
Notes
Article history
The research reported in this issue of the journal was commissioned by the HTA programme as project number 08/13/38. The contractual start date was in February 2010. The draft report began editorial review in November 2011 and was accepted for publication in March 2012. As the funder, by devising a commissioning brief, the HTA programme specified the research question and study design. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the referees for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
none
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2012. This work was produced by Scawn et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to NETSCC. This journal is a member of and subscribes to the principles of the Committee on Publication Ethics (COPE) (http://www.publicationethics.org/). This journal may be freely reproduced for the purposes of private research and study and may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NETSCC, Health Technology Assessment, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2012 Queen’s Printer and Controller of HMSO
Chapter 1 Introduction
Background
Patients in intensive care units (ICUs) are at higher risk of hospital-acquired infections and sepsis than those in non-critical care areas. 1 Hospital-acquired sepsis is reported to occur in 10–70% of patients undergoing invasive mechanical ventilation, the rate varying with the patient population studied and diagnostic criteria used. 2 Despite the major advances in intensive care management, sepsis and its complications remain the leading cause of mortality in ICUs. 3
Bloodstream infections (BSIs), pneumonias and urinary tract infections are the most common hospital-acquired infections and are most often associated with the use of invasive devices. 4 Coagulase-negative Staphylococcus BSIs have recently increased in frequency, and enterococci such as S. aureus (B. S. Weeks & E. Alcamo) Jones & Bartlett International Publishers, 2008, have also been reported as causing BSIs in an increasing numbers of ICUs. Recently, Gram-negative bacilli have been reported more frequently than Gram-positive bacilli in this setting. The incidence of fungal urinary tract sepsis has also increased. 5
Management of ICU sepsis is also complicated by the high incidence of systemic inflammatory response syndrome (SIRS), which mimics many of the signs of sepsis but is often without an infective cause. This is particularly true in ICUs that have a high proportion of patients following major surgery as the surgery alone may precipitate a SIRS episode. A good example of this is the cardiac surgical ICU as cardiopulmonary bypass is a strong trigger for SIRS, generating, for example, pyrexia and a neutrophilia in the absence of an infective cause.
The potential difficulty in differentiating sepsis from SIRS in these high-risk patients makes it inevitable that intensivists often have a low threshold for commencing antibiotics to ‘cover’ the potential of an infection – even though a definite infective cause has not been proven. Indeed, many patients with suspected sepsis in ICU may be given antibiotics for a significant proportion of their stay to reduce the risk of septic complications, even in cases in which there are no compelling positive microbiological results.
To date, most studies have focused on optimising antibiotic treatment either for ventilator-acquired pneumonia, which accounts for approximately 50% of antibiotics use in ICU,6–8 or for treatment of suspected sepsis, often of unknown origin.
In these patients with apparent sepsis of unknown origin, clinical decisions for empirical antibiotic treatment are usually based on fever, excessive tracheal aspirates, increased white cell count and heart rate, even if no radiographic changes are apparent. We hypothesise that prolonged treatments with antibiotics in these patients is unnecessary, particularly if there are no confirmed organisms grown in blood cultures.
Other markers of sepsis may guide early diagnosis and decision-making on the necessity and duration of antibiotic treatment. Existing evidence from a recent retrospective study by Aarts et al. 6 suggests that patients without proof of nosocomial infection receiving empirical antibiotics for > 4 days had higher 28-day mortality (32.1%) than those whose antibiotics were discontinued (7.7%). We hypothesise that, in fact, a 2-day regime with broad-spectrum antibiotics is sufficiently potent to eliminate any potential microbial threat in these patients.
This is consistent with current international recommendations and guidelines that there is a need for continuous reassessment of antibiotic therapy with microbiology and clinical data to reduce duration when appropriate from the traditional 7–10 days of antibiotic therapy. Although early identification and treatment of sepsis can have a major impact on the outcome of these patients,9 diagnosis of sepsis is generally difficult, particularly in cases in which there is no positive isolated microbiological growth.
Although there has been no shortage of proposed markers of sepsis,10 two assays have emerged as increasingly relevant in recent years. These are the biphasic activated partial thromboplastin test (APTT) waveform and procalcitonin (PCT) concentration. The APTT waveform reflects light transmittance changes in plasma. Septic patients have been found by several investigators to show an abnormal biphasic pattern. Increasing abnormality of this waveform correlates with real-time clinical progression; the biphasic waveform is due to the formation of calcium-dependent complexes between C-reactive protein (CRP) and very low-density lipoprotein. 11 This has also been shown to be superior to CRP in the diagnosis of sepsis and the risk of mortality. 9
In a previous small trial it was reported that APTT waveform analysis may be of benefit in differentiating between SIRS and sepsis in the difficult postcardiopulmonary bypass group of patients. 12 However, although it remains a relatively novel technique we did not use the results as a basis for recruitment into the trial, which was based on intention to treat.
For PCT, the degree of the rise in concentration can help differentiate between infectious and non-infectious trigger of sepsis markers. For example, PCT has been shown to be effective in differentiating infectious from non-infectious causes of acute respiratory distress syndrome. 13 Most recent work has shown that the use of PCT tests in combination with the biphasic APTT waveform can increase the specificity of the latter test in identifying sepsis. 14 Indeed, it has recently been shown that serial measurement of PCT may allow monitoring of a reduction in antibiotic treatment duration and exposure in patients with severe sepsis and septic shock, without apparent harm. 15
Evidence from randomised trials about the duration of antibiotic use is absent. In this pilot randomised trial we investigated whether, in the ICU, 48 hours of antibiotic treatment is adequate to safely treat suspected sepsis when it is of unknown and unproven origin compared with a more traditional week-long course. In this pilot study we did not use biomarkers of sepsis as part of the entry criteria as we do not believe that this is currently routine practice in most UK ICUs. However, in the study we had the opportunity to collect samples for the APTT waveform and PCT concentration, and these data are presented.
Study objectives
The main objective of this pilot study was to provide preliminary data on the likely safety and efficacy of a reduced course of antibiotics for the treatment of ICU infections of unknown origin. In addition, we wished to identify the likely barriers to effective recruitment to a full study and the appropriateness and reliability of outcome measures and the data collection methods.
Structure of this report
The main body of this report begins with a description of the methods of investigation used (see Chapter 2). This is followed by the results, economic analysis, discussion, conclusions and synopsis of the findings, and suggestions for future research (see Chapters 3–7, respectively).
Chapter 2 Methods
Trial setting
This study was carried out in the intensive care and postoperative critical care units at Liverpool Heart and Chest NHS Foundation Trust between May 2010 and July 2011. Institutional, ethics and national competent authority (Medicines and Healthcare products Regulatory Agency, MHRA) approvals were obtained before commencing recruitment.
Trial design
This is a feasibility, pilot, open-label, single-centre randomised trial on the impact on safety and efficacy of a reduced course of antibiotics for the treatment of ICU infections of unknown origin (48 hours vs 7 days). The secondary feasibility outcomes of the pilot trial included the assessment of the ratio of patients screened as eligible to the number randomised; the incidence of crossover between the randomised treatment groups; and the accuracy of data collection assessed by a 20% source data verification check. In addition, this pilot study wished to identify the likely barriers to effective recruitment into a main definitive trial, and whether or not the outcome measures and data collection methods were appropriate and reliable.
Selection of patients
Patients being treated within the ICU were recruited into the trial if they were being commenced on the ‘Surviving Sepsis’ Care Bundle antibiotics by the intensivist in the absence of an actual known cause for that potential sepsis. To trigger the bundle, patients needed to have at least two of the four markers of SIRS [i.e. temperature > 38°C or < 36°C, tachycardia (> 90 beats per minute), tachypnoea (≥ 20 breaths per minute) and a white blood cell count > 12 × 109/l or < 4 × 109/l] and a suspected but not proven infection. In other words, patients were recruited if the intensivist was planning to commence antibiotics because of evidence of SIRS and a strong suspicion of infection – but there was no actual known source for that infection.
Inclusion/exclusion criteria
Patients were excluded if they had positive microbiological cultures before randomisation, were < 18 years of age or were enrolled in another study such that randomisation in the trial would result in deviation from either protocol. They were also excluded if they had an allergy to trial antibiotics or if consent/assent was declined or could not be obtained.
Once the decision to start antibiotic treatment was made by the intensivist, a referral was made to the study team who would assess the patient’s eligibility for recruitment. If the intensivist making the decision to start the antibiotics was one of the investigators, a second opinion was required from an independent consultant colleague to assess eligibility. Once the patient was considered a suitable candidate for recruitment, consent or assent was taken depending on the clinical state of the patient. If the next of kin was not present for the assent process, he or she was contacted by telephone to discuss participation in the study. When telephone assent was taken, the next of kin was asked to sign the assent form on his or her earliest visit to the hospital. In all cases of assent, a formal consent was taken from the patient once he or she had sufficiently regained capacity to give consent. Patients who declined consent were withdrawn from the study, and data were used for analysis only after consent had been given.
Randomisation
Eligible patients were randomised in equal proportions between the two trial groups:
-
antibiotic treatment administered for 48 hours
-
antibiotic treatment administered for 7 days.
Treatment assignment was based on the block randomisation method using randomly varying block sizes of 2, 4 and 6 to ensure numerical balance between the groups. An independent statistician provided the randomisation tables. Only trial staff with a unique user identification and password could log onto the bespoke, encrypted database. The allocation was revealed after entering unique patient data, and access to any lists of previously randomised patients was not permitted.
Treatment
After randomisation, a baseline Acute Physiological and Chronic Health Evaluation II (APACHE II) and Sequential Organ Failure Assessment (SOFA) score16 (see Appendix 1) was recorded, two sets of blood culture were taken, at least 15 minutes apart, and blood samples were sent for baseline biphasic APTT waveform and PCT analysis. These samples were centrifuged; serum was separated and frozen for analysis at a later stage. This was followed by the administration of the study antibiotics (Table 1).
| Patient weight (kg) | Teicoplanina | Meropenema |
|---|---|---|
| < 85 |
400 mg twice a day on day 1 400 mg once a day thereafter |
1 g three times a day |
| ≥ 85 |
6 mg/kg rounded to nearest 50 mg twice a day on day 1 6 mg/kg rounded to nearest 50 mg once a day thereafter |
1 g three times a day |
After completion of the treatment regime allocated at randomisation, additional antibiotic use constituted an outcome measure and the reason for the initiation was documented in the trial case record forms (CRFs). Similarly, if the antibiotics were stopped or changed before the scheduled completion of the course, the reason was recorded. Decisions to change, restart or stop antibiotics were made by consultant intensivists or microbiologists who were guided by evidence of positive cultures, radiography and other diagnostic imaging information or poor physiological status believed to be related to infection. The reasons for any deviation from the protocol were also documented.
The trial patients were followed up for a period of 10 days by the research team. SOFA scores were calculated and documented in CRFs. Blood samples were taken at baseline, at 48 hours and on initiation of additional antibiotics beyond the randomised schedule for measurement of prospective biomarkers of sepsis (biphasic APTT and PCT; see Appendix 2). 17–19 Trial antibiotics were prepared, packaged, stored and dispensed in accordance with good manufacturing practice for investigational medicinal products.
Biphasic APTT waveform and PCT concentrations were used as markers for sepsis. A biphasic APTT profile occurs when light transmission decreases before clot formation in the first part of the curve. Slope = –0.05%T/second was considered as biphasic waveform and the value of transmittance at 18 seconds (TL18) was measured. To quantify this abnormality, light transmission at time 0 was set to 100%, and the value recorded 18 seconds later (TL18) was taken as the index of the abnormality. 20–23 TL18 of 100% was considered as normal and TL18 was < 99% when biphasic waveform occurred. PCT levels were measured using a standard enzyme immunoassay available commercially.
Outcome measures
Primary outcome measures were defined by either initiation of antibiotic therapy after the completion of the treatment schedule allocated at randomisation or by trial mortality. Secondary outcome measures were defined in terms of duration of ICU stay, duration of hospital stay, duration of mechanical ventilation and incidence of infection with methicillin-resistant S. aureus (MRSA) and Clostridium difficile (B. S. Weeks & E. Alcamo) Jones & Bartlett International Publishers, 2008.
A small proportion of postcardiac surgery patients do return to theatre, usually for bleeding. It is common to use haemofiltration on patients who have a presurgery low estimated glomerular filtration rate (eGFR). Arrhythmia, usually atrial fibrillation, is also common, especially in heart valve patients, which may be treated with amiodarone infusion or in some cases by cardioversion shock.
Barriers to recruitment to a larger full study were reported as observations made during the recruitment to the trial. Reliability of data collection methods was monitored using a 13-point CRF validation check. An audit clerk who was independent to the trial performed this check. This individual randomly chose 10 CRFs and checked their recorded data against the patient clinical notes and the generic database.
Statistical analysis
The continuous numerical data were analysed using chi-squared, Mann–Whitney U-test and Fisher’s exact tests. The adverse events were analysed using chi-squared and Fisher’s exact tests and outcome measures were analysed using the Mann–Whitney U-test. Logistic regression was used to determine whether or not biomarkers were predictors of clinical outcomes and the outcomes were presented as adjusted odds ratios. Sensitivity and specificity analyses were performed to assess the diagnostic accuracy of APTT measurements.
Chapter 3 Results
Introduction
This chapter starts with a Consolidated Standards of Reporting Trials (CONSORT) diagram that describes the flow of participants through each stage of the trial (Figure 1). This is followed by demographic information, summary findings, trial outcomes, trial adverse events, biomarker results and key findings.
FIGURE 1.
Participant flow chart.
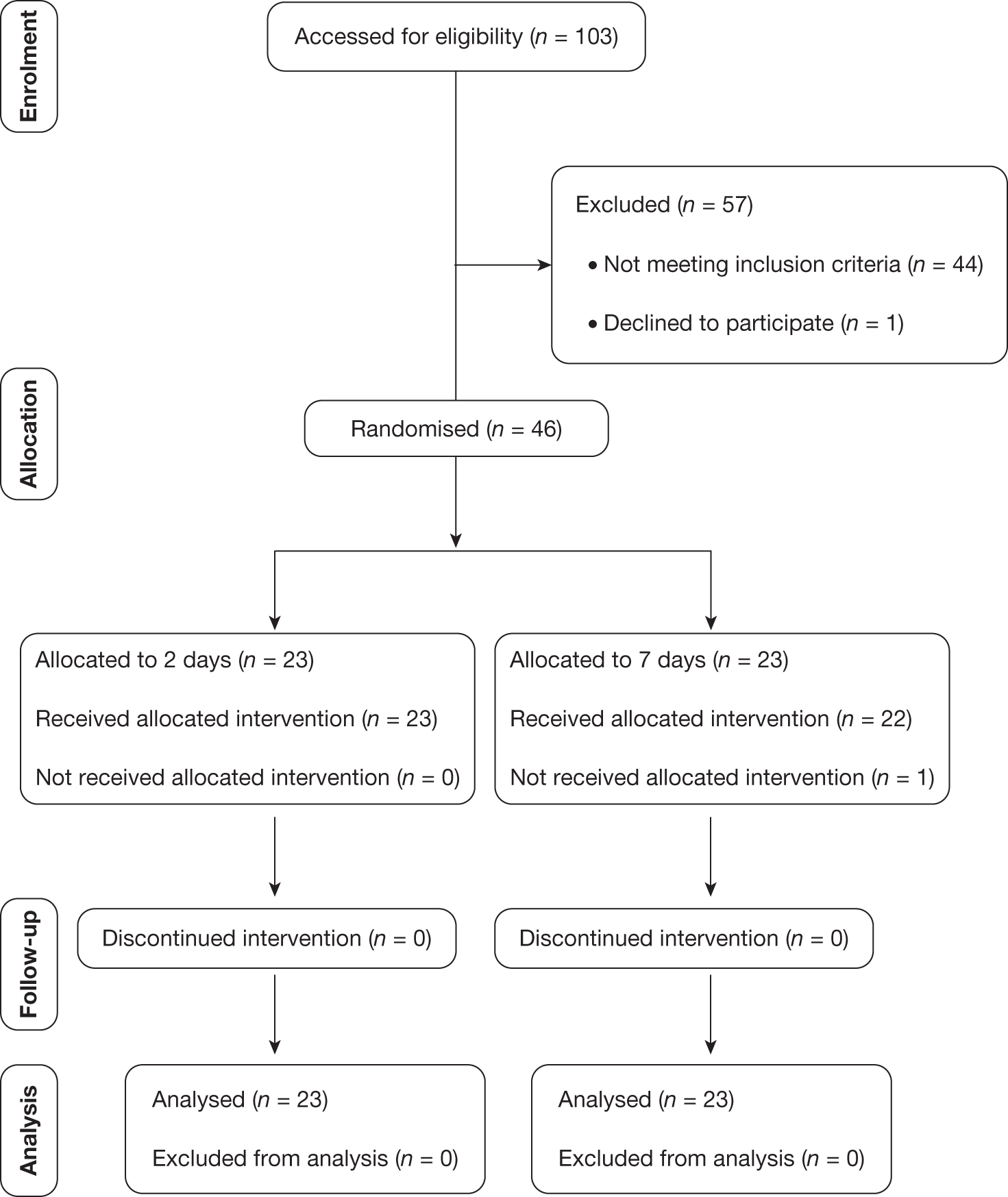
A total of 103 patients were screened for eligibility, of whom 46 were successfully recruited into the trial. Of the 57 patients excluded, 44 did not meet the entry criteria, of whom the majority had an infection for which the likely cause was known (e.g. chest infection as signified by chest radiography changes and a change in sputum); the others were already on an antibiotic but were still showing signs of sepsis. One patient declined to participate in the trial. A further 12 patients were deemed to have been missed.
Of the 12 patients who were deemed to be suitable for the trial but were not recruited, four were not recruited because their families refused to give assent because they were too anxious, three were missed but were then subsequently found to have been suitable for consideration for the trial and a further one was missed because no member of the trial team was available to recruit them. One patient fulfilled the entry criteria but was not recruited because it was 0100 and it was felt inappropriate to approach their family at that time, and a further patient, although eligible, was deemed not to be an ICU patient as he was sufficiently well that he was transferred out of the ICU to the ward on the same day. In addition, one patient was recruited but was then found to be taking immunosuppressant drugs for breast cancer, which are deemed to be an exclusion factor, and a further patient, although recruited, did not actually fulfil the eligibility criteria.
Screening appropriate patients was a weakness of the trial, with reliance on ICU staff to flag up potential patients to the trial team.
Demographic information
As shown in Table 2, the majority of the patients recruited into the trial were postsurgical (mostly cardiac bypass or aortic/mitral valve surgery). All patients in the 2-day group received the allocated antibiotics for at least 2 days.
| Reason | No. of patients | Group 1 (2 days) | Group 2 (7 days) |
|---|---|---|---|
| CABG | 15 | 6 | 9 |
| CABG + heart valve surgery | 9 | 3 | 6 |
| Heart valve surgery | 10 | 8 | 2 |
| Great vessels | 3 | 1 | 2 |
| MI and/or PCI | 5 | 2 | 3 |
| Oesophagogastrectomy | 2 | 2 | 0 |
| Lung surgery for cancer | 2 | 1 | 1 |
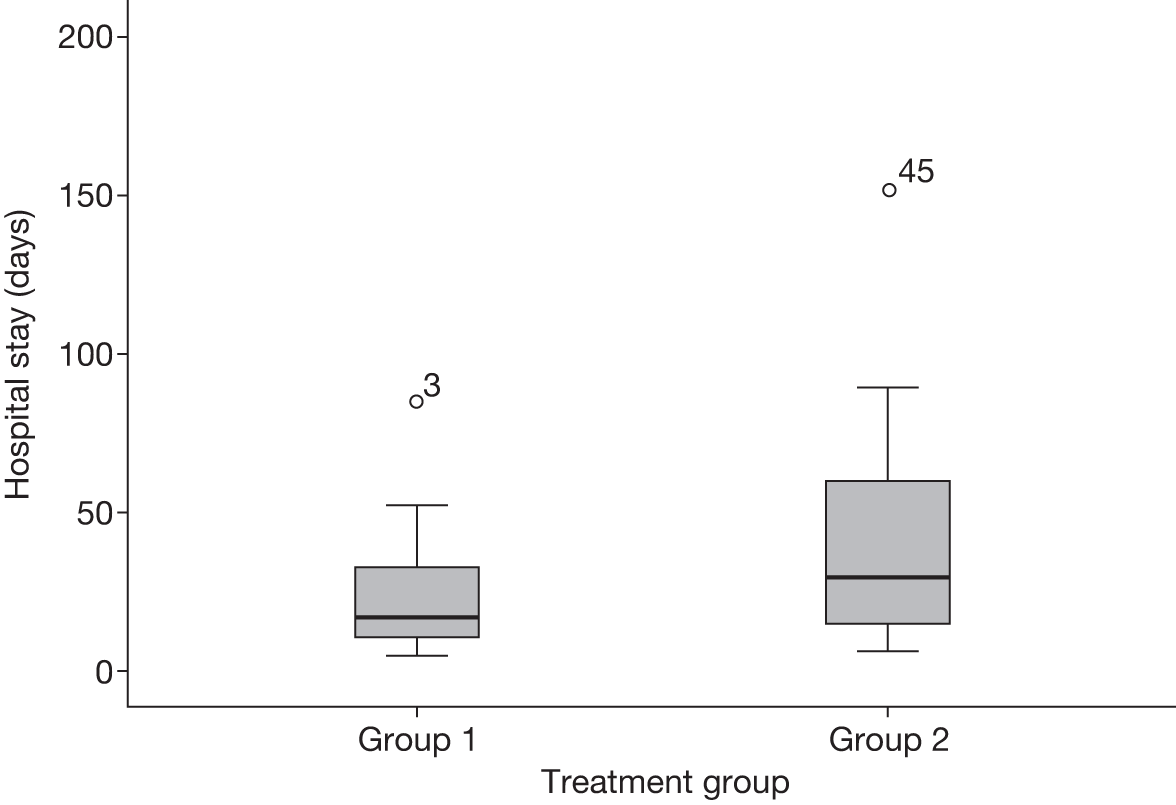
Recruitment took placed over 14 months. A total of 46 patients were recruited instead of the planned target of 60 patients within 12 months. Figures 2 and 3 show the distribution patterns of ICU and hospital stays, which were skewed towards the right. Table 3 shows the baseline demographic data and blood test results of the 46 patients who were recruited into the trial.
FIGURE 2.
The distribution of ICU stay for the two treatment groups. Group 1: 2 days of treatment with antibiotics; group 2: 7 days of treatment with antibiotics.
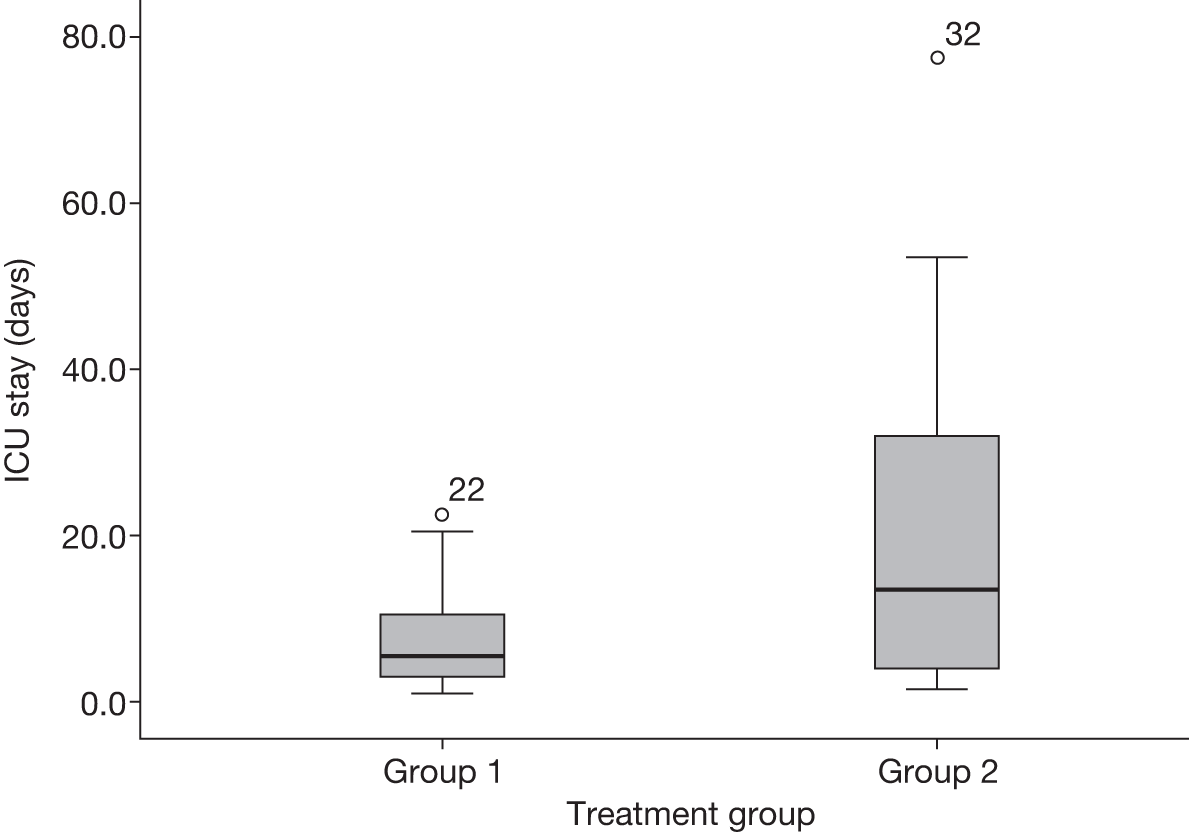
FIGURE 3.
The distribution of hospital stay for the two treatment groups. Group 1: 2 days of treatment with antibiotics; group 2: 7 days of treatment with antibiotics.
| Variable | Group 1 (2 days, n = 23) | Group 2 (7 days, n = 23) | p-value | |
|---|---|---|---|---|
| Gender, n (%) | Female | 9 (39.1) | 7 (30.4) | 0.54a |
| Male | 14 (60.9) | 16 (69.6) | ||
| Age (years) | Mean (SD) | 68.5 (9.8) | 65.3 (11.5) | 0.31 |
| Range | 53–86 | 39–81 | ||
| Ethnicity, n (%) | White British | 23 (100) | 22 (95.7) | 0.99a |
| Asian/Asian British | 0 | 1 (4.3) | ||
| Smoking status, n (%) | Never | 6 (26.1) | 3 (13.3)b | 0.31 |
| Ex-smoker | 15 (65.2) | 13 (61.9) | ||
| Current | 2 (8.7) | 5 (23.8) | ||
| Weight (kg) | Median (IQR) | 78 (28)c | 80.5 (28)c | 0.72d |
| Diabetes, n (%) | None | 14 (60.9) | 18 (81.8)c | NS |
| Type I | 1 (4.3) | 1 (4.5) | ||
| Type 2 | 8 (34.8) | 3 (13.6) | ||
| Preoperatively | ||||
| White blood count (× 109/l) | Median (IQR) | 8.9 (8) | 9.8 (5) | 0.60d |
| Urea (mmol/l) | Median (IQR) | 7.5 (6.3) | 8.6 (7.7) | 0.24d |
| Creatine (μmol/l) | Median (IQR) | 102 (64) | 104 (75) | 0.93d |
| eGFR (ml/minute) | Median (IQR) | 58 (53) | 58 (38) | 0.66d |
| Haemoglobin (g/l) | Median (IQR) | 11.8 (4) | 11.25 (3.3) | 0.64d |
Table 4 shows the comparison between the two groups with regard to their initial physiological state and their clinical course during the trial period.
| Variable | Group 1 (2 days, n = 23) | Group 2 (7 days, n = 23) | p-value | |
|---|---|---|---|---|
| APACHE II score | Median (IQR) | 13 (6) | 14 (8)a | 0.72b |
| Baseline SOFA score | Median (IQR) | 8 (7) | 11 (6)a | 0.15b |
| 48-hour SOFA score | Median (IQR) | 5 (7)a | 11 (7)c | 0.08b |
| Total time vented (hours) | Median (IQR) | 28 (155)a | 108 (218)c | 0.08b |
| Inotropes required (adrenaline and/or noradrenaline), n (%) | No | 13 (56.5) | 10 (43.5) | 0.38d |
| Yes | 10 (43.5) | 13 (56.5) | ||
| Teicoplanin (Targocid®, Sanofi-Aventis) doses given | Median (IQR) | 3 (17) | 8 (12) | < 0.001b |
| Meropenem (Meronem®, AstraZeneca) doses given | Median | 6 | 21 | |
| Positive cultures, n (%) | No | 20 (87.0) | 20 (87.0) | 0.99e |
| Yes | 3 (13.0) | 3 (13.0) | ||
| Length of ICU stay (days) | Median (IQR) | 5.5 (8.5) | 13.5 (30.5) | 0.53b |
| No. of patients by length of stay in ICU | < 3 days | 4 | 4 | 0.047e |
| 3–10 days | 13 | 5 | ||
| > 10 days | 6 | 13 | ||
| No. of patients by length of stay in ICU | ≤ 10 days | 17 | 9 | 0.036d |
| > 10 days | 6 | 13 | ||
| Hospital length of stay (days) | Median (IQR) | 17 (24) | 29.5 (46) | 0.30b |
| Death, n (%) | No | 20 (87) | 22 (95.7) | 0.30e |
| Yes | 3 (13) | 1 (4.3) | ||
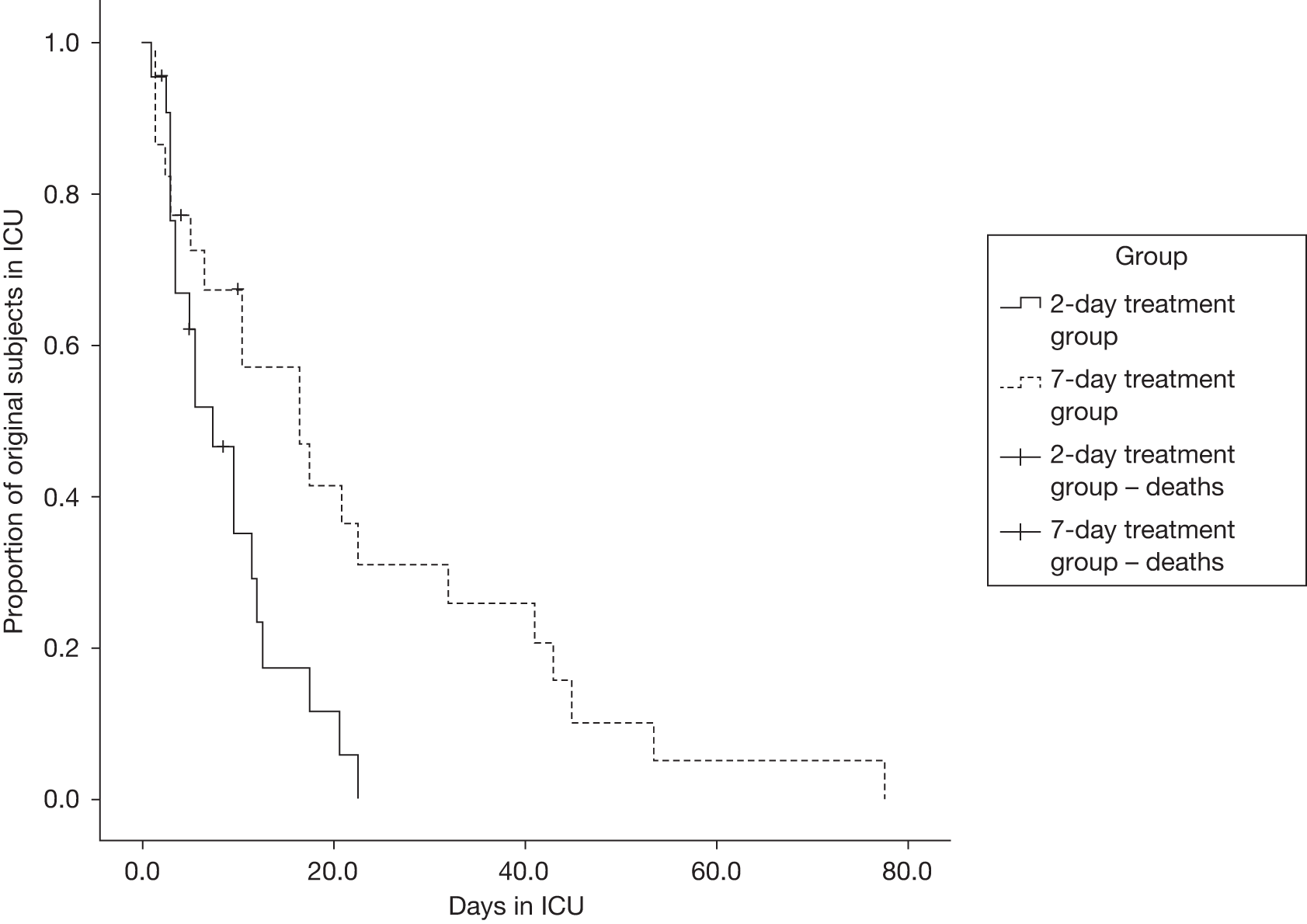
As an event of interest, we compared the cumulative number of patients leaving the ICU at any given time in the two treatment groups using log-rank test and Kaplan–Meier analysis (Figure 4). This produced a log-rank test p = 0.012, which implies that there is a significant difference between the two groups.
FIGURE 4.
Kaplan–Meier curves for days on ICU.
Summary findings
The primary analysis was intention to treat and involved all patients who were randomised. One patient was randomised inappropriately and no data were collected but this patient was counted in the numbers. Twenty-three patients were randomised in each of the two groups. There were no significant differences between the two groups with regard to APACHE II scores, SOFA scores, time on ventilator, inotropes required, positive cultures, days on ICU or death.
One patient in the control arm (7-day group) had meropenem stopped on the third day because of isolated positive cultures from a drain site for S. aureus and was instead treated with rifampicin (Rifadin®, Sanofi-Aventis) (600 mg) antibiotics for the remainder of the time. Of the patients in group 1 (2 days of antibiotics), four (17.4%) required further antibiotics; three (13.0%) patients in group 2 required further antibiotics. All patients had blood cultures taken at the time of randomisation and some patients were swabbed and had samples of tracheal aspirate taken. Of these, six (13.0%) returned positive, three in each arm of the trial. These were for S. aureus (one, 2-day group), Escherichia coli (B. S. Weeks & E. Alcamo) Jones & Bartlett International Publishers, 2008, (one, 2-day group), non-lactose-fermenting coliform (one, 7-day group), coagulase-negative staphylococci (one, 7-day group) and yeasts (two, both 7-day group).
Three patients in the control group failed to complete their 7-day course of trial antibiotics. One of these had their antibiotics stopped after 72 hours because of deranged liver function tests. These tests returned to normal values following cessation of the antibiotics. One patient died before completing the course and one patient was discharged from hospital after 4 days of the antibiotics.
In total, four patients died within the 10-day trial period. The trial events adjudication panel confirmed that none of the deaths was because of the trial intervention. The causes of death were (1) sepsis and gastrointestinal bleed, pulmonary abscess and oesophageal carcinoma, (2) multiorgan failure, sepsis, ischaemic bowel and ischaemic heart disease, (3) cerebrovascular accidents and thoracic aneurysm and (4) multiorgan failure and coronary artery disease. No patients who died during the trial period had received further antibiotics in addition to the trial antibiotics.
Trial outcomes
The risk difference between the two groups for the composite outcome was 0.12 [95% confidence interval (CI) 0.11 to 0.13; p = 0.3]. Tracheotomy was performed on three patients who required a respiratory wean from the ventilator. Table 5 shows the comparison of the need for further antibiotics and the composite outcome between the two groups.
| Outcome | Group 1 | Group 2 | p-value | |
|---|---|---|---|---|
| Need for further antibiotics above those allocated at randomisation, n (%) | No | 19 (82.6) | 20 (87) | 0.68a |
| Yes | 4 (17.4) | 3 (13.0) | ||
| Composite outcome of death and need for further antibiotics above those allocated at randomisation, n (%) | No | 16 (69.6) | 19 (82.6) | 0.30b |
| Yes | 7 (30.4) | 4 (17.4) | ||
Trial adverse events
The trial adverse events are summarised in Table 6. There was a single serious adverse reaction reported. This was a patient whose liver function tests became abnormal after commencement of antibiotic therapy. Alanine transaminase was 506 U/l (normal range 3–35 U/l), gamma-glutamyl transferase was 449 U/l (normal range < 50 U/l ) and alkaline phosphatase was 627 U/l (normal range 35–125 U/l). These values began to return to normal in the days following cessation of antibiotics.
| Variable | Group1 (2 days, n = 23) | Group 2 (7 days, n = 23) | p-value | |
|---|---|---|---|---|
| Re-explore in theatre, n (%) | No | 22 (95.7) | 21 (91.3) | 0.55a |
| Yes | 1 (5.9) | 2 (8.7) | ||
| Other adverse events, n (%) | No | 23 (100) | 20 (87.0) | 0.31a |
| Yes | 3 (13.0) | |||
| HF, n (%) | No | 18 (78.3) | 17 (73.9) | 0.73b |
| Yes | 5 (21.7) | 6 (26.1) | ||
| HF due to anuria or oliguria, n (%) | No | 20 (87.0) | 18 (78.3) | 0.73a |
| Yes | 3 (13.0) | 5 (21.7) | ||
| HF due to abnormal electrolytes, n (%) | No | 21 (91.3) | 22 (95.7) | 0.55a |
| Yes | 2 (8.7) | 1 (4.3) | ||
| Other arrhythmias, n (%) | No | 21 (91.3) | 23 (100) | 0.15a |
| Yes | 2 (8.7) | |||
| Anaemia (defined as haemoglobin < 8 g/dl), n (%) | No | 15 (65.2) | 15 (65.2) | 0.99b |
| Yes | 8 (34.8) | 8 (34.8) | ||
| Standard tracheotomy, n (%) | No | 23 (100) | 20 (87.0) | 0.07a |
| Yes | 3 (13.0) | |||
Biomarker results
Two biomarker blood tests, PCT and APTT, were used as an indication of sepsis. These were measured at baseline (after randomisation), at 48 hours and at 10 days or discharge.
Procalcitonin levels were measured in 45 out of the 46 (98%) patients at the time of randomisation, in 36 (78%) patients at 48 hours and in 26 (57%) patients at 10 days/discharge, with results summarised in Figure 5.
FIGURE 5.
Median (IQR) PCT levels with IQR error bars.
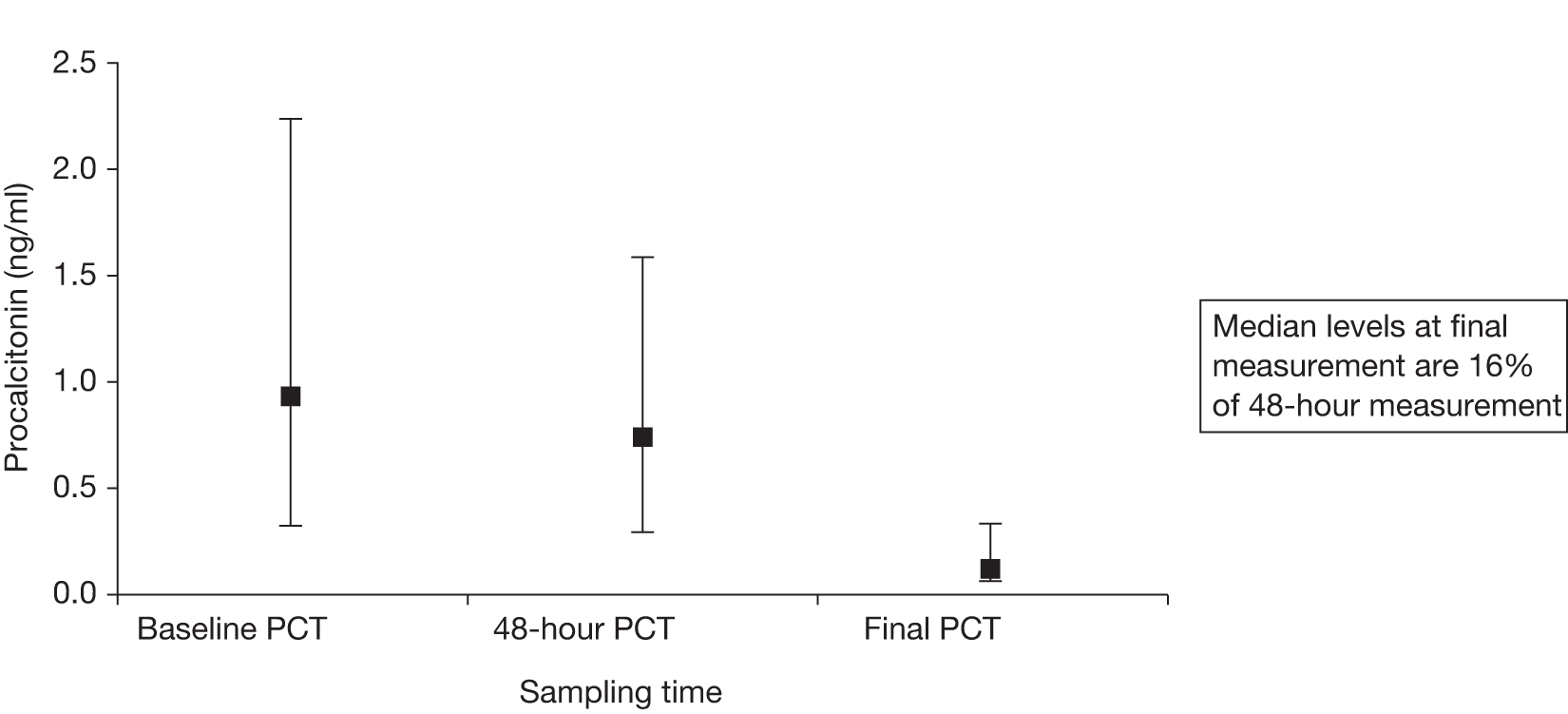
The median [interquartile range (IQR)] PCT for patients who restarted antibiotics was 2.4 (6.2) ng/ml, compared with 0.6 (2.7) ng/ml for those who did not receive further antibiotics in excess of their trial drugs (p = 0.06).
Despite the low numbers of patients, a logistic regression analysis of baseline PCT indicated a trend towards it being a predictor of restarting antibiotics, with an odds ratio of 1.45 (95% CI 1.04 to 2.02; p = 0.01). Similarly, a logistic regression analysis of the composite outcome (death and needing further antibiotics) also showed PCT to be a predictive risk factor, with an odds ratio of 1.79 (95% CI 1.20 to 2.67; p = 0.005).
With regards to the APTT results, unfortunately there is a lack of consensus whether there is a strong relationship between abnormal APTT waveforms and the underlying pathology of sepsis. Matsumoto et al. 24 pointed out that in patients with disseminated intravascular coagulation and sepsis the test was effective in patients who did not have solid cancers. They used a ‘cut-off’ of any waveform slope below –0.25%T/second to indicate biphasic waveform. We decided to use two values as cut-offs (Table 7) as discussed previously by Delannoy et al. ,12 who used a comparable study population to the one in this study. The values that Delannoy et al. 12 used were –0.25 and –0.465%T/second.
| Group and time | –0.25%T/second | –0.465%T/second |
|---|---|---|
| Group 1 (2 hours) baseline | 11 | 9 |
| Group 2 (7 days) baseline | 8 | 6 |
| Group 1 (2 hours) 48 hours | 2 | 2 |
| Group 2 (7 days) 48 hours | 4 | 3 |
The analysis of abnormal APTT waveforms indicated a lack of significant difference between the two groups at baseline. As a measure of validity of the APTT waveform tests we considered the APTT results in patients who had known positive blood cultures and then performed tests for sensitivity and specificity looking for the level of agreement. At the two different cut-off points of slope of APTT waveform data (–0.25, –0.465) there was very poor agreement with ‘blood culture-positive sepsis’ [kappa values (95% CI) of –0.048 (–0.26 to 0.17) and –0.05 (–0.24 to 0.25), respectively].
To test whether or not an abnormal APTT waveform has the ability to identify people with sepsis as correctly as can a positive blood culture, sensitivity analyses were performed. The results at the two different cut-off points of slope of APTT waveform data (–025, –0.465) indicated that sensitivity (95% CI) was also very poor [0.105 (0.00 to 0.24) and 0.13 (0.00 to 0.30), respectively].
To test whether or not an abnormal APTT waveform has the ability to recognise people without sepsis, a specificity analysis was performed. The results at the two different cut-off points of slope of APTT waveform data (–025, –0.465) indicated that specificity (95% CI) was quite reasonable [0.85 (0.72 to 0.98) and 0.87 (0.75 to 0.99), respectively]. With a likelihood ratio for a positive test ranging from 0.5 to 1.0, we can interpret the findings as that there is an equal or slightly less chance of finding a positive abnormal APTT waveform test in someone with ‘blood culture-positive’ sepsis than in someone without sepsis.
Key findings
Although there was a preponderance of male patients this was equally spread between both trial groups. There was no significant difference between the groups in terms of age, ethnicity or weight. Diabetes was less prevalent in the 7-day group but the small number of patients prevented statistical analysis. Renal function and APACHE II scoring were comparable in both sets of patients. Relatively few patients were missed for potential enrolment. It is accepted, however, that patients who were informally discussed between clinicians or patients whom the individual clinician did not deem to be eligible were probably missed from the screening log. A relatively high number (nearly 5%) did not meet the full inclusion criteria but the collected data do not allow us to ascertain why this was.
Presenting signs of SIRS were equally common in both groups and an abnormal white blood cell count was present in > 75% of patients in both groups. Only 10% of patients in either group had positive microbiological isolates during the trial period.
Adverse events were few in both groups and not in excess of expected postoperative complications following major cardiac and thoracic surgery in the study population. There was no statistical difference in adverse events between the two groups.
Sequential Organ Failure Assessment scores decreased over the trial period in both groups with the suggestion (not significant) of lower SOFA scores at 2 days in the 48-hour antibiotics group. This difference was significant at 10 days but data were missing in some patients. Inotrope requirements were unchanged following antibiotic use in either group. Length of stay on ICU was shorter for those who received only 2 days of antibiotics and mortality was comparable between groups. There was a suggestion of longer periods of invasive ventilation for those patients in the 7-day group, although this was not statistically significant.
Less than 10% of patients receiving only 2 days of antibiotics required further antibiotics during the trial period. Only three of these had positive microbiological culture resultsm with two patients receiving an extended course of antibiotics for reasons based on clinician preference alone and one having antifungal therapy added based on clinician suspicion alone. One patient in the 7-day group was on long-term steroids. This patient did not require a longer course of antibiotics but was started on antifungal therapy for yeasts (tracheal aspirate) on day 6. Of those receiving 7 days of antibiotics, three had additions made to their antimicrobial regime based on positive microbiological results, with two patients receiving further doses of teicoplanin based on a clinician decision. No patients who died required antibiotics in excess of those they received as part of the trial. There were no documented incidences of MRSA or C. difficile infection in either group.
Barriers to recruitment
One of the objectives of the trial was to obtain an understanding about what the barriers to the recruitment of potentially septic patients would be. The recruitment of a relatively small number of patients during the trial period highlights that such barriers did exist.
There were a number of major barriers:
-
Probably the single biggest barrier was the screening process used to identify prospective patients. The system we employed for the trial was that the ICU clinical staff would ‘flag up’ appropriate patients to the trial team; however, despite frequent reminders and regular advertising attempts, it is likely that several patients who would have been eligible for inclusion either were not identified to the trial team or were discovered after alternative treatment plans had been established. It would be crucial for a full study to have a larger number of clinical researchers so that all ICU patients could be monitored for signs of sepsis on a more frequent basis.
-
Another barrier was the definition of ‘sepsis of unknown origin’. The commonest ICU infection is respiratory in origin and is often diagnosed primarily by changes on chest radiography. However, in our patient population, which is primarily patients who have undergone cardiac or thoracic surgical procedures, abnormal chest radiography is common and caused by numerous non-infective causes. Because patients with a known ‘new’ chest infection were excluded from randomisation, it seems likely that some patients were missed because they were deemed to have new chest radiography changes suggestive of infection, but in fact had those changes because of other causes, such as contusion.
-
Because this trial was carried out specifically in ICU patients, many of the patients who were eligible for the trial were unable to give their own consent at the time of their recruitment. Because of the need for antibiotics to be prescribed as soon as possible for septic patients, it was important to obtain urgent ‘assent’ from the next of kin. Despite careful and empathetic counselling, several families found that the situation of their family member being in intensive care was sufficiently stressful that they were unable to agree to them being entered into the trial.
-
Further, because most of the patients were elderly and often had elderly relatives, it was decided that it was not appropriate to contact these elderly relatives to gain assent in the middle of the night; therefore, patients were usually not recruited if their sepsis symptoms became apparent after 2200 and before 0600.
-
In addition, many relatives felt that they needed some time to read the trial information documentation and to discuss the trial with other family members. Often this introduced a significant delay, which risked these patients having to be excluded from the trial so that they could receive their antibiotics without this delay.
-
At the time of this trial the cardiothoracic ICU was not a ‘closed’ unit in which patients are cared for by a single intensive care team, but instead is a unit in which patient care is ‘shared’ between the intensivists and the cardiac surgical teams. Within the cardiac surgical teams during the period of the trial, there was significant difficulty in the recruitment and retention of registrar-level doctors. The consequence of this is that it proved much harder than anticipated to control the prescribing habits of this group of staff, resulting in several patients being commenced on antibiotics without prior referral to the research team and not in keeping with the trial protocol.
-
A lack of adequate clinical staff to recruit patients. Other than the primary investigator, all recruiting was carried out by two clinical fellows. Regrettably there were occasions when one of the investigators was not available to obtain assent/consent.
Other smaller barriers were:
-
A small number of patients were referred as fulfilling recruitment criteria but on more detailed screening they had to be excluded either because they had finished a previous course of antibiotics within the previous 24 hours or because they were concurrently taking medication that could alter their immunological response (immunomodulators). For example, one patient was taking treatment for breast cancer, which could also have affected their ability to mount an immune response to infection.
-
During the middle of the recruitment period there was a national H1N1 flu pandemic. The consequence of this was that our cardiac unit stopped all elective operating for several weeks and had reduced operating for several further weeks. The aim was to provide beds for general ICU admissions or flu patients, but in fact these referrals did not arise and instead, for about 6 weeks, we had a virtually empty cardiothoracic ICU with minimal patients eligible for recruitment.
-
Only after the official start of the trial recruitment period were we made aware of the MHRA standards around drug storage and labelling, which resulted in a 6-week period of lost recruitment during which six patients were screened and would have been suitable for recruitment had we been compliant with the appropriate standards at the trial start date.
-
The delayed start described above also resulted in a ‘false start’ for the trial publicity, which meant that once the trial restarted after the initial delay there was more confusion than would be ideal and a sluggish initial referral rate.
Reliability of data collection methods
Data collection was the responsibility of the trial research nurse, who visited the unit daily (Monday to Friday). The research nurse then entered the data onto the trial database. The accuracy and reliability of this method of data collection were assessed by an audit clerk, who was independent of the trial, using a 13-point CRF validation check. The audit clerk randomly chose 10 CRFs and checked their recorded data against the patient clinical notes and the generic database. Because 13 events were checked in each of 10 CRFs, the total number of events checked was 130. From this check there were found to be two data entry errors (1.5%). Both of these errors were in the SOFA score calculations – one at baseline and one at 48 hours. On both occasions, when the data point was corrected and the SOFA score recalculated, there were no differences in the two scores.
Chapter 4 Economic analysis
For the full economic analysis see Appendix 3.
The major findings were as follows:
-
Cost minimisation analysis (CMA) was used as there were no significant differences between the trial and control groups.
-
The mean antibiotic cost per patient for the 2-day group was £168.97 and for the 7-day group was £375.86. The average cost difference was therefore > £200 per patient.
-
We obtained data detailing the number of patients seen in our ICU department for 9 months and the number of these patients who had blood cultures taken and the number who returned a positive result. These data were then extrapolated for a full year. Interestingly, of all the patients who had cultures taken over the 9-month period, only 10.4% returned a positive result.
-
The potential per annum cost saving for the ICU was estimated to range from £108,140 to £126,060, assuming that, at 48 hours, if the cultures were negative, the antibiotics were stopped in all cases.
Chapter 5 Discussion
Sepsis is a potentially serious medical condition that is characterised by an inflammatory response to an infective agent that may affect the whole body. 1,25,26 The patient may develop an inflammatory response to microbes in their blood, urine, lungs, skin or other tissues. 1,26 If left untreated sepsis may progress to severe sepsis and septic shock, which is associated with a high mortality rate.
Most patients who develop suspected sepsis either requiring ICU admission or during an ICU admission are given antibiotics. 1,3–5,26 In cardiothoracic ICUs clinical decisions are often taken to treat patients with suspected sepsis of unknown origin for a week or longer with broad-spectrum antibiotics, usually on the basis of onset of fever, increased tracheal aspirates, increased white cell count and heart rate, even if no radiographic changes are apparent. However, there is increasing concern that this practice may be detrimental to patients, as several observers have highlighted in the medical literature over the last 30 years. 6,27–29 In particular, the rise in reported C. difficile cases linked to mortality30 and the increasing levels of antibiotic-resistant bacterial strains is of concern.
Indeed, current evidence from a recent retrospective study by Aarts et al. 6 has suggested that patients without proof of nosocomial infection receiving empirical antibiotics for longer than 4 days had higher 28-day mortality (32.1%) than those whose antibiotics were discontinued (7.7%). International recommendations and guidelines have suggested that there should be continuous reassessment of antibiotic therapy guided by clinical response, microbiology and clinical data and also a reduction in the duration of therapy when it is appropriate (from the usual 7–10 days). 31 However, in the absence of strong evidence for optimal duration of antibiotic use in the ICU from randomised trials, these recommendations and guidelines have had little impact on current practice. Other evidence in the literature, also from small studies, is suggestive that a reduction in antibiotic use may be cost saving and may reduce the rates of antibiotic resistance. 21
With regard to the primary outcome measures for this pilot trial there was no significant difference in the need for additional antimicrobial therapy or an increased risk of mortality for patients treated with only 2 days of empirical antibiotics compared with those treated for 7 days. This suggests that further investigation by means of a multicentre trial would be safe and advantageous using our approach to treatment of suspected nosocomial infections in critical care areas.
Prolonged ICU stay in those patients receiving antibiotics for 7 days may suggest that the 7-day patients were sicker. This was not borne out, however, by the APACHE and the initial or 48-hour SOFA scores. Alternatively, the continuation of potent, broad-spectrum antibiotics may have led clinicians to perceive these patients as being ‘critically unwell’ for longer periods of time. Continuing antibiotics may have delayed decisions to remove central access catheters and arterial pressure lines. All of these factors may have contributed to prolonged ICU stay.
Although not clinically significant, the difference in total time invasively ventilated is intriguing (although there is significant crossover between the IQRs). Again, if these values did represent a significant difference one might question if the 7-day group were a ‘sicker’ cohort of patients despite the illness severity scores suggesting otherwise. It is difficult to hypothesise why patients on longer courses of antibiotics might need longer periods of ventilation other than because of a clinician perception of patients being ‘critically unwell’ while still on broad-spectrum antibiotics. This may have led clinicians to prolong advanced organ support in these patients for fear of them relapsing should that support be withdrawn too quickly. Additionally, the effect of individual clinicians’ weaning strategies on this result cannot be accounted for from these data.
In the case of both ICU length of stay and length of invasive ventilation, both groups had spent a similar time in ICU before being enrolled. This again refutes the suggestion that those patients who received 7 days of antibiotics were sicker because they had spent more time in ICU before they were involved in the trial.
Several studies exist in which the rate of decline of PCT concentration has been used to guide length of course of antibiotics, but in this trial it was not used as part of the intention-to-treat protocol. However, despite the relatively small trial numbers, the trend for high initial PCT levels to predict the need for further antibiotics was significant. It would be useful in a future, larger trial to try and quantify a concentration of PCT above which the necessity of further antibiotics would be assured. The obvious corollary of this would be that this would give an initial PCT concentration below which it could be assumed that it would be safe to stop the antibiotics after an initial 48-hour course.
Interestingly, the comparison of secondary health economic outcomes – determined by assessing resource utilisation and costs associated with each of the two pilot arms – indicated that there was a significant cost saving in the 2-day arm of up to £126,060 per annum, ranging from £191.06 to £222.72 per patient, a finding that is in agreement with a previous small study. 21 If this cost saving is extrapolated nationally, in England alone it represents a substantial saving to the NHS health-care budget.
As described in the results section, the main obstacles to recruitment in this pilot trial were adequate screening of all of the ICU patients on a frequent basis, excluding sepsis of known origin and factors related to the strategy for obtaining consent. Differences in interpretation of what constituted a ‘probable’ chest infection, particularly when patients had ‘increased sputum production’, in which case they would be considered ineligible for recruitment (because this would constitute sepsis of known origin), undoubtedly resulted in an excess number of patients receiving prolonged courses of antibiotics to treat non-existent chest infections. This was even more the case when there was the appearance of basal or lobar collapse on chest radiography. However, the nature of cardiothoracic surgery (thoracic wall incisions, lung collapse intraoperatively, high incidence of chronic lung disease in the patient population) means that the appearance of abnormalities on chest radiography is more frequent but does not necessarily constitute a pneumonic process. In the future it is recommended that these patients should be considered for inclusion in the trial and it is in this group of patients that PCT may prove to be a beneficial agent for distinguishing infected patients.
The need for expedited initiation of antibiotic treatment once a patient is suspected of having sepsis significantly limits the amount of time that patients or their next of kin have to decide whether or not to participate in the trial. In some cases this was the basis for several patients or their legal representatives declining permission for enrolment in the trial – they had insufficient time to consider their decisions. In addition, difficulties in contacting patients’ legal representatives could result in delays to the commencement of antibiotic treatment. Outcomes have been shown to be worse in septic patients when antibiotics were delayed10,32,33 and so in most instances these patients were excluded.
Although uncommon in our sample group, the onset of possible sepsis during antisocial hours meant that it was inappropriate to contact next of kin for these patients to discuss enrolment. In a few cases, attending clinicians were of the opinion that the illness was too severe and that it would be unethical to delay antibiotics while assent was being sought. It could have been inappropriate to approach relatives/legal representatives in such a situation when objective decisions regarding involvement in a clinical trial would have been difficult. For these reasons some of these patients were not recruited into the trial and therefore may potentially represent a source of bias. It is suggested that these recruitment issues could be addressed by the use of preoperative consent of patients in the outpatient setting. Alternatively, because both arms of the trial are the same for the first 48 hours it might be prudent to agree with the ethical committee that recruitment of these patients could occur at any time within the first 48 hours rather than before any antibiotics are given.
A significant change was seen in clinician behaviour towards antibiotic use during the latter part of the pilot period. Closer scrutiny of antibiotic use brought about by the trial led to clinicians increasing their threshold for starting antibiotics in postoperative patients. Although commendable in reducing antibiotic exposure for patients, this approach led to reduced recruitment to the trial and may have exposed truly ‘septic’ individuals to a delay in treatment.
Another difficulty with the trial in our particular unit is that we are not a ‘closed’ unit. For many patients developing signs of sepsis, their first medical review was by a cardiac surgical registrar. Despite a great deal of publicity and attempts at education there remained several patients who were simply commenced on antibiotics by their ‘team’ doctors before being given a chance to be recruited into the trial. This particular limitation was made worse by the limited number of investigators undertaking this trial. Primarily in order to remain within the governance of good clinical practice, all assents/consents for the trial were undertaken by one of only three individuals. In any future trial it would be important to ensure an adequate number of appropriately qualified researchers to be available to enrol appropriate patients into the trial.
An additional obstacle to recruitment during the trial period was a 6-week closure of the ICU for H1N1 epidemic national planning purposes.
Another area that needs further consideration when designing the larger definitive trial is the dispensing of trial antibiotics, which would in the future require unique packaging with specific tracking numbers different from those for existing medications intended for routine use. This would avoid confusion with stock drugs used in the unit for non-trial patients. In addition, careful consideration of the choice of antibiotic regime when involving general and cardiothoracic ICUs is required. Some units may routinely use third-generation cephalosporins or beta-lactam/beta-lactamase inhibitor drugs as opposed to carbapenems. The choice of a regime for a future trial may also be hindered by varying local susceptibility patterns. Finally, although the use of a glycopeptide drug is becoming a routine part of empirical sepsis cover, having an outcome measure for MRSA infections in which one of the treatment options is the trial drug may not be logical.
The small size of this pilot study prevents any kind of definitive answers being derived or even suggested with any degree of certainty. It is important to note that these were all postoperative patients and that the incidence of pure sepsis episodes as opposed to SIRS in critically ill medical patients may not be as pronounced. This calls into question the external validity of future trial results for general ICU patients.
Measurement of height to allow calculation of basal metabolic index (BMI) would allow for better matching of patient demographics and should be included in future trial data collection. Future trial data would also benefit from more detail being collected around why apparently suitable patients were not included in the trial.
Although the trial protocol took into account the effect of patient weight on antibiotic dosage, it did not address potential changes in dose necessitated by renal and hepatic impairment or the effect of haemofiltration and potential disruptions in this therapy. In practice, this affected only one patient who transferred from haemofiltration to haemodialysis. The use of steroids in sepsis was not addressed as a possible confounding factor (the potential immunocompromise may increase the risk of ongoing infection).
Chapter 6 Conclusion
The preliminary data from this study are suggestive that, if there are no differences in clinical outcomes, there may be significant benefits of reducing broad-spectrum antibiotic use in the ICU without undermining patient safety. In cost terms alone, there would be a potential saving in our unit of > £100,000 per year, which would potentially extrapolate to a massive national overall health economy saving. However, evidence from this pilot trial is not definitive and hence further investigation is warranted using a large randomised trial with greater patient numbers to explore further efficacy and cost implications of reduced antibiotic use in critical care units (general and cardiothoracic), both nationally and internationally.
It must be clarified that we are not of the opinion that all patients can be treated with reduced courses of antibiotics. Invariably, some patients will be experiencing true infective episodes and will require longer periods of antibiotic therapy. From our trial we would predict that such patients are those who have a high baseline PCT concentration. This pilot merely highlights that the distinction between infective and inflammatory processes in critically ill patients is a difficult one. Even the use of PCT and biphasic waveform APTT to identify those patients who truly have sepsis has been questioned by analysis of available studies. 34 Of particular interest in this trial is the observation that baseline PCT levels were strongly predictive of both the need for restarting antibiotics and the composite outcome of death and need for further antibiotics, a feature that requires further investigation in a large trial.
Clinical reassessment of the need for antimicrobial therapy at 48 hours allows those patients experiencing a SIRS response to be exposed to broad-spectrum antimicrobials for as short a time as possible.
Chapter 7 Recommendations for future research
-
A larger multicentre trial needs to be undertaken to confirm the benefits of reducing courses of broad-spectrum antibiotics in ICU patients who have signs of sepsis but with no known cause.
-
A future trial would be designed to provide a better estimate of the savings that could be made to the whole health economy rather than extrapolating from this relatively small trial.
-
Inclusion of general ICU patients as well as cardiothoracic patients would be required to ensure that the assumptions made in this trial about the incidence of aseptic SIRS being the underlying diagnosis in most ICU sepsis patients is applicable in all ICUs and not just those with a primarily postoperative population.
-
The observation that baseline PCT concentrations are predictive of the need for longer courses of antibiotics must be included in the design of a future trial.
The outcomes of this pilot study are very encouraging and suggest that it is feasible to design a binary non-inferiority trial with the need for further antibiotics above those allocated at randomisation as the primary outcome measure. In this pilot study we observed that the need for further antibiotic use in the 2-day treatment (group 1) was 17% compared with 13% in the standard 7-day treatment (group 2). The null hypothesis is that the percentage of patients requiring further antibiotic use in group 2 is better than the percentage of patients requiring further antibiotic use in group 1 by an amount d (non-inferiority limit). Assuming different values of d, Table 8 shows estimated sample sizes at different alpha and power levels.
| Alpha | Power (%) | d (%) | Sample size |
|---|---|---|---|
| 0.05 | 80 | 5 | 31,500 |
| 0.05 | 80 | 10 | 880 |
| 0.05 | 80 | 12.5 | 436 |
| 0.05 | 80 | 15 | 260 |
| 0.05 | 90 | 10 | 1204 |
| 0.05 | 90 | 12.5 | 604 |
| 0.05 | 90 | 15 | 360 |
Secondary outcome measures could include initial PCT as a predictor of the need for an extended course of antibiotics, death, duration of mechanical ventilation, duration of ICU stay, and health economics outcomes.
Acknowledgements
The authors of this report would like to acknowledge the contributions of the following individuals: Professor Paulo Lisboa (Chair of the Trial Steering Committee), Dr Nagesh Kalakonda (Trial Steering Committee Member), Mr Nathan Howes (Trial Steering Committee Member), Dr Mark Jackson (Trial Steering Committee Member), Professor Cheng Hock Toh (Trial Steering Committee Member), Dr Carlos Nistal De Paz (Trial Steering Committee Member), Mr Keith Wilson (Trial Steering Committee Member), Professor William Fraser (analysis of PCT levels), Mr Colin Downey (analysis of APTT levels), Dr Peter Booker (Chair of the Data Monitoring and Safety Committee), Dr Richard Wenstone (Data Monitoring and Safety Committee), Dr Robert Harris (Data Monitoring and Safety Committee).
Contribution of authors
Dr Nigel Scawn (Clinical Lead for Critical Care, Primary Investigator) was the lead for the trial’s inception and co-ordination, co-author of the report and primary responder to referees comments.
Dr Dan Saul (Clinical Fellow) recruited patients into the trial and was a co-author of the report.
Dr Darshan Pathak (Clinical Fellow) recruited patients into the trial and was a co-author of the report.
Dr Bashir Matata (Head of Clinical Trial Department) assisted with the grant application and trial co-ordination, was a coauthor on the report and assisted with responses to referees comments.
Mr Ian Kemp (Research Nurse, Trial Manager) collected data into CRFs and maintained the trial database and was a co-author of the report.
Dr Rod Stables (Consultant Cardiologist) provided advice about the trial protocol and process and was co-author of the final report.
Dr Steven Lane (Statistician, University of Liverpool) was responsible for the statistical component of the trial protocol and report.
Dr Alan Haycox (Reader in Health Economics, University of Liverpool) was jointly responsible for the health economics section of the report.
Ms Rachel Houten (Health Economist, University of Liverpool) was jointly responsible for the health economics section of the report.
Disclaimers
The views expressed in this publication are those of the authors and not necessarily those of the HTA programme or the Department of Health.
References
- Bochud PA, Calandra T. Pathogenesis of sepsis: new concepts and implications for future treatment. BMJ 2003;326:262-6.
- Rello J, Diaz E. Pneumonia in the intensive care unit. Crit Care Med 2003;31:2544-51.
- Gross PA, Neu HC, Aswapokee P, Van Antwerpen C, Aswapokee N. Deaths from nosocomial infections: experience in a university hospital and a community hospital. Am J Med 1980;68:219-23.
- Richards M, Thursky K, Buising K. Epidemiology, prevalence and sites of infections in intensive care units. Semin Respir Crit Care Med 2003;24:3-22.
- Combes A, Figliolini C, Trouillet JL, Kassis N, Dombret MC, Wolff M, et al. Factors predicting ventilator-associated pneumonia recurrence. Crit Care Med 2003;31:1102-7.
- Aarts MA, Brun-Buisson C, Cook DJ, Kumar A, Opal S, Rocker G, et al. Antibiotic management of suspected nosocomial ICU-acquired infection: does prolonged empiric therapy improve outcome?. Intensive Care Med 2007;33:1369-78.
- Micek ST, Ward S, Fraser VJ, Kollef MH. A randomised controlled trial of an antibiotic discontinuation policy for clinically suspected ventilator-associated pneumonia. Chest 2004;125:1791-9.
- Singh N, Rogers P, Atwood CW, Wagener MM, Yu VL. Short-course empiric antibiotic therapy for patients with pulmonary infiltrates in the intensive care unit. Am J Respir Crit Care Med 2000;162:505-11.
- Dellinger RP, Levy MM, Carlet JM, Bion J, Parker MM, Jaeschke R, et al. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock. Intensive Care Med 2008;34:17-60.
- Thomas K. Transmitting and absorbing new information on the early identification of sepsis patients: the partial thromboplastin time biphasic waveform. Crit Care Med 2003;34:1829-31.
- Aouifi A, Piriou V, Blanc P, Bouvier H, Bastein O, Chiari P, et al. Effect of cardiopulmonary bypass on serum procalcitonin and C-reactive protein concentratioins. Br J Anaesth 1999;83:602-7.
- Delannoy B, Guye ML, Slaiman DH, Lehot JJ, Cannesson M. Effect of cardiopulmonary bypass on activated partial thromboplastin time waveform analysis, serum procalcitonin and C-reactive protein concentrations. Crit Care 2009;13:1-10.
- Brunckhorst FM, Eberhard OK, Brunkhorst R. Discrimination of infectious and non-infectious causes of early acute respiratory distress syndrome by procalcitonin. Crit Care Med 1999;27:2172-6.
- Nobre V, Harbarth S, Graf JD, Rohner P, Pugin J. Use of procalcitonin to shorten antibiotic treatment duration in septic patients. Am J Respir Crit Care Med 2008;177:498-505.
- Muckart DJ, Bhagwanjee S. American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference definitions of the systemic inflammatory response syndrome and allied disorders in relation to critically injured patients. Crit Care Med 1997;25:1789-95.
- Vincent J, Moreno R, Takala J, Willatts S, De Mendonca A, Bruining H, et al. The SOFA (Sepsis Related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med 1996;22:707-10.
- Brahms . Guide for the Clinical Use of Procalcitonin (PCT) in Diagnosis and Monitoring of Sepsis 2008.
- Downey C, Kazmi R, Toh CH. Novel and diagnostically applicable information from optical wave-form analysis of blood coagulation in disseminated intravascular coagulation. Br J Haematol 1997;98:68-73.
- O’Grady NP, Barie PS, Bartlett JG, Bleck T, Carroll K, Kalil AC. Guidelines for evaluation of new fever in critically ill patients. Crit Care Med 2008;36:1330-49.
- Downey C, Kazmi R, Toh CH. Early identification and prognostic implications in disseminated intra-vascular coagulatiuon through transmittance waveform analysis. Thromb Haemost 1998;809:65-9.
- Gaieski DF, Mikkelsen ME, Band RA, Pines JM, Massone R, Furia FF, et al. Impact of time to antibiotics on survival in patients with severe sepsis or septic shock in whom early goal-directed therapy was initiated in the emergency department. Crit Care Med 2010;38:1045-53.
- Toh CH, Downey C, Dwyre L. Thromboplastin sensitivity in waveform analysis. Thromb Haemost 2000;84:517-18.
- Toh CH, Samis J, Downey C, Walker J, Becker L, Brufatto N, et al. Biphasic transmittance waveform in the APTT coagulation assay is due to the formation of a Ca(++)-dependent complex of C-reactive protein with very low-density lipoprotein and is a novel marker of impending disseminated intravascular coagulation. Blood 2002;100:2522-9.
- Matsumoto T, Wada H, Nishioka Y, Nishio M, Abe Y, Nishioka J, et al. Frequency of abnormal biphasic aPTT clot waveforms in patients with underlying disorders associated with disseminated intravascular coagulation. Clin Appl Thromb Hemost 2006;12:185-92.
- Bone RC, Sibbald WJ, Sprung CL. The ACCP-SCCM consensus conference on sepsis and organ failure. Chest 1992;101:1481-3.
- Weinstein RA. Nosocomial infection update. Emerg Infect Dis 1998;4:416-20.
- Keuleyan E, Gould M. Key issues in developing antibiotic policies: from an institutional level to Europe-wide. Clin Microbiol Infect 2001;7:16-21.
- Mayer KH. The epidemiology of antibiotic resistance in hospitals. J Antimicrob Chemother 1986;18:223-33.
- Siddiqui S, Hussein K, Manasia R, Samad A, Salahuddin N, Zafar A, et al. Impact of antibiotic restriction on broad spectrum antibiotic usage in the ICU of a developing country. J Pak Med Assoc 2007;57:484-7.
- Office for National Statistics . Deaths Involving Clostridium Difficile: England and Wales, 2003–07. Health Stat Q 2008:67-76.
- Moerer O, Burchardi H. The cost of sepsis. Anaesthetist 2006;55:36-42.
- Kumar A, Roberts D, Wood KE, Light B, Parrillo JE, Sharma S, et al. Duration of hypotension before initiation of effective antimicobial therapy is the critical determinant of survival in human septic shock. Crit Care Med 2006;34.
- Toh CH, Ticknor LO, Downey C, Giles AR, Paton RC, Wenstone R. Early identification of sepsis and mortality risks through simple rapid clot-waveform analysis. Intensive Care Med 2003;29:56-61.
- Tang BM, Eslick GD, Craig JC, McLean AS. Accuracy of procalcitonin for sepsis diagnosis in critically ill patients: systematic review and meta-analysis. Lancet Infect Dis 2007;7:210-17.
- British Medical Association and Royal Pharmaceutical Society of Great Britain . British National Formulary. 2007.
- Arnold RJG. Pharmacoeconomics: from theory to practice. Boca Raton, FL: CRC Press; 2010.
- European Centre for Disease Prevention and Control (ECDC) . Annual Epidemiological Report on Communicable Diseases in Europe 2010 2010.
- Trouillet JL, Chastre J, Vuagnat A, Joly-Guillou ML, Combaux D, Dombret MC, et al. Ventilator-associated pneumonia caused by potentially drug-resistant bacteria. Am J Respir Crit Care Med 1998;157:531-9.
- Opal SM, Calandra T. Antibiotic usage and resistance: gaining or losing ground on infections in critically ill patients?. JAMA 2009;302:2367-8.
- Kollef MH, Fraser VJ. Antibiotic resistance in the intensive care unit. Ann Intern Med 2001;134:298-314.
- Claxton KP, Sculpher MJ. Using value of information analysis to prioritise health research: some lessons from recent UK experience. Pharmacoeconomics 2006;24:1055-68.
- Claxton K, Ginnelly L, Sculpher M, Philips Z, Palmer S. A pilot study on the use of decision theory and value of information analysis as part of the NHS Health Technology Assessment programme. Health Technol Assess 2004;8.
- Donaldson C, Hundley V, McIntosh E. Using economics alongside clinical trials: why we cannot choose the evaluation technique in advance. Health Econ 1996;5:267-9.
- Newby D, Hill S. Use of pharmacoeconomics in prescribing research. Part 2: cost minimisation analysis –when are two therapies equal?. J Clin Pharm Ther 2003;28:145-50.
- Tarnow-Mordi WO, Healy MJ. Distinguishing between ‘no evidence of effect’ and ‘evidence of no effect’ in randomised controlled trials and other comparisons. Arch Dis Child 1999;80:210-11.
- O’Brien BJ, Briggs AH. Analysis of uncertainty in health care cost-effectiveness studies: an introduction to statistical issues and methods. Stat Methods Med Res 2002;11:455-68.
- Hatala R, Holbrook A, Goldsmith CH. Therapeutic equivalence: all studies are not created equal. Can J Clin Pharmacol 1999;6:9-11.
- Pater C. Equivalence and noninferiority trials – are they viable alternatives for registration of new drugs? (III). Curr Control Trials Cardiovasc Med 2004;5.
- Span MM, TenVergert EM, van der Hilst CS, Stolk RP. Noninferiority testing in cost-minimisation studies: practical issues concerning power analysis. Int J Technol Assess Health Care 2006;22:261-6.
Appendix 1 The Sequential Organ Failure Assessment score
| Variable | SOFA score | |||
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |
| Respiration: PaO2/FiO2 (mmHg) | < 400 | < 300 | < 200 (with respiratory support) | < 100 (with respiratory support) |
| Coagulation: platelets (×103/mm3) | < 150 | < 100 | < 50 | < 20 |
| Liver: bilirubin [mg/dl (µmol/l)] | 1.2–1.9 (20–32) | 2.0–5.9 (33–101) | 6.0–11.9 (102–204) | > 12.0 (> 204) |
| Cardiovascular: hypotension | Mean arterial pressure < 70 mmHg | Dopamine ≤ 5 or dobutamine (any dose)a | Dopamine > 5 or adrenaline ≤ 0.01 or noradrenaline ≤ 0.1a | Dopamine > 15 or adrenaline > 0.01 or noradrenaline > 0.1a |
| Central nervous system: Glasgow Coma Scale | 13–14 | 10–12 | 6–9 | < 6 |
| Renal: creatinine [mg/dl (µmol/l)] or urine output | 1.2–1.9 (110–170) | 2.0–3.4 (171–299) | 3.5–4.9 (300–440) or < 500 ml/day | > 5.0 (> 440) or < 200 ml/day |
Appendix 2 Interpretation of procalcitonin levels
| PCT (ng/ml) | Interpretation |
|---|---|
| < 0.05 | Normal values. Local inflammation or infection is possible: systemic inflammatory response unlikely |
| < 0.5 | On first day of ICU admission this indicates a low risk for progression to severe sepsis and/or septic shock. Local inflammation or infection is possible: systemic inflammatory response unlikely |
| ≥ 0.5 and < 2 | Systemic inflammatory response present due to infection, severe trauma, major surgery or cardiogenic shock. If the patient has a proven infection it could be sepsis |
| ≥ 2 and < 10 | Likely to be sepsis (SIRS associated with infection). On first day of ICU admission this indicates a high risk for progression to severe sepsis and/or septic shock |
| ≥ 10 | Severe sepsis or septic shock. Organ dysfunction. High risk of death |
Appendix 3 Economic analysis
READ-ICU (RANDOMISED EVALUATION OF ANTIBIOTIC TREATMENT DURATION IN THE INTENSIVE CARE UNIT) TRIAL: PILOT STUDY ECONOMIC ANALYSIS
Dr Alan Haycox, Reader in Health Economics, University of Liverpool.
Miss Rachel Houten, Health Economist, University of Liverpool.
The economic analysis is conducted in two parts. In the first part analysis of the results of the pilot study will be conducted with consideration for the fact that the study is not powered to obtain any statistical differences and recommendations for data collection requirements to enhance a larger trial. In the second part two lessons learnt from the pilot study will be discussed paying particular attention to economic methodology.
Part I
Introduction
Resource utilisation and costs associated with the comparative antibiotic regimens were measured and valued up until the end of the follow-up period. The economic value of conducting further research [e.g. in the form of a larger randomised controlled trial (RCT)] was addressed in relation to the clinical and economic information collected as part of the pilot study. Unit costs were derived from data on acquisition costs and staff time obtained in the pilot study and combined with other costs derived from the Liverpool Heart and Chest finance department and national sources [the British National Formulary35 (BNF)]. Comparative resource use is provided in both physical (amount of resources consumed in both arms of the trial) and financial (comparative costs in both arms of the trial) terms to facilitate understanding and generalisation of the results obtained to alternative settings and as a form of reference for external model validation of our analyses. The small sample size inhibits the ability of the feasibility study to undertake robust statistical significance testing; however, comparisons between the two antibiotic regimens have been made whenever possible, utilising a range of summary statistics.
Economic outcomes evaluated
The focus of the economic analysis was entirely determined by the clinical objectives of the study, which were to assess the impact of the shorter duration of antibiotic use on the duration of ICU stay, mechanical ventilation and overall hospital stay and overall resource use within the hospital environment. Given this focus, resource utilisation and costs associated with each of the two pilot arms, specifically ICU stay, hospital stay, mechanical ventilation, antibiotics and other medications, tests and procedures, were measured and valued up until the end of the follow-up period. Because this was primarily a feasibility study the economic analysis also sought evidence of any potential tendency towards reduced levels of health outcome (increased incidence of sepsis, increased need to reinitiate antibiotic therapy or increased levels of infection-related mortality) that could be explored further in a larger trial.
Resource use and cost information were collected from an NHS hospital-level perspective. This perspective was chosen for the feasibility study as the health and resource impact associated with the change in hospital antibiotic regimen was expected to fall almost entirely within the secondary care environment. No evidence was obtained within the feasibility study that caused us to question the limited perspective undertaken for the analysis. Along with costing information to support the clinical outcomes, the use of antibiotics, other medications, tests and procedures was also recorded. Both outcome and resource use data were collected on day 10 and on hospital discharge, whichever is sooner.
From a resource perspective the comparison of the two antibiotic regimes may impose changes at a number of levels. First (and most visibly), the a priori expectation was that there would be an immediate and substantial cost saving arising from the shortening of the initial antibiotic regime. The initiation of an expensive 7-day antibiotic regimen based on a clinical suspicion of infection risk may lead to unnecessary treatment and wastage of scarce NHS resources. The initiation of antibiotic therapy is likely to provide no benefit to a substantial proportion of patients who apparently have no infection. In addition, the provision of such treatment is likely to contribute towards developing levels of antibiotic resistance in the UK. Although it is important for clinicians to immediately initiate antibiotic therapy once infection is reasonably suspected, it is equally important that such therapy is reassessed as soon as the results of the laboratory cultures are available. Needless to say, antibiotic therapy should be continued only in patients in whom infection has been confirmed. A further resource saving may appear on identification of lack of infection in patients if it is then possible that they can be relocated outside the ICU setting.
The perceived benefit of reduced antibiotic treatment does not lie with the cost improvement alone. Although treatment with antibiotics without the patient having an underlying infection is not directly harmful, it is unnecessary and can cause resistance to antibiotics in the long run. Antibiotic-resistant bacteria can then spread making infection more expensive and difficult to treat. The short-term focus of the feasibility study will not enable estimates of how the more effective targeting of therapy will impact on antibiotic resistance; this, perhaps, is one element that should be explored further in any larger study.
The economic methodology applied
The choice of economic methodology is entirely derived from the purpose and context of the trial. Our prior clinical expectation is that the removal of treatment with antibiotics in the absence of infection (when infection is initially suspected but unconfirmed by results from blood cultures) does not pose any health threat to the patients whilst simultaneously conserving significant levels of valuable resources for the NHS. As the pilot study was never intended to be sufficiently powered to show statistically significant differences between the outcomes of the two groups we are unable to make definitive recommendations on the cost and outcome implications of the introduction of the protocol. We can, however, look for any large variances between the two treated groups of patients in terms of outcomes, adverse events and survival. If we have a reasonable basis derived from the results of the feasibility study to believe that outcomes appear to be equivalent across the two groups then the analysis collapses to a simple question of which antibiotic regimen achieves this equivalent outcome at the lowest resource cost to the NHS. By posing the question in this manner it becomes apparent that the most appropriate methodology to employ in analysing data from this study is CMA,36 which is the economic framework that deals with cost analysis in the presence of evidence of equivalent outcomes. However, it is important to re-emphasise the fundamental premise on which this choice of methodology is made, which is that it assumes that the outcomes for patients in both of the groups are equivalent and unaffected by the duration of antibiotic treatment. If this is the case then the groups can be compared on a cost basis alone with the key cost saving to the NHS of the 2-day antibiotic treatment group arising from the reduction in the length of antibiotic therapy provided to patients in cases in which this was not clinically required.
In applying the CMA methodology it is crucial to check that the occurrence of adverse events and survival are similar across the two groups to ensure that our assumption of equal outcomes is not being violated. Such variances would have to be closely monitored in a larger trial and outcome variation incorporated to offset any cost saving of antibiotic treatment in the economic analysis. Such issues are addressed in much greater detail later in this appendix.
If variation is apparent between the two groups then a cost-effectiveness methodology would be applied. Cost–utility analysis is the gold standard health economic framework in which outcomes are expressed in terms of quality and quantity of life and evaluated alongside resource use and cost to ensure an effective use of resources. A cost–utility analysis would be inappropriate in the context of the trial as any quality-of-life measurement of the patients within this cohort is unlikely to be sensitive enough to the changes experienced. As the difference between infected and non-infected patients is not symptom related (and why the trial is necessary), it is likely that the short-term quality of life of these patients would in fact be the same.
Results
Three patients whose treatment deviated from the protocol have been excluded from the economic analysis to ensure comparability across groups. The results of the blood cultures taken at the time of initiation of antibiotic therapy are provided in Table 11.
Hours of ventilation
The mean hours that patients in each arm of the trial spent on ventilation were substantially different although this was not statistically significant, an observation that may be related to inclusion of outliers by chance alone. Patients in the 2-day antibiotic arm of the trial (treatment group 1) spent an average of 80 hours on ventilation in comparison to an average of 136 hours for patients in the 7-day arm of the trial (treatment group 2). The results and CIs for both arms of the trial are provided in Table 9.
| Treatment group | No. of patients | Mean hours vented | 95% CI |
|---|---|---|---|
| 1 (48 hours) | 23 | 79.57 | 38.31 to 120.82 |
| 2 (7 days) | 20 | 136.00 | 78.93 to 193.07 |
Patients on 2-day antibiotic regimens spent on average only 55% of the time ventilated that patients on 7-day antibiotics were ventilated for. However, care needs to be taken in estimating the additional cost associated with the additional time spent on ventilators by patients in the 7-day arm of the trial. The cost of ventilation is included in the per day cost of stay in ICU (postoperative coronary care unit) and therefore including an additional per hour cost of ventilation would double count some elements of cost difference between the two groups. However, given the apparent importance of this element, both the reasons for and the cost implications of variations in ventilator use in the critical care context should be explored in greater detail in any subsequent trial.
Duration of intensive care unit stay
The duration of stay in critical care is an important determinant underlying the comparative cost for the patients in this trial. Variations in length of stay may indicate a difference in the speed of recovery being experienced between the patients in each of the treatment groups. This would have important resource use implications.
As the follow-up period is 10 days the data are truncated at one end. This is, however, justified as the influence of the infection and any treatment would not be expected to go beyond this period. Thus, any variation in stay past this point is more likely to be driven by other factors than the length of antibiotic regimen provided to the patients. Patients who died were given their duration in the critical care area as the total number of days that they were in the trial. The results and CIs for mean days in critical care for both arms of the trial are provided in Table 10.
| Treatment group | No. of patients | Mean days on ICU | 95% CIs |
|---|---|---|---|
| 1 (48 hours) | 23 | 6.07 | 4.58 to 7.55 |
| 2 (7 days) | 20 | 8.13 | 6.63 to 9.62 |
| Treatment group | Positive cultures | No. of patients |
|---|---|---|
| 1 (48 hours) | No | 20 |
| Yes | 3 | |
| 2 (7 days) | No | 20 |
| Yes | 3 |
Patients on 2-day antibiotic regimens spent on average 75% of the time on ICU that patients on 7-day antibiotic regimens spent on ICU. An average cost per day for the ICU in the host hospital for the trial was obtained from the finance department of the Liverpool Heart and Chest Hospital. The data provided emphasised that the ICU cost will vary from £1350 to £1600 per day depending on a patient’s requirements, for example the need for ventilation adds substantially to the per day cost in ICU, but unfortunately more detailed costing data were not, at this stage, available.
Results of the blood cultures
The proportion of patients who had been initiated on antibiotic therapy because of clinical suspicion of infection but in whom infection was not identified in the blood culture was remarkably high (87%). Such a result highlights the importance of this trial as it emphasises the potential for resource saving and prevention of unnecessary treatment arising from early identification of negative results. However, interpretation of the results is fundamentally linked to the sensitivity and specificity of the blood culture tests in accurately dichotomising at an early stage between patients with and without infection. Arguably a false-positive is of less concern as it will simply result in having resources wasted on patients through the continuation of antibiotic therapy; however, a false-negative could have huge implications for the health of the patient if treatment is withdrawn as a result and the infection is allowed to develop unchecked. Factors underpinning the accuracy with which the blood culture tests can minimise false-positive and (especially) false-negative results should be a fundamental aspect of the clinical and economic analysis undertaken in a larger trial. A follow-up blood test to ensure patient safety may be one way to circumvent any sensitivity and specificity variation in blood culture results in a larger trial.
Cost analysis
The main costing analysis has been conducted using patient-level data on the duration of antibiotic therapy. Per patient costing for ICU duration could not be calculated as the data set did not contain enough information to correctly allocate the actual cost incurred from the broad range of estimates obtained from the Liverpool Heart and Chest Hospital (from £1350 to £1600). Using the mean difference in days on ICU between the two treatment groups (2.06), the potential cost savings from a reduction in ICU stay can be estimated to range between £2781 and £3296. Using the CIs to estimate the largest and smallest differences we can derive a broader range of potential cost savings of between £1242 and £8064.
As the number of blood cultures obtained as part of the trial should not differ between groups their costs have not been calculated in the economic analysis.
The antibiotic therapy costing analysis undertaken was based on a number of assumptions that were made in collaboration with the clinical investigators. The nature and implications of these assumptions are outlined in the following sections.
Intention-to-treat analysis
The analysis has been conducted on an intention-to-treat basis; however, there were four patients in the 2-day treatment arm who in practice received ≥ 7 days of antibiotic therapy. This will obviously have a negative impact on the cost difference associated with the antibiotic therapy between the treatment groups; however, because of the clinicians’ autonomy to treat their patients as they see fit this cannot be discounted from the analysis as our aim is to produce estimates that best reflect a real-life application of the protocol.
When patient weight was missing an average was imputed for each group for use in the calculation of antibiotic dosage
The appropriate amount of one of the antibiotics routinely prescribed (teicoplanin) is weight dependent. Thus, in estimating the amount and cost associated with the prescribing of teicoplanin, accurate recording of a patient’s weight is fundamentally important. Unfortunately, in a small number of cases (n = 2) it proved impossible to ascertain the weight of the patient as they were emergency admissions and hence in such circumstances an average weight (and hence cost) was attributed to the patient. Patients who weigh < 85 kg are given 400 mg of teicoplanin twice a day for the first day and 400 mg per day thereafter; patients who weigh > 85 kg are given 6 mg/kg twice a day for the first day and 6 mg/kg per day thereafter.
Vials are assumed to be indivisible
For the purposes of the cost analysis it was assumed (following clinical guidance) that the splitting of vials of antibiotics is not permitted. As such, as soon as a new vial is opened, irrespective of how much of the vial is used, the full cost of the vial is allocated to the patient.
The antibiotic wastage over the course of the trial was estimated to quantify the additional cost of being unable to divide a vial of teicoplanin. Between the 46 patients in the trial population, 87 vials’ worth of teicoplanin was wasted, which at a cost of £6.10 per vial equates to £530.70.
The dosage of antibiotics was allocated according to the protocol and multiplied by the number of hours on antibiotic therapy as recorded in the summary data
The cost of the antibiotics calculated included only the antibiotics given as part of randomisation. The use and costs of other antibiotics may be an important factor in the roll-out of such a protocol; however, in such a small sample the costs for the seven patients who needed further antibiotic therapy above that allocated at randomisation would have large implications for the average costs of treatment and would likely disguise the direct cost saving by not providing unnecessary treatment.
The number of hours spent on antibiotic therapy was recorded for each patient. These data were used to estimate the total number of doses of antibiotics provided to each patient. In all cases the BNF costs35 were applied to the estimated quantity of antibiotics used. In the case of teicoplanin the cost per vial (400 mg) was £6.10. The dosage of meropenem is not weight dependent; therefore, along with their teicoplanin treatment patients will be given 1 g three times per day for the duration of the treatment. The cost per 500-mg vial of meropenem is £8.60.
The antibiotic costs in both arms of the trial are provided in Table 12.
| Treatment group | No. of patients | Mean antibiotic cost per patient (£) | 95% CIs (£) |
|---|---|---|---|
| 1 (48 hours) | 23 | 168.97 | 105.76 to 232.18 |
| 2 (7 days) | 20 | 375.86 | 328.48 to 423.24 |
| Average cost difference | 206.89 |
The results of the antibiotic cost analysis emphasise the significant potential savings in antibiotic utilisation and cost that arise from use of an initial 2-day regimen. The cost of antibiotic use in the 2-day group is only 45% of the cost of antibiotic use in the standard care group, leading to an average antibiotic cost saving of over £200 per patient.
Although the difference in antibiotic cost between the two arms of the trial is statistically significant it is important to remember the limitations of the current trial. The patient-level costing was conducted to estimate the cost saving of the protocol in a practical environment and as the 95% CIs show there is variation in the treatment costs that cannot be solely explained by the difference in the average weight of the two treatment arms (group 1: 77.7 kg, group 2: 80.41 kg). We would expect, because of the very nature of the protocol, that the 7-day treatment arm would cost more in terms of antibiotic therapy. However, the exact magnitude of this cost is likely to fluctuate in line with variations in clinical practice. As the sample is currently small the reported cost difference could be largely driven by a few key outliers in the data and the practice of individual consultants who choose to continue or cut short antibiotic therapy on the basis of the blood culture results. A larger trial would enable greater confidence to be placed on the cost difference as the variation due to individualistic factors would be lost and the outcomes could be interpreted as being more robust and generalisable.
It is widely recognised that hospitals are usually able to negotiate significant discounts for the drug therapies that they purchase. However, the baseline analysis follows National Institute for Health and Clinical Excellence guidance in using published costs as the basis for the costing of drug use. To assess the impact of drug price discounts, a sensitivity analysis around the cost of the drugs has been conducted. The results of the sensitivity analysis show that a 30% reduction in the price of both antibiotic therapies would result in the average cost difference between the two groups being reduced to £144.82 per patient.
A larger trial of 2-day compared with 7-day use of antibiotics in this patient group would benefit from resource use data on the outcomes and adverse event profiles of the individuals as well as the calculation of costs that could be allocated to the resource use. This would enable cost-effectiveness estimates to be produced if the evidence suggested that the outcomes of the two treatment arms were not equivalent. The ability to delineate the ICU stay cost from its association with ventilation would also allow any variation in ventilation to be captured as a distinct category.
Potential cost savings
Antibiotic use is one of the major cost elements in most hospitals within the UK. As such, to illustrate the potential cost savings arising through moving to an initial 2-day antibiotic regimen the analysis has estimated the cost saving that would arise if this protocol was applied at a broader level. The first aggregated analysis estimates the potential savings in antibiotic costs if the 2-day policy was applied across the whole of the ICU at Liverpool Heart and Chest Hospital.
The analysis was based on figures obtained from the hospital’s infection control database and relate to the 9-month period from November 2010 to July 2011 (Table 13).
| Description | No. of patients |
|---|---|
| Patient throughput in ICU | 1570 |
| Cultures requested | 473 |
| Positive cultures | 49 |
The most striking aspect arising from these figures is that only 10.4% of those patients for whom blood samples were obtained because they were suspected of infection returned positive cultures indicating that they did indeed have an infection. From a face validity perspective it is perhaps heartening that this figure closely correlates with the 13% observed in the trial. To estimate the potential annual savings in antibiotics that could be made by the ICU the results obtained in the 9-month period analysed were extrapolated into an annual equivalent. This assumption therefore ignores any seasonal fluctuations that may have affected antibiotic use in this 3-month extrapolation.
The annual equivalent analysis estimates that 2093 patients were treated in the ICU over the 12-month period. Of these it is estimated that 631 patients would initially be treated with antibiotics as a consequence of suspected infection and, of these, only 66 (10.4%) would be expected to have this diagnosis confirmed by the blood test. The anticipated cost savings per patient associated with the 95% CIs range between £191.06 and £222.72. Imputing these values into the annual estimates outlined above results in antibiotic cost savings ranging between £108,140 and £126,060 for the ICU at Liverpool Heart and Chest Hospital. These results emphasise that a roll-out of the 2-day antibiotic regimen across all units in the local area or nationally would promote significant cost savings to the NHS without detrimental impact on patient health.
To acknowledge some of the uncertainty surrounding the projection estimates, namely the extrapolation of the recorded values from 9 to 12 months, and to show the impact of each of the parameters a simple sensitivity analysis has been conducted. Table 14 uses the average cost difference per patient observed in the trial of £206.89 and displays the annual cost savings that would be observed in the ICU under a range of alternative assumptions. The original value scenario uses annual throughput estimates and the proportion of patients suspected of sepsis and the proportion of positive cultures obtained from an audit of the ICU in which the trial has been conducted. In this case the savings made from a roll-out of the 2-day protocol would be £117,000 per year. The estimated value scenarios look to quantify the impact of variation of the key assumptions that we have made in the original value scenario. In each scenario only one assumption is altered while all other assumptions remain constant at the original values. For example, if the proportion of patients suspected of sepsis increases to 40% (instead of the current 30%) and the patient annual throughput in the ICU and the proportion of positive cultures observed remain the same then the annual cost saving for the ICU unit would be £156,000.
| Assumption | Original value | Estimated value | ||||
|---|---|---|---|---|---|---|
| Total throughput of patients | 2099 | 70% | 80% | 90% | 110% | 120% |
| Cost saving (£) | 117,100 | 81,970 | 93,680 | 105,390 | 128,810 | 140,520 |
| Proportion of sepsis-suspected patients (%) | 30.1 | 5 | 10 | 20 | 40 | 50 |
| Cost saving (£) | 117,100 | 19,446 | 38,891 | 77,783 | 155,565 | 194,456 |
| Proportion of positive cultures observed (%) | 10.4 | 5 | 15 | 20 | 25 | 30 |
| Cost saving (£) | 117,100 | 124,217 | 111,142 | 104,604 | 98,066 | 91,528 |
This sensitivity analysis shows how the cost-saving potential is dependent on the number of individuals who would be eligible for a reduction in the duration of their antibiotic therapy. If the number of patients suspected of sepsis or eligible for a possible reduction in antibiotic use was in fact only 5% of the total throughput, and holding the total throughput and proportion of individuals with positive cultures constant, the annual cost saving is estimated at around £19,466. If, however, the number of patients displaying symptoms of sepsis was in fact 50% of the total throughput, holding everything else constant, the cost saving could be around £194,456.
Outcomes
Sequential Organ Failure Assessment scores
Sequential Organ Failure Assessment scores were used to measure the change in outcomes associated with 2- and 7-day antibiotic regimens. The SOFA score is a summary score used to monitor the severity of a patient’s health. The summary score is made up from six different observations concerned with the functioning of different systems within the body. These are the respiratory, renal, hepatic, cardiovascular, coagulation and neurological systems. The higher the score the more severe the patient’s condition is deemed to be. The expectation is that patients in both arms of the trial would experience improving SOFA scores as a result of antibiotic treatment or as the symptoms of SIRS diminish. If patients from one arm of the trial improved to a greater extent than those in the comparator arm it would be indicative of the clinical superiority of this arm of the trial.
The collection of quality-of-life information is inappropriate in this trial. Both patients with sepsis and those without display the same symptoms even though the cause of the symptoms is different, and the finite time frame in which the change in symptoms occurs means that the quality of life of patients would be unlikely to display any change.
Only the SOFA scores of patients who have scores at three time points are used. The repeated measurement enables changes and improvements to be recorded over time. In a full trial we can use such scores to ensure equivalence of results between the two groups.
If randomisation has been accurately put into practice we would not, given a larger sample, expect to see a significant difference between the two groups from baseline to 48 hours as there should be no systematic variations in treatment patterns between the two groups until some patients are removed from the antibiotic therapy pathway after 2 days (as a result of negative blood cultures). The results obtained from the SOFA score analysis are provided in Tables 15–17.
| Treatment group | No. of patients | Mean score | 95% CI |
|---|---|---|---|
| 1 (48 hours) | 11 | 9.36 | 5.95 to 12.78 |
| 2 (7 days) | 13 | 12.08 | 9.40 to 14.75 |
| Treatment group | No. of patients | Mean score | 95% CI |
|---|---|---|---|
| 1 (48 hours) | 11 | 6.36 | 3.80 to 8.93 |
| 2 (7 days) | 13 | 10.00 | 7.34 to 12.66 |
| Treatment group | No. of patients | Mean score | 95% CI |
|---|---|---|---|
| 1 (48 hours) | 11 | 2.00 | 0.39 to 3.61 |
| 2 (7 days) | 13 | 4.08 | 1.92 to 6.23 |
Given that the final SOFA score is taken when the patient is discharged from hospital the exact period from commencement on antibiotic therapy is likely to vary from patient to patient. This is, however, true of all of the measurements taken at the end point and is unlikely to skew the results as (particularly in a larger trial) the randomisation should account for variation such as this in both arms of the trial. As we are measuring a process change in which quality-of-life assessment tools are unlikely to be sensitive to those patients suspected of sepsis but in fact have SIRS or some other condition in which antibiotic therapy will not cure, the SOFA scores would enable a check on the clinical comparability of the severity of the condition of each of the groups at the end point of the trial.
Given the comparatively small number of patients with a complete set of SOFA scores (24) it is difficult to derive any firm conclusions from the analysis. However, it would appear that at baseline the patients in treatment group 2 were more severely ill (had higher SOFA scores) than those in treatment group 1. However, the number of patients who have SOFA scores at all time periods recorded is less than half of the total sample. This is because, to obtain a reading for some of the elements, a blood sample needs to be taken. It was deemed unethical to obtain such a sample purely to complete the SOFA score and therefore those patients who no longer had an indwelling cannula fitted at 10 days or those who had left the ICU do not have a SOFA score. In light of this, in any larger trial, the aim should be to collect all elements of the SOFA score that are deemed ethical and, in patients who have recovered sufficiently such that they do not require any intravenous treatments, perhaps a zero score could be allocated for that element.
Preliminary analysis of the change in SOFA scores in the two arms of the trial show a 32% reduction between baseline and 48 hours in treatment arm 1 and a 17% reduction in treatment arm 2. The percentage reduction between 48 hours and discharge is 69% in treatment arm 1 and 59% in treatment arm 2.
Mortality
The death rate observed in the trial (three deaths in treatment arm 1 and one death in treatment arm 2) is far too small to draw any significant conclusions. Analysis of the cause of death indicated that in all cases it appeared to be unrelated to the trial. However, it is crucial that this element is assessed in detail in any larger trial as any variation in this element identified in a longer follow-up period would allow survival to also be incorporated as an outcome in the economic analysis.
Prevention of the development of antibiotic resistance
The European Centre for Disease Prevention and Control highlighted the potential development of antibiotic-resistant disease as Europe’s biggest disease threat. 37 A significant association has been observed between the extent of previous antibiotic usage and development of antibiotic resistance. 38 In addition, the more vulnerable the patient, with many catheter lines and ongoing treatments, the more at risk they become of infection by antibiotic-resistant bacteria. 39 This fact coupled with the routine use of multiple classes of strong antibiotics in critical care environments to treat the critically ill means that these areas become a primary breeding ground for both the development and spread of antibiotic-resistant bacteria. 39 It would also appear that the duration of mechanical ventilation is a particular risk factor for the development of antibiotic resistance. 38
The benefits of reducing antibiotic resistance, although difficult to quantify accurately, especially because of its variability by locality,40 should not be underestimated. In addressing long-term antibiotic resistance it is essential that unnecessary use of antibiotics is minimised. As such, discontinuation of antibiotic treatment in cases in which such treatment is not required and providing the shortest course of antibiotics required to achieve maximum patient benefit are crucial in preventing the development of future antibiotic resistance. 39
The simple adjustment in antibiotic utilisation addressed in this feasibility study (providing that a similar trend was apparent in any larger study) would significantly assist in preventing the future development of antibiotic resistance, through a reduction in both antibiotic use and the length of time that a patient remains on mechanical ventilation.
Applying value of information theory to the results obtained from the feasibility study
Bayesian value of information analysis seeks to quantify the value of research produced to ensure that it exceeds the costs of acquiring the information. The theory is based on the premise that the cost-effectiveness of an intervention is quantified as its net benefit (NB):
where j is the intervention, Q are the expected outcomes, C are the expected costs and λ is the cost-effectiveness threshold. 41
The expected value of perfect information (EVPI) is therefore:
where σ are the parameters in the model. 41
The NB of an intervention is uncertain and, with only current information available, a decision must be made with the presence of uncertainty, with the intervention that provides the maximum expected benefit being the optimal choice. 41
With perfect information there is no uncertainty surrounding the parameter values, and therefore there is no chance of the wrong decision being made, which would have consequences and costs for the health service. 42 However, this is unobtainable in reality and therefore the EVPI is estimated by averaging the maximum NB over the joint distribution of the parameters. 41
The value of further information for each parameter can be calculated separately by estimating the value of perfect information in this factor and leaving everything else constant, at the value of current information. 41
The commencement of this feasibility study was preceded by the thinking that a reduction in the use of unnecessary antibiotic therapy would result in large savings to the NHS in terms of the direct treatment costs. To conduct a Bayesian value of information analysis some certainty over the parameters contained within the model is required. What this feasibility study has discovered, however, is that not only are these direct savings from antibiotic reduction likely to vary in a practical environment as a result of clinical decision-making but also there may in fact be other unforeseen impacts – namely a change in the duration of ICU stay and ventilation use. Producing estimates of the value of additional information through Bayesian methodology when the model structure is uncertain would produce a range of scenarios that are unlikely to offer any meaningful contribution until the justification for the inclusion of each parameter is obtained. A further trial, for example, may contradict the trend that we have seen in this feasibility study for more ventilation use within the 7-day group and find no difference, therefore providing justification for exclusion of this parameter in the model of cost-effectiveness, or adverse event profiles may show between-group variation, which would therefore warrant the inclusion of these adverse events within the health economic model. In light of the questions raised about the possible factors influenced by the change in antibiotic usage within this feasibility study, the potential value of obtaining additional information will be discussed in a more pragmatic sense.
With current assumptions about the equivalence of outcomes in the READ-ICU pilot, the difference between the two treatment groups is simply related to costs. The most obvious cost difference is associated with antibiotic usage; however, there may also be cost differences associated with the length of stay in ICU. Such a value of information model is easily applied using sensitivity analysis when the measurement of outcome can be quantified in monetary terms. However, as the collection of quality-of-life information is inappropriate in this trial, as it would be unlikely to show any difference between the two groups because of the short time frame of the trial, and the fact that the patients in both cases are symptomatic and it is only the cause of the symptoms that differs, the allocation of an outcome threshold becomes somewhat abstract.
The relationship between duration of ventilation and ICU stay and duration of antibiotic therapy needs to be explored in a further trial to confidently estimate its likely magnitude. However, if the trends in the feasibility study are seen in a larger trial its impact on NHS resources is likely to be substantial.
The sensitivity and specificity of the blood cultures used to diagnose sepsis are of paramount importance to both the safety of patients and the likelihood of a cost-effective change in practice. The value of this information would likely be huge and can be estimated by conducting more frequent blood cultures in a future trial setting.
A further study should also collect data on additional variables such as the need to reinitiate antibiotic therapy during the trial period and the monitoring of patients in each arm of the trial in order to account for all variation in outcomes and resource use as a result of the change in duration of antibiotic therapy.
What is evident from the preliminary analysis of the pilot trial data is that there appears to be a significant cost saving to be made from the incorporation of the suggested process change. Although the current data do not sound any alarm bells in terms of adverse event profiles, additional information from a further trial to confirm this would be of immense value. There remains uncertainty around the duration of ICU stay and duration of ventilation because, although there were no statistically significant differences between the groups because of the small sample size, there appears to be a pattern of difference emerging between the two groups. This could have large implications for the cost savings to be made by the introduction of the protocol. Quantification of this difference, if it is in fact a true difference, in a larger sample would enable greater precision when estimating the economic impact of the protocol.
Part II
What lessons can be learnt from the feasibility study for the design of the major study?
Introduction
The principal economic issues that have been addressed in this feasibility study are:
-
What is the most appropriate economic methodology to apply in the context of the clinical results obtained in the study?
-
What lessons have been learnt concerning the structure of any future trials that would improve their reliability as a basis for health-care decision-making?
Each of these issues are explored in further detail in the following sections.
What is the most appropriate structure of economic analysis for comparing 2- and 7-day antibiotic regimens?
The appropriateness of any economic methodology depends on the nature and context of the underlying clinical analysis. Evaluations based on inappropriate or poor-quality clinical data will fail to provide a reliable basis for health-care decision-making. The primacy of clinical data is particularly evident in the choice of economic methodology that is appropriate in any context. In the case of this trial two potential options were available. First, if the feasibility study indicated that there were significant differences in outcome between 2- and 7-day antibiotic regimens then the analysis would be most appropriately undertaken in the context of a cost-effectiveness analysis (CEA). Such an analytical structure would identify, measure and value variations in both costs and outcomes arising between the two antibiotic regimens to generate a cost per unit of outcome. The second potential methodology is to apply a CMA in which, conditional on health benefits between the two competing antibiotic regimens being equivalent, the least expensive option is preferred. Analysis of the clinical data obtained in the trial does not appear to indicate the presence of significant variations in clinical outcome between the two arms of the trial and therefore the approach of the feasibility study to the economic analysis is CMA. However, it is important to emphasise that should significant adverse events arising from the shortening of the antibiotic regimen become apparent then the structure of our analysis would alter.
Many sources of clinical evidence can be used to support economic analyses; however, the ‘gold standard’ is normally considered to be the RCT, which holds everything constant with the exception of the variation being evaluated. The value of having access to the results of the feasibility study arises from the fact that it effectively informs decisions with regard to any further economic study that is undertaken. By definition, the results of clinical trials cannot be known in advance and therefore in the absence of a feasibility study it would be impossible to plan to undertake a CMA alongside an RCT because there would be no prior evidence to determine whether or not the health outcomes being compared could be considered to be equivalent. 43 Therefore, no prospective economic evaluation should be initiated as a CMA unless there are strong theoretical reasons or (as in this case) empirically generated evidence that the outcomes generated by both arms of the trial are expected to be ‘identical or similar’. When such evidence has been generated in a feasibility study CMA can be adopted as an appropriate methodology for subsequent health economic analysis.
The apparent simplicity of the CMA approach belies complex theoretical underpinnings that are just as rigorous as those underpinning other methods of economic evaluation. Before applying this methodology it is essential that the clinical trial evidence obtained in the feasibility study is sufficient to justify the assumption of clinical equivalence between the 2- and the 7-day antibiotic regimens. Extreme rigour is required in ensuring equivalence in health benefits before deciding on the appropriateness of employing CMA as an economic methodology. In the case of our feasibility study, underpinning this choice of methodology was a detailed analysis of clinical data, which indicated that in all crucial characteristics the interventions being compared lead to equivalent health outcomes. If this crucial and indispensable element underpinning the decision to use CMA is found to be erroneous in the light of further evidence the analysis would revert to CEA. 44
What evidence has been obtained from the feasibility study to inform the structure and focus of future trials comparing 2- and 7-day antibiotic regimens?
It is perhaps surprising that the exact nature of the evidence base required to prove ‘therapeutic equivalence’ has not been subject to more intense scrutiny. The inability of a health intervention to prove superiority in a superiority trial in no way indicates that this necessarily implies clinical equivalence. Recent advances in clinical trial design have made it easier to directly compare clinical equivalence in a more meaningful manner, with the development of non-inferiority trials allowing this issue to be directly addressed. Alternatively, when a trial is initially designed as a superiority trial but such superiority remains unproven the analysis can be switched from superiority to non-inferiority in appropriate cases. The implications of adopting an inappropriate clinical trial design or misinterpreting the results of a clinical trial are often considerable: ‘wrongly discounting treatments as ineffective will deprive patients of better care. Wrongly accepting treatments as effective exposes patients to needless risks and wastes.’45
If it is perceived that the results obtained in the feasibility study support the therapeutic equivalence of the antibiotic regimens then this has important implications for the structure of any subsequent trials. RCTs can be structured to evaluate superiority, therapeutic equivalence or therapeutic non-inferiority. The greatest support for the use of CMA occurs when an equivalence trial unambiguously proves that two health-care technologies are clinically equivalent. Such certainty in trial outcomes is rare, however, and in practice there exists a myriad of ‘grey’ areas that may be indicative of therapeutic equivalence but which require more careful analytical consideration and judgement.
The superiority trial estimates the probability that the effect exists when the null hypothesis is true using the test statistic (p-value). However, p-values obtained in superiority trials may be inadequate to interpret the results of clinical trials. The use of CIs and personal judgement may be a more accurate method of determining clinical equivalence before accepting or rejecting an equivalence claim: ‘leaving it up to the reader to decide whether the CI includes or excludes potentially clinically important differences between two treatments. If it does not exclude differences. . . assume that the two drugs are not the same.’44
Superiority trials are specifically designed to demonstrate that there is, indeed, a difference and, thus, to reject the null hypothesis of equivalence in favour of the alternative hypothesis (i.e. that there is a difference). Evidence from well-designed superiority trials obtained in the context of a feasibility study may be of value in generating evidence of health equivalence for use in structuring further clinical trials; however, future trials should be designed in accordance with such prior evidence of equivalence. A short examination of the two suggested structures for a larger and more definitive trial of 2-day compared with 7-day antibiotic regimens is outlined in the following sections.
Structure 1: equivalence trials
Equivalence trials are specifically designed to demonstrate that the effect of one intervention (a 2-day antibiotic regimen) is not worse than the effect of the current intervention (a 7-day antibiotic regimen) by more than a specified equivalence margin. The aim of an equivalence trial is therefore to specifically rule out significant clinical differences between the treatments by directly evaluating the extent to which two health-care interventions have equivalent therapeutic effects. Briggs and O’Brien46 argue that CMA should be employed as an economic methodology only when clinical evidence has been obtained from an equivalence trial. However, even in the case in which an equivalence trial indicates clinical equivalence in primary outcomes (adverse events), scrutiny of secondary outcomes may reveal significant differences in safety, cost or convenience: ‘one therapy may offer clinical benefits such as a more convenient administration schedule, less potential for drug interaction or lower cost’. 47
In the current trial, for example, the duration of stay on the ICU and the duration for which a patient requires ventilation show some differences between the two groups, although statistical significance is not apparent because of the small sample size. Such secondary outcomes have significant implications both for the patients and for the resources required to treat them and therefore need to be considered alongside the adverse event profiles to ensure equivalence on all fronts.
A crucial step in the design of an equivalence trial is the definition of clinical equivalence. The equivalence margin attempts to incorporate all values that represent unimportant clinical differences in treatment and must be stipulated in advance of the clinical trial. The equivalence range, therefore, includes the largest difference between treatments that is clinically acceptable before treatments become defined as providing significantly different benefits. Clinical equivalence can be claimed if the 95% CI around the difference in treatments is found to lie entirely within the predetermined clinical equivalence margin. The setting of the equivalence margin communicates a judgement about what is and what is not clinically and statistically acceptable48 and it is important that good clinical judgement is employed to ensure that the chosen margin is clinically relevant and statistically feasible.
Structure 2: non-inferiority trials
The rationale behind a non-inferiority trial is to demonstrate that the 2-day antibiotic regimen is not worse than the current 7-day antibiotic regimen by a prestated clinical margin. This type of trial is particularly useful when the issue being evaluated relates to the extent to which the new antibiotic regimen is as ‘good’ as the current antibiotic regimen. In non-inferiority trials analysis is focused entirely in one direction – typically that the new treatment is not worse than the established therapy by more than the non-inferiority margin that has been prespecified. An improvement of any size fits within the definition of non-inferiority. Span et al. 49 emphasise the potential value of non-inferiority trials in informing the results of CMAs: ‘the most efficient analysis of the clinical effect in a cost minimisation study is the non-inferiority analysis’. 49
The non-inferiority range should be set in relation to the clinical notion of a minimally important effect. An acceptable non-inferiority margin depends on defining a difference that has previously been identified as being not clinically significant. Non-inferiority is demonstrated when the CI around the treatment difference lies entirely to the right of the lower bound of the non-inferiority margin.
Other issues to be addressed in evaluating ‘equivalence’ in future trials of 2 compared with 7 days of antibiotics
Distinguishing between statistical and clinical significance
One of the failings of statistical analyses undertaken in the context of a superiority trial is that statistical significance may differ from clinical significance. Variables that are identified as exhibiting statistically significant differences may be entirely unimportant from a clinical perspective whereas clinically crucial differences remain crucial even if they fail to achieve statistical significance. In contrast, in equivalence trials and non-inferiority trials, statistical and clinical significance are inextricably linked through the setting of equivalence and non-inferiority margins.
Evaluating equivalence in single or multiple outcomes
In clinical practice it is highly unlikely that two health-care interventions will yield exactly the same health benefits in all dimensions of clinical and patient outcomes. Typically, the design of equivalence trials and non-inferiority trials identifies a single end point for comparison despite the perception that one of the treatments is likely to offer significant advantages in another area. For example, when two treatments have equal efficacy yet one is more convenient to patients, then the extent to which CMA can be appropriately utilised depends largely on the perspective adopted by the analysis. When equivalence is not demonstrated for all important outcomes, the analyst must provide explicit justification for using CMA in light of the study question and perspective. In large part, the interpretation of clinical equivalence will depend on the specific circumstances of the clinical trial, the range of outcomes being measured and the judgement of the analyst. In such cases it is difficult to provide specific guidance that would be appropriate in all cases.
In any clinical trial it is necessary to identify a primary health outcome that is common to the competing alternative interventions. Choice and measurement of such an outcome measure are crucial in determining the appropriateness of the trial as an evidence source on which to undertake CMAs. To be of value, the primary health outcome must be the dominant outcome from the perspective of both patients and clinicians and capture the most clinically relevant benefits of the competing treatments. If not, claims of clinical equivalence, even when based on equivalence trials, are not sufficient to support the use of CMA.
Conclusions
It is essential that health economists and decision-makers are clear on what is meant by the concept of clinical equivalence and acknowledge that, given the heterogeneous nature of patient populations and treatment outcomes, it is likely to prove impossible to achieve exact equivalence between competing health-care interventions.
The appropriateness of using CMA must be judged in the light of the totality of the clinical evidence supporting or refuting the hypothesis of therapeutic equivalence between 2- and 7-day antibiotic regimens. However, certain limited guidance can be provided with regard to the appropriateness of undertaking CMA analysis.
First, the most appropriate design for a clinical trial to generate evidence that two health-care technologies are ‘identical or similar’ is the equivalence trial. Such trials are specifically designed for this purpose and therefore any differences that are identified between the health interventions being compared are neither clinically nor statistically significant. Therefore, clinical evidence from a well-designed equivalence trial represents the gold standard in supporting claims of clinical equivalence between a 2- and a 7-day antibiotic regimen. However, even when data are available from an equivalence trial it still remains important to consider the extent to which the primary health outcome fully captures the benefits being derived from the antibiotic regimens being compared. If other benefits are clinically meaningful to patients and clinicians, additional comparisons of clinical equivalence may be required.
Second, failure to prove clinical superiority should not be interpreted as providing evidence of clinical equivalence. In certain circumstances data from a superiority trial may be reanalysed to assess clinical equivalence. However, such reinterpretation of the data set must be justified through further analysis to show that there is indeed a therapeutic equivalence between 2- and 7-day antibiotic regimens.
Third, the extent to which data from non-inferiority trials can be used to justify the use of CMAs is currently subject to a great amount of uncertainty. In particular, to what extent proof of non-inferiority represents an acceptable approximation of ‘therapeutic equivalence’ in comparing 2- and 7-day antibiotic regimens remains uncertain.
Appendix 4 Trial protocol
A PILOT RANDOMISED CONTROL TRIAL, IN INTENSIVE CARE PATIENTS, COMPARING SEVEN DAYS VERSUS TWO DAYS TREATMENT WITH EMPIRICAL ANTIBIOTICS TO TREAT HOSPITAL ACQUIRED INFECTION OF UNKNOWN ORIGIN
RANDOMISED EVALUATION OF ANTIBIOTIC TREATMENT DURATION IN THE INTENSIVE CARE UNIT – READ-ICU
TRIAL – HTA Ref 08/13/38
Protocol version 5.1
Dated 29th September 2010
Contact Details and Key Personnel
Sponsor:
The Liverpool Heart & Chest Hospital NHS Trust
Research team contact details:
INDEPENDENT TSC MEMBERS:
Chair of the Steering Committee
Professor Paulo Lisboa
Liverpool John Moore’s University, Liverpool
Dr Nagesh Kalakonda
University of Liverpool, Liverpool
Mr Nathan Howes
The Royal and Broadgreen University Hospitals NHS Trust
Other Trial Steering Committee Members:
Chief Investigator
Dr Nigel Scawn, Liverpool Heart & Chest Hospital NHS Foundation Trust
Project Coordinator and Trials Unit Leader
Dr Bashir Matata, Liverpool Heart & Chest Hospital NHS Foundation Trust
Steering Committee Member and Health Economist
Dr Alan Haycox, University of Liverpool, Liverpool
Steering Committee Member
Dr Rod Stables, Liverpool Heart & Chest Hospital NHS Foundation Trust
Statistician
Dr Steven Lane, University of Liverpool, Liverpool
Steering Committee Member
Dr Mark Jackson, Liverpool Heart & Chest Hospital NHS Foundation Trust
Steering Committee Member
Professor Cheng Hock Toh, Royal Liverpool University Hospital NHS Trust
Steering Committee Member
Dr Carlos Nistal De Paz, Royal Liverpool & Broadgreen University Hospitals NHS Trust, Liverpool
Lay Member of the Steering Committee
Mr Keith Wilson, Liverpool
STUDY SUMMARY
| Title of study | A pilot randomised control trial, in intensive care patients, comparing seven days versus two days treatment with empirical antibiotics to treat hospital acquired infection of unknown origin |
| Trial acronym |
Randomised Evaluation of Antibiotic Treatment Duration in the Intensive Care Unit READ-ICU |
| Study design | Single-centre, randomised, prospective clinical trial |
| No of subjects | 60 |
| Study timelines |
Planning, ethics and start-up Sep 2009 – Dec 2009 Recruitment Jan 2010 – Dec 2010 End of follow-up Jan 2011 Analysis and reporting Jan 2011 – Mar 2011 Final report Apr 2011 |
| Inclusion criteria | Patients in the intensive care unit with signs suggestive of new infection in the absence of positive microbiological cultures |
| and | |
| At least two of the four markers of systemic inflammatory response syndrome (SIRS): | |
| temperature of > 38°C or < 36°C | |
| tachycardia (> 90 beats per minute) | |
| tachypnoea (≥ 20 breaths per minute) | |
| white blood count > 12 x 109/L or < 4 x 109/L | |
| Exclusion Criteria |
|
| Primary outcome measure | The rate of death or initiation of antibiotic therapy after the completion of the treatment schedule allocated at randomisation |
| Secondary outcome measures | Clinical
|
| Follow-up | Outcome measures will be assessed at day 10 or hospital discharge, whichever is sooner |
STUDY FLOW CHART
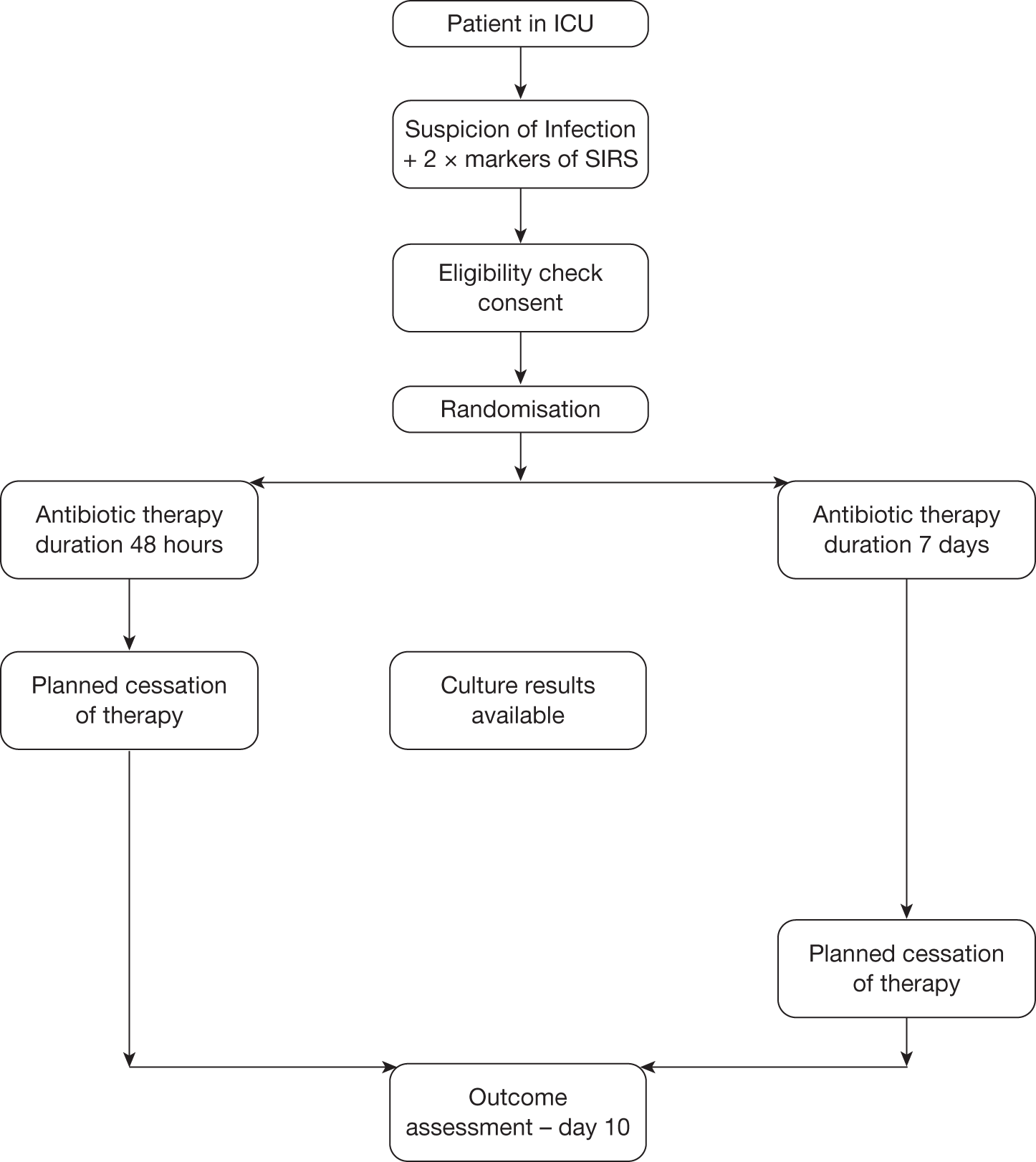
BACKGROUND
Hospital Acquired Infection in the Intensive Care Unit
Patients in intensive care units (ICUs) are at higher risk of hospital-acquired infections and sepsis than those in non-critical care areas [1]. Hospital-acquired sepsis is reported to occur in 10% to 70% of patients undergoing invasive mechanical ventilation, the rate varying with the patient population studied and diagnostic criteria used [5]. Despite the major advances in intensive care management sepsis and its complications remain the leading cause of mortality in ICUs [2]. Bloodstream infections (BSIs), pneumonias, and urinary tract infections (UTIs) are the most common hospital-acquired infections and are most often associated with the use of invasive devices [3]. Coagulase-negative Staphylococcus BSIs have recently increased in frequency, and enterococci such as Staphylococcus aureus have also been reported as causing BSIs in increasing numbers of ICUs. Recently, gram-negative bacilli have been reported more frequently than gram-positives in this setting. Fungal urinary tract sepsis has also increased [4].
Why New Treatment Strategies are Needed?
Many patients with suspected sepsis in ICU are given antibiotics for the entire duration of stay to reduce the risk of complications, even in cases where there are no compelling positive microbiological results. To date most studies have focused on optimising antibiotic treatment for ventilator acquired pneumonia (VAP) that accounts for approximately 50% of antibiotics use in ICU [6–8] and the other proportion is for treatment of suspected sepsis often of unknown origin. Since clinical decisions for empirical antibiotic treatment are usually based on fever, purulent tracheal aspirates, increased white cell counts and heart rate, even if no x-ray changes are apparent, we hypothesise that prolonged treatments with antibiotics is unnecessary in cases where there are no confirmed organisms grown in blood cultures. In addition, other markers of sepsis may guide early diagnosis and decision making on necessity and duration of antibiotic treatment. On the basis of existing evidence from a recent retrospective study by Arts et al 2007 [9], suggesting that patients without proof of nosocomial infection receiving empirical antibiotics for longer than 4-days had higher 28-day mortality (32.1%) than those whose antibiotics were discontinued (7.7%), we hypothesised that 2-day antibiotic regime is sufficiently potent to eliminate any potential microbial threat in these patients. This is consistent with current international recommendations and guidelines that there is a need for continuous reassessment of antibiotic therapy with microbiology and clinical data to reduce duration when appropriate from the usual 7–10 days of antibiotic therapy guided by clinical response [10].
Current Dilemma
Although early identification and treatment of sepsis can have a major impact on the outcome of these patients [11], diagnosis of sepsis is generally difficult particularly in cases where there is no positive isolated microbiological growth. Whilst there has been no shortage of proposed markers of sepsis [12], two assays have emerged as increasingly relevant in recent years. These are the biphasic activated partial thromboplastin test (APTT) waveform and procalcitonin (PCT). The APTT waveform reflects light transmittance changes in plasma and septic patients have been found by several investigators to show an abnormal biphasic pattern. Increasing abnormality of this waveform correlates with real time clinical progression and its molecular mechanism is due to calcium dependent complexes between C-reactive protein (CRP) and very low density lipoprotein [13]. This has also been shown to be superior to CRP in the diagnosis of sepsis and the risk of mortality [9]. For PCT, the degree of rise in concentration can help differentiate between infectious and non-infectious complications in these patients, and indeed, PCT has been shown to be effective in differentiating infectious from non-infectious causes of acute respiratory distress syndrome [14]. Most recent work has shown that the use of PCT tests in combination with the biphasic APTT waveform can increase the specificity of the latter test in identifying sepsis [15]. Indeed, it has recently been shown that serial measurement of PCT may allow monitoring of a reduction in antibiotic treatment duration and exposure in patients with severe sepsis and septic shock without apparent harm [16].
Search For Evidence
We have completed a review of current trials registered in the ISRCTN Register, NHS Trusts Clinical Trials Register, MRC UK and National Institutes of Health (NIH) randomised trial records held on NIH ClinicalTrials.gov website. This yielded no present or past randomised trials of this nature. In addition, we conducted an extensive literature search of the NIH Pubmed, MEDLINE and EMBASE electronic databases between 1990 and January 2008.
Terms that were used for the search were ‘hospital-acquired infection’, ‘antibiotics regimen in intensive care units’ and ‘biphasic transmittance waveform APTT coagulation assay’. The searches were limited to ‘human’ and ‘English language’. Reference lists of identified articles were scanned for additional potentially relevant publications in the Web of Science version 4.1.1, Institute for Scientific Information 2000 which identified all articles that cited the index publication.
However, evidence from randomised trials about the duration of antibiotic use is absent. This pilot randomised trial will investigate whether 48 hours of antibiotic treatment is adequate can safely treat suspected sepsis in the ICU as compared with the traditional week-long course. In this pilot study, we will not be using biomarkers of sepsis as part of the entry criteria as this is not currently routine practice in most UK intensive care units. However, this study presents us with the opportunity to collect samples for procalcitonin and the APTT waveform to perform a retrospective analysis of their potential utility in a future full study.
Potential Benefits of the Trial
At our centre the monthly cost attributed to antibiotics use in our ICU is estimated to be £22,000. A substantial amount of savings of up to approx. £10,000 per month could be realised in our centre if treatment was limited to the first 48 hours of ICU care in cases where no infecting organisms can be isolated. This translates to even bigger savings to the NHS as a whole running into millions every year.
Study Objectives
The main objectives of this pilot study will be to provide preliminary data on the likely safety and efficacy of a reduced course of antibiotics for the treatment of ICU infections of unknown origin. In addition, we wish to identify the likely barriers to an effective recruitment to a full study, the appropriateness and reliability of outcome measures and the data collection methods.
Study Design
This is a pilot, single-centre, randomised, prospective study designed to compare safety and efficacy of a reduced course of antibiotics with a more traditional seven day prescription, for the treatment of ICU infections of unknown origin. This study will be carried out in an intensive care unit (ICU) setting of a Tertiary Heart and Chest Hospital. Approximately 60 patients will be randomised to receive either 48 hours or 7 days of antibiotic treatment.
Selection of Patients
We will screen for trial entry, all ICU patients suspected of having an infection of unknown origin. Samples will be taken for microbiological culture testing at baseline. Initial patient status will be assessed by using APACHE II scoring system and documented in the case record form.
Inclusion Criteria
Patients in the intensive care unit with signs suggestive of new infection in the absence of positive microbiological cultures
and
At least two of the four markers of systemic inflammatory response syndrome (SIRS):
-
temperature of > 38°C or < 36°C
-
tachycardia (> 90 beats per minute)
-
tachypnoea (≥ 20 breaths per minute)
-
white blood count > 12 × 109 /L or < 4 × 109 /L.
Exclusion Criteria
-
Positive microbiological cultures before randomisation
-
Patients < 18 years of age
-
Unable to obtain assent or consent
-
Patients enrolled in another study such that randomisation in READ-ICU would result in deviation from either protocol
-
Known allergy to treatment antibiotics.
Randomisation
Eligible patients with appropriate assent or consent will be randomised in equal proportions between the two trial groups:
-
Antibiotic treatment administered for 48 hours
-
Antibiotic treatment administered for 7 days.
Treatment assignment is based on the block method using randomly varying block sizes of 2, 4 and 6 to ensure numerical balance between the groups. An independent statistician will provide the randomisation tables. Randomisation will be revealed by telephone contact with the clinical trial unit. Investigators will be asked to confirm patient’s initials, date of birth and eligibility criteria before randomisation occurs. The randomisation service will be available 09:00–17:00 (UK time). Outside of these hours urgent randomisation will be performed by opening a sealed, opaque, serial numbered envelope. Once randomised, the patient will be enrolled into the study and will be followed for outcome measures.
Antibiotic Therapy
Patient less than 85 kg will be given a combination of Teicoplanin 400 mg twice a day for day 1, then 400 mg daily thereafter and Meropenem, 1 g three times a day for 2 days or 7 days, as allocated at randomisation. Patients over 85 kg will receive 6 mg/kg Teicoplanin twice a day for day 1, then 6 mg/kg daily thereafter. Meropenem dose remains the same independently of patient weight.
After completion of the treatment regime allocated at randomisation, additional antibiotic use will constitute an outcome measure and the reason for initiation will be documented in the trial case record forms. Antibiotic choice in this setting will be guided by culture information and clinical opinion. Anticipated reasons for extended therapy will include:
-
Proven new or ongoing infection episode with positive microbiology
-
X-ray or other imaging diagnostic information
-
Poor physiological status believed to be related to infection.
Sub-study Protocol
Blood samples for the analysis of biphasic APPT and procalcitonin levels will be taken at:
-
Baseline
-
48 hours
-
On the initiation of additional antibiotic therapy beyond the randomised schedule
-
At day 10 or discharge whichever is the sooner.
Withdrawal from the Trial
Patients can elect to withdraw from the trial at any time without prejudice to their care but every effort will be made to seek permission to track outcome measures for the normal duration of follow-up.
OUTCOME MEASURES
Timing of Outcome Measure Assessment
Outcomes will be assessed at 10 days after randomisation or hospital discharge, whichever is the sooner.
Primary Clinical Outcome Measure
The rate of death or initiation of antibiotic therapy after the completion of the treatment schedule allocated at randomisation.
Secondary Clinical Outcomes
-
Duration of ICU stay
-
Duration of hospital stay
-
Duration of mechanical ventilation
-
Incidence of infection with Clostridium difficile
-
Incidence of infection with MRSA.
Secondary Economic Outcomes
Resource utilisation and costs associated with each of the two pilot arms specifically ICU stay, hospital stay, mechanical ventilation, antibiotics and other medications, tests and procedures measured and valued up until the end of the follow-up period.
Secondary Feasibility Outcomes (Pilot Study Objectives)
-
The ratio of patients – screened : eligible : randomised
-
The incidence of crossover between the randomised treatment groups
-
The accuracy of data collection assessed by a 20% source data verification check.
SAMPLE SIZE
In common with most pilot studies, calculation of an accurate samples size is not possible due to the paucity of existing data. We will, however, comply with previous recommendation for good practice that pilot randomised control trials should recruit a minimum number of 60 patients [19]. A preliminary audit of our ICU database suggests that on average about 10 patients/month in our ICU are treated for suspected infection. We aim to recruit at least 5 patients/month (50% recruitment rate) within the duration of 12 months.
Data Collection
A Manual of Operation containing relevant procedural instructions and definitions will be produced. Structured Case Record Forms (CRFs) will be used to record data at each stage of the patient journey through the trial. Trial documentation will be completed by specific Research Nurses working on the project (Claire Prince and Sandra Roberts). In addition, a medically qualified Clinical Research Fellow will be responsible for day-to-day monitoring of recruitment activity and assist in maintaining the screening log and obtaining trial consent/assent. Trial-related data will be transcribed into a bespoke, secure, password protected database in the Clinical Trials Unit.
Prospective monitoring of adverse and clinical events will start at randomisation and will continue until the end of the trial follow-up period. The Research Nurses will be responsible for ‘tracking’ each patient during their hospital stay to ensure that all tests are carried out and blood samples have been taken at the designated time. The reasons why an eligible patient does not proceed to randomisation will be recorded in the trial specific screening documentation and database.
Resource Utilisation Data Collection
Costs associated with each of the two pilot arms, length of intubation, ICU and hospital ward stay and medications will be estimated to the end of the follow-up period. The cost of antibiotics including the number of regimes used, and length of time on each regime in each arm will be calculated. A preliminary measure of key cost drivers will be estimated by applying routinely collected unit cost figures (NHS Reference costs and PSSRU unit costs), for ICU, ward and BNF prices for medications, to quantify resource utilisation over the length of the follow-up period of the study.
STATISTICAL ANALYSIS
The clinical and economic impact of 7 days versus 2 days antibiotic treatment will be examined. Categorical outcome measures will be examined using a Chi-square test or Fisher’s exact test as required. Length of ICU stay will be compared using the independent sample t-test or Mann–Whitney non-parametric test if necessary. The potential cost differences per patient will be estimated with confidence intervals. Exploratory analysis will be undertaken using Bayesian Value of Information methods described by Tan and Smith 1998 [20] that balance the benefit of detecting a minimally significant difference with at least a given power against the costs of the patient sample size and/or the risk that the research poses to patients (e.g. the probability of incompletely treating sepsis in the intensive care unit). The result of this analysis will provide guidance on the optimal sample size to use in a future RCT that would seek to evaluate the antibiotic regimen studied in this pilot study.
ETHICAL ARRANGEMENTS
The study will be conducted according to the principles of the Declaration of Helsinki (www.wma.net) and Good Clinical Practice, NHS Research Governance (www.doh.gov.uk), EU and NHS Governance Framework. The study will be sponsored by the Liverpool Heart & Chest Hospital NHS Trust. The trial protocol will be approved by an internal review board and the local Research Ethics Committees via the Integrated Research Application System. Approval from the ethics committee will be obtained if the consent form is updated or amended whenever new information becomes available that may be relevant to the patient. Patient’s right to privacy will be respected at all times to comply with the Data Protection Act 1998 and Caldicott Principle. Medical records may be inspected for monitoring auditing purposes by individuals from the Clinical Trials Unit, Liverpool Heart & Chest Hospital NHS Trust. Patients consent to this as part of the written informed consent process. All information will be stored in a password protected NHS computer.
Risks and Anticipated Benefits for Trial Participants and Society
It is common practice to administer broad-spectrum empirical antibiotics to ICU patients who are suspected on clinical grounds of developing generalised nosocomial infection of unknown origin. However, no evidence from prospective randomised studies is available to demonstrate risks associated with duration of antibiotic usage. The possible risks of taking part are common to all patients with suspected sepsis/infection. In addition, there is a possible small risk of recurrence of nosocomial infections associated with a reduced antibiotic treatment regimen. We anticipate that the risks associated with the trial are outweighed by potential benefits to the patients and society as whole as follows:
-
Reduction in NHS costs by cutting overall ICU treatment costs
-
Reduced risk of patients developing antibiotics resistant organisms
-
e.g. MRSA infection rate in ICU is currently at 10% of all admissions [21]
-
-
Reduced risk of patients acquiring other infections
-
e.g. Clostridium difficile, estimated incidences at 2.2–3% of admissions [22]
-
-
Reduced exposure of patients to unnecessary treatment with risk of allergic reactions.
Informing Potential Trial Participants of Possible Benefits and Known Risks
The patient and family will be given information sheets, describing the nature of the study and a consideration of risks, benefits and implications for care. The content of the information sheets will have been approved by the ethics review process and internal mechanisms for oversight by our Trust Service Users Research Group.
Potential participants will be allowed some time for consideration but the nature of the clinical setting and the perceived imperative for early intervention means that the period for reflection may be limited to an hour. There will of course be opportunities for questions and dialogue with trial personnel. If a decision about trial participation cannot be made in this time scale the patient will be excluded from randomisation (a key aspect of the secondary feasibility outcome measures).
Obtaining Informed Consent from Participants Whenever Possible and Proposed Action When Fully Informed Consent is not Possible
In line with Directive 2001/20/EC of the European Parliament of 2001 all research patients are required to provide written informed consent before enrolment in a trial. However, since most of the potential participants in this study will be under sedation or under the influence of anaesthetic agents, and therefore incompetent, in terms of understanding a research protocol and decision-making capacity an ‘assent’ will be obtained from surrogates such as from a legal representative (next of kin or independent professional doctor/nurse). We have experience of conducting similar studies in Liverpool and procedures for obtaining assent are in place [11].
In summary, the research protocol will be approved in advance by our institutional Research & Development Committee. Before obtaining informed assent, information will be given in a language and at a level of complexity understandable to the patient’s legal representative in both oral and written form by the investigator or designee. Legal representatives will not be coerced or unduly influenced in order for the patient to participate or remain in the trial. A legal representative will be given ample time and opportunity to inquire about details of the trial and all questions about the trial should be answered to the satisfaction of the representative. If the legal representative is unable to read the consent form, a witness should be present during the entire informed assent discussion. After the informed consent form is read to and signed by the legal representative, the witness should also sign the consent form, attesting that informed assent was freely given by the patient’s legal representative. The patient’s legal representative must receive a copy of the signed and dated informed consent form. When the patient gets better they will then be asked either in person or in writing if they are happy with this decision retrospectively and whether the information gathered on them as part of the study can be used. Patients that decline consent at this stage will not be included in the study and their results will not be used. Patient will be informed that they may withdraw or discontinue from the study anytime without giving an explanation and that their action will not affect their standard of care. Patient’s that die after randomisation will have their data included in the final analysis, unless legal representatives raise objections.
TRIAL ADMINISTRATION AND DOCUMENTATION
Retention of Trial Documentation
The trial documentation and data will be stored in a secure storage facility within the Clinical Trials Unit for a period for at least 7 years after study completion.
Proposed Action to Comply with ‘The Medicines for Human Use (Clinical Trials) Regulations 2004
Although the trial is not testing a new medicinal product but only comparing the duration of treatment with existing drugs not the type of antibiotics to be used for patient with sepsis, ‘the medicines for human use Regulations 2004’ still applies. A request for authorisation to conduct this clinical trial was made to the licensing authority (i.e. the MHRA) by the sponsor of the trial which was granted in 2010.
Safety Reporting
The study procedures adopted here are part of normal clinical practice. Safety will be assessed by tracking the number and percentage of adverse events (AEs) up to discharge from hospital. Serious and other adverse events will be recorded and reported in accordance with the International Conference for Harmonisation of Good Clinical Practice (ICH GCP) guidelines/the European Clinical Trials Directive 2001/20/EC and the Sponsor’s Research Related Adverse Event Reporting Policy. ICH GCP requires that both investigators and sponsors follow specific procedures when reporting adverse/reactions in clinical trials. All serious adverse events must be reported to the steering committee and documented in CRFs. Such events result in death or are life-threatening, require hospitalisation or prolongation of existing hospitalisation, result in persistent or significant disability or incapacity or may have created a congenital anomaly or birth defect.
Examples would include, but are not limited to:
-
Deaths related or unrelated to infection/antibiotic treatment for healthcare-acquired infection.
-
Life-threatening bleeding.
-
Intracranial haemorrhage.
-
Cerebrovascular accident.
-
Profound thrombocytopenia (platelet counts ≤ 50,000/mm3).
-
Occurrence of MRSA isolation.
-
Occurrence of Clostridium difficile infection.
-
Allergic reactions.
Adverse Event
Any untoward medical occurrence in a patient or clinical trial subject to whom a medicinal product has been administered including occurrences which are not necessarily caused by or related to the product.
Adverse Reaction
Any untoward and unintended response to an investigational product related to any dose administered.
Unexpected Adverse Reaction
An adverse reaction, the nature or severity of which is not consistent with the information about the medicinal product in question set out in the summary of product characteristics (or investigator brochure) for that product).
Research Governance
The Liverpool Heart & Chest Hospital NHS Trust as the sponsor for this trial will ensure that the rights, safety, and wellbeing of participants will be safe guarded. Issues of consent and confidentiality are paramount in line with the MRC Guidelines for Good Clinical Practice in Clinical Trials. Individual patient medical information obtained as a result of this study is considered confidential and disclosure to third parties is prohibited. Patient confidentiality will be further ensured by utilising patient-identification code numbers to correspond to treatment data in the computer files. With appropriate patient authorisation, medical information may be given to the patient’s personal physician or to other appropriate medical personnel responsible for his/her treatment. Data generated as a result of this trial are to be made available for inspection on request by the participating physicians, by the Ethics Committee and the regulatory authorities.
Interim Analysis and Stopping Rules
There is no planned interim analysis or stopping rules for the primary outcome measure, because by the time sufficient data has been accrued, the recruitment will almost be complete.
Major Protocol Violation
Major protocol violations will be documented including: failure to ensure adequate informed consent, recruitment of ineligible patient into the study on the basis of the inclusion and exclusion criteria and incorrect randomisation of a patient such that the patients are entered into the wrong treatment arm for clinical reasons. During the course of the trial, protocol deviations will be tracked.
Indemnity and Insurance
The Liverpool Heart & Chest Hospital NHS Trust is covered under the standard NHS indemnity sponsorship for the study.
Trial Organisation
Steering Committee
Professor Paulo Lisboa, Dr Nigel Scawn, Dr Rod Stables, Mr Nathan Howes, Dr Nagesh Kalakonda, Dr Carlos Nistal De Paz, Dr Bashir Matata, Dr Mark Jackson, Dr Alan Haycox, Professor Cheng-Hock Toh, Mr Keith Wilson, Dr Steven Lane.
The Steering Committee will be responsible for finalising the protocol, discussing any required amendments, monitoring recruitment rates, ensuring the study runs to time and generally overseeing the running of the study. The TSC will include the principal investigators, lay patient representative in the TSC, expert TSC members, trial statisticians and trial co-ordinators. The TSC have responsibility for the day-to-day conduct of the trial.
Data Monitoring Committee
Dr Peter Booker, Dr Richard Wenstone, Dr Robert Harris
It is the only body involved in a trial that has access to the unblinded comparative data. The role of its members is to monitor these data and make recommendations to the TSC on whether there are any ethical or safety reasons why the trial should not continue. The safety, rights and well-being of the trial participants are paramount. The DMC considers the need for any interim analysis advising the TSC regarding the release of data and/or information. The DMC may be asked by the TSC, Trial Sponsor or Trial Funder to consider data emerging from other related studies. If funding is required above the level originally requested, the DMC may be asked by the Chief Investigator, TSC, Trial Sponsor or Trial Funder to provide advice and, where appropriate, information on the data gathered to date in a way that will not compromise the trial. Membership of the DMC should be completely independent, small (3 members) and comprise experts in the field, e.g. a clinician with experience in the relevant area and expert trial statistician. [Independence, in respect of the DMC, is defined as independent from the Chief Investigator, TSC and Host Institution.]
Study Director
Dr Nigel Scawn.
Local Institution Governance
The Research Governance Department, LHCH NHS Trust.
Independent Monitoring
The Research Governance Department, LHCH NHS Trust.
The Clinical Trials Unit at the Liverpool Heart & Chest Hospital NHS Trust will undertake day-to-day management and co-ordination of the trial and are responsible for the collection, management, storage and analysis of all patient information.
Database Coordinator
Ian Kemp.
Research Coordinators
Ian Kemp.
Trial Statistician
Dr Steven Lane.
Publication Policy
The investigators are committed to the publication and widespread dissemination of the results of the study. There is an agreed policy that the recommendation of any party concerning manuscripts or text shall be taken into consideration in the final preparation of scientific documents for publication and presentation. The Steering Committee will be responsible for finalising the protocol, discussing any required amendments, monitoring recruitment rates, ensuring the study runs to time and generally overseeing the running of the study. The trial protocol will be ISRCTN registered before the start of recruitment.
SERVICE USERS INVOLVEMENT
Our institution has established a Service Users Research Endeavour (SURE) group that has been active for more than 10 years. The SURE group is actively involved in our research as follows;
-
Helps researchers to identify and ask the right questions in their project proposals.
-
Makes sure that the research questions are relevant to patients, people using the service and the public in general.
-
Gets involved in the research process itself, in terms of designing and managing service user-led projects.
-
Helps in analysis and dissemination of study results.
-
Assists final internal R&D study approval.
This proposal has been reviewed by our patient service user group (SURE) and any opinions and comments incorporated. A patient representative will attend TSC meetings and be directly involved in decision making of trial process and then relay back information to the SURE groups on a regular basis.
TRIAL FUNDING
The pilot trial costs are £169,821.32 to be funded by a grant from the Health Technology Assessment programme. For the justification of costs and roles of team members please see the Finance Form for details of specific costs.
REFERENCES
-
Weinstein RA. Nosocomial infection update. Emerg Infect Dis 1998;4:416–420.
-
Bochud PY, Calandra T. Pathogenesis of sepsis: new concepts and implications for future treatment. BMJ 2003;326:262–266.
-
Gross PA, Neu HC, Aswapokee P, Van Antwerpen C, Aswapokee N. Deaths from nosocomial infections: experience in a university hospital and a community hospital. Am J Med 1980;68:219–23.
-
Richards M, Thursky K, Buising K. Epidemiology, prevalence, and sites of infections in intensive care units. Semin Respir Crit Care Med 2003;24:3–22.
-
Combes A, Figliolini C, Trouillet JL, Kassis N, Dombret MC, Wolff M, Gibert C, Chastre J. Factors predicting ventilator-associated pneumonia recurrence. Crit Care Med 2003;31:1102–7.
-
Rello J, Diaz E. Pneumonia in the intensive care unit. Crit Care Med 2003;31:2544–51.
-
Micek ST, Ward S, Fraser VJ, Kollef MH. A Randomized Controlled Trial of an antibiotic Discontinuation Policy for clinically suspected ventilator-associated pneumonia. Chest 2004;125:1791–1799.
-
Singh N, Rogers P, Atwood CW, Wagener MM, Yu VL. Short-course Empiric Antibiotic Therapy for Patients with Pulmonary Infiltrates in the Intensive Care Unit – A Proposed Solution for Indiscriminate Antibiotic Prescription. Am J Respir Crit Care Med 2000;162:505–511.
-
Aarts MA, Brun-Buisson C, Cook DJ, Kumar A, Opal S, Rocker G, Smith T, Vincent JL, Marshall JC. Antibiotic management of suspected nosocomial ICU-acquired infection: does prolonged empiric therapy improve outcome? Intensive Care Med 2007;33:1369–78.
-
Dellinger RP, Levy MM, Carlet JM, Bion J, Parker MM, Jaeschke R, Reinhart K, Angus DC, Brun-Buisson C, Beale R, Calandra T, Dhainaut JF, Gerlach H, Harvey M, Marini JJ, Marshall J, Ranieri M, Ramsay G, Sevransky J, Thompson BT, Townsend S, Vender JS, Zimmerman JL, Vincent JL. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Intensive Care Med 2008;34:17–60.
-
Toh CH, Ticknor LO, Downey C, Giles AR, Paton RC, Wenstone R. Early identification of sepsis and mortality risks through simple, rapid clot-waveform analysis. Intensive Care Med 2003;29:56–61.
-
Thomas K. Transmitting and absorbing new information on the early identification of sepsis patients: the partial thromboplastin time biphasic waveform. Crit Care Med 2006;34:1829–31.
-
Dempfle CE, Lorenz S, Smolinski M, Wurst M, West S, Houdijk WP, Quintel M, Borggrefe M. Utility of activated partial thromboplastin time waveform analysis for identification of sepsis and overt disseminated intravascular coagulation in patients admitted to a surgical intensive care unit. Crit Care Med 2004;32:520–4.
-
Aouifi A, Piriou V, Blanc P, Bouvier H, Bastien O, Chiari P, Rousson R, Evans R, Lehot JJ. Effect of cardiopulmonary bypass on serum procalcitonin and C-reactive protein concentrations. Br J Anaesth 1999;83:602–607.
-
Brunckhorst FM, Eberhard OK, Brunkhorst R. Discrimination of infectious and non-infectious causes of early acute respiratory distress syndrome by procalcitonin. Crit Care Med 1999;27:2172–2176.
-
Nobre V, Harbarth S, Graf JD, Rohner P, Pugin J. Use of procalcitonin to shorten antibiotic treatment duration in septic patients. Am J Resp Crit Care Med 2008;177:498–505.
-
Muckart DJ, Bhagwanjee S. American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference definitions of the systemic inflammatory response syndrome and allied disorders in relation to critically injured patients. Crit Care Med 1997;25:1789–95.
-
Napolitano LM, Ferrer T, McCarter RJ, Jr, Scalea TM. Systemic Inflammatory Response Syndrome Score at Admission Independently Predicts Mortality and Length of Stay in Trauma Patients. J Trauma-Injury Infection & Critical Care 2000;49:647–653.
-
Lancaster GA, Dodd SR and Williamson PR. Design and analysis of pilot studies: recommendations for good practice. J Eval Clin Pract 2004;10:307–312.
-
Tan SB, Smith AFM. Exploratory thoughts on clinical trials with utilities. Stat Med 1998;17:2771–2791.
-
Warren DK, Guth RM, Coopersmith CM, Merz LR, Zack JE, Fraser VJ. Epidemiology of methicillin-resistant Staphylococcus aureus colonization in a surgical intensive care unit. Infect Control Hosp Epidemiol 2006;27:1032–1040.
-
Stone SP, Beric V, Quick A, Balestrini AA, Kibbler CC. The effect of an enhanced infection-control policy on the incidence of Clostridium difficile infection and methicillin-resistant Staphylococcus aureus colonization in acute elderly medical patients. Age Ageing 1998;27:561–8.
List of abbreviations
- APACHE II
- Acute Physiology and Chronic Health Evaluation II
- APTT
- activated partial thromboplastin time
- BSI
- bloodstream infection
- CEA
- cost-effectiveness analysis
- CI
- confidence interval
- CMA
- cost minimisation analysis
- CRF
- case record form
- CRP
- C-reactive protein
- eGFR
- estimated glomerular filtration rate
- EVPI
- expected value of perfect information
- ICU
- intensive care unit
- IQR
- interquartile range
- MHRA
- Medicines and Healthcare products Regulatory Agency
- MRSA
- methicillin-resistant Staphylococcus aureus
- NB
- net benefit
- PCT
- procalcitonin
- RCT
- randomised controlled trial
- READ-ICU
- Randomised Evaluation of Antibiotic Treatment Duration in the Intensive Care Unit
- SIRS
- systematic inflammatory response syndrome
- SOFA
- Sequential Organ Failure Assessment
All abbreviations that have been used in this report are listed here unless the abbreviation is well known (e.g. NHS), or it has been used only once, or it is a non-standard abbreviation used only in figures/tables/appendices, in which case the abbreviation is defined in the figure legend or in the notes at the end of the table.
Notes
Health Technology Assessment programme
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, Department of Pharmacology and Therapeutics, University of Liverpool
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
Prioritisation Group
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, Department of Pharmacology and Therapeutics, University of Liverpool
-
Professor Imti Choonara, Professor in Child Health, Academic Division of Child Health, University of Nottingham
Chair – Pharmaceuticals Panel
-
Dr Bob Coates, Consultant Advisor – Disease Prevention Panel
-
Dr Andrew Cook, Consultant Advisor – Intervention Procedures Panel
-
Dr Peter Davidson, Director of NETSCC, Health Technology Assessment
-
Dr Nick Hicks, Consultant Adviser – Diagnostic Technologies and Screening Panel, Consultant Advisor–Psychological and Community Therapies Panel
-
Ms Susan Hird, Consultant Advisor, External Devices and Physical Therapies Panel
-
Professor Sallie Lamb, Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick
Chair – HTA Clinical Evaluation and Trials Board
-
Professor Jonathan Michaels, Professor of Vascular Surgery, Sheffield Vascular Institute, University of Sheffield
Chair – Interventional Procedures Panel
-
Professor Ruairidh Milne, Director – External Relations
-
Dr John Pounsford, Consultant Physician, Directorate of Medical Services, North Bristol NHS Trust
Chair – External Devices and Physical Therapies Panel
-
Dr Vaughan Thomas, Consultant Advisor – Pharmaceuticals Panel, Clinical
Lead – Clinical Evaluation Trials Prioritisation Group
-
Professor Margaret Thorogood, Professor of Epidemiology, Health Sciences Research Institute, University of Warwick
Chair – Disease Prevention Panel
-
Professor Lindsay Turnbull, Professor of Radiology, Centre for the MR Investigations, University of Hull
Chair – Diagnostic Technologies and Screening Panel
-
Professor Scott Weich, Professor of Psychiatry, Health Sciences Research Institute, University of Warwick
Chair – Psychological and Community Therapies Panel
-
Professor Hywel Williams, Director of Nottingham Clinical Trials Unit, Centre of Evidence-Based Dermatology, University of Nottingham
Chair – HTA Commissioning Board
Deputy HTA Programme Director
HTA Commissioning Board
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
-
Professor of Bio-Statistics, Department of Public Health and Epidemiology, University of Birmingham
-
Professor of Clinical Pharmacology, Director, NIHR HTA programme, Department of Pharmacology and Therapeutics, University of Liverpool
-
Professor Zarko Alfirevic, Head of Department for Women’s and Children’s Health, Institute of Translational Medicine, University of Liverpool
-
Professor Judith Bliss, Director of ICR-Clinical Trials and Statistics Unit, The Institute of Cancer Research
-
Professor David Fitzmaurice, Professor of Primary Care Research, Department of Primary Care Clinical Sciences, University of Birmingham
-
Professor John W Gregory, Professor in Paediatric Endocrinology, Department of Child Health, Wales School of Medicine, Cardiff University
-
Professor Steve Halligan, Professor of Gastrointestinal Radiology, Department of Specialist Radiology, University College Hospital, London
-
Professor Angela Harden, Professor of Community and Family Health, Institute for Health and Human Development, University of East London
-
Dr Joanne Lord, Reader, Health Economics Research Group, Brunel University
-
Professor Stephen Morris, Professor of Health Economics, University College London, Research Department of Epidemiology and Public Health, University College London
-
Professor Dion Morton, Professor of Surgery, Academic Department of Surgery, University of Birmingham
-
Professor Gail Mountain, Professor of Health Services Research, Rehabilitation and Assistive Technologies Group, University of Sheffield
-
Professor Irwin Nazareth, Professor of Primary Care and Head of Department, Department of Primary Care and Population Sciences, University College London
-
Professor E Andrea Nelson, Professor of Wound Healing and Director of Research, School of Healthcare, University of Leeds
-
Professor John David Norrie, Director, Centre for Healthcare Randomised Trials, Health Services Research Unit, University of Aberdeen
-
Professor Barney Reeves, Professorial Research Fellow in Health Services Research, Department of Clinical Science, University of Bristol
-
Professor Peter Tyrer, Professor of Community Psychiatry, Centre for Mental Health, Imperial College London
-
Professor Martin Underwood, Professor of Primary Care Research, Warwick Medical School, University of Warwick
-
Professor Caroline Watkins, Professor of Stroke and Older People’s Care, Chair of UK Forum for Stroke Training, Stroke Practice Research Unit, University of Central Lancashire
-
Dr Duncan Young, Senior Clinical Lecturer and Consultant, Nuffield Department of Anaesthetics, University of Oxford
-
Dr Tom Foulks, Medical Research Council
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
HTA Clinical Evaluation and Trials Board
-
Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick and Professor of Rehabilitation, Nuffield Department of Orthopaedic, Rheumatology and Musculoskeletal Sciences, University of Oxford
-
Professor of the Psychology of Health Care, Leeds Institute of Health Sciences, University of Leeds
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Keith Abrams, Professor of Medical Statistics, Department of Health Sciences, University of Leicester
-
Professor Martin Bland, Professor of Health Statistics, Department of Health Sciences, University of York
-
Professor Jane Blazeby, Professor of Surgery and Consultant Upper GI Surgeon, Department of Social Medicine, University of Bristol
-
Professor Julia M Brown, Director, Clinical Trials Research Unit, University of Leeds
-
Professor Alistair Burns, Professor of Old Age Psychiatry, Psychiatry Research Group, School of Community-Based Medicine, The University of Manchester & National Clinical Director for Dementia, Department of Health
-
Dr Jennifer Burr, Director, Centre for Healthcare Randomised trials (CHART), University of Aberdeen
-
Professor Linda Davies, Professor of Health Economics, Health Sciences Research Group, University of Manchester
-
Professor Simon Gilbody, Prof of Psych Medicine and Health Services Research, Department of Health Sciences, University of York
-
Professor Steven Goodacre, Professor and Consultant in Emergency Medicine, School of Health and Related Research, University of Sheffield
-
Professor Dyfrig Hughes, Professor of Pharmacoeconomics, Centre for Economics and Policy in Health, Institute of Medical and Social Care Research, Bangor University
-
Professor Paul Jones, Professor of Respiratory Medicine, Department of Cardiac and Vascular Science, St George‘s Hospital Medical School, University of London
-
Professor Khalid Khan, Professor of Women’s Health and Clinical Epidemiology, Barts and the London School of Medicine, Queen Mary, University of London
-
Professor Richard J McManus, Professor of Primary Care Cardiovascular Research, Primary Care Clinical Sciences Building, University of Birmingham
-
Professor Helen Rodgers, Professor of Stroke Care, Institute for Ageing and Health, Newcastle University
-
Professor Ken Stein, Professor of Public Health, Peninsula Technology Assessment Group, Peninsula College of Medicine and Dentistry, Universities of Exeter and Plymouth
-
Professor Jonathan Sterne, Professor of Medical Statistics and Epidemiology, Department of Social Medicine, University of Bristol
-
Mr Andy Vail, Senior Lecturer, Health Sciences Research Group, University of Manchester
-
Professor Clare Wilkinson, Professor of General Practice and Director of Research North Wales Clinical School, Department of Primary Care and Public Health, Cardiff University
-
Dr Ian B Wilkinson, Senior Lecturer and Honorary Consultant, Clinical Pharmacology Unit, Department of Medicine, University of Cambridge
-
Ms Kate Law, Director of Clinical Trials, Cancer Research UK
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
Diagnostic Technologies and Screening Panel
-
Scientific Director of the Centre for Magnetic Resonance Investigations and YCR Professor of Radiology, Hull Royal Infirmary
-
Professor Judith E Adams, Consultant Radiologist, Manchester Royal Infirmary, Central Manchester & Manchester Children’s University Hospitals NHS Trust, and Professor of Diagnostic Radiology, University of Manchester
-
Mr Angus S Arunkalaivanan, Honorary Senior Lecturer, University of Birmingham and Consultant Urogynaecologist and Obstetrician, City Hospital, Birmingham
-
Dr Diana Baralle, Consultant and Senior Lecturer in Clinical Genetics, University of Southampton
-
Dr Stephanie Dancer, Consultant Microbiologist, Hairmyres Hospital, East Kilbride
-
Dr Diane Eccles, Professor of Cancer Genetics, Wessex Clinical Genetics Service, Princess Anne Hospital
-
Dr Trevor Friedman, Consultant Liason Psychiatrist, Brandon Unit, Leicester General Hospital
-
Dr Ron Gray, Consultant, National Perinatal Epidemiology Unit, Institute of Health Sciences, University of Oxford
-
Professor Paul D Griffiths, Professor of Radiology, Academic Unit of Radiology, University of Sheffield
-
Mr Martin Hooper, Public contributor
-
Professor Anthony Robert Kendrick, Associate Dean for Clinical Research and Professor of Primary Medical Care, University of Southampton
-
Dr Nicola Lennard, Senior Medical Officer, MHRA
-
Dr Anne Mackie, Director of Programmes, UK National Screening Committee, London
-
Mr David Mathew, Public contributor
-
Dr Michael Millar, Consultant Senior Lecturer in Microbiology, Department of Pathology & Microbiology, Barts and The London NHS Trust, Royal London Hospital
-
Mrs Una Rennard, Public contributor
-
Dr Stuart Smellie, Consultant in Clinical Pathology, Bishop Auckland General Hospital
-
Ms Jane Smith, Consultant Ultrasound Practitioner, Leeds Teaching Hospital NHS Trust, Leeds
-
Dr Allison Streetly, Programme Director, NHS Sickle Cell and Thalassaemia Screening Programme, King’s College School of Medicine
-
Dr Matthew Thompson, Senior Clinical Scientist and GP, Department of Primary Health Care, University of Oxford
-
Dr Alan J Williams, Consultant Physician, General and Respiratory Medicine, The Royal Bournemouth Hospital
-
Dr Tim Elliott, Team Leader, Cancer Screening, Department of Health
-
Dr Joanna Jenkinson, Board Secretary, Neurosciences and Mental Health Board (NMHB), Medical Research Council
-
Professor Julietta Patrick, Director, NHS Cancer Screening Programme, Sheffield
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Disease Prevention Panel
-
Professor of Epidemiology, University of Warwick Medical School, Coventry
-
Dr Robert Cook, Clinical Programmes Director, Bazian Ltd, London
-
Dr Colin Greaves, Senior Research Fellow, Peninsula Medical School (Primary Care)
-
Mr Michael Head, Public contributor
-
Professor Cathy Jackson, Professor of Primary Care Medicine, Bute Medical School, University of St Andrews
-
Dr Russell Jago, Senior Lecturer in Exercise, Nutrition and Health, Centre for Sport, Exercise and Health, University of Bristol
-
Dr Julie Mytton, Consultant in Child Public Health, NHS Bristol
-
Professor Irwin Nazareth, Professor of Primary Care and Director, Department of Primary Care and Population Sciences, University College London
-
Dr Richard Richards, Assistant Director of Public Health, Derbyshire County Primary Care Trust
-
Professor Ian Roberts, Professor of Epidemiology and Public Health, London School of Hygiene & Tropical Medicine
-
Dr Kenneth Robertson, Consultant Paediatrician, Royal Hospital for Sick Children, Glasgow
-
Dr Catherine Swann, Associate Director, Centre for Public Health Excellence, NICE
-
Mrs Jean Thurston, Public contributor
-
Professor David Weller, Head, School of Clinical Science and Community Health, University of Edinburgh
-
Ms Christine McGuire, Research & Development, Department of Health
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
External Devices and Physical Therapies Panel
-
Consultant Physician North Bristol NHS Trust
-
Reader in Wound Healing and Director of Research, University of Leeds
-
Professor Bipin Bhakta, Charterhouse Professor in Rehabilitation Medicine, University of Leeds
-
Mrs Penny Calder, Public contributor
-
Dr Dawn Carnes, Senior Research Fellow, Barts and the London School of Medicine and Dentistry
-
Dr Emma Clark, Clinician Scientist Fellow & Cons. Rheumatologist, University of Bristol
-
Mrs Anthea De Barton-Watson, Public contributor
-
Professor Nadine Foster, Professor of Musculoskeletal Health in Primary Care Arthritis Research, Keele University
-
Dr Shaheen Hamdy, Clinical Senior Lecturer and Consultant Physician, University of Manchester
-
Professor Christine Norton, Professor of Clinical Nursing Innovation, Bucks New University and Imperial College Healthcare NHS Trust
-
Dr Lorraine Pinnigton, Associate Professor in Rehabilitation, University of Nottingham
-
Dr Kate Radford, Senior Lecturer (Research), University of Central Lancashire
-
Mr Jim Reece, Public contributor
-
Professor Maria Stokes, Professor of Neuromusculoskeletal Rehabilitation, University of Southampton
-
Dr Pippa Tyrrell, Senior Lecturer/Consultant, Salford Royal Foundation Hospitals’ Trust and University of Manchester
-
Dr Nefyn Williams, Clinical Senior Lecturer, Cardiff University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Interventional Procedures Panel
-
Professor of Vascular Surgery, University of Sheffield
-
Consultant Colorectal Surgeon, Bristol Royal Infirmary
-
Mrs Isabel Boyer, Public contributor
-
Mr Sankaran Chandra Sekharan, Consultant Surgeon, Breast Surgery, Colchester Hospital University NHS Foundation Trust
-
Professor Nicholas Clarke, Consultant Orthopaedic Surgeon, Southampton University Hospitals NHS Trust
-
Ms Leonie Cooke, Public contributor
-
Mr Seumas Eckford, Consultant in Obstetrics & Gynaecology, North Devon District Hospital
-
Professor Sam Eljamel, Consultant Neurosurgeon, Ninewells Hospital and Medical School, Dundee
-
Dr Adele Fielding, Senior Lecturer and Honorary Consultant in Haematology, University College London Medical School
-
Dr Matthew Hatton, Consultant in Clinical Oncology, Sheffield Teaching Hospital Foundation Trust
-
Dr John Holden, General Practitioner, Garswood Surgery, Wigan
-
Dr Fiona Lecky, Senior Lecturer/Honorary Consultant in Emergency Medicine, University of Manchester/Salford Royal Hospitals NHS Foundation Trust
-
Dr Nadim Malik, Consultant Cardiologist/Honorary Lecturer, University of Manchester
-
Mr Hisham Mehanna, Consultant & Honorary Associate Professor, University Hospitals Coventry & Warwickshire NHS Trust
-
Dr Jane Montgomery, Consultant in Anaesthetics and Critical Care, South Devon Healthcare NHS Foundation Trust
-
Professor Jon Moss, Consultant Interventional Radiologist, North Glasgow Hospitals University NHS Trust
-
Dr Simon Padley, Consultant Radiologist, Chelsea & Westminster Hospital
-
Dr Ashish Paul, Medical Director, Bedfordshire PCT
-
Dr Sarah Purdy, Consultant Senior Lecturer, University of Bristol
-
Dr Matthew Wilson, Consultant Anaesthetist, Sheffield Teaching Hospitals NHS Foundation Trust
-
Professor Yit Chiun Yang, Consultant Ophthalmologist, Royal Wolverhampton Hospitals NHS Trust
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Pharmaceuticals Panel
-
Professor in Child Health, University of Nottingham
-
Senior Lecturer in Clinical Pharmacology, University of East Anglia
-
Dr Martin Ashton-Key, Medical Advisor, National Commissioning Group, NHS London
-
Dr Peter Elton, Director of Public Health, Bury Primary Care Trust
-
Dr Ben Goldacre, Research Fellow, Division of Psychological Medicine and Psychiatry, King’s College London
-
Dr James Gray, Consultant Microbiologist, Department of Microbiology, Birmingham Children’s Hospital NHS Foundation Trust
-
Dr Jurjees Hasan, Consultant in Medical Oncology, The Christie, Manchester
-
Dr Carl Heneghan, Deputy Director Centre for Evidence-Based Medicine and Clinical Lecturer, Department of Primary Health Care, University of Oxford
-
Dr Dyfrig Hughes, Reader in Pharmacoeconomics and Deputy Director, Centre for Economics and Policy in Health, IMSCaR, Bangor University
-
Dr Maria Kouimtzi, Pharmacy and Informatics Director, Global Clinical Solutions, Wiley-Blackwell
-
Professor Femi Oyebode, Consultant Psychiatrist and Head of Department, University of Birmingham
-
Dr Andrew Prentice, Senior Lecturer and Consultant Obstetrician and Gynaecologist, The Rosie Hospital, University of Cambridge
-
Ms Amanda Roberts, Public contributor
-
Dr Gillian Shepherd, Director, Health and Clinical Excellence, Merck Serono Ltd
-
Mrs Katrina Simister, Assistant Director New Medicines, National Prescribing Centre, Liverpool
-
Professor Donald Singer, Professor of Clinical Pharmacology and Therapeutics, Clinical Sciences Research Institute, CSB, University of Warwick Medical School
-
Mr David Symes, Public contributor
-
Dr Arnold Zermansky, General Practitioner, Senior Research Fellow, Pharmacy Practice and Medicines Management Group, Leeds University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Mr Simon Reeve, Head of Clinical and Cost-Effectiveness, Medicines, Pharmacy and Industry Group, Department of Health
-
Dr Heike Weber, Programme Manager, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Psychological and Community Therapies Panel
-
Professor of Psychiatry, University of Warwick, Coventry
-
Consultant & University Lecturer in Psychiatry, University of Cambridge
-
Professor Jane Barlow, Professor of Public Health in the Early Years, Health Sciences Research Institute, Warwick Medical School
-
Dr Sabyasachi Bhaumik, Consultant Psychiatrist, Leicestershire Partnership NHS Trust
-
Mrs Val Carlill, Public contributor
-
Dr Steve Cunningham, Consultant Respiratory Paediatrician, Lothian Health Board
-
Dr Anne Hesketh, Senior Clinical Lecturer in Speech and Language Therapy, University of Manchester
-
Dr Peter Langdon, Senior Clinical Lecturer, School of Medicine, Health Policy and Practice, University of East Anglia
-
Dr Yann Lefeuvre, GP Partner, Burrage Road Surgery, London
-
Dr Jeremy J Murphy, Consultant Physician and Cardiologist, County Durham and Darlington Foundation Trust
-
Dr Richard Neal, Clinical Senior Lecturer in General Practice, Cardiff University
-
Mr John Needham, Public contributor
-
Ms Mary Nettle, Mental Health User Consultant
-
Professor John Potter, Professor of Ageing and Stroke Medicine, University of East Anglia
-
Dr Greta Rait, Senior Clinical Lecturer and General Practitioner, University College London
-
Dr Paul Ramchandani, Senior Research Fellow/Cons. Child Psychiatrist, University of Oxford
-
Dr Karen Roberts, Nurse/Consultant, Dunston Hill Hospital, Tyne and Wear
-
Dr Karim Saad, Consultant in Old Age Psychiatry, Coventry and Warwickshire Partnership Trust
-
Dr Lesley Stockton, Lecturer, School of Health Sciences, University of Liverpool
-
Dr Simon Wright, GP Partner, Walkden Medical Centre, Manchester
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health