
Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 07/50/05. The contractual start date was in July 2011. The draft report began editorial review in January 2014 and was accepted for publication in April 2014. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Tjeerd-Pieter van Staa was previously employed (now with the University of Manchester), and Gerard McCann, Shivani Padmanabhan and Rabah Belatri are currently employed by the Clinical Practice Research Datalink (CPRD). CPRD operates within the Medicines and Healthcare products Regulatory Agency (MHRA; the UK regulatory authority for medicine, medical devices and trials and a UK Trading Fund organisation). CPRD provides data and trial services on a commercial basis for both academic and pharmaceutical industry researchers. Neither CPRD nor MHRA had any role in writing the report, or had any input into the content of the report. The authors Gerard McCann, Shivani Padmanabhan and Rabah Belatri were not involved in the review and analysis of research governance challenges and obstacles with the trials. Tjeerd-Pieter van Staa reports grants from the Wellcome Trust, during the conduct of the study; grants from National Institute for Health Research (NIHR), grants from pharmaceutical companies, grants from FP7 Innovative Medicines Initiative (IMI), outside the submitted work. Ben Goldacre reports grants from the Wellcome Trust, during the conduct of the study, and receives income from speaking and writing about problems in medicine, including our failure to conduct trials efficiently where there is uncertainty about treatments. Liam Smeeth reports grants from the Wellcome Trust, during the conduct of the study; grants from the Medical Research Council (MRC), grants from NIHR, and personal fees from GlaxoSmithKline (GSK), outside the submitted work. Munir Pirmohamed is a NIHR Senior Investigator, and is a Commissioner on Human Medicines, and chairs its Pharmacovigilance Expert Advisory Group. Martin Gulliford was member of the CPRD Independent Scientific Advisory Committee (ISAC) throughout the period of this report. None of the other authors has any competing interests to declare.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2014. This work was produced by van Staa et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Background, aims of project and exemplar trials
Explanatory and pragmatic randomised trials
The health-care system aims to provide evidence-based medicine with care informed by research, in addition to that reflecting patients’ values and preferences. Randomised trials are considered the best approach to learn about the effects of many health-care interventions. However, routinely used interventions often lack evidence to guide clinicians to recommend an intervention for a particular patient. This uncertainty can relate to the lack of trial evidence of comparative effectiveness or to the fact that the patient would not have been eligible for previous trials (e.g. due to comorbidity). Many clinicians agree that they have a duty to work with colleagues and patients to help resolve uncertainties about the effects of treatments. This project outlines a possible method to help clinicians and patients to conduct simple randomised trials using routinely collected data.
The development of new health-care interventions (such as medicines and devices) is typically based on randomised trials which compare the new intervention with placebos. These trials often use well-trained investigators in specialist centres with close monitoring of study patients in order to minimise withdrawal rates and non-compliance. Strict eligibility criteria often restrict the study population to those most likely to respond beneficially to treatment and less likely to develop adverse effects. 1 The objective of these trials (also known as explanatory trials) is to test whether or not a new intervention can work in principle. As Schwartz and Lellouch1 outlined, these types of trials are aimed at the understanding of biological effects. These trials use biologically meaningful criteria to assess effects and they are typically applied to a homogeneous group of patients, with selection based on the likelihood of responding to the treatment of interest. A different type of trial (known as a pragmatic trial) is aimed at the decision about which of two clinical decisions should be preferred. 1 An important difference between explanatory and pragmatic trials concerns the handling of interventions with multiple components. For example, an explanatory trial would compare statin with placebo, with lifestyle advice and periodic nurse consultation provided to all participants; the comparison in a pragmatic trial could be statin, lifestyle advice plus periodic nurse consultation compared with withholding all of these. The explanatory trial typically tests for biological effects, whereas the pragmatic trial tests the effects of different clinical decisions (each with its myriad of related actions). A typical characteristics of pragmatic trials is that the management of patients is left to the discretion of the clinicians, as was done, for example, in a trial of dyspepsia management. 2
Explanatory trials often select narrow ranges of patients based on age, sex, comorbidity and concomitant treatments, and then monitor patients carefully. In contrast, patients in routine clinical practice are more diverse, with varying disease histories and comedications and they do not always comply with instructions and persist with treatment over time. Selective cyclo-oxygenase 2 (COX-2) inhibitors provide an example of the challenges in generalising evidence from explanatory trials to routine clinical practice. The main explanatory trials of rofecoxib and celecoxib restricted study eligibility to patients with severe osteoarthritis or rheumatoid arthritis who were expected to use the study drug daily over the long term. 3,4 However, the large majority of patients using selective COX-2 inhibitors in routine clinical practice would not have been eligible for these explanatory trials as they did not have severe osteoarthritis or rheumatoid arthritis and did not use these medicines long term. 5 The compliance and adherence by patients to recommended dosage instructions often also vary substantially between explanatory trials and routine clinical practice. As an example, 89% of study participants were still using alendronate after 3 years in an explanatory trial,6 whereas in real life this number was only about 35%. 7
Ten key domains have been identified that describe the degree to which a trial is pragmatic or explanatory. These relate to the strictness of inclusion criteria for the trial, the extent to which clinicians have flexibility in how to manage the study interventions, range of expertise of the clinicians, study setting, intensity of follow-up procedures, extent of monitoring of compliance by patients and clinicians to the study protocol, and the types of statistical analyses. 8 Trials can also vary in the extent that data are being collected specifically for the trial. 9
Electronic point-of-care trials
Electronic information systems in health care are evolving and increasing, with primary care (general practice) in the forefront in the UK. Initial use of electronic health-care data mostly consisted of aggregate analyses of administrative data, such as hospital admission data. When clinicians started to use computers for record keeping, the first research databases collating anonymised electronic health records (EHRs) were created. One of the first of these was the Value Added Information Medical Products (VAMP) research database that started in 1987 and collated the EHRs of general practitioners (GPs) in the UK. This database eventually developed into the Clinical Practice Research Datalink (CPRD), formerly known as the General Practice Research Database. The richness and completeness of many EHR research databases have been increasing over time as more information is being shared electronically between different parts of the health-care system and as paper records are replaced by electronic ones. Laboratory data provide an example in which test results are increasingly being communicated electronically and loaded automatically into the EHR. An important development is the increased linkage between different health-care databases. In the UK, the EHRs of GPs, hospital admission records, death certificates and disease registries [including cancer and cardiovascular disease (CVD)] have now been linked, providing a more complete picture of the health-care provision. 10
This report focuses on electronic point-of-care trials, defined as pragmatic randomised trials conducted in usual clinical care conditions, with data collection mostly based on routinely collected electronic records. The study interventions compared fall within accepted professional standards but as yet have uncertain comparative effectiveness. Ideally, the recruitment and follow-up procedures would be naturalistic and mimic actual clinical decisions and practices (except for the random allocation of the treatments). Point-of-care trials may concern low-risk interventions but could also involve higher-risk interventions, such as intravenous corticosteroids for major head injury [as was studied in the Corticosteroid Randomisation After Significant Head Injury (CRASH) trial]. 11 Point-of-care trials could also be named large simple trials,12 practical clinical trials,13 testing through randomisation uncertain comparative effectiveness (TRUCE), randomised evaluations of accepted choices in treatment (REACT) trials,14 clinically integrated randomised trials15 or streamlined trials. 16
Electronic health records can facilitate the conduct of these trials as patients can be pre-identified and followed using routinely based collected data. Very few point-of-care trials have been conducted to date. Dutch researchers recently conducted a point-of-care trial using the Integrated Primary Care Information Database. This study found that it was possible to collect high-quality data in this setting but patient recruitment was poor because of multiple inclusion and exclusion criteria making eligibility assessment too time-consuming for routine GP appointments. 17–20 A feasibility trial is currently ongoing using the US Department of Veterans Affairs computerised patient record system. Clinicians are randomising patients with diabetes between sliding scale and weight-based insulin. The clinical end points of interest are based on the EHRs including episodes of suspected hypoglycaemia, length of hospital stay and rates of infection and renal injury. 21 The Standard Care Versus Celecoxib Outcome Trial (SCOT) is an example of an ongoing trial that uses EHRs for patient identification and follow-up but dedicated research staff rather than clinic staff to assess eligibility and recruit patients. 16 The second Leicester Intravenous Magnesium Intervention Trial (LIMIT-2) is an early example of a trial that used a disease registry linked to death certificate records. 22
Electronic health records have been used previously to supplement data collection in randomised trials. One example is the Scandinavian Simvastatin Survival Study (4S), which used death certificates and cancer registries for the end points. 23 The potential for using routinely collected EHRs for trials was highlighted 10 years ago by two studies. Although both studies reported substantial deficiencies in the routinely collected data, the potential of EHRs to support research was recognised. 9,24 The recent Thrombus Aspiration in ST-Elevation myocardial infarction in Scandinavia (TASTE) trial identified patients from the national comprehensive Swedish Coronary Angiography and Angioplasty Registry and evaluated all-cause mortality through national registries. 25
Potential consequences of lack of trial evidence
Delays in starting or not conducting trials can lead to variability in care and suboptimal care, possibly causing preventable disease and harm. One example is antibiotic use. These drugs have been in use for several decades and there are major concerns about overuse and the development of resistance. Despite this, it remains uncertain whether or not clinicians should prescribe these drugs to patients with mild to moderate chronic obstructive pulmonary disease (COPD) exacerbations. A recent study reported that the rate of antibiotic prescribing varied substantially between practices in patients with COPD exacerbations. 26
Another example concerns the lack of evidence of what type of statin would be most effective. The type of statin with which patients start treatment has changed substantially over time in the UK. In 1995, 82.2% of patients started on simvastatin, 11.7% on pravastatin and 6.2% on fluvastatin. After marketing approval of atorvastatin, the lowest use of simvastatin occurred in 2000: 35.7% simvastatin, 37.0% atorvastatin, 11.9% pravastatin, 10.7% cerivastatin and 4.8% fluvastatin. After introduction of cheaper generic versions of simvastatin, its use increased sharply. During 2008–11, over 95% of patients prescribed statins started with simvastatin. In 2012, the use of atorvastatin increased again after launch of generic versions. We estimated the effects of substituting simvastatin for atorvastatin using different hypothetical estimates of differential effects on CVD. As shown in Table 1 , atorvastatin substitution in 2012 would have prevented an additional 2456 CVD cases in the UK over the following 5 years (in the case that atorvastatin was 10% more effective).
| Assumption for relative rate of CVD with atorvastatin compared with simvastatin | Number of CVD cases prevented over the next 5 years |
|---|---|
| 0.60 | 9660 |
| 0.65 | 8472 |
| 0.70 | 7312 |
| 0.75 | 6075 |
| 0.80 | 4901 |
| 0.85 | 3671 |
| 0.90 | 2456 |
| 0.95 | 1239 |
Learning health-care system
There are significant challenges in the current health-care system, with large variation in the intensity of services, failure to deliver recommended services and a delay in the uptake of care innovations. 27 Also, the evidence base for many interventions is limited. A recent review found that the recommendations in cardiology guidelines were rarely based on evidence from multiple trials or meta-analyses (only 12% of recommendations). 28 A recent analysis by John Ioannidis found that only 1 of the 24 blockbuster medicines (with sales > US$1B) had been studied in a trial with more than 10,000 participants. Few of the randomised trials with blockbuster medicines included death as an outcome, so it is unknown whether or not these widely used medicines increase the risk of death as a side effect. 29 A systematic review of all drugs approved for marketing between 2000 and 2010 found that only 42% of the new drugs for chronic use were studied in more than 1000 patients followed for at least 1 year. It concluded that the evidence base for many long-term drugs is insufficient at the time of their approval. 30
The ‘learning health-care system’ has been proposed to improve the quality of health care with research embedded within clinical care. Such a system would continuously monitor for suboptimal care or uncertainty with routinely used interventions. Research is then done to test possible strategies to improve health care and its evidence base, followed by implementation. The aim of the learning health-care system is to generate and apply the best evidence for collaborative health-care choices of each patient and provider. 27 It follows a ‘test, learn, adapt’ methodology, which focuses on gaining a better understanding of what works and continually improving interventions. 31 EHRs and other electronic data could play an important role in this quality improvement system. 32
Aims of project
This research had the following aims:
-
to evaluate the feasibility of point-of-care trials
-
to identify the barriers and facilitators for clinicians and patients who wish to participate or not in point-of-care trials and to document the experiences of trial participants
-
to ascertain perceptions and attitudes among primary care staff concerning the process of computerised trial recruitment (CTR) and opinions about the software used to flag potentially eligible trial participants.
Table 2 lists the deliverables of this project, as agreed with the Trial Steering Committee. One deliverable was the development of a information technology (IT) system ‘piggybacked’ onto the clinical EHR system, which would facilitate the identification of potentially eligible trial patients, clinician’s notification during consultation of trial eligibility, confirmation of eligibility and randomisation on the study website, daily monitoring during the trial for side effects and long-term follow-up of major clinical outcomes. Other deliverables related to qualitative research on the perspectives of clinicians and patients and to the documentation of the experiences with the implementation of the exemplar trials. This report describes the implementation experiences with the two exemplar point-of-care trials. The analysis of follow-up information of the two trials is not included in this report.
| Topic | Deliverable |
|---|---|
| IT system for daily data transfer from practices, daily processing of patient eligibility and side effect monitoring | System works according to specifications and can be used for much larger trials |
| IT system for point-of-care recruitment using flagging software during consultation | System works according to specifications and can be used for much larger trials |
| Standard operating procedures to meet GCP guidelines | No major adverse findings in GCP audit by independent auditor |
| Conduct of two pilot trials |
|
| Research governance challenges | A review of the challenges, delays and lessons learned in obtaining approvals for the trials |
| Qualitative studies | Analysis of feedback from GPs and patients |
| VOI and health economic analyses | Completion of analyses |
| Development of scientific methods to conduct and analyse point-of-care trials | Reviews of rationale of point-of-care trials, fraud detection and GCP |
Chapter 2 Trial methodology used in this project
Electronic health record database
This project concerned the conduct of two feasibility trials Retropro and eLung. 14 This study used data from CPRD. CPRD comprises the computerised medical records maintained by GPs. GPs play a key role in the UK health-care system, as they are responsible for primary health care and specialist referrals. Patients are affiliated with a practice which centralises the medical information from the GPs, specialist referrals and hospitalisations. The data recorded in the CPRD include demographic information, prescription details, clinical events, preventative care provided, specialist referrals, hospital admissions and their major outcomes. Information on the 459 practices that were contributing EHRs to CPRD at the time of the project was used. The primary care data of CPRD have been linked to hospital admission records (Hospital Episode Statistics database), disease registries (including the Myocardial Ischaemia National Audit Project register with data on patients admitted to a hospital with a myocardial infarction) and the Office for National Statistics mortality register (with death certificate data).
Design of exemplar trials
Retropro
Retropro was a pragmatic point-of-care trial funded by the Wellcome Trust, randomising patients with high CVD risk to simvastatin or atorvastatin. Inclusion criteria included age over 40 years, a ≥ 20% or 10-year risk of developing CVD, primary hypercholesterolaemia and consent to participation. The primary objective was to evaluate the feasibility of conducting point-of-care trials using routinely collected data. The secondary objective was to measure laboratory and clinical outcomes in patients prescribed simvastatin or atorvastatin. CPRD was used to identify patients aged ≥ 40 years who had been registered for over 6 months with a practice. Patients with prior CVD or prescription of statins or recent history of liver disease or pregnancy were excluded. The CVD risks as predicted by QRISK®2 and Framingham risk score were then estimated. 33,34 The Framingham risk score uses an algorithm based on age, sex, ethnicity, smoking status, systolic blood pressure, ratio of cholesterol to high-density lipoprotein and history of diabetes mellitus and left ventricular hypertrophy. The QRISK2 score uses information on age, sex, ethnicity, Townsend score for socioeconomic status, body mass index, smoking status, systolic blood pressure, ratio of cholesterol to high-density lipoprotein, family CVD history, history of diabetes mellitus, treated hypertension, rheumatoid arthritis, chronic renal disease and atrial fibrillation. The Framingham risk scores were based on the publicly available algorithm. The Framingham predicted risks were adjusted by 1.4 for South Asians as recommended by the National Institute for Health and Care Excellence (NICE). 35 QRISK2 risk predictions were calculated using the commercial software program provided by CLINRISK Limited, using the 2012 version. Missing values for smoking status, systolic blood pressure and lipid levels and body mass index were imputed. 36 The statin daily dose was determined by the GPs based on clinical need or contraindications. End points of interest included repeat statin prescribing, death and incident CVD (as recorded in the EHRs) and the collection of a blood sample for genetic analysis. The blood sample was to be taken at the same time as the routine liver function test as recommended 3 months after starting a statin. The study team determined the trial topic in Retropro. This was based on the consideration that statins are widely used and on an assessment of NICE recommending research in the comparative effectiveness of different types of statins. 37 The target number for recruitment was 300 patients with, originally, a planned start date of August 2010 and end date of December 2012.
eLung
eLung was funded by the UK National Institute for Health Research (NIHR) Health Technology Assessment programme. It included patients aged ≥ 40 years with a medical history of COPD who, in the opinion of their GP, had an acute exacerbation of COPD with an increase of non-purulent sputum volume, who did not require immediate referral to specialist care for treatment of COPD exacerbation and consented to participation. Patients were randomised between immediate (prophylactic) versus deferred or non-use of antibiotics. The primary objective was to evaluate the feasibility of conducting randomised trials that use routinely collected data for follow-up. The secondary objective was to measure clinical and quality-of-life outcomes and forced expiratory volume in 1 second (FEV1). CPRD was used to identify patients aged ≥ 40 years who had been registered for over 6 months with a practice and had a history of COPD [as defined by the Quality and Outcomes Framework (QOF) diagnostic Read codes38]. Patients were recruited during a consultation with their GP. Prescription of an antibiotic during the previous 2 weeks was an exclusion criterion (the protocol was amended from a period of 3 months). The choice of antibiotic was left to the GP, but they were advised to prescribe whichever antibiotic they would usually use as first line for acute bronchitis in a patient with similar characteristics. The rationale for not stipulating one particular antibiotic is that prescribing of antibiotic type shows marked regional and temporal variation. End points of interest included hospital admission for COPD exacerbation and prescribing of oral corticosteroids (as recorded in the EHRs). The original plan was for the GP to provide the participant with an electronic device with a daily measurement over 4 weeks of disease-specific symptoms and FEV1 (eDiary; Vitalograph In2itive eDiary, 2012 version). During the conduct of the trial, participants were also offered the opportunity to complete by paper two quality-of-life questionnaires. These questionnaires were administered at baseline and at 4 weeks; both included the European Quality of Life-5 Dimensions-3 Level (EQ-5D-3L) and COPD Assessment Test questionnaires. The EQ-5D-3L provides a generic measure of health status over five domains (mobility, self-care, usual activities, pain/discomfort and anxiety/depression) with three levels of response (no problems, moderate problems or severe problems). The 4-week follow-up questionnaire also included a brief section regarding adverse events and any productivity loss in the previous 4 weeks. The funder of eLung had determined the trial topic of evaluating the effectiveness of antibiotics in COPD exacerbations and of measuring both clinical- and patient-reported end points. The target number for recruitment was 150 patients with, originally, a planned start date of August 2010 and end date of December 2012 (covering two winter seasons).
Overall design
In both trials, the GPs were expected to provide the general health care to the trial participants following their normal practice. The treatment allocation schedule was blinded to patients and clinicians, but they knew which treatment the patient was randomised to. The GPs had to confirm all inclusion and exclusion criteria and the consent of patients on the study website before randomisation.
Trial initiation procedures
All practices in England and Scotland submitting data to CPRD were sent a letter about the two studies, including a synopsis of the study and a reply slip for sites to use to express their interest or decline participation. Practices that returned positive responses were then sent the protocol, a synopsis of the main steps required to set-up and conduct the study, and a site contract. Sites that did not reply initially were sent letter and e-mail reminders and also received follow-up telephone calls.
European Union Drug Regulating Authorities Clinical Trials (EudraCT) numbers were obtained for both studies and applications were submitted to the Medicines and Healthcare products Regulatory Agency (MHRA) for regulatory approval and to the National Information Governance Board for governance review. Applications for both studies were simultaneously submitted to the London South East Research Ethics Committee for ethics review. Following this, overarching research and development (R&D) applications were submitted to the lead R&D organisation (Wandsworth NHS Trust) to meet governance requirements, and subsequently to the local R&D department for each site. Site-specific information forms were completed by the Trial Management Team for each site and sent to the relevant GPs for review and signature. GPs were requested to supply a signed and dated curriculum vitae and evidence of Good Clinical Practice (GCP) and protocol training. Bespoke training courses in GCP and protocol, monitored by Brookwood Academy, Surrey, UK were provided online. The GCP module provided an overview of those elements of GCP pertaining to these studies. The protocol modules for Retropro and eLung instructed the GP to read the protocol, but also highlighted the study design, clinical responsibilities, assessments and data to collect. GP responsibilities included the review of patient eligibility, appropriate informed consent, collection of blood samples (in Retropro), provision of usual health care and the recording of major clinical outcomes and suspected side effects. The patient opt-out process (for patients refusing further data collection) was also explained. The average duration of each module was 15 minutes followed by 10 multiple-choice questions to ensure comprehension of responsibilities. For scores of 8 or more out of 10, a certificate of successful completion was issued to the participant and copied to the Trial Master File. The lead GP at each practice was required to successfully complete this GCP training; other GPs at a practice were allowed to recruit after the lead GP had explained the trial and had registered with the study team. On local NHS organisation approval for each site, an internal process was triggered to check completion of all governance requirements followed by activation of the site, allowing access to the study website and activation of the flagging software.
Patients were recruited into the trial and randomly assigned once the GP had confirmed eligibility for the trial and had obtained the patient’s consent. Randomisation was balanced by centre, with an allocation sequence based on a block size of eight, generated with a computer random number generator. Research staff did not visit the sites to facilitate recruitment (with the exception that a few sites that were helped with loading and testing the flagging software).
Monitoring of side effects
The monitoring of side effects was done using three different methods:
-
The EHR research database was updated monthly and used to compare rates of selected outcomes (such as liver failure) in trial and non-trial patients.
-
Clinicians were requested to record suspected adverse drug reactions (ADRs) in the dedicated side effect screen of the EHR, which were transferred expeditiously and extracted and reviewed by the principal investigator.
-
Suspected ADRs also required completion of forms on the trial website.
Qualitative studies
Two qualitative studies were conducted as part of this project. The first study, conducted prior to the start of the two trials, was done in order to ascertain perceptions and attitudes among primary care staff concerning the process of CTR and opinions about the software used to flag potentially eligible trial participants. Further details on this study are provided in Chapter 4 . The second qualitative study was conducted in order to identify the perspectives of patients and GPs on the reasons for participating in eLung or not and to document the actual experiences of trial participants (see Chapter 8 ).
Trial steering and data monitoring committees and funding
A Trial Steering Committee was appointed to provide oversight for Retropro and eLung. Its role included reviewing the practical aspects of the trials, the barriers and conduct of trials, so that these were run informatively and safely. The Committee included two patient representatives who had replied to an advertisement and were appointed after successful interview. The Data Monitoring Committee reviewed the data management and monitoring procedures, recruitment figures, extent of protocol deviations and safety data. Charters for the committees were agreed by both committees before patient recruitment started ( Appendix 1 provides the charters).
eLung was funded by the UK NIHR Health Technology Assessment programme and Retropro by the Wellcome Trust. The funders of the study had no role in study design, data collection, analysis, interpretation or writing of the report. The Writing Committee had full access to all data in the study and had final responsibility for the decision to submit for publication.
Chapter 3 Good Clinical Practice guidelines and point-of-care trials
Scope of review
The key guideline for the conduct of randomised trials is GCP, an international quality standard developed by the International Conference on Harmonisation. GCP must be observed for designing, conducting, recording and reporting clinical trials of drugs. 39 The CPRD database was, at the time of the project, managed by the MHRA. MHRA’s management requested that their GCP inspectors reviewed the proposed procedures for the two exemplar trials prior to the start of the trials in order to assess GCP compliance for the two exemplar trials. The terms of reference for this review stated:
This exercise would identify risk factors and gaps at various stages of the CPRD randomised clinical trial [RCT] process; this should include trial design, protocol, safety reporting process, site selection process, regulatory and ethics approval application and site specific assessment, process to prevent patient recruitment on site level before patient recruitment, consent process, records keeping and documentation, data management processes, monitoring, data quality assurance, interfaces with third parties, trial conclusion, data handling and manipulation, statistical analysis and reporting. The risk assessment exercise will identify the risk factors, their impacts and proposed ways to minimise the risks.
The study team was requested to provide:
-
Exemplar protocol: the protocol should include trial design, inclusion and exclusion criteria, screening, trial conduct (including sampling) visit schedule, data collection points, safety data collection and reporting procedures, monitoring plan
-
GP site selection and assessment of suitability including site-specific sssessment and training of site personnel
-
Consenting procedure and training
-
Regulatory and ethics approval application process
-
Data management plan
-
Charters for the following committees:
-
-
Independent Scientific Advisory Committee [committee that reviews all protocols for CPRD studies]
-
Data Monitoring Committee
-
GP Advisory Committee.
-
Following this request, the study team provided a 47-page document outlining the trial system and procedures. A series of meetings between the principal investigators and GCP inspectors were held to discuss these.
Results
The following sections describe key points of discussions between the study team and GCP inspectors. Key comments by the GCP inspectors and the responses by the study team are provided ( Table 3 provides a summary of these).
| Area | GCP issue | Response |
|---|---|---|
| Training of clinicians | Inappropriate delegation to other staff with no training | Key trial activities (such as randomisation) require registration |
| Consent process | Requirement for ample time and continual consent for long-term follow-up | No need to redeem prescription; anonymous follow-up |
| Site visits | Central monitoring insufficient with large number of sites | Comparison with non-sites; site visit in case of suspected fraud |
| Data integrity | No control of data quality at sites or rest of NHS | Prospective, randomised, open, blinded, end point design40 |
| Computer systems validation | No control over third-party software, data quality, interfaces and validation of database; third parties not legally involved | Use of linked data from different sources |
| Verification of source data | Lack of source document as data directly entered into trial database; impossible to reconstruct trial data | Paper records are being replaced by EHRs |
| Suspected side effects | Requirement for urgent notification | Reporting requirements should be similar to usual practice |
| Loss to follow-up | Need to define contingency for patients who transfer out | Use of linked data from different sources |
| Drug accountability | Not possible to monitor compliance | Follows routine process in usual care |
| Fraud detection | Central monitoring not sensitive to identify data irregularities | No evidence for improved sensitivity of site visits; EHR databases provides rich source for comparisons |
Training of clinicians
Good Clinical Practice requires that every clinician involved in recruiting participants into a trial shall be qualified by education, training and experience to perform his or her tasks. The GCP inspectors commented:
Compliance of GPs may be an issue. Since there is an incentive for the GP to enrol subjects, then a busy GP investigator may not adhere to study procedure. Inappropriate delegation to other member of staff with no protocol or GCP training may lead to protocol and GCP non-compliance.
The study team addressed this by highlighting that the GP EHR systems are password controlled with a record of who entered the information. With respect to the key activities specifically done for point-of-care trials (i.e. patient recruitment and randomisation), these can be done only by GPs approved by the research team. In Retropro and eLung, the lead local investigator in each practice will need to complete web-based training on the protocol and relevant aspects of GCP. Other study-specific data may be collected by other staff members (e.g. nurses) on the web-based secure clinical trial management system (which is password controlled).
Consent process
Point-of-care trials can use different recruitment strategies including recruitment during an unscheduled consultation. The GCP inspectors commented that recruitment at the same consultation as the screening may be less acceptable as ‘Subjects should be given ample time to understand the implications of the study’. GCP inspectors also commented, ‘Continual consent may be required especially when the duration of follow-up is prolonged . . . subjects may have forgotten they are involved in a clinical trial’. In point-of-care trials, participants could ‘opt out’ of the trial by not redeeming the prescription and can return for a consultation. In order to address the concern of continual consent, the trial follow-up was divided into periods with active and passive follow-up. In the latter period, patients were followed only using the routinely collected anonymous medical data and this is considered an observational study. This approach is similar to that of the long-term follow-up of the West of Scotland Coronary Prevention Study. 41
Monitoring of study sites
Good Clinical Practice guidelines state that site visits should routinely be conducted (in order to initiate the site and collect data) and that central monitoring should be used only in exceptional circumstances. 42 The study team proposed the use of central monitoring without site visits. The GCP inspectors stated, ‘Considering a large number of sites are involved, central monitoring is insufficient to ensure GCP compliance at site level’. The study team responded:
Central monitoring may have superior power in detecting abnormalities (or serious GCP non-compliance) compared with site visits for studies that evaluate major clinical outcomes. A central assessment would be more efficient in picking up the poor quality as all the GPs data (outside the RCT) would be compared with that of others and as central monitoring would use data before and after the trial. A site visit could be useful for proxy outcomes measures that are measured in all patients, but it may be less efficient for outcomes that occur in only few patients.
The study team proposed that the trial populations would be monitored for differences with other patients in the EHR database. Also, it was agreed that a site visit could occur in the event of suspected fraud.
Data integrity
The trial data in the point-of-care trials consist of routinely collected data. The GCP inspectors stated:
There is no control on data quality either at the GP surgery or from third parties. These include incomplete data, entry error and omission. In addition, since data are not captured in a case report form, unstructured data are recorded by third parties.
The study team proposed to utilise the prospective, randomised, open, blinded, end point design. In this design, clinicians and patients are aware of the treatment allocation, but independent clinicians blinded to the randomised treatment adjudicate each end point of interest. 40 In case of any questions or discrepancies between the various data sources, a case validation approach could be used requesting the GP investigator to confirm the outcome.
Computer system validation
The trial data in point-of-care trials rely on electronic information systems. The GCP inspectors stated, ‘CPRD has no control on third parties software, data quality, interfaces and validation of databases. There is an inherent doubt on the quality of the data which cannot be qualified or quantified’. Furthermore, it was considered, ‘Third-party data providers are not legally involved in the study. The investigator should have formal agreements with these organisations to allow access by auditor when it is necessary.’ In conventional trials, GCP auditors typically have access to documentation outlining the testing of the information systems which often are developed specifically for the conduct of trials. In point-of-care trials, trial data are obtained from several computer systems, including the GP EHR systems, various hospital information systems that feed data into the national hospital registry and death certificate data. Numerous parties are involved in the recording and processing of these data, which are subject to detailed quality control by the NHS. It was agreed that the study team would seek and collect documentation on the main information systems used.
Verification of source data
The GCP inspectors commented, ‘There is a lack of source documents; data are directly entered in the database. It is not possible for an auditor to reconstruct reported data against source data’. In conventional trials, GCP typically compares the information in paper medical records (i.e. source data) with data in the trial database. The study team disagreed with the notion that electronic data do not constitute source data and that such data cannot be audited. Within several years, it is likely that most medical data will be collected electronically and it would be inefficient to maintain a system that would be based on transcription of electronic data to paper and then back from paper to electronic data. The GP EHR systems incorporate a full audit history of every record including time stamps.
Suspected side effects
Many countries require serious unexpected suspected adverse drug reactions (SUSARs) to be reported to the regulatory authority within 2 weeks of notification to the site. The original proposal consisted of requesting the GPs to expeditiously document any suspected ADR in the side effect fields of the EHR. The study team would review these events for reportability following a weekly electronic transfer of the EHR to the central research database. The GCP inspectors responded, ‘Weekly data extraction from sites to CPRD database might not meet the requirement of SI 2004 1031 Reg 33 that SUSARs are reported no later than 7 days after the sponsor was first aware of the reaction’. Following this discussion, two changes were made, at substantial cost, to the IT system. The first one concerned a change to daily transfer of the EHRs to the central database. The second change consisted of requesting GPs to also complete an ADR form on the study website.
Another concern of the GCP inspectors was, ‘The proposed protocol fails to meet the requirement to inform concerned investigators of any SUSARs which occurs in relation to the investigational product’. The study team tried to argue that the information provision should be similar to patients and clinicians inside or outside a point-of-care trial. Furthermore, SUSARs may include events that have already been reported to the regulatory authorities but did not warrant inclusion into the drug’s summary of product characteristics (e.g. their databases of reported suspected ADRs include many unlabelled events). The study team then proposed to report to regulatory authorities (and GP investigators) only SUSARs that were not listed in the summary of product characteristics and with less than five reports in the Yellow Card database. The GCP inspectors responded, ‘It is unacceptable to use such an imprecise definition of expectedness . . . it does not take into account of the population exposure . . . and there is no process to determine whether the Yellow Card submissions are relevant or not’. Following this discussion, the summary of product characteristics was being used to assess expectedness and procedures were implemented in order to report SUSARs to the regulatory authority, local NHS organisations and GP investigators.
Loss to follow-up
Study participants may leave the practice and be lost to follow-up. The point-of-care trials do not impose any restrictions on the use of the study medication if a patient leaves the practice. The GCP inspectors commented, ‘Some contingency should be defined for patients transferred from CPRD centres to non-CPRD centres. It is unclear how patient’s ADRs are captured in this situation’. Loss to follow-up occurs in all long-term trials. The ability to link various health-care data sets will ensure that outcomes that lead to hospitalisation or death can be captured for trial participants, even after the participant has left the practice (unless the patient has emigrated).
Drug accountability
Drug accountability allows the reconstruction of the trial and documents what medication was received by the site and what medication was received by the patient. 42 In point-of-care trials, the drugs will be provided by any UK pharmacy based on the routine process for dispensing licensed medicines following a prescription by a clinician. The GCP inspectors considered, ‘The lack of drug accountability may not be a problem, but it is not possible to monitor compliance’. The study team responded by stating that prescription records do allow an assessment of compliance (for long-term therapies) by estimating the medication-to-possession ratio. This ratio is the number of days covered by the prescribed therapy divided by the total period of follow-up. This is routinely done in studies that use health-care records. However, this method does not measure whether or not the medication was actually taken.
Fraud detection
One part of fraud prevention in point-of-care trials will be to statistically monitor for unusual data patterns. Currently, site monitoring is seen as the most important approach to fraud detection. The GCP inspectors stated, ‘The statistical method for detecting fraud needs to be validated to ensure the methodology is sufficiently sensitive to identify data irregularities’. The study team replied that there is indeed no evidence to substantiate the sensitivity of statistical monitoring for fraud detection, but such evidence is also lacking for the effectiveness of site visits. Site visits may not be effective in detecting fraud for studies that measure only major clinical outcomes as most sites will not have any records of the end point of interest. Furthermore, trials within existing EHR research databases may minimise the risks of fraud as each GP is not able to recruit ‘fake’ patients (only patients permanently registered for at least 6 months will be eligible), and as the data for the participants prior to and after the trial can be used for fraud detection. The open access of the EHRs to other health-care professionals in the same practice may also minimise false data entry.
Conclusions of the Good Clinical Practice review
The general concluding comments of the GCP review were:
The regulatory environment has moved on . . . The Risk Adaptive model, launched in April 2011, has proposed a reduction in the level of monitoring and pharmacovigilance requirements for the lower-risk studies. This model is well suited for the pragmatic randomised trial proposed by the CPRD . . . It is recommended that CPRD should risk assess its trials and categorises them based on their risk and included in the Clinical Trial Authorisation Applications. Once approved by the Clinical Trial Unit, Category A studies would be deemed as lower risk.
Discussion
Good Clinical Practice was originally developed for the pharmaceutical industry to provide an unified standard for trials that ‘Are intended to be submitted to regulatory authorities’ in order to ‘Facilitate the mutual acceptance of clinical data’. 43 It is unclear why point-of-care trials not intended for regulatory submission also need to comply with GCP. The fundamental question is why point-of-care trials are viewed as an activity that requires elaborate governance procedures rather than as quality improvement that is an intrinsic part of routine clinical care. Clinicians who embark on collective guesswork do not face any paperwork, even it leads to huge variability in care, in contrast to those who want to resolve guesswork. 44,45
Good Clinical Practice imposed a significant burden on the clinicians in our point-of-care trials ( Table 4 ). Rather than providing the relevant treatment guideline, clinicians had to read a lengthy protocol with content more relevant to higher-risk trials. Also, they were required to submit various forms and complete protocol and GCP training even if they had already prescribed (e.g. statins to hundreds of patients). The approvals for the trials required adherence to numerous conditions, including frequent audits. Suspected side effects needed to be reported urgently, not only in the EHR but also on the study website, despite the fact that, for example, statins have already been used by millions of patients. One could argue that the key standards for clinicians in point-of-care trials should be whether or not they obtain informed consent and apply diligence in the eligibility assessment and monitoring of patients. The General Medical Council’s Good Medical Practice guidelines clearly outline the expectations to which clinicians need to adhere. It is unclear why these guidelines are not considered sufficient for clinicians in point-of-care trials.
| Area | Barrier | Proposed risk-proportionate approach by study team for point-of-care trials |
|---|---|---|
| Protocol | Lengthy document (eLung: 15 pages plus several attachments) as format and content need to follow ICH guideline | Short protocol restricted to summary of relevant treatment guidelines and outline of study procedures (i.e. how to initiate recruitment) |
| Local site contract | Legally binding contract between clinician and sponsor (three pages for each trial) | Outline of responsibilities of principal investigator provided to clinician. Clinical care remains responsibility of clinician, any deviations addressed through Good Medical Practice procedures |
| Training of investigators | Requirement for lead clinicians to undertake GCP and protocol training (taking about 45 minutes); proof of completed training explicit requirement for local site approval | No protocol training unless clinician has never/rarely used study medication |
| Informed consent form | Information disclosure in trial much more detailed and onerous compared with that outside trial | Evidence-based consent form based on patient preferences rather than expert/legal opinion |
| Informed consent procedures | Consent during the consultation when patient ill (eLung) | Patients in practice are asked for consent some time before randomisation |
| Local site approval procedures by local NHS organisations | GP to provide curriculum vitae, complete and sign forms, and to name all staff involved in trial. Some NHS organisations also required the curriculum vitae of the nurse taking a routine blood sample | No need for local site approval; any deviations addressed through Good Medical Practice procedures |
| Conditions following local site approval | GP expected to adhere to a series of conditions set by local NHS organisation including patient contact restrictions, informed consent form archiving, data protection, all local health and safety regulations, reporting of adverse events or suspected misconduct, regular project updates, duty to inform about publication, notification of amendment to ethics approval, monthly and annual progress report, monthly recording of recruitment and provision of record access for audit (10% of studies selected for audit) | No need for local site approval; any deviations addressed through Good Medical Practice procedures |
| Study inclusion criteria | Retropro inclusion restricted to high cholesterol levels (licensed indication of statins); NHS guidelines focus on screening for high CVD risk (rather than cholesterol) | Inclusion criteria concern authorised medicinal products, used in accordance with the terms of the marketing authorisation or their use is a standard treatment |
| Reporting of suspected side effects | Clinicians need to enter side effects not only in the EHR but also urgently on the study website | Periodic review analysis of side effects as recorded in the review of EHRs; comparison of rates in trial and non-trial patients only |
| Communication of SUSARs | Urgent notification of SUSARs to regulatory authorities, ethics committee, local investigators and local NHS organisations | Periodic communication of SUSARs and safety analyses to regulatory authorities; communication to clinicians and others restricted to substantive changes in the summary of product characteristics |
The current trial regulations have been criticised by several authors. 44–49 Even the European Commission has criticised its own legislation by stating, ‘It appeared to have hampered the conduct of clinical trials’. 50 Yusuf et al. 46 stated, ‘There is no good evidence that the layers of complexity, approvals, processes and laws to protect subjects entering RCTs have actually achieved their purpose’ and asked, ‘What is the basis for asserting that the procedures enshrined in these GCPs are indeed good, clinically relevant, or even practical?’46 The compromises we had to make in order to get the two trials going clearly undermined the concept of ‘simple’ trials.
Regulatory authorities have recently introduced the concept of risk proportionality in trial governance. 50,51 The 2012 proposed revision of the European Trial Directive includes the definition of ‘low-intervention clinical trial’ which would apply to point-of-care trials. 50 However, the level of risk stratification and simplification appears limited in the proposed revision; the text around ‘low-intervention clinical trials’ appears open to a range of interpretations and the implications optional. The regulatory approach to risk proportionality51 is useful but addresses only 1 of the 10 barriers experienced by clinicians in Retropro and eLung, namely the inclusion criteria for Retropro (see Table 4 ).
The core objectives of GCP are to ensure that trials safeguard the rights, well-being and safety of participants and that the results are credible. 39 We indeed should not compromise on these principles. However, we need to develop a better evidence base for research governance of what improves the quality and what hinders the conduct of trials. A culture of quality improvement can be achieved only if our best method for achieving this, randomised trials, is considered part of clinical care and routinely done by all clinicians rather than by a small minority of clinicians. The General Medical Council’s Good Medical Practice guidelines should be the standard for clinicians in point-of-care trials with trial regulations and approval procedures restricted to higher-risk trials.
Chapter 4 Qualitative research of views on computerised trial recruitment for the information technology system design
Introduction
Systematic reviews have attempted to identify potential physician and patient factors that can affect recruitment of trial participants. 52,53 This is an area where qualitative research may prove beneficial, in providing insight into areas that are complex to study. 54 There has been limited qualitative research to ascertain the attitudes of primary care clinicians towards conducting trials and, without increasing understanding in this area, the success of future trials in primary care is uncertain. 55
If computer flagging is to be a successful trial recruitment tool, primary care clinicians must be amenable to its use within consultations. Thus, it is important to understand the attitudes of potential users prior to incorporating flagging software into the EHR system. 56 Two studies have attempted to investigate clinicians’ attitudes towards CTR tools. 57,58 However, the scope and breadth of the analyses in both studies was limited with the use of only closed questions58 and a focus on ease of use. 57 Neither study investigated attitudes towards recruitment generally. Evidence is lacking on the clinicians’ perceptions to CTR. Our study aimed to elicit the attitudes of both GPs and practice nurses. There are only a few studies that looked at nurses’ attitudes towards recruiting for trials. 55,59 The nurses’ attitudes towards CTR could be important given the increasing proportion of nurse-led primary care patient encounters,59 and that nurses may be a more cost-effective means of recruiting. 60
Methods
A qualitative study was initiated at the time of development of the flagging software (prior to the start of the trials) and conducted by King’s College London. As described in Chapter 5 , this software was to be used for notifying clinicians of eligible trial patients during consultation [this software was called Local Eligible Patient Identification Service (LEPIS)]. The primary aim of this qualitative study was to ascertain perceptions of and attitudes towards recruiting patients to trials and use of CTR tools by primary care clinicians (both GPs and nurses). The secondary aim was to gauge opinions on the use of the LEPIS prototype. A purposive non-probability sample11 of GPs and primary care nurses was sought. Academic GPs within the department of Primary Care and Public Health Sciences, King’s College London, had links with a number of general practices in east and south London. Participants were initially approached by e-mail. Those who expressed interest in participating were provided with information about the study and a consent form. Given time and resource constraints, a sample of 8–10 GPs and five practice nurses was sought.
A questionnaire was completed by all participants to collect demographic data. A semistructured interview technique12 was utilised, allowing the interview to be guided towards the aims and objectives but also to maintain flexibility to reveal emergent themes. 13 An interview schedule informed by literature review was devised ( Appendix 2 provides the topic guide). 14 Open-ended questions were used to avoid leading participants towards predetermined answers. All participants who agreed to participate underwent a one-to-one, face-to-face semistructured interview at their place of work. Interviews were conducted by one researcher between July and August 2011. Semistructured interviews were digitally recorded and transcribed verbatim and written field notes were kept in case problems with the recording emerged. The first part of the interviews incorporated questions pertaining to attitudes surrounding recruitment to trials and the second part focused on attitudes towards the use of CTR tools. The demonstration of the LEPIS prototype involved asking participants to read through the LEPIS Process Map and guiding them through the interface on the researcher’s laptop. Unfortunately, the prototype was still in development and, thus, participants were unable to fully interact with the interface.
Data were analysed using the framework approach in which recurrent themes are identified and charted within a matrix which is used for data interpretation. 15 Framework analysis was developed as a means of analysing qualitative data for applied research,11,12,16–18 which attempts to produce outcomes that are actionable17 within relatively short time frames. 16
Results
Semistructured interviews were conducted with 13 primary care clinicians: nine GPs and four practice nurses. Table 5 lists participant characteristics. All participants were recruited from the south and/or east of London.
| Characteristic | n | Mean age in years (range) | Men, n | Women, n | Partner, n | Salaried, n |
|---|---|---|---|---|---|---|
| GPs | ||||||
| Academica | 3 | 2 | 1 | 2 | 1 | |
| Non-academic | 6 | 3 | 3 | 3 | 3 | |
| Total GPs | 9 | 40.2 (29–55) | 5 | 4 | 5 | 4 |
| Practice nurses | ||||||
| Previous research experienceb | 2 | 0 | 2 | N/A | N/A | |
| No previous research experienceb | 1 | 0 | 1 | N/A | N/A | |
| Total practice nurses | 4 | 41.7c (34–49) | 0 | 4 | N/A | N/A |
Barriers
Time was perceived to be the biggest barrier to recruiting patients into trials. Time seemed to be a universal barrier in that all 13 participants mentioned time at least once:
I think probably the main challenge is time.
AGP-1
I’m someone who finds it hard ever to run to ten minutes . . . even just covering what I’m trying to do now . . . my average is 13 or 14, if I had another five minutes, that just wouldn’t work over the course of a three hour surgery, I’d be three hours late.
NAGP-5
Some participants discussed ways in which the barrier of time could be overcome. Examples included the establishment of dedicated research clinics for recruitment, additional blank appointments to allow for delays, protected research time, and/or lengthening consultations. Others felt that clinicians could merely refer potentially eligible patients to researchers for recruitment, thereby minimising the impact of time on the clinician. In addition, a number of GPs talked about how money could be used to ‘create’ more time:
It might be that the money is used to increase time for the other team members who are involved. Like if they use the money to have extra appointments or blank slots . . . that’s fine . . . So I think it’s a practical thing because it’s whether the money can be used to make the thing happen.
AGP-1
Yeah, but that’s still, when I mean committing more time, that’s still practice time . . . and . . . creating extra appointments purely for people to come back and discuss taking part in research feels like quite a generous use of time. There might need to be some money involved . . . You know, people’s time is money, sadly.
NAGP-2
Another GP thought financial incentives would compensate only to a limited degree as, ultimately, time is of short supply. This implies that, regardless of financial rewards, the time taken to recruit patients still needs to be minimised.
Anything that takes longer than 30 seconds . . . If this would be just as long as choose and book, I’m sure GP’s would say no I’m not bothered. Because it just takes too long. Even if the financial incentive would be there.
NAGP-1
Interestingly, although all four nurses discussed time as a significant barrier, none mentioned using financial incentives to overcome time. Instead the nurses appeared to focus on minimising the impact of time by recruiting patients during dedicated clinics.
Six participants alluded to the fact they may be liable to select patients. Not offering the trial equitably to all potentially eligible patients could introduce selection bias and limit the trial’s external validity:
I think in a GP setting I would worry that I wasn’t being completely random.
NAGP-4
It was commonly reported that a non-random selection of patients was a way of protecting potentially vulnerable patients who, for example, lacked understanding or for whom it was thought a trial would be too burdensome due to pre-existing comorbidities.
The second biggest reported barrier after time was GP concerns about patients not understanding the information:
I suppose it’s whether they can understand, you know, how many of them will be able to truly understand and, you know, give informed consent to a trial . . . That’s quite an issue. And I think of the patients I see, I think some of them it will be quite difficult to explain a trial to them and to understand what it really meant . . . Just lots of, you know, language, literacy skills, you know, a whole host of things.
AGP-2
Some GPs felt that patients’ cultural beliefs may prevent them from participating in a trial:
I mean particularly in an area like this, you’ve got language, cultural . . . issues as well to think about. You know, what’s acceptable and what isn’t.
PN-2
As previously stated, these barriers (culture, understanding and language) may be compounded by selection bias:
There are big problems with language in inner-city general practice. And again, that’s going to deter you because you know any research study’s going to be a battery of questionnaires, you’ve got to be pretty literate to go through most of those questionnaires, at least certainly in the mental health studies. And then who do you recruit to your studies? Surprise, surprise, it’s the middle class, literate people.
AGP-3
The two biggest barriers (time and understanding) were also found to interact in that, for some participants, insufficient time meant that they did not feel they would be able to adequately explain a trial to patients who had difficulty understanding:
We should have sufficient time to create that level of awareness and understanding.
PN-1
Facilitators
Clinicians reported being more inclined to recruit patients if they perceived either a potential benefit to their own patients or to the wider population:
I think people might find it difficult to ask people do they want to be the subject of research. Unless it’s offering them a benefit.
AGP-2
Well we’re sort of doing research perhaps a bit for our own interest, perhaps a bit for our career promotion, but actually it’s because of the greater good for mankind.
AGP-3
The importance of perceiving a benefit also highlighted issues of clinical uncertainty; that there is no known difference in effectiveness between trial treatment interventions. Three GPs did demonstrate an understanding that uncertainty had to exist for a trial to be deemed ethical. However, despite this, some participants described feeling uneasy:
If it was a randomised trial and some people might not get treatment . . . I think that would be quite difficult . . . I think it would conflict with your clinical role of doing the best you could for that particular patient.
AGP-2
Many organisational factors thought to facilitate recruitment were concerned with separating research and clinical roles. These included establishing dedicated research recruitment clinics; employing someone purely to recruit; and having a separate research team solely responsible for recruiting. Other organisational factors that could facilitate recruitment were concerned with making the process easier and ensuring adequate support systems were in place. Some felt that it was important that advertising or promotion of a trial was conducted in order to facilitate recruitment.
Modifying factors
Nine participants talked about seeking informed consent from patients for trial participation. Interestingly, there appeared to be a difference between GPs and nurses. Specifically, most GPs felt that consent would be too difficult to obtain within a consultation and/or they were unwilling to obtain consent from patients:
No, no, no, no, no. That should be done by the study group. In fact the whole consenting and giving more information should be done by the research team.
NAGP-1
Reasons for concern included a lack of research experience, feeling it would be unethical to ask patients for an immediate decision, and insufficient time. Two out of four nurses discussed consent and neither thought that this was an issue, with one stating it should be easy. The nurses appeared to liken it to taking consent in other routine clinical situations. GPs seek consent from patients routinely within their clinical practice but yet felt there was something different about taking informed consent for trial participation which seemed not just due to insufficient time. The unwillingness to seek consent was linked with a feeling that it was something the researchers, and not clinicians, should be doing.
Financial constraints were mentioned by eight participants. Some GPs felt that financial incentives for clinicians and/or practices would be important in facilitating recruitment. Some thought that financial reimbursement would be required to offset additional costs, and may be an important motivator, or at the very least mitigate logistical problems. Interestingly, only one nurse mentioned financial incentives as a facilitator of recruitment.
Ten participants felt that forgetting could impede recruitment and/or reminders could facilitate recruitment. All the GPs mentioned memory as important, in contrast to only one nurse. However, one GP did comment that forgetting was by no means the biggest barrier. Others also mentioned how heightening personal involvement could prevent forgetting:
Because I think if I’ve got an interest in it, I’m more likely to think of it.
NAGP-6
Concern for the clinician–patient relationship was recognised as a potential issue by some participants. However, only one GP was worried about a coercive role which could prevent them from recruiting:
I would hate to feel . . . that my patients felt obliged in any way because I was asking them because I think you have quite a powerful influence as a GP on what patients do or don’t do and I think they find it quite hard to say no to their own GP . . . it’s just a feeling . . . it’s not based on experience, really . . . But I do think . . . if I were sitting in a room with a patient saying . . . etc. etc. . . . I’d worry that that’s a bit of a coercive role to be playing.
NAGP-2
In contrast, clinicians seemed to be aware of the fact their relationship with patients may actually facilitate recruitment, and this appeared to be universal among GPs and nurses:
I mean, the advantage is that they have a relationship with us, they trust us, so that’s presumably why the researchers want to get us involved . . . it might help recruitment.
NAGP-3
I think it’s about the relationship between me and the patient . . . the new patient, perhaps they might be a bit more hesitant, but if it’s a patient that I’ve been seeing and developed a relationship with over a long time, and they have confidence in me . . . I think they’d be more willing to participate in something that I recommend to them.
PN-4
Eleven participants felt that having a research question of personal importance or interest to them was the key to facilitating recruitment. Equally, an uninteresting or unimportant topic would act as a barrier to recruitment. Ownership seemed to be particularly important. Linked with ownership, clinicians reported the importance of personally understanding the trial, as well as having experience and knowledge of recruiting to trials generally. Interestingly, this seemed to be more significant for all four nurses. For GPs, knowledge and understanding was associated with being able to seek consent appropriately. For nurses, it was about having adequate (ideally face-to-face) training.
Attitudes towards computerised trial recruitment
Just under half the participants felt the use of CTR tools would be a good thing, with eight participants liking the idea of using prompts and reminders during consultations. One GP and one nurse also thought CTR tools could add validity to recruitment during consultations:
I think people . . . know that we are always using the tools on the computer, so I think . . . computerised tools as a part of their consultation . . . will be more effective and it will sound more real to the patient.
PN-1
Negative attitudes towards CTR tools were centred on concerns that pop-ups could disrupt the consultation, although others felt this would not be the case given how used primary care clinicians are to interacting with EHR and QOF alert pop-ups during consultations.
Two participants were unsure how they felt about CTR tools and two GPs expressed overtly ambivalent views. Perhaps surprisingly, only one GP expressed concern for patient confidentiality.
Attitudes towards Local Eligible Patient Identification Service
Just over half of the participants liked LEPIS. One GP had neutral feelings and two participants stated that their attitude depended on their motivation or the amount of time they had. The interface was largely seen as user-friendly and easy to use, although two GPs thought that the icons were too small or similar, and two GPs felt that LEPIS was too simplistic. Two GPs liked the fact that LEPIS could be ignored, although one GP saw this as a potential design flaw as LEPIS could be ignored consistently. Some participants liked the fact there were different options to choose within the consultation, but one GP felt that having so many options could be confusing for the clinician.
Four participants felt that LEPIS was too slow with too many steps. Five GPs felt that having to log on would be burdensome, take too much time and be prone to forgetting passwords. Interestingly, this was not a concern for nurses who saw the requirement for logging on as important to protect patient confidentiality. This disparity between GPs and nurses may reflect the fact the latter have longer consultation times. In addition, four GPs felt that a direct link to patient information sheets would be required rather than additionally having to log on.
Feasibility of proposed Retropro and eLung trials
The consensus was that complete trial recruitment within a consultation would not be feasible and that patients would probably need the opportunity to think about participation outside consultation:
I think that would be a bit too quick . . . And I think you’d want the patient to go away and read about it and think about it . . . mean it wouldn’t be ethical to do it like that.
AGP-2
Retropro was felt by some to fit nicely with routine patient consultations:
Well I mean it is only a statin and I’m quite often trying to coerce patients into not taking their expensive atorvastatin and having the cheaper simvastatin so maybe actually it would be rather easier to say we’re really trying to find out which one’s better . . . and maybe have some evidence and what about being randomised between the two? . . . But I’d still worry about having enough time even to do that on top of everything else.
NAGP-2
In addition, there were fewer concerns surrounding Retropro because patients could have the opportunity to go away and think about participating prior to making a decision. In contrast, eLung gave more cause for concern as patients would present acutely unwell, rendering them more vulnerable and less able to give fully informed consent, and in more time-pressured emergency consultations:
If someone’s that ill with COPD exacerbation maybe they’re not mentally . . . in the right place to decide whether they need antibiotics or not.
NAGP-3
As a result, eLung was generally felt to be less feasible than Retropro. However, the pragmatic nature of the proposed trials, particularly Retropro, led to slightly less hesitancy and more positive attitudes from clinicians:
Research which is sort of GP friendly, it’s stuff that we deal with that’s relevant to us . . . and it’s medical knowledge that is our territory . . . so therefore that isn’t too bad.
NAGP-3
The key findings of this qualitative study of views on CTR were as follows:
-
Time was the biggest barrier to recruitment.
-
Financial incentives could be used to minimise the impact of time; financial incentives may motivate personal involvement; financial incentives appear less important to practice nurses.
-
Understanding (or lack of) was the second biggest barrier to recruitment.
-
Selection bias (linked with understanding, language barriers, culture and vulnerability) acts as a barrier to recruitment.
-
Presence of clinical uncertainty would facilitate recruitment; personal involvement and interest were important facilitators.
-
Informed consent acted as a modifying factor; nurses were more amenable to seeking consent.
-
Concern for the clinician–patient relationship could act as a barrier, but most felt it would facilitate recruitment.
-
Attitudes towards CTR tools were favourable.
-
Attitudes towards LEPIS were generally favourable with positive feedback.
-
Point-of-care trials seemed feasible; Retropro is easier to conduct than eLung.
Discussion
Recruitment into trials
In line with literature, time was identified as the prominent barrier for GPs and nurses to recruiting patients into trials. Many of the other barriers, facilitating factors and/or modifiers identified within this study have also been identified in prior literature. However, some important differences and novel themes emerged, perhaps because this study examined attitudes towards clinicians recruiting patients themselves rather than, as with the majority of studies to date, clinicians determining eligibility and then referring patients to researchers for recruitment.
For example, it has previously been demonstrated that seeking informed consent is a barrier to recruitment. 53,61,62 However, this study identified a difference between nurses and GPs in that consent was a barrier for the latter but not the former. Similarly, all 10 nurses interviewed by Potter and Dale59 felt comfortable seeking consent from patients for participation in a trial, even though only four had done so previously. 59 This discrepancy between nurses and GPs requires further research, but may suggest nurses could play more of a role in recruiting patients into trials than has traditionally been the case.
One reason gaining informed consent may be problematic is that:
It may not be easy to forsake the role of the confident prescriber and adopt the attitude of the researcher who is uncertain about the most effective treatment available.
Van der Windt et al. 63
This relates to clinical uncertainty and, as with previous studies,63–67 this was thought (if present) to facilitate recruitment. Thus, providing clinicians with information to demonstrate the need for a trial may aid recruitment. 64,68 The negative impact of a lack of clinical uncertainty was demonstrated by concerns expressed regarding eLung – participants were reluctant to randomise patients to no antibiotics even though no convincing evidence for antibiotic efficacy exists when patients have non-purulent sputum. Similar unease was found in a study investigating the management of urinary tract infections without antibiotics. 65
Concern for the doctor (or clinician)–patient relationship has frequently been cited as an important barrier. 52,53,61,62,67,69 In contrast, such concern was not demonstrated in this study and, in fact, it was thought that such a relationship might facilitate recruitment. Some authors have previously suggested that the existence of a relationship, and thus understanding and trust, may aid trial recruitment. 58,70,71 The results of this study support the importance of the clinician–patient relationship in helping trial recruitment.
Computerised trial recruitment and Local Eligible Patient Identification Service
Participants generally reported favourable attitudes towards CTR, with LEPIS largely perceived to be user-friendly and intuitive. 72 The fact that primary care consultations already utilise computer-based prompts and pop-ups was taken by some to demonstrate that further prompts would be excessive, although others saw this as positive, in that they were already an accepted part of consultations. Overall, as long as issues such as speed and ease of use are dealt with, it would seem that primary care clinicians are, in principle, amenable to the use of CTR tools.
Methodological limitations
Conclusions of this study may be limited because of the small sample size. However, emergent themes were identified and data saturation has previously been demonstrated with similar numbers. 73 Initially it was intended to obtain a stratified purposive sample of academic versus non-academic GPs and nurses to determine if these subgroups held disparate beliefs and attitudes, and to assist comparisons within and between groups. 74 Given resource and time constraints, a convenience purposive sampling technique was used. However, only four practice nurses were included within this final sample, which will not be sufficient for data saturation, although many recurrent themes did emerge. This methodological limitation was recognised and efforts were made to minimise any negative impact by ensuring a range of primary care clinicians were interviewed. No participants worked within a practice that currently employs Vision EHR. This should have limited impact as the early LEPIS prototype had not been integrated with an EHR and the principles demonstrated should be equivalent regardless of EHR. The participants worked within inner-city, deprived and ethnically diverse areas and the identified barriers may have been unique to local concerns – the latter was probably reflected by the frequency with which participants mentioned language barriers.
Conclusion
As with previous literature, insufficient time was perceived to be the biggest barrier to recruitment. Lack of clinical uncertainty was also a significant barrier, which could be easily remedied by providing clinicians with information pertaining to the importance of a trial. The fact that nurses appeared more comfortable than GPs in seeking informed consent for trial participation requires further investigation, but demonstrates that nurses may be underutilised in trial recruitment. In contrast to previous literature, the clinician–patient relationship was perceived to potentially facilitate recruitment. Employing clinicians to recruit patients could improve trial recruitment rates, and could be further aided by the use of CTR tools to enable such recruitment to be embedded within consultations. The fact that primary care clinicians held generally positive attitudes towards the use of CTR tools and LEPIS in particular, suggests that recruitment within consultations is feasible. Indeed, the pragmatic nature of the proposed point-of-care trials appeared reassuring to many of the clinicians, although trials which did not include patients with acute illness were favoured. In order to further aid understanding of trial recruitment, qualitative analyses should be embedded within proposed point-of-care trials.
Chapter 5 Information technology system for flagging and data processing in point-of-care trials
Recruitment strategies in point-of-care trials
The study team discussed, prior to the development of the IT system, the possible strategies to recruit patients in point-of-care trials. We identified the following possible patient recruitment strategies:
-
Study team searches the EHR research database for potentially eligible patients and then sends (e.g. by e-mail) a list of eligible patients to the clinician who then reviews eligibility at a convenient time.
-
Flag at computer screen at start of consultation when a potentially eligible patient visits the clinician (for any reason).
-
Flag during consultation when the clinician enters in the EHR a medical code of interest (such as COPD exacerbation).
-
Flag during consultation when the clinician enters in the EHR a prescription of interest (such as simvastatin).
The first strategy was considered cold recruitment; the clinician can decide when trial eligibility is assessed and can invite patients to a dedicated consultation. The other three strategies could be used for hot recruitment; the clinician recruits the patient during an unscheduled consultation. Retropro could be an example of cold recruitment, whereas eLung required hot recruitment. The study team considered the flagging at the time of EHR entry of a prescription to be least attractive. This flagging appeared to have been used in a previous point-of-care trial that did not succeed. 19 Prescriptions are typically entered in the EHR by clinicians after seeking consent by patients and recruitment into a trial would mean that the clinician has to tell the patient that they changed their mind and were now uncertain about what drug to prescribe. The study team also noted some uncertainty around the timing of data entry in the HER; some clinicians enter information only after the end of the consultation (this would affect the last two strategies).
Functional requirements for the information technology system
The study team outlined the functional requirements of the IT system.
-
The identification of potentially eligible patients could be done centrally by the principal investigator using the EHR research database and/or done by the clinician. Eligibility of patients could be based on simple criteria (such as age) or on more complex criteria, such as a disease risk score (which uses information from multiple places in the EHR and from different time periods).
-
The eligibility of patients can change over time, requiring regular updating. For example, a COPD patient prescribed an antibiotic is not eligible for eLung for a period of 2 weeks.
-
The flagging during consultation and the actual recruitment of patients may need to be restricted in some studies to clinicians who were approved by the principal investigator. In addition, clinicians should be able to opt out of flagging.
-
The ability to turn the flagging and recruitment on and off quickly. As an example, no flags should appear if a site reaches their recruitment target.
-
The actual eligibility of patients may need to be confirmed and documented by the clinician outside the EHR.
-
Ability to enter study-specific information by clinicians and/or patients.
-
Ability to randomise patients, including more complex methods, such as block randomisation.
-
The system should allow cluster and individual randomised trials. In cluster trials, practices are randomised.
-
Clinicians may need access to study documentation (such the protocol and consent forms). Documentation may need to be localised (e.g. some sites may require the practice logo and contact details to be added to the consent form).
-
The system should be secure and password controlled where needed. In addition, there should be log files of all activities.
-
The system will need to be integrated with anonymous patient and staff codes shared between the different components.
-
Scalability of the IT system (i.e. use in other studies with different recruitment strategies and roll-out to many more practices) and easy study initiation.
-
Rapid identification and processing of suspected side effects, allowing review of reportability by the principal investigator and automated reporting to the regulatory authority.
Design of the information technology system
This section outlines the design of the IT system in this project ( Figure 1 ).
FIGURE 1.
Flow of information and IT system in point-of-care trials.
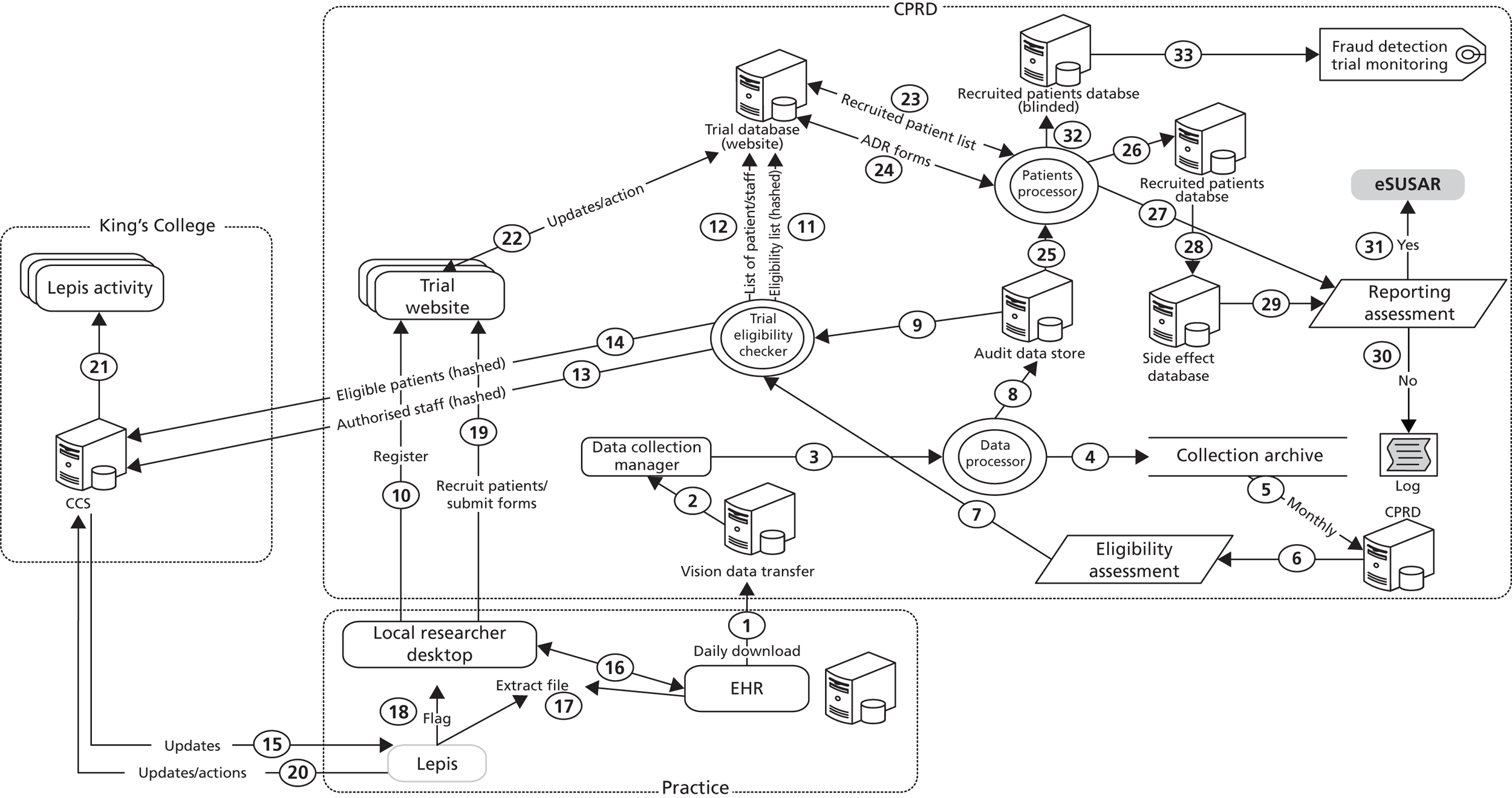
Electronic health record system
There are a number of EHR systems currently used by UK general practices (including EMIS, System One TPP and Vision). CPRD was working only with the Vision system at the time of this project. The clinicians use these system to enter administrative, clinical and prescription data for patients (see process 16 in Figure 1 ). The Vision platform, managed by In Practice System is enabled to anonymise and download patient data, and send incremental data collections to CPRD on a regular basis (every 4 weeks). As part of this project, a daily automated transfer of EHR was implemented between participating study sites and CPRD (see process 1 in Figure 1 ). The Vision system also generates an extract file in real time, which is readable by the flagging technical LEPIS (see below).
Local Eligible Patient Identification Service
Local Eligible Patient Identification Service is a programme developed by the study team residing on clinicians’ computers. It receives patient information from the local extract file (see process 17 in Figure 1 ) and interacts with an external server [central control service (CCS)] in order to download the list of active studies, authorised staff, and the fully encrypted patient eligibility lists (see process 15 in Figure 1 ). It uses this information to compare patients against the eligibility criteria of active clinical trials to find suitable participants. If a patient is found to be (potentially) eligible, a graphical pop-up is generated on the clinician’s computer screen (see process 18 in Figure 1 ). If the clinician decides that the patient is eligible, LEPIS triggers a web link for accessing the study website. This link automatically initiates the local web browser with the unique webpage for the study and the hashed (encrypted) patient identifier is then sent to the study website (see process 10 in Figure 1 ). The clinician can then log onto the study website and confirm the patient’s eligibility. The website will then retrieve the randomisation result and inform the clinician of the outcome. LEPIS can be configured to pop-up only for clinicians authorised take part in the study. Figure 2 shows the LEPIS pop-up boxes in Retropro and eLung. The following buttons are present in the LEPIS pop-up box:
-
More information: this opens a new window with further information about the trial.
-
Recruit patient: this opens a webpage, which allows the patient to be recruited in real time.
-
Patient wants time to think: this will set the notification to be generated again when the patient next returns to the clinic.
-
Patient not interested: this will set the patient as not interested in the specific trial; the practitioner will not be asked about recruiting the patient to this trial again.
-
Patient not eligible: this will set the patient as being not eligible (i.e. the automated eligibility check was inaccurate); the practitioner will not be asked about recruiting the patient to this trial again.
-
Do not disturb me for 2 hours: this will temporarily deactivate LEPIS for 2 hours; no notifications will be generated during that time.
-
Close for this patient: this will close the window; this can be done with or without first registering a response.
-
Other actions: this is a list of other actions that can be performed. Currently, this consists of only one action.
-
Patient not interested in ever participating in trials: no notification will ever be generated for this patient again.
-
FIGURE 2.
Local Eligible Patient Identification Service pop-up boxes for (a) Retropro; and (b) eLung.
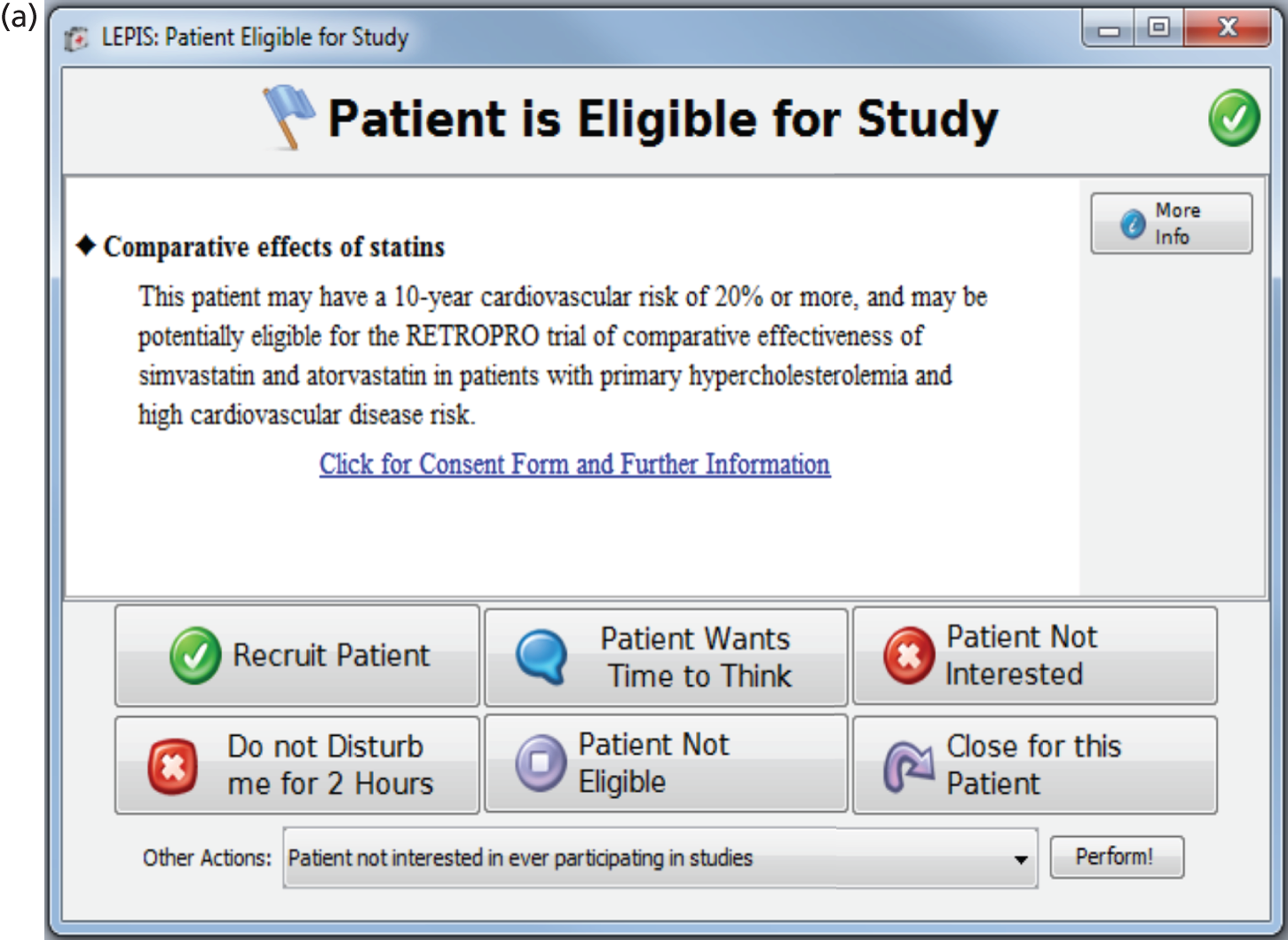
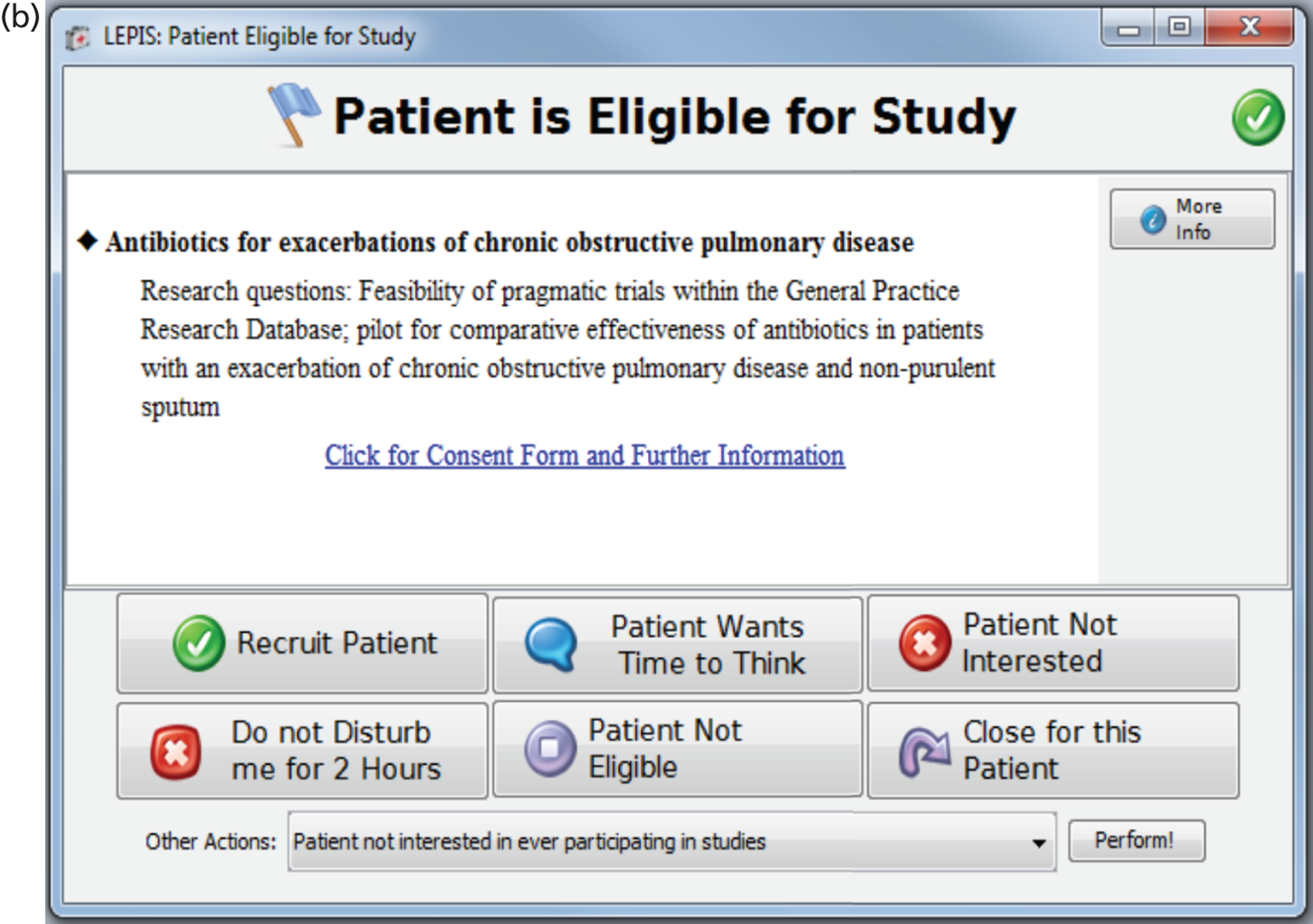
Extraction file
As soon as a new consultation is initiated by a health-care professional (nurse or doctor), a text file (extract file) is created by Vision (see process 17 in Figure 1 ). This file holds limited information for the patient currently in consultation. To ensure that this information cannot compromise patient privacy, no identifiable information is stored. Once it has been read by LEPIS, it is automatically deleted. This file is held in the local file system and acts as a bridge between the EHR system and LEPIS. This extract file will not be created for clinicians who have opted-out from LEPIS and/or for practices that have not activated LEPIS within the EHR system.
The vendor of the Vision system proposed this process of LEPIS searching an extraction file, as this would not adversely impact the performance of Vision. Complex searches can be done centrally with the extraction file providing the latest EHR information which can be analysed by LEPIS. This extraction file can inform LEPIS of a patient’s record being in consultation or a modification to the patient’s record taking place (e.g. a new Read code being added). Through this, LEPIS operates independently of the EHR.
Central control service
The CCS is a central management service for handling the construction and distribution of study information to LEPIS. The lists of eligible patients (see process 14 in Figure 1 ) and authorised clinicians (see process 13 in Figure 1 ) are updated daily and sent through an automatic process to this repository, which LEPIS uses to compare in real time with the patient and staff identifiers at the time of the consultation. The CCS can be considered as a bridge between the EHR research database and LEPIS at the practice level. The CCS also holds information about the study and limited non-confidential details about eligible and recruited patients (stored in an encrypted manner). Furthermore, connections between LEPIS and the CCS are encrypted using Secure Sockets Layer (SSL) and only hosts within the national broadband network for the NHS (N3) are authorised to access CCS. The lists of eligible patients and staff and study details are sent daily to the clinician’s desktop for use by LEPIS (see process 15 in Figure 1 ). LEPIS also sends information regularly to the CCS on the flagging and actions taken following flagging at each clinic (see processes 21 and 22 in Figure 1 ).
Study website
This is a dedicated website used to recruit patients, conduct the randomisation and collect data on/from recruited patients. The use of the secure pages of the study website is available only to authorised users; therefore, registration is mandatory followed by an approval after further checks before the site can be used. The study website allows a user to securely log in and recruit eligible patients to the relevant study or record further information. Clinicians can either access the study website through LEPIS or directly access the website by typing in the web address. In the latter situation, the clinician needs to provide the patient identifier as stored in the EHR, year of birth and sex. The study website will then check whether or nor this patient identifier is recorded in the study database and if the year of birth and sex match with the information in the EHR research database (see process 22 in Figure 1 ).
The study website also acts as the repository for study information (such as the protocol and consent forms). Clinicians can also access the study website in order to submit study-specific data, such as the ADR form (see process 19 in Figure 1 ). There is also the possibility to record that the patient wishes to opt out of further data collections (i.e. the future EHR information will not be processed for the study).
Study database
The study database is a central management service for handling all the forms submitted through the study website. It is the main storage of all data used by the study website, such as registered users, practices approved to recruit, available studies, eligibility lists of patients, lists of approved staff, maximum number of patients who can be recruited at each site and administrative management of the website (including details of who accessed the study website at what time and records of any changes to forms). The study database is also the main source data for internal analysis regarding the various studies. Information on recruited patients (see process 12 in Figure 1 ) and approved staff (see process 11 in Figure 1 ) is shared daily between the study website and the study eligibility checker.
Data processing system
The data processing system is responsible for managing and processing EHR collections received from participating trial practices on a daily basis. Practice data are processed to generate lists of patients eligible for each trial. The system consists of four main components: data collection manager, data processor, trial eligibility checker and patients processor. A brief description of each is provided below.
Data collection manager
This service tracks all data collections received on the vision data transfer server (see process 2 in Figure 1 ). Received data are checked for validity and content. Valid data collections – those that contain mandatory data files and are received in the right sequence with no prior gaps in data will be passed on (see process 3 in Figure 1 ) to the data processor component so that they can be processed. A log report is generated for unsuccessful collections with details of the practice, data collection and reason for failure. Such collections are then reviewed by CPRD staff who then liaises with practices to rectify issues.
Data processor
This component processes all valid data collections received from participating practices into the audit data store or practice data repository. Audit data store files are not trial specific and contain practice data on all patients who may be potentially eligible for recruitment to any trial. Incremental data collections contain a mixture of new records and updates on existing records and a log to enable record deletions. The data processor manages the additions, edits and deletes, and updates the practice audit data store files (see process 8 in Figure 1 ), such that the data held is an accurate representation of the EHR within the Vision system.
All processed collections are securely archived (see process 4 in Figure 1 ) to a ‘collection archive’. All data collections received from a practice in the preceding month are collated (see process 5 in Figure 1 ) and integrated monthly into the EHR research database.
Trial eligibility checker
This component is run on a daily basis generating the list of eligible patients and staff members authorised to recruit for each practice. Practice data for a particular trial will not be processed if the practice has met its recruitment target or the trial has been inactivated. The list of staff authorised to recruit is dependent on the type of trial. In cluster trials, all active staff members at the practice are authorised to recruit whereas in individual randomised trials only those staff members that have registered on the study website (see process 12 in Figure 1 ) may be authorised to recruit.
A trial-specific parameter file is setup when a trial is initiated. The file records information on the type of trial (cluster/individual) as well as patient- and event-level inclusion and exclusion criteria which specify how eligible patients for the trial are to be identified. Patient-level criteria typically consist of conditions on the patient’s demographic or registration information, such as the patient should be at least 30 years of age, should be currently registered at the practice, and should have at least 5 years of follow-up time. Event-level criteria are typically specified as the presence or absence of medical and/or product codes in the patient’s records relative to a time window. For example, patients should have no prescriptions for statins, or should have no prescriptions for antibiotics in the previous 3 months. Data from the CPRD research database can be analysed for more complex eligibility criteria (see process 6 in Figure 1 ). Complex algorithms can assess eligibility and generate a pool of potential patients who could be recruited. The trial-specific parameter file allows the specification and usage of such a look-up file (see process 7 in Figure 1 ).
For each active trial, the latest data in the audit data store are used (see process 9 in Figure 1 ) to identify the list of currently registered patients who satisfy both the patient- and event-level trial-specific criteria. The study database is queried for patients who have already been recruited into the trial or judged to be ineligible by the clinician (see process 12 in Figure 1 ), and these are removed from the eligibility list. The complete set of patients who could be recruited into a given trial is uploaded to the study website (see process 11 in Figure 1 ). At the same time, the derived set of eligible patients (see process 14 in Figure 1 ) and authorised staff (see process 13 in Figure 1 ) is uploaded encrypted to the CCS. This component is run daily as practice data collections are received on a daily basis and patient eligibility can change over time.
Patient processor
This component is run on a daily basis as a scheduled system task to maintain trial databases containing current data on recruited patients and populate the ADR database. This database is maintained for all recruited patients for up to a certain number of days (trial-specific) after the patient has been recruited. The patient processor component is deactivated for a participating practice once its recruitment target for that trial has been met and the last patient to be recruited by the practice for the trial has been followed for the specified number of days.
The lists of patients who recruited into or opted out of the trial are extracted from the study website (see process 23 in Figure 1 ). The data in the audit data store are utilised (see process 25 in Figure 1 ) to extract the latest information on these patients. The trial database for recruited patients is updated on a daily basis – two versions of the database are maintained. The first is a blinded database (see process 32 in Figure 1 ) that has trial-specific fields. This database is used for data monitoring while the trial/study is active. The second version represents complete data on all recruited patients (see process 26 in Figure 1 ) and is used to detect the occurrence of ADRs. Patients who have opted out of the trial have data in the trial databases (see processes 26 and 32 in Figure 1 ) until the date that the patient opted out. A trial-specific file is maintained that logs the list of patients who have been lost to follow-up, along with the date and reason for opt out.
Handling of suspected adverse drug reactions
Clinicians can enter data into the side effect fields within Vision or use a medical code to represent a suspected ADR. The latest EHR information is checked daily for both kinds of entries for recruited patients. The trial side effect database (see process 28 in Figure 1 ) is populated if a potential ADR is detected and an ADR report generated containing details of the potential ADR and the patient’s medical and prescription history for the previous 6 months. This report is then immediately sent to the principal investigator (see process 29 in Figure 1 ) by e-mail for review. Trial-specific guidelines stated that a clinician should fill and submit a suspected ADR form on the study website. Forms submitted on the website are processed daily (see process 24 in Figure 1 ) and an e-mail is sent to the principal investigator (see process27 in Figure 1 ). The ADRs are reviewed by the principal investigator for reportability to the regulatory authority. Suspected ADRs as entered in free text into the EHR were not analysed because of the variability in the recording of this information.
Regulatory electronic submission of serious unexpected suspected adverse drug reactions
If an ADR is considered serious and unexpected, this report is then submitted electronically to the regulatory authority (see process 31 in Figure 1 ). The study website uses a specially designed tool which communicates with the electronic SUSAR reporting system of the UK regulatory authority eliminating the need for further manual input or manipulation of existing data. Log files are maintained of this reportability assessment (see process 30 in Figure 1 ).
Data monitoring during study
The EHR information on recruited patients (see process 32 in Figure 1 ) can be used for monthly analyses of the characteristics of recruited patients and comparisons with patients not recruited into the study. Fraud detection and evaluation of data irregularities can also use this information (see process 33 in Figure 1 ).
Performance of the information technology system
Before rolling out the IT system, the complete IT system was extensively tested on development servers with a replica of data. We also identified a few practices, installed the software and carried out tests with GPs (although this was not as extensive as intended due to the short timelines). A software company also tested LEPIS in terms of performance, scalability and security. The biggest challenge with the roll-out of the IT system was the limited time which was allowed for the testing given the substantial delays in obtaining research approvals. The initial plan was to implement and test the IT system in a few practices locally, but this was replaced by a larger and quicker roll-out across the country in order to start patient recruitment. Table 6 illustrates the challenges in the implementation of the software system. Updates in Vision were required for LEPIS and the daily downloads of EHRs followed by reconfiguration of Vision at the practice. Although this process was not complex, some practices were behind in uploading Vision updates. The implementation of LEPIS provided several major challenges. In some practices, staff were not authorised to load software and detailed negotiations with third parties were required. The network set-up, network restrictions and the clinician’s computer specifications varied between practices. One of the issues encountered was low performance of Vision after LEPIS installation. This was found to be related to the low memory space on local desktops and LEPIS loading of eligibility lists into the memory. This was encountered when starting a new study that required much larger eligibility lists (i.e. all patients in a practice). This was addressed by reconfiguring the patient eligibility lists and respective parts of LEPIS in order to handle larger eligibility lists with small memory usage. LEPIS was also modified in order to provide more diagnostic tools including tests of the network status. Log files could also be sent automatically during a limited period of time to the CCS rather than the practice sending the log files. Even after a successful instalment and test, LEPIS sometimes did not pop-up at the time of consultation in some practices. It was established that LEPIS generally functioned but that the notification did not occur due to a step being inadvertently missed or procedures not being followed (e.g. the extraction file generation was not enabled within Vision, the right Read code or description was not entered in the new consultation, or the desktop had not been rebooted after LEPIS installation).
| Description of issue | Cause of issue |
|---|---|
| Java® Virtual Machine [software needed for LEPIS (Version 1.6.0.0, Oracle, Redwood Shores, California, CA, USA)] | Java Virtual Machine not installed or older version |
| C drive of local desktops is hidden/not visible | Staff at the practice do not have access to the C drive |
| LEPIS software cannot contact the central server to retrieve trial information |
|
| Vision does not interact with LEPIS or extract file not generated |
|
| LEPIS not triggering at the time of consultation |
|
| Low performance of Vision after LEPIS installation |
|
| No daily download of EHRs |
|
| Vision is hosted (located on a remote server) |
|
| Vision consultation manager crashes when copying of acute prescriptions to repeats | Error in creation of extraction file by Vision |
The initial approach by the study team was to send practices a DVD with the flagging software and installation instructions. This was later simplified by sending links for the software installation. However, many practices were unable to load LEPIS successfully, partly because installation steps were not followed and partly because of technical challenges. The study team visited several practices to provide support in loading and testing LEPIS (typically, this was done in one visit). The study team explored the possibility that support in loading and testing LEPIS could be provided by the vendor of Vision, but this was not adopted given that most of the installation challenges were related to settings of local desktops not controlled by the vendor of Vision. Although the site visits were laborious, the study team did consider them to be very useful for practices that had encountered issues in loading LEPIS. Once LEPIS was installed successfully, further visits were not needed.
No major issues were encountered in the development and implementation of the systems other than the flagging software. The study website allowed staff registration and completion of the recruitment, ADR and opt-out forms. Daily and monthly processing of eligibility, the automated identification and e-mailing of suspected ADRs and reporting of recruitment numbers were also implemented successfully. An issue was the perceived disagreement in the assessment of patient eligibility between the clinician and the flagging software. Clinicians were expecting a LEPIS pop-up or wanted to recruit directly on the website but the patient was not found on the website and/or LEPIS did not pop up. In some instances, this was related to EHR information not being noted by the clinician (e.g. a patient received statins previously many years ago, which was an exclusion criterion for Retropro). In other instances, this was related to differences in the eligibility assessment (e.g. differences in the estimates of CVD risk owing to incomplete EHRs). However, it was possible for patients to be added on an ad hoc basis to the eligibility list.
Discussion
The study team believes that all functional requirements for the IT system, as described previously, were successfully met with the exception of scalability of LEPIS. The IT system allows complex search criteria for eligibility, daily review of eligibility, identification and monitoring of side effects, long-term follow-up at minimal cost and pop-ups during consultation. Although the installation process was improved, the roll-out to more practices will require resources as desktop settings vary between practices. Site visits to practices that experienced difficulties were found to be very useful but require resources both for the practices and study team. As outlined in Chapter 8 , the clinicians involved with LEPIS initially expressed serious reservations about the installation process (especially in the early phases), but were very appreciative of the system once LEPIS was successfully installed. A recently started study has utilised LEPIS to flag during consultation and collect additional data (the FLU-CAT study aims to evaluate and refine pandemic inFLUenza Community Assessment Tools). This study has now recruited over 300 patients with flu in 24 practices (ISRCTN87130712). However, future work will need to address the ability of the IT support to support multiple simultaneous trials and users (i.e. stress testing for scalability).
The initial plan (as proposed in the grant application) was to ask the software provider of the EHRs to extend their existing flagging system in order to facilitate trial recruitment. However, the discussions with the vendor of Vision highlighted the difficulties in achieving a flexible flagging system which could be controlled centrally. One requirement was that flagging could be restricted to approved clinicians and turned on–off instantaneously. In addition, there was a need for complex eligibility assessments. The need to monitor flagging was also a requirement, as was the need for simple procedures to initiate a new study. For these reasons, the study team decided to develop the LEPIS system integrating it with systems around the EHR research database.
Our IT system used an external website to document eligibility, recruit patients and collect additional study data. An alternative approach could have been to develop case report forms within the Vision system, directly integrating trial data in the EHR [this approach is now being developed in the Transform project (www.transformproject.eu/)]. However, the study team did not proceed given the substantial costs and timelines required. In addition, the website approach could more easily facilitate emerging IT technologies (such as remote monitoring of patients) as these do not need be built within a complex EHR system that cannot be easily amended. The study website also allows data entry by patients (as long as they are provided with log-in details and patient ID). The need for easy modification was another reason for opting with the study website approach (without the need to update the EHR software for hundreds of practices). The study website approach also facilitated the central control of recruitment with the ability to instantaneously turn on or off recruitment at a site. The study website does allow the option to copy and paste completed forms in the EHR [as portable document formats (PDFs)], so trial information can be stored in the EHR.
Chapter 6 Implementation experiences with the exemplar trials
Practice drop-out rates and timelines
All practices submitting data to CPRD, including practices that had not been involved previously in research, were sent brief information regarding the aims of the two studies and asked to respond by completing an expression of interest form. In addition, the Primary Care Research Networks (PCRNs) in England and Scotland sent the information including an invitation to join CPRD; this was sent to practices using Vision or to all practices in their region if the EHR systems of practices were unknown (about 2000 practices). The practices that expressed interest were then sent an information pack including study protocol and a site contract. The first draft of the site contract was brief, but regulatory requirements mandated a more legalistic document. After signing the contract, the next step for GPs was to complete site-specific information forms and supply further paperwork including a curriculum vitae highlighting their clinical research experience and qualifications, a staff delegation log and certificates of completion GCP and protocol training. Enthusiasm of GPs in participation dropped markedly at each stage of the recruitment and approval process. Table 7 provides the number of practices at each of the different steps. Of the 270 practices that expressed interest in participating, only 30 practices in Retropro and 31 practices in eLung completed the paperwork and mandatory protocol and GCP training. Of those approved practices, 17 practices actually recruited patients in Retropro and six in eLung (3.7% and 1.3% of all practices respectively).
| Step in site recruitment | Retropro | eLung | ||
|---|---|---|---|---|
| n practices | % | n practices | % | |
| Practices contacted | 459 | 100 | 459 | 100 |
| Practices returned letter of interest | 377 | 82.1 | 377 | 82.1 |
| Interested | 270 | 58.8 | 270 | 58.8 |
| Declined | 107 | 23.3 | 107 | 23.3 |
| Site agreement received | 63 | 13.7 | 53 | 11.5 |
| Site-specific submission | 50 | 10.9 | 45 | 9.8 |
| NHS permission granted | 48 | 10.5 | 43 | 9.4 |
| GCP and protocol training complete | 35 | 7.6 | 32 | 7.0 |
| Approved to recruit by study team | 30 | 6.5 | 31 | 6.8 |
| Practices remaining following subsequent withdrawals | 25 | 5.4 | 24 | 5.2 |
| Were seeking to recruit patients | 17 | 3.7 | 8 | 1.7 |
| Recruited patients | 17 | 3.7 | 6 | 1.3 |
Trials can start in the UK only after approval by the MHRA, ethics committee and the NHS R&D organisation locally to a site (local R&D approval is given centrally in Scotland, whereas in England there are 150 organisations for the different regions). With respect to research governance, the clinical trial application to the MHRA was completed within 2 months for both studies. The overarching R&D review took 2 years from original application to approval. This was followed by submission to local NHS organisations that had to give approval for the trial in each of the sites (also known as R&D approvals). It took over 1 year to obtain local R&D approval for 25–30 sites. In Scotland, the R&D approval was received just over 2 months after the review process began and covered all participating sites.
Research governance experiences
Minimal issues were experienced with the regulatory approval and prompt approval for eLung. However, the Retropro application was regarded invalid as the summary of product characteristics for each statin expected to be used (by manufacturer and dose) was initially required. The practicalities of supplying summaries for all marketed statins were discussed and concluded that one for simvastatin and one for atorvastatin would suffice. The ethics committee review encountered greater complexities. During the review meeting, the committee raised concerns that patients would not being given enough time to consider their participation, suggesting patients should be provided with information then return for a separate recruitment appointment. The low-risk nature of these studies was discussed during the meeting to the committee’s satisfaction and they conceded that informed consent could be taken during an unscheduled consultation. The committee insisted that the patient information sheet and consent form be reformatted to fit the standard clinical trial template, which substantially increased the length of the document for potential participants to read.
One of the changes in the consent form concerned private health insurance. The ethics committee required the following amendment to the consent form, ‘Potential participants who hold private medical insurance should contact their insurance companies to inform them that they are considering taking part in the study’. The study team then contacted one of the main private health insurance companies which confirmed that it does not require notification of low-risk point-of-care trials. The ethics committee responded that the opinion of one company can, in no way, be taken as the opinion of all private medical insurance companies. The study team then contacted the Association of British Insurers, which represents UK insurance companies. It provided its information sheet for people who are planning to take part in clinical trials. This stated that there are restrictions on the insurance cover for patients who participate in trials of drugs in the experimental/unproven phase, but no restrictions in cover were imposed for trials that evaluate interventions regarded as safe and which had been accepted for marketing. However, the ethics committee rejected our argument because an insurance company from, for example, China may not be a member of the Association of British Insurers. The consent forms were amended to tell patients to inform their private health-care insurer of participation in the trial.
The reviews by the local NHS organisations produced a multitude and variety of questions often covering the same ground as the ethical review, such as wording of the consent form and the requirement to have local logos to information sheets (adding costs and complexity to the trials). Despite receipts of ‘valid application’ acknowledgement e-mails, the study team was not informed about missing documents. There were significant problems with the document repository system (the central system for gaining NHS permissions). The documents in the system did not follow standard naming conventions, with R&D reviewers unable to find the paper trail of regulatory and ethical reviews. As the study team was not allowed to view their documents in the repository system, this resulted in long and tedious conversations with multiple R&D reviewers to describe documents and try to find the documents.
Both the ethics committee and the MHRA had considerable difficulty in accommodating the mandatory notifications of potential sites. The ethics committee reported that entering details of potential sites was time-consuming and the MHRA requested that, contrary to their amendment guidance, sites information need not be submitted. The administrative procedures for submitting information of potential sites was also complex with the study team having to duplicate information onto different systems.
Established clinical practice and treatment guidelines
Several GPs expressed concerns about eLung because some patients would not get an antibiotic. There also had been changes in recent practice, promoted by local ‘admissions avoidance’ initiatives, in which GPs are providing COPD patients with antibiotics to use with first symptoms of exacerbations before contacting the GP. As a consequence, many patients were ineligible because of recent antibiotic use and GPs were unwilling to randomise. Treatment guidelines by local NHS organisations provided a major barrier to Retropro. At the time of obtaining R&D approvals, the cost of simvastatin was substantially lower than that of atorvastatin. However, atorvastatin was to lose patent protection in about 1 year (May 2012) with expected lowering of prices due to the introduction of generics. Most local NHS organisations restricted atorvastatin use. Participation in Retropro would adversely affect the quality markers of a practice and no exemption was made for prescribing within a trial. Practices declined participation for this reason and others were warned that trial participation would adversely impact this mark of quality of prescribing. The chairperson of the Trial Steering Committee contacted the Chief Medical Officer in England to highlight this paradox of inability to address uncertainty due to guidelines.
Another challenge in Retropro was the discrepancy between the licensed indication of statins and established practice by GPs. The licence restricts use of statins to patients with high cholesterol levels that did not respond to diet. GPs and the latest treatment guidelines indicated that patients with a high CVD risk (20% + over 10 years) are to be treated without dietary advice, even if the cholesterol level is normal. 75 During the conduct of the trial, the risk threshold for statin initiation was further lowered in practice, with many lower-risk patients receiving statins76 and experts advocating indiscriminate use in people aged > 50 years. 77 Retropro’s inclusion criteria retained the requirement of high cholesterol levels, while the need for non-response to diet advice was removed. If Retropro’s inclusion criteria had been aligned with established practice, GCP could have considered the trial to be higher risk because of the unlicensed indications requiring, for example, site visits and more intensive monitoring by clinicians.
Excess treatment costs
Retropro would incur extra costs to the NHS as a result of the use of the more expensive atorvastatin. The study team analysed the level of repeat prescribing in CPRD and predicted the likely level of changes in the cost of atorvastatin after the expiry of its patent. It was estimated, based on price changes with simvastatin and pravastatin, that it would take 7 months for prices to drop substantially after patent expiry. The study team provided a five-page report and offered local NHS organisations to pay for the estimated excess treatment costs of £36.52 for each atorvastatin trial participant. None of the NHS organisations accepted this offer. A senior civil servant in the NIHR commented:
The rejection of your offer seems at least in part to have been due to the difficulties that would be created by the proposed reimbursement as it was to the focus that the local NHS organisation has on its performance targets for statin prescription.
A further comment was:
Local NHS organisation would have been courageous to have accepted such an offer. It is not possible to forecast that following the availability of a generic when and to what level the price of that medicine will fall, nor whether after a fall there will not be a subsequent increase . . . any locality agreeing to this proposal would now be incurring an unnecessary cost against its very tight prescribing budget.
The local NHS organisations are responsible for meeting excess treatment costs for research, unless these are exceptionally large, for which there is a national system. The outcomes of these discussions were that GPs in some regions were required to switch Retropro participants after 3 months from atorvastatin to simvastatin. The study team considered this unethical but the ethics committee approved this. Cheap generic versions of atorvastatin were introduced immediately after patent expiry in May 2012 (rather than the predicted 7 months), resulting in actual excess treatment costs of £2.87 per patient.
Changes in prescribing patterns
Retropro was affected by changes in prescribing patterns. In 2012, the MHRA issued advice that patients taking more than 20 mg daily of simvastatin should not be coprescribed amlodipine (a very commonly used drug for hypertension and ischaemic heart disease). Several GPs indicated that they were switching amlodipine users from simvastatin to atorvastatin and, thus, did not want to include such users into Retropro. An analysis was conducted using a previously compiled cohort of 127,000 statin users with measurements of creatine phosphokinase levels. 78 No difference was found in creatine phosphokinase levels between simvastatin and atorvastatin in amlodipine users. However, the Data Monitoring Committee recommended following the MHRA advice, where appropriate. For this reason, amlodipine users were removed from the eligibility list, although GPs could still enrol these patients by requesting the study team to add an individual patient to the list.
Discussion
Our experiences of complex and lengthy approval procedures in the UK are consistent with previous reports. 48,79 There is a need to fully implement proportionality in research governance, minimising the risks to participants while maximising the feasibility of a study. 48,79 It is essential that this risk proportionality is actually embraced fully in research governance, so that many more clinicians and patients will participate in useful research. Changes in research governance are currently being implemented in England by the new Health Research Authority. Point-of-care trials will be successful only if many more clinicians participate, rather than the 3.7% in Retropro and 1.3% in eLung.
Retropro provided varying perspectives on who is to pay for costs directly related to the trial. The current system of local NHS organisations having to agree to pay for these costs may have caused a prisoner’s dilemma. In ethical literature, this dilemma highlights the tensions between individual rationality and collective benefit. Although it is collectively beneficial to conduct a Retropro-like trial, a local NHS organisation would act rationally if it would just wait for trial results to emerge and for others to incur the costs. A senior civil servant in the NIHR commented:
It seems to me that the pragmatic solution [to excess treatment costs] is relatively straightforward. The research proposals should include the provision of all relevant trial medicines to patients recruited for the duration of the trial, and should not rely on normal prescribing budgets to cover some of the additional costs. This approach has the additional advantage of a fixed cost of the medicines – whereas generic medicines are subject to significant month-to-month variation under their pricing mechanism.
This proposal would undermine the intended naturalistic nature of point-of-care trials, restrict the ability for long-term follow-up, and also would be expensive and hugely inefficient. The costs of quality improvement will be transferred in this proposal from the NHS to research funders, with the NHS passively taking the benefits of research (e.g. reduced expenditure owing to a better evidence base). Our experiences indicate that health-care organisations in the UK may not have been incentivised to proactively initiate research and use research for quality improvement.
Chapter 7 Trial recruitment and data collection
Practice recruitment
Seventeen practices recruited patients in Retropro and six in eLung. Table 8 shows the characteristics of the practices recruiting for Retropro or not. Recruiting and non-recruiting practices had similar percentages of patients aged ≥ 40 years who were prescribed a statin in the year before the start of Retropro.
| Characteristic | Recruiting practices | Non-recruiting practices |
|---|---|---|
| Mean number of patients aged ≥ 40 years registered in the year before start of the trial | 4981 | 4677 |
| Percentage of patients aged ≥ 40 years prescribed a statin in the year before start of the trial | 22.0% | 22.4% |
| Index of Deprivation quintiles for practice postcode | ||
| 1 (most deprived) | 11.8% | 17.2% |
| 2 | 11.8% | 21.4% |
| 3 | 35.3% | 21.4% |
| 4 | 17.6% | 19.5% |
| 5 (least deprived) | 23.5% | 20.5% |
Patient recruitment
Retropro
Figure 3 shows cumulative patient recruitment over time in Retropro. It completed recruitment in November 2013 (301 patients). The 301 Retropro participants were recruited in 17 practices by 25 GPs. One practice recruited 75 patients, nine practices between 10 and 50 patients and seven practices each fewer than 10 patients. Retropro started in September 2012 and recruited steadily over time. Retropro recruited 20.6% of all statin starters in recruiting practices (this number of statin starters included patients with a 10-year CVD risk of < 20% and patients seen by non-trial GPs in the practices). The recruitment rate of Retropro was 1.1% of the statin starters in the EHR database. Several recruitment strategies were used in both trials. Patients could be recruited either through LEPIS flagging during consultation (with provision of a link) or through direct access of the trial website. The criteria for LEPIS flagging were changed over time. Flagging initially occurred in both trials at EHR entry of specific clinical codes and later flagging occurred at start of consultation. Practices varied in their interest in using flagging for recruitment and the preferred criteria for flagging. A challenge in Retropro recruitment was the discrepancy in results between the CVD risk assessment done centrally and that done by the GP. Prediction of high CVD risk varied considerably between the different risk scores. In addition, some GPs considered risk factors not considered by the risk scores. As a consequence, some GPs wanted to recruit while not meeting the CVD threshold according to the assessment done centrally (e.g. one patient had a 12% CVD risk according to the Framingham risk score based on the EHRs).
FIGURE 3.
Cumulative numbers of patients recruited into (a) Retropro; and (b) eLung.
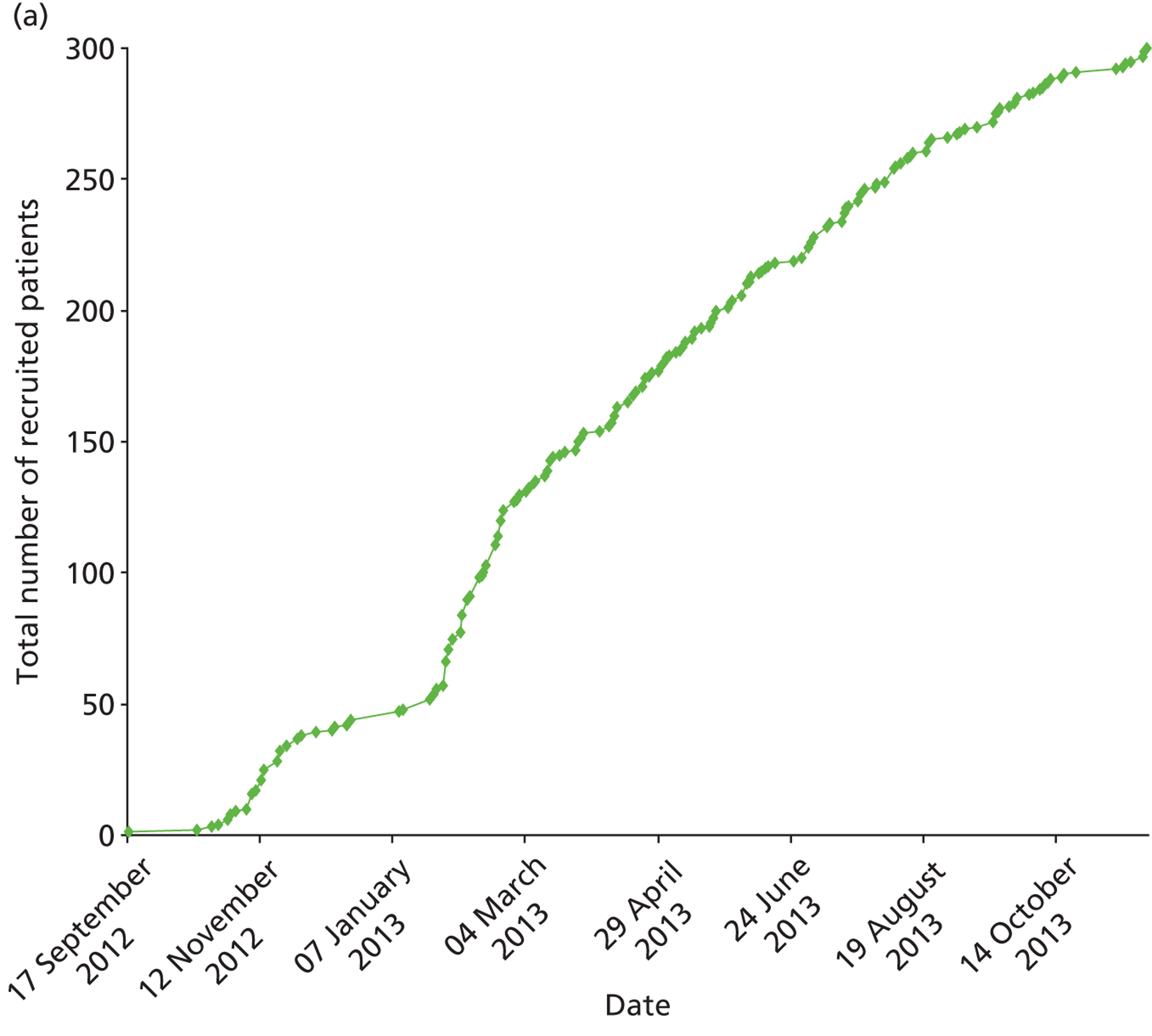
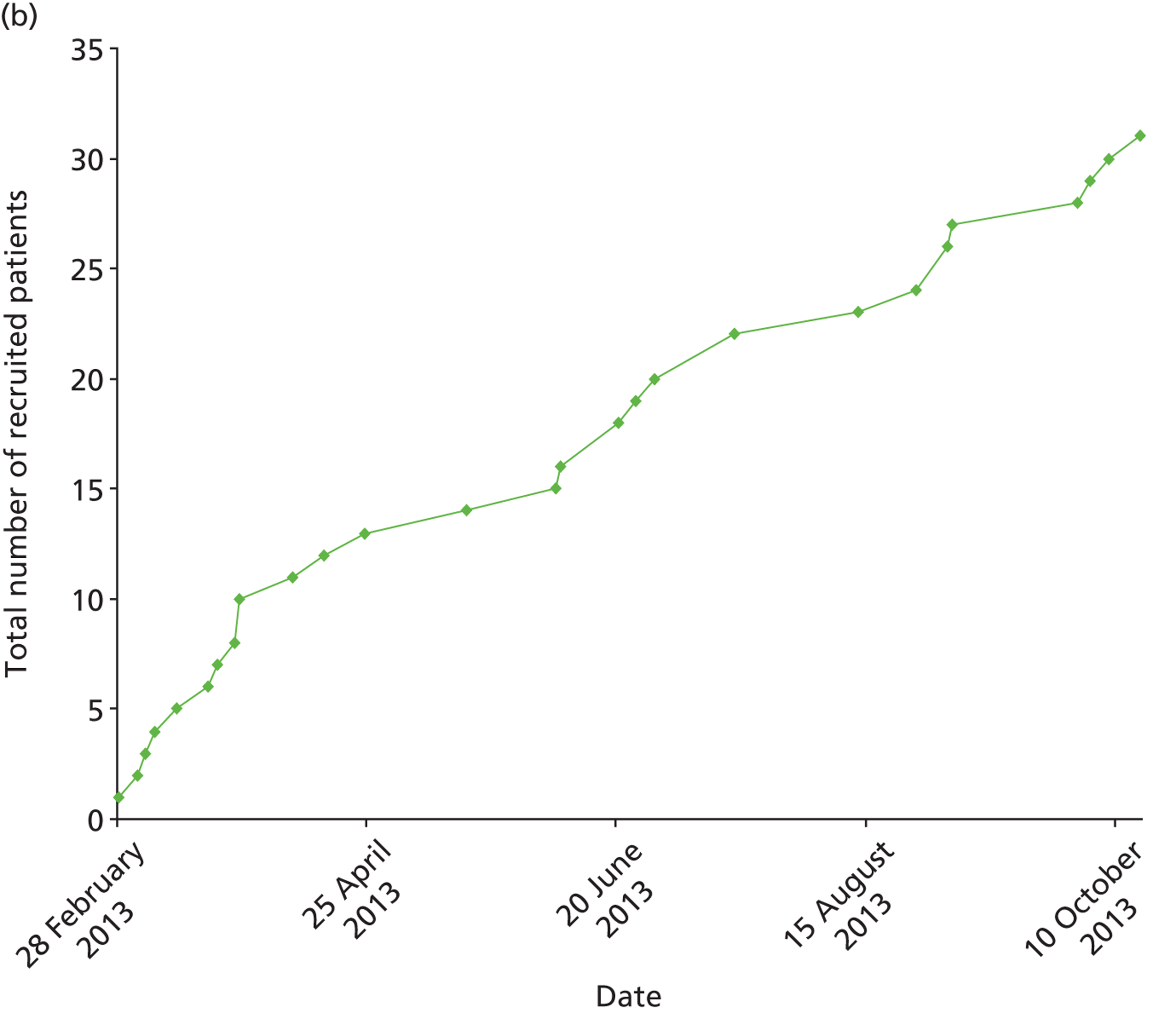
eLung
eLung recruited 31 patients (out of an intended 150). The original plan for eLung was to recruit over two winter seasons, but this could not be done because of the cessation of funding and long delays in study initiation (first patient was recruited in February 2013). The 31 eLung participants were recruited in six practices by seven GPs. One practice recruited 21 patients, whereas the other practices recruited each fewer than five patients.
Data collection and adherence to treatment allocation
Retropro
Table 9 shows the characteristics of the Retropro participants and of non-trial patients who started statin treatment in recruiting and non-recruiting practices. The 10-year CVD risks were found to be higher inRetropro participants than in non-participants (10-year CVD risk was an inclusion criterion). Furthermore, Retropro participants were less likely to have a history of diabetes mellitus than statin starters in the same practices (8.6% and 21.1% respectively). The mean number of patients aged ≥ 40 years was 4981 in thepractices recruiting for Retropro and 4633 in non-recruiting practices. The percentages of patients who had received a statin prescription were comparable in these practices (22.0% and 21.9% respectively). The random treatment allocation was followed by the GP for all participants, except for nine participants who did not have a baseline statin prescription in their EHR (six of these had been allocated to simvastatin and three to atorvastatin). The most frequent daily dose was 40 mg for simvastatin (71.3%) and 10 mg for atorvastatin (58.3%). A blood sample for genetic analyses was provided by 234 participants; this corresponds to a collection rate of 88.6% (at the time of this report). The EHR research database was reviewed monthly for the characteristics of recruited patients compared with non-recruited patients. A monthly analysis reviewed the recording of major clinical outcomes (such as heart attacks) comparing the trial and non-trial populations.
| Characteristic | Retropro participants (n = 301) | Other statin starters in recruiting practicesa (n = 1163) | Statin starters in non-recruiting practicesa (n = 26,898) |
|---|---|---|---|
| Sex | |||
| Woman | 105 (34.9%) | 525 (45.1%) | 12,691 (47%) |
| Mean age (years) | 67.0 | 63.1 | 62.5 |
| Region | |||
| England | 203 (67.4%) | 905 (77.8%) | 23,846 (88.4%) |
| Scotland | 98 (32.6%) | 258 (22.2%) | 3143 (11.6%) |
| QRISK2 10-year CVD risk | |||
| < 5% | 1 (0.3%) | 62 (5.3%) | 2121 (7.9%) |
| 5–10% | 5 (1.7%) | 152 (13.1%) | 3659 (13.6%) |
| 10–15% | 25 (8.3%) | 170 (14.6%) | 3874 (14.4%) |
| 15–20% | 55 (18.3%) | 194 (16.7%) | 3974 (14.7%) |
| > 20% | 215 (71.4%) | 585 (50.3%) | 13,361 (49.5%) |
| Mean baseline cholesterol | 6.2 | 6.2 | 6.3 |
| Medical history | |||
| Treated hypertension | 107 (35.5%) | 420 (36.1%) | 10,331 (38.3%) |
| Diabetes mellitus | 26 (8.6%) | 245 (21.1%) | 5323 (19.7%) |
| Prescribing in 6 months before | |||
| Amlodipine | 34 (11.3%) | 166 (14.3%) | 4570 (16.9%) |
eLung
Table 10 shows the characteristics of the eLung participants and of the potentially eligible non-participants in recruiting and non-recruiting practices. eLung participants received on average more oral corticosteroids in the 6 months before starting the trial compared with the non-trial participants. Furthermore, the rate of prescribing in the year before was considerably higher in eLung participants (73.2%) compared with other COPD exacerbation in the same practices (28.1%). eLung recruited 32.3% of all potentially eligible patients in the recruiting practices (this number includes patients seen by other GPs); the best recruiting practices enrolled over half of their potentially eligible patients (55.2%). There were 3518 potentially eligible patients in all CPRD practices; 0.9% of these were recruited into eLung. The random treatment allocation was followed by the GP for all 31 participants; 11 patients were allocated to immediate antibiotics and 20 to deferred or non-use. The type of antibiotic was amoxicillin for seven, doxycycline for three and clarithromycin for one participant. None of the 31 eLung participants used the eDiary, as originally planned, despite the distribution of 42 eDiaries to 14 practices. Two of these 14 practices recruited three eLung participants, but none were provided with an eDiary. Nineteen eLung participants returned consent forms and were sent the paper EuroQol EQ-5D-3L and COPD Assessment Test questionnaires. Twelve eLung participants then completed the 4-week follow-up questionnaire. The remaining seven participants were sent 4-week questionnaires but did not return them.
| Characteristic | eLung participants (n = 31) | Other patients in recruiting practicesa (n = 65) | Patients in non-recruiting practicesa (n = 3422) |
|---|---|---|---|
| Sex | |||
| Woman | 17 (54.8%) | 23 (35.4%) | 1656 (48.4%) |
| Mean age (years) | 70.2 | 65.8 | 70.5 |
| Region | |||
| England | 10 (32.3%) | 48 (73.8%) | 2861 (83.6%) |
| Scotland | 21 (67.7%) | 17 (26.2%) | 561 (16.4%) |
| Medical history | |||
| Treated hypertension | 18 (58.1%) | 24 (36.9%) | 1346 (39.3%) |
| Prescribing in 6 months before | |||
| Antimuscarinic bronchodilators | 16 (51.6%) | 36 (55.4%) | 2403 (70.2%) |
| Long-acting β2-agonists | 8 (25.8%) | 7 (10.8%) | 212 (6.2%) |
| Short-acting β-agonists | 1 (3.2%) | 9 (13.8%) | 366 (10.7%) |
| Inhaled corticosteroids | 14 (45.2%) | 7 (10.8%) | 315 (9.2%) |
| Oral corticosteroids | 20 (64.5%) | 23 (35.4%) | 1553 (45.4%) |
| Mean number of prescription in 1 year before | |||
| Any medication | 73.2 | 28.1 | 34.9 |
| Antibiotics | 2.9 | 3.3 | 3.1 |
Fraud detection
Review of potential scenarios of fraud
A review was conducted of the risks for fraud in point-of-care trials. A list of potential scenarios for fraud and other GCP violations are outlined in Table 11 .
| Fraud | Example of what is being done | Assessment of potential risk | Approaches to prevent or minimise violations | Approaches to detect violations |
|---|---|---|---|---|
| Inventing patients | GP registers a fake patient into the EHR and fabricates data | Very low: practices are audited for patient registration as NHS payments are related to number of registered patients at practice; patient registration is done by administrative staff | Trials are restricted to patients registered at the practice for at least 6 months | No EHRs are being entered after study termination; no record ever of death or hospitalisation in linked data sets |
| Fabricating data for ineligible patient who is not present at the practice | A healthy patient is being recruited (e.g. into eLung) | Low: EHRs can be seen by other practice staff (unlike the conventional CRF in trials) | Trials are restricted to patients pre-selected within the database (based on pre-recorded medical history) | EHRs prior to and after trial are different from non-trial participants |
| Fabricating data for potentially eligible patient who is not present at the practice | Inclusion criteria and consent form are fabricated | Low: EHRs can be seen by other practice staff (unlike the case report form in conventional trials) | (i) Limited number of patients per site; (ii) alterations and deletions in the EHR are automatically recorded and time-stamped by the EHR software; and (iii) copy of consent form sent centrally | EHRs after study termination are different from non-trial participants |
| No adequate consent obtained | GP recruits patient without explaining trial | Possible | (i) Intervention restricted to low-risk interventions (patient could have received medication outside the trial); (ii) patient can opt out at any time and does not need to redeem GP prescription for study intervention; and (iii) GP needs to complete training in GCP | Trial participants are provided contact details to study website so they can report centrally any issues |
| Fabrication of follow-up data in EHR | GP enters follow-up data | Low: EHRs can be seen by other practice staff (unlike the case report form in conventional trials) | Minimal study-specific data collection | EHRs after study termination are different from non-trial participants |
| Fabrication of patient questionnaires | GP completes questionnaires | Possible | Analyses of patterns of completion |
Monitoring for fraud in point-of-care trials
The EHR research database was used to monitor for data irregularities suggestive of possible fraud at a site. Analyses were conducted to monitor the site with the largest number of recruited patients in Retropro. The first set of analyses evaluated the duration of prior registration with the practice and the frequency of contacts after recruitment. The second set of analyses used statistical techniques as proposed by Stephen Evans for fraud detection. 80 We estimated the Mahalanobis distance, which reflects how extreme a patient is compared with other patients for a large number of variables. It provides a measure of the distance between a patient and the centroid of the total population (i.e. the vector of means of all the variables involved). 81 This technique has been used for the quality control of batches of laboratory analyses, detecting batches with unusual characteristics. 82 Figure 4 shows the distribution of the Mahalanobis distance at the best recruiting Retropro site compared with the other trial sites and a random sample of statin users in non-trial sites. Visual inspection of the distributions of Mahalanobis distance did not suggest any irregularities in the best recruiting Retropro site compared with other sites.
FIGURE 4.
Distribution of the Mahalanobis distance of statin users at the best recruiting (a) Retropro site, other trial sites; and (b) non-trial sites.
Operating procedures and independent audit
Independent regulatory consultants were enlisted to conduct two GCP audits on our systems to assess regulatory compliance. Although we were not subject to any major adverse findings, a number of alterations were suggested to minimise concern in the event of an audit. A full set of standard operating procedures were recommended, detailing the requirements of each process and delegation of responsibilities. It was agreed to reduce the requirement to retain paper copies of all ‘essential documents’ to wet-ink originals only; all other documentation was filed in an electronic trial master file with controlled access rights restricted to key staff and auditors. Evidence of GCP training was ensured by completion of training every other year which was tailored to our specific requirements. Documentation on the specification and validation of the trial tailor-made IT system was created, along with a diagrammatic representation of how each individual module interacts with the others. Following recruitment of the first patient, the sponsor (London School of Hygiene and Tropical Medicine) also conducted an audit. This produced no adverse findings but identified some recommendations to ensure best practice, including the testing of clinical trial systems and the need for a formal statistical plan to be in place.
Discussion
This project used central data monitoring techniques to review data instead of site visits. The main reason for this was that the EHRs in the research database contain copies of the source records (minus the patient identifiers and free text). Central data monitoring is used only infrequently in conventional trials. This type of monitoring was identified in the regulatory review as a possible weakness of point-of-care trials. GCP states that central data monitoring can be used only in ‘exceptional circumstances’. 39 However, a systematic review of on-site monitoring techniques found little empirical evidence to support its widespread and costly use. 83 A detailed analysis of a single international trial found that the large majority of findings as identified during a site visit could have been identified by central monitoring. Only 5% of the findings could only have been identified by manual review of the site records;84 these mostly concerned transcription errors between the source records and the case report forms, which do not apply to point-of-care trials that use EHRs.
There are several advantages of central data monitoring based on an EHR research database. One advantage is that the generalisibility of the trial population and the level of recruitment can be assessed. Retropro and eLung managed to recruit only a small minority of the potentially eligible patients. Furthermore, our review of the possibilities for fraud in point-of-care trials indicates that most scenarios of fraud could be prevented in point-of-care trials as the trial system (i.e. the EHRs) is accessible to other staff at a practice. The examples in this chapter and reports in the literature85 also show that several statistical techniques can be used to monitor for data irregularities. Arguably, central data monitoring techniques should be considered the norm in point-of-care trials rather than the exception, as stated in GCP.
The hot recruitment model, as tested in eLung, was found to be less successful with respect to cold recruitment. There may be several explanations for the low recruitment rate in eLung. First, we were unable to recruit over two winter seasons as planned. Second, many COPD patients regularly received antibiotics already (and there was a recent NHS initiative to provide them prophylactically with antibiotics). It may be less acceptable for clinicians to randomise in case of an established clinical practice. The provision of eDiaries during consultation on top of the consent requirements also failed. GP investigators were critical of the failure to keep things simple in eLung. The study team believes that hot recruitment can work but only if recruitment procedures are kept simple. Study-specific data collection would need to be limited and consent procedures should be conducted some time prior to the actual recruitment (i.e. patients would consent to future randomisation).
Chapter 8 Qualitative process evaluation (QUalitative Evaluation of a trial of ANtibiotics for chronic lung disease study)
Introduction
Employing complementary qualitative research methodologies has been viewed as increasingly relevant in the planning and execution of trials. In particular, it is more common to use qualitative methods to develop the design, conduct, outcomes and efficiency of clinical trials86 or to enable health professionals, service users and other stakeholders to contribute their views and experiences to evaluation of treatments, interventions, policies or methodologies. 87,88
A qualitative process evaluation [QUalitative Evaluation of a trial of ANtibiotics for chronic lung disease (QUEAN)] study was nested within eLung. The primary aim of eLung, as discussed before, was to demonstrate the feasibility of a novel approach of ‘randomisation within the database’ through the modification of GP computer software to randomise patients between treatments and collect data through routinely collected EHRs. Clinician and patient recruitment is one of the most challenging aspects of completing any successful trial. Recruitment in primary care has particular challenges related to the characteristics of primary care practitioners and patients and the dispersed nature of the primary care setting. The resulting recruitment problems have implications for internal and external validity and the timeliness of trials and costs. 89 As Bower et al. 89 describe, the recruitment process in primary care initially has two steps: first, practitioners must agree to provide the context for recruitment by agreeing that their practice will participate (stage 1); and, second, many trials (especially of acute conditions) require practitioners themselves to recruit eligible patients directly within their consultations (stage 2). 89 Regarding stage 1, qualitative interviews with GPs to explore the reasons behind non-participation in research have identified concerns relating to GPs recruiting their own patients into studies (e.g. not being sufficiently recognised as a partner in the research and not having a voice in the research process). 73 Other qualitative research has also highlighted the importance of the clinical relevance of the research question, clarity of practice responsibilities, realistic expectations and support available. Attention to presentation (length, style, clarity) is also important for engaging the attention of GPs. 90
In stage 2 of the recruitment process, recruiting GPs have a great deal of control over the accrual of patients into a trial and make judgements about patient’s preferences, as well as his or her physical and psychological suitability. In addition, scepticism about the benefit of the intervention being offered in the trial has been observed to lead to such ‘gate keeping’ by referrers and resulted in poor recruitment rates. 91 There is also a body of literature that has looked at the role of communication in trial participation and has informed researchers regarding the process of informed consent, understanding of trial terminology and reasons for participation and non-participation. 92
The QUEAN study aimed to ascertain valuable detailed information about this novel recruitment process in eLung to understand the mechanisms behind its success or failure in UK primary care settings. In particular, the qualitative process evaluation aimed to:
-
examine stages 1 and 2 of the recruitment process from the views and experiences of GPs who declined or agreed to participate in eLung
-
understand the experience of patients who agreed or declined to participate within this trial
-
summarise the key challenges to, and recommendations to improve, recruitment into these trials in order to inform best practice.
Methods
The QUEAN study was conducted by the University of York on behalf of the NIHR Health Technology Assessment programme.
Sampling of general practitioner and patient participants
Based on previous research, a sample size of 15–20 GPs who declined and 20 GPs who agreed to participate in eLung was estimated as sufficient to reach data saturation. 92 If participant numbers allowed, purposive sampling aimed to include a broad range of GPs by geographical region, type of location (inner city, urban, rural, remote), size of practice, sex and recruitment level (i.e. a mix of high and low recruiters). A sample of 20 trial participants (10 in each arm of the trial) and 10 individuals who declined to participate in eLung was considered sufficient to achieve data saturation. 92 A purposive sampling method intended to aim for maximum variation to include both men and women in a range of locations. However, due to delays in initiating eLung, it was no longer possible to purposively sample participants for the QUEAN study.
Recruitment methods for general practitioner participants
Practices were initially approached about participation in the QUEAN study by either the CPRD organisation or by PCRN. CPRD’s data protection requirements are the reason for this, as direct contact between CPRD practices and an external organisation, such as the University of York, can occur only after consent by the practice. In the approach by CPRD, the QUEAN study recruitment was integrated with the recruitment process for Retropro and eLung. GPs in the 459 CPRD practices were sent two invitation letters from CPRD for the QUEAN study. The national PCRN co-ordinating centre and several regional offices also contacted practices in order to boost the QUEAN study recruitment (reaching practices that were not part of CPRD). GPs who were interested in receiving more information about the QUEAN study were asked to contact the University of York via e-mail, telephone or an expression of interest form. They were then provided with a full information pack and consent form. Following receipt of written consent, interviews were arranged with individual GP participants by the QUEAN study researcher by telephone or e-mail and confirmed in writing. Remuneration for GPs’ time to participate in the interview was provided at the standard PCRN rate.
Recruitment methods for patient participants
Given the sensitivities of inviting patients who declined to participate in eLung to participate in a related interview study, the initial approach regarding the QUEAN study was conducted by GPs. Following the invitation for eLung during the consultation using LEPIS, GPs could inform their patients about the QUEAN study providing them with a patient information sheet and an expression of interest form. Patients who were interested to receive more information could contact the University of York via telephone, e-mail or returning the completed expression of interest form. Following receipt of written consent, interviews were arranged with individual patient participants by the QUEAN study researcher by telephone or e-mail and confirmed in writing.
Ethics and research governance
The QUEAN study was approved by the South East Research Ethics Committee and over 100 R&D NHS trusts in England. The aim of this approach was to ensure approvals were in place in all NHS trusts in England prior to recruitment of the QUEAN study participants to avoid delay of interviews of newly recruited GP participants. In reality, the approval process continued for 18 months. The need for individual GP practices to obtain additional site-specific approvals instead of being listed as a patient identification centre resulted in further substantive delays. As a result, GPs who had declined to participate in eLung were not interviewed until a year or more after their decision and some patients were interviewed within 6 months of their invitation to participate in eLung. In Scotland, generic and local approval processes were completed for seven health boards and local practices within a month.
Data collection of general practitioner participants
In-depth interviews with GPs and patient participants used a semistructured topic guide (see Appendix 3 ). The questions were as open-ended as possible to enable participants to raise issues important to them. The wording and order of topics covered also varied considerably between interviews to ensure each interview was informal and open-ended to suit individual participants. 93 With the exception of one GP, all participants provided consent for interviews to be digitally recorded. The one GP telephoned the University of York and asked to be interviewed at that time, resulting in the interviewer taking notes while conducting the interview. Interviews with GPs were conducted over the telephone at a time of the GP’s convenience and lasted approximately 1 hour. Interviews with patient participants were conducted over the telephone or face to face at a location. Patient interviews lasted between 30 and 45 minutes.
Data analysis
Audio recordings were fully transcribed with anonymisation of all personal data. Interim analysis was carried out to develop the topic guide in the early stages of data collection to reflect emerging and important topics. 94 Framework method of analysis95 was adopted to organise the data. This approach was developed specifically for applied qualitative research in which the objectives of the study are often known in advance in agreement with a funding body. Although the framework approach starts from pre-set aims and objectives, as reflected in the QUEAN study topic guide, it adapts to reflect the original accounts and observations of the people studied based on a grounded and inductive approach. 96 The stages of framework analysis followed for the QUEAN study were those detailed by Pope et al. 96 The first stage was familiarisation, during which the researcher becomes immersed in the raw data by listening to the recordings and reading transcripts to list key ideas and recurrent themes. Those ideas and themes were then developed into a thematic framework which draws on the questions based on the study objectives and the novel views and experiences raised by the participants themselves. 96 The data in the transcripts were systematically indexed by annotating all the data with numerical codes from the thematic framework. This includes the constant comparison method to check and compare each item with the rest of the data to establish appropriate analytical categories. 96 This process is inclusive to ensure that categories are added to reflect as many as possible of the nuances or outlier views in the data. A charting process then organises the data according to the relevant part of the thematic framework to form charts. One chart per theme per participant was initially developed for theme by case analysis for the QUEAN study containing summaries of the participant views and experiences and references to verbatim text in the transcript. The individual charts were then grouped to include data entries for all participants in each subgroup. This dual process enabled comparative analysis to be carried out to allow data from individual participants and subgroups to be compared and contrasted. The final stage of framework analysis was mapping and interpretation to examine the associations between themes to provide explanations for the findings.
Characteristics of general practitioner sample
Thirty-nine GPs agreed to participate in the QUEAN study. The 39 GP participants comprised 17 GPs who had declined and 22 GPs who had agreed to participate in eLung. Twelve GPs subsequently withdrew from the QUEAN study prior to interview, including five GPs who had declined and seven GPs who had accepted participation in eLung. Twenty-seven GP participants completed an interview for the QUEAN study, a shortfall of eight GPs compared with the target number of 35–40 GP participants ( Table 12 ).
| Group | Target number | Number recruited | Number withdrew | Reasons for withdrawal | Number interviewed |
|---|---|---|---|---|---|
| GP trial decliner or accepter | 35–40 | 39 | 12 | No response n = 10 | 27 |
| Retired n = 1 | |||||
| Maternity leave n = 1 |
The total of 27 GP participants comprised 12 GPs who declined and 15 GPs who accepted participation in eLung (as detailed in Table 13 ). The 12 GPs who declined participation in eLung comprised nine GPs who had declined from the outset (GP decliner) and three GPs who initially accepted participation in eLung but later withdrew following problems during the study set-up phase (GP withdrawal). The 15 GPs who accepted participation in eLung comprised eight GPs who had not yet completed all trial initiation procedures and seven GPs who were recruiting. The seven GPs who were recruiting included a mix of high and low recruiters: three GPs had not recruited any participants and four GPs had recruited between 1 and 12 participants. This sample included GPs who had also been simultaneously invited to participate in Retropro. Most GPs declined or agreed to take part in both Retropro and eLung except for two GPs who declined eLung but accepted Retropro and three GPs who accepted eLung but declined Retropro.
| Number of participants (total n = 27) | eLung status | Retropro status |
|---|---|---|
| 9 | GP decliner | 7 GP decliners |
| 2 GP accepters | ||
| 3 | GP decliner (accepter-withdrawal) | 2 GP decliners |
| 1 GP accepter | ||
| 8 | GP accepter (incomplete set-up) | 1 GP decliner |
| 7 GP accepters | ||
| 7 | GP accepter (recruiting/recruited) | 7 GP accepters |
The sampling characteristics of GP participants within the QUEAN study are presented in Table 14 .
| Characteristic | GP decliners (n = 9) | GP withdrawals (n = 3) | GP accepter – incomplete set-up (n = 8) | GP accepter – recruiting (n = 7) | All GP participants (n = 27) |
|---|---|---|---|---|---|
| Men/women | 8/1 | 1/2 | 3/5 | 6/1 | 18/9 |
| Size of practicea | |||||
| Small | 4 | – | 3 | 4 | 11 |
| Medium | 2 | 3 | 2 | 2 | 9 |
| Large | 3 | – | 3 | 1 | 7 |
| England/Scotland | 9/– | 2/1 | 7/1 | 4/3 | 22/5 |
| England: region | |||||
| North | – | 2 | 3 | 1 | 6 |
| South | 4 | – | 3 | 3 | 10 |
| Central | 2 | – | 1 | – | 3 |
| London | 3 | – | – | – | 3 |
| England: type of location | |||||
| Inner city | 3 | – | 1 | – | 4 |
| Urban | 3 | 1 | 2 | 2 | 8 |
| Rural | 2 | – | 2 | – | 4 |
| Mixed | 1 | 1 | 2 | 2 | 6 |
| Level of deprivation in practice catchment area (compared with national average) | |||||
| High | 3 | – | 3 | 4 | 10 |
| Average | – | 2 | – | 2 | 4 |
| Low | 2 | – | 1 | 1 | 4 |
| Mixed extremes | 4 | 1 | 4 | – | 9 |
| Main ethnic group(s) in practice catchment area | |||||
| Nearly all white | 4 | 3 | 3 | 2 | 12 |
| Some ethnic groups | – | – | 3 | 3 | 6 |
| Diverse mix | 5 | – | 2 | 2 | 9 |
Two-thirds of the GPs who provided their views on and experiences of eLung were men. This is higher than would be expected based on the proportion of male and female GPs registered in general practice across the UK (51% vs. 49% respectively). The GP participants were practising within a variety of sizes of general practice with generally a relatively even spread between the four GP categories of eLung status. The convenience sample of GP participants met the study criteria of including GP views from England and Scotland, a variety of regions in both countries, and a diversity of practices in terms of size and location in inner-city, urban, rural or mixed urban rural areas. In most cases, this diversity was seen across the four categories of GPs who had ‘declined’, ‘withdrawn’, ‘accepter – incomplete set-up’ and ‘accepter – recruiting’. The sample achieved representation from GPs who provide care to population groups experiencing a wide range of deprivation levels and from a variety of ethnic backgrounds. This diversity was seen within the four categories of GPs by eLung status. Limitations of the sample include the relatively smaller numbers of GPs in Scotland than England and the absence of any GPs in Scotland who had declined to participate in eLung from the outset or who are located in an inner-city location.
General practitioner theme 1: factors influencing the general practitioner decision to participate in research
There are several factors which influence the GP decision on whether or not to participate in a research study, across all groups of GP decliner, withdrawal, incomplete set-up and recruiter. The main influencing factors (based on the total number of GPs who identified that factor as influential) were:
-
time and capacity to implement a study (18 GPs)
-
benefit of research for local patients or population (13 GPs)
-
adequate remuneration to cover costs or generate a profit (13 GPs)
-
positive attitudes of patients to research (12 GPs)
-
interest in research in primary care and/or CPRD research (11 GPs).
The most important factors influencing the decision on whether or not to participate in research (based on the total number of GPs who identified that factor as the most important) are slightly different, however, and were:
-
time and capacity to implement a study (11 GPs)
-
interest in research in primary care and/or CPRD research (seven GPs)
-
benefit of research for local patients or population (four GPs)
-
adequate remuneration to cover costs or generate a profit (four GPs).
Positive attitudes of patients to research
It is interesting to note that, although the positive attitudes of patients to research was identified as a factor influencing the GP decision on participation in research (12 GPs), this was not identified as the most important factor by any GP. Descriptive data from the 12 GPs suggest that GPs are confident that their doctor–patient relationship will usually achieve a positive response from patients to the research if the GP takes the time to explain the study properly. This includes six GPs who provide care to populations experiencing high levels of deprivation. One exception to this view came from a GP based in a deprived area in the north who felt the patients are ‘too busy dealing with life stresses to be interested in research’. This GP had accepted participation in eLung, supporting the finding that this single factor is less likely to influence her decision to participate or not in a research study, but rather their ability to recruit among patients experiencing high levels of deprivation might be more difficult.
Time and capacity to implement a study
Nearly all GPs in this sample described a negatively changing context for research in primary care over recent decades due to significant increases in workload, reporting requirements and patient demand:
I don’t think we’ve ever worked so hard as at the moment.
GP 1012 (GP decliner)
The increasing workload and the extra QOF requirements have caused instant increases in our anti-depressant dosages. It’s grim.
GP 1009 (GP decliner)
The workload has quadrupled over the last 20 years, mainly due to the endless contract box ticking, non-contract box ticking, more administration and more patient demand.
GP 1019 (GP recruiting)
It’s all the QOF, all the tick boxes that you have to tick off . . . rather than deal with the individual, it is far more, a collection of diseases and you have to do x, y and z for each disease, it’s much more difficult to take the holistic view.
GP 1036 (incomplete set-up)
Within this context, the amount of time and ability a GP has to conduct a study has been identified as the most common and most important factor influencing the decision to participate in research. The amount of time a GP has may depend on the timing of when the GP is invited to participate in a research study. This is illustrated by a GP who had accepted participation in eLung but would have declined if asked at the point of completing the QUEAN interview because of the particular high level of clinical demand at that time. As indicated above, this suggests the timing of any recruitment activities to GPs should be considered within the context of their routine reporting or clinical commitments factoring in, for example, seasonal variations. The capacity of a GP to conduct a research study has been clearly identified by several GPs as determined by the simplicity of the study design and how easy it will be for the GP to incorporate it into their work:
I just don’t have enough time, even just an extra minute per consultation is not feasible at the moment.
GP 1036 (incomplete set-up)
If it doesn’t look too complicated and easy to understand and incorporate into daily business, it’s more attractive.
GP 1038 (withdrawal)
It depends how difficult the study is and how much it messes up the way we work.
GP 1029 (decliner)
Interest in research
General practitioners identified a personal interest in research as the second most important factor influencing their decision to participate in research. A strong level of personal interest in research appeared to be a predominant characteristic of 17 GPs who initially agreed to participate in eLung:
There’s so much bureaucracy and red tape with research it can put people off, some GPs are like, ‘Oh I’m not interested, I just can’ think about it because it’s just too much extra work. It’s got worse in the last 3 or 4 years, we’re getting used to QOF now although that keeps changing and there’s insurance reports, reports of the benefits agency, people having to appeal because their benefits have stopped so they need a report about their health for the last 20 years. There’s so much paperwork. Then the GP commissioning pulls them away from the practice for meetings, time is tight. You’ve got to be interested in research to overcome all that.
GP 1025 (recruited)
Nearly half of the GPs who identified an interest in research as an influencing (4/11) and important (4/7) factor identified a specific interest in EHR research and particularly EHR trials:
Personally, I want to be part of the national CPRD research.
GP 1005 (incomplete set-up)
I have a very strong interest theoretically in the method of running a trial within the CPRD database.
GP 1036 (incomplete set-up)
I have a personal interest in informing the study design in early stages.
GP 1030 (GP recruiting)
Conversely, three GP decliners who stated that they had no personal interest in research also had strong negative views towards research because of either a fear of litigation (one GP) or feeling extremely stressed and demotivated owing to workload (two GPs). The two GP decliners who expressed an interest in research were either disillusioned with the role of identifying participants for other studies or had a preference for local, practical research and a dislike of national academic-led research. It appears that a lack of strong personal interest in research in primary care enables the influence of other negative pressures or barriers to result in a decision to decline participation in a study. More positively, a strong personal interest in research appears to motivate a GP to accept participation in a study and potentially overcome those same barriers to research in primary care. This suggests that it is important to target recruitment for future trials to lead GPs for research or other GPs with a specific interest combined with broader educational and awareness strategies regarding trials within the database to generate interest in EHR research.
Benefit for local patients or population groups
The potential benefit of research to local patients or population groups was identified as an influential factor by approximately half of all GPs (n = 13), and as of primary importance by four GPs. Potential benefits to patients valued by GPs included improvements in the general standard of care received for a particular condition and an opportunity to take part in research which is often associated with improved health. Most GPs also identified the need for the research to be locally relevant and clinically important for the local population.
Adequate financial remuneration
Thirteen GPs also identified financial remuneration as an influencing factor. Five GPs identified the need for the study to generate a small profit including three GPs across different subgroups who considered this the most important factor influencing their decision to participate. This equates to 11% of GPs within the study sample (3/27) who identified making a profit as the most important factor influencing their decision. Of the eight GPs who identified the need for the study to be adequately remunerated to cover study costs, only one considered this the most important factor influencing his participation decision. The need to cover study costs is a consideration when a practice is involved in several studies, to cover staff costs or when motivation to conduct research is low because of the type of research or pressures of increasing workload:
Financial gain is helpful across some of your portfolio of studies as you would run into difficulty if they all ran at a loss.
GP 1031 (incomplete set-up)
Remuneration from [other organisations] has not been adequate, primarily because small numbers of patients were identified for the study. We don’t expect to make a profit but we’re not happy to run at a loss.
GP 1003 (decliner)
Previously I did research for nothing but it’s important to generate a small income now as it’s less motivating doing research identifying patients for [other organisations] rather than doing our own studies.
GP 1029 (decliner)
The remuneration needs to be adequate to cover the work it entails as we all have to think about staff costs these days and have we got the resources to fulfil the requirements.
GP 1025 (recruiter)
It’s important to make a profit for income as well as cover costs.
GP 1016 (withdrawal)
If the study’s not paid, it’s a definite no. Generous funding makes it more attractive.
GP 1037 (recruiter)
The remuneration should definitely cover costs and be enough to make a profit.
GP 1019 (recruiter)
One GP highlighted the importance of an appropriate payment to allow for the small number of patients who are likely to be recruited in some types of trial or practices.
Package of factors
As the relatively even spread of GP counts for each of the four influencing factors on the GP decision suggests, several factors appear to work in combination to result in an overall decision on whether or not the GP will participate in research generally. There appear to be various combinations of the factors identified as the most important, suggesting they are all important to some degree and need to be considered as a package to promote GP participation in future point-of-care trials:
Studies need to be locally relevant and practical, not academic and all too difficult and not worth the effort.
GP 1012 (decliner)
How feasible it is, how time consuming and awkward it looks and whether it’ll be worth the rewards.
GP 1019 (recruiter)
If the CPRD trials continue and provide the funds and make research interesting and not too labour intensive, then they might get some interest.
GP 1037 (recruiter)
CPRD offers a very attractive package, interesting studies for patient benefit and well renumerated.
GP 1038 (recruiter)
Notably, these factors were found to be similar for all GPs across groups of decliners, withdrawals, incomplete set-up and recruiters. This supports the earlier finding that a strong interest in research appears to be a key motivational factor within which the package of factors is considered as barriers or facilitators to research. The level of remuneration provided for Retropro and eLung was regarded as appropriate by the majority of GPs within each subgroup by eLung status.
General practitioner theme 2: factors influencing decision to participate in eLung
The following results are GP responses to an open question about the main factors which influenced their decision to participate in eLung. These results need to be considered in the context of the most important factors previously identified as influencing the GP decision for research in general, which are (in order of importance) time/capacity to implement the study, personal interest in research, benefit to local patients or population groups, adequate remuneration, improving clinical practice in long term for general population, negative attitude of patients to research and ethics and patient safety. Within this general context, the following practical factors were identified by GPs as influencing factors, positively or negatively as indicated, when deciding whether or not to take part in eLung:
-
Positive factors:
-
consistency with local prescribing guidance (n = 18)
-
relevance of topic (n = 14)
-
feasibility to recruit (n = 13)
-
– number of patients (n = 9)
-
– minimal impact of rescue packs (n = 4)
-
-
computer-based pop-up alerts (n = 10)
-
hot recruitment method (n = 6)
-
demands of study design on GP time (n = 4)
-
benefit to patients (n = 3).
-
-
Negative factors:
-
feasibility to recruit (n = 10)
-
– high impact of rescue packs (n = 4)
-
– patient views on ‘no antibiotics’ (n = 4)
-
– number of patients (n = 2)
-
– lack of referrals from other GPs (n = 1)
-
-
hot recruitment method (n = 9)
-
relevance of topic (n = 4)
-
time to conduct study at point of invitation (n = 4)
-
demands of study design on GP time (n = 4)
-
computer-based pop-up alerts (n = 3)
-
consistency with local prescribing guidance (n = 2).
-
The factors which had a positive influence on some GPs in their decision-making process were the same factors which had a negative influence on other GPs. Based on earlier findings, this is likely to be due to some extent on the underpinning motivation and attitude of the GP based on his or her level of personal interest in research combined with local barriers to research within their working environment. This suggests strategies to enhance the ease, simplicity or relevance of all the influencing factors, combined with strategies to increase or facilitate a high level of interest in the particular study, is likely to have a positive effect across all GPs.
Consistency with local prescribing guidance
Consistency of the eLung protocol with local prescribing guidance was the most frequently cited positive influence on the decision-making process for participation. In these cases, most GPs considered the study protocol to be consistent with their local prescribing guidelines on no antibiotics for a non-infective exacerbation of COPD. In two cases, the protocol provided a strong rationale to justify any contradiction to local prescribing guidelines. Several GPs identified the need to explain the rationale clearly to patients to overcome potential patient concerns and a small number considered the study would facilitate implementation of their ‘no antibiotic’ local prescribing guidelines. These views were expressed by GPs across all groups of decliner, withdrawal, incomplete set-up and recruiting. Two GPs expressed negative views that the protocol contradicted national and local guidelines on management of COPD exacerbations for patients who have previously been provided with antibiotics and corticosteroids for self-treatment at home. 97 The majority of GPs expressed a clear view that it is important for a trial protocol to be consistent with local prescribing guidelines.
Relevance of topic
The relevance of the study topic for eLung was the second most commonly cited positive influencing factor (n = 14) and the third most commonly cited negative factor (n = 4). Fourteen GPs, including GPs across all groups, to varying degrees, considered the eLung research topic as highly relevant and important clinically in the local population. One GP who subsequently withdrew because of problems with set-up considered the topic highly relevant to inform useful and needed advice to standardise practice in this area. Four GPs, all of whom were in the GP decliner group, did not find the topic relevant. The reason for this was the low incidence of COPD in their population (n = 3) or lack of interest in comparing one drug to doing nothing (n = 1). This GP did participate in Retropro because of a personal interest in comparing the side effects of different drugs, again highlighting the importance of personal interest as a key motivating factor in the decision-making process.
Feasibility to recruit
The feasibility to recruit patients into eLung was commonly cited as a positive (13 GPs) and a negative (10 GPs) influencing factor in the decision-making process for participation. Indeed, this was the most frequently cited negative factor influencing the GP decision on whether or not to take part in eLung with 6 of the 10 negative concerns being expressed by GP decliners. Specific concerns regarding feasibility to recruit included the impact of rescue packs on reducing patient numbers and potential confusion for patients, patient concerns regarding no antibiotics, lack of potentially eligible patients due to low incidence of COPD, and a lack of support in identifying and referring patients from other GPs:
We have a very low incidence of smoking and COPD so it’s not a priority issue and not worth the effort. It’s 20 years out of date as well, now we have rescue packs and micro-manage patients on the basis of need. It’s not very practical.
GP 1012
We had difficulty recruiting and following up in a previous trial so that put us off. The low number of people with COPD in the practice may have made it difficult to recruit.
GP 1003
It would have been difficult to recruit given so many patients would have been excluded having used their rescue packs or it would have contradicted local guidance and potentially go against good practice.
GP 1004
Patients without antibiotics are not likely to participate or comply. It would just cause disruption and argument.
GP 1009
In direct contrast, the positive factors underpinning the feasibility to recruit were based on the view that there will be a large number of eligible patients to recruit from within the local GP practice (nine GPs) and a minimal impact of rescue packs on participant numbers. The latter is based on the perceived views of GPs who have not started recruiting or completed set-up (three GPs) and a GP who has recruited four patients.
Computer-based pop-up alerts
The use of computer-based pop-up alerts to identify, screen and recruit patient participants was the fourth most influential factor both for GPs who considered this a positively influencing factor (n = 10) and for GPs who considered this a negatively influencing factor (n = 3). The negative views towards computer-based pop-up alerts appear to be based on a particularly strong dislike of this method from existing use in routine care and reporting. None of the GPs decliners who held these particularly strong negative views cited this as the main factor influencing their decision to decline participation in eLung. In contrast, the GPs’ positive views regarding pop-up alerts include excitement about their potential use in trials and, in particular, their time-saving attributes and efficiencies to reduce workload:
the alerts for QOF feel like brainwashing asking the same questions all the time.
GP 1009 (decliner)
the partners and GPs are not happy with additional pop ups, they’re clutter and an aggravation factor.
GP 1012 (decliner)
I’m not interested in a study with extra pop ups, there are too many already which interfere with the doctor-patient relationship.
GP 1034 (decliner)
I liked LEPIS to reduce the workload element of identifying patients through searches and getting a timely reminder removes the stress of having to remember the study all the time.
GP 1016 (withdrawal)
I thought of the trial as the ‘pop-up trial’, it was an interesting and memorable feature.
GP 1018 (incomplete set-up)
It’s useful to have an alert for an eligible patient.
GP 1017 (recruiter)
Hot recruitment method
Opportunistic recruitment of an eligible patient during the routine GP consultation was the most controversial element of eLung and generated the most frequently cited negative views, primarily from GP decliners (n = 6) but also from a small number of accepters (n = 3). The over-riding concern expressed by nine GPs regarding the concept of the hot recruitment method is a lack of time to include an additional task in the routine patient consultation and the subsequent lack of support from GPs to assist with recruitment:
Surgeries are already long and gruelling so only two GPs in the practice were willing to add an extra 5 minutes into their consultations for another study using hot recruitment.
GP 1029
Time is a fundamental problem.
GP 1032
Surgeries are always running late. Colleagues just won’t agree.
GP 1009
We’re a small busy practice and always running behind.
GP 1012
In contrast, GPs citing this as a positively influencing factor expressed their support for the concept in principle and a general ‘willingness to give it a go’. Only one of the six GPs expressing a positive view had used the hot recruitment method. The need for the recruitment method to be as simple and efficient as possible in practice was identified by GPs as a way of addressing their concerns and increasing motivation:
The hot method using pop-ups sounds like the best method. The truth is, for me, I prefer whatever is most efficient for a practice or as a group as a whole. Usually if I can do it there and then, that is going to be most efficient. If the study is then going to require 20–30 minutes at one time, I would say it’s going to be virtually impossible to do that. I get 10 minutes and I can allow 12–15 minutes so I’d much prefer to pass them onto someone else. If it’s going to take an extra 2–3 minutes on what I’ve already done, then I don’t see much point in passing it on to someone else.
GP 1004 (decliner)
Demands of study design on general practitioner time
Three of the GP decliners cited the study design for eLung as too demanding on GP time. These GPs had also expressed negative views regarding the hot recruitment method, pop-up alerts, patient concerns over no antibiotics, lack of local relevance of the topic, in addition to a lack of time to conduct the study. The same GPs also stated their lack of interest in research or eLung due to it not being locally practical and ‘academic’ or due to a feeling of being overwhelmed by the stress of daily clinical practice. None of these GPs had prior experience in research except one GP who had been involved in a small number of local projects. These three GPs provide care to population groups typically experiencing high levels of deprivation and from diverse ethnic minority backgrounds, including newly arrived immigrants. All three GPs are located in inner-city locations, two of which are in London. The following provides insight into the stresses of one of these busy, inner-city clinics and the interplay between several factors influencing GPs negative views in their decision-making process regarding participation in research:
I’m too set in my ways to change my antibiotic prescribing habits and I’m more than aware that a lot of the time that antibiotics are being prescribed for viral illnesses but because of expectations of patients or by habit from society that we’ve taught them to expect antibiotics for viral illnesses, I knew that one way or another, there’s a rod been made for own backs with prescribing and to break out of it would be extraordinarily difficult.
Just yesterday I was on triage which means no set appointments but all the incoming phone calls are put up on a screen, with a chesty cough etc. Some of that is antibiotic prescribing over the phone to patients you know but some cropped up where they’d contacted a colleague the day before with a child with ear-ache or something, and they’d been given advice but not given antibiotics and the next day the child is worse, well to actually bring that child in and check if they can just persevere and doesn’t need antibiotics, well, it would take time to give an explanation that we haven’t got and the next thing is they’d get them anyway through out-of-hours so, I’m well aware of the problems, if antibiotics had been a lot more unsafe to give, they wouldn’t have been overused in the way they are.
When we’re getting lots of our contacts with sore throats and minor upper respiratory tract infections or whatever, we can deal with those in a few moments, the patient’s happy, we’ve quite likely used an antibiotic NHS resource unnecessarily, the immediate problem is solved and to actually get through a triage session requires a lot of adrenalin, firing on all cylinders, flat out work for whichever of us is doing it whether its morning or afternoon and to actually take something that’s straight forward and make it more difficult it and lengthen it and make it harder would be intolerable! You’re running backwards for the first hour and a half as the calls come in faster than you can cope with but we’ve usually caught up by 12 o’clock.
GP 1034 (decliner)
In contrast, the four GPs within the ‘incomplete set-up’ group who considered the demands of the study design as a positive factor perceived the workload would be minimal using LEPIS alerts, EHRs to collect outcome data and the small numbers of patients. These GPs also expressed positive views on at least two other influencing factors, including the topic, use of computer-based alerts, the feasibility to recruit, hot recruitment or consistency with local prescribing plans. All four GPs were supportive of or had a strong interest in research, particularly studies which are within the EHR database or utilise a potentially quick and efficient method. Three of the GPs had previous experience in research. One GP described the particularly high levels of stress and workload in current primary care practice but had not experienced personal ill health associated as a result of that as had been the case with the two GP decliners who described similar demands. All four GPs provided primary care to predominantly white Caucasian population groups with some pockets of deprivation in mixed urban/rural locations.
These contrasting case studies further illustrate the emerging link between an underpinning interest in, and motivation towards, research in primary care with a highly positive and open attitude towards the novel aspects of a trial using computer-based alerts at the point of care. The need for additional support to encourage and support GPs working in particularly demanding practices to participate in research are discussed in Appendix 4 .
Summary of the factors influencing GP decision on participation in eLung
There are two sets of individual factors which influenced eLung participation. The first one was the decision to participate in any research study and the second one was the specifics of eLung as an example of a trial within the EHR database (see General practitioner theme 3: general practitioner views on hot recruitment using computer flagging and General practitioner theme 4: views of outcome data collection via electronic health records). These factors were consistent across subgroups of GPs who declined or accepted participation in eLung and/or Retropro. Two subsets of the most important factors influencing the decision to participate in any research project and eLung are illustrated in Figure 5 . For the majority of GPs, several influencing factors will work in combination within each of the two levels of the decision-making process. The findings (see Table 32 in Appendix 4 ) suggest that at least two and usually three generic factors on participation in any research project are viewed positively by each GP as a prerequisite before considering the more practical, project-specific factors (see Table 33 in Appendix 4 ). The inter-relationship of factors between the two sets was not explored explicitly within this study.
FIGURE 5.
Two subsets of most important factors influencing the GP decision to accept or decline participation in research. Outer circle: generic factors for any research project. Inner circle: project-specific factors.
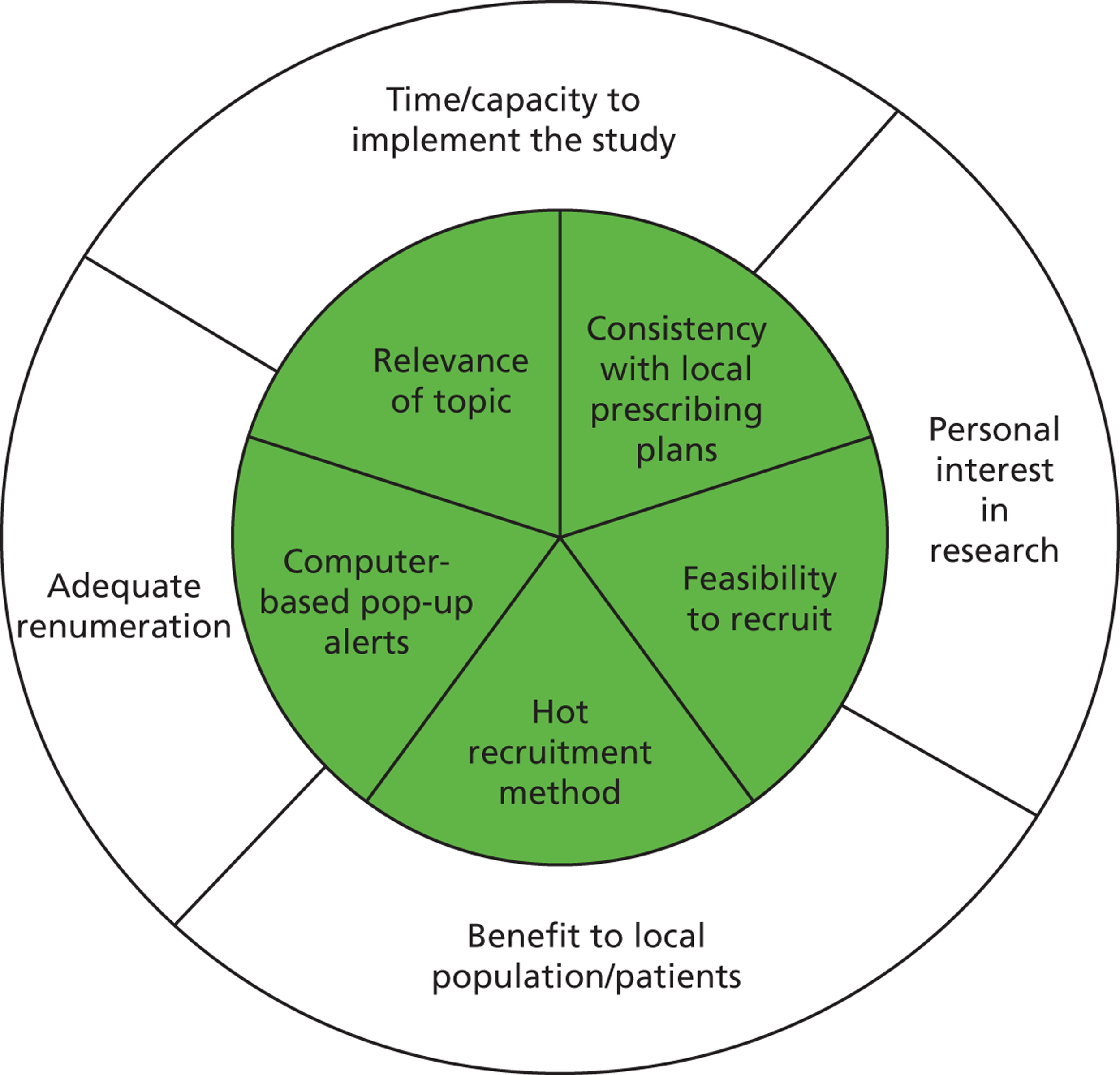
The likelihood of a GP having a positive view towards several individual factors appears to be underpinned by a high level of personal interest and motivation towards research within the context of increasing pressures on GP time in primary care. Conversely, a complete lack of interest in, or negative view towards, research appears to be linked with a generally negative view towards a number of individual factors. This attitudinal factor appears to be a key driver influencing the decision of some GP decliners over and above the companion generic factors of ‘time/capacity’, ‘patient benefit’ or ‘financial remuneration’.
General practitioner theme 3: general practitioner views on hot recruitment using computer flagging
A total of 19 GPs (6/9 decliners; 2/3 withdrawals; 8/8 incomplete set-up; 3/7 recruiters) provided in-depth data on their hypothetical views of recruitment using computer-based pop-ups at the point of care (i.e. hot recruitment). Four GP recruiters had recruited at least one patient into eLung. Lack of time to recruit during an unscheduled consultation was identified as a negative factor for a number of GPs, including GP decliners and accepters. As indicated in Table 15 , GPs identified six further subthemes regarding the proposed point-of-care recruitment method for eLung:
-
time to achieve informed consent
-
time to recruit patients with comorbidities
-
recruiting patients with an acute condition
-
flexibility to opt out of hot recruitment
-
flexible approach to hot and cold recruitment
-
simple and efficient design to give confidence to recruit.
| GP by eLung status | Informed consent | Comorbidities | Acute condition | Flexibility to opt out | Flexible approach to hot and/or cold recruitment method(s) | Simple and efficient design to give confidence and motivation |
|---|---|---|---|---|---|---|
| Decliner (n = 6/9) | 6 | 5 | 0 | 1 | 5 | 4 |
| Withdrawal (n = 2/3) | 1 | 1 | 2 | – | 1 | 2 |
| Incomplete set-up (n = 8/8) | 4 | 1 | 2 | 3 | 5 | 9 |
| Recruiters (n = 3/7) | 1 | 1 | 1 | 1 | 1 | 2 |
| Total (n = 19) | 12 | 7 | 5 | 5 | 12 | 17 |
Views of hot recruitment
The lack of time to discuss the study and achieve informed consent within an unscheduled consultation was a serious concern expressed by nearly two-thirds of GPs (see Table 15 ). This included concerns about the ability to discuss the study properly, particularly with patients from non-English-speaking backgrounds or patients with concerns regarding anonymity of data and data confidentiality, need for the patient to have 24 hours to consider their decision to participate or not, and irritation at the impracticality of having to sign three paper-based consent forms. The acute nature of a COPD exacerbation raised concerns among a small number of GPs that the patient would be too sick to be able to give informed consent. Two GPs felt that it was more appropriate and potentially easier to achieve informed consent with a face-to-face discussion rather than a postal recruitment pack. The suggestions by GPs to adopt a more flexible approach to the recruitment method were primarily aimed at trying to overcome the concerns regarding informed consent. Other hypothetical concerns expressed by GPs regarding hot recruitment were likely difficulties recruiting patients who presented with several comorbidities, compounding the lack of time to combine recruitment with clinical care. One GP suggested a potential solution was to explain to the patient that the focus of the consultation that day would be limited to their COPD and to ask them to book another appointment to discuss any other concerns. Flexibility to opt out of recruiting a particular patient was identified as an important and positive function of LEPIS. This appeared to allay concerns of some GPs regarding the intrinsic difficulties of a lack of time.
Installation of Local Eligible Patient Identification Service software and its functionality
All 17 GPs who tried to have LEPIS installed onto their practice computer, including all three GPs who withdrew, seven of the eight GPs who failed to complete the set-up phase for eLung and all seven GPs who were recruiting patients into eLung, experienced significant problems with the installation, primarily due to local firewalls within the primary care trust or Clinical Commissioning Group. An inability to successfully install LEPIS was the reason for two GPs withdrawing from eLung and for seven of eight GPs failing to complete set-up and progress onto recruitment of patients. The delay to install LEPIS was the main reason why another GP withdrew from eLung, combined with its subsequent poor functionality and the lack of eligible patients owing to home use of antibiotics from ‘rescue packs’ in the last 2 weeks.
General practitioner recruiters (7/7) also experienced delays of 1–9 months for LEPIS installation. Three GPs found LEPIS worked satisfactorily following installation, two GPs had a mixed experience where it was working but incorrectly and two GPs did not consider it reliable enough to use for eLung. Problems regarding functionality included incorrect timing of the flag to alert the GP of an eligible patient (n = 4), no eligibility criteria (n = 1) and causing problems within the functionality of Vision software (n = 2). Examples of the GP feedback on the installation and functionality of LEPIS are:
At several points in it we were thinking, ‘Oh God, do we really want to carry on with this [LEPIS installation], it’s like wading through treacle’. When it was eventually working, the flag appeared to be triggered by the diagnosis which was after you’d given the patient a prescription and they’d left.
GP 1025
I had an awful bother with setting it [LEPIS] up.
GP 1038
LEPIS wasn’t working for a long time but it has improved, it’s still very glitchy but is now a useable tool. It only flags up on diagnosis for eLung and doesn’t go away. It was also removing functionality within VISION for repeat or acute prescriptions on the designated computer. LEPIS can be a tool which can deliver what you want it to but there’s still work to be done.
GP 1030
It would have been nice if LEPIS had been working reliably right at the start and we could just rely on opportunistic recruitment.
GP 1037
Experiences of hot recruitment
The actual experiences of GPs to recruit patients in the routine appointment were generally more positive than the hypothetical views of GPs. As shown in Table 16 , most GPs (3/4) reported the process took 5 minutes and was straightforward and feasible on most occasions. One GP found the process took 20 minutes and did not feel able to recruit when the surgery was running late. One GP experienced no problems in achieving informed consent. The GP who did find it difficult to gain informed consent within the time available suggested a preferred approach of presenting the study to patients at their annual COPD review. Examples of GP’s experiences of the consent process are:
The COPD population tend to be more deprived but no-one’s turned me down for it once I’ve explained it. This is why it is so important for the GP to believe in the ethos of the CPRD to be comfortable to tell patients about the study. I can put my hand on my heart and say I think it is a good idea to do the research.
GP 1038
Once you’ve got your head around the three consent forms and having to explain it all in the time, it’s not too bad. I have to select patients who will be interested and can understand in the time.
GP 1025
I have no concerns about the consenting process. We’re not talking about putting anyone on a horrendous drug which on-one knows about, we’re talking about everyday practice. Does the patient really need to go away and think about that when the doctor will put them on one treatment and another doctor will put them on another every day so why do we have an extra 24 hours to think about it for example?
GP 1030
| GP by eLung status | Amount of time | Informed consent | Comorbidities | Acute condition | Flexibility to opt out | Flexible approach to hot and/or cold recruitment method(s) | Simple and efficient design to give confidence and motivation |
|---|---|---|---|---|---|---|---|
| Recruiters (n = 4) | 3 ✓ | 1 ✓ | 3 ✓ | 2 ✓ | 4 ✓ | 4 ✓ | |
| 1 ✗ | 1 ✗ | 2 ✗ |
Two GPs found the acute nature of the condition did limit the number of patients they felt able to recruit by half. In contrast, a similar number of GPs considered a treatment for an acute condition easier to administer in a hot recruitment situation than a change in a long-term drug. Three of the four GPs had used the study website instead of LEPIS to recruit owing to difficulties with the functionality of LEPIS. This was considered to work well and a useful back-up to LEPIS for two GPs. One GP experienced difficulties when the website would not accept the patient details. The GP’s experiences of hot recruitment in eLung included:
We created our own hot recruitment method by entering a code into the VISION pop-up for important messages so it comes up when we are consulting a patient. We then enter the data and randomise via the website which worked fine.
GP 1037
I was pleasantly surprised how easy it was when I actually did it. As a model of how these trials ought to be done, I think it’s quite good.
GP 1038
It’s impossible to do in the time available for both studies [eLung and Retropro] so if you’re very busy and running late in a surgery, you would probably ignore the recruitment opportunity. eLung takes 20 minutes. Once I got the hang of getting onto the website, it was very easy to use and randomisation is easy.
GP 1037
It’s not been easy, I started recruiting in summer so not many patients were presenting and the ones who do come in tend to need antibiotics or hospital. One eligible patient wanted antibiotics so didn’t consent. I have recruited one though and it was fine within the consultation, it just takes another five minutes to get the consent and randomise. Retropro was easier as there were more patients.
GP 1052
Some patients will be excluded due to mental illness or not being interested but I still would hope to recruit more typical, real world patients than the traditional clinical trials
GP 1019
Ease and efficiency of recruitment method to promote confidence
All four GPs who had experienced hot recruitment using computer-based pop-ups via LEPIS or the website expressed a clear view that, when the system is working, the recruitment process is fine and easy to do. A consistent and emerging theme from several GPs across all groups is a lack of confidence about whether or not it will work and the need for a simple and efficient process which will increase confidence to take the step of recruiting the first and most difficult patients:
I wasn’t sure how easy it would be but I thought I’d see how it went.
GP 1018
It’s all about the confidence to do the first one [recruit the first participant] and overcome the fear factor of ‘Am I going to be able to do it?’ Especially if you’re busy and the numbers are so small, is it worth the effort if you’re unsure? Retropro was bigger numbers and so more motivating in that way. I suspect some places [elung study sites] haven’t broken their duck [recruited] because they are not really sure what is going to happen.
GP 1038
Some studies are harder to start than others so we need to unpick what makes it hard. If the number of steps when you want to go [recruit] are complicated, then you may be less likely to start but if they’re done very slickly, then you are not . . . To start with, eLung failed on all these things as it was complicated. Even after you’ve done the training for eLung including the use of how to use the electronic diaries, I was already unsure what I was doing. If I was going to have to teach it to a patient, that meant I would need a substantial investment of my time to do it again, then my chances of remembering if I wasn’t going to do it for one or two months’ time, were close to zero. So we need to try and keep it simple, so what fell down here was, it’s a very common fault, everyone’s trying to come up with the perfect study but sometimes you make it so perfect to answer every question that comes along, it fails to achieve anything because it never gets off the ground.
The first or second times are the least likely times you are to recruit. What happens is when you first have a go to recruit, it either works well or is a total disaster and that determines how likely you are pre-test to recruit the next person. So where eLung has not been a success in my practice, because I’ve never recruited into eLung yet, therefore I’m thinking I don’t have a good familiarity, I’m aware the first time takes longer because I don’t know what I’m doing. I’ve also got all this in the background and so you never quite get started. So it’s self-fulfilling so that’s why it’s so important to actually get started in all these studies. The way that the study is set up, if it’s complicated or takes time to get set up or is very time consuming or you only see someone infrequently, then these are the reasons why you never get started and these studies often fail.
GP1030
I’d used it [LEPIS] in other studies so was confident it was doable.
GP 1031
The recruitment gets better as we get familiar with the study. So once we had done a few of them, you get to recruit ad hoc.
GP1052
To start with, it puts you off because you think ‘Oh God, I don’t know what to do and I don’t know how long it’s going to take’ but I think we’ve got over that now. Once you’ve enrolled a couple of patients you realise that it’s not going to take forever.
GP 1025
The delays in getting set up knock your confidence to start but when the system works, it’s actually very simple. It’s so important to get LEPIS working properly so it can be used more widely and reduce the workload involved in identifying and recruiting patients into trials.
GP 1037
General practitioner theme 4: views of outcome data collection via electronic health records
Twenty-six out of 27 GPs expressed their strong support for the use of EHRs to collect outcome data for trials within the database ( Table 17 ). Primarily, this approach is regarded as an efficient use of an existing, highly valuable resource of patient information within the primary care database to facilitate much needed research on real patients within real primary care settings. GPs across all groups prefer the remote electronic method for data collection over clinician-led questionnaires to reduce practice workload:
It’s the way forward.
GP 1017 (recruiter)
primary care has been computerised for about 30 years so there’s a huge volume of information in GP computer systems.
GP 1019 (recruiter)
I certainly felt EHR was a sensible and interesting way of getting useful outcome data of real world stuff. We need to be using our data from our patients, we have a strong data base, we have a strong history of computerised records and reasonably good data which we really should be making better use of.
GP 1036 (incomplete set-up)
EHR is intriguing as a modern method to replace subjectivity of patient self-report with hard end-points as well as reducing the clinician’s workload.
GP 1031 (incomplete set-up)
EHR is an interesting method and patient questionnaires have value for different purposes too.
GP 1005 (incomplete set-up)
It’s still in its infancy but it looks to have great potential.
GP 1037 (recruited)
EHR is a preferred method as its difficult to get questionnaires back and is a lot of administrative work to chase them up.
GP 1024 (incomplete set-up)
Use of EHR could help to achieve participation in research from other GPs as it’s less work and tick boxing.
GP 1011 (incomplete set-up)
The use of EHR was one of the things we liked about it.
GP 1048 (withdrawal)
It [EHR] sounds like a good idea as it would reduce the workload.
GP 1029 (decliner)
It’s [EHR] much better than questionnaires.
GP 1032 (decliner)
An integrated approach using routine data is more efficient.
GP 1004 (decliner)
I have no concerns with EHR and it’s less work for GPs.
GP 1052 (recruited)
EHR sounds efficient as the amount of time spent extracting data for study questionnaires is demanding and burdensome.
GP 1025 (recruiter)
EHR is massively advantageous as it removes the need for the GP to be involved.
GP 1030 (recruiter)
| GP by eLung status | Support for EHR | Lack of support for EHR |
|---|---|---|
| Decliner (n = 9) | 9 | 0 |
| Withdrawal (n = 3) | 3 | 0 |
| Incomplete set-up (n = 8) | 7 | 1 |
| Recruiters (n = 7) | 7 | 0 |
| Total (n = 27) | 26 | 1 |
One GP decliner particularly liked the potential for longitudinal data collection using routinely collected EHRs and one GP who had not completed set-up for eLung liked the scope for hard data end points compared with patient-reported outcome measures. One GP believed that EHRs would motivate other GPs to participate, suggesting this might be an important methodological element in potential education and/or recruitment strategies.
The one GP (incomplete set-up) who did not support use of EHRs to collect trial outcome data was very concerned about confidentiality of patient data and similar potential concerns by patients:
If the patient is fine and they’re OK for the data to go [to CPRD], I don’t have any problems with that. But if the patient comes all the time and the data is being downloaded [by CPRD], if they have a problem with that, then I don’t know about EHR. As a GP if you need any information and the patient consents, that’s fine but if the patient says no, that’s it. I don’t know what misconceptions they might have. As long as it specifies what is the reason it’s being collected and what benefits it is and what future outcomes it will be and it’s just for this condition and its confidential and all that. And as long as it says all this and the patient agrees, I don’t see any reason why it can’t be done. But if the patient says no, I have reservations, I don’t want my data being collected and if he’s not very sure about what it involves, then the explanation might take time, it might take time so as long as the leaflet is clear.
GP 1022 (incomplete set-up)
This GP identified the need for patient information to be explicit about how and when the data would be accessed and by whom. Two GP decliners who were supportive of EHR in principle also expressed some concerns regarding patient confidentiality. One GP suggested data should be anonymised at all times and to exclude patients who have signed a disclaimer and another GP highlighted the need for more information from the EHR database on this issue if they had taken part in the pilot trial.
General practitioners across all groups of eLung status generally agreed that most patient data would be available to search remotely via free text with a small number of GPs in each group being willing to enter study data into pre-defined study-specific codes. Free text was generally considered more appropriate to increase access to non-practice data, such as out-of-hours and hospital discharge data which are scanned rather than coded into patient practice records. Most GPs believed that the EHR database would need to consult with individual GPs on a case-by-case basis to assist with interpretation of ambiguous clinical data:
The quality of coding and variability of coding is a major concern. I disagree with up to 50% of hospital codes and valid interpretation is tricky, for example, was it a chest infection, exacerbation of COPD, chest pain, an upper respiratory traction infection? The headline code from the hospital is often not what seems to have been the major reason why the person is in hospital so you have to look at the sub-coding and treatment pathway to try to work it out. CPRD have assessed their coding of episodes per prescription at 95% accurate but 85% for acute prescriptions. This may be a problem statistically and there is huge variation between practices. I would recommend we keep the coding as simple as possible or use free text to avoid missing data on searching.
GP 1005 (incomplete set-up)
This is a result of variation in codes and quality of coding between practices combined with the need for clinical expertise to interpret more complex cases. The need to avoid double entry into the patient and trial records and a preference for using codes consistent with local and national guidance was stated by several GPs across several subgroups. Several GPs flagged the need for financial remuneration to compensate for back-fill of GP time to provide this role. One GP recruiter (GP 1030) suggested a funding formula could be built into the remuneration package for every query answered to achieve quality data in a way that does not waste people’s time.
General practitioner theme 5: views on point-of-care trials
Table 18 summarises the level of support of GPs on four key characteristics of point-of-care trials. These views do not relate directly to the views expressed by GPs on the specific factors influencing their decision to participate in eLung but rather their general viewpoint following their experience in Retropro and eLung. These findings show that GPs widely support the concept of remote collection of outcome data via routinely collected EHRs (26/27). Three-quarters of GPs who provided data (19/25) expressed support for use of the LEPIS computer-based alert system to aid identification, recruitment and randomisation of patients into a trial. This assumes that outstanding developmental problems are resolved and LEPIS can be installed and function effectively. GPs who declined to participate in eLung were the main exception to this with slightly more GPs in this group expressing a lack of support for the use of an alert system. Approximately one-third of GPs who provided data (10/16) were supportive in principle of hot recruitment in point-of-care trials. The remainder expressed a preference for a broader, more flexible, approach which enables use of additional cold recruitment methods, for example, to enable informed consent to be sought prior to the consultation. Twenty-three out of 24 GPs expressed a preference for a small number of patient participants per trial site (in the region of 8–10), with the option to recruit more, as recommended for eLung. This increased GP confidence and motivation in the feasibility to complete recruitment.
| GP eLung status | Hot recruitment | Hot and cold recruitment | Use of LEPIS alert system | Remote collection of outcome data via EHR | Small number of patients per site | |||
|---|---|---|---|---|---|---|---|---|
| ✓ | ✗ | ✓ | ✗ | ✓ | ✗ | |||
| Decliners (n = 9) | 2 | 6 | 3 | 5 | 9 | – | 8 | – |
| Withdrawals (n = 3) | – | 3 | 2 | 1 | 3 | – | 3 | – |
| Incomplete set-up (n = 8) | 6 | 2 | 7 | – | 7 | 1 | 7 | – |
| Recruiters (n = 7) | 2 | 5 | 7 | – | 7 | – | 5 | 1 |
| Total (n =2 7) | 10 | 16 | 19 | 6 | 26 | 1 | 23 | 1 |
General practitioner theme 6: recommendations to improve point-of-care trials
A range of recommendations on how to improve potential recruitment of GPs into future point-of-care trials emerged from GPs during the QUEAN study interviews. Most recommendations were put forward by GP participants across all eLung subgroups of decliner, withdrawal, incomplete set-up and recruiting. These were based on GPs direct experiences of recruitment and/or participation in eLung as well as hypothetical views on how simple trials using EHRs in primary care could be improved. Several of the recommendations are considered important by many GPs given the novelty of these trials and an associated lack of GP confidence to take on new challenges within the context of highly demanding busy surgeries. The need for confidence giving strategies is likely to reduce over time as more GPs become familiar and confident with the trial methodology. GP recommendations fall into the following areas:
-
study documentation and communication with GPs
-
input from GPs to inform future trials
-
flexible strategies to recruit patients
-
computer-based alert system
-
additional support for vulnerable practices
-
incentives for GPs.
Appendix 4 provides a detailed description of GP recommendations within each of these areas.
Characteristics of patient sample
A total of 12 patients agreed to participate in the QUEAN study to conduct an interview to discuss their reasons for, and experiences of, accepting to participate in eLung. Two of the 12 patients withdrew from the QUEAN study prior to interview as a result of a failure to respond to postal or telephone contact or due to having been hospitalised on the day of the interview. A total of 10 patient trial-accepters completed an interview for the QUEAN study as per the study minimum target to achieve likely data saturation. No patient trial-decliners were recruited into the QUEAN study, a shortfall of 10 participants based on the study minimum target ( Table 19 ).
| Type of participant | Target number | Number recruited | Number withdrawn | Reasons for withdrawal | Number interviewed |
|---|---|---|---|---|---|
| Patient: eLung trial-decliners | 10 | 0 | 0 | N/A | 0 |
| Patient: eLung trial-accepters | 20 | 12 | 2 | No response
n = 1 Hospitalised n = 1 |
10 |
The 10 patient participants (eLung-accepters) included an equal number of participants in both the control (5/10) and intervention (5/10) arms and of women (5/10) and men (5/10). Nine of the 10 patient participants were white Caucasian and one patient participant was African Caribbean. The majority of patient participants (6/10) described their health status as typically unwell to very unwell owing to ongoing breathing difficulties and regular flare ups of their COPD. Nine of 10 patients had at least one comorbidity (e.g. asthma, diabetes, Ménière’s disease, sleep apnoea) and three of the patient participants had at least three comorbidities (e.g. tuberculosis, heart failure, kidney disease). Six of 10 participants lived in Ayrshire in Scotland and were registered with the same GP. The remaining four participants lived in England including patients in London (n = 1), Devon (n = 2) and Manchester (n = 1). More than half of the patients described themselves as living in a relatively deprived area and two patients in affluent areas.
Patient views on point-of-care recruitment and electronic health records
All 10 patients in the QUEAN study reported their views and experiences of being recruited into eLung within the previous 6 months. All patients had visited their GP at the date of recruitment primarily due to symptoms related to a COPD exacerbation (bad chest 7/10, bad cough 2/10, or breathing problems 1/10). The main reason for agreeing to take part in eLung was the hope that it might improve their own health (6/10) or other people’s health in the future (4/10). Seven of the 10 patients cited their excellent doctor–patient relationship as a key influencing factor in their decision-making process and their trust that it will be in their best interest to participate in the study if their GP has asked them. All 10 patients (100%) considered it acceptable to be recruited during routine GP consultation, despite their ill health. This included one patient who routinely has a family member present at the clinical consultation for advice and support and would also want the family member present during discussions about participating in a research study. One patient would prefer to receive the study information in advance to allow more time to consider it prior to seeing the doctor during the consultation. Nine of the 10 patients were not aware of the screening and randomisation process using the computer-based alerts during the consultation. One patient was able to clearly describe the computerised process of random allocation into the usual care group and expressed an interest in the novelty of this research method. These findings suggest the computer alert recruitment system at the point of care did not interfere with the doctor–patient interaction from the patients’ perspective.
All the patient trial-accepters could recall receiving the first quality-of-life questionnaire at the time of being recruited into eLung. Four patients could clearly recall receiving the patient information sheet for eLung and two could recall signing the consent forms for eLung. The lack of recollection about the consenting process was a concern for two patients, both of whom had been recruited into eLung nearly 6 months prior to the QUEAN study interview. Eight of 10 patients were satisfied with the consenting procedure for participation in eLung during the consultation. This includes one patient who would want a family member present to assist with consenting, consistent with the need for support when attending a routine clinician consultation.
All 10 patients considered use of EHRs to collect trial outcome data to be acceptable. This included one patient who thought routine medical records would provide more useful information than patient-reported information and one patient who preferred not to complete too many questionnaires due to suffering from arthritis. All patients preferred the data to be anonymised for use by a research team:
I’ve been on steroids and antibiotics before so I’m quite happy to be asked at the time to take part in a study which is familiar.
Patient ID 1049
If I didn’t want to do it, I could have pulled out of it, I didn’t have to do it.
Patient ID 1051
As long as it’s [patient data] being used, you know, for that [research] purpose then, then I’m not concerned about it one little bit.
Patient ID 1051
One patient expressed their explicit support for longitudinal data analysis if the data would not be used for any other purpose. This supports the findings of GP views to ensure clear information is provided on how the data will be accessed, by whom and how often.
All five patients in the intervention arm were satisfied with their allocation, compared with two of the five patients in the control arm. The other participants would have preferred not to have had steroids again (2/5) or had wished to receive antibiotics (1/5). Two patients allocated to the control arm were subsequently hospitalised because of their COPD exacerbation, compared with none in the intervention arm. Two patients in both groups returned to the practice for further treatment.
Summary of qualitative research on patients
The sample does not include views of any participants who declined to participate in eLung. The sample of trial-accepting patients achieved diversity of characteristics in terms of men and women in England and one region of Scotland, participants in both arms of the trial and patients experiencing at least one or several comorbidities in addition to COPD. These findings demonstrate that it is feasible for a GP to use computerised recruitment at the point of care for patients who are often very unwell and who reflect the reality of primary care settings where patients typically have comorbidities alongside the condition of research interest. Improvements in health at a personal or societal level were the main reasons for patients agreeing to take part in the research, underpinned in most cases by a high level of trust in their clinician who the patient believes is acting in their best interest. This is consistent with the views of some GPs who described their need to believe in the study to sell it to their patients. Computerised point-of-care recruitment and use of EHRs for outcome data collection is considered acceptable by all patients in this sample. One patient who routinely takes a family member to any clinical consultation expressed a wish for a family member to also be present during any discussions regarding recruitment into a research study. Another patient expressed a wish for the patient study information to be provided in advance of the invitation to participate at the point of care. Several patients could not clearly recall the consenting process in terms of receiving an information sheet and signing a consent form, including two who were concerned about their lack of recollection. This may be as a result of the time lapse between the date of recruitment and the interview to discuss their views and experience, ill health of the patient and/or the limited time available to complete the consenting process. These findings, consistent with views of several GP participants, highlight the need to further examine the most appropriate methods to achieve informed consent as part of a simple trial using computerised point-of-care recruitment and EHRs.
Strengths and limitations of the qualitative process evaluation (the QUalitative Evaluation of a trial of ANtibiotics for chronic lung disease study)
The QUEAN study achieved its overall aim of ascertaining valuable detailed information from GPs and patients about a pilot point-of-care trial using EHR to better understand the mechanisms behind its success or failure in UK primary care settings. There were several barriers to the successful recruitment of GPs and patients into the QUEAN study. The main one was the length of time needed for research governance approvals in England, leading to delays in initiating eLung. Although 39 participants were recruited into the QUEAN study, 12 GPs withdrew during the time taken to obtain NHS governance approvals. Recruitment of GPs and patients who had declined to take part in eLung was anticipated to be difficult, particularly as the recruitment process would take place during an unscheduled consultation. It is likely therefore that time pressures, sensitivities of providing information about another study to a sick patient who had already declined eLung and the small number of GPs recruiting patients into eLung contributed towards the failure to recruit any patient trial-decliners. Successful data collection from 12 GP decliners is considered a key strength of the QUEAN study as it provides valuable insight into the reasons for declining or withdrawing participation for a point-of-care trial. As described by Salmon et al. ,98 GPs who have declined to participate in a study are particularly hard to recruit to discuss their views on recruitment and yet they are the very group who are ‘information-rich about the attitudes or concerns that deter doctors from participation’. 98 Data collection from 17 GP trial-accepters in the QUEAN study provided detailed insight into GP views and experiences at all stages of the recruitment and trial process. The four GPs who had recruited patients into eLung represented 57% of the all GP recruiters.
The role of nested qualitative studies within pilot trials to collect empirical evidence on recruitment of practitioners has been identified as the most effective intervention to identify and overcome barriers to clinician recruitment activity. 99 This is due to the strengths of in-depth interview techniques, which collect data grounded in the actual experience of the clinician’s recruitment decision. 89,98 Furthermore, Fletcher et al. 99 found a bias in existing research towards investigating barriers to recruitment and recommended ‘future work should also encompass a focus on successfully recruiting trials’. 99 The appropriateness of the QUEAN study research method and dual focus on barriers and enablers among a subsample of GPs and patients who have direct experience of the pilot trial gives confidence therefore in the validity of the findings and their potential to inform recommendations for future point-of-care trials.
The 10 trial-accepting patient participants interviewed in the QUEAN study represent 32% of all patients recruited into eLung. A broad range of issues were identified by individual patient participants on the key topics of interest which appeared to have reached data saturation within the sample. Despite this, the small number of participants raises concerns regarding generalisability of patient findings. The sample of trial-accepting patients achieved diversity of characteristics including equal numbers of men and women, participants in both arms of the trial, several locations in England and one region of Scotland, and patients experiencing at least one or several comorbidities in addition to COPD.
The diverse sample of GPs, locations and population groups and the achievement of data saturation provides confidence in the generalisability of findings in the four key areas of the research study among GPs in England and, to a lesser degree, in Scotland. 100,101 Despite apparent data saturation between participants and the diversity of patient characteristics within the sample, the small sample size warrants caution in generalising findings from this group.
The consistency of a single, experienced qualitative researcher to adhere to the protocol and maintain an approach which is inductive and iterative at all stages of data collection and analysis increases confidence that data and study findings are grounded in GP and patient views and experiences. 96,102 Reliability of participant recall may have been affected by the period of time between recruitment and interviews. The main areas where this appeared to be problematic were recall of the study documents for GP trial-decliners when discussing their views on their experience of being invited to participate in eLung and recall of whether or not patients signed a consent form when being recruited into eLung. Other factors influencing the ability for the patients to recall the information at the GP visit may have been age, level of education or the volume of information being given at the single consultation. 103
In summary, findings on the key areas of the qualitative research study from GP and patient participants appear to be valid and potentially generalisable to GPs with similar characteristics and locations. The quality of the data provides a solid foundation for recommendations for best practice in future point-of-care trials. Key exceptions to this are data from GPs on practical experience of point-of-care recruitment of patients into eLung which has been identified as valid but potentially incomplete, and a lack of data for patient decliners on their reasons for declining to participate in point-of-care trials.
Discussion of findings of the qualitative process evaluation
The QUEAN study identified widespread and strong support for the concept of conducting trials using EHRs to collect outcome data. This is regarded as an efficient use of a highly valued but underutilised resource to facilitate much needed research on real patients within primary care settings. This is supported by the finding that most GPs expressed concern regarding the lack of evidence-based guidance informed by primary care research. Indeed, GPs in all subgroups of trial-decliners and trial-accepters believed the application of most guidance in primary care was not fit-for-purpose and had a negative impact on the quality of care. Randomised trials using EHRs were also considered to be an efficient method for outcome data collection in terms of GP and/or practice time and resource. This is important within a context of perceived increased workload for GPs, particularly reporting requirements, less scope for holistic individualised care and increased financial pressures. GPs also identified a reduction in their role in research in the last decade or so, from running their own studies to identifying participants for other, externally managed studies. All these contextual factors have resulted in, at best, a lack of GP time and capacity for research and, at worst, demotivating GPs from participation in research. Within this context, the strength of personal interest in research appears to be a crucial underpinning motivational factor, which in combination with other more practical factors, is likely to shift the decision in favour or against participation in any given study for GPs in the QUEAN study. An association between an interest in research and recruitment was also found by Wilson et al. 104 A cross-sectional qualitative study among GPs in Germany also found an interest in research to improve the evidence base in primary care was the most influential factor for GPs to participate in research. 105 Conversely, a nested qualitative study identified a lack of interest in research as the key factor influencing the GP decision to decline participation in a trial. 98 The authors stated, ‘GPs described general practice and research as alien fields. Research lacked intrinsic, clinical or professional value’. 98
The QUEAN study identified two sets of factors which influence the decision-making process for GPs on recruitment: first, generic factors relevant to assessing the suitability of any research study; and second, project-specific factors which enable the assessment of the study in question. Interestingly, the same influencing factors were identified by both GPs who had declined or accepted participation in eLung although the emphasis on whether the factor was a positive or negative influence varied between groups. This observation was also made by Hunt et al. 106 who described the presence of the specific factors which have been identified by some GPs as resulting in successful recruitment, in trials which fail to recruit. 106 This complexity led Bower et al. 89 to suggest that factors such as ‘culture, attitudes and leadership’ are potentially over-riding the influence of the same specific factors in different directions for different GPs. 89 This further supports the findings of the QUEAN study regarding the underpinning influence of GP attitudes to research in the recruitment decision within the context of limited time and capacity for research.
Strategies to improve GPs’ attitudes to research and the cultural environment in which GPs conduct research may therefore be potentially more crucial than strategies to address specific barriers to recruitment. Future research to evaluate complex strategies, such as policy and infrastructure support aimed at creating a positive shift in attitudes, culture and leadership, could utilise normalisation theory. This theoretical model has recognised validity in ‘assessing the potential for complex interventions to become routinely embedded in everyday clinical work, and evaluating the factors that promote or inhibit their success and failure in practice’. 107
General practitioners in the QUEAN study identified the following generic factors as the most important influence on participation in eLung: time/capacity to implement the study; personal interest in research; benefit to local patients or population groups; and adequate financial remuneration. The most important project-specific factors influencing the second level of GP decision to participate in eLung were consistency with local prescribing guidance, relevance of topic, feasibility to recruit, computer-based pop-up alerts and hot recruitment method. These findings are consistent with findings of several studies including systematic reviews53,108 and subsequent individual studies68,89,109,110
A qualitative evaluation of GPs’ views of point-of-care recruitment in a randomised trial in Wales also identified emerging themes of difficulties recruiting in the consultation owing to lack of time. 55 Based on their findings, Prout et al. 55 also recommended the ingredients of successful trial implementation using point-of-care recruitment include good organisation, simple documentation and study procedures, and the ability to allay patient concerns about patient safety on the particular intervention. 55 Concerns about use of EHRs were identified in a study among GPs in Germany, specifically regarding the intended sharing of patients summary records to other services. 73 This concern was not shared by the QUEAN study GPs and patients. One study supported findings of the QUEAN study regarding the particular difficulties experienced by GPs in London, often resulting in lower rates of recruitment. 111
Strategies to improve recruitment in research which have been identified in the literature but for which there is little evidence base were summarised by Bower et al. 89 These include the following strategies consistent with those recommended by GPs within the QUEAN study: promotion of the research method; financial incentives; focus on clinicians with an interest in research; clinician involvement in trial design; practice visits by the research team; practical information to inform GPs about the trial; feedback on recruitment rates; and assistance with patient travel. A recent systematic review identified nested qualitative studies within pilot trials to collect empirical evidence on recruitment of practitioners (such as the QUEAN study) as the most effective intervention to identify and overcome barriers to clinician recruitment activity. 99 This evidence base provides further support for the methods and validity of findings of the QUEAN study.
The main factor influencing the decision to participate for the sample of 10 trial-accepting patients in the QUEAN study was the hope of improvements in health at a personal or societal level. This was largely underpinned by a high level of trust in their clinician. This is consistent with the literature as demonstrated by the Cochrane systematic review. 112 The 10 QUEAN study patient participants found recruitment at the point of care and use of EHRs to collect outcome data as acceptable including one patient who expressed a preference for receiving the patient study information prior to recruitment at the consultation. Several patients could not clearly recall the consenting process, a potential concern which was echoed by some GPs within the context of point-of-care recruitment. Research to test strategies on appropriate consenting methods within these trials should therefore be a priority.
Chapter 9 Value of information analysis for eLung
Introduction
When deciding how to allocate limited health-care budgets to different interventions, decision-makers use the evidence that is currently available. However, there can be a paucity of evidence in certain areas, yet decisions still need to be made regarding the optimal use of scarce resources. It is possible to undertake analyses in areas where there is a limited evidence base to determine the value of further information (i.e. the value for money associated with carrying out further research). It is important to assess the uncertainty surrounding the adoption decision and to evaluate if further evidence is required to support this decision in the future. 113 Value of information (VOI) analysis provides an analytical framework to address these questions. 114 Such VOI analyses have firm foundations in statistical decision theory,115,116 have been used successfully in other research areas117,118 and, more recently, have been increasingly used for the evaluation of health-care technologies. 119,120 VOI analysis can aid decision-makers in setting research priorities for health-care technologies114,121,122 and help to inform research recommendations. 123
A VOI analysis is presented here to illustrate the type of analysis that can be conducted using data from EHRs and point-of-care trials. There is currently no clear consensus on whether or not antibiotics should be prescribed for COPD exacerbations; a VOI analysis can demonstrate the value of conducting further research in this area. The findings from trials such as eLung can be used to evaluate the VOI in terms of the expected value of perfect information (EVPI). The EVPI represents the expected costs of uncertainty surrounding a decision problem; when we have perfect information, the possibility of making the wrong decision is eliminated. It is also useful to identify the parameters for which it would be valuable to have more precise estimates to help focus the type of evidence that should be generated. The expected value of perfect parameter information (EVPPI) can be used for this purpose.
Methods
Value of information analysis steps
The VOI analysis followed the following three steps, as proposed by Briggs et al. :113 (i) to construct a decision-analytic model to represent the decision problem; (ii) to characterise the current decision uncertainty by undertaking a probabilistic analysis of this model; and (iii) to establish the value of additional information. 113
Model structure
A decision-analytic model was developed for the VOI analysis in order to investigate the cost-effectiveness of prescribing antibiotics for COPD exacerbations in terms of the expected costs and effects for the two groups. The model, developed in Microsoft Excel (2010; Microsoft Corporation, Redmond, WA, USA), followed a decision tree approach and was based on the structure from Effing et al. ,124 as shown in Figure 6 . A cohort of 10,000 hypothetical COPD patients who present at their general practices with COPD exacerbations was followed in the model. The two alternatives considered by the analysis were ‘immediate antibiotics’ or ‘no immediate antibiotics’; there is a paucity of evidence regarding which is the more cost-effective option. We have taken the perspective of the UK NHS for the analysis125 (i.e. only direct costs to the NHS have been included). Owing to the 12-month time horizon of the model, we did not discount costs and health outcomes.
FIGURE 6.
Schematic of the decision tree model for management of COPD exacerbation.
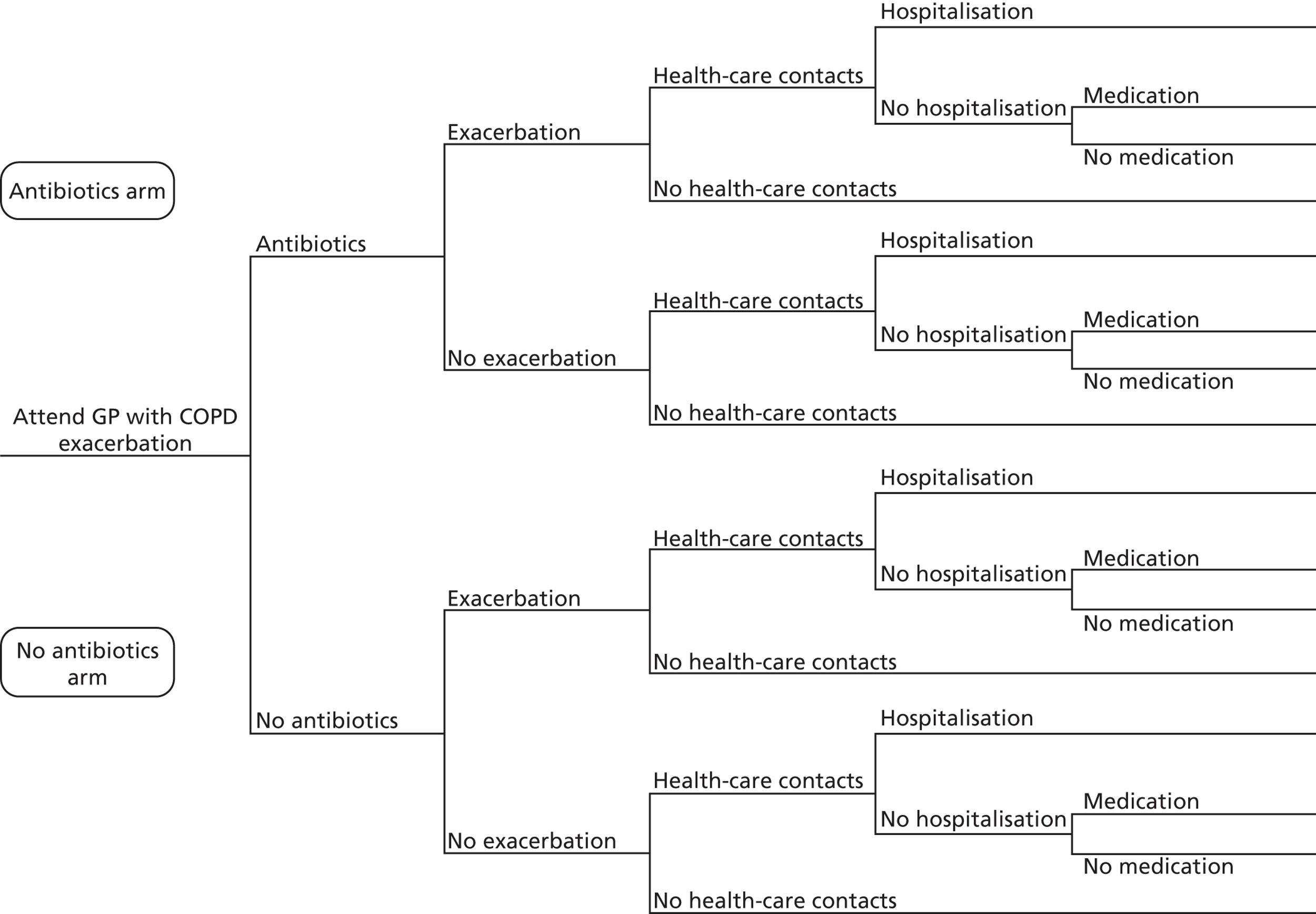
The VOI analysis used data from literature and an observational cost-effectiveness analysis (CEA) which used data from CPRD (as described in Chapter 10 ). The probabilities used in the decision tree were derived from the observational CEA, as shown in Table 20 .
| Probability | Antibiotics group | No antibiotics group | ||
|---|---|---|---|---|
| Mean valuea | Prior distribution | Mean valuea | Prior distribution | |
| Hospitalised COPD exacerbation | 0.044 | Beta (α = 1087, β = 23,641) | 0.169 | Beta (α = 2229, β = 10,953) |
| Health-care contact following COPD exacerbationb | 1.00 | N/A | 1.00 | N/A |
| Health-care contact without COPD exacerbation | 0.993 | Beta (α = 23,483, β = 158) | 0.998 | Beta (α = 10,937, β = 17) |
| Any hospitalisation following COPD exacerbation | 0.810 | Beta (α = 880, β = 206) | 0.751 | Beta (α =1673, β = 555) |
| Any hospitalisation without COPD exacerbation | 0.159 | Beta (α = 3760, β = 19,881) | 0.188 | Beta (α = 2062, β = 8891) |
| Requiring medication following outpatient COPD exacerbation and no hospitalisation | 0.835 | Beta (α = 171, β = 34) | 0.766 | Beta (α = 424, β = 130) |
| Requiring medication without COPD exacerbation | 0.781 | Beta (α = 15,536, β = 4345) | 0.743 | Beta (α = 6604, β = 2287) |
| Being prescribed antibiotics | 0.652 | Beta (α = 24,729, β = 13,183) | N/A | N/A |
Quality-of-life outcomes
Health-related quality-of-life (HRQoL) data were derived from the literature. 126,127 The utility values used in the model are displayed in Table 21 and were applied similarly to both arms (i.e. to both the ‘antibiotics’ and the ‘no antibiotics’ arms). A sensitivity analysis was undertaken using utility values derived from the EuroQol EQ-5D-3L questionnaires completed by eLung participants.
| Utility parameter | Mean value | Prior distribution |
|---|---|---|
| Exacerbation and hospitalisation | 0.690a | Beta (α = 30.3, β = 13.6) |
| Exacerbation and no hospitalisation | 0.722b | Beta (α = 27.1, β = 10.4) |
| No exacerbation and hospitalisation | 0.732c | Beta (α = 26.1, β = 9.5) |
| No exacerbation and no hospitalisation | 0.732c | Beta (α = 26.1, β = 9.5) |
Costs
Cost data were obtained from established national costing sources comprising NHS Reference Costs 2011/2012,128 the British National Formulary 129 and the Personal Social Services Research Unit Unit Costs of Health and Social Care 2012 130 as shown in Table 22 .
| Cost parameter | Mean value (£) | Prior distribution | Details |
|---|---|---|---|
| GP visit | 43130 | Gamma | |
| Hospitalisation | 1235128 | Gamma | Average of 10 COPD-related Healthcare Resource Group codes |
| Antibiotic prescription | 8129 | Gamma | Average of 10 antibiotics reported by observational CEA |
| Referral to community respiratory team | 76128 | Gamma | Community and outreach nursing services: specialist nursing (CN203DAF) |
| Health-care contact | 43130 | Gamma |
Incremental analysis
The results from the model were presented in terms of the incremental cost-effectiveness ratio (ICER). Specifically, the ICER was estimated in terms of the incremental cost per quality-adjusted life-years (QALY) (for cost–utility analysis). A cost-effectiveness acceptability curve (CEAC) was produced to explore the possibility that the use of antibiotics for COPD exacerbations will be cost-effective at different thresholds. 131 Net health benefit (NHB) was used to calculate the EVPI, where the NHB investigates if additional cost associated with a treatment is justified by the additional health gains. The NHB measure is the increase in effectiveness multiplied by the amount that the decision-maker is willing to pay per unit of increased effectiveness (i.e. the cost-effectiveness threshold), minus the increase in cost. The cost-effectiveness results were presented on a cost-effectiveness plane. 132
Expected value of perfect information calculations
The (individual patient) EVPI was calculated as the difference between the expected NHB given full information and the expected NHB given current information. The population EVPI was estimated by multiplying the individual per patient EVPI by the effective population, i.e. the annual population of patients who experience COPD exacerbations discounted over the lifetime of the treatment (assumed to be 10 years). The EVPI represents the maximum amount that a decision-maker should be willing to pay for further information to guide the adoption decision in the future; additional research should be considered if the EVPI exceeds the cost of research. The EVPPI was also calculated in order to provide more focus for further research, by identifying groups of inputs where it would be valuable to have more accurate estimates. Specifically, EVPPI estimates were generated for three sets of parameters: cost parameters, utility parameters and probabilities. The model used for the EVPPI was assumed to be linear for ease of computation.
Probabilistic analysis
In order to represent the decision uncertainty, a probabilistic sensitivity analysis was undertaken using 10,000 Monte Carlo simulations. Probabilistic analysis enables identification of the impact of different parameter values on the costs and effects of the different treatment options. Distributions were fitted to key parameters within the model; beta distributions (bound by zero and one) were used to capture the impact on uncertainty around probabilities and utilities. Cost parameters were modelled using the gamma distribution (producing non-negative values).
Results
Cost-effectiveness results
The cost-effectiveness results from the decision tree model are presented in Table 23 . Patients who immediately received antibiotics experienced slightly more QALYs compared with patients who received no antibiotics. Each ‘antibiotics’ patient in the model was estimated to experience 0.730 QALYs [95% confidence interval (CI) 0.604 to 0.840 QALYs] for the 12-month time horizon, as opposed to 0.726 QALYs (95% CI 0.617 to 0.819 QALYs); an incremental effectiveness of 0.004 QALYs per patient. Hence, the QALYs gains were minimal.
| Measure | Antibiotics | No antibiotics | Incremental |
|---|---|---|---|
| Expected cost (£) per patient (95% CI)a | 329 (291 to 371) | 448 (398 to 501) | −119 (−144 to 94) |
| Expected QALYs per patient (95% CI)b | 0.730 (0.604 to 0.840) | 0.726 (0.617 to 0.819) | 0.004 (−0.152 to 0.163) |
| ICER | Dominantc |
The total cost per patient for COPD exacerbation management for the ‘antibiotics’ and ‘no antibiotics’ groups were estimated to be £329 (95% CI £291 to £371) and £448 (95% CI £398 to £501) respectively. Therefore, the use of antibiotics was associated with a cost saving of £119 per patient. Hence the analysis demonstrated that the use of antibiotics for COPD exacerbations was the dominant management strategy. As the QALYs were only marginally more favourable for the ‘antibiotics’ group, this finding should be interpreted with caution.
Figure 7 shows the cost-effectiveness plane for antibiotics compared with no antibiotics, obtained from the probabilistic sensitivity analysis. The sample estimates are spread entirely across the south-east and south-west quadrants, which demonstrates that antibiotic use was cost saving. The marginal QALY differences can be seen by the iteration results being concentrated close to zero for the incremental QALYs.
FIGURE 7.
Cost-effectiveness plane for antibiotics compared with no antibiotics.
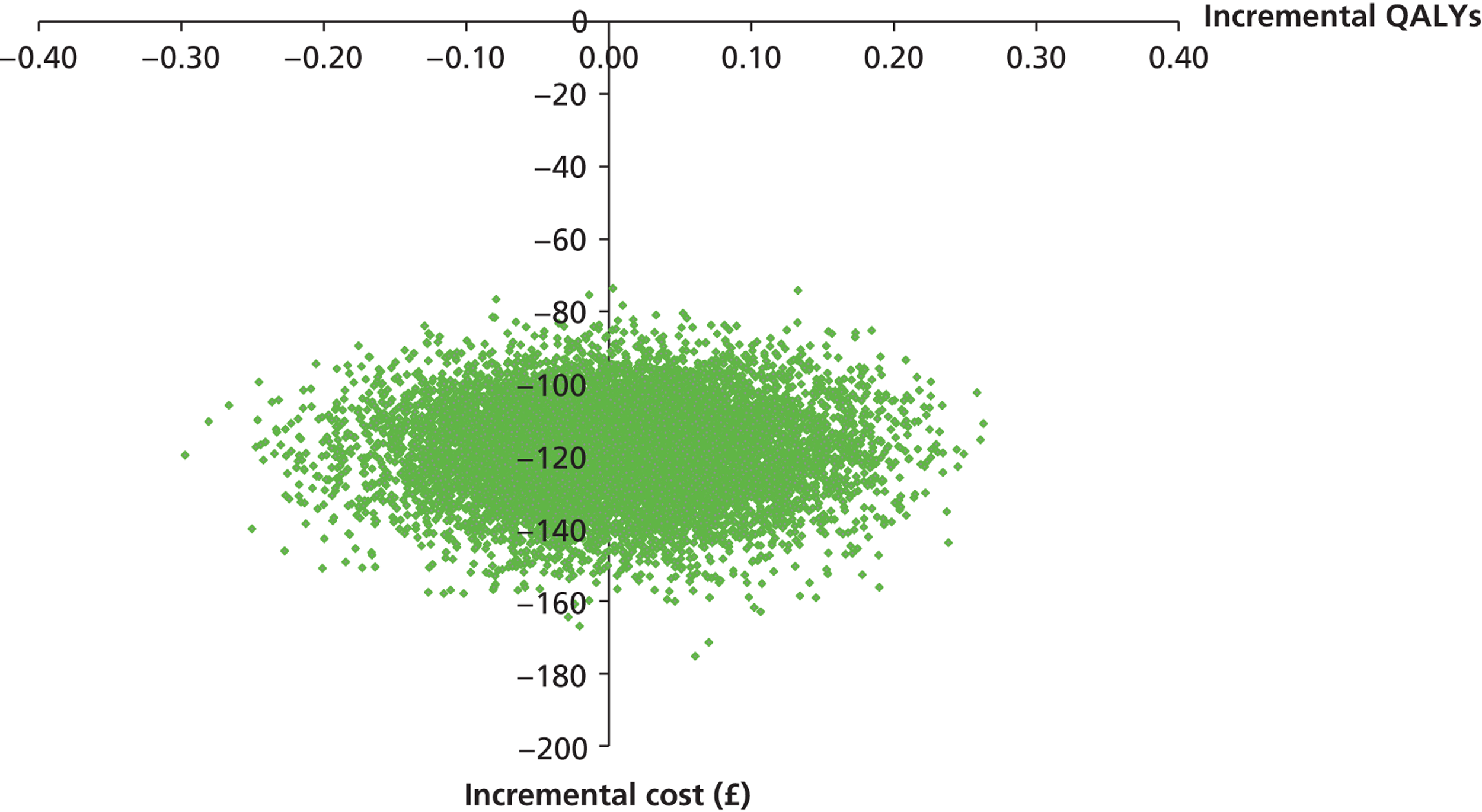
The CEAC in Figure 8 illustrates the probability of antibiotic use and of no antibiotic use being cost-effective for different willingness-to-pay thresholds. For the NICE threshold of £20,000 per QALY, the probability of antibiotics being cost-effective is 0.56 compared with no antibiotics. For low thresholds, antibiotic use is likely to be cost-effective, with the curve for no antibiotic use showing the converse (i.e. that it is unlikely to be cost-effective for low thresholds).
FIGURE 8.
Cost-effectiveness acceptability curve.
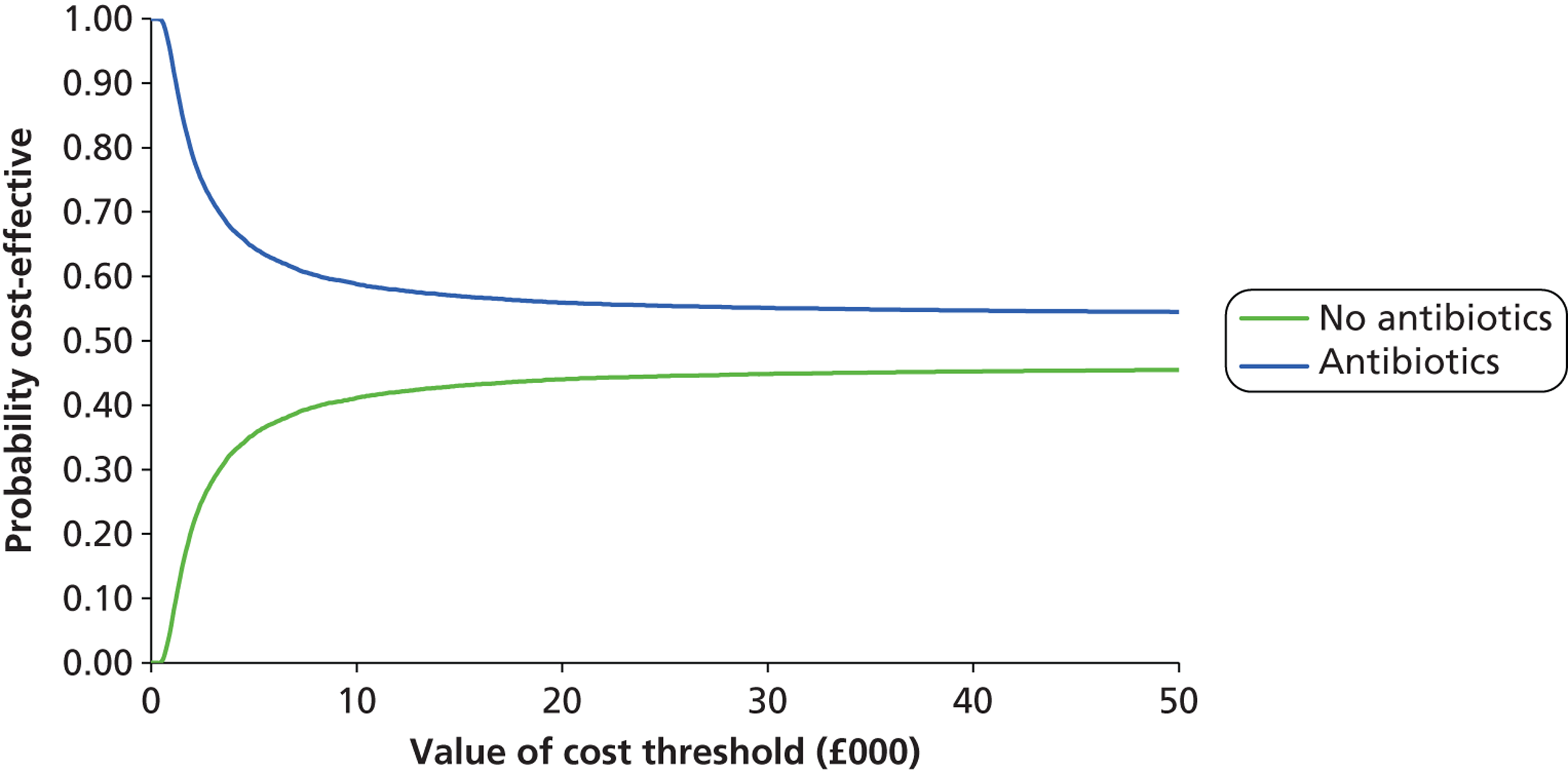
Value of information results
The VOI analysis suggested that further research could be worthwhile ( Table 24 ). At a threshold of £20,000 per QALY, the individual total per patient EVPI was £567. The population EVPI was £3,979,601,769, which far exceeds the cost of a trial in this area (estimated to be £1.5M). Hence, this indicates that further research to inform the decision around use of antibiotics for COPD exacerbations could be worthwhile.
| Measure | Cost-effectiveness at thresholds | ||
|---|---|---|---|
| £10,000 per QALY | £20,000 per QALY | £30,000 per QALY | |
| Net benefits | |||
| Antibiotics | £6985 | £14,299 | £21,613 |
| No antibiotics | £6810 | £14,068 | £21,325 |
| Probability of antibiotics being cost-effective | 0.59 | 0.56 | 0.55 |
| Maximum net benefits under | |||
| Current information | £6985 | £14,299 | £21,613 |
| Perfect information | £7243 | £14,866 | £22,490 |
| EVPI per patient | £258 | £567 | £877 |
| Population EVPI | £1,812,870,246 | £3,979,601,769 | £6,154,966,136 |
Specifying a minimum clinical difference in outcomes required to change clinical practice is one way to incorporate concerns that the results of the research may not be implemented perfectly (i.e. with 100% utilisation). For example, a larger clinical difference in effectiveness may need to be seen before the research has an impact on clinical practice. Requiring that further research must demonstrate larger differences in expected health benefits between antibiotics and no antibiotics reduces the value of research as larger differences are less likely to be found. This is illustrated in Figure 9 , which shows how the value of additional evidence falls as larger differences in NHBs between the treatments is needed to be demonstrated. When the minimum clinical difference between treatments is zero, the expected benefits of additional research is equal to the EVPI (i.e. all the uncertainty is resolved and it is expected that the results of research are implemented perfectly).
FIGURE 9.
Value of additional evidence and minimum clinical difference required.
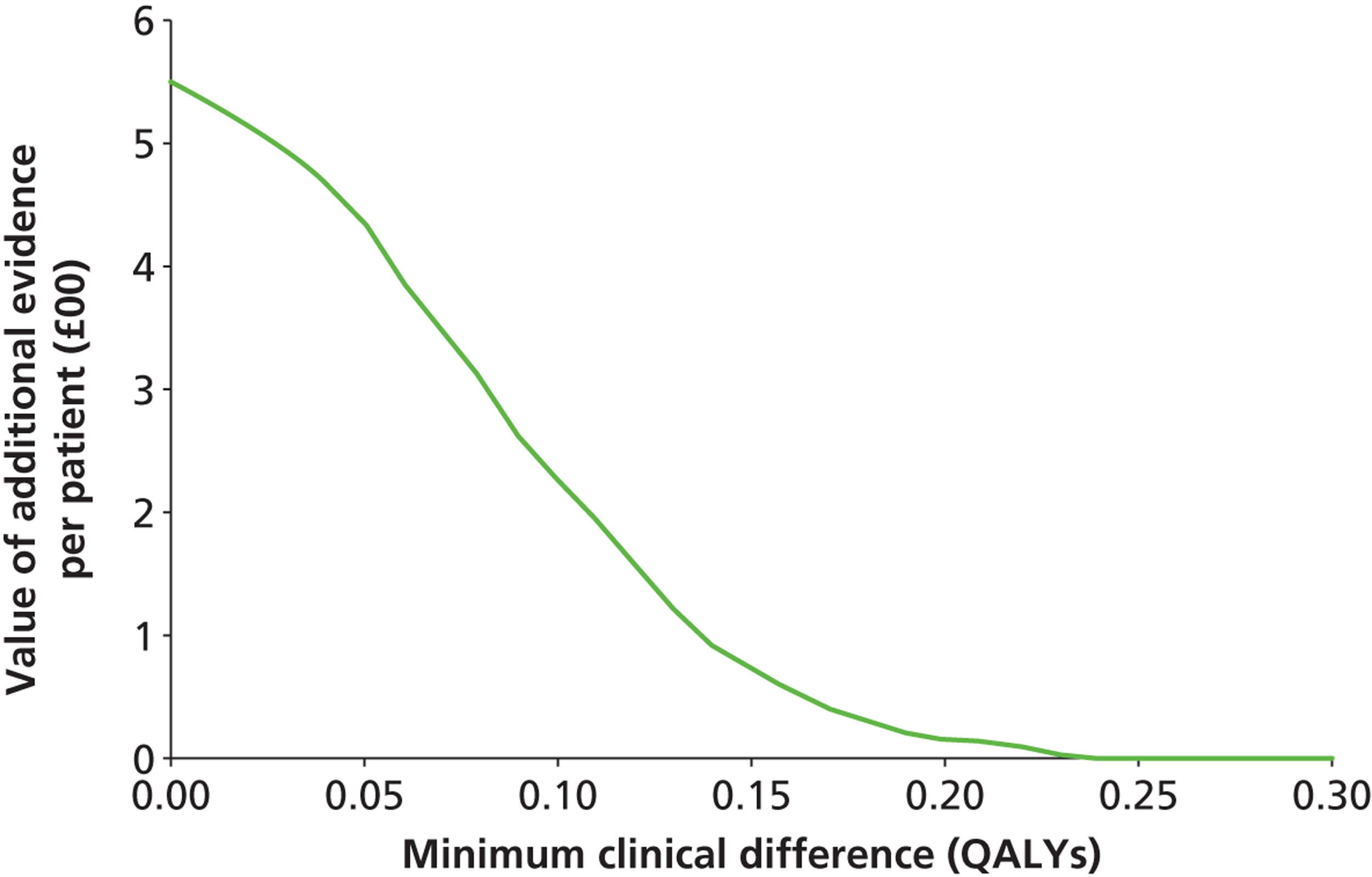
The EVPPI explored the uncertainty of different parameters in the model. The uncertainty associated with the model derives from the utility (i.e. quality-of-life) values; the individual per patient EVPPI associated with utilities was £510. The VOI associated with cost parameters and probabilities was found to be negligible.
eLung quality-of-life data
We investigated the impact of using utility values estimated from eLung (based on the EQ-5D responses reported by the 12 participants who completed both the baseline and 4-week follow-up quality-of-life questionnaires). The mean utility score at baseline was lower for the ‘antibiotics’ group at 0.46 compared with 0.55 for the ‘no antibiotics’ group. At 4-weeks follow-up, the HRQoL had reduced for both groups: mean EQ-5D scores were 0.45 in the ‘antibiotics’ group compared with 0.52 in the ‘no antibiotics’ group. Using these scores, it was found that patients in the ‘antibiotics’ group experienced 0.075 fewer QALYs compared with those in the ‘no antibiotics’ group. Individual patient EVPI decreased to £39, population EVPI decreased to £274,525,916 and individual patient EVPPI for utilities fell to £5, for a threshold of £20,000 per QALY. Therefore, the results still indicate that further research in the area could be worthwhile.
Discussion
The findings from the VOI model represent the considerable variation in the cost-effectiveness results. The resulting high EVPI estimates represent this significant cost of uncertainty, with the high population EVPI estimate indicating that further research is likely to be of value. The EVPI results for individual model parameters indicate that it would be of most value to target future research regarding the extraction of more precise estimates of HRQoL for patients with a COPD exacerbation receiving either antibiotics or no antibiotics.
It was necessary to make a series of assumptions in the construction of the model. In particular, due to a paucity of evidence, assumptions were made about the values used for utilities in the model, which is a source of model uncertainty, as demonstrated by the EVPPI findings. Therefore, the model findings should be interpreted with caution. The model results indicate that further research into the cost-effectiveness of immediate use of antibiotics for COPD exacerbations is likely to be worthwhile. The EVPPI analysis highlighted that a potential area on which to focus research is the impact of antibiotic use on patients’ quality of life. However, we note that these findings are dependent on the data included in the model.
In conclusion, the substantial population EVPI estimated by the model indicates further research into the use of antibiotics for patients with COPD exacerbations might be worthwhile. The key area of uncertainty in the VOI model was indicated to be around quality-of-life estimates; hence, further research appears most valuable in this area. The model aimed to illustrate the type of analysis that can be undertaken using trial data obtained from EHRs, in terms of evaluating whether or not there is value in generating further information in an area, thereby guiding potential future research. It would be valuable to input trial data into our model to generate more evidence-based results.
Chapter 10 Cost-effectiveness analysis of antibiotics for chronic obstructive pulmonary disease exacerbations
Introduction
The aim of this analysis was to illustrate the type of CEA that could be conducted using EHRs. We originally planned to use data obtained from eLung participants to conduct a CEA of antibiotic use compared with usual care for the management of COPD exacerbations. However, owing to lower than anticipated recruitment, we undertook an alternative analysis: a retrospective, observational CEA using EHR data in order to explore the impact of prescribing antibiotics to patients with COPD exacerbations, in terms of the main eLung end points. The following outcomes were analysed: further GP visits; hospital admissions; community respiratory team referrals; antibiotic prescriptions; infections; and all referrals, for up to a 12-month period after the initial exacerbation. Hence, we were able to use similar CPRD data to the type of data that would have been available via eLung for our analysis.
Methods
Patient selection and inclusion
A cohort of patients was identified from CPRD by searching for patients who attended at their general practice for a COPD exacerbation between the year 2000 and April 2013. Individuals who were aged ≥ 40 years and had COPD exacerbation at least 1 year after the start of data collection were selected. In cases of multiple COPD exacerbations that occurred at 6 weeks apart, one was randomly selected. For records of COPD exacerbations within a 6-week period, the first one was used. The date of attendance for COPD exacerbation was defined as the index date. Using these data, two groups were formed based on whether or not antibiotics were prescribed during the index GP consultation. Patients who died before the cut-off point (i.e. at 4 weeks after the index visit) were excluded from the analysis.
Economic evaluation
We conducted a CEA to evaluate the use of antibiotics compared with no antibiotics for COPD exacerbation management. The analysis was undertaken from the perspective of the UK NHS and only direct health-care costs were included. In the base case, the time horizon of the evaluation was 4 weeks after the index visit for the COPD exacerbation. Owing to the short time horizon of the base-case analysis, discounting of costs or outcomes was not necessary. The following resource use areas were included in the analysis: GP visits, hospital admissions, community respiratory team referrals and all referrals. Antibiotics prescriptions and infections data were also assessed. Unit costs were applied based on established costing sources (see Table 22 ),128–130 with costs reported in 2012 figures.
Statistical analysis
Demographic and clinical variables were compared according to group. Poisson regression was used in the first instance to analyse the factors that determined the number of resource use counts over the analysis period. However, the data were found to significantly deviate from a Poisson distribution, hence negative binomial regression was used. Differences in resource use were expressed as rate ratios. For the mean difference in costs, a linear regression was used. We adjusted for covariates that could influence the results: age, sex, smoking history, body mass index and ethnicity.
Results
A total of 45,375 patients aged 40–104 years were selected: 27,904 (61.5%) in the ‘antibiotics’ group and 17,471 (38.5%) in the ‘no antibiotics’ group. The mean age of this population was 71.0 years and 50% were women. The two groups were comparable in terms of age, sex, BMI, ethnicity, smoking history and number of cigarettes smoked per day ( Table 25 ). COPD status varied slightly between the two groups. The ‘antibiotics’ group tended to have a slightly larger proportion with normal and mild COPD, and fewer with moderate and severe COPD than the ‘no antibiotics’ group.
| Characteristic | Antibiotic group (n = 27,904) | No antibiotic group (n = 17,471) |
|---|---|---|
| Sex, no. (%) | ||
| Women | 13,960 (50.0) | 8750 (50.1) |
| Mean age (years) | 70.0 | 72.6 |
| Mean body mass index (kg/m2) | 26.7 | 25.9 |
| Mean no. of cigarettes/day | 14.4 | 14.0 |
| Smoking history, no. (%) | n = 27,847 | n = 17,355 |
| Non-smoker | 3011 (10.8) | 1975 (11.3) |
| Past-smoker | 13,550 (48.7) | 8747 (50.4) |
| Current smoker | 11,286 (40.5) | 6633 (38.2) |
| COPD status, no. (%) | n = 20,213 | n = 10,474 |
| Normal | 4236 (21.0) | 1851 (17.7) |
| Mild | 6653 (32.9) | 2838 (27.1) |
| Moderate | 6676 (33.0) | 3668 (35.0) |
| Severe | 2648 (13.1) | 2117 (20.2) |
Resource use
The resource utilisation data for the two groups are presented in Table 26 . For all outcomes, the antibiotics group used the least number of resources.
| Resource use item | Antibiotics, mean (95% CI) (n = 26,822) | No antibiotics, mean (95% CI) (n = 15,903) |
|---|---|---|
| GP visits | 4.971 (4.921 to 5.021) | 7.733 (7.637 to 7.830) |
| Referrals to community respiratory team | 0.001 (0.001 to 0.002) | 0.002 (0.001 to 0.002) |
| Referrals | 0.091 (0.087 to 0.095) | 0.095 (0.090 to 0.101) |
| Hospitalisations | 0.326 (0.302 to 0.350) | 1.138 (1.032 to 1.243) |
| Antibiotics prescriptions | 0.352 (0.344 to 0.359) | 0.353 (0.343 to 0.363) |
| Infections | 0.073 (0.696 to 0.767) | 0.081 (0.076 to 0.086) |
The rate ratio for GP consultations was 0.65 (95% CI 0.64 to 0.66) for the ‘antibiotics’ group compared with the ‘no antibiotics’ group ( Table 27 ). This indicates a significantly lower number of GP consultations in the ‘antibiotics’ group than in the ‘no antibiotics’ group. The ‘antibiotics’ group also had over three times fewer hospitalisations for the 4-week period (rate ratio 0.30, 95% CI 0.27 to 0.33). The infections analysis showed a slight decrease in the number of infections for the ‘antibiotics’ group, compared with the ‘no antibiotics’ group (rate ratio 0.91, 95% CI 0.84 to 0.99).
| Resource use item | Rate ratio (95% CI) |
|---|---|
| GP consultations | 0.65 (0.64 to 0.66) |
| Referrals to community respiratory team | 0.75 (0.42 to 1.31) |
| Referrals | 0.96 (0.89 to 1.03) |
| Hospitalisations | 0.30 (0.27 to 0.33) |
| Antibiotics prescriptions | 0.99 (0.96 to 1.03) |
| Infections | 0.91 (0.84 to 0.99) |
Costs
The mean adjusted cost per patient with a COPD exacerbation for the ‘antibiotics’ group was estimated to be £748, compared with £1911 for the ‘no antibiotics’ group ( Table 28 ).
| Cost | Antibiotics, mean (95% CI) (n = 26,822) | No antibiotics, mean (95% CI) (n = 15,903) | Incremental, mean (95% CI) |
|---|---|---|---|
| Adjusted cost per patient (£) | 748 (700 to 797) | 1911 (1847 to 1974) | −1162 (−1243 to −1083) |
| Unadjusted cost per patient (£) | 726 (677 to 775) | 1948 (1885 to 2011) | −1222 (−1302 to −1142) |
The results indicate that the use of antibiotics for COPD exacerbations resulted in cost savings and an improvement in all outcomes analysed ( Table 29 ). Hence, the use of antibiotics was dominant over no antibiotics.
| Measure | Antibiotics, mean (95% CI) | No antibiotics, mean (95% CI) | Incremental, mean (95% CI) | ICER |
|---|---|---|---|---|
| Costs (£)a | 748 (700 to 797) | 1911 (1847 to 1974) | −1162 (−1243 to −1083) | |
| GP visitsb | 5.00 (4.95 to 5.05) | 7.67 (7.57 to 7.76) | −2.67 (−2.77 to −2.57) | Dominantc |
| CRT referralsb | 0.0012 (0.0007 to 0.0016) | 0.0016 (0.0009 to 0.0023) | −0.0004 (−0.0012 to 0.0004) | Dominantc |
| Referralsb | 0.091 (0.087 to 0.095) | 0.09 (0.090 to 0.100) | −0.004 (−0.011 to 0.003) | Dominantc |
| Hospitalisationsb | 0.330 (0.305 to 0.355) | 1.111 (1.001 to 1.216) | −0.781 (−0867 to −0.695) | Dominantc |
| Antibiotic prescriptionsb | 0.351 (0.343 to 0.359) | 0.354 (0.344 to 0.364) | −0.003 (−0.016 to −0.010) | Dominantc |
| Infectionsb | 0.073 (0.070 to 0.077) | 0.081 (0.076 to 0.085) | −0.008 (−0.014 to −0.002) | Dominantc |
Discussion
This observational CEA using EHRs found that antibiotic prescribing for COPD exacerbations was associated with lower resource use. The purpose of our CEA was to illustrate the type of data that would be available from a point-of-care trial statistically powered to measure the effects on clinical outcomes.
There were several limitations with our analyses. Residual confounding may have been present in the comparisons of the ‘antibiotics’ and ‘no antibiotics’ groups. This highlights the importance of conducting a trial which would remove such bias from an analysis. Ideally, a cost–utility analysis would be conducted to generate the cost per QALY associated with antibiotics use. However, QALYs were not available for the population under analysis, and hence a cost–utility analysis was not undertaken. Future collection of quality-of-life values for such COPD exacerbation patients would be worthwhile.
In conclusion, a large observational patient data set derived from EHRs was used to illustrate the type of analysis that can be undertaken fort point-of-care trials. The use of antibiotics for patients who present to their GP with a COPD exacerbation was found to be dominant over (i.e. compared favourably with) the use of no antibiotics, as it resulted in cost savings and improved outcomes.
Chapter 11 Scientific challenges in point-of-care trials
This chapter outlines scientific challenges in point-of-care trials. It is based on the feedback from reviewers and responses in relation to an article we published in the British Medical Journal (BMJ) in 2012. 14
Data quality
The key characteristics of good data quality have been outlined by the UK Audit Commission, which can also apply to EHRs. 133 These characteristics concern validity, accuracy, reliability, timeliness, relevance and completeness. A pivotal consideration for the feasibility of an electronic point-of-care trial is if the end points of interest can be measured with reasonable quality in the EHRs and if the criteria used for flagging during consultation are valid. We propose four steps, of which three could be conducted prior to the start of a trial.
Step 1: development of an algorithm for electronic health record definition of end point of interest (in a non-trial population)
An algorithm for the EHR definition of the end point of interest will need to be developed, which could be challenging in EHR research databases given the heterogeneity in recording of data and varying levels of clinical details. Researchers have often used different definitions to extract data,134 ranging from very detailed case algorithms to broad definitions based on the presence of a single code. Researchers have also differed in which code lists should be used to extract data. 135 Rather than trying to classify each patient as a definite case or not, a better approach to dealing with heterogeneity in information may be to estimate the probability that a patient was correctly classified (i.e. positive predictive value). Several methods could be used to estimate the probability that the EHR algorithm for identifying cases was correct. It could be based on clinical review of possible cases providing the probability that cases as identified by the algorithm were confirmed in the review. A sample of cases could be reviewed by the clinicians caring for the relevant patients who are asked to confirm and validate the case outcomes and, ideally, provide supporting evidence independent of the EHR. Alternatively, the complete EHRs (from all linked data sets) could be extracted and reviewed by independent adjudicators. The positive predictive value for cases with, for example, heart attack that are consistently recorded in linked data sets (e.g. primary care records, hospital admissions and disease registry) would be much larger than that for cases recorded in a single source only. The availability of a linked database may be important for increasing the capture of outcomes and data quality. A recent English study that compared the recording in primary care, hospital admission and disease registry records found that each data source missed a substantial proportion (25–50%) of myocardial infarction events. It concluded that the failure to use linked EHRs may lead to biased estimates of the incidence and outcome of myocardial infarction. 136 This approach of the development of an algorithm for data extraction from EHRs has been used before, as was done, for example, in a study that looked at using EHRs for trial recruitment. 137
Step 2: validation of algorithm in a non-trial population
The validity of the algorithm used to identify the end points of interest in the EHRs could be evaluated by the analysis of incidence rates. As an example, the rate of heart attacks as estimated in the EHRs using the algorithm could be compared with estimates from literature. Such comparisons would also provide some evidence in the specificity of the case algorithm. An algorithm that would require extensive corroborating evidence in the EHRs would likely provide an underestimate of the true incidence rate as cases would be misclassified due to incomplete records. On the other hand, a non-specific algorithm could overestimate the incidence. Another analysis could consist of the evaluation of well-known associations, such as the Framingham risk score and CVD risk.
Step 3: identification of sites with unusual patterns of data quality in a non-trial population
Selection of sites with better quality of EHRs could be the third step. Sites with unusually low incidence of the end point of interest or with a pattern of unusual recording could be excluded from point-of-care trials. Figure 10 shows the results of cluster analysis conducted to identify practices with unusual patterns in data recording of CVD. The cluster analysis identified among the 295 CPRD practices, four groups of practices with statistically different patterns of data. The groups included 150 (50.8%), 52 (17.6%), 91 (30.8%) and 2 (0.7%) practices respectively. The two practices in the fourth group were found to have the lowest CVD incidence relative to the practices in the first group (adjusted relative rate 0.67, 95% CI 0.51 to 0.88).
FIGURE 10.
Incidence rates of CVD by calendar year in different groups of practices, stratified by cluster analysis. (a) Group 1; (b) group 2; (c) group 3; and (d) group 4. Each line represents the CVD incidence over time in a practice.

Step 4: application of algorithm to the trial population
The validated algorithm could then be applied to the trial population as long as the recording of data for trial patients would be similar to that outside the trial (i.e. the trial imposes no changes in data recording). In addition, the prospective, randomised, open, blinded, end point design could be used to review recorded end points of interest in a trial. 40 Adjudicators blinded to the randomised treatment would review all relevant information as extracted from the EHRs. In case of any questions or discrepancies between the various data sources, a case validation approach could be used. The EHR information could be summarised in a text report and then sent to the clinician at the site.
Lack of blinding to treatment and use of placebo
Point-of-care trials typically will have allocation concealment of the randomisation schedule (i.e. the person randomising the patient does not know what the next treatment allocation will be). However, patients and clinicians will typically not be blinded to the outcome of randomisation and know which treatment the patient was randomised to. Bias in unblinded trials may occur if perspectives vary between the treatments in the trial resulting in behavioural differences or the placebo effect. 138 The placebo effect has been described as a physiological effect caused by the meaning that the brain associates with the placebo. 139 An example of this is a study that reported substantive differences in symptoms of inflammatory bowel disease between an attentive patient–clinician relationship compared with being on the waiting list. 140 Bias may also occur if the measurements and recording of the trial outcomes vary between allocated treatments. Clinicians may refer patients selectively or outcome assessors may assess patients treated with the experimental intervention more favourably. 138 Blinding to treatment allocation is considered by many researchers to be a crucial method for reducing such observer bias. Tool for assessing risk of bias in randomised trials consider lack of blinding as a limitation. 141
Blinding would also not mimic routine care. These trials aim to randomise between different clinical decisions. In routine care, a clinician may choose between, for example, a statin or no treatment (placebo interventions are rarely used). Placebo effects and behavioural changes as a result of starting a treatment may be relevant for a clinical decision rather than being nuisance factors, as in an explanatory trial. As an example, a clinical decision to prescribe a statin should take into account any behavioural changes leading to a worsening diet, as this may impact on its effectiveness. Enck et al. 142 discussed how the placebo effect should be managed. They argued that the placebo effect should be minimised in explanatory trials in order to optimise drug–placebo differences, thus ensuring that the efficacy of the investigational drug can be truly evaluated. Once the drug is in clinical use, placebo effects should be maximised by harnessing patients’ expectations and learning mechanisms to improve treatment outcomes. 142
Measurement bias in the trial outcomes may also affect point-of-care trials. The degree of measurement bias will probably depend on the type of outcome. Diagnosis of death is unlikely to be influenced by awareness of trial medication. On the other hand, pain recording may be quite sensitive to lack of blinding. 138 A review of 146 meta-analyses found that effect estimates were exaggerated for subjective outcomes when there was inadequate or unclear allocation concealment or lack of blinding, whereas there was little evidence of bias in trials with objective outcomes. 143
The main end points of point-of-care trials could be major clinical outcomes, such as death or heart attacks. The prospective, randomised, open, blinded, end point design could then be implemented. 40 A point-of-care trial that obtains data from different data sets through linkage would further minimise bias due to differential diagnosis and recording of major clinical outcomes. A trial that uses data from various linked health-care data sets would not rely on the diagnosis, coding and recording of a single clinician. Careful consideration will need to be given to the choice of the outcome. The use of non-specific or composite outcomes could increase the proportion of events that are not causally related to the intervention, thus diluting the estimated effect towards the null. Conclusions made on the basis of the effect on a composite outcome may not be readily generalisable to other populations. 144 It is also important that the statistical analyses are prespecified and that the statistical analysis plan is published. EHRs may provide an opportunity for data dredging and selection of most favourable results. As reported previously, bias can be introduced if outcomes are not prespecified. 145
Patient-reported outcomes
The collection of patient-reported outcomes (such as quality of life) may be challenging in point-of-care trials. The possibility of measurement bias, because of the lack of blinding of patients, may affect the validity of more subjective end points, especially when patients have different perspectives on the randomised treatments (in eLung, many patients will have used antibiotics before). The feasibility of collecting these data during unscheduled consultations may be another challenge. As an example, the eDiaries were not used in eLung and the response rate for paper questionnaires was suboptimal. The lack of use of the eDiaries was likely related to the need to instruct trial participants about the device, which was not possible due to the lack of time during the unscheduled consultation. An alternative approach could be to utilise EHRs to develop surrogate measure for patient-reported outcomes. A study by Hutchings et al. 146 found that EHRs may have the capacity to develop these measures but that currently there was a wide under-reporting of symptoms in EHRs. 146 A future approach may be to collect patient-reported outcomes systematically during routine clinical care. For example, these data could be collected in the waiting rooms of clinics.
Assessment of trial eligibility
Screening for trial eligibility is the first step in the patient recruitment for point-of-care trials. One reviewer commented:
I tried to think about this in terms of a clinical question and trial that I would understand and settled on a comparison of sumatriptan versus non-steroidal anti-inflammatory drugs for patients with episodic migraine. Here’s where the problems begin. Diagnosis of migraine in primary care is notoriously inaccurate. The clinical question that would end up being answered is not sumatriptan versus non-steroidal anti-inflammatory drugs for migraine but rather sumatriptan versus non-steroidal anti-inflammatory drugs for patients GPs *think* have migraine.
A related challenge was experienced in Retropro: the calculation of CVD risk was found to be subjective and to vary substantially between different risk scores, without consistent guidance by NHS prescribing guidelines. 76 As a consequence, there were large differences for some patients between the assessments of CVD risk by the GP and principal investigator. There is no single answer to the ideal scope of eligibility criteria for point-of-care trials. One answer is that recruitment should be restricted to patients with a specific indication in which a biological effect has been observed in explanatory trials for the study intervention. An alternative answer is that point-of-care trials should be conducted if a substantial number of clinicians are already using the study intervention for a non-specific condition in order to address the uncertainty.
Heterogeneity of effects
Randomised trials typically address the question of whether or not an intervention is effective in an average patient. Average results can hide variations in individual responses. Analyses of subgroups of the trial population could evaluate any heterogeneity in effect, but they have limitations due to the possibility of random variation of effects across subgroups. Large trials that use EHRs could also provide an opportunity for data fishing expeditions, given the size of the population and the multitude of variables. It has been recommended that subgroups should be defined based on biological plausibility, or validated. 147 Heterogeneity of effects may be a large challenge in point-of-care trials compared with explanatory trials, given the deliberate intent to include broad patient populations.
Ioannidis and Lau148 proposed an interesting approach to systematically assessing heterogeneity which could be applied to point-of-care trials. A predictive statistical model would be built using known risk factors for trial outcomes. The variability in baseline risk can then be assessed and differences in risk evaluated. A homogeneous trial (i.e. with little variation in risk) may not yield results that are generalisable to more diverse population. On the other hand, substantive heterogeneity in baseline risk would indicate the need for evaluating the consistency of treatment effects at different levels of risk and subgroup analyses. 148 EHRs could also be used to compare the distribution of baseline risks in the population likely to be treated with the intervention with the distribution of risk among trial participants.
Generalisability of findings
Penston replied to our BMJ publication on the rationale for point-of-care trials14 with the following comment:
The authors make much of their contention that pragmatic trials will allow the data to be generalised. So much so that, in their conclusions, they imply that the external validity of the results will not be open to question. Given that there will be differences between doctors who participate in research and those who do not, that not all of the eligible patients will be recruited, and that the trial sample will not be drawn randomly from the underlying population, this is very hard to accept.
Sacristán et al. 149
Point-of-care trials should indeed be naturalistic and aim to enrol a ‘random’ sample of clinicians and patients. The original objective of this project was to have many clinicians each recruiting a few patients but we ended up with a few clinicians recruiting many patients. The critique by Preston is valid: point-of-care trials will provide generalisable findings only if clinicians and patients are a representative sample of the population of interest. EHR databases may provide an opportunity to measure characteristics of patients not recruited into a trial.
Lack of effect due to non-compliance
Bergmann supported the concept of point-of-care trials, but mentioned the following limitation:
The necessary intention to treat analysis may often conclude to an absence of difference between the two therapeutic strategies not because of an evidence-based clinical equivalence but because a weak evaluation of the efficacy or safety due to a too large variability linked to the erratic therapeutics compliance in usual conditions. This variability reduces the possibility to demonstrate the superiority of one of the tested treatments even if this superiority exists.
Sacristán et al. 149
Non-compliance is a nuisance in explanatory trials and needs to be minimised there. However, it is important to know for decision-making if a biologically effective medication is not being used by patients for whom it is appropriate. Erratic therapeutic compliance is therefore an end point of interest in pragmatic trials. Furthermore, the artificially high compliance achieved in explanatory trials by careful selection and regular instructions for patients may hide an improved performance in a normal practice setting for a less toxic intervention with fewer instructions.
Treatment contamination over time
Patients may change their treatment over time as highlighted by Bergmann:
After randomisation, the various doses, durations, stops, resumes, cross-over and individual modalities of the studied treatments as the different follows-up of the patients may trouble the demonstration of differences between the two compared treatments.
Sacristán et al. 149
Treatment contamination can indeed bias the study of biological effects as the exposures of interest are changed. However, it would be relevant to know that many patients switch from, for example, simvastatin to atorvastatin due to side effects. Treatment contamination is of direct interest in pragmatic trials as long as it reflects normal practice. Of course, it will be challenging to evaluate long-term side effects in pragmatic trials if there is treatment contamination. Statistical techniques, such as contamination-adjusted intention-to-treat analysis, may reduce bias due to treatment contamination. This method uses the statistical technique of instrumental variable analysis to address the contamination. 150
Concept of large trials
The concept of large trials has been described as deeply flawed by Penston. 151 His main reason for saying this is that the magnitude of treatment effects is often small in absolute rather than relative terms. Penston showed that less than 5% of patients in the CVD prevention trials derived any benefit from taking the medication for years (i.e. by not experiencing CVD). 151 Trials should be targeted at very high-risk people, thereby keeping the trials smaller. Clearly, the magnitude of absolute risk reductions with a treatment should be important for decision-making rather than only relative effects. However, low absolute event rates in trials are indicative not of flaws of randomisation but rather of imperfect identification of high-risk patients and targeting of treatment. Furthermore, patient preferences should be taken into account in deciding worthwhile levels of risk reduction.
Design of point-of-care trials
Retropro and eLung randomised individual patients to different treatments. An alternative design could have been the cluster trial. In cluster trials, entire areas or health service organisational units are randomly allocated to intervention or control groups with outcomes evaluated for individuals within each cluster. 152 Cluster randomised trials are increasingly used in public health and health services research. The main advantage of a cluster trial may be the easier implementation. For example, local prescribing guidelines currently restrict the choice of type of statin to simvastatin. Such policies could be amended to allow random allocation of each practice to the type of statin, which could be easily communicated to all practice GPs and monitored for adherence. However, there are several limitations with cluster trials. 153,154 One limitation occurs if there is variability between practices. For example, the use of antibiotics for COPD exacerbations substantially varies between practices, and this could reduce the statistical power of a cluster trial. Clinicians may also not allocate all of their patients to the intended treatment (e.g. withholding antibiotic treatment to patients with mild to moderate COPD exacerbations given their established clinical practice). However, cluster trials may be useful to compare the effects of different policies that are applied consistently (such as a policy of how to educate patients when to use their spare bottle of antibiotics).
Chapter 12 Discussion, recommendations and guidance
Main lessons of this project
This project provided several lessons with respect to the opportunities and challenges of research using routinely collected data ( Table 30 ). Routinely collected data can provide detailed information on medical and prescribing histories of trial participants and allow long-term follow-up for major clinical outcomes, even after completion of the trial. Data on trial participants and sites can be compared with data from those not participating in the trial. Retropro has shown that a trial can be conducted without site visits and with minimal interference by the research team providing opportunities for more representative trial settings. The challenges in conducting point-of-care trials relate to the complexities in obtaining research governance approvals, recruitment and retainment of GPs and consent procedures for recruiting patients.
| Area | Lesson | Comment |
|---|---|---|
| Opportunity | Clinicians showed interest in participating in simple trials | Two-thirds of practices that were contacted expressed interest; qualitative research also indicated support in principle |
| Patients showed interest in participating in simple trials | Qualitative research indicated support and patient representatives on the Trial Steering Committee advocate further progress (see Appendix 5 ) | |
| Routinely collected EHRs can provide long-term information on major clinical outcomes; linked databases enhance the capture of major clinical outcomes | Data quality can be measured in EHR databases prior to start of trial; end points in trials should be simple to define; linkages can be important in enhancing capture of end points | |
| Feasible to conduct simple trials without site visits and with minimal interference by research team | Central monitoring techniques are valuable especially if data are available on non-trial participants | |
| IT system can facilitate simple recruitment procedures including flagging | Clinicians found the technical procedures for recruitment simple, having done it once; interest was expressed in tailoring flagging to individual preferences of clinicians | |
| IT system can facilitate central control of recruitment | Patients’ eligibility can be controlled centrally (subject to data availability) and the number of trial participants at a site can be managed | |
| Fraud may be less likely because of the greater chance of detection, greater technical challenges for the fraudulent researcher and ease of routine audit | Availability of EHRs before and after the trial plus information on non-trial participants/sites simplify fraud detection | |
| Challenge | Research governance procedures are complex, leading to substantial attrition of clinicians | Trials of routinely used interventions should be considered quality improvement, regulated under Good Medical Practice guidelines155 |
| Informed consent procedures are legalistic and complex, based on the unsubstantiated belief that long paper forms are more informative | Consent procedures should be informed by preferences of patients; alternative models of consent (of content and timing) should be developed and evaluated | |
| Trial recruitment during an unscheduled consultation is difficult for clinicians | Simplification of trial recruitment procedures is required (including consent procedures and collection of study-specific data) | |
| Study interventions are already well established in routine clinical practice or subjected to treatment guidelines | Trials need to be conducted earlier | |
| Protocols and study procedures should be simple and consistent with routine clinical practice | Collection of eDiaries, for example, should be limited to selected sites that have dedicated research staff | |
| Bias in the measurement of more subjective end points due to preferences (such as patient-reported outcomes) | Placebo-controlled trial should be considered in a subset of sites only |
There are many uncertainties about the relative merits of alternative treatments already in wide use and who to treat with these interventions. Even for statins that have been evaluated in a large number of trials, there are important outstanding research questions. Even small differences in the relative merits of alternative treatments could translate to a substantial number of lives saved if the suboptimal treatment is being widely used. A learning health-care system should consider the continuous optimisation of routinely used interventions as a key task. Clinicians should consider it as their duty to help resolve uncertainties about the effects of their treatments. There is increasing recognition that patients have suffered and died unnecessarily because doctors have failed to recognise and confront uncertainties about the effects of their treatments and to support the research needed to reduce these. 156 The challenge will be to embed research into clinical practice and engender a culture of continuous testing with routinely used treatments in order to find out what works in whom. This will require different models of organising and conducting these trials and of involving patients and seeking consent. Faden et al. 157 recently proposed that a trial like Retropro may be allowed under certain conditions to proceed without consent of patients at the time of randomisation. Patients could be regularly informed about randomisation activities in the clinic and be allowed to opt out from future randomisation. There should be engagement with patients and clinicians about the proper role for randomisation and consent in a learning health-care system. 157 Randomised trials of routinely used treatments should be simple to conduct and conducted as a matter of routine. Clinicians and patients should set the priorities for these trials and conduct them, with researchers merely providing the infrastructure for design and analysis.
This project has shown that electronic point-of-care trials are feasible, although the recruitment of clinicians is a major challenge owing to the complexity in the trial approvals and diverse barriers to recruitment (as outlined in Chapter 8 ). However, these trials will contribute substantially to the quality improvement only if interventions, clinicians and participants of these trials are representative and if trials are simple to conduct. The current research governance system mandates special requirements and conditions to trials, even those that aim to evaluate interventions in routine use. As a consequence, only a selective minority of clinicians participate and trial interventions may no longer reflect usual care (e.g. due to lengthy consent procedures or requirements to switch trial medication for administrative convenience). Point-of-care trials will have limited scientific value if the trial setting or participants are artificial. These trials aim to compare actual clinical decisions (rather than testing biological effects of a molecule) and, thus, will need to replicate these clinical decisions. The concept and practice of research exceptionalism are a fundamental and critical challenge to the use of point-of-care trials.
Several authors have advocated large simple point-of-care trials with minimal study-specific data collection, inclusion criteria consistent with normal clinical practice and collection of information on major clinical outcomes. 158–161 As succinctly put by Tognoni et al. ,162 truly important questions need only simple protocols and data collection. Unfortunately, these trials are currently rarely done although the recent TASTE trial provides an example how simple trials can be done. 25 The call for making trials much simpler and larger clearly has not been heeded. 163
An important limitation of this project was that the study setting was restricted to primary care and that simple pharmacological interventions were evaluated. The GPs in the UK have been using EHRs for over two decades. Secondary care in the UK has had a lower uptake of EHRs. The recent experiences in Sweden of the TASTE trial, conducted in secondary care,25 do support the notion that point-of-care trials do not need to be limited to primary care. Our project was developed and based in CPRD, which currently contains the EHRs of about 8% of the UK population. However, many of the issues raised are likely to apply more generically to EHRs.
Best practice for point-of-care trials
This section outlines a proposal for best practice of point-of-care trials focusing on those aspects specific to point-of-care trials. Best practice should include careful assessment of the research objectives. They should be clinically valid and address important uncertainty based on systematic reviews, and the opinions of clinicians and patients. The need to include patients in developing research agendas is now well established. 164,165 Clinicians and patients should be involved in the development of the study protocol and review the need for any study-specific procedures. The protocol and study instructions should be succinct, ideally merely reflecting treatment guidelines. The study interventions to be compared should be administered in a manner consistent with intended practice (for novel interventions) or with usual practice (for interventions already in use). Clinicians should be asked to adhere to Good Medical Practice guidelines in recruiting patients, seeking consent and providing medical care for trial participants. 155 Clinicians who are not familiar with the study interventions should be offered training prior to the start of the study, based on treatment guidelines (e.g. for newly licensed interventions). The proposed patient recruitment models should be tested with clinicians and patients who should be able to choose the trial recruitment model that is best suited to their practice (e.g. choice of who and when to flag during consultation). The trial should aim to enrol many clinicians each enrolling only a few patients. The follow-up of trial participant and data collection should ideally reflect usual care. Importantly, the recruitment, flagging and data collection models should not vary greatly between point-of-care trials (other than the choice of hot and cold recruitment), ensuring that clinicians are familiar with the procedures.
A pivotal question is if the EHR database can identify potentially eligible patients and/or measure study outcomes to an acceptable standard. Prior to the start of the trial, an algorithm for case identification should be tested and validated and sites with unusual patterns of data should be identified and possibly excluded from the trial. The potential bias in the lack of blinding will need to be considered. Outcomes that may be affected by this (such as patient-reported outcomes) may require that this assessment is done only in a subset of sites with blinding of patients and clinicians. After the end of the trial, the prospective, randomised, open, blinded, end point design should be used to review recorded end points of interest in a trial. Adjudicators blinded to the randomised treatment could review all relevant information as extracted from the EHRs. 40 In case of any questions, the EHR information should be summarised in a text report and then sent to the clinician at the site for clarification. Linkage with other health-care databases may improve the capture of the cases and completeness of information and also facilitate the long-term follow-up.
Central data monitoring should be used to identify irregularities in trial data that might indicate incorrect or fabricated data, using statistical tests to detect unusual patterns within and across centres and assess consistency of variables over time. ADR monitoring should consist of periodic analyses of rates of adverse outcomes in the trial population compared with a non-trial population. The EHRs should also be searched for entries of suspected side effects with the study medications. An important analysis will be the comparison of the trial and non-trial populations in order to assess the generalisability of the trial results and the performance in recruitment.
The analyses of trial end points should be based on a statistical plan defined prior to the start of the study which identifies the key analyses for publication. The statistical plan and the operational definitions for outcomes and variables should be published at the time of the publication of trial results. The research staff involved in the analyses of trial data should be blinded to treatment allocation until the analytical programs have been finalised. Programs for pivotal analyses and EHR data extractions should be done independently by two researchers (i.e. double programming). Consideration should be given to how best to communicate results to trial participants. One option may be to use the flagging software to notify clinicians during consultation of a former trial participant for whom they can print out the trial results at the study website.
Data protection and security is of critical importance for point-of-care trials that use EHR databases. Staff training and standard procedures, and skills and attitudes of staff, are important for treating data with appropriate care. 166 Regular audits by external experts, with publication of the audit findings, should be very useful as they help to maintain a culture of continuous improvement. 167 Open access to research data should minimise any effects of conflicts of interest, including any commercial interests of EHR databases. The Royal Society in the UK has recommended an open data culture: scientists should make data available in appropriate data repositories. 168 Although it would not be appropriate to put all EHR data online for open access, an open data culture would support a model in which researchers can access EHR data following scientific review of the protocol done fully independently of the custodians of the EHR database. 169
Recommendations
To develop evidence and implement risk proportionality in trial governance and conduct
The experiences in this project and that of others46 do highlight the need for evidence of what works best and for risk proportionality in the governance and conduct of trials. GCP has been mostly based on expert opinion. It was also developed at a time when information was processed predominantly on paper. When dealing with the question of how to approach computer validation for EHRs from multiple systems and parties, we obtained advice that the best method for this was to print the EHRs and have the clinician sign the paper. GCP is also based on the notion that the records at the site are the source of trial information: the trial database needs to be consistent with the data at a site (i.e. source verification). However, this notion of a single data source is being replaced by a digital revolution with information coming from multiple places with different pieces of information. Loder et al. 170 have outlined that the ways we create, access and store information is changing rapidly. The challenge will be to develop the methods and approaches of how to extract reliable knowledge out of large amounts of data provided by digital technology. This challenge may only be met if the focus is on developing evidence rather than on following opinions and rules.
To develop strategies to increase recruitment in point-of-care trials
The nested qualitative process evaluation of eLung (the QUEAN study) found that recruitment of clinicians and patients into point-of-care trials was widely supported in principle and considered feasible for both acute and chronic conditions. Further research is needed to test effective strategies to improve the efficiency of point-of-care recruitment, particularly regarding innovative approaches to consent procedures. These challenges are fundamentally practical and potentially open to resolution. The greater challenge is the demanding context in which GPs operate and the underpinning requirement for a GP to have a high level of personal interest in research to address the many barriers to successfully implementing research among their daily clinical workload. Indeed, point-of-care trials provide an exciting opportunity to build on and broaden existing levels of GP interest in research despite limited time and capacity through the involvement in the full implementation of a trial with relatively minimal additional workload.
Based on the evidence of the QUEAN study and related literature, a package of recommendations is proposed to increase recruitment in point of care trials, one of the main barriers to the potential success of future point-of-care trials. The recommendations include strategies to address the specific technical and practical challenges unique to point-of-care recruitment, strategies to involve GPs and commissioners in identifying research priorities and informing design of successful implementation on the ground, combined with the crucial element of strategies to provide broader policy and infrastructural support for the promotion of simple and larger trials using EHRs to GPs ( Table 31 ). A comprehensive approach to both assessing the technical barriers and enabling the promotion and support of successful recruitment and implementation is more likely to improve recruitment at each stage of the process. This is important for point-of-care trials which require wide inclusion of GPs to increase external generalisability and high retention rates to maintain internal validity. 89 It also recognises the influence of several generic and project-specific factors on the recruitment decision for a research study as identified by the QUEAN study. Although this would require investment in the short term, including site-specific support to enable participating GPs to recruit successfully and positively, this would be expected to reduce over time as confidence and familiarity with the trial method increases as an integral part of a learning health-care system.
| Stage of recruitment | Recommended strategy to enable recruitment | Underpinning factor | |
|---|---|---|---|
| Generic | Project specific | ||
| Stage 1: policy and infrastructure support | Government policy to support innovative research methods
using EHRs to increase evidence base in primary
care National and local promotion and training to clinicians of trials using EHRs Review of roles and responsibilities of PCRN to provide relevant promotion, training and infrastructure support |
Personal interest in research in primary care EHR as valuable primary care resource |
|
| Stage 2: design and development | Local clinical input into planning of trials:
Involvement of patients into design of trial |
Benefit to local population/patients Time/capacity to implement study |
Relevance of topic Consistency with local prescribing plans Feasibility to recruit Trial procedures and outcomes relevant to patients |
| Stage 3: clinicians’ participation | Improved information
|
Adequate remuneration Time/capacity to implement study |
Feasibility to recruit Point-of-care hot recruitment method Computerised recruitment via pop-up alerts |
| Stage 4: clinicians’ recruitment of patients and clinicians’ retention | Site-specific support during set-up
Clinicians to assist with interpretation of study data as required |
Time/capacity to implement study | Feasibility to recruit Point-of-care hot recruitment method Computerised recruitment via pop-up alerts |
To increase the involvement of patients and clinicians in point-of-care trials
In addition to the formal evaluation of the factors influencing recruitment into point-of-care trials in the QUEAN study, the two patient representatives on the Trial Steering Committee each provided their views on how to progress with point-of-care trials (see Appendix 5 ). The first necessity is to build a clear sense of common purpose and urgency about facilitating the choice of research questions. Second, expediting the set-up process for point-of-care research, particularly with regard to the burden of obtaining research governance approvals, is also required. The third is to establish a team committed to leadership, capable of sustaining momentum and resourced to do so. Early objectives must be to demonstrate the effectiveness and benefits of point-of-care randomised trial research in resolving high profile unresolved uncertainty dilemmas of common concern to clinicians and patients. Pioneer projects should affect both primary and secondary care and ideally point to cost savings to demonstrate that, over the medium term, such research can be financially self-sustaining, as well as providing evidence resulting in patient benefit. Furthermore, alternative models for engaging patients and seeking their consent should be explored, including waiting room posters or rolling TV advertisements in the waiting room of general practices.
The two lead GP investigators also provided their views on how to increase GP involvement in point-of-care trials (see Appendix 5 ). The challenge in point-of-care trials is to simplify the initiation and conduct and to have research questions that will be of concrete interest to clinicians. Such simplicity should make it easier to recruit clinicians and patients. In the UK, the PCRN’s Research Sites Initiative should be utilised to provide support for local feasibility and to include successful participation in point-of-care studies (and maintaining the software tools) as part of the criteria for participation in research site initiatives schemes. Local Clinical Research networks could be used to engage with Clinical Commissioning Groups, NHS and Commissioning Support Units (who maintain practice IT) to ensure wider and more responsive support for the technical challenges in these studies. One of our qualitative studies found that seeking consent for trial participation was a barrier for GPs but not for nurses. The role of nurses in point-of-care trials should be further explored.
To obtain patient views on how to deal with and communicate uncertainty
It is currently difficult to initiate trials even for routinely used interventions, as we and others have experienced. The trial regulation and governance system are complex and only few clinicians participate. The consequence of this complexity may be that trials are not routinely conducted and uncertainty is not addressed. There is currently a paradox in how the health-care system deals with uncertainty. Richard Smithells noted that he needed permission to give a treatment to half of his patients (to find out whether it did more good than harm), but that he did not need any permission if he decided to give the treatment to all of his patients. 45 Informed consent requirements also are very different with trials requiring lengthy consent forms in contrast to brief discussions in usual care. 171 COPD patients are not informed routinely that their exacerbation would be treated differently by another clinician,26 while many obstacles are put into the way for anyone wanting to initiate a trial in order to resolve this variability. The fundamental question is how patients do view this apparent double standard in the handling and disclosure of uncertainty. A survey of over 1000 adults found that 97% of respondents agreed that the NHS has a duty to determine the safety and effectiveness of the drugs its doctors prescribe. 172
To develop consent procedures informed by patient views
The informed consent procedures should be designed in collaboration with patients and be based on empirical evidence on what kind of process best informs participants. However, the GCP standard, which has come to be viewed as canonical for trial governance, was based on expert opinion and has little basis in empirical evidence. A recent systematic review found that there is limited empirical evidence of what information potential participants want to know about research when they are making the decision to take part. 173 There is a need to understand better the type and amount of information people want in order to support their individual decision-making. It would also be very useful if the consent procedures could take place some time prior to randomisation, especially for hot recruitment of acutely ill patients. COPD patients could have been approached in eLung and asked for consent to randomisation for a future exacerbation rather than at the time when they were acutely ill. Rather than lengthy paper forms with a paternalistic content determined by experts, consent forms should be tailored to individual needs and use modern technology to communicate.
To measure and acknowledge systematically uncertainty in guidelines
It is currently no longer possible to initiate Retropro or eLung. Although there is scientific equipoise in the comparative effectiveness of different types of statins and in the effects of antibiotics in mild and moderate COPD exacerbations, there is no equipoise for clinicians due to recommendation in guidelines. For statins, the proposed 2014 NICE guidelines for primary prevention of CVD now recommend atorvastatin over other types of statins. This is despite the finding that there was no clinical and statistical difference in their meta-analysis of trials between different types of statins (http://guidance.nice.org.uk/CG/WaveR/123). 174 This recommendation appears to have been based on a cost-effectiveness model which ignored this clinical and statistical uncertainty. It is important that uncertainty is systematically measured and acknowledged in guidelines. Otherwise, we will remain in a vicious circle in which a guideline is based on uncertain evidence but research cannot be done because of the guideline.
To develop cost–benefit analysis to assess excess treatment costs
The excess treatment costs were found to be a considerable barrier to point-of-care trials. The question is who should bear the costs of the routine treatments provided for a trial. The NHS would be the main financial beneficiary for a full Retropro trial with clinically significant findings. If atorvastatin was 10% more effective than simvastatin in reducing CVD, the NHS would save the costs of over 2400 heart attacks for all patients starting statins in a single year (as outlined in Chapter 1 ). This saving would clearly offset the excess treatment costs of a full Retropro trial. The handling of excess treatment costs should be based on a comparison of the future cost benefits of likely trial findings with the excess treatment costs of a trial.
To develop statistical models for the measurement of electronic health record data quality
There are currently over 300 EHR research databases in 45 countries with different characteristics. 175 Data quality is of course very important, and not all clinical outcomes can be measured accurately solely from the data recorded in the EHRs. Information in EHR research databases can also change substantially over time. There is a clear research need to develop and adopt systematic, statistically based methods of data quality assessment. 176 Furthermore, the development of common data models and dictionaries could simplify research across different EHR research databases.
To test risk prediction and patient identification strategies in randomised trials
There were large differences for some patients between the assessments of CVD risk by the GP and principal investigator. We initiated research that explored the variability between practices in statin prescribing between practices76 and in the performance of the Framingham, ASSIGN and QRISK2 risk predictions. We found that statin prescribing varied substantially between practices76 and that the existing risk scores did not predict well CVD risk in individual patients. Point-of-care trials will be useful only if the right patients are identified consistently. Therefore, risk prediction strategies should be subjected to randomised trials.
To further experiment with point-of-care trials
Michael Lauer of the National Institutes of Health has recently expressed a willingness to experiment, during a meeting at the US Institute of Medicine about simple trials. Emerging technologies should be embedded into existing projects and be allowed to fail often but inexpensively in smaller experiments. This allows for a stepwise approach to learning what works as long as lessons are captured and shared. 177 Both EHRs and pragmatic trials may be considered emerging technologies. Although the concept of pragmatic trials was described over 40 years ago,1 they are conducted less frequently. 178 The combination of randomisation and simple data collection through EHRs can provide the opportunity for a stepwise change in quality improvement in the health-care system and wider use of point-of-care trials. Researchers will need to share data and knowledge openly with a view to improving health outcomes for all. 170
Reith et al. 179 concluded in their recent review of barriers to trials:
It is becoming increasingly clear that more extensive use of health records and informatics platforms, along with more refined ethical approaches characterised by the expectation that participation in clinical trials is the norm rather than the exception, could support a dramatic increase in the emergence of definite evidence about treatments. 179
Reith C, Landray M, Devereaux PJ, Bosch J, Granger CB, Baigent C, et al. Randomized clinical trials – removing unnecessary obstacles. N Engl J Med 2013;369:1061–5. Quote reproduced with permission
We conclude by quoting the comments by Sacristán et al. :149
The combination of experiments and observations in daily practice requires important regulatory and cultural changes oriented to eliminate the barriers between clinical practice and clinical research, realising that all research and all clinical actions begin at the patient’s bedside and that every medical act is structured like an experiment. The increasing use of EHR should contribute to the disappearance of walls between doctors who carry out research, and doctors who do not, between patients who participate in RCTs and the ‘real’ patients that doctors see every day, between the clinical research form used in RCTs and the electronic medical history. The systematic use of EHR to conduct RCTs could contribute to completing the circle that began with evidence-based medicine with the new concept of medicine-based evidence. The real challenge is not the technical infrastructure to implement randomised database studies, but understanding that, in the context of a patient-centred medicine, clinical research and clinical practice are the two faces of the same coin. 149
Sacristán JA. Rapid Response BMJ Re: Pragmatic randomised trials using routine electronic health records: putting them to the test, 2012; URL: www.bmj.com/content/344/bmj.e55?tab=responses (accessed 2 July 2012). Quote reproduced with permission
Acknowledgements
Contributions of authors
All authors made substantial contributions to conception and design, or acquisition of data, analysis and interpretation of data, all were involved in the drafting of the manuscript or revising it critically for important intellectual content and all authors approved the final version to be published.
Tjeerd-Pieter van Staa Principal investigator and Professor of Pharmacoepidemiology, was involved in the design, conduct, analysis and reporting phases of Retropro and eLung and design of the QUEAN study. He was also employed by the CPRD, MHRA, London, UK at the time of the trials.
Lisa Dyson Research Fellow, was involved in design, conduct, analysis and reporting phases of the QUEAN study.
Gerard McCann Clinical Trials Manager, was involved in the conduct and reporting phases of Retropro and eLung.
Shivani Padmanabhan Software System Developer, was involved in the design, development and implementation of the IT system.
Rabah Belatri Software System Developer, was involved in the design, development and implementation of the IT system.
Ben Goldacre Wellcome Research Fellow in Epidemiology, was involved in the conduct, analysis and reporting phases of Retropro and eLung.
Jackie Cassell Professor of Primary Care Epidemiology, was involved in the design, analysis and reporting phases of Retropro and eLung.
Munir Pirmohamed Professor and Director of Molecular and Clinical Pharmacology, was involved in the design, analysis and reporting phases of Retropro and eLung.
David Torgerson Professor and Director of the York Trials Unit, was involved in the design, conduct, analysis and reporting phases of the economic analyses for eLung.
Sarah Ronaldson Research Fellow in the York Trials Unit, was involved in the design, conduct, analysis and reporting phases of the economic analyses for eLung.
Joy Adamson Principal investigator of the QUEAN study, a senior lecturer in Epidemiology and also deputy director of the York Trials Unit, was involved in design, conduct, analysis and reporting phases of the QUEAN study.
Adel Taweel Lecturer in Software Engineering, was involved in the design, development and implementation of the IT system.
Brendan Delaney GP and Professor of Primary Care Research, was involved in the design, development and implementation of the IT system.
Samhar Mahmood Research Associate in Informatics, was involved in the development and implementation of the IT system.
Simona Baracaia Masters of Public Health Candidate (at time of study) and Public Health Specialty Registrar, was involved in conduct, analysis and reporting phases of the qualitative research of views on CTR.
Thomas Round GP and Academic Research Fellow, was involved in design, conduct, analysis and reporting phases of the qualitative research of views on CTR.
Robin Fox GP and Honorary Senior Clinical Lecturer in General Practice, was involved in patient recruitment and reporting phases of Retropro and eLung and observer of the Trial Steering Committee.
Tommy Hunter GP, was involved in patient recruitment and reporting phases of Retropro and eLung.
Martin Gulliford Professor of Public Health, was involved in the design, conduct, analysis and reporting phases of Retropro and eLung and member of the Data Monitoring Committee.
Liam Smeeth GP and Professor of Clinical Epidemiology, was involved in the design, conduct, analysis and reporting phases of Retropro and eLung and member of the Data Monitoring Committee.
Trial Team organisation
Co-ordinating Team for Retropro and eLung: Tjeerd-Pieter van Staa (principal investigator), Gerard McCann (trial manager), Heather Dorricott (quality control), Geoff Ali (project planning), Jon Ford (operations), Shivani Padmanabhan (IT system), Rachael Boggon (randomisation and secretary of the Data Monitoring Committee), Rabah Belatri (IT system), Kareen Taiwo-Odukoya (IT system) and Zaynah Gurreebun (archivist).
Health Economics Team: Sarah Ronaldson (health economist) and David Torgerson (lead health economist).
The QUEAN study team: Joy Adamson (principal investigator, the QUEAN study), Lisa Dyson (principal researcher, the QUEAN study) and Maggie Gowlett (secretary).
Team for qualitative study on CTR: Simona Baracaia (researcher), Thomas Round (supervisor) and Brendan Delaney (supervisor).
Trial Steering Committee: Iain Chalmers (chair), Chris Cates, Marion Cumbers, Gary Simons, John Williams, Paul Wallace, Kent Woods (observer), Patricia Henley (observer for sponsor) and Robin Fox (observer).
Data Monitoring Committee: Brian Gennery, Liam Smeeth, Martin Gulliford.
Retropro GP Investigators: Karen Bates, Muneeb Choudhry, Paul Dhillon, Kevin Douglas, Mark Farrington, Jim Forrer, Robin Fox, Trevor Gooding, Richard Gordon, Tommy Hunter, Deborah Kerr, Bhavesh Kataria, Caroline Knowlden, Raghu Lall, Aldrich Ma, Chris Martin, Sebastian Moss, Sam Mullick, Jitendra Patel, Atif Sabat, Shamim Taherzadeh, Scott Thomson, Dave Weston, Jon Wimborne and John Winward.
eLung GP Investigators: Karen Bates, Muneeb Choudhry, Jim Forrer, Tommy Hunter, Raghu Lall, Chris Martin and Christian Winkelvoss.
Support provided by others.
Robin May, Maimoona Hashmi, Emma Boyle, Rose Cooney, Riyadh Abdallah, Mark Hobbs, Maureen Chege, Mandisa Moyo (CPRD) for site management and IT support, Edmond Ng (CPRD) for statistics, Patricia Henley and Penny Ireland (London School of Hygiene & Tropical Medicine) for sponsor overview and site contracts, Jeremy Jaffe and Mike Robinson (InPractice Systems) for Vision–LEPIS interface, Gareth Tyson (formerly The King’s College London) for LEPIS, David Hutchinson (Brookwood Academy) for GCP and protocol training and GCP audit, Maggie Elliott (South-West London Primary Care), Niki Sachsinger (Wandsworth and PCRN-Greater London), Tanya Beresford (South West London Primary Care), Pamela Shand (NHS Scotland) and Raymond Hamill (NHS Lanarkshire) for R&D support, Natalie Billington (NIHR PCRN) for GP recruitment, Marie Pitkethly, Frank Sullivan (Scottish PCRN) for GP recruitment, Luke Hammond and Chris Newark (RedAnt) for development of trial website, Accenture for CPRD IT infrastructure setup, Diana Elbourne, Stephen Evans, Julia Langham, David Prieto-Merino (London School of Hygiene & Tropical Medicine) for fraud detection and Claire McKenna (Centre for Health Economics, University of York) for health economics advice.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health.
The views and opinions expressed herein are those of the authors and do not necessarily reflect those of the Department of Health and the MHRA which currently maintains CPRD. The authors Gerald McCann, Shivani Padmanabhan and Rabah Belatri (CPRD) were not involved in the GCP review and analysis of research governance challenges and obstacles with the trials.
Publications
Adamson J, van Staa T, Torgerson D. Assuring the safety and effectiveness of new drugs: rigorous phase IV trials randomizing general practices to delayed access to new drugs. J Health Serv Res Policy 2012;17:56–9.
Baracaia S, Round T, van Staa TP, Delaney B. A qualitative study of primary care clinicians’ attitudes towards recruiting patients to randomised clinical trials and use of computerised clinical trial recruitment tools. Submitted.
Boggon R, Hubbard R, Smeeth L, Gulliford M, Cassell J, Eaton S, et al. Variability of antibiotic prescribing in patients with chronic obstructive pulmonary disease exacerbations: a cohort study. BMC Pulm Med 2013 May 31;13:32. doi: 10.1186/1471-2466-13-32.
Feyisetan S, Tyson G, Taweel A, Vargas-vera M, van Staa T, Delaney B. ePCRN-IDEA2: An Agent-Based System for Large-Scale Clinical Trial Recruitment. VII Workshop on Agents Applied in Health Care, A2HC 2012, AAMAS. 2012 (p. 10). Valencia, Spain.
Langham J, Goldacre B, Evans S, Elbourne D, Smeeth L, van Staa TP. Techniques for prevention and detection of fraud in randomised controlled trials based on routinely collected electronic health records. Submitted.
Prieto-Merino D, Smeeth L, van Staa TP, Roberts I. Dangers of non-specific composite outcome measures in clinical trials. BMJ 2013;347:f6782.
Taweel A, Tyson G, Delaney B, van Staa T. Real-Time Identification and Recruitment of Eligible Patients for Clinical Trials, AMIA 2011, San Francisco, CA, USA.
Tyson G, Taweel A, Miles S, Luck M, van Staa T, Delaney B. An Agent-Based Approach to Real-Time Patient Identification for Clinical Trials. In Proc. 4th Intl. Conference on eHealth 2011, Malaga, Spain.
Tyson G, Taweel A, Zschaler S, van Staa T, Delaney B. A model-driven approach to interoperability and integration in systems of systems. In Proc. 7th European Conference on Modelling Foundations and Applications: Workshop on Model-Based Software and Data Integration (MBSDI) 2011, Birmingham, England.
van Staa TP. Risk Management in the Future: Looking into the Crystal Ball. In Sietsems WM, Sprafka JM, editors. Risk Management Principles for Devices and Pharmaceuticals: Global Perspectives on the Benefit-Risk Assessment of Medicinal Products. 1st edn. Rockville, MD: Regulatory Affairs Professionals Society (RAPS); 2012.
van Staa TP, Goldacre B, Gulliford M, Cassell J, Pirmohamed M, Taweel A, et al. Pragmatic randomised trials using routine electronic health records: putting them to the test. BMJ 2012;344:e55.
van Staa TP, Smeeth L, Ng ES, Goldacre B, Gulliford M. The efficiency of cardiovascular risk assessment: do the right patients get statin treatment? Heart 2013;99:1597–602.
van Staa TP, Gulliford M, Ng ES, Goldacre B, Smeeth L. Prediction of cardiovascular risk using Framingham, ASSIGN and QRISK2: how well do they predict individual rather than population risk? Submitted.
van Staa TP, Klungel O. Real-Life Data and Learning from Practice to Advance Innovation. In Priority Medicines for Europe and the World. World Health Organization. URL: www.who.int/medicines/areas/priority_medicines/BP8_4Data.pdf (accessed 2 December 2013).
van Staa TP, Klungel OH, Smeeth L. Use of electronic healthcare records in large simple randomised trials at the point-of-care for the documentation of value based medicine [published online ahead of print March 2014]. J Intern Med 2014. doi: 10.1111/joim.12211
Wicks P, van Staa TP, Lipset C. How Digital Technology and Patient Empowerment is Influencing the Future Direction of Clinical Trials in Advances in Collating and Using Trial Data. URL: www.future-science.com/ (accessed 2 December 2013).
References
- Schwartz D, Lellouch J. Explanatory and pragmatic attitudes in therapeutical trials. J Chronic Dis 1967;20:637-48. http://dx.doi.org/10.1016/0021-9681(67)90041-0.
- Delaney BC, Qume M, Moayyedi P, Logan RFA, Ford AC, Elliott C, et al. Helicobacter pylori test and treat versus proton pump inhibitor in initial management of dyspepsia in primary care: multicentre randomised controlled trial (MRC-CUBE trial). BMJ 2008;336:651-4. http://dx.doi.org/10.1136/bmj.39479.640486.AE.
- Bombardier C, Laine L, Reicin A, Shapiro D, Burgos-Vargas R, Davis B, et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N Engl J Med 2000;343:1520-8. http://dx.doi.org/10.1056/NEJM200011233432103.
- Silverstein FE, Faich G, Goldstein JL, Simon LS, Pincus T, Whelton A, et al. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: the CLASS study: a randomized controlled trial. Celecoxib Long-term Arthritis Safety Study. JAMA 2000;284:1247-55. http://dx.doi.org/10.1001/jama.284.10.1247.
- Van Staa T-P, Leufkens HG, Zhang B, Smeeth L. A comparison of cost effectiveness using data from randomized trials or actual clinical practice: selective cox-2 inhibitors as an example. PLOS Med 2009;6. http://dx.doi.org/10.1371/journal.pmed.1000194.
- Black DM, Cummings SR, Karpf DB, Cauley JA, Thompson DE, Nevitt MC, et al. Randomised trial of effect of alendronate on risk of fracture in women with existing vertebral fractures. Fracture Intervention Trial Research Group. Lancet 1996;348:1535-41. http://dx.doi.org/10.1016/S0140-6736(96)07088-2.
- Gallagher AM, Rietbrock S, Olson M, Staa TP Van. Fracture outcomes related to persistence and compliance with oral bisphosphonates. J Bone Miner Res 2008;23:1569-75. http://dx.doi.org/10.1359/jbmr.080510.
- Thorpe KE, Zwarenstein M, Oxman AD, Treweek S, Furberg CD, Altman DG, et al. A pragmatic-explanatory continuum indicator summary (PRECIS): a tool to help trial designers. J Clin Epidemiol 2009;62:464-75. http://dx.doi.org/10.1016/j.jclinepi.2008.12.011.
- Williams JG, Cheung WY, Cohen DR, Hutchings HA, Longo MF, Russell IT. Can randomised trials rely on existing electronic data? A feasibility study to explore the value of routine data in health technology assessment. Health Technol Assess 2003;7.
- Williams T, van Staa TP, Puri S ES. Recent advances in the utility and use of the General Practice Research Database as an example of a UK primary care data resource. Ther Adv Drug Saf 2012;3:89-9. http://dx.doi.org/10.1177/2042098611435911.
- Edwards P, Arango M, Balica L, Cottingham R, El-Sayed H, Farrell B, et al. Final results of MRC CRASH, a randomised placebo-controlled trial of intravenous corticosteroid in adults with head injury-outcomes at 6 months. Lancet 2005;365:1957-9. http://dx.doi.org/10.1016/S0140-6736(05)66552-X.
- Peto R, Collins R, Gray R. Large-scale randomized evidence: large, simple trials and overviews of trials. Ann N Y Acad Sci 1993;703:314-40. http://dx.doi.org/10.1111/j.1749-6632.1993.tb26369.x.
- Tunis SR, Stryer DB, Clancy CM. Practical clinical trials: increasing the value of clinical research for decision making in clinical and health policy. JAMA 2003;290:1624-32. http://dx.doi.org/10.1001/jama.290.12.1624.
- Staa T-P van, Goldacre B, Gulliford M, Cassell J, Pirmohamed M, Taweel A, et al. Pragmatic randomised trials using routine electronic health records: putting them to the test. BMJ 2012;344. http://dx.doi.org/10.1136/bmj.e55.
- Vickers AJ, Scardino PT. The clinically-integrated randomized trial: proposed novel method for conducting large trials at low cost. Trials 2009;10. http://dx.doi.org/10.1186/1745-6215-10-14.
- Macdonald TM, Mackenzie IS, Wei L, Hawkey CJ, Ford I. Methodology of a large prospective, randomised, open, blinded endpoint streamlined safety study of celecoxib versus traditional non-steroidal anti-inflammatory drugs in patients with osteoarthritis or rheumatoid arthritis: protocol of the standard care versus celecoxib outcome trial (SCOT). BMJ 2013;3.
- Mosis G, Vlug AE, Mosseveld M, Dieleman JP, Stricker BC, van der Lei J, et al. A technical infrastructure to conduct randomized database studies facilitated by a general practice research database. J Am Med Inform Assoc 2005;12:602-7. http://dx.doi.org/10.1197/jamia.M1803.
- Mosis G. Randomized database studies: a combination of the strengths of observational studies and randomized clinical trials. J Am Coll Cardiol Interv 2006;1:211-17.
- Mosis G, Dieleman JP, Stricker BC, van der Lei J, Sturkenboom MCJM. A randomized database study in general practice yielded quality data but patient recruitment in routine consultation was not practical. J Clin Epidemiol 2006;59:497-502. http://dx.doi.org/10.1016/j.jclinepi.2005.11.007.
- Mosis G, Koes B, Dieleman J, Stricker BC, van der Lei J, Sturkenboom MCJM. Randomised studies in general practice: how to integrate the electronic patient record. Inform Prim Care 2005;13:209-13.
- Fiore LD, Brophy M, Ferguson RE, D’Avolio L, Hermos JA, Lew RA, et al. A point-of-care clinical trial comparing insulin administered using a sliding scale versus a weight-based regimen. Clin Trials 2011;8:183-95. http://dx.doi.org/10.1177/1740774511398368.
- Woods KL, Fletcher S, Roffe C, Haider Y. Intravenous magnesium sulphate in suspected acute myocardial infarction: results of the second Leicester Intravenous Magnesium Intervention Trial (LIMIT-2). Lancet 1992;339:1553-8. http://dx.doi.org/10.1016/0140-6736(92)91828-V.
- Strandberg TE, Pyörälä K, Cook TJ, Wilhelmsen L, Faergeman O, Thorgeirsson G, et al. Mortality and incidence of cancer during 10-year follow-up of the Scandinavian Simvastatin Survival Study (4S). Lancet n.d.;364:771-7. http://dx.doi.org/10.1016/S0140-6736(04)16936-5.
- Lewsey JD, Leyland AH, Murray GD, Boddy FA. Using routine data to complement and enhance the results of randomised controlled trials. Health Technol Assess 2000;4.
- Fröbert O, Lagerqvist B, Olivecrona GK, Omerovic E, Gudnason T, Maeng M, et al. Thrombus aspiration during ST-segment elevation myocardial infarction. N Engl J Med 2013;369:1587-97. http://dx.doi.org/10.1056/NEJMoa1308789.
- Boggon R, Hubbard R, Smeeth L, Gulliford M, Cassell J, Eaton S, et al. Variability of antibiotic prescribing in patients with chronic obstructive pulmonary disease exacerbations: a cohort study. BMC Pulm Med 2013;13. http://dx.doi.org/10.1186/1471-2466-13-32.
- McGinnis JM. Evidence-based medicine – engineering the Learning Healthcare System. Stud Health Technol Inform 2010;153:145-57.
- Tricoci P, Allen JM, Kramer JM, Califf RM, Smith SC. Scientific evidence underlying the ACC/AHA clinical practice guidelines. JAMA 2009;301:831-41. http://dx.doi.org/10.1001/jama.2009.205.
- Ioannidis JPA. Mega-trials for blockbusters. JAMA 2013;309:239-40. http://dx.doi.org/10.1001/jama.2012.168095.
- Duijnhoven RG, Straus SMJM, Raine JM, de Boer A, Hoes AW, De Bruin ML. Number of patients studied prior to approval of new medicines: a database analysis. PLOS Med 2013;10. http://dx.doi.org/10.1371/journal.pmed.1001407.
- Haynes L, Service O, Goldacre B, Torgerson D. Test, Learn, Adapt: Developing Public Policy With Randomized Controlled Trials 2012. www.gov.uk/government/uploads/system/uploads/attachment_data/file/62529/TLA-1906126.pdf (accessed 12 July 2013).
- Digital Data Improvement Priorities for Continuous Learning in Health and Health Care: Workshop Summary. Washington, DC: National Academic Press; 2013.
- Anderson KM, Odell PM, Wilson PWF, Kannel WB, Framingham MPH. Cardiovascular disease risk profiles. Am Heart J 1991;121:293-8. http://dx.doi.org/10.1016/0002-8703(91)90861-B.
- Hippisley-Cox J, Coupland C, Vinogradova Y, Robson J, Minhas R, Sheikh A, et al. Predicting cardiovascular risk in England and Wales: prospective derivation and validation of QRISK2. BMJ 2008;336:1475-82. http://dx.doi.org/10.1136/bmj.39609.449676.25.
- Cooper A, Nherera L, Calvert N, O’Flynn N, Turnbull N, Robson JC, et al. Clinical Guidelines and Evidence Review for Lipid Modification: Cardiovascular Risk Assessment and the Primary and Secondary Prevention of Cardiovascular Disease 2007. www.nice.org.uk/nicemedia/pdf/CG67fullguideline.pdf (accessed 2 December 2013).
- Rubin DB. Multiple Imputation for Nonresponse in Surveys. New York, NY: John Wiley; 1987.
- National Institute for Health and Care Excellence . Statins for the Prevention of Cardiovascular Disease: Technology Appraisal 94 2006. www.nice.org.uk/guidance/index.jsp?action=download&o=33152 (accessed 2 December 2013).
- NHS Information Centre, Business Rules team . Chronic Obstructive Pulmonary Disease (COPD) Indicator Set n.d. www.pcc.nhs.uk/uploads/QOF/Business rules v20.0/copd_ruleset_v20_0.pdf (accessed 25 June 2010).
- Good Clinical Practice Guide. London: The Stationery Office; 2012.
- Hansson L, Hedner T, Dahlöf B. Prospective randomized open blinded end-point (PROBE) study. A novel design for intervention trials. Prospective randomized open blinded end-point. Blood Press 1992;1:113-19. http://dx.doi.org/10.3109/08037059209077502.
- Ford I, Murray H, Packard CJ, Shepherd J, Macfarlane PW, Cobbe SM. Long-term follow-up of the West of Scotland Coronary Prevention Study. N Engl J Med 2007;357:1477-86. http://dx.doi.org/10.1056/NEJMoa065994.
- European Medicines Agency . GCP ICH Topic E 6 (R1). – Guideline for Good Clinical Practice n.d. www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000429.jsp&murl=menus/regulations/regulations.jsp&mid=WC0b01ac0580029590 (accessed 3 May 2011).
- International Conference for Harmonisation . Guidance for Industry. E6 Good Clinical Practice: Consolidated Guidance n.d. www.fda.gov/downloads/Drugs/Guidances/ucm073122.pdf (accessed 3 May 2011).
- Evans I, Thornton H, Chalmers I, Glasziow P. Testing Treatments: Better Research for Better Healthcare. London: Pinter & Martin Ltd; 2011.
- Chalmers I. Regulation of therapeutic research is compromising the interests of patients. Int J Pharm Med 2007;21:395-404. http://dx.doi.org/10.2165/00124363-200721060-00004.
- Yusuf S, Bosch J, Devereaux PJ, Devereaus PJ, Collins R, Baigent C, et al. Sensible guidelines for the conduct of large randomized trials. Clin Trials 2008;5:38-9. http://dx.doi.org/10.1177/1740774507088099.
- Duley L, Antman K, Arena J, Avezum A, Blumenthal M, Bosch J, et al. Specific barriers to the conduct of randomized trials. Clin Trials 2008;5:40-8. http://dx.doi.org/10.1177/1740774507087704.
- Snooks H, Hutchings H, Seagrove A, Stewart-Brown S, Williams J, Russell I. Bureaucracy stifles medical research in Britain: a tale of three trials. BMC Med Res Methodol 2012;12. http://dx.doi.org/10.1186/1471-2288-12-122.
- McMahon AD, Conway DI, Macdonald TM, McInnes GT. The unintended consequences of clinical trials regulations. PLOS Med 2009;3. http://dx.doi.org/10.1371/journal.pmed.1000131.
- European Commission . Revision of the Clinical Trial Directive 2001 20 EC: Concept Paper Submitted for Public Consultation n.d. http://ec.europa.eu/health/human-use/clinical-trials/developments/index_en.htm (accessed 11 February 2011).
- Project MJ. Risk-Adapted Approaches to the Management of Clinical Trials of Investigational Medicinal Products n.d. www.mhra.gov.uk/home/groups/l-ctu/documents/websiteresources/con111784.pdf (accessed 11 February 2011).
- Rendell JM, Merritt RD, Geddes JR. Incentives and disincentives to participation by clinicians in randomised controlled trials. Cochrane Database Syst Rev 2007;2.
- Ross S, Grant A, Counsell C, Gillespie W, Russell I, Prescott R. Barriers to participation in randomised controlled trials: a systematic review. J Clin Epidemiol 1999;52:1143-56. http://dx.doi.org/10.1016/S0895-4356(99)00141-9.
- Chew-Graham CA, May CR, Perry MS. Qualitative research and the problem of judgement: lessons from interviewing fellow professionals. Fam Pract 2002;19:285-9. http://dx.doi.org/10.1093/fampra/19.3.285.
- Prout H, Butler C, Kinnersley P, Robling M, Hood K, Tudor-Jones R. A qualitative evaluation of implementing a randomized controlled trial in general practice. Fam Pract 2003;20:675-81. http://dx.doi.org/10.1093/fampra/cmg609.
- Varonen H, Kortteisto T, Kaila M. What may help or hinder the implementation of computerized decision support systems (CDSSs): a focus group study with physicians. Fam Pract 2008;25:162-7. http://dx.doi.org/10.1093/fampra/cmn020.
- Treweek S, Pearson E, Smith N, Neville R, Sargeant P, Boswell B, et al. Desktop software to identify patients eligible for recruitment into a clinical trial: using SARMA to recruit to the ROAD feasibility trial. Inform Prim Care 2010;18:51-8.
- Embi PJ, Jain A, Harris CM. Physicians’ perceptions of an electronic health record-based clinical trial alert approach to subject recruitment: a survey. BMC Med Inform Decis Mak 2008;8. http://dx.doi.org/10.1186/1472-6947-8-13.
- Potter R, Dale J CI. A qualitative study exploring practice nurses’ experience of participating in a primary care-based randomised controlled trial. J Res Nurs 2009;14:439-47. http://dx.doi.org/10.1177/1744987108098228.
- Donovan JL, Peters TJ, Noble S, Powell P, Gillatt D, Oliver SE, et al. Who can best recruit to randomized trials? Randomized trial comparing surgeons and nurses recruiting patients to a trial of treatments for localized prostate cancer (the ProtecT study). J Clin Epidemiol 2003;56:605-9. http://dx.doi.org/10.1016/S0895-4356(03)00083-0.
- Prescott RJ, Counsell CE, Gillespie WJ, Grant AM, Russell IT, Kiauka S, et al. Factors that limit the quality, number and progress of randomised controlled trials. Health Technol Assess 1999;3.
- Lovato LC, Hill K, Hertert S, Hunninghake DB, Probstfield JL. Recruitment for controlled clinical trials: literature summary and annotated bibliography. Control Clin Trials 1997;18:328-52. http://dx.doi.org/10.1016/S0197-2456(96)00236-X.
- Van der Windt DA, Koes BW, van Aarst M, Heemskerk MA, Bouter LM. Practical aspects of conducting a pragmatic randomised trial in primary care: patient recruitment and outcome assessment. Br J Gen Pract 2000;50:371-4.
- Hetherton J, Matheson A, Robson M. Recruitment by GPs during consultations in a primary care randomised controlled trial comparing computerised psychological therapy with clinical psychology and routine GP care: problems and possible solutions. Prim Heal Care Res Dev 2004;5:5-10. http://dx.doi.org/10.1191/1463423604pc168oa.
- Gágyor I, Bleidorn J, Wegscheider K, Hummers-Pradier E, Kochen MM. Practices, patients and (im)perfect data – feasibility of a randomised controlled clinical drug trial in German general practices. Trials 2011;12. http://dx.doi.org/10.1186/1745-6215-12-91.
- Bell-Syer SEM, Thorpe LN, Thomas K, Macpherson H. GP participation and recruitment of patients to RCTs: lessons from trials of acupuncture and exercise for low back pain in primary care. Evid Based Complement Alternat Med 2011;2011.
- Maeland S, Magnussen LH, Eriksen HR, Malterud K. Why are general practitioners reluctant to enrol patients into a RCT on sick leave? A qualitative study. Scand J Public Health 2011;39:888-93. http://dx.doi.org/10.1177/1403494811424613.
- Ward E, King M, Lloyd M, Bower P, Friedli K. Conducting randomized trials in general practice: methodological and practical issues. Br J Gen Pract 1999;49:919-22.
- Mason VL, Shaw A, Wiles NJ, Mulligan J, Peters TJ, Sharp D, et al. GPs’ experiences of primary care mental health research: a qualitative study of the barriers to recruitment. Fam Pract 2007;24:518-25. http://dx.doi.org/10.1093/fampra/cmm047.
- Morrison-Beedy D, Aronowitz T, Dyne J, Mkandawire L, Murphy C, Martin J. The nurse clinician as research participant recruiter: experience from a longitudinal intervention study. J New York State Nurses Assoc 2001;32:9-13.
- Kinney AY, Richards C, Vernon SW, Vogel VG. The effect of physician recommendation on enrollment in the Breast Cancer Chemoprevention Trial. Prev Med n.d.;27:713-19. http://dx.doi.org/10.1006/pmed.1998.0349.
- Newall N, Miller C, Lewin G, Kapp S, Gliddon T, Carville K, et al. Nurses’ experiences of participating in a randomised controlled trial (RCT) in the community. Wound Pract Res 2009;17:24-3.
- Hummers-Pradier E, Scheidt-Nave C, Martin H, Heinemann S, Kochen MM, Himmel W. Simply no time? Barriers to GPs’ participation in primary health care research. Fam Pract 2008;25:105-12. http://dx.doi.org/10.1093/fampra/cmn015.
- Patton M. Qualitative Research and Evaluation Methods. California, CA: Sage Publications; 2002.
- Department of Health . Putting Prevention First. Vascular Checks: Risk Assessment and Management. Vascular Programme 2008 n.d. www.healthcheck.nhs.uk/document.php?o=227 (accessed 29 June 2013).
- Van Staa T-P, Smeeth L, Ng ES-W, Goldacre B, Gulliford M. The efficiency of cardiovascular risk assessment: do the right patients get statin treatment?. Heart 2013;99:1597-602. http://dx.doi.org/10.1136/heartjnl-2013-303698.
- Ebrahim S, Casas JP. Statins for all by the age of 50 years?. Lancet 2012;380:545-7. http://dx.doi.org/10.1016/S0140-6736(12)60694-1.
- Van Staa TP, Carr DF, O’Meara H, McCann G, Pirmohamed M. Predictors and outcomes of increases in creatine phosphokinase levels or rhabdomyolysis risk during statin treatment [published online ahead of print March 2014]. Br J Clin Pharmacol 2014.
- Fudge N, Redfern J, Wolfe C, McKevitt C. Streamlined research governance: are we there yet?. BMJ 2010;341. http://dx.doi.org/10.1136/bmj.c4625.
- Evans S, Lock S, Wells F. Fraud & Misconduct in Medical Research. London: Wiley-Blackwell; 1993.
- Mahalanobis PC. On the generalised distance in statistics. Proc Natl Inst Sci India 1936;2:49-55.
- Cohen Freue G V, Hollander Z, Shen E, Zamar RH, Balshaw R, Scherer A, et al. MDQC: a new quality assessment method for microarrays based on quality control reports. Bioinformatics 2007;23:3162-9. http://dx.doi.org/10.1093/bioinformatics/btm487.
- Macefield RC, Beswick AD, Blazeby JM, Lane JA. A systematic review of on-site monitoring methods for health-care randomised controlled trials. Clin Trials 2013;10:104-24. http://dx.doi.org/10.1177/1740774512467405.
- Bakobaki JM, Rauchenberger M, Joffe N, McCormack S, Stenning S, Meredith S. The potential for central monitoring techniques to replace on-site monitoring: findings from an international multi-centre clinical trial. Clin Trials 2012;9:257-64. http://dx.doi.org/10.1177/1740774511427325.
- Al-Marzouki S, Evans S, Marshall T, Roberts I. Are these data real? Statistical methods for the detection of data fabrication in clinical trials. BMJ 2005;331:267-70. http://dx.doi.org/10.1136/bmj.331.7511.267.
- Adamson J, Bowling A, Ebrahim S. Handbook of Health Research Methods. Berkshire: Open University Press; 2005.
- Rapport F, Storey M, Porter A, Snooks H, Jones K, Peconi J, et al. Qualitative research within trials: developing a standard operating procedure for a clinical trials unit. Trials 2013;14. http://dx.doi.org/10.1186/1745-6215-14-S1-P98.
- Thomas J, Harden A, Oakley A, Oliver S, Sutcliffe K, Rees R, et al. Integrating qualitative research with trials in systematic reviews. BMJ 2004;328:1010-12. http://dx.doi.org/10.1136/bmj.328.7446.1010.
- Bower P, Wallace P, Ward E, Graffy J, Miller J, Delaney B, et al. Improving recruitment to health research in primary care. Fam Pract 2009;26:391-7. http://dx.doi.org/10.1093/fampra/cmp037.
- Moore M, Smith H. Agreeing to collaborate: a qualitative study of how general practices decide whether to respond positively to an invitation to participate in a research study. Prim Heal Care Res Dev 2007;8. http://dx.doi.org/10.1017/S1463423607000163.
- Westcombe AM, Gambles MA, Wilkinson SM, Barnes K, Fellowes D, Maher EJ, et al. Learning the hard way! Setting up an RCT of aromatherapy massage for patients with advanced cancer. Palliat Med 2003;17:300-7. http://dx.doi.org/10.1191/0269216303pm769rr.
- Featherstone K, Donovan JL. ‘Why don’t they just tell me straight, why allocate it?’ The struggle to make sense of participating in a randomised controlled trial. Soc Sci Med 2002;55:709-19. http://dx.doi.org/10.1016/S0277-9536(01)00197-6.
- Mack N, Woodsong C, MacQueen K, Guest G, Namey E. Qualitative Research Methods a Data Collector’s Field Guide. North Carolina, NC: Family Health International; 2009.
- Miles MB, Huberman AM. Qualitative Data Analysis: An Expanded Sourcebook. Thousand Oaks, CA: Sage Publications; 1994.
- Ritchie J, Spencer L, Bryman A, Burgess R. Analyzing Qualitative Data. Thousand Oaks, CA: Sage Publications; 2004.
- Pope C, Ziebland S, Mays N. Qualitative research in health care. Analysing qualitative data. BMJ 2000;320:114-16. http://dx.doi.org/10.1136/bmj.320.7227.114.
- National Institute for Health and Care Excellence . Chronic Obstructive Pulmonary Disease (COPD) (QS10) 2011. http://guidance.nice.org.uk/QS10 (accessed 2 December 2013).
- Salmon P, Peters S, Rogers A, Gask L, Clifford R, Iredale W, et al. Peering through the barriers in GPs’ explanations for declining to participate in research: the role of professional autonomy and the economy of time. Fam Pract 2007;24:269-75. http://dx.doi.org/10.1093/fampra/cmm015.
- Fletcher B, Gheorghe A, Moore D, Wilson S, Damery S. Improving the recruitment activity of clinicians in randomised controlled trials: a systematic review. BMJ Open 2012;2.
- Donovan J, Sanders C, Bowling A, Ebrahim S. Handbook of Health Research Methods. Berkshire: Open University Press; 2005.
- Green J, Thorogood N. Qualitative Methods for Health Research. Thousand Oaks, CA: Sage; 2009.
- Glaser B, Strauss A. The Discovery of Grounded Theory. Strategies for Qualitative Research. Chicago, IL: Aldine Press; 1967.
- Selic P, Svab I, Repolusk M, Gucek NK. What factors affect patients’ recall of general practitioners’ advice?. BMC Fam Pract 2011;12. http://dx.doi.org/10.1186/1471-2296-12-141.
- Wilson I, McGrath B, Russell G, Bridges-Webb C, Hogan C. General practitioners’ views on patient care research. Aust Fam Physician 2000;29:86-8.
- Rosemann T, Szecsenyi J. General practitioners’ attitudes towards research in primary care: qualitative results of a cross sectional study. BMC Fam Pract 2004;5. http://dx.doi.org/10.1186/1471-2296-5-31.
- Hunt CJ, Shepherd LM, Andrews G. Do doctors know best? Comments on a failed trial. Med J Aust 2001;174:144-6.
- May C. A rational model for assessing and evaluating complex interventions in health care. BMC Health Serv Res 2006;6. http://dx.doi.org/10.1186/1472-6963-6-86.
- Vist GE, Hagen KB, Devereaux PJ, Bryant D, Kristoffersen DT, Oxman AD. Systematic review to determine whether participation in a trial influences outcome. BMJ 2005;330. http://dx.doi.org/10.1136/bmj.330.7501.1175.
- Watson JM, Torgerson DJ. Increasing recruitment to randomised trials: a review of randomised controlled trials. BMC Med Res Methodol 2006;6. http://dx.doi.org/10.1186/1471-2288-6-34.
- Williamson MK, Pirkis J, Pfaff JJ, Tyson O, Sim M, Kerse N, et al. Recruiting and retaining GPs and patients in intervention studies: the DEPS-GP project as a case study. BMC Med Res Methodol 2007;7. http://dx.doi.org/10.1186/1471-2288-7-42.
- Morris CJ, Cantrill JA, Weiss MC. GP survey response rate: a miscellany of influencing factors. Fam Pract 2001;18:454-6. http://dx.doi.org/10.1093/fampra/18.4.454.
- Treweek S, Mitchell E, Pitkethly M, Cook J, Kjeldstrøm M, Taskila T, et al. Strategies to improve recruitment to randomised controlled trials. Cochrane Database Syst Rev 2010;1.
- Briggs A, Sculpher M, Claxton K. Decision Modelling for Health Economic Evaluation. Oxford: Oxford University Press; 2006.
- Claxton K, Sculpher M, Drummond M. A rational framework for decision making by the National Institute For Clinical Excellence (NICE). Lancet 2002;360:711-15. http://dx.doi.org/10.1016/S0140-6736(02)09832-X.
- Pratt J, Raiffa H, Schlaifer R. Statistical Decision Theory. Cambridge, MA: MIT Press; 1995.
- Raiffa H, Schlaifer R. Probability and Statistics for Business Decisions. New York, NY: McGraw-Hill; 1959.
- Thompson KM, Evans JS. The value of improved national exposure information for Perchloroethylene (Perc): a case study for dry cleaners. Risk Anal 1997;17:253-71. http://dx.doi.org/10.1111/j.1539-6924.1997.tb00864.x.
- Howard RA. Information value theory. Syst Sci Cybern IEEE Trans 1966;2:22-6. http://dx.doi.org/10.1109/TSSC.1966.300074.
- Ginnelly L, Claxton K, Sculpher MJ, Golder S. Using value of information analysis to inform publicly funded research priorities. Appl Heal Econ Heal Policy 2005;4:37-46. http://dx.doi.org/10.2165/00148365-200504010-00006.
- Yokota F, Thompson KM. Value of information literature analysis: a review of applications in health risk management. Med Decis Mak 2004;24:287-98. http://dx.doi.org/10.1177/0272989X04263157.
- Claxton K. The irrelevance of inference: a decision-making approach to the stochastic evaluation of health care technologies. J Health Econ 1999;18:341-64. http://dx.doi.org/10.1016/S0167-6296(98)00039-3.
- Claxton K, Posnett J. An economic approach to clinical trial design and research priority-setting. Health Econ 1996;5:513-24. http://dx.doi.org/10.1002/(SICI)1099-1050(199611)5:6<513::AID-HEC237>3.0.CO;2-9.
- Claxton K, Ginnelly L, Sculpher M, Philips Z, Palmer S. A pilot study on the use of decision theory and value of information analysis as part of the NHS Health Technology Assessment programme. Health Technol Assess 2004;8.
- Effing T, Kerstjens H, van der Valk P, Zielhuis G, van der Palen J. (Cost)-effectiveness of self-treatment of exacerbations on the severity of exacerbations in patients with COPD: the COPE II study. Thorax 2009;64:956-62. http://dx.doi.org/10.1136/thx.2008.112243.
- Guide to the Methods of Technology Appraisal. London: NICE; 2008.
- Sullivan PW, Slejko JF, Sculpher MJ, Ghushchyan V. Catalogue of EQ-5D scores for the United Kingdom. Med Decis Making 2011;31:800-4. http://dx.doi.org/10.1177/0272989X11401031.
- Rutten-van Mölken MPMH, Hoogendoorn M, Lamers LM. Holistic preferences for 1-year health profiles describing fluctuations in health: the case of chronic obstructive pulmonary disease. Pharmacoeconomics 2009;27:465-77. http://dx.doi.org/10.2165/00019053-200927060-00003.
- Department of Health . NHS Reference Costs 2011 2012 n.d. https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/213060/2011-12-reference-costs-publication.pdf (accessed 11 December 2013).
- British National Formulary. London: BMA and RPS; 2013.
- Curtis L. Unit Costs of Health Social Care 2012. Canterbury: PSSRU, University of Kent; 2012.
- Fenwick E, Claxton K, Sculpher M. Representing uncertainty: the role of cost-effectiveness acceptability curves. Health Econ 2001;10:779-87. http://dx.doi.org/10.1002/hec.635.
- Drummond MF, Sculpher MJ, Torrance GW, O’Brien BJ, Stoddart GL. Methods for the Economic Evaluation of Health Care Programmes. Oxford: Oxford University Press; 2005.
- Improving Information to Support Decision Making: Standards for Better Quality Data. London: UK Audit Commission; 2007.
- Perrio M, Waller PC, Shakir SAW. An analysis of the exclusion criteria used in observational pharmacoepidemiological studies. Pharmacoepidemiol Drug Saf 2007;16:329-36. http://dx.doi.org/10.1002/pds.1262.
- Gulliford MC, Charlton J, Ashworth M, Rudd AG, Toschke AM. Selection of medical diagnostic codes for analysis of electronic patient records. Application to stroke in a primary care database. PLOS ONE 2009;4. http://dx.doi.org/10.1371/journal.pone.0007168.
- Herrett E, Shah AD, Boggon R, Denaxas S, Smeeth L, Van Staa T, et al. Completeness and diagnostic validity of recording acute myocardial infarction events in primary care, hospital care, disease registry, and national mortality records: cohort study. BMJ Clin Res 2013;346.
- Köpcke F, Lubgan D, Fietkau R, Scholler A, Nau C, Stürzl M, et al. Evaluating predictive modeling algorithms to assess patient eligibility for clinical trials from routine data. BMC Med Inform Decis Mak 2013;13. http://dx.doi.org/10.1186/1472-6947-13-134.
- Hróbjartsson A, Boutron I. Blinding in randomized clinical trials: imposed impartiality. Clin Pharmacol Ther 2011;90:732-6. http://dx.doi.org/10.1038/clpt.2011.207.
- Moerman DE, Jonas WB. Deconstructing the placebo effect and finding the meaning response. Ann Intern Med 2002;136:471-6. http://dx.doi.org/10.7326/0003-4819-136-6-200203190-00011.
- Kaptchuk TJ, Kelley JM, Conboy LA, Davis RB, Kerr CE, Jacobson EE, et al. Components of placebo effect: randomised controlled trial in patients with irritable bowel syndrome. BMJ 2008;336:999-1003. http://dx.doi.org/10.1136/bmj.39524.439618.25.
- Higgins JPT, Altman DG, Gøtzsche PC, Jüni P, Moher D, Oxman AD, et al. The Cochrane Collaboration’s tool for assessing risk of bias in randomised trials. BMJ 2011;343. http://dx.doi.org/10.1136/bmj.d5928.
- Enck P, Bingel U, Schedlowski M, Rief W. The placebo response in medicine: minimize, maximize or personalize?. Nat Rev Drug Discov 2013;12:191-204. http://dx.doi.org/10.1038/nrd3923.
- Wood L, Egger M, Gluud LL, Schulz KF, Jüni P, Altman DG, et al. Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: meta-epidemiological study. BMJ 2008;336:601-5. http://dx.doi.org/10.1136/bmj.39465.451748.AD.
- Prieto-Merino D, Smeeth L, van Staa TP RI. Dangers of non-specific composite outcome measures in clinical trials. BMJ 2013;347. http://dx.doi.org/10.1136/bmj.f6782.
- Chan A-W, Hróbjartsson A, Haahr MT, Gøtzsche PC, Altman DG. Empirical evidence for selective reporting of outcomes in randomized trials: comparison of protocols to published articles. JAMA 2004;291:2457-65. http://dx.doi.org/10.1001/jama.291.20.2457.
- Hutchings HA, Cheung W-Y, Williams JG, Cohen D, Longo MF, Russell I. Can electronic routine data act as a surrogate for patient-assessed outcome measures?. Int J Technol Assess Health Care 2005;21:138-43. http://dx.doi.org/10.1017/S026646230505018X.
- Brookes ST, Whitley E, Peters TJ, Mulheran PA, Egger M, Davey Smith G. Subgroup analyses in randomised controlled trials: quantifying the risks of false-positives and false-negatives. Health Technol Assess 2001;5.
- Ioannidis JP, Lau J. Heterogeneity of the baseline risk within patient populations of clinical trials: a proposed evaluation algorithm. Am J Epidemiol 1998;148:1117-26. http://dx.doi.org/10.1093/oxfordjournals.aje.a009590.
- Sacristán JA, Bergmann J-F, Williams J, Penston J. Pragmatic Randomised Trials Using Routine Electronic Health Records: Putting Them to the Test 2012. www.bmj.com/content/344/bmj.e55?tab=responses (accessed 2 July 2012).
- Sussman JB, Hayward RA. An IV for the RCT: using instrumental variables to adjust for treatment contamination in randomised controlled trials. BMJ 2010;340. http://dx.doi.org/10.1136/bmj.c2073.
- Penston J. Large-scale randomised trials – a misguided approach to clinical research. Med Hypotheses 2005;64:651-7. http://dx.doi.org/10.1016/j.mehy.2004.09.006.
- Mallick U, Bakhai A, Wang D, Marcus FM, Wang D, Bakhai A. Clinical Trials. London: Remedica; 2006.
- Eldridge S, Kerry S, Torgerson DJ. Bias in identifying and recruiting participants in cluster randomised trials: what can be done?. BMJ 2009;339. http://dx.doi.org/10.1136/bmj.b4006.
- Giraudeau B, Ravaud P. Preventing bias in cluster randomised trials. PLOS Med 2009;6. http://dx.doi.org/10.1371/journal.pmed.1000065.
- General Medical Council . Good Medical Practice 2013. www.gmc-uk.org/guidance/good_medical_practice.asp (accessed 12 December 2012).
- Roberts I, Chaudhry B, Chalmers I. New GMC guidance takes a major, ethically flawed, backward step. BMJ 2013;346. http://dx.doi.org/10.1136/bmj.f3879.
- Faden R, Kass N, Whicher D, Stewart W, Tunis S. Ethics and informed consent for comparative effectiveness research with prospective electronic clinical data. Med Care 2013;51:S53-7. http://dx.doi.org/10.1097/MLR.0b013e31829b1e4b.
- Yusuf S, Collins R, Peto R. Why do we need some large, simple randomized trials?. Stat Med 1984;3:409-22. http://dx.doi.org/10.1002/sim.4780030421.
- Buring JE, Hennekens CH. The contribution of large, simple trials to prevention research. Prev Med 1984;23:595-8. http://dx.doi.org/10.1006/pmed.1994.1095.
- Peto R, Collins R, Gray R. Large-scale randomized evidence: large, simple trials and overviews of trials. J Clin Epidemiol 1995;48:23-40. http://dx.doi.org/10.1016/0895-4356(94)00150-O.
- Roehr B. The appeal of large simple trials. BMJ 2013;346. http://dx.doi.org/10.1136/bmj.f1317.
- Tognoni G, Alli C, Avanzini F, Bettelli G, Colombo F, Corso R, et al. Randomised clinical trials in general practice: lessons from a failure. BMJ 1991;303:969-71. http://dx.doi.org/10.1136/bmj.303.6808.969.
- Peto R, Baigent C. Trials: the next 50 years. Large scale randomised evidence of moderate benefits. BMJ 1998;317:1170-1. http://dx.doi.org/10.1136/bmj.317.7167.1170.
- Chalmers I, Atkinson P, Fenton M, Firkins L, Crowe S, Cowan K. Tackling treatment uncertainties together: the evolution of the James Lind Initiative, 2003–2013. J R Soc Med 2013;106:482-91. http://dx.doi.org/10.1177/0141076813493063.
- Oliver S, Clarke-Jones L, Rees R, Milne R, Buchanan P, Gabbay J, et al. Involving consumers in research and development agenda setting for the NHS: developing an evidence-based approach. Health Technol Assess 2004;8.
- Kalra D, Gertz R, Singleton P, Inskip HM. Confidentiality of personal health information used for research. BMJ 2006;333:196-8. http://dx.doi.org/10.1136/bmj.333.7560.196.
- Mackenzie IS, Mantay BJ, McDonnell PG, Wei L, MacDonald TM. Managing security and privacy concerns over data storage in healthcare research. Pharmacoepidemiol Drug Saf 2011;20:885-93. http://dx.doi.org/10.1002/pds.2170.
- The Royal Society Science Policy Centre . Science As an Open Enterprise 2012. http://royalsociety.org/policy/projects/science-public-enterprise/report/ (accessed 2 December 2013).
- Van Staa T, Klungel OH. Priority Medicines for Europe and the World ‘A Public Health Approach to Innovation’. World Health Organizaton; 2013.
- Loder J, Bunt L, Wyatt J. Doctor Know: A Knowledge Commons in Health 2013. www.nesta.org.uk/sites/default/files/doctor_know_a_knowledge_commons_in_health.pdf (accessed 2 December 2013).
- Oxman AD, Chalmers I, Sackett DL. A practical guide to informed consent to treatment. BMJ 2001;323:1464-6. http://dx.doi.org/10.1136/bmj.323.7327.1464.
- Mackenzie IS, Wei L, Paterson KR, Macdonald TM. Cluster randomised trials of prescription medicines or prescribing policy – public and general practitioner opinions in Scotland. Br J Clin Pharmacol 2012;74:354-61. http://dx.doi.org/10.1111/j.1365-2125.2012.04195.x.
- Kirkby HM, Calvert M, Draper H, Keeley T, Wilson S. What potential research participants want to know about research: a systematic review. BMJ 2012;2.
- National Institute for Health and Care Excellence . Lipid Modification: Cardiovascular Risk Assessment and the Modification of Blood Lipids for the Primary and Secondary Prevention of Cardiovascular Disease n.d. http://guidance.nice.org.uk/CG/WaveR/123 (accessed 1 May 2014).
- International Society for Pharmacoeconomics and Outcomes Research . International Digest of Databases n.d. www.ispor.org/digestofintdb/countrylist.aspx (accessed 7 May 2013).
- Weiskopf NG, Weng C. Methods and dimensions of electronic health record data quality assessment: enabling reuse for clinical research. J Am Med Inform Assoc 2013;20:144-51. http://dx.doi.org/10.1136/amiajnl-2011-000681.
- Institute of Medicine . Large Simple Trials and Knowledge Generation in a Learning Health System – Workshop Summary 2013. www.iom.edu/Reports/2013/Large-Simple-Trials-and-Knowledge-Generation-in-a-Learning-Health-System.aspx (accessed 7 October 2013).
- Reynolds RF, Lem JA, Gatto NM, Eng SM. Is the large simple trial design used for comparative, post-approval safety research? A review of a clinical trials registry and the published literature. Drug Saf 2011;34:799-820. http://dx.doi.org/10.2165/11593820-000000000-00000.
- Reith C, Landray M, Devereaux PJ, Bosch J, Granger CB, Baigent C, et al. Randomized clinical trials--removing unnecessary obstacles. N Engl J Med 2013;369:1061-5. http://dx.doi.org/10.1056/NEJMsb1300760.
Appendix 1 Charters for Trial Steering and Data Monitoring Committees
This appendix outlines the charters for the Steering Committee and Data Monitoring Committee for Retropro and eLung. The Chair of the Steering Committee will need to agree to any changes in the charters of the Steering Committee and Data Monitoring Committee.
Steering Committee
Roles and responsibilities
The role of the Steering Committee is to provide oversight of the conduct of the two trials. This includes oversight of the practical aspects of the study as well as ensuring that the study continues to be run in a way which is both safe for the patients and provides appropriate safety and efficacy data to the sponsor and investigators. In discharging its safety role, the Steering Committee will work in conjunction with the Data Monitoring Board that will also be established for the trials.
Specific responsibilities of the Steering Committee include, but are not limited to, the following:
-
to provide overall supervision of the trials
-
to take steps to reduce deviations from the protocol to a minimum
-
periodic review of the progress of the study
-
to review safety data; this review is typically done blinded to treatment allocation (in case of a major safety concern, the Steering Committee can request unblinding and the review then can be done in an unblinded manner)
-
to resolve any differences within the research team or between research team and sponsor (London School of Hygiene & Tropical Medicine) on the data management and monitoring procedures in the trials or any recommendations for modifications to the protocol.
The Steering Committee will have ultimate responsibility for the trials and will assume primacy over the Data Monitoring Committee or principal investigator. The Steering Committee can prematurely terminate the trials. The sponsor and principal investigator will agree, in writing prior to the start of the study, to the charter of the Steering Committee.
Membership
The Steering Committee will consist of at least four members, including three members who are independent of the MHRA. A patient representative will be invited. The sponsor and the funder of eLung will need to approve the nomination of the Steering Committee. Membership consists of persons who have no financial, scientific, or other competing interests with the trials. Three members (including the Chairman or designee) will constitute a quorum. The principal investigator and a representative of the financial funder of the trials may attend the meetings, but will have no voting status. The Steering Committee may request that the principal investigator and the funder representative do not attend all or part of any meeting. Members will be reimbursed for any reasonable travel, accommodation or other costs (e.g. telephone) incurred.
All potential members will have sight of the protocol and be invited to comment before agreeing to join the Steering Committee. If a potential member has major reservations about the trials they should report these to the principal investigator and sponsor and may decide not to accept the invitation to join. Steering Committee members should be independent and constructively critical of the ongoing trials, but also supportive of aims and methods of the trial.
There will be no formal contract between Steering Committee members and the sponsor.
Committee process
The Steering Committee will meet prior to the start of the study and 6–12 months after the start of the study. The Steering Committee may also meet on an ad hoc basis should the need arise. Meetings may take place by teleconference, by videoconference, or face to face, whichever is agreed to be most appropriate. Urgent issues may be communicated by e-mail by the principal investigator to the Steering Committee, where appropriate. Members will be reimbursed for any reasonable travel, accommodation or other costs (e.g. telephone) incurred. There will be no formal legal contract between members and the sponsor.
Minutes
A set of minutes (drafted by a member of the research team) will be produced for each meeting of the Steering Committee. These will be circulated in draft form to all members within 1 week of the meeting date. Members then have up to 2 weeks in which to provide comments or amendments, after which the minutes will be considered to be final. The Chair (or in his/her absence from the meeting a nominated deputy) will sign a copy of the final minutes. A copy of the final minutes will be filed in the trial master file.
Confidentiality
All materials, discussions and proceedings of the Steering Committee are completely confidential. Members and other participants of the Steering Committee meetings are expected to maintain confidentiality.
Data Monitoring Committee
Roles and responsibilities
The role of the Data Monitoring Committee is to safeguard the interests of the trial’s participants and to monitor the data collected in the trials. Specific responsibilities of the Data Monitoring Committee include, but are not limited to, the following:
-
to agree to and evaluate the data management and monitoring procedures of the trials as proposed by the research team
-
to assess recruitment figures and data quality, including completeness
-
to assess the extent of protocol deviations (including a comparison of patients enrolled in the trials and other patients in the research database using the same medication)
-
to review safety data, including line-listings of case reports of suspected ADRs and of iatrogenic conditions and to request further analyses; this review will be done blinded to treatment allocation; the Data Monitoring Committee will inform the Steering Committee of any major safety concerns and request their unblinded review of safety data
-
to review the results of the fraud detection procedures
-
to implement early stopping rules for the trials (e.g. Haybittle–Peto rule).
The Data Monitoring Committee can recommend modifications to the data management and monitoring procedures in the trials or to the protocol. Every effort will be made to reach a consensus within the Data Monitoring Committee and with the principal investigator. In case of disagreement on recommended modifications, the Steering Committee will decide. The sponsor and principal investigator will agree, in writing prior to the start of the study, to the charter of the Data Monitoring Committee.
Membership
The Data Monitoring Committee will consist of three members. Two members will constitute a quorum. In the two trials the members of the Data Monitoring Committee will consist of co-investigators (rather than independent members as will be the case for later studies). The reason for this is that these are feasibility trials lacking statistical power to detect effects on major clinical outcomes; the objective of these studies will be to develop the overall structure and procedures for trials.
Committee process
The Data Monitoring Committee will meet prior to the start of the study and 6–12 months after the start of the study. The Data Monitoring Committee may also meet on an ad hoc basis should the need arise. Meetings may take place by teleconference, by videoconference, or face to face, whichever is agreed to be most appropriate. Urgent issues may be communicated by e-mail. Members will be reimbursed for any reasonable travel, accommodation or other costs (e.g. telephone) incurred. There will be no formal legal contract between members and the sponsor.
Minutes
A set of minutes will be produced for each meeting of the Data Monitoring Committee. These will be circulated in draft form to all members within 1 week of the meeting date. Members then have up to 2 weeks in which to provide comments or amendments, after which the minutes will be considered to be final. The Chair (or in his/her absence from the meeting a nominated deputy) will sign a copy of the final minutes. A copy of the final minutes will be filed in the trial master file.
Confidentiality
All materials, discussions and proceedings of the Data Monitoring Committee are completely confidential. Members and other participants of the Data Monitoring Committee meetings are expected to maintain confidentiality.
Appendix 2 Qualitative research of views on computerised trial recruitment: topic guide
Recruiting to trials
-
What are your thoughts* about recruiting patients to clinical trials generally?
-
What are your thoughts* about recruiting your patients to clinical trials?
-
What are your thoughts* about recruiting patients to clinical trials during routine consultations?
-
[*Incorporating beliefs, concerns and expectations.]
-
Barriers:
-
Do you think there may be potential barriers to recruiting patients to clinical trials during routine consultations? (What would stop participants from recruiting patients to RCT’s?)
Facilitators:
-
Do you think there may be anything that could facilitate recruiting patients to clinical trials during routine consultations? (What would help participants to recruit patients to RCT’s?)
Computerised clinical tools
-
What is your opinion of or attitudes** towards the use of computerised clinical tools (CCTs) for recruiting patients to clinical trials during routine consultations?
-
[**Incorporating concerns, and whether CCT would act in a facilitative manner and/or as a barrier.]
-
LEPIS prototype:
-
Do you have any feedback or comments on the LEPIS prototype (positive and/or negative)?
-
Is there anything you particularly like about the prototype?
-
Is there anything you particularly do not like about the prototype?
Changed/novel beliefs or attitudes:
-
Having seen the LEPIS prototype, has this changed your opinion or beliefs or attitudes regarding recruiting patients to RCT’s during routine consultations?
-
Have any new beliefs or attitudes arisen after seeing the LEPIS prototype?
Appendix 3 The Qualitative process Evaluation of ANtibiotics study: general practitioner andpatientinterview topic guides
General practitioner interview topic guide
-
Background/demographic information.
-
Name of GP practice.
-
Name of PCT.
-
Sex of GP.
-
Size of practice.
-
– Number of registered patients on GP lists.
-
– Number of registered GPs.
-
-
Type of practice (single/partnership).
-
Inner city/urban/rural/remote.
-
Deprivation score(s) for population catchment.
-
Ethnic groups served by practice.
-
-
Status regarding participation in eLung.
-
Declined/interested-declined/accepted-declined.
-
Accepted and setting up.
-
Accepted and recruiting.
-
– Length of time since starting recruitment (months).
-
– Number of participants recruited.
-
-
Closed as completed target recruitment.
-
-
Previous research experience in randomised controlled trials.
-
None/one/more than one randomised trial/other research but no trials.
-
-
Process for decision-making on participation in research trials.
-
By individual GP/all GPs/board/other.
-
-
General influencing factors for participation in research trials (tick any that apply).
-
Other existing research commitments.
-
PCRN research active practice.
-
CPRD research study.
-
Type of study (e.g. trial, observational, interview).
-
Cash incentives.
-
Topic.
-
Related to targets or QOF (positive or negative influence).
-
Staff capacity.
-
Time of year.
-
Organisational changes to PCTs.
-
Other.
-
-
Views on research
-
What do you think about the role of research in the context of general practice? Prompt – think it’s necessary, useful? More general views on evidence based practice.
-
Do you find evidence from RCTs useful to inform your clinical practice in primary care? If not, why not? Prompt – not enough time to read the evidence/often not applicable to your local population.
-
Views on Good Medical Practice 2006 paragraph 14f outlining a clinician’s duty to conduct research.
-
– Awareness of this issue.
-
– Influence of this issue on research/clinical practice/audit.
-
-
-
Reasons influencing decision to decline or agree to take part in eLung.
-
Importance of the research topic.
-
Prescribing issues.
-
– Difficulties in prescribing a drug not routinely used.
-
– Cost issues.
-
-
Distinction between research on existing versus new treatment/intervention.
-
– Testing new intervention/drug versus improving existing clinical practice.
-
– Willingness to deliver an intervention which contradicts current practice versus where no evidence available.
-
-
Distinction between acute versus chronic conditions.
-
– Recruitment method/logistics.
-
-
Research on a trial method versus question of clinical effectiveness.
-
– Aware of this primary focus.
-
– Does it make a difference.
-
-
Involvement of single versus multiple doctors.
-
Influence of related clinical policy and practice.
-
– For example, rescue packs.
-
-
-
Views of/experience of taking part in eLung (and/or Retropro).
-
Set-up.
-
– GCP training.
-
– LEPIS installation.
-
-
Recruitment.
-
– Hot versus cold.
-
-
Consent during consultation.
-
Randomisation.
-
Outcome data.
-
– Quality of coding versus free text.
-
– Collection of data from other service providers.
-
-
Downloading/interpreting outcome data.
-
-
Views on the importance of ‘trials within the database’ (if not covered in point 8).
-
Is it clear how this trial method is different from other trials?
-
Is it seen as advantageous? How?
-
Is it seen as problematic? How?
-
Feasibility of implementing this trial.
-
– Recruiting patients during routine consultation.
-
– Time to explain study and ensure patient consent is informed.
-
-
Commitment to evidence-based practice implemented at scale.
-
– Advantage of small number of patients to increase feasibility or too few to become familiar?
-
– Disadvantage of potential internal recruitment bias through selection process, e.g. likely to comply, not too sick.
-
-
-
Interest in advising CPRD on:
-
Priority research topics for trials in database in primary care.
-
Appropriate outcomes.
-
Standardised coding and interpretation of outcome data.
-
-
Views on information sheets (GP and patients).
-
Views on incentives for eLung.
-
Any advice for researchers.
Patient interview topic guide
All interviewees (agreed or declined eLung pilot trial)
Can you tell us what prompted you to visit the GP that day?
Prompt – brief history of condition and history of exacerbations, how are exacerbations normally treated by GP.
Did you find the GP visit helpful?
Prompt – good and bad aspects of consultation.
Are you feeling better?
Prompt – further visits, treatments, care pathway.
Can you tell me what you can remember about being asked to take part in the trial?
Prompt – perhaps have patient information sheet, who mentioned the trial to you in the first place, who provided the information.
What are your views on why this research is being carried out?
Prompt – ascertain information about how understand medical research evidence and why trial was needed in this particular instance.
eLung pilot trial participants
What were your reasons for agreeing to take part in the COPD trial?
Prompt – hope of getting a better treatment, hope of improving treatments in the future for other patients, relationship with the GP.
What was your experience of being asked to take part in the study?
Prompt – views on how you were approached during the consultation, what information was given, time to consider your decision.
Did you feel well enough to think about your decision and give informed consent about taking part in the study?
Prompt – feel the need to discuss with family or friends, able to understand and consider the information.
Were you happy with the treatment you were given after agreeing to be in the study?
Prompt – did understand they allocation process, did they understand equipoise, how did the allocation make them feel about the trial process.
Did you use the prescription and take the treatment?
Prompt – GP prescription or rescue pack, antibiotics and/or steroids, completed the course?
Are you happy with data being taken from your patient records by a third party in London to find out how you responded to the treatment?
Prompt – anonymised, links to hospital data, prefer to fill in a questionnaire, on one or several occasions for years to come.
What has been your experience of taking part in this study?
Prompt –positive/negative factors and why.
Is there anything you would have changed about the study to make it easier for you?
Do you have any advice for researchers about getting people involved in this type of research?
eLung pilot trial decliners
Can you explain what were your reasons for not wanting to take part in the COPD trial?
Prompt – wanted antibiotic treatment, not interested in this trial, not well enough to think about it, not enough information or time to think about it, not clear what was involved, the relationship with the practitioner, did not believe it would make a difference.
Could the research team have done anything differently which might have made you want to take part in the trial?
What are your thoughts on what researchers could do to get more people involved in this type of research?
Appendix 4 Supporting data for general practitioner views and experiences of point-of-care trials (the Qualitative process Evaluation of ANtibiotics study results)
| GP by eLung status | Interest in research in primary care or EHR research | Capacity to implement research studya | Benefit for local patients or population | Improve clinical practice in long term | Adequate remuneration to cover costs | Adequate remuneration to generate profit | Positive attitudes of patients to research | Negative attitudes of patients to research | Ethics and patient safety |
|---|---|---|---|---|---|---|---|---|---|
| Decliners (n = 9) | 2 (2b) | 3 (1b) | 2 (2b) | 2 (1b) | 3 (1b) | 2 (1b) | 3 | 2 | 1 (1b) |
| Withdrawals (n = 3) | 1 (1b) | 2 | 1 | 1 (1b) | 1 | 1 (1b) | 1 | 1 | – |
| Incomplete set-up (n = 8) | 4 (1b) | 6 (6b) | 5 | 2 | 1 | – | 4 (2b) | – | – |
| Recruiters (n = 7) | 4 (3b) | 7 (4b) | 5 (2b) | – | 3 | 2 (1b) | 4 (2b) | – | 1 (1b) |
| Total | 11 (7b) | 18 (11b) | 13 (4b) | 5 (2b) | 8 (1b) | 5 (3b) | 12 (4b) | 3 | 2 (2b) |
| GP by eLung status | Time for study at point of invitation | Demands of study design on GP time | Benefit to patients | Relevance of topic | Feasibility to recruit patients | Views on computer-based pop-up alerts | Views on hot recruitment method | Views on consistency with local prescribing guidance | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient views of ‘no antibiotics’ group | Number of patients | Impact of rescue packs on number of patients | Referral from GPs | |||||||||||||||||||
| ✗ | ✓ | ✗ | ✓ | ✗ | ✓ | ✗ | ✓ | ✗ | ✓ | ✗ | ✓ | ✗ | ✓ | ✗ | ✓ | ✗ | ✓ | ✗ | ✓ | ✗ | ✓ | |
| Decliners (n = 9) | 3 (1a) | – | 3 | – | – | – | 4 | 1 | 3 | – | 1 | – | 2 | – | 1 | – | 3 | 1 | 6 (1a) | – | 2 | 5 |
| Withdrawals (n = 3) | – | – | – | – | – | – | – | 2 | – | – | – | 1 | – | 2 | – | – | – | 1 | – | – | – | 3 |
| Incomplete set-up (n = 8) | 1 | – | – | 4 (2a) | – | – | – | 8 | – | – | – | 6 | 2 | – | – | – | – | 6 | 1 | 5 | – | 7 |
| Recruiters (n = 7) | – | – | 1 | – | 3 (1a) | – | 3 (1a) | 1 | – | 1 | 2 | – | 2 | – | – | – | 2 | 2 | 1 | – | 3 | |
| Total | 4 (1a) | – | 4 | 4 (2a) | – | 3 (1a) | 4 | 14 (1a) | 4 | – | 2 | 9 | 4 | 4 | 1 | – | 3 | 10 | 9 (1a) | 6 | 2 | 18 |
General practitioner recommendations to improve point-of-caretrials
Study documentation and communication with general practitioners
Most GPs considered the eLung documentation for GPs and patients to be clear, particularly regarding core information on the aims of the pilot trial, inclusion/exclusion criteria of eligible patients and study outcomes. Nearly all GPs were aware the trial was a pilot with the primary aim of exploring a methodological question. Some aspects of the study design were less clear for a handful of GPs including three GPs who incorrectly believed their practice had been allocated as a control site and one GP who was also not clear whether the invitation to participate was targeted to a single or multiple GP(s) within the practice. Many GPs were not clear or confident about how the hot recruitment would work in practice and identified the need for more personalised and/or practical input in the early stages of recruitment. A small number of GPs expressed concerns regarding resending of entire documents by e-mail following specific changes to a document resulting in the GP having to reread entire documents several times. Other concerns included the need for study documents to address data protection requirements for remote collection and analysis of patient data and provide clear information on these issues in the patient information sheet. The main concern expressed by several GPs across all groups was regarding access to timely and appropriate support throughout the set-up phase, particularly during the widespread difficulties experienced with the installation of LEPIS. Site visits by the research team to resolve the technical difficulties were highly valued by all GPs. GP recommendations to improve information and communication strategies for future trials include the following:
-
Avoid GP recruitment during busy periods such as reporting for QOF or winter flu epidemics.
-
E-mail communication with GPs should be used sparingly and new information should be separated out from previous versions of documents.
-
Clear and simple information to GPs including a one-page summary of the study design and exclusion criteria.
-
Clear and simple information for patients, particularly on data protection for remote access of patient data and a single signature for consent. Patients who have signed a disclaimer should be excluded.
-
Study materials should include an online demonstration, via a web-based podcast, for example, to demonstrate how hot recruitment using pop-ups actually works to screen, recruit and randomise patients in different scenarios. This would boost confidence for the lead GP and could be used as a training aid for other GPs.
-
Each practice should receive a telephone call from the research team to explain the entire study alongside the invitation letter as part of the initial recruitment process.
-
The option of a face-to-face visit for GPs who have agreed to take part to explain the study in more detail or address specific concerns would boost confidence and motivation for GPs who are new to this trial method.
-
– A face-to-face meeting would also provide the opportunity to promote the study design to multiple GPs to increase support to the lead GP for improved patient recruitment and increase awareness and interest in trials of this method among a broader base of GPs.
-
Input from general practitioners to inform future trials
Nearly all GPs within all subgroups of eLung status believed it would be extremely useful for practising GPs to have input into the design of trials and how they would work on the ground to reflect the realities and variation of how general practice works, and to identify priority topics of local relevance and related outcome measures for future trials. Most GPs suggested that the lead GP on research would be the most appropriate person to provide input on the practicalities of the study design, with a small number of GPs identifying the potential need for financial remuneration to backfill GP time if this task took the GP out of clinical practice. Strategies to identify an ‘Advisory Group of Lead GPs’ with an interest in this role included contact by the EHR database to all lead GPs on research to invite expressions of interest (via internet-based questionnaires for example) and/or use of informal networks with GPs known by the EHR database to have an interest in this role. A small number of GPs did not consider GPs to be the appropriate choice to identify priority topics of research interest for future trials. Instead, Clinical Commissioning Groups and Local Medical Committees were suggested as having the relevant knowledge and expertise to fulfil this role. GP recommendations on input to improve the design of future trials are as follows:
-
An ‘Advisory Group’ of Lead GPs on research should be identified by the CPRD to provide input on the practical design of a study and how it is implemented on the ground in different practice settings.
-
Lead GPs, Clinical Commissioning Groups and Local Medical Committees could assist with identifying priority research topics and key outcome measures with local clinical relevance.
-
Financial remuneration may be necessary to backfill GP time from clinical practice.
Flexible strategies to recruit patients
The need for flexible recruitment strategies to combine more traditional cold recruitment strategies with hot recruitment within the consultation was recommended by several GPs across all groups. These GP recommendations were primarily aiming to ensure sufficient time was available to achieve informed consent while also removing any elements of the recruitment process from the consultation to increase efficiencies and feasibility of the hot recruitment process. GP recommendations on flexible strategies to improve recruitment of patients are as follows:
-
Flexibility for each GP practice to implement a range of recruitment strategies which support hot recruitment using computer-based alerts where appropriate or a mix of hot and cold recruitment as necessary. Specific strategies include:
-
– introducing patients to study materials at routine annual checks/clinics
-
– screening patients at triage, or appointment booking and allocate a 20 minute appointment
-
– text messaging to patients to check eligibility and for appointment reminders
-
– postal letter to eligible patients to alert them to study and recommend change of usual care (e.g. contacting surgery prior to using rescue pack), if necessary
-
– option for GP to connect patients to a CPRD research nurse via the internet to discuss the patient information and consenting process prior to the GP completing consent and recruitment
-
– option for GP to enter some of the patient data onto the system at the end of surgery, allowing more time in the consultation for consenting
-
– important to keep the flexibility for a GP to opt out of recruiting a particular patient if the GP is too busy or patient appears too unwell, for example.
-
Computer-based alert system
The over-riding concern regarding the computer-based alert system for GPs across all subgroups was the need for it to enhance the efficiency and simplicity of the trial to encourage GPs to be willing to take this method on board and take part in the trial and/or to increase confidence in its feasibility. The study website was seen as a valuable alternative to the LEPIS alert system. GP recommendations on how to increase support for use of computer-based alert systems, such as LEPIS, are as follows:
-
Outstanding problems regarding installation and functionality of LEPIS need to be resolved prior to use in a full trial.
-
Screen-based pop-ups must be extremely quick, easy to use and based on data which is routinely collected during the consultation.
-
Data collected in response to the study alerts must be linked directly to the patient record to avoid duplication of data entry.
-
Option for GPs to recruit via the study website as an alternative to LEPIS.
-
Study data should be entered as free text or standard codes.
-
CPRD would interpret study data via remote download with assistance from GPs as required on individual records.
Additional support for vulnerable practices
Over two-thirds of GPs in the QUEAN study, including those who had declined, accepted or recruited participation in eLung, are located in areas with high levels of deprivation and are from a variety of ethnic backgrounds. All three GP participants who are based in London had declined participation in eLung. It appears that the additional factors of a highly mobile population which is generally not compliant with GP advice, due to a suspicion of authorities in the case of immigrant populations or a consumer-led attitude of moving between services until patient needs are met, may be contributing to the decision-making process on participation in research generally or eLung. GPs employed in small practices can also experience additional difficulties, for example increased variation in quality of data entry and coding due to a greater reliance on locums to fulfil staffing quotas. GP recommendations on strategies to provide additional support for vulnerable practices are as follows:
-
Recruitment of a collaboration of service providers across a London region who would provide consistent advice and practice to patients regardless of mobility and patient demand.
-
Financial incentives for patients to participate in the study.
-
Option of additional administrative support to aid, for example, complementary cold recruitment strategies, checking data quality.
Incentives for general practitioners
The GP recommendations of incentives for GPs including financial, educational and developmental ones are as follows:
-
Promotion of the novel trial method to utilise EHRs for collection of outcome data is likely to generate an underpinning personal interest and motivation for most GPs to encourage participation in future trials.
-
Adequate per participant remuneration, consistent with levels provided for eLung to cover costs and potentially generate a small profit.
-
Formulae to calculate flexible financial remuneration packages to cover costs for combined hot and cold recruitment strategies.
-
Packaging of remuneration of other ‘enhanced services’ like QOF targets which improve income streams.
-
GP training on research topic.
-
Quick and timely feedback to practices on recruitment rates including acknowledgement of high recruiters, for example a certificate to display in the practice to acknowledge valued input of patients.
-
Research can be promoted as an opportunity for revalidation within the Good Medical Practice Framework.
Study findings suggest these recommendations should be considered in their entirety to identify a mix of strategies which will have a bearing on the range of generic and project-specific factors identified as important in influencing the GP decision to participate in future point-of-care trials.
Large simple trials in primary care also provide an opportunity for increased personal enjoyment and satisfaction for GPs who have limited time but enjoy research and being involved in all aspects of implementing a trial. Broad-based promotion of the efficiencies of hot recruitment, to identify patients using computer-based alerts and EHRs to collect outcome data have the potential therefore to increase personal interest in point-of-care trials as an underpinning motivating factor in future decision-making processes.
Appendix 5 Point-of-care trials: the way forward– perspectives from patient representatives and general practitioner investigators
Perspectives from Marion Cumbers (patient representative on the Trial Steering Committee)
The NHS now has a duty to undertake research and ensure drugs and procedures are safe and effective. Although EHRs have existed for many years, they have only been used very rarely for clinical research. The report of these trials will demonstrate some of the reasons. Many of the barriers met by the researchers in this project will have to be removed if such research is to produce useful data without causing extra work for GPs, and extra pressure on surgeries.
Patients
Patients should feel that it is their responsibility to help research for the general good by allowing their records to be used. But will the admission that the GP does not always know which treatment is best affect the faith of patients in their doctor? This may not inspire confidence.
It should at least be possible to ask all patients attending a GP surgery for a routine appointment if they are ‘willing to be contacted’ about possible research projects. Consent could be signified by pressing a button at the appointment monitor point, and then flagged on their EHR. Opportunities to broach the subject of research (e.g. annual check-ups for the chronically ill; ‘well-woman’ clinics; and dedicated ‘flu jab’ clinics) could all be used to capture permission. Waiting room posters or rolling TV advertisements could also encourage bored patients to pick up a leaflet to read while waiting.
When a research opportunity arises, those flagged could be sent a Patent Information Sheet through the post to read before their next appointment. This would give them the required ‘thinking time’ to decide whether or not to take part. Appointment reminders sent by text could also be used to remind patients of the leaflet.
Most medical problems resolve naturally, are self-limiting and do not result in a further visit to the surgery. If a patient does not come back, the doctor and his EHR will have no record of the result of the consultation. It will not be known if the patient cashed any resulting prescription, if they took any of the medicine, or if they completed the course. Patients are rarely asked ‘How did you get on with what I prescribed last time?’. Any comment from the patient is seldom noted on the EHR. When reporting ill effects of medication, the reaction is sometimes ‘That is nothing to do with the medicine’, even if it is mentioned as a possible side effect in the Patient Information Leaflet with the medicine. This may mean that EHR research is limited in practice to the chronically or seriously ill patients who attend GP surgeries frequently or regularly.
Patients participating in a trial of commonly prescribed medicines should not be put in the position of having become accustomed to a trial medicine and then being told they are not allowed to continue with it if they prefer it. However, they could be asked to try a different drug after a wash-out period, to see which works best for them. Likewise, if a GP prescribes a branded drug rather than a generic because the patient reports that the generic does not work as well, this decision should be respected, irrespective of financial or practice target considerations. Within reason, individuals should have the right to choose.
Special efforts may need to be made to involve those with poor literacy skills and those who are hard to reach who attend surgeries rarely. Ethnic minorities, who may be the majority of patients in certain practices and may need forms translated or an interpreter, should not be excluded and deserve to be helped to contribute to research If they wish.
Incentives
In general, altruism is the best incentive for a patient to take part in research. Attendance at a manufacturer’s trial may be rewarded with a £10 shopping voucher. However, cash or tokens are unlikely to aid recruitment unless of a significant amount, which only commercial researchers are likely to be able to afford. Anyone to whom a small amount is significant could be considered vulnerable to coercion.
However, if extra visits are involved, travel expenses should be offered as a matter of course, (and not need to be asked for, as this can be thought of as demeaning). Parking places should be provided and/or paid for, and child or elder care likewise if this is a barrier to participation.
General practitioners
General practitioners personally should also feel bound to take part if their practice will not be adversely affected by extra work and appointments, and unreasonable demands on their patients. If protocols are kept simple and records are captured electronically, with no extra work involved, there can be little reason to refuse to investigate further, even if the eventual decision is not to participate. But the ordinary appointment schedule should not be delayed by explanations of research. Some willing participants may not be able to spend extra time with the GP such as those with children to meet from school, babies to feed, seniors to care for, or those without access to their own transport.
Practices taking part in research should not be penalised by the cost of the trial drugs, or the effect of choices on NHS practice performance targets.
Ethics Committees and research and development
It is illogical that a doctor can prescribe whatever he thinks best, but as soon as it is called research, he has to jump through hoops. Information sheets and consent forms for patients should be brief and use clear simple language with a target reading age similar to that of a red-top national newspaper, with no acronyms or technical terms. If successful, the answer to the question ‘Is there anything you do not understand or would like explained further?’ should be ‘No, thank you. That is all very clear’. This should minimise the time needed for explanation and seeking consent. The standard Patient Information Sheet is not suitable for simple EHR research and Ethics Committees should not ask for it to be in that format.
Delays caused by Ethics Committees and R&D offices are not confined to EHR research. An application should not be needed for each individual GP practice or site involved. Approval could also be granted for all EHR research topics at that site in future. The longer the start of research is delayed, the more likely a practice is to lose enthusiastic skilled staff or staff who have possibly undertaken specific training.
Information technology systems
Practices use a number of different IT systems, and have different degrees of access to their host network and even to their ‘desktops’. Knowledge, skills and enthusiasm of staff for IT is also variable. Much time can be spent getting systems to work together. To limit delays, participation could be limited to practices where similar problems have already been overcome and set-up is judged likely to succeed without an inordinate involvement of skilled support staff.
Data sharing
Out-of-hours practitioners, dentists and mental health professionals should, as a matter of course, be able to access patient EHRs, both to read and to enter information. Indeed, from a patient viewpoint, data sharing should be one of the main purposes of EHRs. However, records of referrals, results of hospital tests or emergency care, etc., do not always appear promptly, or at all, on a patient’s EHR, and this could worsen when Social Services data is integrated into the same record at some point.
The use of EHRs in future pragmatic point-of-care trials would therefore be facilitated by:
-
fast-tracking the ethics and R&D approvals, particularly for multiple sites within a Commissioning Group or area. The longer it takes to get research up and running, the more likely it is that practices will lose the staff committed to carrying it out.
Choosing GP practices, where:
-
IT systems are compatible
-
all GPs are enthusiastic, ensuring a common approach whichever GP is seen by the participant
-
and data recording can be shown to be generally robust and of good quality.
Choosing topics for research:
-
with input from GPs
-
with no implications for NHS targets or NICE restrictions
-
and where the data loop will usually be closed by another visit.
Perspectives from Gary Simons (patient representative on the Trial Steering Committee)
-
It is clear that novel aspects of point-of-care trials do not fit comfortably into existing contexts of doctors’, patients’ and ethics committees’ expectations and practice concerning research proposals.
-
In particular, priority given to informed consent, with the entirely proper object of protecting patients from abuse and detriment where such might occur with new procedures, tends to complicate unnecessarily comparisons of well-established alternative treatments that coexist in a state of unresolved uncertainty as to their relative merits.
-
What follows below attempts to define principles for each of the above groups with the aim of developing a new and more appropriate context for randomising point-of-care trials. However, there appears to be one primary over-riding principle which may be seen as a touchstone for the distinct groups. For ease of reference I shall call this the first principle:
-
EACH PATIENT SHOULD HAVE THE OPPORTUNITY TO RECEIVE TREATMENT AND ADVICE WHICH ARE BASED ON THE BEST REASONABLY AVAILABLE EVIDENCE MOST LIKELY TO BENEFIT MAXIMALLY HIS OR HER CONDITION.
-
-
For doctors and other clinicians this clearly implies an obligation to assist the finding of reasonably available evidence. The practice of medicine is a learned profession, in some senses more so than law or pastoral ministry. In law, customary practice, statute and judicial comment, a relatively stable though growing body of knowledge, determine how particular circumstances are to be addressed. In pastoral ministry, scripture, tradition and reason determine belief, practice and the scope of professional discourse. Medicine is unique among the learned professions in recognising scientific method as the appropriate paradigm for enhancing practice. Thus, careful observation, the forming of hypotheses and their testing by controlled experiment constitute the appropriate and essential process for evaluating practice. Without such there can be no evidence-based medicine and what is taught in medical schools cannot be rated as knowledge.
-
If there is unresolved uncertainty but alternative treatments are established in current practice, claims for medical negligence used generally to fail: ‘A doctor is not guilty of negligence if he has acted in accordance with the practice accepted as proper by a responsible body of medical men skilled in that particular art’. The words are those of Mcnair J in a first instance judgment in the Queen’s Bench Division of the High Court [Bolam v Friern Hospital Management Committee [{1957} 2 All ER at 122].
-
However, this could lead to inertia and the persistence of outmoded, untested and second-rate practice; and it is clear now an obligation to assess practice critically is required of clinicians who hold themselves out as having expertise. Lord Browne Wilkinson in Bolitho made this clear as follows: ‘In particular, in cases involving, as they often do, the weighing of risks against benefits, the judge before accepting a body of opinion as being responsible, reasonable or respectable, will need to be satisfied that, in forming their views, the experts have directed their minds to the question of comparative risks and benefits and have reached a defensible conclusion on the matter’. [Bolitho v City and Hackney Health Authority{1998}. 4All ER at 778.]
-
These words are part of the lead judgment in the House of Lords in the course of a hearing before five Lords Justice of Appeal. None of the others dissented from Lord Browne Wilkinson’s dictum.
-
A principle to incorporate this view might be stated as follows:
-
TO ADVANCE THE FIRST PRINCIPLE IN CIRCUMSTANCES OF UNRESOLVED UNCERTAINTY DOCTORS SHOULD ADDRESS THEIR MINDS TO COMPARATIVE RISKS AND BENEFITS OF EACH ALTERNATIVE AND COMMIT TO REACHING A DEFENSIBLE CONCLUSION ON THE MATTER.
-
-
For patients, where their participation would help to determine best treatment there is a duty to participate in research that carries no foreseeable risk of detriment or of loss of benefit beyond those to be expected from other available treatments. GP practices and other institutions might reasonably invite patients to commit to a principle such as:
-
TO ADVANCE THE FIRST PRINCIPLE, MY TREATMENT SHOULD, AS FAR AS REASONABLY POSSIBLE, HELP OTHER PATIENTS AS WELL AS ME.
-
-
This principle might be given effect by clinicians asking their patients to agree well in advance of any treatment to a statement such as: ‘In order to help discern the best treatment for me and for others, providing my taking part in research will not involve greater risk or greater loss of benefit to me than any other available treatment, and providing the research has been approved by an independent ethics committee, I agree in principle to participate’.
-
This would take off some of the pressure of ‘hot recruitment’. The principle and process of point-of-care research could be explained by a variety of means at a time when patients and possible future patients have opportunity to consider the issues and to ask questions. Information could be given about ethics committees’ work and the special safeguards that the committees have approved for point-of-care research. (See point 13 below.)
-
As to ethics committees, it is important that their approach facilitates the first principle. What is needed from them is commitment to that principle and a declared predisposition amounting to a pragmatic sanction in favour of randomising point-of-care trials such that they will normally be approved subject to safeguards. That favourable predisposition should be defeasible, that is to say it might be overcome or subjected in special circumstances that make randomisation unacceptable to doctor or patient. Such might occur when a doctor believes he or she has experience that makes him/her favour one of the alternatives, or a patient feels a strong preference, or when drug tolerance or comorbidities are issues, or where there is reason to believe one of the alternatives will prove less effective.
-
The relevant principle for ethics committees might be as follows:
-
TO ADVANCE THE FIRST PRINCIPLE WE WILL GENERALLY GIVE APPROVAL TO POINT-OF-CARE TRIALS PROVIDING THE NEED FOR THEM IS SUFFICIENTLY DEMONSTRATED AND AGREED SAFEGUARDS FOR PATIENTS ARE ASSURED.
-
-
Among required safeguards the following might be significant: that the unresolved uncertainty is warranted by a sufficiently independent authority; that that authority will warrant the unresolved uncertainty persists while patients follow any particular course of treatment as part of the research; and that, for the same period, that authority will warrant that no new evidence has emerged to adjudicate among existing alternatives. Such safeguards required by ethics committees should be known to patients when their participation in principle is invited. (See point 10 above.)
-
Manifestly, many more inputs are needed to make what is proposed effective. However, I think something like the first principle is a good starting point for a consensus. Consideration should be given to casting that first principle as a patient right. There are advantages in doing so, but I have not chosen this route because to so may raise complications that will cause delay over legal and financial concerns.
-
What is needed is a major initiative at a senior level to bring groups representative of patients, doctors and ethics committees together. Given the need and the opportunity to structure and manage existing data sources for patient benefit there is a sound basis for broad agreement and a compelling objection to failure to achieve it. As a patient I feel content with the first principle, with that for clinicians, with that for patients and with that for ethics committees. What needs to done is to achieve similar content from other parties and perspectives.
-
The first necessity is to build a clear sense of common purpose and urgency about facilitating and expediting the set-up process for point-of-care research, particularly with regard to the burden of obtaining consents from many research ethics committees. The second is to establish a team committed to a leadership role, capable of sustaining momentum and resourced to do so. Early objectives must be to demonstrate the effectiveness and benefits of point-of-care randomised trial research in resolving high profile unresolved uncertainty dilemmas of common concern to clinicians and patients. Pioneer projects should affect both primary and secondary care and ideally point to cost savings to demonstrate that, over the medium term, such research can be financially self-sustaining, as well as providing evidence to further patient benefit.
Perspectives from Robin Fox (general practitioner investigator in Retropro)
In the UK, GPs are independent contractors and if they are doing research then they are not seeing patients. Locum costs are very high and often are no substitute for the GP partner. There are financial and non-financial drivers to practices and GPs for participation in research. Networking with practices and visibility of researchers at GP conferences will also be important. The NIHR PCRN in the UK with it role in the new Local Clinical Research Networks can play a vital role in expanding recruitment of practices and GPs.
Any point-of-care trials should follow the Keep It Simple, Stupid (KISS) principle. This principle states that most systems work best if they are kept simple rather than made complex; therefore, simplicity should be a key goal in design and unnecessary complexity should be avoided. The following should be considered in point-of-care trials:
-
Lots of effort is often put into the study design and research questions, but little into feasibility of recruitment in primary care. However, a GP experienced in recruitment should be involved very early in the planning stages. This would stop ambitious but impractical ideas from wasting everyone’s time if they are doomed to failure (such as the eLung diaries). Furthermore, it should be explored whether the trial would be feasible for patients.
-
Every item that a GP practice needs to do should be minimised. Researchers should work from the principle that the GP is the busiest person involved in the trial and that it will fail without them. Everything should be done to minimise their workload.
-
Introduce the study with a one- to two-page summary (as is current PCRN practice). Ideally, this will explain what the GP practice has to do, financial remuneration and a time commitment for the study (as well as a brief study summary and inclusion/exclusion).
-
Understand time implications for the GP for recruitment and follow-up. Hot recruitment is invariably more challenging for GPs and practices than cold recruitment.
-
Keep the time from first suggesting the study to first recruitment down to a minimum.
-
Automate and auto populate as much as possible. Prefilling as much information as possible on any documents the GP needs to sign at the setup stage would reduce workload.
-
Reduce unnecessary paperwork. Having to complete consent forms in triplicate is an unnecessary barrier. Using carbon copies would have reduced workload.
-
Make recruiting the first patient as easy as possible. The hardest patient is the first one. If the study is complex in the GP’s mind, then the GP will never recruit. Often a YouTube like clip of a GP recruiting a patient or a simulated walkthrough is very useful. This helps overcome ‘first patient recruitment fear syndrome’!
-
Minimising the GP’s workload, keep everything straightforward at every stage.
-
Make data entry as limited as possible and if it has to be entered on a proforma form, ensure a copy to clipboard function exists, so the GP does not have to write it all back in notes but can paste it into the patient’s record.
-
The reward to the practices and GP should not only be financial but also include professional development credits, authorship on publications and a certificate of recognition. Being involved with research is a selling point for the practice to other doctors coming to work in the practice, trainees, students and patients.
Perspectives from Tommy Hunter (general practitioner investigator in Retropro and eLung)
I believe the main points are:
-
Make the patient information and consent process as brief as is ethically possible. In a point-of-care trial, the patient does not have time to read even a two-sided consent/information sheet. If there are multiple elements to the study as in the genetic testing for Retropro, they need to be incorporated in the primary paperwork.
-
Make a dummy patient available. I think many more patients would have been recruited to eLung if GP’s could have tried a dry run and found how easy it was.
-
Acknowledge reasons other than financial for participation. Notices for waiting room, letters thanking patients for participating and most importantly reporting the results back in a format suitable for patients.
-
Although I agree that financial reward is very important, the level of that reward may not need to be particularly high if these other factors are right. Of fundamental importance to most clinicians is that the research should be answering a question that is important to them. I suspect, in the long run, this is more important than money. I think potential recruiters need to be consulted on the questions to be researched to see if they have any interest it the topic. If so, it is likely that the perceived ‘price’ required will be less.
List of abbreviations
- ADR
- adverse drug reaction
- BMJ
- British Medical Journal
- CCS
- central control service
- CEA
- cost-effectiveness analysis
- CEAC
- cost-effectiveness acceptability curve
- CI
- confidence interval
- COPD
- chronic obstructive pulmonary disease
- COX-2
- cyclo-oxygenase 2
- CPRD
- Clinical Practice Research Datalink
- CTR
- computerised trial recruitment
- CVD
- cardiovascular disease
- EHR
- electronic health record
- EQ-5D-3L
- European Quality of Life-5 Dimensions-3 Level
- EVPI
- expected value of perfect information
- EVPPI
- expected value of perfect parameter information
- FEV1
- forced expiratory volume in 1 second
- GCP
- Good Clinical Practice
- GP
- general practitioner
- HRQoL
- health-related quality of life
- ICER
- incremental cost-effectiveness ratio
- IT
- information technology
- LEPIS
- Local Eligible Patient Identification Service
- MHRA
- Medicines and Healthcare products Regulatory Agency
- NHB
- net health benefit
- NICE
- National Institute for Health and Care Excellence
- NIHR
- National Institute for Health Research
- PCRN
- Primary Care Research Network
- QALY
- quality-adjusted life-year
- QOF
- Quality and Outcomes Framework
- QUEAN
- QUalitative Evaluation of a trial of ANtibiotics for chronic lung disease
- R&D
- research and development
- RCT
- randomised clinical trial
- SUSAR
- serious unexpected suspected adverse drug reaction
- TASTE
- Thrombus Aspiration in ST-Elevation myocardial infarction in Scandinavia
- VOI
- value of information