
Notes
Article history
The research reported in this issue of the journal was commissioned and funded by the HTA programme on behalf of NICE as project number 10/08/01. The protocol was agreed in January 2011. The assessment report began editorial review in April 2012 and was accepted for publication in November 2012. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of clinical advisors/peer reviewers
Dr Keith Brownlee attended a Novartis Pharmaceuticals Advisory board on future developments in cystic fibrosis (CF) in 2010, and received a fee and travel expenses from Forest Laboratories for a lecture given at the East Anglia Cystic Fibrosis Club in 2010. Dr Diana Bilton is the chairperson of the UK CF Registry Committee and is involved in assisting the design of registry surveillance of patients receiving Colobreathe. Dr Diana Bilton is also chief investigator on the Insmed trial of once-daily nebulised Arikace for CF and was chief investigator of a trial of aztreonam compared with tobramycin. Dr Diana Bilton is also chief investigator of a planned study on the effects of TOBI Podhaler on treatment burden, quality of life and adherence.
Note
This monograph is based on the Technology Assessment Report produced for NICE. The full report contained a considerable number of data that were deemed commercial-in-confidence and/or academic-in-confidence. The full report was used by the Appraisal Committee at NICE in its deliberations. The full report with each piece of commercial-in-confidence and/or academic-in-confidence data removed and replaced by the statement ‘commercial-in-confidence and/or academic-in-confidence information (or data) removed’ is available on the NICE website: www.nice.org.uk.
The present monograph presents as full a version of the report as is possible while retaining readability, but some sections, sentences, tables and figures have been removed. Readers should bear in mind that the discussion, conclusions and implications for practice and research are based on all the data considered in the original full NICE report.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2013. This work was produced by Tappenden et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Background
Description of the health problem
Brief statement of the health problem
Cystic fibrosis (CF) is an inherited disease that shortens life expectancy and greatly reduces the health-related quality of life (HRQoL) of patients. CF is characterised by abnormal ion movement across transporting epithelia. This leads to the production of thick sticky mucus in the lungs, pancreas, liver, intestine and reproductive tract, and an increase in the salt content in sweat. People with CF have problems with digestion, which can affect growth and body mass index (BMI), and are prone to lung infections by a range of pathogens, including Staphylococcus aureus, Pseudomonas aeruginosa and Burkholderia cepacia. This is thought to be because the thick mucus makes it difficult for the body to clear inhaled bacteria, and because people with CF have an increased airway inflammatory response to pathogens. 1 Although both digestive problems and lung infections contribute to morbidity and mortality, respiratory tract infections with P. aeruginosa have been shown to be a major risk factor contributing to mortality. 2
In the early stages of disease, management aims to identify and vigorously treat infection,3 and thereby limit structural changes that may predispose a patient to chronic infection with P. aeruginosa. If bacterial infection is not successfully prevented or treated, a chronic infection/colonisation can develop, whereby bacterial microenvironments, known as ‘biofilms’, form within the bronchial tree. Biofilms are difficult for immune cells and antibiotics to penetrate, and once established, are associated with clinical deterioration and ultimately increased mortality. 2 Treatment of chronic infection typically involves regular use of nebulised antibiotics, such as tobramycin [Bramitob® (Chiesi) or TOBI® (Novartis Pharmaceuticals)] and colistimethate sodium [Promixin® (Profile Pharma) or Colistin® (Forest Laboratories)], to suppress bacterial growth and prevent flare-ups (known as exacerbations), and to maintain lung function and quality of life. Treatment can be time-consuming for patients, with administration of nebulised antibiotics taking up to 1 hour per day during good health, and longer during periods of ill health. 1 Newer nebulisers, such as the eFlow® rapid nebuliser (PARI Medical, West Byfleet, Surrey, UK) or the I-neb™ (Philips Respironics, Murrysville, PA, USA) adaptive aerosol delivery (AAD) system, may allow for more rapid administration of treatment. Pulmonary exacerbations may have a substantial negative impact on a patient’s quality of life4 and are usually treated using intravenous (i.v.) antibiotics, either in hospital or at home, or in a combination of these settings. 5
Aetiology and pathology
Cystic fibrosis is an autosomal recessive disorder, for which both copies of the gene that codes for a protein called the cystic fibrosis transmembrane conductance regulator (CFTR) contain a mutation. Over 1600 different mutations of the gene have been identified, causing different changes to the function of the protein, and hence different severities of disease in the individual. The most common mutation is the deletion of phenylalanine at codon 508. This deletion was present in an estimated 91.3% of the mutant alleles in the UK in 2010. 6
Cystic fibrosis transmembrane conductance regulator is a large (∼170-kDa) multidomain protein belonging to the adenosine triphosphate (ATP)-binding cassette family of membrane transporters. 7 It is located in the cell membrane of various cells in the body, including epithelial cells in the respiratory tract, pancreas, liver, intestine and reproductive tract, where it regulates fluid secretion. CFTR acts as an ion channel that utilises the energy released by the binding and hydrolysis of ATP to open. When open, chloride ions pass across the cell membrane by diffusion in the direction of their electrochemical gradient. 8 When functional, this promotes efflux of chloride ions from the cell into the extracellular fluid. Sodium ions and water follow by a paracellular route (between cells rather than through cells) and hence the volume of liquid on the epithelial surface is regulated. In CF, impaired CFTR function is most commonly thought to lead to a decrease in the surface liquid volume of epithelial cells (although there are theories that also consider reduced antibacterial properties of mucus and increased mucin secretion as putative mediators of the characteristics of CF). In the epithelia of the airways, these changes result in a decrease in mucociliary clearance in the respiratory tract, which is the body’s primary defence against invading pathogens. People with CF are more prone to respiratory infections as a result. In addition, people with CF have an excessive inflammatory response. The aetiology of this is unknown,9 but along with the damage caused by respiratory infections it leads to bronchiectasis and obstructive pulmonary disease, the primary causes of death among people with CF.
Expression of the CFTR gene in the body is widespread and symptoms are not confined to lung disease. Reproductive function in both males and females may be disrupted (although there is conflicting evidence in women). Exocrine tissues in the pancreas are also affected, where abnormal mucus can block and damage pancreatic ducts. This process starts in utero and causes a decrease in the secretion of digestive juices, which contain the enzymes, bicarbonate and water that are essential to digestion, which, in turn, leads to malabsorption of ingested food and malnutrition. Ultimately, damage to the pancreatic tissue can also lead to destruction of the pancreatic β cells in the islets of Langerhans. 10 These endocrine cells normally secrete insulin into the bloodstream, and their absence leads to diabetes mellitus. It is thought that this has a negative impact on lung disease, as lung function is affected by maintaining a normal body weight. This is also associated with a negative impact on survival. Insulin replacement therapy improves both lung function and body mass. 10
Children with CF are born without lung infection, but from the moment they are born they are exposed to pathogens and they become infected over time. Common infections include S. aureus, Haemophilus influenzae, P. aeruginosa and B. cepacia complex. P. aeruginosa is the most prevalent infection, with 37.5% of patients of all ages having a chronic infection in 2010. 6 Between the ages 20 and 49 years, between 55% and 65% patients have chronic P. aeruginosa infection. P. aeruginosa infection starts as an intermittent infection with non-mucoid variants of the bacterium. Studies suggest that this phenotype can be eradicated by antipseudomonal antibiotics,11,12 and current practice is to treat all incidents of infection energetically, with the aim of clearing the infection from the respiratory tract using oral or nebulised antibiotics (or both, depending on the UK centre). 3 However, over time, intermittent infections develop into colonisation. Chronic infection is associated with increased mortality and morbidity. 13 The environmental pressures imposed on the bacteria by the conditions within the CF lung are thought to drive the conversion of the non-mucoid phenotype to the mucoid phenotype, which secretes large quantities of alginate exopolysaccharide and forms biofilms. Biofilms are aggregates of cells set in an extracellular matrix composed largely of the alginate secreted by the mucoid phenotype. It is hypothesised that these slippery biofilms grow in microaerophilic or anaerobic environments created by the thick mucus that is characteristic of CF. Other factors present in CF lungs, such as actin, deoxyribonucleic acid (DNA) and decreased bacteriocidal secretions, are also thought to contribute to the formation of the biofilms. 14 The biofilms are very resistant to antibiotic treatment,15 and once a chronic mucoid infection has been established then eradication is not possible. Acquisition of this phenotype is again associated with the worsening of symptoms16 and a considerably worse prognosis. 17
Once bacterial colonisation is established, patients experience a gradual deterioration in lung function as lung tissue is damaged by the infection, which ultimately results in atelectasis (diminished lung volume), severe bronchiectasis, respiratory failure and death. 1 Patients experience increasingly frequent respiratory exacerbations, which severely affect quality of life and are usually treated with i.v. antibiotics and may require admission to hospital. Episodes of haemoptysis and pneumothorax may also occur. Historically, there have been differences in the diagnostic criteria for determining an exacerbation. These events have usually been characterised by an acute worsening of symptoms, such as increased cough, increased expectoration, decreased tolerance to physical activity, loss of weight or appetite and a deterioration in respiratory function. A marked increase in airway bacterial load [in colony-forming units (CFUs)/ml] has been cited as a criterion that may indicate an exacerbation,18 but is subject to some contention. Although forced expiratory volume in first second percentage predicted (FEV1%) usually improves with treatment, Wagener et al. 19 demonstrated a progressive decrease in the best FEV1% recorded in the 180 days after the exacerbation compared with the best FEV1% recorded in the year prior to the exacerbation. The authors interpret this as being suggestive of an overall decline in FEV1% associated with each exacerbation. In 2011 the EuroCare CF Working Group published a proposed definition for exacerbations. 20
Patients in the end stages of lung disease may be assessed for lung or lung and heart transplant, and may be added to the transplant waiting list. Owing to the systemic nature of the disease, transplants for other organs (e.g. liver, kidney) may also be necessary. In the UK, in 2010, 169 patients were evaluated and 82 accepted on to the transplant list. 6 Kidney transplants are sometimes needed as a consequence of the toxicity of the high-dose aminoglycoside antibiotics that are used to treat exacerbations. Once a lung transplant has taken place, the risk of death for people with CF is the same as the risk of death for all lung transplants. However, not all patients are fortunate enough to find an appropriate donor in time, and only 27 patients within the UK CF Registry eventually received a bilateral lung or heart and lung transplant in the UK in 2010. 6
Prognosis
The impact of CF on survival is substantial. In 2010, 103 deaths were recorded in UK patients with CF, details of whom were held within the CF Registry; the median age at death was 29 years (minimum = 0 years; maximum = 61 years). 6 Figure 1 shows the age distribution of deaths in patients with CF, based on 2010 data.
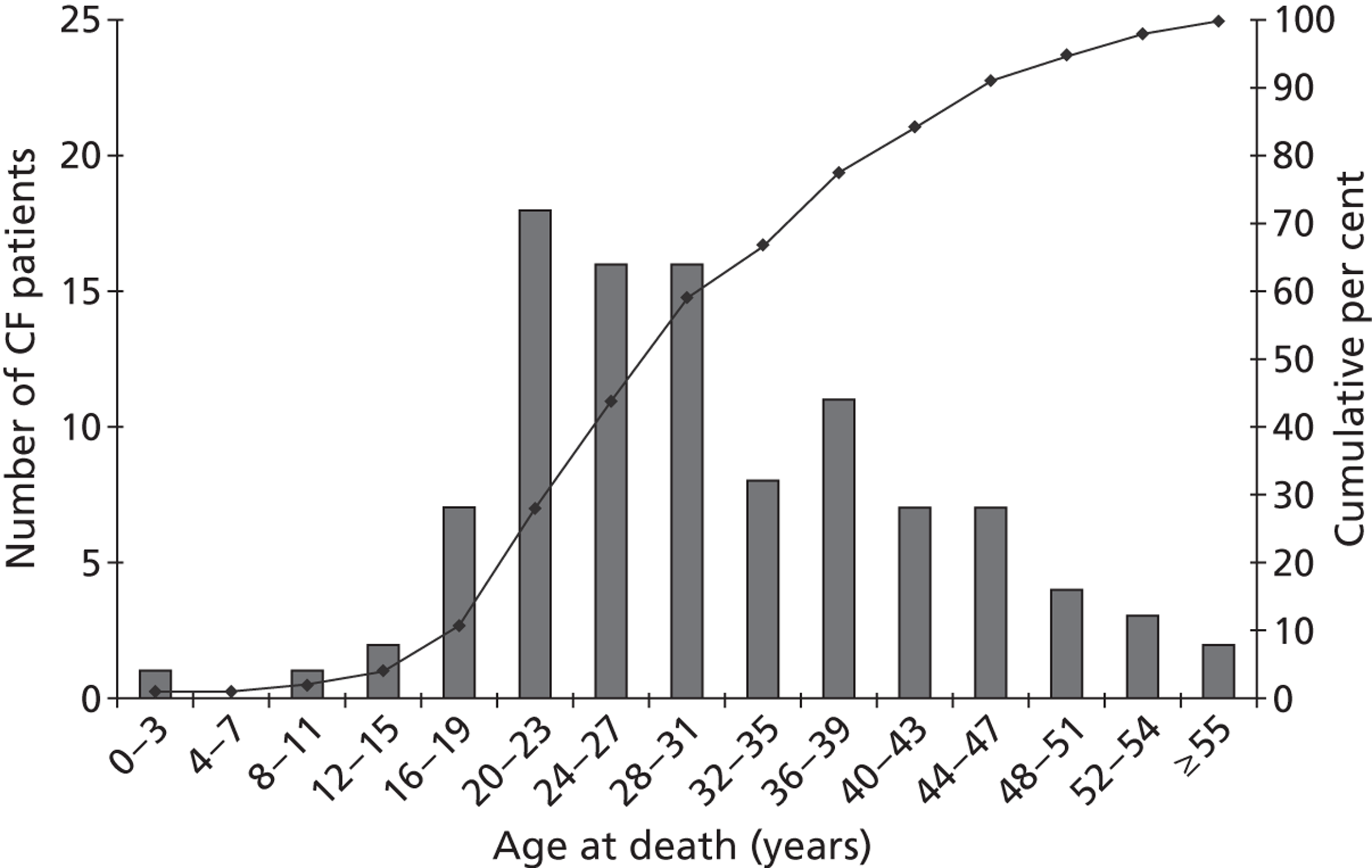
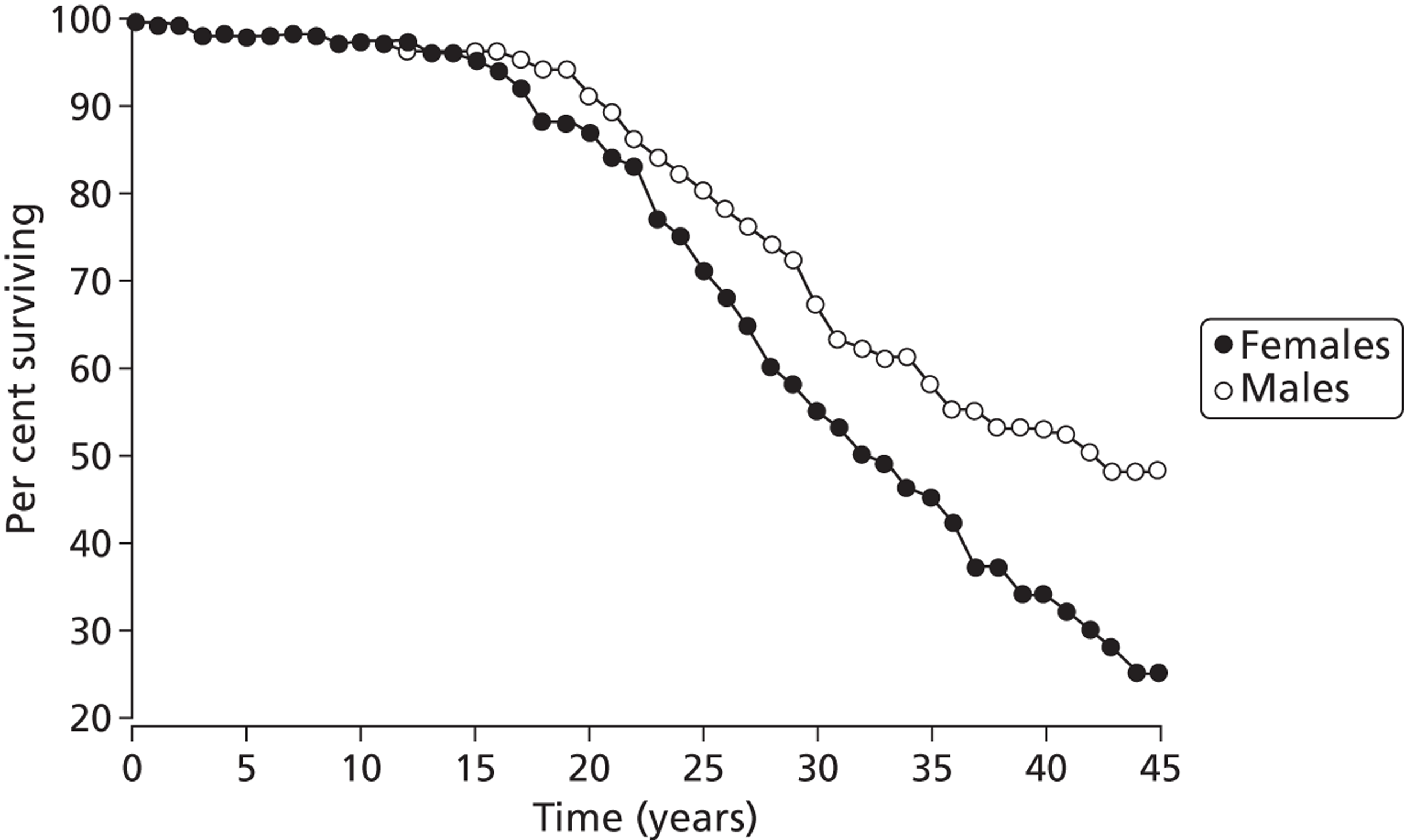
Although more people with the condition are living longer than in previous decades, only half of those patients living with CF are likely to live beyond their late thirties. Figure 2 shows recent estimates of survival for males and females with CF, based on a large UK-based cohort study. 22 Similar actuarial survival estimates are not currently available from the CF Registry.
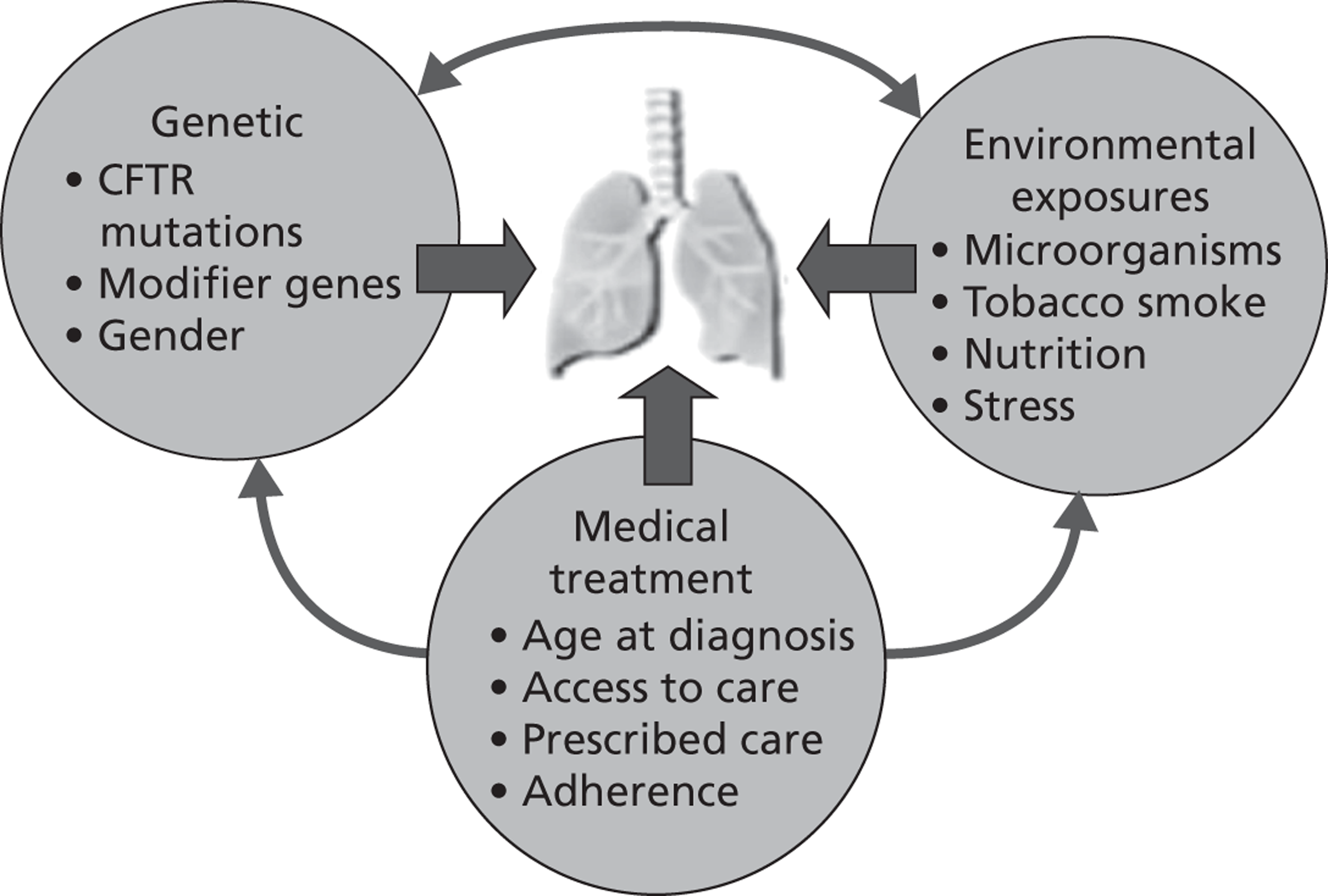
It has been suggested that a number of other factors, such as genetics, medical treatment and environmental exposures, may interdependently influence prognosis, as illustrated in Figure 3.
Epidemiology: incidence and prevalence
According to 2010 estimates from the Cystic Fibrosis Trust, over 9300 people in the UK have CF. Complete data on 7937 of these individuals are available from the CF Registry for 2010. The majority of CF cases are diagnosed by neonatal screening or during early infancy. Around 55.5% of those included in the registry are > 16 years of age and the incidence is spread evenly between males and females. For UK patients registered as having CF, approximately 82.2% are located in England, 3.9% in Wales, 4.7% in Scotland and 9.3% in Northern Ireland (Table 1).
| Location | No. | No. of patients registered at paediatric clinics/centres | Percentage | No. of patients registered at adult clinics/centres | Percentage |
|---|---|---|---|---|---|
| UK | 9336 | 4475 | 47.93 | 4861 | 52.07 |
| England | 7640 | 3627 | 47.47 | 4013 | 52.53 |
| Wales | 366 | 217 | 59.29 | 149 | 40.71 |
| Scotland | 883 | 407 | 46.09 | 476 | 53.91 |
| Northern Ireland | 447 | 224 | 50.11 | 223 | 49.89 |
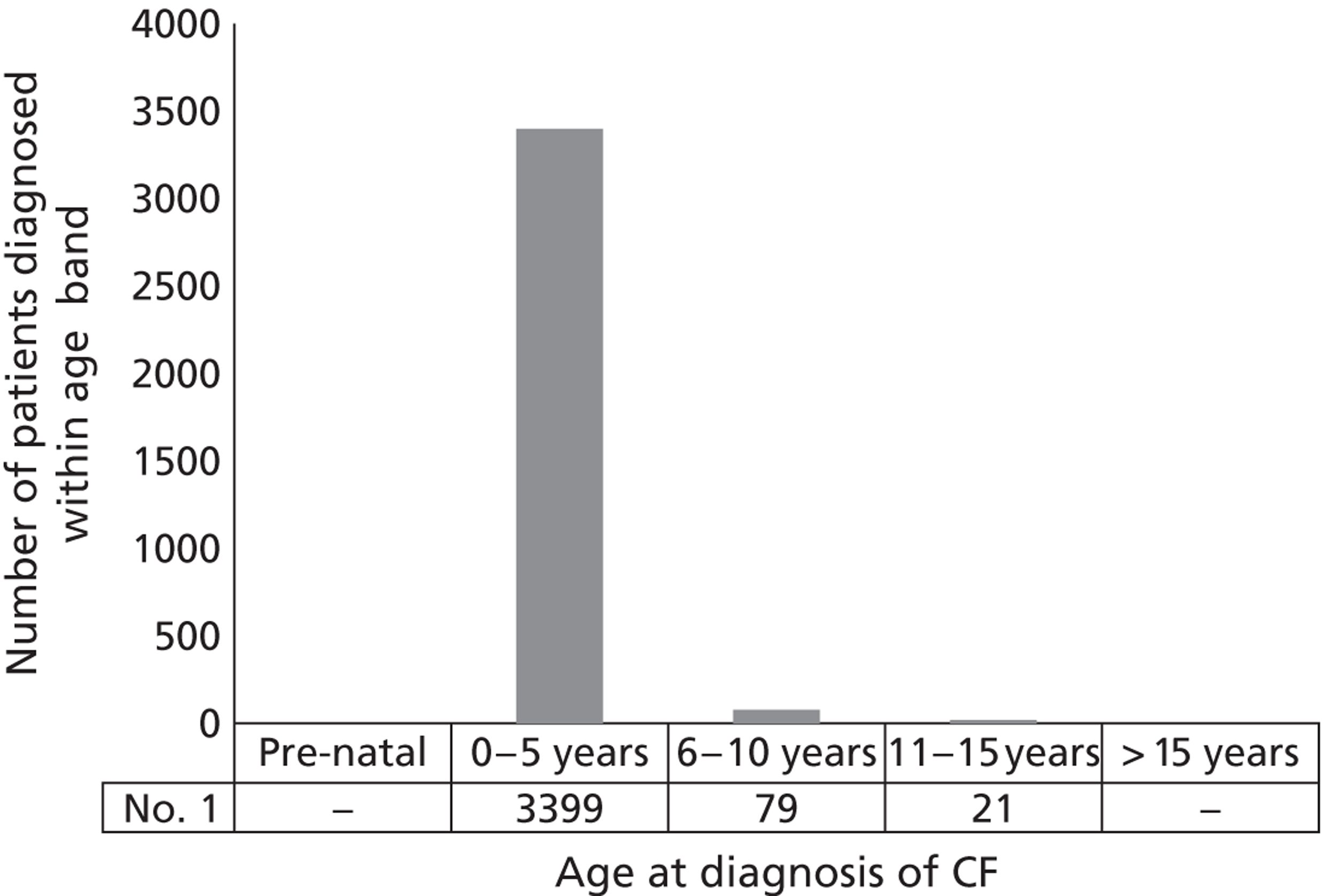
According to the Cystic Fibrosis Trust, approximately five babies are born with CF each week. During the period 2007–10, between 235 and 301 new cases of CF were registered each year. Around 1 in 25 people are thought to be carriers of the CF gene, although this incidence of disease estimate is limited to white people living predominantly in Europe and America. Incidence in other races is lower but CF is increasingly being reported. 1 Figure 4 shows the age distribution of those patients for whom data are available within the 2010 CF Registry report.
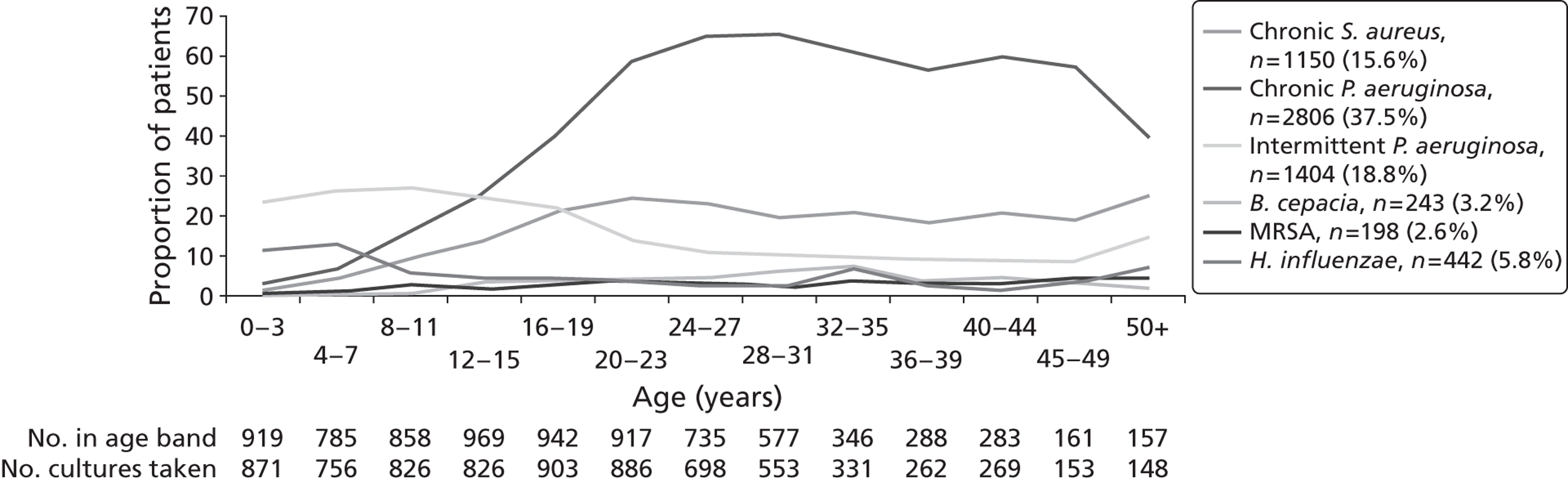
The prevalence of lung infection among the broader CF population is high. Around 37.5% of people living with CF are chronically infected with P. aeruginosa. Age-specific prevalence rates of Pseudomonas infection are shown in Figure 5. The prevalence of P. aeruginosa infection increases markedly with increasing age, up to around 25–30 years of age, with slightly lower rates in older age groups. These lower rates may be due to these patients having less severe mutations, which make them less likely to develop colonisation.
FIGURE 5.
Prevalence of CF lung infections by age group in 2010. 6 MRSA, methicillin-resistant Staphylococcus aureus.
Impact of health problem
Cystic fibrosis has a significant impact on the survival and quality of life of patients. The disease also impacts on carers and requires a considerable commitment of health-care resources. In 2003, an analysis of data from 196 adult patients with CF attending the Manchester Cystic Fibrosis Unit reported that 113 (57.6%) patients were attending work or study; however, 1799 days were lost as a result of sickness. 5 More recently, based on an analysis of complete data records from patients aged > 16 years, the Cystic Fibrosis Trust reported that 69.7% of patients are in work or studying. Although 18.5% were reported to be unemployed, only 5.6% of patients classed themselves as ‘disabled’. 6 Patients require monitoring and treatment by the NHS for the duration of their lives. Additionally, two young lives a week are lost to CF, which represents a significant impact on the families of CF sufferers. As the UK’s most common life-threatening inherited disease, CF continues to present a considerable cost burden for the NHS.
Measuring disease in cystic fibrosis
Cystic fibrosis can be broadly categorised into early stage, intermediate stage and end stage with complications. Patients with early-stage CF are characterised by the absence of infection with P. aeruginosa, or intermittent infection, which can usually be eradicated using antibiotics. Patients with intermediate-stage disease [forced expiratory volume in first second (FEV1)] ∼30–70% predicted are chronically infected with P. aeruginosa or other less common organisms, whereas patients with end-stage disease (FEV1 < 30% predicted) suffer from severe haemoptysis, pneumothorax and respiratory failure. 1 Patients have routine check-ups to monitor the status and stage of their disease. Measurements during these check-ups usually include assessment of bacterial infection and measurement of lung function, both of which contribute to treatment planning and prognosis. In some centres sputum tests are not performed routinely.
In the context of clinical trials, the Committee for Medicinal Products for Human Use (CHMP) research guidelines for the development of new medicinal products for CF18 recommend additional disease measures to gauge the efficacy of interventions. These include measuring rates of microbial resistance, the number of acute exacerbations and the patient’s HRQoL. FEV1 is recommended as the primary end point for studies investigating CF treatments; however, a microbiological primary end point is also considered necessary for confirmatory trials. 18
Measuring microbiological indicators of infection
The presence of a microbial infection is ascertained using sputum colony density. This measurement is also recommended as a secondary end point for clinical trials assessing safety and/or efficacy of antipseudomonal antibiotics. 18
Sputum culture in patients with CF requires the collection of a sample of sputum, which is subsequently cultured and analysed in a clinical laboratory. 25 Sputum samples can either be obtained spontaneously (through expectoration) or induced by the use of throat swabs; nasopharyngeal aspiration (a small catheter through the nostril); or through inhalation of nebulised hypertonic saline to induce expectoration. In spontaneous expectoration the sample may be optimised by chest physiotherapy or by using bronchodilators and/or a recombinant human deoxyribonuclease (rhDNase) aerosol. 26 Clinical analysis of the sputum sample may be assayed for bacterial density, cell count and differential inflammatory markers before and after treatment with antibiotics. These measurements can be quantified in CFUs per gram of sputum and can be used to assess the clinical efficacy of antipseudomonal antibiotics. However, these measurements would not be routinely taken from patients with CF in clinical practice.
Chronic lung colonisation is defined by the European Medicines Agency (EMA) as the ‘presence of P. aeruginosa in the bronchial tree for at least 6 months, based on at least three positive cultures with at least one month between them without direct (inflammation, fever, etc.) or indirect (specific antibody response) signs of infection and tissue damage’. 27
Measuring rates of resistance
Microbial response can also include analyses of resistance through minimum inhibitory concentration (MIC) of isolates or breakpoint analysis. Clinical trials for antipseudomonal antibiotics often use the MIC50 (MIC required to inhibit the growth of 50% of organisms in culture). Sputum samples are analysed for evidence of resistance or susceptibility to the drug in question according to established MIC breakpoints. The British Society for Antimicrobial Chemotherapy (BSAC) publishes breakpoints, which are discriminatory antimicrobial concentrations used in the interpretation of results of susceptibility testing to define isolates as susceptible, intermediate or resistant. Published breakpoints vary from year to year. At the time of the trials, colistimethate breakpoints moved from resistant ≥ 8 mg/l and susceptible ≤ 4 mg/l to a single breakpoint of 4 mg/l. Tobramycin-resistant breakpoints moved from ≥ 8 mg/l to ≥ 4 mg/l, and in 2005 moved to a single breakpoint of 4 mg/l.
Although these breakpoints are well established, and assessment of microbial resistance is recommended in the EMA research guidelines18 and are required by the National Institute for Health and Care Excellence (NICE) for the purpose of this assessment, the relevance of MIC susceptibility breakpoints to inhaled antibiotics is debated. There are two main reasons why the breakpoints may not be relevant:
-
Breakpoints are established primarily in relation to antibiotic concentrations achievable in the bloodstream. Because many antibiotics are toxic above a certain blood concentration, the therapeutic window is necessarily limited by this toxicity, and the breakpoints are correspondingly low. Antibiotics delivered by inhalation can reach far higher concentrations in the lung without causing the same toxic levels in the bloodstream, and the therapeutic window extends to a much higher concentration. Therefore, higher breakpoints may be more relevant in this context.
-
Breakpoints are established by culturing samples in vitro then testing the susceptibility of the organisms. Phenotype (characteristics of the organism in response to their environment) plays a significant part in resistance. Infection with P. aeruginosa in the CF lung often involves the formation of biofilms with the mucoid phenotype (which are more resistant to antibiotics) in response to the environment of the lung. Cultured organisms removed from the environment of the lung display a different phenotype, and therefore a different level of susceptibility to the antibiotic, making the relevance of the cultured organisms’ susceptibility questionable.
Although the phenotype may be different in vivo, and although higher concentrations can be achieved in the lung, an increase in the minimum inhibitory concentration required to inhibit the growth of 50% of organisms in culture (MIC50) may still be an indicator that more resistant genotypes are being selected for by the antibiotic, and may therefore still have some relevance in indicating increased resistance.
Finally, as at present this is the established measure for susceptibility, and it is required by the EMA and listed in the NICE scope, this outcome will be reported for consideration.
Measuring lung function
The widespread availability of spirometers and the availability of standardised methods for assessment28,29 make spirometry the preferred method of measurement of lung function. Spirometry can be reliably performed by children who are aged > 5/6 years, and provides a number of potentially useful measurements. FEV1 is defined as ‘the maximal volume of air exhaled in the first second of a forced expiration from a position of full inspiration, expressed in litres at body temperature (i.e. 37 °C), ambient pressure and saturated with water vapour (BTPS)’. 28 It is converted by use of an equation (e.g. Knudson et al. 30) to a percentage of the normal predicted value for a healthy person of the same age, sex and height to give the ‘FEV1% predicted’ or FEV1%. There are a number of such equations that can be used,30–34 which will affect the FEV1% calculated. There does not appear to be a consensus with respect to which equation should be considered most appropriate.
There are, however, some problems with FEV1 as a measure of the pulmonary health of people with CF. Primarily, FEV1 is a global assessment of lung function, and is largely insensitive to localised disease. Additionally, it is influenced by a number of other (sometimes transitory) factors, including respiratory muscle strength (which, in turn, is sensitive to nutritional status),35 acute exacerbations,36 respiratory viral infections37 and so on. FEV1, like other spirometry tests, relies on volitional motion, and is associated with some degree of error around the mean; one review reports error ranging from 2.2% to 4.7%. 38
There are other spirometry measurements and other technologies that are increasingly being used to assess lung function. Forced vital capacity (FVC) is defined as ‘the maximal volume of air exhaled with maximally forced effort from a maximal inspiration . . . expressed in litres (BTPS)’,28 and the mean forced expiratory flow during the middle half of the FVC is known as forced expiratory flow (at 25–27% of vital capacity; FEF25–75%). Decreases in FEF25–75% are thought to provide the earliest indications of obstructive pulmonary disease. 39 These obstructive changes later become evident in FEV1% readings and will eventually have an impact on FVC. Computerised tomography (CT) and magnetic resonance imaging (MRI) can also be used to assess lung disease. 40 CT is considered the gold standard; however, this exposes the patient to a significant dose of radiation and its use is therefore limited as life expectancy increases. 41 MRI is thought to have lower specificity for small airway disease, but may be comparable or even superior for imaging some other indicators of lung disease. 40
Although there may be a role for FEF25–75%, and CT and MRI may be useful in certain circumstances, FEV1% is currently the recommended primary end point for clinical trials,18 and, owing to the number of studies linking FEV1% (either absolute readings or slope of decline) to prognosis,2,42–46 is a key indicator of disease progression used to monitor patients’ health.
Measuring acute exacerbations
The EMA defines an exacerbation as the onset of an acute episode of clinical deterioration when the patient is in a stable state. The definition of clinical deterioration has recently been revisited by the EuroCare CF Working Group. 20 Clinical deterioration is defined by the EMA27 by the presence of at least three of the following new clinical findings:
-
increased cough
-
increased expectoration (volume and purulence)
-
decreased tolerance to effort or physical activity
-
loss of weight or loss of appetite
-
deterioration of respiratory function (FEV1, FVC), and
-
a marked increase in airway bacterial load (in CFU/ml) during routine monitoring.
There is a lack of clear recommendations for clinical trials with respect to the definition of acute exacerbations and how they should be measured. Frequently, the corresponding measurement for acute exacerbations is ‘mean time to first additional antipseudomonal antibiotic use’ as well as the duration of this reactive treatment and/or whether the rescue medication was i.v. or not. ‘Hospitalisations’ and ‘length of hospital stay’ are also used as measures under the acute exacerbation outcome. More recently there has been a general decrease in hospitalising patients for treatment23 and a trend towards more patients being treated at home. 47 Consequently, the use of ‘hospitalisation’ as a surrogate measure for acute exacerbation may be unreliable. The most robust data at present are likely to be the number of acute exacerbations and the duration of i.v. use, although these measures are also subject to a degree of random error.
There are currently, therefore, several methods of measuring outcomes for acute exacerbation. A clear recommendation regarding how to measure acute exacerbation has yet to be adopted. This judgement requires consensus on reporting the number of acute exacerbation events or the number of patients who experienced an acute exacerbation. Recommendations for measuring acute exacerbations in clinical trials should also consider that these outcomes could be measured as the percentage change from baseline or in terms of absolute event rates.
Measuring health-related quality of life
As CF is incurable, interventions often aim to improve both the quality and duration of a patient’s life. To date, four measures specific to CF have been developed48–51 to overcome a perceived lack of sensitivity of generic HRQoL measures, such as the EQ-5D and SF-6D (Short Form questionnaire-6 Dimensions), to aspects of the disease that are important to people with CF. The Cystic Fibrosis Questionnaire (CFQ) was developed and validated by a French group,49 and exists in different formats for children and adults. A translated version validated in an American cohort52,53 is also available and in common use. This questionnaire is supported by the EMA research guidelines as an outcome measure,18 which should be recorded at least 3–6 months into therapy. These are not preference-based measures and do not allow the calculation of health utility scores. The use of generic health status measures, such as the EQ-5D, have been very limited in the measurement and valuation of different states of health for patients with CF (the available evidence is reviewed in Chapter 4).
Current service provision
Management of disease
The care of most patients in the UK is co-ordinated by a tertiary CF centre, with formal ‘shared care’ with local clinics. Further, primary care teams may also play a role in the surveillance and early treatment of infection, the provision of dietary and nutritional support, and the provision of social and psychological support for patients and their families. 1 A wide range of treatments may be required at various stages of the disease, including physiotherapy, pharmacological therapies, educational advice and surgical interventions for certain complications.
There are two main stages of P. aeruginosa lung infection, each of which requires a different approach to treatment. The first stage is characterised by intermittent growths of both mucoid and non-mucoid P. aeruginosa, and typically develops during infancy and childhood. This can be treated and sometimes eradicated with antibiotics to maintain respiratory function. Colonisation develops subsequently, and may be associated with mucoid change: it is a marker of reduced survival. Chronic infection cannot be eradicated by antibiotics as biofilm formation prevents antibiotics from working effectively. Acute exacerbations characterised by an acute decrease in respiratory function occur and become more frequent as the disease progresses. It is thought that acute exacerbations may contribute to a stepwise decrease in lung function, with FEV1% failing to return to pre-exacerbation baseline values. However, evidence to support this theory remains limited. At this stage, for most patients, continuous antibiotic use will be required.
Current management pathways for patients with chronic Pseudomonas aeruginosa infection
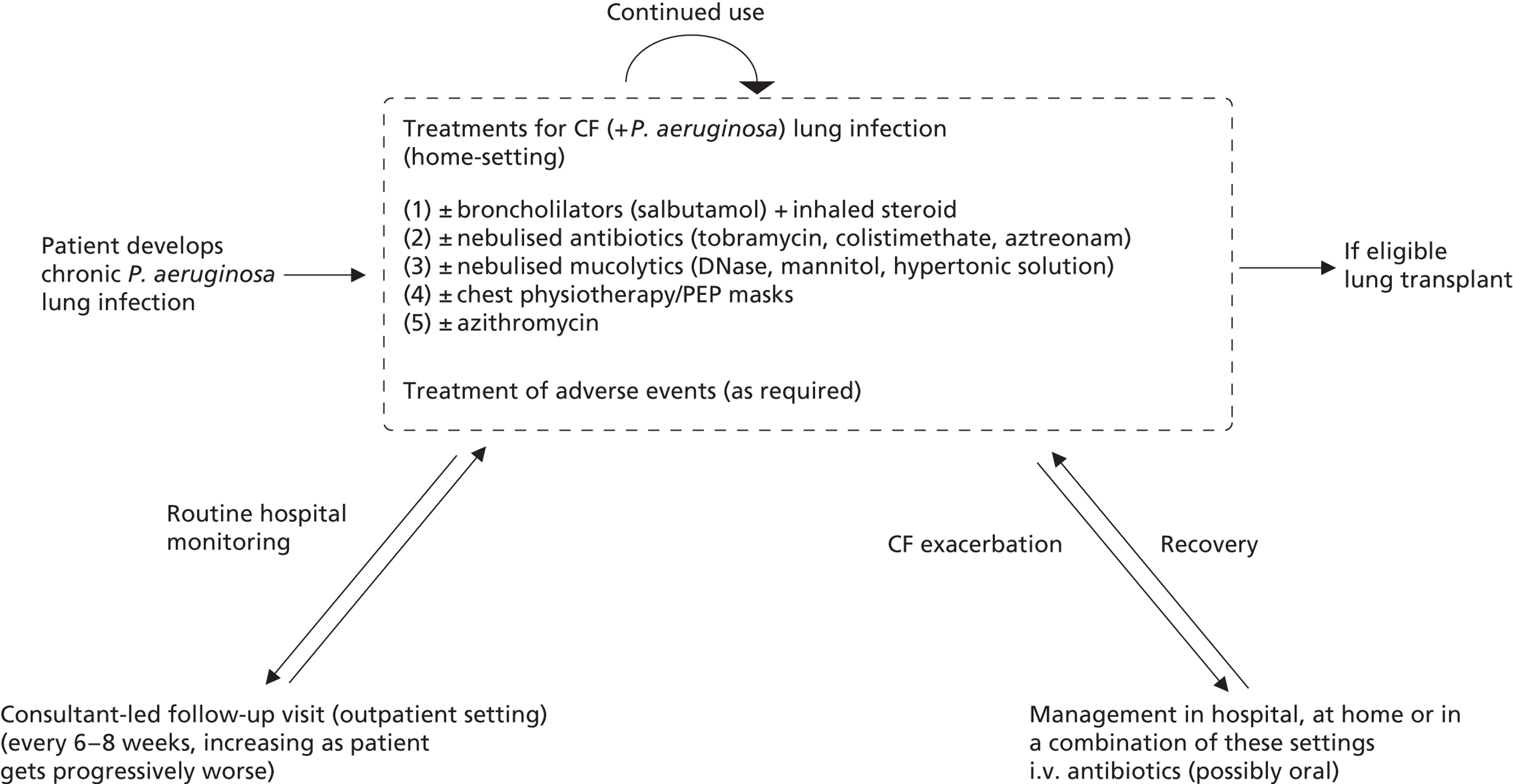
Figure 6 presents a general management pathway for patients with CF with chronic P. aeruginosa lung infection. This is intended to be representative of the UK Cystic Fibrosis Trust guidelines,3 which, in turn, reflect usual clinical practice in the majority of UK CF centres. There is likely to be some variation in practice across some of the smaller centres, and specific antibiotic choices may differ by centre according local bacterial sensitivities. Generally speaking, decisions concerning the use of particular treatments tend to be more related to severity than age, therefore treatment use is broadly similar across both paediatric and adult populations. 6
FIGURE 6.
Treatment pathway for patients with chronic P. aeruginosa. PEP, positive expiratory pressure.
Continuous drug treatments
Following chronic infection with P. aeruginosa, all patients will be offered ongoing nebulised antibiotic treatment, which takes place in the home setting. In a small proportion of patients (around 10–15%) with P. aeruginosa lung infection may not receive nebulised antibiotic therapy (Table 2). Current antibiotic treatment options include colistimethate sodium, tobramycin and, less commonly, aztreonam (Cayston®, Gilead Sciences). Colistimethate sodium is given every day. Tobramycin and aztreonam differ in that each 28-day treatment cycle is followed by a 28-day period that does not include the use of these drugs. The current guidelines from the Cystic Fibrosis Trust recommend initial treatment using Colistin; tobramycin is recommended if Colistin is not tolerated or if clinical progress is unsatisfactory. 3 In practice, some patients whose lung function fails to stabilise on monotherapy may receive 28 days of treatment using colistimethate sodium, followed by 28 days of treatment using tobramycin as an ongoing repeated sequence.
Concomitant therapies
A number of concomitant treatments may be used alongside nebulised antibiotics. A considerable proportion of patients with CF exhibit a degree of airway reversibility (asthma-like changes) and will be treated with bronchodilators (e.g. salbutamol) plus inhaled steroids (e.g. salmeterol xinafoate/fluticasone propionate). This is often administered as a combination inhaler, with up to 50% of patients with CF receiving inhalers for this reason. The inhaled drugs for P. aeruginosa infection result in bronchospasm (narrowing of the airways) in a proportion of patients. In these patients, a bronchodilator is given before the inhaled antibiotics with prophylactic intent. In addition, patients with chronic P. aeruginosa infection typically receive macrolides (most commonly azithromycin). These are given between three and seven times per week on an ongoing basis, and are used for their anti-inflammatory properties, with the intention of arresting the decline in lung function; however, there is conflicting evidence concerning their efficacy. 54–56 Patients may also receive mucolytic drugs [e.g. rhDNase, mannitol (Bronchitol®, Pharmaxis) or hypertonic saline], with the intention of reducing the viscosity, adherence and tenacity of the sputum, and to aid efficient clearance. 57 In addition, many CF centres would advocate some form of airway clearance using either traditional percussion/drainage via chest physiotherapy or using positive expiratory pressure (PEP) devices.
Follow-up
Patients are invited to attend routine follow-up to monitor progression of the disease and to inform decisions regarding treatment. For children, follow-up appointments are usually every 6–8 weeks. However, the frequency of follow-up visits typically increases as the disease progresses. Adults in Band 3 or 4 (see Appendix 1) may be supervised more closely. Band 5 patients may be in hospital more or less continuously.
Adverse events and the management of exacerbations
Adverse events (AEs) should be reported to the CF care team and may be an indication for stopping or modifying therapy. However, patients experiencing exacerbations will require further antibiotic treatment administered intravenously. Many centres now deliver i.v. antibiotics in part at home. Hospital admissions for the management of AEs are more common in adult centres where patients are likely to be more severely ill.
Lung transplantation
A small proportion of patients are eligible for lung transplantation. Most of these patients will no longer require inhaled antibiotics; however, antirejection therapies and treatments for other organs affected by CF will still be required.
Current usage
Table 2 shows current registry estimates of antibiotic use among patients with chronic P. aeruginosa infection. The data suggest that approximately 78.8% of individuals with chronic P. aeruginosa infection receive at least one antibiotic post transplant. The CF Registry states that around 90% of patients with chronic P. aeruginosa should be prescribed one or more of these treatments. 6
| Drug(s) | Overall | Percentage | < 16 years | Percentage | ≥ 16 years | Percentage |
|---|---|---|---|---|---|---|
| Tobramycin solution | 691 | 24.63 | 97 | 22.05 | 594 | 25.11 |
| Other aminoglycoside | 66 | 2.35 | 15 | 3.41 | 51 | 2.16 |
| Colistin | 1237 | 44.08 | 238 | 54.09 | 999 | 42.22 |
| Promixin | 726 | 25.87 | 119 | 27.05 | 607 | 25.66 |
| At least one of the above | 2212 | 78.83 | 383 | 87.05 | 1829 | 77.30 |
| Patients with chronic P. aeruginosa | 2806 | 100.00 | 440 | 100.00 | 2366 | 100.00 |
Recent UK-relevant cost estimates relating to the treatment of CF are limited. A recent UK cost of illness study, undertaken in the east of England, estimated the mean annual cost of treating 174 patients to be £1,040,087 (£5976 per patient). 58 Multiplying this estimate up to the current number of patients in the CF registry yields a crude annual cost of around £57M for patients with CF in England and Wales. However, the true cost to the NHS may be considerably higher (Diana Bilton, Department of Respiratory Medicine, Royal Brompton Hospital, 2012, personal communication).
Variations in services and uncertainties about best practice
It has been noted elsewhere that many aspects of current practice in the management of CF have evolved without being subjected to high-quality clinical trials. 1 This may be partly a result of the rarity of the disease and associated difficulties with recruitment to clinical trials, as well as variations between patients in terms of how the disease manifests and is treated. With respect to interventions for the management of lung infection, evidence relating to the long-term clinical and mortality benefits of treatments is rarely available.
There is currently no NICE guidance relating to the detection, diagnosis or management of patients with CF. A single technology appraisal of mannitol dry powder for inhalation (DPI) for the treatment of CF was completed in 2012. This appraisal did not specifically relate to the management of patients with P. aeruginosa lung infection.
Since 1 April 2011, the Department of Health has adopted a ‘Payment by Results’ (PbR) tariff for patients with CF. This will link ‘activity’ to funding received, whereby money will follow the patient through their hospital journey, paying for treatment and care (excluding drugs) received along the way. 25
Description of technologies under assessment
Summary of interventions and comparators
This assessment includes two interventions that are delivered as a DPI: colistimethate sodium DPI [Colobreathe® (plus Turbospin®), Forest Laboratories] and tobramycin DPI [TOBI® (plus Podhaler®), Novartis Pharmaceuticals]. The antibiotics colistimethate sodium and tobramycin also represent the relevant comparators for the assessment, albeit in nebulised form.
Colistimethate sodium (Colobreathe/Colomycin/Colistin) belongs to the polymyxin group and is a cyclic polypeptide antibiotic derived from Bacillus polymyxa, var. colistinus. Colistimethate sodium works by disrupting the structure of the bacterial cell membrane in a detergent-like way by changing its permeability, leading to bacterial death. It is also thought to act intracellularly to precipitate ribosomes and other cytoplasmic components. Colistimethate sodium is active against aerobic Gram-negative organisms, including P. aeruginosa, Acinetobacter baumanii and Klebsiella pneumoniae. Forest Laboratories currently markets colistin sulphate (Colomycin) as a tablet or syrup, and colistimethate sodium as a powder for injection or nebulisation. Profile Pharma currently markets colistimethate sodium (Promixin) as a powder for i.v. injection or for inhalation specifically using an I-neb device. Colobreathe (which is also colistimethate sodium) is available as 125-mg hard capsules and is administered specifically using the Turbospin inhaler device. It is anticipated that both the treatment and the Turbospin device will be marketed and packaged together.
The summary of product characteristics (SmPC) (www.ema.europa.eu/) lists the following AEs for colistimethate sodium DPI: unpleasant taste (dysgeusia), cough, throat irritation, dyspnoea, dysphonia, coughing, bronchospasm, balance disorder, headache, tinnitus, haemoptysis, asthma, wheezing, chest discomfort, lower respiratory tract infection, productive cough, crackles – lung, vomiting, nausea, arthralgia, pyrexia, asthenia, fatigue, decreased forced expiratory volume, drug hypersensitivity, weight fluctuation, decreased appetite, ear congestion, chest pain, exacerbated dysphonia, pharyngolaryngeal pain, epistaxis, sputum purulent, abnormal chest sound, increased upper airway secretion, diarrhoea, toothache, salivary hypersecretion, flatulence, proteinuria and thirst. Sore throat or mouth (probably due to Candida albicans infection or hypersensitivity) has been reported for nebulised colistimethate sodium and the SmPC states that this may occur with Colobreathe also. AEs listed in the electronic Medicines Compendium (eMC) (www.medicines.org.uk/) for the nebulised form also include bronchospasm, cough, hypersensitivity reactions and skin rash/rashes.
Tobramycin belongs to the aminoglycoside group of antibiotics and is obtained from cultures of Streptomyces tenebrarius. It enters susceptible bacterial cells via a complex active transport mechanism and acts by binding irreversibly to the 30S ribosomal subunit. It is thought that this interferes with essential steps in protein synthesis and consequently affects the permeability of the cell membrane, although there is some suggestion that it may also act directly on the cell membrane. 59 Once the cell envelope becomes compromised, cell death follows. It also acts to induce misreading of the genetic code of the messenger ribonucleic acid (mRNA) template, resulting in incorporation of incorrect amino acids, which can result in cellular malfunction. Two tobramycin nebuliser solution (TNS) products are available: Novartis Pharmaceuticals currently markets TOBI® nebuliser solution, and Chiesi market Bramitob® nebuliser solution. TOBI DPI is available as 28-mg capsules and is administered specifically using the Podhaler device. Both the treatment and device are marketed and packaged together.
Adverse events listed by eMC (www.medicines.org.uk/) for tobramycin DPI include hearing loss, tinnitus, haemoptysis, epistaxis, dyspnoea, dysphonia, productive cough, cough, wheezing, rales, chest discomfort, nasal congestion, bronchospasm, oropharyngeal pain, vomiting, diarrhoea, throat irritation, nausea, dysgeusia, rash, musculoskeletal chest pain and pyrexia. Cough was the most frequent adverse reaction. With respect to nebulised tobramycin, AEs reported in controlled clinical trials include dysphonia and tinnitus. AEs reported in the post-marketing phase include laryngitis, oral candidiasis, fungal infection, lymphadenopathy, hypersensitivity, anorexia, headache, dizziness, aphonia, somnolence, tinnitus, hearing loss, ear disorder, ear pain, dysphonia, dyspnoea, cough, pharyngitis, bronchospasm, chest discomfort, lung disorder, productive cough, haemoptysis, epistaxis, rhinitis, asthma, hyperventilation, hypoxia, sinusitis, dysgeusia, nausea, mouth ulceration, vomiting, diarrhoea, abdominal pain, rash, urticaria, pruritus, back pain, asthenia, pyrexia, chest pain, pain, malaise and pulmonary function test decreased.
Place in the treatment pathway
Both interventions are to be used for the ongoing treatment of chronic P. aeruginosa, as described above (see Current service provision). One of the principal anticipated benefits of the interventions is that they are quicker to use and are portable, which means that they can be self-administered by the patient as indicated, thereby avoiding time required for inhalation using a nebuliser. The DPIs may also result in savings in terms of the time associated with cleaning traditional nebulisers. It is hypothesised that these benefits may lead to improvements in compliance with treatment.
Identification of important subgroups
Specific subgroups have not been identified a priori within this appraisal. Consideration was given within this assessment to evidence relating to those groups of individuals for whom these therapies may be particularly clinically effective or cost-effective.
Current usage in the NHS
The use of TOBI in conjunction with the Podhaler device was granted full marketing authorisation by the EMA in 2011. TOBI Podhaler is indicated for the suppressive therapy of chronic pulmonary infection due to P. aeruginosa in adults and children with CF, aged ≥ 6 years. The Podhaler inhaler device bears an initial date of Conformité Europeénne (CE) marking of 28 July 2005. The Novartis Pharmaceuticals submission states that this date is noted on the European Commission (EC) Declaration of Conformity to the European Union (EU) Medical Device Directive 93/42/EEC as amended for this Class I device. 60
Colobreathe used in conjunction with the Turbospin device was granted full marketing authorisation by the EMA in February 2012. Colobreathe is indicated for the management of chronic pulmonary infections due to P. aeruginosa in patients with CF aged ≥ 6 years.
Anticipated costs associated with the intervention
Table 3 summarises the acquisition costs associated with the interventions and comparators, based on list prices from the British National Formulary (BNF). 61
| Generic name | Trade name | Manufacturer | Indication | Form of administration | Cost per unit | Cost per 28 days of treatment |
|---|---|---|---|---|---|---|
| Colistimethate sodium | Promixin | Profile Pharma | Adult and child > 2 years, 1–2 MU b.i.d.; increased to 2 MU three times daily for subsequent respiratory isolates of P. aeruginosa | Powder for nebuliser solution | 1-MU vial = £4.60 | £257.60 (1 MU per dose b.i.d.) to £772.80 (2 MU per dose three times daily) |
| Colomycin | Forest Laboratories | Powder for injection or nebuliser solution | 1-MU vial = £1.68 2-MU vial = £3.09 |
£94.08 (1 MU per dose b.i.d.) to £259.66 (2 MU per dose three times daily) | ||
| Colobreathe + Turbospin | Forest Laboratories | 125 mg twice daily | DPI | Price not confirmed at the time of the assessment | ||
| Tobramycin | Bramitob | Chiesi | Adult and child > 6 years, 300 mg every 12 hours for 28 days, subsequent courses repeated after 28-day interval without TNS | Powder for nebuliser solution | 75 mg/ml, net price 56 × 4-ml (300-mg) unit = £1187.00 | £1187.00 |
| TOBI | Novartis Pharmaceuticals | Powder for nebuliser solution | 60 mg/ml, net price 56 × 5-ml (300-mg) unit = £1187.20 | £1187.20 | ||
| TOBI + Podhaler | Novartis Pharmaceuticals | Adult and child > 6 years, 112 mg every 12 hours for 28 days, subsequent courses repeated after 28-day interval without tobramycin inhalation powder | DPI | 224 × 28-mg capsules + five Podhalers = £1790.00 56 × 28-mg capsules + one Podhaler = £447.50 |
£1790.00 | |
| Aztreonam | Cayston | Gilead Sciences | Adult > 18 years, 75 mg three times daily (at least 4 hours apart) for 28 days; if additional courses required, a minimum of 28 days without aztreonam nebuliser solution recommended between courses | Powder for nebuliser solution | 84 × 75-mg vials (with solvent and nebuliser handset) = £2566.80 | £2566.80 |
Chapter 2 Definition of the decision problem
Overall aims and objectives of the assessment
This assessment addresses the question ‘what is the clinical effectiveness and cost-effectiveness of colistimethate sodium DPI and tobramycin DPI for the treatment of chronic P. aeruginosa lung infection in CF compared with current treatments?’
Specifically, the objectives of the assessment are to:
-
assess the clinical effectiveness of colistimethate sodium DPI and tobramycin DPI for the treatment of chronic P. aeruginosa lung infection in terms of lung function, microbial response, respiratory symptoms and the frequency/severity of acute exacerbations
-
assess the AE profile associated with colistimethate sodium DPI and tobramycin DPI
-
estimate the incremental cost-effectiveness of colistimethate sodium DPI and tobramycin DPI compared with current standard treatments for the treatment of chronic P. aeruginosa lung infection.
This report contains reference to confidential information provided as part of the NICE appraisal process. This information has been removed from the report and the results, discussions and conclusions of the report do not include the confidential information. These sections are clearly marked in the report.
Decision problem
Interventions
Two interventions are included in this assessment:
-
colistimethate sodium DPI, used in conjunction with the Turbospin device
-
tobramycin DPI, used in conjunction with the TOBI Podhaler device.
Populations and subgroups
The population for the assessment includes people aged ≥ 6 years with CF and chronic P. aeruginosa pulmonary colonisation. Subgroups are considered according to the available evidence.
Relevant comparators
The interventions are compared against each other. Other relevant comparators include antibiotics used for nebulised inhalation, including colistimethate sodium for nebulised inhalation and tobramycin for nebulised inhalation. The availability of evidence of the effectiveness of other less commonly used nebulised antibiotics (e.g. aztreonam) with antipseudomonal activity is also considered within the assessment.
Outcomes
The following outcomes are considered within this assessment:
-
rate and extent of microbial response (e.g. sputum density of P. aeruginosa)
-
lung function measured in terms of FEV1%
-
respiratory symptoms
-
frequency and severity of acute exacerbations
-
HRQoL
-
AEs of treatment (including rate of resistance to antibiotic treatment)
-
cost-effectiveness measured in terms of the incremental cost per quality-adjusted life-year (QALY) gained.
Chapter 3 Clinical effectiveness
This section presents the methods and results of a systematic review of clinical effectiveness of colistimethate sodium DPI and tobramycin DPI in comparison with currently used nebulised treatments.
Methods for reviewing clinical effectiveness
The protocol for this review is registered with PROSPERO (CRD42011001350) and is available from the NICE website (www.nice.org.uk/).
Identification of studies
A comprehensive search was undertaken to systematically identify literature relating to the clinical effectiveness of colistimethate sodium DPI and tobramycin DPI for the treatment of P. aeruginosa in CF. The search strategy comprised the following main elements:
-
searching of electronic databases
-
contact with experts in the field
-
hand-searching of bibliographies of retrieved papers.
The following electronic databases were searched from inception for published trials and systematic reviews:
-
MEDLINE: Ovid. 1950 to present
-
MEDLINE in-Process & Other Non-Indexed Citations: Ovid. 1950 to present
-
EMBASE: Ovid. 1980 to present
-
The Cochrane Library: Wiley Online Library
-
Cochrane Database of Systematic Reviews (CDSR), 1996 to present
-
Database of Abstracts of Reviews of Effects (DARE),1995 to present
-
Cochrane Central Register of Controlled Trials (CCRT), 1995 to present
-
Cochrane Methodology Register, 1904 to present
-
Health Technology Assessment (HTA) database , 1995 to present
-
NHS Economic Evaluation Database (NHS EED), 1995 to present
-
-
Cumulative Index to Nursing and Allied Health Literature (CINAHL): EBSCOhost, 1982 to present
-
Web of Science Citation Index: Web of Knowledge, 1899 to present
-
Conference Proceedings Citation Index (CPCI): Web of Knowledge, 1990 to present
-
Bioscience Information Service (BIOSIS) Previews: Web of Knowledge, 1969 to present.
Additional searches were carried out for unpublished studies (e.g. ongoing, completed):
-
Agency for Healthcare Research and Quality (AHRQ)
-
Bandolier
-
Centre for Health Economics (CHE); University of York
-
ClinicalTrials.gov
-
Current Controlled Trials
-
The National Research Register Archive: NIHR, 2000–7
-
The metaRegister of Controlled Trials: Springer Science + Business Media, 2000 to present.
Manufacturers’ submissions received by NICE, as well as any relevant systematic reviews were also hand-searched in order to identify any further clinical trials.
The MEDLINE search strategy is presented in Appendix 2. The search strategy combined free-text and medical subject heading (MeSH) or thesaurus terms relating to CF with free-text and MeSH or thesaurus terms relating to P. aeruginosa, relevant antibiotics and classes of antibiotics, and the devices and comparator devices of interest. The search strategy was translated across all databases. No date or language restrictions were applied. Literature searches were conducted during February and March 2011. References were collected in a bibliographic management database and duplicates removed.
Inclusion and exclusion criteria
Inclusion and exclusion criteria were based on the scope provided by NICE. 62 These are set out below.
Inclusion criteria
Studies were included if they satisfied the following criteria.
Interventions
Studies assessing the effectiveness of colistimethate sodium DPI (used in conjunction with the Turbospin device) or tobramycin DPI (used in conjunction with the TOBI Podhaler device) were included.
Population
Studies were selected to include only people aged ≥ 6 years with CF and chronic P. aeruginosa pulmonary infection. Children of < 6 years of age were excluded from the assessment, as they are subject to different treatment regimens, methods of assessment of lung function differ, and licensing has not been sought for this age group.
Comparators
Acceptable comparators were (1) the comparator intervention or (2) other antipseudomonal antibiotics for nebulised inhalation, including, as a minimum, colistimethate sodium for nebulised inhalation or tobramycin for nebulised inhalation.
Outcomes
Outcomes to be considered by the review were rate and extent of microbial response (e.g. sputum density of P. aeruginosa); lung function; respiratory symptoms; frequency and severity of acute exacerbations; HRQoL; and AEs of treatment (including rate of resistance to antibiotic treatment). Compliance was also considered as a post hoc addition to the outcomes set out in the NICE scope, as it became evident that this was of relevance to the claims made for the interventions by the manufacturers.
Study types
Randomised controlled trials (RCTs) were included in the assessment. Data from non-randomised studies were considered but were not included, as evidence was available from RCTs.
Systematic reviews were included if they provided additional data for RCTs meeting the inclusion criteria (i.e. unavailable from published trial reports). Other systematic reviews identified were not included but were checked for RCTs that met the inclusion criteria of this review.
Exclusion criteria
The following were excluded: studies based on animal models; preclinical and biological studies; non-RCTs; editorials, opinion pieces; reports published as meeting abstracts only where insufficient details were reported to allow inclusion; studies published only in languages other than English; studies with vasoactive drugs that were not within their licensed indications; studies in which the population was not restricted to CF, unless data for just this population was presented; and studies that did not present data for the included outcomes.
Based on the above inclusion/exclusion criteria, study selection was conducted by one reviewer (SH, CC or LU) and checked by a second reviewer (SH, CC or LU). In the first instance, titles and abstracts were examined for inclusion. The full manuscripts of citations judged to be potentially relevant were retrieved and further assessed for inclusion.
Scoping searches indicated that a head-to-head trial of the two interventions was unlikely to be available. In anticipation of this, studies that could potentially contribute to a network meta-analysis (NMA) were also identified on the basis of their abstract and title. Studies were considered potentially useful if they assessed the efficacy of nebulised antibiotics in the target population for the target condition, and reported relevant outcomes. Key study characteristics of the wider network of evidence were extracted by one reviewer. Based on these characteristics, the available network of evidence was constructed. Were viable networks possible, only studies that could contribute to this network would be included in the review. Were a network not possible, only studies providing direct comparisons with at least one intervention and at least one comparator listed in the inclusion criteria were included in the review.
Data extraction and critical appraisal strategy
Data were extracted without blinding either to authors or journal. Data were extracted by one reviewer using a standardised form and checked by a second reviewer. Where multiple publications of the same study were identified, quality assessment and data extraction were based on all relevant publications, and listed as a single study. The quality of included studies was assessed according to three sets of criteria. The purpose of quality assessment was to provide a narrative account of trial quality for the reader, and to inform subgroup analyses (where data allow). In order to assess the risk of bias, items listed in the NHS Centre for Reviews and Dissemination (CRD) report63 were used and were scored as ‘yes’, ‘no’ or ‘unclear’. To assess the clinical relevance and quality of the studies, items were generated from the EMA research recommendations. 18 Two trials were non-inferiority trials; a separate quality assessment form64 specific to this type of study was also used.
Data synthesis methods
The prespecified outcomes were tabulated and discussed within a descriptive synthesis. Where populations, interventions, outcome measures and available data were comparable and statistical synthesis was considered appropriate, classical meta-analysis or NMA was planned using Bayesian techniques, or Review Manager (RevMan) software version 5 (The Cochrane Collaboration, The Nordic Cochrane Centre, Copenhagen, Denmark; http://ims.cochrane.org/revman). If sufficient trials were available, sensitivity analysis was planned to examine whether the removal of poor-quality trials (in terms of risk of bias or compliance with research guidelines) influenced the results of the meta-analysis. Consideration was also given to subgroup analyses based on study characteristics.
Results
Quantity and quality of research available
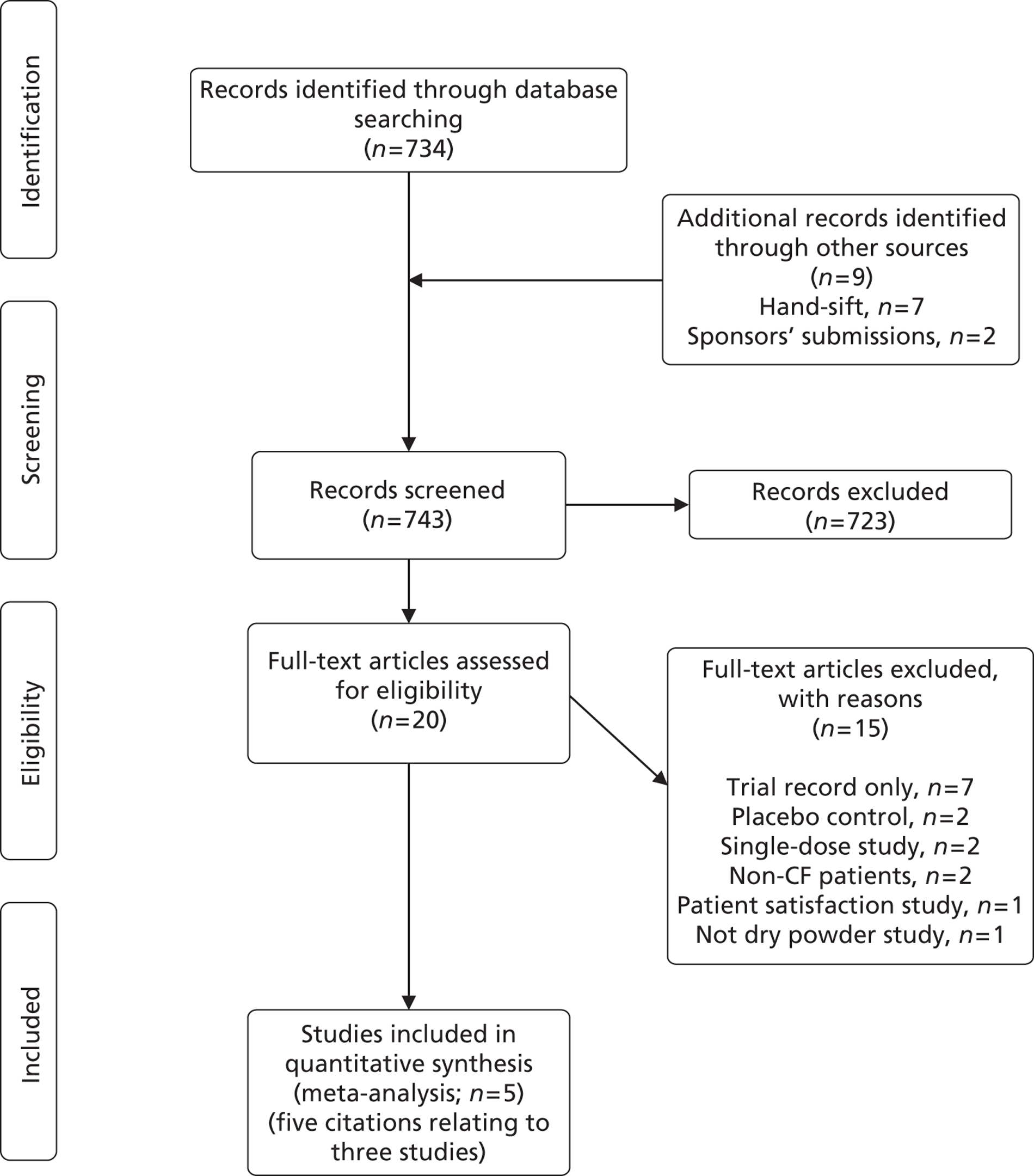
The search retrieved 743 potentially relevant citations (734 from searches of electronic databases, nine from secondary searches of relevant reviews, articles and sponsors submissions). Of these, 723 were excluded at the title and abstract stage, leaving 20 potentially includable citations.
The full texts of the 20 articles were obtained for scrutiny. Fifteen did not meet the inclusion criteria and were excluded (see Appendix 3). Three studies60,65,66 comparing colistimethate sodium DPI or tobramycin DPI with a nebulised antibiotic were included in the review. One study was of tobramycin DPI in combination with the TOBI Podhaler,60,65 and two studies were of colistimethate sodium DPI in combination with the Turbospin device. 66 Information about the three trials included in the systematic review was available from five sources,60,65–68 as indicated in Figure 7. These comprise one published journal article,65 two conference abstracts67,68 and the two manufacturers’ submissions to NICE,60,66 with subsequent clarifications. It should be noted that data for the pivotal colistimethate sodium DPI trial, the COLO/DPI/02/06 trial,66 were available from the manufacturer’s submission,66 the clinical study report (CSR),69 the trial protocol70 and personal communication/clarifications only. None of this information was available in the public domain. The search process is summarised using a PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) diagram in Figure 7.
To assess the viability of a NMA, key study characteristic data were extracted from an additional 13 studies, from 16 citations. 65–67,71–83 Owing to clinical heterogeneity between the studies and incompleteness of the evidence network, a NMA was not performed (see Appendix 4).
Study characteristics
The included trials and the treatments assessed are summarised in Table 4. All studies were open-label, multicentre studies, two of which were multinational studies. 66 The EAGER (Establish A new Gold standard Efficacy and safety with tobramycin in cystic fibrosis) trial65 was a large trial (n = 533) that compared tobramycin DPI with nebulised tobramycin. The COLO/DPI/02/06 trial66 was slightly smaller (n = 380) and compared colistimethate sodium DPI with nebulised tobramycin. Both of these trials were powered to detect clinically relevant changes in FEV1%. The COLO/DPI/02/05 trial66 was much smaller (n = 16) and compared colistimethate sodium DPI with nebulised colistimethate sodium. The EAGER65 and COLO/DPI/02/0666 trials were both of 24 weeks’ duration, whereas COLO/DPI/02/0566 was a crossover trial, which reported outcome data at 4 weeks (before crossover) and 8 weeks (after crossover) only.
FIGURE 7.
Study inclusion (adapted from PRISMA).
Interventions and comparators
Intervention and comparator dosing complied with current UK licensing (www.medicines.org.uk/EMC/medicine/). In the two trials of colistimethate sodium DPI (COLO/DPI/02/0666 and COLO/DPI/02/0566), patients took colistimethate sodium DPI treatment every day throughout the study. In both the EAGER trial65 and the COLO/DPI/02/06 trial,66 the dosing pattern for nebulised tobramycin was cycles of 28 days on treatment, followed by 28 days off treatment, for three cycles. Within the EAGER trial,65 the same treatment approach was used for tobramycin DPI. This administration cycle is standard practice for tobramycin,61 with the aim of preventing antibiotic resistance.
| Study name and sources of information | Source of funding | Study design | Dates study undertaken | Study location | Intervention | Comparator | Duration of trial | Treatment schedule |
|---|---|---|---|---|---|---|---|---|
| EAGER trial Konstan et al. 2011;65 TBM100C2302 manufacturer’s submission;60 manufacturer’s clarifications |
Novartis Pharmaceuticals | RCT, open label (n = 533) |
February 2006 to March 2009 | 127 centres in 15 countries (including North America, Europe, Australia, Israel and Latin America) | Tobramycin DPI T-326 Inhaler 112 mg b.i.d. |
Tobramycin inhalation solution PARI LC Plus jet nebuliser 300 mg/5 ml b.i.d. |
24 weeks | Intervention and comparator: 28 days on treatment followed by 28 days off treatment |
| COLO/DPI/02/06 manufacturer’s submission;66 manufacturer’s clarifications |
Forest Laboratories | RCT, open label (n = 380) |
NR (last patient visit 14 August 2007) | 66 centres in EU countries, Russia and the Ukraine | Colistimethate sodium DPI Turbospin device 125 mg b.i.d. |
Tobramycin inhalation solution PARI LC Plus jet nebuliser 300 mg/5 ml b.i.d. |
24 weeks | Intervention: continuous treatment Comparator: 28 days on treatment followed by 28 days off treatment |
| COLO/DPI/02/05 Davies et al. 2004;67 manufacturer’s submission;66 manufacturer’s clarifications |
Forest Laboratories | RCT, open label with crossover (n = 16) |
NR | Three centres in the UK | Colistimethate sodium DPI Turbospin device 125 mg b.i.d. |
Colistimethate sodium solution Device: NR 2 MU b.i.d. |
8 weeks | Intervention and comparator: continuous treatment |
Inclusion and exclusion criteria
Inclusion and exclusion criteria are summarised and compared in Table 5. Criteria seem largely compatible between the two major trials (EAGER65 and COLO/DPI/02/06),66 although criteria for COLO/DPI/02/0666 were complicated. Inclusion and exclusion criteria are not reported in full here.
The EAGER65 trial and COLO/DPI/02/06 trial66 selected patients with ‘confirmed’ or ‘documented’ CF, who were clinically stable, and aged ≥ 6 years, whereas the COLO/DPI/02/05 trial66 selected patients who were ≥ 8 years. Patients in the EAGER65 trial and COLO/DPI/02/06 trial66 had to have an FEV1% value of ≥ 25%, up to 75%, whereas in the COLO/DPI/02/05 trial66,67 no upper limit for FEV1% was set. Patients in all trials continued with usual CF treatments (except other routine antipseudomonal treatments). Patients in all three trials had a chronic P. aeruginosa infection. The criteria used to define a chronic infection did not meet with EMA recommendations18 in any trial, as all called for only two positive cultures in the last 6 months, rather than three. In the case of the COLO/DPI/02/06 trial,66 three positive cultures were required in the last 6 months, but patients could also qualify with only two in the last 2 months. As such, it is unclear whether or not the trials have truly selected chronically infected patients, and how comparable the degree of infection is between the two trials.
The COLO/DPI/02/06 trial66 had a run-in period whereby participants were required to have received 16 weeks (two cycles) of nebulised tobramycin prior to beginning the trial. Tobramycin has been documented to peak rapidly in efficacy in the first cycle of treatment with the effect not being sustained over time. 76 Therefore, the run-in phase was intended to eliminate this short-term change in FEV1% predicted. In addition, this run-in phase was intended to exclude any patients who could not tolerate tobramycin. In comparison, the EAGER trial65 had a washout period of any systemic or inhaled antipseudomonal antibiotics for 28 days prior to randomisation, which ensured that patients already on tobramycin complied with the standard dosing schedule of 28 days on treatment followed by 28 days off treatment. The difference between these two criteria may result in slightly different populations.
| Study | Inclusion criteria | Exclusion criteria |
|---|---|---|
| All trials | Adequate contraceptive methods for female participants Written informed consent from patient or patient’s guardian Documented diagnosis of CF from a specialist CF unit (genotype and/or positive sweat tests) Current CF condition had to be clinically stable Chronic P. aeruginosa infection |
Pregnant or breastfeeding patients Inability to comply with any of the study procedures or the study regimen (including inability to use study devices, i.e. during dry powder inhaler and nebuliser training) Use of an elective course of i.v. antibiotic therapy or investigational drug within 28 days of screen Acute respiratory exacerbation within 28 days prior to first day of trial medication administration Patients who were colonised with B. cepacia |
| EAGER65 | Aged ≥ 6 years old FEV1 > 25% to < 75% predicted, based on Knudson equations. Patients with chronic P. aeruginosa infection (sputum or throat cultures positive for P. aeruginosa within 6 months of screening and at baseline) |
Use of systemic or inhaled antipseudomonal antibiotics or other drugs that can affect FEV1% within 28 days prior to study drug administration Haemoptysis of > 60 ml within 30 days prior to study Hypersensitivity to aminoglycosides or inhaled antibiotics Serum creatinine ≥ 2 mg/dl, blood urea nitrogen ≥ 40 mg/dl, or an abnormal urinalysis defined as ≥ 2 + proteinuria Clinically relevant history of hearing loss or chronic tinnitus |
| COLO/DPI/02/0666 | Aged ≥ 6 years old FEV1 > 25 to < 75% predicted, based on Knudson equations Run-in inclusion criteria (patients to receive a minimum of two nebulised tobramycin on/off cycles immediately prior to randomisation) Non-smokers or a past smoker who had not smoked within the past 12 months Patients who, on first day of trial medication administration (Visit 1), had ≥ 28 days but ≤ 35 days off tobramycin Patients with chronic P. aeruginosa infection (two or more sputum or throat cultures positive for P. aeruginosa within 6 months of screening) |
|
| COLO/DPI/02/0566 | Aged ≥ 8 years old FEV1 > 25% prediction, based on Knudson equation Non-smokers or a past smoker who had not smoked within the past 12 months prior to the date of entry |
Known sensitivity to colistimethate sodium or salbutamol Existence of any prestudy medical conditions which, in investigator’s judgement, warranted exclusion from the study Inability to communicate/co-operate with investigator due to language problems, poor mental development or impaired cerebral function Laboratory parameters falling outside the expected normal ranges for CF (investigator decision) Patients who, on first day of trial treatment, had < 28 days off tobramycin Patients who had experienced < 72 hours washout from other antipseudomonal agents Patients who were complicated by ABPA Patients who were awaiting heart–lung or lung transplantation |
Patient characteristics
The baseline characteristics of patients in the three trials are presented in Table 6. The patients in the COLO/DPI/02/06 trial66 had a lower mean age than those in the EAGER trial. 65 Mean age was not reported for trial COLO/DPI/02/05. 66 As age and FEV1% status are thought to have an inverse correlation, it might be expected that the patients in the COLO/DPI/02/06 trial66 were earlier in their stage of chronic Pseudomonas infection than those in the EAGER trial. 65 However, the baseline FEV1% predicted values are similar between these two trials, with the FEV1% predicted in COLO/DPI/02/0666 being slightly lower. This may be due to inclusion criteria for chronic infection not being defined according to EMA recommendations. 18 For all trials, this may result in the recruitment of patients with intermittent infections, who may respond differently to treatment than chronically infected patients. It is also probable that some patients recruited to the COLO/DPI/02/06 trial66 may be slightly less well than the EAGER trial65 participants, as criteria were more stringent in this population. In both trials, the lack of consistency and conformity with the EMA guidelines18 may affect generalisability, with the trial populations not being entirely made up of the chronically infected patient population as defined by the EMA and European and French consensus conference. 27
In line with the potentially slightly poorer health (based on FEV1% values) of the COLO/DPI/02/0666 trial patients, the BMI was also, on average, lower than in the EAGER trial. 65 However, it should be noted that these differences have not been subjected to statistical scrutiny and may not be significant. The clinical relevance of differences of this size are also uncertain.
Concomitant medication use could not be compared between trials as few data were provided (after a request for clarification from the Assessment Group) for the EAGER trial. 65 Many allowed medications (e.g. macrolides and bronchodilators) that could affect FEV1% measurements, and their impact on the trial results are unknown, and may be different between studies.
In terms of prior antipseudomonal use, patients in the COLO/DPI/02/06 trial66 had all used nebulised tobramycin immediately before the trial, whereas only around 25% of patients in the EAGER trial65 had used nebulised tobramycin immediately before the trial [with an additional 55% (approximately) having used it within 3 months prior to the trial]. As such, patients in the COLO/DPI/02/0666 trial may have been more tolerant of tobramycin in terms of AEs, and will have experienced the initial peak in tobramycin FEV1% results within the first 4 weeks of the run-in period, rather than during the trial itself. Conversely, the EAGER trial65 had a proportion of patients who were not tobramycin tolerant having never used tobramycin, and a proportion who had not used tobramycin immediately prior to the 28-day washout period. Some or all of these patients may have experienced an initial peak in efficacy (see Table 6) during the trial, and may be more likely to experience AEs associated with tobramycin than patients in the COLO/DPI/02/0666 trial.
Given that age, BMI, concomitant medications, prior exposure to antipseudomonal antibiotics and FEV1% all have prognostic value in CF, it is difficult to determine whether or not these cohorts are comparable in terms of overall health and propensity to benefit from antipseudomonal treatments.
| Study | Age: mean (SD) | Gender: male/total (%) | BMI (kg/m2): mean (SD) | FEV1% predicted mean | Concomitant and previous treatment | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Intervention | Comparator | Intervention | Comparator | Intervention | Comparator | Intervention | Comparator | Intervention | Comparator | |
| EAGER trial65 | 26 (11.4) < 13 years, 9.1%; ≥ 13 years, 90.9% |
25 (10.2) < 13 years, 8.6%; ≥ 13 years, 91.4% |
171/308 (55.5) | 115/209 (55.0) | 20.7 (4.0) | 20.4 (3.5) | 53 (SD 14.2, SE 0.81) | 53 (SD 15.9, SE 1.11) | Chronic macrolide use n = 187 (60.7%) CiC information has been removed Use of antipseudomonal antibiotics prior to first dose (n, %):
|
Chronic macrolide use n = 125 (59.8%) CiC information has been removed Use of antipseudomonal antibiotics prior to first dose (n, %):
|
| ≥ 25 < 50: 41.6 | ≥ 25 < 50: 42.6 | |||||||||
| ≥ 50 ≤ 80:a 58.4 | ≥ 50 ≤ 80:a 57.4 | |||||||||
| COLO/DPI/02/0666 | Mean (SD): 21.3 (9.72) years | Mean (SD): 20.9 (9.30) years | 103/183 (56.3%) | 101/190 (53.2%) | Mean (SD): 18.67 (3.396) | Mean (SD): 18.46 (3.584) | 51.76 (SE 1.02) | 50.82 (SE 0.99) | Any medication (93.4%) Mucolytics (74.3%) Selective β2-adrenoreceptor agonists (76.5%) Macrolides (49.7%) Azithromycin 85 (46.4%) Dornase alpha 94 (51.4%) Glucocorticoids 66/183 (36.1%) Anticholinergics 34/183 (18.6%) |
Any medication (94.2%) Mucolytics (79.1%) Selective β2-adrenoreceptor agonists (71.2%) Macrolides (51.3%) Azithromycin 97 (50.8%) Dornase alpha 105 (55.0%) Glucocorticoids 67/191 (35.1%) Anticholinergics 39/191 (20.4%) |
| COLO/DPI/02/0566 | ≥ 8 to < 13 years; 37.5% ≥ 13 years 62.5% | NR | NR | Overall for participants, mean (SD): 19.99 (4.011) | 75.92 (SE 11.86) | 79.51 (SE 7.707) | Concomitant: NR Patients were permitted to continue with pre-existing non-antipseudomonal CF medications Bronchodilators: refrained from use 4 hours prior to pulmonary function test; salbutamol administered as rescue medication for bronchoconstriction after either intervention or comparator administration Previous: all were on nebulised colistimethate sodium |
|||
Study withdrawals
Table 7 shows the number of participants in each arm of each trial and the numbers of participants who withdrew throughout the study. Both trials saw a relatively high dropout rate, and this was higher in the intervention arm of both major trials. 65,66 In both key trials, there appear to be data missing and unaccounted for in some analyses, with more patients missing than are listed as withdrawals in the intention-to-treat (ITT) analyses for EAGER trial65 [commercial-in-confidence (CiC) information has been removed] (see Table 7). The implications of these missing data points are unknown.
Table 8 describes the reasons for withdrawals. In both trials, more patients withdrew owing to AEs than for any other single reason, with withdrawal of consent/patient request the second most common reason. In the EAGER trial,65 AEs accounted for proportionately more withdrawals in the tobramycin DPI arm than in the nebulised tobramycin arm. Similarly, more patients withdrew consent for the trial in the DPI arm. In the COLO/DPI/02/06 trial,66 the same pattern was seen, with more patients withdrawing from the colistimethate sodium DPI arm than from the nebulised tobramycin arm owing to AEs, although withdrawals owing to patient request were lower in the DPI arm. In this trial, the difference between arms appears larger than in the EAGER trial,65 although the absolute number of withdrawals is smaller in COLO/DPI/02/06. 66 Differences between the two trials in dropout numbers may be attributable to differences between patients’ tolerance to nebulised tobramycin at baseline; patients who tolerated nebulised tobramycin poorly were likely to have been excluded before randomisation in COLO/DPI/02/06. 66 The Forest Laboratories submission to NICE66 reports 16 screening failures but it is unclear if these patients failed during the run-in period because of lack of tolerance for tobramycin. However, if this was the case, it could account for at least some of the difference in withdrawals between arms, and between the two main studies.
| Study | No. randomised | No. who withdrew before medication (%) | No. in ITT analysis: n (%) | No. who withdrew after medication or lost to follow-up:a n (%) | No. in PP analysis | |||
|---|---|---|---|---|---|---|---|---|
| T0 | 28 days | 20 weeks | 24 weeks | |||||
| EAGER trial65 Total | 553 | 36 (6.5) | 517 (93.5) | 462 (83.5) | 398 (72.0) | 391 (70.7) | 121 (21.9)a | CiC information has been removed |
| Intervention | 329 | 21 (6.4) | 308 (93.6) | 268 (81.5) | CiC information has been removed | CiC information has been removed | 83 (25.2)a | CiC information has been removed |
| Control | 224 | 15 (6.7) | 209 (93.3) | 194 (86.6) | CiC information has been removed | CiC information has been removed | 38 (17.0)a | CiC information has been removed |
| COLO/DPI/02/0666 Total |
380 | 7 (1.8) | 373 (98.2) | CiC information has been removed | CiC information has been removed | LOCF:c 373 (98.2) Com: 324 (85.3) |
53 (13.9) | 298 (78.4) |
| Intervention | 187 | 183 (97.9) | CiC information has been removed | CiC information has been removed | LOCF:c 183 (97.9) Com: 153 (81.8) |
32 (17.1) | 141 (75.4) | |
| Control | 193 | 190 (98.4) | CiC information has been removed | CiC information has been removed | LOCF:c 190 (98.4) Com: 171 (88.6) |
21 (10.9) | 157 (81.3) | |
| COLO/DPI/02/0566 | 16b | 0 | 16 | – | – | – | 3 (18.8) | 11 (68.8) |
| Reasons | EAGER trial,65 attrition: n (%) | COLO/DPI/02/06,66 attrition: n (%) | COLO/DPI/02/0566 | ||
|---|---|---|---|---|---|
| Intervention | Control | Interventiona | Controla | Attrition throughout crossover (n = 3/16) | |
| AE | 40 (13.0) | 17 (8.1) | 18 (9.8) | 3 (1.6) | One patient discontinued after receiving dry powder due to cough, throat irritation and unpleasant taste and did not cross over to nebulised treatment. Two patients withdrew owing to AEs, having already completed nebulised treatment |
| Death | 3 (1.0) | 0 (0) | 0 (0) | 2 (1.1) | |
| Consent withdrawnb | 24 (8.0) | 9 (4.3) | NA | NA | |
| Patient request | NA | NA | 9 (5) | 11 (5.8) | |
| Lost to follow-up | 5 (1.6) | 3 (1.4) | NA | NA | |
| Administrative reason | 1 (1.2) | 0 (0) | NA | NA | |
| Protocol violation | 6 (0.3) | 5 (2.4) | 1 (0.5) | 0 (0) | |
| Lack of efficacy | NA | NA | 2 (1.1) | 1 (0.5) | |
| Inappropriate enrolment | 0 (0) | 1 (0.5) | NA | NA | |
| Other | 4 (1.3) | 3 (1.4) | 2 (1.1) | 4 (2.1) | |
| TOTAL | 83/308 (26.9) | 38/209 (18.2) | 32/183 (17.5) | 21/190 (11.1) | 3/16 (18.8) |
Study end points and outcomes
The outcomes reported across the three studies are documented in Table 9,65,66 alongside the outcomes listed in the NICE scope, and the outcomes recommended by the EMA research guidelines. 18
All outcomes requested by NICE were reported in the two major trials; however, where a study reports that an outcome is measured, this does not necessarily indicate that the study was sufficiently powered to detect a clinically meaningful effect or that the outcome was assessed and reported according to EMA guidelines. 18 The COLO/DPI/02/05 trial66 was a Phase II safety trial and therefore the outcomes are more limited in this 8-week trial than the other two larger trials.
| Study | Clinical end point: respiratory function | Microbiological end point: resistance/susceptibility | Clinical end point: exacerbations | Biological end point | Physical end point | Quality-of-life end point | Safety end point | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FEV1 | Sputum density | MIC | No. of exacerbations | Time to exacerbation | No. of hospitalisations | Hospitalisation duration | No. of i.v. treatments | Inflammation or infection markers | BMI changes | HRQoL | AEs | Laboratory safety: haematology biochemistry/urinanalysis | |
| NICE scope | ✓ (lung function) | ✓ | ✓ | ✓ (frequency and severity) | ✓ (preference based) | ✓ | |||||||
| EAGER trial65 | ✓ a | ✓ | ✓ | NR Proxy: lung disorder |
✓ (patients not events) | ✓ | b | NR | ✓ c | ✓ | |||
| COLO/DPI/02/0666 | ✓ c | ✓ | ✓ d | ✓ | b | ✓ | ✓ (CFQ-R) | ✓ | |||||
| COLO/DPI/02/0566 | ✓ | ✓ (CFQ-R) | ✓ c | ✓ | |||||||||
The primary outcome for efficacy trials recommended by the EMA is change in FEV1%. 18 This outcome is reported by all three trials, and although it was not always the primary outcome of the trial, both major trials60,65,66 were powered to detect clinically relevant changes in FEV1%. The COLO/DPI/02/0666 trial followed the American Thoracic Society (ATS) guidelines. The methods by which FEV1% measurement were made (Table 10) were not clear within either the EAGER trial65 or trial COLO/DPI/02/05,66 which may allow a margin for imprecision and/or inaccuracy in the data, and is a potential source of bias in an open-label trial.
The EMA recommends that data on exacerbations, i.v. treatment and hospitalisations (as listed in Table 9) should be reported alongside FEV1% to establish clinical benefit to the patients in terms of harder, more clinically relevant outcomes. The EAGER trial65 did not define an acute exacerbation, and only provided data on a poorly defined AE termed ‘lung disorder’; the European Public Assessment Report (EPAR)84 for DPI states ‘Exacerbations and hospitalizations related to respiratory events were collected to support the data for the relative change from baseline in per cent predicted’, although these data were not provided by the manufacturer on request by the Evidence Review Group (ERG). The data stated only how many patients had at least one event, rather than the overall incidence of events (patients could have multiple events within the timescale of the trial). Incidence data were not provided on request from the Assessment Group. The COLO/DPI/02/0666 trial fully defined an exacerbation, and provided data on the time to the first event, and data on incidence on request from the Assessment Group. Additional antibiotic treatments for exacerbation did not have to be i.v. treatments in either trial reporting this outcome. 65,66
The EMA recommends a microbiological secondary outcome for all trials of bronchopulmonary infection with a clinical primary outcome (e.g. FEV1%),18 and these should include both measures of colony density (e.g. sputum density) and measures of resistance (e.g. MIC values). The two major trials both report measures of resistance but only the EAGER trial65 reports sputum density. Both trials report MIC50 for tobramycin (although it is assumed that the MIC values are MIC50 and not MIC90 in the EAGER trial,65 based on the quoted breakpoints matching MIC50 breakpoints), and COLO/DPI/02/0666 reports MIC50 for colistimethate sodium as well. Both trials provided these data at the old BSAC breakpoint of 8 mg/l for resistance but only COLO/DPI/02/0666 reported this outcome at the new breakpoint issued by BSAC of 4 mg/l. 85 As noted in Chapter 1 (see Measuring disease in cystic fibrosis), the relevance of MIC susceptibility breakpoints to inhaled antibiotics is debated owing to higher than usual concentrations reaching the lungs, and because colonisation usually comprises multiple phenotypes of P. aeruginosa, which have different sensitivities to antibiotics and have different sensitivity when grown in culture. However, the monitoring of breakpoints has relevance in indicating whether or not isolates are becoming more resistant as a population, rather than to indicate whether or not the treatment is likely to be effective at the concentrations delivered. Although the clinical and long-term relevance of increases in MIC50 remain unclear, and as the EMA guidelines recommend their use, the MIC50 concentrations are presented in this report for consideration.
The EMA recommend that BMI is recorded only in studies at least 6 months in duration. A low emphasis is placed on this outcome in both major trials, and this outcome is not listed in the NICE scope. Quality of life was not recorded in the EAGER trial65 and was recorded using a non-preference-based instrument in COLO/DPI/02/06. 66 All trials aimed to measure AEs but it was not clear how this was achieved in either of the major trials (see Table 10).
| Main outcomes | EAGER65 | COLO/DPI/02/0666 |
|---|---|---|
| FEV1% | ||
| Definition | NR (e.g. which equation was used to calculate percentage predicted) | Equations used to calculate FEV1% provided in manufacturer’s submission |
| Method of measurement | Increases in FEV1 from baseline (pre-dose Day 1) at all scheduled post-treatment visits (weeks 2, 5, 9, 13, 17, 21 and 25)a NR: method for measuring FEV1% and equipment used |
β2-Adrenoreceptor agonists at least 2 hours prior to FEV1% measurement. Performed at the same time of day using suitable validated equipment available at the centre Testing performed according to ATS guidelines. FEV1% was calculated as: FEV1% predicted = [highest FEV1/predicted FEV1 (equations given in manufacturer’s submission)] × 100 |
| Acute exacerbation | ||
| Definition | NR Proxy outcomes include:
|
Protocol defined: use of i.v. antibiotics (with or without hospitalisation) plus four of: Change in appearance of sputum Increased productive cough, dyspnoea, or respiratory rate Progressive physical findings (crackles, rhonchi and air exchange) on chest auscultation New (infiltrates) intrusion on chest radiograph Lassitude and decreased exercise tolerance Fever (≥ 38 °C)
|
| Method of measurement | No. of patients requiring new antipseudomonal antibiotics No. of days and type of new antibiotic use No. of patients hospitalised for respiratory-related events and percentage receiving antibiotics (in hospital) |
Time from randomisation to first acute respiratory exacerbation (protocol and non-protocol) using the start date of i.v. antibiotic or visit ID at which reported No. of patients requiring new antipseudomonal antibiotic Time to first new antipseudomonal antibiotic No. of days of new antibiotic use |
| Microbiological | ||
| Definition | Microbial response: change in P. aeruginosa density [log10(CFU)/g sputum] from baseline Resistance: change in P. aeruginosa tobramycin MIC (MIC) susceptibility from baseline |
Microbial response: NR Resistance: MIC that inhibits 50% (MIC50) or 90% (MIC90) of isolates grown on agar For colistimethate sodium the a priori breakpoints applied to MIC50 were ≤ 4 mg/l = susceptible, 6 mg/l = intermediate susceptible, ≥ 8 mg/l = resistant. For tobramycin: ≤ 2 mg/l = susceptible, 4–6 mg/l = intermediate susceptible, ≥ 8 mg/l = resistant. For both, the new BSAC breakpoint (≤ 4 mg/l = susceptible, > 4 mg/l = resistant) was applied post hoc |
| Method of measurement | Microbial response: methods of measurement not reported. Expressed as mean reduction in P. aeruginosa sputum density at 4 and 20 weeks Resistance: P. aeruginosa tobramycin MIC (maximum MIC of all P. aeruginosa biotypes) > 8 μg/ml and ≤ 8 μg/ml |
Microbial response: NR Resistance: determined using the E-test® system (A B Biodisk, Sweden). Pulsed-field gel electrophoresis of DNA determined whether resistance had been selected in the original strain or whether the original organism had been replaced by a resistant variety |
| AEs | ||
| Definition | Categorised by MedDRA organ class and preferred term | Any untoward medical occurrence following the intervention, but which did not necessarily have a causal relationship with this treatment. Categorised by MedDRA organ class and preferred term (Version 7.0) was used by Chiltern to facilitate coding. Classed as definitely, probably or possibly related to study drug or with an ‘unknown’ relationship Severity defined as:
|
| Method of measurement | ‘Patient listings’ (not defined) were provided for AEs, SAEs, deaths and discontinuations due to AEs. Unclear whether reports of AEs were solicited or volunteered. Assume recorded at every study visit | All AEs were recorded in the study CRFs, including those volunteered (unclear whether events were also solicited by investigator) by the patient, as well as clinical or laboratory findings |
Quality assessment
The quality assessment of the three included studies is presented in Table 11. None of the studies performed consistently well for all quality assessment items. Internal validity items (as scored according to the CRD criteria)63 were addressed reasonably well, with the exception of blinding (owing to the open-label trial design). External validity items (scored according to the EMA research guidelines)18 were less well addressed, with omissions in a number of key recommendations. The two non-inferiority trials (EAGER65 and COLO/DPI/02/0666) did not perform well when analysed using the CONSORT (Consolidated Standards of Reporting Trials: www.consort-statement.org) checklist for non-inferiority and equivalence trials as a guide.
Risk of bias assessed using Centre for Reviews and Dissemination criteria63
All three studies included in the review stated that participants were randomised to treatment. 60,65,66 The method of randomisation was acceptable in the EAGER trial60,65 but was not clearly described in COLO/DPI/02/06. 66 The EAGER65 trial also used a modified randomisation method to balance patient characteristics between groups. It is stated in the Forest Laboratories submission66 that the method of randomisation used in the COLO/DPI/02/05 study67 was a randomisation list. The EAGER65 and COLO/DPI/02/0666 trials adopted a method of allocation concealment using an interactive voice system.
Participants were not blinded to treatment arm in two of the trials (EAGER65 and COLO/DPI/02/0666), although the Forest Laboratories submission66 states that FEV1 data were collected by a blinded investigator. The COLO/DPI/02/05 trial67 does not state whether or not any blinding was attempted. 67 Blinding would have been difficult to achieve in all studies because of differences in the mode of delivery of the two interventions. In addition, there may be a difference in taste between inhalations that would have been difficult to mask or simulate. However, failure to blind participants to treatment can still introduce performance bias, even where blinding is not possible. For example, sensitivity to both the potentially positive and negative effects of a novel treatment can be overestimated or underestimated by patients and carers according to their prior beliefs about a treatment. In this case, performance bias could affect outcomes, such as administration of antibiotics for suspected acute exacerbations, as clinicians may be more likely to administer antibiotics to patients undertaking DPI as the efficacy of the intervention was unknown.
Failure to blind the outcome assessor (be this the patient, a member of health-care staff or an independent outcome assessor) can lead to detection bias, whereby systematic differences in how outcomes are determined can arise due to the influence of prior beliefs concerning the effects of the treatment in question. Therefore, subjectively measured and interpreted data (such as AEs) should be interpreted with caution.
| Criteria | EAGER65 | COLO/DPI/02/0566 | COLO/DPI/02/0666 |
|---|---|---|---|
| CRD quality assessment items65 | |||
| Was the method used to generate random allocations adequate? | Y | Y | U |
| Was the allocation adequately concealed? | Y | U | Y |
| Were the groups similar at the outset of the study in terms of prognostic factors, for example severity of disease? | Y | U | Y |
| Were the care providers, participants and outcome assessors blind to treatment allocation? | N | N | N |
| Were there any unexpected imbalances in dropouts between groups? If so, were they explained or adjusted for? | Y, N | N, NA | Y, Y |
| Were all planned outcomes reported? | N | Y | N |
| Did the analysis include an ITT analysis? If so, was this appropriate and were appropriate methods used to account for missing data? | Y, Y, N | Y | Y, Y, Y |
| EMA research guideline items18 | |||
| Were patients stratified by severity at inclusion based on respiratory function tests or was an upper limit for FEV1 at inclusion set? | Y, Y | N | Y |
| Were patients stratified by age in paediatric studies? | NA | Y | NA |
| Was CF diagnosed by combination of sequential approaches? (See EMA18 report for list of acceptable techniques, pp. 11–12) | N | N | N |
| Was chronic lung infection confirmed by presence of P. aeruginosa in the bronchial tree for at least 6 months based on at least three positive cultures, with at least 1 month between them without direct (inflammation, fever, etc.) or indirect (specific antibody response) signs of infection and tissue damage? | N | N | N |
| Was FEV1 measured in a standard way? | U | U | Y |
| Was the primary end point appropriately chosen?a | N | U | Y |
| Was a primary end point of FEV1 supported by a secondary microbiological end point?b | N | N | N |
| If a study end point is the efficacy of respiratory function, was the end point appropriate?c | N | N | N |
| If a study end point is the slowing of rate of decline in respiratory function, was the end point appropriate? – As previous, but end point > 1 year (no consensus on how long) | NA | NA | NA |
| If a study end point was safety, was the end point comprehensive?d | N | N | N |
| If a study end point was quality of life, was the end point appropriate? – 3 months or more – uses CFQR or other measure validated in patients with CF | NA | N | Y |
| If a study end point was microbiological (e.g. sputum density), was the end point appropriate? – 28-day or longer follow-up | Y | NA | NA |
| If study recorded acute exacerbation frequency, was an acute exacerbation defined? | N | NA | Y |
| Is the study classed as a confirmatory trial? | N | N | N |
| Was the comparator an active control? | Y | Y | Y |
Baseline characteristics were reported in all three studies65–67 and were similar between groups in terms of age, gender and severity of disease. The COLO/DPI/02/0567 trial do not provide baseline data separately for intervention and control groups and selection bias cannot be assessed in this trial.
Two of the studies66,67 report that relatively similar numbers of patients in the intervention and control groups dropped out of the trial; however, data do not support this (see Table 7). The EAGER trial65 reports a somewhat higher attrition in the intervention group (26.9%) than in the control group (18.2%). It would seem that more evidence was recorded than was reported for some lung function measurements, for acute exacerbations and for BMI in the EAGER trial,65 which may indicate a degree of reporting bias.
All three included trials65–67 reported FEV1 data for both ITT and per-protocol (PP) populations. The EAGER65 and COLO/DPI/02/0666 trials reported that more participants were randomised than were included in the ITT analysis. For participants to be included in the analysis they had to have received at least one dose of the study drug. This cannot be regarded as true ITT analysis, as not all randomised participants were included. The EAGER trial65 did not perform imputation in its ITT analysis, and is therefore at high risk of attrition bias. The COLO/DPI/02/0567 trial reports ITT data that include all of the participants who were randomised to treatment, but it is not clear if imputation was performed.
Study quality assessed using European Medicines Agency research recommendations
All three studies65–67 met EMA research recommendations18 relating to severity of FEV1% at inclusion. In one study,65 patients were stratified by severity at inclusion based on respiratory function tests, and in two studies65,66 an upper limit for FEV1 at inclusion was set. Two studies also stratified patients by age (COLO/DPI/02/0666 and COLO/DPI/02/0566), although this was not requested by EMA guidelines18 in adult cohorts.
Although none of the studies reported the specific diagnostic strategy called for in the EMA guidelines,18 none of the studies were thought to have included patients who did not have CF; diagnosis of CF was reported as ‘documented from a specialised CF unit’ in two of the studies (COLO/DPI/02/0666 and COLO/DPI/02/0566), whereas one study65 reported the diagnosis as ‘confirmed cystic fibrosis’. The confirmed presence of a chronic P. aeruginosa infection according to EMA guidelines18 was not reported in any trial. All three trials instead used less stringent criteria, which may have led to inclusion of patients with an intermittent infection. The COLO/DPI/02/0666 trial followed the ATS guidelines to measure FEV1% and so scored well for standardisation of method. The methods by which FEV1% measurement were made were not clear within either the EAGER trial65 or trial COLO/DPI/02/05,66 which may allow a margin for imprecision and/or inaccuracy in the data, and is a potential source of bias in an open-label trial.
According to the EMA guidelines18 for CF and for the purpose of this clinical effectiveness review, only one of the studies had an appropriately chosen primary end point (COLO/DPI/02/0666) namely lung function described by FEV1%. The COLO/DPI/02/0566 and EAGER trials65 used the incidence of AEs as the primary end point. However, the EAGER trial65 was powered to detect effects in FEV1%. The COLO/DPI/02/06 and EAGER trials65 aimed to report outcomes at 24 weeks, which is just short of the ≥ 6 months’ follow-up recommended by the EMA for studies of respiratory efficacy, and does not comply with the 1-year follow-up recommended by the EMA for trials aiming to show slowing of rate of decline in respiratory function. Therefore, the studies are unlikely to provide useful data to indicate long-term outcomes. Only one of the studies65 supported the primary FEV1 end point with a microbiological end point (EAGER65). Only the COLO/DPI/02/0666 trial recorded and defined acute exacerbations, whereas the EAGER trial65 referred to ‘lung disorder’, described as ‘generally reported by the investigator as pulmonary or CF exacerbation’ and is therefore not a comprehensive definition of an acute exacerbation. The EPAR84 suggests that data on acute exacerbations were recorded but these were not made available to the ERG on request. All three studies had an active control group.
Study quality assessed using the CONSORT checklist for non-inferiority studies
Two of the three included studies adopted a non-inferiority design (EAGER65 and COLO/DPI/02/0666). An appropriate rationale for this statistical design is not provided within the Novartis Pharmaceuticals submission to NICE60 but this is justified in the corresponding peer-reviewed journal article. 65 The COLO/DPI/02/0666 does provide justification for using a non-inferiority design. One study does not claim to be a non-inferiority trial COLO/DPI/02/05. 66
With respect to the two non-inferiority studies (EAGER65 and COLO/DPI/02/0666), neither explicitly states whether or not its eligibility criteria and, subsequently, its participants, were similar to those in any trial(s) that established efficacy of the reference treatment and the settings and locations where the data were collected. Similarly, neither of the inferiority trials provide precise details of whether or not the interventions intended for each group are identical (or very similar) to that in any trial(s) that established efficacy, and how and when they were actually administered. As such, it is not clear that these trials adequately assess the efficacy and safety of the novel treatment (dry powder formulation), as it is not clear whether or not the population is comparable to the trial that justified the use of the reference standard (nebulised formulation).
Although both non-inferiority studies (EAGER65 and COLO/DPI/02/0666) have clearly defined primary and secondary outcomes, they do not state whether these outcomes are similar or identical to those that established efficacy in the reference treatment. Both studies do provide rationale for sample sizes based on non-inferiority power calculations for FEV1 data. Both studies provide results and confidence intervals (CIs) for the analysis of FEV1 data, which was the primary outcome for COLO/DPI/02/06. 66 However, the EAGER trial65 used safety (in the form of AEs) as the primary outcome and did not perform statistical analysis on these data.
Assessment of effectiveness
Lung function
The most commonly reported measure of lung function across the included studies was FEV1%. Data were sought at 4, 20 and 24 weeks where available. Tobramycin was administered 28 days on and 28 days off, which results in a peak and trough in FEV1% values. 65 This has the potential to bias results, and it would seem appropriate to consider results at both the peak and trough of the efficacy cycle. As such, both 20- and 24-week data are presented within this review.
The presentation of data varied across the studies (see data extraction tables in Appendix 4).
-
The COLO/DPI/02/0666 reported several analyses for FEV1% data. These included:
-
ITT population with last observation carried forward (LOCF) imputation
-
ITT population with no imputation (completers)
-
PP population with LOCF imputation, and
-
PP population with no imputation (completers).
-
Tests specified a priori showed that the data were non-normal in distribution; an additional non-parametric analysis and an analysis using logarithmic transformed data were also performed by the manufacturer to correct for this. 66 As such, 12 analyses were presented for these data. Analysis of covariance (ANCOVA) comparative data were reported, with adjustment for baseline FEV1% and pooled centre.
The EAGER trial65 data were not transformed, nor was a non-parametric test performed, although no test of normality was apparently planned or performed either. No imputation was performed on the Novartis Pharmaceuticals data, and only limited data were presented at 24 weeks. Some adjusted comparative data were presented, with adjustments for main effects treatment, baseline FEV1% predicted and pooled centre.
Where data were not available in the manufacturers’ submissions to NICE or within journal publications, the Assessment Group requested or calculated missing values. However, some values remained missing or unclear. Given the available evidence, a NMA was not possible (see Appendix 4) and, as such, a narrative synthesis is presented for the results.
Although all trials reported ITT analyses, the EAGER trial65 did not perform any imputation. In the COLO/DPI/02/06 trial,66 an analysis of completers is presented (where only those for whom there are data at both baseline and the time point of analysis are analysed) and an analysis with LOCF is presented (which should include all patients at every time point but appears to vary from time point to time point – see Table 12). The differences in exclusion of data in the ‘no-imputation’, ‘LOCF’ and ‘completers’ analyses are likely to affect results but it is unclear in which direction. The most usual direction of effect of attrition is to overestimate efficacy. 86–88 Attrition is most problematic in the EAGER trial65 ‘no imputation’ analysis, as demonstrated by the n numbers in Table 12. As such, results from the trials are not directly comparable. However, to allow some form of simple comparison to be made, data from the ‘no-imputation’ (EAGER trial)60,65 and ‘completers’ (COLO/DPI/06)66 analyses have been collated and synthesised in parts of this section. Note that the data used for COLO/DPI/0666 are from the original analysis, not the transformed or non-parametric analysis. PP analyses are discussed where appropriate. Further results are presented in the data extraction tables in Appendix 5.
Non-inferiority results
Both trials were non-inferiority trials but how comparable their definitions of non-inferiority are is not clear. Both the EAGER trial65 and the COLO/DPI/06 trial66 conclude that tobramycin DPI and colistimethate sodium DPI (respectively) are non-inferior to nebulised tobramycin.
The EAGER trial65 reports non-inferiority for tobramycin DPI at 20 weeks, supported by least-squares mean difference relative change of 1.1% [standard error (SE) 1.75], which has a lower limit of the one-sided 85% CI within the predefined 6% margin for non-inferiority. 65 As noted previously, this analysis was performed with no imputation of data in the ITT population. A non-inferiority analysis was not presented for the 24-week data, where FEV1% measurements are expected to be lower than at 20 weeks.
Trial COLO/DPI/02/0666 reports non-inferiority at 24 weeks (lower bound of the CI of < 3% for ITT and PP populations) for colistimethate sodium DPI under the non-parametric analysis [median difference in change in FEV1%, ITT, LOCF 0.56% (95% CI −2.16% to 1.00%), ITT completers 0.05% (95% CI −1.61% to 1.67%); PP LOCF 0.67% (95% CI −2.57% to 1.16%), PP completers −0.15 (95% CI −2.14% to 1.71%)] but not under the logarithmic analysis or original analysis [adjusted mean difference in change in FEV1%, ITT, LOCF −1.16% (95% CI −3.15% to 0.84%), ITT completers −0.43% (95% CI −2.59% to 1.72%); PP LOCF −1.49% (95% CI −3.79% to 0.81%), PP completers −0.99% (95% CI −3.48% to 1.51%)]. The non-parametric analysis was defined a priori, and all analyses relate to the ITT analysis with LOCF. Similar results were reported for data without imputation and data at 20 weeks.
Mean forced expiratory volume in first second percentage over time
The COLO/DPI/02/0666 and EAGER65 trials both had similar mean FEV1% values at baseline, although the patients in the EAGER trial65 had slightly higher FEV1% values, mean age and BMI. The COLO/DPI/02/05 trial66 started with much higher baseline mean FEV1% values. Mean FEV1% varies over time. The completers analysis presented in COLO/DPI/02/0666 shows a slightly improved FEV1% at 4 and 20 weeks in both treatment arms, with levels falling back to near baseline at 24 weeks (see Table 12). The LOCF analysis is consistently more conservative than the completers analysis, both in terms of absolute FEV1% values, and in terms of relative differences.
| Trial name | Intervention | Baseline | 4 weeks | 20 weeks | 24 weeks |
|---|---|---|---|---|---|
| COLO/DPI/02/0666 | Colistimethate DPI – completers | 51.51 (SE 1.12), n = 153 | 53.01 (SE 1.3), n = 153 | 52.79 (SE 1.42), n = 151 | 51.90 (SE 1.41), n = 153 |
| Colistimethate DPI – LOCF | 51.76 (SE 1.03), n = 183 | 52.60 (SE 1.21), n = 172 | 51.64 (SE 1.269), n = 181 | 50.86 (SE 1.26), n = 183 | |
| Tobramycin nebulised – completers | 50.97 (SE 1.06), n = 171 | 53.58 (SE 1.33), n = 171 | 53.51 (SE 1.418), n = 168 | 51.90 (SE 1.40), n = 171 | |
| Tobramycin nebulised – LOCF | 50.82 (SE 0.99), n = 191 | 53.09 (SE 1.265), n = 187 | 52.53 (SE 1.310), n = 190 | 51.19 (SE 1.30), n = 190 | |
| EAGER65 | Tobramycin DPI – no imputation | 52.9 (SD 14.2, SE 0.81), n = 308 | 54.38 (SE 0.63, SD 10.39), n = 268 | 55.97 (SE and n not reported) | NRa RC: 53.9, n = 222 |
| Tobramycin nebulised – no imputation | 52.8 (SD 15.9, SE 1.11), n = 209 | 54.70 (SE 0.54, SD 7.57), n = 194 | 55.28 (SE and n not reported) | NRa RC: 50.7, n = 161 |
Within the EAGER trial,65 FEV1% values increased at 4 and 20 weeks, although there are no data at 24 weeks. A rough calculation based on reported per cent mean change (provided by Novartis Pharmaceuticals as part of the clarification process) indicates that levels fall back towards or below baseline FEV1% mean values.
It is not possible to determine whether or not the changes seen in the colistimethate DPI arm are significantly different to the changes seen in the tobramycin DPI arm owing to a lack of data at 24 weeks, different population analyses of results and uncertain comparability of patient characteristics at baseline.
Comparative data at 4 weeks
At 4 weeks, three sets of data were available. COLO/DPI/02/0666 compared colistimethate sodium DPI with nebulised tobramycin, COLO/DPI/02/0566 compared colistimethate sodium DPI with nebulised colistimethate sodium, and the EAGER trial65 compared tobramycin DPI with nebulised tobramycin (see Table 13).
The difference in the unadjusted per cent mean change from baseline (Table 13) for colistimethate sodium DPI compared with nebulised tobramycin was −1.67 (SE 1.92) (data calculated by reviewer), whereas for tobramycin DPI compared with nebulised tobramycin this was −0.8 (SE 1.58) (data calculated by reviewer). For COLO/DPI/02/05,66 the difference in the unadjusted mean change from baseline was −3.01 (SE 8.01). In all cases, the intervention (DPI) appeared numerically worse than the comparator (nebulised). Significance statistics were not performed for these outcomes; hence it is not clear if this numerical difference is significant. PP data (not available for COLO/DPI/02/0566) showed a similar trend.
The smaller COLO/DPI/02/0566 trial, which had much higher mean baseline FEV1% values than the other trials, and compared colistimethate sodium DPI to colistimethate sodium nebulised solution, reported simply that there were no significant changes in lung function in either treatment arm. The short duration of the trial, small number of participants and higher mean baseline FEV1% values of this group mean that a meaningful comparison with the other trials cannot be made.
Comparative data at 20 and 24 weeks
Only two studies reported data at 20 and 24 weeks. The COLO/DPI/02/06 trial66 compared colistimethate sodium DPI with nebulised tobramycin, and the EAGER trial65 compared tobramycin DPI with nebulised tobramycin (see Table 13).
A comparison of data within trials at both 20 and 24 weeks is preferred, as tobramycin is given in a cycle of 28 days on and 28 days off, forming peaks and troughs in efficacy. However, there are gaps in the comparative data, as shown in Table 13.
| Time (weeks) | Study | No imputation: difference in per cent mean change from baseline between intervention and comparator (calculated by reviewer) | No imputation: adjusted difference | LOCF: adjusted difference (ANCOVA) |
|---|---|---|---|---|
| 4 | COLO/DPI/02/0566 (colistimethate DPI vs. colistimethate sodium nebulised) | −3.01 (SE 8.01) (RC) | NR | NR |
| COLO/DPI/02/0666 (colistimethate DPI vs. tobramycin nebulised) | −1.67 (SE 1.92) (RC) | NR | NR | |
| EAGER65 (tobramycin DPI vs. tobramycin nebulised) | −0.8 (SE 1.58) (RC) | NR | NR | |
| 20 | COLO/DPI/02/0666 (colistimethate DPI vs. tobramycin nebulised) | −2.63 (SE 2.06) (RC) | NR | −1.40% (95% CI −3.43% to 0.63%) (LOCF analysis) |
| EAGER65 (tobramycin DPI vs. tobramycin nebulised) | 1.1 (SE 1.75) (RC) | 0.59 (SE 0.92) (RC) | NR | |
| 24 | COLO/DPI/02/0666 (colistimethate DPI vs. tobramycin nebulised) | −0.59 (SE 2.23) (RC) | −0.43% (95% CI −2.59% to 1.72%) | −1.16% (95% CI −3.15% to 0.84%) |
| EAGER65 (tobramycin DPI vs. tobramycin nebulised) | 2.2 (SE 1.69) (RC) | NR | NRa |
At 20 weeks, both trials provide adjusted data, but the data for COLO/DPI/02/0666 are available only with a LOCF analysis. The adjusted difference in per cent mean FEV1% (LOCF) from baseline to 20 weeks was −1.40% (95% CI −3.43% to 0.63%) for colistimethate sodium DPI compared with nebulised tobramycin, and the adjusted absolute difference in mean FEV1% (no imputation) from baseline to 20 weeks was 0.59 (SE 0.92) for tobramycin DPI compared with nebulised DPI. The unadjusted difference in per cent mean change from baseline was −2.63 (SE 2.06) (data calculated by reviewer) for colistimethate sodium DPI, and 1.1 (SE 1.75) (data calculated by reviewer) for tobramycin DPI compared with nebulised tobramycin, although this analysis used least mean squares. The unadjusted difference in per cent mean change from baseline was 0.3 (SE 2.31) for tobramycin DPI (calculated by reviewer based on data provided in clarifications). PP analyses were similar at −2.95 (SE 2.40) and 0.3 (SE 2.31), respectively.
At 24 weeks, the per cent mean FEV1% values (see Table 12) show that both treatment arms in both studies fare better at 20 weeks than at 24 weeks. This difference is more pronounced for tobramycin DPI and nebulised tobramycin in both key trials. This is mirrored in the difference in per cent mean change from baseline (see Table 13). However, as the tobramycin DPI group experienced less of a drop in FEV1% than the nebulised tobramycin group at between 20 and 24 weeks (see Table 12), the difference in mean change from baseline appears larger at 24 weeks (see Table 13) than at 20 weeks. For the COLO/DPI/02/0666 trial, the difference in per cent mean change from baseline had narrowed, as the colistimethate sodium DPI arm had deteriorated less between 20 and 24 weeks than the nebulised tobramycin arm.
As discussed above, it is unclear exactly how comparable the baseline characteristics of the two groups are, and how comparable the completer and no-imputation analyses make the data, so between trial comparisons are difficult. It is also difficult to establish whether non-inferiority is maintained in the EAGER trial65 at 24 weeks, although it seems likely that it would be. In the COLO/DPI/02/0666 trial, non-inferiority was demonstrated in the non-parametric analysis at 20 and 24 weeks but not in the original analysis or logarithmic analysis.
Microbiological outcomes (colony density and resistance)
The EMA recommends that microbiological data should support FEV1% efficacy data. 18 The COLO/DPI/02/0566 trial did not report any microbiological data, whereas COLO/DPI/02/0666 reported resistance data (Table 14) and EAGER65 reported both resistance data and sputum density data (Tables 14 and 15).
As noted in Chapter 1 (see Measuring disease in cystic fibrosis), the BSAC breakpoints for resistance have changed since the trials were performed. COLO/DPI/02/0666 reported both the new (4 mg/l) and old (8 mg/l) breakpoints for resistance, whereas only data for the old breakpoint was available for the EAGER trial. In the COLO/DPI/02/06 trial,66 resistance (4 or 8 mg/l breakpoints) to colistimethate sodium remained very low (≤ 1.1%) throughout the 24-week trial in the colistimethate sodium DPI arm, whereas resistance to tobramycin was reported to not change substantially during the study, with values (CiC information has been removed). In the EAGER trial,65 resistance (8 mg/l breakpoint) to tobramycin started at around 20% (the baseline value was at the end of 28 days off treatment) and was lower at 24 weeks (also at the end of 28 days off treatment). Again, it is unclear if there was a trend of change in resistance over time or merely fluctuations around the mean. The high levels of resistance at baseline (CiC information has been removed) is not surprising as 100% of patients in the COLO/DPI/02/0666 trial and more than 90% in the EAGER had received tobramycin before the trial, and are likely to have already developed some level of resistance. As already discussed elsewhere, the significance of increasing resistance to tobramycin is unclear.
| Time point | EAGER65 | CiC information has been removed | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tobramycin | Comparison | CiC information has been removed | CiC information has been removed | CiC information has been removed | ||||||
| DPI: n = 308 | Nebulised: n = 209 | |||||||||
| MIC50 (mg/l) | MIC90 (mg/l) | MIC50 (mg/l) | MIC90 (mg/l) | CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | |||
| Tobramycin | ||||||||||
| Baseline | NR | NR | NR | NR | CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | ||
| Week 4 | NR | NR | NR | NR | CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | ||
| Week 8 | NR | NR | NR | NR | CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | ||
| Week 16 | NR | NR | NR | NR | CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | ||
| Week 20 | NR | NR | NR | NR | CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | ||
| Week 24 | NR | NR | NR | NR | CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | ||
| Colistimethate sodium | ||||||||||
| Baseline | NR | NR | NR | NR | CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | ||
| Week 4 | NR | NR | NR | NR | CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | ||
| Week 8 | NR | NR | NR | NR | CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | ||
| Week 16 | NR | NR | NR | NR | CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | ||
| Week 20 | NR | NR | NR | NR | CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | ||
| Week 24 | NR | NR | NR | NR | CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | ||
| MIC50 tobramycin, breakpoint 4 mg/l | ||||||||||
| 0–24 weeks | NRa | NRa | NR | NA | CiC information has been removed | CiC information has been removed | ||||
| MIC50 colistimethate sodium, breakpoint 4 mg/l | ||||||||||
| 0–24 weeks | NA | NA | NA | Throughout 24 weeks ≤ 1.1% Significance of change from baseline for whole group NR |
CiC information has been removed | CiC information has been removed | ||||
| MIC50 tobramycin, breakpoint 8 mg/l | ||||||||||
| Baseline | 22.1% resistant | 23.0% resistant | NR | NA | CiC information has been removed | CiC information has been removed | ||||
| 24 weeks | 19.1% resistant | 14.9% | ||||||||
| MIC50 colistimethate sodium, breakpoint 8 mg/l | ||||||||||
| NA | NA | CiC information has been removed | NA | CiC information has been removed | ||||||
Table 15 shows the results of the sputum density tests in the EAGER trial. 65 These data were not recorded in the COLO/DPI/02/06 trial. 66 Mean change from baseline log10 values show numerically greater reductions in sputum density are achieved with tobramycin DPI at 20 weeks than with nebulised tobramycin. Results at 24 weeks are not reported. These values are in accordance with the slightly greater increase in FEV1% seen for tobramycin DPI but their statistical and clinical significance is not known.
| Treatment group | Week 4 | Week 20 | ||||
|---|---|---|---|---|---|---|
| Mean change from baseline log10 CFU: unspecified biotype | Mean change from baseline log10 CFU: unspecified biotype | Mean log10 CFU: dry biotype | Mean change from baseline log10 CFU: dry biotype | Mean log10 CFU: mucoid biotype | Mean change from baseline log10 CFU: mucoid biotype | |
| Tobramycin DPI | −1.76 (SD 1.96) | −1.61 (SD 2.03) | 5.17 | −1.77 | 5.40 | −1.60 |
| Tobramycin nebulised | −1.32 (SD 2.03) | −0.77 (SD 1.78) | 6.18 | −0.73 | 6.30 | −0.92 |
Exacerbations
Data on protocol-defined acute exacerbations were not reported in a consistent way across the three included trials. COLO/DPI/02/0666 reported time to event data for acute exacerbations. Despite the EMA research guidelines requesting this outcome, Novartis Pharmaceuticals did not provide any acute exacerbation data, although among the AEs reported by Konstan et al. 65 lung disorder is defined as ‘generally reported by the investigator as pulmonary or CF exacerbation’. This is regarded as a proxy for acute exacerbations for the purpose of this assessment, although is clearly not an entirely specific measure and it is unclear what other events may have also been included in this outcome. The EAGER trial65 data were provided as the number of patients experiencing the event, rather than the number of events. Equally, for the time to event data provided by Forest Laboratories for COLO/DPI/02/0666 to be a useful outcome, it would have to be assumed that the time to the first event is directly related to the overall incidence of events. As one patient could theoretically experience more than one exacerbation through the duration of the trial (24 weeks), the Assessment Group requested data relating to the number of events from both manufacturers. Only Forest Laboratories complied with the request; however, the correct interpretation of these data was unclear. Neither manufacturer was able to provide data on the severity of exacerbations. Data relating to this outcome are presented in Table 16.
Numerically more patients on colistimethate sodium DPI experienced protocol-defined acute exacerbations compared with nebulised tobramycin in COLO/DPI/02/0666 (31.1% vs. 26.1%, respectively), although it is unclear if the same trend would be observed for data relating to the number of events. Conversely, the mean duration of use of antibiotics to treat the exacerbation was slightly less at 13.6 days in the colistimethate sodium DPI arm compared with 14.4 days in the nebulised tobramycin arm. Colistimethate sodium DPI had a more favourable exacerbation rate to nebulised tobramycin when non-protocol defined exacerbations were also considered (38% vs. 39%). The number of patients requiring antipseudomonal treatments was similar in both arms. Within the EAGER trial,65 the number of patients experiencing lung disorders was greater in the tobramycin DPI arm (33.8%) than the nebulised tobramycin arm (30.1%). It is unclear if the same trend would be observed for data relating to the number of events. Mean duration of antipseudomonal antibiotic treatment was also slightly shorter in the tobramycin DPI arm than in the nebulised tobramycin arm (30.9 days vs. 33.4 days). The number of patients receiving additional antipseudomonal treatments was higher in the tobramycin DPI arm.
In both trials there is disparity between the number of exacerbations, the number of days on treatment and the number of treatments given (see Table 16). The clinical advisors to the Assessment Group were unsure why this might be. In both trials the number of patients receiving treatment far exceeds the number of exacerbations. One potential explanation is that treatments are given as soon as an exacerbation is suspected, and stopped when tests do not confirm it. As the trial is open label, performance bias (e.g. being more likely to treat a patient’s symptoms as an exacerbation if they are in the DPI arm) and outcome assessment bias (the criteria for an acute exacerbation were subjective to some degree in the COLO/DPI/02/06 trial,66 and to an unknown degree in the EAGER trial65) could affect results. These types of bias could work to increase or decrease the number of exacerbations and number of patients receiving additional antibiotics, and it is unclear to what extent, and in what direction, they may have affected the outcomes in question.
In the case of the outcome ‘mean duration of antibiotic use’, the direction of effect is opposite to the direction of effect indicated by the number of exacerbations in both trials. As this is a mean value, it does not necessarily indicate that fewer days were spent overall on antibiotics or that DPI treatments reduce duration for which antibiotics are needed for any given exacerbation. This may indicate that the exacerbations were, on average, less severe in the DPI groups, or that exacerbations were not confirmed and treatment terminated more often in the DPI arms. Equally, it may indicate that less severe exacerbations were prevented less often, thus bringing down the mean duration of antibiotic administration. Finally, these small differences may reflect variations in clinical practice rather than demonstrating a real difference in antibiotic use between the two groups.
The influence of bias and the lack of consistency in the direction of effect makes the outcomes relating to acute exacerbations difficult to interpret with any certainty.
It is not possible to draw a comparative conclusion as to the relative efficacy between trials in terms of exacerbations, given the difference in the way they have been reported, and the uncertainty about the comparability of the patient data populations and characteristics.
| Outcome | EAGER65 | COLO/DPI/02/0666 | COLO/DPI/02/0566 | |||
|---|---|---|---|---|---|---|
| Tobramycin | Colistimethate sodium DPI (n = 183) | Tobramycin nebulised (n = 191) | Colistimethate sodium | |||
| DPI (n = 308) | Nebulised (n = 209) | DPI (n = 16) | Nebulised (n = 15) | |||
| No. of patients experiencing at least one (protocol defined) acute exacerbation: n (%) | NR | NR | 57 (31.1) | 50 (26.1) | NR | NR |
| No. of patients experiencing at least one (non-protocol defined) acute exacerbation: n (%) | NR | NR | CiC information has been removed | CiC information has been removed | NR | NR |
| No. of patients experiencing at least one (protocol or non-protocol defined) acute exacerbation: n (%)a | NR | NR | 69 (37.7) | 75 (39.3) | NR | NR |
| No. of patients experiencing at least one episode of ‘Lung disorder’ (sic):bn (%) | 104 (33.8) | 63 (30.1) | NA | NA | NA | NA |
| Time to acute exacerbation: mean no. days (SD) | NR | NR | 63.70 CiC information has been removed |
59.39 CiC information has been removed |
NR | NR |
| No. of patients with at least one hospitalisation:cn (%) | 75 (24.4) | 46 (22.0) | NR | NR | NR | NR |
| Hospitalisation duration: mean no. days (SD) | 15.6 (SE 13.31) | 15.3 (SE 10.23) | NR | NR | NR | NR |
| No. of patients using additional antipseudomonal treatments: n (%) | RC: 200 (64.9) | RC:114 (54.5) | 92 (50.3) | 96 (50.3) | NR | NR |
| Time to first additional antipseudomonal treatment: mean no. days (SD) | NR | NR | 55.28 (43.2) | 51.79 (41.9) | NR | NR |
| Duration of use of additional antipseudomonal treatment: mean no. days (SD) | 30.9 (23.34) | 33.4 (24.42) | 13.6 (5.4) | 14.4 (7.3) | NR | NR |
Body mass index
Table 17 shows the BMI measurements from the EAGER65 and COLO/DPI/02/06 trials. 66 The EAGER trial65 states that BMI is an outcome under investigation; however, the BMI data are not provided for any of the time points after baseline. The COLO/DPI/02/06 trial66 reports BMI at every time point. The data presented in Table 17 are for the ITT population and demonstrate very little change in BMI from baseline. BMI data for the COLO/DPI/02/0566 trial are not presented as BMI was not an outcome under investigation in that trial. It is unlikely that changes in BMI would be seen before 6 months, so the lack of change is unsurprising.
| Outcome | EAGER67 | COLO/DPI/02/0668 | ||
|---|---|---|---|---|
| Tobramycin | Colistimethate sodium DPI (n = 183) | Tobramycin nebulised (n = 191) | ||
| DPI (n = 308) | Nebulised (n = 209) | |||
| Baseline: mean (SD) | 20.77 (5.81) | 20.39 (3.45) | 18.67 (3.39) | 18.46 (3.58) |
| 24 weeks: mean (SD) | NR | NR | 18.70 (3.29) | 18.66 (3.57) |
| Change from baseline, week 24 | NR | NR | 0.08 (0.78) | 0.17 (0.89) |
Health-related quality of life
Table 18 presents the Cystic Fibrosis Questionnaire-Revised (CFQ-R) results for the HRQoL outcome. The two trials that investigated this outcome were COLO/DPI/02/0666 and COLO/DPI/02/05. 66 For the COLO/DPI/02/0666 trial the data presented were the adjusted means in the numerous domains of the CFQ-R from baseline to week 24. Most of the scores tended to be in favour of the dry powder intervention, although none of the differences were statistically significant. It is interesting to note that one of the few negative results is for the respiratory domain, although this does not reach significance. Although the COLO/DPI/02/0566 trial did also use the CFQ-R throughout the trial, the data were not provided. (CiC information has been removed.) As already noted, this quality-of-life measure has not been validated and is not preference based in line with the NICE reference case. It is therefore difficult to interpret the results in terms of impact on HRQoL and relative weight of the individual items in the measure.
One key argument put forward in favour of the DPI formulations is its ease of use. This may have many benefits, including increased patient satisfaction with treatment, reduced treatment burden, and increased compliance with the medication. The results in Table 18 demonstrate a non-significant trend in improvements in treatment burden for colistimethate sodium DPI compared with nebulised tobramycin. In addition, the Treatment Satisfaction Questionnaire for Medication (TSQM) reported in the EAGER trial65 showed higher values in the tobramycin DPI arm (see Appendix 5 for data). It is worth noting that in both of the trials the comparator was administered using a PARI LC Plus jet nebuliser; this device requires approximately 15 minutes to administer the full dose of tobramycin. Nebulisers with quicker delivery time (around 5 minutes), such as the PARI eFlow jet nebuliser, are now on the market and are in widespread use (Diana Bilton, personal communication). However, these quicker nebulisers may still require time to maintain (cleaning) and assemble. With respect to the relative advantages and disadvantages, it remains unclear whether the reduced treatment burden and improved treatment satisfaction scores would remain significant when compared with the newer, quicker nebulisers.
| CFQ-R domain | COLO/DPI/02/0666 | COLO/DPI/02/0566 | ||||
|---|---|---|---|---|---|---|
| Colistimethate sodium DPI (n = 183) | Tobramycin nebulised (n = 191) | Adjusted difference | p-value | Colistimethate sodium | ||
| DPI (n = 16) | Nebulised (n = 15) | |||||
| Physical | 0.26 | −1.56 | 1.82 | 0.353 | NR | NR |
| Vitality | 0.86 | −1.40 | 2.27 | 0.293 | NR | NR |
| Emotion | 2.23 | 0.47 | 1.75 | 0.244 | NR | NR |
| Eating | 0.48 | 0.66 | −0.19 | 0.925 | NR | NR |
| Treatment burden | 5.62 | 2.75 | 2.87 | 0.091 | NR | NR |
| Health perceptions | 0.25 | −2.71 | 2.96 | 0.159 | NR | NR |
| Social | 3.10 | 0.92 | 2.18 | 0.153 | NR | NR |
| Body image | 7.83 | 5.98 | 1.85 | 0.385 | NR | NR |
| Role | 0.65 | 1.87 | −1.22 | 0.607 | NR | NR |
| Weight | 0.88 | −1.93 | 2.81 | 0.461 | NR | NR |
| Respiratory | 2.99 | 3.51 | −0.53 | 0.756 | NR | NR |
| Digestion | 5.06 | 2.89 | 3.22 | 0.077 | NR | NR |
Adverse events
Table 19 shows the number of AEs by severity across the three trials. The percentage of patients experiencing any AE in all three of the trials is high, although this is to be expected in a patient population with CF, who have a high level of baseline AEs. Mild and moderate AEs appear similar between arms within trials. The EAGER trial65 does not state how many events were severe, whereas both of the colistimethate sodium DPI trials report more severe events in the intervention (DPI) arm. Serious adverse events (SAEs; which are internationally defined as AEs that cause death, are life-threatening, require hospitalisation/prolong hospitalisation or result in disability or birth defect89) appear to occur approximately equally but slightly less frequently in the DPI treatments in both key trials. There appears to be a marked difference between the proportion of serious events reported in the EAGER trial65 compared with the colistimethate sodium trials, even when comparing the nebulised tobramycin arms of the EAGER65 and COLO/DPI/02/06 trials. 66 This difference would appear to be too large to be explained by heterogeneity in study characteristics and study populations. It would seem more likely that this is due to a difference in interpretation of the ‘serious’ criteria between trials, rather than an indication that there is a true difference in number of events.
The number of patients withdrawing from the study due to AEs was higher in the dry powder intervention groups across all the trials. As previously discussed, patients in both trials were largely experienced with nebulised tobramycin, and it is likely that this difference in dropout rates is at least in part due to selection bias of patients tolerant to nebulised tobramycin, and desensitisation to its AEs through prior use.
| Outcome | EAGER65 | COLO/DPI/02/0666 | COLO/DPI/02/0566 | |||
|---|---|---|---|---|---|---|
| Tobramycin | Colistimethate sodium DPI | Tobramycin nebulised | Colistimethate sodium | |||
| DPI | Nebulised | DPI | Nebulised | |||
| No. of patients | 308 | 209 | 187 | 193 | 16 | 15 |
| Any AE | 90.3% | 84.2% | 175 (93.6%) | 172 (89.1%) | 16 (100%) | 9 (60%) |
| Mild or moderate AE | 73.4% | 68.5% | 159 (85.0%) | 165 (85.5%) | NR | NR |
| Severe (related) AE | NR | NR | 73 (39.0%) | 12 (6.2%) | 7 (43.75%) | 1 (6.6%) |
| SAE | 27.4% | 29.2% | 8 (4.3%) | 12 (6.2%) | 0 (0%) | 0 (0%) |
| Study drug-related AE | NR | NR | 153 (81.8%) | 90 (46.6%) | 16 (100%) | 9 (60%) |
| Patients withdrawn due to AE | 40 (13.0%) | 17 (8.1%) | 22 (11.8%) | 5 (2.6%) | 2 (12.5%) | 0 (0) |
Table 20 documents the most common AEs (≥ 5% in any group) occurring in any of the three trials. 65,66 The data presented relate to the number of patients who experienced AEs. Data on the actual number of events were available for the COLO/DPI/02/0666 and COLO/DPI/02/0566 trials, but are not presented here. The most common AE in all three trials65,66 is ‘cough’. The percentage of patients experiencing cough was higher in the COLO/DPI/02/0666 and COLO/DPI/02/0566 trials than in the EAGER trial,65 although this may again represent a difference in the definition of cough used in the studies rather than an actual difference in incidence of cough, as the difference persists when comparing the nebulised tobramycin arms of each trial. Cough was more common in the DPI intervention group for all trials. Cough is a known side effect of dry powder formulations and is thought to generally reduce over time, with improved technique, and may be controlled with use of bronchodilators, to some extent, in some patients. Clinical advisors to the Assessment Group were interested to note that haemoptysis did not appear to increase to any great extent in the tobramycin DPI group compared with the nebulised tobramycin group, although there does appear to be a small increase in haemoptysis in the colistimethate sodium DPI group compared with the nebulised tobramycin group. It is unclear whether this difference is clinically or statistically significant.
Although no statistical comparisons have been made, other AEs that appear to be worse in the DPI arm include (CiC information has been removed) chest discomfort and dysphonia in the tobramycin DPI arm in the EAGER trial,65 and throat irritation and dysgeusia in the colistimethate sodium DPI arm in the COLO/DPI/02/06 trial. 66 There are minor improvements in a number of other AEs in the colistimethate sodium DPI arm (see Table 20).
| Outcome | EAGER65 | COLO/DPI/02/0666 | COLO/DPI/02/0566 | |||
|---|---|---|---|---|---|---|
| Tobramycin | Colistimethate sodium DPI | Tobramycin nebulised | Colistimethate sodium | |||
| DPI | Nebulised | DPI | Nebulised | |||
| No. of patients | 308 | 209 | 187 | 193 | 16 | 15 |
| Cough: n (%) | 149 (48.4) | 65 (31.1) | 168 (89.8) | 151 (78.2) | 13 (81.3) | 7 (46.7) |
| Throat irritation: n (%) | AiC information has been removed | AiC information has been removed | 141 (75.4) | 84 (43.5) | 13 (81.3) | 3 (20.0) |
| Productive cough: n (%) | 56 (18.2) | 41 (19.6) | 38 (20.3) | 44 (22.8) | 2 (12.5) | 1 (6.7) |
| Dyspnoea: n (%) | 48 (15.6) | 26 (12.4) | 49 (26.2) | 52 (26.9) | 3 (18.8) | 4 (26.7) |
| Oropharyngeal pain: n (%) | 43 (14.0) | 21 (10.5) | 4 (2.1) | 1 (0.5) | 2 (12.5) | 2 (13.3) |
| Rales: n (%) | 22 (7.1) | 13 (6.2) | 2 (1.1) | 5 (2.6) | NR | NR |
| Rhinorrhoea: n (%) | 22 (7.1) | 15 (7.2) | 1 (0.5) | 2 (1.0) | NR | NR |
| Pulmonary function test decreased: n (%) | 21 (6.8) | 17 (8.1) | 0 (0) | 3 (1.6) | NR | NR |
| Pyrexia: n (%) | 48 (15.6) | 26 (12.4) | 23 (12.3) | 19 (9.8) | 2 (12.5) | 1 (6.7) |
| Dysgeusia: n (%) | AiC information has been removed | AiC information has been removed | 117 (62.6) | 53 (27.5) | 14 (87.5) | 3 (20.0) |
| Respiratory disorders: n (%) | 21 (6.8) | 18 (8.6) | 53 (28.3) | 57 (29.5) | 16 (100) | 7 (46.7) |
| Wheezing: n (%) | 21 (6.8) | 13 (6.2) | 31 (16.6) | 38 (19.7) | 7 (43.8) | 5 (33.3) |
| Chest discomfort: n (%) | 20 (6.5) | 6 (2.9) | 26 (13.9) | 34 (17.6) | 4 (25) | 2 (13.3) |
| Sinusitis: n (%) | 18 (5.8) | 15 (7.2) | 3 (1.6) | 2 (1.0) | NR | NR |
| Pulmonary congestion: n (%) | 17 (5.5) | 9 (4.3) | NR | NR | NR | NR |
| Dysphonia: n (%) | 42 (13.6) | 8 (3.8) | 22 (11.8) | 30 (15.5) | NR | NR |
| Nasal congestion: n (%) | 25 (8.1) | 15 (7.2) | 2 (1.1) | 4 (2.1) | NR | NR |
| Vomiting: n (%) | 19 (6.2) | 12 (5.7) | 6 (3.2) | 8 (4.1) | 2 (12.0) | 0 (0) |
| Haemoptysis: n (%) | 40 (13.0) | 26 (12.4) | 20 (10.7) | 13 (6.7) | NR | NR |
| Nausea: n (%) | 23 (7.5) | 20 (9.6) | 7 (3.7) | 9 (4.7) | NR | NR |
| Headache: n (%) | 35 (11.4) | 25 (12.0) | 9 (4.8) | 16 (8.3) | 1 (6.3) | 2 (13.3) |
| Fatigue: n (%) | 20 (6.5) | 10 (4.8) | 9 (4.8) | 8 (4.1) | NR | NR |
| Serious lung disorder: n (%) | AiC information has been removed | AiC information has been removed | NR | NR | NR | NR |
| Chest pain: n (%) | AiC information has been removed | AiC information has been removed | 13 (7.0) | 16 (8.3) | NR | NR |
| Crackles, lung: n (%) | NR | NR | 13 (7.0) | 14 (7.3) | NR | NR |
| Increased upper airway secretion: n (%) | NR | NR | 12 (6.4) | 13 (6.7) | NR | NR |
| Pharyngitis: n (%) | NR | NR | 10 (5.3) | 14 (7.3) | NR | NR |
| Rhonchi: n (%) | NR | NR | 8 (4.3) | 10 (5.2) | NR | NR |
Mortality
Three patients died in the tobramycin DPI group in the EAGER trial65 (Table 21). Two patients died in the nebulised tobramycin group in the COLO/DPI/02/06 trial. 66 None of the deaths is attributed to the study medication. (CiC information has been removed.) For the COLO/DPI/02/0666 trial there were two deaths (both in the nebulised tobramycin group), both of which were assessed as being unrelated to study medication, and were attributed to the underlying disease, although it is unclear if these deaths were also due to acute exacerbations. One clinical advisor to the Assessment Group noted that the number of deaths seemed high for the size of the cohorts and length of the studies, and may indicate that the selected population were not well defined for the purpose of the study. With the small number of events in all arms and the relatively short time horizon of the trials, it is very difficult to draw firm conclusions regarding mortality.
Compliance
Compliance with study medication was reported in both key trials, but it is not clear whether or not the methods and analyses provided are compatible between trials. In the COLO/DPI/02/0666 trial, fewer patients were compliant with medication in the colistimethate sodium DPI arm than in the nebulised tobramycin arm (66.7% vs. 70.7%, respectively, complied with > 75% of doses). The EAGER trial65 did not define how compliance was judged but simply states it was ‘generally high’, with > 90% compliance in both arms. It is not clear if these data include those who withdrew, but seems likely that it does not as the discontinuation rate was 26.7% in the tobramycin DPI arm and 18.2% in the nebulised tobramycin arm, so values for compliance would be expected to be lower if these patients were counted. In comparison, withdrawals in the COLO/DPI/02/0666 were 17.2% in the colistimethate sodium DPI arm and 14.2% in the nebulised tobramycin arm, and it is unclear if these are counted in the compliance figures reported. Results for both DPI formulations do not appear to support the manufacturer’s claim that the improved delivery time would result in better compliance.
Discussion
Three trials were included in the review of clinical effectiveness. Both colistimethate sodium DPI and tobramycin DPI were reported to be non-inferior to nebulised tobramycin in pivotal Phase III trials, for the outcome FEV1%. 60,66 A small trial comparing colistimethate sodium DPI to nebulised colistimethate sodium in a younger, healthier cohort of patients showed no significant change in lung function in either arm but was primarily a safety trial. 67
The quality of the included studies was generally poor to moderate. None of the trials scored well on all risk of bias items, with blinding and non-adherence to the EMA research guidelines18 being key problems. This could lead to selection bias and reporting bias for subjective outcomes, such as AEs, inaccuracies and imprecision in the results, and may limit the generalisability of the findings.
Specific criticisms of the data analysis for the EAGER trial65 include using an ITT analysis without imputation, and not providing an analysis at both 20 and 24 weeks. Criticisms of the COLO/DPI/02/0666 trial could include the use of a non-parametric analysis to show non-inferiority, although this analysis was defined a priori.
It was not possible to draw any firm conclusions as to the relative efficacy of any intervention compared with any other intervention (except nebulised tobramycin) owing to missing data, uncertain comparability of patient characteristics and incompatible data populations used when analysing the data. Both tobramycin DPI and colistimethate sodium DPI appeared to result in more exacerbations or more people experiencing at least one exacerbation (as indicated by the surrogate outcome ‘lung disorders’ in the EAGER trial65) than nebulised tobramycin, and it is unclear if these results support non-inferiority of the intervention to nebulised tobramycin. AEs were mostly similar between arms within trials, except for cough, which was higher in both DPI arms. More patients in DPI arms withdrew owing to AEs in both trials. 65,66 Resistance of around 20% was reported for tobramycin arms across both key trials; a rate of ≤ 1.1% was reported for colistimethate sodium DPI. The statistical and clinical significance of exacerbation, resistance and AE data is not known.
This review has been conducted to a high standard including comprehensive search strategies with study selection, data extraction and quality assessment checked by a second reviewer. It is limited by the small number of trials available, and methodological weaknesses and incompatibilities within the trials. There are variations in the definition and measurement of the key outcomes, owing to non-compliance with EMA research guidelines. No data that comply with the NICE reference case on quality of life were available from any of the trials.
A number of uncertainties remain, in particular:
-
The comparability of the patients in the two pivotal trials. 65,66
-
The comparability of the definitions of non-inferiority between the two pivotal trials. 65,66
-
The impact on estimates of efficacy of presenting ITT analysis with no imputation. 65
-
The adequacy of short-term data in predicting long-term outcomes.
-
The significance of resistance to tobramycin in terms of long-term outcomes.
-
How many patients would not be able to tolerate the DPI formulations.
-
The long-term impact of these treatments on mortality.
-
The impact of DPI on HRQoL.
-
Whether or not the definitions of ‘acute exacerbation’ used in the trials were generalisable to a wider population, given the lack of an international consensus.
In addition, although the key outcome measure, FEV1%, is the standard measure in CF research, it is considered by some within the research community to be insensitive to small changes, especially in early disease. However, the EMA still recommend that FEV1% should be the primary outcome measure, but should be considered in conjunction with microbial outcomes and ‘harder’ outcomes, such as acute exacerbations. In these trials,65,66 acute exacerbations or their surrogate appeared to be slightly higher in both DPI arms, although it is unclear whether the studies were powered for this outcome, whether these results were clinically or statistically relevant and whether 6 months is long enough to see an effect on exacerbations.
In summary, colistimethate sodium DPI and tobramycin DPI have both been reported to be non-inferior in terms of FEV1% in appropriately powered Phase III non-inferiority trials at 20 or 24 weeks. 65,66 However, it would appear that both DPI interventions may potentially result in more acute exacerbations and possibly more patients would not tolerate the formulation. A significant number of patients in both trials65,66 dropped out from the intervention arms due to AEs, and cough was reported more often in the DPI treatment groups than in the nebulised groups. A comparison of colistimethate sodium DPI to tobramycin DPI was not possible owing to data limitations and study heterogeneity. Both studies can be criticised for statistical analysis techniques and a lack of adherence to EMA research guidelines. 18 The long-term efficacy of either intervention is unknown, and trials recording and powered for non-surrogate outcomes, such as exacerbations and mortality over the longer term, are required.
Chapter 4 Assessment of cost-effectiveness
This section presents evidence concerning the cost-effectiveness of colistimethate sodium DPI and tobramycin DPI for the treatment of P. aeruginosa in patients with CF.
Systematic review of existing economic analyses
Cost-effectiveness review methods
Systematic literature searches were undertaken to identify published economic evaluations of colistimethate sodium and tobramycin for the treatment of CF. Details of the search strategies are reported in Chapter 3 (see Methods for reviewing clinical effectiveness) and the search strategies for the cost-effectiveness review are presented in Appendix 2. Hand-searching of sponsors’ submissions to NICE60,70 was also undertaken in order to identify any further studies missed by the electronic searches. The studies included in the review were critically appraised using the Drummond et al. checklist for economic evaluations. 90
Results of the systematic review
Three published studies were identified by the systematic searches. 5,91,92 None of these three studies relates to either colistimethate sodium or tobramycin in DPI form. However, these studies do provide some information concerning the costs and outcomes of the comparator therapies for this assessment and elucidate some of the key methodological problems surrounding the economic evaluation of treatments for CF. A critical appraisal of these studies is briefly detailed below.
Wolter et al.: Home intravenous therapy in cystic fibrosis: a prospective randomized trial examining clinical, quality of life and costs aspects
Wolter et al. 91 conducted a cost–consequence analysis of home i.v. antibiotic therapy in adult patients with an infective exacerbation of CF compared with hospital i.v. antibiotic therapy. The perspective of the study was not clearly stated; however, the authors appear to include costs incurred by the hospital and costs incurred by patients and families. The main clinical outcome measures assessed within the study were lung function and HRQoL. Seventeen adolescents and adults with CF attending two hospitals in Brisbane, Australia, were randomised between the groups (31 admissions: 13 home and 18 hospital). The median age of study subjects was 22 years (range 19–41 years), with no statistically significant difference between patient characteristics in the two groups regarding age, sex, FEV1 at admission or type of i.v. therapy received. Type of i.v. therapy included peripheral, portacath and central line.
Antibiotic treatment consisted of ceftazidime 2 g, 12-hourly, and tobramycin 4–6 mg/kg daily as a single bolus. Treatment was conducted for a minimum of 10 days and was guided by clinical response. Patients also received twice-daily physiotherapy plus 20 minutes of aerobic exercise. There were no statistically significant differences between the groups in terms of the duration of treatment or use of antibiotics. The median duration of treatment was 12 days (range 10–24 days) for the home group and 11 days (range 7–26 days) for the hospital group (p = 0.2).
Clinical outcomes were presented in terms of HRQoL, FEV1, FVC, weight gain (kg), 12-minute walking distance, sputum production over 12 hours, pulse oximetry, serum creatinine levels, aminoglycoside levels and audiology. HRQoL was measured using the Chronic Respiratory Disease Questionnaire (CRDQ),93 which measures change in dyspnoea, fatigue, emotion and patient’s feeling of control over the disease and its consequences (referred to as ‘mastery’). In addition, non-validated quality-of-life questions were also administered to patients, based on a grade out of seven, to assess the degree of disruption to their family, personal life, sleeping and eating as a result of their illness. The timing of outcome measurement within the study is summarised in Table 22.
| Outcome measure | Time point assessed |
|---|---|
| Spirometry (FEV1, FVC), pulse oximetry, 12-minute walking distance, sputum weight (production over hours), weight gain | Day 0, Day 10, Post Rx |
| Serum creatinine | Day 0, Day 2, Day 7 |
| Aminoglycoside levels | Day 2, Day 7 |
| Audiology | Before and after therapy |
| Dyspnoea score, fatigue score, emotional score, mastery score | Day 0, Post Rx |
| Family disruption, personal disruption, sleep disruption, eating disruption | Post Rx |
| Toxicity and complications: death, short-term readmission, drug attributable events | Unknown |
Costs were valued in Australian dollars (A$) at 1992–3 prices. Hospital costs were calculated using CF inpatient costs from the Prince Charles Hospital and from projected diagnostic-related group reimbursement figures. Home therapy costs were calculated based on hospital acquisition costs and consumption of resource. Staff costs spent on education and home visits were calculated from hourly wages. Travel costs were determined according to a standard cents-per-kilometre fee. Other patient and family costs were determined by interview; however, details are not given within the paper with respect to who undertook the interview. Costs were also compared in terms of mean total cost of therapy including the costs of home physiotherapy, home visits, training, equipment, drugs and bed occupancy.
Results are presented in terms of means and standard deviations (SDs); formal uncertainty analysis was not undertaken. Discounting was also not applied; however, this is reasonable given the short time horizon of the study. The headline results of the study are presented in Table 23.
| Outcome | Home (no. of admissions = 13) | Hospital (no. of admissions = 18) | p-value |
|---|---|---|---|
| Clinical outcomes | |||
| FEV1% predicted value: Day 0, Day 10, Day 21 | 39 (17), 45 (22), 43 (19) | 44 (20), 50 (21), 51 (21) | 0.27 |
| FVC% predicted value: Day 0, Day 10, Day 21 | 56 (19), 58 (21), 58 (22) | 58 (17), 64 (19), 66 (19) | 0.30 |
| CRDQ quality-of-life dimensions | |||
| Change in dyspnoea score | 5.9 (5.5) | 8.2 (5.4) | 0.25 |
| Change in fatigue score | 3.6 (3.4) | 6.8 (4.6) | 0.04 |
| Change in emotional score | 4.4 (5.2) | 8.6 (8.1) | 0.11 |
| Change in mastery score | 2.6 (3.4) | 5.5 (3.8) | 0.03 |
| Change in total score | 16.5 (14.8) | 29.5 (16.5) | 0.03 |
| Other quality-of-life dimensions | |||
| Mean change in family disruption | 6.2 (1.1) | 4.5 (1.3) | 0.001 |
| Mean change in personal disruption | 5.1 (1.0) | 3.8 (1.3) | 0.004 |
| Mean change in sleep disruption | 6.0 (1.3) | 4.4 (1.6) | 0.005 |
| Change in eating disruption | 6.6 (0.6) | 5.9 (1.5) | 0.07 |
| Change in total disruption | 23.9 (3.3) | 18.3 (3.3) | < 0.001 |
| Cost outcomes (A$ 1992–3) | |||
| Cost per day for families | A$15.08 (AUS$13.48) | A$23.77 (AUS$17.77) | NR |
| Crude mean hospital cost per episode | A$2476 | A$5028 | NR |
The authors conclude that home therapy is considerably less expensive for families than hospitalisation per day of hospitalisation (A$15.08 vs. A$23.77). The crude estimated cost saving for managing exacerbations at home compared with hospital was estimated to be A$2552. These estimates should be approached with some caution owing to the small sample size within the study. It is also unclear whether or not these findings would hold in a UK setting.
Thornton et al.: Clinical and economic choices in the treatment of respiratory infections in cystic fibrosis: comparing hospital and home care
Thornton et al. 5 assessed the cost-effectiveness of home-based i.v. antibiotics for respiratory exacerbations in adults with CF (not limited to P. aeruginosa lung infection) compared with hospital i.v. antibiotic therapy. The study was conducted from the perspective of a secondary care provider (NHS trust). The study was a retrospective, observational, 1-year pragmatic study analysed on an ITT basis. The primary clinical outcome was lung function measured in terms of FEV1%. One hundred and sixteen patients in the Manchester Adult Cystic Fibrosis Centre in the UK were recruited, 88.8% of whom had P. aeruginosa lung infection. The authors report that there were no differences in patient characteristics or FEV1% at the start of the study.
Treatment consisted of nebulised tobramycin, nebulised Colistin, nebulised rhDNase, nebulised gentamicin, oral antibiotics, inhaled/nebulised corticosteroids, regular oral corticosteroids, and inhaled/nebulised bronchodilators or oral bronchodilators. Patients received treatment over the course of 1 year and their outcomes were analysed, retrospectively. Patients were categorised as belonging to the ‘home’ or ‘hospital’ group if they received > 60% of treatment at home or in hospital correspondingly. The remaining patients who received 40–60% of treatment at home or in hospital were categorised as belonging to the ‘both’ group. Of the 116 patients, two patients (1.7%) received nebulised tobramycin and 58 patients (50%) received nebulised Colistin. During the study period patients in the ‘home’ group received a mean of 63 days (range 10–182 days) treatment in total, of which 52 days were at home and the remaining 11 days were in hospital. The ‘hospital’ group received a mean of 54 days (range 8–308 days) treatment in total, of which 45 days were in hospital and 9 days were at home. Patients in the ‘both’ group received a mean of 66 days (range 14–166 days) treatment, of which 40 days were at home and 26 days in hospital.
Health outcomes were presented in terms of FEV1%. The frequency of FEV1 measurement was different between the two groups. FEV1 for home-based patients was measured at the start and end of each course of i.v. antibiotics, whereas for hospital-based patients FEV1 was measured at admission, twice weekly, and at discharge. Two baseline FEV1 values were determined for each patient in the 1-year baseline period before the 1-year study period. The ‘best’ FEV1 was the highest FEV1 value recorded during the baseline year, with the ‘average’ FEV1 value being the mean of all FEV1 measurements during the baseline year. Treatment was defined as effective if lung function was maintained at the baseline ‘best’ FEV1 level, i.e. percentage decline in FEV1 was ≤ 0%. Given that it may be more reasonable to expect FEV1 to decline over time, an additional analysis with a less stringent definition of effectiveness of percentage decline in FEV1 of ≤ 2% was also performed. HRQoL was not measured or valued within the study.
Costs were valued in UK pounds sterling at 2002 prices. Unit costs were calculated from the NHS Trust, the CF Unit budget, the BNF and the hospital-supplied catalogue. Resource use and costs were estimated for i.v. antibiotics, disposable equipment, home kits, sputum microbiology, and sensitivity and blood drug level assays. The time spent with each patient was estimated using a time sheet completed by each staff member attending the patient. Staff costs were obtained from the CF Unit budget. Clinical records were used to determine the number of days patients spent in hospital relating to i.v. antibiotic treatment. Fixed costs for the ward and outpatient clinic were calculated from the CF Unit budget; these were used to estimate a fixed cost per hour related to an inpatient stay or clinic visit. A standard time per home visit was determined by interviewing staff. Travel time from the clinic to each patient’s home was estimated using data from the Automobile Association. The cost of travel for each home visit was calculated using a standard mileage allowance obtained from the hospital payroll department. Uncertainty analysis was conducted using non-parametric bootstrapping. Discounting was not applied to either the health outcomes or costs, presumably due to the short time horizon for the analysis. The headline results of the study are reported in Table 24.
| Outcome | Hospital (n = 51) | Both (n = 18) | Home (n = 47) | p-value |
|---|---|---|---|---|
| FEV1 baseline values | ||||
| Mean (SD) FEV1,% predicted ‘best’ | 59.3 (22.1) | 60.6 (19.1) | 64.7 (22.4) | – |
| Mean (SD) FEV1,% predicted ‘average’ | 49.3 (18.6) | 50.4 (16.0) | 54.8 (19.0) | – |
| Effectiveness and costs at end of 1 year | ||||
| Patients with decline in FEV1 ≤ 0% over study period: n (%) | 30 (58.8) | 9 (50.0) | 20 (42.6) | – |
| Patients with decline in FEV1 ≤ 2% over study period: n (%) | 32 (62.7) | 10 (55.6) | 20 (42.6) | 0.045 |
| Mean cost (£) per patient per year: cost (95% CI) | 22,609 (17,648 to 27,569) | 19,927 (13,433 to 26,421) | 13,528 (9989 to 17,068) | |
| Incremental cost-effectiveness (2002 UK pounds sterling) | ||||
| ICER (FEV1 ≤ 0%) | vs. both £10,923 | vs. home: £71,710 | – | – |
| ICER (FEV1 ≤ 2%) | vs. both: £12,878 | vs. home: £39,122 | – | – |
The authors reported that hospital-based treatment was more effective in terms of FEV1 but also more expensive compared with home-based treatment. The authors report that there was a decline in baseline average FEV1 in home-based patients, whereas there was an improvement for hospital-based patients (Tukey’s honesty significant difference mean difference 10.1%, 95% CI 2.9% to 17.2%; p = 0.003). The decline in FEV1 over the study period was significantly different using a criterion of decline in FEV1 of ≤ 2% (p = 0.045); however, it was not statistically significant using FEV1 of ≤ 0% (p-value not reported). Analysis of patients lung function on a course-by-course basis suggests that hospital-based patients had statistically significantly more courses of treatment in which lung function was maintained at baseline ‘average’ (FEV1 ≤ 0%) than home-based patients (17.4% compared with 9.0%; p = 0.001). For each course of treatment the improvement in FEV1 from the baseline ‘best’ was also statistically significantly higher for hospital-based patients than home-based (mean difference 4.6%, 95% CI 1.8% to 7.4%; p = 0.001). The cost of administering i.v. antibiotics at hospital was significantly higher than home-based therapy (mean difference £9005, 95% CI £3507 to £14,700; p < 0.001).
Incremental cost-effectiveness ratios (ICERs) were calculated separately using the two benefit criteria of decline in FEV1 of ≤ 0% and FEV1 of ≤ 2%. Cost-effectiveness planes and cost-effectiveness acceptability curves (CEACs) were also presented. The authors report that hospital-based care may be cost-effective with a 95% probability at a willingness to pay of £262,500 for one extra patient with a decline in FEV1 of ≤ 2%. However, using a stricter definition of lung function (decline in FEV1 of ≤ 0%) the probability that hospital-based care is cost-effective at a willingness to pay of £10M per patient is < 0.05.
Iles et al.: Economic evaluation of tobramycin nebuliser solution in cystic fibrosis
Iles et al. 92 report the methods and results of a cost–consequence analysis of inhaled TNS in children and adults with CF compared with usual therapy (Table 25). Usual therapy is referred to as actual clinical practice in the UK; however, no further details are provided within the paper. The study was conducted from the perspective of the NHS, with other interventions and medications taken on and off the study treatment being recorded. Lung function and body weight were the main dimensions of clinical outcome assessed. Seventy-one patients with P. aeruginosa lung infection were studied; 41 patients received TNS, of which 30 were matched with a paired control on usual therapy. The time horizon for the evaluation was 24 months. Outcomes and costs were not synthesised into an ICER.
Treatment in the TNS group consisted of 300 mg/5 ml TNS twice daily for 28 days, followed by 28 days without treatment. Treatment was conducted for 12 months, with patients monitored for 12 months prior to therapy. Patients in the TNS group were matched at the start of treatment to patients who had not had TNS therapy (the control group). Matching was conducted according to age (within ± 6 months), gender, lung function (within ± 20% FEV1% predicted) and chronic infection with P. aeruginosa. The authors state that the TNS group and control group were ‘well matched’ in terms of age, gender and pre-treatment FEV1.
Clinical outcomes were presented in terms of FEV1% and body weight expressed in the form of the net effect during the 1-year prior to therapy and 1-year following therapy. Impacts on HRQoL were neither measured nor valued. Resource use recorded within the study included days in hospital, length of i.v. infusions, clinics attendances, outpatient visits, i.v. courses, ward admissions and intensive care unit (ICU) admissions. The authors state that illness that occurred during or in between therapy (referred to as intercurrent illness), and surgical procedures were also recorded; however, these are not reported within the paper.
Costs were valued in UK pounds sterling at 2001 prices. Unit costs of ward and ICU stays were ascertained using routinely available NHS reference cost data. The mean unit cost of a hospitalisation day (for general medical paediatric and adult beds) and an ICU day was reported. The authors note that the mean cost of a hospitalisation day used in the analysis may be an underestimate of the true cost of ward care for patients with CF because the estimate does not specifically relate to this patient population. The number of days in hospital was recorded as the authors state that these are expensive and may be expected to degrade HRQoL and reduce patient’s educational possibilities or capacity for wage earning (although these outcome and resource impacts were not assessed within the study). No formal uncertainty analysis was undertaken and the results were not discounted.
Twenty-nine (71%) patients in the tobramycin group used inhaled antibiotics prior to therapy compared with 16 (53%) patients in the control group. In addition, 20 (49%) patients in the tobramycin group received inhaled Colistin during the alternating months off tobramycin. There were also imbalances between the tobramycin group and the control group in terms of the number of days of hospitalisation for the year before therapy (32.1 days compared with 27.0 days, respectively).
| Matched pairs | Year pre | Year post | Change | |
|---|---|---|---|---|
| FEV1% predicted (n = 30) | Tobramycin | 56.3 | 54.9 | −1.36 |
| Control | 57.4 | 55.8 | −1.63 | |
| Weight SD score (n = 27) | Tobramycin | −1.16 | −1.05 | 0.12 |
| Control | −1.27 | −1.24 | 0.03 | |
| Days in hospital (n = 30) | Tobramycin | 32.1 | 21.6 | −10.5 |
| Control | 27.0 | 14.1 | −12.9 | |
| Length of i.v. treatment (days) (n = 30) | Tobramycin | 57.6 | 33.5 | −24.1 |
| Control | 33.6 | 32.9 | −0.8 | |
| No. of clinics (n = 30) | Tobramycin | 10.8 | 8.1 | −2.66 |
| Control | 9.7 | 8.5 | −1.20 | |
| No. of outpatient visits (n = 30) | Tobramycin | 0.9 | 0.7 | −0.23 |
| Control | 0.2 | 0.4 | 0.19 | |
| No. of i.v. courses (n = 30) | Tobramycin | 3.6 | 2.3 | −1.27 |
| Control | 2.3 | 2.3 | 0.00 | |
| No. of ward admissions (n = 30) | Tobramycin | 2.9 | 2.0 | −0.94 |
| Control | 2.1 | 1.5 | −0.59 | |
| No. of ICU admissions (n = 30) | Tobramycin | 0.2 | 0.2 | 0.07 |
| Control | 0.1 | 0.1 | −0.01 | |
Mean FEV1% predicted decreased less in the study group (−1.36) than in the control group (−1.63); however, the authors do not state whether or not the difference was statistically significant. The increase in weight SDs was marginally greater in the treatment group (0.12) than in the control group (0.03); however, this was also not statistically significant (p-value not reported). The mean total number of days of hospitalisation decreased from 32.1 days to 21.6 days in the tobramycin group (a reduction of 10.5 days); however, there was a greater reduction of 12.9 days in the control group, which decreased from 27.0 to 14.1 days. The authors state that the figure before treatment in the control group was considerably increased by an outlier; a patient who was admitted as an inpatient due to pulmonary exacerbation before the period that corresponded to tobramycin treatment of his matched pair. This increase in the mean total number of hospitalisation days in only the control group prior to treatment may therefore have contributed to the greater reduction in hospitalisation days in the control group than in the tobramycin group. There was a statistically significant reduction in the length of i.v. treatment days in the tobramycin group compared with the control group: −24.1 days compared with −0.8 days (p < 0.001). The authors also report that in both the tobramycin and control groups there was a reduction in the number of hospital attendances compared with the year prior to therapy. However, the magnitude of the reduction was slightly greater in the control group and the authors attribute this to the inclusion of an outlier in the control group. There was a statistically significant reduction in the length of i.v. treatment days in the tobramycin group compared with the control group (p < 0.001). The authors report that the mean costs within the tobramycin-treated subgroup (41 patients) increased by £6292 over the study period; the majority of this difference was driven by the higher acquisition cost of tobramycin. However, the results of this study should be interpreted with caution owing to the case-matching design and the imbalances between the treatment groups.
Summary of published economic analyses
The review of the three published economic analyses presented above highlights the lack of relevant economic evidence relating to the cost-effectiveness of tobramycin and colistimethate sodium, in either nebulised or dry powder form, for the treatment of P. aeruginosa in patients with CF. Only one of the three studies is a cost-effectiveness analysis5 and, even in this case, the adopted measure of clinical benefit is difficult to interpret in a policy context. None of the three studies met the NICE reference case owing to their short time horizons. None of the included studies reported final patient outcomes in terms of life-years gained or QALYs gained.
Review of manufacturers’ submissions
This section presents a detailed exposition and critical appraisal of the economic evidence submitted by the manufacturers of colistimethate sodium DPI and tobramycin DPI. 60,66
The Novartis Pharmaceuticals submission (tobramycin dry powder for inhalation)
Novartis Pharmaceuticals60 submitted evidence to NICE relating to the clinical effectiveness of tobramycin DPI for the treatment of P. aeruginosa lung infection in patients with CF. The Novartis Pharmaceuticals submission presents the details of a NMA, a discussion of the difficulties of undertaking economic analyses of treatments for CF, and a brief discussion of three previously published economic analyses of CF treatments. 5,92,94 The submission makes particular note that these three economic analyses have deviated considerably from NICE’s reference case95 with respect to the primary health economic outcome measure adopted, which in each case relates to short-term FEV1 improvements rather than QALYs gained. The Novartis Pharmaceuticals submission does not include any form of de novo economic evaluation. Although the submission states that a cost–utility analysis was explored, this was not pursued owing to data limitations (including the absence of sufficient public domain information relating to the efficacy of colistimethate sodium DPI), a failure to demonstrate statistical significance within the NMA, and the presence of considerable heterogeneity in study design across the trials included in the network. In addition, at the time of writing their submission, Novartis Pharmaceuticals had not proposed a list price, or potential range of list prices, for tobramycin DPI. The submission therefore does not present any economic evidence for tobramycin DPI.
The Forest Laboratories submission (colistimethate sodium dry powder for inhalation)
The Forest Laboratories submission66 reports the methods and results of five clinical studies of colistimethate sodium DPI and an economic analysis of colistimethate sodium DPI compared with nebulised tobramycin using data from the Phase III COLO/DPI/02/06 trial. 96
The review of the Forest Laboratories economic model undertaken by the Assessment Group is divided into three parts: (1) a descriptive exposition of the model’s mathematical structure and the evidence sources used to inform its parameters; (2) a critical appraisal of the Forest Laboratories model including a summary of adherence to, and deviations from, the NICE reference case;95 and (3) a reanalysis of the Forest Laboratories model using assumptions deemed more appropriate by the Assessment Group. This critical review is based on four main evidence sources which were made available to the Assessment Group by Forest Laboratories:
-
a partially executable cost-effectiveness model developed using Microsoft Excel® version 2010 (Microsoft Corporation, Redmond, WA, USA) and Visual Basic for Applications
-
a written description of the methods and results of the economic analysis presented within the Forest Laboratories submission to NICE66
-
an accompanying mapping report detailing methods to estimate health utilities using data from the COLO/DPI/02/06 trial96
-
a detailed spreadsheet showing the translation of FEV1% to expected QALY gains (note: this was not included within the original Forest Laboratories submission, but was later provided by Forest Laboratories as part of the clarification process for the appraisal).
In addition, further clarification regarding the methods of the analysis was sought from Forest Laboratories by the Assessment Group over the course of the technology appraisal.
Exposition of the Forest Laboratories model
The Forest Laboratories submission presents a model-based cost–utility analysis of colistimethate sodium DPI compared with nebulised tobramycin from the perspective of the UK NHS. The model time horizon is short, but is unclear and inconsistent between health outcomes and costs. The primary economic outcome is presented in terms of incremental net monetary benefit, assuming a willingness-to-pay threshold of £30,000 per QALY gained. The form of the model would be most accurately described as a cohort-based decision analysis.
The economic analysis includes an estimate of incremental QALY gains accrued over a lifetime horizon, and the short-term costs associated with antibiotic drug acquisition and the management of exacerbations in each treatment group. The economic analysis draws on seven evidence sources: (1) patient-level data from the COLO/DPI/02/06 trial;66 (2) patient-level data from a study in which individuals completed both the child-friendly EQ-SD, Youth version (EQ-5D-Y) and CFQ-R;96 (3) a mapping study used to map from the CFQ-R instrument to the EQ-5D-Y;97 (4) observational data relating to an assumed relationship between 1- and 2-year mortality risk and FEV1% predicted;43 (5) a fixed estimate of life expectancy from the Cystic Fibrosis Foundation;98 (6) 2009–10 NHS reference costs;99 and (7) the BNF. 100
The general derivation of estimated QALYs for patients receiving either colistimethate sodium DPI or nebulised tobramycin within the Forest Laboratories model is summarised in Figure 8. This approach is based on three mapped relationships: (1) the translation of FEV1% predicted to the probability of mortality at 1 or 2 years; (2) the estimation of remaining life expectancy given the individual patient’s age; and (3) the translation of the CFQ-R to the EQ-5D-Y.
FIGURE 8.
Quality-adjusted life-year derivation within the Forest Laboratories model.
Predicted mortality differences between colistimethate sodium DPI and nebulised tobramycin were estimated by deriving regression equations for mortality at 1 year and 2 years using reported data on FEV1% predicted and death from a retrospective analysis of the risk of mortality by FEV1, FVC, arterial oxygen pressure (PaO2), arterial carbon dioxide tension (PaCO2), sex, weight and height. 43 Forest Laboratories fitted polynomial regression equations to the Kerem et al. data43 for 1 or 2 years by FEV1% group. The derived mortality risk equations are as follows:
All patients in the analysis are assumed to have a fixed survival duration of 37.4 years, based on an estimate from the Cystic Fibrosis Foundation. Remaining expected life-years for each individual patient were calculated as the patient’s remaining life expectancy (maximum survival − current patient age) multiplied by the probability of surviving beyond 1 or 2 years based on the regression equation.
Preference-based health utilities were not collected within the COLO/DPI/02/06 trial. 66 Forest Laboratories undertook a mapping exercise to cross-walk from the CFQ-R to the EQ-5D-Y using patient data from a German study reported by Eidt-Koch et al. 96 This mapping exercise produced a single utility value for patients with CF with chronic P. aeruginosa lung infection within the COLO/DPI/02/06 trial. 66 The calculations used to estimate the total QALYs gained are not entirely clear from the Forest Laboratories submission but appear to adopt the following general logic:
-
The mortality risk equation based on Kerem et al. 43 was applied to the individual patient’s FEV1% predicted score at baseline within COLO/DPI/02/0666 (Visit 0).
-
The individual patient’s remaining life expectancy was calculated as the difference between a fixed life expectancy of 37.4 years and the patient’s current age.
-
Expected QALYs before treatment were calculated as the probability of surviving at 1 or 2 years multiplied by the patient’s remaining life expectancy multiplied by a fixed utility score for patients with CF.
-
The Kerem et al. 43 mortality risk equation was applied to the individual patient’s FEV1% predicted score at 24 weeks (Visit 6).
-
The individual patient’s remaining life expectancy was calculated as the difference between a fixed life expectancy of 37.4 years and the patient’s current age.
-
Expected QALYs after treatment were calculated as the probability of surviving at 1 or 2 years multiplied by the patient’s remaining life expectancy multiplied by a fixed utility score for patients with CF.
-
The mean QALY change for patients receiving a given treatment was calculated as the difference between mean predicted QALYs before treatment and mean predicted QALYs after 24 weeks of treatment (i.e. the difference between Step 6 and Step 3).
-
Steps 1 to 7 were undertaken separately for colistimethate sodium DPI and nebulised tobramycin. Incremental QALYs between the colistimethate sodium DPI and the nebulised tobramycin groups were calculated as the difference in mean QALY change within each treatment group.
Remaining survival was discounted at a rate of 3.5%. The FEV1→QALY analysis excludes all patients already aged > 37.4 years and those for whom FEV1% predicted estimates were not available at both Visit 0 and Visit 6 within COLO/DPI/02/06. 66
Acquisition costs for nebulised tobramycin were taken from the BNF. 100 The model assumes a cost per dose of £21.20. The Forest Laboratories submission states that the annual cost of tobramycin is £7738, which corresponds to a regimen in which two doses of nebulised tobramycin are used each day, and each 28-day treatment period is followed by 28 days without nebulised tobramycin (Forest Laboratories’ submission,66 see Table 8).
Acquisition costs for colistimethate sodium DPI are not yet listed within the BNF. The Forest Laboratories submission states that if colistimethate sodium DPI was priced at parity with nebulised tobramycin it would cost £7738.00 per year (Forest Laboratories’ submission, p. 32). 66 The model, however, includes a parameter called ‘Colobreathe price’, which has a value of £21.20 per dose, which, if used twice daily on a continuous basis, as per the COLO/DPI/02/06 trial,70 would imply an annual cost of £15,476.00 (2 × £21.20 × 365). The Forest Laboratories submission later states that the proposed unit cost for colistimethate sodium DPI is £39.29 per dose (Forest Laboratories’ submission, page 33);66 if used continuously, this would imply an annual treatment cost of £28,681.70 (2 × 39.29 × 365). Forest Laboratories later stated via personal correspondence that the inclusion of this price within the submission was a mistake but is included here for the sake of transparency. Towards the end of the appraisal process, Forest Laboratories stated that their anticipated price for colistimethate sodium DPI was in the range £510.00 and £1100.00 for 56 doses. The range of potential prices for colistimethate sodium DPI are summarised in Table 26. Importantly, the costs of antibiotic treatment are not actually included in Forest Laboratories’ calculations of incremental net benefit. During the peer review process for this assessment (and following EMA approval), Forest Laboratories put forward an anticipated list price of £895 per 56 doses; this corresponds to an annual cost of £11,666.96 per patient.
| No. | Price statement | Annual cost (£) | Cost per dose (£) | Source |
|---|---|---|---|---|
| 1 | Lower end of anticipated price range | 6648.21a | 9.11 | Correspondence with Forest Laboratories during appraisal |
| 2 | Parity with tobramycin | 7738.00 | 10.60a | Forest Laboratories’ submission, p. 32 |
| 3 | Anticipated list price following EMA approval | 11,666.96a | 15.98 | Correspondence with NICE during peer review |
| 4 | Upper end of anticipated price range | 14,339.29 | 19.64 | Correspondence with Forest Laboratories during appraisal |
| 5 | Cost per dose in model | 15,476.00a | 21.20 | Forest Laboratories’ model parameter |
| 6 | Initial price | 28,681.70a | 39.29 | Forest Laboratories’ submission, table 9 |
Rates of CF exacerbations were estimated by calculating the mean time to exacerbation in each treatment group and converting this to the mean number of exacerbations within a 1-year period, thereby assuming a constant exacerbation rate. For colistimethate sodium DPI, the mean time to first exacerbation was estimated to be 63.6 days; the mean number of exacerbations within 1 year was then calculated as 365/63.6 = 5.74 exacerbations. For nebulised tobramycin, the mean time to first exacerbation was estimated to be 59.4 days; the corresponding mean number of exacerbations within 1 year was calculated as 365/59.4 = 6.14 exacerbations. Each exacerbation was assumed to cost £2587 based on NHS reference cost tariffs for the management of asthma with major comorbidities and complications without intubation.
All parameter values used in the Forest Laboratories model are summarised in Table 27.
| Model parameter | Value | Source |
|---|---|---|
| Health outcomes/treatment effectiveness | ||
| Health utility for CF | 0.68 | Jointly derived using data from the Cystic Fibrosis Foundation,98 Rowen and Brazier 201097 Kerem et al. 199243 and the COLO/DPI/02/06 trial66 |
| QALYs | ||
| Colistimethate sodium DPI, 1-year mortality model | 0.194 | |
| Colistimethate sodium DPI, 2-year mortality model | 0.209 | |
| Nebulised tobramycin, 1-year mortality model | 0.163 | |
| Nebulised tobramycin, 2-year mortality model | 0.168 | |
| Time to exacerbation: colistimethate sodium DPI | 63.6 | COLO/DPI/02/0666 |
| Time to exacerbation: nebulised tobramycin | 59.4 | |
| Resource costs (£) | ||
| Cost of managing an exacerbation | 2587.00 | NHS reference costs 2009–1099 |
| Assumed willingness-to-pay threshold (λ) | 30,000 | Forest Laboratories’ submission66 |
The incremental net benefit for colistimethate sodium DPI compared with nebulised tobramycin is simply calculated as:
where λ = assumed willingness-to-pay threshold; Coli = colistimethate sodium, and Tobi neb = nebulised tobramycin.
Health economic results reported within the Forest Laboratories submission
The base-case results from the Forest Laboratories economic analysis are summarised in Tables 28 and 29.
| Outcomes | Colistimethate sodium DPI | Tobramycin nebulised | Incremental |
|---|---|---|---|
| Health outcomes | |||
| QALY gained | 0.194 | 0.163 | 0.031 |
| Costs (£) | |||
| Costs of drug acquisitiona | 7738.00 | 7738.00 | 0 |
| Costs of managing exacerbations | 14,856.95 | 15,907.44 | 1050.49 |
| Total cost | 22,584.78 | 23,634.59 | 1050.49 |
| Cost-effectiveness outcomes (£) | |||
| Net benefit (assuming λ = £20,000/QALY) | −14,821.54 | −15,485.76 | 1670.49 |
| Net benefit (assuming λ = £30,000/QALY) | −14,821.54 | −15,485.76 | 1980.49 |
| ICER | Colistimethate sodium DPI dominates | ||
| Outcomes | Colistimethate sodium DPI | Tobramycin nebulised | Incremental |
|---|---|---|---|
| Health outcomes | |||
| QALY gained | 0.209 | 0.168 | 0.041 |
| Costs (£) | |||
| Costs of drug acquisitiona | 7738.00 | 7738.00 | 0 |
| Costs of managing exacerbations | 14,856.95 | 15,907.44 | 1050.49 |
| Total cost | 22,584.78 | 23,634.59 | 1050.49 |
| Cost-effectiveness outcomes (£) | |||
| Net benefit (assuming λ = £20,000/QALY) | −14,856.95 | −15,907.44 | 1870.49 |
| Net benefit (assuming λ = £30,000/QALY) | −14,856.95 | −15,907.44 | 2280.49 |
| ICER | Colistimethate sodium DPI dominates | ||
Irrespective of whether the 1- or 2-year mortality risk model is assumed, the Forest Laboratories economic analysis suggests that colistimethate sodium DPI dominates nebulised tobramycin. The Forest Laboratories submission also notes the following:
[The] Net benefit approach is very often used in cost-effectiveness analysis of health technologies. If price for the new technology (Colobreathe®) were at parity to TOBI® at £7,738 per annum, Colobreathe® would show a net benefit of £2280.49 per patient per year. This does not reflect the additional benefits compared to TOBI® which have not been modelled:
The more favourable performance of Colobreathe® with respect to antimicrobial sensitivity of respiratory tract isolates of P. aeruginosa. This will have impact both on costs and patient quality of life.
The costs of devices and consumables required for nebulisation
Carer time in relation to nebulisation by a predominantly young patient population
The benefit of the patient experience (ease of use) that is not adequately captured by the quality-of-life instrument.
Taking into account these additional benefits, the proposed price for Colobreathe® is £1,100 per pack66
Critical review of the Forest Laboratories model
Critical appraisal of the Forest Laboratories model
This section presents a detailed critical appraisal of the Forest Laboratories model. This critical appraisal should be interpreted in light of the limitations of the available evidence base surrounding the effectiveness of colistimethate sodium DPI as compared against other antibiotics for the treatment of P. aeruginosa lung infection as well as the context of care within which these treatments are used. Most patients with chronic P. aeruginosa will receive antibiotics for the rest of their lives. However, there is no long-term evidence to demonstrate the efficacy of colistimethate sodium DPI or tobramycin DPI beyond a maximum 24-week trial follow-up period (see Chapter 5) and the short-term trial evidence that is available does not include the direct measurement of HRQoL using a preference-based instrument (e.g. the EQ-5D). There is also only very limited evidence relating to survival benefits for either colistimethate sodium DPI or tobramycin DPI. The implications of these problems are discussed further below (see Methodological issues surrounding the economic evaluation of cystic fibrosis treatments). The use of modelling as a means of translating from intermediate end points to final outcomes, and/or for projecting beyond the termination of a trial, is not a substitute for empirical evidence and should thus be interpreted with an appropriate degree of caution. Given these limitations in the available evidence, the appropriate handling of uncertainty should therefore be considered key.
Despite the limitations of the evidence base, the Forest Laboratories model is subject to a number of methodological problems that are likely to produce considerable bias in the results. These concerns, limitations and biases are summarised in Box 1; specific issues are then discussed in more detail below.
Multiple deviations from the NICE reference case.
Conceptually inconsistent time horizon for costs and health outcomes.
Assumption of intermittent treatment using colistimethate sodium DPI.
Limitations of the CFQ-R→EQ-5D-Y mapping exercise.
Questionable validity of methods for estimating mortality benefits.
Incremental net benefit estimates may not reflect the proposed price of colistimethate sodium DPI.
Potential biases in modelling of exacerbation rates.
Omission of relevant costs and health impacts.
Incorrect application of discounting formula applied to future health gains.
Limited justification of modelling methods and identification, selection and use of evidence.
Multiple deviations from the National Institute for Health and Care Excellence reference case
Table 30 shows the extent to which the Forest Laboratories model adheres to the NICE reference case. The perspective of the economic analysis, namely that of the NHS, is appropriate. The use of discounting is however partial. No discounting is undertaken for costs owing to the short time horizon considered. Future QALY gains were discounted. The justification for presenting economic results in terms of incremental net benefit rather than the incremental cost per QALY gained is unclear. Further, the Forest Laboratories model is entirely deterministic and the submission report does not include any probabilistic sensitivity analysis (PSA). No justification is given regarding this exclusion. Simple sensitivity analysis is presented but this is limited to examining the differential impact of using 1- or 2-year mortality predictions.
| Element of economic analysis | Reference case | Comments |
|---|---|---|
| Defining the decision problem | The scope developed by the Institute | The submission report does not include a description of the scope of the decision problem to be addressed. The scope of the Forest Laboratories economic analysis is narrower than the scope of the appraisal.62 Only colistimethate sodium DPI is included as an intervention |
| Comparator | Therapies routinely used in the NHS, including technologies regarded as current best practice | Nebulised tobramycin is the sole treatment comparator considered within the analysis. The relative cost-effectiveness of colistimethate sodium DPI compared with nebulised colistimethate sodium or combination (cyclical switching) strategies is not considered |
| Perspective on costs | NHS and PSS | An NHS perspective was adopted. Only exacerbation costs over a 1-year period are included within the incremental net benefit calculation |
| Perspective on outcomes | All health effects on individuals | Health benefits for NHS patients are included. Short-term FEV1 changes are translated into QALY gains accruing over the patient’s estimated remaining lifetime |
| Type of economic evaluation | Cost-effectiveness analysis | The economic analysis takes the form of a cost-effectiveness analysis. Economic outcomes are expressed in terms of incremental net monetary benefit rather than the incremental cost per QALY gained |
| Synthesis of evidence on outcomes | Based on a systematic review | The economic analysis is based on one RCT (COLO/DPI/02/06) and other indirect evidence43,66,97,98 |
| Measure of health effects | QALYs | Health outcomes are valued in terms of QALYs gained |
| Source of data for measurement of HRQoL | Reported directly by patients and/or carers | Health utilities were derived from a mapping study to translate the CFQ-R to the child-friendly EQ-5D-Y.97 Preferences were valued using the adult EQ-5D tariff. Carer QALYs and process utilities associated with more convenient treatment are discussed but not included in the analysis |
| Source of preference data for valuation of changes in HRQoL | Representative sample of the public | |
| Discount rate | An annual rate of 3.5% on both costs and health effects | Costs were not discounted. Future QALY gains were discounted although the discount rate applied is incorrect |
| Equity weighting | An additional QALY has the same weight regardless of the other characteristics of the individuals receiving the health benefit | No additional equity weighting is applied to the modelled QALY gains |
Conceptually inconsistent time horizon for costs and health outcomes
As noted above, there currently exists no evidence relating to the long-term costs or health outcomes associated with colistimethate sodium DPI or nebulised tobramycin for the treatment of P. aeruginosa lung infection in patients with CF. The Forest Laboratories submission does not state the intended time horizon for their economic analysis. However, it is evident from the model exposition presented above that the adopted time horizon is inconsistent in terms of the time period considered for costs and that considered for health outcomes. The model uses changes in FEV1% predicted measured from baseline to 24 weeks within the COLO/DPI/02/06 trial66 and ‘extrapolates’ the impact of this shift in FEV1 to lifetime QALY benefits. The economic model therefore reflects the predicted long-term mortality benefits associated with 24 weeks of treatment only. In direct contradiction to this, exacerbation costs are arbitrarily modelled over a 1-year period, without any consideration of longer-term costs. As acquisition costs are excluded entirely from the incremental net benefit calculation, there is an implication underlying the economic analysis that treatment in the intervention and control groups is the same after the first 24 weeks. Given this mismatch between the time horizon for costs and outcomes, it is conceptually unclear how the time horizon for incremental net benefit should be interpreted. The Forest Laboratories submission provides no discussion or justification of this issue.
Assumption of intermittent treatment using colistimethate sodium dry powder for inhalation
There is a significant bias with respect to the options assessed within the Forest Laboratories model. As the model includes equal cost-per-dose parameters for colistimethate sodium DPI and nebulised tobramycin, the model appears to assume that colistimethate sodium DPI is used according to the same treatment schedule as nebulised tobramycin, that is, each 28-day treatment period is followed by a 28-day period without treatment. This reflects the licensed indication for nebulised tobramycin, but does not reflect either the protocol or practice of the COLO/DPI/02/06 trial or the licensed indication for colistimethate sodium DPI. Within the COLO/DPI/02/06 trial, patients allocated to the colistimethate sodium DPI group received treatment on a continuous basis. 70 The consequence of assuming a ‘cycle-on, cycle-off’ regimen is that the modelled treatment benefits reflect those associated with the continuous use of colistimethate sodium DPI at only half of the cost of generating these benefits. Unless colistimethate sodium DPI is priced at parity with the annual cost of nebulised tobramycin, this is inappropriate and produces a substantial bias in favour of colistimethate sodium DPI.
Limitations of the Cystic Fibrosis Questionnaire-Revised to EQ-SD-Y mapping exercise
The COLO/DPI/02/06 trial66 did not include the collection of data on HRQoL using a preference-based instrument. As a consequence, no data were available from the clinical trial to produce direct estimates of QALYs gained for colistimethate sodium DPI or nebulised tobramycin. However, the COLO/DPI/02/06 trial66 did include the use of a disease-specific measure: the CFQ-R. To estimate health utilities associated with colistimethate sodium DPI and nebulised tobramycin, Forest Laboratories undertook a mapping exercise using patient-level data from a published supplementary study in patients with CF. 96 The mapping exercise relates to two distinct patient cohorts: (1) an ‘estimation data set’ – Eidt-Koch et al. 96 and (2) an ‘application data set’ – the COLO/DPI/02/06 trial. 66 Collection of data for the estimation data set96 was undertaken in 2006 across four CF centres in Germany. Within this study, a cohort of 96 patients with CF completed both the German version101 of the CFQ49,53 and the EQ-5D-Y. 102 Patients included in this study were generally of young age (mean = approximately 13 years, range 8–17 years) and mean FEV1% predicted was generally high both in the children and adolescent groups (93.6% and 90.7%, respectively). Patient-level data from the estimation data set were used to produce a series of regression equations to ‘cross-walk’ from the CFQ to the EQ-5D-Y. The selected regression equation was then applied to patient-level data from the COLO/DPI/02/06 trial66 in which quality-of-life data were collected only using the CFQ-R instrument. Details relating to the estimation of alternative mapping functions in the child and adolescent populations were made available by Forest Laboratories as a technical appendix to the main submission. 97
There are a number of problems associated with the mapping exercise and its use within the colistimethate sodium DPI model; these are detailed below.
The NICE Decision Support Unit technical support document103 on the use of mapping in health technology appraisals states the following:
The characteristics of the estimation sample should be similar to the target sample for the mapping analysis, and should contain all variables from the target sample or included in the economic model that are thought to impact on EQ-5D scores. Under some circumstances, it may be appropriate for the estimation sample to include a broader range of people, providing that the target sample is sufficiently represented.
The comparability of the estimation data set96 and the application data set (COLO/DPI/02/06)66 appears to be subject to certain potentially important heterogeneities in terms of basic demographic and clinical variables. A crude comparison of patient characteristics within the Eidt-Koch et al. cohort96 and the baseline characteristics of patients recruited to COLO/DPI/02/0666 is presented in Table 31. In particular, only 65 patients (67.7%) included in Eidt-Koch et al. 96 had a bacterial colonisation of the lung, although it is unclear what proportion of these patients had chronic P. aeruginosa or an alternative type of bacterial infection. In addition, there are noticeable differences in terms of patient age and baseline FEV1 lung function.
| Variable | Estimation data set (Eidt-Koch et al.96) | COLO/DPI/02/06 at baseline66 | |
|---|---|---|---|
| Children (8–13 years) | Adolescents (14–17 years) | ITT population (6–56 years) | |
| Sex (male) | 43.6% (n = 24) | 58.5% (n = 24) | 54.5% (n = 374) |
| Age (mean/SD) | 10.8/1.7 | 15.9/1.80 | 21.1/9.49 58.8% patients were aged > 18 years |
| % vital capacity (mean/SD) | 92.5/11.9 (n = 47) | 97.2/13.1 (n = 34) | NR |
| %FEV1 (mean/SD) | 93.6/15.2 (n = 47) | 90.7/20.3 (n = 34) | Precise values not reported. FEV1 range 25–75% predicted required for eligibility |
| %MEF25 (mean/SD) | 68.4/41.7 (n = 47) | 58.9/37.5 (n = 34) | NR |
| Bacterial colonisation of the lung (%) | 63.6 (n = 35) | 73.2 (n = 30) | 100% infected with chronic P. aeruginosa |
| Pneumothorax (%) | 1.8 (n = 1) | 0 (n = 0) | NR |
| ABPA (%) | 3.6 (n = 2) | 12.2 (n = 5) | Exclusion criteria within trial |
| Pancreatic insufficiency (%) | 80.0 (n = 44) | 78.1 (n = 32) | NR |
| Hepatobiliary complications (%) | 23.6 (n = 13) | 26.8 (n = 11) | 33.3% in the colistimethate sodium DPI group; 40.3% in the nebulised tobramycin group |
| Distal intestinal obstruction (%) | 7.3 (n = 4) | 0 (n = 0) | NR |
| Diabetes mellitus (%) | 0 (n = 0) | 7.3 (n = 3) | NR |
| Nasal polyp (%) | 10.9 (n = 6) | 17.1 (n = 7) | NR |
| Isolation obligation for patient (%) | 1.8 (n = 1) | 9.8 (n = 4) | NR |
The Eidt-Koch et al. study96 recruited only a small number of patients (n = 96). Of these, 93 patients completed both the CFQ and the EQ-5D-Y, and 93 patients were included in the mapping exercise. 97 Inevitably, this leads to considerable uncertainty surrounding the use of the mapping function, none of which is addressed in the health economic analysis.
The Eidt-Koch et al. 96 publication states that 44.6% patients had no problems on any of the dimensions of the EQ-5D-Y. In other words, nearly half of the estimation data set cohort reported an EQ-5D-Y profile of (1,1,1,1,1), which represents a notional state of ‘perfect health’ (health utility = 1.0). This can be a common problem in utility mapping exercises, but is further compounded here by the small sample size of the estimation data set. At the lower ends of the scale, Eidt-Koch et al. 96 report that only ‘one or two patients reported extreme problems (level 3)’ on at least one of the dimensions of the EQ-5D-Y. As a consequence, the limited coverage of the EQ-5D state space within the estimation data set may call to question the validity of applying the mapping function to a cohort of patients with a generally higher level of disease activity.
Eidt-Koch et al. 96 used the child-friendly EQ-5D-Y (see Wille et al. 102). Within the mapping exercise, responses were valued using the UK adult EQ-5D tariff reported by Kind et al. 104 in which the lowest age of respondents was 18 years. A valuation tariff for children below this age does not currently exist.
The Forest Laboratories submission66 presents 12 mapping functions including ordinary least squares (OLS), Tobit regressions and Censored Least Absolute Deviations (CLAD) forms. Seven alternative regression models are presented for children (aged 8–13 years) and five alternative regression models are presented for adolescents and adults (aged 14–17 years). The Forest Laboratories submission states ‘The preferred model was chosen using root mean squared error, mean squared error and mean absolute error’. These selection criteria are appropriate. 103 However, neither the submission report nor the accompanying appendices state which mapping function was actually selected for use in the health economic model analysis. Further, while the Forest Laboratories submission claims favourable benefits in terms of improved ease of use and improved sensitivity for colistimethate sodium DPI, the use of a single health utility score within the model indicates that such potential benefits are not captured in the economic model.
Questionable validity of methods for estimating mortality benefits
Although mortality was recorded within the COLO/DPI/02/06 trial66 as a safety end point, the study was not powered to demonstrate a treatment benefit in terms of survival. Within the COLO/DPI/02/06 trial,66 two patients died during the study follow-up period, both of whom were allocated to the nebulised tobramycin group (see Table 21). Both of these deaths were reported to be unrelated to the study drug and were instead attributed to the underlying disease. Within the economic analysis, modelled differences in survival are captured by deriving and applying regression equations describing a potential relationship between FEV1% predicted and mortality at 1 year and 2 years from a retrospective analysis of the risk of mortality by FEV1%, PaO2, FVC, weight and height43 to patient-level changes in FEV1% observed within the COLO/DPI/02/06 trial. 66 This change in predicted survival is then weighted by a single utility score, discounted, and compared incrementally between treatments. There are a number of problems with this approach, as detailed below.
The Forest Laboratories analysis assumes that all patients have a fixed maximum life expectancy of 37.4 years. In reality, the trial cohort would be expected to follow a survival distribution. Furthermore, the potential QALY gains of individuals with an age greater than 37.4 years within COLO/DPI/02/0666 were excluded from the analysis (n excluded = 32). The impact of this bias on the cost-effectiveness of colistimethate sodium DPI is unclear.
The long-term mortality benefits included in the model are based on data presented within two figures reported by Kerem et al. 43 The Forest Laboratories submission itself notes that this assumed relationship is only ‘a suggestion’. 66 As the model applies a common health utility score for all patients irrespective of treatment group, this predicted survival benefit drives the entire QALY gain attributed to colistimethate sodium DPI. However, scrutiny of the Kerem et al. 43 publication indicates that increased mortality risk was also associated with decreasing PaO2, increasing PaCO2, increasing weight-for-height, and increasing age. Further, a multivariate regression presented within Kerem et al. 43 indicates that all variables except sex were statistically significant at the 5% level. It is therefore reasonable to suggest that the potential for confounding within the proposed FEV1% mortality relationship is substantial. The validity of using FEV1% as a single independent surrogate for mortality is not explored, justified or discussed within the submission.
Although Kerem et al. 43 clearly report categorical data, Forest Laboratories fitted their 1- and 2-year regression equations to the mid-point of each FEV1% category (Figures 9 and 10; refitted by the Assessment Group), thereby inappropriately treating categorical data as if they were continuous. It is unclear why Forest Laboratories needed to apply a regression equation (which, in itself, is an approximation), as it should have been possible to directly apply the Kerem et al. 43 mortality probabilities to the categorical FEV1% bands from COLO/DPI/02/06. 66 The value of the regression equation is thus unclear and is not justified within the submission.
It should also be noted that the FEV1% mean change from baseline value reported in COLO/DPI/02/0666 did not show that colistimethate sodium DPI was superior to nebulised tobramycin. A crude analysis of the raw trial data indicates undertaken by the Assessment Group indicates that the mean FEV1% change is actually more favourable for nebulised tobramycin than colistimethate sodium DPI. It therefore appears counterintuitive that a less favourable FEV1% improvement may lead to a more favourable estimate of QALY gain for colistimethate sodium DPI than with nebulised tobramycin.
FIGURE 9.
Fitted mortality probabilities predicted at 1 year by FEV1% category.
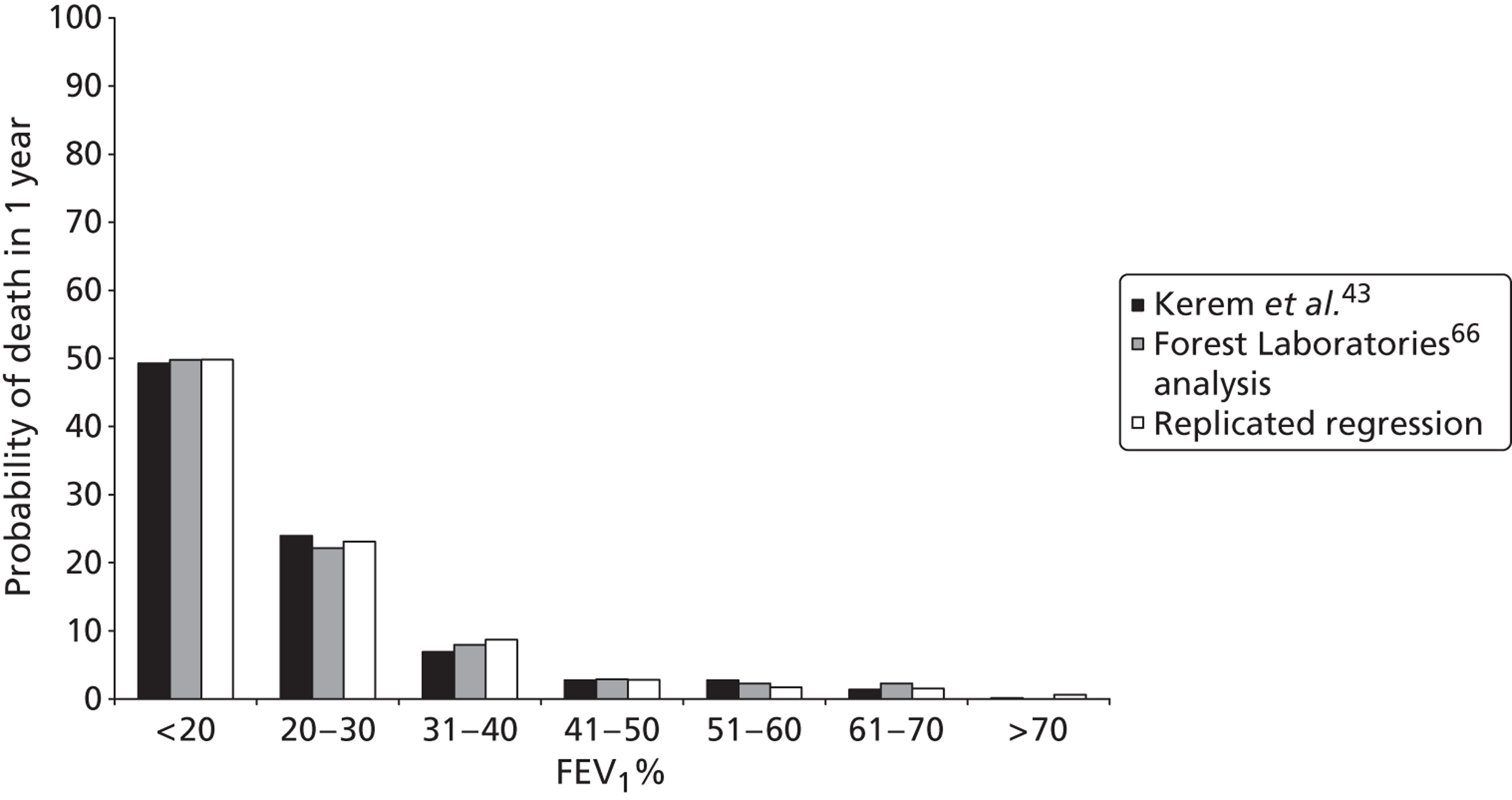
FIGURE 10.
Fitted mortality probabilities predicted at 2 years by FEV1% category.
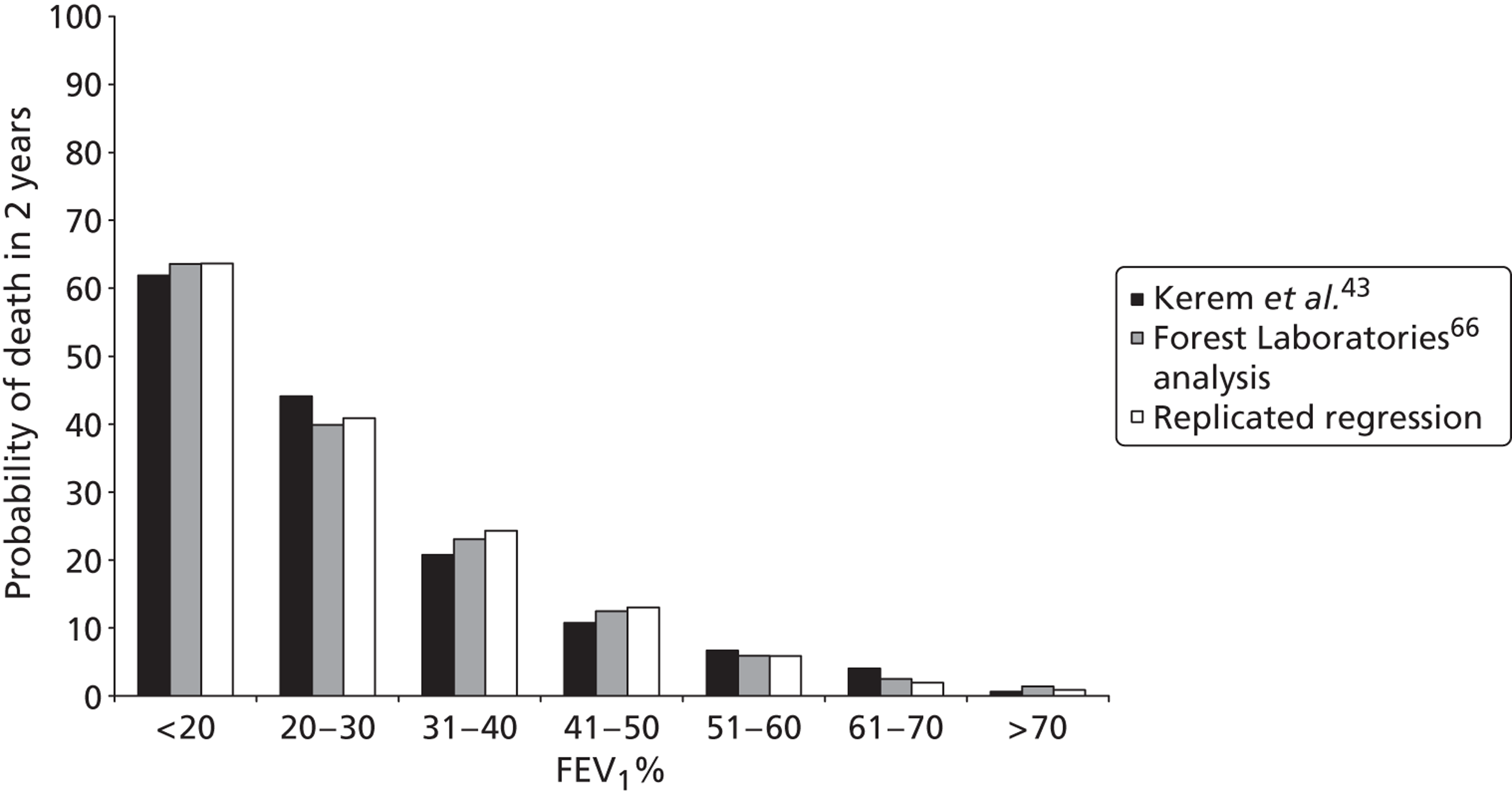
Incremental net benefit estimates do not reflect the price of colistimethate sodium dry powder for inhalation
On p. 32 of the Forest Laboratories submission, there is a suggestion that colistimethate sodium DPI will be priced at parity with nebulised tobramycin on an annual basis (£7738). 66 However, the economic model includes a parameter that indicates that the unit cost per dose of colistimethate sodium DPI is £21.20, which indicates price parity with tobramycin on a per-dose basis. As nebulised tobramycin is used on a ‘cycle-on, cycle-off’ basis, but colistimethate sodium DPI is not, the actual cost of colistimethate sodium priced on this basis would be double that of nebulised tobramycin over a 1-year period. The submission later states that as the model does not capture other benefits of colistimethate sodium DPI (more favourable performance regarding antimicrobial resistance, reduced costs of devices and consumables, reductions in carer time and ease of use), a per-dose price of £39.29 is proposed. This would obviously lead to a higher incremental cost for colistimethate sodium DPI than with nebulised tobramycin. Crucially, the positive incremental net benefits claimed within the submission do not therefore reflect either the appropriate dosage regimen or the actual proposed price of colistimethate sodium DPI; the results presented by Forest Laboratories therefore only reflect the scenario in which colistimethate sodium DPI is priced at parity with tobramycin on an annual basis. This is the lowest price suggested by Forest Laboratories (£9.11 per dose). As noted above, Forest Laboratories later proposed a cost of £895 for 56 doses of colistimethate sodium DPI (see Table 26).
Potential biases in modelling exacerbation rates
The Forest Laboratories model uses data on time to exacerbation for colistimethate sodium DPI and nebulised tobramycin from COLO/DPI/02/0666 to estimate the mean number of expected exacerbations over a 1-year period assuming a constant rate in each group. As the acquisition costs of the intervention and the comparator are not included in the incremental net benefit calculation, all predicted cost savings are driven by this element of the model. In order to produce these time-to-event estimates, Forest Laboratories must have used data on the observed number of exacerbations in each group together with the time at which they occurred. Therefore, the analysis takes the observed number and timing of exacerbations, converts these into time-to-event estimates, and then converts them back to the estimated number of exacerbations over a one year period. No justification of this approach is provided within the Forest Laboratories submission. It would have been more appropriate simply to use annualised exacerbation rates (taking into account censored observations) using the COLO/DPI/02/06 trial data,66 and a relative risk to reflect the differences between treatment groups.
Omission of relevant costs and health impacts
The systematic review of clinical effectiveness (see Chapter 3) reported that across many AEs, incidence was higher for colistimethate sodium DPI than nebulised tobramycin. Many of these AEs may be self-limiting and transient in nature, and the costs of managing them may be minimal. However, the Forest Laboratories model does not include any consideration of these costs or potential impacts on HRQoL and the submission report does not discuss or justify their exclusion.
Incorrect discounting formulae applied to future health benefits
Although the Forest Laboratories analysis includes discounting of future health benefits, the discounting formulae have been applied incorrectly. Within the Forest Laboratories FEV1% → QALY analysis, the discount weight is calculated using the following formula for each additional year of survival:
If this method is used to calculate discount weights, the discount rate r should be converted to (log[1 + r]). This error produces only a minor bias in the results.
Limited justification of modelling methods and identification, selection and use of evidence
The Forest Laboratories submission presents very little justification for the modelling approach adopted. The methods used to identify, select or use particular sources of evidence (e.g. Kerem et al. 43) are not discussed within the Forest Laboratories submission.
Reanalysis of the Forest Laboratories model by the Assessment Group
This section presents some simple reanalyses of the Forest Laboratories model to demonstrate the impact of some of the biases detailed above. These analyses are presented as detailed in Table 32 for both the 1- and 2-year mortality models.
| Revised scenario | Description of model amendment | ||
|---|---|---|---|
| Forest Laboratories’ base case | Lifetime benefits of 24 weeks’ treatment | No drug acquisition costs included | 1-year horizon for exacerbations |
| Scenario 1 | Colistimethate sodium DPI priced at £9.11 per dosea | 24-week horizon for exacerbations | |
| Scenario 2 | Colistimethate sodium DPI priced at £10.60 per dosea | ||
| Scenario 3 | Colistimethate sodium DPI priced at £15.98 per dosea | ||
| Scenario 4 | Colistimethate sodium DPI priced at £19.64 per dosea | ||
| Scenario 5 | Colistimethate sodium DPI priced at £21.20 per dosea | ||
| Scenario 6 | Colistimethate sodium DPI priced at £39.29 per dosea | ||
The results of these alternative analyses are presented in Table 33. It should be noted that this reanalysis does not fully resolve the problems regarding the model time horizon, the health impact of AEs, or the considerable uncertainty surrounding the estimation of QALY benefits for colistimethate sodium DPI. The results of the analysis suggest that the price of colistimethate sodium DPI and the time horizon for costs are highly sensitive within the analysis. The reanalysis suggests that if colistimethate sodium DPI is priced lower than nebulised tobramycin per annum, it may dominate due to modelled cost savings associated with avoided exacerbations and the estimated incremental QALY gains. For the range of higher prices per dose administered, the incremental cost-effectiveness of colistimethate sodium DPI compared with nebulised tobramycin is in the range £42,872–485,550 per QALY gained depending on assumptions regarding time horizon, mortality estimates and drug acquisition costs.
| Revised scenario | Incremental results for colistimethate sodium DPI vs. nebulised tobramycin | ||
|---|---|---|---|
| Incremental QALYs gained | Incremental costs (£) | Incremental cost (£) per QALY gained | |
| 1-year mortality prediction model results | |||
| Forest Laboratories’ base case66 | 0.031 | −1050.49 | Dominating |
| Scenario 1 (£9.11/dose) | 0.031 | −987.20 | Dominating |
| Scenario 2 (£10.60/dose) | 0.031 | −484.84 | Dominating |
| Scenario 3 (£15.98/dose) | 0.031 | 1329.05 | 42,872.44 |
| Scenario 4 (£19.64/dose) | 0.031 | 2563.03 | 82,678.35 |
| Scenario 5 (£21.20/dose) | 0.031 | 3088.99 | 99,644.80 |
| Scenario 6 (£39.29/dose) | 0.031 | 9188.10 | 296,390.38 |
| 2-year mortality prediction model results | |||
| Forest Laboratories’ base case66 | 0.041 | −1050.49 | Dominating |
| Scenario 1 (£9.11/dose) | 0.041 | −2138.94 | Dominating |
| Scenario 2 (£10.60/dose) | 0.041 | −1050.49 | Dominating |
| Scenario 3 (£15.98/dose) | 0.041 | 2879.60 | 70,234.12 |
| Scenario 4 (£19.64/dose) | 0.041 | 5553.23 | 135,444.61 |
| Scenario 5 (£21.20/dose) | 0.041 | 6692.81 | 163,239.24 |
| Scenario 6 (£39.29/dose) | 0.041 | 19,907.55 | 485,550.09 |
Discussion of available economic evidence
There is clearly considerable uncertainty surrounding the cost-effectiveness of alternative antibiotics for the treatment of P. aeruginosa in patients with CF. The review of published economic evaluations did not identify any directly relevant studies that report on the cost-effectiveness of colistimethate sodium DPI or tobramycin DPI compared with current standard treatments.
The Novartis Pharmaceuticals submission did not report any economic results for tobramycin DPI within their submission.
Forest Laboratories did present a simple economic analysis; however, this is subject to a number of methodological weaknesses, as detailed above. The majority of these weaknesses cannot be easily rectified given Forest Laboratories’ adopted model structure. One of the most significant problems within the Forest Laboratories model relates to the direct contradiction between the lifetime context of care and the apparently short time horizon adopted. As a consequence it is unclear how the results of the net benefit analysis should be interpreted. There is no obvious reason why colistimethate sodium DPI would be stopped after 24 weeks, yet Forest Laboratories’ economic analysis appears to imply the use of a ‘stopping rule’ at this point. This reflects the limitations of the trial evidence and the methods for estimating QALYs rather than what would be considered reasonable clinical practice. The disparity between the time horizon for clinical benefit, the cost of managing exacerbations and drug costs mean that it is impossible to interpret Forest Laboratories’ economic analysis in a meaningful way. Further, the methods for translating a lower level of FEV1 benefit for colistimethate sodium DPI into a greater number of QALYs than nebulised tobramycin remains counterintuitive. Given Forest Laboratories’ model structure, if the selected price of colistimethate sodium DPI is set to one of the prices which is higher than nebulised tobramycin, the cost per QALY gained for colistimethate sodium DPI compared with nebulised tobramycin is expected to be in the range £42,872–485,550.
The next section discusses the difficulties in undertaking a robust economic evaluation of colistimethate sodium DPI and tobramycin DPI in order to justify the modelling approach adopted by the Assessment Group and to highlight the uncertainties surrounding the results of the de novo analysis.
Methodological issues surrounding the economic evaluation of cystic fibrosis treatments
Undertaking a robust economic evaluation of alternative treatments for chronic P. aeruginosa in patients with CF represents a considerable challenge. There are a number of methodological issues which make such an evaluation difficult and, in turn, these lead to considerable uncertainty in the cost-effectiveness of colistimethate sodium DPI and tobramycin DPI. The most prominent of these are (1) the absence of any direct comparative evidence of the impact of either colistimethate sodium DPI or tobramycin DPI on HRQoL; (2) the use of a short time horizon within the pivotal trials of colistimethate sodium DPI and tobramycin DPI; (3) the questionable validity of relationships between the available intermediate end points and final outcomes; and (4) the limited availability of evidence on clinical outcome measures for all treatments relevant to the decision problem. These issues and their implications for the health economic analysis are briefly discussed below.
Absence of any direct evidence of the impact of treatment on health-related quality of life
Within the pivotal trials of colistimethate sodium and tobramycin DPI, HRQoL was not directly assessed using a preference-based health utility instrument. As such, it is not possible to directly estimate health utilities for each competing treatment option from these sources. Whilst Forest Laboratories reported a mapping exercise to translate the CFQ-R to the EQ-5D,97 the resulting estimates of health utility were not differentiated by treatment; instead a common mean value was applied to both treatments. If it is plausible that DPI treatment influences quality of life, the only means of quantifying this is by assuming some relationship between other clinical end points measured within the clinical trials and their impact on HRQoL.
The use of a short time horizon within the pivotal trials of colistimethate sodium dry powder for inhalation and tobramycin dry powder for inhalation
Research recommendations from the EMA CHMP state that for interventions which are intended to slow or stop pulmonary disease progression, a 12-month FEV1 end point should be used. 18 Although this 12-month end point would represent only the impact of treatment within a limited proportion of a patient’s lifetime, neither pivotal trial of colistimethate sodium DPI or tobramycin DPI met this criterion, as both trials were less than 6 months in duration. The adoption of such short study durations has three negative consequences: (1) the trial durations are insufficient to assess any treatment benefits in terms of potential mortality reduction; (2) uncertainties surrounding the relevance of intermediate outcome measures such as FEV1% predicted and final end points such as mortality are inflated by the absence of long-term evidence; and (3) evidence surrounding long-term AEs and treatment compliance is absent.
Owing to the short study durations adopted within the EAGER trial65 and the COLO/DPI/02/06 trial,66 mortality estimates are subject to very high levels of censoring (approximately 99% in each trial). Within the COL/DPI/02/06 trial, no patients died in the colistimethate sodium DPI arm, whereas two patients died within the tobramycin arm. 70 Within the EAGER trial,65 three patients died in the tobramycin DPI arm, whereas no patients died within the nebulised tobramycin arm. These event numbers are insufficient for comparative survival extrapolation over a lifetime horizon.
Validity of relationships between intermediate and final end points
As a consequence of the absence of direct comparative evidence of HRQoL impacts and the limited evidence of survival benefits, the economic analysis of treatments for P. aeruginosa requires some proposition and quantification of relationships between other clinical end points which may impact on HRQoL and/or survival. In order for an intermediate end point to be useful, it must represent an end point that can substitute for and be predictive of a final patient relevant clinical outcome. 105 Judgements about the credibility and validity of such relationships may be made on the basis of a range of evidence and may be interpreted within a hierarchy, as suggested by Taylor and Elston105 (Table 34).
| Hierarchical level | Evidence requirement | Source of evidence |
|---|---|---|
| Level 1 | Biological plausibility of relationship between surrogate outcome and final patient-related outcome | Pathophysiological studies and understanding of disease process |
| Level 2 | Consistent association between surrogate outcome and final patient-related outcome | Epidemiological (observational) studies demonstrating an association between the surrogate outcome and final patient-related outcome |
| Level 3 | Treatment effects on the surrogate correspond to effects on the patient-related outcome | Clinical trials showing that change in surrogate outcome with treatment is associated with a commensurate change in the final patient-related outcome |
Potential intermediate outcome measures include one or more of the following: FEV1% predicted, exacerbation rates and the incidence and duration of other AEs. 18 The plausibility and methodological problems of using these relationships to estimate the QALY gains associated with DPI treatment are considered below.
Relationship between forced expiratory volume in first second percentage predicted and health-related quality of life
Systematic searches were undertaken by the Assessment Group to identify any studies which attempted to quantify the relationship between FEV1% predicted and HRQoL in patients with CF (see Appendix 7). Searches were undertaken across MEDLINE, MEDLINE In-Process & Other Non-Indexed Citations, EMBASE, BIOSIS, Citation Indexes and The Cochrane Library. The searches identified just 12 studies, of which only one was relevant to CF. 108 Additional studies were identified by searching for evidence relating to specific symptoms associated with CF and its treatment (see Appendix 6), undertaking ad hoc searches and by hand-searching the manufacturers’ submissions. Four studies were identified which explored the potential relationship between health utility and a range of levels of FEV1% predicted. 108–111 Three of these studies were undertaken in patients with CF,108,110,111 whereas the fourth was undertaken in patients with chronic obstructive pulmonary disease (COPD). 109
Johnson et al. 108 report a prospective observational study assessing the relationship between a number of clinical variables including FEV1%, age, gender, BMI, and hospital admission, with SF-36 and EQ-5D quality of life in patients with CF. Fifty-nine patients were assessed at baseline and HRQoL was reassessed 1 year later by postal questionnaire. The observed mean change in EQ-5D index after 1 year was reported to be 0.000 (95% CI −0.069 to 0.069). The authors also reported the use of a multivariate OLS regression model to examine associations between the clinical variables and EQ-5D index scores. The results of this analysis suggested a statistically significant association between FEV1% and EQ-5D utility; however, the β coefficient for EQ-5D from the regression was reported to be 0.000 (SE = 0.001), which indicates that this relationship is unlikely to be clinically meaningful.
Bradley et al. 111 report a health utility study in patients aged ≥ 16 years, diagnosed with CF P. aeruginosa infections, and who were taking nebulised or oral antibiotics. Study subjects were recruited from specialist clinics across the UK. The results of this analysis were available as a conference poster111 and additional analyses were presented within the Novartis Pharmaceuticals submission. 60 Patients included in the study performed spirometry tests for FEV1 and completed the CFQ-R and the EQ-5D questionnaire. Mean EQ-5D utility across three FEV1% strata (70–99%, 40–69% and < 40%) was presented within the Novartis Pharmaceuticals submission. 60 In addition, this study reports utility decrements associated with minor exacerbations and major exacerbations. EQ-5D values reported within this study are summarised in Table 35. It is worth noting that the mean EQ-5D score for FEV1 < 40% reported by Bradley et al. 111 is higher than the mean cohort value produced by Forest Laboratories’ mapping exercise. 97
| FEV1% stratum/exacerbation severity | Mean EQ-5D (SD) |
|---|---|
| > 70% predicted | 0.864 (0.165) |
| 40–70% predicted | 0.81 (0.216) |
| < 40% predicted | 0.641 (0.319) |
| Major exacerbation | 0.174 decrement (0.341) |
| Minor exacerbation | 0.015 decrement (0.048) |
Yi et al. 110 reported a health utility study in adolescents with CF in order to assess how health status and clinical variables influence their health values. Sixty-five adolescents between the ages of 12 and 18 years completed Child Health Questionnaire (CHQ), the Health Utilities Index Mark 2 (HUI-2) questionnaire in addition to valuing their own current health state using time trade-off (TTO), standard gamble (SG) and visual analogue scale (VAS) elicitation methods. HRQoL estimates were presented according to four strata of FEV1% function (> 79%, 60–79%, 40–59% and < 40%). The results for TTO, SG and HUI-2 suggested only very small differences in health utility between the three strata of FEV1 of > 40%. Across these higher FEV1 strata there was no consistent relationship between worsening lung function and health utility for TTO, SG or HUI-2. In contrast, the VAS scores, which do not involve any form of trade-off between health states, did suggest a consistent decline in health utility with decreasing FEV1. For all instruments, the < 40% strata was associated with a lower level of HRQoL than other FEV1 states; this difference was most pronounced for the VAS but considerably less so for the preference-based methods. Health utility values estimated within this study are summarised in Table 36.
| FEV1 stratum | VAS | TTO | SG | HUI-2 |
|---|---|---|---|---|
| > 79% predicted | 0.85 (0.14) | 0.96 (0.08) | 0.92 (0.16) | 0.82 (0.15) |
| 60–79% predicted | 0.79 (0.12) | 0.97 (0.06) | 0.96 (0.08) | 0.85 (0.15) |
| 40–59% predicted | 0.71 (0.12) | 0.98 (0.03) | 0.96 (0.04) | 0.83 (0.19) |
| < 40% predicted | 0.47 (0.22) | 0.91 (0.09) | 0.80 (0.21) | 0.80 (0.16) |
Stahl et al. 109 undertook a study to assess the relationship between disease severity and HRQoL in patients with COPD. One hundred and sixty-eight patients completed the SF-36, the St George’s Respiratory Questionnaire and the EQ-5D. EQ-5D results were stratified according to FEV1 level. This study suggests that EQ-5D utility declines with worsening lung function. Health utility values estimated within this study are summarised in Table 37.
| FEV1 stratum | EQ-5D (GOLD criteria) | EQ-5D (BTS criteria) |
|---|---|---|
| > 79% predicted | 0.84 (0.15) | 0.84 (0.15) |
| 60–79% predicted | 0.73 (0.23) | 0.74 (0.21) |
| 40–59% predicted | 0.74 (0.25) | 0.72 (0.28) |
| < 40% predicted | 0.52 (0.26) | 0.63 (0.25) |
In summary, only one study identified attempted to examine whether a statistical association exists between FEV1 and EQ-5D utility. 108 This study suggests that such a relationship may exist; however, the size of the coefficient is very small and is unlikely to be clinically meaningful. This indicates that FEV1, at least within the range of scores assessed within Johnson et al. 108 does not represent a good discriminatory indicator of HRQoL. The remaining three studies109–111 are inconsistent with respect to whether or not a relationship exists between FEV1 and HRQoL. The results of two of these studies109,111 appear to support the hypothesis that HRQoL is markedly lower for FEV1 of < 40%; however, this appears to be influenced considerably by the method of preference elicitation. 110 The only EQ-5D study undertaken using patients with CF111 does not suggest a clear distinction in health status for FEV1 of > 40%.
Using the taxonomy presented by Taylor and Elston,105 it is reasonable to argue that there is at best Level 1 evidence to support the hypothesis that FEV1% represents a useful surrogate for HRQoL. Although the evidence does not support a consistent decline in HRQoL with decreasing FEV1%, there is consistent evidence to support the theory that HRQoL is lower for lower FEV1% strata (< 40%).
Relationship between forced expiratory volume in first second percentage predicted and survival
Systematic searches were also undertaken to identify any studies which reported the use of statistical models through which to translate FEV1% to survival/mortality in patients with CF. Searches were undertaken in MEDLINE, MEDLINE In-Process & Other Non-Indexed citations and EMBASE in January 2012. The search strategy is shown in Appendix 7.
A total of 625 citations were identified by the searches, of which 21 studies were examined in further detail. Of these, 14 studies presented regression analyses that included either absolute FEV1 levels or decline in FEV1 as an independent variable and either survival or mortality as a dependent variable. 18,43,45,112–122 The findings of these included studies are summarised in Table 38.
Of the 14 studies identified by the searches,18,43,45,112–122 all considered a large number of other clinical variables alongside FEV1. Most of the studies adopted a Cox proportional hazards model approach, although some used logistic regression or other statistical analyses. Few studies justified why particular covariates had been included in the regression analyses, although in a minority of cases a stepwise approach was used to identify those covariates that significantly predicted survival for inclusion in the model. Some authors commented that predicting survival on the basis of FEV1% alone remains controversial. 112 Within all of these studies, other clinical variables were also found to be statistically significant predictors of survival. Several studies suggested that the rate of decline in FEV1, rather than absolute FEV1, is likely to be a better predictor of survival; however, the regression analyses were not consistent in this finding. It should be noted that decline in FEV1 is also problematic owing to the fluctuating nature of FEV1 measurements. Irrespective of how lung function was characterised within individual studies, there was a broadly consistent finding across the studies that other clinical variables are also important in predicting survival in patients with CF. In some analyses,112,113 FEV1 was not actually a statistically significant predictor of survival at all.
| Study | Study type | Population | Model form (description of FEV1 covariate) | Summary of study findings |
|---|---|---|---|---|
| Simmonds et al. 2010114 | Case control (78 case subjects, 152 control subjects) | Case subjects (long-term survivors) were patients with complete records who had reached 40 years of age without transplantation by 31 December 2004. Control subjects were selected from all patients with complete records who had died before 30 years of age or required transplantation at 30 years of age by 31 December 2004 | Probability-weighted logistic regression to predict survival up to 40 years (absolute FEV1) | A number of factors resulted in increased probabilities of survival, including BMI, FEV1, FVC at transfer to the adult clinic and exclusive use of oral antibiotics. Factors resulting in decreased probabilities of survival included P. aeruginosa acquisition or pneumothorax before transfer to the adult clinic and referral from a paediatric clinic in a deprived area |
| Ketchell et al. 2009112 | Retrospective case review (121 patients) | All adult patients with end-stage CF who died while on the Royal Brompton and Harefield Hospital lung transplant waiting list between July 1988 and June 2004 | Cox proportional hazards model (absolute FEV1) | Significant association found between survival and FVC (p = 0.027), but not FEV1 (p = 0.08) or any other parameter in patients performing the 6-minute walk test |
| Courtney et al. 2007115 | Longitudinal analysis (183 patients) | Adult patients from Belfast and Cork were studied from 1995 to 2005. The patients studied were aged ≥ 17 years in 2000 | Cox proportional hazards model (absolute FEV1) | The patients who died during the study period had a significantly lower mean (SD) FEV1% predicted in 1995 than those who remained alive: 41.5 (15.2)% compared with 69.8 (23.2)%, respectively (p < 0.001) |
| Elaffi et al. 2004113 | Retrospective case review (92 patients) | All patients admitted with severe pulmonary exacerbations to pulmonary department or ICU between 1 January 1997 and 30 June 2001 | Cox proportional hazards model (absolute FEV1 and slope of FEV1 decline) | Clinical characteristics before admission found to influence 1-year mortality were prior colonisation with B. cepacia and a rapid decline in FEV1 (FEV1 was significant only in the univariate analysis). Absolute FEV1 values were not significantly associated with probability of death |
| Schlucter et al. 2002116 | Model development study with validation against registry data (188 patients) | Population-based sample of 188 patients with the delta-F homozygous genotype for CF born after 1 January 1965, followed at the CF Centre at Rainbow Babies and Children’s Hospital, Cleveland, OH, USA | Random effects linear model for FEV1 and Gaussian model for age at death. Parameters estimated using MLE methods (absolute FEV1 and slope of FEV1 decline) | Separate results are presented by age group. The relationship between FEV1 and age at death appears to be non-linear |
| Augarten et al. 2001117 | Retrospective case review (40 patients) | Patients with FEV1% predicted of < 30% and were followed up for at least 3 years between 1985 and 1997 | Kaplan–Meier product method with log-rank test between strata (FEV1 decline) | Rate of change in FEV1 values found to be good predictor of survival. Patients whose slope was above the median (−2.33) were found to have a significantly superior prognosis compared with patients with a slope below the median (p = 0.04) |
| Milla et al. 1998118 | Retrospective case review (61 patients) | Patients who consistently had a FEV of < 30% | Cox proportional hazards model (rate of change in FEV1) | Of the covariates included in the Cox model, only the rate of decline in FEVl was reported to be a significant predictor of death (p = 0.0001) |
| Hayllar et al. 1997119 | Prospective case analysis with split sample validation (403 patients) | All patients with CF seen in the Royal Brompton Hospital between 1969 and 1987 | Cox proportional hazards model (absolute FEV1) | Percentage predicted FEV, percentage predicted FVC, height, white blood cell count, hepatomegaly, serum concentrations of albumin, alkaline phosphatase reported to be significantly associated with survival (p < 0.001) |
| Kerem et al. 199243 | Cohort study (673 patients) | Patients with CF followed up at the Toronto Hospital for Sick Children between 1977 and 1989 | Cox proportional hazards model (absolute FEV1) | All clinical covariates (FEV1, FVC, PaO2, PaCO2 and weight for height) except age were significantly associated with 1- and 2-year mortality rates |
| Liou et al. 200145 | Retrospective analysis of registry data (11,630 patients) | Patients with CF within the US Cystic Fibrosis Foundation Patient Registry who were alive on 1 January 1993, and for whom follow-up data were available through 31 December 1997, were included in the study | Cox proportional hazards model (absolute FEV1 and rate of decline in FEV1) | FEV1 slope was not statistically significant and was therefore excluded from the predictive model. Absolute FEV1 was significant and was included in the final model. The best multiple logistic regression model included nine variables with one interaction (age, gender, FEV1, weight for age score, pancreatic sufficiency, diabetes mellitus, S. aureus, B. cepacia, number of acute exacerbations, and number of acute exacerbations × B. cepacia) |
| Aurora et al. 2000120 | Retrospective case review (181 patients) | Subjects consisted of children with severe CF lung disease referred for transplantation assessment between 1988 and 1998 | Cox proportional hazards model (absolute FEV1) | Univariate Cox model suggests that SaO2 minimum, FEV1, FVC, distance, AAHR, albumin levels, number of courses of i.v. antibiotics administered, and blood haemoglobin concentrations were significantly associated with survival |
| Mayer-Hamblett et al. 2002121 | Analysis of registry data (14,572 patients) | Patients in the Cystic Fibrosis Foundation National Patient Registry who were ≥ 6 years of age in 1996 | Multiple logistic regression (absolute and slope of decline considered) | Significant predictors of mortality in the univariate analyses included number of hospitalisations for acute exacerbations, number of courses of home i.v. antibiotics, respiratory colonisation with B. cepacia, FEV1% predicted, height percentile and age. Multiple logistic regression, each litre increase in FEV1 significantly decreased the odds of dying within 2 years by 9% |
| Belkin et al. 2005122 | Retrospective cohort study (343 patients) | Adult and paediatric patients with CF listed for lung, heart–lung or heart–lung–liver transplant at the University of Pennsylvania Medical Centre | Cox regression (yearly rate of decline in FEV1) | Univariate analyses suggest that FEV1 of < 30% was associated with a higher risk of death (p < 0.01). Other significant variables included decrease in FEV1 and FVC, hypercapnia, rise in PaCO2, place of referral, and time of listing. Multivariate analyses suggested a significant interaction between FEV1 and PaCO2 |
| Henry et al. 199218 | Cohort study (81 patients) | Children with CF who coughed up sputum daily | Cox proportional hazards model (absolute FEV1) | Stepwise survival analysis suggested that FEV1 and younger age were significantly associated with poorer survival (p < 0.05) |
Of the identified studies, only one reported summary data on survival stratified by FEV1% group (albeit in an unadjusted manner43). However, the prognostic value of this study has been criticised elsewhere. In particular, George et al. ,123 highlight that a number of clinical developments in the management of CF over the past 20 years may make the findings of the Kerem et al. 43 study unreliable. These factors include the drive towards intensive nutritional management in CF, the development of new treatments, the increased use of non-invasive ventilation in those with respiratory failure and the push towards multidisciplinary care. 123 Other commentators were further critical of using absolute FEV1 levels owing to measurement error and fluctuations in FEV1 values over time.
On the basis of this review, it is reasonable to suggest that there exists Level 1/2 evidence to support the hypothesis that a change in FEV1% directly leads to a change in mortality, and therefore FEV1% alone is unlikely to represent a valid independent surrogate for patient survival. As such, the assumption of a direct linear relationship between FEV1% alone and mortality risk, without adjustment for other confounding factors, as assumed within the Forest Laboratories analysis, should be approached with considerable caution.
Relationship between exacerbation rates and other incidence of adverse events and health-related quality of life/survival
It is clinically plausible that the incidence of pulmonary exacerbation and other AEs could have meaningful impacts on HRQoL. If HRQoL had been assessed directly within the trials, one may expect such effects to be directly captured. However, without the use of preference-based measures, such as the EQ-5D, the inclusion of such effects becomes reliant on (1) the availability of external valuation studies that assess the impact of all potential AEs and (2) the adequate reporting of the number of AEs experienced within clinical study publications and reports. It should also be noted that many AEs associated with CF and its treatment do not occur in isolation but instead may manifest simultaneously. Ignoring this potential overlap would probably skew the results of an economic analysis and may lead to overestimating the benefits associated with those technologies with more favourable AE profiles.
Systematic searches were undertaken to identify studies that report EQ-5D utility estimates with and without specific AEs associated with a range of AEs associated with CF treatments (Table 39). Searches were undertaken across MEDLINE, MEDLINE In-Process & Other Non-Indexed Citations, EMBASE, BIOSIS, Citation Indexes and the The Cochrane Library. The search strategy is shown in Appendix 6.
| Respiratory | Nasal/mouth/throat | Other |
|---|---|---|
| Cough Lung disorder/exacerbation Dyspnoea Haemoptysis Rales/respiratory noises Respiratory tract infection Wheezing Chest discomfort Pulmonary function test decreased pulmonary congestion/blockage |
Oropharyngeal pain (mouth and pharynx pain) Other pain Dysphonia Nasal congestion/obstruction rhinorrhoea/runny nose Sinusitis |
Pyrexia/fever Hyperthermia Headache Fatigue Nausea Vomiting |
A total of 325 studies were identified by the searches. One additional study was identified by hand-searching the Novartis Pharmaceuticals submission. 60 However, of these, only five studies111,124–127 reported sufficient information through which to directly estimate a utility decrement for specific symptoms (note this figure excludes those studies detailed above which consider report EQ-5D values by FEV1% level as discussed above). Undertaking an economic evaluation which attempts to quantify the HRQoL impact of a selection of AEs but ignores others would inevitably result in bias, however the direction of such bias would be unclear. Given the current availability of evidence relating AEs to EQ-5D utility, this approach should be avoided.
Limited availability of evidence on clinical outcome measures for all relevant treatments
A comprehensive economic analysis of CF treatments would synthesise all relevant evidence on treatment effects within a meta-analytic framework. 103 However, from the perspective of health economic evaluation, this type of evidence synthesis would be useful only if a plausible and quantifiable relationship exists between FEV1%, or other intermediate clinical end points, and HRQoL and/or survival. The majority of clinical trials of colistimethate sodium and tobramycin (in either dry powder or nebulised form) report mean change in FEV1% within the trial cohorts, and very few report FEV1% outcomes beyond 4 weeks. Given the concerns regarding the validity of the relationships between FEV1% and mortality and HRQoL outlined above, this would not be useful, as it would require that the translated relationship has interval properties (e.g. x% change in FEV1% leads to y% change in HRQoL). Further, the systematic reviews presented above suggest that this type of relationship is unlikely to hold; the value of a NMA based on summary data is therefore questionable in this context.
De novo independent economic analysis
This section presents the methods and results of the de novo economic analysis undertaken by the Assessment Group.
Scope of the economic analysis
A number of potential options are relevant to the economic analysis of antibiotic treatments for P. aeruginosa. These include:
-
colistimethate sodium DPI
-
tobramycin DPI
-
colistimethate sodium nebulised
-
tobramycin nebulised
-
aztreonam.
Some patients may switch between tobramycin and colistimethate sodium at some point in their lives. This may be happen due to apparent treatment failure on the current drug, or may be part of a planned treatment regimen whereby colistimethate sodium and tobramycin are alternated every 28 days.
The Assessment Group developed a de novo health economic model to assess the cost-effectiveness of two competing treatment options: (1) colistimethate sodium DPI compared with (2) nebulised tobramycin for the treatment of chronic P. aeruginosa in patients with CF. A number of potentially relevant interventions and comparators were therefore excluded from the analysis (Table 40). In addition, a crude threshold analysis is presented to compare tobramycin DPI with nebulised tobramycin.
| Treatment option | Reasons for inclusion/exclusion |
|---|---|
| Options included in the economic analysis | |
| Colistimethate sodium DPI | Patient-level data on FEV1% from COL/DPI/02/06 available (data held on file) |
| Tobramycin nebulised | |
| Options excluded from the economic analysis | |
| Tobramycin DPI | Patient-level FEV1% data were not available, the price of tobramycin DPI was not determined or suggested until February 2012. The implied incremental QALY requirement given the drug’s incremental cost is considered as part of a threshold analysis |
| Colistimethate sodium nebulised | No relevant studies included in the evidence network, patient-level data not available |
| Aztreonam | Predominantly used third-line and not currently recommended for use in published UK consensus guidelines4 |
| Treatment sequences (switching) | Lack of evidence of clinical efficacy and safety |
Model structure
The model estimates the expected costs and QALY gains associated with colistimethate sodium DPI compared with nebulised tobramycin. The analysis adopts an NHS perspective over a lifetime horizon. The primary economic outcome measure for the analysis is the incremental cost per QALY gained. All costs and health outcomes within the model were discounted using the standard approach at a rate of 3.5%. Costs were valued at 2011 prices.
The model takes the form of a state transition model to estimate transitions between three FEV1% strata [(1) FEV1 70–99%; (2) FEV1 40–69% and (3) FEV1 < 40%]. Twenty-four week transition probabilities are estimated, based on those observed within COL/DPI/02/0666. Different levels of HRQoL are assumed for each health state. Treatment duration, which is assumed to be directly related to survival duration, is assumed to be exactly equivalent between the competing treatment options. During each cycle, patients may remain in their current FEV1% state, transit to an improved or worsened FEV1% state or die. Patients with FEV1 of < 40% may undergo lung transplantation and do not subsequently receive further treatment with colistimethate sodium DPI or tobramycin; other treatments received by these patients are assumed to be identical irrespective of previous antibiotic treatments received. Additional HRQoL decrements are applied for minor and major exacerbations based on treatment-specific rates and data relating to the mean time receiving i.v. antibiotics. Total QALYs are calculated as the total sojourn time in each health state weighted by the respective utility for that health state, less any QALY losses resulting from exacerbations. Costs within each treatment group include drug acquisition costs and the costs of managing exacerbations (either in hospital or at home). Potential cost savings associated with reduced maintenance of nebulisers are also included in the economic analysis. Costs associated with follow-up and concomitant medications are assumed to be related only to treatment time and are therefore assumed to be equivalent between treatment groups. A conceptual form of the implemented health economic model is presented in Figure 11.
FIGURE 11.
Conceptual form of the implemented economic model.
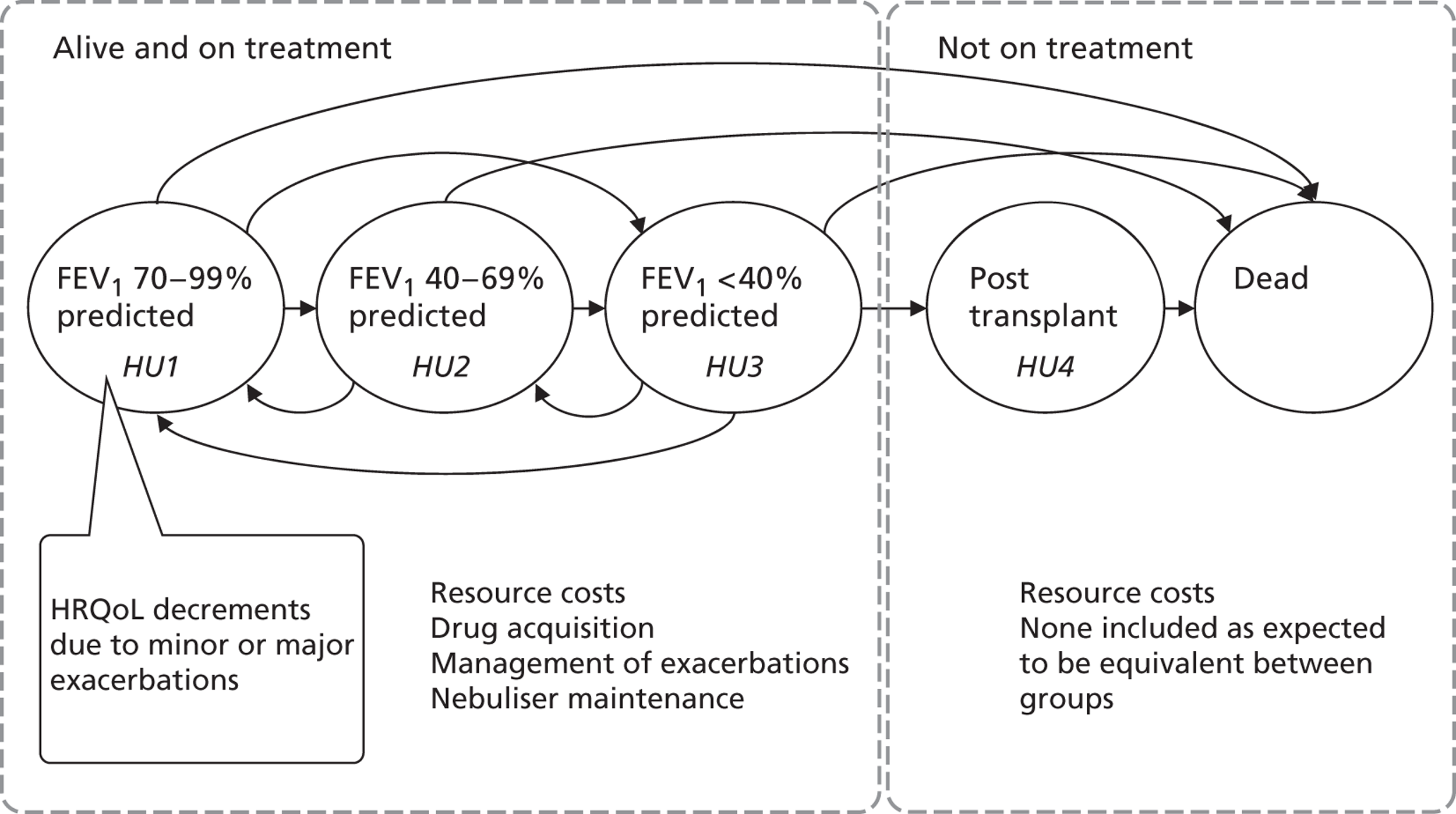
Given the considerable uncertainty surrounding the extrapolation of the 24-week efficacy data to a lifetime horizon, two separate analyses are presented:
-
a reference case analysis based on FEV1% extrapolation over a lifetime horizon
-
a ‘within-trial’ analysis that does not include any extrapolation.
Separate analyses are presented for each of the six prices presented in Table 26.
Evidence used to inform the model parameters
The sources of evidence used to inform the model parameters are detailed below. For the most part, the model parameters have been informed directly using data from the COLO/DPI/02/06 trial;66 these have been augmented using other data from external sources. A summary of model parameter values and distributions used in the base-case analysis is presented in Table 41.
| Parameter description | Distribution | Parameter 1 | Parameter 2 | Mean | Source |
|---|---|---|---|---|---|
| Management variables | |||||
| Initial cohort age | NA | – | – | 21 | COLO/DPI/02/0666 |
| Discount rate: QALYs | NA | – | – | 0.035 | NICE Methods Guide 201195 |
| Discount rate: costs | NA | – | – | 0.035 | |
| Survival parameters | |||||
| Weibull log λ | Multivariate normal | −12.33 | Variance log λ = 0.004, variance γ = 0.0041, covariance = 0.003 | −12.33 | Based on Dodge et al. 200722 |
| Weibull γ | 3.34 | 3.34 | |||
| Transplant parameters | |||||
| Probability of transplant during 24 weeks | Beta | 7.89 | 858.17 | 0.0092 | CF Registry 20106 |
| Initial distribution of patients | |||||
| FEV 60–79% | Dirichlet | CiC information has been removed | CiC information has been removed | 0.09 | COLO/DPI/02/0666 (pooled arms) |
| FEV 40–59% | Dirichlet | CiC information has been removed | CiC information has been removed | 0.65 | |
| FEV < 40% | Dirichlet | CiC information has been removed | CiC information has been removed | 0.26 | |
| Transition probabilities between FEV1% strata | |||||
| FEV 70–99% → FEV 70–99% (Coli) | Dirichlet | CiC information has been removed | CiC information has been removed | 0.63 | COLO/DPI/02/0666 (individual treatment arms) |
| FEV 70–99% → FEV 40–69% (Coli) | Dirichlet | CiC information has been removed | CiC information has been removed | 0.32 | |
| FEV 70–99% → FEV < 40% (Coli) | Dirichlet | CiC information has been removed | CiC information has been removed | 0.05 | |
| FEV 40–69% → FEV 70–99% (Coli) | Dirichlet | CiC information has been removed | CiC information has been removed | 0.14 | |
| FEV 40–69% → FEV 40–69% (Coli) | Dirichlet | CiC information has been removed | CiC information has been removed | 0.71 | |
| FEV 40–69% → FEV < 40% (Coli) | Dirichlet | CiC information has been removed | CiC information has been removed | 0.15 | |
| FEV < 40% → FEV 70–99% (Coli) | Dirichlet | CiC information has been removed | CiC information has been removed | 0.02 | |
| FEV < 40% → FEV 40–69% (Coli) | Dirichlet | CiC information has been removed | CiC information has been removed | 0.17 | |
| FEV < 40% → FEV < 40% (Coli) | Dirichlet | CiC information has been removed | CiC information has been removed | 0.81 | |
| FEV 70–99% → FEV 70–99% (Tobi) | Dirichlet | CiC information has been removed | CiC information has been removed | 0.75 | |
| FEV 70–99% → FEV 40–69% (Tobi) | Dirichlet | CiC information has been removed | CiC information has been removed | 0.20 | |
| FEV 70–99% → FEV < 40% (Tobi) | Dirichlet | CiC information has been removed | CiC information has been removed | 0.05 | |
| FEV 40–69% → FEV 70–99% (Tobi) | Dirichlet | CiC information has been removed | CiC information has been removed | 0.15 | |
| FEV 40–69% → FEV 40–69% (Tobi) | Dirichlet | CiC information has been removed | CiC information has been removed | 0.75 | |
| FEV 40–69% → FEV < 40% (Tobi) | Dirichlet | CiC information has been removed | CiC information has been removed | 0.10 | |
| FEV < 40% → FEV 70–99% (Tobi) | Dirichlet | CiC information has been removed | CiC information has been removed | 0.02 | |
| FEV < 40% → FEV 40–69% (Tobi) | Dirichlet | CiC information has been removed | CiC information has been removed | 0.13 | |
| FEV < 40% → FEV < 40% (Tobi) | Dirichlet | CiC information has been removed | CiC information has been removed | 0.85 | |
| Exacerbation rates | |||||
| Probability of exacerbation (Tobi) | Beta | 75 | 191 | 0.39 | COLO/DPI/02/06 (CSR)69 |
| Probability of exacerbation (Coli) | Beta | 69 | 183 | 0.38 | |
| HRQoL parameters | |||||
| Disutility major exacerbation | Beta | 0.17 | 0.08 | 0.1740 | Bradley et al. 2010111/Novartis Pharmaceuticals’ submission 201160 |
| Disutility minor exacerbation | Beta | 0.02 | 0.01 | 0.0150 | |
| Utility > 70% predicted | Beta | 0.86 | 0.03 | 0.8640 | |
| Utility 40–69% predicted | Beta | 0.81 | 0.04 | 0.8100 | |
| Utility < 40% predicted | Beta | 0.64 | 0.06 | 0.6400 | |
| Probability exacerbation is major | Beta | CiC information has been removed | CiC information has been removed | 0.66 | COLO/DPI/02/06 (CSR)69 |
| Duration exacerbation (Coli) | Beta | CiC information has been removed | CiC information has been removed | 0.0372 | |
| Duration exacerbation (Tobi) | Beta | CiC information has been removed | CiC information has been removed | 0.0394 | |
| Utility post transplant | Beta | 0.83 | 0.02 | 0.8300 | Anyanwu et al. 2001128 |
| Cost parameters | |||||
| Cost per dose (Coli) | See Table 26 (price range = £9.11– £ 39.29) | Forest Laboratories’ submission 201166 | |||
| Cost per dose (Tobi) | NA | £21.20 | NA | £21.20 | BNF 6261 |
| Cost minor exacerbation | Normal | £427.69 | £10.98 | £412.74 | NHS reference costs 2010–11129 |
| Cost major exacerbation | Normal | £1500.14 | £33.06 | £1500.14 | |
| Marginal nebuliser savings | Normal | £200.00 | £10.00 | £200.00 | Personal communicationa |
Patient survival
Mean survival for patients with CF was estimated using data reported by Dodge et al. 22 This study reported survival data up to the end of 2003 for all subjects with CF born in the UK in the period 1968–92, collated via active enquiry of CF clinics and other hospital consultants. Survival curves are reported within this paper separately for males and females (see Figure 2). Data are not available on the number of patients at risk over time.
Engauge® digitising software version 5 (http://digitizer.sourceforge.net/) was used to replicate the published survival data from the graphs assuming a 50 : 50 split between males and females. Parametric survival curves were fitted to these data in order to estimate the mean durations of survival within the cohorts. Exponential, Weibull, linear, Gompertz and log-logistic curves were fitted to the empirical survival curve data. Each of these curves result in different distributions of survival. Information regarding the number of patients at risk and the number of events was not available from Dodge et al. 22 hence curves were fitted using Solver add-in within Microsoft Excel. Each curve was inspected visually with respect to how well the distribution fitted the observed data. The plausibility of the unobserved portion of each curve was considered by comparing the median survival of the fitted curve against the predicted median survival from the 2010 CF Registry report (Table 42).
| Year | 2007 | 2008 | 2009 | 2010 |
|---|---|---|---|---|
| Median predicted survival: years (95% CI) | 35.2 (31.0 to 42.6) | 38.8 (34.2 to 47.3) | 34.4 (30.7 to 37.0) | 41.4 (36.8 to 46.7) |
Figure 12 presents the actual and predicted survival using a range of different curves. The Weibull and log-logistic models appear to provide the best fit to the data. Both of these curves provide a reasonable fit to the median survival as well as the overall distribution. However, the tail of the log-logistic distribution appears to overestimate survival during the later decades of life. Therefore, the Weibull curve was used in the base-case economic analysis.
Uncertainty surrounding the two parameters of the Weibull survivor function was modelled using a multivariate normal distribution. As patient-level data were not available, the variance and covariance of the parameters was assumed rather than estimated. These were fitted against the maximum and minimum median predicted survival data from the CF Registry reports for the years 2007–10.
Forced expiratory volume in first second: transition probabilities
Transition probabilities between the health states were estimated directly using patient-level FEV1% data from Visit 0 and Visit 6 within the COL/DPI/02/06 trial (the same data used by Forest Laboratories to estimate mortality gains). A summary of the number of number of transitions from and to each FEV1% stratum is presented in Tables 43 and 44.
| FEV1% stratum | FEV1 70–99% | FEV1 40–69% | FEV1 < 40% | Total |
|---|---|---|---|---|
| FEV1 70–99% | 11 | 5 | 0 | 16 |
| FEV1 40–69% | 16 | 85 | 17 | 118 |
| FEV1 < 40% | 0 | 7 | 37 | 44 |
| Total | 27 | 97 | 54 | 178 |
| FEV1% stratum | FEV1 70–99% | FEV1 40–69% | FEV1 < 40% | Total |
|---|---|---|---|---|
| FEV1 70–99% | 14 | 3 | 0 | 17 |
| FEV1 40–69% | 17 | 89 | 11 | 117 |
| FEV1 < 40% | 0 | 6 | 44 | 50 |
| Total | 31 | 98 | 55 | 184 |
Uncertainty surrounding these transition probabilities was characterised using Dirichlet distributions with minimally informative priors using the methods reported by Briggs et al. 130
Initial forced expiratory volume in first second distribution
As there are some slight imbalances in the baseline distribution of FEV1% across the two treatment groups, the initial distribution of patients across the three FEV1% strata within the model uses pooled estimates from both treatment groups (FEV1 70–99% = 33, FEV1 40–69% = 235, FEV1 < 40% = 94). Uncertainty surrounding the initial distribution of patients is again characterised using a Dirichlet distribution with minimally informative priors. 130
Exacerbation probabilities
Exacerbations were reported within the CSR for COL/DPI/02/06 as either protocol- or non-protocol-defined. The overall number of exacerbations (the sum of protocol- and non-protocol-defined exacerbations) was used to estimate the baseline risk of exacerbation for nebulised tobramycin group (75/191 = 0.39). The exacerbation rate in the colistimethate sodium DPI group was estimated to be 69/183 = 0.38. 69 As the CSR states the number of patients with documented exacerbations, the assumption underlying these calculations is that only one exacerbation occurred per patient. It should be noted that including ‘overall’ exacerbations rather than the protocol-defined values favours colistimethate sodium DPI. Uncertainty surrounding exacerbation probabilities was characterised using independent beta distributions.
Probability of undergoing lung transplantation
There is limited information concerning the lifetime probability that an individual with CF will undergo lung transplantation. The probability that a patient with FEV1 of < 40% undergoes lung transplant during each cycle was estimated crudely based on data from the CF Registry and data from the US Cystic Fibrosis Foundation (www.cff.org/treatments/LungTransplantation/). The model assumes that the lifetime probability of undergoing lung transplantation is approximately 3%. The probability of undergoing transplantation within each model cycle was assumed to be stable over time and independent of patient age. After transplantation, patients are assumed to no longer require the use of antipseudomonal drugs. Uncertainty surrounding this probability was modelled using a beta distribution.
Health-related quality of life
The base-case scenario uses the utilities for FEV1% strata and disutilities for exacerbations reported by Bradley et al. ,111 as shown in Table 35. The impact of minor/major exacerbations on HRQoL were modelled by applying a disutility based on the event rates reported within the COLO/DPI/02/06 CSR69 and minor/major utility decrements reported by Bradley et al. 111 Uncertainty surrounding these parameters was characterised using beta distributions. The duration of minor/major exacerbations was not available directly but was instead estimated by taking the mean duration of time on additional i.v. antibiotics as a proxy;69 a slightly longer duration was assumed for the tobramycin group based on these data (13.3 days for colistimethate sodium DPI and 14.4 days for nebulised tobramycin). 69 For those patients who undergo lung transplantation, a health utility score of 0.83 was assumed until death;128 further utility decrements relating to exacerbations are not applied to these patients. Uncertainty surrounding this utility score was characterised using a beta distribution.
Resources and costs
The model includes only the acquisition costs associated with colistimethate sodium DPI and nebulised tobramycin as well as the costs associated with managing exacerbations. The range of potential prices of colistimethate sodium DPI was sourced from the Forest Laboratories submission to NICE66 and from further correspondence with Forest Laboratories. A price range of £9.11–39.29 per dose was assumed over six pricing scenarios (see Table 26). Colistimethate sodium DPI was assumed to be used twice every day.
The acquisition cost of nebulised tobramycin was derived from BNF 62. 61 At the time of writing, the cost of 56 5-ml (300-mg) units costed £1187.20. Assuming a treatment regimen in which tobramycin is used for 28 days and then not used for the next 28 days, this corresponds to a price per dose of £21.20.
The model assumes that minor exacerbations incur less cost than major exacerbations, and that the latter require hospitalisation. The 2010–11 NHS Reference Costs129 do not report costs specific to CF exacerbations. Instead, the costs of asthma complications were taken as a proxy. The reference cost for asthma with major complications without intubation (DZ15D, long stay) was assumed to reflect the cost of major exacerbations due to CF (mean = £1500). The cost of asthma complications without intubation (DZ15E, short stay) was assumed to reflect the cost of minor exacerbations (mean = £403). Uncertainty surrounding the costs of exacerbations was characterised using normal distributions.
Costs associated with other treatments and hospital appointments for CF are assumed to be identical between the treatment groups.
The use of nebulisers for the delivery of antibiotic treatments is associated with fixed costs related to equipment purchase and ongoing costs associated with maintenance and replacement parts (e.g. aerosol heads and filters). Maintenance costs may be dependent on the number of drugs being nebulised. Some nebuliser devices are funded separately, whereas others are intended to be used for the administration of specific drugs, for example the I-neb AAD device is provided specifically for use with Promixin (although can be programmed to operate with certain other drugs) and its purchase price and maintenance costs are both covered by Profile Pharma. Purchasing arrangements for these devices in the UK are complex. Some nebulisers funded directly by NHS Trusts, whereas others may be funded by third-party donations from charitable organisations or pharmaceutical companies. Some nebuliser devices are currently privately funded by NHS patients. There is limited information available within the public domain with respect to the proportion of devices funded by the NHS and the uptake of specific devices or the true costs borne by the NHS. It appears likely that arrangements for funding for nebuliser purchasing and maintenance are also subject to geographical variability.
The potential implications of introducing DPIs on the costs of purchasing and maintaining nebulisers borne by the NHS are not straightforward. For some patient subgroups, the introduction of DPIs could lead to a reduction in the costs of nebulisers – whereas some patients with P. aeruginosa may still require nebulisers, a shift to DPIs may lead to a reduction in the costs of nebuliser maintenance. This may or may not also result in some switching behaviour within Trusts to lower cost devices. For those patients who do not require nebulised bronchodilators or mucolytics, nebulisers may not be required at all, thereby leading to savings both on purchase costs and maintenance costs. However, the introduction of DPIs could also lead to some additional costs – for example replacing Promixin with colistimethate sodium DPI in patients who still require a nebuliser for the administration of other drugs would mean that a nebuliser device would have to be funded for the administration of bronchodilators and/or mucolytics where previously the costs were funded by other parties.
Given the uncertainty both with respect to the current costs of nebulisers and the implications of switching to DPIs, the base-case health economic analysis includes a crude estimate of the maintenance costs of nebuliser maintenance. This is assumed to be £200 per year and covers the replacement of aerosol heads and filters; this estimate is based expert opinion (Dr Diana Bilton, personal communication) and information provided by PARI EU. A SE of £10 is assumed. It is likely that this represents an overestimate of the actual cost savings and therefore favours colistimethate sodium DPI.
Key assumptions within the de novo economic analysis
The model makes the following key assumptions
-
FEV1 measurements are stable and not subject to measurement error.
-
HRQoL is assumed to differ by FEV1% strata.
-
Transitions between FEV1% strata over time are assumed to be independent of patient’s previous transitions.
-
Colistimethate sodium DPI has no additional benefit over nebulised tobramycin in terms of patient survival.
-
The costs of follow-up and concomitant medication are equivalent between colistimethate sodium DPI and nebulised tobramycin.
Uncertainty analysis
The model is fully probabilistic. Monte Carlo sampling was used to propagate uncertainty through the model in order to produce distributions of expected costs and outcomes for each treatment option. The model was run over 5000 Monte Carlo samples. The results of the PSA are presented in terms of incremental cost-effectiveness planes and CEACs. Simple sensitivity analyses were undertaken to examine the impact of parameter uncertainty and structural uncertainty on the model results. The following analyses were explored:
-
Scenario 1. The analysis was run using point estimates of parameters rather than the probabilistic means.
-
Scenarios 2–6. Secondary analyses are presented using alternative FEV1% utility estimates reported by Yi et al. 110 and Stahl et al. 109 It should be noted that these studies report health utility using different categories of FEV1% bands (FEV1 > 79%, FEV1 60–79%, FEV1 40–59% and FEV1 < 40%). As such, it was necessary to redefine the structure of the model and re-estimate the transition probabilities between four instead of three states. The general logic of the model, however, remains the same as the base-case analysis.
-
Scenario 7. FEV1% transition probabilities for the nebulised tobramycin group were set equal to those for the colistimethate sodium DPI group.
-
Scenario 8. The HRQoL decrement associated with minor and major exacerbations was doubled.
-
Scenario 9. The cost of hospitalisation for major exacerbations was doubled.
Model validation and verification methods
A number of measures were taken to ensure that the Assessment Group model was credible and not subject to computational errors. First, the methods and results of the health economic model were peer reviewed by three clinical advisors to the project (see Acknowledgements). The executable model and its underlying logic were checked by the model authors and a third modeller who was not involved in its development. The expectations of each model parameter distribution were compared against their deterministic counterpart. All model input parameters were double-checked against the sources from which they were derived. The plausibility of the model results were considered against the model developers’ expectations of those results prior to model development.
In addition to the above activities, a validation exercise was undertaken to examine the plausibility of the extrapolated Markov trace based on the COLO/DPI/02/06 trial66 by deriving equivalent transition matrices using longitudinal panel data from the CF Registry for the period 1997–2008. Transition matrices were generated as follows:
-
The first FEV1% measurement each calendar year was taken as each patient’s observation for year t.
-
As patients entered the registry during different calendar years, the first observation for each patient was transposed to a common starting year.
-
Missing data between observations were imputed according to a LOCF rule (final observations were not imputed).
-
FEV1 scores were mapped to three FEV1% bands (FEV1 > 70%, FEV1 40–69%, FEV1 < 40%).
-
Transition matrices for year t were calculated based on the number of patients transiting between each state between years t and year t + 1.
These transition matrices were then applied to the initial distribution of patients in the model and the resulting Markov trace was compared against the Markov trace for the tobramycin group. The results of this analysis are shown in Figure 13.
FIGURE 13.
Comparison of registry- and trial-derived Markov trace.
This analysis shows that the registry-derived transition matrices suggest a similar shape in the Markov trace as those derived from the COLO/DPI/02/06 trial. 66 There are clearly some differences between the traces generated using the registry data and the trial; however, the FEV1 < 40% state population, which has the greatest impact on HRQoL, appears fairly similar between the two sources. Some of this discrepancy may be caused by differences between the registry and the trial in terms of patient characteristics, for example the registry cohort does not exclusively include those patients with P. aeruginosa. This analysis lends some weight to the credibility of the trial extrapolation.
Simplifications and exclusions from the economic analysis
Potential process utilities resulting from increased convenience and faster treatment delivery
One of the appealing aspects of using the DPIs is the increased convenience afforded by reduced treatment time and increased portability in the administration as compared against nebulised antibiotics. It is plausible that this represents a ‘process utility’ which is not captured in the health economic analysis presented here. However, neither the Novartis Pharmaceuticals submission60 nor the Forest Laboratories submission66 reported any empirical preference-based evidence of the impact of this potential benefit on HRQoL. It should be also noted that that this impact would be lessened by the use of newer faster delivery nebuliser devices such as the eFlow® Rapid Nebuliser and the I-neb AAD system.
Exclusion of disutilities owing to adverse events
The model does not include utility adjustments to account for the incidence of AEs. Although the incidence of cough, productive cough and dysgeusia were markedly higher for colistimethate sodium DPI than nebulised tobramycin, some AEs were less common for colistimethate sodium DPI. As a consequence, it is unclear whether the inclusion of health utility decrements associated with the incidence of AEs would improve or worsen the economic case for colistimethate sodium DPI. Although Forest Laboratories kindly provided detailed AE data for each treatment group at each visit, the considerable gaps in the available EQ-5D evidence (see Methodological issues surrounding the economic evaluation of cystic fibrosis treatments) relating to the disutility of these events precluded the inclusion of these effects within the model.
It should also be noted that the model does not include the potential impact of resistance to tobramycin. This exclusion is reasonable, as it is unclear how this phenomenon would manifest in terms of reduced treatment effect.
Limitations in methods for modelling treatment benefits
The model extrapolates treatment effects in terms of shifts between different health states, each of which is associated with different EQ-5D scores. The definition of health states within the model is ‘blunt’ in that only three FEV1% strata are defined and there appears to be little difference in health utility for FEV1% states of > 40%. Although it could be argued that the EQ-5D is not particularly sensitive in the valuation of health for patients with CF, the study reported by Yi et al. 110 suggests that other preference-based health utility instruments result in a similar relationship between FEV1% and HRQoL. On the basis of the weaknesses in the evidence associated with the potential relationship between FEV1% and mortality (see Methodological issues surrounding the economic evaluation of cystic fibrosis treatments), this relationship was not considered within the Assessment Group model.
Health economic results
Headline cost-effectiveness results
The base-case probabilistic model results are presented in Table 45. The impact of constraining the time horizon to 24 weeks (the duration of the COLO/DPI/02/06 trial66) is shown in Table 46.
| Colistimethate sodium DPI price (£) | QALYs | Costs (£) | ICER | ||||
|---|---|---|---|---|---|---|---|
| Colistimethate sodium DPI | Tobramycin nebulised | Incremental | Colistimethate sodium DPI | Tobramycin nebulised | Incremental | ||
| 9.11 | 9.48 | 9.61 | −0.13 | 93,915.58 | 110,518.68 | −16,603.10 | 126,259 |
| 10.60 | 9.48 | 9.61 | −0.13 | 107,390.59 | 110,518.68 | −3128.08 | 23,788 |
| 15.98 | 9.48 | 9.61 | −0.13 | 156,045.35 | 110,518.68 | 45,526.67 | Dominated |
| 19.64 | 9.48 | 9.61 | −0.13 | 189,145.05 | 110,518.68 | 78,626.38 | Dominated |
| 21.20 | 9.48 | 9.61 | −0.13 | 203,253.12 | 110,518.68 | 92,734.45 | Dominated |
| 39.29 | 9.48 | 9.61 | −0.13 | 366,852.48 | 110,518.68 | 256,333.80 | Dominated |
The results presented in Table 45 suggest that colistimethate sodium DPI is expected to result in a loss of around 0.13 QALYs over the patient’s lifetime compared with nebulised tobramycin. If colistimethate sodium DPI is priced at one of the prices that is higher than that of nebulised tobramycin, it is also expected to have a positive incremental cost compared with nebulised tobramycin. As a consequence, colistimethate sodium DPI is expected to be dominated for these pricing scenarios. If priced at £9.11 per dose or £10.60 per dose, colistimethate sodium DPI is expected to be less expensive and less effective than nebulised tobramycin. The resulting ICERs are around £126,000 and £24,000 per QALY gained. It should be noted that the positive ICER in this instance reflects a QALY loss and cost savings for colistimethate sodium DPI compared with nebulised tobramycin (therefore colistimethate sodium DPI lies in the south-west quadrant of the cost-effectiveness plane).
| Colistimethate sodium DPI price (£) | QALYs | Costs (£) | ICER | ||||
|---|---|---|---|---|---|---|---|
| Colistimethate sodium DPI | Tobramycin nebulised | Incremental | Colistimethate sodium DPI | Tobramycin nebulised | Incremental | ||
| 9.11 | 0.35 | 0.35 | −0.00 | 3469.00 | 4075.35 | −606.35 | 276,814 |
| 10.60 | 0.35 | 0.35 | −0.00 | 3966.71 | 4075.35 | −108.64 | 49,596 |
| 15.98 | 0.35 | 0.35 | −0.00 | 5763.82 | 4075.35 | 1688.48 | Dominated |
| 19.64 | 0.35 | 0.35 | −0.00 | 6986.40 | 4075.35 | 2911.05 | Dominated |
| 21.20 | 0.35 | 0.35 | −0.00 | 7507.49 | 4075.35 | 3432.15 | Dominated |
| 39.29 | 0.35 | 0.35 | −0.00 | 13,550.21 | 4075.35 | 9474.86 | Dominated |
The results of the short-term ‘within-trial’ analysis suggest that colistimethate sodium DPI is expected to result in a small decrease in QALYs compared against nebulised tobramycin (0.002 QALYs lost). If colistimethate sodium DPI is priced at one of the prices that is higher than that of nebulised tobramycin, it is also expected to have a positive incremental cost than nebulised tobramycin and is thus dominated. If priced at £9.11 per dose or £10.60 per dose, colistimethate sodium DPI is expected to be less expensive than nebulised tobramycin (ICER = £277,000 and £50,000 per QALY gained, respectively). Again, the positive ICER reflects a QALY loss and cost savings for colistimethate sodium DPI compared with nebulised tobramycin.
Uncertainty analysis
Results of the long-term reference case economic analysis
Figures 14–25 present cost-effectiveness planes and CEACs for the long-term reference case model over the range of pricing scenarios for colistimethate sodium DPI.
FIGURE 14.
Reference case model: cost-effectiveness plane (price per dose = £9.11).
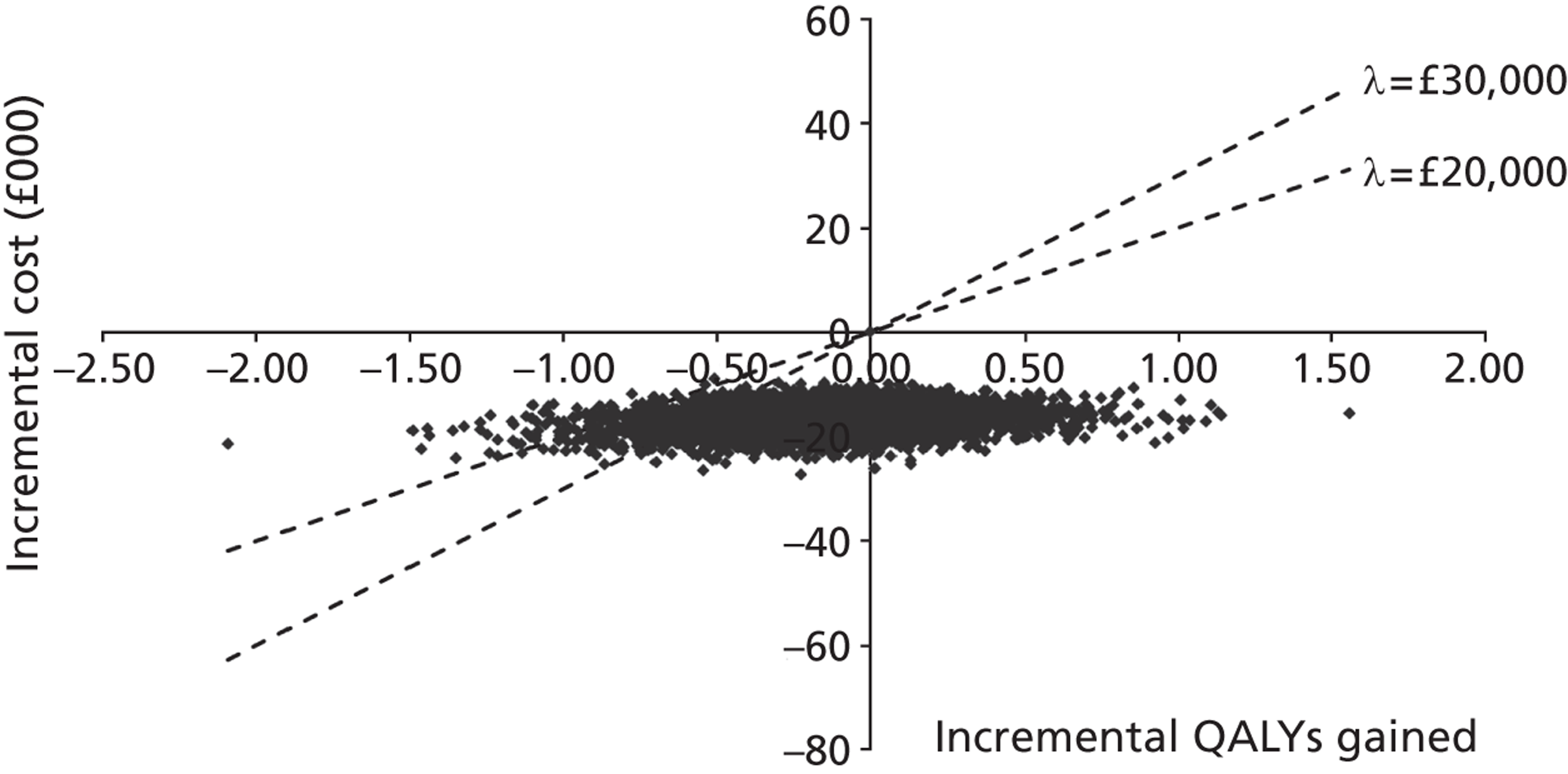
Assuming a price per dose of £9.11, the long-term model suggests that the probability that colistimethate sodium DPI produces a positive QALY gain and has an ICER that is better than £20,000 per QALY gained is around 0.32. The probability that colistimethate sodium DPI produces a positive QALY gain and has an ICER that is better than £30,000 per QALY gained is also approximately 0.32. The probability that colistimethate sodium DPI is dominated by nebulised tobramycin is also approximately zero.
FIGURE 15.
Reference case model: CEAC (price per dose = £9.11).
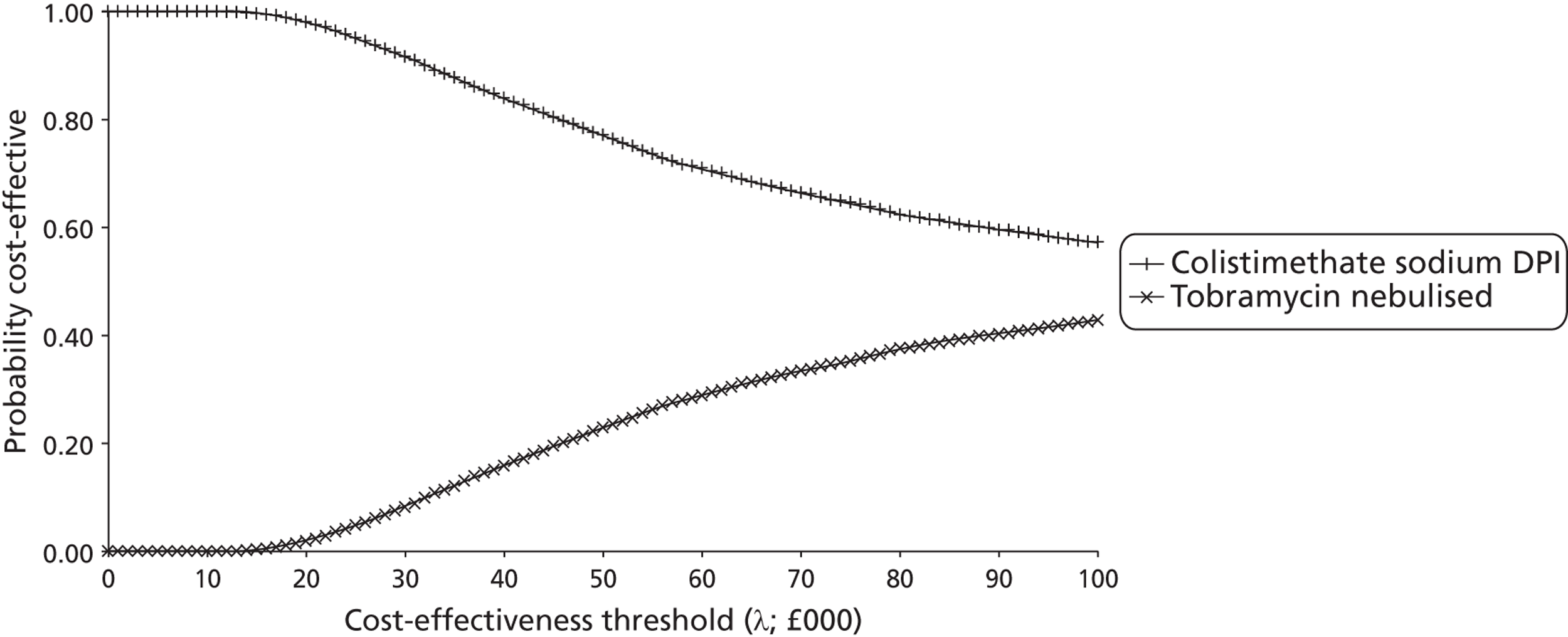
Assuming a willingness-to-pay threshold of £20,000 per QALY gained and a price per dose of £9.11, the probability that colistimethate sodium DPI is optimal is approximately 0.98. At a willingness-to-pay threshold of £30,000 per QALY gained, the probability that colistimethate sodium DPI is optimal is approximately 0.92.
FIGURE 16.
Reference case model: cost-effectiveness plane (price per dose = £10.60).
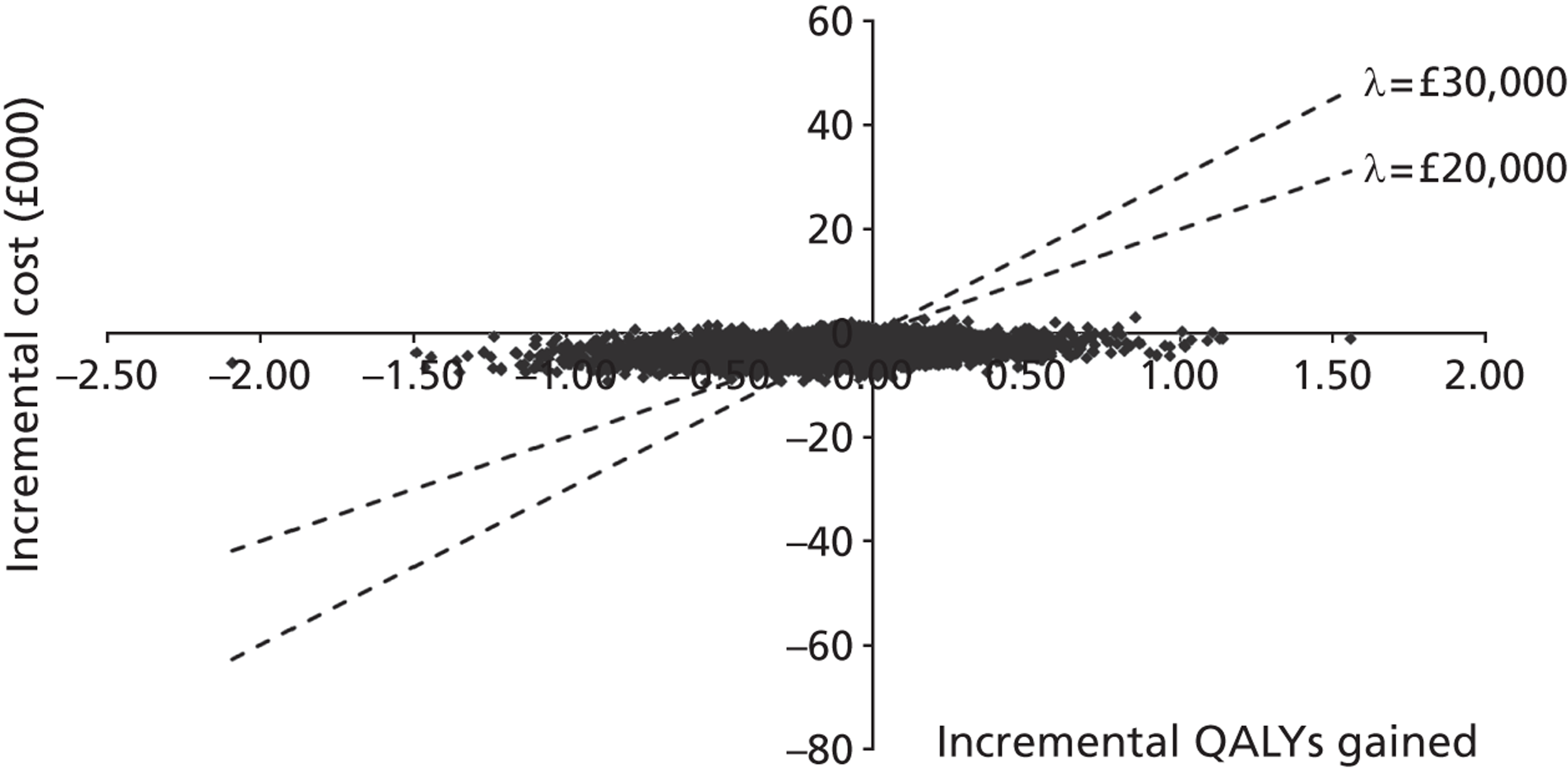
Assuming a price per dose of £10.60, the long-term model suggests that the probability that colistimethate sodium DPI produces a positive QALY gain and has an ICER that is better than £20,000 per QALY gained is around 0.32. The probability that colistimethate sodium DPI produces a positive QALY gain and has an ICER that is better than £30,000 per QALY gained is also approximately 0.32. The probability that colistimethate sodium DPI is dominated by nebulised tobramycin is also approximately 0.01.
FIGURE 17.
Reference case model: CEAC (price per dose = £10.60).
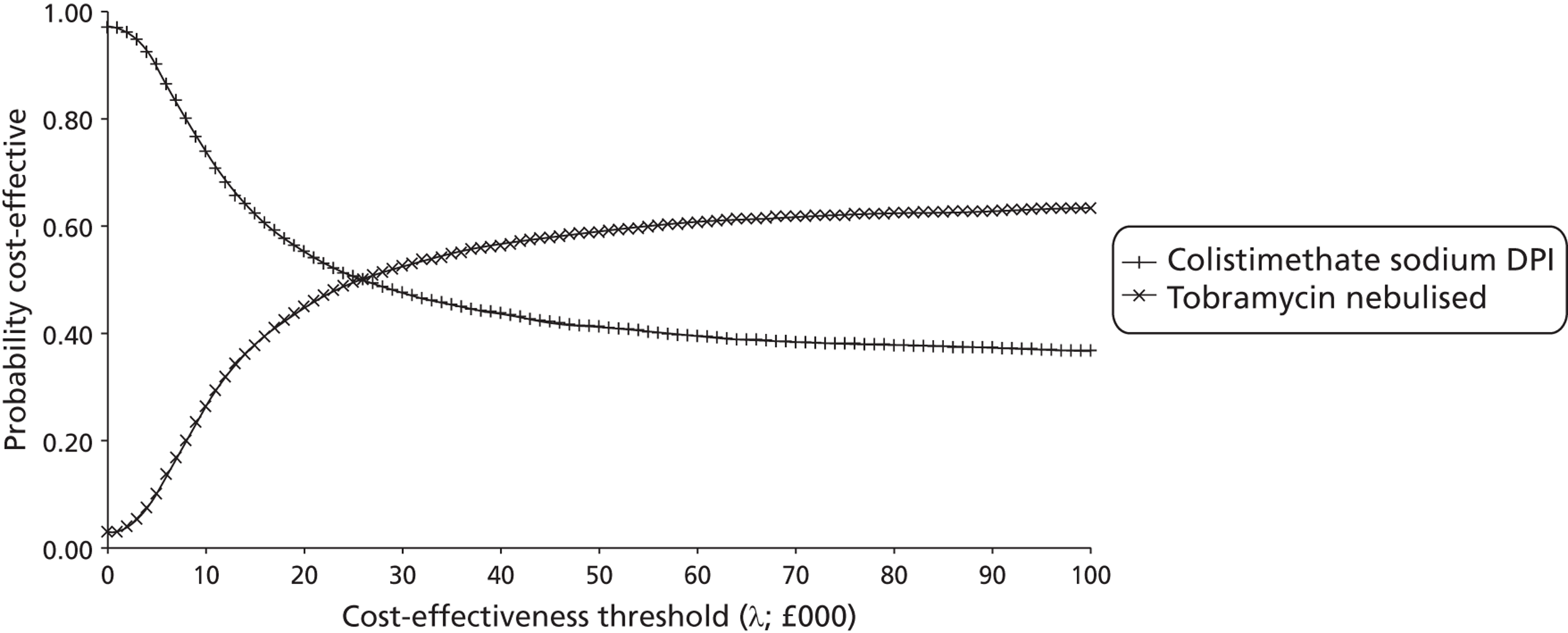
Assuming a willingness-to-pay threshold of £20,000 per QALY gained and a price per dose of £10.60, the probability that colistimethate sodium DPI is optimal is approximately 0.55. At a willingness-to-pay threshold of £30,000 per QALY gained, the probability that colistimethate sodium DPI is optimal is approximately 0.48.
FIGURE 18.
Reference case model: cost-effectiveness plane (price per dose = £15.98).
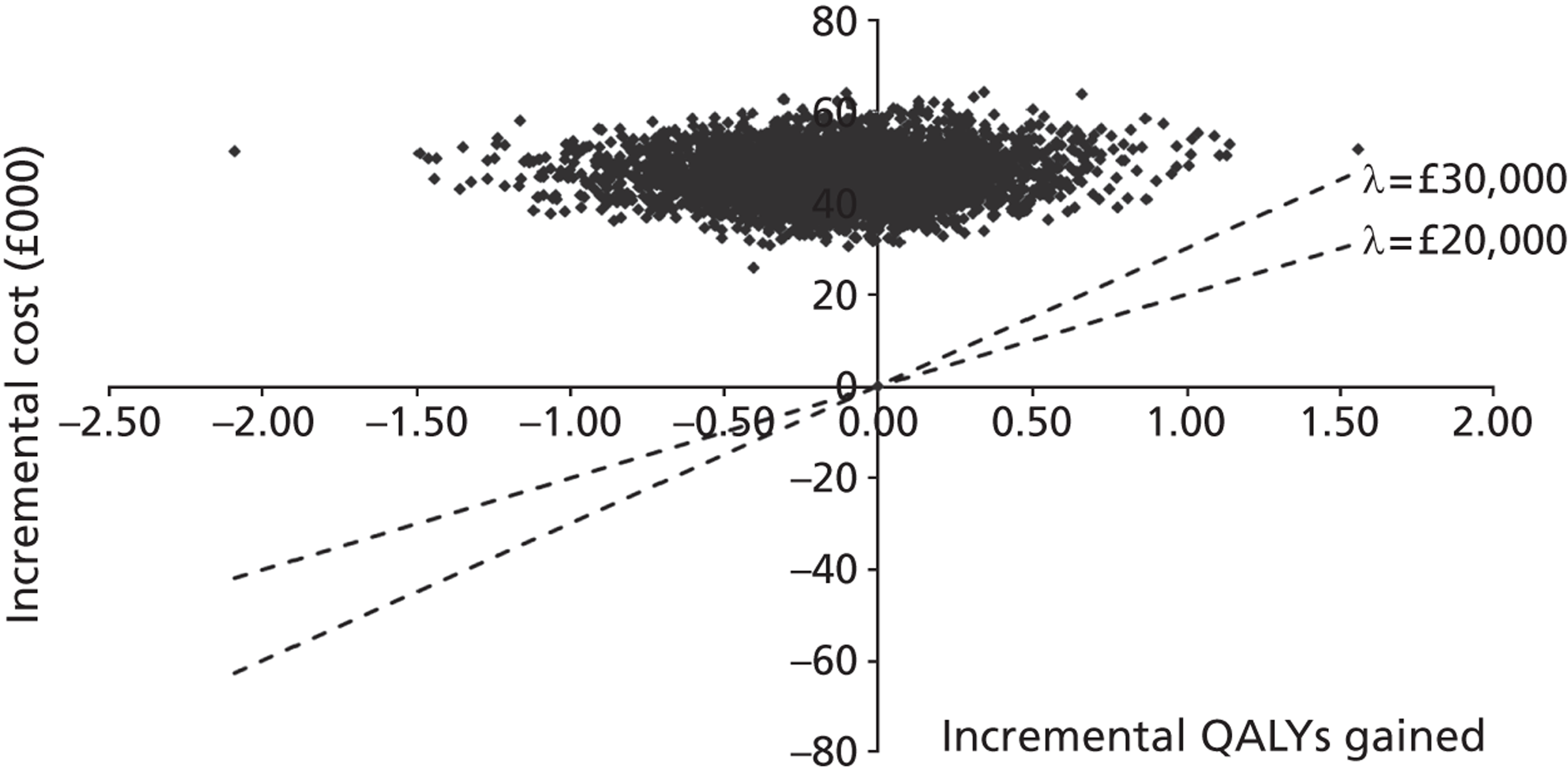
Assuming a price per dose of £15.98, the long-term model suggests that the probability that colistimethate sodium DPI produces a positive QALY gain and has an ICER that is better than £30,000 per QALY gained is zero. The probability that colistimethate sodium DPI is dominated is approximately 0.68.
FIGURE 19.
Reference case model: CEAC (price per dose = £15.98).
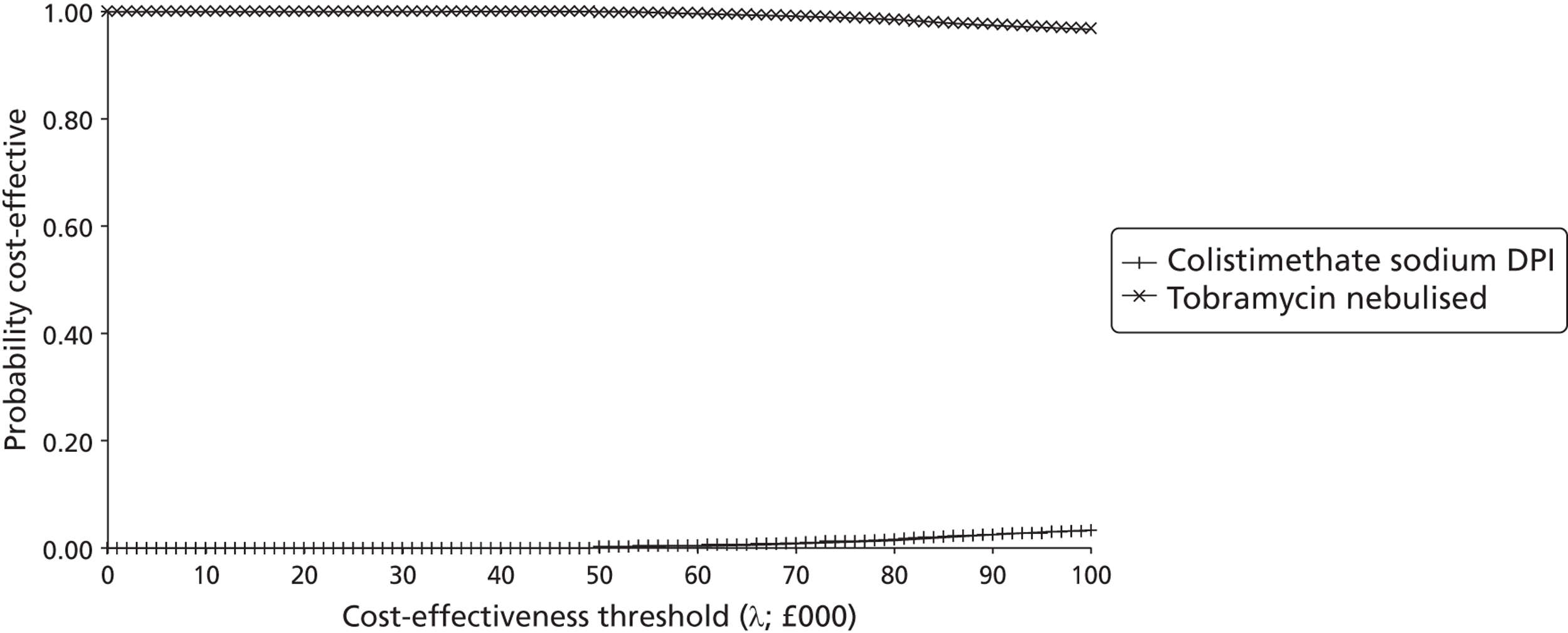
Assuming a willingness-to-pay threshold of £20,000 per QALY gained and a price per dose of £15.98, the probability that colistimethate sodium DPI is optimal is zero. At a willingness-to-pay threshold of £30,000 per QALY gained, the probability that colistimethate sodium DPI is optimal is also zero.
FIGURE 20.
Reference case model: cost-effectiveness plane (price per dose = £19.64).
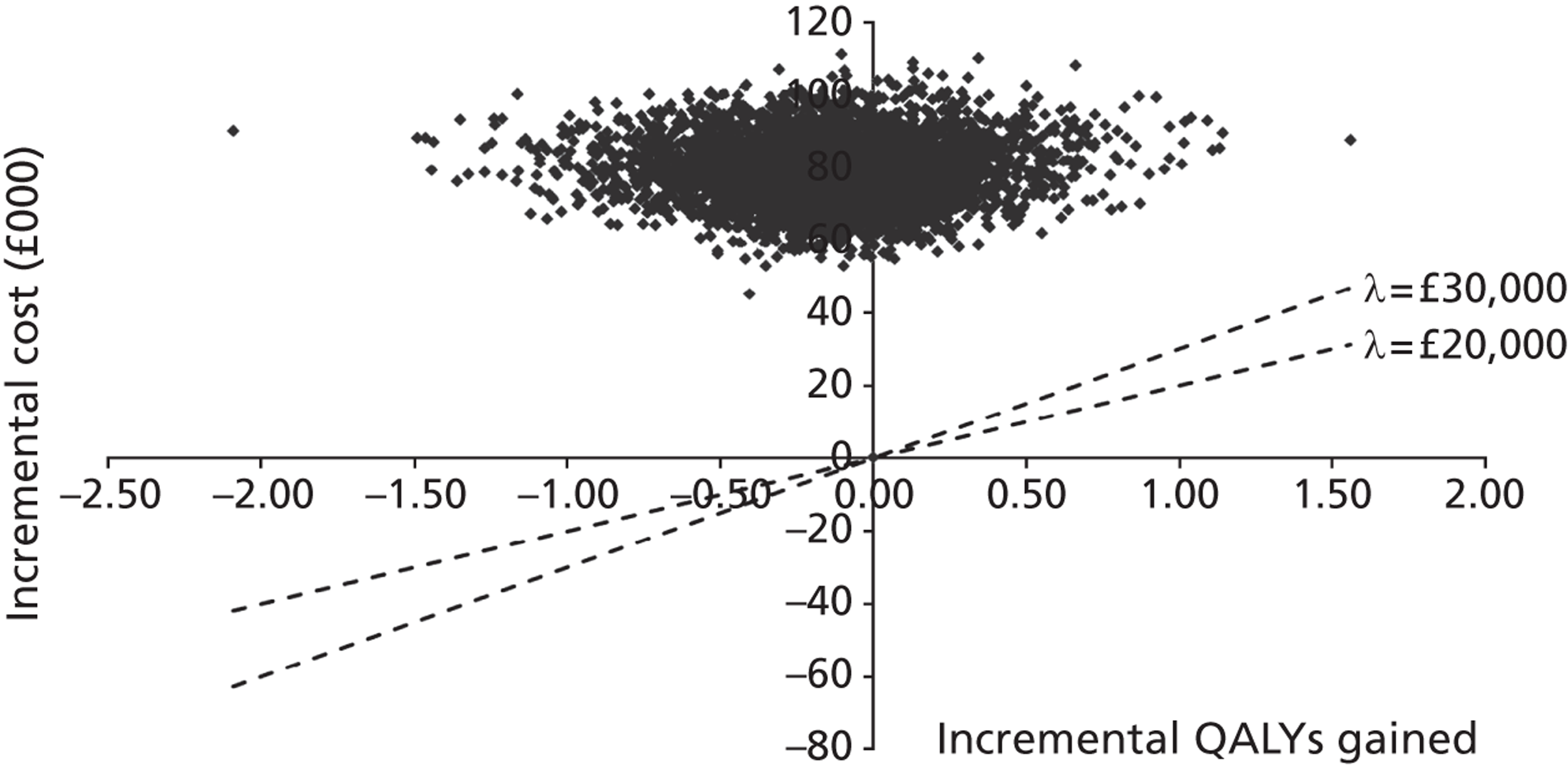
Assuming a price per dose of £19.64, the long-term model suggests that the probability that colistimethate sodium DPI produces a positive QALY gain and has an ICER that is better than £30,000 per QALY gained is zero. The probability that colistimethate sodium DPI is dominated is approximately 0.68.
FIGURE 21.
Reference case model: CEAC (price per dose = £19.64).
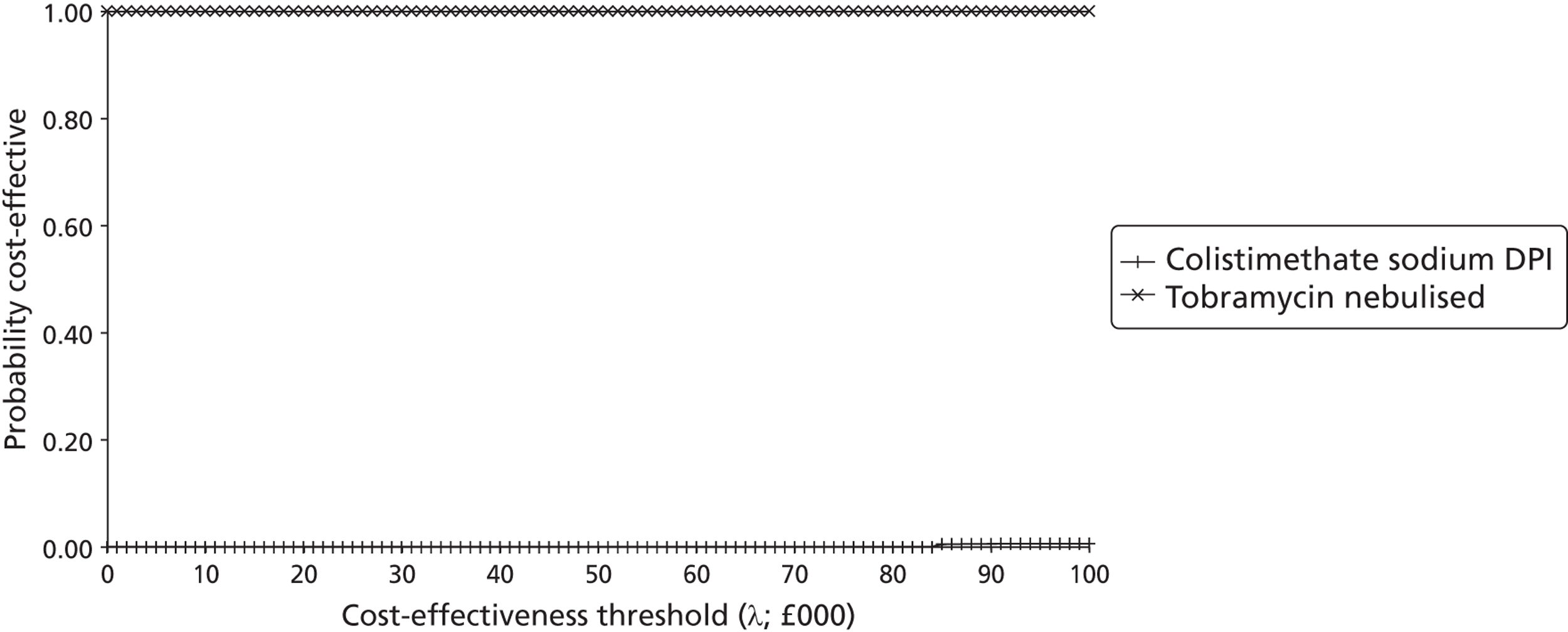
Assuming a willingness-to-pay threshold of £20,000 per QALY gained and a price per dose of £19.64, the probability that colistimethate sodium DPI is optimal is zero. At a willingness-to-pay threshold of £30,000 per QALY gained, the probability that colistimethate sodium DPI is optimal is also zero.
FIGURE 22.
Reference case model: cost-effectiveness plane (price per dose = £21.20).
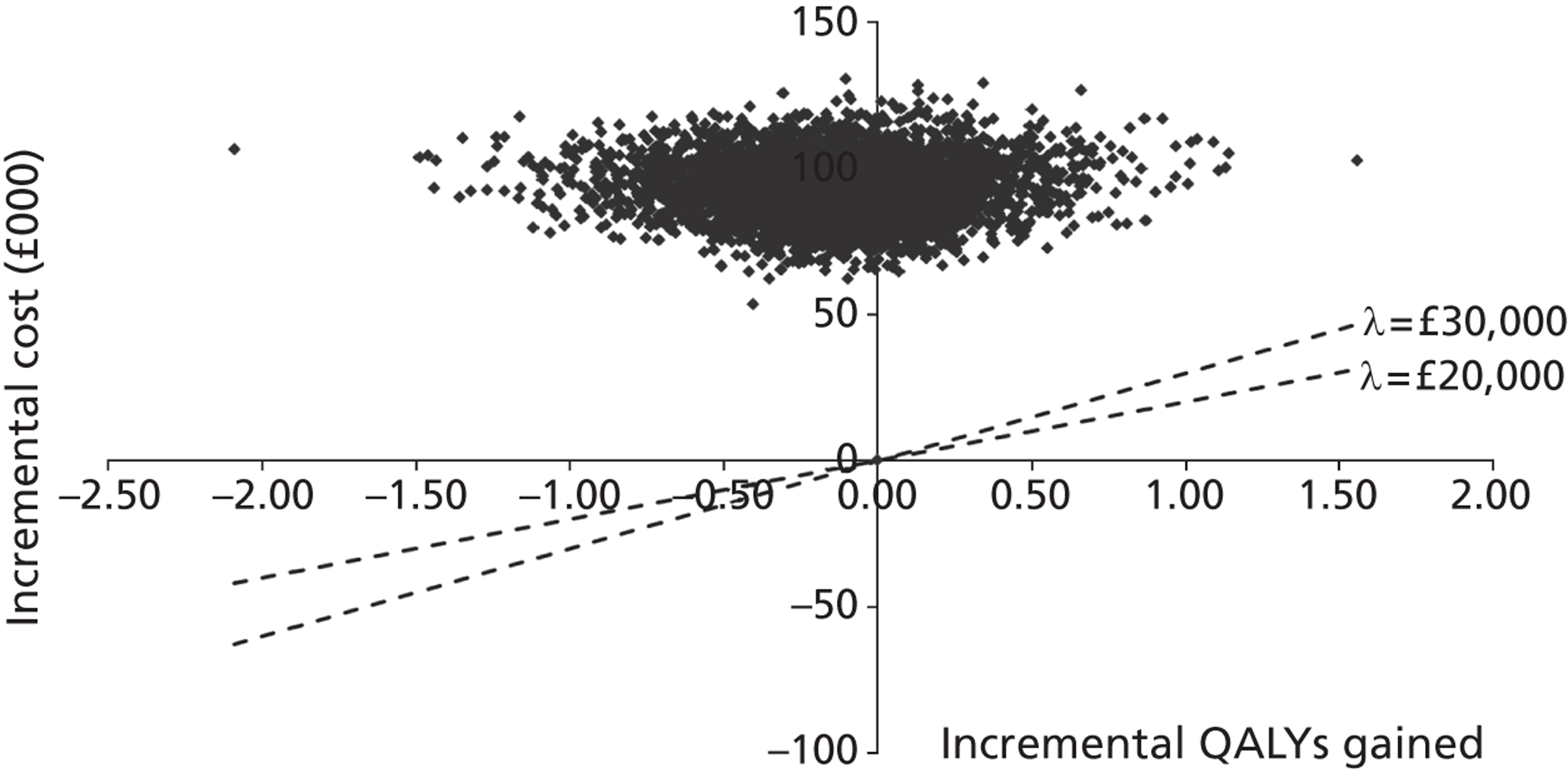
Assuming a price per dose of £21.20, the long-term model suggests that the probability that colistimethate sodium DPI produces a positive QALY gain and has an ICER that is better than £30,000 per QALY gained is zero. The probability that colistimethate sodium DPI is dominated is approximately 0.68.
FIGURE 23.
Reference case model: CEAC (price per dose = £21.20).
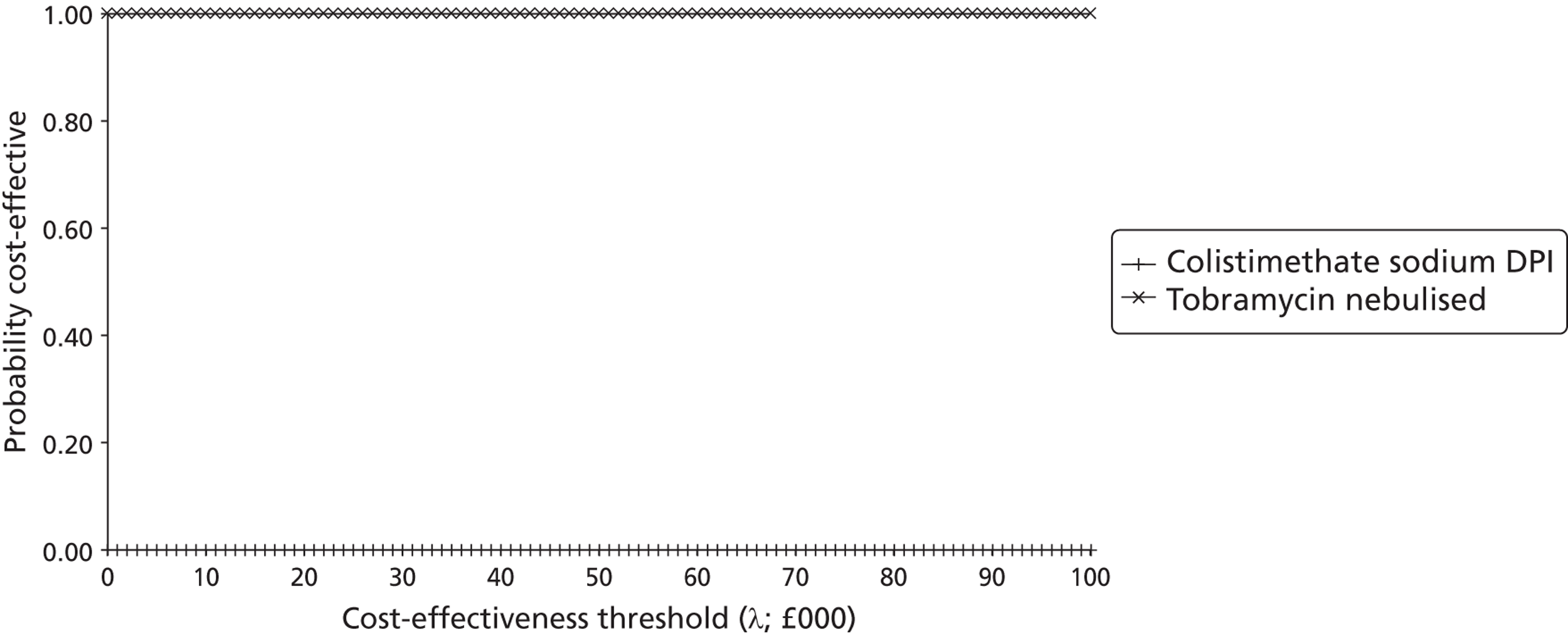
Assuming a willingness-to-pay threshold of £30,000 per QALY gained and a price per dose of £21.20, the probability that colistimethate sodium DPI is optimal is zero.
FIGURE 24.
Reference case model: cost-effectiveness plane (price per dose = £39.29).
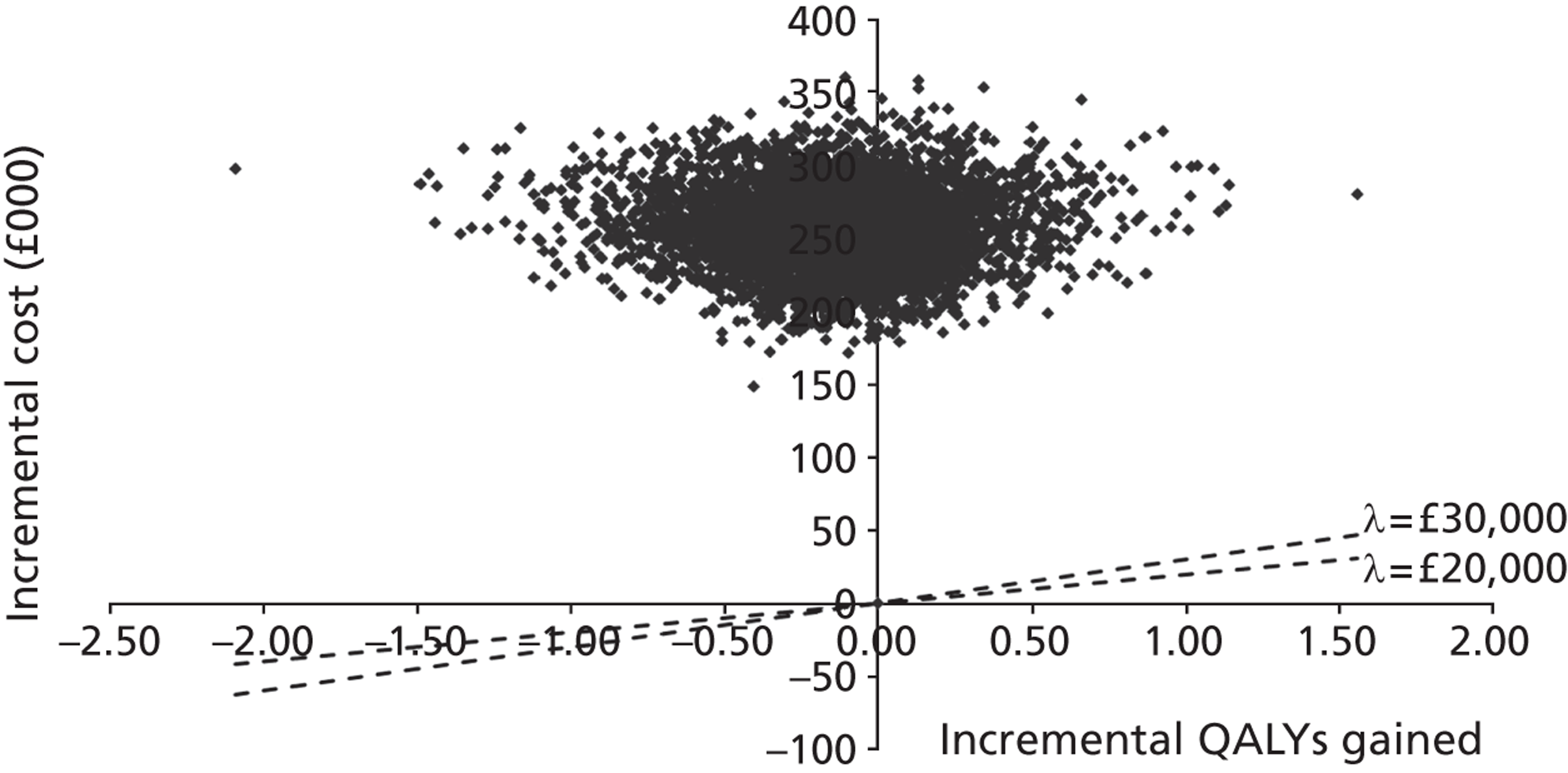
Assuming a price per dose of £39.29, the long-term model suggests that the probability that colistimethate sodium DPI produces a positive QALY gain and has an ICER that is better than £30,000 per QALY gained is zero. The probability that colistimethate sodium DPI is dominated is approximately 0.68.
FIGURE 25.
Reference case model: CEAC (price per dose = £39.29).
Assuming a willingness-to-pay threshold of £30,000 per QALY gained and a price per dose of £39.29, the probability that colistimethate sodium DPI is optimal is zero.
Results of the short-term ‘within-trial’ economic analysis (excluding any extrapolation)
Figures 26–37 present cost-effectiveness planes and CEACs for the short-term model over the six pricing scenarios for colistimethate sodium DPI.
FIGURE 26.
Short-term model: cost-effectiveness plane (price per dose = £9.11).
Assuming a price per dose of £9.11, the short-term model suggests that the probability that colistimethate sodium DPI produces a positive QALY gain and has an ICER that is better than £20,000 per QALY gained is around 0.23. The probability that colistimethate sodium DPI produces a positive QALY gain and has an ICER that is better than £30,000 per QALY gained is also approximately 0.23. The probability that colistimethate sodium DPI is dominated is approximately zero.
FIGURE 27.
Short-term model: CEAC (price per dose = £9.11).
Assuming a willingness-to-pay threshold of £20,000 per QALY gained and a price per dose of £9.11, the probability that colistimethate sodium DPI is optimal within the short-term model is approximately 1.0. At a willingness-to-pay threshold of £30,000 per QALY gained, the probability that colistimethate sodium DPI is optimal is also approximately 1.0.
FIGURE 28.
Short-term model: cost-effectiveness plane (price per dose = £10.60).
Assuming a price per dose of £10.60, the short-term model suggests that the probability that colistimethate sodium DPI produces a positive QALY gain and has an ICER that is better than £20,000 per QALY gained is around 0.23. The probability that colistimethate sodium DPI produces a positive QALY gain and has an ICER that is better than £30,000 per QALY gained is also approximately 0.23. The probability that colistimethate sodium DPI is dominated is approximately 0.03.
FIGURE 29.
Short-term model: CEAC (price per dose = £10.60).
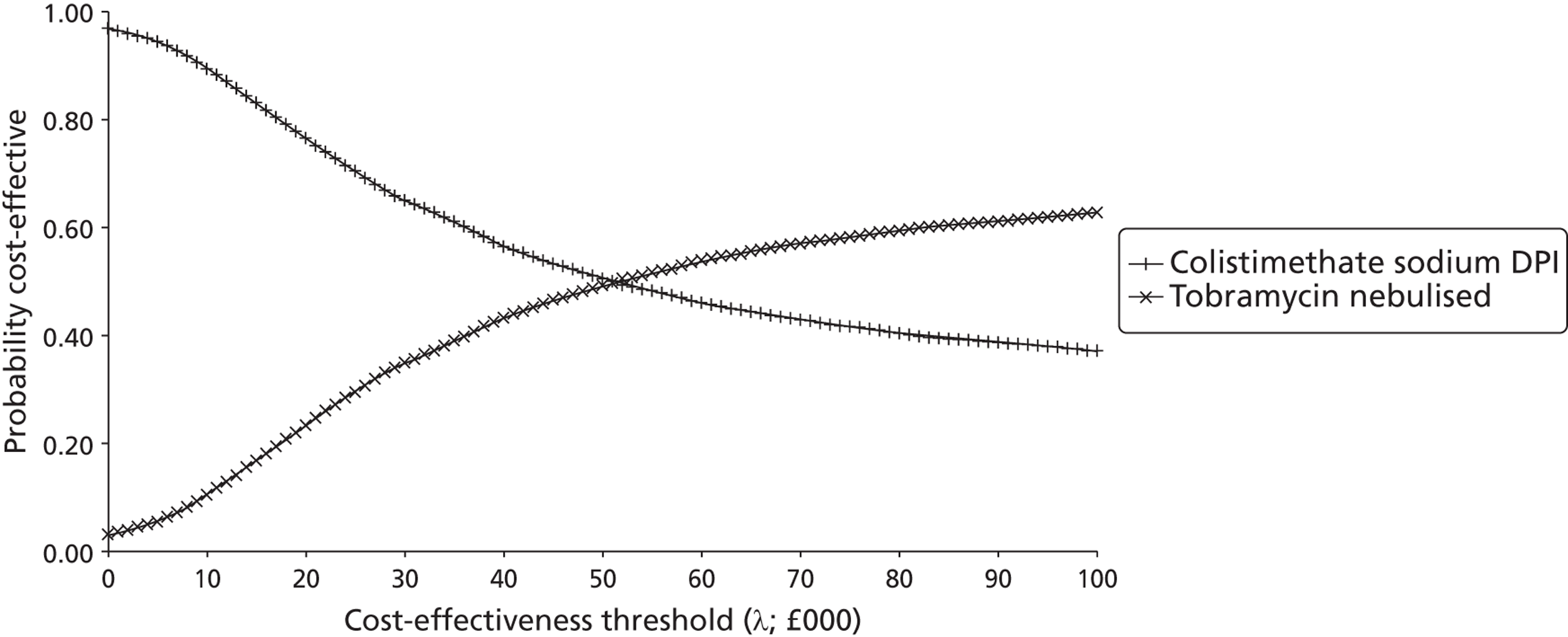
Assuming a willingness-to-pay threshold of £20,000 per QALY gained and a price per dose of £10.60, the probability that colistimethate sodium DPI is optimal within the short-term model is approximately 0.77. At a willingness-to-pay threshold of £30,000 per QALY gained, the probability that colistimethate sodium DPI is optimal is approximately 0.65.
FIGURE 30.
Short-term model: cost-effectiveness plane (price per dose = £15.98).
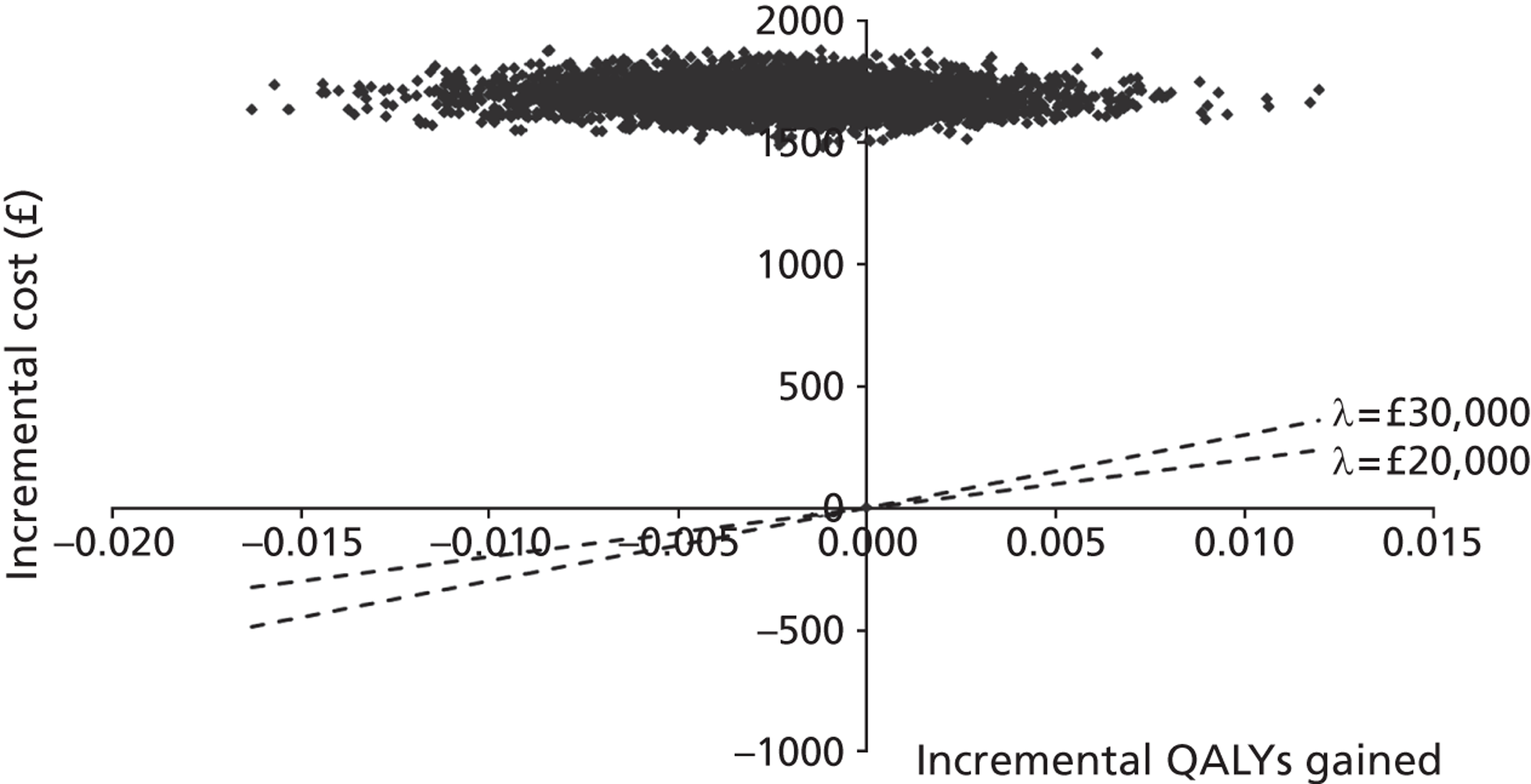
Assuming a price per dose of £15.98, the short-term model suggests that the probability that colistimethate sodium DPI produces a positive QALY gain and has an ICER that is better than £30,000 per QALY gained is zero. The probability that colistimethate sodium DPI is dominated is approximately 0.77.
FIGURE 31.
Short-term model: CEAC (price per dose = £15.98).
Assuming a willingness-to-pay threshold of £30,000 per QALY gained and a price per dose of £15.98, the probability that colistimethate sodium DPI is optimal within the short-term model is zero.
FIGURE 32.
Short-term model: cost-effectiveness plane (price per dose = £19.64).
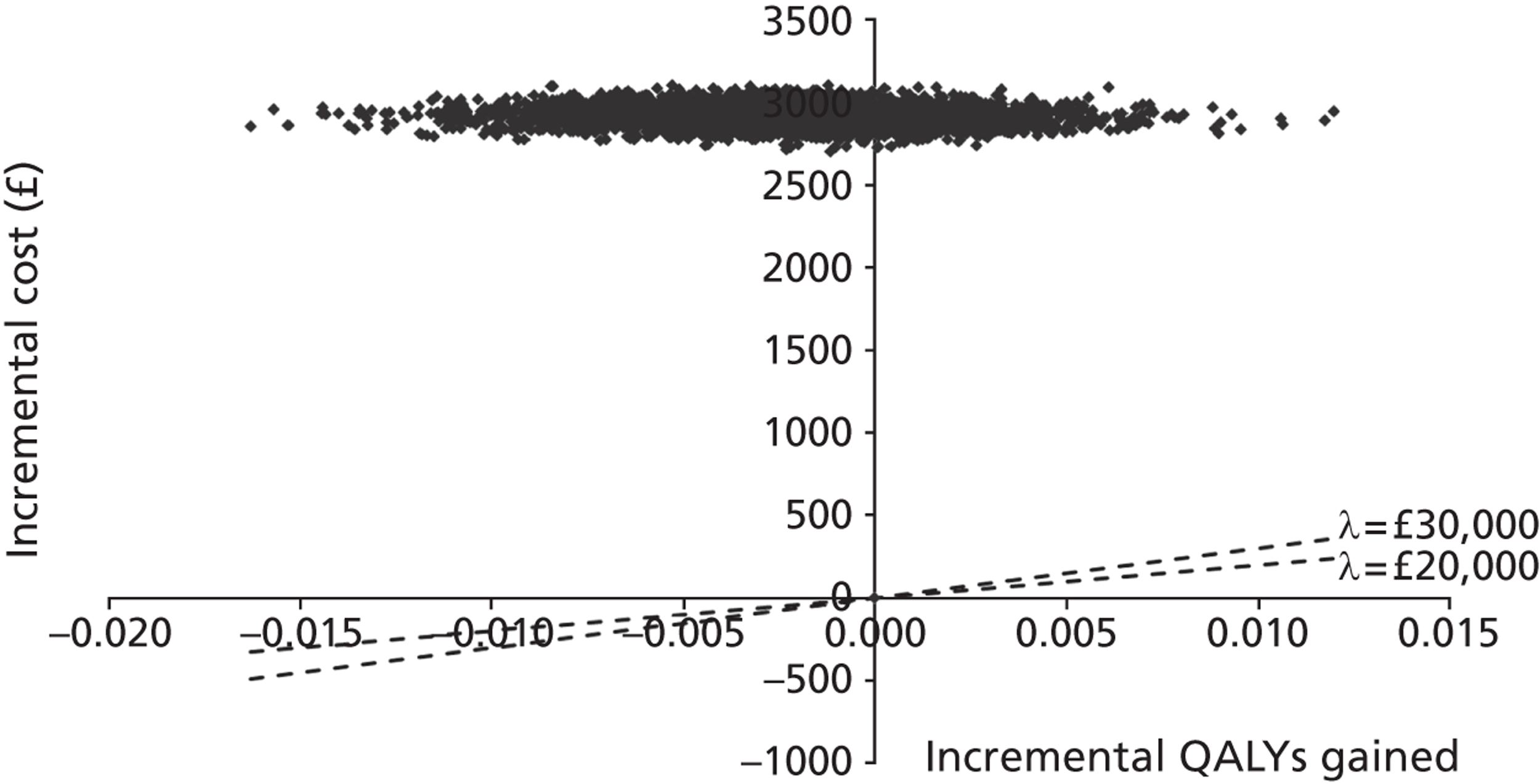
Assuming a price per dose of £19.64, the short-term model suggests that the probability that colistimethate sodium DPI produces a positive QALY gain and has an ICER that is better than £30,000 per QALY gained is zero. The probability that colistimethate sodium DPI is dominated is approximately 0.77.
FIGURE 33.
Short-term model: CEAC (price per dose = £19.64).
Assuming a willingness-to-pay threshold of £30,000 per QALY gained and a price per dose of £19.64, the probability that colistimethate sodium DPI is optimal within the short-term model is zero.
FIGURE 34.
Short-term model: cost-effectiveness plane (price per dose = £21.20).
Assuming a price per dose of £21.20, the short-term model suggests that the probability that colistimethate sodium DPI produces a positive QALY gain and has an ICER that is better than £30,000 per QALY gained is zero. The probability that colistimethate sodium DPI is dominated is approximately 0.77.
FIGURE 35.
Short-term model: CEAC (price per dose = £21.20).
Assuming a willingness-to-pay threshold of £30,000 per QALY gained and a price per dose of £21.20, the probability that colistimethate sodium DPI is optimal within the short-term model is zero.
FIGURE 36.
Short-term model: cost-effectiveness plane (price per dose = £39.29).
Assuming a price per dose of £39.29, the short-term model suggests that the probability that colistimethate sodium DPI produces a positive QALY gain and has an ICER that is better than £30,000 per QALY gained is zero. The probability that colistimethate sodium DPI is dominated is approximately 0.77.
FIGURE 37.
Short-term model: CEAC (price per dose = £39.29).
Assuming a willingness-to-pay threshold of £30,000 per QALY gained and a price per dose of £39.29, the probability that colistimethate sodium DPI is optimal within the short-term model is approximately zero.
The results of the PSA highlight an important aspect of the model: although there is clearly considerable uncertainty surrounding the extrapolation of the COLO/DPI/02/06 trial data,66 this element of the model has virtually no bearing on the economic conclusions of the model, as colistimethate sodium DPI remains dominated when the price is set at one of the higher prices than that of nebulised tobramycin.
Simple (deterministic) sensitivity analysis
Table 47 presents the results of the simple sensitivity analysis; this is presented only for the long-term model. It should be noted that this analysis is deterministic and uses the point estimates of each parameter rather than the expectation of the mean (although the transition probabilities also include the weak priors to allow for comparison against the probabilistic results).
The results of the deterministic sensitivity analysis presented in Table 47 show that the results are particularly sensitive to the choice of utility values used within the model. Where colistimethate sodium DPI produces a positive QALY gain, this is very small and results in ICER ranging from dominating to in excess of £121M per QALY gained.
| Colistimethate sodium DPI price (£) | QALYs | Costs (£) | ICER | ||||
|---|---|---|---|---|---|---|---|
| Colistimethate sodium DPI | Tobramycin nebulised | Incremental | Colistimethate sodium DPI | Tobramycin nebulised | Incremental | ||
| 1. Deterministic point estimates for parameters | |||||||
| 9.11 | 9.46 | 9.57 | −0.12 | 93,720.31 | 110,278.31 | −16,558.00 | 143,325 |
| 10.60 | 9.46 | 9.57 | −0.12 | 107,166.44 | 110,278.31 | −3111.87 | 26,936.14 |
| 15.98 | 9.46 | 9.57 | −0.12 | 155,716.90 | 110,278.31 | 45,438.59 | Dominated |
| 19.64 | 9.46 | 9.57 | −0.12 | 188,745.65 | 110,278.31 | 78,467.34 | Dominated |
| 21.20 | 9.46 | 9.57 | −0.12 | 202,823.48 | 110,278.31 | 92,545.17 | Dominated |
| 39.29 | 9.46 | 9.57 | −0.12 | 366,072.13 | 110,278.31 | 255,793.82 | Dominated |
| 2. TTO utility values from Yi et al.110 | |||||||
| 9.11 | 11.83 | 11.83 | 0.00 | 93,651.08 | 110,086.92 | −16,435.84 | Dominating |
| 10.60 | 11.83 | 11.83 | 0.00 | 107,087.28 | 110,086.92 | −2999.64 | Dominating |
| 15.98 | 11.83 | 11.83 | 0.00 | 155,601.87 | 110,086.92 | 45,514.96 | 21,699,661 |
| 19.64 | 11.83 | 11.83 | 0.00 | 188,606.22 | 110,086.92 | 78,519.31 | 37,434,780 |
| 21.20 | 11.83 | 11.83 | 0.00 | 202,673.65 | 110,086.92 | 92,586.74 | 44,141,552 |
| 39.29 | 11.83 | 11.83 | 0.00 | 365,801.72 | 110,086.92 | 255,714.80 | 121,914,312 |
| 3. SG utility values from Yi et al.110 | |||||||
| 9.11 | 11.15 | 11.15 | 0.00 | 93,651.08 | 110,086.92 | −16,435.84 | Dominating |
| 10.60 | 11.15 | 11.15 | 0.00 | 107,087.28 | 110,086.92 | −2999.64 | Dominating |
| 15.98 | 11.15 | 11.15 | 0.00 | 155,601.87 | 110,086.92 | 45,514.96 | 13,458,617 |
| 19.64 | 11.15 | 11.15 | 0.00 | 188,606.22 | 110,086.92 | 78,519.31 | 23,217,891 |
| 21.20 | 11.15 | 11.15 | 0.00 | 202,673.65 | 110,086.92 | 92,586.74 | 27,377,581 |
| 39.29 | 11.15 | 11.15 | 0.00 | 365,801.72 | 110,086.92 | 255,714.80 | 75,613,992 |
| 4. HUI-2 utility values from Yi et al.110 | |||||||
| 9.11 | 10.25 | 10.24 | 0.01 | 93,651.08 | 110,086.92 | −16,435.84 | Dominating |
| 10.60 | 10.25 | 10.24 | 0.01 | 107,087.28 | 110,086.92 | −2999.64 | Dominating |
| 15.98 | 10.25 | 10.24 | 0.01 | 155,601.87 | 110,086.92 | 45,514.96 | 4,030,094 |
| 19.64 | 10.25 | 10.24 | 0.01 | 188,606.22 | 110,086.92 | 78,519.31 | 6,952,444 |
| 21.20 | 10.25 | 10.24 | 0.01 | 202,673.65 | 110,086.92 | 92,586.74 | 8,198,036 |
| 39.29 | 10.25 | 10.24 | 0.01 | 365,801.72 | 110,086.92 | 255,714.80 | 22,642,110 |
| 5. EQ-5D values from Stahl et al.109 (GOLD criteria) | |||||||
| 9.11 | 8.28 | 8.36 | −0.08 | 93,651.08 | 110,086.92 | −16,435.84 | 213,185 |
| 10.60 | 8.28 | 8.36 | −0.08 | 107,087.28 | 110,086.92 | −2999.64 | 38,907 |
| 15.98 | 8.28 | 8.36 | −0.08 | 155,601.87 | 110,086.92 | 45,514.96 | Dominated |
| 19.64 | 8.28 | 8.36 | −0.08 | 188,606.22 | 110,086.92 | 78,519.31 | Dominated |
| 21.20 | 8.28 | 8.36 | −0.08 | 202,673.65 | 110,086.92 | 92,586.74 | Dominated |
| 39.29 | 8.28 | 8.36 | −0.08 | 365,801.72 | 110,086.92 | 255,714.80 | Dominated |
| 6. EQ-5D values from Stahl et al.109 (BTS criteria) | |||||||
| 9.11 | 8.74 | 8.81 | −0.06 | 93,651.08 | 110,086.92 | −16,435.84 | 264,903 |
| 10.60 | 8.74 | 8.81 | −0.06 | 107,087.28 | 110,086.92 | −2999.64 | 48,346 |
| 15.98 | 8.74 | 8.81 | −0.06 | 155,601.87 | 110,086.92 | 45,514.96 | Dominated |
| 19.64 | 8.74 | 8.81 | −0.06 | 188,606.22 | 110,086.92 | 78,519.31 | Dominated |
| 21.20 | 8.74 | 8.81 | −0.06 | 202,673.65 | 110,086.92 | 92,586.74 | Dominated |
| 39.29 | 8.74 | 8.81 | −0.06 | 365,801.72 | 110,086.92 | 255,714.80 | Dominated |
| 7. Transition probabilities for nebulised tobramycin set equal to those for colistimethate sodium DPI | |||||||
| 9.11 | 9.46 | 9.46 | 0.00 | 93,724.58 | 110,118.99 | −16,394.41 | Dominating |
| 10.60 | 9.46 | 9.46 | 0.00 | 107,171.33 | 110,118.99 | −2947.67 | Dominating |
| 15.98 | 9.46 | 9.46 | 0.00 | 155,724.00 | 110,118.99 | 45,605.00 | 9,793,372 |
| 19.64 | 9.46 | 9.46 | 0.00 | 188,754.25 | 110,118.99 | 78,635.26 | 16,886,400 |
| 21.20 | 9.46 | 9.46 | 0.00 | 202,832.72 | 110,118.99 | 92,713.73 | 19,909,658 |
| 39.29 | 9.46 | 9.46 | 0.00 | 366,088.82 | 110,118.99 | 255,969.82 | 54,967,822 |
| 8. Utility decrement for exacerbations doubled | |||||||
| 9.11 | 9.41 | 9.52 | −0.11 | 93,720.31 | 110,278.31 | −16,558.00 | 149,445 |
| 10.60 | 9.41 | 9.52 | −0.11 | 107,166.44 | 110,278.31 | −3111.87 | 28,086 |
| 15.98 | 9.41 | 9.52 | −0.11 | 155,716.90 | 110,278.31 | 45,438.59 | Dominated |
| 19.64 | 9.41 | 9.52 | −0.11 | 188,745.65 | 110,278.31 | 78,467.34 | Dominated |
| 21.20 | 9.41 | 9.52 | −0.11 | 202,823.48 | 110,278.31 | 92,545.17 | Dominated |
| 39.29 | 9.41 | 9.52 | −0.11 | 366,072.13 | 110,278.31 | 255,793.82 | Dominated |
| 9. Cost of hospitalisation doubled | |||||||
| 9.11 | 9.46 | 9.57 | −0.12 | 103,782.02 | 120,772.51 | −16,990.49 | 147,069 |
| 10.60 | 9.46 | 9.57 | −0.12 | 117,228.15 | 120,772.51 | −3544.36 | 30,680 |
| 15.98 | 9.46 | 9.57 | −0.12 | 165,778.61 | 120,772.51 | 45,006.10 | Dominated |
| 19.64 | 9.46 | 9.57 | −0.12 | 198,807.36 | 120,772.51 | 78,034.85 | Dominated |
| 21.20 | 9.46 | 9.57 | −0.12 | 212,885.19 | 120,772.51 | 92,112.68 | Dominated |
| 39.29 | 9.46 | 9.57 | −0.12 | 376,133.84 | 120,772.51 | 255,361.33 | Dominated |
Commentary on the cost-effectiveness of tobramycin dry powder for inhalation
The de novo Assessment Group model explicitly excludes tobramycin DPI. This decision was taken by the Assessment Group, and NICE were informed of this in December 2011. This exclusion reflects the absence of suitable FEV1% data for tobramycin DPI and the absence of a health economic model within the Novartis Pharmaceuticals submission to NICE. 60 Despite the absence of a model, it is possible to crudely postulate the likely incremental cost-effectiveness of tobramycin DPI compared with nebulised tobramycin on the basis of the following observations regarding the available evidence base.
-
There is no empirical evidence that tobramycin DPI provides an improved or equivalent level of HRQoL as nebulised tobramycin.
-
Within the EAGER trial,65 study follow-up was insufficient to assess any potential benefit in survival duration for tobramycin DPI. Three deaths occurred, all of which were in the tobramycin DPI group. 65
-
There appears to be a small incremental FEV1% predicted associated with tobramycin DPI (see Table 12). However, as above (see Methodological issues surrounding the economic evaluation of CF treatments), assumptions of a simple independent relationship between FEV1% predicted and mortality should be interpreted with caution.
-
Although Novartis Pharmaceuticals60 did not provide data on exacerbations requested by the Assessment Group, results reported by Konstan et al. 65 suggest that the incidence of lung disorder, which remains the best available proxy for exacerbation incidence, was higher in the tobramycin DPI group (relative risk = 1.12). It is likely that the cost of managing exacerbations would therefore be higher for tobramycin DPI than nebulised tobramycin. This would also likely result in a small QALY loss.
-
The EAGER trial65 suggests a less favourable profile for tobramycin DPI across almost all of the common AEs (especially cough and dysphonia) compared with nebulised tobramycin.
-
The incremental drug cost per 28-day treatment cycle for tobramycin DPI is £602.80 higher than that for nebulised tobramycin. Based on the treatment time within the Assessment Group model, this would result in a discounted lifetime drug and nebuliser cost of around £144,442 per patient. Compared against nebulised tobramycin, the discounted lifetime incremental drug cost of tobramycin DPI is around £46,168 per patient. This explicitly excludes any cost disadvantage associated with the apparently higher exacerbation rate for tobramycin DPI.
Given the incremental cost of tobramycin DPI, tobramycin DPI would have to produce 1.54 additional discounted QALYs compared with nebulised tobramycin to achieve a cost–utility ratio of £30,000 per QALY gained. In order to achieve a cost per QALY ratio of £20,000, tobramycin DPI would have to produce 2.31 additional discounted QALYs compared with nebulised tobramycin. Given the nebulised tobramycin transition matrix from COLO/DPI/02/0666 for the comparator, the assumed treatment starting age (21 years) and the use of EQ-5D values for alternative FEV1% bands,60,111 neither of these incremental QALY thresholds is actually possible within the Assessment Group model.
As noted above, there may be a process-related utility benefit associated with tobramycin DPI owing to treatment convenience that has not been considered within the above analysis. However, the trial investigators did not collect any information relating to HRQoL and therefore the plausibility of such an argument cannot be demonstrated empirically.
Following this assessment, the Assessment Group undertook further economic analyses of both colistimethate sodium DPI and tobramycin DPI compared with nebulised tobraymcin. These analyses included Patient Access Schemes for both DPI products. These analyses are not presented here.
Budget impact analysis
Table 48 presents a simple budget impact analysis for colistimethate sodium DPI over the six prices. This analysis assumes that colistimethate sodium DPI and tobramycin DPI would replace only nebulised tobramycin, as this reflects the limitations of the scope of the economic analysis undertaken.
| Parameter | Value | Population value | Budget impact | Source |
|---|---|---|---|---|
| No. of patients with CF with chronic P. aeruginosa | 2806 | CF Registry report 20106 | ||
| Proportion patients receiving tobramycin | 0.24 | |||
| Estimated patients per year eligible for treatment | 673 | |||
| Probability exacerbation/year (tobramycin) | 0.85 | COLO/DPI/02/0669 | ||
| Probability exacerbation/year (colistimethate sodium) | 0.82 | |||
| Probability exacerbation is major | 0.66 | |||
| Cost minor exacerbation (£) | 427.69 | NHS Reference Costs 2010–11129 | ||
| Cost major exacerbation (£) | 1500.14 | |||
| Mean cost exacerbation (£) | 1135.51 | |||
| Marginal cost of nebuliser maintenance (£) | 200.00 | Personal communicationa | ||
| Tobramycin nebulised: drug, nebuliser and exacerbation costs/year (£) | 8898 | 5,991,936 | ||
| Cost colistimethate sodium DPI/year (plus exacerbation costs), price = £9.11 | 7585 | 5,108,179 | −£883,757 | |
| Cost colistimethate sodium DPI/year (plus exacerbation costs), price = £10.60 | 8673 | 5,840,680 | −£151,256 | |
| Cost colistimethate sodium DPI/year (plus exacerbation costs), price = £15.98 | 12,600 | 8,485,548 | £2,493,612 | |
| Cost colistimethate sodium DPI/year (plus exacerbation costs), price = £19.64 | 15,272 | 10,284,845 | £4,292,909 | |
| Cost colistimethate sodium DPI/year (plus exacerbation costs), price = £21.20 | 16,411 | 11,051,758 | £5,059,822 | |
| Cost colistimethate sodium DPI/year (plus exacerbation costs), price = £39.29 | 29,617 | 19,945,005 | £13,953,069 | |
| Cost tobramycin DPI/year (plus exacerbation costs) | 12,742 | 8,580,710 | £2,588,774 |
The estimated budget impact of colistimethate sodium DPI is negative (cost saving) if the price of colistimethate sodium DPI is set at £9.11 per dose or £10.60 per dose. At the top end of the price range, the estimated cost to the NHS is around £14M per year. Tobramycin DPI is expected to have an annual budget impact of around £2.6M.
Discussion
Summary of available evidence
There is a dearth of economic evidence relating to the cost-effectiveness of colistimethate sodium DPI and tobramycin DPI for the treatment of P. aeruginosa in patients with CF. The literature review did not identify any published economic analyses of colistimethate sodium DPI or tobramycin DPI. Novartis Pharmaceuticals60 did not submit any economic evidence relating to the cost-effectiveness of tobramycin DPI. Forest Laboratories did submit an economic model to assess colistimethate sodium DPI compared with nebulised tobramycin, which suggests that colistimethate sodium DPI is expected to dominate nebulised tobramycin. However, this was subject to a number of methodological problems and biases that are likely to produce overly favourable estimates of cost-effectiveness. The basis of this model assumes that an absolute FEV1 measurement is directly associated with survival duration. A review of the literature suggests that the validity of this relationship is dubious and is likely to be subject to considerable confounding. A reanalysis of the Forest Laboratories model using more plausible assumptions suggests that the cost-effectiveness of colistimethate sodium DPI compared with nebulised tobramycin is expected to range from dominating to £485,550 per QALY gained, depending on the price of the intervention.
Summary of the economic analysis undertaken by the Assessment Group
The Assessment Group developed a de novo health economic model based on patient-level data from the COLO/DPI/02/06 trial66 augmented using external sources. This model extrapolates 24-week FEV1% to a lifetime horizon. Although this extrapolation is clearly subject to considerable uncertainty, the conclusions of the analysis appear robust as the short-term 24-week analysis of the COLO/DPI/02/06 trial66 produces consistent results to the lifetime model. An analysis of longitudinal patient-level data from the CF Registry suggests that the probabilities of transition between FEV1% are relatively stable over time, which lends some weight to the credibility of the trial extrapolation.
The results of this analysis suggest that colistimethate sodium DPI is expected to produce fewer QALYs than nebulised tobramycin, both in the short-term and over a lifetime horizon. If the price of colistimethate sodium DPI is set at one of the prices which is higher than that of nebulised tobramycin, it is expected to be more expensive and hence dominated by nebulised tobramycin. If the price of colistimethate sodium DPI is set at £9.11, the incremental cost-effectiveness of nebulised tobramycin compared with colistimethate sodium DPI is expected to be in the range £126,000–277,000 per QALY gained. If the price of colistimethate sodium DPI is set at £10.60, the incremental cost-effectiveness of nebulised tobramycin compared with colistimethate sodium DPI is expected to be in the range £24,000–50,000 per QALY gained.
Insufficient data were available, at the time of the assessment, to produce a full economic evaluation of tobramycin DPI compared with any comparator. Instead, a crude threshold analysis is presented to estimate the necessary QALY gain that tobramycin DPI would need to produce given its incremental lifetime cost. The model structure suggests that given its acquisition cost, it is not possible for tobramycin to have a cost-effectiveness ratio that is better than £30,000 per QALY gained.
Chapter 5 Assessment of factors relevant to the NHS and other parties
The introduction of colistimethate sodium DPI and tobramycin DPI would have a number of other implications for the NHS.
Treatment adherence and convenience
As noted in Chapter 1, one of the key potential benefits of dry powder formulations of these therapies is the reduced burden in treatment administration. However, it is unclear whether this would necessarily lead to improved treatment compliance, as a number of patients may not adhere to treatment due to increased side effects (e.g. cough). In principle, the technology could be used in a variety of settings, including home, hospital, work or at school. It should also be noted that for many patients, the development of newer nebuliser devices that enable faster treatment administration will have already reduced the treatment time compared against traditional nebulisers. It could be argued that the availability of dry powder treatment would increase the burden of treatment as most patients with P. aeruginosa require nebulisers for other treatments, therefore a further inhaler device would involve adding to the equipment needed to treat patients. Research in this area is planned but has not yet commenced.
Training/impact on primary care
The introduction of colistimethate sodium DPI and tobramycin DPI may have implications for NHS staff training. There may be a need to monitor patients closely when they are initially placed on dry powder formulations owing to the increase in some AEs. Support for the patient will be needed from doctors, specialist nurses and physiotherapists. Most staff, however, are likely to already be familiar with dry powder technology formulations for other drugs such as bronchodilators.
Age of patients/appropriateness of use for children
Young children may struggle to use the dry powder technology. However, this is most likely already dealt with by the licensing conditions of the dry powder technologies.
Reduced risk of contamination
Dry powder inhalers are disposable, which reduces the risk of contamination and further infection. Previous nebulisers are prone to this kind of contamination unless regular maintenance is performed, thereby increasing the burden of treatment to the patient and family. Compliance with keeping these devices clean is poor. Cross contamination is less of a problem with single-dose powder capsules as P. aeruginosa can also colonise in bottles of opened solutions for nebulisers.
Chapter 6 Discussion
Statement of principal findings
Principal findings: clinical effectiveness
Three trials were included in the review. 65,66 Both colistimethate sodium DPI and nebulised tobramycin DPI were reported to be non-inferior to nebulised tobramycin in pivotal Phase III non-inferiority trials, for the outcome FEV1%, based on information from two of the trials. However, there are problems with the trials, which indicate that the results should be judged with caution. None of the trials complied with the time horizon of 12 months’ follow-up recommended by the EMA for efficacy trials, with both following patients for 24 weeks only. As such, the existing evidence base does not include information about the long-term efficacy and safety of these treatments. Both of the large trials65,66 could also be criticised for the way they analysed the results, with the COLO/DPI/02/06 trial66 of colistimethate sodium DPI only reaching non-inferiority when analysed non-parametrically, and the EAGER trial65 of tobramycin DPI only presenting results without imputation. The COLO/DPI/02/0566 trial was not powered to detect an effect in FEV1%, was only 4 weeks in duration (prior to crossover), and reported no significant differences in FEV1% between arms and from baseline. It was not possible to draw any firm conclusions as to the relative efficacy as measured by FEV1% of any intervention compared with any other intervention (except nebulised tobramycin) owing to missing data, uncertain comparability of patient characteristics and incompatible methods of analysing the data.
As FEV1% is a surrogate outcome, the EMA recommends that it should be considered alongside ‘harder’ outcomes such as exacerbations, and should be supported with microbiological data. Sputum density data for tobramycin DPI supported the FEV1% values seen with a decrease at week 20. Data on sputum density outcomes were not available for colistimethate sodium DPI. Resistance of around 20% was reported for the tobramycin arms across both Phase III trials, and of ≤ 1.1% for colistimethate sodium DPI. Both tobramycin DPI and colistimethate sodium DPI appeared to be less effective in reducing the frequency of exacerbations, but patients treated with DPIs spent less time on antibiotics. AEs were mostly similar between arms within trials, except for cough, which was higher in both DPI intervention arms. More patients in the DPI intervention arms withdrew owing to AEs in both trials. 65,66 The statistical and clinical significance of differences in sputum density, resistance data, exacerbations and AE data is not known. Insufficient mortality events were recorded and the study follow-up was not long enough to draw conclusions as to the effect of DPI formulations on mortality in comparison with nebulised tobramycin.
Principal findings: cost-effectiveness
The cost-effectiveness of colistimethate sodium DPI and tobramycin DPI are subject to considerable uncertainty. This is driven by a number of factors including (1) an absence of any direct method of HRQoL elicitation within the pivotal clinical trials; (2) the short-term nature of follow-up within these studies and the absence of sufficient survival data to allow extrapolation; (3) the questionable validity of absolute measures of FEV1 as an independent predictor of CF mortality; and (4) gaps in the evidence base concerning the relative effectiveness of competing treatments for P. aeruginosa lung infection. Given current evidence, questions relating to the long-term cost-effectiveness of the colistimethate sodium DPI and tobramycin are therefore inevitably hinged on the credibility of relationships between intermediate and final outcomes.
A systematic review of existing cost-effectiveness studies did not identify any full economic evaluations of colistimethate sodium DPI or tobramycin DPI. Previous analyses were short term and did not involve extrapolation to more relevant time horizons, nor did they involve the translation of intermediate outcomes to more policy-relevant economic outcome measures.
Two submissions were received from the manufacturers of colistimethate sodium DPI and tobramycin DPI. Novartis Pharmaceuticals60 did not submit any economic evidence to support the argument that tobramycin DPI represents a cost-effective use of resources. Forest Laboratories66 did submit an economic model to assess colistimethate sodium DPI compared with nebulised tobramycin, which suggests that colistimethate sodium DPI is expected to dominate nebulised tobramycin. However, this was subject to a number of methodological problems and biases that are likely to produce overly favourable estimates of cost-effectiveness. The Forest Laboratories submission was ambiguous with respect to the actual proposed price of colistimethate sodium DPI, and the net benefit estimates produced from the economic model did not include the acquisition costs of either the intervention or the comparator. This model is underpinned by the assumption that an absolute FEV1 measurement is directly associated with survival duration (either at 1 year or 2 years post measurement). A review of the available literature suggests that the validity of this relationship is dubious and is likely to be subject to confounding owing to other clinically relevant variables. Even if this relationship is considered plausible, and the methods of prediction are considered accurate, a reanalysis of the Forest Laboratories model using more plausible assumptions suggests that the incremental cost–utility of colistimethate sodium DPI compared with nebulised tobramycin is expected to range from dominating to £485,550 per QALY gained, depending on the proposed price of the intervention.
The de novo health economic model developed by the Assessment Group is based on patient-level data from the COLO/DPI/02/06 trial66 augmented using external sources. This model defines differential states of HRQoL by FEV1 strata, and extrapolates the observed FEV1 transitions within COLO/DPI/02/06 trial to a lifetime horizon. No additional survival benefit is assumed. Although this extrapolation is clearly subject to considerable uncertainty, the conclusions of the analysis appear robust as the short-term 24-week analysis of the COLO/DPI/06 trial66 produces consistent results to the lifetime model. The results of this economic analysis suggest that colistimethate sodium DPI is expected to produce fewer QALYs than nebulised tobramycin, both in the short term and over a lifetime horizon. If the price of colistimethate sodium DPI set at one of the prices that is higher than that of nebulised tobramycin, it is expected to be more expensive and hence dominated by nebulised tobramycin. If the price of colistimethate sodium DPI is set at £9.11, the incremental cost-effectiveness of nebulised tobramycin compared with colistimethate sodium DPI is expected to be in the range £126,000–277,000 per QALY gained. If the price of colistimethate sodium DPI is set at £10.60, the incremental cost-effectiveness of nebulised tobramycin compared with colistimethate sodium DPI is expected to be in the range £24,000–50,000 per QALY gained.
At the time of the assessment, insufficient data were available to produce a full economic evaluation of tobramycin DPI compared with any relevant comparator. Instead, a crude threshold analysis is presented to estimate the necessary QALY gain that tobramycin DPI would need to produce in order to achieve a particular cost–utility ratio, given its incremental lifetime cost. In order to achieve a cost–utility ratio of £30,000 per QALY gained, tobramycin DPI would need to produce an estimated 1.54 additional discounted QALYs compared with nebulised tobramycin. In order to achieve a cost per QALY ratio of £20,000, tobramycin DPI would need to produce an estimated 2.31 additional discounted QALYs compared with nebulised tobramycin. The model structure suggests that neither of these QALY thresholds is achievable given the price of tobramycin DPI.
Following this assessment, the Assessment Group undertook further economic analyses of both colistimethate sodium DPI and tobramycin DPI compared with nebulised tobraymcin. These analyses included Patient Access Schemes for both DPI products. These analyses are not presented here.
Strengths and limitations of the assessment
A key strength of this assessment is that the systematic review has been conducted to a high standard including comprehensive search strategies with study selection, data extraction and quality assessment checked by a second reviewer.
The review is limited by the small number of trials available, and methodological weaknesses and incompatibilities within the trials which inevitably limit the comparability of evidence across the trials. There are variations in the definition and measurement of the key outcomes, owing to non-compliance with EMA research guidelines. No data that comply with the NICE reference case on quality of life were available from any of the trials.
The health economic model developed within this assessment was based on clinical opinion regarding current treatment pathways and systematic reviews of evidence relating to the plausibility of relationships between intermediate and final end points (rather than pure assumption). The model was populated using the best available evidence and was peer reviewed by several individuals with clinical and methodological expertise.
The Assessment Group model involves extrapolation of FEV1 estimates within the COLO/DPI/06 trial. 66 Within this analysis, the observable period is 24 weeks in duration, whereas the projected period is around 43 years (when < 1% patients are still alive). The considerable uncertainty surrounding the short-term evidence base inevitably results in uncertainty surrounding the long-term cost-effectiveness of colistimethate sodium DPI. One strength of the assessment is that the model considers the impact of this extrapolation on the cost-effectiveness of treatment. In addition, uncertainty surrounding the appropriate method of health state valuation is explored by applying a variety of health utility estimates within the model.
The key anticipated benefits of colistimethate sodium DPI and nebulised tobramycin concern the increased convenience afforded by reduced treatment administration time as compared against nebulised antibiotics. In addition, the DPIs are more portable than nebulisers. These may represent ‘process utilities’. However, none of the clinical trials attempted to capture these potential effects using a preference-based instrument. As a consequence, this potential effect is not reflected in the health economic analysis. It should be noted, however, that newer nebulisers, such as the I-neb and eFlow devices, also allow for faster treatment delivery than conventional nebulisers. The incremental benefits of this aspect of DPI delivery thus remain unclear.
Uncertainties
The key uncertainties within this assessment are:
-
The relative efficacy and safety profiles of colistimethate sodium DPI and tobramycin DPI.
-
The long-term efficacy of treatment using colistimethate sodium DPI and tobramycin DPI compared with current standard nebulised therapies.
-
The validity of the relationship between short-term impact on lung function and longer-term final patient outcomes (mortality and HRQoL).
-
Whether or not there exists any long-term impact of DPI treatment on patient survival.
-
Long-term treatment compliance.
-
The clinical relevance of resistance to DPIs and its impact upon treatment efficacy.
-
The trade-off between ease/speed of drug administration using the inhaler devices and AEs (and the impact of both on patients’ HRQoL).
Chapter 7 Conclusions
Main conclusions of the assessment
Both DPI formulations have been shown to be non-inferior to nebulised tobramycin as measured by FEV1%. However, the results of these trials should be interpreted with caution owing to means by which the results were analysed, the length of follow-up, and concerns about the ability of FEV1% to accurately represent changes in lung health. The impact of resistance to tobramycin is not known. When considered alongside other outcomes, it would appear possible that patients on DPI formulations experience more exacerbations but spend less time on antibiotics, experience more cough AEs and may be more likely to not tolerate the treatment. As such, the advantages and non-inferiority of DPI treatments compared with nebulised tobramycin remain unclear when all relevant outcomes are considered. Inevitably, the cost-effectiveness of the dry powder formulations is subject to considerable uncertainty. The Assessment Group model suggests that colistimethate sodium is expected to produce fewer QALYs than nebulised tobramycin. Depending on the price adopted for colistimethate sodium DPI, this results either in a situation whereby colistimethate sodium DPI is dominated by nebulised tobramycin, or one whereby the incremental cost-effectiveness of nebulised tobramycin compared with colistimethate sodium DPI is in the range £24,000–277,000 per QALY gained (south-west quadrant). The economic analysis also suggests that given its price, it is highly unlikely that tobramycin DPI has a cost-effectiveness ratio of < £30,000 per QALY gained when compared against nebulised tobramycin.
Implications for service provision
The burden on the NHS of introducing DPIs is generally in terms of the drug acquisition cost. For many of these patients, nebulisers will still be required for administration of mucolytics and bronchodilators; however, there may be some reduction in the requirement for nebuliser maintenance.
Suggested research priorities
A RCT to assess the longer-term (≥ 12 months) efficacy of colistimethate sodium DPI and tobramycin DPI in comparison with nebulised treatments would be beneficial. Such a study should include the direct assessment of HRQoL using a relevant preference-based instrument. Future studies should ensure adherence to the EMA guidelines. 18 In addition, high-quality research concerning the relationship between FEV1 or other measures of lung function and survival/HRQoL would be useful.
Acknowledgements
We would like to thank the following individuals for providing considerable input into this assessment: Professor Chris Taylor (Sheffield Children’s NHS Foundation Trust), Dr Martin Walshaw (Liverpool Heart and Chest Hospital NHS Foundation Trust) and Dr Keith Brownlee (Leeds Teaching Hospitals NHS Trust). We would like to thank Forest Laboratories and Novartis Pharmaceuticals for providing additional data requested throughout the course of the assessment. We thank Dr John Stevens for providing advice on the interpretation of the statistical analysis undertaken within the included studies. Thanks also to Elaine Gunn and the CF Registry for providing patient-level data to inform the economic analysis. We would also like to thank Dr Diana Bilton and Rod Taylor for their helpful comments on the draft report.
Contributions of authors
Paul Tappenden acted as principal investigator for this assessment.
Anna Cantrell developed the electronic search strategies.
Sue Harnan, Lesley Uttley and Chris Carroll undertook the review of clinical effectiveness.
Paul Tappenden and Matthew Mildred undertook the health economic review and developed the Assessment Group model.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health.
The School of Health and Related Research
The School of Health and Related Research (ScHARR) is one of the 12 departments that constitute the Faculty of Medicine, Dentistry and Health at the University of Sheffield. ScHARR specialises in health services and public health research, and the application of health economics and decision science to the development of health services and the improvement of public health.
The ScHARR Technology Assessment Group (ScHARR-TAG) synthesises research on the clinical effectiveness and cost-effectiveness of health-care interventions for the NIHR Health Technology Assessment Programme on behalf of a range of policy-makers, including NICE. ScHARR-TAG is part of a wider collaboration of a number of units from other regions, including Southampton Health Technology Assessment Centre (SHTAC), University of Southampton; Aberdeen Health Technology Assessment Group (Aberdeen HTA Group), University of Aberdeen; Liverpool Reviews & Implementation Group (LRiG), University of Liverpool; Peninsula Technology Assessment Group (PenTAG), University of Exeter; CRD and Centre for Health Economics (CHE), University of York; Warwick Evidence, University of Warwick; and the BMJ Group and Kleijnen Systematic Reviews.
References
- Davies JC, Alton EW, Bush A. Cystic fibrosis. BMJ 2007;335:1255-9. http://dx.doi.org/10.1136/bmj.39391.713229.AD.
- Emerson J, Rosenfeld M, McNamara S, Ramsey B, Gibson RL. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatr Pulmonol 2002;34:91-100. http://dx.doi.org/10.1002/ppul.10127.
- Antibiotic treatment for cystic fibrosis: report of the UK Cystic Fibrosis Antibiotic Working Group. Kent: Cystic Fibrosis Trust; 2009.
- Yi MS, Tsevat J, Wilmott RW, Kotagal UR, Britto MT. The impact of treatment of pulmonary exacerbations on the health-related quality of life of patients with cystic fibrosis: does hospitalization make a difference?. J Pediatr 2004;144:711-18. http://dx.doi.org/10.1016/j.jpeds.2004.02.032.
- Thornton J, Elliott RA, Tully MP, Dodd M, Webb AK. Clinical and economic choices in the treatment of respiratory infections in cystic fibrosis: comparing hospital and home care. J Cyst Fibros 2005;4:239-47. http://dx.doi.org/10.1016/j.jcf.2005.08.003.
- UK CF Registry annual data report 2010. London: Cystic Fibrosis Trust; 2011.
- Gray MA. CFTR is a mechanosensitive anion channel: a real stretch?. J Cell Sci 2010;7:1-7.
- Gadsby DC, Vergani P, Csanady L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature 2006;440:477-83. http://dx.doi.org/10.1038/nature04712.
- Mogayzel PJ, Flume PA. Update in cystic fibrosis. Am J Respir Crit Care Med 2009;181:539-44. http://dx.doi.org/10.1164/rccm.200912-1943UP.
- Moran A, Becker D, Casella SJ, Gottlieb PA, Kirkman MS, Marshall BC, et al. Epidemiology, pathophysiology, and prognostic implications of cystic fibrosis-related diabetes: a technical review. Diabetes Care 2010;33:2677-83. http://dx.doi.org/10.2337/dc10-1279.
- Taccetti G, Campana S, Festini F, Mascherini M, Doring G. Early eradication therapy against Pseudomonas aeruginosa in cystic fibrosis patients. Eur Respir J 2005;26:458-61. http://dx.doi.org/10.1183/09031936.05.00009605.
- Griese M, Muller I, Reinhardt D. Eradication of initial Pseudomonas aeruginosa colonization in patients with cystic fibrosis. Eur J Med Res 2002;7:79-80.
- Kosorok MR, Zeng L, West SH, Rock MJ, Splaingard ML, Laxova A, et al. Acceleration of lung disease in children with cystic fibrosis after Pseudomonas aeruginosa acquisition. Pediatr Pulmonol 2001;32:277-87. http://dx.doi.org/10.1002/ppul.2009.abs.
- Moreau-Marquis S, Stanton BA, O’Toole GA. Pseudomonas aeruginosa biofilm formation in the cystic fibrosis airway. Pulm Pharmacol Ther 2008;21:595-9.
- Costerton JW, Philip S, Stewart E, Reenberg P. Bacterial biofilms: a common cause of persistent infections. Science 1999;284. http://dx.doi.org/10.1126/science.284.5418.1318.
- Li Z, Kosorok MR, Farrell PM, Laxova A, West SE, Green CG, et al. Longitudinal development of mucoid Pseudomonas aeruginosa infection and lung disease progression in children with cystic fibrosis. JAMA 2005;293:581-8.
- Henry RL, Millis CM, Petrovic L. Mucoid Pseudomonas aeruginosa is a marker of poor survival in cystic fibrosis. Pediatr Pulmonol 1992;12:158-61.
- Committee for Medicinal Products for Human Use (CHMP) . Guideline on the Clinical Development of Medicinal Products for the Treatment of Cystic Fibrosis 2009. www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/12/WC500017055.pdf. (accessed June 2011).
- Wagener J, Rasouliyan L, Pasta DJ, Mabie J, Morgan W, Konstan MW. Practice patterns for treating respiratory exacerbations in cystic fibrosis. Pediatr Pulmonol 2008;43.
- Bilton D, Canny GJ, Conway S, Dumcius S, Hjelte L, Proesmans M, et al. Pulmonary exacerbation: towards a definition for use in clinical trials. Report from the EuroCareCF Working Group on outcome parameters in clinical trials. J Cyst Fibros 2011;10:79-81. http://dx.doi.org/10.1016/S1569-1993(11)60012-X.
- Cystic Fibrosis Registry . UK CF Registry Annual Data Report 2008 2008. www.cftrust.org.uk/aboutcf/publications/cfregistryreports/UK_CF_Registry-Annual_Data_Report_2008.pdf (accessed 1 April 2012).
- Dodge JA, Lewis PA, Stanton M, Wilsher J. Cystic fibrosis mortality and survival in the UK: 1947–2003. Eur Respir J 2007;29:522-6. http://dx.doi.org/10.1183/09031936.00099506.
- Zemanick ET, Harris JK, Conway S, Konstan MW, Marshall B, Quittner AL, et al. Measuring and improving respiratory outcomes in cystic fibrosis lung disease: Opportunities and challenges to therapy. J Cyst Fibros 2010;9:1-16. http://dx.doi.org/10.1016/j.jcf.2009.09.003.
- UK CF Registry Annual Data Report 2009. London: CF Registry; 2011.
- Cystic Fibrosis Trust . Funding of Clinical Care for Cystic Fibrosis Patients Is Revolutionised 2011. www.cftrust.org.uk/pressoffice/pressreleases/PR_PbR.doc (accessed July 2011).
- Henderson AJ. Bronchoalveolar lavage. Arch Dis Child 1994;70:167-9. http://dx.doi.org/10.1136/adc.70.3.167.
- Paris; 2002.
- Miller MR, Hankinson J, Brusasco V, Burgos F, Casabur R, Coates A, et al. Standardisation of spirometry. Eur Respir J 2005;26:319-38. http://dx.doi.org/10.1183/09031936.05.00034805.
- Spirometry in practice. A practical guide to using spirometry in primary care. London: British Thoracic Society; 2005.
- Knudson RJ, Slatin RC, Lebowitz MD, Burrows B. The maximal expiratory flow-volume, curve normal standards, variability and effects of age. Am Rev Respir Dis 1976;113:589-90.
- Hankinson JL, Odencrantz JR, Fedan KB. Spirometric reference values from a sample of the general US population. Am J Respir Crit Care Med 1999;159:179-87.
- Morris JF, Koski A, Johnson LC. Spirometric standards for healthy non-smoking adults. Am Rev Respir Dis 1971;103:57-6.
- Crapo R, Morris AH, Clayton PD, Nixon CR. Lung volumes in healthy non-smoking adults. Bull Europ Physiopathol Respir 1982;18:419-25.
- Cherniak RM, Raber MD. Normal standards for ventilatory function using an automated wedge spirometer. Am Rev Respir Dis 1972;106:38-46.
- Dalzell AM, Shepherd RW, Dean B, Cleghorn GJ, Holt TL, Francis PJ. Nutritional rehabilitation in cystic fibrosis: a 5 year follow-up study. J Pediatr Gastroenterol Nutr 1992;15:141-5. http://dx.doi.org/10.1097/00005176-199208000-00007.
- Konstan MW, Morgan WJ, Butler SM, Pasta DJ, Craib ML, Silva SJ, et al. Risk factors for rate of decline in forced expiratory volume in one second in children and adolescents with cystic fibrosis. J Pediatr 2007;151:134-9. http://dx.doi.org/10.1016/j.jpeds.2007.03.006.
- Wang EE, Prober CG, Manson B, Corey M, Levison H. Association of respiratory viral infections with pulmonary deterioration in patients with cystic fibrosis. N Engl J Med 1984;311:1653-8. http://dx.doi.org/10.1056/NEJM198412273112602.
- Kunzli N, Ackermann-Liebrich U, Keller R, Perruchoud AP, Schindler C. Variability of FVC and FEV1 due to technician, team, device and subject in an eight centre study: three quality control studies in SAPALDIA. Swiss Study on Air Pollution and Lung Disease in Adults. Eur Respir J 1995;8:371-6.
- Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med 2003;168:918-51. http://dx.doi.org/10.1164/rccm.200304-505SO.
- Eichinger M, Heussel CP, Kauczor HU, Tiddens H, Puderbach M. Computed tomography and magnetic resonance imaging in cystic fibrosis lung disease. J Magn Reson Imaging 2010;32:1370-8. http://dx.doi.org/10.1002/jmri.22374.
- Brenner DJ, Hall EJ. Computed Tomography: an increasing source of radiation exposure. N Engl J Med 2007;357:2277-84. http://dx.doi.org/10.1056/NEJMra072149.
- Corey M, Levison H, Crozier D. Five- to seven-year course of pulmonary function in cystic fibrosis. Am Rev Respir Dis 1976;114:1085-92.
- Kerem E, Reisman J, Corey M, Canny GJ, Levison H. Prediction of mortality in patients with cystic fibrosis. N Engl J Med 1992;326:1187-91. http://dx.doi.org/10.1056/NEJM199204303261804.
- Corey M, Edwards L, Levison H, Knowles M. Longitudinal analysis of pulmonary function decline in patients with cystic fibrosis. J Pediatr 1997;131:809-14. http://dx.doi.org/10.1016/S0022-3476(97)70025-8.
- Liou TG, Adler FR, FitzSimmons S, Cahill BC, Hibbs JR, Marshall BC. Predictive 5-year survivorship model of cystic fibrosis. Am J Epidemiol 2001;153:345-52. http://dx.doi.org/10.1093/aje/153.4.345.
- Hamblett N, Rosenfeld M, Emerson J, Goss CH, Aitken ML. Developing cystic fibrosis lung transplant referral criteria using predictors of 2-year mortality. Am J Respir Crit Care Med 2002;166:1550-5.
- Nathanson I, Ramirez-Garnicia G, Wiltrout SA. Decreased attendance at cystic fibrosis centers by children covered by managed care insurance. Res Prac 2005;95:1958-63. http://dx.doi.org/10.2105/AJPH.2004.059089.
- Gee L, Abbott J, Conway SP, Etherington C, Webb AK. Development of a disease specific health related quality of life measure for adults and adolescents with cystic fibrosis. Thorax 2000;55:946-54. http://dx.doi.org/10.1136/thorax.55.11.946.
- Henry B, Aussage P, Grosskopf C, Goehrs JM. Development of the Cystic Fibrosis Questionnaire (CFQ) for assessing quality of life in pediatric and adult patients. Qual Life Res 2003;12:63-76.
- Goss CH, Edwards TC, Ramsey BW, Aitken ML, Patrick DL. Patient reported respiratory symptoms in cystic fibrosis. J Cyst Fibros 2009;8:245-52. http://dx.doi.org/10.1016/j.jcf.2009.04.003.
- Goldbeck L, Schmitz TG, Henrich G, Herschbach P. Questions on life satisfaction for adolescents and adults with cystic fibrosis: development of a disease-specific questionnaire. Chest 2003;123:42-8. http://dx.doi.org/10.1378/chest.123.1.42.
- Quittner AL, Sweeney S, Watrous M, Munzenberger P, Bearss K, Gibson NA, et al. Translation and linguistic validation of a disease-specific quality of life measure for cystic fibrosis. J Pediatr Psychol 2000;25:403-14. http://dx.doi.org/10.1093/jpepsy/25.6.403.
- Modi AC, Quittner AL. Validation of a disease-specific measure of health-related quality of life for children with cystic fibrosis. J Pediatr Psychol 2003;28:535-45. http://dx.doi.org/10.1093/jpepsy/jsg044.
- Jaffé A, Francis J, Rosenthal MBA. Long-term azithromycin may improve lung function in children with cystic fibrosis. Lancet 1998;351. http://dx.doi.org/10.1016/S0140-6736(05)78360-4.
- Cai Y, Chai D, Wang R, Bai N, Liang BB, Liu Y. Effectiveness and safety of macrolides in cystic fibrosis patients: a meta-analysis and systematic review. J Antimicrob Chemother 2011;66:968-78. http://dx.doi.org/10.1093/jac/dkr040.
- Høiby N. Recent advances in the treatment of Pseudomonas aeruginosa infections in cystic fibrosis. BMC Med 2011;9. http://dx.doi.org/10.1186/1741-7015-9-32.
- Wallis C. Mucolytic therapy in cystic fibrosis. J Roy Soc Med 2001;94:17-24.
- Jarrett J. The Economics of Cystic Fibrosis Care in the East of England. 2010.
- Lorian V. Antibiotics in laboratory medicine. London: Williams & Wilkins Press; 1996.
- Colistimethate sodium powder (Colobreathe®) and tobramycin powder for inhalation (TOBI Podhaler®) for the treatment of Pseudomonas lung infection in cystic fibrosis – Novartis submission to NICE. Surrey: Novartis Pharmaceuticals; 2011.
- British National Formulary. London: BMA and RPS; 2011.
- National Institute for Health and Care Excellence (NICE) . Colistimethate Sodium Powder and Tobramycin Powder for Inhalation for the Treatment of Pseudomonas Lung Infection in Cystic Fibrosis – Final Scope. 2010. www.nice.org.uk/nicemedia/live/13068/52778/52778.pdf (accessed June 2011).
- CRD’s guidance for undertaking systematic reviews in health care. York: University of York, CRD; 2009.
- Paiggio G, Elbourne DR, Altman DG, Pocock SJ, Evans SJV. Reporting of non-inferiority and equivalence randomized trials. JAMA 2006;295:1152-60.
- Konstan MW, Flume PA, Kappler M, Chiron R, Higgins M, Brockhaus F, et al. Safety, efficacy and convenience of tobramycin inhalation powder in cystic fibrosis patients: The EAGER trial. J Cyst Fibros 2011;10:54-61. http://dx.doi.org/10.1016/j.jcf.2010.10.003.
- Colistimethate sodium powder for inhalation for the treatment of Pseudomonas lung infection in cystic fibrosis – Forest submission to NICE. Kent: Forest Laboratories UK; 2011.
- Davies JC, Hall P, Francis J, Scott S, Geddes DM, Conway S, et al. A dry powder formulation of colistimethate sodium is safe and well tolerated in adults and children with CF. Cystic Fibrosis Conference 2004.
- Konstan M, Flume PA, Brockhaus F, Angyalosi G, He E, Geller D. Safety and efficacy of tobramycin inhalation powder (TIP) in treating CF patients infected with Pseudomonas aeruginosa (Pa). J Cyst Fibros 2010;9. http://dx.doi.org/10.1016/S1569-1993(10)60083-5.
- Forest Laboratories UK . A Randomised, Open Label Study to Compare the Efficacy and Safety of a Dry Powder Formulation of Inhaled Colistimethate Sodium and Nebulised TNSFI (tobramycin Nebuliser Solution for Inhalation, TOBI®) in Cystic Fibrosis Patients With Pseudomonas Aeruginosa Lung Infection – Clinical Study Report 2009:1-8411.
- A randomised, open label study to compare the efficacy and safety of a dry powder formulation of inhaled colistimethate sodium and nebulised TNSFI (tobramycin nebuliser solution for inhalation, TOBI®) in cystic fibrosis patients with Pseudomonas aeruginosa lung infection. Kent: Forest Laboratories UK; 2011.
- Hodson ME, Gallagher CG, Govan JR. A randomised clinical trial of nebulised tobramycin or colistin in cystic fibrosis. Eur Respir J 2002;20:658-64. http://dx.doi.org/10.1183/09031936.02.00248102.
- Jensen T, Pedersen SS, Garne S, Heilmann C, Hoiby N, Koch C. Colistin inhalation therapy in cystic fibrosis patients with chronic Pseudomonas aeruginosa lung infection. J Antimicrob Chemother 1987;19:831-8. http://dx.doi.org/10.1093/jac/19.6.831.
- Chuchalin A, Csiszer E, Gyurkovics K, Bartnicka MT, Sands D, Kapranov N, et al. A formulation of aerosolized tobramycin (Bramitob) in the treatment of patients with cystic fibrosis and Pseudomonas aeruginosa infection: a double-blind, placebo-controlled, multicenter study. Paediatr Drugs 2007;9:21-3. http://dx.doi.org/10.2165/00148581-200709001-00004.
- Lenoir G, Antypkin YG, Miano A, Moretti P, Zanda M, Varoli G, et al. Efficacy, safety, and local pharmacokinetics of highly concentrated nebulized tobramycin in patients with cystic fibrosis colonized with Pseudomonas aeruginosa. Pediatr Drugs 2007;9:11-20. http://dx.doi.org/10.2165/00148581-200709001-00003.
- Maclusky IB, Gold R, Corey M, Levison H. Long-term effects of inhaled tobramycin in patients with cystic fibrosis colonized with Pseudomonas aeruginosa. Pediatr Pulmonol 1989;7:42-8.
- Ramsey BW, Pepe MS, Quan JM, Otto KL, Montgomery AB, Williams-Warren J, et al. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. Cystic Fibrosis Inhaled Tobramycin Study Group. N Engl J Med 1999;340:23-30.
- Moss RB. Administration of aerosolized antibiotics in cystic fibrosis patients. Chest 2001;120:107-13. http://dx.doi.org/10.1378/chest.120.3_suppl.107S.
- Moss RB. Long-term benefits of inhaled tobramycin in adolescent patients with cystic fibrosis. Chest 2002;121:55-63. http://dx.doi.org/10.1378/chest.121.1.55.
- Nasr SZ, Gordon D, Sakma, E, Yu X, Christodoulou E, Eckhardt BP, et al. High resolution computerized tomography of the chest and pulmonary function testing in evaluating the effect of tobramycin solution for inhalation in cystic fibrosis patients. Pediatr Pulmonol 2006;41:1129-37. http://dx.doi.org/10.1002/ppul.20447.
- Nasr SZ, Sakmar E, Christodoulou E, Eckhardt BP, Streetman DS, Strouse PJ. The use of high resolution computerized tomography (HRCT) of the chest in evaluating the effect of tobramycin solution for inhalation in cystic fibrosis lung disease. Pediatr Pulmonol 2010;45:440-9. http://dx.doi.org/10.1002/ppul.21188.
- Murphy TD, Anbar RD, Lester LA, Nasr SZ, Nickerson B, VanDevanter DR, et al. Treatment with tobramycin solution for inhalation reduces hospitalizations in young CF subjects with mild lung disease. Pediatr Pulmonol 2004;38:314-20. http://dx.doi.org/10.1002/ppul.20097.
- Ramsey BW, Dorkin HL, Eisenberg JD, Gibson RL, Harwood IR, Kravitz RM, et al. Efficacy of aerosolized tobramycin in patients with cystic fibrosis. N Engl J Med 1993;328:1740-6. http://dx.doi.org/10.1056/NEJM199306173282403.
- Konstan MW, Geller DE, Minic P, Brockhaus F, Zhang J, Angyalosi G. Tobramycin inhalation powder for P. aeruginosa infection in cystic fibrosis: the EVOLVE trial. Pediatr Pulmonol 2011;46:230-8. http://dx.doi.org/10.1002/ppul.21356.
- European Medicines Agency (EMA) . Assessment Report Tobi Podhaler 2011. www.ema.europa.eu/docs/en_GB/document_library/EPAR__Public_assessment_report/human/002155/WC500110922.pdf (accessed 11 January 2011).
- British Society for Antimicrobial Chemotherapy (BSAC) . BSAC Methods for Antimicrobial Susceptibility Testing. 2011.
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273:408-12. http://dx.doi.org/10.1001/jama.273.5.408.
- Kjaergard LL, Villumsen J, Gluud C. Reported methodologic quality and discrepancies between large and small randomized trials in meta-analyses. Ann Intern Med 2001;135:982-9. http://dx.doi.org/10.7326/0003-4819-135-11-200112040-00010.
- Juni P, Altman DG, Egger M. Systematic reviews in health care: assessing the quality of controlled clinical trials. BMJ 2001;323:42-6. http://dx.doi.org/10.1136/bmj.323.7303.42.
- ICH Expert Working Group . ICH Harmonised Tripartite Guideline: Clinical Safety Data Management – Definitions and Standards for Expedited Reporting E2A. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use 1994. www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E2A/Step4/E2A_Guideline.pdf (accessed 1 April 2012).
- Drummond MF, Sculpher M, Torrance GW, O’Brien BJ, Stoddart GL. Methods for the economic evaluation of health care programmes. New York, NY: Oxford University Press; 2005.
- Wolter JM, Bowler SD, Nolan PJ, McCormack JG. Home intravenous therapy in cystic fibrosis: a prospective randomized trial examining clinical, quality of life and cost aspects. Eur Respir J 1997;10:896-900.
- Iles R, Legh-Smith J, Drummond M, Prevost A, Vowler S. Economic evaluation of Tobramycin nebuliser solution in cystic fibrosis. J Cyst Fibros 2003;2:120-8. http://dx.doi.org/10.1016/S1569-1993(03)00064-X.
- Guyatt GH, Berman LB, Townsend M, Pugsley SO, Chambers LW. A measure of quality of life for clinical trials in chronic lung disease. Thorax 1987;42:773-8. http://dx.doi.org/10.1136/thx.42.10.773.
- Grieve R, Thompson S, Normand C, Suri R, Bush A, Wallis C. A cost-effectiveness analysis of rhDNase in children with CF. Int J Technol Assess Health Care 2003;19:71-9.
- Guide to the methods of technology appraisal. London: NICE; 2011.
- Eidt-Koch D, Mittendorf T, Greiner W. Cross-sectional validity of the EQ-5D-Y as a generic health outcome instrument in children and adolescents with cystic fibrosis in Germany. BMC Pediatr 2009;9.
- Rowen D, Brazier J. Estimation of EQ-5D-Y Utilities Using CFQ n.d.
- Cystic Fibrosis Foundation 2011. www.cff.org/ (accessed September 2011).
- NHS reference costs 2009–2010. London: DoH; 2011.
- British Medical Association and Royal Pharmaceutical Society of Great Britain . British National Formulary. n.d. http://bnf.org/ (accessed July 2011).
- Wenninger K, Aussage P, Wahn U, Staab D. The revised German Cystic Fibrosis Questionnaire: validation of a disease-specific health-related quality of life instrument. Qual Life Res 2003;12:77-85.
- Wille N, Badia X, Bonsel GJ, Burstrom K, Cavrini G, Devlin N, et al. Development of the EQ-5D-Y: a child-friendly version of the EQ-5D. Qual Life Res 2010;19:875-86. http://dx.doi.org/10.1007/s11136-010-9648-y.
- Rowen D, Longworth L. The Use of Mapping Methods to Estimate Health State Utility Values. 2011. www.nicedsu.org.uk/TSD%2010%20mapping%20FINAL.pdf (accessed September 2011).
- Kind P, Hardman G, Macran S. UK Population Norms for EQ-5D. 2011. www.york.ac.uk/media/che/documents/papers/discussionpapers/CHE%20Discussion%20Paper%20172.pdf (accessed August 2011).
- Taylor R, Elston J. The use of surrogate outcomes in model-based cost-effectiveness analyses: a survey of UK Health Technology Assessment reports. Health Technol Assess 2009;13.
- International Conference on Harmonisation guidelines for the conduct of clinical trials for the registration of drugs . E9: Statistical Principles for Clinical Trials 2012. www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E9/Step4/E9_Guideline.pdf (accessed January 2012).
- Biomarkers Definitions Working Group . Biomarkers and surrogate endpoints: preferred and conceptual framework. Clin Pharmacol Ther 2001;22:485-502.
- Johnson JA, Connolly MA, Zuberbuhler P, Brown NE. Health-related quality of life for adults with cystic fibrosis: a regression approach to assessing the impact of recombinant human DNase. Pharmacotherapy 2000;20:1167-74. http://dx.doi.org/10.1592/phco.20.15.1167.34583.
- Stahl E, Lindberg A, Jansson SA, Ronmark E, Svensson K, Andersson F, et al. Health-related quality of life is related to COPD disease severity. Health Qual Life Outcomes 2005;3.
- Yi MS, Britto MT, Wilmott RW, Kotagul UR, Eckman MH, Nielson DW, et al. Health values of adolescents with cystic fibrosis. J Pediatr 2003;142:133-40. http://dx.doi.org/10.1067/mpd.2003.51.
- Bradley J, Blume S, Stafford M, Balp M, Elborn S. n.d.
- Ketchell RI, Roughton M, Agent P, Gyi K, Hodson ME. Predicting survival in end-stage cystic fibrosis. Respir Med 2009;103:1441-7. http://dx.doi.org/10.1016/j.rmed.2009.04.025.
- Ellaffi M, Vinsonneau C, Coste J, Hubert D, Burgel P, Dhainaut J, et al. One-year outcome after severe pulmonary exacerbation in adults with cystic fibrosis. Am J Respir Crit Care Med 2005;171:158-64. http://dx.doi.org/10.1164/rccm.200405-667OC.
- Simmonds NJ, Macneill SJ, Cullinan P, Hodson ME. Cystic fibrosis and survival to 40 years: a case–control study. Eur Respir J 2010;36:1277-83. http://dx.doi.org/10.1183/09031936.00001710.
- Courtney JM, Bradley J, McCaughan J, O’Connor TM, Shortt C, Bredin CP, et al. Predictors of mortality in adults with cystic fibrosis. Pediatr Pulmonol 2007;2:525-32. http://dx.doi.org/10.1002/ppul.20619.
- Schluchter MD, Konstan MW, Davis PB. Jointly modelling the relationship between survival and pulmonary function in cystic fibrosis patients. Stat Med 2002;21:1271-87. http://dx.doi.org/10.1002/sim.1104.
- Augarten A, Akons H, Aviram M, Bentur L, Blau H, Picard E, et al. Prediction of mortality and timing of referral for lung transplantation in cystic fibrosis patients. Pediatr Transplant 2001;5:339-42. http://dx.doi.org/10.1034/j.1399-3046.2001.00019.x.
- Milla CE, Warwick WJ. Risk of death in cystic fibrosis patients with severely compromised lung function. Chest 1998;113:1230-4. http://dx.doi.org/10.1378/chest.113.5.1230.
- Hayllar KM, Williams SG, Wise AE, Pouria S, Lombard M, Hodson ME, et al. A prognostic model for the prediction of survival in cystic fibrosis. Thorax 1997;52:313-17. http://dx.doi.org/10.1136/thx.52.4.313.
- Aurora P, Wade A, Whitmore P, Whitehead B. A model for predicting life expectancy of children with cystic fibrosis. Eur Respir J 2000;16:1056-60. http://dx.doi.org/10.1034/j.1399-3003.2000.16f06.x.
- Mayer-Hamblett N, Rosenfeld M, Emerson J, Goss C, Aitken ML. Developing cystic fibrosis lung transplant referral criteria using predictors of 2-year mortality. Am J Respir Crit Care Med 2002;166:1550-5. http://dx.doi.org/10.1164/rccm.200202-087OC.
- Belkin RA, Henig NR, Singer LG, Chaparro C, Rubenstein RC, Xie SX, et al. Risk factors for death of patients with cystic fibrosis awaiting lung transplantation. Am J Respir Crit Care Med 2005;173:659-66. http://dx.doi.org/10.1164/rccm.200410-1369OC.
- George PM, Banya W, Pareek N, Bilton D, Cullinan P. Improved survival at low lung function in cystic fibrosis: cohort study from 1990 to 2007. BMJ 2011;342. http://dx.doi.org/10.1136/bmj.d1008.
- Grutters J, Joore M, Wiegman E, Langendijk H, De RD, Hochstenbag M, et al. n.d.
- Polley L, Yaman N, Heaney L, Cardwell C, Murtagh E, Ramsey J, et al. Impact of cough across different chronic respiratory diseases: comparison of two cough-specific health-related quality of life questionnaires. Chest 2008;134:295-302. http://dx.doi.org/10.1378/chest.07-0141.
- Punekar YS, Rodriguez-Roisin R, Sculpher M, Jones P, Spencer M. Implications of chronic obstructive pulmonary disease (COPD) on patients’ health status: a western view. Respir Med 2007;101:661-9. http://dx.doi.org/10.1016/j.rmed.2006.06.001.
- Ko Y, Coons SJ. Self-reported chronic conditions and EQ-5D index scores in the US adult population. Curr Med Res Opin 2006;22:2065-71. http://dx.doi.org/10.1185/030079906X132622.
- Anyanwu AC, McGuire A, Rogers CA, Murday AJ. Assessment of quality of life in lung transplantation using a simple generic tool. Thorax 2001;56:218-22. http://dx.doi.org/10.1136/thorax.56.3.218.
- NHS reference costs 2010–2011. London: DoH; 2012.
- Briggs A, Claxton K, Sculpher M. Decision modelling for health economic evaluation. New York, NY: Oxford University Press; 2006.
- Conway S. A randomised, open label study to compare the efficacy and safety of a dry powder formulation of inhaled colistimethate sodium and nebulised TNSFI (Tobramycin Nebuliser Solution for Inhalation, TOBI) in cystic fibrosis patients with Pseudomonas aeruginosa 2006. www.nihr.ac.uk/Profile/Pages/NRRResults.aspx?publication_id=N0436193944.
- Edenborough FP. Assessing total and regional lung deposition of colistin delivered using a dry powder inhaler and a nebuliser device in adult volunteers with cystic fibrosis 2001. www.nihr.ac.uk/Profile/Pages/NRRResults.aspx?publication_id=N0164087242 (accessed 28 February 2011).
- Geller DE, Konstan MW, Smith J, Noonberg SB, Conrad C. Novel tobramycin inhalation powder in cystic fibrosis subjects: pharmacokinetics and safety. Pediatr Pulmonol 2007;42:307-13. http://dx.doi.org/10.1002/ppul.20594.
- Geller DE, Flume PA, Brockhaus F, Zhang J, Angyalosi G, He E, et al. Treatment convenience and satisfaction of tobramycin inhalation powder (TIP) versus TOBI in cystic fibrosis (CF) patients. J Cyst Fibros 2010;9. http://dx.doi.org/10.1016/S1569-1993(10)60084-7.
- Konstan M. Safety of Tobramycin Inhalation Powder (TIP) Vs Tobramycin Solution for Inhalation in Patients With Cystic Fibrosis 2006. http://clinicaltrials.gov/ct2/show/NCT00388505?term=CTBM100C2302&rank=1 (accessed 28 February 2011).
- Konstan MW, Eller DE, Rockhaus F, Hang J, Ngyalosi G. Tobramycin inhalation powder is effective and safe in the treatment of chronic pulmonary Pseudomonas aeruginosa (Pa) infection in patients with cystic fibrosis. Am J Respir Crit Care Med 2009;179.
- Le Brun PPH, De Boer AH, Mannes GPM, de Fraeture DMI, Brimicombe RW, Touw DJ, et al. Dry powder inhalation of antibiotics in cystic fibrosis therapy: part 2: Inhalation of a novel colistin dry powder formulation: a feasibility study in healthy volunteers and patients. Eur J Pharm Biopharm 2002;54:25-32.
- Newhouse MT, Hirst PH, Duddu SP, Walter YH, Tarara TE, Clark AR, et al. Inhalation of a dry powder tobramycin PulmoSphere formulation in healthy volunteers. Chest 2003;124:360-6. http://dx.doi.org/10.1378/chest.124.1.360.
- Novartis Pharmaceuticals . A Randomized, Double-Blind, Placebo-Controlled, Multi-Center Phase III Study in Cystic Fibrosis (CF) Subjects to Assess Efficacy, Safety and Pharmacokinetics of Tobramycin Inhalation Powder from a Modified Manufacturing Process (TIPnew) 2009. http://clinicaltrials.gov/show/NCT00918957 (accessed 28 February 2011).
- A Study of Tobramycin Inhalation Powder from a Modified Manufacturing Process Versus Placebo (EDIT) 2009. http://clinicaltrials.gov/show/NCT00918957 (accessed 28 February 2011).
- Novartis Pharmaceuticals . A Randomized, Double-Blind, Placebo-Controlled, Multicenter, Phase 3 Trial to Assess the Efficacy and Safety of Tobramycin Inhalation Powder (TIP) in Cystic Fibrosis (CF) Subjects 2005. http://clinicaltrials.gov/show/NCT00125346 (accessed 28 February 2011).
- Novartis Pharmaceuticals . Tobramycin Inhalation Powder (tip) in Cystic Fibrosis Subjects 2005. http://clinicaltrials.gov/show/NCT00125346 (accessed 28 February 2011).
- Westerman EM, Le Brun PP, Touw DJ, Frijlink HW, Heijerman HG. Effect of nebulized colistin sulphate and colistin sulphomethate on lung function in patients with cystic fibrosis: a pilot study. J Cyst Fibros 2004;3:23-8. http://dx.doi.org/10.1016/j.jcf.2003.12.005.
- Westerman EM, De Boer AH, Le Brun PP, Touw DJ, Roldaan AC, Frijlink HW, et al. Dry powder inhalation of colistin in cystic fibrosis patients: a single dose pilot study. J Cyst Fibros 2007;6:284-92. http://dx.doi.org/10.1016/j.jcf.2006.10.010.
- Dias S, Welton NJ, Sutton AJ, Ades AE. Technical Support Document 1: Introduction to evidence synthesis for decision making. University of Sheffield; 2011.
- Alothman GA, Alsaadi MM, Ho BL, Ho SL, Dupuis A, Corey M, et al. Evaluation of bronchial constriction in children with cystic fibrosis after inhaling two different preparations of tobramycin. Chest 2002;122:930-4. http://dx.doi.org/10.1378/chest.122.3.930.
- Beringer PM, Vinks AA, Jelliffe RW, Shapiro BJ. Pharmacokinetics of tobramycin in adults with cystic fibrosis: implications for once-daily administration. Antimicrob Agents Chemother 2000;44:809-13. http://dx.doi.org/10.1128/AAC.44.4.809-813.2000.
- Denk O, Coates AL, Keller M, Leung K, Green M, Chan J, et al. Lung delivery of a new tobramycin nebuliser solution (150 mg/1.5 ml) by an investigational eFlow nebuliser is equivalent to TOBI but in a fraction of time. J Cyst Fibros 2009;8. http://dx.doi.org/10.1016/S1569-1993(09)60261-7.
- Mazurek H. A Study Comparing the Efficacy and Tolerability of Tobrineb® Actitob® Bramitob® Versus TOBI® 2009. http://clinicaltrials.gov/show/NCT00885365 (accessed 28 February 2011).
- Keller M, Coates AL, Griese M, Den, O, Schierholz J, Knoch M. In-vivo data support equivalent therapeutic efficacy of a new tobramycin inhalation solution (150 mg/1.5 ml) administered by the eFlow electronic nebuliser compared to TOBI in the PARI LC PLUS. J Cyst Fibros 2010;9. http://dx.doi.org/10.1016/S1569-1993(10)60085-9.
- Nikolaizik WH, Vietzke D, Ratjen F. A pilot study to compare tobramycin 80 mg injectable preparation with 300 mg solution for inhalation in cystic fibrosis patients. Can Respir J 2008;15:259-62.
- Winnie GB, Cooper JA, Witson J, Cowan RG, Mayer D, Lepow M. Comparison of 6 and 8 hourly tobramycin dosing intervals in treatment of pulmonary exacerbations in cystic fibrosis patients. Pediatr Infect Dis J 1991;10:381-6. http://dx.doi.org/10.1097/00006454-199105000-00007.
- Poli G, Acerbi D, Pennini R, Soliani RA, Corrado ME, Eichler HG, et al. Clinical pharmacology study of Bramitob, a tobramycin solution for nebulization, in comparison with Tobi. Paediatr Drugs 2007;9:3-9. http://dx.doi.org/10.2165/00148581-200709001-00002.
- Riethmueller J, Ballmann M, Schroete, TW, Franke P, von BR, Claass A, et al. Tobramycin once- vs. thrice-daily for elective intravenous antipseudomonal therapy in pediatric cystic fibrosis patients. Infection 2009;37:424-31.
- Rietschel E, Staab D, Vn KS, Merkel N, Heuer H-E, Posselt H-G. Pharmacokinetics of continuous treatment with o.d. or b.i.d. inhalation of tobramycin (TOBI). J Cyst Fibros 2010;9. http://dx.doi.org/10.1016/S1569-1993(10)60086-0.
- Spencer D. Efficacy and Safety of 28 or 56 Days Treatment With Inhaled Tobramycin Nebuliser Solution for Pseudomonas Aeruginosa Infection in Children With Cystic Fibrosis 2006. http://clinicaltrials.gov/show/NCT00391976 (accessed 28 February 2011).
- Westerman EM, Boer AH, Touw DJ, Brun PP, Roldaan AC, Frijlink HW, et al. Aerosolization of tobramycin (TOBI) with the PARI LC PLUS reusable nebulizer: which compressor to use? Comparison of the CR60 to the PortaNeb compressor. J Aerosol Med Pulm Drug Deliv 2008;21:269-80. http://dx.doi.org/10.1089/jamp.2007.0674.
- Whitehead A, Conway SP, Etherington C, Caldwell NA, Setchfield N, Bogle S. Once-daily tobramycin in the treatment of adult patients with cystic fibrosis. Eur Respir J 2002;19:303-9. http://dx.doi.org/10.1183/09031936.02.00221602.
- Wood PJ, Ioannides-Demos LL, Li SC, Williams TJ, Hickey B, Spicer WJ, et al. Minimisation of aminoglycoside toxicity in patients with cystic fibrosis. Thorax 1996;51:369-73. http://dx.doi.org/10.1136/thx.51.4.369.
- Adeboyeku D, Scott S, Hodson ME. Open follow-up study of tobramycin nebuliser solution and colistin in patients with cystic fibrosis. J Cyst Fibros 2006;5:261-3. http://dx.doi.org/10.1016/j.jcf.2006.05.009.
- Webb AK. An Open-Label, Randomised Clinical Trial of the Efficacy and Safety of Tobramycin Solution for Inhalation and Aerosolised Colistin Sulphomethate Sodium in Patients With Cystic Fibrosis 1999. www.nihr.ac.uk/Profile/Pages/NRRResults.aspx?publication_id=N0226050348 (accessed 28 February 2011).
- Weller P. An Open Label, Randomised Clinical Trial of the Efficacy and Safety of Tobramycin Solution for Inhalation or Aerosolised Colistin Sulphomethate Sodium (Colomycin) in Patients With Cystic Fibrosis (CF). 1999. www.nihr.ac.uk/Profile/Pages/NRRResults.aspx?publication_id=N0045026034 (accessed 28 February 2011).
- Montgomery AB. Phase III Randomized Study of the Inhalation of Tobramycin in Patients With Cystic Fibrosis 2000. http://clinicaltrials.gov/show/NCT00004829 (accessed 28 February 2011).
- Wientzen R, Prestidge CB, Kramer RI. Acute pulmonary exacerbations in cystic fibrosis. A double-blind trial of tobramycin and placebo therapy. Am J Dis Child 1980;134:1134-8. http://dx.doi.org/10.1001/archpedi.1980.02130240018007.
- Wiesemann HG, Steinkamp G, Ratjen F, Bauernfeind A, Przyklenk B, Doring G, et al. Placebo-controlled, double-blind, randomized study of aerosolized tobramycin for early treatment of Pseudomonas aeruginosa colonization in cystic fibrosis. Pediatr Pulmonol 1998;25:88-92. http://dx.doi.org/10.1002/(SICI)1099-0496(199802)25:2<88::AID-PPUL3>3.3.CO;2-9.
- Al Ansari NA, Foweraker J, Mackeown D, Bilton D. Evaluation of once daily tobramycin versus the traditional three time daily for the treatment of acute pulmonary exacerbations in adult cystic fibrosis patients. Qatar Med J 2006;15:34-8.
- Ramsey BW, Retsch-Bogart G, Treggiari M. Comparison of Two Treatment Regimens to Reduce PA Infection in Children With Cystic Fibrosis 2004. http://clinicaltrials.gov/show/NCT00097773 (accessed 28 February 2011).
- Master V, Roberts GW, Coulthard KP, Baghurst PA, Martin A, Roberts ME, et al. Efficacy of once-daily tobramycin monotherapy for acute pulmonary exacerbations of cystic fibrosis: a preliminary study. Pediatr Pulmonol 2001;31:367-76. http://dx.doi.org/10.1002/ppul.1060.
- Flume P. Trial of Aeroquin Versus Tobramycin Inhalation Solution (TIS) in Cystic Fibrosis (CF) Patients 2011. http://clinicaltrials.gov/show/NCT01270347 (accessed 28 February 2011).
- Kassaa B. Pharmacokinetic Bioequivalence Study of Nebcinal® 150 mg 3 ml Administered by Aeroneb® Idehaler® Versus Tobi® 300 mg 5 ml Administered by Pari LC Plus®. 2011. http://clinicaltrials.gov/show/NCT01288170 (accessed 28 February 2011).
- Smyth A, Tan KH, Hyman-Taylor P, Mulheran M, Lewis S, Stableforth D, et al. Once versus three-times daily regimens of tobramycin treatment for pulmonary exacerbations of cystic fibrosis – the TOPIC study: a randomised controlled trial. Lancet 2005;365:573-8. http://dx.doi.org/10.1016/S0140-6736(05)17906-9.
- Canis F, Trivier D, Vic P, Ategbo S, Turck D, Husson MO. Comparison of the pharmacokinetics of tobramycin administered in a single daily dose or in 3 doses in cystic fibrosis. Pathol Biol 1998;46:449-51.
- Trapnell BC, Rolfe M, McColley S, Montgomery AB, Moorehead L, Geller D. Fosfomycin/tobramycin for inhalation (FTI): efficacy results of a phase 2 placebo controlled trial in patients with cystic fibrosis and Pseudomonas aeruginosa. Pediatr Pulmonol 2010;45.
- Conway SP, Miller MG, Ramsden C, Littlewood JM. Intensive treatment of Pseudomonas chest infection in cystic fibrosis: a comparison of tobramycin and ticarcillin, and netilmicin and ticarcillin. Acta Paediatr Scand 1985;74:107-13. http://dx.doi.org/10.1111/j.1651-2227.1985.tb10929.x.
- De Boeck K, Smet M, Eggermont E. Treatment of Pseudomonas lung infection in cystic fibrosis with piperacillin plus tobramycin versus ceftazidime monotherapy: preliminary communication. Pediatr Pulmonol 1989;7:171-3.
- Martin AJ, Smalley CA, George RH, Gealing DE, Anderson CM. Gentamicin and tobramycin compared in the treatment of mucoid Pseudomonas lung infections in cystic fibrosis. Arch Dis Child 1980;55:604-7. http://dx.doi.org/10.1136/adc.55.8.604.
- McLaughlin FJ, Matthews WJ, Strieder DJ, Sullivan B, Taneja A, Murphy P, et al. Clinical and bacteriological responses to three antibiotic regimens for acute exacerbations of cystic fibrosis ticarcillin tobramycin azlocillin tobramycin and azlocillin placebo. J Infect Dis 1983;147:559-67. http://dx.doi.org/10.1093/infdis/147.3.559.
- Parry MF, Neu HC. A comparative study of ticarcillin plus tobramycin versus carbenicillin plus gentamicin for the treatment of serious infections due to Gram-negative bacilli. Am J Med 1978;64:961-6. http://dx.doi.org/10.1016/0002-9343(78)90450-3.
- Pedersen SS, Pressler T, Pedersen M. Immediate and prolonged clinical efficacy of ceftazidime versus ceftazidime plus tobramycin in chronic Pseudomonas aeruginosa infection in cystic fibrosis. Scand J Infect Dis 1986;18:133-7. http://dx.doi.org/10.3109/00365548609032319.
- Wesley AW, Quested C, Edgar BW, Lennon DR. n.d.
- Taccetti G, Bianchini E, Zavataro L, Campana S, Defilippi G, Ravenni N, et al. Pseudomonas aeruginosa eradication in cystic fibrosis: preliminary data from a randomized multicenter study of two different early antibiotic treatment protocols. Pediatr Pulmonol 2010;45:337-8.
- Zavataro L, Taccetti G, Cariani L, Ravenni N, Braccini G, Bresci S, et al. Epidemiology of first/new Pseudomonas aeruginosa infection in cystic fibrosis patients. J Cyst Fibros 2010;9. http://dx.doi.org/10.1016/S1569-1993(10)60111-7.
- Gilead Sciences . Aztreonam for Inhalation Solution (AZLI) Vs. Tobramycin Inhalation Solution (TOBI®) in Patients With Cystic Fibrosis &Amp; Pseudomonas Aeruginosa 2008. http://clinicaltrials.gov/show/NCT00757237 (accessed 28 February 2011).
- Oermann CM, Assael B, Nakamura C, Boas SR, Bresnik M, Montgomery AB, et al. Aztreonam for inhalation solution (AZLI) vs. tobramycin inhalation solution (TIS), a 6-month comparative trial in cystic fibrosis patients with Pseudomonas aeruginosa. Pediatr Pulmonol 2010;45.
- Gibson RL, Retsch-Bogart GZ, Oermann C, Milla C, Pilewski J, Daines C, et al. Microbiology, safety, and pharmacokinetics of aztreonam lysinate for inhalation in patients with cystic fibrosis. Pediatr Pulmonol 2006;41:656-65. http://dx.doi.org/10.1002/ppul.20429.
- Burns JL, Stapp J, Lofland D, Phase AI. Microbiology results from a phase 2 clinical study of aztreonam lysinate for inhalation (AI): a new inhaled antibiotic to treat CF patients with Pseudomonas aeruginosa (PA). J Cyst Fibros 2005;4.
- McCoy K, Retsch-Bogart G, Gibson RL, Oermann C, Braff M, Montgomery AB. Investigation of susceptibility breakpoints for inhaled antibiotic therapies in cystic fibrosis. Pediatr Pulmonol 2010;341.
- Retsch-Bogart GZ, Burns JL, Otto KL, Liou TG, McCoy K, Oermann C, et al. A phase 2 study of aztreonam lysine for inhalation to treat patients with cystic fibrosis and Pseudomonas aeruginosa infection. Pediatr Pulmonol 2008;43:47-58.
- Retsch-Bogart GZ, Quittner AL, Gibson RL, Oermann CM, McCoy KS, Montgomery AB, et al. Efficacy and safety of inhaled aztreonam lysine for airway Pseudomonas in cystic fibrosis. Chest 2009;135:1223-32. http://dx.doi.org/10.1378/chest.08-1421.
- Wainwright C, Nakamura C, Geller D, Montgomery AB. A double-blind, multinational, randomized, placebo-controlled trial evaluating aztreonam for inhalation solution (AZLI) in patients with cystic fibrosis (CF), mild lung disease and P. aeruginosa (PA). J Cyst Fibros 2010;9. http://dx.doi.org/10.1016/S1569-1993(10)60082-3.
- Salh B, Bilton D, Dodd M, Abbot J, Webb K. A comparison of aztreonam and ceftazidime in the treatment of respiratory infections in adults with cystic fibrosis. Scand J Infect Dis 1992;24:215-18. http://dx.doi.org/10.3109/00365549209052615.
- Schaad UB, Wedgwood-Krucko J, Guenin K, Buehlmann U, Kraemer R. Antipseudomonal therapy in cystic fibrosis: aztreonam and amikacin versus ceftazidime and amikacin administered intravenously followed by oral ciprofloxacin. Eur J Clin Microbiol Infect Dis 1989;8:858-65. http://dx.doi.org/10.1007/BF01963771.
- Bosso JA, Black PG. Controlled trial of aztreonam vs. tobramycin and azlocillin for acute pulmonary exacerbations of cystic fibrosis. Pediatr Infect Dis J 1988;7:171-6. http://dx.doi.org/10.1097/00006454-198803000-00008.
- McCoy K, Quittner AL, Oermann C, Gibson RL, Retsch-Bogart GZ, Montgomery AB, et al. Inhaled Aztreonam Lysine for Chronic Airway Pseudomonas aeruginosa in Cystic Fibrosis. Am J Respir Crit Care Med 2008;178:921-8.
- Signorovitch J, Latremouille-Viau D, Dea K, Wu E, Zhang J. Comparison of drug costs and clinical outcomes with tobramycin solution for inhalation and aztreonam lysine for inhalation in cystic fibrosis. Pediatr Pulmonol 2010;309.
- Blumer JL, Minkwitz M, Saiman L, San GP, Iaconis J, Melnick D. Meropenem (MEM) compared with ceftazidime (CAZ) in combination with tobramycin (TOB) for treatment of acute pulmonary exacerbations (APE) in patients with cystic fibrosis (CF) infected with Pseudomonas aeruginosa (PA) or Burkholderia cepacia (BC). Pediatr Pulmonol 2003.
- Richard DA, Nousia-Arvanitakis S, Sollich V, Hampel BJ, Sommerauer B, Schaad UB. Oral ciprofloxacin vs. intravenous ceftazidime plus tobramycin in pediatric cystic fibrosis patients: comparison of anti-Pseudomonas efficacy and assessment of safety with ultrasonography and magnetic resonance imaging. Cystic Fibrosis Study Group. Pediatr Infect Dis J 1997;16:572-8.
- Church D, Kanga J, Kuhn R, Rubio T, Haverstock D, Perroncel R, et al. Prospective, double-blind, randomized comparison of ciprofloxacin (CIP) vs ceftazidime/tobramycin (C/T) in the treatment of pediatric cystic fibrosis (CF). Abstr Intersci Conf Antimicrob Agents Chemother 1995;35.
- McCarty JM, Tilden SJ, Black P, Craft JC, Blumer J, Waring W, et al. Comparison of piperacillin alone versus piperacillin plus tobramycin for treatment of respiratory infections in children with cystic fibrosis. Pediatr Pulmonol 1988;4:201-4. http://dx.doi.org/10.1002/ppul.1950040403.
- Hoiby N. Supplementary Oral Azithromycin in Treatment of Intermittent Pseudomonas Aeruginosa Colonization in CF-Patients With Inhaled Colistin and Oral Ciprofloxacin; Postponing Next Isolate of Pseudomonas and Prevention of Chronic Infection. A Prospective, Double-Blinded, Placebo-Controlled Scandinavian Multi-Centre Study 2006. http://clinicaltrials.gov/show/NCT00411736 (accessed 28 February 2011).
Appendix 1 Treatment bands for cystic fibrosis (from NHS specialised services)
Treatment bands
Band 1
Patients who receive only outpatient care from doctors, nurses, physiotherapist, dieticians, social workers, etc. No intravenous antibiotics required. No inpatient admissions apart from an annual assessment and review as a day case.
Band 1a
Previously as above BUT require up to 14 days of intravenous antibiotics (at home or in hospital) and spend a maximum of 7 days in hospital over the course of a 12-month period or receive short-term (up to 3 months) nebulised antibiotics for eradication treatment.
Band 2
Patients who require maintenance nebulised antibiotics for Pseudomonas infection or maintenance nebulised dornase alfa. Patients receive up to 28 days of intravenous antibiotics in a year or spend a maximum of 14 days in hospital.
Band 2a
Patients who receive both nebulised antibiotics and dornase alfa and require up to 56 days of antibiotics intravenously at home or in hospital or a maximum of 14 days in hospital.
Band 3
Patients who have more frequent inpatient visits, have up to a maximum of 84 days on intravenous antibiotics (at home or in hospital) or spend up to 57 days in hospital or patients with gastrostomy feeding or any of listed CF complications namely CF-related diabetes, allergic bronchopulmonary aspergillosis (ABPA), massive haemoptysis, pneumothorax.
Band 4
Patients who have severe disease and usually spend up to 112 days in hospital per year, although it is recognised that some patients, at this stage of their illness, prefer to be treated/supported at home with the support of the CF multidisciplinary team. Patients require a minimum of 85 days per year on i.v. antibiotics (at home or in hospital). Patients have CF-related complications of diabetes, pneumothorax or haemoptysis.
Band 5
Patients are severely ill and stay in hospital for greater than 113 days per year, awaiting transplantation or receiving palliative care. As above, it is recognised that some patients, at this stage of their illness, prefer to be treated/supported at home with the support of the CF multidisciplinary team. Patients may be receiving nocturnal ventilation and feeding gastrostomies. Patient’s life expectancy is usually no more than 1 year to 18 months.
Appendix 2 MEDLINE search strategy for clinical effectiveness and cost-effectiveness evidence
-
Cystic Fibrosis/ (24,739)
-
cystic fibrosis.tw. (26,433)
-
fibrosis cystic.tw. (46)
-
1 or 2 or 3 (31,197)
-
Pseudomonas aeruginosa/ (27,224)
-
Pseudomonas Infections/ (14,449)
-
pseudomonas aeruginosa.tw. (31,883)
-
pseudomonas infection$.tw. (728)
-
“P. aeruginosa”.tw. (12,458)
-
10 Respiratory Tract Infections/ (27,457)
-
respiratory tract infection$.tw. (11,475)
-
infection$ respiratory tract.tw. (57)
-
5 or 6 or 7 or 8 or 9 or 10 or 11 or 12 (76,944)
-
4 and 13 (4642)
-
Colistin/ (1843)
-
colistin.tw. (1557)
-
colistimethate sodium.tw. (16)
-
colobreathe.tw. (0)
-
turbospin device.tw. (1)
-
turbospin.tw. (9)
-
pentasodium colistimethanesulfate.tw. (0)
-
1066-17-7.rn. (1843)
-
12705-41-8.rn. (32)
-
polymyxin.tw. (4460)
-
promixin.tw. (0)
-
coly-mycin.tw. (10)
-
colisticin.tw. (0)
-
colimycin.tw. (216)
-
colomycin.tw. (14)
-
colymycin.tw. (12)
-
totazina.tw. (0)
-
or/15-31 (6866)
-
Tobramycin/ (3418)
-
tobramycin.tw. (4880)
-
tip.tw. (33,364)
-
tobi podhaler.tw. (0)
-
podhaler.tw. (0)
-
32986-56-4.rn. (3418)
-
nebicin.tw. (1)
-
nebcin.tw. (7)
-
nebramycin factor 6.tw. (8)
-
brulamycin.tw. (13)
-
obracin.tw. (2)
-
bramitob.tw. (5)
-
tobi.tw. (74)
-
or/33-45 (39,018)
-
Amikacin/ (3191)
-
amikacin.tw. (5581)
-
Gentamicins/ (15,261)
-
gentamicin$.tw. (16,822)
-
Ceftazidime/ (2877)
-
ceftazidime.tw. (5581)
-
Aztreonam/ (1199)
-
aztreonam.tw. (2113)
-
exp Aminoglycosides/ (113,459)
-
aminoglycoside$.tw. (12,835)
-
exp Cephalosporins/ (33,683)
-
cephalosporin$.tw. (14,765)
-
exp Fluoroquinolones/ (21,184)
-
fluoroquinolone$.tw. (8258)
-
or/47-60 (179,078)
-
exp “Nebulizers and Vaporizers”/ (7091)
-
(nebulis$ or nebuliz$).tw. (6454)
-
exp Administration, Inhalation/ (20,759)
-
inhal$.tw. (68,855)
-
exp Aerosols/ (22,745)
-
aerosol$.tw. (24,353)
-
eFlow.tw. (35)
-
eflow.tw. (22)
-
or/62-69 (100,254)
-
61 and 70 (957)
-
32 or 46 or 71 (46,179)
-
14 and 72 (557)
-
randomized controlled trial.pt. (299,024)
-
controlled clinical trial.pt. (81,706)
-
randomized controlled trials/ (70,561)
-
random allocation/ (70,117)
-
double blind method/ (108,074)
-
single blind method/ (14,529)
-
clinical trial.pt. (459,075)
-
exp Clinical Trial/ (625,172)
-
(clin$ adj25 trial$).ti,ab. (182,356)
-
((singl$ or doubl$ or trebl$ or tripl$) adj25 (blind$ or mask$)).ti,ab. (108,653)
-
placebos/ (29,247)
-
placebos.ti,ab. (1512)
-
random.ti,ab. (119,723)
-
research design/ (61,104)
-
or/74-87 (1,008,372)
-
Meta-Analysis/ (26,827)
-
meta analy$.tw. (31,188)
-
metaanaly$.tw. (962)
-
meta analysis.pt. (26,827)
-
(systematic adj (review$1 or overview$1)).tw. (24,154)
-
exp Review Literature/ (1,576,254)
-
or/89-94 (1,602,463)
-
cochrane.ab. (15,057)
-
embase.ab. (12,552)
-
(psychlit or psyclit).ab. (794)
-
(psychinfo or psycinfo).ab. (4051)
-
(cinahl or cinhal).ab. (4919)
-
science citation index.ab. (1201)
-
bids.ab. (284)
-
cancerlit.ab. (480)
-
or/96-103 (23,561)
-
reference list$.ab. (5697)
-
bibliograph$.ab. (8544)
-
hand-search$.ab. (2488)
-
relevant journals.ab. (425)
-
manual search$.ab. (1419)
-
or/105-109 (16,658)
-
selection criteria.ab. (13,294)
-
data extraction.ab. (6142)
-
111 or 112 (18,395)
-
review.pt. (1,574,024)
-
113 and 114 (12,590)
-
comment.pt. (428,431)
-
letter.pt. (699,325)
-
editorial.pt. (268,459)
-
animal/ (4,660,797)
-
human/ (11,509,301)
-
119 not (119 and 120) (3,452,597)
-
or/116-118,121 (4,452,285)
-
95 or 104 or 110 or 115 (1,608,257)
-
123 not 122 (1,462,773)
-
Economics/ (25,932)
-
“costs and cost analysis”/ (38,407)
-
Cost allocation/ (1884)
-
Cost-benefit analysis/ (49,718)
-
Cost control/ (18,516)
-
cost savings/ (6869)
-
Cost of illness/ (13,523)
-
Cost sharing/ (1626)
-
“deductibles and coinsurance”/ (1266)
-
Health care costs/ (20,501)
-
Direct service costs/ (921)
-
Drug costs/ (10,095)
-
Employer health costs/ (1025)
-
Hospital costs/ (6290)
-
Health expenditures/ (11,326)
-
Capital expenditures/ (1887)
-
Value of life/ (5118)
-
exp economics, hospital/ (16,929)
-
exp economics, medical/ (13,069)
-
Economics, nursing/ (3833)
-
Economics, pharmaceutical/ (2189)
-
exp “fees and charges”/ (24,941)
-
exp budgets/ (10,783)
-
(low adj cost).mp. (14,051)
-
(high adj cost).mp. (5970)
-
(health?care adj cost$).mp. (2434)
-
(fiscal or funding or financial or finance).tw. (57,634)
-
(cost adj estimate$).mp. (1049)
-
(cost adj variable).mp. (26)
-
(unit adj cost$).mp. (1107)
-
(economic$ or pharmacoeconomic$ or price$ or pricing).tw. (124,497)
-
or/125-155 (364,158)
-
73 and 88 (166)
-
73 and 124 (89)
-
73 and 156 (12)
Appendix 3 Table of excluded studies
| Author | Year | Reason for exclusion |
|---|---|---|
| Conway131 | 2006 | Trial record only |
| Edenborough132 | 2001 | Trial record only |
| Geller et al.133 | 2007 | Single-dose study |
| Geller et al.134 | 2010 | Satisfaction study |
| Konstan135 | 2006 | Trial record |
| Konstan et al.136 | 2009 | Placebo control |
| Konstan et al.83 | 2011 | Placebo control |
| Le Brun et al.137 | 2002 | Not patients with CF |
| Newhouse et al.138 | 2003 | Healthy population, non-CF |
| Novartis Pharmaceuticals139 | 2009 | Trial record only |
| Novartis Pharmaceuticals140 | 2009 | Trial record only |
| Novartis Pharmaceuticals141 | 2005 | Trial record only |
| Novartis Pharmaceuticals142 | 2005 | Trial record only |
| Westerman et al.143 | 2004 | Nebulised Colistin |
| Westerman et al.144 | 2007 | Single-dose study |
Appendix 4 Evidence network considered for meta-analysis
The purpose of a NMA is to allow comparison of the interventions defined in the scope (tobramycin DPI and colistimethate sodium DPI) with the comparators defined in the scope (antibiotics for nebulisation, defined as tobramycin and colistimethate sodium). These interventions will be referred to collectively as the decision comparator set. 145 Where there is not direct RCT evidence between these, a network can be constructed by introducing an additional treatment or treatments. The decision comparator set plus these additional treatments is known as the synthesis set. The synthesis set is usually not extended beyond studies that include at least one of the treatments in the comparator set,145 and for a study to be included in the review it must include two of the treatments in the synthesis set.
For a network to be possible, a good degree of homogeneity between study characteristics within the synthesis set is needed, or, where heterogeneity exists ,this should be appropriately corrected for (modelled). In the context of this assessment, there are important prognostic factors that are likely to affect estimates of FEV1%. The most significant of these are:
-
the mean age of participants (prognosis worsens with increasing age)
-
the mean FEV1% at baseline (prognosis worsens with decreasing FEV1%)
-
the mean BMI at baseline (prognosis worsens with lower BMI for age).
These factors are associated with life expectancy, and may be associated with treatment efficacy, for example patients with advanced lung damage (low FEV1%) gain less benefit from inhaled antibiotics as bacterial plaques impede the dispersal of treatment throughout the lung. Without knowing the distribution of these factors at a patient level, correcting for them within the analysis would introduce an unacceptable level of uncertainty.
The 20 citations considered for inclusion in a NMA are listed in Table 49. Most citations related to studies of nebulised tobramycin. No head-to-head studies of tobramcyin or colistimethate sodium dry powders compared with each other were identified.
A network was formed from these studies, starting with studies that included colistimethate sodium DPI or tobramycin DPI. Constructing the network involved two stages:
-
identification of which studies used compatible interventions to those already in the network (potentially includable in the synthesis set)
-
data extraction (from the abstract or full text if necessary) of key variables for the studies within the synthesis set.
Figures 30 and 31 of this appendix show the data available at 4 weeks after the commencement of treatment and 20 and 24 weeks after commencement of treatment. These figures also indicate where the network was judged unviable owing to heterogeneity in study variables. The data extraction of potentially includable studies is presented in Table 50 of this appendix.
At 4 weeks (Figure 38), a network could potentially be constructed which included colistimethate sodium DPI to tobramycin DPI via nebulised tobramycin. This network would depend on the published data being compatible in terms of statistical analyses performed (e.g. some data are presented as logarithmic transforms). This analysis was not performed for the following reasons, which became evident during the course of the assessment:
-
Data at 4 weeks are of little use to assess long-term outcomes.
-
There is evidence to suggest that FEV1% measured at 4 weeks in groups treated with tobramycin (DPI or nebulised) may be unrepresentative of true long-term efficacy, as FEV1% usually peaks within the first few days of treatment and may not have levelled at 4 weeks. This would unfairly advantage tobramycin DPI and nebulised tobramycin, and disadvantage colistimethate sodium DPI.
The month-on, month-off dosing of tobramycin leads to peaks and troughs in efficacy (see Chapter 3, Assessment of effectiveness). For this reason, it would see appropriate to look at outcomes at both 20 and 24 weeks, to allow for a best-case and worst-case scenario assessment of efficacy. Data were provided by Novartis Pharmaceuticals at 20 weeks in the initial submission,60 and data at 24 weeks were provided on request (Novartis Pharmaceuticals’ clarifications). The network of evidence at 20 and 24 weeks is presented in Figure 39. Data are available at both 20 and 24 weeks for colistimethate sodium DPI and tobramycin DPI, compared with nebulised tobramycin. However, no data are available for nebulised colistimethate.
Despite there being trial data with common comparators (two trials comparing with nebulised tobramycin at 20/24 weeks, two trials comparing with nebulised tobramycin, and two trials using colistimethate sodium DPI at 4 weeks), an indirect comparison was not performed for the following reasons:
-
EAGER trial data65 were presented only with no imputation. An equivalent set of data was not presented for COLO/DPI/02/06. 66
-
There is a lack of certainty around the comparability of the patient populations, methods for recording FEV1%, and definitions of acute exacerbations (see Chapter 3).
-
There are gaps in the data, especially at 24 weeks.
-
The small number of studies may make an indirect comparison prone to being influenced by the priors and therefore potentially uninformative.
| Comparator | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Tobramycin DPI | Colistimethate DPI | Nebulised tobramycin | Nebulised colistimethate sodium | Placebo | Tobramycin in combination | Colistimethate sodium in combination | Aztreonam | Aztreonam in combination | Other | ||
| Intervention | Tobramycin DPI | Geller et al. 2007133 (90) Konstan et al. 201165 (553) Konstan et al. 2009 abstract136 Trial record of Konstan 2006135 Novartis Pharmaceuticals’ submission 201160 Konstan et al. 2010 abstract68 |
Novartis Pharmaceuticals’ trial records139,140 (NR) Novartis Pharmaceuticals’ trial records141,142 (NR) Novartis Pharmaceuticals’ submission60 Konstan et al. 201183 |
||||||||
| Colistimethate sodium DPI | Conway 2006 et al.131 (360) Forest Laboratories’ submission 201166 |
Westerman et al. 2007144 (10) Davies et al. 200467 (12) |
|||||||||
| Tobramycin nebulised | Alothman et al. 2002146 (19)a Beringer et al. 2000147 (60)a Denk et al. 2009148 (16)b Mazurek 2009149 (NR)b Keller et al. 2010150 (92)b Nikolaizik et al. 2008151 (32)a Winnie et al. 1991152 (NR)a Poli et al. 2007153 (11)b Riethmueller et al. 2010154 (30) Rietschel et al. 2010155 (29)a Spencer 2006156 (EEN = 121)a Westerman et al. 2008157 (10)b Whitehead et al. 2002158 (60)a Wood et al. 1996159 (29)a |
Hodson 200271 (NR) Adeboyeku et al. 2006160 (21) Unpublished study: (Taylor 1998c) (EEN = 3–5) Webb 1999161 (NR) Weller 1999162 (115) |
Chuchalin et al. 200773 (247) Poli et al. 2007153 (396) Montgomery 2000163 (EEN = 200) Moss et al. 200177 (520) Nasr et al. 201080 (32) Ramsey et al. 199382 (71) Ramsey et al. 199976 (520) Wientzen et al. 1980164 (22) Wiesemann et al. 1998165 (22) Moss 200278 (128) Lenoir et al. 200774 Maclusky et al. 198975 |
Al Ansari et al. 2006166 (15)a Ramsey et al. 2004167 (NR)a |
Master et al. 2001168 (98)a | Flume et al. 2011169 TS vs. Aeroquin (NR) Kassaa 2011170 TS vs. Nebcinal (NR) Murphy et al. 200481 TS vs. routine treatment (184) (unclear if chronic P. aeruginosa) |
|||||
| Nebulised colistimethate sodium | Westerman et al. 2004143 (9) | Jensen et al. 198772 | |||||||||
| Tobramycin in combination | Topic Study Group 2005171 (244)a Canis et al. 1998172 (20)a Trapnell et al. 2010173 (135) |
Conway et al. 1985174 (17) De Boeck et al. 1989175 (21) Martin et al. 1980176 (18) McLaughlin et al. 1983177 (NR) Parry and Neu 1978178 (82) Pederson et al. 1986179 (20)c Wesley et al. 1999180 (13) |
|||||||||
| Colistimethate in combination | Taccetti et al. 2010181 (215) Zavatoro et al. 2010182 (198) |
||||||||||
| Aztreonam | Gilead Sciences 2008183 (240) Oermann et al. 2010184 (273) |
Gibson et al. 2006185 (12) Burns et al. 2005186 (105) McCoy et al. 2010187 (NR) Retsch-Bogart et al. 2008188 (131) Retsch-Bogart et al. 2009189 (164) Wainwright et al. 2010190 (157) |
Salh et al. 1992,191 aztreonam vs. ceftazidime Schaad et al. 1989,192 i.v. aztreonam vs. ceftazidime and amikacin (42) Bosso and Black 1988,193 aztreonam vs. tobramycin and azlocillin (15) McCoy et al. 2008194 AS vs. TS vs. placebo (211) |
||||||||
| Aztreonam in combination | Signorovitch et al. 2010195 TS vs. placebo vs. AS vs. placebo (692) | ||||||||||
| Other | Blumer et al. 2003196 (NR) Richard et al. 1997197 (108) Church et al. 1995198 (NR) McCarty et al. 1988199 (17) |
Hoiby 2006200 (NR) | |||||||||
| Study | Trial design | Age range (years) | Mean age (years) | N (ITT) | Baseline FEV1%: mean (SD) | Chronic P. aeruginosa? | Time to outcome | Outcomes reported | Dose dry powder | Dose nebulised solution | Placebo | Exclude reason |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tobramycin, dry powder | ||||||||||||
| Konstan et al. 201183 (EVOLVE) | RCT | 6–21 | Intervention: 13.4 (4.42) Control: 13.2 (3.91) |
517 | Intervention: 54.7 (18.89) Control: 58.5 (20.03) |
Yes | 4, 8 weeks | FEV1%; SpD; i.v. antibiotics; hospitalisations; resistance; AEs; AcEx | 112 mg b.i.d. T-326 inhaler |
NA | Inhaler with four capsules b.i.d. | Placebo control |
| Konstan et al. 201165 (EAGER) | RCT, open label | ≥ 6 | Intervention: 26 (11.4) Control: 25 (10.2) |
373 | Intervention: 53 (14.2) Control: 53 (15.9) |
Yes | 4, 24 weeks | SpD; FEV1%; AcEx; hospitalisations; AEs; HRQoL; compliance | 112 mg b.i.d. T-326 inhaler |
300 mg/ml b.i.d., PARI LC plus | NA | NA |
| Colistimethate sodium, dry powder | ||||||||||||
| COLO/DPI/02/06 Forest Laboratories’ submission66 |
RCT open label | ≥ 6 | Intervention: 21.3 (9.72) Control: 20.9 (9.30) |
396 | Intervention: 51.76 (1.02) Control: 50.82 (0.98) |
Yes | 20, 24 weeks | FEV1%; AcEx; hospitalisations; AEs; HRQoL; resistance; compliance | 125 mg b.i.d. Turbospin |
300 mg/ml Tobramycin b.i.d. | NA | NA |
| COLO/DPI/02/05 Davies et al. 200467 Forest Laboratories’ submission66 |
RCT with crossover | ≥ 8 | 20.3 | 16 | Intervention: 77.14 (6.78) Control: 76.25 (7.32) |
Yes | 4 weeks | FEV1%; AEs | 125 mg b.i.d. Turbospin |
2 MIU Colistimethate sodium b.i.d. | NA | NA |
| Nebulised colistin | ||||||||||||
| Hodson et al. 200271 | RCT with crossover | ≥ 6 | Colistimethate sodium: 30.0 Tobramycin: 30.2 |
115 | Tobramycin: 55 Colistimethate sodium: 59 |
Yes | 4 weeks (poor data for 8, 24 and 44 weeks) | FEV1%; SpD; AEs | Tobramycin: 300 mg/5 ml b.i.d. Colistimethate sodium: 80 mg b.i.d. |
NA | Incompatible Colistin dose | |
| Jensen et al. 198772 | RCT | ≥ 7 | Intervention: 13.6 Control: 14.7 |
40 | Intervention: 71 (25) Control: 79 (29) |
Yes | 12 weeks | FVC, FEV1%; AEs; FEF25–75%; SpD | 1 MIU b.i.d. | Saline b.i.d. | Incompatible Colistin dose; all dosed with i.v. tobramycin 2 weeks before study | |
| Nebulised tobramycin | ||||||||||||
| Hodson et al. 2002 (also listed above)71 | RCT with crossover | ≥ 6 | Colistimethate sodium: 30.0 Tobramycin: 30.2 |
115 | Tobramycin: 55 Colistimethate sodium: 59 |
Yes | 4 weeks (poor data for 8, 24 and 44 weeks) | FEV1%; SpD; AEs | Tobramycin: 300 mg/ml b.i.d. Colistimethate sodium: 80 mg b.i.d. |
n/a | Incompatible Colistin dose | |
| Chuchalin et al. 200773 | RCT | ≥ 6 | Intervention: 14.8 Control: 14.7 |
247 | Intervention: 61 Control: 64 |
Yes | 4 weeks | FVC, FEV1%; AEs; FEF25–75%; susceptibility; MIC; hospitalisations; BMI | 300 mg in 4 ml b.i.d. | Saline b.i.d. | Bramitob device | |
| Lenoir et al. 200774 | RCT | ≥ 6 | Intervention: 11.0 Control: 14.2 |
59 | Intervention: 58 Control: 60 |
No | 4, 8 weeks | FEV1%; FEF25–75%; FVC; susceptibility; MIC; SpD | 300 mg in 4 ml b.i.d. | Saline b.i.d. | Not all chronic P. aeruginosa | |
| MacLusky et al. 198975 | RCT | ≥ 7 | Intervention: 13.9 Control: 14.3 |
27 | Intervention: 70 (22) Control: 78 (21) |
Yes | Up to 32 months | FEV1%; FEF25–75%; FVC; susceptibility; hospitalisations | 80 mg t.i.d. | Saline t.i.d. | Incompatible tobramycin dose | |
| Ramsey et al. 1999;76 Moss 2001,77 200278 | RCT | ≥ 6 | Intervention: 20.8 Control: 20.6 |
520 | Intervention: 49.9 (15.5) Control: 51.2 (16.8) |
Unclear | 20, 24 weeks | FEV1%; SpD; resistance | 300 mg (ml unclear) b.i.d. | Saline b.i.d. | Unclear if chronic infection | |
| Nasr et al. 2010,80 200679 | RCT | ≥ 6 | 11.8 (tobramycin), 15.9 (placebo) | 32 | Intervention: 95.73 (17.21) Control: 83.71 (21.07) |
Yes | 4 weeks | Weight; FEF25%–75%; FEV1%; chest tomography; CFQ-R | 300 mg/5 ml b.i.d. | Saline b.i.d. | Young participants; mild disease by FEV1% | |
| Murphy et al. 200481 | RCT | 6–15 | 10.2 (tobramycin group) 9.9 (placebo) |
184 | Intervention: 85.1 (12.0) Control: 86.3 (9.4) |
Yes | 56 weeks (no 4-week data for FEV1) | Hospitalisations; AEs; i.v. antibiotics | 300 mg b.i.d. | Saline b.i.d. | Young participants; mild disease; large withdrawal numbers | |
| Ramsey et al. 199382 | RCT with crossover | NR | 17.7 16.6 |
71 | 57.5 (3.5) | Unclear | 4 weeks to first crossover | FVC; FEV1%; FEF25–75%; AcEx; i.v. antibiotics; toxicity; resistance | 600 mg t.i.d. | Saline t.i.d. | Incompatible tobramycin dose | |
FIGURE 38.
Network of evidence for colistimethate sodium DPI and tobramycin DPI with outcomes measured 4 weeks after commencement of treatment. Solid bold line, studies with comparable baseline characteristics; dashed line, studies with incomparable baseline characteristics or unknown characteristics. MU, million units.
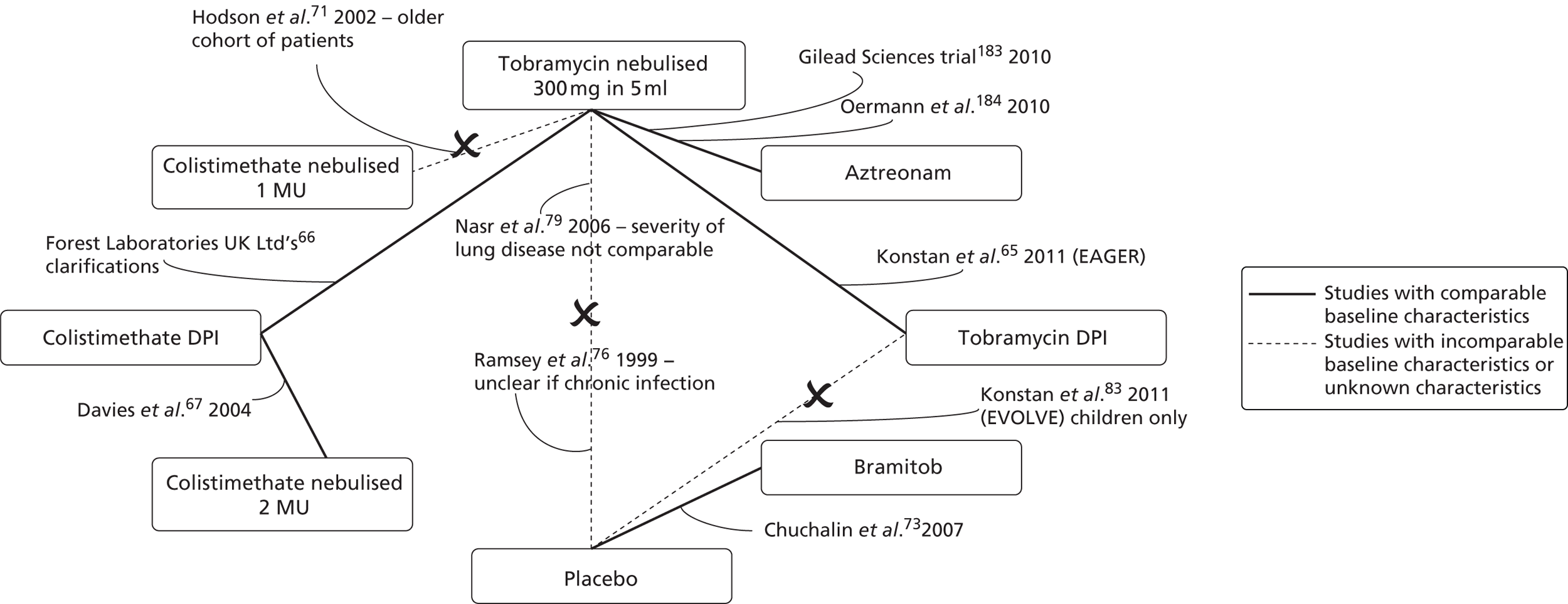
FIGURE 39.
Network of evidence for colistimethate sodium DPI and tobramycin DPI with outcomes measured 20 and 24 weeks after commencement of treatment. Solid bold line, studies with comparable baseline characteristics; dashed line, studies with incomparable baseline characteristics or unknown characteristics. b.i.d., twice daily; NR, not reported.
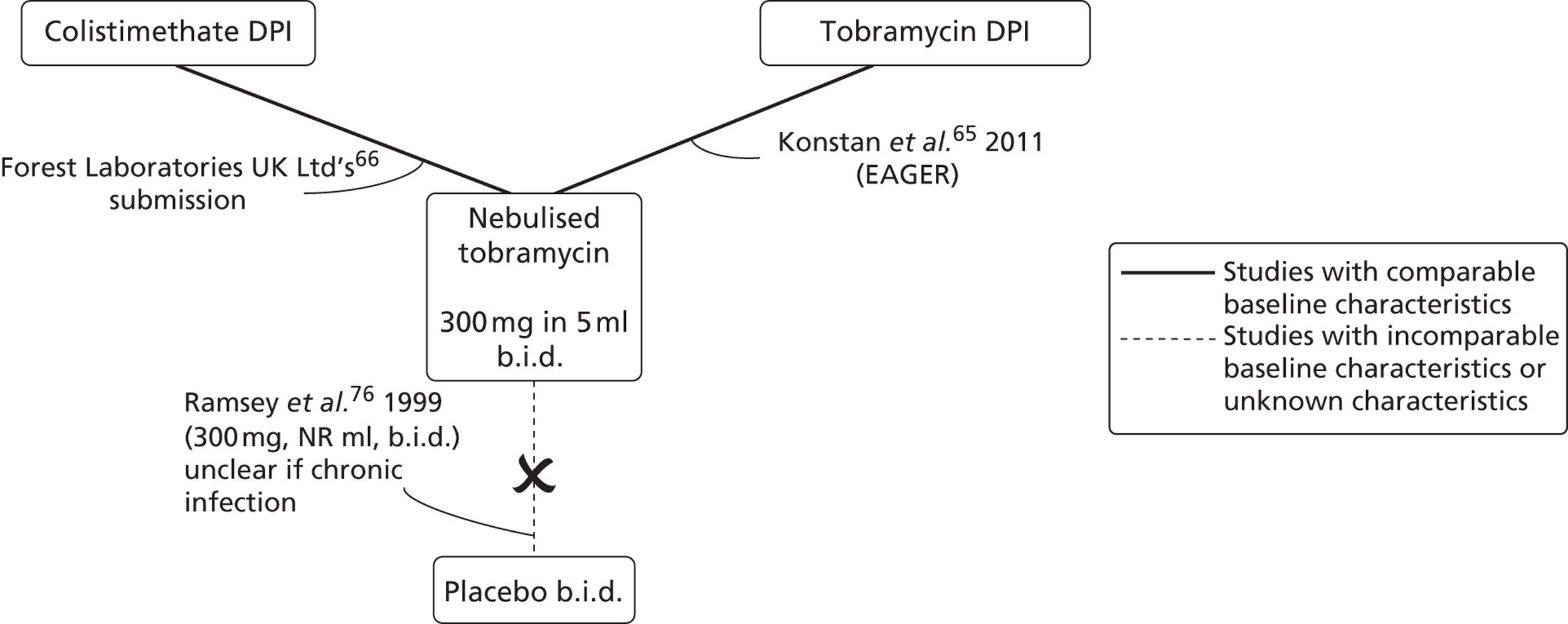
Appendix 5 Data extraction tables
Davies et al. 200467
Note: data in square brackets denote data extracted from the ‘additional sources of data’ listed for the study.
| Study details | ||||
|---|---|---|---|---|
| Publication type | Davies et al.67 Conference abstract from CF conference | |||
| Additional source of data | Industry submission from Forest Laboratories UK COL/DPI/02/0566 (data extracted from this source indicated by square brackets) | |||
| Trial design | RCT with crossover, open-label, multidose tolerability study | |||
| Country | UK | |||
| Dates of participant recruitment | NR | |||
| Source of funding | Forest Laboratories UK Ltd | |||
| Intervention(s) and comparator | ||||
| Treatment groups | Salbutamol followed by micronised Colistin (125 g) via Turbospin DPI Micronised Colistin alone (125 mg) b.i.d. via Turbospin DPI |
|||
| Comparator | Colistimethate sodium (2 MIU) solution in 4 ml 0.9% NaCl b.i.d. via nebuliser | |||
| Run-in phase | [72 hours’ washout] | |||
| Treatment duration | 8 weeks (2 × 28-day cycles) – 4 weeks of powder then crossover to nebulised colistimethate sodium | |||
| Outcome(s) | ||||
| Follow-up | 4 and 8 weeks, although lung function measured at 1, 2, 3 and 4 weeks | |||
| Outcomes and measures | [Clinical tolerability from AEs Laboratory safety from haematology, biochemistry, urinanalysis and renal markers FEV1 Safety confirmed by CFQ for QoL] |
|||
| Population | ||||
| Eligibility criteria |
|
|||
| Concomitant interventions allowed or excluded | Allowed Patients were permitted to continue with pre-existing non antipseudomonal CF medications. Bronchodilators refrained from use 4 hours prior to pulmonary function test. Salbutamol administered as rescue medication for bronchoconstriction after either intervention or comparator administration | |||
| Power calculation | NR [Sample size chosen based on practical considerations rather than formal statistical arguments, as this was a pilot study] | |||
| No. randomised to treatments | [ITT 16; PP 11] 12 |
|||
| Treatment group | DPI colistimethate sodium 125 mg | Nebulised colistimethate sodium 2 MIU | ||
| No. randomised to treatment | 12 [16] | 12 [16] | ||
| Baseline characteristics | Between-group characteristics NR | |||
| Age (years) | [mean 20.3 (SD 12.87)] | |||
| Sex | [M, 50%; F, 50%] | |||
| FEV1 | [mean 76.75 (SD 26.43)] | |||
| FEV1% predicted | 77.14 (6.784) | 76.25 (7.315) | ||
| BMI (kg/m2) | [mean 19.99 (SD 4.01)] | |||
| Withdrawals | n = 3 withdrew early (1 = subject request, 2 = AE) | |||
| Withdrawals/loss to follow-up | One discontinued owing to cough, throat irritation and unpleasant taste [two withdrew owing to AEs, having already completed nebulised treatment] | 0 | ||
| Results | ||||
| Notes on statistics used | [A priori: safety set; all of those who received one dose of study drug PP set: All of those who completed the study and had missed 20% or less of any component of their dosing regimen. No confirmatory testing was performed. Statistical tests were interpreted in a descriptive manner. Descriptive statistics used for missing observations] |
|||
| Microbial response | ||||
| Biochemistry, haematology and urinary N-acetyl-β-Dd-glucosaminidase (NAG) all showed no treatment-related or consistent effects (no statistics provided) | ||||
| Lung function | ||||
| FEV1 | No significant changes in lung function in either treatment arm [Two-sided t-test p < 0.05] |
|||
| Acute exacerbations | ||||
| NR | ||||
| Resistance | ||||
| NA | ||||
| Compliance | ||||
| One discontinued use [Two left the study owing to AEs] |
NR | |||
| Mortality | ||||
| [0] | [0] | |||
| [AEs (solicited and spontaneously reported)a] | ||||
| Treatment group | DPI colistimethate sodium 125 mg, n = 16 | Nebulised solution colistimethate sodium 2 MIU, n = 15 | ||
| Patients (%) | No. of events | Patients (%) | No. of events | |
| Patients with at least one treatment-emergent AE | 16 (100) | 106 | 9 (60.0) | 55 |
| Gastrointestinal disorders | 14 (87.5) | 18 | 3 (20.0) | 3 |
| Gastrointestinal disorderb | 14 (87.5) | 16 | 2 (13.3) | 2 |
| Vomiting | 2 (12.5) | 2 | 0 | 0 |
| General disorders and administration site conditions | 5 (31.3) | 8 | 3 (20.0) | 7 |
| Pyrexia | 2 (12.5) | 2 | 1 (6.7) | 1 |
| Infections and infestation | 2 (12.5) | 2 | 0 | 0 |
| Injury, poisoning and procedural complication | 2 (12.5) | 2 | 1 (6.7) | 1 |
| Investigations | 0 | 0 | 1 (6.7) | 1 |
| Metabolism and nutrition disorders | 1 (6.3) | 1 | 0 | 0 |
| Nervous system disorders | 5 (31.3) | 6 | 3 (20.0) | 6 |
| Headache | 1 (6.3) | 1 | 2 (13.3) | 5 |
| Reproductive system and breast disorders | 1 (6.3) | 1 | 0 | 0 |
| Pharyngolaryngeal pain | 2 (12.5) | 2 | 2 (13.3) | 2 |
| Throat irritation | 13 (81.3) | 19 | 3 (20.0) | 6 |
| Skin and subcutaneous tissue disorders | 0 | 0 | 1 (6.7) | 1 |
| Pruritus | 0 | 0 | 1 (6.7) | 1 |
| Severe symptoms | ||||
| Unpleasant taste | 2 | NR | NR | NR |
| Wheezing | 1 | NR | NR | NR |
| New cough/increased cough | 1 | NR | NR | NR |
| Nasal pain | 3 | NR | NR | NR |
| Sinus pain | NR | NR | 1/55 (1.8) | NR |
| Comparison between groups | Percentage of patients with related TEAEs was higher in the dry powder group than in the nebulised group for most system organ classes and preferred terms. States that unpleasant taste and new cough/increase in cough higher in DPI than nebulised but do not provide the data | |||
EAGER trial65
Note: data in square brackets denote data extracted from the ‘additional sources of data’ listed for the study.
| Study details | ||
|---|---|---|
| Publication type | Konstan et al. 2011,65 full report in peer-reviewed journal | |
| Additional source of data | Novartis Pharmaceuticals’ industry submission60 (data extracted from this source indicated by square brackets) | |
| Trial design | Randomised, multicentre, two-arm, open-label, non-inferiority trial | |
| Country | 127 centres in 15 countries, including North America, Europe, Australia, Israel and Latin America | |
| Dates study undertaken | February 2006 to March 2009 (from clinical trial record) | |
| Source of funding | Novartis Pharmaceuticals | |
| Intervention(s) and comparator | ||
| Treatment group | Tobramycin inhalation powder 112 mg (four capsules) b.i.d. with T-326 Inhaler | |
| Comparator | Tobramycin inhalation solution 300 mg/5 ml tobramycin b.i.d. with PARI LC PLUS jet nebuliser and DeVilbiss PulmoAide compressor | |
| Run-in phase | NR | |
| Treatment duration | 24 weeks (three cycles of 28 days on, 28 days off) | |
| Outcome(s) | ||
| Follow-up | 24 weeks | |
| Outcomes and measures | [Incidence and intensity of all AEs, changes in haematology, blood chemistry, urine protein, audiology, physical condition, body weight, audiology testing, clinical laboratories and vital signs] [Relative change in FEV1% from baseline to all study treatment visits Change in sputum density, tobramycin susceptibility to P. aeruginosa (MIC), antipseudomonal antibiotic use, respiratory related hospitalisations, serum and sputum pharmacokinetics Time to first hospitalisation and duration of hospitalisation Time to first antipseudomonal antibiotic use and duration of treatment] |
|
| Population | ||
| Eligibility criteria | Inclusion > 6 years; [confirmed] patients with CF; FEV1 > 25 to < 75% predicted based on Knudson equations; sputum or throat cultures positive for P. aeruginosa within 6 months of screening [and at the screening visit; ability to comply with all protocol requirements; clinically stable in the opinion of the investigator; contraception – reliable method used (females); consent – written informed consent Exclusion If initiated following drugs within 28 days of study drug administration (if > 28 days, they are eligible for inclusion); chronic macrolide therapy; dornase alpha; inhaled steroids; inhaled hypertonic saline (but where used, must have stable regimen, consistent administration time and not within 30 minutes of conducting pulmonary function tests); sputum culture with B. cepacia within 2 years prior to screening or at screening; haemoptysis more than 60 ml at any time within 30 days prior to study drug administration; Hypersensitivity to aminoglycosides or inhaled antibiotics; serum creatinine level of ≥ 2 mg/dl, blood urea nitrogen level of ≥ 40 mg/dl, or an abnormal urinalysis defined as ≥ 2+ proteinuria; pregnant, attempting to become pregnant or lactating; clinically relevant history of hearing loss or chronic tinnitus; used systemic or inhaled antipseudomonal antibiotics within 28 days prior to study drug administration] |
|
| Concomitant interventions allowed or excluded | Allowed Adrenergics, bile acid preparations, cephalosporins, corticosteroids, enzyme preparations, fluoroquinolones, mucolytics, multivitamins, non-drug therapies, other aminoglycosides, proprionic acid derivatives, proton pump inhibitors, selective β2-adrenoreceptor agonists, dornase alpha, macrolides, anticholinergics, bronchodilators (patients taking short-acting bronchodilators were to take the medication 15–90 minutes before inhalation of study drug; patients taking long-acting bronchodilators were to take the medication as prescribed within the preceding 24 hours) and glucocorticoids | |
| Power calculation | [Based on primary variable of safety, 300 patients provide a 99.8% chance of observing at least one AE with a true incidence of 2% in the tobramycin inhalation powder group. Inclusion of 500 patients (tobramycin inhalation powder: 300; tobramycin inhalation solution: 200) provides 96% power to demonstrate non-inferiority of tobramycin inhalation powder to tobramycin inhalation solution with non-inferiority margin of 6% based on 500 patients for relative change from baseline in FEV after three cycles, with one-sided significance level of 0.15 (assuming 1% true tobramycin inhalation solution–tobramycin inhalation powder treatment difference and 20% SD)] | |
| No. randomised to treatments included in review | 553 | |
| Treatment group | Tobramycin inhalation powder, 112 mg b.i.d. | Tobramycin inhalation solution, 300 mg/5 ml TOBI b.i.d. |
| No. randomised to treatment | [329 randomised] 308 ITT |
[224 randomised] 209 ITT |
| Baseline characteristics | ||
| Age (years) | Mean 26 (SD 11.4) | Mean 25 (SD 10.2) |
| Sex | M, 55.5%; F, 44.5% | M, 55.0%; F, 45.0% |
| FEV1 | Mean 53 (SD 14.2) [SE 0.81] | Mean 53 (SD 15.9) [SE 1.11] |
| BMI (kg/m2) | Mean 20.7 (SD 4.0) | Mean 20.4 (SD 3.5) |
| Withdrawals | ||
| From 553 participants randomised, 36 discontinued prior to receiving study medication, [21 from tobramycin inhalation powder arm, 15 from tobramycin inhalation solution arm]. Reasons with withdrawal given but not extracted here A further 121 discontinued after at least one dose of study medication |
||
| Withdrawals/loss to follow-up | 83 discontinued | 38 discontinued |
| AE 40 (13.0%) | AE 17 (8.1%) | |
| Cough 12/308 | Cough 2/209 | |
| Death 3 (1.0%) | ||
| Consent withdrawn 24 (7.8%) | Consent withdrawn 9 (4.3%) | |
| Lost to follow-up 5 (1.6%) | Lost to follow-up 3 (1.4%) | |
| Administrative reason 1 (0.3%) | Inappropriate enrolment 1 (0.5%) | |
| Protocol violation 6 (1.9%) | Protocol violation 5 (2.4%) | |
| Other 4 (1.3%) | Other 3 (1.4%) | |
| Results | ||
| Notes on statistics | Population used were randomised and treated with no imputation for missing data. Information for each outcome given, but not extracted here. Non-inferiority inferential analysis: The non-inferiority of TOBI Podhaler relative to TOBI was assessed using a CI approach (margin of 6%). Pharmacokinetics based on a subset of patients (30 tobramycin inhalation powder; 14 tobramycin inhalation solution) but do not report the tobramycin inhalation solution statistics. Post hoc sensitivity analyses assessed the impact of patient discontinuation. All randomised patients who received one or more doses of study drug were included in the safety and efficacy (ITT) populations. Efficacy (FEV1%) was measured by least-squares means difference. Efficacy data reported as least-squares mean difference (SE) | |
| [Microbial response at 28 days, mean decrease log10 (SD)] | ||
| [Mean P. aeruginosa sputum density change from baseline, unspecified phenotype] | [−1.76 (SE 0.14, SD 1.96)] | [−1.32 (SE 0.17, SD 2.03)] |
| [Mean P. aeruginosa sputum density change log10 from baseline at week 20] | ||
| [Mean CFU non-mucoid] | [5.17] | [6.18] |
| [Mean CFU mucoid] | [5.40] | [6.30] |
| Mean P. aeruginosa sputum density change from baseline at week 20 | ||
| Non-mucoid phenotype | −1.77 | −0.73 |
| Mucoid phenotype | −1.6 | −0.92 |
| [Unspecified phenotype] | [−1.61 (SE 0.16, SD 2.03)] | [−0.77 (SE 0.16, SD 1.78)] |
| Negative P. aeruginosa culture | 11.6% | 9.9% |
| [Lung function, 28-day data] | ||
| [No. of patients] | [268] | [194] |
| [Mean FEV1% predicted at predose] | [54.381 (SE 0.634643, SD 10.38956)] | [54.7008 (SE 0.543224, SD 7.56624)] |
| [Mean change from baseline (or least-squares mean)] | [1.48 (SE 0.63)] | [1.9 (SE 0.54)] |
| [% mean change from baseline] | [2.80% (SD 19.64%)] | [3.60% (SD 14.33%)] |
| [% mean change from baseline PP, n = 187] | [3.7 (SD 20.31)] | [4.2 (SD 14.40)] |
| [Lung function, 20-week data] | ||
| [Mean FEV1% predicted at pre dose] | [55.9682 (NR)] | [55.2816 (NR)] |
| [Mean change from baseline (or least-squares mean)] | [3.0682 (SE0.6546041)] | [2.4816 (SE 0.65336667)] |
| [% mean change from baseline] | [5.80% (SE1.24%) (least-squares mean)] | [4.70% (SE 1.24%) (least-squares mean)] |
| [Lung function 24-week data] | ||
| [% mean change from baseline] | [1.8 (SD 16.07)] | [−0.4 (16.93)] |
| Lung function comparison between groups | ||
| Reported as ‘similar between groups using least-squares mean difference 1.1% relative change (SE 1.75)’. The lower limit (−0.67%) of the one-sided 85% [CI] (equivalent to 70% two sided) was within the predefined 6% margin for predefined non-inferiority, indicating that tobramycin inhalation powder was non-inferior to tobramycin inhalation solution | ||
| Least-squares mean difference (PP population) | Least-squares mean difference in FEV1 of 1.2%, lower limit of the one-sided 85% CI was −1.02% | |
| [Difference in mean change from baseline (or least-squares mean) at 28 days] | [−0.4196 (SE 0.835383114)] | |
| [Difference in mean change from baseline at 20 weeks] | [0.5866 (SE 0.924875414)] | |
| Acute exacerbations | ||
| Required additional antipseudomonal antibiotic | 64.9% | 54.5% |
| Oral antibiotics used | 55.5% | 39.7% |
| Mean no. of days of antibiotic use | 30.9 (SD 23.34) | 33.4 (SD 24.42) |
| Hospitalised for respiratory-related eventsa | 24.4% | 22.0% |
| Lung disorderb | 104 (33.8%) | 63 (30.1%) |
| [No. of patients using an P. aeruginosa antibiotics at 24 weeks] | [200/308 (64.9%)] | [114/209 (54.5%)] |
| [No. of patients with at least one hospitalisation] | [75 (24.4%)] | [46 (22.0%)] |
| [Mean (SE) days in hospital] | [15.6 (13.31)] | [15.3 (10.23)] |
| Resistance | ||
| P. aeruginosa isolates (all phenotypes) with MIC > 8 µg/ml (resistant) at baseline | 68/308 (22.1%) | |
| P. aeruginosa isolates (all phenotypes) with MIC ≤ 8µg/ml (susceptible) at baseline | 240/308 (77.9%) | |
| MIC > 8 µg/ml at the end of cycle 3 | 19.1% | |
| Increased MIC of tobramycin against P. aeruginosa from baseline to day 28 of cycle 3 | Fourfold or greater increase: 67/199 (33.7%) Twofold or greater increase: 97/199 (48.7%) (unclear which numbers relate to which group) |
|
| Pharmacokinetics of P. aeruginosa isolates | ||
| At baseline MIC at least 20 times lower than the mean sputum concentration observed within 30 minutes of the first dosing in cycle 1 | 91.2% (≤ 64 μg/ml) | NR |
| At the end of cycle 3, (all phenotypes) MIC at least 30 times lower than the mean sputum concentration observed 30 minutes post dose | 86.4% (≤ 64 μg/ml) | NR |
| Compliance | ||
| > 90% | > 90% | |
| Discontinuation rate | 26.9% | 18.2% |
| Mortality | ||
| Three (two are related to acute exacerbations according to clarifications provided by manufacturer in clarifications) | 0 | |
| AEs | ||
| No. of patients | NR | NR |
| Any AE | 90.3% | 84.2% |
| Mild or moderate AE | 73.4% | 68.5% |
| SAEs | 27.4% | 29.2% |
| AEs cycle 1 | 77.9% | 66.5% |
| AEs cycle 2 | 67.0% | 66.3% |
| AEs cycle 3 | 65.8% | 58.5% |
| Cough | 149 (48.4%) | 65 (31.1%) |
| Productive cough | 56 (18.2%) | 41 (19.6%) |
| Severe cough | 2.6% | 1.9% |
| Dyspnoea | 48 (15.6%) | 26 (12.4%) |
| Oropharyngeal pain | 43 (14.0%) | 21 (10.5%) |
| Rales | 22 (7.1%) | 13 (6.2%) |
| Rhinorrhoea | 22 (7.1%) | 15 (7.2%) |
| Pulmonary function test decreased | 21 (6.8%) | 17 (8.1%) |
| Pyrexia | 48 (15.6%) | 26 (12.4%) |
| AiC information has been removed | AiC information has been removed | AiC information has been removed |
| Upper respiratory tract infection | 21 (6.8%) | 18 (8.6%) |
| Wheezing | 21 (6.8%) | 13 (6.2%) |
| Chest discomfort | 20 (6.5%) | 6 (2.9%) |
| Sinusitis | 18 (5.8%) | 15 (7.2%) |
| Pulmonary congestion | 17 (5.5%) | 9 (4.3%) |
| Dysphonia | 42 (13.6%) | 8 (3.8%) |
| Nasal congestion | 25 (8.1%) | 15 (7.2%) |
| Vomiting | 19 (6.2%) | 12 (5.7%) |
| Haemoptysis | 40 (13.0%) | 26 (12.4%) |
| Nausea | 23 (7.5%) | 20 (9.6%) |
| Headache | 35 (11.4%) | 25 (12.0%) |
| Fatigue | 20 (6.5%) | 10 (4.8%) |
| AiC information has been removed | AiC information has been removed | AiC information has been removed |
| Audiology from subgroup | n = 78 (25.3%) | n = 45 (21.5%) |
| Decrease from baseline at any visit | 20 (25.6%) | 7 (15.6%) |
| Clinically significant decrease | 3 (0.97%) | 2 (0.96%) |
| Clinically significant bronchospasm (acute relative change of ≥ 20% decrease in FEV1% from pre dose to 30 minutes post dose) | 5.2% | 5.3% |
Forest Laboratories trial 201166
| Study details | ||||
|---|---|---|---|---|
| Publication type | Industry submission from Forest Laboratories UK COL/DPI/02/0666 | |||
| Additional source of data | None | |||
| Trial design | RCT, multicentre | |||
| Country | EU, Russia and Ukraine | |||
| Dates of participant recruitment | NR, but last patient visit was 14 August 2007 | |||
| Source of funding | Forest Laboratories UK | |||
| Intervention(s) and comparator | ||||
| Treatment groups | Colistimethate sodium dry powder 125 mg b.i.d. with Turbospin device | |||
| Comparator | Tobramycin (TOBI) nebulised solution 300 mg b.i.d. with PARI LC nebuliser | |||
| Run-in phase | 16 weeks, two cycles of TOBI treatment | |||
| Treatment duration | 24 weeks (intervention had continuous treatment; control group had three cycles of 28 days on, 28 days off) | |||
| Outcome(s) | ||||
| Follow-up | 24 weeks, with interim data at 20 weeks | |||
| Outcomes and measures | FEV1% predicted | |||
| Antibiotic sensitivity of respiratory tract P. aeruginosa isolates (MIC and BSAC) | ||||
| FVC | ||||
| Peak expiratory flow rate | ||||
| Forced expiratory flow between 25% and 75% of the FEV (FEF25–75%) | ||||
| Acute exacerbations | ||||
| Sputum Colistin levels | ||||
| Compliance with study medication | ||||
| AEs | ||||
| Dropout rates | ||||
| CFQ-R | ||||
| Population | ||||
| Eligibility criteria | Male or female aged ≥ 6 years. Patients who had received a minimum of two TOBI on/off cycles immediately prior to randomisation. Heterosexually active females had to use adequate effective contraceptive methods. Patients were required to be non-smokers or a past smoker who had not smoked within the past 12 months. Patient or parent/guardian had to be capable of reading and understanding informed consent and clinical trial information leaflet, and to have granted written informed consent. Documented diagnosis of CF from a specialist CF unit (genotype and/or positive sweat tests). Current CF condition had to be clinically stable in the investigator’s opinion, i.e. there was no evidence of a current acute respiratory exacerbation within 28 days prior to the first day of trial medication administration. Patients with P. aeruginosa. Patient’s lung function had to be clinically stable (Investigator’s decision) after completing i.v. therapy (elective or treatment for exacerbation) at visit 1 prior to randomisation. Patients who, on the first day of trial medication administration, had at least 28 days but no more than 35 days off TOBI | |||
| Concomitant interventions allowed or excluded | Allowed Continued chronic use of bronchodilators, hypertonic saline, use of oxygen, nutritional supplements and enzymes. In addition, use of dornase alfa, inhaled steroids and macrolides (if initiated > 28 days before study drug) | |||
| Power calculation | Non-inferiority of CP vs. TS. 95% two-sided CI for the difference between the two groups was computed, and if the lower limit was not < −3.0% then non-inferiority was accepted Based on a two-group t-test with a 0.05 two-sided significance level and a common SD of 16%, and assume a difference of 2% in favour of Colobreathe® against TOBI (using nQuery Advisor® 4.0) Assuming a 10% dropout/non-compliance rate, to obtain 324 evaluable patients approximately 360 patients were to be entered into the study (180 TOBI patients and 180 Colobreathe patients) |
|||
| No. randomised to treatments included in review | 380 | |||
| Treatment group | Colistimethate sodium dry powder 125 mg b.i.d. | (TOBI) nebulised solution 300 mg b.i.d. | ||
| No. randomised to treatment | 187 | 193 | ||
| Baseline characteristics | ||||
| Age (years) | Mean 21.3 (SD 9.72) | Mean 20.9 (SD 9.30) | ||
| Sex | M, 56.3%; F, 43.7% | M, 52.9%; F, 47.1% | ||
| FEV1 | NR | NR | ||
| BMI (kg/m2) | Mean 18.67 (SD 3.39) | Mean 18.46 (SD 3.58) | ||
| Medical history (ITT population) | ||||
| Respiratory, thoracic and mediastinal disorders (%) | 77.0 | 73.3 | ||
| Gastrointestinal disorders (%) | 72.1 | 75.4 | ||
| Hepatobiliary disorders (%) | 29.5 | 37.7 | ||
| Musculoskeletal and connective tissue disorders (%) | 23.0 | 19.4 | ||
| Metabolism and nutrition disorders (%) | 21.3 | 17.8 | ||
| Infections and infestations (%) | 19.1 | 16.2 | ||
| Prior medication: n (%) | ||||
| Fluoroquinolones | 11 patients (6) | 6 patients (3.1) | ||
| Macrolides | 10 patients (5.5) | 10 patients (3.1) | ||
| Withdrawals | ||||
| Withdrawals/loss to follow-up: n (%) | 32 withdrawn (17.1) | 21 withdrawn (14.2) | ||
| AE: 18 (56.3) | AE: 3 (14.3) | |||
| Lack of efficacy: 2 (6.3) | Lack of efficacy: 1 (4.8) | |||
| Patient request: 5 (28.1) | Patient request: 11 (52.4) | |||
| Protocol violation: 1 (3.1) | Death: 2 (9.5) | |||
| Other: 2 (6.3) | Other: 4 (19.0) | |||
| Phase of withdrawal: n (%) | ||||
| Within 4 weeks | 5 (2.7) | 1 (0.5) | ||
| Between 4 and 8 weeks | 12 (6.4) | 6 (3.1) | ||
| Between 8 and 16 weeks | 9 (4.8) | 5 (2.6) | ||
| Between 16 and 20 weeks | 5 (2.7) | 5 (2.6) | ||
| Between 20 and 24 weeks | 1 (0.5) | 4 (2.1) | ||
| Protocol violations resulting in exclusion from ITT analysis | 46 patients | 35 patients | ||
| Results: 24-week data | ||||
| Notes on statistics | Analysed 380 randomised (safety population), 374 patients (ITT population), 298 patients (PP population). ANCOVA model using main effects treatment, baseline FEV% predicted and pooled centre. Adjusted means by treatment presented as well as an estimate of the difference between adjusted means | |||
| Microbial response | ||||
| Mean P. aeruginosa sputum density | No sputum density tests were performed during the trials | |||
| Lung function | ||||
| Baseline FEV1% predicted (SE) | 51.76 (1.029) | 50.82 (0.989) | ||
Lung function: data also available from manufacturer’s submission but not extracted here include:
|
||||
| Lung function: change in FEV1% predicted LOCF ITT population | ||||
| No. of patients | n = 183 | n = 190 | ||
| Mean (SD) | −0.90 (10.015) | 0.35 (10.756), | ||
| Median; range FEV1% | −1.43; minimum −32.9, maximum 43.4 | −1.09; minimum −33.6, maximum 49.3 | ||
| ANCOVA adjusted mean: | −1.28 | −0.13 | ||
| Comparison between groups | ANCOVA adjusted least-squares mean difference between treatments = −1.16% (95% CI −3.15% to 0.84%) | |||
| Lung function: change in FEV1% predicted LOCF PP population | ||||
| No. of patients | n = 141 | n = 157 | ||
| Mean (SD) | −0.30 (10.306) | 1.12 (11.120) | ||
| Median; range FEV1% | −1.28; minimum −29.0, maximum 43.4 | −0.61, minimum −33.6, maximum 49.3 | ||
| ANCOVA adjusted mean | −1.02. | −0.47. | ||
| Comparison between groups | ANCOVA adjusted least-squares mean difference between treatments = −1.49% (95% CI −3.79% to 0.81%) | |||
| Lung function: change in FEV1% predicted: completers ITT population | ||||
| No. of patients | n = 153 | n = 171 | ||
| Mean (SD) | −0.39 (9.715) | 0.78 (10.900) | ||
| Median; range FEV | −0.70, minimum −29.0, maximum 43.4 | −0.58, minimum −33.6, maximum 49.3 | ||
| ANCOVA adjusted mean | −0.36 | 0.08 | ||
| Comparison between groups | ANCOVA adjusted least-squares mean difference between treatments = −0.43% (95% CI −2.59% to 1.72%) | |||
| Lung function: change in FEV1% predicted: completers PP population | ||||
| No. of patients | n = 120 | n = 141 | ||
| Mean (SD) | 0.83 (10.236) | 1.6 (11.260) | ||
| Median; range FEV1% | −0.51, minimum −29.0, maximum 43.4 | −0.26, minimum −33.6, maximum 49.3 | ||
| ANCOVA adjusted mean | −0.26 | 0.73 | ||
| Comparison between groups | ANCOVA adjusted least-squares mean difference between treatments = −0.99 (95% CI −3.48% to 1.51%) | |||
| Lung function FVC: adjusted treatment difference (ITT population) | ||||
| Comparison between groups | 0.01 l (95% CI −0.09 l to 0.10 l) not significant (p = 0.886) | |||
| Lung function peak expiratory flow rate: adjusted treatment difference (ITT population) | ||||
| Comparison between groups | −3.32 l/minute (95% CI −16.31 to 9.67 l/minute) not significant (p = 0.616) | |||
| Lung function FEF25–75%: adjusted treatment difference (ITT population) | ||||
| Comparison between groups | −0.12 l/second (95% CI −0.23 to −0.01 l/second) significant (p = 0.038) | |||
| Lung function FEF25–75%: adjusted treatment difference (PP population) | ||||
| Comparison between groups | −0.12 l/second (95% CI −0.26 to −0.01 l/second) not significant (p = 0.063) | |||
| Acute exacerbations (mean no. of days) | ||||
| Time to acute respiratory exacerbation | CiC information has been removed | CiC information has been removed | ||
| Time to first additional antipseudomonal treatment | 55.28 | 51.79 | ||
| Duration of use of additional antipseudomonal agents | CiC information has been removed | CiC information has been removed | ||
| CiC information has been removed | ||||
| CiC information has been removed | CiC information has been removed | CiC information has been removed | ||
| CiC information has been removed | CiC information has been removed | CiC information has been removed | ||
| Proportion of antibiotic resistant isolates | Colistin: ≤ 1.1% | CiC information has been removed | ||
| CiC information has been removed | CiC information has been removed | |||
| CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | |
| CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed |
| CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed |
| CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed |
| CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed |
| CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed |
| CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed |
| CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed |
| CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed |
| CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed |
| CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed |
| CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed |
| CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed | CiC information has been removed |
| CiC information has been removed | CiC information has been removed | |||
| BSACa for resistant P. aeruginosa isolates | ≤ 1.1% | CiC information has been removed | ||
| BSACa comparison between groups | No significant difference between groups until broken down by age (note: this analysis was not defined a priori and is also underpowered). In the TOBI group, a notable increase between baseline and week 24 in the proportion of tobramycin-resistant isolates was documented in the 6- to 12-year age group (resistant: 10.5–22.2%), as well as in the 13- to 17-year age group (resistant: 11.4–16.0%) | |||
| Compliance | ||||
| Patients > 75% compliant with study medication | 66.7% | 70.7% | ||
| Withdrew | 30 (16.4%) | 20 (10.5%) | ||
| < 75% of doses | 31 (16.9%) | 36 (18.8%) | ||
| > 75% – < 100% of doses | 105 (57.4%) | 120 (62.8%) | ||
| > 100% of doses | 17 (9.3%) | 15 (7.9%) | ||
| Mortality | ||||
| 0 | Two (both deaths reported as being unrelated to study drug: one death was due to multiorgan failure and one was due to lower respiratory tract infection) | |||
| AEs: data by patient | ||||
| Study drug-related AEs | 153/187 (81.8%) | 90/193 (46.6%) | ||
| Patients withdrawn due to AEs | 22/187 (11.8%) | 5/193 (2.6%) | ||
| Severeb (related) | AE 48/187 (25.7%) | 13/193 (6.7%) | ||
| SAEs | 8/187 (4.3%) | 12/193 (6.2%) | ||
| Serious related AEsc | 3/187 (1.6%) | 2/193 (1.0%) | ||
| AEs: data by event | ||||
| Study drug-related AEs | 528 | 325 | ||
| Severe (related?) AEs | 73 | 12 | ||
| Moderate dysgeusia | 10.7% | 5.2% | ||
| Cough | 15.7% | 10.3% | ||
| Dyspnoea | 6.6% | 8.2% | ||
| BMI (kg/m2) mean change from baseline to week 24 | < 1.0 kg | < 0.20 kg | ||
| Audiology | Not tested | Not tested | ||
| Intravenous Colistin administered during ‘off’ periods | NA: no off periods | 7 (3.6%) | ||
| Inhaled Colistin administered during ‘off’ periods | NA: no off periods | 4 (2.1%) | ||
| HRQoL | ||||
| CFQ-R adjusted mean change from baseline to week 24 | ||||
| Physical | 0.26 | −1.56 | ||
| Vitality | 0.86 | −1.40 | ||
| Emotion | 2.23 | 0.47 | ||
| Eating | 0.48 | 0.66 | ||
| Treatment burden | 5.62 | 2.75 | ||
| Health perceptions | 0.25 | −2.71 | ||
| Social | 3.10 | 0.92 | ||
| Body image | 7.83 | 5.98 | ||
| Role | 0.65 | 1.87 | ||
| Weight | 0.88 | −1.93 | ||
| Respiratory | 2.99 | 3.51 | ||
| Digestion | 5.06 | 2.89 | ||
Appendix 6 MEDLINE search strategy for EQ-5D utility data on adverse events related to cystic fibrosis and its treatment
Database: Ovid MEDLINE(R) – 1948 to week 4 April 2011
Search strategy
-
eq-5d.tw. (1422)
-
eq5d.tw. (71)
-
euroqol.tw. (1254)
-
euro qol.tw. (24)
-
or/1-4 (2120)
-
Cough/ (10,389)
-
cough$.tw. (27,671)
-
lung disorder$.tw. (817)
-
pulmonary exacerbation$.tw. (441)
-
cystic fibrosis exacerbation$.tw. (8)
-
cf exacerbation$.tw. (15)
-
Dyspnea/ (12,637)
-
dyspnea.tw. (18,987)
-
(short$ adj2 breath).tw. (3582)
-
Fever/ (27,386)
-
fever.tw. (96,463)
-
pyrexia$.tw. (2791)
-
hyperthermia$.tw. (17,458)
-
oropharyngeal pain.tw. (22)
-
mouth pain.tw. (49)
-
pharynx pain.tw. (1)
-
oropharynx pain.tw. (2)
-
Oropharynx/ (2850)
-
exp Pain/ (263,562)
-
Pain Measurement/ (47,304)
-
24 or 25 (279,815)
-
23 and 26 (35)
-
Dysphonia/ (303)
-
dysphonia.tw. (2274)
-
phonation disorder$.tw. (19)
-
Hemoptysis/ (4409)
-
hemoptys$.tw. (4729)
-
Headache/ (19,831)
-
exp Headache Disorders/ (22,683)
-
headache$.tw. (46,063)
-
Nasal Obstruction/ (2989)
-
nasal congestion.tw. (1103)
-
nasal block$.tw. (355)
-
block$ nasal.tw. (23)
-
nose block$.tw. (18)
-
block$ nose.tw. (104)
-
Nausea/ (11,432)
-
nause$.tw. (33,719)
-
Respiratory Sounds/ (5788)
-
rale$.tw. (1042)
-
rhinorrhea.tw. (2385)
-
rhinorrhoea.tw. (541)
-
runny nose$.tw. (313)
-
exp Respiratory Function Tests/ (176,514)
-
respiratory function test$.tw. (832)
-
pulmonary function test$.tw. (6977)
-
49 or 50 or 51 (178,682)
-
decreas$.tw. (1,351,277)
-
lower$.tw. (985,107)
-
reduc$.tw. (1,673,323)
-
53 or 54 or 55 (3,288,804)
-
52 and 56 (59,579)
-
Respiratory Tract Infections/ (27,721)
-
upper respiratory tract infection$.tw. (3153)
-
infection$ upper respiratory tract.tw. (19)
-
wheez$.tw. (7993)
-
chest discomfort.tw. (719)
-
discomfort chest.tw. (13)
-
Fatigue/ (15,659)
-
fatigue.tw. (42,610)
-
weariness.tw. (105)
-
lassitude.tw. (291)
-
Vomiting/ (17,262)
-
vomit$.tw. (39,561)
-
emesis.tw. (4604)
-
exp Sinusitis/ (13,915)
-
sinusiti$.tw. (10,053)
-
pulmonary congestion.tw. (948)
-
pulmonary obstruction.tw. (145)
-
pulmonary blockage.tw. (0)
-
or/6-23,27-48,57-75 (444,541)
-
5 and 76 (96)
Appendix 7 MEDLINE search strategy for forced expiratory volume in first second and mortality
-
exp Forced Expiratory Volume/ (17,802)
-
forced expiratory volume.tw. (10,738)
-
fev1.tw. (14,038)
-
exp CF/ (25,639)
-
CF.tw. (27,580)
-
exp Mortality/ (240,291)
-
mortality.tw. (357,242)
-
exp Survival/ (3413)
-
survival.tw. (457,634)
-
1 or 2 or 3 (28,225)
-
4 or 5 (32,439)
-
6 or 7 or 8 or 9 (866,700)
-
10 and 11 and 12 (258)
Appendix 8 Final protocol
Glossary
- Dominated (extended)
- Where the incremental cost-effectiveness ratio for a given treatment alternative is higher than that of the next more effective comparator.
- Dominated (simple)
- Where an intervention is less effective and more expensive than its comparator.
- Meta-analysis
- A statistical method by which the results of a number of studies are pooled to give a combined summary statistic.
- Relative risk
- Ratio of the probability of an event occurring in an exposed group relative to a non-exposed or control group.
- Surrogate outcome
- An intermediate outcome that is intended to substitute for, and be predictive of, a final patient-relevant clinical outcome.
List of abbreviations
- AAD
- adaptive aerosol delivery
- ABPA
- allergic bronchopulmonary aspergillosis
- AE
- adverse event
- AHRQ
- Agency for Healthcare Research and Quality
- AiC
- academic-in-confidence
- ANCOVA
- analysis of covariance
- ATP
- adenosine triphosphate
- ATS
- American Thoracic Society
- A$
- Australian dollars
- b.i.d.
- twice daily
- BMI
- body mass index
- BNF
- British National Formulary
- BSAC
- British Society for Antimicrobial Chemotherapy
- CEAC
- cost-effectiveness acceptability curve
- CF
- cystic fibrosis
- CFQ
- Cystic Fibrosis Questionnaire
- CFQ-R
- Cystic Fibrosis Questionnaire-Revised
- CFTR
- cystic fibrosis transmembrane conductance regulator
- CFU
- colony-forming unit
- CHE
- Centre for Health Economics
- CHMP
- Committee for Medicinal Products for Human Use
- CHQ
- Child Health Questionnaire
- CI
- confidence interval
- CiC
- commercial-in-confidence
- CONSORT
- Consolidated Standards of Reporting Trials
- COPD
- chronic obstructive pulmonary disease
- CRD
- Centre for Reviews and Dissemination
- CRDQ
- Chronic Respiratory Disease Questionnaire
- CSR
- clinical study report
- CT
- computerised tomography
- DNA
- deoxyribonucleic acid
- DPI
- dry powder for inhalation
- EAGER
- Establish A new Gold standard Efficacy and safety with tobramycin in cystic fibrosis
- EMA
- European Medicines Agency
- eMC
- electronic Medicines Compendium
- EPAR
- European Public Assessment Report
- EQ-5D-Y
- EQ-5D, Youth version
- ERG
- Evidence Review Group
- EU
- European Union
- FEF25–75%
- forced expiratory flow (at 25–75% of vital capacity)
- FEV1
- forced expiratory volume in first second
- FEV1%
- forced expiratory volume in first second percentage predicted
- FVC
- forced vital capacity
- HRQoL
- health-related quality of life
- HUI-2
- Health Utilities Index Mark 2
- ICER
- incremental cost-effectiveness ratio
- ICU
- intensive care unit
- ITT
- intention to treat
- i.v.
- intravenous
- LOCF
- last observation carried forward
- MeSH
- medical subject heading
- MIC
- minimum inhibitory concentration
- MIC50
- minimum inhibitory concentration required to inhibit the growth of 50% of organisms in culture
- MRI
- magnetic resonance imaging
- NICE
- National Institute for Health and Care Excellence
- NMA
- network meta-analysis
- OLS
- ordinary least squares
- PaCO2
- arterial carbon dioxide tension
- PaO2
- arterial oxygen preasure
- PEP
- positive expiratory pressure
- PP
- per protocol
- PRISMA
- Preferred Reporting Items for Systematic Reviews and Meta-Analyses
- PSA
- probabilistic sensitivity analysis
- QALY
- quality-adjusted life-year
- RCT
- randomised controlled trial
- SAE
- serious adverse event
- SaO2
- saturation level of oxygen in haemoglobin
- SD
- standard deviation
- SE
- standard error
- SF-6D
- Short Form questionnaire-6 Dimensions
- SG
- standard gamble
- SmPC
- summary of product characteristics
- TEAE
- treatment-emergent adverse event
- TNS
- tobramycin nebuliser solution
- TTO
- time trade-off
- VAS
- visual analogue scale