
Notes
Article history paragraph text
The research reported in this issue of the journal was funded by the HTA programme as project number 09/22/16. The contractual start date was in August 2010. The draft report began editorial review in February 2012 and was accepted for publication in June 2012. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Permissions
Copyright statement
© Queen's Printer and Controller of HMSO 2013. This work was produced by Collinsonet al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Background
DESCRIPTION OF THE HEALTH PROBLEM
Introduction
Patients with chest pain constitute the largest single category of patients admitted to hospitals in the UK. 1They are also diagnostically challenging. The majority of patients admitted have either stable ischaemic heart disease or no ischaemic heart disease. 2Such admissions are often short and clinically inappropriate. Conversely, it has been estimated that 2–7% of patients with an acute myocardial infarction (AMI) are inappropriately discharged from the emergency department. 3,4Attempts to improve diagnosis have included risk-scoring systems,5computerised decision support6,7and automated electrocardiographic interpretation. 8Although clinical assessment remains integral to the assessment of patients with chest pain, biomarker measurement has become an essential component.
Current service provision
Development of biomarker measurement in patients with chest pain and suspected acute coronary syndrome
The development of immunoassays for cardiac-specific proteins resulted in a paradigm shift in the role of biomarker measurement in patients presenting with acute chest pain. 9–11Before the development of immunoassays for the cardiac troponins, cardiac troponin T (cTnT) and cardiac troponin I (cTnI), biomarker measurement had been used for retrospective confirmation and was limited by the lack of cardiospecificity of the biomarkers. Diagnosis was orientated towards clinical features and the electrocardiogram (ECG), despite the known limitations of the ECG. The use of rapid serial measurement of creatine kinase (CK) improved the timeliness of diagnosis,12,13and measurement, of the MB isoenzyme of CK (CK-MB) improved cardiospecificity, especially when mass rather than activity measurements were introduced. 14,15The development of rapid assay techniques and the use of serial measurements of CK and CK-MB improved both speed and diagnostic sensitivity. The diagnosis of an AMI was limited only by the ability to detect a significant change between consecutive measurements, the relative change value (RCV). The RCV is determined by the assay imprecision (typically < 5%) and the intra-individual biological variation of the marker. Over short time frames (2–6 hours) this intra-individual variation is small. 12,14Diagnosis within 4–12 hours of admission was possible and diagnostic strategies were developed for early rule-out of an AMI and early discharge of patients when significant ischaemic heart disease could be excluded. 16–20
Measurement of cTnT21and cTnI22,23was initially introduced as a totally cardiospecific marker to replace CK and CK-MB mass measurement, with diagnosis based on equivalence to existing World Health Organization (WHO) criteria for an AMI. Early studies of the diagnostic efficiency of cardiac troponin showed that, when the diagnosis of an AMI was based on WHO criteria utilising CK24or CK-MB25–28measurements, troponin measurement showed excellent diagnostic sensitivity. Specificity was variable with values from 46% to 98% reported in different studies. The paradigm shift occurred when studies were performed that examined not diagnostic efficiency but independent measures such as the major adverse cardiac events (MACE) of death, myocardial infarction, readmission with unstable angina or need for urgent revascularisation. Outcome studies demonstrated that patients with a final diagnosis that excluded an AMI on WHO diagnostic criteria based on clinical and ECG findings and measurement of CK29or CK-MB30–32(hence a final diagnosis of unstable angina), but with detectable cTnT or cTnI, had a significantly higher incidence of a MACE. The finding of elevated cTnT and increased rate of MACE in patients with a WHO diagnosis of unstable angina was a consistent observation and confirmed by meta-analysis. 33The subsequent redefinition of myocardial infarction with troponin as the preferred biomarker represented the acceptance that measurement of cTnT or cTnI is the biochemical arbiter of myocardial injury and a prerequisite for the diagnosis of myocardial infarction. 9–11
The clinical need for other biomarkers of cardiomyocyte necrosis
The first generation of cardiac troponin assays were relatively insensitive. 34Comparison of the time course of release of markers of myocardial necrosis suggested that cytoplasmic markers such as myoglobin, CK and CK-MB were released earlier than cardiac structural proteins such as cTnT and cTnI. 9The apparent earlier release of cytoplasmic markers had two practical clinical consequences. First, the consensus statement on the use of biomarkers in patients for the diagnosis of an AMI, which contributed to the redefinition of AMI, recommended blood sampling at 10–12 hours from admission to achieve optimal diagnostic sensitivity. 9,35Second was a recommendation that measurement of a cytoplasmic marker in the early time period following chest pain admission should be used to cover the period of diagnostic insensitivity of cTnT and cTnI, the time period of ‘troponin blindness’. 35A practical consequence of the latter was the recommendation of measurement of myoglobin and/or CK-MB as early markers with measurement of cTnT and cTnI at 6–9 and 12–24 hours. The combined biomarker strategy led to the production of point-of-care diagnostic devices incorporating panels of markers to cover the different time windows. 36Hence, myoglobin for early sensitivity, CK-MB for early sensitivity and specificity and cardiac troponin for specificity. Progressive improvement of troponin assays showed comparable sensitivity of troponin and myoglobin for early diagnosis, challenging the troponin blindness concept,37whereas the advent of sensitive troponin assays suggests that troponin may be detectable in the early stages of myocardial necrosis before myoglobin. 38
The role of biomarkers in the diagnosis and management of the chest pain patient
The measurement of cardiac biomarkers combined with clinical symptoms and the ECG forms part of the assessment of the patient with chest pain, but the different diagnostic modalities are used in different ways. In the patient presenting with chest pain and ST-segment elevation on the ECG, management is by revascularisation by thrombolysis or preferably by percutaneous coronary intervention and stent placement. 39,40In this patient group, the role of measurement of cardiac biomarkers is to provide confirmation of diagnosis for audit of diagnostic accuracy and to provide a degree of quantitation of infarct size. 41In patients presenting without definitive ECG changes, measurement of cardiac biomarkers is crucial to subsequent management. Elevation of cardiac troponin confirms non-ST-segment elevation myocardial infarction (NSTEMI) and defines subsequent management, including therapy with antiplatelet agents and subsequent angiography. 42As the majority of patients presenting with chest pain do not have a final diagnosis of NSTEMI, the objective is to achieve diagnosis as rapidly as possible.
The role of risk stratification in chest pain diagnosis
The clinical decisions that are required in the assessment of the chest pain patient are different according to whose perspective is taken – that of emergency department clinician or that of the cardiologist. The emergency department clinician is most concerned with the early exclusion of a myocardial infarction – rule-out of a myocardial infarction – whereas the cardiologist is most concerned with confirmation of myocardial infarction – rule-in of a myocardial infarction. In patients with a diagnostic ECG [i.e. ST-segment elevation myocardial infarction (STEMI) patients] there will be immediate assessment and transfer to the coronary care unit. Patients not showing characteristic ECG changes are assessed on the basis of the ECG and clinical features into high-, medium- and low-risk groups. High-risk patients are those with clinical or ECG evidence suggestive of myocardial ischaemia. Such patients will be admitted and further investigated. Medium- or low-risk patients are those without clinical or ECG evidence of ischaemia who require biomarker measurement to exclude an AMI. Confirmation of myocardial infarction requires measurement of troponin levels at a time point when 100% diagnostic sensitivity is obtained. On the basis of previous studies a measurement at 12–24 hours was considered appropriate, and current guidelines recommend 6–9 and 12 hours. Utilising a more sensitive assay should bring this time window forward. 43
In addition to ruling in or ruling out myocardial infarction, it has been proposed that measurement of biomarkers of myocardial necrosis could be combined with measurement of other markers to allow either earlier diagnosis or risk stratification. Earlier diagnosis is based on the concept that myocardial injury will affect myocardial function. Impaired myocardial function could then be assessed either directly by measurement of biomarkers of myocardial function44or indirectly by measurement of biomarkers of circulatory stress. 45The understanding of atherothrombotic disease as a disease of plaque rupture46,47and the appreciation of the role of plaque instability48was a paradigm shift in the understanding of the pathophysiology of acute ischaemic heart disease. The measurement of risk stratification biomarkers that would reflect plaque instability is therefore attractive. 49The idea is that these markers would define high-risk groups requiring further investigation and low-risk groups who could be promptly and safely discharged.
Description of technologies for consideration for this assessment
Biomarkers for the differential diagnosis of the patient presenting with chest pain may be considered under the following categories.
Markers of cardiomyocyte necrosis
Cytoplasmic markers
Myoglobin (molecular weight 16.7 kDa) is a single-chain globular protein containing a haem prosthetic group and is the primary oxygen-carrying pigment of muscle tissues. It is found in the cytoplasm and this, combined with its low molecular weight, means that it would theoretically be released earlier than other cytoplasmic biomarkers following myocyte necrosis. Initial studies showed that this was the case and myoglobin measurement has been proposed as an early marker for an AMI. 50–52Comparison with the kinetics and cardiospecificity of other markers suggested that myoglobin measurement could be combined with other cardiac biomarkers in a panel for early diagnosis of an AMI,53–55especially in the setting of point-of-care testing. 36,56–58
Creatine kinase MB isoenzyme is the more cardiac-specific isoenzyme of CK. It is found in the cytoplasm and comprises 5–50% of the CK found in the myocardium. CK was one of the earliest cardiac biomarkers used for the biochemical detection of an AMI. 59–61Although initially measured by immunoinhibition, mass assays for CK-MB were developed and automated and form the basis of current methodology. 62–64Measurement of CK-MB mass is the most established biomarker of an AMI and is still recognised in the universal definition of myocardial infarction; the only perceived advantage is an earlier rise of CK-MB than of cardiac troponin. 15
Fatty acid-binding proteins (FABPs) are relatively small (15 kDa) proteins of 126–137 amino acids present in tissues with an active fatty acid metabolism, such as heart, liver and intestine. They reversibly bind long-chain fatty acids to facilitate their intracellular translocation. Nine distinct FABP types have been identified. Each type has a characteristic pattern of tissue distribution and a stable intracellular half-life of 2–3 days. 65The myocardial isoform, heart-type fatty acid-binding protein (H-FABP; 132 amino acids), is present predominantly in the heart, but is also found in other tissues including skeletal muscle and the distal tubal cells in the kidney. A number of studies have examined the potential role of H-FABP in the diagnosis of myocardial infarction. 66–68The interest is that H-FABP may be an early cytoplasmic marker of myocardial ischaemia and myocardial injury.
Studies on H-FABP have concentrated on its potential as a very early marker when combined with troponin (Table 1). When compared with a conventional troponin assay, measurement of H-FABP was found to provide additional diagnostic sensitivity for early presentation. 69,71,72However, the reported sensitivity of the cardiac troponin assays are low and the specificity of H-FABP is also low. Two meta-analyses have suggested that H-FABP does not meet the criteria for an early diagnostic test. 74,75H-FABP has been shown to be a prognostic marker in patients with chest pain and suspected acute coronary syndrome (ACS). 70,76,77
| Study | H-FABP | cTnT | |||
|---|---|---|---|---|---|
| Sensitivity (%) | Specificity (%) | H-FABP assay | Sensitivity cTnT (%) | cTnT assay | |
| McCann 2008,69McCann 200970 | 73 | 71 | ELISA | 55 | Fourth-generation cTnT |
| Gururajan 201071 | 87 | 93 | ELISA | 54 | Abbott Diagnostics, Maidenhead, Berkshire |
| Garcia-Valdecasas 201172 | 81 | 53 | ? | Not stated | Unknown |
| Body 201173 | 75 | 89 | ELISA | 42 | Unknown |
Studies using contemporary high-sensitivity troponin assays have suggested that there is no additional value of H-FABP measurement. 78–81Three of these studies used a sensitive H-FABP assay. 79–81
Cardiac structural proteins
The cardiac troponins form part of the cardiac contractile apparatus, the troponin–tropomyosin complex. This is found within the sarcomere of all types of striated muscle but not in smooth muscle. The troponin– tropomyosin complex acts to regulate muscular contraction and comprises three troponins: troponin C (TnC;18 kDa), troponin I (TnI; 22 kDa) and troponin T (TnT; 37 kDa), plus tropomyosin. There are three isoforms of TnT and TnI, found in cardiac muscle, fast-twitch muscle and slow-twitch muscle, encoded by individual genes. There is only one isoform of TnC, which is common to all types of muscle. The cardiac isoforms of troponin, cTnT (chromosome 1q32) and cTnI (chromosome 19q13.3), have unique sequences and hence unique amino acid compositions and structures. After development of the preliminary immunoassays for cTnT21and cTnI,23,82measurement of both troponins was introduced into routine clinical practice and cardiac troponin became the recommended biomarker for diagnosis of an AMI. 9,11,83Progressive assay improvement34has now resulted in the ability to measure cardiac troponin in normal populations, producing the current generation of high-sensitivity troponin assays. 38,84
Myocardial function markers
Natriuretic peptides
The natriuretic peptides form a family of phylogenetically highly conserved bioactive peptides that have effects on sodium and water balance. These effects may be systemic, autocrine or paracrine or a combination of all three depending on the type of natriuretic peptide. Three natriuretic peptides are found in humans. Atrial natriuretic peptide (ANP) is found in storage granules in the atria and release occurs in response to changes in vascular pressure. B-type natriuretic peptide (BNP; 3.5 kDa), originally known as brain-type natriuretic peptide, is found in both atria and ventricles and is produced in response to tension in the atrial and ventricular walls. C-type natriuretic peptide (CNP) is produced by the endothelial cells as a vasodilator. Currently, routine measurement of BNP is performed and, recently, a method for measuring ANP that may be suitable for routine clinical use has been developed. 85
B-type natriuretic peptide is not stored, but undergoes continuous transcription and translation. Increases in wall tension stretch the cardiac myocytes and result in upregulation of BNP production. In addition, BNP responds to a range of other neuroendocrine and inflammatory stimuli. 86BNP is secreted as a prohormone, pro-BNP. This then undergoes cleavage to produce the N-terminal fragment of the prohormone (NTproBNP; 8.5 kDa) and the active BNP. 87The role of BNP measurements is in the differential diagnosis of breathlessness in patients suspected of acute88–90or chronic heart failure. 91,92It has been suggested that BNP is elevated in patients with chest pain and suspected ischaemic heart disease as a consequence of myocardial ischaemia. 44,93In addition, measurement has been shown to have prognostic value in patients presenting with ACS94,95and to be a predictive risk marker for recurrent cardiac events. 96It has been suggested in guidelines that measurement may be useful in patients with chest pain and suspected ACS. 97
Vascular stress markers
Copeptin
In view of the important role of arginine vasopressin (AVP) in acute and chronic disease, knowledge of endogenous plasma AVP concentrations may be helpful in the diagnosis of chest pain and suspected cardiovascular diseases and the monitoring of treatment. Copeptin (5 kDa) is a 39-amino-acid AVP-associated glycopeptide that contains a leucine-rich core segment. Together with AVP, copeptin is derived from a 164-amino-acid precursor termed preprovasopressin, which consists of a signal peptide, AVP, neurophysin II and copeptin; thus, copeptin is the C-terminal part of pro-AVP. 45Copeptin levels are elevated after an AMI98and are associated with left ventricular dysfunction and remodelling and clinical heart failure post AMI. 99Copeptin levels are affected by gender and renal function. 100Copeptin may be elevated as a consequence of ischaemia and studies have been performed which suggest that measurement of copeptin is useful in patients with chest pain to rule in and rule out an AMI. 101,102
Adrenomedullin
Adrenomedullin (AM) is a member of the calcitonin gene-related peptide family. It is synthesised as an immature 53-amino-acid precursor and is modified by amidation into a mature 52-amino-acid peptide with an intramolecular disulphide bond. In the heart, AM is present in ventricular tissue. Although mainly produced by vascular endothelial cells, vascular smooth muscle cells and macrophages, AM can also be produced by fibroblasts, adipocytes and cardiac myocytes. It does not appear to be stored so is probably regulated by transcription triggered by proinflammatory cytokines such as tumour necrosis factor-alpha, interleukin 1 beta (IL-1β), interferon gamma and nitrous oxide. The actions of AM are mediated by the seven transmembrane domain G protein-coupled calcitonin receptor-like receptors. The potential functions of AM include vasodilator, natriuretic, diuretic, antiapoptotic and prosurvival roles, angiogenesis and modulation of inflammation. 103The predominant inotropic effect is on the atria. 104
Adrenomedullin predicts the risk of future cardiovascular events, including heart failure, in an asymptomatic population [in which it was found to be superior to C-reactive protein (CRP) measurement]105and following an AMI. 106–108Prediction of risk and prognosis in heart failure post myocardial infarction seems to be the most effective role. 109AM is raised in the circulation110,111and ventricular tissue111of patients with congestive heart failure and released from the lungs. 112Values are proportional to the degree of heart failure, although elevation does not seem to be marked in New York Heart Association (NYHA) grade I heart failure. 110AM has been studied as a prognostic marker in heart failure and has been compared with BNP. It has been demonstrated to be an independent risk predictor and synergistic with BNP in acute113and chronic heart failure. 114,115
Plaque instability markers
Inflammatory markers
The pentraxins are a superfamily of conserved proteins that are characterised by a cyclic multimeric structure. The classical short pentraxins, CRP and serum amyloid P component, are acute-phase proteins produced in the liver in response to inflammatory mediators. Long pentraxins have an unrelated, long amino-terminal domain coupled to the carboxy-terminal pentraxin domain.
CRP (25 kDa) was originally isolated as a protein that binds to the C (capsular)- polysaccharide of the cell wall of pneumococcus. CRP is a pentraxin composed of five 23-kDa subunits that plays a key role in the innate immune response. 116,117It is produced mainly by hepatocytes after stimulation by cytokines, of which interleukin 6 (IL-6) appears to be the major inducer. CRP levels increase 6 hours after acute stimuli, reaching a peak within 48 hours (up to 100-fold). With abrupt cessation of stimuli, values decrease exponentially at a rate close to the half-life of CRP (18–20 hours). 116Population based cut-offs have been proposed for risk stratification. 118Although no diurnal variation and no age or gender dependence were demonstrated in initial studies,118,119these reports were based on comparisons of CRP concentrations across dissimilar studies with heterogeneous populations. The Dallas Heart Study compared levels of CRP between different race and gender groups, and found race and gender effects. 120
There is a large body of evidence in populations with and without prior cardiovascular disease that CRP measurement predicts risk of cardiovascular events, death and risk of developing cardiac failure. 116Assessment of the additional independent prognostic value of CRP is not easy. In ACS patients, CRP is said to add to risk prediction. 121Routine measurement of CRP has been said to be valuable across the range of cardiac disease122and has been included in guidelines,97but is not routinely used.
Cytokines
Interleukin 6 (24 kDa), a 185-amino-acid polypeptide, is a pleiotropic cytokine with a variety of biological activities. 123,124IL-6 is secreted from several cell types, including endothelial cells, macrophages, lymphocytes and adipocytes. The IL-6 receptor complex consists of two membrane-bound glycoproteins, an 80-kDa ligand-binding component (termed IL-6R) and a 130-kDa signal-transducing component (termed gp130). IL-6 also activates a soluble IL-6R (sIL-6R). The activated IL-6–sIL-6R complex is a potent agonist that binds the signal-transducing component of the membrane-bound receptor, gp130, with high affinity. CRP is a physiological regulator of sIL-6R shedding in human neutrophils and markedly increases formation of the sIL-6R–IL-6 complex. 125Along with adrenergic agonists, cytokines play a major role in inducing cardiac hypertrophy. The main hypertrophic cytokines are all members of the IL-6 family and include IL-6 itself, leukaemia-inhibitory factor (LIF) and cardiotrophin 1 (CT-1). All IL-6 cytokines utilise gp130 in combination with ligand-specific receptors and mediate their effects through intracellular signal transduction pathways. 124As IL-6 is the primary cytokine of the inflammatory process126and inflammatory plaque destabilisation is key to plaque rupture, it has been suggested that IL-6 measurement may be useful in patients with ACS. IL-6 has been shown to be of cardiac origin in ACS127and to relate to other risk factors. 128Studies have shown that IL-6 measurement is prognostic in patients with ACS,129–131but data are not consistent. 132
ST2 (556 amino acids; 63 kDa), also known as IL1RL1, DER4, T1 and FIT-1, is a member of the Toll-like/interleukin 1 (IL-1) receptor superfamily. Four isoforms of ST2 exist: sST2, ST2L, ST2v and ST2Lv. Soluble ST2 (sST2) lacks the transmembrane and cytoplasmic domains contained within the structure of ST2L and includes a unique nine-amino-acid carboxy-terminal sequence. The overall structure of ST2L is similar to the structure of the type I IL-1 receptors. The ligand for ST2 is an 18-kDa protein, interleukin 33 (IL-33; also known as IL-1F11), and a member of the IL-1 family. The mode by which IL-33 exerts its effect has not been fully established but it probably acts in a similar way to other members of the IL-1 family, specifically IL-1β and IL-18,133and appears to be anti-inflammatory. 134IL-33 was originally described as a modulator of inflammation, but the IL-33/ST2 system might also participate in the fibrotic response to tissue injury. Expression of ST2 is markedly upregulated on the application of mechanical strain to cardiac myocytes. 135IL-33/ST2 signalling is a mechanically activated, cardioprotective fibroblast–cardiomyocyte paracrine system. 136,137
ST2 is elevated following myocardial infarction138and elevated levels predict an adverse outcome independently of NTproBNP. 139The role appears to be predominantly prognostic,140but a role in the emergency department population has been questioned,141as has the concept of additional risk prediction over current markers. 142The major role appears to be as a marker of myocardial remodelling143–145and hence dysfunction. In patients with acute heart failure, elevation of ST2 predicts an adverse outcome and is an independent prognostic marker. 146–149Interestingly, ST2 predicts an adverse outcome in patients presenting with acute dyspnoea regardless of whether or not they have heart failure146and in patients with pulmonary disease. 150
Growth differentiation factor 15
Growth differentiation factor 15 (GDF15), also known as MIC-1, is a secreted member of the transforming growth factor-beta superfamily. GDF15 is synthesised as a precursor protein that undergoes disulphide-linked dimerisation. Proteolysis cleaves the correctly folded GDF15 precursor protein to release the N-terminal propeptide from the mature GDF15 peptide, which is then secreted as a disulphide-linked dimer with anMr of approximately 28,000. 151–153GDF15 is detectable only in the liver and placenta,154but can be induced in the heart by myocardial infarction and pressure overload. 155,156It has been proposed that GDF15 is a cytokine released in an auto- or paracrine way that displays antihypertrophic and cardioprotective features; in particular, it protects the heart from ischaemia/reperfusion injury. 156
The preanalytical and population characteristics of GDF15 have been characterised. GDF15 correlates with CRP and cystatin C in the healthy elderly but not with NTproBNP. 157The role of GDF15 measurement has been examined in patients with both NSTEMI and STEMI. In NSTEMI, GDF15 has been proposed as a selection criterion for invasive coronary intervention. 158In prospective trials of ACS [the Global Utilization of Strategies To Open occluded arteries (GUSTO)-IV non-ST-elevation acute coronary syndrome trial159and the PRavastatin OR atorVastatin Evaluation and Infection Therapy – Thrombolysis In Myocardial Infarction 22 (PROVE IT-TIMI 22) trial160] GDF15 has been shown to be a prognostic marker. GDF15 has been shown to be a long-term prognostic marker in patients admitted with NSTEMI161and a prognostic marker in STEMI162,163and in the general AMI population. 164Measurement of GDF15 has been shown to be prognostic in the chest pain population165and to add to conventional risk scores such as the Global Registry of Acute Coronary Events (GRACE) score. 166Although some workers have concluded that GDF15 measurement is useful across the range of coronary artery disease,167when examined as part of a multimarker strategy it was not considered useful above conventional risk factor prediction. 168GDF15 is elevated in patients with chronic heart failure and correlates with NYHA class. Values correlate with NTproBNP, but are independent predictors of prognosis. 169GDF15 may have a role to play in diagnosis and risk stratification in patients with cardiac failure, but more studies are required. The role of GDF15 appears to be associated with the remodelling of the heart;170the mechanism appears to be through the inhibition of integrins. 171
Myeloperoxidase
Myeloperoxidase (MPO) is a peroxidase enzyme (EC 1.11.1.7) that is most abundant in neutrophil granulocytes (a subtype of white blood cells). The 150-kDa MPO protein is a dimer consisting of two 15-kDa light chains and two variable-weight glycosylated heavy chains bound to a prosthetic haem group. Three isoforms have been identified, differing only in the size of the heavy chains. The situation with MPO has been confusing (Table 2). Early studies in patients presenting with chest pain suggested that, in patients with undetectable troponin, MPO measurement predicted short-term risk of myocardial infarction and risk of MACE. 172Studies in chest pain and ACS patients confirmed the prognostic value of MPO measurements,173–175although not in all patients. 176There are some major limitations of these published studies. First, not all studies have used a contemporary high-sensitivity troponin assay. Second, there are some significant preanalytical questions. MPO is present in significant amounts in leucocytes and is released with clotting. Only EDTA (ethylenediaminetetraacetic acid) plasma is a suitable sample matrix and serum should not be used. Mechanistically, one study has shown no relationship between MPO and coronary disease when assessed angiographically. 183
| Study | Early diagnosis | Prognosis | Contemporary troponin assay | Comments |
| Brennan 2003172 | Admission values | Second-generation cTnT (Roche diagnostics, Indianapolis, IN) | Plasma ns, ACS patients | |
| Baldus 2003173 | Baseline samples | Third-generation cTnT (Roche diagnostics, Mannheim) | Serum, CAPTURE trail | |
| Cavusolglu 2007174 | Baseline samples, long-term prognosis | Matrix not stated, ACS patients | ||
| Apple 2007176 | Admission sample, not significant | cTnI Dimension® (Dade-Behring, Glasgow, Delaware) | Lithium-heparin plasma, chest pain patients | |
| Morrow 2008175 | Baseline samples, short-term outcome | Citrated plasma ns, TACTICS TIMI 18 | ||
| McCann 2008,69McCann 200970,76 | Admission sample, not significant | Admission sample, not significant | Third-generation cTnT (Roche diagnostics, Burgess Hill, Sussex) | Serum |
| Apple 2009177 | Admission sample, not significant | cTnI Dimension, third-generation cTnT (Roche diagnostics, Indianapolis, IN) | Lithium-heparin plasma, chest pain patients | |
| Rudolph 2010178 | Diagnosis in the early phase of onset of chest pain in patients negative for cTnI | EDTA plasma | ||
| Scirica 2011179 | Did not add to cTnI and BNP | Seimens Ultra cTnI (Siemens Healthcare Diagnostics, Deerfield, IL) | EDTA plasma, MERLIN-TIMI 36 | |
| Sawicki 2011180 | Diagnosis in the early phase of onset of chest pain in patients negative for cTnI | Architect (Abbot Diagnostics, IL) | EDTA plasma, generalised chest pain | |
| Oemrawsingh 2011181 | Predictive value after NSTEMI | Serum and lithium-heparin plasma, CAPTURE trial | ||
| Apple 2011182 | MPO is an independent risk predictor | EDTA plasma |
Matrix metalloproteinases and their inhibitors
Matrix metalloproteinases (MMPs) are zinc-dependent endopepetidases collectively capable of degrading extracellular matrix proteins and processing bioactive molecules. They are known to be involved in the cleavage of cell surface receptors, the release of apoptotic ligands (such as the Fas ligand) and chemokine activation and inactivation. They are therefore involved in both remodelling and inflammation. There are 28 MMPs (Table 3).
| Abbreviation | Other names |
| MMP1 | Interstitial collagenase |
| MMP2 | Gelatinase-A, 72-kDa gelatinase |
| MMP3 | Stromelysin 1 |
| MMP7 | Matrilysin, PUMP 1 |
| MMP8 | Neutrophil collagenase |
| MMP9 | Gelatinase-B, 92-kDa gelatinase |
| MMP10 | Stromelysin 2 |
| MMP11 | Stromelysin 3 |
| MMP12 | Macrophage metalloelastase |
| MMP13 | Collagenase 3 |
| MMP14 | MT1-MMP |
| MMP15 | MT2-MMP |
| MMP16 | MT3-MMP |
| MMP17 | MT4-MMP |
| MMP18 | Collagenase 4, xcol4,Xenopuscollagenase |
| MMP19 | RASI-1, occasionally referred to as stromelysin-4 |
| MMP20 | Enamelysin |
| MMP21 | X-MMP |
| MMP23A | CA-MMP |
| MMP23B | |
| MMP24 | MT5-MMP |
| MMP25 | MT6-MMP |
| MMP26 | Matrilysin-2, endometase |
| MMP27 | MMP-22, C-MMP |
| MMP28 | Epilysin |
The MMPs are inhibited by specific endogenous tissue inhibitor of metalloproteinases (TIMPs), which comprise a family of four protease inhibitors: TIMP-1, TIMP-2, TIMP-3 and TIMP-4.
Matrix metalloproteinase 9 (MMP9) correlates with the extent of angiographically described coronary disease184and disease progression,185with similar data reported for MMP1,186whereas MMP2 has been reported to be associated with calcified plaque. 187TIMP-1 has been reported to be a predictor of death and myocardial infarction in patients with angiographically demonstrated coronary disease. 188MMPs do not appear to be consistently good tests for myocardial infarction, but do appear to be prognostic markers. 69,177,189,190MMP9 concentrations correlate with the extent of infarction. 191
It is likely that MMP levels reflect remodelling rather than acute disease. 192,193After myocardial infarction, MMP2 levels show an inverse correlation and MMP9 levels a positive correlation with ventricular dysfunction,194so MMPs are related to heart failure rather than ACS. Post-infarct survival and development of heart failure is predicted by MMP3,195MMP9196,197and TIMP-1. 197
Studies in heart failure patients show variable concordance. Reports of changes in MMPs and TIMPs in patients with heart failure are generally consistent; reports of the ability to predict outcome are not. In heart failure, MMP1,198MMP2,199,200MMP9199,200and TIMP-1,198,199,201–203but not MMP3,199are reported as elevated. One large study has reported that MMP9 is not elevated in patients with heart failure after adjustment for other variables. 203In other studies MMP1 levels are reduced,201but TIMP-2 is not elevated in heart failure. 200Noradrenaline correlates with MMP2 levels in heart failure and appears to increase its synthesis. 200MMP1 but not TIMP-1 has been shown to predict outcome. 201TIMP-1 has also been shown to be an outcome predictor. 203MMP2 (but not MMP3, MMP9 or TIMP-1) and BNP correlated with NYHA grade (although not with each other) and were independent outcome predictors. 199Others have found MMP3, but not MMP2, and BNP to be independent outcome predictors. 204MMP9, despite the ability to predict adverse effects after an AMI, appears consistently to be a poor outcome predictor199,203–205in heart failure patients, especially compared with BNP205or TIMP-1. 203
Currently, MMPs and TIMPs remain poorly understood. Until reference interval values of MMPs and TIMPs are defined, including biological determinants of variation, methodological standardisation occurs, the pathophysiology is more clearly defined206and large studies are undertaken directly comparing the individual candidates using agreed and validated methods, it is unlikely that these analytes will achieve clinical application.
Pregnancy-associated plasma protein A
Pregnancy-associated plasma protein A (PAPP-A), also known as pappalysin 1, is a high-molecular-weight (200 kDa) zinc-binding metalloproteinase (EC 3.4.24.79) that cleaves insulin-like growth factor-binding proteins. 207,208It has been suggested that PAPP-A is a marker of plaque instability. The initial study that documented increased levels of PAPP-A in atheromatous plaques also documented elevated serum levels. It was suggested that measurement of PAPP-A could be used as an additional diagnostic and prognostic biomarker in patients presenting with chest pain. 209Early studies showed that there are more patterns in patients with chest pain and it was suggested that it might not prove to be a useful early biomarker for diagnosis210but does appear to be prognostic. 211,212Elevations of PAPP-A were reported to correlate with the extent of both coronary213and peripheral214vascular disease.
There are a number of problems with the measurement of PAPP-A. It has been demonstrated that the molecular form circulating in patients with ACS is different from that measured in pregnancy. The ACS-related form circulates as a monomer not complexed with a proform of eosinophilic major basic protein. The pregnancy-related form circulates as a complex with eosinophilic major basic protein. 215–217Different assays may therefore perform differently. 218It has been shown that free PAPP-A is a better predictor than total PAPP-A. 219
Finally, it has been demonstrated that administration of intravenous heparin causes a rise in PAPP-A due to release from the arterial wall. 220,221
Table 4provides a summary of studies of PAPP-A.
| Study | Early diagnosis of chest pain | Prognostic marker | Population | Comments |
| Dominguez-Rodriguez 2005222 | No elevation in AMI patients | Comparison of levels in AMI patients and age-matched control subjects | ||
| Elesber 2007223 | Predicts a diagnosis of ACS | Chest pain patients | ||
| Sanchis 2008224 | Not prognostic | Chest pain patients | ||
| Iversen 2008225 | Early elevation in STEMI | STEMI patients | ||
| Kavsak 2009226 | Prognostic of long-term outcome | Chest pain patients | ||
| Iversen 2009227 | Risk of myocardial infarction and death in NSTE-ACS and death in STEMI patients | NSTE-ACS and STEMI patients | ||
| Iversen 2010228 | Chest pain, normal ECG, normal biomarkers | |||
| Khan 2011229 | cTnT superior to PAPP-A and MPO | Chest pain patients | High-sensitivity cTnT | |
| Body 2011230 | Not prognostic |
Ischaemia markers
Ischaemia-modified albumin
The N-terminal portion of human serum albumin is known to be a binding site for transition metal ions, binding cobalt, copper and nickel in their (II) forms. 231It is also known that the N-terminal portion of human serum albumin is susceptible to biochemical degradation and is less stable than the albumin of other species. 232Ischaemia-modified albumin (IMA) is a form of human serum albumin in which the N-terminal amino acids have been affected so that they are unable to bind transition metals. Measurement of IMA is with the albumin cobalt-binding (ACB®, Ischemia Technologies, Denver, CO) test. This involves addition of a known amount of cobalt to a serum sample, addition of dithiothreitol to bind the unbound cobalt and measurement of the colorimetric change. As normal albumin will bind cobalt, the amount of free cobalt, hence the absorbance, will be proportional to the amount of IMA present.
The postulated mechanism is that localised ischaemia results in acidosis and release of copper(II) from weak binding sites on circulating proteins and peptides. This is then scavenged by albumin. Copper-bound albumin is then damaged by hydroxyl free radicals, causing removal of the three N-terminal amino acids and release of the copper(II) ion to repeat the process in a chain reaction. 233This has not been confirmed, however. In a study of patients with increased IMA the N-terminal portion of albumin was sequenced and no evidence of N-terminal degradation or truncation was found. 234Recent physicochemical studies of cobalt binding to human serum albumin have suggested a different explanation. Three binding sites for cobalt were identified, two of which showed greater avidity than the N-terminal binding site. 235Fatty acid binding to albumin occurs at one of the additional cobalt binding sites with a negative allosteric interaction. It is hypothesised that in myocardial ischaemia, the release of fatty acids results in binding of fatty acids to albumin. This would then reduce the ability of albumin to take up cobalt and would account for the presence of IMA. 235If this also produced a conformational change in the albumin affecting the N-terminal site, it would also reduce cobalt binding. The most consistent finding across all studies of IMA is of a high negative predictive value. This has been highlighted in a recently published meta-analysis specifically examining the role of IMA as a rule-out test. 236The role of IMA has been reviewed and it is not considered suitable for routine laboratory measurement. 237
Chapter 2 Research objectives and research questions
The objectives of this study were:
-
to test the diagnostic accuracy for an AMI of highly sensitive troponin assays and a range of new cardiac biomarkers of plaque destabilisation, myocardial ischaemia and necrosis
-
to test the prognostic accuracy for adverse cardiac events of highly sensitive troponin assays and this range of new cardiac biomarkers
-
to estimate the potential cost-effectiveness of using highly sensitive troponin assays or this range of new cardiac biomarkers instead of admission and 12-hour troponin measurements.
These objectives were addressed with the following research questions:
-
Is a panel of cardiac markers, as currently available, required for early diagnosis of myocardial infarction?
-
Do novel cytoplasmic markers of myocardial damage contribute to the early differential diagnosis of patients presenting with chest pain?
-
Are all high-sensitivity cardiac troponin markers of equivalent diagnostic efficiency?
-
Do markers of myocardial dysfunction contribute to the early differential diagnosis of patients presenting with chest pain?
-
Do markers of vascular dysfunction contribute to the early differential diagnosis of patients presenting with chest pain?
-
What is the prognostic role of cytoplasmic markers of myocardial damage compared with troponin measurement?
-
What is the prognostic role of high-sensitivity troponin assays?
-
What is the prognostic role of myocardial dysfunction compared with troponin measurement?
-
What is the cost-effectiveness of the identified strategies?
Study rationale
The Randomised Assessment of Treatment using Panel Assay of Cardiac markers (RATPAC) trial was a multicentre pragmatic randomised controlled trial and economic evaluation of a point-of-care cardiac marker panel in the management of patients with a suspected, but not proven, AMI in six emergency departments in the UK. The RATPAC – Contemporary Biomarker Evaluation (RATPAC-CBE) study aimed to examine whether the biomarker panel measured by point-of-care testing was the most appropriate diagnostic strategy or whether other cardiac biomarkers could replace or supplement the point-of-care biomarker panel.
The archived blood samples from the RATPAC study represented an ideal opportunity to extend the findings of the RATPAC trial in a cost-effective way. The enrolled patients were fully characterised and were followed up for MACEs. The population was also unique as it represented one found within the emergency department and selected on the basis of low cardiac risk rather than one enrolled in a clinical trial with a high prior probability of cardiovascular disease. This is a major limitation of many existing biomarker studies and has been highlighted in recent editorials and the consensus statement83on biomarker series of the working group of the European Society of Cardiology. As with other biomarker studies of this type, patient enrolment was prospective, but analysis was retrospective.
Selection of biomarkers for investigation was based on evidence obtained from reviewing the existing literature (seeChapter 1) and from knowledge of current and potential future clinical practice. In selecting the biomarkers for evaluation, the most important criteria were that:
-
a validated automated assay was available for the biomarker, which could be used in the routine clinical laboratory
-
there was already existing evidence suggesting that the biomarker might be of value
-
a comprehensive comparative evaluation of the biomarker had not already been performed
-
an appropriate sample was available and there was adequate sample volume.
The biomarkers finally selected were CK-MB, myoglobin, cTnT and cTnI measured with a range of high-sensitivity assays, H-FABP, copeptin and BNP measured as NTproBNP. Although CRP was initially considered, the lack of clinical use, despite widespread availability of the assay and sample volume limitations, mitigated against its final inclusion.
Chapter 3 Methods
Population
The population was patients presenting to the emergency department with chest pain due to a suspected, but not proven, AMI in which cardiac biomarker measurement by point-of-care testing could potentially rule out an AMI and allow discharge home. All patients with chest pain were considered for participation, but were then excluded if they met any of the following criteria:
-
Diagnostic ECG changes for an AMI or high-risk ACS (> 1 mm ST deviation or > 3 mm inverted T waves). These patients are at high risk of adverse outcome and require inpatient care even if initial cardiac biomarker testing is negative.
-
Known coronary heart disease presenting with prolonged (> 1 hour) or recurrent episodes of typical cardiac-type pain. These patients have unstable angina and require inpatient care for symptom control even if cardiac biomarker testing is negative.
-
Proven or suspected serious non-coronary pathology such as pulmonary embolus that requires inpatient care even if an AMI is ruled out.
-
Comorbidity or social problems that require hospital admission even if an AMI can be ruled out.
-
Patients with an obvious non-cardiac cause of chest pain such as pneumothorax or muscular pain, in whom an AMI can be excluded as a possible cause without resorting to further diagnostic testing.
-
Presentation > 12 hours after the most significant episode of pain. In such patients a single troponin measurement would clearly be more appropriate than panel testing.
-
Previous participation in the RATPAC trial.
-
Inability to understand the trial information because of cognitive impairment.
-
Non-English-speaking patients for whom translation facilities were not available.
For every fourth week of trial recruitment the research nurse at each hospital examined emergency department attendance lists to identify patients attending with chest pain and record basic demographic details and reasons for exclusion. The huge number of attendances with chest pain meant that undertaking this process throughout the whole trial would have produced an excessive workload, whereas monitoring every fourth week achieved the aim of reporting sample selection within acceptable use of resources.
Recruitment and randomisation
Research nurses and emergency department staff identified eligible patients, provided trial information and obtained written consent. Participants were then randomly allocated to receive either (1) diagnostic assessment using the point-of-care biochemical marker panel or (2) conventional diagnostic assessment without the panel.
The Nottingham Clinical Trials Unit (CTU) generated a simple randomisation sequence, stratified by centre, which was not revealed to any person involved in patient recruitment. Recruiting doctors and research nurses accessed a secure website provided by the Nottingham CTU and entered participant details. The CTU revealed each participant's allocated treatment group to the emergency department only after the participant's details were entered, written consent was confirmed and the participant irrevocably entered into the trial.
Planned interventions
Participants were randomised to receive either
-
diagnostic assessment using the point-of-care biochemical marker panel or
-
conventional diagnostic assessment without the panel.
The only difference between the two arms of the trial was that patients in the intervention arm received testing with the point-of-care panel. The use of all other tests and treatments, and decision-making in the emergency department, was at the discretion of the attending clinician.
The point-of-care cardiac marker panel comprised CK-MB (mass), myoglobin and cTnI, measured at presentation and 90 minutes later, using the Stratus® CS analyser (Siemens Healthcare Diagnostics, Camberley, Surrey, UK). Clinical staff were trained to use the test and given guidance in interpretation of the results. A recommended protocol that advised a first panel test immediately after initial emergency department assessment and a second panel test 90 minutes later was used. The protocol then advised hospital admission or discharge on the basis of point-of-care results. Decisions were ultimately at the discretion of clinical staff in patients randomised to use of the point-of-care protocol and its use was not enforced.
In addition to obtaining consent, collecting data and random allocation to use of the point-of-care test, the only change to routine practice was that clinical staff took an additional blood sample for storage (without repeating venepuncture) each time a point-of-care blood sample was required. The additional blood remaining after point-of-care testing was transported to the hospital laboratory to be centrifuged and refrigerated. Batches of samples were then transported quarterly to St George's Hospital for storage and subsequent secondary analysis.
The RATPAC study was a pragmatic trial intended to determine whether or not point-of-care testing should be standard practice for patients presenting to the emergency department with a suspected AMI and was designed to compare two alternatives (management with and without point-of-care testing) under routine conditions. This pragmatic design had the following implications:
-
There was no attempt to blind clinical staff, patients or carers to the allocated treatment group after randomisation.
-
The point-of-care test was provided with a recommended protocol for use, but management decisions were ultimately at the discretion of the clinical staff.
-
All other diagnostic tests and the use of laboratory blood tests in the control group were at the discretion of the clinical staff.
-
Blood samples were taken only for the purposes of clinical management. Additional blood samples to evaluate theoretical management strategies or to evaluate the accuracy of diagnostic assessments were not taken. The additional blood samples taken at the time of blood draw for point-of-care tests were utilised. This allows direct comparison with conventional management strategies.
Outcome measures
The primary outcome in the RATPAC study was the proportion of patients successfully discharged home after emergency department assessment. To be considered successfully discharged the patient had to (1) either have left the hospital or be awaiting transport home with a discharge decision having been made at 4 hours after initial presentation and (2) suffer no adverse event (as defined below) during the following 3 months.
Secondary outcomes were:
-
reattendance at and/or readmission to hospital over the following 3 months
-
adverse events (death, non-fatal AMI, emergency revascularisation or hospitalisation for myocardial ischaemia)
-
the proportion of admitted patients ultimately diagnosed as having an AMI by the universal definition of myocardial infarction. 11
Recruiting staff recorded baseline data, the results of initial assessment (including any biochemical cardiac tests) and admission or discharge from the emergency department. Research nurses then used emergency department and hospital inpatient notes to record management decisions at initial attendance and admission, extract resource-use data and identify subsequent attendances/admissions and adverse events up to 3 months.
Research nurses checked patient status (dead or alive) at 1 and 3 months using hospital information systems. Participants who were not recorded as dead were mailed a questionnaire at 1 and 3 months from the University of Sheffield to identify adverse events and hospital attendances.
Classification of cases of AMI and adverse events was carried out by blind independent review of the relevant data. A single reviewer blinded to treatment group classified emergency department reattendances, subsequent hospital admissions and outpatient reviews as either potentially chest pain related (including non-cardiac conditions that could have initially presented as chest pain) or clearly non-chest pain related.
Ethical arrangements
Ethical approval was granted by Leeds East Research Ethics Committee (07/Q1206/22) and review was provided by the local research ethics committee at each participating centre. The study was performed in accordance with the Declaration of Helsinki. 238The trial was conducted in accordance with Medical Research CouncilGuidelines for Good Clinical Practice in Clinical Trials. 239The University of Sheffield was the sponsor for the trial. The RATPAC trial was registered with the international clinical trials authority (ISRCTN378239293).
All participants were asked to provide written informed consent. Although participants were recruited in an emergency setting and there was only a limited amount of time available for considering trial information, the nature of the selected group (in particular the exclusion of people clearly requiring hospital treatment) ensured that eligible patients would not be incapacitated by their medical condition. No provision was made for recruitment of incapacitated patients by personal or professional legal representatives.
Blood samples for the subsequent analysis were made anonymous as follows. Each patient received a clinical trial number that was used as the prime identifier in all subsequent data analysis. For each participating site, test packs were prepared. One test pack was to be used for each patient entered into the trial. Each test pack contained the following: four primary sample tubes [two lithium-heparin tubes for point-of-care testing plus two serum separator gel tubes for additional sampling (Becton Dickinson, Oxford, UK)], four long-term storage tubes, preprinted barcode labels and a site-specific pro forma. On first presentation, one lithium-heparin tube was taken and used for point-of-care testing and one serum separator gel tube was taken and sent to the laboratory. The serum separator gel tube was allowed to clot, was centrifuged and the supernatant serum separated into two of the long-term storage tubes, which were labelled with a preprinted barcode. The same preprinted barcode was then used to label the site-specific pro forma, which was also labelled with the patient trial number but with no additional information. At 90 minutes the process was repeated. The long-term storage tubes were frozen to –20 ºC and then transferred to St George's Hospital for long-term storage until analysis.
Diagnostic criteria
The universal definition of myocardial infarction11was used to categorise patients into those with or without an AMI utilising clinical, ECG, trial and local laboratory-derived cardiac troponin values and troponin measurements subsequently performed in the trial central laboratory on the admission and 90 minute samples using the Siemens Ultra assay as the predicate troponin method.
The initial working diagnosis and final diagnosis were those recorded in the notes by a senior clinician at the end of the initial emergency department assessment and at the end of hospital admission, respectively, based on available information at that time. This included the results of point-of-care testing as well as the results of local laboratory troponin measurements. Patients were classified as having an AMI on the basis of appropriate clinical features, electrocardiographic changes and the presence of a rise in troponin level above the diagnostic discriminant of the relevant assay in use locally and no alternative clinical cause of a troponin rise. Patients with a troponin rise consistent with an AMI and a final diagnosis of ACS or an AMI were classified as having an AMI. Patients with no troponin rise consistent with an AMI and a final diagnosis that was neither ACS nor an AMI were classified as not having an AMI. Patients with a final diagnosis of ACS or an AMI but no troponin rise were assessed by a single reviewer blind to treatment group who reviewed the initial and next-day ECG and categorised these patients as having an AMI only if an ECG showed ST-segment elevation and coronary reperfusion was performed. Patients with a troponin rise and a final diagnosis other than ACS or an AMI were assessed by two reviewers blinded to treatment group who reviewed case details and decided whether or not an AMI was the most likely diagnosis. Disagreements were resolved by discussion and patients classified as having an AMI or not.
Diagnostic review was then performed by two independent clinicians with access to all of the relevant information, utilising the 99th percentile from the local laboratory and also including troponin measurements performed in the central laboratory and compared with the final diagnosis. The trial admitted patients suspected of having an AMI on the basis of a rise in levels of cTnI, CK-MB or myoglobin measured on the Stratus CS analyser. These measurements were not used for the final diagnostic classification. All patients with a cTnI (measured on the Siemens Ultra assay) exceeding the 99th percentile or a troponin measurement from the local laboratory exceeding the 99th percentile were reviewed and the final diagnosis confirmed. Patients with a troponin rise and a final diagnosis other than ACS or an AMI were reviewed to decide whether or not an AMI was the most likely diagnosis. Disagreements were resolved by discussion and patients classified as having an AMI or not. Patients were categorised as AMI (type 1 MI, primary ischaemic cardiac injury), troponin elevation not due to an AMI but with a probable background of underlying coronary atheroma (type 2 MI, secondary ischaemic cardiac injury) and no myocardial injury.
Data processing
Trial data were collected on the case report form and follow-up form and were then entered by the research nurses into an online database provided on a secure central sever by the Sheffield CTU. The system had a full electronic audit trail. Quality control procedures were applied to validate the trial data. Error reports were generated when data clarification was required. All activities were performed in accordance with Sheffield Clinical Trials Research Unit (CTRU) standard operating procedures.
Core patient data were maintained by the trial co-ordinator using a unique trial number. Demographic, risk factor and diagnostic data were extracted as CSV (comma-separated value) files and transferred for combination with the analytical data. All of the analytical data were stored in a relational database (Microsoft Access, Microsoft Corporation, Redmond, WA, USA) using the combination of trial number and unique sample number as identifiers. Database queries were extracted into Microsoft Excel for statistical analysis.
Analytical methods
Biochemical measurements were performed at trial sites (cardiac troponin measurements for local diagnostic classification), were standardised across sites for point-of-care testing and were performed at the core laboratory.
Trial sites
Trial sites measured cardiac troponin as follows:
-
Siemens cTnI Ultra® assay (three sites: Barnsley, Leeds and Leicester) – the cTnI Ultra measurements were performed using an ADVIA Centaur® XP system (Siemens Healthcare Diagnostics, Camberley, Surrey, UK). The detection limit of the instrument is 6 ng/l and the upper limit is 50,000 ng/l. The claimed 10% coefficient of variation (CV) is 30 ng/l with a 99th percentile of 40 ng/l. Decision limits for diagnosis of an AMI used at the three sites were as follows: Barnsley 200 ng/l, Leeds 50 ng/l and Leicester 60 ng/l.
-
Abbott cTnI (one site: Edinburgh) was measured on an Architect i2000SR system® (Abbott Diagnostics). The detection limit of the instrument is 10 ng/l and the upper limit is 50,000 ng/l. The claimed 10% CV is 32 ng/l and the 99th percentile 12 ng/l. A decision limit for diagnosis of an AMI of 50 ng/l was used.
-
Beckman AccuTnI™enhanced assay (one site: Bristol Frenchay) measurements were performed using an Access®2 system (Beckman-Coulter, High Wycombe, Buckinghamshire, UK). The detection limit of the instrument is 10 ng/l and the upper limit is 100,000 ng/l. The claimed 10% CV is 60 ng/l with a 99th percentile of 40 ng/l. A diagnostic discriminant for an AMI of 60 ng/l was used.
-
Roche cTnT (one site: Plymouth Derriford) measurements were performed using a Modular® E170 system (Roche Diagnostics, Burgess Hill, Sussex). The detection limit of the assay is 10 ng/l with an upper limit of 25,000 ng/l. The claimed 10% CV is 30 ng/l with a 99th percentile of 10 ng/l. The 99th percentile was used for diagnosis.
Point-of-care testing assays (all sites)
The cardiac panel measured was myoglobin, CK-MB and cTnI. Measurements were performed using the Stratus CS analyser. The analytical characteristics of the assays for each analyte were as follows. Myoglobin: detection limit 1μg/l, analytical range 1–900μg/l, interassay CV 1.9–12.7% (56–308μg/l), 95% reference interval, males 21–98μg/l, females 19–56μg/l, combined 20–82μg/l; CK-MB: detection limit 0.3μg/l, analytical range 0.3–150μg/l, interassay CV 0.15–1.27% (3.7–39.3μg/l), 95% reference interval 0.6–3.5μg/l; cTnI: detection limit 0.03μg/l, analytical range 0.03–50μg/l, interassay CV 4.0–8.2% (0.067–0.344μg/l), the 99th percentile of the assay is 0.07μg/l.
Core laboratory assays
Cardiac troponin
Three high-sensitivity cardiac troponin measurements were performed.
Cardiac troponin T
The Roche Diagnostics high-sensitivity cTnT assay was used. The high-sensitivity cTnT measurements were performed using an Elecsys®2010 system (Roche Diagnostics). The detection limit of the assay is 3 ng/l with an upper limit of 10,000 ng/l. The claimed 10% CV is 13 ng/l with a 99th percentile of 14 ng/l.
Cardiac troponin I
The cTnI Ultra measurements were performed using an ADVIA Centaur® XP system. The detection limit of the instrument is 6 ng/l and the upper limit is 50,000 ng/l. The claimed 10% CV is 30 ng/l with a 99th percentile of 40 ng/l. 240
The AccuTnI enhanced measurements were performed using an Access 2 system. The detection limit of the assay is 1 ng/l and the upper limit is 100,000 ng/l. The claimed 10% CV is 30 ng/l with a 99th percentile of 40 ng/l.
Heart-type fatty acid-binding protein
Heart-type fatty acid-binding protein measurements were performed using the Evidence Cardiac Array measured on the Evidence Investigator (Randox Laboratories, Crumlin, County Antrim, UK). The detection limit of the assay is 1.5 mg/l and the upper limit is 100 mg/l, with a CV of 9.1% at 3.1 mg/l, 7.5% at 17.6 mg/l and 9.8% at 44.1 mg/l. The 95th percentile is 2.5 mg/l and the 99th percentile is 3.0 mg/l.
Myoglobin
Myoglobin measurements were performed using the Evidence Cardiac Array measured on the Evidence Investigator. The detection limit of the assay is 1.8 mg/l and the upper limit is 700 mg/l, with a CV of 8.8% at 83 mg/l, 9.4% at 119 mg/l and 9.5% at 125.9 mg/l. The 97.5th percentile is 66 mg/l.
Neurohormones
Copeptin
Copeptin was measured by time-resolved amplified cryptate emission (TRACE), which measures the signal that is emitted from an immunocomplex with time delay, using the KRYPTOR compact system (Brahms, Hennigsdorf, Germany). The detection limit of the assay is 4.8 pmol/l. The analytical range is 4.8–500 pmol/l with a CV of 12–17% at 12–20 pmol/l, 6–12% at 20–50 pmol/l and 6% above 50 pmol/l. The functional sensitivity (20% CV) is < 12 pmol/l and the limit of quantitation (10% CV) is 14.1 pmol/l. The 97.5th percentile is 17.4 pmol/l (19.1 pmol/l male, 12.9 pmol/l female).
B-type natriuretic peptide by N-terminal pro-B-type natriuretic peptide measurement
N-terminal pro-B-type natriuretic peptide was measured using a solid-phase two-site chemiluminescent sandwich immunoassay using an Immulite 2500 (Siemens Healthcare Diagnostics, Camberley, Surrey, UK). The detection limit is 20 ng/l and the measuring range 20–35,000 ng/l. The interassay %CV is 5.0–4.0 in the concentration range 40.9–32,096 ng/l.
Statistical methods
Demographics and patient characteristics were analysed by non-parametric statistics. Diagnostic test comparison was performed using AMI or MACE as the dichotomous variable. Individual markers on admission and at 90 minutes from admission, the peak of the admission or 90-minute value and delta (90-minute value – admission value) values were examined by the construction of receiver operating characteristic (ROC) curves and calculation of the area under the curve (AUC). Integrated strategies utilising prespecified cut-off values plus delta values were compared by construction of contingency tables analysed by Fisher's exact test. In addition, 95% confidence intervals were calculated. All statistical analysis was performed using Analyse-it for Microsoft Excel (version 2.21;www.analyse-it.com).
Economic analysis
Economic analysis was predicated by two different scenarios. In the first scenario it was assumed that a number of biomarkers would have equivalent diagnostic and prognostic efficiency or that one single biomarker would be superior to all other single biomarkers or combinations of biomarkers. Cost minimisation analysis was used for economic modelling for this scenario. In the second scenario it was assumed that a single biomarker or combinations of biomarkers at different time points or combinations of markers at different time points would be used to achieve optimal diagnostic and prognostic patient categorisation. For this scenario a decision-analysis cost-effectiveness approach was utilised.
The decision-analysis model developed for a related Health Technology Assessment (HTA) project ‘Cost-effectiveness of diagnostic strategies for suspected acute coronary syndrome (ACS)’ (HTA 09/22/21) was used. 241Full details of the model are given in the HTA journal publication for this project, but the essential details are as follows.
A decision tree model was developed using Simul8 software (Simul8 Corporation, Boston, MA, USA) to explore the costs and health outcomes associated with different diagnostic strategies. The model took an economic perspective of the NHS in England and Wales and a lifetime horizon with mean life expectancy based on UK interim lifetables. 242The basic model structure is shown inFigure 1.
FIGURE 1.
Basic model structure. MI, myocardial infarction.
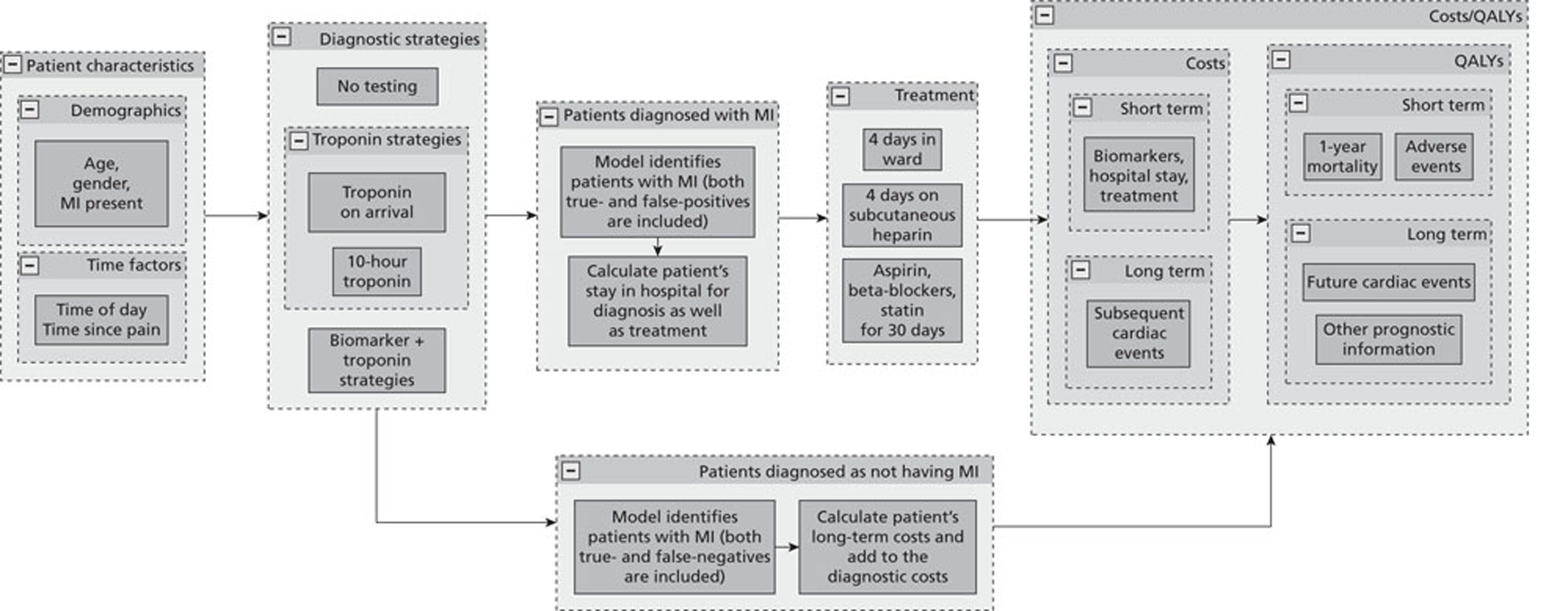
The model applied different testing strategies for myocardial infarction to a hypothetical cohort of patients presenting to hospital with symptoms suggestive of myocardial infarction but with no diagnostic ECG changes (ST deviation > 1 mm or T-wave inversion > 3 mm), no known history of coronary heart disease and no major comorbidities requiring inpatient treatment (such as heart failure or arrhythmia). Each patient entering the model had the following characteristics defined by sampling from the RATPAC trial population: age, gender, myocardial infarction present or not, time delay between onset of worst pain and arrival at hospital, and time of day.
The following diagnostic strategies were applied to each patient:
-
no testing: discharge all patients without treatment
-
high-sensitivity troponin at presentation: discharge home if test is negative or admit to hospital for troponin testing at 10–12 hours if positive
-
high-sensitivity troponin and a combination of cytoplasmic or neurohormone biomarkers at presentation: discharge home if both tests are negative or admit to hospital for troponin testing at 10–12 hours if either test is positive
-
high-sensitivity troponin at presentation and at 90 minutes as in the RATPAC protocol: discharge home if both tests are negative or admit to hospital for troponin testing at 10–12 hours if either test is positive
-
standard troponin testing at 10–12 hours.
Strategy 1 is a theoretical ‘zero option’ strategy designed to test whether or not any of the testing strategies are cost-effective. Strategy 5 is current standard practice as recommended in guidance from the National Institute for Health and Care Excellence (NICE). 243
It was assumed that blood tests performed at presentation were undertaken in the emergency department and that results would be available and a decision made within 2 hours of sampling. Subsequent time delays are likely to depend on the system in place for managing admissions with chest pain and, so, three different scenarios with regard to troponin measurement at 10–12 hours were tested:
-
the ‘doctor-on-demand’ scenario in which medical staff were available 24 hours a day to make a disposition decision within 1 hour of the results being available
-
the twice-daily ward round scenario in which medical staff were available only during twice-daily ward rounds (e.g. 0900 and 1800 hours) to make disposition decisions
-
the once-daily ward round scenario in which medical staff were available only during one daily ward round (e.g. 1400 hours) to make disposition decisions.
It was assumed that standard troponin measurement at 10–12 hours was the reference standard for myocardial infarction and was therefore effectively 100% sensitive and specific. All patients who were not discharged after presentation testing received testing at 10–12 hours to confirm or refute the diagnosis of myocardial infarction. Those with myocardial infarction were admitted to hospital and treated. Those without myocardial infarction were discharged home without treatment.
The sensitivity and specificity of presentation biochemical testing was estimated using data from this study. It was assumed that true-positives would be confirmed at 10–12 hours and admitted for treatment, false-positives would be admitted until a 10- to 12-hour troponin test ruled out myocardial infarction, and true-negatives and false-negatives would be discharged without treatment. Costs were accrued throughout the diagnostic process dependent on length of stay in hospital, number of biochemical tests received and receipt of treatment for myocardial infarction. It was assumed that all biochemical tests would cost £20 per test regardless of current availability or price. Current availability and price depend on current usage, which in turn depends on evidence of effectiveness and cost-effectiveness. It was assumed that any biochemical test that was shown convincingly to be effective and cost-effective would become widely available at a reasonable cost, regardless of current availability and cost.
Following diagnosis and treatment it was assumed that patients would die, suffer reinfarction or survive without reinfarction over the following year, depending on (1) whether or not they had myocardial infarction and (2) whether or not myocardial infarction was treated. The rates of death and reinfarction up to 1 year for patients with treated myocardial infarction, untreated myocardial infarction and no myocardial infarction were estimated using data from a cohort study of patients before and after implementation of a change of operational threshold. 244The parameters used in the model are shown inTable 5.
| Parameter | Estimate | Distribution | Source |
| Population characteristics | |||
|---|---|---|---|
| Age (years), mean (SD) | 53.0 (13.5) | SE 0.30 | Goodacreet al.245 |
| Male (%) | 58.1 | n/N = 1138/1958 | |
| MI prevalence (%) | 7.0 | n/N = 137/1958 | |
| Time delay (minutes), median (IQR) | 132 (80–255) | ||
| 1-year probabilities of death and non-fatal MI (%) | |||
| Death, treated MI | 11 | n/N = 9/80 | Millset al.244 |
| Death, untreated MI | 21 | n/N = 19/90 | |
| Reinfarction, treated MI | 11 | n/N = 9/80 | |
| Reinfarction, untreated MI | 29 | n/N = 26/90 | |
| Costs of tests, hospital stay and treatment (£) | |||
| Treatment of MI (index or reinfarction) | 3587 | (3000,4000) | NHS reference costs246 |
| Hospital stay (per hour) for testing | 22 | (20,30) | NHS reference costs for general medical ward246 |
| Biochemical testing (per test) | 20 | (18,25) | Goodacreet al.245 |
It was assumed that survival and cardiac events after the first year would be independent of the diagnostic testing strategy at initial hospital admission, but that additional health-care costs and quality-adjusted life-years (QALYs) would be accrued by survivors and influenced by whether or not they suffered myocardial infarction and reinfarction. Table 6shows lifetime costs and QALYs for those with myocardial infarction. Patients without myocardial infarction were assumed to have normal quality-adjusted life expectancy and no additional health-care costs.
| Age (years) | Cost (£) | QALYs | QALYs with reinfarction |
|---|---|---|---|
| 30–44 | 4012.5 | 12.20 | 9.76 |
| 45–54 | 3115 | 9.47 | 7.58 |
| 55–64 | 2215 | 6.73 | 5.39 |
| 65–74 | 1530 | 4.65 | 3.72 |
| ≥ 75 | 800 | 2.43 | 1.95 |
Chapter 4 Results
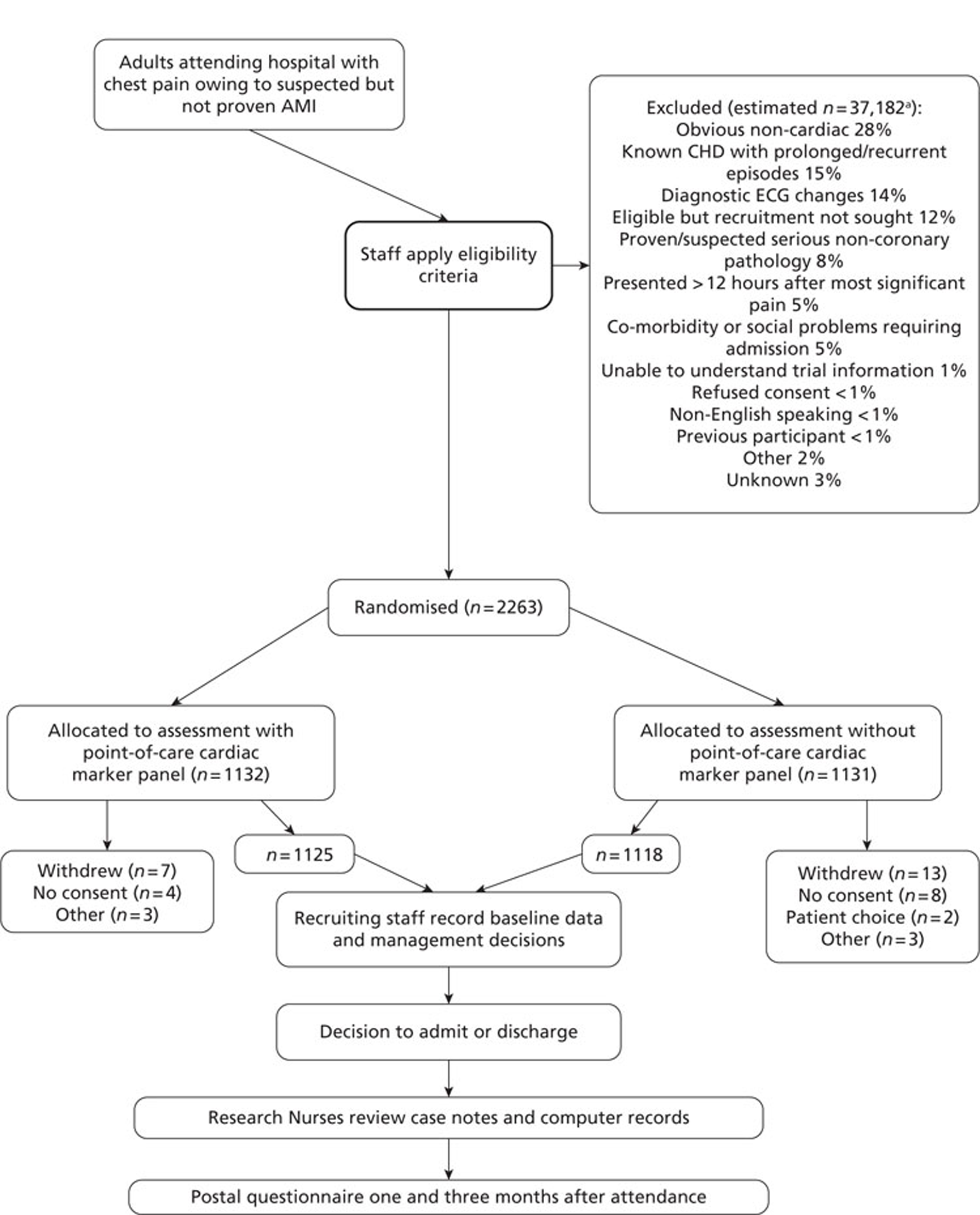
A total of 2263 participants were successfully recruited between 30 January 2007 and 2 June 2008; 1125 patients were successfully randomised to the point-of-care testing arm and 18 did not complete the 3-month follow-up. In the point-of-care testing arm there were 36 patients with events (3%): death 6 (1%); non-fatal myocardial infarction 5 (< 1%); hospitalisation for ACS (without myocardial infarction) 18 (2%); life-threatening arrhythmia 6 (1%), emergency revascularisation 10 (1%). Event rates between the point-of-care testing arm and the central laboratory testing arm were not statistically different, although slightly more patients with an AMI were detected in the point-of-care testing arm (90/1125 vs 72/1118). Study enrolment is summarised inFigure 2.
FIGURE 2.
Trial enrolment. CHD, coronary heart disease. a, Patients were sampled on pre-determined screening days to assess the number of patients not recruited.
Estimated number of patients not recruited=number not recruited on screening days×total days recruitingtotal days screening.
Percentages are out of the total number of non-recruited patient notes screened (n = 9109).
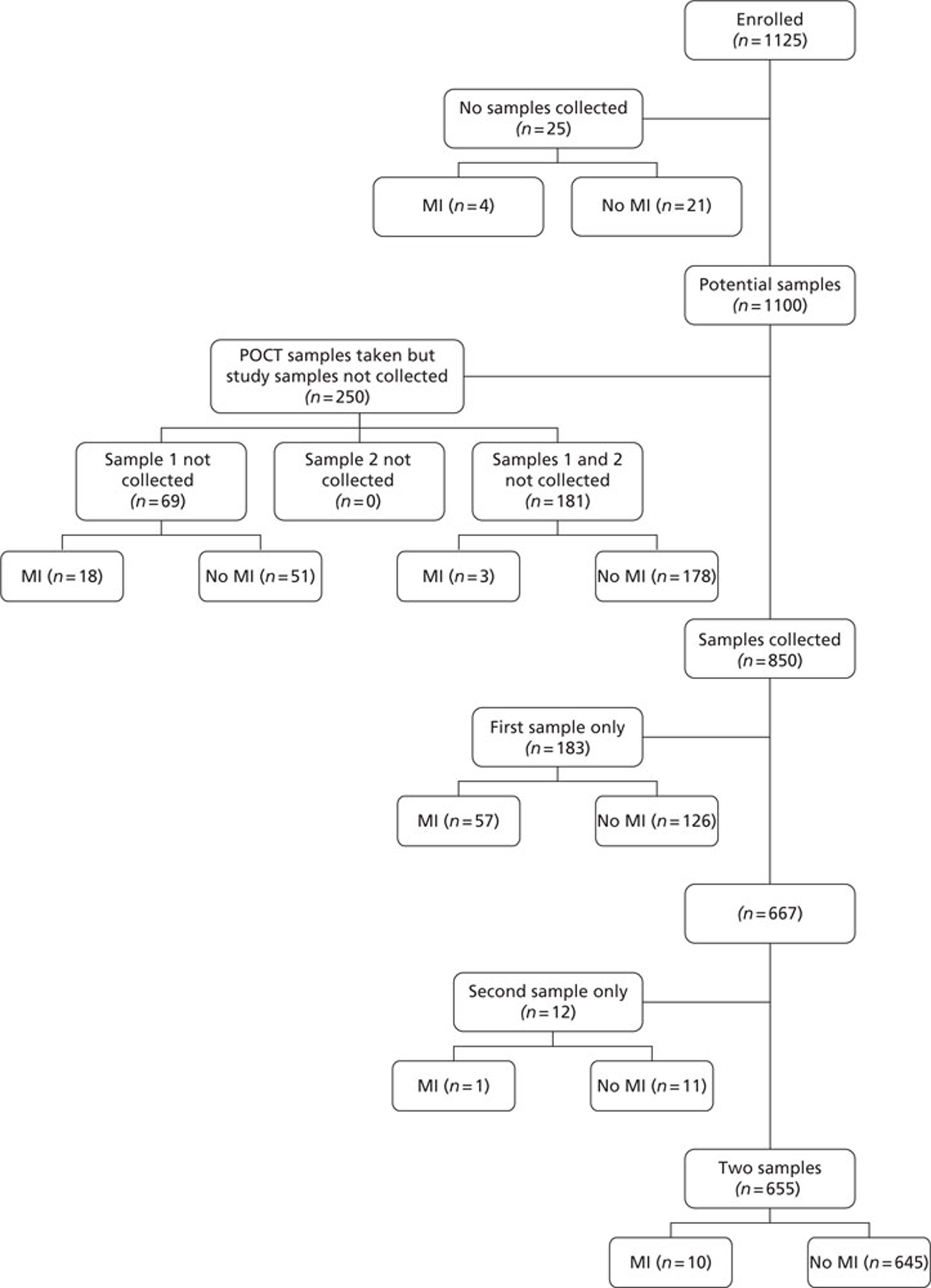
Sample and result availability for the point-of-care arm and samples available from the subsequent extended biomarker phase are summarised inFigures 3and4respectively.
FIGURE 3.
Sample and result availability for patients enrolled in the point-of-care arm of the study. MI, myocardial infarction.
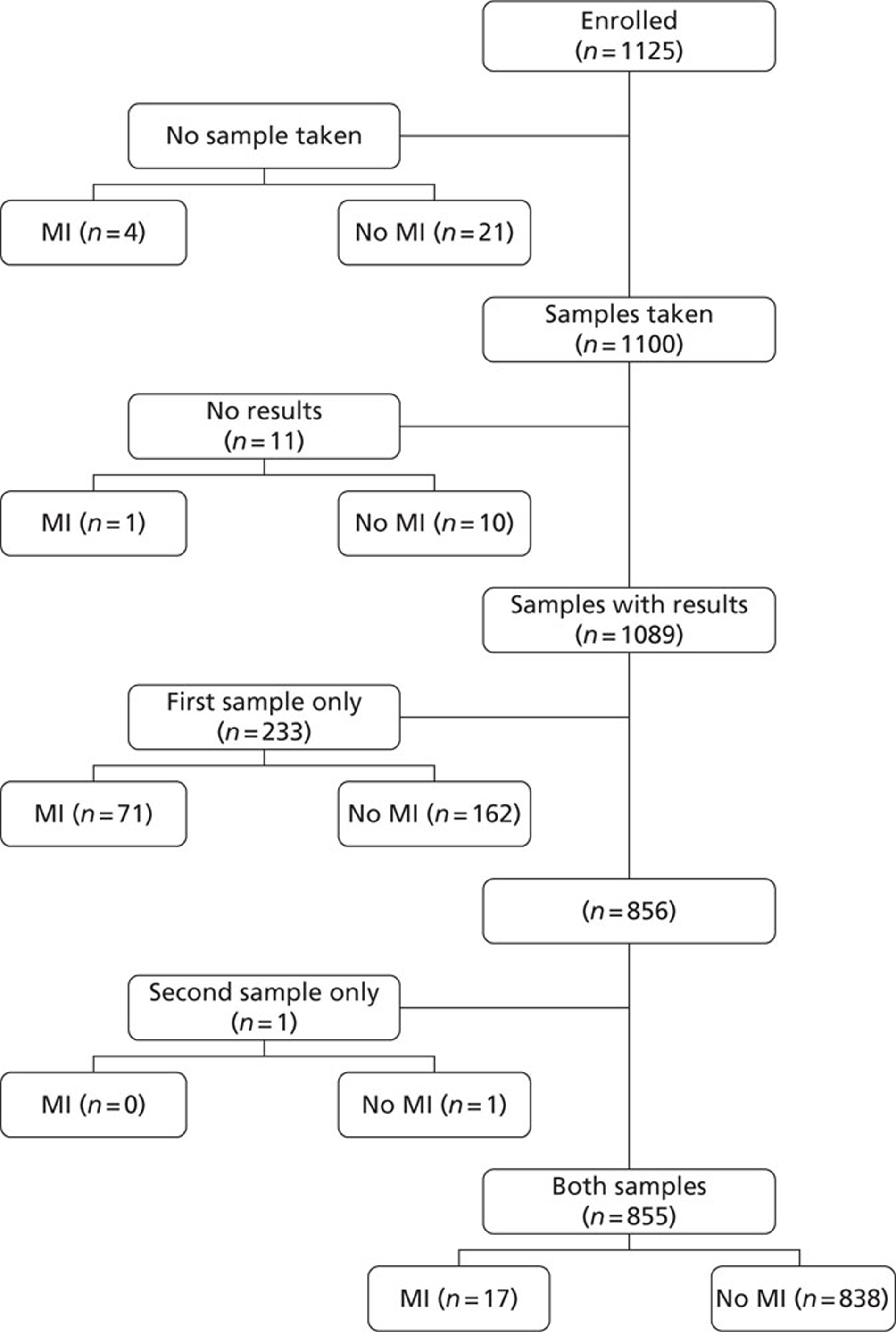
FIGURE 4.
Samples available from the extended biomarker phase of the study. MI, myocardial infarction; POCT, point-of-care testing.
Demographics and patient characteristics in the point-of-care testing arm are summarised inTable 7.
The patient characteristics of the original data and the biomarker subset were not statistically significantly different. The diagnostic categorisation of patients on admission, following final diagnostic categorisation in the point-of-care arm of the RATPAC study and following review using the 99th percentile from the local laboratory and core laboratory troponin data are summarised inTable 8. In total, 847 out of 850 patients had laboratory troponin measurements performed. Median time from onset of chest pain to the last troponin measurement performed in the laboratory was 495 minutes [range 95–46,600 minutes, interquartile range (IQR) 310–738 minutes]. In total, 285 out of 850 of these samples (33.5%) were taken < 6 hours from onset of chest pain, 556 (65.4%) were taken ≥ 6 hours from onset of chest pain and 364 (42.8%) were taken ≥ 10 hours from onset of chest pain. Hence, the majority of patients had a troponin measurement performed in accordance with the current recommendations of the European Society of Cardiology. Following a review of the laboratory troponin results and measurement of the admission and 90-minute samples using the Siemens Ultra assay three patients were reclassified as having an AMI.
| Point-of-care testing arm | All patients (N = 1125),n(%) | Biomarker subset (N = 850),n(%) |
|---|---|---|
| Median age (IQR) (years) | 53.4 (44–64) | 53.7 (44–64) |
| Min.–max. age (years) | 21–92 | 23–92 |
| Male | 683 (61) | 507 (60) |
| Female | 442 (39) | 343 (40) |
| Previous myocardial infarction | 60 (5) | 49 (6) |
| Angina + positive diagnostic test | 46 (4) | 32 (4) |
| Previous coronary artery bypass surgery | 12 (1) | 8 (1) |
| Angioplasty | 37 (3) | 32 (4) |
| Stenosis > 50% on angiography | 14 (1) | 8 (1) |
| Unproven clinical label of coronary heart disease | 36 (3) | 29 (3) |
| Diabetes | 86 (8) | 69 (8) |
| Hypertension | 376 (33) | 301 (35) |
| Hyperlipidaemia | 271 (24) | 201 (24) |
| Present smoker | 310 (28) | 242 (28) |
| Ex-smoker (last 10 years) | 144 (13) | 101 (12) |
| Cocaine abuse | 6 (1) | 6 (1) |
| First-degree relative with angina/myocardial infarction, onset age < 60 years | 344 (31) | 271 (32) |
| Use of aspirin in previous 7 days | 207 (18) | 162 (19) |
| More than one episode of rest angina in < 24 hours | 75 (7) | 52 (6) |
| IQR, interquartile range; max., maximum; min., minimum. | ||
| Diagnostic categorisation | All patients (N = 1125) | Biomarker subset (N = 850) | |
|---|---|---|---|
| Admission diagnosis,n(%) | Final diagnosis,n(%) | Review diagnosis,n(%) | |
| Non-specific chest pain | 233 (21) | 361 (32) | 279 (33) |
| Anxiety | 51 (5) | 36 (3) | 31 (4) |
| Angina, no ACS | 173 (15) | 83 (7) | 72 (9) |
| ACS (suspected AMI) | 334 (30) | 90 (8) | 68 (8) |
| Gastro-oesophageal pain | 117 (10) | 124 (11) | 100 (12) |
| Musculoskeletal pain | 108 (10) | 143 (13) | 116 (14) |
| Other | 86 (8) | 228 (20) | 154 (18) |
| Unknown | 23 (2) | 60 (5) | 30 (3.5) |
The distribution of diagnoses in the final and review diagnosis groups was not statistically different.
Evaluation of the diagnostic accuracy for acute myocardial infarction of highly sensitive troponin assays and a range of new cardiac biomarkers of plaque destabilisation, myocardial ischaemia and necrosis
Is a panel of cardiac markers required for the early diagnosis of chest pain?
The cardiac troponin assay used in the RATPAC trial was the Stratus CS analyser. Although this is not considered to be a high-sensitivity troponin assay, it meets the criteria proposed as a guideline-acceptable assay. 84The 10% CV is below the 99th percentile according to the manufacturer's data sheet. The need for additional cytoplasmic biomarkers as well as troponin is based on the argument that these will rise earlier than troponin as they are more readily released. The ability to detect troponin values around the 99th percentile may supersede the need for earlier markers as the time point for diagnostic insensitivity of troponin (the period of ‘troponin blindness’) may no longer occur. The markers traditionally suggested have been myoglobin and CK-MB. The first phase of the evaluation was therefore to assess the comparative diagnostic efficiency of cTnI measured on the Stratus CS alone utilising all of the available data for simultaneous real-time measurement of cTnI, myoglobin and CK-MB. The diagnostic efficiency of the panel approach was compared with that of single markers and single marker combinations utilising the final clinical diagnosis derived from the RATPAC study to allow comparison against the independent diagnostic standard used for patient management during the trial. The following diagnostic strategies were compared: individual marker values (cTnI > 99th percentile, CK-MB > 5μg/l20and myoglobin > 95th percentile), delta CK-MB > 1.5μg/l20and myoglobin (defined as percentage change from admission measurement > 25%). These were then compared with the combination of individual markers at presentation, the combination of individual markers at 90 minutes and the combination of admission and 90-minute marker values or delta values. For each combination, rule-in diagnosis of an AMI (specificity and positive predictive value) and rule-out diagnosis of an AMI (sensitivity and negative predictive value) were calculated.
Full data were available for 84 out of 90 patients of AMI and for 987 out of 1035 patients in whom an AMI was excluded. There were no interpretable results for cTnI in 45 patients (five AMI), for CK-MB in 47 patients (six AMI) and for myoglobin in 40 patients (six AMI). In the admission sample measurement of cTnI was the most diagnostically efficient, with an area under the ROC curve (95% confidence intervals in parentheses) of 0.96 (0.93 to 0.98) compared with 0.85 (0.80 to 0.90) for CK-MB and 0.75 (0.68 to 0.81) for myoglobin, and was statistically significantly greater than all other analytes, with a sensitivity of 0.845 (0.750 to 0.915) and a specificity of 0.976 (0.964 to 0.984). At 90 minutes, cTnI measurement had the highest AUC – 0.95 (0.87 to 1.00) compared with 0.86 (0.77 to 0.94) for CK-MB, 0.78 (0.65 to 0.91) for myoglobin, 0.83 (0.65 to 1.00) for delta CK-MB and 0.58 (0.35 to 0.81) for delta myoglobin – but was statistically significantly different only from delta myoglobin (p = 0.0035) and delta CK-MB (p = 0.0064). Comparison of final diagnoses showed that in one case there was no diagnostic elevation of cTnI, CK-MB or myoglobin on admission or at 90 minutes, although there was a clinical diagnosis of myocardial infarction and the patient was admitted. In one case CK-MB and myoglobin were elevated, but cTnI was not. The data are summarised inTable 9.
| Diagnosis | Positive test | cTnI | CK-MB | Myoglobin | cTnI or CK-MB | cTnI or myoglobin | Triple test | CK-MB or myoglobin |
| On admission | ||||||||
|---|---|---|---|---|---|---|---|---|
| Ml (n) | Y | 71 | 33 | 30 | 72 | 74 | 74 | |
| N | 13 | 51 | 54 | 11 | 9 | 8 | ||
| No Ml (n) | Y | 24 | 31 | 50 | 47 | 67 | 82 | |
| N | 966 | 959 | 942 | 933 | 912 | 889 | ||
| Sensitivity (95% Cl) | 0.845 (0.750 to 0.915) | 0.393 (0.288 to 0.505) | 0.357 (0.256 to 0.469) | 0.867 (0.775 to 0.932) | 0.892 (0.804 to 0.949) | 0.902 (0.817 to 0.957) | ||
| p-value | < 0.0001 | < 0.0001 | NS | NS | NS | |||
| Specificity (95% Cl) | 0.976 (0.964 to 0.984) | 0.969 (0.956 to 0.979) | 0.950 (0.934 to 0.962) | 0.952 (0.937 to 0.965) | 0.932 (0.914 to 0.947) | 0.916 (0.896 to 0.932) | ||
| p-value | NS | 0.003 | 0.0069 | < 0.0001 | < 0.0001 | |||
| PPV (95% Cl) | 0.747 (0.648 to 0.831) | 0.516 (0.387 to 0.642) | 0.375 (0.269 to 0.490) | 0.605 (0.517 to 0.693) | 0.525 (0.442 to 0.607) | 0.474 (0.396 to 0.553) | ||
| NPV (95% Cl) | 0.987 (0.977 to 0.993) | 0.950 (0.934 to 0.962) | 0.946 (0.930 to 0.959) | 0.988 (0.979 to 0.994) | 0.990 (0.982 to 0.996) | 0.991 (0.982 to 0.996) | ||
| At 90 minutes | ||||||||
| MI (n) | Y | 16 | 3 | 4 | 12 | 11 | 11 | |
| N | 1 | 9 | 7 | 0 | 0 | 0 | ||
| No MI (n) | Y | 13 | 16 | 28 | 27 | 41 | 51 | |
| N | 814 | 809 | 798 | 789 | 775 | 756 | ||
| Sensitivity (95% CI) | 0.941 (0.713 to 0.999) | 0.250 (0.055 to 0.572) | 0.364 (0.109 to 0.692) | 1.000 (0.735 to 1.000) | 1.000 (0.715 to 1.000) | 1.000 (0.715 to 1.000) | ||
| p-value | 0.0004 | 0.004 | NS | NS | NS | |||
| Specificity (95% Cl) | 0.984 (0.973 to 0.992) | 0.981 (0.969 to 0.989) | 0.966 (0.951 to 0.977) | 0.967 (0.952 to 0.978) | 0.950 (0.932 to 0.964) | 0.937 (0.918 to 0.953) | ||
| p-value | NS | 0.03 | 0.0337 | 0.0002 | < 0.0001 | |||
| PPV (95% CI) | 0.552 (0.357 to 0.736) | 0.158 (0.034 to 0.396) | 0.125 (0.035 to 0.290) | 0.308 (0.170 to 0.476) | 0.212 (0.111 to 0.347) | 0.177 (0.092 to 0.295) | ||
| NPV (95% CI) | 0.999 (0.993 to 1.000) | 0.989 (0.979 to 0.995) | 0.991 (0.982 to 0.996) | 1.000 (0.995 to 1.000) | 1.000 (0.995 to 1.000) | 1.000 (0.995 to 1.000) | ||
| Peak or delta | ||||||||
| MI (n) | Y | 83 | 33 | 33 | 84 | 84 | 84 | 43 |
| N | 2 | 51 | 51 | 1 | 1 | 1 | 42 | |
| No MI (n) | Y | 37 | 31 | 127 | 60 | 155 | 170 | 145 |
| N | 958 | 963 | 874 | 941 | 849 | 834 | 857 | |
| Sensitivity (95% CI) | 0.976 (0.918 to 0.997) | 0.393 (0.288 to 0.505) | 0.393 (0.288 to 0.505) | 0.988 (0.936 to 1.000) | 0.988 (0.936 to 1.000) | 0.988 (0.936 to 1.0) | 0.506 (0.395 to 0.616) | |
| p-value | < 0.0001 | < 0.0001 | NS | NS | NS | < 0.0001 | ||
| Specificity (95% CI) | 0.963 (0.949 to 0.974) | 0.969 (0.969 to 0.979) | 0.873 (0.853 to 0.894) | 0.940 (0.924 to 0.954) | 0.846 (0.823 to 0.868) | 0.831 (0.807 to 0.861) | 0.855 (0.834 to 0.877) | |
| p-value | NS | 0.003 | 0.0239 | < 0.0001 | < 0.0001 | < 0.0001 | ||
| PPV (95% CI) | 0.692 (0.609 to 0.774) | 0.516 (0.387 to 0.643) | 0.206 (0.144 to 0.269) | 0.583 (0.503 to 0.664) | 0.351 (0.291 to 0.412) | 0.331 (0.273 to 0.389) | 0.229 (0.169 to 0.289) | |
| NPV (95% CI) | 0.998 (0.992 to 1.000) | 0.950 (0.934 to 0.962) | 0.945 (0.928 to 0.959) | 0.999 (0.994 to 1.000) | 0.999 (0.994 to 1.000) | 0.999 (0.993 to 1.000) | 0.953 (0.937 to 0.966) | |
Optimal diagnostic performance for all markers was achieved by 90 minutes. Both on admission and at 90 minutes, measurement of cTnI was diagnostically significantly more sensitive (hence, would allow rule-out of an AMI) than CK-MB or myoglobin but was more specific (rule-in of an AMI) only than myoglobin. Examining the combination of peak values and peak values plus change (delta) showed that cTnI was superior to both CK-MB and myoglobin alone or in combination. Delta cTnI was less sensitive (12/16) than peak cTnI (83/85), but was more sensitive than CK-MB (p = 0.018) or myoglobin (p = 0.018).
Do cytoplasmic markers of myocardial damage contribute to the early differential diagnosis of patients presenting with chest pain?
The measurement of H-FABP has been proposed as a more sensitive marker because of the cytoplasmic location of H-FABP and its low molecular weight. However, the molecular weight (15 kDa) is not substantially less than that of myoglobin (16.7 kDa), although the tissue concentration is higher. The diagnostic performance of H-FABP was therefore compared with that of the other cytoplasmic markers of myocardial necrosis and cTnI measured using the Stratus CS analyser. H-FABP measurement was also compared with the other high-sensitivity cTnI and cTnT methods. ROC analysis used the diagnosis of myocardial infarction based on the review diagnosis utilising the cTnI Ultra assay as the reference standard to provide a degree of independence of the diagnostic classification from the biomarkers undergoing evaluation. ROC curves were constructed for markers measured on admission and for peak values (admission or 90 minutes from admission) and are shown inFigures 5–8. AUCs and comparison of AUCs are provided inTables 10and11.
FIGURE 5.
Receiver operating characteristic curves for cytoplasmic biomarkers compared with cTnI for the diagnosis of an AMI: markers measured on admission. cTnI CS 1, Stratus CS admission sample; CK-MB 1, CK-MB admission sample; Myo 1, myoglobin admission sample; FABP 1, H-FABP admission sample.
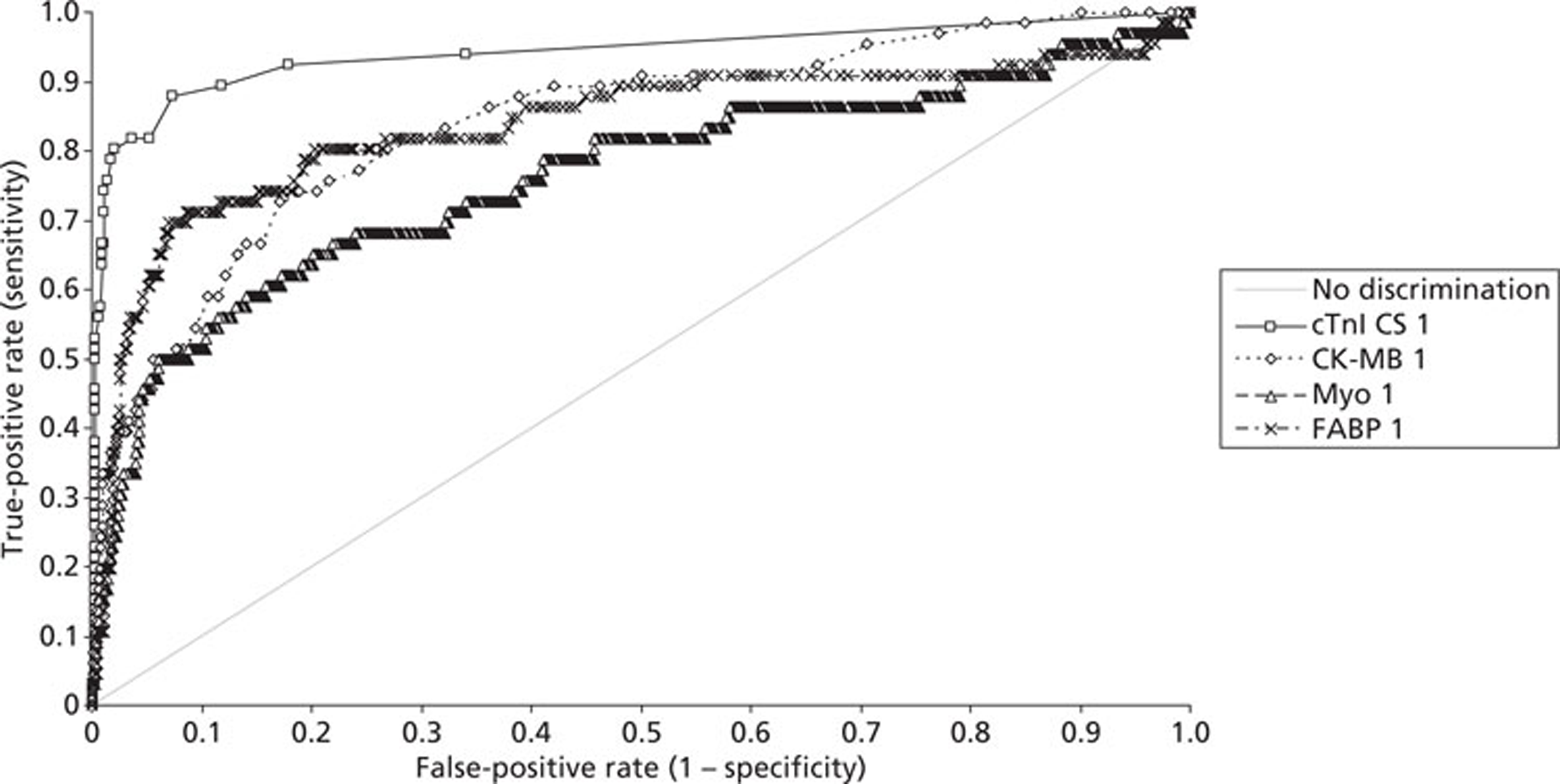
FIGURE 6.
Receiver operating characteristic curves for cytoplasmic biomarkers compared with cTnI for the diagnosis of an AMI. Markers are peak values of measurement on admission or 90 minutes from admission. cTnI CS peak, Stratus CS peak value; CK-MB peak, CK-MB peak value; Myo peak, myoglobin peak value; FABP peak, H-FABP peak value.
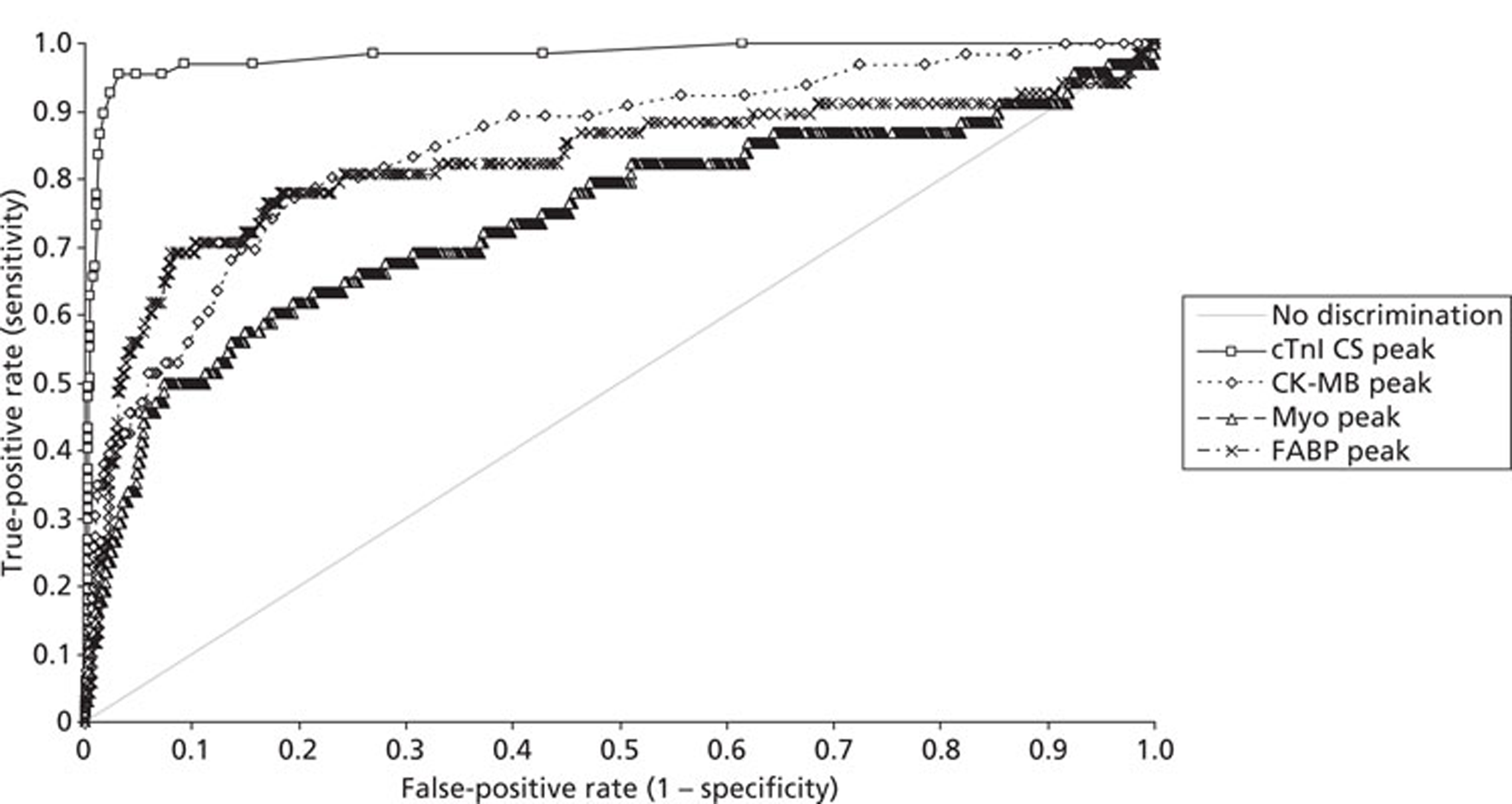
FIGURE 7.
Receiver operating characteristic curves for cytoplasmic biomarkers compared with cTnI and cTnT for the diagnosis of an AMI: markers measured on admission. cTnI CS 1, Stratus CS admission sample; cTnT 1, Roche high-sensitivity cTnT admission value; cTnI B1, Beckman AccuTnl admission sample; CK-MB 1, CK-MB admission sample; Myo 1, myoglobin admission sample; FABP 1, H-FABP admission sample.
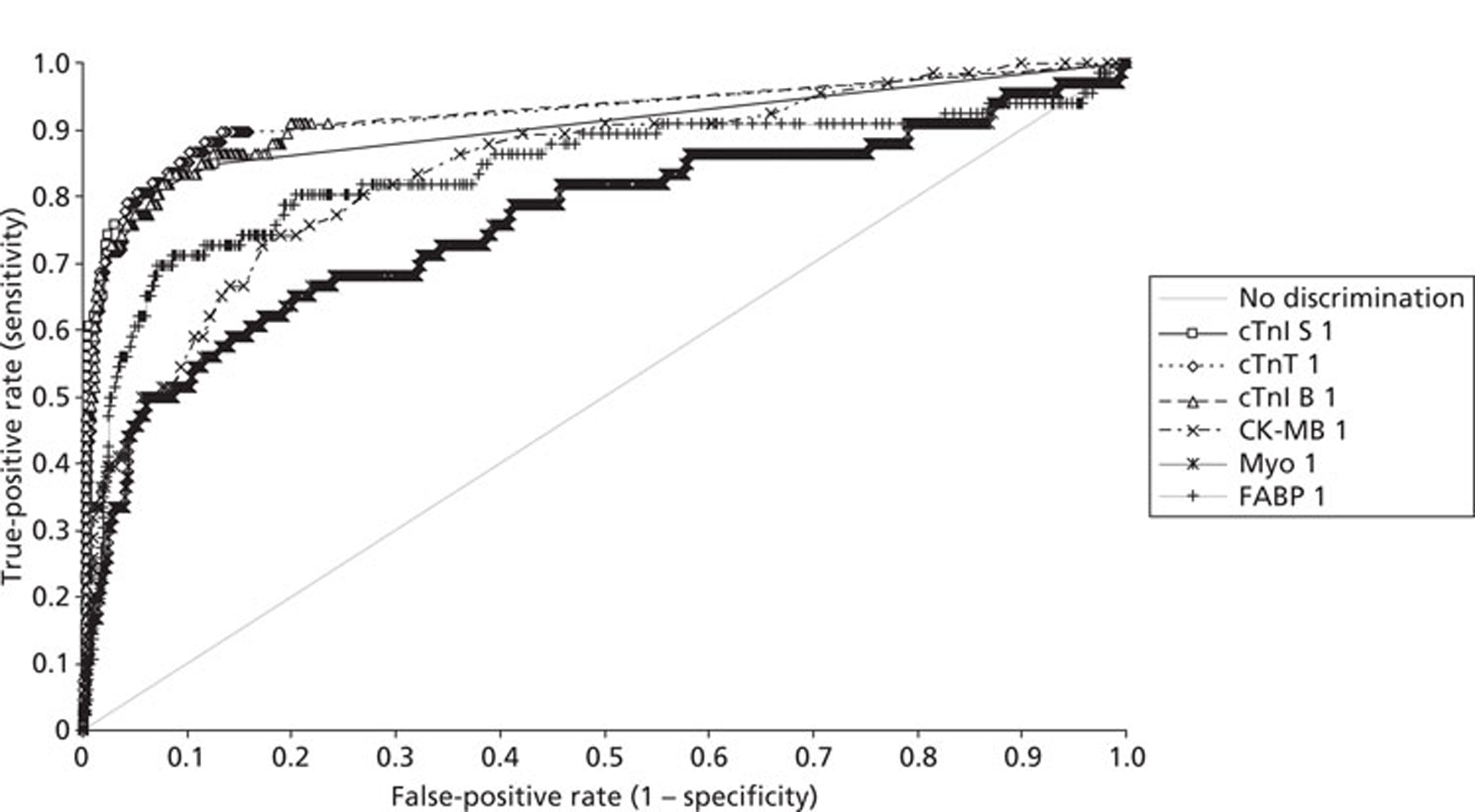
FIGURE 8.
Receiver operating characteristic curves for cytoplasmic biomarkers compared with cTnI and cTnT for the diagnosis of an AMI. Markers are peak values of measurement on admission or 90 minutes from admission. cTnI B peak, Beckman AccuTnI peak value; cTnI S peak, Siemens Ultra peak value; cTnT peak, Roche high-sensitivity cTnT peak value; CK-MB peak, CK-MB peak value; Myo peak, myoglobin peak value; FABP peak, H-FABP peak value.
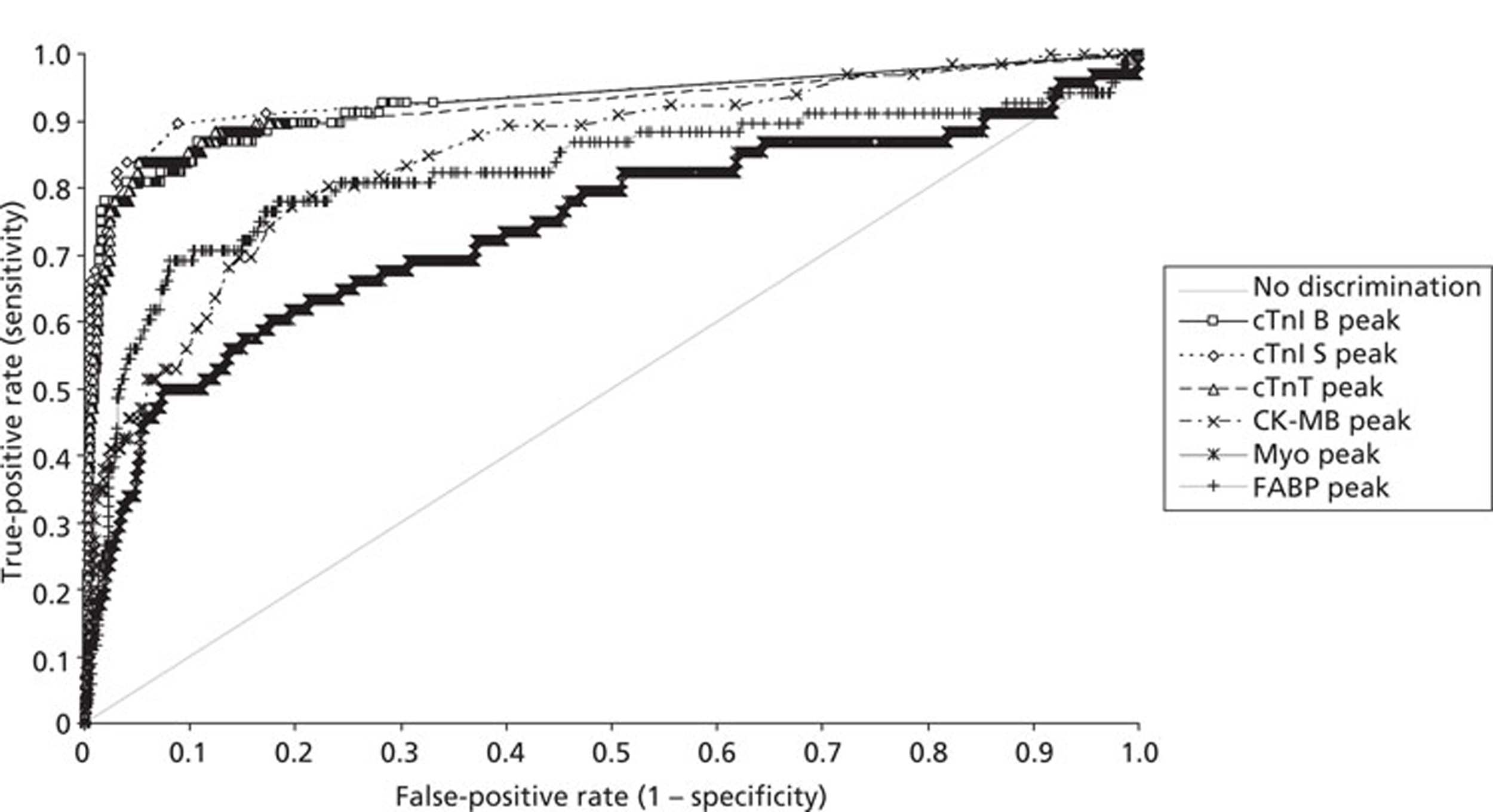
| Test | Area (95% CI) | Contrast | Difference (95% CI) | p-value |
| cTnI CS 1 | 0.94 (0.90 to 0.98) | cTnI CS 1 vs CK-MB 1 | 0.10 (0.04 to 0.16) | 0.0023 |
| CK-MB 1 | 0.84 (0.79 to 0.90) | cTnI CS 1 vs Myo 1 | 0.18 (0.10 to 0.26) | < 0.0001 |
| Myo 1 | 0.76 (0.69 to 0.84) | cTnI CS 1 vs FABP 1 | 0.10 (0.02 to 0.18) | 0.0115 |
| FABP 1 | 0.84 (0.77 to 0.90) | CK-MB 1 vs Myo 1 | 0.08 (0.02 to 0.14) | 0.0107 |
| cTnI B 1 | 0.92 (0.88 to 0.96) | CK-MB 1 vs FABP 1 | 0.00 (−0.05 to 0.05) | 0.8579 |
| cTnT 1 | 0.92 (0.88 to 0.96) | Myo 1 vs FABP 1 | −0.08 (−0.14 to −0.01) | 0.0146 |
| cTnI S 1 | 0.90 (0.85 to 0.95) | cTnI B 1 vs CK-MB 1 | 0.08 (0.02 to 0.13) | 0.0049 |
| cTnI B 1 vs Myo 1 | 0.16 (0.09 to 0.23) | < 0.0001 | ||
| cTnI B 1 vs FABP 1 | 0.08 (0.02 to 0.14) | 0.0054 | ||
| cTnI S 1 vs CK-MB 1 | 0.06 (0.01 to 0.11) | 0.0267 | ||
| cTnI S 1 vs Myo 1 | 0.14 (0.07 to 0.21) | 0.0002 | ||
| cTnI S 1 vs FABP 1 | 0.07 (0.01 to 0.12) | 0.0317 | ||
| cTnT 1 vs CK-MB 1 | 0.08 (0.03 to 0.13) | 0.0018 | ||
| cTnT 1 vs Myo 1 | 0.16 (0.09 to 0.23) | < 0.0001 | ||
| cTnT 1 vs FABP 1 | 0.08 (0.03 to 0.14) | 0.0036 |
| Test | Area (95% CI) | Contrast | Difference (95% CI) | p-value |
| cTnI CS peak | 0.98 (0.96 to 1.00) | cTnI CS peak vs CK-MB peak | 0.13 (0.08 to 0.19) | < 0.0001 |
| CK-MB peak | 0.85 (0.80 to 0.90) | cTnI CS peak vs Myo peak | 0.24 (0.16 to 0.32) | < 0.0001 |
| Myo peak | 0.74 (0.67 to 0.82) | cTnI CS peak vs FABP peak | 0.16 (0.09 to 0.23) | < 0.0001 |
| FABP peak | 0.82 (0.75 to 0.89) | CK-MB peak vs Myo peak | 0.11 (0.04 to 0.17) | 0.0008 |
| cTnI B peak | 0.93 (0.88 to 0.97) | CK-MB peak vs FABP peak | 0.03 (–0.03 to 0.08) | 0.3328 |
| cTnI S peak | 0.94 (0.90 to 0.97) | Myo peak vs FABP peak | –0.08 (–0.15 to –0.02) | 0.0109 |
| cTnT peak | 0.92 (0.88 to 0.97) | cTnI B peak vs CK-MB peak | 0.08 (0.02 to 0.13) | 0.0041 |
| cTnI B peak vs Myo peak | 0.18 (0.11 to 0.26) | < 0.0001 | ||
| cTnI B peak vs FABP peak | 0.10 (0.04 to 0.16) | 0.0005 | ||
| cTnI S peak vs CK-MB peak | 0.09 (0.04 to 0.13) | 0.0006 | ||
| cTnI S peak vs Myo peak | 0.19 (0.12 to 0.26) | < 0.0001 | ||
| cTnI S peak vs FABP peak | 0.11 (0.06 to 0.17) | < 0.0001 | ||
| cTnT peak vs CK-MB peak | 0.07 (0.02 to 0.12) | 0.0046 | ||
| cTnT peak vs Myo peak | 0.18 (0.11 to 0.25) | < 0.0001 | ||
| cTnT peak vs FABP peak | 0.10 (0.04 to 0.16) | 0.0019 |
Cardiac troponin measurement was diagnostically superior to all of the other markers on admission as assessed by comparison of areas under the ROC curve. Both H-FABP and CK-MB were superior to myoglobin on admission. The diagnostic efficiencies of CK-MB and H-FABP were equivalent. The following diagnostic strategies were also compared: individual marker values [(cTnI > 99th percentile (0.07μg/l), CK-MB > 5μg/l20myoglobin > 95th percentile (66 mg/l), hFABP > 95th percentile (2.5 mg/l)]; delta CK-MB > 1.6μg/l20and myoglobin (defined as % change from admission measurement > 25%); the combination of presentation or 90-minute value and the combination of presentation or 90-minute value plus delta value. These results are summarised inTable 12. As found previously, the diagnostic sensitivity of troponin was superior to that of all of the other biomarkers examined but its diagnostic specificity was not significantly different from that of CK-MB, although it was superior to that of the other two biomarkers. The use of a delta value for CK-MB, myoglobin and H-FABP did not significantly improve sensitivity but significantly worsened the specificity for myoglobin and H-FABP.
| Diagnosis | Positive test | cTnl | CK-MB | Myoglobin | H-FABP |
| On admission | |||||
|---|---|---|---|---|---|
| MI (n) | Y | 53 | 26 | 36 | 43 |
| N | 13 | 40 | 30 | 23 | |
| No MI (n) | Y | 16 | 23 | 81 | 48 |
| N | 749 | 739 | 684 | 717 | |
| Sensitivity (95% CI) | 0.803 (0.6867 to 0.891) | 0.394 (0.276 to 0.522) | 0.546 (0.418 to 0.669) | 0.652 (0.524 to 0.765) | |
| p-valuea | < 0.0001 | 0.0027 | NS | ||
| Specificity (95% Cl) | 0.979 (0.966 to 0.988) | 0.970 (0.955 to 0.981) | 0.894 (0.870 to 0.915) | 0.937 (0.918 to 0.953) | |
| p-valuea | NS | < 0.0001 | < 0.0001 | ||
| NPV (95% Cl) | 0.983 (0.971 to 0.991) | 0.949 (0.931 to 0.963) | 0.958 (0.941 to 0.971) | 0.969 (0.954 to 0.980) | |
| At 90 minutes | |||||
| MI (n) | Y | 13 | 2 | 6 | 7 |
| N | 1 | 8 | 5 | 4 | |
| No MI (n) | Y | 9 | 14 | 57 | 20 |
| N | 646 | 638 | 589 | 626 | |
| Sensitivity (95% CI) | 0.929 (0.661 to 0.998) | 0.200 (0.0252 to 0.556) | 0.546 (0.234 to 0.833) | 0.636 (0.308 to 0.891) | |
| p-valuea | 0.0010 | NS | NS | ||
| Specificity (95% CI) | 0.986 (0.974 to 0.994) | 0.979 (0.964 to 0.988) | 0.912 (0.887 to 0.933) | 0.969 (0.953 to 0.981) | |
| p-valuea | NS | < 0.0001 | NS | ||
| NPV (95% CI) | 0.998 (0.991 to 1.000) | 0.988 (0.976 to 0.995) | 0.992 (0.980 to 0.997) | 0.994 (0.984 to 0.998) | |
| Peak value | |||||
| MI (n) | Y | 64 | 27 | 38 | 44 |
| N | 3 | 39 | 30 | 24 | |
| No MI (n) | Y | 24 | 25 | 107 | 58 |
| N | 742 | 740 | 673 | 722 | |
| Sensitivity (95% CI) | 0.955 (0.875 to 0.991) | 0.409 (0.289 to 0.537) | 0.559 (0.433 to 0.679) | 0.647 (0.522 to 0.759) | |
| p-valuea | < 0.0001 | < 0.0001 | < 0.0001 | ||
| Specificity (95% CI) | 0.969 (0.954 to 0.980) | 0.967 (0.952 to 0.979) | 0.863 (0.839 to 0.887) | 0.926 (0.905 to 0.943) | |
| p-valuea | NS | < 0.0001 | 0.0003 | ||
| Peak value or delta | |||||
| MI (n) | Y | 64 | 27 | 38 | 46 |
| N | 3 | 39 | 30 | 22 | |
| No MI (n) | Y | 24 | 25 | 246 | 213 |
| N | 742 | 740 | 534 | 567 | |
| Sensitivity (95% CI) | 0.955 (0.875 to 0.991) | 0.409 (0.289 to 0.537) | 0.559 (0.433 to 0.679) | 0.676 (0.552 to 0.785) | |
| p-valuea | < 0.0001 | < 0.0001 | < 0.0001 | ||
| Specificity (95% CI) | 0.969 (0.954 to 0.980) | 0.967 (0.952 to 0.979) | 0.680 (0.647 to 0.713) | 0.727 (0.696 to 0.758) | |
| p-valuea | NS | < 0.0001 | < 0.0001 | ||
Are all high-sensitivity cardiac troponin methods of equivalent diagnostic efficiency?
The diagnostic performance of four methods of measuring cardiac troponin was compared utilising the review diagnosis as the gold standard. The four methods were cTnI measured using the Stratus CS analyser, the Beckman AccuTnI method and the Siemens Ultra assay and cTnT measured using the Roche high-sensitivity assay. Diagnosis was initially compared using the admission sample alone and then using the peak value of the admission or the 90-minute sample. The ROC curves obtained are shown inFigures 9and10with comparison of the AUCs inTable 13. Diagnosis based on classification using the 99th percentile value is summarised inTable 14.
FIGURE 9.
Receiver operating characteristic curves for troponin measurement for the diagnosis of an AMI: markers measured on admission. cTnI CS 1, Stratus CS admission sample; cTnI B 1, Beckman AccuTnI admission sample; cTnI S 1, Siemens Ultra admission sample; cTnT 1, Roche high-sensitivity cTnT admission sample.
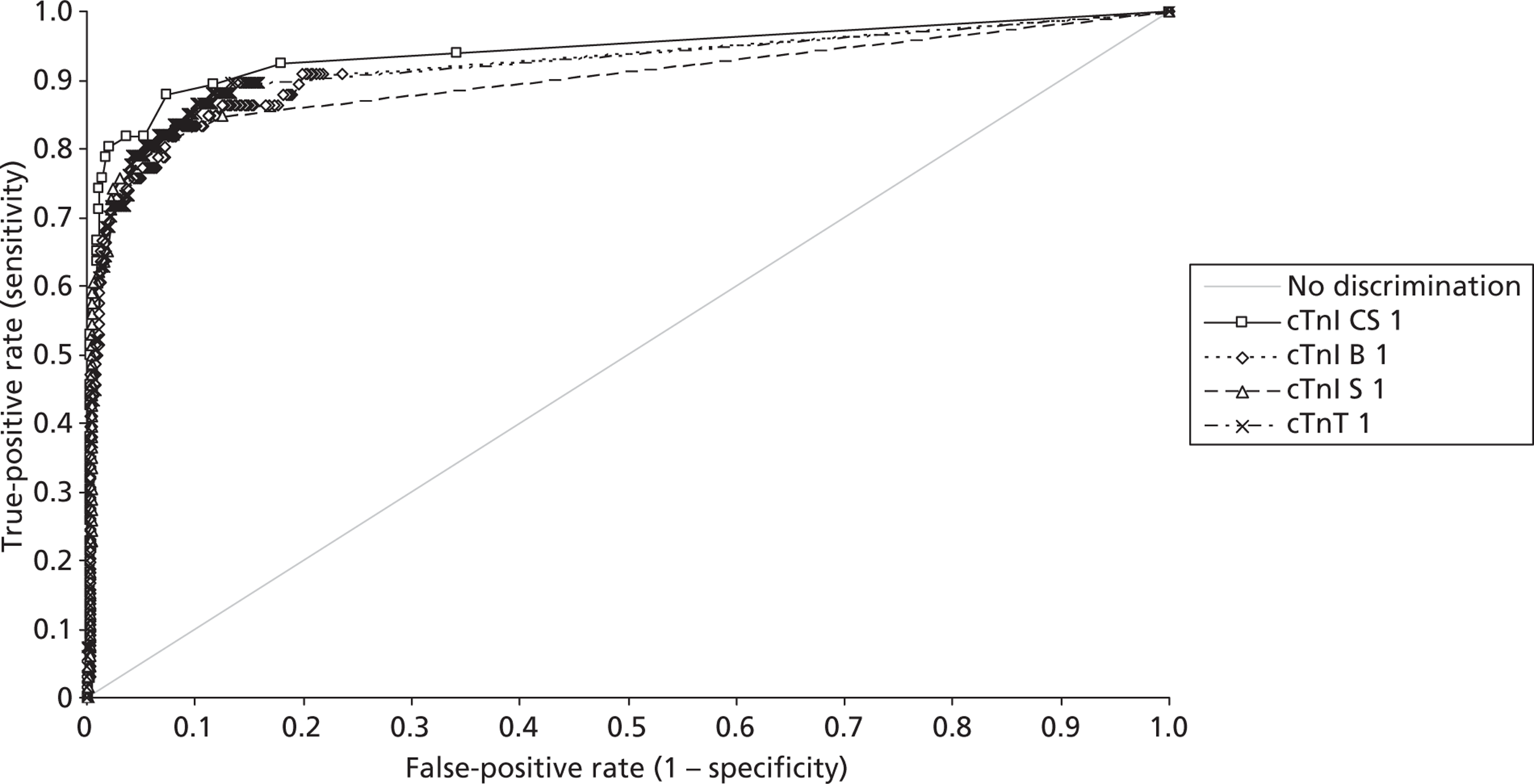
FIGURE 10.
Receiver operating characteristic curves for troponin measurement for the diagnosis of an AMI: peak values on admission or at 90 minutes from admission. cTnI CS peak, Stratus CS peak value; cTnI B peak, Beckman AccuTnI peak value; cTnI S peak, Siemens Ultra peak value; cTnT peak, Roche high-sensitivity cTnT peak value.
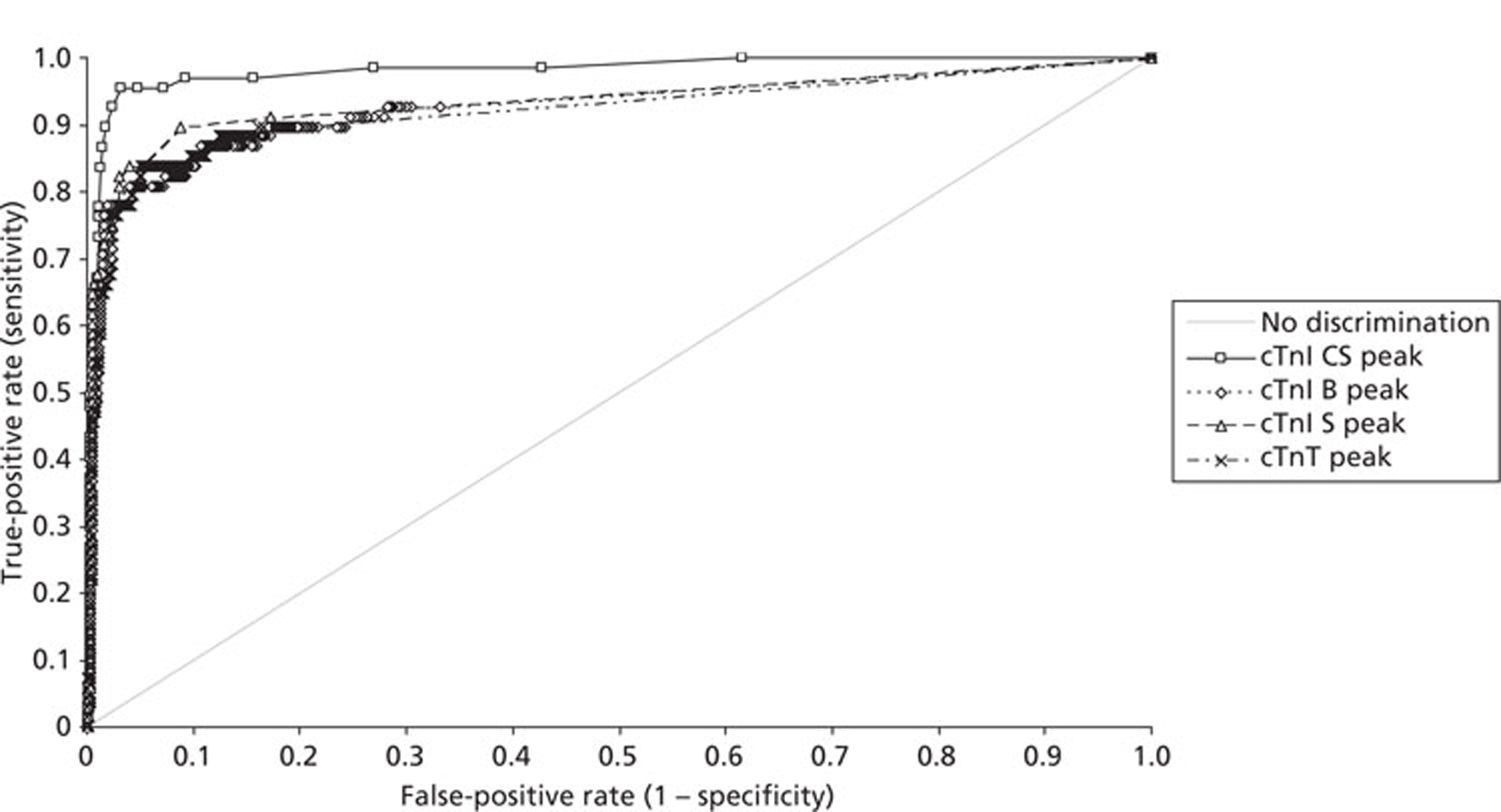
| Test | Area (95% CI) | Contrast | Difference (95% CI) | p-value |
| cTnI CS 1 | 0.94 (0.90 to 0.98) | cTnI CS 1 vs cTnI B 1 | 0.02 (−0.02 to 0.07) | 0.3752 |
| cTnI B 1 | 0.92 (0.88 to 0.96) | cTnI CS 1 vs cTnI S 1 | 0.04 (−0.01 to 0.08) | 0.0947 |
| cTnI S 1 | 0.90 (0.85 to 0.95) | cTnI CS 1 vs cTnT 1 | 0.02 (−0.03 to 0.07) | 0.4610 |
| cTnT 1 | 0.92 (0.88 to 0.96) | cTnI B 1 vs cTnI S 1 | 0.02 (−0.01 to 0.05) | 0.2117 |
| cTnI B 1 vs cTnT 1 | 0.00 (−0.04 to 0.04) | 0.9317 | ||
| cTnI S 1 vs cTnT 1 | −0.02 (−0.05 to 0.01) | 0.2494 | ||
| cTnI CS peak | 0.98 (0.96 to 1.00) | cTnI CS peak vs cTnI B peak | 0.05 (0.02 to 0.09) | 0.0056 |
| cTnI B peak | 0.93 (0.88 to 0.97) | cTnI CS peak vs cTnI S peak | 0.05 (0.01 to 0.08) | 0.0152 |
| cTnI S peak | 0.94 (0.90 to 0.97) | cTnI CS peak vs cTnT peak | 0.06 (0.01 to 0.11) | 0.0134 |
| cTnT peak | 0.92 (0.88 to 0.97) | cTnI B peak vs cTnI S peak | −0.01 (−0.02 to 0.01) | 0.2340 |
| cTnI B peak vs cTnT peak | 0.00 (−0.04 to 0.04) | 0.8454 | ||
| cTnI S peak vs cTnT peak | 0.01 (−0.02 to 0.05) | 0.4760 |
| Diagnosis | cTnl CS | cTnl B | cTnl S | cTnT |
|---|---|---|---|---|
| On admission | ||||
| MI (n) | 53 | 44 | 49 | 53 |
| 13 | 22 | 17 | 14 | |
| No MI (n) | 16 | 11 | 19 | 33 |
| 749 | 758 | 751 | 733 | |
| Sensitivity (95% CI) | 0.803 (0.687 to 0.891) | 0.667 (0.540 to 0.778) | 0.742 (0.620 to 0.842) | 0.791 (0.674 to 0.881) |
| p-valuea | NS | NS | NS | |
| Specificity (95% CI) | 0.979 (0.966 to 0.988) | 0.986 (0.975 to 0.993) | 0.975 (0.962 to 0.985) | 0.957 (0.940 to 0.970) |
| p-valuea | NS | NS | 0.021 | |
| NPV (95% CI) | 0.983 (0.971 to 0.991) | 0.972 (0.958 to 0.982) | 0.978 (0.965 to 0.987) | 0.981 (0.969 to 0.990) |
| Peak value | ||||
| MI (n) | 64 | 53 | 57 | 57 |
| 3 | 15 | 11 | 11 | |
| No MI (n) | 24 | 15 | 31 | 43 |
| 742 | 766 | 751 | 736 | |
| Sensitivity (95% CI) | 0.955 (0.875 to 0.991) | 0.779 (0.662 to 0.871) | 0.838 (0.729 to 0.916) | 0.838 (0.729 to 0.916) |
| p-valuea | 0.005 | 0.048 | 0.048 | |
| Specificity (95% CI) | 0.969 (0.954 to 0.980) | 0.981 (0.969 to 0.989) | 0.960 (0.944 to 0.973) | 0.945 (0.926 to 0.960) |
| p-valuea | NS | NS | 0.029 | |
There were no statistically significant differences between the AUCs or diagnostic categorisation for the admission samples for any of these troponin methods. For peak value, the Stratus CS seemed to be diagnostically superior to all of the other troponin methods. There were no differences between any of the other methods. This may well represent a selection bias in the analysis as patients with an elevated Stratus CS troponin level were admitted and a second sample was not taken.
The individual methods missed varying numbers of patients with a final diagnosis of myocardial infarction. The Stratus CS cTnI missed three patients. One was diagnosed on ECG criteria, but had an elevated cTnT. The other two patients had elevated cTnI by the Siemens assay and elevated cTnT. The Beckman cTnI assay missed 15 patients. In 13 patients there was no second sample but six showed elevation of cTnI measured by the Siemens assay and seven showed elevation of cTnT. The Siemens cTnI assay missed 11 patients, eight of whom had no second sample, but showed elevation of cTnI on the Beckman assay (two patients) and elevation of cTnT (four patients). Measurement of cTnT missed 11 patients, 10 of whom did not have a second sample. Three patients had an elevated cTnI on the Beckman and Siemens assays and one had an elevated cTnI on the Siemens assay alone.
Increased sensitivity of troponin methods is at the expense of specificity due to the detection of myocardial injury in a range of other non-ACS conditions. In the population where AMI was excluded, elevation of all four troponins occurred in three cases, two of which had myocarditis. Three patients showed elevation of all three of the troponin methods under investigation but not of cTnI measured by the Stratus CS. One of these patients had myocarditis. Elevation of cTnI measured by the Stratus CS occurred in 24/766 (3.1%) of cases with a final diagnosis that excluded myocardial infarction, two also showed elevation of cTnI by the Siemens Ultra, 1 by the Beckman method and eight showed elevation of cTnT. Elevation of troponin in patients without a final diagnosis of myocardial infarction occurred in 15/781 (1.9%) patients for cTnI measured by the Beckman method, 31 (31/782, 4.0%) patients for cTnI measured by the Siemens Ultra assay and 43/779 (5.5%) patients for cTnT. One in 15 with elevated cTnI by the Beckman method had elevation by the Stratus CS alone and 9/15 by the Siemens Ultra alone and 11/15 had an elevated cTnT. For the Siemens method, 16/31 also had elevation of cTnT. Detailed information on missed diagnosis of myocardial infarction for all four troponin methods and elevations in patients without a final diagnosis of myocardial infarction is summarised inAppendices 1and2.
Do markers of vascular dysfunction contribute to the early differential diagnosis of patients presenting with chest pain?
Measurement of cardiac troponin was compared with measurement of copeptin and NTproBNP on admission and for the peak of the admission or 90 minutes from admission value. The diagnostic efficiency was compared by ROC curve analysis using the review final diagnosis as above. The ROC curves are shown inFigures 11and12with the areas under the ROC curve and comparisons inTable 15. Measurement of NTproBNP was superior to copeptin measurement in the admission and peak samples. Measurement of cardiac troponin was superior to both NTproBNP and copeptin measurement on admission and in the peak sample.
FIGURE 11.
Receiver operating characteristic curves for vascular dysfunction markers compared with troponin measurements for the diagnosis of an AMI: markers measured on admission. cTnI CS 1, Stratus CS admission sample; cTnI B1, Beckman AccuTnl admission sample; cTnI S 1, Siemens Ultra admission sample; cTnT 1, Roche high-sensitivity cTnT admission value; copeptin 1, copeptin admission sample; NTproBNP 1, NTproBNP admission sample
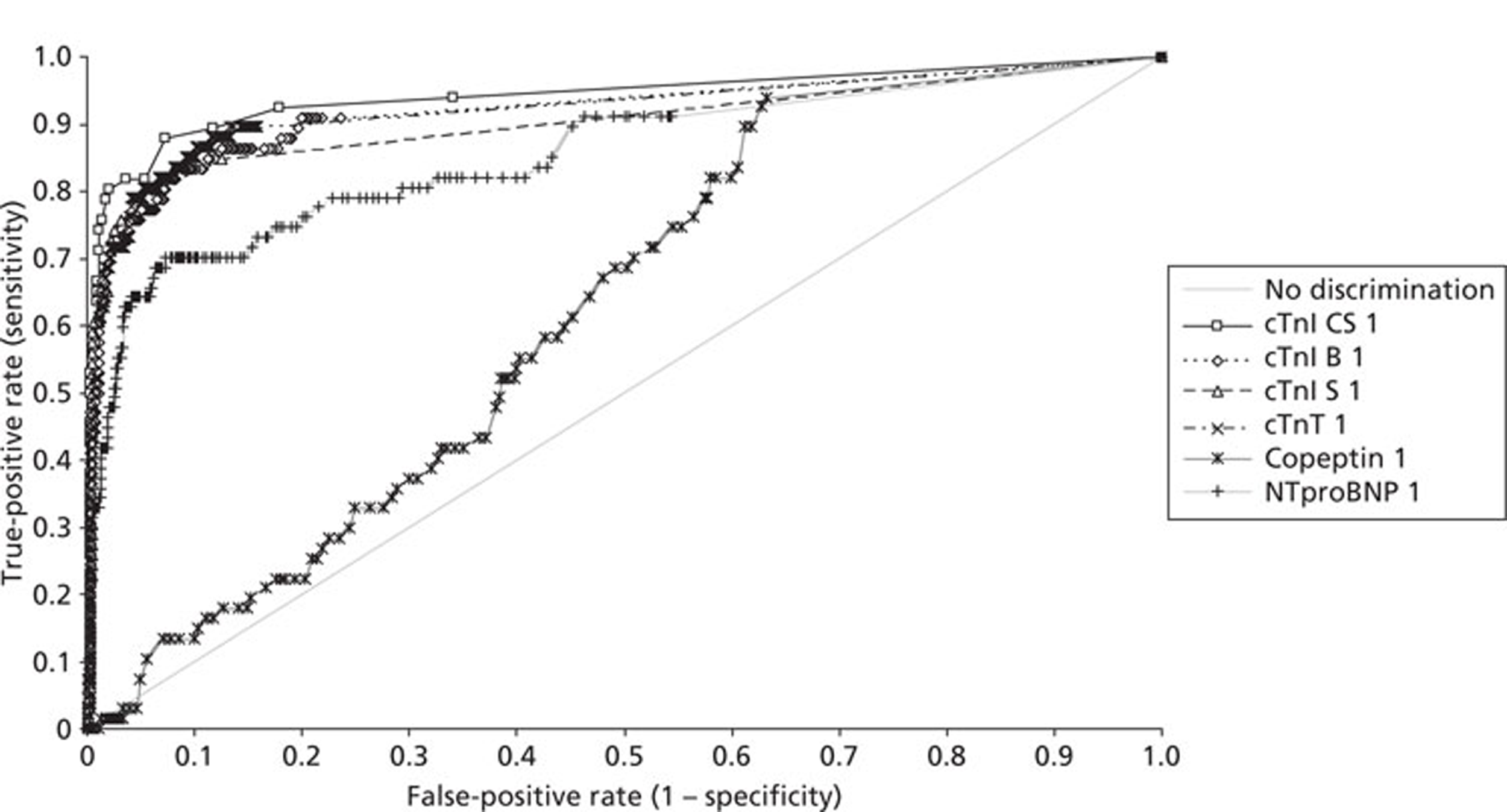
FIGURE 12.
Receiver operating characteristic curves for vascular dysfunction markers compared with troponin measurements for the diagnosis of an AMI. Markers are peak values of measurement on admission or 90 minutes from admission. cTnI CS peak, Stratus CS peak sample; cTnI B peak, Beckman AccuTnI peak value; cTnI S peak, Siemens Ultra peak value; cTnT peak, Roche high-sensitivity cardiac cTnT peak value; copeptin peak, copeptin peak value; NTproBNP peak, NTproBNP peak value
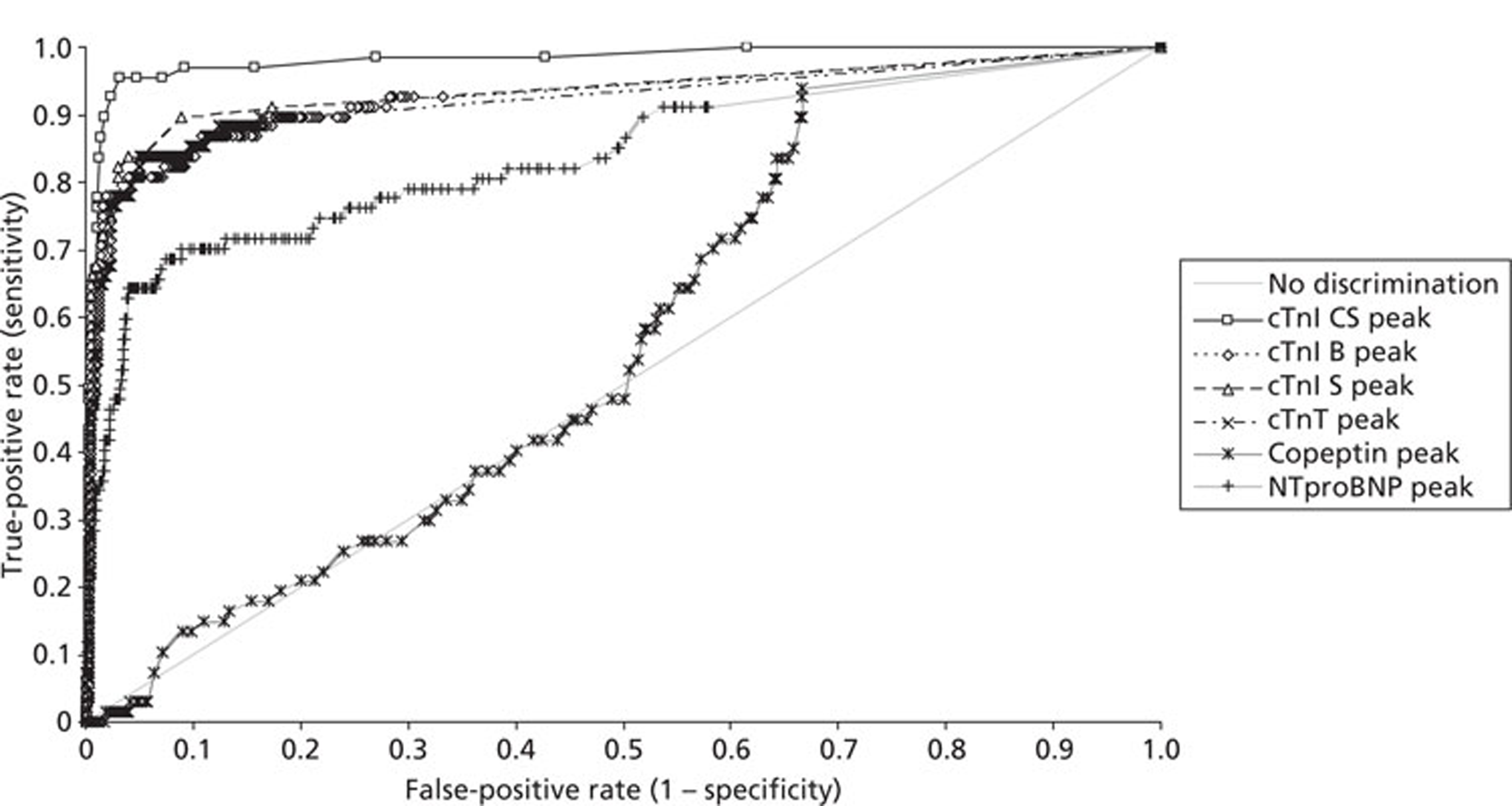
| Test | Area (95% CI) | Contrast | Difference (95% CI) | p-value |
| cTnI CS 1 | 0.94 (0.90 to 0.98) | cTnI CS 1 vs copeptin 1 | 0.32 (0.26 to 0.38) | < 0.0001 |
| cTnI B 1 | 0.92 (0.88 to 0.96) | cTnI CS 1 vs NTproBNP 1 | 0.09 (0.03 to 0.14) | 0.0029 |
| cTnI S 1 | 0.90 (0.85 to 0.95) | cTnI B 1 vs copeptin 1 | 0.30 (0.23 to 0.36) | < 0.0001 |
| cTnT 1 | 0.92 (0.88 to 0.96) | cTnI B 1 vs NTproBNP 1 | 0.07 (0.02 to 0.12) | 0.0101 |
| Copeptin 1 | 0.62 (0.57 to 0.68) | cTnI S 1 vs copeptin 1 | 0.28 (0.22 to 0.34) | < 0.0001 |
| NTproBNP 1 | 0.85 (0.80 to 0.9) | cTnI S 1 vs NTproBNP 1 | 0.05 (0.01 to 0.09) | 0.0273 |
| cTnT 1 vs copeptin 1 | 0.30 (0.24 to 0.36) | < 0.0001 | ||
| cTnT 1 vs NTproBNP 1 | 0.07 (0.03 to 0.11) | 0.0010 | ||
| Copeptin 1 vs NTproBNP 1 | −0.23 (−0.30 to −0.16) | < 0.0001 | ||
| cTnI CS peak | 0.98 (0.96 to 1.00) | cTnI CS peak vs copeptin peak | 0.42 (0.35 to 0.48) | < 0.0001 |
| cTnI B peak | 0.93 (0.88 to 0.97) | cTnI CS peak vs NTproBNP peak | 0.14 (0.08 to 0.21) | < 0.0001 |
| cTnI S peak | 0.94 (0.90 to 0.97) | cTnI B peak vs copeptin peak | 0.36 (0.29 to 0.43) | < 0.0001 |
| cTnT peak | 0.92 (0.88 to 0.97) | cTnI B peak vs NTproBNP peak | 0.09 (0.03 to 0.15) | 0.0044 |
| Copeptin peak | 0.56 (0.50 to 0.62) | cTnI S peak vs copeptin peak | 0.37 (0.30 to 0.44) | < 0.0001 |
| NTproBNP peak | 0.84 (0.78 to 0.90) | cTnI S peak vs NTproBNP peak | 0.10 (0.04 to 0.16) | 0.0014 |
| cTnT peak vs copeptin peak | 0.36 (0.30 to 0.42) | < 0.0001 | ||
| cTnT peak vs NTproBNP peak | 0.08 (0.04 to 0.13) | 0.0002 | ||
| Copeptin peak vs NTproBNP peak | −0.27 (−0.35 to −0.20) | < 0.0001 |
Do combinations of biomarkers allow earlier rule-in or rule-out of an acute myocardial infarction in patients presenting with chest pain?
The rationale for the selection of other cytoplasmic biomarkers of cardiac necrosis or neurohormones is that they may provide earlier information or supplementary information to measurement of cardiac troponin alone. A number of studies have suggested that the combination of either H-FABP or copeptin measurement with measurement of cardiac troponin on admission would allow very early diagnostic categorisation and potentially discharge solely on the basis of admission measurement. The combination of the different troponins with H-FABP or copeptin for rule-in or rule-out diagnosis was examined by construction of contingency tables utilising 99th percentile cut-offs for troponin and the 95th percentile cut-off for H-FABP. For copeptin, none of the values obtained exceeded the 95th percentile. In view of this an ROC optimised cut-off of 7.4 mg/l was used. The results of the analysis are summarised inTable 16. Measurement of H-FABP improved diagnostic sensitivity of all of the troponin measurements, but significantly reduced specificity. The increase in sensitivity obtained from combined measurement was equivalent to the sensitivity obtained from the combination of the measurement of troponin alone on admission and at 90 minutes from admission.
| Diagnosis | Positive test | Admission sample | H-FABP | Combined | Copeptin | Combined | NTproBNP | Combined | Peak troponin sample |
| cTnl CS | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| MI (n) | Y | 50 | 41 | 58 | 34 | 57 | 44 | 56 | 60 |
| N | 13 | 22 | 5 | 29 | 6 | 19 | 7 | 3 | |
| No MI (n) | Y | 15 | 46 | 57 | 291 | 303 | 48 | 71 | 23 |
| N | 725 | 694 | 683 | 449 | 437 | 692 | 669 | 717 | |
| Sensitivity (95% CI) | 0.794 (0.673 to 0.885) | 0.651 (0.520 to 0.767) | 0.921 (0.824 to 0.974) | 0.540 (0.409 to 0.666) | 0.905 (0.804 to 0.964) | 0.698 (0.570 to 0.808) | 0.889 (0.784 to 0.954) | 0.952 (0.867 to 0.990) | |
| p-value | NS | NS | 0.0043 | NS | NS | NS | |||
| Specificity (95% CI) | 0.980 (0.967 to 0.989) | 0.938 (0.918 to 0.954) | 0.923 (0.901 to 0.941) | 0.607 (0.571 to 0.642) | 0.591 (0.555 to 0.626) | 0.935 (0.915 to 0.952) | 0.904 (0.881 to 0.924) | 0.969 (0.954 to 0.980) | |
| p-value | < 0.0001 | < 0.0001 | < 0.0001 | < 0.0001 | < 0.0001 | ||||
| NPV (95% CI) | 0.982 (0.970 to 0.991) | 0.969 (0.954 to 0.981) | 0.993 (0.983 to 0.998) | 0.939 (0.914 to 0.959) | 0.986 (0.971 to 0.995) | 0.973 (0.959 to 0.984) | 0.990 (0.979 to 0.996) | 0.996 (0.988 to 0.999) | |
| cTnl S | |||||||||
| MI (n) | Y | 46 | 54 | 52 | 52 | 52 | |||
| N | 17 | 9 | 11 | 11 | 11 | ||||
| No MI (n) | Y | 14 | 52 | 297 | 55 | 26 | |||
| N | 726 | 688 | 443 | 685 | 714 | ||||
| Sensitivity (95% CI) | 0.730 (0.603 to 0.834) | 0.857 (0.746 to 0.933) | 0.825 (0.709 to 0.909) | 0.825 (0.709 to 0.909) | 0.825 (0.709 to 0.909) | ||||
| p-value | NS | NS | NS | NS | |||||
| Specificity (95% CI) | 0.981 (0.968 to 0.990) | 0.930 (0.909 to 0.947) | 0.599 (0.563 to 0.634) | 0.926 (0.904 to 0.944) | 0.965 (0.949 to 0.977) | ||||
| p-value | < 0.0001 | < 0.0001 | < 0.0001 | < 0.0001 | |||||
| NPV (95% CI) | 0.977 (0.964 to 0.987) | 0.987 (0.976 to 0.994) | 0.976 (0.957 to 0.988) | 0.984 (0.972 to 0.992) | 0.985 (0.973 to 0.992) | ||||
| cTnT | |||||||||
| MI (n) | Y | 49 | 54 | 53 | 53 | 52 | |||
| N | 14 | 9 | 10 | 10 | 11 | ||||
| No MI (n) | Y | 28 | 62 | 299 | 65 | 38 | |||
| N | 712 | 678 | 441 | 675 | 702 | ||||
| Sensitivity (95% CI) | 0.778 (0.655 to 0.873) | 0.857 (0.746 to 0.933) | 0.841 (0.727 to 0.921) | 0.841 (0.727 to 0.921) | 0.825 (0.709 to 0.909) | ||||
| p-value | NS | NS | NS | NS | |||||
| Specificity (95% CI) | 0.962 (0.946 to 0.975) | 0.916 (0.894 to 0.935) | 0.596 (0.561 to 0.631) | 0.912 (0.889 to 0.932) | 0.949 (0.930 to 0.963) | ||||
| p-value | 0.0003 | < 0.0001 | < 0.0001 | < 0.0001 | |||||
| NPV (95% CI) | 0.981 (0.968 to 0.989) | 0.987 (0.975 to 0.994) | 0.978 (0.969 to 0.989) | 0.985 (0.973 to 0.993) | 0.985 (0.974 to 0.993) | ||||
| cTnl B | |||||||||
| MI (n) | Y | 42 | 49 | 51 | 49 | 49 | |||
| N | 21 | 14 | 12 | 14 | 14 | ||||
| No MI (n) | Y | 11 | 53 | 296 | 53 | 15 | |||
| N | 729 | 687 | 444 | 687 | 725 | ||||
| Sensitivity (95% CI) | 0.667 (0.537 to 0.780) | 0.778 (0.655 to 0.873) | 0.841 (0.727 to 0.921) | 0.778 (0.665 to 0.873) | 0.778 (0.655 to 0.873) | ||||
| p-value | NS | NS | NS | NS | |||||
| Specificity (95% CI) | 0.985 (0.974 to 0.993) | 0.928 (0.907 to 0.946) | 0.596 (0.561 to 0.631) | 0.928 (0.907 to 0.946) | 0.980 (0.980 to 0.989) | ||||
| p-value | < 0.0001 | < 0.0001 | < 0.0001 | < 0.0001 | |||||
| NPV (95% CI) | 0.980 (0.967 to 0.989) | 0.974 (0.955 to 0.986) | 0.981 (0.968 to 0.990) | ||||||
The prognostic accuracy for adverse cardiac events of highly sensitive troponin assays and the range of new cardiac biomarkers
What is the prognostic role of cytoplasmic markers of myocardial damage when compared with troponin measurement?
Receiver operating characteristic curves using the combined MACE of death, readmission with myocardial infarction, readmission with unstable angina or the need for urgent revascularisation as the dichotomous variable were constructed using the admission sample measurement and peak of the admission or 90-minute sample measurement comparing cytoplasmic markers with the four troponin measurement methods. The results are summarised inFigures 13–16with the AUCs and comparison of the AUCs inTables 17and18.
FIGURE 13.
Receiver operating characteristic curves for cytoplasmic markers and troponin measurements for the prediction of MACE: markers measured on admission cTnI CS 1, Stratus CS admission sample; CK-MB 1, CK-MB admission sample; Myo 1, myoglobin admission sample; FABP 1, H-FABP admission sample.
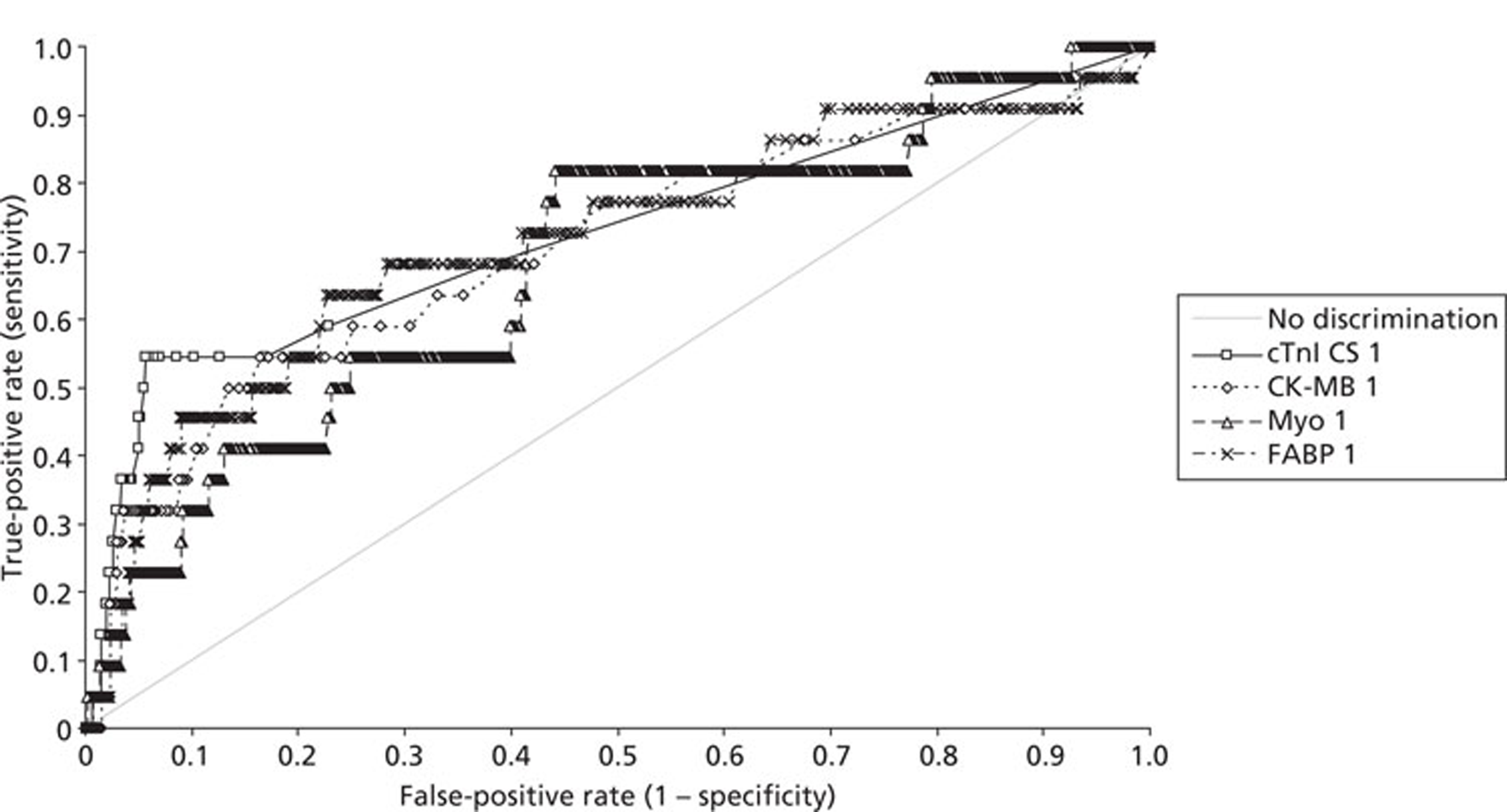
FIGURE 14.
Receiver operating characteristic curves for troponin measurements for the prediction of MACE: markers measured on admission. cTnI CS 1, Stratus CS admission sample; cTnI B 1, Beckman AccuTnI admission sample; cTnI S 1, Siemens Ultra admission sample; cTnT 1, Roche high-sensitivity cTnT admission sample.
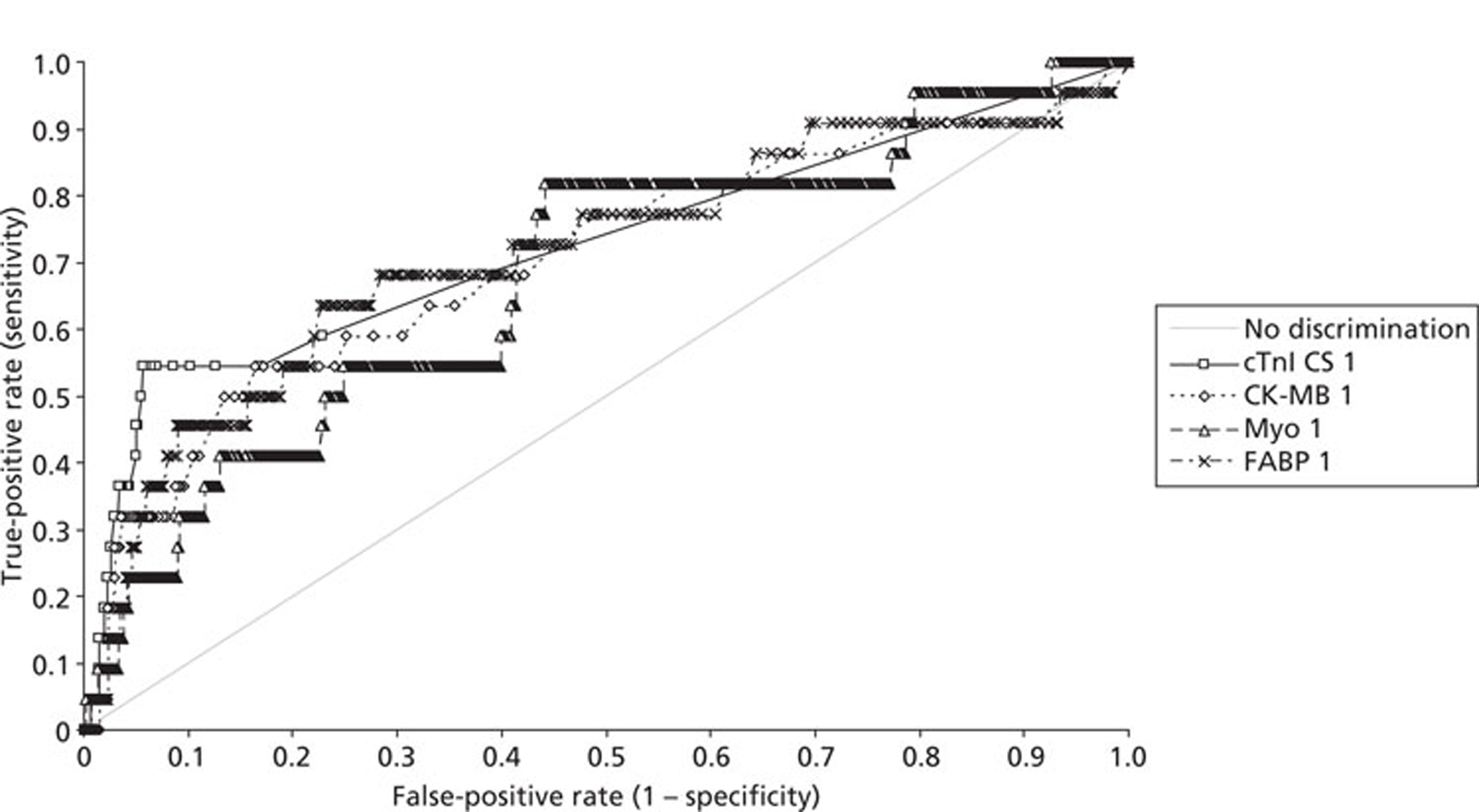
FIGURE 15.
Receiver operating characteristic curves for cytoplasmic markers and troponin measurements for the prediction of MACE. Markers are peak values of measurement on admission or 90 minutes from admission. cTnI CS peak, Stratus CS peak value; CK-MB peak, CK-MB peak value; Myo peak, myoglobin peak value; FABP peak, H-FABP peak value.
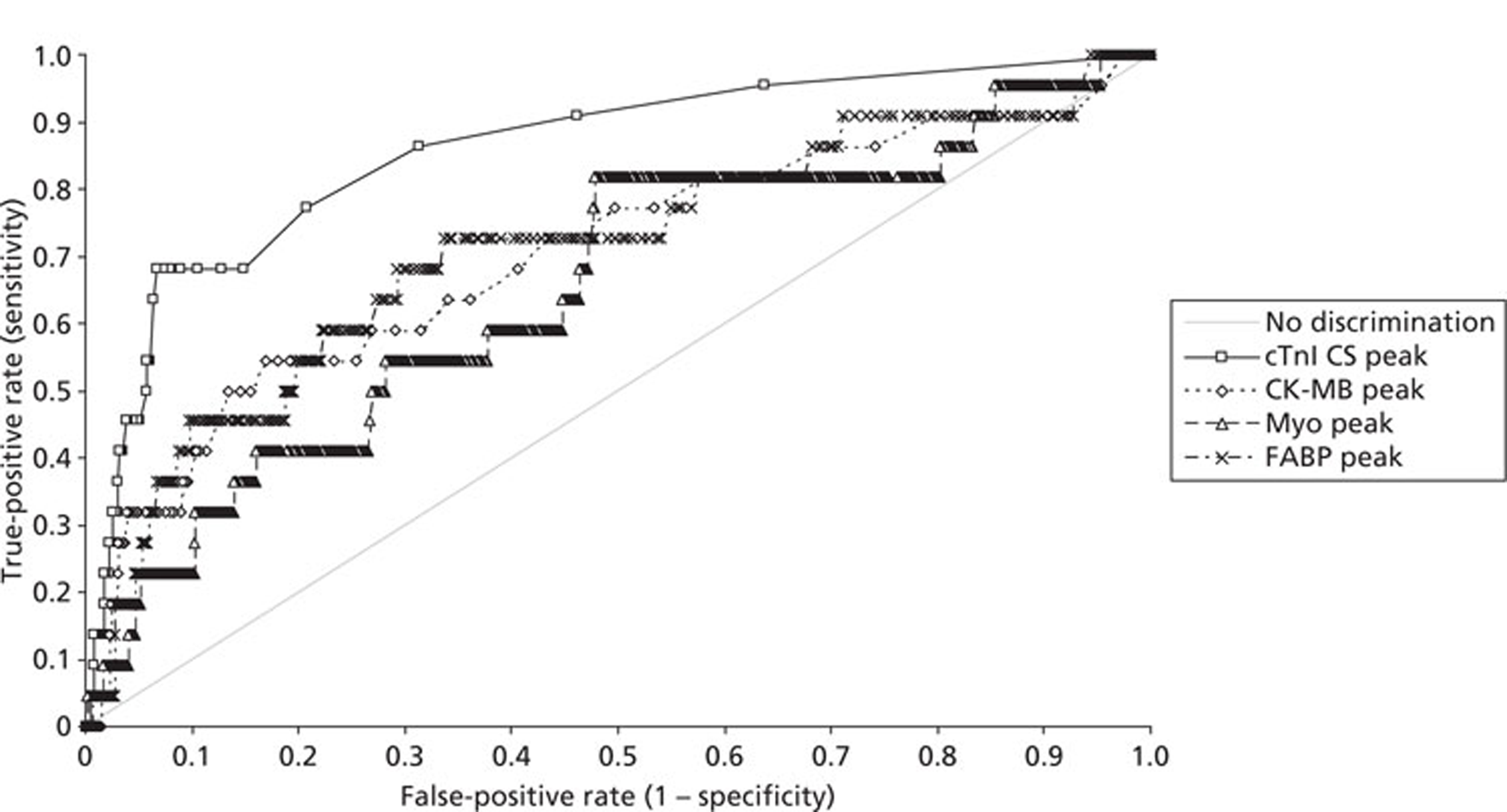
FIGURE 16.
Receiver operating characteristic curves for troponin measurements for the prediction of MACE. Markers are peak values of measurement on admission or 90 minutes from admission. cTnI CS peak, Stratus CS peak value; cTnI B peak, Beckman AccuTnI peak value; cTnI S peak, Siemens Ultra peak value; cTnT peak, Roche high-sensitivity cTnT peak value.
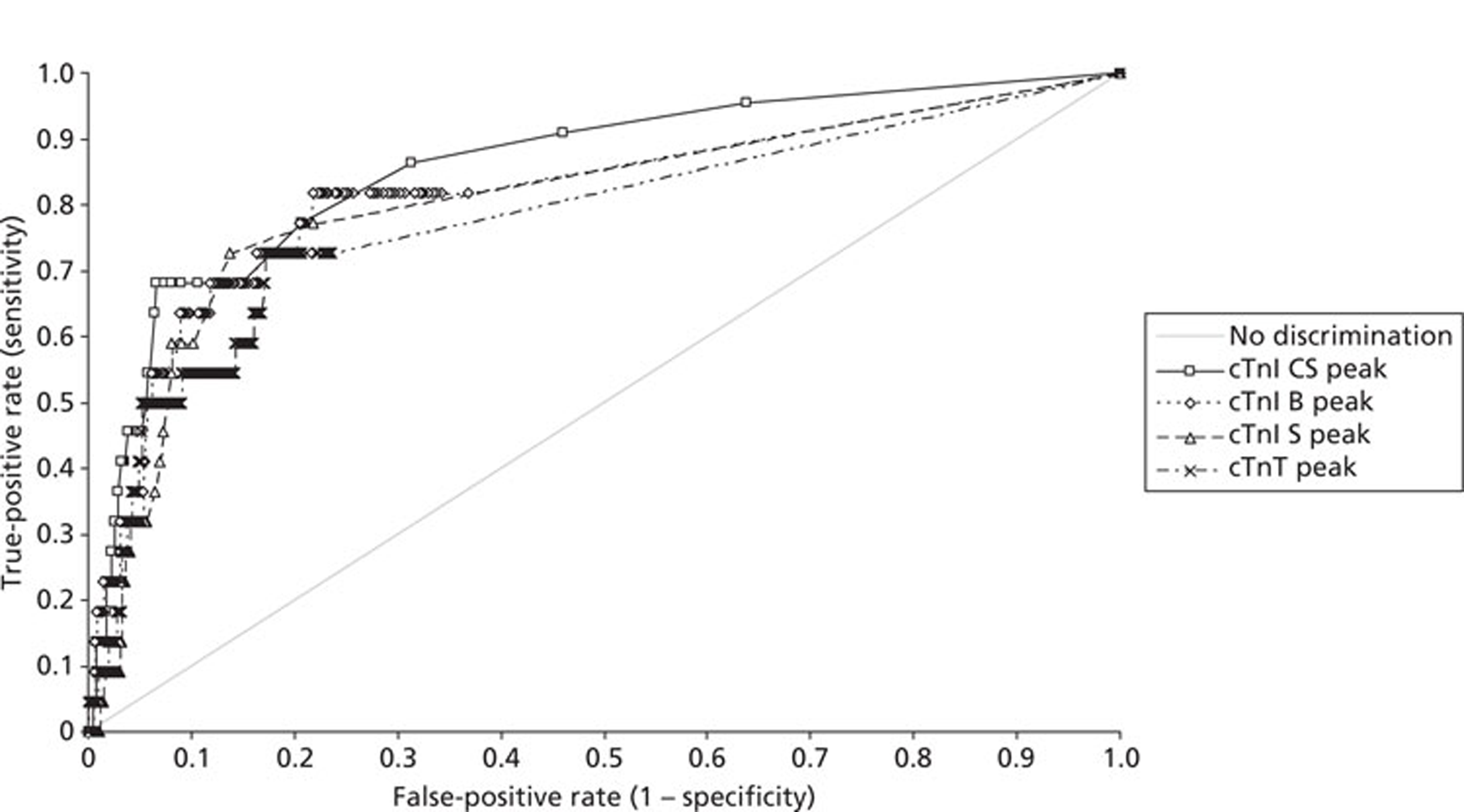
| Test | Area (95% CI) | Contrast | Difference (95% CI) | p-value |
| cTnI CS 1 | 0.73 (0.60 to 0.85) | cTnI CS 1 vs CK-MB 1 | 0.02 (−0.08 to 0.12) | 0.6798 |
| CK-MB 1 | 0.71(0.58 to 0.84) | cTnI CS 1 vs Myo 1 | 0.05 (−0.07 to 0.17) | 0.4471 |
| Myo 1 | 0.68 (0.56 to 0.80) | cTnI CS 1 vs FABP 1 | 0.01 (−0.10 to 0.12) | 0.8484 |
| FABP 1 | 0.72 (0.59 to 0.84) | CK-MB 1 vs Myo 1 | 0.03 (−0.07 to 0.12) | 0.5908 |
| cTnI B 1 | 0.83 (0.74 to 0.93) | CK-MB 1 vs FABP 1 | −0.01 (−0.08 to 0.06) | 0.7965 |
| cTnI S 1 | 0.80 (0.70 to 0.90) | Myo 1 vs FABP 1 | −0.04 (−0.12 to 0.05) | 0.4295 |
| cTnT 1 | 0.79 (0.68 to 0.89) | cTnI B 1 vs CK-MB 1 | 0.13 (0.06 to 0.19) | 0.0001 |
| cTnI B 1 vs Myo 1 | 0.15 (0.07 to 0.24) | 0.0004 | ||
| cTnI B 1 vs FABP 1 | 0.12 (0.05 to 0.19) | 0.0012 | ||
| cTnI S 1 vs CK-MB 1 | 0.09 (0.02 to 0.16) | 0.0093 | ||
| cTnI S 1 vs Myo 1 | 0.12 (0.03 to 0.21) | 0.0126 | ||
| cTnI S 1 vs FABP 1 | 0.08 (0.01 to 0.16) | 0.0337 | ||
| cTnT 1 vs CK-MB 1 | 0.08 (−0.01 to 0.18) | 0.0810 | ||
| cTnT 1 vs FABP 1 | 0.07 (−0.01 to 0.16) | 0.0982 | ||
| cTnT 1 vs Myo 1 | 0.11 (0.01 to 0.21) | 0.0363 |
| Test | Area (95% CI) | Contrast | Difference (95% CI) | p-value |
| cTnI CS peak | 0.86 (0.77 to 0.94) | cTnI CS peak vs CK-MB peak | 0.15 (0.07 to 0.24) | 0.0003 |
| CK-MB peak | 0.70 (0.57 to 0.83) | cTnI CS peak vs Myo peak | 0.20 (0.09 to 0.32) | 0.0003 |
| Myo peak | 0.65 (0.53 to 0.78) | cTnI CS peak vs FABP peak | 0.15 (0.06 to 0.23) | 0.0009 |
| FABP peak | 0.71 (0.58 to 0.84) | CK-MB peak vs Myo peak | 0.05 (−0.05 to 0.15) | 0.3486 |
| cTnI B peak | 0.82 (0.71 to 0.92) | CK-MB peak vs FABP peak | −0.01 (−0.08 to 0.06) | 0.8263 |
| cTnI S peak | 0.81 (0.71 to 0.91) | Myo peak vs FABP peak | −0.06 (−0.15 to 0.03) | 0.2153 |
| cTnT peak | 0.78 (0.67 to 0.89) | cTnI B peak vs CK-MB peak | 0.12 (0.05 to 0.18) | 0.0004 |
| cTnI B peak vs Myo peak | 0.17 (0.08 to 0.25) | 0.0003 | ||
| cTnI B peak vs FABP peak | 0.11 (0.05 to 0.17) | 0.0005 | ||
| cTnI S peak vs CK-MB peak | 0.11 (0.04 to 0.17) | 0.0021 | ||
| cTnI S peak vs Myo peak | 0.16 (0.06 to 0.25) | 0.0011 | ||
| cTnI S peak vs FABP peak | 0.10 (0.04 to 0.16) | 0.0018 | ||
| cTnT peak vs CK-MB peak | 0.08 (−0.01 to 0.17) | 0.0936 | ||
| cTnT peak vs Myo peak | 0.13 (0.02 to 0.23) | 0.0168 | ||
| cTnT peak vs FABP peak | 0.07 (−0.02 to 0.16) | 0.1108 |
Differences between the admission value and the peak value were observed. In the case of the admission measurement, cTnI measured on the Stratus CS was equivalent to all of the cytoplasmic markers (CK-MB, myoglobin and H-FABP) for outcome prediction. The three sensitive troponin methods demonstrated a greater AUC than the cytoplasmic markers, which was statistically significant for both cTnI methods but just failed to reach significance for cTnT for CK-MB and H-FABP. All of the cytoplasmic markers had equivalent ability to predict outcome on the admission sample measurement. When peak values were examined, troponin measurement was overall a significantly better outcome predictor than the cytoplasmic markers. Measurement of cTnI on the Stratus CS for the peak values was significantly better than measurement of all three cytoplasmic markers. The other cTnI methods also demonstrated improved outcome prediction with an increase in significance. Measurement of cTnT showed improvement in diagnostic performance only when compared with measurement of myoglobin when peak values were examined.
What is the prognostic role of high-sensitivity troponin assays?
Receiver operating characteristic curves using the combined MACEs of death, readmission with myocardial infarction, readmission with unstable angina or the need for urgent revascularisation as the dichotomous variable were constructed using the admission sample measurement and peak of the admission or 90-minute sample measurement comparing the four troponin measurement methods. The results are summarised inFigures 14and16with the AUCs and comparison of the AUCs inTable 19.
| Test | Area (95% CI) | Contrast | Difference (95% CI) | p-value |
| cTnI CS 1 | 0.73 (0.60 to 0.85) | cTnI CS 1 vs cTnI B 1 | −0.11 (−0.22 to 0.00) | 0.0523 |
| cTnI B 1 | 0.83 (0.74 to 0.93) | cTnI CS 1 vs cTnI S 1 | −0.07 (−0.16 to 0.01) | 0.1054 |
| cTnI S 1 | 0.80 (0.70 to 0.90) | cTnI CS 1 vs cTnT 1 | −0.06 (−0.16 to 0.04) | 0.2112 |
| cTnT 1 | 0.79 (0.68 to 0.89) | cTnI B 1 vs cTnI S 1 | 0.04 (−0.02 to 0.10) | 0.2231 |
| cTnI B 1 vs cTnT 1 | 0.04 (−0.04 to 0.13) | 0.3295 | ||
| cTnI S 1 vs cTnT 1 | 0.01 (−0.06 to 0.07) | 0.8215 | ||
| cTnI CS peak | 0.86 (0.77 to 0.94) | cTnI CS peak vs cTnI B peak | 0.04 (−0.02 to 0.10) | 0.1791 |
| cTnI B peak | 0.82 (0.71 to 0.92) | cTnI CS peak vs cTnI S peak | 0.05 (−0.01 to 0.11) | 0.1109 |
| cTnI S peak | 0.81 (0.71 to 0.91) | cTnI CS peak vs cTnT peak | 0.08 (−0.03 to 0.18) | 0.1454 |
| cTnT peak | 0.78 (0.67 to 0.89) | cTnI B peak vs cTnI S peak | 0.01 (−0.04 to 0.06) | 0.6676 |
| cTnI B peak vs cTnT peak | 0.04 (−0.06 to 0.13) | 0.4538 | ||
| cTnI S peak vs cTnT peak | 0.03 (−0.06 to 0.11) | 0.5406 |
There was no statistically significant difference between the ability of the different troponin methods to predict MACEs on either admission or for peak sample measurements. Although cTnI measurement using the Stratus CS showed superior performance for the diagnosis of an AMI when assessed using peak values, the ability of the different troponin methods to predict MACEs was not statistically significantly different between admission values and peak values. The apparent superiority of the Stratus CS method for detecting an AMI may reflect selection bias in the population. A positive cTnI measured using the Stratus CS was used to select patients for admission. When an independent measure of performance was utilised, the ability to predict an adverse prognosis, this apparent discrepancy between the methods disappears.
What is the prognostic role of myocardial dysfunction markers compared with troponin measurement?
Receiver operating characteristic curves using the combined MACE of death, readmission with myocardial infarction, readmission with unstable angina or the need for urgent revascularisation as the dichotomous variable were constructed using the admission sample measurement and the peak of the admission or 90-minute sample measurement comparing the four troponin methods with neurohormone measurement. The results are summarised inFigures 17and18with the AUCs and comparison of the AUCs inTable 20.
FIGURE 17.
Receiver operating characteristic curves for troponin and neurohormone measurements for the prediction of MACE: markers measured on admission cTnI CS 1, Stratus CS admission sample; cTnI B1, Beckman AccuTnl admission sample; cTnI S 1, Siemens Ultra admission sample; cTnT 1, Roche high-sensitivity cTnT admission value; copeptin 1, copeptin admission sample; NTproBNP 1, NTproBNP admission sample.
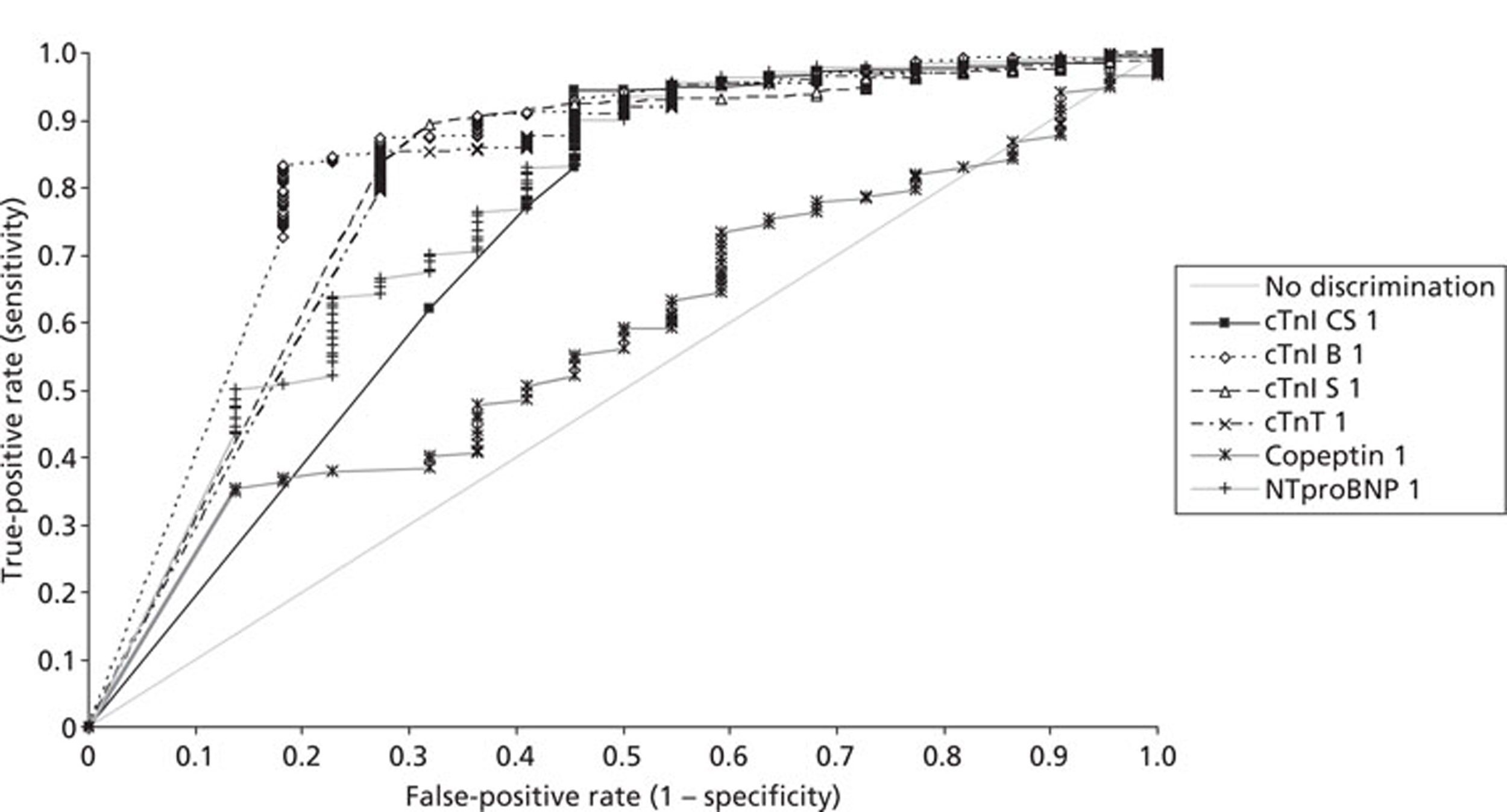
FIGURE 18.
Receiver operating characteristic curves for troponin and neurohormone measurements for the prediction of MACE. Markers are peak values of measurement on admission or 90 minutes from admission. cTnI CS peak, Stratus CS peak sample; cTnI B peak, Beckman AccuTnI peak value; cTnI S peak, Siemens Ultra peak value; cTnT peak, Roche high-sensitivity cardiac cTnT peak value; copeptin peak, copeptin peak value; NTproBNP peak, NTproBNP peak value
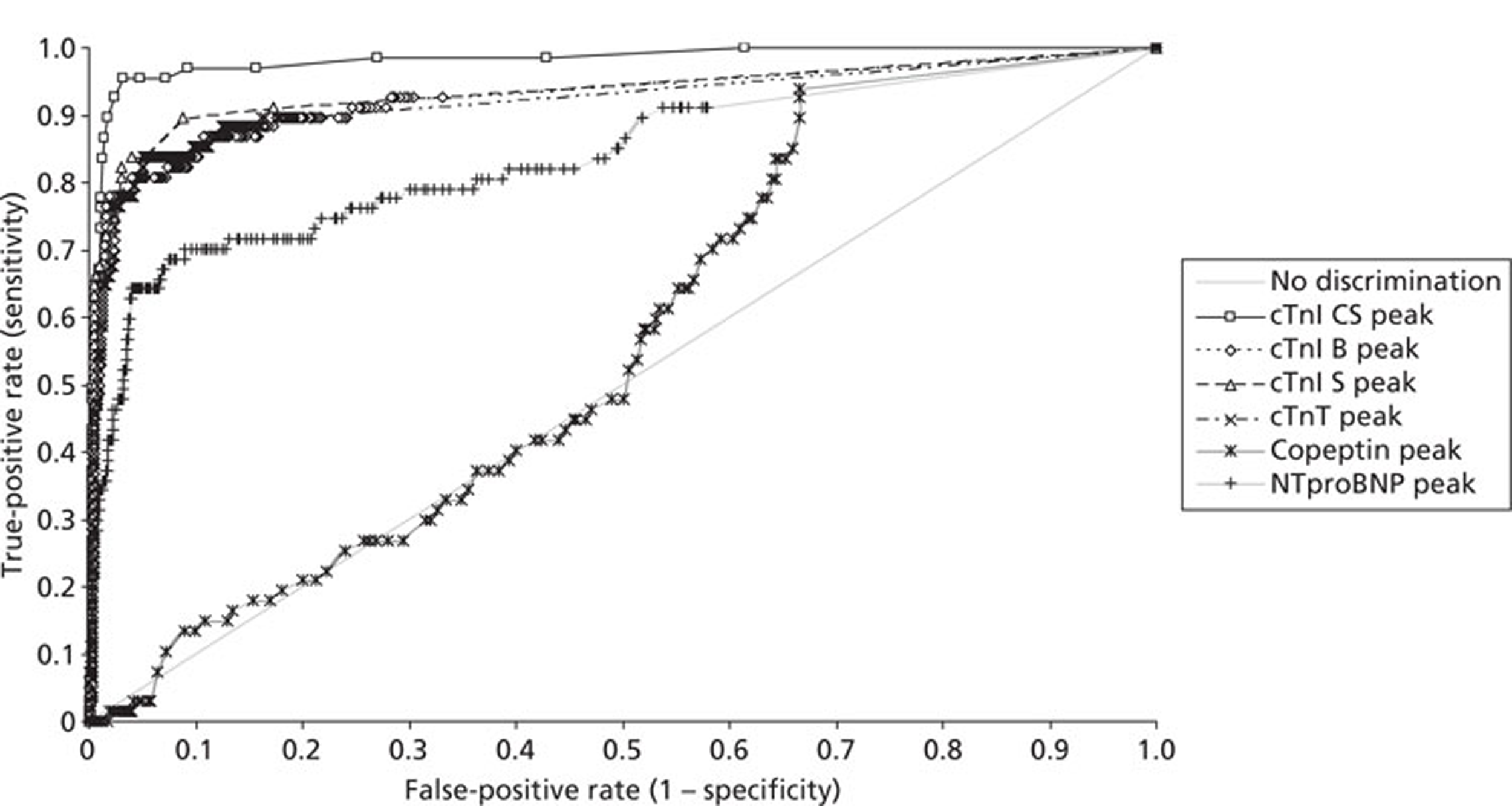
| Test | Area (95% CI) | Contrast | Difference (95% CI) | p-value |
| cTnI CS 1 | 0.73 (0.60 to 0.85) | cTnI CS 1 vs copeptin 1 | 0.15 (0.02 to 0.28) | 0.0291 |
| cTnI B 1 | 0.83 (0.74 to 0.93) | cTnI CS 1 vs NTproBNP 1 | −0.04 (−0.15 to 0.07) | 0.4431 |
| cTnI S 1 | 0.80 (0.70 to 0.90) | cTnI B 1 vs copeptin 1 | 0.25 (0.13 to 0.37) | < 0.0001 |
| cTnT 1 | 0.79 (0.68 to 0.89) | cTnI B 1 vs NTproBNP 1 | 0.06 (−0.04 to 0.17) | 0.2496 |
| Copeptin 1 | 0.58 (0.47 to 0.69) | cTnI S 1 vs copeptin 1 | 0.22 (0.10 to 0.34) | 0.0004 |
| NTproBNP 1 | 0.77 (0.65 to 0.89) | cTnI S 1 vs NTproBNP 1 | 0.03 (−0.07 to 0.13) | 0.5951 |
| cTnT 1 vs copeptin 1 | 0.21 (0.09 to 0.33) | 0.0006 | ||
| cTnT 1 vs NTproBNP 1 | 0.02 (−0.08 to 0.12) | 0.6860 | ||
| Copeptin 1 vs NTproBNP 1 | −0.19 (−0.30 to −0.09) | 0.0004 | ||
| cTnI CS peak | 0.98 (0.96 to 1.00) | cTnI CS peak vs copeptin peak | 0.42 (0.35 to 0.48) | < 0.0001 |
| cTnI B peak | 0.93 (0.88 to 0.97) | cTnI CS peak vs NTproBNP peak | 0.14 (0.08 to 0.21) | < 0.0001 |
| cTnI S peak | 0.94 (0.90 to 0.97) | cTnI B peak vs copeptin peak | 0.36 (0.29 to 0.43) | < 0.0001 |
| cTnT peak | 0.92 (0.88 to 0.97) | cTnI B peak vs NTproBNP peak | 0.09 (0.03 to 0.15) | 0.0044 |
| Copeptin peak | 0.56 (0.50 to 0.62) | cTnI S peak vs copeptin peak | 0.37 (0.30 to 0.44) | < 0.0001 |
| NTproBNP peak | 0.84 (0.78 to 0.90) | cTnI S peak vs NTproBNP peak | 0.10 (0.04 to 0.16) | 0.0014 |
| cTnT peak vs copeptin peak | 0.36 (0.30 to 0.42) | < 0.0001 | ||
| cTnT peak vs NTproBNP peak | 0.08 (0.04 to 0.13) | 0.0002 | ||
| Copeptin peak vs NTproBNP peak | −0.27 (−0.35 to −0.20) | < 0.0001 |
When measured on admission, all four troponin methods and NTproBNP were significantly better outcome predictors than copeptin. On the admission sample, none of the four troponin methods predicted adverse events significantly better than measurement of NTproBNP. A different pattern was seen when examining measurements of peak value. All four troponin methods were significantly better outcome predictors than NTproBNP when measured as the peak of the admission or 90-minute sample. Again, copeptin performed poorly and was significantly worse than NTproBNP.
Estimation of the potential economic impact (clinical effectiveness and cost-effectiveness) of using highly sensitive troponin assays or the range of new cardiac biomarkers instead of an admission and 12-hour troponin measurement
Economic impact of measuring a panel of contemporary cardiac markers compared with the measurement of cardiac troponin alone for early differential diagnosis in chest pain patients
Currently, the costs of performing troponin, CK-MB and myoglobin measurements are equivalent. All utilise the same equipment and measurement is performed by immunoassay. The measurement of troponin is required by the universal definition of myocardial infarction. Measurement of CK-MB and myoglobin in addition could be justified only if there was superior diagnostic performance of the panel of assays. As there was no diagnostic efficiency to be gained in measuring a panel compared with a single test, Cost minimisation analysis shows that measurement of cardiac troponin alone was the most cost-effective diagnostic strategy.
Economic impact of combining a novel biomarker of myocardial injury with cardiac troponin measurement
Objective
The objective was to estimate the potential cost-effectiveness of using highly sensitive troponin assays (at presentation alone or at presentation and 90 minutes later) and new cardiac biomarkers instead of a 10- to 12-hour troponin measurement.
After review of the results of the analytical part of the project, the following diagnostic strategies were applied to each patient:
-
no testing: discharge all patients without treatment
-
high-sensitivity troponin at presentation: discharge home if test is negative or admit to hospital for troponin testing at 10–12 hours if positive
-
high-sensitivity troponin and H-FABP at presentation: discharge home if both tests are negative or admit to hospital for troponin testing at 10–12 hours if either test is positive
-
high-sensitivity troponin at presentation and at 90 minutes: discharge home if both tests are negative or admit to hospital for troponin testing at 10–12 hours if either test is positive
-
standard troponin testing at 10–12 hours.
The cTnT and H-FABP data from this study were used to estimate the sensitivity and specificity of strategies 2–4. Only H-FABP in combination with troponin was tested because this combination was both more sensitive and more specific than other biomarker combinations (troponin with copeptin or troponin with NTproBNP) and so it would clearly dominate them if the costs of all biomarkers were assumed to be the same.
The main analysis used data from cTnT to estimate the sensitivity and specificity of cTnT at presentation, cTnT and H-FABP at presentation and cTnT at presentation and at 90 minutes. In a secondary analysis the same strategies were tested but using data from cTnI (Stratus CS), which analysis suggested may have superior diagnostic accuracy. The estimates used are presented inTables 21and22.
| Strategy | Sensitivity (95% CI) | Specificity (95% CI) |
|---|---|---|
| No testing | 0 | 1 |
| High-sensitivity cTnT at presentation | 0.778 (0.655 to 0.873) | 0.962 (0.946 to 0.975) |
| High-sensitivity cTnT and H-FABP at presentation | 0.857 (0.746 to 0.933) | 0.916 (0.894 to 0.935) |
| High-sensitivity cTnT at presentation and at 90 minutes | 0.825 (0.709 to 0.909) | 0.949 (0.930 to 0.963) |
| Troponin at 10–12 hours | 1 | 1 |
| Strategy | Sensitivity (95% CI) | Specificity (95% CI) |
|---|---|---|
| No testing | 0 | 1 |
| High-sensitivity cTnI at presentation | 0.794 (0.673 to 0.885) | 0.980 (0.967 to 0.989) |
| High-sensitivity cTnI and H-FABP at presentation | 0.921 (0.824 to 0.974) | 0.923 (0.901 to 0.941) |
| High-sensitivity cTnI at presentation and at 90 minutes | 0.952(0.867 to 0.990) | 0.969 (0.954 to 0.980) |
| Troponin at 10–12 hours | 1 | 1 |
Main analysis deterministic results
The results of the main deterministic analysis for cTnT strategies in a hypothetical cohort of patients (n = 2240) in the three scenarios tested, that is, doctor on demand, twice-daily ward round and once-daily ward round, are shown inTable 23. As expected, the effectiveness of the strategies (as measured by the total QALYs) increases in accordance with the strategy sensitivity, as an increase in sensitivity results in more patients of myocardial infarction being detected and treated. The total costs for each testing strategy increase as the specificity decreases and more patients require 10-hour troponin testing. However, the direct costs of measuring troponin at baseline and at 90 minutes exceed the costs of measuring troponin and H-FABP at baseline, so the former strategy is more expensive and thus dominated by the latter. The incremental cost-effectiveness ratio (ICER) reports the additional cost required using the strategy to accrue 1 additional QALY compared with the next most effective alternative. NICE decision-making suggests that a threshold of £20,000–30,000 per QALY is usually used, so a strategy is unlikely to be considered cost-effective if the ICER exceeds £20,000–30,000 per QALY.
| Doctor-on-demand scenario | Twice-daily ward round scenario | Once-daily ward round scenario | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Total costs (£) | Total QALYs | ICER (£) | Total costs (£) | Total QALYs | ICER (£) | Total costs (£) | Total QALYs | ICER (£) | |
| No testing | 965,994 | 26,226.68 | – | 965,994 | 26,226.68 | – | 965,994 | 26,226.68 | – |
| High-sensitivity cTnT at presentation | 1,581,263 | 26,349.42 | 5012 | 1,614,048 | 26,349.42 | 5280 | 1,638,450 | 26,349.42 | 5479 |
| High-sensitivity cTnT at presentation and at 90 minutes | 1,715,256 | 26,354.37 | Dominated | 1,755,642 | 26,354.37 | Dominated | 1,785,754 | 26,354.37 | Dominated |
| High-sensitivity cTnT and H-FABP at presentation | 1,682,362 | 26,358.72 | 11,026 | 1,736,440 | 26,358.72 | 13,160 | 1,776,146 | 26,358.72 | 14,806 |
| 10-hour troponin | 2,016,540 | 26,386.36 | 12,090 | 2,416,409 | 26,386.36 | 24,600 | 2,705,696 | 26,386.36 | 33,630 |
At the £20,000 per QALY threshold, 10-hour troponin testing is cost-effective (£12,090 per QALY) in the doctor-on-demand scenario (if a decision can be made and the patient discharged as soon as a negative troponin result is available) but not in the other scenarios, where the ICER for 10-hour troponin, compared with high-sensitivity cTnT and H-FABP at presentation, exceeds £20,000 per QALY. In the other two scenarios (once-daily ward round and twice-daily ward rounds), the analysis shows that the strategies based on high-sensitivity cTnT and H-FABP at presentation are likely to be considered cost-effective compared with the next most effective alternative using a £20,000 per QALY threshold.
If a £30,000 per QALY threshold is used then 10-hour troponin testing is cost-effective in the doctor-on-demand scenario (£12,090 per QALY) and the twice-daily ward round scenario (£24,600 per QALY), whereas the high-sensitivity cTnT and H-FABP at presentation strategy is considered cost-effective in the once-daily ward round scenario (£14,806 per QALY).
Secondary analysis deterministic results
The results of the secondary analysis using cTnI are reported inTable 24. High-sensitivity cTnI at presentation and at 90 minutes was cost-effective in all three scenarios at the £20,000 per QALY threshold. At the £30,000 per QALY threshold, the 10-hour troponin strategy is cost-effective only in the doctor-on-demand scenario (£24,327 per QALY), with its ICER substantially exceeding the threshold in the other scenarios. High-sensitivity cTnI at presentation and at 90 minutes is cost-effective in the once-daily ward round scenario (£8310 per QALY) and the twice-daily ward round scenario (£7929 per QALY) at the £30,000 per QALY threshold.
| Doctor-on-demand scenario | Twice-daily ward round scenario | Once-daily ward round scenario | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Total costs (£) | Total QALYs | ICER (£) | Total costs (£) | Total QALYs | ICER (£) | Total costs (£) | Total QALYs | ICER (£) | |
| No testing | 965,994 | 26,226.68 | – | 965,994 | 26,226.68 | – | 965,994 | 26,226.68 | – |
| High-sensitivity cTnI at presentation | 1,576,820 | 26,350.78 | 4922 | 1,609,240 | 26,350.78 | 5183 | 1,628,460 | 26,350.78 | 5338 |
| High-sensitivity cTnI and H-FABP at presentation | 1,711,921 | 26,370.19 | 6960 | 1,764,695 | 26,370.19 | Extendedly dominated | 1,803,022 | 26,370.19 | Extendedly dominated |
| High-sensitivity cTnI at presentation and at 90 minutes | 1,773,748 | 26,376.38 | 9988 | 1,812,245 | 26,376.38 | 7929 | 1,841,210 | 26,376.38 | 8310 |
| 10-hour troponin | 2,016,540 | 26,386.36 | 24,327 | 2,416,409 | 26,386.36 | 60,537 | 2,705,696 | 26,386.36 | 86,621 |
Main probabilistic results
Probabilistic analysis incorporated uncertainty in the parameter estimates to provide estimates of the probability that each cTnT strategy would be cost-effective at different thresholds for willingness to pay for health gain. Figure 19shows the probability in the probabilistic sensitivity analyses that each strategy is optimal at thresholds of willingness to pay ranging from zero to £50,000 per QALY in the doctor-on-demand, twice-daily ward round and once-daily ward round scenarios. Tables containing the probabilities of cost-effectiveness of each cTnT strategy for the three scenarios at different willingness-to-pay thresholds are presented inAppendix 3.
FIGURE 19.
Cost-effectiveness acceptability curves of cTnT strategies. (a) Doctor-on-demand scenario; (b) twice-daily ward round scenario; (c) once-daily ward round scenario. MAICER, maximum acceptable incremental cost-effectiveness ratio; High Sens Trop T, high-sensitivity TnT at presentation; High Sens Trop T + H-FABP, high-sensitivity TnT and H-FABP at presentation; High Sens Trop T + 90 minutes, high-sensitivity TnT at presentation and 90 minutes; 10 hour Trop, 10-hour troponin.
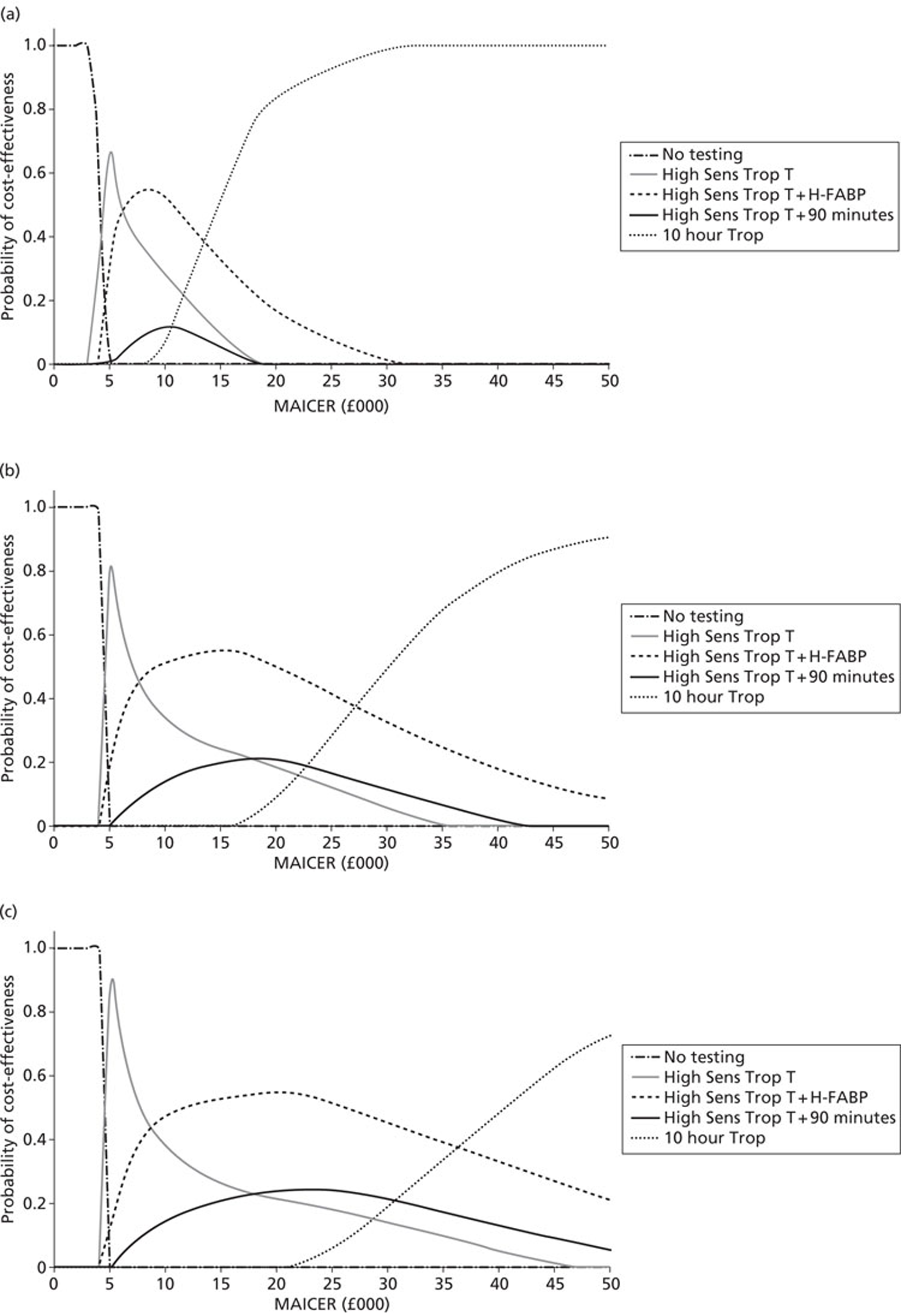
In the doctor-on-demand scenario the high-sensitivity cTnT and H-FABP at presentation strategy has the highest probability of being optimal at thresholds between £6000 and £14,000 per QALY, whereas above £14,000 per QALY 10-hour troponin testing has the highest probability of being optimal. In the twice-daily ward round scenario, high-sensitivity cTnT and H-FABP at presentation has the highest probability of being optimal at thresholds between £8000 and £27,000 per QALY, whereas above £27,000 per QALY 10-hour troponin testing has the highest probability of being optimal. In the once-daily ward round scenario, high-sensitivity cTnT and H-FABP at presentation has the highest probability of being optimal at thresholds between £8000 and £37,000 per QALY, whereas 10-hour troponin testing is optimal only above thresholds of £37,000 per QALY. These results reflect the deterministic analysis and suggest that high-sensitivity cTnT and H-FABP at presentation has the highest probability of being cost-effective in most scenarios and at typically used thresholds for willingness to pay.
Secondary analysis probabilistic results
The secondary probabilistic analysis for cTnI strategies using the doctor-on-demand, twice-daily ward round and once-daily ward round scenarios is shown inFigure 20. Tables containing the probabilities of cost-effectiveness of each cTnI strategy for the three scenarios at different willingness-to-pay thresholds are presented inAppendix 4.
FIGURE 20.
Cost-effectiveness acceptability curves of cTnI strategies. (a) Doctor-on-demand scenario; (b) twice-daily ward round scenario; (c) once-daily ward round scenario. MAICER, maximum acceptable incremental cost-effectiveness ratio; High Sens Trop I, high-sensitivity TnI at presentation; High Sens Trop I + H-FABP, high-sensitivity TnI and H-FABP at presentation; High Sens Trop I + 90 minutes, high-sensitivity TnI at presentation and 90 minutes; 10 hour Trop, troponin at 10–12 hours.
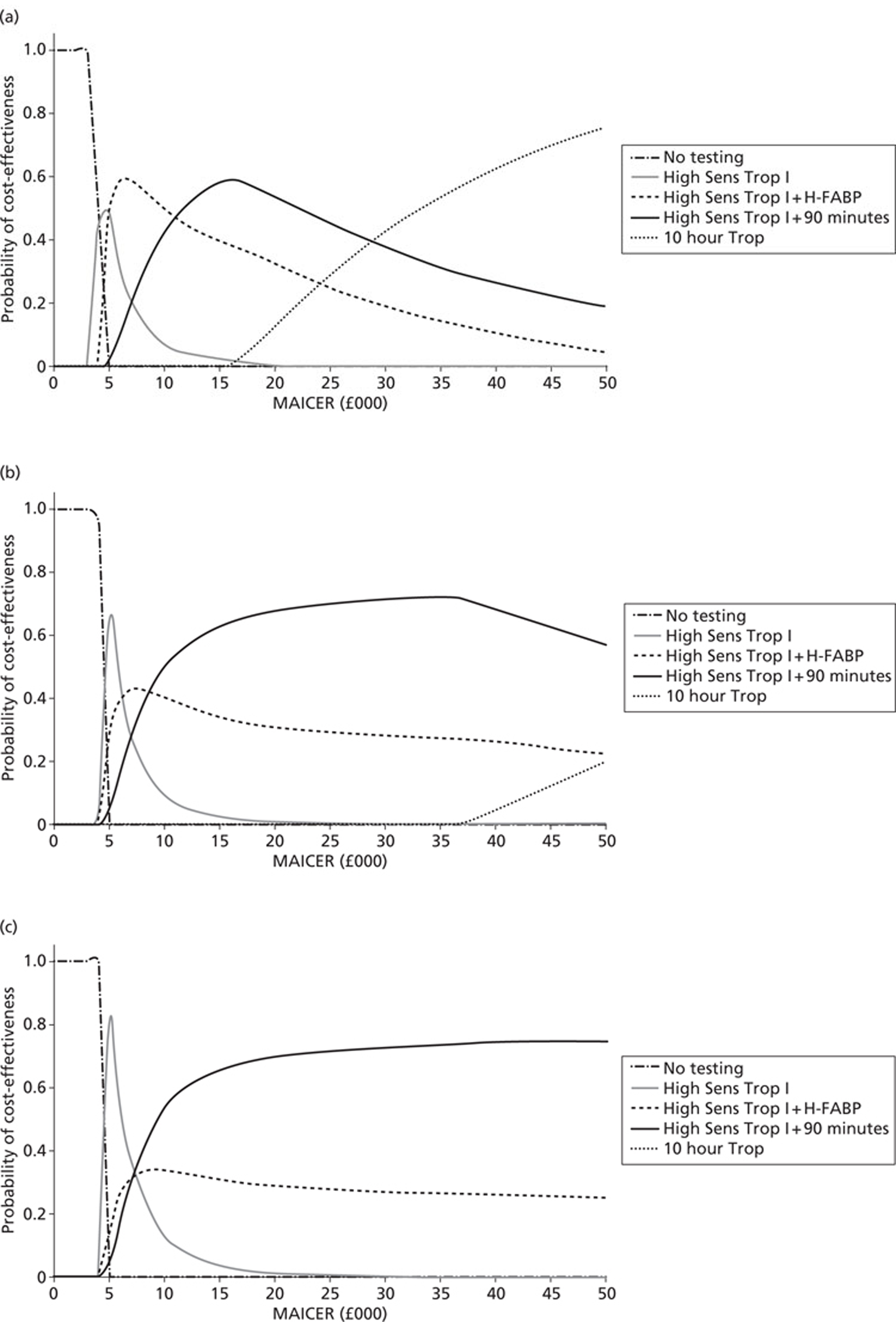
In the doctor-on-demand scenario the high-sensitivity cTnI and H-FABP at presentation strategy has the highest probability of being optimal at thresholds between £5000 and £12,000 per QALY; the high-sensitivity cTnI at presentation and at 90 minutes strategy is optimal between £12,000 and £28,000 per QALY; and 10-hour troponin testing is optimal above thresholds of £28,000 per QALY. In the twice-daily ward round and once-daily ward round scenarios, the high-sensitivity cTnI at presentation and at 90 minutes strategy has the highest probability of being optimal at all thresholds tested above £8000 per QALY and £7000 per QALY respectively. Again, these results reflect the deterministic analysis results inTable 24and suggest that the strategy using high-sensitivity cTnI on presentation and at 90 minutes has the highest probability of being cost-effective in most scenarios and at typically used thresholds for willingness to pay.
Chapter 5 Discussion
Role of conventional biomarkers for diagnosis
The measurement of myoglobin or CK-MB as part of a cardiac marker panel did not contribute to the diagnostic efficiency in a population of low-risk chest pain patients. Previous studies which have demonstrated that the combination of cTnI, myoglobin and CK-MB is diagnostically efficient combined the two cytoplasmic markers with a low-sensitivity troponin assay. 36,56A prospective study of chest pain patients with a third-generation cTnT assay utilising a 99th percentile cut-off demonstrated superior diagnostic sensitivity of cTnT measurement compared with CK-MB measurement for both rule-in and rule-out of an AMI. 247The findings in RATPAC-CBE are in agreement with the findings of studies in which an appropriately sensitive cTnI method has been used and combined with a 99th percentile cut-off. In these studies cTnI measurement is superior to the combination of CK-MB and myoglobin measurement. 248,249The use of rate of change (delta value) for myoglobin has been claimed to improve diagnostic sensitivity. 250The addition of delta value served only to reduce the specificity of myoglobin measurement without a concomitant increase in sensitivity. Sensitive troponin measurement alone appears superior to measurement of CK-MB and myoglobin. 251A recent systematic review has also shown that myoglobin and CK-MB do not have incremental value for early diagnosis of an AMI. 74The most recently reported study of low-risk chest pain patients demonstrated that, even with a relatively low sensitivity cTnI measurement, additional measurement of CK-MB and myoglobin was not clinically useful.252
Role of novel cytoplasmic biomarkers
The measurement of H-FABP has been proposed to be a superior diagnostic marker to both CK-MB and myoglobin on the basis of its high tissue concentration and small molecular size. Admission measurement of H-FABP was diagnostically superior to measurement of myoglobin but not to measurement of CK-MB when assessed by ROC curve analysis. When prespecified diagnostic thresholds were used, H-FABP was superior to CK-MB but at the cost of a significantly lower specificity. When compared with all four troponin methods, measurement of H-FABP was diagnostically inferior. Measurement was statistically equivalent when using prespecified diagnostic thresholds; the failure to demonstrate diagnostic inferiority was due to the relatively small sample size of the population with a final diagnosis of myocardial infarction. As a single diagnostic test, measurement of H-FABP is not reliable for the diagnosis of patients presenting with acute chest pain and a suspected AMI. In RATPAC-CBE the diagnostic sensitivity of H-FABP and the AUC by ROC curve analysis are comparable to those reported previously69,71–73,79although there is one report that found much higher values. 71
The role of H-FABP is therefore as a supplementary test. In the RATPAC-CBE population the combination of admission measurement of H-FABP and troponin by a high-sensitivity method improved diagnostic sensitivity when prespecified diagnostic thresholds were used. The improvement was not statistically significantly different from single marker measurement or from the diagnostic sensitivity achieved by combining admission and 90-minute troponin measurements. When compared by ROC curve analysis, sensitive troponin measurement was diagnostically and prognostically superior to that of H-FABP. This suggests that the combination of H-FABP with sensitive troponin measurement may not be diagnostically useful compared with repeating troponin measurement after a short time interval.
Assessment of the value of measurement of H-FABP as a supplementary test on the basis of the existing literature is difficult. Published studies showing improved diagnostic performance over the combination of H-FABP plus troponin have a number of methodological factors that make interpretation difficult. Some have included a mixed population of patients, including those with ST elevation myocardial infarction,69,71and in some the population is not completely defined. 72,73These studies have also used a relatively insensitive troponin assay rather than a current generation or high-sensitivity assay. 69,71–73
The data from studies that have compared a high-sensitivity troponin assay with H-FABP measurement are also difficult to evaluate. All are in agreement that measurement of H-FABP does not add value above measurement of a high-sensitivity troponin alone. One study enrolled patients presenting with ST-segment elevation, for whom early diagnosis using biomarkers is not clinically useful. 81Three studies used an appropriate chest pain population. In one, a semiquantitative method for measuring H-FABP was used. 78The diagnostic threshold was 7 ng/l, a relatively high cut-off. Although evidence for the diagnostic performance of H-FABP measurement is presented in the paper, the power of combining troponin with H-FABP measurement is explored only for the diagnosis of non-ST-segment elevation ACS and not for NSTEMI. When high-sensitivity troponin and measurement of H-FABP by a sensitive method has been used, the diagnostic performance of the combination is not analysed. 79A multicentre study of a high-sensitivity troponin assay compared with emerging biomarkers used an appropriate population and looked at the ability of new biomarkers to improve on diagnostic classification, but again a final diagnostic classification of ACS rather than myocardial infarction was used. 80Only one study has reported both an appropriate population and the diagnostic ability of marker combinations. This study demonstrated that the addition of H-FABP to troponin measurement did not improve diagnostic performance. 253
Role of high-sensitivity troponin assays
In RATPAC-CBE, slightly lower AUCs and diagnostic sensitivities for troponin measurement were obtained than in previously published studies. Studies published to date have used mixed populations that included patients with ST-segment elevation and have utilised a final diagnosis based on a less sensitive troponin method. Diagnostic sensitivities of 94–100% for troponin measured on admission have been reported in one study. 254Diagnostic sensitivities of 82–84%, comparable with data from this study, have also been reported. 251,255Neither of these two studies specifically addressed the problem of patients with NSTEMI. It is notable that, when patients considered to have undetectable troponin by conventional methods but detectable troponin by high-sensitivity methods are followed up, elevation of a sensitive troponin on admission is associated with an adverse prognosis. 256,257This finding suggests that patients with NSTEMI are being missed when diagnosis is based on conventional troponin methods. The analogy would be the early literature on the evaluation of cTnT and cTnI in which diagnosis was based on measurement of CK-MB or CK, in which a high sensitivity but poor specificity were reported for troponin measurement. When a high-sensitivity cardiac troponin assay is used as the reference test for myocardial injury, the diagnostic sensitivity of previous-generation troponin assays is reduced. 258When a different high-sensitivity troponin assay is used as the diagnostic gold standard biochemical test, the diagnostic sensitivity of a new high-sensitivity troponin assay may not be demonstrably statistically significantly different. 259Moving the diagnostic threshold for cardiac troponin towards a lower diagnostic discriminant significantly improves clinical outcomes, supporting the view that lower is better and the diagnostic discriminant should be the 99th percentile. 244
The optimal time point to undertake troponin measurement for diagnosis also remains undefined. Troponin measured on admission alone was not adequately sensitive; however, when combined with early serial measurement, rapid exclusion of myocardial infarction can be safely achieved within 2 hours in the low-risk chest pain population, as was demonstrated in RATPAC and other published studies. 252,260Recent publications have suggested that, in a higher-risk population, diagnosis can be made with acceptable sensitivity by 3 hours from admission255,261Questions remain as only one study was identified in the literature that specifically utilised a non-ST-segment elevation population and examined patients initially with a troponin below the 99th percentile on presentation. 262This study demonstrated that follow-up to 6 hours was required before all patients with a final diagnosis of an AMI showed elevation of cTnT above the 99th percentile.
Role of neurohormones
Copeptin performed poorly both as a diagnostic test for myocardial infarction and as an outcome predictor. Copeptin was first developed as a stable peptide derived from the precursor of vasopressin that could be used to measure vasopressin levels without the problems of sample stability. 263It was first systematically examined in a large series of patients following an AMI. The peak values of copeptin occurred at 24 hours and copeptin was elevated to a greater extent in patients with STEMI than in patients with NSTEMI. The study also found that copeptin levels predicted heart failure and death but not recurrent AMI. 98Copeptin has been shown not to detect myocardial ischaemia. 102There have been a number of reports on the role of copeptin for very early diagnosis of an AMI, in particular as a rule-out test. The results are contradictory, with some studies claiming high diagnostic sensitivity and others no diagnostic utility at all. The reasons for the discrepancies may relate to the populations studied and the troponin method used as the biochemical gold standard. The first group to report the value of copeptin examined consecutive chest pain admissions and utilised the fourth-generation cTnT assay as the biochemical gold standard at a diagnostic discriminant of 40 ng/l. 101Copeptin was useful only in patients presenting < 10 hours of onset of chest pain. The AUC of copeptin for the diagnosis of an AMI was low (0.75) and was less than that of troponin alone (0.86). The combination of cTnT and copeptin improved the AUC for diagnosis to 0.97. Levels of copeptin were significantly lower in the NSTEMI population. Exclusion of patients with ST-segment elevation did not affect the diagnostic performance. A detailed examination of the data reported in this study shows that 362 of the 487 patients examined corresponded to a group comparable with that in the RATPAC study. In this group, 23 patients had a final diagnosis of myocardial infarction, of whom five (22%) had an undetectable cTnT (< 10 ng/l) and were detected by copeptin elevation. The study did not include any outcome data. The second large study that examined the role of copeptin again used a mixed population of patients with and without ST-segment elevation on presentation. 264Diagnosis was based on a contemporary troponin method (not a high-sensitivity assay). Results were similar with troponin overall performing better than copeptin. The study reported that copeptin showed higher earlier diagnostic sensitivity than the other markers studied. When patients with ST-segment elevation were excluded, the admission AUC for troponin was 0.87, which improved to 0.93 on addition of copeptin. Data for a high-sensitivity assay were also reported. This yielded an admission AUC of 0.96, which improved to 0.97 on addition of copeptin. This improvement in AUC just achieved statistical significance. The reported experience with high-sensitivity troponin assays has been more mixed. A study of patients presenting to the emergency department with acute chest pain showed copeptin levels comparable to those found in the RATPAC study. 265An additive value of copeptin and high-sensitivity cTnT was not demonstrated. A criticism of this study is that only a minority of patients had myocardial infarction. A second study examining patients presenting to a chest pain unit showed that the AUC for copeptin for diagnosis of an AMI was low (0.70) compared with that for high-sensitivity cTnT (0.90). 266Exclusion of patients with ST-segment elevation showed no added benefit of using copeptin in combination with high-sensitivity cTnT when ROC curve analysis was used to assess diagnostic performance. Utilising a prespecified cut-off of the 99th percentile for cTnT (14 ng/l) and 14 pmol/l for copeptin improved diagnostic sensitivity but specificity was low at 0.56. It is possible that the poor sensitivity of copeptin was due to sample degradation; however, copeptin is known to be stable and no comparable problems were seen with other markers and the diagnostic sensitivity is comparable with that found in other studies. The role of copeptin to predict outcome is not well studied. In patients with established coronary artery disease, copeptin was not a strong predictor of cardiovascular outcome168and is probably a better predictor in patients with heart failure than in patients with chest pain. 267
Measurement of BNP has been proposed as an early diagnostic test for patients presenting with chest pain. 44Although studies have shown an additive value of simultaneous measurement of BNP and cardiac troponin,268–270increased sensitivity is achieved at the expense of diagnostic specificity. 268,269Measurement of BNP is known to predict mortality across the entire spectrum, from normal populations271to patients with stable coronary artery disease272or presenting with ACS. 140,273Similarly, in chest pain patients, BNP measurement is a risk predictor but is not useful for early diagnosis. 274–279
Biomarkers for prognosis
As a prognostic marker, even in this low-risk population with a relatively small number of events, troponin measurement was superior to the cytoplasmic markers. This finding is in agreement with previous meta-analysis in the higher-risk ACS population. 33All four troponin methods were equally efficient in predicting outcome. Copeptin was a poor outcome predictor. NTproBNP measurement is known to be a better predictor of mortality than troponin measurement, whereas troponin predicts risk of a repeat myocardial infarction. 280In the RATPAC study, mortality was very low and cardiovascular events rather than cardiovascular death was the most common adverse event, which would account for the fact that the peak troponin value was the best predictor of MACEs.
Cost-effectiveness of biomarker strategies
The results showed that, as expected, effectiveness (QALYs) increased with increasing sensitivity and costs increased with decreasing specificity. At the £20,000 per QALY threshold, in all but one scenario a strategy of measuring high-sensitivity cTnT and H-FABP at presentation (with admission for 10-hour troponin testing if positive and discharge home if negative) was the optimal strategy; the 10-hour troponin strategy may have been optimal in the doctor-on-demand scenario. However, at the £30,000 per QALY threshold, 10-hour troponin testing is cost-effective in both the doctor-on-demand scenario and the twice-daily ward round scenario.
Sensitivity analyses considered alternative strategies using data from cTnI (Stratus CS), which our analysis suggested may have superior diagnostic accuracy. These analyses showed that delayed troponin testing is likely to be cost-effective only if patients with a negative test can be discharged as soon as the result is available and if a £30,000 per QALY threshold for willingness to pay is used. Thus, delayed troponin testing at 10 hours after symptom onset is unlikely to be cost-effective in most scenarios at commonly used thresholds of £20,000–30,000 per QALY. The implication of our analysis is that NICE guidance recommending 10- to 12-hour troponin testing, compared with high-sensitivity troponin at presentation combined with H-FABP or repeated at 90 minutes, does not appear to promote cost-effective use of NHS resources unless services are in place to allow rapid decision-making once delayed test results are available. However, there are a number of assumptions in the model that need to be taken into account when interpreting these findings.
Any modelling process involves simplifications and assumptions that may not accurately reflect clinical practice. It was assumed that delayed troponin testing is 100% sensitive and specific, reflecting its role as the reference standard for myocardial infarction. However, studies of presentation troponin testing report specificities of < 100%. It is known that not all troponin elevations represent myocardial infarction; however, modelling the effect of treating false-positive patients would be complex and require substantial assumptions with little supporting data. It was therefore assumed that sustained false-positive troponin elevations would affect presentation and delayed troponin testing equally, and that the only consequence of false-positive presentation troponin was the requirement for subsequent delayed testing. It was also assumed that a patient with a false-negative troponin at presentation would have the same prognosis (and thus ability to benefit from treatment) as a patient with a true-positive at presentation. This assumption may not hold if those patients with false-negative troponin at presentation have a smaller infarct and better prognosis. However, there was inadequate data available to test this assumption.
Also, our model assumes that patients waiting for troponin testing are cared for in hospital (even if not formally admitted) and therefore incur hospital costs. It could be argued that the benefits of delayed troponin testing could be accrued without most of the costs if patients were discharged home and asked to return for delayed testing. However, the feasibility and acceptability of this practice has not been tested and it is not routinely used.
This analysis is based on a model developed for a related project, ‘Cost-effectiveness of diagnostic strategies for suspected acute coronary syndrome (ACS)’ (HTA09/22/21), which used estimates of sensitivity and specificity for high-sensitivity troponin and H-FABP derived from a systematic literature review and meta-analysis. 241The findings were similar, suggesting that a 10-hour troponin measurement is likely to be cost-effective only if a £30,000 per QALY threshold is used and the patient can be discharged as soon as the results are available. The combination of troponin with H-FABP or measurement of high-sensitivity troponin at baseline and at 90 minutes were both potentially cost-effective, but estimates of sensitivity and specificity in each case were derived from one study. The estimates of sensitivity for cTnT and cTnI derived from the meta-analysis were the opposite of the estimates in this study, with cTnT having higher sensitivity than cTnI. As a result, the secondary analysis with cTnI from the meta-analysis suggested that 10-hour troponin measurement was more cost-effective, whereas secondary analysis using cTnI in this study suggested that 10-hour troponin measurement was less cost-effective.
Overall, it appears that the sensitivity of the rapid rule-out strategy is the key determinant of whether or not 10-hour troponin measurement is, by comparison, cost-effective. If analysis includes a rapid rule-out strategy with a sensitivity of around 95% then a 10-hour troponin measurement is not cost-effective in most scenarios. If analysis includes a rapid rule-out strategy with a sensitivity of around 85% then a 10-hour troponin measurement is cost-effective in several scenarios. This should not be interpreted to mean that a rapid rule-out strategy with a sensitivity of 95% is ‘acceptable’ or ‘safe’, as both of these considerations involve value judgements that are separate from cost-effectiveness. What it does mean, however, is that if a rapid rule-out strategy is available with 95% sensitivity (and specificity of around 90%) then a 10-hour troponin strategy does not in comparison represent a worthwhile use of health-care resources.
The findings from this part of the study suggest that the following areas of future research may be worthwhile:
-
The sensitivity of troponin at baseline and at 90 minutes, and in combination with H-FABP, needs to be confirmed in further studies. Measurement at other time points for the optimisation of sensitivity without the requirement for hospital admission (such as at 120 minutes after presentation or 6 hours after symptom onset) could be explored.
-
Further modelling could be used to estimate the relative importance of sensitivity and specificity in determining cost-effectiveness. Alternative strategies, such as using the limit of detection to define a positive troponin test at baseline instead of the 99th percentile, could be used to optimise sensitivity at the expense of specificity. However, it is not clear how much specificity can be sacrificed to optimise sensitivity without adversely affecting cost-effectiveness. Modelling could be used to determine the relative effects of varying sensitivity and specificity of a hypothetical rapid rule-out strategy on cost-effectiveness.
General observations
Any biomarker must fulfil a number of criteria to achieve routine clinical use. These can be divided into three categories: analytical suitability, plausibility for clinical application and treatment impact.
Analytical suitability has three components. The population aspects of the test must be known, that is, the effect of age, gender and ethnicity and the influence of physiological and comorbid conditions. The preanalytical factors that may affect the results, such as collection conditions, anticoagulant requirements, preanalytical sample handling and stability in storage, need to be defined. A marker suitable for routine clinical use must be measurable in the routine clinical laboratory without special handling conditions. Finally, test performance needs to be adequate. In addition to the ability to measure the biomarker with precision and accuracy, the analysis must be simple and have a rapid turnaround time. Ideally, it should be implemented on existing laboratory equipment rather than requiring additional apparatus. In practice, this means that a colorimetric or more likely an immunoassay for the marker is available.
Plausibility of a biomarker will involve the biological and clinical plausibility for the putative clinical role. Biological plausibility means that the pathobiology of the marker needs to be understood. This means an understanding of the genesis of the biomarker and of the relationship of the biomarker to the medical condition of interest. It is not sufficient to simply state that a negative result allows rule-out of the condition of interest. The positive results must also be explicable. Clinical plausibility for the putative clinical role requires sensitivity and specificity for the medical condition of interest in clinically appropriate populations, the patient groups in whom the test will be used in routine clinical practice. Many studies on biomarkers have evaluated diagnostic performance in clinical trial sample banks or highly selected patient groups. These populations do not constitute an appropriate environment to evaluate test performance as the disease prevalence is inappropriately high, often close to 100%. Such studies allow proof of concept but need to be followed by prospective evaluation in a clinically representative population. There is a lack of prospective observational studies of biomarkers performed using routine clinical populations with no exclusion criteria and well-documented final diagnoses and outcomes. Such studies are essential before any biomarker can be used in routine clinical practice.
Treatment effectiveness (evidence-based medicine) is one of the most important of the criteria. Any new biomarker must either offer a significant improvement in diagnostic efficiency or result in a change in treatment. In addition, it should demonstrate cost-effectiveness.
Strengths and weaknesses of the study
The RATPAC-CBE trial was a retrospective observational study of the point-of-care arm of a prospective randomised controlled trial. It therefore shares the same limitations of all such diagnostic studies – that although patient enrolment is prospective, biomarker analysis is retrospective. The strength of this design is that it represented real clinical practice. The population selected was a low-risk group that was managed according to a biomarker-based protocol. The population studied is therefore representative of the majority of patients who present for assessment of acute chest pain and a suspected AMI. Patient selection was randomised between conventional management and a rapid rule-out protocol. The equivalent rate of major adverse events between the point-of-care arm and the conventional management arm suggests that the population was well matched. The nature of the protocol means that an admission sample was obtained for the majority of patients but a follow-up 90-minute sample was obtained only when there was a much lower likelihood that the patient had an AMI. There is therefore a selection bias as patients with a final diagnosis of an AMI may be over-represented in the assessment of the 90-minute samples for diagnostic and prognostic ability. This was mitigated by analysing the peak of the presentation and 90-minute samples, both to increase numbers and to reduce the immediate temporal bias.
Ideally, the study would have had a 10- to 12-hour sample taken on all patients in the point-of-care arm for analysis in the core laboratory to provide a definitive diagnosis of myocardial injury. Three of the trial sites utilised a high-sensitivity troponin assay, whereas the other three used a contemporary assay but with a decision limit close to optimal. Nearly two-thirds of the patients studied had a troponin measurement at ≥ 6 hours from onset of chest pain and 42.8% had a troponin measurement at ≥ 10 hours from onset of chest pain. Samples were measured at presentation and at 90 minutes with a high-sensitivity assay. In addition, the Stratus CS analyser has analytical performance appropriate for a high-sensitivity assay. This means that the final diagnosis of myocardial infarction was based largely on a sensitive troponin measurement rather than on a conventional but lower-sensitivity troponin assay.
The use of a cohort study for the modelling also means that unanticipated effects cannot be evaluated.
Implications for practice
The findings of RATPAC-CBE support the widespread implementation of high-sensitivity troponin assays. They also support the use of troponin alone as the gold standard diagnostic test and suggest that additional measurement of myoglobin and CK-MB is not required. Copeptin cannot be recommended as a useful additional test for diagnostic or prognostic purposes. Although measurement of NTproBNP was confirmed as a useful prognostic test, it is no more useful than measurement of troponin and the measurement of both NTproBNP and cTnT or cTnI should not be undertaken.
The role of H-FABP remains unclear. Measurement of both a cardiac troponin and H-FABP does improve diagnostic performance but does not, on the basis of the evidence from RATPAC-CBE and the literature published to date, permit ruling out myocardial infarction based on a single measurement of both markers on admission.
Implications for future research
High-sensitivity troponin measurement is the best test for the assessment of patients with chest pain and for diagnosis of myocardial infarction. Recent guidelines have recommended the use of admission and 3-hour high-sensitivity troponin measurements for the diagnosis of an AMI,281but this recommendation is not truly evidence based. To date, no studies have been published that have combined clinical data and ECG data with a high-sensitivity troponin measurement performed at 10–12 hours from admission as the biochemical diagnostic gold standard to provide the definitive final diagnosis. Such a population would allow assessment of the earliest time point when it can be guaranteed that a rise in troponin can be detected with close to 100% diagnostic accuracy and definition of what would constitute an early diagnostic strategy.
A prospective observational study is required, combining clinical and ECG data with a high-sensitivity troponin measurement performed at 10–12 hours from admission as the biochemical diagnostic gold standard to define the kinetics of troponin release. Such a study would also support assessment of the diagnostic efficiency of H-FABP as well as any other putative new markers. This study should also address the prognostic utility of small troponin elevations between the 99th percentile and the limits determined from previous studies. Although it is assumed that low-level elevations of troponin predict an adverse short-term prognosis, the clinical effect of minor elevations on short-term prognosis is not known. In addition, the study should address to what extent short-term changes in troponin levels can be used to distinguish between acute and chronic elevations.
Elevations of troponin occur in the general population, associated with previous history of ischaemic disease, requirement for cardiac medication, coexistent conditions and evidence of echocardiographic abnormalities. These minor elevations of cardiac troponin in the general population are associated with an adverse long-term prognosis. In patients with chest pain they are associated with abnormalities in cardiac anatomy and evidence of atherosclerotic disease. The use of high-sensitivity troponin assays will inevitably detect small elevations in the population presenting to the emergency department that are representative of chronic rather than acute disease. Although data from RATPAC-CBE show that the number of such patients is relatively small, this number is significant. It is currently thought that change in troponin levels over a short time frame would allow the distinction between acute myocardial injury seen in AMI, associated with a poor prognosis, and more chronic elevations.
Studies on biological variation of troponin have been performed. There is some published evidence on change in troponin levels in patients presenting with suspected ACS, but again the studies have contained a mixed population of STEMI and NSTEMI patients. Data to determine what would be a discriminatory level of rate of change of troponin to distinguish between chronic elevation and acute NSTEMI in a more general chest pain population are currently lacking.
Conclusions
The measurement of cardiac troponin as cTnT or cTnI over a short time frame offers the best strategy for early confirmation or exclusion of an AMI. In this study, a low-risk group was successfully discharged on the basis of admission and 90-minute measurements. Questions remain as to what is the optimal timing for troponin measurement. In addition, troponin measurement needs to be incorporated within a clinical decision-making strategy that utilises clinical and ECG findings. Of all markers studied, only H-FABP appears to offer some improvement in diagnostic efficiency that might also be cost-effective. However, as yet, measurement of H-FABP is not carried out on routine clinical laboratory equipment suitable for a 24-hour diagnostic service.
Acknowledgements
Contributions of authors
Paul Collinson(Professor of Cardiovascular Biomarkers, St George's University of London) – chief investigator, conception and design of the study, analysis and interpretation of the data, writing and revising the report and final approval of the version to be published.
David Gaze(Clinical Research Scientist, St George's Hospital, London) – design of the study, analytical measurement of the biomarkers, data entry and data checking, drafting and revising the report and final approval of the version to be published.
Praveen Thokala(Research Fellow, Health Services Research, University of Sheffield) – health economic modelling and analysis, drafting and revising the report and final approval of the version to be published.
Steve Goodacre(Professor of Emergency Medicine, Health Services Research, University of Sheffield) – conception and design of the study, analysis and interpretation of the data, drafting and revising the report and final approval of the version to be published.
Publications
Collinson P, Goodacre S, Gaze D, Gray A. Very early diagnosis of chest pain by point-of-care testing: comparison of the diagnostic efficiency of a panel of cardiac biomarkers compared with troponin measurement alone in the RATPAC trial. Heart2012;98:312–18.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health.
References
- Goodacre S, Cross E, Arnold J, Angelini K, Capewell S, Nicholl J. The health care burden of acute chest pain. Heart 2005;91:229-30. http://dx.doi.org/10.1136/hrt.2003.027599.
- Collinson PO, Rao AC, Canepa-Anson R, Joseph S. Impact of European Society of Cardiology/American College of Cardiology guidelines on diagnostic classification of patients with suspected acute coronary syndromes. Ann Clin Biochem 2003;40:156-60. http://dx.doi.org/10.1258/000456303763046085.
- Pope JH, Aufderheide TP, Ruthazer R, Woolard RH, Feldman JA, Beshansky JR, et al. Missed diagnoses of acute cardiac ischemia in the emergency department. N Engl J Med 2000;342:1163-70. http://dx.doi.org/10.1056/NEJM200004203421603.
- Collinson PO, Premachandram S, Hashemi K. Prospective audit of incidence of prognostically important myocardial damage in patients discharged from emergency department. BMJ 2000;320:1702-5. http://dx.doi.org/10.1136/bmj.320.7251.1702.
- Pozen MW, D’Agostino RB, Selker HP, Sytkowski PA, Hood WB. A predictive instrument to improve coronary-care-unit admission practices in acute ischemic heart disease. A prospective multicenter clinical trial. N Engl J Med 1984;310:1273-8. http://dx.doi.org/10.1056/NEJM198405173102001.
- de Dombal FT, Clamp SE, Softley A, Unwin BJ, Staniland JR. Prediction of individual patient prognosis: value of computer-aided systems. Med Decis Making 1986;6:18-22. http://dx.doi.org/10.1177/0272989X8600600104.
- Goldman L, Cook EF, Brand DA, Lee TH, Rouan GW, Weisberg MC, et al. A computer protocol to predict myocardial infarction in emergency department patients with chest pain. N Engl J Med 1988;318:797-803. http://dx.doi.org/10.1056/NEJM198803313181301.
- Willems JL, Abreu-Lima C, Arnaud P, van Bemmel JH, Brohet C, Degani R, et al. The diagnostic performance of computer programs for the interpretation of electrocardiograms. N Engl J Med 1991;325:1767-73. http://dx.doi.org/10.1056/NEJM199112193252503.
- Wu AH, Apple FS, Gibler WB, Jesse RL, Warshaw MM, Valdes R. National Academy of Clinical Biochemistry Standards of Laboratory Practice: recommendations for the use of cardiac markers in coronary artery diseases. Clin Chem 1999;45:1104-21.
- Myocardial infarction redefined – a consensus document of The Joint European Society of Cardiology/American College of Cardiology Committee for the redefinition of myocardial infarction. Eur Heart J 2000;21:1502-13. http://dx.doi.org/10.1053/euhj.2000.2305.
- Thygesen K, Alpert JS, White HD, Jaffe AS, Apple FS, Galvani M, et al. Universal definition of myocardial infarction. Circulation 2007;116:2634-53. http://dx.doi.org/10.1161/CIRCULATIONAHA.107.187397.
- Collinson PO, Rosalki SB, Flather M, Wolman R, Evans T. Early diagnosis of myocardial infarction by timed sequential enzyme measurements. Ann Clin Biochem 1988;25:376-82.
- Collinson PO, Ramhamadamy EM, Stubbs PJ, Rosalki SB, Garrat HM, Mosely D, et al. Rapid enzyme diagnosis of patients with acute chest pain reduces patient stay in the coronary care unit. Ann Clin Biochem 1993;30:17-22.
- Collinson PO, Rosalki SB, Kuwana T, Garratt HM, Ramhamadamy EM, Baird IM, et al. Early diagnosis of acute myocardial infarction by CK-MB mass measurements. Ann Clin Biochem 1992;29:43-7.
- Bakker AJ, Gorgels JP, van Vlies B, Haagen FD, Smits R. The mass concentrations of serum troponin T and creatine kinase-MB are elevated before creatine kinase and creatine kinase-MB activities in acute myocardial infarction. Eur J Clin Chem Clin Biochem 1993;31:715-24.
- Gibler WB, Lewis LM, Erb RE, Makens PK, Kaplan BC, Vaughn RH, et al. Early detection of acute myocardial infarction in patients presenting with chest pain and nondiagnostic ECGs: serial CK-MB sampling in the emergency department. Ann Emerg Med 1990;19:1359-66. http://dx.doi.org/10.1016/S0196-0644(05)82598-3.
- Hedges JR, Young GP, Henkel GF, Gibler WB, Green TR, Swanson JR. Serial ECGs are less accurate than serial CK-MB results for emergency department diagnosis of myocardial infarction. Ann Emerg Med 1992;21:1445-50. http://dx.doi.org/10.1016/S0196-0644(05)80057-5.
- Hedges JR, Young GP, Henkel GF, Gibler WB, Green TR, Swanson JR. Early CK-MB elevations predict ischemic events in stable chest pain patients. Acad Emerg Med 1994;1:9-16. http://dx.doi.org/10.1111/j.1553-2712.1994.tb02794.x.
- Hedges JR, Gibler WB, Young GP, Hoekstra JW, Slovis C, Aghababian R, et al. Multicenter study of creatine kinase-MB use: effect on chest pain clinical decision making. Acad Emerg Med 1996;3:7-15. http://dx.doi.org/10.1111/j.1553-2712.1996.tb03295.x.
- Goodacre SW, Morris FM, Campbell S, Arnold J, Angelini K. A prospective, observational study of a chest pain observation unit in a British hospital. Emerg Med J 2002;19:117-21. http://dx.doi.org/10.1136/emj.19.2.117.
- Katus HA, Remppis A, Looser S, Hallermeier K, Scheffold T, Kubler W. Enzyme linked immunoassay of cardiac troponin T for the detection of acute myocardial infarction in patients. J Mol Cell Cardiol 1989;21:1349-53. http://dx.doi.org/10.1016/0022-2828(89)90680-9.
- Cummins B, Auckland ML, Cummins P. Cardiac-specific troponin-I radioimmunoassay in the diagnosis of acute myocardial infarction. Am Heart J 1987;113:1333-44. http://dx.doi.org/10.1016/0002-8703(87)90645-4.
- Cummins P, Young A, Auckland ML, Michie CA, Stone PC, Shepstone BJ. Comparison of serum cardiac specific troponin-I with creatine kinase, creatine kinase-MB isoenzyme, tropomyosin, myoglobin and C-reactive protein release in marathon runners: cardiac or skeletal muscle trauma?. Eur J Clin Invest 1987;17:317-24. http://dx.doi.org/10.1111/j.1365-2362.1987.tb02194.x.
- Collinson PO, Moseley D, Stubbs PJ, Carter GD. Troponin T for the differential diagnosis of ischaemic myocardial damage. Ann Clin Biochem 1993;30:11-6.
- Katus HA, Remppis A, Neumann FJ, Scheffold T, Diederich KW, Vinar G, et al. Diagnostic efficiency of troponin T measurements in acute myocardial infarction. Circulation 1991;83:902-12. http://dx.doi.org/10.1161/01.CIR.83.3.902.
- Gerhardt W, Katus H, Ravkilde J, Hamm C, Jorgensen PJ, Peheim E, et al. S-troponin T in suspected ischemic myocardial injury compared with mass and catalytic concentrations of S-creatine kinase isoenzyme MB. Clin Chem 1991;37:1405-11.
- Gerhardt W, Ljungdahl L, Herbert AK. Troponin-T and CK MB (mass) in early diagnosis of ischemic myocardial injury. The Helsingborg Study, 1992. Clin Biochem 1993;26:231-40. http://dx.doi.org/10.1016/0009-9120(93)90122-M.
- Wu AH, Valdes R, Apple FS, Gornet T, Stone MA, Mayfield-Stokes S, et al. Cardiac troponin-T immunoassay for diagnosis of acute myocardial infarction. Clin Chem 1994;40:900-7.
- Stubbs P, Collinson P, Moseley D, Greenwood T, Noble M. Prospective study of the role of cardiac troponin T in patients admitted with unstable angina. BMJ 1996;313:262-4. http://dx.doi.org/10.1136/bmj.313.7052.262.
- Hamm CW, Ravkilde J, Gerhardt W, Jorgensen P, Peheim E, Ljungdahl L, et al. The prognostic value of serum troponin T in unstable angina. N Engl J Med 1992;327:146-50. http://dx.doi.org/10.1056/NEJM199207163270302.
- Antman EM, Tanasijevic MJ, Thompson B, Schactman M, McCabe CH, Cannon CP, et al. Cardiac-specific troponin I levels to predict the risk of mortality in patients with acute coronary syndromes. N Engl J Med 1996;335:1342-9. http://dx.doi.org/10.1056/NEJM199610313351802.
- Galvani M, Ottani F, Ferrini D, Ladenson JH, Destro A, Baccos D, et al. Prognostic influence of elevated values of cardiac troponin I in patients with unstable angina. Circulation 1997;95:2053-9. http://dx.doi.org/10.1161/01.CIR.95.8.2053.
- Heidenreich PA, Alloggiamento T, Melsop K, McDonald KM, Go AS, Hlatky MA. The prognostic value of troponin in patients with non-ST elevation acute coronary syndromes: a meta-analysis. J Am Coll Cardiol 2001;38:478-85. http://dx.doi.org/10.1016/S0735-1097(01)01388-2.
- Collinson PO, Boa FG, Gaze DC. Measurement of cardiac troponins. Ann Clin Biochem 2001;38:423-49. http://dx.doi.org/10.1258/0004563011901109.
- Panteghini M, Apple FS, Christenson RH, Dati F, Mair J, Wu AH. Use of biochemical markers in acute coronary syndromes. IFCC Scientific Division, Committee on Standardization of Markers of Cardiac Damage. International Federation of Clinical Chemistry. Clin Chem Lab Med 1999;37:687-93. http://dx.doi.org/10.1515/CCLM.1999.107.
- McCord J, Nowak RM, McCullough PA, Foreback C, Borzak S, Tokarski G, et al. Ninety-minute exclusion of acute myocardial infarction by use of quantitative point-of-care testing of myoglobin and troponin I. Circulation 2001;104:1483-8. http://dx.doi.org/10.1161/hc3801.096336.
- Collinson PO, Stubbs PJ, Kessler AC. Multicentre evaluation of the diagnostic value of cardiac troponin T, CK-MB mass, and myoglobin for assessing patients with suspected acute coronary syndromes in routine clinical practice. Heart 2003;89:280-6. http://dx.doi.org/10.1136/heart.89.3.280.
- Collinson PO. Sensitive troponin assays. J Clin Pathol 2011;66:845-9. http://dx.doi.org/10.1136/jclinpath-2011-200164.
- Van de Werf F, Bax J, Betriu A, Blomstrom-Lundqvist C, Crea F, Falk V, et al. Management of acute myocardial infarction in patients presenting with persistent ST-segment elevation: the Task Force on the Management of ST-Segment Elevation Acute Myocardial Infarction of the European Society of Cardiology. Eur Heart J 2008;29:2909-45. http://dx.doi.org/10.1093/eurheartj/ehn416.
- Kushner FG, Hand M, Smith SC, King SB, Anderson JL, Antman EM, et al. 2009 Focused Updates: ACC/AHA Guidelines for the Management of Patients With ST-Elevation Myocardial Infarction (updating the 2004 Guideline and 2007 Focused Update) and ACC/AHA/SCAI Guidelines on Percutaneous Coronary Intervention (updating the 2005 Guideline and 2007 Focused Update): a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2009;120:2271-306. http://dx.doi.org/10.1161/CIRCULATIONAHA.109.192663.
- Collinson PO. Biochemical estimation of infarct size. Heart 2011;97:169-70. http://dx.doi.org/10.1136/hrt.2010.212175.
- Bassand JP, Hamm CW, Ardissino D, Boersma E, Budaj A, Fernandez-Aviles F, et al. Guidelines for the diagnosis and treatment of non-ST-segment elevation acute coronary syndromes. The Task Force for the Diagnosis and Treatment of Non-ST-Segment Elevation Acute Coronary Syndromes of the European Society of Cardiology. Eur Heart J 2007;28:1598-660. http://dx.doi.org/10.1093/eurheartj/ehm161.
- Melanson SE, Morrow DA, Jarolim P. Earlier detection of myocardial injury in a preliminary evaluation using a new troponin I assay with improved sensitivity. Am J Clin Pathol 2007;128:282-6. http://dx.doi.org/10.1309/Q9W5HJTT24GQCXXX.
- Bassan R, Potsch A, Maisel A, Tura B, Villacorta H, Nogueira MV, et al. B-type natriuretic peptide: a novel early blood marker of acute myocardial infarction in patients with chest pain and no ST-segment elevation. Eur Heart J 2005;26:234-40. http://dx.doi.org/10.1093/eurheartj/ehi033.
- Morgenthaler NG, Struck J, Jochberger S, Dunser MW. Copeptin: clinical use of a new biomarker. Trends Endocrinol Metab 2008;19:43-9. http://dx.doi.org/10.1016/j.tem.2007.11.001.
- Davies MJ, Thomas AC, Knapman PA, Hangartner JR. Intramyocardial platelet aggregation in patients with unstable angina suffering sudden ischemic cardiac death. Circulation 1986;73:418-27. http://dx.doi.org/10.1161/01.CIR.73.3.418.
- Falk E. Unstable angina with fatal outcome: dynamic coronary thrombosis leading to infarction and/or sudden death. Autopsy evidence of recurrent mural thrombosis with peripheral embolization culminating in total vascular occlusion. Circulation 1985;71:699-708. http://dx.doi.org/10.1161/01.CIR.71.4.699.
- Davies MJ. Stability and instability: two faces of coronary atherosclerosis. The Paul Dudley White Lecture 1995. Circulation 1996;94:2013-20. http://dx.doi.org/10.1161/01.CIR.94.8.2013.
- Apple FS, Wu AH, Mair J, Ravkilde J, Panteghini M, Tate J, et al. Future biomarkers for detection of ischemia and risk stratification in acute coronary syndrome. Clin Chem 2005;51:810-24. http://dx.doi.org/10.1373/clinchem.2004.046292.
- Kubasik NP, Guiney W, Warren K, D’Souza JP, Sine HE, Brody BB. Radioimmunoassay of serum myoglobin: evaluation and modification of a commercial kit and assessment of its usefulness for detecting acute myocardial infarction. Clin Chem 1978;24:2047-9.
- Roxin LE, Venge P, Friman G, Hallgren R. Radioimmunoassays of human myoglobin in serum and urine. Scand J Clin Lab Invest 1979;39:37-46. http://dx.doi.org/10.3109/00365517909104937.
- Roxin LE, Venge P, Wide L. A fast and sensitive radioimmunoassay of human myoglobin for use in the early diagnosis of heart infarction. Clin Chim Acta 1980;107:129-34. http://dx.doi.org/10.1016/0009-8981(80)90422-2.
- Van Steirteghem AC, Zweig MH, Robertson EA, Bernard RM, Putzeys GA, Bieva CJ. Comparison of the effectiveness of four clinical chemical assays in classifying patients with chest pain. Clin Chem 1982;28:1319-24.
- Gibler WB, Gibler CD, Weinshenker E, Abbottsmith C, Hedges JR, Barsan WG, et al. Myoglobin as an early indicator of acute myocardial infarction. Ann Emerg Med 1987;16:851-6. http://dx.doi.org/10.1016/S0196-0644(87)80521-8.
- Kontos MC, Anderson FP, Hanbury CM, Roberts CS, Miller WG, Jesse RL. Use of the combination of myoglobin and CK-MB mass for the rapid diagnosis of acute myocardial infarction. Am J Emerg Med 1997;15:14-9. http://dx.doi.org/10.1016/S0735-6757(97)90040-1.
- Ng SM, Krishnaswamy P, Morissey R, Clopton P, Fitzgerald R, Maisel AS. Ninety-minute accelerated critical pathway for chest pain evaluation. Am J Cardiol 2001;88:611-17. http://dx.doi.org/10.1016/S0002-9149(01)01801-X.
- Schouten Y, de Winter RJ, Gorgels JP, Koster RW, Adams R, Sanders GT. Clinical evaluation of the CARDIAC STATus, a rapid immunochromatographic assay for simultaneous detection of elevated concentrations of CK-MB and myoglobin in whole blood. Clin Chem Lab Med 1998;36:469-73. http://dx.doi.org/10.1515/CCLM.1998.079.
- Schwartz JG, Gage CL, Farley NJ, Prihoda TJ. Evaluation of the cardiac STATus CK-MB/myoglobin card test to diagnose acute myocardial infarctions in the ED. Am J Emerg Med 1997;15:303-7. http://dx.doi.org/10.1016/S0735-6757(97)90021-8.
- Duma RJ, Siegel AL. Serum creatine phosphokinase in acute myocardial infarction. Arch Intern Med 1960;115:443-51. http://dx.doi.org/10.1001/archinte.1965.03860160069011.
- Rosalki SB. An improved procedure for serum creatine phosphokinase determination. J Lab Clin Med 1967;69:696-705.
- Rokos JA, Rosalki SB, Tarlow D. Automated fluorometric procedure for measurement of creatine phosphokinase activity. Clin Chem 1972;18:193-8.
- Vaidya HC, Maynard Y, Dietzler DN, Ladenson JH. Direct measurement of creatine kinase-MB activity in serum after extraction with a monoclonal antibody specific to the MB isoenzyme. Clin Chem 1986;32:657-63.
- Youens JE, Calvin J, Price CP. Clinical and analytical validation of an enzymometric assay for creatine kinase-MB isoenzyme. Ann Clin Biochem 1986;23:463-9.
- Rosalki SB, Roberts R, Katus HA, Giannitsis E, Ladenson JH, Apple FS. Cardiac biomarkers for detection of myocardial infarction: perspectives from past to present. Clin Chem 2004;50:2205-13. http://dx.doi.org/10.1373/clinchem.2004.041749.
- Azzazy HM, Pelsers MM, Christenson RH. Unbound free fatty acids and heart-type fatty acid-binding protein: diagnostic assays and clinical applications. Clin Chem 2006;52:19-2. http://dx.doi.org/10.1373/clinchem.2005.056143.
- Pagani F, Bonora R, Bonetti G, Panteghini M. Evaluation of a sandwich enzyme-linked immunosorbent assay for the measurement of serum heart fatty acid-binding protein. Ann Clin Biochem 2002;39:404-5. http://dx.doi.org/10.1258/000456302760042173.
- Alansari SE, Croal BL. Diagnostic value of heart fatty acid binding protein and myoglobin in patients admitted with chest pain. Ann Clin Biochem 2004;41:391-6. http://dx.doi.org/10.1258/0004563041731565.
- Colli A, Josa M, Pomar JL, Mestres CA, Gherli T. Heart fatty acid binding protein in the diagnosis of myocardial infarction: where do we stand today?. Cardiology 2007;108:4-10. http://dx.doi.org/10.1159/000095594.
- McCann CJ, Glover BM, Menown IB, Moore MJ, McEneny J, Owens CG, et al. Novel biomarkers in early diagnosis of acute myocardial infarction compared with cardiac troponin T. Eur Heart J 2008;29:2843-50. http://dx.doi.org/10.1093/eurheartj/ehn363.
- McCann CJ, Glover BM, Menown IB, Moore MJ, McEneny J, Owens CG, et al. Investigation of a multimarker approach to the initial assessment of patients with acute chest pain. Adv Ther 2009;26:531-4. http://dx.doi.org/10.1007/s12325-009-0032-7.
- Gururajan P, Gurumurthy P, Nayar P, Srinivasa Nageswara RG, Babu S, Cherian KM. Heart fatty acid binding protein (H-FABP) as a diagnostic biomarker in patients with acute coronary syndrome. Heart Lung Circ 2010;19:660-4. http://dx.doi.org/10.1016/j.hlc.2010.06.665.
- Garcia-Valdecasas S, Ruiz-Alvarez MJ, Garcia DT, De Pablo R, Huerta I, Barrionuevo M, et al. Diagnostic and prognostic value of heart-type fatty acid-binding protein in the early hours of acute myocardial infarction. Acta Cardiol 2011;66:315-21.
- Body R, McDowell G, Carley S, Wibberley C, Ferguson J, Mackway–Jones K. A FABP-ulous ‘rule out’ strategy? Heart fatty acid binding protein and troponin for rapid exclusion of acute myocardial infarction. Resuscitation 2011;82:1041-6. http://dx.doi.org/10.1016/j.resuscitation.2011.03.015.
- Dekker MS, Mosterd A, van ‘t Hof AW, Hoes AW. Novel biochemical markers in suspected acute coronary syndrome: systematic review and critical appraisal. Heart 2010;96:1001-10. http://dx.doi.org/10.1136/hrt.2009.189886.
- Bruins Slot MH, Reitsma JB, Rutten FH, Hoes AW, van der Heijden GJ. Heart-type fatty acid-binding protein in the early diagnosis of acute myocardial infarction: a systematic review and meta-analysis. Heart 2010;96:1957-63. http://dx.doi.org/10.1136/hrt.2010.208272.
- McCann CJ, Glover BM, Menown IB, Moore MJ, McEneny J, Owens CG, et al. Prognostic value of a multimarker approach for patients presenting to hospital with acute chest pain. Am J Cardiol 2009;103:22-8. http://dx.doi.org/10.1016/j.amjcard.2008.08.026.
- Viswanathan K, Kilcullen N, Morrell C, Thistlethwaite SJ, Sivananthan MU, Hassan TB, et al. Heart-type fatty acid-binding protein predicts long-term mortality and re-infarction in consecutive patients with suspected acute coronary syndrome who are troponin-negative. J Am Coll Cardiol 2010;55:2590-8. http://dx.doi.org/10.1016/j.jacc.2009.12.062.
- Charpentier S, Ducasse JL, Cournot M, Maupas-Schwalm F, Elbaz M, Baixas C, et al. Clinical assessment of ischemia-modified albumin and heart fatty acid-binding protein in the early diagnosis of non-ST-elevation acute coronary syndrome in the emergency department. Acad Emerg Med 2010;17:27-35. http://dx.doi.org/10.1111/j.1553-2712.2009.00614.x.
- Kurz K, Giannitsis E, Becker M, Hess G, Zdunek D, Katus HA. Comparison of the new high sensitive cardiac troponin T with myoglobin, h-FABP and cTnT for early identification of myocardial necrosis in the acute coronary syndrome. Clin Res Cardiol 2011;100:209-15. http://dx.doi.org/10.1007/s00392-010-0230-y.
- Bhardwaj A, Truong QA, Peacock WF, Yeo KT, Storrow A, Thomas S, et al. A multicenter comparison of established and emerging cardiac biomarkers for the diagnostic evaluation of chest pain in the emergency department. Am Heart J 2011;162:276-82. http://dx.doi.org/10.1016/j.ahj.2011.05.022.
- Ilva T, Lund J, Porela P, Mustonen H, Voipio-Pulkki LM, Eriksson S, et al. Early markers of myocardial injury: cTnI is enough. Clin Chim Acta 2009;400:82-5.
- Bodor GS, Porter S, Landt Y, Ladenson JH. Development of monoclonal antibodies for an assay of cardiac troponin-I and preliminary results in suspected cases of myocardial infarction. Clin Chem 1992;38:2203-14.
- Thygesen K, Mair J, Katus H, Plebani M, Venge P, Collinson P, et al. Recommendations for the use of cardiac troponin measurement in acute cardiac care. Eur Heart J 2010;31:2197-204. http://dx.doi.org/10.1093/eurheartj/ehq251.
- Apple FS. A new season for cardiac troponin assays: it's time to keep a scorecard. Clin Chem 2009;55:1303-6. http://dx.doi.org/10.1373/clinchem.2009.128363.
- Ghosh N, Haddad H. Atrial natriuretic peptides in heart failure: pathophysiological significance, diagnostic and prognostic value. Can J Physiol Pharmacol 2011;89:587-91. http://dx.doi.org/10.1139/y11-040.
- Collinson PO, Bayes-Genis A, Jannuzzi J. NT-proBNP as a biomarker in cardiovascular disease. Barcelona: Thomson Reuters; 2008.
- Mair J. Clinical significance of pro-B-type natriuretic peptide glycosylation and processing. Clin Chem 2009;55:394-7. http://dx.doi.org/10.1373/clinchem.2008.119271.
- Maisel AS, Krishnaswamy P, Nowak RM, McCord J, Hollander JE, Duc P, et al. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med 2002;347:161-7. http://dx.doi.org/10.1056/NEJMoa020233.
- Januzzi JL, van Kimmenade R, Lainchbury J, Bayes-Genis A, Ordonez-Llanos J, Santalo-Bel M, et al. NT-proBNP testing for diagnosis and short-term prognosis in acute destabilized heart failure: an international pooled analysis of 1256 patients: the International Collaborative of NT-proBNP Study. Eur Heart J 2006;27:330-7. http://dx.doi.org/10.1093/eurheartj/ehi631.
- Januzzi JL, Camargo CA, Anwaruddin S, Baggish AL, Chen AA, Krauser DG, et al. The N-terminal Pro-BNP Investigation of Dyspnea in the Emergency Department (PRIDE) study. Am J Cardiol 2005;95:948-54. http://dx.doi.org/10.1016/j.amjcard.2004.12.032.
- Hildebrandt P, Collinson PO, Doughty RN, Fuat A, Gaze DC, Gustafsson F, et al. Age-dependent values of N-terminal pro-B-type natriuretic peptide are superior to a single cut-point for ruling out suspected systolic dysfunction in primary care. Eur Heart J 2010;31:1881-9. http://dx.doi.org/10.1093/eurheartj/ehq163.
- Hildebrandt P, Collinson PO. Amino-terminal pro-B-type natriuretic peptide testing to assist the diagnostic evaluation of heart failure in symptomatic primary care patients. Am J Cardiol 2008;101:25-8. http://dx.doi.org/10.1016/j.amjcard.2007.11.016.
- Pascual-Figal DA, Antolinos MJ, Bayes-Genis A, Casas T, Nicolas F, Valdes M. B-type natriuretic peptide release in the coronary effluent after acute transient ischaemia in humans. Heart 2007;93:1077-80. http://dx.doi.org/10.1136/hrt.2006.101303.
- Sabatine MS, Morrow DA, de Lemos JA, Gibson CM, Murphy SA, Rifai N, et al. Multimarker approach to risk stratification in non-ST elevation acute coronary syndromes: simultaneous assessment of troponin I, C-reactive protein, and B-type natriuretic peptide. Circulation 2002;105:1760-3. http://dx.doi.org/10.1161/01.CIR.0000015464.18023.0A.
- Jernberg T, Stridsberg M, Venge P, Lindahl B. N-terminal pro brain natriuretic peptide on admission for early risk stratification of patients with chest pain and no ST-segment elevation. J Am Coll Cardiol 2002;40:437-45. http://dx.doi.org/10.1016/S0735-1097(02)01986-1.
- Blankenberg S, McQueen MJ, Smieja M, Pogue J, Balion C, Lonn E, et al. Comparative impact of multiple biomarkers and N-terminal pro-brain natriuretic peptide in the context of conventional risk factors for the prediction of recurrent cardiovascular events in the Heart Outcomes Prevention Evaluation (HOPE) Study. Circulation 2006;114:201-8. http://dx.doi.org/10.1161/CIRCULATIONAHA.105.590927.
- Morrow DA, Cannon CP, Jesse RL, Newby LK, Ravkilde J, Storrow AB, et al. National Academy of Clinical Biochemistry Laboratory Medicine Practice Guidelines: clinical characteristics and utilization of biochemical markers in acute coronary syndromes. Circulation 2007;115:e356-75. http://dx.doi.org/10.1161/CIRCULATIONAHA.107.182882.
- Khan SQ, Dhillon OS, O’Brien RJ, Struck J, Quinn PA, Morgenthaler NG, et al. C-terminal provasopressin (copeptin) as a novel and prognostic marker in acute myocardial infarction: Leicester Acute Myocardial Infarction Peptide (LAMP) study. Circulation 2007;115:2103-10. http://dx.doi.org/10.1161/CIRCULATIONAHA.106.685503.
- Kelly D, Squire IB, Khan SQ, Quinn P, Struck J, Morgenthaler NG, et al. C-terminal provasopressin (copeptin) is associated with left ventricular dysfunction, remodeling, and clinical heart failure in survivors of myocardial infarction. J Card Fail 2008;14:739-45. http://dx.doi.org/10.1016/j.cardfail.2008.07.231.
- Bhandari SS, Loke I, Davies JE, Squire IB, Struck J, Ng LL. Gender and renal function influence plasma levels of copeptin in healthy individuals. Clin Sci 2009;116:257-63. http://dx.doi.org/10.1042/CS20080140.
- Reichlin T, Hochholzer W, Stelzig C, Laule K, Freidank H, Morgenthaler NG, et al. Incremental value of copeptin for rapid rule out of acute myocardial infarction. J Am Coll Cardiol 2009;54:60-8. http://dx.doi.org/10.1016/j.jacc.2009.01.076.
- Staub D, Morgenthaler NG, Buser C, Breidthardt T, Potocki M, Noveanu M, et al. Use of copeptin in the detection of myocardial ischemia. Clin Chim Acta 2009;399:69-73. http://dx.doi.org/10.1016/j.cca.2008.09.016.
- Yanagawa B, Nagaya N. Adrenomedullin: molecular mechanisms and its role in cardiac disease. Amino Acids 2007;32:157-64. http://dx.doi.org/10.1007/s00726-005-0279-5.
- Bisping E, Tenderich G, Barckhausen P, Stumme B, Bruns S, von Lewinski D, et al. Atrial myocardium is the predominant inotropic target of adrenomedullin in the human heart. Am J Physiol Heart Circ Physiol 2007;293:H3001-7. http://dx.doi.org/10.1152/ajpheart.01276.2006.
- Nishida H, Horio T, Suzuki Y, Iwashima Y, Kamide K, Kangawa K, et al. Plasma adrenomedullin as an independent predictor of future cardiovascular events in high-risk patients: comparison with C-reactive protein and adiponectin. Peptides 2008;29:599-605. http://dx.doi.org/10.1016/j.peptides.2007.12.006.
- Behnes M, Papassotiriou J, Walter T, Fiedler E, Sauer T, Lang S, et al. Long-term prognostic value of mid-regional pro-adrenomedullin and C-terminal pro-endothelin-1 in patients with acute myocardial infarction. Clin Chem Lab Med 2008;46:204-11. http://dx.doi.org/10.1515/CCLM.2008.040.
- Walter T, Brueckmann M, Lang S, Sauer T, Fiedler E, Papassotiriou J, et al. Comparison of long-term prognostic value of N-terminal-proBNP and midregional-pro-adrenomedullin in patients with acute myocardial infarction. Clin Lab 2010;56:303-9.
- Dhillon OS, Khan SQ, Narayan HK, Ng KH, Struck J, Quinn PA, et al. Prognostic value of mid-regional pro-adrenomedullin levels taken on admission and discharge in non-ST-elevation myocardial infarction: the LAMP (Leicester Acute Myocardial Infarction Peptide) II study. J Am Coll Cardiol 2010;56:125-33. http://dx.doi.org/10.1016/j.jacc.2010.01.060.
- Klip IT, Voors AA, Anker SD, Hillege HL, Struck J, Squire I, et al. Prognostic value of mid-regional pro-adrenomedullin in patients with heart failure after an acute myocardial infarction. Heart 2011;97:892-8. http://dx.doi.org/10.1136/hrt.2010.210948.
- Nishikimi T, Saito Y, Kitamura K, Ishimitsu T, Eto T, Kangawa K, et al. Increased plasma levels of adrenomedullin in patients with heart failure. J Am Coll Cardiol 1995;26:1424-31. http://dx.doi.org/10.1016/0735-1097(95)00338-X.
- Jougasaki M, Wei CM, McKinley LJ, Burnett JC. Circulation 1995;92:286-9. http://dx.doi.org/10.1161/01.CIR.92.3.286.
- Oie E, Ahmed MS, Ueland T, Sikkeland LI, Dahl CP, Hagelin EM, et al. Adrenomedullin is increased in alveolar macrophages and released from the lungs into the circulation in severe heart failure. Basic Res Cardiol 2010;105:89-98. http://dx.doi.org/10.1007/s00395-009-0070-y.
- Gegenhuber A, Struck J, Dieplinger B, Poelz W, Pacher R, Morgenthaler NG, et al. Comparative evaluation of B-type natriuretic peptide, mid-regional pro-A-type natriuretic peptide, mid-regional pro-adrenomedullin, and copeptin to predict 1-year mortality in patients with acute destabilized heart failure. J Card Fail 2007;13:42-9. http://dx.doi.org/10.1016/j.cardfail.2006.09.004.
- Pousset F, Masson F, Chavirovskaia O, Isnard R, Carayon A, Golmard JL, et al. Plasma adrenomedullin, a new independent predictor of prognosis in patients with chronic heart failure. Eur Heart J 2000;21:1009-14. http://dx.doi.org/10.1053/euhj.1999.1904.
- Richards AM, Doughty R, Nicholls MG, MacMahon S, Sharpe N, Murphy J, et al. Plasma N-terminal pro-brain natriuretic peptide and adrenomedullin: prognostic utility and prediction of benefit from carvedilol in chronic ischemic left ventricular dysfunction. Australia–New Zealand Heart Failure Group. J Am Coll Cardiol 2001;37:1781-7. http://dx.doi.org/10.1016/S0735-1097(01)01269-4.
- Ridker PM. Clinical application of C-reactive protein for cardiovascular disease detection and prevention. Circulation 2003;107:363-9. http://dx.doi.org/10.1161/01.CIR.0000053730.47739.3C.
- Osman R, L’Allier PL, Elgharib N, Tardif JC. Critical appraisal of C-reactive protein throughout the spectrum of cardiovascular disease. Vasc Health Risk Manag 2006;2:221-37. http://dx.doi.org/10.2147/vhrm.2006.2.3.221.
- Rifai N, Ridker PM. Population distributions of C-reactive protein in apparently healthy men and women in the United States: implication for clinical interpretation. Clin Chem 2003;49:666-9. http://dx.doi.org/10.1373/49.4.666.
- Meier-Ewert HK, Ridker PM, Rifai N, Price N, Dinges DF, Mullington JM. Absence of diurnal variation of C-reactive protein concentrations in healthy human subjects. Clin Chem 2001;47:426-30.
- Khera A, de Lemos JA, Peshock RM, Lo HS, Stanek HG, Murphy SA, et al. Relationship between C-reactive protein and subclinical atherosclerosis: the Dallas Heart Study. Circulation 2006;113:38-43. http://dx.doi.org/10.1161/CIRCULATIONAHA.105.575241.
- Sabatine MS, Morrow DA, Jablonski KA, Rice MM, Warnica JW, Domanski MJ, et al. Prognostic significance of the Centers for Disease Control/American Heart Association high-sensitivity C-reactive protein cut points for cardiovascular and other outcomes in patients with stable coronary artery disease. Circulation 2007;115:1528-36. http://dx.doi.org/10.1161/CIRCULATIONAHA.106.649939.
- Scirica BM, Morrow DA, Cannon CP, de Lemos JA, Murphy S, Sabatine MS, et al. Clinical application of C-reactive protein across the spectrum of acute coronary syndromes. Clin Chem 2007;53:1800-7. http://dx.doi.org/10.1373/clinchem.2007.087957.
- Kanda T, Takahashi T. Interleukin-6 and cardiovascular diseases. Jpn Heart J 2004;45:183-93. http://dx.doi.org/10.1536/jhj.45.183.
- Barry SP, Davidson SM, Townsend PA. Molecular regulation of cardiac hypertrophy. Int J Biochem Cell Biol 2008;40:2023-39. http://dx.doi.org/10.1016/j.biocel.2008.02.020.
- Modur V, Li Y, Zimmerman GA, Prescott SM, McIntyre TM. Retrograde inflammatory signaling from neutrophils to endothelial cells by soluble interleukin-6 receptor alpha. J Clin Invest 1997;100:2752-6. http://dx.doi.org/10.1172/JCI119821.
- Schuett H, Luchtefeld M, Grothusen C, Grote K, Schieffer B. How much is too much? Interleukin-6 and its signalling in atherosclerosis. Thromb Haemost 2009;102:215-22.
- Shu J, Ren N, Du JB, Zhang M, Cong HL, Huang TG. Increased levels of interleukin-6 and matrix metalloproteinase-9 are of cardiac origin in acute coronary syndrome. Scand Cardiovasc J 2007;41:149-54. http://dx.doi.org/10.1080/14017430601164263.
- Picciotto S, Forastiere F, Pistelli R, Koenig W, Lanki T, Ljungman P, et al. Determinants of plasma interleukin-6 levels among survivors of myocardial infarction. Eur J Cardiovasc Prev Rehabil 2008;15:631-8. http://dx.doi.org/10.1097/HJR.0b013e3283069d9a.
- Ferroni P, Rosa A, Di Franco M, Palmirotta R, Guadagni F, Davi G, et al. Prognostic significance of interleukin-6 measurement in the diagnosis of acute myocardial infarction in emergency department. Clin Chim Acta 2007;381:151-6. http://dx.doi.org/10.1016/j.cca.2007.03.002.
- Tan J, Hua Q, Li J, Fan Z. Prognostic value of interleukin-6 during a 3-year follow-up in patients with acute ST-segment elevation myocardial infarction. Heart Vessels 2009;24:329-34. http://dx.doi.org/10.1007/s00380-008-1128-8.
- Borrayo-Sanchez G, Pacheco-Bouthillier A, Mendoza-Valdez L, Isordia-Salas I, Arguero-Sanchez R, Careaga RG. Prognostic value of serum levels of interleukin-6 in patients with ST-segment elevation acute myocardial infarction. Cir Cir 2010;78:25-30.
- Fernandez-Berges D, Bertomeu-Gonzalez V, Sanchez PL, Cruz-Fernandez JM, Arroyo R, Barriales Alvarez V, et al. Clinical scores and patient risk stratification in non-ST elevation acute coronary syndrome. Int J Cardiol 2011;146:219-24. http://dx.doi.org/10.1016/j.ijcard.2010.04.016.
- Kakkar R, Lee RT. The IL–33/ST2 pathway: therapeutic target and novel biomarker. Nat Rev Drug Discov 2008;7:827-40. http://dx.doi.org/10.1038/nrd2660.
- Miller AM, Xu D, Asquith DL, Denby L, Li Y, Sattar N, et al. IL-33 reduces the development of atherosclerosis. J Exp Med 2008;205:339-46. http://dx.doi.org/10.1084/jem.20071868.
- Weinberg EO, Shimpo M, De Keulenaer GW, MacGillivray C, Tominaga S, Solomon SD, et al. Expression and regulation of ST2, an interleukin-1 receptor family member, in cardiomyocytes and myocardial infarction. Circulation 2002;106:2961-6. http://dx.doi.org/10.1161/01. CIR.0000038705.69871.D9.
- Sanada S, Hakuno D, Higgins LJ, Schreiter ER, McKenzie AN, Lee RT. IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system. J Clin Invest 2007;117:1538-49. http://dx.doi.org/10.1172/JCI30634.
- Seki K, Sanada S, Kudinova AY, Steinhauser ML, Handa V, Gannon J, et al. Interleukin-33 prevents apoptosis and improves survival after experimental myocardial infarction through ST2 signaling. Circ Heart Fail 2009;2:684-91. http://dx.doi.org/10.1161/CIRCHEARTFAILURE.109.873240.
- Shimpo M, Morrow DA, Weinberg EO, Sabatine MS, Murphy SA, Antman EM, et al. Serum levels of the interleukin-1 receptor family member ST2 predict mortality and clinical outcome in acute myocardial infarction. Circulation 2004;109:2186-90. http://dx.doi.org/10.1161/01.CIR.0000127958.21003.5A.
- Sabatine MS, Morrow DA, Higgins LJ, MacGillivray C, Guo W, Bode C, et al. Complementary roles for biomarkers of biomechanical strain ST2 and N-terminal prohormone B-type natriuretic peptide in patients with ST-elevation myocardial infarction. Circulation 2008;117:1936-44. http://dx.doi.org/10.1161/CIRCULATIONAHA.107.728022.
- Eggers KM, Armstrong PW, Califf RM, Simoons ML, Venge P, Wallentin L, et al. ST2 and mortality in non-ST-segment elevation acute coronary syndrome. Am Heart J 2010;159:788-94. http://dx.doi.org/10.1016/j.ahj.2010.02.022.
- Brown AM, Wu AH, Clopton P, Robey JL, Hollander JE. ST2 in emergency department chest pain patients with potential acute coronary syndromes. Ann Emerg Med 2007;50:153-8. http://dx.doi.org/10.1016/j.annemergmed.2007.02.015.
- Dhillon OS, Narayan HK, Quinn PA, Squire IB, Davies JE, Ng LL. Interleukin 33 and ST2 in non- ST-elevation myocardial infarction: comparison with Global Registry of Acute Coronary Events Risk Scoring and NT-proBNP. Am Heart J 2011;161:1163-70. http://dx.doi.org/10.1016/j.ahj.2011.03.025.
- Weir RA, Miller AM, Murphy GE, Clements S, Steedman T, Connell JM, et al. Serum soluble ST2: a potential novel mediator in left ventricular and infarct remodeling after acute myocardial infarction. J Am Coll Cardiol 2010;55:243-50. http://dx.doi.org/10.1016/j.jacc.2009.08.047.
- Shah RV, Januzzi JL. ST2: a novel remodeling biomarker in acute and chronic heart failure. Curr Heart Fail Rep 2010;7:9-14. http://dx.doi.org/10.1007/s11897-010-0005-9.
- Daniels LB, Clopton P, Iqbal N, Tran K, Maisel AS. Association of ST2 levels with cardiac structure and function and mortality in outpatients. Am Heart J 2010;160:721-8. http://dx.doi.org/10.1016/j.ahj.2010.06.033.
- Januzzi JL, Peacock WF, Maisel AS, Chae CU, Jesse RL, Baggish AL, et al. Measurement of the interleukin family member ST2 in patients with acute dyspnea: results from the PRIDE (Pro-Brain Natriuretic Peptide Investigation of Dyspnea in the Emergency Department) study. J Am Coll Cardiol 2007;50:607-13. http://dx.doi.org/10.1016/j.jacc.2007.05.014.
- Rehman SU, Martinez-Rumayor A, Mueller T, Januzzi JL. Independent and incremental prognostic value of multimarker testing in acute dyspnea: results from the ProBNP Investigation of Dyspnea in the Emergency Department (PRIDE) study. Clin Chim Acta 2008;392:41-5. http://dx.doi.org/10.1016/j.cca.2008.03.002.
- Mueller T, Dieplinger B, Gegenhuber A, Poelz W, Pacher R, Haltmayer M. Increased plasma concentrations of soluble ST2 are predictive for 1-year mortality in patients with acute destabilized heart failure. Clin Chem 2008;54:752-6. http://dx.doi.org/10.1373/clinchem.2007.096560.
- Boisot S, Beede J, Isakson S, Chiu A, Clopton P, Januzzi J, et al. Serial sampling of ST2 predicts 90-day mortality following destabilized heart failure. J Card Fail 2008;14:732-8. http://dx.doi.org/10.1016/j.cardfail.2008.06.415.
- Martinez-Rumayor A, Camargo CA, Green SM, Baggish AL, O’Donoghue M, Januzzi JL. Soluble ST2 plasma concentrations predict 1-year mortality in acutely dyspneic emergency department patients with pulmonary disease. Am J Clin Pathol 2008;130:578-84. http://dx.doi.org/10.1309/WMG2BFRC97MKKQKP.
- Bootcov MR, Bauskin AR, Valenzuela SM, Moore AG, Bansal M, He XY, et al. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-beta superfamily. Proc Natl Acad Sci USA 1997;94:11514-19. http://dx.doi.org/10.1073/pnas.94.21.11514.
- Bauskin AR, Zhang HP, Fairlie WD, He XY, Russell PK, Moore AG, et al. The propeptide of macrophage inhibitory cytokine (MIC-1), a TGF-beta superfamily member, acts as a quality control determinant for correctly folded MIC-1. EMBO J. 2000;19:2212-20. http://dx.doi.org/10.1093/emboj/19.10.2212.
- Fairlie WD, Moore AG, Bauskin AR, Russell PK, Zhang HP, Breit SN. MIC-1 is a novel TGF-beta superfamily cytokine associated with macrophage activation. J Leukoc Biol 1999;65:2-5.
- Paralkar VM, Vail AL, Grasser WA, Brown TA, Xu H, Vukicevic S, et al. Cloning and characterization of a novel member of the transforming growth factor-beta/bone morphogenetic protein family. J Biol Chem 1998;273:13760-7. http://dx.doi.org/10.1074/jbc.273.22.13760.
- Xu J, Kimball TR, Lorenz JN, Brown DA, Bauskin AR, Klevitsky R, et al. GDF15/MIC-1 functions as a protective and antihypertrophic factor released from the myocardium in association with SMAD protein activation. Circ Res 2006;98:342-50. http://dx.doi.org/10.1161/01.RES.0000202804.84885.d0.
- Kempf T, Eden M, Strelau J, Naguib M, Willenbockel C, Tongers J, et al. The transforming growth factor-beta superfamily member growth-differentiation factor-15 protects the heart from ischemia/reperfusion injury. Circ Res 2006;98:351-60. http://dx.doi.org/10.1161/01.RES.0000202805.73038.48.
- Kempf T, Horn-Wichmann R, Brabant G, Peter T, Allhoff T, Klein G, et al. Circulating concentrations of growth-differentiation factor 15 in apparently healthy elderly individuals and patients with chronic heart failure as assessed by a new immunoradiometric sandwich assay. Clin Chem 2007;53:284-91. http://dx.doi.org/10.1373/clinchem.2006.076828.
- Wollert KC, Kempf T, Lagerqvist B, Lindahl B, Olofsson S, Allhoff T, et al. Growth differentiation factor 15 for risk stratification and selection of an invasive treatment strategy in non ST-elevation acute coronary syndrome. Circulation 2007;116:1540-8. http://dx.doi.org/10.1161/CIRCULATIONAHA.107.697714.
- Wollert KC, Kempf T, Peter T, Olofsson S, James S, Johnston N, et al. Prognostic value of growth-differentiation factor-15 in patients with non-ST-elevation acute coronary syndrome. Circulation 2007;115:962-71. http://dx.doi.org/10.1161/CIRCULATIONAHA.106.650846.
- Bonaca MP, Morrow DA, Braunwald E, Cannon CP, Jiang S, Breher S, et al. Growth differentiation factor-15 and risk of recurrent events in patients stabilized after acute coronary syndrome: observations from PROVE IT-TIMI 22. Arterioscler Thromb Vasc Biol 2011;31:203-10. http://dx.doi.org/10.1161/ATVBAHA.110.213512.
- Eggers KM, Kempf T, Lagerqvist B, Lindahl B, Olofsson S, Jantzen F, et al. Growth-differentiation factor-15 for long-term risk prediction in patients stabilized after an episode of non-ST-segment-elevation acute coronary syndrome. Circ Cardiovasc Genet 2010;3:88-96. http://dx.doi.org/10.1161/CIRCGENETICS.109.877456.
- Kempf T, Bjorklund E, Olofsson S, Lindahl B, Allhoff T, Peter T, et al. Growth-differentiation factor-15 improves risk stratification in ST-segment elevation myocardial infarction. Eur Heart J 2007;28:2858-65. http://dx.doi.org/10.1093/eurheartj/ehm465.
- Eitel I, Blase P, Adams V, Hildebrand L, Desch S, Schuler G, et al. Growth-differentiation factor 15 as predictor of mortality in acute reperfused ST-elevation myocardial infarction: insights from cardiovascular magnetic resonance. Heart 2011;97:632-40. http://dx.doi.org/10.1136/hrt.2010.219543.
- Khan SQ, Ng K, Dhillon O, Kelly D, Quinn P, Squire IB, et al. Growth differentiation factor-15 as a prognostic marker in patients with acute myocardial infarction. Eur Heart J 2009;30:1057-65. http://dx.doi.org/10.1093/eurheartj/ehn600.
- Eggers KM, Kempf T, Allhoff T, Lindahl B, Wallentin L, Wollert KC. Growth-differentiation factor-15 for early risk stratification in patients with acute chest pain. Eur Heart J 2008;29:2327-35. http://dx.doi.org/10.1093/eurheartj/ehn339.
- Eggers KM, Kempf T, Venge P, Wallentin L, Wollert KC, Lindahl B. Improving long-term risk prediction in patients with acute chest pain: the Global Registry of Acute Coronary Events (GRACE) risk score is enhanced by selected nonnecrosis biomarkers. Am Heart J 2010;160:88-94. http://dx.doi.org/10.1016/j.ahj.2010.05.002.
- Kempf T, Sinning JM, Quint A, Bickel C, Sinning C, Wild PS, et al. Growth-differentiation factor- 15 for risk stratification in patients with stable and unstable coronary heart disease: results from the AtheroGene study. Circ Cardiovasc Genet 2009;2:286-92. http://dx.doi.org/10.1161/CIRCGENETICS.108.824870.
- Schnabel RB, Schulz A, Messow CM, Lubos E, Wild PS, Zeller T, et al. Multiple marker approach to risk stratification in patients with stable coronary artery disease. Eur Heart J 2010;31:3024-31. http://dx.doi.org/10.1093/eurheartj/ehq322.
- Kempf T, von Haehling S, Peter T, Allhoff T, Cicoira M, Doehner W, et al. Prognostic utility of growth differentiation factor-15 in patients with chronic heart failure. J Am Coll Cardiol 2007;50:1054-60. http://dx.doi.org/10.1016/j.jacc.2007.04.091.
- Dominguez-Rodriguez A, Abreu-Gonzalez P, Avanzas P. Relation of growth-differentiation factor 15 to left ventricular remodeling in st-segment elevation myocardial infarction. Am J Cardiol 2011;108:955-8. http://dx.doi.org/10.1016/j.amjcard.2011.05.028.
- Kempf T, Zarbock A, Widera C, Butz S, Stadtmann A, Rossaint J, et al. GDF-15 is an inhibitor of leukocyte integrin activation required for survival after myocardial infarction in mice. Nat Med 2011;17:581-8. http://dx.doi.org/10.1038/nm.2354.
- Brennan ML, Penn MS, Van Lente F, Nambi V, Shishehbor MH, Aviles RJ, et al. Prognostic value of myeloperoxidase in patients with chest pain. N Engl J Med 2003;349:1595-604. http://dx.doi.org/10.1056/NEJMoa035003.
- Baldus S, Heeschen C, Meinertz T, Zeiher AM, Eiserich JP, Munzel T, et al. Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation 2003;108:1440-5. http://dx.doi.org/10.1161/01.CIR.0000090690.67322.51.
- Cavusoglu E, Ruwende C, Eng C, Chopra V, Yanamadala S, Clark LT, et al. Usefulness of baseline plasma myeloperoxidase levels as an independent predictor of myocardial infarction at two years in patients presenting with acute coronary syndrome. Am J Cardiol 2007;99:1364-8. http://dx.doi.org/10.1016/j.amjcard.2006.12.060.
- Morrow DA, Sabatine MS, Brennan ML, de Lemos JA, Murphy SA, Ruff CT, et al. Concurrent evaluation of novel cardiac biomarkers in acute coronary syndrome: myeloperoxidase and soluble CD40 ligand and the risk of recurrent ischaemic events in TACTICS-TIMI 18. Eur Heart J 2008;29:1096-102. http://dx.doi.org/10.1093/eurheartj/ehn071.
- Apple FS, Pearce LA, Chung A, Ler R, Murakami MM. Multiple biomarker use for detection of adverse events in patients presenting with symptoms suggestive of acute coronary syndrome. Clin Chem 2007;53:874-81. http://dx.doi.org/10.1373/clinchem.2006.080192.
- Apple FS, Smith SW, Pearce LA, Murakami MM. Assessment of the multiple-biomarker approach for diagnosis of myocardial infarction in patients presenting with symptoms suggestive of acute coronary syndrome. Clin Chem 2009;55:93-100. http://dx.doi.org/10.1373/clinchem.2008.102905.
- Rudolph V, Goldmann BU, Bos C, Rudolph TK, Klinke A, Friedrichs K, et al. Diagnostic value of MPO plasma levels in patients admitted for suspected myocardial infarction. Int J Cardiol 2011;153:267-71. http://dx.doi.org/10.1016/j.ijcard.2010.08.015.
- Scirica BM, Sabatine MS, Jarolim P, Murphy SA, de Lemos JL, Braunwald E, et al. Assessment of multiple cardiac biomarkers in non-ST-segment elevation acute coronary syndromes: observations from the MERLIN-TIMI 36 trial. Eur Heart J 2011;32:697-705. http://dx.doi.org/10.1093/eurheartj/ehq468.
- Sawicki M, Sypniewska G, Kozinski M, Gruszka M, Krintus M, Obonska K, et al. Diagnostic efficacy of myeloperoxidase for the detection of acute coronary syndromes. Eur J Clin Invest 2011;41:667-71. http://dx.doi.org/10.1111/j.1365-2362.2010.02457.x.
- Oemrawsingh RM, Lenderink T, Akkerhuis KM, Heeschen C, Baldus S, Fichtlscherer S, et al. Multimarker risk model containing troponin-T, interleukin 10, myeloperoxidase and placental growth factor predicts long-term cardiovascular risk after non-ST-segment elevation acute coronary syndrome. Heart 2011;97:1061-6. http://dx.doi.org/10.1136/hrt.2010.197392.
- Apple FS, Smith SW, Pearce LA, Schulz KM, Ler R, Murakami MM. Myeloperoxidase improves risk stratification in patients with ischemia and normal cardiac troponin I concentrations. Clin Chem 2011;57:603-8. http://dx.doi.org/10.1373/clinchem.2010.158014.
- de Azevedo LE, Goncalves SC, Ribeiro JP, Nunes GL, de Oliveira JR, Araujo GN, et al. Lack of association between plasma myeloperoxidase levels and angiographic severity of coronary artery disease in patients with acute coronary syndrome. Inflamm Res 2011;60:137-42. http://dx.doi.org/10.1007/s00011-010-0247-8.
- Kalela A, Koivu TA, Sisto T, Kanervisto J, Hoyhtya M, Sillanaukee P, et al. Serum matrix metalloproteinase–9 concentration in angiographically assessed coronary artery disease. Scand J Clin Lab Invest 2002;62:337-42. http://dx.doi.org/10.1080/00365510260296483.
- Brunner S, Kim JO, Methe H. Relation of matrix metalloproteinase-9/tissue inhibitor of metalloproteinase-1 ratio in peripheral circulating CD14+ monocytes to progression of coronary artery disease. Am J Cardiol 2010;105:429-34. http://dx.doi.org/10.1016/j.amjcard.2009.10.013.
- Lehrke M, Greif M, Broedl UC, Lebherz C, Laubender RP, Becker A, et al. MMP-1 serum levels predict coronary atherosclerosis in humans. Cardiovasc Diabetol 2009;8. http://dx.doi.org/10.1186/1475-2840-8-50.
- Bamberg F, Truong QA, Koenig W, Schlett CL, Nasir K, Butler J, et al. Differential associations between blood biomarkers of inflammation, oxidation, and lipid metabolism with varying forms of coronary atherosclerotic plaque as quantified by coronary CT angiography. Int J Cardiovasc Imaging 2012;28:183-92.
- Cavusoglu E, Ruwende C, Chopra V, Yanamadala S, Eng C, Clark LT, et al. Tissue inhibitor of metalloproteinase-1 (TIMP-1) is an independent predictor of all-cause mortality, cardiac mortality, and myocardial infarction. Am Heart J 2006;151:1101-8. http://dx.doi.org/10.1016/j.ahj.2006.02.029.
- Dhillon OS, Khan SQ, Narayan HK, Ng KH, Mohammed N, Quinn PA, et al. Matrix metalloproteinase-2 predicts mortality in patients with acute coronary syndrome. Clin Sci (Lond) 2010;118:249-57. http://dx.doi.org/10.1042/CS20090226.
- Beygui F, Silvain J, Pena A, Bellemain-Appaix A, Collet JP, Drexler H, et al. Usefulness of biomarker strategy to improve GRACE score's prediction performance in patients with non-ST-segment elevation acute coronary syndrome and low event rates. Am J Cardiol 2010;106:650-8. http://dx.doi.org/10.1016/j.amjcard.2010.04.019.
- Kaden JJ, Dempfle CE, Sueselbeck T, Brueckmann M, Poerner TC, Haghi D, et al. Time-dependent changes in the plasma concentration of matrix metalloproteinase 9 after acute myocardial infarction. Cardiology 2003;99:140-4. http://dx.doi.org/10.1159/000070670.
- Matsunaga T, Abe N, Kameda K, Hagii J, Fujita N, Onodera H, et al. Circulating level of gelatinase activity predicts ventricular remodeling in patients with acute myocardial infarction. Int J Cardiol 2005;105:203-8. http://dx.doi.org/10.1016/j.ijcard.2005.01.011.
- Onai Y, Suzuki J, Maejima Y, Haraguchi G, Muto S, Itai A, et al. Inhibition of NF-KB improves left ventricular remodeling and cardiac dysfunction after myocardial infarction. Am J Physiol Heart Circ Physiol 2007;292:H530-8. http://dx.doi.org/10.1152/ajpheart.00549.2006.
- Squire IB, Evans J, Ng LL, Loftus IM, Thompson MM. Plasma MMP-9 and MMP-2 following acute myocardial infarction in man: correlation with echocardiographic and neurohumoral parameters of left ventricular dysfunction. J Card Fail 2004;10:328-33. http://dx.doi.org/10.1016/j.cardfail.2003.11.003.
- Kelly D, Khan S, Cockerill G, Ng LL, Thompson M, Samani NJ, et al. Circulating stromelysin-1 (MMP-3): a novel predictor of LV dysfunction, remodelling and all-cause mortality after acute myocardial infarction. Eur J Heart Fail 2008;10:133-9. http://dx.doi.org/10.1016/j.ejheart.2007.12.009.
- Wagner DR, Delagardelle C, Ernens I, Rouy D, Vaillant M, Beissel J. Matrix metalloproteinase-9 is a marker of heart failure after acute myocardial infarction. J Card Fail 2006;12:66-72. http://dx.doi.org/10.1016/j.cardfail.2005.08.002.
- Kelly D, Khan SQ, Thompson M, Cockerill G, Ng LL, Samani N, et al. Plasma tissue inhibitor of metalloproteinase-1 and matrix metalloproteinase-9: novel indicators of left ventricular remodelling and prognosis after acute myocardial infarction. Eur Heart J 2008;29:2116-24. http://dx.doi.org/10.1093/eurheartj/ehn315.
- Schwartzkopff B, Fassbach M, Pelzer B, Brehm M, Strauer BE. Elevated serum markers of collagen degradation in patients with mild to moderate dilated cardiomyopathy. Eur J Heart Fail 2002;4:439-4. http://dx.doi.org/10.1016/S1388-9842(02)00092-2.
- George J, Patal S, Wexler D, Roth A, Sheps D, Keren G. Circulating matrix metalloproteinase-2 but not matrix metalloproteinase-3, matrix metalloproteinase-9, or tissue inhibitor of metalloproteinase-1 predicts outcome in patients with congestive heart failure. Am Heart J 2005;150:484-7. http://dx.doi.org/10.1016/j.ahj.2004.11.016.
- Banfi C, Cavalca V, Veglia F, Brioschi M, Barcella S, Mussoni L, et al. Neurohormonal activation is associated with increased levels of plasma matrix metalloproteinase-2 in human heart failure. Eur Heart J 2005;26:481-8. http://dx.doi.org/10.1093/eurheartj/ehi073.
- Jordan A, Roldan V, Garcia M, Monmeneu J, de Burgos FG, Lip GY, et al. Matrix metalloproteinase-1 and its inhibitor, TIMP-1, in systolic heart failure: relation to functional data and prognosis. J Intern Med 2007;262:385-92. http://dx.doi.org/10.1111/j.1365-2796.2007.01823.x.
- Milting H, Ellinghaus P, Seewald M, Cakar H, Bohms B, Kassner A, et al. Plasma biomarkers of myocardial fibrosis and remodeling in terminal heart failure patients supported by mechanical circulatory support devices. J Heart Lung Transplant 2008;27:589-96. http://dx.doi.org/10.1016/j.healun.2008.02.018.
- Frantz S, Stork S, Michels K, Eigenthaler M, Ertl G, Bauersachs J, et al. Tissue inhibitor of metalloproteinases levels in patients with chronic heart failure: an independent predictor of mortality. Eur J Heart Fail 2008;10:388-95. http://dx.doi.org/10.1016/j.ejheart.2008.02.015.
- Ohtsuka T, Nishimura K, Kurata A, Ogimoto A, Okayama H, Higaki J. Serum matrix metalloproteinase-3 as a novel marker for risk stratification of patients with nonischemic dilated cardiomyopathy. J Card Fail 2007;13:752-8. http://dx.doi.org/10.1016/j.cardfail.2007.06.730.
- Vorovich EE, Chuai S, Li M, Averna J, Marwin V, Wolfe D, et al. Comparison of matrix metalloproteinase 9 and brain natriuretic peptide as clinical biomarkers in chronic heart failure. Am Heart J 2008;155:992-7. http://dx.doi.org/10.1016/j.ahj.2008.01.007.
- Agostoni P, Banfi C. Matrix metalloproteinase and heart failure: is it time to move from research to clinical laboratories?. Eur Heart J 2007;28:659-60. http://dx.doi.org/10.1093/eurheartj/ehl574.
- Kristensen T, Oxvig C, Sand O, Moller NP, Sottrup-Jensen L. Amino acid sequence of human pregnancy-associated plasma protein-A derived from cloned cDNA. Biochemistry 1994;33:1592-8. http://dx.doi.org/10.1021/bi00172a040.
- Oxvig C, Sand O, Kristensen T, Kristensen L, Sottrup-Jensen L. Isolation and characterization of circulating complex between human pregnancy-associated plasma protein-A and proform of eosinophil major basic protein. Biochim Biophys Acta 1994;1201:415-23. http://dx.doi.org/10.1016/0304-4165(94)90071-X.
- Bayes-Genis A, Conover CA, Overgaard MT, Bailey KR, Christiansen M, Holmes DR, et al. Pregnancy-associated plasma protein A as a marker of acute coronary syndromes. N Engl J Med 2001;345:1022-9. http://dx.doi.org/10.1056/NEJMoa003147.
- Qin QP, Laitinen P, Majamaa-Voltti K, Eriksson S, Kumpula EK, Pettersson K. Release patterns of pregnancy associated plasma protein A (PAPP-A) in patients with acute coronary syndromes. Scand Cardiovasc J 2002;36:358-61. http://dx.doi.org/10.1080/140174302762659085.
- Lund J, Qin QP, Ilva T, Pettersson K, Voipio-Pulkki LM, Porela P, et al. Circulating pregnancy-associated plasma protein A predicts outcome in patients with acute coronary syndrome but no troponin I elevation. Circulation 2003;108:1924-6. http://dx.doi.org/10.1161/01.CIR.0000096054.18485.07.
- Heeschen C, Dimmeler S, Hamm CW, Fichtlscherer S, Simoons ML, Zeiher AM. Pregnancy-associated plasma protein-A levels in patients with acute coronary syndromes: comparison with markers of systemic inflammation, platelet activation, and myocardial necrosis. J Am Coll Cardiol 2005;45:229-37. http://dx.doi.org/10.1016/j.jacc.2004.09.060.
- Cosin-Sales J, Kaski JC, Christiansen M, Kaminski P, Oxvig C, Overgaard MT, et al. Relationship among pregnancy associated plasma protein-A levels, clinical characteristics, and coronary artery disease extent in patients with chronic stable angina pectoris. Eur Heart J 2005;26:2093-8. http://dx.doi.org/10.1093/eurheartj/ehi433.
- Mueller T, Dieplinger B, Poelz W, Haltmayer M. Increased pregnancy-associated plasma protein-A as a marker for peripheral atherosclerosis: results from the Linz Peripheral Arterial Disease Study. Clin Chem 2006;52:1096-103. http://dx.doi.org/10.1373/clinchem.2005.065763.
- Qin QP, Kokkala S, Lund J, Tamm N, Voipio-Pulkki LM, Pettersson K. Molecular distinction of circulating pregnancy-associated plasma protein A in myocardial infarction and pregnancy. Clin Chem 2005;51:75-83. http://dx.doi.org/10.1373/clinchem.2004.036467.
- Wittfooth S, Qin QP, Lund J, Tierala I, Pulkki K, Takalo H, et al. Immunofluorometric point-of-care assays for the detection of acute coronary syndrome-related noncomplexed pregnancy-associated plasma protein A. Clin Chem 2006;52:1794-801. http://dx.doi.org/10.1373/clinchem.2006.070607.
- Qin QP, Kokkala S, Lund J, Tamm N, Qin X, Lepantalo M, et al. Immunoassays developed for pregnancy-associated plasma protein-A (PAPP-A) in pregnancy may not recognize PAPP-A in acute coronary syndromes. Clin Chem 2006;52:398-404. http://dx.doi.org/10.1373/clinchem.2005.058396.
- Fredericks S, Bertomeu-Gonzalez V, Petrovic I, Holt DW, Kaski JC. Comment on immunoassays developed for pregnancy-associated plasma protein-A (PAPP-A) in pregnancy may not recognize PAPP-A in acute coronary syndromes. Clin Chem 2006;52:1619-20. http://dx.doi.org/10.1373/clinchem.2006.074138.
- Body R, Pemberton P, Ali F, McDowell G, Carley S, Smith A, et al. Low soluble P-selectin may facilitate early exclusion of acute myocardial infarction. Clin Chim Acta 2011;412:614-18. http://dx.doi.org/10.1016/j.cca.2010.12.016.
- Tertti R, Wittfooth S, Porela P, Airaksinen KE, Metsarinne K, Pettersson K. Intravenous administration of low molecular weight and unfractionated heparin elicits a rapid increase in serum pregnancy-associated plasma protein A. Clin Chem 2009;55:1214-17. http://dx.doi.org/10.1373/clinchem.2008.108738.
- Lund J, Wittfooth S, Qin QP, Ilva T, Porela P, Pulkki K, et al. Free vs total pregnancy-associated plasma protein A (PAPP-A) as a predictor of 1-year outcome in patients presenting with non-ST-elevation acute coronary syndrome. Clin Chem 2010;56:1158-65. http://dx.doi.org/10.1373/clinchem.2009.136960.
- Dominguez-Rodriguez A, Abreu-Gonzalez P, Garcia-Gonzalez M, Ferrer J, Vargas M. Circulating pregnancy-associated plasma protein A is not an early marker of acute myocardial infarction. Clin Biochem 2005;38:180-2. http://dx.doi.org/10.1016/j.clinbiochem.2004.10.015.
- Elesber AA, Lerman A, Denktas AE, Resch ZT, Jared BT, Schwartz RS, et al. Pregnancy associated plasma protein-A and risk stratification of patients presenting with chest pain in the emergency department. Int J Cardiol 2007;117:365-9. http://dx.doi.org/10.1016/j.ijcard.2006.05.021.
- Sanchis J, Bosch X, Bodi V, Bellera N, Nunez J, Benito B, et al. Combination of clinical risk profile, early exercise testing and circulating biomarkers for evaluation of patients with acute chest pain without ST-segment deviation or troponin elevation. Heart 2008;94:311-15. http://dx.doi.org/10.1136/hrt.2007.115626.
- Iversen KK, Teisner AS, Teisner B, Kliem A, Thanning P, Grande P, et al. Pregnancy associated plasma protein A, a novel, quick, and sensitive marker in ST-elevation myocardial infarction. Am J Cardiol 2008;101:1389-94. http://dx.doi.org/10.1016/j.amjcard.2008.01.015.
- Kavsak PA, Wang X, Henderson M, Ko DT, MacRae AR, Jaffe AS. PAPP-A as a marker of increased long-term risk in patients with chest pain. Clin Biochem 2009;42:1012-18. http://dx.doi.org/10.1016/j.clinbiochem.2009.03.015.
- Iversen KK, Dalsgaard M, Teisner AS, Schoos M, Teisner B, Nielsen H, et al. Usefulness of pregnancy-associated plasma protein A in patients with acute coronary syndrome. Am J Cardiol 2009;104:1465-71. http://dx.doi.org/10.1016/j.amjcard.2009.07.017.
- Iversen KK, Dalsgaard M, Teisner AS, Schoos M, Teisner B, Nielsen H, et al. Pregnancy-associated plasma protein-A, a marker for outcome in patients suspected for acute coronary syndrome. Clin Biochem 2010;43:851-7. http://dx.doi.org/10.1016/j.clinbiochem.2010.03.018.
- Khan DA, Sharif MS, Khan FA. Diagnostic performance of high-sensitivity troponin T, myeloperoxidase, and pregnancy-associated plasma protein A assays for triage of patients with acute myocardial infarction. Korean J Lab Med 2011;31:172-8. http://dx.doi.org/10.3343/kjlm.2011.31.3.172.
- Iversen K, Teisner A, Dalager S, Olsen KE, Floridon C, Teisner B. Pregnancy associated plasma protein-A (PAPP-A) is not a marker of the vulnerable atherosclerotic plaque. Clin Biochem 2011;44:312-18. http://dx.doi.org/10.1016/j.clinbiochem.2010.12.010.
- Sadler PJ, Tucker A, Viles JH. Involvement of a lysine residue in the N-terminal Ni2+ and Cu2+ binding site of serum albumins. Comparison with Co2+, Cd2+ and Al3+. Eur J Biochem 1994;220:193-200. http://dx.doi.org/10.1111/j.1432-1033.1994.tb18614.x.
- Chan B, Dodsworth N, Woodrow J, Tucker A, Harris R. Site-specific N-terminal auto-degradation of human serum albumin. Eur J Biochem 1995;227:524-8. http://dx.doi.org/10.1111/j.1432-1033.1995.tb20419.x.
- Marx G, Chevion M. Site-specific modification of albumin by free radicals. Reaction with copper(II) and ascorbate. Biochem J 1986;236:397-400.
- Bhagavan NV, Lai EM, Rios PA, Yang J, Ortega-Lopez AM, Shinoda H, et al. Evaluation of human serum albumin cobalt binding assay for the assessment of myocardial ischemia and myocardial infarction. Clin Chem 2003;49:581-5. http://dx.doi.org/10.1373/49.4.581.
- Mothes E, Faller P. Evidence that the principal CoII-binding site in human serum albumin is not at the N-terminus: implication on the albumin cobalt binding test for detecting myocardial ischemia. Biochemistry 2007;46:2267-74. http://dx.doi.org/10.1021/bi061783p.
- Peacock F, Morris DL, Anwaruddin S, Christenson RH, Collinson PO, Goodacre SW, et al. Meta-analysis of ischemia-modified albumin to rule out acute coronary syndromes in the emergency department. Am Heart J 2006;152:253-62. http://dx.doi.org/10.1016/j.ahj.2005.12.024.
- Collinson PO, Gaze DC. Ischaemia-modified albumin: clinical utility and pitfalls in measurement. J Clin Pathol 2008;61:1025-8. http://dx.doi.org/10.1136/jcp.2007.053363.
- WMA Declaration of Helsinki – Ethical Principles for Medical Research Involving Human Subjects n.d. www.wma.net/en/30publications/10policies/b3/.
- Guidelines for Good Clinical Practice in Clinical Trials 1998. www.mrc.ac.uk/Utilities/Documentrecord/index.htm?d=MRC002416.
- Collinson PO, Clifford-Mobley O, Gaze D, Boa F, Senior R. Assay imprecision and 99th-percentile reference value of a high-sensitivity cardiac troponin I assay. Clin Chem 2009;55:1433-4. http://dx.doi.org/10.1373/clinchem.2009.124925.
- Goodacre S, Thokala P, Carroll C, Stevens J, Leaviss J, Al Khalaf M, et al. Systematic review, meta-analysis and economic modelling of diagnostic strategies for suspected acute coronary syndrome. Health Technol Assess 2013;17.
- Office for National Statistics (ONS) . Interim Life Tables 2006–08 2008. www.ons.gov.uk/ons/dcp171780_234021.pdf.
- National Institute for Health and Care Excellence . CG95 Chest Pain of Recent Onset: NICE Guideline n.d. http://guidance.nice.org.uk/CG95/NICEGuidance/pdf/English.
- Mills NL, Churchhouse AM, Lee KK, Anand A, Gamble D, Shah AS, et al. Implementation of a sensitive troponin I assay and risk of recurrent myocardial infarction and death in patients with suspected acute coronary syndrome. JAMA 2011;305:1210-16. http://dx.doi.org/10.1001/jama.2011.338.
- Goodacre S, Bradburn M, Fitzgerald P, Cross E, Collinson P, Gray A, et al. The RATPAC (Randomised Assessment of Treatment using Panel Assay of Cardiac markers) trial: a randomised controlled trial of point-of-care cardiac markers in the emergency department. Health Technol Assess 2011;15.
- Tariff information: confirmation of payment by results (PbR) arrangements for 2009–10. London: Department of Health; 2010.
- Collinson PO, Gaze DC, Morris F, Morris B, Price A, Goodacre S. Comparison of biomarker strategies for rapid rule out of myocardial infarction in the emergency department using ACC/ESC diagnostic criteria. Ann Clin Biochem 2006;43:273-80. http://dx.doi.org/10.1258/000456306777695555.
- Eggers KM, Oldgren J, Nordenskjold A, Lindahl B. Diagnostic value of serial measurement of cardiac markers in patients with chest pain: limited value of adding myoglobin to troponin I for exclusion of myocardial infarction. Am Heart J 2004;148:574-81. http://dx.doi.org/10.1016/j.ahj.2004.04.030.
- Lee-Lewandrowski E, Januzzi JL, Grisson R, Mohammed AA, Lewandrowski G, Lewandrowski K. Evaluation of first-draw whole blood, point-of-care cardiac markers in the context of the universal definition of myocardial infarction: a comparison of a multimarker panel to troponin alone and to testing in the central laboratory. Arch Pathol Lab Med 2011;135:459-63.
- Straface AL, Myers JH, Kirchick HJ, Blick KE. A rapid point-of-care cardiac marker testing strategy facilitates the rapid diagnosis and management of chest pain patients in the emergency department. Am J Clin Pathol 2008;129:788-95. http://dx.doi.org/10.1309/9GGNMURLJWJD88W3.
- Keller T, Zeller T, Peetz D, Tzikas S, Roth A, Czyz E, et al. Sensitive troponin I assay in early diagnosis of acute myocardial infarction. N Engl J Med 2009;361:868-77. http://dx.doi.org/10.1056/NEJMoa0903515.
- Than M, Cullen L, Reid CM, Lim SH, Aldous S, Ardagh MW, et al. A 2-h diagnostic protocol to assess patients with chest pain symptoms in the Asia–Pacific region (ASPECT): a prospective observational validation study. Lancet 2011;377:1077-84. http://dx.doi.org/10.1016/S0140-6736(11)60310-3.
- Aldous S, Pemberton C, Troughton R, Than M, Mark RA. Heart fatty acid binding protein and myoglobin do not improve early rule out of acute myocardial infarction when highly sensitive troponin assays are used. Resuscitation 2012;83:e27-e28.
- Reichlin T, Hochholzer W, Bassetti S, Steuer S, Stelzig C, Hartwiger S, et al. Early diagnosis of myocardial infarction with sensitive cardiac troponin assays. N Engl J Med 2009;361:858-67. http://dx.doi.org/10.1056/NEJMoa0900428.
- Keller T, Zeller T, Ojeda F, Tzikas S, Lillpopp L, Sinning C, et al. Serial changes in highly sensitive troponin I assay and early diagnosis of myocardial infarction. JAMA 2011;306:2684-93. http://dx.doi.org/10.1001/jama.2011.1896.
- Reiter M, Twerenbold R, Reichlin T, Haaf P, Peter F, Meissner J, et al. Early diagnosis of acute myocardial infarction in the elderly using more sensitive cardiac troponin assays. Eur Heart J 2011;32:1379-89. http://dx.doi.org/10.1093/eurheartj/ehr033.
- Weber M, Bazzino O, Navarro Estrada JL, de Miguel R, Salzberg S, Fuselli JJ, et al. Improved diagnostic and prognostic performance of a new high-sensitive troponin T assay in patients with acute coronary syndrome. Am Heart J 2011;162:81-8. http://dx.doi.org/10.1016/j.ahj.2011.04.007.
- Melki D, Lind S, Agewall S, Jernberg T. Diagnostic value of high sensitive troponin T in chest pain patients with no persistent ST-elevations. Scand Cardiovasc J 2011;45:198-204. http://dx.doi.org/10.3109/14017431.2011.565792.
- Freund Y, Chenevier-Gobeaux C, Bonnet P, Claessens YE, Allo JC, Doumenc B, et al. High-sensitive versus conventional troponin in the emergency department for the diagnosis of acute myocardial infarction. Crit Care 2011;15. http://dx.doi.org/10.1186/cc10270.
- Goodacre SW, Bradburn M, Cross E, Collinson P, Gray A, Hall AS. The Randomised Assessment of Treatment using Panel Assay of Cardiac Markers (RATPAC) trial: a randomised controlled trial of point-of-care cardiac markers in the emergency department. Heart 2011;97:190-6. http://dx.doi.org/10.1136/hrt.2010.203166.
- Reichlin T, Irfan A, Twerenbold R, Reiter M, Hochholzer W, Burkhalter H, et al. Utility of absolute and relative changes in cardiac troponin concentrations in the early diagnosis of acute myocardial infarction. Circulation 2011;129:136-45. http://dx.doi.org/10.1161/CIRCULATIONAHA.111.023937.
- Giannitsis E, Becker M, Kurz K, Hess G, Zdunek D, Katus HA. High-sensitivity cardiac troponin T for early prediction of evolving non-ST-segment elevation myocardial infarction in patients with suspected acute coronary syndrome and negative troponin results on admission. Clin Chem 2010;56:642-50. http://dx.doi.org/10.1373/clinchem.2009.134460.
- Morgenthaler NG, Struck J, Alonso C, Bergmann A. Assay for the measurement of copeptin, a stable peptide derived from the precursor of vasopressin. Clin Chem 2006;52:112-19. http://dx.doi.org/10.1373/clinchem.2005.060038.
- Keller T, Tzikas S, Zeller T, Czyz E, Lillpopp L, Ojeda FM, et al. Copeptin improves early diagnosis of acute myocardial infarction. J Am Coll Cardiol 2010;55:2096-106. http://dx.doi.org/10.1016/j.jacc.2010.01.029.
- Karakas M, Januzzi JL, Meyer J, Lee H, Schlett CL, Truong QA, et al. Copeptin does not add diagnostic information to high-sensitivity troponin T in low- to intermediate-risk patients with acute chest pain: results from the rule out myocardial infarction by computed tomography (ROMICAT) study. Clin Chem 2011;57:1137-45. http://dx.doi.org/10.1373/clinchem.2010.160192.
- Giannitsis E, Kehayova T, Vafaie M, Katus HA. Combined testing of high-sensitivity troponin T and copeptin on presentation at prespecified cutoffs improves rapid rule-out of non-ST-segment elevation myocardial infarction. Clin Chem 2011;57:1452-5. http://dx.doi.org/10.1373/clinchem.2010.161265.
- Maisel A, Xue Y, Shah K, Mueller C, Nowak R, Peacock WF, et al. Increased 90-day mortality in patients with acute heart failure with elevated copeptin: secondary results from the Biomarkers in Acute Heart Failure (BACH) study. Circ Heart Fail 2011;4:613-20. http://dx.doi.org/10.1161/CIRCHEARTFAILURE.110.960096.
- Brown A, George J, Murphy MJ, Struthers A. Could BNP screening of acute chest pain cases lead to safe earlier discharge of patients with non-cardiac causes? A pilot study. QJM 2007;100:755-61. http://dx.doi.org/10.1093/qjmed/hcm116.
- Brown AM, Sease KL, Robey JL, Shofer FS, Hollander JE. The impact of B-type natriuretic peptide in addition to troponin I, creatine kinase-MB, and myoglobin on the risk stratification of emergency department chest pain patients with potential acute coronary syndrome. Ann Emerg Med 2007;49:153-63. http://dx.doi.org/10.1016/j.annemergmed.2006.08.024.
- Connolly SB, Collier T, Khugputh R, Tataree D, Kyereme K, Merritt S, et al. Brain natriuretic peptide testing for angina in a rapid-access chest pain clinic. QJM 2007;100:779-83. http://dx.doi.org/10.1093/qjmed/hcm098.
- Wang TJ, Larson MG, Levy D, Benjamin EJ, Leip EP, Omland T, et al. Plasma natriuretic peptide levels and the risk of cardiovascular events and death. N Engl J Med 2004;350:655-63. http://dx.doi.org/10.1056/NEJMoa031994.
- Kragelund C, Gronning B, Kober L, Hildebrandt P, Steffensen R. N-terminal pro-B-type natriuretic peptide and long-term mortality in stable coronary heart disease. N Engl J Med 2005;352:666-75. http://dx.doi.org/10.1056/NEJMoa042330.
- de Lemos JA, Morrow DA, Bentley JH, Omland T, Sabatine MS, McCabe CH, et al. The prognostic value of B-type natriuretic peptide in patients with acute coronary syndromes. N Engl J Med 2001;345:1014-21. http://dx.doi.org/10.1056/NEJMoa011053.
- Weber M, Bazzino O, Navarro Estrada JL, Fuselli JJ, Botto F, Perez de Arenaza D, et al. N-terminal B-type natriuretic peptide assessment provides incremental prognostic information in patients with acute coronary syndromes and normal troponin T values upon admission. J Am Coll Cardiol 2008;51:1188-95. http://dx.doi.org/10.1016/j.jacc.2007.11.054.
- Brugger-Andersen T, Ponitz V, Staines H, Pritchard D, Grundt H, Nilsen DW. B-type natriuretic peptide is a long-term predictor of all-cause mortality, whereas high-sensitive C-reactive protein predicts recurrent short-term troponin T positive cardiac events in chest pain patients: a prognostic study. BMC Cardiovasc Disord 2008;8. http://dx.doi.org/10.1186/1471-2261-8-34.
- Bassan R, Tura BR, Maisel AS. B-type natriuretic peptide: a strong predictor of early and late mortality in patients with acute chest pain without ST-segment elevation in the emergency department. Coron Artery Dis 2009;20:143-9. http://dx.doi.org/10.1097/MCA.0b013e3283292ac6.
- van der Zee PM, Cornel JH, Bholasingh R, Fischer JC, van Straalen JP, de Winter RJ. N-terminal pro B-type natriuretic peptide identifies patients with chest pain at high long-term cardiovascular risk. Am J Med 2011;124:961-9. http://dx.doi.org/10.1016/j.amjmed.2011.05.026.
- Haaf P, Reichlin T, Corson N, Twerenbold R, Reiter M, Steuer S, et al. B-type natriuretic peptide in the early diagnosis and risk stratification of acute chest pain. Am J Med 2011;124:444-52. http://dx.doi.org/10.1016/j.amjmed.2010.11.012.
- Schaub N, Reichlin T, Twerenbold R, Reiter M, Steuer S, Bassetti S, et al. Growth differentiation factor-15 in the early diagnosis and risk stratification of patients with acute chest pain. Clin Chem 2012;58:441-9. http://dx.doi.org/10.1373/clinchem.2011.173310.
- James SK, Lindahl B, Siegbahn A, Stridsberg M, Venge P, Armstrong P, et al. N-terminal pro-brain natriuretic peptide and other risk markers for the separate prediction of mortality and subsequent myocardial infarction in patients with unstable coronary artery disease: a Global Utilization of Strategies To Open occluded arteries (GUSTO)-IV substudy. Circulation 2003;108:275-81. http://dx.doi.org/10.1161/01.CIR.0000079170.10579.DC.
- Hamm CW, Bassand JP, Agewall S, Bax J, Boersma E, Bueno H, et al. ESC guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: The Task Force for the management of acute coronary syndromes (ACS) in patients presenting without persistent ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J 2011;32:2999-3054. http://dx.doi.org/10.1093/eurheartj/ehr236.
Appendix 1 Detailed results of missed acute myocardial infarction cases by troponin measurement method
Detailed results of missed acute myocardial infarction cases by troponin measurement method (PDF download)
Appendix 2 Detailed results of troponin elevation in non-acute coronary syndrome patients
Detailed results of troponin elevation in non-acute coronary syndrome patients (PDF download)
Appendix 3 Probabilistic sensitivity analysis results for troponin T strategies
Probabilistic sensitivity analysis results for troponin T strategies (PDF download)
Appendix 4 Probabilistic sensitivity analysis results for troponin I strategies
Probabilistic sensitivity analysis results for troponin I strategies (PDF download)
Appendix 5 Study protocol
Glossary
- Area under the curve
- The calculated integral underneath a plot of test sensitivity against (1 – test specificity).
- Biomarker
- A metabolic intermediary molecule, signal molecule, enzyme or protein that significantly changes in response to a disease state and can be used to monitor onset or progress of disease, or predict outcome in response to treatment.
- Cost-effectiveness acceptability curve
- A way of illustrating cost-effectiveness results by plotting the probability that the intervention is cost-effective (y-axis) against the maximum that society is willing to pay for an improvement in health (x-axis).
- Cost-effectiveness plane
- A way of illustrating cost-effectiveness results by plotting the mean incremental cost and effectiveness on a four-quadrant graph. Interventions that are more costly and more effective fall in the north-east quadrant.
- Dalton
- Unit of atomic mass.
- False-negative
- A test result erroneously indicating that a patient with a condition does not have that condition.
- False-positive
- A test result erroneously indicating that a patient without a condition does have that condition.
- Incremental cost-effectiveness ratio
- The difference in costs between one intervention and an alternative, divided by the difference in outcomes.
- Likelihood ratio
- Describes how many times more likely a person with a disease is to receive a particular test result than a person without the disease. A likelihood ratio of a positive test result is usually a number > 1; a likelihood ratio of a negative test result usually lies between 0 and 1.
- Quality-adjusted life-year
- A measure of the benefit of health care combining the impact of both expected length of life and quality of life.
- Receiver operating characteristic curve
- Graphical representation of the relationship between ‘true-positive fraction’ (sensitivity) and ‘false-positive fraction’ (1 – specificity). It displays the trade-offs between sensitivity and specificity as a result of varying the cut-off value for positivity in case of a continuous test result.
- Reference standard
- Established test(s) against which the accuracy of a new test for detecting a particular condition can be evaluated.
- Sensitivity (true-positive rate)
- The proportion of individuals with the target condition in a population who are correctly identified by a diagnostic test.
- Specificity (true-negative rate)
- The proportion of individuals free of the target condition in a population who are correctly identified by a diagnostic test.
- Test accuracy
- The proportion of test results that are correctly identified by the test.
- True-negative
- A test result correctly identifying that a patient without a condition does not have that condition.
- True-positive
- A test result correctly identifying that a patient with a condition has that condition.
List of abbreviations
- ACS
- acute coronary syndrome
- AM
- adrenomedullin
- AMI
- acute myocardial infarction
- ANP
- atrial natriuretic peptide
- AUC
- area under the curve
- AVP
- arginine vasopressin
- BNP
- B-type natriuretic peptide
- CK
- creatine kinase
- CK-MB
- creatine kinase MB isoenzyme
- CRP
- C-reactive protein
- cTnI
- cardiac troponin I
- cTnT
- cardiac troponin T
- CTU
- Clinical Trials Unit
- CV
- coefficient of variation
- ECG
- electrocardiogram
- ED
- emergency department
- EDTA
- ethylenediaminetetraacetic acid
- FABP
- fatty acid-binding protein
- GDF15
- growth differentiation factor 15
- GRACE
- Global Registry of Acute Coronary Events
- H-FABP
- heart-type fatty acid-binding protein
- HTA
- Health Technology Assessment
- ICER
- incremental cost-effectiveness ratio
- IL
- interleukin
- IL-1
- interleukin 1
- IL-1β
- interleukin 1 beta
- IL-33
- interleukin 33
- IL-6
- interleukin 6
- IL-6R
- interleukin-6 receptor
- IMA
- ischaemia-modified albumin
- IQR
- interquartile range
- MACE
- major adverse cardiac event
- MMP
- matrix metalloproteinase
- MPO
- myeloperoxidase
- NICE
- National Institute for Health and Care Excellence
- NSTEMI
- non-ST-segment elevation myocardial infarction
- NTproBNP
- N-terminal pro-B-type natriuretic peptide
- NYHA
- New York Heart Association
- PAPP-A
- pregnancy-associated plasma protein A
- QALY
- quality-adjusted life-year
- RATPAC
- Randomised Assessment of Treatment using Panel Assay of Cardiac markers
- RATPAC-CBE
- Randomised Assessment of Treatment using Panel Assay of Cardiac markers – Contemporary Biomarker Evaluation
- RCV
- relative change value
- ROC
- receiver operating characteristic
- sIL-6R
- soluble interleukin-6 receptor
- STEMI
- ST-segment elevation myocardial infarction
- TIMP
- tissue inhibitor of metalloproteinase
- TnC
- troponin C
- TnI
- troponin I
- TnT
- troponin T
- WHO
- World Health Organization
All abbreviations that have been used in this report are listed here unless the abbreviation is well known (e.g. NHS), or it has been used only once, or it is a non-standard abbreviation used only in figures/tables/appendices in which case the abbreviation is defined in the figure legend or at the end of the table.