
Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 08/13/35. The contractual start date was in April 2010. The draft report began editorial review in September 2013 and was accepted for publication in January 2014. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
The institution of DWC and TEAP received per-case funding from Optimer Pharmaceuticals to support fidaxomicin trial patient expenses for a trial in Clostridium difficile infection. DWC and TEAP also received honoraria from Optimer Pharmaceuticals for participation in additional meetings related to investigative planning for fidaxomicin. MHW has received consulting fees from Actelion, Astellas, AstraZeneca, Cerexa, Cubist, Durata, Merck, Nabriva, Novacta, Novartis, Optimer, Paratek, Pfizer, Roche, Sanofi Pasteur, Summit, Synthetic Biologics, The Medicines Company and VHsquared; lecture fees from Abbott, Astellas, AstraZeneca and Pfizer; grant support from Abbott, Actelion, Astellas, bioMérieux, Cubist, Da Volterra, The European Tissue Symposium, Merck and Summit; and a lecture fee from Alere paid to his department. The Medicines Company and VH Squared; lecture fees from Abbott, Astellas, AstraZeneca and Pfizer; grant support from Abbott, Actelion, Astellas, bioMerieux, Cubist, Da Volterra, The European Tissue Symposium, Merck and Summit; and a lecture fee from Alere paid to his department. Unrelated to C. difficile or any gastrointestinal pathogen, DWC has received research funding from BioMérieux for Staphylococcus aureus bacterial genome wide association studies, from Pfizer for Streptococcus pneumoniae surveillance, and from Microsoft Azure for cloud-based access for bacterial genomic analysis. The institution where ASW holds a part-time post (not involved or acknowledged in this study) has received funding from Gilead Sciences and ViiV Healthcare/GlaxoSmithKline for additional assays and/or analyses within clinical trials in human immunodeficiency virus (HIV) infection; from Gilead Sciences for her lecturing on educational workshops, and from Janssen and Janssen (formerly Tibotec) for her Data and Safety Monitoring Board membership. No other author has a conflict of interest.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2014. This work was produced by Pankhurst et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Introduction
The clinical problem
Every year a large 1500-bed hospital can expect to see approximately 9000 patients with potentially infectious diarrhoea necessitating isolation under the statutory 2006 Hygiene Code (now incorporated with The Health and Social Care Act 2008). 1 An infecting organism will be identified in as few as 1 in 10 of these cases, necessitating substantial isolation capacity primarily for diagnostic reasons. The NHS currently has insufficient single rooms to effectively accommodate all such patients. 2 Single rooms may, therefore, be ‘blocked’ by patients with diarrhoea not yet confirmed as infectious, while other patients with infectious diarrhoea are still in open bays because of a lack of free side rooms. A rapid test for identifying cases of infectious diarrhoea could provide major benefits to the smooth running of hospitals by promoting efficient use of isolation beds.
The microbiological diagnostic problem
Much current research in microbiological diagnostics is focused on developing simple and rapid molecular tests to identify the aetiology of infectious conditions such as diarrhoea. However, most molecular tests focus on identifying one organism and a major hindrance is that many different pathogens can cause infectious syndromes. For example, the cause of a case of infectious diarrhoea is likely to be one of approximately 4–6 common pathogens, or possibly one of a further approximately 20 rarer pathogens, or, extremely rarely, one of hundreds of uncommon organisms. Symptoms are generally similar regardless of the specific causative pathogen, meaning diagnostic tests are the only way to identify appropriate treatment and management. There are excellent nucleic acid amplification tests (NAATs) for individual pathogens, but combining these tests into a multiplex with 10 or more pathogens brings significant challenges in terms of cross-reactivity (primer dimers), random products and inhibition. Primer dimers are potential by-products from polymerase chain reactions (PCRs), in which a primer molecule (a piece of single-stranded nucleic acid designed to match that in one pathogen) attaches (hybridises) to a primer molecule for a different pathogen, because they share complementary bases. As a result, in the PCR, the polymerase amplifies the primer dimer, outcompeting any original pathogen nucleic acid for the PCR reagents, and inhibiting amplification and detection of the original pathogen nucleic acid. Clearly, the more pathogens that are to be detected in a multiplex reaction, the more primers that are included and the greater potential for primer dimers to form – primer dimers may lead to false positives or false negatives. Random (PCR) products form when a primer attaches randomly to a non-target pathogen nucleic acid sequence leading to false positives. Inhibition occurs when other molecules in the stool sample prevent the PCR enzyme from amplifying the target nucleic acids, leading to false negatives. Thus, multiplexing tests may produce substantial numbers of false positives and false negatives, even compared with the original single PCR, and may also have decreased sensitivity in terms of the number of copies present in a sample required for the pathogen to be detected. At present, achieving relative certainty over which patients with diarrhoea are infected with enteropathogens can take up to 3 days, since numerous different time-consuming individual processes are needed to test for each pathogen (Figure 1). It is this delay in turnaround time from sample collection to test result which has a major impact on bed management and patient pathways. In the case of many bacteria (e.g. Campylobacter spp. and Salmonella spp.), a minimum of 48–72 hours is needed for growth and identification (or to confirm the absence of growth). In the case of Clostridium difficile, the gold standard cytotoxin test takes 1–2 days to provide a result because it relies on cell culture and a negative result is not issued until the test has been reread at 48 hours, whereas the substantially faster enzyme-linked immunosorbent assay (ELISA)-based tests have the recognised drawback of lower sensitivity (≈ 50–85%),3–5 thus leading to repeat testing over a few days for a substantial minority of cases. As diarrhoea in most hospitalised patients does not have an infectious cause, a better approach would be same-day (< 24 hours) differentiation of non-infectious diarrhoea from infectious diarrhoea caused by the most common enteropathogens: C. difficile, Campylobacter spp., Salmonella spp., Shigella spp., Escherichia coli, rotavirus and norovirus. This would address the current widespread lack of sufficient isolation capacity in the NHS by providing either an almost immediate negative result or a causative pathogen for the vast majority of patients with diarrhoea. In turn, this would allow the instigation of individualised patient treatment and appropriate infection precautions to avoid exposing other patients to the risk of acquiring enteropathogens and to limit the dissemination of epidemic bacteria (such as C. difficile) and viruses (such as norovirus). Not only would such a test radically change the patient pathway for many infectious syndromes, but, in particular, it should also alter the urgent need for rapidly increasing the isolation capacity of the NHS to meet the needs of infectious diarrhoea (e.g. that caused by C. difficile). This would enable the prioritisation of high-cost rebuild/refurbishment projects to yield more single rooms to be revisited in many hospitals.
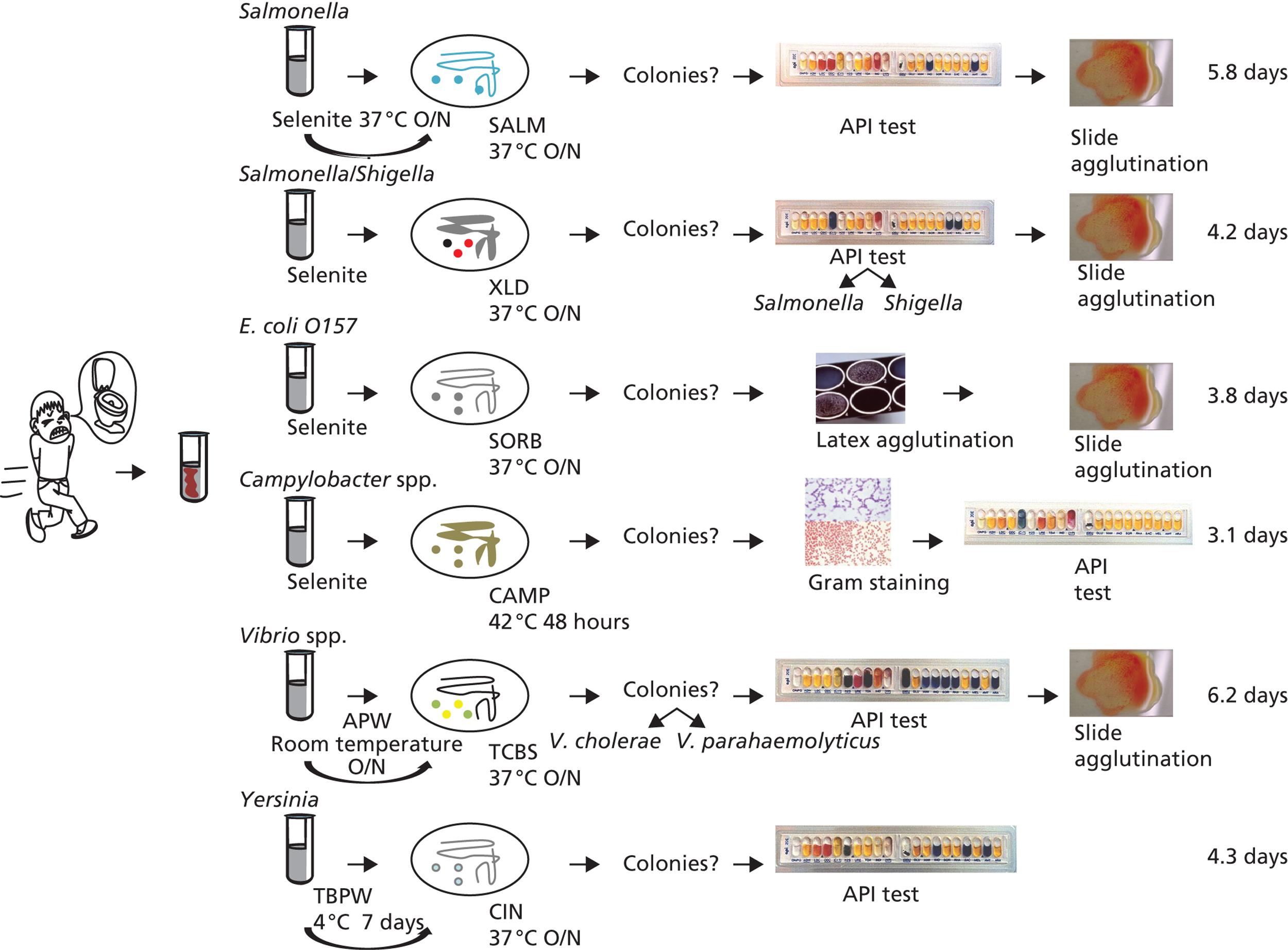
FIGURE 1.
Current diagnostic workflow to identify common pathogens causing infectious diarrhoea. API, analytical profile index test; APW, alkaline peptone water; CAMP, Campylobacter-free blood agar; CIN, cefsulodin irgasan novobiocin agar; O/N, overnight; SALM, chromogenic agar; SORB, sorbitol MacConkey agar; XLD, xylose lysine deoxycholate agar; TBPW, tris-buffered peptone water; TCBS, thiosulfate citrate bile salts sucrose agar.
MassCode multiplex polymerase chain reaction-based diagnostics
This project was originally designed to investigate a newly developed multiplex PCR, called MassCode,6,7 which was designed to provide a rapid (3–4 hours) test for up to 30 pathogens simultaneously in a single reaction, potentially offering a cost-effective and rapid mechanism of testing one single stool sample for multiple organisms in close to real time. This would also have offered the opportunity to effectively rule out infectious causes of many syndromes, not only infectious diarrhoea, but also meningitis, infectious arthritis, empyema, etc. In contrast, conventional PCR tests are usually limited to detecting one pathogen per test with effectively a maximum of six, based on the number of PCRs that can be reliably multiplexed together. Consequently, MassCode multiplex PCR-based diagnostics avoid the need to conduct multiple individual tests (whether antigen testing, culture or conventional PCR) on samples (Figures 1 and 2) while (theoretically) delivering comparably high sensitivity and specificity to conventional single-pathogen PCR tests.
FIGURE 2.
MassCode diagnostic workflow.
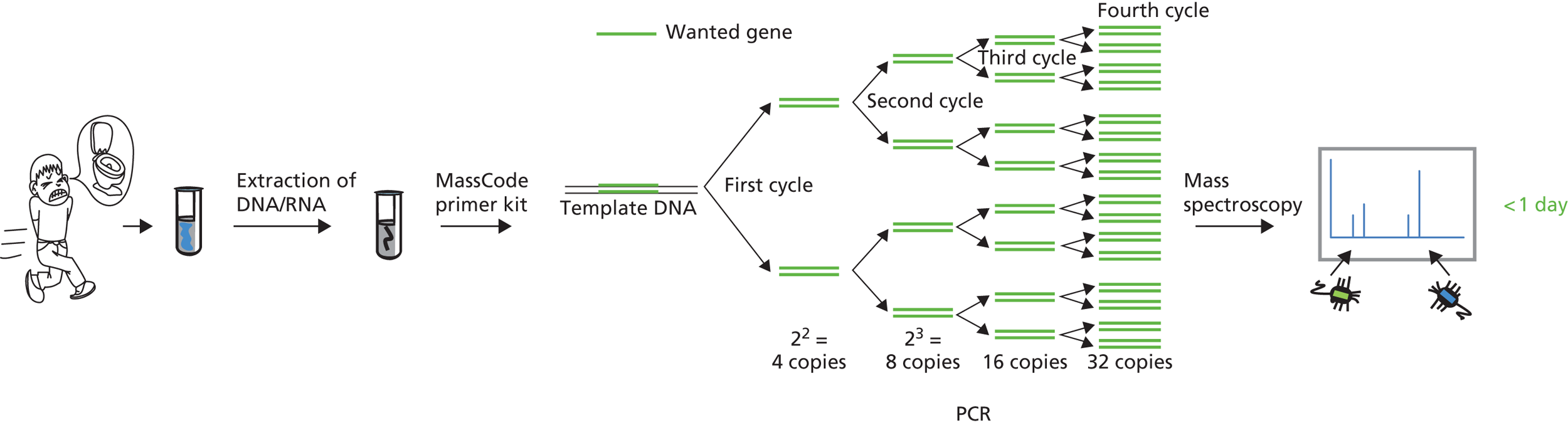
For suspected infectious diarrhoea, the technology tests a stool sample against a panel of pre-specified gastrointestinal pathogens. The panel consists of a pair of primers (short runs of nucleotides unique to each pathogen) for each of the multiple pathogens (Figure 3). Each primer is coded up with a unique tag of different mass; thus, there are up to 60 tagged primers in each reaction. The panel is combined with the patient sample, and rapid endpoint PCR then selectively amplifies any pathogen deoxyribonucleic acid (DNA) in the original patient sample. The amplified DNA is identified by reading the corresponding mass tags (which remain attached to the primers incorporated in the amplified DNA) in a mass spectrometer. These increasingly simple instruments are very sensitive and can easily detect and differentiate between the specially designed mass tags, in a matter of seconds. Simple software analyses the instrument’s output and reveals the presence of any pathogens in the original sample. A sample is positive for a pathogen when both the tags (from the forward and reverse primers) are detected above the threshold value (Figure 4). The low concentrations that can theoretically be detected in this system means that fewer rounds of PCR amplification are needed, which substantially reduces the reaction time and leads to a very rapid turnaround.
FIGURE 3.
MassCode tagged primers. UV, ultraviolet.
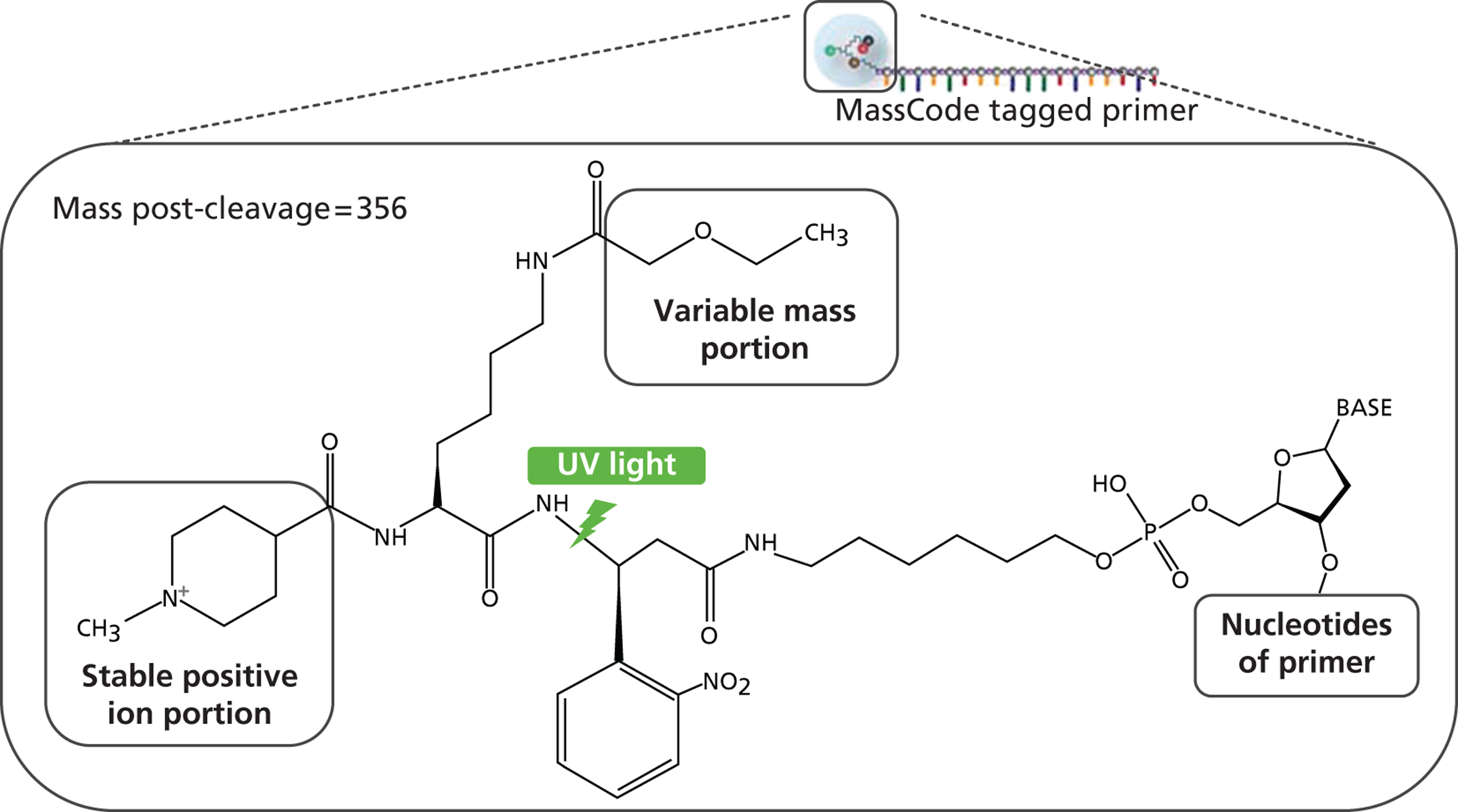
FIGURE 4.
Output from MassCode. Note: where detection of both forward and reverse primers exceeds pre-specified limits (blue lines), the sample is positive for that pathogen. Ccoli, C. coli; Cdiff, C. difficile; Cjej, C. jejuni; Crypto, Cryptosporidium; IAC-MS2, internal control MS2 bacteriophage; Noro2, norovirus; Rota, rotavirus; Sent, S. enterica; Shig, Shigella.
The gastrointestinal/food poisoning panel evaluated in this project included 11 primer pairs targeted at four major pathogens (the primary focus of the evaluation):
-
Clostridium difficile
-
Campylobacter jejuni and Campylobacter coli
-
Salmonella enterica
-
norovirus.
Plus an additional six less common pathogens:
-
Salmonella Typhi/Paratyphi
-
Giardia lamblia
-
Cryptosporidium
-
rotavirus A/B/C
-
Shigella spp.
-
Escherichia coli O157.
This panel is modifiable and could have been updated to include new pathogen strains as they were discovered. It already included many of the key high-burden or -impact pathogens targeted for surveillance by the Health Protection Agency (adenovirus, astrovirus, Clostridium botulinum, calicivirus, Campylobacter jejuni and C. coli, Cryptosporidium, E. coli O157, Giardia lamblia, norovirus, Salmonella enteritidis, S. Typhimurium, S. Typhi, S. Paratyphi A and S. Paratyphi B).
The product for MassCode PCR is a kit containing the primer mix for incorporation in the PCR. DNA extraction, and preparation of the sample for amplification including, where necessary, reverse transcription for ribonucleic acid (RNA) viruses needs to be undertaken as preliminary steps.
Wider impact on the NHS
Accurate multiplexed assays for diagnosing gastrointestinal infections could have a major impact on patients in NHS hospitals by achieving the early recognition of infectious diarrhoea. This would substantially improve the care of patients by ensuring that only appropriate patients are kept in isolation and, as a consequence, could not only reduce the transmission of enteropathogens but also greatly improve the use of single rooms by reducing their unnecessary use for non-infectious diarrhoea cases awaiting results of tests to rule out infectious diarrhoea. However, despite their potential advantages, whether or not such a test will actually deliver cost-effective improvements in patient management and outcomes, and whether or not it can be generalised across the NHS is unknown. In particular, the key risk is that the PCR test identifies a high proportion of patients with colonisation rather than infection (true ‘colonisation positive’, false ‘infection positive’) which could lead to unnecessarily increased anxiety for patients, considerable additional unnecessary treatment costs, and also increase (rather than decrease) pressure on side rooms. Rolling out such new technology across the NHS requires an evidence base covering both costs to microbiology service and benefits to patients.
Objectives
This diagnostic test study was therefore designed to evaluate a newly developed technology – MassCode multiplex PCR – for the simultaneous diagnosis of multiple enteropathogens directly from stool, in terms of core metrics of performance:
-
sensitivity/specificity and real-time predictive values to detect a range of pathogens and overall to rule out any infectious causative agent
-
turnaround time (speed of diagnosis), net health-care costs and utilisation of isolation resources to assess whether or not it can improve hospital management of patients with suspected infectious diarrhoea, in particular by avoiding/reducing isolation of patients with non-infectious diarrhoea.
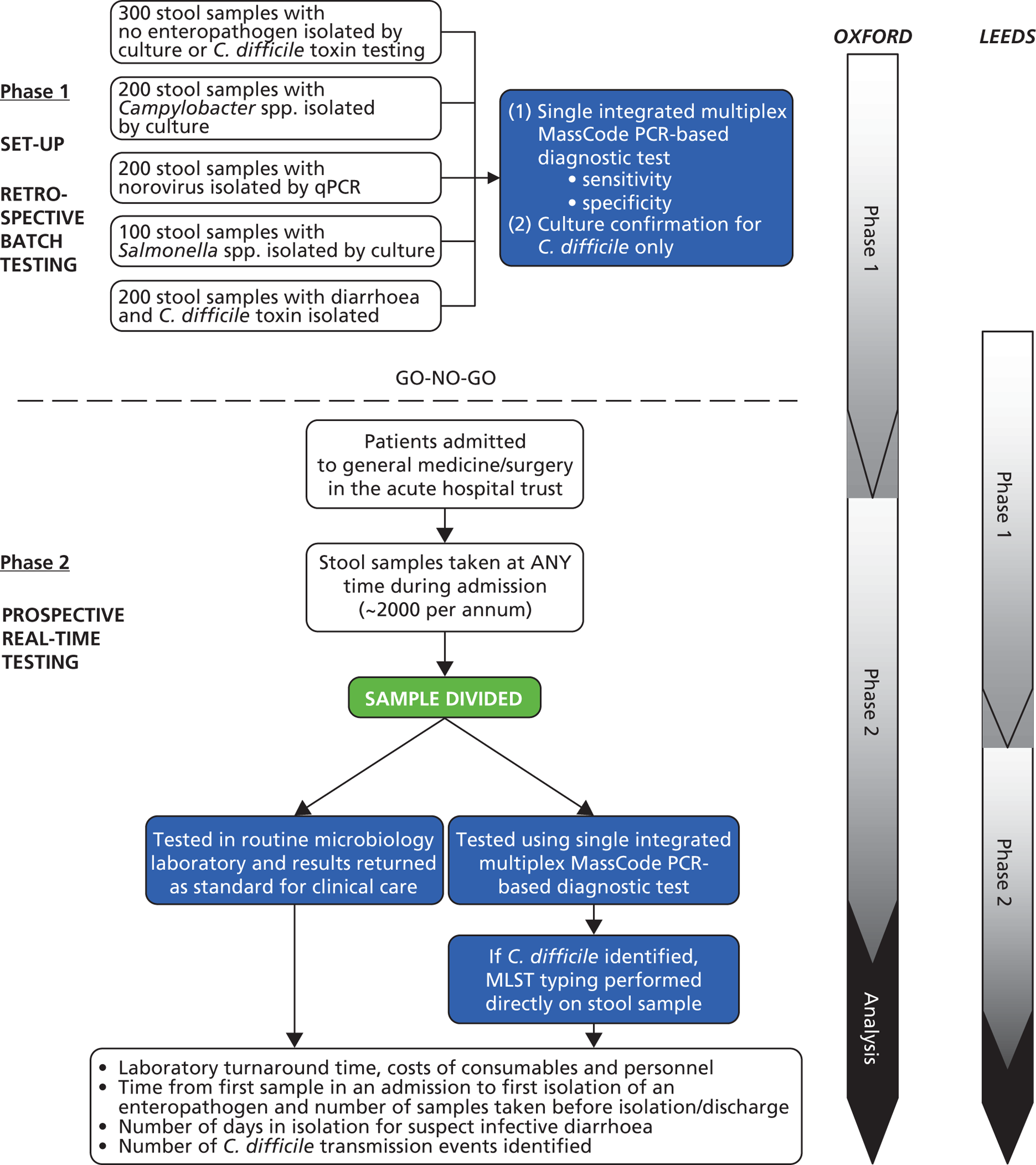
These objectives were to be addressed by a two-stage study in two hospitals (Leeds Teaching and Oxford University Hospitals NHS Trusts) (Figure 5). Phase 1 was a retrospective batch study based on fixed numbers of samples positive and negative for C. difficile, Campylobacter spp., Salmonella spp. and norovirus to estimate sensitivity/specificity of the MassCode test against these major pathogens. If MassCode met pre-specified criteria indicating it had the potential to be a successful test for the NHS, phase 2 was intended to be a prospective real-time parallel-group study testing the same stool samples from general medicine and surgery in both the routine microbiology laboratory and by the new technology to estimate positive/negative predictive values in a real-world setting and to directly compare turnaround time, net health-care costs and patient-centred outcomes (utilisation of isolation resources, detection of outbreaks). Phase 1 was supplemented by a parallel health economic study examining current practice and costs of managing infectious diarrhoea for infection control teams and microbiology laboratories; health economic analyses in phase 2 were intended to estimate the cost-effectiveness of MassCode multiplex PCR using a semi-Markov model and diagnostic decision tree.
FIGURE 5.
Planned study. MLST, multilocus sequence type.
Patient and public involvement
Patient and public involvement in this diagnostic study for gastrointestinal pathogens was via the public representative on the independent oversight committee, Katherine Innes Ker. As no individual patient data were collected, ethical approval was granted for the study without requiring individual patient consent.
Chapter 2 MassCode characterisation and optimisation
Introduction
Traditional methods for the detection of pathogens from faecal samples are time-consuming and may have poor sensitivities. 8,9 Owing to the varied aetiology of diarrhoea, a wide range of investigative methods are sometimes required. This includes culture, microscopy and serology. Subsequent testing, for example after initial culture, is also sometimes necessary for phenotypic classification and to gain antimicrobial sensitivity profiles. Molecular methods provide an alternative with the potential to reduce time taken to identify the causative agent of diarrhoea, and to increase the sensitivity of testing for some micro-organisms. For this reason, recent years have seen an increase in the number of large-scale multiplex PCR-based diagnostic methods being developed for enteric pathogens, including the MassCode assay, as well as a range of novel molecular methods aimed at improving diagnosis. 8–10
All laboratory tests for infectious organisms have a lower limit at which the target organism can be detected, required to be lower than reported concentrations of pathogens in stool samples. Liu et al. 10 report concentrations of 103–109 colony-forming units (CFUs)/gram of stool for bacterial pathogens, 103–105 CFU/g for protozoa and 104–1011 CFU/g for viruses. The limit of detection of the specific test will affect overall sensitivity of the assay compared with reference standard. Molecular tests also have a dynamic range, meaning that efficiency of detection of each target may alter according to the concentration of the target or the ratio of each target if more than one is present. Users must be aware of these assay characteristics, so they can interpret results as accurately as possible in light of the clinical presentation of the patient. Quantification of the limits of detection for MassCode was, therefore, one of the first aims of this investigation.
Faecal samples are acknowledged as among the most challenging to handle for nucleic acid-based assays, as they contain nucleic acids from many other sources, including human DNA and non-target bacteria, and contain many PCR inhibitors. 11,12 Interactions between non-target DNA and primers within the panel also increase as more primers are added to construct a large-scale multiplex such as MassCode. These influences may cause both false-positive and false-negative results when analysed by PCR-based methods. Testing faecal samples under controlled conditions and with known positive samples was, therefore, the first step in using the MassCode gastric primer panel with clinical samples.
The nucleic acid extraction method chosen for use with the MassCode assay should minimise inhibition and be suitable for isolation of bacterial and viral DNA and RNA in one reaction. This is of particular importance where, as with MassCode, nucleic acid extraction methods are user defined rather than being part of a single machine which processes and tests samples in one system. Development of standardised workflows for MassCode, which could be used in its wider rollout across the NHS, were therefore one important goal of this project. Other considerations affecting selection of nucleic acid extraction methods included the basic cost and time input to process the samples and whether or not any specialist equipment would be required by clinical laboratories. Validation of the optimised method was also performed, to ensure that when adopted for diagnosis the assay is robust.
The protocol for evaluating the large-scale multiplex PCR-based MassCode gastric panel included a retrospective investigation of faecal samples (phase 1), followed by a prospective real-time investigation (phase 2). Before implementing phase 1, pre-phase testing aimed to characterise the MassCode assay and standardise its workflow, taking into account the factors discussed above. These tests provided the opportunity to feedback the performance of the MassCode assay to Agilent, and optimise the sample preparation method and assay primer panel before instigating phases 1 and 2 of the investigation.
Methods
Participants
Samples were collected according to the MassCode standard operating procedures (SOPs) (see Appendix 1). In brief, all samples collected were initially sent for faecal culture and/or C. difficile toxin testing at the Oxford University Hospitals microbiology laboratory by hospital-based doctors or general practitioners (GPs) as a result of a suspected enteric infection. For negative samples, no pathogens were found using standard microbiological workflows and the samples were sent for discard. For known positive samples, one or more pathogens were found and the samples were sent for discard once testing was complete. The service microbiology laboratory in the Oxford University Hospitals uses an enzyme immunoassay (EIA) to identify C. difficile, which is well recognised to have suboptimal sensitivity and specificity. Separately to the MassCode study, all EIA-positive C. difficile samples were cultured in the parallel research laboratory, and only those positive for C. difficile on both EIA and culture were used as reference positives in this study. At this point, both sample groups were completely anonymised and collected for this investigation. As norovirus testing was not carried out by the microbiology laboratory unless an outbreak was suspected, positive norovirus samples were obtained through a separate investigation conducted by a National Institute for Health Research (NIHR) clinical fellow. These samples had been confirmed positive by PCR13,14 and were anonymised prior to collection. All samples were stored at 4 °C. For limit of detection testing, negative samples containing adequate quantities of faecal matter were selected at random and homogenised.
Reference standard
Initial diagnosis of faecal pathogens was performed according to Health Protection Agency (now Public Health England) guidelines. These are the current gold standard methods for investigating faecal specimens for enteric pathogens in the UK. Readers of the reference standard tests were qualified laboratory staff. Throughout all reference standard and index tests normal aseptic microbiological laboratory working practices were followed.
Limits of detection
Limit of detection experiments were designed and implemented for the four main bacterial targets of interest in the MassCode panel: C. difficile, S. enterica, C. jejuni and C. coli. As norovirus cannot be cultured it was not included in these experiments. National Collection of Type Cultures (NCTC) strains of each of the bacterial organisms were obtained. These were C. difficile (NCTC 13307), C. jejuni (NCTC 11168), C. coli (NCTC 11353) and S. enterica serovar Enteritidis (NCTC 13349). Each organism was cultured on Columbia blood agar (E&O Laboratories Ltd, Bonnybridge, UK). C. difficile was incubated for 48 hours at 37 °C under anaerobic conditions; S. enterica for 24 hours at 37 °C under aerobic conditions; and C. jejuni and C. coli for 24 hours at 42 °C under microaerophilic conditions. Resulting plates of growth were scraped into 200 µl of nutrient broth with 10% glycerol (E&O Laboratories Ltd). Serial 1 : 10 dilutions were made from the neat sample to 10–15. Aliquots of 50 µl of each serial dilution were spread onto blood agar plates and incubated under the conditions previously described for each organism. Colonies were enumerated and the cultivable range selected for subsequent limit of detection experiments. Culture aliquots were stored at –80 °C prior to use.
Negative stool samples from five patients were initially chosen for use in the limit of detection testing across the different pathogens. Aliquots of 200 µl or 0.2 mg of faecal matter were placed into clean 2-ml vials and spiked with a known volume of culture stock from the previously described serial dilutions. In accordance with the MassCode SOPs (see Appendix 1), 8 µg of yeast transfer RNA (tRNA; Invitrogen, Carlsbad, CA, USA) carrier RNA and 105 MS2 bacteriophage internal control (IC) were also added at this stage. Following preparation of the sample, nucleic acids were extracted and purified with the QIAamp DNA Stool Mini Kit (Qiagen, Limburg, the Netherlands) following the manufacturers protocol. This kit was chosen because of its specific design for processing faecal samples, and ability to isolate both DNA and RNA from bacteria and viruses, since in clinical practice whether a stool sample will contain a bacteria or a virus is unknown. Thus, processing workflows have to be sufficiently robust and sensitive for both types of pathogen.
Eluted DNA (10 µl) was reverse transcribed using MassCode Single Strand complementary DNA (cDNA) Synthesis Kits (Agilent, Santa Clara, CA) to enable detection of the MS2 IC; as well as to ensure any other RNAs were converted to cDNA in preparation for the method being used to detect pathogens such as norovirus in the future. All samples were stored at –20 °C prior to testing. Control samples included the patient stool samples without spiked culture for all organisms, and samples negative for contaminating human and other DNA (molecular-grade water).
MassCode sample processing and analysis was performed according to the SOPs described (see Appendix 1). A 14-plex primer mix targeting MS2, C. difficile, S. enterica, C. jejuni, C. coli, norovirus (two targets), E. coli O157 (two targets), Shigella spp., S. Typhi/Paratyphi, Giardia spp., Cryptosporidium spp. and rotavirus was used. Quantitative PCR (qPCR) assays using the same primer sequences as the MassCode panel, with the inclusion of a dual-labelled probe (Table 1), were used to verify unexpected results.
| Organism | Target gene | Primer or probe | Sequence (5’ → 3’) | Tm (°C) | Amplicon size (bp) | Source |
|---|---|---|---|---|---|---|
| MS2 (IC) | lys/rep | Forward primer | CGCGATCTTTCTCTCGAAAT | 49.7 | 317 | Agilent |
| Reverse primer | GACGATCGGTAGCCAGAGAG | 55.9 | Agilent | |||
| Probe | FAM-TCGCTACTGGAAGCGGTGATCCGCAC-BHQ1 | 64.3 | In-house | |||
| C. difficile | tcdB | Forward primer | ACTTCCTACATTATCTGAAGGATTACCT | 55.5 | 110 | Agilent |
| Reverse primer | GTCTTAATAATGGGTCACTHGTTTCACT | 55.5–57.0 | Agilent | |||
| Probe | ROX-ATAGATGGTGTAAGTTTAGGTGCAGCAATCAAAGAGCT-BHQ2 | 63.4 | In-house | |||
| S. enterica | invA | Forward primer | GTTGAGGATGTTATTCGCAAAGG | 53.5 | 75 | Suo et al.15 |
| Reverse primer | GGAGGCTTCCGGGTCAAG | 54.9 | Suo et al.15 | |||
| Probe | JOE-CCGTCAGACCTCTGGCAGTACCTTCCTC-BHQ1 | 65.8 | Suo et al.15 | |||
| C. jejuni | Cj0414 | Forward primer | CTGAATTTGATACCTTAAGTGCAGC | 54.4 | 86 | Nogva et al.16 |
| Reverse primer | AGGCACGCCTAAACCTATAGCT | 54.8 | Nogva et al.16 | |||
| Probe | ROX-TCTCCTTGCTCATCTTTAGGATAAATTCTTTCACA-BHQ2 | 59.7 | Nogva et al.16 | |||
| C. coli | ceuE | Forward primer | ACGCGCACAAGGCATACTT | 51.1 | 91 | Fukushima et al.17 |
| Reverse primer | CCAGTATTCAGGATCAAGATAAATGATTT | 54.4 | Fukushima et al.17 | |||
| Probe | Cy5-TGCACTTGTAACTAAAACCAACGCTGCTACAAAT-BBQ650 | 60.8 | In-house | |||
| Norovirus 1 | ORF1 | Forward primer | TCGTGGCTGAGTAGGAGAAT | 51.8 | 311 | Agilent |
| Reverse primer | GCAATCATCGCAGACACATC | 51.8 | Agilent | |||
| Probe | JOE-AGAGTTCTGCCAAGAGTATCAGGGACTCCACTATGCC-BHQ1 | 67.8 | In-house | |||
| Norovirus 2 | RdRP/VP1 | Forward primer | ATCGCAATCTGGCTCCCAGTTT | 54.8 | 119 | Agilent |
| Reverse primer | GGCTCCAAAGCCATAACCTCAT | 54.8 | Agilent | |||
| Probe | Cy5-TCGAGTGACGCCAACCCATCTGATGGGTCCACAGCC-BBQ650 | 71.3 | In-house | |||
| E. coli | uidA | Forward primer | AAAACGGCAAGAAAAAGCAG | 47.7 | 147 | Bej et al.18 |
| Reverse primer | AYGCGTGGTTACAGTCTTGCG | 54.4–56.3 | Bej et al.,18 modified in-house | |||
| Probe | ROX-TGGACGATATCACCGTGGTGACGCA-BHQ2 | 61 | In-house | |||
| Klebsiella pneumoniae | khe | Forward primer | GATGAAACGACCTGATTGCATTC | 53.5 | 77 | Hartman et al.19 |
| Reverse primer | CCGGGCTGTCGGGATAAG | 54.9 | Hartman et al.19 | |||
| Probe | Cy5-CGCGAACTGGAAGGGCCCG-BBQ650 | 80.7 | Hartman et al.19 |
Quantitative PCRs were performed with Brilliant Multiplex MasterMix (Agilent). Each target organism was tested for in a duplex reaction with MS2. In addition, C. jejuni and C. coli could be tested for in triplex with MS2; the two norovirus gene targets could also be tested for in triplex with MS2. In-house optimisation for each qPCR assay was performed and the optimised primer and probe concentrations were used for each reaction. All qPCR runs included a 1 : 10 standard curve from 108 copies of the target sequence to one copy, plus a positive control for MS2 (104 copies of target sequence) and no template controls.
Product (2 µl) from the cDNA reaction was amplified in a reaction mixture consisting of target primers (forward and reverse primers), target probes, 12.5 µl of Multiplex Mastermix and nuclease-free water to a final volume of 25 µl. Amplification and reading was carried out with the Stratagene MX3005P® (Agilent) under the following conditions:
95 °C for 10 minutes95 °C for 15 seconds60 °C for 1 minute}× 40
Following qPCR analysis, copy numbers for each culture serial dilution were generated and averaged across the five faecal samples. For MassCode, the lower limit of detection was defined as the sample CFU concentration at which more than half of the spiked stool samples (i.e. ≥ 3/5) could be detected, although the concentration at which all five of the spiked samples could be detected was also estimated. For qPCR, the lower limit of detection was defined as the sample CFU concentration at which copy numbers could be reliably calculated according to the standard curve included on each assay.
Optimisation of primer panel
Concurrently with limit of detection testing, the MassCode primer panel was tested against a set of anonymised clinical samples, collected as described above, retrieved essentially at random from laboratory discards. The laboratory worker was unblinded to the expected pathogens present in each sample to allow parallel processing of the samples by MassCode and the appropriate single qPCR assay(s). Samples were extracted with the QIAamp DNA Stool Mini Kit and reverse transcribed according to the MassCode SOP; samples were then stored at –20 °C prior to analysis. A total of 57 samples were S. enterica positive, 122 C. difficile positive, 119 Campylobacter spp. positive and 109 norovirus positive; 127 samples were negative.
MassCode testing was performed with the 14-plex primer mix. Samples that were unexpectedly positive or negative for target organisms were retested by qPCR. Where dual-labelled probe assays were not available, samples were tested with SYBR® Green (Life Technologies, Carlsbad, CA, USA) qPCR under the following conditions: Product (2 µl) from the cDNA reaction was amplified in a reaction mixture consisting of target primers (forward and reverse primers), 10 µl SYBR Green MasterMix, and nuclease-free water to a final volume of 20 µl. Amplification and reading was carried out with the Stratagene MX3005 under the following conditions:
95 °C for 10 minutes95 °C for 15 seconds60 °C for 1 minute}× 40
Dissociation curve 95 °C for 1 minute followed by 55 °C (for 30 seconds) ramping to 95 °C (for 30 seconds).
The sensitivity of the MassCode assay was determined through calculation of the percentage of positive samples found by MassCode compared with the expected number of positives as previously determined by conventional laboratory methods. Specificity was determined by calculating the percentage of genuine negatives that were called as negative by MassCode.
A subinvestigation was also carried out to explore other options for S. enterica targets. The primer sets and conditions used were as published by Liu et al. ,9 targeting invA, and Malorny et al. ,20 targeting ttrRSBCA. Twenty S. enterica-positive samples, collected according to the protocol above, were tested by the Liu et al. 9 primer set; and 64 by the Malorny et al. 20 primer set.
Extraction optimisation
Following initial limit of detection and primer panel optimisation testing as performed above, a series of experiments designed to optimise nucleic acid extraction and purification were performed. The organisms chosen for optimisation experiments were C. difficile and S. enterica.
A homogenised negative stool sample, collected as described above, was used in 200-µl aliquots. Stool aliquots were spiked with 1 : 10 serial dilutions of culture above, at and below the limit of detection, as previously determined. The prevalent methods for nucleic acid purification were explored with a variety of pre-steps to facilitate lysis. Chemical lysis was carried out using Qiagen’s QIAamp DNA Stool Kit buffer ATL, and Stool Transport and Recovery (STAR) buffer plus chloroform (Roche, Basel, Switzerland). Physical lysis was carried out through heating, freeze–thaw, bead beating and sonication. Purification focused on the three major techniques of silica membrane spin column (QIAamp DNA Stool Kit), magnetic bead (MagMax, Life Technologies) and precipitation-based purification (Masterpure, Epicentre, Madison, WI, USA). In addition, two automated extraction platforms were tested: the magnetic bead-based QIAsymphony (Qiagen) and silica membrane Corbett (QIAxtractor) systems (Qiagen).
All samples were reverse transcribed according to the MassCode SOP post extraction and stored at –20 °C before qPCR analysis as described above (see Table 1). Control samples included stool samples without spiked culture for all extraction methods and negative samples (molecular-grade water) for all extraction methods. Quantitative PCRs were performed with the assays and controls described in Limits of detection.
QIAamp protocol
Aliquots of 200 µl of stool were spiked with C. difficile or S. enterica, carrier RNA and IC. As well as following the supplier’s protocol, the following adjustments were implemented. For pre-treatment by freeze–thaw, samples were placed at –80 °C until frozen and then allowed to thaw before proceeding with the extraction. For pre-treatment by bead beating, samples were placed into Lysing Matrix E tubes (MP Biomedicals, Calsbad, CA, USA) and beaten twice at 6 m/s for 40 seconds. For pre-treatment by sonication samples were sonicated (Ultrawave sonicator bath, Cardiff, UK) for 5 minutes on the maximum setting at room temperature. Samples processed by all pre-treatments were also subject to the supplier’s protocol steps of chemical (buffer ASL, Qiagen) and heat (5 minutes at 95 °C) lysis. The standard protocol plus bead beating pre-treatment was also tested both with and without the InhibitEX tablet (Qiagen) step.
Following lysis and the manufacturer’s protein and inhibitor removal steps, the nucleic acids isolated via this kit were purified using the supplied silica filter spin columns. In one further modification to the manufacturer’s protocol, nucleic acids were eluted in 100 µl of molecular-grade water at 55 °C for 10 minutes prior to centrifugation and collection of the eluate.
MagMax protocol
Aliquots of 200 µl of stool were spiked with C. difficile or S. enterica, carrier RNA and IC. The supplier’s protocol was followed in addition to the following adjustments. For pre-treatment by freeze–thaw, samples were placed at –80 °C until frozen and then allowed to thaw before proceeding with the extraction. For pre-treatment by sonication, samples were treated for 5 minutes on the maximum setting. For pre-treatment by heating, samples were heated at 99 °C for 10 minutes. Samples processed by all pre-treatments were also subject to the supplier’s protocol lysis steps, which were chemical (Lysis/Binding solution, Life Technologies) and physical (bead beating). Bead beating was carried out according to the manufacturer’s protocol with two rounds of 6.5 m/s beating for 1 minute.
Following lysis, samples were processed following the manufacturer’s purification process, using the supplied nucleic acid-binding beads. Nucleic acids were eluted in 50 µl of molecular-grade water heated to 65 °C, in accordance with to the manufacturer’s instructions.
Masterpure protocol
Aliquots of 200 µl of stool were spiked with C. difficile or S. enterica, carrier RNA and IC. The supplier’s protocol was followed in addition to the following adjustments. For pre-treatment by freeze–thaw, samples were placed at –80 °C until frozen and then allowed to thaw before proceeding with the extraction. For pre-treatment by bead beating, samples were placed into Lysing Matrix E tubes (Epicentre) and beaten twice at 6 m/s for 40 seconds. For pre-treatment by sonication, samples were sonicated for 5 minutes on the maximum setting. For pre-treatment by heating, samples were heated at 99 °C for 10 minutes. Samples processed by all pre-treatments were also subject to the supplier’s protocol chemical lysis step with Tissue and Cell Lysis Solution (Epicentre).
Following the manufacturer’s protocol, nucleic acid purification was carried out via DNA precipitation, pelleting and wash steps prior to resuspension of the nucleic acid pellet in 35 µl of molecular-grade water.
Automated extractions
Aliquots of 200 µl of stool were spiked with C. difficile or S. enterica and IC, carrier RNA was also spiked into samples extracted with the Corbett system (Qiagen). For the QIAsymphony, STAR buffer with chloroform or ASL buffer, followed by bead beating in Lysing Matrix E was used as a pre-treatment prior to extraction with the virus/pathogen midi kit under the Complex 400 protocol (Qiagen). Final elution volume was 60 µl. Yeast tRNA was not used for QIAsymphony samples, as the kit is provided with carrier RNA. For the Corbett automated method, STAR buffer with chloroform was used as a pre-treatment step, in accordance with an existing protocol for extraction with the DX reagent pack (Qiagen).
Following the pre-step, samples were centrifuged and the supernatant transferred to the corresponding automated system.
Method validation
After optimisation of the primer panel and extraction methods, the MassCode assay was challenged against a batch of known positive clinical samples. All samples were reverse transcribed according to the MassCode SOP post extraction and stored at −20 °C before qPCR analysis as described above (see Table 1). A total of 20 samples positive for Campylobacter spp., 20 positive for S. enterica, 20 positive for C. difficile and 40 norovirus-positive samples were processed by the MassCode assay and by qPCR, where sufficient material remained.
Extraction efficiency
Further experiments investigating the extraction efficiency of target organisms alongside other Enterobacteriaceae were performed. American Type Culture Collection (ATCC) specimens of E. coli (ATCC 700928) and Klebsiella pneumoniae (ATCC 700721) were obtained. Both were plated on blood agar and incubated for 24 hours at 37 °C. Serial dilutions were made and enumerated as described previously. Known volume spikes of serially diluted culture were extracted following the optimised protocol, with K. pneumoniae also spiked into homogenised negative stool as described above. Culture spiking extraction experiments were also repeated with C. difficile, S. enterica and C. jejuni using the optimised extraction protocol.
All samples were reverse transcribed according to the MassCode SOP post extraction and stored at −20 °C before qPCR analysis as described above (see Table 1).
Results
Limits of detection
Limit of detection testing was performed between December 2011 and March 2012. The negative stool samples used were collected in October 2011 immediately after reference standard testing from which no pathogens were found. Samples were completely anonymised, no patient information was obtained.
The results for limit of detection testing are shown in Table 2. MassCode limits of detection for spiked stool were determined to be 2.5 × 103 CFU/µl for C. difficile, 2.5 × 104 CFU/µl for S. enterica, 2.5 × 103 CFU/µl for C. jejuni and 1.2 × 102 CFU/µl for C. coli. Quantitative PCR limits of detection for spiked stool were one order of magnitude lower for all organisms other than C. jejuni, where limits of detection were two orders of magnitude lower with qPCR. Fewer target copies were found and limits of detection were typically higher for pure culture than for spiked stools. Some evidence of contamination was observed within the S. enterica qPCR results.
| Organism | Spike (total CFU) | qPCR | MassCode | ||
|---|---|---|---|---|---|
| Spiked stool (mean copies) (n = 5) | SD | No. of positives (n = 5) | No. of positives; including indeterminate positive samples (n = 5) | ||
| C. difficile | 250,000 | 45,120 | 15,934 | 5 | 5 |
| 25,000 | 4408 | 1148 | 5 | 5 | |
| 2500 | 371 | 138 | 5 | 5 | |
| 250 | 24 | 4 | 0 | 0 | |
| 65 | 2 | 0.4 | 0 | 0 | |
| 8 | 0.2 | 0.5 | 0 | 0 | |
| 1 | 0.1 | 0.2 | 0 | 0 | |
| 0.5 | BDL | 0 | 0 | 0 | |
| 0.5 | BDL | 0 | 0 | 0 | |
| 0 | 2 | 4 | 0 | 0 | |
| S. enterica | 2,500,000 | 2998 | 1176 | 5 | 5 |
| 250,000 | 305 | 150 | 5 | 5 | |
| 25,000 | 32 | 24 | 3 | 3 | |
| 2500 | 3 | 2 | 0 | 0 | |
| 252 | 0.5 | 0.6 | 0 | 0 | |
| 20 | 0.1 | 0.2 | 0 | 0 | |
| 2.5 | 0.03 | 0.06 | 0 | 0 | |
| 1 | BDL | 0 | 0 | 0 | |
| 0 | BDL | 0 | 0 | 0 | |
| 0 | BDL | 0 | 0 | 0 | |
| C. jejuni | 25,000,000 | 965,600 | 366,897 | 5 | 5 |
| 2,500,000 | 120,580 | 39,190 | 5 | 5 | |
| 250,000 | 10,152 | 4552 | 5 | 5 | |
| 25,000 | 1348 | 681 | 5 | 5 | |
| 2500 | 137 | 83 | 3 | 4 | |
| 205 | 17 | 6 | 0 | 1 | |
| 24 | 2 | 2 | 0 | 0 | |
| 6 | BDL | 0 | 0 | 0 | |
| 0 | 0.5 | 1 | 0 | 0 | |
| 0 | BDL | 0 | 0 | 0 | |
| C. coli | 2,500,000 | 714,800 | 105,046 | 5 | 5 |
| 250,000 | 93,460 | 23,969 | 5 | 5 | |
| 25,000 | 9046 | 1785 | 5 | 5 | |
| 2500 | 1045 | 191 | 5 | 5 | |
| 122 | 60 | 24 | 5 | 5 | |
| 27 | 7 | 5 | 0 | 0 | |
| 0.5 | 0.2 | 0.5 | 0 | 0 | |
| 0 | 0 | 1 | 0 | 0 | |
| 0 | BDL | 0 | 0 | 0 | |
| 0 | BDL | 0 | 0 | 0 | |
Optimisation of primer panel
Processing and testing of known positive anonymised clinical samples was carried out between August 2011 and May 2012. The stool samples used were collected between November 2008 and October 2011. Collection parameters were as described earlier; no patient information was obtained.
Table 3 provides a summary of the results from initial clinical sample testing in August 2011. The percentage positive calls (sensitivity) varied between MassCode and qPCR. For MassCode, sensitivities varied between 40% for S. enterica and 83% for Campylobacter spp. For qPCR, sensitivities varied between 72% for S. enterica and 97% for C. difficile.
| Organism | qPCR | MassCode | MassCode (including indeterminate positive samples) |
|---|---|---|---|
| C. difficile | |||
| Number tested | 122 | 122 | 122 |
| Number positive | 118 | 57 | 62 |
| Number negative | 4 | 65 | 60 |
| % positive (sensitivity) | 97 | 47 | 51 |
| S. enterica | |||
| Number tested | 57 | 57 | 57 |
| Number positive | 41 | 23 | 25 |
| Number negative | 16 | 34 | 32 |
| % positive (sensitivity) | 72 | 40 | 44 |
| Campylobacter spp. | |||
| Number tested | 119 | 119 | 119 |
| Number positive | 114 | 99 | 103 |
| Number negative | 5 | 20 | 16 |
| % positive (sensitivity) | 96 | 83 | 87 |
| Norovirus | |||
| Number tested | 109 | 109 | 109 |
| Number positive | 98 | 67 | 68 |
| Number negative | 11 | 42 | 41 |
| % positive (sensitivity) | 90 | 62 | 62 |
Table 4 breaks down these results by copy number as determined by qPCR. For C. difficile, 95% (62/65) of the samples undetected by MassCode had copy numbers below 100, but only 6% (4) were below detection limits (BDLs) for qPCR. For Campylobacter spp. and norovirus, all samples undetected by MassCode had copy numbers below 100, while 85% (17/20) and 90% (38/42) of samples, respectively, had copy numbers below 10; however, 25% (5) and 26% (11) were also not detected by qPCR, respectively. For S. enterica, 91% (31/34) of samples undetected by MassCode had copy numbers below 100 and 47% were also BDLs for qPCR.
| Organism | qPCR categories (copies) | No. (column %) | MassCode+ (row %) | MassCode+ including indeterminate positive samples (row %) | MassCode– (row %) | MassCode– excluding indeterminate positive samples (row %) |
|---|---|---|---|---|---|---|
| C. difficile | BDL | 4 (3) | 0 (0) | 0 (0) | 4 (100) | 4 (100) |
| 0–9 | 16 (13) | 0 (0) | 0 (0) | 16 (100) | 16 (100) | |
| 10–99 | 51 (42) | 9 (18) | 14 (27) | 42 (82) | 37 (73) | |
| 100–999 | 34 (28) | 31 (91) | 31 (91) | 3 (9) | 3 (9) | |
| 1000–9999 | 13 (11) | 13 (100) | 13 (100) | 0 (0) | 0 (0) | |
| 10,000–99,999 | 4 (3) | 4 (100) | 4 (100) | 0 (0) | 0 (0) | |
| > 100,000 | 0 (0) | |||||
| S. enterica | BDL | 16 (28) | 0 (0) | 0 (0) | 16 (100) | 16 (100) |
| 0–9 | 9 (16) | 0 (0) | 0 (0) | 9 (100) | 9 (100) | |
| 10–99 | 9 (16) | 3 (33) | 4 (44) | 6 (67) | 5 (56) | |
| 100–999 | 8 (14) | 7 (88) | 7 (88) | 1 (12) | 1 (12) | |
| 1000–9999 | 7 (12) | 7 (100) | 7 (100) | 0 (0) | 0 (0) | |
| 10,000–99,999 | 7 (12) | 5 (71) | 6 (86) | 2 (29) | 1 (14) | |
| > 100,000 | 1 (2) | 1 (100) | 1 (100) | 0 (0) | 0 (0) | |
| Campylobacter spp. | BDL | 5 (4) | 0 (0) | 0 (0) | 5 (100) | 5 (100) |
| 0–9 | 14 (12) | 2 (14) | 3 (21) | 12 (86) | 11 (79) | |
| 10–99 | 11 (9) | 8 (73) | 11 (100) | 3 (27) | 0 (0) | |
| 100–999 | 28 (24) | 28 (100) | 28 (100) | 0 (0) | 0 (0) | |
| 1000–9999 | 30 (25) | 30 (100) | 30 (100) | 0 (0) | 0 (0) | |
| 10,000–99,999 | 20 (17) | 20 (100) | 20 (100) | 0 (0) | 0 (0) | |
| > 100,000 | 11 (9) | 11 (100) | 11 (100) | 0 (0) | 0 (0) | |
| Norovirus | BDL | 11 (10) | 0 (0) | 0 (0) | 11 (100) | 11 (100) |
| 0–9 | 27 (25) | 0 (0) | 1 (4) | 27 (100) | 26 (96) | |
| 10–99 | 11 (10) | 7 (64) | 7 (64) | 4 (36) | 4 (36) | |
| 100–999 | 18 (17) | 18 (100) | 18 (100) | 0 (0) | 0 (0) | |
| 1000–9999 | 10 (9) | 10 (100) | 10 (100) | 0 (0) | 0 (0) | |
| 10,000–99,999 | 14 (13) | 14 (100) | 14 (100) | 0 (0) | 0 (0) | |
| > 100,000 | 18 (17) | 18 (100) | 18 (100) | 0 (0) | 0 (0) |
Testing clinical samples also led to some unexpected positives, particularly among additional targets not included in phase 1 testing (Table 5). These were retested by qPCR where possible (if sufficient sample remained). The organisms most commonly found were Giardia spp., S. Typhi, and norovirus, with 61, 20, and 27 unexpected positives, respectively, (including indeterminate results) and most of these positives were found to be negative by qPCR testing, with 55, 20, and 22 of the samples confirmed as false positives, respectively.
| Organism (no. positive based on routine laboratory results) | No. of unexpected positives | No. of unexpected positives (including indeterminate positive samples) | No. confirmed positive by qPCR | No. confirmed negative by qPCR |
|---|---|---|---|---|
| C. difficile (119) | 3 | 8 | 3 | 5 |
| S. enterica (57) | 0 | 0 | 0 | 0 |
| C. jejuni (Campylobacter spp. 119) | 7 | 8 | 7 | 1 |
| C. coli (Campylobacter spp. 119) | 5 | 5 | 4 | 1 |
| Norovirus (108) | 12 | 27 | 5 | 22 |
| E. coli stx 1 (0) | 1 | 1 | 0 | 1 |
| E. coli stx 2 (0) | 0 | 1 | 0 | 1 |
| Shigella spp. (0) | 3 | 3 | 2 | 1 |
| S. Typhi (0) | 1 | 20 | 0 | 20 |
| Cryptosporidium spp. (0) | 7 | 12 | 3 | 9 |
| Giardia spp. (0) | 38 | 61 | 6 | 55 |
| Rotavirus (0) | 0 | 0 | 0 | 0 |
A number of samples were also found to be positive by MassCode and qPCR, but not by the reference standard tests. This included 11 samples positive for Campylobacter spp., five positive for norovirus and six positive for Giardia spp.
During the course of these experiments, it was also observed that the ‘norovirus 1’ (see Table 1) primer set underperformed compared with the ‘norovirus 2’ primer set. Norovirus 1 amplification was achieved if norovirus 2 was also positive, while norovirus 2 amplification could also occur alone.
Testing of S. enterica-positive samples by alternative primer sets revealed little difference between detection using the MassCode primer set and the published alternatives. In particular, the alternative invA assay by Liu et al. 9 produced near-identical results to those obtained by qPCR with MassCode primers. The Malorny et al. 20 2ttrRSBCA primer set detected 43 out of 64 samples, whereas the MassCode primer set detected 46 of the same sample batch. Of the 18 samples not detected by MassCode, all were also negative by the Malorny et al. 20 assay.
Extraction optimisation
Following limit of detection testing and primer panel optimisation, experiments were performed between May 2012 and January 2013 to optimise the isolation of nucleic acids from faecal samples. The organisms chosen for investigation were C. difficile and S. enterica. Anonymised negative stool samples were collected between October 2011 and December 2012, immediately after reference standard testing from which no pathogens were found. No patient information was obtained.
Recovery (copy number compared with CFU spiked into the stool sample) for C. difficile increased from mean 5% following the QIAamp DNA Stool Kit with standard protocol to mean 689% using the QIAsymphony with STAR buffer and bead beating pre-steps (Table 6). (Bead beating plausibly increases yields to > 100% because it liberates DNA from spores.) Extraction efficiencies saw less improvement for S. enterica across all methods tested. However, some improvement in copy number yield was found, increasing from mean 0.2% using the QIAamp DNA Stool kit with standard protocol to mean 1% with the QIAsymphony plus STAR buffer and bead beating pre-steps. Other protocols also saw increases in yield to levels up to and above 1%, but these were less consistent across the CFU concentrations of S. enterica used.
| Protocol (no. of repeats) | C. difficile mean % return (% SD) | S. enterica mean % return (% SD) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| QIAamp | MagMax | MasterPure | QIAsymphony | Corbett | QIAamp | MagMax | MasterPure | QIAsymphony | Corbett | |
| Standard protocol (15) | 5 (3) | 29 (26) | 0 (0) | N/A | N/A | 0.2 (0.2) | 0.23 (0.25) | 3.24 (5.61) | N/A | N/A |
| Bead beating (15) | 191 (166) | N/A | 0 (0) | N/A | N/A | 0.86 (0.53) | N/A | 0 (0) | N/A | N/A |
| Bead beating no InhibitEX (15) | 215 (336) | N/A | N/A | N/A | N/A | 0.49 (0.42) | N/A | N/A | N/A | N/A |
| Boiling (15) | N/A | 2 (2) | 4 (10) | N/A | N/A | N/A | 0.09 (0.08) | 0.55 (1.22) | N/A | N/A |
| Freeze–thaw (15) | 25 (21) | 73 (65) | 0 (0) | N/A | N/A | 0.31 (0.22) | 0.16 (0.18) | 0 (0) | N/A | N/A |
| Sonication (15) | 333 (216) | 81 (151) | 0 (0) | N/A | N/A | 0.39 (0.31) | 0.52 (0.62) | 0 (0) | N/A | N/A |
| STAR buffer bead beating (15) | N/A | N/A | N/A | 689 (371) | N/A | N/A | N/A | N/A | 0.96 (0.70) | N/A |
| ASL buffer bead beating (15) | N/A | N/A | N/A | 105 (120) | N/A | N/A | N/A | N/A | 0.21 (0.27) | N/A |
| STAR buffer (6) | N/A | N/A | N/A | N/A | 48 (7) | N/A | N/A | N/A | N/A | 0.01 (0.19) |
Method validation
Following optimisation of extraction methods, a small batch of anonymised positive clinical samples was used to validate the new extraction method. These samples were collected between July 2009 and June 2012, immediately after reference standard testing. Samples were processed and tested during June and July 2012. No patient details were collected.
The percentage of positive samples (sensitivity) determined by MassCode increased for Campylobacter spp. and C. difficile (Table 7). In particular, C. difficile sensitivity increased from 47% to 95%. The MassCode sensitivity for S. enterica was slightly lower. A second batch of norovirus samples was processed as a result of concerns regarding the validity of the positive reference standard test on the initial batch. Although it was not possible to process the second norovirus batch by MassCode, all samples were positive by qPCR.
| Organism (n) | Previous MassCode % positive (excluding indeterminate positive samples) | qPCR | MassCode | ||
|---|---|---|---|---|---|
| No. positive | % positive | No. positive | % positive (excluding indeterminate positive samples) | ||
| C. difficile (20) | 47 | 20 | 100 | 19 | 95 |
| S. enterica (20) | 40 | 12 | 60 | 1 | 5 |
| Campylobacter spp. (20) | 83 | 20 | 100 | 18 | 95 |
| Norovirus (20) | 62 | 16 | 80 | 13 | 65 |
| Norovirus 2 (20) | 62 | 20 | 100 | N/A | N/A |
Copy number comparisons between the QIAamp DNA Stool Kit and optimised extraction method were performed, where possible (Table 8). Mean copy number change was positive for each organism.
| Organism (no. of samples) | No. of samples with increased copy number | No. decreased copy number | No. newly detected | Mean % change |
|---|---|---|---|---|
| C. difficile (11) | 7 | 4 | 1 | 594 |
| S. enterica (19) | 19 | 0 | 1 | 2260 |
| Campylobacter spp. (18) | 12 | 6 | 0 | 69 |
| Norovirus (11) | 5 | 6 | 2 | 1539 |
Extraction efficiency
Extraction efficiencies of a panel of bacteria (C. difficile, C. jejuni, S. enterica, E. coli and K. pneumoniae) were evaluated through repeating limit of detection testing using 1 : 10 serial dilutions of culture and the optimised extraction method. Anonymised negative stool samples were collected between October 2011 and December 2012, immediately after reference standard testing from which no pathogens were found. No patient information was obtained.
All Gram-negative Enterobacteriaceae (S. enterica, E. coli and K. pneumoniae) had mean extraction efficiencies below 1%, whereas C. jejuni had an average extraction efficiency of 5% (Table 9). The Gram-positive bacterium, C. difficile, had the highest extraction efficiency at 625% on average.
| Organism (no. of samples) | Median | Mean | Maximum | Minimum | SD |
|---|---|---|---|---|---|
| C. difficile (24) | 557 | 625 | 1646 | 0 | 444 |
| S. enterica (23) | 0.57 | 0.76 | 3 | 0 | 0.65 |
| C. jejuni (18) | 5 | 5 | 11 | 0 | 3 |
| E. coli (6) | 0.92 | 0.92 | 3 | 0.50 | 1 |
| K. pneumoniae (15) | 0.95 | 0.81 | 2 | 0 | 0.76 |
Discussion
The experiments described formed part of the initial evaluation of the performance of the MassCode assay in detecting C. difficile, S. enterica, Campylobacter spp. and norovirus; and development of the standardised workflows necessary for its wider implementation in the NHS, prior to the implementation of phase 1 testing. Although phase 1 testing focused on five key pathogens (C. difficile, C. coli and jejuni, norovirus and S. enterica), the 14-plex MassCode primer mix was used, as detection of the additional organisms added value to the experiments performed.
Limits of detection
Limit of detection testing was performed for all four cultivable organisms targeted as part of the phase 1 testing. Limits of detection were one to two orders of magnitude higher for the MassCode assay than for qPCR. This finding was anticipated as amplification efficiency of individual targets decreases when performed as part of a large multiplex reaction. Nevertheless, the limits of detection observed were higher than expected.
Compared with other published data of limits of detection for enteric multiplex reactions, the limits found here are varied. Liu et al. 9 developed an in-house Luminex panel and presented limit of detection in CFU/g. Assuming a stool sample input of 0.2 g, the lowest concentration at which all five samples were MassCode positive were, therefore, 1.3 × 104 CFU/g, 1.3 × 106 CFU/g, 1.3 × 105 CFU/g and 6 × 102 CFU/g for C. difficile, S. enterica, C. jejuni and C. coli, respectively. This is compared with 103 CFU/g for Salmonella spp. and C. jejuni, and 105 CFU/g for C. coli as reported previously. 9 However, the reported investigation used a different commercial DNA extraction kit than was used here.
In order to be detected by MassCode, S. enterica had to be spiked into stool at a CFU level one to two orders of magnitude higher than required for the other organisms tested. For S. enterica, qPCR copy numbers were also three orders of magnitude lower than the CFU of the spike, whereas for the majority of other samples copy numbers were one order of magnitude lower than the CFU of the spike. These findings suggested there may be a specific problem with S. enterica nucleic acid isolation and detection. Pathogen load has been reported at 103 to 109 CFU/g of stool, suggesting a single order of magnitude improvement in limits of detection for MassCode would detect most bacterial pathogens. 9,10
Other than for C. coli, for which an average copy number of 60 led to all five samples being detected by MassCode, all five samples were detected by MassCode when copy numbers averaged over 100. Therefore, copy number yield needed to be over 100 in order to reliably detect clinically positive samples. Overall, the data strongly indicated that, although limits of detection were insufficient with the currently implemented protocol, an increase in nucleic acid yield would significantly improve limits of detection for MassCode.
Optimisation of primer panel
Concurrently with limit of detection testing, a collection of 407 known positive clinical samples was processed by QIAamp DNA Stool Kit, MassCode 14-plex panel and the appropriate qPCR assay.
Three out of four targets fell below the 75% sensitivity required for the MassCode study to progress through phase 1 to phase 2. Validated duplex/triplex qPCR assays using the same primer pairs as were used in the MassCode multiplex mix (see Table 1) were also more sensitive than the MassCode multiplex, with only S. enterica falling below 75% sensitivity. Investigating how yield of nucleic acids was affecting the MassCode assay sensitivity showed that the majority of samples undetected by MassCode fell below the limits of detection for this assay (> 100 copies).
The exception to this was S. enterica: in 47% of samples that tested negative for S. enterica by MassCode, S. enterica was also below the limits of detection of qPCR. To confirm that the target sequence for S. enterica was not limiting its detection, some S. enterica samples were retested by qPCR with alternative primer sets. Use of these alternative qPCR assays did not lead to any additional detection of S. enterica or a decrease in qPCR cycle threshold, suggesting no additional target copies were detected. These data supported limit of detection testing, suggesting that yield of nucleic acids was the main limitation to MassCode sensitivity.
Although only C. difficile-, S. enterica-, Campylobacter spp-. and norovirus-positive samples were obtained for this pre-phase 1 investigation, use of the 14-plex MassCode primer panel yielded some unexpected positives (Table 5). Retesting these positive samples by qPCR suggested that some of the primer choices for the 14-plex panel may be resulting in false positives; in particular for norovirus, S. Typhi and Giardia spp. A proportion of unexpected positive samples were found to be genuinely positive by qPCR, illustrating how molecular methods may detect positive samples missed by reference standard tests. However, as shown previously, known positive samples in which nucleic acid yield was low were not detected by MassCode.
These data, along with the observation that the norovirus 1 primer set did not contribute to any additional diagnosis of norovirus, led to a recommendation that the norovirus 1 primer set should be removed from the primer panel in an effort to reduce false positives, creating a 13-plex primer panel. As only C. difficile, S. enterica, Campylobacter spp. and norovirus were directly under investigation, no recommendations were made regarding the additional primer sets in the panel.
The two main outcomes from the processing of the known positive sample collection were a recommended adjustment to the MassCode primer panel and the identification of nucleic acid yield as a major limitation to the sensitivity of MassCode. In order to improve yield of nucleic acids, a comprehensive investigation into nucleic acid isolation methods was instigated.
Extraction optimisation
In the light of previous results with a commercial extraction kit, a comparison between the dominant methods for nucleic acid extraction was performed to ensure the method was optimal for the MassCode assay. The results showed that through addition of extra lysis steps to standardised extraction protocols, nucleic acid yield could be dramatically increased (see Appendix 1, Table 48).
The most successful lysis method was bead beating. The addition of bead beating to the QIAamp DNA Stool Kit standard protocol resulted in C. difficile yields increasing from 5% to 191% and S. enterica yields increasing from 0.2% to 0.9% (see Table 6). Recent studies have also employed the QIAamp protocol modified to include additional lysis steps,10 implying that the increased nucleic acid yield has independently been found to improve detection assay sensitivity. The InhibitEX tablet step of the QIAamp protocol was also removed to test whether or not its use was resulting in DNA loss for S. enterica. The results suggested that use of the InhibitEX tablet did not improve nucleic acid yield for this organism. However, standard deviations (SDs) were higher without the InhibitEX tablet, suggesting that its use did improve the consistency of yields.
The comparison of a Gram-positive organism (C. difficile) and a Gram-negative organism (S. enterica) illustrated how the physiology of organisms affected what was the most efficient lysis method. However, this also varied across nucleic acid purification method. For example, coupled with QIAamp purification, sonication was one of the most efficient lysis methods for C. difficile. However, for S. enterica sonication was only the most efficient lysis method when coupled with purification by MagMax, which included bead beating as part of the standard protocol.
For both organisms, the MasterPure purification method failed to consistently yield any nucleic acids. This is because the method was not designed for stool extractions, although efforts were made to adapt the method for this purpose.
A comparison was also performed using QIAsymphony, an automated magnetic bead-based extraction system. STAR buffer, which includes chloroform compared with ASL buffer, and the standard lysis buffer included in the QIAamp DNA Stool Kit were compared. Both chemical lysis methods were combined with bead beating. Automated extraction methods are purported to be more efficient and consistent than manual methods. This was reflected in the results, with STAR buffer, bead beating and purification by the QIAsymphony found to be the most efficient extraction method, and also one of the most consistent, as shown by SDs.
Although a large increase in extraction efficiency for C. difficile was achieved, improvements were marginal for S. enterica. C. difficile yields improved dramatically as sporulation is likely to occur within the sample, and the increasingly vigorous lysis methods access the spore DNA. Spores are uncultivable through normal plating and culture methods; hence, copy number yield was higher than CFU input for the more efficient lysis methods. 21 The poor efficiency for Salmonella spp. suggests that their nucleic acids may be subject to effects that have failed to be identified through these experiments. For example, their nucleic acids may be susceptible to specific DNase degradation, or may bind to proteins and be discarded through the extraction process. Poor sensitivities for molecular-based tests for Salmonella spp. compared with culture-based tests have been noted previously. Schuurman et al. 12 tested a range of extraction methods for S. enterica in faecal samples, and found sensitivities using molecular methods were significantly lower than culture diagnosis. It has been reported that a manual extraction method developed by Boom et al. 22 is the most efficient; however, this method is prohibitively laborious for use in the clinical laboratory. 12
Furthermore, the wide range of pathogens targeted by MassCode and other large multiplex methods demands an extraction method suitable for all organisms. The introduction of more vigorous lysis methods increased the potential that more fragile nucleic acids, such as norovirus’ single-stranded RNA, may be lost during the process. As shown by Yang et al. 23 for respiratory pathogens, no single method is likely to be superior for all targeted organisms. The next stage of experiments aimed to validate the optimised extraction method for MassCode with the other phase 1 target organisms.
Method validation
In order to validate the optimised extraction method (STAR buffer with bead beating and purification with QIAsymphony) a batch of 80 positive clinical samples were extracted and analysed with qPCR and MassCode. An additional 20 norovirus samples were tested as, for the initial batch, the diagnosis of norovirus was found to be problematic (see below).
Overall, sensitivities for the MassCode assay improved for all organisms other than S. enterica. Norovirus also remained below the target 75% minimum sensitivity. However, only 80% of the first batch of norovirus samples were found to be positive by qPCR, most likely because of a faulty initial diagnosis of the infection. The second batch of norovirus samples were 100% positive by qPCR, leading to the confident prediction that more than 75% would be positive by MassCode.
The underperformance of S. enterica was anticipated, given the data presented above. The decrease in sensitivity compared with the original extraction method is likely because of the heterogeneity of stool samples, meaning that two separate extractions of the same clinical sample cannot be guaranteed to contain the same concentration of target organism.
In order to better predict how detection may improve using the new extraction method with a larger sample set, copy numbers of samples extracted by both the original and new extraction methods were compared. When newly detected samples are included, more samples increased in copy number than decreased with the new extraction method. Decreases in copy number are also likely to result from stool sample heterogeneity. Although some samples decreased in copy number, net percentage copy number change was calculated to be positive for all organisms. The data suggest that with a larger sample set the optimised extraction method would result in an increase in sensitivity for all organisms, as the improved nucleic acid yield would produce more samples above detection limits.
Although nucleic acid yields improved for S. enterica, the data still suggested that overall yield would be poor, and many samples would remain below the limits of detection. These results implied that poor extraction efficiency was intrinsic to Salmonella spp., potentially as a result of an unidentified biochemical or biological process. Limit of detection testing was repeated with other Enterobacteriaceae alongside non-Enterobacteriaceae to explore whether the problem was bacterial family wide or restricted to Salmonellae.
Extraction efficiency
The data suggest that members of the Enterobacteriaceae family are a challenging group of organisms to analyse directly from clinical samples by molecular methods. Although extraction efficiency of C. jejuni also appears poor at 5%, the other data suggest this yield provides sufficient DNA for molecular analysis, as MassCode sensitivity was good for Campylobacter spp.
Evidence regarding the efficiency of extraction methods is limited within the available literature, but it is possible to compare the limits of detection found here with other investigations. For example, a large multiplex PCR assay for E. coli directly from stool samples reported limits of detection of 104–105 CFU/ml. 24 The qPCR assay used as part of this investigation produced limits of detection of 4 × 103; this limit of detection would be expected to increase by one to two orders of magnitude within a large multiplex assay.
Summary
The data presented above illustrate how optimisation of extraction methods is crucial to the success of large multiplex assays. A major challenge is to identify an extraction method that successfully isolates DNA and RNA from a wide range of physiologically diverse organisms. Although the method presented here has successfully improved DNA yield, the improvement was not sufficient for S. enterica. Further exploration of this issue has suggested that a biochemical or biological mechanism may result in poor DNA yield from Enterobacteriaceae; however, these investigations have not been able to identify what mechanism this may be. This has serious implications for the future use of these tests, as these organisms are an important source of gastrointestinal infections with or without increasing antimicrobial resistance.
Despite the relatively poor performance of S. enterica, the optimised extraction method and adjusted primer mix were taken forward to phase 1 of the MassCode investigation. Progression to phase 1 was intended to provide sensitivities with sufficiently narrow 95% confidence interval (CI) to decide whether or not the MassCode assay was suitable for further evaluation in phase 2, with a view to rollout across the NHS.
Chapter 3 Phase 1 blinded investigation
Introduction
The phase 1 investigation of the MassCode assay aimed to establish the sensitivity and specificity of the assay to detect target pathogens and/or rule out any infectious causative agent compared with the reference standard tests performed in the service microbiology laboratory.
Methods
Participants and sample collection
Phase 1 evaluation was conducted in the Oxford University Hospitals NHS Trust. All samples collected were initially sent to the service microbiology laboratory for faecal culture and/or C. difficile toxin testing by hospital-based doctors or GPs as a result of a suspected enteric infection. Consecutive samples positive for one of the three key pathogens being tested routinely (C. coli, C. jejuni and S. enterica) were put aside by routine microbiology staff rather than being sent for discard, as were samples that had been tested for C. difficile by toxin EIA in the service microbiology laboratory and that were negative for all pathogens. The only exclusion criterion was insufficient sample remaining after standard microbiological testing. During the period of the study, the service microbiology laboratory in the Oxford University Hospitals used an EIA to identify C. difficile manufactured by Meridian Bioscience, Inc. (Cincinnati, OH, USA). A large study conducted in 2011 independently of the manufacturer5 demonstrated that this EIA test has particularly poor sensitivity compared with the gold standard cell cytotoxicity assay (69.2%), considerably lower than an alternative EIA test (82.3%) which has been used in Oxford University Hospitals since April 2012 as part of a ‘two-step’ testing algorithm mandated by the UK Department of Health. 25 There are two reasons why EIA tests have been used historically by many trusts, and continue to be used within the ‘two-step’ testing algorithm to detect C. difficile: first, their turnaround time is shorter (≈ 1 day compared with 3 days for the cell cytotoxicity assay) and, second, their cost (consumables and staff) is lower, particularly important given the large number of tests performed (≈ 10,000 tests/year in Oxford University Hospitals). Separately to the MassCode study, all EIA-positive C. difficile samples were cultured in the parallel research laboratory under a separate research protocol. Samples positive for C. difficile on both EIA and culture were retrieved from this sample collection as reference positives for use in the MassCode study. As norovirus testing was not carried out by the microbiology laboratory unless an outbreak was suspected, qPCR-positive norovirus samples were obtained through a separate investigation conducted by a NIHR clinical fellow.
A research assistant not involved in the MassCode study therefore collected samples from the three sources (culture-positive C. difficile samples from the research laboratory, PCR-positive norovirus samples from a different study in the research laboratory and samples positive for Campylobacter spp., Salmonella spp. or negative for all pathogens from the service microbiology laboratory). The independent research assistant assigned each sample to one of 1000 pre-generated study numbers at random and maintained a list of which sample corresponded to which study number and what pathogen (if any) had been identified by the service microbiology laboratory. The research assistant produced a blinded random order of samples from the various sources for testing using the MassCode assay from this list (so that not all C. difficile- and norovirus-positive samples were processed in the same batches). As numbers of S. enterica-positive samples were lower than predicted, additional S. enterica-positive samples were sourced from Leeds, sent to the research assistant not involved with the MassCode study, assigned anonymous study numbers and periodically inserted into the workflow as for C. difficile and norovirus-positive samples. All samples were stored at 4 °C prior to processing.
All samples used in the phase 1 blinded evaluation were collected independently to those samples used to determine the extraction protocol (see Chapter 3).
Reference standard
Initial diagnosis of the target faecal pathogens was performed in accordance with Public Health England guidelines in the service microbiology laboratory. Approximately 1 g of faecal sample was inoculated into selenite broth and the broth inoculated onto xylose lysine deoxycholate (XLD) agar for culture of Salmonella spp. and Shigella spp., sorbitol MacConkey (SORB) agar for culture of E. coli O157 and Campylobacter-free blood (CAMP) agar for culture of Campylobacter spp. CAMP agar was incubated microaerophilically at 42 °C for 48 hours, XLD, SORB and the selenite broth, for culture of Salmonella spp. was incubated at 37 °C for 24 hours. After the 24-hour period, the selenite broth was inoculated onto chromogenic agar (SALM) for culture of Salmonella spp. This was incubated for a further 24 hours at 37 °C. Suspect colonies on XLD or SALM were inoculated onto analytical profile index (API®) 10S, for identification of Salmonella spp., a Columbia agar slope for slide agglutination tests, and a MacConkey agar purity plate. If C. difficile infection was suspected, samples were subject to EIA testing for toxins A and B. Subsequent serological and sensitivity testing was performed as required for each organism identified. Throughout all reference standard and index tests, normal aseptic microbiological laboratory working practices were followed. Campylobacter was identified only to the species level, i.e. C. jejuni and C. coli were not distinguished by the reference standard testing.
Reference standard testing was performed by trainees and state-registered biomedical scientists (BMSs), but all results were confirmed by an experienced state-registered BMS before being passed onto a doctor. As reference tests were carried out before the MassCode assay was run, staff performing the reference assays did not know the results of the MassCode assay.
Blinded investigation
The full SOP for sample preparation and processing is detailed elsewhere (see Appendix 1). A total of 948 clinical samples were collected and extracted using the optimised protocol. This included 200 Campylobacter spp., 199 C. difficile, 60 S. enterica, 199 norovirus and 295 negative samples (some samples had more than one pathogen), compared with targets of 200, 200, 100, 200 and 300, respectively. Insufficient S. enterica-positive samples accrued during the period of the study. Samples were reverse transcribed and stored at −20 °C prior to amplification with the MassCode 13-plex primer mix. Following amplification, samples were cleaned according to the MassCode SOP and analysed by mass spectrometry (MS). All extraction batches included a water extraction as a control and all MS plates included MassCode calibrant controls.
All MassCode assays were conducted by a postdoctoral fellow and a research assistant who had received training on the MassCode assay from Agilent Technologies, and had jointly conducted the pre-phase 1 evaluations (see Chapter 2). The output of the MassCode assay was a number, indicating that the sample was positive on both PCR products for a pathogen (positive), near threshold values or positive on one product and near threshold value for the second product (indeterminate), or was not positive on either (negative). No expert interpretation was, therefore, required to read the MassCode outputs, which were also exported electronically from the spectrometer. The cut-offs defining positivity were set by Agilent Technologies, based on their in-house development and were not altered for this evaluation.
Researchers conducting MassCode assays were blinded to results of the reference test and all other clinical information; samples were identified only by their unique study number (assigned by a different laboratory researcher not involved with the MassCode study), i.e. were completely anonymised with regards to patient and microbiological characteristics (what pathogen, if any, had had been isolated).
MassCode results were used only for the research study and were not returned for patient management.
Sample size
In phase 1 testing, 200 positive detections of C. difficile, norovirus and Campylobacter spp. with reference standard culture/toxin detection and new PCR-based tests would produce a 95% CI around sensitivities of 90% and 96% of ± 4.2% and ± 2.7%, respectively. Testing 100 positive isolations of Salmonella spp. would produce a 95% CI around a sensitivity of 90% of approximately ± 5.9%. Samples positive for each pathogen would act as negative controls for other pathogens, together with 300 samples negative for all pathogens (total 1000 samples to be tested in Oxford and Leeds hospitals).
Analysis
Analysis of the MassCode assay results was conducted by an independent member of the team. All microbiological results were extracted from the microbiology database to confirm the reference standard test results, and identify any co-infections with other pathogens in the 13-plex MassCode panel which had not been part of original sample retrieval procedures. Sensitivity and specificity were calculated compared with the microbiological reference standard for each of the main organisms (primary reference standard), counting identification of either Campylobacter species as correct (as reference microbiological testing did not speciate). Primary analysis did not count indeterminate results (either forward or reverse primer above pre-specified threshold, but not both) as a positive: secondary analysis included these indeterminates as positive. Uncertainty was quantified using 95% CIs. All analysis were conducted in Stata 11.2 (StataCorp LP, College Station, TX, USA).
Samples which were either false negative for any of the five key pathogens or false positive for any of the 11 pathogens on MassCode were then retested using single qPCRs. This testing was not done blinded as the researchers needed to know which qPCR to run. The qPCR assays used are described elsewhere (see Chapter 2 and Appendix 1). Each qPCR assay was run in duplicate and any discrepancies confirmed by additional qPCRs.
As several unexpected positives were confirmed by qPCR results (implying the target pathogen was genuinely present in the sample but missed in the original microbiology work-up), and as some missed positives could not be detected in the original sample, even using single qPCR, a secondary analysis was carried out in which the MassCode-positive results were compared with a combined standard of reference microbiological assay plus single qPCR testing. In this secondary reference standard, samples positive on standard reference microbiology but negative on qPCR were considered negative, and samples negative on standard reference microbiology but positive on qPCR were considered positive. Possible explanations for the former are sample degradation or labelling errors. This secondary reference standard does not reflect a gold standard, but rather a standard whereby single qPCR demonstrates that, respectively, either it is unrealistic to expect the multiplex MassCode assay to detect the nucleic acid target (because it cannot be detected even with a single qPCR) or that the target was present and should have been detected by MassCode even if not identified on original microbiological testing. Effectively this standard addresses the impact on diagnostic capability of multiplexing the MassCode assay rather than running multiple single qPCRs.
Phase 2
Analysis of sensitivity/specificity of MassCode compared with standard reference methods was to be conducted at the end of phase 1 in each trust.
A small steering group was set up to review results of phase 1 (in each hospital and overall) in order to determine whether or not phase 2 should proceed. Criteria for not moving to phase 2 were pre-specified as extremely poor estimated sensitivity for detecting any of the key organisms (under 75%) such that the test would not be likely to be adopted in routine NHS practice. The choice of a threshold of 75% was based on estimated sensitivity of the currently widely used ELISAs for detecting C. difficile. The steering group comprised investigators (DWC, TEAP, MHW and ASW) plus two independent researchers – Professor Ajit Lalvani (Imperial College, expertise in development, assessment and implementation of interferon gamma release assays) and (as chairperson) Dr Christine McCartney (Director of the Health Protection Agency Regional Microbiology Network) and one independent member from outside academia, Ms Katherine Innes Ker, who has extensive commercial experience and will also represent the patient community. Inclusion of investigators and independent researchers in one oversight committee was considered appropriate because there was no patient management which could be influenced by knowledge of results to date, as no results were returned for clinical care.
Sequencing Cryptosporidium product
Single SYBR Green PCR results for Cryptosporidium, performed as part of the phase 1 analysis, suggested that the primer pair targeting Cryptosporidium was resulting in amplification of non-specific targets. To investigate, sequencing of the Cryptosporidium PCR product was performed after the primary analysis of sensitivity and specificity had been conducted. Eight samples were chosen for sequencing, including Cryptosporidium Positive Template Control, C. parvum oocysts, samples found to be positive for Cryptosporidium spp. by reference standard testing, and samples negative/not tested by reference standard testing and found to be positive by MassCode. Extracted samples were subject to conventional PCR following the MassCode SOP (see Appendix 1), but with Cryptosporidium spp. primers in singleplex; forward (5′-GAGGTAGTGACAAGAAATAACAATACAGG-3′) and reverse (5′-CTGCTTTAAGCACTCTAATTTTCTCAAAG-3′) primers were both used at 250 nm. Both primers were designed by Agilent, targeted small subunit ribosomal RNA (SSU rRNA) and were included in the MassCode 13-plex panel.
Following amplification, the PCR product was cleaned to remove unused primers and other ingredients. AMPure XP beads (Agencourt, Beckman Coulter, High Wycombe, UK) were added in a ratio of 1.8 : 1 (18 µl of beads added to 10 µl of PCR product), mixed well and incubated for 3 minutes at room temperature. The beads were separated from the supernatant by placing sample tubes on a magnetic stand and the supernatant removed and discarded. The beads were then washed twice with 200 µl of 70% molecular-grade ethanol. All remaining ethanol was removed and the beads left to air dry for 15 minutes. DNA was eluted in 40 µl of 1 × TE [tris-ethylenediaminetetraacetic acid (EDTA)] buffer by adding the buffer to each sample, mixing well, and incubating at room temperature for 2 minutes. Finally, the beads were separated from the eluate by placing the samples on a magnetic stand and the eluate transferred to new tubes.
A sequencing reaction was performed on the cleaned PCR product using the Big Dye Terminator Cycle Sequencing Kit (Life Technologies). The reaction comprised 1.75 µl of 5 × Sequencing Buffer, 0.5 µl of Big Dye, 0.5 µl of either the forward or reverse primer,and 8.75 µl of molecular-grade water per sample. A total volume of 2 µl of each sample was added to the reaction mix, and each sample was subject to a sequencing reaction with both the forward and reverse primer. Cycling conditions were as follows:
96 °C for 2 minutes96 °C for 10 seconds50 °C for 5 seconds60 °C for 3 minutes}× 30
Post-sequencing reaction products were cleaned using CleanSEQ beads (Agencourt). A volume of 10 µl of CleanSEQ beads was added to 10 µl of sequencing reaction product and mixed well. A volume of 42 µl of 85% molecular-grade ethanol was added to each bead sample mixture and, again, mixed well. Sample mixtures were placed on a magnetic stand and incubated for 3 minutes at room temperature to allow the beads to separate from the supernatant. The supernatant was then removed and discarded. The beads were subsequently washed twice with 100 µl of 85% ethanol. Once all ethanol was removed, the beads were air dried for 15 minutes and the DNA eluted from the beads in 80 µl of 0.05 mM EDTA.
The final eluate was sequenced by Applied Biosystems 3730 DNA Analyser (Life Technologies). Forward and reverse sequences for each sample were aligned in Geneious v5.6.6 (Biomatters Ltd, Auckland, New Zealand) and the consensus sequences submitted to Basic Local Alignment Search Tool (BLAST; NCBI) for identification.
Results
A total of 948 samples were collected between June 2009 and September 2012, in accordance with the MassCode SOP and protocol (Table 10). As samples were discarded from the routine laboratory, information on age, sex and other demographics was not collected. Five samples had both Campylobacter spp. and S. enterica isolated in the service laboratory. One Campylobacter-positive sample also had Cryptosporidium spp. isolated, and one S. enterica-positive sample also had Shigella spp. isolated in the service microbiology laboratory. No patient data were obtained. Testing of samples was performed between July 2012 and November 2012, with all extractions for MassCode testing done between July and August 2012.
| Pathogen | n | Collection date | ||
|---|---|---|---|---|
| Minimum | Median | Maximum | ||
| Campylobacter spp. | 200 | 13 December 2010 | 5 June 2011 | 26 August 2012 |
| C. difficile | 199 | 11 June 2009 | 9 December 2009 | 19 March 2012 |
| Norovirus | 199 | 1 January 2010 | 10 February 2012 | 31 August 2012 |
| S. enterica | 60 | 14 April 2011 | 13 January 2012 | 25 August 2012 |
| Negative for all these pathogens | 295 | 16 October 2011 | 6 November 2011 | 21 September 2012 |
Sensitivity and specificity for the main organisms
Sensitivities of each organism varied for the MassCode assay (Figures 6–9 and Table 11), ranging from 43% to 94%. Including indeterminate sample calls (where the MassCode result is near threshold values for a positive call) increased sensitivities only very slightly to 48–96%. Specificities for the MassCode assay were 95–98% or 89–96% including indeterminates. Including qPCR results also led to an increase in sensitivity and specificity, which ranged from 60% to 95% and from 97% to 100%, respectively.
FIGURE 6.
MassCode assay sensitivity and specificity for Campylobacter spp. excluding (a) and including (b) qPCR results. Sensitivity = 100 × called positive/true positive; Specificity = 100 × called negative/true negative.
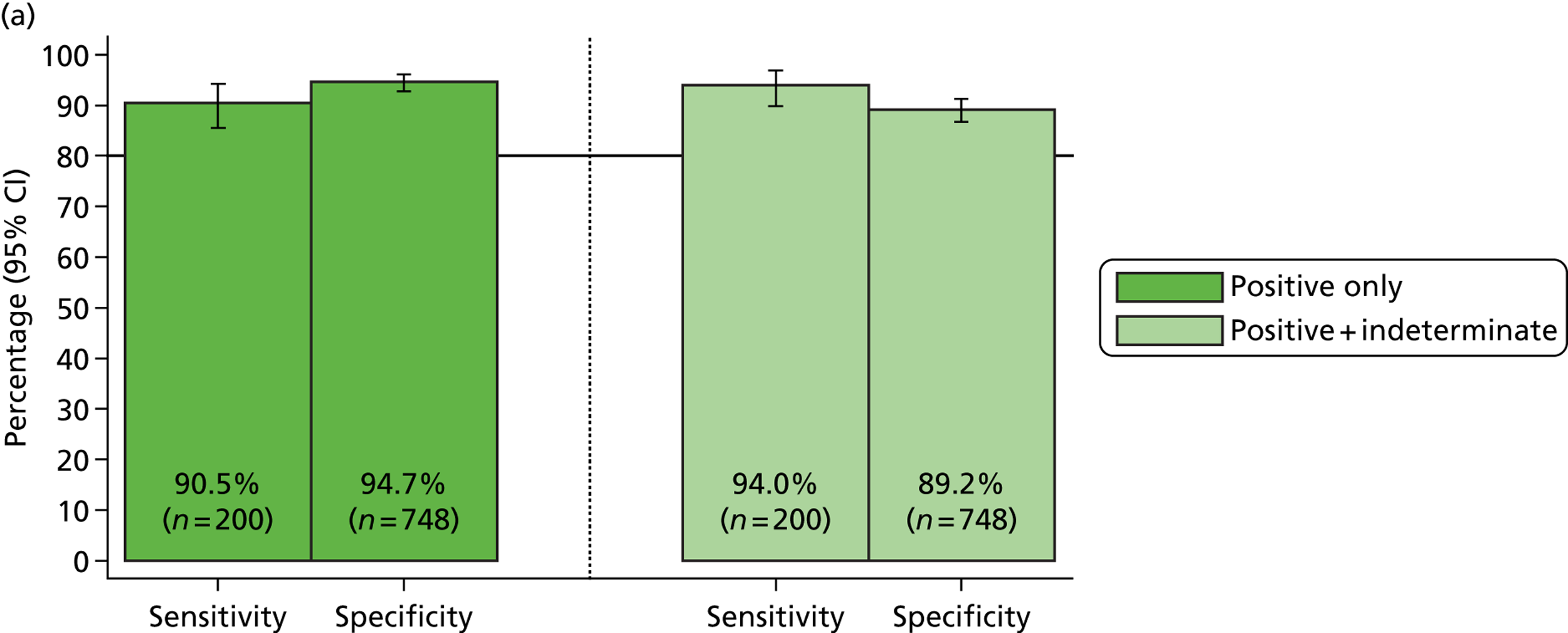
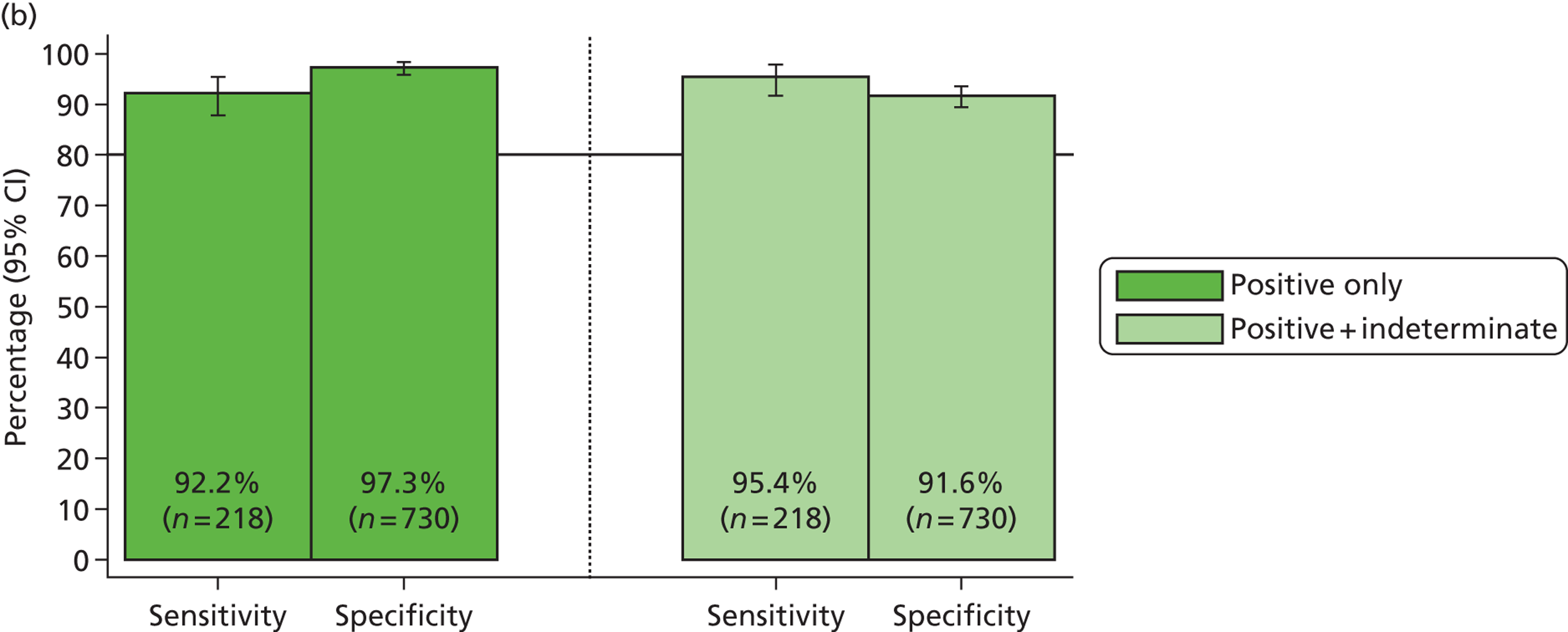
FIGURE 7.
MassCode assay sensitivity and specificity for C. difficile excluding (a) and including (b) qPCR results. Sensitivity = 100 × called positive/true positive; Specificity = 100 × called negative/true negative.
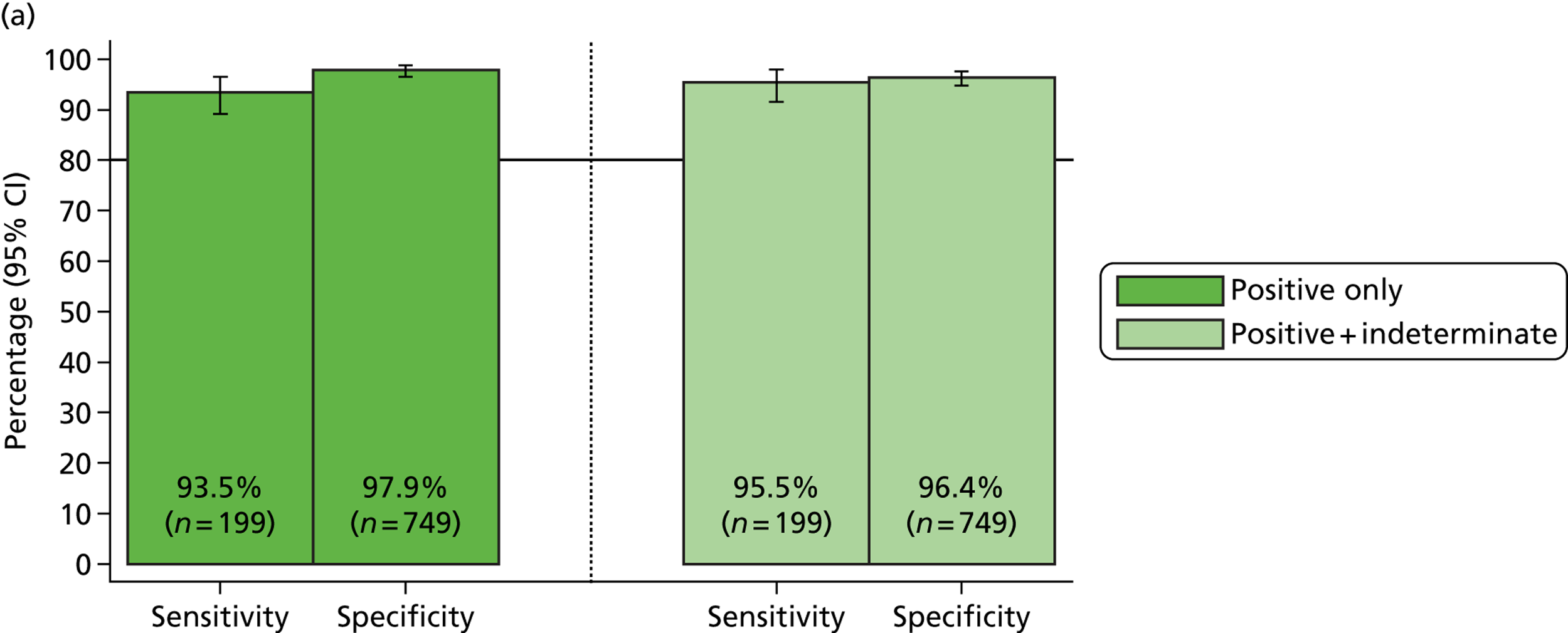
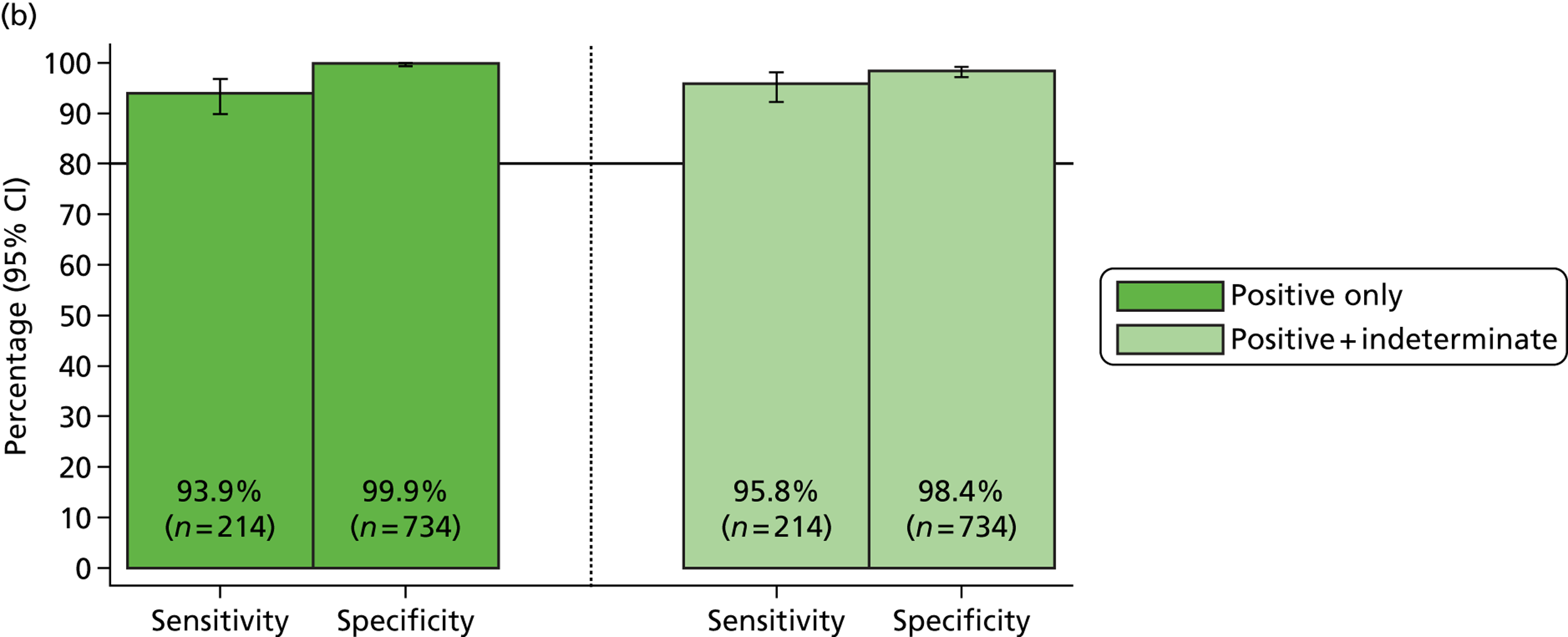
FIGURE 8.
MassCode assay sensitivity and specificity for norovirus excluding (a) and including (b) qPCR results. Sensitivity = 100 × called positive/true positive; Specificity = 100 × called negative/true negative.
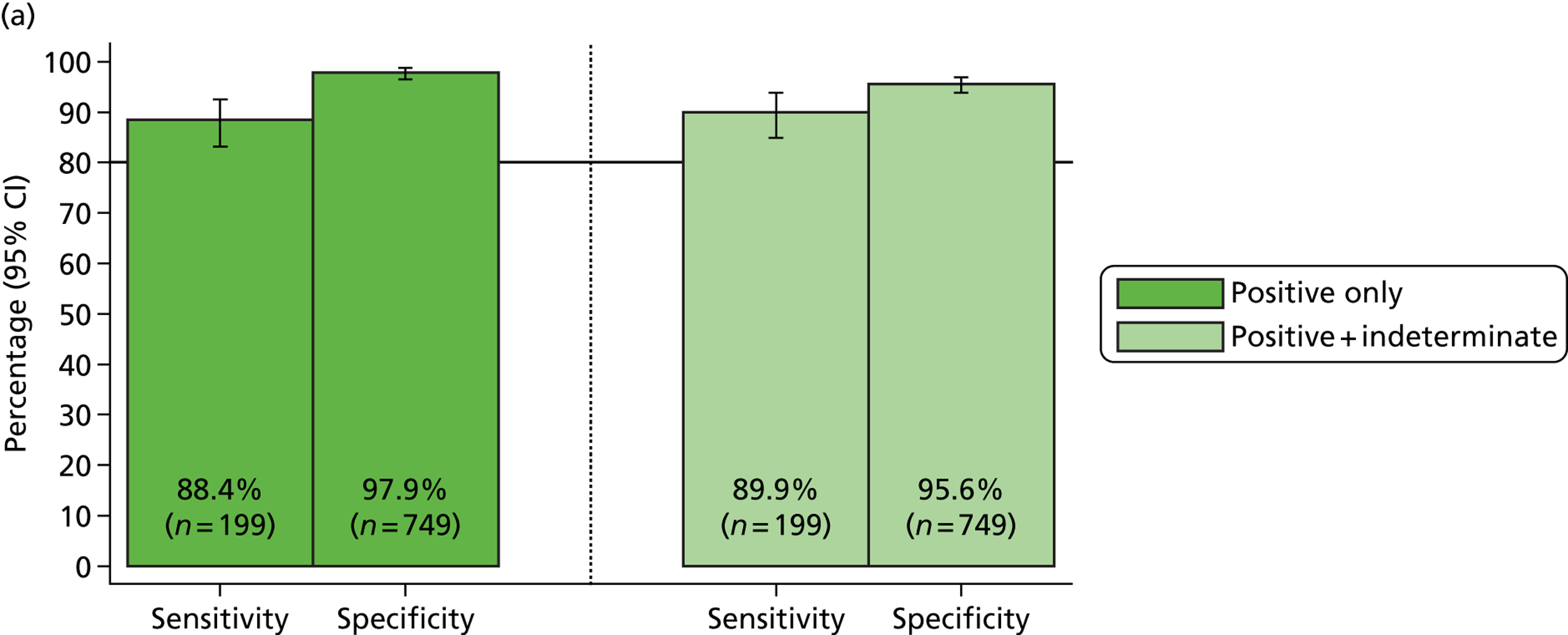
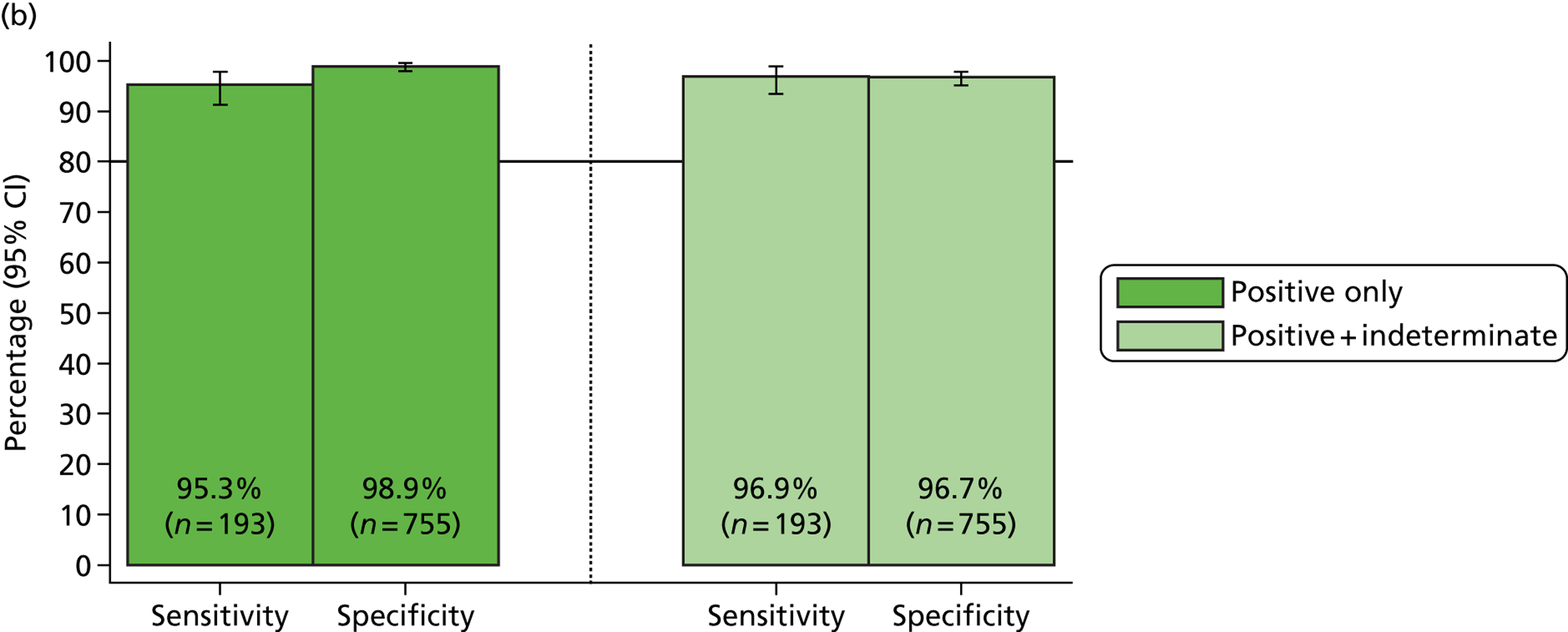
FIGURE 9.
MassCode assay sensitivity and specificity for S. enterica excluding (a) and including (b) qPCR results. Sensitivity = 100 × called positive/true positive; Specificity = 100 × called negative/true negative.
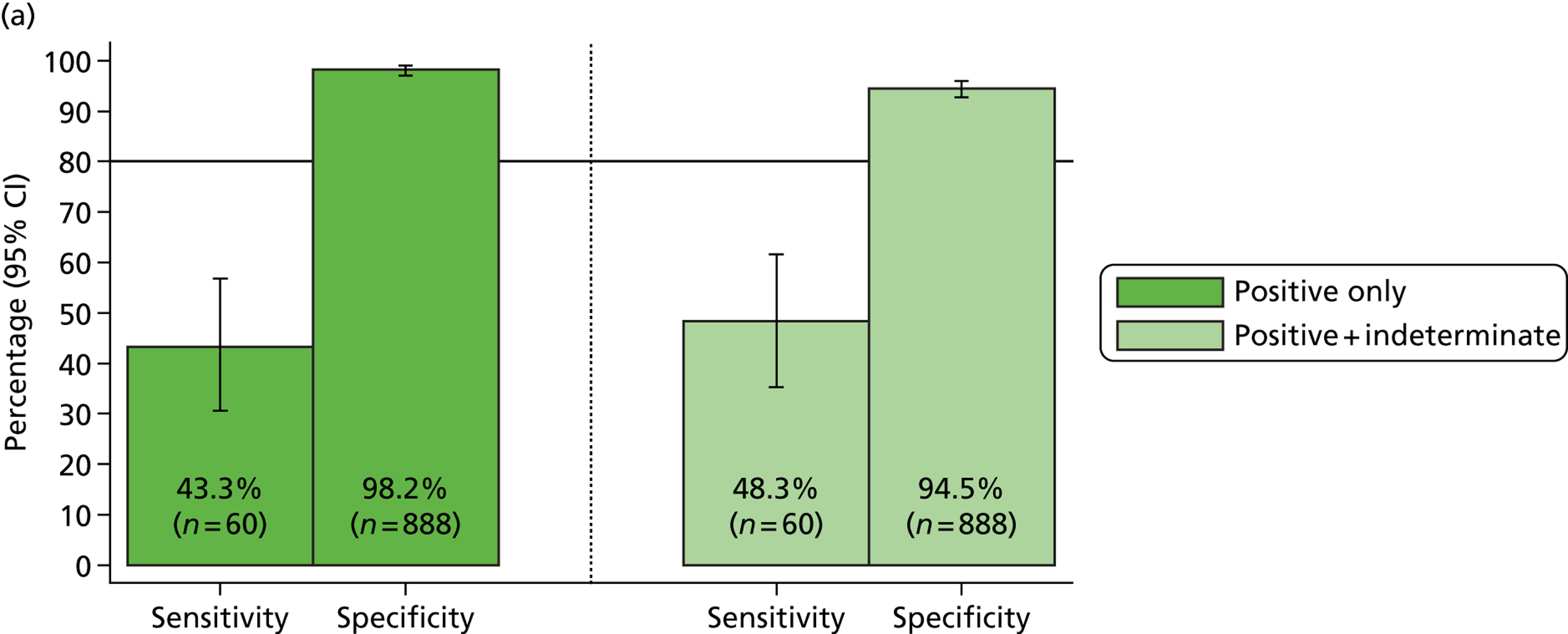
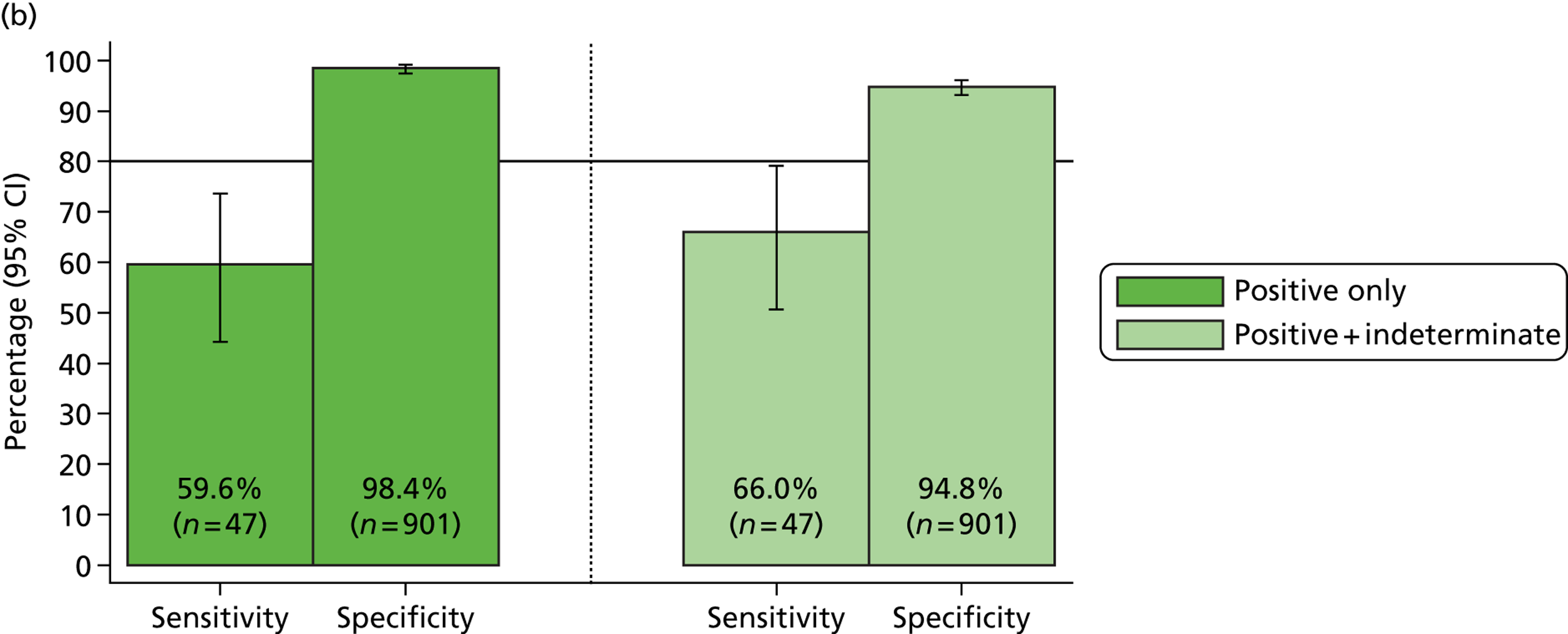
| Organism | MassCode positive | MassCode positive + indeterminate | ||||||
|---|---|---|---|---|---|---|---|---|
| Microbiology positive | Microbiology negative | Microbiology positive | Microbiology negative | |||||
| MassCode positive | MassCode negative | MassCode positive | MassCode negative | MassCode positive/indeterminate | MassCode negative | MassCode positive/indeterminate | MassCode negative | |
| Campylobacter | 181 | 19 | 40 | 708 | 188 | 12 | 81 | 667 |
| C. difficile | 186 | 13 | 16 | 733 | 190 | 9 | 27 | 72 |
| Norovirus | 176 | 23 | 16 | 733 | 179 | 20 | 33 | 716 |
| S. enterica | 26 | 34 | 16 | 872 | 29 | 31 | 49 | 839 |
The best-performing organism was C. difficile, although Campylobacter spp. and norovirus also had sensitivities and specificities well above the 75% threshold required by the MassCode protocol in order to proceed to phase 2, with the lower limits of the 95% CI exceeding 83% (sensitivity) and 92% (specificity). However, the sensitivity of S. enterica remained well below this threshold; even including qPCR results the upper limit of the 95% CI around the estimated sensitivity of 60% was just below 75%.
Inspection of the copy numbers returned by qPCR for missed positive samples revealed that 59% of samples positive for Campylobacter spp., 77% of C. difficile-positive samples and 89% of norovirus-positive samples had copy numbers < 100, the previously defined limit of detection. For S. enterica, 61% of the missed positives that were recovered by qPCR had copy numbers < 10, suggesting very low nucleic acid yields.
Tables 12a-c show more detail about the discrepancies between MassCode positives and reference microbiology. C. difficile was the most accurate target, with all but one of the unexpected positives confirmed positive by qPCR (see Table 12c). These samples may reflect isolates that were not producing toxin and so not causing disease at the time of the diarrhoea, i.e. may have been carried toxigenic strains (colonisation). The primer targets the tcdB toxin gene, so would not have identified non-toxigenic strains. The reference laboratory test for C. difficile is an EIA-based test for toxin; however, as this test is known to report false positives and false negatives, the samples chosen for this investigation had been confirmed C. difficile positive by culture.
| Organism | Reference: microbiology | Reference: microbiology + qPCR | ||||||
|---|---|---|---|---|---|---|---|---|
| Positive | Positive + indeterminate | Positive | Positive + indeterminate | |||||
| Sensitivity | 95% CI | Sensitivity | 95% CI | Sensitivity | 95% CI | Sensitivity | 95% CI | |
| Campylobacter | 90.5% | 86.5% to 94.2% | 94.0% | 89.9% to 96.9% | 92.2% | 87.8% to 95.4% | 95.4% | 91.7% to 97.8% |
| C. difficile | 93.5% | 89.1% to 96.5% | 95.5% | 91.6% to 97.9% | 93.9% | 89.8% to 96.7% | 95.8% | 92.2% to 98.1% |
| Norovirus | 88.4% | 83.2% to 92.5% | 89.9% | 84.9% to 93.8% | 95.3% | 91.3% to 97.8% | 96.9% | 93.4% to 98.9% |
| S. enterica | 43.3% | 30.6% to 56.8% | 48.3% | 35.2% to 61.6% | 59.6% | 44.3% to 73.6% | 66.0% | 50.7% to 79.1% |
| Organism | Reference: microbiology | Reference: microbiology + qPCR | ||||||
|---|---|---|---|---|---|---|---|---|
| Positive | Positive + indeterminate | Positive | Positive + indeterminate | |||||
| Specificity | 95% CI | Specificity | 95% CI | Specificity | 95% CI | Specificity | 95% CI | |
| Campylobacter | 94.7% | 92.8% to 96.2% | 89.2% | 86.7% to 91.3% | 97.3% | 95.8% to 98.3% | 91.6% | 89.4% to 93.5% |
| C. difficile | 97.9% | 96.6% to 98.8% | 96.4% | 94.8% to 97.6% | 99.9% | 99.2% to 100.0% | 98.4% | 97.2% to 99.2% |
| Norovirus | 97.9% | 96.6% to 98.8% | 95.6% | 93.9% to 96.9% | 98.9% | 97.9% to 99.5% | 96.7% | 95.2% to 97.8% |
| S. enterica | 98.2% | 97.1% to 99.0% | 94.5% | 92.8% to 95.9% | 98.4% | 97.4% to 99.1% | 94.8% | 93.1% to 96.1% |
| Organism | Positive on MassCode, not identified in laboratory (false positive) | Confirmed as positive on PCR (either carried organisms or missed by original reference test) | Confirmed positive on PCR, but evidence of test negative on microbiology systema | Negative on MassCode, identified as positive in laboratory (false negative) | Confirmed as positive on single PCR | Indeterminate on MassCode |
|---|---|---|---|---|---|---|
| Campylobacter | 40/748 | 20/40 | 16/20 | 19/200 | 17/19 | 7/17 |
| C. difficile | 16/749 | 15/16 | 9/15 | 13/199 | 13/13 | 4/13 |
| Norovirus | 16/749 | 8/16 | n/a | 23/199 | 9/23 | 3/9 |
| S. enterica | 16/888 | 2/16 | 2/2 | 34/60 | 19/34 | 3/19 |
Higher rates of identification of C. difficile on moving from toxin-based tests to NAAT- or PCR-based tests have been widely reported, and identification of C. difficile on the basis of PCR alone is not recommended, as outcomes are similar in PCR-positive toxin-negative and PCR-negative toxin-negative individuals. 5 All 13 samples testing postive for C. difficile in the laboratory but missed by MassCode were identified by qPCR, suggesting that these errors were simply due to the multiplex assay and the loss of sensitivity seen in large multiplex assays, rather than to the target primers or the extraction method.
For the other species, fewer of the MassCode-positive reference-negative samples were confirmed as containing the target organism using qPCR (50% for Campylobacter spp. and norovirus, 12% for S. enterica). S. enterica had a particularly high rate of false-positive calls by MassCode among the main target organisms, with only two of the unexpected positives being confirmed by qPCR. Most of those positive on PCR had evidence of a test negative on the microbiology system, i.e. not isolated rather than not tested for in error. Although this could, in theory, represent detection of lower levels of organisms than required to cause disease by MassCode, the relatively high limits of detection for the MassCode assay for S. enterica suggest that it is more likely these are genuinely false-positive calls by the multiplex assay.
For Campylobacter spp., most (17/19) of the reference laboratory-positive MassCode-negative samples were positive on qPCR, again, suggesting that these errors were simply because of the multiplex assay, not the target primers or the extraction method. Just over half (19/34) the S. enterica laboratory-positive, MassCode-negative samples were positive using a single PCR, suggesting that both primers or extraction method and multiplexing the assays were playing important roles in the lower sensitivity for this organism. Lower PCR positivity for norovirus may reflect degradation of the single-stranded RNA even with a single assay or possibly sample degradation with storage.
Interestingly, of the 181 samples positive for Campylobacter spp. using both reference laboratory and MassCode assays, MassCode identified 130 C. jejuni infections, 10 C. coli infections and 41 co-infections with both C. jejuni and C. coli. It is not known whether these represent genuine co-infections or cross-reactive primer pairs, as Campylobacter is not routinely speciated in the service laboratory.
Specificity of MassCode for additional organisms
It was possible to calculate the specificity of the MassCode assay for the additional targets (Figure 10). Known co-infections identified by the routine laboratory were excluded from this primary analysis. All unexpected positives were tested with singleplex assays targeting the MassCode primers: secondary analyses considered any confirmed positives as a positive standard (i.e. excluded them from analyses of specificity). The results are summarised in Tables 13 and 14. Specificity was lowest for Giardia spp. and Cryptosporidium spp. at 87.8% and 87.9%, respectively, after excluding qPCR positives, and highest for E. coli O157 at 97.8%.
FIGURE 10.
Specificity of MassCode assay across all organisms, excluding (a) and including (b) qPCR results. Specificity = 100 × called negative/true negative.
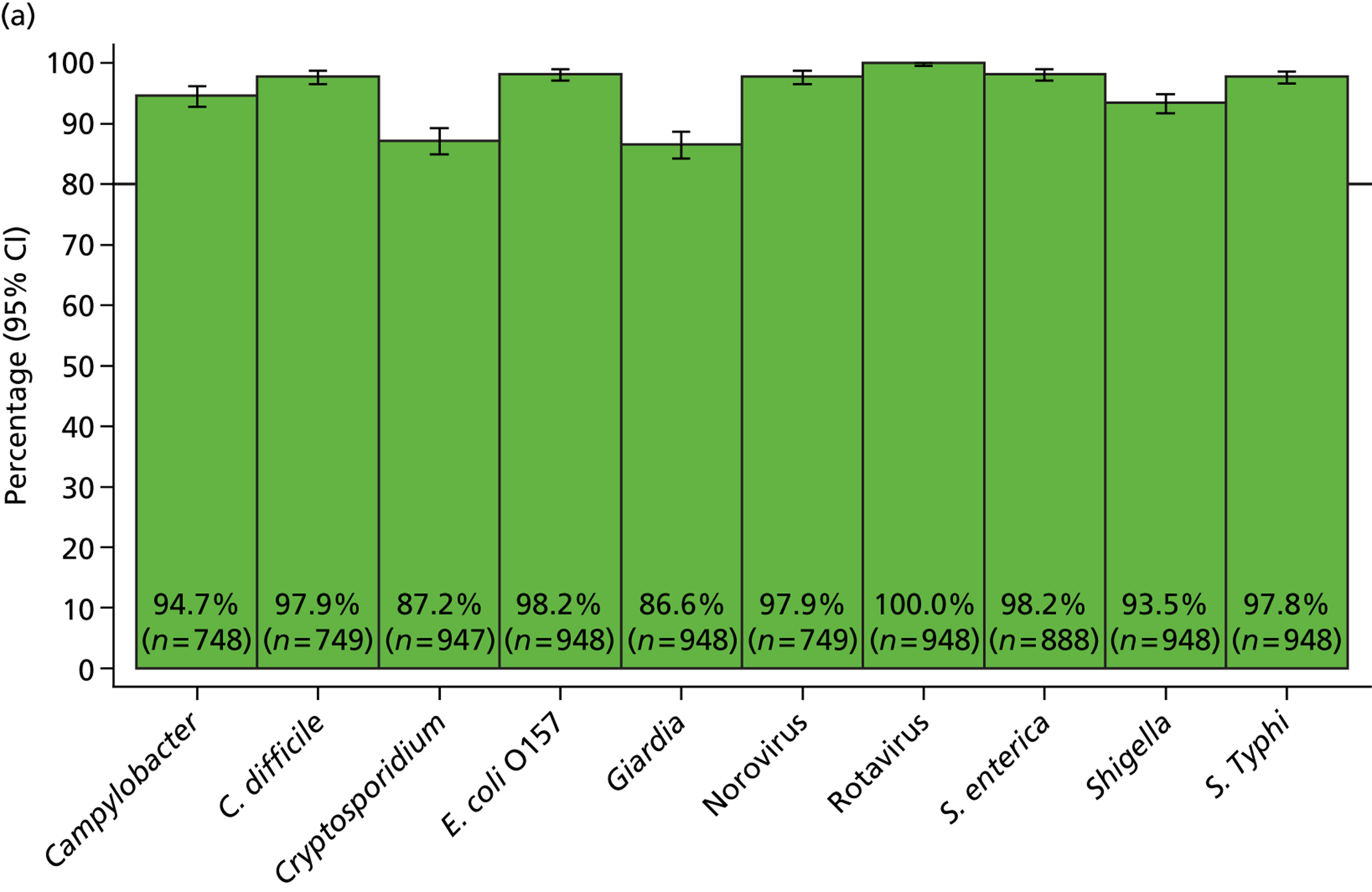
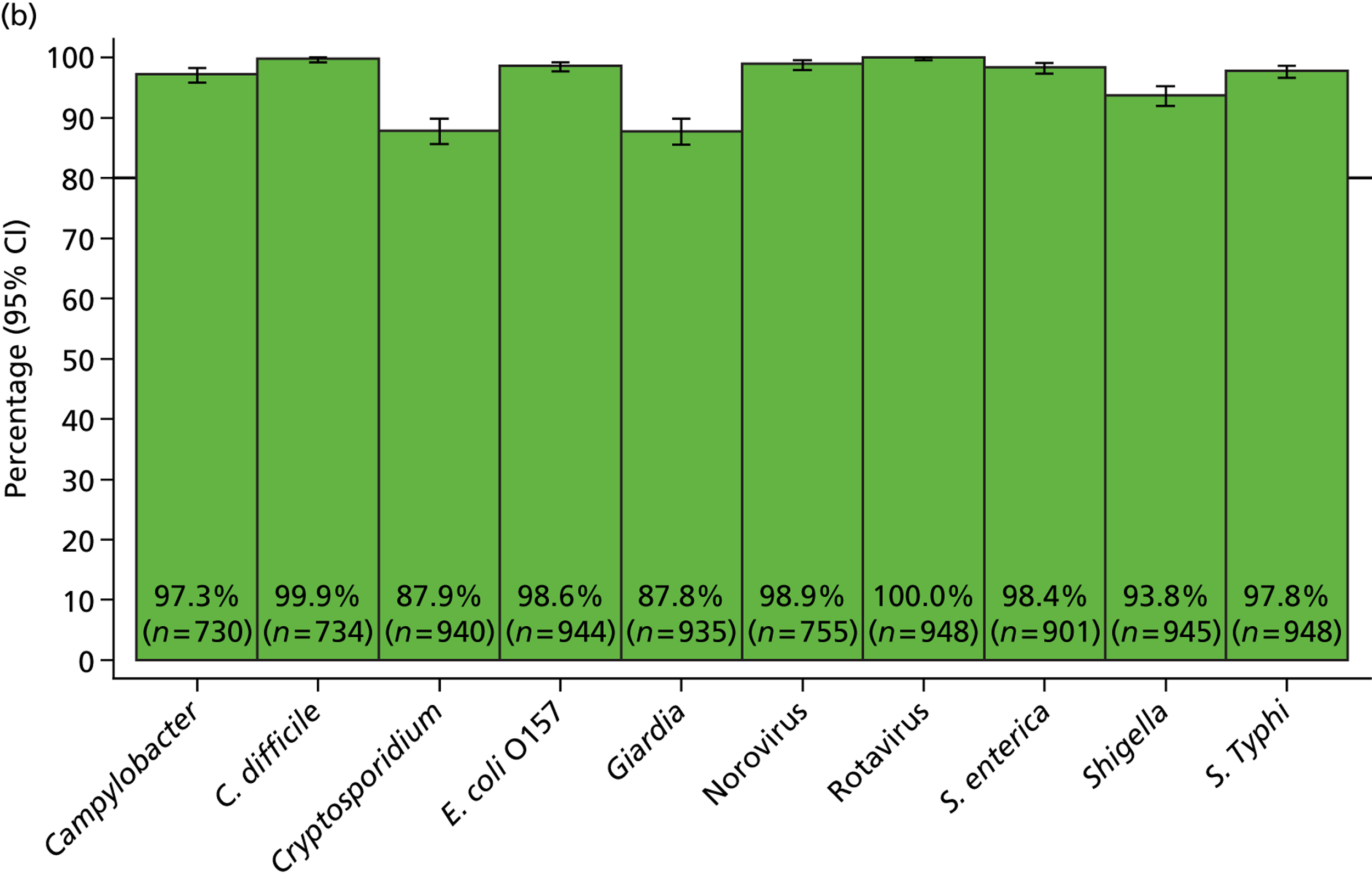
| Organism | Reference: microbiology | Reference: microbiology + PCR | ||
|---|---|---|---|---|
| Specificity (%) | 95% CI (%) | Specificity (%) | 95% CI (%) | |
| E. coli O157 | 98.2 | 97.1 to 99.0 | N/A | |
| S. Typhi | 97.8 | 96.6 to 98.6 | 97.8 | 96.6 to 98.6 |
| Shigella spp. | 93.5 | 91.7 to 94.9 | 93.8 | 92.0 to 95.2 |
| Cryptosporidium spp. | 87.2 | 84.9 to 89.3 | 87.9 | 85.6 to 89.9 |
| Giardia spp. | 86.6 | 84.3 to 88.7 | 87.8 | 85.5 to 89.8 |
| Rotavirus | 100.0 | 99.6 to 100.0 | 100 | 99.6 to 100.0 |
| Organism | Missed positives, n | Missed positives qPCR positive, n (%) | Unexpected positives, n | Unexpected positives qPCR positive, n (%) |
|---|---|---|---|---|
| Campylobacter spp. | 19 | 17 (89) | 40 | 20 (50) |
| C. difficile | 13 | 13 (100) | 16 | 15 (94) |
| Norovirus | 23 | 9 (39) | 16 | 8 (50) |
| S. enterica | 34 | 19 (56) | 16 | 2 (12) |
| E. coli O157 | 0 | 0 | 17 | 4 (24)a |
| S. Typhi | 0 | 0 | 21 | 0 (0) |
| Shigella spp. | 0 | 0 | 62 | 3 (5) |
| Cryptosporidium spp. | 0 | 0 | 121 | 7 (6) |
| Giardia spp. | 0 | 0 | 127 | 13 (10) |
| Rotavirus | 0 | 0 | 0 | 0 |
Discrepant results for additional organisms
Unexpected positive samples were all retested by the appropriate PCR assay, as for the main organisms. E. coli, S. Typhi, Shigella spp., Cryptosporidium spp., and Giardia spp. all also showed a high rate of false-positive calling (Table 14). However, some unexpected positives were also confirmed by PCR, including four (0.4% of all samples) E. coli O157 positives (three of which had definitely had faecal culture without identifying this pathogen), 13 (1.4%) Giardia spp. (none tested for parasites in the service microbiology laboratory), seven (0.7%) Cryptosporidium (one sample tested for this and no oocysts seen) and three (0.3%) Shigella spp. (two had faecal culture without identifying this pathogen). Nevertheless, these unexpected true positives were identified at the cost of a substantial number of false positives, particularly for rarer species, Cryptosporidium spp. and Giardia spp., with 7% and 13%, respectively, of the total 948 samples testing positive for these species by MassCode.
Co-infections
Co-infections were defined by either expected positives from reference standard testing or unexpected positives by MassCode followed by qPCR to confirm the unexpected positive. C. jejuni and C. coli were included as Campylobacter spp. for this analysis. Of the 948 samples, 32 (3.4%) were confirmed positive for two of the four main organisms (Table 15). Overall, 46 (4.9%) of the 948 samples were confirmed positive for more than one organism, only two of which were confirmed to be positive for more than two organisms. Co-infections with parasites (Cryptosporidium spp. and Giardia spp.) accounted for 11 of the co-infections, whereas co-infections with C. difficile and Campylobacter spp. or norovirus were most common. Of note, only eight Cryptosporidium-positive samples were found in total (one known co-infection, seven unexpected positives), but six out of eight organisms were present with another pathogen, suggesting this organism might be more commonly carried than previously suspected. All of the four unexpected E. coli O157 organisms found were also found with other, more common species, as were five of the unexpected 13 Giardia spp. organisms.
| First organism | Second organism | Third organism | No occurrences |
|---|---|---|---|
| C. difficile | Campylobacter spp. | 12 | |
| C. difficile | Campylobacter spp. | Cryptosporidium spp. | 1 |
| C. difficile | Norovirus | 9 | |
| C. difficile | Cryptosporidium spp. | 1 | |
| C. difficile | Giardia spp. | 2 | |
| S. enterica | Campylobacter spp. | 6 | |
| S. enterica | Campylobacter spp. | Giardia spp. | 1 |
| S. enterica | E. coli O157 | 1 | |
| S. enterica | Shigella spp. | 1 | |
| S. enterica | Norovirus | 1 | |
| Campylobacter spp. | Cryptosporidium spp. | 1 | |
| Campylobacter spp. | E. coli O157 | 1 | |
| Campylobacter spp. | Giardia spp. | 1 | |
| Campylobacter spp. | Norovirus | 2 | |
| Norovirus | Cryptosporidium spp. | 3 | |
| Norovirus | E. coli O157 | 2 | |
| Norovirus | Giardia spp. | 1 |
As a consequence of the relatively low sensitivity and specificity of the MassCode assay, far more samples were identified as having co-infections with enteric pathogens using these results alone (Figure 11). A total of 48 (5.1%) were MassCode positive for two of the four main organisms; eight samples were microbiologically negative. Of the 948 tested samples, only 241 (25%) had no organism of any type identified by MassCode, 181/295 (61%) of those microbiologically negative compared with 60/653 (9%) positive for any of the main four pathogens. In one sample up to six organisms were identified by MassCode; 159 samples contained two organisms, 55 samples contained three organisms, 17 contained four organisms and four samples contained five organisms. As expected from the results above, Giardia spp. and Cryptosporidium spp. were commonly incorrectly identified as additional co-infecting organisms.
FIGURE 11.
Number of pathogens in samples by MassCode assay. (a) Including only main pathogens (Campylobacter spp., C. difficile, norovirus and S. enterica); and (b) including all 11 pathogens.
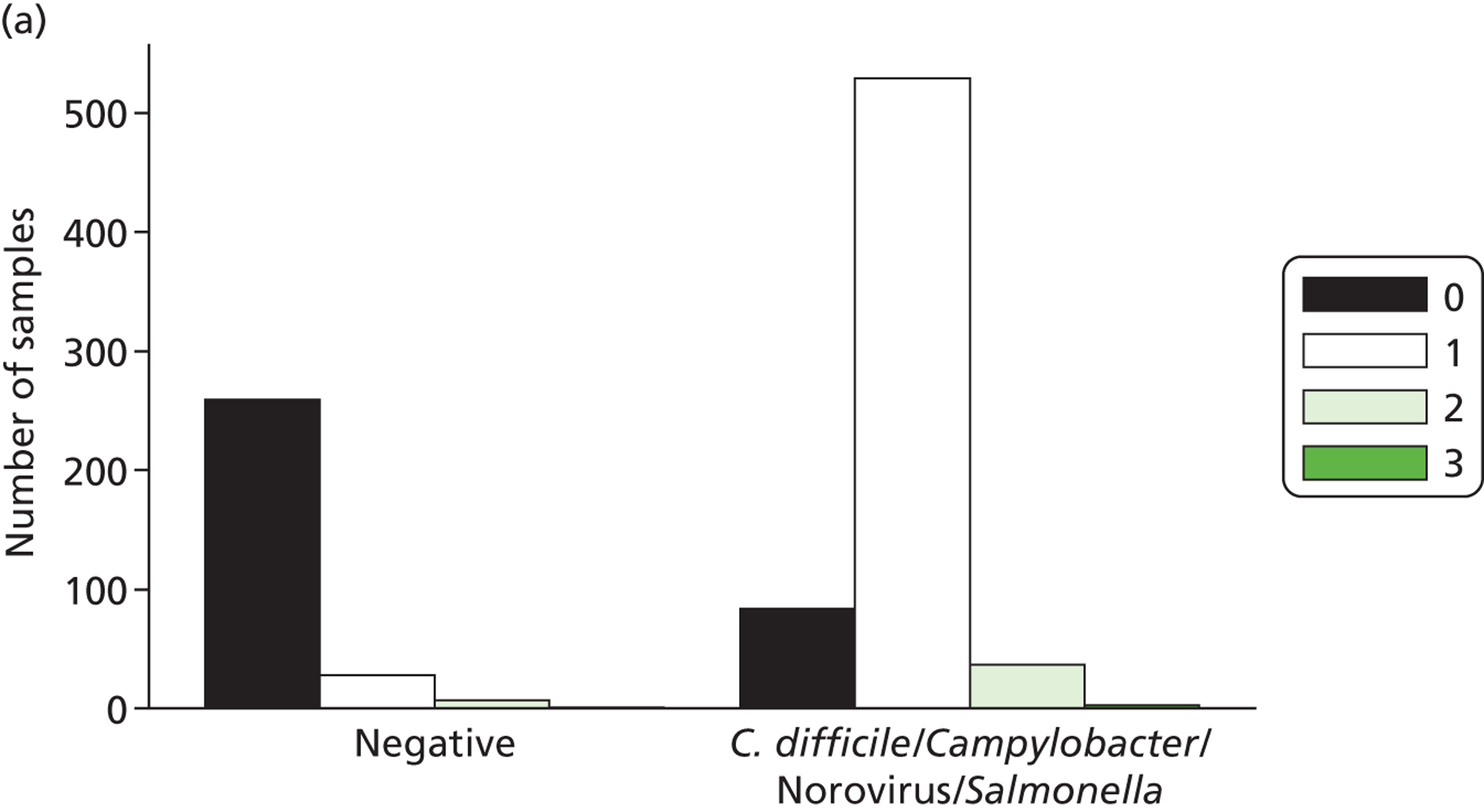
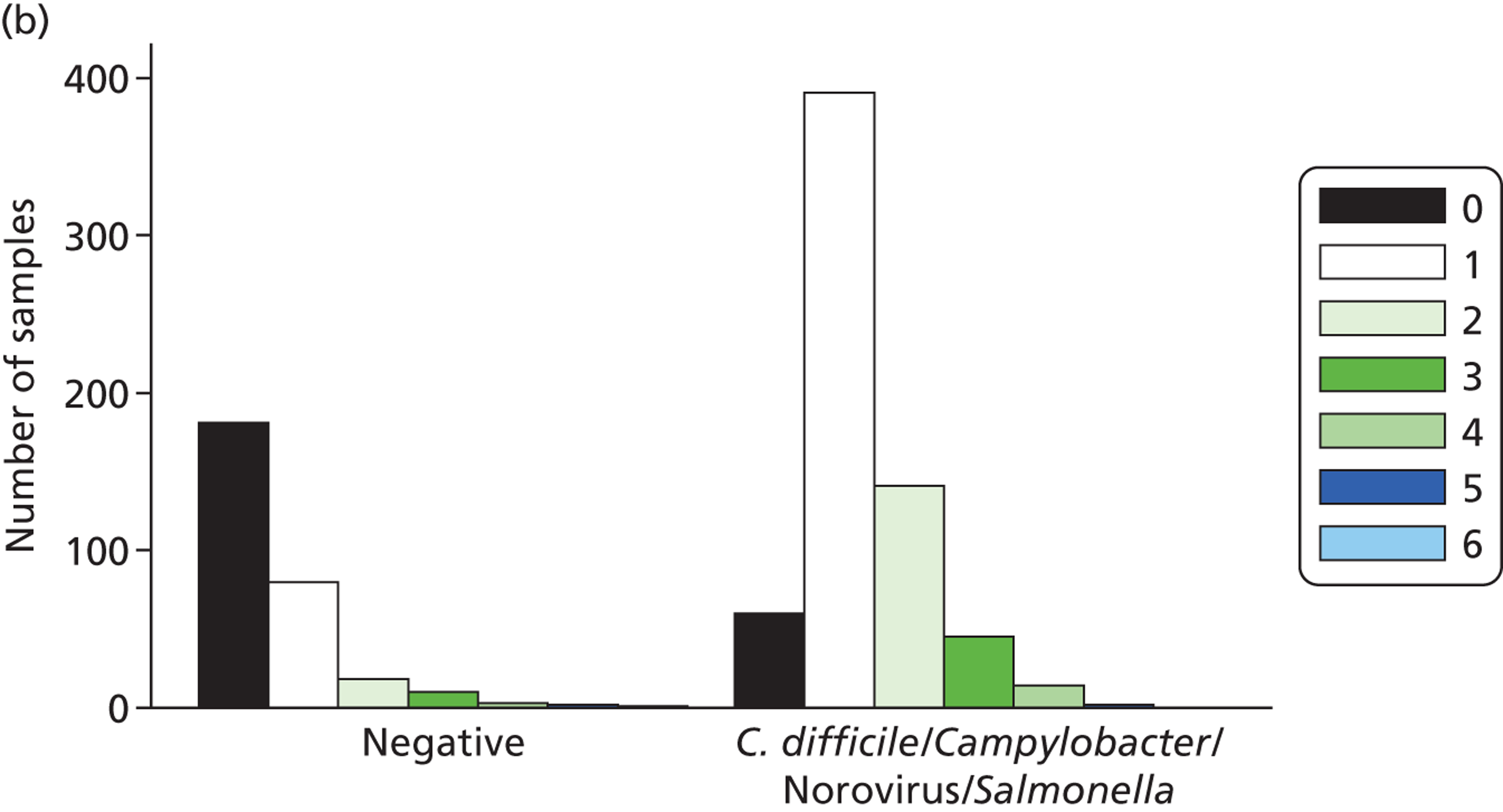
Sequencing Cryptosporidium product
The sections Specificity of MassCode for additional organisms and Discrepant results for additional organisms demonstrated that, although Cryptosporidium spp. were detected in high numbers by MassCode, 94% of these were confirmed to be false positive by singleplex PCR. Results from the sequencing of the Cryptosporidium product confirmed that the primers targeting Cryptosporidium SSU rRNA were also amplifying Candida spp. Of the four clinical samples tested, two that were called positive for Cryptosporidium spp. by MassCode were found to have produced Candida spp. products rather than Cryptosporidium products. This result helps to explain the high false-positive call rate by MassCode, as Candida spp. could be distinguished by melt curve analysis when tested by singleplex SYBR Green PCR. Although not confirmed through sequencing, it is possible that the high false-positive rate seen in Giardia (only 10% of MassCode positives were confirmed positive by singleplex PCR) may also be as a result of unspecific primer pairs being used, as the Giardia primers selected for use in MassCode also target SSU rRNA.
Discussion
The results of the blinded phase 1 testing of the MassCode assay were mixed, with both advantages and disadvantages readily apparent. In particular, the assay showed sensitivities and specificities over 75% for C. difficile, Campylobacter spp. and norovirus, but failed to achieve this cut-off sensitivity for S. enterica by a considerable margin. The sensitivities found for MassCode are also lower than those reported for a competing enteric multiplex panel, the Luminex assay, albeit in studies conducted and reported by the manufacturer rather than independent studies. Interestingly, Salmonella spp. were also found to have the lowest sensitivity in the Luminex assay, at 85% compared with 98% for C. difficile and Campylobacter spp., and 94% for norovirus. 26 However, although the extraction method recommended for Luminex processing is similar to that used for MassCode, the source of clinically positive samples and criteria for including them in testing remain unknown. Further analysis of qPCR results revealed that over half (59%) of samples missed by MassCode but detectable by qPCR had copy numbers < 100, the limit of detection at which samples can be reliably detected by MassCode. However, for S. enterica fewer than half of the missed positive samples were detectable by qPCR. These data support previous findings from the pre-phase 1 phase (see Chapter 2) that S. enterica is being inefficiently isolated from stool samples.
Perhaps as importantly, among the additional targets of the MassCode assay a high number of false-positive results were found, particularly for Giardia spp. and Cryptosporidium spp. This may be because of the target selected for the detection of these organisms by MassCode; both primer sets targeted SSU rRNA, or part of the 18S gene region common to all eukaryotes. This may increase the opportunity for non-target amplification because of the presence of other eukaryotic and human DNA within the sample matrix, leading to MassCode calling a false-positive result. This was demonstrated through sequencing of the Cryptosporidium PCR product, which confirmed that the SSU rRNA primers also amplified Candida spp. In clinical practice, such a high false-positive rate for rare but serious pathogens could seriously limit an assays utility, as it would require either large amounts of confirmatory testing or substantial additional treatment without a confident diagnosis.
There were also 21 false-positive calls for S. Typhi. Although this is a lower false-positive rate than for Giardia spp. and Cryptosporidium spp., this is also of concern with regards to the MassCode assay, as S. Typhi infection would be regarded as serious, and is also uncommon in the UK. A positive result would lead to (potentially intensive) public health investigations; in this context, false-positive results are extremely undesirable. This result also indicates the primer set chosen for S. Typhi may lead to random product or non-target amplification and positive calling by MassCode.
Conversely, a number of unexpected positives were confirmed positive by qPCR retesting. In particular, the MassCode assay was highly sensitive and specific for C. difficile, finding a number of C. difficile-positive samples that were not found through reference standard testing based on toxin detection by EIA. Of these samples, the majority were collected prior to the introduction in April 2012 of GDH (glutamate dehydrogenase) EIA testing as a first step in a two-stage C. difficile testing algorithm, followed by detecting the presence of toxin using EIA as a second test. 5 Therefore, these additional positives were either toxin negative or were missed by the original EIA. Although it is well known that the particular EIA test used prior to April 2012 has relatively poor sensitivity for C. difficile (≈ 80%),27 it is also known that toxigenic C. difficile can be carried without necessarily causing disease. In the largest study of C. difficile diagnostics to date, patients with PCR-positive toxin-negative C. difficile (carriers) in fact had very similar mortality to PCR-negative toxin-negative patients, with increased mortality associated only with PCR-positive toxin-positive cases. 5 Thus, at least some of these unexpected positives may represent carriage rather than disease isolates. Similarly, it is possible that many of the rarer pathogens identified were coincidentally carried, rather than causing disease, as they were disproportionately represented in co-infections. Sensitivity and specificity were also high for Campylobacter spp. In addition, several of the unexpected positive samples that were confirmed positive by qPCR had been cultured for bacterial pathogens by reference standard testing and none was found. The sensitivity of MassCode in these instances illustrates how PCR-based methods can provide rapid and accurate diagnosis of enteric pathogens.
The independent oversight committee reviewed these results and, as the MassCode assay had clearly failed the pre-specified threshold sensitivity to proceed to phase 2, further investigation of the assay was abandoned. However, as the results indicated that detection of S. enterica might provide generic challenges to other multiplex assays for gastrointestinal pathogens, and given the lack of a large independent validation of the Luminex panel, Health Technology Assessment (HTA) agreed that the Luminex assay should also be run on the same set of samples as for MassCode.
Chapter 4 Blinded investigation of phase 1 samples using the Luminex assay
Introduction
The results of the phase 1 investigation of the MassCode assay raised a generic issue as to the sensitivity and specificity of multiplex tests for gastrointestinal infections, particularly with regards to S. enterica. The phase 1 collection of a large panel of well-characterised stool samples provided an opportunity to independently compare and validate the only marketed multiplex assay, the Luminex xTAG® Gastrointestinal Pathogen Panel (GPP). As the MassCode assay did not meet the required threshold level of sensitivity and specificity, and given the lack of a large independent validation of this different Luminex assay, the funders agreed that this should also be run in Leeds on the same set of samples.
Methods
xTAG Gastrointestinal Pathogen Panel
Nucleic acid extracts were screened with the xTAG GPP according to the manufacturers’ protocol (www.luminexcorp.com/Products/Assays/ClinicalDiagnostics/xTAGGPP/).
The GPP is a qualitative multiplex test intended for the simultaneous detection and identification of nucleic acids from multiple gastroenteritis-causing viruses, parasites and bacteria (including toxin gene detection) in human stool samples that are fresh, frozen or in a holding medium, from individuals with signs and symptoms of infectious colitis or gastroenteritis.
The following pathogen types and subtypes are identified using the xTAG GPP:
-
adenovirus 40/41
-
Campylobacter
-
Clostridium difficile toxin A/B
-
Cryptosporidium
-
Entamoeba histolytica
-
Escherichia coli O157
-
enterotoxin-producing E. coli (ETEC) – heat-labile toxin/heat-stable toxin
-
Giardia
-
norovirus GI/GII
-
rotavirus A
-
Salmonella
-
Shiga-like toxin-producing E. coli (STEC) stx 1/stx 2
-
Shigella
-
Vibrio cholerae
-
Yersinia enterocolitica.
The xTAG Gastrointestinal Pathogen Panel (xTAG GPP) incorporates a multiplex reverse transcriptase-polymerase chain reaction (RT-PCR) with Luminex’s proprietary universal tag sorting system on the Luminex platform. Each target or IC in the sample results in PCR amplicons ranging from 58 to 293 base pair (bp) (not including the 24-mer tag). The RT-PCR product is added to a hybridisation/detection reaction containing the universal tag and the streptavidin, R–phycoerythrin conjugate. Each Luminex bead population detects a specific bacterial, viral or parasitic target or assay control through a specific antitag/tag hybridisation.
Briefly, for each sample, 10 µl of extracted nucleic acid was amplified in a single multiplex RT-PCR containing xTAG GPP primer mix, and xTAG® OneStep buffer and enzyme mix (Luminex). Two positive controls (norovirus GI RNA and S. enterica DNA) and three negative controls (molecular-grade water) were included per PCR run (96 reactions). The bacteriophage MS2 added prior to nucleic acid extraction acted as an internal/inhibition control for the assay. Amplification was carried out on a Mastercycler® Pro thermal cycler (Eppendorf UK Ltd, Stevenage, UK), using the following cycling conditions:
1 × 53 °C, for 20 minutes
1 × 95 °C for 15 minutes
36 × 95 °C for 30 seconds
58 °C for 30 seconds
72 °C for 30 seconds
1 × 72 °C for 2 minutes
1 × hold at 4 °C.
For the hybridisation/detection reaction, 5 µl of RT-PCR product was added to the appropriate well of a 96-well plate containing xTAG GPP bead mix (20 µl). xTAG reporter solution (75 µl; xTAG 0.22 streptavidin, R–phycoerythrin conjugate and xTAG® Reporter Buffer) was then added to the wells. Hybridisation was performed on the Mastercycler Pro thermal cycler, programmed for 60 °C for 3 minutes, followed by 45 °C for 45 minutes.
Following hybridisation, the median fluorescence intensity (MFI) was generated for each xTAG bead population using the Magpix® System (Luminex; pre-heated to 45 °C). Data were analysed with the xTAG Data Analysis Software for the GPP (TDAS; Luminex) to establish the presence or absence of bacterial, viral or parasitic targets and/or controls in each sample.
Blinded investigation
Two sets of Luminex assays were run, blinded to pathogens isolated from the original sample using the same anonymised study numbers (see Chapter 3). The sensitivity and specificity of the Luminex test, based on the same standardised nucleic acid extraction (see Appendix 1) as had been done for MassCode, was investigated first. All remaining DNA/RNA extracts made in Oxford for phase 1 (which had been stored at –20 °C) were shipped to Leeds (delay of 26 weeks between completion of MassCode testing and beginning of Luminex testing) (denoted ‘MassCode’ and/or ‘Oxford’ extracts). Comparison of MassCode with Luminex results on the same extracts removes one source of variability.
However, the MassCode extraction SOP does not follow the manufacturer’s recommendations for the Luminex assay, and, therefore, all original stool samples (stored at 4 °C) were also shipped to Leeds (see Table 10 for dates of collection of samples), and in June–July 2013 all samples with sufficient material remaining were re-extracted using the QIAsymphony® sample preparation (SP) (Qiagen) automated nucleic acid extraction platform following the manufacturer’s recommendations, and tested in a separate run (denoted ‘fresh’ and/or ‘Leeds’ extracts). Prior to extraction, stool samples (200 µl/pea-sized sample) were emulsified in a pre-treatment solution [900 µl of L6 lysis buffer (Severn Biotech) and 20 µl of isoamyl alcohol], and spiked with 10 µl of MS2 bacteriophage (109 copies/µl; Luminex). Following a bead beating step with Lysing Matrix E (3000 r.p.m. for 1 minute; MP Biomedicals, Santa Ana, CA, USA), samples were centrifuged at 12000 r.p.m. for 15 seconds. The resultant supernatant (250 µl) was transferred to a fresh tube and diluted 1 : 1 with sterile phosphate-buffered saline (PBS). Nucleic acid was extracted using the Virus/Pathogen Mini Kit (Qiagen) and the QIAsymphony® ‘Pathogen Complex 200 with IC’ programme. The final elution volume was set at 110 µl. Negative controls (sterile PBS) were included in each extraction run. Extracts were stored at –20 °C prior to testing.
All Luminex assays were conducted by one pre-registration clinical scientist, who had received training on the assay from Luminex. The output of the xTAG data analysis software for the GPP was a presence or absence call for each pathogen alongside MFI value. No interpretation was therefore required to read the Luminex outputs, which were also exported electronically.
The pre-registration clinical scientist conducting Luminex assays was blinded to results of the reference microbiology test and the results of previous qPCRs done in Oxford, and all other clinical information; samples were identified only by their unique study number and were completely anonymised with regards to patient and microbiological characteristics (what pathogen, if any, had been isolated). Luminex results were only used for the research study and were not returned for patient management.
As detailed previously (see Chapter 3), samples that were unexpectedly positive or negative for target organisms were retested in duplicate using qPCR assays with either dual-labelled probes or SYBR Green master mix. This testing was not done blinded, as the researcher needed to know which qPCR to run. The qPCR assays used are described elsewhere (see Chapter 2 and Appendix 1).
Analysis
Analysis of the Luminex assay results was conducted by an independent member of the team, using an identical analysis plan to the MassCode assay. The Luminex assay has two targets for C. difficile (tcdA and tcdB gene) and includes two primers against two major groups of noroviruses (GI and GII strains). A positive call on either target was counted as indicating the presence of the organism. However, the single qPCRs targeted only the tcdB gene or the GII strain. As only one A+B– strain of C. difficile has ever been described, missed or unexpected positives on either C. difficile target were retested. However, as GI and GII norovirus strains can co-circulate during an epidemic, only norovirus missed or unexpected positives based on the GII strain were retested with single qPCR.
Sensitivity and specificity were calculated compared with the microbiological reference standard for each of the main organisms (primary reference standard). As for the MassCode assay, several unexpected positives on the Luminex assay were confirmed by qPCR results (implying the target pathogen was genuinely present in the sample, but missed in the original microbiology work-up). In secondary analysis we therefore compared the Luminex-positive results with a combined standard of reference microbiological assay plus single qPCR testing of unexpected and missed positives from the specific Luminex assay. In these analyses, additional qPCR results that had been generated from MassCode missed/unexpected positives were ignored, because they would not have been known had only the Luminex assay been evaluated. Thus, these analyses aimed to consider each assay/extraction method on a like-for-like basis. Thus, samples positive on standard reference microbiology but negative on the Luminex assay and confirmed negative on qPCR were considered negative, and samples negative on standard reference microbiology but positive Luminex and confirmed positive on qPCR were considered positive.
See Chapter 5 for a comparison of the results across MassCode and Luminex assays.
Results
Of the original 948 DNA/RNA MassCode/Oxford extracts, 937 had sufficient material remaining to be tested using the Luminex assay in a direct comparison. A total of 839 of the original 948 samples had sufficient material remaining for re-extraction (Table 16). Testing of samples was performed between June and July 2013.
| Pathogen | MassCode | Luminex | |
|---|---|---|---|
| Oxford/MassCode extract (n = 948) | Oxford/MassCode extract (n = 937) | Leeds/fresh extract (n = 839) | |
| Campylobacter spp. | 200 | 199 | 117 |
| C. difficile | 199 | 195 | 199 |
| Norovirus | 199 | 198 | 199 |
| S. enterica | 60 | 58 | 33 |
| Negative for all these pathogens | 295 | 292 | 295 |
Sensitivity and specificity for the main organisms
For Campylobacter spp., C. difficile and norovirus, high sensitivities (> 92%, most > 97%) and specificities (> 96%) were observed, regardless of extraction method and regardless of whether comparisons were made with microbiology alone or microbiology plus qPCR results (Figures 12–14 and Table 17). Interestingly, the lower sensitivities were observed on the Luminex test of freshly extracted material (Campylobacter spp. 98%, C. difficile 96%, norovirus 92%).
FIGURE 12.
Luminex assay sensitivity and specificity for Campylobacter spp., using original MassCode/Oxford and fresh/Leeds extracts, and excluding (a) and including (b) qPCR results. Sensitivity = 100 × called positive/true positive; specificity = 100 × called negative/true negative.
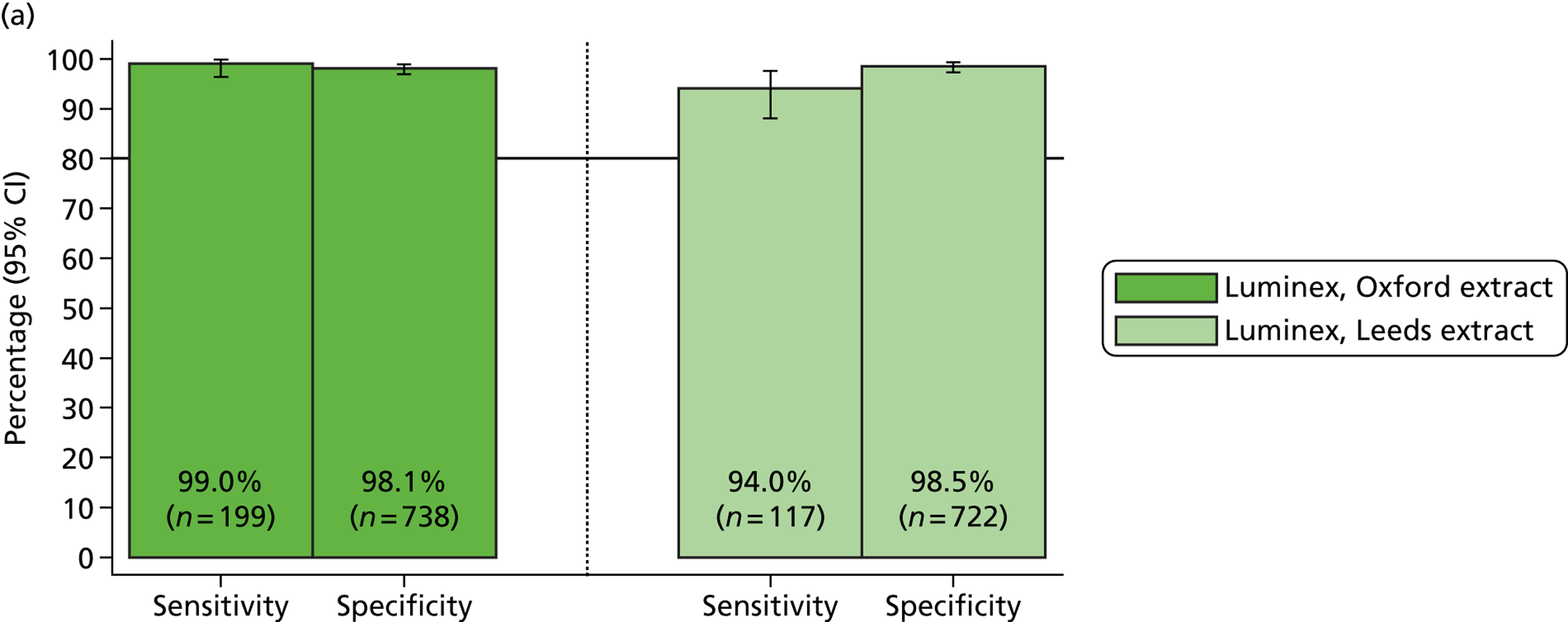
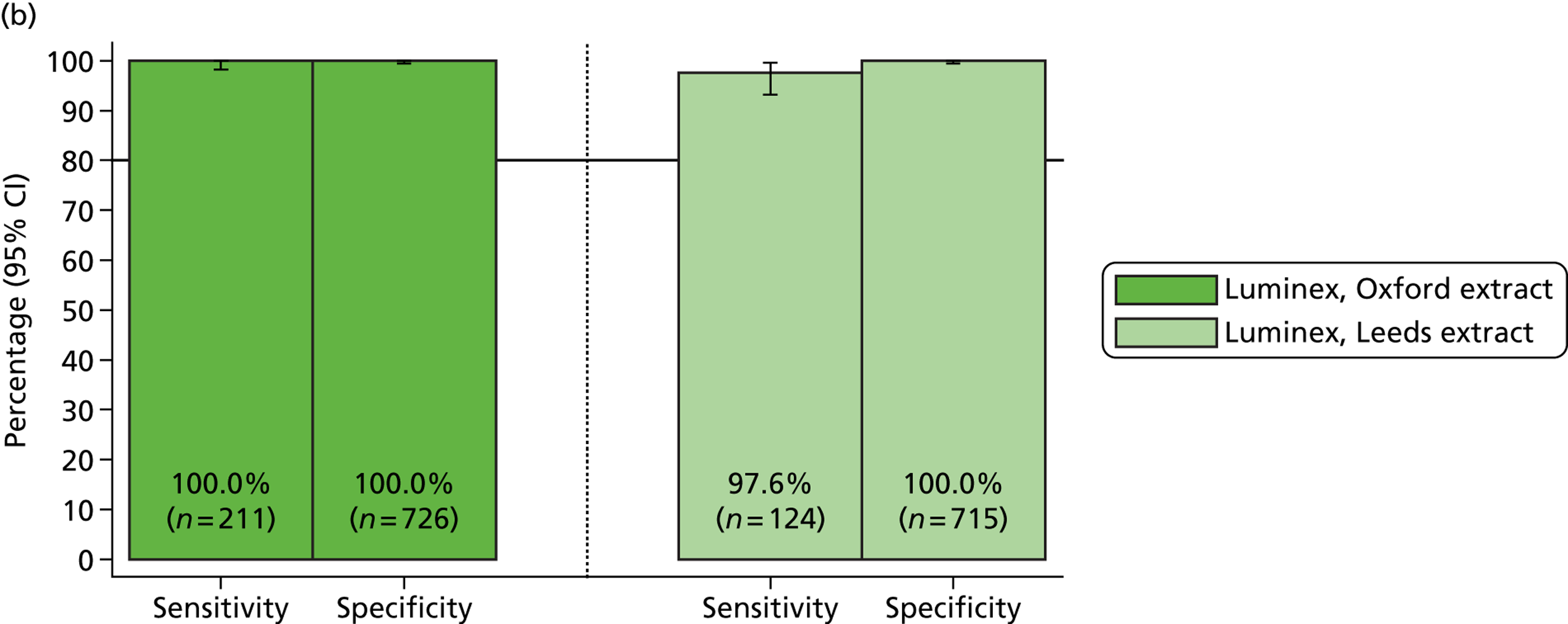
FIGURE 13.
Luminex assay sensitivity and specificity for C. difficile, using original MassCode/Oxford and fresh/Leeds extracts, and excluding (a) and including (b) qPCR results. Sensitivity = 100 × called positive/true positive; specificity = 100 × called negative/true negative.
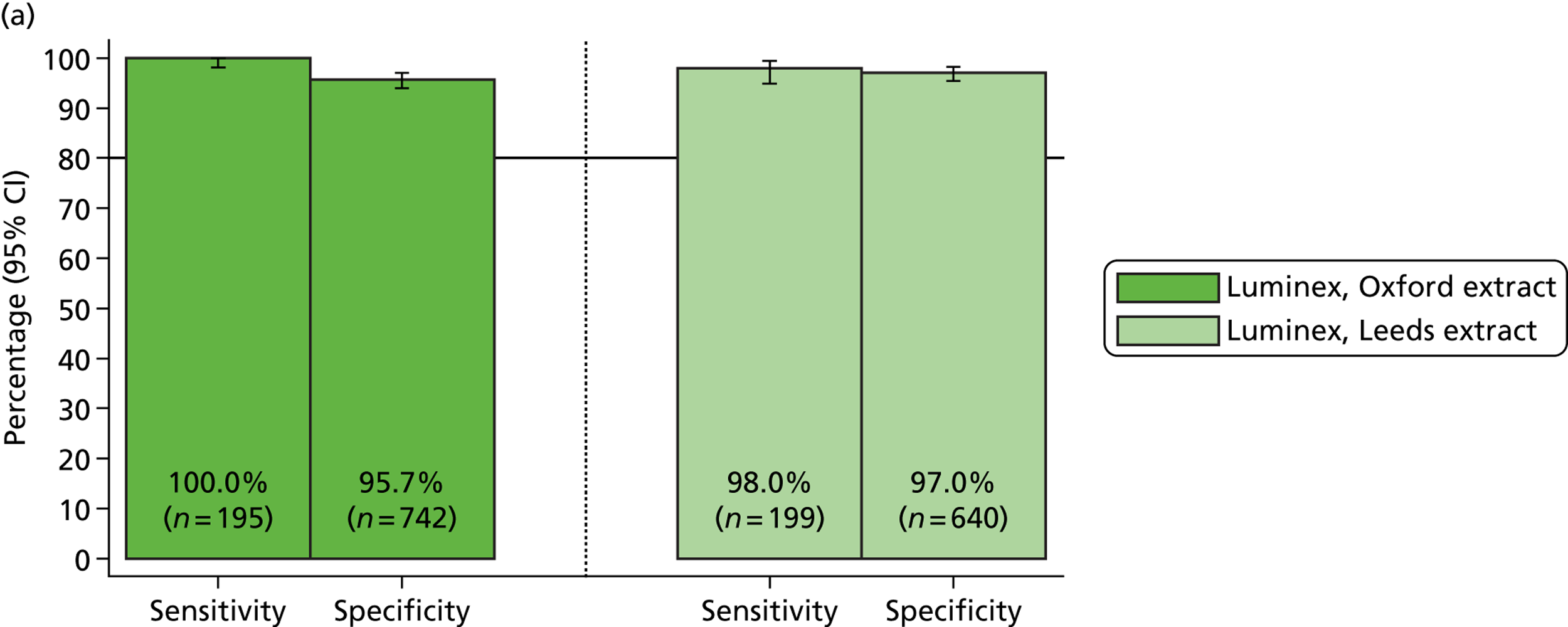
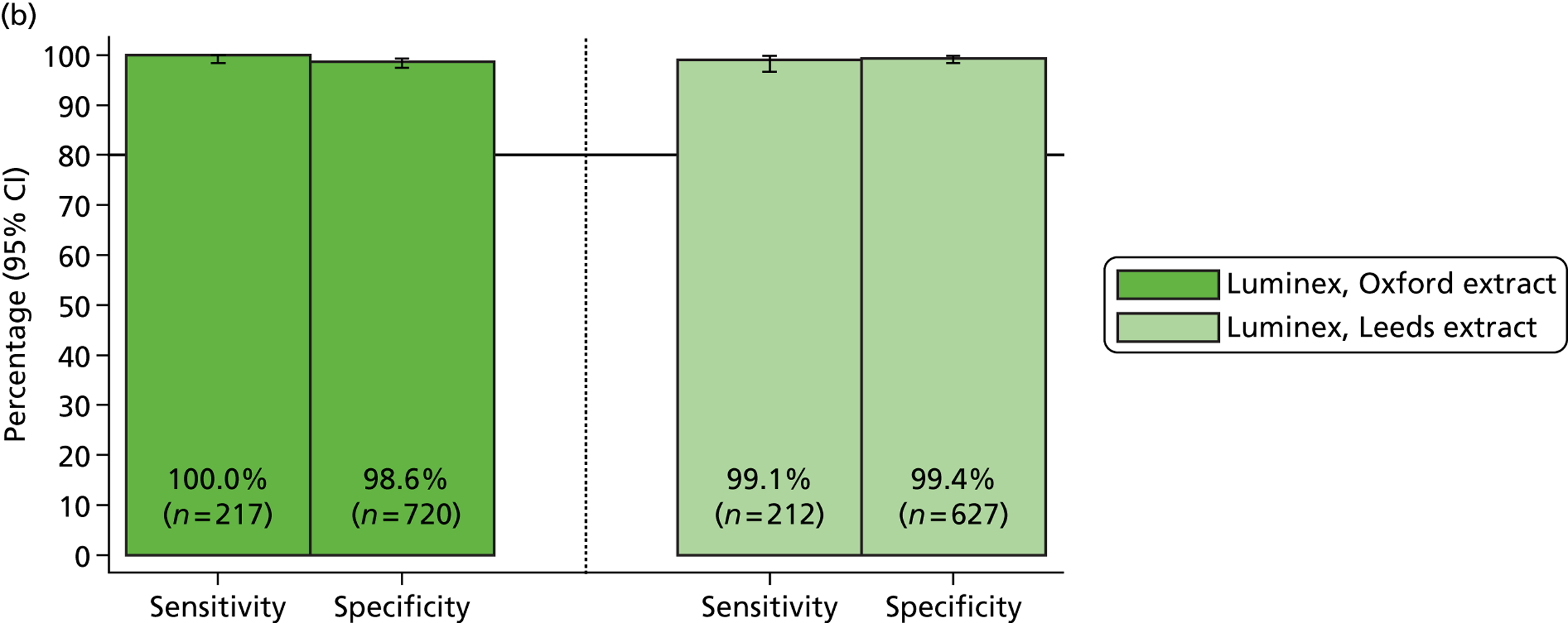
FIGURE 14.
Luminex assay sensitivity and specificity for norovirus, using original MassCode/Oxford and fresh/Leeds extracts, and excluding (a) and including (b) qPCR results. Sensitivity = 100 × called positive/true positive; specificity = 100 × called negative/true negative.
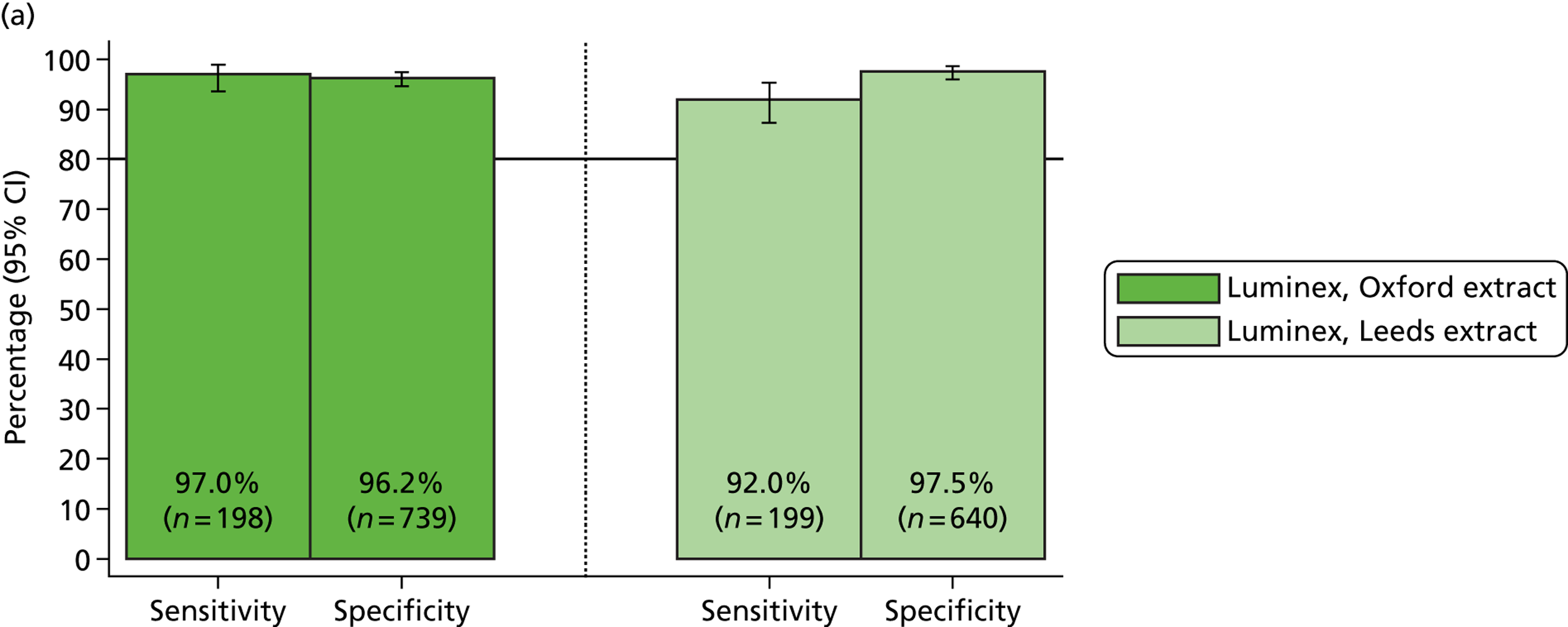
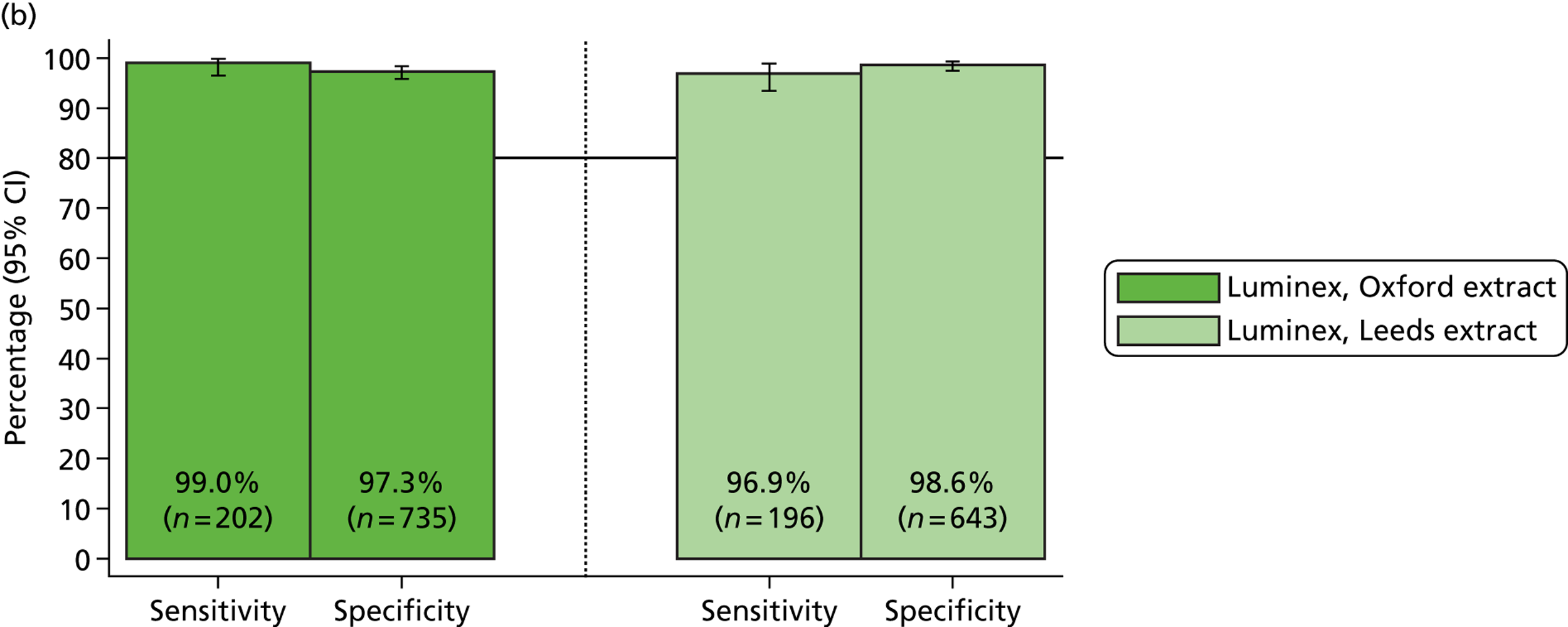
| Organism | Reference: microbiology | Reference: microbiology + qPCR | ||||||
|---|---|---|---|---|---|---|---|---|
| MassCode/Oxford extracts | Fresh/Leeds re-extractions | MassCode/Oxford extracts | Fresh/Leeds re-extractions | |||||
| Sensitivity | 95% CI | Sensitivity | 95% CI | Sensitivity | 95% CI | Sensitivity | 95% CI | |
| Campylobacter | 99.0% | 96.4% to 99.9% | 94.0% | 88.1% to 97.6% | 100.0% | 98.3% to 100.0% | 97.6% | 93.1% to 99.5% |
| C. difficile | 100% | 98.1% to 100.0% | 98.0% | 94.9% to 99.4% | 100.0% | 98.3% to 100.0% | 99.1% | 96.6% to 99.9% |
| Norovirus | 97.0% | 93.5% to 98.9% | 92.0% | 87.3% to 95.4% | 99.0% | 96.5% to 99.9% | 96.9% | 93.5% to 98.9% |
| S. enterica | 84.5% | 72.6% to 92.7% | 45.5% | 28.1% to 63.6% | 98.1% | 89.7% to 100.0% | 60.0% | 38.7% to 78.9% |
| Organism | Reference: microbiology | Reference: microbiology + qPCR | ||||||
|---|---|---|---|---|---|---|---|---|
| MassCode/Oxford extracts | Fresh/Leeds re-extractions | MassCode/Oxford extracts | Fresh/Leeds re-extractions | |||||
| Specificity | 95% CI | Specificity | 95% CI | Specificity | 95% CI | Specificity | 95% CI | |
| Campylobacter | 98.1% | 96.8% to 99.0% | 98.5% | 97.3% to 99.2% | 100.0% | 99.5% to 100.0% | 100.0% | 99.5% to 100.0% |
| C. difficile | 95.7% | 94.0% to 97.0% | 97.0% | 95.4% to 98.2% | 98.2% | 96.9% to 99.0% | 99.4% | 98.4% to 99.8% |
| Norovirus | 96.2% | 94.6% to 97.5% | 97.5% | 96.0% to 98.6% | 97.3% | 95.8% to 98.3% | 98.6% | 97.4% to 99.4% |
| S. enterica | 91.7% | 89.7% to 93.4% | 98.9% | 97.9% to 99.5% | 92.0% | 90.0% to 93.7% | 98.9% | 97.9% to 99.5% |
| Organism | Positive on Luminex, not identified in laboratory – ‘false positive’/ standard negative | Confirmed as positive on PCR (either carried organisms or missed by original reference test) [not testeda] | Confirmed positive on PCR, but evidence of test negative on microbiology systemb | Negative on Luminex, identified as positive in laboratory – ‘false negative’/ standard positive | Confirmed as positive on single PCR |
|---|---|---|---|---|---|
| Campylobacter | 14/738 | 14/14 | 14/14 | 2/199 | 0/2 |
| C. difficile | 32/742 (12 A+B–) | 22/32 [5c] | 14/22 | 0 | N/A |
| Norovirus | 28/739 (8 GI) | 8/22 [6d] | N/A | 6/198 | 2/6 |
| S. enterica | 73/879 | 2/73 | 0/2 | 9/58 | 1/9 |
| Organism | Positive on Luminex, not identified in laboratory – ‘false positive’/ standard negative | Confirmed as positive on PCR (either carried organisms or missed by original reference test) [not testeda] | Confirmed positive on PCR, but evidence of test negative on microbiology systemb | Negative on Luminex, identified as positive in laboratory – ‘false negative’/ standard positive | Confirmed as positive on single PCR |
|---|---|---|---|---|---|
| Campylobacter | 11/722 | 11/11 | 11/11 | 7/117 | 3/7 |
| C. difficile | 19/640 (5 A+B–) | 15/19 [2c] | 9/15 | 4/199 | 2/4 |
| Norovirus | 16/640 (2 GI) | 7/16d | N/A | 16/199 | 6/16 |
| S. enterica | 9/806 | 0/9 | N/A | 18/33 | 10/18 |
However, major differences in results with the same Luminex assay were found for S. enterica (Figure 15). On the original MassCode/Oxford extracts, sensitivity against microbiological testing was 84% (95% CI 73% to 93%). Although this clearly would have some limitations in clinical practice, it was a substantial improvement over the MassCode sensitivity. However, sensitivity when assayed using fresh/Leeds extracts dropped to 46%, very similar to the MassCode sensitivity, with a corresponding increase in specificity from 92% to 99%. Results were similar including qPCR results, with sensitivity for detecting S. enterica from freshly extracted material of only 60% with the upper limit of the 95% CI around the estimated sensitivity just below 79%.
FIGURE 15.
Luminex assay sensitivity and specificity for S. enterica, using original MassCode/Oxford and fresh/Leeds extracts, and excluding (a) and including (b) qPCR results. Sensitivity = 100 × called positive/true positive; specificity = 100 × called negative/true negative.
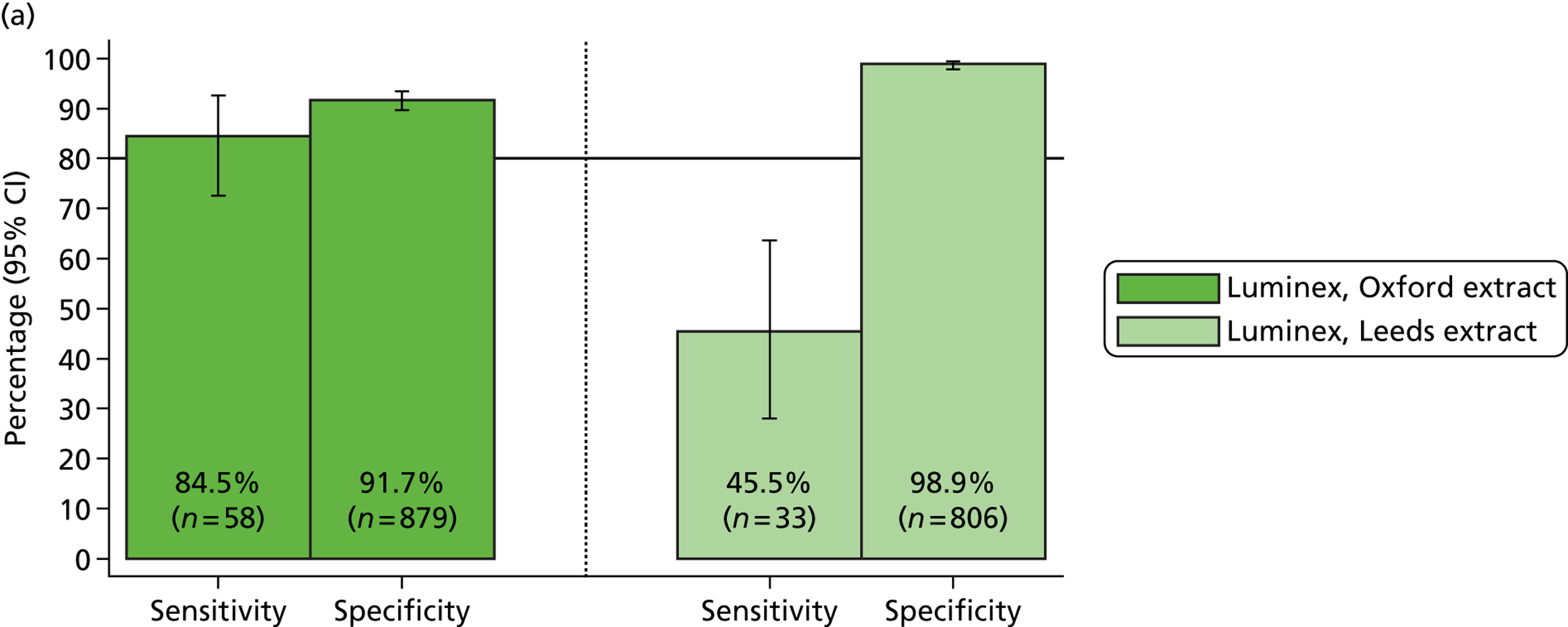
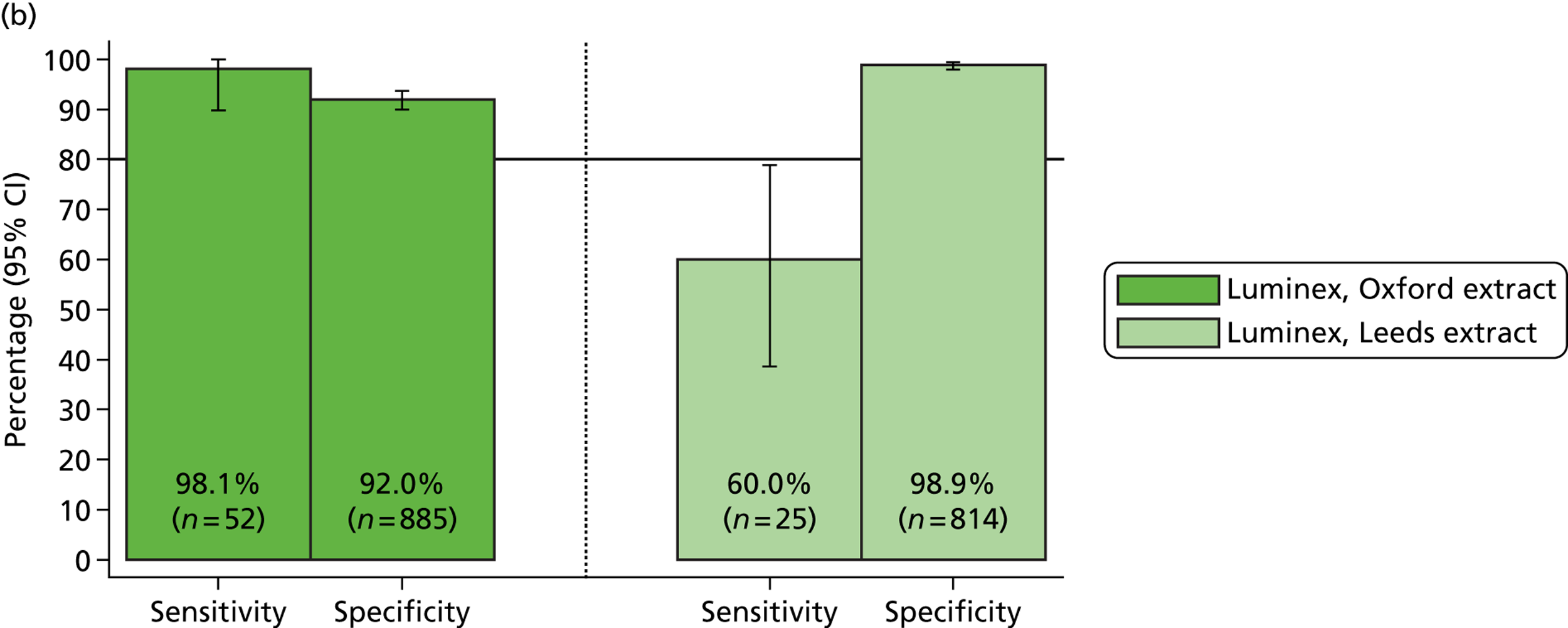
Discrepant results for the main organisms
Table 17(c) and (d) shows more detail about the discrepancies between Luminex positives and reference microbiology, together with results of confirmatory qPCR testing.
Campylobacter was the most accurate target, with all unexpected positives on Luminex confirmed positive by qPCR on both original MassCode/Oxford extracts and freshly extracted material. All samples had undergone faecal culture in the service laboratory, so this organism had been missed. However, it is still possible that the Campylobacter spp. identified by Luminex were present only as carriage and were not present in sufficient numbers to be the cause of disease. The Luminex assay also missed very low numbers of microbiologically identified Campylobacter on both assay runs (two and seven, respectively); neither call on the first run could be confirmed with qPCR, and four out of seven calls on the second run could not be confirmed, suggesting either sample degradation or laboratory error, but demonstrating that the Luminex multiplex assay was highly specific for Campylobacter spp. For Campylobacter spp., the Luminex assay performed similarly on original MassCode/Oxford extracts and material freshly extracted in Leeds.
For C. difficile and norovirus, the Luminex assay had slightly lower sensitivity, more so with the original MassCode/Oxford extracts than the fresh/Leeds extracts, with a corresponding reduction in specificity with the fresh/Leeds extracts compared with the MassCode/Oxford extracts.
For C. difficile, most of the unexpected positives identified by Luminex on both runs were confirmed by qPCR, even in the minority of samples that were positive based on the tcdA gene only (Table 18). In contrast to the MassCode assay and the single qPCR primers for C. difficile, the Luminex assay targets both the C. difficile tcdA and tcdB toxin genes. The majority of toxin-producing strains are A+B+, but there is one important clade which is A–B+ (identified only using the MassCode/Oxford extracts, not the freshly extracted material). A+B– strains have not been confirmed to occur in vivo, although they have been engineered in vitro in knockout studies. Therefore, where Luminex identified a strain as being A+B–, it was either actually an A+B+ strain (a false negative for the tcdB gene) or the sample was actually negative (a false positive for the tcdA gene and a false positive for the pathogen). Both occurred – there were instances of unexpected A+B+ strains called by Luminex that could not be confirmed by qPCR for the tcdB gene and instances of A+B– samples called by Luminex that could be confirmed by tcdB gene qPCR. Very few samples had C. difficile identified by the laboratory but not by Luminex (based on either gene target).
| Toxin genes | MassCode/Oxford extracts | Fresh/Leeds extracts | ||
|---|---|---|---|---|
| Microbiology positive | Microbiology negative | Microbiology positive | Microbiology negative | |
| A+B+ | 193 | 17 | 185 | 14 |
| A–B+ | 1 | 3 | 0 | 0 |
| A+B– | 1 | 12 | 10 | 5 |
| Not found | 0 | 710 | 4 | 621 |
| Total | 195 | 742 | 199 | 640 |
For norovirus, a lower proportion (just under half) of unexpected positives identified in either Luminex assay were confirmed by qPCR. Lower PCR positivity for norovirus may reflect degradation of the single-stranded RNA even with a single assay, or possibly sample degradation with storage. Proportionately, unexpected positives were more likely to be identified as the GI strain than as the widely circulating GII strain (Table 19). No sample was identified with both GI and GII strains. As qPCR testing was based on GII, it was not possible to assess whether or not these GI strain calls reflect false positives, and difficult to assess whether or not these strains are now more likely to be carried without causing disease.
| Genogroup | MassCode/Oxford extracts | Fresh/Leeds extracts | ||
|---|---|---|---|---|
| Microbiology positive | Microbiology negative | Microbiology positive | Microbiology negative | |
| GII | 189 | 20 | 181 | 14 |
| GI | 3 | 8 | 2 | 2 |
| Not found | 6 | 711 | 16 | 624 |
| Total | 198 | 739 | 18 | 640 |
The most marked mismatch between the Luminex assay run on MassCode DNA/RNA extractions compared with freshly extracted samples was in S. enterica. Of the microbiology-negative MassCode/Oxford extracts, 8.3% (73/879) produced unexpected S. enterica positives, but only 16% (9/58) of microbiology-positives were missed. Of these 73 unexpected and nine missed S. enterica positives, only two and one, respectively, were confirmed on qPCR. Both of the two samples unexpectedly confirmed positive for S. enterica with the MassCode/Oxford extracts run on the Luminex assay had definitely been cultured originally in the service microbiology laboratory without identifying S. enterica; one was also positive for S. enterica using the MassCode assay. These two unconfirmed unexpected positives are particularly concerning for this potentially virulent pathogen. Unconfirmed unexpected positives occurred in similar numbers using the Luminex assay on MassCode/Oxford extracts and using MassCode assay on MassCode/Oxford extracts.
In contrast, running the same assay on freshly extracted samples substantially reduced the proportion of unexpected positives to 1.1% (9/806), but correspondingly far more microbiology positives were missed (55%;18/33). Thus, the sensitivity of the Luminex assay on fresh/Leeds extracts was similar to that of the MassCode assay. None of the nine unexpected positives, but 10 of the 18 missed positives using Luminex, were confirmed by qPCR, suggesting that both the primers/extraction method and multiplexing the assays make important contributions to the reduced sensitivity for this organism; as for MassCode.
Specificity of Luminex for additional organisms
It was possible to calculate the specificity of the Luminex assay for the additional targets. Known co-infections identified by the routine laboratory were excluded from each specificity analysis (one co-infection with Shigella spp. and one with Cryptosporidium spp. as for MassCode, plus one with Entamoeba histolytica). All unexpected positives for organisms in common with the MassCode assay were tested with singleplex assays using the MassCode primers: secondary analyses considered any confirmed positives as a positive standard (i.e. excluded them from analyses of specificity). However, for those targets not included in the MassCode primer set, additional PCRs were not available and so were not carried out. For these pathogens, only known microbiology positives were excluded from analyses of specificity.
The results are summarised in Tables 20 and 21 and Figures 16 and 17. Considering the Luminex assay run on MassCode/Oxford extracts, specificity was lowest for Giardia spp., at 97%, after excluding qPCR positives from the reference standard (compared with 88% for MassCode), and equal to or more than 99% for E. coli O157, Shigella spp. and rotavirus. Specificities were considerably higher than for MassCode, particularly for Cryptosporidium spp. (88% specificity with MassCode excluding qPCR-positives). Specificities were slightly higher for the Luminex assay run on fresh/Leeds extracts.
| Organism | Reference: microbiology | Reference: microbiology + PCR | ||
|---|---|---|---|---|
| Specificity (%) | 95% CI (%) | Specificity % | 95% CI (%) | |
| In MassCode panel | ||||
| E. coli O157 | 99.5 | 98.8 to 99.8 | N/A | |
| S. Typhi | N/A | |||
| Shigella spp. | 99.0 | 98.2 to 99.6 | 99.6 | 98.9 to 99.9 |
| Cryptosporidium spp. | 98.7 | 97.8 to 99.3 | 99.9 | 99.4 to 100 |
| Giardia spp. | 95.2 | 93.6 to 96.5 | 96.6 | 95.3 to 97.7 |
| Rotavirus | 99.8 | 99.2 to 100.0 | 99.8 | 99.2 to 100.0 |
| In Luminex panel only | ||||
| Entamoeba histolytica | 97.6 | 96.5 to 98. | N/A | |
| Yersinia enterocolitica | 99.6 | 98.9 to 99.9 | N/A | |
| Vibrio cholerae | 100.0 | 99.6 to 100.0 | N/A | |
| Adenovirus | 99.8 | 99.2 to 100.0 | N/A | |
| ETEC | 98.3 | 97.2 to 99.0 | N/A | |
| STEC | 99.6 | 98.9 to 99.9 | 99.7 | 99.1 to 99.9 |
| Organism | Reference: microbiology | Reference: microbiology + PCR | ||
|---|---|---|---|---|
| Specificity % | (95% CI) | Specificity % | (95% CI) | |
| In MassCode panel | ||||
| E. coli O157 | 99.6 | 99.0 to 99.9 | N/A | |
| S. Typhi | N/A | |||
| Shigella spp. | 99.5 | 98.8 to 99.9 | 100.0 | 99.6 to 100 |
| Cryptosporidium spp. | 99.6 | 99.0 to 99.9 | 100.0 | 99.6 to 100 |
| Giardia spp. | 98.0 | 96.8 to 98.8 | 99.4 | 98.6 to 99.8 |
| Rotavirus | 99.9 | 99.3–100.0 | 99.9 | 99.3 to 100.0 |
| In Luminex panel only | ||||
| Entamoeba histolytica | 99.8 | 99.1 to 100.0 | N/A | |
| Yersinia enterocolitica | 99.5 | 98.8 to 99.9 | N/A | |
| Vibrio cholerae | 100.0 | 99.6 to 100.0 | N/A | |
| Adenovirus | 99.8 | 99.1 to 100.0 | N/A | |
| ETEC | 98.7 | 97.7 to 99.3 | N/A | |
| STEC | 99.8 | 99.1 to 100.0 | 99.8 | 99.1 to 100.0 |
| Organism | Missed positives, n | Missed positives qPCR positive, n (%) | Unexpected positives, n | Unexpected positives qPCR positive, n (%) |
|---|---|---|---|---|
| Main pathogens | ||||
| Campylobacter spp. | 2 | 0 (0) | 14 | 14 (100) |
| C. difficile | 0 | N/A | 32 (12 A+B–) | 22 (68)a |
| Norovirus | 6 | 2 (33) | 28 (8 GI) | 8 (29)b |
| S. enterica | 9 | 1 (11) | 73 | 2 (3) |
| Additional pathogens in common with MassCode | ||||
| E. coli O157 | – | 5 | N/A | |
| Shigella spp. | 0 | 9 | 5 (56) | |
| Cryptosporidium spp. | 0 | 12 | 11 (92) | |
| Giardia spp. | – | 45 | 14 (31) | |
| Rotavirus | – | 2 | 0 (0) | |
| Additional pathogens in Luminex only | ||||
| Entamoeba histolytica | 1 | 22 | ||
| Yersinia enterocolitica | – | 4 | ||
| Vibrio cholerae | – | 0 | ||
| Adenovirus | – | 2 | ||
| ETEC | – | 16 | ||
| STEC | – | 4 | 1 (25) | |
| Organism | Missed positives, n | Missed positives qPCR positive, n (%) | Unexpected positives, n | Unexpected positives qPCR positive, n (%) |
|---|---|---|---|---|
| Main pathogens | ||||
| Campylobacter spp. | 7 | 3 (43) | 11 | 11 (100) |
| C. difficile | 4 | 2 (50) | 19 (5 A+B–) | 15 (79)a |
| Norovirus | 16 | 6 (38) | 16 (2 GI) | 7 (44)b |
| S. enterica | 18 | 10 (56) | 9 | 0 (0) |
| Additional pathogens in common with MassCode | ||||
| E. coli O157 | – | 3 | N/A | |
| Shigella spp. | 0 | 4 | 4 (100) | |
| Cryptosporidium spp. | 0 | 3 | 3 (100) | |
| Giardia spp. | – | 17 | 12 (71) | |
| Rotavirus | – | 1 | 0 (0) | |
| Additional pathogens in Luminex only | ||||
| Entamoeba histolytica | 1 | 2 | ||
| Yersinia enterocolitica | – | 4 | ||
| Vibrio cholerae | – | 0 | ||
| Adenovirus | – | 2 | ||
| ETEC | – | 11 | ||
| STEC | – | 2 | Not tested | |
FIGURE 16.
Specificity of the Luminex assay run on MassCode/Oxford extracts across all organisms. (a) Reference microbiology; and (b) reference microbiology + PCR. Specificity = 100 × called negative/true negative. Light green bars are tests where no qPCR was done to exclude positives missed by original microbiology.
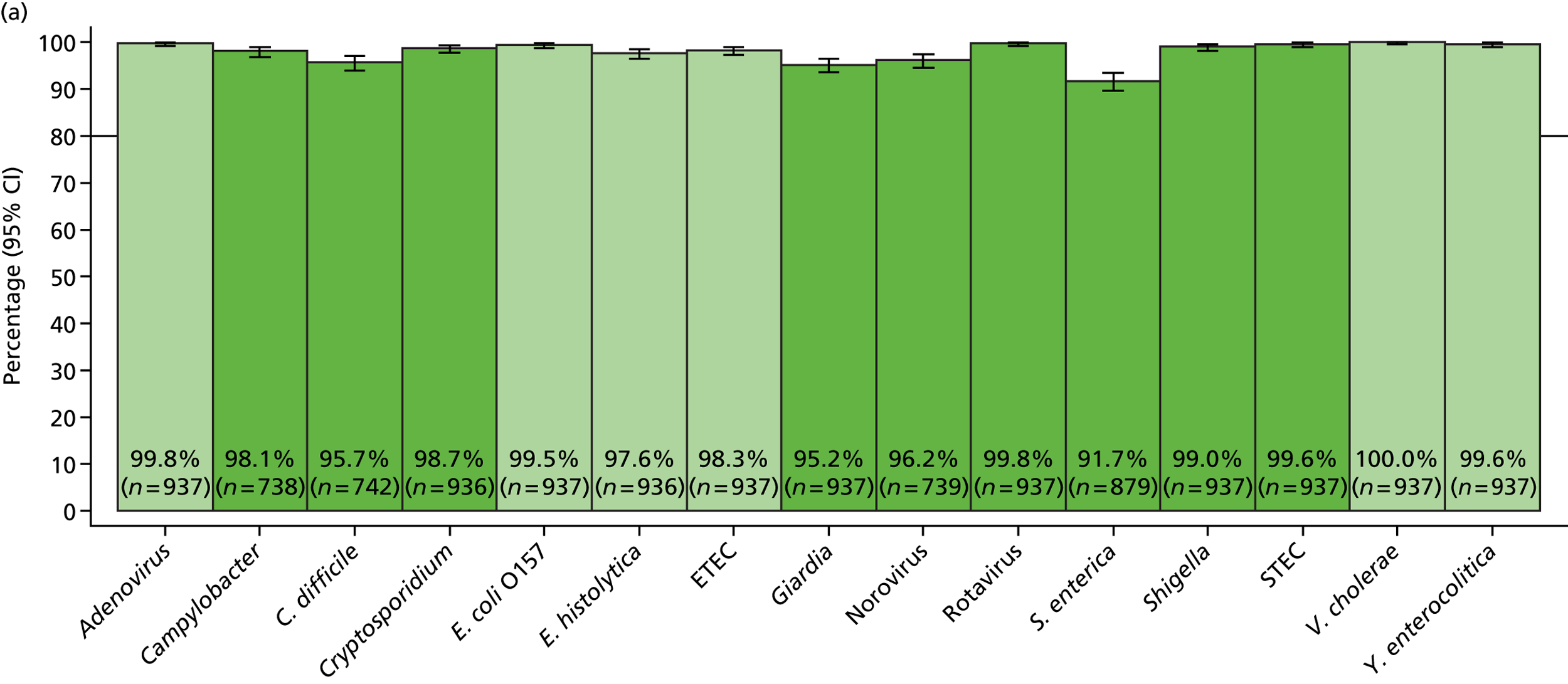
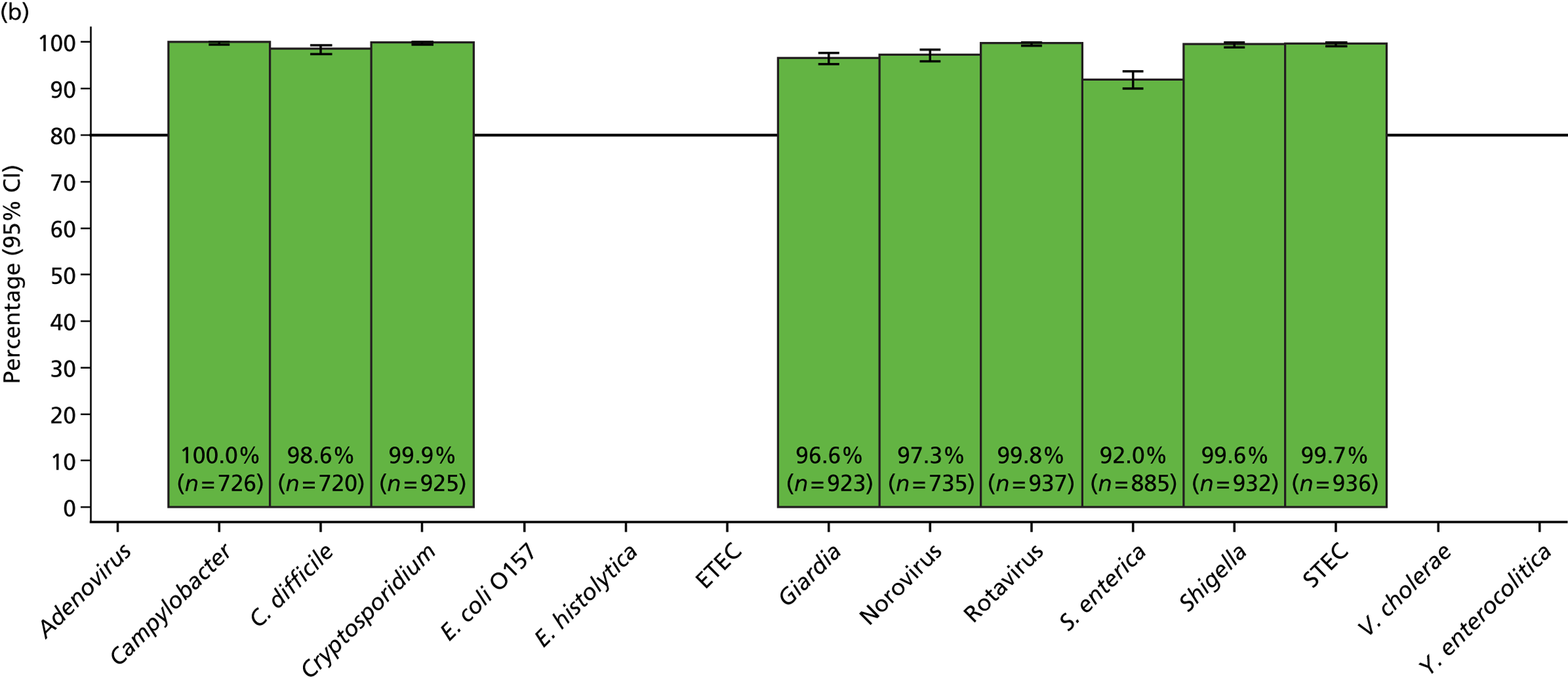
FIGURE 17.
Specificity of the Luminex assay run on fresh/Leeds extracts across all organisms. (a) Reference microbiology; and (b) reference microbiology + PCR. Specificity = 100 × called negative/true negative. Light green bars are tests where no qPCR was done to exclude positives missed by original microbiology.
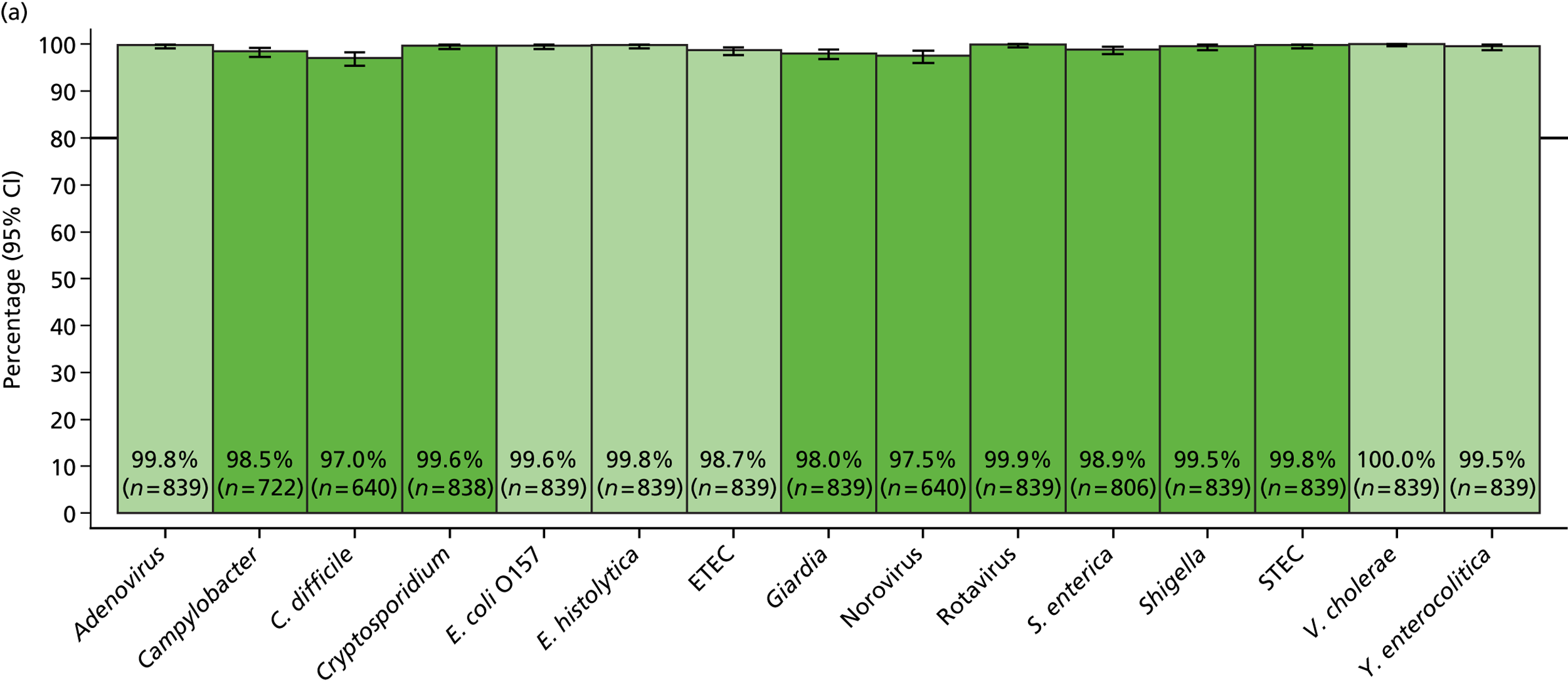
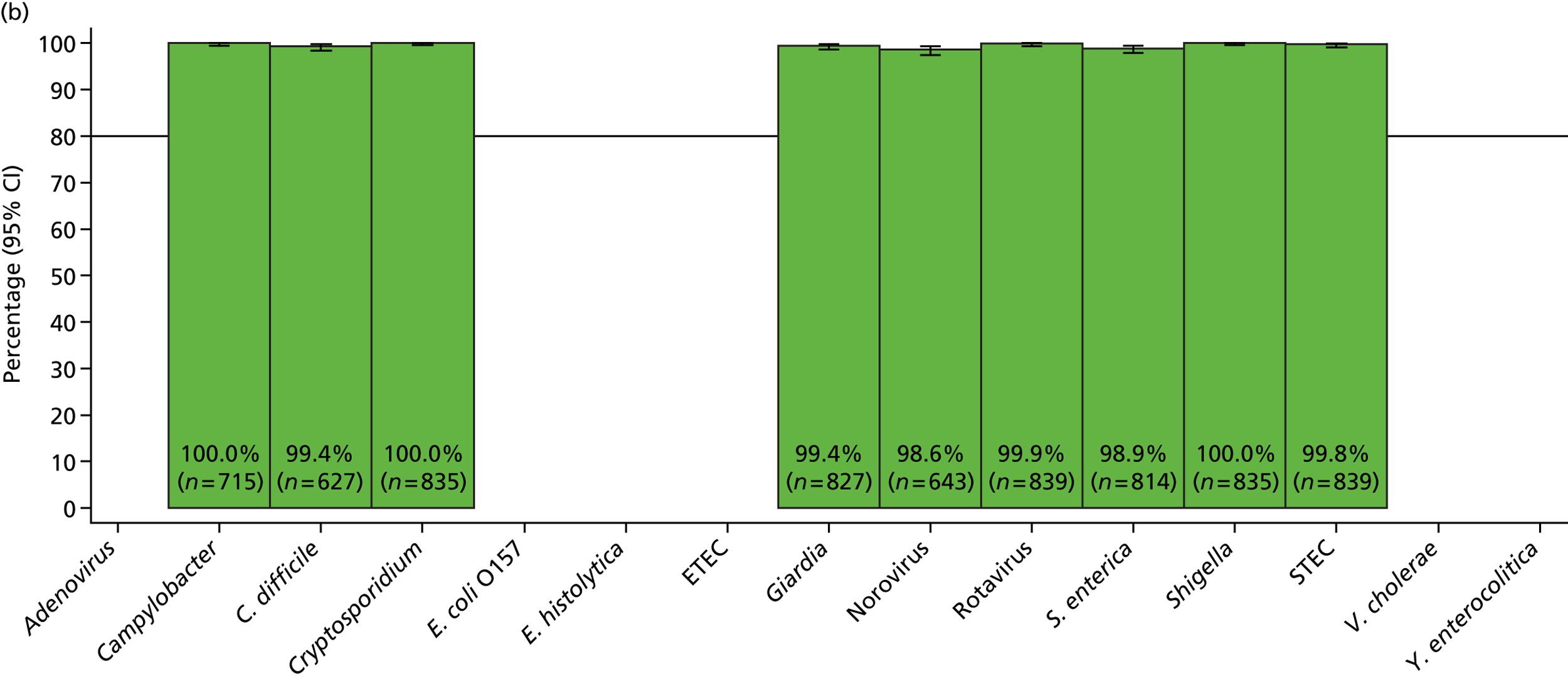
Discrepant results for additional organisms
Unexpected positive samples were retested using the same PCR primers/probes as used to investigate unexpected positives found by the MassCode assay (see Discrepant results for the main organisms), where these were available (Table 21).
Although false-positive call rates were substantially lower than for the MassCode assay, there were still a reasonable number (45) for Giardia using the MassCode/Oxford extracts, of which only 31% were confirmed on qPCR. In contrast, all but one of the unexpected Cryptosporidium spp. calls using the Luminex assay on the MassCode/Oxford extracts were confirmed by qPCR. We were not able to confirm what proportion of the substantial minority of calls for Entamoeba (n = 22) and ETEC (n = 16) were genuine. However, there were few false positives for Shigella spp., and rotavirus. In contrast to the MassCode assay, using MassCode/Oxford extracts with the Luminex assay, these unexpected true positives were identified without increasing the number of false negatives.
In contrast, performing the identical assay on fresh/Leeds extracts, the number of unexpected positives (false positives vs. microbiology) dropped substantially, most notably for S. enterica and Giardia spp. Correspondingly, a greater proportion of these unexpected positives were confirmed on qPCR. However, this was accompanied by a larger number of false negatives for S. enterica.
Co-infections
Co-infections were defined by either expected positives from reference standard testing or unexpected positives by Luminex followed by qPCR to confirm the unexpected positive. Each assay was considered separately.
Using the original MassCode/Oxford extracts, 47 (5.0%) samples had two organisms identified (none had three or more), with 26 (2.8%) having two of the main four pathogens. The proportion identified with co-infections was smaller using the fresh/Leeds extracts; 26 (3.1%) samples had two organisms identified (none had three or more), with 16 (1.9%) having two of the main four pathogens (Table 22).
| First organism | Second organism | Luminex, MassCode/Oxford extract, n | Luminex, fresh/Leeds extract, n |
|---|---|---|---|
| Total co-infections | 47 | 26 | |
| C. difficile | Campylobacter spp. | 3 | 2 |
| C. difficile | Norovirus | 14 | 8 |
| C. difficile | Giardia spp. | 2 | 2 |
| S. enterica | Campylobacter spp. | 5 | 4 |
| S. enterica | STEC E. coli | 1 | 0 |
| S. enterica | Shigella spp. | 2 | 1 |
| S. enterica | Norovirus | 1 | 0 |
| S. enterica | Entamoeba histolytica | 1 | 0 |
| Campylobacter spp. | Cryptosporidium spp. | 1 | 1 |
| Campylobacter spp. | Giardia spp. | 1 | 1 |
| Campylobacter spp. | Norovirus | 3 | 2 |
| Campylobacter spp. | Shigella spp. | 1 | 1 |
| Norovirus | Cryptosporidium spp. | 10 | 2 |
| Norovirus | Giardia spp. | 2 | 2 |
Co-infections with parasites (Cryptosporidium spp. and Giardia spp.) accounted for 14 and 6 of the co-infections, respectively, while co-infections with C. difficile and Campylobacter spp. or norovirus were most common overall. Using the MassCode/Oxford extract, there were 10 co-infections with norovirus and Cryptosporidium spp. Of note, only 12 and three Cryptosporidium-positive samples were found in total, but 11 and three were present with another pathogen, suggesting this might be more commonly carried than previously suspected, as suggested by the MassCode results. Many of the unexpected Shigella spp. and Giardia spp. were also found as co-infections with other more common species.
As a consequence of the higher sensitivity and specificity of the Luminex compared with the MassCode assay, fewer samples were identified as having co-infections with enteric pathogens. Nevertheless, because sensitivity/specificity is effectively cumulated across the large number of tests in the multiplex panel, using the MassCode/Oxford extracts (Figure 18), 79 (8.4%) samples were positive for two of the four main organisms (12 microbiologically negative samples) and 6 (0.9%) were positive for three of the four on Luminex assay. Of the 937 tested samples, only 202 (22%) had no organism of any type identified by the Luminex assay on MassCode/Oxford extracts, 191 out of 292 (65%) of those microbiologically negative compared with 11 out of 645 (2%) of those positive for any of the four main pathogens. Up to four organisms were identified by Luminex assay in one sample based on the MassCode/Oxford extracts, with 127 samples having two organisms identified by Luminex, 25 samples having three organisms identified and two samples having four organisms.
FIGURE 18.
Number of pathogens in samples by Luminex assay on MassCode/Oxford extracts. (a) Including only main pathogens (Campylobacter spp., C. difficile, norovirus, S. enterica); and (b) including all 11 pathogens.
Using the fresh/Leeds extracts (Figure 19), only 20 (2.4%) samples were positive for two of the four main organisms (just one microbiologically negative sample), and one (0.2%) sample was positive for all four on Luminex assay. Of the 839 tested samples, only 278 (33%) had no organism of any type identified by the Luminex assay on fresh/Leeds extracts, 243 out of 295 (82%) of those microbiologically negative compared with 35 out of 544 (6%) positive for any of the main four pathogens. Similarly to the MassCode/Oxford extracts, up to four organisms were identified by Luminex assay in one sample based on the fresh/Leeds extracts, but in far fewer cases, with only 42 samples with two organisms identified by Luminex on fresh/Leeds extracts, two samples with three organisms, and one sample with four organisms.
FIGURE 19.
Number of pathogens in samples by Luminex assay on fresh/Leeds extracts. (a) Including only main pathogens (Campylobacter spp., C. difficile, norovirus, S. enterica); and (b) including all 11 pathogens.
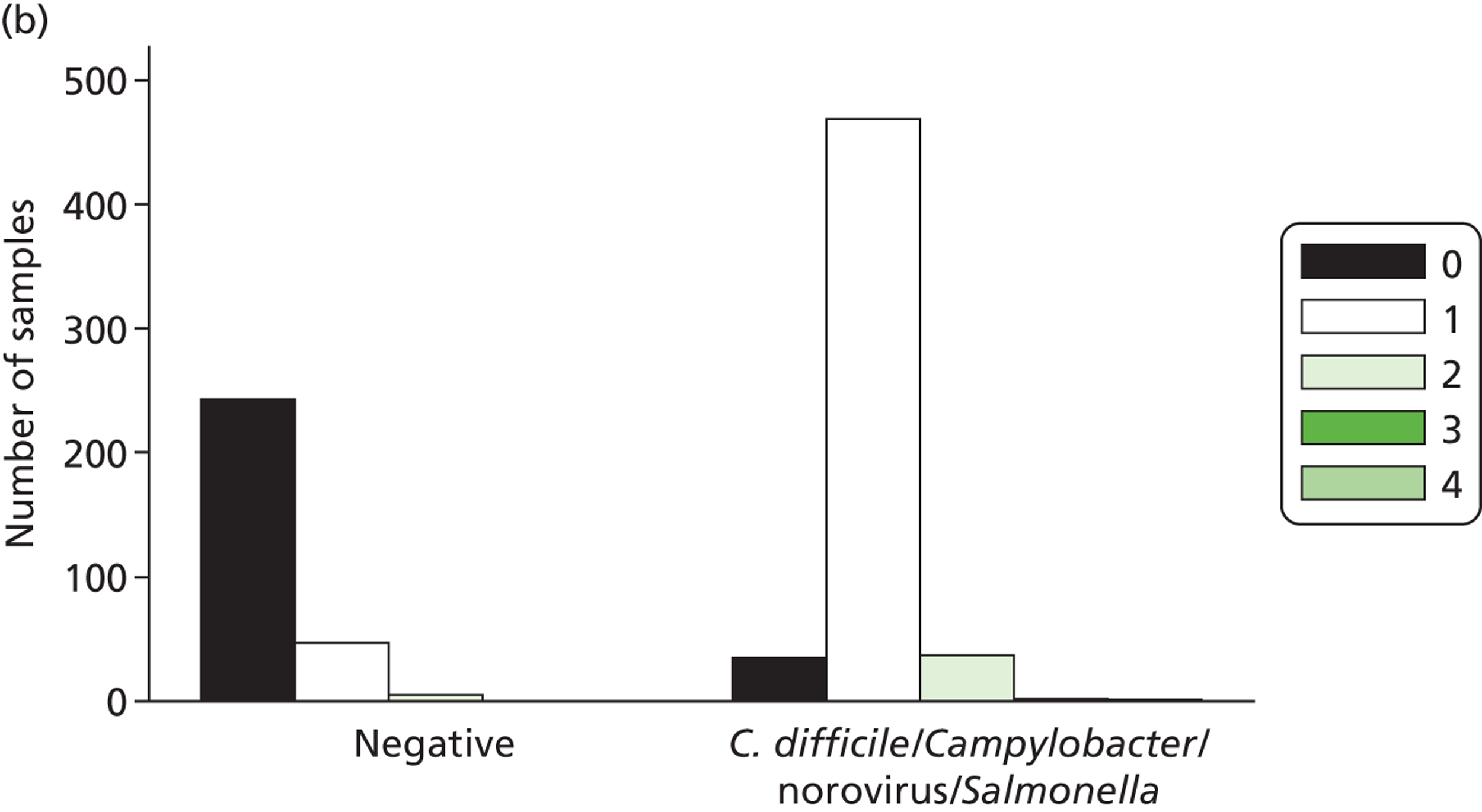
Discussion
Additional testing of the same set of stool samples with the Luminex assay based on two different extractions illustrates the complexity of multiplex tests for gastrointestinal pathogens.
First, overall the Luminex xTag panel showed similar or superior sensitivity and specificity to the MassCode panel. In particular, using fresh/Leeds extracts, the number of unexpected positives using the Luminex extract was very small across all organisms. However, these high specificities came at the cost of low sensitivity to detect a key enteric pathogen, S. enterica; such test sensitivity is too low for this test to be a realistic option for many microbiology laboratories. Interestingly, this low sensitivity was very similar to that observed for the MassCode assay, suggesting that extraction efficiency is genuinely a major obstacle for nucleic acid-based tests for this organism, regardless of platform. However, whereas for the MassCode assay this low sensitivity was also paralleled by relatively low specificity and a substantial number of false positives, for the Luminex assay using fresh/Leeds extracts the number of false positives was also low. Nevertheless, it should be highlighted that the reactions and nucleic acid extracts were kept on ice or in cool blocks at all times and were tested as 4 × 24 reactions (i.e. three strips of eight PCR tubes), using four PCR machines to reduce the possibility of false positives. However, this may not be practical in a service microbiology laboratory where robust assays are required and high numbers of false positives are intolerable.
The storage time of samples between extraction for MassCode and fresh extractions for Luminex testing may have led to degradation of organisms and nucleic acids. This may resolve some of the discrepancy between the Luminex assay results (e.g. 85% and 46% sensitivities for S. enterica for original MassCode/Oxford and fresh/Leeds extracts, respectively). However, it is unknown whether or not this would account for the entire disparity. In-house experiments in Oxford for pre-phase 1 testing led to repeated extraction and testing of S. enterica-positive samples by qPCR, and no significant reduction in copy numbers over approximately 12 months (data not shown) was observed. Nevertheless, the original MassCode/Oxford extracts were approximately 1 year old when they were tested using the Luminex assay, and the stool samples were 1–3 years old when DNA/RNA was extracted in Oxford and freshly re-extracted in Leeds (although they had been stored at 4 °C). Furthermore, the phenomenon of increased detection of S. enterica from stool sample extracts after long-term (> 12-month) storage has been previously reported. Schuurman et al. 12 attribute this to degradation of unstable PCR inhibitors during freeze–thaw cycles. Also striking, was the fact that running the identical assay on original MassCode/Oxford extractions from the same samples also produced a higher number of ‘false-positives’ using original MassCode/Oxford versus fresh/Leeds extractions. Although some of these ‘false-positives’ were subsequently confirmed by qPCR, those for S. enterica and Giardia spp. in particular were rarely confirmed, a problem also demonstrated by the MassCode assay. Extraction efficiency is unlikely to be the cause of this discrepancy. Differences between the two extractions on the same samples suggest that non-target amplification is less likely to be the underlying cause, although we had originally hypothesised this on the basis of the MassCode results, particularly regarding Cryptosporidium spp. and Giardia spp. However, it is possible that each individual extraction would result in carry-over of differing volumes and types of contaminating DNA and inhibitors. This may result in unique amplification of non-specific targets on a per extraction basis. This may be particularly the case where no genuine target is present to compete for PCR resources with non-target products. It is interesting that there were 11 confirmed Cryptosporidium spp. according to the Luminex assay using the original MassCode/Oxford extracts, but only three using fresh/Leeds extracts. The greater isolation with older extracts suggest that some organisms might be present only at low levels, which is consistent with co-carriage with another gastrointestinal pathogen (or even no pathogen being present and diarrhoea being antibiotic associated), rather than the cause of disease itself. Although the OneStep enzyme mix included in this kit is very sensitive, it was used for both Luminex runs and, therefore, cannot be the cause of the differences between the Luminex results.
The product literature for the Luminex assay, the Multi-Site Clinical Summary for CE-marked product, provides the following sensitivity and specificity for the four target organisms:
-
Campylobacter spp.: sensitivity, 97.5%; specificity, 97.8%.
-
C. difficile: sensitivity, 97.7%; specificity, 94.9%.
-
Norovirus GI/GII: sensitivity, 93.5%; specificity, 98.0%.
-
Salmonella spp.: sensitivity, 82.1%; specificity, 99.1%.
For Campylobacter spp., C. difficile and norovirus, these are similar to what was found in this study. For Salmonella spp., this sensitivity is similar to what was found with fresh/Leeds extracts, but the specificity is much higher than our study found using this approach. Rather, the specificity quoted for Salmonella spp. is more similar to that using original MassCode/Oxford extracts with the Luminex assay, where we found considerably lower sensitivity (45.5%).
Chapter 5 Combined analysis of MassCode and Luminex assay results
Introduction
To our knowledge, this is the largest systematic evaluation of multiplex gastrointestinal pathogen diagnostics on the same comprehensively characterised set of stool samples. Here we compare and contrast the MassCode and Luminex assays in terms of their overall ability to identify which pathogens are present in the original sample based on results from combined microbiological and qPCR testing.
Methods
Analysis
Results from the three assays (MassCode panel on MassCode/Oxford extracts, Luminex panel on MassCode/Oxford extracts, Luminex panel on fresh/Leeds extracts) were as described above. Pathogens were the four main pathogens common to both assays (Campylobacter spp., C. difficile, norovirus, S. enterica), additional pathogens common to both assays (Giardia spp., Cryptosporidium spp., E. coli O157, rotavirus, Shigella spp.) plus pathogens only in the MassCode (S. Typhi/Paratyphi) or Luminex (adenovirus, E. histolytica, Y. enterocolitica, V. cholerae and ETEC and STEC E. coli).
The results reported previously (see Chapters 3 and 4) considered each assay (MassCode and Luminex) separately, and used single qPCRs to confirm or refute unexpected and missed positives identified on that specific assay. In order to compare results across the different assays/extraction methods, one single common reference standard is needed. For all samples, a microbiological diagnosis of infecting pathogens was available, and so this could have been used. However, each assay found some unexpected positives that were confirmed by qPCR results, implying that the target pathogen was genuinely present in the sample but had been missed in the original microbiology work-up, and it is reasonable to expect that each multiplex assay should have detected this pathogen if it could have been identified using single qPCRs. However, because of time and financial constraints, not every sample was tested with every qPCR, for each assay/extraction method, single PCRs had been performed only for missed and unexpected positives with that assay/extraction method. Further, the comparison with qPCR-confirmed diagnosis on the individual assays was designed to investigate whether there were specific challenges with the primers being used in the multiplex and not to assess the ability of each assay to provide an NHS-relevant diagnostic tool. For example, all samples included with microbiologically confirmed C. difficile had been tested with an EIA and cultured, suggesting that it was highly likely that the sample contained C. difficile even if subsequently this could not be confirmed by single PCR.
Therefore, for this combined analysis, we used a reference standard designed to reflect the best information about whether or not a pathogen was present in the original specimen, defining a sample to be positive for an organism if (i) the organism had been identified microbiologically, regardless of whether or not its subsequent single PCR was positive or negative, or (ii) the organism had not been identified microbiologically, but one or more single PCRs were positive for the organism.
Samples negative for each organism had no microbiological isolation, and had either not been tested by PCR or all PCRs done were consistently negative (note: single PCRs were all done in duplicate, i.e. were confirmed presence of an organism).
Based on the pathogens common to the Luminex and MassCode assays, Table 23 shows the composition of the phase 1 sample panel. Two pathogens were identified in a total of 60 (6.3%) and three pathogens were identified in two samples (0.2%). In only seven of the samples were co-infections identified by microbiological testing (five S. enterica + Campylobacter spp., one Campylobacter spp. + Cryptosporidium spp., one S. enterica + Shigella spp.). Of the 16 samples confirmed to contain Cryptosporidium spp., 13 represented co-infections as did 10 out of 19 confirmed Giardia-positive samples and three out of five confirmed Shigella spp.-positive samples.
| First organism | Second organism | Third organism | Phase 1 panel (n = 948) | Luminex assay, MassCode extract (n = 937) | Luminex assay, fresh extract (n = 839) |
|---|---|---|---|---|---|
| Campylobacter spp. | 197 (20.8%) | 196 (20.9%) | 117 (14.0%) | ||
| Campylobacter spp. | Cryptosporidium spp. | 1 (0.1%) | 1 (0.1%) | 1 (0.1%) | |
| Campylobacter spp. | Giardia spp. | 1 (0.1%) | 1 (0.1%) | 1 (0.1%) | |
| Campylobacter spp. | Norovirus | 3 (0.3%) | 3 (0.3%) | 3 (0.3%) | |
| Campylobacter spp. | Shigella spp. | 1 (0.1%) | 1 (0.1%) | 1 (0.1%) | |
| C. difficile | 190 (20.0%) | 185 (19.7%) | 190 (22.6%) | ||
| C. difficile | Campylobacter spp. | 14 (1.5%) | 14 (1.5%) | 12 (1.4%) | |
| C. difficile | Campylobacter spp. | Cryptosporidium spp. | 1 (0.1%) | 1 (0.1%) | 1 (0.1%) |
| C. difficile | Cryptosporidium spp. | 1 (0.1%) | 1 (0.1%) | 1 (0.1%) | |
| C. difficile | Giardia spp. | 5 (0.5%) | 5 (0.5%) | 5 (0.6%) | |
| C. difficile | Norovirus | 15 (1.6%) | 15 (1.6%) | 15 (1.8%) | |
| Norovirus | 176 (18.6%) | 175 (18.7%) | 176 (21.0%) | ||
| Norovirus | Cryptosporidium spp. | 10 (1.1%) | 10 (1.1%) | 10 (1.2%) | |
| Norovirus | Giardia spp. | 2 (0.2%) | 2 (0.2%) | 2 (0.2%) | |
| S. enterica | 53 (5.6%) | 51 (5.4%) | 28 (3.3%) | ||
| S. enterica | Campylobacter spp. | 6 (0.6%) | 6 (0.6%) | 4 (0.5%) | |
| S. enterica | Campylobacter spp. | Giardia spp. | 1 (0.1%) | 1 (0.1%) | 1 (0.1%) |
| S. enterica | Norovirus | 1 (0.1%) | 1 (0.1%) | 1 (0.1%) | |
| S. enterica | Shigella spp. | 2 (0.2%) | 2 (0.2%) | 2 (0.2%) | |
| Cryptosporidium spp. | 3 (0.3%) | 3 (0.3%) | 3 (0.2%) | ||
| Giardia spp. | 10 (1.1%) | 10 (1.1%) | 10 (1.2%) | ||
| Shigella spp. | 2 (0.2%) | 2 (0.2%) | 2 (0.2%) | ||
| None of these pathogens | 253 (26.7%) | 251 (26.8%) | 253 (30.2%) | ||
Results
Sensitivity and specificity compared with a combined microbiology plus polymerase chain reaction-positive reference standard
Figures 20–23 show sensitivity and specificity across the three assay runs for the four key pathogens. All 948 phase 1 samples were tested with the MassCode assay; because varying amounts of MassCode/Oxford extract and original sample remained, only 937 samples were tested using the Luminex assay on MassCode/Oxford extracts, and 839 samples were tested using the Luminex assay on fresh extracts. In total, 830 samples were tested using all three methods. Pathogens identified varied across the different assays, and even between the Luminex assays based on different extracts, with only moderate to reasonable within-assay agreement for positive calls (42% for S. enterica, 86–94% for other pathogens), and discrepancies within Luminex run for negative calls for S. enterica (91%).
FIGURE 20.
Campylobacter species (including all PCRs). Sensitivity = 100 × called positive/true positive (including microbiology positive plus any PCR positive). Specificity = 100 × called negative/true negative (excluding microbiology negatives with any PCR positive). Not all samples had sufficient volume to be included in all test evaluations, denominators as shown.
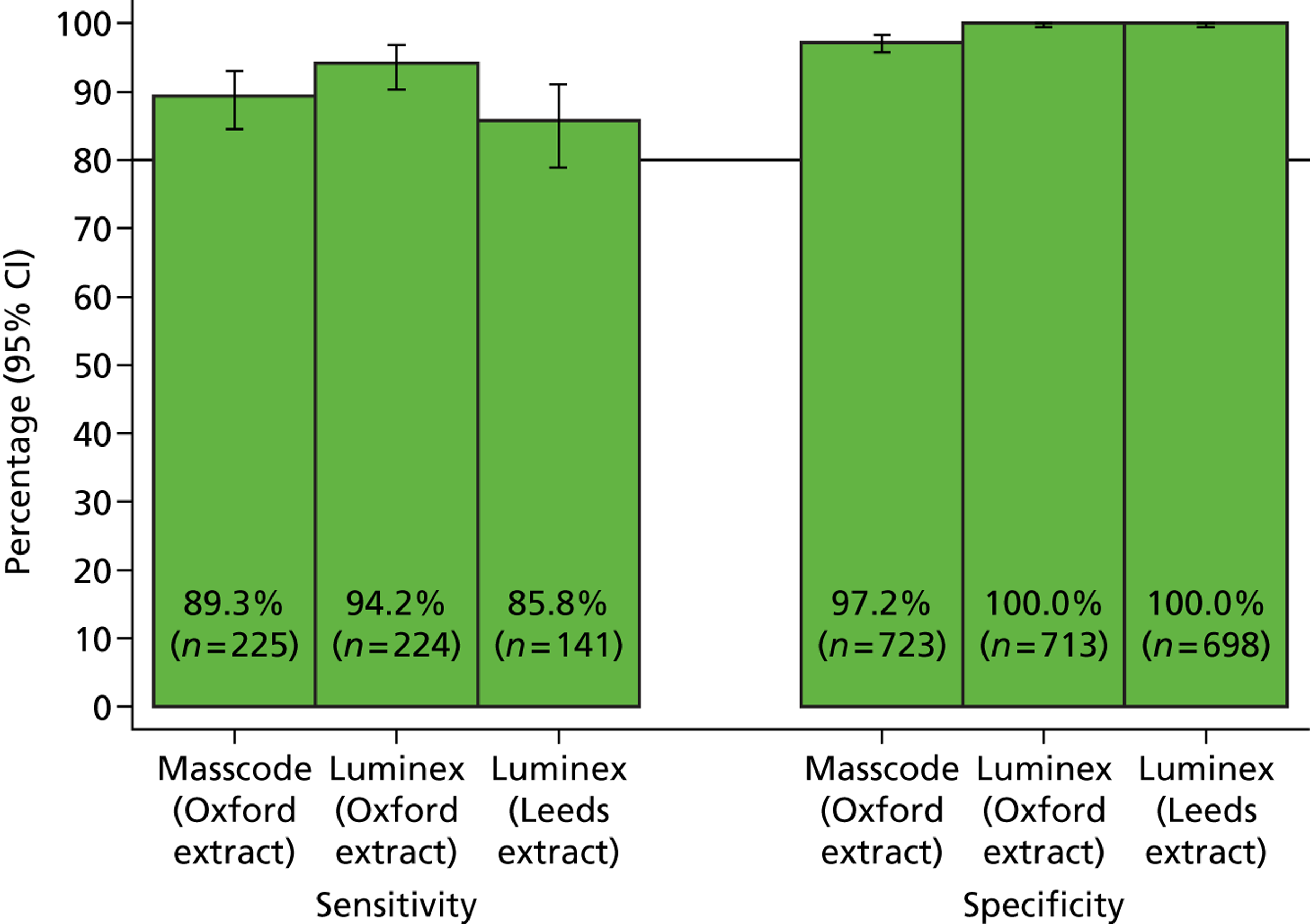
FIGURE 21.
C. difficile (including all PCRs). Sensitivity = 100 × called positive/true positive (including microbiology positive plus any PCR positive). Specificity = 100 × called negative/true negative (excluding microbiology negatives with any PCR positive). Not all samples had sufficient volume to be included in all test evaluations, denominators as shown.
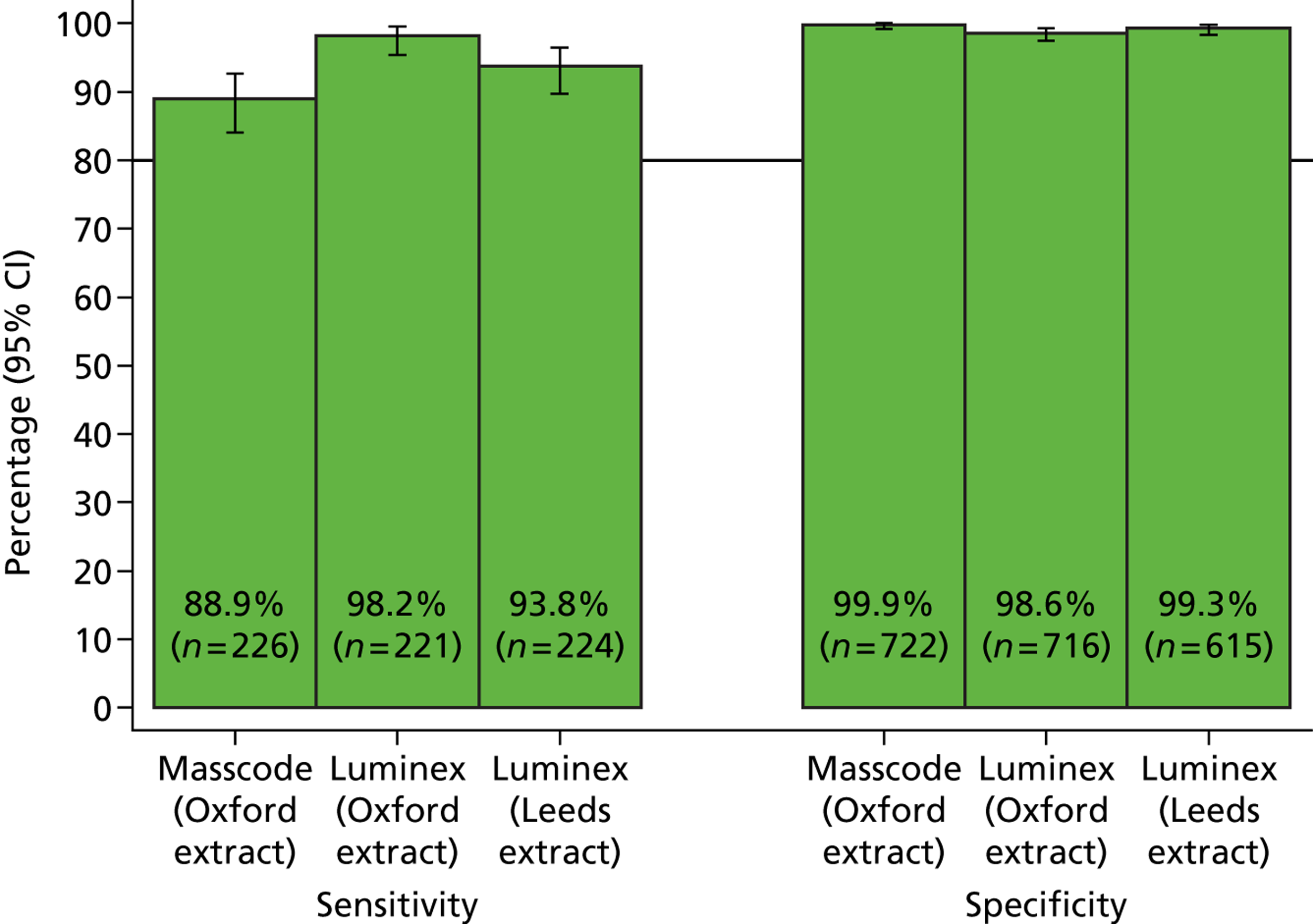
FIGURE 22.
Norovirus (including all PCRs). Sensitivity = 100 × called positive/true positive (including microbiology positive plus any PCR positive). Specificity = 100 × called negative/true negative (excluding microbiology negatives with any PCR positive). Not all samples had sufficient volume to be included in all test evaluations, denominators as shown.
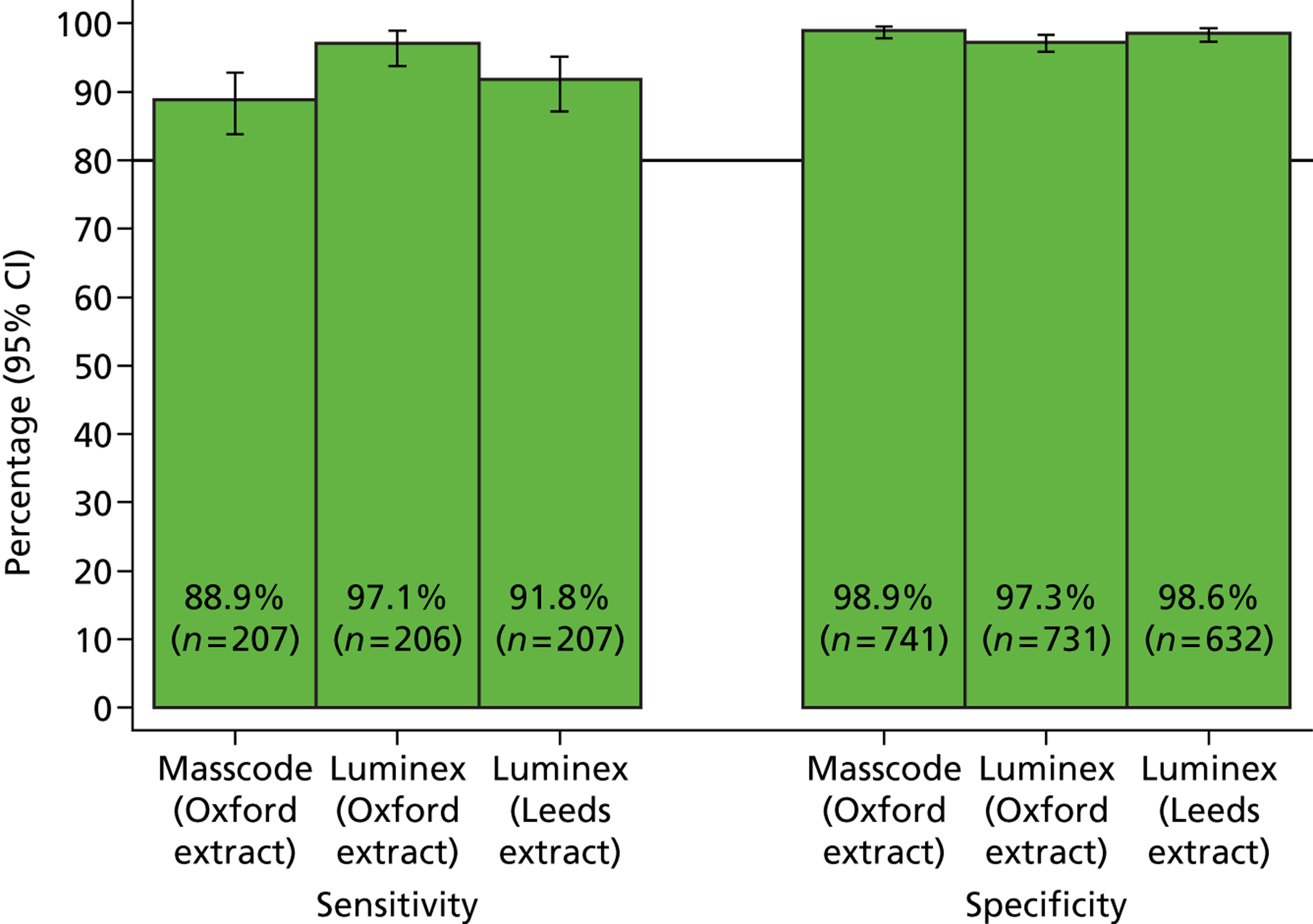
FIGURE 23.
S. enterica (including all PCRs). Sensitivity = 100 × called positive/true positive (including microbiology positive plus any PCR positive). Specificity = 100 × called negative/true negative (excluding microbiology negatives with any PCR positive). Not all samples had sufficient volume to be included in all test evaluations, denominators as shown.
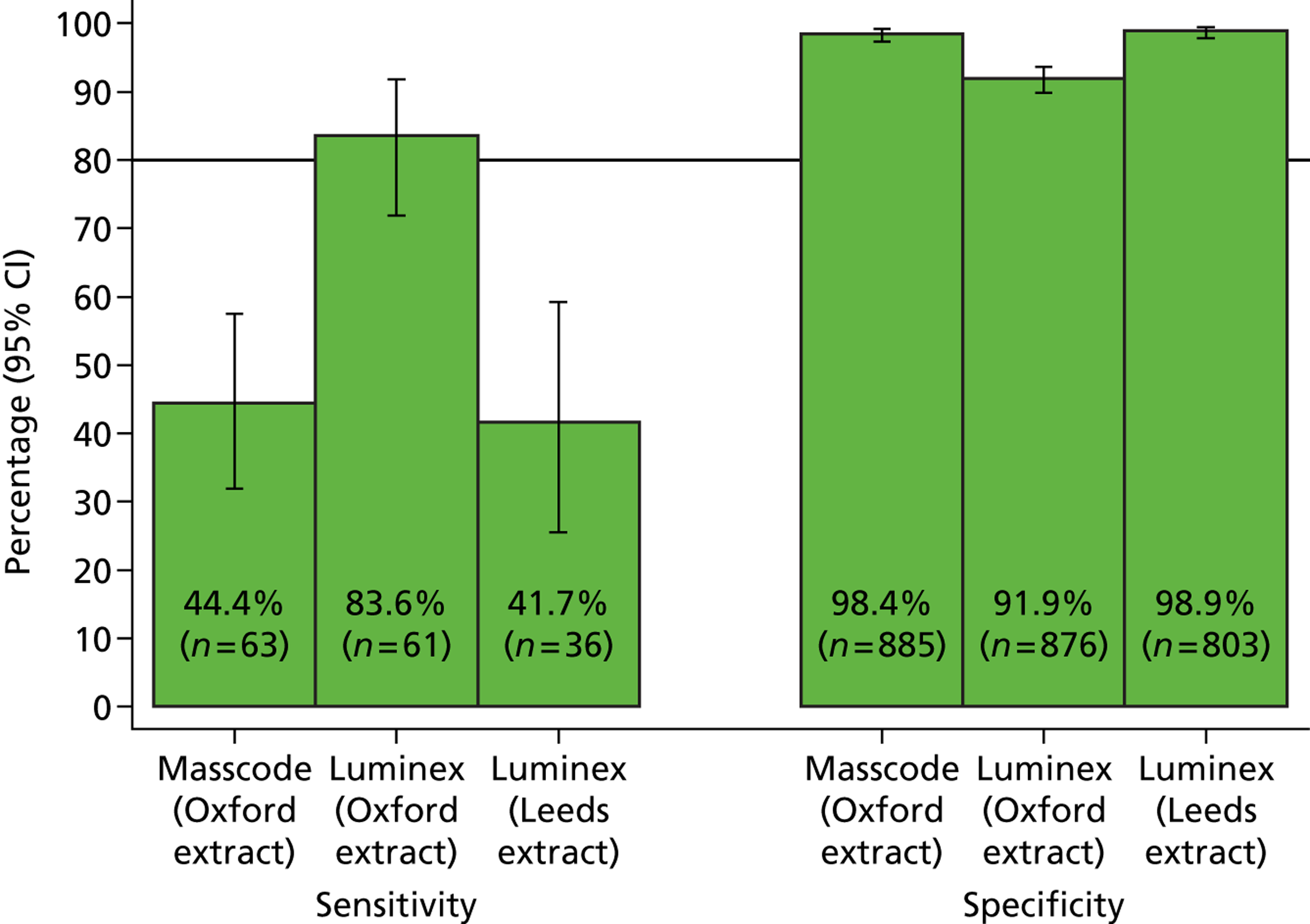
Campylobacter species
A total of 20 additional Campylobacter spp.-positive samples were identified by PCR testing of unexpected positives from one of the three assays. Overall, therefore, Campylobacter spp. were identified in 225 samples by microbiology or PCR, and in 723 samples Campylobacter spp. were not identified by either method (see Figure 20).
-
Two (0.9%) positive samples did not have Campylobacter spp. identified by at least one of the assays (one negative on all three assays, one negative on two assays). Of 140 positive samples tested with all three assays, 109 (78%) were positive on all three: 120 (86%) were positive on both MassCode/Oxford and fresh/Leeds extracts tested by Luminex.
-
Of 723 negative samples, no Campylobacter spp. were identified on any assay run in 703 (97%) of them. Of 690 negative samples tested with all three assays, 670 (97%) were negative on all three: all 690 (100%) were negative on both MassCode/Oxford and fresh/Leeds extracts tested by Luminex (20 false positive only on MassCode).
Clostridium difficile
A total of 27 additional C. difficile-positive samples were identified by PCR testing of unexpected positive samples from one of the three assays. Overall, therefore, C. difficile was identified by microbiology or PCR in 226 samples, and was not identified by either method in 722 samples (see Figure 21).
-
All positive samples had C. difficile identified by at least one of the assays. Of 219 positive samples tested with all three assays, 188 (86%) were positive on all three: 203 (93%) were positive on both MassCode/Oxford and fresh/Leeds extracts tested by Luminex.
-
Microbiological plus PCR positives identified by the Luminex assay on MassCode/Oxford extracts were 210 A+B+, one A–B+ and three A+B– (falsely B– on Luminex), compared with 198 A+B+ and 12 A+B– (falsely B– on Luminex) for fresh/Leeds extracts.
-
-
Out of 725 negative samples, 710 (98%) never had C. difficile identified on any assay run. Of 614 negative samples tested with all three assays, 599 (98%) were negative on all three; 600 (98%) were negative on both MassCode/Oxford and fresh/Leeds extracts tested by Luminex.
-
Out of 13 Luminex false positives using MassCode/Oxford extracts, 10 could be explained by strains being called as A+B–, as could three out of four Luminex false positives using fresh/Leeds extracts. Three of these strains were in common, i.e. were identified as A+B– strain C. difficile in both Luminex assays but had not been identified as containing C. difficile by standard microbiological testing and had a negative tcdB gene PCR. The remainder were either A+B+/A–B+ on the other Luminex assay but not confirmed by tcdB gene PCR, or had not been detected on the other Luminex assay originally.
-
Norovirus
Eight additional norovirus positives were identified by PCR testing of unexpected positives from one of the three assays. Overall, therefore, 207 samples had norovirus identified by microbiology or PCR and 741 did not have norovirus identified by either (see Figure 22).
-
Out of 207 positive samples, six (3%) did not have norovirus identified by any of the assays. Of 206 positive samples tested with all three assays, 178 (86%) were positive on all three; 190 (92%) were positive on both MassCode/Oxford and fresh/Leeds extracts tested by Luminex.
-
Norovirus positives identified by the Luminex assay on MassCode/Oxford extracts were 197 GII and three GI strains, compared with 188 GII and two GI strains for fresh/Leeds extracts. Two strains were identified as GI on both Luminex panels (and not detected by MassCode); one was identified as GI on the Luminex assay of the MassCode/Oxford extract but not identified as norovirus on the Luminex assay of the fresh/Leeds extracts.
-
-
Out of 741 negative samples, 717 (97%) never had norovirus identified on any assay run performed. Of 623 negative samples tested with all three assays, 597 (96%) were negative on all three; 611 (98%) were negative on both MassCode/Oxford and fresh/Leeds extracts tested by Luminex.
-
Out of 20 Luminex false positives using MassCode/Oxford extracts, eight could be explained by GI strains and two out of nine Luminex false positives using fresh/Leeds extracts. Two of these strains were in common, i.e. were identified as GI norovirus in both Luminex assays, but had not been identified as containing norovirus by standard testing (and were negative for GII norovirus). The remainder had not been detected on the other Luminex assay originally.
-
Salmonella enterica
Three additional S. enterica positives were identified by PCR testing of unexpected positives from one of the three assays. Overall, therefore, 63 samples had S. enterica identified by microbiology or PCR, and 885 did not have S. enterica identified by either (Figure 23).
-
Ten (16%) positive samples did not have S. enterica identified by at least one of the assays [five were negative on all three assays, four on two, and one was only tested (negative) with MassCode]. Conversely 53 out of 63 did have S. enterica identified at least once, 29 in at least two assays. Of 36 positive samples tested with all three assays, 12 (33%) were positive on all three; 15 (42%) were positive on both MassCode/Oxford and fresh/Leeds extracts tested by Luminex.
-
A total of 25 positive samples did not have sufficient material remaining to assay fresh/Leeds extracts using the Luminex assay. Of these ,11 were positive only using Luminex on MassCode/Oxford extracts and 10 were positive on both other assays, a similar ratio to the 14 and 16, respectively, where all three assays had been run.
-
-
Out of 885 negative samples, 794 (90%) never had S. enterica identified on any assay run. Of 794 negative samples tested with all three assays, 712 (90%) were negative on all three; 725 (91%) were negative on both MassCode/Oxford and fresh/Leeds extracts tested by Luminex.
Figures 24–26 show the specificities across all organisms tested, based on all microbiological results and whatever PCR results were available. Overall, specificity of the MassCode assay is clearly suboptimal (see Figure 24), particularly for Giardia and Cryptosporidium spp. In contrast, Luminex achieves better specificity on the MassCode/Oxford extracts (see Figure 25), but would really be suitable only for testing thousands of samples based on results from the fresh/Leeds extracts (see Figure 26).
FIGURE 24.
Specificity: MassCode assay on MassCode (Oxford) extract. Specificity = 100 × called negative/true negative (excluding microbiology negatives with any PCR positive). Light green bars are tests where no qPCR was done to exclude positives missed by original microbiology.
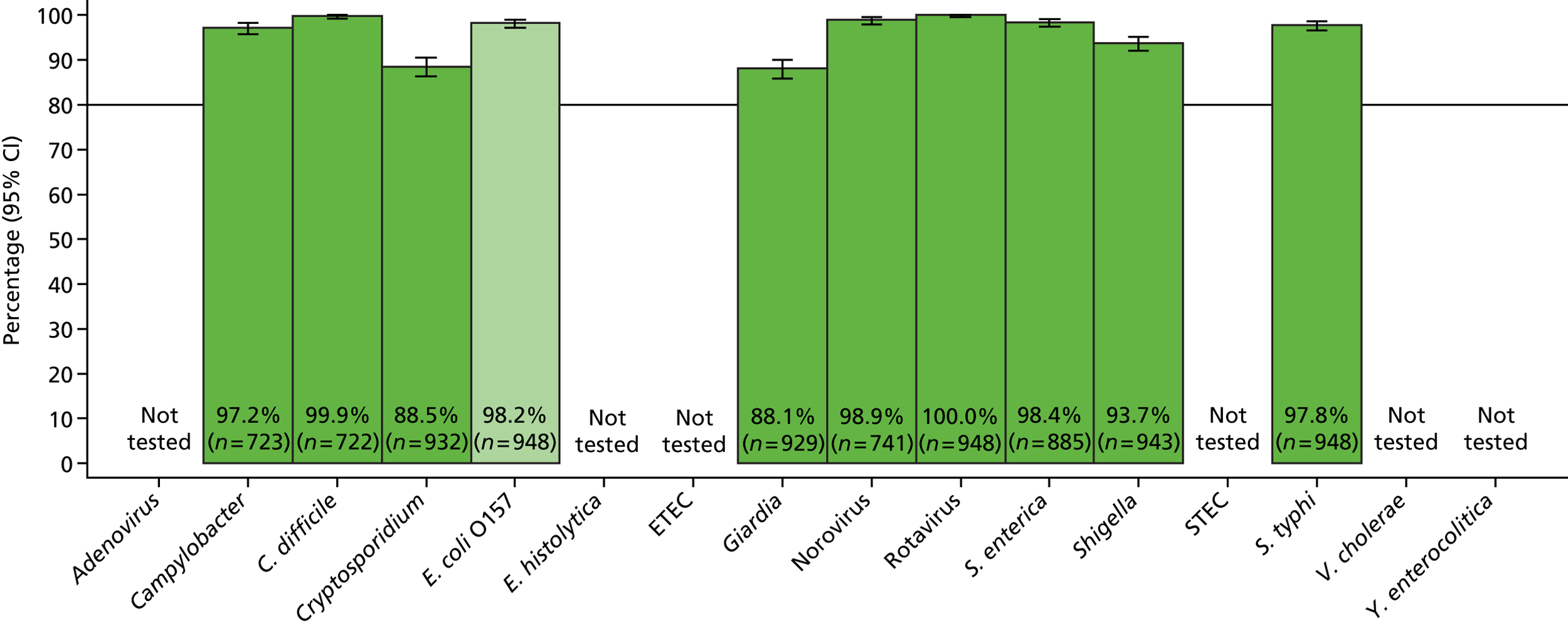
FIGURE 25.
Specificity: Luminex assay on MassCode (Oxford) extract. Specificity = 100 × called negative/true negative (excluding microbiology negatives with any PCR positive). Light green bars are tests where no qPCR was done to exclude positives missed by original microbiology.
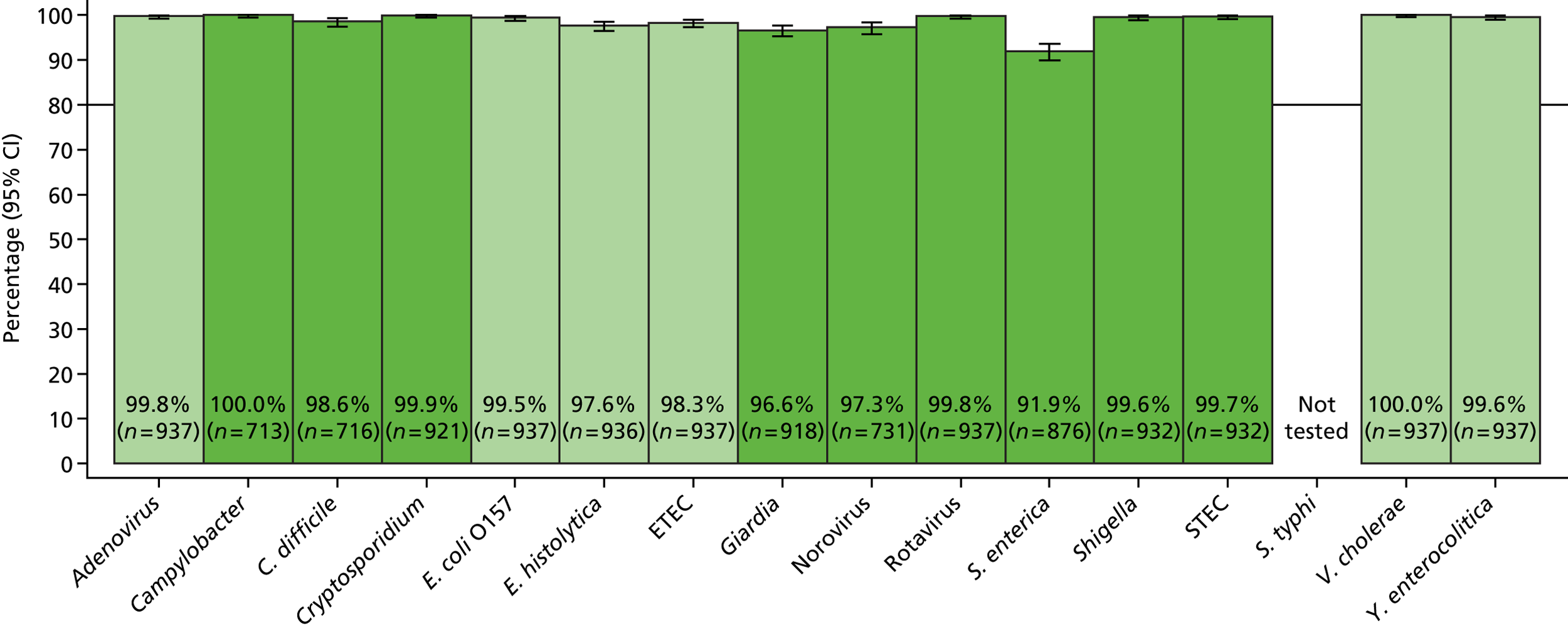
FIGURE 26.
Specificity: Luminex assay on fresh (Leeds) extract. Specificity = 100 × called negative/true negative (excluding microbiology negatives with any PCR positive). Light green bars are tests where no qPCR was done to exclude positives missed by original microbiology.
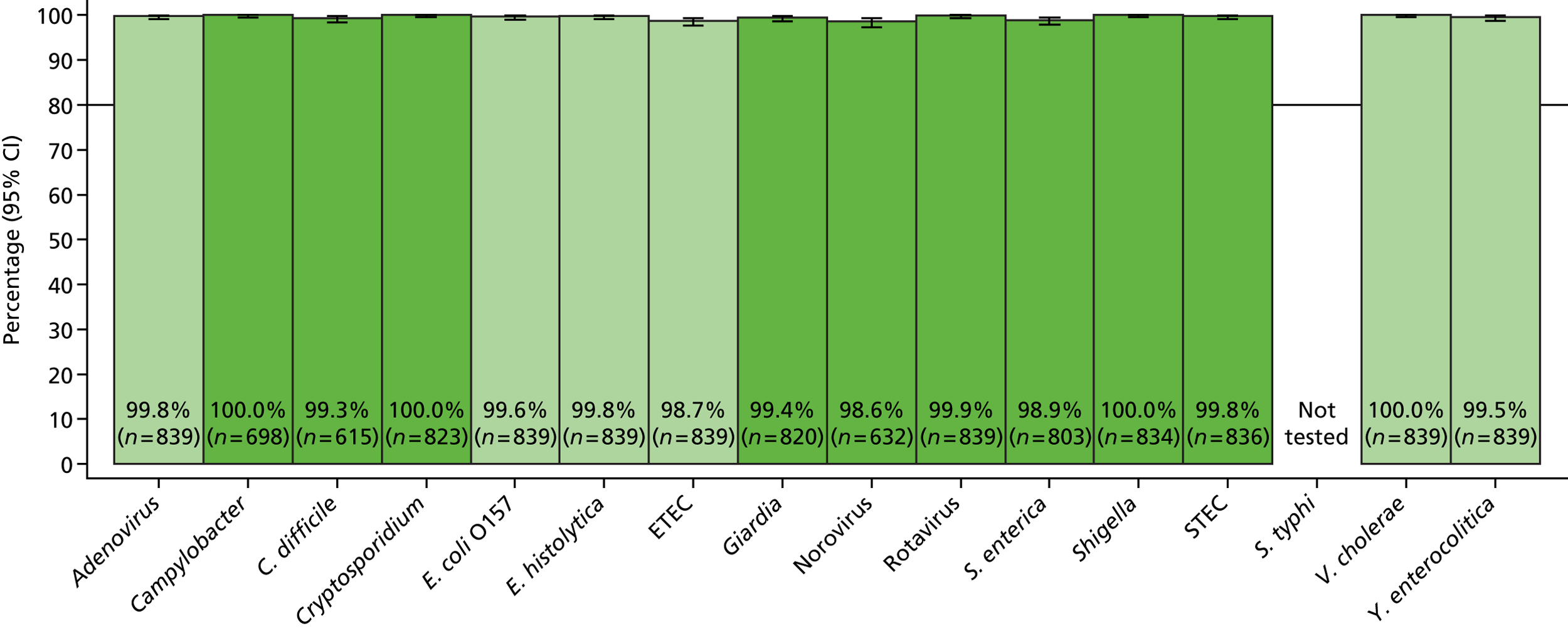
Table 24 shows the trade-off between sensitivity and specificity, which is particularly apparent for Cryptosporidium and Giardia spp. Cryptosporidium spp. were identified in 16 samples in total (one known to be microbiology positive). Four samples were identified as containing Cryptosporidium spp. in all three assays, seven only in MassCode and Luminex assays run on MassCode/Oxford extracts, and four and one in each of these assays singly. However, the increased sensitivity of MassCode came at the cost of 107 false positives, and it is unclear whether these organisms really were causing disease or simply co-carried. Similarly, Giardia spp. were identified in 19 samples (none known from microbiology). Ten samples were identified as containing Giardia spp. in all three assays, another three in two of the three assays, and six in only one assay. Again, the increased sensitivity of MassCode came at the cost of 111 false positives.
| Organism | Missed positivesa (number missed/number positive) | Additional false positives | ||||
|---|---|---|---|---|---|---|
| MassCode, MassCode/Oxford extracts | Luminex, MassCode/Oxford extractsb | Luminex, fresh/Leeds extractsb | MassCode, MassCode/Oxford extracts | Luminex, MassCode/Oxford extractsb | Luminex, fresh/Leeds extractsb | |
| Main pathogens | ||||||
| Campylobacter spp. | 24/225 | 13/224 | 20/141 | 20 | 0 | 0 |
| C. difficile | 25/226 | 4/221 | 14/224 | 1 | 10 | 4 |
| Norovirus | 23/207 | 6/206 | 17/207 | 8 | 20 | 9 |
| S. enterica | 35/63 | 10/61 | 21/36 | 14 | 71 | 9 |
| Additional pathogens in common with MassCode | ||||||
| E. coli O157c | N/A | N/A | N/A | 17 | 5 | 3 |
| Shigella spp. | 2/5 | 0/5 | 1/5 | 59 | 4 | 0 |
| Cryptosporidium | 1/16 | 4/16 | 12/16 | 107 | 1 | 0 |
| Giardia spp. | 3/19 | 5/19 | 7/19 | 111 | 31 | 5 |
| Rotavirus | 0/0 | 0/0 | 0/0 | 0 | 2 | 1 |
| Additional pathogens in MassCode only | ||||||
| S. Typhi | 0/0 | N/A | N/A | 21 | N/A | N/A |
| Additional pathogens in Luminex only | ||||||
| E. histolyticac | N/A | 1/1 | N/A | N/A | 22 | 2 |
| Y. enterocoliticac | N/A | 0/0 | 0/0 | N/A | 4 | 4 |
| Vibrio choleraec | N/A | 0/0 | 0/0 | N/A | 0 | 0 |
| Adenovirusc | N/A | 0/0 | 0/0 | N/A | 2 | 2 |
| ETECc | N/A | 0/0 | 0/0 | N/A | 16 | 11 |
| STECd | N/A | 4/5 | 3/3 | N/A | 3 | 2 |
Main four pathogens
Even combining results across the four main pathogens, there was reasonable variety in what would have been concluded about each sample (Table 25). Overall agreement with the combined microbiology and all PCRs performed standard was 85.6% (κ = 0.81), 87.0% (κ = 0.84) and 89.8% (κ = 0.87) for the MassCode assay, Luminex assay/MassCode extract and Luminex assay/fresh extract, respectively. Most discrepancies compared with the conclusion based on microbiology and all PCRs performed were finding no organism in reference-positive samples, or identifying one or more organisms in reference-negative samples (Table 26 and Figures 27–29). Falsely identifying additional organisms in reference-positive samples occurred relatively rarely. Tests of symmetry for the number of main pathogens identified (all p < 0.0001) suggested that the MassCode assay and the Luminex assay on fresh/Leeds extracts (i.e. assays where extracts were recent) tended to systematically underestimate the number of main pathogens in each sample, whereas the Luminex assay on MassCode/Oxford extracts tended to systematically overestimate the number of main pathogens in each sample.
| First organism | Second organism | Third organism | Phase 1 panel (n = 948) | MassCode, MassCode/Oxford extract (n = 948) | Luminex, MassCode/Oxford extract (n = 937) | Luminex, fresh/Leeds extract (n = 839) |
|---|---|---|---|---|---|---|
| Campylobacter | 200 (21.1%) | 184 (19.4%) | 177 (18.9%) | 115 (13.7%) | ||
| Campylobacter | Norovirus | 3 (0.3%) | 8 (0.8%) | 7 (0.8%) | 2 (0.2%) | |
| C. difficile | 196 (20.7%) | 175 (18.5%) | 187 (20.0%) | 198 (23.6%) | ||
| C. difficile | Campylobacter | 15 (1.6%) | 14 (1.5%) | 3 (0.3%) | 2 (0.2%) | |
| C. difficile | Campylobacter | Norovirus | – | 3 (0.3%) | – | – |
| C. difficile | Norovirus | 15 (1.6%) | 8 (0.8%) | 18 (1.9%) | 11 (1.3%) | |
| C. difficile | S. enterica | – | 2 (0.2%) | 14 (1.5%) | 2 (0.2%) | |
| C. difficile | S. enterica | Campylobacter | – | – | 2 (0.2%) | – |
| C. difficile | S. enterica | Norovirus | – | – | 3 (0.3%) | – |
| Norovirus | 188 (19.8%) | 171 (18.0%) | 175 (18.7%) | 183 (21.8%) | ||
| S. enterica | 55 (5.8%) | 27 (2.8%) | 65 (6.9%) | 18 (2.2%) | ||
| S. enterica | Campylobacter | 7 (0.7%) | 11 (1.2%) | 21 (2.2%) | 1 (0.1%) | |
| S. enterica | Campylobacter | Norovirus | – | 1 (0.1%) | 1 (0.1%) | – |
| S. enterica | Norovirus | 1 (0.1%) | 1 (0.1%) | 16 (1.7%) | 2 (0.2%) | |
| All four | – | – | – | 1 (0.1%) | ||
| None | 268 (28.3%) | 343 (36.2%) | 248 (26.5%) | 304 (36.2%) |
| Versus combined microbiology + PCR (four pathogens) | MassCode, MassCode/Oxford extract (n = 948) | Luminex, MassCode/Oxford extract (n = 937) | Luminex, fresh/Leeds extract (n = 839) |
|---|---|---|---|
| Agree | 811 (85.6%) | 815 (87.0%) | 753 (89.7%) |
| Different number of organisms, extra organism in standard missed by assay | 11 (1.2%) | 13 (1.4%) | 22 (2.6%) |
| Different number of organisms, extra organism not in standard found by assay | 14 (1.5%) | 54 (5.8%) | 8 (1.0%) |
| Different number, no overlap | 1 (0.1%) | 0 | 0 |
| Same number, no overlap | 0 | 3 (0.3%) | 4 (0.5%) |
| Negative on standard, organisms found by assay | 18 (1.9%) | 35 (3.7%) | 8 (1.0%) |
| Positive on standard, no organisms found by assay | 93 (9.8%) | 17 (1.8%) | 44 (5.2%) |
FIGURE 27.
Agreement between MassCode assay and combined microbiology plus PCR for the four main pathogens. Note: size of circle proportional to number of isolates. White indicates either identical pathogen(s) identified, or only additional pathogens were identified in one assay. Black indicates incorrect pathogens identified.

FIGURE 28.
Agreement between Luminex assay using MassCode (Oxford) extracts and combined microbiology plus PCR for the four main pathogens. Note: size of circle proportional to number of isolates. White indicates either identical pathogen(s) identified, or only additional pathogens were identified in one assay. Black indicates incorrect pathogens identified.
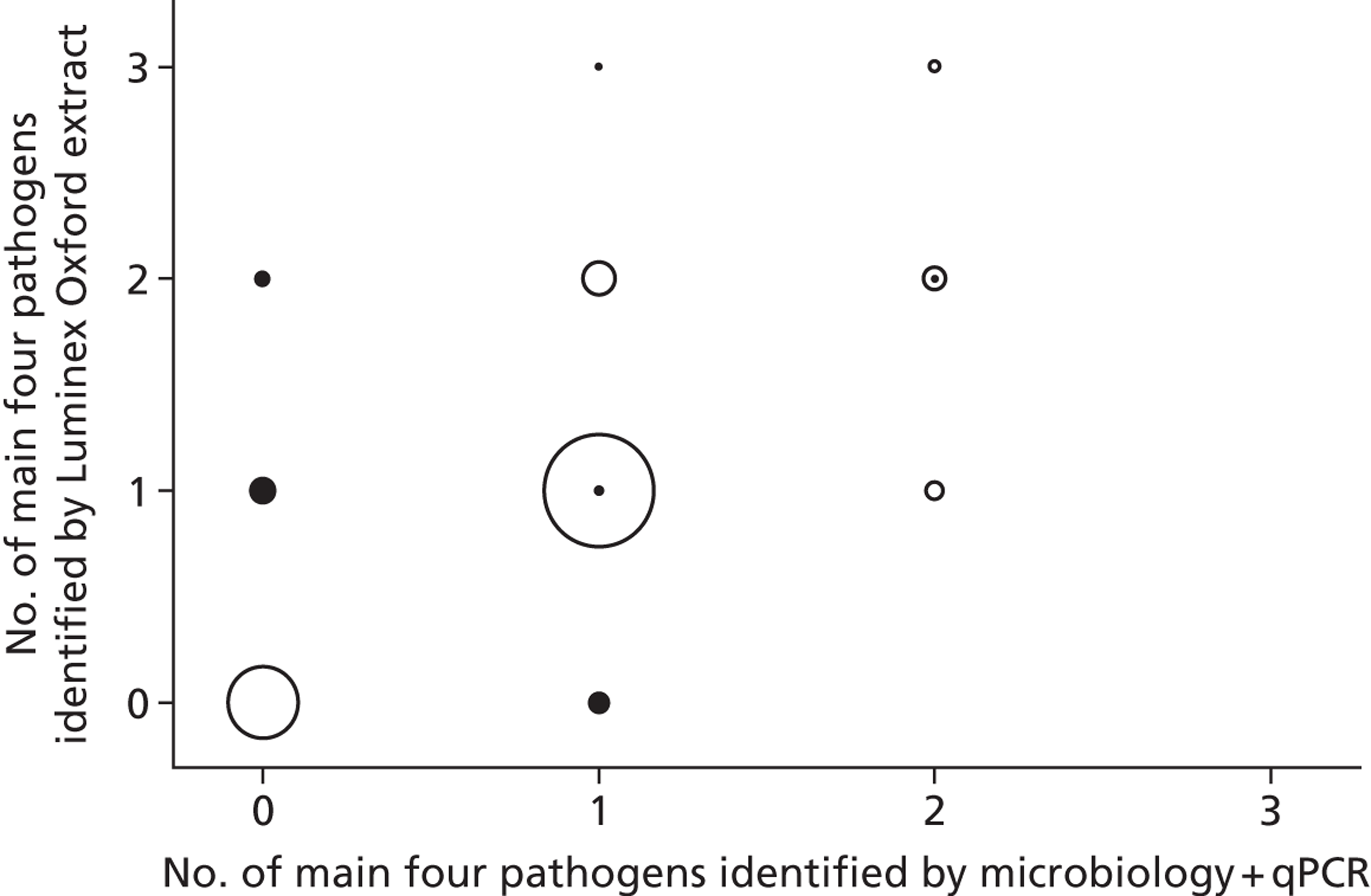
FIGURE 29.
Agreement between Luminex assay using fresh (Leeds) extracts and combined microbiology plus PCR for the four main pathogens. Note: size of circle proportional to number of isolates. White indicates either identical pathogen(s) identified, or only additional pathogens were identified in one assay. Black indicates incorrect pathogens identified.
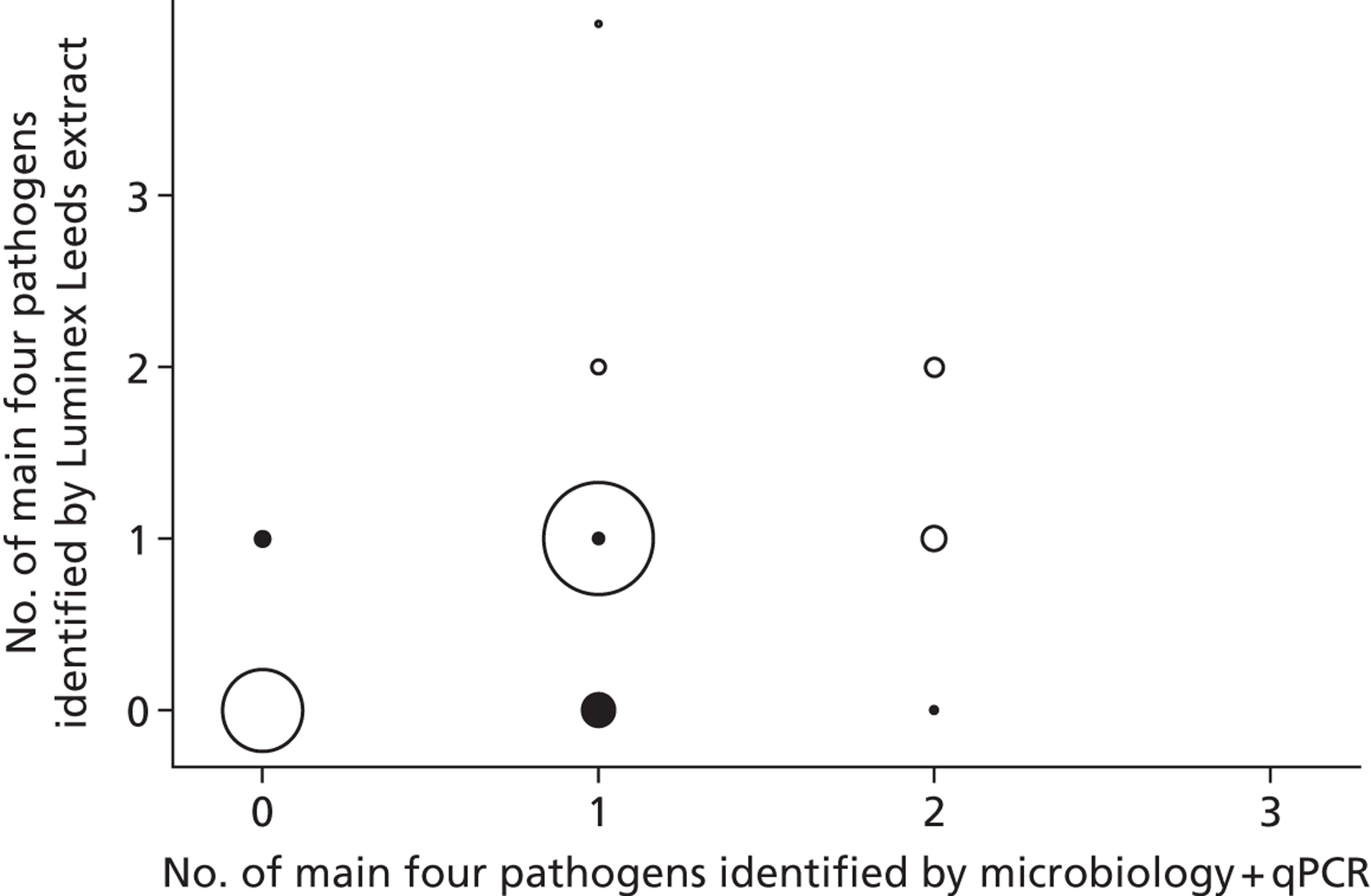
Although the Luminex assay on freshly extracted samples was most accurate overall (89.8%), nevertheless it would have concluded that 5.2% of samples did not represent infectious diarrhoea caused by the four main pathogens, even though these pathogens were genuinely present in the sample.
Common nine pathogens
Of the 948 samples, based on combined microbiology and PCR results, 631 (66.6%), 62 (6.5%) and 2 (0.2%) had one, two or three of the pathogens tested by both the MassCode and the Luminex assays identified. There were 22 different combinations of pathogens (not shown).
Overall, agreement with the combined result from microbiology and all PCRs performed as the standard reference was 64.7% (κ = 0.58), 82.9% (κ = 0.79) and 86.8% (κ = 0.83) for the MassCode assay, Luminex assay/MassCode extract and Luminex assay/fresh extract, respectively. Most discrepancies compared with the conclusion based on microbiology and all PCRs performed were missing any organism in reference-positive samples, or identifying organisms in reference-negative samples (see Table 27 and Figures 30–32). In contrast to the four main pathogens, tests of symmetry for the number of common pathogens identified (all p < 0.0001) suggested that while the Luminex assay on fresh/Leeds extracts still tended to systematically underestimate the number of main pathogens in each sample, the MassCode and Luminex assay on MassCode/Oxford extracts tended to systematically overestimate the number of common pathogens in each sample (as a consequence of their higher false-positive rates).
| Versus combined microbiology + PCR (nine pathogens) | MassCode, MassCode/Oxford extract (n = 948) | Luminex, MassCode/Oxford extract (n = 937) | Luminex, fresh/Leeds extract (n = 839) |
|---|---|---|---|
| Agree | 613 (64.7%) | 777 (82.9%) | 728 (86.8%) |
| Different number of organisms, extra organism in standard missed by assay | 12 (1.3%) | 17 (1.8%) | 36 (4.3%) |
| Different number of organisms, extra organism not in standard found by assay | 144 (15.2%) | 70 (7.5%) | 14 (1.7%) |
| Different number, no overlap | 4 (0.4%) | 0 | 0 |
| Same number, no overlap | 25 (2.6%) | 6 (0.6%) | 5 (0.6%) |
| Negative on standard, organisms found by assay | 81 (8.5%) | 50 (5.3%) | 10 (1.2%) |
| Positive on standard, no organisms found by assay | 69 (7.3%) | 17 (1.8%) | 46 (5.5%) |
FIGURE 30.
Agreement between MassCode assay and combined microbiology plus PCR for the nine common pathogens. Note: size of circle proportional to number of isolates. White indicates either identical pathogen(s) identified, or only additional pathogens were identified in one assay. Black indicates incorrect pathogens identified.
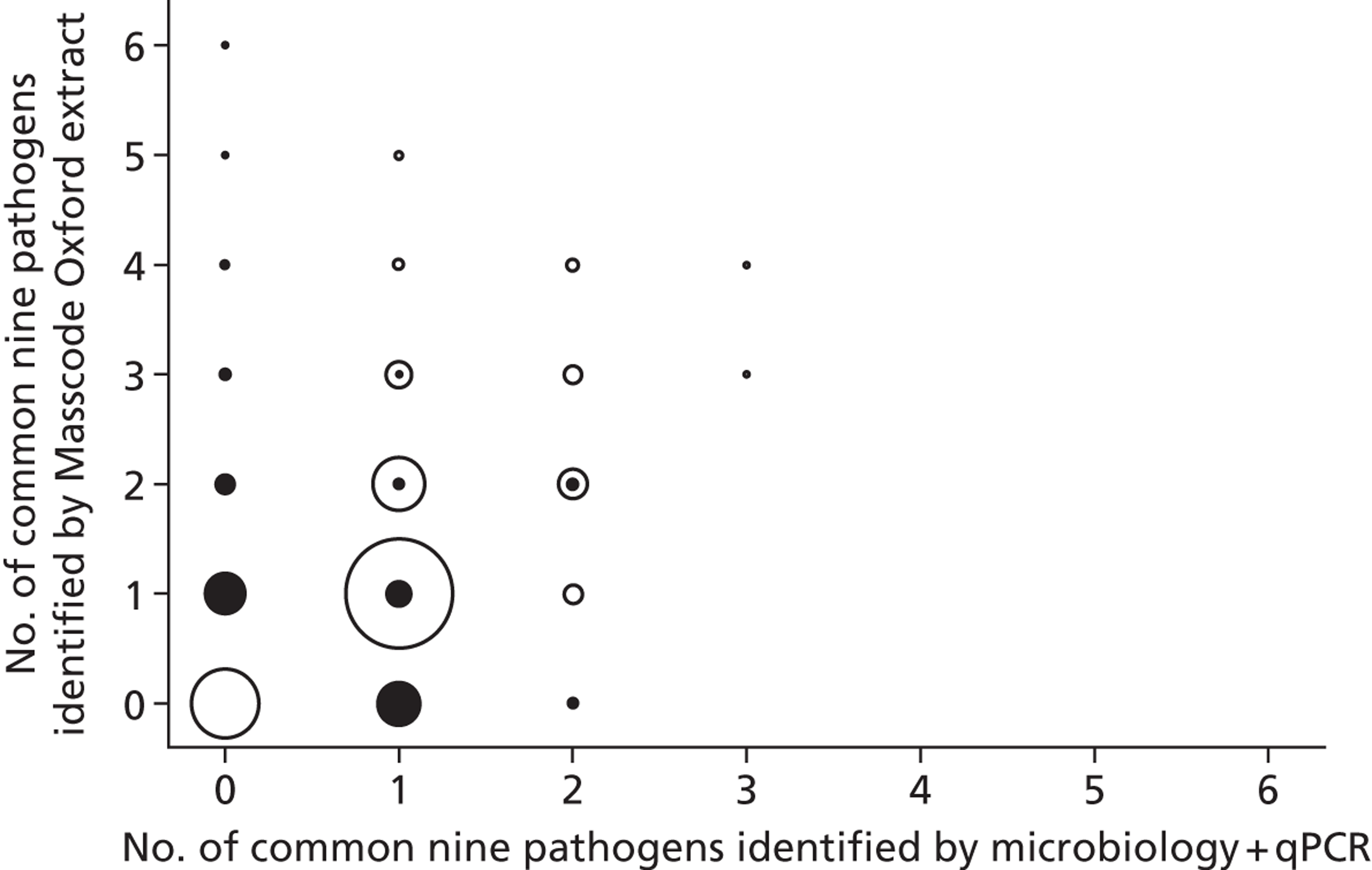
FIGURE 31.
Agreement between Luminex assay using MassCode (Oxford) extracts and combined microbiology plus PCR for the nine common pathogens. Note: size of circle proportional to number of isolates. White indicates either identical pathogen(s) identified, or only additional pathogens were identified in one assay. Black indicates incorrect pathogens identified.
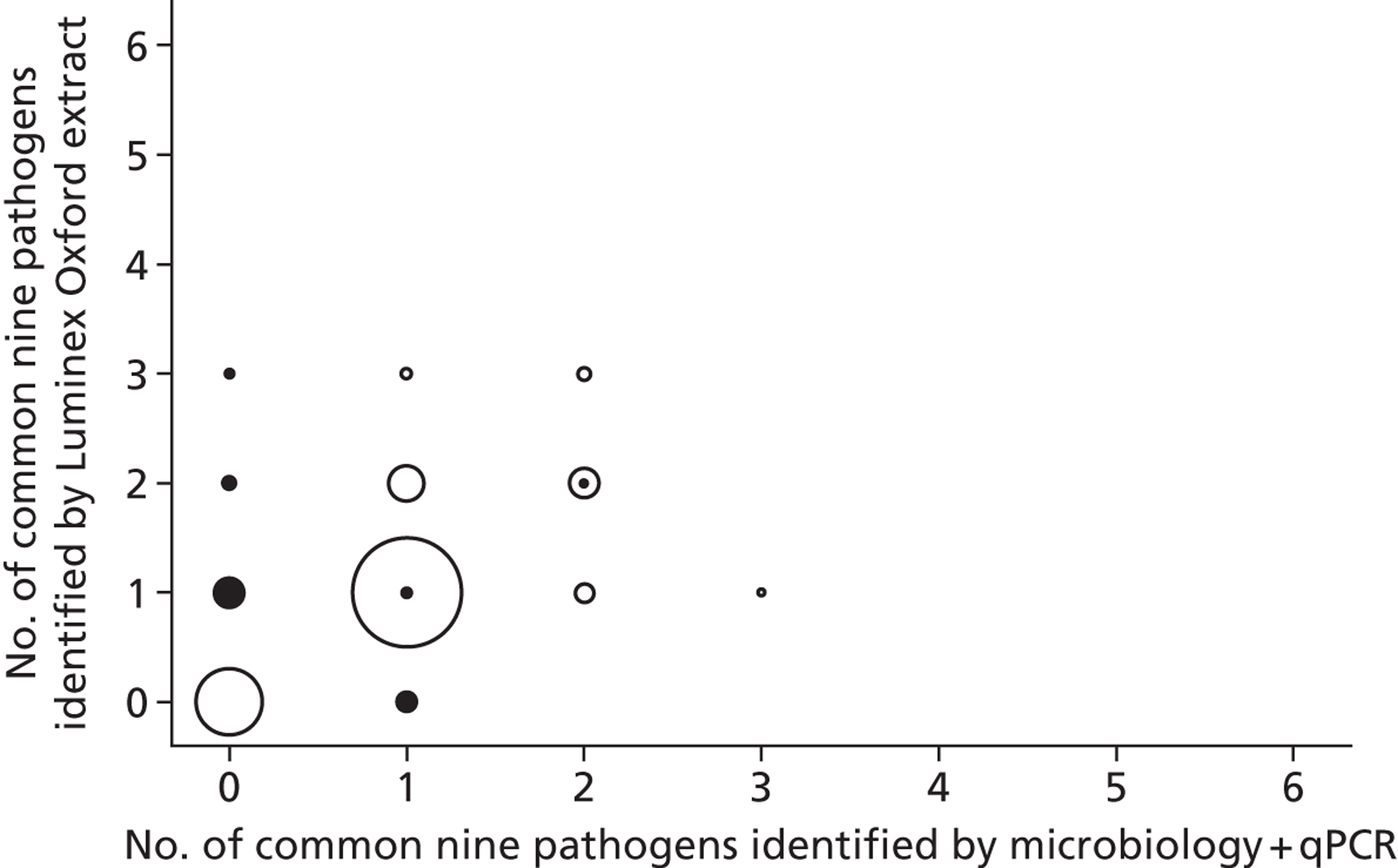
FIGURE 32.
Agreement between Luminex assay using fresh (Leeds) extracts and combined microbiology plus PCR for the nine common pathogens. Note: size of circle proportional to number of isolates. White indicates either identical pathogen(s) identified, or only additional pathogens were identified in one assay. Black indicates incorrect pathogens identified.
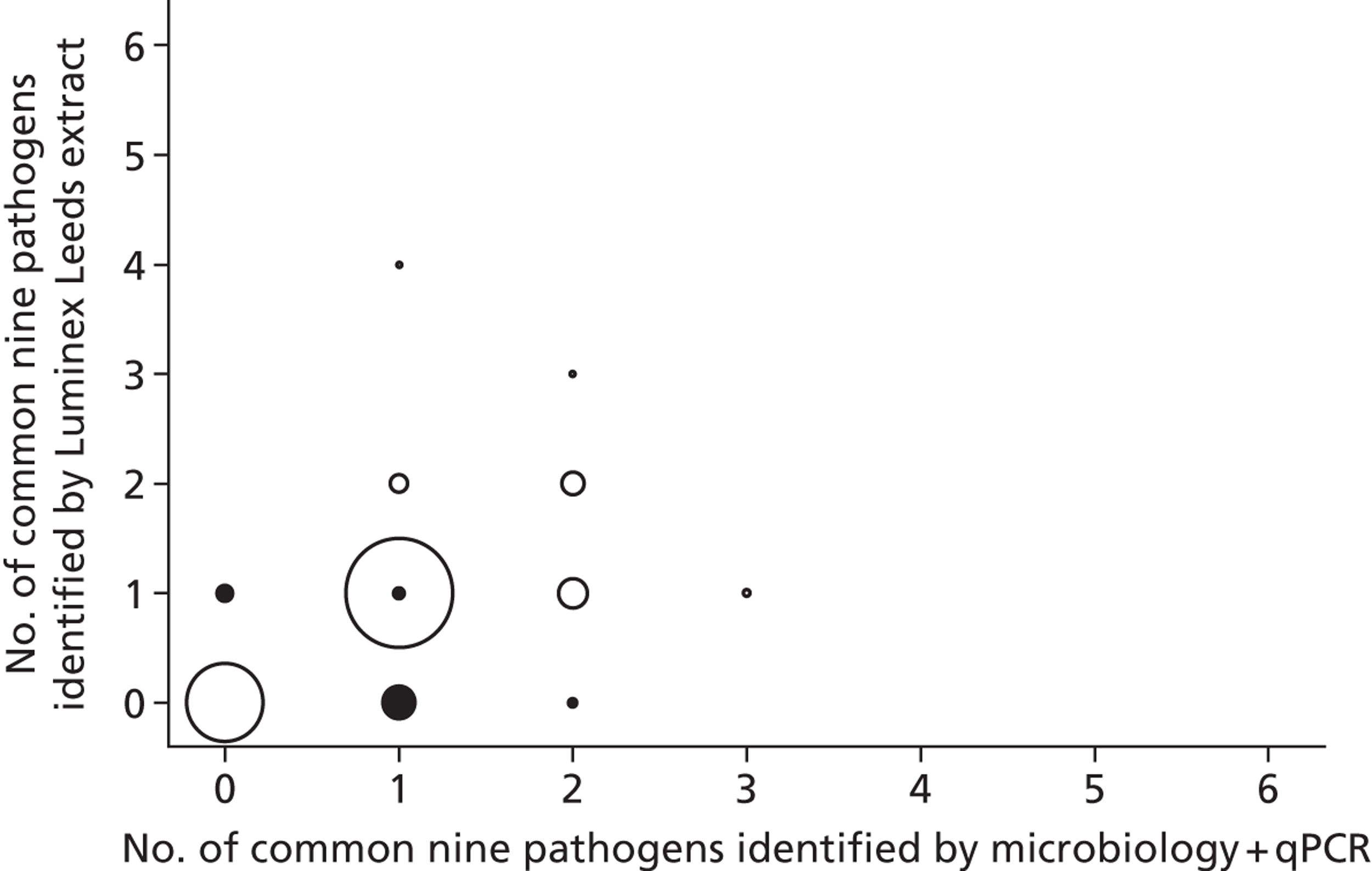
Although the Luminex assay on freshly extracted samples remained most accurate overall (86.8%, compared with 89.8% for the main four pathogens), nevertheless it would still have concluded that 5.5% of samples did not represent infectious diarrhoea caused by these common pathogens, even though at least one was genuinely present in the sample.
Comparing Luminex assay with different extracts
Overall, agreement between the two Luminex assays on the common nine pathogens was only 81.1% (κ = 0.76), with most discrepancies because of extra organisms being identified using the Luminex assay with the MassCode/Oxford extract compared with no organisms being identified with the fresh/Leeds extract [76 (9.2%) samples] or in addition to other organisms identified in both [68 (8.2%) samples] (Figure 33).
FIGURE 33.
Agreement between Luminex assay using MassCode (Oxford) vs. fresh (Leeds) extracts for the nine common pathogens. Note: size of circle proportional to number of isolates. White indicates either identical pathogen(s) identified, or only additional pathogens were identified in one assay. Black indicates incorrect pathogens identified.
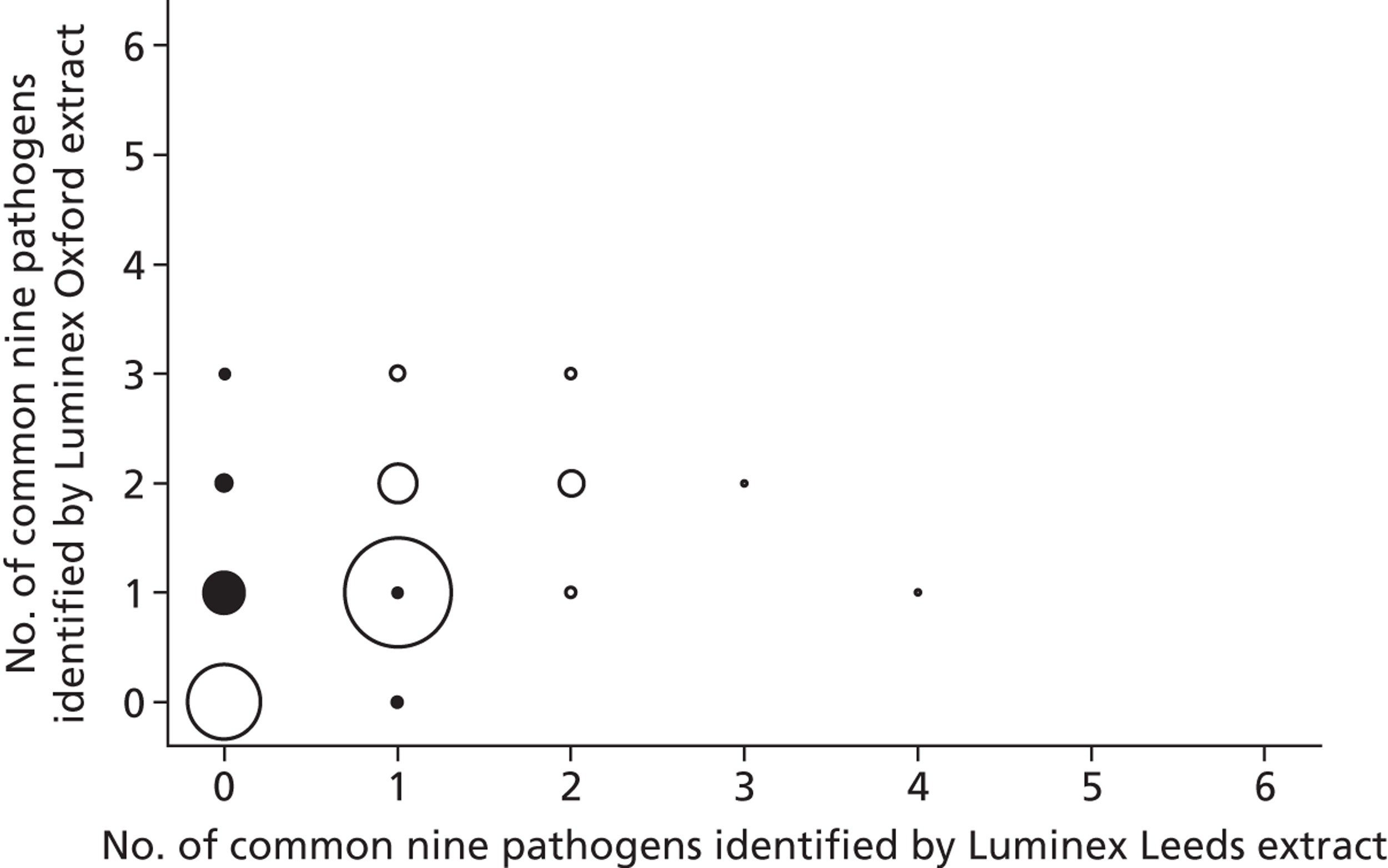
Considering all organisms present in the Luminex assay, with the original MassCode/Oxford extracts 581 (62.0%) samples would have been declared to have a single organism present, and 127 (13.6%), 25 (2.7%) and 2 (0.2%) would have been declared to have two, three or four organisms, respectively. With the fresh (Leeds) extracts, only 516 (61.5%), 42 (5.0%), 2 (0.2%) and 1 (0.1%) would have been declared to have one, two, three or four organisms present, respectively.
Discussion
Direct comparison of the three assays against the same microbiological plus qPCR standard identified the same problems with suboptimal sensitivity for detection of S. enterica using fresh extracts with either the MassCode or the Luminex assay, and suboptimal specificity for the detection of S. enterica using MassCode/Oxford extracts with the Luminex assay, as the individual analyses (see Chapters 3 and 4). The Luminex assay clearly outperformed the MassCode assay.
Considering the best information as to what pathogens were actually present in each sample according to combined microbiological plus qPCR testing, however, overall agreement with what each assay would have reported as infecting organisms from the main four pathogens was moderate, at 85.6%, 87.0% and 89.8% for the MassCode assay, Luminex assay/MassCode extract and Luminex assay/fresh extract, respectively. That is, each assay would have reported incorrect results for around 1 in 10 samples. Although the Luminex assay on freshly extracted samples was most accurate overall (89.8%), nevertheless it would still have concluded that 5.2% of samples did not represent infectious diarrhoea caused by the main four pathogens, even though these pathogens were genuinely present in the sample. Reflecting the poor performance of the MassCode assay, agreement was much worse considering all nine pathogens tested for in all three assays, at 64.7%, compared with 82.9% and 86.8% for the Luminex assay/MassCode extract and Luminex assay/fresh extract, respectively. Considering that a NHS service microbiology laboratory might process 15,000–20,000 faecal specimens per year, the sensitivities, specificities and overall agreement from all these assays suggest none are ready for widespread deployment in the NHS.
Chapter 6 Impact of gastrointestinal infections on infection control practice
Introduction
Aims and research questions
The main aims of the health economic component of this study were to evaluate MassCode multiplex PCR in terms of net health-care costs and utilisation of isolation resources, and to determine whether or not the use of these tests can improve the hospital management of patients with suspected infectious diarrhoea. The specific objectives were, therefore, to:
-
identify current practice for the management of patients with suspected infectious diarrhoea, in order to determine the likely impact of these new testing practices on factors such as isolation procedures and test turnaround times
-
estimate the cost-effectiveness of MassCode relative to current diagnostic methods and the net cost to the NHS of the new technology.
For the first objective, surveys were planned to map current infection control, and microbiologist and laboratory practice across England, in terms of how patients with suspected infectious diarrhoea are managed. With advances in molecular and genomic testing on the horizon in infection control and microbiology departments, it is important to understand how these departments currently operate, so that the implications of more widespread genetic and genomic testing can be assessed. Three surveys were, therefore, designed to provide data on how typical the study centres were in relation to the rest of the country, making it possible to generalise our findings across the wider NHS. This information would also be useful in terms of developing a base-case scenario within the health economic evaluations.
For the second objective, our planned analytical approach (as specified in the study protocol) was to conduct a cost-effectiveness analysis of the MassCode technology relative to other diagnostic tests, combining information on diagnostic procedures and short-term consequences collected within the phase 2 study with longer-term survival payoffs modelled from the study and a structured literature review. However, as the project was stopped before reaching the phase 2 study, the clinical and laboratory evidence required for the planned health economic analyses was not produced. Therefore, economic evaluations of the technologies under consideration could not be conducted, and we are unable to report any findings for this objective.
This chapter presents the results of the three initial surveys, summarising current infection control, and microbiologist and laboratory practice in terms of how patients with suspected infectious diarrhoea across England are managed. With advances in genetic and genomic testing on the horizon in this context, it is important to understand how infection control departments currently manage this patient group, so that the implications of more widespread genetic and genomic testing can be assessed for both patients and hospital trusts. These surveys provide answers pertaining to this information, which can be incorporated into future health economic evaluations of genetic and genomic interventions in infection control.
Methods
Survey design
To inform the design of the surveys, current infection control, and microbiologist and laboratory practice within the Oxford University Hospitals NHS Trust with respect to the management of patients with suspected infectious diarrhoea, was mapped between November 2010 and January 2011. Several approaches were used. First, a number of locations across this NHS trust were visited in order to observe and interview key infection control team members to gain an insight into current practice. These locations included infection control departments, laboratories and multiple wards at both the John Radcliffe Hospital and the Horton General Hospital (including the Emergency Departments, the Medical Assessment Unit, the Medical Short Stay Unit and the Surgical Emergency Unit). To supplement the information gathered during these visits, all relevant SOPs were examined, including infection control (for C. difficile: isolation, management of patients with diarrhoea and vomiting, and standard precautions) and microbiology SOPs (for C. difficile toxins and faeces: culture, see Figure 1 for examples).
The information gathered within this mapping exercise was used to design three surveys to be completed by NHS trusts across England. In all three surveys, it was specified that respondents should only consider infection control practice in relation to adult patients, not children. Copies of the surveys are supplied in Appendices 2–4 of this report.
Survey 1 (‘infection control survey’) was designed to be completed by infection control managers, collecting information on the infection control team, the monitoring of patients with infectious diarrhoea, infection control training and practice, and the management of outbreaks. Participants were also asked to consider how two potential future scenarios might impact on the management of patients with suspected infectious diarrhoea.
Scenario 1 was:
A consolidation of microbiology laboratory services has been proposed. The current model, with smaller microbiology laboratories based in hospitals and serving particular trusts may be replaced by a model which requires samples to be sent for testing to a small number of regional microbiology centres spread at regular intervals throughout the UK, each serving multiple trusts.
Scenario 2 was:
This survey is part of a larger study investigating the feasibility of introducing a new diagnostic test in microbiology laboratories across the UK. Using a stool sample taken from a patient with suspected infectious diarrhoea, this test can accurately detect 30 pathogens in a single reaction and rule in or out an infectious causative agent within 24 hours of the stool sample being taken.
Survey 2 (‘laboratory manager survey’) was designed to be completed by laboratory staff, collecting information on the testing process (e.g. which factors and patient characteristics drive testing decisions) and the cost of current testing practice in this context, for four types of test (PCR, ELISA, microscopy, culture).
Survey 3 (‘microbiologist survey’) was designed to be completed by microbiologists, collecting information on commonly requested tests, standard treatment practice for patients with positive C. difficile, Campylobacter spp. or Salmonella spp. tests, and patient management following discharge. Participants were also asked to consider the same two potential future scenarios as in the infection control survey.
Conducting the survey
The surveys were piloted in three trusts during February 2012: Leeds Teaching Hospitals NHS Trust, Brighton and Sussex University Hospitals NHS Trust and St George’s Healthcare NHS Trust, London. Following some minor formatting changes, the final versions of the surveys were sent to a sample of 51 acute NHS trusts (around one-third of all acute NHS trusts) in May 2012. A sample of all NHS trusts was used because of time and resource restrictions. Trusts were categorised by size (small/medium/large or teaching) and a weighted random sample was chosen to reflect the number of trusts of each type across the country: 10 small trusts, 18 medium trusts and 23 large or teaching trusts were contacted.
Four staff contact points in each trust were identified before the survey began [Director of Infection, Prevention and Control (DIPC), the senior infection control nurse (ICN), the lead microbiologist and the microbiology laboratory manager]. The DIPC was initially contacted by e-mail, which contained an introduction letter and examples of the questionnaires. Staff were offered a £20 Amazon voucher as an incentive for completion.
If approval was declined, the trust was not contacted again. When approval was provided (or no response from the DIPC was received) the remaining three staff were initially e-mailed, followed up by a telephone call. If no response was received, a second e-mail was sent in June 2012, again followed up by a telephone call. If no response was received again, respondents were contacted for a third time in September 2012. A final attempt to recruit respondents was made in January 2013.
Results
A total of 26 survey replies were received from 21 NHS trusts across England (17 infection control surveys, six microbiologist surveys and three laboratory surveys). Of the trusts contacted, 41% (21/51) responded to at least one of the surveys. The overall response rate across all surveys was 17% (26/153 surveys). The results of the three surveys are presented in the following sections.
Infection control survey results
Tables 28 and 29 report the characteristics of the 17 NHS trusts which completed the infection control survey. NHS trusts of all sizes and regions completed this survey (with the exception of London), although there were proportionally more responses from small and medium trusts. The average number of whole-time equivalent (WTE) infection control staff per trust is around seven (range 2.5–16.0), and most of these are grade 6 staff.
| Characteristic | Category | Number of NHS trusts |
|---|---|---|
| Type of NHS trust | Small | 6 |
| Medium | 6 | |
| Large/teaching | 5 | |
| Region | East Midlands | 1 |
| East of England | 3 | |
| London | 0 | |
| North east | 1 | |
| North west | 2 | |
| South east | 3 | |
| South west | 3 | |
| West Midlands | 3 | |
| Yorkshire and the Humber | 1 |
| Type of staff | Mean number of staff per NHS trust (SD) |
|---|---|
| Administrative staff | 1.19 (0.61) |
| Grade 6 nurses | 2.15 (1.58) |
| Grade 7 nurses | 1.69 (1.55) |
| Grade 8 nurses | 1.16 (0.57) |
| Doctors | 0.79 (0.61) |
| Total | 6.98 (3.48) |
Monitoring of patients with suspected infectious diarrhoea
Tables 30 and 31 provide information on the burden of suspected infectious diarrhoea and the monitoring of patients with infectious diarrhoea across the trusts. On average, 21% of the time of each infection control team is spent on the routine management of diarrhoea. In around two-thirds of cases, infection control staff are informed about patients with suspected infectious diarrhoea by ward staff or laboratory result. The mean number of patients with suspected infectious diarrhoea admitted to each trust per month is 96 (minimum 18; maximum 450). The average is greatest in small trusts. However, one small trust reported that 450 patients were admitted each month. Disregarding this trust, the average for small trusts was 74.50 (SD 50.74). Infection control teams spend around 1 hour 40 minutes per day tracking patients with suspected or confirmed diarrhoea of infectious origin, with most infection control teams (14/17) tracking both bed and ward moves. This translates to 17.5 minutes per day for each infection control team member. A mixture of manual paper-based and computer systems are used for monitoring: 12 out of 17 trusts use a combination of both. A variety of different computer systems are used, and most of these provide fairly comprehensive information to assist with tracking. However, only two trusts noted that their computer system could provide automatic alerts to notify them of patients with potentially infectious diarrhoea. A total of 80% of patients with suspected infectious diarrhoea enter monitoring systems on the same day that symptoms are initiated (taking on average 3.5 hours to do so) and 53% of all trusts share monitoring information outside of the infection control team.
| Variable | Category | Valuea |
|---|---|---|
| Mean percentage of infection control team time spent on the routine management of diarrhoea (SD) | 20.71 (12.42) | |
| Method by which infection control staff are informed about patients with suspected diarrhoea: % of cases (SD) | Member of infection control team visits ward | 22.42 (18.64) |
| Ward staff contact infection control team | 35.10 (25.99) | |
| Laboratory result received by infection control team | 31.30 (27.90) | |
| Other | 11.12 (26.03)b | |
| Mean number of patients with suspected infectious diarrhoea admitted per month (SD) | Small trust | 149.60 (173.58) |
| Medium trust | 75.00 (27.84) | |
| Large/teaching trust | 43.75 (37.72) | |
| All trusts | 95.67 (117.92)c | |
| Mean number of hours per day spent by the infection control team tracking patients (SD) | 1.66 (0.75)d | |
| Mean number of hours per day spent by each infection control team member tracking patients (SD) | 0.29 (0.19)d | |
| Median percentage of patients with suspected infectious diarrhoea who enter monitoring systems on the same day as initiation of symptoms (width of the interquartile range) | 80.00 (50.00)e | |
| Mean number of hours taken for patients to enter monitoring systems, for those patients who enter this system on the same day as initiation of symptoms (SD) | 3.50 (2.37)e | |
| Percentage of trusts in which access to the monitoring system is limited to the infection control team | 53.33e,f |
| Variable | Category | Number of trustsa |
|---|---|---|
| Method used to track the movements of patients with suspected or confirmed diarrhoea of infectious origin | Only bed moves are tracked | 1 |
| Only ward moves are tracked | 2 | |
| Both bed and ward moves are tracked | 14 | |
| Systems used to monitor patients with suspected or confirmed infectious diarrhoea | Manual paper-based system | 3 |
| Computer-based system | 2 | |
| Both manual and computer-based systems | 12 | |
| Type of computer system used to monitor patientsb,c | CRS/PAS | 8 |
| ICN net | 5 | |
| Other | 8d | |
| Information provided by computer systemc,e | Automatic alerts to notify infection control staff of patients with potentially infectious diarrhoea | 2 |
| Automatic alerts to notify infection control staff of patients with confirmed infectious diarrhoea | 11 | |
| Identifies patients who have previously been admitted with infectious diarrhoea | 7 | |
| Tracks patients with suspected or confirmed infectious diarrhoea through hospital system | 7 | |
| Collects regular data on incidence of infectious diarrhoea | 5 | |
| Provides automated electronic transfer of test results from local microbiology laboratory | 7 | |
| Other | 1f |
Infection control staff training and practice
Table 32 provides information on the training of infection control staff. A ‘standard precautions policy’ operates in 94% of trusts and median compliance with this policy is 80%. All trusts undertake terminal cleans of side rooms or bed spaces in bays when these are vacated by patients with suspected or confirmed infectious diarrhoea. Two-thirds of trusts would undertake a terminal clean of a whole bay in the same situation. All trusts would change the curtains in a side room in this situation, while 88% would change the curtains in a bed space in a bay and and 63% would change the curtains in a whole bay. Glove use increases in 88% of trusts in cases of suspected or confirmed infectious diarrhoea, and in 29% of trusts the cleaning policy varies depending on whether a diagnosis of infectious diarrhoea is suspected or confirmed. In 94% of trusts, the cleaning policy extends to cover other locations that an affected patient may visit, but only 6% of trusts carry out routine environmental testing.
| Variable | Category | Valuea |
|---|---|---|
| Percentage of trusts with a ‘standard precautions policy’ | 94.12 | |
| Median compliance with standard precautions: percentage (width of the interquartile range) | 80.00 (15.00)b | |
| Percentage of trusts in which a terminal clean is undertaken when a patient with suspected or confirmed infectious diarrhoea vacates a space within a ward, by type of spaced | Side room | 100.00 |
| Bed space in bay | 100.00 | |
| Whole bay | 68.75c | |
| Percentage of trusts in which curtains are changed as part of this terminal clean, by type of spaced | Side room | 100.00c |
| Bed space in bay | 88.24 | |
| Whole bay | 62.50c | |
| Percentage of trusts in which glove use increases in cases of suspected or confirmed infectious diarrhoea | 88.24 | |
| Percentage of trusts in which the cleaning policy varies depending on whether a diagnosis of infectious diarrhoea is suspected or confirmed | 29.41 | |
| Percentage of trusts in which the cleaning policy is extended to cover other locations in the hospital which the affected patient may visit | 94.12 | |
| Percentage of trusts that carry out routine environmental testing | 5.88 | |
| Percentage of trusts with a policy for the ‘management of patients with diarrhoea and vomiting’ | 94.12 | |
| Percentage of trusts with an ‘isolation policy’ | 94.12 | |
| Median percentage of patients with suspected infectious diarrhoea isolated in a side room (width of the interquartile range)c | 99.00 (10.00) | |
| Percentage of trusts in which all patients (with suspected infectious diarrhoea) are isolated in a side roomc | 43.75 | |
| When insufficient side rooms are available to manage multiple patients with suspected infectious diarrhoea, how are patients prioritised? (Mean rankinge) | The most severely ill patients are prioritised | 2.33 |
| Older patients are prioritised | 3.50 | |
| Particular pathogens or strains are prioritised | 1.00 | |
| Patients who have been sick for longer are prioritised | 4.00 | |
| Other | 1.60 | |
| Median length of time (hours) for symptomatic patients to be isolated in a side room or cohorted in a closed bay width of the (interquartile range) | 2.00 (2.60) | |
| Mean number of bed moves a typical patient with suspected or confirmed infectious diarrhoea will make across the entire duration of their inpatient stay (SD) | 2.12 (0.86) | |
| Percentage of trusts with a ‘Clostridium difficile policy’ | 100.00 | |
| Percentage of trusts in which staff receive training on the management of patients with potentially infectious diarrhoea, over and above that which is provided in standard operating procedures and policies | 94.12 |
All trusts have a ‘Clostridium difficile policy’, while 94% of trusts have a policy for the ‘management of patients with diarrhoea and vomiting’. The same percentage have an ‘isolation policy’. A total of 16 trusts provided information on the circumstances that would lead to the isolation of a patient with suspected infectious diarrhoea. In 10 trusts, respondents said that all patients with diarrhoea were isolated. Three trusts had criteria based on the frequency and type of stool as classified by the Bristol Stool Chart (‘type 5 stool or above on more than one occasion’; ‘A risk assessment is completed for all cases of type 5–7 stool and a pathway assigned following assessment’; ‘After three or more type 5, 6 or 7 stools in 24 hours’). The other three trusts provided differing criteria (‘prioritised for single room isolation in all cases’; ‘if we have an outbreak or clinically suspect norovirus or C. difficile infection’; ‘if infective diarrhoea suspected, i.e. no other cause’).
The median percentage of patients with suspected infectious diarrhoea who are isolated in a side room is 99%, with 44% of all trusts isolating all such patients in a side room (see Table 32). When insufficient side rooms are available to manage multiple patients with suspected infectious diarrhoea, most trusts prioritise patients by isolating those with particular pathogens/strains, or the most severely ill (see Table 32).
The median length of time it takes to isolate a patient is 2 hours, with patients making two bed moves, on average, during their inpatient stay. In 94% of trusts, staff receive training on the management of patients with potentially infectious diarrhoea over and above that which is provided in SOPs and policies. This extra training can take a number of different forms: Table 33 provides further details.
| Trust | Form of extra training |
|---|---|
| 1 | Included in mandatory clinical updates |
| 2 | Induction and mandatory training face-to-face and e-learning, study days, drop-in sessions |
| 3 | Clostridium difficile drop in sessions/ad-hoc training on wards |
| 4 | Currently face-to-face as part of annual infection control update. E-learning being developed |
| 5 | Ward training based on audit findings and following issues of concern or cases of infection |
| 6 | Via Infection Prevention and Control link practitioner scheme and ward ad-hoc training |
| 7 | Specifically part of clinical mandatory training. There is also a commode cleaning objectively structured clinical examination (OSCE) and additional study days and ‘road shows’ |
| 8 | ‘Getting Ready for Winter – Think Norovirus’ campaign, each winter |
| 9 | Covered in annual updates specifically addressing the need for isolation and personal protective equipment |
| 10 | Covered in mandatory training and induction. Also ad-hoc by ICN if situation arises where knowledge needs updating |
| 11 | Mandatory training and link networker sessions pre-winter season |
| 12 | Ad-hoc ward-based training. ICN attendance at ward training days. Infection control link days |
| 13 | Ward-based training sessions |
See Figure 34 for information on how infection control policy documents are distributed to staff. These documents are predominantly made available via local intranet and targeted training sessions.
FIGURE 34.
Methods by which policy documents are made available to staff.
Management of outbreaks
Table 34 provides information on the management of outbreaks. Most trusts (14/17) classified multiple cases of infectious diarrhoea as an outbreak when a threshold level of cases had been reached. The median annual number of outbreaks of infectious diarrhoea recorded across respondent trusts was 1.5 (viral gastroenteritis), 0.0 (C. difficile) and 6.0 (norovirus). Most trusts (12/17) would consider closing a ward because of outbreak of infectious diarrhoea once a threshold number of cases had been reached. The mean number of wards closed annually, per trust, as a consequence of outbreaks of infectious diarrhoea was approximately 12. Almost two-thirds of trusts had a policy of cohorting multiple patients with infectious diarrhoea in the same ward, if tests indicated that these patients shared the same causative agent. Table 35 provides further information about cohorting policies in seven of the trusts which responded.
| Variable | Category | Valuea |
|---|---|---|
| Conditions under which a trust would class multiple cases of infectious diarrhoea as an outbreak: number of trustsb | For particular strains of pathogen | 6 |
| Once a threshold level of cases has been reached | 14 | |
| Once an attributable death has been recorded | 2 | |
| Other | 4c | |
| Median number of outbreaks of infectious diarrhoea recorded across a trust between 1 April 2010 and 31 March 2011 (width of the interquartile range)d | Viral gastroenteritis | 1.50 (4.75) |
| C. difficile | 0.00 (2.25) | |
| Norovirus | 6.00 (11.75) | |
| Other causes | 0.00 (0.00) | |
| Conditions under which a trust would consider closing a ward as a result of an outbreak of infectious diarrhoea: number of trustsb | Once a single case has been positively identified | 1 |
| For particular strains of pathogen | 6 | |
| Once a threshold level of cases has been reached | 12 | |
| Once an attributable death has been recorded | 0 | |
| Other | 7e | |
| Mean number of wards closed as a consequence of outbreaks of infectious diarrhoea across a trust between 1 April 2010 and 31 March 2011 (SD) | Small trust | 8.00 (5.57) |
| Medium trust | 10.20 (5.40) | |
| Large/teaching trust | 16.25 (11.15) | |
| All trusts | 11.67 (7.90)d,f | |
| Percentage of trusts with a policy of cohorting multiple patients with infectious diarrhoea in the same ward if tests indicate that these patients share the same causative agent | 64.71 |
| Trust | Further information about cohorting policies |
|---|---|
| 1 | Towards the end of an outbreak, patients with prolonged symptoms may be cohorted to enable wards to be reopened |
| 2 | We cohort when wards are closed with suspected norovirus and MRSA patients to free-up side rooms for diarrhoea and vomiting patients |
| 3 | (We only cohort) for viral gastroenteritis |
| 4 | We cohort using single room isolation, where all the rooms have en-suite facilities and are cared for by a specialist doctor and separate nursing staff |
| 5 | Dependent on capacity we will cohort same pathogens based on the length of time that patients have been symptomatic |
| 6 | We don’t create an outbreak ward. Patients are cohorted within their base ward, but we don’t stipulate the need for testing – we would rely on the clinical picture, as per Department of Health guidance |
| 7 | We cohort in a bay with doors |
Microbiologist survey results
Table 36 reports the characteristics of the six NHS trusts who completed the microbiologist survey. No small NHS trusts completed this survey and only a partial geographic spread was achieved.
| Characteristic | Category | Number of NHS trusts |
|---|---|---|
| Type of NHS trust | Small | 0 |
| Medium | 2 | |
| Large/teaching | 4 | |
| Region | East Midlands | 0 |
| East of England | 0 | |
| London | 2 | |
| North east | 0 | |
| North west | 0 | |
| South east | 1 | |
| South west | 1 | |
| West Midlands | 1 | |
| Yorkshire and the Humber | 1 |
Trusts were asked a number of questions about stool sample testing, focusing on tests for seven pathogens (C. difficile, Shigella spp., E. coli O157, Campylobacter spp., norovirus, Salmonella spp. and Cryptosporidium spp.). Table 37 and Figures 35 and 36 detail trust responses to these questions. Pathogens are specified on stool test requests only once every five requests, usually either norovirus or C. difficile. The patient characteristics that influence testing decisions vary by pathogen. Patient age is said to be important for C. difficile testing (probably reflecting Department of Health guidance on mandatory testing), while length of stay is noted as being important for Shigella spp., E. coli, Campylobacter spp. and Salmonella spp. testing (probably reflecting the fact that these pathogens are typically acquired in the community and bought into hospital, rather than being acquired in the hospital). Symptoms/clinical details are important for E. coli, norovirus and Cryptosporidium spp. testing, other patient diagnoses in same ward/hospital are important for Salmonella spp. testing, and the immunocompromised status of a patient is important for Cryptosporidium spp. testing. As expected, the average length of time between taking a stool sample and receiving test results was around 1 day for C. difficile and norovirus, but around 2 days for Cryptosporidium spp. and Campylobacter spp., and 2.5 days for Shigella spp., E. coli and Salmonella spp.
| Variable | Category | Valuea |
|---|---|---|
| Percentage of cases in which pathogens are specified on stool test requests (SD) | 20.17 (16.97) | |
| Pathogens commonly specified: number of trusts | Salmonella spp. | 0 |
| Campylobacter spp. | 0 | |
| Shigella spp. | 0 | |
| Cryptosporidium spp. | 0 | |
| Norovirus | 4 | |
| C. difficile | 5 | |
| E. coli O157 | 2 | |
| Other | 1 | |
| Median length of time (hours) between taking a stool sample and receiving test results, by pathogen (width of the interquartile range) | C. difficile | 21.00 (6.00) |
| Shigella spp. | 56.00 (22.00) | |
| E. coli O157 | 60.00 (20.00) | |
| Campylobacter spp. | 48.00 (12.00) | |
| Norovirus | 24.00 (16.50) | |
| Salmonella spp. | 56.00 (22.00) | |
| Cryptosporidium spp. | 42.00 (30.00) |
FIGURE 35.
Which patient characteristics influence the decision to test for particular pathogens?
FIGURE 36.
What actions are taken if specific pathogens are not identified in a patient sample?
Trusts were also asked about the predicted sensitivity of tests for the seven pathogens, but most stated that predicted sensitivities were unknown. The exceptions were C. difficile (predicted sensitivity 90.50%, SD 0.00, n = 6), norovirus (predicted sensitivity 90.50%, SD 0.00, n = 5) and Cryptosporidium spp. (predicted sensitivity 80.50%, SD 20.00, n = 4). For each of these three pathogens, most trusts (four, five and three, respectively) stated that, because sensitivity was known (to be reasonably high), a negative result was sufficient to rule out an infection. [A reviewer queried the wording of this question: if the sensitivity of a test is low, then practitioners will be concerned that just having a negative result does not rule out infection. This is more than the negative predictive value, because if there are many more negatives than positive, then the negative predictive value may be high. But where the consequences of a missed positive are very severe (e.g. death), practitioners will still be extremely worried that, in an individual patient, a negative could still be an important missed positive because of the low sensitivity.] Most infection control staff remove patients from isolation in the event of a negative result. Antibiotics are stopped in most cases of C. difficile and norovirus, whereas only one trust noted that additional cleaning was stopped following negative results from all seven pathogens.
Around 5% of C. difficile cases were estimated to fall into a potential outbreak situation (Table 38). Strain typing information is requested in 96% of such cases, falling to 66% in cases that fall outside a potential outbreak situation. Strain typing information is used to inform clinical management in a number of ways, including:
-
to identify potentially linked cases and map the extent of outbreaks
-
to detect evidence of cross-transmission over longer periods
-
to demonstrate to commissioners absence of outbreak.
| Variable | Mean (SD) |
|---|---|
| Percentage of cases of C. difficile infection that are considered to fall into a potential outbreak situation | 5.25 (5.75) |
| Percentage of cases of C. difficile infection in which strain typing information is requested in potential outbreaks | 95.50 (0.00) |
| Percentage of cases of C. difficile infection in which strain typing information is requested outside of potential outbreaks | 65.50 (46.48) |
| Percentage of cases of C. difficile associated infection in which further tests are required to confirm a diagnosis | 18.58 (19.87) |
Table 39 summarises treatment practice for suspected or confirmed infectious diarrhoea. Two-thirds of trusts routinely empirically treat patients following a positive C. difficile test. In the majority of cases, first-line treatment is metronidazole and second-line treatment is vancomycin. Standard practice differs on whether treatment is started before samples are sent for testing or once a causative pathogen has been identified. No trusts routinely empirically treat patients with positive Campylobacter spp. or Salmonella spp. tests, focusing any treatment on only the most severe cases. About 47% of all patients complete antibiotic therapy in hospital. Of those discharged before treatment has been completed, 10% will be readmitted within 14 days of discharge. In 27% of cases, causative pathogens are identified after discharge. In these circumstances, all trusts inform the clinical team, advising them to contact the patients’s GP or the patient directly. However, only 40% of trusts ever follow-up patients with diagnoses of infectious diarrhoea in primary care.
| Variable | Category (if applicable) | Valuea |
|---|---|---|
| Percentage of trusts which routinely empirically treat patients with positive C. difficile tests | 66.67 | |
| First-line antibiotic treatment for patients with diarrhoea caused by a C. difficile infection: percentage of trusts which use each treatment optionb | Metronidazole (400 mg, 10–14 days, oral) | 80.00 |
| Fidaxomicin (200 mg, 10 days, oral) | 20.00 | |
| Second-line antibiotic treatment for patients with diarrhoea caused by a C. difficile infection: percentage of trusts which use each treatment optionb | Vancomycin (125 mg, 10–14 days, oral) | 100.00 |
| When is antibiotic treatment initiated in patients with diarrhoea caused by a C. difficile infection: percentage of trusts | When a sample is sent for testing | 0.00 |
| When a causative pathogen is identified | 50.00 | |
| Other | 50.00c | |
| Percentage of trusts which routinely empirically treat patients with positive Campylobacter spp. tests | 0.00 | |
| Percentage of trusts which routinely empirically treat patients with positive Salmonella spp. tests | 0.00 | |
| Mean percentage of patients who complete antibiotic therapy in hospital (SD) | 47.17 (40.82) | |
| Of those patients discharged on antibiotics for infectious diarrhoea before a full treatment course has been completed, the mean percentage who are readmitted within 14 days of discharge (SD) | 10.00 (0.00) | |
| Percentage of trusts reporting that there are regular circumstances in which antibiotic treatment given to treat suspected infectious diarrhoea impacts on other antibiotics that patients may be receiving | 16.67 | |
| Mean percentage of cases in which causative pathogens are identified after discharge (SD) | 26.83 (26.83) | |
| Actions taken if causative pathogens are identified after discharge: percentage of trusts | Clinical team informed and advised to inform patients’ GP or patient | 100.00 |
| Percentage of trusts that follow up patients with diagnoses of infectious diarrhoea in primary care | 40.00d | |
| Percentage of trusts with procedures in place to identify patients who have been readmitted within 14 days of discharge, again with infectious diarrhoea | 60.00 |
Laboratory survey results
Only three trusts responded to the laboratory survey. Furthermore, these three surveys all contained incomplete information: only three questions were answered by all three trusts. The level of missing information was such that we determined that no insight would be gained from presenting this incomplete information in this report.
Questions common to multiple surveys
Two questions were common to both the infection control and microbiologist surveys: trusts were asked to consider how two potential future scenarios might impact on the management of patients with suspected infectious diarrhoea. Table 40 presents the responses to scenario 1, which asked respondents to consider the impact of a consolidation of microbiology laboratory services. This scenario is being discussed, but has not currently been implemented. Typically, at present, each large NHS trust runs its own service microbiology laboratory; smaller NHS trusts would send specimens to the closest large trust microbiology laboratory. Centralising services further might provide economies of scale, enabling more rapid uptake of new technologies such as the MassCode assay, and also making them more cost-efficient, particularly if such ‘super-laboratories’ ran 24 hours a day. The responses from trusts indicated a number of concerns. These included:
-
increased length of time to receive test results (because of greater transportation costs, loss of specimens);
-
greater transmission and more frequent outbreaks;
-
slower decision-making (leading to delayed treatment, bed-blocking, and delayed discharges);
-
loss of local epidemiology data and responsiveness to local needs.
| Trust | Survey type | Response |
|---|---|---|
| 1 | InfC | It would lengthen the time taken to establish whether or not norovirus is confirmed and slow down the decision-making process re: restrictions on wards, for example [by] delay[ing] discharges |
| 2 | InfC | Delays in specimen transport and delays in results. Unlikely to happen here as we would be the central laboratory, but increased workload without additional staff might compromise our existing service |
| 3 | InfC | Would take longer to receive results |
| 4 | InfC | Concerned that this will lead to a lack of timely results, which in turn will cause delays in treatment, greater transmission, and increased operational pressures |
| 5 | InfC | This should have no impact if the processes are developed well, e.g. transport of samples, communication with laboratory and effective systems for receiving results in real time (not waiting for authorisation to release results) |
| 6 | InfC | None if system and processes are well defined and developed [this is occurring at (trust)] |
| 7 | InfC | Patients with suspected diarrhoea must be treated using the DH SIGHT protocol (Suspect that a case may be infective when there is no clear alternative cause for diarrhoea; Isolate the patient within 2 hours; Gloves and aprons must be used for all contacts with the patient and their environment; Hand washing with soap and water should be carried out before and after each contact with the patient and the patient’s environment; Test the stool for C. difficile by sending a specimen immediately); however, if there was a delay in sending or receiving results, i.e. if the laboratory was off-site then this may delay patient treatment and cause bed-blocking if norovirus is suspected. Also delay in swift identification of outbreaks |
| 8 | InfC | For C. difficile, this will be detrimental as it will delay results. While there is an expectation that wards inform us of cases of diarrhoea, this does not always happen. Therefore, the positive result is the first instance that the infection control team are aware and can ensure compliance. Having regional units will increase transportation time. We currently have two sites with a microbiology laboratory on one site. The site without a microbiology laboratory already sees a 24-hour delay in results in general because of this. Regional centres will just make this worse. I think this is less of a case for viral diarrhoea, as we are more likely to be made aware early (as multiple cases) and take action before results are available |
| 9 | InfC | Possibly none if all testing based on PCR with a service-level agreement of quick turnaround and courier service. Impact will be based on current level of provision and change |
| 10 | InfC | Currently all inpatients with confirmed C. difficile are transferred to the isolation unit within 2 hours of their diagnosis being made. Any delay in receiving this diagnosis (currently usually same day as specimen is received) could result in further transmission of infection and potentially delay appropriate treatment for the patient |
| 11 | InfC | We have our own laboratory within the hospital. I think any delay in obtaining results will be detrimental |
| 12 | InfC | Increased time to gain result. Loss of specimens (breakages/spillages/misplacement). Loss of local epidemiology data – strains/antibiotic resistance patterns/identification of periods of increased incidence. Loss of responsiveness to local needs – hours of working/changes to testing frequency/changes to testing because of resistance patterns/lack of suppression of antibiotic data – increased antibiotic resistance |
| 13 | InfC | None if a system of timely results is put in place |
| 14 | InfC | Delays in confirmation/samples going missing |
| 15 | Inf | There may be a delay in the time it takes to send a sample to the laboratory to be processed and results reported back. There will be a loss of local knowledge to manage each ward independently. Central laboratories will not know the hospital or its population/cohort of patient on each speciality ward. The flip side to this is the advice should be generic and the same precautions taken, the advice to close wards will be more difficult and may not happen as it currently does |
| 16 | InfC | Possible higher risk of samples being lost and longer time for results to be available |
| 17 | InfC | Delay in results, more logistical issues, loss of personalised service, less learning and teaching using shared knowledge, more bureaucracy and less flexibility. Larger hospitals may drive the changes and this may compromise smaller hospitals |
| 18 | M | Serious negative impact on norovirus outbreak management inevitable (whatever the powers that be claim) with significant length of ward closures. Negative impact on centres that have a C. difficile problem. In both cases, results that become available after c. 4 p.m. are worse than useless as ICN and ward sisters will have gone home and this is not the sort of thing that can be managed by telephone from home |
| 19 | M | None. We have already merged two trusts’ microbiology laboratories into one (on one site) and are planning to move the whole laboratory off-site, with no deterioration in service |
| 20 | M | Delay in diagnosis resulting from increased turnaround times (because of transportation), subsequent reduction in volume of tests requested |
| 21 | M | If samples are having to be sent to off-site laboratories for testing this may increase turnaround times. It would be a particular concern for C. difficile testing on inpatient samples |
| 22 | M | This is all right as long as the results are at least as reliable and rapid as the current model |
| 23 | M | None as we are a regional centre |
Successful implementation was said to require well-defined processes and good communication systems, and could lead to increased consistency in infection control advice offered.
Table 41 presents the responses to scenario 2, which asked respondents to consider the impact of a new diagnostic test which could detect 30 pathogens in a single reaction. Most trusts were positive about the consequences of this scenario. A number of potential benefits were identified, which included:
-
more informed and faster decisions regarding the need for isolation and de-isolation;
-
more effective use of limited side room space and reduced bed-blocking;
-
improved patient treatment outcomes;
-
earlier identification of outbreaks and implementation of cohorting.
| Trust | Survey typea | Response |
|---|---|---|
| 1 | InfC | This would help enormously with deciding whether patients require isolation or not and would improve the use of our limited number of side rooms |
| 2 | InfC | Enables rapid isolation if not already done and enables de-isolation if no pathogens detected, to free-up side rooms for other cases |
| 3 | InfC | Quicker results |
| 4 | InfC | Sounds very helpful. At least then you would know what you were dealing with |
| 5 | InfC | If results for all pathogens were known it would be easier to discontinue isolation and prioritise single rooms. Early identification of outbreaks |
| 6 | InfC | Ability to discontinue isolation if all negative, will it identify C. difficile carriers? And cohort if appropriate |
| 7 | InfC | Improve patient management and treatment, prevent or reduce bed blocking. Aid in the allocation of available side rooms, improved outcomes for patient |
| 8 | InfC | It would really depend on accuracy, primarily around C. difficile (if this is part of it). There have been issues nationally until recently on ensuring the testing algorithm is the same in all institutions. The accuracy is important for patient management but additionally for targets. We cannot as an organisation accept any level of false positives as the financial implications are too great. If the test was introduced we would review against data and current testing systems |
| 9 | InfC | Most likely impact would be release of contact bays quickly. Currently when patient moved to a cubicle the rest of bay is closed as a contact for 72 hours. This measure would only be taken if norovirus was confirmed |
| 10 | InfC | The biggest advantage would be the increased evidence for prioritising single room allocation since they are an overused resource |
| 11 | InfC | We already get results within 24 hours. I think there are already near patient testing kits which will have a turnaround time of 30 minutes for norovirus – this is the way forward |
| 12 | InfC | Early identification of non-infectious causes will allow more flexible use of side rooms and earlier referral to gastrointestinal services. Some benefit would be achieved if the 30 pathogens detected included viral causes, e.g. norovirus, rotavirus. Most benefit would be achieved if sample could be gained on a swab – major difficulties with gaining suitable specimens from incontinent patients because of the use of incontinence pads. If this is a laboratory-based test, then there is still reliance on 24-hour laboratory cover (not achieved in this trust). Near-patient testing would be more beneficial |
| 13 | InfC | As we isolate all patients on onset of diarrhoea, not on receiving a positive sample, then little impact would be seen. It would however allow assessment onto the correct treatment quicker |
| 14 | InfC | Free-up isolation capacity/better management of patients/fewer pseudoviral gastroenteritis outbreaks |
| 15 | InfC | A useful tool if virology pathogens are included. Virology results can take up to 72 hours when sent to external laboratories for testing, blocking side rooms for some time and putting other patients at risk, as diarrhoea and vomiting will take precedence over some other pathogens such as MRSA colonisation. IC teams will always restrict movement/flow until they are satisfied that the risk of an infectious cause has been ruled out. Early results help patient flow and bed management for an organisation |
| 16 | InfC | Quicker treatment and fewer in-patient days. Isolation would happen anyway because of the number of side rooms we have |
| 17 | InfC | None, we already have this service |
| 18 | M | Clostridium difficile is a common coloniser (estimated at 2% plus). Therefore, a possible C. difficile result will be meaningless in the absence of clear-cut evidence of infection (disease may have developed of all manner of reasons, e.g. too much Weetabix for breakfast). Similarly, we do not know how long Salmonella continues to be detectable after the summer barbeque. Norovirus remains positive for weeks after acute infection. I rest my case |
| 19 | M | This would be of enormous benefit. Currently we use a mixture of fast tests (PCR, antigen) and slow tests (culture). However even having a negative ‘fast test result’ does not rule out other causes of infectious diarrhoea. The combination of rapid results for a wide range of pathogens will mean that, within 24 hours, we will have reliable results which can give confidence to bring patients out of isolation, or send them home, stop antibiotics (or initiate new antibiotics) |
| 20 | M | The majority of patients with hospital-acquired diarrhoea will not need a panel of 30 targets, probably just norovirus and C. difficile. Potentially useful for community-onset diarrhoea. Samples positive for Salmonella [spp.], Shigella [spp.] and E. coli O157 will still need culture for sensitivity/typing/epidemiology |
| 21 | M | Reduced turnaround time leading to more rapid diagnosis. The knock-on benefits would include earlier initiation of appropriate antimicrobial therapy and more rapid removal of patients without infectious diarrhoea from source isolation facilities, improving infection control management |
| 22 | M | A reliable rapid single test for multiple pathogens would streamline laboratory processing and simplify clinical decision-making and infection control. There would need to be provision for further investigation of positive results to allow for typing for outbreak investigation and public health |
| 23 | M | It will not affect initial clinical management but may affect choice of antimicrobial where indicated. It will not affect C.difficile, norovirus, or Cryptosporidium/Giardia as results are already known within this time scale. Susceptibility testing of bacterial pathogens will still be needed for optimal therapy |
Concerns were, however, raised about the need for such tests to be accurate and the requirement for samples to be taken as simply as possible.
Discussion
With advances in molecular and genomic testing on the horizon in infection control and microbiology departments, it is important to understand how these departments currently operate, so that the implications of more widespread genetic and genomic testing can be assessed. This chapter reports the results of three surveys conducted to map current infection control, microbiologist and laboratory practice across England with respect to the management of patients with suspected infectious diarrhoea.
A significant proportion (21%) of infection control staff time is devoted to the management of this patient group. A variety of monitoring systems are used, most of which provide fairly comprehensive information to assist with tracking. Training of infection control staff in the management of these patients is generally good, although alternative methods of disseminating training information could be considered, and mean compliance with ‘standard precautions’ policy documents is only 87%. Reasons may be cultural or structural (e.g. high level of agency staff) or reflect a near-continuous emergency situation in some trusts facing acute pressures. There is some variation in both cleaning and isolation policies across different trusts, suggesting either that these policies are not evidence based or that the evidence base is weak in this area. There is more agreement on outbreak definitions and management, as well as cohorting policies.
In the absence of evidence describing the transmission routes of these organisms, it is not clear that each trust should follow the same infection control protocols. Having said that, the clinical condition of the patient and the ward design often dictates how the patient is managed, with people working with what they have.
The microbiologist survey revealed that pathogens are specified on stool samples on a minority of occasions, with different patient characteristics driving this decision for each pathogen. Time from sampling to test result also varies considerably by pathogen, but negative results do generally lead to patients being removed from isolation. Strain typing information is commonly used if a C. difficile outbreak is suspected, probably reflecting the easy access to ribotyping through the Clastridium Difficile Ribotyping Network (CDRN),28 but is used less so outside of these situations. Many patients complete antibiotic therapy outside of hospital and a significant minority are subsequently readmitted for additional treatment.
The comments in response to the two potential future scenarios (and the free-text comments throughout the rest of the surveys) indicate a clear need for the type of interventions that have been considered in this study. Respondents identified a number of difficulties currently faced in this clinical context, including the lack of side room capacity and the existence of bed-blocking. Respondents also revealed a clear appetite for molecular and genomic testing to assist with the management of patients with suspected infectious diarrhoea, highlighting a variety of potential benefits, including more informed and faster decisions regarding the need for isolation and de-isolation, improved use of limited side room space and reduced bed-blocking, improved patient treatment outcomes, and earlier identification of outbreaks and implementation of cohorting. However, respondents identified several issues that should be addressed when planning the future introduction of such interventions, particularly if this leads to a more centralised molecular testing service. These issues included the likely increased length of time to receive test results, slower decision-making and the creation of a service which is less responsive to local needs. This was particularly the case regarding the potential for such interventions to be introduced as part of a broader move towards consolidation of laboratory services in 10 or so ‘super-laboratories’ across England, rather than the current state in which most large NHS trusts maintain their own service microbiology laboratory. Discussions around introduction of such ‘super-laboratories’ are independent of the specific assays that would be used in them. Nevertheless, placing new technologies such as MassCode or Luminex that require significant capital infrastructure might be a more cost-effective mechanism for rolling out these technologies across the NHS, were they to be effective, particularly if such ‘super-laboratories’ ran 24 hours a day, 7 days a week, to maximise use of the fixed-cost infrastructure. Respondents identified key concerns about time from sample collection to return of results. Whether this would be a real or only a perceived problem is unclear, since centralised laboratory systems run successfully in several countries in Europe.
Although the questionnaire was reviewed by multiple individuals from different disciplines before being sent out, one possible limitation is that, nevertheless, some questions contained potential ambiguities. For example, a reviewer noted that the question ‘please estimate the average number of patients with suspected infectious diarrhoea admitted to your trust per month’ could be interpreted as ‘estimate the average number of patients who are reported to have suspected infectious diarrhoea each month’, or as ‘estimate the average number of patients admitted each month who already have suspected infectious diarrhoea’. Given that all NHS staff reviewing the questionnaire pre-implementation interpreted it as the former, we consider that this is the most likely interpretation, but this still highlights challenges in developing survey questionnaires.
The response rate for these surveys was low and somewhat disappointing, particularly for the microbiologist and laboratory manager surveys. This may, in part, be as a result of the length of the surveys and the detailed questions that were asked, designed to provide as accurate a picture as possible of the issues in current practice. In addition, some of the questions in the surveys requested information which some respondents could have viewed as being sensitive, potentially dissuading some people from responding (even though all respondents were told that their responses would be anonymised). Furthermore, it should be noted that several questions required respondents to self-report adherence to trust policies. Only very limited conclusions can be drawn from these surveys as a result. Although this could limit the generalisability of the results to all NHS trusts, our view is that they are generalisable given that the responses that were provided were internally consistent, and are consistent with requests for advice regarding management made to those in the research team and reflect anecdotal comments. Future studies in this area should consider alternative methods to incentivise participation in surveys which request potentially sensitive information and also explore other approaches for collecting potentially sensitive data in this context.
To place these results into context and quantify the potential benefit of improved molecular/genetic diagnostics in infection control, it is informative to consider the costs associated with microbiological testing and isolation measures. However, there is very little information available within the literature on such costs, particularly in the UK. Wiegand et al. 29 identified three studies that have quantified the economic burden of C. difficile infection, noting that the incremental cost of infection ranged from £4577 in Ireland to £8843 in Germany. The only cost estimates for the UK were based on historical data from 1996. 30 Estimates for the USA lie within a broadly similar range, depending on the analytical perspective adopted. 31 No studies were identified which provided cost data that could be used to estimate the potential monetary benefits of improved molecular/genetic diagnostics in infection control.
Conclusions
This chapter summarises current infection control, microbiologist and laboratory practice across England with respect to the management of patients with suspected infectious diarrhoea. A significant proportion of NHS time is devoted to the management of these patients, and improvements in the quantity and quality of molecular and genomic information relating to diagnosis of gastrointestinal pathogens could have significant clinical and economic impacts in this context.
Chapter 7 Conclusions
Overall, this large and comprehensive assessment of two multiplex assays (MassCode and Luminex) for gastrointestinal pathogens has demonstrated that neither is currently ready for deployment in the NHS. However, the substantial burden of, and difficulties in, dealing with infective diarrhoea, as evidenced by the survey of infection control practice (see Chapter 6), and the clear desire for improved molecular/genetic diagnostics to become available demonstrate that better and faster diagnostics for gastrointestinal pathogens in this extremely challenging but common clinical specimen type remain a critical unmet need. The eventual implementation of such diagnostics must be carefully planned to minimise the potentially negative effects of a probably more centralised diagnostic service, including potentially slower decision-making and the creation of a service which is less responsive to local needs.
The yield of nucleic acids was one of two main challenges for the MassCode assay, proving insufficient for S. enterica and suboptimal for other species. Although this problem was not demonstrated to the same limiting degree for Luminex, its sensitivity for detecting S. enterica on fresh extracts (i.e. precisely how it would be used in a service microbiology laboratory) is still a major concern given the clinical importance of this organism. The specific problem with DNA extraction from Salmonella spp. remains unclear; it is possible that their nucleic acids may be susceptible to specific DNase degradation, or may bind to proteins and be discarded through the extraction process, but there is no direct support for such hypotheses at present. However, poor sensitivities for molecular-based tests for Salmonella spp. compared with culture-based tests have been noted previously,12 demonstrating that this is an area requiring more research. The small study of extraction efficiency that we were able to conduct across other members of the Enterobacteriaceae family suggests this is a challenging group of organisms to assay with molecular methods directly on stool samples.
It was interesting that using the Luminex assay on extracts originally done for the MassCode assay approximately 6 months previously substantially increased the sensitivity of the Luminex assay. This suggests that inhibitors present in stool samples may be contributing to the problem; however, the corresponding substantial increase in false positives using these original extracts means that leaving samples for a period of time would not improve the overall assay performance. In any case, this would negate the potential benefits from faster diagnostics, which remains a key goal. The lack of agreement between an identical assay performed by the same clinical scientist on two different extracts from the same samples is concerning. It is important to appreciate that one major challenge for multiplex panels including bacteria, parasites and viruses is that extraction methods have to be generic and suitable for both DNA and RNA, since the pathogen that will eventually be isolated is unknown at the time the extraction is performed. It is unrealistic to expect one single method to be optimal for all types of genetic material, probably necessitating trade-offs.
The second challenge for both MassCode and Luminex multiplex assays on the MassCode/Oxford extracts was the considerable number of false positives, illustrating major issues with multiplexing large numbers of PCR primers together, and the major development work that would be needed to add just one single new primer set into a reaction mix. For S. enterica, in particular, a positive result would lead to potentially intensive public health investigations; in this context, false-positive results are extremely undesirable. Interestingly, the Luminex assay using fresh samples did not seem to suffer from this problem to the same degree. Anecdotal reports from the clinical scientist operating the Luminex instrument suggested that particular efforts had to be made during sample preparation to reduce false positives (i.e. samples have to be kept very cold during the entire process). This suggests that the assay may lack the robustness required to roll out to NHS laboratories, although it does not explain differences between the same Luminex assay run on original MassCode/Oxford versus fresh/Leeds extracts. Nevertheless, given the large number of stool samples tested per year in large service laboratories (≈ 12,000/year in Oxford and Leeds laboratories), even 99% specificity would still result in an additional 100–200 false positives per year for infections which are rarely identified microbiologically. This highlights the very large sample sizes needed to evaluate whether or not assays really are NHS ready and the challenges that workflows will need to address to ensure patient management is not compromised. For example, if specificity (sensitivity) is expected to be 99%, sample size calculations suggest that 1567 negative (positive) samples would need to be tested to provide > 0.95 probability that the estimated lower 95% confidence limit is above 98%. 32 In terms of workflows, at minimum, unexpected positives for rare pathogens should probably be cultured or tested for using single qPCR assays and reports of pathogens to physicians should be considered only preliminary pending confirmation. However, this means that service laboratories would have to continue to operate a standard ‘faeces’ bench, and provide a large number of tests, in conflict with the goal of multiplex assays to reduce the number of workflows operating (see Figure 1).
Nevertheless, unexpected positives by both the MassCode and the Luminex assays were confirmed by single qPCR, illustrating that co-infections and/or undetected infections may be occurring in a small but important minority of diarrhoea cases. Whether these represent coincidental carriage, or whether the organisms are actually causing disease in these patients is unclear, as the majority of unexpected positives (particularly Cryptosporidium spp. and Giardia) were identified as co-infections with other important pathogens, rather than as single organisms (which might be more plausibly the cause of infection). Disentangling the contribution of a rare organism to disease is extremely difficult when asymptomatic carriage is possible, and where diarrhoea can be because of a number of non-infective causes. This remains an area that should be addressed in future studies, for example by looking at recovery rates without specific treatment for the co-infecting/co-carried but rare organisms.
When the study was designed, a major concern about the MassCode assay was this potential to identify a high proportion of patients with colonisation rather than infection even with the main four pathogens considered (true ‘colonisation positive’, false ‘infection positive’). This could lead to unnecessarily increased anxiety for patients and considerable additional unnecessary treatment costs. This risk was envisaged to be magnified by the fact that the MassCode technology simultaneously tests for multiple organisms – any of which could be either infecting or colonising. In contrast, the expected benefits were that the PCR test would identify a pathogen faster. In fact we did not see strong evidence for this, although cocolonisation may have been responsible for a small number of the unexpected positives with the four main pathogens; rather, we encountered generalised problems with false positives probably relating to challenges including multiple primers in one reaction. Nevertheless, the results do highlight an intrinsic issue with identifying which organisms in a case of potentially infective diarrhoea may be causing the disease, compared with merely being co-carried and, hence, detected. However, it might also plausibly be argued that even if only carried, a patient with diarrhoea might nevertheless have the potential to transmit pathogen(s) onwards, making their identification still relevant clinically.
Although the Luminex assay did perform better than the MassCode assay, nevertheless several features raise concerns about its widespread adoption in the NHS. First, as discussed above, is the lack of reproducibility between results using original and fresh extracts. In routine service, however, it would be used on fresh extracts – here, its sensitivity for detecting S. enterica is likely to be a key barrier to widespread adoption. As pointed out by a reviewer, this strongly supports further diagnostic studies in this area being powered to detect a lower limit of the 95% CI around sensitivity above a specific threshold, rather than on the basis of the width of the 95% CI, as in our study. Further, it is interesting that a reasonable minority of C. difficile-positive samples (n = 10) were identified as A+B– strains using the Luminex assay on fresh/Leeds extracts, more than with original MassCode/Oxford extracts (n = 3), suggesting some issues with sensitivity for detecting the tcdB gene since A+B– strains have never been confirmed clinically, only as laboratory-generated mutants. The fact that a similar number of C. difficile-negative samples were also identified as A+B– would make distinguishing these plausible false negatives from false positives impossible without further testing. This could be problematic in a busy laboratory where C. difficile is one of the key organisms that many samples are tested for.
One important limitation of the diagnostic study raised by one of the reviewers is that it is not strictly speaking statistically orthogonal; that is the two tests (MassCode and Luminex) were not applied in identical conditions. There were several reasons for this. The original study was designed based only on MassCode assay, as at the time the Luminex assay was not available for comparison. The challenges with extraction (see Chapter 2) and failure of the MassCode assay to proceed to phase 2 (see Chapter 3) led to concerns over the potential for similarly poor performance of the Luminex assay. The funders agreed and enabled some of the remaining funding to be used for a direct comparison with Luminex on as many samples as possible. However, only the Leeds and not the Oxford service laboratories had a Luminex machine already installed, necessitating testing of the two different assays in two different locations. We aimed to at least partially address this limitation by firstly testing material extracted in Oxford on the Luminex machine in Leeds, albeit with a time delay from storage and, then, also to test material freshly extracted in Leeds. Unfortunately, not all samples had sufficient material remaining to be tested with all three approaches.
As the phase 1 study did not meet its pre-defined objectives, the originally planned phase 2 study, directly comparing turnaround times from sample submission to pathogen identification between the standard service microbiology laboratories in Leeds and Oxford versus the MassCode assay, did not proceed. Phase 1 was a standard diagnostic study performed on batches of discarded specimens, and so turnaround times were not a relevant outcome measure. Based on phase 1, however, an assay run of 84 samples plus 12 controls on a 96-well plate would take approximately 2 hours using the MassCode assay, but DNA extraction and preparation would take as much as four times longer. In all, 24 samples could be run from specimen to MassCode result in approximately 7 hours (2-hour extraction, 1.5-hour cDNA synthesis, 1.5-hour PCR, 1-hour clean-up and 1-hour MassCode). Luminex turnaround times were similar (as might be expected from a commercially available assay); in total, the pre-extraction step and QIAsymphony extraction took approximately 2.5 hours per batch of 23 clinical samples plus one extraction control. The Luminex assay then took approximately 7 hours to set up and run 91 samples and five controls in a 96-well plate. Variations to the workflow are possible: certain steps can be run overnight or stored at 4 °C for short periods to fit in with the working day. Many standard microbiological processes take at least 1 day per organism to be identified, and a negative faecal culture typically 3 days, demonstrating the enormous potential gains that remain to be realised from better multiplex PCR-based assays for gastrointestinal pathogens.
Despite all the challenges with the assays, the survey of infection control practitioners demonstrated the importance of this area to the NHS. Respondents identified a number of difficulties currently faced in this clinical context, including the lack of side room capacity and the existence of bed-blocking. Managing infectious diarrhoea was a significant burden for infection control teams (taking 21% of their time), with patients with suspected infectious diarrhoea making two bed moves on average during their inpatient stay. As expected, the average length of time between taking a stool sample and receiving test results from the service microbiology laboratory was around 1 day for C. difficile and norovirus, but around 2 days for Cryptosporidium spp. and 2.5 days for Shigella spp., E. coli, Campylobacter spp. and Salmonella spp. Better molecular diagnostics were identified as having major potential benefits for patients, including more informed and faster decisions regarding the need for isolation and de-isolation; more effective use of limited side room space and reduced bed-blocking; improved patient treatment outcomes, and earlier identification of outbreaks and implementation of cohorting. Concerns were, however, raised about the need for such tests to be accurate and the requirement for samples to be taken as simply as possible. The possibility of such tests being introduced into the NHS through wide-scale laboratory consolidation also raised substantial concerns, particularly regarding time from sample collection to receiving test results, and the impact that delays would have on patient management and onward transmission. One major limitation was the very low response rate of microbiologists and laboratory managers to the survey, despite offering a modest financial incentive. This might reflect the complexity of the questionnaire, which by necessity included detailed questions on all the workflows relevant to the MassCode assay (see Figure 1). Alternatively, it could reflect sensitivity to the issue of outsourcing diagnostic services, which would have negative implications for local staff even if it led to greater efficiency across the NHS as a whole.
Independently of the phase 1 study, the company marketing the MassCode assay (Agilent Technologies) decided to stop development of the assay, citing the fact that whole-genome sequencing approaches were likely to supersede this technology over the coming decade. While not denying the enormous potential of whole-genome sequencing, the challenges encountered in this evaluation (low extraction efficiency, inhibition) are likely to also apply to any whole genomic approach to sequencing direct from stool, or following 16S- or other PCR-based amplification. Issues around identification of multiple organisms seem likely to only increase with these approaches. As a consequence of the large amount of human genetic material, inhibitors, other non-infectious bacterial species, etc., stool samples are widely recognised to be the most challenging clinical specimen and, any DNA-based multiplex gastrointestinal assay therefore has to try to identify multiple targets in the most challenging possible human sample type. Nevertheless, gastrointestinal infections place a huge diagnostic and management burden on the NHS. To improve workflows in service microbiology laboratories, to reduce workload for infection control practitioners, and to improve outcomes for patients with potentially infective diarrhoea and at risk from contracting it, further research on multiplex gastrointestinal diagnostics is urgently needed.
The issues encountered in this study provide some pointers as to what form such research might usefully take. Obtaining high-quality DNA from stool samples was one major challenge, particularly for Salmonella spp. Although it may appear unexciting, and would not fall naturally under the remit of any funding body in the UK, research investigating methods of optimising nucleic acid extraction from different sample types would probably have an enormous impact on the field of diagnostics, since all molecular testing relies on the amount of input pathogen material. Our literature searches following initial problems, as described previously (see Chapter 2), found very little literature on this topic. Any future whole-genome sequence-based approaches, in particular, are likely to require far more nucleic acid than the PCR-based methods evaluated here. Given that DNA extraction and sample processing took nearly four times longer than running the assay itself on both MassCode and Luminex platforms, research into reducing durations for these critical steps would also be of immense value. Research developing PCR enzymes would also be beneficial, potentially increasing the fidelity of large multiplex reactions. However, the view of the investigators is that multiplex PCR-based approaches are unlikely to provide a sufficiently accurate solution to enteropathogen diagnostics in the long term given the challenges inherent in human faecal specimens. Multiplexing even 5–10 different primer sets together in the MassCode and Luminex assays appears to affect sensitivity and specificity to a degree which would not be acceptable in routine clinical practice. Furthermore, a major challenge with PCR multiplexes is that adding even one additional primer set can lead to unforeseen cross-reactions and completely change the test performance of the previous primers, necessitating a new round of large-scale evaluation. Multiplex PCR-based approaches are therefore relatively inflexible. Whole-genomic sequencing may well turn out to provide a better strategy for simultaneous diagnosis of multiple enteropathogens, but the field is in its infancy and many technical challenges remain, particularly in terms of sequencing direct from samples. One particularly attractive option for future research would be to retain the multiplex PCR, but sequence the entirety of the PCR products, rather than just trying to detect their presence/absence using MS (MassCode) or fluorescent probes (Luminex). This should address the issue of non-specificity, since amplicons which did not match the product sequence expected from the species targeted by the primer pair would simply be discarded. More importantly, it would retain the ability to detect low-level or minority species, through the PCR amplification step of specific target sequence from these organisms, which could simply be present at too low a frequency to be detected from direct sequencing of the ‘bulk’ sample. However, using next-generation sequencing approaches as a read-out of multiplex reactions can only increase specificity while retaining sensitivity: it cannot address the problem of low extraction yields for Enterobacteriaceae, which remains the most important challenge and area for research in our opinion. With regards to evaluation, the differences between results from our (blinded) study and those reported by the manufacturer and manufacturer-sponsored studies highlights the critical importance of large-scale evaluations funded by independent organisations, ideally comparing multiple assays on the same samples, as was recently done for C. difficile. 5 For implementation, we still believe that a large parallel study directly comparing turnaround time and accuracy with standard microbiological processes, as we had proposed for phase 2, is the best design, since this directly answers the questions relevant for rollout as well as providing a framework for the necessary health economic evaluations. As proposed, we had also intended to quantify downstream consequences in terms of changes in isolation capacity; we consider that providing information on such clinical outcomes, and particularly demonstrating that the new technology does not lead to adverse unintended consequences, will be key to increasing uptake. However, the extremely low response rate to the laboratory manager survey, designed to elucidate and cost current workflows and pathways for specimen processing, illustrates the challenges in reliably costing current practice and estimating cost–benefit ratios from introducing new technologies, which are typically associated with greater upfront costs, and are potentially disruptive for existing staff.
Acknowledgements
Contributions of authors
Louise Pankhurst (senior research scientist, molecular diagnostics) validated MassCode assay performance, conducted blinded MassCode assays, conducted confirmatory PCR testing and prepared the publication.
Louissa Macfarlane-Smith (pre-registration clinical scientist, microbiology) conducted Luminex testing on MassCode/Oxford and fresh/Leeds extracts, conducted confirmatory PCR testing and prepared the publication.
James Buchanan (researcher, health economics) developed and tested the infection control, microbiologist and laboratory manager questionnaires, prepared the results for publication and wrote the chapter describing these results.
Luke Anson (research assistant, molecular diagnostics) validated MassCode assay performance, conducted blinded MassCode assays and confirmatory PCR testing.
Kerrie Davies (clinical scientist, microbiology) managed the Luminex testing.
Lily O’Connor (consultant nurse, infection control manager) contributed to the development of infection control, microbiologist and laboratory manager questionnaires and to their completion by different NHS trusts.
Helen Ashwin (research technician, microbiology) extracted samples for Luminex testing.
Graham Pike (senior infection control nurse) contributed to the development of infection control, microbiologist and laboratory manager questionnaires and to their completion by different NHS trusts.
Kate E Dingle (senior research scientist, microbiology) contributed to the assessment of MassCode assay performance.
Timothy EA Peto (professor and consultant, infectious diseases) reviewed and contributed to the publication.
Sarah Wordsworth (senior researcher, health economics) contributed to the development of infection control, microbiologist and laboratory manager questionnaires and prepared the publication.
A Sarah Walker (senior research fellow, medical statistics) analysed the MassCode and Luminex test results and prepared the publication.
Mark H Wilcox (professor and consultant, microbiology) provided oversight of Luminex testing in Leeds, and prepared the publication.
Derrick W Crook (professor and consultant, microbiology) provided oversight for the whole project, including the MassCode testing in Oxford, and prepared the publication.
Contributions of others
We are grateful to the staff in the routine service laboratories at the Oxford University Hospitals NHS Trust for assistance in sourcing specimens, and to Kirsti Morris from the Leeds Teaching Hospital NHS Trust for assistance in sourcing additional S. enterica positive specimens. We would also like to thank Alison Vaughan who anonymised the samples for blinded testing with the MassCode assay, and Jessica Evans, Tonya Votintseva and Teresa Street for assistance with experiments to assess different extraction methods. Finally, we particularly appreciate the contribution of the staff from the different NHS trusts who completed the infection control, laboratory manager and microbiologist questionnaires.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health.
References
- Department of Health . The Health and Social Care Act 2008 n.d. www.gov.uk/government/uploads/system/uploads/attachment_data/file/216227/dh_123923.pdf (accessed May 2014).
- Wigglesworth N, Wilcox MH. Prospective evaluation of hospital isolation room capacity. J Hosp Infect 2006;63:156-61. http://dx.doi.org/10.1016/j.jhin.2006.02.008.
- Planche T, Aghaizu A, Holliman R, Riley P, Poloniecki J, Breathnach A, et al. Diagnosis of Clostridium difficile infection by toxin detection kits: a systematic review. Lancet Infect Dis 2008;8:777-84. http://dx.doi.org/10.1016/S1473-3099(08)70233-0.
- Planche T, Wilcox M. Reference assays for Clostridium difficile infection: one or two gold standards?. J Clin Pathol 2011;64:1-5. http://dx.doi.org/10.1136/jcp.2010.080135.
- Planche TD, Davies KA, Coen PG, Finney JM, Monahan IM, Morris KA, et al. Differences in outcome according to C. difficile detection: a multicentre study of C. difficile infection. Lancet Infect Dis 2013;13. http://dx.doi.org/10.1016/S1473-3099(13)70200-7.
- Briese T, Palacios G, Kokoris M, Jabado O, Liu Z, Renwick N, et al. Diagnostic system for rapid and sensitive differential detection of pathogens. Emerg Infect Dis 2005;1:310-13. http://dx.doi.org/10.3201/eid1102.040492.
- Palacios G, Briese T, Kapoor V, Jabado O, Liu Z, Venter M, et al. MassTag polymerase chain reaction for differential diagnosis of viral hemorrhagic fever. Emerg Infect Dis 2006;12:692-5. http://dx.doi.org/10.3201/eid1204.051515.
- Jex AR, Stanley KK, Lo W, Littman R, Verweij JJ, Campbell BE, et al. Detection of diarrhoeal pathogens in human faeces using an automated, robotic platform. Mol Cell Probes 2012;26:11-5. http://dx.doi.org/10.1016/j.mcp.2011.10.004.
- Liu J, Gratz J, Maro A, Kumburu H, Kibiki G, Taniuchi M, et al. Simultaneous detection of six diarrhea-causing bacterial pathogens with an in-house PCR-luminex assay. J Clin Microbiol 2012;50:98-103. http://dx.doi.org/10.1128/JCM.05416-11.
- Liu J, Gratz J, Amour C, Kibiki G, Becker S, Janaki L, et al. A laboratory-developed TaqMan Array Card for simultaneous detection of 19 enteropathogens. J Clin Microbiol 2013;51:472-80. http://dx.doi.org/10.1128/JCM.02658-12.
- Persson S, de Boer RF, Kooistra-Smid AM, Olsen KE. Five commercial DNA extraction systems tested and compared on a stool sample collection. Diagn Microbiol Infect Dis 2011;69:240-4. http://dx.doi.org/10.1016/j.diagmicrobio.2010.09.023.
- Schuurman T, de Boer R, Patty R, Kooistra-Smid M, van Zwet A. Comparative evaluation of in-house manual, and commercial semi-automated and automated DNA extraction platforms in the sample preparation of human stool specimens for a Salmonella enterica 5′-nuclease assay. J Microbiol Methods 2007;71:238-45. http://dx.doi.org/10.1016/j.mimet.2007.09.003.
- Kageyama T, Kojima S, Shinohara M, Uchida K, Fukushi S, Hoshino FB, et al. Broadly reactive and highly sensitive assay for Norwalk-like viruses based on real-time quantitative reverse transcription-PCR. J Clin Microbiol 2003;41:1548-57. http://dx.doi.org/10.1128/JCM.41.4.1548-1557.2003.
- Vennema H, de Bruin E, Koopmans M. Rational optimization of generic primers used for Norwalk-like virus detection by reverse transcriptase polymerase chain reaction. J Clin Virol 2002;25:233-5. http://dx.doi.org/10.1016/S1386-6532(02)00126-9.
- Suo B, He Y, Tu SI, Shi X. A multiplex real-time polymerase chain reaction for simultaneous detection of Salmonella spp. Foodborne Pathog Dis 2010;7:619-28. http://dx.doi.org/10.1089/fpd.2009.0430.
- Nogva HK, Bergh A, Holck A, Rudi K. Application of the 5′-nuclease PCR assay in evaluation and development of methods for quantitative detection of Campylobacter jejuni. Appl Environ Microbiol 2000;66:4029-36. http://dx.doi.org/10.1128/AEM.66.9.4029-4036.2000.
- Fukushima H, Katsube K, Tsunomori Y, Kishi R, Atsuta J, Akiba Y. Comprehensive and rapid real-time PCR analysis of 21 foodborne outbreaks. Int J Microbiol 2009.
- Bej AK, DiCesare JL, Haff L, Atlas RM. Detection of Escherichia coli and Shigella spp. in water by using the polymerase chain reaction and gene probes for uid. Appl Environ Microbiol 1991;57:1013-17.
- Hartman LJ, Selby EB, Whitehouse CA, Coyne SR, Jaissle JG, Twenhafel NA, et al. Rapid real-time PCR assays for detection of Klebsiella pneumoniae with the rmpA or magA genes associated with the hypermucoviscosity phenotype: screening of nonhuman primates. J Mol Diagn 2009;11:464-71. http://dx.doi.org/10.2353/jmoldx.2009.080136.
- Malorny B, Paccassoni E, Fach P, Bunge C, Martin A, Helmuth R. Diagnostic real-time PCR for detection of Salmonella in food. Appl Environ Microbiol 2004;70:7046-52. http://dx.doi.org/10.1128/AEM.70.12.7046-7052.2004.
- Naaber P, Stsepetova J, Smidt I, Ratsep M, Koljalg S, Loivukene K, et al. Quantification of Clostridium difficile in antibiotic-associated-diarrhea patients 2011;49:3656-8. http://dx.doi.org/10.1128/JCM.05115-11.
- Boom R, Sol CJ, Salimans MM, Jansen CL, Wertheim-van Dillen PM, van der Noordaa J. Rapid and simple method for purification of nucleic acids. J Clin Microbiol 1990;28:495-503.
- Yang G, Erdman DE, Kodani M, Kools J, Bowen MD, Fields BS. Comparison of commercial systems for extraction of nucleic acids from DNA/RNA respiratory pathogens. J Virol Methods 2011;171:195-9. http://dx.doi.org/10.1016/j.jviromet.2010.10.024.
- Antikainen J, Tarkka E, Haukka K, Siitonen A, Vaara M, Kirveskari J. New 16-plex PCR method for rapid detection of diarrheagenic Escherichia coli directly from stool samples. Eur J Clin Microbiol Infect Dis 2009;28:899-908. http://dx.doi.org/10.1007/s10096-009-0720-x.
- Department of Health . Updated Guidance on the Diagnosis and Reporting of n.d. www.gov.uk/government/uploads/system/uploads/attachment_data/file/215135/dh_133016.pdf (accessed May 2014).
- Luminex corporation . XTAG Gastrointestinal Pathogen Panel (GPP) for In Vitro Diagnostic Use n.d. www.theradiag.com/en/files/2013/07/xTAG-GPP-leaflet.pdf (accessed May 2014).
- Wilcox MH. Policy development for Clostridium difficile. J Antimicrob Chemother 2012;67:i19-22. http://dx.doi.org/10.1093/jac/dks203.
- Wilcox MH, Shetty N, Fawley WN, Shemko M, Coen P, Birtles A, et al. Changing epidemiology of Clostridium difficile infection following the introduction of a national ribotyping-based surveillance scheme in England. Clin Infect Dis 2012;55:1056-63. http://dx.doi.org/10.1093/cid/cis614.
- Wiegand PN, Nathwani D, Wilcox MH, Stephens J, Shelbaya A, Haider S. Clinical and economic burden of Clostridium difficile infection in Europe: a systematic review of healthcare-facility-acquired infection. J Hosp Infect 2012;81:1-14. http://dx.doi.org/10.1016/j.jhin.2012.02.004.
- Wilcox MH, Cunniffe JG, Trundle C, Redpath C. Financial burden of hospital-acquired Clostridium difficile infection. J Hosp Infect 1996;34:23-30. http://dx.doi.org/10.1016/S0195-6701(96)90122-X.
- McGlone SM, Bailey RR, Zimmer SM, Popovich MJ, Tian Y, Ufberg P, et al. The economic burden of Clostridium difficile. Clin Microbiol Infect 2012;18:282-9. http://dx.doi.org/10.1111/j.1469-0691.2011.03571.x.
- Flahault A, Cadilhac M, Thomas G. Sample size calculation should be performed for design accuracy in diagnostic test studies. J Clin Epidemiol 2005;58:859-62. http://dx.doi.org/10.1016/j.jclinepi.2004.12.009.
- Jothikumar N, Griffiths MW. Rapid detection of Escherichia coli O157:H7 with multiplex real-time PCR assays. Appl Environ Microbiol 2002;68:3169-71. http://dx.doi.org/10.1128/AEM.68.6.3169-3171.2002.
- Villalobo E, Torres A. PCR for detection of Shigella spp. in mayonnaise. Appl Environ Microiol 1998;64:1242-45.
- Verweij JJ, Blangé RA, Templeton K, Schinkel J, Brienen ETA, van Rooyen MAA, et al. Simultaneous detection of Entamoeba histolytica, Giardia lamblia, and Cryptosporidium parvum in fecal samples using multiplex real-time PCR. J Clin Microbiol 2004;43:1220-23. http://dx.doi.org/10.1128/JCM.42.3.1220-1223.2004.
- Hadfield SJ, Robinson G, Elwin K, Chalmers RM. Detection and differentiation of Cryptosporidium spp. in human clinical samples by use of real-time PCR. J Clin Microbiol 2011;49:918-24. http://dx.doi.org/10.1128/JCM.01733-10.
- Logan C, O’Leary JJ, O’Sullival N. Real-time reverse transcription-PCR for detection of rotavirus and adenovirus as causative agents of acute viral gastroenteritis in children. J Clin Microbiol 2006;44:3189-95. http://dx.doi.org/10.1128/JCM.00915-06.
Appendix 1 Laboratory standard operating procedures for MassCode polymerase chain reaction for enteric samples
First-strand complementary DNA synthesis protocol: using Agilent MassCode complementary DNA synthesis kit. Cat# 5190–3553
Note: wear gloves at all times during the first-strand cDNA synthesis and PCR amplification procedures and while handling materials and equipment to prevent contamination by ribonucleases (RNases).
-
Prepare the required amount of master mix 1 with 10% overage for first-strand cDNA synthesis reaction:
-
Note: mix each component and spin down before use.
-
Master mix 1 (per sample).
-
3 µl of random primers (0.1 µg/µl).
-
1.7 µl of RNase-free water.
-
-
Prepare the cDNA synthesis reaction in a microcentrifuge tube:
-
5.7 µl of master mix 1 per microcentrifuge tube.
-
10 µl of nucleic acids isolated from clinical sample or positive control.
-
-
Incubate the reaction at 70 °C for 10 minutes.
-
Flash cool the reaction by transferring directly to ice.
-
Prepare the required amount of master mix 2 with 10% overage for first-strand cDNA synthesis reaction:
-
Master mix 2 (per sample + 10% overage).
-
2.0 µl of 10 × MassCode Reverse Transcriptase Buffer.
-
0.8 µl of dNTP mix (25 mM of each dNTP).
-
0.5 µl of RNase Block Ribonuclease Inhibitor (40 units/µl).*
-
1 µl of MassCode Multiple Temperature Reverse Transcriptase.*
-
-
Add 4.3 µl of master mix 2 per reaction.*
-
Note: To prevent heat inactivation, MassCode Reverse Transcriptase and RNase Block must be added after the 70 °C incubation is completed and the reaction has cooled.
-
-
Transfer tubes to thermal cycler and run on following cycle:
-
25 °C, 10 minutes, primer extension.
-
42 °C, 60 minutes, strand synthesis.
-
70 °C, 15 minutes, terminate reaction.
-
[*DO NOT VORTEX OR CENTRIFUGE. KEEP ON ICE]
Polymerase chain reaction protocol: using 2x MassCode polymerase chain reaction master mix. Cat# 5190–3745 and Multiplex Tagged Primer Mix
MassCode Tags are light sensitive. Minimise exposure to light as much as possible!
If using ready-made Primer mix go to step 6; or to make Primer mix:
-
Reconstitute each lyophilised primer with RNase/DNase-free water to make a final concentration of 100 µM.
-
Mix an equal volume of each primer to make a master mix. For example, to make a primer mix for 20-plex assay combine 100 µl of each primer × 40 primers = 4000 µl, then aliquot them 500 µl × 8 tubes (see Table 46).
-
The final concentration of each primer in PCR is 250 nM. You have to add 0.05 µl of each primer per reaction or 0.1 µl of primer set (two primers).
-
For example, for 20-plex assay you have to add 0.05 µl × 40 primers = 2 µl of primer mix (or 0.1 µl × 20-plex = 2 µl) per 1 reaction. For 100 reactions (1 plate) 2 µl × 100 = 200 µl of primer mix.
-
For example, for 25-plex assay (50 primers) add 0.1 × 25-plex= 2.5 µl/reaction or 250 µl per plate (100 reactions).
-
Prepare the required amount of PCR master mix with 10% overage and aliquot 16 µl into each PCR tube or 96-well plate well (Table 42).
-
Add 4 µl of the sample template, control template (provided), or water (for no template control) according to plate layout (Table 43).
-
Transfer to thermal cycler and run on following cycle (Table 44).
| Temperature (°C) | Time | Cycles |
|---|---|---|
| 95 | 3 minutes | Hold |
| 95 | 20 seconds | 15 |
| 56 | 50 seconds | |
| 95 | 20 seconds | 10 |
| 56 | 80 seconds | |
| 95 | 20 seconds | 10 |
| 56 | 120 seconds | |
| 72 | 5 minutes | Hold |
| 4 | ∞ | Hold |
Protocol for MassTag amplicon purification using a vacuum manifold. Cat# 5190–3745
Reminder: MassCode Tags are light sensitive. Minimise exposure to light as much as possible.
-
Prepare the DNA binding solution by adding an equal volume of 30% ethanol (final ethanol concentration is 15%). Put 20 ml per 96-well plate of DNA binding solution/ethanol mixture into a reservoir tray for a multichannel pipettor.
-
Using a multichannel pipettor, add 200 µl of DNA binding solution/ethanol mixture to each 20 µl PCR product in the PCR plate.
-
Place a MassCode 96-well binding plate on a vacuum manifold that contains a deep-well waste plate at the bottom. Using a multichannel pipette set for 250 µl, mix the contents of each well and then transfer the PCR product/DNA binding solution/ethanol mixtures into the wells of the binding plate.
Note: If some of the 96 wells do not contain samples, seal the tops of the empty wells with tape.
-
Apply 400 mbar of vacuum to the binding plate until each well is dry. The vacuum force may decrease as the wells become dry (≈ 1 minute). Continue the vacuum for an additional 1 minute after the wells appear dry.
-
Release the vacuum and remove the binding plate from the manifold. Blot the bottom of the binding plate onto clean paper towels. Place the binding plate on top of a 96-well collection plate. Place a plate sealer on top of the binding plate. Tape the plate’s sides or use a rubber band to keep the two plates together to prevent an accidental spill. Centrifuge the plates at 1500 × g for 3 minutes at room temperature. Inspect the filter plate. Make sure the plate wells have been centrifuged until dry, repeat the centrifugation if there is any liquid still seen on the filters. Any buffer remaining will contribute to background noise in the mass spectrometer.
-
Wash #1. Add 500 µl of DNA Binding solution/ethanol mixture (from step #1) into each well of the DNA binding plate.
-
Apply 400 mbar of vacuum until each well is dry (≈ 1 minute). Continue the vacuum for an additional 1 minute after the wells appear dry.
-
Release the vacuum and remove the binding plate from the manifold. Blot the bottom of the binding plate onto clean paper towels. Place the binding plate on top of a 96-well collection plate. Place a plate sealer on top of the binding plate. Tape the plate’s sides or rubber band the two plates together to prevent an accidental spill. Centrifuge the plates at 1500 × g for 3 minutes at room temperature.
-
Open the vacuum manifold and discard the wash solution from the waste tray. Replace the waste tray inside and place the binding plate on top of the vacuum manifold.
-
Wash #2. Prepare 1 × PCR wash buffer by adding four volumes of 100% (v/v) ethanol to the 5 × PCR wash buffer container. After adding the ethanol, check the box on the label: [ □ ] 1 × (Ethanol Added). Store the 1 × PCR wash buffer tightly sealed at room temperature.
-
Add 700 µl of 1 × PCR wash buffer to each well of the binding plate.
-
Apply 400 mbar of vacuum until each well is dry (≈ 1 minute). Continue the vacuum for an additional 1 minute after the wells appear dry.
-
Release the vacuum and remove the binding plate from the manifold. Blot the bottom of the binding plate onto clean paper towels. Place the binding plate on top of a 96-well collection plate. Place a plate sealer on top of the binding plate. Tape the plate’s sides or rubber band the two plates together to prevent an accidental spill. Centrifuge the plates at 1500 × g for 3 minutes at room temperature.
-
Open the vacuum manifold and discard the wash solution from the waste tray. Replace the waste tray inside and place the binding plate on top of the vacuum manifold.
-
Wash #3. Prepare 80% ethanol wash solution by mixing 160 ml of 100% ethanol with 40 ml of nuclease-free water.
-
Add 700 µl of 80% ethanol wash solution to each well of the binding plate.
-
Apply 400 mbar of vacuum until each well is dry (≈ 1 minute). Continue the vacuum for an additional 1 minute after the wells appear dry.
-
Release the vacuum and remove the binding plate from the manifold. Blot the bottom of the binding plate onto clean paper towels. Place the binding plate on top of a 96-well collection plate. Place a plate sealer on top of the binding plate. Tape the plate’s sides or rubber band the two plates together to prevent an accidental spill. Centrifuge the plates at 1500 × g for 5 minutes at room temperature.
Note: This step is to ensure that any ethanol from the wash solution is removed prior to adding elution buffer. Ethanol contamination in the eluted PCR product can cause quantification errors.
-
Remove the binding plate and 96-well collection plate from the centrifuge. Remove the 96-well collection plate and place the binding plate on top of a fresh 96-well collection plate that is suitable for the mass spectrometer autosampler (i.e. Agilent P/N 5042–1386). Make sure both plates are correctly oriented A1 to A1. Remove the plate sealer from the binding plate. Add 140 µl* of nuclease-free water directly onto the top of the fiber matrix at the bottom of each well using a multichannel pipette. Replace the plate sealer. Tape the plate’s sides or rubber band the two plates together to prevent an accidental spill.
-
Incubate the binding plate for 2 minutes at room temperature.
-
Centrifuge the binding plate and 96-well collection plate together at 1500 × g for 5 minutes at room temperature.
-
The purified MassCode amplicons are in the bottom of each well of the 96-well collection plate. Place a pre-slit well cap (Agilent P/N 5042–1389) on top of the 96-well collection plate for transfer to the mass spectrometer autosampler.
[*70 µl is the minimum volume required. Eluting in twice the volume (140 µl) allows for a duplicate mass spectrometric run of the samples.]
Protocol for mass spectrometer run
Ensure pre-run checks carried out and maintenance performed as detailed in MassCode maintenance guide.
-
Turn on UV unit – allow to warm up for 20–30 minutes until both lights are green.
-
Prepare mobile phase (1 l). Weigh out 0.46248 g liquid chromatography/MS-grade ammonium acetate (6 mM) and add to clean 1 l glass bottle. Add 500 ml liquid chromatography/MS-grade water and 500 ml liquid chromatography/MS-grade methanol. Swirl to mix.
-
Prepare system suitability calibrant. Defrost neat system suitability calibrant add 950 µl of liquid chromatography/MS-grade water and mix well. Transfer mixture to polypropylene vial suitable for MS autosampler.
-
Add 1 ml liquid chromatography/MS-grade water to glass vial suitable for MS autosampler.
-
Prepare run by placing water vial into vial slot 1 of autosampler (lid off), system suitability calibrant into vial slot 2 (lid off), and prepared 96-well plate into plate slot 1.
-
Set up plate details in MassCode software.
-
Check method set to ‘Enteric_13plex’ and run plate.
Target ions and calibrant mixes are detailed below and should be previously set up in ‘Target panel manager’ for ‘Enteric_13plex’.
Enteric 13-plex target panel as set up in ‘Target panel manager’ (Table 45).
| Target | Short name | Ion 1 | Ion 2 |
|---|---|---|---|
| MS2 bacteriophage | IAC-MS2 | 394 | 506 |
| C. coli (ceuE) | Ccoli | 645 | 709 |
| C. difficile (tcdB) | Cdiff | 486 | 438 |
| C. jejuni (Cj0414) | Cjeju | 705 | 729 |
| Norovirus (RdRp/VP1) | Noro2 | 426 | 526 |
| S. enterica (invA) | Sent | 641 | 733 |
| E. coli O157 (stx1) | Stx1 | 677 | 697 |
| E. coli O157 (stx2) | Stx2 | 494 | 402 |
| S. Typhi (sty4220) | Typhi | 406 | 609 |
| Shigella spp. (virA) | Shig | 498 | 653 |
| Giardia spp. (18S) | Gia | 458 | 565 |
| Rotavirus (vp6) | Rota | 466 | 685 |
| Cryptosporidium spp. (18S) | Crypto | 518 | 557 |
| CAL-PTC 1 (C) | CAL-PTC 2 (K) | CAL-PTC 3 (M) | CAL-PTC 4 (N) |
| Ccoli | Noro2 | Noro2 | Cjeju |
| Cdiff | Stx2 | Sent | Stx1 |
| Gia | Typhi | Typhi | Shig |
| Rota | Crypto | Cypto | Rota |
| Organism | Target gene | Primer | Sequence (5′ → 3′) | Tm (°C) | Amplicon size (bp) | Source |
|---|---|---|---|---|---|---|
| MS2 (IC) | lys/rep | Forward | CGCGATCTTTCTCTCGAAAT | 49.7 | 317 | In-house |
| Reverse | GACGATCGGTAGCCAGAGAG | 55.9 | In-house | |||
| C. difficile | tcdB | Forward | ACTTCCTACATTATCTGAAGGATTACCT | 55.5 | 110 | In-house |
| Reverse | GTCTTAATAATGGGTCACTHGTTTCACT | 55.5 – 57 | In-house | |||
| S. enterica | invA | Forward | GTTGAGGATGTTATTCGCAAAGG | 53.5 | 75 | Suo et al.15 |
| Reverse | GGAGGCTTCCGGGTCAAG | 54.9 | Suo et al.15 | |||
| C. jejuni | Cj0414 | Forward | CTGAATTTGATACCTTAAGTGCAGC | 54.4 | 86 | Nogva et al.16 |
| Reverse | AGGCACGCCTAAACCTATAGCT | 54.8 | Nogva et al.16 | |||
| C. coli | ceuE | Forward | ACGCGCACAAGGCATACTT | 51.1 | 91 | Fukushima et al.17 |
| Reverse | CCAGTATTCAGGATCAAGATAAATGATTT | 54.4 | Fukushima et al.17 | |||
| Norovirus 1 | ORF1 | Forward | TCGTGGCTGAGTAGGAGAAT | 51.8 | 311 | In-house |
| Reverse | GCAATCATCGCAGACACATC | 51.8 | In-house | |||
| Norovirus 2 | RdRP/VP1 | Forward | ATCGCAATCTGGCTCCCAGTTT | 54.8 | 119 | In-house |
| Reverse | GGCTCCAAAGCCATAACCTCAT | 54.8 | In-house | |||
| E. coli O157 | stx1 | Forward | TCGTTGACTACTTCTTATCTGGA | 51.7 | 95 | Jothikumar and Griffiths33 |
| Reverse | GTCACAGTAACAAACCGTAACA | 51.1 | Jothikumar and Griffiths33 | |||
| E. coli O157 | stx2 | Forward | CGACCCCTCTTGAACATA | 48 | 108 | Jothikumar and Griffiths33 |
| Reverse | GATAGACATCAAGCCCTCGT | 51.8 | Jothikumar and Griffiths33 | |||
| Shigella flexneri | virA | Forward | TGATGAGCTAACTTCGTAAGCCCTCC | 59.5 | 215 | Villalobo and Torres34 |
| Reverse | CTGCATTCTGGCAATCTCTTCACA | 55.7 | Villalobo and Torres34 | |||
| S. Typhi/Paratyphi | sty4220 | Forward | GGCAGCAATTGGCTCATACA | 51.8 | 175 | In-house |
| Reverse | AGTATCACCGCCTGCCATCT | 53.8 | In-house | |||
| Giardia lamblia | SSU rRNA | Forward | GCTCTCCCCAAGGACACAAG | 55.9 | 118 | In-house |
| Reverse | CGGGTTGCCAGCGGTGTCCG | 62 | Verweij et al.35 modified in-house | |||
| Cryptosporidium spp. | SSU rRNA | Forward | GAGGTAGTGACAAGAAATAACAATACAGG | 57.3 | 299 | Hadfield et al.36 |
| Reverse | CTGCTTTAAGCACTCTAATTTTCTCAAAG | 55.9 | Hadfield et al.36 | |||
| Rotavirus | vp6 | Forward | GGATGTCCTGTACTCCTTGTCAAAA | 56 | 145 | Logan et al.37 |
| Reverse | TCCAGTTTGGAACTCATTTCCA | 51.1 | Logan et al.37 |
Nucleic acid extraction: using bead beating pre-step and QiaSymphony virus/pathogen kit. Cat#937055
Additional materials required:
-
MS2 phage ssRNA – control for nucleic acids extraction and amplification (provided).
-
STAR buffer (Roche; cat no 03335208001).
-
Chloroform.
-
Lysing matrix E (MPBiomedicals; cat no 116914500).
Note: MS2 is currently provided at two different concentrations:
-
5 × 105 copies MS2/µl.
-
5 × 106 copies/µl.
Amount of MS2 for extraction can vary depending on a sample type and the volume of the Elution Buffer recommended in the nucleic acids extraction protocol (50–200 µl).
We recommend using 5 × 104 copies of MS2 for 50 µl of Elution Buffer, then 10 µl of extracted nucleic acids per RT cDNA synthesis reaction (104 copies of MS2 per RT) and 4 µl of cDNA per 20 µl PCR. Based on our experience, 2 × 103 copies of MS2 per PCR are optimal for detection of a good positive signal on MS. If a higher volume of Elution Buffer is required by an extraction protocol increase MS2 amount proportionally (e.g. 1 × 105 copies of MS2 for 100 µl elution volume). Input amount of MS2 phage ssRNA can be increased or decreased depending on MassCode results.
Sample prep and bead bashing
-
For each extraction, add 1 ml of Roche stool transport and recovery (STAR) buffer to a tube of MPBiomedicals Lysing matrix E.
-
Add 1 µl of MS2 ssRNA (105 copies) to each sample.
-
For each sample add 200 µl or one 10 µl loop full of stool sample to a tube.
-
Add 100 µl of chloroform to each tube.
-
Bead bash for 40 s at 6.0 m/s on the MPBiomedicals Fastprep 24 then store sample at 4 °C for 5 minutes.
-
Repeat bead bash for 40 s at 6.0 m/s on the MPBBio Fastprep 24.
-
Centrifuge samples for 1 minute at 1000 × g
-
Transfer at least 500 µl of supernatant to a labelled sterile 2 ml microcentrifuge tube (suitable for use in Qiasymphony).
Symphony loading
Waste Draw:
-
Affix waste bag to waste draw undercarriage.
-
Insert tip station, tip chute and waste vessels into waste draw ensuring that the tip station is fully inserted then close the waste draw.
-
On touchscreen select ‘scan now’ or ‘scan later’.
Elution Draw:
-
Place black elution rack onto stainless carrier and place on slot 1 (cooled) in the elution draw.
-
On touchscreen select the rack and press ‘rack id’ button.
-
Scan the barcode on the Qiagen elution plate and place in elution rack in correct orientation and close the eluate draw.
-
On touchscreen press next and the Qiasymphony will briefly lock the draws and hood while scanning the elution plate. Once scanning is complete and the machine is unlocked, proceed to the next step.
Reagents and Consumables Draw:
-
Remove the magnetic beads container from the reagents cassette and vortex for 3 minutes or until beads are evenly suspended and reinsert into reagent cassette.
-
Lift the reagent cartridges out of the white plastic stand and place into a grey cassette rack.
-
If the reagent cassette is being used for the first time then proceed to steps 11–15. If the cassette has been used before then go to steps 16–19.
-
Place a piercing lid on top of the cassette with the curved side of the lid on the opposite side of the cassette to the magnetic beads cartridge. Do not press down on the piercing lid or otherwise pierce the top of the reagent cartridges as the QiaSymphony will do this automatically.
-
Remove the seal from the magnetic beads trough ensuring that all parts of the seal are fully detached.
-
Using some clean tissue paper wipe away all droplets of the magnetic bead solution from around the rim of the bead container.
-
Slide the white enzyme rack onto the side of the reagents cassette and unscrew the proteinase K lids (the lids can be placed in the lid holders between the proteinase K rack and the cassette rack).
-
Proceed to step 20.
-
If the reagents cassette has been used before then remove the lids from the individual reagent cartridges and from the magnetic beads cartridge.
-
Using some clean tissue paper wipe away all droplets of the magnetic bead solution around the rim of the bead container.
-
Unscrew the proteinase K lids (the lids can be placed in the lid holders between the proteinase K rack and the cassette rack).
-
Proceed to step 20.
-
Load black 1500 µl and blue 200 µl tips, 8 rod covers and sample prep cartridges into reagents and consumables draw. For one run consisting of 24 samples the QIAsymphony will require 85 1500 µl tips, 24 200 µl tips, 3 8-rod covers and 18 sample prep cartridges. These quantities do not have to be exact but are the minimum amounts required.
-
On touchscreen press ‘bottle id’ button and scan ATL buffer bottle barcode and insert bottle into rear of reagents and consumables draw.
-
Load the reagents cassette that has been setup as described in steps 10–17.
-
Close the reagents and consumables draw and on touchscreen press ‘scan now’.
-
While the Qiasymphony is scanning, proceed to step 25.
Samples Draw:
-
Make up carrier RNA in 2 ml microcentrifuge tubes suitable for the Qiasymphony using AVE buffer and carrier RNA stock. The amount required for any number of samples (up to 24) can be found in Table 47.
-
Unscrew the lids of the carrier RNA microcentrifuge tubes and place into a sample rack ensuring that the insert type in the rack matches the tube type. For 2 ml microcentrifuge tubes the insert has a red number above the barcode.
-
Unscrew the lids of the sample tubes and place them into a sample rack ensuring that the insert type in the rack matches the tube type. For 2 ml microcentrifuge tubes the insert has a red number above the barcode.
-
Only when the Qiasymphony has finished the scan will the sample drawer be unlocked. Once open, slide the samples rack up to the line in one of the first four slots and wait for the barcode scanner to align. Wait for the slot light to flash and then slide the rack past the barcode scanner until it clicks into place at the back of the draw. If correctly inserted the light will change from green to yellow. Do not place samples in the slot marked ‘A’ as this is for carrier RNA or internal controls only.
-
Slide the carrier RNA rack up to the line in the fourth slot and wait for the barcode scanner to align. Wait for the slot light to flash and then slide the rack past the barcode scanner until it clicks into place at the back of the draw. If correctly inserted the light will change to yellow.
-
Gently close the samples draw.
| Vial 1 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Carrier (µl) | 10 | 12.5 | 15 | 17.5 | 20 | 22.5 | 25 | 27.5 | 30 | 32.5 | 35 | 37.5 | 40 |
| AVE (µl) | 470 | 587.5 | 705 | 822.5 | 940 | 1057.5 | 1175 | 1292.5 | 1410 | 1527.5 | 1645 | 1762.5 | 1880 |
| Total | 480 | 600 | 720 | 840 | 960 | 1080 | 1200 | 1320 | 1440 | 1560 | 1680 | 1800 | 1920 |
| Vial 2 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | ||
| Carrier (µl) | 10 | 12.5 | 15 | 17.5 | 20 | 22.5 | 25 | 27.5 | 30 | 32.5 | 35 | ||
| AVE (µl) | 470 | 587.5 | 705 | 822.5 | 940 | 1057.5 | 1175 | 1292.5 | 1410 | 1527.5 | 1645 | ||
| Total (µl) | 480 | 600 | 720 | 840 | 960 | 1080 | 1200 | 1320 | 1440 | 1560 | 1680 | ||
Setting up the run:
-
On touchscreen select the samples display, press the ‘batch’ button and then press the ID button. As barcodes are not used for these samples, a unique identifier must be manually assigned to each tube in the samples rack (e.g. 1, 2, 3, 4, etc.). To do this click on the first sample and press the ‘edit ID’ button and then click ‘1’ and then ‘enter’. Repeat this for all samples in the run and then press ‘next’.
-
To assign the correct assay to the samples press ‘select all’ then find the ‘Pathogen’ folder and select the program ‘virus pathogen complex 400 DSP’ from the list. Press ‘next’.
-
To select the elution volume press on the elution rack in slot 1 and then press ‘60’.
-
Press the ‘Queue’ button.
-
Go back to the samples menu and press the carrier button. Select all carrier tubes and then assign the carrier status to them (‘virus pathogen complex 400 DSP’).
-
Press the ‘Run’ button.
-
If prompted to scan any draw again press ‘scan’ and the run will start automatically after completion.
| C. difficile | |||||||
|---|---|---|---|---|---|---|---|
| Culture dilution (no.) | av. % recovery | % SD | MS2 av. Ct | MS2 SD Ct | Missed MS2 (n) | False positives (n) | |
| Qiagen | |||||||
| Protocol | 10–3 (5) | 10 | 1 | 1 | 0 | ||
| 10–4 (5) | 3 | 1 | 1 | 0 | |||
| 10–5 (5) | 3 | 7 | 1 | 0 | |||
| Culture negative (3) | |||||||
| Water negative (3) | |||||||
| Bead beating | 10–3 (5) | 176 | 19 | 37 | 2.3 | 3 | 0 |
| 10–4 (5) | 169 | 54 | 38 | 1.7 | 2 | 0 | |
| 10–5 (5) | 226 | 424 | 37 | 1.3 | 2 | 0 | |
| Culture negative (3) | > 40 | – | 3 | 0 | |||
| Water negative (3) | 32 | 0.6 | 0 | 0 | |||
| Bead beating – no INHIBIT EX tablet | 10–3 (5) | 83 | 65 | 1 | 0 | ||
| 10–4 (5) | 516 | 902 | 2 | 0 | |||
| 10–5 (5) | 47 | 41 | 1 | 0 | |||
| Culture negative (3) | |||||||
| Water negative (3) | |||||||
| Freeze–thaw | 10–3 (5) | 41 | 31 | 38 | 1.1 | 1 | 0 |
| 10–4 (5) | 35 | 33 | 38 | 0.8 | 1 | 0 | |
| 10–5 (5) | 0 | 0 | 36 | 1.1 | 1 | 0 | |
| Culture negative (3) | 35 | 3.4 | 1 | 0 | |||
| Water negative (3) | 31 | 1.3 | 0 | 0 | |||
| Sonication | 10–3 (5) | 338 | 156 | 36 | 3.0 | 1 | 0 |
| 10–4 (5) | 315 | 136 | 36 | 2.0 | 1 | 0 | |
| 10–5 (5) | 346 | 357 | 37 | 2.7 | 1 | 0 | |
| Culture negative (3) | 35 | 2.0 | 0 | 0 | |||
| Water negative (3) | 32 | 0.5 | 0 | 0 | |||
| Boiling | 10–3 (5) | Standard protocol | |||||
| 10–4 (5) | |||||||
| 10–5 (5) | |||||||
| Culture negative (3) | |||||||
| Water negative (3) | |||||||
| Masterpure | |||||||
| Protocol | 10–3 (5) | 0 | 0 | > 40 | – | 5 | 0 |
| 10–4 (5) | 0 | 0 | > 40 | – | 5 | 0 | |
| 10–5 (5) | 0 | 0 | > 40 | – | 5 | 0 | |
| Culture negative (3) | > 40 | – | 3 | 0 | |||
| Water negative (3) | 33 | 5.9 | 0 | 0 | |||
| Bead beating | 10–3 (5) | 0 | 0 | > 40 | – | 5 | 0 |
| 10–4 (5) | 0 | 0 | > 40 | – | 5 | 0 | |
| 10–5 (5) | 0 | 0 | > 40 | – | 5 | 0 | |
| Culture negative (3) | > 40 | – | 3 | 1 | |||
| Water negative (3) | 33 | 0.2 | 0 | 0 | |||
| Freeze–thaw | 10–3 (5) | 0 | 0 | > 40 | – | 5 | 0 |
| 10–4 (5) | 0 | 0 | > 40 | – | 5 | 0 | |
| 10–5 (5) | 0 | 0 | 35 | – | 4 | 1 | |
| Culture negative (3) | > 40 | – | 3 | 0 | |||
| Water negative (3) | 31 | 2.7 | 0 | 0 | |||
| Sonication | 10–3 (5) | 0 | 0 | 38 | – | 4 | 1 |
| 10–4 (5) | 0 | 0 | > 40 | – | 5 | 0 | |
| 10–5 (5) | 0 | 0 | > 40 | – | 5 | 0 | |
| Culture negative (3) | > 40 | – | 3 | 0 | |||
| Water negative (3) | 31 | 1.8 | 0 | 0 | |||
| Boiling | 10–3 (5) | 13 | 28 | > 40 | – | 5 | 0 |
| 10–4 (5) | 0 | 0 | > 40 | – | 5 | 0 | |
| 10–5 (5) | 0 | 0 | > 40 | – | 5 | 0 | |
| Culture negative (3) | > 40 | – | 3 | 0 | |||
| Water negative (3) | 33 | 5.0 | 0 | 0 | |||
| MagMax | |||||||
| Protocol | 10–3 (5) | 30 | 9 | 35 | 1.5 | 0 | 0 |
| 10–4 (5) | 29 | 29 | 36 | 1.4 | 0 | 0 | |
| 10–5 (5) | 29 | 41 | 36 | 1.5 | 0 | 0 | |
| Culture negative (3) | 35 | 1.8 | 0 | 0 | |||
| Water negative (3) | 31 | 1.0 | 0 | 0 | |||
| Bead beating | 10–3 (5) | Standard protocol | |||||
| 10–4 (5) | |||||||
| 10–5 (5) | |||||||
| Culture negative (3) | |||||||
| Water negative (3) | |||||||
| Freeze–thaw | 10–3 (5) | 71 | 46 | 35 | 2.3 | 0 | 0 |
| 10–4 (5) | 56 | 62 | 35 | 1.2 | 0 | 0 | |
| 10–5 (5) | 94 | 88 | 36 | 1.2 | 0 | 0 | |
| Culture negative (3) | 35 | 0.3 | 0 | 0 | |||
| Water negative (3) | 30 | 0.5 | 0 | 0 | |||
| Sonication | 10–3 (5) | 38 | 32 | 35 | 1.2 | 0 | 0 |
| 10–4 (5) | 32 | 36 | 35 | 0.5 | 0 | 0 | |
| 10–5 (5) | 172 | 385 | 35 | 0.8 | 0 | 0 | |
| Culture negative (3) | 34 | 1.2 | 0 | 0 | |||
| Water negative (3) | 31 | 1.8 | 0 | 0 | |||
| Boiling | 10–3 (5) | 5 | 4 | 39 | 0.2 | 3 | 0 |
| 10–4 (5) | 1 | 2 | > 40 | – | 4 | 1 | |
| 10–5 (5) | 0 | 0 | 38 | 1.1 | 1 | 0 | |
| Culture negative (3) | > 40 | – | 3 | 0 | |||
| Water negative (3) | 32 | 1.2 | 0 | 0 | |||
| QIAsymphony | |||||||
| ASL buffer + bead beating | 10–3 (5) | 160 | 243 | 36 | 0.5 | 2 | 0 |
| 10–4 (5) | 55 | 34 | 37 | 1.5 | 0 | 0 | |
| 10–5 (5) | 99 | 82 | 36 | 0.6 | 0 | 0 | |
| Culture negative (3) | 35 | 0.5 | 0 | 0 | |||
| Water negative (3) | 31 | 1.4 | 0 | 0 | |||
| STAR buffer + bead beating | 10–3 (5) | 1023 | 469 | 33 | 0.5 | 2 | 0 |
| 10–4 (5) | 761 | 323 | 34 | 0.7 | 1 | 0 | |
| 10–5 (5) | 283 | 320 | 35 | 0.2 | 0 | 0 | |
| Culture negative (3) | 34 | 0.6 | 0 | 0 | |||
| Water negative (3) | 31 | 0.2 | 0 | 0 | |||
| Corbett | |||||||
| Protocol (STAR buffer) | 10–3 (2) | 75 | 19 | 0 | 0 | ||
| 10–4 (2) | 70 | 1 | 0 | 0 | |||
| 10–5 (2) | 0 | 0 | 0 | 0 | |||
| Culture negative (2) | |||||||
| Water negative (2) | |||||||
| S. enterica | |||||||
| Culture dilution (no.) | av. % recovery | % SD | MS2 av. Ct | MS2 SD | No. missed MS2 | No. false positives | |
| Qiagen | |||||||
| Protocol | 10–4 (5) | 0.13 | 0.10 | 2 | 0 | ||
| 10–5 (5) | 0.11 | 0.07 | 1 | 0 | |||
| 10–6 (5) | 0.18 | 0.25 | 2 | 0 | |||
| Culture negative (3) | |||||||
| Water negative (3) | |||||||
| Bead beating | 10–4 (5) | 0.57 | 0.11 | 35 | 3.9 | 3 | 0 |
| 10–5 (5) | 0.84 | 0.25 | 37 | 1.1 | 0 | 0 | |
| 10–6 (5) | 1.17 | 1.24 | 38 | 1.5 | 1 | 0 | |
| Culture negative (3) | 38 | 0.4 | 1 | 1 | |||
| Water negative (3) | 31 | 5.9 | 0 | 1 | |||
| Bead beating – no InhibitEX tablet | 10–4 (2) | 0.35 | 0.15 | 0 | 0 | ||
| 10–5 (2) | 0.55 | 0.46 | 2 | 0 | |||
| 10–6 (2) | 0.56 | 0.66 | 1 | 0 | |||
| Culture negative (2) | |||||||
| Water negative (2) | |||||||
| Freeze–thaw | 10–4 (5) | 0.28 | 0.06 | 37 | 2.0 | 2 | 0 |
| 10–5 (5) | 0.39 | 0.13 | 38 | 1.6 | 1 | 0 | |
| 10–6 (5) | 0.26 | 0.47 | 39 | 0.9 | 1 | 0 | |
| Culture negative (3) | 39 | 1.3 | 1 | 3 | |||
| Water negative (3) | 32 | 1.7 | 0 | 0 | |||
| Sonication | 10–4 (5) | 0.32 | 0.07 | 36 | 1.5 | 1 | 0 |
| 10–5 (5) | 0.32 | 0.23 | 37 | 1.7 | 0 | 0 | |
| 10–6 (5) | 0.52 | 0.63 | 35 | 0.4 | 1 | 0 | |
| Culture negative (3) | 35 | 1.8 | 0 | 0 | |||
| Water negative (3) | 32 | 0.4 | 0 | 1 | |||
| Boiling | 10–4 (5) | Standard protocol | |||||
| 10–5 (5) | |||||||
| 10–6 (5) | |||||||
| Culture negative (3) | |||||||
| Water negative (3) | |||||||
| Masterpure | |||||||
| Protocol | 10–4 (5) | 0.00 | 0.00 | > 40 | – | 5 | 0 |
| 10–5 (5) | 9.72 | 16.83 | > 40 | – | 5 | 2 | |
| 10–6 (5) | 0.00 | 0.00 | > 40 | – | 5 | 0 | |
| Culture negative (3) | > 40 | – | 3 | 0 | |||
| Water negative (3) | 32 | 6.4 | 0 | 0 | |||
| Bead beating | 10–4 (5) | 0.00 | 0.00 | > 40 | – | 5 | 1 |
| 10–5 (5) | 0.00 | 0.00 | > 40 | – | 5 | 0 | |
| 10–6 (5) | 0.00 | 0.00 | > 40 | – | 5 | 2 | |
| Culture negative (3) | > 40 | – | 3 | 1 | |||
| Water negative (3) | 33 | 0.5 | 0 | 0 | |||
| Freeze–thaw | 10–4 (5) | 0.00 | 0.00 | > 40 | – | 5 | 0 |
| 10–5 (5) | 0.00 | 0.00 | > 40 | – | 5 | 0 | |
| 10–6 (5) | 0.00 | 0.00 | > 40 | – | 5 | 0 | |
| Culture negative (3) | > 40 | – | 3 | 0 | |||
| Water negative (3) | 30 | 3.0 | 0 | 0 | |||
| Sonication | 10–4 (5) | 0.00 | 0.00 | > 40 | – | 5 | 1 |
| 10–5 (5) | 0.00 | 0.00 | > 40 | – | 5 | 1 | |
| 10–6 (5) | 0.00 | 0.00 | > 40 | – | 5 | 1 | |
| Culture negative (3) | > 40 | – | 3 | 0 | |||
| Water negative (3) | 31 | 2.2 | 0 | 1 | |||
| Boiling | 10–4 (5) | 0.00 | 0.00 | > 40 | – | 5 | 2 |
| 10–5 (5) | 0.00 | 0.00 | > 40 | – | 5 | 1 | |
| 10–6 (5) | 1.64 | 3.66 | > 40 | – | 5 | 0 | |
| Culture negative (3) | > 40 | – | 3 | 0 | |||
| Water negative (3) | 29 | 0.5 | 1 | 0 | |||
| MagMax | |||||||
| Protocol | 10–4 (5) | 0.16 | 0.15 | 35 | 2.5 | 1 | 0 |
| 10–5 (5) | 0.29 | 0.34 | 36 | 2.2 | 0 | 1 | |
| 10–6 (5) | 0.23 | 0.26 | 35 | 2.1 | 1 | 0 | |
| Culture negative (3) | 34 | 1.9 | 0 | 0 | |||
| Water negative (3) | 30 | 1.3 | 0 | 0 | |||
| Bead beating | 10–4 (5) | Standard protocol | |||||
| 10–5 (5) | |||||||
| 10–6 (5) | |||||||
| Culture negative (3) | |||||||
| Water negative (3) | |||||||
| Freeze–thaw | 10–4 (5) | 0.15 | 0.16 | 35 | 2.0 | 0 | 0 |
| 10–5 (5) | 0.17 | 0.13 | 36 | 1.4 | 0 | 0 | |
| 10–6 (5) | 0.15 | 0.24 | 36 | 1.6 | 0 | 0 | |
| Culture negative (3) | 36 | 1.2 | 0 | 0 | |||
| Water negative (3) | 31 | 1.9 | 0 | 0 | |||
| Sonication | 10–4 (5) | 0.30 | 0.30 | 35 | 1.3 | 0 | 0 |
| 10–5 (5) | 0.37 | 0.30 | 35 | 1.5 | 0 | 0 | |
| 10–6 (5) | 0.88 | 1.26 | 35 | 1.9 | 0 | 0 | |
| Culture negative (3) | 35 | 0.2 | 0 | 0 | |||
| Water negative (3) | 31 | 1.5 | 0 | 0 | |||
| Boiling | 10–4 (5) | 0.10 | 0.08 | > 40 | – | 5 | 0 |
| 10–5 (5) | 0.17 | 0.15 | 39 | – | 4 | 1 | |
| 10–6 (5) | 0.00 | 0.00 | 39 | 0.2 | 3 | 3 | |
| Culture negative (3) | > 40 | – | 3 | 0 | |||
| Water negative (3) | 31 | 0.7 | 0 | 1 | |||
| QIAsymphony | |||||||
| ASL buffer + bead beating | 10–4 (5) | 0.14 | 0.05 | 37 | 1.0 | 0 | 0 |
| 10–5 (5) | 0.24 | 0.37 | 36 | 3.0 | 1 | 0 | |
| 10–6 (5) | 0.26 | 0.39 | 37 | 1.3 | 0 | 0 | |
| Culture negative (3) | 36 | 1.2 | 0 | 1 | |||
| Water negative (3) | 32 | 1.7 | 0 | 2 | |||
| STAR buffer + bead beating | 10–4 (5) | 1.00 | 0.37 | 37 | 1.0 | 0 | 0 |
| 10–5 (5) | 0.87 | 0.37 | 36 | 2.3 | 0 | 0 | |
| 10–6 (5) | 1.01 | 1.37 | 37 | 1.2 | 0 | 2 | |
| Culture negative (3) | 38 | 1.1 | 0 | 1 | |||
| Water negative (3) | 34 | 0.1 | 0 | 1 | |||
| Corbett | |||||||
| Protocol (STAR buffer) | 10–4 (2) | 0.03 | 0.57 | 0 | 0 | ||
| 10–5 (2) | 0 | 0 | 0 | 0 | |||
| 10–6 (2) | 0 | 0 | 0 | 0 | |||
| Culture negative (2) | |||||||
| Water negative (2) | |||||||
Appendix 2 Infection control team questionnaire
Appendix 3 Laboratory manager questionnaire
Appendix 4 Microbiologist questionnaire
List of abbreviations
- ATCC
- American Type Culture Collection
- BMS
- biomedical scientist
- cDNA
- complementary deoxyribonucleic acid
- CFU
- colony-forming unit
- CI
- confidence interval
- DIPC
- Director of Infection Prevention and Control
- DNA
- deoxyribonucleic acid
- EIA
- enzyme immunoassay
- ELISA
- enzyme-linked immunosorbent assay
- ETEC
- enterotoxin-producing Escherichia coli
- GP
- general practitioner
- GPP
- Gastrointestinal Pathogen Panel
- HTA
- Health Technology Assessment
- IC
- internal control
- ICN
- infection control nurse
- MFI
- median fluorescence intensity
- MS
- mass spectrometry
- NAAT
- nucleic acid amplification test
- NCTC
- National Collection of Type Cultures
- NIHR
- National Institute for Health Research
- PBS
- phosphate-buffered saline
- PCR
- polymerase chain reaction
- qPCR
- quantitative polymerase chain reaction
- RNA
- ribonucleic acid
- RT-PCR
- reverse transcriptase-polymerase chain reaction
- SD
- standard deviation
- SOP
- standard operating procedure
- SSU rRNA
- small subunit ribosomal RNA
- STAR
- stool transport and recovery
- STEC
- Shiga-like toxin-producing E. coli
- tRNA
- transfer ribonucleic acid
- WTE
- whole-time equivalent