
Notes
Article history
The research reported in this issue of the journal was commissioned by the HTA programme as project number 05/14/02. The contractual start date was in May 2007. The draft report began editorial review in June 2011 and was accepted for publication in February 2012. As the funder, by devising a commissioning brief, the HTA programme specified the research question and study design.The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the referees for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
none
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2012. This work was produced by Appleton et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to NETSCC. This journal is a member of and subscribes to the principles of the Committee on Publication Ethics (COPE) (http://www.publicationethics.org/). This journal may be freely reproduced for the purposes of private research and study and may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NETSCC, Health Technology Assessment, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Queen’s Printer and Controller of HMSO 2012
Abstract
Background
Difficulties in initiating and maintaining sleep are common in children with neurodevelopmental disorders. Melatonin is unlicensed in children yet widely prescribed for sleep problems.
Objective
To determine whether or not immediate-release melatonin is beneficial compared with placebo in improving total duration of night-time sleep in children with neurodevelopmental problems.
Design
Randomised, double-blind, placebo-controlled, parallel study.
Setting
Hospitals throughout England and Wales recruited patients referred by community paediatricians and other clinical colleagues.
Participants
Children with neurodevelopmental problems aged from 3 years to 15 years 8 months who did not fall asleep within 1 hour of lights out or who had < 6 hours of continuous sleep. Before randomisation, patients meeting eligibility criteria entered a 4- to 6-week behaviour therapy period in which a behaviour therapy advice booklet was provided. Sleep was measured using sleep diaries and actigraphy. After this period the sleep diaries were reviewed to determine if the sleep problem fulfilled the eligibility criteria. Eligible participants were randomised and followed for 12 weeks.
Interventions
Melatonin or placebo capsules in doses of 0.5 mg, 2 mg, 6 mg and 12 mg for a period of 12 weeks. The starting dose was 0.5 mg and the dose could be escalated through 2 mg and 6 mg to 12 mg during the first 4 weeks, at the end of which the child was maintained on that dose.
Main outcome measures
The primary outcome was total night-time sleep time (TST) calculated using sleep diaries at 12 weeks compared with baseline. Secondary outcome measures included TST calculated using actigraphy data, sleep-onset latency (SOL) (time taken to fall asleep), sleep efficiency, Composite Sleep Disturbance Index score, global measure of child’s sleep quality, Aberrant Behaviour Checklist, Family Impact Module of the Pediatric Quality of Life Inventory (PedsQL™), the Epworth Sleepiness Scale, number and severity of seizures and adverse events. Salivary melatonin concentrations and association of genetic variants with abnormal melatonin production were also investigated.
Results
A total of 275 children were screened to enter the trial; 263 (96%) children were registered and completed the 4- to 6-week behaviour therapy period and 146 (56%) children were randomised, of whom 110 (75%) contributed data for the primary outcome. The difference in TST time between the melatonin and placebo groups adjusted for baseline was 22.43 minutes [95% confidence interval (CI) 0.52 to 44.34 minutes; p = 0.04] measured using sleep diaries. A reduction in SOL, adjusted for baseline, was seen for melatonin compared with placebo when measured by sleep diaries (–37.49 minutes, 95% CI –55.27 to –19.71 minutes; p < 0.0001) and actigraphy (–45.34 minutes, 95% CI –68.75 to –21.93 minutes; p = 0.0003). There were no significant differences between the two groups in terms of the reporting of adverse events. The results of other secondary outcomes favoured melatonin but were not statistically significant.
Conclusions
On average, the children treated with melatonin slept 23 minutes longer than those in the placebo group; however, the upper limit of the confidence interval was less than 1 hour, the minimum clinically worthwhile difference specified at the outset of the trial. Melatonin is effective in reducing SOL in children with neurodevelopmental delay by a mean of 45 minutes; a value of 30 minutes was specified a priori to be clinically important. Future studies should be conducted over longer periods and directly compare different formulations of melatonin with conventional hypnotic and sedative medications. It would also be important to study groups of children with specific neurological disorders.
Trial registration
Current Controlled Trials ISRCTN05534585.
Funding
This project was funded by the NIHR Health Technology Assessment programme and will be published in full in Health Technology Assessment; Vol. 16, No. 40. See the HTA programme website for further project information.
Chapter 1 Introduction
Background
Circadian rhythms, including the sleep–wake cycle, are entrained by the transmission of light from the retina to the circadian pacemaker, situated in the suprachiasmatic nucleus (SCN) of the hypothalamus. Light perception is all that is required for synchronisation with the SCN. 1
Melatonin (N-acetyl-5-methoxytryptamine) is a natural substance produced by the pineal gland in the evening in response to SCN signals, with concentrations peaking at approximately midnight and secretion being extremely low during daylight hours. The melatonin signal forms part of the system that can influence sleep-promoting and sleep–wake rhythm-regulating actions through the specific activation of MT1 (melatonin 1a) and MT2 (melatonin 1b) receptors, the two major melatonin receptor subtypes found in the SCN and retinae of mammals.
Abnormalities in melatonin production can potentially arise secondary to dysfunction of the SCN or abnormalities of the pineal gland. 1,2 In addition, receptor abnormalities in the retina or SCN may lead to receptors that are unable to respond appropriately to increased concentrations of melatonin produced by the pineal gland in response to dim-light stimulation. 3
Considerable work undertaken in healthy adult volunteers has evaluated the pharmacology and pharmacokinetics of both endogenous and prescribed exogenous melatonin. Early results, subsequently confirmed, suggested that melatonin is of value in treating sleep disturbances in blind or severely visually impaired people in whom endogenous melatonin secretion may be altered or deficient. Melatonin has also been suggested to be useful in inducing sleep in groups at specific risk of insomnia, including shift workers4 and those with jet lag. 5
The circadian clock is entrained not only by light but also by behavioural and social cues (zeitgebers). 6 An inability to correctly interpret these zeitgebers in children with neurodevelopmental disorders can lead to abnormalities in circadian rhythms. 7 Children with neurological and/or developmental disorders have a higher prevalence of sleep disturbances, which are frequently chronic and are usually far more difficult to treat than those in their ‘normally’ developing peers. 8–11 These sleep disorders may result in additional learning and behaviour problems. Further, disturbed sleep, and specifically discontinuous sleep with frequent awakenings, commonly results in disturbed sleep in their parents and siblings. This may have secondary detrimental effects on families, which may be physical, emotional and social – and, if chronic, may impair their ability to continue in employment or further education. Finally, chronic sleep disturbance in multiply disabled children is a frequent cause of families giving up their care.
Ensuring adequate sleep hygiene and, when appropriate, the use of specific behaviour therapy to improve sleep are first-line treatments for many sleep problems in children with developmental disorders. Although these approaches to management might be sufficient in themselves for some children, or should at least be considered as a component of melatonin therapy, it is worth noting that behavioural approaches can be difficult to apply, are time-consuming and usually require skilled and scarce manpower. Treatment with commonly used hypnotic sedative drugs is often ineffective and can result in both side effects and tolerance, and may even be contraindicated in certain situations. There is considerable evidence that many chronic sleep–wake disorders in children with neurodevelopmental disorders are associated with an inability to synchronise their sleep–wake cycle-generating system with environmental zeitgebers, resulting in abnormal melatonin secretion. 7,8 Following early results suggesting that melatonin may be effective in improving sleep in these children,8–14 together with the observation that melatonin appeared to have neither short- nor long-term side effects, melatonin was (and continues to be) increasingly used in open studies in the treatment of sleep disorders in children with a range of neurological disabilities and disorders. Furthermore, because children with a range of neurodevelopmental disorders will be seen by many different disciplines and specialists, including general (hospital- and community-based) paediatricians, paediatric neurologists and child and adolescent psychiatrists, there has been a predictable enthusiasm to find an intervention or drug that is both effective and ‘safe’ in treating the sleep impairment that is typically seen in these children. This would, at least in part, explain the dramatic increase in the prescription of melatonin (and its many formulations) for this population throughout the UK.
Rationale
Several reports have suggested that melatonin is beneficial in children with developmental delay and in particular in those with visual problems8,14–17 and autism,18 and also in more specific neurogenetic syndromes, including fragile X syndrome,18 Rett syndrome,19 Angelman syndrome20 and tuberous sclerosis. 21 Importantly, melatonin appears to be effective in both reducing the time it takes children to fall asleep (time to sleep onset or sleep latency) and increasing the total duration of continuous sleep throughout the night. 8,16,21,22 Since the commencement of the MENDS (MElatonin in children with Neurodevelopmental Disorders and impaired Sleep) study, increasing numbers of small placebo-controlled trials have spawned the publication of two meta-analyses,23,24 both of which are relevant to our study population in that they included children with intellectual disabilities and autism spectrum difficulties. Both meta-analyses indicated that melatonin reduces sleep latency and increases total sleep duration. Unfortunately, there are methodological problems with many of the studies and, in their conclusions, both meta-analyses emphasise the need for larger placebo-controlled and, ideally, dose-ranging trials.
Other placebo-controlled trials have demonstrated that melatonin appears to be effective in elementary (primary) school children without neurodevelopmental delay or neurological disorders and idiopathic chronic sleep-onset insomnia20,21 as well as in some children with epilepsy. 25–27 The drug has also been used with some success in inducing sleep in children undergoing a range of medical procedures, including sedation electroencephalograms and even brain scans. 28,29
Melatonin levels in both saliva and blood vary from person to person for a number of reasons, some of which are known and some of which are unknown; these may include the person’s age and any underlying neurological or visual impairment. Consequently, neither therapeutic levels nor physiological or pharmacological doses have been established. There is some evidence that there may be a dose–response relationship for both melatonin30–32 and melatonin agonists (beta-methyl-6-chlormelatonin). 33 There is no convincing evidence that tolerance develops to exogenous melatonin. 15,17 It has been suggested that some children who respond poorly to melatonin over time are slow metabolisers [through decreased cytochrome P450 1A2 (CYP1A2) enzyme activity] and that consequently levels of melatonin accumulate throughout the daytime, thereby limiting its effectiveness. 34
Melatonin is unlicensed for this clinical use in children (improving sleep in children whether or not the child has neurodevelopmental problems) and it is estimated that in the UK there are currently well in excess of 6000 children being treated with melatonin. In some countries, including the USA, melatonin is considered to be a food supplement and is not subject to the regulations governing medicinal agents. There are at least 50 preparations that are either imported into or manufactured within the UK, including immediate-release capsules and tablets, sustained-release capsules and tablets and at least one liquid formulation. The majority of these formulations are health foods/dietary supplements with no guarantee of quality or preparations manufactured to the standards of good manufacturing practice (GMP). Since the start of MENDS, a commercial tablet preparation of sustained-release melatonin (Circadin®, Lundbeck) has become available but its current license is for the short-term treatment of primary insomnia in individuals aged ≥ 55 years.
Current, and predominantly anecdotal, evidence, together with the rapidly increasing and largely haphazard use of melatonin, justified the need to undertake a multicentre, randomised, placebo-controlled, parallel study of melatonin in children with neurodevelopmental delay and a range of neurological disorders and impaired sleep to confirm (or refute) the findings that the drug may reduce the time taken to fall asleep and increase the total duration of night-time sleep.
Potential risks and benefits
Potential risks
Clinical studies in humans (adult volunteers and patients of both sexes and all ages) have not shown any consistent or serious short- or long-term adverse side effects. 35 Most of the reported adverse side effects have been described in very small numbers of patients. 36,37 Although the chronic use of exogenous melatonin for sleep problems in paediatrics appears widespread, there is a paucity of data on its safety. Melatonin is widely distributed at different densities throughout the body and appears to be implicated in various physiological functions other than sleep. There are therefore theoretical risks to the chronic administration of exogenous melatonin to children, and particularly to children with a range of neurological problems, including epilepsy and behavioural problems. The most significant theoretical risks in this population are related to:
-
sexual development
-
nocturnal asthma
-
growth
-
seizures.
With age, nocturnal melatonin levels appear to decrease with the most striking falls occurring around puberty. Nocturnal melatonin levels have been assessed in children at various pubertal stages and it is observed that they are higher in the earlier than in the later stages. 38 Whether this is cause or effect is not known but there is a potential risk that exogenous melatonin may delay sexual maturity.
Elevated endogenous melatonin levels have been associated with an increased incidence of nocturnal asthma,39 although there is at least one study in adults that demonstrated an improvement in sleep in adults with asthma following administration of 3 mg of melatonin with no apparent worsening of their asthma symptoms. 40
Melatonin has been observed to have a direct effect on growth hormone. 41 Eight male volunteers received single doses of 0.05 mg, 0.5 mg and 5 mg of melatonin or placebo with serum growth hormone levels measured for up to 150 minutes afterwards. Compared with placebo, growth hormone levels were found to increase for doses of 0.5 mg and 5 mg of melatonin. The exact mechanism is not clear and the effect of increases in growth hormone of this magnitude on longitudinal bone growth in children is not known.
One study has suggested that seizure control may deteriorate in some children with epilepsy42 but this observation has not been confirmed in a number of anecdotal8,17 and limited randomised controlled trials (RCTs). 25–27 There is some anecdotal evidence that seizure control or seizure severity may actually improve as a secondary effect of improved sleep and increased seizure threshold. 8 There have been two spontaneous reports to the Medicines and Healthcare products Regulatory Agency of seizures associated with exogenous melatonin and responders to the survey by Waldron et al. 37 reported an increase in seizure activity or new-onset seizures.
Melatonin oral capsules contain melatonin, lactose and magnesium stearate. Placebo oral capsules contain lactose and magnesium stearate. Individuals with lactose intolerance are able to consume significant quantities of dairy products without manifesting any symptoms of lactose malabsorption and therefore individuals with lactose intolerance are able to consume capsules without adverse effects and were eligible for inclusion into the study.
Potential benefits
There are very few meta-analyses of RCTs of melatonin. 35,36 Those reported have indicated that exogenous melatonin may improve sleep in a number of clinical situations, including:
-
children with autism and intellectual disability
-
patients with visual impairment [particularly when the visual impairment is due to an abnormality within the anterior visual pathway (specifically in patients with micro-ophthalmia or anophthalmia) rather than cortical visual impairment]
-
elderly patients with insomnia.
Reported benefits include a reduced sleep latency time (i.e. reduced time to fall asleep), reduced number of awakenings throughout the night (i.e. increased periods of continuous, uninterrupted sleep throughout the night) and improved behaviour and performance during the day. Seizure frequency and seizure control may also improve, probably as a secondary or indirect effect of improved quality of sleep. 8,43
It is important to emphasise that all of the reported studies show a marked heterogeneity in inclusion and exclusion criteria, the types and causes of impaired sleep in the populations studied, the doses and formulations of melatonin used, the methods of assessment and the reported outcomes. Although the current study includes a heterogeneous group of children with neurodevelopmental delay, all were treated according to a strict protocol and within a dose-escalation framework. Finally, the patient population in this RCT is almost as large as that of the combined studies that were assessed in a recent meta-analysis. 23
Chapter 2 Methods
Objective
The primary objective of the trial was to confirm (or refute) that immediate-release melatonin is beneficial compared with placebo in improving total duration of night-time sleep in children with neurodevelopmental problems.
Design
This was a randomised, double-blind, placebo-controlled, parallel-group, multicentre clinical trial that compared the effects of melatonin with placebo in children with neurodevelopmental disorders and impaired sleep from sites throughout England and Wales.
The trial was designed to have a 4- to 6-week behaviour therapy period during which eligible participants were provided with a behaviour therapy advice booklet (see Appendix 1) and had their sleep monitored using both parent-completed sleep diaries (see Appendix 2) and actigraphy. At the end of this period participants who continued to fulfil eligibility criteria were randomised to receive melatonin or placebo (randomisation ratio 1 : 1).
At randomisation each child was given 0.5 mg of melatonin and was kept on that dose for a minimum of 7 days. For the next 3 weeks at 1-week intervals the child’s sleep pattern was reviewed and the medication either left unchanged or increased to the next dose increment. There were a maximum of three dose increments after the starting dose of 0.5 mg (2 mg, 6 mg and 12 mg).
Participants
The population studied was a heterogeneous group comprising a large number of children with a wide range of neurological and developmental disorders, including those with specific genetic disorders but also those without a specific diagnosis. This group was chosen because it reflects the typical population that is currently prescribed melatonin in the UK.
Eligibility criteria for entry to the behaviour therapy period and randomised trial were consistent; however, the sleep disorder criteria for the behaviour therapy period were based on parental perception whereas sleep diaries completed during the behavioural therapy period were used to determine whether or not the sleep disorder fulfilled the same criteria prior to randomisation.
The inclusion and exclusion criteria were as follows.
Inclusion criteria
-
Children aged from 3 years to 15 years and 8 months at screening (the age at screening was set to ensure that all those enrolled in the study were minors, because of the implications for consent in incapacitated adults).
-
Diagnosis of a neurodevelopmental disorder by a community paediatrician, paediatric neurologist or paediatric neurodisability consultant, categorised as:
-
– developmental delay alone
-
– developmental delay and epilepsy
-
– developmental delay and autistic spectrum disorder (ASD) (in coding the presence of epilepsy and ASD diagnoses we required sight of documentation from relevant services which demonstrated that appropriate diagnostic assessments and investigations have been used)
-
– developmental delay with ‘other’ (‘other’ is defined as the child having a specific genetic/chromosomal disorder)
-
– any combination of the above.
-
-
Adaptive Behaviour Assessment System (ABAS) questionnaire score with a percentile rank < 7.
-
Minimum 5 months’ history of impaired sleep at screening as defined by:
-
– not falling asleep within 1 hour of ‘lights off’ or ‘snuggling down to sleep’ at age-appropriate times for the child [this was the child’s usual bedtime (recorded in the sleep diary) based upon the family’s normal routine; ‘age appropriate’ was defined as a sensible target sleep onset time earlier than 20:30 for children at age 6 years and 15 minutes later per year for older children44] in three nights out of five and/or
-
– < 6 hours of continuous sleep in three nights out of five .
-
-
Children whose parents were likely to be able to use the actigraph and complete sleep diaries.
-
Children who were able to comply with taking the study drug.
-
English speaking.
-
Children whose parents had completed sleep diaries for an average of 5 out of 7 nights at baseline (T0W).
Exclusion criteria
-
Children treated with melatonin within 5 months of screening (T–4W).
-
Children who had been taking the following medication for < 2 months:
-
– any benzodiazepines
-
– amisulpride (Solian®, Sanofi-Aventis)
-
– chlorpromazine (Largactil®, Sanofi-Aventis)
-
– haloperidol (Haldol®, Janssen)
-
– olanzapine (Zyprexa®, Lilly)
-
– risperidone (Risperdal®, Janssen)
-
– sertindole (Serdolect®, Lundbeck)
-
– sulpiride (Sulpor®, Rosemont)
-
– thioridazine (Melleril®, Novartis)
-
– trifluoperazine (Stelazine®, Goldshield).
-
-
Current use of beta-blockers (minimum of 7 days’ washout required).
-
Current use of sedative or hypnotic drugs, including chloral hydrate, triclofos, and alimemazine tartrate (Vallergan®, Sanofi-Aventis) (minimum of 14 days’ washout required).
-
Children with a known allergy to melatonin.
-
Regular consumption of alcohol (more than three times per week).
-
Children for whom there are suggestive symptoms of obstructive sleep apnoea syndrome (OSAS) (including combinations of snoring, gasping, excessive sweating or stopping breathing during sleep), physical signs supportive of OSAS (such as very large tonsils/very small chin) or results of investigations suggesting OSAS (such as overnight pulse oximetry or polysomnography), for which the child should be referred to appropriate respiratory or ear, nose and throat colleagues for specific assessment and treatment.
-
Girls or young women who were pregnant at the time of screening (T–4W).
-
Currently participating in a conflicting clinical study or participation in a clinical study involving a medicinal product within the last 3 months.
Behaviour therapy advice booklet
The intention underlying the use of the behaviour therapy advice booklet was to ensure that children progressing to the randomisation phase did not include those whose sleep disorder would be amenable to treatment with a brief non-pharmacological intervention. The behaviour therapy advice booklet used during the baseline period was one previously shown to be effective for reducing sleep problems in children with neurodevelopmental disorders. 45 The booklet advises about some key principles underlying behaviour therapy (i.e. use of operant and classical conditioning, the need for consistency and persistence), explains general sleep hygiene principles and offers specific behavioural strategies for dealing with problems of settling to sleep, night waking and sleeping in the parents’ bed and for changing the timing of children’s sleep periods. Research nurses introduced the booklet to families using a script to ensure that the nature and the scope of the booklet were fully explained, the key principles were emphasised and common parental concerns about the use of behaviour therapy were addressed.
Interventions
The active compound (melatonin, Alliance Pharmaceuticals) and the placebo (matching in package and appearance) were administered 45 minutes before the child’s usual bedtime; whenever possible, this time remained the same throughout the study. The study treatment was administered orally or, if the patient was not able to feed orally, through a nasogastric feeding tube or gastrostomy feeding tube. In these last two situations the capsule was opened and the study treatment suspended in an appropriate vehicle for administration. These vehicles had been identified following formal pharmacokinetic and stability studies before the study and included water, orange juice, semi-skimmed milk, strawberry yoghurt and strawberry jam. 46
The starting dose was 0.5 mg and following this there was a 4-week Dose escalation phase in which children meeting the following criteria for a dose increment could progress through 2 mg and 6 mg to 12 mg:
-
absence of serious adverse events
-
a minimum of five of seven days completed in the sleep diary in the preceding week
-
no ‘significant increase’ (defined as a doubling in seizure activity over the preceding 4 weeks) in seizure activity (where applicable)
-
child had received at least five of the possible seven doses in the current week
-
child not falling asleep within 1 hour of ‘lights off’ or ‘snuggling down to sleep’ at age-appropriate times for the child in three nights out of five and/or child having less than 6 hours of continuous sleep in three nights out of five .
Study procedures
Eligible patients for whom informed consent was obtained were registered onto the behaviour therapy phase of the trial. This was a period (minimum of 4 weeks and maximum of 6 weeks) during which the parents were asked to follow the recommendations of a behaviour therapy advice booklet (see Appendix 1) and to complete nightly sleep diaries (see Appendix 2) to record their child’s sleep. The children were asked to wear an actigraphy watch to monitor their sleep behaviour during the behaviour therapy phase. After the behaviour therapy phase the patients returned to clinic where their sleep diaries were reviewed. Patients who continued to meet the entry criteria (see inclusion and exclusion criteria) and whose parents/carers and, when possible, the patients themselves were able to give informed consent were randomly allocated to receive either melatonin or matching placebo capsules. Each randomised participant was followed up for 12 weeks from the date of randomisation with a combination of home visits, telephone calls and attendance at clinic. The schedule of study procedures is provided in Table 1.
| Procedure | Time (T) (weeks) | Premature discontinuation | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| –4 | –2 | 0a | 1 | 2 | 3 | 4 | 5 and 6 | 7–9 | 10 | 11 | 12 | ||
| Screening (clinic visit) | Home visit | Clinic visit | Home visit | Home visit | Home visit | Home visit | Tel. call | Tel. call | Tel. call | Home visit | Study completion (clinic visit) | ||
| Signed informed consent | ✗ b | ✗ c | ✗ d | ✗ b | ✗ c | ✗ d | |||||||
| Adaptive Behaviour Assessment System | ✗ | ||||||||||||
| Assessment of eligibility criteria | ✗ | ✗ | ✗ | ||||||||||
| Review of medical history | ✗ | ✗ | |||||||||||
| Registration for 4-week behaviour therapy | ✗ | ||||||||||||
| Review of concomitant medications | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||
| Discussion and issue of behaviour therapy booklet | ✗ | ||||||||||||
| Social Communication Questionnaire | ✗ | ||||||||||||
| Behaviour therapy booklet evaluation form | ✗ | ||||||||||||
| Randomisation | ✗ | ||||||||||||
| Children’s Sleep Habits Questionnaire | ✗ | ✗ | |||||||||||
| Composite Sleep Disturbance Index | ✗ | ✗ | ✗ | ||||||||||
| Family Impact Module of PedsQL | ✗ | ✗ | |||||||||||
| Epworth Sleepiness Scale | ✗ | ✗ | |||||||||||
| Aberrant Behaviour Checklist | ✗ | ✗ | |||||||||||
| Sleep and seizure diary (if applicable) | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||
| Actigraph watch is worn (actigraphy) | ✗ | ✗ | ✗ | ✗ | |||||||||
| Study intervention | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ||||
| Stepwise increase in treatment dose | (✗) | (✗) | (✗) | (✗) | (✗) | ||||||||
| Physical examination | |||||||||||||
| Complete | ✗ | ✗ | ✗ | (✗) | |||||||||
| Symptom directed | (✗) | (✗) | (✗) | (✗) | (✗) | (✗) | (✗) | (✗) | |||||
| Vital signs, weight, height | ✗ | ✗ | (✗) | (✗) | (✗) | (✗) | (✗) | (✗) | (✗) | (✗) | ✗ | (✗) | |
| Occipitofrontal head circumference | ✗ | ||||||||||||
| Assessment of adverse events | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | |||
| Special assay or procedure | |||||||||||||
| Salivary melatonin | ✗ | ||||||||||||
| DNA (salivary sample) | ✗ | ||||||||||||
Data collection tools
Sleep outcomes were measured using subjective (sleep diaries) and objective (actigraph) methods as has been recommended. 47
Sleep diaries
Each week parents were asked to complete a two-sided A4 sleep diary that covered a period of 7 days, with one column per day (see Appendix 2). Parents recorded the time that their child went to bed, fell asleep and woke up the next morning. They also recorded any daytime naps, night-time awakenings and the time and duration of any actigraphy removal. Sleep diaries were completed continuously between T–4W and review at T0W, and also continuously throughout the study until study completion (T+12W). A sleep diary records the parental observation and perception of the child’s sleep. Parents were not required to differentiate between periods when the child was actually asleep and periods when the child was awake but quiet (i.e. not disturbing the rest of the household). Consequently, parents did not have to stay awake to complete the sleep diary and were not requested or expected to repeatedly check their child throughout the night.
Sleep diaries were used to calculate TST, SOL (the time taken to fall asleep) and daily global sleep quality in a subjective manner from the parents’ perceptions.
Actigraphy
Actigraphy is the use of accelerometers to measure human movement. The actigraph is worn on the wrist and the movement of the wrist is monitored continuously whilst it is being worn. The actigraph is very lightweight and can be used on individuals of all ages for long periods of time. Wrist movement data are stored within the unit and processed using software programmes to give an indication of the activity levels of the wearer. Analysis of frequency and pattern of movement by means of validated algorithms permits detection of basic sleep–wake patterns. 48
The actigraph was worn continuously day and night for the behaviour therapy phase and for the final week of the study period. The actigraph could be removed or worn during bathing or showering. It could be worn on either wrist but it was emphasised that the same wrist should be used throughout the study. The actigraph used in this study was the MicroMini-Motionlogger®, supplied by Ambulatory Monitoring Inc.
The actigraph measures and stores data on movements. Frequency of movements above a preset threshold are scored in 1-minute epochs; all epochs that are scored above a preset threshold (sensitivity level) are scored as ‘wake’ and those that are below this threshold are scored as ‘sleep’. The threshold is not set on an individual basis.
In line with existing guidance,49 interpretation of the actigraphy data was informed by the sleep diaries. The sleep diaries recorded the child’s ‘snuggle down to sleep’ time, and the start of sleep was determined from the actigraph as the first 10-minute interval after ‘snuggle-down’ time when there was no more than one epoch that was above the threshold (automatically calculated by the software) for determining wakefulness. The software then considered the first minute of this 10-minute period as the time of sleep onset.
Any sleep interruptions were determined from the actigraph by searching for 10-minute intervals in which activity in more than one epoch was above the threshold set automatically for determining wakefulness or ‘wake’. Final wake-up time was recorded by parents in the sleep diary to the nearest minute. Sleep offset was determined to be the last 10-minute period before final wake-up time in which there was no more than one epoch that was above the threshold for determining ‘wake’.
Total night-time sleep was calculated as the sum of all epochs scored as sleep from sleep onset to sleep offset.
As the actigraphy watch defines periods of sleep as periods with little/no activity, it is acknowledged that those periods of restless sleep may be interpreted by the unit as periods of ‘wake’. This may be a particular issue for children with motor problems, including cerebral palsy.
Treatment-emergent signs and symptoms
Assessment of adverse effects was undertaken weekly between weeks T0W to T+12W. These reviews were performed by the investigator at clinic attendance or the research practitioner during home visits or by telephone assessment. Adverse effects were assessed using treatment-emergent signs and symptoms (TESS). The TESS evaluation included the following specific signs and symptoms:
-
somnolence (drowsiness)
-
increased excitability
-
mood swings
-
seizures (de novo presentation of epilepsy in a child with no pre-existing diagnosis of epilepsy or an exacerbation of seizures in a child with a pre-existing diagnosis of epilepsy) (A seizure diary was given to the parents of those children who had an established diagnosis of epilepsy, whether or not they were receiving any antiepileptic medication. Seizure diaries were also to be completed for any child who experienced a seizure post registration.)
-
rash
-
hypothermia
-
cough
-
other adverse effects not listed were also documented; the Investigator’s Brochure was referred to when assessing causality and expectedness.
Signs and symptoms were graded and reported as no symptoms, mild symptoms, moderate symptoms and severe symptoms. Seriousness and causality were also assessed by the reporting researcher (principal investigator).
Seizure diaries
Seizure diaries were completed between T–4W and T0W and reviewed at randomisation (T0W). Post randomisation they were reviewed at weekly intervals for the first 4 weeks during home visits by the research practitioner (T+1W, T+2W, T+3W, T+4W), at the final home visit (T+11W) and at the clinic visit at week 12 (T+12W). Seizure status was also discussed during telephone review by the research practitioner in weeks T+5W to T+10W. Information was collected on the number and type of seizures and whether the child was asleep or awake at the time of the seizure. No attempt was made to grade the severity of the seizures because this is not routine practice in the assessment and management of children with epilepsy.
Questionnaires
Parents were asked to complete a questionnaire booklet. The details of the scoring methods for each questionnaire are provided within the statistical analysis plan (see Appendix 3). The following questionnaires were completed.
Children’s Sleep Habits Questionnaire
A comprehensive, parent-reported sleep-screening instrument designed for school-age children, the Children’s Sleep Habits Questionnaire (CSHQ)50 yields both a total score and eight subscale scores, reflecting key sleep domains that encompass the major medical and behavioural sleep disorders in this age group. The questionnaire takes 10 minutes to complete. It was undertaken at T–4W and T0W.
Pediatric Quality of Life Inventory Family Impact Module
The PedsQL Family Impact Module51 is designed to measure the impact of paediatric chronic health conditions on parents and the family. It measures parent self-reported physical, emotional, social and cognitive functioning, communication and worry. The module also measures parent-reported family daily activities and family relationships. Scores range between 0 and 100 and higher scores indicate better functioning. The questionnaire takes approximately 5 minutes to complete. It was undertaken at T0W and T+12W.
Epworth Sleepiness Scale
The Epworth Sleepiness Scale (ESS)52 is a simple, self-administered questionnaire that provides a measurement of the caregiver’s general level of daytime sleepiness. Scores range between 0 and 24 and higher scores indicate poorer functioning. The questionnaire takes approximately 3 minutes to complete. It was undertaken by one caregiver and the same caregiver completed the questionnaire at T0W and T+12W.
Aberrant Behaviour Checklist
The Aberrant Behaviour Checklist (ABC)53,54 is an instrument for assessing individual baseline behaviour and for evaluating behavioural change. The ABC contains five subscales and higher scores indicate poorer functioning. The checklist takes approximately 20 minutes to complete. It was undertaken at T0W and T+12W.
Composite Sleep Disturbance Index
A Composite Sleep Disturbance Index (CSDI), based on allocating scores according to the frequency and duration of sleep problems reported by parents in questionnaires, was first used by Richman and Graham55 and has since been used in many other studies, including that by Quine,10 who reported high internal reliability and showed that the measure was sensitive to change. 56 The questionnaire takes 3 minutes to complete and was undertaken at T–4W, T0W and T+12W.
Applying the scoring criteria of Table 2 the CSDI was calculated as follows: settling problems, night waking, early waking (before 0500) and co-sleeping were each assigned a score of 0–2 based upon their reported weekly frequency; settling and night-waking problems were also assigned a score of 0–2 based upon the reported duration of the problem, when it occurred. Total scores were derived by adding the scores assigned for these six items. The scores ranged from 0 to 12 and higher scores indicate greater sleep disturbance.
| Night waking | Score | ||
|---|---|---|---|
| 0 | 1 | 2 | |
| Frequency | Less than once per week | One to two times per week | Three or more times per week |
| Duration | Few minutes | ≤ 30 minutes | 31+ minutes |
Biochemical and genetic investigations
Salivary melatonin assay
Salivary melatonin levels were measured for each patient at two time points. Saliva samples were collected hourly from 17:00 until the child’s usual bedtime at:
-
T–1W, on the night before the randomisation clinic visit
-
T+10W, on the night after a dose of trial treatment had been omitted and on which night no trial medication was given (i.e. two doses were missed at the beginning of the eleventh week of study treatment).
This is a very similar methodology to that described by Keijzer et al. 57 in which five evening collections were effective in the majority of cases when evaluating dim-light melatonin onset (DLMO) timing for patients with circadian rhythm disorders. A minimum of 2 ml of saliva was obtained by asking the child to spit into a tube or by placing a saliva sponge in the buccal cavity (cheek pouch) of the child’s mouth (the space between the gums and the inner cheek).
Salivary samples were collected and stored by the parent in a domestic freezer at a maximum temperature of –18°C. Samples were collected by the research practitioner for storage until trial completion, when they were placed in dry ice and transported to the School of Biomedical and Molecular Sciences, University of Surrey, Guildford for blinded analysis. The theoretical basis for these measurements is that they should allow accurate categorisation of which children are physiologically phase delayed at the beginning of the study, which may prove to be an important variable when comparing responders to non-responders in any secondary analysis.
Baseline salivary melatonin levels are at their lowest during the day. During the evening, as light levels decrease, there is a natural rise in melatonin levels (usually between 2000 and 2200 depending on age) that starts to peak around midnight. The time when the melatonin levels naturally start to rise from the low daytime baseline is called the DLMO period. This is measured biochemically as the time when melatonin levels first start to rise by at least two standard deviations above the mean baseline level. An 8-year-old child who has a DLMO of midnight, for example, would be classified as having a delayed sleep phase and, according to recent research, should respond better to exogenous melatonin than another child with a normal DMLO of 2000. 44
If sampling is taken regularly for a 24-hour period the DLMO should be measurable, whenever it occurs. However, for practical reasons, it is common in paediatric populations to take five swabs before bedtime. It is therefore possible to ‘miss’ a DLMO that precedes sampling or occurs when sampling has finished.
Deoxyribonucleic acid analysis
Salivary deoxyribonucleic acid (DNA) was collected from 186 patients [with some samples taken from children who participated only in the behaviour therapy part of the study and not in the interventional (randomised controlled) part of the trial] using the DNA collection kit from DNA Genotek (OG-250®, DNA Genotek, Kanata, ON, Canada). DNA was extracted according to the manufacturer’s instruction in a small volume of 200 µl of tris–ethylenediaminetetraacetic acid (EDTA; TE) buffer 10 : 1 to increase the quality of the high-throughput DNA genotyping. Samples passing DNA quality control were subjected to genome-wide genotyping using several different Illumina single nucleotide polymorphism (SNP) arrays and sample processing was in accordance with the manufacturer’s protocol. Each array contained a minimum of 600,000 SNPs. Genotype data were generated using Illumina’s® BeadStudio software (Illumina Inc., Chesterford, UK). All copy-number variants (CNVs) were detected using the QuantiSNP algorithm based on the signal intensity and the B allele frequency values of each SNP. Visualisation was undertaken using the SnipPeep software.
After fully blinded genotyping had taken place, each genetic variant was tested for association with each outcome of interest. Full details of the outcomes investigated and the statistical methods used in the genetics substudy are provided in the statistical analysis plan (see Appendix 3). These methods were agreed in advance before undertaking any analyses.
In addition to genome-wide genotyping, all coding exons of AANAT (arylalkylamine N-acetyltransferase) and ASMT (N-acetylserotonin O-methyltransferase), the two genes of the melatonin synthesis pathway, were also sequenced. Sequencing conditions and primers have been described previously. 58 The impact of mutation on protein function was addressed in silico using the Polyphen2 algorithm (http://genetics.bwh.harvard.edu/pph2/) and/or according to previously published in vitro experiments. 59
A partial duplication of the ASMT gene, involving exons 1–7, has been described as occurring more frequently in patients with autism spectrum disorders than in the general population. 60 As this CNV cannot be detected using genotyping arrays, it was necessary to use a polymerase chain reaction (PCR)-based genotyping test. The CNV breakpoint was amplified together with a positive control PCR using the Qiagen Multiplex PCR kit (Crawley, UK) according to the manufacturer’s instruction. The annealing temperature was 66°C. The CNV-specific primers were as follows: forward primer: 5′–GTGGTGACAGATCTCGGCTCCCTTCAA–3′; reverse primer: 5′–GTCTGGCAGGACGGTTTCAG–3′. The positive control primers were as follows: forward primer: 5′–TGGTGCAATCTCATTTGACTCTG–3′.; reverse primer: 5′–GGGTTCATGCCATTCTCCTG–3′. The presence of PCR products was assessed by migration on 2% agarose gels.
Outcomes
Primary outcome
The primary outcome was TST, calculated using parentally completed diaries. The total amount of sleep for 1 night was calculated as the amount of time between the time that the child went to sleep and the time that the child woke up the following morning minus any night-time awakenings. The baseline measurement was calculated using the average total amount of night-time sleep in the 7 days before randomisation and the post-treatment measurement was calculated as the average total amount of night-time sleep from day 77 to day 84 post randomisation (this corresponds to the final 7 days of treatment because patients received enough drug supply only for 84 days).
A minimum of 5 nights of sleep from each time period was required for the data to contribute to the primary outcome. If a child had less than 5 out of 7 nights completed the data were regarded as missing and were not included in the primary analysis.
Secondary outcomes
-
TST calculated using actigraphy data.
-
SOL (the time taken to fall asleep) calculated using actigraphy.
-
SOL (the time taken to fall asleep) calculated using sleep diaries (the number of minutes between lights out/‘snuggle-down’ time and sleep start time).
-
Sleep efficiency calculated using actigraphy: (number of minutes spent sleeping in bed/total number of minutes spent in bed) × 100.
-
CSDI score.
-
Daily global measure of parental perception of child’s sleep quality (a ‘smiley face’ scale).
-
Behavioural problems assessed using the ABC.
-
Quality of life of the parent assessed using the Family Impact Module of the PedsQL.
-
Level of daytime sleepiness in caregivers assessed using the ESS.
-
Number and severity of seizures evaluated using seizure diaries throughout trial follow-up.
-
Adverse effects of melatonin treatment assessed weekly between weeks T0W and T+12W using TESS.
-
Salivary melatonin concentration.
-
Associations between genetic variants and abnormal melatonin production.
Sample size calculations
Sample size calculations were undertaken using nQuery Advisor® software version 4.0 (Statistical Solutions Ltd, Cork, Ireland). The decision on the magnitude of clinically relevant outcomes was based on:
-
(a) parent involvement (the parent was a co-applicant and a member of the Trial Management Group)
-
(b) informal discussion with parents presenting at clinics and
-
(c) the results from a number of parent/carer focus groups undertaken a few years earlier by two co-applicants of MENDS with many years’ experience in sleep studies in children.
The trial was originally designed with two primary outcomes assumed to be independent: TST calculated using the data recorded in sleep diaries and SOL calculated using actigraphy. Bonferoni’s adjustment was used in the sample size calculation (2.5% significance level) to allow for the multiplicity of the two primary outcomes. During the recruitment phase of the trial, high rates of missing data (66%) were observed for actigraphy. A proposal was discussed and agreed by the Trial Management Group to amend the protocol to move the end point SOL measured using actigraphy to a secondary outcome. The integrity of the trial was protected as the decision was based solely on the proportion of missing actigraphy data and was taken before carrying out any comparative analysis for any outcome. Independent advisors outside of the Independent Data Safety Monitoring Committee (IDSMC) and Trial Steering Committee (TSC) were also consulted as suggested in Evans. 61
The original and revised sample size calculations are presented in full in the following sections.
Original sample size calculations
For the outcome total night-time sleep, the change between the total amount of sleep before randomisation and the total amount of sleep following randomisation will be calculated for each child. The titration period will not be used for the analysis of change. The null hypothesis is that there is no difference in the total amount of sleep between the melatonin and the placebo groups. The alternative hypothesis is that there is a difference in the total amount of sleep between the groups. The study is designed to detect a difference of 1 hour TST between the melatonin group and the placebo group. Assuming a common standard deviation of 1.7 (based on published data in similar populations/settings8,21), a sample size of 57 per group, increasing to 63 per group to allow for an estimated 10% loss to follow-up, will be required to provide 80% power using a t-test with a 0.025 two-sided significance level (adjusted to allow for multiple outcomes).
For the outcome SOL, the null hypothesis is that there is no difference in the time to sleep onset between the melatonin and the placebo groups. The alternative hypothesis is that there is a difference in the time to sleep onset. A sample size of 78 in each group (86 per group with estimated loss to follow-up of 10%) will have 80% power to detect a difference in means of 30 minutes, assuming a common standard deviation of 60 minutes using a two-group t-test with a 0.025 two-sided significance level.
Randomising a total of 172 children, 86 into each of the study arms, satisfies both sample size calculations. The sample size calculations are based on the use of nightly sleep diaries for total night-time sleep and actigraphy for time to sleep onset. Both outcomes will be analysed using analysis of covariance (ANCOVA), which will give an additional increase in statistical power.
Sample size calculation revision
The original trial recruitment target was 172 randomised patients, which was the maximum of the sample size calculations for the outcomes SOL (n = 172) and TST (n = 126). The original sample size calculation for TST was powered at 80% to detect a difference of 1 hour between the melatonin and the placebo groups using a common standard deviation of 1.7. Using a t-test with a 0.025 two-sided significance level, a sample size of 57 per group, increasing to 63 per group to allow for estimated 10% loss to follow-up, was required. Following the amendment to move SOL from a primary to a secondary outcome the sample size for the trial was recalculated based on the TST outcome. The revised calculation required 47 per group based on a 0.05 two-sided significance level as the multiplicity adjustment was no longer required; this was increased to 57 per group to allow for 20% missing data based on observed rates at the time of the amendment.
Randomisation and blinding
Randomisation lists were generated in Stata release 9 (StataCorp LP, College Station, TX, USA) using block randomisation with random variable block length. Randomisation was stratified by centre. The study drugs were identical in external and internal appearance and identically packaged. The treatment packs were numbered sequentially and held within each site pharmacy. Each treatment pack held sufficient drugs for the 12-week period following randomisation and allowed for potential dose escalation. The pharmacy dispensed the treatment packs in sequence and the unique number on each treatment pack was then used as the participant’s randomisation number. All trial personnel were blinded to treatment allocation throughout the trial.
Data management
Each site research practitioner was provided with a MENDS laptop that was installed with a copy of InferMed MACRO™ version 3 (InferMed, London, UK). At each clinic and home visit the research practitioners would enter data directly onto the laptop and then securely synchronise the contents of the local database with the central database held on the server. Electronic files from the actigraphy were attached within MACRO. Research practitioners were instructed to synchronise their laptops within 24 hours of a participant visit. Data that were completed by the parent or care provider on hard copy first were reviewed by the research practitioner in the presence of the parent/care provider to identify and resolve any apparent discrepancies. Hard copies of participant sleep diaries and questionnaires were sent to the Medicines for Children Research Network Clinical Trials Unit (MCRN CTU) and 100% source data verification completed on data for the primary outcome with a random 10% completed on secondary outcomes. A helpdesk was provided to assist the research practitioners with any technical difficulties and hard copy case report forms were provided as an emergency backup.
MACRO was used by the Trial Coordinator within the CTU to raise data queries and these were responded to and resolved within MACRO by the research practitioner.
Statistical methods
Interim monitoring
The estimate of the common standard deviation used in the sample size calculation was checked after the first 20 participants had been randomised and completed follow-up. This blinded internal pilot is not deemed to have any significant impact on the final analysis and no between-group comparisons were made. If the standard deviation had been found to be smaller than that used in the sample size calculation, suggesting that fewer patients were required than initially proposed, then no action would have been taken and the size of the study would have remained as originally planned. If the standard deviation was found to be larger than assumed, suggesting the need for more patients, then, on the advice of the Data Monitoring Committee (DMC), the TSC would have aimed to increase recruitment and consider implications for funding and existing resources. The DMC was presented with the results of the blinded internal pilot and recommended no change to the sample size based on these results.
Levels of missing data were monitored throughout and strategies developed to minimise its occurrence; however, as much information as possible was collected about the reasons for missing data.
Analysis plan
All analyses were conducted according to the statistical analysis plan (see Appendix 3), which provides a detailed and comprehensive description of the main, preplanned analyses for the study. Analyses were performed with standard statistical software [Statistical Analysis Software (SAS®) 9.1.3; SAS Institute Inc., Cary, NC, USA] apart from those in the genetic substudy, which were undertaken using specialist genetic association software (see Appendix 3 for details).
The main features of the analysis plan are summarised below.
The Consolidated Standards of Reporting Trials (CONSORT) flow diagram is used to summarise representativeness of the study sample and patient throughput. Baseline characteristics are presented by treatment group and overall, with continuous variables presented with means and standard deviations and categorical variables with numbers and percentages.
The intention-to-treat principle is used as far as practically possible, with a two-sided p-value of 0.05 (5% level) used to declare statistical significance and 95% CIs reported throughout.
All continuous study outcomes are presented with means and standard deviations at T0 and T+12 and for the change over baseline (T+12 – T0) for each treatment group. ANCOVA is used to present results adjusted for baseline values. Reasons for missing data are provided (see Appendix 5). Sensitivity analyses are used to investigate the robustness of the primary outcome results to missing data (see Appendix 6).
Protocol amendments
The protocol amendments are provided in Appendix 4.
In summary, the main amendments were to the sample size calculation as described above, lowering the age limit in the inclusion criteria from 5 years to 3 years and the removal of the electronic games ‘MARS’ and ‘DENEM’. The reason for lowering the age limit from 5 to 3 years was to increase recruitment and the generalisability of the results across the age range currently being prescribed melatonin in the UK. There were a number of reasons for removing the MARS and DENEM electronic games: first, because of the limited ability of many patients to play the Maudsley Attention and Response Suppression Task Battery Items (MARS) game because of the degree of neurodevelopmental delay and additional comorbid impairments (e.g. attention deficit–hyperactivity disorder and autism spectrum disorder) (this also applied to some of the patients’ carers, who were unable to play the DENEM game); second, there was a degree of equipment failure (the hardware failing to download the completed games).
Chapter 3 Results
Participant flow and recruitment
The first patient registered was on 11 December 2007, the first patient randomised was on 28 January 2008, the last patient registered was on 7 May 2010 and the last patient randomised was on 4 June 2010. Table 3 shows all of the 19 recruiting centres and for each site the date that the site was initiated, the target recruitment, the number of participants registered, the number of participants randomised, the date of the first randomisation and the date of the last randomisation. All 19 centres registered at least one patient and 18 centres randomised at least one participant.
| Centre | Date site initiated | Target recruitment | Registered | Number randomised (% of registered) | Date of first randomisation | Date of last randomisation |
|---|---|---|---|---|---|---|
| Evelina Children’s Hospital, London | 18 January 2008 | 17 | 24 | 15 (63) | 26 February 2008 | 8 April 2010 |
| Royal Liverpool Children’s Hospital | 10 December 2007 | 15 | 25 | 13 (52) | 22 February 2008 | 20 May 2010 |
| University College London Hospitals | 14 December 2007 | 12 | 49 | 19 (39) | 28 January 2008 | 4 June 2010 |
| John Radcliffe Hospital, Oxford | 11 September 2008 | 14 | 26 | 15 (58) | 21 October 2008 | 25 May 2010 |
| Birmingham Children’s Hospital | 5 December 2007 | 15 | 27 | 11 (41) | 13 March 2008 | 30 July 2009 |
| Queen Mary’s Hospital, London | 26 June 2008 | 7 | 4 | 2 (50) | 31 July 2008 | 24 March 2009 |
| Royal Manchester Children’s Hospital | 3 January 2008 | 10 | 23 | 16 (70) | 04 February 2008 | 3 June 2010 |
| Derbyshire Children’s Hospital | 11 April 2008 | 5 | 4 | 2 (50) | 28 May 2008 | 20 January 2010 |
| Nottingham City Hospital | 11 April 2008 | 5 | 13 | 4 (31) | 10 October 2008 | 26 April 2010 |
| Southmead Hospital, Bristol | 4 January 2008 | 7 | 15 | 9 (60) | 08 July 2008 | 17 May 2010 |
| Chesterfield Royal Hospital | 6 February 2008 | 8 | 5 | 4 (80) | 19 March 2008 | 16 April 2010 |
| Torbay Hospital | 5 January 2009 | 3 | 2 | 2 (100) | 31 March 2009 | 16 February 2010 |
| Royal Devon & Exeter Hospital | 5 September 2008 | 6 | 16 | 12 (75) | 22 October 2008 | 16 December 2009 |
| Arrowe Park Hospital, Wirral | 19 August 2009 | 7 | 1 | 1 (100) | 23 September 2009 | 23 September 2009 |
| Blackpool Victoria Hospital | 16 June 2009 | 5 | 16 | 11 (69) | 22 July 2009 | 2 June 2010 |
| Leicester Royal Infirmary | 11 December 2009 | 6 | 1 | 0 (0) | N/A | N/A |
| Sheffield Children’s Hospital | 3 November 2009 | 6 | 3 | 3 (100) | 9 February 2010 | 30 March 2010 |
| Southampton General Hospital | 20 November 2009 | 10 | 8 | 6 (75) | 4 January 2010 | 24 May 2010 |
| Children’s Hospital for Wales, Cardiff | 12 November 2009 | 8 | 1 | 1 (100) | 23 December 2009 | 23 December 2009 |
Recruitment rates
The initial target sample size of the trial (172 participants) was expected to be achieved within a 12-month recruitment period. This had been based on estimates provided from each centre that had agreed to participate in the trial. The actual rates of recruitment were much lower (Figure 1). Suggested reasons for the slower than expected recruitment rates included availability of a marketed pharmaceutical grade of melatonin that was not available at the planning stage of the trial, the parental perception of the severity of the child’s sleep disorder at registration not being evident within the sleep diaries used to determine eligibility at randomisation, the potential impact of the 4- to 6-week behavioural phase of the trial on reducing the number of eligible participants, and a restrictive lower age limit of 5 years specified in the eligibility criteria of the protocol.
FIGURE 1.
Expected vs actual recruitment rates.
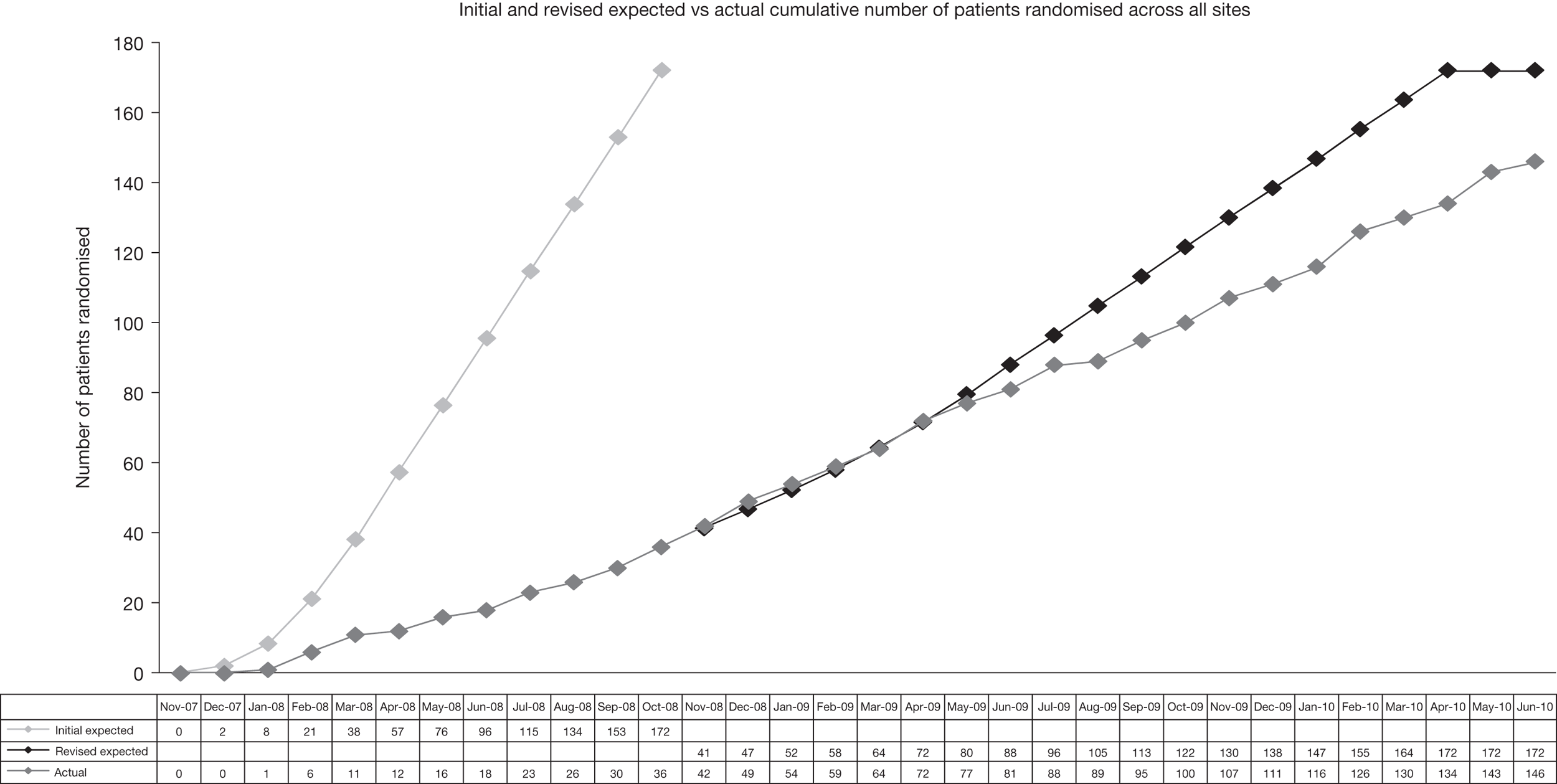
The recruitment period of the trial was extended and recruitment rates improved following intervention of the MCRN Local Research Networks (LRNs), which conducted a feasibility survey to identify additional recruiting centres.
The protocol amendment that removed the need to adjust the level of statistical significance for multiplicity of primary outcomes (see sample size calculations) reduced the required sample size from 172 to 114; this number was achieved within the extended timeline for the study.
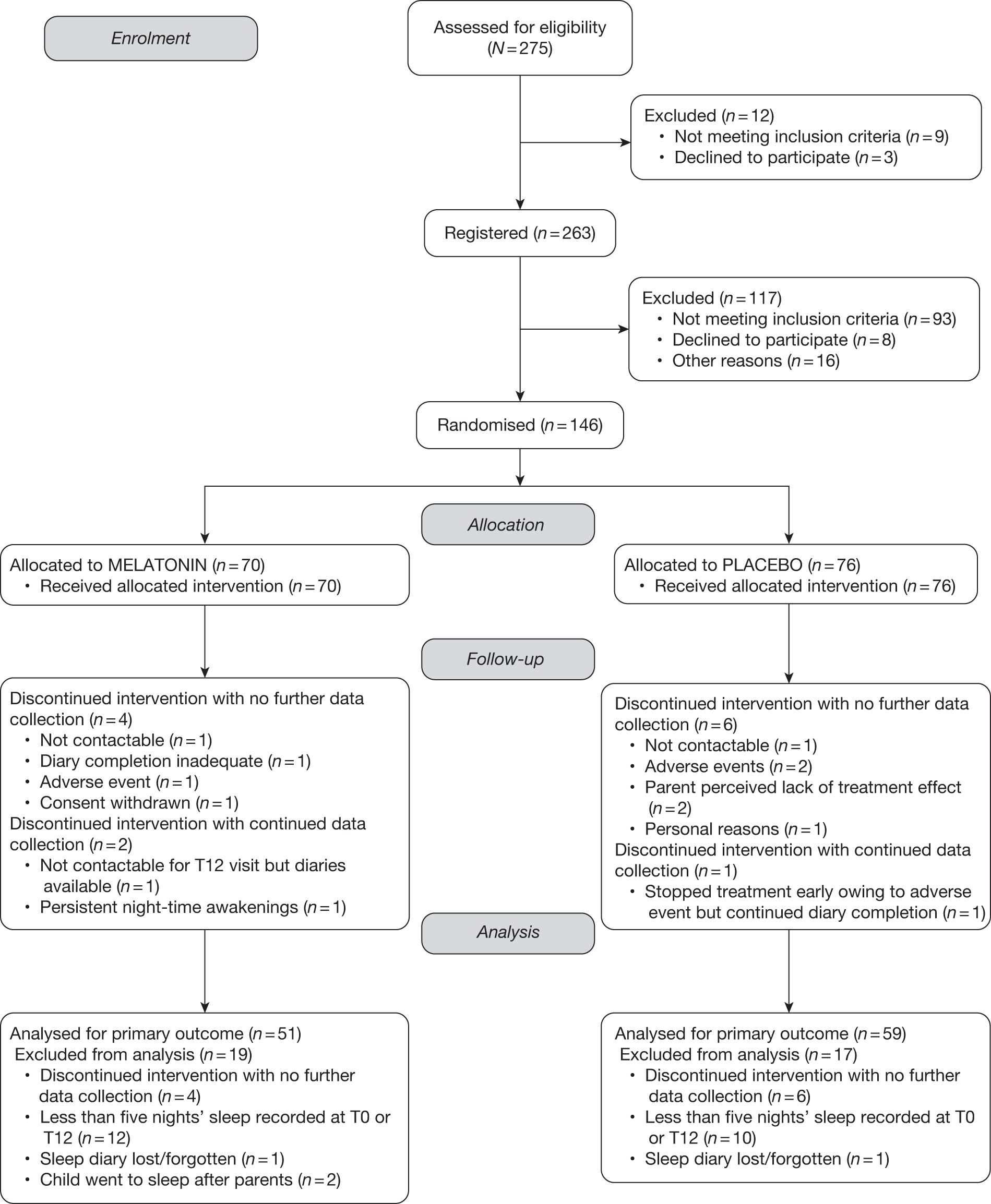
The flow of participants through the trial is represented in the CONSORT flow diagram in Figure 2. A total of 275 patients were assessed for eligibility to the trial of whom 12 (4%) were not registered [nine (75%) did not meet the inclusion criteria and three (25%) declined to participate]. A total of 263 participants entered the behaviour therapy phase and at the end of this period were assessed for eligibility to be randomised to receive melatonin or placebo. In total, 117 (45%) participants were not registered [93 (79%) did not meet the inclusion criteria, of whom 66 did not meet the definition of a sleep disorder according to the sleep diaries, 8 declined to participate and, of the remaining 16, there was a variety of reasons for non-randomisation]. A total of 146 patients were randomised, 70 (48%) to the melatonin group and 76 (52%) to the placebo group. In total, six (9%) participants withdrew in the melatonin arm: four discontinued the intervention and did not provide any further data and two continued to provide data following withdrawal. Seven (9%) participants withdrew from the placebo group: six provided no further data and one discontinued the intervention but continued to provide data for the primary outcome. In total, 19 (27%) participants on melatonin and 17 (22%) participants on placebo did not have data to contribute to the primary outcome analysis. Consequently, 51 (73%) participants were analysed for the primary outcome in the melatonin group and 59 (78%) participants were analysed for the primary outcome in the placebo group.
FIGURE 2.
Consolidated Standards of Reporting Trials flow diagram.
Baseline comparability of randomised groups
Table 4 shows that the baseline characteristics of the 146 randomised participants were comparable. Participants ranged in age between 37 and 186 months, with the mean age being slightly lower in the placebo group. There were five categories of neurodevelopmental delay; the numbers in each of these categories were similar in both treatment groups. The mean ABAS General Adaptive Composite (GAC) score and the number of males in each treatment group were also almost identical.
| Baseline characteristic | Melatonin (n = 70) | Placebo (n = 76) | Total (n = 146) |
|---|---|---|---|
| Age (months), mean (SD), range | 106 (34.8), 44 to 181 | 100.7 (37.4), 37 to 186 | 103.2 (36.2), 37 to 186 |
| Neurodevelopmental delay, n (%) | |||
| Developmental delay (DD) alone | 13 (19) | 9 (12) | 22 (15) |
| DD and epilepsy | 8 (11) | 5 (7) | 13 (9) |
| DD and ASD | 30a (43) | 30 (39) | 60 (41) |
| DD, ASD and epilepsy | – | 3 (4) | 3 (2) |
| DD and ‘other’ | 19a (27) | 29 (38) | 48 (33) |
| ABAS GAC score, mean (SD), range | 50.8 (9.9), 40 to 73 | 51.9 (11.29), 10 to 74 | 51.4 (10.6), 10 to 74 |
| Male, n (%) | 49 (70) | 48 (63) | 97 (66) |
Following the protocol amendment aimed at increasing recruitment rates by lowering the age limit from 5 to 3 years, 10 children were randomised who were under the age of 5 years (four in the melatonin arm and six in the placebo arm).
Description of dose escalation
At randomisation, each child was given 0.5 mg of melatonin or placebo and kept on that dose for a minimum of 7 days. For the next 3 weeks at 1-week intervals, each child’s sleep pattern was reviewed using set criteria and the medication either left unchanged or increased to the next dose increment. There were a maximum of three dose increments after the starting dose of 0.5 mg, through 2 mg and 6 mg up to a maximum of 12 mg.
Table 5 shows dose escalation for participants included in the primary analysis and Table 6 provides the same information for all randomised participants. There were no differences in dose escalation between the populations contained in Tables 5 and 6, supporting the generalisability of the results across all randomised participants. The tables show that participants randomised to placebo titrated more rapidly up to the maximum dose; by week 12, 38% of participants on melatonin were receiving 12 mg compared with 83% on placebo.
| Time point from randomisation (in weeks) | No. of participants | Melatonin | Placebo | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n M | n P | n T | wd | 0.5 mg | % | 2 mg | % | 6 mg | % | 12 mg | % | 0.5 mg | % | 2 mg | % | 6 mg | % | 12 mg | % | |
| Dose escalation phase | ||||||||||||||||||||
| T0 | 51 | 59 | 110 | 51 | 100 | 0 | 0 | 0 | 59 | 100 | 0 | 0 | 0 | |||||||
| T1 | 51 | 59 | 110 | 19 | 37.3 | 32 | 62.7 | 0 | 0 | 10 | 16.9 | 49 | 83.1 | 0 | 0 | |||||
| T2 | 51 | 59 | 110 | 13 | 25.5 | 15 | 29.4 | 23 | 45.1 | 0 | 4 | 6.8 | 15 | 25.4 | 40 | 67.8 | 0 | |||
| T3 | 51 | 59 | 110 | 10 | 19.6 | 15 | 29.4 | 8 | 15.7 | 18 | 35.3 | 1 | 1.7 | 9 | 15.3 | 18 | 30.5 | 31 | 52.5 | |
| T4 | 51 | 59 | 110 | 9 | 17.6 | 13 | 25.5 | 10 | 19.6 | 19 | 37.3 | 1 | 1.7 | 4 | 6.8 | 12 | 20.3 | 42 | 71.2 | |
| Dose maintenance phase | ||||||||||||||||||||
| T5 | 51 | 59 | 110 | 9 | 17.6 | 13 | 25.5 | 10 | 19.6 | 19 | 37.3 | 1 | 1.7 | 5 | 8.5 | 10 | 16.9 | 43 | 72.9 | |
| T6 | 51 | 59 | 110 | 9 | 17.6 | 12 | 23.5 | 10 | 19.6 | 20 | 39.2 | 1 | 1.7 | 3 | 5.1 | 9 | 15.3 | 46 | 78.0 | |
| T7 | 51 | 59 | 110 | 8 | 15.7 | 11 | 21.6 | 12 | 23.5 | 20 | 39.2 | 1 | 1.7 | 3 | 5.1 | 5 | 8.5 | 50 | 84.7 | |
| T8 | 50 | 59 | 109 | 1 | 8 | 16.0 | 11 | 22.0 | 12 | 24.0 | 19 | 38.0 | 1 | 1.7 | 3 | 5.1 | 6 | 10.2 | 49 | 83.1 |
| T9 | 50 | 59 | 109 | 1 | 8 | 16.0 | 11 | 22.0 | 11 | 22.0 | 20 | 40.0 | 1 | 1.7 | 3 | 5.1 | 6 | 10.2 | 49 | 83.1 |
| T10 | 50 | 59 | 109 | 1 | 9 | 18.0 | 10 | 20.0 | 12 | 24.0 | 19 | 38.0 | 1 | 1.7 | 4 | 6.8 | 5 | 8.5 | 49 | 83.1 |
| T11 | 50 | 59 | 109 | 1 | 9 | 18.0 | 10 | 20.0 | 12 | 24.0 | 19 | 38.0 | 1 | 1.7 | 5 | 8.5 | 4 | 6.8 | 49 | 83.1 |
| T12 | 50 | 59 | 109 | 1 | 9 | 18.0 | 10 | 20.0 | 12 | 24.0 | 19 | 38.0 | 1 | 1.7 | 4 | 6.8 | 5 | 8.5 | 49 | 83.1 |
| Time point from randomisation (in weeks) | No. of participants | Melatonin | Placebo | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n M | n P | n T | wd | 0.5 mg | % | 2 mg | % | 6 mg | % | 12 mg | % | 0.5 mg | % | 2 mg | % | 6 mg | % | 12 mg | % | |
| Dose escalation phase | ||||||||||||||||||||
| T0 | 70 | 76 | 146 | 70 | 100 | 0 | 0 | 0 | 76 | 100 | 0 | 0 | 0 | |||||||
| T1 | 68 | 76 | 144 | 2 | 29 | 42.6 | 39 | 57.4 | 0 | 0 | 20 | 26.3 | 56 | 73.7 | 0 | 0 | ||||
| T2 | 67 | 75 | 142 | 4 | 18 | 26.9 | 23 | 34.3 | 26 | 38.8 | 0 | 10 | 13.3 | 18 | 24.0 | 47 | 62.7 | 0 | ||
| T3 | 67 | 73 | 140 | 6 | 13 | 19.4 | 19 | 28.4 | 16 | 23.9 | 19 | 28.4 | 4 | 5.5 | 11 | 15.1 | 21 | 28.8 | 37 | 50.7 |
| T4 | 67 | 72 | 139 | 7 | 11 | 16.4 | 17 | 25.4 | 14 | 20.9 | 25 | 37.3 | 3 | 4.2 | 6 | 8.3 | 14 | 19.4 | 49 | 68.1 |
| Dose maintenance phase | ||||||||||||||||||||
| T5 | 67 | 71 | 138 | 8 | 11 | 16.4 | 17 | 25.4 | 14 | 20.9 | 25 | 37.3 | 2 | 2.8 | 7 | 9.9 | 12 | 16.9 | 50 | 70.4 |
| T6 | 67 | 71 | 138 | 8 | 11 | 16.4 | 15 | 22.4 | 15 | 22.4 | 26 | 38.8 | 2 | 2.8 | 4 | 5.6 | 11 | 15.5 | 54 | 76.1 |
| T7 | 67 | 71 | 138 | 8 | 9 | 13.4 | 14 | 20.9 | 18 | 26.9 | 26 | 38.8 | 2 | 2.8 | 4 | 5.6 | 6 | 8.5 | 59 | 83.1 |
| T8 | 65 | 70 | 135 | 11 | 9 | 13.8 | 14 | 21.5 | 17 | 26.2 | 25 | 38.5 | 2 | 2.9 | 4 | 5.7 | 7 | 10.0 | 57 | 81.4 |
| T9 | 65 | 70 | 135 | 11 | 9 | 13.8 | 14 | 21.5 | 16 | 24.6 | 26 | 40.0 | 2 | 2.9 | 4 | 5.7 | 7 | 10.0 | 57 | 81.4 |
| T10 | 65 | 70 | 135 | 11 | 10 | 15.4 | 13 | 20.0 | 17 | 26.2 | 25 | 38.5 | 2 | 2.9 | 5 | 7.1 | 6 | 8.6 | 57 | 81.4 |
| T11 | 65 | 70 | 135 | 11 | 10 | 15.4 | 13 | 20.0 | 17 | 26.2 | 25 | 38.5 | 2 | 2.9 | 6 | 8.6 | 5 | 7.1 | 57 | 81.4 |
| T12 | 65 | 70 | 135 | 11 | 10 | 15.4 | 13 | 20.0 | 17 | 26.2 | 25 | 38.5 | 2 | 2.9 | 5 | 7.1 | 6 | 8.6 | 57 | 81.4 |
Unblinding of randomised treatments
The treatment allocation for two participants was unblinded during the course of the trial (one in the melatonin group and one in the placebo group) to facilitate treatment of a suspected unexpected serious adverse reaction.
Protocol deviation
One participant was randomised but was ineligible because the participant did not produce sleep diaries at the T0 visit. This participant was not contactable after T0 and did not provide data for inclusion in the final analysis. There were no occurrences of participants who did not take any medication, no reported overdoses and no reports of patients taking any supplementary sleep-inducing medications.
Sleep outcomes
The results for the sleep outcomes are presented in Table 7. For each outcome each participant needed to have had at least 5 out of 7 nights’ completed sleep diary data at both the baseline assessment and during the final week of treatment.
| Sleep measures and outcomes | Melatonin | Placebo | Estimate (95% CI) | |||||
|---|---|---|---|---|---|---|---|---|
| Baseline mean (SD) | T12 mean (SD) | Change mean (SD) | Baseline mean (SD) | T12 mean (SD) | Change mean (SD) | Difference in mean change over baseline | Adjusted difference | |
| Sleep diary | ||||||||
| TST (minutes) (nM = 51, nP = 59) | 530.81 (64.84) | 571.26 (71.98) | 40.45 (71.75) | 545.49 (66.01) | 558.03 (68.94) | 12.54 (52.54) | 27.91 (4.35 to 51.48) (p = 0.0207) | 22.43 (0.52 to 44.34) (p = 0.0449) |
| SOL (minutes) (nM = 54, nP = 59) | 101.98 (72.56) | 54.82 (51.91) | –47.16 (64.38) | 102.09 (57.72) | 92.36 (63.02) | –9.72 (49.64) | –37.44 (–58.77 to –16.11) (p = 0.007) | –37.49 (–55.27 to –19.71) (p < 0.0001) |
| Actigraphy | ||||||||
| TST (minutes) (nM = 30, nP = 29) | 434.21 (72.30) | 449.88 (73.82) | 15.67 (63.60) | 412.27 (83.18) | 420.57 (82.90) | 8.30 (51.97) | 7.37 (–22.97 to 37.71) (p = 0.6285) | 13.33 (–15.48 to 42.15) (p = 0.3579) |
| SOL (minutes) (nM = 24, nP = 25) | 126.75 (71.45) | 68.42 (41.03) | –58.32 (53.65) | 107.83 (54.88) | 104.12 (59.53) | –3.71 (47.37) | –54.61 (–83.67 to –25.56) (p = 0.0004) | –45.34 (–68.75 to –21.93) (p = 0.0003) |
| Sleep efficiency (%) (nM = 30, nP = 28) | 65.42 (11.28) | 70.23 (11.28) | 4.81 (9.82) | 63.27 (12.34) | 64.83 (11.72) | 1.56 (9.52) | 3.25 (–1.84 to 8.35) (p = 0.2064) | 4.03 (–0.6 to 8.67) (p = 0.0869) |
The mean difference in TST between the two treatment groups adjusting for baseline mean TST was 22.43 minutes (95% CI 0.52 to 44.34 minutes) more in the melatonin group when using the sleep diaries and slightly less when using actigraphy (13.33 minutes, 95% CI –15.48 to 42.15 minutes). Although the difference between the treatment groups was statistically significant when diaries were used, the 95% CI does not contain the minimum clinically important difference of 60 minutes.
The outcome of SOL measured the time taken for a child to go to sleep from ‘snuggle-down’ time. This was calculated using both actigraphy and sleep diary data. The mean difference between treatment groups, adjusting for the mean baseline SOL, was –37.49 minutes (95% CI –55.27 to –19.71 minutes) using the sleep diary and –45.34 minutes (95% CI –68.75 to –21.93 minutes) using actigraphy in favour of the melatonin group. Both measures showed that the time taken to fall asleep by children in the melatonin group was statistically and clinically significantly less than that in the placebo group. The adjusted difference in sleep efficiency between the two treatment groups was not statistically significant, with an average improvement of 4.03% in the melatonin group (95% CI –0.6% to 8.67%).
A chi-squared test was used to test for differences between the groups in the number of patients with ≥ 5 days of sleep diary data (T0 and T12) contributing to the final analysis of TST. In the melatonin group, 51/70 (73%) had ≥ 5 days of sleep diary data at T0 and T12, and in the placebo group, 59/76 (78%) had ≥ 5 days of sleep diary data at T0 and T12. There was no difference between the groups [χ2 = 0.4471, p = 0.5037, relative risk 0.94 (95% CI 0.78 to 1.13)].
The reasons for the exclusion of participants from sleep outcome analyses are provided in Appendix 5, with the results of the sensitivity analyses and treatment interaction analyses given in Appendix 6. Plots of the mean change from baseline against the mean baseline TST for participants whose final dose was 0.5 mg, 2 mg, 6 mg and 12 mg are presented in Appendix 7.
Questionnaires
There were four questionnaires that were completed at baseline (T0) and at the final study visit (T12). These were the CSDI, the ABC (to assess behavioural problems), the Family Impact Module of the PedsQL (to assess the quality of life of the caregiver) and the ESS (to assess the level of daytime sleepiness of the caregiver). Note that higher scores are worse for the CSDI, ABC and ESS and lower scores are worse for the PedsQL. The results are shown in Table 8.
| Secondary measure | Melatonin | Placebo | Difference in mean change over baseline, mean (95% CI), p-value | Adjusted difference, mean (95% CI), p-value | ||||
|---|---|---|---|---|---|---|---|---|
| Baseline, mean (SD), range | T12, mean (SD), range | Change, mean (SD), range | Baseline, mean (SD), range | T12, mean (SD), range | Change, mean (SD), range | |||
| CSDI (0–12) (nM = 60, np = 65) | 7.48 (2.36), 3 to 12 | 5.05 (2.91), 1 to 12 | –2.43 (2.84), –9 to 5 | 7.03 (2.13), 2 to 12 | 5.77 (2.52), 1 to 12 | –1.26 (2.15), –6 to 2 | –1.17 (–2.06 to –0.29), p = 0.01 | –1.00 (–1.83 to –0.16), p = 0.02 |
| ABC | ||||||||
| Irritability, agitation, crying (0–45) (nM = 64, nP = 68) | 16.64 (10.27), 0 to 36 | 13.52 (10.10), 1 to 37 | –3.13 (6.62), –23 to 11 | 15.53 (10.28), 0 to 36 | 13.62 (9.98), 0 to 40 | –1.91 (6.74), –21 to 20 | –1.21 (–3.52 to 1.09), p = 0.30 | –0.95 (–3.11 to 1.21), p = 0.38 |
| Lethargy, social withdrawal (0–48) (nM = 60, nP = 67) | 12.42 (9.55), 0 to 36 | 9.33 (8.31), 0 to 29 | –3.08 (6.15), –20 to 9 | 10.76 (8.77), 0 to 37 | 7.97 (7.45), 0 to 36 | –2.79 (6.01), –20 to 16 | –0.29 (–2.43 to 1.85), p = 0.79 | 0.29 (–1.54 to 2.12), p = 0.76 |
| Stereotypical behaviour (0–21) (nM = 64, nP = 69) | 6.06 (4.86), 0 to 18 | 5.06 (4.5), 0 to 16 | –1.00 (3.63), –8 to 10 | 5.03 (4.79), 0 to 18 | 4.29 (4.12), 0 to 17 | –0.74 (3.42), –11 to 12 | –0.26 (–1.47 to 0.95), p = 0.67 | 0.12 (–0.93 to 1.17), p = 0.82 |
| Hyperactivity, non- compliance (0–48) (nM = 64, nP = 68) | 23.48 (9.94), 0 to 46 | 18.55 (10.44), 1 to 42 | –4.94 (7.65), –23 to 11 | 21.94 (11.07), 0 to 42 | 18.90 (11.17), 0 to 44 | –3.04 (8.51), –29 to 25 | –1.89 (–4.69 to 0.90), p = 0.18 | –1.48 (–4.11 to 1.15), p = 0.27 |
| Inappropriate speech (0–12) (nM = 64, nP = 67) | 4.78 (3.17), 0 to 12 | 3.47 (2.74), 0 to 12 | –1.31 (2.70), –8 to 5 | 3.72 (3.20), 0 to 11 | 3.15 (3.24), 0 to 12 | –0.57 (2.17), –7 to 4 | –0.75 (–1.59 to 0.10), p = 0.08 | –0.37 (–1.14 to 0.39), p = 0.33 |
| PedsQL Family Impact Module | ||||||||
| HRQoL (0–100) (nM = 64, nP = 69) | 53.33 (17.46), 10.00 to 86.25 | 58.72 (20.84), 7.50 to 100 | 5.39 (14.70), –32.50 to 38.75 | 56.16 (17.97), 21.25 to 95.00 | 57.50 (20.60), 6.25 to 100 | 1.34 (15.72), –43.75 to 35.00 | 4.05 (–1.18 to 9.28), p = 0.13 | 3.52 (–1.62 to 8.66), p = 0.18 |
| Family functioning (0–100) (nM = 64, nP = 69) | 50.24 (21.31), 6.25 to 100 | 56.64 (23.60), 3.13 to 100 | 6.40 (16.88), –28.13 to 56.25 | 50.14 (22.78), 0 to 100 | 52.13 (23.70), 0 to 100 | 1.99 (14.56), –37.50 to 31.25 | 4.40 (–0.99 to 9.80), p = 0.11 | 4.42 (–0.82 to 9.67), p = 0.10 |
| Total (0–100) (nM = 64, nP = 69) | 51.53 (16.98), 8.33 to 86.81 | 56.85 (20.04), 11.11 to 98.61 | 5.32 (13.17), –25.00 to 36.11 | 54.05 (17.85), 17.36 to 94.44 | 55.40 (19.01), 21.53 to 96.53 | 1.36 (13.03), –38.19 to 33.33 | 3.96 (–0.53 to 8.46), p = 0.08 | 3.57 (–0.86 to 8.00), p = 0.11 |
| ESS (0–24) (nM = 62, nP = 66) | 6.68 (5.36), 0 to 24 | 5.42 (4.53), 0 to 17 | –1.26 (5.04), –16 to 9 | 6.85 (5.27), 0 to 21 | 7.11 (5.01), 0 to 21 | 0.26 (3.78), –12 to 9 | –1.52 (–3.07 to 0.04), p = 0.056 | –1.59 (–2.91 to –0.27), p = 0.019 |
The results of the CSDI showed that there was a statistically significant difference between the two treatment groups, with a small reduction favouring those children in the melatonin group. The adjusted difference (adjusting for baseline CSDI) was –1.00 (95% CI –1.83 to –0.16), indicating that parents thought that the frequency and duration of sleep problems had reduced after treatment with melatonin. However, it is questionable whether or not a 1-point reduction on a 12-point scale is clinically important; previous work has suggested that at least a 50% reduction in problems is considered by parents to be worthwhile. 45
The unadjusted results of the other instruments tended to favour melatonin but improvements were small. On average, a reduction of 4 points on the 100-point scale was estimated for each domain in the PedsQL Family Impact Module. The ESS demonstrated an average improvement of 1.5 points on the 24-point scale for melatonin compared with placebo, which was borderline for statistical significance in the unadjusted analysis but reached statistical significance in the adjusted analysis. The importance of the change is unknown although a change in scores of < 2 points has been used previously to indicate clinically insignificant change. 62 Of note is that the mean ESS scores of parents in the two treatment groups at baseline and T12 were all within the normal range, indicating that excessive daytime sleepiness was not prominent in this sample of parents. Consideration should be given to whether or not the size of the observed effect in part reflects the sensitivity of the instruments to detect a change within the short 12-week time frame between assessments.
Results of the daily global measure of parental perception of child’s sleep quality (the ‘smiley face’ scale) are provided in Table 9. Results are expressed as the percentage of night sleeps with which the parent was dissatisfied (faces 5–7) and the mean score. The results from this analysis showed that there was a reduction in the mean percentage of dissatisfied nights’ sleep and in the mean score for the melatonin group compared with the placebo group, but this difference failed to reach statistical significance.
| Global measure summary [‘smiley face’] | Melatonin (n = 52) | Placebo (n = 55) | Unadjusted difference, mean (95% CI), p-value | Adjusted difference, mean (95% CI), p-value | ||||
|---|---|---|---|---|---|---|---|---|
| Baseline, mean (SD), range | T12, mean (SD), range | Change, mean (SD), range | Baseline, mean (SD), range | T12, mean (SD), range | Change, mean (SD), range | |||
| % of night sleeps with which the parent was dissatisfied | 31.25 (34.25), 0 to 100 | 23.97 (33.22), 0 to 100 | –7.27 (35.45), –100 to 83.33 | 33.74 (35.26), 0 to 100 | 31.01 (36.30), 0 to 100 | –2.73 (36.94) | –4.54 (–18.44 to 9.35), p = 0.52 | –5.89 (–17.83 to 6.05), p = 0.33 |
| Mean score | 3.81 (1.21), 1.00 to 6.57 | 3.41 (1.41), 1.00 to 6.86 | –0.40 (1.39), –3.57 to 2.52 | 4.02 (1.27), 1.00 to 7.00 | 3.74 (1.24), 1.00 to 6.14 | –0.28 (1.37), –4.00 to 2.37 | –0.12 (–0.65 to 0.41), p = 0.65 | –0.24 (–0.70 to 0.23), p = 0.32 |
Biochemical and genetic investigations
Salivary melatonin assay
Tables 10 and 11 provide a summary of the results obtained for DLMO.
| Laboratory result | Melatonin | Placebo | Total |
|---|---|---|---|
| No samples | 15 | 8 | 23 |
| Insufficient samples | 12 | 11 | 23 |
| DLMO calculated exact time | 10 | 13 | 23 |
| ‘Probably’ between two times | 6 | 7 | 13 |
| ‘Not before’ a particular time | 18 | 29 | 47 |
| None | 9 | 8 | 17 |
| Laboratory result | Melatonin | Placebo | Total |
|---|---|---|---|
| No samples | 23 | 16 | 39 |
| Insufficient samples | 15 | 12 | 27 |
| DLMO calculated exact time | 2 | 18 | 20 |
| ‘Probably’ between two times | 0 | 3 | 3 |
| ‘Not before’ a particular time | 5 | 20 | 25 |
| None | 25 | 7 | 32 |
At T–1W the time to reach DLMO is shown in Figure 3 (excludes ‘none’ and ‘no samples’).
FIGURE 3.
Kaplan–Meier graph of time to reach DLMO at T–1W.
When producing the Kaplan–Meier curve, in those patients with results between two times, the latest time was taken.
It is of note that, at T+10W, only seven participants on melatonin provided samples from which DLMO time could be calculated compared with 41 for placebo. This difference between the two groups was not detected at T–1W. Of the samples classified as ‘none’ for DLMO at T+10W, 26 were possibly contaminated, two had a high baseline and four had a low volume (Table 12).
| Sample status | Melatonin | Placebo |
|---|---|---|
| Possibly contaminated | 23 | 3 |
| High baseline | 2 | 0 |
| Low volume | 0 | 4 |
Some of the contamination and high baseline values in the melatonin arm are likely to reflect children who are poor metabolisers and whose levels of exogenous melatonin had accumulated during the study. Unfortunately, limited numbers at T+10W prevent meaningful analysis of the impact of this phenomenon on treatment response.
For the genetic substudy we defined DLMO categories using quartiles [1: DLMO ≤ 1930; 2: 1930 < DLMO ≤ 2030; 3: 2030 < DLMO < 2200; 4: DLMO ≥ 2200 (or no melatonin peak)] or medians [1: DLMO < 2100; 2: DLMO ≥ 2100 (or no melatonin peak)].
Genetic investigations
After applying the quality control filters, genotype data were available for 125 individuals. The principal component analysis indicated that the genotyped cohort was relatively homogeneous, with only eight outliers (Figure 4). All analyses were performed both with and without the outliers, and this did not affect the results.
FIGURE 4.
Multidimensional scaling plot of the MENDS sample to identify outliers (x) according to their identity by state with their nearest neighbour.
Outcome 1(i): association between genetic variations and sleep-onset latency (by sleep diary at T0)
It was possible to assess this outcome for 106 of the genotyped patients. A Manhattan plot showing results for association with this outcome is given in Figure 5. The different colours in the Manhattan plots are used to distinguish between chromosomes. Each point on the plot represents the p-value for an individual SNP. The p-values (indicated on the y-axis) are log-transformed to base 10 for ease of interpretation. Thus, a p-value of 10–8 (which is typically taken as the threshold for significance in a genome-wide study) would have a value of 8 when log-transformed. None of the SNPs reached genome-wide significance.
FIGURE 5.
Manhattan plot for association with SOL at T0W.
Outcome 1(ii): association between genetic variations and amount of total sleep (by sleep diary at T0W)
It was possible to assess this outcome for 107 of the genotyped patients. A Manhattan plot showing results for association with this outcome is given in Figure 6. None of the SNPs reached genome-wide significance.
FIGURE 6.
Manhattan plot for association with amount of total sleep at T0W.
Outcome 2: association between genetic variations and melatonin levels and synthesis
Analyses as well as verification are still ongoing in these domains. There is still debate about the security of absolute melatonin levels based on single salivary melatonin assays.
Outcome 3: association between genetic variations and melatonin synthesis
These analyses are currently ongoing.
Outcome 4(i): association between genetic variations and difference in sleep-onset latency between T0W and T+12W
A Manhattan plot showing results for association with this outcome is given in Figure 7. One SNP [chromosome 4, rs17580458 in gene ANK2 (ankyrin 2)] reached genome-wide significance (p = 1.05 × 10–9).
FIGURE 7.
Manhattan plot for association between genetic variations and difference in SOL between T0W and T+12W.
Outcome 4(ii): association between genetic variations and difference in amount of total sleep between T0W and T+12W
A Manhattan plot showing results for association with this outcome is given in Figure 8. None of the SNPs reached genome-wide significance.
FIGURE 8.
Manhattan plot for association between genetic variations and difference in amount of total sleep between T0W and T+12W.
Investigation of association between melatonin levels and sleep disorders
A significant, positive correlation was found between DLMO, either classified in quartiles or in two categories as defined earlier, and SOL (Figures 9a and b, p = 0.04 and p = 0.02, respectively, Wilcoxon test). Children with a later DLMO displayed a longer SOL. In line with this finding, there was correlation between salivary melatonin concentration at 20:00 and SOL (Figure 9c, r2 = 0.16, p = 0.0006).
FIGURE 9.
Relationship between melatonin levels and sleep disorders. (a) DLMO quartiles against SOL displayed as box plots and histograms at baseline; (b) DLMO medians against SOL displayed as box plots and histograms at baseline; (c) salivary melatonin levels (ng/l) at 20:00 hours plotted against SOL at baseline with least squares regression line; (d) DLMO quartiles against sleep duration displayed as box plots and histograms at baseline; (e) DLMO medians against sleep duration displayed as box plots and histograms at baseline; and (f) salivary melatonin levels (ng/l) at 20:00 hours plotted against sleep duration at baseline with least squares regression line.
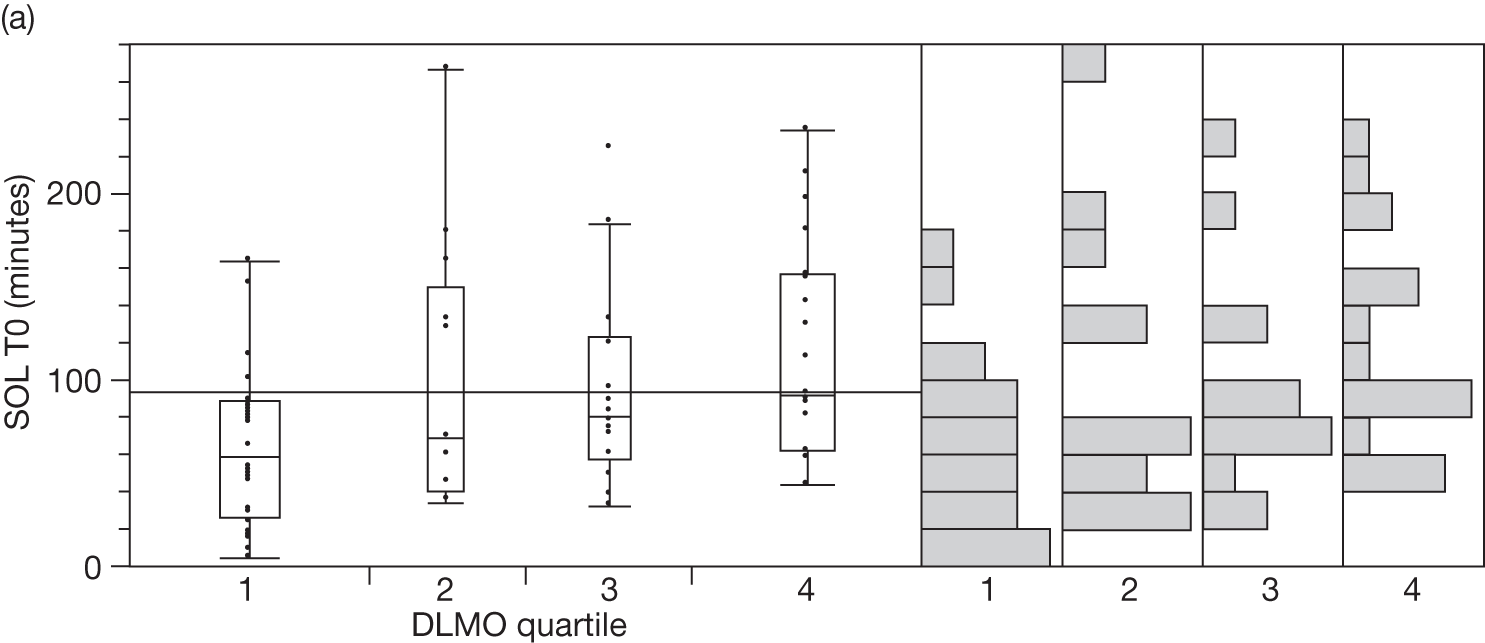
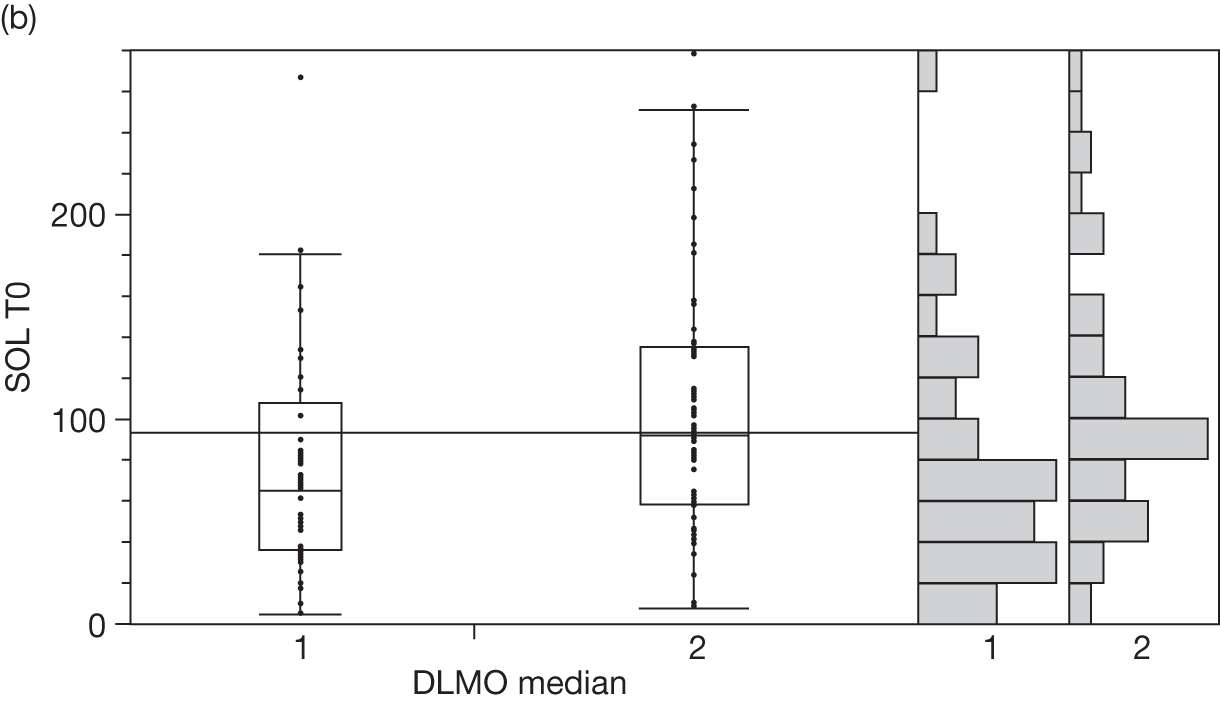
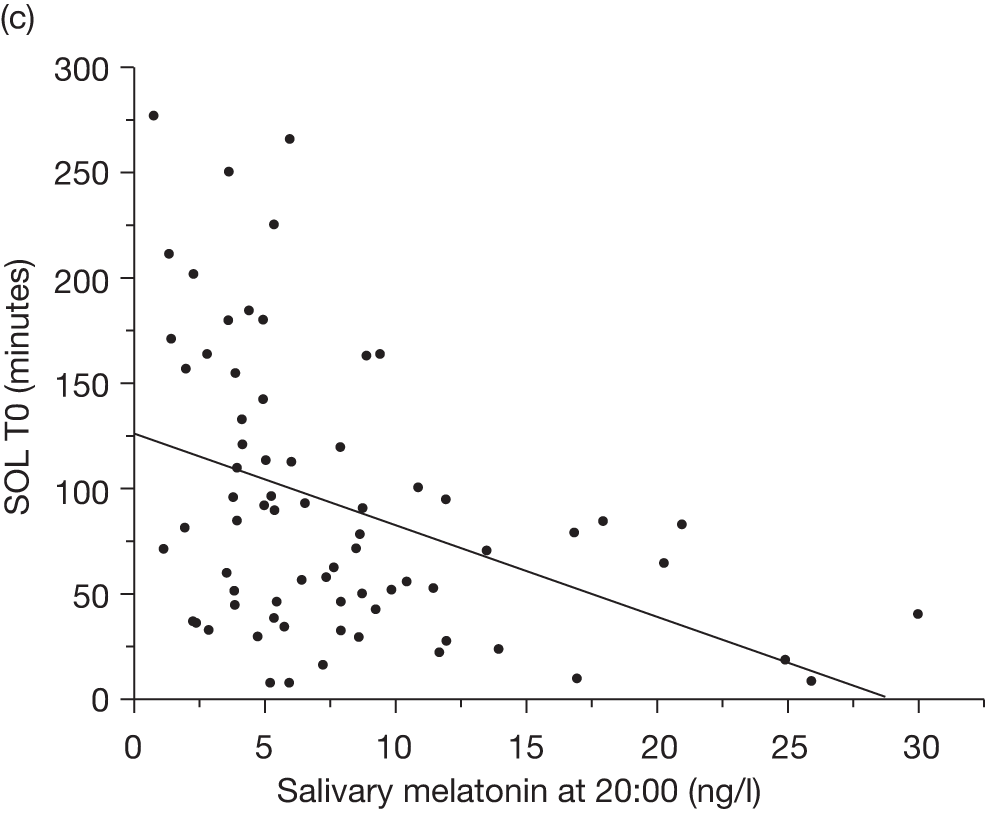
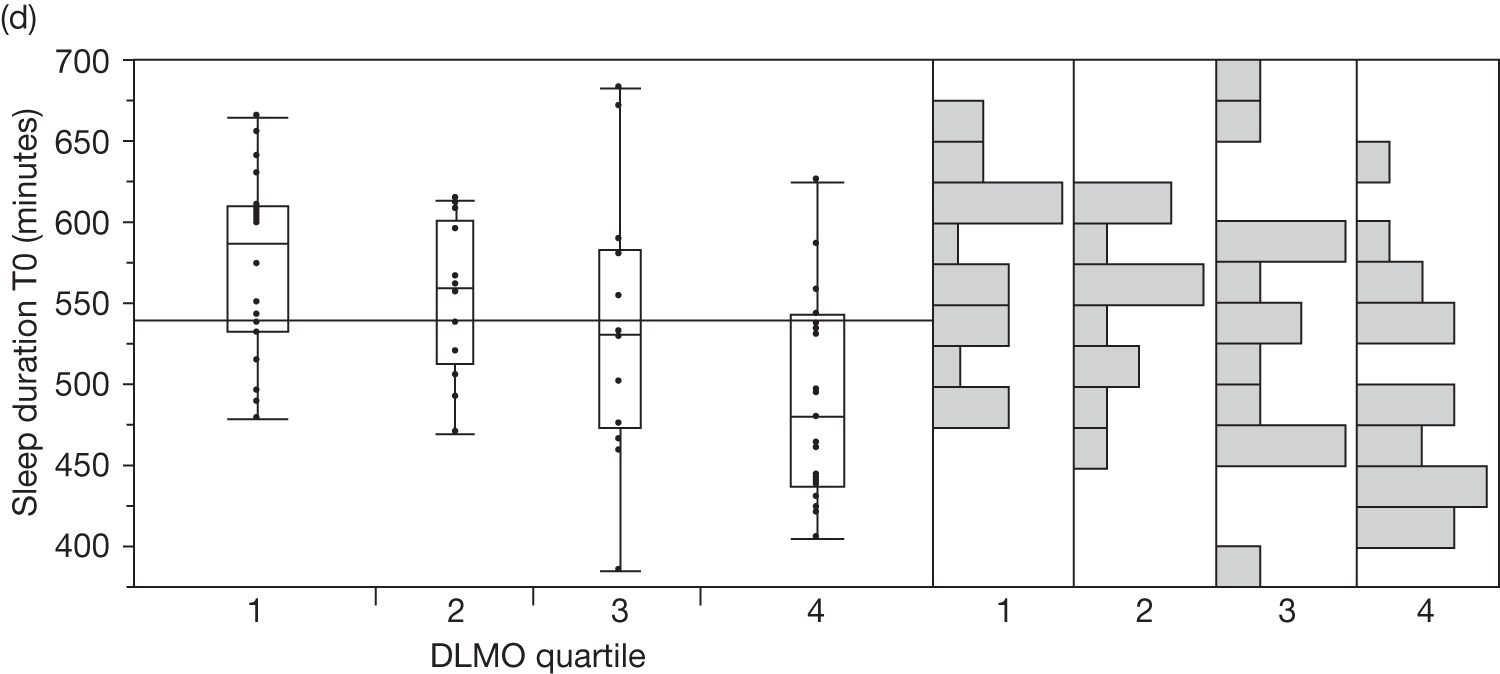
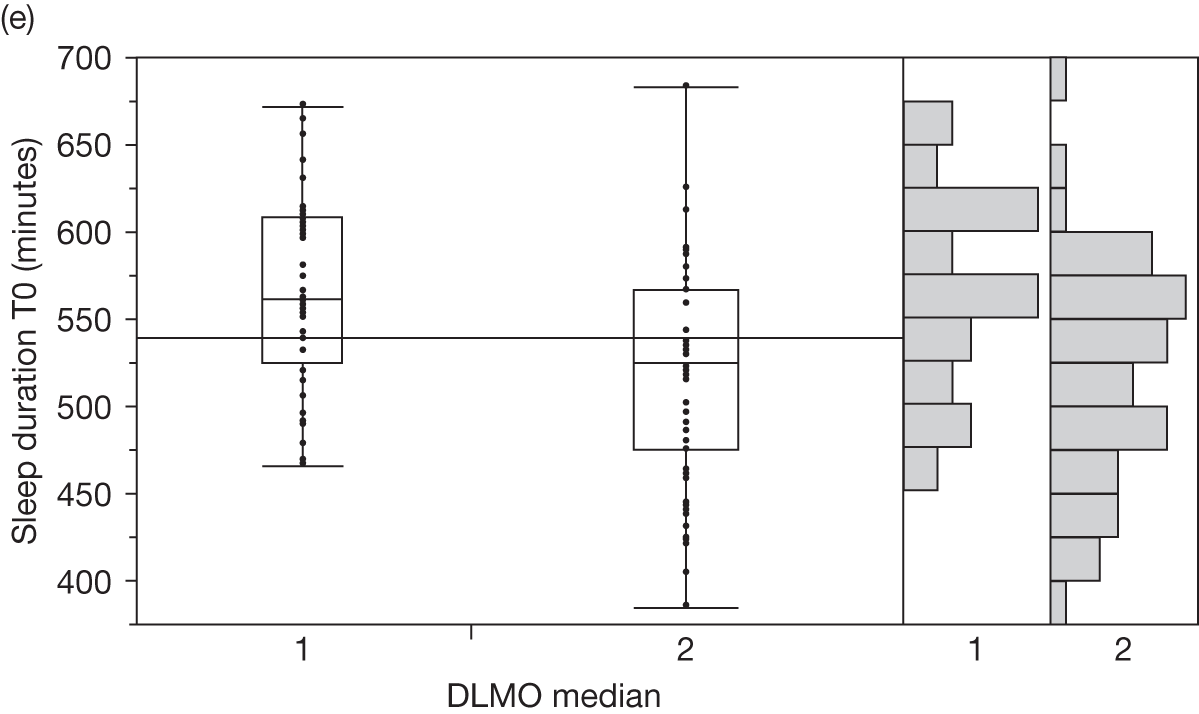
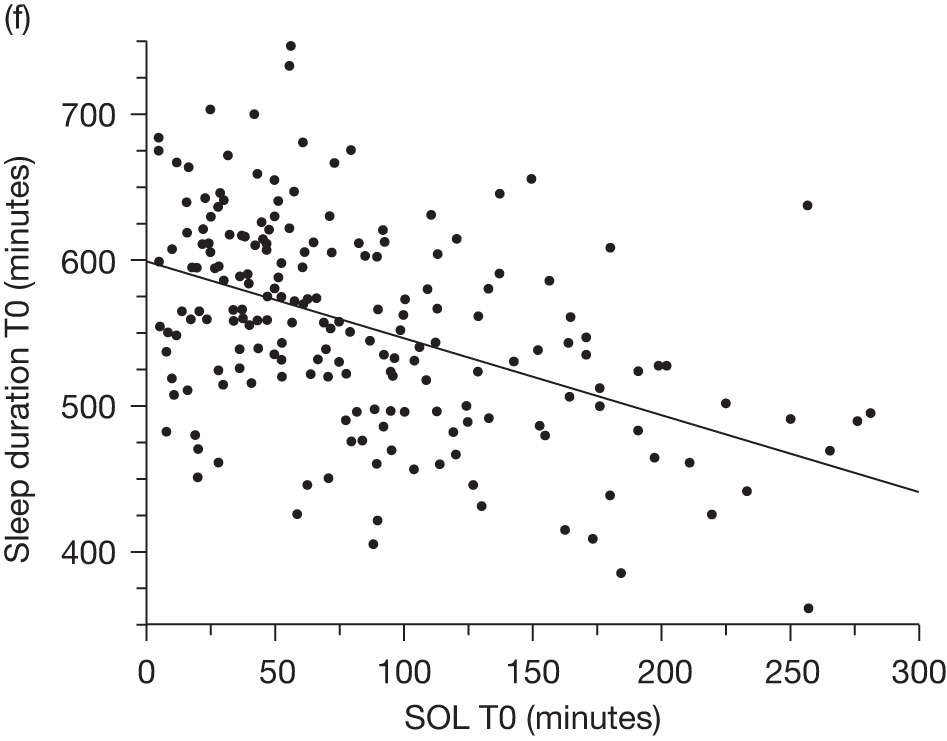
Conversely, DLMO was significantly negatively correlated with amount of total sleep (Figures 9d and e, p = 0.003 and p = 0.001, respectively, Wilcoxon test). It has to be noted that SOL and sleep duration are two correlated parameters (Figure 9f, r2 = 0.22, p < 0.0001).
Copy-number variant detection
Detection of CNVs was highly variable, depending on the genotyping array used. Chromosomal aneuploidy and known deleterious CNVs were present in several patients. Among the main findings, five patients had Down syndrome, one had DiGeorge syndrome, one had Cornelia de Lange syndrome and one had Smith–Magenis syndrome. Several CNVs were identified that affect genes known to be associated with developmental delay and/or ASD as well as genes involved in the clock/circadian pathway. Validation of these CNVs is in progress.
Exomic sequencing of AANAT and ASMT
Deoxyribonucleic acid of sufficient quality for sequencing was available for 134 individuals. The sequencing phase is completed for the ASMT gene. Four individuals were identified as carrying non-synonymous variations (Table 13). Two novel variations, G32C and H264D, were identified and a previously reported damaging mutation N17K was observed in two female patients. The G32C mutation is predicted to be ‘probably damaging’ whereas the H246D mutation is predicted to be benign. The overall frequency of ASMT mutations among the MENDS patients (0.029) does not seem to be different from that observed in the general population.
| Patient ID | Sex | Gene | Mutation | Validation of mutation | Predicted impact of mutation (Polyphen2 or in vitro) | Mutation previously described | Diagnostic | Trial arm |
|---|---|---|---|---|---|---|---|---|
| 056040 | Male | AANAT | T3M | In progress | Benign | Yes | Developmental delay | Melatonin |
| 056041 | N/A | AANAT | T3M/A163V | In progress | Benign | Yes | N/A | Not in RCT |
| 002002 | Male | AANAT | A163V | In progress | Benign | Yes | Developmental delay | Melatonin |
| 133011 | Female | ASMT | N17K | Validated | Damaging | Yes | Developmental delay | Placebo |
| 056014 | Female | ASMT | N17K | In progress | Damaging | Yes | Developmental delay and ASD | Melatonin |
| 056004 | Male | ASMT | G32C | In progress | Damaging | No | Developmental delay with ‘other’ | Melatonin |
| 230003 | Female | ASMT | H264D | In progress | Benign | No | Developmental delay with ‘other’ | Melatonin |
The sequencing phase is in progress for the AANAT gene but preliminary results are available for 77 patients. Three individuals were identified as carrying non-synonymous variations (see Table 13). Two patients were both carriers of a single variation, either T3M or A163V. A further patient was carrying two variations, T3M and A163V. The A163V mutation has been previously described and shown to moderately affect protein function. The T3M mutation is predicted to be benign. All patients with ASMT or AANAT mutations and treated with melatonin responded to treatment (Table 14).
| Patient | Variant | DLMO | Sleep disorders | Allocation | Melatonin response | ||
|---|---|---|---|---|---|---|---|
| Delayed sleep onseta | Short sleep durationb | Sleep onset | Sleep duration | ||||
| 133011 | ASMT N17K | 1830 | Yes | No | Placebo | N/A | N/A |
| 056014 | ASMT N17K | Not detected, last sample 2000 | Yes | Yes | Melatonin | Yes | Yes |
| 230003 | ASMT H264D | Not measured | No | Yes | Melatonin | N/A | Yes |
| 082007 | ASMT CNV | Not detected, last sample 2100 | Yes | No | Melatonin | Yes | N/A |
| 056040 | AANAT T3M | Not measured | Yes | Yes | Melatonin | Yes | Yes |
Detection of ASMT copy-number variants
The presence of the ASMT CNV was assessed in 126 patients. Three of them were carriers of the CNV (Table 15). The overall frequency of this CNV among the MENDS patients (0.024) does not seem to be different from that observed in the general population (0.036, unpublished data, p = 0.80). One patient carrying the ASMT CNV was treated with melatonin and responded to treatment (see Table 14).
| Patient ID | Sex | Diagnosis | Treatment allocation |
|---|---|---|---|
| 082007 | Male | Developmental delay with ‘other’ | Melatonin |
| 056043 | Female | Developmental delay with ‘other’ | Not in RCT |
| 056007 | Male | Developmental delay with ‘other’ | Not in RCT |
Safety outcomes
Number and severity of seizures evaluated using seizure diaries throughout trial follow-up
A total of 16 children (eight melatonin and eight placebo) had a diagnosis of epilepsy before randomisation. Thirteen children experienced seizures in the period between randomisation and the end of the study. There were a total of 411 seizures post randomisation and 123 seizures pre randomisation.
Table 16 shows the number of seizures by type and the number of children experiencing the seizure for each treatment group for both pre and post randomisation. Severity data were not recorded. The pre-randomisation data were based on seizure activity in a 4-week period and the post-randomisation data were based on seizure activity in a 12-week period. No formal statistical analyses were undertaken because of the limited data, particularly on the different seizure types.
| Participant allocation | Seizure type | Pre randomisation | Post randomisation | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Absence | Myoclonic | Other | Partial/focal | Tonic–clonic | Absence | Myoclonic | Other | Partial/focal | Tonic–clonic | ||
| 1. Melatonin | PWGTC | 31 | 0 | 0 | 0 | 0 | 152 | 0 | 0 | 0 | 0 |
| 2. Melatonin | PWGTC | 9 | 0 | 0 | 0 | 0 | 43 | 0 | 0 | 0 | 0 |
| 3. Melatonin | Generalised: atypical absence | 8 | 0 | 0 | 0 | 0 | 11 | 0 | 0 | 0 | 0 |
| 4. Placeboa | PWGTC | 1 | 1 | 0 | 0 | 0 | 0 | 2 | 9 | 0 | 0 |
| 5. Placebo | Generalised: atypical absence, atonic, tonic–clonic | 1 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 1 | 5 |
| 6. Melatonin | PWGTC | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 |
| 7. Placebo | Partial | 0 | 0 | 0 | 56 | 0 | 0 | 0 | 0 | 138 | 0 |
| 8. Placebo | Generalised: tonic–clonic | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 8 |
| 9. Melatonin | Generalised: tonic–clonic | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 |
| 10. Placebo | Information missing | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 23 | 0 | 0 |
| 11. Melatonin | PWGTC | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 |
| 12. Placebo | Generalised: atypical absence, atonic, tonic–clonic | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 1 |
| 13. Placebob | Generalised: atypical absence, atonic, tonic–clonic | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 |
| 14. Melatonin | Generalised: tonic–clonic | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 15. Melatonin | Generalised: tonic–clonic | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 16. Placebo | PWGTC | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ||
Treatment-emergent signs and symptoms
Adverse effects were assessed weekly between baseline and the final visit using TESS.
The numbers (and percentages) of participants experiencing each aspect of TESS are presented for each treatment arm in Table 17. Table 18 presents TESS categorised by severity. For each participant only the maximum severity experienced of each symptom is displayed. Originally there were 14 aspects of TESS (somnolence, increased excitability, mood swings, seizures, rash, hypothermia, cough, increased activity, dizziness, hung-over feeling, tremor, vomiting, nausea and breathlessness); however, on 27 April 2009 TESS was reduced to seven domains (somnolence, increased excitability, mood swings, seizures, rash, hypothermia and cough).
| Event | Melatonin (n = 70) | Placebo (n = 76) | Total (n = 146) | |||
|---|---|---|---|---|---|---|
| No. (%) of patients | Events | No. (%) of patients | Events | No. (%) of patients | Events | |
| Prompted adverse events reported (TESS) | ||||||
| Somnolence | 9 (12.9) | 14 | 10 (13.2) | 13 | 19 (13) | 27 |
| Increased excitability | 13 (18.6) | 23 | 16 (21.1) | 19 | 29 (19.9) | 42 |
| Mood swings | 16 (22.9) | 34 | 17 (22.4) | 25 | 33 (22.6) | 59 |
| Seizures | 0 (0) | 0 | 1 (1.3) | 1 | 1 (0.7) | 1 |
| Rash | 11 (15.7) | 17 | 8 (10.5) | 10 | 19 (13) | 27 |
| Hypothermia | 6 (8.6) | 8 | 4 (5.3) | 4 | 10 (6.8) | 12 |
| Coughing | 22 (31.4) | 36 | 28 (36.8) | 42 | 50 (34.2) | 78 |
| Increased activitya | 6 (8.6) | 12 | 9 (11.8) | 13 | 15 (10.3) | 25 |
| Dizzinessa | 1 (1.4) | 2 | 5 (6.6) | 6 | 6 (4.1) | 8 |
| Hung-over feelinga | 1 (1.4) | 1 | 0 (0) | 0 | 1 (0.7) | 1 |
| Tremora | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 |
| Nauseaa | 3 (4.3) | 3 | 11 (14.5) | 13 | 14 (9.6) | 16 |
| Vomitinga | 15 (21.4) | 29 | 18 (23.7) | 32 | 33 (22.6) | 61 |
| Breathlessnessa | 1 (1.4) | 2 | 1 (1.3) | 1 | 2 (1.4) | 3 |
| Unprompted adverse events spontaneously reported | ||||||
| Fatigue | 8 (11.4) | 14 | 8 (10.5) | 10 | 16 (11) | 24 |
| Headache | 10 (14.3) | 12 | 7 (9.2) | 14 | 17 (11.6) | 26 |
| Other | 31 (44.3) | 82 | 40 (52.6) | 107 | 71 (48.6) | 189 |
| Event | Severity | No. of events | No. (%) of patientsa | ||||
|---|---|---|---|---|---|---|---|
| Melatonin | Placebo | Total | Melatonin | Placebo | Total | ||
| Somnolence | Mild | 7 | 7 | 14 | 5 (7.1) | 5 (6.6) | 10 (6.8) |
| Moderate | 5 | 5 | 10 | 3 (4.3) | 4 (5.3) | 7 (4.8) | |
| Severe | 1 | 1 | 2 | 1 (1.4) | 1 (1.3) | 2 (1.4) | |
| Increased excitability | Mild | 13 | 15 | 28 | 8 (11.4) | 12 (15.8) | 20 (13.7) |
| Moderate | 8 | 4 | 12 | 3 (4.3) | 4 (5.3) | 7 (4.8) | |
| Severe | 2 | 0 | 2 | 2 (2.9) | 0 (0) | 2 (1.4) | |
| Mood swings | Mild | 15 | 14 | 29 | 8 (11.4) | 10 (13.2) | 18 (12.3) |
| Moderate | 14 | 8 | 22 | 5 (7.1) | 5(6.6) | 10 (6.8) | |
| Severe | 5 | 2 | 7 | 3 (4.3) | 2 (2.6) | 5 (3.4) | |
| Seizures | Mild | 0 | 0 | 0 | 0 (0) | 0 (0) | 0 (0) |
| Moderate | 0 | 1 | 1 | 0 (0) | 1 (1.3) | 1 (0.7) | |
| Severe | 0 | 0 | 0 | 0 (0) | 0 (0) | 0 (0) | |
| Rash | Mild | 15 | 10 | 25 | 9 (12.9) | 8 (10.5) | 17 (11.6) |
| Moderate | 2 | 0 | 2 | 2 (2.9) | 0 (0) | 2 (1.4) | |
| Severe | 0 | 0 | 0 | 0 (0) | 0 (0) | 0 (0) | |
| Hypothermia | Mild | 8 | 4 | 12 | 6 (8.6) | 4 (5.3) | 10 (6.8) |
| Moderate | 0 | 0 | 0 | 0 (0) | 0 (0) | 0 (0) | |
| Severe | 0 | 0 | 0 | 0 (0) | 0 (0) | 0 (0) | |
| Coughing | Mild | 28 | 35 | 63 | 15 (21.4) | 22 (28.9) | 37 (25.3) |
| Moderate | 6 | 7 | 13 | 5 (7.1) | 6 (7.9) | 11 (7.5) | |
| Severe | 2 | 0 | 2 | 2 (2.9) | 0 (0) | 2 (1.4) | |
| Increased activitya | Mild | 5 | 11 | 16 | 3 (4.3) | 7 (9.2) | 10 (6.8) |
| Moderate | 6 | 2 | 8 | 2 (2.9) | 2 (2.6) | 4 (2.7) | |
| Severe | 1 | 0 | 1 | 1 (1.4) | 0 (0) | 1 (0.7) | |
| Dizzinessa | Mild | 1 | 5 | 6 | 0 (0) | 4 (5.3) | 4 (2.7) |
| Moderate | 0 | 1 | 1 | 0 (0) | 1 (1.3) | 1 (0.7) | |
| Severe | 1 | 0 | 1 | 1 (1.4) | 0 (0) | 1 (0.7) | |
| Hung-over feelinga | Mild | 1 | 0 | 1 | 1 (1.4) | 0 (0) | 1 (0.7) |
| Moderate | 0 | 0 | 0 | 0 (0) | 0 (0) | 0 (0) | |
| Severe | 0 | 0 | 0 | 0 (0) | 0 (0) | 0 (0) | |
| Tremora | Mild | 0 | 0 | 0 | 0 (0) | 0 (0) | 0 (0) |
| Moderate | 0 | 0 | 0 | 0 (0) | 0 (0) | 0 (0) | |
| Severe | 0 | 0 | 0 | 0 (0) | 0 (0) | 0 (0) | |
| Nauseaa | Mild | 3 | 12 | 15 | 3 (4.3) | 10 (13.2) | 13 (8.9) |
| Moderate | 0 | 0 | 0 | 0 (0) | 0 (0) | 0 (0) | |
| Severe | 0 | 1 | 1 | 0 (0) | 1(1.3) | 1 (0.7) | |
| Vomitinga | Mild | 29 | 31 | 60 | 15 (21.4) | 17 (22.4) | 32 (21.9) |
| Moderate | 0 | 1 | 1 | 0 (0) | 1 (1.3) | 1 (0.7) | |
| Severe | 0 | 0 | 0 | 0 (0) | 0 (0) | 0 (0) | |
| Breathlessnessa | Mild | 2 | 1 | 3 | 1 (1.4) | 1 (1.3) | 2 (1.4) |
| Moderate | 0 | 0 | 0 | 0 (0) | 0 (0) | 0 (0) | |
| Severe | 0 | 0 | 0 | 0 (0) | 0 (0) | 0 (0) | |
| Unprompted adverse events spontaneously reported | |||||||
| Fatigue | Mild | 11 | 9 | 20 | 6 (8.6) | 7 (9.2) | 13 (8.9) |
| Moderate | 3 | 1 | 4 | 2 (2.9) | 1 (1.3) | 3 (2.1) | |
| Severe | 0 | 0 | 0 | 0 (0) | 0 (0) | 0 (0) | |
| Headache | Mild | 11 | 14 | 25 | 9 (12.9) | 7 (9.2) | 16 (11) |
| Moderate | 1 | 0 | 1 | 1 (1.4) | 0 (0) | 1 (0.7) | |
| Severe | 0 | 0 | 0 | 0 (0) | 0 (0) | 0 (0) | |
| Other | Mild | 60 | 87 | 147 | 17 (24.3) | 30 (39.5) | 47 (32.2) |
| Moderate | 19 | 16 | 35 | 12 (17.1) | 8 (10.5) | 20 (13.7) | |
| Severe | 3 | 2 | 5 | 2 (2.9) | 2 (2.6) | 4 (2.7) | |
No formal statistical testing was undertaken on these data.
From a careful evaluation of the data in Tables 17 and 18, there did not appear to be any major events in either of the treatment groups and no obvious differences between the two treatment groups.
Serious adverse events and suspected unexpected serious adverse reactions
There were five serious adverse events and two suspected unexpected serious adverse reactions during the course of the trial. Two serious adverse events were deemed to be unrelated and three unlikely to be related. There was one suspected unexpected serious adverse reaction in each treatment group. Details are given in Table 19.
| Treatment allocation | Visit | Description | Seriousness | Severity | Expectedness | Relationship | Cause | Outcome | Patient status | Unblinded | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Melatonin | T+4W home visit | Waking up in the night because of nightmares | Medically significant/important | Moderate | Unexpected | Probably | – | Resolved | Patient continuing in trial | SUSAR was unblinded at the CTU but site was not unblinded |
| 2 | Placebo | T+12W | Dislocated elbow in accident at school | Required hospitalisation | Moderate | Unexpected | Unrelated | Other illness | Ongoing at final follow-up | Patient continuing in trial | No |
| 3 | Placebo | T+12W | Petechiae covering the dorsum of the right hand | Medically significant/important | Mild | Unexpected | Possibly | – | Ongoing at final follow-up | Patient withdrew from trail | SUSAR was unblinded at the CTU but site was not unblinded |
| 4 | Melatonin | Unscheduled telephone call (between T+9W and T+10W) | Severe irritation to skin | Medically significant/important | Severe | Unexpected | Unlikely | Other illness | Resolved | Patient continuing in trial | No |
| 5 | Placebo | T+3W home visit | Choking on dinner | Medically significant/important | Moderate | Unexpected | Unrelated | Other illness | Resolved | Patient continuing in trial | No |
| 6 | Placebo | T+11W home visit | Vomiting caused by viral illness, which caused dehydration | Required hospitalisation | Moderate | Expected | Unlikely | Other illness | Resolved | Patient continuing in trial | No |
| 7 | Melatonin | T+11W home visit | Seizure | Required hospitalisation | Moderate | Unexpected | Unlikely | Other illness | Resolved | Patient continuing in trial | No |
Withdrawals
There were a total of 13 withdrawals from the trial: seven in the placebo group and six in the melatonin group. The reasons for withdrawal are shown in Table 20 by time point. Three participants provided data following withdrawal; these are indicated by an asterisk.
| Treatment allocation | Reason for withdrawal from the study | Time point (weeks) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| T0 | T+1 | T+2 | T+3 | T+4 | T+5 | T+6 | T+7 to T+10 | T+11 | T+12 | ||
| Melatonin | Persistent intractable night-time awakenings | ✗ | |||||||||
| Placebo | Medication not working | ✗ | |||||||||
| Melatonin | Did not attend T+12W visit on more than one occasion | ✗ | |||||||||
| Placebo | Serious adverse event: petechiae with lowered platelet count | ✗ | |||||||||
| Melatonin | Concerns regarding diary completion | ✗ | |||||||||
| Placebo | Too much going on at home | ✗ | |||||||||
| Placebo | Adverse event: severe mood swings | ✗ | |||||||||
| Placebo | Adverse event: increased excitability | ✗ | |||||||||
| Melatonin | Patient withdrew consent completely at T+1W. This patient was not contactable at T–2W or in the time between T–2W and T0W. Patient arrived at clinic at T0W with diaries for T–4W to T–2W, which were mistaken for diaries for T–2W to T0W. Patient was randomised but then could not be contacted for further visits | ✗ | |||||||||
| Placebo | Patient stopped at T+8W as the parents felt that sleep was not improving. They agreed to fill in the last week of sleep diaries for T+11W to T+12W, which would then provide data for the primary outcome | ✗ | |||||||||
| Melatonin | Father did not want child taking the medication | ✗ | |||||||||
| Placebo | No contact could be made after the T+4W visit | ✗ | |||||||||
| Melatonin | Adverse event: hallucinations | ✗ | |||||||||
Chapter 4 Discussion
The results of a systematic review published in 200436 and a meta-analysis published in 200923 recommended that a methodologically sound, adequately powered and placebo-controlled randomised trial of melatonin should be undertaken in children with ‘neurological problems, neurodevelopmental disabilities or intellectual disability’. 23 A more recent meta-analysis of children with ASD has further endorsed this earlier recommendation. 24
Main findings
Primary outcome
The MENDS study is the largest RCT of melatonin in children with neurodevelopmental disabilities, including children with ASD, which was powered to detect a minimum clinically worthwhile change in TST of 1 hour. The main findings based on a blinded evaluation of the primary end point of mean change in TST at 12 weeks compared with baseline, measured using sleep diaries, showed that melatonin does increase TST but the increase is not clinically worthwhile. The upper limit of the CI was < 1 hour, the minimum clinically worthwhile difference specified at the outset of the trial.
The trial included a heterogeneous group of children covering a wide age range. Of the 10 children who did achieve a 1-hour increase in TST there were six on placebo and four on melatonin.
Other outcomes
Sleep-onset latency measured using actigraphy was a primary outcome at the outset of the trial but became a secondary outcome because of the large proportion of missing data; however, SOL remains an important end point for which a reduction of > 30 minutes at 12 weeks compared with baseline was considered to be clinically worthwhile. The results were both clinically important and statistically significant; however, the mean size of the effect was approximately 10 minutes larger when SOL was measured using actigraphy than when it was measured using sleep diaries. The CIs of SOL measured using actigraphy and measured using sleep diaries largely overlapped and each contained clinically worthwhile values. The differences in the results may be a reflection of the subgroup of children who were able to wear actigraphy monitors or differences between the subjective and objective methods of measuring sleep, or both.
Changes in the 12-week scores for each of the sleep and behaviour questionnaires were small but the direction of the change tended to favour melatonin. The magnitude of the change may be a reflection of the sensitivity of the instruments to change over a 12-week period, or the improvements in sleep seen were not sufficient to impact on the domains being assessed. With each of the instruments there is a lack of guidance on the size of change that would be considered worthwhile.
This study raised no safety concerns in relation to the medication, at least in the short term, with only five serious and two suspected unexpected serious adverse reactions, none of which was considered to be related to the active medication (melatonin). Seizure exacerbation was not seen in the children with an established diagnosis of epilepsy. However, because of the small numbers involved in the trial no firm conclusions can be drawn from the study.
Genetic investigations
Among the main findings of the genetic study, specific genetic disorders were identified in five patients with Down syndrome and one each with DiGeorge syndrome, Cornelia de Lange syndrome and Smith–Magenis syndrome; none of these disorders had been diagnosed in these patients before enrolling in MENDS. Several CNVs that affect genes known to be associated with developmental delay and/or ASD as well as genes involved in the clock/circadian pathway were detected and the validation of these CNVs is in progress. Partial deletions in the ASMT gene involving exons 1–7 have been described as occurring more frequently in patients with ASD than in the general population. 60 The sequencing phase for ASMT is complete and four individuals were identified as carrying non-synonymous variations. The sequencing phase for AANAT is still in progress with three non-synonymous variations identified thus far. The rates of these findings in our study population did not exceed those in the general population. It is of interest that all patients with ASMT or AANAT mutations treated with melatonin responded to treatment.
Strengths and weaknesses
Design
The MENDS study was designed as a parallel RCT, although many of the previous RCTs that had assessed the use of melatonin (against placebo) in children had utilised a crossover design. We believe that the possible residual effect of either a behavioural or a melatonin intervention on sleep patterns and circadian rhythm make this particular intervention poorly suited for a crossover study.
The MENDS study incorporated a Dose escalation phase during the first 4 weeks of the study following randomisation. Although this cannot be considered a dose-ranging study, it demonstrated that patients who received placebo titrated more rapidly up to the maximum dose and also remained on the maximum dose at the end of the double-blind phase of the study. However, these results do not allow us to conclude which dose is the most effective.
The definition of sleep disorder used within MENDS was broad and did not make allowances for the age of the child in determining whether or not they had a sleep disorder. Data were not collected on the specific nature of the clinical diagnosis of the sleep disorder at baseline (delayed sleep onset or poor TST, or a combination of both) and this is being collected retrospectively to facilitate secondary analyses.
The length of follow-up post randomisation was short and this may have impacted on the ability of the quality-of-life instruments to detect change. Longer-term follow-up should be considered for future studies and use appropriate quality-of-life tools as well as qualitative interviews to fully explore the impact of impaired (and improved) sleep on family life.
Recruitment and retention
Recruitment into the trial was slower than expected. This may have reflected the widespread availability and prescribing of melatonin outside of the trial and its perceived excellent safety profile. The sample size target for the primary outcome TST was achieved over an extended recruitment period. The trial was supported by the MCRN LRNs and this aided trial recruitment, motivation at sites and data collection. The trial showed a low withdrawal rate such that once participants were randomised into the trial they continued until the completion of the follow-up period. This is considered to reflect not only the commitment of the families and the motivation and hard work of the research practitioners, but also the design of the study, which maintained weekly contact with the families, an important activity that was supported by the LRNs.
Outcomes
The study was ambitious in its goals in that the initial design had two primary outcomes and a large number of secondary outcomes. The decision to use two primary outcomes reflected the perceived characteristic sleep problems in children with neurodevelopmental delay, namely their difficulty in falling asleep (measured by actigraphy) but also their difficulty in sleeping continuously throughout the night (measured by sleep diaries); many children experience both problems.
The trial used both subjective and objective measures of sleep as recommended by Sadeh. 47 There were potential benefits of each approach and underlying reasons why the results would not be expected to be concordant. These included that the sleep diaries would not detect periods when the child was awake but not disturbing the household (a particular concern for determining SOL), and that the actigraph would interpret restless sleep as being awake (a concern for children with motor problems, including cerebral palsy). Dayyat et al. 63 have also considered these objective and subjective measures and their findings concur with our observed differences when measuring SOL and TST using actigraphy and sleep diaries.
It was clear that many children were unable to co-operate with wearing actigraphs and these missing actigraphy data were compounded by a significant rate of actigraph failure. Consequently, SOL measured using actigraphy was moved to a secondary outcome.
The impact on generalisability of the actigraphy outcomes needs to be considered when interpreting results; however, the results were largely consistent with the results for the same outcomes calculated using data from sleep diaries.
The ability of the potentially sleep-deprived families to complete the sleep diaries continuously over the 16-week period required was a concern. The completion rates of the diaries were monitored, and improvements made to the sleep diary to highlight key fields led to improvements in their completion. Overall, completion of sleep diaries following the amendment was very good and parents did complete diaries for the full 16 weeks. This trial further demonstrates the acceptability of such data collection to parents and carers, as has been reported previously. 64
Parents and carers were requested to obtain saliva samples from the children for salivary melatonin assays to determine the time of DMLO. The salivary melatonin analysis was undertaken primarily as an exploratory or hypothesis-generating approach. This was an attempt to enable biochemical phenotyping of those children with a genuinely delayed sleep phase and who might be expected to be better responders to melatonin. However, the limitations of trying to collect saliva in the home and without causing the children any distress were reflected in the limited data obtained. Therefore, it is very difficult to interpret our salivary melatonin data. The salivary melatonin results of those children who do not display a DLMO before sampling stopped may reflect a delayed DLMO or a lack of melatonin production or a combination of both; unfortunately, we are not able to say which is more likely.
The number of sleep awakenings was not a prespecified end point of the study as the impact of night awakenings was expected to impact on TST. However, this end point will be considered in secondary analyses.
We did not measure ‘before and after’ cognitive abilities of the children in this study. Experimentally, sleep restriction can affect certain cognitive processes and we might arguably have been expected to have found changes in these abilities. However, many of the children in this study were low or very low functioning academically, and IQ tests are less reliable for this population and require experienced testers to perform reliable and interpretable assessments. Our compromise was to try to use the computer-based, picture-formatted and child-specific MARS test but even this was too difficult to undertake for most of the children. Participation (compliance) was so poor that the MARS test was omitted from the battery of secondary outcomes.
Genotyping
Because of the use of several different genotyping arrays, it was only possible to undertake analyses of association on approximately 200,000 SNPs that were common to all arrays. Genotype imputation is due to start shortly in order to increase the number of SNPs investigated, and thus coverage of the genome.
Comparison with other studies
A number of systematic reviews with meta-analyses are available that include trials relevant to the MENDS population. 23,24,28,35,36,65 There are many differences between these systematic reviews, including the robustness of the eligibility criteria for inclusion of the trials and heterogeneity between the trials. A relevant protocol for a systematic review of randomised evidence of melatonin in children with neurodevelopmental disorders,66 poignant to the MENDS trial, is published on The Cochrane Library and, following publication, the MENDS trial will be included in an update.
The most relevant systematic reviews with meta-analyses23,24 have been published since the inception of MENDS. Although MENDS had a primary focus on children with neurodevelopmental disorders, approximately one-third had ASD. We have therefore also carefully studied the recently published meta-analysis by Rossignol and Frye24 of melatonin use in individuals with ASDs. It is important to note that the meta-analyses by Braam et al. 23 and Rossignol and Frye24 contain overlapping studies, and the total number of participants in the included trials is small.
The MENDS study has shown that, with melatonin, total night-time sleep duration on average increased by 23 minutes, a clinically unimportant increase based on our prior power calculations, and SOL was reduced on average by 38 minutes when measured using sleep diaries. The meta-analysis of the effect of melatonin in individuals with intellectual disability23 found that melatonin increased TST by a mean of 50 minutes and reduced SOL by 34 minutes. Rossignol and Frye24 found a mean increase in TST of 44 minutes and a reduction in SOL of 39 minutes. The size of the effect for SOL is similar and consistent between MENDS and these meta-analyses; however, it is estimated that there is a larger average effect for TST in the meta-analyses than in MENDS.
We have some concerns about the robustness of these meta-analyses because of a number of shared methodological issues. Many of the issues of concern have been discussed in a critique of the Buscemi review67 and are applicable to the two meta-analyses. This included concerns around the reasons why studies were excluded, the differences between studies, the information reported, the quality/risk of bias of included studies, and outcome reporting bias.
All of the included studies in the Rossignol and Frye meta-analysis24 and seven of nine studies in the Braam et al. meta-analysis23 were crossover designs. It is likely that the primary reason for choosing this design was to reduce sample size. Unfortunately, the possible residual effects of either a behavioural or a melatonin intervention on sleep patterns and circadian rhythm make this particular intervention poorly suited for a crossover study. The reasons for the persistence of the effect of melatonin on body clock synchronisation are summarised in the meta-analysis of Braams et al. 23 The difficulties that this posed to the researchers are exemplified in their choice of variable washout periods of just 3 days to 1 month. There are simply inadequate data to be confident that a change in timing of sleep onset and offset will securely ‘wash out’ to baseline timings after any specified time period.
The actual definition of a sleep problem (nature, persistence or severity) justifying treatment with melatonin also varies widely between studies. In the study by Wirojanan et al. ,18 entry depended exclusively on parents reporting a sleep disorder. In the study by Wright et al. ,68 children were included who manifested any type of sleep problem that persisted after a behavioural intervention. In the study by Garstang et al. ,69 children were required to have a sleep latency of > 1 hour for inclusion. In addition, this last study was suspended after only seven children completed, because of the discovery that some placebo capsules were empty. Consequently, little can be concluded and therefore generalised about the nature of the underlying sleep problem from these few studies.
Different doses of different preparations of melatonin were used in most of the studies, with some relying on a fixed dose (from 2 mg to 9 mg) and others allowing dose escalation.
Very few of the studies included objective outcome measures such as actigraphy. In most studies outcome measures were largely based on subjective parental diaries. The lack of any objective measures in most studies, and differences between objective and subjective measures, was a problem acknowledged by the reviewers.
It is interesting that we found a similar magnitude of change in SOL as did a robust study on ‘normally’ developing children with sleep-onset insomnia. 70 This study also failed to demonstrate any significant increase for total sleep duration. The correlation between sleep latency and sleep duration identified in MENDS is important and supports a hypothesis that melatonin exerts its main effects by reducing sleep latency, and that this on its own would increase sleep duration if sleep offset (time of wakening) remains the same.
A recently published, randomised, placebo-controlled trial explored the dose of melatonin and response in typically (‘normally’) developing children aged from 6 to 12 years with sleep-onset insomnia. 68 The children received either melatonin (0.05 mg/kg, 0.1 mg/kg and 0.15 mg/kg) or placebo for 1 week (to allow some comparison of dose ranges, an average 9-year-old boy in MENDS on this regime weighing 30 kg would receive 1.5 mg, 3 mg or 4.5 mg, and no child would have received the 6-mg or 12-mg MENDS dose). The authors did not include sleep duration in their results having previously shown that this does not change in this group of children. 70 Even with these relatively low doses of melatonin, the authors did find that melatonin significantly advanced DLMO by approximately 60 minutes and decreased SOL by 35 minutes. They were unable to find any dose–response relationship, but did show that the circadian time of administration played a significant role. The fact that this study did not report on sleep duration, and all children were typically (‘normally’) developing, limits our ability to directly compare this study with MENDS. Clearly, the 6-mg and 12-mg doses in MENDS were much higher than the doses in this recent study. 68 In addition, their study predominantly focused on melatonin’s ability to phase shift DLMO and sleep onset, rather than its soporific effects, although these were briefly discussed. It is possible that, for children with often profound developmental delay, higher doses are required based on their sedating rather than phase-shifting role.
Generalisability
It is important to appreciate that, to date, MENDS is the largest RCT undertaken in children with neurodevelopmental delay and, although the population studied was heterogeneous, the results for both SOL and TST are similar to those reported for a total of 183 patients in the meta-analysis carried out in 2009. 23 These observations would suggest that the results of MENDS could reasonably be extrapolated to a much larger paediatric population with a range of neurological disorders, including neurodevelopmental delay.
The MENDS study was intended to be pragmatic and, as far as possible, to reflect usual clinical practice for this group of children. This strategy has resulted in strengths and weaknesses. We have attempted to faithfully mirror current clinical practice and believe our results are generalisable. However, the sheer heterogeneity of the population studied has inevitably limited our ability to accurately estimate the impact of melatonin treatment for individual groups of patients with specific clinical (genetic), behavioural or developmental presentations. To a certain extent, this also applies to the different preparations (formulations) of melatonin that are currently in use throughout the UK. At the time that this study was under design, no licensed slow-release preparations of melatonin were available. Reports at the time cast significant doubt and concern over the ‘slow-release’ properties of the available non-pharmaceutical-grade products. Although there is now one slow-release preparation available (Circadin), its slow-release properties rely on the tablet being swallowed whole, which was not a skill possessed by many of the children in the study population. In the future, more flexible slow-release preparations might be available. Head-to-head trials comparing fast- and slow-release preparations are warranted.
Conclusion
Interpretation
The results of MENDS have confirmed the results of previous studies which have shown that melatonin is effective in reducing SOL (the time taken to fall asleep) in children with neurodevelopmental delay, reducing this time by a mean of 45 minutes. The results of meta-analysis demonstrated that melatonin reduced SOL by a mean of 34 minutes. 29
A clinically significant increase in TST remains an important target for sleep interventions. Although the total sleep duration did not increase by 1 hour, we acknowledge that an increase in sleep duration of < 1 hour may still be clinically important. There is a lack of robust information to guide a clinical decision in this context about the importance of a small increase in total duration of night-time sleep. Much of the data that argue about the importance of sleep duration are based on experimental situations in which sleep duration is acutely and artificially reduced. In these experimental models, sleep has an impact on daytime learning and behaviours, although the size of this impact is unclear and inconsistent. 48 At a neurobiological level, some researchers speculate that, over ‘time’ (unspecified), sleep loss may cause actual neuronal loss. 71 If this hypothesis of the effect of cumulative sleep loss on neuronal loss is confirmed, then even a small increase in TST that is achieved consistently, night after night, week after week, may be worthwhile.
Our findings show that the strongest effects of exogenous melatonin are on SOL. In exploratory analyses we have found a strong correlation between those children with later DLMO peaks and those children who fall asleep later. We have also found that the amplitude of treatment response is strongly correlated with the initial severity of the sleep disorder. Thus, as has been described in typically developing children, but not replicated in this population, children who have later DLMO times fall asleep later and respond better to exogenous melatonin. As SOL and sleep duration are related, so an improvement in SOL may also lengthen sleep duration, but this depends on whether or not sleep offset (the time that the child wakes up) alters. This does, however, support the possible utility of pretreatment DLMO measurement to predict the better treatment responders. However, this will clearly require further evaluation as well as a full economic assessment, which was beyond the scope of MENDS. As far as we are aware, no controlled trial has demonstrated a reduction in night awakenings on active treatment, and, although this was not one of our specified secondary outcome measures, it is an important question that should be included in any future study.
The genetic analysis substudy has allowed high-throughput genome-wide genotyping to be completed for all 125 samples that passed the quality control for DNA genotyping. We were fortunate to be able to use the latest high-density Illumina arrays measuring up to 2.5 million SNPs. SNP association analysis was undertaken with several important outcomes, including severity of sleep disorder, melatonin levels and response to melatonin treatment. The analysis also allowed an investigation of the incidence of rare CNVs in the MENDS sample in comparison to the general population control rate. Specific sequencing was undertaken of all exons of AANAT and ASMT, two important and putative enzymes for which rare deletions (seen more commonly in children with autism) have been reported to account for reduced melatonin levels. Although preliminary results for this part of the analysis are available, the volume and the complexity of the data demand that analysis needs to be carefully undertaken by specific software and there are limited resources for such work. Consequently, this is currently still ‘work in progress’, both in Liverpool and at the Pasteur Institute (Paris).
There were only five serious and two suspected unexpected serious adverse reactions during the study period, none of which was considered to be related to the active medication (melatonin). In the 16 patients with epilepsy, equally distributed between the two treatment groups, none showed any deterioration in seizure control, emergence of a new seizure type or de novo epilepsy. This concurs with the vast majority of previous reports, including one which suggested that melatonin may have an inherent anticonvulsant effect. 72 Only one report published in 1998 has suggested that it may have a proconvulsant effect. 42
Implications for health care
Sleep disorders are a common presentation in children with a wide variety of neurodevelopmental conditions. Medication should not be the first-line intervention and, as has been shown previously, our behavioural run-in period was successful, with many children no longer meeting the inclusion criteria for the study. This effect was seen after a relatively short period using a specific evidence-based behaviour therapy advice booklet and monitoring but with no direct contact with psychology or other sleep behavioural specialists. However, it is possible that the relatively large ‘dropout’ of patients in the 4- to 6-week behaviour intervention (therapy) phase may also reflect parental perceptions of the child’s sleep problem. The process of formally observing and documenting their child’s sleep pattern in sleep diaries may have unmasked a significant gap between their perceived interpretation of their child’s sleep problem and their child’s actual sleep problem. It would be relatively easy to test this hypothesis in a future RCT of a behavioural intervention compared with no intervention in this type of population. The results of MENDS would also suggest that in routine clinical practice families should be asked to complete a 2- to 4-week sleep diary before melatonin is prescribed.
Melatonin is more effective than placebo for children with neurodevelopmental delay who have trouble falling asleep. This is a common presenting complaint and melatonin reduces this period by an average of 37 minutes. This is helpful for parents desperate to settle their child with neurodevelopmental delay and may result in a calmer evening for themselves or for siblings (and other family members) or both. However, we found no evidence that this reduction in sleep latency measurably improved the family’s quality of life or children’s behaviour over the 3-month period. It did seem to reduce parents’ reports of daytime fatigue, which is an interesting finding that should be explored in future studies.
Although the children fell asleep earlier, they gained very little extra sleep when measured over the whole night. An extra 23 minutes of sleep over a whole night is small and was deemed not to be clinically significant for our study. The increase does of course vary with individuals and is likely to be cumulative. Experimentally, an artificial reduction of this amount of sleep every night has been shown over the very short term to affect daytime behaviour and cognition. 73 However, in the 3-month study period of MENDS we were unable to identify any improvement in children’s behaviour. Cognition was not formally assessed in MENDS.
Implications for research
-
Our trial compared melatonin only with placebo. There are a number of other licensed and unlicensed medications (including hypnotics and sedatives) for children with sleep problems. 74 Head-to-head trials are needed to help clinicians and families decide which medication(s) is (are) likely to be the safest and most helpful.
-
Further studies need to be undertaken to try and establish the most appropriate dose of melatonin, incorporating the child’s age, weight, 24-hour endogenous melatonin profile and DLMO and whether they are a fast or slow metaboliser. Both fast- and slow-release preparations should be considered. A dose-ranging study in children with sleep-onset disorder has recently been published;75 however, no dose–response relationship of melatonin in advancing DLMO or reducing SOL was found within a dosage range of 0.05–0.15 mg/kg.
-
Patients’ cognition was not directly measured in MENDS. Given that cognitive function may reflect important end points around learning potential, this outcome will be important to explore in future intervention trials, however difficult this is likely to be.
-
Future RCTs that assess the effectiveness and safety of melatonin should be undertaken over a longer period and should include both appropriate quality of life and economic evaluations.
Acknowledgements
The MENDS Trial Management Group is very grateful to all of the principal investigators, research practitioners, site pharmacists, referring paediatricians, children and their carers and families without whose commitment, energy and patience MENDS would not have been the successful study it has been. The MENDS Trial Management Group is also grateful to the following: Alliance and Penn Pharmaceuticals for financial and other support for the manufacture of melatonin and the matching placebo; the LRNs in England whose work and support were invaluable in the early stages of MENDS; Dr Thomas Bourgeron of the Pasteur Institute, Paris, France (genetics analyses), and Professor Debra Skene and Dr Benita Middleton of the University of Guildford, Surrey, UK (salivary melatonin measurements); Joanna Milton, Charlotte Stockton and Hannah Short, MENDS trial co-ordinators; the Trial Steering Committee (chaired by Professor Stuart Logan) and the Data Monitoring and Ethics Committee (chaired by Professor Anthony Marson) for their support and work throughout the study.
Contributions of authors
Richard Appleton (Co-chief Investigator) co-led the clinical developmental of the protocol and ongoing oversight and management of the study and prepared the report for publication.
Paul Gringras (Co-chief Investigator) co-led the clinical developmental of the protocol and ongoing oversight and management of the study and prepared the report for publication.
Ashley Jones (Trial Statistician) performed the statistical analyses and prepared the report for publication.
Carrol Gamble (Reader in Medical Statistics) led the statistical team and contributed to the design of the study, its conduct and analysis and prepared the report for publication.
Paula Williamson (Director of the Clinical Trials Unit and Professor of Medical Statistics) contributed to the design and conduct of the study and prepared the report for publication.
Luci Wiggs was a member of the Trial Management Group and provided expertise in actigraphy, behavioural interventions and assessments and prepared the report for publication.
Paul Montgomery was a member of the Trial Management Group and provided expertise in actigraphy and reviewed a draft of the report.
Alistair Sutcliffe was a member of the Trial Management Group and a principal investigator and reviewed a draft of the report.
Catrin Barker (Pharmacist) was a member of the Trial Management Group and provided expertise on packaging, storage, accountability and distribution of the investigational medicinal product and reviewed a draft of the report.
The MENDS study group
| Name | Role/contribution |
|---|---|
| Tom Allport | Principal investigator, Southmead Hospital, Bristol |
| Fiona Atkin | Research nurse, Royal Liverpool Children’s Hospital |
| Louise Barnes | Research nurse, Royal Devon & Exeter Hospital/Torbay Hospital |
| Emily Benson | Research nurse, Birmingham Children’s Hospital |
| Jackie Bradley | Research nurse, Blackpool Victoria Hospital |
| Lucy Bray | Research nurse, Royal Liverpool Children’s Hospital |
| Imti Choonara | Principal investigator, Derbyshire Children’s Hospital, Derby |
| Helen Clark | Research nurse, Southmead Hospital, Bristol |
| Michelle Cooper | Research nurse, Royal Devon & Exeter Hospital/Torbay Hospital |
| Christina Daines | Research nurse, Leicester Royal Infirmary |
| Jacqui Dalglish | Research nurse, Birmingham Children’s Hospital |
| Sarah Dyas | Research nurse, Arrowe Park Hospital, Wirral |
| Penny Erskine | Research nurse, Nottingham City Hospital |
| Melanie Farman | Research nurse, Royal Manchester Children’s Hospital |
| Fabien Fauchereau | Statistical analyses of genetic data |
| Sheila Fox | Research nurse, Southmead Hospital, Bristol |
| Jayaprakash Gosalakkal | Principal investigator, Leicester Royal Infirmary |
| Hany Goubran Botros | Generation of genetic data and their sequenced analyses |
| Val Harpin | Principal investigator, Sheffield Children’s Hospital |
| Jasmine Heslop | Research nurse, Royal Devon & Exeter Hospital |
| Cathy Hill | Principal investigator, Southampton General Hospital |
| Lorraine Hodson | Research nurse, Evelina Children’s Hospital/University College London Hospitals |
| Simone Holley | Research nurse, Southampton General Hospital |
| Anwen Howells | Research nurse, Children’s Hospital for Wales, Cardiff |
| Adrian Hughes | Principal investigator, Arrowe Park Hospital, Wirral |
| Guillaume Huguet | Generation of genetic data and statistical analyses |
| Lisa Hydes | Research nurse, Birmingham Children’s Hospital |
| Samantha Jones | Research nurse, Derbyshire Children’s Hospital/Chesterfield Royal Hospital/Nottingham City Hospital |
| Andrea Jorgensen | Co-writing of protocol amendment and statistical analysis plan for genetic substudy, advice regarding analysis of genetic data, integration of genetic results into report |
| Xenya Kantaris | Research nurse, University College London Hospitals |
| Mark Lathrop | Generation of high-throughput genetic data |
| Natalie Lemière | Generation of candidate gene sequences |
| Ros Loxton | Research nurse, Queen Mary’s Hospital, London |
| Cilla Long | Research nurse, Southampton General Hospital |
| Emma Macleod | Research nurse, Southampton General Hospital |
| Tim Martland | Principal investigator, Royal Manchester Children’s Hospital |
| Andrew McKay | Statistical analyses of secondary outcomes, Liverpool |
| Alison McQueen | Research nurse, Children’s Hospital for Wales, Cardiff |
| Julie Menzies | Research nurse, Birmingham Children’s Hospital |
| Teresa Moorcroft | Research nurse, Arrowe Park Hospital, Wirral |
| Tracey Oakden | Research nurse, University College London Hospitals |
| Cécile Pagan | Generation of genetic data and statistical analyses |
| Vicky Payne | Research nurse, Southmead Hospital, Bristol |
| Karen Pratt | Research nurse, Evelina Children’s Hospital, London |
| Philip Preece | Principal investigator, Chesterfield Royal Hospital |
| Béatrice Regnault | Generation of high-throughput genetic data |
| Claire Roberts | Research nurse, Southmead Hospital, Bristol |
| Heather Rostron | Research nurse, Royal Manchester Children’s Hospital/Blackpool Victoria Hospital |
| Ann Russell | Research nurse, Children’s Hospital for Wales, Cardiff |
| Clive Sainsbury | Principal investigator, Torbay Hospital |
| Penny Scardifield | Research nurse, Nottingham City Hospital |
| Heather Smee | Research nurse, Southmead Hospital, Bristol |
| Coral Smith | Research nurse, Derbyshire Children’s Hospital, Derby |
| Joanna Smith | Research nurse, Chesterfield Royal Hospital/Nottingham City Hospital |
| Ron Smith | Principal investigator, Royal Devon & Exeter Hospital |
| Masako Sparrowhawk | Research nurse, John Radcliffe Hospital, Oxford |
| Louise Spencer Walsh | Research nurse, University College London Hospitals |
| Louise Strickland | Research nurse, Royal Devon & Exeter Hospital/Torbay Hospital |
| Jacqui Tahari | Research nurse, Royal Liverpool Children’s Hospital (Alder Hey)/Arrowe Park Hospital, Wirral |
| Johann te Water Naude | Principal investigator, Children’s Hospital for Wales, Cardiff |
| Amanda Thobinson | Research nurse, Royal Liverpool Children’s Hospital/Royal Manchester Children’s Hospital |
| Megan Thomas | Principal investigator, Blackpool Victoria Hospital |
| Jeremy Turk | Principal investigator, Queen Mary’s Hospital, London |
| Vanessa Unsworth | Research nurse, Derbyshire Children’s Hospital/Chesterfield Royal Hospital/Nottingham City Hospital |
| Catherine Upton | Research nurse, Southmead Hospital, Bristol |
| Lynda Viles | Research nurse, Chesterfield Royal Hospital/Nottingham City Hospital/Sheffield Children’s Hospital |
| Rachel Voce | Research nurse, Leicester Royal Infirmary |
| Evangeline Wassmer | Principal investigator, Birmingham Children’s Hospital |
| William Whitehouse | Principal investigator, Nottingham City Hospital |
| Helen Wilde | Research nurse, Royal Liverpool Children’s Hospital (Alder Hey)/Arrowe Park Hospital, Wirral |
| Su Wilkins | Research nurse, Royal Devon & Exeter Hospital/Torbay Hospital |
| Jacqui Woods | Research nurse, Royal Manchester Children’s Hospital/Blackpool Victoria Hospital |
| Zenobia Zaiwalla | Principal investigator, John Radcliffe Hospital, Oxford |
| Diana Zelenica | Generation of high-throughput genetic data |
Disclaimers
The views expressed in this publication are those of the authors and not necessarily those of the HTA programme or the Department of Health.
References
- Claustrat B, Brun J, Chazgot G. The basic physiology and pathophysiology of melatonin. Sleep Med Rev 2005;9:11-24.
- Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature 2002;418:935-41.
- Rivkees SA. Rest–activity patterns in children with hypopituitarism. Pediatrics 2003;111:e720-4.
- Arendt J, Middleton B, Lockley SW, Deacon S, Skene D. Efficacy of melatonin treatment in jet lag, shift work and blindness. J Biol Rhythms 1997;12:604-17.
- Herxheimer A, Petrie KJ. Melatonin for the prevention and treatment of jet lag. Cochrane Database Syst Rev 2002;2.
- Arendt J, Bojkowski C, Folkard S, Franey C, Marks V, Minors D, et al. Some effects of melatonin and the control of its secretion in humans. Ciba Found Symp 1985;117:266-83.
- Arendt J. Melatonin and the mammalian pineal gland. London: Chapman and Hall; 1994.
- Jan JE, Espezel H, Appleton RE. The treatment of sleep disorders with melatonin. Dev Med Child 1994;36:97-107.
- Wiggs L. Sleep problems in children with developmental disorders. J R Soc Med 2001;94:177-9.
- Quine L. Sleep problems in children with mental handicap. J Ment Defic Res 1991;35:269-90.
- Wasdell MB, Jan JE, Bomben MM, Freeman RD, Rietveld WJ, Tai J, et al. A randomised, placebo-controlled trial of controlled release melatonin treatment of delayed sleep phase syndrome and impaired sleep maintenance in children with neurodevelopmental disabilities. J Pineal Res 2008;44:57-64.
- Miyamoto A, Oki J, Takahashi S, Okuno A. Serum melatonin kinetics and long-term melatonin treatment for sleep disorders in Rett syndrome. Brain Dev 1999;21:59-62.
- Alvarez B, Dahlitz MJ, Vignau J, Parkes JD. The delayed sleep phase syndrome: clinical and investigative findings in 14 subjects. J Neurol Neurosurg Psychiatry 1992;55:665-70.
- Palm L, Blennow G, Wetterberg L. Correction of non-24-hour sleep/wake cycle by melatonin in a blind retarded boy. Ann Neurol 1991;29:336-9.
- Palm L, Blennow G, Wetterberg L. Long-term melatonin treatment in blind children and young adults with circadian sleep–wake disturbances. Dev Med Child Neurol 1997;39:319-25.
- Jan MJ. Melatonin for the treatment of handicapped children with severe sleep disorders. J PediatricNeurol 2000;23:229-32.
- Hung J, Appleton RE, Nunn AJ, Rosenbloom L. The use of melatonin in the treatment of sleep disturbances in children with neurological and behavioural disorders. J Pediatr Pharm 1998;3:250-6.
- Wirojanan J, Jacquement S, Diaz R, Bacalman S, Anders TF, Hagerman RJ, et al. The efficacy of melatonin for sleep problems in children with autism, fragile X syndrome, or autism and fragile X syndrome. J Clin Sleep Med 2009;5:145-50.
- McArthur AJ, Budden SS. Sleep dysfunction in Rett syndrome: a trial of exogenous melatonin treatment. Dev Med Child Neurol 1998;40:186-92.
- Braam W, Didden R, Smits MG, Curfs MG. Melatonin for chronic insomnia in Angelman syndrome: a randomised placebo-controlled trial. J Child Neurol 2009;23:649-54.
- O’Callaghan FJ, Clarke AA, Hancock E, Hunt A, Osborne JP. Use of melatonin to treat sleep disorders in tuberous sclerosis. Dev Med Child Neurol 1999;41:123-6.
- Ayyash HF, Morton R, Alfanek H, Preece P. Melatonin for the treatment of children with sleep disorders. Arch Dis Child 2005;90:A27-8.
- Braam W, Smits MG, Didden R, Korzilius H, van Geijlswijk M, Curfs LMG. Exogenous melatonin for sleep problems in individuals with intellectual disability: a meta-analysis. Dev Med Child Neurol 2009;51:340-9.
- Rossignol DA, Frye RE. Melatonin in autism spectrum disorders: a systematic review and meta-analysis. Dev Med Child Neurol 2011;53:783-92.
- Coppola G, Iervolino G, Mastrosimone M, La Torre G, Ruiu F, Pascotto A. Melatonin in wake–sleep disorders in children, adolescents and young adults with mental retardation with or without epilepsy: a double-blind, cross-over, placebo-controlled trial. Brain Dev 2004;26:373-6.
- Gupta M, Aneja S, Kohli K. Add-on melatonin improves sleep behavior in children with epilepsy: randomized, double-blind, placebo-controlled trial. J Child Neurol 2005;20:112-15.
- Gupta M, Aneja S, Kohli K. Add-on melatonin improves quality of life in epileptic children on valproate monotherapy: a randomized, double-blind, placebo-controlled trial. Epilepsy Behav 2004;5:316-21.
- Buscemi N, Vandermeer B, Hooton N, Pandya R, Tjosvold L, Hartling L, et al. The efficacy and safety of exogenous melatonin for primary sleep disorders. A meta-analysis. J Gen Intern Med 2005;20:1151-8.
- Wassmer E, Ross C, Whitehouse W. Therapeutic options for melatonin use in children. Paediatr Perinat Drug Ther 2000;4:14-20.
- Cavallo A, Good WV, Douglas RM, Succop P. Dose response to melatonin treatment for disordered sleep rhythm in a blind child. Sleep Med 2002;3:159-61.
- Jan JE, O’Donnell ME. Use of melatonin in the treatment of paediatric sleep disorders. J Pineal Res 1996;21:193-9.
- Defrance R, Quera-Salva MA. Therapeutic applications of melatonin and related compounds. Horm Res 1998;19:142-6.
- Zemlan FP, Mulchahey JJ, Scharf MB, Mayleben DW, Rosenberg R, Lankford A. The efficacy and safety of the melatonin agonist beta-methyl-6-chloromelatonin in primary insomnia: a randomized, placebo-controlled, crossover clinical trial. J Clin Psychiatry 2005;66:384-90.
- Braam W, van Geijlswijk I, Keijzer H, Smits MG, Didden R, Curfs LM. Loss of response to melatonin treatment is associated with slow melatonin metabolism. J Intellect Disabil Res 2010;54:547-55.
- Buscemi N, Vandermeer B, Hooton N, Pandya R, Tjosvold L, Hartling L, et al. Efficacy and safety of exogenous melatonin for secondary sleep disorders and sleep disorders accompanying sleep restriction: meta-analysis. BMJ 2006;332:385-93.
- Phillips L, Appleton RE. Systematic review of melatonin treatment in children with neurodevelopmental disabilities and sleep impairment. Dev Med Child Neurol 2004;46:771-5.
- Waldron DL, Bramble D, Gringras P. Melatonin: prescribing practices and adverse events. Arch Dis Child 2005;90:1206-7.
- Salti R, Galluzzi F, Bindi G, Perfetto F, Tarquini R, Halberg F, et al. Nocturnal melatonin patterns in children. J Clin Endocrinol Metab 2000;85:2137-44.
- Sutherland ER, Ellison MC, Kraft M, Martin RJ. Elevated serum melatonin is associated with the nocturnal worsening of asthma. J Allergy Clin Immunol 2003;112:513-17.
- Campos FL, da Silva-Junior FP, de Bruin VMS, de Bruin PFC. Melatonin improves sleep in asthma: a randomized, double-blind, placebo-controlled study. Am J Respir Crit Care Med 2004;170:947-51.
- Forsling ML, Wheeler MJ, Williams AJ. The effect of melatonin administration on pituitary hormone secretion in man. Clin Endocrinol 1999;51:637-42.
- Sheldon SH. Pro-convulsant effects of oral melatonin in neurologically disabled children. Lancet 1998;351.
- Elkhayat HA, Hassanein SM, Tomoum HY, Abd-Elhamid IA, Asaad T, Elwakkad AS. Melatonin and sleep-related problems in children with intractable epilepsy. Pediatr Neurol n.d.:42-54.
- van der Heijden KB, Smits MG, van Someren EJ, Boudewijn Gunning W. Prediction of melatonin efficacy by pretreatment dim light melatonin onset in children with idiopathic chronic sleep onset insomnia. J Sleep Res 2005;14:187-94.
- Montgomery P, Stores G, Wiggs L. The relative efficacy of two brief treatments for sleep problems in young learning disabled (mentally retarded) children: a randomised controlled tria. Arch Dis Child 2004;89:125-30.
- Shah T, Tse PYA, Gill H, Wong ICK, Sutcliffe A, Gringras P, et al. Administration of melatonin mixed with common soft food and liquids for children with neurodevelopmental difficulties. Dev Med Child Neurol 2008;50:845-9.
- Sadeh A. The role and validity of actigraphy in sleep medicine: an update. Sleep Med Rev 2011;15:259-67.
- Sadeh A, Hauri P, Kripke D, Lavie P. The role of actigraphy in the evaluation of sleep disorders. Sleep 1995;18:288-302.
- Littner M, Kushida CA, Anderson WM, Bailey D, Berry RB, Davila DG, et al. Standards of Practice Committee of the American Academy of Sleep Medicine. Practice parameters for the role of actigraphy in the study of sleep and circadian rhythms: an update for 2002. Sleep 2003;26:337-41.
- Owens JA, Spirito A, McGuinn M. The Children’s Sleep Habits Questionnaire (CSHQ): psychometric properties of a survey instrument for school-aged children. Sleep 2000;23:1043-51.
- Varni JW, Sherman SA, Burwinkle TM, Dickinson PE, Dixon P. The PedsQL Family Impact Module: preliminary reliability and validity. Health Qual Life Outcomes 2004;2.
- Johns MW. A new method for measuring daytime sleepiness: the Epworth Sleepiness Scale. Sleep 1991;14:540-5.
- Aman MG, Singh NN. Aberrant Behavior Checklist manual. New York, NY: Slosson Educational Publications; 1986.
- Aman MG, Singh NN. Aberrant Behavior Checklist – community supplementary manual. New York, NY: Slosson Educational Publications; 1994.
- Richman N, Graham PJ. A behaviour screening questionnaire for use with three year old children. J Child Psychol Psychiatry 1971;12:5-33.
- Quine L, Gibbons J. The Children Act 1989 and family support: principles into practice. London: HMSO; n.d.
- Keijzer H, Smits MG, van Someren EJ, Boudewijn Gunning W. Evaluation of salivary melatonin measurements for dim light melatonin onset calculations in patients with possible sleep–wake rhythm disorders. Clin Chim Acta 2011;412:1616-20.
- Chaste P, Clement N, Botros H, Guillaume JL, Konyukh M, Pagan C, et al. Genetic variations of the melatonin pathway in patients with attention-deficit and hyperactivity disorders. J Pineal Res 2011;51:394-9.
- Melke J, Goubran-Botros HG, Chaste P, Betancur C, Nygren G, Anckarsäter H, et al. Abnormal melatonin synthesis in autism spectrum disorders. Mol Psychiatry 2008;13:90-8.
- Cai G, Endelmann L, Goldsmith JE, Cohen N, Nakamine A, Reichert JG, et al. Multiplex ligation-dependent probe amplification for genetic screening in autism spectrum disorders: efficient identification of known micoduplications and identification of a novel microduplication in. ASMT BMC Med Genomics 2008;16:1-50.
- Evans S. When and how can endpoints be changed after initiation of a randomized clinical trial. PLOS Clin Trial 2007;2.
- Antic NA, Buchan C, Esterman A, Hensley M, Naughton MT, Rowland S, et al. A randomized controlled trial of nurse-led care for symptomatic moderate-severe obstructive sleep apnea. Am J Respir Crit Care Med 2009;179:501-8.
- Dayyat E, Spruyt K, Molfese DL, Goza D. Sleep estimates in children: parental versus actigraphic assessments. Nat Sci Sleep 2011;3:115-23.
- Thomas M, Hunt A, Hurley M, Robertson S, Carter B. Time-use diaries are acceptable to parents with a disabled preschool child and are helpful in understanding families’ daily lives. Child Care Health Dev 2011;37:168-74.
- Brzezinski A, Vangel M, Wurtman R, Norrie G, Zhdanova I, Ben-Shushan A, et al. Effects of exogenous melatonin on sleep: a meta-analysis. Sleep Med Rev 2005;9:41-50.
- Khan S, Heussler H, McGuire T, Dakin C, Pache D, Cooper D, et al. Melatonin for non-respiratory sleep disorders in children with neurodevelopmental disorders (protocol). Cochrane Database Syst Rev 2011;5.
- Dwan K, Gamble C, Kolamunnage-Dona R, Mohammed S, Powell C, Williamson PR. Assessing the potential for outcome reporting bias in a review: a tutorial. Trials 2010;11.
- Wright B, Sims D, Smart S, Alwazeer A, Alderson-Day B, Allgar V, et al. Melatonin versus placebo in children with autism spectrum conditions and severe sleep problems not amenable to behavior management strategies: a randomized controlled crossover trial. J Autism Dev Disord 2010;41:175-84.
- Garstang J, Wallis M. Randomized controlled trial of melatonin for children with autistic spectrum disorders and sleep problems. Child Care Health Dev 2006;32:585-9.
- Smits MG, van Stel HF, van der Heijden K, Meijer AM, Coenen AM, Kerkhof GA. Melatonin improves health status and sleep in children with idiopathic chronic sleep-onset insomnia: a randomized placebo-controlled trial. J Am Acad Child Adolesc Psychiatry 2003;42:1286-93.
- Jan JE, Reiter RJ, Bax MCO, Ribary U, Freeman RD, Wasdell MB. Long-term sleep disturbance in children: a cause of neuronal loss. Eur J Neurol 2010;14:380-90.
- Peled N, Shorer Z, Peled E, Pillar G. Melatonin effect on seizures in children with severe neurologic deficit disorders. Epilepsia 2001;42:1208-10.
- Sadeh A, Gruber R, Raviv A. The effects of sleep restriction and extension on school-age children: what a difference an hour makes. Child Dev 2003;7:444-5.
- Gringras P. When to use drugs to help sleep. Arch Dis Child 2008;93:976-81.
- van Geijlswijk I, van der Heijden K, Egberts A, Hubert P, Korzilius L, Smits M. Dose finding of melatonin for chronic idiopathic childhood sleep onset insomnia: an RCT. Psychopharmacology 2010;212:379-91.
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. J Hum Genet 2011;81:559-75.
Appendix 1 Behavioural booklet
Appendix 2 Sleep diary
Appendix 3 Statistical analysis plan
Patient groups for analysis
To provide a pragmatic comparison of the different interventions, the principle of intention to treat, as far as is practically possible, will be the main strategy of the analysis adopted for the primary outcome and all of the secondary outcomes. These analyses will be conducted on all patients randomised to the treatment groups. For the sleep outcomes calculated using sleep diaries and actigraphy, a minimum of 5 nights of data from the 7 days before the randomisation visit date and a minimum of 5 nights of data from day 77 to day 84 from the randomisation visit date are required. The sleep diary is provided in Appendix 2 and Table 21 details the fields that must be completed for calculation of each sleep outcome using the sleep diary or actigraphy. For example, for SOL calculated using sleep diaries, ‘snuggle-down’ and sleep start times must be recorded; for SOL calculated using actigraphy only ‘snuggle-down’ time is required.
| Outcome | Outcome based on sleep diaries | Outcome based on actigraphy |
|---|---|---|
| TST | Sleep start time; wake-up time; night-time awakenings | |
| SOL | Snuggle down to sleep time; sleep start time | Snuggle down to sleep time |
| Sleep efficiency | N/A | Snuggle down to sleep time, wake-up time |
No imputation methods will be used for any missing primary outcome data for the primary analyses. Sensitivity analyses will be carried out on the primary outcome (see Analysis of missing data) but no sensitivity analyses will be carried out on secondary outcomes.
The membership of the analysis set for each outcome will be determined and documented and reasons for participant exclusion will be given prior to the blind being broken and the randomisation lists being requested (the analysis set may be refined under review before the final statistical analysis). Reasons for exclusion may include missing data, loss to follow-up and treatment withdrawal.
The safety analysis data set will contain all participants who are randomised and who received at least one dose of trial medication.
Description of safety outcomes
All adverse events and serious adverse events reported by the clinical investigator will be presented, identified by treatment group. The number (and percentage) of patients experiencing each adverse event/serious adverse event will be presented for each treatment arm categorised by severity. For each participant, only the maximum severity experienced of each type of adverse event will be displayed. The number (and percentage) of occurrences of each adverse event/serious adverse event will also be presented for each treatment arm. No formal statistical testing will be undertaken.
Analysis of primary outcome
Total night-time sleep calculated using sleep diaries
The total amount of sleep for 1 night will be calculated in minutes using the amount of time between the time that the child went to sleep and the time that the child woke up the following morning minus any night-time awakenings that the child has had. The baseline measurement will be calculated using the average total amount of sleep in the 7 days before randomisation and the post-treatment measurement will be the average total amount of sleep from day 77 to day 84 post randomisation (this corresponds to the final 7 days of treatment as patients received enough drug supply only for 84 days).
An example of the data used to calculate TST for a participant randomised on 20 January 2010 is given in Box 1.
(Wake-up time on 20 Jan – sleep start time 19 Jan – night awakenings 19 Jan) + (Wake-up time on 19 Jan – sleep start time 18 Jan – night awakenings 18 Jan) + (Wake-up time on 18 Jan – sleep start time 17 Jan – night awakenings 17 Jan) + (Wake-up time on 17 Jan – sleep start time 16 Jan – night awakenings 16 Jan) + (Wake-up time on 16 Jan – sleep start time 15 Jan – night awakenings 15 Jan) + (Wake-up time on 15 Jan – sleep start time 14 Jan – night awakenings 14 Jan) + (Wake-up time on 14 Jan – sleep start time 13 Jan – night awakenings 13 Jan)
The sum of the total night sleep for these 7 nights is then divided by seven to give the mean total night sleep for this patient at baseline.
If the patient was randomised on the 20 January 2010 they will receive enough drug supply for 84 days. Their final visit should then take place on the 24 March 2010.
Mean total night-time sleep for the 7 days before the T+12W visit(Wake-up time on 24 Mar – sleep start time 23 Mar – night awakenings 23 Mar) + (Wake-up time on 23 Mar – sleep start time 22 Mar – night awakenings 22 Mar) + (Wake-up time on 22 Mar – sleep start time 21 Mar – night awakenings 21 Mar) + (Wake-up time on 21 Mar – sleep start time 20 Mar – night awakenings 20 Mar) + (Wake-up time on 20 Mar – sleep start time 19 Mar – night awakenings 19 Mar) + (Wake-up time on 19 Mar – sleep start time 18 Mar – night awakenings 18 Mar) + (Wake-up time on 18 Mar – sleep start time 17 Mar – night awakenings 17 Mar)
The sum of the total night sleep for these 7 nights is then divided by seven to give the mean total night sleep for the final week of treatment for this patient.
A minimum of 5 nights of sleep from each time period is required for the data to contribute to the primary outcome. If a child has < 5 out of 7 nights completed the data will be regarded as missing and the remaining data will not be included in the primary analysis. Methods for handling missing data are discussed in Analysis of missing data.
The mean total night sleep (and standard deviation) for the week before randomisation (T0W) and the final 7 days of treatment (days 77–84, T+12W) and the mean change over baseline (T+12W – T0W) will be presented by treatment group. The mean difference in change over baseline between the two groups will be reported with 95% CIs. A two-sided p-value of 0.05 (5% level) will be used to declare statistical significance and will be reported alongside the CI.
The method of ANCOVA will be used to adjust for baseline sleep, the outcome measure being the average total amount of sleep in the final week of treatment (days 77–84) and the covariates that will be used in the model being treatment group (melatonin or placebo) and the baseline measurement (the average total amount of sleep in the 7 days before randomisation).
Randomisation is stratified by centre. However, because of the large number of small centres, the centre will not be included in the model as a covariate. Including a large number of small centres may lead to unreliable estimates of the treatment effect and p-values that may be too large or too small.
Analysis of missing data
Analysis of missing data will be restricted to the primary outcome only; no imputation methods will be used on any of the secondary outcomes.
The number of completed sleep diary days available for the 7-day period before T0W and T+12W will be reported for each group. A chi-squared test will be used to test for differences between the groups.
The primary analyses for the primary outcome will contain data only on participants who have ≥ 5 days of complete data during the 7 days immediately before randomisation and ≥ 5 days during days 77–84 post randomisation. Those participants who have < 5 days at either time period will not be included in the primary analyses.
The sensitivity of the results to those with missing data will be assessed within two population groups:
-
those contributing data to the primary outcome with ≥ 5 nights observed (missing data for 1 or 2 nights only) at T0W and T+12W
-
those contributing data to the primary outcome with ≥ 2 nights observed (missing data for up to 5 nights) at T0W and T+12W.
For each missing data point that a person has at both T0W and T+12W the worst recorded night of sleep from baseline will be imputed.
Data available from all sources will be considered to inform any further imputation strategies. This includes use of actigraphy for total night-time sleep and the daily global measure. The imputation strategies will be within patient rather than averages across patients. The potential for this approach will be informed by completion rates and the demonstration of a relationship but it will be performed only if the robustness of the results to the approach above is questionable.
Further sensitivity analyses were identified after the preliminary results were presented. These were to include:
-
An analysis of those patients who had ≥ 5 completed sleep diary days at T+12W only. This will compare the mean TST between the treatment groups at T+12W only. A t-test will be used to compare the difference between the two groups and a p-value and 95% CI will be presented.
-
An analysis that will be same as the primary analysis but which will include patients who have a minimum of 4 out of 7 completed sleep diary days. The analysis will be the same as that described in Analysis of primary outcome and the results will be presented similarly.
Analysis of secondary outcomes
The null hypothesis for each secondary outcome (for which statistical tests are being performed) will be that there is no difference in outcome between the melatonin and the placebo groups. The alternative hypothesis is that there is a difference between the two groups.
Total night-time sleep calculated using actigraphy data
The analysis will use the method of ANCOVA and will not adjust for any missing data and the model will contain only two covariates: the baseline average total amount of sleep and the treatment group. Those participants who have < 5 days of data will not be included in the analysis. The adjusted and unadjusted results will be presented as well as means and standard deviations for T0W, T+12W and the change over baseline (T+12 – T0W) for each treatment group.
Reasons for missing data will be documented and the results interpreted as appropriate.
Sleep efficiency calculated using actigraphy data [(number of minutes spent sleeping in bed/total number of minutes spent in bed) × 100]
The analysis will use the method of ANCOVA and will not adjust for any missing data and the model will contain only two covariates: the baseline average total amount of sleep and the treatment group. Those participants who have < 5 days of data will not be included in the analysis. The adjusted and unadjusted results will be presented as well as means and standard deviations for T0W, T+12 and the change over baseline (T+12 – T0W) for each treatment group.
Reasons for missing data will be documented and the results interpreted as appropriate.
Sleep-onset latency (the time taken to fall asleep) calculated using actigraphy data
The baseline measurement will be calculated using the average sleep latency in the 7 days before randomisation and the post-treatment measurement will be the average sleep latency from day 77 to day 84 post randomisation (this corresponds to the final 7 days of treatment as the patients received enough drug supply only for 84 days).
The analysis will use the method of ANCOVA and will not adjust for any missing data and the model will contain only two covariates: the baseline average total amount of sleep and the treatment group. Those participants who have < 5 days of data will not be included in the analysis. The adjusted and unadjusted results will be presented as well as means and standard deviations for T0W, T+12W and the change over baseline (T+12 – T0W) for each treatment group.
Sleep-onset latency (the time taken to fall asleep) calculated using sleep diaries
The SOL will be calculated using the sleep diary that has been completed. The baseline measurement will be calculated using the average sleep latency in the 7 days before randomisation and the post-treatment measurement will be the average sleep latency from day 77 to day 84 post randomisation (this corresponds to the final 7 days of treatment as the patients received enough drug supply only for 84 days).
The analysis will use the method of ANCOVA and will not adjust for any missing data and the model will contain only two covariates: the baseline average total amount of sleep and the treatment group. Those participants who have < 5 days of data will not be included in the analysis. The adjusted and unadjusted results will be presented as well as means and standard deviations for T0W, T+12W and the change over baseline (T+12 – T0W) for each treatment group.
Composite Sleep Disturbance Index
The CSDI is based on six items and includes measures on settling frequency and duration, night-waking frequency and duration, frequency of early waking and frequency of co-sleeping. Settling problems, night waking, early waking and co-sleeping are measured in terms of weekly frequency, and settling and night waking problems are also assessed in terms of nightly duration of the problem. The questions are scored as in Table 22.
The analysis will use the method of ANCOVA and will not adjust for any missing data and the model will contain only two covariates: the baseline average and the treatment group. The adjusted and unadjusted results will be presented as well as means and standard deviations for T0W, T+12W and the change over baseline (T+12W – T0W) for each treatment group. If the data are found not to be normally distributed, then the equivalent non-parametric test will be used.
| Night waking | Score | ||
|---|---|---|---|
| 0 | 1 | 2 | |
| Frequency | Less than once per week | One to two times per week | Three or more times per week |
| Duration | Few minutes | < 31 minutes | 31+ minutes |
Daily global measure of parental perception of child’s sleep quality
The daily global measure of parental perception of a child’s sleep quality is measured on the sleep diary with the use of ‘smiley faces’ on a scale of 1–7, with 1 being a very good night’s sleep and 7 being a very bad night’s sleep. To be included in the analysis the global measure must be completed for a minimum of 5 out of 7 nights.
The score will be calculated in two ways at T0W and T+12W:
-
the percentage of night sleeps with which the parent was dissatisfied (faces 5–7)
-
the mean of the scores for each night.
The change between T0W and T+12W will be calculated and presented with 95% CIs; for analysis 1 the mean percentage and standard deviation of night sleeps with which the parent was dissatisfied will be reported for T0, T+12W and the change over baseline (T+12W – T0W) for each treatment group, and for analysis 2 the mean score and standard deviation for T0, T12 and the change over baseline (T+12W – T0W) will be reported for each treatment group.
Behavioural problems assessed using the Aberrant Behaviour Checklist
The ABC consists of 58 items, each scored on a 4-point scale (0 = not a problem to 3 = problem is severe in degree). The items fall into five subscales: (1) irritability, agitation, crying (15 items), (2) lethargy, social withdrawal (16 items), (3) stereotypical behaviour (7 items), (4) hyperactivity, non-compliance (16 items) and (5) inappropriate speech (4 items).
The ABC manual does not describe methods of analysis of data collected using this tool, nor does it discuss methods for handling missing data. The manual does present the results of data in subscales and not as a total and therefore we will adopt this method for the presentation of the results of this tool.
No imputation will be made for missing items within a subscale and therefore subscales must be complete. The analysis will use the method of ANCOVA and will not adjust for any missing data and the model will contain only two covariates: the baseline average and the treatment group. The adjusted and unadjusted results will be presented as well as means and standard deviations for T0W, T+12W and the change over baseline (T+12W – T0W) for each treatment group. If the data are found not to be normally distributed then the non-parametric equivalent test will be used.
Quality of life of the caregiver assessed using the Family Impact Module of the Pediatric Quality of Life Inventory
The scoring instructions for the PedsQL Family Impact Module were taken from Varni et al. 51
The PedsQL Family Impact Module consists of 36 items. The module is split into two sections: six scales measure parent self-reported functioning and two scales measure parent-reported family functioning. The six scales that measure parent self-reported functioning are as follows:
-
physical functioning (6 items)
-
emotional functioning (5 items)
-
social functioning (4 items)
-
cognitive functioning (5 items)
-
communication (3 items)
-
worry (5 items).
The two scales that measure parent reported family functioning are as follows:
-
daily activities (3 items)
-
family relationships (5 items).
To score the module, a 5-point scale is used (0 = if it is never a problem, 1 = if it is almost never a problem, 2 = if it is sometimes a problem, 3 = if it is often a problem and 4 = if it is almost always a problem) and items are reverse scored and linearly transformed to a 0–100 scale (0 = 100, 1 = 75, 2 = 50, 3 = 25 and 4 = 0); therefore, higher scores indicate better functioning.
Each scale is scored and the scale score is calculated as the sum of the items divided by the number of items answered. If > 50% of the items in the scale have not been answered then the scale score should not be computed. A sensitivity analysis will impute the mean of the completed items in the scale (if the scale does not contain > 50% missing data).
The PedsQL Family Impact Module total scale score is calculated as the sum of all 36 items divided by the number of items answered. If more than 50% of the items have not been answered then the total scale score should not be computed. A sensitivity analysis will impute the mean of the completed items in the scale (if the total scale score does not contain > 50% missing data). The parent health-related quality-of-life summary score (20 items) is computed as the sum of the items divided by the number of items answered in the physical, emotional, social and cognitive functioning scales. The family functioning summary score (8 items) is computed as the sum of the items divided by the number of items completed in the daily activities and family relationships scales. The same procedure for handling missing data will used for these scales as described above.
The analysis will use the method of ANCOVA and will not adjust for any missing data and the model will contain only two covariates: the baseline average and the treatment group. The adjusted and unadjusted results will be presented as well as means and standard deviations for T0W, T+12W and the change over baseline (T+12W – T0W) for each treatment group. If the data are found not to be normally distributed then the non-parametric equivalent test will be used.
Level of daytime sleepiness of caregiver assessed using the Epworth Sleepiness Scale
The ESS is used to determine the parent’s (not the child’s) level of daytime sleepiness. There are eight questions and a 4-point scale for each question. The total ESS score is the sum of the eight questions and can range between 0 and 24. If a question has been left blank then the ESS is not valid and no methods of imputation will be used; this is as recommended by the author of the ESS (http://epworthsleepinessscale.com/1997-version-ess/).
The analysis will use the method of ANCOVA and will not adjust for any missing data and the model will contain only two covariates: the baseline average and the treatment group. The adjusted and unadjusted results will be presented as well as means and standard deviations for T0W, T+12W and the change over baseline (T+12W – T0W) for each treatment group. If the data are found not to be normally distributed then the non-parametric equivalent test will be used.
Number and severity of seizures evaluated using seizure diaries throughout trial follow-up
The numbers and percentages of patients (who have epilepsy) experiencing a seizure will be presented for each treatment arm pre and post randomisation. The type of epilepsy that each patient had at the initial screening visit will also be presented. For each patient, only the maximum frequency experienced of each seizure will be displayed. No formal statistical testing will be undertaken.
Adverse effects assessed weekly between weeks T0W and T12+W using treatment-emergent signs and symptoms
The numbers (and percentages) of patients experiencing each aspect of TESS will be presented for each treatment arm categorised by severity. Originally there were 14 aspects of TESS (somnolence, increased excitability, mood swings, seizures, rash, hypothermia, cough, increased activity, dizziness, hangover feeling, tremor, vomiting, nausea and breathlessness). On 27 April 2009 TESS was reduced to seven aspects (somnolence, increased excitability, mood swings, seizures, rash, hypothermia and cough). Results will be presented for all data that have been collected during the study period.
For each patient, only the maximum severity experienced of each type of TESS will be displayed. The numbers (and percentages) of occurrences of each type will also be presented for each treatment arm. No formal statistical testing will be undertaken.
Salivary melatonin concentrations
From the saliva concentrations measured, the DLMO time will be calculated for T0W and T+12W as the time when saliva melatonin levels reach 2 × SD of baseline values.
Kaplan–Meier curves and the log-rank test will be used to compare time to DLMO between the melatonin and the placebo groups at T0W and T+12W.
Biochemical and genetic investigations
Outcomes
For the genetic substudy, the associations between genetic variants and the following outcomes were of interest:
-
severity of sleep disorder, assessed using two different measures:
-
SOL (as assessed by sleep diary at T0W)
-
TST (as assessed by sleep diary at T0W)
-
-
melatonin level, defined as salivary melatonin concentration at 2000 at T–1W
-
ability to synthesise melatonin
-
response to melatonin treatment, assessed using two different measures:
-
difference in SOL between T0W and T+12W (as assessed by sleep diaries at these two time points)
-
difference in TST between T0W and T+12W (as assessed by sleep diaries at these two time points).
-
For the biochemical investigation, the relationship between melatonin levels and sleep disorders was of interest. The variables of interest here were SOL and TST, both as defined above, and DLMO, which was represented as a categorical covariate in two different ways:
-
using quartiles: 1: DLMO ≤ 1930; 2: 1930 < DLMO ≤ 2030; 3: 2030 < DLMO < 2200; 4: DLMO ≥ 2200 (or no melatonin peak)
-
using medians: 1: DLMO < 2100; 2: DLMO ≥ 2100 (or no melatonin peak).
Genotyping
Before the analyses of association, the genotype data were subjected to a number of quality control filters. Patient samples with a call rate < 98% were excluded from further analysis, as were SNPs with call rates < 98% and SNPs showing departure from the Hardy–Weinberg equilibrium (p < 10–6). To check for genetic diversity within the genotyped cohort, a principal component analysis was undertaken using the identity by state metrics, which estimates genetic distances between individuals. 76 To identify outliers, the distance of each individual to their fifth nearest neighbour was calculated and those having the largest distance flagged as outliers. When population outliers were detected, all analyses were performed both with and without the outliers, and unless otherwise stated this did not affect the results.
Analyses of association
The association analyses between genetic variants and each outcome were performed using the software package PLINK. 76 For the outcomes of severity of sleep disorder, a linear regression model was fitted for each SNP in turn. As the outcomes are age dependent, a covariate to represent age was included in the models. An additive mode of inheritance was assumed, with SNP represented by a single covariate. For the outcomes of response to melatonin treatment, a similar approach was taken; however, rather than adjusting for age the regression models were this time adjusted for severity of sleep disorder at baseline (i.e. SOL at T0W was taken as a covariate when outcome was difference in SOL, and sleep duration at T0W was taken as a covariate when outcome was difference in sleep duration). For the outcome of melatonin levels, an unadjusted linear regression model was fitted for each SNP in turn.
Results were presented as Manhattan plots, which are plots of –log10 of the p-value from the test of association with each SNP against its chromosomal location. A p-value ≤ 1 × 10–8 [or –log10(p-value) ≤ 8] is widely accepted as representing statistical significance at the genome-wide level.
To investigate the association between melatonin levels and sleep disorders, JMP statistical software (version 9; SAS, NC, USA) was used. The Wilcoxon test was used when the outcome was categorical and linear regression was used when the outcome was continuous.
Appendix 4 Details of protocol amendments
Version 7.0 (1 April 2010)
Seventh substantial amendment version 6.2 (23 June 2009) to version 7.0 (1 April 2010)
| Page no. | Comment |
|---|---|
| Throughout | Updated version and date; updated cross-referencing to subsections and page numbers where appropriate and updated sponsor’s name and email address (formerly known as Royal Liverpool Children’s NHS Trust – now known as Alder Hey Children’s NHS Foundation Trust) |
| Throughout | Removed all references to Royal Liverpool Children’s Hospital (RLCH) as this is now known as Alder Hey Hospital |
| 4, 5, 22, 70, 83 | Updated email address for trial co-ordinator and chief investigator and job titles for Statistics Team Leader, Trial Statistician and DMC Chair |
| 22 and 83 | Updated address for Royal Manchester Children’s Hospital |
| 10, 18, 48–50 | Changes to statistical considerations, namely sample size calculation and recruitment target |
| 10, 18, 48–50 | SOL calculated using actigraphy has been moved from a primary to a secondary outcome. SOL calculated using sleep diaries has been added as a secondary outcome |
| 14 | SOL has been removed from the objective of the trial |
| 22, 23, 84, 86 | Removed Great Ormond Street Hospital (GOSH) and Northampton from lists of pharmacies and centres as these centres were never initiated |
| 7, 37, 39, 45 | References to ESS put back into their original locations in the protocol. In the previous substantial amendment, the removal of the ESS was described in the substantial amendment form in error. There was never an intention to remove the scale, which has been used throughout the trial |
| 49 | Targets for centres updated |
Version 6.2 (10 July 2009)
Substantial amendment version 6.1 (4 March 2009) to version 6.2 (10 July 2009)
| Page no. | Comment |
|---|---|
| Throughout | Updated version and date; updated cross-referencing to subsections and references |
| 4, 21, 69 | Change to the trial co-ordinator – Charlotte Stockton has replaced Joanne Milton as the trial co-ordinator |
| 5 and 69 | Dr Megan Thomas is no longer an independent member of the TSC but she remains a non-independent member of the TSC |
| 11 | Amendment of text in trial summary and Figure 1 to reduce the age of inclusion to 3 years |
| 18 | Reduction in age of inclusion to 3 years at the time of registration. A number of sites have raised the current age of inclusion (5 years) as a barrier to recruitment, because children with severe sleep problems have often been prescribed melatonin before 5 years of age. We expect this amendment to increase the number of registrations by approximately 20%. The decision has been made not to produce a patient information sheet specifically for the 3–5 age range, particularly as children in the MENDS trial have moderate to severe developmental delay. If a child under 5 years is considered to have sufficient understanding they can be provided with the patient information sheet for the 5–10 age group for their caregiver to read to them. An ABAS questionnaire is available for this age group to confirm developmental delay. The cut-off for inclusion into the trial will remain as a percentile rank below 7 |
| 31 | The drug alimemazine tartrate (Vallergan) has been moved from the list of exclusion medications (section 5.2.2) to the medications that require a 14-day washout (section 5.2.4). In addition, the text relating to exclusion drugs has been amended. This reflects the decision that children who have been taking exclusion drugs for < 2 months must be excluded; however, those children who have been taking exclusion drugs for > 2 months can still be included in the trial as it is considered that they will have adjusted to their medication after 2 months |
| 77 | References updated as appropriate to include supporting documents for age reduction |
Version 6.0 (27 January 2009)
Fifth substantial amendment version 5.0 (9 July 2008) to version 6.1 (4 March 2009)
Version 6.0 (27 January 2009) was submitted to the Multicentre Research Ethics Committee and required additional amendments before approval.
| Page no. | Comment |
|---|---|
| 10–11 | Amendment to the number of participating sites in the trial summary |
| Amendment of Figure 1 to remove the reference to the completion of the neuropsychological electronic tests (DENEM and MARS) at T0W and T+12W. Amendment to Figure 1 to remove the reference to actigraphy data collection at T+1W to T+4W | |
| 18 | Removal of secondary outcomes: attention and vigilance assessed in caregivers using the car game from the DENEM project and attention and vigilance assessed in children using the ‘Go/no go’ game from the MARS battery |
| 22–3 | Table 1 – addition of pharmacy contact details for new centres |
| 37–8 | Removal of reference to completion of neuropsychological electronic tests at T0W and T+12W and reference to actigraphy data collection at T+1W to T+4W |
| 39 | Table of schedule of study procedures – removal of neuropsychological electronic tests (DENEM and MARS) and removal of actigraphy data collection from weeks T+1W to T+4W |
| 40–1 | Amendment of text to reflect the removal of actigraphy data collection from T+1W to T+4W |
| 41–2 | Reduction in the number of TESS specifically enquired about at each visit. If one of the removed TESS is reported spontaneously by a child or their caregiver it will still be reported as expected (based on its presence in the Investigator’s Brochure) and reviewed for relationship to study drug, severity and seriousness |
| 44–5 | Removal of obsolete text and instructions relating to the completion of the neuropsychological electronic tests |
| 48 | Removal of obsolete secondary outcomes: attention and vigilance assessed in caregivers using the car game from the DENEM project and attention and vigilance assessed in children using the ‘Go/no go’ game from the MARS battery |
| 51 | Table 4 – planned recruitment targets at each centre amended to reflect the addition of new centres and revised targets for existing centres based on performance to date |
| 55 | Reduction in the number of TESS specifically enquired about at each visit |
| 74 | Updated amendment summary |
| 84–7 | Appendix A – addition of contact details for the principal investigators at new centres and change to contact telephone number for Dr Tom Allport (Bristol principal investigator) |
| 88–116 | Patient information sheets and consent forms amended to remove the reference to completion of the neuropsychological electronic tests at T0W and T+12W and to remove the reference to the collection of actigraphy data from T+1W to T+4W. The list of TESS recorded in the patient information sheets was reduced to reflect the above change to the protocol |
Version 5.0 (9 July 2008)
Forth substantial amendment version 4.0 (6 May 2008) to version 5.0 (9 July 2008)
| Page no. | Comment |
|---|---|
| Throughout | Updated version and date |
| 21–2 | Table 1 – addition of pharmacy contact details for additional sites in Exeter and Torbay. Change of site name from St George’s Hospital to Queen Mary’s Hospital (London) and change to fax number for pharmacy department |
| 49 | Table 4 – change of St George’s Hospital as a collaborating and recruiting centre to Queen Mary’s Hospital. Addition of Royal Devon & Exeter Hospital and Torbay Hospital as collaborating and recruiting centres and reduction of Bristol’s recruitment target from 19 to 10 patients |
| 81–2 | Appendix A – addition of Royal Devon & Exeter Hospital and Torbay Hospital as participating sites |
Version 4.0 (6 May 2008)
Third substantial amendment version 3.0 (25 January 2008) to version 4.0 (6 May 2008)
| Page no. | Comment |
|---|---|
| Throughout | Updated version and date |
| 21 | Table 1 – updated pharmacy contact details for Nottingham and deletion of Nicola Cuff as one of the pharmacy contacts for Bristol as Nicola has left this post |
| 48 | Addition of Gulson Hospital in Coventry as a collaborating and recruiting centre and addition of Bristol Royal Hospital for Children as a recruiting centre |
| 79–80 | Addition of two health centres in Bristol and Gulson Hospital in Coventry as participating sites and change to Professor Turk’s contact details |
Version 3.0 (25 January 2008)
Second substantial amendment version 2.3 (3 December 2007) to version 3.0 (25 January 2008)
| Page no. | Comment |
|---|---|
| Throughout | Updated version and date |
| 18 | Addition to exclusion criteria of current use of sedative or hypnotic drugs, including chloral hydrate and triclofos |
| 30 | Addition of sedative and hypnotic drugs as prohibited medications throughout the trial |
| 33 | Clarification that the behaviour therapy period should be a minimum of 4 weeks’ duration, but that it can be extended to a maximum of 6 weeks if required to allow flexibility in the scheduling of the randomisation visit |
| 60–1 | Clarification that, if children are unable to provide assent, this should be documented in the medical notes and recorded on the age and stage of development specific Patient Information Sheet and Consent form |
| 64 | Clarification that date of conducting the assent (as well as the consent) process should be recorded in the medical notes |
| 85 | Addition to the parent PISC of sedative and hypnotic drugs as prohibited medications for the duration of the trial |
Version 2.0 (17 August 2007)
First substantial amendment Version 1.0 (26 April 2007) to Version 2.0 (17 August 2007)
| Page no. | Comment |
|---|---|
| Throughout | Updated version and date; correction of typographical and grammatical errors; reference to ‘ASD questionnaire’ replaced with correct name of ‘Social Communication Questionnaire; addition of email and telephone number when MCRN CTU referred to; updated cross-referencing to subsections and references |
| 9 | Updated list of abbreviations |
| 10–11 | Clarification of text in trial summary |
| 13 | Clarification of patient pack allocation |
| 17 | Clarification of outcome measures, removal of Kidscreen-10 questionnaire and addition of evaluation form for behaviour therapy booklet and addition of ESS |
| 18–19 | Inclusion/exclusion criteria revised to replace Vineland assessment with ABAS; criteria text reworded to provide easier reference; presence/absence of sleep apnoea no longer determined using Children’s Sleep Habits Questionnaire because of limited validation of cut-offs; addition of compliance check with sleep diaries as an inclusion criteria at T0W; addition of use of beta-blockers within 7 days, allergy to melatonin and regular alcohol consumption as exclusion criteria |
| 20 | Clarification of screening procedure and documentation |
| 20 | Replacement of Vineland assessment with ABAS |
| 20 | Consent/assent forms from T-4W to be sent to MCRN CTU within 7 days of registration |
| 21 | Updated contact details for pharmacy contact. Replacement of Bristol Royal Hospital for Children with Southmead Hospital |
| 23 | Label description amended to reflect replacement of Health Technology Assessment reference with EudraCT number. Process for ordering and delivery of trial supplies amended |
| 24–5 | Expanded details relating to storage and destruction of trial supplies |
| 25–6 | Clarified procedures for mixing capsule contents in a vehicle for administration |
| 28 | Clarification of unblinding process |
| 29–30 | Destruction details added |
| 30 | Trade name of alimemazine tartrate added |
| 33 | Clarification of procedure for dose increments |
| 34–6 | Replacement of Vineland assessment with ABAS; clarification of questionnaires to be completed; addition of sleep habits booklet evaluation form and CSHQ at T0W; volume of trial medication supplied updated and explained; timing of obtaining salivary samples amended |
| 37 | Table of schedule of study procedures: – replacement of Vineland assessment with ABAS, removal of Kidscreen-10 questionnaire, addition of behaviour therapy evaluation form, ESS and CSHQ at T0W |
| 38 | Sleep diaries have been piloted and amended, therefore text updated to reflect amended diary |
| 39 | Schedule for downloading actigraph data clarified |
| 39–40 | Bulleted TESS criteria simplified; ‘somnolence’ and ‘fatigue’ defined |
| 40–1 | Revision of genetic substudy section to clarify that the research will involve a genome-wide association study |
| 43 | Vineland assessment changed to ABAS; addition of CSHQ at T0W |
| 44 | Removal of Kidscreen-10 assessment; addition of ESS; clarification of CSDI scoring |
| 45 | Clarification of time points for salivary sampling |
| 46 | Primary outcome statistical analysis amended to reflect changes to sleep and seizure diaries |
| 47 | Secondary outcome measures amended as per page 17 update of end points |
| 48 | Replacement of Bristol Royal Hospital for Children with Southmead Hospital; change in recruitment target at Evelina Children’s Hospital and St George’s Hospital |
| 49–50 | Revision of genetic substudy analysis section to reflect genome-wide association study |
| 53 | Rewording of outcomes for serious adverse events and suspected unexpected serious adverse reactions |
| 54 | Additional detail on procedures by which research practitioners report serious adverse reactions, serious adverse events and suspected unexpected serious adverse reactions to the MCRN CTU |
| 59 | Clarification that all substantial amendments will be submitted for review |
| 62 | Clarification that notification of substantial amendments will be submitted to the Medicines and Healthcare products Regulatory Agency |
| 63 | Amended details of how source data will be indicated in electronic case report forms |
| 71 | Amendment summary |
| 73–5 | Updated references |
| 76–7 | Change to Principal Investigator for Derbyshire Children’s Hospital; change to Bristol site details |
| 78–110 | Patient Information Sheets and Consent forms amended to clarify when and how saliva samples are to be collected; stage of consent (registration at T-4W and randomisation at T0W) and table of procedures updated in parent Patient Information Sheets and Consent form |
| 112–14 | Amended instructions for collection of salivary samples and addition of version control to documentation |
| 119 | Removed block sizes from shipment request and addition of version control to documentation |
| 120–1 | Addition of version control to nurse’s script for providing sleep booklet |
| 122–3 | Amended drug accountability log and addition of version control to documentation |
| 125 | Addition of instructions for collection of DNA samples |
Appendix 5 Reasons for exclusion of participants from sleep outcome analyses
There were 19 patients on melatonin who did not contribute data for the analysis of the primary outcome of TST calculated using sleep diaries (five patients could not contribute data at either time point). There were 17 patients on placebo who did not contribute data for the analysis of the primary outcome (one patient did not contribute data at either time point).
| Reason for missing data | Melatonin, n | Placebo, n | ||
|---|---|---|---|---|
| T0W | T+12W | T0W | T+12W | |
| Child only had 1/7 completed sleep times | 1 | 1 | 0 | 1 |
| Child only had 2/7 completed sleep times | 1 | 1 | 1 | 0 |
| Child only had 3/7 completed sleep times | 1 | 3 | 0 | 2 |
| Child only had 4/7 completed sleep times | 5 | 1 | 0 | 5 |
| Child withdrew from study | 0 | 4 | 0 | 5 |
| Unsure of sleep start time | 2 | 1 | 1 | 1 |
| Diary lost/forgotten and not brought to clinic | 2 | 1 | 0 | 1 |
| No sleep diary for final week of treatment | 0 | 0 | 0 | 1 |
| Total | 12 | 12 | 2 | 16 |
| Reason for missing data | Placebo, n | Melatonin, n | ||
|---|---|---|---|---|
| T0W | T+12W | T0W | T+12W | |
| Child only had 1/7 completed sleep times | 2 | 1 | 3 | 2 |
| Child only had 2/7 completed sleep times | 2 | 0 | 3 | 1 |
| Child only had 3/7 completed sleep times | 2 | 4 | 2 | 2 |
| Child only had 4/7 completed sleep times | 1 | 4 | 1 | 2 |
| No file attached at time point | 2 | 1 | 3 | 1 |
| Error with watch or in download | 8 | 6 | 5 | 3 |
| Watch refused or not tolerated | 11 | 10 | 12 | 12 |
| Child broke watch | 1 | 0 | 0 | 0 |
| Watch full | 0 | 0 | 1 | 0 |
| No sleep diary information | 0 | 1 | 1 | 2 |
| Withdrew | 0 | 6 | 0 | 5 |
| Not worn at correct time | 1 | 3 | 0 | 0 |
| Watch not given out at T+11W | 0 | 2 | 0 | 2 |
| Lost watch | 0 | 1 | 0 | 0 |
| Total | 30 | 39 | 31 | 32 |
| Reason for missing data | Placebo, n | Melatonin, n | ||
|---|---|---|---|---|
| T0W | T+12W | T0W | T+12W | |
| Child only had 1/7 nights completed SOL | 0 | 0 | 0 | 1 |
| Child only had 2/7 nights completed SOL | 1 | 1 | 3 | 0 |
| Child only had 3/7 nights completed SOL | 0 | 1 | 0 | 2 |
| Child only had 4/7 nights completed SOL | 0 | 6 | 4 | 1 |
| Diary lost/forgotten and not brought to clinic | 0 | 1 | 2 | 1 |
| Unsure of sleep start time | 1 | 1 | 2 | 1 |
| No lights-out data entered | 0 | 0 | 0 | 1 |
| No sleep diary for the final week | 0 | 1 | 0 | 0 |
| Withdrew | 0 | 5 | 0 | 4 |
| Total | 2 | 16 | 11 | 11 |
| Reason for missing data | Placebo, n | Melatonin, n | ||
|---|---|---|---|---|
| T0W | T+12W | T0W | T+12W | |
| Child only had 1/7 nights data completed for SOL | 1 | 1 | 4 | 3 |
| Child only had 2/7 nights data completed for SOL | 3 | 2 | 4 | 2 |
| Child only had 3/7 nights data completed for SOL | 1 | 2 | 1 | 3 |
| Child only had 4/7 nights data completed for SOL | 5 | 5 | 3 | 6 |
| No file attached at time point | 2 | 0 | 3 | 1 |
| Error with watch or in download | 8 | 6 | 5 | 3 |
| Watch put on after ‘snuggle down to sleep time’ | 2 | 0 | 2 | 0 |
| Watch refused or not tolerated | 11 | 10 | 12 | 12 |
| Child broke watch | 1 | 0 | 0 | 0 |
| Watch full | 0 | 0 | 1 | 0 |
| No sleep diary information | 0 | 0 | 1 | 1 |
| Withdrew | 0 | 6 | 0 | 5 |
| Not worn at correct time | 1 | 3 | 0 | 0 |
| Watch not given out at T+11W | 0 | 2 | 0 | 2 |
| Lost watch | 0 | 1 | 0 | 0 |
| Total | 35 | 38 | 36 | 38 |
| Reason for missing data | Placebo | Melatonin | ||
|---|---|---|---|---|
| T0W | T+12W | T0W | T+12W | |
| Child only had 1/7 nights data completed for sleep efficiency | 3 | 0 | 4 | 2 |
| Child only had 2/7 nights data completed for sleep efficiency | 1 | 1 | 3 | 2 |
| Child only had 3/7 nights data completed for sleep efficiency | 2 | 4 | 1 | 2 |
| Child only had 4/7 nights data completed for sleep efficiency | 2 | 3 | 1 | 1 |
| No file attached at time point | 2 | 1 | 3 | 1 |
| Error with watch or in download | 8 | 6 | 5 | 3 |
| Watch refused or not tolerated | 11 | 10 | 12 | 12 |
| Child broke watch | 1 | 0 | 0 | 0 |
| Watch full | 0 | 0 | 1 | 0 |
| No sleep diary information | 0 | 2 | 2 | 3 |
| Withdrew | 0 | 6 | 0 | 5 |
| Not worn at correct time | 1 | 3 | 0 | 0 |
| Watch not given out at T+11W | 0 | 2 | 0 | 2 |
| Lost watch | 0 | 1 | 0 | 0 |
| Total | 31 | 39 | 32 | 33 |
Appendix 6 Sensitivity analyses and treatment–covariate interactions
Sensitivity analyses were carried out to investigate the robustness of the conclusions of the primary outcome analysis to missing data. The sensitivity of the results to those with missing data for the primary outcome was assessed within two population groups [total sleep A – those contributing data to the primary outcome with ≥ 5 nights observed (missing data for 1 or 2 nights only) at T0W and T+12W; total sleep B – those contributing as in total sleep A and those excluded from the primary analysis because they had > 2 nights of missing data at T0W and T+12W]. For each missing data point that a person had at both T0W and T+12W, the worst recorded night of sleep from baseline was imputed. Although the statistical significance of the results changed from statistically significant to non-significant, the clinical significance of the results did not change, with the minimum clinically importance difference of 60 minutes not contained within the 95% CIs (Table 28).
| Sensitivity analysis | Melatonin | Placebo | Difference in mean change over baseline, (95% CI), p-value | Adjusted difference (95% CI), p-value | ||||
|---|---|---|---|---|---|---|---|---|
| Baseline mean (SD) | T+12W mean (SD) | Change mean (SD) | Baseline mean (SD) | T+12 W mean (SD) | Change mean (SD) | |||
| Sleep diary (minutes) | ||||||||
| Total sleep A (nM = 51, nP = 59) | 516.12 (68.58) | 562.50 (73.28) | 46.38 (72.11) | 530.59 (68.09) | 553.54 (69.31) | 22.96 (53.42) | 23.42 (–0.37 to 47.21), p = 0.0536 | 17.93 (–3.96 to 39.82), p = 0.1074 |
| Total sleep B (nM = 61, nP = 67) | 515.71 (68.98) | 556.67 (73.21) | 40.96 (76.55) | 527.05 (66.68) | 547.30 (71.46) | 20.25 (53.58) | 20.71 (–2.24 to 43.66), p = 0.0765 | 16.19 (–4.87 to 37.26), p = 0.1307 |
| Total sleep C (nM = 58 nP = 60) | 568.86 (69.64) | 558.13 (68.35) | 10.73 (–14.43 to 35.89), p = 0.4001 | |||||
| Total sleep D (nM = 55, nP = 64) | 530.73 (65.23) | 573.14 (71.28) | 42.40 (71.74) | 542.07 (65.54) | 560.67 (70.38) | 18.60 (57.20) | 23.81 (0.39 to 47.23), p = 0.0464 | 19.30 (–2.30 to 40.90), p = 0.0794 |
Two post hoc sensitivity analyses were requested following the presentation of the preliminary results. The first was an analysis of those patients who had ≥ 5 nights observed at T+12W only (total sleep C, see Table 28). This will compare the mean TST only for days 77–84 (T12+W) between the treatment groups. The second analysis was the same as the primary analysis except that it comprised patients who had a minimum of 4 out of 7 nights observed (total sleep D, see Table 28).
The statistical significance of both of the additional analyses changed from statistically significant to non-significant but the clinical significance did not change, as in the previous sensitivity analysis.
Treatment–covariate interactions
A treatment–covariate interaction was requested following the presentation of the preliminary results. This was to investigate whether the treatment effect was greater or less in those children who had autism. The results from three models are presented below. The first model adjusts for treatment group and the baseline mean TST, the second model adjusts for the same two variables as well as whether or not the child had autism, and the third model investigates whether or not there was a treatment–covariate interaction for autism.
Model 1: mean TST at T+12W = intercept + treatment group + baseline mean TST:
| Parameter | Estimate | Standard error | p-value |
|---|---|---|---|
| Intercept | 216.26 | 46.65 | |
| Mean T0W | 0.63 | 0.08 | < 0.0001 |
| Melatonin | 22.43 | 11.05 | 0.0449 |
Model 2: mean TST at T+12W = intercept + treatment group + baseline mean TST + autism group:
| Parameter | Estimate | Standard error | p-value |
|---|---|---|---|
| Intercept | 211.45 | 47.42 | |
| Mean T0W | 0.63 | 0.08 | < 0.0001 |
| Melatonin | 22.76 | 11.12 | 0.0427 |
| Autism | 6.88 | 11.12 | 0.5372 |
Model 3: mean TST at T+12W = intercept + treatment group + baseline mean TST + autism group + treatment group*autism group:
| Parameter | Estimate | Standard error | p-value |
|---|---|---|---|
| Intercept | 209.85 | 48.42 | |
| Mean T0W | 0.63 | 0.09 | < 0.0001 |
| Melatonin | 25.32 | 17.16 | 0.0432 |
| Autism | 8.83 | 15.36 | 0.5469 |
| Melatonin*autism | –4.13 | 22.40 | 0.8539 |
The results from the models indicate that there is no statistically significant difference for the treatment–covariate interaction and inclusion of autism as the main effect and interaction did not improve the fit of the model.
Appendix 7 Mean change from baseline plotted against baseline mean total sleep time (TST) for each dose group
FIGURE 10.
Mean difference in TST (T+12W – T0W) plotted against baseline mean TST by treatment group for participants whose final dose was 0.5 mg.
FIGURE 11.
Mean difference in TST (T+12W – T0W) plotted against baseline mean TST by treatment group for participants whose final dose was 2 mg.
FIGURE 12.
Mean difference TST (T+12W – T0W) plotted against baseline mean TST by treatment group for participants whose final dose was 6 mg.
FIGURE 13.
Mean difference TST (T+12W – T0W) plotted against baseline mean TST by treatment group for participants whose final dose was 12 mg.
Appendix 8 Protocol
List of abbreviations
- AANAT
- arylalkylamine N-acetyltransferase
- ABAS
- Adaptive Behaviour Assessment System
- ABC
- Aberrant Behaviour Checklist
- ANCOVA
- analysis of covariance
- ASD
- autistic spectrum disorder
- ASMT
- N-acetylserotonin O-methyltransferase
- CNV
- copy-number variant
- CONSORT
- Consolidated Standards of Reporting Trials
- CSHQ
- Children’s Sleep Habits Questionnaire
- CTU
- Clinical Trials Unit (refers to MCRN CTU)
- DNA
- deoxyribonucleic acid
- DLMO
- dim-light melatonin onset
- DMC
- Data Monitoring Committee
- MCRN CTU
- Medicines for Children Research Network Clinical Trials Unit
- MENDS
- MElatonin in children with Neurodevelopmental Disorders and impaired Sleep
- OSAS
- obstructive sleep apnoea syndrome
- PCR
- polymerase chain reaction
- PedsQL
- Paediatric Quality of Life Inventory
- RCT
- randomised controlled trial
- SCN
- suprachiasmatic nucleus
- SNP
- single nucleotide polymorphism
- SOL
- sleep-onset latency
- TESS
- treatment-emergent signs and symptoms
- TSC
- Trial Steering Committee
- TST
- total sleep time
All abbreviations that have been used in this report are listed here unless the abbreviation is well known (e.g. NHS), or it has been used only once, or it is a non-standard abbreviation used only in figures/tables/appendices, in which case the abbreviation is defined in the figure legend or in the notes at the end of the table.
Notes
Health Technology Assessment programme
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, Department of Pharmacology and Therapeutics, University of Liverpool
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
Prioritisation Group
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, Department of Pharmacology and Therapeutics, University of Liverpool
-
Professor Imti Choonara, Professor in Child Health, Academic Division of Child Health, University of Nottingham
Chair – Pharmaceuticals Panel
-
Dr Bob Coates, Consultant Advisor – Disease Prevention Panel
-
Dr Andrew Cook, Consultant Advisor – Intervention Procedures Panel
-
Dr Peter Davidson, Director of NETSCC, Health Technology Assessment
-
Dr Nick Hicks, Consultant Adviser – Diagnostic Technologies and Screening Panel, Consultant Advisor–Psychological and Community Therapies Panel
-
Ms Susan Hird, Consultant Advisor, External Devices and Physical Therapies Panel
-
Professor Sallie Lamb, Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick
Chair – HTA Clinical Evaluation and Trials Board
-
Professor Jonathan Michaels, Professor of Vascular Surgery, Sheffield Vascular Institute, University of Sheffield
Chair – Interventional Procedures Panel
-
Professor Ruairidh Milne, Director – External Relations
-
Dr John Pounsford, Consultant Physician, Directorate of Medical Services, North Bristol NHS Trust
Chair – External Devices and Physical Therapies Panel
-
Dr Vaughan Thomas, Consultant Advisor – Pharmaceuticals Panel, Clinical
Lead – Clinical Evaluation Trials Prioritisation Group
-
Professor Margaret Thorogood, Professor of Epidemiology, Health Sciences Research Institute, University of Warwick
Chair – Disease Prevention Panel
-
Professor Lindsay Turnbull, Professor of Radiology, Centre for the MR Investigations, University of Hull
Chair – Diagnostic Technologies and Screening Panel
-
Professor Scott Weich, Professor of Psychiatry, Health Sciences Research Institute, University of Warwick
Chair – Psychological and Community Therapies Panel
-
Professor Hywel Williams, Director of Nottingham Clinical Trials Unit, Centre of Evidence-Based Dermatology, University of Nottingham
Chair – HTA Commissioning Board
Deputy HTA Programme Director
HTA Commissioning Board
-
Professor of Dermato-Epidemiology, Centre of Evidence-Based Dermatology, University of Nottingham
-
Professor of Bio-Statistics, Department of Public Health and Epidemiology, University of Birmingham
-
Professor of Clinical Pharmacology, Director, NIHR HTA programme, Department of Pharmacology and Therapeutics, University of Liverpool
-
Professor Zarko Alfirevic, Head of Department for Women’s and Children’s Health, Institute of Translational Medicine, University of Liverpool
-
Professor Judith Bliss, Director of ICR-Clinical Trials and Statistics Unit, The Institute of Cancer Research
-
Professor David Fitzmaurice, Professor of Primary Care Research, Department of Primary Care Clinical Sciences, University of Birmingham
-
Professor John W Gregory, Professor in Paediatric Endocrinology, Department of Child Health, Wales School of Medicine, Cardiff University
-
Professor Steve Halligan, Professor of Gastrointestinal Radiology, Department of Specialist Radiology, University College Hospital, London
-
Professor Angela Harden, Professor of Community and Family Health, Institute for Health and Human Development, University of East London
-
Dr Joanne Lord, Reader, Health Economics Research Group, Brunel University
-
Professor Stephen Morris, Professor of Health Economics, University College London, Research Department of Epidemiology and Public Health, University College London
-
Professor Dion Morton, Professor of Surgery, Academic Department of Surgery, University of Birmingham
-
Professor Gail Mountain, Professor of Health Services Research, Rehabilitation and Assistive Technologies Group, University of Sheffield
-
Professor Irwin Nazareth, Professor of Primary Care and Head of Department, Department of Primary Care and Population Sciences, University College London
-
Professor E Andrea Nelson, Professor of Wound Healing and Director of Research, School of Healthcare, University of Leeds
-
Professor John David Norrie, Director, Centre for Healthcare Randomised Trials, Health Services Research Unit, University of Aberdeen
-
Professor Barney Reeves, Professorial Research Fellow in Health Services Research, Department of Clinical Science, University of Bristol
-
Professor Peter Tyrer, Professor of Community Psychiatry, Centre for Mental Health, Imperial College London
-
Professor Martin Underwood, Professor of Primary Care Research, Warwick Medical School, University of Warwick
-
Professor Caroline Watkins, Professor of Stroke and Older People’s Care, Chair of UK Forum for Stroke Training, Stroke Practice Research Unit, University of Central Lancashire
-
Dr Duncan Young, Senior Clinical Lecturer and Consultant, Nuffield Department of Anaesthetics, University of Oxford
-
Dr Tom Foulks, Medical Research Council
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
HTA Clinical Evaluation and Trials Board
-
Director, Warwick Clinical Trials Unit, Warwick Medical School, University of Warwick and Professor of Rehabilitation, Nuffield Department of Orthopaedic, Rheumatology and Musculoskeletal Sciences, University of Oxford
-
Professor of the Psychology of Health Care, Leeds Institute of Health Sciences, University of Leeds
-
Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Professor Keith Abrams, Professor of Medical Statistics, Department of Health Sciences, University of Leicester
-
Professor Martin Bland, Professor of Health Statistics, Department of Health Sciences, University of York
-
Professor Jane Blazeby, Professor of Surgery and Consultant Upper GI Surgeon, Department of Social Medicine, University of Bristol
-
Professor Julia M Brown, Director, Clinical Trials Research Unit, University of Leeds
-
Professor Alistair Burns, Professor of Old Age Psychiatry, Psychiatry Research Group, School of Community-Based Medicine, The University of Manchester & National Clinical Director for Dementia, Department of Health
-
Dr Jennifer Burr, Director, Centre for Healthcare Randomised trials (CHART), University of Aberdeen
-
Professor Linda Davies, Professor of Health Economics, Health Sciences Research Group, University of Manchester
-
Professor Simon Gilbody, Prof of Psych Medicine and Health Services Research, Department of Health Sciences, University of York
-
Professor Steven Goodacre, Professor and Consultant in Emergency Medicine, School of Health and Related Research, University of Sheffield
-
Professor Dyfrig Hughes, Professor of Pharmacoeconomics, Centre for Economics and Policy in Health, Institute of Medical and Social Care Research, Bangor University
-
Professor Paul Jones, Professor of Respiratory Medicine, Department of Cardiac and Vascular Science, St George‘s Hospital Medical School, University of London
-
Professor Khalid Khan, Professor of Women’s Health and Clinical Epidemiology, Barts and the London School of Medicine, Queen Mary, University of London
-
Professor Richard J McManus, Professor of Primary Care Cardiovascular Research, Primary Care Clinical Sciences Building, University of Birmingham
-
Professor Helen Rodgers, Professor of Stroke Care, Institute for Ageing and Health, Newcastle University
-
Professor Ken Stein, Professor of Public Health, Peninsula Technology Assessment Group, Peninsula College of Medicine and Dentistry, Universities of Exeter and Plymouth
-
Professor Jonathan Sterne, Professor of Medical Statistics and Epidemiology, Department of Social Medicine, University of Bristol
-
Mr Andy Vail, Senior Lecturer, Health Sciences Research Group, University of Manchester
-
Professor Clare Wilkinson, Professor of General Practice and Director of Research North Wales Clinical School, Department of Primary Care and Public Health, Cardiff University
-
Dr Ian B Wilkinson, Senior Lecturer and Honorary Consultant, Clinical Pharmacology Unit, Department of Medicine, University of Cambridge
-
Ms Kate Law, Director of Clinical Trials, Cancer Research UK
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
Diagnostic Technologies and Screening Panel
-
Scientific Director of the Centre for Magnetic Resonance Investigations and YCR Professor of Radiology, Hull Royal Infirmary
-
Professor Judith E Adams, Consultant Radiologist, Manchester Royal Infirmary, Central Manchester & Manchester Children’s University Hospitals NHS Trust, and Professor of Diagnostic Radiology, University of Manchester
-
Mr Angus S Arunkalaivanan, Honorary Senior Lecturer, University of Birmingham and Consultant Urogynaecologist and Obstetrician, City Hospital, Birmingham
-
Dr Diana Baralle, Consultant and Senior Lecturer in Clinical Genetics, University of Southampton
-
Dr Stephanie Dancer, Consultant Microbiologist, Hairmyres Hospital, East Kilbride
-
Dr Diane Eccles, Professor of Cancer Genetics, Wessex Clinical Genetics Service, Princess Anne Hospital
-
Dr Trevor Friedman, Consultant Liason Psychiatrist, Brandon Unit, Leicester General Hospital
-
Dr Ron Gray, Consultant, National Perinatal Epidemiology Unit, Institute of Health Sciences, University of Oxford
-
Professor Paul D Griffiths, Professor of Radiology, Academic Unit of Radiology, University of Sheffield
-
Mr Martin Hooper, Public contributor
-
Professor Anthony Robert Kendrick, Associate Dean for Clinical Research and Professor of Primary Medical Care, University of Southampton
-
Dr Nicola Lennard, Senior Medical Officer, MHRA
-
Dr Anne Mackie, Director of Programmes, UK National Screening Committee, London
-
Mr David Mathew, Public contributor
-
Dr Michael Millar, Consultant Senior Lecturer in Microbiology, Department of Pathology & Microbiology, Barts and The London NHS Trust, Royal London Hospital
-
Mrs Una Rennard, Public contributor
-
Dr Stuart Smellie, Consultant in Clinical Pathology, Bishop Auckland General Hospital
-
Ms Jane Smith, Consultant Ultrasound Practitioner, Leeds Teaching Hospital NHS Trust, Leeds
-
Dr Allison Streetly, Programme Director, NHS Sickle Cell and Thalassaemia Screening Programme, King’s College School of Medicine
-
Dr Matthew Thompson, Senior Clinical Scientist and GP, Department of Primary Health Care, University of Oxford
-
Dr Alan J Williams, Consultant Physician, General and Respiratory Medicine, The Royal Bournemouth Hospital
-
Dr Tim Elliott, Team Leader, Cancer Screening, Department of Health
-
Dr Joanna Jenkinson, Board Secretary, Neurosciences and Mental Health Board (NMHB), Medical Research Council
-
Professor Julietta Patrick, Director, NHS Cancer Screening Programme, Sheffield
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Disease Prevention Panel
-
Professor of Epidemiology, University of Warwick Medical School, Coventry
-
Dr Robert Cook, Clinical Programmes Director, Bazian Ltd, London
-
Dr Colin Greaves, Senior Research Fellow, Peninsula Medical School (Primary Care)
-
Mr Michael Head, Public contributor
-
Professor Cathy Jackson, Professor of Primary Care Medicine, Bute Medical School, University of St Andrews
-
Dr Russell Jago, Senior Lecturer in Exercise, Nutrition and Health, Centre for Sport, Exercise and Health, University of Bristol
-
Dr Julie Mytton, Consultant in Child Public Health, NHS Bristol
-
Professor Irwin Nazareth, Professor of Primary Care and Director, Department of Primary Care and Population Sciences, University College London
-
Dr Richard Richards, Assistant Director of Public Health, Derbyshire County Primary Care Trust
-
Professor Ian Roberts, Professor of Epidemiology and Public Health, London School of Hygiene & Tropical Medicine
-
Dr Kenneth Robertson, Consultant Paediatrician, Royal Hospital for Sick Children, Glasgow
-
Dr Catherine Swann, Associate Director, Centre for Public Health Excellence, NICE
-
Mrs Jean Thurston, Public contributor
-
Professor David Weller, Head, School of Clinical Science and Community Health, University of Edinburgh
-
Ms Christine McGuire, Research & Development, Department of Health
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
External Devices and Physical Therapies Panel
-
Consultant Physician North Bristol NHS Trust
-
Reader in Wound Healing and Director of Research, University of Leeds
-
Professor Bipin Bhakta, Charterhouse Professor in Rehabilitation Medicine, University of Leeds
-
Mrs Penny Calder, Public contributor
-
Dr Dawn Carnes, Senior Research Fellow, Barts and the London School of Medicine and Dentistry
-
Dr Emma Clark, Clinician Scientist Fellow & Cons. Rheumatologist, University of Bristol
-
Mrs Anthea De Barton-Watson, Public contributor
-
Professor Nadine Foster, Professor of Musculoskeletal Health in Primary Care Arthritis Research, Keele University
-
Dr Shaheen Hamdy, Clinical Senior Lecturer and Consultant Physician, University of Manchester
-
Professor Christine Norton, Professor of Clinical Nursing Innovation, Bucks New University and Imperial College Healthcare NHS Trust
-
Dr Lorraine Pinnigton, Associate Professor in Rehabilitation, University of Nottingham
-
Dr Kate Radford, Senior Lecturer (Research), University of Central Lancashire
-
Mr Jim Reece, Public contributor
-
Professor Maria Stokes, Professor of Neuromusculoskeletal Rehabilitation, University of Southampton
-
Dr Pippa Tyrrell, Senior Lecturer/Consultant, Salford Royal Foundation Hospitals’ Trust and University of Manchester
-
Dr Nefyn Williams, Clinical Senior Lecturer, Cardiff University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Interventional Procedures Panel
-
Professor of Vascular Surgery, University of Sheffield
-
Consultant Colorectal Surgeon, Bristol Royal Infirmary
-
Mrs Isabel Boyer, Public contributor
-
Mr Sankaran Chandra Sekharan, Consultant Surgeon, Breast Surgery, Colchester Hospital University NHS Foundation Trust
-
Professor Nicholas Clarke, Consultant Orthopaedic Surgeon, Southampton University Hospitals NHS Trust
-
Ms Leonie Cooke, Public contributor
-
Mr Seumas Eckford, Consultant in Obstetrics & Gynaecology, North Devon District Hospital
-
Professor Sam Eljamel, Consultant Neurosurgeon, Ninewells Hospital and Medical School, Dundee
-
Dr Adele Fielding, Senior Lecturer and Honorary Consultant in Haematology, University College London Medical School
-
Dr Matthew Hatton, Consultant in Clinical Oncology, Sheffield Teaching Hospital Foundation Trust
-
Dr John Holden, General Practitioner, Garswood Surgery, Wigan
-
Dr Fiona Lecky, Senior Lecturer/Honorary Consultant in Emergency Medicine, University of Manchester/Salford Royal Hospitals NHS Foundation Trust
-
Dr Nadim Malik, Consultant Cardiologist/Honorary Lecturer, University of Manchester
-
Mr Hisham Mehanna, Consultant & Honorary Associate Professor, University Hospitals Coventry & Warwickshire NHS Trust
-
Dr Jane Montgomery, Consultant in Anaesthetics and Critical Care, South Devon Healthcare NHS Foundation Trust
-
Professor Jon Moss, Consultant Interventional Radiologist, North Glasgow Hospitals University NHS Trust
-
Dr Simon Padley, Consultant Radiologist, Chelsea & Westminster Hospital
-
Dr Ashish Paul, Medical Director, Bedfordshire PCT
-
Dr Sarah Purdy, Consultant Senior Lecturer, University of Bristol
-
Dr Matthew Wilson, Consultant Anaesthetist, Sheffield Teaching Hospitals NHS Foundation Trust
-
Professor Yit Chiun Yang, Consultant Ophthalmologist, Royal Wolverhampton Hospitals NHS Trust
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Pharmaceuticals Panel
-
Professor in Child Health, University of Nottingham
-
Senior Lecturer in Clinical Pharmacology, University of East Anglia
-
Dr Martin Ashton-Key, Medical Advisor, National Commissioning Group, NHS London
-
Dr Peter Elton, Director of Public Health, Bury Primary Care Trust
-
Dr Ben Goldacre, Research Fellow, Division of Psychological Medicine and Psychiatry, King’s College London
-
Dr James Gray, Consultant Microbiologist, Department of Microbiology, Birmingham Children’s Hospital NHS Foundation Trust
-
Dr Jurjees Hasan, Consultant in Medical Oncology, The Christie, Manchester
-
Dr Carl Heneghan, Deputy Director Centre for Evidence-Based Medicine and Clinical Lecturer, Department of Primary Health Care, University of Oxford
-
Dr Dyfrig Hughes, Reader in Pharmacoeconomics and Deputy Director, Centre for Economics and Policy in Health, IMSCaR, Bangor University
-
Dr Maria Kouimtzi, Pharmacy and Informatics Director, Global Clinical Solutions, Wiley-Blackwell
-
Professor Femi Oyebode, Consultant Psychiatrist and Head of Department, University of Birmingham
-
Dr Andrew Prentice, Senior Lecturer and Consultant Obstetrician and Gynaecologist, The Rosie Hospital, University of Cambridge
-
Ms Amanda Roberts, Public contributor
-
Dr Gillian Shepherd, Director, Health and Clinical Excellence, Merck Serono Ltd
-
Mrs Katrina Simister, Assistant Director New Medicines, National Prescribing Centre, Liverpool
-
Professor Donald Singer, Professor of Clinical Pharmacology and Therapeutics, Clinical Sciences Research Institute, CSB, University of Warwick Medical School
-
Mr David Symes, Public contributor
-
Dr Arnold Zermansky, General Practitioner, Senior Research Fellow, Pharmacy Practice and Medicines Management Group, Leeds University
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Mr Simon Reeve, Head of Clinical and Cost-Effectiveness, Medicines, Pharmacy and Industry Group, Department of Health
-
Dr Heike Weber, Programme Manager, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health
Psychological and Community Therapies Panel
-
Professor of Psychiatry, University of Warwick, Coventry
-
Consultant & University Lecturer in Psychiatry, University of Cambridge
-
Professor Jane Barlow, Professor of Public Health in the Early Years, Health Sciences Research Institute, Warwick Medical School
-
Dr Sabyasachi Bhaumik, Consultant Psychiatrist, Leicestershire Partnership NHS Trust
-
Mrs Val Carlill, Public contributor
-
Dr Steve Cunningham, Consultant Respiratory Paediatrician, Lothian Health Board
-
Dr Anne Hesketh, Senior Clinical Lecturer in Speech and Language Therapy, University of Manchester
-
Dr Peter Langdon, Senior Clinical Lecturer, School of Medicine, Health Policy and Practice, University of East Anglia
-
Dr Yann Lefeuvre, GP Partner, Burrage Road Surgery, London
-
Dr Jeremy J Murphy, Consultant Physician and Cardiologist, County Durham and Darlington Foundation Trust
-
Dr Richard Neal, Clinical Senior Lecturer in General Practice, Cardiff University
-
Mr John Needham, Public contributor
-
Ms Mary Nettle, Mental Health User Consultant
-
Professor John Potter, Professor of Ageing and Stroke Medicine, University of East Anglia
-
Dr Greta Rait, Senior Clinical Lecturer and General Practitioner, University College London
-
Dr Paul Ramchandani, Senior Research Fellow/Cons. Child Psychiatrist, University of Oxford
-
Dr Karen Roberts, Nurse/Consultant, Dunston Hill Hospital, Tyne and Wear
-
Dr Karim Saad, Consultant in Old Age Psychiatry, Coventry and Warwickshire Partnership Trust
-
Dr Lesley Stockton, Lecturer, School of Health Sciences, University of Liverpool
-
Dr Simon Wright, GP Partner, Walkden Medical Centre, Manchester
-
Dr Kay Pattison, Senior NIHR Programme Manager, Department of Health
-
Dr Morven Roberts, Clinical Trials Manager, Health Services and Public Health Services Board, Medical Research Council
-
Professor Tom Walley, CBE, Director, NIHR HTA programme, Professor of Clinical Pharmacology, University of Liverpool
-
Dr Ursula Wells, Principal Research Officer, Policy Research Programme, Department of Health