*.mp.
-
Reminyl*.mp.
-
or/10-18
-
donepezil*.mp.
-
donezepil*.mp.
-
aricept*.mp.
-
Memac*.mp.
-
Memorit*.mp.
-
Eranz*.mp.
-
or/20-25
-
rivastigmin*.mp.
-
exelon*.mp.
-
prometax*.mp.
-
or/27-29
-
30 or 26 or 19 or 9
-
3 and 31
-
Randomized controlled trial.pt.
-
randomized controlled trial/
-
(random$or placebo$).ti,ab,sh.
-
((singl$or double$or triple$or treble$) and (blind$or mask$)).tw,sh.
-
or/33-36
-
clinical trial/
-
“controlled clinical trial”.pt.
-
(retraction of publication or retracted publication).pt.
-
37 or 38 or 39 or 40
-
32 and 41
-
(animals not humans).sh.
-
42 not 43
-
limit 44 to (english language and yr = “2004 -Current”)
Cost-effectiveness search strategy
This following MEDLINE search strategy was translated and run in:
| Database | Search date |
|---|---|
| MEDLINE (Ovid) and MEDLINE In-Process & Other Non-Indexed Citation: 1950 to present | 5 February 2010 |
| EMBASE (Ovid): 1980 to 2009 week 46 | |
| PsycINFO (Ovid): 2002 to November week 2 2009 | 4 February 2010 |
| CCTR: 2009 Issue 4 | |
| CDSR: 2009 Issue 4 | 13 November 2009 |
| CRD databases: NHS EED, HTA, DARE | 5 February 2010 |
| ISI Web of Science: SCI | |
| ISI Web of Science: CPCI | |
| BIOSIS – via ISI Web of Science | |
| EconLit |
MEDLINE Ovid 1950 to present
Searched 4 February 2010.
-
exp Alzheimer Disease/
-
alzheimer$.ti,ab.
-
1 or 2
-
Economics, Medical/
-
Economics, Nursing/
-
exp economics, hospital/
-
economics pharmaceutical/
-
ec.fs.
-
exp “Costs and Cost Analysis”/
-
exp Cost-benefit Analysis/
-
“Value of Life”/
-
exp Models, Economic/
-
exp “Fees and Charges”/
-
Resource Allocation/
-
exp Budgets/
-
budget*.tw.
-
(economic$or price$or pricing or financ$or fee$or pharmacoeconomic$or pharma economic$).tw.
-
(expenditure$not energy).tw.
-
(value$5 adj2 (money or monetary or life or lives or cost$2)).tw.
-
(economic adj2 burden).tw.
-
(resource$2 adj2 (use* or utili* or allocat*)).tw.
-
(cost$2 adj2 (benefit$or consequence* or analys* or saving* or breakdown* or lowering or estimat* or variable* or allocation* or control* or illness* or affordable* or instrument* or technolog* or fee* or charge$2 or utilit$or minim$or effective$or effective* or efficac*)).ab.
-
cost.ti.
-
22 or 23
-
or/4-24
-
Memantine/
-
Memantine.mp.
-
ebixa.mp.
-
axura.mp.
-
namenda*.mp.
-
Galantamine/
-
galantamin*.mp.
-
galanthamine.mp.
-
Epigalanthamin.mp.
-
Jilkon*.mp.
-
Lycoremin*.mp.
-
Nivalin*.mp.
-
Razadyne*.mp.
-
Reminyl*.mp.
-
donepezil*.mp.
-
donezepil*.mp.
-
aricept*.mp.
-
Memac*.mp.
-
Memorit*.mp.
-
Eranz*.mp.
-
rivastigmin*.mp.
-
exelon*.mp.
-
prometax*.mp.
-
or/26-48
-
3 and 25 and 49
-
limit 50 to (english language and yr = “2004 -Current”)
Quality of life and utilities search strategy
The following MEDLINE search strategy was translated and run in:
| Database | Search date |
|---|---|
| MEDLINE (Ovid) and MEDLINE In-Process & Other Non-Indexed Citations: 1950 to present | 6 January 2010 |
| EMBASE (Ovid): 1980 to 2009 week 46 | 5 February 2010 |
| PsycINFO (Ovid): 2002 to November week 2 2009 | 4 February 2010 |
| CCTR: 2009 Issue 4 | |
| CDSR: 2009 Issue 4 | 13 November 2009 |
| CRD databases: NHS EED, HTA, DARE | 5 February 2010 |
| ISI Web of Science: SCI | |
| ISI Web of Science: CPCI | |
| BIOSIS – via ISI Web of Science | |
| EconLit |
-
“Quality of Life”/
-
“Value of Life”/
-
((qualit$3 or value) adj2 life).tw.
-
quality-adjusted life years/
-
quality adjusted.tw.
-
(qaly* or qald* or qale* or qtime* or qualy).tw.
-
sickness impact profile/
-
(disabilit$3 adj2 life).tw.
-
daly.tw.
-
Health Status Indicators/
-
(sf36 or sf 36 or short form 36 or shortform 36 or sf thirtysix or sf thirty six or shortform thirstysix or shortform thirty six or short form thirty six or short form thirtysix or short form thirty six).tw.
-
(sf6 or sf 6 or short form 6 or shortform 6 or sf six or sfsix or shortform six or short form six).tw.
-
(sf12 or sf 12 or short form 12 or shortform 12 or sf twelve of sftwelve or shortform twelve or short form twelve).tw.
-
(sf16 or sf 16 or short form 16 or shortform 16 or sf sixteen or sfsixteen or shortform sixteen or short form sixteen).tw.
-
(sf20 or sf 20 or short form 20 or shortform 20 or sf twenty of sftwenty or shortform twenty of short form twenty).tw.
-
(euroqol or euro qol or eq5d or eq 5d).tw.
-
(hql or hqol or qol or hrqol).tw.
-
(hye or hyes).tw.
-
health$year$equivalent$.tw.
-
health utilit* or utilities or utility value*).tw.
-
hui$1.tw.
-
disutil$.tw.
-
rosser.tw.
-
(quality adj3 well).tw.
-
quality of wellbeing.tw.
-
qwb.tw.
-
willingness to pay.tw.
-
standard gamble$.tw.
-
(time trade off or time tradeoff or tto).tw.
-
(health adj3 (utilit$3 or value$2 or preference$2)).tw.
-
(visual analog$3 scale or VAS).tw.
-
(health adj2 (utilit$3 or value$2 or preference$2)).tw.
-
patient preference$2.tw.
-
or/1-33
-
mini mental state exam$.ti,ab.
-
((mmse or mmmse) adj5 alzheimer*).ti,ab.
-
modified mmse.ti,ab.
-
alzheimer$disease assessment scale$.ti,ab.
-
adas.ti,ab.
-
adas cog$.ti,ab.
-
cibic$.ti,ab.
-
progressive deterioration scale$.ti,ab.
-
(pds adj5 alzheimer*).ti,ab.
-
(clinical global impression of change or CGIC).tw.
-
clinic* interview based impression of change.tw.
-
(CDR or clinical dementia rating).tw.
-
alzheimer$.tw.
-
Alzheimer Disease/
-
47 or 48
-
34 and 49
-
(cognitive adj (scale* or rating or rate)).tw.
-
49 and 51
-
or/35-46
-
49 and 53
-
50 or 52 or 54
-
limit 55 to (english language and yr = “2004 -Current”)
Additional searches for economic modelling parameters
The following MEDLINE search strategy was translated and run in:
| Databases | Search date |
|---|---|
| Ovid MEDLINE: 1950 to present | 7 January 2010 |
| Ovid MEDLINE In-Process & Other Non-Indexed Citations | |
| BIOSIS via Web of Science | 8 January 2010 |
| EMBASE 1980 to 2009 week 46 | 7 January 2010 |
| ISI Web of Science: SCI-EXPANDED | 8 January 2010 |
| ISI Web of Science: CPCI-S | |
| NHS EED via CRD databases | |
| EconLit via First Search |
Ovid MEDLINE(R) 1950 to October week 2 2007
Searched: 24 October 2007.
-
Alzheimer Disease/
-
alzheimer$.tw.
-
1 or 2
-
exp Models, Economic/
-
*Models, Theoretical/
-
*Models, Organizational/
-
economic model$.ti,ab.
-
Markov Chains/
-
markov$.ti,ab.
-
Monte Carlo Method/
-
monte carlo.ti,ab.
-
exp Decision Theory/
-
(decision$adj2 (tree$or analy$or model$)).ti,ab.
-
or/4-13
-
3 and 14
-
limit 15 to (english language and yr = “2004 -Current”)
Additional searches for dementia model parameter, quality of life and utilities
The following MEDLINE search strategy was translated and run in:
| Databases | Search date |
|---|---|
| Ovid MEDLINE 1950 to present | 19 February 2010 |
| Ovid MEDLINE In-Process & Other Non-Indexed Citations | |
| EMBASE – 1980 to 2009 week 46 | |
| PsycINFO (Ovid): 2002 to November week 2 2009 | |
| NHS EED via CRD databases |
Ovid MEDLINE(R) 1950 to October week 2 2007
Search date: 19 February 2010.
-
Dementia/ (29,095)
-
*Dementia/ (22,077)
-
dementia.ti. (22,047)
-
2 or 3 (30,348)
-
exp Models, Economic/ (6944)
-
(economic next model* or markov* or monte next carlo).ti. (1847)
-
(economic next model* or markov* or monte next carlo).ab. (7968)
-
or/5–7 (14,883)
-
4 and 8 (28)
-
1 and 8 (29)
-
9 or 10 (33)
-
“Quality of Life”/ (79,428)
-
(quality adj2 life).ti. (26,019)
-
(quality adj2 life).ab. (87,761)
-
quality-adjusted life years/ (4171)
-
(sf36 or sf 36 or short form 36 or shortform 36 or sf thirtysix or sf thirty six or shortform thirstysix or shortform thirty six or short form thirty six or short form thirtysix or short form thirty six).tw. (9883)
-
(sf6 or sf 6 or short form 6 or shortform 6 or sf six or sfsix or shortform six or short form six).tw. (1012)
-
(sf12 or sf 12 or short form 12 or shortform 12 or sf twelve of sftwelve or shortform twelve or short form twelve).tw. (1382)
-
(sf16 or sf 16 or short form 16 or shortform 16 or sf sixteen or sfsixteen or shortform sixteen or short form sixteen).tw. (19)
-
(sf20 or sf 20 or short form 20 or shortform 20 or sf twenty of sftwenty or shortform twenty of short form twenty).tw. (288)
-
(euroqol or euro qol or eq5d or eq 5d).tw. (1832)
-
utilit*.ti. (12,510)
-
or/12-22 (141,512)
-
4 and 23 (1064)
-
9 or 24 (1085)
-
limit 25 to english language (905)
-
from 26 keep 1-905 (905)
Additional citation searching and ad hoc searches were performed for model parameters.
Appendix 3 Data extraction forms
Brodaty et al.96
| Design | Participants | Arms | Outcomes |
|---|---|---|---|
|
Brodaty et al . (2005) 96 Study design: parallel double-blind RCT Countries: USA, Australia, Canada, South Africa and New Zealand No. of centres: 93 Funding: none reported Length of follow-up (weeks): 26 Notes: |
No. randomised: 971 MMSE min: 10 MMSE max: 24 Inclusion criteria: mild-to-moderate probable AD (NINCDS-ADRDA) MMSE 10–24 ADAS-cog/11 ≥ 18 History of cognitive decline that was gradual in onset and progressive over a period of ≥ 6 months Living with or regular daily visits from a responsible caregiver (≥ 5 days/weeks) Exclusion criteria: other neurodegenerative disorders or cognitive impairment due to acute cerebral trauma, hypoxic cerebral damage, vitamin deficiency states, infection, primary or metastatic cerebral neoplasia, significant endocrine or metabolic disease, or mental retardation Vascular dementia or evidence of clinically active cerebrovascular disease History of epilepsy or convulsions; current clinically significant psychiatric disease; active peptic ulcer; clinically significant hepatic, renal, pulmonary, metabolic, or endocrine disturbances; clinically significant urinary outflow obstruction; clinically significant cardiovascular disease Use of any agent for the treatment of dementia (approved, experimental, or over the counter) including, but not limited to nootropic agents, cholinomimetic agents, oestrogens taken without medical need, chronic non-steroidal anti-inflammatory agents or cyclo-oxygenase-2 inhibitors (> 30 consecutive days, regardless of indication), and vitamin E (unless a stable dose had been taken for ≥ 6 months prior to trial initiation) Therapy common to all participants: 1-month placebo run-in prior to treatment allocation Sample attrition/dropout: 768 of 971 completed study. 203 withdrew after allocation: did not receive treatment (n = 6); AE (n = 67); withdrew consent (n = 62); non-compliance (n = 29); lost to follow-up (n = 10); insufficent response (n = 10); death (n = 5); other reasons (n = 3). No differences between groups |
Arm no.: 1 Name: galantamine prolonged-release o.d. N: 320 Drug: galantamine Starting daily dose (mg): 8 Dosage details: prolonged-release formulation Titrated from an initial dosage of 8 mg/day for the first 4 weeks, up to a maximum of 24 mg/day in increments of 8 mg/day every 4 weeks after the placebo run-in Whole dose given in single capsule am; placebo given pm Arm no.: 2 Name: galantamine b.i.d. N: 327 Drug: galantamine Starting daily dose (mg): 8 Dosage details: titrated from an initial dosage of 8 mg/day for the first 4 weeks, up to a maximum of 24 mg/day in increments of 8 mg/day every 4 weeks after the placebo run-in Single capsules am and pm Arm no.: 3 Name: placebo N: 324 Drug: placebo Starting daily dose (mg): Dosage details: single placebo dose am and pm |
Participants attended clinic visits scheduled for day 0 (baseline) and weeks 4, 8, 12 and 26 Cognitive ADAS-cog (assessment of 11 items on the cognitive subscale of the ADAS) Functional ADCS-ADL (measured using a 23-item subscale of the ADCS-ADL appropriate for subjects in the mild-to-moderate category of AD) Behavioural NPI (severity and frequency of each symptom rated on the basis of scripted questions administered to the subject’s caregiver) Global severity CIBIC-plus AEs |
Baseline characteristics
| Type | Galantamine prolonged-release o.d. | Placebo | ||||||
|---|---|---|---|---|---|---|---|---|
| N | K | Mean | N | K | Mean | p-value | ||
| Demographics | ||||||||
| Age | C | 319 | 76.6 (SD 7.64) | 320 | 76.3 (SD 8.03) | 0.629a | ||
| Gender (n male) | D | 319 | 114 | 35.7% | 320 | 115 | 35.9% | 0.976b |
| Weight (kg) | C | 318 | 68.6 (SD 14.2) | 319 | 67.8 (SD 14.6) | 0.472a | ||
| Race (n white) | D | 319 | 297 | 93.1% | 320 | 289 | 90.3% | 0.256b |
| Cognitive | ||||||||
| MMSE | C | 319 | 18 (SD 3.97) | 320 | 18.1 (SD 4.08) | |||
| C, continuous; D, dichotomous; o.d., once a day. | ||||||||
| Type | Galantamine b.i.d. | Placebo | ||||||
| N | K | Mean | N | K | Mean | p-value | ||
| Demographics | ||||||||
| Age (years) | C | 326 | 76.5 (SD 7.77) | 320 | 76.3 (SD 8.03) | 0.748a | ||
| Gender (n male) | D | 326 | 36.2% | 320 | 115 | 35.9% | 0.989b | |
| Weight (kg) | C | 326 | 118 | 68.3 (SD 15.9) | 319 | 67.8 (SD 14.6) | 0.671a | |
| Race (n white) | D | 326 | 89.9% | 320 | 289 | 90.3% | 0.957b | |
| Cognitive | ||||||||
| MMSE | C | 326 | 293 | 17.8 (SD 4.14) | 320 | 18.1 (SD 4.08) | ||
Results
| Type | Galantamine prolonged-release o.d. | Placebo | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| n | K | Mean | n | K | Mean | |||
| Study medication: duration of treatment – 26 weeks | C | 319 | 152 (SD 46.9) | 320 | 161 (SD 46.9) | |||
| ITT population | ||||||||
| Disposition of participants | ||||||||
| Discontinued treatment due to AEs – 26 weeks | D | 320 | 28 | 8.8% | 324 | 15 | 4.6% | |
| Discontinued treatment before end of trial – 26 weeks | D | 320 | 68 | 21.3% | 324 | 54 | 16.7% | |
| LOCF analysis | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 8 weeks | MC | 287 | –1.5 (SD 5.08) | 293 | 0 (SD 5.14) | |||
| ADAS-cog – 12 weeks | MC | 290 | –2 (SD 5.28) | 296 | 0.2 (SD 5.33) | |||
| ADAS-cog – 26 weeks | MC | 240 | –1.3 (SD 5.29) | 248 | 1.2 (SD 5.68) | < 0.001a | ||
| ADAS-cog – 26 weeks | MC | 291 | –1.3 (SD 5.29) | 296 | 1.2 (SD 5.68) | < 0.001a | ||
| Functional | ||||||||
| ADCS-ADL – 26 weeksb | MC | 245 | 0 (SD 7.51) | 258 | –2.7 (SD 8.99) | < 0.001a | ||
| Behavioural | ||||||||
| NPI – 26 weeksb | MC | 245 | –0.6 (SD 10.3) | 258 | 0.6 (SD 9.96) | 0.941a | ||
| Global severity | ||||||||
| CIBIC-plus score – 26 weeks | C | 291 | 4.21 (SD 1.1) | 301 | 4.35 (SD 1.14) | NSc | ||
| CIBIC-plus: markedly improved – 26 weeks | D | 291 | 3 | 1.0% | 301 | 3 | 1.0% | 0.712d |
| CIBIC-plus: moderately improved – 26 weeks | D | 291 | 14 | 4.8% | 301 | 11 | 3.7% | 0.621d |
| CIBIC-plus: minimally improved – 26 weeks | D | 291 | 49 | 16.8% | 301 | 48 | 15.9% | 0.856d |
| CIBIC-plus: no change – 26 weeks | D | 291 | 114 | 39.2% | 301 | 111 | 36.9% | 0.623d |
| CIBIC-plus: minimally worse – 26 weeks | D | 291 | 81 | 27.8% | 301 | 80 | 26.6% | 0.802d |
| CIBIC-plus: moderately worse – 26 weeks | D | 291 | 24 | 8.2% | 301 | 41 | 13.6% | 0.050d |
| CIBIC-plus: markedly worse – 26 weeks | D | 291 | 6 | 2.1% | 301 | 7 | 2.3% | 0.951d |
| OC population | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 8 weeks | MC | 284 | –1.5 (SD 5.06) | 289 | 0 (SD 5.1) | |||
| ADAS-cog – 12 weeks | MC | 269 | –2.2 (SD 5.25) | 275 | 0 (SD 5.14) | |||
| ADAS-cog – 26 weeks | MC | 240 | –1.4 (SD 5.27) | 248 | 1.3 (SD 5.67) | < 0.001a | ||
| Functional | ||||||||
| ADCS-ADL – 8 weeks | MC | 280 | 0.8 (SD 6.86) | 294 | –0.7 (SD 7.72) | |||
| ADCS-ADL – 12 weeks | MC | 276 | 0.4 (SD 6.65) | 281 | –0.3 (SD 7.71) | |||
| ADCS-ADL – 26 weeks | MC | 245 | 0 (SD 8.61) | 258 | –2.4 (SD 9.64) | 0.003a | ||
| Behavioural | ||||||||
| NPI – 26 weeks | MC | 245 | –0.6 (SD 10.8) | 258 | 0.1 (SD 13.2) | 0.451a | ||
| Global severity | ||||||||
| CIBIC-plus score – 26 weeks | C | 246 | 4.19 (SD 1.13) | 259 | 4.36 (SD 1.15) | NSc | ||
| CIBIC-plus: markedly improved – 26 weeks | D | 246 | 3 | 1.2% | 259 | 3 | 1.2% | 0.728d |
| CIBIC-plus: moderately improved – 26 weeks | D | 246 | 14 | 5.7% | 259 | 9 | 3.5% | 0.327d |
| CIBIC-plus: minimally improved – 26 weeks | D | 246 | 43 | 17.5% | 259 | 41 | 15.8% | 0.705d |
| CIBIC-plus: no change – 26 weeks | D | 246 | 90 | 36.6% | 259 | 94 | 36.3% | 0.981d |
| CIBIC-plus: minimally worse – 26 weeks | D | 246 | 69 | 28.0% | 259 | 70 | 27.0% | 0.875d |
| CIBIC-plus: moderately worse – 26 weeks | D | 246 | 23 | 9.3% | 259 | 36 | 13.9% | 0.146d |
| CIBIC-plus: markedly worse – 26 weeks | D | 246 | 4 | 1.6% | 259 | 6 | 2.3% | 0.812d |
| Safety population | ||||||||
| AEs | ||||||||
| Any AE – 0 weeks | D | 319 | 253 | 79.3% | 320 | 224 | 70.0% | 0.009d |
| Any gastrointestinal – 0 weeks | D | 319 | 111 | 34.8% | 320 | 80 | 25.0% | 0.009d |
| Any psychiatric – 0 weeks | D | 319 | 73 | 22.9% | 320 | 66 | 20.6% | 0.551d |
| Any general – 0 weeks | D | 319 | 76 | 23.8% | 320 | 60 | 18.8% | 0.141d |
| Any central/peripheral nervous system – 0 weeks | D | 319 | 77 | 24.1% | 320 | 52 | 16.3% | 0.017d |
| Any respiratory – 0 weeks | D | 319 | 45 | 14.1% | 320 | 43 | 13.4% | |
| Any metabolic/nutritional – 0 weeks | D | 319 | 42 | 13.2% | 320 | 36 | 11.3% | |
| Any urinary – 0 weeks | D | 319 | 40 | 12.5% | 320 | 38 | 11.9% | |
| Any secondary term – 0 weeks | D | 319 | 28 | 8.8% | 320 | 39 | 12.2% | |
| Anorexia – 0 weeks | D | 319 | 19 | 6.0% | 320 | 8 | 2.5% | |
| Nausea – 0 weeks | D | 319 | 54 | 16.9% | 320 | 16 | 5.0% | |
| Diarrhoea – 0 weeks | D | 319 | 15 | 4.7% | 320 | 22 | 6.9% | |
| Vomiting – 0 weeks | D | 319 | 21 | 6.6% | 320 | 7 | 2.2% | |
| Agitation – 0 weeks | D | 319 | 22 | 6.9% | 320 | 21 | 6.6% | |
| Depression – 0 weeks | D | 319 | 18 | 5.6% | 320 | 8 | 2.5% | |
| Injury – 0 weeks | D | 319 | 24 | 7.5% | 320 | 18 | 5.6% | |
| Dizziness – 0 weeks | D | 319 | 33 | 10.3% | 320 | 14 | 4.4% | |
| Headache – 0 weeks | D | 319 | 29 | 9.1% | 320 | 18 | 5.6% | |
| Upper respiratory tract infection – 0 weeks | D | 319 | 15 | 4.7% | 320 | 16 | 5.0% | |
| Weight decrease – 0 weeks | D | 319 | 14 | 4.4% | 320 | 4 | 1.3% | |
| Urinary tract infection – 0 weeks | D | 319 | 22 | 6.9% | 320 | 26 | 8.1% | |
| Fall – 0 weeks | D | 319 | 20 | 6.3% | 320 | 19 | 5.9% | |
Methodological issues
|
Randomisation and allocation: Randomisation to treatment was determined by calling an interactive voice-response system. The subject number and treatment code (which corresponded to a specific medication kit) were randomly generated after the caller at the site provided the requested subject details. All treatments were supplied in opaque, size-0 gelatin capsules that were identical in appearance, taste and smell. All subjects received one capsule twice daily Data analysis:* ADAS-cog/11, ADCS-ADL, NPI, ADAS-cog/13, non-memory ADAS-cog and memory ADAS-cog scores: ANOVA model with factors for treatment and pooled country (USA vs non-USA) *CIBIC-plus: Cochran–Mantel–Haenszel statistic using modified ridit scores, derived from rank score (the van%%Elteren test) and controlling for country effect (USA vs non-USA) was used to compare the distribution of subjects with scores on the seven-point scale between groups as well as subgroups *Percentage of responders for ADAS-cog/11 and CIBIC-plus were analysed via Cochran–Mantel–Haenszel test using modified ridit scores derived from rank scores The primary efficacy analyses were based on the OC population at week 26. The ITT population was defined as all randomised subjects who received ≥ 1 dose of study medication and who provided ≥ 1 post-baseline primary efficacy measurement (ADAS-cog or CIBIC-plus). OC data were defined as data slotted into the last scheduled time interval. Analyses based on ITT LOCF method for missing data also were performed to demonstrate the robustness of results Power calculation: Powered at > 95% to detect a 2.5-point (SD 6.2) difference in ADAS-cog/11 score and at 90% to detect a 15% difference between active and placebo groups in their CIBIC-plus responder rates, assuming a 55% placebo responder rate (no change/improved CIBIC-plus score). Required sample size not explicitly reported Conflicts of interest: Lead author declares consultancy fees, a grant and sponsored speaking engagements from Janssen |
Quality appraisal
|
Bullock et al.101
| Design | Participants | Arms | Outcomes |
|---|---|---|---|
|
Bullock et al . (2004) 101 Study design: parallel double-blind RCT Countries: ‘including’ Canada, Denmark, Finland, France, Germany, Israel, the Netherlands, Poland and the UK No. of centres: 62 Funding: none reported Length of follow-up (weeks): 26 Notes: Follow-up also at 32 and 52 weeks during the open-label phase of the trial Unable to calculate attrition n, as using percentages quoted in the text gives non-whole numbers |
No. randomised: 285 MMSE min: 10 MMSE max: 25 Inclusion criteria: Probable vascular dementia (NINDS-AIREN definition) or AD + CVD (NINCDS-ADRDA definition) (with CVD evidenced by CT or MRI) Mild-to-moderate dementia (MMSE 10–25) Score ≥ 12 on 11-item subscale of of AD assessment scale Presence of focal neurological signs Disease onset at between 40 and 90 years of age Exclusion criteria: Neurogenerative disorders Cognitive impairmentresulting from other cerebral trauma Cerebral neoplasia Mental retardation Vitamin deficiency Significant endocrine or metabolic disease Clinically significant co-existing medical conditions Significant cardiovascular disease that would likely limit the patient’s ability to complete the study Current use of agents for the treatment of dementia Recent history (within 30 days) of treatment with other investigational agents History of alcohol or drug abuse Therapy common to all participants: 1-month single-blind placebo run-in prior to treatment allocation Sample attrition/dropout: 230 of 285 completed study |
Arm no.: 1 Name: galantamine N: 152 Drug: galantamine Starting daily dose (mg): 4 Dosage details: titrated upwards in weekly 4-mg increments over a period of 6 weeks, and then continued at this maintenance dose (24 mg/day) for an additional 4.5 months Arm no.: 2 Name: placebo N: 86 Drug: placebo Starting daily dose (mg): – Dosage details: single placebo dose AM and PM |
Cognitive ADAS-cog (not defined) ADAS-cog/13 (methods note as secondary efficacy variable, but outcome data not reported) Functional DAD (outcome data only available from study including IPD in a pooled analysis (Feldman et al.)130 Behavioural NPI (methods note as secondary efficacy variable, but outcome data not reported) |
Baseline characteristics
| Galantamine | Placebo | |||||||
|---|---|---|---|---|---|---|---|---|
| Type | N | K | Mean | N | K | Mean | p-value | |
| Demographics | ||||||||
| Age (years) | C | 152 | 75.8 (SD 6.78) | 86 | 77.6 (SD 6.12) | 0.043a | ||
| Gender (n male) | D | 152 | 73 | 48.0% | 86 | 42 | 48.8% | 0.988b |
| Height (cm) | C | 152 | 164 (SD 10.4) | 86 | 164 (SD 10.6) | 0.943a | ||
| Weight (kg) | C | 152 | 69.9 (SD 12.9) | 86 | 67 (SD 13) | 0.099a | ||
| Cognitive | ||||||||
| ADAS-cog – 0 weeks | C | 148 | 22.7 (SD 9.25) | 85 | 23.9 (SD 9.86) | 0.358a | ||
| MMSE | C | 152 | 20.5 (SD 3.95) | 86 | 20.2 (SD 3.52) | 0.559a | ||
Results
| Galantamine | Placebo | |||||||
|---|---|---|---|---|---|---|---|---|
| Type | n | K | Mean | n | K | Mean | p-value | |
| ITT population | ||||||||
| Disposition of participants | ||||||||
| Discontinued treatment due to AEsa | D | 188 | 49 | 26.1% | 97 | 16 | 16.5% | |
| LOCF analysis | ||||||||
| Functional | ||||||||
| DAD – 26 weeks | MC | 188 | –1 (SD 15.8) | 97 | –6 (SD 14.5) | < 0.01b | ||
| OC population | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 6 weeksc | MC | 148 | –0.5 (SD 4.62) | 85 | 0.15 (SD 6.26) | 0.366d | ||
| ADAS-cog – 13 weeksc | MC | 148 | –1.48 (SD 4.32) | 85 | 0 (SD 6.03) | 0.031d | ||
| ADAS-cog – 26 weeks | C | 147 | 21.5 (SD 10.5) | 83 | 25.7 (SD 12) | 0.006e | ||
| ADAS-cog – 26 weeks | MC | 147 | –1.1 (SD 5.79) | 83 | 2 (SD 5.56) | < 0.001e | ||
Methodological issues
|
Randomisation and allocation: Randomisation was conducted using a ‘computer-generated code’ (no further details provided). No details provided about appearance, taste or smell of placebo Data analysis: ADAS-cog/11 change from baseline with treatment and country as factors, treatment groups compared using two-way ANOVA. Paired t-test for comparisons within treatment groups (baseline vs each visit) of ADAS-COG/11, vital signs, ECG results and body weight. Wilcoxon’s signed-rank test used for within-group comparisons if data not distributed normally. Primary efficacy analysis based on OC population at 26 weeks. Reported as ITT analysis, but no further details about this or how missing data were handled are reported Power calculation: Not reported Conflicts of interest: None reported |
Quality appraisal
|
Bullock et al.148
| Design | Participants | Arms | Outcomes |
|---|---|---|---|
|
Bullock et al . (2005) 148 Study design: parallel double-blind RCT Countries: Australia, Canada, France, Germany, Italy, Spain and the UK No. of centres: 94 Funding: study supported by Novartis Pharma AG Four of the study authors (YH, JN, GR, RL) are employees of Novartis The remaining four authors (RB, JT, HB, GG) did not receive remuneration for taking part in the study or writing the manuscript Length of follow-up (weeks): 104 Notes: |
No. randomised: 998 MMSE min: 10 MMSE max: 20 Inclusion criteria: Male or female outpatients aged 50–85 years AD (DSM-IV criteria) or probable AD (NINCDS-ADRDA criteria) MMSE 10–20 Contact with a responsible caregiver at least once a day [Patients with AD who also had symptoms suggestive of concomitant Lewy body disease (McKeith criteria) were also permitted to enter the study] Exclusion criteria: Current diagnosis of any primary neurodegenerative disorder other than AD (including Parkinson’s disease) Any advance, severe, progressive or unstable disease or disability A major depressive episode Active, uncontrolled seizure disorder or peptic ulceration Acute, severe or unstable asthmatic conditions Severe or unstable cardiovascular disease History or diagnosis of cerebrovascular disease Known hyperensitivity to drugs similar to rivastigmine or donepezil in structure or pharmacological action Use of any cholinesterase inhibitor or other approved treatment for AD in the 6 weeks prior to randomisation Use of any investigational drug, any drug or treatment known to cause major organ system toxicity, or any new psychotropic medication during the 4 weeks prior to randomisation Anticholinergic drugs at randomisation Therapy common to all participants: None Sample attrition/dropout: 578 of 994 (58.1%) completed study (rivastigmine 261 of 495 (52.7%), donepezil 317 of 499 (63.5%) (998 were randomised, four withdrew before receiving treatment) Reasons for non-completion: Rivastigmine – AEs (n = 129); abnormal lab values (n = 1); unsatisfactory therapeutic effect (n = 19); protocol violation (n = 12); withdrawn consent (n = 34); lost to follow-up (n = 10); administrative problems (n = 4); death (n = 26) Donepezil – AEs (n = 80); abnormal lab values (n = 1); unsatisfactory therapeutic effect (n = 17); protocol violation (n = 9); withdrawn consent (n = 22); lost to follow-up (n = 13); administrative problems (n = 6); death (n = 34) |
Arm no.: 1 Name: rivastigmine N: 498 Drug: rivastigmine Starting daily dose (mg): 3 Dosage details: titrated from an initial dosage of 3 mg/day for the first 4 weeks up to a maximum of 12 mg/day in increments of 3 mg/day every 4 weeks Arm no.: 2 Name: donepezil N: 499 Drug: donepezil Starting daily dose (mg): 5 Dosage details: titrated from an initial dosage of 5 mg/day for the first 8 weeks up to 10 mg/day in weeks 9–16 Notes: for patients who did not achieve the maximum dose during the titration period, investigators were asked to make at least one attempt during the maintenance period to increase the dose to the next highest dose level; the overall dosing strategy was to treat patients at the highest doses that were individually well tolerated, but dose adjustments were permitted |
Cognitive MMSE (not defined) SIB (consists of six subscales (attention, orientation, language, memory, visuoperception and construction), including brief assessments of social skills, praxis and responding to name (score range 0–100, lower scores indicating a greater degree of cognitive impairment) Functional ADCS-ADL (not defined) Behavioural NPI (not defined) Global severity GDS (not defined) AEs An AE was defined as any undesirable sign, symptom or medical condition occurring after starting study drug, even if the event was not considered to be related to study drug. A serious AE was classed as one that was considered one of the following: fatal, life-threatening, necessitating prolonged hospitalisation, resulting in significant disability or requiring medical intervention to prevent any of these outcomes. Information about all AEs was recorded at each follow-up visit, whether or not volunteered by the subject or carer, or discovered through investigator questioning or examination, laboratory test, ECG or other means. AEs were coded with a standard glossary |
Baseline characteristics
| Rivastigmine | Donepezil | |||||||
|---|---|---|---|---|---|---|---|---|
| Type | N | K | Mean | N | K | Mean | p-value | |
| Demographics | ||||||||
| Age (years) | C | 495 | 75.9 (SD 6.6) | 499 | 75.8 (SD 6.8) | 0.814a | ||
| Age ≥ 75 years | D | 495 | 318 | 64.2% | 499 | 314 | 62.9% | 0.715b |
| Gender (n male) | D | 495 | 154 | 31.1% | 499 | 157 | 31.5% | 0.959b |
| Disease characteristics | ||||||||
| Duration of dementia (months) | C | 495 | 33.6 (SD 22.2) | 499 | 34.2 (SD 26.5) | 0.699a | ||
| Probable concomitant Lewy body dementia | D | 495 | 18 | 3.6% | 499 | 22 | 4.4% | 0.647b |
| Family history: mother | D | 495 | 55 | 11.1% | 499 | 63 | 12.6% | 0.522b |
| Family history: father | D | 495 | 17 | 3.4% | 499 | 18 | 3.6% | 0.981b |
| Family history: sibling | D | 495 | 37 | 7.5% | 499 | 50 | 10.0% | 0.191b |
| Domestic circumstances | ||||||||
| Living alone | D | 495 | 92 | 18.6% | 499 | 85 | 17.0% | 0.578b |
| Living with caregiver or other | D | 495 | 370 | 74.7% | 499 | 393 | 78.8% | 0.155b |
| Assisted living/group home | D | 495 | 33 | 6.7% | 499 | 21 | 4.2% | 0.116b |
| Cognitive | ||||||||
| MMSE – 0 weeks | C | 495 | 15.1 (SD 3) | 499 | 15.1 (SD 2.9) | 1.000a | ||
| MMSE – ≥15 | D | 495 | 280 | 56.6% | 499 | 283 | 56.7% | 0.986b |
| LOCF analysis | ||||||||
| Cognitive | ||||||||
| MMSE – 0 weeks | C | 471 | 15.2 (SD 3) | 484 | 15.1 (SD 2.9) | 0.917a | ||
| SIB – 0 weeks | C | 471 | 87.8 (SD 10.9) | 483 | 87.8 (SD 11.2) | |||
| Functional | ||||||||
| ADCS-ADL – 0 weeks | C | 454 | 46.6 (SD 17.2) | 475 | 48.4 (SD 16.6) | |||
| Behavioural | ||||||||
| NPI – 0 weeks | C | 471 | 14.5 (SD 12.9) | 484 | 14.4 (SD 13.9) | |||
| Global severity | ||||||||
| GDS – 0 weeks | C | 471 | 4.39 (SD 0.7) | 483 | 4.27 (SD 0.8) | |||
Results
| Rivastigmine | Donepezil | |||||||
|---|---|---|---|---|---|---|---|---|
| Type | n | K | Mean | n | K | Mean | p-value | |
| ITT population | ||||||||
| Disposition of participants | ||||||||
| Discontinued treatment due to AEs | D | 498 | 128 | 25.7% | 500 | 80 | 16.0% | < 0.001a |
| Discontinued treatment before end of trial | D | 498 | 237 | 47.6% | 500 | 183 | 36.6% | < 0.001a |
| LOCF analysis | ||||||||
| Cognitive | ||||||||
| MMSE – 104 weeks | MC | 471 | –2.35 (SD 6.51) | 484 | –2.85 (SD 6.6) | 0.089b | ||
| MMSE – 104 weeks | MC | 471 | –2.35 (SD 6.51) | 484 | –2.85 (SD 6.6) | 0.106c | ||
| SIB – 104 weeks | MC | 471 | –9.3 (SD 23.9) | 483 | –9.91 (SD 24.2) | 0.609b | ||
| SIB – 104 weeks | MC | 471 | –9.3 (SD 23.9) | 483 | –9.91 (SD 24.2) | 0.738c | ||
| Functional | ||||||||
| ADCS-ADL – 104 weeks | MC | 454 | –12.8 (SD 19.2) | 475 | –14.9 (SD 19.6) | 0.007c | ||
| ADCS-ADL – 104 weeks | MC | 454 | –12.8 (SD 19.2) | 475 | –14.9 (SD 19.6) | 0.047b | ||
| Behavioural | ||||||||
| NPI – 104 weeks | MC | 471 | 2.4 (SD 17.4) | 484 | 2.94 (SD 17.6) | 0.505c | ||
| NPI – 104 weeks | MC | 471 | 2.4 (SD 17.4) | 484 | 2.94 (SD 17.6) | 0.554b | ||
| Global severity | ||||||||
| GDS – 104 weeks | MC | 471 | 0.58 (SD 0.9) | 483 | 0.69 (SD 0.9) | 0.049c | ||
| Safety population | ||||||||
| AEs | ||||||||
| Any serious AE – 104 weeks | D | 495 | 157 | 31.7% | 499 | 162 | 32.5% | 0.854a |
| Safety population – titration phase | ||||||||
| AEs | ||||||||
| Any AE – 16 weeks | D | 495 | 406 | 82.0% | 499 | 323 | 64.7% | < 0.001a |
| Anorexia – 16 weeks | D | 495 | 45 | 9.1% | 499 | 20 | 4.0% | 0.002a |
| Nausea – 16 weeks | D | 495 | 163 | 32.9% | 499 | 76 | 15.2% | < 0.001a |
| Diarrhoea – 16 weeks | D | 495 | 41 | 8.3% | 499 | 34 | 6.8% | 0.449a |
| Vomiting – 16 weeks | D | 495 | 138 | 27.9% | 499 | 29 | 5.8% | < 0.001a |
| Agitation – 16 weeks | D | 495 | 35 | 7.1% | 499 | 50 | 10.0% | 0.121a |
| Depression – 16 weeks | D | 495 | 19 | 3.8% | 499 | 10 | 2.0% | 0.126a |
| Headache – 16 weeks | D | 495 | 27 | 5.5% | 499 | 23 | 4.6% | 0.642a |
| Weight decrease – 16 weeks | D | 495 | 30 | 6.1% | 499 | 9 | 1.8% | < 0.001a |
| Urinary tract infection – 16 weeks | D | 495 | 8 | 1.6% | 499 | 13 | 2.6% | 0.388a |
| Fall – 16 weeks | D | 495 | 25 | 5.1% | 499 | 10 | 2.0% | 0.015a |
| Hypertension – 16 weeks | D | 495 | 20 | 4.0% | 499 | 7 | 1.4% | 0.018a |
| Aggression – 16 weeks | D | 495 | 7 | 1.4% | 499 | 11 | 2.2% | 0.486a |
| Safety population – maintenance phase | ||||||||
| AEs | ||||||||
| Any AE – 104 weeks | D | 404 | 318 | 78.7% | 453 | 349 | 77.0% | 0.613a |
| Anorexia – 104 weeks | D | 404 | 26 | 6.4% | 453 | 14 | 3.1% | 0.031a |
| Nausea – 104 weeks | D | 404 | 52 | 12.9% | 453 | 24 | 5.3% | < 0.001a |
| Diarrhoea – 104 weeks | D | 404 | 26 | 6.4% | 453 | 30 | 6.6% | 0.978a |
| Vomiting – 104 weeks | D | 404 | 62 | 15.3% | 453 | 20 | 4.4% | < 0.001a |
| Agitation – 104 weeks | D | 404 | 34 | 8.4% | 453 | 47 | 10.4% | 0.389a |
| Depression – 104 weeks | D | 404 | 21 | 5.2% | 453 | 16 | 3.5% | 0.303a |
| Headache – 104 weeks | D | 404 | 13 | 3.2% | 453 | 12 | 2.6% | 0.771a |
| Weight decrease – 104 weeks | D | 404 | 36 | 8.9% | 453 | 43 | 9.5% | 0.861a |
| Urinary tract infection – 104 weeks | D | 404 | 18 | 4.5% | 453 | 26 | 5.7% | 0.487a |
| Fall – 104 weeks | D | 404 | 33 | 8.2% | 453 | 44 | 9.7% | 0.503a |
| Hypertension – 104 weeks | D | 404 | 21 | 5.2% | 453 | 18 | 4.0% | 0.487a |
| Aggression – 104 weeks | D | 404 | 19 | 4.7% | 453 | 25 | 5.5% | 0.700a |
Methodological issues
|
Randomisation and allocation: Performed using an interactive voice-response system that automated the random assignment of treatment groups to randomisation numbers. Randomisation was stratified with respect to severity, i.e. was done separately with MMSE scores of 10–14 and 15–20. All treatments were supplied as capsules that were identical in size, shape and colour, and all patients received the same number of capsules per day Data analysis: Primary – SIB; secondary – GDS, ADCS-ADL, MMSE, NPI. ANCOVA and/or Wilcoxon’s rank-sum test conducted with treatment, country, MMSE category and baseline scores as explanatory variables. Additional analyses on SIB, NPI, ADCS-ADL where patients had different baseline disease severities, gender, ages and vascular risk profiles. Exploratory analyses conducted on pharmacogenetic subpopulation [for BuChE – the more common BuChE wild type (wt/wt) and those with one or two BuChE-K variants – and by apolipoprotein E (APOE) E4 carrier status]. Additional secondary analysis conducted in patients with AD who had symptoms suggestive of concomitant Lewy body disease (dementia lewy body diagnosed according to McKeith criteria, or receiving parkinsonian medication, but not formally diagnosed with Parkinson’s disease). ANCOVA and/or Wilcoxon’s rank-sum test conducted with treatment, country, MMSE category and baseline scores as explanatory variables. Exploratory analyses of pharmacogenetic data assessed by ANCOVA with age, gender and baseline values as explanatory variables. ITT population defined as all randomised patinets who received study medication and from whom at least one efficacy measurement was obtained while on treatment. Missing values were imputed with LOCF data. In addition, supportive analyses comprised an evaluable patients population of all patients who were treated with study medication for at least 16 weeks (with a LOCF imputation), and an OC population of patients who had evaluations on treatment at designated assessment times, with no imputation of missing values, whether or not they had completed the study or not Power calculation: Powered at 85% to detect a statistically significant (significance level 5%, two sided) difference in SIB of four points between the two groups (assuming a SD of 20 on change from baseline in mean SIB scores, as observed in previous trials), sample size of 450 patients per treatment group was required Conflicts of interest: Study supported by Novartis Pharma AG. Four of the study authors (YH, JN, GR, RL) are employees of Novartis. The remaining four authors (RB, JT, HB, GG) did not receive remuneration for taking part in the study or writing the manuscript |
Quality appraisal
|
Cumbo99
| Design | Participants | Arms | Outcomes |
|---|---|---|---|
|
Cumbo (2005) 99 Study design: – Country: funded by an Italian health agency, but not stated whether study conducted in Italy or elsewhere No. of centres: not stated; small sample size suggests single centre Funding: Supported by Department of Neuroscience (NHS District of Caltanissetta) Novartis Farma SpA supported the English editing of the manuscript Length of follow-up (weeks): 78 Notes: |
No. randomised: 101 MMSE min: 10 MMSE max: 27 Inclusion criteria: Probable AD (NINCD-ARDRA) MMSE 10–27 ≥ 3-year duration of disease No behavioural symptoms Carer who could ensure compliance to treatment and attendance and provide the information required for psychometric and behavioural assessments Exclusion criteria: History of primary neurological or psychiatric disease other than AD Drug or alcohol abuse Clinically significant medical or surgical disorders independently of stability Previous therapy for dementia Concomitant treatment with cholinomimetic or anticholinergic drugs, investigational drugs, tricyclic antidepressants or neuroleptic drugs Refusal to give informed consent in writing Therapy common to all participants: None Sample attrition/dropout: None |
Arm no.: 1 Name: rivastigmine n: 37 Drug: rivastigmine Starting daily dose (mg): 9 Dosage details: no details reported of titration. Notes: starting daily dose is only reported as the mean for the whole arm No maximum dose reported Arm no.: 2 Name: galantamine n: 33 Drug: galantamine Starting daily dose (mg): 16 Dosage details: no details reported of titration Notes: starting daily dose is only reported as the mean for the whole arm No maximum dose reported Arm no.: 3 Name: donepezil n: 31 Drug: donepezil Starting daily dose (mg): 10 Dosage details: no details reported of titration Notes: starting daily dose is only reported as the mean for the whole arm No maximum dose reported |
Behavioural NPI Developing BPSD Time to BPSD BEHAVE-AD AEs |
Baseline characteristics
| All study participants | |||
|---|---|---|---|
| N | k | Mean | |
| Demographics | |||
| Age (years) | 101 | 76.35 (range 66–83) | |
| Gender (n male) | 101 | 43 | 42.6% |
| Education (years) | 101 | 5 (range 3–12) | |
| Disease characteristics | |||
| Duration of dementia (months) | 101 | 61.08 (range 36–108) | |
| Cognitive | |||
| MMSE | 101 | 16.6 | |
| Functional | |||
| ADL | 101 | 3.7 | |
| IADL | 101 | 5.3 | |
| Behavioural | |||
| NPI | 101 | 0 | |
| NPI – caregiver distress | 101 | 0 | |
| BEHAVE-AD | 101 | 0 | |
| Global severity | |||
| GDS | 101 | 5 | |
Results
| Rivastigmine | Galantamine | |||||||
|---|---|---|---|---|---|---|---|---|
| Type | n | K | Mean | n | K | Mean | p-value | |
| Behavioural | ||||||||
| NPI – delusions – 78 weeks | D | 37 | 1 | 2.7% | 33 | 4 | 12.1% | 0.288a |
| NPI – hallucinations – 78 weeks | D | 37 | 0 | 0.0% | 33 | 0 | 0.0% | 0.341a |
| NPI – agitation/aggression – 78 weeks | D | 37 | 4 | 10.8% | 33 | 9 | 27.3% | 0.144a |
| NPI – depression/dysphoria – 78 weeks | D | 37 | 13 | 35.1% | 33 | 10 | 30.3% | 0.861a |
| NPI – anxiety – 78 weeks | D | 37 | 14 | 37.8% | 33 | 15 | 45.5% | 0.687a |
| NPI – elation/euphoria – 78 weeks | D | 37 | 0 | 0.0% | 33 | 0 | 0.0% | 0.341a |
| NPI – apathy/indifference – 78 weeks | D | 37 | 7 | 18.9% | 33 | 7 | 21.2% | 0.952a |
| NPI – disinhibition – 78 weeks | D | 37 | 0 | 0.0% | 33 | 3 | 9.1% | 0.252a |
| NPI – irritability/lability – 78 weeks | D | 37 | 12 | 32.4% | 33 | 14 | 42.4% | 0.538a |
| NPI – aberrant motor behaviour – 78 weeks | D | 37 | 0 | 0.0% | 33 | 0 | 0.0% | 0.341a |
| NPI – night-time behaviour – 78 weeks | D | 37 | 1 | 2.7% | 33 | 9 | 27.3% | 0.010a |
| NPI – appetite/eating change – 78 weeks | D | 37 | 0 | 0.0% | 33 | 1 | 3.0% | 0.936a |
| Developing BPSD – 78 weeks | D | 37 | 14 | 37.8% | 33 | 15 | 45.5% | 0.687a |
| BEHAVE-AD – delusional and paranoid ideation – 78 weeks | D | 37 | 1 | 2.7% | 33 | 4 | 12.1% | 0.288a |
| BEHAVE-AD – hallucinations – 78 weeks | D | 37 | 0 | 0.0% | 33 | 0 | 0.0% | 0.341a |
| BEHAVE-AD – activity disturbances – 78 weeks | D | 37 | 0 | 0.0% | 33 | 0 | 0.0% | 0.341a |
| BEHAVE-AD – aggression – 78 weeks | D | 37 | 4 | 10.8% | 33 | 9 | 27.3% | 0.144a |
| BEHAVE-AD – diurnal cycle disturbances – 78 weeks | D | 37 | 1 | 2.7% | 33 | 9 | 27.3% | 0.010a |
| BEHAVE-AD – affective disturbances – 78 weeks | D | 37 | 13 | 35.1% | 33 | 10 | 30.3% | 0.861a |
| BEHAVE-AD – anxiety and phobias – 78 weeks | D | 37 | 14 | 37.8% | 33 | 15 | 45.5% | 0.687a |
| AEs | ||||||||
| Anorexia – 78 weeks | D | 37 | 1 | 2.7% | 33 | 1 | 3.0% | 0.524a |
| Nausea – 78 weeks | D | 37 | 3 | 8.1% | 33 | 2 | 6.1% | 0.894a |
| Vomiting – 78 weeks | D | 37 | 1 | 2.7% | 33 | 1 | 3.0% | 0.524a |
| Headache – 78 weeks | D | 37 | 1 | 2.7% | 33 | 0 | 0.0% | 0.936a |
| Weight decrease – 78 weeks | D | 37 | 0 | 0.0% | 33 | 1 | 3.0% | 0.936a |
| Disposition of participants | ||||||||
| Discontinued treatment due to AEs – 1 week | D | 37 | 0 | 0.0% | 33 | 0 | 0.0% | 0.341a |
| Discontinued treatment before end of trial – 1 week | D | 37 | 0 | 0.0% | 33 | 0 | 0.0% | 0.341a |
| Rivastigmine | Donepezil | |||||||
|---|---|---|---|---|---|---|---|---|
| Type | n | K | Mean | n | K | Mean | p-value | |
| Behavioural | ||||||||
| NPI – delusions – 78 weeks | D | 37 | 1 | 2.7% | 31 | 5 | 16.1% | 0.130a |
| NPI – hallucinations – 78 weeks | D | 37 | 0 | 0.0% | 31 | 3 | 9.7% | 0.226a |
| NPI – agitation/aggression – 78 weeks | D | 37 | 4 | 10.8% | 31 | 7 | 22.6% | 0.326a |
| NPI – depression/dysphoria – 78 weeks | D | 37 | 13 | 35.1% | 31 | 13 | 41.9% | 0.746a |
| NPI – anxiety – 78 weeks | D | 37 | 14 | 37.8% | 31 | 14 | 45.2% | 0.716a |
| NPI – elation/euphoria – 78 weeks | D | 37 | 0 | 0.0% | 31 | 1 | 3.2% | 0.902a |
| NPI – apathy/indifference – 78 weeks | D | 37 | 7 | 18.9% | 31 | 8 | 25.8% | 0.698a |
| NPI – disinhibition – 78 weeks | D | 37 | 0 | 0.0% | 31 | 1 | 3.2% | 0.902a |
| NPI – irritability/lability – 78 weeks | D | 37 | 12 | 32.4% | 31 | 15 | 48.4% | 0.276a |
| NPI – aberrant motor behaviour – 78 weeks | D | 37 | 0 | 0.0% | 31 | 0 | 0.0% | 0.355a |
| NPI – night-time behaviour – 78 weeks | D | 37 | 1 | 2.7% | 31 | 0 | 0.0% | 0.902a |
| NPI – appetite/eating change – 78 weeks | D | 37 | 0 | 0.0% | 31 | 1 | 3.2% | 0.902a |
| Developing BPSD – 78 weeks | D | 37 | 14 | 37.8% | 31 | 16 | 51.6% | 0.371a |
| BEHAVE-AD – delusional and paranoid ideation – 78 weeks | D | 37 | 1 | 2.7% | 31 | 5 | 16.1% | 0.130a |
| BEHAVE-AD – hallucinations – 78 weeks | D | 37 | 0 | 0.0% | 31 | 3 | 9.7% | 0.226a |
| BEHAVE-AD – activity disturbances – 78 weeks | D | 37 | 0 | 0.0% | 31 | 0 | 0.0% | 0.355a |
| BEHAVE-AD – aggression – 78 weeks | D | 37 | 4 | 10.8% | 31 | 7 | 22.6% | 0.326a |
| BEHAVE-AD – diurnal cycle disturbances – 78 weeks | D | 37 | 1 | 2.7% | 31 | 10 | 32.3% | 0.003a |
| BEHAVE-AD – affective disturbances – 78 weeks | D | 37 | 13 | 35.1% | 31 | 13 | 41.9% | 0.746a |
| BEHAVE-AD – anxiety and phobias – 78 weeks | D | 37 | 14 | 37.8% | 31 | 15 | 48.4% | 0.529a |
| AEs | ||||||||
| Anorexia – 78 weeks | D | 37 | 1 | 2.7% | 31 | 0 | 0.0% | 0.902a |
| Nausea – 78 weeks | D | 37 | 3 | 8.1% | 31 | 2 | 6.5% | 0.837a |
| Vomiting – 78 weeks | D | 37 | 1 | 2.7% | 31 | 0 | 0.0% | 0.902a |
| Headache – 78 weeks | D | 37 | 1 | 2.7% | 31 | 2 | 6.5% | 0.875a |
| Weight decrease – 78 weeks | D | 37 | 0 | 0.0% | 31 | 0 | 0.0% | 0.355a |
| Disposition of participants | ||||||||
| Discontinued treatment due to AEs – 1 week | D | 37 | 0 | 0.0% | 31 | 0 | 0.0% | 0.355a |
| Discontinued treatment before end of trial – 1 week | D | 37 | 0 | 0.0% | 31 | 0 | 0.0% | 0.355a |
| Galantamine | Donepezil | |||||||
|---|---|---|---|---|---|---|---|---|
| Type | n | K | Mean | n | K | Mean | p-value | |
| Behavioural | ||||||||
| NPI – delusions – 78 weeks | D | 33 | 4 | 12.1% | 31 | 5 | 16.1% | 0.919a |
| NPI – hallucinations – 78 weeks | D | 33 | 0 | 0.0% | 31 | 3 | 9.7% | 0.274a |
| NPI – agitation/aggression – 78 weeks | D | 33 | 9 | 27.3% | 31 | 7 | 22.6% | 0.885a |
| NPI – depression/dysphoria – 78 weeks | D | 33 | 10 | 30.3% | 31 | 13 | 41.9% | 0.479a |
| NPI – anxiety – 78 weeks | D | 33 | 15 | 45.5% | 31 | 14 | 45.2% | 0.820a |
| NPI – elation/euphoria – 78 weeks | D | 33 | 0 | 0.0% | 31 | 1 | 3.2% | 0.965a |
| NPI – apathy/indifference – 78 weeks | D | 33 | 7 | 21.2% | 31 | 8 | 25.8% | 0.890a |
| NPI – disinhibition – 78 weeks | D | 33 | 3 | 9.1% | 31 | 1 | 3.2% | 0.651a |
| NPI – irritability/lability – 78 weeks | D | 33 | 14 | 42.4% | 31 | 15 | 48.4% | 0.820a |
| NPI – aberrant motor behaviour – 78 weeks | D | 33 | 0 | 0.0% | 31 | 0 | 0.0% | 0.328a |
| NPI – night-time behaviour – 78 weeks | D | 33 | 9 | 27.3% | 31 | 0 | 0.0% | 0.008a |
| NPI – appetite/eating change – 78 weeks | D | 33 | 1 | 3.0% | 31 | 1 | 3.2% | 0.500a |
| Developing BPSD – 78 weeks | D | 33 | 15 | 45.5% | 31 | 16 | 51.6% | 0.808a |
| BEHAVE-AD – delusional and paranoid ideation – 78 weeks | D | 33 | 4 | 12.1% | 31 | 5 | 16.1% | 0.919a |
| BEHAVE-AD – hallucinations – 78 weeks | D | 33 | 0 | 0.0% | 31 | 3 | 9.7% | 0.274a |
| BEHAVE-AD – activity disturbances – 78 weeks | D | 33 | 0 | 0.0% | 31 | 0 | 0.0% | 0.328a |
| BEHAVE-AD – aggression – 78 weeks | D | 33 | 9 | 27.3% | 31 | 7 | 22.6% | 0.885a |
| BEHAVE-AD – diurnal cycle disturbances – 78 weeks | D | 33 | 9 | 27.3% | 31 | 10 | 32.3% | 0.871a |
| BEHAVE-AD – affective disturbances – 78 weeks | D | 33 | 10 | 30.3% | 31 | 13 | 41.9% | 0.479a |
| BEHAVE-AD – anxiety and phobias – 78 weeks | D | 33 | 15 | 45.5% | 31 | 15 | 48.4% | 0.988a |
| AEs | ||||||||
| Anorexia – 78 weeks | D | 33 | 1 | 3.0% | 31 | 0 | 0.0% | 0.965a |
| Nausea – 78 weeks | D | 33 | 2 | 6.1% | 31 | 2 | 6.5% | 0.651a |
| Vomiting – 78 weeks | D | 33 | 1 | 3.0% | 31 | 0 | 0.0% | 0.965a |
| Headache – 78 weeks | D | 33 | 0 | 0.0% | 31 | 2 | 6.5% | 0.519a |
| Weight decrease – 78 weeks | D | 33 | 1 | 3.0% | 31 | 0 | 0.0% | 0.965a |
| Disposition of participants | ||||||||
| Discontinued treatment due to AEs – 1 week | D | 33 | 0 | 0.0% | 31 | 0 | 0.0% | 0.328a |
| Discontinued treatment before end of trial – 1 week | D | 33 | 0 | 0.0% | 31 | 0 | 0.0% | 0.328a |
Methodological issues
|
Randomisation and allocation: No details of randomisation procedure reported. Open-label trial Data analysis: Primary outcome: Time to onset of BPSD, analysed using survival analysis according to the actuarial method, grouping events with onset in the same predefined time interval. The first time interval comprised the first 6 months; thereafter, the intervals were monthly. Curves related to the probability of survival without BPSD were compared using Wilcoxon’s test between pairs of treatments. The remaining parameters were analysed descriptively in view of the small sample size Power calculation: None reported Conflicts of interest: Supported by Department of Neuroscience (NHS District of Caltanissetta). Novartis Farma SpA supported the English editing of the manuscript |
Quality appraisal
|
dos Santos Moraes et al.119
| Design | Participants | Arms | Outcomes |
|---|---|---|---|
|
dos Santos Moraes et al . (2006) 119 Study design: parallel double-blind RCT Country: Brazil No. of centres: 1 Funding: FAPESP (Fundação de%%Amparo à Pesquisa do Estado de%%São Paulo) AFIP (Associação Fundo de%%Incentivo à Psicofarmacologia) Length of follow-up (weeks): 26 Notes: |
No. randomised: 35 MMSE min: – MMSE max: – Inclusion criteria: Probable AD (ADRDA criteria) CDR (Brazilian version) 1–2 (mild to moderate) Exclusion criteria: Other causes of dementia Other current severe medical or psychiatric disease Evidence of moderate-to-severe sleep disorders, based on medical, sleep and psychiatric interviews Apnoea–hypoapnoea index > 10/hour and periodic leg movement index > 5/hour at baseline polysomnographic recording Psychoactive drugs in the month prior to entering the study Therapy common to all participants: two nights of polysomnographic recording (for purposes of habituation) Sample attrition/dropout: eight patients left the study because of technical difficulties in polysomnography recordings |
Arm no.: 1 Name: donepezil n: 17 Drug: donepezil Starting daily dose (mg): 5 Dosage details: starting daily dose of 5 mg for the first month, increased to 10 mg/day in the second month Arm no.: 2 Name: placebo n: 18 Drug: placebo Starting daily dose (mg): – Dosage details: single daily dose |
ADAS-cog (selected aspects of cognitive performance, including elements of memory, orientation, reasoning, language and praxis) |
Baseline characteristics
| Donepezil | Placebo | |||||||
|---|---|---|---|---|---|---|---|---|
| Type | N | K | Mean | N | K | Mean | p-value | |
| OC population | ||||||||
| Demographics | ||||||||
| Age (years) | C | 17 | 77.4 (SD 6.6) | 18 | 74.5 (SD 9.8) | 0.32a | ||
| Gender (n male) | D | 17 | 4 | 23.5% | 18 | 7 | 38.9% | 0.34a |
| Body mass index (kg/m2) | C | 17 | 26 (SD 4.8) | 18 | 24.9 (SD 4.5) | 0.48a | ||
| Education (years) | C | 17 | 4.4 (SD 3.6) | 18 | 6 (SD 5.2) | 0.30a | ||
| Cognitive | ||||||||
| ADAS-cog – 0 weeks | C | 17 | 35.6 (SD 13.7) | 18 | 39 (SD 18.5) | 0.543b | ||
| Global severity | ||||||||
| CDR | C | 17 | 1.2 (SD 0.4) | 18 | 1.5 (SD 0.5) | 0.11a | ||
Results
| Donepezil | Placebo | |||||||
|---|---|---|---|---|---|---|---|---|
| Type | N | K | Mean | N | K | Mean | p-value | |
| OC population | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 13 weeks | C | 17 | 30.7 (SD 13.9) | 18 | 40.9 (SD 19.4) | 0.085a | ||
| ADAS-cog – 26 weeks | C | 17 | 28.3 (SD 12.3) | 18 | 42.8 (SD 18.7) | < 0.01b | ||
Methodological issues
|
Randomisation and allocation: Randomisation process not reported. Individual responsible for the random allocation of patients to the trial arms was blind to the treatment code (how blinding was attained is not reported). Appearance of donepezil and placebo tablets is not described Data analysis: Polysomnographic and cognitive data were analysed using two-way ANOVA for repeated measures with treatment group and treatment time as the main factors and time/treatment interaction effect. Post hoc Duncan multiple-range test performed, with p-level set at ≤ 0.01. Spearman test to assess correlation between cognitive improvement rate and REM sleep and electroencephalography parameters Power calculation: Data from 10 patients was initially analysed for sample size estimation (procedure not reported). Based on this analysis, a sample size of 15 subjects in each group was calculated to set out a difference of 8 percentage points in REM sleep percentage (significance level of 1% and power of 95%). To assess the interaction term in the ANOVA model, 27 subjects were required in each group (sample size not attained) – power of 80% was possible with the sample size analysed Conflicts of interest: Authors state no financial conflicts of interest. No financial support from industry for study |
Quality appraisal
|
Feldman and Lane138
| Design | Participants | Arms | Outcomes |
|---|---|---|---|
|
Feldman and Lane (2007) 138 Study design: parallel double-blind RCT Countries: Australia, Canada, Ireland, Italy, South Africa and the UK No. of centres: 37 Funding: commissioned by Novartis Pharma AG (Switzerland) Length of follow-up (weeks): 26 Notes: |
No. randomised: 678 MMSE min: 10 MMSE max: 26 Inclusion criteria: AD (DSM-IV criteria) and probable AD (NINCDS-ADRDA) MMSE: 10–26 Responsible caregiver Exclusion criteria: Severe and unstable cardiac disease Severe and obstructive pulmonary disease Other life-threatening conditions Use of anticholinergic drugs, health food supplements containing ACh precursors, putative memory enhancers or insulin Use of psychotropic drugs, with the exception of chloral hydrate, short-acting benzodiazepines and haloperidol (≤ 3 days in succession and not < 72 hours before any efficacy assessment) Therapy common to all participants: None Sample attrition/dropout: 553 of 678 completed study. 125 withdrew after allocation: AEs (n = 83); ECG abnormalities (n = 4); laboratory abnormalities (n = 1); withdrawn consent (n = 14); protocol violation (n = 8); treatment failure (n = 2); failure to attend (n = 7); other reasons (n = 6). Differences between groups was only on AEs (rivastigmine t.i.d. 11%; rivastigmine b.i.d. 17%; placebo 9%) |
Arm no.: 1 Name: rivastigmine t.i.d. N: 227 Drug: rivastigmine Starting daily dose (mg): 2 Dosage details: Dose administered three times a day. Titrated from an initial dose of 2 mg/day for the first week up to a maximum of 12 mg in 1 mg/day steps at weekly intervals. Patients unable to tolerate 2 mg/day by day 10 were withdrawn from the study. Tolerability could be optimised by maintaining a dose level for periods of up to 2 weeks Arm no.: 2 Name: rivastigmine b.i.d. N: 229 Drug: rivastigmine Starting daily dose (mg): 2 Dosage details: Dose administered two times a day (plus one placebo tablet). Titrated from an initial dose of 2 mg/day for the first week up to a maximum of 12 mg in 1 mg/day steps at weekly intervals. Patients unable to tolerate 2 mg/day by day 10 were withdrawn from the study. Tolerability could be optimised by maintaining a dose level for periods of up to 2 weeks Arm no.: 3 Name: placebo N: 222 Drug: placebo Starting daily dose (mg): Dosage details: |
Cognitive ADAS-cog (11-item assessment of memory, language, praxis, orientation, total score range 0–70, with decreasing score indicating improved cognitive function) ADAS-cogA (ADAS-cog with an added item of attention (concentration/distractability), total score range 0–75, where decreasing score indicated improved cognitive function) MMSE (recent memory, attention, concentration, naming, repetition, comprehension and ability to formulate a sentence (10-item assessment, with a range of 0–30 points, with higher score representing better cognitive function) Functional PDS (activities of daily living, 29-item score on a VAS scale 0–100, where an increase in score indicated improvement in the patient’s ability to perform activities of daily living) Global severity CIBIC-plus score (overall global assessment of patient response on seven-point Likert scale where 1 = markedly improved and 7 = markedly worsened) GDS (overall staging of AD severity, seven-stage scale where a higher stage indicates more advanced AD) AEs |
Baseline characteristics
| Rivastigmine t.i.d. | Placebo | |||||||
|---|---|---|---|---|---|---|---|---|
| Type | N | K | Mean | N | K | Mean | p-value | |
| Demographics | ||||||||
| Age (years) | C | 227 | 71.4 (SD 7.9) | 222 | 71.7 (SD 8.7) | 0.702a | ||
| Gender (n male)b | D | 227 | 91 | 40.1% | 222 | 89 | 40.1% | 1.000c |
| Height (cm) | C | 227 | 164 (SD 10.7) | 222 | 164 (SD 10.3) | 1.000a | ||
| Weight (kg) | C | 227 | 65.9 (SD 12.9) | 222 | 65.9 (SD 12.3) | 1.000a | ||
| Disease characteristics | ||||||||
| Duration of dementia (months) | C | 227 | 38.4 (SD 25.5) | 222 | 39.7 (SD 28.2) | 0.608a | ||
| Disease severity (NINCDS-ADRDA): mild | D | 227 | 43 | 18.9% | 222 | 45 | 20.3% | 0.723c |
| Disease severity (NINCDS-ADRDA): moderate | D | 227 | 55 | 24.2% | 222 | 52 | 23.4% | 0.841c |
| Disease severity (NINCDS-ADRDA): severe | D | 227 | 3 | 1.3% | 222 | 3 | 1.4% | 0.978c |
| Cognitive | ||||||||
| MMSE – 0 weeks | C | 227 | 18.3 (SD 4.5) | 222 | 18.7 (SD 4.6) | 0.352a | ||
| Global severity | ||||||||
| GDS – 0 weeks | C | 227 | 4.1 (SD 0.8) | 222 | 4.1 (SD 0.9) | 1.000a | ||
| ITT population | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 0 weeks | C | 227 | 28.1 (SD 12.5) | 220 | 28.5 (SD 12.3) | 0.733a | ||
| ADAS-cogA – 0 weeks | C | 227 | 29.1 (SD 13.1) | 220 | 29.4 (SD 13) | 0.808a | ||
| MMSE – 0 weeks | C | 227 | 18.1 (SD 4.7) | 220 | 18.8 (SD 4.6) | 0.112a | ||
| Functional | ||||||||
| PDS – 0 weeks | C | 225 | 49.2 (SD 19.8) | 221 | 49 (SD 19.6) | 0.915a | ||
| Global severity | ||||||||
| GDS – 0 weeks | C | 227 | 4.1 (SD 0.9) | 222 | 4.1 (SD 0.9) | 1.000a | ||
| LOCF analysis | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 0 weeks | C | 209 | 28.3 (SD 12.2) | 208 | 28.5 (SD 12.2) | 0.867a | ||
| ADAS-cogA – 0 weeks | C | 209 | 29.2 (SD 12.9) | 208 | 29.4 (SD 12.8) | 0.874a | ||
| MMSE – 0 weeks | C | 193 | 18.1 (SD 4.5) | 198 | 18.8 (SD 4.6) | 0.129a | ||
| Functional | ||||||||
| PDS – 0 weeks | C | 207 | 49 (SD 19.6) | 209 | 48.9 (SD 19.4) | 0.958a | ||
| Global severity | ||||||||
| GDS – 0 weeks | C | 195 | 4.1 (SD 0.9) | 202 | 4.1 (SD 0.9) | 1.000a | ||
| OC population | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 0 weeks | C | 180 | 27.9 (SD 11.8) | 183 | 27.7 (SD 11.9) | 0.872a | ||
| Rivastigmine b.i.d. | Placebo | |||||||
|---|---|---|---|---|---|---|---|---|
| Type | N | K | Mean | N | K | Mean | p-value | |
| Demographics | ||||||||
| Age (years) | C | 229 | 71 (SD 8.2) | 222 | 71.7 (SD 8.7) | 0.380a | ||
| Gender (n male)b | D | 229 | 98 | 42.8% | 222 | 89 | 40.1% | 0.560c |
| Height (cm) | C | 229 | 164 (SD 10.7) | 222 | 164 (SD 10.3) | 0.480a | ||
| Weight (kg) | C | 229 | 66.7 (SD 12.2) | 222 | 65.9 (SD 12.3) | 0.488a | ||
| Disease characteristics | ||||||||
| Duration of dementia (months) | C | 229 | 40.6 (SD 31.2) | 222 | 39.7 (SD 28.2) | 0.748a | ||
| Disease severity (NINCDS-ADRDA): mild | D | 229 | 45 | 19.7% | 222 | 45 | 20.3% | 0.869c |
| Disease severity (NINCDS-ADRDA): moderate | D | 229 | 53 | 23.1% | 222 | 52 | 23.4% | 0.944c |
| Disease severity (NINCDS-ADRDA): severe | D | 229 | 2 | 0.9% | 222 | 3 | 1.4% | 0.628c |
| Cognitive | ||||||||
| MMSE – 0 weeks | C | 229 | 18.8 (SD 4.6) | 222 | 18.7 (SD 4.6) | 0.818a | ||
| Global severity | ||||||||
| GDS – 0 weeks | C | 229 | 4 (SD 0.9) | 222 | 4.1 (SD 0.9) | 0.239a | ||
| ITT population | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 0 weeks | C | 228 | 27.7 (SD 12.3) | 220 | 28.5 (SD 12.3) | 0.492a | ||
| ADAS-cogA – 0 weeks | C | 228 | 28.6 (SD 13) | 220 | 29.4 (SD 13) | 0.515a | ||
| MMSE – 0 weeks | C | 227 | 18.7 (SD 4.6) | 220 | 18.8 (SD 4.6) | 0.818a | ||
| Functional | ||||||||
| PDS – 0 weeks | C | 227 | 48.7 (SD 19.5) | 221 | 49 (SD 19.6) | 0.871a | ||
| Global severity | ||||||||
| GDS – 0 weeks | C | 229 | 4 (SD 0.9) | 222 | 4.1 (SD 0.9) | 0.239a | ||
| LOCF analysis | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 0 weeks | C | 199 | 27.7 (SD 12.3) | 208 | 28.5 (SD 12.2) | 0.510a | ||
| ADAS-cogA – 0 weeks | C | 199 | 28.5 (SD 13) | 208 | 29.4 (SD 12.8) | 0.482a | ||
| MMSE – 0 weeks | C | 186 | 18.7 (SD 4.6) | 198 | 18.8 (SD 4.6) | 0.832a | ||
| Functional | ||||||||
| PDS – 0 weeks | C | 195 | 48.6 (SD 19.7) | 209 | 48.9 (SD 19.4) | 0.878a | ||
| Global severity | ||||||||
| GDS – 0 weeks | C | 188 | 4 (SD 0.9) | 202 | 4.1 (SD 0.9) | 0.274a | ||
| OC population | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 0 weeks | C | 173 | 28.6 (SD 12.1) | 183 | 27.7 (SD 11.9) | 0.480a | ||
| Rivastigmine t.i.d | Placebo | |||||||
|---|---|---|---|---|---|---|---|---|
| Type | n | K | Mean | n | K | Mean | p-value | |
| ITT population | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 12 weeksa | MC | 227 | –1.9 (SD 6.66) | 220 | 0.9 (SD 5.93) | < 0.001b | ||
| ADAS-cog – 18 weeksa | MC | 227 | –1.6 (SD 6.66) | 220 | 1.8 (SD 6.67) | < 0.001b | ||
| ADAS-cog – 26 weeks | MC | 227 | –0.2 (SD 7.3) | 220 | 2.8 (SD 7.2) | ≤ 0.001c | ||
| ADAS-cog: any improvement – 12 weeksa | D | 227 | 68 | 30.0% | 220 | 36 | 16.4% | ≤ 0.001d |
| ADAS-cog: any improvement – 18 weeksa | D | 227 | 75 | 33.0% | 220 | 28 | 12.7% | ≤ 0.001d |
| ADAS-cog: any improvement – 26 weeksa | D | 227 | 52 | 22.9% | 220 | 28 | 12.7% | |
| ADAS-cogA – 26 weeks | MC | 227 | –0.1 (SD 7.9) | 220 | 3.2 (SD 7.8) | ≤ 0.001c | ||
| MMSE – 26 weeks | MC | 227 | 0.3 (SD 3.6) | 220 | –1.4 (SD 3.6) | ≤ 0.001b | ||
| Functional | ||||||||
| PDS – 26 weeks | MC | 225 | –1.5 (SD 11.3) | 221 | –4.9 (SD 11.2) | ≤ 0.001c | ||
| Global severity | ||||||||
| CIBIC-plus score – 12 weeksa | C | 220 | 3.9 | 213 | 4.3 | ≤ 0.001b | ||
| CIBIC-plus score – 18 weeksa | C | 220 | 3.9 (SD 1.04) | 213 | 4.5 (SD 1.02) | ≤ 0.001b | ||
| CIBIC-plus score – 26 weeks | C | 222 | 3.9 (SD 1.3) | 216 | 4.5 (SD 1.3) | ≤ 0.001e | ||
| CIBIC-plus: any improvement – 12 weeksa | D | 220 | 66 | 30.0% | 213 | 34 | 16.0% | ≤ 0.001d |
| CIBIC-plus: any improvement – 18 weeksa | D | 220 | 68 | 30.9% | 213 | 40 | 18.8% | ≤ 0.001d |
| CIBIC-plus: any improvement – 26 weeksa | D | 220 | 68 | 30.9% | 213 | 40 | 18.8% | < 0.05d |
| GDS – 26 weeks | MC | 227 | 0 (SD 0.7) | 222 | –0.3 (SD 0.7) | < 0.05b | ||
| Disposition of participants | ||||||||
| Discontinued treatment due to AEs – 26 weeks | D | 227 | 24 | 10.6% | 222 | 20 | 9.0% | |
| Discontinued treatment before end of trial – 26 weeks | D | 227 | 38 | 16.7% | 222 | 33 | 14.9% | |
| LOCF analysis | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 26 weeks | MC | 209 | –0.7 (SD 6.9) | 208 | 2.7 (SD 6.8) | ≤ 0.001c | ||
| ADAS-cogA – 26 weeks | MC | 209 | –0.6 (SD 7.5) | 208 | 3.1 (SD 7.4) | ≤ 0.001c | ||
| MMSE – 26 weeks | MC | 193 | 0.4 (SD 3.4) | 198 | –1.4 (SD 3.5) | ≤ 0.001b | ||
| Functional | ||||||||
| PDS – 26 weeks | MC | 207 | –1 (SD 11.4) | 209 | –4.7 (SD 11.3) | ≤ 0.001c | ||
| Global severity | ||||||||
| CIBIC-plus score – 26 weeks | C | 206 | 3.9 (SD 1.2) | 205 | 4.5 (SD 1.2) | ≤ 0.001e | ||
| GDS – 26 weeks | MC | 195 | 0 (SD 0.7) | 202 | –0.3 (SD 0.7) | < 0.05b | ||
| OC population | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 26 weeks | MC | 180 | –0.9 (SD 6.8) | 183 | 2.1 (SD 6.8) | ≤ 0.001c | ||
| Global severity | ||||||||
| CIBIC-plus score – 26 weeks | C | 177 | 3.9 (SD 1.2) | 179 | 4.4 (SD 1.2) | ≤ 0.001e | ||
| Safety population | ||||||||
| AEs | ||||||||
| Any AE – 0 weeks | D | 227 | 208 | 91.6% | 222 | 169 | 76.1% | < 0.05f |
| Any serious AE – 0 weeks | D | 227 | 40 | 17.6% | 222 | 33 | 14.9% | NSf |
| Anorexia – 0 weeks | D | 227 | 42 | 18.5% | 222 | 6 | 2.7% | < 0.05f |
| Nausea – 0 weeks | D | 227 | 109 | 48.0% | 222 | 31 | 14.0% | < 0.05f |
| Diarrhoea – 0 weeks | D | 227 | 38 | 16.7% | 222 | 20 | 9.0% | < 0.05f |
| Vomiting – 0 weeks | D | 227 | 68 | 30.0% | 222 | 14 | 6.3% | < 0.05f |
| Abdominal pain – 0 weeks | D | 227 | 26 | 11.5% | 222 | 12 | 5.4% | < 0.05f |
| Agitation – 0 weeks | D | 227 | 14 | 6.2% | 222 | 26 | 11.7% | < 0.05f |
| Anxiety – 0 weeks | D | 227 | 8 | 3.5% | 222 | 3 | 1.4% | NSf |
| Dizziness – 0 weeks | D | 227 | 39 | 17.2% | 222 | 16 | 7.2% | < 0.05f |
| Headache – 0 weeks | D | 227 | 36 | 15.9% | 222 | 23 | 10.4% | NSf |
| Flatulence – 0 weeks | D | 227 | 15 | 6.6% | 222 | 4 | 1.8% | < 0.05f |
| Haemorrhoids – 0 weeks | D | 227 | 2 | 0.9% | 222 | 6 | 2.7% | NSf |
Results
| Rivastigmine b.i.d. | Placebo | |||||||
|---|---|---|---|---|---|---|---|---|
| Type | n | K | Mean | n | K | Mean | p-value | |
| ITT population | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 12 weeksa | MC | 228 | –0.8 (SD 6.04) | 220 | 0.9 (SD 5.93) | < 0.05b | ||
| ADAS-cog – 18 weeksa | MC | 228 | –0.1 (SD 6.79) | 220 | 1.8 (SD 6.67) | < 0.001b | ||
| ADAS-cog – 26 weeks | MC | 228 | 1.2 (SD 7.2) | 220 | 2.8 (SD 7.2) | < 0.05c | ||
| ADAS-cog: any improvement – 12 weeksa | D | 228 | 52 | 22.8% | 220 | 36 | 16.4% | < 0.05d |
| ADAS-cog: any improvement – 18 weeksa | D | 228 | 57 | 25.0% | 220 | 28 | 12.7% | ≤ 0.001d |
| ADAS-cog: any improvement – 26 weeksa | D | 228 | 41 | 18.0% | 220 | 28 | 12.7% | NSd |
| ADAS-cogA – 26 weeks | MC | 228 | 1.5 (SD 7.8) | 220 | 3.2 (SD 7.8) | < 0.05c | ||
| MMSE – 26 weeks | MC | 227 | –0.6 (SD 3.6) | 220 | –1.4 (SD 3.6) | < 0.05b | ||
| Functional | ||||||||
| PDS – 26 weeks | MC | 227 | –2.6 (SD 11.1) | 221 | –4.9 (SD 11.2) | < 0.05c | ||
| Global severity | ||||||||
| CIBIC-plus score – 12 weeksa | C | 215 | 3.9 | 213 | 4.3 | ≤ 0.001b | ||
| CIBIC-plus score – 18 weeksa | C | 215 | 4.1 (SD 1.03) | 213 | 4.5 (SD 1.02) | ≤ 0.001b | ||
| CIBIC-plus score – 26 weeks | C | 222 | 4.1 (SD 1.3) | 216 | 4.5 (SD 1.3) | < 0.05e | ||
| CIBIC-plus: any improvement – 12 weeksa | D | 215 | 62 | 28.8% | 213 | 34 | 16.0% | < 0.05d |
| CIBIC-plus: any improvement – 18 weeksa | D | 215 | 47 | 21.9% | 213 | 40 | 18.8% | NSd |
| CIBIC-plus: any improvement – 26 weeksa | D | 215 | 49 | 22.8% | 213 | 40 | 18.8% | NSd |
| GDS – 26 weeks | MC | 229 | –0.2 (SD 0.7) | 222 | –0.3 (SD 0.7) | NSb | ||
| Disposition of participants | ||||||||
| Discontinued treatment due to AEs – 26 weeks | D | 229 | 39 | 17.0% | 222 | 20 | 9.0% | |
| Discontinued treatment before end of trial – 26 weeks | D | 229 | 54 | 23.6% | 222 | 33 | 14.9% | |
| LOCF analysis | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 26 weeks | MC | 199 | 0.8 (SD 6.9) | 208 | 2.7 (SD 6.8) | < 0.05c | ||
| ADAS-cogA – 26 weeks | MC | 199 | 1 (SD 7.5) | 208 | 3.1 (SD 7.4) | < 0.05c | ||
| MMSE – 26 weeks | MC | 186 | –0.4 (SD 3.5) | 198 | –1.4 (SD 3.5) | < 0.05b | ||
| Functional | ||||||||
| PDS – 26 weeks | MC | 195 | –2.3 (SD 11.5) | 209 | –4.7 (SD 11.3) | < 0.05c | ||
| Global severity | ||||||||
| CIBIC-plus score – 26 weeks | C | 198 | 4.1 (SD 1.2) | 205 | 4.5 (SD 1.2) | < 0.05e | ||
| GDS – 26 weeks | MC | 188 | –0.1 (SD 0.7) | 202 | –0.3 (SD 0.7) | NSb | ||
| OC population | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 26 weeks | MC | 173 | 0.9 (SD 7) | 183 | 2.1 (SD 6.8) | NSc | ||
| Global severity | ||||||||
| CIBIC-plus score – 26 weeks | C | 167 | 4.1 (SD 1.2) | 179 | 4.4 (SD 1.2) | < 0.05e | ||
| Safety population | ||||||||
| AEs | ||||||||
| Any AE – 0 weeks | D | 228 | 208 | 91.2% | 222 | 169 | 76.1% | < 0.05f |
| Any serious AE – 0 weeks | D | 228 | 40 | 17.5% | 222 | 33 | 14.9% | NSf |
| Anorexia – 0 weeks | D | 228 | 47 | 20.6% | 222 | 6 | 2.7% | < 0.05f |
| Nausea – 0 weeks | D | 228 | 123 | 53.9% | 222 | 31 | 14.0% | < 0.05f |
| Diarrhoea – 0 weeks | D | 228 | 40 | 17.5% | 222 | 20 | 9.0% | < 0.05f |
| Vomiting – 0 weeks | D | 228 | 88 | 38.6% | 222 | 14 | 6.3% | < 0.05f |
| Abdominal pain – 0 weeks | D | 228 | 34 | 14.9% | 222 | 12 | 5.4% | < 0.05f |
| Agitation – 0 weeks | D | 228 | 21 | 9.2% | 222 | 26 | 11.7% | NSf |
| Anxiety – 0 weeks | D | 228 | 13 | 5.7% | 222 | 3 | 1.4% | < 0.05f |
| Dizziness – 0 weeks | D | 228 | 42 | 18.4% | 222 | 16 | 7.2% | < 0.05f |
| Headache – 0 weeks | D | 228 | 40 | 17.5% | 222 | 23 | 10.4% | < 0.05f |
| Flatulence – 0 weeks | D | 228 | 11 | 4.8% | 222 | 4 | 1.8% | NSf |
| Haemorrhoids – 0 weeks | D | 228 | 0 | 0.0% | 222 | 6 | 2.7% | < 0.05f |
Methodological issues
|
Randomisation and allocation: Randomisation procedure not described. Rivastigmine and placebo tablets were identical and the number taken was the same at each dose in all groups Data analysis: ADAS-cog – two-way treatment by centre ANOVA and ANCOVA (SAS type III analysis) on changes from baseline for each time point (12, 18 and 26 weeks), using the baseline score as covariate ADAS-cog – categorical analysis to determine the proportion of patinets showing at least a 4.0-point score at 26 weeks, with Mantel–Haenszel blocking for centre CIBIC-plus improvers – categorical analysis to determine proportion showing imporvements vs those showing no change or worsening, with Mantel–Haenszel blocking for centre CIBIC-plus – two-way ANOVA (SAS type III analysis) PDS and ADAS-cogA – ANCOVA on changesd from baseline to week 26, and post hoc Cohen’s D effect sizes calculated at each visit for the ADAS-cog and CIBIC-plus by dividing mean differences by pooled SDs Comparisons with placebo were two-tailed, with the critical significance level set at p < 0.05. In order to control for multiplicity in the analyses of efficacy data, the primary comparison was specified as rivastigmine administered b.i.d. against placebo. If this test was statistically significant at the 0.05 level then the rivastigmine administered t.i.d. against placebo was tested at the 0.05 level subsequently. As both primary efficacy variables were required to be significant, no further correction of the size of the tests for the multiplicity of variables was required Power calculation: The study sample size was determined on the basis of an estimated 3.0-point difference between rivastigmine administered b.i.d. and placebo on the ADAS-cog, an estimated 0.4-point difference between b.i.d. and placebo on the CIBIC-plus and an increased proportion of responders with CIBIC-plus ratings of 0.4 of 20% within the b.i.d. rivastigmine group (35% rivastigmine vs 15% placebo). Sample sizes of 192 per group were required. For practical reasons the sample size was chosen as 200 (ITT population). An individual power of 90% guaranteed protection of the global power in view of the requirement that both ADAS-cog and CIBIC-plus analyses should be significant at the 0.0499 level Conflicts of interest: HF has received honoraria for consulting, advisory boards and for participation in CME programmes sponsored by Novartis. He has also received grant-in-aid funding for research from Novartis. RL is an employee of Novartis. The study was commissioned by Novartis Pharma AG in Switzerland |
Quality appraisal
|
Mazza et al.118
| Design | Participants | Arms | Outcomes |
|---|---|---|---|
|
Mazza et al. (2006) 118 Study design: parallel double-blind RCT Country: Italy? No. of centres: 1 Funding: not reported Length of follow-up (weeks): 24 Notes: |
No. randomised: 76 MMSE min: 13 MMSE max: 25 Inclusion criteria: AD (DSM-IV criteria) Brief Cognitive Rating Scale mean score 3–5 Hachinski Ischaemic Score < 4 Adequate level of premorbid intelligence (IG > 80, global assessment) Exclusion criteria: Dementia of other aetiology Severe organic diseases (tumours, severe infectious diseases, brain trauma, epilepsy, cerebrovascular malformations, alcohol or drug abuse) Pseudodementia or a histiory of schizophrenic or affective psychoses (Geriatric Depression Scale, 15-item version, total score < 9) Vasoactive drugs, nootropics and long-term treatment with other drugs were proscribed during the study, with the exception of low doses of benzodiazepines and neuroleptic drugs in the treatment of behavioural disturbances Therapy common to all participants: Single-blind placebo 4-week run-in period (in order to exclude placebo responders) Sample attrition/dropout: 60 of 76 randomised patients completed the study (a further 41 were excluded during the run-in period; reasons not reported) |
Arm no.: 1 Name: donepezil n: 25 Drug: donepezil Starting daily dose (mg): 5 Dosage details: 5 mg daily Arm no.: 2 Name: placebo n: 26 Drug: placebo Starting daily dose (mg): Dosage details: not reported |
Cognitive MMSE SKT [psychometric test battery for assessment of memory and attention, consisting of nine 1-minute subtests that are partly speed orientated and partly span orientated: total score ranged from 1 (very good) to 27 (very poor)] CGI-2 (cognitive) [global change in observable cognitive functioning, transitional scale ranging from 1 (very much improved) to 7 (very much deteriorated)] |
Baseline characteristics
| Type | Donepezil | Placebo | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| N | K | Mean | N | K | Mean | |||
| ITT population | ||||||||
| Demographics | ||||||||
| Age (years) | C | 25 | 64.5 (SD 6) | 26 | 69.8 (SD 3) | < 0.001a | ||
| Gender (n male) | D | 25 | 13 | 52.0% | 26 | 10 | 38.5% | 0.490b |
| Cognitive | ||||||||
| MMSE – 0 weeks | C | 25 | 18.6 (SD 3.47) | 26 | 18.8 (SD 3.63) | |||
| SKT – 0 weeks | C | 25 | 15.2 (SD 3.48) | 26 | 15.9 (SD 3.86) | |||
| CGI-2 (cognitive) – 0 weeks | C | 25 | 4.5 (SD 0.76) | 26 | 5.05 (SD 0.99) | |||
Results
| Type | Donepezil | Placebo | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| N | K | Mean | N | K | Mean | |||
| ITT population | ||||||||
| Cognitive | ||||||||
| MMSE – 24 weeks | C | 25 | 19.8 (SD 3.16) | 26 | 18.6 (SD 3.66) | NSa | ||
| MMSE – 24 weeks | MC | 25 | 1.2 (SD 12.2) | 26 | –0.25 (SD 5)b | 0.06a | ||
| SKT – 24 weeks | C | 25 | 11.8 (SD 2.9) | 26 | 16.9 (SD 3.9) | 0.01a | ||
| SKT – 24 weeks | MC | 25 | –3.3 (SD –2.55) | 26 | 0.9 (SD 1.3) | < 0.001a | ||
| CGI-2 (cognitive) – 24 weeks | C | 25 | 3.6 (SD 0.94) | 26 | 5.2 (SD 0.95) | 0.01a | ||
| CGI-2 (cognitive) – 24 weeks | MC | 25 | –0.9 (SD 1.02) | 26 | 0.15 (SD 0.338) | < 0.001a | ||
| Disposition of participants | ||||||||
| Discontinued treatment due to AEs – 24 weeks | D | 25 | 4 | 16.0% | 26 | 0 | 0.0% | |
| Discontinued treatment before end of trial – 24 weeks | D | 25 | 4 | 16.0% | 26 | 6c | 23.1% | |
Methodological issues
| Randomisation and allocation: Randomisation computer generated (whether or not unreadable before allocation is not stated). Appearance of pills and placebo not reported |
| Data analysis: MMSE, SKT, CGI-2 – t-test for paired samples was used to compare each group from baseline to 24 weeks of treatment. ANOVA to detect difference between groups (age, gender, and severity of cognitive impairment at baseline were factors of ANOVA model) |
| Power calculation: Not reported |
| Conflicts of interest: Not reported |
Quality appraisal
|
Moraes et al.120
| Design | Participants | Arms | Outcomes |
|---|---|---|---|
|
Moraes et al. (2008) 120 Study design: parallel double-blind RCT Country: Brazil No. of centres: 1 Funding: FAPESP, AFIP Length of follow-up (weeks): 12 Notes: |
No. randomised: 23 MMSE min: 6 MMSE max: 27 Inclusion criteria: AD (ADRDA criteria) Rating of 1–2 (mild to moderate) on Brazilian version of CDR Exclusion criteria: Rating of ≥ 3 on Brazilian version of CDR Other causes of dementia Other current severe medical or psychiatric disease Psychoactive drugs in the month prior to entering the study Therapy common to all participants: two nights of polysomnographic recording (for purposes of habituation) Sample attrition/dropout: not reported |
Arm no.: 1 Name: donepezil n: 11 Drug: donepezil Starting daily dose (mg): 5 Dosage details: single dose of 5 mg (administered at bedtime) in the first month, increased to single dose of 10 mg in second month Arm no.: 2 Name: placebo n: 12 Drug: placebo Starting daily dose (mg): Dosage details: single dose administered at bedtime |
ADAS-cog (multiple cognitive functions including word evocation, verbal fluency, understanding of simple commands, constructive praxis, ideational praxis, temporospatial orientation, word recognition, verbal fluency, vocabulary, and understanding: scores range from 0 to 70, with higher scores indicating more cognitive deterioration) |
Baseline characteristics
| Type | Donepezil | Placebo | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| N | K | Mean | N | K | Mean | |||
| OC population | ||||||||
| Demographics | ||||||||
| Age (years) | C | 11 | 76.8 (SD 6.2) | 12 | 72.6 (SD 11) | 0.27a | ||
| Gender (n male) | D | 11 | 3 | 27.3% | 12 | 5 | 41.7% | 0.49a |
| BMI (kg/m2) | C | 11 | 26.3 (SD 4.8) | 12 | 26.6 (SD 4.1) | 0.85a | ||
| Cognitive | ||||||||
| ADAS-cog – 0 weeks | C | 11 | 34.5 (SD 15.8) | 12 | 29.3 (SD 17.3) | |||
| MMSE | C | 11 | 19 (SD 3.6) | 12 | 17.2 (SD 7.8) | 0.50a | ||
| Global severity | ||||||||
| CDR | C | 11 | 1.3 (SD 0.5) | 12 | 1.3 (SD 0.5) | 0.76a | ||
Results
| Type | Donepezil | Placebo | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| n | K | Mean | n | K | Mean | |||
| OC population | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 13 weeks | C | 11 | 29.7 (SD 15.7) | 12 | 31.8 (SD 18.5) | < 0.05a | ||
Methodological issues
| Randomisation and allocation: Randomisation performed using computer-generated random number list (0–1) with uniform distribution, with patients consecutively allocated to the two treatment groups (≤ 0.5 to group A, > 0.5 to group B). Donepezil and placebo pills were ‘packed in the same fashion’, but precise appearance of pills not reported |
| Data analysis: One-way ANOVA was used to compare all variables for donepezil and placebo groups during the baseline recording night. Polysomnographic and cognitive data at baseline and after 3 months of treatment were analysed using two-way ANOVA for repeated measures with treatment group and treatment time as the main factors and time/treatment interaction effect followed by the Bonferroni test, with p ≤ 0.01 comparing data |
| Power calculation: Not reported |
| Conflicts of interest: Authors state no conflicts of interest to disclose |
Quality appraisal
|
Mowla et al.139
| Design | Participants | Arms | Outcomes |
|---|---|---|---|
|
Mowla et al. (2007) 139 Study design: parallel double-blind RCT Country: not reported; lead author based in the Islamic Republic of Iran No. of centres: not reported Funding: Shiraz University of Medical Sciences Length of follow-up (weeks): 12 Notes: 12-week mean MMSE/WMS/ADL/HAM scores in the fluoxetine plus rivastigmine arm were much lower than in the other arms – potential error? |
No. randomised: 122 MMSE min: 10 MMSE max: 24 Inclusion criteria: AD (DSM-IV criteria) Brief Cognitive Rating Score mean 3–5 Hachinski Ischaemic Score < 4 Adequate level of premorbid intelligence (IG > 80, global assessment) Exclusion criteria: Dementia of other aetiology Severe organic disease (tumours, severe infectious disease, brain trauma, epilepsy, cerebrovascular malformations, alcohol or drug abuse) Other psychiatric disorders (Hamilton Depression Scale, 17-item version, total score < 10) Therapy common to all participants: single-blind placebo 6-week run-in period to exclude placebo responders Sample attrition/dropout: 98 of 122 completed study. Dropouts: rivastigmine arm n = 7; fluoxetine plus rivastigmine n = 9; and placebo n = 8. Major cause of withdrawal in fluoxetine plus rivastigmine arm was AEs, in placebo arm it was loss of efficacy |
Arm no.: 1 Name: rivastigmine n: 41 Drug: rivastigmine Starting daily dose (mg): 3 Dosage details: titrated from initial dose of 1.5 mg twice a day, doubled every 2 weeks until maximum dose of 6 mg twice a day reached (or dose which patient could tolerate) Notes: no details of placebo fluoxetine administration Arm no.: 2 Name: rivastigmine plus fluoxetine n: 41 Drug: rivastigmine Starting daily dose (mg): 3 Dosage details: titrated from initial dose of 1.5 mg b.i.d., doubled every 2 weeks until maximum dose of 6 mg b.i.d. reached (or dose which patient could tolerate) Notes: fluoxetine 20 mg/day Arm no.: 3 Name: placebo n: 40 Drug: placebo Starting daily dose (mg): Dosage details: |
Cognitive MMSE WMS-III (immediate and delayed logical memory, digit span forward and backward, and family pictures I and II from Persian standardised WMS-III) CGI-2 (cognitive) [global change in observable cognitive functioning, scale from 1 (very much improved) to 7 (very much deteriorated)] Functional ADL (Lawton and Brody scale, eight items in Instrumental ADL and six items in Basic ADL, subtest scores aggregated to give a total functional assessment (ADL) score [scale in subtests from 1 (being completely capable of doing the activity) to 5 (being thoroughly unable to perform the activity)] Behavioural Hamilton Depression Scale (not reported) |
Baseline characteristics
| All study participants | |||
|---|---|---|---|
| N | k | Mean | |
| Demographics | |||
| Age (years) | 122 | 69.2 | |
| Gender (n male) | 122 | 65a | 53.3% |
| Type | Rivastigmine | Placebo | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| n | K | Mean | n | K | Mean | |||
| Cognitive | ||||||||
| MMSE – 0 weeks | C | 41 | 16.3 (SD 4.1) | 40 | 16.5 (SD 3.6) | 0.816a | ||
| WMS-III – 0 weeks | C | 41 | 7.7 (SD 2.2) | 40 | 8.3 (SD 2) | 0.203a | ||
| Functional | ||||||||
| ADL – 0 weeks | C | 41 | 26.5 (SD 7.7) | 40 | 26.8 (SD 7.5) | 0.860a | ||
| Behavioural | ||||||||
| Hamilton Depression Scale – 0 weeks | C | 41 | 8.06 (SD 1.7) | 40 | 7.33 (SD 1.39) | 0.038a | ||
| Type | Rivastigmine + fluoxetine | Placebo | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| n | K | Mean | n | K | Mean | |||
| Cognitive | ||||||||
| MMSE – 0 weeks | C | 41 | 15.6 (SD 0.73) | 40 | 16.5 (SD 3.6) | 0.121a | ||
| WMS-III – 0 weeks | C | 41 | 8 (SD 0.32) | 40 | 8.3 (SD 2) | 0.346a | ||
| Functional | ||||||||
| ADL – 0 weeks | C | 41 | 27.4 (SD 1.3) | 40 | 26.8 (SD 7.5) | 0.615a | ||
| Behavioural | ||||||||
| Hamilton Depression Scale – 0 weeks | C | 41 | 8.17 (SD 0.32) | 40 | 7.33 (SD 1.39) | < 0.001a | ||
Results
| Type | Rivastigmine | Placebo | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| n | K | Mean | n | K | Mean | |||
| ITT population | ||||||||
| Disposition of participants | ||||||||
| Discontinued treatment due to AEs – 12 weeks | D | 41 | 3 | 7.3% | 40 | 0a | 0.0% | |
| Discontinued treatment before end of trial – 12 weeks | D | 41 | 7 | 17.1% | 40 | 8 | 20.0% | |
| OC population | ||||||||
| Cognitive | ||||||||
| MMSE – 12 weeks | MC | 41 | 1.1 (SD 1.4) | 40 | –0.5 (SD 0.5) | < 0.001b | ||
| MMSE – 12 weeks | C | 34 | 17.4 (SD 3.7) | 32 | 16 (SD 3.7) | 0.129c | ||
| MMSE – 12 weeks | MC | 34 | 1.1 (SD 1.4) | 32 | –0.5 (SD 0.5) | < 0.001b | ||
| WMS-III – 12 weeks | MC | 41 | 0.97 (SD 1.7) | 40 | –0.66 (SD 1.1) | < 0.001b | ||
| WMS-III – 12 weeks | C | 34 | 8.7 (SD 2.2) | 32 | 7.5 (SD 1.4) | 0.011c | ||
| WMS-III – 12 weeks | MC | 34 | 0.97 (SD 1.7) | 32 | –0.66 (SD 1.1) | < 0.001b | ||
| Clinical Global Impression-item 2 (cognitive) – 12 weeks | C | 34 | 3.1 (SD 0.96) | 32 | 3.7 (SD 0.67) | 0.005c | ||
| Functional | ||||||||
| ADL – 12 weeks | MC | 41 | 1.2 (SD 2.6) | 40 | –0.68 (SD 1.3) | 0.58d | ||
| ADL – 12 weeks | C | 34 | 25.3 (SD 6.6) | 32 | 27.1 (SD 6.9) | 0.283c | ||
| ADL – 12 weeks | MC | 34 | 1.2 (SD 2.6) | 32 | –0.68 (SD 1.3) | 0.58d | ||
| Behavioural | ||||||||
| Hamilton Depression Scale – 12 weeks | C | 34 | 6.26 (SD 2.9) | 32 | 8.33 (SD 1.12) | < 0.001c | ||
| Type | Rivastigmine + fluoxetine | Placebo | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| n | K | Mean | n | K | Mean | |||
| ITT population | ||||||||
| Disposition of participants | ||||||||
| Discontinued treatment due to AEs – 12 weeks | D | 41 | 5 | 12.2% | 40 | 0a | 0.0% | |
| Discontinued treatment before end of trial – 12 weeks | D | 41 | 9 | 22.0% | 40 | 8 | 20.0% | |
| OC population | ||||||||
| Cognitive | ||||||||
| MMSE – 12 weeks | MC | 41 | 1.6 (SD 2.7) | 40 | –0.5 (SD 0.5) | 0.002b | ||
| MMSE – 12 weeks | C | 32 | 17.2 (SD 0.63) | 32 | 16 (SD 3.7) | |||
| MMSE – 12 weeks | MC | 32 | 1.6 (SD 2.7) | 32 | –0.5 (SD 0.5) | 0.002b | ||
| WMS-III – 12 weeks | MC | 41 | 0.96 (SD 2.1) | 40 | –0.66 (SD 1.1) | < 0.001b | ||
| WMS-III – 12 weeks | C | 32 | 8.9 (SD 0.54) | 32 | 7.5 (SD 1.4) | |||
| WMS-III – 12 weeks | MC | 32 | 0.96 (SD 2.1) | 32 | –0.66 (SD 1.1) | < 0.001b | ||
| Clinical Global Impression-item 2 (cognitive) – 12 weeks | C | 32 | 2.5 (SD 1.2) | 32 | 3.7 (SD 0.67) | |||
| Functional | ||||||||
| ADL – 12 weeks | MC | 41 | 3.2 (SD 3.2) | 40 | –0.68 (SD 1.3) | 0.001b | ||
| ADL – 12 weeks | C | 32 | 24.2 (SD 0.95) | 32 | 27.1 (SD 6.9) | |||
| ADL – 12 weeks | MC | 32 | 3.2 (SD 3.2) | 32 | –0.68 (SD 1.3) | 0.001b | ||
| Behavioural | ||||||||
| Hamilton Depression Scale – 12 weeks | C | 32 | 6.55 (SD 0.32) | 32 | 8.33 (SD 1.12) | |||
Methodological issues
| Randomisation and allocation: Computer-generated (on-site) randomisation – whether or not researchers were able to view randomisation sequence prior to allocation is not reported. Same number of pills for all trial arms, but appearance of these pills not reported (simply described as ‘similar’) |
| Data analysis: MMSE/WMS/ADL/HAM: t-test for paired samples (within-group comparisons). MMSE/WMS/ADL/CGI-2: ANOVA followed by Tukey post hoc comparison when significant effects present |
| Power calculation: not reported |
| Conflicts of interest: not reported |
Quality appraisal
|
Ancoli-Israel et al.149
| Design | Participants | Arms | Outcomes |
|---|---|---|---|
|
Ancoli-Israel et al. (2005) 149 Study design: parallel double-blind RCT Country: not reported; all study authors based in the USA No. of centres: not reported Funding: Janssen Medical Affairs Length of follow-up (weeks): 8 Notes: |
No. randomised: 63 MMSE min: 10 MMSE max: 24 Inclusion criteria: Mild to moderate AD (criteria not reported) MMSE 10–24 ≥ 60 years of age Resident with a responsible caregiver who agreed to participate and monitor sleep and answer questionnaires Exclusion criteria: Other neurodegenerative disease contributing to dementia (including multi-infarct dementia or clinically active cerebrovascular disease) Other medical condittions causing cognitive impairment Clinically significant co-existing medical conditions (psychiatric, cardiovascular, or active peptic ulcer disease; urinary outflow obstruction; hepatic, renal, pulmonary, metabolic or endocrine disturbances) Use of a muscarinic-1 agonist or AChEI within 30 days prior to involvement Therapy common to all participants: 2-week, single-blind, placebo run-in Sample attrition/dropout: 54 of 63 completed study; discontinued due to AE (n = 3 in galantamine arm; n = 4 in donepezil arm); discontinued due to severe AE possibly related to trial drug (hepatic failure, n = 1 in donepezil arm); and death (judged to be unrelated to trial drug, n = 1) |
Arm no.: 1 Name: donepezil n: 32 Drug: donepezil Starting daily dose (mg): 5 Dosage details: dose titrated from 5 mg o.d. at night for the first 4 weeks up to 10 mg o.d. at night for remainder of study Arm no.: 2 Name: galantamine n: 31 Drug: galantamine Starting daily dose (mg): 8 Dosage details: dose titrated from 4 mg b.i.d. for the first 4 weeks up to 8 mg b.i.d. for remainder of study |
Global severity CIBIC-plus (Clinician’s assessment of patient’s general functioning, cognition, behaviour, and performance of daily living activities) AEs |
Baseline characteristics
| Type | Donepezil | Galantamine | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| N | K | Mean | N | K | Mean | |||
| Demographics | ||||||||
| Age (years) | C | 32 | 77.8 (SD 6.2) | 31 | 76.5 (SD 7.7) | 0.463a | ||
| Gender (n male) | D | 32 | 14 | 43.8% | 31 | 10 | 32.3% | 0.497b |
| Education (at least high school) | D | 32 | 26 | 81.3% | 31 | 22 | 71.0% | 0.508b |
| Race (n white) | D | 32 | 26 | 81.3% | 31 | 25 | 80.6% | 0.795b |
| Race (n black) | D | 32 | 2 | 6.3% | 31 | 3 | 9.7% | 0.970b |
| Race (n hispanic) | D | 32 | 1 | 3.1% | 31 | 2 | 6.5% | 0.978b |
| Race (n Asian) | D | 32 | 1 | 3.1% | 31 | 1 | 3.2% | 0.487b |
| Race (n other) | D | 32 | 2 | 6.3% | 31 | 0 | 0.0% | 0.573b |
| Caregiver characteristics | ||||||||
| Age (years) | C | 32 | 69.4 (SD 11.4) | 31 | 67.7 (SD 15.9) | 0.627a | ||
| Gender (n male) | D | 32 | 15 | 46.9% | 31 | 15 | 48.4% | 0.895b |
| Race (n white) | D | 32 | 26 | 81.3% | 31 | 25 | 80.6% | 0.795b |
| Race (n black) | D | 32 | 2 | 6.3% | 31 | 3 | 9.7% | 0.970b |
| Race (n Hispanic) | D | 32 | 1 | 3.1% | 31 | 2 | 6.5% | 0.978b |
| Race (n Asian) | D | 32 | 1 | 3.1% | 31 | 1 | 3.2% | 0.487b |
| Race (n other) | D | 32 | 2 | 6.3% | 31 | 0 | 0.0% | 0.573b |
| Education: at least high school | D | 32 | 26 | 81.3% | 31 | 24 | 77.4% | 0.949b |
| Relationship to participant: spouse | D | 32 | 24 | 75.0% | 31 | 22 | 71.0% | 0.939b |
| Relationship to participant: child | D | 32 | 7 | 21.9% | 31 | 5 | 16.1% | 0.795b |
| Relationship to participant: relative/friend | D | 32 | 0 | 0.0% | 31 | 3 | 9.7% | 0.287b |
| Relationship to participant: other | D | 32 | 1 | 3.1% | 31 | 1 | 3.2% | 0.487b |
| Cognitive | ||||||||
| MMSE | C | 32 | 19.4 (range 13–24) | 31 | 19.3 (range 11–24) | NSc | ||
Results
| Type | Donepezil | Galantamine | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| n | K | Mean | n | K | Mean | |||
| Global severity | ||||||||
| CIBIC-plus score – 8 weeks | C | 29 | 3.97 (SD 1.02) | 27 | 3.59 (SD 0.636) | 0.106a | ||
| CIBIC-plus: markedly improved – 8 weeks | D | 29 | 0 | 0.0% | 27 | 0 | 0.0% | 0.330b |
| CIBIC-plus: moderately improved – 8 weeks | D | 29 | 3 | 10.3% | 27 | 2 | 7.4% | 0.933b |
| CIBIC-plus: minimally improved – 8 weeks | D | 29 | 4 | 13.8% | 27 | 7 | 25.9% | 0.421b |
| CIBIC-plus: no change – 8 weeks | D | 29 | 18 | 62.1% | 27 | 18 | 66.7% | 0.936b |
| CIBIC-plus: minimally worse – 8 weeks | D | 29 | 3 | 10.3% | 27 | 0 | 0.0% | 0.334b |
| CIBIC-plus: moderately worse – 8 weeks | D | 29 | 3 | 10.3% | 27 | 0 | 0.0% | 0.334b |
| CIBIC-plus: markedly worse – 8 weeks | D | 29 | 0 | 0.0% | 27 | 0 | 0.0% | 0.330b |
| AEs | ||||||||
| Nausea – 8 weeks | D | 32 | 1 | 3.1% | 31 | 3 | 9.7% | 0.583b |
| Diarrhoea – 8 weeks | D | 32 | 5 | 15.6% | 31 | 1 | 3.2% | 0.212b |
| Injury – 8 weeks | D | 32 | 2 | 6.3% | 31 | 2 | 6.5% | 0.628b |
| Headache – 8 weeks | D | 32 | 3 | 9.4% | 31 | 2 | 6.5% | 0.970b |
| Constipation – 8 weeks | D | 32 | 3 | 9.4% | 31 | 0 | 0.0% | 0.317b |
| Pain – 8 weeksc | D | 32 | 3 | 9.4% | 31 | 2 | 6.5% | 0.970b |
| Bronchitis – 8 weeks | D | 32 | 0 | 0.0% | 31 | 3 | 9.7% | 0.287b |
| Disposition of participants | ||||||||
| Discontinued treatment due to AEs – 1 week | D | 32 | 4 | 12.5% | 31 | 3 | 9.7% | 0.964b |
| Discontinued treatment before end of trial – 1 week | D | 32 | 4 | 12.5% | 31 | 5 | 16.1% | 0.959b |
Methodological issues
| Randomisation and allocation: Randomisation procedure not described |
| Data analysis: Per cent sleep [MC from baseline (SE)] |
| Actigraphy measured [mean (SE)] |
| Pittsburgh Sleep Quality Index (PSQI) [mean (SE) and Pearson correlation coefficient] |
| CIBIC-plus, descriptive statistics only (%) |
| Power calculation: None |
| Conflicts of interest: Lead author declares no financial disclosure; co-authors are employees of funder (Janssen Medical Affairs) |
Quality appraisal
|
Nordberg et al.147
| Design | Participants | Arms | Outcomes |
|---|---|---|---|
|
Nordberg et al. (2009) 147 Study design: Country: not reported No. of centres : not reported Funding: Novartis Pharmaceuticals; Swedish Research Council; KI foundations, L-H Osterman and Stohne’s Foundations supported two co-authors (AN, TDS). Alpha-Plus provided editorial assistance with the production of the manuscript Length of follow-up (weeks): 13 Notes: |
No. randomised: 63 MMSE min: 10 MMSE max: 20 Inclusion criteria: AD (DSM-IV criteria) and probable or possible AD (NINCDS-ADRDA criteria) Age 50–85 years MMSE 10–20 Provided the dose had been stabilised for the past month, treatment with psychotropics was permitted Exclusion criteria: Prior exposure to rivastigmine, donepezil or galantamine Advance, severe or unstable disease of any type that might interfere with study evaluation or put the patient at special risk Imaging findings consistent with a condition other than AD that would explain the patient’s dementia Current treatment with coumarin derivatives Blood clotting abnormalities or inadequate platelet function Therapy common to all participants: none Sample attrition/dropout: 53 of 63 completed study. 10 withdrew after allocation; AEs (n = 8), withdrew consent (n = 1), lost to follow-up (n = 1) |
Arm no.: 1 Name: donepezil n: 20 Drug: donepezil Starting daily dose (mg): 5 Dosage details: starting dose 5 mg q.d.; after ≥ 4 weeks, if tolerated, up-titrated to 10 mg q.d.; no subsequent up-titrations Arm no.: 2 Name: galantamine n: 21 Drug: galantamine Starting daily dose (mg): 8 Dosage details: starting dose 4 mg b.i.d.; after ≥ 4 weeks, if tolerated, up-titrated to 8 mg b.i.d.; subsequent up-titrations could be made after ≥ 4 weeks at each dose, based upon the patient’s well-being and tolerability, to a maximum of 12 mg b.i.d. Arm no.: 3 Name: rivastigmine n: 22 Drug: rivastigmine Starting daily dose (mg): 3 Dosage details: starting dose 1.5 mg b.i.d.; after ≥ 4 weeks, if tolerated, up-titrated to 3 mg b.i.d.; subsequent up-titrations could be made after ≥ 4 weeks at each dose, based upon the patient’s well-being and tolerability, to a maximum of 6 mg b.i.d. |
AEs only |
Baseline characteristics
| Type | Donepezil | Galantamine | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| N | K | Mean | N | K | Mean | |||
| Demographics | ||||||||
| Age (years) | C | 20 | 74 (SD 8) | 21 | 73.7 (SD 6.5) | 0.896a | ||
| Gender (n male) | D | 20 | 9 | 45.0% | 21 | 5 | 23.8% | 0.271b |
| Weight (kg) | C | 20 | 65.2 (SD 8) | 21 | 65.7 (SD 11.5) | 0.873a | ||
| Race (n white) | D | 20 | 20 | 100.0% | 21 | 21 | 100.0% | 0.323b |
| Race (n other) | D | 20 | 0 | 0.0% | 21 | 0 | 0.0% | 0.323b |
| Disease characteristics | ||||||||
| Duration of dementia (months) | C | 20 | 32.4 (SD 19.2) | 21 | 39.6 (SD 25.2) | 0.312a | ||
| Family history of AD | D | 20 | 7 | 35.0% | 21 | 9 | 42.9% | 0.845b |
| Cognitive | ||||||||
| MMSE | C | 20 | 20 (SD 3.5) | 21 | 19.2 (SD 3.1) | 0.443a | ||
| Type | Donepezil | Rivastigmine | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| N | K | Mean | N | K | Mean | |||
| Demographics | ||||||||
| Age (years) | C | 20 | 74 (SD 8) | 22 | 76.8 (SD 8.9) | 0.292a | ||
| Gender (n male) | D | 20 | 9 | 45.0% | 22 | 5 | 22.7% | 0.230b |
| Weight (kg) | C | 20 | 65.2 (SD 8) | 22 | 65.1 (SD 9.7) | 0.971a | ||
| Race (n white) | D | 20 | 20 | 100.0% | 22 | 21 | 95.5% | 0.947b |
| Race (n other) | D | 20 | 0 | 0.0% | 22 | 1 | 4.5% | 0.947b |
| Disease characteristics | ||||||||
| Duration of dementia (months) | C | 20 | 32.4 (SD 19.2) | 22 | 34.8 (SD 25.2) | 0.732a | ||
| Family history of AD | D | 20 | 7 | 35.0% | 22 | 9 | 40.9% | 0.940b |
| Cognitive | ||||||||
| MMSE | C | 20 | 20 (SD 3.5) | 22 | 18.8 (SD 3.8) | 0.295a | ||
| Type | Galantamine | Rivastigmine | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| N | k | Mean | N | k | Mean | |||
| Demographics | ||||||||
| Age (years) | C | 21 | 73.7 (SD 6.5) | 22 | 76.8 (SD 8.9) | 0.201a | ||
| Gender (n male) | D | 21 | 5 | 23.8% | 22 | 5 | 22.7% | 0.782b |
| Weight (kg) | C | 21 | 65.7 (SD 11.5) | 22 | 65.1 (SD 9.7) | 0.854a | ||
| Race (n white) | D | 21 | 21 | 100.0% | 22 | 21 | 95.5% | 0.974b |
| Race (n other) | D | 21 | 0 | 0.0% | 22 | 1 | 4.5% | 0.974b |
| Disease characteristics | ||||||||
| Duration of dementia (months) | C | 21 | 39.6 (SD 25.2) | 22 | 34.8 (SD 25.2) | 0.536a | ||
| Family history of AD | D | 21 | 9 | 42.9% | 22 | 9 | 40.9% | 0.857b |
| Cognitive | ||||||||
| MMSE | C | 21 | 19.2 (SD 3.1) | 22 | 18.8 (SD 3.8) | 0.708a | ||
Results
| Type | Donepezil | Galantamine | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| n | K | Mean | n | K | Mean | |||
| Safety population | ||||||||
| AEs | ||||||||
| Nausea – 13 weeks | D | 20 | 2 | 10.0% | 21 | 6 | 28.6% | 0.269a |
| Diarrhoea – 13 weeks | D | 20 | 0 | 0.0% | 21 | 6 | 28.6% | 0.046a |
| Vomiting – 13 weeks | D | 20 | 0 | 0.0% | 21 | 3 | 14.3% | 0.317a |
| Abdominal pain – 13 weeks | D | 20 | 2 | 10.0% | 21 | 0 | 0.0% | 0.522a |
| Dizziness – 13 weeks | D | 20 | 1 | 5.0% | 21 | 3 | 14.3% | 0.635a |
| Headache – 13 weeks | D | 20 | 2 | 10.0% | 21 | 2 | 9.5% | 0.635a |
| Upper respiratory tract infection – 13 weeks | D | 20 | 1 | 5.0% | 21 | 0 | 0.0% | 0.973a |
| Weight loss – 13 weeks | D | 20 | 1 | 5.0% | 21 | 1 | 4.8% | 0.490a |
| Insomnia – 13 weeks | D | 20 | 2 | 10.0% | 21 | 2 | 9.5% | 0.635a |
| Influenza – 13 weeks | D | 20 | 0 | 0.0% | 21 | 2 | 9.5% | 0.578a |
| Muscle spasms – 13 weeks | D | 20 | 3 | 15.0% | 21 | 1 | 4.8% | 0.563a |
| Disposition of participants | ||||||||
| Discontinued treatment due to AEs – 1 week | D | 20 | 1 | 5.0% | 21 | 4 | 19.0% | 0.370a |
| Discontinued treatment before end of trial – 1 week | D | 20 | 1 | 5.0% | 21 | 5 | 23.8% | 0.207a |
| Type | Donepezil | Rivastigmine | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| n | K | Mean | n | K | Mean | |||
| Safety population | ||||||||
| AEs | ||||||||
| Nausea – 13 weeks | D | 20 | 2 | 10.0% | 22 | 10 | 45.5% | 0.028a |
| Diarrhoea – 13 weeks | D | 20 | 0 | 0.0% | 22 | 2 | 9.1% | 0.605a |
| Vomiting – 13 weeks | D | 20 | 0 | 0.0% | 22 | 4 | 18.2% | 0.187a |
| Abdominal pain – 13 weeks | D | 20 | 2 | 10.0% | 22 | 0 | 0.0% | 0.496a |
| Dizziness – 13 weeks | D | 20 | 1 | 5.0% | 22 | 3 | 13.6% | 0.670a |
| Headache – 13 weeks | D | 20 | 2 | 10.0% | 22 | 3 | 13.6% | 0.910a |
| Upper respiratory tract infection – 13 weeks | D | 20 | 1 | 5.0% | 22 | 2 | 9.1% | 0.932a |
| Weight loss – 13 weeks | D | 20 | 1 | 5.0% | 22 | 2 | 9.1% | 0.932a |
| Insomnia – 13 weeks | D | 20 | 2 | 10.0% | 22 | 1 | 4.5% | 0.932a |
| Influenza – 13 weeks | D | 20 | 0 | 0.0% | 22 | 1 | 4.5% | 0.947a |
| Muscle spasms – 13 weeks | D | 20 | 3 | 15.0% | 22 | 0 | 0.0% | 0.252a |
| Disposition of participants | ||||||||
| Discontinued treatment due to AEs – 1 week | D | 20 | 1 | 5.0% | 22 | 3 | 13.6% | 0.670a |
| Discontinued treatment before end of trial – 1 week | D | 20 | 1 | 5.0% | 22 | 4 | 18.2% | 0.401a |
| Type | Galantamine | Rivastigmine | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| n | K | Mean | n | K | Mean | |||
| Safety population | ||||||||
| AEs | ||||||||
| Nausea – 13 weeks | D | 21 | 6 | 28.6% | 22 | 10 | 45.5% | 0.407a |
| Diarrhoea – 13 weeks | D | 21 | 6 | 28.6% | 22 | 2 | 9.1% | 0.212a |
| Vomiting – 13 weeks | D | 21 | 3 | 14.3% | 22 | 4 | 18.2% | 0.946a |
| Abdominal pain – 13 weeks | D | 21 | 0 | 0.0% | 22 | 0 | 0.0% | 0.323a |
| Dizziness – 13 weeks | D | 21 | 3 | 14.3% | 22 | 3 | 13.6% | 0.705a |
| Headache – 13 weeks | D | 21 | 2 | 9.5% | 22 | 3 | 13.6% | 0.956a |
| Upper respiratory tract infection – 13 weeks | D | 21 | 0 | 0.0% | 22 | 2 | 9.1% | 0.577a |
| Weight loss – 13 weeks | D | 21 | 1 | 4.8% | 22 | 2 | 9.1% | 0.967a |
| Insomnia – 13 weeks | D | 21 | 2 | 9.5% | 22 | 1 | 4.5% | 0.967a |
| Influenza – 13 weeks | D | 21 | 2 | 9.5% | 22 | 1 | 4.5% | 0.967a |
| Muscle spasms – 13 weeks | D | 21 | 1 | 4.8% | 22 | 0 | 0.0% | 0.974a |
| Disposition of participants | ||||||||
| Discontinued treatment due to AEs – 1 week | D | 21 | 4 | 19.0% | 22 | 3 | 13.6% | 0.946a |
| Discontinued treatment before end of trial – 1 week | D | 21 | 5 | 23.8% | 22 | 4 | 18.2% | 0.937a |
Methodological issues
| Randomisation and allocation: Randomisation procedure not described. Open-label trial (although laboratory personnel who processed CSF samples were blinded) |
| Data analysis: Changes from baseline compared between treatment groups using ANCOVA with baseline and treatment as factors. Correction factor for multiplicity applied for primary outcome, but not for secondary outcomes (intended to be hypothesis generating only). All statistical tests were conducted against a two-sided alternative hypothesis, employing a significance level of 0.05. Primary efficacy analyses were based on the completer population. Secondary analyses were based on ITT population (all randomised patients who received at least one dose of study medication and provided at least one postbaseline efficacy measurement) |
| Power calculation: Assuming a mean treatment difference of 0.3 U/l (primary outcome variable), SD 0.28 and two-sided significance level of 0.025, z-test showed approximately 20 patients per treatment group were required to achieve a power of 0.85 for detecting a significant pairwise treatment difference |
| Conflicts of interest: Three co-authors (AN, TD-S, MM) were responsible for the enzyme analysis and received research sponsorship from Novartis. One co-author’s (HS) institute received research sponsorship from Novartis for this study. Two co-authors (GE, RL) are full-time employees of Novartis |
Quality appraisal
|
Peng et al.121
| Design | Participants | Arms | Outcomes |
|---|---|---|---|
|
Peng et al. (2005) 121 Study design: parallel double-blind RCT Country: China No. of centres: 15 hospitals in Beijing, Shanghai and Guangzhou Funding: not reported Length of follow-up (weeks): 12 Notes: |
No. randomised: 90 MMSE min: 10 MMSE max: 24 Inclusion criteria: AD (NINCDS-ADRDA and DSM-IVR criteria) ≥ 55 years old In female patients, menopause ≥ 2 years MMSE 10–24 Sufficient vision and hearing to complete assessments Exclusion criteria: Other disease that may lead to dementia Severe heart or kidney dysfunction, active peptic ulcer, or active epilepsy Allergy to cholinergic drugs Therapy common to all participants: none Sample attrition/dropout: 89 of 90 completed the study; 1 dropped out due to AE (dizziness) – not stated from which arm |
Arm no. : 1 Name: donepezil n: 46 Drug: donepezil Starting daily dose (mg): 5 Dosage details: Same dose administered throughout duration of study Arm no. : 2 Name: placebo n: 43 Drug: placebo Starting daily dose (mg): Dosage details: |
Cognitive MMSE (cognitive functions (direction, memory, calculation, language) Functional ADL (described as ‘testing daily living abilities’) Global severity CDR (not defined) |
| Type | Donepezil | Placebo | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| N | K | Mean | N | K | Mean | |||
| OC population | ||||||||
| Demographics | ||||||||
| Age (years) | C | 46 | 72.6 (SD 6.8) | 43 | 71.8 (SD 8.2) | 0.617a | ||
| Gender (n male) | D | 46 | 21 | 45.7% | 43 | 19 | 44.2% | 0.941b |
| Cognitive | ||||||||
| MMSE – 0 weeks | C | 46 | 17.8 (SD 2.3) | 43 | 18.2 (SD 2.7) | 0.453a | ||
| Functional | ||||||||
| ADL – 0 weeks | C | 46 | 47.2 (SD 7.9) | 43 | 47.2 (SD 7.9) | 1.000a | ||
| Global severity | ||||||||
| CDR – 0 weeks | C | 46 | 1.9 (SD 0.3) | 43 | 2 (SD 0.2) | 0.070a | ||
Results
| Type | Donepezil | Placebo | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| n | K | Mean | n | K | Mean | |||
| OC population | ||||||||
| Cognitive | ||||||||
| MMSE – 12 weeks | C | 46 | 22.1 (SD 2) | 43 | 18.7 (SD 2.4) | < 0.01a | ||
| Functional | ||||||||
| ADL – 12 weeks | C | 46 | 40.5 (SD 7.6) | 43 | 49.5 (SD 6.3) | < 0.01a | ||
| Global severity | ||||||||
| CDR – 12 weeks | C | 46 | 1.2 (SD 0.2) | 43 | 2 (SD 0.2) | < 0.05a | ||
Methodological issues
| Randomisation and allocation: Randomisation procedure not described. Placebo described as having the same colour, shape, flavour and size as donepezil |
| Data analysis: MMSE/CDR/ADL – t-test |
| Power calculation: Not reported |
| Conflicts of interest: Not reported |
Quality appraisal
|
Porsteinsson et al.150
| Design | Participants | Arms | Outcomes |
|---|---|---|---|
|
Porsteinsson et al. (2008)150 Study design: parallel double-blind RCT Country: USA No. of centres: 38 Funding: Forest Laboratories, Inc. (New York, NY) provided all financial and material support for research and analyses – and assisted the Memantine Study Group in the development of the trial design, implementation, data collection, post hoc analyses, and manuscript development Length of follow-up (weeks): 24 Notes: |
No. randomised: 433 MMSE min: 10 MMSE max: 22 Inclusion criteria: Probable AD (NINCDS-ADRDA criteria) Age ≥ 50 years MRI or CT scan results consistent with AD diagnosis and acquired within 1 year of study MMSE 10–22 at screening and baseline Treatment with cholinesterase inhibitors for ≥ 6 months, and a stable dosing regimen for ≥ 3 months (donepezil 5 or 10 mg/day; rivastigmine 6, 9 or 12 mg/day; galantamine 16 or 24 mg/day) A knowledgable and reliable caregiver to accompany the participant to all study visits and supervise administraton of study drug Ability to ambulate Vision and hearing sufficient to permit compliance with assessments Montgomery–Åsberg Depression Rating Scale (MADRS) score < 22 Medically stable Postmenopausal for ≥ 2 years, or surgically sterile (female participants) Exclusion criteria: Clinically significant and active pulmonary, gastrointestinal, renal, hepatic, endocrine or cardiovascular disease Clinically significant vitamin B12 or folate deficiency Evidence (including CT/MRI) of other psychiatric or neurological disorders Dementia complicated by organic disease or AD with delusions or delirium Undergoing treatment for an oncology diagnosis, or completion of treatment within 6 months of screening Modified Hachinski Ischaemic Score, score > 4 Poorly controlled hypertension Substance abuse Participation in an investigational drug study or use of an investigational drug within 30 days (or five half-lives, whichever is longer) of screening Depot neuroleptic drug use within 6 months of screening Positive urine drug test Likely institutionalisation during trial Previous memantine treatment or participation in an investgational study of memantine Likely cessation of cholinesterase inhibitors during the trial Therapy common to all participants: all participants continued to take cholinesterase inhibitor (donepezil, galantamine or rivastigmine) 1- to 2-week single-blind placebo lead-in phase completed before randomisation to assess compliance Sample attrition/dropout: 385 of 433 completed study. Dropouts in memantine arm: AEs n = 13, withdrew consent n = 4, protocol violation n = 5, insufficient therapeutic response n = 1; dropouts in placebo arm: AEs n = 17, withdrew consent n = 4, protocol violation n = 1, insufficient therapeutic response n = 1, other n = 2. No differences between groups |
Arm no. : 1 Name: memantine + AChEI N: 217 Drug: memantine+AChEI Starting daily dose (mg): 5 Dosage details: titrated from an initial dosage of 5 mg/day in 5-mg weekly increments to a maximum dose of 20 mg/day (administered as four 5-mg tablets o.d. at bedtime) Notes: tablets dispensed in blister packs to allow assessment of compliance (inventory of returned blister packs): 97.2% of participants received at least 75% of the memantine doses Arm no. : 2 Name: placebo + AChEI N: 216 Drug: placebo + AChEI Starting daily dose (mg): Dosage details: Notes: tablets dispensed in blister packs to allow assessment of compliance (inventory of returned blister packs): 97.2% of participants received at least 75% of the placebo doses |
Cognitive ADAS-cog (not defined) MMSE (not defined) Functional ADCS-ADL (not defined) Behavioural NPI (not defined) Global severity CIBIC-plus score (not defined) AEs |
Baseline characteristics
| Type | Memantine + AChEI | Placebo + AChEI | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| N | K | Mean | N | K | Mean | |||
| Demographics | ||||||||
| Age (years) | C | 217 | 74.9 (SD 7.64) | 216 | 76 (SD 8.43) | 0.156a | ||
| Gender (n male) | D | 217 | 100 | 46.1% | 216 | 107 | 49.5% | 0.533b |
| Weight (kg) | C | 217 | 70 (SD 14.9) | 216 | 72.2 (SD 14.7) | 0.123a | ||
| Disease characteristics: | ||||||||
| Hachinski Ischaemic Score | C | 217 | 0.6 (SD 0.76) | 216 | 0.6 (SD 0.68) | 1.000a | ||
| Cognitive | ||||||||
| MMSE – 0 weeks | C | 217 | 16.7 (SD 3.67) | 216 | 17 (SD 3.64) | 0.394a | ||
| Behavioural | ||||||||
| Montgomery–Åsberg Depression Rating Scale | C | 217 | 5.7 (SD 4.65) | 216 | 5.3 (SD 4.1) | 0.343a | ||
| LOCF analysis | ||||||||
| Cognitive | ||||||||
| ADAS-cogA | C | 212 | 27.9 (SD 11) | 212 | 26.8 (SD 9.88) | 0.279a | ||
| MMSE – 0 weeks | C | 213 | 16.7 (SD 3.68) | 213 | 17 (SD 3.63) | 0.397a | ||
| Functional | ||||||||
| ADCS-ADL – 0 weeks | C | 214 | 54.7 (SD 14.4) | 213 | 54.8 (SD 13.1) | 0.940a | ||
| Behavioural | ||||||||
| NPI – 0 weeks | C | 214 | 11.8 (SD 13.1) | 213 | 12.3 (SD 13.3) | 0.696a | ||
Results
| Type | Memantine + AChEI | Placebo + AChEI | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| N | K | Mean | N | K | Mean | |||
| ITT population | ||||||||
| Disposition of participants | ||||||||
| Discontinued treatment due to AEs – 24 weeks | D | 217 | 13 | 6.0% | 216 | 17 | 7.9% | |
| Discontinued treatment before end of trial – 24 weeks | D | 217 | 26 | 12.0% | 216 | 25 | 11.6% | |
| Study medication | ||||||||
| Dose (mg/day) – 24 weeks | C | 217 | 19.5 (SD 1.2) | 216 | 19.6 (SD 1) | |||
| LOCF analysis | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 24 weeks | C | 214 | 28.5 (SD 12.8) | 213 | 28 (SD 11.9) | 0.184a | ||
| MMSE – 24 weeks | C | 210 | 16.5 (SD 5.38) | 198 | 16.4 (SD 5.08) | 0.123a | ||
| Functional | ||||||||
| ADCS-ADL – 24 weeks | C | 214 | 51.8 (SD 15.9) | 213 | 52 (SD 15.7) | 0.816a | ||
| Behavioural | ||||||||
| NPI – 24 weeks | MC | 212 | 0.7 (SD 12) | 209 | 0.4 (SD 12.3) | |||
| NPI – 24 weeks | C | 212 | 12.9 (SD 14.5) | 209 | 12.6 (SD 14.6) | 0.743a | ||
| Global severity | ||||||||
| CIBIC-plus score – 24 weeks | C | 214 | 4.38 (SD 1) | 213 | 4.42 (SD 0.96) | 0.843b | ||
| OC population | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 24 weeks | C | 192 | 28.2 (SD 12.8) | 188 | 27.6 (SD 11.7) | 0.186a | ||
| MMSE – 24 weeks | C | 193 | 16.6 (SD 5.41) | 188 | 16.4 (SD 5.08) | 0.190a | ||
| Functional | ||||||||
| ADCS-ADL – 24 weeks | C | 193 | 51.8 (SD 16) | 189 | 53.6 (SD 14.6) | 0.741a | ||
| Behavioural | ||||||||
| NPI – 12 weeksc | MC | 193 | 0.8 (SD 10.8) | 189 | 0.3 (SD 10.6) | NSa | ||
| NPI – 24 weeks | C | 193 | 12.3 (SD 13.7) | 189 | 11.9 (SD 13.5) | 0.985a | ||
| NPI – 24 weeks | MC | 193 | 0 (SD 11.8) | 189 | 0 (SD 11.7) | NSa | ||
| Global severity | ||||||||
| CIBIC-plus score – 24 weeks | C | 192 | 4.36 (SD 1.01) | 189 | 4.4 (SD 0.96) | 0.650b | ||
| Safety population | ||||||||
| AEs | ||||||||
| Any serious AE – 24 weeks | D | 217 | 27 | 12.4% | 216 | 30 | 13.9% | 0.762d |
| Diarrhoea – 24 weeks | D | 217 | 12 | 5.5% | 216 | 14 | 6.5% | 0.830d |
| Agitation – 24 weeks | D | 217 | 17 | 7.8% | 216 | 17 | 7.9% | 0.869d |
| Depression – 24 weeks | D | 217 | 14 | 6.5% | 216 | 15 | 6.9% | 0.990d |
| Injury – 24 weeks | D | 217 | 20 | 9.2% | 216 | 16 | 7.4% | 0.612d |
| Dizziness – 24 weeks | D | 217 | 16 | 7.4% | 216 | 16 | 7.4% | 0.865d |
| Upper respiratory tract infection – 24 weeks | D | 217 | 12 | 5.5% | 216 | 6 | 2.8% | 0.233d |
| Fall – 24 weeks | D | 217 | 22 | 10.1% | 216 | 15 | 6.9% | 0.309d |
| Influenza-like symptoms – 24 weeks | D | 217 | 15 | 6.9% | 216 | 12 | 5.6% | 0.700d |
| Abnormal gait – 24 weeks | D | 217 | 14 | 6.5% | 216 | 9 | 4.2% | 0.398d |
| Confusion – 24 weeks | D | 217 | 12 | 5.5% | 216 | 9 | 4.2% | 0.662d |
| Fatigue – 24 weeks | D | 217 | 11 | 5.1% | 216 | 7 | 3.2% | 0.476d |
| Hypertension – 24 weeks | D | 217 | 11 | 5.1% | 216 | 6 | 2.8% | 0.327d |
Methodological issues
| Randomisation and allocation: Randomised in permuted blocks of four in accordance with randomisation list generated and retained by Forest Research Institute, Department of Statistical Programming. Participants were sequentially assigned randomisation numbers at the baseline visit. No individual participant randomisation code was revealed during the trial. Memantine and placebo tablets described as being identical in appearance |
| Data analysis: Primary efficacy analyses (ADAS-cog and CIBIC-plus) based on the ITT population with LOCF for missing data imputation with only post-baseline data carried forward. Secondary efficacy analyses (ADCS-ASL, NPI, MMSE) used the OCs approach |
| ADAS-cog (inlcuding post hoc analyses of items and subscales), ADCS-ADL, NPI, and MMSE: two-way ANCOVA with treatment group and centre as main effects and baseline as covariate (least square means) for differences between memantine and placebo groups on change from baseline |
| CIBIC-plus: Cochran–Mantel–Haenszel statistic using modified ridit scores (van Elteren test) controlling for study centre was used to compare distributions between groups |
| Power calculation: Assuming an effect size [defined as difference of mean scores between treatment groups on ADAS-cog at end point (LOCF), relative to pooled SD] of 0.325, at least 400 participants were needed to provide 90% power at an alpha level of 0.05 (two sided), based on a two-sided t-test. The total patient population, consisting of all participants randomised into the study (n = 433) was identical to the safety population, which consusted of randomised participants who received at least one dose of double-blind study medication. The ITT population (n = 427) comprised participants in the safety population who completed at least one postbaseline ADAS-cog or CIBIC-plus assessment |
| Conflicts of interest: One co-author’s (JO) affilliation is Novartis, Inc. |
Quality appraisal
|
Rockwood et al.97
| Design | Participants | Arms | Outcomes |
|---|---|---|---|
|
Rockwood et al. (2006) 97 Study design: parallel double-blind RCT Country: Canada No. of centres : 10 Funding: Janssen-Ortho Canada (80%) and the Canadian Institutes of Health Research (20%) (grant no. DCT-49981). The sponsor provided all medications and matching placebos, conducted on-site monitoring and gathered and electronically coded the case report forms. All data are held by the principal investigator (Kenneth Rockwood), who initiated and supervised all analyses. Janssen-Ortho received the paper 45 days before submission to verify protocol details. At the authors’ request, Janssen-Ortho statisticians answered questions about the use of the mixed-effects model, but had no other input in the analyses Length of follow-up (weeks): 16 Notes: Five patients (two in galantamine group, three in placebo group) had MMSE scores that were outside the 10–25 range stipulated in the inclusion criteria; one had a MMSE score of < 10, the other four had MMSE scores of > 25. Seven patients (four in galantamine group, three in placebo group) had ADAS-cog scores that were outside the > 17 range stipulated in the inclusion criteria; in each case the score was below the lower limit, which indicated milder impairment |
No. randomised: 130 MMSE min: 10 MMSE max: 25 Inclusion criteria: Probable AD (NINCDS-ADRDA criteria) MMSE score 10–25 inclusive ADAS-cof score ≥ 18 Daily contact with a responsible caregiver Exclusion criteria: Resident in nursing home Disabling communication difficulties (problems in language, speech, vision or hearing) Other active medical issues or competing causes of dementia Patients who had taken anti-dementia medications within 30 days before screening for study enrolment Hypersensitivity to cholinomimetic agents or bromide Participation in other galantamine trials Therapy common to all participants: none reported Sample attrition/dropout: 109 of 130 completed study. 21 withdrew after allocation: AEs n = 7; noncompliance n = 6; insufficient response n = 4; lost to follow-up n = 1; withdrew consent n = 2; died n = 1. More patients in the galantamine group (n = 5 withdrew due to AEs than in the placebo group (n = 2), otherwise no difference between groups |
Arm no. : 1 Name: galantamine N: 64 Drug: galantamine Starting daily dose (mg): 8 Dosage details: Initial dose of 8 mg/day (4 mg b.i.d.) for 4 weeks, followed by 16 mg/day for another 4 weeks. At the end of week 8, dose could be increased to 24 mg/day depending on tolerability. At week 12, patients were re-evaluated; the dose could then be reduced to 16 mg/day if necessary, after which time it could not be changed. Arm no.: 2 Name: placebo N: 66 Drug: placebo Starting daily dose (mg): Dosage details: Notes: Sham titration schedule |
Cognitive ADAS-cog [assessed memory, language, and praxis, scores ranging from 0 (no impairment) to 70 (severe impairment)] Functional GAS (individualised outcome measure in which goals are set and then followed over the course of a trial. The goals are personalised (i.e. people set goals according to their own needs). What is standardised is the extent of their attainment, which can be either ‘no change’ or ‘much better’ (or ‘much worse’) than expected. Two independent GAS assessments were completed: one by physicians, after interviewing patients and caregivers and completing all study procedures, and the other by patients and caregivers, in a separate interview facilitated by an experienced, independent health professional (usually a research nurse) who was blinded to all other outcomes and AEs except for the CIBIC-plus, which the health professional also scored. GAS raters completed a 4-hour training session. Blinded qualitative raters from the coordinating study site coded every video-recorded interview and made domain assignments; this step provided quality assurance for how goals were set but did not influence scoring Global severity CIBIC-plus score [score range from 1 (very much improved) through 4 (no change) to 7 (very much worse)] AEs |
Baseline characteristics
| Type | Galantamine | Placebo | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| N | K | mean | N | K | mean | |||
| Demographics | ||||||||
| Age (years) | C | 64 | 77 (SD 8) | 66 | 78 (SD 8) | 0.477a | ||
| Gender (n male) | D | 64 | 23 | 35.9% | 66 | 25 | 37.9% | 0.962b |
| Education (years) | C | 64 | 11 (SD 3) | 66 | 11 (SD 3) | 1.000a | ||
| Cognitive | ||||||||
| ADAS-cog – 0 weeks | C | 64 | 24.2 (SD 6.4) | 66 | 27.9 (SD 8.4) | 0.006a | ||
| MMSE | C | 64 | 20.8 (SD 3.3) | 66 | 19.9 (SD 4.2) | 0.178a | ||
| MMSE: 10–19 | D | 64 | 17 | 26.6% | 66 | 26 | 39.4% | 0.171b |
| MMSE: 20–25 | D | 64 | 47 | 73.4% | 66 | 40 | 60.6% | 0.171b |
| Functional | ||||||||
| DAD | C | 64 | 76.4 (SD 19.7) | 66 | 70.6 (SD 21.4) | 0.111a | ||
| Caregiver Burden Scale | C | 64 | 29 (SD 10) | 66 | 29 (SD 10) | 1.000a | ||
| Global severity | ||||||||
| CIBIC-plus score – 0 weeksc | C | 64 | 3.4 (SD 0.7) | 66 | 3.7 (SD 0.9) | 0.036a | ||
| Data extracted from secondary publication reporting subgroup with verbal repetition goals | ||||||||
| Demographics | ||||||||
| Age (years) | C | 24 | 77.3 (SD 6.1) | 33 | 79.1 (SD 7.2) | 0.325a | ||
| Gender (n male) | D | 24 | 10 | 41.7% | 33 | 12 | 36.4% | 0.896b |
| Education (years) | C | 24 | 10.4 (SD 2.8) | 33 | 11.9 (SD 3) | 0.061a | ||
| Cognitive | ||||||||
| ADAS-cog – 0 weeks | C | 24 | 23.8 (SD 5.9) | 33 | 27.2 (SD 8) | 0.084a | ||
| MMSE | C | 24 | 21.8 (SD 2.5) | 33 | 19.9 (SD 4.5) | 0.067a | ||
| MMSE: 10–19 | D | 24 | 4 | 16.7% | 33 | 12 | 36.4% | 0.182b |
| MMSE: 20–25 | D | 24 | 20 | 83.3% | 33 | 21 | 63.6% | 0.182b |
| Functional | ||||||||
| DAD | C | 24 | 72.1 (SD 18.7) | 33 | 70.1 (SD 21.6) | 0.717a | ||
| Caregiver Burden Scale | C | 24 | 30.9 (SD 10.4) | 33 | 31 (SD 9.4) | 0.970a | ||
| Global severity | ||||||||
| CIBIC-plus score – 0 weeksc | C | 24 | 3.3 (SD 0.8) | 33 | 3.7 (SD 0.9) | 0.088a | ||
Results
| Type | Galantamine | Placebo | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| n | K | Mean | n | K | Mean | |||
| ITT population | ||||||||
| Disposition of participants | ||||||||
| Discontinued treatment due to AEs – 16 weeks | D | 64 | 5 | 7.8% | 66 | 2 | 3.0% | |
| Discontinued treatment before end of trial – 16 weeks | D | 64 | 11 | 17.2% | 66 | 10 | 15.2% | |
| LOCF analysis | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 8 weeks | MC | 62 | –1.85 (SD 4.18) | 65 | –0.25 (SD 4.97) | |||
| ADAS-cog – 16 weeks | MC | 62 | –1.6 (SD 5.38) | 65 | 0.325 (SD 5.49) | |||
| Functional | ||||||||
| Goal Attainment Scaling (clinician-rated) – 8 weeks | C | 61 | 52.5 (SD 9.12) | 66 | 52.2 (SD 6.97) | |||
| Goal Attainment Scaling (clinician-rated) – 16 weeks | C | 61 | 54.8 (SD 9.36) | 66 | 50.9 (SD 9.74) | 0.02a | ||
| Goal Attainment Scaling (patient/caregiver rated) – 8 weeks | C | 61 | 54.6 (SD 7.97) | 66 | 52.5 (SD 8.57) | |||
| Goal Attainment Scaling (patient/caregiver rated) – 16 weeks | C | 61 | 54.2 (SD 10.8) | 66 | 52.3 (SD 9.12) | 0.27a | ||
| Global severity | ||||||||
| CIBIC-plus score – 8 weeks | C | 61 | 3.64 (SD 0.797) | 65 | 4.17 (SD 0.905) | |||
| CIBIC-plus score – 16 weeks | C | 61 | 3.67 (SD 0.996) | 65 | 4.12 (SD 0.987) | 0.03b | ||
| Safety population | ||||||||
| AEs | ||||||||
| Any AE – 0 weeks | D | 64 | 54 | 84.4% | 66 | 41 | 62.1% | |
| Anorexia – 0 weeks | D | 64 | 7 | 10.9% | 66 | 1 | 1.5% | |
| Nausea – 0 weeks | D | 64 | 15 | 23.4% | 66 | 4 | 6.1% | |
| Vomiting – 0 weeks | D | 64 | 11 | 17.2% | 66 | 2 | 3.0% | |
| Upper respiratory tract infection – 0 weeks | D | 64 | 8 | 12.5% | 66 | 2 | 3.0% | |
| Data extracted from secondary publication reporting subgroup with verbal repetition goals | ||||||||
| Functional | ||||||||
| GAS – verbal repetition: improved – 16 weeks | D | 20 | 14 | 70.0% | 30 | 8 | 26.7% | < 0.01c |
| GAS – verbal repetition: no change – 16 weeks | D | 20 | 4 | 20.0% | 30 | 12 | 40.0% | |
| GAS – verbal repetition: worsened – 16 weeks | D | 20 | 2 | 10.0% | 30 | 10 | 33.3% | |
Methodological issues
| Randomisation and allocation: Randomisation was determined immediately before medication was administered by research nurse telephoning into a contracted, interactive voice-response system for an assignment number. Nurse was blind to the number’s meaning in terms of treatment assignment. Randomisation was in blocks of two, by site, to decrease the chance of incomplete blocks (the GAS instrument was new to investigators at the study sites and that some sites might have had to withdraw if investigators did not know how to complete it) |
| Data analysis: GAS (clinician rated and patient/caregiver rated); ADAS-cog; CIBIC-plus; DAD; CBS – effect sizes estimated as standardised response means, derived as the mean difference between groups divided by the pooled SD of their change; GAS; CIBIC-plus. Secondary analysis were conducted using a mixed-effects model (to allow the effects of dropout to be assessed and adjust for dementia severity at baseline) |
| All of the patients who were randomly assigned were included in analyses of safety, demographic and baseline characteristics. The ITT analysis included all randomly assigned patients who took at least one dose (treatment drug or placebo) during the placebo-controlled phase and who provided any follow-up GAS. Missing data were imputed based on the LOCF (excluding baseline data) during the placebo-controlled phase. The OC analysis included only data from scheduled time points |
| Power calculation: Authors state that on the basis that the GAS instrument can be more responsive than standard measures because it is personalised; this attribute had not been tested in a controlled trial in dementia. For the exploratory analysis, the sample size was estimated from the authors’ limited experience with GAS in anti-dementia drug trials. Assuming a moderate effect size of about 0.524 and a 15% dropout at 4 months, it was determined that 152 subjects would be required to detect differences at the 5% significance level (two tailed) with 80% power. Authors recognised that this might not result in statistically significant results for the secondary outcomes, which were used to compare with the primary outcomes and with results from other studies |
| Conflicts of interest: Lead author has undertaken consultancies and received honoraria from Janssen-Ortho, the study’s co-sponsor, and from Pfizer, Novartis and Merck, and was also lead author of an earlier galantamine study. Lead author owns no stock in pharmaceutical companies. Lead author is part owner of DementiaGuide, which is developing a website to aid in goal setting for people with dementia. Co-authors: CM has received research grants from Janssen-Ortho, Pfizer, Lundbeck and Novartis, but has received no personal payments; MG has received honoraria and travel grants from Janssen-Ortho, Pfizer and Merck; SF and XS have no conflicts of interest to declare |
Quality appraisal
|
Van Dyck et al.143
| Design | Participants | Arms | Outcomes |
|---|---|---|---|
|
Van Dyck et al. (2007)143 Study design: parallel double-blind RCT Country: USA No. of centres : 35 Funding: Forest Laboratories, Inc. provided all financial and material support for the study, as well as statistical and editorial support for the manuscript Length of follow-up (weeks): 24 Notes: |
No. randomised: 350 MMSE min: 5 MMSE max: 14 Inclusion criteria: Probable AD (NINCDS-ADRDA criteria) MMSE score 5–14 at screening and baseline Age ≥ 50 years Brain imaging evaluation (CT or MRI performed within 12 months before study entry) consistent with probable AD A knowledgable and reliable caregiver to accompany the participant to all study visits and supervise administration of the study drug Ability to ambulate Sufficient vision and hearing to comply with assessments Medical stability Stable doses of the following medications were allowed: antihypertensives, anti-inflammatories, diuretics, laxatives, antidepressants, atypical antipsychotic drugs, tocopherol Exclusion criteria: Significant and active pulmonary, gastrointestinal, renal, hepatic, endocrine or cardiovascular disease Clinically significant vitamin B12 or folate deficiency Evidence of any psychiatric or neurological disorder other than AD Hachinski Ischaemic Score > 4 Delusions or delirium (DSM-IV criteria) Active malignancy History of substance abuse within 10 years Likelihood of nursing home placement within 6 months Previous memantine treatment Treatment with an investigational drug within 30 days (or five drug half-lives, whichever was longer) of screening Postmenopausal > 2 years, or surgically sterile (female participants) Therapy common to all participants: 1 to 2 weeks’ single-blind placebo lead-in phase to assess compliance and minimise treatment response at baseline Sample attrition/dropout: 260 of 350 completed study. 90 withdrew after allocation: AEs (n = 45), consent withdrawn (n = 26), protocol violation (n = 8), insufficient therapeutic response (n = 3), other (n = 8). No differences between groups |
Arm no. : 1 Name: memantine N: 178 Drug: memantine Starting daily dose (mg): 5 Dosage details: Initial dosage of 5 mg/day with titration in 5-mg weekly increments to a final dosage of 20 mg/day (administered as two 5-mg tablets b.i.d.). Dose adjustments were permitted between weeks 3 and 8 for participants with AEs. Participants unable to tolerate 20 mg/day by the end of week 8 were discontinued from the study Notes: Compliance monitored by inventory of returned individual blister packs, and protocol adherence by routine assessment of concomitant medication use Arm no. : 2 Name: placebo N: 172 Drug: placebo Starting daily dose (mg): Dosage details: |
Cognitive SIB (100-point, 40-item test to evaluate cognitive dysfunction [memory, language, social interaction, visuospatial ability, attention, praxis, construction) in patients with moderate-to-severe AD (higher score indicates better performance)] Functional ADCS-ADL [modified 54-point, assesses function in patients with moderate and severe dementia (higher scores reflect better functional ability)] ADCS-ADL-19 FAST (not defined) Behavioural NPI (not defined) BGP: total (35-item rating scale, not defined) BGP: care dependency (not defined) Global severity CIBIC-plus score (not defined) AEs |
Baseline characteristics
| Type | Memantine | Placebo | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| N | K | Mean | N | K | Mean | |||
| Demographics | ||||||||
| Age (years) | C | 178 | 78.1 (SD 8.2) | 172 | 78.3 (SD 7.6) | 0.813a | ||
| Gender (n male) | D | 178 | 49 | 27.5% | 172 | 51 | 29.7% | 0.748b |
| Weight (kg) | C | 176 | 64.4 (SD 13.5) | 172 | 65.8 (SD 12.8) | 0.322a | ||
| Race (n white) | D | 178 | 142 | 79.8% | 172 | 141 | 82.0% | 0.698b |
| Cognitive | ||||||||
| MMSE | C | 178 | 10 (SD 2.8) | 172 | 10.3 (SD 3.1) | 0.342a | ||
| SIB – 0 weeks | C | 170 | 77.2 (SD 16.5) | 165 | 75.6 (SD 19.7) | 0.420a | ||
| Functional | ||||||||
| ADCS-ADL – 0 weeks | C | 171 | 33.1 (SD 11) | 165 | 33.6 (SD 10.6) | 0.672a | ||
| FAST – 0 weeks | C | 171 | 1.4 (SD 2) | 165 | 1.2 (SD 2) | 0.360a | ||
| Behavioural | ||||||||
| NPI – 0 weeks | C | 171 | 20.3 (SD 15.7) | 165 | 17.5 (SD 16.4) | 0.111a | ||
| BGP: total – 0 weeks | C | 171 | 17.3 (SD 8.9) | 165 | 16.7 (SD 8.8) | 0.535a | ||
| BGP: care dependency – 0 weeks | C | 171 | 11.5 (SD 7) | 165 | 11 (SD 6.7) | 0.504a | ||
Results
| Type | Memantine | Placebo | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| n | K | Mean | n | K | Mean | |||
| ITT population | ||||||||
| Disposition of participants | ||||||||
| Discontinued treatment due to AEs – 24 weeks | D | 178 | 22 | 12.4% | 172 | 23 | 13.4% | 0.902a |
| Discontinued treatment before end of trial – 24 weeks | D | 178 | 44 | 24.7% | 172 | 46 | 26.7% | 0.756a |
| LOCF analysis | ||||||||
| Cognitive | ||||||||
| SIB – 24 weeks | MC | 170 | –2 (SD 13) | 165 | –2.5 (SD 12.8) | 0.616b | ||
| Functional | ||||||||
| ADCS-ADL-19 – 24 weeks | MC | 171 | –2 (SD 7.85) | 165 | –2.7 (SD 7.71) | 0.282b | ||
| FAST – 24 weeks | MC | 151 | 0.3 (SD 1.23) | 141 | 0.6 (SD 1.19) | 0.093b | ||
| Behavioural | ||||||||
| NPI – 24 weeks | MC | 161 | 1 (SD 16.5) | 154 | 1.1 (SD 17.4) | 0.963b | ||
| BGP: total – 24 weeks | MC | 151 | 0.6 (SD 6.14) | 141 | 1.5 (SD 7.12) | 0.197b | ||
| BGP: care dependency – 24 weeks | MC | 151 | 0.5 (SD 4.92) | 141 | 1.4 (SD 4.75) | 0.076b | ||
| Global severity | ||||||||
| CIBIC-plus score – 24 weeks | C | 171 | 4.3 (SD 1) | 163 | 4.6 (SD 1) | 0.182c | ||
| OC population | ||||||||
| Cognitive | ||||||||
| SIB – 4 weeksd | MC | 167 | 0.875 (SD 7.43) | 164 | –0.3 (SD 6.4) | 0.146b | ||
| SIB – 8 weeksd | MC | 158 | 2.08 (SD 7.86) | 155 | 0.375 (SD 7.16) | 0.064b | ||
| SIB – 12 weeksd | MC | 146 | 1.65 (SD 9.06) | 150 | –0.825 (SD 8.27) | 0.008b | ||
| SIB – 18 weeksd | MC | 140 | 0 (SD 8.28) | 139 | –2.12 (SD 9.14) | 0.065b | ||
| SIB – 24 weeks | MC | 131 | –1.8 (SD 12.6) | 126 | –2.4 (SD 13.5) | 0.617b | ||
| Functional | ||||||||
| ADCS-ADL-19 – 4 weeksd | MC | 168 | 0.312 (SD 4.37) | 164 | 0.512 (SD 4) | 0.801b | ||
| ADCS-ADL-19 – 8 weeksd | MC | 159 | –0.0875 (SD 5.2) | 156 | –0.188 (SD 4.84) | 0.665b | ||
| ADCS-ADL-19 – 12 weeksd | MC | 147 | 0 (SD 5.46) | 150 | –0.488 (SD 5.05) | 0.155b | ||
| ADCS-ADL-19 – 18 weeksd | MC | 142 | –0.688 (SD 7.3) | 140 | –1.38 (SD 5.62) | 0.357b | ||
| ADCS-ADL-19 – 24 weeks | MC | 133 | –1.3 (SD 6.92) | 127 | –2.3 (SD 6.76) | 0.188b | ||
| FAST – 24 weeks | MC | 133 | 0.3 (SD 1.15) | 127 | 0.6 (SD 1.13) | 0.074b | ||
| Behavioural | ||||||||
| NPI – 24 weeks | MC | 133 | 0.5 (SD 15) | 127 | 1 (SD 15.8) | 0.782b | ||
| BGP: total – 24 weeks | MC | 133 | 0.4 (SD 6.92) | 127 | 1.1 (SD 6.76) | 0.312b | ||
| BGP: care dependency – 24 weeks | MC | 133 | 0.4 (SD 4.61) | 127 | 1.2 (SD 5.63) | 0.138b | ||
| Global severity | ||||||||
| CIBIC-plus score – 24 weeks | C | 134 | 4.3 (SD 1.1) | 127 | 4.6 (SD 1) | 0.089c | ||
| Safety population | ||||||||
| AEs | ||||||||
| Any AE – 24 weeks | D | 178 | 131 | 73.6% | 172 | 125 | 72.7% | 0.941a |
| Any serious AE – 24 weeks | D | 178 | 26 | 14.6% | 172 | 29 | 16.9% | 0.666a |
| Diarrhoea – 24 weeks | D | 178 | 10 | 5.6% | 172 | 8 | 4.7% | 0.867a |
| Agitation – 24 weeks | D | 178 | 16 | 9.0% | 172 | 24 | 14.0% | 0.197a |
| Anxiety – 24 weeks | D | 178 | 10 | 5.6% | 172 | 6 | 3.5% | 0.485a |
| Depression – 24 weeks | D | 178 | 9 | 5.1% | 172 | 5 | 2.9% | 0.451a |
| Injury – 24 weeks | D | 178 | 10 | 5.6% | 172 | 13 | 7.6% | 0.605a |
| Dizziness – 24 weeks | D | 178 | 12 | 6.7% | 172 | 11 | 6.4% | 0.932a |
| Headache – 24 weeks | D | 178 | 3 | 1.7% | 172 | 11 | 6.4% | 0.048a |
| Urinary tract infection – 24 weeks | D | 178 | 9 | 5.1% | 172 | 9 | 5.2% | 0.867a |
| Fall – 24 weeks | D | 178 | 10 | 5.6% | 172 | 17 | 9.9% | 0.195a |
| Influenza-like symptoms – 24 weeks | D | 178 | 10 | 5.6% | 172 | 8 | 4.7% | 0.867a |
| Confusion – 24 weeks | D | 178 | 9 | 5.1% | 172 | 8 | 4.7% | 0.942a |
| Hypertension – 24 weeks | D | 178 | 14 | 7.9% | 172 | 4 | 2.3% | 0.035a |
| Peripheral oedema – 24 weeks | D | 178 | 12 | 6.7% | 172 | 8 | 4.7% | 0.541a |
| Constipation – 24 weeks | D | 178 | 11 | 6.2% | 172 | 8 | 4.7% | 0.693a |
| Insomnia – 24 weeks | D | 178 | 4 | 2.2% | 172 | 9 | 5.2% | 0.233a |
Methodological issues
| Randomisation and allocation: Randomisation procedure not reported |
| Data analysis: SIB, BGP, ADCS-ADL, FAST, NPI, change from baseline compared between memantine and placebo groups: two-way ANCOVA with treatment group and centre as main effects and baseline as covariate |
| CIBIC-plus: Cochran–Mantel–Haenszel test using modified Ridit score (van Elteren test) controlling for study centre to compare distribution between groups |
| Post-hoc analyses: |
| SIB, ADCS-ADL, NPI, CIBIC-plus: ANCOVA analyses repeated adding previous AChEI use or age as covariates. For CIBIC-plus, additional Cochran–Mantel–Haenszel tests were performed controlling either for prior AChEI use or age group (≤ 64 years, 65–74 years, 75–84 years, ≥ 85 years) in addition to study centre |
| SIB, ADCS-ADL: assumption of normality was violated at week 24 (when tested using Shapiro–Wilk test), therefore Wilcoxon’s rank-sum test performed on the change from baseline scores at each timepoint using LOCF and OC approaches |
| SIB, ADCS-ADL: reanalysed using mixed-effects model repeated measures (as LOCF may introduce biases, including favouring the treatment group with the higher dropout rate in a deteriorating illness) – change from baseline with treatment group, time from baseline, centre, and interaction of treatment group by time as fixed effects, and baseline score as covariate, with an unstructured covariance matrix to model the correlations of residuals over time |
| Power calculation: Assuming an effect size of 0.35, at least 340 participants were needed to provide 90% power at an alpha level of 0.05 (two-sided) on the basis of a two-sample t-test for change from baseline to week 24 in SIB and ADCS-ADL scores |
| Conflicts of interest: Lead author (CD) and 2 co-authors (PT, BM) have received grant support and honoraria from Forest Laboratories, Inc. One co-author (PT) has given expert testimony related to memantine. One author (EM) is an employee of Forest Laboratories, Inc. |
Quality appraisal
|
Winblad et al.140
| Design | Participants | Arms | Outcomes |
|---|---|---|---|
|
Winblad et al . (2007) 140 Study design: parallel double-blind RCT Countries: Chile, Czech Republic, Denmark, Finland, Germany, Guatemala, Israel, Italy, Republic of Korea, Mexico, Norway, Peru, Poland, Portugal, Russian Federation, Slovak Republic, Sweden, Taiwan (Province of China), USA, Uruguay and Venezuela No. of centres: 100 Funding: Novartis Pharma AG, Basel, Switzerland Length of follow-up (weeks): 24 Notes: |
No. randomised: 1195 MMSE min: 10 MMSE max: 20 Inclusion criteria: AD (DSM-IV criteria) and probable AD (NINCDS/ADRDA criteria) (brain scan (MRI or CT) used for establishing these criteria must have been done within 1 year prior to randomisation) Age 50–85 years MMSE 10–20 Living with someone in the community or, if living alone, in daily contact with a responsible caregiver Exclusion criteria: Advanced, severe, progressive, or unstable disease of any type that could interfere with study assessments or put the patient at special risk Any condition other than AD that could explain the dementia Use of any investigational drugs, new psychotropic or dopaminergic agents, cholinesterase inhibitors or anticholinergic agents during the 4 weeks prior to randomisation Therapy common to all participants: None reported Sample attrition/dropout: 970 of 1195 patients completed study. Reasons for dropout: AEs, withdrawn consent, lost to follow-up, death, unsatisfactory therapeutic effect. No difference between groups |
Arm no. : 1 Name: rivastigmine patch (10 cm2) n: 293 Drug: rivastigmine Starting daily dose (mg): 4.75 Dosage details: 10-cm2 patch group: titrated from initial 5-cm2 dose (starting dose above calculated by review team as half the daily dose delivered by 10-cm2 patch) up to 10-cm2 patch in 5-cm2 step at 4 weeks’ interval, followed by an 8 weeks’ maintenance phase Notes: Dose adjustments (interruptions or down-titrations) were permitted to address perceived safety or tolerability issues. If the target dose was not achieved during the titration period the investigator could resume titration during the maintenance period. Patients were maintained at their highest well tolerated doses until the end of the study The patch was applied by caregivers to clean, dry, hairless skin on the patient’s upper back every morning and worn for 24 hours, during which normal activities, including bathing, were allowed. To minimise possible skin irritation, patch placement on the upper back was alternated between the left and right sides, daily Arm no. : 2 Name: rivastigmine patch (20 cm2) n: 303 Drug: rivastigmine Starting daily dose (mg): 4.75 Dosage details: 20-cm2 patch group: titrated from initial 5-cm2 dose (starting dose above calculated by review team as half the daily dose delivered by 10-cm2 patch) up to 20-cm2 patch in 5-cm2 steps at 4-week intervals, followed by an 8 weeks’ maintenance phase Notes: Dose adjustments (interruptions or down-titrations) were permitted to address perceived safety or tolerability issues. If the target dose was not achieved during the titration period the investigator could resume titration during the maintenance period. Patients were maintained at their highest well tolerated doses until the end of the study The patch was applied by caregivers to clean, dry, hairless skin on the patient’s upper back every morning and worn for 24 hours, during which normal activities including bathing were allowed. To minimise possible skin irritation, patch placement on the upper back was alternated between the left and right sides, daily Arm no. : 3 Name: rivastigmine capsules n: 297 Drug: rivastigmine Starting daily dose (mg): 3 Dosage details: Tablet group: initial dosage of 3 mg/day titrated upwards in steps of 3 mg/day up to a maximum of 12 mg/day Notes: Dose adjustments (interruptions or down-titrations) were permitted to address perceived safety or tolerability issues. If the target dose was not achieved during the titration period the investigator could resume titration during the maintenance period. Patients were maintained at their highest well-tolerated doses until the end of the study Arm no. : 4 Name: placebo n: 302 Drug: placebo Starting daily dose (mg): Dosage details: Notes: The placebo patch was applied by caregivers to clean, dry, hairless skin on the patient’s upper back every morning and worn for 24 hours, during which normal activities including bathing were allowed. To minimise possible skin irritation, patch placement on the upper back was alternated between the left and right sides, daily |
Cognitive ADAS-cog (to assess orientation, memory, language, visuospatial and praxis functions) MMSE (not defined) Ten-point clock-drawing test (for assessment of visuospatial and executive functions) Trail-making test (for assessment of attention, visual tracking and motor processing speed) Functional ADCS-ADL (not defined) Behavioural NPI (for assessment of behaviour and psychiatric symptoms) NPI – caregiver distress (not defined) Global severity ADCS-CGIC: score (for assessment of orientation, memory, language, visuospatial and praxis functions) AEs |
Baseline characteristics
| Rivastigmine patch (10 cm2) | Placebo | |||||||
|---|---|---|---|---|---|---|---|---|
| Type | N | K | Mean | N | K | Mean | p-value | |
| Demographics | ||||||||
| Age (years) | C | 291 | 73.6 (SD 7.9) | 302 | 73.9 (SD 7.3) | 0.631a | ||
| Gender (n male) | D | 291 | 93 | 32.0% | 302 | 101 | 33.4% | 0.766b |
| Education (years) | C | 291 | 9.9 (SD 4.3) | 302 | 9.9 (SD 4.3) | 1.000a | ||
| Race (n white) | D | 291 | 220 | 75.6% | 302 | 227 | 75.2% | 0.978b |
| Race (n black) | D | 291 | 1 | 0.3% | 302 | 2 | 0.7% | 0.974b |
| Race (n oriental) | D | 291 | 25 | 8.6% | 302 | 27 | 8.9% | 0.996b |
| Race (n other) | D | 291 | 45 | 15.5% | 302 | 46 | 15.2% | 0.972b |
| Disease characteristics | ||||||||
| Duration of dementia (months) | C | 291 | 13.2 (SD 16.8) | 302 | 13.2 (SD 16.8) | 1.000a | ||
| Domestic circumstances | ||||||||
| Living alone | D | 291 | 43 | 14.8% | 302 | 27 | 8.9% | 0.038b |
| Living with caregiver or other | D | 291 | 240 | 82.5% | 302 | 264 | 87.4% | 0.116b |
| Assisted living/group home | D | 291 | 8 | 2.7% | 302 | 11 | 3.6% | 0.701b |
| Cognitive | ||||||||
| MMSE – 0 weeks | C | 291 | 16.6 (SD 3.1) | 302 | 16.4 (SD 3) | 0.425a | ||
| LOCF analysis | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 0 weeks | C | 248 | 27 (SD 10.3) | 281 | 28.6 (SD 9.9) | 0.069a | ||
| MMSE – 0 weeks | C | 250 | 16.7 (SD 3) | 281 | 16.4 (SD 3) | 0.251a | ||
| Ten-point clock-drawing test – 0 weeks | C | 251 | 4.5 (SD 3.6) | 269 | 4.3 (SD 3.6) | 0.527a | ||
| Trail-making test – 0 weeksc | C | 241 | 183 (SD 85.5) | 258 | 178 (SD 85.6) | 0.514a | ||
| Functional | ||||||||
| ADCS-ADL – 0 weeks | C | 247 | 50.1 (SD 16.3) | 281 | 49.2 (SD 16) | 0.523a | ||
| Behavioural | ||||||||
| NPI – 0 weeks | C | 248 | 13.9 (SD 14.1) | 281 | 14.9 (SD 15.7) | 0.444a | ||
| NPI – caregiver distress – 0 weeks | C | 248 | 7.4 (SD 7.1) | 281 | 7.8 (SD 7.7) | 0.537a | ||
| Rivastigmine patch (20 cm2) | Placebo | |||||||
|---|---|---|---|---|---|---|---|---|
| Type | N | K | Mean | N | K | Mean | p-value | |
| Demographics | ||||||||
| Age (years) | C | 302 | 74.2 (SD 7.7) | 302 | 73.9 (SD 7.3) | 0.623a | ||
| Gender (n male) | D | 302 | 103b | 34.1% | 302 | 101 | 33.4% | 0.931c |
| Education (years) | C | 302 | 9.9 (SD 4.4) | 302 | 9.9 (SD 4.3) | 1.000a | ||
| Race (n white) | D | 302 | 227 | 75.2% | 302 | 227 | 75.2% | 0.925c |
| Race (n black) | D | 302 | 3 | 1.0% | 302 | 2 | 0.7% | 1.000c |
| Race (n oriental) | D | 302 | 27 | 8.9% | 302 | 27 | 8.9% | 0.887c |
| Race (n other) | D | 303 | 46 | 15.2% | 302 | 46 | 15.2% | 0.924c |
| Disease characteristics | ||||||||
| Duration of dementia (months) | C | 302 | 13.2 (SD 16.8) | 302 | 13.2 (SD 16.8) | 1.000a | ||
| Domestic circumstances | ||||||||
| Living alone | D | 302 | 30 | 9.9% | 302 | 27 | 8.9% | 0.781c |
| Living with caregiver or other | D | 302 | 265 | 87.7% | 302 | 264 | 87.4% | 1.000c |
| Assisted living/group home | D | 302 | 8 | 2.6% | 302 | 11 | 3.6% | 0.641c |
| Cognitive | ||||||||
| MMSE – 0 weeks | C | 302 | 16.6 (SD 2.9) | 302 | 16.4 (SD 3) | 0.405a | ||
| LOCF analysis | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 0 weeks | C | 262 | 27.4 (SD 9.7) | 281 | 28.6 (SD 9.9) | 0.155a | ||
| MMSE – 0 weeks | C | 262 | 16.6 (SD 2.9) | 281 | 16.4 (SD 3) | 0.431a | ||
| Ten-point clock-drawing test – 0 weeks | C | 245 | 4.7 (SD 3.8) | 269 | 4.3 (SD 3.6) | 0.221a | ||
| Trail-making test – 0 weeksd | C | 238 | 176 (SD 84) | 258 | 178 (SD 85.6) | 0.813a | ||
| Functional | ||||||||
| ADCS-ADL – 0 weeks | C | 263 | 47.6 (SD 15.7) | 281 | 49.2 (SD 16) | 0.240a | ||
| Behavioural | ||||||||
| NPI – 0 weeks | C | 263 | 15.1 (SD 13.4) | 281 | 14.9 (SD 15.7) | 0.873a | ||
| NPI – caregiver distress – 0 weeks | C | 263 | 8.4 (SD 7.6) | 281 | 7.8 (SD 7.7) | 0.361a | ||
| Rivastigmine capsules | Placebo | |||||||
|---|---|---|---|---|---|---|---|---|
| Type | N | K | Mean | N | K | Mean | p-value | |
| Demographics | ||||||||
| Age (years) | C | 294 | 72.8 (SD 8.2) | 302 | 73.9 (SD 7.3) | 0.084a | ||
| Gender (n male) | D | 294 | 101 | 34.4% | 302 | 101 | 33.4% | 0.882b |
| Education (years) | C | 294 | 9.9 (SD 4.4) | 302 | 9.9 (SD 4.3) | 1.000a | ||
| Race (n white) | D | 294 | 219 | 74.5% | 302 | 227 | 75.2% | 0.924b |
| Race (n black) | D | 294 | 5 | 1.7% | 302 | 2 | 0.7% | 0.426b |
| Race (n oriental) | D | 294 | 29 | 9.9% | 302 | 27 | 8.9% | 0.806b |
| Race (n other) | D | 297 | 41 | 13.8% | 302 | 46 | 15.2% | 0.704b |
| Disease characteristics | ||||||||
| Duration of dementia (months) | C | 294 | 13.2 (SD 16.8) | 302 | 13.2 (SD 16.8) | 1.000a | ||
| Domestic circumstances | ||||||||
| Living alone | D | 294 | 35 | 11.9% | 302 | 27 | 8.9% | 0.293b |
| Living with caregiver or other | D | 294 | 255 | 86.7% | 302 | 264 | 87.4% | 0.900b |
| Assisted living/group home | D | 294 | 4 | 1.4% | 302 | 11 | 3.6% | 0.129b |
| Cognitive | ||||||||
| MMSE – 0 weeks | C | 294 | 16.4 (SD 3.1) | 302 | 16.4 (SD 3) | 1.000a | ||
| LOCF analysis | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 0 weeks | C | 253 | 27.9 (SD 9.4) | 281 | 28.6 (SD 9.9) | 0.404a | ||
| MMSE – 0 weeks | C | 256 | 16.4 (SD 3) | 281 | 16.4 (SD 3) | 1.000a | ||
| Ten-point clock-drawing test – 0 weeks | C | 246 | 4.4 (SD 3.6) | 269 | 4.3 (SD 3.6) | 0.753a | ||
| Trail-making test – 0 weeksc | C | 240 | 177 (SD 86.2) | 258 | 178 (SD 85.6) | 0.886a | ||
| Functional | ||||||||
| ADCS-ADL – 0 weeks | C | 254 | 49.3 (SD 15.8) | 281 | 49.2 (SD 16) | 0.942a | ||
| Behavioural | ||||||||
| NPI – 0 weeks | C | 253 | 15.1 (SD 14.1) | 281 | 14.9 (SD 15.7) | 0.877a | ||
| NPI – caregiver distress – 0 weeks | C | 253 | 8.2 (SD 7.6) | 281 | 7.8 (SD 7.7) | 0.547a | ||
Results
| Rivastigmine patch (10 cm2) | Placebo | |||||||
|---|---|---|---|---|---|---|---|---|
| Type | n | K | Mean | n | K | Mean | p-value | |
| ITT population | ||||||||
| Disposition of participants | ||||||||
| Discontinued treatment due to AEs – 24 weeks | D | 293 | 28 | 9.6% | 302 | 15 | 5.0% | |
| Discontinued treatment before end of trial – 24 weeks | D | 293 | 64 | 21.8% | 302 | 36 | 11.9% | |
| LOCF analysis | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 16 weeksa | MC | 248 | –0.825 (SD 6.3) | 281 | 0 (SD 6.71) | 0.09b | ||
| ADAS-cog – 24 weeks | MC | 248 | –0.6 (SD 6.4) | 281 | 1 (SD 6.8) | 0.005b | ||
| MMSE – 24 weeks | MC | 250 | 1.1 (SD 3.3) | 281 | 0 (SD 3.5) | 0.002c | ||
| Ten-point clock-drawing test – 24 weeks | MC | 251 | 0.1 (SD 3.1) | 269 | –0.1 (SD 3.2) | 0.08c | ||
| Trail-making test – 24 weeks | MC | 241 | –12.3 (SD 55.1) | 258 | 7.7 (SD 56.6) | < 0.001b | ||
| Functional | ||||||||
| ADCS-ADL – 16 weeksa | MC | 247 | –0.6 (SD 9.43) | 281 | –1.6 (SD 7.96) | NSb | ||
| ADCS-ADL – 24 weeks | MC | 247 | –0.1 (SD 9.1) | 281 | –2.3 (SD 9.4) | 0.01b | ||
| Behavioural | ||||||||
| NPI – 24 weeks | MC | 248 | –1.7 (SD 11.5) | 281 | –1.7 (SD 13.8) | 0.74b | ||
| NPI – caregiver distress – 24 weeks | MC | 248 | –1 (SD 5.5) | 281 | –1.1 (SD 6.3) | 0.37b | ||
| Global severity | ||||||||
| ADCS-CGIC: score – 16 weeksa | C | 248 | 3.9 (SD 1.14) | 278 | 4.35 (SD 1.25) | NSc | ||
| ADCS-CGIC: score – 24 weeks | C | 248 | 3.9 (SD 1.2) | 278 | 4.2 (SD 1.3) | 0.01c | ||
| ADCS-CGIC: markedly improved – 24 weeks | D | 248 | 5 | 2.0% | 278 | 2 | 0.7% | 0.361d |
| ADCS-CGIC: moderately improved – 24 weeks | D | 248 | 29 | 11.7% | 278 | 26 | 9.4% | 0.463d |
| ADCS-CGIC: minimally improved – 24 weeks | D | 248 | 43 | 17.3% | 278 | 50 | 18.0% | 0.937d |
| ADCS-CGIC: unchanged – 24 weeks | D | 248 | 105 | 42.3% | 278 | 91 | 32.7% | 0.029d |
| ADCS-CGIC: minimally worse – 24 weeks | D | 248 | 41 | 16.5% | 278 | 65 | 23.4% | 0.065d |
| ADCS-CGIC: moderately worse – 24 weeks | D | 248 | 22 | 8.9% | 278 | 36 | 12.9% | 0.177d |
| ADCS-CGIC: markedly worse – 24 weeks | D | 248 | 3 | 1.2% | 278 | 8 | 2.9% | 0.303d |
| Safety population | ||||||||
| AEs | ||||||||
| Any AE – 0 weeks | D | 291 | 147 | 50.5% | 302 | 139 | 46.0% | NSe |
| Nausea – 0 weeks | D | 291 | 21 | 7.2% | 302 | 15 | 5.0% | NSe |
| Diarrhoea – 0 weeks | D | 291 | 18 | 6.2% | 302 | 10 | 3.3% | NSe |
| Vomiting – 0 weeks | D | 291 | 18 | 6.2% | 302 | 10 | 3.3% | NSe |
| Dizziness – 0 weeks | D | 291 | 7 | 2.4% | 302 | 7 | 2.3% | NSe |
| Headache – 0 weeks | D | 291 | 10 | 3.4% | 302 | 5 | 1.7% | NSe |
| Weight loss – 0 weeks | D | 291 | 8 | 2.7% | 302 | 4 | 1.3% | NSe |
| Decreased appetite – 0 weeks | D | 291 | 2 | 0.7% | 302 | 3 | 1.0% | NSe |
| Asthenia – 0 weeks | D | 291 | 5 | 1.7% | 302 | 3 | 1.0% | NSe |
| Rivastigmine patch (20 cm2) | Placebo | |||||||
|---|---|---|---|---|---|---|---|---|
| Type | n | K | Mean | n | K | Mean | p-value | |
| ITT population | ||||||||
| Disposition of participants | ||||||||
| Discontinued treatment due to AEs – 24 weeks | D | 303 | 26 | 8.6% | 302 | 15 | 5.0% | |
| Discontinued treatment before end of trial – 24 weeks | D | 303 | 62 | 20.5% | 302 | 36 | 11.9% | |
| LOCF analysis | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 16 weeksa | MC | 262 | –1.39 (SD 6.47) | 281 | 0 (SD 6.71) | < 0.05b | ||
| ADAS-cog – 24 weeks | MC | 262 | –1.6 (SD 6.5) | 281 | 1 (SD 6.8) | < 0.001b | ||
| MMSE – 24 weeks | MC | 262 | 0.9 (SD 3.4) | 281 | 0 (SD 3.5) | < 0.001c | ||
| Ten-point clock-drawing test – 24 weeks | MC | 245 | 0.3 (SD 3.4) | 269 | –0.1 (SD 3.2) | 0.08c | ||
| Trail-making test – 24 weeks | MC | 238 | –6.5 (SD 55.9) | 258 | 7.7 (SD 56.6) | 0.005b | ||
| Functional | ||||||||
| ADCS-ADL – 16 weeksa | MC | 263 | 0.4 (SD 9.73) | 281 | –1.6 (SD 7.96) | < 0.05b | ||
| ADCS-ADL – 24 weeks | MC | 263 | 0 (SD 11.6) | 281 | –2.3 (SD 9.4) | 0.02b | ||
| Behavioural | ||||||||
| NPI – 24 weeks | MC | 263 | –2.3 (SD 13.3) | 281 | –1.7 (SD 13.8) | 0.69b | ||
| NPI – caregiver distress – 24 weeks | MC | 263 | –1.1 (SD 6.4) | 281 | –1.1 (SD 6.3) | 0.98b | ||
| Global severity | ||||||||
| ADCS-CGIC: score – 16 weeksa | C | 260 | 3.93 (SD 1.17) | 278 | 4.35 (SD 1.25) | NSc | ||
| ADCS-CGIC: score – 24 weeks | C | 260 | 4 (SD 1.3) | 278 | 4.2 (SD 1.3) | 0.054c | ||
| ADCS-CGIC: markedly improved – 24 weeks | D | 260 | 5 | 1.9% | 278 | 2 | 0.7% | 0.395d |
| ADCS-CGIC: moderately improved – 24 weeks | D | 260 | 32 | 12.3% | 278 | 26 | 9.4% | 0.334d |
| ADCS-CGIC: minimally improved – 24 weeks | D | 260 | 48 | 18.5% | 278 | 50 | 18.0% | 0.975d |
| ADCS-CGIC: unchanged – 24 weeks | D | 260 | 94 | 36.2% | 278 | 91 | 32.7% | 0.457d |
| ADCS-CGIC: minimally worse – 24 weeks | D | 260 | 50 | 19.2% | 278 | 65 | 23.4% | 0.285d |
| ADCS-CGIC: moderately worse – 24 weeks | D | 260 | 27 | 10.4% | 278 | 36 | 12.9% | 0.429d |
| ADCS-CGIC: markedly worse – 24 weeks | D | 260 | 4 | 1.5% | 278 | 8 | 2.9% | 0.448d |
| Safety population | ||||||||
| AEs | ||||||||
| Any AE – 0 weeks | D | 303 | 200 | 66.0% | 302 | 139 | 46.0% | ≤ 0.001e |
| Nausea – 0 weeks | D | 303 | 64 | 21.1% | 302 | 15 | 5.0% | ≤ 0.001e |
| Diarrhoea – 0 weeks | D | 303 | 31 | 10.2% | 302 | 10 | 3.3% | ≤ 0.001e |
| Vomiting – 0 weeks | D | 303 | 57 | 18.8% | 302 | 10 | 3.3% | ≤ 0.001e |
| Dizziness – 0 weeks | D | 303 | 21 | 6.9% | 302 | 7 | 2.3% | ≤ 0.05e |
| Headache – 0 weeks | D | 303 | 13 | 4.3% | 302 | 5 | 1.7% | NSe |
| Weight loss – 0 weeks | D | 303 | 23 | 7.6% | 302 | 4 | 1.3% | ≤ 0.001e |
| Decreased appetite – 0 weeks | D | 303 | 15 | 5.0% | 302 | 3 | 1.0% | ≤ 0.01e |
| Asthenia – 0 weeks | D | 303 | 9 | 3.0% | 302 | 3 | 1.0% | NSe |
| Rivastigmine capsules | Placebo | |||||||
|---|---|---|---|---|---|---|---|---|
| Type | n | K | Mean | n | K | Mean | p-value | |
| ITT population | ||||||||
| Disposition of participants | ||||||||
| Discontinued treatment due to AEs – 24 weeks | D | 297 | 24 | 8.1% | 302 | 15 | 5.0% | |
| Discontinued treatment before end of trial – 24 weeks | D | 297 | 63 | 21.2% | 302 | 36 | 11.9% | |
| LOCF analysis | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 16 weeksa | MC | 253 | –0.5 (SD 6.36) | 281 | 0 (SD 6.71) | NSb | ||
| ADAS-cog – 24 weeks | MC | 253 | –0.6 (SD 6.2) | 281 | 1 (SD 6.8) | 0.003b | ||
| MMSE – 24 weeks | MC | 256 | 0.8 (SD 3.2) | 281 | 0 (SD 3.5) | 0.002c | ||
| Ten-point clock-drawing test – 24 weeks | MC | 246 | 0.2 (SD 2.9) | 269 | –0.1 (SD 3.2) | 0.15c | ||
| Trail-making test – 24 weeks | MC | 240 | –9.8 (SD 66.1) | 258 | 7.7 (SD 56.6) | < 0.001b | ||
| Functional | ||||||||
| ADCS-ADL – 16 weeksa | MC | 254 | –0.4 (SD 7.97) | 281 | –1.6 (SD 7.96) | NSb | ||
| ADCS-ADL – 24 weeks | MC | 254 | –0.5 (SD 9.5) | 281 | –2.3 (SD 9.4) | 0.04b | ||
| Behavioural | ||||||||
| NPI – 24 weeks | MC | 253 | –2.2 (SD 11.9) | 281 | –1.7 (SD 13.8) | 0.51b | ||
| NPI – caregiver distress – 24 weeks | MC | 253 | –1.1 (SD 6.6) | 281 | –1.1 (SD 6.3) | 0.12b | ||
| Global severity | ||||||||
| ADCS-CGIC: score – 16 weeksa | C | 253 | 4.25 (SD 1.11) | 278 | 4.35 (SD 1.25) | NSc | ||
| ADCS-Clinical Global Impression of Change: score – 24 weeks | C | 253 | 3.9 (SD 1.3) | 278 | 4.2 (SD 1.3) | 0.009c | ||
| ADCS-CGIC: markedly improved – 24 weeks | D | 253 | 3 | 1.2% | 278 | 2 | 0.7% | 0.916d |
| ADCS-CGIC: moderately improved – 24 weeks | D | 253 | 29 | 11.5% | 278 | 26 | 9.4% | 0.513d |
| ADCS-CGIC: minimally improved – 24 weeks | D | 253 | 60 | 23.7% | 278 | 50 | 18.0% | 0.129d |
| ADCS-CGIC: unchanged – 24 weeks | D | 253 | 96 | 37.9% | 278 | 91 | 32.7% | 0.244d |
| ADCS-CGIC: minimally worse – 24 weeks | D | 253 | 30 | 11.9% | 278 | 65 | 23.4% | < 0.001d |
| ADCS-CGIC: moderately worse – 24 weeks | D | 253 | 30 | 11.9% | 278 | 36 | 12.9% | 0.803d |
| ADCS-CGIC: markedly worse – 24 weeks | D | 253 | 5 | 2.0% | 278 | 8 | 2.9% | 0.696d |
| Safety population | ||||||||
| AEs | ||||||||
| Any AE – 0 weeks | D | 294 | 186 | 63.3% | 302 | 139 | 46.0% | ≤ 0.001e |
| Nausea – 0 weeks | D | 294 | 68 | 23.1% | 302 | 15 | 5.0% | ≤ 0.001e |
| Diarrhoea – 0 weeks | D | 294 | 16 | 5.4% | 302 | 10 | 3.3% | NSe |
| Vomiting – 0 weeks | D | 294 | 50 | 17.0% | 302 | 10 | 3.3% | ≤ 0.001e |
| Dizziness – 0 weeks | D | 294 | 22 | 7.5% | 302 | 7 | 2.3% | ≤ 0.01e |
| Headache – 0 weeks | D | 294 | 18 | 6.1% | 302 | 5 | 1.7% | ≤ 0.01e |
| Weight loss – 0 weeks | D | 294 | 16 | 5.4% | 302 | 4 | 1.3% | ≤ 0.01e |
| Decreased appetite – 0 weeks | D | 294 | 12 | 4.1% | 302 | 3 | 1.0% | ≤ 0.05e |
| Asthenia – 0 weeks | D | 294 | 17 | 5.8% | 302 | 3 | 1.0% | ≤ 0.001e |
Methodological issues
|
Randomisation and allocation: Automated random assignment of treatment using an interactive voice-response system. Blocking was done on a study centre basis. All personnel directly involved in the conduct of the study remained unaware of the active treatment groups until all data had been retrieved and finalised for analysis. Appearance of tablets, patches and placebo not reported Data analysis: A hierarchical testing strategy was applied to adjust for multiplicity. Study objectives were assessed according to four hypotheses tested in sequence. If any of the four tests failed to show statistical significance, testing of subsequent hypotheses would be stopped in order to control the type 1 error. These hypotheses were that, based on changes from baseline at week 24: (1) on the ADAS-cog and ADCS-CGIC, the rivastigmine 20-cm2 patch would show superiority over placebo; (2) on the ADAS-cog, the rivastigmine 20-cm2 patch would show non-inferiority to 12 mg/day rivastigmine capsules; (3) on the ADAS-cog and ADCS-CGIC, the rivastigmine 10-cm2 patch would show superiority over placebo; (4) on the ADCS-ADL, the rivastigmine 20-cm2 patch would show superiority over placebo. The second hypothesis, which tested for non-inferiority, was a one-sided hypothesis. The remaining three hypotheses were two-sided hypotheses ADAS-cog: changes from baseline assessed by ANCOVA, with baseline values as covariates and treatment groups and countries as factors ADCS-CGIC: analysis was the treatment comparison based on a stratified Wilcoxon’s rank-sum test using country as a blocking factor. Robustness analyses using a proportional odds model were prospectively planned ADCS-ADL, NPI-12, NPI distress, MMSE, ten-point clock-drawing score, trail-making Test A score: Changes from baseline analysed using an ANCOVA model with treatment, country, and the corresponding baseline measurement as covariates, or a Cochran–Mantel–Haenszel test A prospective categorical analysis was conducted to determine percentages of patients demonstrating clinically significant improvements on the ADAS-cog (defined as ≥ 4-point improvement over baseline at 24 weeks); a Cochran–Mantel–Haenszel test blocking for country was performed to compare treatment groups The main efficacy analysis was based on the ITT population using a LOCF imputation. This ITT-LOCF population was pre-defined as all randomised patients who received at least one dose of study medication and had at least a pre- and postbaseline assessment for one of the primary efficacy variables on treatment (i.e. not more than 2 days after the last known date of study drug). Additional supportive analyses were included to confirm whether or not imputations and early discontinuations influenced the results. Among others, these included the ITT population without imputation (OC, ITT-OC), the ITT-retrieved dropout population (all randomised patients who received at least one dose of study medication and had at least a pre- and postbaseline assessment for one of the primary efficacy variables, either under treatment or not), and a population that included all randomised patients |
|
Power calculation: In previous placebo-controlled trials of the rivastigmine capsule in AD patients, a treatment difference to placebo in the ADAS-cog change from baseline of approximately 2.5 points was observed in the ITT analysis. In the current trial, a non-inferiority margin was pre-defined as 1.25 points on the ADAS-cog to preserve 50% of this effect, which was considered the smallest value that could represent a clinically meaningful difference. To determine the power of this study, the assumptions on delta (difference in means) and SD for the change in ADAS-cog and ADCS-CGIC from baseline were based on 24-week data from the rivastigmine capsule studies that used the ADAS-cog and CIBIC-plus. The ADCS-CGIC scale is comparable with the CIBIC-plus, which was used in previous rivastigmine capsule studies. To ensure that the study had adequate power, 1040 evaluable patients were needed. In order to reach an overall power of 80% for all of the first three hypotheses (which is defined as the product of the individual powers), the sample size was 260 patients per treatment group Conflicts of interest: Three co-authors (SZ, JN, RL) are employees of Novartis. Remaining authors were investigators (BW, NA, GG, MO, CS) and/or Study Publication Committee members (BW, JC, NA, GG, MO, SZ, JN, RL). BW, JC, NA, GG, MO and CS have provided consultation services to many pharmaceutical companies that develop dementia drugs, including Novartis. A writing committee prepared an initial draft of the manuscript, based on a report provided by Novartis, and all authors contributed to its finalisation through interactive review Data were collected by investigators and co-investigators, entered into a central database using electronic data capture software, and analysed by Novartis Pharma AG, which vouches for the data and the analysis |
Quality appraisal
|
Winstein et al.122
| Design | Participants | Arms | Outcomes |
|---|---|---|---|
|
Winstein et al. (2007)122 Study design: parallel double-blind RCT Country: USA No. of centres : 1 Funding: USC Alzheimer’s Disease Research Centre, Alzheimer’s Disease Research Centres of California, and Pfizer, Inc. Length of follow-up (weeks): 4 Notes: |
No. randomised: 10 MMSE min: 11 MMSE max: 26 Inclusion criteria: Probable AD diagnosis (criteria not reported) Independent in ambulation Alert Able to follow simple instructions MMSE 11–26 Exclusion criteria: Delirium Familial tremor Parkinson’s disease Stroke Peripheral neuropathy Dementia due to other than probable AD Use of any concurrent pharmaceutical treatment for cognitive dysfunction Therapy common to all participants: None Sample attrition/dropout: 10 of 10 completed study |
Arm no.: 1 Name: donepezil n: 5 Drug: donepezil Starting daily dose (mg): 5 Dosage details: one tablet taken nightly Arm no.: 2 Name: placebo n: 5 Drug: placebo Starting daily dose (mg): Dosage details: |
ADAS-cog [assessment of comprehension, spoken language, word finding, and praxis (score 0–70)] Serial Reaction Time Task [assessment of implicit (non-declarative) learning through comparing median response times to a coloured light stimulus] |
Baseline characteristics
| Type | Donepezil | Placebo | p-value | |||||
|---|---|---|---|---|---|---|---|---|
| N | K | Mean | N | K | Mean | |||
| ITT population | ||||||||
| Demographics | ||||||||
| Age (years) | C | 5 | 84.2 (SD 8.67) | 5 | 88 (SD 7.62) | 0.483a | ||
| Gender (n male) | D | 5 | 2 | 40.0% | 5 | 1 | 20.0% | 1.000b |
| Cognitive | ||||||||
| ADAS-cog – 0 weeks | C | 5 | 24 (SD 3.08) | 5 | 26 (SD 11.6) | 0.720a | ||
| MMSE | C | 5 | 19.2 (SD 3.35) | 5 | 20.2 (SD 4.09) | 0.683a | ||
Results
| Donepezil | Placebo | |||||||
|---|---|---|---|---|---|---|---|---|
| Type | n | K | Mean | n | K | Mean | p-value | |
| ITT population | ||||||||
| Cognitive | ||||||||
| ADAS-cog – 4 weeks | MC | 5 | –5 (SD 2) | 5 | 0 (SD 4.85) | 0.066a | ||
| Serial Reaction Time Task – 4 weeks | MC | 5 | 3.32 (SD 8.39) | 5 | 1.65 (SD 10.1) | 0.782a | ||
Methodological issues
|
Randomisation and allocation: Randomisation procedure not described. Placebo described as identical in appearance to donepezil Data analysis: Serial Reaction Time Test and ADAS-cog: multivariate between group test (Hotelling’s trace statistic) Power calculation: Not reported Conflicts of interest: None reported |
Quality appraisal
|
Appendix 4 Funnel plots from the synthesis with existing evidence
Donepezil versus placebo
FIGURE 96.
Cognitive outcomes (SMD) at 24–26 weeks – donepezil (all dosages) vs placebo: funnel plot.
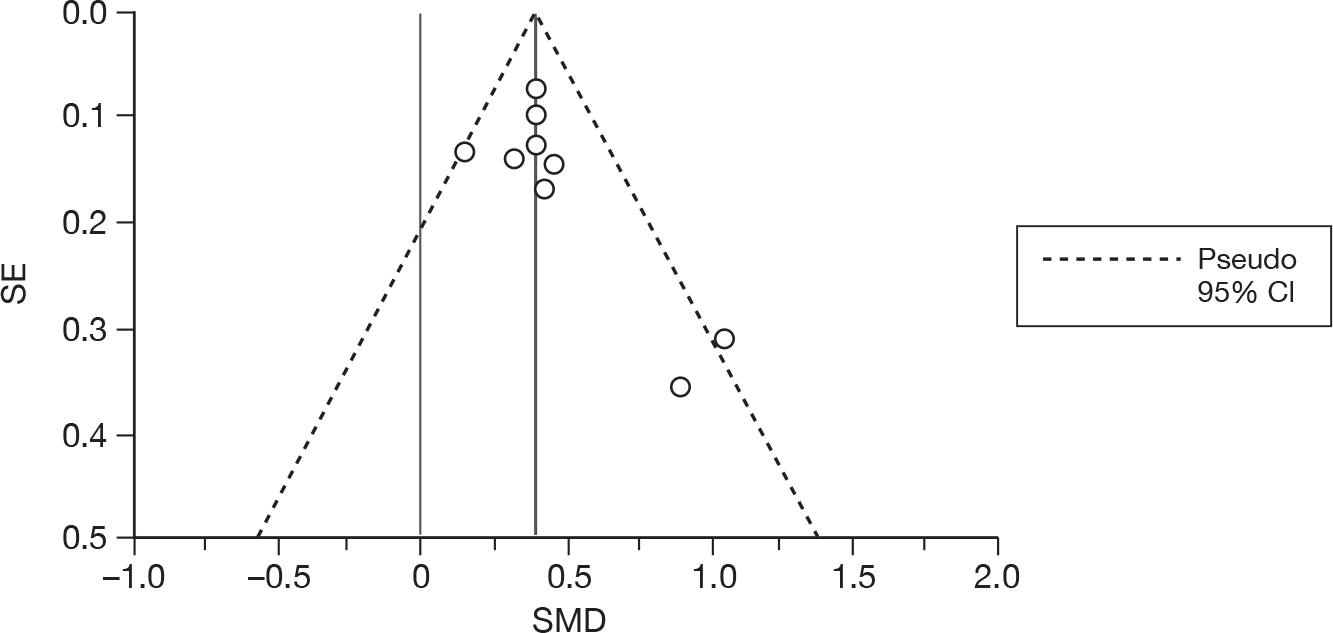
FIGURE 97.
Functional outcomes (SMD) at 24 weeks – donepezil (all dosages) vs placebo: funnel plot.
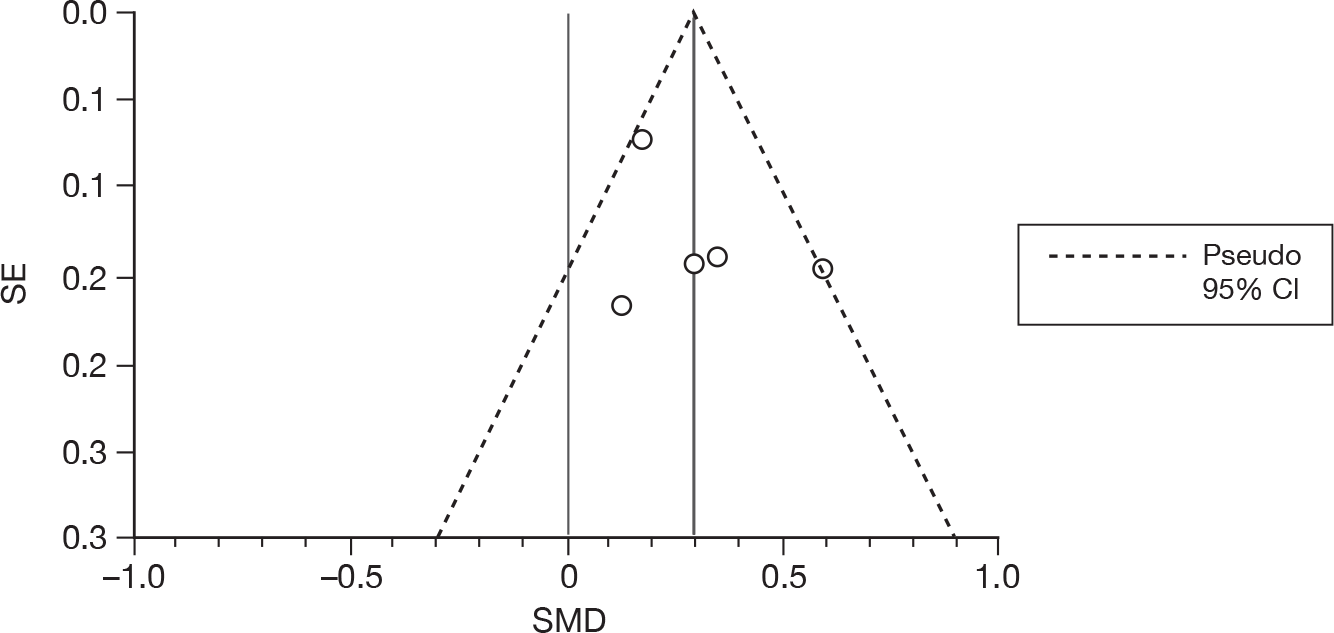
FIGURE 98.
Global outcomes (SMD) at 24 weeks – donepezil (all dosages) vs placebo: funnel plot.
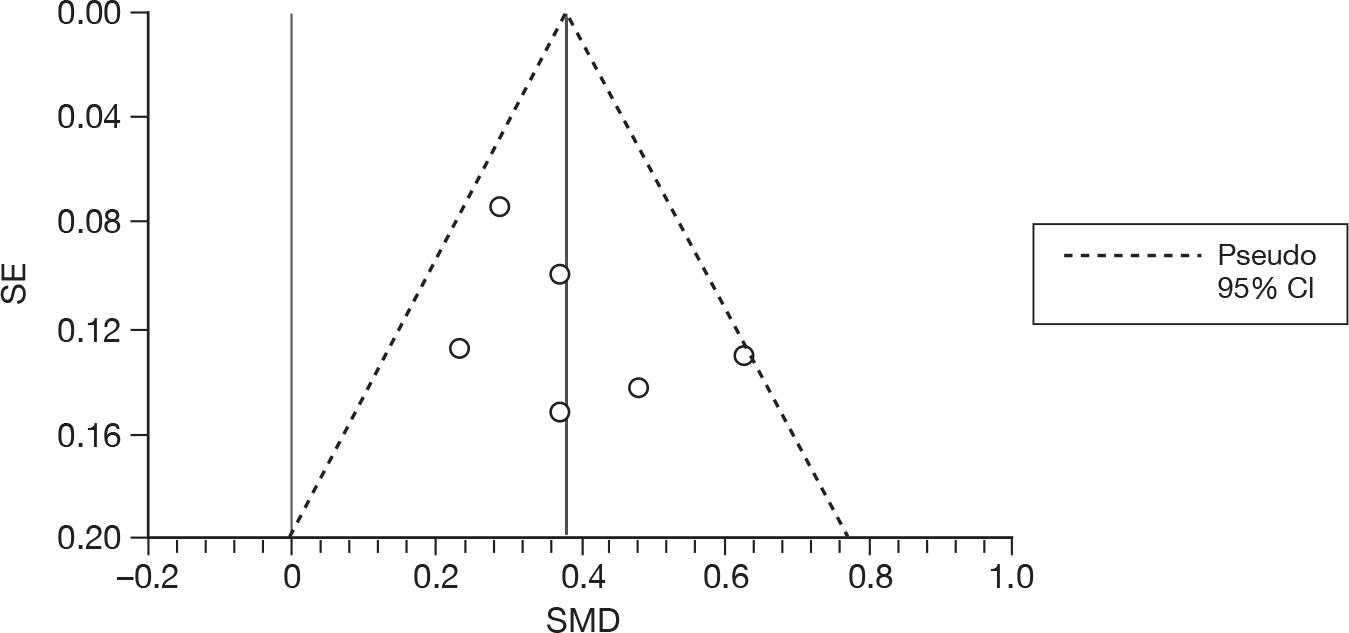
Appendix 5 Combined-dose and dose-specific meta-analyses
Donepezil
Donepezil 5 mg/day
FIGURE 99.
Random-effects meta-analysis – ADAS-cog at 12 weeks (mean change from baseline): donepezil (5 mg/day) vs placebo.
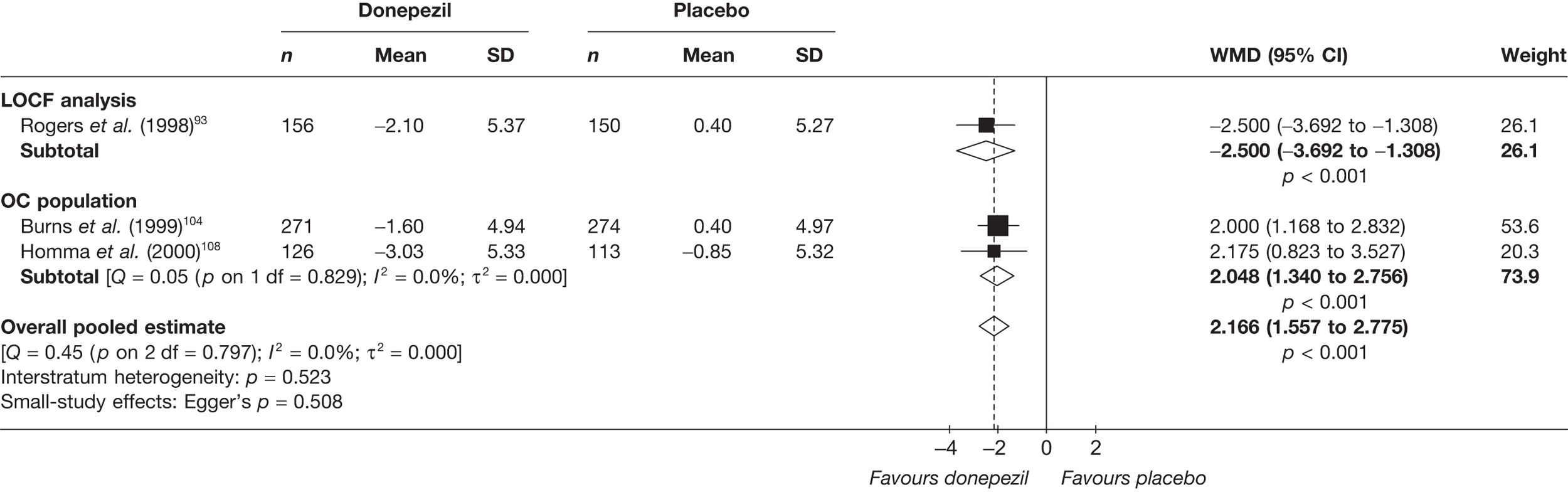
FIGURE 100.
Random-effects meta-analysis – ADAS-cog at 24 weeks (mean change from baseline): donepezil (5 mg/day) vs placebo.
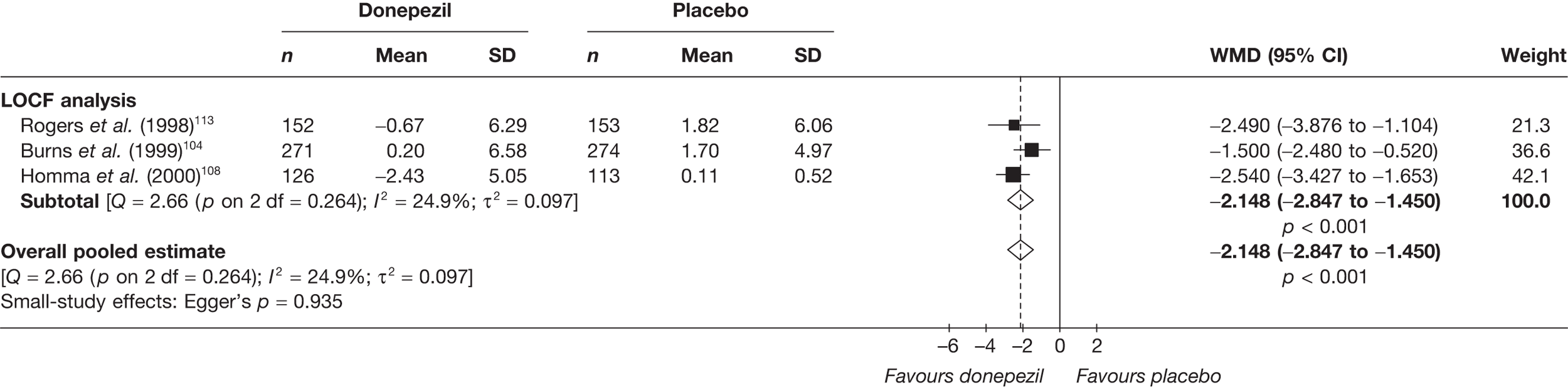
FIGURE 101.
Random-effects meta-analysis – MMSE at 24 weeks (mean change from baseline): donepezil (5 mg/day) vs placebo.
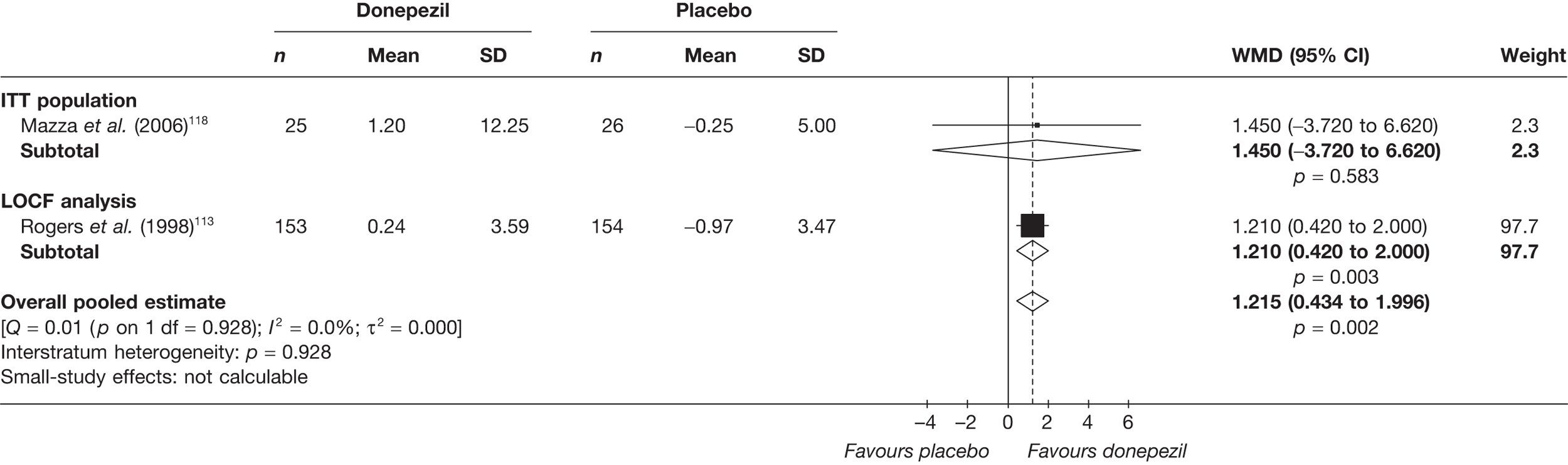
FIGURE 102.
Random-effects meta-analysis – CIBIC-plus at 12 weeks (mean change from baseline): donepezil (5 mg/day) vs placebo.
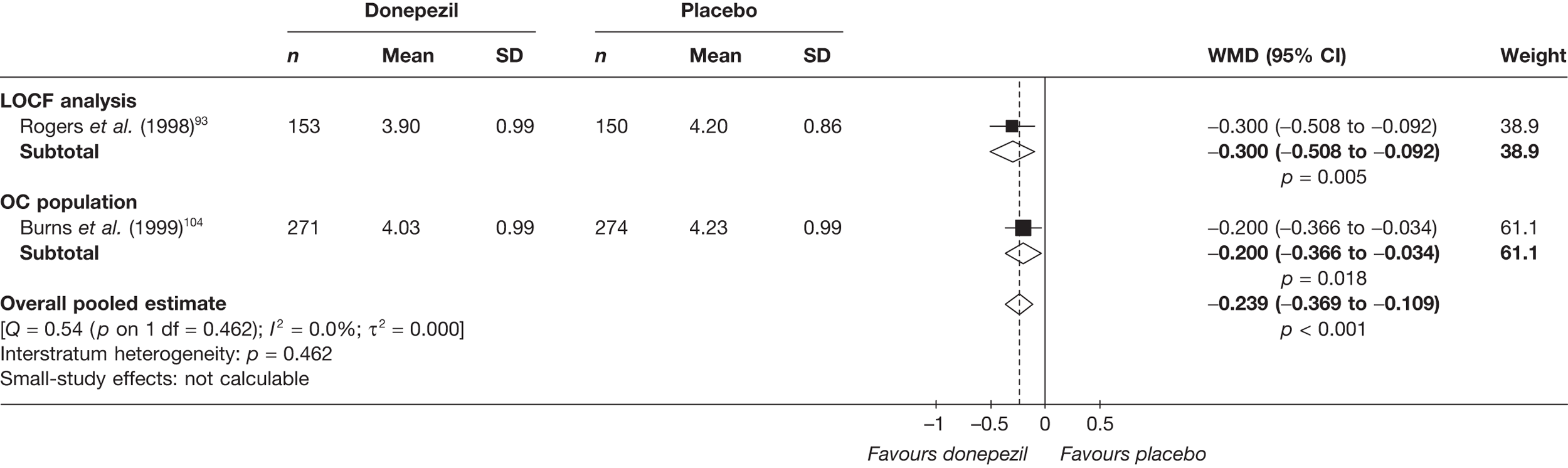
FIGURE 103.
Random-effects meta-analysis – CIBIC-plus at 24 weeks (mean change from baseline): donepezil (5 mg/day) vs placebo.
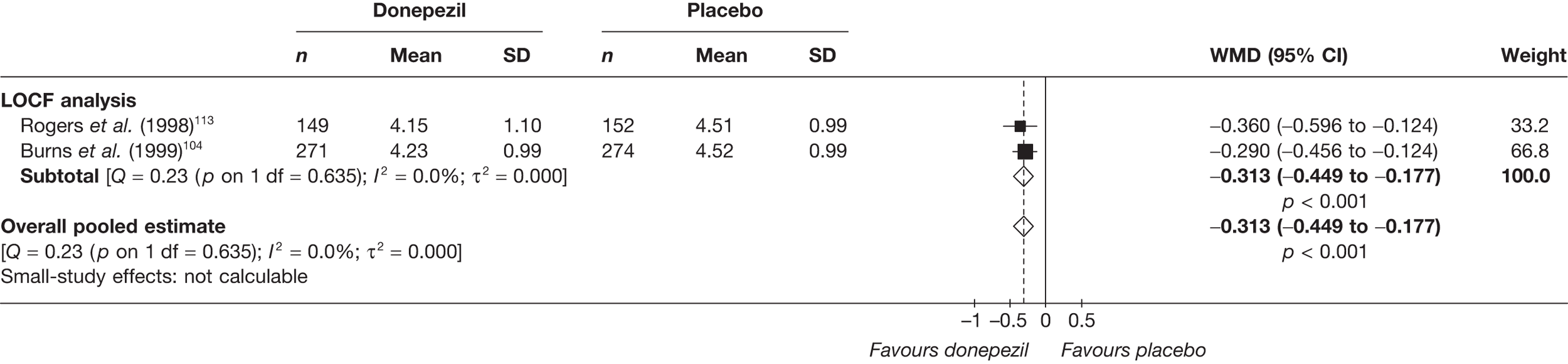
FIGURE 104.
Random-effects meta-analysis – CDR at 12 weeks (mean change from baseline): donepezil (5 mg/day) vs placebo.
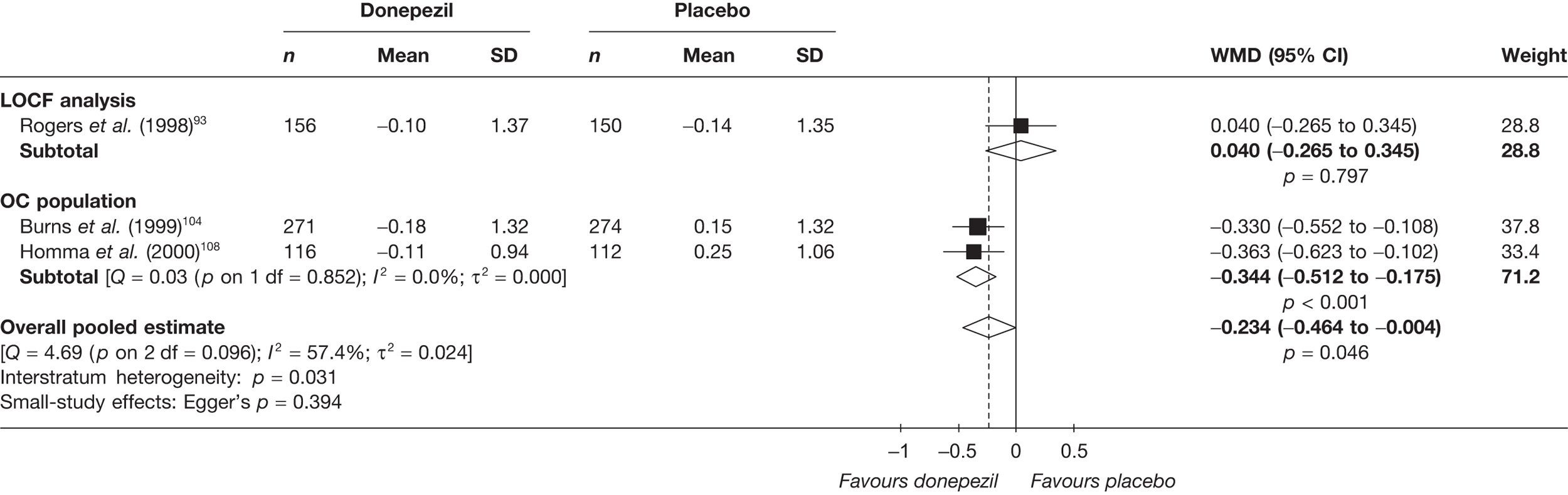
FIGURE 105.
Random-effects meta-analysis – CDR at 24 weeks (mean change from baseline): donepezil (5 mg/day) vs placebo.
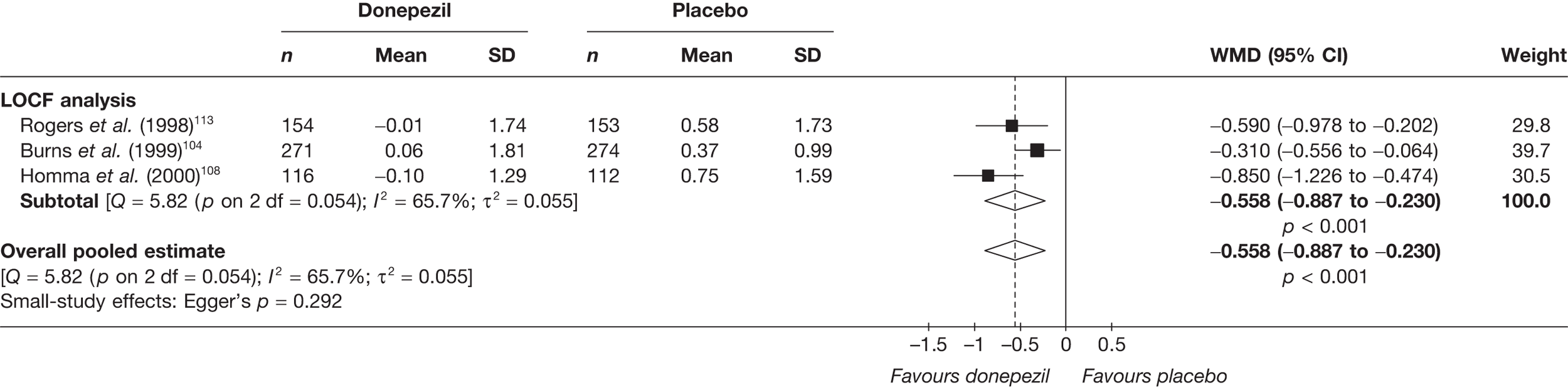
Donepezil all doses combined
FIGURE 106.
Random-effects meta-analysis – ADAS-cog at 12 weeks (mean change from baseline): donepezil (all dosages) vs placebo. apooled 5 mg/day and 10 mg/day arms.
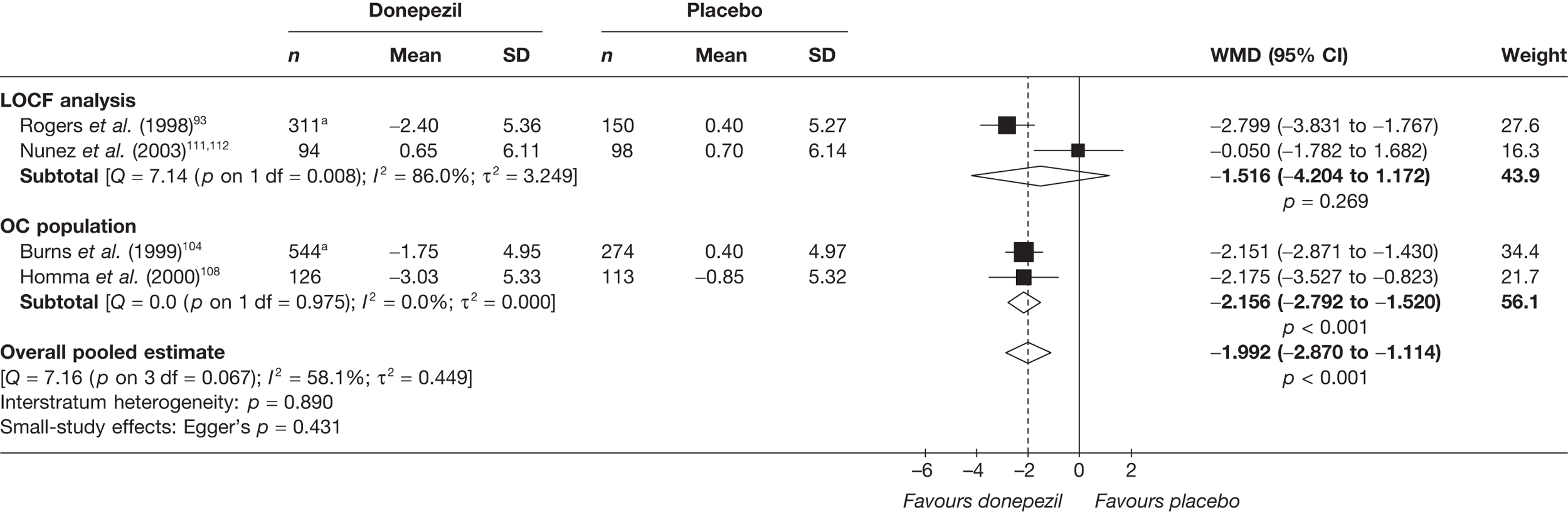
FIGURE 107.
Random-effects meta-analysis – ADAS-cog at 24 weeks (mean change from baseline): donepezil (all dosages) vs placebo. apooled 5 mg/day and 10 mg/day arms.
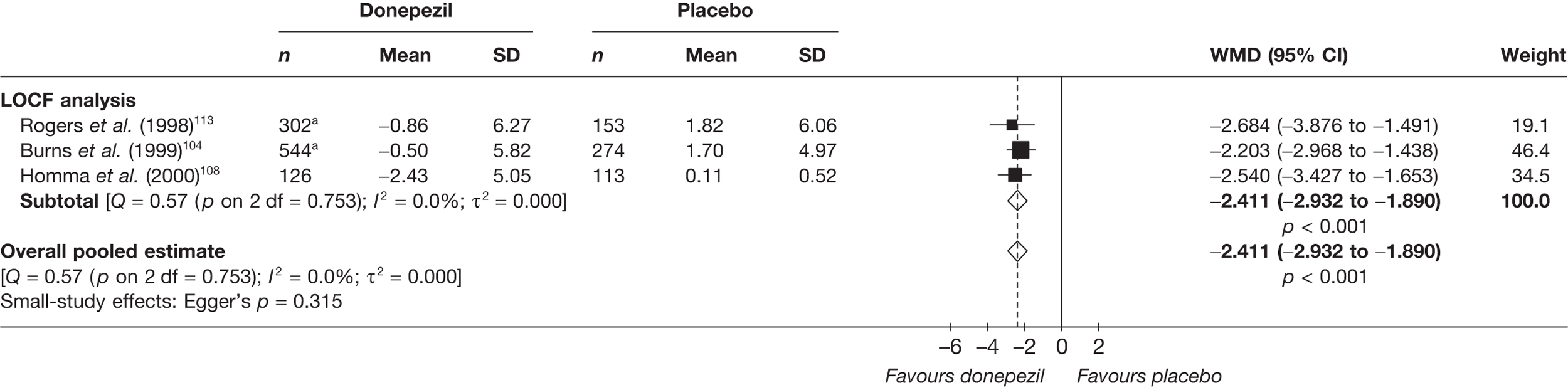
FIGURE 108.
Random-effects meta-analysis – MMSE at 12 weeks (mean change from baseline): donepezil (all dosages) vs placebo. apooled 5 mg/day and 10 mg/day arms.
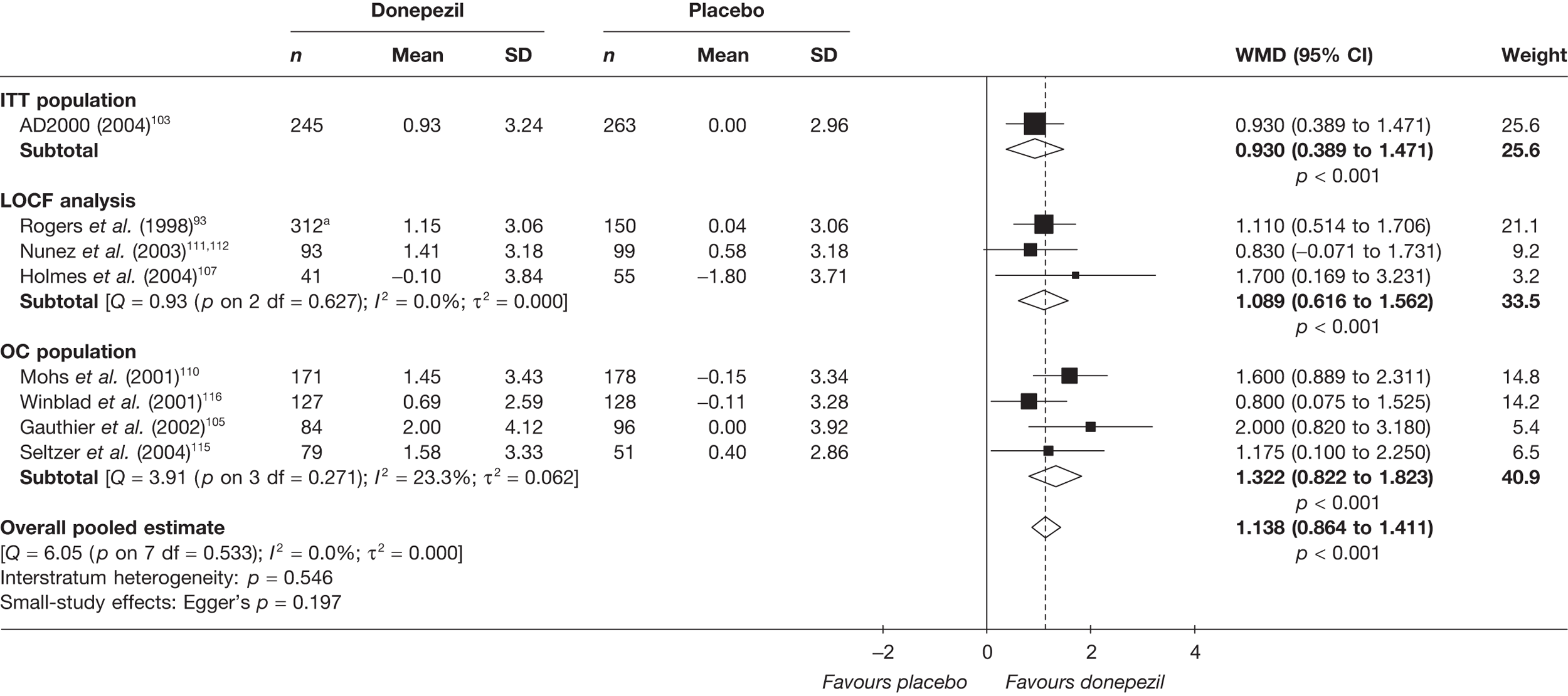
FIGURE 109.
Random-effects meta-analysis – MMSE at 24 weeks (mean change from baseline): donepezil (all dosages) vs placebo. aWMD and error bars provided in publication; SE estimated on assumption that error bars represent 95% CIs. bpooled 5 mg/day and 10 mg/day arms.
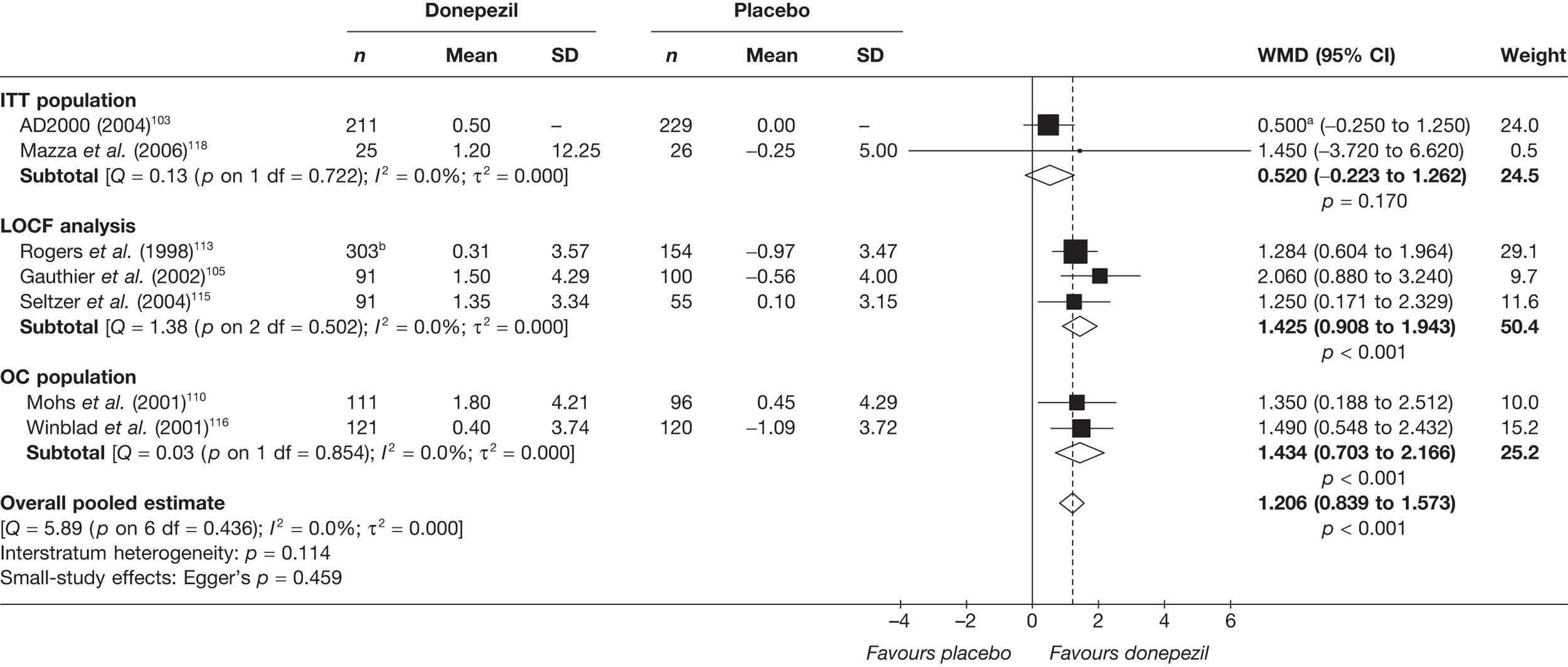
FIGURE 110.
Random-effects meta-analysis – CIBIC-plus at 12 weeks (mean change from baseline): donepezil (all dosages) vs placebo. a, Pooled 5 and 10 mg/day arms.
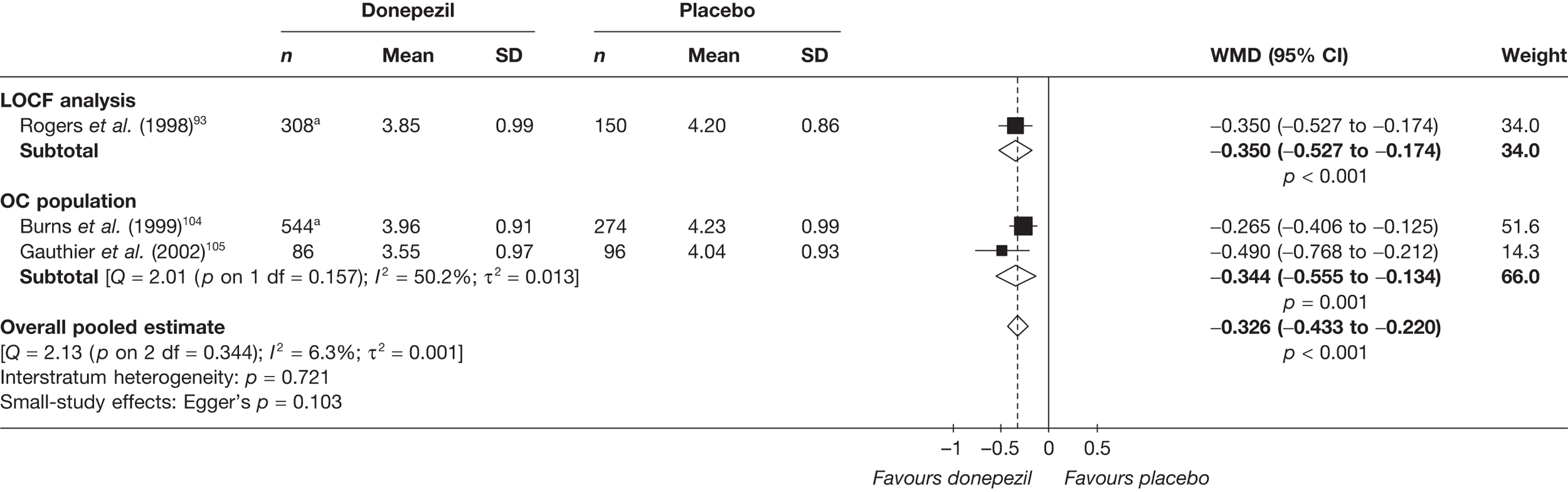
FIGURE 111.
Random-effects meta-analysis – CIBIC-plus at 24 weeks (mean change from baseline): donepezil (all dosages) vs placebo. a, Pooled 5 and 10 mg/day arms.
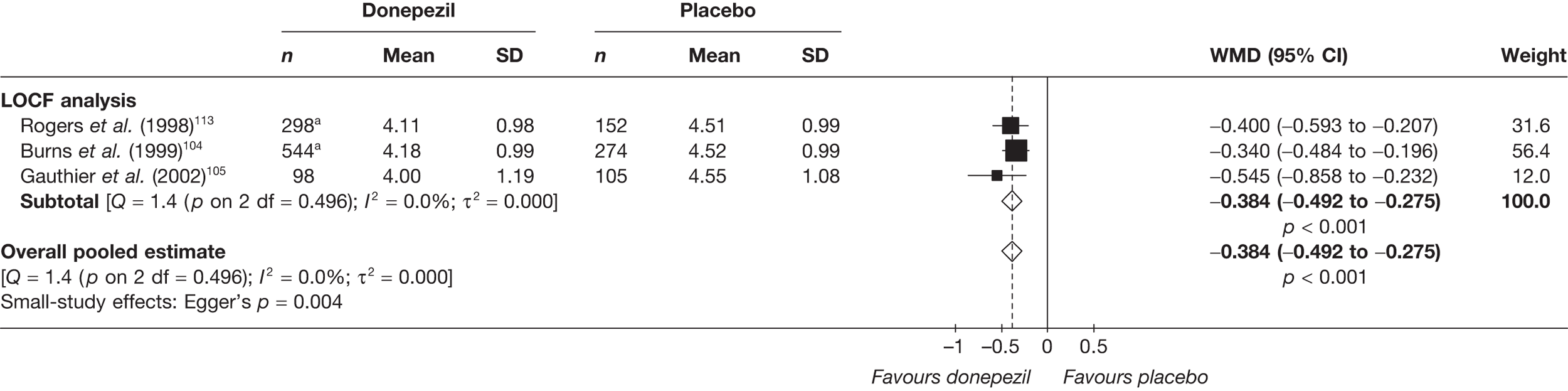
Galantamine
Galantamine > 24 mg/day
FIGURE 112.
Random-effects meta-analysis – ADAS-cog at 12–16 weeks (mean change from baseline): galantamine (maximum dose > 24 mg/day) vs placebo.
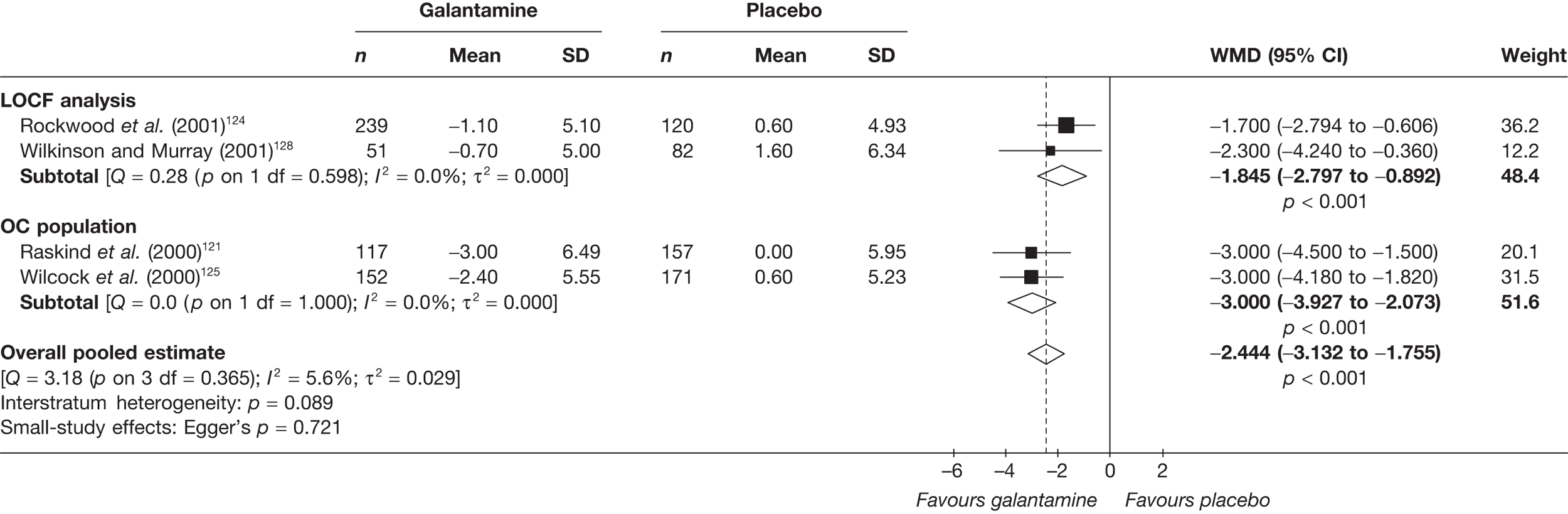
FIGURE 113.
Random-effects meta-analysis – ADAS-cog at 21–26 weeks (mean change from baseline): galantamine (maximum dose > 24 mg/day) vs placebo.
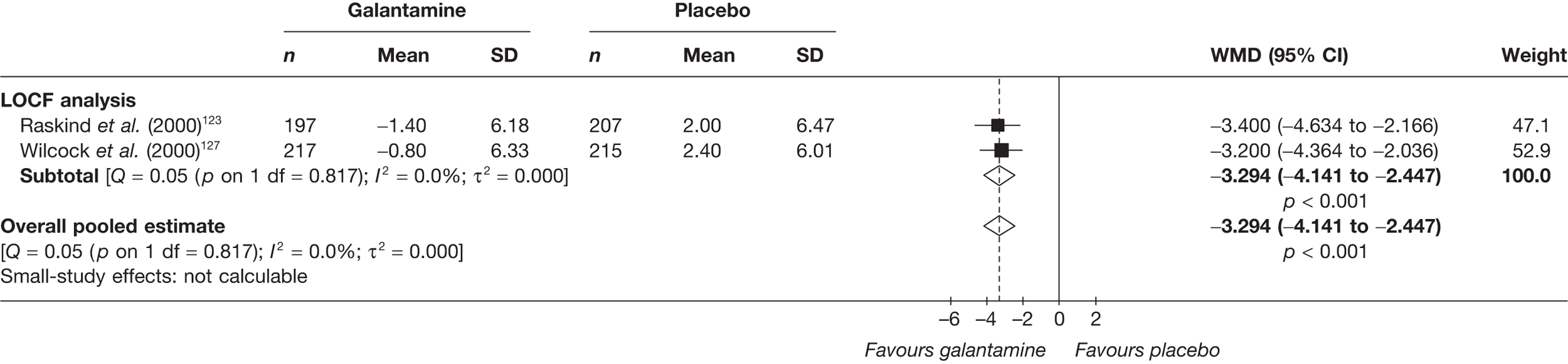
FIGURE 114.
Random-effects meta-analysis – CIBIC-plus at 26 weeks: galantamine (maximum dose > 24 mg/day) vs placebo.
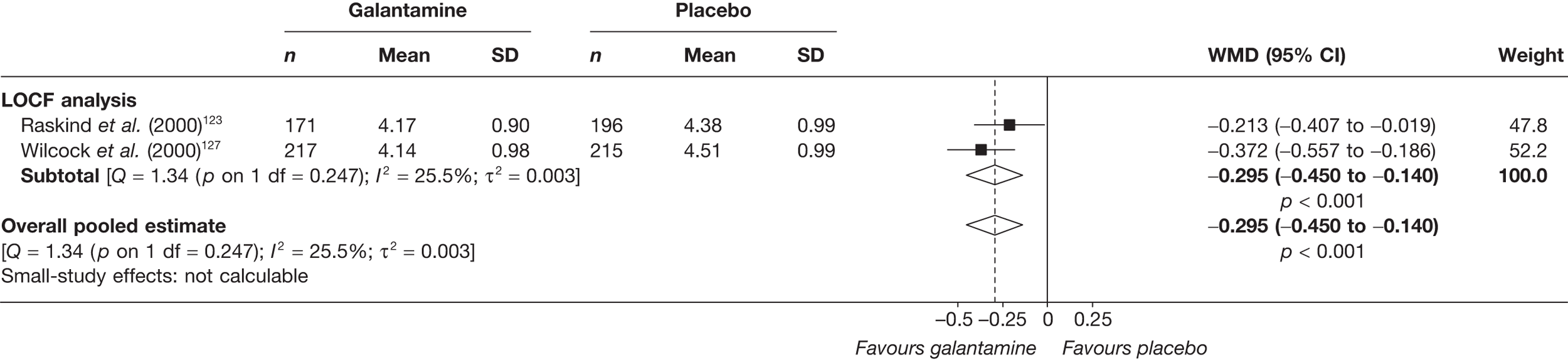
Galantamine all doses
FIGURE 115.
Random-effects meta-analysis – CIBIC-plus at 13–16 weeks: galantamine (all dosages) vs placebo.
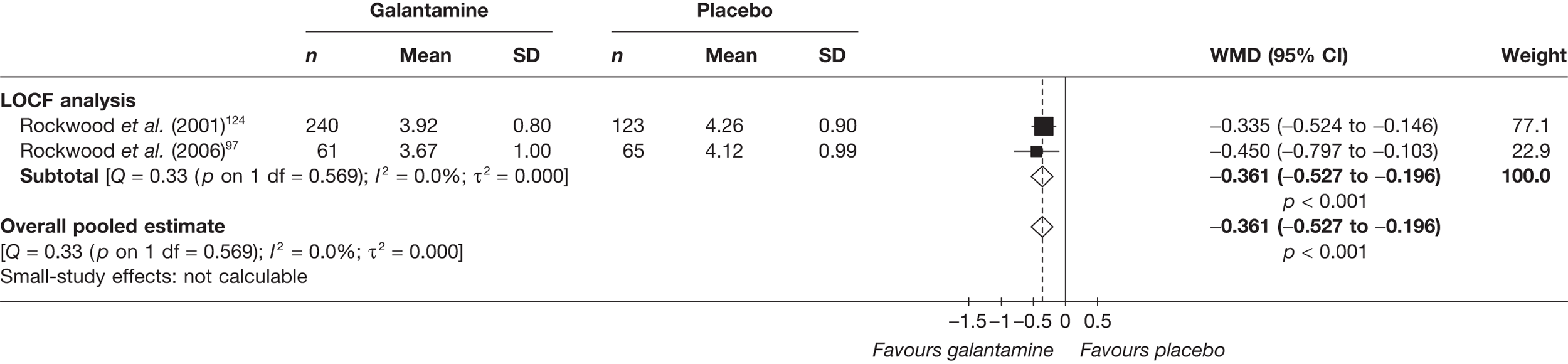
FIGURE 116.
Random-effects meta-analysis – CIBIC-plus at 26 weeks: galantamine (all dosages) vs placebo. a, 24 and 32 mg/day arms pooled; b, once daily prolonged-release formulation and twice daily standard formulation pooled.
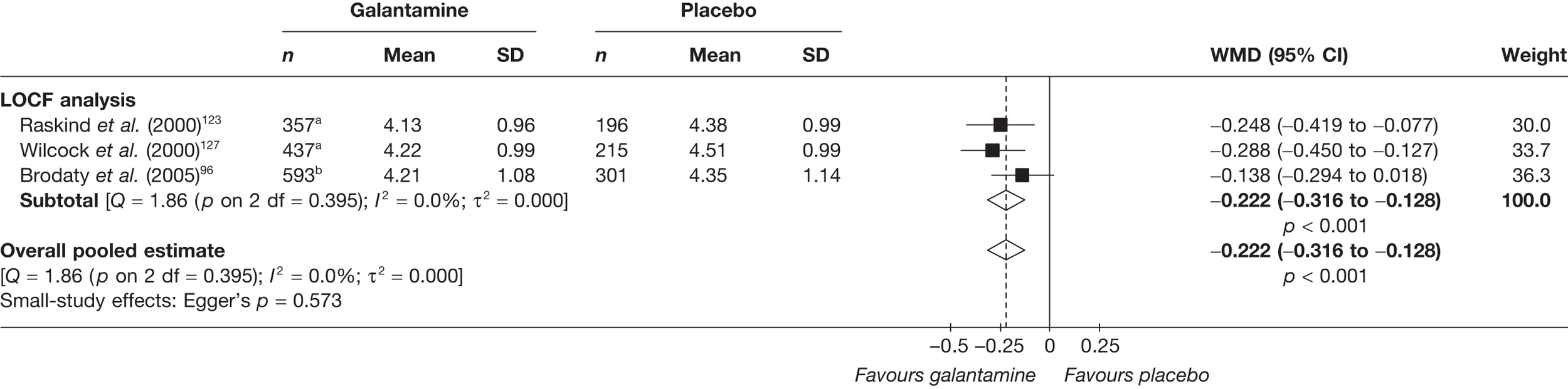
FIGURE 117.
Random-effects meta-analysis – ADAS-cog at 12–16 weeks (mean change from baseline) – galantamine (all dosages) vs placebo. a, 18, 24 and 36 mg/day arms pooled; b, once daily prolonged-release formulation and twice daily standard formulation pooled; c, 24 and 36 mg/day arms pooled; d, 8, 16 and 24 mg/day arms pooled. e, 24 and 32 mg/day arms pooled.
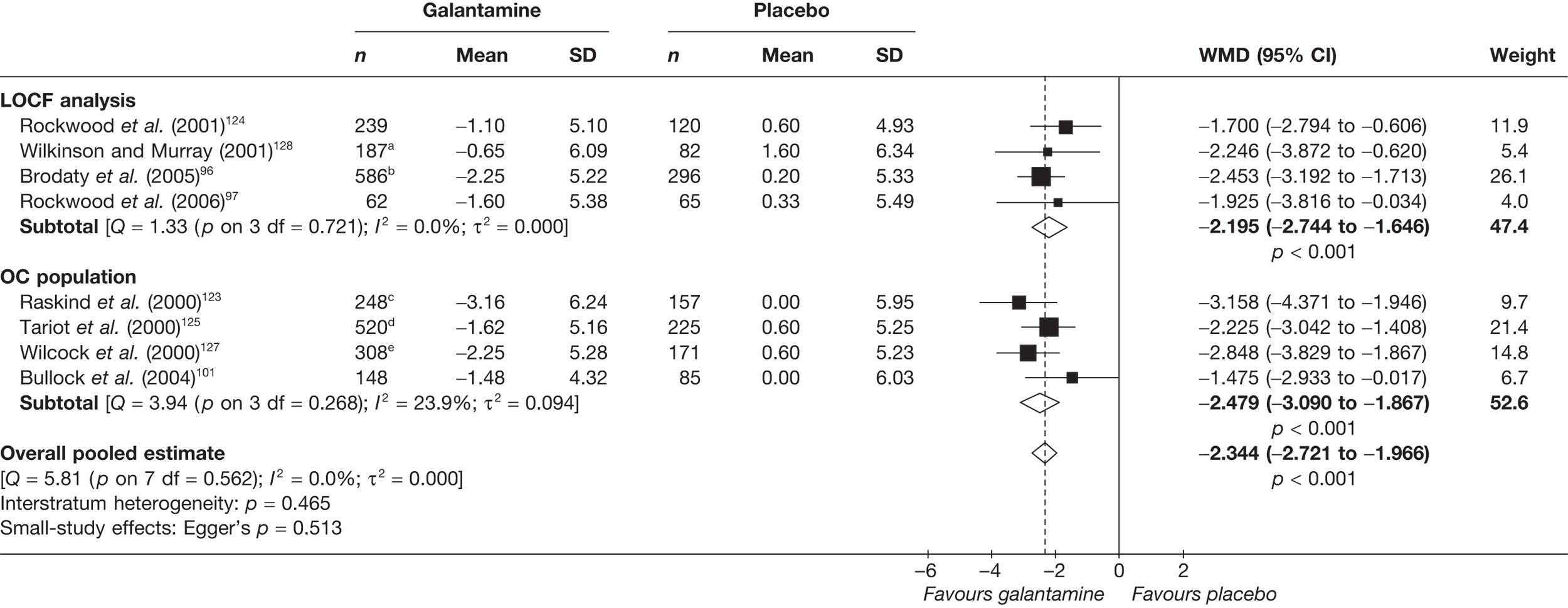
FIGURE 118.
Random-effects meta-analysis – ADAS-cog at 21–26 weeks (mean change from baseline) – galantamine (all dosages) vs placebo. a, 24 and 36 mg/day arms pooled; b, 8, 16 and 24 mg/day arms pooled. c, 24 and 32 mg/day arms pooled. d, once daily prolonged-release formulation and twice daily standard formulation pooled.
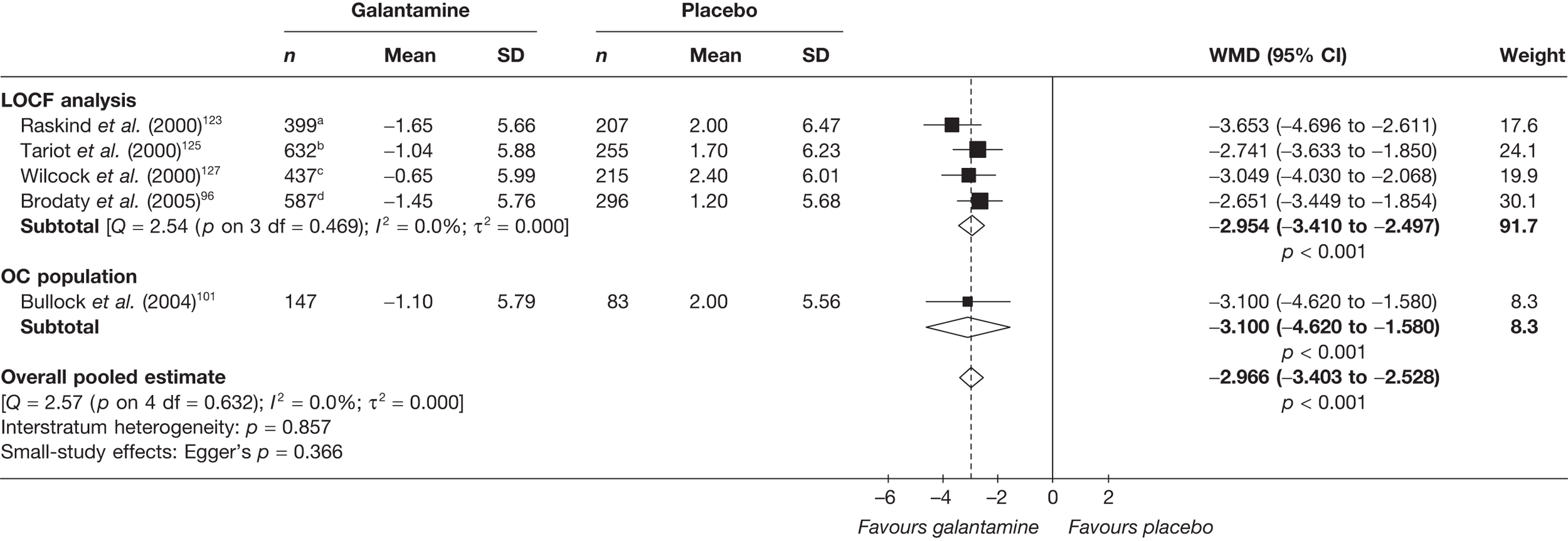
Rivastigmine
Rivastigmine ≤10 mg/day
FIGURE 119.
Random-effects meta-analysis – ADAS-cog at 12–16 weeks (mean change from baseline): rivastigmine (maximum dose ≤ 10 mg/day) vs placebo. a, twice a day and three times a day arms pooled; b, 20-cm2 patch and 12 mg/day capsules arms pooled.
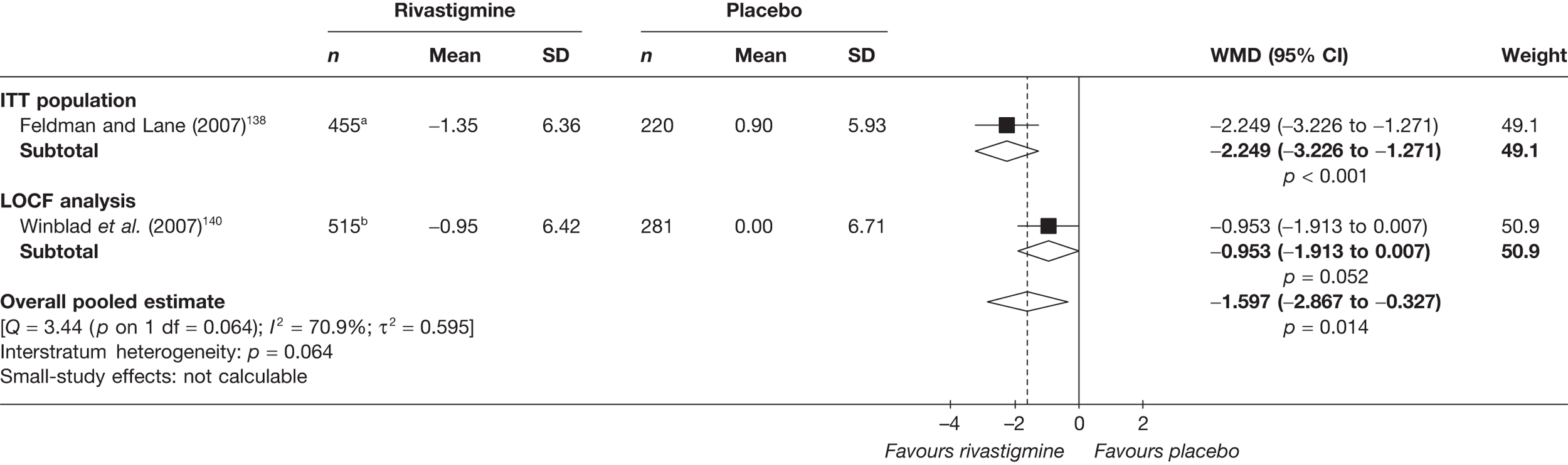
FIGURE 120.
Random-effects meta-analysis – ADAS-cog at 24–26 weeks (mean change from baseline): rivastigmine (≤ 10 mg/day) vs placebo.
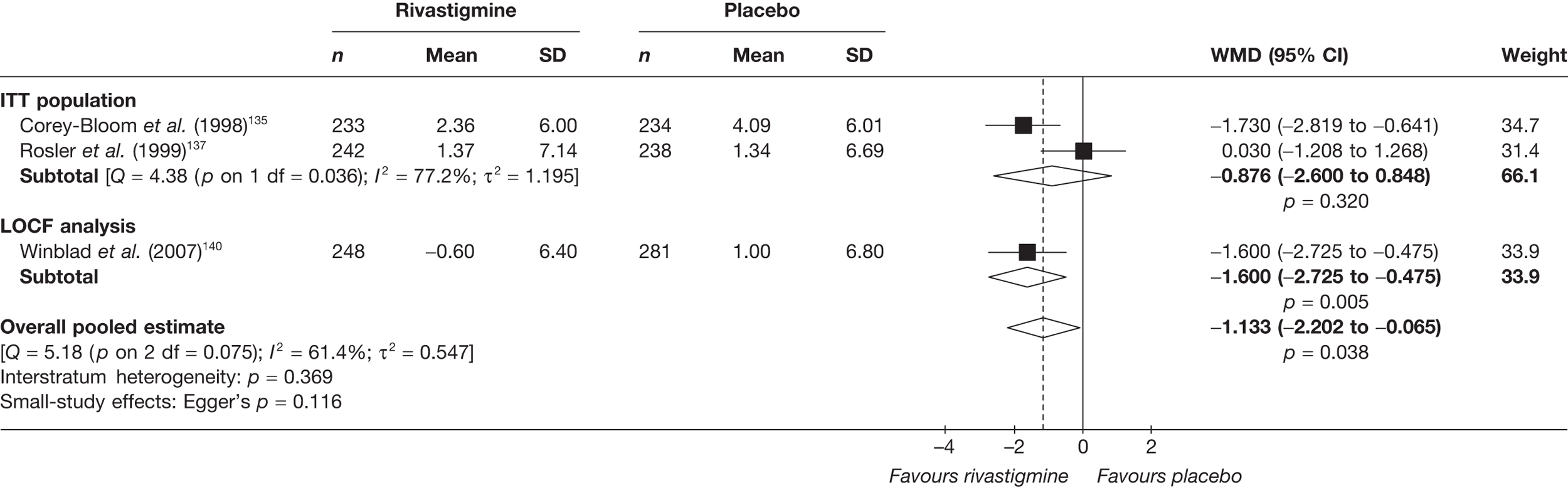
FIGURE 121.
Random-effects meta-analysis – CIBIC-plus at 26 weeks: rivastigmine (4 mg/day) vs placebo.
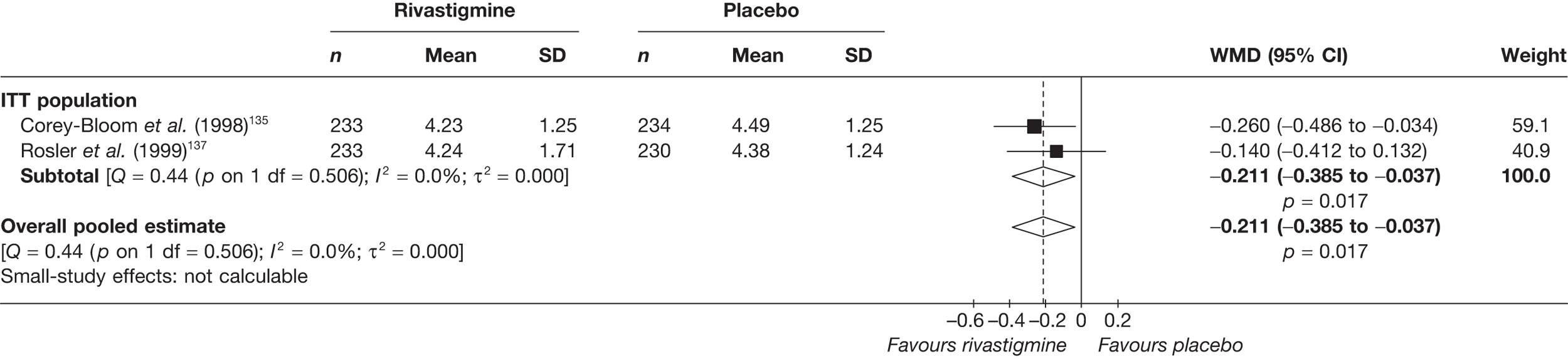
Rivastigmine all doses
FIGURE 122.
Random-effects meta-analysis – ADAS-cog at 12–16 weeks (mean change from baseline): rivastigmine (all dosages) vs placebo. a, twice a day and three times a day arms pooled; b,10-cm2 patch, 20-cm2 patch and 12 mg/day capsules arms pooled.
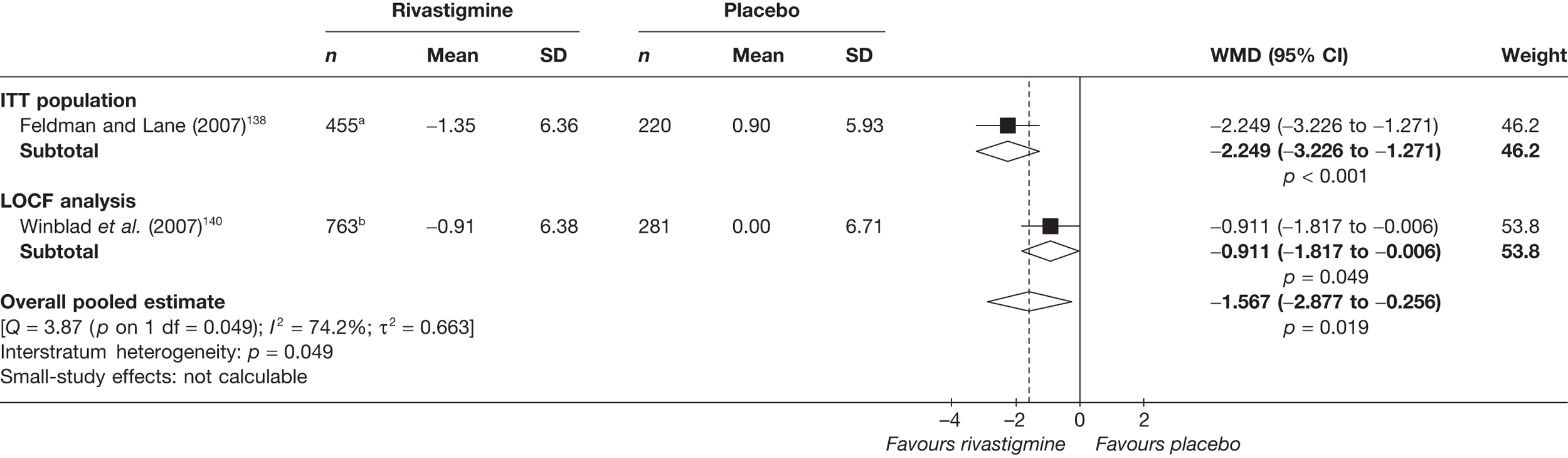
FIGURE 123.
Random-effects meta-analysis – ADAS-cog at 24–26 weeks (mean change from baseline): rivastigmine (all dosages) vs placebo. a, 4 and 12 mg/day arms pooled; b, twice a day and three times a day arms pooled; c, 10-cm2 patch, 20-cm2 patch and 12 mg/day capsules arms pooled.
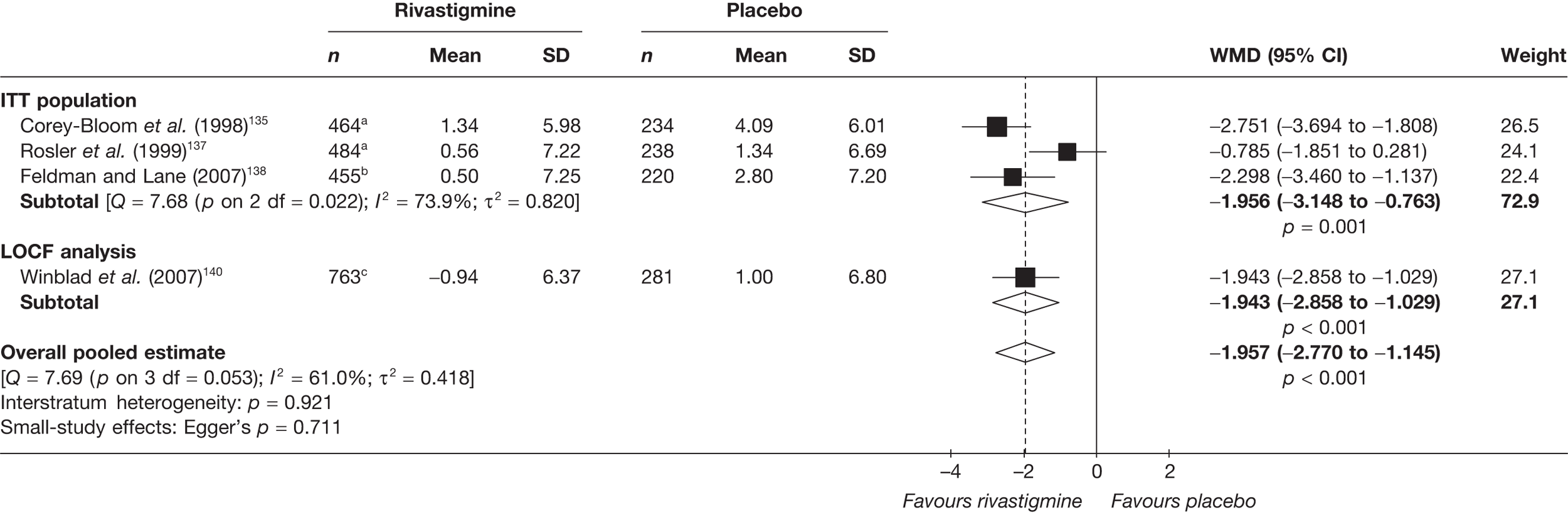
FIGURE 124.
Random-effects meta-analysis – MMSE at 12–13 weeks (mean change from baseline): rivastigmine (all dosages) vs placebo. a. 4 and 6 mg/day arms pooled.
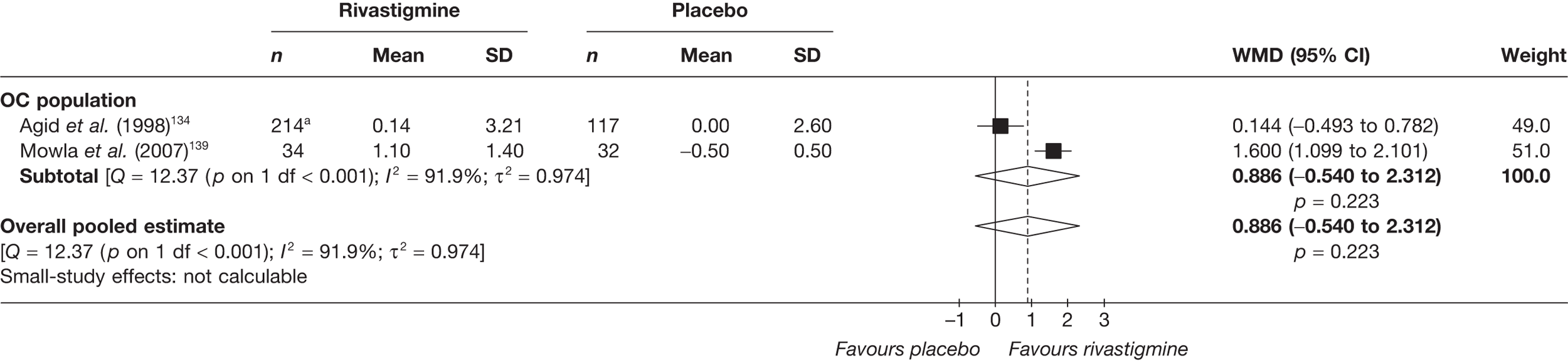
FIGURE 125.
Random-effects meta-analysis – MMSE at 24–26 weeks (mean change from baseline): rivastigmine (all dosages) vs placebo. a, b.i.d. and t.i.d. arms pooled; b, 10-cm2 patch, 20-cm2 patch and 12 mg/day capsules arms pooled.
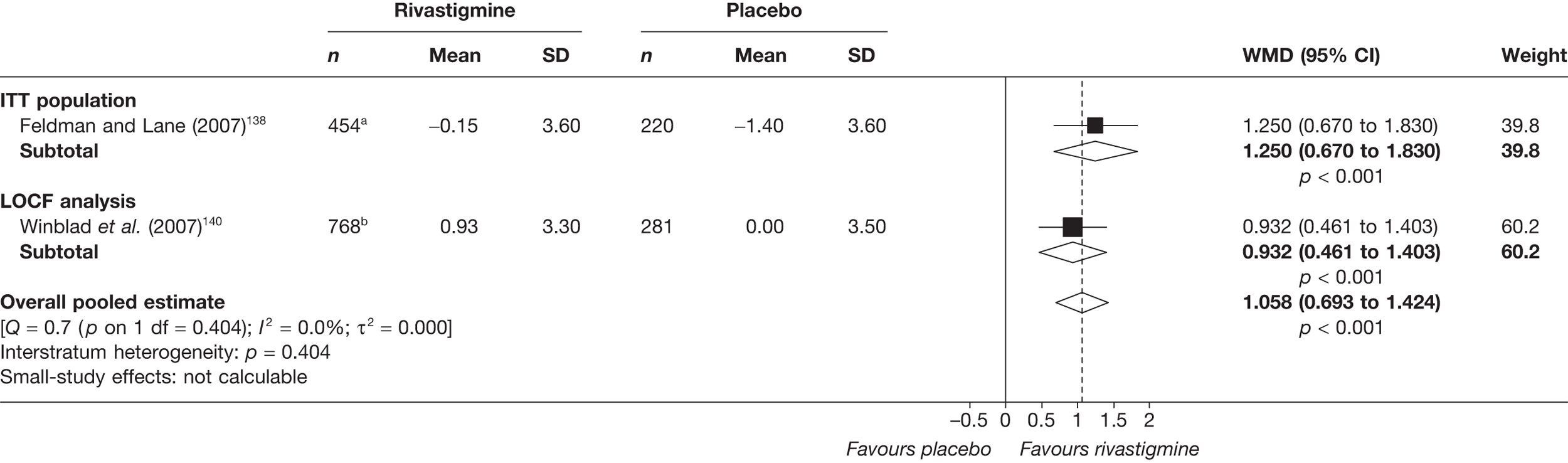
FIGURE 126.
Random-effects meta-analysis – PDS at 24–26 weeks (mean change from baseline): rivastigmine (all dosages) vs placebo. a, 4 and 12 mg/day arms pooled; b, twice a day and three times a day arms pooled.
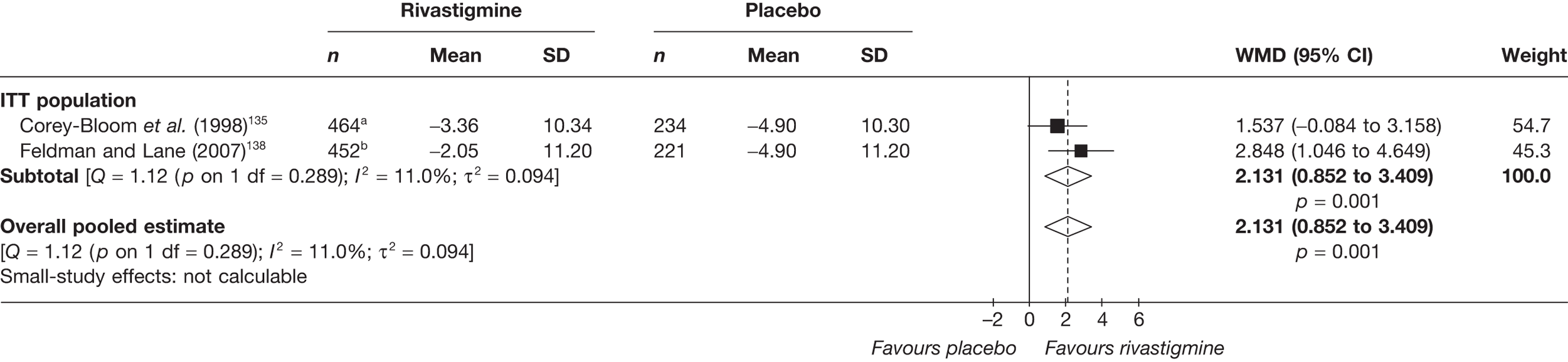
FIGURE 127.
Random-effects meta-analysis – CIBIC-plus at 26 weeks; rivastigmine (all dosages) vs placebo. a. 4 and 12 mg/day arms pooled; b, twice a day and three times a day arms pooled.
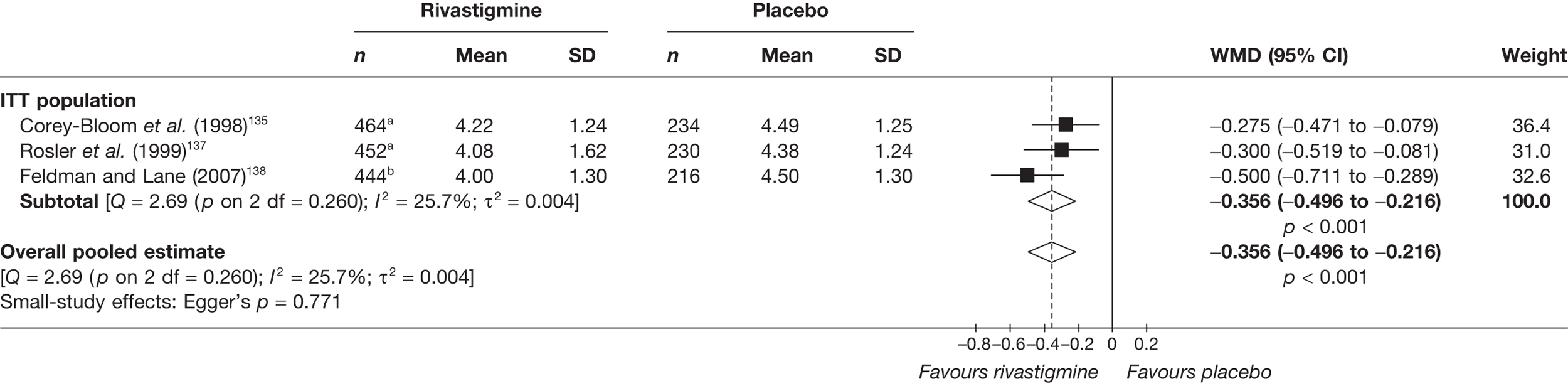
FIGURE 128.
Random-effects meta-analysis – CIBIC-plus at 26 weeks: rivastigmine (12 mg/day) vs placebo. a, twice a day and three times a day arms pooled.
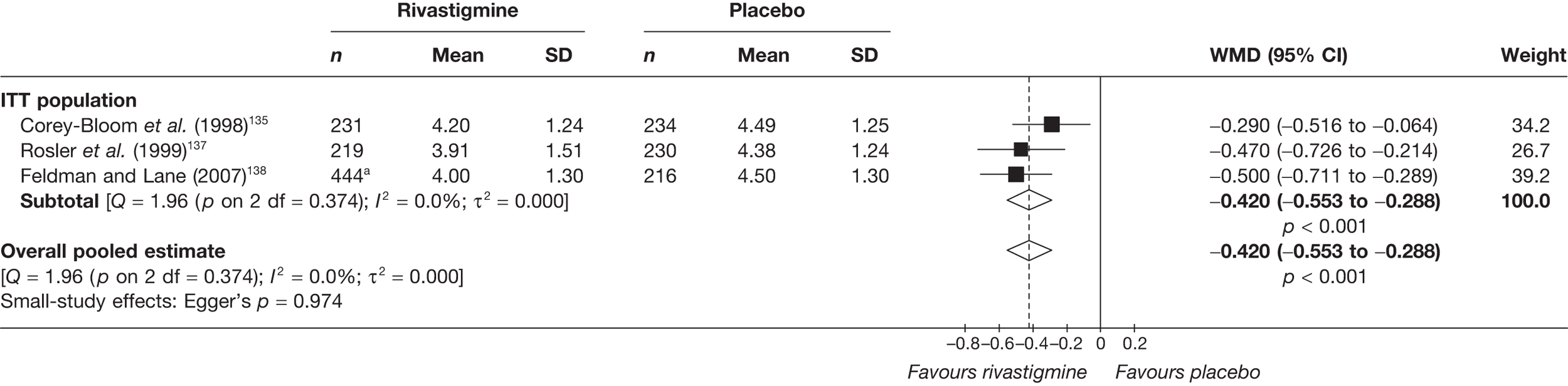
FIGURE 129.
Random-effects meta-analysis – GDS at 26 weeks (mean change from baseline): rivastigmine (all dosages) vs placebo. a, 4 and 12 mg/day arms pooled; b, twice a day and three times a day arms pooled.
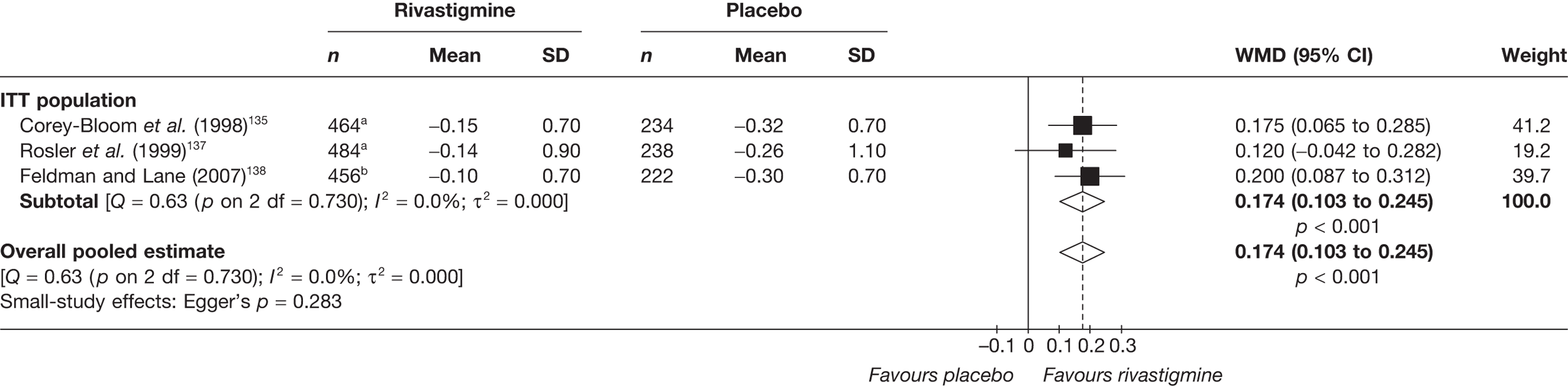
Appendix 6 Data sets used in meta-analysis of pooled multiple outcome measures
Donepezil
| Study | Outcome | Type | +/– | Donepezil | Placebo | SMD | 95% CI | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| n | Mean | SD | n | Mean | SD | ||||||
| ITT population | |||||||||||
| Mazza et al. (2006)118 | MMSE | MC | + | 25 | 1.20 | 12.25 | 26 | –0.25 | 5.00 | 1.059 | 0.445 to 1.673 |
| Syndrom Kurztest | MC | – | 25 | –3.30 | 2.55 | 26 | 0.90 | 1.30 | |||
| CGI-item 2 | MC | – | 25 | –0.90 | 1.02 | 26 | 0.15 | 0.34 | |||
| LOCF analysis | |||||||||||
| Rogers et al. (1998)113 | MMSE | MC | + | 303a | 0.31 | 3.57 | 154 | –0.97 | 3.47 | 0.398 | 0.202 to 0.594 |
| ADAS-cog | MC | – | 302a | –0.86 | 6.27 | 153 | 1.82 | 6.06 | |||
| Burns et al. (1999)106 | ADAS-cog | MC | – | 544a | –0.50 | 5.82 | 274 | 1.70 | 4.97 | 0.397 | 0.250 to 0.543 |
| Homma et al. (2000)108 | MENFIS | MC | + | 116 | –0.72 | 5.71 | 112 | 1.84 | 7.30 | 0.150 | –0.112 to 0.412 |
| ADAS-cog | MC | – | 126 | –2.43 | 5.05 | 113 | 0.11 | 0.52 | |||
| Gauthier et al. (2002)105 | SIB | MC | + | 98 | 1.58 | 11.14 | 104 | –2.85 | 11.22 | 0.445 | 0.161 to 0.728 |
| MMSE | MC | + | 91 | 1.50 | 4.29 | 100 | –0.56 | 4.00 | |||
| Seltzer et al. (2004)115 | MMSE | MC | + | 91 | 1.35 | 3.34 | 55 | 0.10 | 3.15 | 0.427 | 0.089 to 0.766 |
| ADAS-cog/13 | MC | – | 91 | –1.65 | 4.77 | 55 | 0.58 | 4.64 | |||
| OC population | |||||||||||
| Mohs et al. (2001)110 | MMSE | MC | + | 111 | 1.80 | 4.21 | 96 | 0.45 | 4.29 | 0.318 | 0.043 to 0.593 |
| Winblad et al. (2001)116 | MMSE | MC | + | 121 | 0.40 | 3.74 | 120 | –1.09 | 3.72 | 0.399 | 0.144 to 0.654 |
| dos Santos Moraes et al. (2006)119 | ADAS-cog | A | – | 17 | 28.30 | 12.30 | 18 | 42.80 | 18.70 | 0.911 | 0.212 to 1.609 |
| Study | Outcome | +/– | Galantamine | Placebo | SMD | 95% CI | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | Meana | SD | n | Meana | SD | |||||
| LOCF analysis | ||||||||||
| Burns et al. (1999)104 | IDDD – complex tasks | – | 544b | 69.90c | 6.60 | 274 | 71.10c | 6.62 | 0.182 | 0.036 to 0.327 |
| Homma et al. (2000)108 | CMCS | – | 103 | 1.03 | 6.70 | 99 | 3.45 | 7.06 | 0.352 | 0.074 to 0.630 |
| Gauthier et al. (2002)105 | DAD | + | 92 | 0.00 | 15.35 | 101 | –9.25 | 15.58 | 0.598 | 0.309 to 0.887 |
| OC population | ||||||||||
| Mohs et al. (2001)110 | ADFACS | – | 97 | –0.30 | 4.19 | 94 | 0.90 | 4.00 | 0.293 | 0.008 to 0.578 |
| Winblad et al. (2001)116 | Caregiver time (m/d) | – | 69 | –11.40 | 161.98 | 74 | 10.80 | 163.44 | 0.136 | –0.192 to 0.465 |
| Study | Outcome | +/– | Donepezil | Placebo | SMD | 95% CI | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | Meana | SD | n | Meana | SD | |||||
| LOCF analysis | ||||||||||
| Rogers et al. (1998)113 | CDR-SB | – | 305b | –0.01 | 1.73 | 153 | 0.58 | 1.73 | 0.375 | 0.178 to 0.571 |
| CIBIC-plus | – | 298b | 4.11c | 0.98 | 152 | 4.51c | 0.99 | |||
| Burns et al. (1999)104 | CDR-SB | – | 544b | 0.00 | 1.81 | 274 | 0.37 | 0.99 | 0.288 | 0.142 to 0.434 |
| CIBIC-plus | – | 544b | 4.18c | 0.99 | 274 | 4.52c | 0.99 | |||
| Homma et al. (2000)108 | ADCS-CGIC | – | 133 | 3.58c | 1.08 | 128 | 4.40c | 1.39 | 0.626 | 0.370 to 0.883 |
| CDR-SB | – | 116 | –0.10 | 1.29 | 112 | 0.75 | 1.59 | |||
| Gauthier et al. (2002)105 | CIBIC-plus | – | 98 | 4.00c | 1.19 | 105 | 4.55c | 1.08 | 0.482 | 0.202 to 0.761 |
| OC population | ||||||||||
| Winblad et al. (2001)116 | Gottfries–Bråne–Steen scale | – | 122 | 1.70 | 13.25 | 121 | 5.00 | 15.40 | 0.236 | –0.017 to 0.488 |
| GDS | – | 122 | 0.01 | 0.66 | 121 | 0.17 | 0.66 | |||
| Gauthier et al. (2002)105 | CIBIC-plus | – | 83 | 3.95c | 1.14 | 93 | 4.40c | 1.25 | 0.375 | 0.076 to 0.673 |
Galantamine
| Study | Outcome | +/– | Galantamine | Placebo | SMD | 95% CI | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | Meana | SD | n | Meana | SD | |||||
| LOCF analysis | ||||||||||
| Tariot et al. (2000)125 | ADCS-ADL | + | 637b | –1.52 | 9.47 | 262 | –3.80 | 9.71 | 0.239 | 0.094 to 0.383 |
| Wilcock et al. (2000)127 | DAD | + | 426c | –2.85 | 15.26 | 210 | –6.00 | 15.65 | 0.205 | 0.039 to 0.370 |
| Bullock et al. (2004)101 | DAD | + | 188 | –1.00 | 15.77 | 97 | –6.00 | 14.48 | 0.326 | 0.079 to 0.572 |
| Brodaty et al. (2005)96 | ADCS-ADL | + | 487d | –0.50 | 5.36 | 258 | –2.70 | 8.99 | 0.322 | 0.170 to 0.474 |
Rivastigmine
| Study | Outcome | +/– | Rivastigmine | Placebo | SMD | 95% CI | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | Meana | SD | n | Meana | SD | |||||
| ITT population | ||||||||||
| Corey-Bloom et al. (1998)135 | ADAS-cog | – | 464b | 1.34 | 5.98 | 234 | 4.09 | 6.01 | 0.459 | 0.300 to 0.618 |
| Rosler et al. (1999)137 | ADAS-cog | – | 484b | 0.56 | 7.22 | 238 | 1.34 | 6.69 | 0.111 | –0.044 to 0.267 |
| Feldman and Lane (2007)138 | MMSE | + | 454c | –0.15 | 3.60 | 220 | –1.40 | 3.60 | 0.328 | 0.166 to 0.490 |
| ADAS-cog | – | 455c | 0.50 | 7.25 | 220 | 2.80 | 7.20 | |||
| ADAS-cogA | – | 455c | 0.70 | 7.85 | 220 | 3.20 | 7.80 | |||
| LOCF analysis | ||||||||||
| Winblad et al. (2007)140 | Ten-point clock-drawing test | + | 742d | 0.20 | 3.14 | 269 | –0.10 | 3.20 | 0.242 | 0.103 to 0.381 |
| ADAS-cog | – | 763d | –0.94 | 6.37 | 281 | 1.00 | 6.80 | |||
| MMSE | + | 768d | 0.93 | 3.30 | 281 | 0.00 | 3.50 | |||
| Trail-making test | – | 719d | –9.55 | 59.25 | 258 | 7.70 | 56.60 | |||
| Study | Outcome | +/– | Rivastigmine | Placebo | SMD | 95% CI | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | Meana | SD | n | Meana | SD | |||||
| ITT population | ||||||||||
| Corey-Bloom et al. (1998)135 | PDS | + | 464b | –3.36 | 10.34 | 234 | –4.90 | 10.30 | 0.149 | –0.008 to 0.306 |
| Feldman and Lane (2007)138 | PDS | + | 452c | –2.05 | 11.20 | 221 | –4.90 | 11.20 | 0.254 | 0.093 to 0.416 |
| LOCF analysis | ||||||||||
| Winblad et al. (2007)140 | ADCS-ADL | + | 764d | –0.20 | 10.15 | 281 | –2.30 | 9.40 | 0.211 | 0.074 to 0.348 |
| Study | Outcome | +/– | Rivastigmine | Placebo | SMD | 95% CI | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | Meana | SD | n | Meana | SD | |||||
| ITT population | ||||||||||
| Corey-Bloom et al. (1998)135 | GDS | + | 464b | –0.15 | 0.70 | 234 | –0.32 | 0.70 | 0.235 | 0.078 to 0.393 |
| CIBIC-plus score | – | 464b | 4.22c | 1.24 | 234 | 4.49c | 1.25 | |||
| Rosler et al. (1999)137 | GDS | + | 484b | –0.14 | 0.90 | 238 | –0.26 | 1.10 | 0.161 | 0.003 to 0.318 |
| CIBIC-plus score | – | 452b | 4.08c | 1.62 | 230 | 4.38c | 1.24 | |||
| Feldman and Lane (2007)138 | GDS | + | 456d | –0.10 | 0.70 | 222 | –0.30 | 0.70 | 0.334 | 0.171 to 0.496 |
| CIBIC-plus score | – | 444d | 4.00c | 1.30 | 216 | 4.50c | 1.30 | |||
| LOCF analysis | ||||||||||
| Winblad et al. (2007)140 | ADCS−CGIC | – | 761e | 3.93c | 1.27 | 278 | 4.20c | 1.30 | 0.208 | 0.071 to 0.346 |
Appendix 7 Metaregression figures
Donepezil versus placebo: cognitive
FIGURE 130.
Mini Mental State Examination at 12 weeks (mean change from baseline) – donepezil (all dosages) vs placebo: association of treatment effect with average age of population. Area of circles is proportional to weight of each study in random-effects meta-analysis; dashed line shows univariate metaregression estimate (α = –7.447; β = 0.115; p = 0.253).
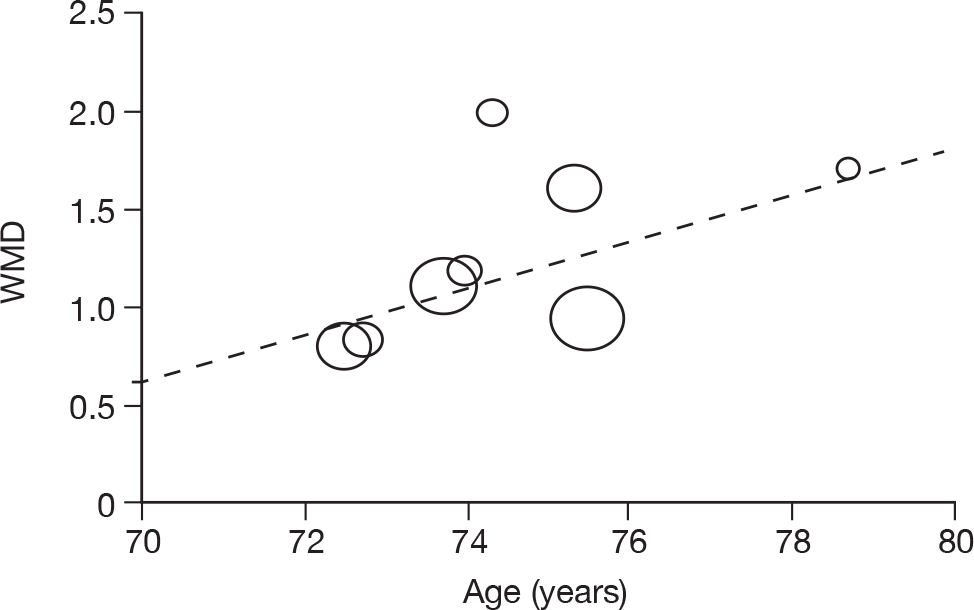
FIGURE 131.
Mini Mental State Examination at 12 weeks (mean change from baseline) – donepezil (all dosages) vs placebo: association of treatment effect with average baseline MMSE score of population. Area of circles is proportional to weight of each study in random-effects meta-analysis; dashed line shows univariate metaregression estimate (α = 2.743; β = –0.085; p = 0.227).
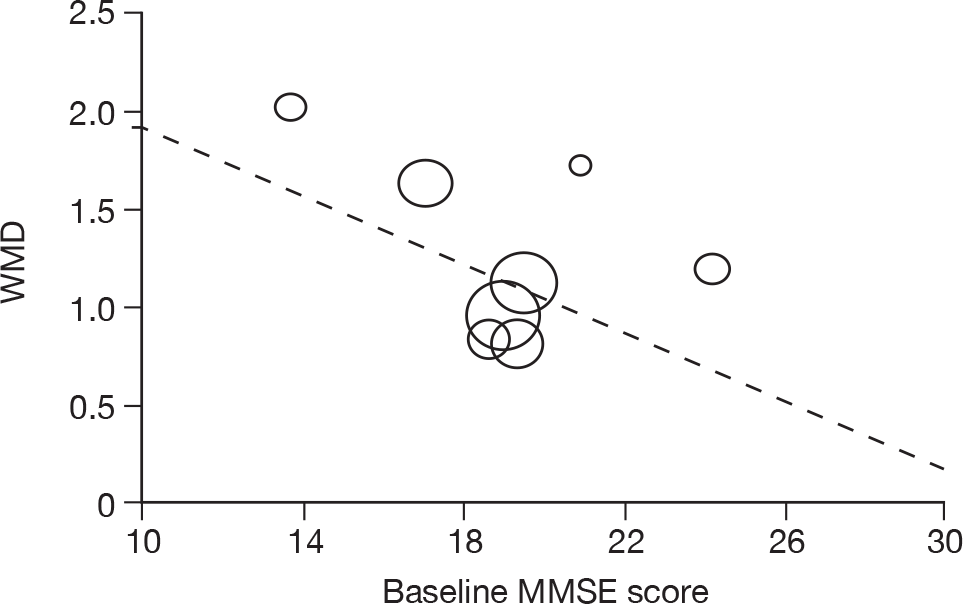
FIGURE 132.
Mini Mental State Examination at 12 weeks (mean change from baseline) – donepezil (all dosages) vs placebo: association of treatment effect with gender of population. Area of circles is proportional to weight of each study in random-effects meta-analysis; dashed line shows univariate metaregression estimate (α = 1.701; β = –1.463; p = 0.771).
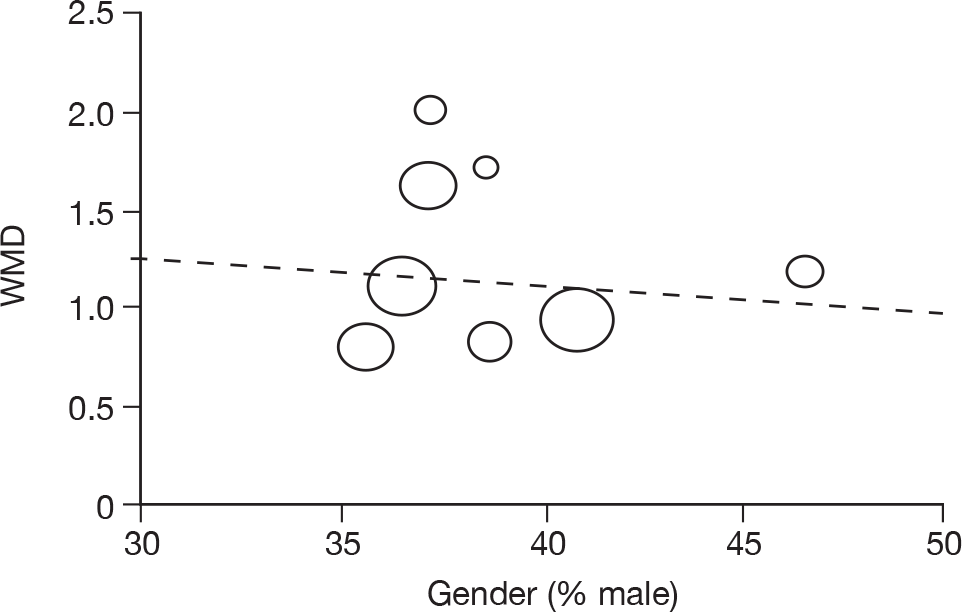
FIGURE 133.
Mini Mental State Examination at 24 weeks (mean change from baseline) – donepezil (all dosages) vs placebo: association of treatment effect with average age of population. Area of circles is proportional to weight of each study in random-effects meta-analysis; dashed line shows univariate metaregression estimate (α = 19.302; β = –0.244; p = 0.157).
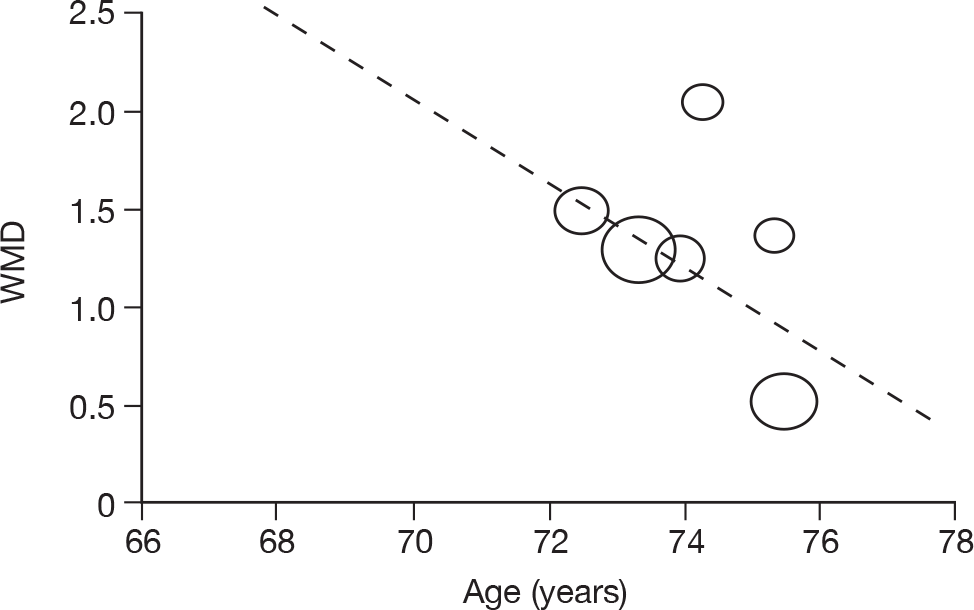
FIGURE 134.
Mini Mental State Examination at 24 weeks (mean change from baseline) – donepezil (all dosages) vs placebo: association of treatment effect with average baseline MMSE score of population. Area of circles is proportional to weight of each study in random-effects meta-analysis; dashed line shows univariate metaregression estimate (α = 2.489; β = –0.067; p = 0.373).
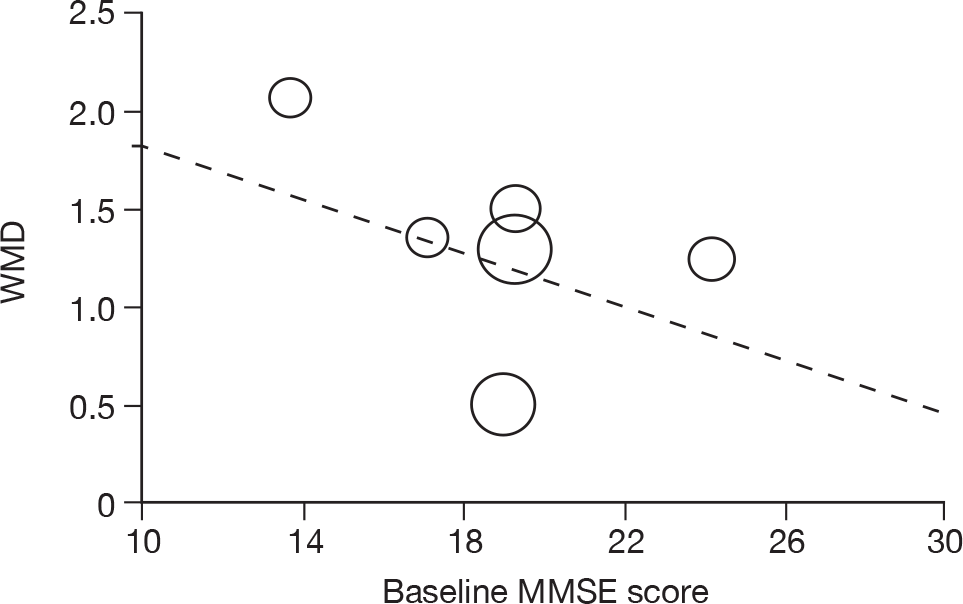
FIGURE 135.
Mini Mental State Examination at 12 weeks (mean change from baseline) – donepezil (all dosages) vs placebo: association of treatment effect with gender of population. Area of circles is proportional to weight of each study in random-effects meta-analysis; dashed line shows univariate metaregression estimate (α = 3.582; β = –6.066; p = 0.308).
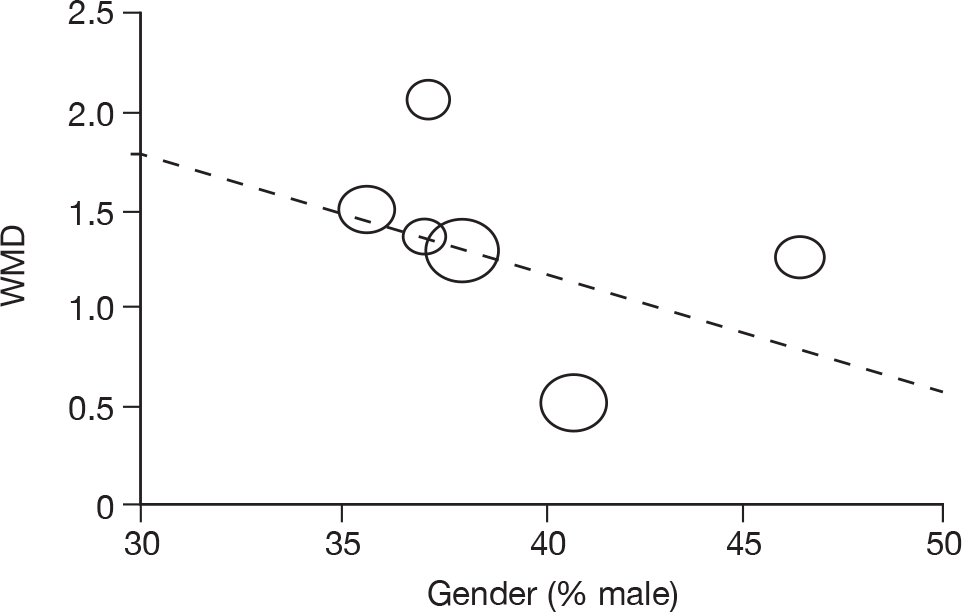
Pooled multiple outcomes
FIGURE 136.
Cognitive outcomes (SMD) at 24–26 weeks – donepezil (all dosages) vs placebo: association of standardised treatment effect with average age of population. Area of circles is proportional to weight of each study in random-effects meta-analysis; dashed line shows univariate metaregression estimate (α = –0.073; β = 0.006; p = 0.796).
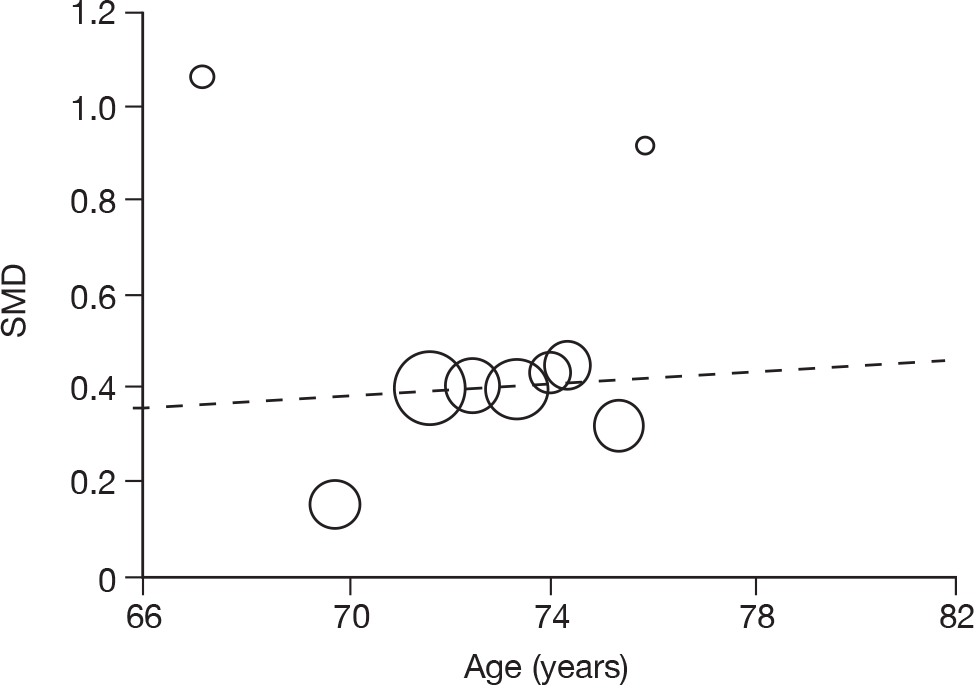
FIGURE 137.
Cognitive outcomes (SMD) at 24–26 weeks – donepezil (all dosages) vs placebo: association of standardised treatment effect with average baseline MMSE score of population. Area of circles is proportional to weight of each study in random-effects meta-analysis; dashed line shows univariate metaregression estimate (α = 0.229; β = 0.008; p = 0.668).
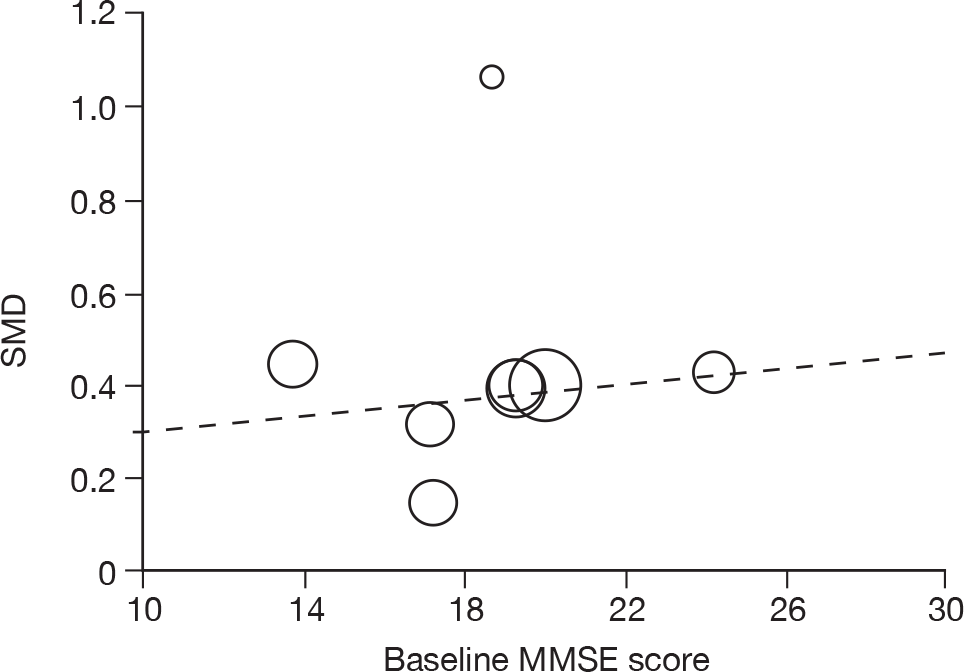
FIGURE 138.
Cognitive outcomes (SMD) at 24–26 weeks – donepezil (all dosages) vs placebo: association of standardised treatment effect with gender of population. Area of circles is proportional to weight of each study in random-effects meta-analysis; dashed line shows univariate metaregression estimate (α = –0.121; β = 1.307; p = 0.240).
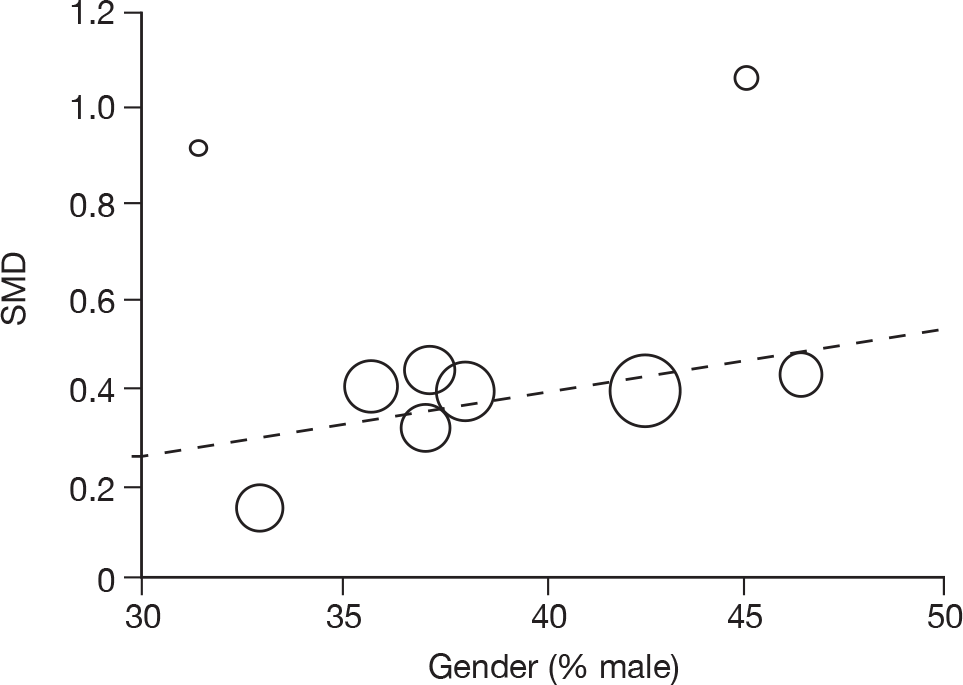
Donepezil versus placebo: functional
FIGURE 139.
Functional outcomes at 24 weeks – donepezil (all dosages) vs placebo: association of standardised treatment effect with average age of population. Area of circles is proportional to weight of each study in random-effects meta-analysis; dashed line shows univariate metaregression estimate (α = –1.593; β = 0.026; p = 0.552).
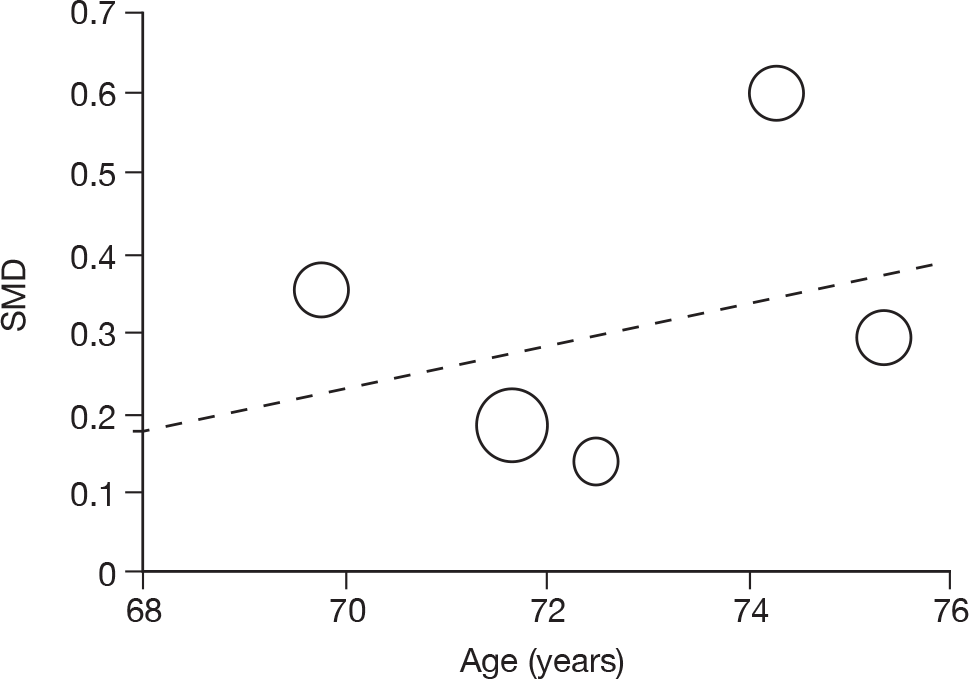
FIGURE 140.
Functional outcomes at 24 weeks – donepezil (all dosages) vs placebo: association of standardised treatment effect with average baseline MMSE score of population. Area of circles is proportional to weight of each study in random-effects meta-analysis; dashed line shows univariate metaregression estimate (α = 1.456; β = –0.065; p = 0.009).
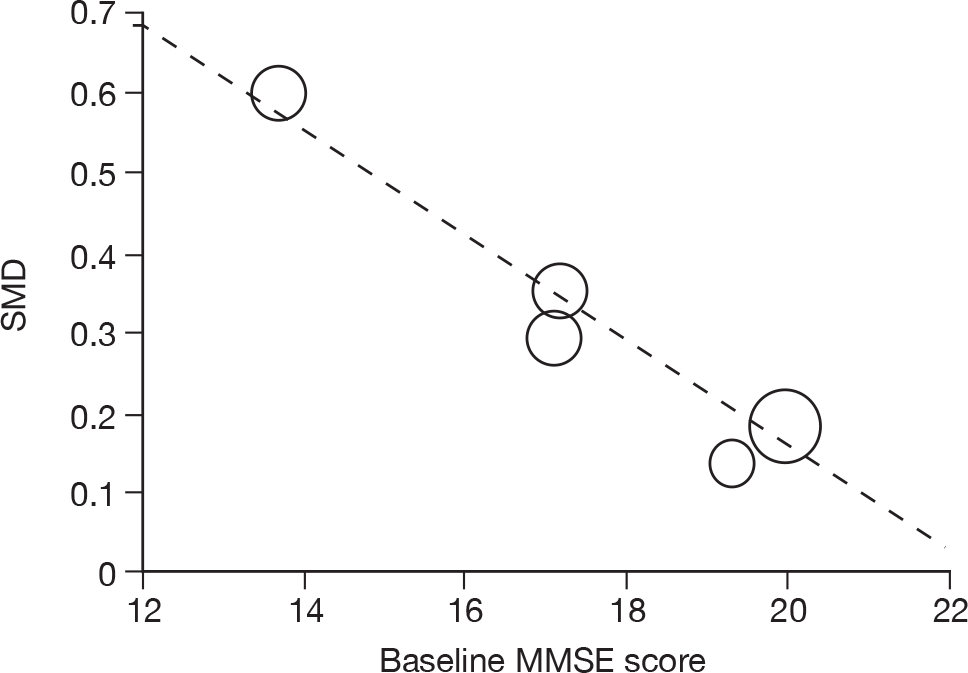
FIGURE 141.
Functional outcomes at 24 weeks – donepezil (all dosages) vs placebo: association of standardised treatment effect with gender of population. Area of circles is proportional to weight of each study in random-effects meta-analysis; dashed line shows univariate metaregression estimate (α = 0.932; β = –1.673; p = 0.435).
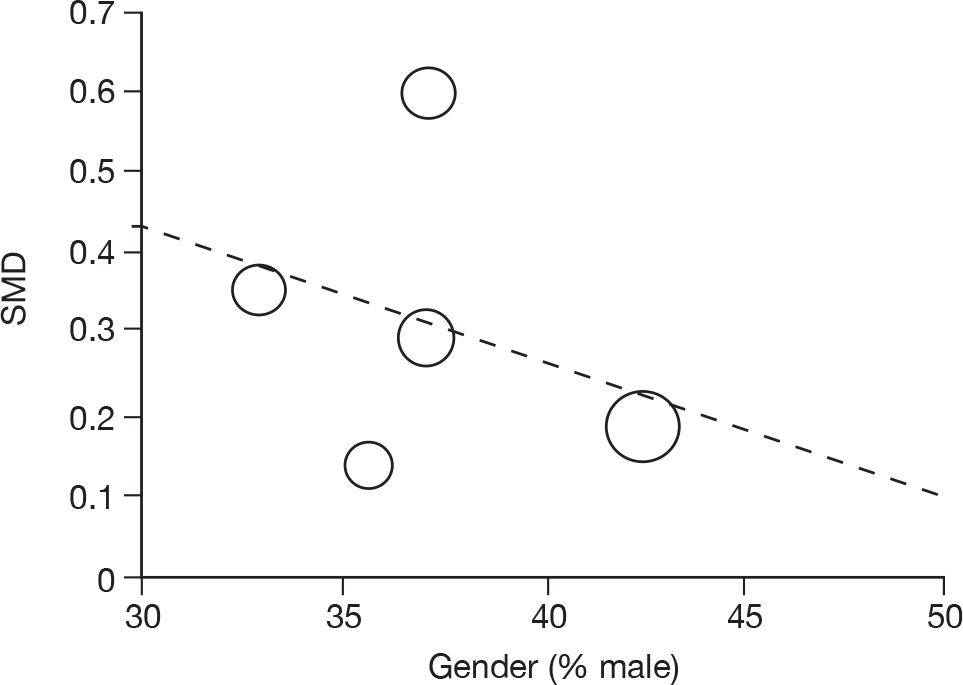
Donepezil versus placebo: global
FIGURE 142.
Global outcomes (SMD) at 24 weeks – donepezil (all dosages) vs placebo: association of standardised treatment effect with average age of population. Area of circles is proportional to weight of each study in random-effects meta-analysis; dashed line shows univariate metaregression estimate (α = 2.191; β = –0.025; p = 0.536).
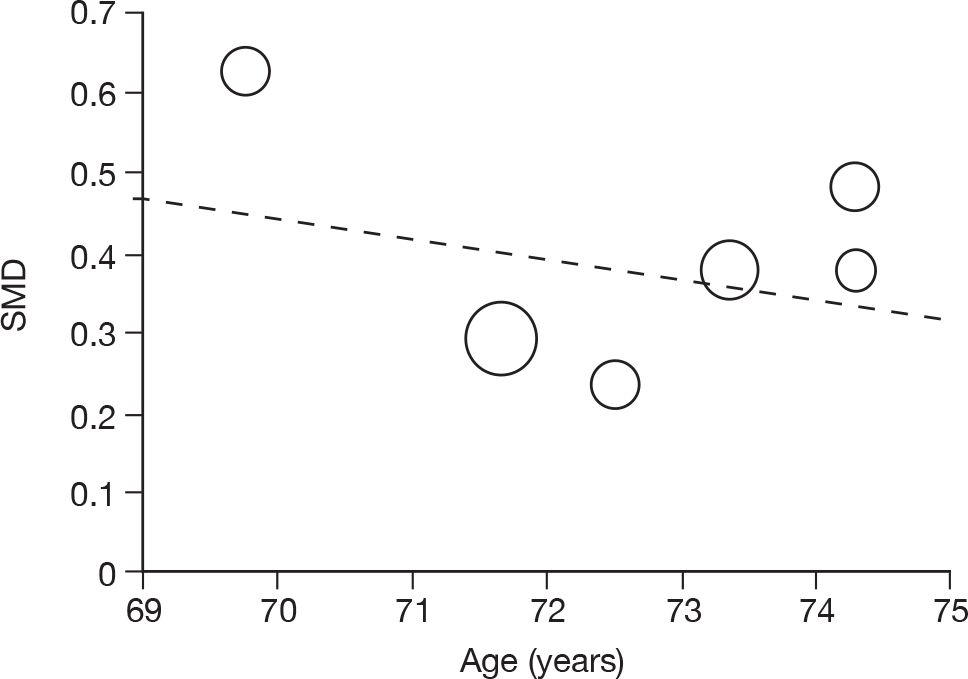
FIGURE 143.
Global outcomes (SMD) at 24 weeks – donepezil (all dosages) vs placebo: association of standardised treatment effect with average baseline MMSE score of population. Area of circles is proportional to weight of each study in random-effects meta-analysis; dashed line shows univariate metaregression estimate (α = 0.876; β = –0.028; p = 0.147).
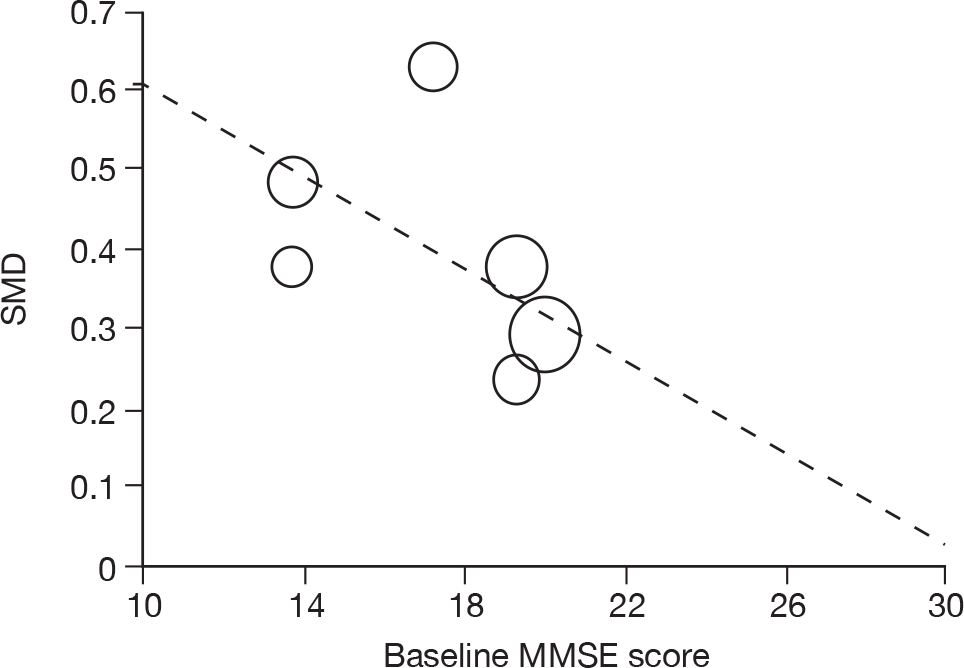
FIGURE 144.
Global outcomes (SMD) at 24 weeks – donepezil (all dosages) vs placebo: association of standardised treatment effect with gender of population. Area of circles is proportional to weight of each study in random-effects meta-analysis; dashed line shows univariate metaregression estimate (α = 1.277; β = –2.357; p = 0.082).
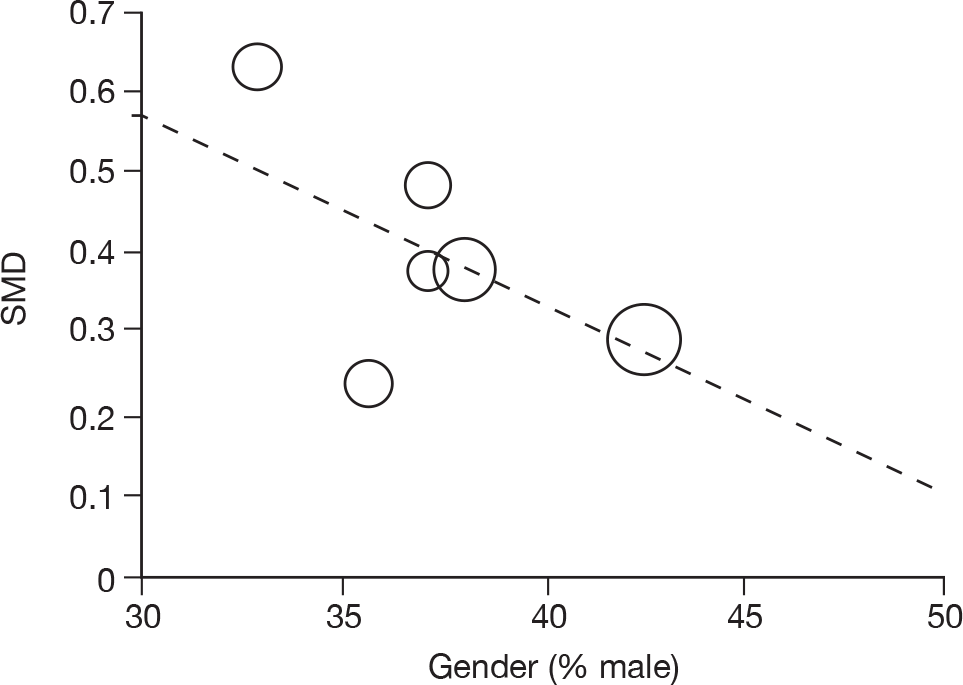
Galantamine versus placebo: cognitive
FIGURE 145.
Alzheimer’s Disease Assessment Scale – cognitive subscale at 12–16 weeks (mean change from baseline) – galantamine (all dosages) vs placebo: association of treatment effect with average age of population. Area of circles is proportional to weight of each study in random-effects meta-analysis; dashed line shows univariate metaregression estimate (α = –10.938; β = 0.114; p = 0.335).
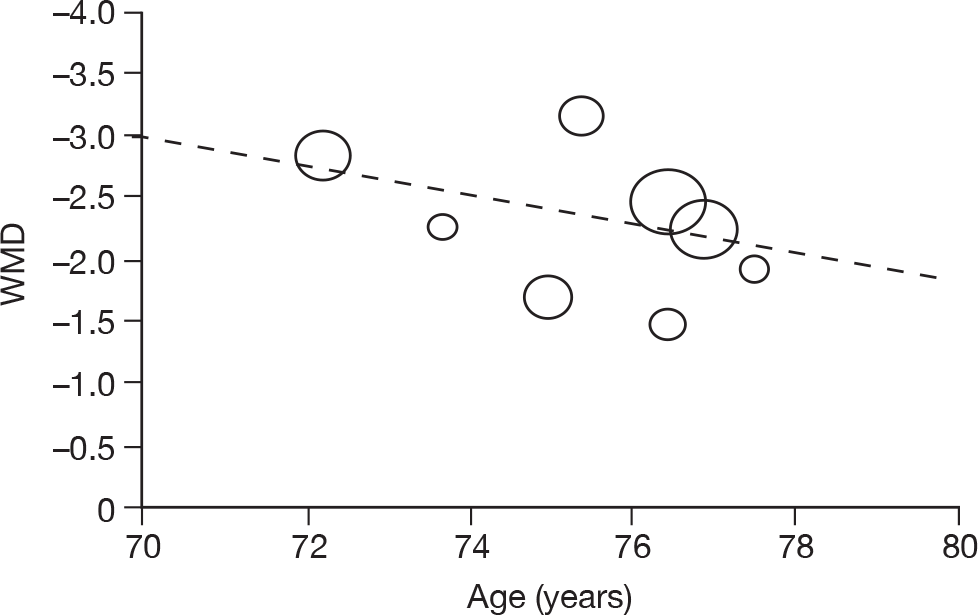
FIGURE 146.
Alzheimer’s Disease Assessment Scale – cognitive subscale at 12.16 weeks (mean change from baseline) – galantamine (all dosages) vs placebo: association of treatment effect with average baseline MMSE score of population. Area of circles is proportional to weight of each study in random-effects meta-analysis; dashed line shows univariate metaregression estimate (α = –4.851; β = 0.134; p = 0.529).
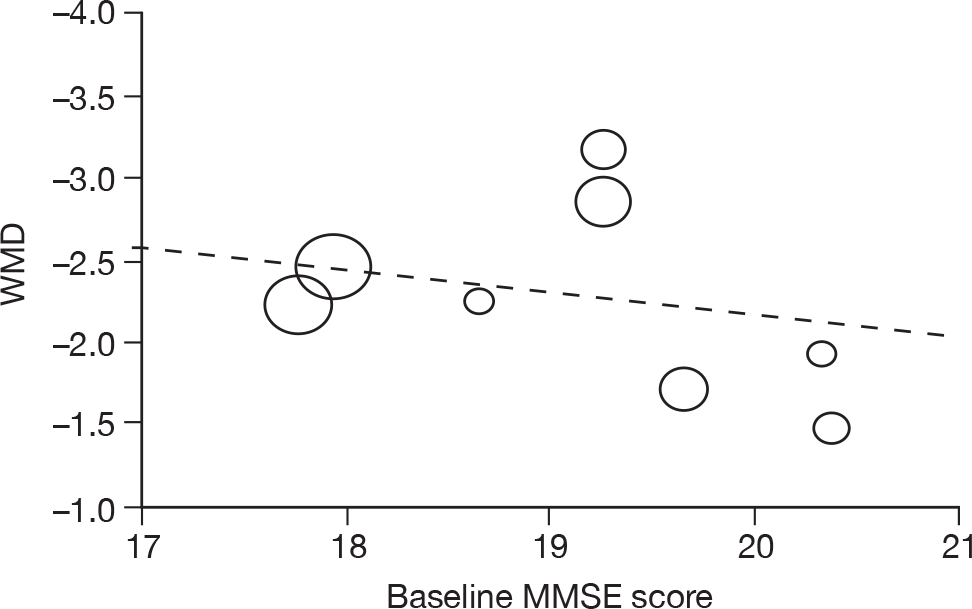
FIGURE 147.
Alzheimer’s Disease Assessment Scale – cognitive subscale at 12–16 weeks (mean change from baseline) – galantamine (all dosages) vs placebo: association of treatment effect with gender of population. Area of circles is proportional to weight of each study in random-effects meta-analysis; dashed line shows univariate metaregression estimate (α = –5.372; β = 7.845; p = 0.120).
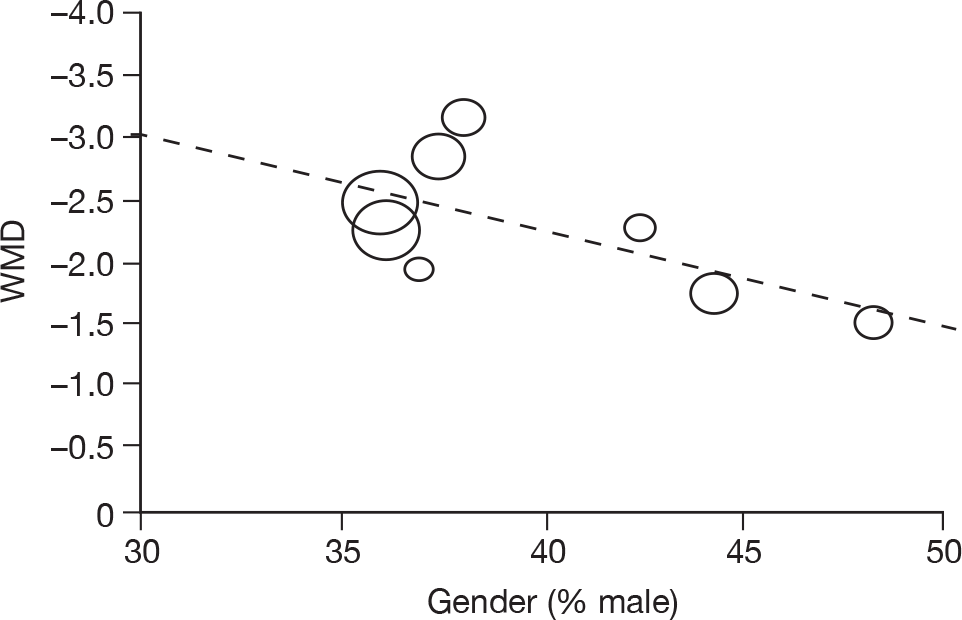
FIGURE 148.
Alzheimer’s Disease Assessment Scale – cognitive subscale at 21–26 weeks (mean change from baseline) – galantamine (all dosages) vs placebo: association of treatment effect with average age of population. Area of circles is proportional to weight of each study in random-effects meta-analysis; dashed line shows univariate metaregression estimate (α = –8.677; β = 0.076; p = 0.561).
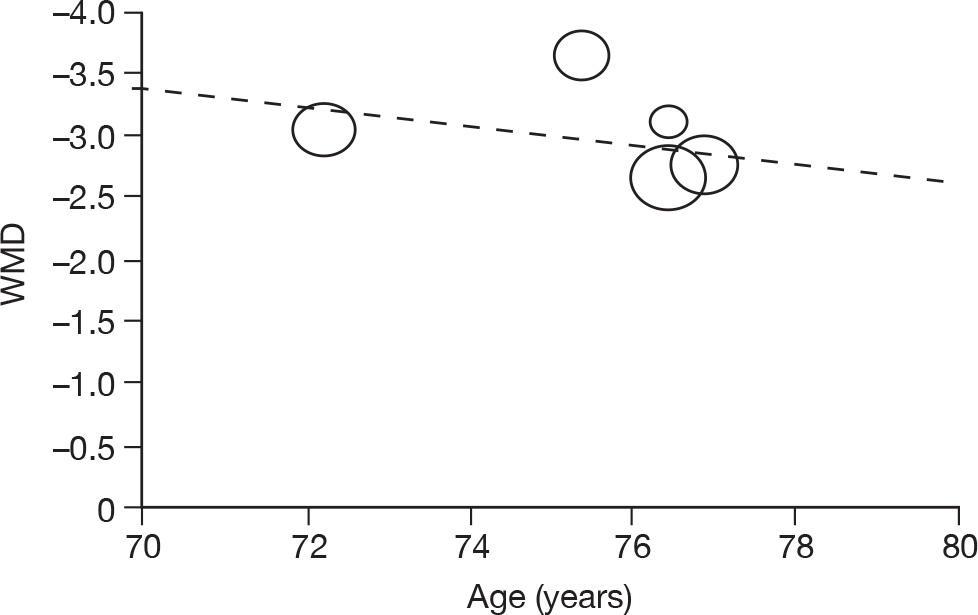
FIGURE 149.
Alzheimer’s Disease Assessment Scale – cognitive subscale at 21–26 weeks (mean change from baseline) – galantamine (all dosages) vs placebo: association of treatment effect with average baseline MMSE score of population. Area of circles is proportional to weight of each study in random-effects meta-analysis; dashed line shows univariate metaregression estimate (α = 2.623; β = –0.300; p = 0.251).
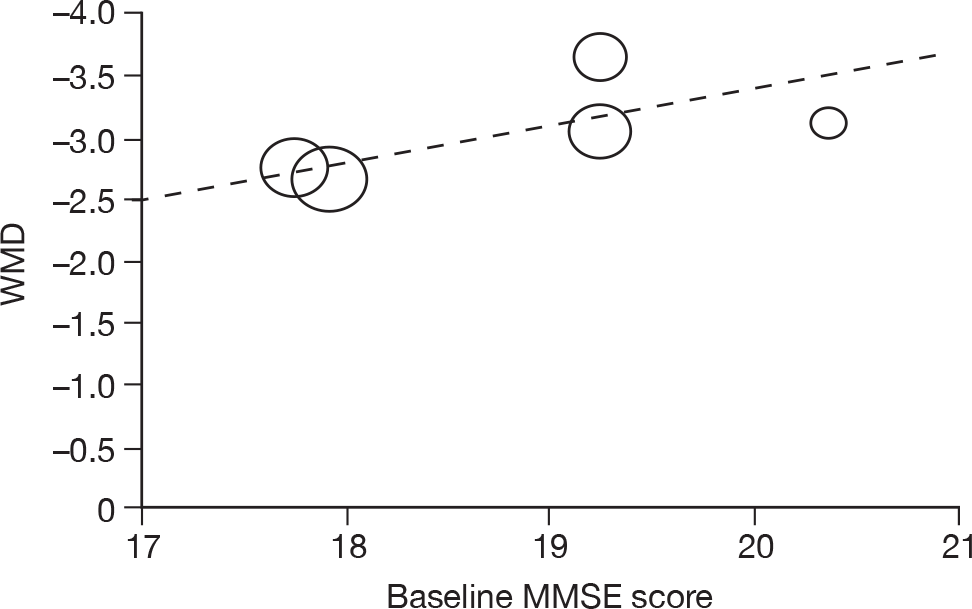
FIGURE 150.
Alzheimer’s Disease Assessment Scale – cognitive subscale at 21–26 weeks (mean change from baseline) – galantamine (all dosages) vs placebo: association of treatment effect with gender of population. Area of circles is proportional to weight of each study in random-effects meta-analysis; dashed line shows univariate metaregression estimate (α = –1.562; β = –3.725; p = 0.581).
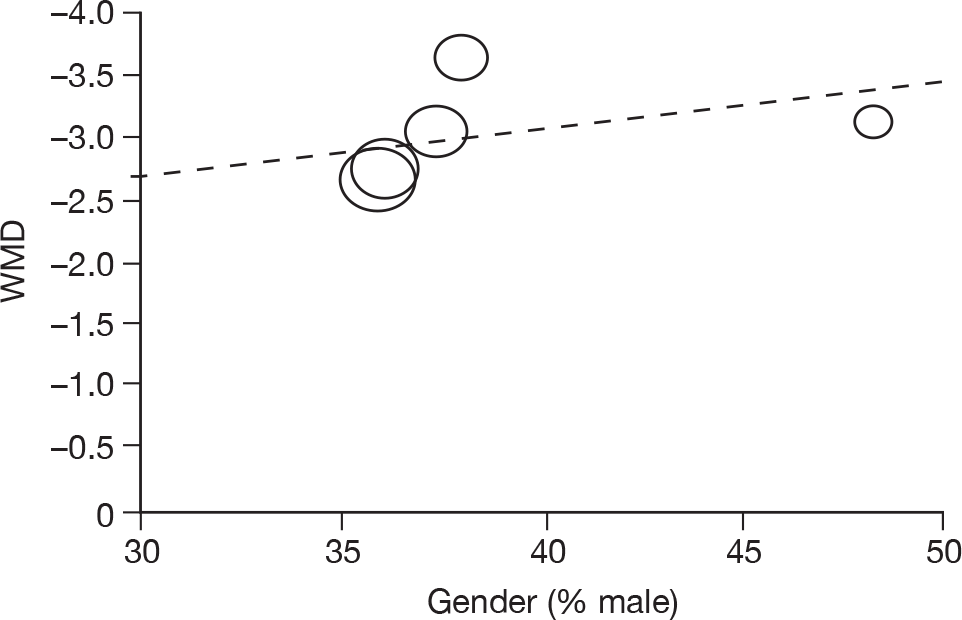
Appendix 8 WinBUGS code for mixed-treatment comparisons
Appendix 9 Mixed-treatment comparisons performed in specified measurement populations
Cognitive
Alzheimer’s Disease Assessment Scale – cognitive subscale
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Rogers et al. (1998)113 | –2.799 | –3.831 to –1.767 |
| Nunez et al. (2003)111,112 | –0.050 | –1.782 to 1.682 | ||
| Galantamine vs placebo | Rockwood et al. (2001)124 | –1.700 | –2.794 to –0.606 | |
| Wilkinson and Murray (2001)128 | –2.246 | –3.872 to –0.620 | ||
| Brodaty et al. (2005)96 | –2.453 | –3.192 to –1.713 | ||
| Rockwood et al. (2006)97 | –1.925 | –3.816 to –0.034 | ||
| Rivastigmine vs placebo | Jones et al. (2004)146 | –2.225 | –4.131 to –0.319 | |
| Donepezil vs galantamine | Winblad et al. (2007)140 | –0.911 | –1.817 to –0.006 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.000 |
| Donepezil | –2.350 | –3.887 to –0.684 | 0.995 | 0.681 |
| Galantamine | –1.840 | –2.951 to –0.489 | 0.995 | 0.212 |
| Rivastigmine | –0.901 | –3.390 to 1.573 | 0.814 | 0.107 |
| Memantine | – | – | – | – |
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Rogers et al. (1998)113 | –2.799 | –3.831 to –1.767 |
| Nunez et al. (2003)111,112 | –0.050 | –1.782 to 1.682 | ||
| Galantamine vs placebo | Rockwood et al. (2001)124 | –1.700 | –2.794 to –0.606 | |
| Wilkinson and Murray (2001)128 | –2.246 | –3.872 to –0.620 | ||
| Brodaty et al. (2005)96 | –2.453 | –3.192 to –1.713 | ||
| Rockwood et al. (2006)97 | –1.925 | –3.816 to –0.034 | ||
| Rivastigmine vs placebo | Feldman and Lane (2007)138 | –2.249 | –3.226 to –1.271 | |
| Winblad et al. (2007)140 | –0.911 | –1.817 to –0.006 | ||
| Donepezil vs galantamine | Jones et al. (2004)146 | –2.225 | –4.131 to –0.319 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.000 |
| Donepezil | –2.334 | –3.907 to –0.714 | 0.996 | 0.630 |
| Galantamine | –1.833 | –2.980 to –0.540 | 0.996 | 0.190 |
| Rivastigmine | –1.567 | –3.290 to 0.133 | 0.968 | 0.180 |
| Memantine | – | – | – | – |
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Burns et al. (1999)104 | –2.151 | –2.871 to –1.430 |
| Homma et al. (2000)108 | –2.175 | –3.527 to –0.823 | ||
| Nunez et al. (2003)111,112 | –0.570 | –2.497 to 1.357 | ||
| Galantamine vs placebo | Raskind et al. (2000)123 | –3.158 | –4.371 to –1.946 | |
| Tariot et al. (2000)125 | –2.225 | –3.042 to –1.408 | ||
| Wilcock et al. (2000)127 | –2.848 | –3.829 to –1.867 | ||
| Rockwood et al. (2001)124 | –1.900 | –3.037 to –0.763 | ||
| Bullock et al. (2004)101 | –1.475 | –2.933 to –0.017 | ||
| Brodaty et al. (2005)96 | –2.400 | –3.148 to –1.652 | ||
| Donepezil vs rivastigmine | Wilkinson et al. (2002)145 | 0.150 | –1.561 to 1.861 | |
| Donepezil vs galantamine | Jones et al. (2004)146 | –2.550 | –4.490 to –0.610 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.000 |
| Donepezil | –2.287 | –3.306 to –1.344 | 1.000 | 0.251 |
| Galantamine | –2.208 | –2.829 to –1.425 | 1.000 | 0.252 |
| Rivastigmine | –2.433 | –4.851 to –0.079 | 0.978 | 0.497 |
| Memantine | – | – | – | – |
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Rogers et al. (1998)113 | –2.684 | –3.876 to –1.491 |
| Burns et al. (1999)104 | –2.203 | –2.968 to –1.438 | ||
| Homma et al. (2000)108 | –2.540 | –3.427 to –1.653 | ||
| Galantamine vs placebo | Raskind et al. (2000)123 | –3.653 | –4.696 to –2.611 | |
| Tariot et al. (2000)125 | –2.741 | –3.633 to –1.850 | ||
| Wilcock et al. (2000)127 | –3.049 | –4.030 to –2.068 | ||
| Brodaty et al. (2005)96 | –2.651 | –3.449 to –1.854 | ||
| Rivastigmine vs placebo | Rosler et al. (1999)137 | –1.179 | –2.310 to –0.048 | |
| Feldman and Lane (2007)138 | –2.668 | –3.810 to –1.527 | ||
| Winblad et al. (2007)140 | –1.943 | –2.858 to –1.029 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.000 |
| Donepezil | –2.430 | –3.134 to –1.739 | 1.000 | 0.106 |
| Galantamine | –2.974 | –3.593 to –2.371 | 1.000 | 0.882 |
| Rivastigmine | –1.929 | –2.678 to –1.177 | 1.000 | 0.012 |
| Memantine | – | – | – | – |
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Rogers et al. (1998)113 | –2.684 | –3.876 to –1.491 |
| Burns et al. (1999)104 | –2.203 | –2.968 to –1.438 | ||
| Homma et al. (2000)108 | –2.540 | –3.427 to –1.653 | ||
| Galantamine vs placebo | Raskind et al. (2000)123 | –3.653 | –4.696 to –2.611 | |
| Tariot et al. (2000)125 | –2.741 | –3.633 to –1.850 | ||
| Wilcock et al. (2000)127 | –3.049 | –4.030 to –2.068 | ||
| Brodaty et al. (2005)96 | –2.651 | –3.449 to –1.854 | ||
| Rivastigmine vs placebo | Corey-Bloom et al. (1998)135 | –2.751 | –3.694 to –1.808 | |
| Rosler et al. (1999)137 | –0.785 | –1.851 to 0.281 | ||
| Feldman and Lane (2007)138 | –2.298 | –3.460 to –1.137 | ||
| Winblad et al. (2007)140 | –1.943 | –2.858 to –1.029 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.000 |
| Donepezil | –2.427 | –3.213 to –1.686 | 1.000 | 0.120 |
| Galantamine | –2.972 | –3.648 to –2.327 | 1.000 | 0.867 |
| Rivastigmine | –1.971 | –2.657 to –1.271 | 1.000 | 0.012 |
| Memantine | – | – | – | – |
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Burns et al. (1999)104 | –2.003 | –2.811 to –1.195 |
| Galantamine vs placebo | Raskind et al. (2000)123 | –3.853 | –5.129 to –2.577 | |
| Tariot et al. (2000)125 | –3.111 | –4.101 to –2.121 | ||
| Wilcock et al. (2000)127 | –3.594 | –4.679 to –2.508 | ||
| Bullock et al. (2004)101 | –3.100 | –4.620 to –1.580 | ||
| Brodaty et al. (2005)96 | –2.894 | –3.775 to –2.014 | ||
| Rivastigmine vs placebo | Corey-Bloom et al. (1998)135 | –3.189 | –4.280 to –2.098 | |
| Rosler et al. (1999)137 | –1.224 | –2.527 to 0.079 | ||
| Feldman and Lane (2007)138 | –2.118 | –3.338 to –0.898 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.000 |
| Donepezil | –2.002 | –3.502 to –0.518 | 0.991 | 0.048 |
| Galantamine | –3.267 | –4.027 to –2.546 | 1.000 | 0.913 |
| Rivastigmine | –2.267 | –3.221 to –1.245 | 1.000 | 0.039 |
| Memantine | – | – | – | – |
Mini Mental State Examination
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Rogers et al. (1998)113 | 1.110 | 0.514 to 1.706 |
| Nunez et al. (2003)111,112 | 0.830 | –0.071 to 1.731 | ||
| Holmes et al. (2004)107 | 1.700 | 0.169 to 3.231 | ||
| Donepezil vs galantamine | Jones et al. (2004)146 | 0.888 | 0.004 to 1.771 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.017 |
| Donepezil | 1.115 | 0.060 to 2.286 | 0.979 | 0.866 |
| Galantamine | 0.236 | –1.911 to 2.466 | 0.618 | 0.117 |
| Rivastigmine | – | – | – | – |
| Memantine | – | – | – | – |
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Rogers et al. (1998)113 | 1.110 | 0.514 to 1.706 |
| Nunez et al. (2003)111,112 | 0.830 | –0.071 to 1.731 | ||
| AD2000 (2004)103 | 0.930 | 0.389 to 1.471 | ||
| Holmes et al. (2004)107 | 1.700 | 0.169 to 3.231 | ||
| Donepezil vs galantamine | Jones et al. (2004)146 | 0.888 | 0.004 to 1.771 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.005 |
| Donepezil | 1.038 | 0.394 to 1.775 | 0.994 | 0.915 |
| Galantamine | 0.159 | –1.366 to 1.763 | 0.600 | 0.081 |
| Rivastigmine | – | – | – | – |
| Memantine | – | – | – | – |
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Mohs et al. (2001)110 | 1.600 | 0.889 to 2.311 |
| Winblad et al. (2001)116 | 0.800 | 0.075 to 1.525 | ||
| Gauthier et al. (2002)105 | 2.000 | 0.820 to 3.180 | ||
| Nunez et al. (2003)111,112 | 1.130 | 0.146 to 2.114 | ||
| Seltzer et al. (2004)115 | 1.175 | 0.100 to 2.250 | ||
| Rivastigmine vs placebo | Agid et al. (1998)134 | 0.144 | –0.493 to 0.782 | |
| Mowla et al. (2007)136 | 1.600 | 1.099 to 2.101 | ||
| Donepezil vs rivastigmine | Wilkinson et al. (2002)142 | –0.490 | –1.825 to 0.845 | |
| Donepezil vs galantamine | Jones et al. (2004)146 | 0.753 | –0.215 to 1.720 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.001 |
| Donepezil | 1.222 | 0.468 to 1.988 | 0.997 | 0.505 |
| Galantamine | 0.469 | –1.487 to 2.449 | 0.704 | 0.149 |
| Rivastigmine | 1.079 | 0.075 to 2.144 | 0.980 | 0.346 |
| Memantine | – | – | – | – |
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Rogers et al. (1998)113 | 1.284 | 0.604 to 1.964 |
| Gauthier et al. (2002)105 | 2.060 | 0.880 to 3.240 | ||
| Seltzer et al. (2004)115 | 1.250 | 0.171 to 2.329 | ||
| Rivastigmine vs placebo | Feldman and Lane (2007)138 | 1.407 | 0.809 to 2.006 | |
| Winblad et al. (2007)140 | 0.932 | 0.461 to 1.403 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.001 |
| Donepezil | 1.460 | 0.581 to 2.420 | 0.995 | 0.741 |
| Galantamine | – | – | – | – |
| Rivastigmine | 1.137 | 0.152 to 2.160 | 0.982 | 0.258 |
| Memantine | – | – | – | – |
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
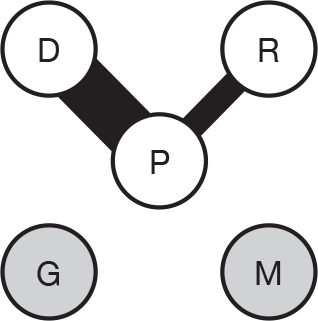
|
Donepezil vs placebo | Rogers et al. (1998)113 | 1.284 | 0.604 to 1.964 |
| Gauthier et al. (2002)105 | 2.060 | 0.880 to 3.240 | ||
| AD2000 (2004)103 | 0.500 | –0.250 to 1.250 | ||
| Seltzer et al. (2004)115 | 1.250 | 0.171 to 2.329 | ||
| Mazza et al. (2006)118 | 1.450 | –3.720 to 6.620 | ||
| Rivastigmine vs placebo | Feldman and Lane (2007)138 | 1.250 | 0.670 to 1.830 | |
| Winblad et al. (2007)140 | 0.932 | 0.461 to 1.403 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.001 |
| Donepezil | 1.169 | 0.476 to 1.978 | 0.996 | 0.582 |
| Galantamine | – | – | – | – |
| Rivastigmine | 1.076 | 0.102 to 2.059 | 0.981 | 0.418 |
| Memantine | – | – | – | – |
| Evidence Network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
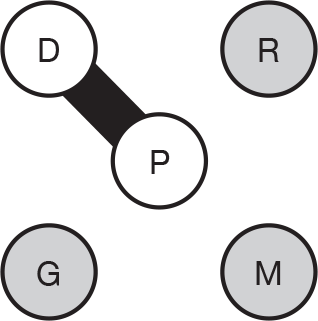
|
Donepezil vs placebo | Mohs et al. (2001)110 | 1.350 | 0.188 to 2.512 |
| Winblad et al. (2001)116 | 1.490 | 0.548 to 2.432 | ||
| Gauthier et al. (2002)105 | 2.000 | 0.787 to 3.213 | ||
| Seltzer et al. (2004)115 | 1.200 | –0.086 to 2.486 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.003 |
| Donepezil | 1.507 | 0.637 to 2.371 | 0.997 | 0.997 |
| Galantamine | – | – | – | – |
| Rivastigmine | – | – | – | – |
| Memantine | – | – | – | – |
Severe Impairment Battery
| Evidence network | Comparison | Pair-wise meta-analysis | Study | WMD | 95% CI |
|---|---|---|---|---|---|
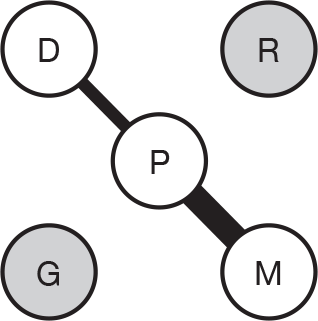
|
Donepezil vs placebo | – | Gauthier et al. (2002)105 | 3.900 | 1.474 to 6.326 |
| Memantine vs placebo | Reisberg et al. (2003)142 | 6.200 | 3.138 to 9.262 | ||
| Van Dyck et al. (2007)143 | 2.475 | 0.497 to 4.453 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.000 |
| Donepezil | 3.884 | 0.343 to 7.414 | 0.983 | 0.506 |
| Galantamine | – | – | – | – |
| Rivastigmine | – | – | – | – |
| Memantine | 3.849 | 1.416 to 6.509 | 0.998 | 0.494 |
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
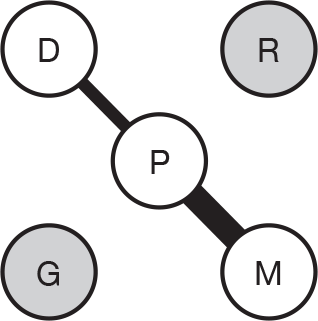
|
Donepezil vs placebo | Gauthier et al. (2002)105 | 4.425 | 1.341 to 7.509 |
| Memantine vs placebo | Reisberg et al. (2003)142 | 6.100 | 2.989 to 9.211 | |
| Van Dyck et al. (2007)143 | 0.500 | –2.272 to 3.272 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.001 |
| Donepezil | 4.420 | 0.268 to 8.572 | 0.981 | 0.701 |
| Galantamine | – | – | – | – |
| Rivastigmine | – | – | – | – |
| Memantine | 3.104 | 0.263 to 5.985 | 0.983 | 0.298 |
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Gauthier et al. (2002)105 | 5.325 | 1.895 to 8.755 |
| Memantine vs placebo | Reisberg et al. (2003)142 | 5.700 | 2.137 to 9.263 | |
| Van Dyck et al. (2007)143 | 0.600 | –2.591 to 3.791 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.000 |
| Donepezil | 5.327 | 1.061 to 9.583 | 0.992 | 0.821 |
| Galantamine | – | – | – | – |
| Rivastigmine | – | – | – | – |
| Memantine | 2.949 | –0.041 to 5.957 | 0.974 | 0.179 |
Behavioural
Neuropsychiatric Inventory
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Gauthier et al. (2002)105 | –2.900 | –6.783 to 0.983 |
| Nunez et al. (2003)111,112 | –3.160 | –5.947 to –0.373 | ||
| Galantamine vs placebo | Tariot et al. (2000)125 | –0.719 | –2.056 to 0.618 | |
| Rockwood et al. (2001)124 | –0.700 | –2.675 to 1.275 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.003 |
| Donepezil | –3.073 | –5.678 to –0.458 | 0.988 | 0.931 |
| Galantamine | –0.713 | –2.525 to 1.079 | 0.815 | 0.066 |
| Rivastigmine | – | – | – | – |
| Memantine | – | – | – | – |
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Nunez et al. (2003)111,112 | –2.870 | –5.406 to –0.334 |
| AD2000 (2004)103 | 1.250 | 1.500 to 4.000 | ||
| Holmes et al. (2004)107 | –6.200 | –11.374 to –1.026 | ||
| Galantamine vs placebo | Rockwood et al. (2001)124 | –0.900 | –2.688 to 0.888 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.020 |
| Donepezil | –1.780 | –4.299 to 0.602 | 0.930 | 0.663 |
| Galantamine | –0.886 | –4.237 to 2.413 | 0.720 | 0.316 |
| Rivastigmine | – | – | – | – |
| Memantine | – | – | – | – |
Global
Clinician’s Interview-based Impression of Change plus Caregiver Input
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Rogers et al. (1998)93 | –0.350 | –0.527 to –0.174 |
| Galantamine vs placebo | Rockwood et al. (2001)124 | –0.335 | –0.524 to –0.146 | |
| Rockwood et al. (2006)97 | –0.450 | –0.797 to –0.103 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.047 |
| Donepezil | –0.352 | –2.125 to 1.417 | 0.808 | 0.458 |
| Galantamine | –0.374 | –1.663 to 0.866 | 0.863 | 0.496 |
| Rivastigmine | – | – | – | – |
| Memantine | – | – | – | – |
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Burns et al. (1999)104 | –0.265 | –0.406 to –0.125 |
| Gauthier et al. (2002)105 | –0.490 | –0.768 to –0.212 | ||
| Galantamine vs placebo | Rockwood et al. (2001)124 | –0.367 | –0.582 to –0.152 | |
| Rivastigmine vs placebo | Rosler et al. (1999)137 | –0.007 | –0.186 to 0.172 | |
| Memantine vs placebo | Reisberg et al. (2003)142 | –0.070 | –0.347 to 0.20 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.013 |
| Donepezil | –0.351 | –1.697 to 0.934 | 0.843 | 0.330 |
| Galantamine | –0.369 | –2.249 to 1.522 | 0.791 | 0.403 |
| Rivastigmine | –0.007 | –1.871 to 1.890 | 0.510 | 0.113 |
| Memantine | –0.072 | –1.958 to 1.808 | 0.578 | 0.142 |
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Rogers et al. (1998)113 | –0.400 | –0.593 to –0.207 |
| Burns et al. (1999)104 | –0.340 | –0.484 to –0.196 | ||
| Gauthier et al. (2002)105 | –0.545 | –0.858 to –0.232 | ||
| Galantamine vs placebo | Raskind et al. (2000)123 | –0.248 | –0.419 to –0.077 | |
| Wilcock et al. (2000)127 | –0.288 | –0.450 to –0.127 | ||
| Brodaty et al. (2005)96 | –0.138 | –0.294 to 0.018 | ||
| Rivastigmine vs placebo | Rosler et al. (1999)137 | –0.284 | –0.538 to –0.030 | |
| Feldman and Lane (2007)138 | –0.502 | –0.704 to –0.300 | ||
| Memantine vs placebo | Reisberg et al. (2003)142 | –0.300 | –0.582 to –0.018 | |
| Van Dyck et al. (2007)143 | –0.300 | –0.515 to –0.085 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.000 |
| Donepezil | –0.393 | –0.558 to –0.247 | 1.000 | 0.367 |
| Galantamine | –0.223 | –0.364 to –0.086 | 0.995 | 0.008 |
| Rivastigmine | –0.414 | –0.611 to –0.205 | 0.999 | 0.514 |
| Memantine | –0.300 | –0.518 to –0.086 | 0.994 | 0.111 |
| Evidence Network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Rogers et al. (1998)113 | –0.400 | –0.593 to –0.207 |
| Burns et al. (1999)104 | –0.340 | –0.484 to –0.196 | ||
| Gauthier et al. (2002)105 | –0.545 | –0.858 to –0.232 | ||
| Galantamine vs placebo | Raskind et al. (2000)123 | –0.248 | –0.419 to –0.077 | |
| Wilcock et al. (2000)127 | –0.288 | –0.450 to –0.127 | ||
| Brodaty et al. (2005)96 | –0.138 | –0.294 to 0.018 | ||
| Rivastigmine vs placebo | Corey–Bloom et al. (1998)135 | –0.275 | –0.471 to –0.079 | |
| Rosler et al. (1999)137 | –0.300 | –0.519 to –0.081 | ||
| Feldman and Lane (2007)138 | –0.500 | –0.711 to –0.289 | ||
| Memantine vs placebo | Reisberg et al. (2003)142 | –0.300 | –0.582 to –0.018 | |
| Van Dyck et al. (2007)143 | –0.300 | –0.515 to –0.085 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.000 |
| Donepezil | –0.392 | –0.549 to –0.251 | 1.000 | 0.546 |
| Galantamine | –0.222 | –0.356 to –0.091 | 0.997 | 0.010 |
| Rivastigmine | –0.354 | –0.508 to –0.203 | 1.000 | 0.285 |
| Memantine | –0.300 | –0.507 to –0.100 | 0.996 | 0.159 |
| Evidence network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Burns et al. (1999)104 | –0.335 | –0.497 to –0.174 |
| Gauthier et al. (2002)105 | –0.450 | –0.803 to –0.097 | ||
| Galantamine vs placebo | Raskind et al. (2000)123 | –0.281 | –0.480 to –0.082 | |
| Wilcock et al. (2000)127 | –0.407 | –0.592 to –0.223 | ||
| Brodaty et al. (2005)96 | –0.156 | –0.327 to 0.016 | ||
| Rivastigmine vs placebo | Corey–Bloom et al. (1998)135 | –0.333 | –0.547 to –0.119 | |
| Rosler et al. (1999)137 | –0.259 | –0.558 to 0.040 | ||
| Feldman and Lane (2007)138 | –0.403 | –0.620 to –0.186 | ||
| Memantine vs placebo | Reisberg et al. (2003)142 | –0.300 | –0.629 to 0.029 | |
| Van Dyck et al. (2007)143 | –0.300 | –0.555 to –0.045 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.000 |
| Donepezil | –0.363 | –0.593 to –0.151 | 0.997 | 0.413 |
| Galantamine | –0.277 | –0.439 to –0.118 | 0.997 | 0.077 |
| Rivastigmine | –0.341 | –0.523 to –0.157 | 0.998 | 0.293 |
| Memantine | –0.300 | –0.556 to –0.048 | 0.988 | 0.218 |
Global Deterioration Scale
| Evidence Network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Rivastigmine vs placebo | Corey-Bloom et al. (1998)135 | 0.175 | 0.065 to 0.285 |
| Rosler et al. (1999)137 | 0.120 | –0.042 to 0.282 | ||
| Feldman and Lane (2007)138 | 0.200 | 0.087 to 0.312 | ||
| Memantine vs placebo | Reisberg et al. (2003)142 | –0.100 | –0.220 to 0.020 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.034 |
| Donepezil | – | – | – | – |
| Galantamine | – | – | – | – |
| Rivastigmine | 0.171 | –0.145 to 0.471 | 0.943 | 0.901 |
| Memantine | –0.101 | –0.638 to 0.434 | 0.187 | 0.065 |
| Evidence Network | Comparison | Study | WMD | 95% CI |
|---|---|---|---|---|
|
Donepezil vs placebo | Winblad et al. (2001)116 | 0.160 | –0.006 to 0.326 |
| Rivastigmine vs placebo | Corey-Bloom et al. (1998)135 | 0.184 | 0.068 to 0.301 | |
| Memantine vs Placebo | Reisberg et al. (2003)142 | –0.100 | –0.242 to 0.042 |
| Technology | Vs placebo | Probability most effective | ||
|---|---|---|---|---|
| Effect | 95% CI | Probability more effective than placebo | ||
| Placebo | – | – | – | 0.087 |
| Donepezil | 0.159 | –2.347 to 2.677 | 0.608 | 0.347 |
| Galantamine | – | – | – | – |
| Rivastigmine | 0.181 | –2.344 to 2.690 | 0.623 | 0.367 |
| Memantine | –0.101 | –2.607 to 2.420 | 0.424 | 0.199 |
Appendix 10 Studies included by industry, but excluded from the PenTAG clinical effectiveness systematic review
Eisai/Pfizer submission
| Studies included in their systematic review | Reason for exclusion from PenTAG systematic review |
|---|---|
| Bentham R, Gray J, Raftery R, Hills E, Sellwood C, Courtney et al. and A.D.C. Grp. Long-term donepezil treatment in 565 patients with Alzheimer’s disease (AD2000): randomised double-blind trial. Lancet 2004;363:2105–15 | Included in the previous review |
| Burns A, Gauthier S, Perdomo C. Efficacy and safety of donepezil over 3 years: an open-label, multicentre study in patients with Alzheimer’s disease. Int J Geriatr Psychiatry 2007;22:806–12 | Secondary study to studies included in the 2004 review |
| Cummings JL, McRae T, Zhang R. Effects of donepezil on neuropsychiatric symptoms in patients with dementia and severe behavioral disorders. Am J Geriatr Psychiatry 2006;14:605–12 | Observational |
| Feldman HH, Schmitt FA, Olin JT. Activities of daily living in moderate-to-severe Alzheimer disease: An analysis of the treatment effects of memantine in patients receiving stable donepezil treatment. Alzheimer Dis Assoc Disord 2006;20:263–8 | Secondary study to studies included in the 2004 review |
| Gasper MC, Ott BR. Is donepezil therapy associated with reduced mortality in nursing home residents with dementia? Am J Geriatr Pharmacother 2005;3:1–7 | Observational |
| Persson CM, Wallin AK, Levander S, Minthon L, Persson CM, Wallin AK et al. Changes in cognitive domains during three years in patients with Alzheimer’s disease treated with donepezil. BMC Neurology 2009;9:7 | Observational |
| Riepe MW, Kohler J, Horn R. Donepezil in Alzheimer’s disease: a clinical observational study evaluating individual treatment response. Curr Med Res Opin 2007;23:1829–35 | Observational |
| Schmitt FA, Van%%Dyck CH, Wichems CH, Olin JT. Cognitive response to memantine in moderate to severe Alzheimer disease patients already receiving donepezil: An exploratory reanalysis. Alzheimer Dis Assoc Disord 2006;20:255–62 | Secondary study to studies included in the 2004 review |
| Tariot PN, Farlow MR, Grossberg GT, Graham SM, McDonald S, Gergel I. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomised controlled trial. JAMA 2004;291:317–24 | Included in the previous review |
| Wallin AK, Andreasen N, Eriksson S, Batsman S, Nasman B, Ekdahl A, et al. and Swedish Alzheimer Treatment Study Group. Donepezil in Alzheimer’s disease: what to expect after 3 years of treatment in a routine clinical setting. Dement Geriatr Cogn Disord 2007;23:150–60 | Observational |
| Wimo A, Winblad B, Shah SN, Chin W, Zhang R, McRae T. Impact of donepezil treatment for Alzheimer’s disease on caregiver time. Curr Med Res Opin 2004;20:1221–5 | Secondary study to studies included in the 2004 review |
Lundbeck submission
| Study ID: Lundbeck | Studies included in their systematic review | Reason for exclusion from PenTAG systematic review |
|---|---|---|
| 10158 | A randomised, double-blind, parallel group study examining the efficacy and safety of memantine on behavioural symptoms in patients with moderate to severe dementia of the Alzheimer’s type | Ongoing study |
| 10252 | Open-label extension to study 10158 (effect of memantine on behavioral symptoms in patients with moderate to severe dementia of the Alzheimer’s type) | Observational |
| 10112 | A 1-year multicentre, double-blind placebo-controlled study to evaluate the disease-modifying effects of memantine in patients with AD of moderate severity | Poster presentation |
| 10116 | A randomised, double-blind, placebo controlled evaluation of the efficacy and tolerability of memantine in Chinese patients with dementia of Alzheimer’s type (including extension) | Not English language (Chinese) |
| 10113 | A randomised, double-blind study to evaluate the safety and tolerability of once-daily vs twice-daily memantine treatment in patients with moderate to severe dementia of the Alzheimer’s type | No relevant comparators |
| 10114 | Evaluation of the safety and tolerability of randomised, double-blind switching of treatment from donepezil to memantine in patients with moderate to severe dementia of the Alzheimer’s type | Commentary |
| 99679 | A randomised, double-blind, placebo-controlled evaluation of the efficacy and safety of memantine in patients with mild to moderate dementia of the Alzheimer’s type | Wrong population – mild |
| 99819 | A long-term open-label extension study evaluating the safety and tolerability of memantine in patients with mild to moderate dementia of the Alzheimer’s type. Extension of 99679 | |
| 99817 | A double-blind, randomised, placebo-controlled study to evaluate the safety and efficacy of memantine in patients with dementia of the Alzheimer’s type | Conference abstract |
| Asubio MA-2101 | Late phase II clinical study of sun Y7017 (memantine hydrochloride) in patients with moderately severe to severe dementia of the Alzheimer’s type – evaluation of recommended dose and long-term safety (extension study for dose-finding and long term safety): double-blind period | Poster presentation |
| (CiC information has been removed) | (CiC information has been removed) | Unpublished study prior to 2004 |
| (CiC information has been removed) | (CiC information has been removed) | Unpublished open-label extension study |
| (CiC information has been removed) | (CiC information has been removed) | Unpublished Japanese study |
| (CiC information has been removed) | (CiC information has been removed) | Wrong population – mild |
| (CiC information has been removed) | (CiC information has been removed) | Observational |
| MRZ 90001–0608 | Prospective, open-label, single-arm, multicentre study to investigate the efficacy and safety of the once-daily (OD) memantine treatment | Observational |
| MRZ 90001–0716 | Prospective, single-arm, multicentre, open-label study to investigate the potential to reduce concomitant antipsychotics use in patients with moderate dementia of Alzheimer’s type (DAT) treated with memantine | Observational |
| MRZ 90001–9605/1 | Efficacy and long term tolerability of memantine in patients with moderately severe-to-severe AD | Included in the previous review |
| MRZ 90001–9605/2 | A randomised, placebo-controlled study of memantine in patients with moderate-to-severe AD. Phase III open-label extension | Observational |
| MRZ 90001-AD-3001 | Open-label, single-arm, multi-centre validation study of the ROSA-Scale (Relevant Outcome Scale for Alzheimer Patients) in patients with dementia of Alzheimer’s type (DAT) treated with memantine over a 3 months’ period | Observational |
| MRZ 9403 | Efficacy and long-term tolerability of memantine in care-dependent patients with moderately severe-to-severe primary dementia | Excluded from previous review due to population |
| MRZ 9104 | Multicentre, randomised double blind, comparative study of the efficacy and tolerabilty of akatinol memantine and placebo in patients suffering from senile dementia, Alzheimer type | No publications, date 1999 |
| Forest MEM-MD-02 | A randomised, double-blind, placebo-controlled evaluation of the safety and efficacy of memantine in patients with moderate to severe dementia of the Alzheimer’s type (on ≥ 6 months’ Aricept therapy) | Included in previous review |
| Forest MEM-MD-03 A/B | A long-term extension study evaluating the safety and tolerability of four memantine dosing regimens in patients with moderate to severe dementia of the Alzheimer’s type. Extension of MEM-MD-01 and MEM-MD-02 Phase A/B = 4 weeks double blind + 24 weeks open | Observational |
| Forest MEM-MD-03 C | Extension of MEM-MD-01 and MEM-MD-02. Phase C = 52 weeks open | Observational |
| Forest MEM-MD-03 D | Extension of MEM-MD-01 and MEM-MD-02. Phase D = open continuation until memantine is commercially available | Observational |
| Forest MEM-MD-10 | A randomised, double-blind, placebo-controlled evaluation of the safety and efficacy of memantine in patients with mild to moderate dementia of the Alzheimer’s type (monotherapy) | Population – mild Alzheimer’s |
| Forest MEM-MD-11 A/B | A long-term extension study evaluating the safety and tolerability of b.i.d. and q.d. administration of memantine in patients with mild to moderate dementia of the Alzheimer’s type. Extension of MEM-MD-10. Phase A/B = 8 weeks double blind + 20 weeks open | |
| Forest MEM-MD-11 C | Extension of MEM-MD-10. Phase C = 52 weeks open | Observational |
| Forest MEM-MD-11 D | Extension of MEM-MD-10. Phase D = open continuation until memantine is commercially available | Observational |
| Forest MEM-MD-12 A | Open extension of MEM-MD-12: 28 weeks | Observational |
| Forest MEM-MD-12 B | Open extension of MEM-MD-12 A: continuation until memantine is commercially available | Observational |
| Forest MEM-MD-22 | A randomised, double-blind, placebo-controlled evaluation of the safety and efficacy of namenda in nursing home patients with moderate-to-severe AD | Summary |
| Forest MEM-MD-23 | A randomised, double-blind, placebo controlled evaluation of the safety and efficacy of memantine in patients with moderate-to-severe AD with behavioral disturbances | Summary |
| Forest MEM-MD-71 | A randomised, double-blind, placebo-controlled evaluation of the effectiveness of memantine on functional communication in patients with moderate dementia of the Alzheimer’s type | Summary |
| Lundbeck 11267 | Memantine for agitation and aggression in severe AD – open-label, explorative study | Observational |
| Lundbeck 11875A | An open-label, post-marketing, naturalistic, multicentre study evaluating the safety and efficacy of Ebixa (memantine) in the treatment of Chinese patients with AD | Ongoing |
| Lundbeck 12292A | Memantine on aggression and agitation of AD – open-label study | Ongoing |
| Lundbeck 12484A | Memantine and changes of biological markers and brain PET imaging in AD – double blind, randomised, placebo controlled | Ongoing |
| Lundbeck 12484A | Memantine and changes of biological markers and brain PET imaging in AD – double blind, randomised, placebo controlled | Ongoing |
| Lundbeck 12732A | An open-label, observational, multicentre study evaluating efficacy and safety profile of Memantine in Chinese patients with AD | Not started |
| Lundbeck 11784A | Psychiatric symptoms and caregiver distress in patients with moderate-to-severe AD treated with memantine – study design: pre/post-treatment study (no randomisation, no blinding, no groups) | Observational |
| Lundbeck 11232 | A randomised, double-blind placebo-controlled trial of memantine in the treatment of the agitation in Alzheimer’s dementia | Ongoing |
| Lundbeck 11786A | Impact on aggressive behaviour and cognition of switching from donepezil to memantine in patients with moderate-to-severe AD – design: open label, pilot, observational, head to head | Ongoing |
| Lundbeck 11829A | Memantine for the maintenance treatment of neuropsychiatric symptoms in people with AD living in care facilities: a double-blind, controlled comparison to neuroleptic medication (Maintenance of Neuropsychiatric Symptoms in AD: MAIN-AD) | Ongoing |
| Lundbeck 11967A | Donepezil and memantine in moderate-to-severe AD (DOMINO Study) – design: pragmatic, multi-centre, double-blind, randomised, placebo controlled (double-dummy), parallel group, 2 × 2 factorial clinical trial | Ongoing |
| Lundbeck 10710 | Memantine effects on cortical excitability and its neurophysiological/neuropsychological effects on AD patients in combination with AChEI: a pilot study – design: first phase open label, second phase partial blind | No publication or report |
| Lundbeck 10997 | Behaviour and cognition in AD patients treated with the NMDA receptor antagonist memantine: correlation with the apoptotic mechanism | Ongoing |
| Lundbeck 10998 | Effect of memantine treatment on brain function and morphological structure in patients with moderate-to-severe AD: a structural MR and FMRI study. Experimental design | Wrong outcomes |
| Lundbeck 10712 | Effectiveness and tolerability of memantine treatment in outpatients with AD of mixed dementia. Multicentre, open-label trial | Observational |
| Lundbeck 11198 | Memantine therapy for treatment of AD | Commentary |
| Lundbeck 11830A | Investigating the effects of treatment on neurotrophic factors by means of functional magnetic resonance imaging (FMRI) in patients with Alzheimer’s Disease – design: double blind, prospective, randomised | Not started |
| MRZ 10001–0207 | A randomised double-blind controlled trial to evaluate the efficacy and safety of an antidementive combination therapy (galantamine and memantine) in subjects with mild-to-moderate stage of probable AD. ‘MEGA-COMBI-2’ | Ongoing |
| MRZ-9605 MD-01 MD-02 MD-10 MD-12 Lu-99679 | The meta-analysis population comprised the subgroup of patients from these studies (n = 1826) with a baseline MMSE score < 20 (i.e. moderate-to-severe AD). Assessments were made in the key domains of global response, function, cognition and behaviour | Pooled secondary analysis |
| As above | Data from six randomised, double-blind, placebo-controlled, 6-month studies were pooled and a subgroup of patients (867 on placebo, 959 on memantine) with moderate-to-severe AD (MMSE < 20) was analysed | Pooled secondary analysis |
| As above | Data were pooled from six 24/28-week, randomised, placebo-controlled, double-blind studies. Of the 2311 patients included in these studies, 1826 patients with moderate-to-severe AD (MMSE < 20) were included in this analysis. In this subgroup, 959 patients received memantine 20 mg/day and 867 received placebo. Behavioural symptoms were rated using the NPI total and single-item scores at weeks 12 and 24/28 | Pooled secondary analysis |
| As above | Data from six multicentre, randomised, placebo-controlled, parallel-group, double-blind, 6-month studies were used as the basis for these post hoc analyses. All patients with a MMSE score of < 20 were included. Analyses of patients with moderate AD (MMSE 10–19), evaluated with the ADAS-cog and analyses of patients with moderate-to-severe AD (MMSE 3–14), evaluated using the SIB, were performed separately | Pooled secondary analysis |
| As above | The current analysis combined data from six previously published studies and assessed the effect of memantine on various cognitive functions in 1826 patients (867 on placebo and 959 on memantine) with moderate-to-severe AD (MMSE < 20). The ADAS-cog and the SIB scores from all six studies were pooled and combined into three clusters representing discrete cognitive domains: language, memory and praxis | Pooled secondary analysis |
| As above | Data were pooled from patients with moderate-to-severe AD (MMSE score < 20 at baseline) from six randomised, double-blind, placebo-controlled, 6-month clinical trials on the efficacy and safety of memantine in AD | Pooled secondary analysis |
| MRZ 9403 MRZ-9605 | The aim of this additional analysis was to investigate how the global benefit reported in these earlier publications translates into specific functional effects, and the impact that these findings may have on AD patients and their caregivers | Pooled secondary analysis |
| Farlow et al. Memantine for the Treatment of Alzheimer’s Disease Tolerability and Safety Data from Clinical Trials. Drug Safety 2008;31 | Pooled secondary analysis | |
| Wilcock et al. Memantine for agitation/aggression and psychosis in moderately severe to severe AD: a pooled analysis of three studies. J Clin Psychiatry 2008;69 | Pooled secondary analysis | |
| Ferris et al. Treatment effects of memantine on language in moderate-to-severe AD patients. Alzheimers Dement 2009;5:369–74 | Pooled secondary analysis | |
| McKeage. Memantine: A review of its use in moderate-to-severe AD. ADIS drug evaluation. CNS Drugs 2009;23:881–97 | Review | |
| Grossberg et al. Memantine therapy of behavioral symptoms in community-dwelling patients with moderate-to-severe AD. Dement Geriatr Cogn Disord 2009;27:164–72 | Review | |
| Merz Pharma Ltd, a partner, has initiated two projects on the analyses of the prescription databases, General Practice Research Database (GPRD) in the UK and Insight Health in Germany and. The projects aim to analyse prescription patterns in AD, including use of memantine, AChEIs and concomitant use of antipsychotic medications in AD patients. In addition, GPRD data presents an opportunity to estimate a risk of hip fractures and of implantation of cardiac pacemakers by treatment group | Ongoing | |
|
Livingston G, Katona C, Roch B, Guilhaume C, Rive B. A dependency model for patients with AD: its validation and relationship to the costs of care – the LASER-AD Study. Curr Med Res Opin 2004;20:1007–16 Ryu SH, Katona C, Rive B, Livingston G. Persistence of and changes in neuropsychiatric symptoms in Alzheimer disease over 6 months: the LASER-AD study. Am J Geriatr Psychiatry 2005;13:976–83 Livingston G, Katona C, François C, Guilhaume C, Cochran J, Sapin C. Characteristics and health status change over 6 months in people with moderately severe to severe Alzheimer’s disease in the U.K. Int Psychogeriatr 2006;18:527–38 Habermann S, Cooper C, Katona C, Livingston G. Predictors of entering 24-h care for people with Alzheimer’s disease: results from the LASER-AD study. Int J Geriatr Psychiatry 2009;24:1291–8 |
Epidemiological | |
| Lopez et al. Long-term effects of the concomitant use of memantine with cholinesterase inhibition in Alzheimer disease. J. Neurol Neurosurg Psychiatry 2009;80;600–7 | Observational | |
| Atri et al. Long-term course and effectiveness of combination therapy in Alzheimer disease. Alzheimer Dis Assoc Disord 2008;22:209–21 | Observational | |
|
Vidal et al. Evaluation of the impact of memantine treatment – initiation on psychotropics use: a study from the French National Health Care Database. Neuroepidemiology 2008;31:193–200. Vidal et al. Memantine therapy for Alzheimer disease in real-world practice: An observational study in a large representative sample of French patients. Alzheimer Dis Assoc Disord 2008;22:125–30 |
Observational | |
| Rountree et al. Persistent treatment with cholinesterase inhibitors and/or memantine slows clinical progression of Alzheimer disease. Alzheimers Res Ther 2009;1:7 | Observational | |
| Clerici F et al., Memantine in moderately-severe-to-severe Alzheimer’s disease. Drugs Aging 2009;26:321–32 | Observational | |
| Orgogozo et al. Alzheimer’s disease behavioural symptoms increase resource utilisation. Poster ICAD 2008 | Poster | |
| Martinez-Rivera et al., Psychiatric symptoms and caregiver distress in patients with moderate to severe Alzheimer’s disease treated with memantine, Poster EFNS 2008; Eur J Neurol 2008;15(Suppl. 3):222–390 | Poster | |
| Clerici et al. Adverse Events in a Cohort of Alzheimer’s Disease Patients treated with Memantine, Poster ISoP 2007 | Poster | |
| Shaughnessy et al. Real-world clinical effectiveness of combination therapy with ChEI and Memantine in AD. Poster AAN 2007 | Poster | |
| Calabrese et al. Memantine in clinical practice – results of an observational study. Dement Geriatr Cogn Disord 2007;24:111–17 | Observational | |
| Hartmann S et al. Memantine in moderately-severe-to-severe Alzheimer’s disease. Int Clin Psychopharmacol 2003;1:81–5 | Observational |
Appendix 11 Ongoing trials
| Register/identifier number (if not available then study ID cited) | Sponsor/collaborators | Trial name | Investigator | Country | Established/anticipated sample size | Phase | Status |
|---|---|---|---|---|---|---|---|
| ISRCTH96337233 | West Midlands NHS Research & Development Executive | A reliable assessment of the efficacy and safety of donepezil and aspirin in Alzheimer’s disease (AD2000) | Professor Richard Gray (University of Birmingham Clinical Trials Unit) | UK | 310 | Completed – 2004 | |
| NCT00843518 | Eli Lilly & Company | Treatment for aggression and agitation in patients with Alzheimer’s disease | Not specified | USA | Not specified | Phase II | Recruiting |
| NCT00035204 | J&J | A double-blind, randomized pilot study to evaluate the effects of galantamine and donepezil on sleep and attention and gastrointestinal (GI) tolerance in patients with mild to moderate Alzheimer’s disease | Not specified | Not specified | Not specified | Phase IV | Completed |
| NCT00523666 | Ludwig-Maximilians – University of Munich | Diffusion tensor weighted MRI in Alzheimer’s disease: prediction and mapping of symptomatic and disease modifying treatment effects of galantamine (Reminyl®) | Stefan Teipel | Germany | Not specified | Phase IV | Recruiting |
| NCT01024660 | AstraZeneca | The effect of cognitive function as measured by repeated cognitive measures after 12 weeks treatment with donepezil | Malene Jensen | Canada, Peru, South Africa, Poland | 155 | NA | Recruiting |
| NCT00693004 | Epix Pharmaceuticals Inc. | A phase II, multicenter, randomized, double-blind, placebo-controlled, parallel group study to evaluate the efficacy and safety of PRX-03140 as monotherapy in subjects with Alzheimer’s disease | Not specified | US | 236 | Phase II | Terminated |
| NCT00645190 | Xian-Janssen Pharmaceutical Ltd | A randomized, double blind, active control, flexible dose, multicentre study to evaluate galantamine HBr in the treatment of Alzheimer’s disease: safety and effectiveness of an immediate-release table formulation | Not specified | Not specified | 215 | Phase III | Completed |
| NCT00100334 | Praecis Pharmaceuticals Inc. | Multiple dose safety and preliminary pharmacodynamic study of PPI-1019 in subjects with mild–moderate Alzheimer’s disease | Not specified | USA | 24 | Phase I/II | Completed |
| NCT00645190 | Xian-Janssen Pharmaceutical Ltd | A randomized, double blind, active control, flexible dose, multicenter study to evaluate galantamine HBr in the treatment of Alzheimer’s disease: safety and effectiveness of an immediate-release table formulation | Not specified | Not specified | 215 | Phase III | Completed |
| NCT00190021 | Beersheva Mental Health Center | Donepezil as add-on treatment of psychotic symptoms in patients with dementia of the Alzheimer’s type | Vladimir Lerner | Israel | 80 | Phase III | Not yet recruiting |
| NCT00099242 | Novartis | Efficacy and safety of the rivastigmine transdermal patch in patients with probable Alzheimer’s disease | Not specified | USA, Chile, Czech Republic, Denmark, Finland, Guatemala, Israel, Italy Korea (Republic of), Mexico, Norway, Peru, Poland, Portugal, Russian Federation, Slovakia, Sweden, Taiwan (Province of China), Venezuela | 1040 | Phase III | Completed |
| NCT00096473 | Eisai Inc./Pfizer | A 24 week, multicenter, randomized, double-blind, placebo-controlled evaluation of the safety and efficacy of donepezil hydrochloride (E2020) in patients with severe Alzheimer’s disease followed by a 12 week open-label extension period | Sharon Richardson, Honglan Li | USA | Not specified | Phase III | Completed |
| NCT00916383 | Teikoku Pharma USA | A randomized, placebo-controlled study in elderly Alzheimer’s subjects on an established and well tolerated dose of aricept to assess skin tolerability, skin irritation and adhesion with three consecutive seven-day applications of the 350 mg donepezil transdermal patch-system | Not specified | USA | 48 | Phase II | Ongoing but not recruiting |
| NCT00711204 | Eisai Inc./Pfizer | A 12-week, double-blind, placebo-controlled study to evaluate the impact of donepezil hydrochloride (Aricept) on behavioral and psychological symptoms in patients with severe Alzheimer’s disease | Thomas McRae (Pfizer) | USA | 200 | Phase IV | Terminated |
| NCT00478205 | Eisai Inc./Eisai Ltd | Double-blind, parallel-group comparison of 23 mg donepezil sustained release (SR) to 10 mg donepezil immediate release (IR) in patients with moderate to severe Alzheimer’s disease | Jane Yardley, Eisai Ltd | USA | 1200 | Phase III | Completed |
| NCT00216593 | Janssen Pharmaceutica NV, Belgium | Treatment of severe Alzheimer’s disease in a residential home, nursing home, or geriatric residential setting: evaluation of efficacy and safety of galantamine hydrobromide in a randomised, doubleblind, placebo-controlled study | Janssen Pharmaceutica N.VS Clinical Trial Janssen Pharmaceutica N.V. | Not specified | 415 | Phase III | Completed |
| NCT00235716 | Department of Veterans Affairs/Forest Laboratories/DSM Nutritional Products, Inc. | CSP #546 – A randomised, clinical trial of vitamin E and memantine in Alzheimer’s disease (TEAM-AD) | Maurice Dysken (Minneapolis Veterans Affairs Medical Center) | USA, Puerto Rico | 840 | Phase III | Recruiting |
| NCT00216593 | Janssen Pharmaceutica NV, Belgium | Treatment of severe Alzheimer’s disease in a residential home, nursing home, or geriatric residential setting: evaluation of efficacy and safety of galantamine hydrobromide in a randomised, double-blind, placebo-controlled study |
Janssen Pharmaceutica NV Clinical Trial Janssen Pharmaceutica NV |
Not specified | 415 | Phase III | Completed |
| NCT00814801 | Janssen Pharmaceutical KK | Placebo-controlled confirmatory study of galantamine (R113675) for Alzheimer’s type dementia |
Janssen Pharmaceutical KK Clinical Trial, Study Director, Janssen Pharmaceutical KK |
Not specified | 580 | Phase III | Completed |
| NCT00183729 | National Institute of Mental Health (NIMH) | Memantine for enhancement of rehabilitation efficacy and prevention of major depressive disorder in older adults | Eric J. Lenze, MD (University of Pittsburgh) | USA | 40 | Phase IV | Active, not recruiting |
| ISRCTN24953404 | East Kent Hospitals Research and Development Committee (UK) (funded by Lundbeck Pharmaceuticals UK) | A randomized, double-blind, placebo-controlled trial of memantine in the treatment of agitation in dementia (MAGD) | Dr Chris Fox (Folkestone Health Centre), Dr Art Artionou (Buckland Hospital, Dover) | UK | 154 | Not specified | Ongoing |
| ISRCTN55568578 | Department of Health, London (funded by Avon and Wiltshire Mental Health Partnership NHS Trust) | Making evidence-based decisions using Alzheimer therapy (MEDUSA Therapy) | Dr Roger Bullock, (Kingshill Research Centre, Victoria Hospital, Swindon) | UK | 75 | Not specified | Completed |
| ISRCTN49545035 | Institute of Psychiatry (UK) [funded by Medical Research Council (UK) (grant ref: G0600989)] | Donepezil and memantine IN moderate to severe Alzheimer’s disease (DOMINO-AD) | Professor Robert Howard (Institute of Psychiatry, London, UK) | UK | 800 | Not specified | Ongoing |
| ISRCTN68407918 | Kings College London (UK) (funded by Lundbeck Pharmaceuticals Ltd) | Memantine for the long term management of neuropsychiatric symptoms in Alzheimer’s disease (MAIN-AD) | Professor Clive Ballard (King’s College, London) | UK | 300 | Not specified | Ongoing |
| ISRCTN62185868 | Kings College London (UK) [funded by Medical Research Council (UK)] | A randomized placebo controlled trial of a cholinesterase inhibitor in the management of agitation in dementia that is unresponsive to a psychological intervention (CALM-AD) | Professor Robert Howard (Institute of Psychiatry, London, UK) | UK | 285 | Not specified | Completed |
| NCT00857649 | H Lundbeck A/S | A randomised, double-blind, parallel-group study examining the efficacy and safety of memantine in patients with moderate to severe dementia of the Alzheimer’s type | Dr Sauge Gauthier and Dr Nathan Hermann | Canada | 450 | Phase III | Ongoing, not recruiting |
| NCT00857233 | H Lundbeck A/S | An open-label extension study examining the safety and tolerability of memantine in patients with moderate to severe dementia of the Alzheimer’s type having completed Study 10158 | Dr Sauge Gauthier and Dr Nathan Hermann | Canada | 450 | Phase III | Ongoing, not recruiting |
| NCT00862940 | H Lundbeck A/S | A 1-year randomised, double-blind placebo-controlled study to evaluate the effects of memantine on rate of brain atrophy in patients with Alzheimer’s disease | Dr David Wilkinson | Not specified | 278 | Phase IV | Completed |
| (Lundbeck 99819) | H Lundbeck A/S | A long-term open-label extension study evaluating the safety and tolerability of memantine in patients with mild to moderate dementia of the Alzheimer’s type | Professor Serge Bakchine | Not specified | Not specified | Phase III | Not specified |
| (Lundbeck 99817) | H Lundbeck A/S | A double-blind, randomized, placebo-controlled study to evaluate the safety and efficacy of memantine in patients with dementia of the Alzheimer’s type | Dr Pei-Ning Wang, Dr Sui-Hing Yan | Not specified | Not specified | Phase III | Not specified |
| (Asubio IE-2101) | Late phase II clinical study of sun Y7017 (memantine hydrochloride) in patients with moderately severe to severe dementia of the Alzheimer’s type: evaluation of recommended dose and long-term safety (extension study for dose-finding and long-term safety) | Professor Akira Homma | Not specified | Not specified | Phase II | Not specified | |
| (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | |
| (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | |
| (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | |
| (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | |
| (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | (CiC information has been removed) | |
| NCT00624026 | Merz Pharmaceuticals GmbH | Prospective, single-arm, multicenter, open-label study to investigate the efficacy and tolerability of the once daily (OD) memantine treatment | Professor Joerg Schulz | Germany | 107 | Phase IIIb | Completed |
| NCT00649220 | Merz Pharmaceuticals GmbH | Prospective, single-arm, multi-centre, open-label study to investigate the potential to reduce concomitant antipsychotics use in patients with moderate to severe dementia of Alzheimer’s type (DAT) treated with memantine | Professor Ralf Ihl | Germany | 27 | Phase IV | Completed |
| (MRZ 90001-AD-3001) | Merz Pharmaceuticals GmbH | Open-label, single-arm, multicenter validation study of the ROSA-Scale (Relevant Outcome Scale for Alzheimer Patients) in patients with dementia of Alzheimer’s type (DAT) treated with memantine over a 3-month period | Professor Vjera Holthoff | Not specified | Not specified | Phase IIIb | Not specified |
| MRZ 9104 | Merz Pharmaceuticals GmbH | Multicentre, randomised, double-blind, comparative study of the efficacy and tolerability of akatinol memantine and placebo in patients suffering from senile dementia, Alzheimer type | Professor Derouesne | Not specified | Not specified | Phase II | Not specified |
| (Forest MEM-MD-03 C) | Forest Laboratories | Extension of MEM-MD-01 and MEM-MD-02 Phase C = 52 weeks open | Not specified | Not specified | Not specified | Phase III | Not specified |
| (Forest MEM-MD-03 D) | Forest Laboratories | Extension of MEM-MD-01 and MEM-MD-02 Phase D = open continuation until memantine is commercially available | Not specified | Not specified | Not specified | Phase III | Not specified |
| (Forest MEM-MD-11 A/B) | Forest Laboratories | A long-term extension study evaluating the safety and tolerability of b.i.d. and q.d. administration of memantine in patients with mild to moderate dementia of the Alzheimer’s type. Extension of MEM-MD-10. Phase A/B = 8 weeks Double-blind + 20 weeks open | Not specified | Not specified | Not specified | Phase III | Not specified |
| (Forest MEM-MD-11 C) | Forest Laboratories | Extension of MEM-MD-10. Phase C = 52 weeks open | Not specified | Not specified | Not specified | Phase III | Not specified |
| (Forest MEM-MD-11 D) | Forest Laboratories | Extension of MEM-MD-10. Phase D = open continuation until memantine is commercially available | Not specified | Not specified | Not specified | Phase III | Not specified |
| (Forest MEM-MD-12 A) | Forest Laboratories | Open extension of MEM-MD-12. 28 weeks | Not specified | Not specified | Not specified | Phase III | Not specified |
| (Forest MEM-MD-12 B) | Forest Laboratories | Open extension of MEM-MD-12 A. A continuation until memantine is commercially available | Not specified | Not specified | Not specified | Phase III | Not specified |
| (Forest MEM-MD-22) | Forest Laboratories | A randomized, double-blind, placebo-controlled evaluation of the safety and efficacy of namenda in nursing home patients with moderate to severe Alzheimer’s disease | Not specified | Not specified | Not specified | Phase IV | Not specified |
| (Forest MEM-MD-23) | Forest Laboratories | A randomised, double-blind, placebo-controlled, evaluation of the safety and efficacy of memantine in patients with moderate to severe Alzheimer’s disease with behavioral disturbances | Not specified | Not specified | Not specified | Phase III | Not specified |
| NCT00401167 | Sunnybrook Health Sciences Centre/H Lundbeck A/S | Phase IV – an open-label prospective study of memantine in institutionalized patients with severe Alzheimer’s disease and significant behavioural and psychological symptoms of dementia | Nathan Herrmann MD | Canada | 32 | Phase IV | Completed |
| (Lundbeck 11875A) | Lundbeck A/S | An open-label, post-marketing, naturalistic, multi-centre study evaluating the safety and efficacy of Ebixa (memantine) in the treatment of Chinese patients with Alzheimer’s disease | Hong Zhen | Not specified | Not specified | Not specified | Ongoing |
| (Lundbeck 12292A) | Lundbeck A/S | Memantine on aggression and agitation of AD – open-label study | Xin Yu, Wang Hu | Not specified | Not specified | Not specified | Ongoing |
| NCT00800709 | Shanghai Mental Health Center/Lundbeck A/S | Memantine and changes of biological markers and brain PET imaging in Alzheimer’s disease – double-blind, randomized, placebo-controlled | Xiao Shi Fu | China | 26 | Phase IV | Recruiting |
| (Lundbeck 12732A) | Lundbeck A/S | An open-label, observational, multicentre study evaluating efficacy and safety profile of memantine in Chinese patients with Alzheimer’s disease | Yinhua Wang | Not specified | Not specified | Not specified | Ongoing |
| (Lundbeck 13143A) | Lundbeck A/S | A randomised, double-blind, placebo-controlled study to investigate the improvement of language function in Chinese AD patients with memantine | Dantao Peng | Not specified | Not specified | Not specified | Not yet initiated |
| (Lundbeck 11232) | Lundbeck A/S | A randomised, double-blind, placebo-controlled trial of memantine in the treatment of the agitation in Alzheimer’s dementia | Fox | Not specified | Not specified | Not specified | Ongoing |
| (Lundbeck 11786A) | Lundbeck A/S |
Impact on aggressive behaviour and cognition of switching from donepezil to memantine in patients with moderate-to-severe AD Design: open-label, pilot, observational, head-to-head |
Huertas | Not specified | Not specified | Not specified | Ongoing |
| (Lundbeck 10710) | Lundbeck A/S |
Memantine effects on cortical excitability and its neurophysiological/neuropsychological effects on AD patients in combination with AChEI: a pilot study Design: first phase open-label, second phase partial blind |
Stefani | Not specified | Not specified | Not specified | Completed |
| (Lundbeck 10997) | Lundbeck A/S | Behaviour and cognition in ad patients treated with the NMDA receptor antagonist memantine: correlation with apoptotic mechanism | Spalleta | Not specified | Not specified | Not specified | Ongoing |
| (Lundbeck 11830A) | Lundbeck A/S |
Investigating the effect of treatment on neurotrophic factors by means of functional magnetic resonance imaging (FMRI) in patients with Alzheimer’s disease Design: double-blind, prospective randomised |
Tamer Aker | Not specified | Not specified | Not specified | Not yet initiated |
| (MRZ 10001–0207) | Merz Pharmaceuticals GmbH | A randomized, double-blind, controlled trial to evaluate the efficacy and safety of an antidementive combination therapy (galantamine and memantine) in subjects with mild-to-moderate stage of probable AD (MEGA-COMBI-2) | Heuser | Not specified | Not specified | Not specified | Ongoing |
Appendix 12 Preferred Reporting Items for Systematic Reviews and Meta-analyses statement checklist
Preferred Reporting Items for Systematic Reviews and Meta-analyses statement checklist comparison of the quality of included clinical effectiveness systematic reviews a–d
| Section/topic | Item | Checklist item | a | b | c | d |
|---|---|---|---|---|---|---|
| Title | ||||||
| Title | 1 | Identify the report as a systematic review, meta-analysis or both | ✓ | ✓ | × | ✓ |
| Abstract | ||||||
| Structured summary | 2 | Provide a structured summary including, as applicable, background, objectives, data sources, study eligibility criteria, participants, interventions, study appraisal and synthesis methods, results, limitations, conclusions and implications of key findings, systematic review registration number | × | × | × | × |
| Introduction | ||||||
| Rationale | 3 | Describe the rationale for the review in the context of what is already known | × | × | × | × |
| Objectives | 4 | Provide an explicit statement of questions being addressed with reference to participants, interventions, comparisons, outcomes and study design | × | ~ | ~ | × |
| Methods | ||||||
| Protocol and registration | 5 | Indicate if a review protocol exists, if and where it can be accessed and if available, provide registration information including registration number | ~ | ✓ | ✓ | × |
| Eligibility criteria | 6 | Specify study characteristics and report characteristics used as criteria for eligibility, giving rationale | × | × | × | × |
| Information sources | 7 | Describe all information sources in the search and date last searched | × | × | × | × |
| Search | 8 | Present full electronic search strategy for at least one database, including any limits used, such that it could be repeated | ✓e | ✓ | ✓ | × |
| Study selection | 9 | State the process for selecting studies | × | ~ | × | × |
| Data collection process | 10 | Describe method of data extraction from reports and any processes for obtaining and confirming data from investigators | × | × | × | × |
| Data items | 11 | List and define all variables for which data are sort and any assumptions and simplifications made | × | × | × | × |
| Risk of bias in individual studies | 12 | Describe methods used for assessing risk of bias of individual studies and how this information is to be used in any data synthesis | × | ✓ | ✓ | × |
| Summary measures | 13 | State the principal summary measures | × | × | × | × |
| Synthesis of results | 14 | Describe the methods of handling data and combining results of studies, if done, including measure of consistency for each meta-analysis | × | × | × | × |
| Risk of bias across studies | 15 | Specify any assessment of risk of bias that may affect the cumulative evidence | ✓ | ✓ | × | ✓ |
| Additional analyses | 16 | Describe methods of additional analyses, if done, indicating which were pre-specified | ✓ | – | – | – |
| Results | ||||||
| Study selection | 17 | Give numbers of studies screened, assessed for eligibility, and included in the review, with reasons for exclusions at each stage, ideally from a flow diagram | ✓ | × | × | × |
| Study characteristics | 18 | For each study, present characteristics for which data were extracted and provide the citations | × | ×f | × | × |
| Risk of bias within studies | 19 | Present data on risk of bias of each study and, if available, any outcome-level assessments | × | ✓ | ✓ | × |
| Results of individual studies | 20 | For all outcomes considered, present for each study (a) simple summary data for each intervention group and (b) effect estimates and CIs, ideally with a forest plot | × | × | ~ | × |
| Synthesis of results | 21 | Present results of each meta-analysis done, including CIs and measure of consistency | × | × | ~ | × |
| Risk of bias across studies | 22 | Present results of any assessment of risk of bias across studies | ✓ | ✓ | ✓ | ✓ |
| Additional analysis | 23 | Give results of additional analyses, if done | × | – | – | – |
| Discussion | ||||||
| Summary of evidence | 24 | Summarise the main findings including the strength of evidence for each main outcome: consider their relevance for key groups | × | × | × | × |
| Limitations | 25 | Discuss limitation at study and outcome level and at review level | ~ | ~ | × | × |
| Conclusions | 26 | Provide a general interpretation of the results in the context of other evidence, and implication for future research | × | ×g | ×g | × |
| Funding | ||||||
| Funding | 27 | Describe sources of funding for the systematic review and other support and role of funders for the systematic review | ~ | × | × | ✓ |
Appendix 13 Summary tables of results from the Institute of Quality and Efficiency in Health Care
| Therapy | Donepezil | Galantamine | Rivastigmine |
|---|---|---|---|
| Patient-relevant therapy goals | |||
| ADL | ↑ | ↑ | ↑ |
| Psychological symptoms | ↔ | ↑ | No data available |
| Cognitive function | ↑↑ | ↑↑ | ↑↑ |
| HRQoL | ↔ | No data available | No data available |
| Nursing home care (institutionalisation) | No data available | No data available | No data available |
| Mortality | (↔) | (↔) | (↔) |
| AEs | ↓↓ | ↓↓ | ↓↓ |
| Therapy goals relevant to relatives | |||
| QoL of (caregiving) relatives | ↔ | ↑ | No data (or only uncertain data) available |
| Degree of care provided | ↔ | ↑ | No data available |
| Additional information | |||
| Clinical disease stage | ↑↑ | ↑↑ | ↑↑ |
| Dose–effect relationship | Lower efficacy (cognition) and fewer AEs for low (5 mg) or flexible dose | No favourable effect, and not consistently more AEs with the 8-mg dose; otherwise no differences | Uncertain effect for 1–4 mg |
| Therapy goal | Donepezil vs galantamine | Donepezil vs rivastigmine | Galamantine vs rivastigmine |
|---|---|---|---|
| Patient-relevant therapy goals | |||
| ADL | (↔) | (↓)a | No data available |
| Psychopathological symptoms | (↔) | ↔ | (↔) |
| Cognitive function | (↔) | ↔ | No data available |
| HRQoL | No data available | No data available | No data available |
| Placement in a nursing home (institutionalisation) | No data (or only uncertain data) available | No data available | No data available |
| Mortality | (↔) | ↔ | No data available |
| AEs | (↔) | ↑↑ | (↔) |
| Therapy goals relevant to relatives | |||
| QoL (caregiving relatives) | No data available | No data available | No data available |
| Degree of care provided | No data available | No data available | No data available |
| Additional information | |||
| Clinical disease stage | No data available | No data available | No data available |
| Comments | In the largest study, possibly less favourable dose for donepezil | Possibly less favourable dose for donepezil | |
Appendix 14 Memantine ± acetylcholinesterase inhibitor versus placebo ± acetylcholinesterase inhibitor
Memantine ± acetylcholinesterase inhibitor versus placebo ± acetylcholinesterase inhibitor
If, as per the 2004 review, it is assumed that evidence on memantine monotherapy is equivalent to that detailing combination therapy including memantine, a larger evidence base can be assembled. The following analysis combines evidence on memantine monotherapy versus placebo (as detailed and explored in Chapter 3: see Results/Memantine versus placebo) with that on memantine + AChEIs versus placebo + AChEIs (Chapter 3: see Combination therapy).
Cognition
New data
Data from newly identified RCTs are presented in Chapter 3 (see Memantine versus placebo: memantine monotherapy versus placebo, and Combination therapy (memantine + AChEI vs placebo + AChEI).
Synthesis with existing evidence base
Alzheimer’s Disease Assessment Scale – cognitive subscale
Because ADAS-cog scores are only reported by one relevant study (Porsteinsson and colleagues150; see Chapter 3, Combination therapy), it is not possible to undertake any synthesis on this outcome.
An additional source of data is Mecocci and colleagues’ pooled IPD study,234 which includes the participants from Porsteinsson and colleagues’ RCT150 and also relevant individuals from two trials that could not be included in this review because the primary publications also reported participants from beyond the UK licensed indication of memantine). 184,185 This analysis suggests that, following 24–28 weeks of treatment with memantine ± AChEIs, a benefit of 1.55 points (95% CI 0.487 to 2.613 points) over individuals taking placebo ± AChEIs is seen.
Mini Mental State Examination
A synthesis of data from the existing evidence with the new study showed there was no significant cognitive benefit from memantine either combined with an AChEI or on its own compared with placebo, either on its own or with an AChEI, when measured by the MMSE at 24–28 weeks’ follow-up (Figure 151).
FIGURE 151.
Random-effects meta-analysis – MMSE at 24–28 weeks (mean change from baseline): memantine ± AChEI vs placebo ± AChEI.
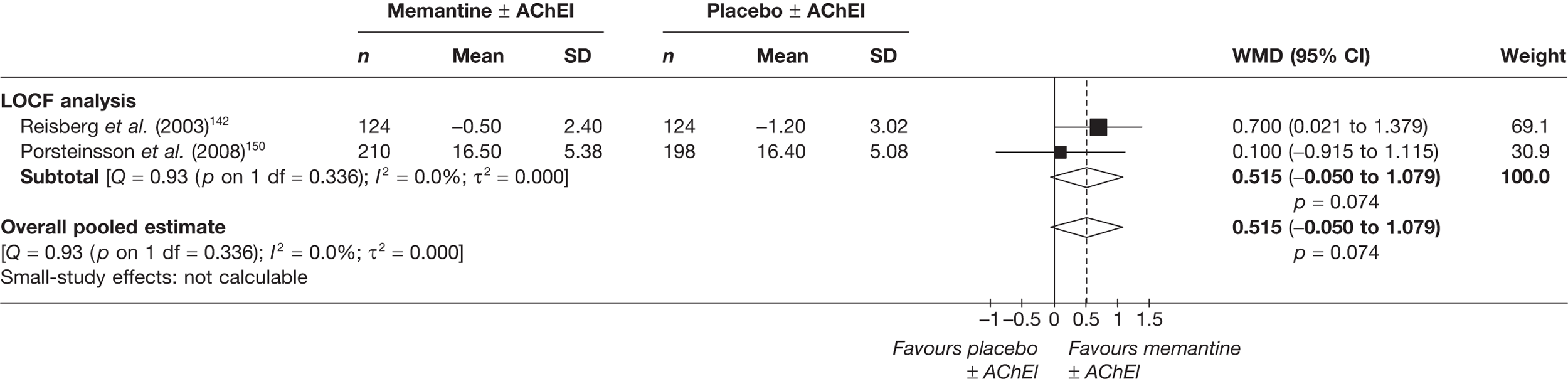
Severe impairment battery
In contrast, a significant benefit was seen when cognitive outcomes were measured with the SIB. The overall pooled estimate has been calculated as WMD = 3.27 (95% CI 0.55 to 6.04), p = 0.021 (Figure 152).
FIGURE 152.
Random-effects meta-analysis – SIB at 24–28 weeks (mean change from baseline): memantine ± AChEI vs placebo ± AChEI.
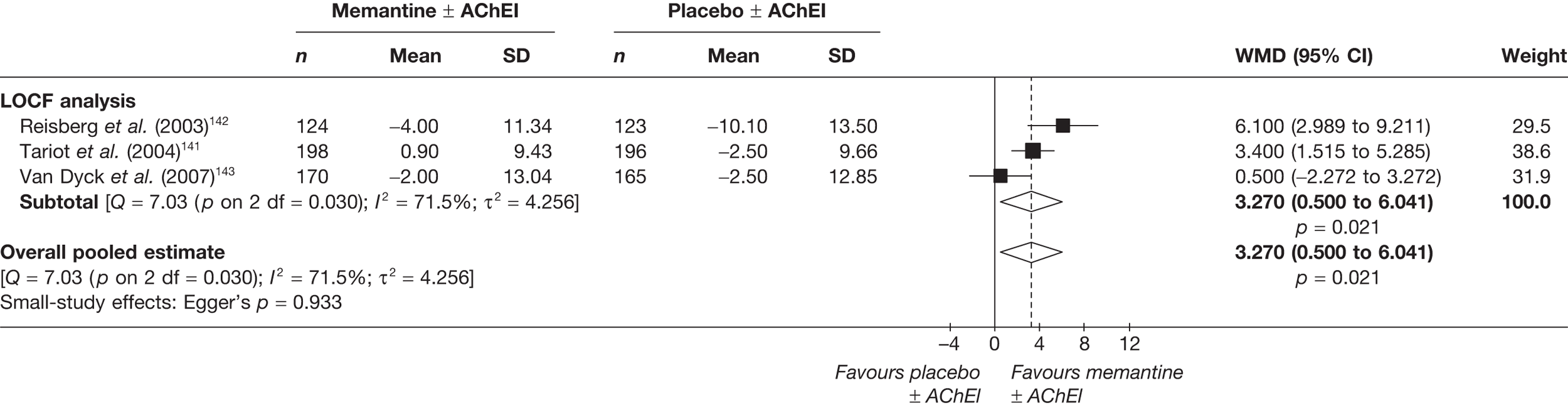
IPD: Mecocci et al. 234 3.175 (95% CI 1.566 to 4.784).
Functional
New data
For data on functional outcomes in newly identified studies of memantine ± AChEIs versus placebo ± AChEIs, see Chapter 3, Combination therapy/Evidence of Clinical effectiveness/Functional and Table 44.
Synthesis with existing evidence base
Alzheimer’s Disease Cooperative Study – Activities of Daily Living Index
When we meta-analysed the data for function outcome measures from new and existing studies we found more favourable results for memantine when considered on its own and in combination with an AChEI. When measured with the ADCS-ADL at 12 weeks and 24–28 weeks, the overall pooled estimates showed significant gain from memantine: 12 weeks, WMD = 1.03 (95% CI 0.29 to 1.77), p = 0.006; 24–28 weeks, WMD = 1.41 (95% CI 0.51 to 2.30), p = 0.002 (Figures 153 and 154).
FIGURE 153.
Random-effects meta-analysis – ADCS-ADL19 at 12 weeks (mean change from baseline): memantine ± AChEI vs placebo ± AChEI.
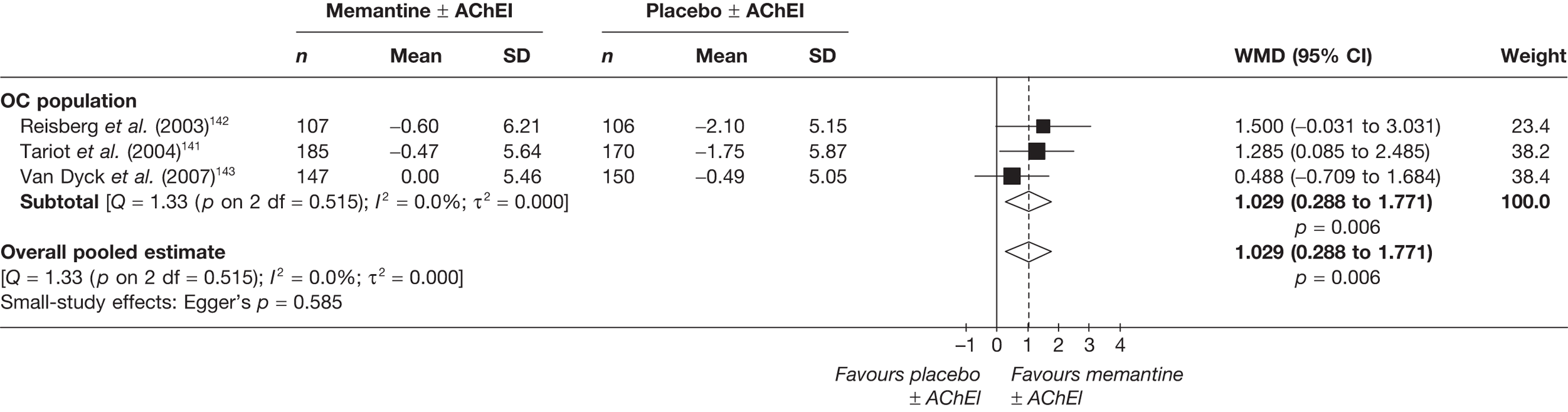
FIGURE 154.
Random-effects meta-analysis – ADCS-ADL19 at 24–28 weeks (mean change from baseline): memantine ± AChEI vs placebo ± AChEI.
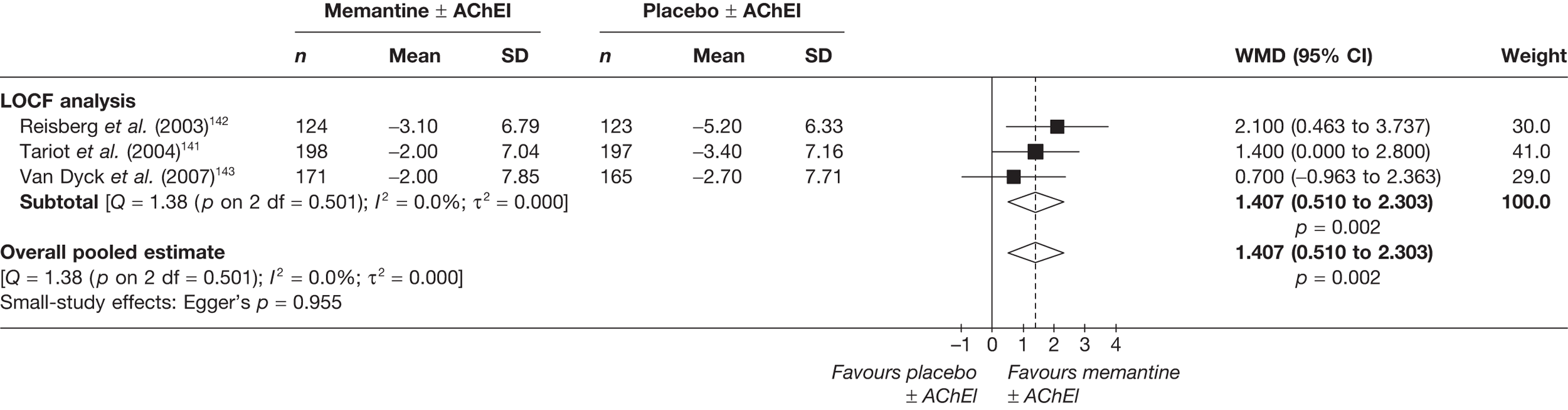
Behavioural and mood
New data
Behavioural outcome data reported in included RCTs of memantine ± AChEIs in comparison with placebo ± AChEIs are tabulated in Tables 27 and 45.
Synthesis with existing evidence base
Neuropsychiatric Inventory
A meta-analysis of data from new and existing studies using the NPI at 24–28 weeks showed no significant gain from memantine (Figure 155).
FIGURE 155.
Random-effects meta-analysis – NPI at 24–28 weeks (mean change from baseline): memantine ± AChEI vs placebo ± AChEI.
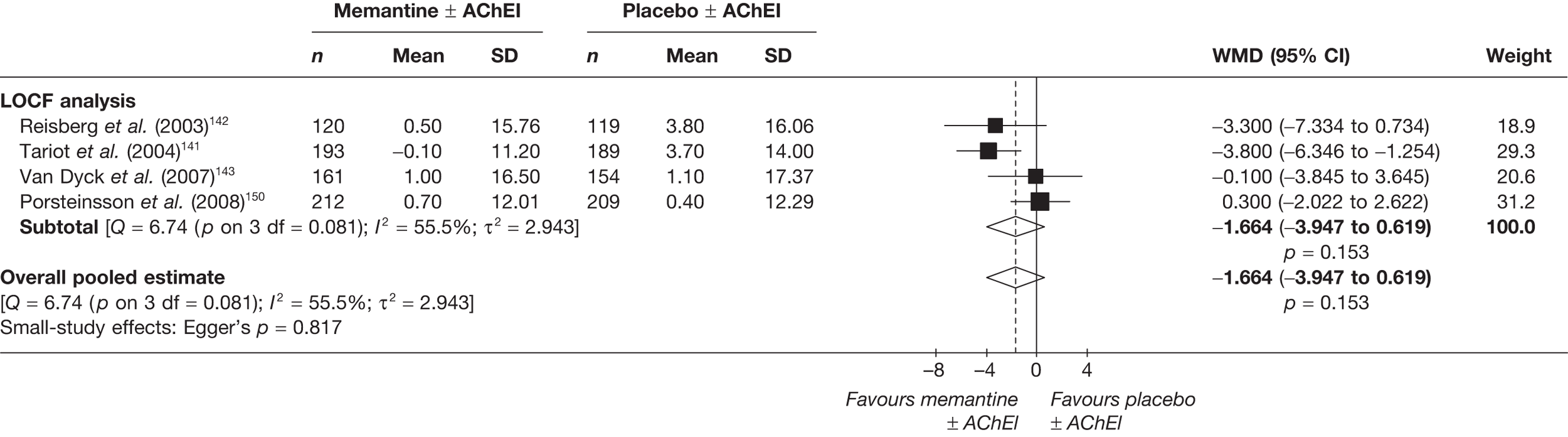
This result closely reflects the findings of Gauthier and colleagues’ analysis235 of pooled IPD from six trials (including the four included here), in which the WMD at 24–28 weeks (LOCF analysis) was –1.675 (95% CI: –3.270 to –0.080). This publication also provides information on the individual items making up the NPI. At 24 weeks, participants taking memantine ± AChEIs showed more improvement (or less deterioration) than those taking placebo ± AChEIs on all 12 single items of the NPI, with the difference achieving conventional levels of statistical significance (p < 0.05 by Kruskal–Wallis test without adjustment for multiplicity of testing) on three items: delusions, agitation/aggression and irritability.
An additional pooled IPD analysis236 concentrates on treatment effect of memantine ± AChEIs on agitation and psychotic symptoms, concluding that therapy with memantine confers benefit on the NPI cluster (agitation/aggression, delusions and hallucinations) score at both 12 weeks (–0.8 points vs 0.5 points; p = 0.0014) and 24–28 weeks (–0.7 points vs 0.7 points; p = 0.0004). This effect was substantially driven by a large difference on the agitation item: whereas the proportions of responders in the single items, delusions and hallucinations, were numerically higher for participants receiving memantine, the difference from placebo did not reach statistical significance.
Global effect
New data
Data from newly identified RCTs are presented in Table 28 (memantine monotherapy vs placebo) and Chapter 3 (see Global effect: memantine + AChEI vs placebo + AChEI).
Synthesis with existing evidence base
Clinician’s Interview-based Impression of Change
When the new data from mono and combined therapies were synthesised with the existing data, the overall pooled estimate showed a significant gain from memantine: WMD = –0.21 (95% CI –0.34 to –0.080), p = 0.002 (Figure 156).
FIGURE 156.
Random-effects meta-analysis: CIBIC-plus at 24–28 weeks (mean change from baseline) – memantine ± AChEI vs placebo ± AChEI.
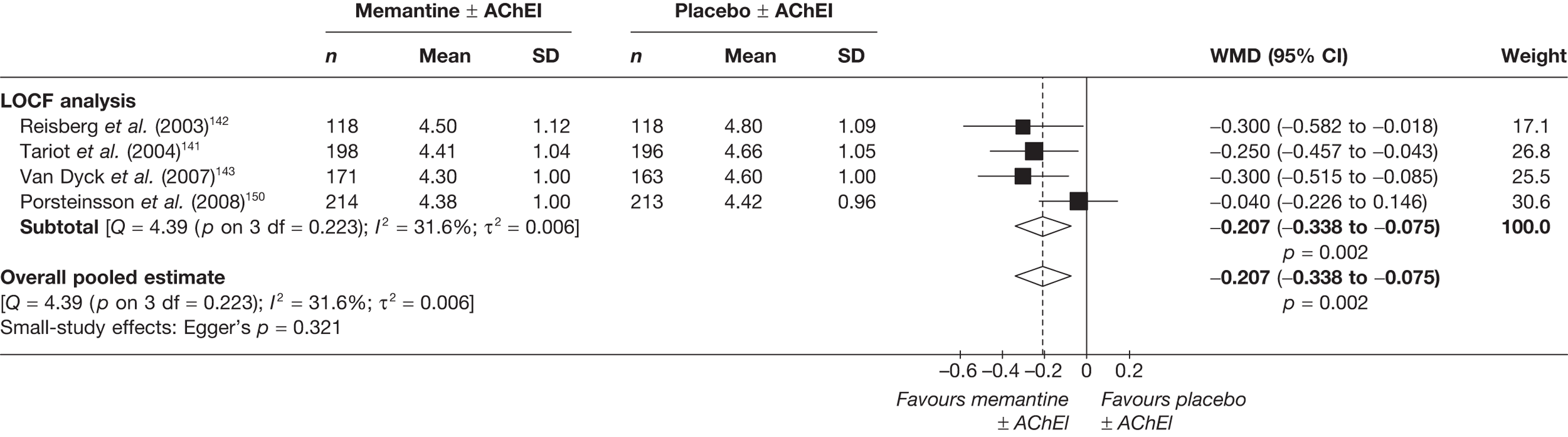
Safety
A pooled IPD paper by Farlow and colleagues237 provides extensive detail on the safety profile of memantine ± AChEI, as investigated in trials with placebo ± AChEI control arms. In total, 1242 individuals who received memantine are compared with 1242 who did not. Their findings showed that overall the proportion of AEs in those with moderate-to-severe AD was the same in treatment and control arms (68%). Agitation (12%) and falls (7%) caused the greatest percentage of AEs in the memantine group, with agitation being the most frequently cited cause for discontinuation due to an AE, n = 51 (2%). Agitation (18%) and falls (8%) were also the most frequent AE reported by the control group; again agitation was the most likely cause of AE-related discontinuation, n = 72 (14%). 237
Summary: memantine + acetylcholinesterase inhibitor versus placebo + acetylcholinesterase inhibitor
When data from monotherapy and combination therapy were combined in meta-analysis, the results from cognitive outcomes varied. Analyses using the ADAS-cog and the SIB showed significant benefits from memantine ± AChEI, while that using the MMSE did not. Functional and global outcomes were also shown to favour memantine ± AChEI, although, there was no similar benefit shown from behavioural outcomes.
Graphical summary of memantine + acetylcholinesterase inhibitor versus placebo + acetylcholinesterase inhibitor
The summary graphic in Figure 157 clearly shows the difference in results in studies included in the new and previous reviews. The main difference between these two groups of studies is that those in the 2004 review were not analysed by full ITT and those included in the current review were. The lack of ITT analysis may introduce bias.
FIGURE 157.
Summary of all studies included in the 2004 and 2010 reviews – memantine ± AChEI vs placebo ± AChEI. Analy, analysis; be, behavioural; Blind, blinding; Char, characteristics of patients; cog, cognitive; F, female; func, functional; glo, global effect; M, male; Rand, randomisation.
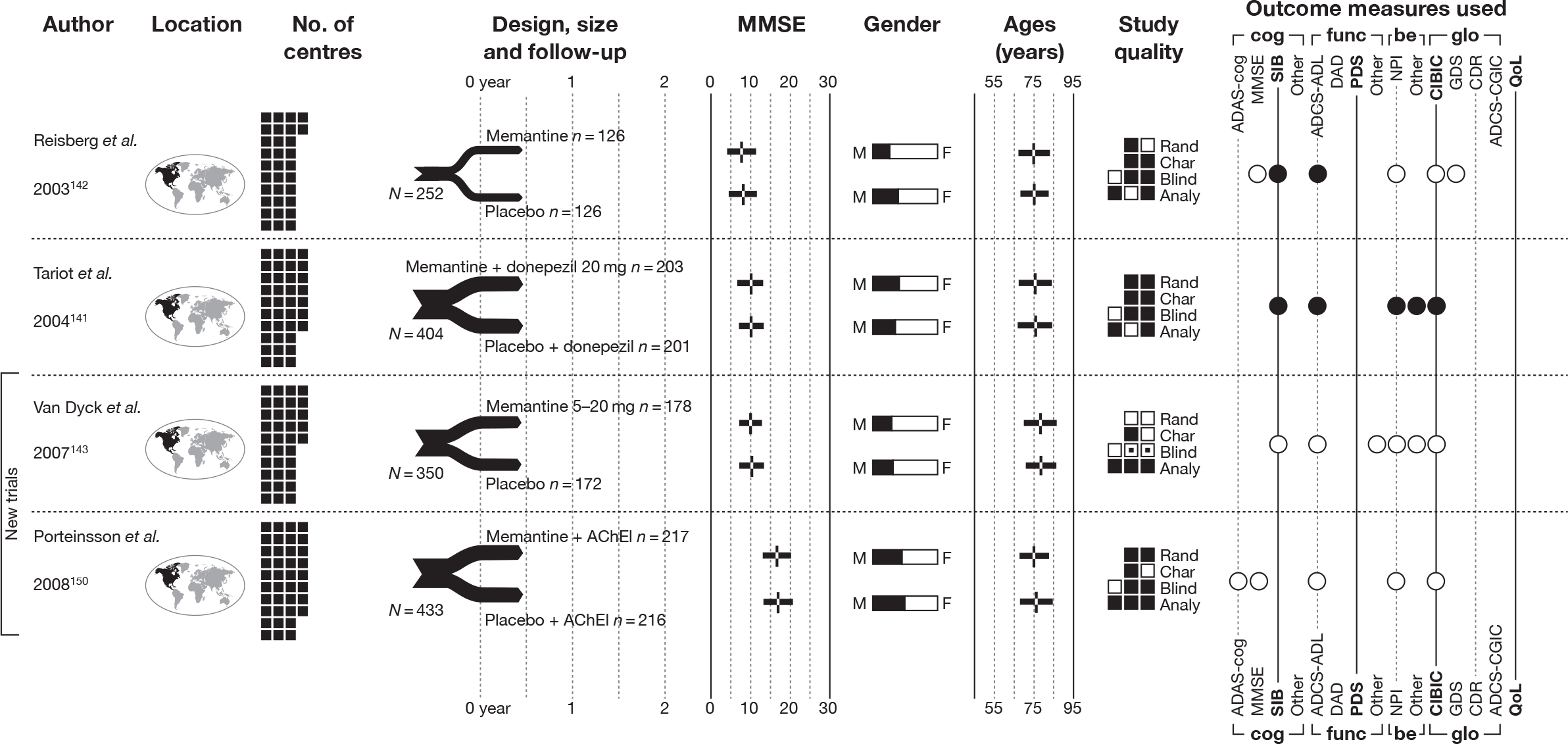
Appendix 15 Update on evidence about the care cost of Alzheimer’s disease in the UK
In relation to patients with AD in the UK, there have been three major reports published since 2004 that contain care cost estimates: the Dementia UK report in 2007 (by the PSSRU at the London School of Economics, the Institute of Psychiatry and the Alzheimer’s Society),8 a report by the NAO in 20079 on improving services for people with dementia, and a more recent (2010) cost of illness study by a health economics. 10 The 2010 study estimates that dementia will cost the UK economy £23B this year – and approximately 60% of this cost would be attributable to AD. 10 This translates to approximately £27,600 per patient per year.
We also reviewed a number of recent papers about the cost of AD for patients outside the UK, including a recent systematic review of cost-of-illness studies that focused on the stage dependency of costs238 and a recent systematic review of the cost of dementia in Europe. 156
1. Which clinical events – or main stages of Alzheimer’s disease progression or changes in a patient’s living situation – lead to a step-change in health or social care costs?
In the UK, the main markers of AD progression that leads to a step-change in health-social-care costs appears to be the events that trigger the transition from home or community care to institutional care (Dementia UK report8). When deterioration in the condition necessitates a move into long-term institutional care, the cost of care then shifts to the state – either via the NHS or social services (NAO report9); this shift in cost carrying is shown below (see Figure 159, showing the annual cost of services in the UK used by people with late-onset dementia by disease severity and care setting). 8 Although still living in the community, care for individuals with severe AD, informal-care costs are estimated at £27,096 per annum, compared with combined NHS, Social Services Department (SSD) and accommodation costs of £10,377. When community care moves to residential care, informal-care costs drop to an estimated £938 per annum, compared with combined NHS, SSD and accommodation costs of £30,358 per annum, of which accommodation costs constitute the majority at £28,646 per annum.
The transition from community care to institutional care is clearly related to an increase in disease severity and this increase in severity is related to a rise in costs; however, the relationship between disease progression and increase in costs is not clear cut. 239,240 A report on AD and dementia by the Parliamentary Office of Science and Technology (POST)241 stated that the greatest impact caused by AD and dementia on sufferers, carers and society is concentrated in individuals in the severe stages of disease progression, that is between 17% and 28% of people with dementia over 65 years old. The POST report also highlighted that in 2007, 62–75% of residents in care institutions had dementia. 242
Kavanagh and Knapp242 showed that cognitive disability, in the context of its cost-raising impact, needs to be understood in the context of comorbid disabilities and their complex interactions rather than viewed in isolation. Specifically, when analysing cognitive disability alongside non-disability variables, cognitive disability is strongly significant (p < 0.001) and the coefficient (4.286, R2 = 0.062) is three times larger than when analysed with individual disability domains (continence disability, hearing morbidity, summary mental disability, summary physical disability, summary physical ability, living alone and whether or not patients had been ill with a recent underlying condition) as independent variables (1.438, R2 = 0.136). However, the overall goodness of fit is worse when analysing cognitive disability with non-disability variables, as can be seen from the R2-values.
2. Which markers or measures of Alzheimer’s disease progression (e.g. cognitive function, functional ability, behavioural or psychotic symptoms, physical health), either individually or in combination, are most predictive of health- and/or social-care costs?
Patients are commonly assessed for cognitive function using the MMSE and are allocated into distinct severity groups. A less commonly used measure of cognitive and behavioural function is the Office of Population Censuses and Surveys disability instrument. 243 In this instance, the researchers reviewed survey data already gathered for a 1988 study (Martin et al. 243), which measured disability across 13 domains, including locomotion, dexterity, continence, intellectual functioning, consciousness and disfigurement. Kavanagh and Knapp242 reported that the instrument has good inter-rater reliability and is highly correlated with the Barthel ADL Index, although more comprehensive. They found that the link between cognitive disability and cost was sensitive to the inclusion or exclusion of behavioural disability.
The Barthel ADL Index is used to assess functional status on a scale of 0–20, with zero indicating the greatest impairment. There has been a more detailed scale developed, which rates 10 items individually on a 0–10 scale (with a maximum score of 100). Wolstenholme et al. 190 report that both the MMSE and the Barthel ADL Index are significant predictors of time to institutionalisation and cost of care, but changes in the Barthel ADL Index are particularly important in predicting costs outside institutional care.
Wolstenholme et al. 196 also examined associations between costs and cognitive assessment scores, reporting from a regression-based analysis that each one-point decline in the MMSE score was associated with a cost of care increase of £56 every 4 months, whereas each one-point decline in the Barthel score was associated with a cost of care increase of £586 every 4 months.
On a neurological level, structural imaging (magnetic resonance imaging or computed tomography scanning) and functional imaging [position emission tomography (PET) and single-photon emission computed tomography (SPET) scans] are sometimes carried out in order to exclude other cerebral pathologies and to help establish the type of dementia. Individual monitoring over time can indicate disease progression and PET scanning with the use of a dye can indicate amyloid plaques in AD, again allowing monitoring of disease progression. 237 However, access to resources is limited and NICE estimates that the additional national cost of implementing its recommendation on structural imaging will be £20.22M (Improving services and support for people with dementia7).
3. In England and Wales, what are the typical stages or pathways of care for people with Alzheimer’s disease?
This has been largely summarised in the background section of the main report:
-
Q5: In England and Wales, to what extent are the costs of caring for people with AD borne by (i) the NHS (ii) PSS (iii) local authorities (iv)other organisations such as voluntary organisations?
Within the community, informal care costs are typically borne by the patient and/or carers and these make up the majority of the financial burden for mild, moderate and severe late-onset dementia. 8 In their 2007 document ‘Dementia UK: the full report’, Knapp and colleagues8 assessed mean annual informal care costs for those with late-onset dementia in 2005–6 as rising from £9246 for individuals with mild impairment to £17,223 for people with moderate symptoms, and, finally, to £27,096 for people with severe impairment.
Although informal care costs reduce when individuals with AD move into residential care,8 only Wolstenholme and colleagues196 were able to attach a clear accommodation and care cost increase of around £8000 per 4-month period for patients in institutional care, assuming that all other cost variables hold constant. This is at least partly owing to the lack of a ‘single assessment process’ (POST 278, February 2007) with a clear care pathway catering for people with AD throughout their disease progression and across all the agencies involved at various stages.
However, Figure 158 gives a clear picture of the split between the NHS (13%), social services (care-home costs at 44%), local authorities and other organisations, such as voluntary organisations (community social services costs at 24%) and individuals (self-funded care-home costs at 19%) in caring for dementia in 2007. 8
Further breakdown of individual costs is given in Table 198, although the allocation of these costs is by type of resource (e.g. health-care costs, social-care costs) rather than by funding organisation. 10
FIGURE 158.
The total estimated direct cost of dementia is £9.1B, the bulk of which relates to the cost of care home places. Reproduced from Improving services and support for people with dementia, NAO, 2007. 9 Source: Knapp et al. (2007). 8 Dementia UK: Report to the Alzheimer’s Society, King’s College London and London School of Economics and Political Science. Note: £5.76B, equivalent to almost two-thirds of the direct costs of dementia (i.e. excluding informal care costs), borne by families, the NHS and social services, relates to the provision of care home places for people with dementia. NHS costs and social services costs account for the remaining one-third of direct costs.
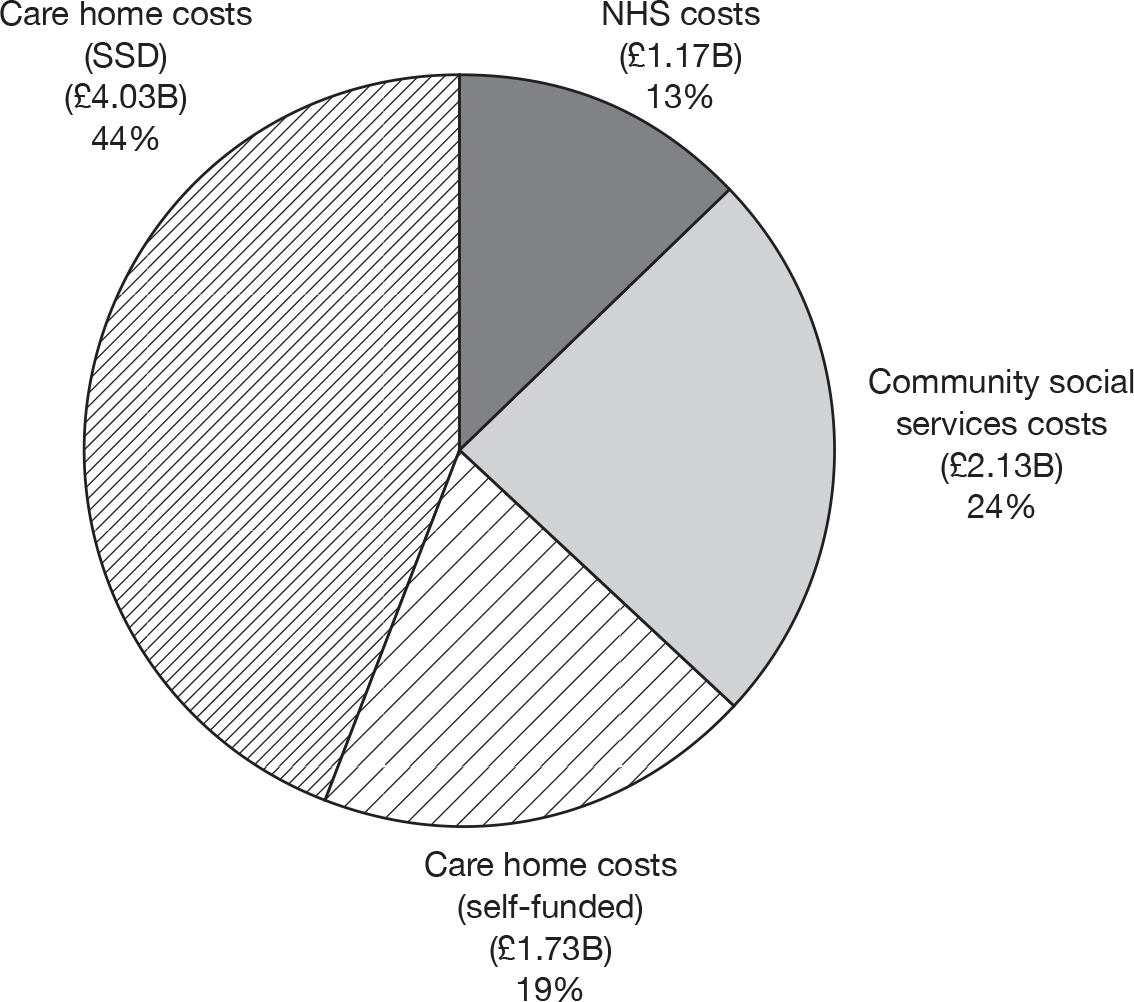
FIGURE 159.
Annual cost of services in the UK used by people with late-onset dementia. Source: Dementia UK: the full report by the Alzheimer’s Society 2007. 8
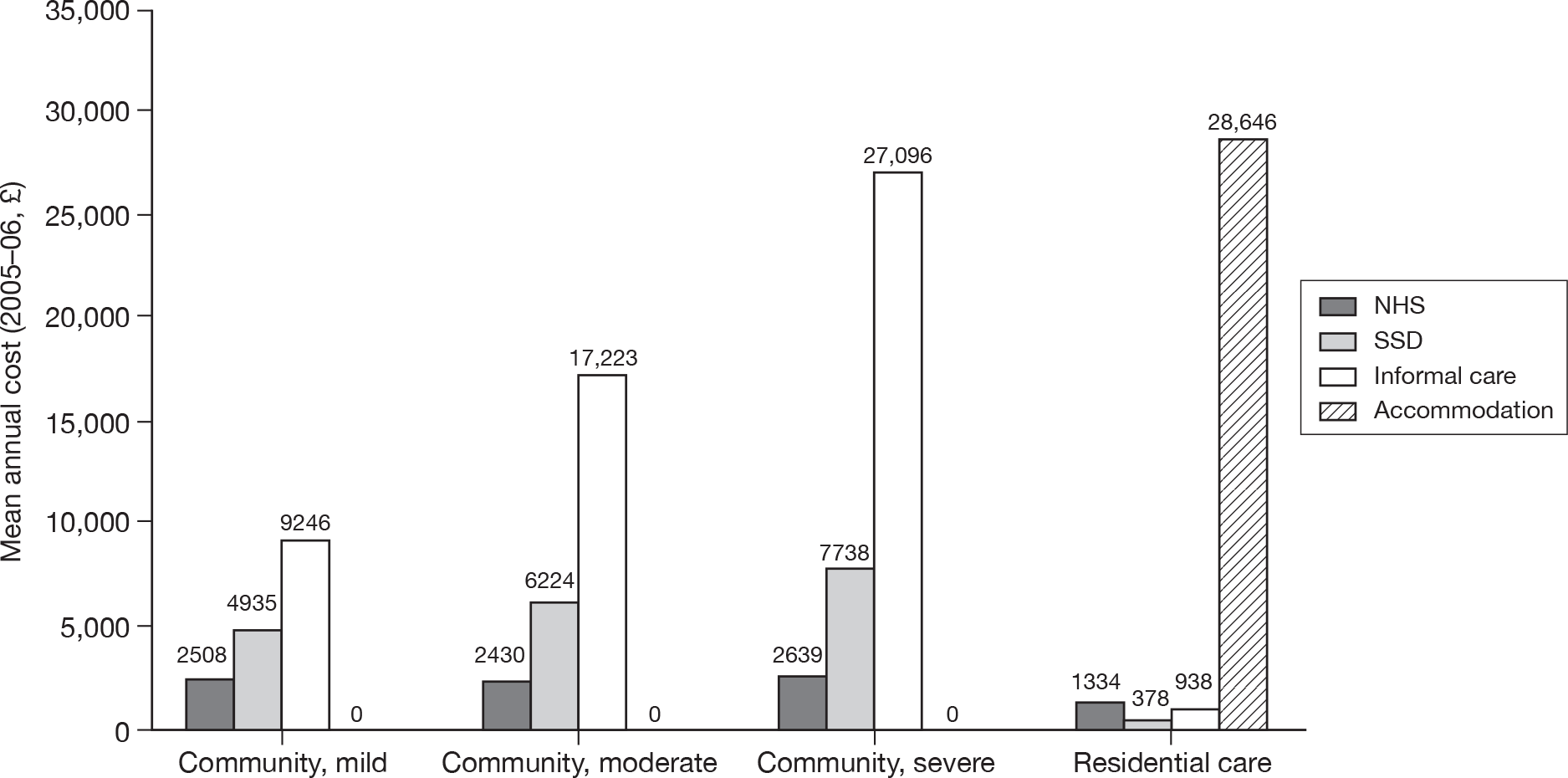
FIGURE 160.
Total annual cost of care for people aged 65 years and over with dementia in the UK. Source: Dementia UK: the full report by the Alzheimer’s Society 2010. 8
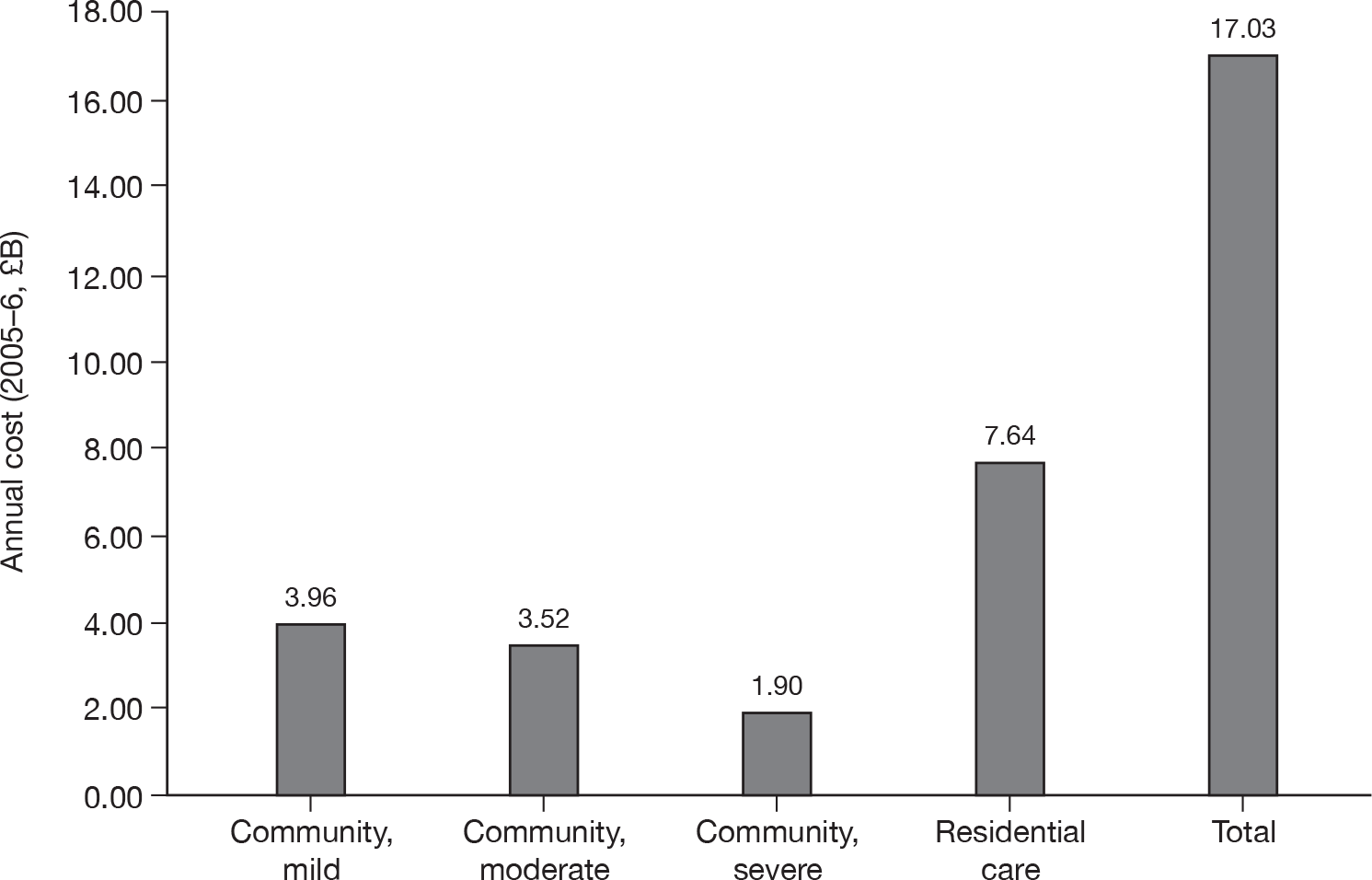
| Type of resource used | Unit of measurement | Units of resources consumed | Average unit cost (£) | Total cost (£000) |
|---|---|---|---|---|
| Health care | ||||
| Primary care | Nurse home visits | 2,492,220 | 26 | 64,798 |
| Nurse surgery visits | 186,753 | 9 | 1681 | |
| GP home visits | 3,567,046 | 58 | 206,889 | |
| GP surgery visits | 1,161,197 | 36 | 41,803 | |
| GP telephone visits | 83,939 | 22 | 1847 | |
| Total | ||||
| A&E | Attendances | 298,867 | 89 | 26,737 |
| Outpatient care | Attendances | 489,766 | 112 | 55,044 |
| Inpatient care | Hospital bed-days | 1,485,471 | 311 | 462,590 |
| Hospital day cases | 209 | 2755 | 576 | |
| Medications | 228,399 | |||
| Private care | Private part of total health expenditure | 12.7% | 109,469 | |
| Health-care cost subtotal | 1,199,832 | |||
| Social care | ||||
| Long-term care | Years in long-term care accommodation | 304,850 | 29,822 | 9,091,177 |
| Social care cost subtotal | 9,091,177 | |||
| Non-health/social care | ||||
| Informal care | Hours of care provided by economically active carers | 512,457,980 | 13 | 6,671,816 |
| Hours of care provided by economically inactive carers | 996,638,065 | 6 | 5,710,736 | |
| Mortality | Working-years lost (men) | 2025 | 32,838a | 22,515 |
| Working-years lost (women) | 1933 | 18,958 | 5994 | |
| Morbidity (friction adjusted) | Certified incapacity days | 160,603 | 104 | 16,743 |
| Work days lost | 38,380 | 104 | 4001 | |
| Non-health/social care subtotal (friction adjusted) | 12,431,804 | |||
| Total economic burden (friction adjusted) | 22,722,813 | |||
Appendix 16 Consideration of a two-dimensional Markov model for Alzheimer’s disease
The feasibility of a two-dimensional Markov model has been considered. Limitations for the development of such a model include structural uncertainty (such as how to translate the treatment effect measured and reported in RCTs to transition probabilities and/or state occupancy proportions for the Markov model) in addition to the limitations of data availability.
Background
Important predictors of QoL and cost were assessed to identify the variables most likely to be considered for the two-dimensional model, with institutionalisation the variable associated with largest cost changes, but unclear evidence as to the role of cognition, function and behaviour on the QoL of someone with AD (with behaviour and carer-related variables being found to be related to probability of institutionalisation). Further investigation reviewed the relationships between cognition, behaviour and function and the different measures used to reflect these variables. The review suggested some evidence for a correlation between cognition and functional status, whereas for cognition and behavioural status the evidence was unclear. Thus, leading to cognition and behavioural status as prime candidates for the two-dimensional model, although functional status was not totally ruled out.
Two-dimensional Markov model: cognitive status versus behavioural or functional status
Best supportive care cohort – Alzheimer’s disease progression
Individual personal development data for control groups were requested from manufacturers to model disease progression along two dimensions. IPD was also requested from two UK longitudinal studies: the LASER-AD study and the Oxfordshire data set. The majority of people in the LASER-AD study were treated with cholinesterase inhibitors; however, the data are of use for characterising disease progression in more severe patients.
Treatment effect
As noted below, the majority of available evidence on treatment effect is reported as mean difference between untreated and treated at a particular time point. There are very few, if any, data reported by cognition and another variable, for example only mean difference in MMSE score of 0.4 at 6 months, mean difference in NPI of 0.3, rather than of those with poor functional/behavioural status the mean difference in MMSE was 0.3, while for those with good functional/behavioural status the mean difference in MMSE was 0.6. We therefore have the problem of translating these mean differences into transition probabilities or state occupancy proportions (as in the one-dimensional model), but also have the added problem of coinciding treatment effects on cognition with treatment effects on functional or behavioural status.
Assuming the one-dimensional model, there are many questions in assuming how this measure of effectiveness is incorporated into transition probabilities for the treated cohort. One approach is to calculate the expected MMSE score at time t for a treated individual (point b on Figure 161) which is the expected score for an untreated individual plus the mean difference, (see Figure 161), assuming that decline between start of treatment and time t is constant (see line ab in Figure 161). It is then assumed that decline after time t continues at the same rate as that in the untreated individual, but that the treated individual is constantly x points above the untreated individual (see explanation of treatment effect for the one-dimensional Markov model below for discussion of this assumption if the Mendiondo and colleagues244 disease progression equation is used). The time to one-point change in the treated individual is then calculated as the time to a one-point change in the untreated individual plus z, the additional time spent at that MMSE score due to the treatment effect. Thus, allowing treatment to slow progression.
FIGURE 161.
Alzheimer’s disease progression based on MMSE for an untreated individual (thin line) and for a treated individual (thick line).
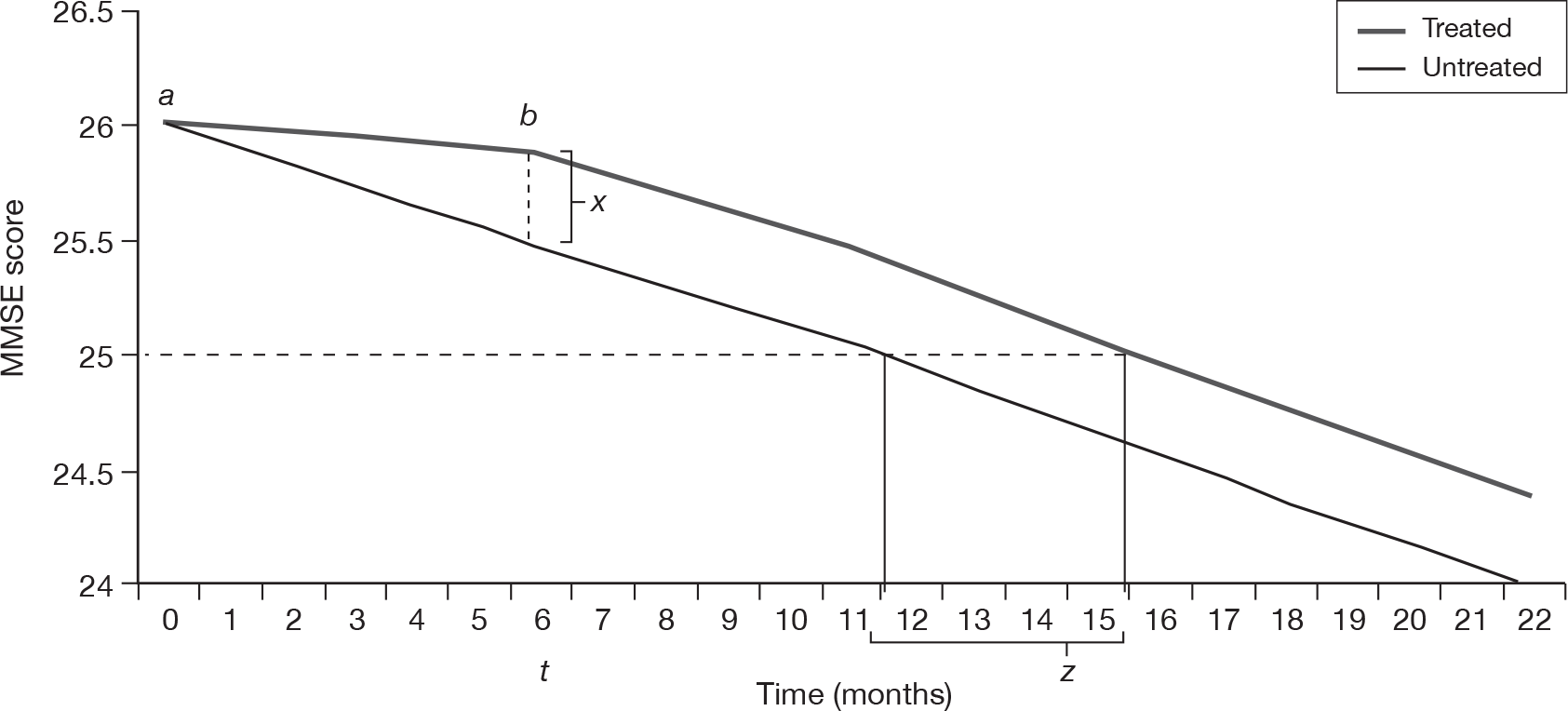
However, this extended time at MMSE scores only applies to earlier transitions, therefore some ‘memory’ has to be built into the model, where already there are 32 states. Of course, for a two-dimensional model, the number of states is twofold, although aggregation of cognition states may be possible if not using the Mendiondo and colleagues244 equation for disease progression.
It is also important to note that in applying the treatment effect to baseline data from elsewhere (e.g. IPD from UK study or the Mendiondo and colleagues244 equation), it is quite possible that an improvement in MMSE score is modelled rather than just allowing for a slowing of decline. It is unclear whether or not the evidence base agrees with an assumption than treatment can increase MMSE score, rather than delay decline.
Utilities
Utility data for MMSE is available. Utility data for functional status are also available, but are not independent of cognition score. Only utility data concerning depression can be identified for any type of behavioural symptom.
One-dimensional Markov model: cognitive status
Great deal of evidence to suggest that MMSE alone is not a good basis for summarising AD progression. Has MMSE been validated for AD?
Best supportive care cohort: Alzheimer’s disease progression
The Mendiondo and colleagues244 model can be used to inform AD progression in terms of the time to next point change on MMSE scale. Assuming a constant rate and an exponential function, the time-dependent probabilities for transition across MMSE scores can be obtained (Figure 162).
FIGURE 162.
Probability of time spent at a particular MMSE score.
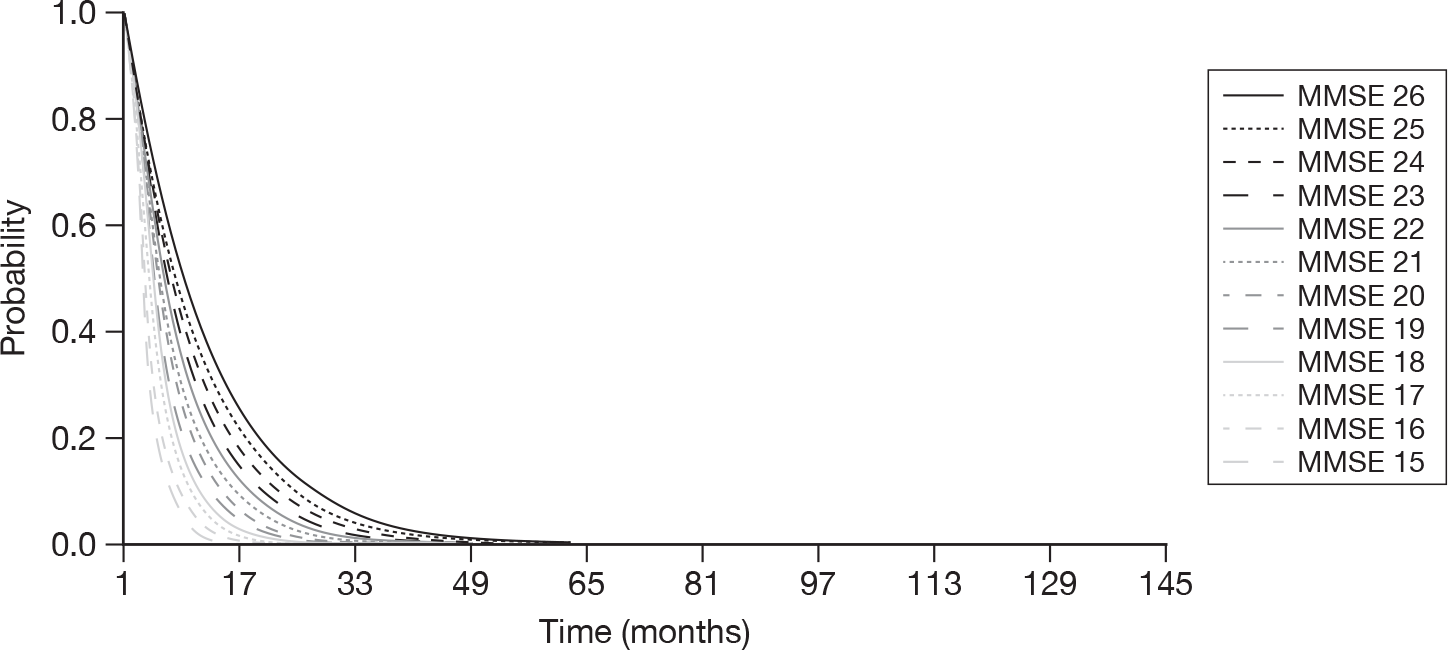
Treatment effect
Treatment effects are commonly reported as mean difference in MMSE between treated and untreated people with AD, for example at 6 months the mean difference is 0.4 point. See above for a description of the issues associated with translating the treatment effect into the decision model.
Additionally, as Figure 162 demonstrates, the probability of moving to the next MMSE score depends upon severity, and therefore assuming a decline of the same rate as the untreated individual for a treated individual after time t does not follow the equation of Mediondo and colleagues. 244
Utilities
Utility data by MMSE are available, including EQ-5D.
Costs
Cost data by MMSE are available.
Appendix 17 Published utility values for Alzheimer’s disease
| Source | Health-state utility scale | Sample | Factor | Category | Utility |
|---|---|---|---|---|---|
| Kerner et al.212 | QWB | Spousal proxy | 0.51 (SD 0.06) | ||
| Miller et al. 2008202 | HUI-3 | Carer proxy | Time | Baseline | 0.184 (range –0.291 to 1) |
| 3 months | 0.162 | ||||
| 6 months | 0.148 | ||||
| 9 months | 0.123 | ||||
| Sano et al. 1999213 | TTO | AD experts | CDR | Mild (CDR = 1) | 0.67 (SD 0.32) |
| Severe (CDR = 3) | 0.31 (SD 0.27) | ||||
| Students | Mild (CDR = 1) | 0.58 (SD 0.23) | |||
| Severe (CDR = 3) | 0.29 (SD 0.21) | ||||
| VAS | AD experts | CDR | Mild (CDR = 1) | 0.75 (SD 0.14) | |
| Severe (CDR = 3) | 0.26 (SD 0.18) | ||||
| Students | Mild (CDR = 1) | 0.65 (SD 0.17) | |||
| Severe (CDR = 3) | 0.30 (SD 0.13) | ||||
| Ekman et al. 2007204 | TTO | Members of public in Sweden aged 45–84 years | CDR | Mild cognitive impairment (CDR = 0.5) | 0.82 (SD 0.21) |
| Mild (CDR = 2) | 0.62 (SD 0.25) | ||||
| Moderate (CDR = 3) | 0.4 (SD 0.26) | ||||
| Severe (CDR = 3) | 0.25 (SD 0.28) | ||||
| Naglie et al. 2006203 | Patient-utility scores | Health status tool | EQ-5D | 0.86 | |
| QWB | 0.60 | ||||
| HUI-3 | 0.73 | ||||
| VAS (from EQ-5D) | 0.81 | ||||
| Carer-proxy scores | EQ-5D | 0.62 | |||
| QWB | 0.42 | ||||
| HUI-3 | 0.23 | ||||
| VAS (from EQ-5D) | 0.59 | ||||
| Andersen et al.206 | EQ-5D mapped from health status and ADL | Obtained from interviews with patients and carer | MMSE | MMSE > 20 | 0.636 (SD 0.2109) |
| 9 < MMSE < 20 | 0.596 (SD 0.2152) | ||||
| MMSE < 10 | 0.486 (SD 0.2191) | ||||
| Dependency | Independent | 0.641 (SD 0.1952) | |||
| Dependent | 0.343 (SD 0.2324) | ||||
| Residential status | Community | 0.621 (SD 0.2173) | |||
| Institution | 0.564 (SD 0.1861) | ||||
| Wlodarczyk et al. 2004205 | AQoL (extracted from figures 1 and 2) (95% CIs available and yet to be extracted) | Carer proxy | MMSE | 0–10 | 0.4 |
| 10–15 | 0.46 | ||||
| 15–20 | 0.475 | ||||
| 20–25 | 0.52 | ||||
| 25+ | 0.59 | ||||
| IADL | 0–2 | 0.36 | |||
| 3–5 | 0.5 | ||||
| 6–8 | 0.62 | ||||
| Patient | MMSE | 0–10 | 0.52 | ||
| 10–15 | 0.54 | ||||
| 15–20 | 0.61 | ||||
| 20–25 | 0.68 | ||||
| 25+ | 0.71 | ||||
| IADL | 0–2 | 0.53 | |||
| 3–5 | 0.62 | ||||
| 6–8 | 0.77 | ||||
| Karlawish et al.207 | EQ-5D | Patient self-ratings | MMSE | 24–29 | 0.78 (SD 0.261) |
| 20–23 | 0.8 (SD 0.228) | ||||
| 11–19 | 0.885 (SD 0.132) | ||||
| IADL | 8–10 | 0.885 (SD 0.136) | |||
| 11–14 | 0.835 (SD 0.249) | ||||
| 15–27 | 0.744 (SD 0.233) | ||||
| BADLS | 6 | 0.851 (SD 0.21) | |||
| 7–14 | 0.761 (SD 0.226) | ||||
| HUI-2 | Patient self-ratings | MMSE | 24–29 | 0.886 (SD 0.133) | |
| 20–23 | 0.846 (SD 0.19) | ||||
| 11–19 | 0.916 (SD 0.105) | ||||
| IADL | 8–10 | 0.941 (SD 0.084) | |||
| 11–14 | 0.894 (SD 0.129) | ||||
| 15–27 | 0.811 (SD 0.191) | ||||
| BADLS | 6 | 0.928 (SD 0.087) | |||
| 7–14 | 0.795 (SD 0.20) | ||||
| EQ-5D | Carer-proxy ratings | MMSE | 24–29 | 0.72 (SD 0.202) | |
| 20–23 | 0.63 (SD 0.251) | ||||
| 11–19 | 0.604 (SD 0.233) | ||||
| IADL | 8–18 | 0.753 (SD 0.219) | |||
| 19–24 | 0.7 (SD 0.183) | ||||
| 25–31 | 0.476 (SD 0.208) | ||||
| BADLS | 6 | 0.789 (SD 0.116) | |||
| 7–8 | 0.646 (SD 0.247) | ||||
| 9–22 | 0.519 (SD 0.233) | ||||
| HUI-2 | Carer-proxy ratings | MMSE | 24–29 | 0.763 (SD 0.158) | |
| 20–23 | 0.703 (SD 0.201) | ||||
| 11–19 | 0.707 (SD 0.172) | ||||
| IADL | 8–18 | 0.791 (SD 0.164) | |||
| 19–24 | 0.77 (SD 0.123) | ||||
| 25–31 | 0.595 (SD 0.185) | ||||
| BADLS | 6 | 0.791 (SD 0.144) | |||
| 7–8 | 0.752 (SD 0.154) | ||||
| 9–22 | 0.635 (SD 0.196) | ||||
| Neuman et al. 1999214 | HUI-2 | Carer-proxy | CDR | 0.5 | 0.73 |
| 1 | 0.69 | ||||
| 2 | 0.53 | ||||
| 3 | 0.38 | ||||
| 4 | 0.27 | ||||
| 5 | 0.14 | ||||
| Jonsson et al. 2006187 | EQ-5D | Self-ratings (both carer and self-ratings available) | MMSE | 26–30 | 0.84 |
| 21–25 | 0.85 | ||||
| 15–20 | 0.83 | ||||
| 10–15 | 0.73 | ||||
| 0–9 | 0.78 | ||||
| Carer proxy (both carer and self-ratings available) | 26–30 | 0.7 | |||
| 21–25 | 0.65 | ||||
| 15–20 | 0.52 | ||||
| 10–15 | 0.51 | ||||
| 0–9 | 0.4 | ||||
| Only carer-proxy ratings available | 26–30 | 0.5 | |||
| 21–25 | 0.19 | ||||
| 15–20 | 0.21 | ||||
| 10–15 | 0.39 | ||||
| 0–9 | 0.22 | ||||
| Only self-ratings available | 26–30 | 0.81 | |||
| 21–25 | 0.78 | ||||
| 15–20 | 0.82 | ||||
| 10–15 | 1 | ||||
| 0–9 | 0.94 |
Appendix 18 Figures from the statistical analysis of individual patient data from Wolstenholme and colleagues196
FIGURE 163.
Inflated cost per month as a function of time until pre-institutionalisation for each of 92 patients with AD.

FIGURE 164.
Mini Mental State Examination as a function of time until end of pre-institutionalisation for each of 92 patients with AD.
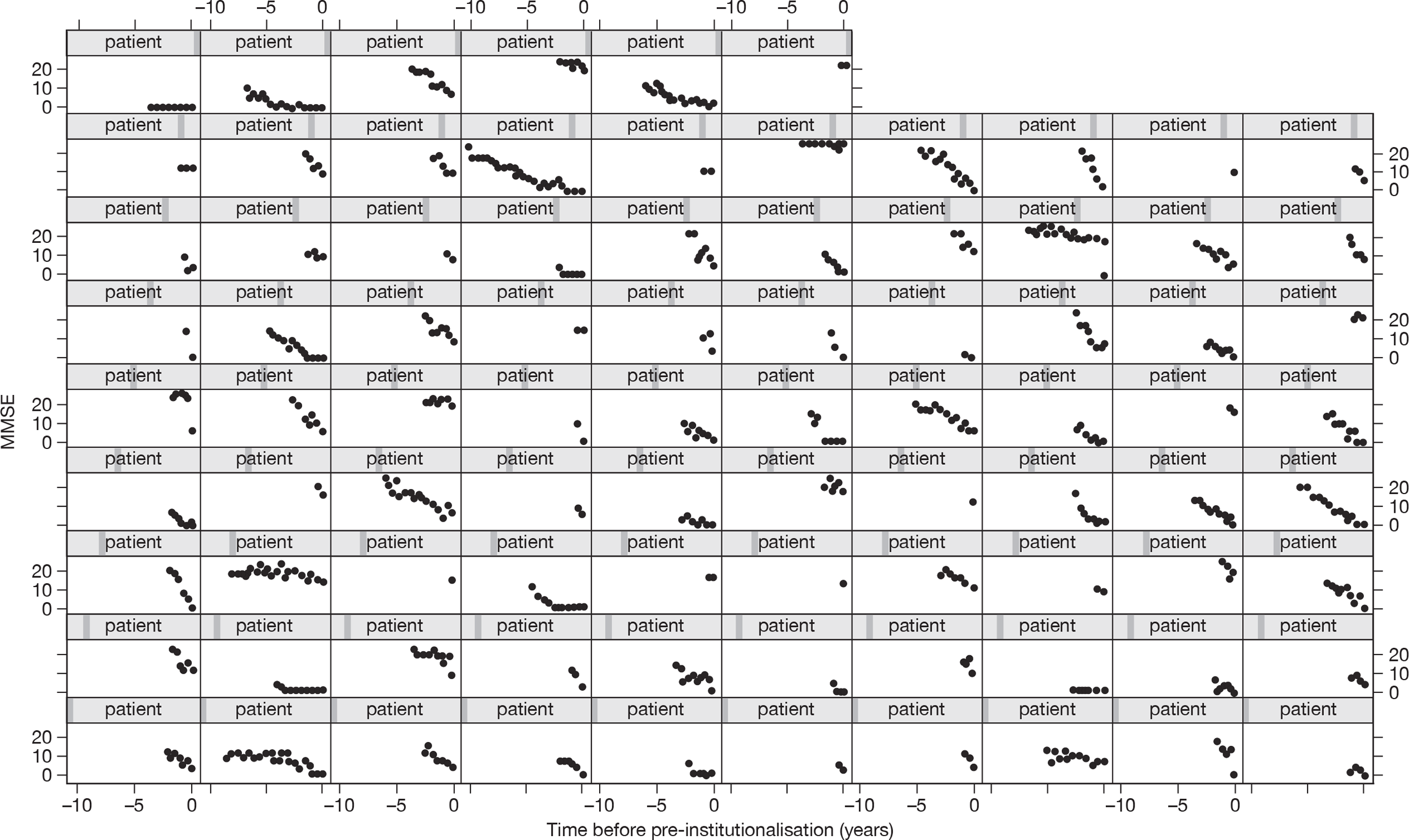
Appendix 19 Graphical presentation of distributions for probabilistic sensitivity analysis
FIGURE 165.
Density plots of the effectiveness parameters included in the PSAs (refer to Chapter 6).
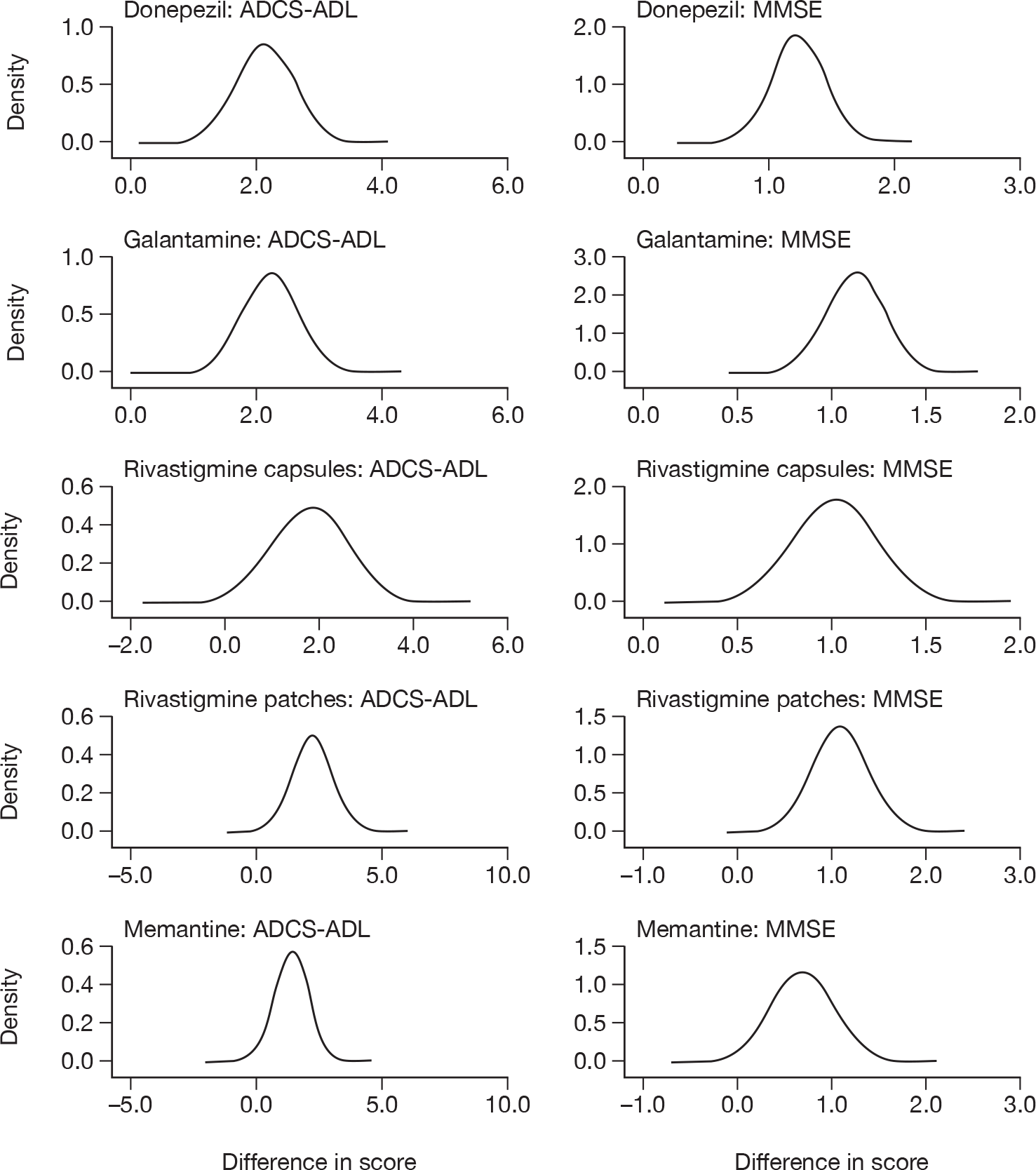
FIGURE 166.
Density plots of the uncertain cost parameters included in the PSAs.
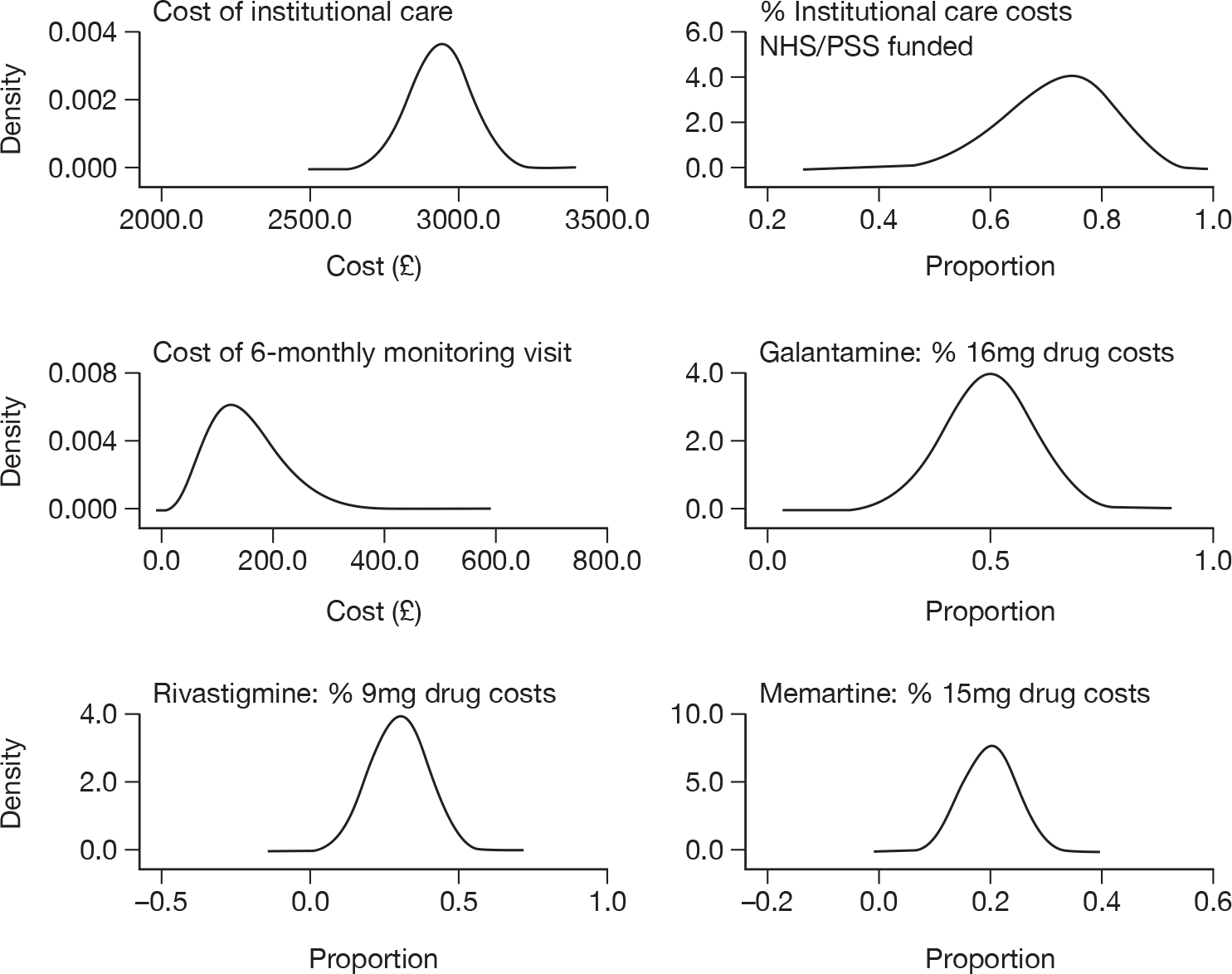
FIGURE 167.
Density plots of the utility estimates included in the PSAs.
FIGURE 168.
Density plots of other uncertain parameters included in the PSAs.
Appendix 20 Tornado plots for acetylcholinesterase inhibitor versus best supportive care
Tornado plots for comparisons between BSC and galantamine (Figure 169), rivastigmine capsules (Figure 170) and rivastigmine patches (Figure 171) in the base-case analyses for people with mild to moderate AD.
FIGURE 169.
Galantamine vs BSC.
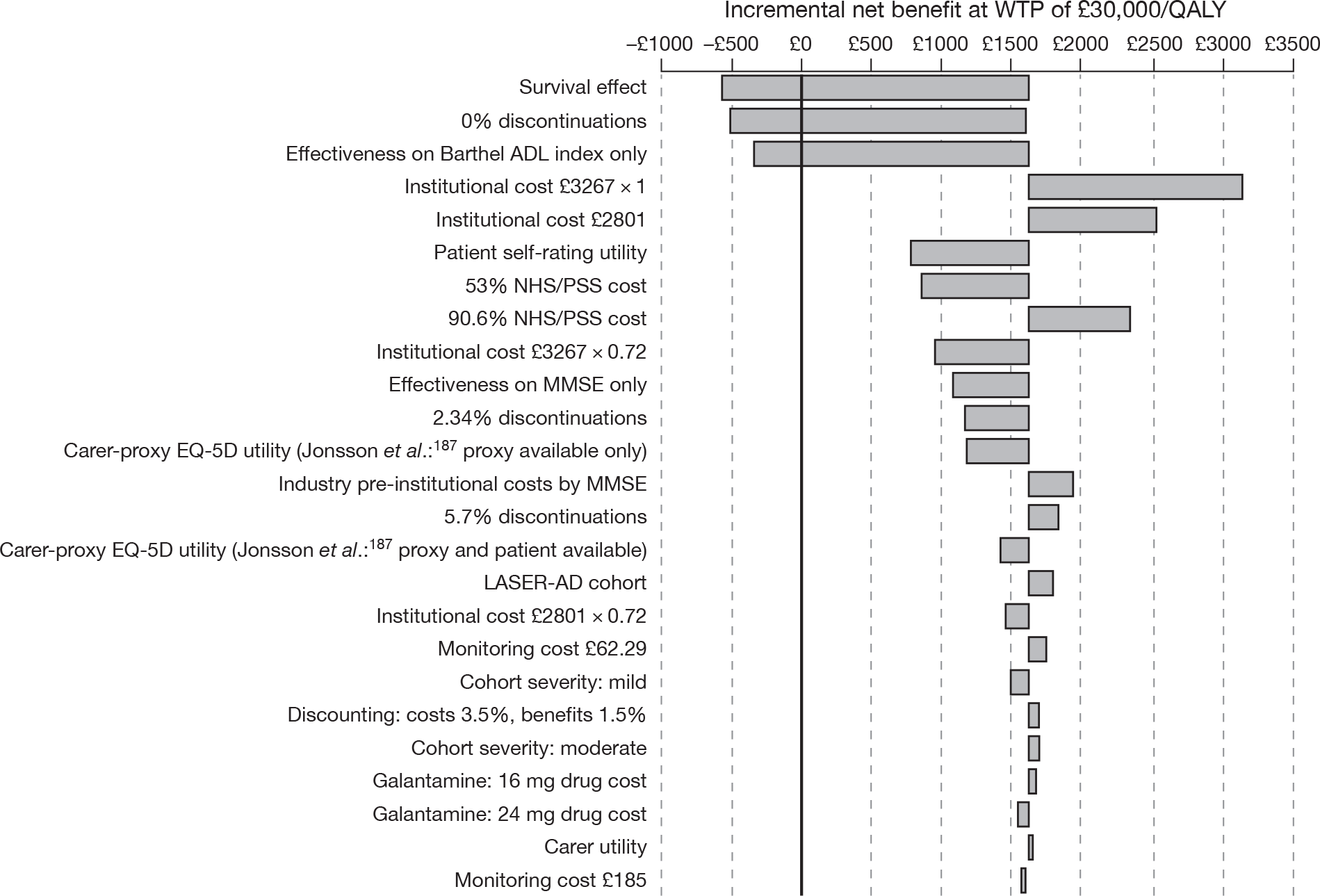
FIGURE 170.
Rivastigmine capsules vs BSC.
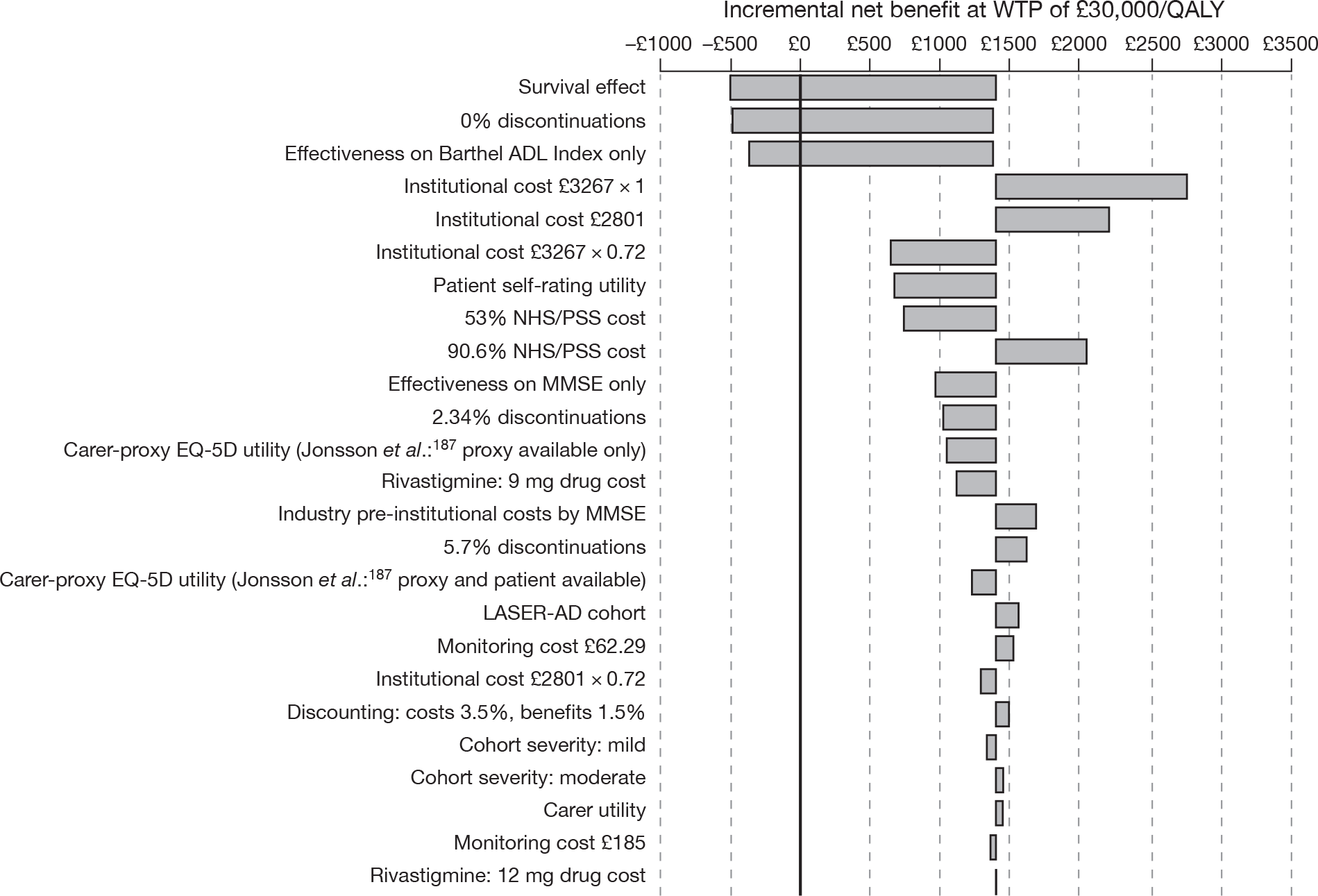
FIGURE 171.
Rivastigmine patches vs BSC.
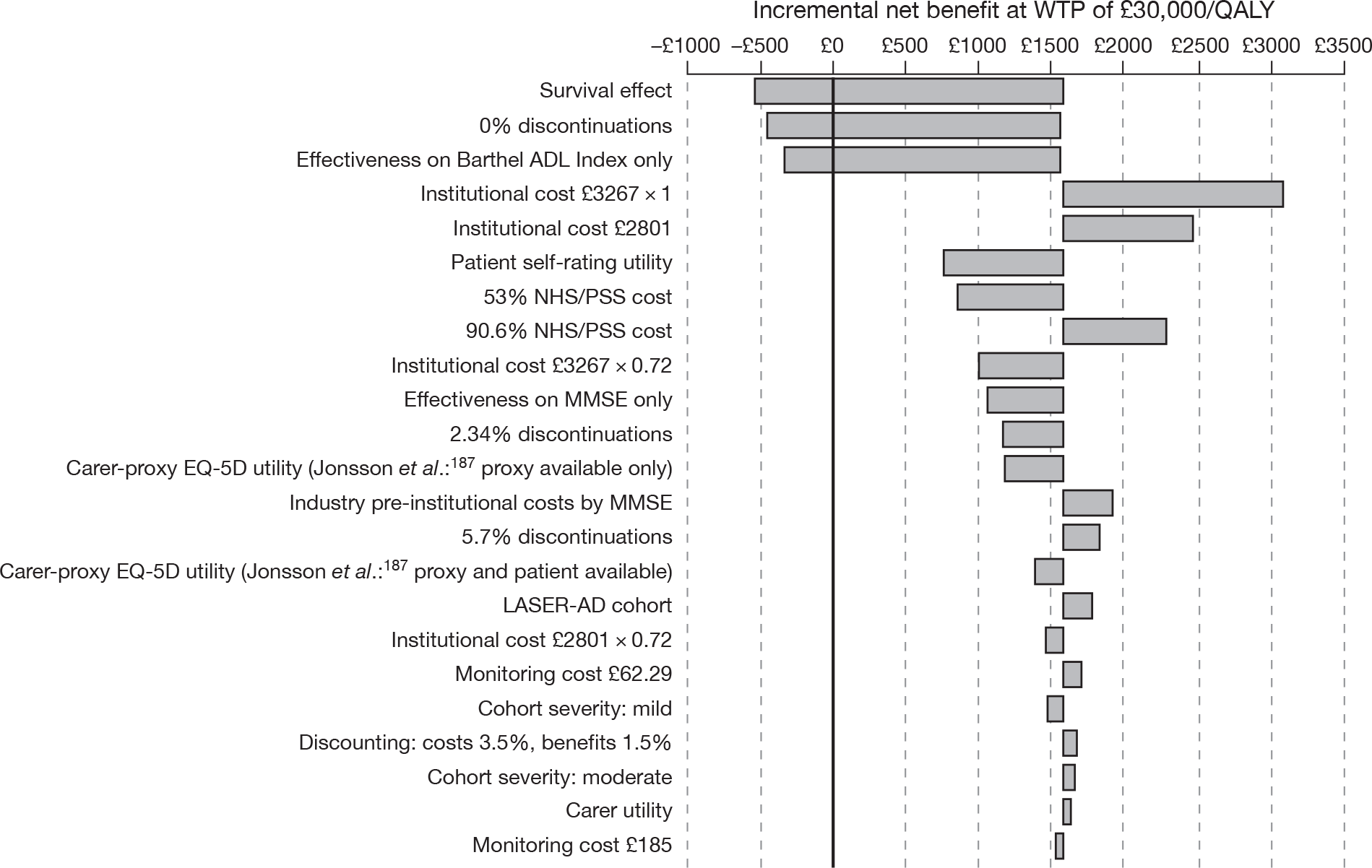
List of abbreviations
- AChEI
- acetylcholinesterase inhibitor
- AD
- Alzheimer’s disease
- ADAS-cog
- Alzheimer’s Disease Assessment Scale – Cognitive Subscale
- ADCS-ADL
- Alzheimer’s Disease Cooperative Study – Activities of Daily Living Index
- ADCS-CGIC
- Alzheimer’s Disease Cooperative Study – Clinical Global Impression of Change
- ADL
- Activities of Daily Living Scale
- AE
- adverse event
- AHEAD
- Assessment of Health Economics in Alzheimer’s disease
- APS
- agitation/aggression and/or psychotic symptoms
- AQoL
- Assessment of Quality of Life
- BADLS
- Bristol Activities of Daily Living Scale
- BEHAVE-AD
- Behaviour Pathology in Alzheimer’s Disease
- BGP
- Behavioural Rating Scale for Geriatric Patients
- BNF
- British National Formulary
- BPSD
- behavioural and psychological symptoms of dementia
- BSC
- best supportive care
- CDN
- Canadian
- CDR
- Clinical Dementia Rating Scale
- CEA
- cost-effectiveness analysis
- CHEC
- City Health Economics Centre
- CI
- confidence interval
- CIBIC
- Clinician’s Interview-based Impression of Change
- CIBIC-plus
- Clinician’s Interview-based Impression of Change – plus Caregiver Input
- CiC
- commercial-in-confidence
- CRD
- Centre for Reviews and Dissemination
- CSRI
- Client Service Receipt Inventory
- CUA
- cost–utility analysis
- DAD
- Disability Assessment for Dementia scale
- DES
- discrete-event simulation
- DOMINO-AD
- Donepezil and Memantine in Moderate-to-Severe Alzheimer’s Disease
- DSM-IV
- Diagnostic and Statistical Manual of Mental Disorders-Fourth Edition
- DSU
- Decision Support Unit
- EQ-5D
- European Quality of Life-5 Dimensions
- FAST
- Functioning Assessment Staging Tool
- FTC
- full-time care
- GAS
- Goal Attainment Scale
- GDS
- Global Deterioration Scale
- HR
- hazard ratio
- HRQoL
- health-related quality of life
- HTA
- health technology assessment
- HUI-2
- Health Utilities Index version 2
- HUI-3
- Health Utilities Index version 3
- IADL
- Instrumental Activities of Daily Living
- ICD-10
- International Classification of Diseases, 10th Revision
- ICER
- incremental cost-effectiveness ratio
- IFTC
- institutionalised full-time care
- IPD
- individual patient data
- IQR
- interquartile range
- IQWiG
- Institute for Quality and Efficiency in Health Care
- ISPOR
- International Society for Pharmacoeconomics and Outcomes Research
- ISRCTN
- International Standard Randomised Controlled Trial Number
- ITT
- intention to treat
- LOCF
- last observation carried forward
- MCMC
- Markov Chain Monte Carlo
- MMSE
- Mini Mental State Examination
- MRC CFAS
- Medical Research Council's Cognitive Function and Ageing Study
- MRI
- magnetic resonance imaging
- MTA
- multiple technology appraisal
- MTC
- mixed-treatment comparison
- NAO
- National Audit Office
- NHS CRD
- NHS Centre for Reviews and Dissemination
- NHS EED
- NHS Economic Evaluation Database
- NICE
- National Institute for Health and Clinical Excellence
- NMDA
- N-methyl-d-aspartate
- NPI
- Neuropsychiatric Inventory
- OC
- observed case
- ONS
- Office for National Statistics
- PCT
- primary care trust
- PDS
- Progressive Deterioration Scale
- PenTAG
- Peninsula Technology Assessment Group
- PET
- positron emission tomography
- POST
- Parliamentary Office of Science and Technology
- PRC
- prolonged-release capsule
- PRISMA
- Preferred Reporting Items for Systematic Reviews and Meta-Analyses
- PSA
- probabilistic sensitivity analysis
- PSS
- Personal Social Services
- PSSRU
- Personal Social Services Research Unit
- QALY
- quality-adjusted life-year
- QoL
- quality of life
- QoL-AD
- Quality of Life in Alzheimer's Disease Scale
- QWB
- Quality of Well-being
- RCT
- randomised controlled trial
- SD
- standard deviation
- SE
- standard error
- SF-12
- Short Form questionnaire-12 items
- SF-36
- Short Form questionnaire-36 items
- SHTAC
- Southampton Health Technology Assessment Centre
- SIB
- Severe Impairment Battery
- SMD
- standardised mean difference
- SSD
- Social Services Department
- STA
- single technology appraisal
- TAG
- Technology Assessment Group
- TA
- technology appraisal
- TAR
- technology assessment report
- TTE
- time to event
- TTO
- time trade-off
- VAS
- visual analogue scale
- WMD
- weighted mean difference
- WTP
- willingness to pay
All abbreviations that have been used in this report are listed here unless the abbreviation is well known (e.g. NHS), or it has been used only once, or it is a non-standard abbreviation used only in figures/tables/appendices, in which case the abbreviation is defined in the figure legend or in the notes at the end of the table.
NoteThis monograph is based on the Technology Assessment Report produced for NICE. The full report contained a considerable number of data that were deemed commercial-in-confidence. The full report was used by the Appraisal Committee at NICE in their deliberations. The full report with each piece of commercial-in-confidence data removed and replaced by the statement ‘commercial-in-confidence information (or data) removed’ is available on the NICE website: www.nice.org.uk.
The present monograph presents as full a version of the report as is possible while retaining readability, but some sections, sentences, tables and figures have been removed. Readers should bear in mind that the discussion, conclusions and implications for practice and research are based on all the data considered in the original full NICE report.
Notes
Health Technology Assessment programme
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
Prioritisation Group
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Imti Choonara, Professor in Child Health, Academic Division of Child Health, University of Nottingham
Chair – Pharmaceuticals Panel
-
Dr Bob Coates, Consultant Advisor – Disease Prevention Panel
-
Dr Andrew Cook, Consultant Advisor – Intervention Procedures Panel
-
Dr Peter Davidson, Director of NETSCC, Health Technology Assessment
-
Dr Nick Hicks, Consultant Adviser – Diagnostic Technologies and Screening Panel, Consultant Advisor–Psychological and Community Therapies Panel
-
Ms Susan Hird, Consultant Advisor, External Devices and Physical Therapies Panel
-
Professor Sallie Lamb, Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick
Chair – HTA Clinical Evaluation and Trials Board
-
Professor Jonathan Michaels, Professor of Vascular Surgery, Sheffield Vascular Institute, University of Sheffield
Chair – Interventional Procedures Panel
-
Professor Ruairidh Milne, Director – External Relations
-
Dr John Pounsford, Consultant Physician, Directorate of Medical Services, North Bristol NHS Trust
Chair – External Devices and Physical Therapies Panel
-
Dr Vaughan Thomas, Consultant Advisor – Pharmaceuticals Panel, Clinical
Lead – Clinical Evaluation Trials Prioritisation Group
-
Professor Margaret Thorogood, Professor of Epidemiology, Health Sciences Research Institute, University of Warwick
Chair – Disease Prevention Panel
-
Professor Lindsay Turnbull, Professor of Radiology, Centre for the MR Investigations, University of Hull
Chair – Diagnostic Technologies and Screening Panel
-
Professor Scott Weich, Professor of Psychiatry, Health Sciences Research Institute, University of Warwick
Chair – Psychological and Community Therapies Panel
-
Professor Hywel Williams, Director of Nottingham Clinical Trials Unit, Centre of Evidence-Based Dermatology, University of Nottingham
Chair – HTA Commissioning Board
Deputy HTA Programme Director
HTA Commissioning Board
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
-
Department of Public Health and Epidemiology, University of Birmingham
-
Professor of Clinical Pharmacology, Director, NIHR HTA programme, University of Liverpool
-
Professor Ann Ashburn, Professor of Rehabilitation and Head of Research, Southampton General Hospital
-
Professor Judith Bliss, Director of ICR-Clinical Trials and Statistics Unit, The Institute of Cancer Research
-
Professor Peter Brocklehurst, Professor of Women’s Health, Institute for Women’s Health, University College London
-
Professor David Fitzmaurice, Professor of Primary Care Research, Department of Primary Care Clinical Sciences, University of Birmingham
-
Professor John W Gregory, Professor in Paediatric Endocrinology, Department of Child Health, Wales School of Medicine, Cardiff University
-
Professor Steve Halligan, Professor of Gastrointestinal Radiology, University College Hospital, London
-
Professor Angela Harden, Professor of Community and Family Health, Institute for Health and Human Development, University of East London
-
Dr Martin J Landray, Reader in Epidemiology, Honorary Consultant Physician, Clinical Trial Service Unit, University of Oxford
-
Dr Joanne Lord, Reader, Health Economics Research Group, Brunel University
-
Professor Stephen Morris, Professor of Health Economics, University College London, Research Department of Epidemiology and Public Health, University College London
-
Professor Dion Morton, Professor of Surgery, Academic Department of Surgery, University of Birmingham
-
Professor Gail Mountain, Professor of Health Services Research, Rehabilitation and Assistive Technologies Group, University of Sheffield
-
Professor Irwin Nazareth, Professor of Primary Care and Head of Department, Department of Primary Care and Population Sciences, University College London
-
Professor E Andrea Nelson, Professor of Wound Healing and Director of Research, School of Healthcare, University of Leeds
-
Professor John David Norrie, Chair in Clinical Trials and Biostatistics, Robertson Centre for Biostatistics, University of Glasgow
-
Dr Rafael Perera, Lecturer in Medical Statisitics, Department of Primary Health Care, University of Oxford
-
Professor Barney Reeves, Professorial Research Fellow in Health Services Research, Department of Clinical Science, University of Bristol
-
Professor Peter Tyrer, Professor of Community Psychiatry, Centre for Mental Health, Imperial College London
-
Professor Martin Underwood, Professor of Primary Care Research, Warwick Medical School, University of Warwick
-
Professor Caroline Watkins, Professor of Stroke and Older People’s Care, Chair of UK Forum for Stroke Training, Stroke Practice Research Unit, University of Central Lancashire
-
Dr Duncan Young, Senior Clinical Lecturer and Consultant, Nuffield Department of Anaesthetics, University of Oxford
-
Dr Tom Foulks, Medical Research Council
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
HTA Clinical Evaluation and Trials Board
-
Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick and Professor of Rehabilitation, Nuffield Department of Orthopaedic, Rheumatology and Musculoskeletal Sciences, University of Oxford
-
Professor of the Psychology of Health Care, Leeds Institute of Health Sciences, University of Leeds
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Keith Abrams, Professor of Medical Statistics, Department of Health Sciences, University of Leicester
-
Professor Martin Bland, Professor of Health Statistics, Department of Health Sciences, University of York
-
Professor Jane Blazeby, Professor of Surgery and Consultant Upper GI Surgeon, Department of Social Medicine, University of Bristol
-
Professor Julia M Brown, Director, Clinical Trials Research Unit, University of Leeds
-
Professor Alistair Burns, Professor of Old Age Psychiatry, Psychiatry Research Group, School of Community-Based Medicine, The University of Manchester & National Clinical Director for Dementia, Department of Health
-
Dr Jennifer Burr, Director, Centre for Healthcare Randomised trials (CHART), University of Aberdeen
-
Professor Linda Davies, Professor of Health Economics, Health Sciences Research Group, University of Manchester
-
Professor Simon Gilbody, Prof of Psych Medicine and Health Services Research, Department of Health Sciences, University of York
-
Professor Steven Goodacre, Professor and Consultant in Emergency Medicine, School of Health and Related Research, University of Sheffield
-
Professor Dyfrig Hughes, Professor of Pharmacoeconomics, Centre for Economics and Policy in Health, Institute of Medical and Social Care Research, Bangor University
-
Professor Paul Jones, Professor of Respiratory Medicine, Department of Cardiac and Vascular Science, St George‘s Hospital Medical School, University of London
-
Professor Khalid Khan, Professor of Women’s Health and Clinical Epidemiology, Barts and the London School of Medicine, Queen Mary, University of London
-
Professor Richard J McManus, Professor of Primary Care Cardiovascular Research, Primary Care Clinical Sciences Building, University of Birmingham
-
Professor Helen Rodgers, Professor of Stroke Care, Institute for Ageing and Health, Newcastle University
-
Professor Ken Stein, Professor of Public Health, Peninsula Technology Assessment Group, Peninsula College of Medicine and Dentistry, Universities of Exeter and Plymouth
-
Professor Jonathan Sterne, Professor of Medical Statistics and Epidemiology, Department of Social Medicine, University of Bristol
-
Mr Andy Vail, Senior Lecturer, Health Sciences Research Group, University of Manchester
-
Professor Clare Wilkinson, Professor of General Practice and Director of Research North Wales Clinical School, Department of Primary Care and Public Health, Cardiff University
-
Dr Ian B Wilkinson, Senior Lecturer and Honorary Consultant, Clinical Pharmacology Unit, Department of Medicine, University of Cambridge
-
Ms Kate Law, Director of Clinical Trials, Cancer Research UK
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
Diagnostic Technologies and Screening Panel
-
Scientific Director of the Centre for Magnetic Resonance Investigations and YCR Professor of Radiology, Hull Royal Infirmary
-
Professor Judith E Adams, Consultant Radiologist, Manchester Royal Infirmary, Central Manchester & Manchester Children’s University Hospitals NHS Trust, and Professor of Diagnostic Radiology, University of Manchester
-
Mr Angus S Arunkalaivanan, Honorary Senior Lecturer, University of Birmingham and Consultant Urogynaecologist and Obstetrician, City Hospital, Birmingham
-
Dr Diana Baralle, Consultant and Senior Lecturer in Clinical Genetics, University of Southampton
-
Dr Stephanie Dancer, Consultant Microbiologist, Hairmyres Hospital, East Kilbride
-
Dr Diane Eccles, Professor of Cancer Genetics, Wessex Clinical Genetics Service, Princess Anne Hospital
-
Dr Trevor Friedman, Consultant Liason Psychiatrist, Brandon Unit, Leicester General Hospital
-
Dr Ron Gray, Consultant, National Perinatal Epidemiology Unit, Institute of Health Sciences, University of Oxford
-
Professor Paul D Griffiths, Professor of Radiology, Academic Unit of Radiology, University of Sheffield
-
Mr Martin Hooper, Public contributor
-
Professor Anthony Robert Kendrick, Associate Dean for Clinical Research and Professor of Primary Medical Care, University of Southampton
-
Dr Nicola Lennard, Senior Medical Officer, MHRA
-
Dr Anne Mackie, Director of Programmes, UK National Screening Committee, London
-
Mr David Mathew, Public contributor
-
Dr Michael Millar, Consultant Senior Lecturer in Microbiology, Department of Pathology & Microbiology, Barts and The London NHS Trust, Royal London Hospital
-
Mrs Una Rennard, Public contributor
-
Dr Stuart Smellie, Consultant in Clinical Pathology, Bishop Auckland General Hospital
-
Ms Jane Smith, Consultant Ultrasound Practitioner, Leeds Teaching Hospital NHS Trust, Leeds
-
Dr Allison Streetly, Programme Director, NHS Sickle Cell and Thalassaemia Screening Programme, King’s College School of Medicine
-
Dr Matthew Thompson, Senior Clinical Scientist and GP, Department of Primary Health Care, University of Oxford
-
Dr Alan J Williams, Consultant Physician, General and Respiratory Medicine, The Royal Bournemouth Hospital
-
Dr Tim Elliott, Team Leader, Cancer Screening, Department of Health
-
Dr Joanna Jenkinson, Board Secretary, Neurosciences and Mental Health Board (NMHB), Medical Research Council
-
Professor Julietta Patrick, Director, NHS Cancer Screening Programme, Sheffield
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Disease Prevention Panel
-
Professor of Epidemiology, University of Warwick Medical School, Coventry
-
Dr Robert Cook, Clinical Programmes Director, Bazian Ltd, London
-
Dr Colin Greaves, Senior Research Fellow, Peninsula Medical School (Primary Care)
-
Mr Michael Head, Public contributor
-
Professor Cathy Jackson, Professor of Primary Care Medicine, Bute Medical School, University of St Andrews
-
Dr Russell Jago, Senior Lecturer in Exercise, Nutrition and Health, Centre for Sport, Exercise and Health, University of Bristol
-
Dr Julie Mytton, Consultant in Child Public Health, NHS Bristol
-
Professor Irwin Nazareth, Professor of Primary Care and Director, Department of Primary Care and Population Sciences, University College London
-
Dr Richard Richards, Assistant Director of Public Health, Derbyshire County Primary Care Trust
-
Professor Ian Roberts, Professor of Epidemiology and Public Health, London School of Hygiene & Tropical Medicine
-
Dr Kenneth Robertson, Consultant Paediatrician, Royal Hospital for Sick Children, Glasgow
-
Dr Catherine Swann, Associate Director, Centre for Public Health Excellence, NICE
-
Mrs Jean Thurston, Public contributor
-
Professor David Weller, Head, School of Clinical Science and Community Health, University of Edinburgh
-
Ms Christine McGuire, Research & Development, Department of Health
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
External Devices and Physical Therapies Panel
-
Consultant Physician North Bristol NHS Trust
-
Reader in Wound Healing and Director of Research, University of Leeds
-
Professor Bipin Bhakta, Charterhouse Professor in Rehabilitation Medicine, University of Leeds
-
Mrs Penny Calder, Public contributor
-
Dr Dawn Carnes, Senior Research Fellow, Barts and the London School of Medicine and Dentistry
-
Dr Emma Clark, Clinician Scientist Fellow & Cons. Rheumatologist, University of Bristol
-
Mrs Anthea De Barton-Watson, Public contributor
-
Professor Nadine Foster, Professor of Musculoskeletal Health in Primary Care Arthritis Research, Keele University
-
Dr Shaheen Hamdy, Clinical Senior Lecturer and Consultant Physician, University of Manchester
-
Professor Christine Norton, Professor of Clinical Nursing Innovation, Bucks New University and Imperial College Healthcare NHS Trust
-
Dr Lorraine Pinnigton, Associate Professor in Rehabilitation, University of Nottingham
-
Dr Kate Radford, Senior Lecturer (Research), University of Central Lancashire
-
Mr Jim Reece, Public contributor
-
Professor Maria Stokes, Professor of Neuromusculoskeletal Rehabilitation, University of Southampton
-
Dr Pippa Tyrrell, Senior Lecturer/Consultant, Salford Royal Foundation Hospitals’ Trust and University of Manchester
-
Dr Nefyn Williams, Clinical Senior Lecturer, Cardiff University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Interventional Procedures Panel
-
Professor of Vascular Surgery, University of Sheffield
-
Consultant Colorectal Surgeon, Bristol Royal Infirmary
-
Mrs Isabel Boyer, Public contributor
-
Mr Sankaran Chandra Sekharan, Consultant Surgeon, Breast Surgery, Colchester Hospital University NHS Foundation Trust
-
Professor Nicholas Clarke, Consultant Orthopaedic Surgeon, Southampton University Hospitals NHS Trust
-
Ms Leonie Cooke, Public contributor
-
Mr Seumas Eckford, Consultant in Obstetrics & Gynaecology, North Devon District Hospital
-
Professor Sam Eljamel, Consultant Neurosurgeon, Ninewells Hospital and Medical School, Dundee
-
Dr Adele Fielding, Senior Lecturer and Honorary Consultant in Haematology, University College London Medical School
-
Dr Matthew Hatton, Consultant in Clinical Oncology, Sheffield Teaching Hospital Foundation Trust
-
Dr John Holden, General Practitioner, Garswood Surgery, Wigan
-
Dr Fiona Lecky, Senior Lecturer/Honorary Consultant in Emergency Medicine, University of Manchester/Salford Royal Hospitals NHS Foundation Trust
-
Dr Nadim Malik, Consultant Cardiologist/Honorary Lecturer, University of Manchester
-
Mr Hisham Mehanna, Consultant & Honorary Associate Professor, University Hospitals Coventry & Warwickshire NHS Trust
-
Dr Jane Montgomery, Consultant in Anaesthetics and Critical Care, South Devon Healthcare NHS Foundation Trust
-
Professor Jon Moss, Consultant Interventional Radiologist, North Glasgow Hospitals University NHS Trust
-
Dr Simon Padley, Consultant Radiologist, Chelsea & Westminster Hospital
-
Dr Ashish Paul, Medical Director, Bedfordshire PCT
-
Dr Sarah Purdy, Consultant Senior Lecturer, University of Bristol
-
Dr Matthew Wilson, Consultant Anaesthetist, Sheffield Teaching Hospitals NHS Foundation Trust
-
Professor Yit Chiun Yang, Consultant Ophthalmologist, Royal Wolverhampton Hospitals NHS Trust
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Pharmaceuticals Panel
-
Professor in Child Health, University of Nottingham
-
Senior Lecturer in Clinical Pharmacology, University of East Anglia
-
Dr Martin Ashton-Key, Medical Advisor, National Commissioning Group, NHS London
-
Dr Peter Elton, Director of Public Health, Bury Primary Care Trust
-
Dr Ben Goldacre, Research Fellow, Division of Psychological Medicine and Psychiatry, King’s College London
-
Dr James Gray, Consultant Microbiologist, Department of Microbiology, Birmingham Children’s Hospital NHS Foundation Trust
-
Dr Jurjees Hasan, Consultant in Medical Oncology, The Christie, Manchester
-
Dr Carl Heneghan, Deputy Director Centre for Evidence-Based Medicine and Clinical Lecturer, Department of Primary Health Care, University of Oxford
-
Dr Dyfrig Hughes, Reader in Pharmacoeconomics and Deputy Director, Centre for Economics and Policy in Health, IMSCaR, Bangor University
-
Dr Maria Kouimtzi, Pharmacy and Informatics Director, Global Clinical Solutions, Wiley-Blackwell
-
Professor Femi Oyebode, Consultant Psychiatrist and Head of Department, University of Birmingham
-
Dr Andrew Prentice, Senior Lecturer and Consultant Obstetrician and Gynaecologist, The Rosie Hospital, University of Cambridge
-
Ms Amanda Roberts, Public contributor
-
Dr Gillian Shepherd, Director, Health and Clinical Excellence, Merck Serono Ltd
-
Mrs Katrina Simister, Assistant Director New Medicines, National Prescribing Centre, Liverpool
-
Professor Donald Singer, Professor of Clinical Pharmacology and Therapeutics, Clinical Sciences Research Institute, CSB, University of Warwick Medical School
-
Mr David Symes, Public contributor
-
Dr Arnold Zermansky, General Practitioner, Senior Research Fellow, Pharmacy Practice and Medicines Management Group, Leeds University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Mr Simon Reeve, Head of Clinical and Cost-Effectiveness, Medicines, Pharmacy and Industry Group, Department of Health
-
Dr Heike Weber, Programme Manager, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Psychological and Community Therapies Panel
-
Professor of Psychiatry, University of Warwick, Coventry
-
Consultant & University Lecturer in Psychiatry, University of Cambridge
-
Professor Jane Barlow, Professor of Public Health in the Early Years, Health Sciences Research Institute, Warwick Medical School
-
Dr Sabyasachi Bhaumik, Consultant Psychiatrist, Leicestershire Partnership NHS Trust
-
Mrs Val Carlill, Public contributor
-
Dr Steve Cunningham, Consultant Respiratory Paediatrician, Lothian Health Board
-
Dr Anne Hesketh, Senior Clinical Lecturer in Speech and Language Therapy, University of Manchester
-
Dr Peter Langdon, Senior Clinical Lecturer, School of Medicine, Health Policy and Practice, University of East Anglia
-
Dr Yann Lefeuvre, GP Partner, Burrage Road Surgery, London
-
Dr Jeremy J Murphy, Consultant Physician and Cardiologist, County Durham and Darlington Foundation Trust
-
Dr Richard Neal, Clinical Senior Lecturer in General Practice, Cardiff University
-
Mr John Needham, Public contributor
-
Ms Mary Nettle, Mental Health User Consultant
-
Professor John Potter, Professor of Ageing and Stroke Medicine, University of East Anglia
-
Dr Greta Rait, Senior Clinical Lecturer and General Practitioner, University College London
-
Dr Paul Ramchandani, Senior Research Fellow/Cons. Child Psychiatrist, University of Oxford
-
Dr Karen Roberts, Nurse/Consultant, Dunston Hill Hospital, Tyne and Wear
-
Dr Karim Saad, Consultant in Old Age Psychiatry, Coventry and Warwickshire Partnership Trust
-
Dr Lesley Stockton, Lecturer, School of Health Sciences, University of Liverpool
-
Dr Simon Wright, GP Partner, Walkden Medical Centre, Manchester
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health