
Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 10/28/01. The contractual start date was in May 2012. The draft report began editorial review in June 2013 and was accepted for publication in November 2013. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
none
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2014. This work was produced by Snowsill et al. under the terms of a commissioning contract issued by the Secretary of State for Health. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
Chapter 1 Background
Nature of disease
Lynch syndrome (LS) is the most common form of genetically defined, hereditary colorectal cancer (CRC), accounting for 1–3% of all such tumours. Historically, a variety of names have been used for the disease, originally identified by Aldred Scott Warthin in 1913 and then rediscovered by Henry T Lynch in 1966. Lynch coined the terms ‘site-specific colon cancer’ and ‘family cancer’ syndromes. During a workshop in Amsterdam in 1989, the participants agreed upon the name hereditary non-polyposis colorectal cancer (HNPCC), as at that time the syndrome was unknown to most doctors. 1 The appropriateness of the name was discussed again at the international collaborative group on HNPCC meeting in Bethesda, MD, in 2004 where, as the syndrome is also associated with many other tumours, it was proposed that the name ‘Lynch syndrome’ should be reintroduced. 1
Lynch syndrome is inherited as an autosomal dominant disorder, whereby if one parent has the disease, there is a 50% chance that each of his or her children will inherit it. It is characterised by an increased risk of CRC and cancers of the endometrium, ovary, stomach, small intestine, hepatobiliary tract, urinary tract, brain and skin among others, with the lifetime cancer risk highest for CRC (Table 1).
| Cancer | Estimated lifetime cancer risk for individuals with LS (%) | Estimated lifetime cancer risk in the general population (%) |
|---|---|---|
| Colorectal by age 70 years | Men: 382 | 5–63 |
| Women: 312 | ||
| Endometrial | Women: 332 | Women: 2–33 |
| Gastric | 0.72 | 13 |
| Ovarian | Women: 92 | Women: 1–23 |
| Small bowel | 0.62 | 0.013 |
| Bladder | 43 | 1–33 |
| Urinary tract | 1.9–8.45 | 46,7 |
| Brain | 43 | 0.63 |
| Kidney, renal pelvis | 33 | 13 |
| Biliary tract | 0.62 | 0.53 |
| Pancreas | 0.4–3.75 | 1.48 |
| Prostate | Men: 9.1–30.05 | Men: 13.28 |
| Breast | Women: 5.4–14.45 | Women: 12.98 |
Overall, LS accounts for between 0.3% and 2.4% of CRCs, and its prevalence in the general population is of the order of 1 : 3100 (although this may be subject to underestimation due to the current lack of systematic testing). 9,10 The risk of a second primary CRC in individuals with LS is high (estimated at 16% within 10 years) and the risk of a LS cancer in a first- or second-degree family member is approximately 45% for men and 35% for women by age 70 years. 1
Lynch syndrome is caused by mutations in deoxyribonucleic acid (DNA) mismatch repair (MMR) genes, namely MutL homologue 1 (MLH1), MutS homologues 2 and 6 (MSH2 and MSH6) and postmeiotic segregation increased 2 (PMS2). 4,11 Loss of DNA MMR activity in a cell, due to mutations in both alleles of one of the MMR genes, leads to an inability to repair base–base mismatches and small insertions and deletions, resulting in genetic mutations which may then progress to cancer. 12 Mutations occur all over the genome, but especially in repetitive DNA sequences, such as microsatellites. These cause abnormal patterns of microsatellite repeats to be observed when DNA is amplified from a tumour with defective MMR compared with DNA amplified from surrounding normal tissue. This phenomenon is known as microsatellite instability (MSI).
Based on data from 12,624 observations worldwide, MLH1 accounts for 39%, MSH2 34%, MSH6 20% and PMS2 8% of entries in the International Society for Gastrointestinal Hereditary Tumours (InSiGHT) database (www.insight-group.org/mutations/). However, all such estimates are subject to bias, because these are generally mutations found in families referred to genetics clinics, subject to fulfilment of local referral guidelines.
Diagnosis/testing
Currently, the Amsterdam criteria (AC) II and Revised Bethesda criteria, as seen in Table 2, may be used to assist with diagnosis of LS. In 1989, the AC were proposed in order to provide uniform family material required for international collaborative research studies. In 1999, these criteria were revised to include extracolonic tumours. 1 However, with the development of techniques to investigate tumours, such as MSI and MMR immunohistochemistry (IHC), in 1997 the Bethesda guidelines were developed to aid selection of tumours for testing and subsequently identifying individuals with LS. These guidelines were revised in 2004. It should be noted that all AC must be met whereas only one Bethesda criterion is necessary.
| AC II | Revised Bethesda guidelines |
|---|---|
| At least three separate relatives with CRC or a LS-associated cancer | CRC diagnosed in a patient aged < 50 years |
| One relative must be a FDR of the other two | Presence of synchronous, metachronous colorectal or other LS-related tumours, regardless of age |
| At least two successive generations affected | CRC with MSI-H phenotype diagnosed in a patient aged < 60 years |
| At least one tumour should be diagnosed before the age of 50 years | Patient with CRC and a FDR with a LS-related tumour, with one of the cancers diagnosed at age < 50 years |
| FAP excluded in CRC case(s) | Patient with CRC with two or more FDRs or SDRs with a LS-related tumour, regardless of age |
| Tumours pathologically verified |
The Bethesda criteria include MSI-high (MSI-H). This refers to MSI testing where the National Cancer Institute (NCI) has recommended a panel of five markers, known as Bethesda (or NCI) markers, which include two mononucleotides (BAT25 and BAT26) and three dinucleotide repeats (D2S123, D5S346 and D17S250). Tumours with no instability in any of the markers are considered to be microsatellite stable (MSS). When one reference marker is mutated, a tumour is considered to be MSI-low (MSI-L), and if two or more markers are altered, it is considered to be MSI-H. 12 In some cases an additional panel of five markers is used; if 3 out of 10 show instability then it is classified as MSI-H, and if two or fewer, MSI-L.
Unfortunately, there are limitations to MSI testing due to MLH1 silencing commonly occurring in non-hereditary cancers. Thus, MSI is found in approximately 15% of sporadic CRC cases (i.e. CRC with no apparent hereditary component),1 and according to Umar and colleagues, as many as 50% of suspected cases of LS are not confirmed by a genetic defect (that is, mutation in one of the known MMR genes). 12 Hence, the Bethesda criteria have been criticised as being insensitive and non-specific, because strictly applied they would result in approximately 25% of all CRC being tested. In turn, this has stimulated the development of additional tests for the diagnosis of LS, as presented in Table 3.
| Test | Description |
|---|---|
| MSI | Preliminary test performed on tumour tissue. Those with high instability proceed to either DNA analysis or IHC. However, the presence of MSI in the tumour alone is not sufficient to diagnose LS as sporadic CRC may exhibit MSI |
| IHC | Preliminary test performed on tumour tissue to identify one of four MMR proteins (MLH1, MSH2, MSH6 and PMS2). Those with negative staining proceed to DNA analysis of the gene/genes indicated IHC testing helps to identify the MMR gene that most likely harbours a constitutional (‘germline’) mutation, as abnormal expression of a MMR protein points to a mutation in that gene |
| Methylation of MLH1 and/or BRAF V600E testing of tumour tissue | Preliminary molecular genetic test performed on tumour tissue of patients with negative staining for MLH1 on IHC The presence of BRAF V600E mutation or hypermethylation of MLH1 make LS unlikely |
| DNA analysis of MMR genes (MLH1, MSH2, MSH6, PMS2) | Diagnostic test, typically performed on blood. DNA analysis (gene sequencing, deletion/duplication testing) of MLH1, MSH2, MSH6, PMS2 |
Current evidence supports genetic testing for LS to include:13
-
evaluation of tumour tissue for MSI through molecular MSI testing and/or IHC of the four MMR proteins (MLH1, MSH2, MSH6 and PMS2)
-
molecular genetic testing of the tumour for MLH1 gene methylation and/or somatic BRAF V600E mutation to help identify those tumours more likely to be sporadic than hereditary, as the presence of a BRAF V600E mutation makes LS very unlikely1
-
molecular genetic testing of the MMR genes to identify a constitutional (germline) mutation when findings are consistent with LS.
Prognosis
Colorectal tumours in LS appear to evolve through the adenoma–carcinoma sequence. However, this progression is accelerated compared with CRC in sporadic and other familial settings, i.e. 2–3 years as opposed to 8–10 years. 12,14 Furthermore, adenomas in LS often occur in younger individuals and tend to be larger and more severely dysplastic than in sporadic cases. 14 That said, recent studies have confirmed early suspicions that patients with CRC from LS families survive longer than sporadic CRC patients with same-stage tumours. 15 The reasons for the favourable survival rate with CRC in this syndrome remain unclear, but are likely related to a reduced propensity to metastasise. Explanations include that immunological host defence mechanisms may be more active in tumours of the MSI Pathology Research International 3 phenotype, and that the relatively high mutational load that occurs in tumours with defective DNA repair systems is detrimental to their survival. 14
Furthermore, there is definite evidence for a genotype–phenotype correlation in LS; for example, one study found that MSH6 mutation carriers had markedly lower cancer risks overall than MLH1 or MSH2 mutation carriers. 2 Carriers of a MMR gene mutation have a very high risk of developing CRC (25–70%) and endometrial cancer (EC) (30–70%) and an increased risk of developing other tumours. 5
Management of disease
Surveillance
As LS is a hereditary condition, identification of family members carrying a MMR gene defect is desirable, as colonoscopic surveillance, and possibly prophylactic and/or altered surgical management, may be offered to high-risk individuals.
Given that screening for a mutation is time-consuming and expensive – largely because four genes may have to be analysed and their mutational spectra are wide (Vasen 20071) – the British Society of Gastroenterology (BSG) and the Association of Coloproctology of Great Britain and Ireland (ACPGBI)9 recommend that individuals with a substantially elevated personal risk of gastrointestinal malignancy be offered surveillance on the basis of one or more of the following criteria:
-
a family history (FH) consistent with an autosomal dominant cancer syndrome
-
pathognomonic features of a characterised polyposis syndrome personally or in a close relative
-
the presence of a constitutional (‘germline’) pathogenic mutation in a CRC susceptibility gene
-
molecular features of a familial syndrome in a CRC arising in a first-degree relative (FDR).
Individuals fulfilling at least one of the above criteria should be referred to a NHS regional genetics centre for assessment, genetic counselling and mutation analysis of relevant genes, where appropriate.
Vasen and colleagues (2007)1 highlight a study in which 10-year surveillance of 22 LS families reduced the development of CRC by 60% and also decreased mortality. 10,16 Appropriately targeted surveillance also means that those without a gene defect may be spared intensified surveillance, which is costly and carries not insignificant risks of morbidity and mortality. 1
If LS has been identified, large bowel surveillance is recommended by the BSG and ACPGBI for probands and family members as follows:9
Total colonic surveillance (at least biennial) should commence at age 25 years. Surveillance colonoscopy every 18 months may be appropriate because of the occurrence of interval cancers in some series. Surveillance should continue to age 70–75 years or until co-morbidity makes it clinically inappropriate. If a causative mutation is identified in a relative and the consultand is a non-carrier, surveillance should cease and measures to counter general population risk should be applied.
Reproduced from Gut, Cairns SR, Scholefield JH, Steele RJ, Dunlop MG, Thomas HJ, Evans GD, et al. , Volume 59, pp. 666–89, 2010 with permission from BMJ Publishing Group Ltd.
-
Families fulfilling Amsterdam criteria, but without evidence of DNA mismatch repair gene defects (following negative analysis of constitutional DNA and negative tumour analysis by microsatellite instability testing/immunohistochemistry), require less frequent colonoscopic surveillance.
-
Gastrointestinal surveillance should cease for people tested negative by an accredited genetics laboratory for a characterised pathogenic germ-line mutation shown to be present in the family, unless there was a significant, coincidental finding on prior colonoscopy.
-
Reproduced from Gut, Cairns SR, Scholefield JH, Steele RJ, Dunlop MG, Thomas HJ, Evans GD, et al. , Volume 59, pp. 666–89, 2010 with permission from BMJ Publishing Group Ltd.
The evidence for upper gastrointestinal surveillance in all of these disorders is weak, but limited evidence suggests it may be beneficial.
Debate continues regarding the appropriate age for and frequency of surveillance, but the above criteria are in agreement with further published data. 1 However, the situation becomes more complex when the proband does not have a detectable DNA alteration associated with LS, or when an alteration with an unclear significance is identified. 4 Vasen and colleagues (2007)1 suggest that this is the case for approximately 30% of families meeting the AC I, for whom a less intensive surveillance protocol may be recommended (i.e. colonoscopy at 3–5 year intervals, starting 5–10 years before the first diagnosis of CRC or at > 45 years).
Surgical management
Several studies have shown that patients with LS have an increased risk of developing multiple (synchronous and metachronous) CRCs. 1 The type of surgery received, i.e. total or subtotal colectomy, depends on the location of the tumour and the stage of the cancer. Studies have shown that adenomas in patients with LS are located mainly in the proximal colon (ascending and transverse);14 therefore, a subtotal colectomy is favoured, which involves removal of most of the colon, leaving a small amount to be reattached to the rectum. Clinicians may also discuss prophylactic colectomy as a reasonable option in mutation carriers for whom colonoscopy is painful or difficult, or for a patient with adenomas that cannot be removed easily; however, this remains controversial.
Chemotherapy
At least three chemotherapeutic agents have been proven to be effective in the treatment of CRC – 5-FU (also known as fluorouracil) with or without leucovorin (also known as folinic acid), oxaliplatin and irinotecan – although experimental and clinical studies suggest that MSI-H tumours are resistant to 5-FU-based chemotherapy. Therefore, according to Vasen and colleagues (2007),1 prospective clinical trials are needed before definitive recommendations can be given.
Epidemiological studies have demonstrated that non-steroidal anti-inflammatory drugs (e.g. aspirin) reduce the risk of CRC. 14 A recent study showed that a daily dose of aspirin reduced the incidence of CRC in carriers of LS after 56 months’ follow-up. 17 The mechanisms by which aspirin prevents the development of cancer are unknown, though some have suggested that aspirin may be proapoptotic in the early stages of CRC development. Importantly, the Colorectal Adenoma/Carcinoma Prevention Programme 2 (CAPP2) trial of aspirin prophylaxis in LS has demonstrated that aspirin treatment for up to 3 years reduces, a decade later, the overall incidence of LS-associated cancers, including CRC, by 63%. 17 A further dosage determination trial (CAPP3) is therefore planned in LS patients worldwide (www.capp3.org) and highlights the importance of identifying individuals and families with LS.
Description of technologies under assessment
The major laboratory tests used in the evaluation of patients suspected of having LS include testing of tumour tissue using IHC, MSI testing and constitutional testing for MMR mutations (generally from peripheral blood mononuclear cells). Family members undergo predictive genetic testing for the pathogenic mutation identified in the proband (unless they have also developed a relevant cancer). 4 Other tests which may be carried out on tumours include BRAF V600E and methylation of MLH1.
Immunohistochemistry
In families with an increased probability of a MMR gene mutation, IHC analysis for MMR proteins MSH2, MLH1 and MSH6 in tumour tissue may be used as the first step to confirm the presence of MMR deficiency. Pathogenic mutations in MMR proteins frequently lead to the absence of a detectable gene product, or expression of the protein in an abnormal location, for example in the cytoplasm rather than the cell nucleus. Therefore, when tumour tissue from patients suspected of having LS is stained for MMR proteins, a negative or less intense nuclear staining may be visible as compared with the surrounding normal colonic tissue used as a positive control. 4,18
The advantage of IHC, as opposed to MSI, is that abnormal staining of a specific MMR protein is related to the underlying gene defect and can therefore direct further genetic mutation analysis. 1 IHC is a well-established technique widely available in cell pathology laboratories; however, when used to analyse MMR proteins in the setting of LS diagnosis, it must be performed to an adequate standard. Hence, at a workshop in 2006 it was decided that MMR IHC should only be available within the NHS via a laboratory accredited to Clinical Pathology Accreditation standards, obliged to participate in the UK National External Quality Assessment Scheme Immunocytochemistry (NEQAS ICC) for MMR proteins. 19 This workshop made a number of recommendations, including that MMR IHC should be performed for all four main MMR proteins, in part to address the issue of tissue fixation artefact. Care must also be taken in histopathological interpretation of MMR IHC that an adequate and representative tissue sample has been analysed.
Although MMR IHC can give useful and informative results, its sensitivity is limited by a number of factors, for example tissue fixation, the variety and different performance characteristics of primary antibodies and the fact that some pathogenic mutations may result in catalytically inactive but antigenically intact proteins. 15,20–23 Hence, there is a place for MSI analysis in cases with a high prior probability of LS, but with apparently normal expression of the MMR proteins. 1
A particular issue with IHC is that approximately 15% of sporadic colon cancers lose expression of MLH1 because of somatic hypermethylation of the gene’s promoter. Therefore, whereas abnormal expression of MSH2, MSH6 or PMS2 is in itself reasonably good evidence that a tumour was due to LS, loss of MLH1 in itself is not. Other evidence must be used in interpretation in these circumstances, and thus testing for BRAF V600E and/or MLH1 promoter methylation may also be performed. The presence of the BRAF V600E mutation indicates a sporadic rather than LS-associated CRC, but the absence of BRAF V600E does not distinguish between sporadic tumours and those caused by LS. Similarly, MLH1 promoter methylation is highly correlated with a sporadic origin for a tumour, but is not absolutely conclusive, because individuals and families are described with constitutional MLH1 promoter methylation defects. 24
Microsatellite instability testing
Microsatellite instability refers to the variety of patterns of microsatellite repeats observed when DNA is amplified from a tumour with defective MMR compared with DNA amplified from surrounding normal colonic tissue. Repetitive mono- or dinucleotide DNA sequences (microsatellites) are particularly vulnerable to defective MMR. 4 MSI is prevalent in tumours from patients with MMR mutations, and in patients meeting either AC. 4 Therefore, microsatellite analysis is commonly used as the first diagnostic screening test for LS. 18
Microsatellite instability testing involves amplification of a standardised panel of DNA markers (Bethesda/National Institutes of Health markers), although laboratories may use 10 or more markers and, more recently, a commercially available kit based on five mononucleotide markers has become popular as mononucleotide microsatellites may be the most sensitive markers for use in detecting MSI. 4 The process involves microdissection of tumour tissue, followed by extraction of DNA which is then amplified and run on a DNA fragment length analyser. Using such microsatellite markers, additional peaks in tumour tissue DNA in comparison with normal tissue DNA indicate MSI. 25 Instability in 30% or more of the markers is considered MSI-H, less than 30% MSI-L and no shifts or additional peaks MSS. However, if instability is observed at any mononucleotide markers, MSI may be diagnosed. For this reason, MSI testing is moving to a smaller panel of mononucleotide markers, making the process more efficient and cheaper.
As for any molecular pathological analysis, tissue to be selected for MSI analysis must be first assessed by a histopathologist, prior to some degree of microdissection, which aids in maximising sensitivity. There is debate regarding the relative costs of MMR IHC and MSI testing, but NHS service laboratory costings indicate there is little to choose between the two. MSI may be more reproducible and can be performed with smaller amounts of tissue. 26 As there is not yet a UK NEQAS scheme for MSI, the reproducibility of MSI compared with IHC is not established.
BRAF V600E and methylation testing
The presence of MSI in the tumour by itself is not sufficient to diagnose LS because 10–15% of sporadic CRCs exhibit MSI. 25 MSI in non-LS tumours is usually caused by hypermethylation of the MLH1 gene. This acquired epigenetic inactivation of MLH1 is typically associated with mutations in the BRAF gene (specifically the V600E mutation), which has been described in ≈ 35% of sporadic MSI-H CRCs. 25 Therefore, identification of hypermethylation of MLH1 and/or BRAF V600E is an indication that a patient does not have the LS germline mutation.
Ideally, tests would be performed together as the presence of the BRAF V600E mutation theoretically reduces the chance of LS as the cause of that tumour; however, because any test has a finite false negative (FN) rate, it is still a possibility. Additionally, if MLH1 promoter methylation is present but the BRAF V600E mutation is not, this would highlight the small possibility that the patient may have LS due to a constitutional MLH1 methylation defect. It is also possible that he or she could have an inherited MLH1 genetic mutation and could have acquired MLH1 promoter methylation as the ‘second hit’ in the tumour. In these cases, loss of heterozygosity of chromosome 3p (where MLH1 is located) is observed. 25
Constitutional genetic testing
Multiple methods have been used for constitutional genetic testing (tests for mutations that affect all cells in the body and have been there since conception) in LS, in order to find inherited or, if de novo, potentially inheritable MMR gene mutations. The method(s) used should ideally be able to detect any possible mutation associated with LS, for example nonsense, missense and frameshift mutations, genomic deletions, duplications and rearrangements, as explained in Tables 4 and 5. 4
| Mutation | Description |
|---|---|
| Missense | A change in one DNA base pair that results in the substitution of one amino acid for another in the protein made by a gene |
| Nonsense | A change in one DNA base pair that results in a premature signal to stop building a protein. This type of mutation results in a shortened protein that may function improperly or not at all |
| Insertion | Changes the number of DNA bases in a gene by adding a piece of DNA. As a result, the protein made by the gene may not function properly |
| Deletion | Changes the number of DNA bases by removing a piece of DNA. Small deletions may remove one or a few base pairs within a gene, while larger deletions can remove an entire gene or several neighbouring genes. The deleted DNA may alter the function of the resulting protein(s) |
| Duplication | Consists of a piece of DNA that is abnormally copied one or more times. This type of mutation may alter the function of the resulting protein |
| Frameshift mutation | Occurs when the addition or loss of DNA bases changes a gene’s reading frame. A reading frame consists of groups of three bases that each code for one amino acid. A frameshift mutation shifts the grouping of these bases and changes the code for amino acids. The resulting protein is usually nonfunctional. Insertions, deletions and duplications can all be frameshift mutations |
| Splice site | Causes abnormal mRNA processing, generally leading to in-frame deletions of whole exons or out-of-frame mRNA mutations leading to nonsense-mediated decay of mRNA. Mutations may be located deep in intronic sequences |
| Promoter | Mutations in the controlling region of a gene leading to its non-expression. Epigenetic mutations, i.e. abnormal methylation of CpG sites may give rise to the same effect |
| Test | Description | Comments |
|---|---|---|
| High-output screening techniques | SSCP CSGE DGGE DHPLC |
These methods all take advantage of the observation that alteration of DNA confers chemical properties that allow it to be differentiated from normal DNA (now considered obsolescent/obsolete in the UK) |
| DNA sequencing | This can be used following a high-output screening technique or as a primary approach when IHC patterns allow for targeting of a MMR gene | The main method used in the UK for detecting most MMR gene mutations. However, it does not reliably allow for detection of deletions or rearrangements, which are also important in LS. DNA sequencing has become automated in recent years, greatly reducing the required time, costs and expertise4 |
| Methods to detect large structural DNA abnormalities | MLPA is the preferred technique in the UK | Large structural DNA abnormalities are an important cause of LS (5–25% of cases, depending on the gene) but are not generally detected by high-output screening techniques or DNA sequencing. There are several methods for detecting these defects. MLPA, which involves measurement of the relative copy number of DNA sequences, has evolved to become a standard approach for analysing MMR genes for deletions4 |
| Conversion analysis | Only a single allele is analysed at a time. This can increase the yield of genetic testing but is technically complicated, expensive and not widely available |
Measuring the accuracy of diagnostic tests for Lynch syndrome
One aspect of the evaluation of new tests is measuring their accuracy by calculating their sensitivity and specificity. This requires specification of the best available method of identifying the target condition of interest, known as the reference standard. Most mutations causing LS are point mutations or small insertions or deletions, suitably detected by DNA sequencing. However, some LS-associated mutations are deletions/duplications of exons in MLH1 and MSH2. These are more difficult to detect and, currently, the most appropriate technology available is multiplex ligation-dependent probe amplification (MLPA), which is a multiplex polymerase chain reaction (PCR) method able to simultaneously detect copy number changes across multiple DNA sequences within one sample. Therefore, the ideal reference standard is considered to be sequencing plus MLPA.
Chapter 2 Definition of the decision problem and review question
The question addressed by this health technology assessment (HTA) is as set out in the final scope published by the National Institute for Health Research (NIHR), and is reproduced here for reader convenience.
A protocol was developed a priori by the authors to address the decision problem.
The methods used to address specific aspects of the decision problem are detailed at the beginning of each of the relevant chapters which follow.
Test accuracy review question
What is the accuracy of tumour-based tests for LS in all newly diagnosed persons with CRC under 50 years of age, and those considered according to clinical criteria to be at high risk?
Population
-
All newly diagnosed patients under the age of 50 years with CRC.
-
Participants considered to be at high risk of LS, i.e. those fulfilling AC II or Bethesda criteria.
-
Individuals with personal cancer history or FH indicators.
Intervention
Tumour-based tests for evidence of mutations in the genes encoding the MLH1, MSH2, MSH6 and PMS2 DNA MMR enzymes. These tests include MSI, IHC, BRAF and methylation.
Comparators
Genetic testing by sequencing followed by MLPA is considered the gold standard.
Design
An evidence synthesis by systematic review to determine the accuracy of tumour-based tests.
Health-care setting
Primary and secondary care settings.
Test outcomes
The outcomes of interest include measures of:
-
diagnostic test accuracy
-
test failure rate
-
discordant test results.
Decision problem
We will compare genetic testing of all identifiable close relatives with no genetic testing (extreme case analysis) and with a level of genetic testing similar to that carried out in the local health-care setting, which we believe is reasonably typical of current practice across the NHS.
For clarity we would restate and define the suggested specific outcomes contributing to the general aim of assessing effectiveness, cost-effectiveness and cost–utility, as follows:
-
diagnostic accuracy of identifying LS in those presenting with CRC < 50 years of age
-
patient outcome, considering both quantity and quality of life, in those presenting with CRC < 50 years of age
-
diagnostic accuracy of identifying LS in close family members of those presenting with CRC < 50 years of age
-
patient outcome, considering both quantity and quality of life, in close family members of those presenting with CRC < 50 years of age
-
contributing to patient outcome, the number of cancers, particularly CRCs detected, their severity and their age at onset
-
cost of alternative strategies
-
contributing to cost (and patient outcome), the number of surveillance investigations, particularly check colonoscopies, undertaken.
Outcomes of interest are the cost-effectiveness and cost–utility of different strategies for testing probands and their close relatives, the diagnostic accuracy and yield of different strategies for high-risk subjects, and cases of surveillance avoided. Data on these outcomes are likely to be used along with clinical utility scores to estimate quality-adjusted life-years (QALYs).
Modelling will be employed to identify the cost-effectiveness of strategies for the investigation of all new cases of CRC in individuals < 50 years of age for markers of HNPCC. The models will explore the yield of individuals at high risk of HNPCC among the close relatives of probands and identify to what extent unnecessary surveillance (by colonoscopy or other methods) can be avoided. The analysis will also briefly examine whether or not it could be more cost-effective to undertake genetic testing alone without IHC or MSI.
Cost considerations
The cost analysis will be based on the UK NHS setting and will be from an NHS and Personal Social Services (PSS) perspective.
The costs for consideration include:
-
cost of equipment, any additional tests (pre screening), reagents and consumables, participation in NEQAS
-
staff and training of staff
-
maintenance of equipment
-
costs associated with surgeon time and the management of operating theatre time
-
medical costs arising from ongoing care following test results, including those associated with clinical genetics, surgery, time spent in hospital and treatment of cancer.
Chapter 3 Assessment of test accuracy
Methods for reviewing test accuracy
The diagnostic accuracy of the tests IHC and MSI was assessed by a systematic review of research evidence. The review was undertaken following the principles published by the NHS Centre for Reviews and Dissemination. 28
Identification of studies
The search used clusters for LS and HNPCC, joined together using the Boolean connector OR for sensitivity. The following databases were searched: MEDLINE, MEDLINE In-Process & Other Non-Indexed Citations, EMBASE, PsycINFO, Health Management Information Consortium (HMIC) (all via Ovid), The Cochrane Library (all), Cumulative Index to Nursing and Allied Health Literature (CINAHL) (via EBSCOhost), Applied Social Sciences Index and Abstracts (ASSIA) [via Cambridge Scientific Abstracts (CSA)] and Web of Science [via Institute for Scientific Information (ISI)]. The search was limited to human-only populations and to the English language, but did not use any methodological search filters. The National Research Register (NRR), Current Controlled Trials, ClinicalTrials.gov, the Food and Drug Administration (FDA) website, the European Medicines Agency (EMEA) website and Google were also searched. The search is recorded in Appendix 1.
Searches were deduplicated and managed using EndNote X5 (Thomson Reuters, CA, USA). Relevant studies were then identified in two stages. Titles and abstracts returned by the search strategy were examined independently by two researchers (TJH and HC) and screened for possible inclusion. Disagreements were resolved by discussion. Full texts of the studies which could not be excluded were obtained. Two researchers (TJH and HC) examined these independently for inclusion or exclusion, and disagreements were again resolved by discussion.
Inclusion and exclusion criteria
Population
Persons at risk of LS according to any of the following clinical or FH indicators:
-
age < 50 years at diagnosis
-
clinical criteria (e.g. AC II or Bethesda criteria)
-
FH indicators
-
personal cancer history indicators
-
combinations of the above.
In the case of two-gate diagnostic accuracy studies, the population could be persons with any CRC, but must have included a subsample with a known mutation in the genes encoding the MLH1, MSH2, MSH6 and PMS2 DNA MMR enzymes.
Interventions and comparators
The use of tumour-based tests, such as MSI and IHC, to look for evidence of mutations in the genes encoding the MLH1, MSH2, MSH6 and PMS2 DNA MMR enzymes.
The assessment of test accuracy assumed a genetic definition of LS. The reference standard for test accuracy studies was, therefore, genetic testing by sequencing.
Outcomes
Studies were included if outcomes were relevant to diagnostic test accuracy, i.e. if data were available to populate a 2 × 2 table and/or sensitivities and specificities were provided. Additionally, data on test failure rates were included in the review.
Study design
For the review of test accuracy, the protocol allowed inclusion of all study designs, unless evidence on the intervention and outcome of interest was already available from more rigorous study designs (as judged with reference to standard hierarchies of evidence).
Systematic reviews were used as a source for finding further studies and to compare with our systematic review. For the purpose of this review, a systematic review was defined as one that has:
-
a focused research question
-
explicit search criteria that are available to review, either in the document or on application
-
explicit inclusion/exclusion criteria, defining the population(s), intervention(s), comparator(s) and outcome(s) of interest
-
a critical appraisal of included studies, including consideration of internal and external validity of the research
-
a synthesis of the included evidence, whether narrative or quantitative.
Studies were excluded if they did not match the inclusion criteria, and in particular if they were:
-
pre-clinical or in animals
-
reviews, editorials and opinion pieces
-
case reports
-
studies with < 10 participants.
Data extraction strategy
Data were extracted by one reviewer (TJH) using a standardised data extraction form and checked by a second reviewer (HC). Disagreements were resolved by discussion, with involvement of a third reviewer if necessary. Appendix 2 shows the blank data extraction forms used.
Critical appraisal strategy
The methodological quality of the studies was assessed according to criteria specified by the Quality Assessment of Diagnostic Accuracy Studies-2 (QUADAS-2) tool for test accuracy studies. 29
Quality was assessed by one reviewer and judgements were checked by a second. Any disagreement was resolved by discussion, with involvement of a third reviewer as necessary. The two instruments are summarised below. Results were tabulated and the relevant aspects described in the data extraction forms.
Internal validity
The QUADAS-2 quality appraisal tool sought to assess the following considerations:
-
Description of patient selection.
-
Was a consecutive or random sample of patients enrolled?
-
Was a case–control design avoided?
-
Did the study avoid inappropriate exclusions?
-
Could the selection of patients have introduced bias?
-
Are there concerns that the included patients do not match the review question?
-
Description of index and reference tests.
-
Was the index test assessor blind to the results of the reference standard and vice versa?
-
Was a threshold pre-specified?
-
Could the conduct or interpretation of the index test or reference standard have introduced bias?
-
Are there concerns that the conduct or interpretation of the question have introduced bias for the index test or reference standard?
-
Is the reference standard likely to classify the target condition?
-
Description of patient flow and timing.
-
Did all patients receive a reference standard and was it the same test for each?
-
Were all patients included in the analysis?
-
Could the patient flow have introduced bias?
External validity
External validity was judged according to the ability of a reader to consider the applicability of findings to a particular patient group and service setting. Study findings can only be generalisable if they provide enough information to consider whether or not a cohort is representative of the affected population at large. Therefore, studies that appeared to be typical of the UK CRC population with regard to these considerations were judged to be externally valid.
Methods of data synthesis
Details of the extracted data and quality assessment for each individual study are presented in structured tables and as a narrative description. Any possible effects of study quality on the effectiveness data are discussed. Data on test accuracy are presented as sensitivity and specificity, where available.
In most of the studies, the accuracy of the interventions has been evaluated against the reference (gold) standard of constitutional genetic testing and thus, for the purpose of this assessment of test accuracy, a genetic definition of LS is assumed. The results are generally reported as follows:
-
Sensitivity: true positive (TP)/(TP + FN). This is the probability of detecting LS in someone with LS.
-
Specificity: true negative (TN)/[false positive (FP) + TN). This is the probability of not detecting LS in someone without LS.
-
Positive predictive value (PPV): TP/(TP + FP). This is the probability of someone with a positive result actually having LS.
-
Negative predictive value (NPV): TN/(TN + FN). This is the probability of someone with a negative test result actually not having LS.
-
Accuracy or concordance with reference standard: (TP + TN)/(TP + TN + FP + FN). This is the percentage of test results correctly identified by the test, i.e. the rate of agreement with the reference standard.
-
Discordance: cases of disagreement between the reference and index test.
Results
The results of the assessment of test accuracy will be presented as follows:
-
an overview of the quantity and quality of available evidence together with a table summarising all included trials (see Table 9), a table of patient characteristics (see Table 10) and a summary table of key quality indicators (see Table 11)
-
a critical review of the available evidence, covering:
-
the quantity and quality of available evidence
-
a summary table of the study characteristics
-
a summary table of the population characteristics
-
study results in terms of sensitivity and specificity analysis, presented in narrative and tabular form
-
quantity and quality of research available.
-
Number of studies identified
The electronic searches retrieved a total of 3713 titles and abstracts. A total of 3640 papers were excluded, based on screening of title and abstract. As a relevant technology assessment (TA) was retrieved in the search [Bonis and colleagues (2007)4], rather than duplicate effort, we included studies dated from 2005 onwards, and provide a summary of findings by the previous TA. Full text of the remaining 73 papers was requested for more in-depth screening, to give a total of 10 published papers included in the review. The process of study selection is shown in Figure 1.
FIGURE 1.
Summary of study selection. a, It is unclear whether or not two of the included studies are from the same population.
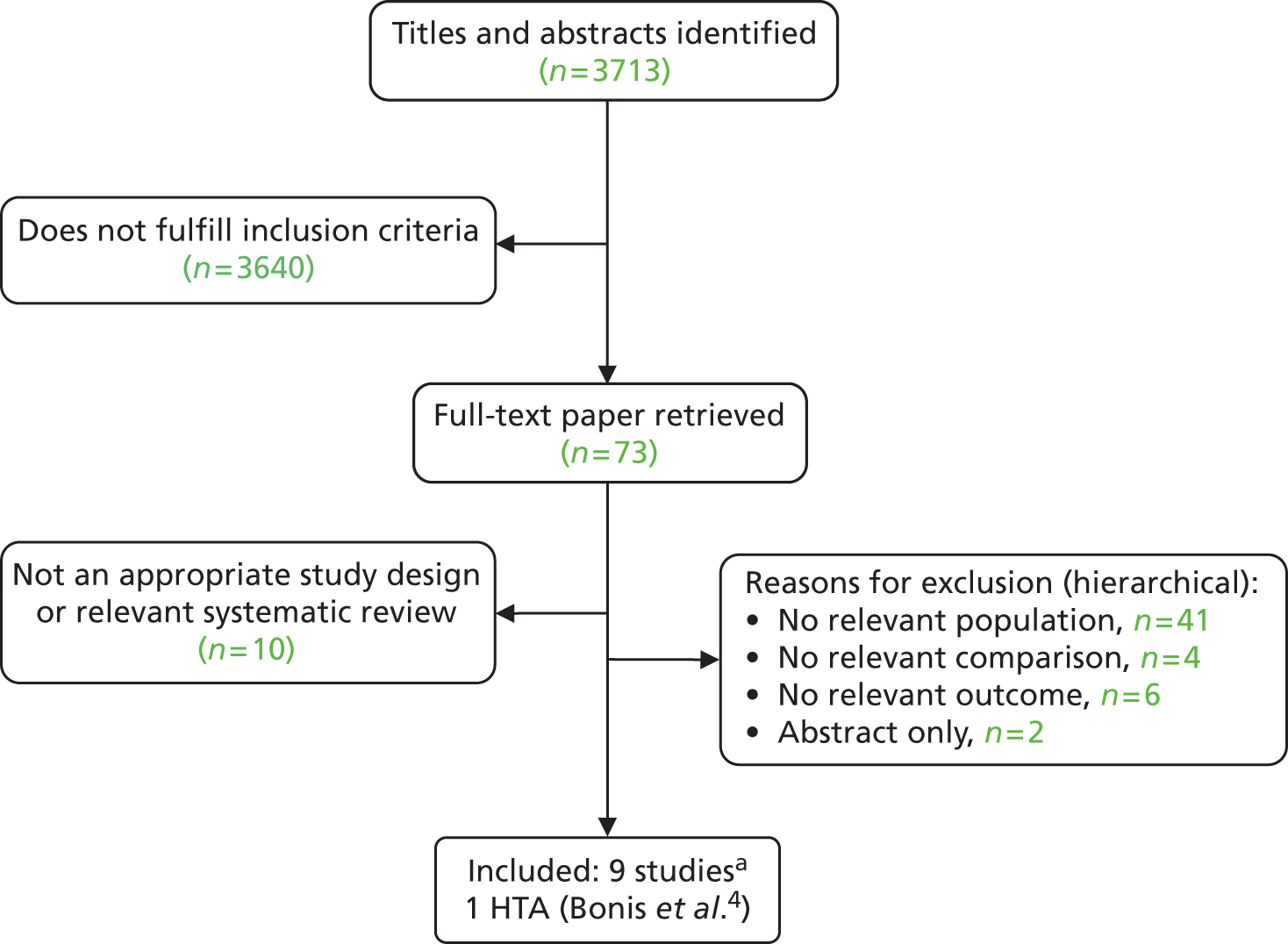
Number of excluded studies
Papers were excluded for at least one of the following reasons: duplicate publication; narrative review; and publication (systematic review or individual primary study) not considering the relevant intervention, population, comparison or outcomes. The bibliographic details of the 73 studies retrieved as full papers and subsequently excluded, along with the reasons for their exclusion, are detailed in Appendix 3.
Number and description of included studies
Bonis and colleagues (2007)
This review continues from a well-presented and thorough systematic review produced by Bonis and colleagues (2007), which is a TA commissioned by the US Department of Health and Human Services. 4 As such, an overview of the findings is discussed. The assessment of multiple systematic reviews (AMSTAR) quality assessment criteria for systematic reviews are displayed below (Table 6). 30 The one concern regarding the quality of the Bonis and colleagues (2007) TA is that MEDLINE was the only database searched. However, other sources included clinical experts and bibliographies of reviews.
| AMSTAR criterion | Response | |
|---|---|---|
| Was an ‘a priori’ design provided? | Yes | ✓ |
| No | ||
| Cannot answer | ||
| Not applicable | ||
| Was there duplicate study selection and data extraction? | Yes | ✓ |
| No | ||
| Cannot answer | ||
| Not applicable | ||
| Was a comprehensive literature search performed? | Yes | |
| No | ✓a | |
| Cannot answer | ||
| Not applicable | ||
| Was the status of publication (i.e. grey literature) used as an inclusion criterion? | Yes | |
| No | ✓ | |
| Cannot answer | ||
| Not applicable | ||
| Was a list of studies (included and excluded) provided? | Yes | ✓ |
| No | ||
| Cannot answer | ||
| Not applicable | ||
| Were the characteristics of the included studies provided? | Yes | ✓ |
| No | ||
| Cannot answer | ||
| Not applicable | ||
| Was the scientific quality of the included studies assessed and documented? | Yes | ✓ |
| No | ||
| Cannot answer | ||
| Not applicable | ||
| Was the scientific quality of the included studies used appropriately in formulating conclusions? | Yes | ✓ |
| No | ||
| Cannot answer | ||
| Not applicable | ||
| Were the methods used to combine the findings of studies appropriate? | Yes | ✓ |
| No | ||
| Cannot answer | ||
| Not applicable | ||
| Was the likelihood of publication bias assessed? | Yes | |
| No | ✓ | |
| Cannot answer | ||
| Not applicable | ||
| Was the conflict of interest stated? | Yes | ✓ |
| No | ||
| Cannot answer | ||
| Not applicable | ||
The characteristics of the studies relevant to this review which were included in Bonis and colleagues (2007) are presented in Table 7.
| Author and location | Population | Analysis |
|---|---|---|
| Calistri 200031 Italy Multicentre |
45 unrelated patients with CRC either fulfilling AC; from families meeting 2/3 AC; diagnosed with CRC at age < 50 years but with no FH; having at least one FDR with CRC; or having multiple neoplasms | Tissue samples from cancer analysed for MSI. DNA from peripheral blood samples analysed for MSH2 and MLH2 |
| Christensen 200232 and Katballe 200233 Denmark Single centre |
42 patients with CRC selected based upon clinical and FH meeting either AC I (n = 11) or a suggestive FH | MSH2 and MLH1 genes sequenced in 31 patients. MSI obtained in 35 patients; IHC performed in 40 patients. Compared sensitivity/specificity of these tests against sequencing as the reference standard |
| Debniak 200034 Poland Single centre |
168 consecutive patients with CRC in whom FAP was excluded Group A: 43/143 patients apparently sporadic, i.e. late onset (age > 40 years), no FH of LS-related tumours and no synchronous or metachronous cancer Group B: 25 were LS based on age ≤ 40 years, familial LS-related cancer or synchronous or metachronous cancer. The remainder were apparently sporadic |
IHC performed in all patients. MSI examined in all. Sequencing performed in all from group B and those from group A who showed abnormal IHC or MSI |
| Dieumegard 200035 France Multicentre |
34 patients with CRC who represented one of three groups: (1) AC I, (2) incomplete AC I (missing at least one criterion but strong FH), (3) age < 50 years and absence of LS-related cancers in family | All patients tested for MSI. Patients with MSI-H and nine MSS cases underwent germline testing for MMR. IHC was performed in all but four patients |
| Durno 200536 Canada Multicentre |
Patients with CRC at age ≤ 24 years (selected from a total of 1382 patients in a cancer registry) | Tumours analysed for MSI, IHC and blood for MMR |
| Farrington 199837 Scotland Multicentre |
50 unrelated patients with CRC at age < 30 years. Identified retrospectively from cancer registrations since 1970 compared with 26 age-matched volunteers without cancer | Detailed FH obtained from cases, paraffin-embedded archival tumour material obtained along with matched normal tissue from 42 patients. Tumour and normal tissue analysed for MSI. Genomic sequencing done on all patients and controls using peripheral blood |
| Peel 200038 USA Multicentre |
Referral of LS-case families (AC). These were from the ICG HNPCC group database (but they give MSI-HL vs. MSS which is not covered in the other papers) | MSI testing was performed in 10 families; diagnostic mutation testing of MSH2 and MLH1 was performed in 11 families |
| Shia 200511 USA Single centre |
A group of 112 colorectal adenocarcinomas (n = 83) or adenomas (n = 29) obtained from 110 patients treated at the cancer centre. These cases had a FH that fulfilled one of the following criteria: (1) AC I or II, (2) a set of relaxed AC that we referred to as ‘HNPCC-like’ and (3) Bethesda criteria | All patients started with MSI testing, followed by mutation analysis |
| Southey 200539 Australia Single centre |
Men and women from the Victorian Colorectal Cancer Family Study who were younger than age 45 years when diagnosed with a histologically confirmed, first primary adenocarcinoma of the colon or rectum. A random selection of 222 patients were asked to participate | Patients answered a risk factor questionnaire, and received IHC and MSI screening. Germline MMR mutation testing was conducted for all patients with one or more of the following characteristics: a FH that fulfilled the AC for LS; a tumour that was MSI-H, MSI-L, or that lacked expression of at least one MMR protein; and presence in a random sample of 23 patients selected from those who had tumours that were MSS and did not lack expression of any MMR protein |
| Terdiman 200140 USA Single centre |
Eligible families had to have two or more FDRs with CRC at any age, an individual with CRC diagnosed before 50 years of age or a single individual with synchronous or metachronous CRCs. Probands were selected based on convenience and age at cancer diagnosis. When multiple family members were available for molecular testing, the individual with cancer diagnosed at the youngest age was selected as proband | Paraffin-embedded tumour samples were obtained from all probands for MSI analysis and MSH2/MLH1 immunostaining. Subjects found to have tumours demonstrating MSI-H (n = 47) were invited for germline genetic testing of MSH2 and MLH1. Gene testing was carried out in 32 of the 47 eligible families. Eight probands refused testing for fear of insurance discrimination. In seven instances, the proband was deceased (n = 4) or could not be recontacted (n = 3) |
| Wahlberg 200241 USA Single centre |
Families were identified by self- or health-care provider referral and were enrolled on the basis of multiple cases of CRC, early age of CRC diagnosis or the familial association of CRC with other LS-associated tumours | Sequencing and MSI analysis of tumour samples from 48 families. IHC analysis of 24 tumour samples (subset of 48 for MSI analysis) of sufficient quality for IHC |
In terms of test accuracy results, Bonis and colleagues (2007)4 found very little published information related to the analytic validity of laboratory testing for LS and there was some concern that there may be variability between testing facilities. They found that genomic rearrangements and large deletions were missed when only sequencing and gene screening was performed, with limited evidence to suggest that approximately one-quarter to one-third of the identified MMR mutations were large genomic deletions/rearrangements. Most studies identified cases from cancer registries or used other selection strategies to target patients at risk of LS. Although this is a valid recruitment technique from a clinically relevant population, the definition of high risk differs from study to study (among the selected populations) and this may or may not reflect the criteria used to refer for testing in clinical practice.
A summary of the relevant test accuracy results from Bonis and colleagues (2007)4 is presented in Table 8. The sample sizes of the studies were generally small. Sensitivity and specificity for both MSI and IHC appear variable with very broad confidence intervals (CIs).
| Author and location | Reference test | Index test | n | Sensitivity, % (95% CI, %) | Specificity, % (95% CI, %) | Risk of bias according to Bonis and colleagues | Qualitya |
|---|---|---|---|---|---|---|---|
| Calistri 200031 Italy Multicentre |
PCR → SSCP | MSI | 56 | 100 (59 to 100)b | 44 (14 to 79)b | Comment: Study sample assembled with unclear selection process Verification bias: No |
C |
| Christensen 200232 and Katballe 200233 Denmark Single centre |
PCR → SSCP and HD → sequencing of abnormal patterns | IHC | 42 | 69 (39 to 91)c | 83 (59 to 96)c | Comment: Selection from a population of 1514 incident CRCs Verification bias: No |
B |
| 11 | 50 (7 to 93)d | 100 (48 to 100)d | |||||
| MSI | 45 | 100 (69 to 100)b | 87 (66 to 97)b | ||||
| Debniak 200034 Poland Single centree |
PCR → sequencing | IHC | 168 | 18 (2 to 51)c | 100 (94 to 100)c | Comment: Sampled from consecutive CRCs, selection process not transparent Verification bias: Yes; only 43/143 apparently sporadic CRCs were tested, but it is unclear how they were selected |
C |
| MSI | 168 | 83 (36 to 100)b | 87 (76 to 94)b | ||||
| Dieumegard 200035 France Multicentre |
PCR → SSCP → sequencing of abnormal patterns | IHC | 34 | 57 (18 to 90)c | 64 (35 to 87)c | Comment: Sampled with unclear selection process Verification bias: Yes; only seven sporadic CRCs underwent genetic testing |
B |
| 10 | 50 (7 to 93)d | 50 (7 to 93)d | |||||
| MSI | 34 | 100 (66 to 100)b | 60 (32 to 84)b | ||||
| 10 | 100 (54 to 100)f | 25 (0 to 81)f | |||||
| Durno 200536 Canada Multicentre |
PTT and sequencing | IHC | 16 | 75 (19 to 99)c | 75 (19 to 99)c | Comment: Retrospective cohort of CRC patients aged < 24 years at diagnosis who were still alive (since 1970) Verification bias: No |
C |
| MSI | 16 | 100 (48 to 100)b | 25 (0 to 81)b | ||||
| Farrington 199837 Scotland Single centre |
PCR → IVSP → sequencing PCR → sequencing |
MSI | 50 | 86 (57 to 98)b | 73 (52 to 88)b | Comment: Retrospective cohort of CRC patients aged < 30 years at diagnosis who were still alive (since 1970) Verification bias: No |
B |
| Peel 200038 USA Multicentre |
PCR → sequencing | MSI | 11 | 100 (29 to 100)b | 83 (36 to 100)b | Comment: Referral HNPCC cases, other than the 1134 CRC probands who were also included but were not assessed with laboratory tests Verification bias: No |
C |
| 100 (29 to 100)f | 83 (36 to 100)f | ||||||
| Terdiman 200140 USA Single centre |
MSI → PCR → DGGE → sequencing | IHC | 114 | 94 (71 to 100)g | 13 (4 to 30)g | Comment: Retrospective cohort of CRC probands with ≥ 2 CRCs in FDRs, age < 50 years at diagnosis or multiple tumours in same patient Verification bias: Yes; only patients with MSI-H were assessed |
B |
| Wahlberg 200241 USA Single centre |
PCR → sequencing | IHC | 70 | 55 (23 to 83)c | 88 (69 to 97)c | Comment: Selection among referrals to a specialised centre Verification bias: No |
B |
| MSI | 70 | 100 (77 to 100)b | 59 (41 to 75)b |
Included primary research studies
Nine test accuracy papers were included, five investigating IHC as the index test, one MSI and three studying both IHC and MSI. No papers were identified on tests for BRAF V600E and methylation of MLH1. All included citations are summarised in Table 9.
| Author and year | Patients (n) | Test | Centre and country | Design | Outcomes |
|---|---|---|---|---|---|
| aBarrow 201120 | Sample, 36 Control, 6 |
IHC | Single centre, UK | Two-gate Sample patients retrospectively identified with mutation Control patients consecutively recruited Supported in part by grants from the Bowel Disease Research Foundation and Central Manchester and Manchester Children’s University Hospitals NHS Trust research grant scheme. This study group is supported by the NIHR Manchester Biomedical Research Centre |
ROC curves, sensitivity and specificity at optimum cut-offs |
| aBarrow 201042 | Sample, 51 Control, 17 |
IHC | Single centre, UK | Two-gate Sample patients retrospectively identified with mutation Control patients consecutively recruited Supported by MAHSC and the NIHR Manchester Biomedical Research Centre and by a grant from the Bowel Disease Research Foundation |
ROC curves, sensitivity and specificity at optimum cut-offs |
| Becouarn 200543 | 197 | IHC | Unclear, France | Single-gate Recruitment not described Funded by PHRC from the Délégation Régionale à la Recherche Clinique d’Aquitaine |
Sensitivity, specificity |
| Limburg 201144 | 195 | IHC | Unclear, USA/Canada/Australia | Single-gate Recruitment described as random, but no further details Funded by Myriad Genetic Laboratories, Salt Lake City, UT. The Colon Cancer Family Registry is supported by NIH National Cancer Institute grants |
Sensitivity, specificity, NPV, PPV |
| Niessen 201221 | 281 | IHC and MSI | Unclear, the Netherlands | Single-gate Recruitment not described Funded by the Dutch Cancer Society |
Sensitivity, specificity, NPV, PPV |
| Shia 200511 | 110 | IHC and MSI | Single centre, USA | Single-gate Recruitment not described Funded in part by the Kleber Foundation, the Sloan Kettering Institute, the Byrne Foundation and the Tavel-Reznik Fund for Colon Cancer Research |
Sensitivity, specificity |
| Southey 200539 | 131 | IHC and MSI | Single centre, Australia | Single-gate Random recruitment Funded by grants from the National Health and Medical Research Council (Australia) and the Victorian Health Promotion Foundation |
Sensitivity, specificity, NPV, PPV |
| Stomorken 200545 | 250b | IHC | Single centre, Norway | Single-gate Consecutive recruitment Funded by the Norwegian Cancer Society |
Sensitivity, specificity |
| Wolf 200646 | 81 | MSI | Single centre, Austria | Single-gate Recruitment not described Funded by the Medical Scientific Fund of the University of Vienna Medical School and the Medical Scientific Fund of the Mayor of Vienna |
Sensitivity, specificity |
Study characteristics
The majority of included studies employed a single-gate design where one sample of individuals was assessed by both the index test and reference standard. Only Barrow and colleagues (2010 and 2011) used a two-gate design, where the index test was performed on a group of participants with a known (positive) mutation status and a smaller group of controls with no applicable mutation status. 20,42 However, despite most studies being of a single-gate design, not all participants received the reference standard. Many studies cited cost as a reason for not performing genetic testing on participants who appeared to be MSS (i.e. those with no evidence of abnormal patterns of microsatellite repeats). This often led to confusing patient numbers and apparent missing data. In general, sample sizes were relatively small with poor reporting of patient characteristics. Details on robustness of testing were often lacking (e.g. results being checked by a second assessor), particularly for IHC, which may be prone to interobserver variability.
Barrow and colleagues (2010)42 present a two-gate, single-centre UK study investigating the semi-quantitative assessment of IHC for MMR proteins in LS. Patients with LS which had already been confirmed by germline mutation in one of the MMR genes – MLH1, MSH2 or MSH6 – and previous histologically proven CRC were identified through the North West Regional Genetics Lynch Syndrome Database. The control cases were consecutive unselected patients aged > 60 years with histologically proven left-sided colonic or rectal cancer (i.e. considered to be sporadic CRC as opposed to LS). A relatively small LS sample (n = 51) was recruited, with an even smaller control group (n = 17). IHC methods were described in detail; sections of tumour tissue were incubated with antibodies against the MMR proteins or antigens MLH1, MSH2, PMS2 and MSH6. The intensity of immunoreactivity (a measure of the reaction between the antibody and antigens of the tumour cells) was measured on a 0–3 scale, based on comparison of intensity of reactivity of the tumour cells with the positive control cells. A score of 0 = no tumour cell immunopositivity; 1 = 1–10% positive tumour cells; 2 = 11–50% positive tumour cells; 3 = 51–80% positive tumour cells; and 4 = ≥ 80% positive tumour cells. Information on the reference standard was not provided. Outcomes were receiver operating characteristic (ROC) curves and sensitivity and specificity at optimum cut-offs (the cut-off score which demonstrates the best trade-off between sensitivity and specificity). However, raw data were not provided to populate a 2 × 2 table of positive and negative results.
The second paper by Barrow and colleagues (2011)20 appears to use the same pool of participants as the previous study, although with lower numbers. In this instance, the aim was to compare two novel methodologies: quantitative 3,3′-diaminobenzidine IHC (DAB-IHC) and quantitative quantum dot IHC (QD-IHC) in the identification of MMR mutation carriers. Also a two-gate design, this study had an LS sample size of 36, and a control group of only six. As per the previous study, participants had already received germline mutation testing, although details were not provided.
With regard to the DAB-IHC, sections were incubated with antibodies against the MMR proteins MLH1, MSH2, MSH6 and PMS2. With control positive tissue (normal colon), the protocol was optimised to a level that maximised specific nuclear immunoreactivity, while minimising non-specific background reactivity. For the QD-IHC, only staining for MLH1 and MSH2 was performed.
Again, relevant outcomes include ROC curves, and sensitivity and specificity at optimum cut-off, yet the raw data were not provided to populate a 2 × 2 table of positive and negative results.
A French study presented by Becouarn and colleagues (2005)43 examines a strategy to detect LS in patients by combining clinical selection (patient age at onset of cancer < 50 years or FH of HNPCC tumours) and MSI testing plus IHC, leading to MMR germline mutation analysis. It should be noted that only IHC was considered to be the index test, with MSI the prior test.
It is not clear how many centres were involved; however, the sample size was reported to be 197. Recruited participants were diagnosed with CRC between 1998 and 2001, and deemed high risk for LS owing to young age at onset or FH. Patient flow throughout the study is somewhat unclear and complex. It appears that testing by MSI took place in order to group participants according to MSI-H, MSI-L and MSS. Only patients who were MSI-H and MSI-L and those with valid IHC results for MLH1 and MSH2 received germline testing.
For IHC, the search for MMR proteins MLH1 and MSH2 was conducted on fixed tissue embedded in paraffin. Loss of MLH1 or MSH2 expression was defined as the absence of nuclear staining in tumour cells, in the presence of positive controls; preservation of protein expression was defined as the presence of nuclear staining in tumour cells and internal controls; non-interpretable staining was defined as the absence of staining in tumour cells and internal controls or slice detachment. For the reference standard, the search for mutations of the MSH2 and MLH1 genes was performed in MSI-H and MSI-L tumour tissues. The search for germline mutations and their characterisation was based on denaturing high-performance liquid chromatography (DHPLC) screening and/or direct sequencing using an automatic sequencer. The search for large MSH2 and MLH1 gene rearrangements was performed in certain patients when point mutations were not identified. For IHC, data were provided to populate a 2 × 2 table, therefore sensitivity and specificity could be calculated.
Limburg and colleagues (2011)44 present a study examining the prevalence of mutations in MLH1, MSH2 and MSH6 via IHC in a population-based sample of patients with young-onset (age at onset < 50 years) CRC. Employing six centres across Canada, the USA and Australia, a random sample of 195 CRC cases were recruited during phase 1 of the Colon Cancer Family Registry collaboration (1997–2002). No prior testing was performed and no preselection based on FH, so high risk for LS was based on age criterion alone. Minimal details are given for the index test, with MMR protein expression reported as present, absent or inconclusive.
The reference standard uses extracted DNA samples from peripheral blood for full mutation analyses of MLH1, MSH2 and MSH6. DNA was amplified by PCR and then directly sequenced. Large rearrangement testing for MLH1 and MSH2 was performed by Southern blot analysis in conjunction with MLPA. Germline alterations were categorised as deleterious/suspected deleterious, likely neutral or variant of uncertain significance. Reported outcomes were sensitivity, specificity, NPV and PPV, with raw data available to populate a 2 × 2 table.
The study by Niessen and colleagues (2006)21 investigated the sensitivity and specificity of IHC and MSI, the aim being to analyse the value of FH, MSI analysis and MMR protein staining in the tumour to predict the presence of a MMR gene mutation in such patients. Performed in the Netherlands (although it is unclear how many centres were involved), 281 individuals with CRC, who were high risk for LS according to young onset or personal cancer history, were recruited.
Microsatellite instability markers included two mononucleotide repeats (BAT25 and BAT26) and three dinucleotide repeats (D2S123, D5S346 and D17S250). For MSI analysis, control DNA was obtained from normal tissue or from peripheral blood lymphocytes from the same patient. Cancers were classified as MSI-H when two or more markers showed MSI and as MSI-L when no more than one marker showed MSI. The authors state that as a limited number of markers were analysed, the classification MSS was not used. IHC for the MLH1, MSH2 and MSH6 proteins was also carried out. The sections were scored as either negative (i.e. absence of detectable nuclear staining of cancer cells) or positive for MLH1, MSH2 and MSH6 staining. Protein expression in normal tissue adjacent to the cancer served as an internal positive control.
Mutation analysis of the MLH1, MSH2 and MSH6 genes was carried out on DNA isolated from peripheral blood lymphocytes by denaturing gradient gel electrophoresis, followed by direct sequencing. For the detection of large deletions (exonic deletions or deletions of a complete gene) and duplications, MLH1/MSH2 exon deletion MLPA was used. Cases that had deletions of more than one exon in the MLH1 or MSH2 gene were confirmed by Southern blot analysis. Sensitivity, specificity, PPV and NPV were reported along with data to populate a 2 × 2 table.
Shia and colleagues (2005)11 report a single-centre study performed in the USA with 110 participants. The study objective is not clearly described. Participants were recruited from 1995 to 2003 by a FH questionnaire administered in gastrointestinal endoscopy and oncology clinics, by personal interview of persons undergoing surgery for CRC or by referrals to the clinical genetics service. In order to be included, participants had to have a FH that fulfilled one of the following criteria: (1) AC I or II, (2) a set of relaxed AC (three or more CRCs among first- and second-degree relatives of a family) or (3) Bethesda criteria.
The study started with MSI testing, followed by germline analysis of MLH1 and MSH2 in all cases that exhibited MSI and cases with carcinoma that did not exhibit MSI. Cases that showed no mutation in MLH1 or MSH2 were tested for mutation in MSH6.
Microsatellite instability testing was performed on microdissected DNA from paraffin-embedded tissue blocks using a standard PCR method. All tissue was tested with seven markers: four mononucleotide markers (BAT25, BAT26, BAT40, PAX6); two dinucleotide markers (D2S123, D17S250); and one mixed dinucleotide and trinucleotide marker (MYCLI). IHC was performed using antibodies against MLH1, MSH2 and MSH6. Normal colon mucosa were used as a positive control and MSI tumours known to lack MLH1 or MSH2 protein expression were used as a negative control. Tumours displaying a total absence of nuclear staining while adjacent normal mucosa or stromal/lymphoid cells showed presence of nuclear staining were scored ‘negative’ for expression of protein. Tumours were scored according to staining intensity:
-
weak if < 10% of the tumour was stained and the intensity was weak
-
heterogeneous if two or more of the following staining patterns were identified, each present in at least 20% of the tumour: (1) no nuclear staining, (2) weak nuclear staining, (3) moderate nuclear staining and (4) strong nuclear staining.
Mutation analysis was performed using DHPLC and direct sequencing. Cases with tumours that exhibited MSI but in which a point mutation in MLH1, MSH2 or MSH6 was not detected were analysed for large deletions in MLH1 and MSH2. Mutations were determined to be disease-causing based on sequencing results, segregation analysis and published data and mutation databases. Outcomes include sensitivity and specificity with raw data available.
An investigation into the relationship between MMR protein expression, MSI, FH and germline MMR status was performed by Southey and colleagues (2005). 39 The study took place in a single centre in Australia. One hundred and thirty-one patients with young-onset CRC were randomly recruited, i.e. patients who were younger than 45 years when diagnosed with a histologically confirmed, first primary adenocarcinoma of the colon or rectum. Sensitivity and specificity results for both IHC and MSI were reported.
For IHC, the expression of MLH1, MSH2, MSH6 and PMS2 was assessed on paraffin-embedded sections using antibodies MLH1, MSH2, MSH6 and PMS2. Normal colonic epithelium adjacent to tumour and lymphocytes served as positive controls. A gastrointestinal pathologist scored the tumours as positive when nuclear staining in tumour tissue was present, or negative when staining was absent.
Microsatellite instability testing was performed on invasive tumour cells microdissected from 5-µm sections of paraffin-embedded archival tumour tissue. DNA extracted from histologically normal cells microdissected from colonic or lymph node tissue, or DNA extracted from peripheral-blood lymphocytes, was used as a negative control. Ten microsatellite markers were assessed: three dinucleotide repeats (D5S346, D17S250 and D2S123) and seven mononucleotide repeats (BAT25, BAT26, BAT40, MYB, TGFβRII, IGFIIR and BAX). The degree of instability in each tumour was scored as MSS, MSI-L and MSI-H when zero to one, two to five and six to 10 markers, respectively, were identified as unstable.
For the reference standard, MLH1, MSH2, MSH6 and PMS2 genes were screened for germline mutations using sequencing approaches, except for exon 4 of MSH6, which was screened in eight overlapping fragments using DHPLC. Putative mutations were confirmed via direct automated sequencing. Variants were defined as deleterious if they could be predicted to produce a shortened or truncated protein product, or if they were missense mutations that have been reported previously to be deleterious. The MLPA assay to detect large genomic alterations in MLH1 and MSH2 was performed on samples from 10 patients who had tumours lacking at least one MMR protein expression and for which no previous mutation had been identified by sequencing. Mutation testing was conducted on participants with one or more of the following characteristics: a FH that fulfilled the AC for HNPCC; a tumour that was MSI-H, MSI-L or that lacked expression of at least one MMR protein; and a random sample of 23 patients selected from those who had tumours that were MSS.
Stomorken and colleagues (2005)45 report on a single-centre study based in Norway. Two hundred and fifty families were consecutively recruited according to their FH of CRC and other cancers. Inclusion criteria consisted of AC I or II, aggregation of four or more LS-related cancers on one side of the family, patients with ‘very early onset’ CRC and those with multiple primaries including colorectal or endometrial cancers. It should be noted that the participants with CRC provided a subsample of 105 families. The aim of the study was to validate the sensitivity, specificity and predictive value of IHC, compared with various clinical criteria, to select LS relatives for mutation testing.
Immunohistochemistry of all tumours for the presence of MLH1, MSH2 and MSH6 MMR proteins was performed using a formalin-fixed, paraffin-embedded tissue block containing tumour tissue and normal adjacent mucosa. Staining of tumours was evaluated using normal epithelial cells, stromal cells or lymphocytes in the same slide as controls. The percentage of nuclear staining was graded as follows: complete absence of detectable nuclear staining (0); positive staining in < 30% of the tumour cells (1+); positive staining in 30–60% of the tumour cells (2+); or positive staining in > 60% of the tumour cells (3+).
MLH1 and MLH2 genes were sequenced by Myriad Genetics Inc. (Salt Lake City, UT). All index persons without a mutation demonstrated by sequencing and with a lack of MMR protein expression were subjected to analyses for large rearrangements in the MLH1 and MSH2 genes. The remaining individuals who were lacking gene products of MSH2 and/or MSH6 genes were subjected to mutation analysis of the MSH6 gene by sequencing. Large rearrangements in MSH6 were not tested for. Data were available to populate a 2 × 2 table, although sensitivity and specificity were not reported.
Wolf and colleagues (2006)46 report an Austrian study where participants were selected retrospectively from among individuals with suspected hereditary CRC from 2000 to 2003. The sample size was 81, with all tumours obtained by surgical resection. The aim of the study was to evaluate the revised AC and Bethesda guidelines (therefore the index test under scrutiny was MSI).
Nuclear DNA was isolated from paraffin-embedded tissue after histological verification by an experienced pathologist, prior to PCR amplification. DNA was also taken from blood samples.
For MSI, two groups of five markers each were selected: group 1 consisted of D5S346, HSCAP53L, D2S123, BAT26 and D18S34, while group 2 consisted of D5S82, D2S134, D13S175, D11S904 and BAT25. In the event of instability, additional smaller fragments were identified in the tumour sample compared with the corresponding normal tissue. If only one of the markers in the first group showed instability, five further markers (group 2) were used. The degree of instability was evaluated according to the percentage of markers showing band shifts. MSI-H was considered to exist when at least 30% of the analysed markers were unstable; any lower degree of instability, or no instability, was interpreted as MSS.
The exons of MLH1 and MSH2 as well as the promoter regions of each gene underwent sequence analysis. If DNA from tumour tissue was available, analysis of DNA from corresponding normal tissue or peripheral blood was performed on fragments containing a mutation. If no mutation was found in the tumour, sequence analysis was performed with DNA from normal tissue or peripheral blood.
Appropriate outcomes for this review were sensitivity and specificity, with raw data provided.
Population characteristics
In general, patient characteristics were poorly reported, as were inclusion and exclusion criteria, although patients were often filtered by prior testing. Comparable characteristics are presented in Table 10.
| Characteristic | Becouarn 200543 | Limburg 201144 | Niessen 200621 | Shia 200511 | Stomorken 200545 | Wolf 200646 |
|---|---|---|---|---|---|---|
| Number of patients | 197 | 195 | 281 | 110 | 105a (50)b | 81 |
| Description | Patient age at onset of cancer < 50 years or FH of HNPCC tumours | Population-based sample of patients with young-onset (age < 50 years) CRC | Individuals with CRC who were high risk for LS according to young onset or personal cancer history were recruited | Participants recruited from 1995 to 2003 by a FH questionnaire, by personal interview prior to surgery for CRC, or by referrals to the clinical genetics service | Families recruited according to FH of CRC and other cancers. Inclusion criteria consisted of AC I or II, four or more LS-related cancers on one side of the family, early-onset CRC, multiple primaries including CRC or EC | Participants selected retrospectively from individuals with suspected hereditary CRC |
| Age, years | ||||||
| 0–30, n | 13 | |||||
| 31–40, n | 40 | |||||
| 41–50, n | 111 | |||||
| ≥ 51, n | 33 | |||||
| < 50, n | 224 | |||||
| Mean age, years (SD) | 42.9 (6.1) | 45 (11)c | 50.5d | |||
| Sex | ||||||
| Male, n (%) | 115 (58) | 128 (46) | 48 (44) | 39 (48) | ||
| Female, n (%) | 82 (42) | 104 (53.3) | 153 (54) | 62 (56) | 42 (52) | |
| Cancer location | ||||||
| Rectum, n | 41 | |||||
| Left colon, n (%) | 75 | 130 (74.7) | ||||
| Right colon, n (%) | 68 | 44 (25.3) | ||||
| Transverse colon, n | 13 | |||||
| Number meeting AC II (%) | 10 (5.1) | 50 | 38 | 43.2 | ||
| Number meeting Bethesda criteria | 12 | 43 | 72 | |||
| Number HNPCC-like | 40 | |||||
| Number with ≥ 2 LS cancers | 79 |
Barrow and colleagues (2010 and 2011)20,42 did not report participants’ characteristics. Southey and colleagues (2005)39 provided no information on characteristics other than stating that participants were adult men and women who were younger than 45 years when diagnosed with a histologically confirmed, first primary adenocarcinoma of the colon or rectum.
Overall, owing to the sparseness of the data, it is difficult to draw a conclusion regarding the heterogeneity of the participants both within and between studies.
Assessment of study quality
A summary of the quality assessment of studies included in this review is shown in Table 11.
| QUADAS 2 domain | Question | Barrow 201042 | Barrow 201120 | Becouarn 200543 | Limburg 201144 | Niessen 200621 | Shia 200511 | Southey 200539 | Stomorken 200545 | Wolf 200646 |
|---|---|---|---|---|---|---|---|---|---|---|
| Patient selection | Was a consecutive or random sample of patients enrolled? (Y/N/U) | U | U | U | Y | U | U | Y | Y | U |
| Was a case–control study design avoided?a (Y/N/U) | N | N | Y | Y | Y | Y | Y | Y | Y | |
| Did the study avoid inappropriate exclusions? (Y/N/U) | U | U | Y | Y | U | Y | Y | U | Y | |
| Could the selection of patients have introduced bias? (H/L/U) | U | U | U | L | U | L | L | L | L | |
| Are there concerns that the included patients do not match the review question? (H/L/U) | L | L | L | L | L | L | L | L | L | |
| Index test | Were the index test results interpreted without knowledge of the results of the reference standard? (Y/N/U) | Y | U | U | U | U | U | U | Y | U |
| If a threshold was used, was it pre-specified? (Y/N/U) | Y | U | Y | U | Y | N | Y | Y | Y | |
| Could the conduct or interpretation of the index test have introduced bias? (H/L/U)b | L | L | L | L | L | L | L | L | L | |
| Are there concerns that the index test, its conduct or interpretation differ from the review question? (H/L/U) | L | L | L | L | L | L | L | L | L | |
| Reference standard | Is the reference standard likely to correctly classify the target condition? (Y/N/U) | Y | Y | Y | Y | Y | Y | Y | Y | Y |
| Were the reference standard results interpreted without knowledge of the results of the index test? (Y/N/U) | Y | Y | U | Y | U | U | U | U | U | |
| Could the reference standard, its conduct or its interpretation have introduced bias? (H/L/U) | L | L | U | L | U | U | U | U | U | |
| Are there concerns that the target condition as defined by the reference standard does not match the review question? | L | L | L | L | L | L | L | L | L | |
| Flow and timing | Was there an appropriate interval between index test(s) and reference standard? | Y | Y | Y | U | Y | U | U | U | U |
| Did all patients receive a reference standard? (Y/N/U) | N | N | N | Y | Y | Y | N | N | N | |
| Did all patients receive the same reference standard? (Y/N/U) | U | U | Y | U | N | Y | Y | N | Y | |
| Were all samples (that should have been) included in the analysis? (Y/N/U) | U | U | N | N | N | N | Y | N | N | |
| Could the patient flow have introduced bias? (H/L/U) | H | H | H | H | H | H | H | H | H |
Concerns included lack of detail on patient recruitment, minimal information on patient characteristics and unclear tissue sampling methods; for example, little evidence was given of sample replicates and reproducibility for molecular analysis. Furthermore, test failures, such as operator or instrumental error, were rarely reported. It was also often not mentioned whether or not the outcome assessors were blinded. Papers were also unclear on robustness of IHC, for example whether or not results were checked by a second party. Often, not all patients received both the index and reference test, likely owing to the cost of constitutional genetic testing. This frequently caused a lack of clarity regarding patient flow and resulted in all studies being at high risk of bias.
Two-gate studies
The study by Barrow and colleagues (2010)42 used a two-gate design, i.e. a group with LS confirmed by germline testing and a control group. Methods of recruitment were unclear, with no patient characteristics described. The LS sample was small (n = 51), with an even smaller control group (n = 17). No technical details were provided for the reference standard. More positively, to minimise interobserver variation, a semiquantitative scoring system was used. The slides were scored by an experienced consultant gastrointestinal pathologist who was blinded to the mutational status of the participants. Test failures and attrition were not reported.
The second study by Barrow and colleagues (2011)20 appeared to use consecutively recruited participants from the previous study’s population. Again, this was a two-gate design with a small LS sample (n = 36) and much smaller control (n = 6). No details were provided on participant characteristics, outcome assessors or blinding. Test failures and attrition were not reported.
Single-gate studies
The study reported by Becouarn and colleagues (2005)43 only performed the reference standard on MSI-H and MSI-L patients. Therefore, there are insufficient data to populate a 2 × 2 table for MSI versus reference standard, and only IHC can be deemed to be an index test for the purposes of this review. Moreover, this means that the data for IHC versus reference standard are based upon a sample who had received prior MSI testing (with a MSI-H/MSI-L result). There also appear to be missing data; of the 33 patients receiving the reference standard, 10 did not receive IHC, and the reasons for this are unclear. Dropouts were only reported for the reference standard. Additionally, there was one IHC test failure. No blinding or outcome assessor details were reported, and the recruitment method was unclear.
Limburg and colleagues (2011)44 recruited participants randomly, although the method was not given. Details of the outcome assessor for IHC were unclear; however, Southern blot and MLPA were subjected to dual reviews involving technical personnel and at least one laboratory director for confirmation. The testing centre was blinded to all clinical data associated with the specimens. On receipt, samples were assigned a unique bar code for robotic specimen tracking. From the initial sample of 201 cases, six were excluded based on tumour location (appendix, n = 4; anus, n = 2). Not all patients received IHC because tumour phenotype was not considered a primary study end point. As such, complete IHC data were only available for 155 (79%) of 195 subjects. No test failures were reported. Results for sensitivity and specificity were given, but no raw data were available.
The study reported by Niessen and colleagues (2006)21 lacked clarity in a number of areas, including number of centres and method of recruitment. Although IHC staining was scored blinded to the MSI or mutation status, no further details were provided on the outcome assessor. Unfortunately, data provided for IHC were inconsistent, so a 2 × 2 table could not be populated. The data for MSI appear more reliable. However, not all participants received mutation analysis for MLH1 and MSH2, and this was on the basis of MSI results (i.e. for the final third of patients, mutations in MLH1 and MSH2 were sought only for those with an MSI-H tumour).
Shia and colleagues (2005)11 report on a single-gate, single-centre study, with an exceptionally confusing patient flow. The number of test failures was unclear as were excluded data. It appears that five samples for IHC could not be interpreted (4 of 108 for MSH6 and 1 of 110 for MSH2), and only results for 81 IHC samples and 73 MSI samples are reported (of 110 and 104 samples, respectively). It was not clear why the numbers used for IHC and MSI versus germline were fewer than the numbers reported to have received the tests. Individuals with adenomas met AC I or II, or Bethesda criteria, but it is possible that they did not have CRC themselves and are therefore only includable in this review as relatives of those with CRC. Methods of recruitment, blinding and checking of outcome assessor were not reported.
The study by Southey and colleagues (2005)39 was single gate and single centre, with unspecified randomised recruitment. An assessment via MSI testing was not successful for 13 (12%) tumour samples because of technical reasons related to DNA quality, which left 105 tumours tested. No blinding or patient characteristics were reported. Tumour samples were obtained for 118 of 131 patients (six did not consent to release tissue to the study and two laboratories had not agreed to release the remaining seven samples); however, of these, not all patients received germline testing. Instead, germline testing was conducted only on those with a FH that fulfilled the AC for LS; those who were MSI-H, MSI-L or lacked expression of at least one MMR protein; and a random sample (n = 23) of individuals who were MSS and did not lack expression of any MMR protein. For untested persons, data were imputed based upon the results of the randomly sampled individuals who were MSS and did not lack expression of any MMR protein.
Stomorken and colleagues (2005)45 report a study of 250 consecutively recruited families, although the relevant population with CRC reduces this number to 105, as other cancers are included in the paper. No participant characteristics were given. Furthermore, the study flow was very unclear, contributing to bias, as participants were selected for the reference test based upon their index test results. No test failures were reported and there appeared to be missing data (e.g. from 21 families, blocks for IHC were not available and from 30 families blocks were not asked for). Scoring of the tumour was performed by pathologists without any knowledge of patients’ FH or results of mutation analyses.
The study reported by Wolf and colleagues (2006)46 was single centre with no mention of recruitment method, blinding or outcome assessors. It was also unclear whether or not some patients had already had MSI testing. Sample size was small (n = 81), with limited patient characteristics provided. No test failures were reported. Twenty-six patients had no tumour tissue of suitable quality for DNA examination; however, all patients received index test (IHC) and germline testing.
Assessment of test accuracy
Individual results for this review from 2005 to 2012 are presented alongside a narrative description below. Summary tables are displayed at the end of this section (see Tables 23 and 24).
Immunohistochemistry
Two-gate studies
Unfortunately, neither of the papers by Barrow and colleagues (2010) provide data to populate a 2 × 2 table; however, results for sensitivity and specificity are presented. 42 For the identification of MLH1 germline mutation, at optimum cut-off (where the trade-off between sensitivity and specificity is optimum) sensitivity was 100.0% (95% CI 84.0% to 100.0%) and specificity was 91.5% (95% CI 79.6% to 97.6%); for PMS2 stain, sensitivity was 95.2% (95% CI 76.2% to 99.9%) and specificity was 91.5% (95% CI 79.6% to 97.6%).
For the identification of MSH2 germline mutation, MSH2 staining was less sensitive and specific (sensitivity 87.5%, 95% CI 61.7% to 98.4%; specificity 88.5%, 95% CI 76.5% to 95.6%), and MSH6 staining had an even lower performance (sensitivity 81.3%, 95% CI 54.4% to 96.05%; specificity 80.8%, 95% CI 67.5% to 90.4%).
The authors discuss that staining does not show a binary response to the presence or absence of a mutation; for example, a pathogenic nonsense mutation may theoretically lead to a shorter, non-functional protein. Where this happens, in theory, immunoreactivity may be preserved.
In this study, sensitivity and specificity were greater for MLH1 than for MSH2. The authors acknowledge this may be a reflection of small sample size and use of different antibodies. Other variables include poor fixation and degraded tissue in archived paraffin blocks.
In the second paper, Barrow and colleagues (2011)20 examine two forms of IHC: QD-IHC and DAB-IHC. At the optimum cut-offs, MLH1 DAB-IHC had a sensitivity of 89.5% (95% CI 66.9% to 98.7%) and a specificity of 78.3% (95% CI 56.3% to 92.5%). MSH2 DAB-IHC had a sensitivity of 86.7% (95% CI 59.5% to 98.3%) and a specificity of 77.8% (95% CI 57.7% to 91.4%). Sensitivity and specificity were not given for the other test under investigation (QD-IHC); however, the authors state that DAB-IHC demonstrated superior results. Suggested reasons for this include variability in manual staining for QD-IHC and practical difficulties in fluorescence imaging.
Single-gate studies
As mentioned in Assessment of study quality above, the study reported by Becouarn and colleagues (2007)43 investigated IHC and MSI; however, the reference standard is only performed on MSI-H and MSI-L patients. Therefore, there are insufficient data to populate a 2 × 2 table for MSI. Table 12 shows the data for IHC, which give a sensitivity of 73.3% (95% CI 44.8% to 92.2%) and a specificity of 28.5% (95% CI 3.7% to 71.0%).
| IHC | Reference standard | ||
|---|---|---|---|
| Positive | Negative | Total | |
| Positive | 11 | 5 | 16 |
| Negative | 4 | 2 | 6 |
| Total | 15 | 7 | 22 |
Both the sensitivity and specificity are poor, although the small sample size should be noted and the authors mention failures due to defective tissue fixation and detachment of the histological slice during antigenic revelation. IHC was also limited to proteins MLH1 and MSH2, whereas the reference standard may have detected additional mutations. Furthermore, there are also some concerns regarding missing data with this study because, of the 33 patients receiving the reference standard, 10 did not receive IHC and the reasons for this are unclear.
The authors discuss that IHC demonstrated poor sensitivity in four patients with LS in whom a causal mutation was detected. Two of these had missense mutations, which possibly alter protein function but not translation and therefore would not be detected by IHC, which evaluates protein expression. The other two patients had a germline splicing mutation or large rearrangement which could lead to either (i) preserved reading frame with persistent expression by IHC; or (ii) total loss of expression of the mutated allele. Again, the authors consider the produced protein to have deficient function but not a total loss of expression.
Sensitivity and specificity for MLH1, MSH2 and MSH6 by IHC were reported by Limburg and colleagues (2011)44 as 85.7% (95% CI 42.1% to 99.6%) and 91.9% (95% CI 86.3% to 99.7%), respectively. The sample size was reasonable for a study in this area, with randomly recruited participants without prior testing. Data for IHC are shown in Table 13.
| IHC | Reference standard | ||
|---|---|---|---|
| Positive | Negative | Total | |
| Positive | 6 | 12 | 18 |
| Negative | 1 | 136 | 137 |
| Total | 7 | 148 | 155 |
The authors consider that the 12 FPs may be due to epigenetic modification (i.e. MLH1 promoter hypermethylation), recently described mutations not detectable by the applied methodology or mutations in other, non-analysed genes that interact with the MMR proteins tested.
The results presented for IHC by Niessen and colleagues (2006)21 appear inconsistent, and therefore a 2 × 2 table was not possible. However, according to the text the sensitivity and specificity of IHC staining for MLH1 were 80% (95% CI 38% to 96%) and 89% (95% CI 84% to 93%), respectively; for MSH2 the sensitivity and specificity were 100% (95% CI 57% to 100%) and 96% (92% to 98%), respectively; and for MSH6 the sensitivity and specificity were 86% (95% CI 49% to 97%) and 93% (95% CI 88% to 96%), respectively. The sample size was 174; however, despite this, the CIs for MLH1 and MSH6 sensitivities were wide.
Shia and colleagues (2005)11 report on IHC results for 83 adenocarcinomas and 29 adenomas. Negative (i.e. abnormal) IHC staining was observed in 25 patients with carcinoma, for a sensitivity of 81% (95% CI 61% to 93%), but false abnormal IHC staining was present in four tumours, for a specificity of 89% (95% CI 75% to 97%).
The sensitivity of IHC was particularly low in detecting MLH1 gene mutation, which the authors attribute partly to missense mutations that result in mutant proteins that are catalytically inactive but antigenically active. In addition, truncating mutations in MLH1 may result in a protein that reacts with the MLH1 antibody. IHC was more favourable in detecting MSH2 (21 of 23) and MSH6 (four of five) gene mutations. The difference in sensitivities between adenomas and carcinomas was reported as not significant (p = 1.00). Data for participants with carcinomas and adenomas are shown in Tables 14 and 15, respectively.
| IHC | Reference standard | ||
|---|---|---|---|
| Positive | Negative | Total | |
| Positive | 21 | 4 | 25 |
| Negative | 5 | 34 | 39 |
| Total | 26 | 38 | 64 |
| IHC | Reference standard | ||
|---|---|---|---|
| Positive | Negative | Total | |
| Positive | 8 | 0 | 8 |
| Negative | 4 | 5 | 9 |
| Total | 12 | 5 | 17 |
The results could not be interpreted in five samples owing to a lack of definitive staining in normal cells on the same slide and diffuse and strong cytoplasmic staining in the tumour cells and normal cells. It was also noted that not all IHC positive cases showed uniform positivity throughout the tumour, and weak or focal expression of a MMR protein may be associated with MSI or gene mutation, or both.
The study presented by Southey and colleagues (2005)39 gives a sensitivity of 100% (95% CI 82% to 100%) and a specificity of 91% (95% CI 83% to 96%). Data for IHC are shown in Table 16.
| IHC | Reference standard | ||
|---|---|---|---|
| Positive | Negative | Total | |
| Positive | 18 | 8 | 26 |
| Negative | 0 | 79 | 79 |
| Total | 18 | 87 | 105 |
The authors suggest that if they had not conducted IHC testing for all four proteins, evidence would have been missed for some carriers; for example, by not testing for MSH6 or PMS2, two patients with large deletions in MLH1 and all four patients with MSH6 mutations would not have been detected. It should be noted that not all patients received germline testing.
Owing to the inclusion of extra colonic cancers, the number of participants for calculating sensitivity and specificity for IHC is relatively small for the study presented by Stomorken and colleagues (2005). 45 Proteins tested for were MLH1, MSH2 and MSH6, to give a sensitivity of 100.0% (95% CI 75.2% to 100.0%) and a very low specificity of 12.5% (95% CI 0.3% to 52.7%), due to seven FPs (four FPs were MLH1, which might possibly be due to methylation) and only one TN. When investigating all cancers, a specificity of 82% was reported. Table 17 shows the correlation between IHC and germline testing.
| IHC | Reference standard | ||
|---|---|---|---|
| Positive | Negative | Total | |
| Positive | 13 | 7 | 20 |
| Negative | 0 | 1 | 1 |
| Total | 13 | 8 | 21 |
Microsatellite instability
Niessen and colleagues (2006)
The sensitivity and specificity calculated using the results in the 2 × 2 table are 88.0% (95% CI 68.7% to 97.4%) and 68.1% (95% CI 61.7% to 74.1%), respectively, for the study reported by Niessen and colleagues (2006)21 (Table 18).
| MSI | Reference standard | ||
|---|---|---|---|
| Positive | Negative | Total | |
| Positive | 22 | 75 | 97 |
| Negative | 3 | 160 | 163 |
| Total | 25 | 235 | 260 |
The authors mention that interobserver variation can occur when scoring MSI and that sporadic CRCs show MSI as a result of hypermethylation of the promoter region of MLH1, hence the high number of FPs.
Shia and colleagues (2005)
Shia and colleagues (2005)11 produced a sensitivity and specificity for participants with carcinoma investigated by MSI of 100% (95% CI 86% to 100%) and 84% (95% CI 68% to 94%), respectively. The sensitivity for adenomas is lower at 86% and the specificity is 80%. Data for participants with carcinomas and adenomas are shown in Tables 19 and 20.
| MSI | Reference standard | ||
|---|---|---|---|
| Positive | Negative | Total | |
| Positive | 24 | 6 | 30 |
| Negative | 0 | 31 | 31 |
| Total | 24 | 37 | 61 |
| MSI | Reference standard | ||
|---|---|---|---|
| Positive | Negative | Total | |
| Positive | 6 | 1 | 7 |
| Negative | 1 | 4 | 5 |
| Total | 7 | 5 | 12 |
Presence of MSI was also seen in 7 of 42 carcinomas and adenomas that did not harbour any detectable germline mutations. The seven false MSI-present cases included the four false-negative IHC cases.
The difference in sensitivities between adenomas and carcinomas were reported as not significant for IHC or MSI (IHC, p = 1.00; MSI, p = 0.25).
Southey and colleagues (2005)
Southey and colleagues (2005)39 report a sensitivity and specificity of 94% (95% CI 73% to 100%) and 80% (95% CI 71% to 88%), respectively. Table 21 shows that for every TP, MSI testing produces a FP. It should be noted that this study used a panel of 10 markers, unlike the NCI panel, which tests five markers.
| MSI | Reference standard | ||
|---|---|---|---|
| Positive | Negative | Total | |
| Positive | 17 | 17 | 34 |
| Negative | 1 | 70 | 71 |
| Total | 18 | 87 | 105 |
Wolf and colleagues (2006)
Wolf and colleagues (2006)46 state that MSI was associated with a high sensitivity (100.0%; 95% CI 71.7% to 100.0%), although specificity is lower, with nine FPs (78.6%; 95% CI 62.8% to 89.2%). Data are shown in Table 22.
| MSI | Reference standard | ||
|---|---|---|---|
| Positive | Negative | Total | |
| Positive | 13 | 9 | 22 |
| Negative | 0 | 33 | 33 |
| Total | 13 | 42 | 55 |
Summary of test accuracy studies
Nine primary research studies and one technology appraisal were identified for inclusion in this review. Four studies investigated MSI testing and eight investigated IHC. As tests to identify LS are a rapidly evolving area, and in the knowledge that a TA was already in existence which had identified studies up to 2005, only studies from 2005 onwards were included in this review.
Overall, the quality of the studies was mixed and, as many studies were not directly comparable owing to variations in design, the data could not be pooled. The results are summarised in Tables 23 and 24.
| First author | Patients (n) | Reference standard | Protein | TP | TN | FP | FN | Sensitivity, % (95% CI, %) | Specificity, % (95% CI, %) |
|---|---|---|---|---|---|---|---|---|---|
| Barrow 201042 | 68 | No details Germline testing performed before recruitment |
MLH1 | NR | NR | NR | NR | 100 (84 to 100) | 91.5 (79.6 to 97.6) |
| PMS2 | NR | NR | NR | NR | 95.2 (76.2 to 99.9) | 91.5 (79.6 to 97.6) | |||
| MSH2 | NR | NR | NR | NR | 87.5 (61.7 to 98.4) | 88.5 (76.5 to 95.6) | |||
| MSH6 | NR | NR | NR | NR | 81.3 (54.4 to 96.0) | 80.8 (67.5 to 90.4) | |||
| Barrow 201120 | 42 | No details Germline testing performed before recruitment |
MLH1 | NR | NR | NR | NR | 89.5 (66.9 to 98.7) | 78.3 (56.3 to 92.5) |
| MSH2 | NR | NR | NR | NR | 86.7 (59.5 to 98.3) | 77.8 (57.7 to 91.4) | |||
| Becouarn 200543 | 197a | Direct sequencing with search for large mutations if point mutation not identified | MLH1 and MSH2 | 11 | 2 | 5 | 4 | 73.3 (44.9 to 92.2)b | 28.6 (3.7 to 71.0)b |
| Limburg 201144 | 195 | Direct sequencing with Southern blot and MLPA | MLH1, MSH2 and MSH6 | 6c | 136c | 12c | 1c | 85.7 (42.1 to 99.6)b | 91.9 (86.3 to 95.7)b |
| Niessen 200621 | 281 | Direct sequencing and MLPA | MLH1 | NR | NR | NR | NR | 80 (38 to 96) | 89 (84 to 93) |
| MSH2 | NR | NR | NR | NR | 100 (57 to 100) | 96 (92 to 98) | |||
| MSH6 | NR | NR | NR | NR | 86 (46 to 97) | 93 (88 to 96) | |||
| Shia 200511 | 110a | Direct sequencing and large deletion analysis | MLH1, MSH2 and MSH6 | 21 | 34 | 4 | 5 | 80.8 (60.6 to 93.4)b | 89.5 (75.2 to 97.1)b |
| Southey 200539 | 131d | Direct sequencing and MLPA | MLH1, MSH2 and MSH6 | 18 | 79 | 8 | 0 | 100 (82 to 100) | 91 (83 to 96) |
| Stomorken 200545 | 250e | Direct sequencing and large rearrangement analysis | MLH1, MSH2 and MSH6 | 13 | 1 | 7 | 0 | 100 (75.3 to 100.0)b | 12.5 (0.3 to 52.7)b |
| First author | Patients (n) | Reference standard | Panel of markers | TP | TN | FP | FN | Sensitivity, % (95% CI, %) | Specificity, % (95% CI, %) |
|---|---|---|---|---|---|---|---|---|---|
| Niessen 200621 | 281a | Direct sequencing and MLPA |
BAT25
BAT26 D2S123 D5S346 D17S250 |
22 | 160 | 75 | 3 | 88.0 (68.8 to 97.5)b | 68.1 (61.7 to 74.0)b |
| Shia 200511 | 110c | Direct sequencing and large deletion analysis |
BAT25
BAT26 BAT40 PAX6 D2S123 D17S250 MYCL1 |
24 | 31 | 6 | 0 | 100 (85.8 to 100.0)b | 84 (68.0 to 93.8)b |
| Southey 200539 | 131a | Direct sequencing and MLPA |
D5S346
D17S250 D2S123 BAT25 BAT26 BAT40 MYB TGFβRII IGFIIR BAX |
17 | 70 | 17 | 1 | 94 (73 to 100) | 80 (71 to 88) |
| Wolf 200646 | 55 | Sequence analysis |
D5S346
HSCAP53L D2S123 BAT26 D18S34 Or D5S82 D2S134 D13S175 D11S904 BAT25 |
13 | 33 | 9 | 0 | 100 (71.7 to 100.0) | 78.6 (62.8 to 89.2) |
Immunohistochemistry
Immunohistochemistry is a technique in which the abnormal staining of a specific MMR protein is related to the underlying gene defect. As such, this test has the advantage of directing further genetic mutation analysis. Its sensitivity is, however, limited by tissue fixation, the variability of staining and the problem of inactive but intact proteins (e.g. caused by methylation of MLH1 in sporadic CRC). 1
The sensitivity and specificity for IHC are wide-ranging, varying from 73.3% to 100.0% and from 12.5% to 100.0%, respectively, although not all studies searched for all proteins and some of the results were combined. The raw data were not supplied for all studies and therefore could not be verified. CIs were generally wide and sample sizes small. Clearly, specificity is the greatest concern with IHC, as the high number of FPs means that individuals may be told they have LS when they do not. Furthermore, those studies recruiting from a population that had no prior testing may include an increased number of FPs due to MLH1 methylation found in sporadic CRC. A test is available to detect methylation of MLH1. However, no studies that tested for MLH1 methylation were identified as includable for this review.
A major methodological concern was that often the reference standard was applied according to the results of the index test. Therefore, because of attempts to minimise costs, only patients testing positive for LS by IHC or MSI received germline testing. As a result, this introduced an element of bias and often caused a confusing flow of patients through the study and difficulties creating a 2 × 2 table. Another concern was that IHC is dependent on the quality of the staining and the experience of the pathologist; however, there was rarely any indication of initial results being confirmed by a second party.
Microsatellite instability
The MSI test uses preselected markers to identify the existence of defective MMR within vulnerable DNA sequences. Although there is a standardised panel of five markers, many laboratories may use 10 or more mono- or dinucleotide sequences. As displayed in Table 24, no two studies included in this review have used the same panel and therefore a comparison is difficult and results vary.
The sensitivity for MSI ranged from 88% to 100% and specificity from 68 to 84%. The study by Niessen and colleagues (2006)21 used only five markers and displayed the lowest sensitivity and specificity (88% and 68%, respectively); however, the study reported by Wolf and colleagues (2006)46 also used five markers with improved results (sensitivity 100% and specificity 79%). In all cases the CIs were wide, signifying heterogeneity. As with IHC, not all patients received the reference standard and no information was given on robustness of the test, for example number of sample replicates.
Chapter 4 Assessment of cost-effectiveness: systematic review
Systematic review of existing cost-effectiveness evidence
The aim of this section is to identify and assess cost-effectiveness studies related to the identification and management of persons with LS. The assessment of cost-effectiveness comprises a systematic review of the literature on the cost-effectiveness of identification and management of persons with LS.
Methods
Searches of electronic databases were devised by a trained information specialist (CC) and applied to the following databases:
-
MEDLINE (via Ovid)
-
MEDLINE In-Process & Other Non-Indexed Citations (via Ovid)
-
EMBASE (via Ovid)
-
PsycINFO (via Ovid)
-
HMIC (via Ovid)
-
EconLit (via EBSCOhost)
-
CINAHL (via EBSCOhost)
-
Web of Science (via ISI)
-
The Cochrane Library
-
NRR
-
Web of Science Proceedings
-
Current Controlled Trials
-
ClinicalTrials.gov
-
FDA website
-
EMEA website.
A search filter was used to identify economic evaluations, as used for identifying studies for inclusion in the NHS Economic Evaluation Database (NHS EED). No publication date limit was imposed. Searching was limited to the English language and to human-only populations.
Study selection criteria and procedures
Titles and abstracts obtained from searching were screened by four reviewers (NH, CH, RM and TS) using inclusion criteria described in Table 25, with disagreements resolved by discussion between two reviewers (NH and TS). An inclusion criterion relating to guidelines was included to assist in the understanding of LS. Full papers of citations that met these criteria were obtained and assessed for inclusion in the review by two reviewers (NH and TS), with disagreements resolved by discussion by the same two reviewers.
| PICO criteria | Inclusion criteria |
|---|---|
| Population | Persons who have or may have LS |
| Intervention | Any of the following (including combinations):
|
| Comparator | Current clinical practice (may or may not include efforts to identify LS) |
| Outcomes | Any of the following:
|
| Study type | Any of the following:
|
Study quality assessment
A quality appraisal was conducted on cost-effectiveness studies or other economic evaluations, using the well-established Drummond checklist,47 following advice from CH.
Data extraction strategy
For those studies which were of relevance to the current decision problem, data were extracted by two researchers (NH, TS) into three data extraction tables: one to describe the study design of each economic evaluation, one to describe the main results, and the third a checklist of review-specific criteria that the two reviewers had previously agreed upon. Examples of the study design and results tables are provided in Tables 26 and 27, with the review-specific criteria checklists presented in Tables 28–30, respectively.
| Study | Ladabaum et al. 201148 |
| Publication type | Peer-reviewed journal paper |
| Setting, perspective | USA, third-party payer |
| Industry role/conflicts of interest | Primary funding source: NIH Authors’ potential conflicts of interest: Epigenomics, Quest Diagnostics, Abbott Molecular, Given Imaging, Roche Diagnostics, GE Healthcare, GeneNews, Archimedes |
| Population | All persons with newly diagnosed CRC and their relatives |
| Study purpose | Cost-effectiveness analysis of strategies to identify LS |
| Outcomes measured | Life-years, cancer cases and deaths, costs and ICERs |
| Diagnostic strategies | Referent strategy (no attempt to identify LS) Clinical criteria strategies
|
| Treatment strategies | Surveillance: annual colonoscopy, TVU and endometrial sampling Prophylactic surgery: TAHBSO at age 40 years, subtotal colectomy (unclear when) |
| Study approach | Decision tree with Markov subtrees |
| Health states | Unclear (includes at least healthy, CRC, EC, OC and dead) |
| Model duration (cycle length) | Lifetime (1 year) |
| Uncertainty analysis | Scenario analyses Univariate sensitivity analyses Partial PSA (not all parameters varied) |
| Base year prices | 2010 US dollars |
| Discount rate | 3% |
| Study | Ladabaum et al. 201148 |
| Analysis year | 2010 |
| Summary of findings | Key findings If clinical criteria strategies are excluded then IHC with BRAF is a clinically effective and cost-effective measure with ICER of $36,206 per LY gained (0.2248 LYs gained at a cost of $8139) Referent strategy resulted in 23.5071 discounted LYs per person at a discounted cost of $11,242 Clinical criteria strategies resulted in 23.5565–23.7292 discounted LYs per person at a discounted cost of $12,933–18,737 per person. Strategies using IHC as a second-line test were generally cheaper but provided fewer discounted LYs than those going direct from clinical criteria to germline testing. Only Bethesda and MMRpro strategies are on the cost-effectiveness frontier (others dominated or extended dominated) Tumour-testing strategies resulted in 23.7319–23.7711 discounted LYs per person at a discounted cost of $19,551–23,642. Strategies involving BRAF testing dominated equivalent strategies without BRAF testing Up-front germline testing resulted in 23.8047 LYs per person at a cost of $33,492 per person (ICER $293,155 per LY gained) |
| Results of sensitivity analysis | Scenario analyses
Cost-effectiveness of IHC with BRAF testing vs. the referent strategy was sensitive to (i.e. ICER exceeded $50,000 per LY for values within the analysis):
(Note: not all parameters about which there was uncertainty were varied in the PSA) CEACs for individual strategies vs. the referent strategy are overlaid in figure 3, showing that apart from (MSI + IHC, MSI + IHC/BRAF testing, up-front germline testing) all strategies were cost-effective (ICER ≤ $50,000/LY with p > 0.5) compared to referent strategy The authors indicate that at a threshold of $50,000 per LY gained, IHC/BRAF testing is the optimal strategy in 53% of iterations |
| Criteria | Brown et al. 199622 | Ramsey et al. 200123 | Ramsey et al. 200349 | Kievit et al. 200550 | Breheny et al. 200651 | DACEHTA 200752 | Olsen et al. 200753 | Mvundura et al. 201054 | Dinh et al. 201155 | Kwon et al. 201156 | Ladabaum et al. 201148 | Wang et al. 201257 | Wang et al. 201258 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diagnosis | |||||||||||||
| Input population contains CRC patients | ✗ | ✓ | ✓ | ✓ | N/A | ? | ? | ✓ | ✓ | ✗ | ✓ | ✓ | N/A |
| Input population contains EC patients | ✗ | ✗ | ✗ | ✗ | N/A | ? | ? | ✗ | ✗ | ✓ | ✗ | ✗ | N/A |
| Input population contains OC patients | ✗ | ✗ | ✗ | ✗ | N/A | ? | ? | ✗ | ✗ | ✗ | ✗ | ✗ | N/A |
| Input population contains other LS-associated cancer patients | ✗ | ✗ | ✗ | ✗ | N/A | ? | ? | ✗ | ✗ | ✗ | ✗ | ✗ | N/A |
| There are diagnostic strategies which do not include FH | ✓ | ✗ | ✓ | ✗ | N/A | ? | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | N/A |
| Patients are tested for MLH1 mutations | ✓ | ✓ | ✓ | ✓ | ? | ? | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ? |
| Patients are tested for MSH2 mutations | ✓ | ✓ | ✓ | ✓ | ? | ? | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ? |
| Patients are tested for MSH6 mutations | ? | ✗ | ✗ | ✗ | ? | ? | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ? |
| Patients are tested for PMS2 mutations | ? | ✗ | ✗ | ✗ | ? | ? | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ? |
| Appropriate informed consent and counselling is included | ✓ | ✓ | ✓ | ✓ | ✓ | ? | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ |
| The study considers patients declining counselling | ✗ | ✓ | ? | ? | ✗ | ? | ✗ | ✓ | ✗ | ✓ | ? | ? | ✗ |
| The study considers patients declining genetic testing | ✗ | ✓ | ✓ | ✓ | ✗ | ? | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ |
| The effect of diagnostic errors is considered | ✓ | ✓ | ✓ | ✓ | ✗ | ? | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ |
| The study considers the impact of a national strategy on the proportion of patients who do not already know their LS status | ✗ | ✗ | ✗ | ✗ | ✗ | ? | ✗ | ✗ | ? | ✗ | ✗ | ✗ | ✗ |
| Management | |||||||||||||
| The study considers CRC | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| The study considers EC | ✗ | ✗ | ✗ | ✗ | * | ✗ | ✗ | ✗ | ✓ | ✗ | ✓ | ✓ | ✗ |
| The study considers OC | ✗ | ✗ | ✗ | ✗ | * | ✗ | ✗ | ✗ | ✗ | ✗ | ✓ | ✓ | ✗ |
| The study considers other LS-associated cancers | ✗ | ✗ | ✗ | ✗ | * | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| The study considers interactions of cancers appropriately | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | ✓ | N/A | ? | ? | N/A |
| Colonoscopic surveillance in the study is explicitly justified (e.g. by reference to guidelines or clinical practice) | ✗ | ✓ | ✓ | ✗ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ | ✗ | ✓ |
| The study considers patients declining recommended surveillance | ✗ | ✓ | ✓ | ✗ | ✗ | ? | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| The study considers the difference in incidence of CRC between males and females | ✗ | ✗ | ✗ | ? | ? | ? | ✗ | ✗ | ✓ | N/A | ✓ | ✓ | ✗ |
| The study considers the difference in incidence of LS-associated cancers between mutations of different MMR genes | ✗ | ✗ | ✗ | ✗ | ✗ | ? | ✗ | ✗ | ✓ | ? | ✗ | ✗ | ✗ |
| The study accounts for the improved survival of LS CRCs relative to sporadic CRCs | ✗ | ✓ | ? | ✗ | ✗ | ? | ✓ | ✓ | ✓ | ? | ✓ | ✓ | ✗ |
| The study considers the potential psychological impact of genetic testing | ✗ | ✗ | ✗ | ✗ | ✗ | ? | ✗ | ✓ | ✓ | ✗ | ✗ | ✓ | ✗ |
| Criteria | Debniak et al. 200034 | Reyes et al. 200259 | Pigatto et al. 200460 | Pinol et al. 200561 | Engel et al. 200662 | Bessa et al. 200863 | Yan et al. 200864 | Palomaki et al. 200965 | Ramsoekh et al. 200966 | Resnick et al. 200967 | Horwitz et al. 201068 | Gudgeon et al. 201169 | Perez-Carbonell et al. 201170 | Williams et al. 201171 | Gausachs et al. 201272 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Input population contains CRC patients | ✓ | ✓ | ✓ | ✓ | ? | ✓ | ✓ | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Input population contains EC patients | ✗ | ✗ | ✗ | ✗ | ? | ✗ | ✗ | ✗ | ✗ | ✓ | ✗ | ✗ | ✗ | ✗ | ✗ |
| Input population contains OC patients | ✗ | ✗ | ✗ | ✗ | ? | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| Input population contains other LS-associated cancer patients | ✗ | ✗ | ✗ | ✗ | ? | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| There are diagnostic strategies which do not include FH | ✗ | ✓ | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Patients are tested for MLH1 mutations | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Patients are tested for MSH2 mutations | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ |
| Patients are tested for MSH6 mutations | ✗ | ✗ | ✗ | ✗ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ |
| Patients are tested for PMS2 mutations | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✓ | ✗ | ✗ | ✓ | ✓ | ✓ | ✓ | ✗ |
| Appropriate informed consent and counselling is included | ✗ | ✓ | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ | ? | ? |
| The study considers patients declining counselling | ✗ | ✗ | ✗ | ? | ✗ | ✗ | ✗ | ✓ | ? | ✗ | ✓ | ✗a | ✗ | ? | ✗ |
| The study considers patients declining genetic testing | ✗ | ✓b | ✗ | ✓ | ✗ | ✗ | ✗ | ✓ | ? | ✗ | ✓ | ✓ | ✗ | ? | ✗ |
| The effect of diagnostic errors is considered | ✗ | ✓ | ✗ | ✓ | ✗ | ✗ | ✓ | ✓ | ✓ | ✗ | ✗ | ✓ | ✗ | ? | ✓ |
| The study considers the impact of a national strategy on the proportion of patients who do not already know their LS status | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ | ✗ |
| Criteria | Vasen et al. 199873 | Dunlop et al. 200274 | Kwon et al. 200875 | Yang et al. 201176 |
|---|---|---|---|---|
| The study considers CRC | ✓ | ✓ | ✓ | ✗ |
| The study considers EC | N/A | ✗ | ✓ | ✓ |
| The study considers OC | N/A | ✗ | ✓ | ✓ |
| The study considers other LS-associated cancers | ✗ | ✗ | ✗ | ✗ |
| The study considers interactions of cancers appropriately | N/A | N/A | ? | ? |
| Colonoscopic surveillance in the study is explicitly justified (e.g. with reference to guidelines or clinical practice) | ✓ | ✓ | N/A | N/A |
| The study considers patients declining recommended surveillance | ✗ | ✗ | ✗ | ✗ |
| The study considers the difference in incidence of CRC between males and females | N/A | ✗ | N/A | N/A |
| The study considers the difference in incidence of LS-associated cancers between mutations of different MMR genes | ✗ | ✗ | ✗ | ✗ |
| The study accounts for the improved survival of LS CRCs relative to sporadic CRCs | ? | ✗ | ✓ | N/A |
| The study considers the potential psychological impact of genetic testing | ✗ | ✗ | ✗ | ✗ |
In the study design table, the sections included study, publication type, setting/perspective, industry role/conflicts of interest, population, study purpose, outcomes measured, diagnostic strategies, treatment strategies, study approach, health states, model duration/cycle length, the approach to uncertainty analysis, base-year prices and discount rate.
In the results table, the components were the analysis year, the base-case results and the main sensitivity analysis results.
Items on the review-specific checklist included the types of cancers included in the analysis (both in the input population and in the management section); whether or not the interactions of these cancers in the long term were considered appropriately; whether or not the diagnostic strategies used FH; which MMR gene mutations were tested for; whether or not differences in LS cancer incidence due to these MMR gene mutations, and the improved survival of LS CRCs relative to sporadic CRCs, were accounted for; whether or not the study considered the difference in CRC incidence between males and females; whether or not adherence to counselling, genetic testing and management strategies was included; whether or not the psychological impact of genetic testing was accounted for; and whether or not the study assessed the impact of diagnostic errors.
Synthesis of extracted evidence
The evidence base was assessed using narrative synthesis supported by abridged data extraction tables.
Results
The flow diagram of papers is summarised in Figure 2. In summary, 2036 citations were identified, 227 of which were ordered in full. Three of these could not be retrieved but the information available from the titles and abstracts did not suggest that they would have a high probability of remaining included studies. Of the 224 which were retrieved, 7 were duplicates and 119 were excluded. Of the remaining 98, 1 was an opinion piece, 55 were guidelines only, 3 were previous reviews of cost-effectiveness studies, 5 were purely effectiveness models (kept for their insight into modelling this problem) and the remaining 34 included papers represented cost-effectiveness studies of some form. An additional three cost-effectiveness papers, one of which was an update of a previously identified paper, and one additional review were identified during the update search. Of the final 37 cost-effectiveness papers, 16 looked at the short-term cost-effectiveness of identifying LS, 5 looked only at the long-term cost-effectiveness of management of LS and 16 examined the long-term impact on cost-effectiveness of both the strategies to identify and manage LS. Of the 16 sources that only looked at short-term cost-effectiveness, there were 15 distinct studies (one of the papers was an abstract of a study written up in full in another paper). Of the five that looked purely at management of LS, only four were distinct studies (as one paper was an abstract of preliminary results which were reported in full in another paper). Of the 16 sources that looked at the cost-effectiveness of both the strategies to identify and manage LS, there were two papers on the same study, one abstract of another paper and a commentary on a paper, making the total number of distinct studies 13. The most common reasons for exclusion were on the basis of a study design or population not relevant to the review.
FIGURE 2.
Flow diagram of the search, retrieval and inclusion of articles in the systematic review of evidence on the economic evaluations to identify and manage LS.
We did not formally review the papers that included guidelines, but they were generally consistent in their suggestions for the management of LS, i.e. routine surveillance for CRC by 1- to 2-yearly colonoscopy. As our criteria for guidance were quite inclusive, we included a wide range of sources for this, from different study groups including working groups and HTA groups, among others.
Summary of previous cost-effectiveness reviews
There were four papers identified whose cost-effectiveness section consisted purely of a review of previous cost-effectiveness studies,77–80 as well as two cost-effectiveness model papers65,69 that contained a review section. The purpose of reviewing these studies was to investigate whether or not a systematic review had been conducted previously, and also to check for any additional studies that our search may have missed.
Only two reviews77,78 were studies from the last 10 years (2006 and 2012, respectively) and were clear on their search strategy and methods, but neither of them looked exclusively at LS. Furthermore, neither review identified more than four studies related to LS. Between them they identified only seven separate reviews,23,49,50,59,61,81,82 of which all but one had already been identified by our review. The one study we had not identified, by Hagen and colleagues,82 was written in German and therefore excluded from our review. The only results presented by Antonanzas and colleagues were from the Barrow and colleagues (2008)81 paper, which we excluded on the basis of population. The four studies23,49,50,59 reported by Rogowski and colleagues77 were all included in our review, which had a more comprehensive set of studies.
Of the other two papers with a review of previous cost-effectiveness studies, only one could be considered an actual review. The other,79 from 2001, mentions the findings of the 1998 study by Vasen and colleagues,73 but no further details are given. This study was excluded from our review on the basis of study design. The other review,80 from 2008, identified six studies, two of which were not identified by Rogowski and colleagues. 53,73
Two of the included cost-effectiveness studies also conducted reviews of previous cost-effectiveness studies. 65,69 Again, the details of these reviews are not in depth and did not identify any additional papers that we had not already screened.
In the reports where authors made conclusions about the studies they had identified, the main conclusion was that strategies to identify and manage LS could be cost-effective, depending on the cost-effectiveness threshold of the relevant health-care provider, but that there was no conclusive evidence as to which strategies would be most cost-effective. Phillips and colleagues (2008)80 suggested that further research was needed, particularly as the understanding of LS has changed in recent years. This assumption also explained the conclusion of Palomaki and colleagues (2009),65 who criticised the studies they had included51,53,73,83 for being inadequate as they did not include certain tests and did not address the differences in performance of these tests for different MMR gene mutations. These reviews identified 12 studies that were relevant to our review; this was significantly fewer than the number our search had identified, suggesting that there might be further work that these previous reviews had missed and justifying the need for our own systematic review. As our search had already identified the 12 studies that these reviews had also included, we were confident of the completeness of our review.
Summary of previous effectiveness models
As part of our review, we identified and assessed previous studies that included effectiveness models. This was to provide insight into what modelling had previously been done in this area and to help guide our own modelling efforts. We identified five previous studies that included some attempt at modelling without costs. 83–87 Four of these studies83,85–87 looked exclusively at management strategies for people with LS; three related to the management (such as use of aggressive colorectal surgery to prevent further CRCs) and prevention of CRC, and one examined the prevention of EC. The study on EC85 appeared to have been later updated to include costs, in a paper by Yang and colleagues (2011). 76 The general finding of these studies was that management strategies were effective for LS patients and that aggressive prophylactic surgery [total abdominal hysterectomy with bilateral salpingo-oophorectomy (TAHBSO) for EC or proctocolectomy/total colectomy for CRC] was generally the most effective strategy. All of the CRC studies employed some form of Markov modelling and the EC model was quoted to be a decision model built in TreeAge 2004 (TreeAge Software, Inc., Williamstown, MA, USA). The final paper84 was a resource minimisation study employing a test accuracy study and a decision tree to compare strategies of testing for LS, and finding that a strategy that included BRAF testing was more effective than one without (reducing the number of genetic tests by 17%, for the same number of mutations identified).
These models were not of great use as they each focused on one aspect of LS and so did not give much insight into how to model both the diagnostic and management pathways for LS, including more than one cancer, as was required of our model. They did, however, indicate that a new model may be necessary.
Summary of cost-effectiveness studies
For the purposes of the review, we split the 32 cost-effectiveness studies into the three subgroups: those that looked at the short-term cost-effectiveness of identifying LS; those that looked only at the long-term cost-effectiveness of management of LS; and those that examined the long-term impact on cost-effectiveness of both the strategies to identify and manage LS.
Given the large number of studies which our review identified, we do not report each study in great detail, preferring to identify important similarities and differences and potential areas of improvement for which our model was intended to provide information. The summary tables of characteristics and results are shown below (see Tables 31–36). Our model was required to look at the long-term consequences of diagnosis and management of LS, so these studies were the main focus of our review. As such, the other two subgroups are reported in less depth, though their summary tables are reported in similar detail.
Following on from the summaries, we also present the results of our quality appraisal for these studies.
Lynch syndrome diagnosis-only studies
This was the largest subgroup of cost-effectiveness studies identified, including 15 studies looking exclusively at the short-term cost-effectiveness of strategies to diagnose LS. 34,59–72 All studies were published between 2000 and 2012 and summary details of each study can be found in Tables 31 and 32.
| Author and year | Setting, perspective | Population | Study purpose | Study approach | Diagnostic comparators | ||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Debniak et al. 200034 | Poland, health-care provider | Newly diagnosed CRC patients (FAP excluded) | Cost-effectiveness analysis of strategies to identify MLH1/MSH2 mutations | Prospective population-based study Decision tree |
Test 1Test 2Test 3Test 4PCDGTPCDMSIGTPCDIHCGTPCDIHCGT (IHC positive)MSIGT | Test 1 | Test 2 | Test 3 | Test 4 | PCD | GT | PCD | MSI | GT | PCD | IHC | GT | PCD | IHC | GT (IHC positive) | MSI | GT | |||||||||||||||||||||||||||
| Test 1 | Test 2 | Test 3 | Test 4 | ||||||||||||||||||||||||||||||||||||||||||||||
| PCD | GT | ||||||||||||||||||||||||||||||||||||||||||||||||
| PCD | MSI | GT | |||||||||||||||||||||||||||||||||||||||||||||||
| PCD | IHC | GT | |||||||||||||||||||||||||||||||||||||||||||||||
| PCD | IHC | GT (IHC positive) | |||||||||||||||||||||||||||||||||||||||||||||||
| MSI | GT | ||||||||||||||||||||||||||||||||||||||||||||||||
| Reyes et al. 200259 | USA, health-care system | Newly diagnosed CRC patients who agree to GT and their FDRs | Cost-effectiveness analysis of strategies to identify LS | Decision model | StrategyTest 1Test 2Test 31AC IIGT2mCCMSIGT3MSIGTmCC, modified guidelines (personal or family history of CRC/EC, or diagnosed < 50 years old).GT here is only for MLH1 and MSH2. A fourth ‘mixed’ strategy uses both sets of clinical guidelines and pathway is decided by which the patient fulfils FDRs of mutation-positive patients receive GT |
Strategy | Test 1 | Test 2 | Test 3 | 1 | AC II | GT | 2 | mCC | MSI | GT | 3 | MSI | GT | mCC, modified guidelines (personal or family history of CRC/EC, or diagnosed < 50 years old). | GT here is only for MLH1 and MSH2. | ||||||||||||||||||||||||||||
| Strategy | Test 1 | Test 2 | Test 3 | ||||||||||||||||||||||||||||||||||||||||||||||
| 1 | AC II | GT | |||||||||||||||||||||||||||||||||||||||||||||||
| 2 | mCC | MSI | GT | ||||||||||||||||||||||||||||||||||||||||||||||
| 3 | MSI | GT | |||||||||||||||||||||||||||||||||||||||||||||||
| mCC, modified guidelines (personal or family history of CRC/EC, or diagnosed < 50 years old). | |||||||||||||||||||||||||||||||||||||||||||||||||
| GT here is only for MLH1 and MSH2. | |||||||||||||||||||||||||||||||||||||||||||||||||
| Pigatto et al. 200460 | UK, health-care provider | Families referred to regional genetics service | Cost-effectiveness analysis of strategies to identify LS mutations | Clinical study Decision tree |
Probands Test 1Test 2Test 3Test 4CCGT2CCIHC2GT1CCMSI1GT2CCIHC2GT1MSI1GT1GT1, directed genetic test; GT2, genetic test for mutations in MLH1 and MSH2; IHC2, immunohistochemical analysis of MLH1 and MSH2; MSI1, microsatellite instability analysis of single marker (BAT26).CC: AC, modified Amsterdam (MA), Bethesda criteria 1–3 (B1–3), Bethesda (B), all patients (Any CC). |
Test 1 | Test 2 | Test 3 | Test 4 | CC | GT2 | CC | IHC2 | GT1 | CC | MSI1 | GT2 | CC | IHC2 | GT1 | MSI1 | GT1 | GT1, directed genetic test; GT2, genetic test for mutations in MLH1 and MSH2; IHC2, immunohistochemical analysis of MLH1 and MSH2; MSI1, microsatellite instability analysis of single marker (BAT26). | CC: AC, modified Amsterdam (MA), Bethesda criteria 1–3 (B1–3), Bethesda (B), all patients (Any CC). | |||||||||||||||||||||||||
| Test 1 | Test 2 | Test 3 | Test 4 | ||||||||||||||||||||||||||||||||||||||||||||||
| CC | GT2 | ||||||||||||||||||||||||||||||||||||||||||||||||
| CC | IHC2 | GT1 | |||||||||||||||||||||||||||||||||||||||||||||||
| CC | MSI1 | GT2 | |||||||||||||||||||||||||||||||||||||||||||||||
| CC | IHC2 | GT1 | |||||||||||||||||||||||||||||||||||||||||||||||
| MSI1 | GT1 | ||||||||||||||||||||||||||||||||||||||||||||||||
| GT1, directed genetic test; GT2, genetic test for mutations in MLH1 and MSH2; IHC2, immunohistochemical analysis of MLH1 and MSH2; MSI1, microsatellite instability analysis of single marker (BAT26). | |||||||||||||||||||||||||||||||||||||||||||||||||
| CC: AC, modified Amsterdam (MA), Bethesda criteria 1–3 (B1–3), Bethesda (B), all patients (Any CC). | |||||||||||||||||||||||||||||||||||||||||||||||||
| Pinol et al. 200561 | Spain, national health-care system | Newly diagnosed CRC patients (FAP and inflammatory bowel patients excluded) | Cost-effectiveness analysis of testing for MLH1 and MSH2 genes | Prospective, multicentre population-based study in community hospitals Decision tree |
Combinations of clinical criteria, FH criteria (AC II and Revised Bethesda), MSI testing, IHC and gene testing (MLH1 and MSH2) | ||||||||||||||||||||||||||||||||||||||||||||
| Engel et al. 200662 | Germany, health-care provider | Families selected by the German HNPCC consortium | Cost-minimisation analysis of strategies to identify LS | Clinical study Decision tree with logistic regression |
Current practice Simultaneous IHC and MSI analysis, with any abnormalities triggering GT Proposed strategy Sequential IHC and MSI analysis, order determined by prediction of results from clinical criteria to minimise expected cost |
||||||||||||||||||||||||||||||||||||||||||||
| Bessa et al. 200863 | Spain, health-care provider | Newly diagnosed CRC patients | Cost-minimisation analysis of strategies to identify LS | Prospective, multicentre population-based study Decision tree |
Test 1Test 2Test 3Test 4MSIGTMSIBRAFGTIHCGTIHCBRAFGTCCMSIGTCCMSIBRAFGTCCIHCGTCCIHCBRAFGTBRAF if MSI-H or MLH1 absent on IHC.CC: Revised Bethesda guidelines. | Test 1 | Test 2 | Test 3 | Test 4 | MSI | GT | MSI | BRAF | GT | IHC | GT | IHC | BRAF | GT | CC | MSI | GT | CC | MSI | BRAF | GT | CC | IHC | GT | CC | IHC | BRAF | GT | BRAF if MSI-H or MLH1 absent on IHC. | CC: Revised Bethesda guidelines. | ||||||||||||||
| Test 1 | Test 2 | Test 3 | Test 4 | ||||||||||||||||||||||||||||||||||||||||||||||
| MSI | GT | ||||||||||||||||||||||||||||||||||||||||||||||||
| MSI | BRAF | GT | |||||||||||||||||||||||||||||||||||||||||||||||
| IHC | GT | ||||||||||||||||||||||||||||||||||||||||||||||||
| IHC | BRAF | GT | |||||||||||||||||||||||||||||||||||||||||||||||
| CC | MSI | GT | |||||||||||||||||||||||||||||||||||||||||||||||
| CC | MSI | BRAF | GT | ||||||||||||||||||||||||||||||||||||||||||||||
| CC | IHC | GT | |||||||||||||||||||||||||||||||||||||||||||||||
| CC | IHC | BRAF | GT | ||||||||||||||||||||||||||||||||||||||||||||||
| BRAF if MSI-H or MLH1 absent on IHC. | |||||||||||||||||||||||||||||||||||||||||||||||||
| CC: Revised Bethesda guidelines. | |||||||||||||||||||||||||||||||||||||||||||||||||
| Yan et al. 200864 | China, health-care system (not stated) | CRC patients taken from Hereditary CRC Registry Center of Changhai hospital | Cost-effectiveness analysis of strategies to identify LS | Prospective population-based study | Four clinical/FH criteria: AC II, S-HNPCC (suspected LS criteria: at least two FDRs with LS-related cancers and multiple colorectal tumours, or at least one HNPCC-associated cancer diagnosed at < 50 years of age), E-CRC and L-CRC. These followed by MSI, IHC (MLH1 methylation testing included where MLH1 staining is absent and there is insufficient FH), GT | ||||||||||||||||||||||||||||||||||||||||||||
| Palomaki et al. 200965 | USA, health-care provider | Newly diagnosed CRC patients and their relatives | Cost-effectiveness analysis of strategies to identify LS | Decision model (decision tree) | Probands Test 1Test 2Test 3GT3MSIGT3IHCGT1+IHCBRAF (MLH1 absent)GT1+GT1+GT1+, directed genetic testing of one or more genes according to IHC results; GT3, sequential genetic testing of MSH2, MLH1 and MSH6. Relatives Predictive testing if mutation identified in proband |
Test 1 | Test 2 | Test 3 | GT3 | MSI | GT3 | IHC | GT1+ | IHC | BRAF (MLH1 absent) | GT1+ | GT1+ | GT1+, directed genetic testing of one or more genes according to IHC results; GT3, sequential genetic testing of MSH2, MLH1 and MSH6. | |||||||||||||||||||||||||||||||
| Test 1 | Test 2 | Test 3 | |||||||||||||||||||||||||||||||||||||||||||||||
| GT3 | |||||||||||||||||||||||||||||||||||||||||||||||||
| MSI | GT3 | ||||||||||||||||||||||||||||||||||||||||||||||||
| IHC | GT1+ | ||||||||||||||||||||||||||||||||||||||||||||||||
| IHC | BRAF (MLH1 absent) | GT1+ | |||||||||||||||||||||||||||||||||||||||||||||||
| GT1+ | |||||||||||||||||||||||||||||||||||||||||||||||||
| GT1+, directed genetic testing of one or more genes according to IHC results; GT3, sequential genetic testing of MSH2, MLH1 and MSH6. | |||||||||||||||||||||||||||||||||||||||||||||||||
| Ramsoekh et al. 200966 | Netherlands, health-care system (not stated) | Families referred to genetics laboratory. Index probands identified as youngest family member with CRC, who underwent MSI, IHC and/or GT | To analyse the cost-effectiveness of five prediction models for LS | Nationwide, single-centre study using patients referred from general practitioners Decision tree |
Prediction models: Leiden, PREMM1,2 (for MLH1, MSH2), Edinburgh, UK-Ams, UK-Alt (for MLH1, MSH2, MSH6) Strategies:
|
||||||||||||||||||||||||||||||||||||||||||||
| Resnick et al. 200967 | USA, third-party payer | Newly diagnosed EC patients | Cost-effectiveness analysis of strategies to identify LS | Decision model (decision tree) | Test 1Test 2AC IIGT3GT3Age < 60 yearsGT3IHCGT1+GT1+, test for mutations in MLH1 if MLH1 absent on IHC and patient aged < 60 years, or, test for mutations in MSH6 then MSH2 if either absent on IHC (regardless of age); GT3, test for mutations in MLH1, MSH2 and MSH6. | Test 1 | Test 2 | AC II | GT3 | GT3 | Age < 60 years | GT3 | IHC | GT1+ | GT1+, test for mutations in MLH1 if MLH1 absent on IHC and patient aged < 60 years, or, test for mutations in MSH6 then MSH2 if either absent on IHC (regardless of age); GT3, test for mutations in MLH1, MSH2 and MSH6. | ||||||||||||||||||||||||||||||||||
| Test 1 | Test 2 | ||||||||||||||||||||||||||||||||||||||||||||||||
| AC II | GT3 | ||||||||||||||||||||||||||||||||||||||||||||||||
| GT3 | |||||||||||||||||||||||||||||||||||||||||||||||||
| Age < 60 years | GT3 | ||||||||||||||||||||||||||||||||||||||||||||||||
| IHC | GT1+ | ||||||||||||||||||||||||||||||||||||||||||||||||
| GT1+, test for mutations in MLH1 if MLH1 absent on IHC and patient aged < 60 years, or, test for mutations in MSH6 then MSH2 if either absent on IHC (regardless of age); GT3, test for mutations in MLH1, MSH2 and MSH6. | |||||||||||||||||||||||||||||||||||||||||||||||||
| Horwitz et al. 201068 | USA, health-care provider | CRC patients undergoing resection meeting any of first three Bethesda criteria | Cost-effectiveness of reflex MSI testing for LS | Clinical study | MSI → (IHC) → GT | ||||||||||||||||||||||||||||||||||||||||||||
| Gudgeon et al. 201169 | USA, health-care organisation (Intermountain Healthcare) | Newly diagnosed CRC patients | Cost-effectiveness analysis of strategies to identify LS and budget impact analysis | Decision model | Test 1Test 2Test 3Test 4IHCGTIHCBRAFGTIHCMethylGTIHCBRAFMethylGTIHCMethylBRAFGTGT, genetic testing by sequencing; Methyl, methylation.BRAF and/or methylation follow IHC when abnormal on MLH1, other abnormalities go to GT. | Test 1 | Test 2 | Test 3 | Test 4 | IHC | GT | IHC | BRAF | GT | IHC | Methyl | GT | IHC | BRAF | Methyl | GT | IHC | Methyl | BRAF | GT | GT, genetic testing by sequencing; Methyl, methylation. | BRAF and/or methylation follow IHC when abnormal on MLH1, other abnormalities go to GT. | ||||||||||||||||||||||
| Test 1 | Test 2 | Test 3 | Test 4 | ||||||||||||||||||||||||||||||||||||||||||||||
| IHC | GT | ||||||||||||||||||||||||||||||||||||||||||||||||
| IHC | BRAF | GT | |||||||||||||||||||||||||||||||||||||||||||||||
| IHC | Methyl | GT | |||||||||||||||||||||||||||||||||||||||||||||||
| IHC | BRAF | Methyl | GT | ||||||||||||||||||||||||||||||||||||||||||||||
| IHC | Methyl | BRAF | GT | ||||||||||||||||||||||||||||||||||||||||||||||
| GT, genetic testing by sequencing; Methyl, methylation. | |||||||||||||||||||||||||||||||||||||||||||||||||
| BRAF and/or methylation follow IHC when abnormal on MLH1, other abnormalities go to GT. | |||||||||||||||||||||||||||||||||||||||||||||||||
| Perez-Carbonell et al. 201170 | Setting not stated, health-care provider | Newly diagnosed CRC patients | Cost-effectiveness of strategies to identify LS mutations | Population-based study | Test 1Test 2Test 3Test 4IHCMethylation (MLH1 absent)GTGTRevised Bethesda criteriaIHCMethylation (MLH1 absent)GTGT | Test 1 | Test 2 | Test 3 | Test 4 | IHC | Methylation (MLH1 absent) | GT | GT | Revised Bethesda criteria | IHC | Methylation (MLH1 absent) | GT | GT | |||||||||||||||||||||||||||||||
| Test 1 | Test 2 | Test 3 | Test 4 | ||||||||||||||||||||||||||||||||||||||||||||||
| IHC | Methylation (MLH1 absent) | GT | |||||||||||||||||||||||||||||||||||||||||||||||
| GT | |||||||||||||||||||||||||||||||||||||||||||||||||
| Revised Bethesda criteria | IHC | Methylation (MLH1 absent) | GT | ||||||||||||||||||||||||||||||||||||||||||||||
| GT | |||||||||||||||||||||||||||||||||||||||||||||||||
| Williams et al. 201171 | USA, health-care organisation (Intermountain Healthcare) | Newly diagnosed CRC patients | Follow-up to Gudgeon 201169 to analyse the cost-effectiveness of an age cut-off for testing of LS | Decision model | IHC followed by GT (GT not stated) | ||||||||||||||||||||||||||||||||||||||||||||
| Gausachs et al. 201272 | Spain? (not explicitly stated), health-care system | Newly diagnosed CRC patients with loss of MLH1 expression and their FDRs and SDRs | Cost-effectiveness analysis of using methylation in the diagnosis of LS | Decision tree | Probands
Germline testing if proband positive for LS |
||||||||||||||||||||||||||||||||||||||||||||
| Author and year | Outcomes measured | Base results | Sensitivity analysis approach | Main sensitivity analysis resultsa |
|---|---|---|---|---|
| Debniak et al. 200034 | Mutations detected Costs |
Dominated PCD → GT PCD → MSI → GT Not dominated PCD → IHC → GT detected five mutations at total cost of €4400 (ICER €880 per mutation) PCD → IHC → (MSI) → GT detected one additional mutation (ICER $6202 per additional mutation detected) |
N/A | N/A |
| Reyes et al. 200259 | Mutations detected Costs Incremental cost per mutation detected |
In order of efficiency (strategy 1 least effective, strategy 3 most effective) Strategy 2 vs. strategy 1 had ICER of $6832 per case detected, but strategy 2 extended dominated by strategy 4, which had an ICER of £6441 per case detected compared with strategy 1. Strategy 3 compared with strategy 4 had ICER of $51,151 per case detected |
Univariate sensitivity analyses | When AC II positive rate greater than 0.025, strategy 4 (mixed) is most effective and dominates strategy 3 Increasing the numbers of FDRs tested improves cost-effectiveness of all strategies |
| Pigatto et al. 200460 | Mutations detected Cost |
‘B’ strategies dominate ‘Any CC’ IHC2 strategies dominate MSI1 and IHC2 + MSI1 strategies Cost-effectiveness frontier: AC + IHC2: 10 mutations, €7150 B1–3 + IHC2: 14 mutations, ICER €1237 per mutation MA: 37 mutations, ICER €2441 per mutation B1–3: 45 mutations, ICER €3847 per mutation B: 49 mutations, €136,024, ICER €9256 per mutation |
N/A | N/A |
| Pinol et al. 200561 | Cost per mutation detected Accuracy |
Using Revised Bethesda guidelines before IHC ($13,837 per case detected) or MSI ($15,586 per case detected) was more cost-effective than directly using the tumour tests themselves (IHC $49,343 per case detected; MSI $41,782 per case detected) | N/A | N/A |
| Engel et al. 200662 | Mutations detected Costs |
Strategies equally sensitive and specific Using proposed strategy would achieve cost reduction of ≈ 25% |
Univariate sensitivity analysis | Cost reductions of 25–27% are achieved as the ratio of the cost of MSI to IHC is varied from 3.25 to 5.00 |
| Bessa et al. 200863 | Mutations detected Costs |
All strategies equally sensitive MSI + BRAF + GT cheapest if CC not used (€35,230 per mutation detected) CC + IHC + BRAF + GT cheapest if CC used (€11,450 per mutation detected) |
N/A | N/A |
| Yan et al. 200864 | Cost per mutation detected | Regardless of FH, the IHC + methylation testing strategy had the lowest cost per mutation detected (from $653 using AC II to $3600 using L-CRC). MSI had the highest cost per mutation detected as it did not inform gene sequencing (from $2220 using AC II to $22,800 using L-CRC). AC II were the most cost-effective FH criteria | N/A | N/A |
| Palomaki et al. 200965 | Mutations detected Cost |
Universal genetic testing most effective (4041 mutations detected) but most expensive ($71,800 per mutation) IHC with BRAF as preliminary tests detected fewest mutations and had lowest cost per mutation detected ($12,600) Incremental cost of detecting mutations for other strategies was over $300,000 per mutation |
Probabilistic sensitivity analysis | 90% CIs for average cost per mutation detected Universal genetic testing $49,000–101,000 MSI as preliminary test $20,000–44,000 IHC as preliminary test $9000–20,000 IHC with BRAF as preliminary test $8000–18,000 |
| Ramsoekh et al. 200966 | Mutations detected Costs Cost per mutation detected |
Direct GT most expensive (€9100 per mutation detected). TT less expensive (€3800 per mutation detected). PMs lower costs, but fewer mutations detected | Threshold analysis, adjusting probability cut-offs for PMs to optimise them | Leiden and UK-Ams optimised at cut-off of < 5%, UK-Alt and PREMM1,2 at ≥ 5%, Edinburgh at ≥ 10% |
| Resnick et al. 200967 | Mutations detected Cost ICERs |
Universal genetic testing most effective (920 mutations detected) but not cost-effective (ICER $1.4M per mutation) Genetic testing for all aged < 60 years dominated AC II strategy detects 83 at cost of $7M IHC strategy detects 858 (ICER $13,812 per mutation) (deemed cost-effective) |
Threshold analysis | Cost of sequencing three genes from $2474 to $600 → age < 60 years, strategy cost-effective (ICER $12,318 per mutation) Age cut-off 50 years → no longer dominated (ICER $16,931 per mutation) |
| Horwitz et al. 201068 | Mutations detected Costs |
Average cost per mutation detected $8492 | N/A | N/A |
| Gudgeon et al. 201169 | Mutations detected Costs Cost per case detected Sensitivity of screening protocol |
IHC with methylation with BRAF cheapest ($10,369 per case detected), second-to-last sensitivity (91.26% compared with IHC with methylation 91.25%) IHC straight to GT had highest sensitivity (92.90%) |
Univariate sensitivity analyses | Most sensitive to prevalence of LS, sensitivity and specificity of IHC |
| Perez-Carbonell et al. 201170 | Mutations detected Cost |
Both strategies identified 11 germline mutations; universal IHC identified more that were ‘probably LS’ Universal IHC cost €43,114 per mutation detected Revised Bethesda followed by IHC cost €16,109 per mutation detected |
N/A | N/A |
| Williams et al. 201171 | Cases detected Costs Cost per case detected Cases missed |
Cost per LS case detected (age limit): $10,684 (no age cut-off); $9673 (age 80 years); $7952 (age 70 years); $5656 (age 60 years); $3353 (age 50 years) Percentage of LS cases missed increased as age limit lowered. At age 50 years, 54.5% cases missed |
N/A | N/A |
| Gausachs et al. 201272 | Cases detected Costs Cost per case detected Incremental cost per case detected |
Strategy 1 dominated by strategy 2 ICER for strategy 3 vs. strategy 2 was €27,220 per additional case detected for probands and €7991 per additional case detected for FDRs/SDRs |
Univariate sensitivity analysis | All results most sensitive to mutation prevalence and unit cost of GT |
There was consistency in the perspective of these studies, as all but one were from a health-care perspective; the other67 was from the perspective of a third-party payer. Most other aspects of the studies were quite varied.
There were six studies from the USA,59,65,67–69,71 six from European countries,34,60–63,66 one from China64 and two where the setting was not clear. 70,72 Most studies were not specific about the ‘current practice’ in their respective countries, but their strategies were generally compared against a no testing strategy, implying that there was no standard ‘current practice’ in each of the countries or that no testing for LS was the ‘current practice’. Relevant to our assessment, one of the European studies was based in the UK,60 but this was 9 years old at the time of writing and was not consistent with our input population; Pigatto and colleagues60 investigated families referred for genetic testing, rather than newly diagnosed CRC patients aged < 50 years, who were the focus of our investigation.
With regards to the other studies’ input populations, one looked at EC patients67 and three (including the UK study) examined families who were referred to genetic testing centres,60,62,66 neither of which were the target population of our TA. The other 11 studies followed newly diagnosed CRC patients, and three of these59,65,72 included the impact on relatives of the CRC patients.
As well as having different study populations, the different studies also assessed different tests, which included the following subsets: clinical criteria/FH,34,59–61,63,64,67,68,70 prediction models,66 tumour testing65,34,59–64,66–72 and genetic testing. 65,34,59–64,66–72 Genetic testing was split into gene sequencing for probands and targeted genetic testing for relatives. Not all genetic testing was conducted on all four known LS genes, primarily because MSH6 and PMS2 are more recent discoveries. All of the studies had strategies that included universal genetic testing and at least one that included a tumour-based test followed by genetic testing. Most also included some variant of clinical criteria, FH criteria or a prediction model based on these criteria.
In addition to the different individual tests, each of the studies evaluated a different combination and sequence of tests. In general, these combinations fell into the pattern of clinical criteria/FH/prediction model followed by tumour testing followed by genetic testing, though in some strategies certain steps in this sequence included multiple tests or were missed out entirely. Most studies included at least one strategy that did not include FH. Our assessment attempted to cover all plausible, well-recognised strategies and therefore included strategies similar to those presented in some of these studies.
As well as the testing strategies, we were interested in the acceptance of genetic testing and genetic counselling. Ten of the studies from this section59,61–69 mentioned appropriate informed genetic counselling (two were unclear71,72), but only four61,65,68,69 explicitly modelled the acceptance associated with it or genetic testing. This indicated that this was an area where our model could provide added value.
With regards to the study design, nine papers34,60–64,66,68,70 included primary research (prospective or clinical) and 1234,59–63,65–67,69,71,72 included some form of decision modelling, usually using a decision tree approach. Six papers34,60–63,66 used a combination, using the study to inform the decision tree results. In every study, the costs and numbers of people with mutations detected were reported, and therefore the cost per mutation detected and the related incremental cost-effectiveness ratios (ICERs) could be calculated if they were not reported. In general, genetic testing alone had larger ICERs than strategies with other forms of testing included, and was stated to be more cost-effective, but as these ICERs are based on cost per case detected and not on life-years or QALYs of patients, this is not a measure of cost-effectiveness that has any meaning within our TA framework. For the other tests, there were mixed results for which were most effective, but, in general, when tumour-based tests were tested against FH or clinical criteria (CC) they were found to have lower ICERs compared with no testing. Given that the measure of cost-effectiveness has little meaning to our decision process – and that there are several different tests within the subgroups tumour-based tests, FH and CC – the results do not indicate which individual test is the most cost-effective.
Of the 12 studies34,59–63,65–67,69,71,72 that included models, seven59,62,65–67,69,72 conducted a sensitivity analysis (four univariate sensitivity analyses,59,62,69,72 one probabilistic sensitivity analysis,65 two threshold analyses66,67). The only consistent results from these sensitivity analyses were that the most influential parameters were the prevalence of LS and cost of testing for LS.
Lynch syndrome management-only studies
This group of studies, which only assessed the long-term cost-effectiveness of management strategies for LS, was the smallest subgroup of studies identified by our systematic review. The earliest study was published in May 199873 and the most recent in May 2011. 76 Details of these studies can be found in Tables 33 and 34. There were four included studies,73–76 three from a health-care provider perspective73,74,76 (one of which claimed to be societal) and one from a societal perspective. 75 Again there was a range of settings: two US,75,76 two European. 73,74 As with the diagnostic studies, there was one study based in the UK,74 which assessed the cost-effectiveness of biennial colonoscopic surveillance in families meeting the AC or with confirmed MMR gene mutations. This was also the only study with a mixed-sex cohort and was therefore more relevant to our assessment. However, it only considered colonoscopic surveillance, and therefore CRC, as long-term concerns for LS patients. Of the other three studies, two investigated the cost-effectiveness of gynaecological surveillance and prophylactic gynaecologic surgery on an all-female cohort aged 30 years,75,76 and one investigated the cost-effectiveness of colonoscopic surveillance (annually or 2.5-yearly) for an all-male cohort aged 25 years. 73 All populations were assumed to be asymptomatic at the start of each model.
| Author and year | Setting, perspective | Population | Study purpose | Study approach | Treatment strategies |
|---|---|---|---|---|---|
| Vasen et al. 199873 | Western population (data from the Netherlands), international health-care systems | Male LS carriers aged 25 years | Cost-effectiveness analysis of colonoscopic surveillance | Decision tree | Colonoscopy every 2.5 years Annual colonoscopy No surveillance |
| Dunlop et al. 200274 | UK hospital | Families meeting AC or with MMR gene mutations | Cost-effectiveness analysis of colonoscopic surveillance | Decision tree (implicit) | Biennial colonoscopy from age 25 to 75 years |
| Kwon et al. 200875 | USA, societal | LS women aged 30 years without gynaecological cancer | Cost–utility analysis of surveillance and PS for gynaecological cancers | Markov cohort model Markov microsimulation |
No prevention PS at age 30 years PS at age 40 years Annual screening from age 30 years Combined (annual screening from age 30 years and PS at 40 years) |
| Yang et al. 201176 | USA, societal stated (health-care system assumed) | Women with LS aged 30 years | Cost–utility analysis of gynaecological surveillance vs. PS | Decision model using TreeAge | Annual gynaecological examination (with possibility of TAHBSO) Annual gynaecological surveillance including TVU, endometrial biopsy and serum CA125 testing (with possibility of TAHBSO) No surveillance, prophylactic TAHBSO at age 30 years |
| Author and year | Outcomes measured | Discount rate | Base results | Sensitivity analysis approach | Main sensitivity analysis resultsa |
|---|---|---|---|---|---|
| Vasen et al. 199873 | Life expectancy Costs Number of colonoscopies and polypectomies |
5% costs | Both surveillance arms dominate no surveillance Annual surveillance vs. 2.5-yearly surveillance has an ICER of $39 per LYG |
Univariate sensitivity analysis | Cost-effectiveness of 2.5-yearly surveillance increased as CRC risk during surveillance decreased Reducing the proportion of early CRC diagnosed during surveillance reduced LE and increased costs for surveillance, but surveillance still dominated no surveillance |
| Dunlop et al. 200274 | Lives saved Costs |
Not discounted | Cost per life saved £14,925 | N/A | N/A |
| Kwon et al. 200875 | QALYs Costs ICERs |
3% | Combined strategy most effective (18.9766 QALYs per patient) but not cost-effective (ICER $194,650/QALY) PS at age 40 years cost-effective (18.9430 QALYs at $11,477/QALY relative to no prevention) PS at age 30 years extended dominated by no prevention and PS at 40 years Annual screening dominated by all interventions |
Univariate sensitivity analysis Threshold analysis |
Univariate sensitivity analysis Combined strategy remains not cost-effective as surveillance start age varied from 30 years to 35 years Threshold analysis Age of PS in combined strategy > 42 years → PS at age 40 years most effective Utility of PS < 0.88 → PS at age 40 years dominates PS at 30 years |
| Yang et al. 201176 | QALYs Costs Cost per QALY |
3% | For average cost per QALY, PS strategy was most cost-effective at $904 per QALY. The next most cost-effective was annual gynaecological surveillance at $2718 per QALY; annual examination strategy was $4085 per QALY | Threshold analysis Univariate and multivariate sensitivity analysis |
Threshold analysis When prophylactic surgery cost less than $90,000 per patient, it was always most cost-effective Univariate sensitivity analysis Model most sensitive to cost and surgical mortality of PS, but at WTP of $50,000 PS still an optimal strategy Multivariate sensitivity analysis Monte Carlo simulation showed PS dominant in 99.97% of trials. At a WTP of $50,000 per QALY, surgery was cost-effective compared with surveillance in 100% of trials and surveillance was cost-effective compared with examination in 99.98% of trials |
The modelling approach in these studies varied: both studies investigating colonoscopic surveillance used decision tree modelling;73,74 Kwon and colleagues’ study75 on gynaecological surveillance/prophylactic surgery used a Markov cohort model and Markov microsimulation approach; and Yang and colleagues’ similar study76 used a decision model built in TreeAge.
All studies reported costs, but they reported different measures of effectiveness. Both studies investigating gynaecological management reported QALYs,75,76 Vasen and colleagues73 reported life expectancy and Dunlop and colleagues74 reported lives saved. Generally in these studies, the authors concluded that any management strategies could be cost-effective. For EC, prophylactic surgery was seen as more cost-effective than gynaecological surveillance, even under sensitivity analysis. For preventing CRC, colonoscopic surveillance dominated no surveillance in the Vasen and colleagues study,73 even under sensitivity analysis, and the cost per life saved in the Dunlop and colleagues study74 was £14,925, which they concluded was a ‘favourable’ result.
Studies that incorporated both diagnosis and management of Lynch syndrome
This subgroup contained 13 studies22,23,48–58,88 focused on the cost-effectiveness of strategies to first identify and then manage LS, published over a period of 18 years (1995–2012). Six of these were published between 2010 and February 2013. As Ladabaum and colleagues (2011)48 and Wang and colleagues (2012)57 were based on the same model, they are discussed as the same study in this section of the review.
Details of all these studies can be found in Tables 35 and 36.
| Author and year | Setting, perspective | Population | Study purpose | Study approach | Diagnostic comparators | Treatment strategies | |||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Brown et al. 199588 and 199622 | USA, health-care system | US population | Cost-effectiveness analysis of primary care screening for LS in unaffected persons | Ad hoc calculations | Probands ArmTestControlNone – not statedTestGenetic test at age 25 years |
Arm | Test | Control | None – not stated | Test | Genetic test at age 25 years |
Confirmed mutation
|
|||||||||||||||||||||||||
| Arm | Test | ||||||||||||||||||||||||||||||||||||
| Control | None – not stated | ||||||||||||||||||||||||||||||||||||
| Test | Genetic test at age 25 years | ||||||||||||||||||||||||||||||||||||
| Ramsey et al. 200123 | USA, health-care system (stated societal) | Newly diagnosed CRC patients and their siblings and children | Cost-effectiveness of strategies to identify LS | Decision model Survival modelled using Weibull distribution |
Probands Test 1Test 2Test 3NoneBethesdaMSIGTBethesda: Revised Bethesda criteria. Relatives (proband confirmed mutation/indeterminate result) Germline testing for siblings and children |
Test 1 | Test 2 | Test 3 | None | Bethesda | MSI | GT | Bethesda: Revised Bethesda criteria. |
Confirmed mutation
|
|||||||||||||||||||||||
| Test 1 | Test 2 | Test 3 | |||||||||||||||||||||||||||||||||||
| None | |||||||||||||||||||||||||||||||||||||
| Bethesda | MSI | GT | |||||||||||||||||||||||||||||||||||
| Bethesda: Revised Bethesda criteria. | |||||||||||||||||||||||||||||||||||||
| Ramsey et al. 200349 | USA, health-care system | Newly diagnosed CRC patients and their siblings and children | Cost-effectiveness of strategies to identify LS | Decision model Ad hoc |
Probands Test 1Test 2Test 3NoneBethesdaMSIGTMSIGTBethesdaGTGTBethesda: Revised Bethesda criteria. Relatives (proband confirmed mutation/indeterminate result) Germline testing for siblings and children |
Test 1 | Test 2 | Test 3 | None | Bethesda | MSI | GT | MSI | GT | Bethesda | GT | GT | Bethesda: Revised Bethesda criteria. |
Confirmed mutation
|
||||||||||||||||||
| Test 1 | Test 2 | Test 3 | |||||||||||||||||||||||||||||||||||
| None | |||||||||||||||||||||||||||||||||||||
| Bethesda | MSI | GT | |||||||||||||||||||||||||||||||||||
| MSI | GT | ||||||||||||||||||||||||||||||||||||
| Bethesda | GT | ||||||||||||||||||||||||||||||||||||
| GT | |||||||||||||||||||||||||||||||||||||
| Bethesda: Revised Bethesda criteria. | |||||||||||||||||||||||||||||||||||||
| Kievit et al. 200550 | Netherlands, health-care system | Newly diagnosed CRC patients and their siblings and children | Cost-effectiveness of strategies to identify LS | Decision model Decision tree and Markov subtrees |
Probands Test 1Test 2Test 3Test 4Test 5Selection criteriaIHC + MSIGT (IHC or MSI positive)FHACGTFHACGT (AC positive)IHC + MSIGTShading indicates experimental arm; selection criteria = (CRC < 50 years of age) or (second CRC) or (CRC and LS-associated cancer) or (adenoma with high-grade dysplasia < 40 years of age). Relatives Test 1Test 2AC family and/or positive GT in probandGT (positive GT in proband)Surveillance (no mutation found in proband) |
Test 1 | Test 2 | Test 3 | Test 4 | Test 5 | Selection criteria | IHC + MSI | GT (IHC or MSI positive) | FH | AC | GT | FH | AC | GT (AC positive) | IHC + MSI | GT | Shading indicates experimental arm; selection criteria = (CRC < 50 years of age) or (second CRC) or (CRC and LS-associated cancer) or (adenoma with high-grade dysplasia < 40 years of age). | Test 1 | Test 2 | AC family and/or positive GT in proband | GT (positive GT in proband) | Surveillance (no mutation found in proband) | HNPCC mutation carrier or AC family
No treatment strategy |
|||||||||
| Test 1 | Test 2 | Test 3 | Test 4 | Test 5 | |||||||||||||||||||||||||||||||||
| Selection criteria | IHC + MSI | GT (IHC or MSI positive) | |||||||||||||||||||||||||||||||||||
| FH | AC | GT | |||||||||||||||||||||||||||||||||||
| FH | AC | GT (AC positive) | |||||||||||||||||||||||||||||||||||
| IHC + MSI | GT | ||||||||||||||||||||||||||||||||||||
| Shading indicates experimental arm; selection criteria = (CRC < 50 years of age) or (second CRC) or (CRC and LS-associated cancer) or (adenoma with high-grade dysplasia < 40 years of age). | |||||||||||||||||||||||||||||||||||||
| Test 1 | Test 2 | ||||||||||||||||||||||||||||||||||||
| AC family and/or positive GT in proband | GT (positive GT in proband) | ||||||||||||||||||||||||||||||||||||
| Surveillance (no mutation found in proband) | |||||||||||||||||||||||||||||||||||||
| Breheny et al. 200651 | Western Australia, government | FDRs of known LS mutation carriers | Cost-effectiveness analysis of predictive testing, surveillance and surgical management for LS | Decision model (Markov model) | Intervention arm: Predictive testing for family mutation Control arms 1 and 2: No testing |
Intervention arm Intensive surveillance (colonoscopy, upper gastric surveillance, gynaecological surveillance) and colorectal surgery for mutation-positive individuals; population surveillance for non-carriers Control arm 1 Intensive surveillance and surgery for all FDRs Control arm 2 Population surveillance for all FDRs |
|||||||||||||||||||||||||||||||
| DACEHTA 200752 | Denmark, health-care system | Persons referred to Danish HNPCC registry | Cost-effectiveness of surveillance programme for persons referred | Not stated | Not stated | Not explicitly stated, includes colonoscopy | |||||||||||||||||||||||||||||||
| Olsen et al. 200753 | Denmark, health-care system | Persons referred to Danish HNPCC registry | Cost-effectiveness analysis of LS diagnosis and surveillance | Decision model Markov model |
Test 1Test 2Risk categoryAC IIGT (AC II positive)High (GT positive/negative)FH (AmsII–)Moderate (FH positive)Low (FH–)LowShading indicates experimental arm; FH = FH suggestive of LS but not AC II. | Test 1 | Test 2 | Risk category | AC II | GT (AC II positive) | High (GT positive/negative) | FH (AmsII–) | Moderate (FH positive) | Low (FH–) | Low | Shading indicates experimental arm; FH = FH suggestive of LS but not AC II. | High/moderate risk Biennial colonoscopy from age 25 years (high risk) or 45 years (moderate risk); colectomy or proctocolectomy on CRC diagnosis Low risk No surveillance; segmental resection on CRC diagnosis |
||||||||||||||||||||
| Test 1 | Test 2 | Risk category | |||||||||||||||||||||||||||||||||||
| AC II | GT (AC II positive) | High (GT positive/negative) | |||||||||||||||||||||||||||||||||||
| FH (AmsII–) | Moderate (FH positive) | ||||||||||||||||||||||||||||||||||||
| Low (FH–) | |||||||||||||||||||||||||||||||||||||
| Low | |||||||||||||||||||||||||||||||||||||
| Shading indicates experimental arm; FH = FH suggestive of LS but not AC II. | |||||||||||||||||||||||||||||||||||||
| Mvundura et al. 201054 | USA, health-care system | Newly diagnosed CRC patients and relatives | Cost-effectiveness and cost–utility of strategies to identify LS | Decision model Ad hoc |
Probands Test 1Test 2Test 3NoneIHCBRAF (abnormal IHC for MLH1)GT (MLH1)GT (all other abnormal IHC)IHCGTMSIGTGT Relatives (proband confirmed mutation) GT |
Test 1 | Test 2 | Test 3 | None | IHC | BRAF (abnormal IHC for MLH1) | GT (MLH1) | GT (all other abnormal IHC) | IHC | GT | MSI | GT | GT |
Relatives with confirmed mutation
|
||||||||||||||||||
| Test 1 | Test 2 | Test 3 | |||||||||||||||||||||||||||||||||||
| None | |||||||||||||||||||||||||||||||||||||
| IHC | BRAF (abnormal IHC for MLH1) | GT (MLH1) | |||||||||||||||||||||||||||||||||||
| GT (all other abnormal IHC) | |||||||||||||||||||||||||||||||||||||
| IHC | GT | ||||||||||||||||||||||||||||||||||||
| MSI | GT | ||||||||||||||||||||||||||||||||||||
| GT | |||||||||||||||||||||||||||||||||||||
| Dinh et al. 201155 | USA, health-care system (not stated) | US population aged 20 years or over | Cost–utility analysis of primary care screening for LS in unaffected persons | Decision model Ad hoc |
Probands ArmTest 1Test 2Test 3Test 4ControlCRC/ECCCbIHCGT (single)CRC/ECCCbGTTestPREMMGT Relatives (at risk) GT |
Arm | Test 1 | Test 2 | Test 3 | Test 4 | Control | CRC/EC | CCb | IHC | GT (single) | CRC/EC | CCb | GT | Test | PREMM | GT |
Confirmed mutation
|
|||||||||||||||
| Arm | Test 1 | Test 2 | Test 3 | Test 4 | |||||||||||||||||||||||||||||||||
| Control | CRC/EC | CCb | IHC | GT (single) | |||||||||||||||||||||||||||||||||
| CRC/EC | CCb | GT | |||||||||||||||||||||||||||||||||||
| Test | PREMM | GT | |||||||||||||||||||||||||||||||||||
| Kwon et al. 201156 | USA, health care | Newly diagnosed EC patients | Cost-effectiveness analysis of strategies to identify LS | Decision model Decision tree with Markov subtrees |
Test 1Test 2Test 3Age < 50 years, at least one FDR with LS-associated cancer at any ageGTAge < 50 yearsIHCGTAge < 60 yearsIHCGTAC IIGTIHCGT1+ FDRIHCGT | Test 1 | Test 2 | Test 3 | Age < 50 years, at least one FDR with LS-associated cancer at any age | GT | Age < 50 years | IHC | GT | Age < 60 years | IHC | GT | AC II | GT | IHC | GT | 1+ FDR | IHC | GT | Confirmed mutation Annual colonoscopy GT negative or not tested 50% receive one or more colonoscopy in next 10 years |
|||||||||||||
| Test 1 | Test 2 | Test 3 | |||||||||||||||||||||||||||||||||||
| Age < 50 years, at least one FDR with LS-associated cancer at any age | GT | ||||||||||||||||||||||||||||||||||||
| Age < 50 years | IHC | GT | |||||||||||||||||||||||||||||||||||
| Age < 60 years | IHC | GT | |||||||||||||||||||||||||||||||||||
| AC II | GT | ||||||||||||||||||||||||||||||||||||
| IHC | GT | ||||||||||||||||||||||||||||||||||||
| 1+ FDR | IHC | GT | |||||||||||||||||||||||||||||||||||
| Ladabaum et al. 201148 | USA, third-party payer (NIH) | Newly diagnosed CRC patients and relatives | Cost-effectiveness of strategies to identify LS | Decision model Decision tree model, Markov subtrees |
Probands Test 1Test 2Test 3NoneCCaGTCCaIHCGTTBTGTGT Relatives (proband confirmed mutation) Germline testing |
Test 1 | Test 2 | Test 3 | None | CCa | GT | CCa | IHC | GT | TBT | GT | GT |
Persons with confirmed LS mutations or assumed LS and their FDRs
|
|||||||||||||||||||
| Test 1 | Test 2 | Test 3 | |||||||||||||||||||||||||||||||||||
| None | |||||||||||||||||||||||||||||||||||||
| CCa | GT | ||||||||||||||||||||||||||||||||||||
| CCa | IHC | GT | |||||||||||||||||||||||||||||||||||
| TBT | GT | ||||||||||||||||||||||||||||||||||||
| GT | |||||||||||||||||||||||||||||||||||||
| Wang G et al. 201257 | USA, third-party payer (NIH) | Newly diagnosed CRC patients and relatives | Cost–utility of strategies to identify LS (update of Ladabaum et al. 201148 to include HRQoL) | Decision model Decision tree model, Markov subtrees |
Probands Test 1Test 2Test 3NoneCC1GTCC1IHCGTTBTGTGT Relatives (proband confirmed mutation) Germline testing |
Test 1 | Test 2 | Test 3 | None | CC1 | GT | CC1 | IHC | GT | TBT | GT | GT |
Persons with confirmed LS mutations or assumed LS and their FDRs
|
|||||||||||||||||||
| Test 1 | Test 2 | Test 3 | |||||||||||||||||||||||||||||||||||
| None | |||||||||||||||||||||||||||||||||||||
| CC1 | GT | ||||||||||||||||||||||||||||||||||||
| CC1 | IHC | GT | |||||||||||||||||||||||||||||||||||
| TBT | GT | ||||||||||||||||||||||||||||||||||||
| GT | |||||||||||||||||||||||||||||||||||||
| Wang VW et al. 201258 | Singapore, health-care system | 21-year-old FDRs of mutation-confirmed LS cases | Cost-effectiveness analysis of targeted genetic testing for LS to determine surveillance programmes | Decision model Decision tree model, Markov subtrees |
|
Biannual colonoscopy surveillance for all in strategy 2 and those who test positive in strategy 1. Those in strategy 1 who test negative receive general population-level surveillance | |||||||||||||||||||||||||||||||
| Author and year | Outcomes measured | Discount rate | Base results | Sensitivity analysis approach | Main sensitivity analysis resultsa | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Brown et al. 199622 | LYs Costs Cost per LYS Cost per mutation identified |
5% | Cost-effective compared with current practice (threshold $50,000):
|
Univariate sensitivity analysis | Sensitive to cost of genetic testing, average life expectancy of LS treated patient, penetrance of LS | ||||||||||||||||||||||||||||||
| Ramsey et al. 200123 | LYs Costs |
3% | Added cost per LYG for probands $42,210, FDRs have a cost saving; ICER for probands and relatives compared with no screening $7556 | Univariate sensitivity analysis PSA |
Univariate sensitivity analysis Sensitive to relative mortality risk associated with increased surveillance in LS siblings and children; the overall prevalence of HNPCC mutation carriers among patients with newly diagnosed CRC; and the discount rate PSA 90% CI $4874 to $21,576 per LYG |
||||||||||||||||||||||||||||||
| Ramsey et al. 200349 | LYs Costs ICERs |
3% | Compared with no screening, all strategies cost-effective ($50,000 threshold) apart from universal testing ICERs StrategyICER ($/LY)Bethesda/MSI11,865MSI394,067Bethesda441,172Universal2,553,345 |
Strategy | ICER ($/LY) | Bethesda/MSI | 11,865 | MSI | 394,067 | Bethesda | 441,172 | Universal | 2,553,345 | Univariate sensitivity analysis PSA |
Univariate Sensitive to survival benefit for increased surveillance in LS positive, specificity of FH/MSI, prevalence of LS in probands PSA Unclear which strategy is most cost-effective, but universal testing is the least cost-effective |
||||||||||||||||||||
| Strategy | ICER ($/LY) | ||||||||||||||||||||||||||||||||||
| Bethesda/MSI | 11,865 | ||||||||||||||||||||||||||||||||||
| MSI | 394,067 | ||||||||||||||||||||||||||||||||||
| Bethesda | 441,172 | ||||||||||||||||||||||||||||||||||
| Universal | 2,553,345 | ||||||||||||||||||||||||||||||||||
| Kievit et al. 200550 | LYs Costs ICERs |
4% | Discounted cost per LYG €3801 if testing limited to probands Discounted cost per LYG €2184 if testing extended to relatives |
Univariate sensitivity analysis | Univariate Sensitive to adherence with selection criteria, positive predictive value of MSI and discount rates for costs and benefits |
||||||||||||||||||||||||||||||
| Breheny et al. 200651 | CRC-free years Costs |
5% (costs) 0% (CRC-free years) |
Intervention arm dominates control arm 1 (surveillance for all FDRs) and is more costly but more effective than control arm 2 (no surveillance); ICER = AU$1518/CRC-free year for males, AU$1575/CRC-free year for females | Univariate sensitivity analysis | Adjusting efficacy of surveillance and surgery from 30% to 70% (base value 50%) reduced ICER of intervention arm vs. control arm 2 for women from AU$2007 to AU$1320/CRC-free year | ||||||||||||||||||||||||||||||
| DACEHTA 200752 | LYs Costs |
5% | Discounted cost per LYG = DKK 7294 | Univariate sensitivity analysis | Varying the following led to price per LYG of between DKK 2696 and DKK 17,600: effectiveness of colonoscopy, costs of molecular diagnosis, incidence of CRC, incidence of metachronous CRC | ||||||||||||||||||||||||||||||
| Olsen et al. 200753 | LYs Costs ICERs |
5% | All outcomes discounted at 5% Experimental arm gives 0.51 LYs at cost of €500 (ICER €980/LY) High-risk patients gain 2.01 LYs at cost of €1021 (ICER €508/LY) Moderate-risk patients gain 0.21 LYs at cost of €336 (ICER €1600/LY) |
Univariate sensitivity analysis Scenario analysis |
Univariate Cost of colonoscopy (base case €349; −50% → experimental arm dominates; +50% → ICER €2365/LY) Reduction in CRC incidence owing to surveillance (base case 62%; 25% → ICER €2262/LY; 100% → ICER €162/LY) Lifetime risk of CRC for moderate risk (base case 10.8%; 7.2% → ICER €2189/LY; 18.0% → ICER €2233/LY) Therefore, cost-effectiveness worsened when risk was either lowered or increased Scenario analysis Improve quality of referrals (proportion referred who are low risk reduced from 31% to 10%) → ICER €605/LY No surveillance of moderate-risk families (but retain counselling costs and surgical management) → ICER €1947/LY Improve detection rate of mutation analysis from 22% to 50% → ICER €946/LY No mutation analysis → ICER €946/LY |
||||||||||||||||||||||||||||||
| Mvundura et al. 201054 | LYs Costs ICERs QALYs |
3% | All strategies with preliminary tests cost-effective relative to no testing IHC and BRAF as preliminary were optimal (ICER $22,552/LY) |
Univariate sensitivity analysis Scenario analyses |
Univariate Most sensitive to CRC risk among relatives, number of relatives per proband and adherence to surveillance Scenario analyses Using median laboratory prices increased costs and meant that age-targeted IHC and BRAF was most cost-effective Cascade testing reduced all ICERs relative to no testing Using QALYs on average scaled ICERs by 1.18 LY/QALY |
||||||||||||||||||||||||||||||
| Dinh et al. 201155 | QALYs Costs ICERs |
3% | Cost-effective compared with current practice:
|
Univariate sensitivity analysis | Most sensitive to discount rate, mutation prevalence in the general population and FDR adherence to testing Screening at age 30 years with PREMM threshold 5% remains cost-effective relative to current practice |
||||||||||||||||||||||||||||||
| Kwon et al. 201156 | LYs Costs ICERs |
3% | Dominated: AC II → GT, age < 50/60 years → IHC → GT IHC → GT most effective (14.53077 QALYs) but not cost-effective (ICER $648,494/LY) At least one FDR with LS-associated cancer at any age → IHC → GT cost-effective (ICER $9126/LY) |
Scenario analysis Threshold analyses |
Scenario analysis 50% adherence with GT and surveillance (down from 100%) had little effect on ICERs Threshold analyses Sensitivity and specificity of AC II > 95% makes AC II → GT cost-effective Sensitivity of IHC < 70% makes age < 50 years, 1+ FDR → GT cost-effective |
||||||||||||||||||||||||||||||
| Ladabaum et al. 201148 | LYs Costs ICERs Cancer cases Cancer deaths |
3% | StrategyICER ($/LY)ICER excluding CC strategiesMMRpro/IHC30,600–Bethesda/IHC39,600–MMRpro41,400–Bethesda50,200–IHC (+ BRAF)–36,200MSI + IHC (+ BRAF)117,000108,000Universal GT293,000293,000All other strategies dominated or extended dominated. | Strategy | ICER ($/LY) | ICER excluding CC strategies | MMRpro/IHC | 30,600 | – | Bethesda/IHC | 39,600 | – | MMRpro | 41,400 | – | Bethesda | 50,200 | – | IHC (+ BRAF) | – | 36,200 | MSI + IHC (+ BRAF) | 117,000 | 108,000 | Universal GT | 293,000 | 293,000 | All other strategies dominated or extended dominated. | Univariate sensitivity analysis PSA Scenario analyses |
Univariate Most sensitive to age of relative, effectiveness of LS surveillance for CRC and prevalence of LS PSA IHC (+ BRAF) optimal strategy in 53% of iterations Scenario analyses Age limit for probands improves cost-effectiveness Three to four relatives needed for most strategies to be cost-effective compared with doing nothing |
|||||
| Strategy | ICER ($/LY) | ICER excluding CC strategies | |||||||||||||||||||||||||||||||||
| MMRpro/IHC | 30,600 | – | |||||||||||||||||||||||||||||||||
| Bethesda/IHC | 39,600 | – | |||||||||||||||||||||||||||||||||
| MMRpro | 41,400 | – | |||||||||||||||||||||||||||||||||
| Bethesda | 50,200 | – | |||||||||||||||||||||||||||||||||
| IHC (+ BRAF) | – | 36,200 | |||||||||||||||||||||||||||||||||
| MSI + IHC (+ BRAF) | 117,000 | 108,000 | |||||||||||||||||||||||||||||||||
| Universal GT | 293,000 | 293,000 | |||||||||||||||||||||||||||||||||
| All other strategies dominated or extended dominated. | |||||||||||||||||||||||||||||||||||
| Wang G et al. 201257 | QALYs Costs ICERs |
3% | StrategyICER ($/QALY)ICER excluding CC strategiesMMRpro/IHC50,562–Bethesda/IHC65,347–MMRpro68,384–Bethesda82,864–IHC (+ BRAF)–59,719MSI + IHC–179,576MSI + IHC (+ BRAF)193,343–Universal GT393,303271,219All other strategies dominated or extended dominated. | Strategy | ICER ($/QALY) | ICER excluding CC strategies | MMRpro/IHC | 50,562 | – | Bethesda/IHC | 65,347 | – | MMRpro | 68,384 | – | Bethesda | 82,864 | – | IHC (+ BRAF) | – | 59,719 | MSI + IHC | – | 179,576 | MSI + IHC (+ BRAF) | 193,343 | – | Universal GT | 393,303 | 271,219 | All other strategies dominated or extended dominated. | Univariate sensitivity analysis PSA Scenario analyses |
Univariate Results consistent with Ladabaum et al. 201148 PSA IQRs narrow and within cost-effective ranges, ICERs had wide 95% CIs, reflecting wide distributions of utility estimates Scenario analyses Length of effect from disutility associated with GT or surveillance affected ICERs; longer than 12 months and the ICERs exponentially increased |
||
| Strategy | ICER ($/QALY) | ICER excluding CC strategies | |||||||||||||||||||||||||||||||||
| MMRpro/IHC | 50,562 | – | |||||||||||||||||||||||||||||||||
| Bethesda/IHC | 65,347 | – | |||||||||||||||||||||||||||||||||
| MMRpro | 68,384 | – | |||||||||||||||||||||||||||||||||
| Bethesda | 82,864 | – | |||||||||||||||||||||||||||||||||
| IHC (+ BRAF) | – | 59,719 | |||||||||||||||||||||||||||||||||
| MSI + IHC | – | 179,576 | |||||||||||||||||||||||||||||||||
| MSI + IHC (+ BRAF) | 193,343 | – | |||||||||||||||||||||||||||||||||
| Universal GT | 393,303 | 271,219 | |||||||||||||||||||||||||||||||||
| All other strategies dominated or extended dominated. | |||||||||||||||||||||||||||||||||||
| Wang VW et al. 201258 | LYs Costs ICERs |
3% | Targeted GT dominates intensive surveillance for everyone, with lower costs (−$13,588) and higher LYG (0.01) | Univariate sensitivity analysis Scenario analyses |
Univariate Strategy A remains dominant for all univariate analyses Scenario analyses When changing the adherence to surveillance in both strategies from the base case 100%:
|
||||||||||||||||||||||||||||||
As with the previous two groups of studies, the majority of these studies were from a health sector perspective. There was also one which was conducted from a government perspective51 and one from a third-party payer perspective. 48,57 The majority of studies were set in the USA, with one from Singapore,58 one from Australia,51 two from Denmark52,53 and one from the Netherlands. 50 No UK studies were reported. The most common input population in the studies, occurring in five separate studies, was newly diagnosed CRC patients and their relatives. 23,48,49,50,54,57 These studies were particularly relevant to our assessment as our input population lay within this group of patients. Other input populations included healthy, unaffected members of the general population;22,55,88 FDRs of known LS mutation carriers;51,58 families referred to a genetics registry;52,53 and, in one study, newly diagnosed EC patients. 56
Similar to the diagnostic-only studies, the majority of the diagnostic strategies in these studies included some form of CC/FH, tumour testing and genetic testing. Steps such as FH were sometimes missed out entirely, or strategies included several tests. CC/FH included the Bethesda and Revised Bethesda guidelines and AC I/II (and selected criteria from either guideline), as well as prediction models such as PREMM (prediction of mismatch repair gene mutations), MMRPro and MMRpredict. These prediction models were algorithms that used specified patient history, CC and FH to determine the likelihood of LS for a patient. Tumour testing in these studies included MSI, IHC, BRAF and methylation. The level of testing was not always the same; MSI was conducted on different numbers of markers in different studies, and sometimes IHC was conducted only for specific MMR proteins rather than all four. Genetic testing was divided into sequencing tests for probands and predictive tests for their relatives. Each of the studies used a different set and combination of these tests, making it difficult to decide which strategies were most commonly used.
Across the studies, the management strategies for CRC were fairly consistent. Colonoscopy every 1–2 years from age 25 years, with more aggressive colorectal surgery on diagnosis of CRC to prevent further CRCs, was the management strategy for patients diagnosed with LS in the majority of studies. This was consistent with the current published guidance identified by our systematic review. For the studies that considered EC, the management strategies included gynaecological surveillance/screening with prophylactic TAHBSO at an appropriate age and were once again fairly consistent across the studies.
The design of these studies was predominantly decision modelling with a mix of decision trees, Markov cohort modelling and a variety of ad hoc modelling.
As knowledge about LS has altered over recent years, we thought it prudent to assess how up to date the various studies were with regards to this knowledge. One parameter we thought particularly influential to the cost-effectiveness analysis was the risk of CRC in LS patients, as a lower estimate of CRC risk was likely to make strategies for diagnosing and managing LS less cost-effective. We summarise the values (and sources) for this parameter used in the studies in Table 37. There was a divide between those studies that were more than 5 years old, where lifetime CRC risk in LS patients was generally around 80%, and the more recent studies, in which lifetime CRC risk for LS patients was reported to be closer to 40–50%. This divide also provided us with information on which studies’ results were more likely to be comparable with ours; only 6 of the 13 studies48,54–58 used a lower (40–50%) estimate of CRC risk. The risk of CRC is also known to be different in males and females, which was only modelled explicitly in two of the studies. 48,55 These were also the only two studies to consider more than CRC in the long term, suggesting that this was an area where further modelling could add value.
| Author and year | CRC risk and source |
|---|---|
| Diagnosis and management | |
| Brown et al. 199622 | Not stated |
| Ramsey et al. 200123 | Not explicitly stated, but:
|
| Ramsey et al. 200349 | Above holds, and: |
| Kievit et al. 200550 | CRC risk in LS patients with/without surveillance not stated (Jarvinen et al. 200094) Sporadic CRC risk also not stated |
| Breheny et al. 200651 | Age-related population risk of CRC in 2000 (Australian Institute of Health and Welfare 200395) Age-specific LS CRC risk based on 72% penetrance by age 50 years and 80% by age 75 years (Renkonen-Sinisalo et al. 2000,96 Green et al. 200297) |
| DACEHTA 200752 (summary) | The risk of metachronous CRC is four times greater for CRC patients with LS than those with sporadic CRC Lifetime risk of CRC for FDRs of patients with LS mutation confirmed, 80%; for FDRs of patients with LS mutation unknown (pedigree-based assumption), 40% (no sources stated) |
| Olsen et al. 200753 | Lifetime risk of CRC for LS carriers: low risk, 3.3%; moderate risk, 10.8%; high risk (unknown mutation), 46%; high risk (known mutation), 80–90% (Jarvinen et al. 1995,16 Statens Institut for Medicinsk Teknologivurdering 200198) |
| Mvundura et al. 201054 | Risk of developing CRC for LS carriers, 40% Risk of second CRC diagnosis for LS carriers, 16% (Palomaki et al. 200965) |
| Dinh et al. 201155 | Lifetime risk for female carriers of MLH1 or MSH2 mutations: almost 50%; separate risks for males and females and for carriers of MSH6 mutations and PMS2 mutations (meta-analysis of results from Hendriks et al. 2004,99 Buttin et al. 2004,100 Dunlop et al. 1997,101 Quehenberger et al. 2005,102 Hampel et al. 2005,103 Wagner et al. 2001,104 Senter et al. 2008,105 Barrow et al. 2008,81 Stoffel et al. 2009106) |
| Kwon et al. 201156 | LS, surveillance,a 15% (Syngal et al. 1998,83 Vasen et al. 1998,73 Jarvinen et al. 2009,107 Jarvinen et al. 2000,94 Vasen et al. 2010108) LS, no surveillance, 40% (Jarvinen et al. 2000,94 de Jong et al. 2006109) Sporadic surveillance,a 3% (Cotterchio et al. 2005,110 Boursi et al. 2009,111 Rundle et al. 2008112) Sporadic, no surveillance, 5% (National Cancer Institute/SEER 2010113) |
| Ladabaum et al. 201148 | CRC risk by age 70 years:
|
| Wang G et al. 201257 | See Ladabaum et al. 201148 |
| Wang VW et al. 201258 | Lifetime CRC risk for LS carriers, 43% (Jenkins et al. 2006,114 Altekruse et al. 2010115) Sporadic CRC risk taken from SEER (Altekruse et al. 2010115) |
| Management only | |
| Vasen et al. 199873 | CRC risk for LS carriers by age 75 years (for men), 80–85% (Vasen et al. 1996116) |
| Dunlop 200274 | Lifetime CRC risk for LS carriers diagnosed by FH, 80% (Vasen et al. 1995,117 Aarnio et al. 199515); diagnosed systematic analyses, 74% men, 30% women (Dunlop et al. 1997101) |
| Kwon et al. 200875 | Lifetime CRC risk for LS carriers, 42% (Aarnio et al. 1999,118 Dunlop et al. 1997101) |
| Yang et al. 201176 | N/A |
Modelling of imperfect adherence to testing and management of LS appears to occur in over half of the studies; however, it is not always clear what patients are exactly complying with. In the genetic testing process, there is adherence to both genetic counselling and genetic testing. Some studies were explicit about the two separate sections; some appeared to treat this as one adherence issue; and some did not state it at all. Adherence to colonoscopic surveillance was included in a much clearer manner and was modelled in most studies, so that those who complied initially with surveillance continued to do so.
The majority of studies reported life-years and costs (and the respective ICERs) as their main outcomes. Two studies, the update to Ladabaum and colleagues (2011)57 and Dinh and colleagues (2011),55 reported QALYs rather than life-years and one study51 reported CRC-free years as the main outcome. Mvundura and colleagues (2010)54 reported QALYs as a scenario analysis, scaling their ICERs by 1.18 life-years per QALY, which is an approach that demands that a number of assumptions be made. One early study22 reported the short-term outcome of cost per mutation identified, as well as the long-term cost per life-year saved. A range of discount rates were used in the long-term calculations, generally ranging from 3–5% per year for both costs and benefits.
In general, the studies concluded that strategies that screened for LS were cost-effective compared with no screening, with all finding at least one strategy that fell below a pre-specified threshold. However, given the different strategies and costs for each country, there was little consistency over which strategies or individual tests were the most cost-effective. In two studies,48,54 IHC with BRAF appeared to be the most cost-effective strategy. In all studies where strategies with additional tests were included, universal genetic testing was not cost-effective. 48,49,54,56,57
The minimum uncertainty analysis performed by these studies was a univariate sensitivity analysis. Most studies looked at a large number of parameters in their univariate analysis, but only reported those that were most influential on the cost-effectiveness results, which made comparison between them difficult. Influential parameters that were identified by more than one study included age of population, number of relatives, cost of testing, effectiveness of the diagnostic and management strategies, and prevalence of LS in the population.
Quality appraisal of cost-effectiveness studies
Though the studies were assessed using all of the criteria from the Drummond checklist,47 specific criteria are reported in Table 38.
| Author and year | The viewpoint(s) of the analysis are clearly stated and justified | The source(s) of effectiveness estimates are stated | Methods for the estimation of quantities and unit costs are described | Currency and price date are recorded | Details of any models used are given | Time horizon of costs and benefits is stated | The ranges over which the variables are varied are justified |
|---|---|---|---|---|---|---|---|
| Diagnosis and management | |||||||
| Brown et al. 199622 | ✗ | ✓ | ✓ | ✓ | ✗ | ✓ | ✗ |
| Ramsey et al. 200123 | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Ramsey et al. 200349 | ✗ | ✓ | ✓ | ✓ | ✗ | ✓ | ✓ |
| Kievit et al. 200550 | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Breheny et al. 200651 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ |
| DACEHTA 200752 | ✗ | ✗ | ✗ | ✗ | ✗ | ✓ | ✗ |
| Olsen et al. 200753 | ✗ | ✓ | ✓ | ✓ | ✓ | ✗ | ✓ |
| Mvundura et al. 201054 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Dinh et al. 201155 | ✗ | ✓ | ✓ | ✓ | ✓ | ✗ | ✗ |
| Kwon et al. 201156 | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ |
| Ladabaum et al. 201148 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Wang G et al. 201257 | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Wang VW et al. 201258 | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Diagnosis only | |||||||
| Debniak et al. 200034 | ✗ | ✓ | ✗ | ✗ | ✗ | N/A | |
| Reyes et al. 200259 | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | |
| Pigatto et al. 200460 | ✗ | ✓ | ✓ | ✗ | ✗ | N/A | |
| Pinol et al. 200561 | ✓ | ✓ | ✓ | ✗ | ✗ | N/A | |
| Engel et al. 200662 | ✗ | ✓ | ✓ | ✗ | ✓ | ✓ | ✓ |
| Bessa et al. 200863 | ✗ | ✓ | ✓ | ✗ | ✗ | N/A | |
| Yan et al. 200864 | ✗ | ✓ | ✓ | ✗ | N/A | N/A | |
| Palomaki et al. 200965 | ✗ | ✓ | ✓ | ✗ | ✗ | ✗ | |
| Ramsoekh et al. 200966 | ✗ | ✓ | ✓ | ✗ | ✗ | ✗ | |
| Resnick et al. 200967 | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ | |
| Horwitz et al. 201068 | ✗ | ✓ | ✗ | ✗ | N/A | N/A | |
| Gudgeon et al. 201169 | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | |
| Perez-Carbonell et al. 201170 | ✗ | ✓ | ✗ | ✗ | N/A | N/A | |
| Williams et al. 201171 | ✗ | ✓ | ✓ | ✗ | ✗ | N/A | |
| Gausachs et al. 201272 | ✗ | ✓ | ✓ | ✗ | ✓ | ✓ | |
| Management only | |||||||
| Vasen et al. 199873 | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ |
| Dunlop et al. 200274 | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ | ✗ |
| Kwon et al. 200875 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✗ |
| Yang et al. 201176 | ✗ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
One consistent problem in terms of the quality of the studies was that the reporting of perspective was poorly done, with 25 studies not stating and justifying their perspective. 22,23,34,49,50,52,53,55–60,62–66,68–73,76 This included three studies55,64,66 not stating a perspective, two70,72 not stating a setting and two23,76 incorrectly stating a health-care perspective as societal.
The depth of detail related to modelling (when it was reported) was mixed and ranged from brief descriptions to full details. Sensitivity analysis was conducted in seven of the diagnostic papers,59,62,65–67,69,72 three of the management papers73,75,76 and all of the diagnostic-plus-management papers. 22,23,48–58 Justification for ranges used in sensitivity analyses was poorly given, if at all. Reporting of sources was generally well done, as was the reporting of sources and methods of estimation for unit costs and quantities.
Of the three groups of studies (diagnostic, management and diagnostic plus management), diagnostic studies seem to be the least well reported; as well as only two61,67 clearly stating and justifying their setting and viewpoint, only three59,67,69 reported a currency and/or price date and only 5 of the 12 models59,62,67,69,72 gave details of the modelling.
As one of our included papers was only a summary,52 it did poorly in the quality assessment. The accompanying full report was in Danish and could not be quality assessed as it was not included in our report.
The small number of management-only papers made it difficult to draw overall conclusions about their quality, but again it appeared to be mixed, with similar problems to the other two groups of studies.
Conclusions
Despite the large number of studies identified by this review, there was still the need for a new model to be developed to address our decision problem.
Firstly, the majority of studies identified by this review addressed either diagnosis or management of LS only, and were further divided by their disease focus (predominantly CRC or EC). Therefore, each of these studies could only attempt to answer part of our study question.
The remaining studies that looked at both diagnosis and management were hindered by the advancements in understanding of LS, and populations and country settings not relevant to our analysis. On the basis of population and the parameter used for CRC risk, the studies could therefore be narrowed down to two that would be directly comparable with our model: Ladabaum and colleagues (2011)48 [plus Wang and colleagues (2012)57] and Mvundura and colleagues (2010). 54 Neither of these were UK-based studies and therefore the results could not necessarily be translated to the NHS.
The wide range of diagnostic strategies across all the studies did not make it clear which tests or combinations thereof would be most cost-effective, particularly in a NHS setting. This therefore justified further modelling.
There had also been little investigation into the modelling of more than one cancer in the long term; in those studies that did investigate more than one cancer, the methods were not clear in terms of whether or not the modelling of interactions between them was actually appropriate. Additionally, adherence to genetic counselling was also something rarely touched upon in detail in the studies. Both of these concerns provided other areas where our model could add value to those that had come before.
Our review agreed with the conclusions of those reviews that had been conducted previously, and this supported our justification for conducting further research into this area.
Chapter 5 Assessment of cost-effectiveness: description of the economic model
Summary of the Peninsula Technology Assessment Group cost-effectiveness analysis
Our model of the cost-effectiveness of systematic screening for LS is comprised of two distinct components: a decision tree model of the short-term outcomes of diagnosis of probands and relatives, and an individual patient simulation model to assess the survival of these patients (the ‘survival’ or ‘long-term outcomes’ model).
The cost-effectiveness of the testing strategies is calculated by comparing the total discounted costs and QALYs across strategies. In keeping with the National Institute for Health and Care Excellence (NICE) reference case, the economic perspective is NHS and PSS, and costs and benefits are discounted at 3.5% per annum.
Costs of interventions were estimated from Department of Health reference costs 2011–12 with inflation to 2013–14 prices or, where this was not possible, from published literature with appropriate conversion and inflation.
Parameters of the natural histories of diseases, the effectiveness of interventions and the impact on quality of life of diseases and interventions were sourced, where possible, from national statistics and published literature (where such parameters were not available, expert opinion and grey data were used).
For each testing strategy, the total discounted cost is calculated as the total cost of testing plus the total discounted cost of treatment after testing. For each testing strategy, the total discounted QALYs are calculated for the patients after testing.
Diagnostic model
The diagnosis of LS is performed for probands with newly diagnosed CRC and their relatives. It is assumed that all diagnosis occurs at time zero.
We consider the following strategies to identify LS in probands:
-
No genetic testing, subdivided:
-
1(1) no testing
-
1(2) AC only.
-
-
IHC four-panel test for MLH1, MSH2, MSH6 and PMS2, followed by genetic testing if IHC result abnormal.
-
IHC four-panel test, followed by BRAF testing for abnormal MLH1 results. Genetic testing is done for any other (not MLH1) abnormal IHC result or for a negative BRAF test (negative for V600E).
-
MSI testing, followed by genetic testing for MSI result.
-
MSI testing, followed by BRAF testing for MSI result, followed by genetic testing for negative BRAF test.
-
MSI testing, followed by BRAF testing for MSI result, followed by IHC testing for negative BRAF test. Genetic testing occurs regardless of IHC result.
-
IHC testing, followed by genetic testing if result abnormal. For normal IHC results: MSI testing, followed by BRAF testing for MSI result followed by genetic testing for negative BRAF test.
-
Universal genetic testing.
There are three possible diagnoses that result from each of these testing strategies: LS mutation positive, LS assumed and LS negative. LS mutation positive occurs when a proband receives a positive genetic test result and LS negative occurs when a proband is ruled out by one of the tests prior to genetic testing, or by FH when genetic testing is either declined or uninformative. ‘LS assumed’ can occur when genetic testing is either uninformative or simply not done (the proband declines testing) and is informed by the proband’s FH. Probands who test mutation positive or LS assumed are offered LS management. It is assumed that only a proportion of probands diagnosed as LS positive or LS assumed accept an offer of LS surveillance.
The diagnosis of LS in relatives of a newly diagnosed CRC proband directly depends on the diagnosis of the proband. Relatives of probands who test mutation positive can be identified as LS positive with a predictive genetic test. FDRs of probands diagnosed as LS assumed cannot be tested, but instead are also assumed to have LS and are offered LS management. Relatives of a higher degree are assumed to be LS negative, as are relatives of probands diagnosed LS negative.
The main outcomes for each testing strategy are:
-
the overall sensitivity and specificity for probands alone
-
the overall sensitivity and specificity for probands and relatives combined
-
the numbers of probands and relatives according to true LS status, diagnosis and management strategy.
The accuracy of individual tests, given by sensitivity and specificity, with regard to either LS in general or to particular genes, were taken from published literature, particularly the Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group Review. 65 Owing to a lack of evidence, the sensitivities and specificities of individual tests are treated as independent of tests performed previously, except for the BRAF test. In common with other cost-effectiveness models [Mvundura and colleagues (2010)54 and Ladabaum and colleagues (2011)48], we assume that the test accuracies apply to all qualities of tumour/blood sample and that tests are always available and successfully produce a result. The only known exception to this is the IHC test, where the failure rate is specifically included in the sensitivity.
There is little published evidence on the rate of acceptance of testing. The following assumptions were made: that the rate of acceptance of a test was independent of any previous tests, and acceptance of one genetic test implied acceptance of all genetic testing. The rate of acceptance of IHC and BRAF was assumed to be the same as for MSI, which was the only tumour-based test for which an estimate was available. The rate of acceptance of genetic counselling allowed for the rate of acceptance of FH screening. For patients who decline genetic counselling and for strategies where genetic testing is not offered, rate of acceptance of FH is estimated separately.
The number of probands aged < 50 years per year in England, 1699, was taken from the Office for National Statistics (ONS). 119 Of these, 8.4%120 were assumed to have LS. These probands were then subdivided into the number with each gene associated with LS.
The number of relatives per proband is set to five in the base case to balance the values from two main UK sources: Barrow and colleagues121 and unpublished data supplied by Ian Frayling (Cardiff University, 2012). Based on two published studies114,120 and these unpublished data, the proportion of relatives that are first degree is estimated at 42%. Next, we estimate that the proportion of relatives that test positive for LS is 44%, based on a random-effects meta-analysis.
The costs of the preliminary tumour tests were obtained either directly from laboratories in the UK or from experts. Costs for genetic tests (for probands and relatives) are taken directly from genetic testing laboratories, identified from the UK Genetic Testing Network (UKGTN). As genetic testing becomes routine, these costs are expected to decrease. We performed a sensitivity analysis to reflect this. The cost of genetic counselling was also included.
The psychological impacts of testing for LS and prophylactic TAHBSO were incorporated into overall health-related quality of life (HRQoL), using data from the literature. All disutilities were assumed to apply for 4 months.
-
A disutility of 0.04 was applied to people declining testing.
-
A disutility of 0.02 was applied to people accepting testing and subsequently being diagnosed with LS.
-
A disutility was applied to people offered TAHBSO and a further disutility was applied if TAHBSO was declined.
Survival model
Survival model structure
The survival/management section of the model takes the form of an individual patient simulation (individual sampling model), in which thousands of hypothetical patients are simulated from the time of LS diagnosis to death or age 100 years (whichever occurs first).
Patients with LS are assumed to have an increased risk of CRC and EC compared with the general population. Other cancers associated with LS, such as ovarian cancer (OC), are not modelled.
The patient state at any time is defined by the following properties, which collectively provide all the information necessary to simulate appropriate treatment pathways and calculate risks of events:
-
whether or not the patient is alive
-
the patient’s age (at the start of the year)
-
the patient’s sex
-
previous bowel surgery
-
whether or not the patient has CRC, and if so, the Dukes’ stage
-
whether or not the patient has had TAHBSO
-
whether or not the patient has EC
-
the patient’s LS status and diagnosis status
-
the patient’s acceptance of LS surveillance if it is offered.
The following clinically important events are simulated:
-
mortality events
-
mortality due to CRC
-
mortality due to EC for women with LS
-
mortality due to causes other than CRC or EC (general mortality)
-
mortality (and morbidity) due to adverse events from surveillance colonoscopies
-
mortality due to adverse events from prophylactic TAHBSO
-
-
non-mortality events
-
index (primary) CRC incidence for relatives only (all probands enter model with recently diagnosed index CRC)
-
metachronous CRC incidence for probands and relatives
-
CRC surgery
-
EC incidence for women with LS
-
TAHBSO for women diagnosed with EC
-
prophylactic TAHBSO for women diagnosed with LS
-
biennial surveillance colonoscopies for patients diagnosed with LS accepting surveillance
-
adverse events from surveillance colonoscopies (bleeding and perforation).
-
The events determine costs incurred and HRQoL for each simulated patient. These are used to estimate the total discounted costs and QALYs for each testing strategy.
Twenty-four patient groups are simulated in the management section of the model, according to:
-
whether proband or relative
-
true LS status
-
whether or not LS has been diagnosed
-
whether or not increased surveillance is accepted as a result of a diagnosis of LS
-
whether male or female.
In the base-case analysis, the maximum age of probands is 50 years. In sensitivity analyses, this is increased to 60 years and separately to 70 years.
Age at entry is a function of sex, true LS status and whether proband or relative. The median age at onset of CRC was 45 years in male probands and 70–74 years for males in the general population.
Colorectal cancer
The rate of incidence of CRC is dependent on the following patient characteristics:
-
age
-
sex
-
whether or not the patient has had a previous CRC
-
time since first CRC
-
LS status
-
whether or not the patient is receiving LS colonoscopic surveillance
-
previous colorectal surgery.
The cumulative risk of CRC to age 70 years is 38% for males with LS and 31% for females with LS, compared with 2.8% for males without LS and 1.8% for females without LS.
The Dukes’ stage of index and metachronous CRCs was recorded. The site of CRCs (colon or rectum) is dependent on sex, LS status and any previous surgery. No disutility was assumed for CRC at Dukes’ stages A–C, but a disutility of 0.13 was assumed for Dukes’ stage D CRC.
People diagnosed with LS are offered 2-yearly colonoscopies. The cost of a colonoscopy was adjusted from £553 to £395 to allow for the fact that the effectiveness of colonoscopy was taken from a regime of 3-yearly colonoscopy. People with CRC who are not diagnosed with LS are offered 5-yearly colonoscopies.
The regime of colonoscopies for people either tested LS positive or assumed to have LS was assumed to reduce the incidence of CRC and to improve the stage distribution of incident CRCs.
-
A hazard ratio (HR) of 0.387 was applied to the incidence of index (first) CRC given LS surveillance colonoscopies compared with no surveillance, and a HR of 0.533 was applied to incidence of metachronous CRC given LS surveillance colonoscopies compared with non-LS surveillance.
-
The CRC stage distribution was improved (69% Dukes’ A, 11% Dukes’ B, 13% Dukes’ C, 8% Dukes’ D) versus individuals not receiving LS surveillance (16% Dukes’ A, 32% Dukes’ B, 27% Dukes’ C, 25% Dukes’ D).
-
The initial rate of adherence to enhanced CRC surveillance depends on whether the person is a proband or relative and whether he or she has tested LS positive or is LS assumed. It is assumed that patients who initially accept LS surveillance continue to receive biennial colonoscopies, and that patients initially declining LS surveillance do not take it up subsequently.
-
No disutility was assumed for people receiving LS surveillance colonoscopies.
Mortality due to CRC is assumed
-
to be higher for more advanced stages of the disease (e.g. Dukes’ D).
-
to be higher for older patients
-
to depend on time since diagnosis
-
to be lower for patients with Dukes’ A or B CRC with LS than for patients without LS. Mortality for Dukes’ C and D CRC is not altered.
The following surgical treatments for CRC are modelled: segmental resection, subtotal colectomy, anterior resection and proctocolectomy. The type of surgery depends on the location of the CRC, the nature of previous surgery and whether or not the patient has been diagnosed with LS. The incidence of CRC was assumed to reduce according to the type of surgery. In the base-case analysis, no additional disutility was assumed for surgery that reduces the risk of CRC. Disutilities were assumed in sensitivity analyses.
The costs of the following treatments for CRC were included: primary surgery, chemotherapy and radiotherapy for primary CRC, follow-up surveillance, stoma care, surgery and chemotherapy for CRC recurrence and palliative care.
Endometrial cancer
Most published models of the cost-effectiveness of testing for LS consider only CRC, not EC and OC. In our model, in addition to CRC we also consider EC, but not OC. If OC were included in our model, it is likely that our estimates of the cost-effectiveness of testing would improve. This is because, though we already cost for prophylactic TAHBSO surgery, which eliminates the risk not just of EC but also of OC, we do not model the associated reduction in the risk of OC.
We do not subdivide EC by stage. Surveillance for EC was not modelled as evidence for its effectiveness is lacking and as it seems unreasonable to include the substantial cost of surveillance with no associated benefit. The cumulative risk of EC at age 70 years was assumed to be 34% for women with LS and 0% for women without LS.
All women are assumed to have a TAHBSO on diagnosis of EC, at a cost of £3900. Prophylactic TAHBSO, at a cost of £3300, is offered to women diagnosed with LS when they reach age 45 years or upon diagnosis of LS if older, with 55% of women accepting TAHBSO. TAHBSO is assumed to eliminate the risk of EC completely. As in previous cost-effectiveness analyses, we do not model complications of TAHBSO, owing to their low incidence. However, we model surgical mortality because it clearly has an impact on total QALYs.
Mortality from EC was assumed to be dependent on time since diagnosis and independent of LS status. Costs for chemotherapy and radiotherapy for EC were modelled.
In the base-case analysis, no disutility was assumed for having EC, nor after TAHBSO. Disutilities were assumed in sensitivity analyses. The cost-effectiveness of genetic testing was found to be very sensitive to the assumed disutility after prophylactic TAHBSO.
Uncertainty
Cost-effectiveness analyses often employ probabilistic sensitivity analysis, whereby uncertainty in many of the parameters is simultaneously considered. We did not use probabilistic sensitivity analysis. Instead, we investigated uncertainty as follows. First, we performed the following scenario analyses.
-
EC and prophylactic TAHBSO were excluded to assess their impact on the cost-effectiveness results.
-
The maximum age of probands was increased from the base case of 50 years to 60 years, and separately to 70 years.
-
BRAF testing, which was assumed in some testing strategies in the base case, was replaced by methylation testing.
Second, the sensitivity of cost-effectiveness to individual parameters was assessed either by doubling and halving the parameter value (e.g. the cost of all genetic tests), or by selecting a different plausible data source (e.g. for the disutilities due to CRC).
Model structure
To model the impact of systematically screening for LS, we created a model with two distinct sections: a decision tree model to investigate the short-term outcomes of strategies to identify LS patients, and an individual patient simulation model to assess the long-term implications of strategies to identify and manage LS. These are discussed in detail in the following sections.
Diagnostic testing model
Here, we consider two groups: the newly diagnosed CRC patients aged under 50 years (the probands), and their biological relatives. These groups are connected, as shown in Figure 3. This section of the model was built as a decision tree and has no time component. It therefore assumes that diagnosis occurs instantaneously, though the diagnosis of LS may actually take up to several months or even years; laboratories report waiting times of around 20 working days for tumour-based tests, 40 working days for gene sequencing for probands and 10–20 working days for predictive genetic testing for relatives. 122–126 This also does not account for the time taken to collect a tissue/blood sample, transport the sample and arrange counselling sessions, nor when in a proband’s treatment this process is begun. According to our clinical experts (Carole Brewer and Ian Daniels, Royal Devon and Exeter NHS Trust), it is also not standard practice to have LS results available before the proband’s treatment is given. Therefore, to emulate standard practice, where the proband’s LS status is not yet known, we assume that he or she receives standard treatment for CRC. In most cases this is surgical resection with the possibility of chemotherapy or radiotherapy, depending on the stage of the cancer.
FIGURE 3.
Overview decision tree for diagnosing LS.
Diagnostic strategies for probands
Probands follow one of nine diagnostic strategies, chosen on the basis of available tests, existent cost-effectiveness models, expert advice and requirements of the project scope. As the only conclusive test for LS is constitutional (‘germline’) genetic testing, this is used in every testing strategy. We use three well-documented preliminary tumour-based tests: IHC, MSI and BRAF. There is little consistency in the literature about the usage of these tests, either in sequence or individually, so we include a variety of testing strategies. Although FH, for example assessed by the AC, is no longer recommended as a selection tool for the diagnosis of LS, we include it as one of the testing strategies for comparison with the genetic testing strategies. 13 We compare the following eight strategies:
-
No systematic testing to identify LS
-
No testing to identify LS
-
AC
-
-
IHC four-panel test for MLH1, MSH2, MSH6 and PMS2, followed by genetic testing if IHC result abnormal. 54
-
IHC four-panel test, followed by BRAF testing for abnormal MLH1 results. Genetic testing is done for any other (not MLH1) abnormal IHC result or for a negative BRAF test (negative for V600E). 54
-
MSI testing followed by genetic testing for MSI result. 54
-
MSI testing, followed by BRAF testing for MSI result, followed by genetic testing for negative BRAF test (advised by Ian Frayling).
-
MSI testing, followed by BRAF testing for MSI result, followed by IHC testing for negative BRAF test. Genetic testing occurs regardless of IHC result (advised by Ian Frayling).
-
IHC testing, followed by genetic testing if result abnormal. For normal IHC results: MSI testing, followed by BRAF testing for MSI result followed by genetic testing for negative BRAF test (advised by Ian Frayling).
-
Universal genetic testing (i.e. as first and only test for all) (asked for in the project scope).
Figures 4–11 show further details of these eight strategies.
FIGURE 4.
Strategy 1: no testing.
FIGURE 5.
Strategy 2: IHC test followed by genetic testing.
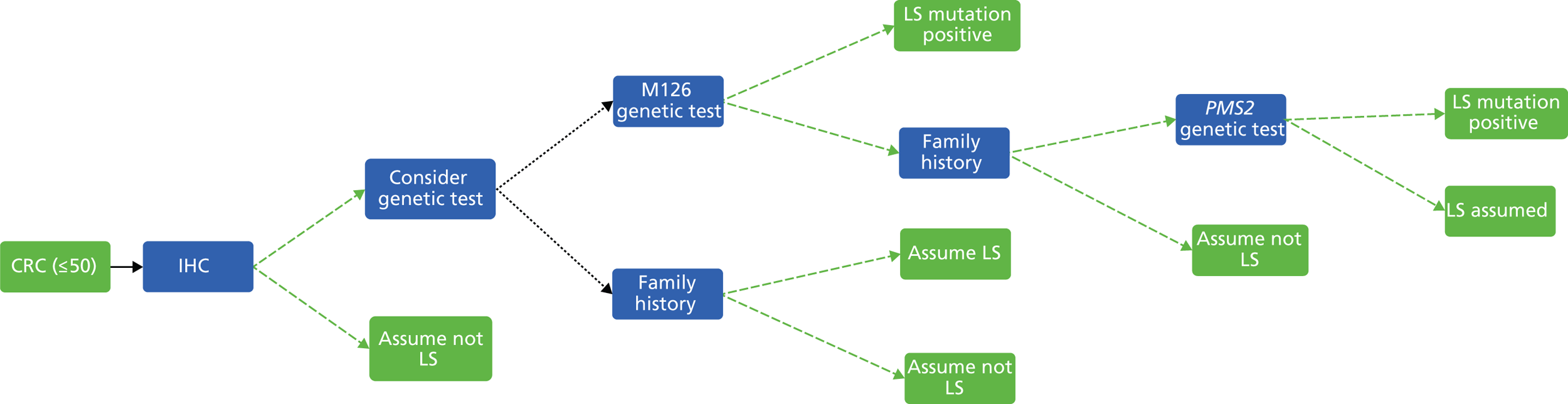
FIGURE 6.
Strategy 3: IHC test followed by BRAF testing and genetic testing.
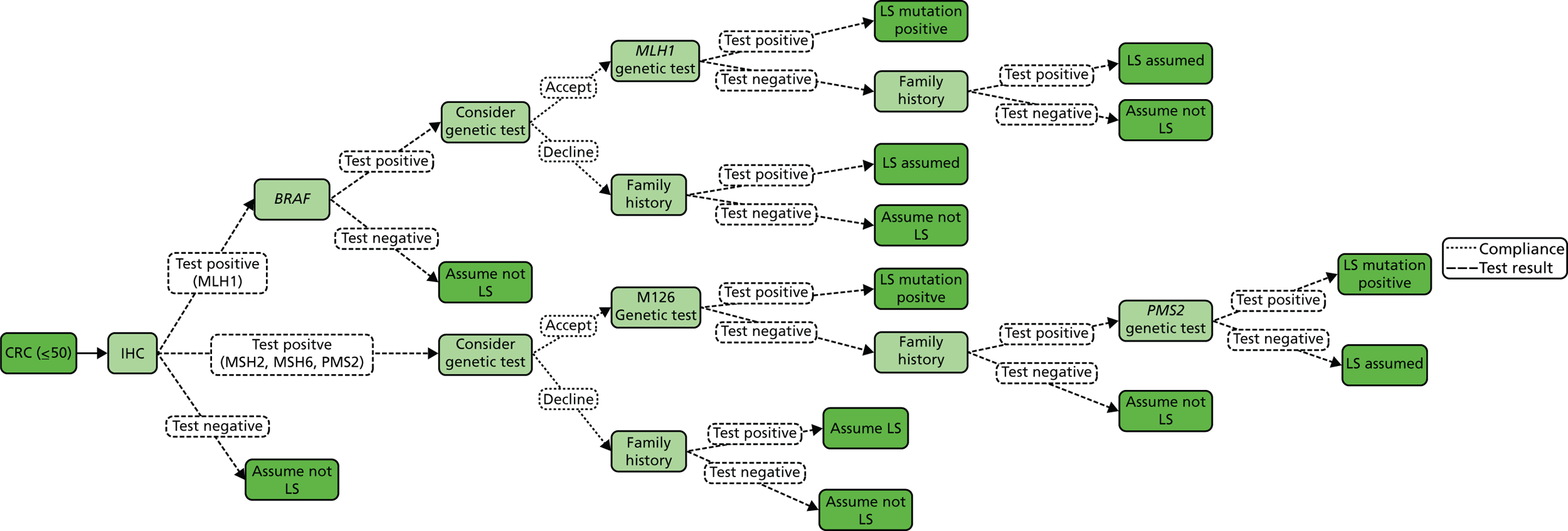
FIGURE 7.
Strategy 4: MSI test followed by genetic testing.
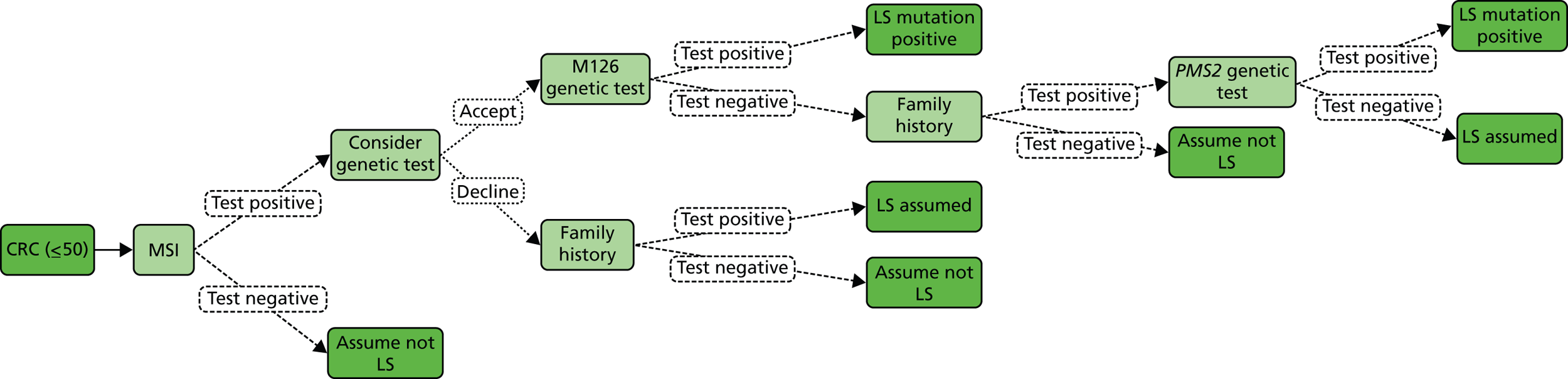
FIGURE 8.
Strategy 5: MSI test followed by BRAF test and genetic testing.
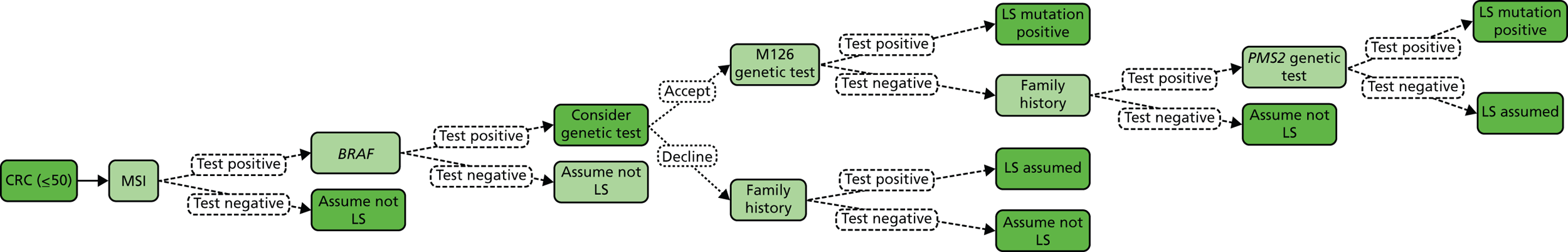
FIGURE 9.
Strategy 6: MSI test followed by BRAF, IHC and genetic testing.
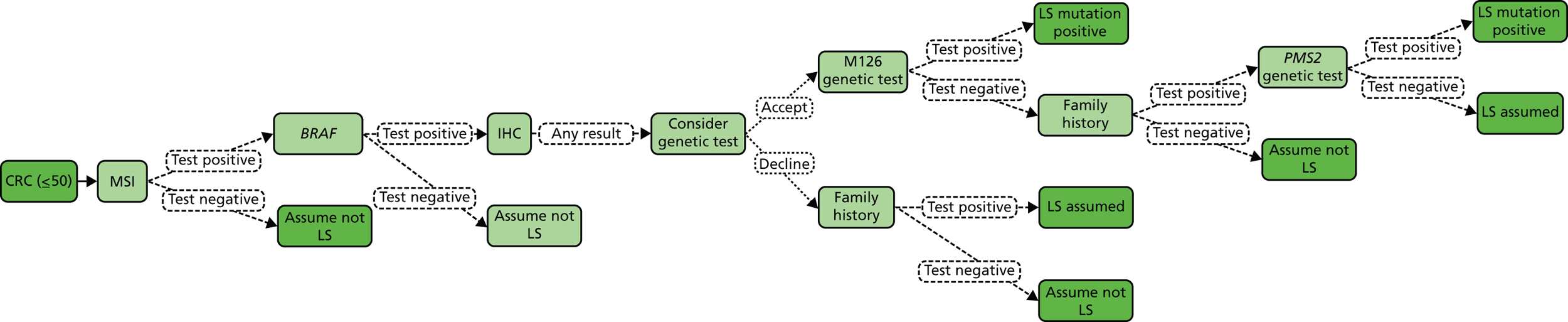
FIGURE 10.
Strategy 7: IHC test followed by MSI, BRAF and/or genetic testing.
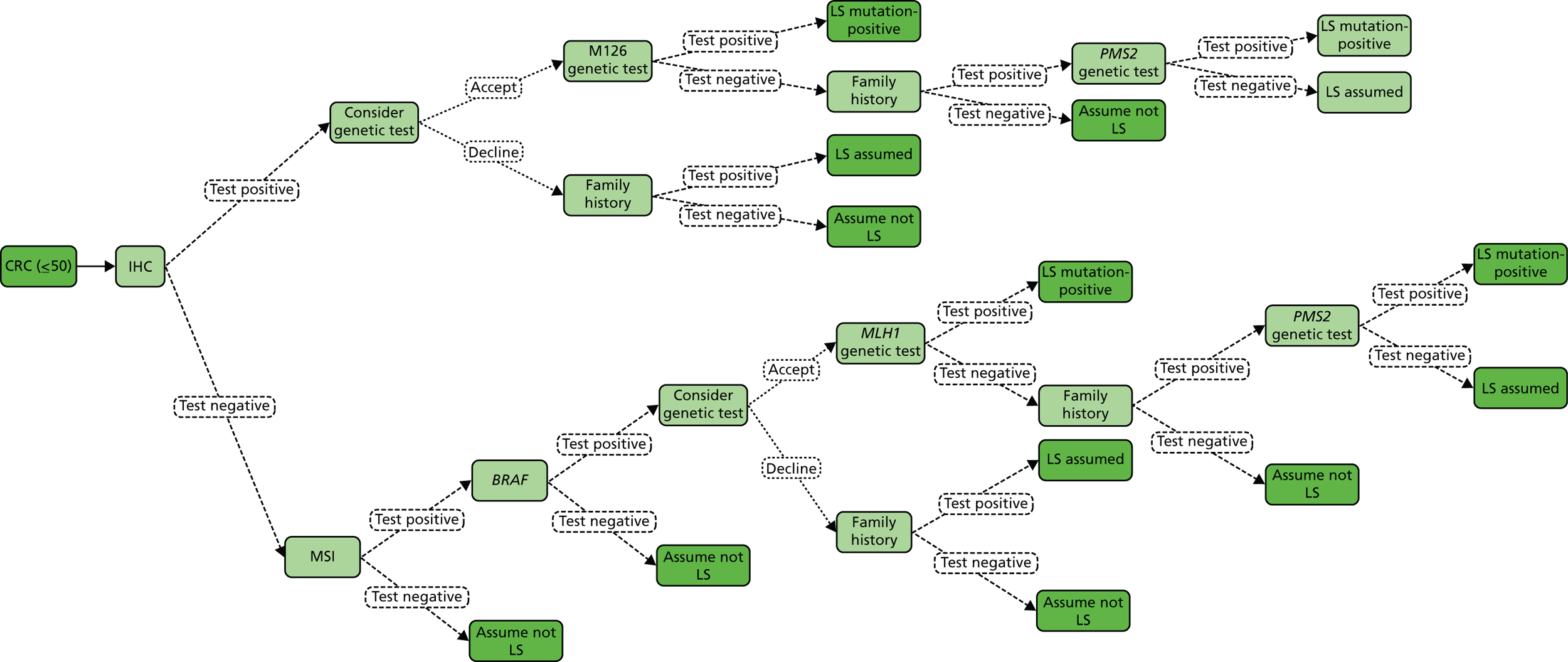
FIGURE 11.
Strategy 8: universal genetic testing.
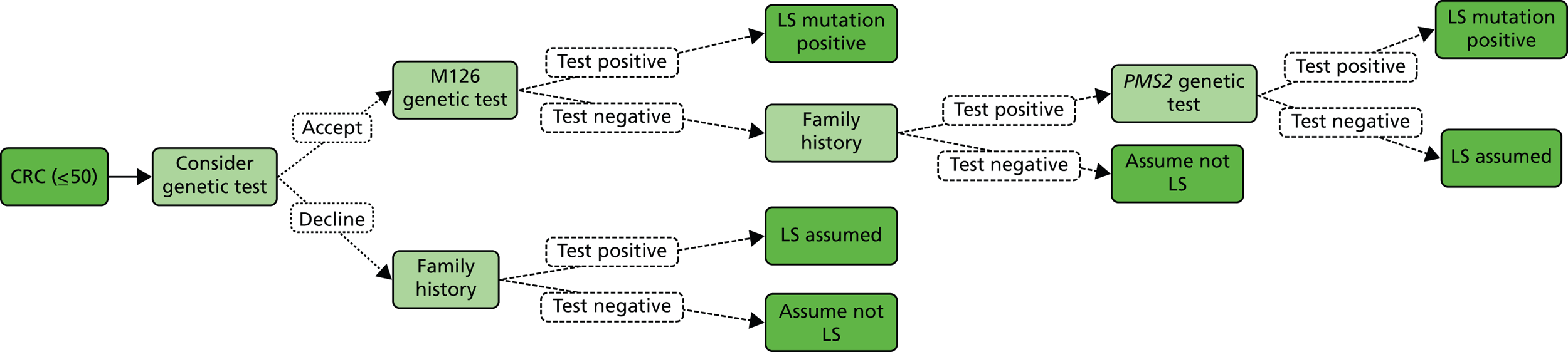
There are three possible diagnoses given by these testing strategies: LS mutation positive, LS assumed and LS negative. LS mutation positive occurs when a proband receives a positive genetic test result and LS negative occurs when a proband is ruled out by one of the tests prior to genetic testing, or by FH when genetic testing is either declined or uninformative.
‘LS assumed’ can occur when genetic testing is either uninformative or simply not done (the proband declines testing). For example, we assume that a negative genetic test result is uninformative for probands, meaning that although the test did not detect LS, it does not rule it out. This is because there is still debate over which genes and mutations cause LS, some of which will not be identified by current genetic tests. Our experts agree that this is a plausible interpretation as current practice does not necessarily rule out LS on the basis of a genetic test, especially with regard to LS management strategies. The outcome allows us to reflect clinical practice, whereby management strategies are offered to both LS mutation-positive and LS-assumed probands (regular surveillance) and with regard to strategies identifying LS in relatives of both probands diagnosed as LS mutation positive and LS assumed. In practice, the decision to diagnose a proband as LS assumed is informed by FH and we model it similarly. As our probands already fulfil the Revised Bethesda guidelines, we model this based on the AC II. We also use this approach to decide how many of the probands who tested negative for mutations in MLH1, MSH2 and MSH6 should be put forward for PMS2 testing. We do this to reflect the smaller number of PMS2 tests carried out in current practice, compared with testing for MLH1, MSH2 and MSH6. This occurs because testing PMS2 is technically challenging and hence more expensive, and there are correspondingly fewer laboratories capable of testing PMS2 (in the UK there is currently only one126). Using the AC II, instead of a fixed proportion, allows the flexibility of a changing population; the proportion of patients at diagnosis of CRC with a FH will not be the same as the proportion of patients with a FH after tumour testing.
Figures 5–11 show that we split genetic testing into simultaneous testing for MLH1, MSH2 and MSH6 (abbreviated to M126), and a separate test for PMS2 where necessary. This strategy has been adopted in current practice where available, and is likely to become standard as three-gene sequencing becomes more widely available, according to our expert advisors. The only strategy where this differs slightly is strategy 3, where probands with a MLH1 (with or without a PMS2) abnormal IHC result, followed by a BRAF test where V600E is not found, have a single genetic test for MLH1. This is to reflect the phenomenon that a mutation in the MLH1 gene can cause abnormal MLH1 with abnormal PMS2 results in IHC; PMS2 mutations can only cause abnormal IHC results for PMS2; MSH2 mutations can cause abnormal MSH2 and MSH2 with MSH6 IHC results; and MSH6 mutations can only cause abnormal MSH6 IHC results. 127 Those with an abnormal IHC result in any of the other genes follow the genetic testing route of the other strategies, M126 followed by PMS2. In all other strategies, we do not attempt to identify the gene responsible before the genetic testing process, as there can be discrepancies in the quality and interpretation of tumour testing (particularly IHC) between laboratories. 128 It is also assumed that if a proband complies with some genetic testing, he or she agrees to all of it.
It is assumed that only a proportion of probands diagnosed as LS positive or LS assumed accept an offer of LS surveillance.
In Figures 5–10, IHC refers to IHC testing associated with all four known LS genes (MLH1, MSH2, MSH6 and PMS2). Though MSI is simply the initialised form of ‘microsatellite instability’, in our case we use it to mean the associated test for microsatellite instability rather than the state itself. In the diagrams, all positive test results indicate (do not rule out) LS. In other words, the test results reflect the LS diagnosis; so, for example, a positive BRAF test result for V600E is indicated as a negative test result for LS.
In the strategies using MSI, test sensitivities are higher for MLH1 and MSH2 than for MSH6 and PMS2, to reflect the known reduced sensitivity associated with the last two genes. 65 Thus, the MSI section of the model is split into MLH1 with MSH2 (M12) and MSH6 with PMS2 (M6P2), with results reported for both individual genes and overall (as it is actually only one test). The individual gene results are then incorporated into the prevalence at the next stage of the strategy by combining them appropriately. For example, in the case of strategies using MSI, the prevalence after MSI is split between M12 and M6P2 (given the information we have for the sensitivity and specificity of MSI), but as the genetic test for M126 only looks at three of those genes, the input prevalence for the M126 genetic test must only include the prevalences of mutations in MLH1, MSH2 and MSH6. The prevalence for P2 is carried through in the negative population so that it is later used in PMS2 genetic testing.
Testing outcomes for probands
The primary outputs from the short-term model for each testing strategy that lead into the survival (i.e. long-term) section of the model are:
-
number of probands with LS receiving LS surveillance
-
number of probands with LS not receiving LS surveillance (probands will receive some surveillance in line with BSG guidelines9); these are split into those identified as LS positive but who declined surveillance, and those who were diagnosed LS negative
-
number of probands without LS receiving LS surveillance
-
number of probands without LS who do not receive LS surveillance (probands will receive some surveillance in line with BSG guidelines9); these are split into those identified as LS positive but who declined surveillance, and those who were diagnosed LS negative.
Other outcomes include overall sensitivity and specificity of each diagnostic strategy. Probands who are diagnosed as LS (either mutation positive or assumed), but who refuse surveillance may still be offered prophylactic surgery for metachronous CRC.
Diagnostic strategies for relatives
Testing of relatives depends on whether the proband was diagnosed as LS positive, LS assumed or LS negative, as follows.
Relatives of probands diagnosed as Lynch syndrome mutation positive
The decision tree for relatives of these probands is shown in Figure 12.
FIGURE 12.
Decision tree for relatives of probands diagnosed LS mutation positive.
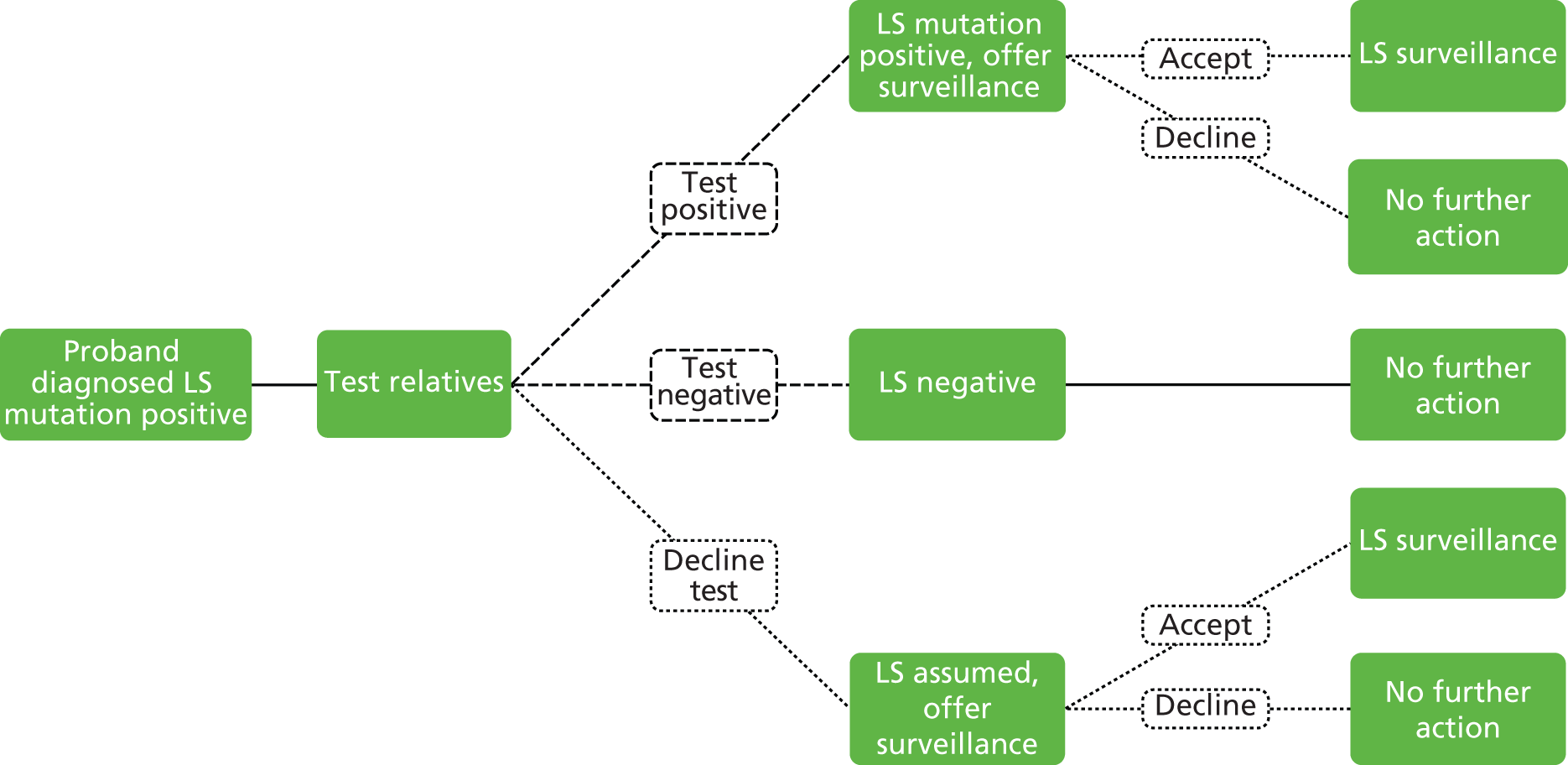
Relatives of probands who receive a positive genetic test result are offered a predictive genetic test which targets the particular gene (MLH1, MSH2, MSH6 or PMS2) found to be mutated in the proband. Relatives who accept are then diagnosed as LS mutation positive or LS negative depending on their test result. We assume that the targeted testing for relatives is 100% accurate, so we do not include an option for inconclusive tests. This assumption is confirmed as current best practice by our expert advisors and is also made in other models. 54 Additionally, on advice from our clinical experts, the small numbers of relatives at high risk of LS, but who decline targeted genetic testing, are diagnosed as LS assumed and are offered LS surveillance. Relatives diagnosed as LS mutation positive or LS assumed are offered risk-reducing surgery if they develop CRC, regardless of their surveillance status.
Relatives of probands diagnosed as Lynch syndrome assumed
The decision tree for relatives of these probands is shown in Figure 13.
FIGURE 13.
Decision tree for relatives of probands diagnosed LS assumed.

As agreed by our advisors, FDRs of probands who are diagnosed as LS assumed are also assumed to have LS and are therefore at sufficiently high risk of LS to be offered LS surveillance, as targeted genetic testing for relatives cannot be done without a specific mutation for which to test. Higher-degree relatives (e.g. cousins, grandchildren, nieces and nephews, etc.) of this group are assumed to be LS negative, and therefore do not warrant being offered LS surveillance.
Relatives of probands diagnosed as Lynch syndrome negative
All relatives of probands who are diagnosed LS negative are assumed to be LS negative also, and these relatives are subject to no LS tests or LS surveillance (Figure 14).
FIGURE 14.
Decision tree for relatives of probands diagnosed as LS negative.

Testing outcomes for relatives
The primary short-term model outputs are:
-
number of relatives with LS receiving LS surveillance
-
number of relatives with LS not receiving LS surveillance (split into those identified as LS positive but who declined surveillance, and those who were diagnosed LS negative)
-
number of relatives without LS receiving LS surveillance
-
number of relatives without LS who do not receive surveillance (split into those identified as LS positive but who declined surveillance, and those who were diagnosed LS negative).
The sensitivity and specificity for each testing strategy for relatives is recorded.
Long-term management model
All patients continue from the diagnostic section of the model to the survival section (this includes all probands and their relatives, irrespective of their adherence and diagnosis), in which a number of outcomes are simulated.
Outcomes are simulated for 24 patient groups, which are all combinations of:
-
male/female
-
proband/relative
-
truly LS positive/negative
-
LS diagnosed and surveillance accepted/LS diagnosed and surveillance declined/LS not diagnosed.
The survival section of the model is an individual sampling model. Each patient is simulated for 1 year at a time and starts each year in a particular health state. A cycle length of 1 year was chosen because it was judged reasonable to assume constant hazard from various risks for a year, and indeed most parameters informing the model are based on annual measurements of risk.
During each year there are a number of competing risks (Table 39) to which the patient may be exposed, and these are all assumed to have a constant hazard rate throughout the year (with the exception of colonoscopies, which have a different mechanism; see Morbidity and mortality of Lynch syndrome surveillance colonoscopy). For each competing risk, a time to event is drawn randomly according to the hazard rate of that event. If none of the times is within a year, then no event has occurred and the patient commences the next year in the same state. If at least one of the times is within a year, then the earliest of the times determines which event occurs and the state is updated accordingly for the next year. Figure 15 gives a graphical representation of the events and states in the model, in the form of an influence diagram.
| Patient group | Competing events | Non-competing events |
|---|---|---|
| All patients | General mortality | |
| Patients undergoing LS surveillance (aged 25–75 years) | Mortality following colonoscopy | Adverse events (includes bleeding and perforation) following colonoscopy |
| Patients with CRC (aged < 75 years) | Mortality following colonoscopy | Adverse events (includes bleeding and perforation) following colonoscopy |
| Patients with CRC | CRC mortality | |
| Patients with an index CRC (without metachronous CRC) | Metachronous CRC incidence | |
| Patients without CRC | CRC incidence | |
| LS females without EC | EC incidence | |
| LS females with EC | EC mortality | |
| Females diagnosed with LS without EC | Mortality following prophylactic TAHBSO | Prophylactic TAHBSO |
FIGURE 15.
Influence diagram of management model. (M)CRC, (metachronous) CRC.
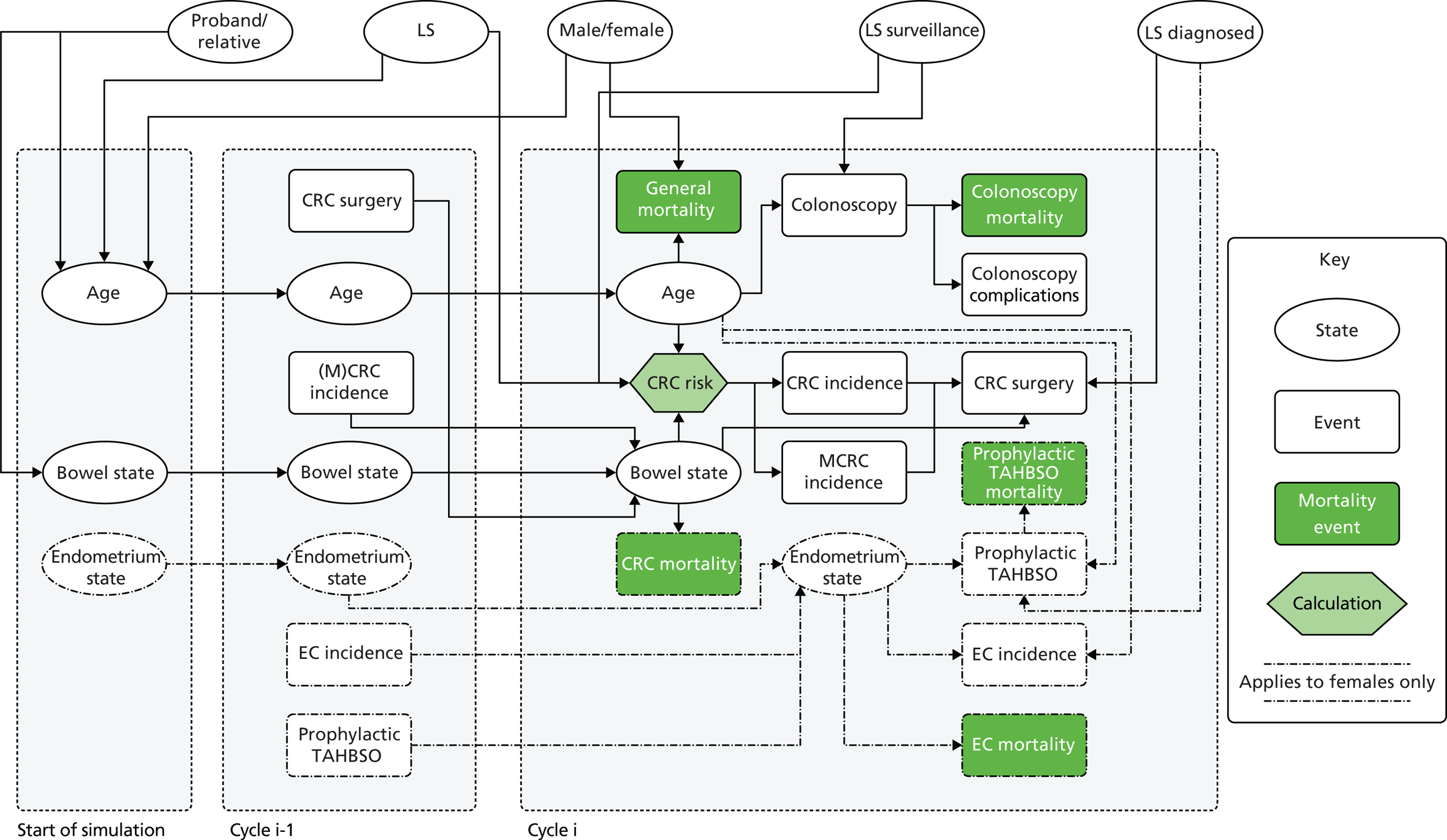
Description of individual sampling model
The structure of the economic model has been described as an ‘individual sampling model’ in two review articles,129,130 defined as a model which tracks individual patients independently. This is to be contrasted with models which allow for interactions between patients, for example for infectious diseases, and models that track groups or cohorts of patients simultaneously. Barton and colleagues (2004)129 recommend individual sampling models when there are no interactions between patients, and when a cohort-based model would require an excessive number of health states.
Indeed, we chose an individual sampling model structure for precisely these two reasons. Patient interactions are assumed to be completed during the diagnostic testing and do not factor into long-term management. A cohort-based model would require an excessive number of health states to represent the complexity of the natural histories of multiple diseases (CRC and EC) and the health-care processes to prevent and treat those diseases (colonoscopy, risk-reducing surgery). For example, if we chose to construct a cohort model in which up to two CRCs were modelled (including their Dukes’ stages), with four surgery types and EC and TAHBSO modelled (which would approach the complexity of the individual sampling model), we would require almost 150 different health states (Table 40). Even with this many health states, we would need to make simplifications to fit the Markov memory-less property for transitions between states, and the model would need to be replicated 24 times to cover the different subgroups of patients.
| Adaptation | Additional states required | Total number of Markov states |
|---|---|---|
| Basic model: healthy/CRC/dead | 3 | 3 |
| MCRC | +1 | 4 |
| Dukes’ stages (four first CRC, 16 first/second CRC pairs) | +18 | 22 |
| Surgery types (none,a segmental resection,b anterior resection,b subtotal colectomy, proctocolectomy) | +28 | 50 |
| EC | +49 | 99 |
| TAHBSO | +49 | 148 |
More specifically, our model structure is denoted ‘D2’ in the notation of Brennan and colleagues (2006),130 as it is at the patient level, does not simulate patient interactions and does not assume the Markov memory-less property for transitions between health states.
The model uses a hybrid approach between discrete and continuous time periods, in which there are discrete cycles of length 1 year but events may occur at any point during that year, rather than being assumed to occur at the beginning, end or midpoint, as is the case in general discrete time models. Figures 16–18 demonstrate this.
FIGURE 16.
No event occurs.
FIGURE 17.
Colorectal cancer incidence occurs.
FIGURE 18.
Colorectal cancer mortality occurs.
In Figure 16 a time to event of 9.0 years is drawn for CRC incidence and a time to event of 5.3 years is drawn for general mortality. Neither of these events falls within 1 year, so no event occurs and time in the model moves on by a year. In Figure 17 a time to event of 0.8 years is drawn for CRC incidence and a time to event of 8.8 years is drawn for general mortality. CRC incidence falls within 1 year, and hence CRC incidence occurs at a time of 0.8 years. In Figure 18 a time to event of 0.6 years is drawn for CRC mortality and a time to event of 0.9 years is drawn for general mortality. Both times fall within 1 year but 0.6 years is the earliest, and hence CRC mortality occurs.
Tables 41–44 below give example individual patient simulation (IPS) traces from the model. Note that many of the times generated seem implausible, for example general mortality is predicted to be thousands of years in the future; this is because in each cycle the incidence rate is constant, and note that the only important aspect in these instances is that the time generated is greater than 1 year. Events occur when a simulated time to event is generated which is < 1 year (see shaded areas).
| Cycle | Age (years) | Alive (yes/no) | Bowel state | Times to competing events (years) | Times to non-competing events (years) | |
|---|---|---|---|---|---|---|
| General mortality | CRC mortality | CRC incidence | ||||
| 0 | 30 | Yes | Healthy | 1502 | N/A | 703.1 |
| 1 | 31 | Yes | Healthy | 4297 | N/A | 583.0 |
| 2 | 32 | Yes | Healthy | 151.8 | N/A | 2.719 |
| . . . | . . . | . . . | . . . | . . . | . . . | . . . |
| 57 | 87 | Yes | Healthy | 9.423 | N/A | 1159 |
| 58 | 88 | Yes | Healthy | 0.279 | N/A | 150.7 |
| 59 | 89 | No | N/A | N/A | N/A | N/A |
| Total life-years | 58.279 | |||||
| Cycle | Age (years) | Alive (yes/no) | Bowel state | Times to competing events (years) | Times to non-competing events (years) | |
|---|---|---|---|---|---|---|
| General mortality | CRC mortality | MCRC incidence | ||||
| 0 | 41 | Yes | Dukes’ A | 1918 | 178.6 | 5165 |
| 1 | 42 | Yes | Dukes’ A | 515.0 | 27.55 | 17,602 |
| 2 | 43 | Yes | Dukes’ A | 6.666 | 14.22 | 16,369 |
| . . . | . . . | . . . | . . . | . . . | . . . | . . . |
| 25 | 66 | Yes | Dukes’ A | 36.49 | 1.174 | 1320 |
| 26 | 67 | Yes | Dukes’ A | 50.85 | 0.287 | 69.67 |
| 27 | 68 | No | N/A | N/A | N/A | N/A |
| Total life-years | 26.287 | |||||
| Cycle | Age (years) | Alive (yes/no) | Bowel state | Endometrium state | Number of colonoscopies | Times to competing events (years) | Times to non-competing events (years) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| General mortality | CRC mortality | EC mortality | Colonoscopy mortality | CRC incidence | EC incidence | ||||||
| 0 | 50 | Yes | Healthy | Healthy | 1 | 420.8 | N/A | N/A | M | 15.51 | 30.56 |
| 1 | 51 | Yes | Healthy | Healthy | 0 | 103.1 | N/A | N/A | N/A | 552.1 | 21.47 |
| 2 | 52 | Yes | Healthy | Healthy | 1 | 395.4 | N/A | N/A | M | 701.5 | 32.05 |
| . . . | . . . | . . . | . . . | . . . | . . . | . . . | . . . | . . . | . . . | . . . | . . . |
| 21 | 71 | Yes | Healthy | Healthy | 0 | 56.37 | N/A | N/A | N/A | 73.44 | 98.68 |
| 22 | 72 | Yes | Healthy | Healthy | 1 | 0.646 | N/A | N/A | M | 242.9 | 409.2 |
| 23 | 73 | No | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| Total life-years | 22.646 | ||||||||||
| Cycle | Age (years) | Alive (yes/no) | Bowel state | Number of colonoscopies | Times to competing events (years) | Times to non-competing events (years) | |||
|---|---|---|---|---|---|---|---|---|---|
| General mortality | CRC mortality | Colonoscopy mortality | CRC incidence | MCRC incidence | |||||
| 0 | 45 | Yes | Healthy | 1 | 647.5 | N/A | M | 5.847 | N/A |
| 1 | 46 | Yes | Healthy | 0 | 71.86 | N/A | N/A | 48.04 | N/A |
| 2 | 47 | Yes | Healthy | 1 | 268.2 | N/A | M | 264.7 | N/A |
| . . . | . . . | . . . | . . . | . . . | . . . | . . . | . . . | . . . | . . . |
| 48 | 93 | Yes | Healthy | 0 | 2.107 | N/A | N/A | 0.014 | N/A |
| 49 | 94 | Yes | Dukes’ A | 0 | 0.272 | 12.09 | N/A | N/A | 30.39 |
| 50 | 95 | No | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| Total life-years | 49.272 | ||||||||
Patient state
The patient state at any time is defined by a number of properties, which collectively provide all the information necessary to select appropriate treatment pathways and calculate risks of events. The patient state is composed of:
-
whether or not the patient is alive
-
the patient’s age (at the start of the year)
-
the patient’s sex
-
the patient’s bowel state (defined below)
-
whether or not the patient has had TAHBSO
-
whether or not the patient has EC
-
the patient’s LS status and diagnosis status
-
the patient’s acceptance of LS surveillance if it is offered.
Patient bowel state
The patient bowel state encapsulates whether or not the patient has a clinically diagnosed CRC and the extent of any bowel surgery. Though it is possible within an individual sampling model to track a number of primary CRCs and their properties, we make the simplifying assumption that each patient will have no more than two primary CRCs throughout his or her life (as in other decision models, e.g. Mvundura and colleagues54).
The Dukes’ CRC stage is used as a measure of how advanced a CRC is, from A to D, as described in Table 45. More recently the tumour node metastasis (TNM) system has been used to stage cancers, but we did not identify data sources for incidence or survival according to TNM stage and so use Dukes’ stage.
| Dukes’ stage | AJCC stage | TNM equivalent | Description | ||
|---|---|---|---|---|---|
| A | I | T1 T2 |
N0 N0 |
M0 M0 |
Tumour invades submucosa or tumour invades muscularis propria; no regional lymph node metastasis; no distant metastasis |
| B | IIA | T3 | N0 | M0 | Tumour invades through the muscularis propria into pericolorectal tissues or tumour penetrates to the surface of the visceral peritoneum or tumour directly invades or is adherent to other organs or structures; no regional lymph node metastasis; no distant metastasis |
| IIB | T4a | N0 | M0 | ||
| IIIC | T4b | N0 | M0 | ||
| C | IIIA | T1–T2 T1 |
N1/N1c N2a |
M0 M0 |
Tumour invades submucosa or tumour invades muscularis propria or tumour invades through the muscularis propria into pericolorectal tissues or tumour penetrates to the surface of the visceral peritoneum or tumour directly invades or is adherent to other organs or structures; metastasis in at least one regional lymph node; no distant metastasis |
| IIIB | T3–T4a T2–T3 T1–T2 |
N1/N1c N2a N2b |
M0 M0 M0 |
||
| IIIC | T4a T3–T4a T4b |
N2a N2b N1–N2 |
M0 M0 M0 |
||
| D | IVA | Any T | Any N | M1a | Distant metastasis (in one or more organ or site) |
| IVB | Any T | Any N | M1b | ||
In the model we keep track of the Dukes’ stages of both primary cancers (as appropriate) and how long it has been since the cancer was diagnosed. For example, a patient may have experienced a first CRC Dukes’ A 12 years ago and a metachronous CRC with Dukes’ stage C 2 years ago.
We model two portions of the bowel: the colon and the rectum. CRCs can develop in any portion of the bowel still intact. We model four surgery types, based on the extent of bowel removed (Table 46). This is a small extension to the three surgery types used in Maeda and colleagues86 to account for the fact that rectal cancer can be the first primary cancer in our cohort. See Colorectal cancer surgical management pathways for details on surgical management.
| Surgery | Bowel removed |
|---|---|
| Segmental colon resection | Part (but not all) of the colon |
| Subtotal colectomy | All of the colon |
| Anterior resection | All of the rectum |
| Proctocolectomy | All of the colon and rectum |
Outcomes
For each diagnostic testing strategy, the primary outcomes of the survival model are the QALYs lived and the total care costs accrued, both discounted and undiscounted. These are then combined with the outcomes from each diagnostic testing strategy to obtain the overall expected QALYs and costs for each strategy.
The following secondary outcomes are also reported:
-
life-years lived (discounted and undiscounted)
-
number of incident CRCs
-
number of incident ECs
-
number of colonoscopies performed
-
disaggregated costs (costs of surveillance, colonoscopy complications, CRC diagnosis, surgery, chemotherapy and follow-up, EC surgery and treatment), discounted and undiscounted.
Perspective, discounting, time horizon
The perspective of the analysis is that of the NHS and PSS. In keeping with the NICE reference case,132 costs and benefits are discounted at 3.5% per annum and the time horizon is lifetime or age 100 years (whichever is earlier).
Surveillance pathways
Surveillance pathways are based on published guidelines by the BSG and ACPGBI,9 NICE guidance133 and a report commissioned by the Department of Health. 134
Surveillance pathways are different for patients affected and unaffected by CRC, as patients affected by CRC are at risk of cancer recurrence and metastatic disease. Surveillance pathways are also different for patients diagnosed with LS or suspected/assumed to have LS because the risk of CRC incidence is greatly increased.
There are no evidence-based standard follow-up pathways for CRC,9,133,134 although there is some evidence that follow-up improves overall survival. NICE guidelines make some minimum recommendations:133
A minimum of two CTs [computerised tomography scans] of the chest, abdomen and pelvis in the first 3 years.
Regular serum carcinoembryonic antigen tests (at least every 6 months in the first 3 years).
Offer a surveillance colonoscopy at 1 year after initial treatment. If this investigation is normal consider further colonoscopic follow-up after 5 years, and thereafter as determined by cancer networks.
Trueman and colleagues134 describe 3-monthly carcinoembryonic antigen (CEA) tests as common for the first 2 years, followed by 6-monthly tests for the following 3 years. The BSG/ACPGBI guidelines suggest that 5-yearly colonoscopies should continue until comorbidities outweigh the benefits. 9
The BSG/ACPGBI guidelines suggest that patients diagnosed with LS should receive 2-yearly colonoscopy commencing at age 25 years until the age of 70–75 years, or until comorbidity makes colonoscopy clinically inappropriate, and also suggest that surveillance every 18 months may be appropriate. 9
Table 47 describes the surveillance pathways as included in the model, which are an attempt by the Peninsula Technology Assessment Group (PenTAG) to unify the various recommendations.
| Patient type | Surveillance | Source |
|---|---|---|
| Affected by CRC; diagnosed with LS | CEA test every 3 months for 2 years after resection CEA test every 6 months from 2 years after resection until 5 years after resection CT scan of chest, abdomen and pelvis 1 and 2 years after resection Colonoscopy 1 year after resection 2-yearly colonoscopy to age 75 years |
NICE 2011133 BSG/ACPGBI 20109 Trueman et al. 2007134 |
| Affected by CRC; not diagnosed with LS | CEA test every 3 months for 2 years after resection CEA test every 6 months from 2 years after resection until 5 years after resection CT scan of chest, abdomen and pelvis 1 and 2 years after resection Colonoscopy 1 year after resection 5-yearly colonoscopy to age 75 years |
NICE 2011133 BSG/ACPGBI 20109 Trueman et al. 2007134 |
| Unaffected by CRC; diagnosed with LS | 2-yearly colonoscopy from age 25 to age 75 years | BSG/ACPGBI 20109 |
| Unaffected by CRC; not diagnosed with LS | None |
Colorectal cancer surgical management pathways
Colorectal cancer surgical pathways are based on published guidelines9 with input from clinical experts.
Patients undergo surgical management if they are diagnosed with CRC and the cancer is deemed to be operable (this includes surgery where intent is palliative rather than curative). We make the simplifying assumption that all patients diagnosed with CRC undergo surgical management [over 75% of patients in the National Bowel Cancer Audit (2011)135 were treated surgically]. In each case, surgery would remove the bowel portion affected by the cancer, and in some cases additional portions, depending on previous surgery and whether or not LS had been diagnosed [Figure 19, adapted from Maeda and colleagues (2010)86]. Our clinical expert advice is that, in general, surgery for patients without LS tends to be conservative, without a risk-reducing element.
FIGURE 19.
Surgical management pathways for CRC.
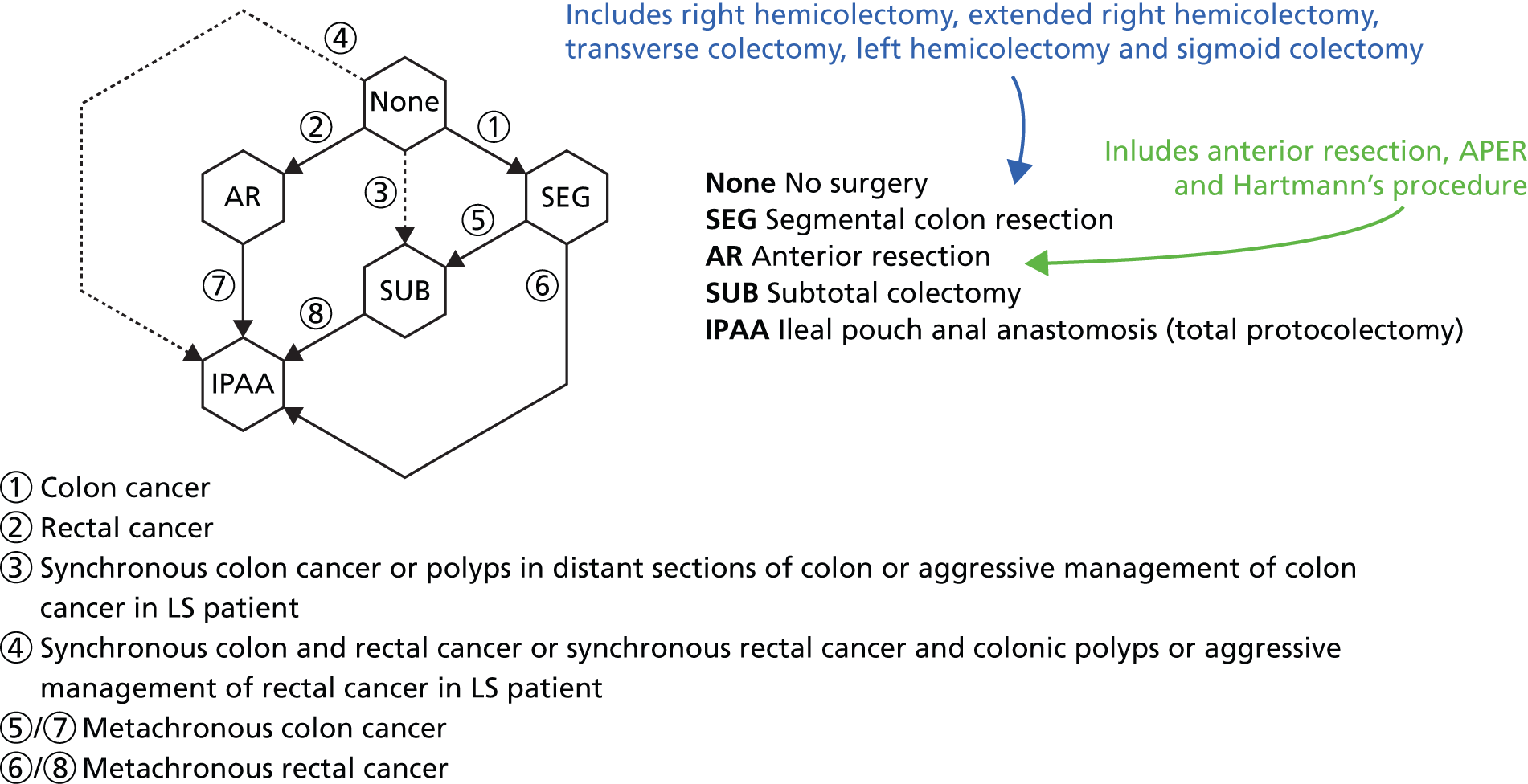
Clinical guidelines indicate that there is a place for more aggressive surgery, with a risk-reducing element, for patients known to have LS upon CRC diagnosis, in particular that ‘For patients with proximal tumours, colectomy and ileorectal anastomosis is most relevant.’9 Input from our clinical expert has suggested that this particular guidance would rarely be followed as it is from a low category of evidence (evidence obtained from expert committee reports or opinions, or clinical experiences of respected authorities) and colonoscopic surveillance is deemed effective enough to negate the need for aggressive surgery. To resolve this disagreement, we include a parameter in the model which defines the probability that more aggressive surgery would be used for LS patients, which can be varied from 0 (ignore guidelines; surgical treatment not affected by LS diagnosis) to 1 (full adherence to guidelines; aggressive surgery always used).
When surgery removes the rectum because of cancer in the rectum there are two common operations: anterior resections which preserve the anus and abdominoperineal excisions of the rectum (APERs) which result in permanent stoma. We group these operations together and assume that they are equally effective at preventing metachronous rectal cancer. Some patients would require a permanent stoma, which would affect HRQoL and costs. Rather than modelling this on an individual patient basis, we assume an average effect across all patients.
Any subsequent surgery depends on the location of the CRC, the nature of previous surgery and whether or not the patient has been diagnosed with LS (unless the parameter described above is 0) (Tables 48 and 49).
| Previous surgery | Segmental resection | Subtotal colectomy | Anterior resection | Proctocolectomy | Source |
|---|---|---|---|---|---|
| None | 96%a | 4%b | 0% | 0% | NHS Bowel Cancer Audit report 2011135 |
| Segmental resection | 0% | 100% | 0% | 0% | Assumption |
| Subtotal colectomy | N/A | N/A | N/A | N/A | Assumption |
| Anterior resection | 0% | 0% | 0% | 100% | Assumption |
| Proctocolectomy | N/A | N/A | N/A | N/A | Assumption |
| Previous surgery | Segmental resection | Subtotal colectomy | Anterior resection | Proctocolectomy | Source |
|---|---|---|---|---|---|
| None | 0% | 0% | 98%a | 2%b | NHS Bowel Cancer Audit report 2011135 |
| Segmental resection | 0% | 0% | 0% | 100% | Assumption |
| Subtotal colectomy | 0% | 0% | 0% | 100% | Assumption |
| Anterior resection | N/A | N/A | N/A | N/A | Assumption |
| Proctocolectomy | N/A | N/A | N/A | N/A | Assumption |
Surgery distributions for CRC patients diagnosed with LS are adjusted by the parameter representing the probability of aggressive surgery. If we denote this probability as p then for colon cancer patients:
where Pr is probability, SEG is segmental resection and SUB is subtotal colectomy. For rectal cancer patients:
where AR is anterior resection and IPAA is ileal pouch–anal anastomosis (total proctocolectomy). Our estimate of this parameter is given and justified in More aggressive colorectal cancer surgery for individuals diagnosed with Lynch syndrome.
Characteristics of the model population
Age on entry
In the base case, probands enter the model below the age of 50 years as a result of the age targeting of testing, and all relatives enter the model below the age of 75 years to reflect the small number of interventions after this age. In scenario analyses, we investigate the impact of increasing the maximum age of probands to 60 and 70 years separately.
The ages of probands without LS in the simulation are distributed to reflect the ages observed in CRC registration statistics for England between 2006 and 2010 inclusive. 119,136–139 The distribution of ages of probands with LS is approximated using our estimated rates of incidence of CRC for individuals with LS not receiving risk-reducing interventions (see Incidence rates in patients with Lynch syndrome).
The difference in the age distributions between individuals with and without LS (Figure 20) is accounted for by the earlier average age at onset of CRC in individuals with LS [in the study by Bonadona and colleagues (2011)2 the median age at onset of CRC was 45 years, whereas it is 70–74 years in the general population119]. We do not show the analogous age distribution for females as it shows the same pattern.
FIGURE 20.
Age distribution of male probands on diagnosis of CRC.
It was assumed that all relatives would be aged between 18 and 75 years because it would be unusual to offer predictive testing for LS to individuals under the age of 18 years (and concomitantly, intervention would be unlikely to take place until age 20–25 years), and because intervention is rarely continued after age 75 years. Age distributions for relatives with and without LS were adjusted accordingly.
The ages of relatives without LS are distributed according to the ages of the general population of England and Wales. 140,141 The ages of relatives with LS are estimated similarly, but with an adjustment to reflect substantially greater mortality (currently only from CRC) for individuals with LS compared with the general population. Although we have some limited data on age of relatives who are tested for LS, these are from a relatively small sample size, and we were unable to find data on age distributions in the literature. This adjustment used incidence rates of CRC for individuals with LS as described in Incidence rates in patients with Lynch syndrome, Dukes’ stage distributions as described in Dukes’ stage on diagnosis and 1- to 10-year survival, calculated according to parameters described in Mortality due to colorectal cancer for patients with Lynch syndrome to estimate the proportion of individuals with LS that would survive to a certain age if CRC was the only cause of death. This was then applied to the age distribution of the population of England and Wales, resulting in LS relatives being more likely to be younger (Figure 21).
FIGURE 21.
Age distribution of male relatives when the proband is diagnosed with CRC.
The proportion of relatives with LS is relatively higher for younger ages owing to the additional mortality experienced by individuals with LS from CRC. We do not show the analogous figure for females as it follows the same pattern.
Bowel state on entry
All probands enter the model with an index CRC (i.e. without a metachronous CRC). The Dukes’ stage for probands is sampled randomly using the distribution described in Dukes’ stage on diagnosis.
Probands entering the model were randomly assigned a surgical state in accordance with the estimated probability that they had colon cancer versus rectal cancer (Table 50) and the probabilities of different types of surgery for those cancers (Table 51). Table 52 gives the resulting distribution of initial surgical states for probands entering the model, according to their sex and LS status.
| Proband type | Male | Female | Source |
|---|---|---|---|
| With LS | 0.94 | 0.94 | Dinh online appendix55 |
| Without LS | 0.58 | 0.61 | ONS Cancer Registration Statistics, England119 |
| Location of CRC | Surgery (% of cases) | Source | Notes |
|---|---|---|---|
| Colon | Segmental resection (96) Subtotal colectomy (4) |
NHS Bowel Cancer Audit 2011135 | Included in segmental resection:a right hemicolectomy, transverse colectomy, left hemicolectomy, sigmoid colectomy Included in subtotal colectomy:a total/subtotal colectomy Excluded:a anterior resection, APER, Hartmann procedure |
| Rectum | Anterior resection (98) Proctocolectomy (2) |
NHS Bowel Cancer Audit 2011135 | Included in anterior resection:b anterior resection, APER, Hartmann procedure Included in proctocolectomy:b total/subtotal colectomy Excluded:b right hemicolectomy, transverse colectomy, left hemicolectomy, sigmoid colectomy |
| Surgery | With LS | Without LS | ||
|---|---|---|---|---|
| Male | Female | Male | Female | |
| Segmental resection | 0.907 | 0.907 | 0.564 | 0.587 |
| Subtotal colectomy | 0.033 | 0.033 | 0.021 | 0.022 |
| Anterior resection | 0.059 | 0.059 | 0.408 | 0.384 |
| Proctocolectomy | 0.001 | 0.001 | 0.008 | 0.007 |
Relatives enter the model without CRC, i.e. they are at risk of up to two CRCs (index and metachronous). In reality some relatives would be survivors of previous CRC.
Table 53 gives an estimate of what proportion of relatives would be survivors of previous CRC. Estimates for relatives without LS are based on 10-year CRC prevalence published by the (UK) National Cancer Intelligence Network (NCIN),142 assuming that the proportion of CRC survivors with colon cancer is the same as the proportion of incident CRCs which are colon cancer. Prevalence of previous CRC for relatives with LS is estimated by multiplying by a scale factor of 45%/2.6% = 17.4 for males and 35%/1.7% = 20.3 for females, where 45% and 35% are estimates of the cumulative risk of CRC to age 70 years for males and females with LS, respectively,65 and 2.6% and 1.7% are estimates of the cumulative risk of CRC to age 70 years for males and females without LS, respectively, calculated using population, CRC incidence and CRC mortality statistics for England and Wales in 2010. 119,143–145 Again, it was assumed that the proportion of survivors with colon cancer would match the proportion of incident cases, this time estimated by Dinh and colleagues. 55
| CRC prevalence | With LS | Without LS | ||
|---|---|---|---|---|
| Male | Female | Male | Female | |
| None | 0.9628 | 0.9717 | 0.9979 | 0.9986 |
| Colon cancer | 0.0350 | 0.0266 | 0.0012 | 0.0009 |
| Rectal cancer | 0.0022 | 0.0017 | 0.0009 | 0.0005 |
Relatives with previous CRC would experience a higher mortality rate and, therefore, preventing a further CRC would be expected to give a smaller life-year gain than in relatives without previous CRC. These CRC survivors would be likely to have early-stage CRC, to have undergone segmental resection and to be followed up for recurrence or metachronous cancer.
The model incorporates initial surgical states for relatives entering the model (Table 54; proportions based on surgical choice for people not known to have LS and prevalence of colon and rectal cancer as in Table 53). Most relatives have no previous surgery (as most relatives have no previous CRC), and of those with previous surgery, the majority have a previous segmental resection which imparts no risk reduction in the model. A very small number (<< 1%) of relatives enter the model with previous surgery which does impart a risk reduction. As all these relatives enter with risk reduction irrespective of the diagnostic strategy, this would decrease the potential life-year gain of correctly identifying relatives as having LS; we therefore expect that including initial surgical states has a very small (probably negligible) negative impact on cost-effectiveness of strategies identifying LS (i.e. strategies made less cost-effective).
| Surgery | With LS | Without LS | ||
|---|---|---|---|---|
| Male | Female | Male | Female | |
| None | 0.9628 | 0.9717 | 0.9979 | 0.9986 |
| Segmental resection | 0.0338 | 0.0256 | 0.0012 | 0.0009 |
| Subtotal colectomy | 0.0012 | 0.0009 | 0.0000 | 0.0000 |
| Anterior resection | 0.0022 | 0.0017 | 0.0009 | 0.0005 |
| Proctocolectomy | 0.0000 | 0.0000 | 0.0000 | 0.0000 |
Gynaecological cancers
Gynaecological cancers in models of testing for Lynch syndrome
Most published models of the cost-effectiveness of testing for LS consider only CRC, not EC and OC (Table 55). Two analyses, Dinh and colleagues (2011)55 and Kwon and colleagues (2011),56 consider only EC in addition to CRC, and five analyses consider both EC and OC in addition to CRC: Chen and colleagues (2007)85 (clinical effectiveness only), Kwon and colleagues (2008),75 Ladabaum and colleagues (2011),48 Wang and colleagues (2012)57 (extension of analysis by Ladabaum and colleagues48) and Yang and colleagues (2011)76 (extension of Chen and colleagues85 analysis).
| Cost-effectiveness analysis | EC | OC |
|---|---|---|
| Breheny et al. 200651 | No | No |
| Brown et al. 199622 | No | No |
| Chen et al. 200785 | Clinical effectiveness model (no costs). Stages I to IV of both EC and OC modelled. Screening assumed to yield clinical benefits. TAHBSO assumed | |
| DACEHTA 200752 | No | No |
| Dinh et al. 201155 | Endometrial biopsy and TVU, assumed to incur costs but yield no benefits. TAHBSO offered to women with EC and those LS positive without. TAHBSO assumed to eliminate risk of EC. Stage of cancer not modelled | No |
| Kievit et al. 200550 | No | No |
| Kwon et al. 200875 | Stages I to IV of both EC and OC modelled. Screening assumed to yield clinical benefits. Prophylactic gynaecological surgery assumed to remove risk of gynaecological cancers | |
| Kwon et al. 201156 | EC modelled, but very little detail concerning incidence, screening or surgery | No |
| Ladabaum et al. 201148 | Gynaecological screening starting at age 35 years, assumed to incur costs but yield no benefits. Prophylactic TAHBSO at age 40 years, assumed to eliminate risk of gynaecological cancers. Stage of cancer not modelled | |
| Mvundura et al. 201054 | No | No |
| Olsen et al. 200753 | No | No |
| Ramsey et al. 200123 | No | No |
| Ramsey et al. 200349 | No | No |
| Reyes et al. 200259 | No | No |
| Wang et al. 201257 | Same as Ladabaum et al. 201148 | |
| Yang et al. 201176 | Similar to clinical effectiveness model of Chen et al. 200785 with addition of utilities and costs for screening, gynaecological surgery, cancer care, chemotherapy and radiotherapy | |
Ladabaum and colleagues (2011)48 modelled the occurrence of EC and OC. They modelled annual gynaecological screening with transvaginal ultrasonography and endometrial sampling, starting at age 35 years. Gynaecological screening was assumed to incur costs but yield no benefits, given that no proven benefit was found. The authors note that this biases cost-effectiveness against genetic testing for LS.
Ladabaum and colleagues (2011) also modelled prophylactic TAHBSO at age 40 years, after completion of childbearing,48 and they assumed that this eliminates the risk of EC and OC, citing Schmeler and colleagues (2006). 146 It was predicted that 19% of probands and 18% of relatives with LS respectively would accept TAHBSO. The death rate from TAHBSO was estimated at 0.0003.
This screening and surgical regime was also offered to (a) relatives whose status was uncertain because they had declined genetic testing, but who had a 50% risk for carrying a mutation; (b) probands in whom LS was diagnosed on the basis of AC II and tumour features, despite the lack of a detectable mutation; and (c) their FDRs. 48 Individuals whose tumours showed abnormal IHC or MSI results, but who had normal genetic results and did not meet the AC II, were not offered the regime, because such cases were deemed most likely to represent FP results on tumour tests rather than FN results on genetic tests. 48
Ladabaum and colleagues (2011) modelled the probabilities that women with and without LS develop EC and OC. For example, the probability that a woman with LS develops EC was estimated as 37% by age 70 years, and the probability of dying from EC within 5 years from diagnosis was estimated as 0.17.
In the base case, deaths from EC and OC were reduced by between 1% and 6% according to the testing strategy for LS. Testing was predicted to benefit women more than men. For example, given universal acceptance of genetic testing and perfect adherence to screening, female probands were predicted to survive an additional 0.47 years given genetic testing versus the referent strategy, compared with an additional 0.22 years for male probands. The sex difference was attributed to prevention of gynaecological cancers by TAHBSO and to women having a greater life expectancy than men.
Dinh and colleagues (2011)55 modelled the occurrence of EC, but not OC. The incidence of EC in individuals with LS was estimated from a meta-analysis conducted by the authors, but unfortunately the incidence rates are not published.
In common with Ladabaum and colleagues (2011),48 in their survey of the literature Dinh and colleagues (2011) found that the clinical benefit of surveillance for EC for women with LS is not clear. Although Dinh and colleagues (2011) assumed that women with positive genetic test results for LS were screened by endometrial biopsy and transvaginal ultrasound (TVU), in common with Ladabaum and colleagues (2011) they also assumed that surveillance has no effect on the incidence of and survival from EC.
Dinh and colleagues (2011) assumed that women with positive genetic test results for LS were offered TAHBSO, assuming a mortality of 0.02% for this operation. The adherence rate for TAHBSO for women without EC and without CRC was estimated to increase with age, from 0% for women aged under 30 years to 75% for those aged 80 years. Slightly higher rates were assumed for women without EC but with CRC. The adherence rate for women with EC was estimated at 100%. As in the study by Ladabaum and colleagues (2011),48 the chance of developing EC after hysterectomy was assumed to be zero. Also in common with Ladabaum and colleagues (2011),48 any costs and morbidities due to oestrogen therapy following TAHBSO were ignored.
Dinh and colleagues (2011) assumed that mortality due to EC for individuals with LS affected by EC is the same as for individuals without LS, and this was taken from the US Surveillance, Epidemiology, and End Results (SEER) database. However, the authors do not publish these rates.
Kwon and colleagues (2008)75 modelled the occurrence of both EC and OC. Five treatment strategies of various combinations of screening and risk-reducing surgery were modelled. Unlike Ladabaum and colleagues (2011)48 and Dinh and colleagues (2011),55 the stages I to IV of both cancers were modelled explicitly.
A lifetime risk of 50% for EC and 10% for OC was assumed.
In common with Ladabaum and colleagues (2011)48 and Dinh and colleagues (2011),55 Kwon and colleagues (2008) also modelled gynaecological screening, and found that the evidence on the clinical benefit of screening for EC was mixed. Unlike Ladabaum and colleagues (2011)48 and Dinh and colleagues (2011),55 Kwon and colleagues (2008) assumed that screening for gynaecological cancers leads to clinical benefits, albeit at substantial cost. Screening is assumed to give an improved stage distribution for gynaecological cancers at detection.
In common with Ladabaum and colleagues (2011)48 and Dinh and colleagues (2011),55 Kwon and colleagues (2008) assumed that prophylactic gynaecological surgery removes the risk of gynaecological cancers. Unlike Ladabaum and colleagues (2011)48 and Dinh and colleagues (2011),55 100% adherence to surveillance and surgery was assumed.
As in the studies by Ladabaum and colleagues (2011)48 and Dinh and colleagues (2011),55 costs of adjuvant radiotherapy or chemotherapy were not modelled.
In their model of the cost-effectiveness of testing for LS in women with EC, Kwon and colleagues (2011)56 modelled the occurrence of EC, but not OC. Unfortunately, no details are given concerning screening or surgery for EC, and only outline data are provided on incidence of EC.
In their analysis of the clinical (as opposed to cost-) effectiveness of prophylactic TAHBSO surgery versus surveillance for women with LS, Chen and colleagues (2007)85 modelled both EC and OC. As in the study by Kwon and colleagues (2008),75 the stages I to IV of both cancers were modelled explicitly, in particular at diagnosis and at the time of surgery. Two types of surveillance were compared: lifetime annual gynaecological examination only versus annual gynaecological TVU, endometrial biopsy and serum CA125 testing. All interventions were assumed to start at age 30 years. Unlike Ladabaum and colleagues (2011)48 and Dinh and colleagues (2011),55 but in common with Kwon and colleagues (2008),75 it was assumed that screening for gynaecological cancers yields clinical benefits. Screen-detected EC was assumed to have a similar stage distribution as for EC diagnosed at risk-reducing surgery. Survival from gynaecological cancers was taken from the US SEER database.
The analysis of the cost-effectiveness of prophylactic TAHBSO surgery versus surveillance for women with LS by Yang and colleagues (2011)76 was performed by the same authors as the clinical effectiveness model of Chen and colleagues (2007). 85 The model of Yang and colleagues (2011) builds on the model of Chen and colleagues (2007)85 by additionally incorporating utilities and the following costs: screening, gynaecological surgery, cancer care, chemotherapy and radiotherapy.
Ovarian cancer in the Peninsula Technology Assessment Group model
Only 7 of the 16 models of LS (see Table 89) consider EC, and fewer still (five) consider OC [Chen and colleagues (2007),85 Kwon and colleagues (2008),75 Ladabaum and colleagues (2011),48 Wang and colleagues (2012)57 and Yang and colleagues (2011)76]. Further, two of these models are simply extensions of previous models [Wang and colleagues (2012)57 is an extension of Ladabaum and colleagues (2011),48 and Yang and colleagues (2011)76 is an extension of Chen and colleagues (2007)85].
Lynch syndrome is associated with an increased incidence of many cancers, including colorectal, endometrial, ovarian, stomach, small intestine, hepatobiliary tract, urinary tract, brain and skin. 5 Given time and data constraints, it has not been possible to model all LS-associated cancers. Therefore, it is judicial to model only those cancers which are likely to substantially affect the cost-effectiveness of genetic testing for probands with CRC with LS. Of the gynaecological cancers, we model EC, but not OC. This is because the incidence of OC in women with LS is substantially lower than that of EC. In particular, as stated below (see Incidence rates in patients with Lynch syndrome), a recent study of 10,283 individuals from several French clinics,2 which appears well conducted, estimated the cumulative risk of EC by the age of 70 years as 34% and that of OC as 9%. We approximated the cumulative risk of death from each cancer by age 75 years by multiplying the cumulative risk to age 70 years by the proportion of affected individuals not surviving to 5 years from diagnosis (i.e. 100% minus 5-year survival). This gives a cumulative risk of death of 34% × (100% − 76.7%) = 7.9% for EC and 9% × (100% − 44.0%) = 5.0% for OC. 147 This justifies modelling EC ahead of OC.
If OC were included in our model, it is likely that our estimates of the cost-effectiveness of testing would improve, as follows. We already cost for TAHBSO prophylactic surgery, which eliminates the risk not just of EC, but also of OC. If we were to model OC, then testing for LS would eliminate the incidence of OC for those female probands who present with CRC and who accept TAHBSO, and for those female relatives who accept TAHBSO. Therefore, the estimated QALYs for those strategies that test for LS would increase. If we were to assume no or relatively low costs for treating OC, the estimated total cost of all testing strategies would change little.
Endometrial cancer in the Peninsula Technology Assessment Group model
Table 56 gives a summary of the parameters concerning EC in the PenTAG model. The model assumes that neither the relatives nor probands have previously had EC (or TAHBSO) before entering the model. This would be an additional complexity that would be unlikely to have a large impact on cost-effectiveness, because patients with a prior EC or TAHBSO would have both reduced costs and reduced benefits in our model.
| Parameter | Base-case value | Source | Alternative values |
|---|---|---|---|
| Cumulative incidence of EC | 0% by age 30 years, 2% by age 40 years, 8% by age 50 years, 23% by age 60 years, 34% by age 70 years, 35% by age 80 years | Bonadona et al. 20112 | N/A |
| Screening for EC | Not modelled | Assumption; see Screening for endometrial cancer | N/A |
| Probability of dying of EC each year after diagnosis | 9.9% in first year, 5.0% per annum in years 2–3, 2.6% per annum in years 4–5, 0.7% per annum in years 5–10, 0% thereafter | ONS,148 NCIN149 and Cancer Research UK150 | N/A |
| Surgery | |||
| Use of TAHBSO on diagnosis of EC | 100% | Expert opinion | N/A |
| Age when given prophylactic TAHBSO | 45 years for individuals aged ≤ 45 years, otherwise age when tested positive for LS | Expert opinion | N/A |
| Acceptance of prophylactic TAHBSO for LS-positive probands | 55% | Lorraine Cowley, Northern Genetics Service, 2012, personal communication | 20%, 90% |
| Acceptance of prophylactic TAHBSO for LS-positive relative | 55% | Lorraine Cowley, personal communication | 20%, 90% |
| Probability of mortality from TAHBSO | 0.0002 | Average over studies reported in Palomaki et al. 200965 | N/A |
| Incidence of EC after TAHBSO | 0% | Schmeler et al. 2006146 | N/A |
| Cost of prophylactic TAHBSO | £3322 | £3104 (Department of Health reference costs 2011/12:151 MA07, MA08) inflated over 2 years; see Cost of endometrial cancer prevention | N/A |
| Cost of TAHBSO for EC | £3877 | £3622 (Department of Health reference costs 2011/12:151 MA06) inflated over 2 years; see Cost of surgery for endometrial cancer | N/A |
| Other treatment for EC | |||
| EC stage at diagnosis | 73% stage I, 18% stages II and III, 9% stage IV | Based on US SEER database (http://seer.cancer.gov) via Havrilesky et al. 2009152 | N/A |
| Use of radiotherapy | 33% of stage I patients, 100% of stage II and III patients, 50% of stage IV patients | Havrilesky et al. 2009152 | N/A |
| Cost of course of radiotherapy | £5856 | Havrilesky et al. 2009152 adjusted for purchasing power and inflation | N/A |
| Chemotherapy for EC | Six cycles of TAP (paclitaxel, doxorubicin, cisplatin) | Havrilesky et al. 2009152 | N/A |
| Use of chemotherapy | 50% of stage II and III patients, 100% of stage IV patients | Havrilesky et al. 2009152 | N/A |
| Cost of course of chemotherapy (TAP) | £2974 | Drug costs from eMit database,153 drug administration costs from NHS reference costs 2008–9154 | £1487 and £5947 |
| Cost of follow-up management for EC | Zero | See Cost of follow-up management of endometrial cancer | N/A |
Of the seven analyses that include EC,48,55–57,75,76,85 six assume that some patients will receive either a hysterectomy or TAHBSO (see Table 55). It is not clear whether or not the seventh analysis, by Kwon and colleagues (2011)56 included such surgery. In the PenTAG base-case analysis, we assume TAHBSO, rather than just hysterectomy, as this is the usual treatment for EC155 (see Table 56). Further, our clinical experts advise that either total laparoscopic hysterectomy (TLH) or laparoscopic-assisted vaginal hysterectomy (LAVH) is now preferred. As we are not able to differentiate the costs of hysterectomy with bilateral salpingo-oophorectomy from hysterectomy alone, we do not perform any sensitivity analysis in which hysterectomy alone is performed. We assume that surgery may be performed in either of two cases: on diagnosis of EC, and prophylactically for women who do not have EC, but have tested positive for LS.
The main treatment for EC in the UK is hysterectomy. 156 For stage I EC, patients usually have a TAHBSO; a radical hysterectomy is possible for stages II or III cancer, and for stage IV cancer, debulking surgery (removal of as much cancer as possible). 156 We assume 100% adherence to TAHBSO for all stages of disease (see Table 56).
It is assumed that female probands who either test positive for LS or are assumed LS, who present with CRC and are younger than 45 years of age, are offered a TAHBSO at age 45 years, on advice from our clinical experts. Those aged 45 years or older are offered an immediate TAHBSO. The age of 45 years is chosen to coincide with the end of child-rearing.
Next, it is assumed that female relatives who either test positive for LS or are assumed LS, and who are younger than 45 years, are offered a TAHBSO at age 45 years, again as suggested by our clinical experts. Those aged 45 years or older are offered an immediate TAHBSO.
Risks from hysterectomy vary by surgical technique (abdominal, laparoscopic, vaginal) and include infection (3–33%), bleeding (3–9%), organ injury (1–3%) and rarely death (0.00–0.04%). 65
Death is the only complication following surgery modelled by any of the seven previous cost-effectiveness analyses that considered endometrial cancer (see Table 55). Chen and colleagues85 report a mortality rate of approximately 0.2% within 30 days of surgery for women aged 40–49 years for EC, which they took from the US SEER database. Dinh and colleagues55 assumed a mortality rate of 0.02% for TAHBSO, which they state is a weighted average of the rates reported in nine studies. 65 Ladabaum and colleagues48 assumed a mortality rate of 0.03% for TAHBSO, citing Palomaki and colleagues. 65 It appears that Kwon and colleagues75 modelled no complications from prophylactic surgery.
As in previous cost-effectiveness analyses, we do not model infection, bleeding or organ injury owing to low incidence. However, we model surgical mortality because it clearly has an impact on total QALYs. As in the study by Dinh and colleagues,55 we assume a probability of death of 0.0002, which is a weighted average of the rates reported in nine studies,65 although this only applies to prophylactic TAHBSO because surgical mortality for EC patients is already included in the first year of EC mortality (see Mortality due to endometrial cancer).
Oophorectomy in premenopausal women induces the sudden onset of menopause. 65,146 However, we do not model the effect of this because premature menopause can usually be managed with hormonal or non-hormonal medication (which is inexpensive)146 and because we model oophorectomy at a minimum age of 45 years, which is at the early stages of natural menopause.
The approach to modelling treatments for EC varies widely among the seven previous cost-effectiveness analyses that considered EC (see Table 55).
Ladabaum and colleagues (2011)48 assumed a cost of care for EC of $31,027, citing a study of the cost-effectiveness of screening for EC. 152 However, this figure is not given in the source publication and Ladabaum and colleagues48 do not explain how they derived the figure from the publication. Further, although not stated, we assume that this refers to the total mean lifetime cost of care for EC per person. In addition, no indication is given of the nature of the treatment.
Yang and colleagues (2011)76 is the only study to model the treatment of EC in detail. They assumed the following cumulative care costs: $127,711 for 5-year survival, $218,497 for 10-year survival and $164,685 for a cure, citing a US Medicare study157 published in 1989. They also assumed a cost of $8418 for radiation treatment for EC, citing the University of California, San Francisco Department of Radiation Oncology billing office. Patients with stage III or IV EC were assumed to receive six cycles of chemotherapy.
It appears that Chen and colleagues (2007)85 did not model care for EC. Kwon and colleagues (2008)75 say that they did not include costs for adjuvant radiotherapy or chemotherapy. Indeed, it appears that they do not include any costs for care for EC. Dinh and colleagues (2011)55 assume a total cost of $24,291 for treatment for EC, citing Kwon and colleagues (2008). 75 However, we do not see this figure in Kwon and colleagues (2008). 75
Ideally, we would cost for the treatment of EC using published clinical guidelines specifically for the UK. However, NICE have not published such guidelines. 158 Instead, treatment is described in two other sources. 156,159 The main treatment for EC in the UK is hysterectomy. 156 This is sometimes followed by radiotherapy, depending on the stage and grade of the cancer. 156 Chemotherapy may also be used for stage III or IV EC. 156
Radiotherapy is recommended if there is a significant risk that the cancer could return in the pelvis, and may also be used to slow the spread of cancer when a surgical cure is not possible. 156 Two types of radiotherapy are used to treat EC: internal radiotherapy (brachytherapy) and external radiotherapy. A course of external radiotherapy is usually given as an outpatient for 5 days a week, over approximately 4 weeks. 156 Most women with stage II or III EC have radiotherapy after surgery. 159
Chemotherapy for stage III or IV EC can be used after surgery to try to prevent the return of the cancer or, in cases of advanced cancer, to slow the spread of the cancer and relieve symptoms. 159 Chemotherapy is usually given in an outpatient setting. There is a range of possible chemotherapies for EC, including single-agent carboplatin or paclitaxel, combination carboplatin plus paclitaxel and combination paclitaxel, doxorubicin and cisplatin (TAP). 160
In a US study of the cost-effectiveness of screening for EC,152 the following costs were assumed:
-
external beam pelvic radiotherapy for 33% of stage I cancer patients, 100% of stage II and III patients, and 50% of stage IV patients at a total cost of $7895 per patient
-
six cycles of TAP (paclitaxel, doxorubicin, cisplatin) chemotherapy for 50% of stage II and III patients and 100% of stage IV patients at a cost of $19,462 for six cycles.
The following distribution of stages of EC at diagnosis was assumed in this study: 70% stage I, 17% stages II and III, 9% stage IV, 4% unknown (taken from the US SEER database). 152
In our model, we assume the same treatment regime and distribution of stages of cancer as Havrilesky and colleagues152 because it is consistent with the treatment of EC in the UK, described above. More precisely, the following split across stages at diagnosis is assumed: 73% stage I, 18% stages II and III, 9% stage IV, where the 4% unknowns are spread across the stages.
Disease natural history parameters and assumptions
Colorectal cancer
Colorectal cancer incidence
Colorectal cancer incidence rates
Colorectal cancer incidence rates in the model are dependent on the following patient characteristics:
-
age
-
sex
-
whether or not the patient has had a previous CRC
-
time since first CRC
-
LS status.
These are in addition to risk-reducing measures, i.e. regular colonoscopies, as described in Effect of colonoscopy on index colorectal cancer incidence rates and Effectiveness of colorectal cancer surgery. Different annual incidence rates are provided for the eight combinations of sex, previous cancer (yes/no) and LS status, and then risk-reducing measures are incorporated as HRs which have a simple multiplicative effect on the incidence rate.
The incidence rates for males and females without previous cancer and without LS were estimated from pooled registration statistics for CRC in England between 2006 and 2010 inclusive119,136–139 and the estimated population in the midpoints of those years. 144 Following the methodology adopted by the ONS,119 we calculate the age-specific rate of CRC incidence by dividing the number of CRC registrations within a time period by an estimate of the person-years lived during that period. Incidence figures were pooled across 5 years to achieve a large sample size, but not further back than 2006 as such data may not reflect more recent developments in cancer detection and registration.
Cancer registration statistics are not provided for each year of age but for age groups, generally of 5 years. We assumed that within each of these age groups the incidence rate would remain constant. The resulting cumulative risk of CRC for individuals without LS is shown in Figure 22.
FIGURE 22.
Cumulative risk of CRC for persons without LS (note: does not account for non-CRC mortality).
We estimated the incidence of metachronous CRC (i.e. the incidence in individuals who had a previous CRC) for individuals without LS by adjusting the incidence of first CRC by a HR of 1.4 for the first 3 years after first CRC and 1.3 for the following 7 years, from Mulder and colleagues. 161 Mulder and colleagues studied 10,283 Dutch patients with CRC undergoing standard follow-up. After 10 years no additional hazard was applied.
The estimation of CRC incidence rates in patients with LS is complicated by a number of factors, as detailed in Table 57. Estimated incidence rates are also much less often reported than estimated cumulative risks (normally to age 70 years).
| Source of bias | Explanation | Methods to avoid/overcome bias | References |
|---|---|---|---|
| Ascertainment bias | If LS is only sought in CRC patients below a certain age or with a particular FH, this will result in a biased identification of LS towards families with more penetrance | Use genotype-restricted likelihood methods conditioned on the likelihood of ascertaining the proband (including the phenotype of relatives if necessary) | Carayol and Bonaïti-Pellie (2004)162 |
| MMR genes included | It has been observed in many studies that incidence rates are higher for mutations of the MLH1 and MSH2 genes than for the MSH6 and PMS2 genes. Studies which only include a subset of the genes may overestimate the average risk in LS patients | Screen for mutations in all four genes If MMR functionality is assessed through MSI or IHC as gatekeepers for germline mutation testing, ensure that these are sufficiently sensitive for all four genes |
Bonadona et al. 20112 Senter et al. 2008105 |
| Mutation detection method | Sequencing techniques are less able to detect large deletions and rearrangements | Screen for mutations using sequencing and large deletions and rearrangements (usually by MLPA) | Bonis et al. 20074 |
| Inclusion of VUS | Some mutations (usually missense mutations) cannot be instantly classified as polymorphic or deleterious without detailed investigation of functional effect and segregation analysis. Excluding or including all VUS may bias the penetrance estimate | Make fullest attempts to classify VUS correctly | Rasmussen et al. 2012163 |
| Presence of founder mutations | In countries with a significant prevalence of founder mutations, the penetrance of the founder mutation may be over-represented | Separate analyses with/without founder mutations included | |
| Risk-reducing interventions | Incidence should be reduced when risk-reducing interventions are implemented | Censor at the earliest risk-reducing intervention | Bonadona et al. 20112 |
| Follow-up period | If cancers informing the cumulative risk analysis are from many decades ago there may be differences in baseline incidence due to changes in diet, etc. | Restriction to recent cancers or adjustment for period |
We identified three papers – Palomaki and colleagues,65 Dinh and colleagues55 and Bonadona and colleagues2 – which had searched the literature for papers reporting cumulative risk to age 70 years. We also examined previous cost-effectiveness studies as identified in the systematic review of cost-effectiveness for evidence on the risk of CRC in LS. The findings of these studies and their characteristics with regard to possible biases are presented in Appendix 5.
Estimates of cumulative risk vary substantially according to whether or not there is proper correction for ascertainment bias. Table 58 shows central estimates for cumulative risks from studies with appropriate correction for ascertainment bias [excluding van Vliet and colleagues (2011)164 as parent-of-origin effects are beyond the scope of this project]. A picture emerges that cumulative risks are generally higher for males and may be higher for MSH2 carriers than for MLH1 carriers, but that cumulative risks for both of these are significantly higher than those for MSH6 and PMS2 carriers, which are similar to each other. Although some studies estimated cumulative risks for combined sets of MMR genes, these will ultimately be less useful than risk estimates for individual genes because different case mixes will be observed and less intense risk-reducing strategies may be suggested for MSH6 and PMS2. 167
| Genes included | Individuals (male and female) | Males | Females |
|---|---|---|---|
| MLH1, MSH2, MSH6, PMS2 | 45% (Jenkins et al. 2006114) | 38% (Jenkins et al. 2006114) | |
| MLH1, MSH2, MSH6 | 35% (Bonadona et al. 20112) | 38% (Bonadona et al. 20112) 34% (Stoffel et al. 2009106) |
31% (Bonadona et al. 20112) 32% (Stoffel et al. 2009106) |
| MLH1, MSH2 | 47% (Alarcon et al. 2007165) 56% (Jenkins et al. 2006114) 27% (Quehenberger et al. 2005102) |
33% (Alarcon et al. 2007165) 48% (Jenkins et al. 2006114) 22% (Quehenberger et al. 2005102) |
|
| MLH1 | 41% (Bonadona et al. 20112) | 22% (Quehenberger et al. 2005102) | 18% (Quehenberger et al. 2005102) |
| MSH2 | 48% (Bonadona et al. 20112) | 30% (Quehenberger et al. 2005102) | 25% (Quehenberger et al. 2005102) |
| MSH6 | 12% (Bonadona et al. 20112) | 22% (Baglietto et al. 2010166) | 10% (Baglietto et al. 2010166) |
| PMS2 | 20% (Senter et al. 2008105) | 15% (Senter et al. 2008105) |
In cost-effectiveness analysis it is important not only to model the correct lifetime risk of a disease but also to correctly model the cumulative risk over a range of ages to get the appropriate incidence rate at each age. Of the studies presented in Appendix 5, which adjust appropriately for ascertainment bias, seven report the cumulative risk of CRC to different ages, and these are summarised in Table 59.
| Study (number of families) | MMR genes included | Cumulative risk of CRC for males | Cumulative risk of CRC for females |
|---|---|---|---|
| Bonadona et al. 20112 (n = 537) | MLH1, MSH2, MSH6 | (From figure) | (From figure) |
| 20y 0.3% | 20y 0.3% | ||
| 30y 2.2% | 30y 0.8% | ||
| 40y 7.5% | 40y 4.2% | ||
| 50y 17.3% | 50y 10.1% | ||
| 60y 28.0% | 60y 20.0% | ||
| 70y 38.4% | 70y 31.3% | ||
| 80y 43.4% | 80y 38.4% | ||
| van Vliet et al. 2011164 (n = 17)a | MLH1, MSH2, MSH6, PMS2 | Paternal origin | Paternal origin |
| 30y 3% | 30y 5% | ||
| 40y 7% | 40y 12% | ||
| 50y 16% | 50y 22% | ||
| 60y 29% | 60y 34% | ||
| 70y 41% | 70y 42% | ||
| 80y 48% | 80y 47% | ||
| Maternal origin | Maternal origin | ||
| 30y 9% | 30y 4% | ||
| 40y 22% | 40y 9% | ||
| 50y 44% | 50y 17% | ||
| 60y 67% | 60y 27% | ||
| 70y 81% | 70y 34% | ||
| 80y 88% | 80y 38% | ||
| Baglietto et al. 2010166 (n = 113) | MSH6 | 50y 3% | 50y 2% |
| 60y 9% | 60y 5% | ||
| 70y 22% | 70y 10% | ||
| 80y 44% | 80y 20% | ||
| Senter et al. 2008105 (n = 55) | PMS2 | 50y 2% | 50y 2% |
| 60y 8% | 60y 6% | ||
| 70y 20% | 70y 15% | ||
| Alarcon et al. 2007165 (n = 36) | MLH1, MSH2 | (From figure 2) | (From figure 2) |
| 40y 7% | 40y 5% | ||
| 50y 16% | 50y 14% | ||
| 60y 30% | 60y 25% | ||
| (From abstract) | (From abstract) | ||
| 70y 47% | 70y 34% | ||
| Jenkins et al. 2006114 (n = 17) | MLH1, MSH2, MSH6, PMS2 | 50y 41% | 50y 22% |
| 70y 45% | 70y 38% | ||
| Jenkins et al. 2006114 (n = 12) | MLH1, MSH2 | 50y 51% | 50y 36% |
| 70y 56% | 70y 48% | ||
| Quehenberger et al. 2005102 (n = 84) | MLH1, MSH2 | 30y 0.5% | 30y 0.6% |
| 40y 2.3% | 40y 2.5% | ||
| 50y 8.9% | 50y 9.0% | ||
| 60y 19.8% | 60y 17.6% | ||
| 70y 26.7% | 70y 22.4% | ||
| 80y 28.5% | 80y 23.7% |
As the study by Bonadona and colleagues (2011)2 is recent and large [537 families compared with the next largest study by Baglietto and colleagues (2010)166 with 113 families], and uses statistically rigorous techniques to adjust for ascertainment bias and interventions, we decided that this was the most relevant study and used its estimates of cumulative risk to inform CRC incidence in our model.
A logistic model for cumulative risk was fitted to this as in Dunlop and colleagues,101 with the following parameterisation:
These parameters are interpreted as follows: β0 is the hypothetical maximum risk (the proportion of individuals who would be affected by CRC if there were no competing risks such as general mortality); β1 controls the gradient of cumulative risk around β2; β2 is the time by which half the maximum risk has been experienced. The parameter values were obtained by ordinary least squares regression and are shown in Table 60. Bonadona and colleagues2 provide 95% CIs for the cumulative risks at age 70 years for males and females (males 25–59%, females 19–50%), and these are used to inform the parameters of our model in sensitivity analysis, such that we scale β0 to set the cumulative risk at age 70 years equal to the lower and upper bounds of the CI (see Table 60; Figures 23 and 24). We do not conduct sensitivity analyses on the parameters β1 and β2.
| Parameter | Base case | Sensitivity analysis |
|---|---|---|
| β 0 | Male 0.464 | Male 0.303–0.715 |
| Female 0.435 | Female 0.265–0.697 | |
| β 1 | Male 0.107 | None |
| Female 0.108 | ||
| β 2 | Male 55.5 | None |
| Female 61.3 |
FIGURE 23.
Model of cumulative risk of CRC for male MMR mutation carriers.
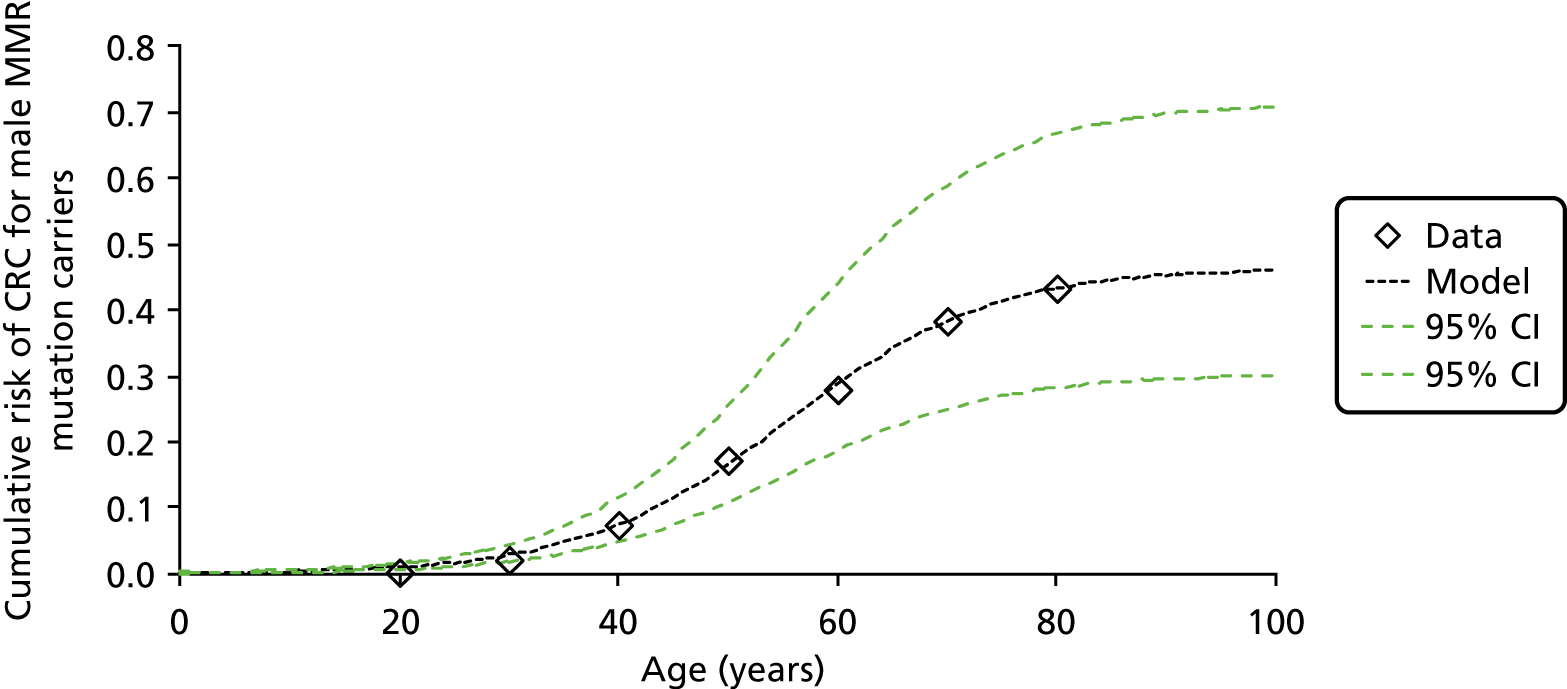
FIGURE 24.
Model of cumulative risk of CRC for female MMR mutation carriers.
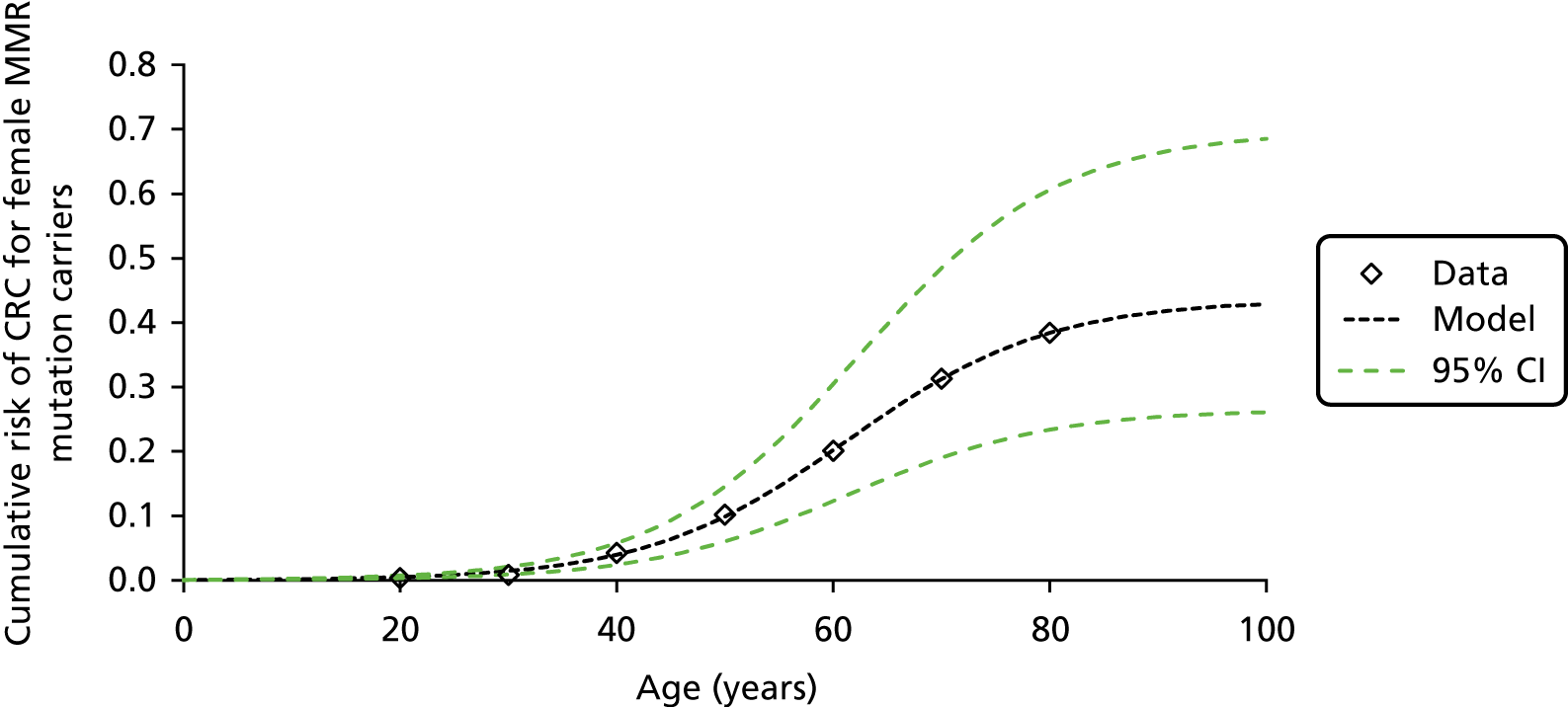
Figure 25 gives a comparison of the estimated cumulative risks of CRC for people with and without MMR mutations. As can be seen, the lifetime cumulative risk is substantially greater for people with LS.
FIGURE 25.
Cumulative risks of CRC for people with and without MMR mutations.
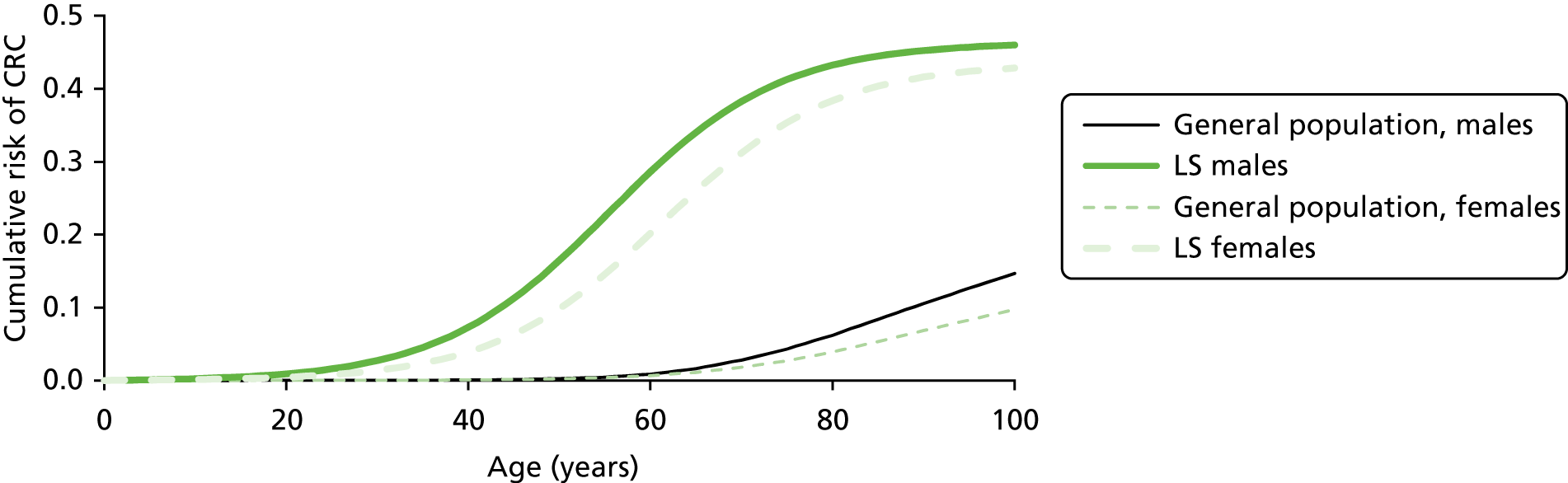
Figure 26 shows the impact of surveillance colonoscopy (as described in Effect of colonoscopy on index colorectal cancer incidence rates) in reducing CRC incidence.
FIGURE 26.
Impact of colonoscopy (age 25–75 years) in reducing cumulative risk of CRC for people with LS.
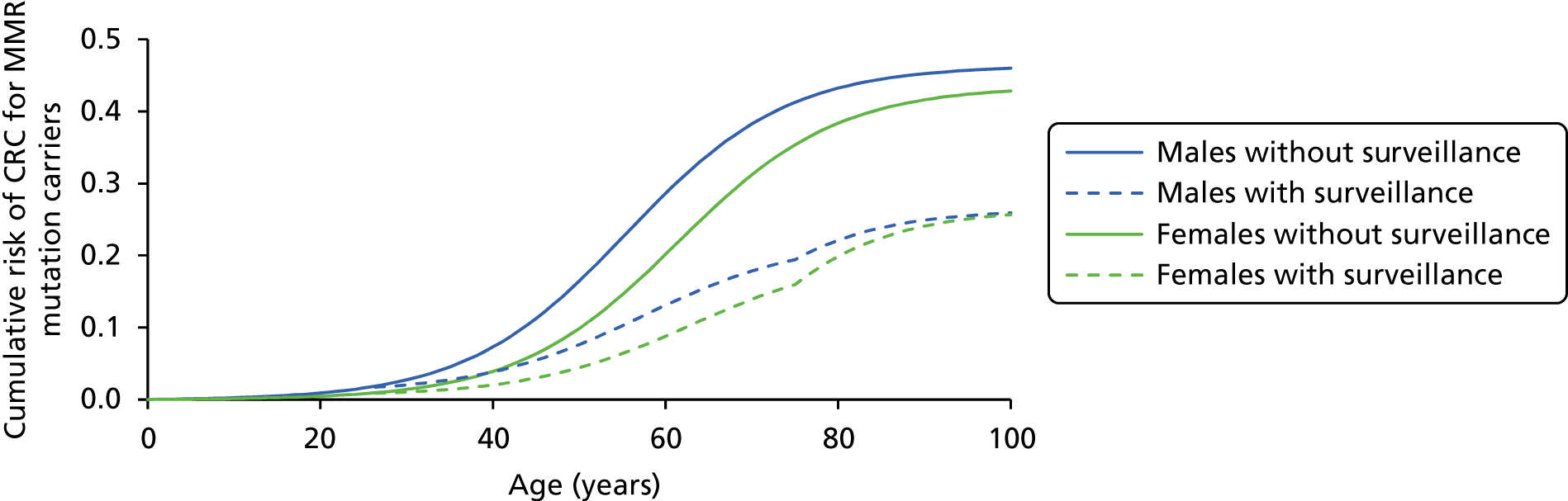
We estimated the risk of metachronous CRC for individuals with LS not undergoing regular surveillance jointly for men and women, according to cumulative risk estimates from Parry and colleagues. 168 As patients in the study by Parry and colleagues168 underwent regular colonoscopic surveillance, and given that we require incidence rates of metachronous CRC without regular surveillance, we apply a HR equal to the inverse of the HR for colonoscopic surveillance as described in Effect of colonoscopy on index colorectal cancer incidence rates. As a result, if these individuals do undergo colonoscopic surveillance, the incidence of metachronous CRC will be equal to that reported by Parry and colleagues. Figure 27 shows the cumulative risk of metachronous CRC in the PenTAG model for individuals with LS according to whether or not they received regular colonoscopic surveillance.
FIGURE 27.
Cumulative risk of metachronous CRC for individuals with LS.
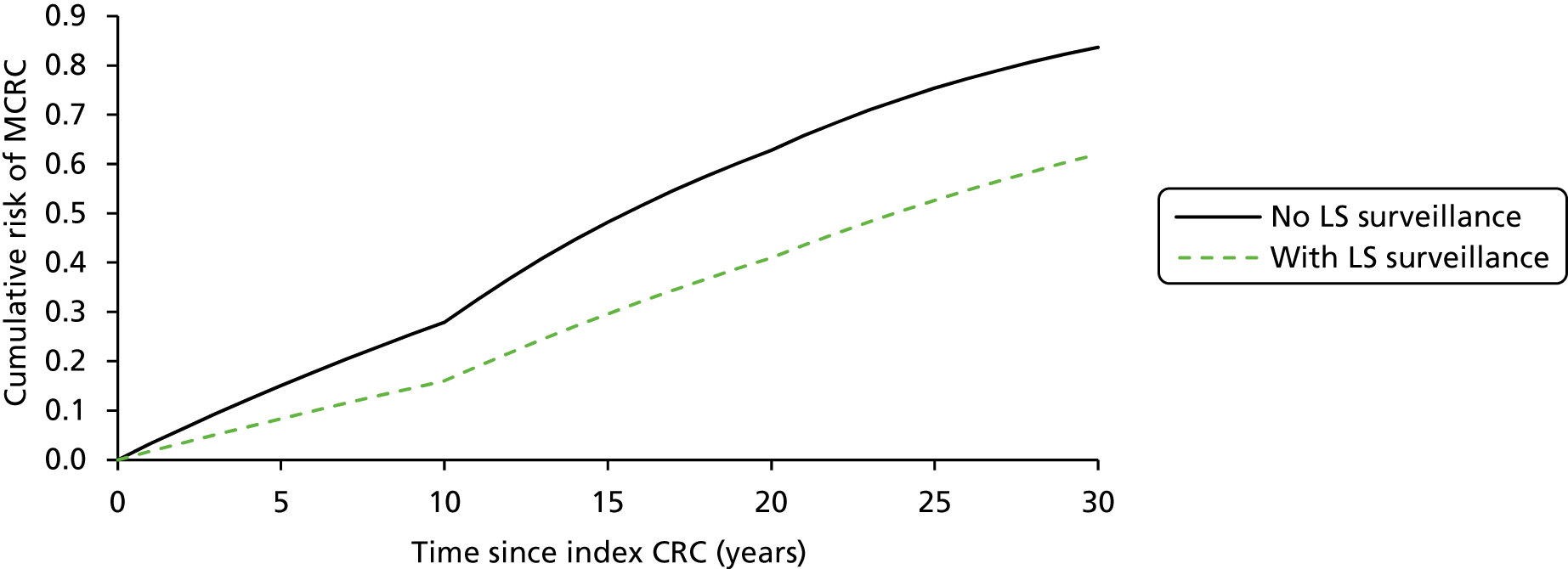
Dukes’ stage on diagnosis
The Dukes’ stage on diagnosis is an important predictor of survival. Here we describe our assumptions and data sources for the Dukes’ stage on diagnosis where there are no LS surveillance colonoscopies. The effect of colonoscopy on Dukes’ stage is addressed below (see Effect of colonoscopy on Dukes’ stage of incident colorectal cancers).
We assumed that the Dukes’ stage on diagnosis would be independent of age, sex, LS status and whether it was the first (index) CRC or a metachronous CRC (Fajobi and colleagues169 conclude that Dukes’ stages for metachronous CRC are no worse than for index CRC, and there was no consensus on whether or not they might be better). We also assumed that Dukes’ stage on diagnosis of metachronous CRC was independent of the stage of the index CRC and that the Dukes’ stage was independent of the CRC site. Dukes’ stage was assumed to depend only on whether or not the person was undergoing LS surveillance colonoscopies.
We estimated the Dukes’ stage distribution for individuals not undergoing LS surveillance colonoscopies by using data published in the National Bowel Cancer Audit Report (2011),135 as shown in Table 61. This report is based on data collected in England and Wales relating to patients diagnosed with bowel cancer between 1 August 2009 and 31 July 2010. We did not use data published in the 2012 report because it did not give stages in Dukes’ classification but in the TNM classification, and it was not possible to map these to Dukes’ stages because the incident population is not described by full TNM codes (e.g. T1N0M0), but is characterised by tumour, lymph nodes and distant metastases separately, with a significant number of data missing.
| Dukes’ stage | Colon | Rectosigmoid | Rectum | All sites | Proportion with Dukes’ stage (excluding unknown) |
|---|---|---|---|---|---|
| Dukes’ A | 1771 | 206 | 1495 | 3472 | 0.164 |
| Dukes’ B | 4831 | 349 | 1518 | 6698 | 0.317 |
| Dukes’ C | 3789 | 317 | 1617 | 5723 | 0.271 |
| Dukes’ D | 3482 | 331 | 1416 | 5229 | 0.248 |
| Unknown | 3875 | 388 | 2727 | 6990 | |
| Total (excluding unknown) | 13,873 | 1203 | 6046 | 21,122 | |
| Total (including unknown) | 17,748 | 1591 | 8773 | 28,112 |
Colorectal cancer incident site
As stated previously (see Patient bowel state), we model two sections of the bowel: the colon and the rectum. We grouped rectosigmoid cancer [International Classification of Diseases, Tenth Edition (ICD-10) code C19] into rectal cancer. The site of incident CRCs was dependent on sex, whether or not the person has LS and any previous surgery (Table 62).
| Previous surgery | With LS | Without LS | |
|---|---|---|---|
| Male | Female | ||
| None | 0.94 | 0.58 | 0.61 |
| Segmental resection | 0.94 | 0.58 | 0.61 |
| Subtotal colectomy | 0.00 | 0.00 | 0.00 |
| Anterior resection | 1.00 | 1.00 | 1.00 |
| Proctocolectomy | N/A | N/A | N/A |
It is assumed that all CRCs are colon cancers following anterior resection, and that all CRCs are rectal cancers following subtotal colectomy. If there is no previous surgery or a previous segmental resection, the probability of the CRC being situated in the colon for a person with LS is estimated as 0.94, based on Dinh and colleagues. 55 For males and females without LS, the probability of colon cancer is estimated from ONS cancer registration statistics for patients aged under 50 years. 119 Estimating from patients of all ages would not significantly affect the proportion estimated to be situated in the colon.
Mortality due to colorectal cancer
We assume that mortality due to CRC depends on the following:
-
Dukes’ stage at diagnosis
-
years since diagnosis
-
age at diagnosis
-
LS status (see Mortality due to colorectal cancer for patients with Lynch syndrome).
We did not consider the effect of the following on mortality due to CRC:
-
patient’s sex
-
site of CRC
-
surgery for CRC.
The baseline annual rate of mortality due to CRC was derived from data provided by the NCIN170 by extracting 1-, 2-, 3-, 4- and 5-year relative survival from survival curves and assuming constant rates of mortality within each year (Table 63 and Figure 28). It was assumed that the mortality rate for 4–5 years since diagnosis also applies after 5 years (Table 64).
| Years since diagnosis | Dukes’ A | Dukes’ B | Dukes’ C | Dukes’ D |
|---|---|---|---|---|
| 1 | 0.969 | 0.917 | 0.815 | 0.380 |
| 2 | 0.965 | 0.872 | 0.681 | 0.193 |
| 3 | 0.957 | 0.831 | 0.583 | 0.116 |
| 4 | 0.945 | 0.799 | 0.522 | 0.083 |
| 5 | 0.932 | 0.770 | 0.477 | 0.066 |
FIGURE 28.
Colorectal cancer survival in model.
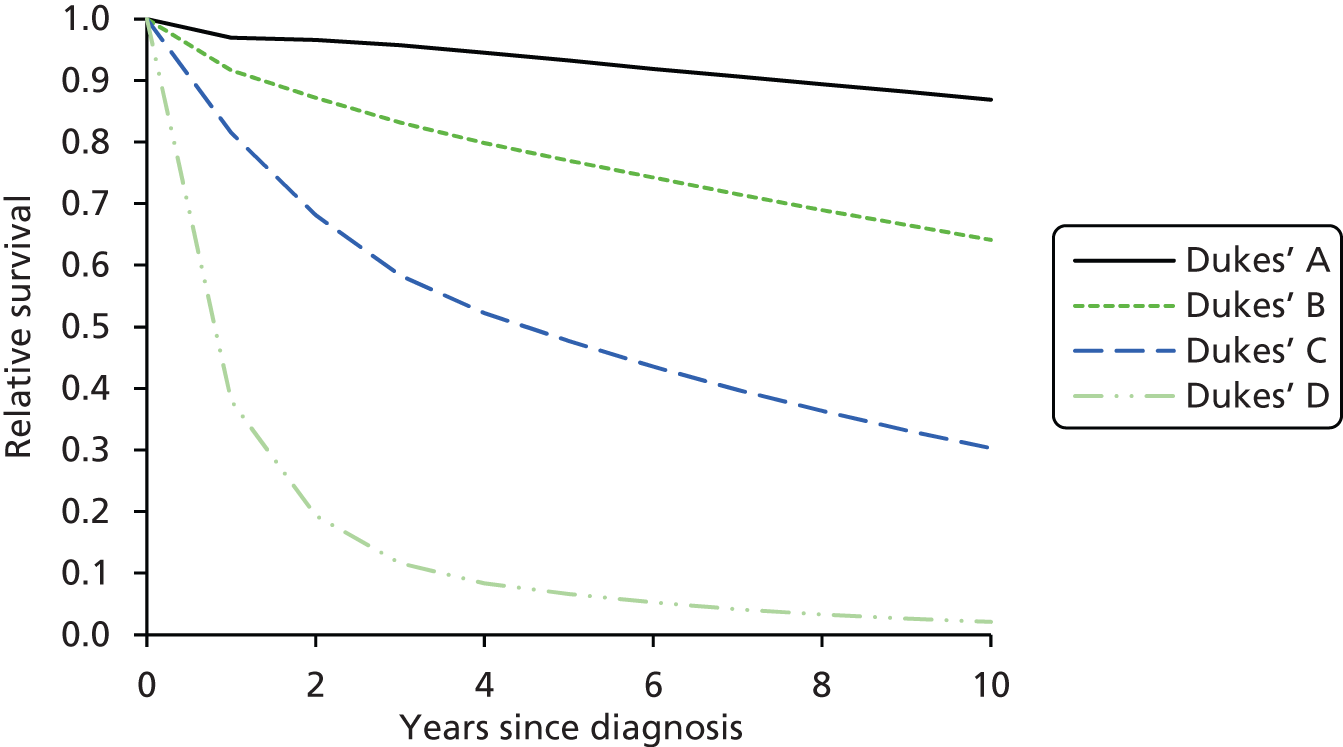
| Years since diagnosis | Dukes’ A | Dukes’ B | Dukes’ C | Dukes’ D |
|---|---|---|---|---|
| 0–1 | 3102 | 8709 | 20,460 | 96,729 |
| 1–2 | 419 | 5000 | 17,971 | 67,733 |
| 2–3 | 843 | 4761 | 15,465 | 51,116 |
| 3–4 | 1279 | 4000 | 11,060 | 32,857 |
| > 4 | 1400 | 3667 | 9068 | 23,375 |
The assumption that the mortality rate after 5 years is equal to the mortality rate for 4–5 years is likely to be a slight overestimate of CRC mortality (Table 65). The result of a slight overestimate of CRC mortality in the model would be a slight improvement in the cost-effectiveness of strategies with high yield of LS mutations.
| Years since diagnosis | Male colon cancer | Female colon cancer | Male rectal cancer | Female rectal cancer | Model CRC |
|---|---|---|---|---|---|
| 1 | 0.730 | 0.722 | 0.788 | 0.788 | 0.764 |
| 5 | 0.544 | 0.551 | 0.546 | 0.575 | 0.544 |
| 10 | 0.501 | 0.508 | 0.473 | 0.521 | 0.436 |
The HRs for CRC mortality by age, compared with CRC mortality across all ages, were estimated using net survival statistics from the ONS,147 and are shown in Table 66. Details of calculations are given in Appendix 6.
| Age group | HR for CRC mortality | ||
|---|---|---|---|
| First year | Following 4 years | Thereafter | |
| < 70 years | 0.599 | 0.972 | 1 |
| 70–79 years | 0.956 | 0.966 | 1 |
| ≥ 80 years | 1.797 | 1.116 | 1 |
Mortality due to metachronous colorectal cancer
Mortality due to metachronous CRC was modelled by adding together the mortality rates for both the index and metachronous CRC as calculated above, assuming that mortality from the metachronous cancer would be no different from mortality from the index cancer for the same Dukes’ stage [as assumed by, for example, Mvundura and colleagues (2010)54 and Dinh and colleagues (2011)55]. The same approach is used independent of the LS status of the patient. As the mortality rates are dependent on the time since diagnosis in the model, we keep track of time since diagnosis of the index cancer and the metachronous cancer.
Although the data informing mortality due to CRC for people without LS will include additional mortality due to metachronous CRC, the incidence of metachronous CRC in the data is likely to be very small so would have a negligible effect on observed survival rates. Similarly, it is possible that mortality due to CRC for patients with LS (see Mortality due to colorectal cancer for patients with Lynch syndrome, below) would also include additional mortality due to metachronous CRC. However, we believe that this is unlikely to have a significant impact, and indeed our estimate is that mortality due to CRC is lower for some patients with LS.
Mortality due to colorectal cancer for patients with Lynch syndrome
Although it has long been believed that CRC patients with LS have better survival than those with sporadic CRC, recent evidence is not unanimous in support of this. 172 In a previous model of the cost-effectiveness of testing for LS, Mvundura and colleagues54 cite a systematic review by Popat and colleagues (2005)173 to make adjustments to survival for individuals with CRC and LS. Popat and colleagues173 derive HRs for survival according to the MSI phenotype, which is observed in most LS CRCs and around 15% of all CRCs (these correspond to the sensitivity and specificity of MSI). In another model of the cost-effectiveness of testing for LS, Dinh and colleagues (2011)55 perform a meta-analysis and estimate a HR of 0.53 for mortality due to CRC between individuals with and without LS.
There are a number of considerations to make when estimating such a HR. Firstly, many of the studies cited investigate patients with tumours showing the MSI phenotype, rather than LS. Whereas most LS CRCs show the MSI phenotype, it is not the case that most tumours showing the MSI phenotype are LS CRCs. 174 Secondly, survival may be reported as being better in LS CRC patients because of other predictive factors such as age and stage at diagnosis, as LS CRCs are characterised by early onset and surveillance may be in place to identify CRCs at an earlier stage. These factors should be accounted for when estimating a HR for mortality due to CRC between individuals with and without LS. Thirdly, as LS CRC patients are more likely to develop metachronous CRC, there may be differences in how these are treated statistically; for example, if patients are not censored when they develop metachronous CRC, then mortality would be confounded by additional mortality from the metachronous CRC. Additionally, if patients are retrospectively recruited to studies on the basis of FH, it is more likely that less penetrant alleles will be included. 175
As Popat and colleagues (2005)173 investigate the HR for the MSI phenotype and not for LS, we do not use these results. We followed citations from Coate and colleagues (2010)172 and summarise results in Appendix 7.
Of the studies we identified, only one [Barnetson and colleagues (2006)175] was in a similar setting to that in our model. In this study, 870 patients diagnosed with CRC in Scotland under the age of 55 years were tested for mutations in MLH1, MSH2 and MSH6 and were subsequently followed up to investigate the effect of LS on CRC survival. This is a similar setting to that in our model as patients were selected by age alone, and not according to FH, and were genotyped. The conclusion of this study is that after adjusting for tumour stage [split by localised (stages I–II)/metastatic (stages III–IV)] there is no significant effect on survival of LS status. The Kaplan–Meier curves for survival in metastatic cancer seem relatively convincing of the argument that there is no effect, but those for survival in localised cancer are less convincing; it seems that a lack of statistical power may have led to the conclusion that there is no survival benefit, and certainly there is not enough evidence to suggest that there is no survival benefit in localised disease from having LS.
To obtain an alternative estimate of the effect of LS status on survival for localised CRC, we use results from Lin and colleagues (1998). 176 Lin and colleagues176 retrospectively identified 75 CRC cases with confirmed MLH1/MSH2 mutations and compared their survival to 820 CRC cases believed to be from the general population. Although Lin and colleagues recruited LS families in a different way from that proposed in our model, we believed that this was the next most appropriate study as it includes a large number of CRC cases with proven MMR mutations and does not include non-genotyped cases, adjusts for the CRC stage and reports a HR from a Cox regression. Lin and colleagues do not censor at the time of metachronous CRC incidence, which could lead to a HR estimate biased towards 1, but the rate of metachronous cancer was relatively low (around 2% annually) so we believe that this would not have a significant impact. It is also notable that the data in Lin and colleagues’ study are quite old, with LS CRC diagnosed between 1945 and 1991. The comparator cohort included patients diagnosed between 1965 and 1996. The control cohort could therefore be expected to have slightly improved survival due to improved medical and surgical techniques; this would also bias the HR estimate towards 1, although this would likely have a minor effect as patients deemed to have suffered from postoperative mortality were excluded.
In our model we assume that the HR for survival for LS carriers is 0.57 for Dukes’ stages A and B and 1 for Dukes’ stages C and D (Figure 29). We perform two scenario analyses in which these HRs are altered. In the first we use a HR of 1 for all Dukes’ stages and in the second we use a HR of 0.57 for all Dukes’ stages.
FIGURE 29.
Survival for patients with CRC according to LS status and Dukes’ stage.
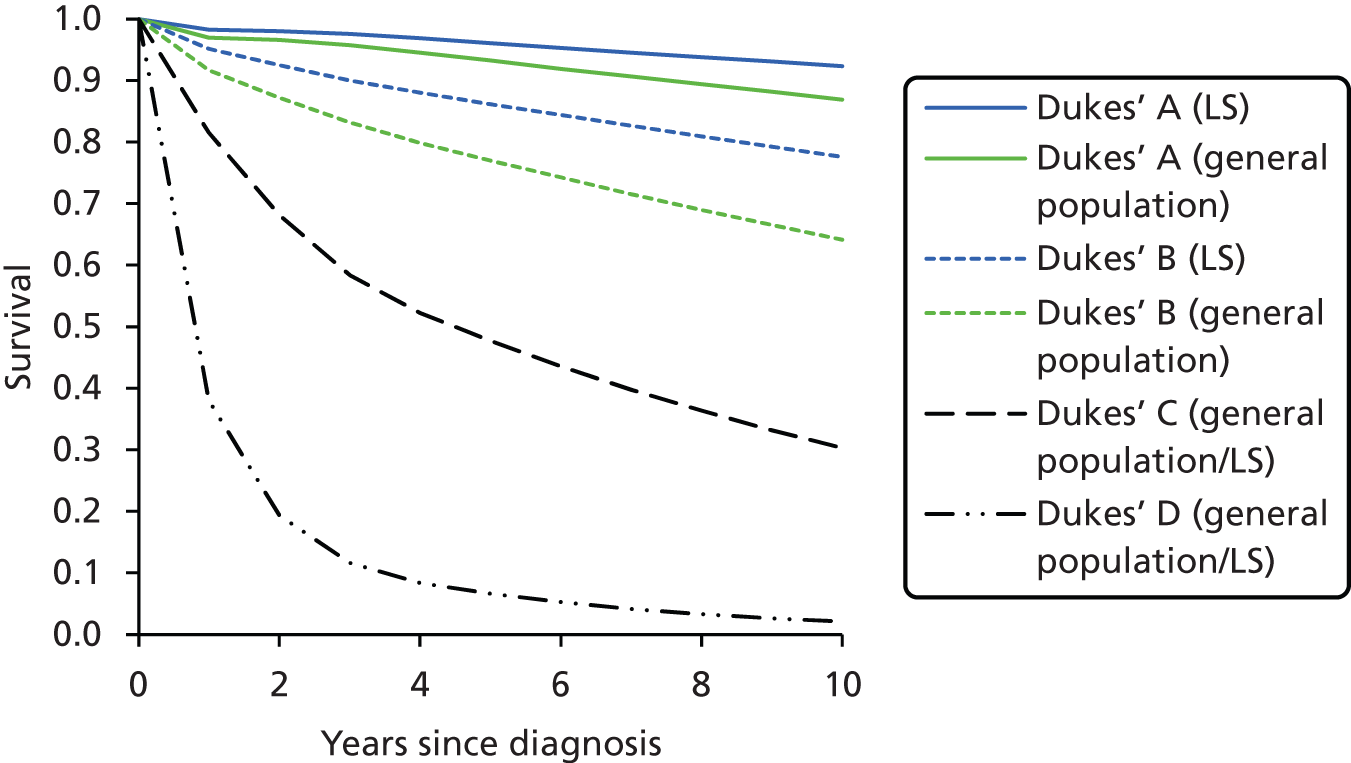
Gynaecological cancers
Endometrial cancer
Endometrial cancer incidence
We assume that the incidence of EC is zero for women without LS, because it is much lower than the incidence rate for women with LS, which is given below. In particular, given that the lifetime risk of uterine cancer for women in the UK is 2.4%,8 and that EC accounts for most cases of uterine cancer, the lifetime risk of EC for women in the UK is nearly 2.4%. This compares with the estimate of 1.7% obtained by Ladabaum and colleagues (2011)48 from the US SEER database. 177 By contrast, we estimate a lifetime risk of about 34% for women with LS.
As explained previously (see Surgery for endometrial cancer), we also assume that prophylactic surgery for EC completely eliminates the risk of this cancer. Henceforth, we discuss the incidence of EC for women with LS who have not had prophylactic surgery for EC. This applies to both female probands who are diagnosed with CRC and their female relatives who have LS.
Of the seven analyses that incorporate EC,48,55–57,75,76,85 only three [Chen and colleagues (2007),85 Kwon and colleagues (2008)75 and Yang and colleagues (2011)76] model the specific stage (I to IV) of cancer, and Yang and colleagues (2011)76 is an extension of Chen and colleagues (2007)85 (see Table 89). The stage of EC is not incorporated in the PenTAG model. Instead, individuals are deemed to either have the cancer or not. Indeed, given that we assume no screening for EC (see Screening for endometrial cancer), we believe that there is no reason to model the stage of the disease.
A recent study by Bonadona and colleagues2 of 10,283 individuals from several French clinics investigated the risks of various cancers for individuals with LS. All these participants were from 537 families in which MMR mutations were confirmed. In total, 2662 people were genotyped and, of these, 1633 were found to have mutations. Most families had the MSH1 or MSH2 mutation, and a small number had the MSH6 mutation. For EC and OC, women were censored at the time of gynaecological surgery. The estimated cumulative risk of EC by the age of 70 years was 34% and for OC 9%. The estimated cumulative risk of EC was 0% by age 30 years, 2% by age 40 years, 8% by age 50 years, 23% by age 60 years, 34% by age 70 years and 35% by age 80 years.
We use the rates of incidence of EC from the study by Bonadona and colleagues (2011)2 in our model because (1) the study was large; (2) as far as we can tell, it appears methodologically sound; (3) it was conducted recently; (4) a statistical technique was used to correct for ascertainment bias due to recruitment of families with multiple cases of cancer; and (5) the rates are consistent with those found in other studies, particularly as explained in the literature review of Palomaki and colleagues (2009),65 as discussed below. However, as acknowledged by the authors, the cumulative risk of EC becomes rather uncertain above about 55 years of age.
In their cost-effectiveness model, Ladabaum and colleagues (2011)48 assumed that the cumulative incidence of EC by age 70 years is 37%, which is similar to our estimate of 34%. Ladabaum and colleagues (2011) cite Palomaki and colleagues (2009)65 and Horner and colleagues (2009)177 as the sources for their estimate. In their review of the literature, Palomaki and colleagues (2009)65 found that estimates are variable, ranging from 31% to 64% across five studies. They suspect that some of the higher estimates may be subject to FH bias.
In a study of 147 US families with LS, Stoffel and colleagues (2009)106 used a statistical method to control for ascertainment bias to estimate a risk of EC of 39% by age 70 years. This estimate is also similar to our estimate of 34%.
Mortality due to endometrial cancer
For women with EC, the rate of mortality due to EC was assumed to vary according to time since diagnosis. As the stage of EC is not modelled, the rate of mortality was assumed to be the same for all affected patients. In addition, mortality was assumed to be equal for women with and without LS. 55,178
Three sources of EC survival data were identified for women in the general population of England. 148–150 The Cancer Research UK data150 included ONS data148 and unpublished research commissioned by Cancer Research UK (Table 67). Cancer Research UK and ONS data are both reported as age-standardised relative survival, which is calculated by weighting age-specific relative survival estimates by the age distribution of uterine cancer in England and Wales in 1996–9. ONS data also report relative survival according to age groups and overall relative survival not age standardised (see Table 67). 148 Survival in the NCIN UK cancer e-Atlas149 is reported at 1, 3 and 5 years after diagnosis without age standardisation (Table 68).
| Years since diagnosis | Age-standardised relative survival (%) | Non-standardised relative survival (%) |
|---|---|---|
| 1 | 90.1 | 91.0 |
| 5 | 77.3 | 79.2 |
| 10 | 74.5 | NR |
| Years since diagnosis | Relative survival (%) |
|---|---|
| 1 | 90.5 |
| 3 | 81.4 |
| 5 | 77.9 |
We assumed that survival data for uterine cancer were appropriate to use for EC on the basis that EC comprises the vast majority of uterine cancer cases. For example, in 2010, of 6834 registrations of uterine cancer (ICD-10 codes C54–C55), 6409 (93.8%) were EC, while only 44 (0.6%) had a specified location other than the endometrium and 381 (5.6%) were of an unspecified location in the uterus. 119
A piecewise constant hazard rate was assumed for death from EC which was fitted to survival data points at 1, 3, 5 and 10 years (Tables 69 and 70). Age-standardised survival data were used when available (1, 5 and 10 years). For 3-year survival, non-standardised relative survival was used because age-standardised relative survival was not available and because the effect of age standardisation appears to be small at 1 and 5 years. For survival beyond 10 years, a hazard rate of zero was assumed for death from EC. Figure 30 shows the result of our modelling.
| Years since diagnosis | Relative survival (%) | Source |
|---|---|---|
| 1 | 90.1 | ONS148 |
| 3 | 81.4 | NCIN UK Cancer e-Atlas149 |
| 5 | 77.3 | ONS148 |
| 10 | 74.5 | Cancer Research UK150 |
| Years since diagnosis | Hazard of death from EC (deaths per 1000 EC patients per year) |
|---|---|
| 0–1 | 104.25 |
| 1–3 | 50.77 |
| 3–5 | 25.84 |
| 5–10 | 7.38 |
| 10+ | 0.00 |
FIGURE 30.
Relative survival of women with EC in the UK.
Mortality due to other causes
Death from other causes was modelled using separate mortality rates for men and women, provided in life tables for England and Wales (2008–10),179 adjusted to remove the proportion of mortality due to CRC. The proportion of mortality caused by CRC was estimated by dividing the number of deaths from CRC in each age group by the total number of deaths in that age group, using mortality data for England in 2010. 143 We did not adjust for mortality from EC as this accounted for < 1% of deaths in the general population (whereas CRC accounted for 2.8%), and we did not adjust for mortality from other LS-associated cancers as these are not included in our model.
Effectiveness parameters and assumptions
Diagnostic effectiveness parameters and assumptions
Diagnostic effectiveness is made up of the following three key components, which we discuss below:
-
test accuracy
-
adherence to testing
-
numbers of probands and relatives per year in England, and proportion of probands and relatives with LS.
These are important to both short- and long-term outcomes. For instance, adherence can affect the accuracy of a strategy; a higher adherence to genetic testing will make a strategy more accurate as genetic testing has a higher accuracy than diagnosing a patient using FH. Diagnostic accuracy and the proportion of patients diagnosed with (and without) LS have important implications for the long-term cost-effectiveness of a strategy.
Test accuracy
Estimates of test accuracy (sensitivity and specificity) were taken from the available literature (Table 71). Accuracies of tests (e.g. IHC, MSI) are almost always available only for tests in isolation. One exception is the BRAF test, for which we have accuracy information after an IHC test with abnormal MLH1 staining [Palomaki and colleagues (2009)65] and after an MSI test [Domingo and colleagues (2005)183]. Given that information on the accuracy of tests in sequence is generally lacking, the accuracy of such tests is assumed as if it were applied in isolation.
| Test | Sensitivity | Specificity | Source |
|---|---|---|---|
| MSI | |||
| MLH1 and MSH2 | 0.89 | 0.902 | Palomaki et al. 200965 Assumed |
| MSH6 | 0.77 | ||
| PMS2 | 0.77 | ||
| IHC | 0.770 | 0.888 | Palomaki et al. 200965 |
| BRAF after IHC MLH1 abnormal | 1.00 | 0.69 | Palomaki et al. 200965 |
| BRAF after MSI | 1.00 | 0.40 | Domingo et al. 200484 |
| AC II | 0.39 | 0.98 | Hampel et al. 2005,103 Salovaara et al. 2000,180 Green et al. 2009,181 Barnetson et al. 2006,175 Balmana et al. 2008182 |
| Genetic test MLH1, MSH2 and MSH6 (for probands) | 0.90 | 0.9997 | Dinh et al. 2011,55 Palomaki et al. 200965 |
| Genetic test PMS2 (for probands) | 0.62 | 0.9997 | Dinh et al. 2011,55 Palomaki et al. 200965 |
| Targeted genetic test (for relatives) | 1 | 1 | Assumed |
Some test accuracies refer to specific genes; the EGAPP supplementary evidence review reports a different sensitivity for genetic testing of PMS2 compared with MLH1, MSH2 and MSH6, and there are different sensitivities for each of MLH1, MSH2 and MSH6 for MSI. The sensitivity for PMS2 is not reported owing to lack of evidence, but the indication is that it is lower than those for MLH1 and MSH2. Therefore, as in Palomaki’s review,65 we assume it is the same as for MSH6. BRAF has a sensitivity specifically reported for abnormal MLH1 IHC results. For tests and accuracies where genes are not specified, we have assumed that the accuracies are the same for every gene and therefore can refer to LS as a whole, rather than to individual genes. In some cases, where accuracies are not available for all genes, there may be evidence that this can be extrapolated to all LS genes. One example concerns the accuracy of a BRAF test after MSI testing. A study by Domingo and colleagues (2004)84 on BRAF after MSI only considered MLH1 and MSH2; their follow-up [Domingo and colleagues (2005)183] additionally considered MSH6 and suspected LS cases where a genetic mutation had not been identified. Domingo’s study indicates that V600E is only found in sporadic tumours, and so a BRAF test will not behave differently for the various LS genes. We therefore extrapolated the sensitivity and specificity of BRAF after MSI to PMS2.
As already stated, the assumed accuracy of FH is that attributed to the AC II (as our probands already fulfil the Revised Bethesda criteria), which has sensitivity 39% and specificity 98% according to sources identified by Ladabaum. 48 This value is taken from the original sources: Hampel and colleagues (2005),103 Salovaara and colleagues (2000),180 Green and colleagues (2009),181 Barnetson and colleagues (2006)175 and Balmana and colleagues (2008). 182 These papers compare the AC II to genetic testing as a gold standard, where a variety of genes (at least MLH1 and MSH2) are assessed. Furthermore, they are from a variety of populations: one US, one Canadian and three European, of which one was UK-based. The sensitivity and specificity of the AC II does not seem to be related to population or number of genes, with mixed values coming from each individual study. The specificity estimates are more consistent than the estimates of sensitivity; however, the UK study, which seems to be most relevant and looks at all genes except PMS2, has a sensitivity of 42% and a specificity of 98%, suggesting that the average values of sensitivity and specificity from Ladabaum48 (39% and 98%, respectively) are acceptable.
For simplicity, and in common with other cost-effectiveness models [Mvundura and colleagues (2010),54 Ladabaum and colleagues (2011)48], we assume that the test accuracies apply to all qualities of tumour sample and that all tests are available and always successfully produce a result. In reality, some tests may not be available, depending on the laboratory or provision of usable tumour tissue, and some tests may ‘fail’. There is one exception made in the model: the failure rate is specifically included in the sensitivity of the IHC test [Palomaki and colleagues (2009)65] because it is the only test to state it explicitly.
The sensitivity of diagnostic genetic testing for genes MLH1, MSH2 and MSH6 in probands is conservatively set to 90% as it was in Dinh and colleagues’ report in 2011. 55 This is to reflect genetic variants of uncertain significance and other such factors. We consider targeted genetic testing in relatives to be 100% accurate.
The sensitivities and specificities of these individual tests for probands are plotted in Figure 31. This shows that BRAF has the highest sensitivity for any individual test, regardless of which test it is conducted after, though this does affect the specificity. Genetic testing has the highest specificity, but different sensitivity depending upon whether testing for PMS2 or MLH1, MSH2 and MSH6. In this figure, MSI’s difference in accuracy for genes MLH1 and MSH2 compared with MSH6 and PMS2 is illustrated, rather than the overall values of sensitivity and specificity for MSI. It is important to remember that in this instance MSI is one test, influenced by the proband’s characteristics, not two separate tests as is the case for genetic testing. As is typical, there appears to be a general pattern that those tests that are more sensitive are less specific and vice versa.
FIGURE 31.
Receiver operating characteristic plot of individual tests. Here ‘|’ means ‘given’ or ‘after’, for example ‘BRAF|MSI’ means ‘BRAF given MSI’ or ‘BRAF after MSI’. GT, genetic testing; GT M126, combined diagnostic mutation testing for genes MLH1, MSH2 and MSH6; GT PMS2, diagnostic mutation testing for PMS2 gene; MSI M12, MSI test in individuals with MLH1 or MSH2 constitutional mutations; MSI M6P2, MSI test in individuals with MSH6 or PMS2 constitutional mutations.
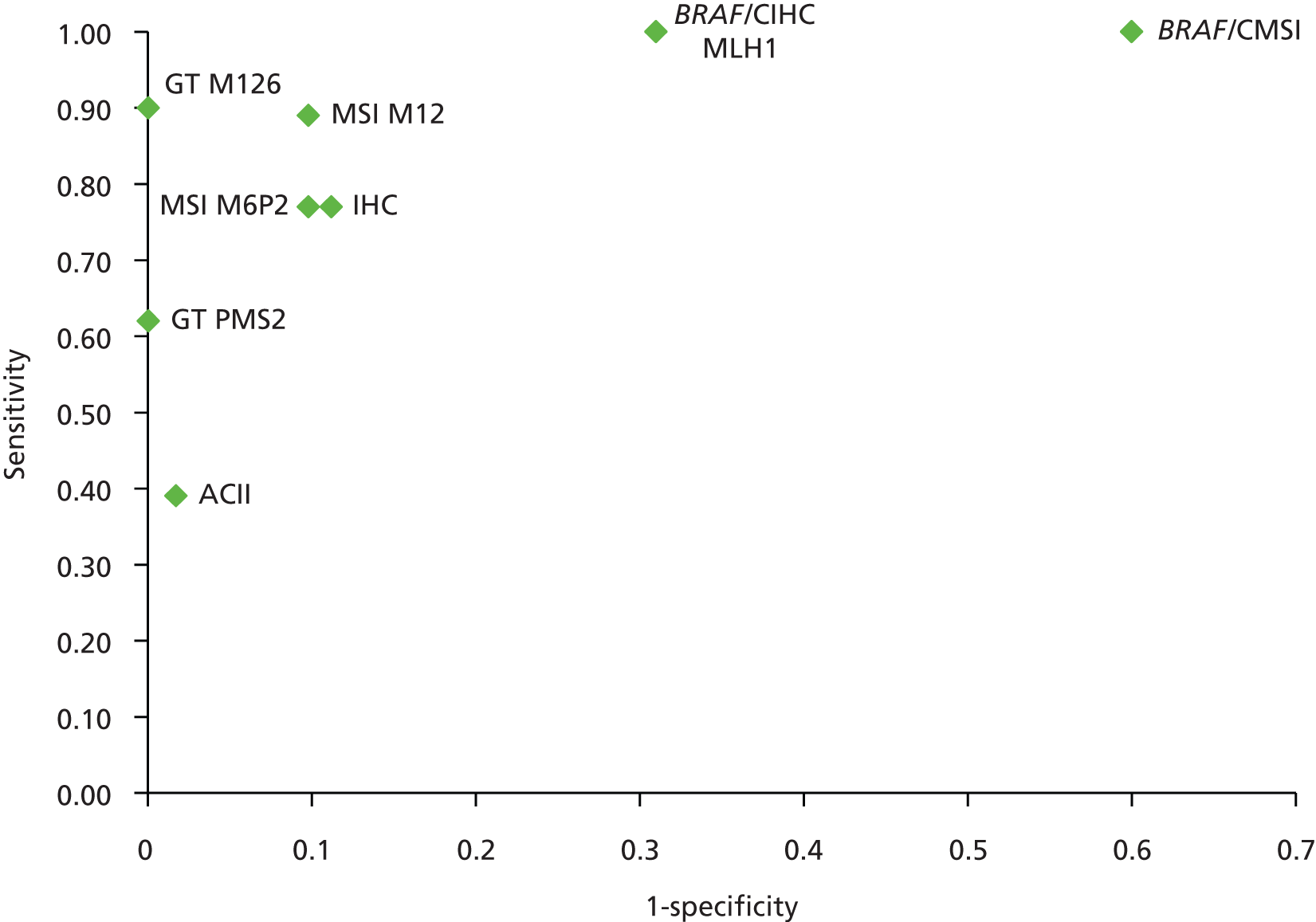
Acceptance of testing and surveillance
Given that there is little published information on acceptance of testing, and that what evidence there is seems variable, we have made several assumptions:
-
The rate of acceptance of a test is independent of any previous tests the patient has taken.
-
Acceptance of one genetic test implies acceptance of all.
-
The rate of acceptance of IHC and BRAF is assumed equal to that of MSI (100%49) as they are all preliminary tumour tests. Some clinicians might consider the results of the BRAF test enough of an indicator for familial cancer that genetic counselling should be carried out prior to the test. If so, this might influence acceptance of the BRAF test.
-
Genetic counselling includes an investigation into FH and therefore, for strategies using genetic counselling, the rate of acceptance of FH screening is included in the rate of acceptance of genetic counselling. For patients who decline genetic counselling and for strategies where genetic testing is not offered, there is a separate rate of acceptance of FH screening, initially set to 1.
Assumed acceptance rates are given in Table 72. Where possible, and if applicable, we checked the original sources, and if necessary, recalculated the values.
| Test | Proband/relative | Acceptance rate (%) | Source |
|---|---|---|---|
| MSI | Proband | 100 | Ramsey et al. 200349 confirmed by expert Ian Frayling (2012, personal communication) |
| IHC | Proband | 100 | Assumed, see text |
| BRAF | Proband | 100 | Assumed, see text |
| First genetic test (proband) | Proband | 90 | Ladabaum et al. 201148 |
| Second genetic test (proband) | Proband | 100 | Assumed |
| Genetic counselling (proband) | Proband | 92.5 | Clinical experts (IMF) gave range 90–95% |
| FH assessment after genetic test (proband) | Proband | 100 | Assumed (as included in counselling) |
| FH assessment when genetic counselling or test declined (proband) | Proband | 100 | Assumed |
| Genetic test (relative) | Relative | 96a | Calculated from Palomaki et al. 200965 |
| Genetic counselling (relative) | Relative | 45a | Calculated from Palomaki et al. 200965 |
Initial adherence to the surveillance strategy for CRC offered to the probands and relatives who test LS mutation positive or LS assumed are given in Table 73. Of these adherence estimates, there was substantial evidence only for relatives who test mutation positive. Probands who test mutation positive are assumed to have a similar level of adherence given lack of evidence to the contrary. The rates of adherence for probands and relatives assumed to have LS are taken from Ladabaum and colleagues (2011)48 and based on expert opinion.
| Patient characteristic | Initial adherence to surveillance (%) | Source |
|---|---|---|
| Proband tested LS mutation positive | 80 | Ladabaum et al. 201148 |
| Proband LS assumed | 70 | Expert opinion from Ladabaum et al. 201148 |
| Relative tested LS mutation positive | 80 | Ladabaum et al. 201148 |
| Relative LS assumed | 50 | Expert opinion from Ladabaum et al. 201148 |
Numbers of probands and relatives with and without Lynch syndrome
Numbers of probands
The total number of probands aged < 50 years in England, 1699, was extracted from the latest edition of the ONS cancer registration statistics. 119 As the number of familial adenomatous polyposis (FAP) patients in this group will be minimal (current practice indicates that nearly all FAP patients would now be identified and managed before developing CRC), we believed it appropriate to assume that this value was 0, and that all probands would therefore have CRC as a result of sporadic cancer or due to LS. Other hereditary CRCs are possible, but are not well defined and make up a small proportion of those diagnosed with CRC, and as such are not considered here.
The number of TP LS probands is calculated using the prevalence of LS among CRC patients under age 50 years (taken from Hampel and colleagues120) of 8.4%. These are then subdivided into the number of each gene using the breakdown from the supplementary evidence in the EGAPP review, which states that for all true LS-positive patients, 32% will have a MLH1 mutation, 39% a MSH2 mutation, 14% a MSH6 mutation and 15% a PMS2 mutation. 65 These values are consistent with those reported currently by UK genetics centres, where there is currently no systematic testing for LS. Countries where systematic testing already occurs appear to have a different distribution of genes, for instance Sjursen and colleagues184 report 18% MLH1, 50% MSH2, 26% MSH6 and 6% PMS2 mutations in Norway. We use these values in a sensitivity analysis.
In strategy 3, outcomes of the IHC test need to correspond to the relevant genes, so the false-positive IHC results are also split by gene, using values from Mvundura and colleagues54 (90% MLH1, 6% MSH2, 2% MSH6, 2% PMS2).
Numbers of relatives
Many published studies of LS contain data regarding the number of probands identified, the number of relatives identified (and, of these, how many are FDRs of the proband), the number of relatives counselled, the number of relatives receiving predictive testing for LS and the number of relatives testing positive for the family mutation (although in many cases not all of these are reported).
We collected a number of studies which provide these data and noticed substantial heterogeneity, which suggested a meta-analysis would not be appropriate. We therefore attempted to identify studies with particular relevance to the NHS.
Barrow and colleagues (2009)121 recruited LS patients referred to the Manchester Regional Genetics Service and performed statistical analyses to estimate the risk of extracolonic cancers in mutation carriers (in a previous study81 they estimated the risk of CRC). Although their methodology for risk estimation has been criticised by Bonadona and colleagues (2011),2 their recruitment statistics are not questioned and are valuable in this setting. The authors report that there were 121 families on the database, which means there were up to 121 probands referred (some probands may have been discovered to be from the same family, but this is not reported). The total number of family members on the database was 1420, which means that at least 1299 relatives were identified. This gives an estimate that an expected 1299/121 = 10.7 relatives would be identified for each proband. Of the 1420 family members, 249 were proven mutation carriers. If we assume that 121 probands are accounted for in the 249, this leaves 128 relatives testing positive. If we assume that 0.45 of those tested are found to be carriers, this suggests that 284 of the 1299 relatives identified would be tested. This corresponds to 0.218 of the relatives identified. We estimate that the ratio of relatives tested to probands identified from this study is 284/121 = 2.35.
As in this study a FDR of a proven mutation carrier with a LS-associated cancer is labelled a ‘putative carrier’ (n = 331) and a number of relatives were obligate carriers (n = 90), it is possible that the number of relatives being recommended for testing would be lower.
Hampel and colleagues (2008)120 attempted to identify LS in 1566 CRC patients (unselected for age or FH) and identified 44 patients with deleterious mutations. They subsequently attempted to identify, counsel and test relatives appropriately, and tested 249 relatives. This gives a ratio of 5.66 relatives tested to each proband. If we assume that 46% of relatives identified would be counselled, of which 95% would be tested,65 then this suggests that around 582 relatives would have been identified, with a ratio of 13.24 relatives identified to each proband.
Palomaki and colleagues (2009)65 identified seven studies (none of them from the UK) to estimate what proportion of relatives would be counselled and tested. They performed a meta-analysis on these two proportions and eliminated one study from the meta-analysis for the proportion counselled as it was a significant outlier. They concluded that the proportion of relatives who would be counselled was 0.46 (95% CI 0.41 to 0.50) and the proportion of those counselled who would be tested was 0.95 (95% CI 0.93 to 0.97). The authors did not suggest a figure for how many relatives would be identified for each proband, but they did note that two studies focused on large families and that after excluding these the range of ratios was 2.1–12 relatives identified per proband.
Data from Ian Frayling suggest that within 5 years of a proband receiving a positive diagnosis with a specific mutation, on average just over three relatives will have been tested for the family mutation (see Appendix 8). We assume that the rate of acceptance of testing in this instance will equal Pr(agrees to counselling) × Pr(agrees to testing|accepted counselling) = 0.45 × 0.96 = 0.436. Therefore, if 0.436 of the total number of relatives identified are tested, 0.436 × number of relatives identified = 3.04, i.e. the number of relatives identified = 6.98.
We assume a base-case value of five relatives being identified per proband. This was chosen to balance between the values of 2.35 estimated from Barrow and colleagues (2009)121 and 6.98 estimated from unpublished data supplied by Ian Frayling. Given a total of 1699 probands aged < 50 years per year in England, this implies 8495 relatives per year in England. There is uncertainty in both of these estimates, but both are from UK sources. We conducted a univariate sensitivity analysis on this parameter, varying it from 0 to 12, which includes the range of values suggested by Palomaki and colleagues (2009). 65
Number of first-degree relatives
When probands are diagnosed LS assumed, their relatives cannot be offered genetic testing. Instead, LS surveillance colonoscopies are offered to their FDRs. As such, it is important to know how many relatives this is likely to affect.
As described above (see Numbers of relatives) we identified a number of published studies reporting the number of relatives identified by strategies to identify LS. The significant level of heterogeneity led us to look for studies which were most relevant to the NHS.
Jenkins and colleagues114 studied the risk of CRC and other LS-associated cancers in people with LS. They recruited 131 men and women diagnosed with CRC under the age of 45 years in the Melbourne metropolitan area, Australia, performed mutation testing on 59 (all with MSI-H or MSI-L tumours, all with protein expression lacking on IHC and all from families meeting the AC, and a random sample of 23 with MSS tumours and full protein expression on IHC), and identified 18 as carriers of germline MMR gene mutations. 39 One proband was discovered to have a de novo mutation. Jenkins and colleagues sought FDRs and second-degree relatives (SDRs) aged > 18 years for all 131 initially recruited patients (Table 74).
| Relatives | Number of relatives identified | |
|---|---|---|
| Mutation carriers (n = 17)114 | All early-onset CRC (n = 131)185 | |
| FDRs | 79a | 672 |
| SDRs | 100a | 1333 |
| Total | 179a | 2005 |
Of the 79 FDRs of mutation carriers, 51 (65%) received predictive testing, whereas among the 100 SDRs of mutation carriers only 18 (18%) received predictive testing. However, as it seems the predictive tests were not done to inform the relatives but solely for research purposes, it is not clear whether or not the rate of predictive testing is informative for our analysis. We may, however, choose to use the result that 79 (44%) of 179 relatives identified (by the proband during interview) were FDRs.
Hampel and colleagues120 tested 249 relatives of 44 probands identified from an unselected series of CRC patients. Of the 249 relatives, 99 were first degree, which corresponds to 39.8% (95% CI 33.8% to 45.9%) of relatives tested being first degree.
Data from Ian Frayling show that 33 of 70 relatives tested (whose relationship to the proband was known) were first degree. This corresponds to 47.1% (95% CI 35.8% to 58.8%) of relatives tested being first degree.
We assume a base-case value of 42% of relatives being first degree as an approximate midpoint between the results from the studies by Jenkins and colleagues114 and Hampel and colleagues. 120 We conducted a univariate sensitivity analysis of this proportion, varying it between 35% and 55%. Figure 32 shows published and unpublished estimates of the proportion, our base-case value and sensitivity analysis range for comparison.
FIGURE 32.
Proportion of relatives who are first degree.
Given our base case of five relatives identified per proband, a proportion of 42% being FDRs leads to an estimate of 2.1 FDRs per proband.
Proportion of relatives testing positive
Although theoretically it would be expected that the probability that the FDR of a known carrier of a gene is also a carrier is 50% (unless the relative is an obligate carrier or non-carrier), in practice there are a number of reasons why the observed probability is lower:
-
de novo mutations can occur, which mean that no relatives of the index case will have the mutation [in the study by Jenkins and colleagues (2006),114 1 of 18 probands had a de novo mutation]
-
non-paternity can occur
-
mortality bias can occur, meaning that mutation carriers are more likely to have died before being able to receive predictive testing.
Almost all previous studies of cost-effectiveness which model diagnosis and management assume that 50% of FDRs of mutation carriers will also be carriers when tested (Table 75). Mvundura and colleagues54 are the only exception, assuming 45%.
| Study | Probability that a FDR of a LS mutation carrier is also a carrier |
|---|---|
| Ramsey et al. 200123 | 0.50 |
| Ramsey et al. 200349 | 0.50 |
| Kievit et al. 200550 | 0.50 |
| Breheny et al. 200651 | 0.50 |
| Olsen et al. 200753 | 0.50 |
| Mvundura et al. 201054 | 0.45 |
| VW Wang et al. 201258 | 0.50 |
| Ladabaum et al. 201148 and G Wang et al. 201257 | 0.50 |
Jenkins and colleagues114 tested blood samples from 179 relatives, which resulted in 79 mutation carriers (44.1%; 95% CI 37.0% to 51.5%). Hampel and colleagues120 tested 249 relatives, which resulted in 109 mutation carriers (43.8%; 95% CI 37.7% to 50.0%).
Unpublished data provided by Ian Frayling (see Appendix 8) showed that 44 of 109 relatives tested were mutation carriers (40.4%; 95% CI 31.5% to 49.7%). Unpublished data provided by Munaza Ahmed (Wessex Clinical Genetics Service, 2013) (see Appendix 9) showed that 145 of 319 relatives tested were mutation carriers (45.5%; 95% CI 40.1% to 50.9%).
A simple random-effects meta-analysis of the proportion of relatives testing positive was conducted. This gave a base-case estimate of 44.0% (Table 76). We conducted a sensitivity analysis varying the proportion from 40% to 48% (slightly larger than the 95% CI from the meta-analysis to include the proportion in data from Ian Frayling).
Lynch syndrome surveillance colonoscopy
Effect of colonoscopy on index colorectal cancer incidence rates
Colonoscopies (and accompanying polypectomies) reduce the incidence of CRC by removing premalignant lesions from the bowel. Colonoscopies also allow asymptomatic CRCs to be detected, which improves the Dukes’ stage distribution when CRC is diagnosed. Dukes’ stage at diagnosis affects survival (as described in Mortality due to colorectal cancer), meaning that colonoscopies will also reduce the mortality from CRC in the screened group. These factors are included in our model.
It is also documented by Jarvinen and colleagues (2009)107 that some patients will have colorectal surgery upon discovery of large adenomas (this has been confirmed by one of our clinical experts). This occurred in 7 out of 242 (2.9%) of participants undergoing surveillance. Additionally, three patients opted for prophylactic surgery because surveillance was too painful. Neither of these factors is included in our model.
The reduction in index CRC incidence due to polypectomies is estimated from results from Jarvinen and colleagues (2000),94 and is implemented as a HR applied when a person accepts colonoscopic surveillance. This study was chosen as it reported a HR for CRC incidence and was identified by Palomaki and colleagues (2009)65 as the key study on the effectiveness of colonoscopic surveillance.
The HR was estimated by extracting CRC incidence events from figure 1 (p. 831) of Jarvinen and colleagues (2000)94 and conducting a Cox proportional hazards regression (see Appendix 10). This gave an estimate of the HR of 0.387 (95% CI 0.169 to 0.885). As this is a very important parameter, sensitivity analyses at one standard error above (0.590) and below (0.254) the central estimate were conducted. A ‘back of the envelope’ calculation based on the 15-year CRC-free survival reported in figure 1 of the study by Jarvinen and colleagues (0.817 in screening group, 0.578 in control group) gives a HR of ln(0.817)/ln(0.578) = 0.369, which validates our base case of 0.387 from the Cox proportional hazards regression.
The interval between colonoscopies in the study was 5 years between 1982 and 1986 and 3 years from 1986 onwards (the results are based on follow-up to the end of 1998). This is less frequent than the 2 years which we model for intensive surveillance, and it is therefore likely that the estimated HR is an underestimate of the effectiveness of biennial colonoscopy. In our base-case analysis, we allow for this by adjusting the cost of colonoscopies (see Costs of Lynch syndrome surveillance colonoscopies). The authors also acknowledge that there may be selection bias as the study and control groups were self-selecting.
Effect of colonoscopy on metachronous colorectal cancer incidence
The effectiveness of biennial colonoscopy in reducing the incidence of metachronous CRC is not well established. Parry and colleagues (2011)168 showed no statistically significant difference in the surveillance of patients initially affected by colon cancer who were subsequently affected by metachronous CRC and those who were not affected, a finding also made by Win and colleagues (2013)186 in relation to patients initially affected by rectal cancer. Cirillo and colleagues (2012)187 found that biennial colonoscopy was not associated with a significant reduction in metachronous CRC in patients meeting the AC.
Most existing models of cost-effectiveness do not model metachronous CRC or do not report how it is modelled in sufficient detail for reproducibility (Table 77). The two studies in which the methodology for modelling the effectiveness in reducing metachronous CRC incidence is reported or was made available to us [Dinh and colleagues (2011)55 and Mvundura and colleagues (2010)54] differ completely in their implementation. Dinh and colleagues (2011)55 assume the same effectiveness in reducing metachronous CRC incidence as for index CRC, whereas Mvundura and colleagues (2010)54 assume no reduction in metachronous CRC incidence.
| Study | Effectiveness of LS surveillance in reducing metachronous CRC incidence |
|---|---|
| Vasen et al. 199873 | Metachronous CRC not modelled |
| Ramsey et al. 200123 | NR |
| Dunlop 200274 | Metachronous CRC not modelled |
| Ramsey et al. 200349 | NR |
| Kievit et al. 200550 | Metachronous CRC not modelled |
| Breheny et al. 200651 | Metachronous CRC not modelled |
| Olsen et al. 200753 | NR |
| Mvundura et al. 201054 | NR in paper; no effectiveness (Scott Grosse, Centers for Disease Control and Prevention, 2012, personal communication) |
| Dinh et al. 201155 | Equally effective as for index CRC |
| VW Wang et al. 201258 | Metachronous CRC not modelled |
| Ladabaum et al. 201148 and G Wang et al. 201257 | NR |
In the PenTAG model, we estimated effectiveness from Cirillo and colleagues,187 making a proportional hazards assumption and assuming that there was similar follow-up for the patients receiving ‘appropriate’ (up to 24 months between colonoscopies) and ‘inappropriate’ (> 24 months between colonoscopies) surveillance. Ten of 46 patients receiving appropriate surveillance and 7 of 19 patients receiving inappropriate surveillance developed metachronous CRC. 187 We accordingly estimated the HR as 0.533, as (12/19)0.533 = (36/46). Note that in our base case surveillance is effective at reducing metachronous CRC incidence, albeit less effective than at reducing index CRC incidence. We also performed a sensitivity analysis in which the HR was 1 (i.e. no reduction in metachronous CRC incidence due to LS surveillance).
Effect of colonoscopy on Dukes’ stage of incident colorectal cancers
We assumed that a Dukes’ stage distribution different to that observed in the general population would apply to CRC diagnoses in individuals undergoing LS surveillance colonoscopies, independent of LS status, age, sex and any other variable. Mecklin and colleagues188 reported that between 1982 and 2005 (during which time colonoscopic surveillance was 5-yearly between 1982 and 1989, 3-yearly between 1989 and 1994 and, from 1994 onwards, 3-yearly up to age 35 years then 2-yearly) 41 carcinomas were detected, with the distribution 29 × Dukes’ A, 4 × Dukes’ B, 5 × Dukes’ C and 3 × Dukes’ D. We chose this as an appropriate study as it has surveillance intervals matching those in Jarvinen and colleagues (2000),94 from which the effect of colonoscopy on index CRC incidence is drawn, and there were no CRC cases of unknown Dukes’ stage. The Dukes’ stage distribution in our model is the expected proportion from the Dirichlet posterior distribution where a Jeffreys prior is used, and is shown in Table 78 and compared with the distribution when patients do not receive surveillance in Figure 33.
| Dukes’ stage | Number of carcinomas (simple proportion) | Dirichlet posterior hyperparameters (prior distribution of Jeffreys prior) | Expected proportion used in model |
|---|---|---|---|
| A | 29 (0.707) | 29.5 | 0.686 |
| B | 4 (0.098) | 4.5 | 0.105 |
| C | 5 (0.122) | 5.5 | 0.128 |
| D | 3 (0.073) | 3.5 | 0.081 |
FIGURE 33.
Stage distribution of CRCs for people with and without LS surveillance.
Adherence to Lynch syndrome surveillance colonoscopy
The initial acceptance of LS surveillance is detailed above (see Acceptance of testing and surveillance). In the model it is assumed that patients who initially accept LS surveillance continue to receive biennial colonoscopies as recommended. It is also assumed that patients who initially decline LS surveillance do not subsequently take up surveillance.
Effectiveness of surveillance colonoscopy in preventing index CRC in the model is based on the study by Jarvinen and colleagues (2000),94 in which long-term adherence was reported as 93% (although 3% deemed not to adhere long term had negative gene test results, so 95% is an alternative estimate of adherence). Additionally, in this study 63% of patients who initially declined surveillance colonoscopy were participating in surveillance by the end of the study. The effectiveness of surveillance colonoscopy in this study is based on an intention-to-treat analysis, which means it may be biased to appear less effective than it really is (as there was greater crossover into the surveillance programme than out of it).
There are issues with applying the results of Jarvinen and colleagues directly to the UK setting. It is proposed that surveillance would consist of 2-yearly colonoscopy, rather than the 3-yearly colonoscopy described by Jarvinen and colleagues (note that for consistency we adjust the cost of colonoscopy in our model to reflect this; see Costs of Lynch syndrome surveillance colonoscopies). Our clinical expert advice suggests that decreasing the interval between colonoscopies can lead to better or worse adherence due to the competing factors of putting patients off (as colonoscopies are uncomfortable procedures) and giving the impression that intensive surveillance is needed (if surveillance is infrequent patients may assume it is not important). It may also be the case that cultural differences mean that Finnish patients are more likely to accept and adhere to surveillance than UK patients.
As we did not find data relating to long-term adherence of UK patients to biennial colonoscopy, we judged that assuming no crossover was reasonable and as consistent as possible with the effectiveness estimates from Jarvinen and colleagues. 94 It may be that adherence falls off for patients, but this may be balanced by delayed uptake of surveillance by patients initially declining surveillance.
Morbidity and mortality of Lynch syndrome surveillance colonoscopy
In our model we consider morbidity from colonoscopy in the form of bleeding requiring admission (mild, moderate and severe) and perforation. These events incur treatment costs but are assumed not to impact on QALYs as they are acute events from which a full recovery should be possible. Mortality from colonoscopy is also included, with an associated cost. Table 79 gives the probabilities of these events in the model.
| Event | Probability (per colonoscopy) |
|---|---|
| Mortality | 0.83 per 10,000 |
| Perforation | 4 per 10,000 |
| Bleeding | 26 per 10,000 |
| Of which required admission | 21% |
| Of which were mild | 73% |
| Of which were moderate | 18% |
| Of which were severe | 9% |
Our estimates of the proportion of colonoscopies resulting in morbidity come from the UK National Colonoscopy Audit of 2011 as reported by Gavin and colleagues. 189 In this audit, 0.26% (95% CI 0.20% to 0.36%) of colonoscopies led to bleeding, with 21% (95% CI 12.2% to 34.0%) of these requiring admission. Of those requiring admission, 18% (95% CI 5.1% to 47.7%) suffered moderate bleeding and 9% (95% CI 1.6% to 37.7%) suffered severe bleeding. These estimates are from very small populations, which accounts for the large CIs.
Perforations occurred in 0.04% (95% CI 0.02% to 0.08%) of colonoscopies in the study by Gavin and colleagues,189 with all of them requiring admission.
Cairns and colleagues9 report that the mortality rate of colonoscopy is 0.83 per 10,000 procedures. Colonoscopy mortality is assumed to occur at the beginning of the cycle.
Effectiveness of colorectal cancer surgery
Effectiveness of CRC surgery is incorporated by applying a HR to the CRC incidence rate according to the surgery type. Segmental resection was assumed to have no effect on the incidence of CRC (HR of 1). Proctocolectomy was assumed to completely eliminate the risk of CRC (HR of 0). The HR for subtotal colectomy on CRC incidence was estimated as 0.06 as that is the result of subtracting the relevant proportion of incident CRCs in patients with LS (see Colorectal cancer incident site) from 1 (1 – 0.94 = 0.06). Similarly, the HR for anterior resection was estimated as 0.94.
The effectiveness was assumed to be independent of whether the person has LS or not. If additional complexity were introduced to include a dependency on LS status, then subtotal colectomy would be modelled as less effective for persons without LS than for those with LS and anterior resection would be modelled as more effective. As very few people without LS develop CRC in the model, we judged that this was unlikely to have an impact on cost-effectiveness.
Morbidity and mortality due to colorectal cancer surgery
We assumed that mortality of CRC surgery was already included in the 1-year survival of CRC and hence did not model it separately. We assumed that any long-term morbidity of CRC surgery would be included in our estimates for HRQoL for CRC and did not include any acute morbidity of CRC surgery.
More aggressive colorectal cancer surgery for individuals diagnosed with Lynch syndrome
As previously described (see Colorectal cancer surgical management pathways), we include a parameter representing the probability that a CRC patient receives more aggressive surgery than they otherwise would because they have LS. When this parameter is set to 0 there is no difference between the surgery choice for LS patients and that for general population patients.
There is a difference of opinion between surgical guidelines, for example BSG/ACPGBI guidelines9 (which appear to recommend aggressive surgery for patients with known LS mutations), and our clinical expert opinion, which is that the surgery would not be affected by the presence of a LS mutation.
For our base case we set the parameter to 0, for the following reasons:
-
The BSG/ACPGBI recommendations are not based on high-quality evidence.
-
In our base case we do not assume any disutility from different surgery types (see Impact of colorectal cancer surgeryon quality of life), even though this is a major consideration when deciding which surgery to perform. If we set the parameter to anything other than 0 we would introduce a bias, as the reduction in metachronous CRC incidence (see Effectiveness of colorectal cancer surgery) would not be matched by a disutility for more aggressive surgery.
-
We do not include different surgical mortality for different surgery types (see Morbidity and mortality due to colorectal cancer surgery), even though 30-day surgical mortality following total/subtotal colectomy is significantly greater than mortality following right hemicolectomy (a typical segmental resection procedure) [odds ratio 1.52 (95% CI 1.15 to 2.01)]. 135 If we set the parameter to anything other than 0 we would introduce a bias.
We vary the parameter in the sensitivity analyses.
Screening for endometrial cancer
Of the seven analyses that include EC, six assume screening for EC (see Table 89). It is not clear whether or not the seventh analysis, by Kwon (2011),56 models screening. However, screening is assumed to yield clinical benefits in only three of the six analyses: Chen and colleagues (2007),85 Kwon and colleagues (2008)75 and Yang and colleagues (2011). 76 No benefit is assumed in the remaining analyses: Dinh and colleagues (2011),55 Ladabaum and colleagues (2011)48 and Wang and colleagues (2012). 57
Gynaecological cancer surveillance in the Peninsula Technology Assessment Group model
We assume no costs or benefits related to surveillance for EC in our model. This is because we found insufficient evidence to suggest that gynaecological cancer surveillance reduces the incidence of gynaecological cancers or improves survival when individuals are affected (see Literature review of the effectiveness of surveillance for endometrial cancer), and because it seems unreasonable to include the substantial cost of surveillance with no benefit. In this sense, our approach differs from the three US analyses that assumed costs, but no clinical benefit of surveillance. 48,55,57
However, we note that our clinical advisors suggest that some surveillance occurs in clinical practice and the Northern Genetic Service audit of LS carriers suggests that 28 out of 69 (41%) of women without previous cancer and with intervention recorded were referred to or offered surveillance (Lorraine Cowley, personal communication).
Literature review of the effectiveness of surveillance for endometrial cancer
We searched the literature for evidence of the effectiveness of gynaecological surveillance for patients with LS (see Appendix 11). We identified three primary studies107,190,191 and four reviews65,167,192,193 which were critically appraised in relation to the UK context and the decision problem.
None of the three primary studies was a randomised controlled trial (RCT). Two of the studies107,190 were not intended to have a control group, but a selection of patients did not undergo the recommended surveillance and we considered them a ‘control group’ in each case. Renkonen-Sinisalo and colleagues191 describe two cohorts, one comprising 83 women identified retrospectively with LS who had a previous EC, and the other 175 women with LS in a surveillance programme.
As detailed in Appendix 11, we performed statistical analyses on the three primary studies to determine the relative risk of gynaecological cancer and prophylactic hysterectomy for those receiving gynaecological surveillance, and we did not find statistically significant results. We also performed statistical analysis to determine the effect of surveillance on the surgical stage of gynaecological cancers and again did not find statistically significant results.
Existing reviews
Auranen and Joutsiniemi (2011)192 conducted a systematic review of gynaecological cancer screening in women belonging to LS families, which had five included studies. 190,191,194–196 In our review, the study by Lécuru and colleagues (2008)195 was excluded because the abstract suggested that there was no comparator for the surveillance strategy. Auranen and Joutsiniemi (2011) conclude that the studies they included do not allow for evidence-based decision-making.
Koornstra and colleagues (2009)193 conducted a review of extracolonic cancers, including gynaecological cancer screening, and included three studies relevant to gynaecological cancer screening. 190,191,196 They conclude that the only evidence of surveillance benefit is that TVU and endometrial sampling detect endometrial tumours in early stages based on the study by Renkonen-Sinisalo and colleagues (2007),191 despite this study not reporting a statistical difference by the Pearson chi-squared method.
Lindor and colleagues167 included two studies190,196 and concluded that there was insufficient evidence to argue for or against endometrial sampling or TVU.
Palomaki and colleagues (2009)65 included three studies190,191,196 and concluded that TVU is not highly effective at identifying ECs in women with LS, but that endometrial biopsy is effective at identifying both premalignant and malignant lesions, according to the study by Renkonen-Sinisalo and colleagues (2007). 191 Palomaki and colleagues recommend surveillance for women with LS but say that ‘Inadequate data are available to document that transvaginal ultrasound and endometrial biopsy can reduce the incidence of endometrial cancer.’65
Surgery for endometrial cancer
We assume that TAHBSO completely eliminates the risk of EC, for the following reason. In a retrospective study of prophylactic surgery to reduce the risk of gynaecological cancer,146 0 out of 61 women with LS who underwent either prophylactic hysterectomy or TAHBSO developed EC, whereas 69 out of 210, i.e. 33% of women who did not undergo either surgery developed EC. Each woman who had undergone hysterectomy or TAHBSO was matched (by age, treated at the same institution) with one or more control women. The median age at surgery was 41 years, and at diagnosis of EC, 46 years. All women were followed until the occurrence of gynaecological cancer, death or censorship. Although a randomised trial would be preferable to a retrospective study, the study appears to give compelling evidence that prophylactic surgery eliminates the risk of EC.
Prophylactic total abdominal hysterectomy with bilateral salpingo-oophorectomy for women diagnosed with Lynch syndrome
Acceptance of prophylactic total abdominal hysterectomy with bilateral salpingo-oophorectomy
The acceptance rate for TAHBSO for both probands and relatives is assumed to be 55%, i.e. 21 out of 38 women over 45 years of age without previous cancer, based on data provided by Lorraine Cowley (personal communication) from the Northern Genetics Service audit of LS carriers in the UK (see Table 56).
By comparison, Dinh and colleagues55 assumed similar rates at the appropriate age: for relatives with LS without CRC, acceptance was assumed to be 0% at age 30 years, 40% at age 40 years, 60% at age 50 years and 75% at age 80 years. Slightly higher rates were assumed for probands with CRC. By contrast, Ladabaum and colleagues48 assumed substantially lower acceptance rates: 19% and 18% for probands and relatives, respectively, citing a US study. 146 Given that our estimate was taken from a small sample size of 38 women, that there is substantial variability in this parameter in previous cost-effectiveness analyses, and that this is an important parameter, we assumed wide variation (20% and 90%) in acceptance rates for both probands and relatives in sensitivity analyses.
Utility parameters and assumptions
In this section, we follow the principles for the identification, review and synthesis of health state utility values from the literature, as recommended recently by the NICE Decision Support Unit in the UK. 197 There are no agreed reporting standards for studies of utilities, but the following information is key to understanding the nature and the quantity and quality of evidence:197
-
the population describing the health state (e.g. age, sex, disease severity)
-
the approach used to describe the health state
-
utility value elicitation technique, for example time trade-off, standard gamble, visual analogue score
-
sample size
-
respondent selection and recruitment, inclusion and exclusion criteria
-
survey response rates, numbers lost to follow-up (and reasons), methods of handling missing data.
Clearly, the relevance of the data to the decision model, and to the agency to which the model will be submitted, is important. In the current project, the NICE reference case is used. 198 Modification of utility values from the literature for use in economic models, and sensitivity analyses using less relevant utility values, should be considered. 197
A systematic search for studies reporting utilities should be undertaken. 197 For the current project, the search method is given in Appendix 4. In addition, sources of utility values were obtained from published models on the cost-effectiveness of testing for LS.
If disutilities are included in the model for CRC, then strategies which prevent the greatest numbers of CRCs will experience a relative improvement in cost-effectiveness (likewise for EC). In addition, if greater disutilities are included for later CRC stages then strategies with the most surveillance will experience a relative improvement in cost-effectiveness, as surveillance reduces the proportion of CRCs diagnosed in later stages. If disutilities are included for people undergoing colonoscopy surveillance (e.g. as this can be painful and uncomfortable), this would result in a worsening of cost-effectiveness for strategies with more surveillance. Therefore, there could be a balance between disutility of CRC and surveillance, in which strategies with the fewest diagnostic errors would fare better. Similar considerations apply to risk-reducing surgery. If disutilities are included for the psychological impact of LS germline testing (and separately according to the results), this could worsen the cost-effectiveness of strategies involving extensive LS testing, particularly strategy 8, in which all CRC patients are offered LS testing.
Utilities in cost-effectiveness models particularly for testing for Lynch syndrome
In Table 80, we outline approaches taken to incorporate utilities in previous models of the cost-effectiveness of testing for LS. Only a few analyses – Kwon and colleagues (2008),75 Wang and colleagues (2012),57 Dinh and colleagues (2011),55 Mvundura and colleagues (2010)54 and Yang and colleagues (2011)76 – considered utilities.
| Cost-effectiveness analysis | Utilities included? | Utilities age specific? | Psychological effects of genetic testing | CRC stage | CRC surgery | Gynaecological cancers | Gynaecological surgery |
|---|---|---|---|---|---|---|---|
| Breheny et al. 200651 | No | – | – | – | – | – | – |
| Brown et al. 199622 | No | – | – | – | – | – | – |
| DACEHTA 200752 | No | – | – | – | – | – | – |
| Dinh et al. 201155 | Yes | No | No, as ‘Decrease in quality of life due to detection of MMR mutation in unaffected carriers was found to be transient’199 | 0.74 stage I, 0.67 stage II, 0.50 stage III, 0.25 stage IV (Ness et al. 1999200) | 0.84 for total colectomy with IRA or proctocolectomy and IPAA for individuals with advanced adenoma,201 0.83 proctocolectomy202 | 0.83 for EC (Kwon et al. 200875) | 0.86 for TAHBSO (Kwon et al. 200875) |
| Kievit et al. 200550 | No | – | – | – | – | – | – |
| Kwon et al. 200875 | Yes | No | No | No | No | 0.83 EC, citing five references,203–207 0.75 OC, citing three references208–210 | 0.86 prophylactic surgery208,211 |
| Kwon et al. 201156 | No | – | – | – | – | – | – |
| Ladabaum et al. 201148 | No | – | – | – | – | – | – |
| Mvundura et al. 201054 | In sensitivity analysis, not in base case | Yes | No | Disutility of 0.10 for 2 years for ‘survivors of non-metastatic CRC’, citing two references21,44 | No | No | No |
| Olsen et al. 200753 | No | – | – | – | – | – | – |
| Ramsey et al. 200123 | No | – | – | – | – | – | – |
| Ramsey et al. 200349 | No | – | – | – | – | – | – |
| Reyes et al. 200259 | No | – | – | – | – | – | – |
| Wang et al. 201257 | Yes | Yes | Assume disutilities related to testing for LS last 1 year (from Kuppermann et al.212) | Utility 0.601 for CRC (Kuppermann et al.212), applied for 5 years only | No | Utility 0.728 for EC, 0.593 for OC,212 applied for 5 years only | No |
| Yang et al. 201176 | Yes | No | No | No | No | 0.83 for EC, 0.75 for OC, 0.59 for metastatic (stage IV) cancer | No |
Owing to limitations of empirical data identified in an earlier review,213 the authors chose to use European Quality of Life-5 Dimensions (EQ-5D) values from the Health Survey for England, using the absolute utility decrement for individuals with cancer versus individuals of a similar age without cancer. 214,215 It should be noted that the cancer population subgroup included individuals with benign lumps and cysts and was not specifically limited to CRC. 215 Their estimates were taken simply as the utilities for age-matched individuals in England without cancer (0.80) and individuals with cancer in general (0.70). Although we appreciate the effort to find some method to allow for disutility due to CRC, we believe that it is unreasonable to use a figure based on all cancers.
Baseline quality of life
Baseline quality of life (assumed to apply to patients without CRC, EC or other events which might have an impact on HRQoL) was modelled as a function of age and sex, using the model for calculating EQ-5D utility values described by Ara and Brazier:216
We used the age at the beginning of each cycle to determine the baseline utility for that year.
Impact of colorectal cancer on quality of life
Impact of colorectal cancer on quality of life according to Dukes’ stage
A systematic review was recently undertaken for the NIHR to identify utility values for CRC by a group mostly based at the University of Sheffield, UK. 213 This review identified six relevant studies (Table 81). 200,217,219–222 Cooper and colleagues (2010)213 concluded that these studies do not demonstrate a clear relationship between HRQoL and stage of cancer, treatment, phase of disease or time since diagnosis. They further concluded that, at the time of their review, only two studies, Ness and colleagues (1999)200 and Ramsey and colleagues (2000),217 had attempted to estimate utilities for patients according to stage of CRC. They noted that Ness and colleagues (1999)200 estimated that utility decreased substantially with more advanced cancer, from 0.74 at stage I to 0.24 at stage IV (see Table 81). They noted that this contrasted with the results from Ramsey and colleagues (2000),217 which estimated that utility is approximately independent of CRC stage (utility = 0.84 at stage I and stage IV), and also contrasted with the results from the MABEL study,222 which estimated a utility of 0.73 for individuals with metastatic CRC who had failed one prior line of chemotherapy. Cooper and colleagues (2010)213 based their estimates of utilities by stage of CRC on those from Ness and colleagues (1999). 200 Their reasoning was as follows:213
the study by Ness and colleagues involved eliciting preferences for hypothetical health states from individuals who had previously undergone polypectomy, while the other studies involved eliciting preferences from patients currently experiencing the health state. As NICE recommend that utilities should be based upon public preferences, the study by Ness was used to estimate utilities associated with CRC.
Cooper and colleagues (2010)213 adjusted the utilities from Ness and colleagues (1999)200 to allow for the age-related utilities of the general population of England. They estimated the ratio of the utility of someone with CRC to the utility of a member of the general population, independent of age, as follows: 0.88 Dukes’ A, 0.70 Dukes’ B, 0.70 Dukes’ C, 0.30 Dukes’ D. For example, their utility estimates for someone with CRC aged 60 years, given that the utility of a member of the general population of England at this age is approximately 0.83,216 are: 0.73 Dukes’ A, 0.58 Dukes’ B, 0.58 Dukes’ C, 0.25 Dukes’ D.
| Study | Study population | Preference elicitation | Results | Criticisms of study |
|---|---|---|---|---|
| aNess et al. 1999200 | 81 participants from the USA who had previously undergone removal of colorectal adenoma | Seven health states describing various stages of colon and rectal cancer. Preferences elicited using standard gamble | Stage I rectal or stage I/II colon cancer (mean 0.74); stage III colon cancer (mean 0.67); stage II/III rectal cancer without ostomy (mean 0.59); stage II/III rectal cancer with ostomy (mean 0.50); stage IV rectal or colon cancer (mean 0.25) | Although health states rated by individuals who had previously undergone removal of colorectal adenoma, health states described were not experienced by these individuals A standard quality of life questionnaire, such as the EQ-5D or SF-36, was not used Standard gamble is not preferred Valuation performed by 81 participants, not by large sample of general public Sample size of 81 people is very small |
| aRamsey et al. 2000217 | 173 long-term survivors of CRC, mean age 70 years, sampled from US SEER database | HUI3 questionnaire | Stage I = 0.84, stage II = 0.86, stage III = 0.85, stage IV = 0.84 | Although a recognised quality of life questionnaire, the HUI3, was used, the EQ-5D is preferred Small sample size Possible response bias in favour of healthier subjects |
| Ramsey et al. 2002218 | 227 individuals at least 5 years from diagnosis of CRC, sampled from US SEER database, average age 74 years | HUI3 questionnaire | Stage I = 0.83, stage II = 0.86, stage III = 0.87, stage IV = 0.81 | Although a recognised quality of life questionnaire, the HUI3, was used, the EQ-5D is preferred Possible response bias in favour of healthier subjects Biased sample of individuals who survived at least 5 years from diagnosis |
| aKo et al. 2003219 | 169 patients from USA with colon cancer | The Health and Activities Limitation Index was mapped on to a utility scale | Mean utility shortly after diagnosis 0.67 (n = 32), not divided by disease stage, mean utility after 5 years 0.71 (n = 80) | CRC stage not measured The Health and Activities Limitation Index is not preferred by NICE198 Small sample sizes |
| aPetrou and Campbell 1997220 | 30 nurses experienced in oncology careb | Utilities for six chemotherapy-specific scenarios elicited using standard gambleb | CRC stage not measured Health states not experienced by population Extremely small sample sizes of patients Standard gamble is not preferred |
|
| aMRC FOCUS trial, Seymour et al. 2007221 | 2135 patients with inoperable metastatic or locoregional disease | EORTC QLQ-C30 | Utilities not obtained | Utilities not derived, as quality of life assessed by EORTC QLQ-C30 cancer-specific questionnaire |
| aMABEL trial, Wilke et al. 2008222 | Utilities for 125 patients taking cetuximab (Erbitux®, Merck Serono) + irinotecan with metastatic CRC | EQ-5D questionnaire | Mean utility = 0.75223 | Utilities for cetuximab treatment not relevant, as cetuximab not recommended by NICE in UK |
| Jonker et al. 2007224 | 260 patients receiving BSC in RCT of cetuximab vs. BSC for metastatic CRC | HUI3 questionnaire, valued by Canadian general population | For BSC arm, mean utility = 0.71 (n = 260) at baseline, 0.68 (n = 184) at week 4, 0.66 (n = 149) at week 8, 0.63 (n = 72) at week 16, 0.70 (n = 36) at week 24225 | Although a recognised quality of life questionnaire, the HUI3, was used, the EQ-5D is preferred |
| Best et al. 2010226 | Convenience samples of 49 CRC patients | Seven health states described, reflecting stage III CRC EQ-5D questionnaire |
Adjusted utility values ranged from remission 0.83 to metastatic progressive 0.37 | Although individuals who rated health states had CRC, health states described were not experienced by these people Small sample size |
We find five serious criticisms with the study by Ness and colleagues (1999). 200 In all cases, the methods used are not preferred by NICE. 198 First, although health states were rated by individuals who had previously undergone removal of colorectal adenoma, the health states described were not experienced by these individuals. Second, a standard quality of life questionnaire, such as the EQ-5D or SF-36 (Short Form questionnaire-36 items), was not used. Third, the standard gamble technique was used to value health states, rather than the preferred time trade-off method. Fourth, valuation was performed by the participants, not by a large sample of the general public. Fifth, the sample size of 81 participants is very small.
In a study of 173 long-term survivors of CRC, sampled from US SEER database, Ramsey and colleagues (2000)217 found almost no difference in mean utilities by disease stage at diagnosis (see Table 81). This is in contrast to the findings of Ness and colleagues (1999). 200 For any given disease stage at diagnosis, Ramsey and colleagues (2000)217 found much variability in utilities in the first 4 or so years after diagnosis, and much less variability thereafter. The mean utility increased over time for some stages at diagnosis, and decreased for others. Individuals who were in the terminal phase of their illness had much lower quality of life.
The findings of a study of long-term survivors of CRC by Ramsey and colleagues (2002)218 agree with those of Ramsey and colleagues (2000). 217 Two hundred and twenty-seven individuals, mostly diagnosed with stages I, II and III CRC, who were at least 5 years from diagnosis were sampled from the US SEER database. As in the study by Ramsey and colleagues (2000),217 mean utilities were high and similar across disease stage. However, we consider the study by Ramsey and colleagues (2000)217 in preference to that of Ramsey and colleagues (2002),218 because in the latter study, patients were a biased sample of long-term survivors.
Although we find criticisms with the study by Ramsey and colleagues (2000),217 unlike Cooper and colleagues (2010),213 we believe that this is the most reliable and useful of all studies for our purposes because (a) it is one of only two studies that estimated utilities as a function of CRC stage [the other being Ness and colleagues (1999)200]; (b) unlike Ness and colleagues (1999),200 a standard quality of life questionnaire, the Health Utilities Index Mark 3 (HUI3), was used, as required by NICE;198 (c) unlike Ness and colleagues (1999),200 the individuals scoring the health states were those actually experiencing the health state, as required by NICE; and (d) unlike Ness and colleagues (1999),200 valuation was performed by a large sample of the general public, not by those scoring the health states, as required by NICE. Criticisms of the study by Ramsey and colleagues (2000)217 are as follows. First, the sample size of 173 patients, though twice that of Ness and colleagues (1999),200 is still small, especially to detect differences in utilities between disease stages. Second, Ramsey and colleagues (2000)217 admit that there may have been response bias in favour of healthier subjects, as less than half of all individuals contacted completed the questionnaire. Third, although the HUI3 is a recognised preference-based generic quality of life questionnaire, the EQ-5D is preferred by NICE. 198
Like Cooper and colleagues (2010),213 we consider the study by Ko and colleagues (2003)219 to be of limited relevance, mostly because CRC stage was not reported, but also because the Health and Activities Limitation Index used in the study is not preferred by NICE,198 and because of the small sample size (see Table 81).
We are unable to source the full text of the study by Petrou and Campbell (1997). 220 However, Cooper and colleagues (2010)213 say they did not consider the results of this study for their purposes because the study does not provide utilities by disease stage. Further, Cooper and colleagues (2010)213 state that the study population consisted of an extremely small sample of 30 nurses experienced in oncology care. In addition, the health states were not experienced by the population. For all these reasons, we do not consider this study further.
We do not consider in any detail the results of the Medical Research Council FOCUS [Fluorouracil, Oxaliplatin and CPT11 (irinotecan) – Use and Sequencing] trial221 for patients with inoperable metastatic or locoregional CRC because utilities were not estimated, although HRQoL was measured using the European Organization for Research and Treatment of Cancer Quality of Life Questionnaire-C30 (EORTC QLQ-C30) cancer-specific questionnaire.
Utilities were assessed by the HUI3 questionnaire estimated in a RCT of cetuximab versus best supportive care for patients with advanced CRC who had typically had three or four previous lines of drug treatment. 224 Detailed utilities by time from randomisation are given in Table 81, with an average value, weighted by the number of observations, of 0.68. We attach importance to this value because it is derived from a large sample of patients using a recognised HRQoL questionnaire.
As noted by Cooper and colleagues (2010),213 the results from the Monoclonal Antibody Erbitux in a European License (MABEL) study by Wilke and colleagues (2008)222 of patients taking cetuximab plus irinotecan with metastatic CRC are more consistent with those of Ramsey and colleagues (2000)217 than Ness and colleagues (1999). 200 The mean utility for 125 of these patients was estimated as 0.75. 223 Unfortunately, although this study used the EQ-5D questionnaire, which is preferred, the relevance of the study is limited by the fact that patients took cetuximab plus irinotecan, which is not recommended by NICE.
Best and colleagues (2010)226 elicited utilities from a convenience sample of 49 CRC patients in the USA. Patients completed the EQ-5D questionnaire for seven different health state descriptions consistent with stage III CRC. Estimated utilities varied greatly, ranging from 0.83 for remission to 0.48–0.67 for individuals taking adjuvant chemotherapy (according to the degree of neuropathy), to 0.37 for metastatic progressive disease. Although it is interesting to note the great variability in utilities, we do not consider the results further because of the very small sample size and because the health states described were not experienced by the participants (although they had CRC).
Peninsula Technology Assessment Group estimated utilities by stage of colorectal cancer
A highly detailed model for individuals with CRC would allow for increased HRQoL during remission, when patients can lead a reasonably normal life, and decreased quality of life while taking chemotherapy, when individuals may experience a range of problems such as fatigue, anxiety and reduced social activity. 226 However, given the lack of high-quality relevant data, we simply apply a disutility from the normal utility of the general population, unique for each Dukes’ stage (Table 82).
| CRC stage | Base case | Sensitivity analysis | ||||
|---|---|---|---|---|---|---|
| Disutility | Based on study | Utility for 60-year-old male | Utility for 60-year-old female | Disutility | Based on study | |
| Dukes’ A | 0.00 | Ramsey et al. 2000217 | 0.84 | 0.82 | 0.11 | Ness et al. 1999200 |
| Dukes’ B | 0.00 | Ramsey et al. 2000 | 0.84 | 0.82 | 0.23 | Ness et al. 1999 |
| Dukes’ C | 0.00 | Ramsey et al. 2000 | 0.84 | 0.82 | 0.26 | Ness et al. 1999 |
| Dukes’ D | 0.13 | Mittmann et al. 2009225 | 0.71 | 0.69 | 0.60 | Ness et al. 1999 |
We base our estimated disutility for Dukes’ stages A, B and C on the study by Ramsey and colleagues (2000)217 because, as we note above, we judge this study to be the most relevant. In this study, the mean age of respondents was 71 years. The mean utilities for members of the general population of England and Wales at this age are approximately 0.79 and 0.77 for men and women, respectively. 216 Ramsey and colleagues estimated utilities slightly in excess of these values for individuals with CRC of all stages (see Table 81). Therefore, in our base-case analysis, we estimate no disutility for individuals with stages A, B and C CRC.
For Dukes’ stage D, we estimate a disutility of 0.13 as follows. In the RCT of cetuximab versus BSC,224 the mean utility for patients in the BSC arm was 0.68,225 and the median age of patients in this arm at baseline was 64 years. Given that the utility for males (constituting 64% of patients in the trial) in the general population at this age is 0.82216 and that for females is 0.80, this implies a disutility of 0.81 – 0.68 = 0.13.
We also perform a sensitivity analysis, in which we base estimates of utilities on the study by Ness and colleagues (1999),200 because this is the only other study which reports utilities for all Dukes’ stages and because it has been used in the cost-effectiveness analyses by Dinh and colleagues (2011)55 and Cooper and colleagues (2010). 213 The mean age of participants in Ness and colleagues (1999)200 was 54 years, with approximately 50% male and 50% female. The utility for a general member of the population of England and Wales at this age is 0.85. 216 We estimate the disutility for Dukes’ A as 0.85 – 0.74 (stage I rectal or stage I/II colon cancer) = 0.11; the disutility for Dukes’ B as 0.85 – 0.62 = 0.23, where 0.62 is the average of 0.74 for stage I rectal or stage I/II colon cancer and 0.50 for stage II/III rectal cancer with ostomy; the disutility for Dukes’ C as 0.85 – 0.59 = 0.26, where 0.59 is the average of 0.67 for stage III colon cancer and 0.50 for stage II/III rectal cancer with ostomy; and the disutility for Dukes’ D as 0.85 – 0.25 (stage IV rectal or colon cancer) = 0.60.
Impact of colorectal cancer surgery on quality of life
In the base-case analysis, no additional disutility for surgery that reduces the risk of CRC was assumed. Our base-case disutilities for Dukes’ stages A, B and C are taken from Ramsey and colleagues (2000)217 (see Table 82), a study of 173 long-term survivors of CRC, sampled from the US SEER database. Although not explicitly stated, we assume that a substantial proportion of the patients had undergone surgery for CRC. Next, our base-case disutility for Dukes’ stage D is taken from a RCT of cetuximab versus best supportive care for patients with metastatic CRC. 224 Although the nature of previous surgery in patients included in this trial is not stated,224 it is likely that a large proportion had undergone surgery for CRC given that patients were heavily pre-treated. Finally, in a sensitivity analysis, we estimate disutilities for all Dukes’ stages from Ness and colleagues (1999). 200 In this study, descriptions of health states for various stages of colon and rectal cancer included resection and use of ostomy (see Table 82). Given this, it could be argued that any disutilities related to surgery for CRC are already incorporated in our chosen disutilities according to Dukes’ stage. Nonetheless, we now present an overview of the relevant literature on the HRQoL related to surgery for CRC, summarised in Table 83.
| Study | Study population | Preference elicitation | Results | Criticisms of study |
|---|---|---|---|---|
| van Duijvendijk et al. 2000201 | 183 patients with FAP in the Netherlands with colectomy and IRA, 140 patients with FAP with proctocolectomy and IPAA, 279 individuals from general Dutch population. Mean age 40 years | SF-36 questionnaire | HRQoL similar for individuals after two types of surgery, but significantly poorer than for individuals in general population | No utilities quoted |
| Fazio et al. 1999202 | Patients with proctocolectomy with stapled anastomosis mostly for colitis. Median age 37 years at surgery | Cleveland Global Quality of Life instrument. Also SF-36 questionnaire in subgroup of 163 patients | Postoperative quality of life as measured by the SF-36 was excellent and compared well with norms for the general US population. Quality of life increased in the 2 years after surgery, and did not deteriorate thereafter | Although a recognised quality of life questionnaire, the SF-36, was used, the EQ-5D is preferred |
| Kuppermann et al. 2013212 | 71 highly educated individuals based in USA ‘with a range of understanding about and experience with Lynch Syndrome’ | Health state vignettes | Mean utility 0.68 for probands tested positive for LS who undergo colectomy, 0.65 for probands tested positive who decline colectomy, 0.66 for probands who decline testing (see Table 84) | Health states not experienced by population Health state questionnaires, especially EQ-5D, are preferred to health state vignettes |
| Kalady et al. 2011227 | 95 patients, aged approximately 55 years, meeting AC for LS 50 patients total proctocolectomy and 45 partial colectomy |
SF-36 questionnaire | 1 year after surgery, no significant difference in any of the eight domains of the SF-36 between two types of surgery | Although a recognised quality of life questionnaire, the SF-36, was used, the EQ-5D is preferred Study published in abstract form only No utilities quoted |
| Syngal et al. 199883 | Seven gastroenterologists and three genetic counsellors | Standard gamble | Utilities of 0.95 for patients with subtotal colectomy, 0.89 for proctocolectomy | Very small sample size Patients not scoring questionnaire Time trade-off preferred to standard gamble |
The only study on the cost-effectiveness of genetic testing for LS that has incorporated disutilities after surgery for CRC is that of Dinh and colleagues (2011). 55 First, a utility of 0.84 was assumed for total colectomy with ileorectal anastomosis (IRA) or proctocolectomy and IPAA for individuals with advanced adenoma, which Dinh and colleagues (2011) cite from van Duijvendijk and colleagues (2000). 201 This study compared the HRQoL, as measured by the SF-36 questionnaire, of 183 patients with FAP in the Netherlands who underwent colectomy and IRA with 140 patients, also with FAP, who underwent proctocolectomy and IPAA, and with 279 people from the general Dutch population (see Table 83). HRQoL was found to be similar for participants after the two types of surgery, but significantly poorer than for people in the general population. However, we find no utilities reported in the study by van Duijvendijk and colleagues (2000);201 in particular, the value of 0.84 used by Dinh and colleagues (2011)55 is not cited. If we assume that none of the patients with FAP in the study had CRC, the study suggests a disutility from having either of the operations; however, this is not quantified. The study by van Duijvendijk and colleagues does report mean scores and standard deviations in the eight main SF-36 categories (plus mean scores for the health transition category), and following the methodology of Ara and Brazier (2008)228 we were able to map these mean scores to a set of mean EQ-5D preference-based scores [a different score for each model in the study by Ara and Brazier (2008)] and compute a disutility of 0.208–0.290 for subtotal colectomy with IRA and 0.214–0.299 for proctocolectomy with IPAA (see Appendix 12 for results and details of the calculation method). These do not correspond to the disutility of 0.16 implied in the study by Dinh and colleagues (2011),55 although Dinh and colleagues cite Ara and Brazier (2008). 228
Second, in their cost-effectiveness analysis, Dinh and colleagues (2011)55 estimated a utility of 0.83 after proctocolectomy, which they cite from Fazio and colleagues (1999). 202 Patients who underwent proctocolectomy with stapled anastomosis, mostly for colitis, were included in the study (see Table 83). HRQoL was measured using the Cleveland Global Quality of Life instrument, and in a subgroup of 163 patients, with the SF-36 form. Postoperative quality of life as measured by the SF-36 was excellent and compared well with norms for the general US population. Quality of life increased in the 2 years after surgery, and did not deteriorate thereafter.
In summary, in one study201 HRQoL was found to be substantially lower for FAP patients who underwent colectomy and IRA or proctocolectomy and IPAA compared with individuals from the general Dutch population, although the difference in utilities was not reported. Conversely, in the other study202 HRQoL was found to be similar in colitis patients who underwent proctocolectomy with stapled anastomosis as in individuals from the general US population. Therefore, it seems reasonable to assume no additional disutility after proctocolectomy, given that (a) as mentioned above, arguably any disutilities related to surgery for CRC are already incorporated in our chosen disutilities according to Dukes’ stage, (b) one study found no disutility compared with a general population, and (c) another study found an unquantified disutility compared with a general population.
The only study of which we are aware that measured the psychological impact of risk-reducing surgery for CRC is by Kuppermann and colleagues (2013). 212 In this study, based on health state descriptions assessed by individuals with a range of familiarity with LS, the mean utility was 0.68 for probands tested positive for LS who undergo colectomy, 0.65 for probands tested positive who decline colectomy and 0.66 for probands who decline testing (see Table 84). Given that these utilities are similar, this suggests no substantial impact or psychological impact of colectomy on HRQoL.
Kalady and colleagues (2011)227 used the SF-36 questionnaire to measure the HRQoL of 95 patients who underwent surgery for colon cancer meeting the AC for LS at a single institution. One year after surgery, there was no significant difference in any of the eight domains of the questionnaire between the 50 patients who underwent a total proctocolectomy and the 45 who underwent a partial colectomy.
In another study,83 a panel of 10 experts (seven gastroenterologists and three genetic counsellors) used the standard gamble approach to estimate the utility of patients with subtotal colectomy as 0.95 and that of those with proctocolectomy as 0.89. Although this is low-quality evidence (very small sample size, patients not scoring questionnaire, time trade-off not used), it suggests little disutility after these surgeries.
Given this additional evidence, and that (a) arguably any disutilities related to surgery for CRC are already incorporated in our chosen disutilities according to Dukes’ stage and (b) we assume no additional disutility after proctocolectomy (which is the most substantial of all the risk-reducing surgeries for CRC), we also assume no additional disutility for patients who have had any CRC risk-reducing surgery, including partial colectomy, rectal excision and subtotal colectomy.
We performed a sensitivity analysis in which a disutility of 0.1 was applied to all surgeries except segmental resection, on the basis that they could conceivably have an impact on quality of life.
Impact of endometrial cancer on quality of life
There are contradictory results in the literature regarding the impact of EC and its treatment on HRQoL. 229 Some studies report that most women are cured but that treatment may induce alterations in functional status, activity and family relationships and cause much psychological distress, whereas other studies report good quality of life despite physical symptoms and effects of treatment. 229
In their cost-effectiveness model, Kwon and colleagues (2008)75 assume a utility of 0.83 for women with EC, citing five references. 203–207 Unfortunately, the authors provide no detail as to how they derived their estimate from these sources. In their cost-effectiveness models, both Dinh and colleagues (2011)55 and Yang and colleagues (2011)76 also assume a utility of 0.83 for women with EC, both citing Kwon (2008). 75 Wang and colleagues (2012)57 assume a utility of 0.73 for women with EC, citing Kuppermann and colleagues (2013). 212 However, as explained below (see Psychological impacts of Lynch syndrome testing and management on quality of life), we believe the methodology of this source study to be flawed, and we therefore disregard this value.
There appears to be scant literature on the long-term quality of life of women with EC. Our systematic search for HRQoL for women living with EC revealed only the following two studies of interest.
A study of 79 Norwegian women 3–4 years after radiotherapy for cancer of the endometrium and cervix used the European Organization for Research and Treatment of Cancer Quality of Life Questionnaire-C36 (EORTC QLQ-C36) to measure HRQoL. 229 Of these women, 57% had undergone surgery prior to radiotherapy. Although the nature of surgery is not specified, we assumed that it included hysterectomy. The stages of the cancers were I and II. These women suffered few treatment and/or disease-related side effects. Compared to the general Norwegian population, the women scored lower on role functioning and experienced more diarrhoea. Scores on the social functioning and global health status were similar to those in the general population. 229
A study of 264 Dutch, 5- to 10-year survivors of stage I–II EC found that the HRQoL of women treated with surgery alone or surgery plus radiotherapy was similar to that of an age-matched general population. 230 The authors therefore concluded that the HRQoL of women with stage I–II EC is generally good.
In our base case, we assume no disutility for EC. This is because (1) we can find no high-quality data on relevant HRQoL, (2) the two studies discussed above of women with stages I and II EC found that the HRQoL of women with EC was similar to that of age-matched general populations,229,230 and (3) although there is likely to be a disutility for women with advanced EC, we assume that only a small proportion of women have advanced cancer at diagnosis (73% stage I, 18% stages II and III, 9% stage IV). However, in a sensitivity analysis, we assume a lifetime disutility of 0.10 for all women with EC.
Impact of prophylactic total abdominal hysterectomy with bilateral salpingo-oophorectomy on quality of life
In our model, we assume that a large proportion of female probands and relatives who test positive for LS will have prophylactic TAHBSO at a minimum age of 45 years. It seems plausible to include a long-term disutility after TAHBSO for the following reasons. Hysterectomy is a major operation from which it takes about 6 to 8 weeks to fully recover. 231 Depression may be triggered, there may be some changes to bowel and bladder function and some women develop urinary tract infection or constipation. 231 After oophorectomy, severe menopausal symptoms are likely, which may be treated with hormone replacement therapy. 231
In their cost-effectiveness model, Kwon 200875 assumed a utility of 0.86 for premenopausal women who have prophylactic surgery, citing Grann and colleagues (2002)208 and van Roosmalen and colleagues (2002). 211 Grann and colleagues (2002)208 cite a utility of 0.82 for women who have had prophylactic oophorectomy. However, we are unable to trace this value from their source references. Next, van Roosmalen and colleagues (2002)211 do not give the utility for women who have had just prophylactic oophorectomy or hysterectomy or both.
In their cost-effectiveness model, Dinh and colleagues (2011)55 also assumed a utility of 0.86 for women after TAHBSO, citing Kwon (2008). 75 No other existing models explicitly allowed for a disutility for prophylactic TAHBSO.
We assume no disutility after TAHBSO for the following reasons: (1) we can find no high-quality data on relevant HRQoL; (2) the two studies of women with stages I and II EC (which we discuss in the next section), most of whom had undergone surgery (which we presume included hysterectomy), found that women suffer few treatment and/or disease-related side effects;229 and (3) we assume prophylactic surgery at a minimum age of 45 years, coinciding with the end of childbearing. However, given that long-term disutility after TAHBSO seems plausible, we assume a lifetime disutility of 0.10 in a sensitivity analysis.
In Chapter 6, Utilities associated with endometrial cancer, we demonstrate that overall cost-effectiveness is very sensitive to this assumption.
Impact of Lynch syndrome surveillance colonoscopies on quality of life
In the cost-effectiveness model that underpins the current UK NHS Bowel Cancer Screening Programme,214 no disutilities are assumed for having screening (including flexible sigmoidoscopy). The authors justify their decision by reasoning that any reduction in QALYs would be negligible because the time period over which any disutility would apply would be very small. We also assume no disutility for colonoscopy for the same reason.
Psychological impacts of Lynch syndrome testing and management on quality of life
Background
Although diagnosis of LS can lead to interventions to reduce the chance of developing colorectal, gynaecological and other cancers, it can also lead to anxiety about developing these cancers and the need to make difficult decisions about whether or not to undergo risk-reducing surgeries. Furthermore, those diagnosed with LS must decide whether or not and how to inform relatives about their test results so that these relatives can consider whether or not they wish to be tested themselves. Given that anxiety is one aspect of HRQoL, such effects should be considered in the estimation of health-state utilities of probands and relatives.
We identified just a single study of the cost-effectiveness of strategies for testing for LS [Wang and colleagues (2012)57] that incorporates disutilities associated with the psychological impact of testing. In this study it was assumed that such disutilities are transient, lasting 1 year in the base-case analysis. This assumption was based on several empirical studies. 12,199,232–238 Disutilities due to testing itself and the test results were taken from the empirical study by Kuppermann and colleagues (2013). 212
Kuppermann and colleagues (2013)
Based on our survey of the literature, we agree with Kuppermann and colleagues (2013)212 that their study is the first to measure disutilities associated with genetic testing for LS. They elicited utilities using the time trade-off method, which is recommended by NICE in the UK. 198 Seventy-one highly educated individuals based in the USA, ‘with a range of understanding about and experience with LS’, were given a range of health-state vignettes to value. Most of this sample (n = 49) were neither knowledgeable about nor at high risk of LS. Health-state vignettes covered a range of possible testing scenarios, where participants had to imagine first that their sibling had CRC and was to be tested for LS, and second, that they themselves had CRC that was suspected to be related to LS. Scenarios included whether or not testing for LS was accepted and whether or not risk-reducing surgery was undertaken.
Unfortunately, we question the reliability of the utilities from this study for several reasons. First, NICE’s preferred method of obtaining utilities in the UK is for patients (in this case probands and their relatives) to complete the EQ-5D questionnaire, and then for these responses to be valued by a representative sample of the population of the UK. 198 However, in the study by Kuppermann and colleagues (2013),212 utilities were elicited not by probands and their relatives, but by individuals ‘with a range of understanding about and experience with LS’, and not using the EQ-5D questionnaire, but using health-state vignettes. Next, the sample was from the USA, not the UK, as preferred in our case. Furthermore, the sample of individuals valuing the vignettes was biased as they were highly educated. For these reasons, we believe that the utilities from Kuppermann and colleagues (2013)212 should be considered only as indicative. The results were as shown in Table 84.
| States | Mean (SE)a |
|---|---|
| Siblings | |
| Undergo testing, LS negative | 0.76 (0.03) |
| Undergo testing, LS positive, no surgery offered (men) | 0.74 (0.03) |
| Undergo testing, LS positive, undergo TAHBSO (women) | 0.70 (0.03) |
| Undergo testing, LS positive, decline TAHBSO (women) | 0.67 (0.03) |
| Decline testing | 0.72 (0.03) |
| Probands | |
| Undergo testing, LS positive, accept surgery | |
| Colectomy (men) | 0.68 (0.03) |
| TAHBSO (women) | 0.67 (0.03) |
| Undergo testing, LS positive, decline surgery | |
| Colectomy (men) | 0.65 (0.03) |
| TAHBSO (women) | 0.61 (0.03) |
| Decline testing | 0.66 (0.03) |
As expected, for relatives there is a disutility associated with testing positive for LS compared with testing negative, with utilities of 0.74, 0.70 and 0.67 for testing positive versus 0.76 for testing negative. In our model, we assume that relatives who accept testing and who are tested negative suffer no disutility compared with members of the general population of England and Wales (see Table 85). As explained below, in our base-case analysis we assume that all test-related disutilities apply for only 4 months, as the literature suggests that the psychological impact of testing is transient. Next, in our model we assume that relatives who decline testing incur a disutility over 4 months of 0.04, equal to the utility of 0.76 (siblings who undergo testing, and test negative) minus 0.72 (siblings who decline testing). This disutility reflects anxiety the relative may feel in not knowing whether or not he or she has LS, with the corresponding substantial risk of developing cancer. Next, we assume a disutility of 0.02 for male relatives who are diagnosed with LS, equal to 0.76 for siblings who undergo testing and test negative, minus 0.74 for males who are tested positive for LS. Similarly, we assume a disutility of 0.06 for women who test positive and undergo TAHBSO, equal to 0.76 minus 0.70, and a disutility of 0.09 for women who test positive and decline TAHBSO, equal to 0.76 minus 0.67. The disutility is greater for women who decline TAHBSO presumably because they know that there remains a chance that they will develop gynaecological cancers. For women who test positive but are not offered TAHBSO as they are not at the appropriate age, we assume that the disutility of testing positive will be the same as for men who test positive, i.e. 0.02. Although women are at risk of more cancers than men (i.e. the additional risks of EC and OC), women are at lower risk of CRC,65 and so we assumed that their anxiety related to future cancer risk should not differ greatly from that of men.
With regard to the disutilities for probands, unfortunately Kuppermann and colleagues212 did not measure the utility for probands who accepted testing and were diagnosed as LS negative. In the absence of this information, we assume that these individuals have no associated disutility due to genetic testing (Table 85). Next, we estimate the disutility for probands of declining testing as 0.04, equal to the corresponding value for relatives. In our analysis, risk-reducing colorectal surgery is not offered to probands because the surgical treatment of their CRC is assumed to be completed before genetic testing takes place and our clinical experts advised that colectomy solely for prophylaxis would not be offered. Kuppermann and colleagues212 do not measure the utility for probands who accepted testing and were diagnosed with LS but were not offered any risk-reducing surgery, so we assume that the disutility for testing positive for male probands is the same as the disutility for male relatives, i.e. 0.02. For female probands who test positive and are not offered any risk-reducing surgery, we again assume the same disutility as for males, i.e. 0.02. For female probands who test positive and are offered prophylactic TAHBSO, we assume disutilities of 0.03 for those accepting surgery and 0.09 for those declining it. These disutilities are estimated by subtracting the utilities of 0.67 and 0.61 reported in Kuppermann and colleagues212 from the imagined utility of probands testing negative, which we estimate as the utility of probands declining testing (0.66) plus a utility of 0.04 for not declining testing taken from the relatives, to give a utility for probands testing negative for LS of 0.70. If a proband or relative declines testing but is still diagnosed with LS (by FH for probands or on account of being a FDR of a known carrier for relatives) and offered TAHBSO, we assume a disutility of 0.01 for probands and 0.04 for relatives (i.e. the same disutility as for probands or relatives testing positive), with an additional disutility of 0.06 for probands declining TAHBSO and 0.03 for relatives declining TAHBSO. For example, the total disutility for a proband declining testing and accepting TAHBSO would be 0.04 (declined testing) + 0.01 (offered TAHBSO) = 0.05, while the total disutility for a relative declining testing and declining TAHBSO would be 0.04 (declined testing) + 0.04 (offered TAHBSO) + 0.03 (declined TAHBSO) = 0.11.
| Result of genetic testing | Disutility | |
|---|---|---|
| Males | Females | |
| Proband | ||
| Test declined, surgery not offered | 0.04 | 0.04 |
| Test declined, accept TAHBSO | N/A | 0.05 |
| Test declined, decline TAHBSO | N/A | 0.11 |
| Test accepted, LS negative | 0.00 | 0.00 |
| Test accepted, LS positive, surgery not offered | 0.02 | 0.02 |
| Test accepted, LS positive, accept TAHBSO | N/A | 0.03 |
| Test accepted, LS positive, decline TAHBSO | N/A | 0.09 |
| Relative | ||
| Test declined, surgery not offered | 0.04 | 0.04 |
| Test declined, accept TAHBSO | N/A | 0.08 |
| Test declined, decline TAHBSO | N/A | 0.11 |
| Test accepted, LS negative | 0.00 | 0.00 |
| Test accepted, LS positive, surgery not offered | 0.02 | 0.02 |
| Test accepted, LS positive, accept TAHBSO | N/A | 0.06 |
| Test accepted, LS positive, decline TAHBSO | N/A | 0.09 |
Other empirical studies
Heshka and colleagues (2008)239 performed a systematic review of studies that measured the psychological impact of genetic testing on a range of hereditary conditions. Twelve of these studies concerned LS. Most psychological outcomes were measured in one of the following four domains: general distress, specific distress, anxiety and depression. In summary, most studies reported negative effects on those diagnosed with LS, but these were short-lived, typically lasting up to 4 months. In our base-case analysis, we assume that any disutilities concerning the psychological impact of genetic testing last only 4 months. Detailed findings for each of the four domains are as follows:
-
Most studies found no effect of genetic testing on general distress for either those diagnosed LS positive or those diagnosed negative, and most studies found no difference in general distress between those diagnosed positive or negative after disclosure of test results.
-
Studies found either that genetic testing had no effect on cancer-specific distress, or that distress increased for about 2 weeks and then returned to baseline levels. For individuals tested negative, cancer-specific distress either decreased after test result disclosure or did not change. Those tested negative generally had lower cancer-specific distress than carriers.
-
Anxiety generally increased in those tested positive at results disclosure and 2 weeks later, but had returned to baseline levels or lower after 1 year. Anxiety in those tested negative decreased at results disclosure or 1 month after the test, but also returned to baseline levels or lower by 1 year. Those tested positive generally had higher anxiety than those tested negative at test result disclosure, but later anxiety was similar.
-
Most studies reported no effect of genetic testing on depression in either those tested positive or those tested negative.
Heshka and colleagues (2008)239 noted that most studies in the review acknowledged several limitations. Most study populations were small and self-selected, not representative of individuals eligible for testing. Participants were typically white and highly educated. Additionally, most studies included extensive pre- and post-test counselling and education, which may not always generalise to the routine clinical setting, although in our model we assume that individuals must receive genetic counselling if they are to have genetic testing.
Four other studies not considered in the review by Heshka and colleagues (2008)239 also found no long-term psychological impact of testing for LS:
-
In a study of 92 individuals in the Netherlands,240 cancer distress and worry increased slightly in those diagnosed LS positive after test disclosure and decreased at the next time point measured (6 months after disclosure). Those tested negative remained at the same level of distress shortly after disclosure, but distress had decreased by the 6-month reporting time.
-
In a study of 116 individuals in the Netherlands,241 4 years after genetic counselling for LS only a small minority of participants reported significant levels of distress, or significant family or social problems.
-
A Canadian study242 found that 49 individuals diagnosed with LS who did not have CRC had adapted psychologically to their test result at the mean follow-up time of approximately 6 years.
-
An Australian study232 of 19 individuals diagnosed with LS and 54 without LS also found no long-term evidence (at 1 and 3 years after testing) of undue psychological distress in those diagnosed with LS.
Cost parameters and assumptions
Many of the published models use cost data from countries other than the UK. However, it was important to choose values relevant to a UK and NHS perspective.
Adjustments to 2013–14 prices
All costs and prices in the model are inflated to 2011–12 prices using the Hospital and Community Health Services (HCHS) Pay and Prices index,243 and then further inflated by 3.46% per annum for 2 years to 2013–14 prices, where 3.46% is the average (geometric mean) inflation of the index between 2000–1 and 2011–12.
Costs of tumour testing
Costs of the preliminary tumour tests have been obtained directly from laboratories in the UK (see Table 86). Where possible, we also included the additional costs of administration, additional wear and tear on machinery, training time and repeat tests.
Cost of genetic testing
Costs for genetic tests (for probands and relatives) are taken directly from genetic testing laboratories, identified from the UKGTN. 244 Only costs applicable to the NHS are collected (we were careful to exclude private fees) and overall values for each cost are calculated across the laboratories that supply each given test. Available genetic tests are individual sequencing tests for probands for MLH1, MSH2, MSH6 and PMS2, with PMS2 testing currently only available in one UK laboratory in Leeds; individual targeted tests for MLH1, MSH2, MSH6 and PMS2 for relatives; a combined MLH1, MSH2 sequencing test for probands; and a combined MLH1, MSH2 and MSH6 sequencing test for probands. We decided to model using the combined MLH1, MSH2 and MSH6 test for probands, because it is cheaper than doing all three individual tests. This is followed by an individual sequencing test for PMS2. Relevant individual targeted tests are conducted for relatives. The cost for PMS2 tests (for probands or relatives) was taken from the laboratory in Leeds, as this is the only laboratory that provides this test. The cost for the combined M126 test was taken as the mean cost from the four laboratories that offer this test and provide costs on their websites (Yorkshire Regional Genetics Service,126 East Anglian Medical Genetics Laboratories,125 All Wales Molecular Genetics Laboratory123 and Oxford Medical Genetics Laboratories122). Similarly, the cost of the individual targeted genetic tests for relatives is calculated as the mean cost across laboratories. The listed costs of genetic tests are currently all-inclusive for the NHS; that is, there are no additional charges for laboratory, processing or transportation costs as they are accounted for through core funding.
Genetic testing costs reduce as technology enables greater efficiency, hence the laboratory costs are expected to decrease. We performed a sensitivity analysis with reduced costs to reflect this.
Cost of genetic counselling
Genetic counselling is assumed to occur after initial tumour testing and before genetic testing. Genetic counselling also includes taking a FH (according to our experts), so for all strategies that include genetic testing, the cost of taking a FH is included in the cost of genetic counselling. A separate cost is given for FH assessment in strategy 1 and for those who decline genetic counselling (Table 86 and Figure 34).
| Test | Patient | Base-case cost | Base-case source |
|---|---|---|---|
| MSI | Proband | £202 | Average of £221 (Oxford Medical Genetics Laboratories),122 £150 (All Wales Molecular Genetics Laboratory),123 £215 (West Midlands Regional Genetics Laboratory, via the UKGTN),244 2012 costs updated to 2013–14 costs |
| IHC | Proband | £238 | Dr Mark Arends (Department of Pathology, University of Cambridge, 2012, personal communication) and Dr Ian Frayling (All Wales Genetics Service, 2012, personal communication) £220–240, 2012 costs updated to 2013–14 costs |
| BRAF | Proband | £118 | Average of £140 [Mr Michael Gandy, University College London (UCL)-Advanced Diagnostics, 2012, personal communication], £117 (East of Scotland Regional Genetic Service),124 £85 (All Wales Molecular Genetics Laboratory),123 2012 costs updated to 2013–14 costs |
| Proband genetic test (M126) | Proband | £812 | Average of £860 (Oxford Medical Genetics Laboratories),122 £900 (All Wales Molecular Genetics Laboratory),123 £850 (East Anglian Medical Genetics Laboratories),125 £530 (Yorkshire Regional Genetics Service),126 2012 costs updated to 2013–14 costs |
| Proband genetic test (PMS2) | Proband | £735 | £710 (Yorkshire Regional Genetics Service),126 2012 costs updated to 2013–14 costs |
| Proband genetic test (MLH1) | Proband | £464 | Average of £400 (All Wales Molecular Genetics Laboratory),123 £390 (East of Scotland Regional Genetic Service),124 £475 (East Anglian Medical Genetics Laboratories),125 £530 (Yorkshire Regional Genetics Service),126 2012 costs updated to 2013–14 costs |
| Proband genetic counselling | Proband | £67 | PSSRU243 and personal communication with Professor Mary Porteous (South East Scotland Genetic Service) updated to 2013–14 costs |
| Taking FH | Proband | £22 | PSSRU243 and personal communication with Professor Mary Porteous (South East Scotland Genetic Service) updated to 2013–14 costs |
| Targeted genetic test for relatives (MLH1) | Relative | £169 | Average of £170 (Oxford Medical Genetics Laboratories),122 £160 (All Wales Molecular Genetics Laboratory),123 £143 (East of Scotland Regional Genetic Service),124 £175 (East Anglian Medical Genetics Laboratories),125 £170 (Yorkshire Regional Genetics Service),126 2012 costs updated to 2013–14 costs |
| Targeted genetic test for relatives (MSH2) | Relative | £172 | Average of £170 (Oxford Medical Genetics Laboratories),122 £160 (All Wales Molecular Genetics Laboratory),123 £170 (Yorkshire Regional Genetics Service),126 2012 costs updated to 2013–14 costs |
| Targeted genetic test for relatives (MSH6) | Relative | £172 | Average of £170 (Oxford Medical Genetics Laboratories),122 £160 (All Wales Molecular Genetics Laboratory),123 £170 (Yorkshire Regional Genetics Service),126 2012 costs updated to 2013–14 costs |
| Targeted genetic test for relatives (PMS2) | Relative | £176 | £170 (Yorkshire Regional Genetics Service),126 2012 costs updated to 2013–14 costs |
| Relative genetic counselling | Relative | £67 | PSSRU243 and personal communication with Professor Mary Porteous (South East Scotland Genetic Service) |
FIGURE 34.
Costs of genetic counselling and FH assessment in strategies to identify LS in probands and their relatives.
Genetic counselling was a parameter without a standard unit cost. As such, it was calculated using estimates of time, staff involved and associated unit costs.
We found no standardised approach to genetic counselling (some places may provide two or three times more counselling per patient than others) and, as such, the estimates we detail may not reflect the practice of all genetics centres. To calculate the time and staff involved in genetic counselling we corresponded with Professor Mary Porteous of the South East Scotland Genetic Service, based at the Western General Hospital in Edinburgh (2013, personal communication).
In this centre genetic counselling occurred, where applicable, after the tumour tests (IHC and/or MSI) for the proband. Generally, probands received a maximum of a single 45-minute session with a band 7 counsellor before gene testing, and a 30-minute session to discuss the results. The same was also true of the relatives, though in practice the total 75 minutes could be split in various ways (for example, sometimes relatives would have a group session then return for a shorter individual session before they were tested). In this centre the cost of genetic counselling incurred for a relative of a proband was therefore the same as that for the proband.
Using the costs from the Personal Social Services Research Unit (PSSRU) 2012,243 we could only find costs for counselling services in primary medical care, which seemed more applicable to band 7 mental health counsellors. However, no specific costs for genetic counsellors were stated and it seemed plausible that the staff and overhead costs would be similar, so we concluded that this cost would be appropriate. The cost therefore worked out to be £50 per hour (which included wages; salary oncosts; overheads including management, administration and estates staff and non-staff; and capital overheads); £65 per hour of client contact (including wages; salary oncosts; overheads including management, administration and estates staff and non-staff; and capital overheads); or £59 per consultation. We chose to use the ‘per hour’ cost, which resulted in a cost of £62.50 per patient for 75 minutes of counselling. We updated this to 2013 costs, by inflating at 3.46% per annum over 2 years, from 2011–12 to 2013–14. This increased the cost to £67.
The South East Scotland Genetic Service was, at the time of writing, moving towards ward staff being responsible for taking FH, a role for which a band 5 clinic nurse would be deemed appropriate. In this setting the intention was to reduce the time spent with the counsellor. Again, this does not reflect all practice across UK genetics services; some centres have, for example, clinical pedigree support workers at similar banding levels, who are neither nurses nor counsellors. Owing to a paucity of alternative information, we considered that this could also be used to cost for patients who do not visit a genetic counsellor. The amount of the nurses’ time this was expected to take was 30 minutes.
Again using the PSSRU 2012, we found that the costs for a band 5 staff nurse were £35 (£41) per hour, and £85 (£100) per hour of patient contact, where the costs in brackets included qualifications. Assuming that training for taking FH could be included in the qualifications, we took the hourly cost of £41, which worked out at £20.50 per patient. Using the same approach as before we updated this cost to 2013–14 costs, giving £22.
Given that different genetic centres have different approaches to genetic counselling, we performed a sensitivity analysis on the cost of counselling, halving and doubling the time spent on each patient.
Figure 35 shows the costs of each test for probands and relatives. The cost of gene testing for relatives appears much reduced compared with that for probands, as ‘predictive testing’ is less costly than full gene sequencing. As the predictive gene testing only occurs as a result of finding a particular gene mutation in probands, gene testing in relatives only ever tests for one gene, hence relatives cannot have a combined gene test (such as M126). As MSI, IHC and BRAF are tumour tests, they are not conducted on asymptomatic relatives.
FIGURE 35.
Costs of individual tests to identify LS in probands and relatives.
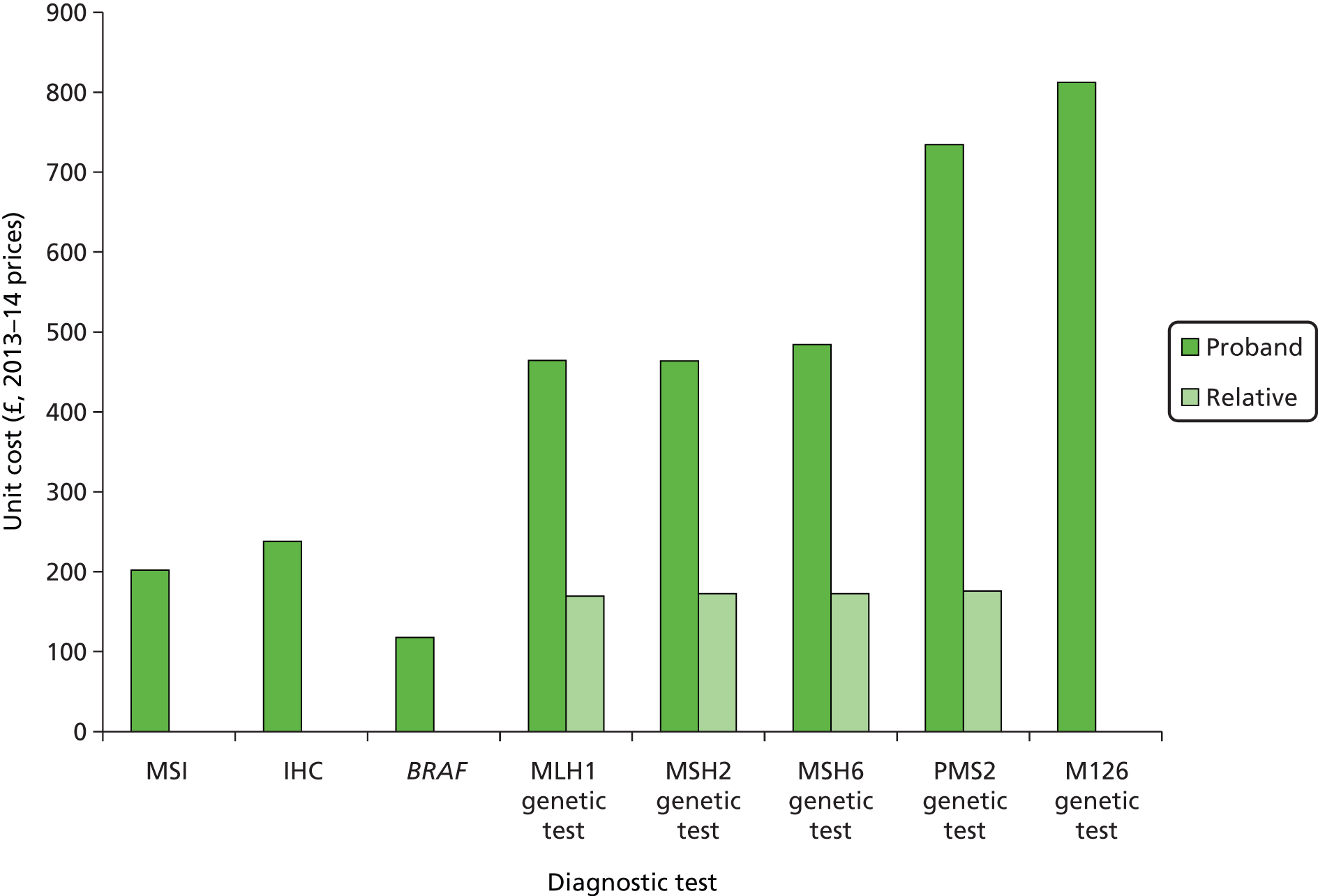
Costs of Lynch syndrome surveillance colonoscopies
We estimated the cost of colonoscopy from Department of Health reference costs 2011–12151 using Healthcare Resource Groups (HRGs) FZ51Z (diagnostic colonoscopy, 19 years and over), FZ52Z (diagnostic colonoscopy with biopsy, 19 years and over) and FZ53Z (therapeutic colonoscopy, 19 years and over). The reference costs for these HRGs are shown in Table 87, as well as the average cost weighted by activity (£553 in 2011–12 prices). We uprated this to 2013–14 prices as described in Adjustments to 2013–14 prices to obtain a cost of £592.
| HRG | Description | Number of colonoscopies | Unit cost (£) | Total cost (£) |
|---|---|---|---|---|
| FZ51Z | Diagnostic colonoscopy, 19 years and over | 143,080 | 528 | 75,555,599 |
| FZ52Z | Diagnostic colonoscopy with biopsy, 19 years and over | 129,405 | 572 | 73,975,538 |
| FZ53Z | Therapeutic colonoscopy, 19 years and over | 90,181 | 567 | 51,164,816 |
| Weighted average | 362,666 | 553 | 200,695,953 | |
Finally, the cost of colonoscopy was adjusted to reflect the fact that the estimate of the effectiveness of colonoscopy comes from a population in which colonoscopies were performed 3-yearly rather than 2-yearly. 94 We could adjust for this by only including 3-yearly colonoscopies in the model, but given that the number of additional colonoscopies required for each strategy was an outcome of interest, we preferred instead to adjust the cost of colonoscopy. Jarvinen and colleagues (2000)94 report that over 600 colonoscopies were performed in the study group, with a total follow up of 1854 person-years. This means that the average number of colonoscopies per person-year was at least 0.324, or a rate of at least one colonoscopy per 3.09 years. As the surveillance protocol dictated 3-yearly colonoscopies and the observed rate is very similar to this, we assumed a rate of one colonoscopy per 3 years. The cost of colonoscopy was adjusted from £592 to £395.
Costs of morbidity and mortality due to Lynch syndrome surveillance colonoscopies
Table 88 summarises the unit costs included for morbidity and mortality due to LS surveillance colonoscopies.
| Event | Cost (£) | Source |
|---|---|---|
| Mortality | 5134 | Assumed same as perforation following Whyte et al. 2012214 |
| Perforation | 5134 | Department of Health reference costs,151 FZ77 (Major large intestine procedure) |
| Mild bleeding not requiring admission | 0 | Assumption |
| Mild bleeding requiring admission | 318 | Whyte et al. 2012214 |
| Moderate bleeding | 490 | Department of Health reference costs,151 FZ38F (Gastrointestinal bleed with length of stay 1 day or less) |
| Severe bleeding | 1984 | Department of Health reference costs,151 FZ38D/FZ38E (Gastrointestinal bleed with length of stay 2 days or more) |
We assumed that bleeding not requiring admission would incur no cost. We estimated the cost of mild bleeding requiring admission by uprating the cost of this item, reported by Whyte and colleagues214 as £278 in 2008–9 prices, using the HCHS Pay and Prices inflation index, to £318 in 2013–14 prices (as described in Adjustments to 2013–14 prices). We estimated the cost of moderate bleeding by using the Department of Health reference costs 2011–12,151 using HRG FZ38F (gastrointestinal bleed with length of stay 1 day or less) and including only non-elective inpatient activity, resulting in a cost of £458 in 2011–12 prices, which we uprated to £490 in 2013–14 prices. We estimated the cost of severe bleeding by using HRGs FZ38D and FZ38E (gastrointestinal bleed with length of stay 2 days or more) and including only non-elective inpatient activity, resulting in a cost of £1853 in 2011–12 prices, which we uprated to £1984 in 2013–14 prices.
We estimated the cost of treating perforation from colonoscopy from Department of Health reference costs 2011–12,151 using HRG FZ77 (major large intestine procedures, 19 years and over) and including only non-elective inpatient activity, resulting in a cost of £4797 in 2011–12 prices, which was uprated to £5134 in 2013–14 prices.
We estimated that the cost of colonoscopy mortality would be the same as for perforation, i.e. £5134 in 2013–14 prices, following the assumption of Whyte and colleagues214 that mortality only follows perforation and does not include any additional costs.
Costs of colorectal cancer treatment
Costs of colorectal cancer treatment in published models of cost-effectiveness of testing for Lynch syndrome
There is much variability in the assumed nature and magnitude of costs for treating CRC across studies of the cost-effectiveness of testing for LS (Table 89). Approaches are:
-
a single total lifetime cost over all Dukes’ stages22,50,51,56
-
a single lifetime cost specific to each disease stage, separately for a first and second CRC23,49,54
-
costs according to whether incurred initially, regularly or at end of life48,57
-
costs split by stage, whether incurred initially, regularly or at end of life55
-
cost for chemotherapy only. 53
| Cost-effectiveness analysis | Chemotherapy for CRC | Medical management for CRC | End of life for CRC |
|---|---|---|---|
| Breheny et al. 200651 | AU$18,358 in 2001–2 prices for total cost over lifetime to treat CRC. Evidence appears lacking | ||
| Brown and Kessler 199622 | $40,000 in mid-1990s prices for total cost over lifetime to treat CRC, citing a US study published in 1994244 | ||
| DACEHTA 200752 | Not clear from publication | ||
| Dinh et al. 201155 | Stage I $27,221 initially, $2166 continuing | Stage I $48,791 | |
| Stage II $37,563 initially, $2024 continuing | Stage II $48,662 | ||
| Stage III $45,804 initially, $2883 continuing | Stage III $51,276 | ||
| Stage IV $59,812 initially, $8945 continuing | Stage IV $68,809 | ||
| aAll from US Medicare 1998–2003246 | All from US Medicare 1998–2003 | ||
| Kievit et al. 200550 | €1634 for follow-up for 3 years after subtotal colectomy in 2002 prices | ||
| Kwon et al. 200875 | None | ||
| Kwon et al. 201156 | $35,000 in 2010 prices for total cost over lifetime to treat CRC, citing US studies247–249 | ||
| Ladabaum et al. 201148 | $34,000–44,000 initially, decreasing from $2500–3500 in year 1 to $2400–2800 in year 5, depending on whether LS positive or negative and whether or not screened. In 2010 US$, from US data source246 | $49,000–52,000, depending on whether LS positive or negative and whether or not screened. In 2010 US$, from US data source | |
| Mvundura et al. 201054 | See Ramsey et al. 200123 | ||
| Olsen et al. 200753 | €12,320 for Dukes’ C and D based on Danish practice in 2004 | None | None |
| Ramsey et al. 200123 | First CRC diagnosis: stage I $25,516, stage II $28,166, stage III $31,907, stage IV $45,393 Second CRC diagnosis: stage I $27,794, stage II $28,872, stage III $33,658, stage IV $49,352b 1999 prices; source: US Medicare249 |
||
| Ramsey et al. 200349 | See Ramsey et al. 200123 | ||
| Reyes et al. 200259 | None | ||
| Wang et al. 201257 | See Ladabaum et al. 201148 | ||
| Yang et al. 201176 | None | ||
Costs of colorectal cancer treatment in the Peninsula Technology Assessment Group model
There are many stages in the management of patients with CRC from the time of diagnosis [see Trueman and colleagues (2007),134 pp. 28–9, for detailed diagrams showing treatment pathways for colon and rectal cancer]. Furthermore, there is substantial variation across England in the management of CRC patients. 133,134,251 For these reasons, costing the treatment of patients with CRC from the time of diagnosis is a very substantial project in its own right.
The data sources used in previous models of LS to inform the cost of treatment for CRC (see Table 89) were not used because they are not UK based and are rather old. However, fortunately, two UK-based studies have recently done just this.
The UK NHS Bowel Cancer Screening Programme, which started in 2006, and which currently recommends biennial screening with the guaiac faecal occult blood test for individuals aged 60–74 years, was informed by a cost-effectiveness analysis that currently assumes the following lifetime costs for treating CRC: £12,455 for Dukes’ A, £17,137 for Dukes’ B, £23,502 for Dukes’ C and £25,703 for Dukes’ D. 214 These estimates were taken from a 2009 UK study that quantified the activities, costs and outcomes associated with the treatment of bowel cancer,251 and inflated to the year 2010. Unfortunately, though the source publication251 gives some of the parameter values that underlie these figures, it contains insufficient data to allow the reader to recreate these lifetime costs.
Another study was published in 2007 on the costs and benefits of bowel cancer services in England. 134 Numerous parameters were used to estimate the costs of treating CRC patients. Sources for resource use included the Hospital Episode Statistics, ONS, nationally published audits, locally published data, published literature and expert opinion. 134 Unit cost data were taken from NHS reference costs published by the Department of Health, the PSSRU at the University of Kent, published literature, and local sources and expert opinion. 134 The costs associated with treatment of patients with CRC are shown in Table 90. All costs relate to those patients who incur the particular cost item. For example, the cost of £11,209 for primary chemotherapy and radiotherapy for patients with colon cancer relates only to those patients who receive either chemotherapy or radiotherapy. Some patients diagnosed with colon cancer may receive neither chemotherapy nor radiotherapy. All values are in 2004–5 prices.
| Cost | Components | Cost per patient with colon cancer (£) | Cost per patient with rectal cancer (£) |
|---|---|---|---|
| Diagnosis | Referral and diagnosis. Presenting via GP, A&E or elsewhere in secondary care. Diagnostic tests include colonoscopy, flexible sigmoidoscopy, double contrast barium enema | 379 | 379 |
| Primary surgery | MRI and pathology, stoma, stoma closure, complications and stenting | 4616 | 5980 |
| Primary chemotherapy and radiotherapy | Pre- and postoperative chemoradiation, adjuvant chemotherapy | 11,209 | 7726 |
| Chemotherapy is a mix of capecitabine and oxaliplatin plus 5-FU/leucovorin (folinic acid) (FOLFOX) | |||
| Follow-up surveillance (mean annual cost) | CEA test, CT scan, colonoscopy, consultation. Assumed for 5 years following primary treatment | 204 | 195 |
| Recurrence surgery and chemotherapy | Almost all due to metastatic recurrence. Assumed to occur within 5 years of primary treatment | 9554 | 9279 |
| Stoma care (annual cost) | Assumed mix of ileostomy and colostomy | 1279 | 1279 |
| Palliative care | Interventions (almost all chemotherapy, mix of 5-FU followed by 5-FU and irinotecan and FOLFIRI/FOLFOX), and end-of-life care (symptom management, hospital and hospice stays) | 7703 | 7016 |
With the exception of primary surgery (the costs of which are estimated in Colorectal cancer surgery costs), we based the costs for CRC treatment in the PenTAG model on the costs in Table 90, and our adapted costs are shown in Table 91.
| Cost | When incurred | Colon cancer (by Dukes’ stage at diagnosis) | Rectal cancer (by Dukes’ stage at diagnosis) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| A | B | C | D | A | B | C | D | ||
| Diagnosis | Diagnosis | £499 all Dukes’ | £499 all Dukes’ | ||||||
| Primary chemotherapy and radiotherapy | Diagnosis | £0 | £5755 | £13,133 | £13,133 | £0 | £2848 | £7628 | £7628 |
| Follow-up surveillance (mean annual cost) | Maximum 5 years from index CRC | £269 all Dukes’ | £256 all Dukes’ | ||||||
| Recurrence surgery and chemotherapy | In last year of life if patient dies of CRC within 5 years of diagnosis | £12,578 all Dukes’ | £12,216 all Dukes’ | ||||||
| Stoma care (annual cost) | Every year after surgery | £1684 all Dukes’ for 11% of patients | £1684 all Dukes’ for 49% of patients | ||||||
| Palliative care | In last year of life if patient dies of CRC | £10,141 all Dukes’ | £9236 all Dukes’ | ||||||
In our model we distinguish between colon and rectal cancer only in the year of diagnosis (as it is used in the logic to decide which surgery is appropriate). Therefore, costs incurred in the year of diagnosis (diagnosis costs, surgery costs, primary chemotherapy and radiotherapy) have costs dependent on both the stage at diagnosis and the site of the CRC (colon or rectum). Costs incurred later (surveillance, treatment for recurrence, stoma care and palliative care) have costs dependent on the stage at diagnosis and whether or not the patient has LS, on the basis that 94% of LS CRC patients have colon cancer55 and 65% of sporadic CRC patients have colon cancer. 119
All costs were inflated over 9 years, to 2013–14 prices, as described in Adjustments to 2013–14 prices. Next, these unit costs were adjusted for the proportions of patients receiving the cost item according to Dukes’ stage at diagnosis. The resulting cost of £499 for diagnosis clearly applies regardless of Dukes’ stage at diagnosis.
The proportions of patients who receive chemotherapy and radiotherapy according to Dukes’ stage at diagnosis were estimated for colon cancer patients as 0% Dukes’ A, 39% Dukes’ B, 89% Dukes’ C and 89% Dukes’ D. The corresponding proportions for rectal cancer patients were estimated as 0% Dukes’ A, 28% Dukes’ B, 75% Dukes’ C and 75% Dukes’ D. The values for Dukes’ A, B and C are taken from Trueman and colleagues (2007). 134 We assumed that the proportions for Dukes’ D equal the proportions for Dukes’ C. These assumptions appear consistent with the recent NICE clinical guidelines for the treatment of CRC. 133
In common with Trueman and colleagues (2007),134 follow-up surveillance was assumed to occur up to a maximum of 5 years after diagnosis of the index CRC. Further, the same resource use was assumed regardless of cancer stage. Although this may be unlikely, this simplifying assumption is unimportant given the relatively small costs.
The York Health Economics Consortium (YHEC) estimates of the cost of treatment for CRC recurrence were assumed to be due almost totally to metastatic recurrence. 134 Unfortunately, YHEC do not report the proportion of patients who experience metastatic recurrence. However, given that recurrence was assumed to occur within the first 5 years after primary treatment,134 and given that life expectancy for patients who have metastatic CRC is short, in the PenTAG model it is assumed that a patient who dies from CRC (i.e. not general mortality or EC) within 5 years from diagnosis of the index CRC does so owing to metastatic recurrence, thus incurring the cost of treatment for metastatic recurrence. The effect of this assumption is that this cost is more likely to be incurred for those patients diagnosed with a more advanced CRC, because these patients are more likely to die within 5 years of diagnosis.
In common with Trueman and colleagues (2007),134 the annual cost of stoma care of £1684 in 2013–14 prices was assumed to be incurred in each year after surgery for those patients who received a stoma and for whom the stoma was not subsequently closed. The YHEC study estimated that 67.0% of rectal cancer patients require a stoma, with 26.6% of these subsequently closed, and that 14.5% of colon cancer patients require a stoma, again with 26.6% of these subsequently closed. Together this implies that 49% of rectal cancer patients and 11% of colon cancer patients require a stoma which is not subsequently closed. Resource use was assumed independent of cancer stage at diagnosis.
The cost of palliative care is assumed to be incurred in the last year of life only if the patient dies of CRC (as opposed to other causes, including EC). Although resource use was assumed independent of cancer stage at diagnosis, the cost of palliative care is more likely to be incurred for patients diagnosed with more advanced disease, because they are more likely to die of CRC.
No biological agents are assumed for treating CRC as NICE has not recommended first-line treatment with bevacizumab in combination with oxaliplatin and either 5-FU plus folinic acid or capecitabine for the treatment of metastatic CRC (www.nice.org.uk/nicemedia/live/13291/52091/52091.pdf), nor has it recommended treatment with bevacizumab (Avastin®, Roche) in combination with fluoropyrimidine-based chemotherapy, cetuximab chemotherapy or monotherapy, and panitumumab (Vectibix®, Amgen) monotherapy for metastatic CRC that has progressed following first-line treatment with chemotherapy. 133 NICE was also unable to recommend panitumumab in combination with FOLFIRI (FOLinic acid, Fluorouracil, IRInotecan) for the treatment of metastatic CRC that has progressed following first-line treatment with chemotherapy,133 and second-line use of bevacizumab, panitumumab or cetuximab (http://guidance.nice.org.uk/TA/WaveR/102/Consultation/EvaluationReport/AssessmentReport/pdf/English).
Colorectal cancer surgery costs
We assumed that the cost of surgery would depend on the nature of surgery, independent of the Dukes’ stage of the CRC. As LS CRCs have a greater tendency to be proximal than sporadic,55 we estimated costs separately for LS CRCs and sporadic CRCs (although there is only a difference for segmental resections).
To estimate the cost of colorectal surgery we chose the most relevant Office of Population, Censuses and Surveys Classification of Surgical Operations and Procedures (4th revision) (OPCS-4.6) codes for the procedures and used the Healthcare Resource Groups 4 (HRG4) 2011/12 Reference Costs Grouper Code to Group workbook252 to map these to the appropriate HRGs, which were then located in reference costs to obtain 2011–12 costs for the procedures (see Appendix 13). All costs were then uprated to 2013–14 prices as described in Adjustments to 2013–14 prices. The cost of stoma reversal was then added for the appropriate proportion of patients, and for segmental resections the proportion of proximal/distal procedures was based on LS status.
The estimated costs of surgery for CRC are summarised in Table 92.
| Surgery | LS CRC (£) | Sporadic CRC (£) |
|---|---|---|
| Segmental resection | 6154 | 6104 |
| Subtotal colectomy IRA | 7331 | 7331 |
| Anterior resection | 7399 | 7399 |
| Proctocolectomy IPAA | 7441 | 7441 |
Cost of endometrial cancer prevention
The cost of prophylactic hysterectomy and bilateral salpingo-oophorectomy was estimated as £3322 in 2013–14 prices (inflated from £3104 in 2011–12 prices). This estimate is based on Department of Health reference costs 2011–12151 and the HRGs MA07 (major open upper genital tract procedures) and MA08 (major laparoscopic or endoscopic, upper genital tract procedures), weighted by activity recorded in the reference costs (Table 93). These HRGs were determined by mapping the relevant OPCS-4.6 codes for the procedures253 using the HRG4 Code to Group workbook distributed with the Department of Health HRG4 2011/12 Reference Cost Grouper,252 as shown in Table 94.
| HRG | Number of procedures | Unit cost (£) | Total cost (£) |
|---|---|---|---|
| MA07C Major open upper genital tract procedures with major CC | 1381 | 4535 | 6,262,304 |
| MA07D Major open upper genital tract procedures without major CC | 33,863 | 3269 | 110,692,366 |
| MA08Z Major laparoscopic or endoscopic, upper genital tract procedures | 15,225 | 2606 | 39,680,196 |
| Weighted average | 50,469 | 3104 | 156,634,867 |
| Procedure | OPCS codes | HRG |
|---|---|---|
| TAHBSO | Q07.4 + Q22.1 | MA07 |
| TLHBSO | Q07.4 + Q22.1 + Y75.2 | MA08 |
| LAVHBSO | Q08.9 + Q22.1 + Y75.1 | MA08 |
Cost of endometrial cancer treatment
Cost of surgery for endometrial cancer
The cost of hysterectomy and bilateral salpingo-oophorectomy as a treatment for EC was estimated as £3877 in 2013–14 prices (inflated from £3622 in 2011–12 prices). This estimate is based on Department of Health reference costs 2011–12151 HRG MA06Z (major open or laparoscopic, upper or lower genital tract procedures for malignancy). As before, we confirmed that this was the correct HRG using the HRG4 Code to Group workbook distributed with the Department of Health HRG4 2011/12 Reference Cost Grouper. 252
Cost of radiotherapy for endometrial cancer
We estimate the cost of radiotherapy per patient as £5856. We base our estimate on the value of $7895 from the study by Havrilesky and colleagues (2009),152 which is similar to the estimate of $8418 used in the model of Yang and colleagues (2011). 76 First, the $7895 is adjusted from US dollars to pounds sterling by purchasing power parities and from year 2009 to 2011,254 to yield £5520. Next, this is inflated by 2 years, from 2011 to 2013–14 prices (see Adjustments to 2013–14 prices) to give £5909.
Given the estimated split of stages of EC at diagnosis and the proportions of patients at each stage who receive radiotherapy, this implies that 47% = (73% × 33%) + (18% × 100%) + (9% × 50%) of patients diagnosed with EC will receive radiotherapy. Therefore, the cost of radiotherapy per EC patient is 47% × £5909 = £2753. In our model, this cost is assumed to occur immediately on diagnosis of EC.
Cost of chemotherapy for endometrial cancer
There is no general agreement as to what currently constitutes the best chemotherapy for EC, as very few phase III studies have been done comparing different chemotherapy regimens. 255 Treatments include:255
-
single-agent therapy
-
cisplatin
-
carboplatin
-
paclitaxel
-
doxorubicin
-
liposomal doxorubicin
-
-
combination therapy
-
doxorubicin 60 mg/m2 intravenous (IV) plus cisplatin 50 mg/m2
-
doxorubicin 45 mg/m2 IV plus cisplatin 50 mg/m2 plus paclitaxel 160 mg/m2
-
cisplatin 50 mg/m2 IV plus doxorubicin 50 mg/m2 or
-
doxorubicin 45 mg/m2 IV plus cisplatin 50 mg/m2 IV plus paclitaxel 160 mg/m2 IV plus filgrastim (Neupogen®, Amgen) 5 µg/kg subcutaneous
-
carboplatin area under the curve 5–7 plus paclitaxel 175 mg/m2.
-
Next, we assume the treatment regime above of doxorubicin 45 mg/m2 IV plus cisplatin 50 mg/m2 IV on day 1, plus paclitaxel 160 mg/m2 on day 2, repeated every 21 days, as it is representative of the above treatments and it has been used in a RCT. 256 Unlike the RCT,256 we do not include the acquisition and administration costs of filgrastim, because this drug appears to be given rarely and because it is given on 10 days in each treatment cycle, hence incurring a very high administration cost, as it is given subcutaneously. The costs of doxorubicin, cisplatin and paclitaxel were taken from the electronic Market Information Tool (eMit) database. 153 These data include an estimate of the average prices paid for generic drugs in the UK in the 4 months up to May 2012.
Given the prices doxorubicin 50 mg/25 ml injection £6.38, cisplatin 50 mg/50 ml injection £7.60 and paclitaxel 300 mg/50 ml injection £21.00 from the eMit database,153 this implies a total drug cost over six cycles of £272, assuming a mean body surface area of 1.85 m2 and weight of 70 kg.
Next, the estimated total cost of 12 days of administration of chemotherapy (over six cycles) is £2732, given a cost per administration of £228. The cost per administration is calculated as £206 in 2010–11 prices, corresponding to the HRG SB15Z ‘Deliver subsequent elements of a chemotherapy cycle’ in the sheet ‘Chemotherapy delivery: outpatients’ from the NHS reference costs 2010–11,257 inflated over 3 years to 2013–14 prices as described in Adjustments to 2013–14 prices.
Together, the cost of drugs and administration for a course of six cycles is £272 + £2732 = £3005.
Given the estimated split of stages of EC at diagnosis described above and the proportions of patients at each stage who receive chemotherapy, this implies that 18% = (73% × 0%) + (18% × 50%) + (9% × 100%) of patients diagnosed with EC will receive chemotherapy. Therefore, the cost of chemotherapy per patient is 18% × £3005 = £541. In our model, this cost is assumed to occur immediately on diagnosis of EC.
To acknowledge the lack of agreement as to what constitutes best chemotherapy, we perform two sensitivity analyses, in which the total cost of chemotherapy of £3005 is halved and doubled, respectively.
Cost of follow-up management of endometrial cancer
A review of the routine follow-up management of patients after treatment for EC identified 11 studies258–268 from a range of countries, including one study from England263 and one from Scotland,262 covering the period 1977–97. 269 The management was designed to identify recurrence of EC and included clinical examination, smear of the vaginal vault, chest X-ray, ultrasound and CT scans, although the study from England reported only occasional clinical examinations and the study from Scotland only occasional clinical examinations and occasional smear. In our model, we do not include the costs for any such follow-up management, because of the very low cost relative to other costs, such as the cost of hysterectomy, radiotherapy and chemotherapy.
Assessing convergence of the Peninsula Technology Assessment Group model
As the PenTAG model is a patient-level simulation, it reflects patient-level variability (sometimes termed first order or stochastic uncertainty) and heterogeneity. 270,271 The uncertainty due to these factors (sometimes termed variability) is generally not of interest in a national decision-making framework, where there is more interest in the exploration of the parameter uncertainty and perhaps structural uncertainty. Nevertheless, such uncertainty does result from the PenTAG model, so it is necessary to quantify the amount of uncertainty in the decision problem and how this relates to the number of patients simulated using the model.
Given an infinite number of iterations, the results of the model would converge to a stable estimate of cost-effectiveness for all strategies, but time and computer power are limited and it is important to assess the effect of parameter uncertainty on key outcomes of the model.
For each patient group we simulated n patients and calculated the sample means of the costs, life-years and QALYs (all discounted and undiscounted) along with the standard errors of those sample means. The sample means of the disaggregated costs across several cost groups (discounted and undiscounted) were also calculated. This allowed estimation of the incremental effect of a correct diagnosis of LS and acceptance of LS surveillance on costs and benefits, i.e. the incremental effect of moving a patient from the FN group to the TP with surveillance group. It also allowed estimation of the incremental effects of other changes in patient diagnosis.
The overall decision problem is to find which strategy (if any) for diagnosing LS in CRC patients under 50 years of age is cost-effective, so it is important to assess how the uncertainty due to patient variability affects the uncertainty in the decision problem.
The net health benefit (NHB) framework270 is very useful in this context, and Appendix 14 details how the uncertainty in the outcomes for the patient groups is carried through into the uncertainty in the incremental net health benefit (INHB) of each strategy (relative to a reference strategy).
We judged that the PenTAG model had converged when the 95% CI for the INHB of the seemingly optimal strategy versus strategy 1(1) (no testing) did not cross 0 (i.e. we are confident that the optimal strategy would give a positive INHB). We were also able to assess the level of confidence that the seemingly optimal strategy would have a positive INHB versus other strategies which may be employed through the same methodology.
This convergence criterion would adequately ensure that the basic decision problem was answered, but would not necessarily allow for an exploration of parameter uncertainty, which is dealt with in Exploring parameter uncertainty in the Peninsula Technology Assessment Group model.
Checking the Peninsula Technology Assessment Group model for wiring errors
The PenTAG model was checked for wiring errors in the following ways:
-
All model formulae written by one modeller were checked by a different modeller.
-
The reasonableness of outputs given extreme input values was checked. For example, no deaths due to CRC when the mortality rate from CRC was set to zero.
-
Base-case model results were checked for reasonableness using numerous graphs.
-
Model results were checked for reasonableness given numerous univariate sensitivity analyses.
Exploring parameter uncertainty in the Peninsula Technology Assessment Group model
Some parameters in the PenTAG model could be explored without simulating a new set of patients. Simulating a new set of patients is undesirable because of the added time taken for simulation and because different simulated sets of patients would lead to different results unless an extremely large number of simulations was run.
Parameters relating to the diagnostic section of the model are easily explored without simulating a new set of patients.
Cost parameters for the long-term management component of the model can be explored where they are included in the disaggregated costs. For example, the cost of colonoscopies is estimated for each strategy and the effect of changing the cost of an individual colonoscopy can therefore be very simply incorporated into the total cost for each strategy. Some costs are bundled together in disaggregated costs, for example the total cost of colonoscopy complications is estimated for each strategy, but not the total cost of mild bleeding complications requiring admission. We judged that the level of cost disaggregation was appropriate for exploring parameter uncertainty, and hence all explorations of cost parameter uncertainty were conducted without simulating new sets of patients.
Chapter 6 Assessment of cost-effectiveness: results of the cost-effectiveness model
Summary of Peninsula Technology Assessment Group cost-effectiveness results
Here we present a summary of our findings, details of which are provided later in the chapter.
Base-case results
Diagnostic results
The sensitivity and specificity of each strategy are lower for the relatives than they are for the probands except where no testing occurs. As there are more relatives than probands, relatives have a greater weight on the test accuracy for probands and relatives combined, and therefore the overall test accuracies are more similar to those of the relatives.
Diagnostic results for probands
-
The specificity of all strategies for probands is > 98%.
-
Sensitivities are far lower than specificities for all strategies. Sensitivities vary considerably across strategies, from 39% based on AC alone to 78% when all probands are initially offered genetic testing.
-
The sensitivity and specificity of strategies 2 and 3 are virtually identical as the strategies are very similar. The same is true of strategies 4, 5 and 6, though they have higher sensitivities than strategies 2 and 3 (67% compared with 60%).
-
The overall test accuracies of strategies are influenced by the test accuracies of the individual tests within the strategies and the orders of the tests in the strategies.
-
Additional tests in a strategy appear to either improve the overall sensitivity and worsen the specificity, or vice versa.
-
The prevalence of LS in the proband population is assumed to be 8.4%. Strategies using genetic testing diagnose around 5–7% of probands correctly as LS positive (TP). Around 0.1–0.3% are diagnosed incorrectly as LS positive (FP) when tumour-based tests are included in the strategies, increasing to 1.6% for the strategy that uses only genetic testing. In all testing strategies, 90–92% of probands are diagnosed correctly as LS negative; 1.9–3.4% of probands are incorrectly diagnosed as LS negative if genetic testing is used, increasing to 5.1% without gene testing.
Diagnostic results for relatives
-
The test accuracies for relatives are similar to, but slightly lower than, those for probands.
-
The specificity of all strategies for relatives is > 97%.
-
The sensitivity varies considerably across strategies, from 16% based on AC alone to 71% where all probands are initially offered genetic testing.
-
The prevalence of LS in relatives of probands with LS is roughly 44%, resulting in an overall prevalence in the relatives’ population (relatives of all probands) of 3.7%. For all strategies, there is a greater proportion of TNs for relatives than probands because of the lower prevalence of LS in relatives (3.7% vs. 8.4%).
Costs of diagnostic test for probands
-
The total costs per proband of testing comprise the initial test in a strategy (e.g. IHC in strategy 2) and the subsequent tests in a strategy (e.g. genetic tests, genetic counselling/FH) multiplied by the proportion of probands undergoing these tests.
-
The total cost of testing per proband varies across the testing strategies in the range £300–800.
-
The largest component costs are in respect of IHC, MSI and the genetic test M126. The contributions to total costs are far smaller for BRAF, genetic counselling and FH, and PMS2 and MLH1 genetic testing.
-
The most expensive strategy for probands is direct genetic testing and the least expensive are the no testing strategies.
Costs of diagnostic test for relatives
-
The great majority (approximately 92–95%) of probands are diagnosed LS negative, and the relatives of these probands incur no testing costs. Instead, only those relatives of probands who are diagnosed with LS incur testing costs. For these relatives, there are just two component costs: counselling and predictive genetic tests for individual gene mutations. Hence, the average cost of diagnosis per relative of a CRC proband (£5–7) is significantly less than for probands (£300–800).
-
The costs of testing per relative for those relatives with probands who test positive are much higher, at about £80–90.
-
Total costs per annum in England of diagnostic tests for probands and relatives combined are in the range £600,000–1,400,000.
-
The great majority of the costs are in respect of diagnosis of probands, rather than relatives. This is because, even though we assume five relatives per proband, the average cost of diagnosis per relative of a CRC proband (£5–7) is far less than for probands (£300–800).
Life expectancy
-
Life expectancy for probands is similar across the strategies, with females having a higher life expectancy than males. The same is true for relatives.
-
Despite the fact that relatives are assumed to be slightly older than probands when probands are diagnosed with CRC (e.g. mean age of male LS-negative relatives is 44 years compared with 41 years for probands), the life expectancy of relatives is much greater than for probands (approximately 37 vs. 14 years). This is because all probands, but very few relatives, have CRC.
-
The life expectancy of probands without LS is 13.9 years for all strategies, whereas the life expectancy of probands with LS increases from 12.9 years with no testing to 14.5 years for strategy 8 (all probands offered genetic testing), an improvement of 1.6 years.
-
For relatives without LS, life expectancy is 37.5 years in all strategies, and for relatives with LS, life expectancy increases from 34.0 years for no testing to 35.6 years in strategy 8, also an improvement of 1.6 years.
-
For female probands, life expectancy is lowest for patients diagnosed FN (12.9 years) and highest for patients diagnosed TP with surveillance (15.8 years), a difference of 2.9 years. For female relatives, life expectancy is lowest for patients diagnosed FN (34.4 years) and highest for patients diagnosed FP with surveillance (38.7 years), a difference of 4.3 years. The results for males follow a similar pattern.
Costs of treating cancers
-
The long-term costs are substantially greater than the diagnostic costs.
-
Strategies 1(2) (AC only) and 8 (direct genetic testing) have the highest long-term costs. Strategy 8 has a higher cost of colonoscopies. Strategy 1(2) has higher CRC treatment costs.
-
Treatment costs are substantially greater for probands than for relatives because all probands have CRC, and thus incur the substantial costs of treatment for CRC, whereas only some relatives develop CRC.
-
Costs are higher for patients who are LS positive rather than LS negative because of the greater chance of primary and metachronous CRC.
-
Patients diagnosed FN have the highest costs owing to the increased risks of CRC and EC from having LS without any treatment to improve survival.
-
Prevention costs are greater for relatives than for probands.
-
The cost of CRC treatment is far greater than the cost of EC treatment because a much greater proportion of patients are diagnosed with CRC. The cost of CRC prevention is higher than the cost of EC prevention because a greater proportion of the cohort undergo colonoscopies than TAHBSO.
Cost-effectiveness
-
The ICERs of all strategies compared with no testing are < £10,000 per QALY, which is considerably lower than the £20,000-per-QALY threshold routinely used by NICE in England and Wales.
-
As the cost of LS testing increases across testing strategies, the additional costs of CRC and EC prevention are not all recouped by reduced costs of treatment.
-
The main components of the savings in the costs of treatment are in respect of chemotherapy and radiotherapy for primary CRC, chemotherapy and radiotherapy for recurrence CRC, palliative care for CRC and surgery for CRC. Savings in the remaining components of treatment are much smaller: CRC diagnosis, CRC stoma care, CRC follow-up surveillance, TAHBSO on diagnosis of EC, chemotherapy and radiotherapy for EC.
-
Most, but not all, of the additional costs of surveillance for CRC are offset by savings in treatment for CRC. Some of the costs of prophylactic TAHBSO are offset by savings in treatment for EC, though increased costs of prophylactic TAHBSO are far greater than the decreased costs of treatment for EC.
-
The differences in QALYs across testing strategies related to the psychological impact of diagnosis are far smaller than all other sources of QALYs (i.e. life expectancy adjusted for disutility of Dukes’ D CRC and age-related quality of life) because it is assumed that the disutilities due to the psychological impact of testing have an effect only in the very short term (4 months), whereas all other sources of QALYs are accrued over the future life expectancy of probands and relatives.
-
Total QALYs increase with the sensitivity of the testing strategy because as the sensitivity increases, the number of LS-positive people who are treated increases, and such treatment can substantially improve life expectancy.
-
All ICERs versus no testing are similar, varying from £5491 per QALY for strategy 5 to £9571 per QALY for strategy 8.
-
The testing strategies on the efficiency frontier are strategies 1(1), 5, 7 and 8. The remaining strategies are either dominated (less effective and more expensive than at least one other strategy) (strategies 2, 4 and 6) or extended dominated (less effective and more expensive than some combination of two other strategies) (strategies 1(2) and 3). On the efficiency frontier, the ICER of strategy 5 versus no testing is £5491 per QALY. The ICER of strategy 7 versus strategy 5 is £25,106 and therefore marginally greater than NICE’s cost-effectiveness threshold. The ICER of strategy 8 versus strategy 7 is £82,962 per QALY, and therefore far greater than the threshold.
-
At a willingness-to-pay threshold of £20,000 per QALY, strategies 4, 5, 6 and 7 offer the best value for money, and the cost-effectiveness of these strategies is similar. These strategies are predicted to result in an additional 130 discounted QALYs per year in England compared with no testing. This equates to the total discounted QALYs accrued over the lives of approximately five people.
Numbers of colonoscopies, colorectal cancers and endometrial cancers
-
The number of colonoscopies per patient is greater for those testing strategies that diagnose a larger proportion of patients with LS, because these patients are offered enhanced surveillance.
-
For probands with LS, the number of colonoscopies increases from 2.1 given no testing to 5.6 in strategy 8 (direct genetic testing), as a greater proportion of probands with LS are diagnosed as such under genetic testing, and hence a greater proportion receive enhanced surveillance. The number of colonoscopies per relative with LS increases from 0.3 given no testing to 7.0 in strategy 8, for the same reason.
-
The expected total number of colonoscopies performed related to probands aged < 50 years and their relatives in England per year increases from approximately 4200 given no testing to 8600 in strategy 8.
-
Among male relatives and probands, the proportion developing CRC is significantly higher than among their female counterparts and a higher proportion of relatives develop index CRC compared with metachronous CRC in probands.
-
The expected number of new cases of CRC for the entire cohort of 1699 probands (metachronous CRCs) and 8495 relatives (primary and metachronous CRCs), in England per year, is approximately 665 given no testing, reducing to a minimum of 633 in strategy 8 (a difference of 32).
-
Given no testing, 79 metachronous cancers are expected in the proband population and 586 index and metachronous cancers in the relative population. Given direct genetic testing, 72 metachronous CRCs are expected in the probands and 561 index and metachronous CRCs in the relatives.
-
The greater the sensitivity of the test strategy, the lower the probability that probands and relatives develop CRC. For example, the probability of metachronous CRC among probands with LS falls from 40% (no testing) to 35% (direct genetic testing), and the probability of primary and metachronous CRC among relatives with LS falls from 30.2% (no testing) to 24.1% (direct genetic testing).
-
The lifetime risk of EC decreases as more LS-positive relatives and probands are identified by a strategy because these people receive prophylactic TAHBSOs. For example, the probability of EC among probands with LS falls from 11.2% (no testing) to 7.4% (direct genetic testing), and the probability of EC among relatives with LS falls from 23.7% (no testing) to 15.5% (direct genetic testing).
-
The expected annual number of ECs related to probands aged < 50 years and their relatives in England in the no testing strategy is 54, with 46 occurring in female relatives and 8 in female probands. This reduces to a minimum of 35 given direct genetic testing, with 30 in female relatives and 5 in the probands.
Increasing the maximum age of probands
-
In an extension of the scope of our work, we examine the consequences of extending the testing of LS to a wider population. We raise the age limit of proband testing from 50 years in our base case to age 60 years, and then to age 70 years.
-
When the age limit of probands is raised, the prevalence of LS in probands falls from 8.4% at a maximum age of 50 years (base case) to 5.7% at an age limit of 60 years and 3.8% at an age limit of 70 years. This is because the incidence of CRC in the general population rises more rapidly than the incidence of CRC in people with LS.
-
The total annual incidence of cases of CRC in England increases from 1699 at maximum proband age 50 years, to 5018 at maximum age 60 years and 13,900 at maximum age 70 years. The corresponding number of relatives increases from 8495 in the base case to 25,090 when the age limit of the probands is raised to 60 years and 69,500 when the age limit is raised to 70 years.
-
Some other parameters are also changed when the maximum age for probands is increased.
Increasing the maximum age of probands from 50 to 60 years
-
The life expectancy of relatives remains fairly similar to the base case, regardless of LS status, as the relatives remain mostly unaffected by the change in the proband age limit. The life expectancy of probands, however, falls regardless of LS status, owing to the higher mean age of probands and consequently shorter life expectancy.
-
As expected, total costs increase dramatically and approximately in proportion to the increase in the number of probands and relatives. The distribution of these costs across strategies is very similar to those in the base case.
-
Similar to the costs, the overall number of colonoscopies increases as the number of patients increases. But the proportional increase in the number of colonoscopies is lower than the proportional increase in the number of probands and relatives because a smaller proportion of probands are LS positive.
-
The proportion of relatives developing a CRC remains similar to the base case and the proportion of probands reduces because their life expectancy reduces.
-
The lifetime risk of EC remains similar to that in the base case for both probands and relatives.
-
All test strategies are now slightly worse value for money versus no testing compared with the base case. However, the ICERs are all well below the £20,000-per-QALY threshold. The lowest ICER is approximately £7700 per QALY, again for strategy 5.
-
Despite a slight deterioration in cost-effectiveness, the INHB at the population level compared with no testing increases in all strategies except universal genetic testing, compared with the base case. This increase is expected, as the diagnostic testing now benefits a larger group of patients. Strategy 5 again gives the greatest INHB at a willingness to pay of £20,000 per QALY of 193 discounted QALYs for the population of England per year. This compares with 130 discounted QALYs for this strategy in the base-case analysis.
Increasing the maximum age of probands from 50 to 70 years
-
As with the 60-years age limit, we find a marked increase in the incremental diagnostic costs for all strategies compared with no testing. Furthermore, the proportional increase in these costs is greater than the proportional changes in the other costs (CRC prevention and treatment, and EC prevention and treatment). Again, this is because of an increase in the proband population and a reduction in the prevalence of LS. In all genetic testing strategies, the incremental cost of LS diagnosis is now higher than the incremental cost of CRC prevention.
-
As with the age limit of 60 years, the ICERs of each strategy compared with no testing are larger than in the base case. All ICERs are now > £10,000 per QALY gained. Strategy 5 again has the lowest ICER, at £10,800 per QALY, and the ICER for strategy 8, universal genetic testing, is now above the £20,000-per-QALY threshold, which implies that it is no longer cost-effective versus no testing.
-
The INHB compared with no testing for all probands and relatives in England per year increases from the base case for all strategies except strategies 7 and 8. Strategy 5 again gives the maximum INHB, now at approximately 271 discounted QALYs. This compares with 130 discounted QALYs for this strategy in the base-case analysis.
Endometrial cancer excluded
-
In this scenario, we remove EC from the model. First, this allows us to examine the impact of EC on our base-case analysis. Additionally, it could be argued that it would be more appropriate to exclude EC from our base-case analysis, given that we do not model OC. This is because the genetic testing strategies incur substantial costs for prophylactic TAHBSO in our base case, but we do not allow for the associated reduction in incidence of OC.
-
The life expectancies of all patient groups change only very slightly when EC is removed, which shows that there is little mortality from EC.
-
For female relatives there is a slight increase in life expectancy, but most noticeably for those diagnosed TP and FN, as mortality due to EC for females with LS is removed. This pattern is repeated for female probands, though the effect is less pronounced.
-
As expected, total costs are reduced when EC is excluded, as there are no longer costs for the prevention or treatment of EC.
-
Costs for females diagnosed as having LS (TP, FP) are reduced because the substantial costs of prophylactic TAHBSO are avoided. Costs for females with LS (FN, TP) are also reduced given that treatment for EC (TAHBSO, chemotherapy and radiotherapy) is avoided.
-
Given the reduced costs and slight increase in life expectancy, plus no disutility from EC, the costs for all strategies are reduced from the base case and the QALYs increased. The incremental QALY gains compared with no testing are lower for all testing strategies than in the base case, but given the much larger reductions in costs due to the lack of prophylactic TAHBSO, the ICERs compared with no testing are reduced compared with the base case, i.e. all strategies become more cost-effective compared with no testing. This may seem counter-intuitive, as one might expect the cost-effectiveness of testing to improve as more people with LS are correctly identified. In a sense, the result is more a reflection of the cost-effectiveness of treatment for EC. It is important to reiterate that our base-case analysis does not account for the benefit gained from prophylactic TAHBSO in the prevention of OC.
-
The ranking of cost-effectiveness among strategies remains the same, with strategy 5 still giving the largest net health benefit.
BRAF replaced by methylation testing
-
In this scenario BRAF testing is replaced by methylation testing in strategies 3, 5, 6 and 7.
-
Methylation testing was assumed to have sensitivity 93% and specificity 80% (compared with sensitivity 100% and specificity 40% or 69% for BRAF testing following MSI or IHC, respectively).
-
Methylation testing was assumed to cost £166 (compared with £118 for BRAF testing).
-
Sensitivities of strategies 3, 5, 6 and 7 were reduced slightly and specificities were increased marginally.
-
Diagnostic costs of strategies 3, 5, 6 and 7 were reduced slightly because fewer patients received further testing (in particular genetic testing).
-
Long-term costs and QALYs were reduced, which appears to be because fewer patients test positive (with more patients falsely testing negative).
-
The cost-effectiveness of strategies 3, 5, 6 and 7 changed marginally, with the INHB of all four strategies decreasing versus no testing at a threshold of £20,000 per QALY.
-
The scenario analysis suggests that based on current evidence, BRAF testing is more cost-effective than methylation testing, but the difference is small.
Univariate sensitivity analyses
-
Several univariate sensitivity analyses were conducted to investigate the impact of various parameters on the cost-effectiveness results.
-
In the majority of cases, the values used in the sensitivity analyses were not meant to represent plausible alternative values, but were chosen to demonstrate the impact on cost-effectiveness of changing the parameter value.
-
The disutility after prophylactic TAHBSO was the only parameter which was found to result in the ICER of any testing versus no testing exceeding the £20,000-per-QALY threshold, as described below (see Utilities associated with endometrial cancer).
-
Table 95 lists parameters with a minimal impact on cost-effectiveness.
-
Table 96 lists parameters with a moderate impact on cost-effectiveness.
-
Table 97 lists parameters with a substantial impact on cost-effectiveness.
-
The incidence of CRC for individuals with LS has a substantial impact on cost-effectiveness for the values chosen, which are the 95% confidence limits from Bonadona and colleagues (2011),2 which is the study used in our base-case analysis. The confidence limits are wide because individuals with LS become uninformative for incidence estimates once they receive surveillance and surveillance is widespread. In reality, we may have greater confidence in the incidence of CRC for individuals with LS because other studies have been conducted with appropriate methodology and have obtained central estimates within these confidence limits. It was expected that incidence of CRC would have a significant impact on cost-effectiveness because the greater the incidence of CRC, the more potential there is to prevent CRCs and achieve QALY gains. It is notable that all strategies still remain cost-effective at a threshold of £20,000 per QALY.
-
The mean number of relatives identified per proband has a substantial impact on cost-effectiveness, although it is notable that even when we assume that no relatives would be identified, all strategies still remain cost-effective at a threshold of £20,000 per QALY. It was anticipated that cost-effectiveness would improve as more relatives are identified, as there should be a greater potential for QALY gains in relatives with LS without CRC, and as predictive genetic testing is less costly than diagnostic testing. However, we do not find that increasing the number of relatives results in cost savings.
-
In the base-case analysis, the HR for colonoscopy in the prevention of CRC is assumed to be 0.533. We find that the effectiveness of colonoscopy in preventing metachronous CRC has a substantial impact on cost-effectiveness, although it is notable that when no benefit is assumed (HR = 1), all strategies still remain cost-effective at a threshold of £20,000 per QALY. Metachronous CRC results in additional costs for treatment of CRC and reduces life expectancy. Therefore, it is to be expected that reducing the effectiveness of a measure to prevent metachronous CRC would result in a worsening of cost-effectiveness.
-
The cost of colonoscopy has a substantial impact on cost-effectiveness, although it is notable that even when the cost is doubled, all strategies remain cost-effective at a threshold of £20,000 per QALY. As colonoscopy is the principal intervention for reducing CRC incidence, it is not surprising that the cost of colonoscopy has a substantial impact on cost-effectiveness. The base-case estimate of cost of colonoscopy is likely to be accurate, as it is based on a weighted average of 360,000 colonoscopies. In our base case, however, we did reduce the cost of colonoscopy to balance the efficacy of colonoscopy, taken from Jarvinen and colleagues (2000),94 in which colonoscopies were 3 yearly. Reversing this adjustment would scale the cost of colonoscopy by a factor of 1.5, i.e. less than the doubling which is examined in our univariate sensitivity analysis.
-
The disutility from prophylactic TAHBSO has a very significant impact on cost-effectiveness. When a disutility of 0.1, which is an arbitrary value, is assumed, all strategies become dominated by no testing, i.e. testing results in greater costs and reduced QALYs. This is a very important result, and results from 55% of women diagnosed with LS accepting prophylactic TAHBSO and incurring a substantial lifetime disutility. This is the only univariate sensitivity analysis in which any strategy becomes not cost-effective versus no testing. Were such a disutility to be considered reasonable, our model would suggest that prophylactic TAHBSO should not be offered as the health benefits from preventing EC are outweighed by the loss in HRQoL owing to the prophylactic surgery. It is also worth noting that even when no disutility is assumed for prophylactic TAHBSO, increasing the acceptance of prophylactic TAHBSO worsens cost-effectiveness.
-
The length of time for which psychological disutilities are applied has a substantial impact on cost-effectiveness. In the base case, psychological disutilities relating to genetic testing are assumed to apply for 4 months. In our sensitivity analysis we assume they apply for 1 year (i.e. three times as long), which is an arbitrary value. This worsens the cost-effectiveness of all strategies but they all remain cost-effective at a threshold of £20,000 per QALY.
| Parameter | How varied |
|---|---|
| Diagnosis | |
| Relative prevalence of four MMR genes | Published data from Norway184 |
| Adherence of probands to genetic counselling | Non-adherence halved |
| Adherence of probands and relatives to genetic testing | Non-adherence halved |
| Probability that a relative of a LS-positive proband has LS | Alternative source: All Wales Medical Genetics Service (Ian Frayling, 2013, personal communication) |
| Proportion of relatives identified who are first degree | Varied from 35% to 55% |
| Effectiveness | |
| Probability of aggressive surgery for LS | Set to 1 (subtotal colectomy always used instead of segmental resection; proctocolectomy always used instead of anterior resection) |
| Colonoscopy mortality rate | Set to rate for polypectomies |
| Costs | |
| Cost of BRAF tumour testing | Halved and doubled |
| Cost of applying FH | Halved and doubled |
| Cost of genetic counselling | Halved and doubled |
| Cost of predictive genetic testing (for relatives) | Halved and doubled |
| Cost of colonoscopy complications | Halved and doubled |
| Cost of CRC diagnosis | Halved and doubled |
| Cost of CRC surgery | Halved and doubled |
| Cost of CRC stoma care | Halved and doubled |
| Cost of CRC follow-up surveillance | Halved and doubled |
| Cost of CRC recurrence | Halved and doubled |
| Cost of CRC palliative care | Halved and doubled |
| Cost of EC surgery | Halved and doubled |
| Cost of EC radiotherapy | Halved and doubled |
| Cost of EC chemotherapy | Halved and doubled |
| HRQoL | |
| EC disutility | Set to 0.1 (base case 0) |
| Parameter | How varied |
|---|---|
| CRC natural history | |
| HR of LS CRC survival | HR 0.57 HR 1 |
| EC natural history | |
| EC incidence given LS positive | Halved and doubled |
| Diagnosis | |
| Prevalence of LS in probands | Halved and doubled |
| Adherence to genetic counselling and testing (separately for probands and relatives) | Adherence halved 100% adherence |
| Adherence to genetic counselling for relatives | Non-adherence halved |
| Effectiveness | |
| HR for colonoscopy for preventing index CRC | 1 SE above and below base case |
| Acceptance of prophylactic TAHBSO | Set to 20% and 90% |
| Initial adherence to LS surveillance colonoscopy | Adherence halved Non-adherence halved 100% adherence |
| Costs | |
| Cost of IHC tumour testing | Halved and doubled |
| Cost of MSI tumour testing | Halved and doubled |
| Cost of diagnostic genetic testing (for probands) | Halved and doubled |
| Cost of CRC chemotherapy and radiation | Halved and doubled |
| Cost of prophylactic TAHBSO | Halved and doubled |
| HRQoL | |
| CRC disutilities | Based on Ness et al. (1999)200 |
| CRC surgery disutility | Set to 0.1 except for segmental resection (base case 0) |
| Psychological disutilities relating to genetic testing and offering prophylactic TAHBSO | Set to 0 Doubled |
| Parameter | How varied |
|---|---|
| CRC natural history | |
| CRC incidence for individuals with LS | Set to 95% confidence limits from Bonadona et al. (2011)2 |
| Diagnosis | |
| Mean number of relatives per proband | Set to 0 and 12 (base case 5) |
| Effectiveness | |
| HR for colonoscopy for preventing metachronous CRC | Set to 1 (no effectiveness) (0.533 in base case) |
| Costs | |
| Cost of colonoscopy | Halved and doubled |
| HRQoL | |
| Prophylactic TAHBSO disutility | Set to 0.1 (0 in base case) |
| Length of psychological disutility | Set to 1 year (base case 4 months) |
Peninsula Technology Assessment Group cost-effectiveness results
We first present the base-case cost-effectiveness results. In our base case, the maximum age of probands is 50 years, EC is modelled and our best estimates are used for parameters in the model. Next we present the cost-effectiveness results under a number of scenarios, in which we either change parameters or change the model structure from the base case. Finally, we present a number of sensitivity analyses in which one or two parameters are individually varied.
Throughout this section we use diagnostic strategy identifiers as shown in Table 98, and as shown graphically in Chapter 5, Diagnostic strategies for probands (see Figures 4–11).
| Identifier | Test 1 | Test 2 | Test 3 | Test 4 | Test 5 | Test 6 |
|---|---|---|---|---|---|---|
| 1(1) | None Assume not LS |
|||||
| 1(2) | FH Normal → assume not LS Abnormal → assume LS |
|||||
| 2 | IHC Normal → assume not LS Abnormal → proceed to test 2 (determined by acceptance of GT) |
GT MLH1 + MSH2 + MSH6 Mutation found → diagnose LS No mutation found → proceed to test 3 |
FH Normal → assume not LS Abnormal → proceed to test 4 |
GT PMS2 Mutation found → diagnose LS No mutation found → assume LS |
||
| FH Normal → assume not LS Abnormal → assume LS |
||||||
| 3 | IHC Normal → assume not LS MLH1 abnormal → proceed to test 2 (BRAF) Abnormal (except MLH1) → proceed to test 2 (determined by acceptance of GT) |
BRAF BRAF V600E mutation → assume not LS BRAF wild type → proceed to test 3 |
GT MLH1 Mutation found → diagnose LS No mutation found → proceed to test 4 |
FH Normal → assume not LS Abnormal → assume LS |
||
| GT MLH1 + MSH2 + MSH6 Mutation found → diagnose LS No mutation found → proceed to test 3 |
FH Normal → assume not LS Abnormal → proceed to test 4 |
GT PMS2 Mutation found → diagnose LS No mutation found → assume LS |
||||
| FH Normal → assume not LS Abnormal → assume LS |
||||||
| 4 | MSI MSI-L/MSS → assume not LS MSI-H → proceed to test 2 (determined by acceptance of GT) |
GT MLH1 + MSH2 + MSH6 Mutation found → diagnose LS No mutation found → proceed to test 3 |
FH Normal → assume not LS Abnormal → proceed to test 4 |
GT PMS2 Mutation found → diagnose LS No mutation found → assume LS |
||
| FH Normal → assume not LS Abnormal → assume LS |
||||||
| 5 | MSI MSI-L/MSS → assume not LS MSI-H → proceed to test 2 |
BRAF BRAF V600E mutation → assume not LS BRAF wild type → proceed to test 3 (determined by acceptance of GT) |
GT MLH1 + MSH2 + MSH6 Mutation found → diagnose LS No mutation found → proceed to test 4 |
FH Normal → assume not LS Abnormal → proceed to test 5 |
GT PMS2 Mutation found → diagnose LS No mutation found → assume LS |
|
| FH Normal → assume not LS Abnormal → assume LS |
||||||
| 6 | MSI MSI-L/MSS → assume not LS MSI-H → proceed to test 2 |
BRAF BRAF V600E mutation → assume not LS BRAF wild type → perform IHC then proceed to test 3 (determined by acceptance of GT) |
GT MLH1 + MSH2 + MSH6 Mutation found → diagnose LS No mutation found → proceed to test 4 |
FH Normal → assume not LS Abnormal → proceed to test 5 |
GT PMS2 Mutation found → diagnose LS No mutation found → assume LS |
|
| FH Normal → assume not LS Abnormal → assume LS |
||||||
| 7 | IHC Abnormal → proceed to test 2 (GT or FH, determined by acceptance of GT) Normal → proceed to test 2 (MSI) |
GT MLH1 + MSH2 + MSH6 Mutation found → diagnose LS No mutation found → proceed to test 3 |
FH Normal → assume not LS Abnormal → proceed to test 4 |
GT PMS2 Mutation found → diagnose LS No mutation found → assume LS |
||
| FH Normal → assume not LS Abnormal → assume LS |
||||||
| MSI MSI-L/MSS → assume not LS MSI-H → proceed to test 3 |
BRAF BRAF V600E mutation → assume not LS BRAF wild type → perform IHC then proceed to test 4 (determined by acceptance of GT) |
GT MLH1 + MSH2 + MSH6 Mutation found → diagnose LS No mutation found → proceed to test 5 |
FH Normal → assume not LS Abnormal → proceed to test 6 |
GT PMS2 Mutation found → diagnose LS No mutation found → assume LS |
||
| FH Normal → assume not LS Abnormal → assume LS |
||||||
| 8 | GT MLH1 + MSH2 + MSH6 (if accepted) Mutation found → diagnose LS No mutation found → proceed to test 2 |
FH Normal → assume not LS Abnormal → proceed to test 3 |
GT PMS2 Mutation found → diagnose LS No mutation found → assume LS |
|||
| FH (if GT not accepted) Normal → assume not LS Abnormal → assume LS |
Base-case results
Details of the base-case inputs are given in Chapter 5. In summary, the base case models the estimated 1699 probands with newly diagnosed CRC under 50 years old per year in England, and their 8495 relatives between 18 and 75 years old. Both CRC and EC are modelled for proband and relative populations. The base-case results are presented in the following order: short-term diagnostic outcomes, long-term survival outcomes and, finally, cost-effectiveness.
Diagnostic results (base case)
The base-case diagnostic results are shown in Table 99.
| Population | Strategy | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1(1) | 1(2) | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
| Probands, na (%)b | |||||||||
| Total | 1699 (100.0) | 1699 (100.0) | 1699 (100.0) | 1699 (100.0) | 1699 (100.0) | 1699 (100.0) | 1699 (100.0) | 1699 (100.0) | 1699 (100.0) |
| Without LS | 1556 (91.6) | 1556 (91.6) | 1556 (91.6) | 1556 (91.6) | 1556 (91.6) | 1556 (91.6) | 1556 (91.6) | 1556 (91.6) | 1556 (91.6) |
| Correctly diagnosed as not LS | 1556 (91.6) | 1530 (90.1) | 1553 (91.4) | 1555 (91.5) | 1554 (91.5) | 1555 (91.5) | 1555 (91.5) | 1552 (91.3) | 1529 (90.0) |
| Incorrectly diagnosed as LS | 0 (0.0) | 27 (1.6) | 3 (0.2) | 1 (0.1) | 3 (0.2) | 2 (0.1) | 2 (0.1) | 4 (0.2) | 27 (1.6) |
| Declined surveillance | 0 (0.0) | 8 (0.5) | 1 (0.1) | 0 (0.0) | 1 (0.1) | 0 (0.0) | 0 (0.0) | 1 (0.1) | 8 (0.5) |
| Accepted surveillance | 0 (0.0) | 19 (1.1) | 2 (0.1) | 1 (0.1) | 2 (0.1) | 1 (0.1) | 1 (0.1) | 3 (0.2) | 19 (1.1) |
| With LS | 143 (8.4) | 143 (8.4) | 143 (8.4) | 143 (8.4) | 143 (8.4) | 143 (8.4) | 143 (8.4) | 143 (8.4) | 143 (8.4) |
| Incorrectly diagnosed as not LS | 143 (8.4) | 87 (5.1) | 57 (3.4) | 57 (3.4) | 47 (2.8) | 47 (2.8) | 47 (2.8) | 35 (2.1) | 32 (1.9) |
| Correctly diagnosed as LS | 0 (0.0) | 56 (3.3) | 86 (5.1) | 86 (5.1) | 96 (5.7) | 96 (5.7) | 96 (5.7) | 108 (6.4) | 111 (6.5) |
| Declined surveillance | 0 (0.0) | 17 (1.0) | 18 (1.1) | 18 (1.1) | 21 (1.2) | 21 (1.2) | 21 (1.2) | 23 (1.4) | 24 (1.4) |
| Accepted surveillance | 0 (0.0) | 39 (2.3) | 67 (3.9) | 67 (3.9) | 75 (4.4) | 75 (4.4) | 75 (4.4) | 85 (5.0) | 87 (5.1) |
| Relatives, na (%)b | |||||||||
| Total | 8495 (100.0) | 8495 (100.0) | 8495 (100.0) | 8495 (100.0) | 8495 (100.0) | 8495 (100.0) | 8495 (100.0) | 8495 (100.0) | 8495 (100.0) |
| Without LS | 8181 (96.3) | 8181 (96.3) | 8181 (96.3) | 8181 (96.3) | 8181 (96.3) | 8181 (96.3) | 8181 (96.3) | 8181 (96.3) | 8181 (96.3) |
| Correctly diagnosed as not LS | 8181 (96.3) | 8060 (94.9) | 8044 (94.7) | 8048 (94.7) | 8029 (94.5) | 8032 (94.5) | 8032 (94.5) | 8008 (94.3) | 7955 (93.6) |
| Incorrectly diagnosed as LS | 0 (0.0) | 122 (1.4) | 137 (1.6) | 133 (1.6) | 152 (1.8) | 149 (1.8) | 149 (1.8) | 173 (2.0) | 226 (2.7) |
| Declined surveillance | 0 (0.0) | 61 (0.7) | 68 (0.8) | 66 (0.8) | 76 (0.9) | 75 (0.9) | 75 (0.9) | 87 (1.0) | 113 (1.3) |
| Accepted surveillance | 0 (0.0) | 61 (0.7) | 68 (0.8) | 66 (0.8) | 76 (0.9) | 75 (0.9) | 75 (0.9) | 87 (1.0) | 113 (1.3) |
| With LS | 314 (3.7) | 314 (3.7) | 314 (3.7) | 314 (3.7) | 314 (3.7) | 314 (3.7) | 314 (3.7) | 314 (3.7) | 314 (3.7) |
| Incorrectly diagnosed as not LS | 314 (3.7) | 262 (3.1) | 141 (1.7) | 141 (1.7) | 120 (1.4) | 120 (1.4) | 120 (1.4) | 97 (1.1) | 90 (1.1) |
| Correctly diagnosed as LS | 0 (0.0) | 51 (0.6) | 173 (2.0) | 173 (2.0) | 194 (2.3) | 194 (2.3) | 194 (2.3) | 217 (2.6) | 224 (2.6) |
| Declined surveillance | 0 (0.0) | 26 (0.3) | 65 (0.8) | 65 (0.8) | 73 (0.9) | 73 (0.9) | 73 (0.9) | 82 (1.0) | 85 (1.0) |
| Accepted surveillance | 0 (0.0) | 26 (0.3) | 107 (1.3) | 107 (1.3) | 121 (1.4) | 121 (1.4) | 121 (1.4) | 135 (1.6) | 139 (1.6) |
To assess the impact of the testing strategies in the short term, we examined the results each strategy had, firstly on diagnosing the probands, then the relatives and, finally, the combined results. We present the total cost of diagnosis per person and across the expected annual population of probands and relatives in England.
We first present the sensitivities and specificities of the strategies in identifying the probands, shown in Figure 36 and the associated ROC plot in Figure 37. These figures demonstrate that specificity is consistently higher than sensitivity across the strategies. This occurs because the sensitivities of the individual tests are generally lower than the specificities and because conducting further tests on only the proportion of the cohort that test positive (as is the case in most of the strategies) can only lead to an increase in specificity. This phenomenon is discussed in greater detail later in this section. We now attempt to account for the differences in sensitivities and specificities across the strategies.
FIGURE 36.
Sensitivities and specificities of the proband diagnostic strategies (probands only).
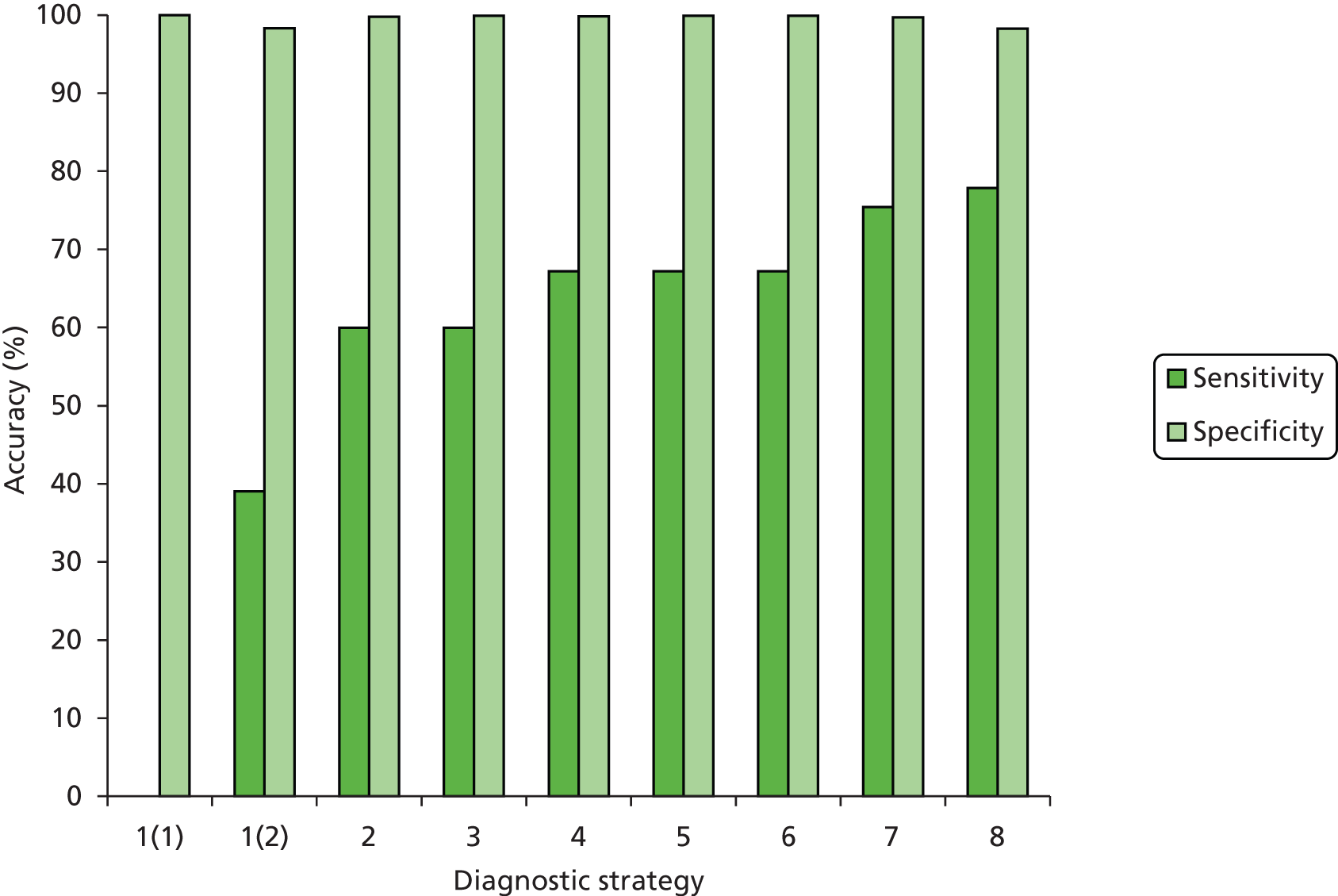
FIGURE 37.
Receiver operating characteristic plot of the diagnostic strategies (probands only).
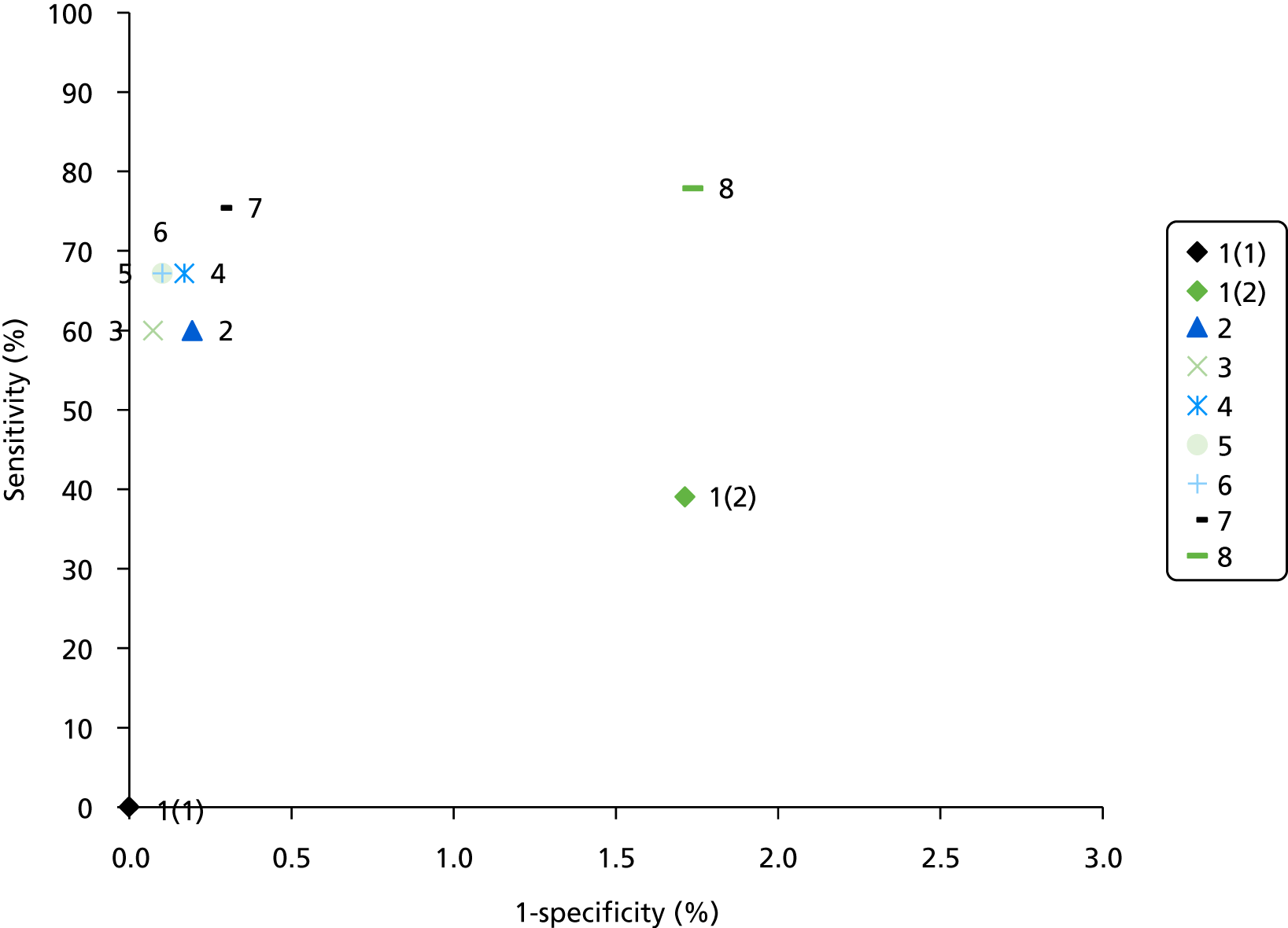
The sensitivity of strategy 1(2) is particularly low as it is based purely on FH, which has a much lower sensitivity than tumour-based tests and gene tests. The sensitivities and specificities of strategies 2 and 3 are virtually identical as the strategies themselves are very similar. Specifically, in both, probands all undertake IHC testing, and further down the testing sequence some are offered gene testing. Strategy 3 separates out the probands who are found to have MLH1 missing on their IHC result and performs a BRAF test before the offer of a gene test. As these represent a small proportion of probands, this causes the two strategies to be very similar in sensitivity and specificity, and the slightly higher specificity of strategy 3 is related to the additional BRAF test. The sensitivities and specificities of strategies 4 and 5 are virtually identical for the same reason; the strategies are fairly similar, with all probands undergoing MSI and the addition of BRAF in strategy 5. Again, the specificity of strategy 5 is slightly higher as a consequence of the additional BRAF test. The sensitivities of strategies 4 and 5 are higher than those of strategies 2 and 3 because the overall sensitivity of the MSI test is higher than the sensitivity of IHC.
These results also demonstrate that the sensitivity of some strategies is lower than that of an individual test in the sequence. In the case of strategy 2, where the overall sensitivity for probands is 60% but the sensitivity for IHC testing is 77%, this occurs mainly because the diagnosis of some probands who decline genetic testing is based upon the FH, which has a much lower sensitivity than IHC. Additionally, when a test which does not have 100% sensitivity is followed by another test which also does not have 100% sensitivity, the overall sensitivity of the sequence will be reduced. This happens for all strategies that include both tumour testing and gene testing, as gene testing does not have 100% sensitivity (90% sensitivity for MLH1, MSH2 and MSH6 mutations, and 60% for PMS2 mutations). The same pattern occurs in strategy 8, where the overall sensitivity is lower than the sensitivity for M126 gene testing. Again, this is partly due to the sensitivity of PMS2 testing being lower than that of M126, and also because approximately 17% of people decline genetic testing and are assessed by their FH, which has a much lower sensitivity (39%).
The accuracy of test strategies is also altered by both the number of tests and the order in which they were conducted. Strategies 3 and 5 demonstrate that a BRAF test, either after an IHC MLH1 abnormal or a MSI result, can improve the specificity of a strategy; however, this improvement is less noticeable when conducted on a MSI cohort with IHC-normal results, as in strategy 7. In general, adding tests to a strategy whose previous test result was indicative of LS increases the specificity of the strategy, as demonstrated by the fact that the specificity of strategy 3 is slightly higher than that of strategy 2, and the specificity of strategy 5 is slightly higher than that of strategy 4; and the result of lower specificity in strategy 8 (which uses only gene testing) compared with all other testing strategies which include not only gene testing but also tumour-based tests.
Strategy 8 has the lowest specificity (98.3%), mostly because it relies on only one type of test (gene testing, plus some decisions based on FH), and partly because a larger proportion of probands decline genetic testing and subsequently undergo FH screening. The probands that decline genetic testing are therefore assessed only by their FH, so that the maximum specificity for them is equal to that of the FH assessment: 98%. The overall effect of adding tests in this manner is to enrich the cohort by eliminating more FPs from further testing. Depending on the characteristics of the additional tests, it may also lessen the sensitivity; but this does not happen in strategies 3 and 5 (compared with strategies 2 and 4, respectively) as BRAF has a sensitivity of 100%, the impact of which was previously discussed.
Additional tests in a strategy appear to either improve the overall sensitivity and worsen the specificity, or vice versa. Additional tests on those probands who test negative by a previous test appear to increase the overall sensitivity, but slightly reduce the specificity of the strategy. For example, strategy 7 has a higher sensitivity than strategy 2 because the IHC-normal probands are not ruled out immediately; instead, further tests (MSI followed by BRAF and gene testing if appropriate) are undertaken. Additionally, strategy 7 has a higher sensitivity than strategy 5 because the use of MSI and BRAF after IHC normal gives a higher sensitivity than MSI and BRAF alone, as the additional TPs identified in the IHC-normal cohort combined with the TPs identified by the IHC-abnormal result mean that a higher proportion of TPs are identified, compared with either IHC or MSI and BRAF alone.
The test accuracies for relatives are similar to those for probands because the approach to diagnosing relatives is the same across all strategies: genetic testing is offered to relatives of probands who test positive for LS; of those relatives who decline testing, FDRs are assumed to have LS if probands are assumed to have LS (so-called ‘LS assumed’), and all other relatives are assumed not to have LS.Differences in test accuracies between probands and relatives across the testing strategies are driven by differences in the proportions of probands diagnosed LS mutation positive, LS assumed and LS negative. In general, the sensitivity and specificity of each strategy are lower for relatives than for probands as, aside from the proportion who undergo genetic testing, the relatives cannot be allocated a diagnosis based on a testing procedure. As such, the methods used are less accurate for relatives than for probands. The exceptions to this are the two no-testing strategies. Strategy 1(1) has the same sensitivity (0%) and specificity (100%) for relatives as for probands, as both assume that no one has LS. Strategy 1(2) has a much lower sensitivity for relatives than for probands, because LS relatives are misdiagnosed as LS negative when the proband is also misdiagnosed as LS negative, and when the higher-degree relatives of probands assumed to have LS are treated as LS negative. The higher-degree relatives of probands assumed to have LS who are treated as LS negative are also related to the slight increase in specificity for relatives in strategy 1(2), as there will be probands in the group who were misdiagnosed as LS assumed, for whom the majority of relatives will be treated as LS negative, therefore lowering the false positivity rate. This occurs because the majority of probands’ relatives are not FDRs; out of the specified five relatives per proband, roughly 2.1 are FDRs. The sensitivities and specificities of the strategies for relatives are shown in Figure 38; Figure 39 presents the associated ROC plot.
FIGURE 38.
Sensitivities and specificities of the proband diagnostic strategies (relatives only).
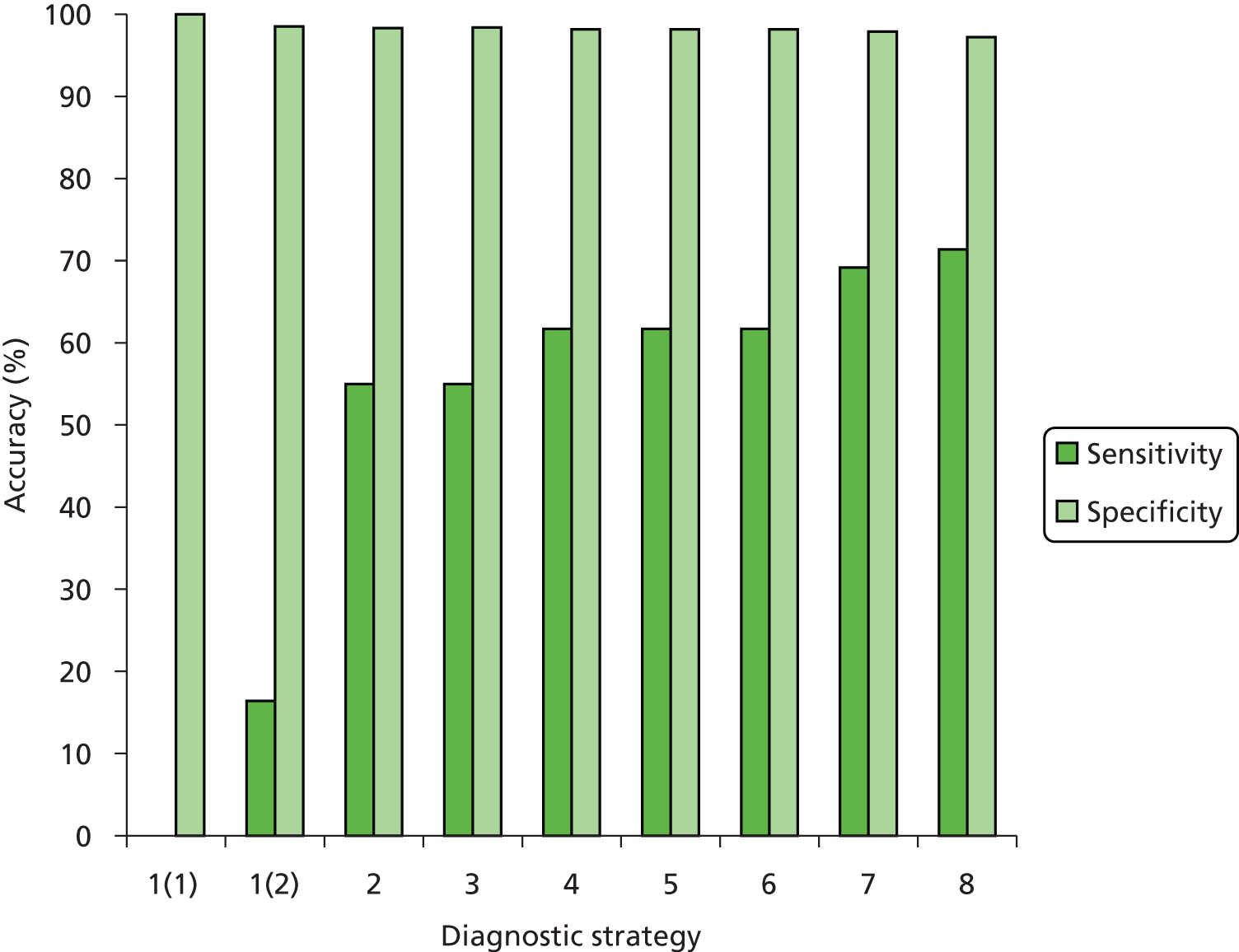
FIGURE 39.
Receiver operating characteristic plot of the diagnostic strategies (relatives only).
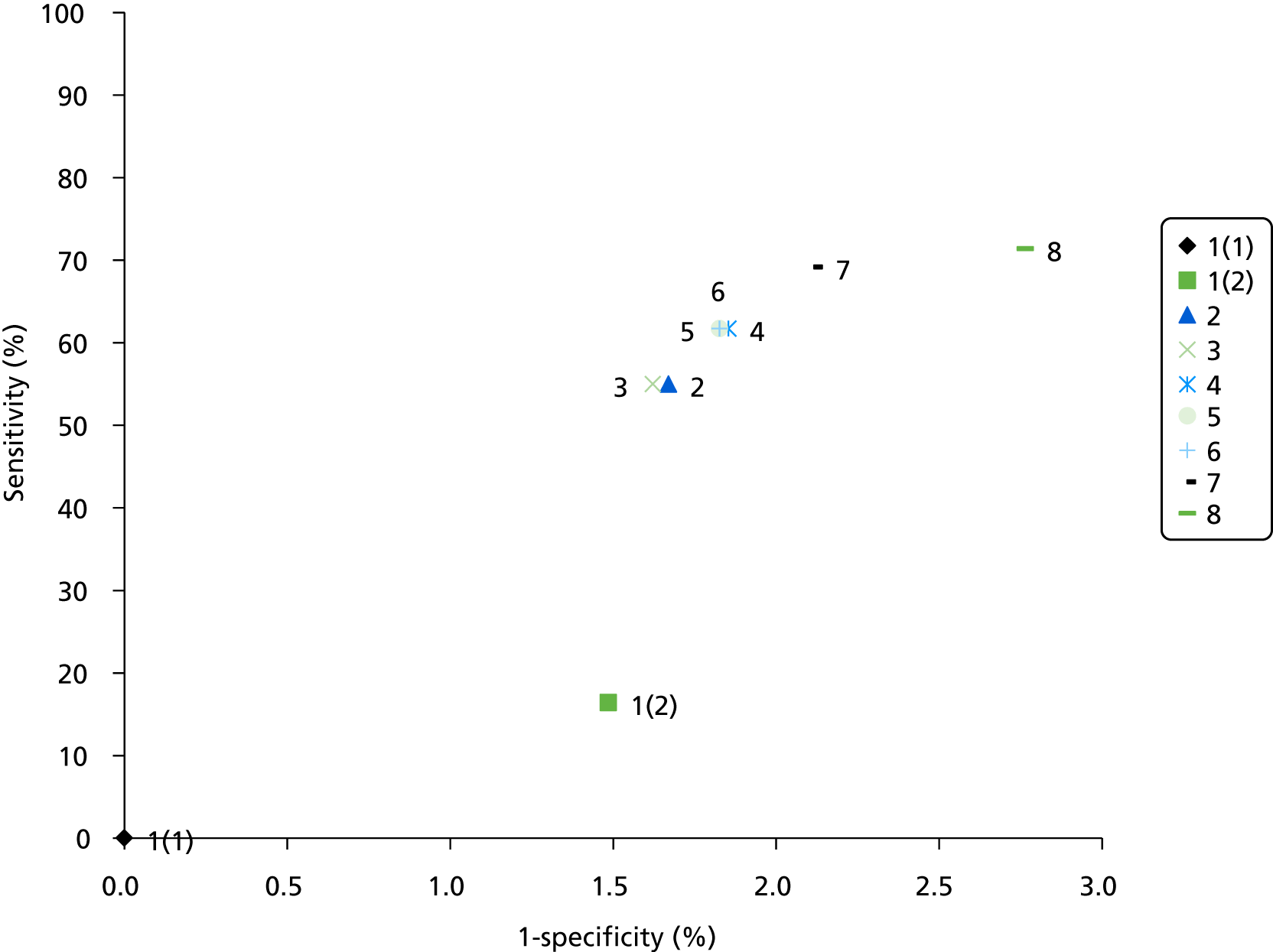
The test accuracy results for all probands and relatives combined are a weighted average of the test accuracies for probands and relatives. The weighting is greater for relatives as there are more relatives than probands (Figure 40). Here, all strategies that identify some patients as LS positive have a specificity of 97% or above, with a sensitivity ranging from 23% to 74%.
FIGURE 40.
Receiver operating characteristic plot of the diagnostic strategies (probands and relatives combined).
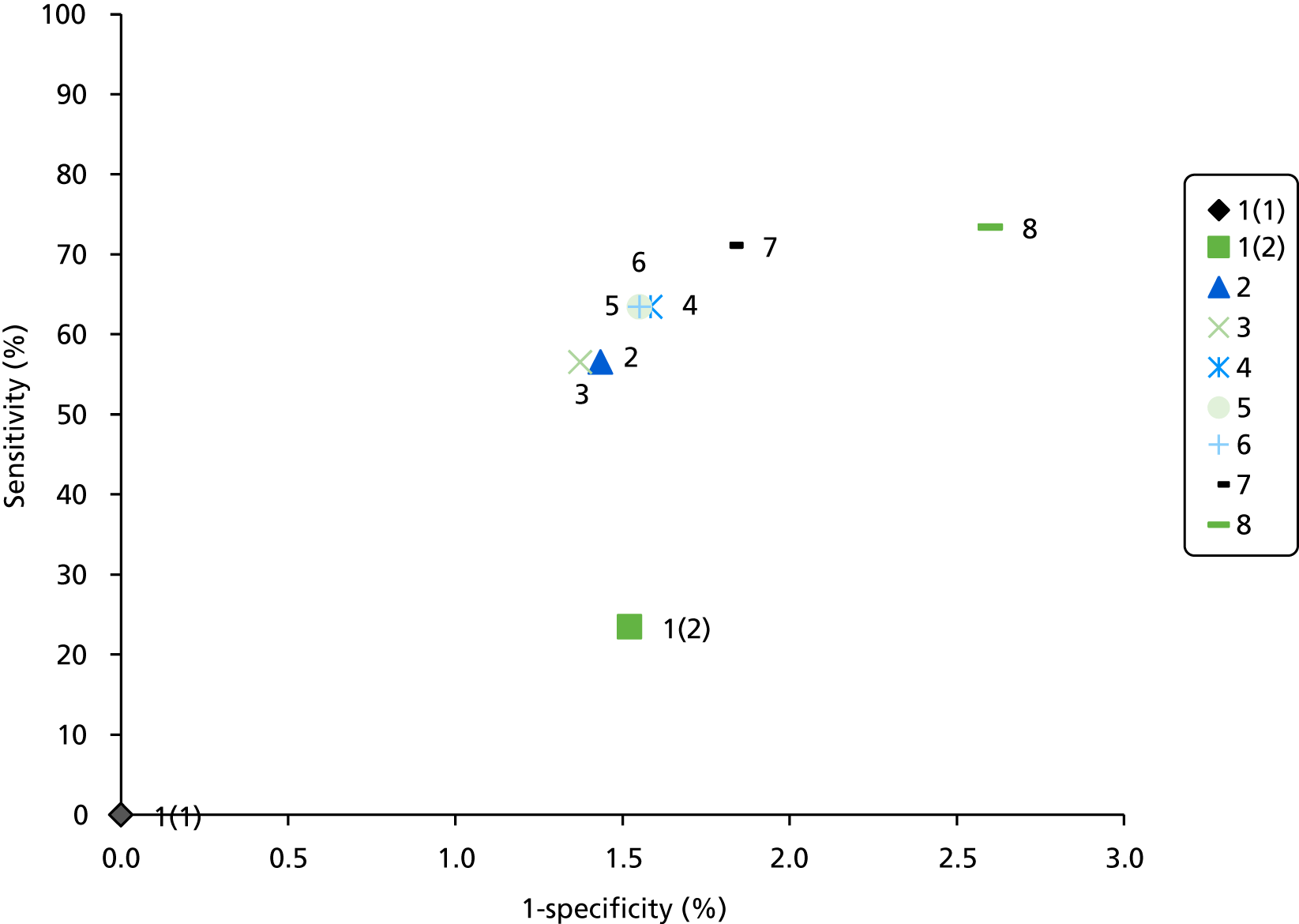
To improve our understanding of the test accuracy results, we now discuss the numbers of TPs, FPs, TNs and FNs for all strategies.
The prevalence of LS in the proband population is assumed to be 8.4%. In general, strategies using genetic testing diagnose around 5–7% of the probands correctly as LS positive (TP), with 79% of these accepting colonoscopic surveillance (Figures 41 and 42). Around 0.1–0.3% are diagnosed incorrectly as LS positive (FP) when tumour-based tests are included in the strategies (with similar levels of acceptance of surveillance), increasing to 1.6% for strategy 8, which uses only genetic testing. In strategies 2–8 where testing is done, 90–92% of probands are diagnosed TN and 1.9–3.4% are diagnosed FN if genetic testing is used, increasing to 5.1% without genetic testing.
FIGURE 41.
Proportions of probands diagnosed TP, FP, TN and FN, by strategy.
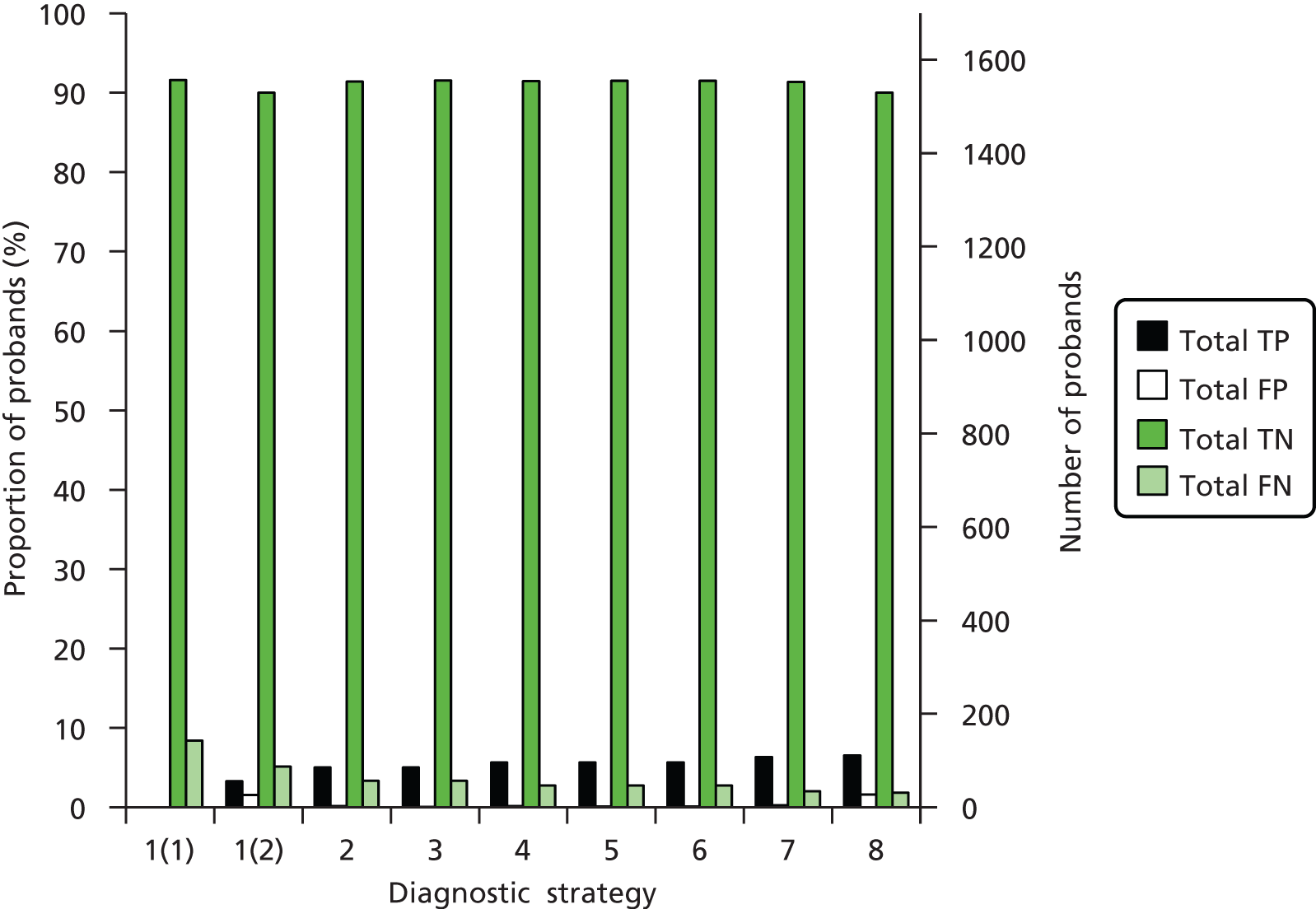
FIGURE 42.
Proportions of probands diagnosed TP, FP, TN and FN, by strategy, rescaled to 10%. Note, total TN exceed bar limits.
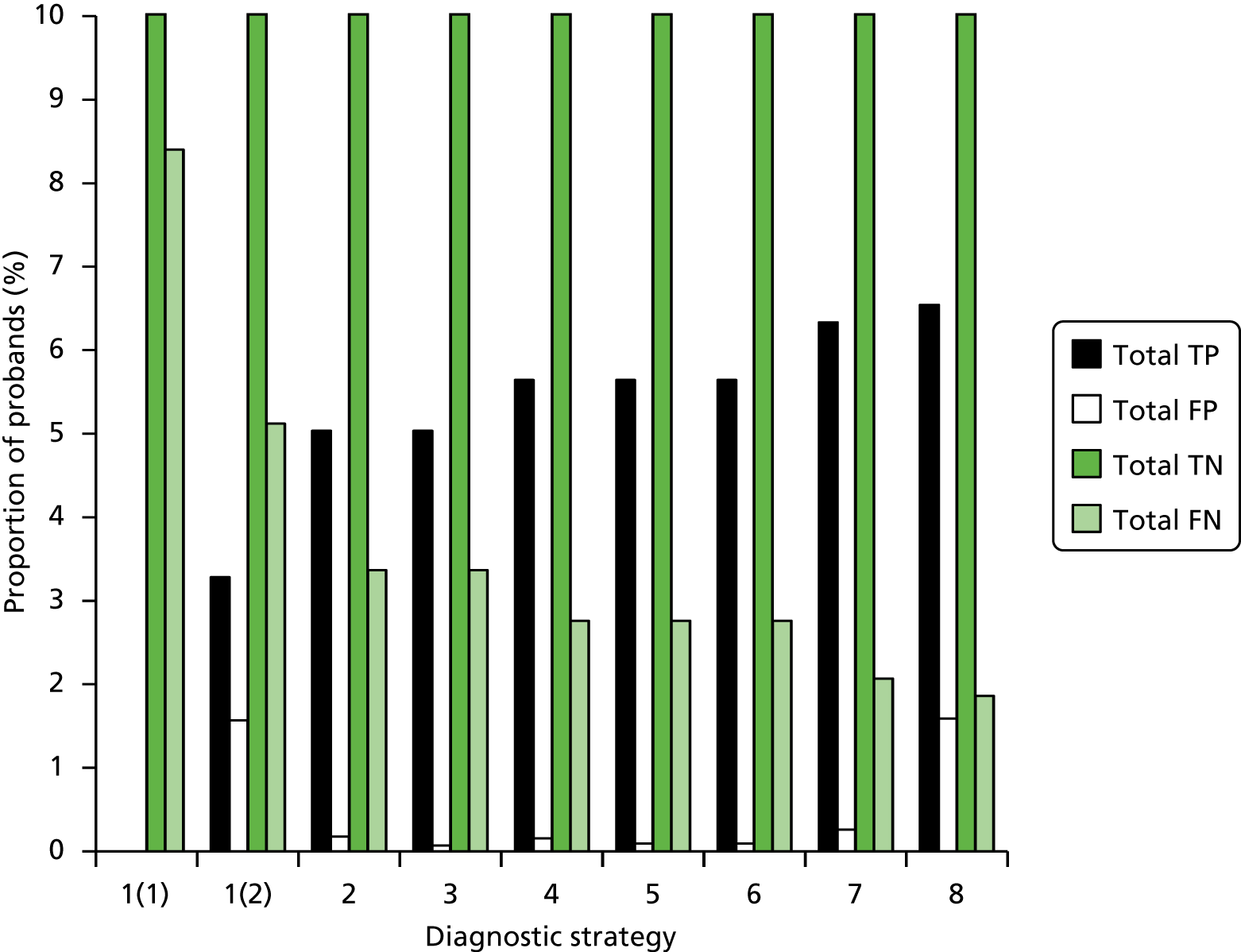
The prevalence of LS in relatives of probands with LS is roughly 44%, resulting in an overall prevalence in the relatives’ population (relatives of all probands) of 3.7%. Strategies that use gene testing identify 2.0–2.6% of relatives as LS positive correctly (1.3–1.6% accepting surveillance), and this drops to 0.6% in strategy 1(2) (0.3% accepting surveillance), which is based on FH assessment of the probands only (Figures 43 and 44). Between 1.4% and 2.7% of relatives are incorrectly diagnosed LS positive, with half of these (0.7–1.3%) accepting unnecessary surveillance. Most probands and their relatives fall into the category of TN. There is a greater proportion of TNs for all strategies for relatives, as shown in Figure 43, than for probands (see Figure 41) because of the lower prevalence of LS in relatives (3.7% vs. 8.4%). Furthermore, any relatives of FP probands will be correctly diagnosed as TN as the predictive genetic test for relatives has specificity 100%. The lower prevalence of LS in the relatives’ population also results in a smaller proportion of TPs for relatives, as well as a smaller proportion of FN relatives than FN probands. There is a greater proportion of FPs for all strategies for relatives compared with probands, as relatives who decline testing are assumed to have LS (over half of whom are actually LS negative).
FIGURE 43.
Proportions of relatives diagnosed TP, FP, TN and FN, by strategy.
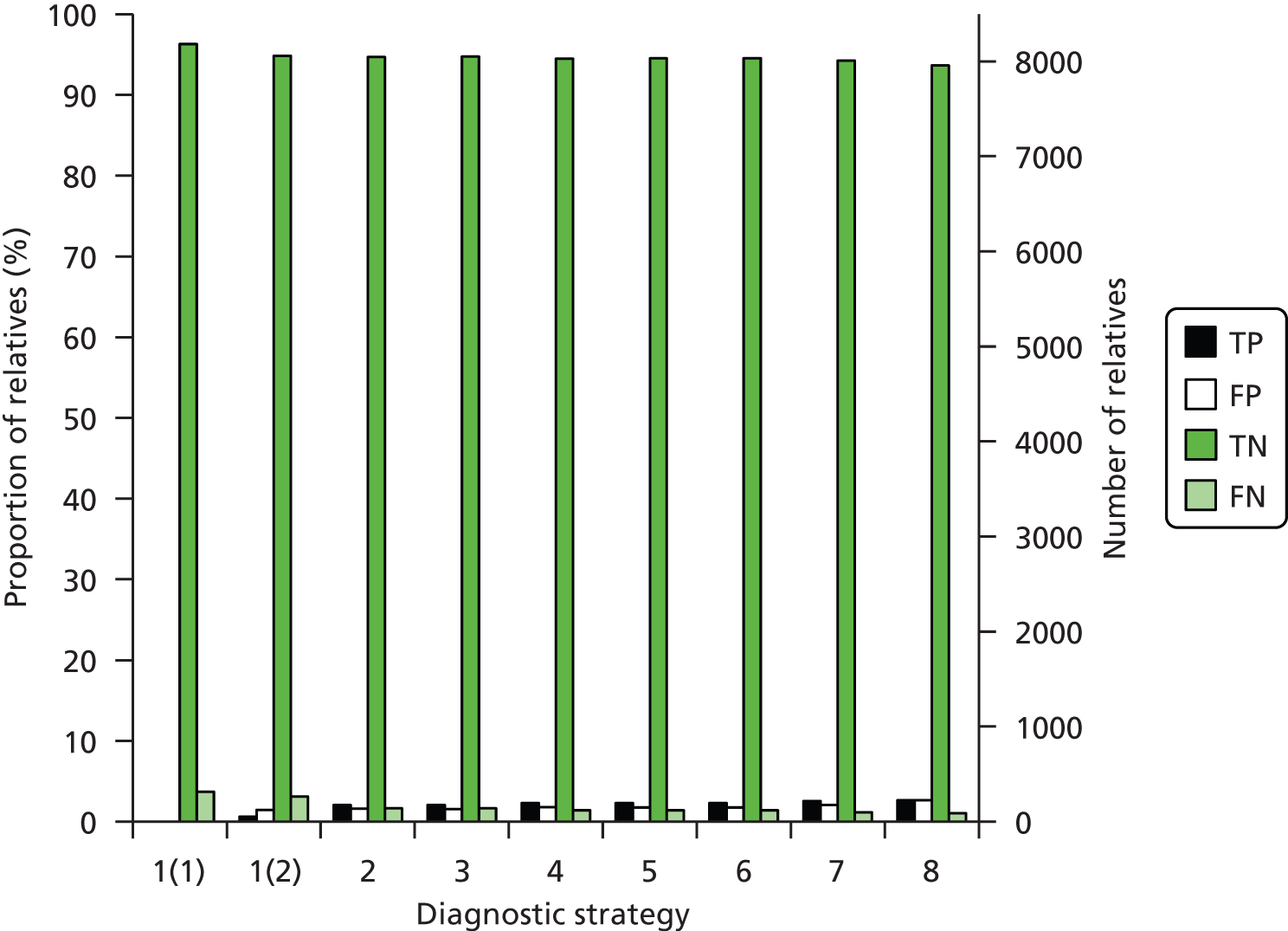
FIGURE 44.
Proportions of relatives diagnosed TP, FP, TN and FN, by strategy, rescaled to 5%. Note, total TN exceed bar limits.
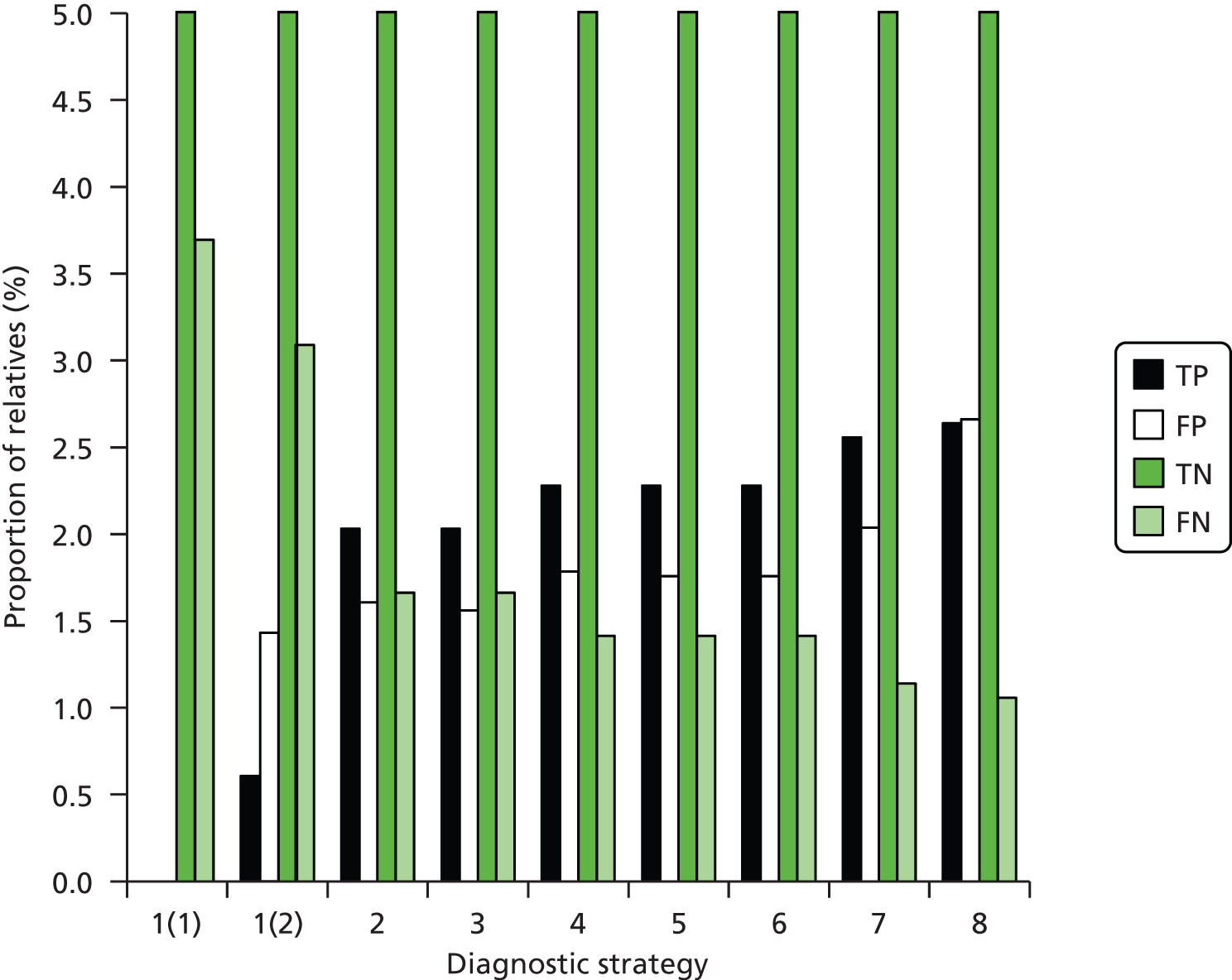
We now investigate the proportions of probands and relatives diagnosed with LS, by test result and true LS status (Figures 45 and 46). Most probands judged to be LS positive are tested mutation positive, with a smaller proportion correctly assumed LS positive. The strategies always identify fewer than the true proportion of probands who are LS positive, even including FPs (see Figure 45). By contrast, most strategies identify more relatives as LS positive than there actually are (see Figure 46). The main driver appears to be the greater proportion of relatives assumed to have LS, mostly incorrectly. The proportion of probands assumed to have LS incorrectly is much smaller as it is based on strict family criteria rules, rather than assumptions as for relatives.
FIGURE 45.
Proportions of probands diagnosed with LS, by test result and true LS status.
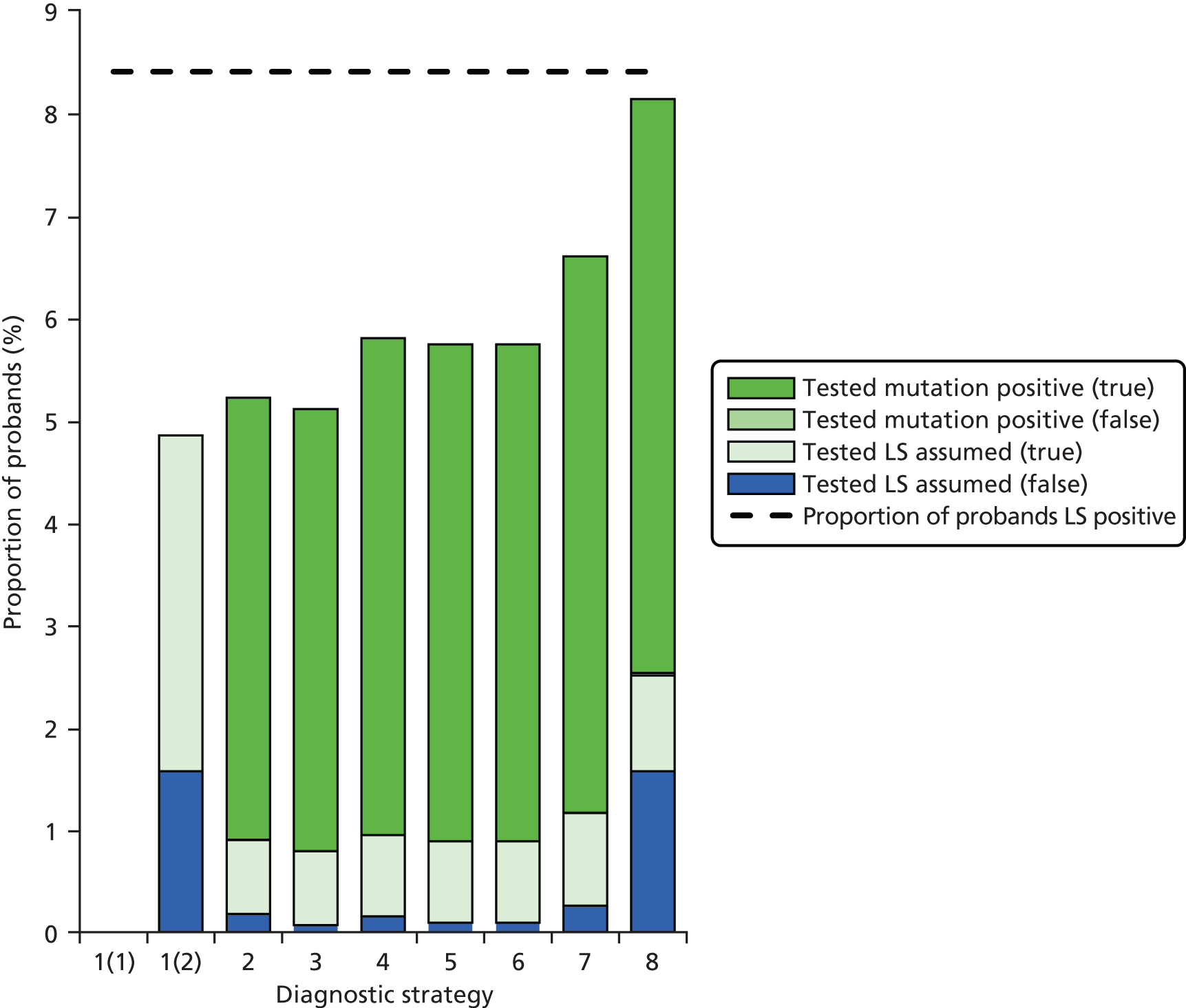
FIGURE 46.
Proportions of relatives diagnosed with LS, by test result and true LS status.
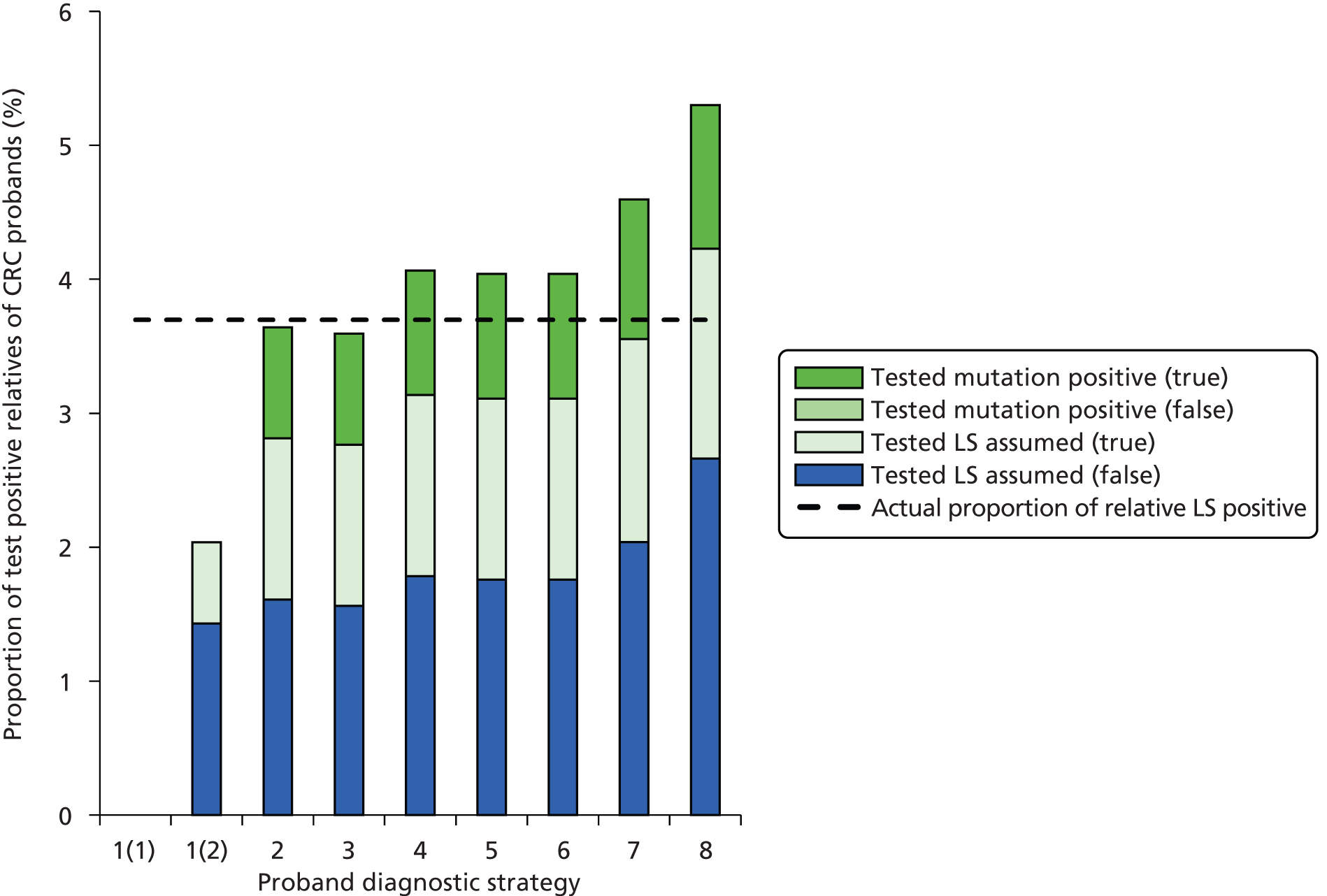
Costs of diagnostic tests (base case)
Here we examine the costs of the diagnostic strategies. The total cost per proband for all strategies is roughly £300–800 (Figure 47). Costs comprise the initial test in a strategy (e.g. IHC in strategy 2) and the subsequent tests in a strategy (e.g. genetic tests, genetic counselling/FH) multiplied by the proportion of probands undergoing these tests. The largest component costs are in respect of IHC, MSI and the genetic test M126. The contributions to total costs are far smaller for BRAF, genetic counselling and FH, and PMS2 and MLH1 genetic testing. We now account for the differences in the costs across strategies.
FIGURE 47.
Total testing cost per CRC proband by strategy.
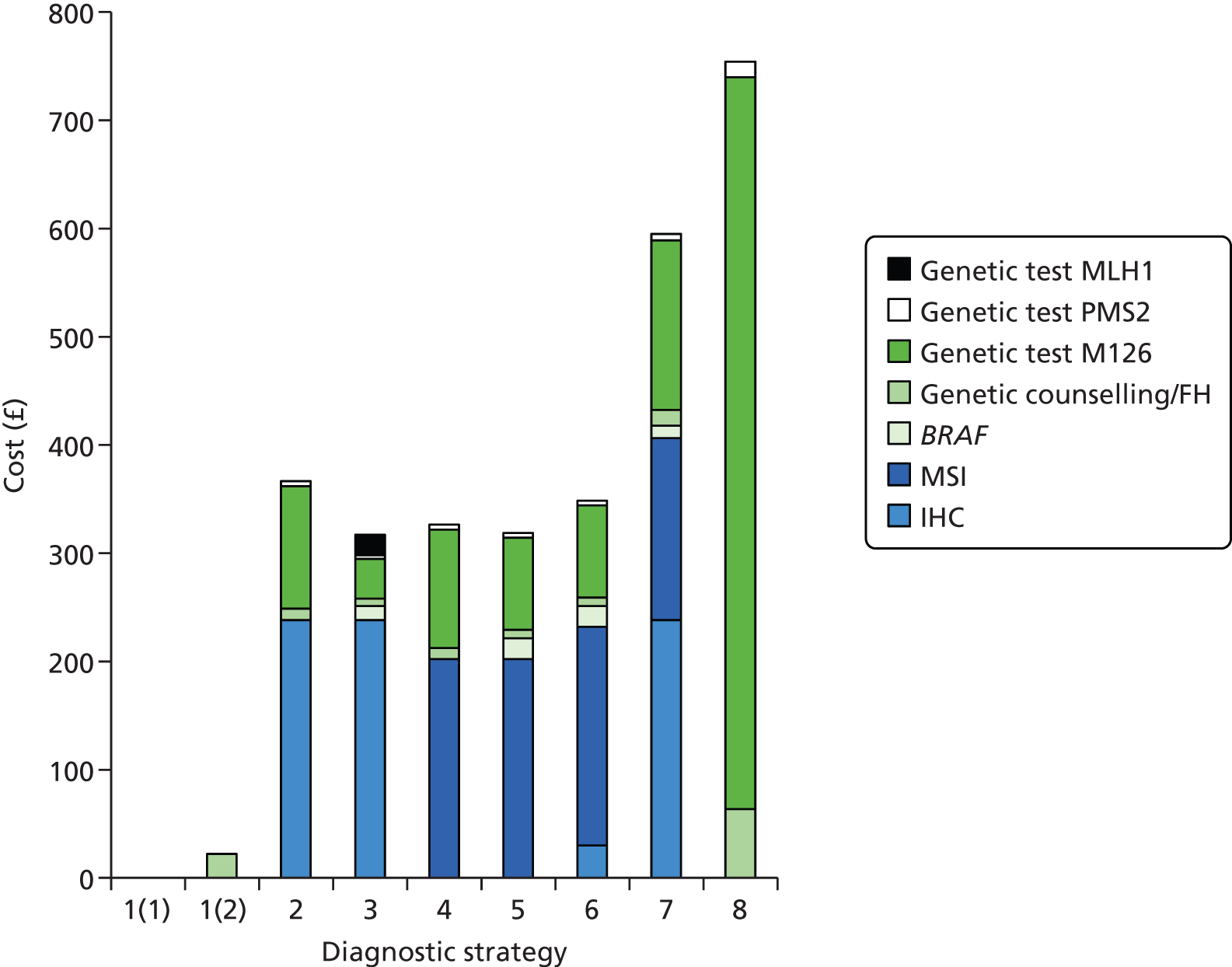
The most expensive strategy for probands is strategy 8 and the least expensive are the no testing strategies. Strategy 8 is particularly expensive because genetic testing is the first test in the strategy, and genetic testing is far more expensive than IHC and MSI testing, both of which are initial tests in other strategies.
There is no set relationship between the number of tumour tests and the cost of the strategy. In strategies with an additional component test (e.g. strategies 3 and 5 additionally include BRAF testing compared with strategies 2 and 4, respectively), the overall strategy cost is lowered, as the additional test reduces the proportion of probands undergoing genetic testing, which is relatively expensive. In strategies where an additional test occurs in a proportion of probands who received a previous test result that was not indicative of LS (e.g. the MSI test in strategy 7 but not in strategy 2), total costs are greatly increased as more of the cohort are exposed to further testing, both tumour based and genetic.
Note that the cost of strategy 6 is the same as that of strategy 5, with the addition of a cost for IHC. This is because IHC in strategy 6 is assumed not to alter the proportion of probands undergoing further tests. This IHC does, however, represent the additional cost if genetics centres request an IHC result in order to understand the results of the genetic tests. Indeed, our expert advisor, Ian Frayling, suggests that the IHC is routinely performed, although the cost of additional tests is generally borne by genetics centres.
The total cost of M126 gene testing is lower in strategy 3 than in strategy 2, because a large proportion of the abnormal IHC results are abnormal for MLH1 and these probands proceed to BRAF and then genetic testing for MLH1, rather than testing for M126.
Despite the high cost of the PMS2 test (£735), the total cost of PMS2 testing per proband is very small in all strategies as very few probands undergo this test. This is because most probands are either ruled out for LS (or have been given a diagnosis for one of the other mutations and are thus ruled out from further testing) before the test for PMS2 occurs; patients who are negative on the M126 genetic test must still have a positive FH before they are offered PMS2 testing.
In strategy 7, the cost of MSI is only slightly lower than in strategy 6 (resource use of MSI is 86% of that in strategy 6), whereas the cost of BRAF is much lower (resource use of MSI is 60% of that in strategy 6). Here the cost of MSI is only slightly lower in strategy 7 because the majority of the cohort (83%) are previously tested as IHC normal and therefore proceed to a MSI test. However, in this new cohort, the prevalence of LS is reduced from 8% to 2%, which means that fewer people test positive after MSI (a reduction from 16% to 10% of the total cohort) and so proceed to BRAF testing. Strategy 7 has a much higher cost of M126 than strategies 1–5. As previously explained, in strategy 7, IHC alone is not used to rule out LS, but probands who do test IHC abnormal proceed directly to M126 testing (if they accept). The remaining probands continue to further testing, being excluded by MSI or BRAF. Therefore, the proportion of patients undergoing M126 is the same as in strategy 2, plus those who are MSI positive and BRAF V600E negative and who accept genetic testing.
The per-proband costs of genetic counselling and FH are lower than testing costs in all strategies because the unit costs are lower.
We now turn to the costs of diagnosis for relatives. First, the great majority (approximately 92–95%) of probands are diagnosed LS negative, and the relatives of these probands incur no testing costs. Instead, only those relatives of probands who are diagnosed with LS incur testing costs. For these relatives, there are just two component costs: counselling and predictive genetic tests for individual gene mutations. Hence, the average cost of diagnosis per relative of a CRC proband (£5–7) (Figure 48) is significantly lower than the cost per proband (£300–800). The cost for relatives appears to be primarily driven by the cost of counselling; there is a greater variation in the cost of counselling across strategies that offer it than in the cost of any single gene test. This is because counselling is given to most relatives of CRC probands who test either LS mutation positive or LS assumed. Therefore, the cost of counselling for relatives in strategies that identify more probands as LS mutation positive or LS assumed will consequently be higher. The cost for relatives is highest in strategy 8 because this has the highest proportion of probands diagnosed LS positive, and therefore the highest proportion of relatives offered testing or counselling.
FIGURE 48.
Total strategy costs per relative of a CRC proband.
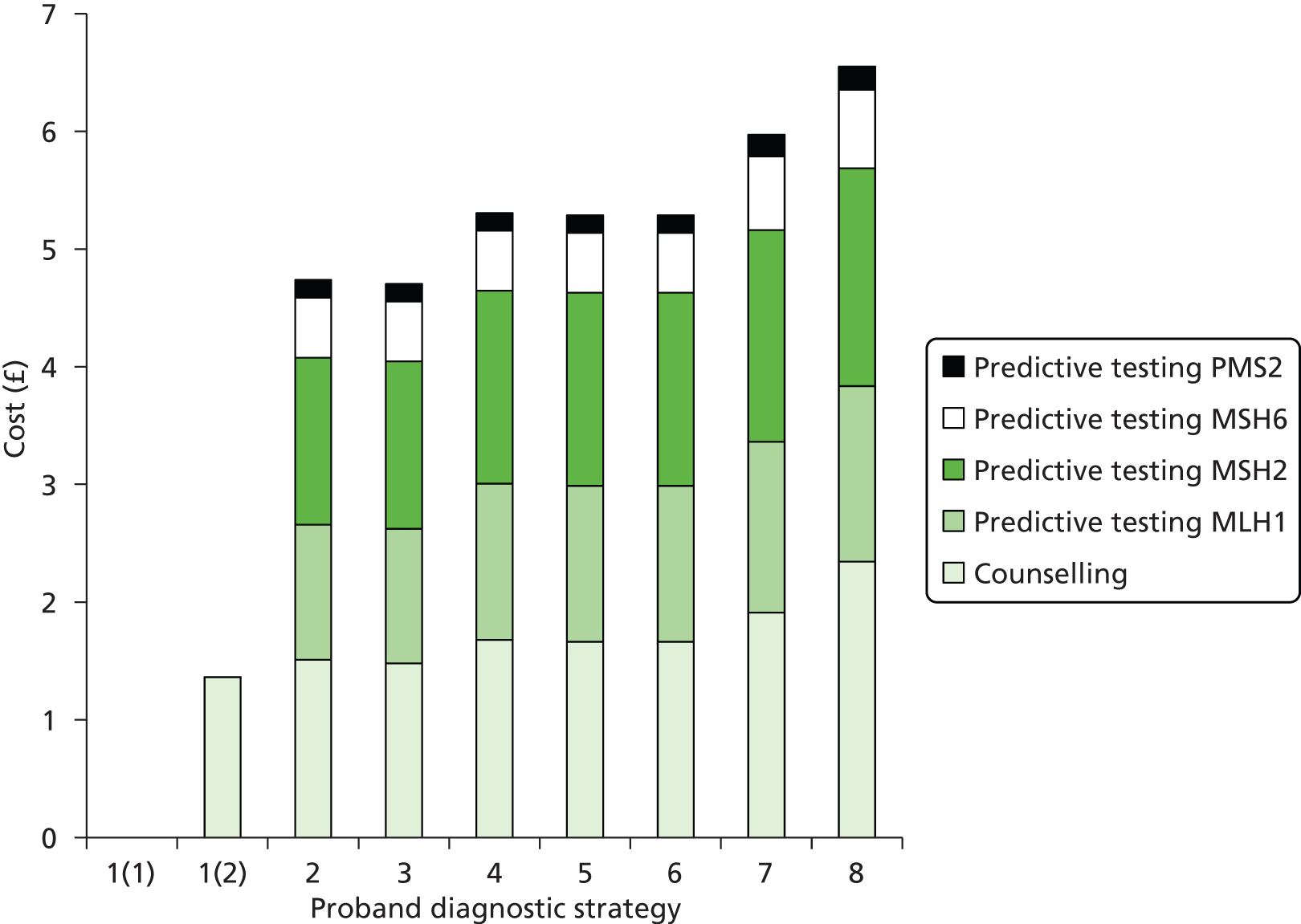
For those relatives with probands who test positive, the costs of testing per relative are much higher, at about £80–90 (Figure 49). The counselling costs are now very similar, as these are no longer affected by differences in the proportions of probands diagnosed LS positive across strategies. The cost per relative of a LS-diagnosed proband is lowest for strategy 8 among the strategies that include genetic testing. This is because strategy 8 has the highest proportion of probands testing LS mutation positive or LS assumed. The larger proportion of LS-assumed probands (in comparison with the mutation positives) compared with the other strategies reduces the cost per relative of a LS-diagnosed proband, as proportionally less genetic testing is undertaken.
FIGURE 49.
Total strategy costs per relative of a LS-diagnosed proband.
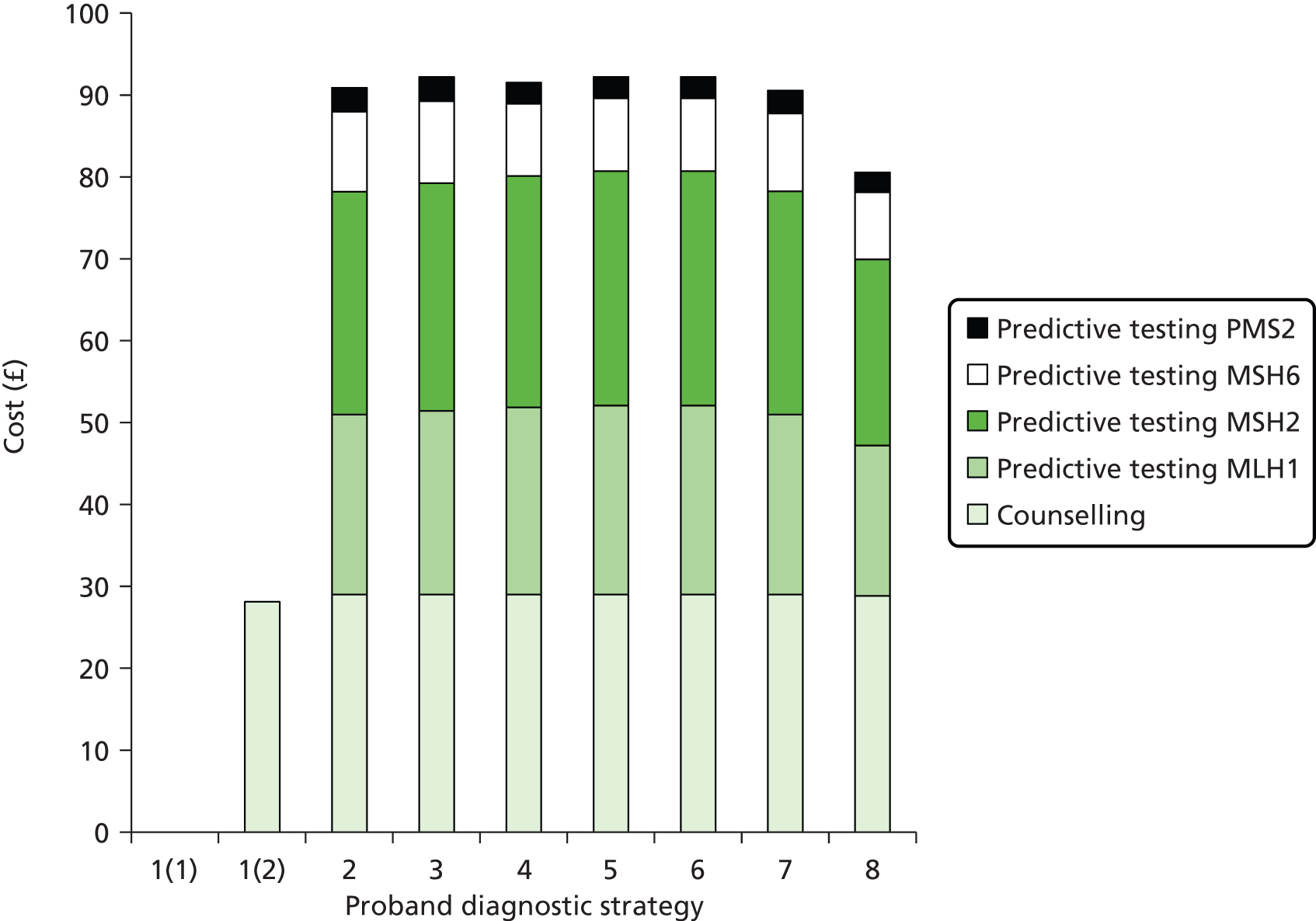
Total costs per annum in England of diagnostic tests for probands and relatives combined are in the range £600,000–1,400,000 (Figure 50). The great majority of the costs are in respect of diagnosis of probands, rather than relatives. This is because, even though we assume five relatives per proband, the average cost of diagnosis per relative of a CRC proband (£5–7) is far lower than that for probands (£300–800). The pattern of differences in total cost across strategies is therefore similar to the pattern for probands alone (see Figure 47).
FIGURE 50.
Total costs per annum in England of diagnostic tests for probands and relatives combined.
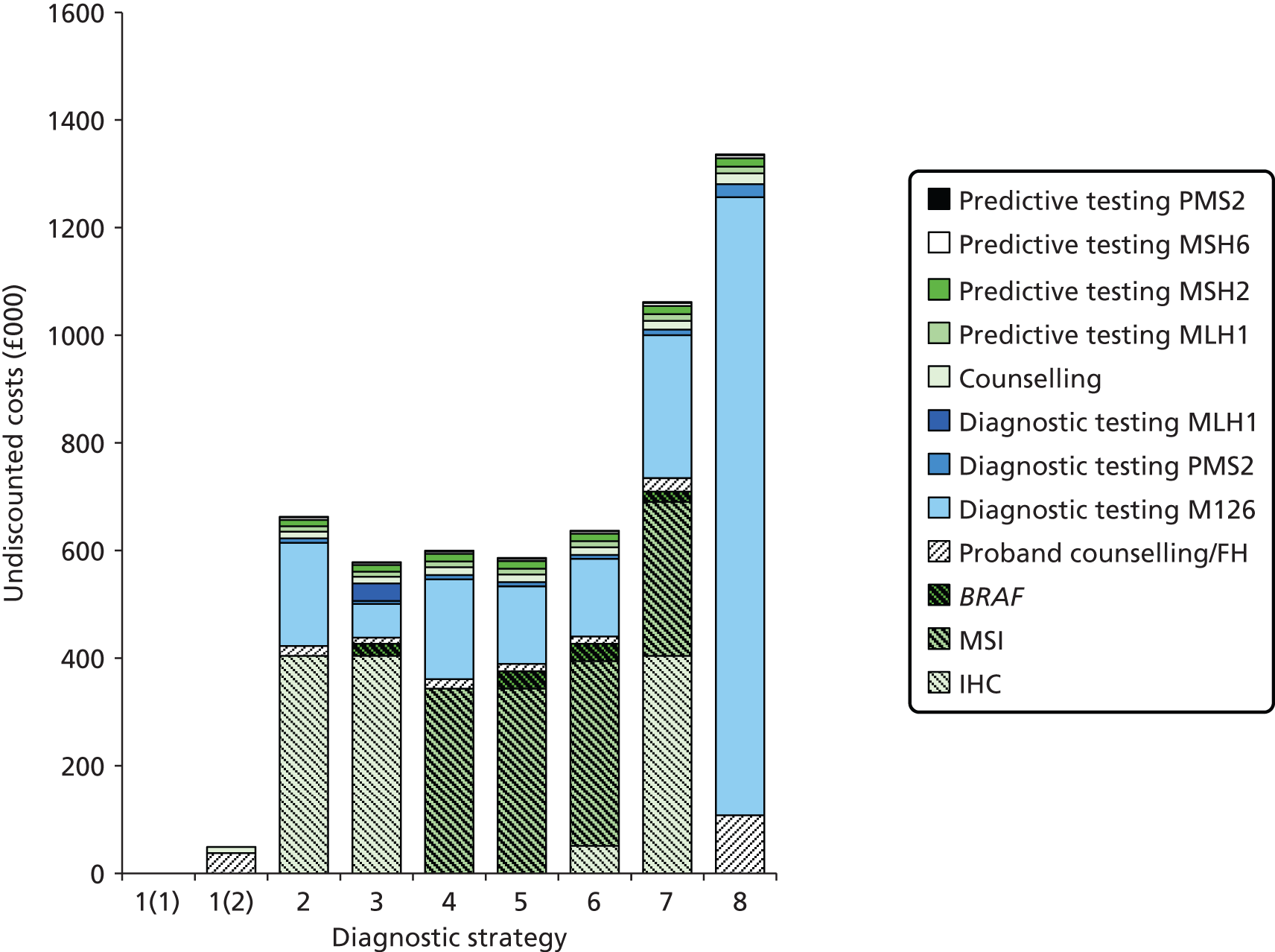
Life expectancy (base case)
Here we present the life expectancies for probands and relatives according to sex, LS status, test result and testing strategy.
Firstly, life expectancy for probands is similar across the strategies, with females having a higher life expectancy than males (Figure 51). The same is true for relatives (Figure 52). This is expected as general population data demonstrate a greater life expectancy for females than males. 179 Despite the fact that relatives are assumed to be slightly older than probands when probands are diagnosed with CRC (e.g. mean age of male LS-negative relatives is 44 years compared with 41 years for probands), the life expectancy of relatives is much greater than that of probands (approximately 37 vs. 14 years). This is because all probands, but very few relatives, have CRC.
FIGURE 51.
Life expectancy of probands by testing strategy.
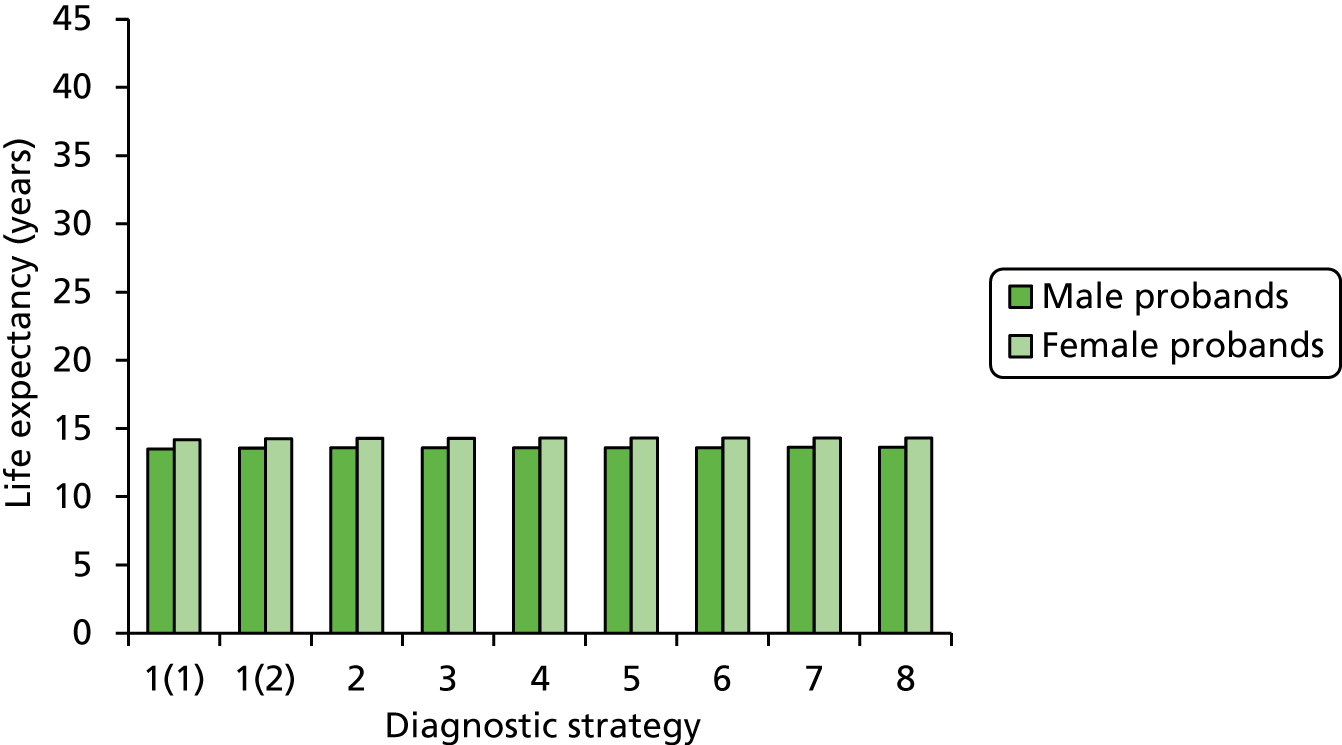
FIGURE 52.
Life expectancy of relatives by testing strategy.
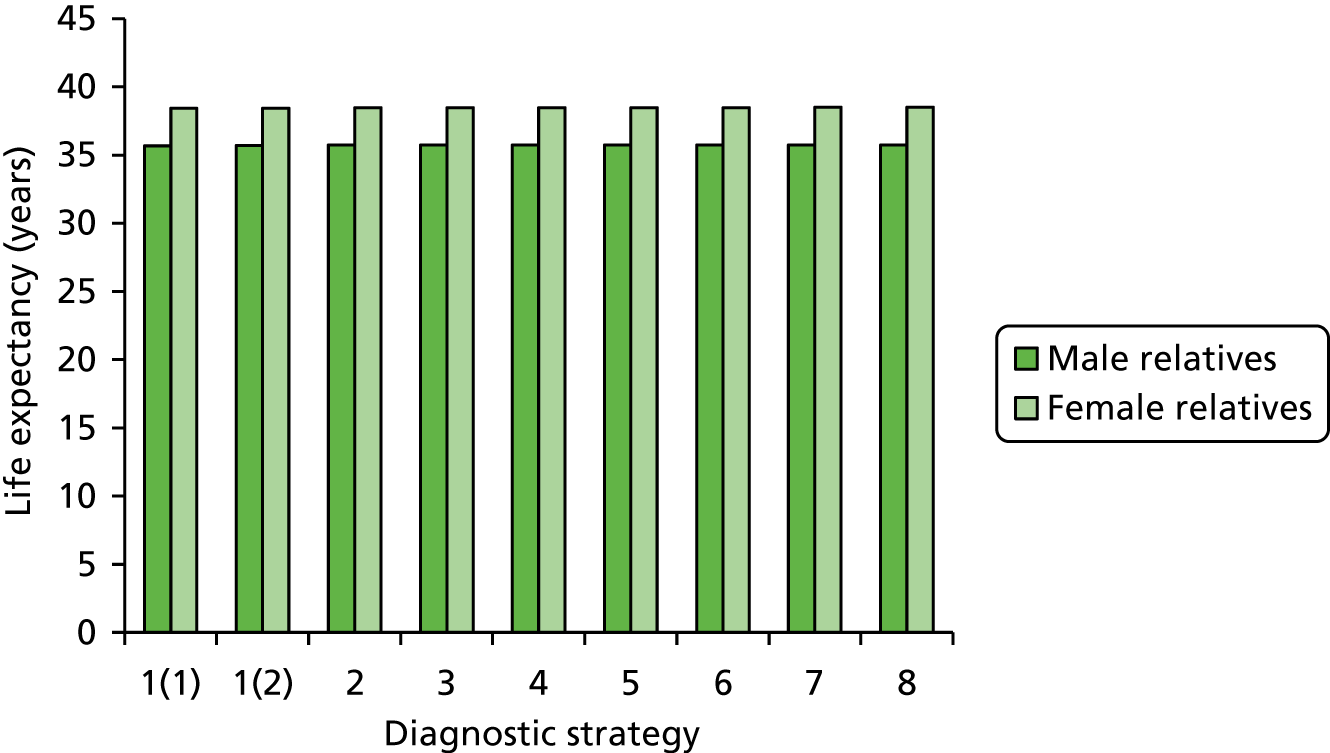
We now consider the impact of testing on people with and without LS. The life expectancy of probands without LS is 13.9 years for all strategies, whereas the life expectancy of probands with LS increases from 12.9 years with no testing to 14.5 years for strategy 8 (all probands offered genetic testing), an improvement of 1.6 years (Figure 53). For relatives without LS, life expectancy is 37.5 years in all strategies, and for relatives with LS, life expectancy increases from 34.0 years for no testing to 35.6 years in strategy 8, also an improvement of 1.6 years (Figure 54). This demonstrates that the impact on life expectancy of testing for LS may be similar for both probands and relatives (this has not been investigated in previous models). Life expectancy for probands and relatives improves under genetic testing as more patients are identified as LS positive and hence proceed to enhanced colonoscopic surveillance and prophylactic TAHBSO. Both of these reduce mortality, as surveillance colonoscopies reduce the risk and stage of CRC, and TAHBSO eliminates the risk of EC.
FIGURE 53.
Life expectancy of probands, by LS status and testing strategy.
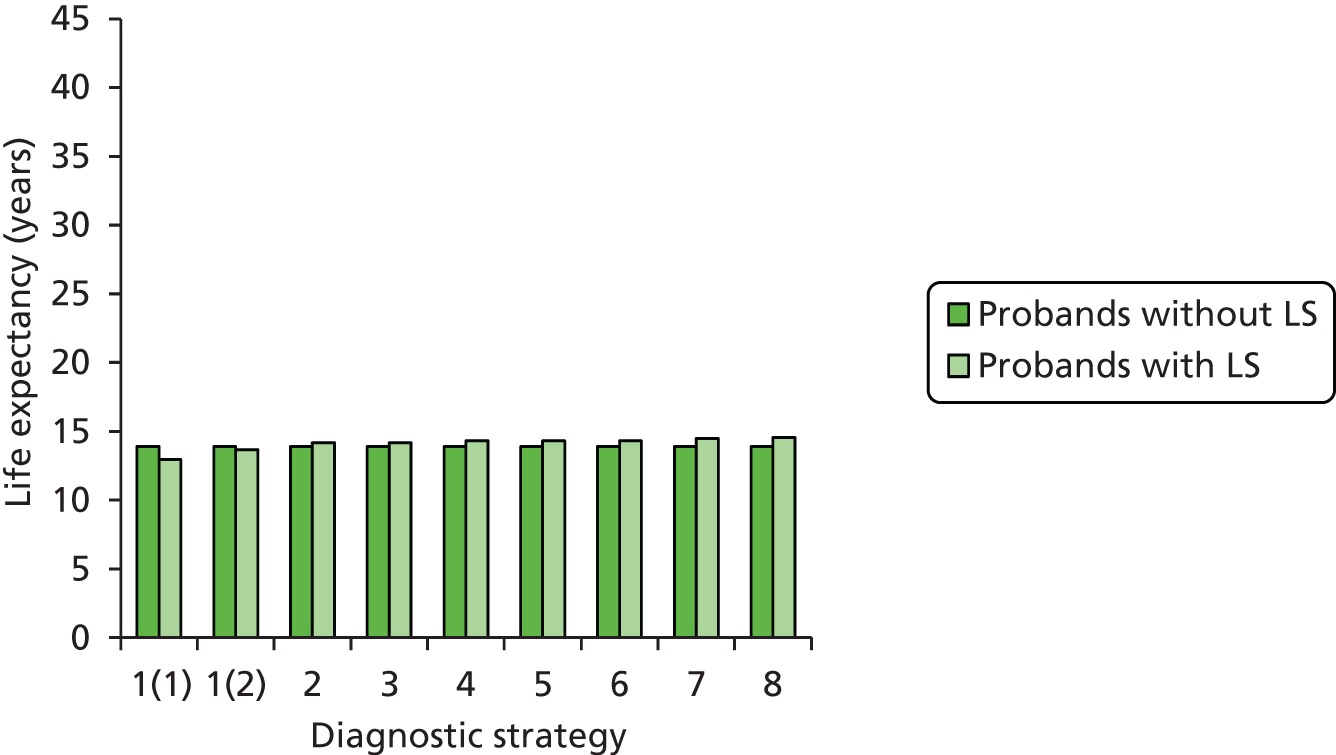
FIGURE 54.
Life expectancy of relatives, by LS status and testing strategy.
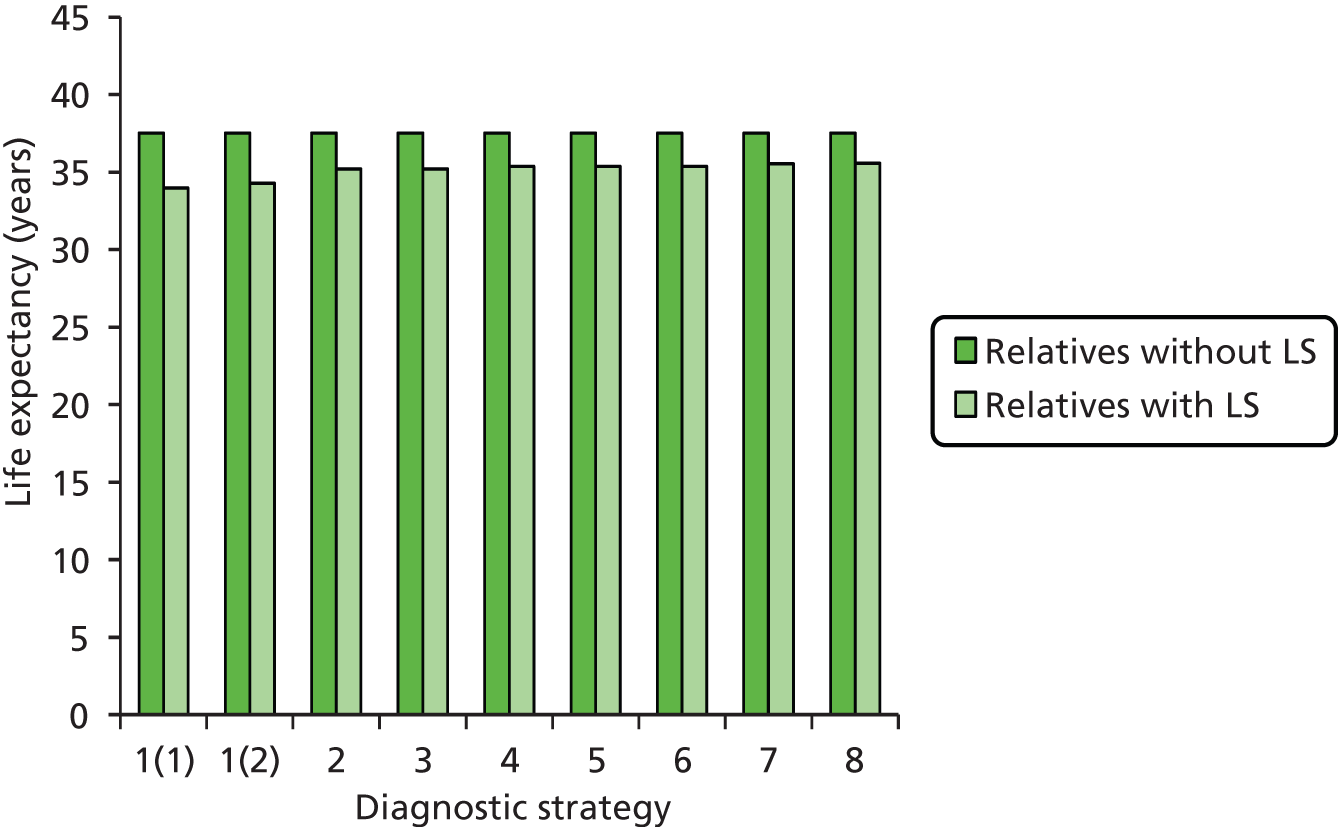
The life expectancies of patients who are TP, FP, FN and TN with/without surveillance help to explain the results above (see Figures 55 and 56).
FIGURE 55.
Life expectancy of male probands by test result and surveillance.
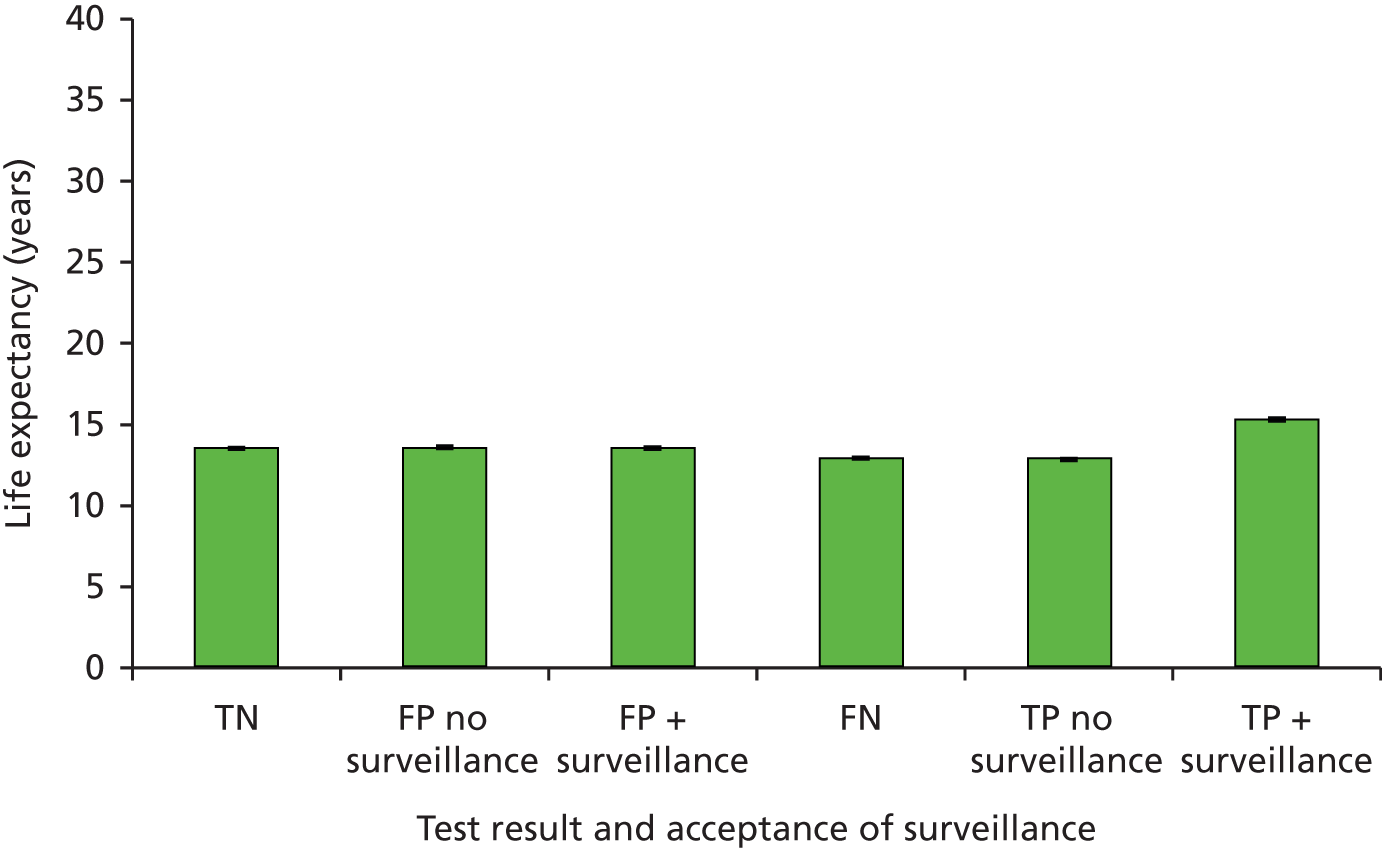
FIGURE 56.
Life expectancy of male relatives by test result and surveillance.
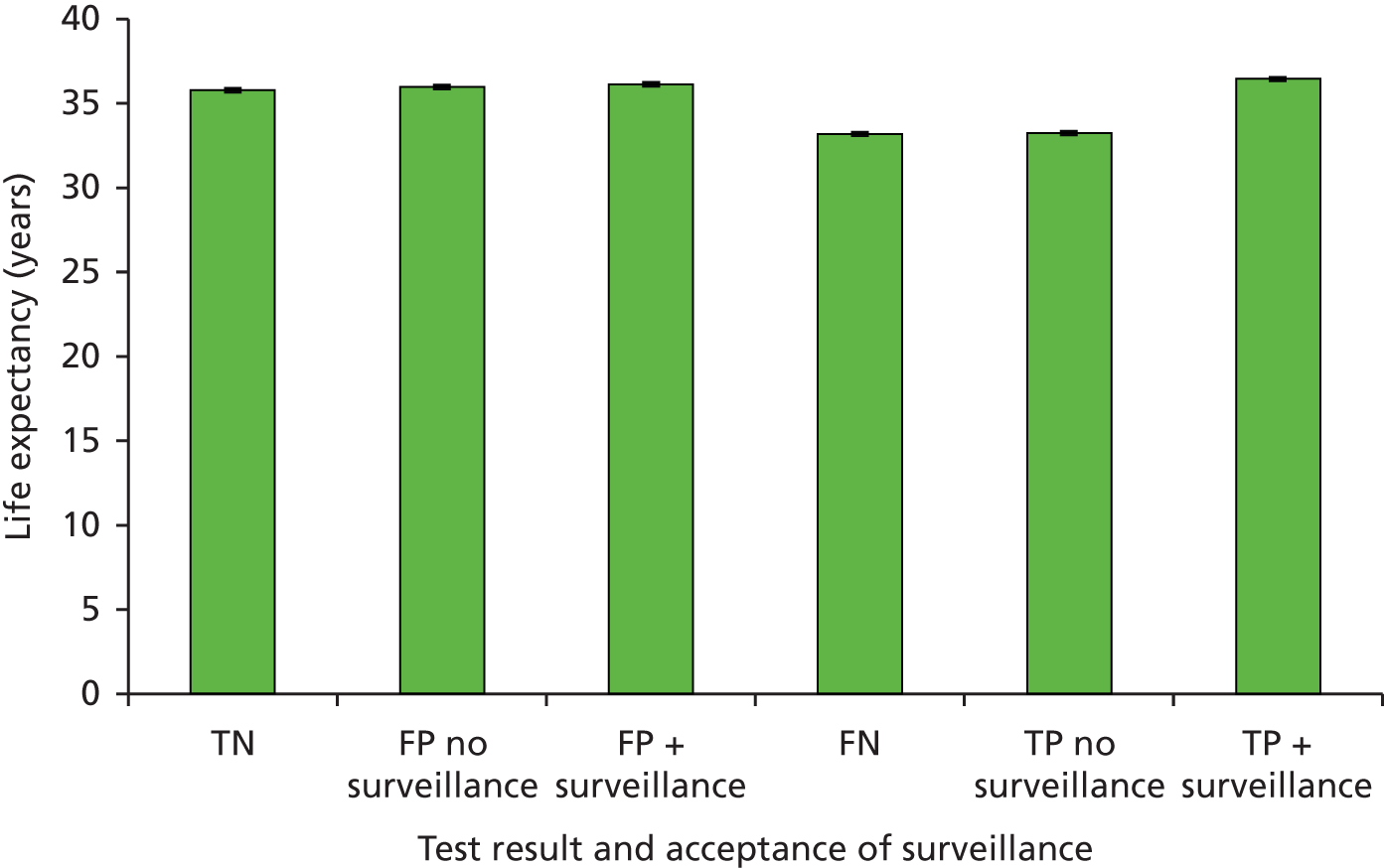
For female probands, life expectancy is lowest for patients diagnosed FN (12.9 years) and highest for patients diagnosed TP with surveillance (15.8 years), a difference of 2.9 years (Figure 57). For female relatives, life expectancy is lowest for patients diagnosed FN (34.4 years) and highest for patients diagnosed FP with surveillance (38.7 years), a difference of 4.3 years (Figure 58). Here we note that females (probands and relatives) benefit from LS diagnosis in reducing both CRC and EC mortality, whereas males benefit from reduced CRC mortality only.
FIGURE 57.
Life expectancy of female probands by test result and surveillance
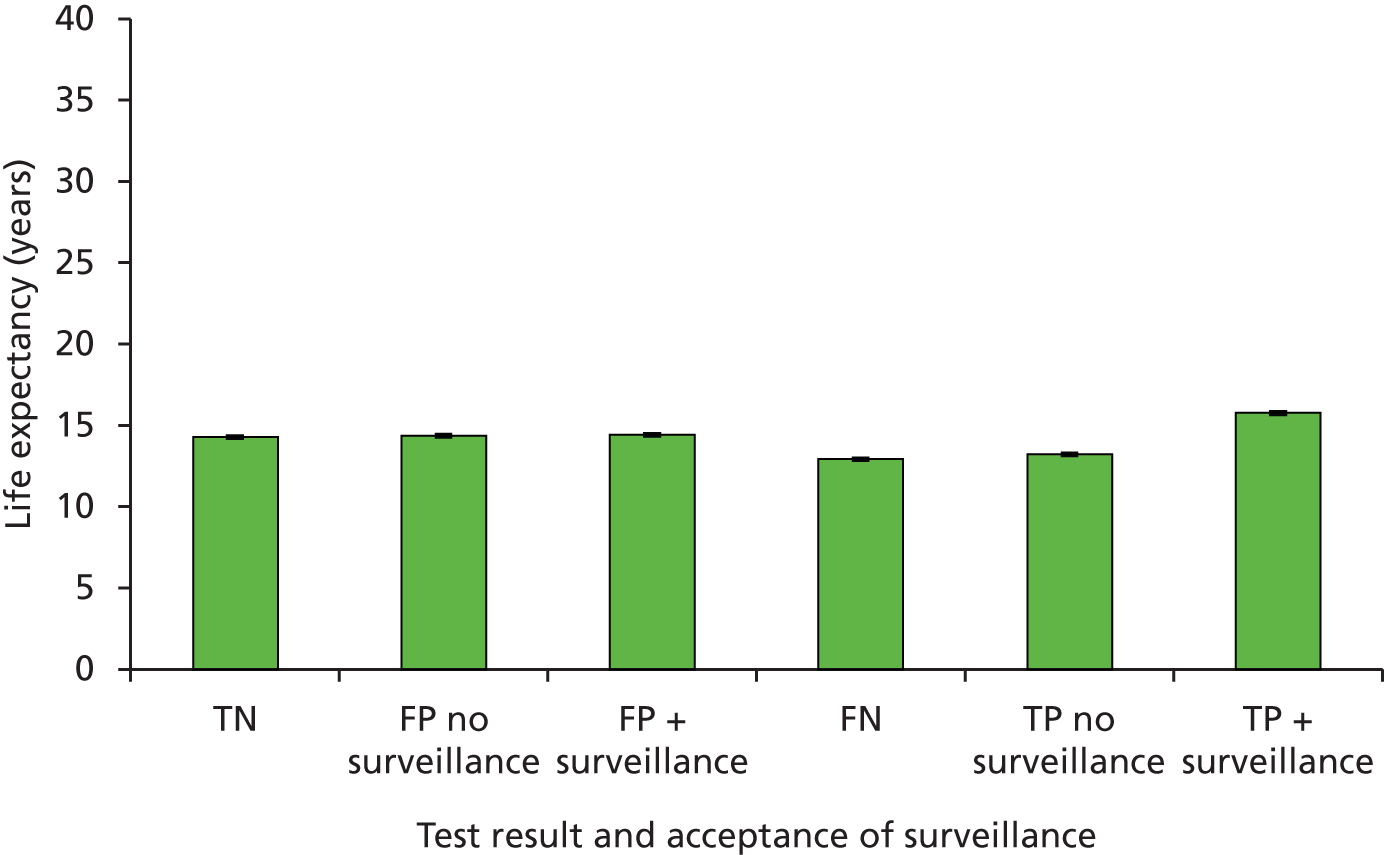
FIGURE 58.
Life expectancy of female relatives by test result and surveillance.
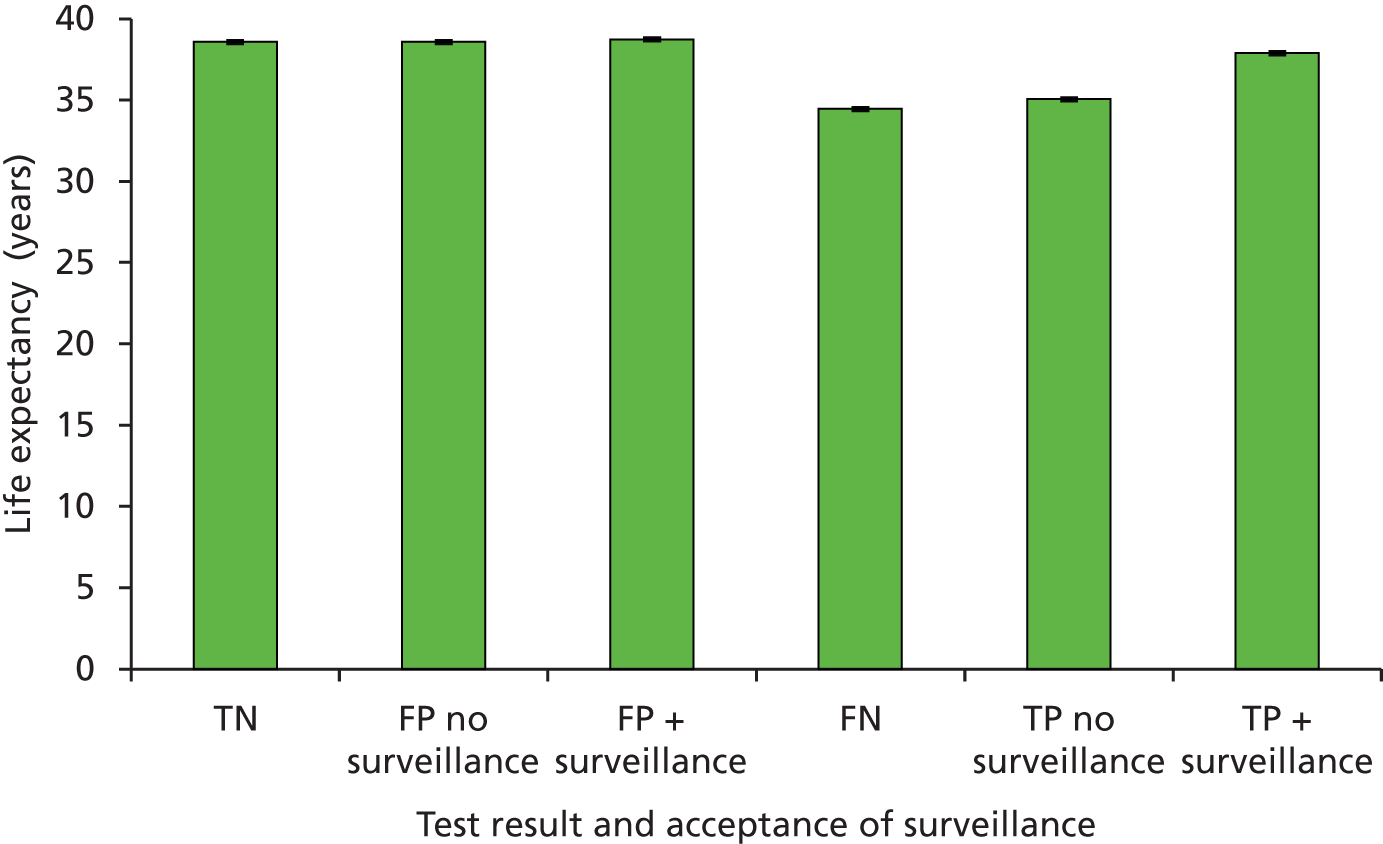
Probands can benefit from LS diagnosis by reducing the incidence of metachronous CRC. As such, TP probands who undergo surveillance have the highest life expectancy, as the surveillance reduces CRC risk (and, in general, the CRC is caught at an earlier stage than in probands not undergoing surveillance who develop a metachronous CRC). They also have a higher life expectancy than FP probands with surveillance, because CRC mortality rates are generally lower for people with LS. For probands and relatives, regardless of sex, life expectancy is lowest for patients who are LS positive and receive no surveillance, i.e. those who are diagnosed FN or TP with no surveillance. Therefore, testing strategies which result in larger numbers in these categories will have lower life expectancies overall.
Life expectancies of relatives who are TN, FP or FP with surveillance and those who are TP are very similar. Relatives benefit from LS diagnosis by avoiding primary CRC. Relatives who undergo surveillance but are FP have a slightly higher survival than TN and FP relatives, as there is some additional benefit from the colonoscopic surveillance. For male relatives, those who are TP with surveillance have the highest life expectancy, as a result of the improved survival from surveillance, plus the lower CRC mortality associated with LS. Given that only female relatives with LS benefit from EC prevention and that all female relatives have a lower risk of CRC than male relatives, the female relatives with LS who undergo surveillance (TP with surveillance) have a lower life expectancy than the TN, FP and FP-with-surveillance female relatives.
The small 95% error bars on Figures 55–58 demonstrate that the results are not subject to significant Monte Carlo variation.
Cost results (base case)
We have previously presented the costs of diagnostic testing [see Costs of diagnostic tests (base case)]. Total diagnostic costs are far smaller than total long-term costs of treating CRC and EC across all strategies (Figure 59). However, as we will explain below (see Incremental costs), the differences in diagnostic costs between strategies constitute a substantial proportion of the differences in total costs, and hence strongly influence cost-effectiveness. Discounting does not affect diagnostic costs, as they are assumed to be incurred at time zero, but reduces total costs by about 33%, as it reduces long-term costs (Figure 60).
FIGURE 59.
Undiscounted long-term and diagnostic costs by test strategy.
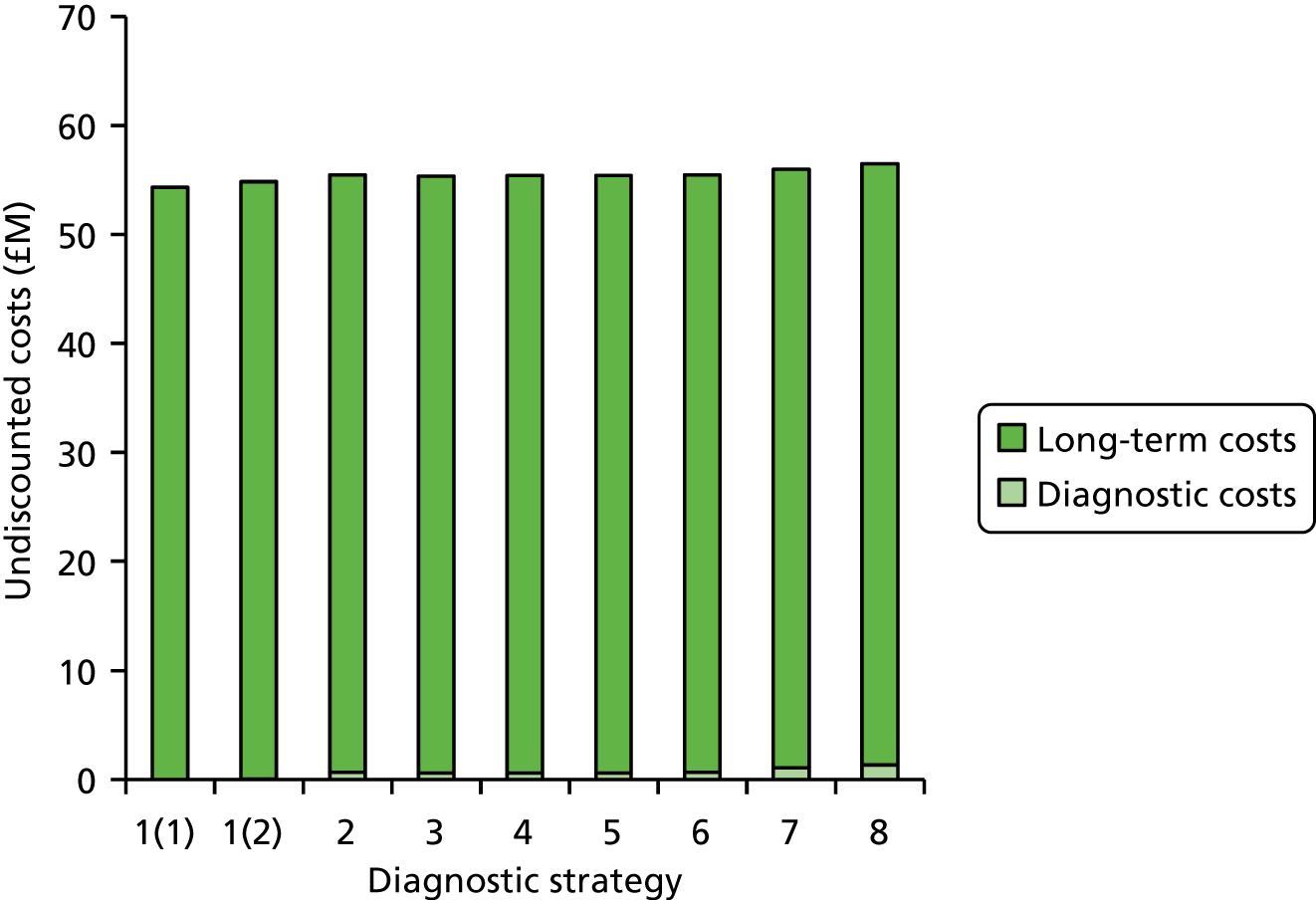
FIGURE 60.
Discounted long-term and diagnostic costs by test strategy.
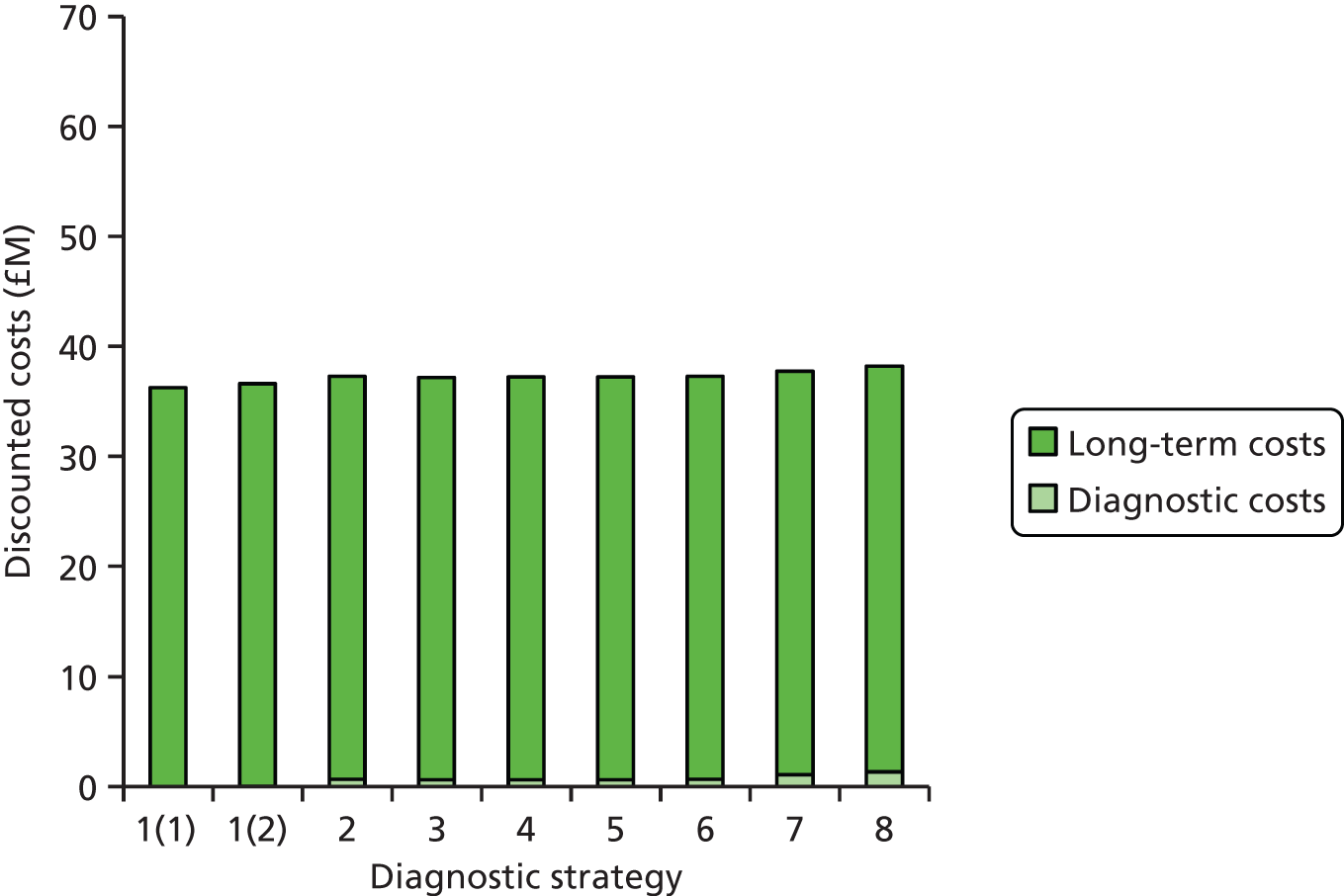
Total long-term costs are similar across strategies (Figure 61); however, the differences between strategies are large enough to strongly influence their cost-effectiveness (see Incremental costs). Strategy 8, direct genetic testing, has the greatest total long-term cost, due to particularly high costs of colonoscopies and prophylactic TAHSBO, which in turn are due to the large number of patients tested positive.
FIGURE 61.
Undiscounted long-term costs disaggregated by test strategy.
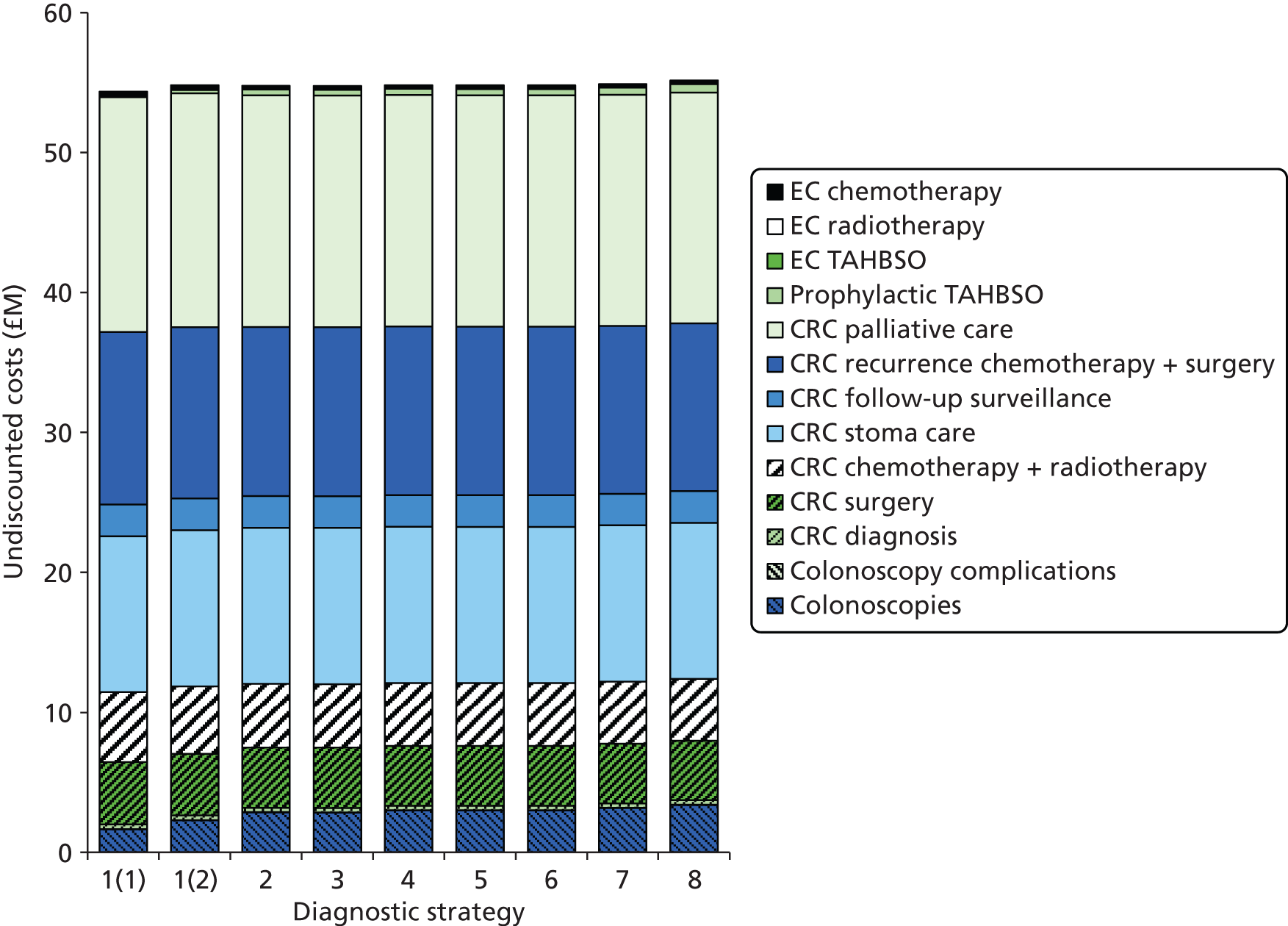
Examining Figure 61, we see that the largest cost items in all strategies are the costs for CRC palliative care, CRC recurrence chemotherapy and surgery, and CRC stoma care. This is a result of both the large unit costs for each of these items, plus the fact that they are accrued over time. The endometrial prevention and management costs are far smaller than the equivalent costs for CRC, as they affect a much smaller proportion of the total cohort; only female patients with LS can get EC and only female patients identified as LS are offered prophylactic surgery for EC (and not all adhere to this offer).
In Figures 62–65, it is shown that treatment costs are higher for probands than for relatives. This is because all probands have CRC, and thus incur the substantial costs of treatment for CRC, whereas only some relatives develop CRC. Costs are higher for patients who are LS positive rather than LS negative because of the greater chance of primary and metachronous CRC (see Expected number of colorectal cancers). Costs are lowest for patients who are TN and FP with no surveillance, as these subgroups do not incur the cost of surveillance and have general population risk of CRC. For female patients, there is a slight increase in costs for FP with no surveillance over TN, as patients who are FP with no surveillance are offered prophylactic TAHBSO, whereas TN patients are not. The cost for probands and relatives diagnosed FP with surveillance is higher owing to the substantial cost of colonoscopies. Patients diagnosed FN have the highest costs owing to the increased risks of CRC and EC from having LS without any treatment to improve survival. TP patients who do not undergo surveillance incur similar costs to FN patients, as they both have LS and do not undergo surveillance. However, the costs for male relatives are slightly lower as they may still benefit from the more aggressive colorectal surgery given to patients with LS, reducing further instances of CRC and therefore costs. For female relatives and probands, patients diagnosed TP with no surveillance incur higher costs than patients diagnosed FN, as some will undergo prophylactic TAHBSO. For all subgroups, patients diagnosed TP with surveillance incur the third highest costs, as they incur surveillance costs but reduced CRC treatment costs, owing to the enhanced surveillance.
FIGURE 62.
Undiscounted treatment and prevention costs for male probands.
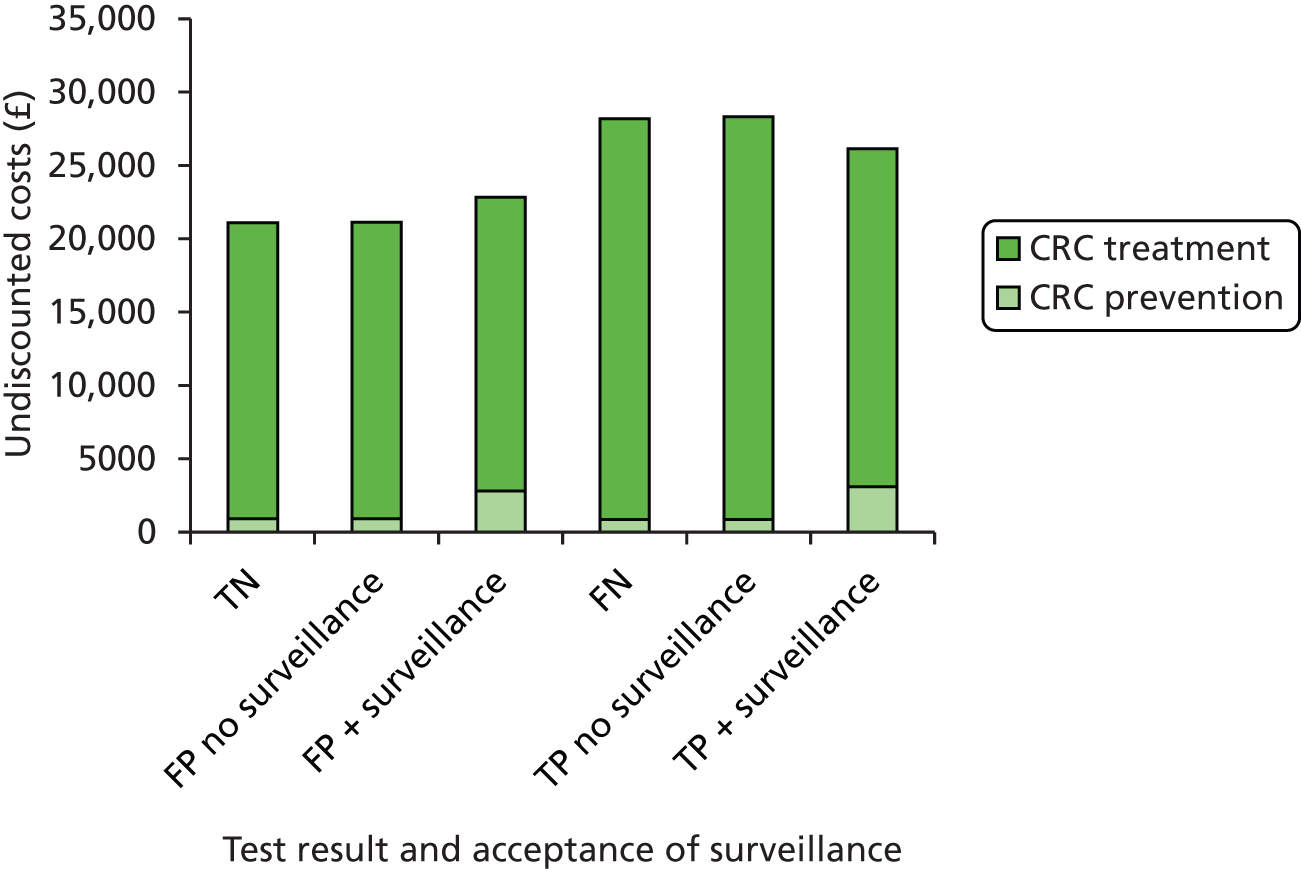
FIGURE 63.
Undiscounted treatment and prevention costs for male relatives.
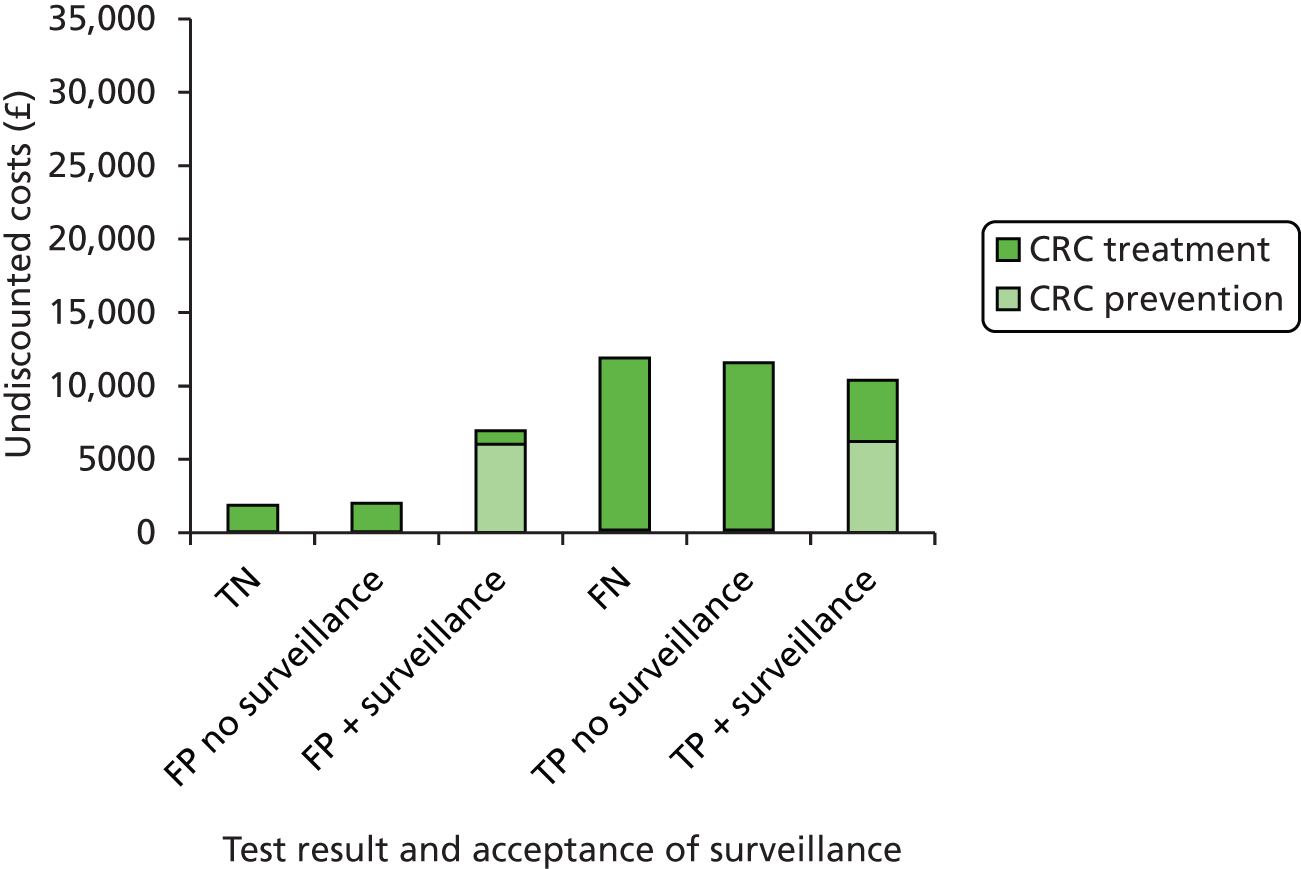
FIGURE 64.
Undiscounted treatment and prevention costs for female probands.
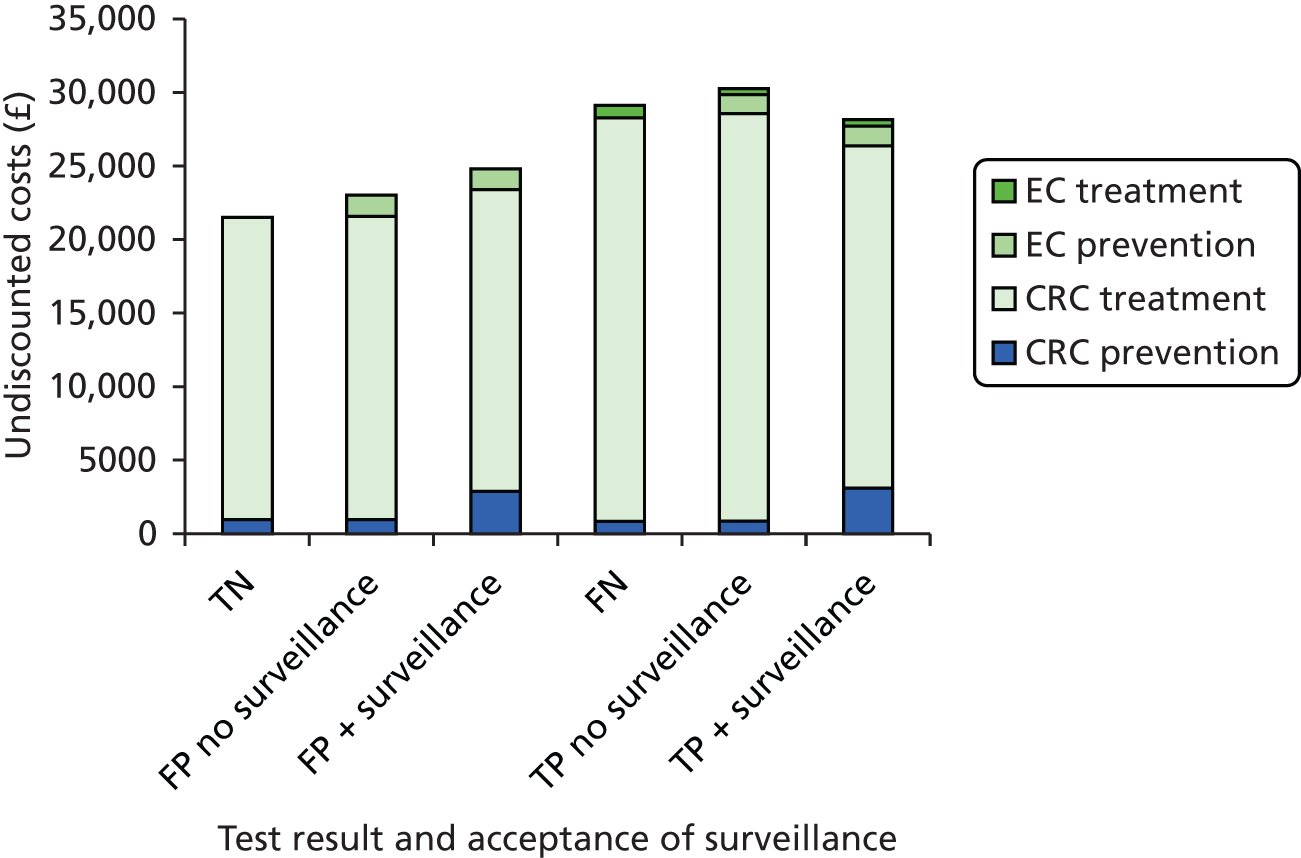
FIGURE 65.
Undiscounted treatment and prevention costs for female relatives.
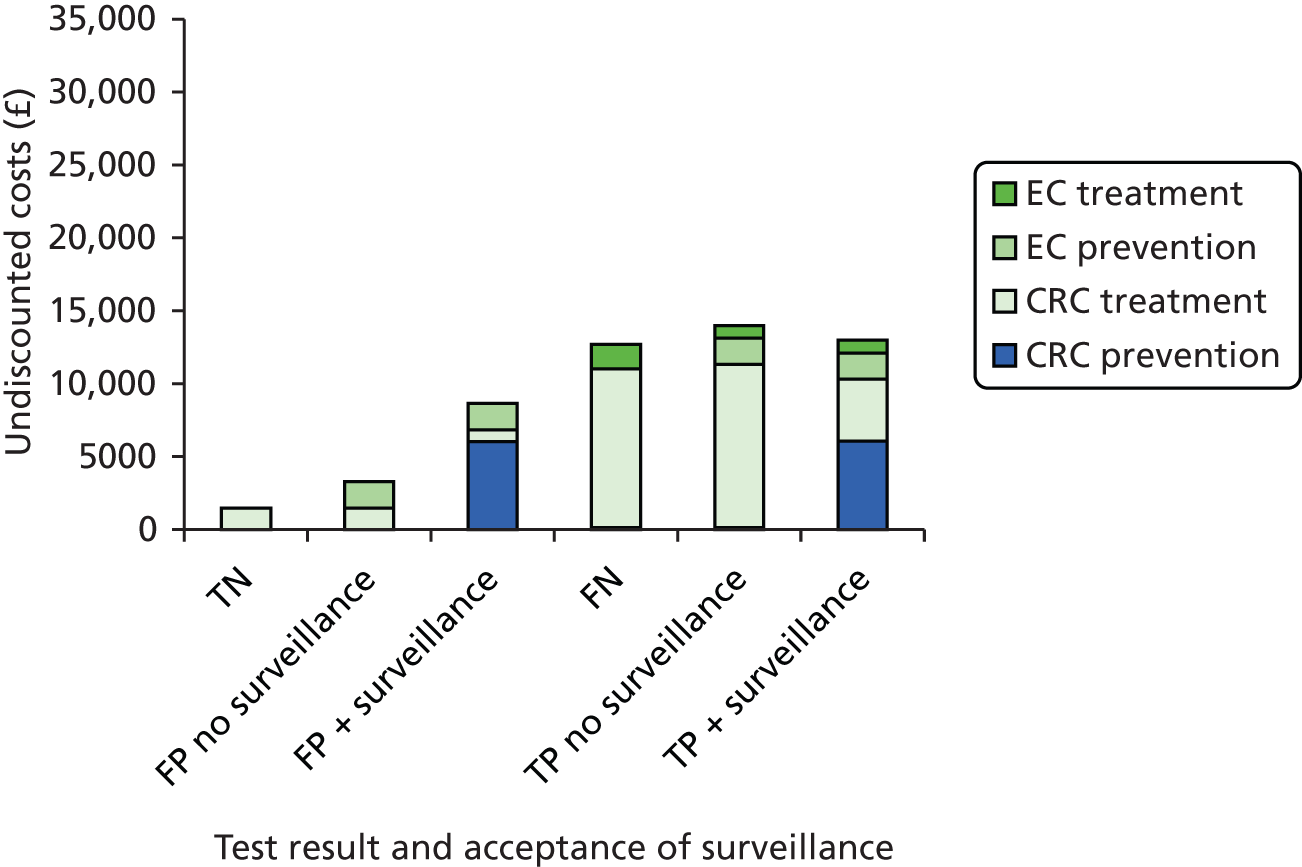
Figures 62–65 also demonstrate that the finding that the costs of EC prevention and management are lower than the equivalent CRC costs is replicated when the cohort is divided into the subgroups represented by the figures. Once again, the cost of CRC treatment is higher than the cost of treatment for EC as a much greater proportion of patients are diagnosed with CRC. The cost of CRC prevention is higher than the cost of EC prevention because a greater proportion of the cohort undergo colonoscopies than TAHBSO. In general, prevention costs are higher for relatives than for probands when the colonoscopies are based on LS diagnosis (there are CRC prevention costs for all probands as they receive colonoscopies as standard follow-up to their CRC, though these are not as frequent as in the case of LS).
Cost-effectiveness results (base case)
In order to understand the cost-effectiveness of the various testing strategies, we first examine the disaggregated incremental costs and QALYs.
Incremental costs
We consider five main groups of costs: LS testing, CRC prevention, CRC treatment, EC prevention and EC treatment (Figure 66). These are related as follows: as the cost of LS testing increases across testing strategies, the costs of CRC and EC prevention also tend to increase. As these in turn increase, the costs of CRC and EC treatment decrease. However, the net effect is that as the total cost of prevention increases, so too does the total cost of prevention and treatment, i.e. the additional costs of prevention are not all recouped by reduced costs of treatment.
FIGURE 66.
Disaggregated incremental discounted costs vs. strategy 1(1).
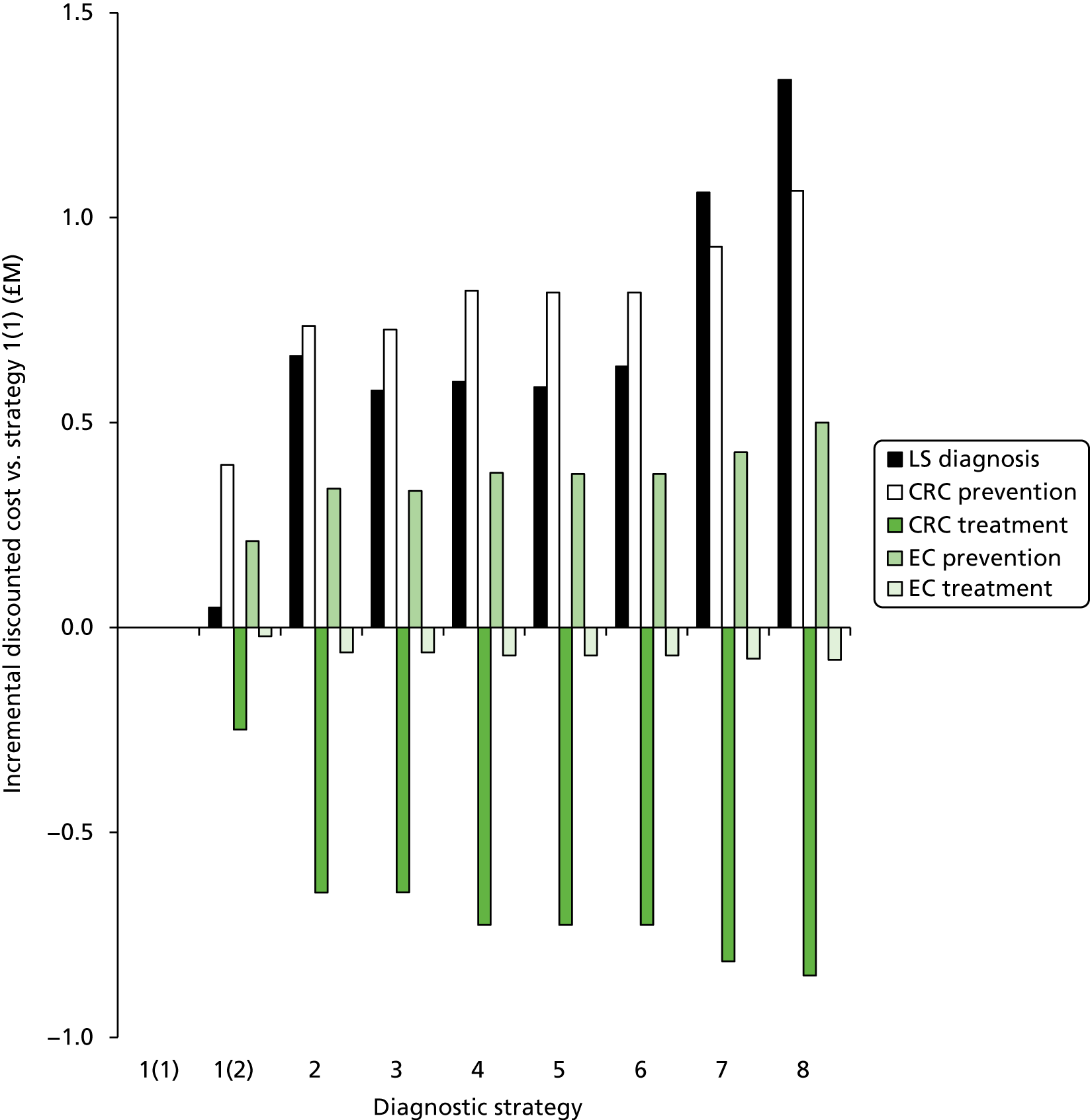
The main components of the savings in the costs of treatment are in respect of chemotherapy and radiotherapy for primary CRC, chemotherapy and radiotherapy for recurrence CRC, palliative care for CRC and surgery for CRC (Figure 67). Savings are much smaller in the remaining components of treatment: CRC diagnosis, CRC stoma care, CRC follow-up surveillance, TAHBSO on diagnosis of EC, and chemotherapy and radiotherapy for EC.
FIGURE 67.
Long-term disaggregated incremental discounted costs versus strategy 1(1).
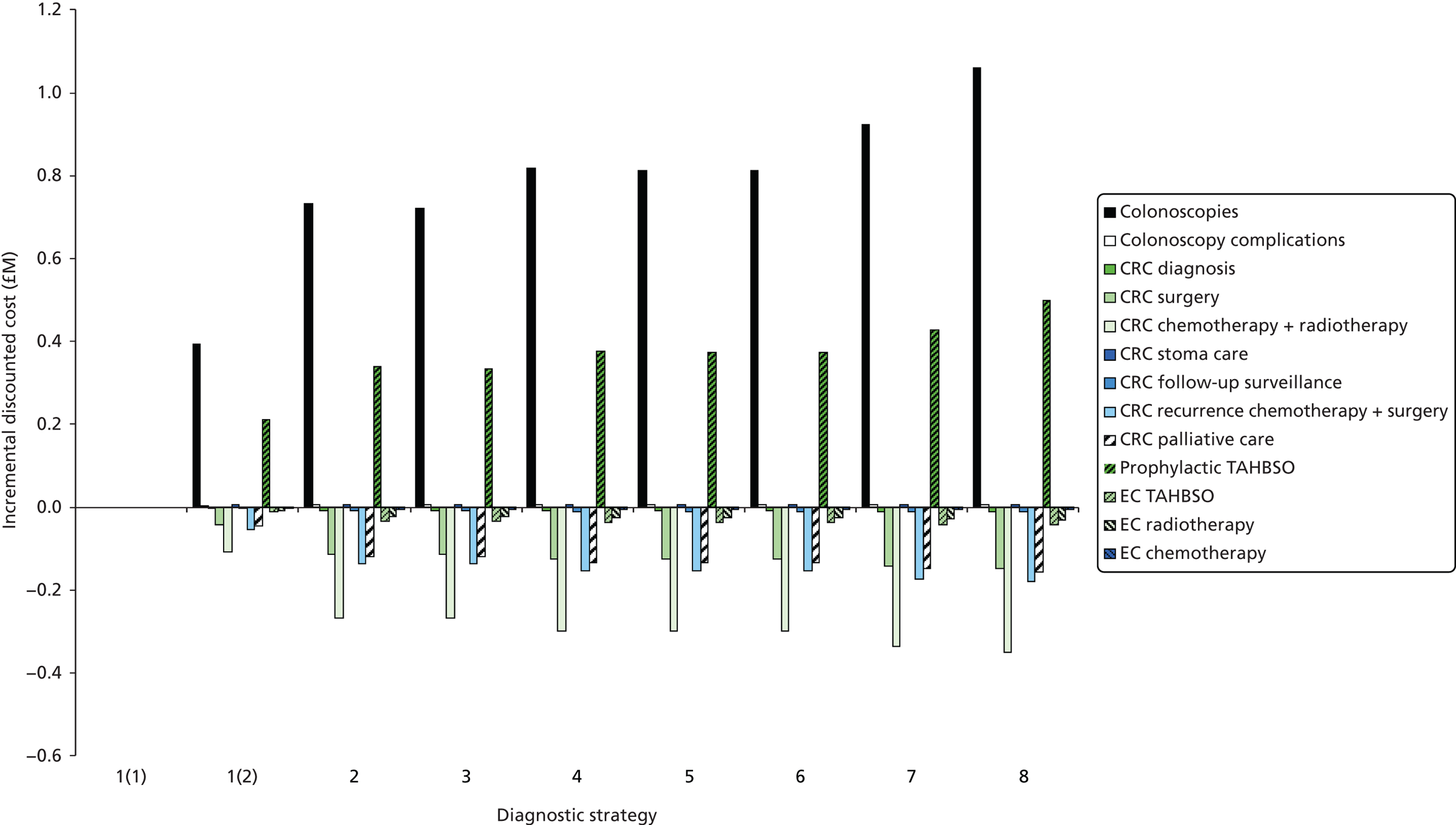
Most, but not all of the additional costs of surveillance for CRC are offset by savings in treatment for CRC (see Figure 66), though the additional costs are approximately £100,000 greater than the savings. Strategy 3 has the smallest difference in these costs (£80,760) and strategy 8 has the largest difference (£217,000).
Similarly, some of the costs of prophylactic TAHBSO are offset by savings in treatment for EC (see Figure 66), though increased costs of prophylactic TAHBSO are far greater than the decreased costs of treatment for EC; the net cost rises from a minimum of £188,682 in strategy 1(2) (AC only) to a maximum of £420,948 in strategy 8 (direct genetic testing). As the sensitivity of the strategies increases, the net cost also increases. When sensitivities are similar between strategies, the net cost increases with specificity. As an individual prophylactic TAHBSO is less expensive than treatment for EC (£3322 compared with £7171), the higher cost of EC prevention compared with the savings from EC treatment avoided must be due to the smaller number of ECs compared with prophylactic TAHBSOs.
Incremental quality-adjusted life-years
The differences in total QALYs across testing strategies related to the psychological impact of diagnosis are far smaller than all other sources of QALYs (i.e. life expectancy, adjusted for disutility of Dukes’ D CRC, and age-related quality of life) (Figure 68). This is because it is assumed that the disutilities due to the psychological impact of testing have an effect only in the very short term (4 months), whereas all other sources of QALYs are accrued over the future life expectancy of probands and relatives.
FIGURE 68.
Incremental discounted QALYs vs. strategy 1(1), by diagnostic and long-term outcomes.
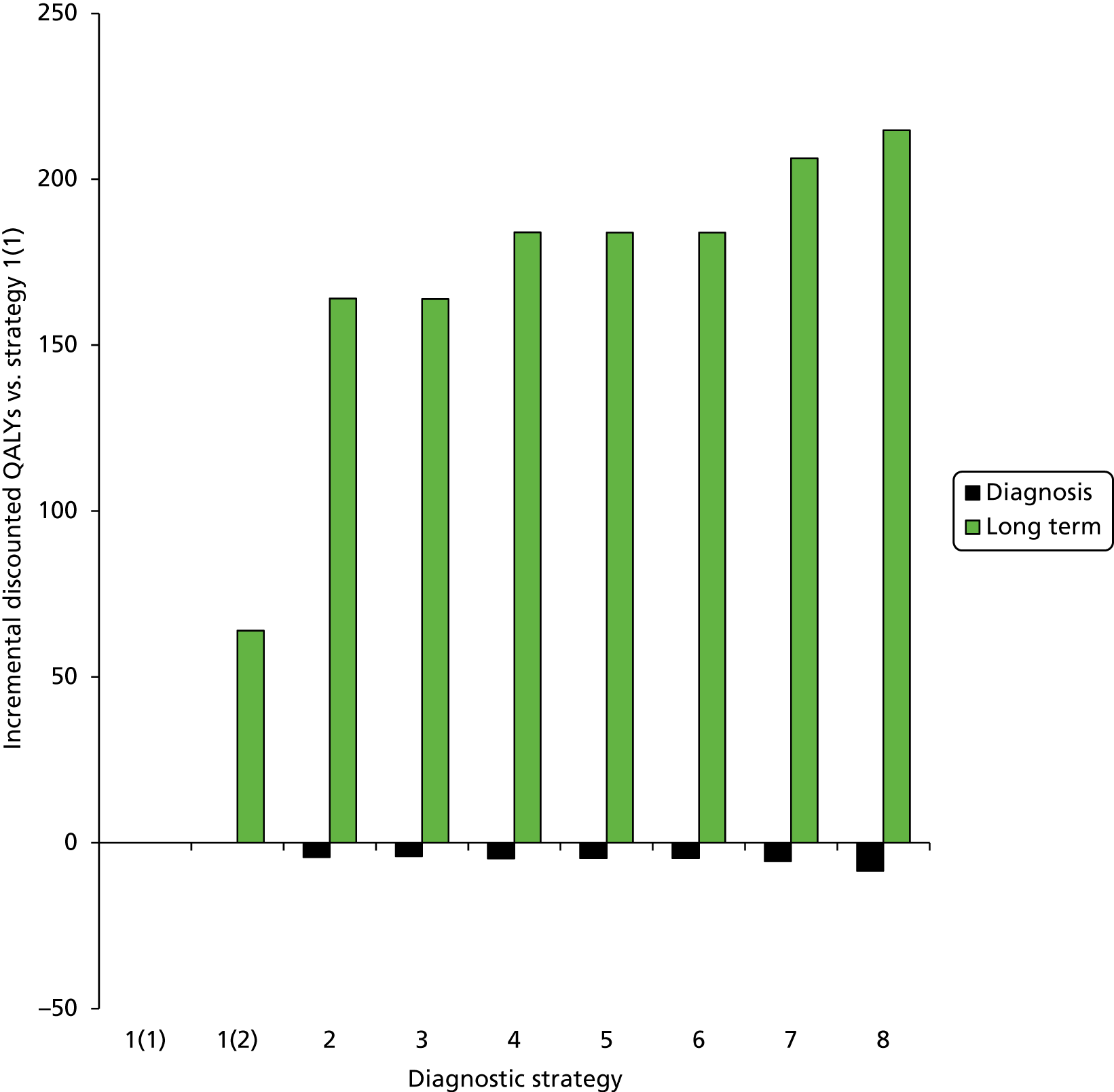
Total QALYs increase with the sensitivity of the testing strategy because as the sensitivity increases, the number of LS-positive people who are treated increases, and such treatment can substantially improve life expectancy [see Life expectancy (base case)]. Total QALYs are far less sensitive to the number of FPs because increased surveillance for these people has little benefit for life expectancy.
Incremental cost-effectiveness ratios
Total discounted costs and QALYs, per strategy, over all probands and relatives per annum in England are given in Figures 69 and 70. Figure 69 demonstrates that the difference in costs and QALYs across the strategies is not large but, in general, as Table 100 demonstrates, as the QALYs increase so do the costs. Discounted QALYs increase from 151,793 given no testing to 152,000 in strategy 8 (direct genetic testing). The total discounted cost increases from £36,224,000 given no testing to £38,198,000 in strategy 8. Total discounted QALYs are the same for strategies 2 and 3, as are those for strategies 4, 5 and 6. Strategies 5 and 6 are expected to have the same total QALYs, as the addition of IHC in strategy 6 is assumed to affect only costs. In strategies 3 and 5, the addition of BRAF appears only to reduce the cost of the strategy compared with strategies 2 and 4, respectively, with no effect on QALYs. Taking strategies 4 and 5 as an example, we see that they have identical sensitivities, with only a slight improvement in specificity. The decrease in FPs does not significantly improve the results, other than reducing the costs by reducing the number of colonoscopies.
FIGURE 69.
Total discounted costs and QALYs for all probands and relatives per annum in England, by testing strategy.
FIGURE 70.
Total discounted costs and QALYs for all probands and relatives per annum in England, by testing strategy, rescaled.
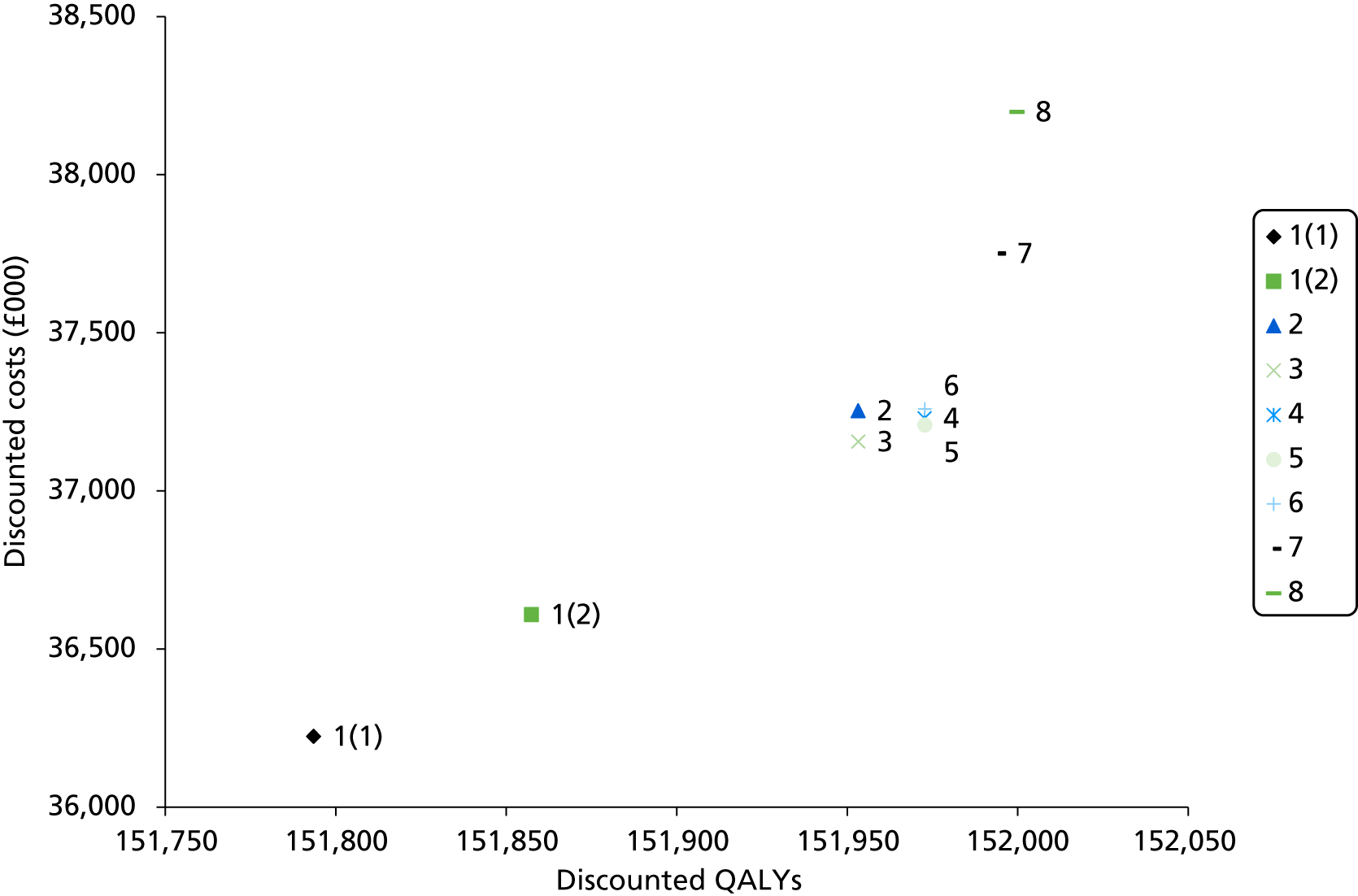
| Strategy | Total discounted QALYs | Total discounted cost (£) | Total NHB at £20,000 per QALY | Incremental discounted QALYs vs. strategy 1(1) | Incremental discounted cost vs. strategy 1(1) (£) | INHB at £20,000 per QALY vs. strategy 1(1) | ICER vs. strategy 1(1) (£/QALY) | ICER (£/QALY) |
|---|---|---|---|---|---|---|---|---|
| 1(1) | 151,793 | 36,223,787 | 149,982 | – | – | – | – | – |
| 1(2) | 151,857 | 36,608,672 | 150,027 | 64 | 384,885 | 44.7 | 6021 | Extended dominated by strategies 1(1) and 3 |
| 2 | 151,953 | 37,253,017 | 150,091 | 160 | 1,029,230 | 108.3 | 6444 | Dominated by strategy 5 |
| 3 | 151,953 | 37,155,626 | 150,096 | 160 | 931,839 | 113.2 | 5831 | Extended dominated by strategies 1(2) and 5 |
| 4 | 151,973 | 37,229,210 | 150,111 | 179 | 1,005,423 | 129.0 | 5610 | Dominated by strategy 5 |
| 5 | 151,973 | 37,208,248 | 150,112 | 179 | 984,461 | 130.1 | 5491 | 5491 |
| 6 | 151,973 | 37,259,067 | 150,110 | 179 | 1,035,280 | 127.5 | 5774 | Dominated by strategy 5 |
| 7 | 151,994 | 37,750,884 | 150,107 | 201 | 1,527,097 | 124.5 | 7601 | 25,106 |
| 8 | 152,000 | 38,198,324 | 150,090 | 206 | 1,974,537 | 107.6 | 9571 | 82,962 |
Accounting for both the discounted costs and QALYs, we investigate the ICERs of the strategies compared with both no testing and each other. The ICERs of all strategies compared with no testing are < £10,000 per QALY (see Table 100), which is considerably less than the £20,000-per-QALY threshold routinely used by NICE in England and Wales132 (Figure 71). All ICERs versus no testing are similar, varying from £5491 per QALY for strategy 5 to £9571 per QALY for strategy 8.
FIGURE 71.
Incremental discounted costs and QALYs for all probands and relatives per annum in England, by testing strategy.
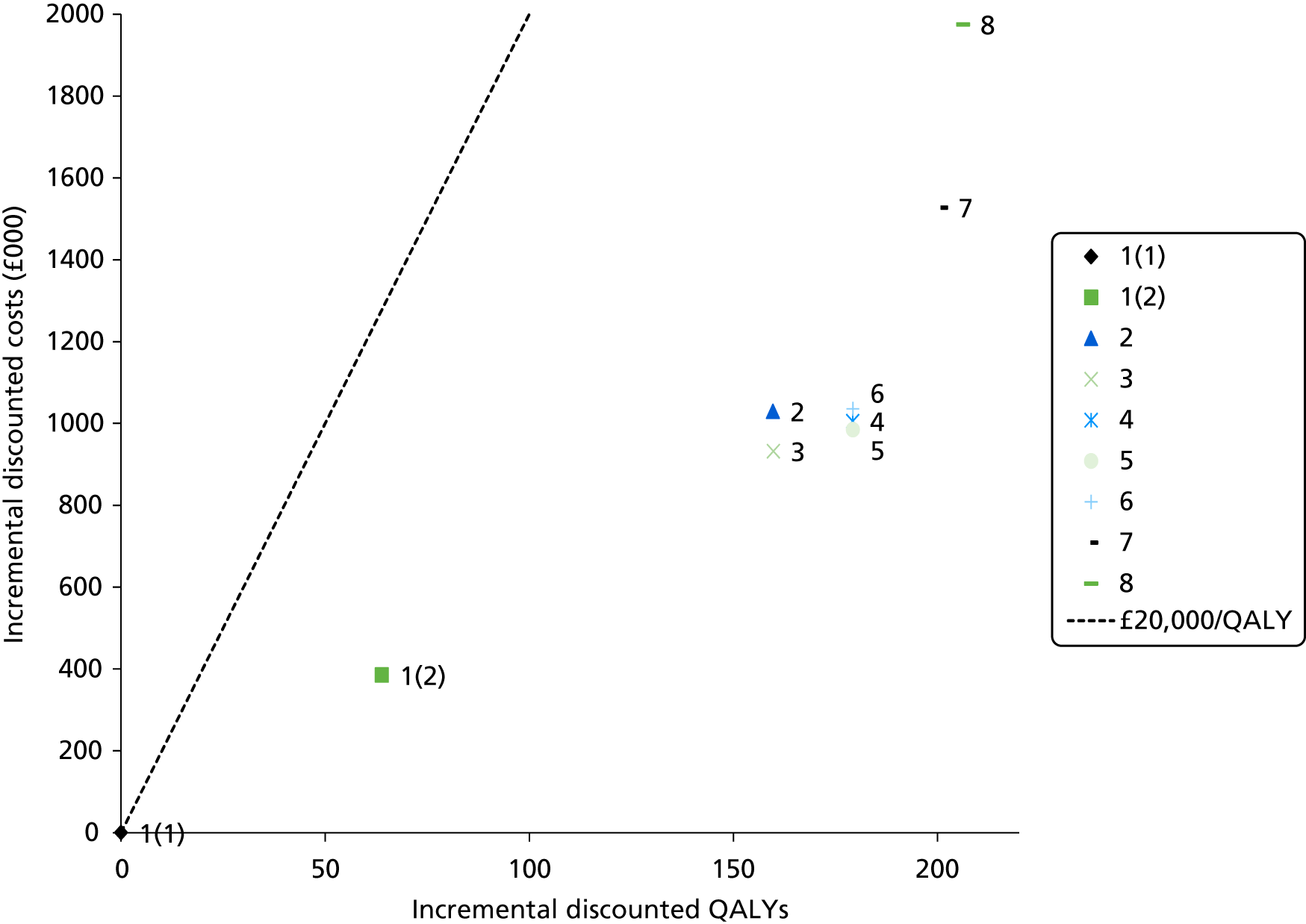
The testing strategies on the efficiency frontier are strategies 1(1), 5, 7 and 8 (see Table 97). The remaining strategies are either dominated (less effective and more expensive than at least one other strategy) (strategies 2, 4 and 6) or extended dominated (less effective and more expensive than some combination of two other strategies) [strategies 1(2) and 3].
On the efficiency frontier, the ICER of strategy 5 versus no testing is £5491 per QALY. The ICER of strategy 7 versus strategy 5 is £25,106, and therefore marginally greater than NICE’s cost-effectiveness threshold. The ICER of strategy 8 versus strategy 7 is £82,962 per QALY, and therefore far greater than the threshold.
Strategy 6 is always dominated by strategy 5 because they have the same test accuracies, but strategy 6 additionally includes the cost of IHC testing.
We now consider NHBs, defined as total QALYs minus (total costs divided by the willingness-to-pay threshold) of each strategy. Strategy 5 has the highest NHB at a willingness-to-pay threshold of £20,000 per QALY (Figures 72 and 73). In Figures 72 and 73, error bars represent the 95% CI over the 32,000 simulated patients. In Figure 72, the 95% CIs overlap, particularly for strategies 4, 5 and 6. This suggests that there is uncertainty about which strategy is the most cost-effective at a willingness to pay of £20,000 per QALY. However, in Figure 73 these error bars are much smaller as they compare the INHB of each strategy with strategy 5, rather than with strategy 1(1). This demonstrates that strategy 5 is the most cost-effective at a willingness to pay of £20,000 per QALY. In summary, at a willingness-to-pay threshold of £20,000 per QALY, strategies 4, 5, 6 and 7 offer the best value for money, and the cost-effectiveness of these strategies is similar. These strategies are predicted to result in an additional 130 discounted QALYs per year in England compared with no testing. By comparison, the total discounted QALYs for a man who lives to age 80 years, allowing for age-related quality of life, is approximately 25. The 130 discounted QALYs therefore equate to the total discounted QALYs accrued over the lives of approximately five people.
FIGURE 72.
Incremental net health benefit compared with strategy 1(1) at a willingness to pay of £20,000 per QALY.
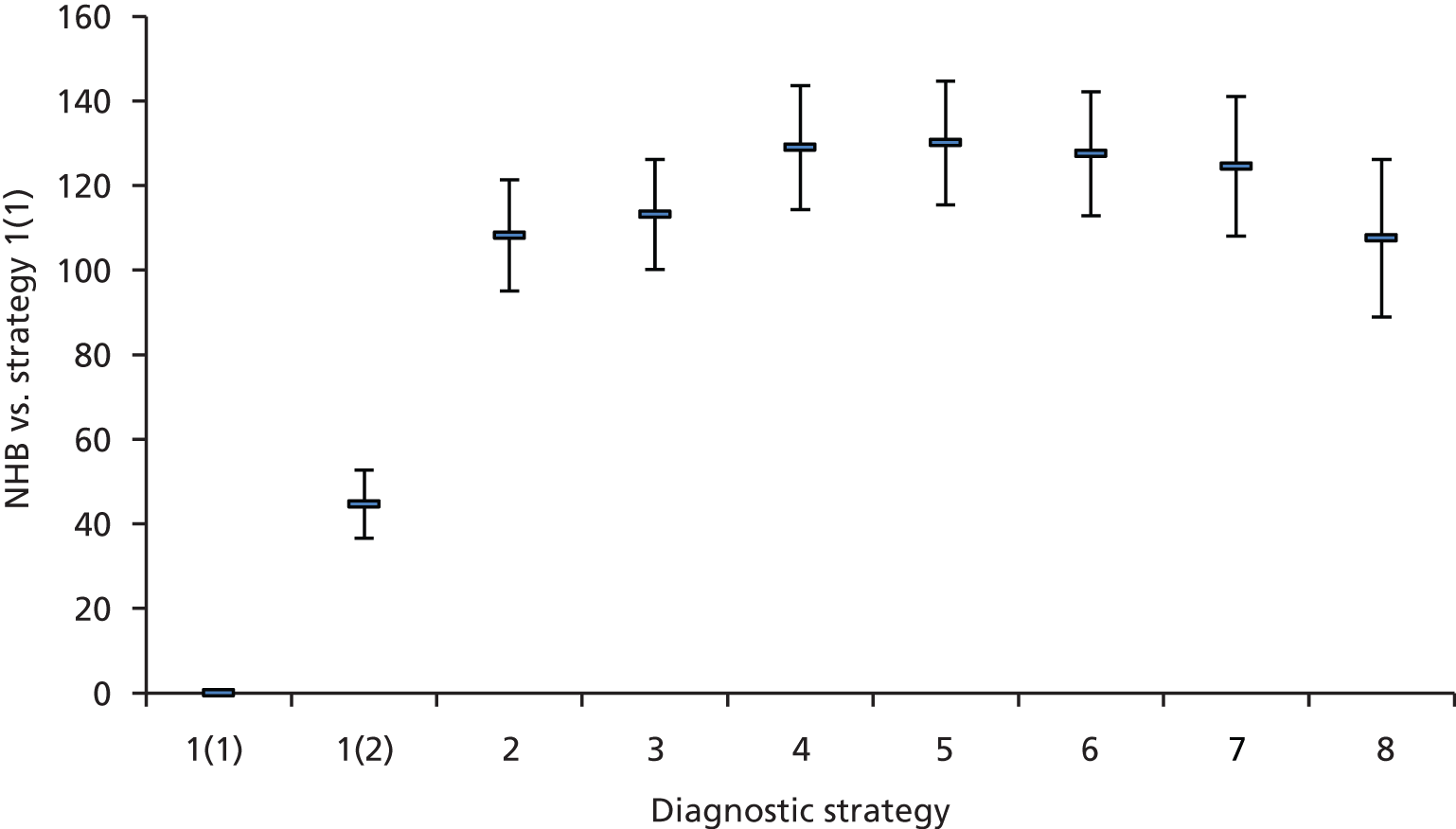
FIGURE 73.
Incremental net health benefit compared with strategy 5 at a willingness to pay of £20,000 per QALY.
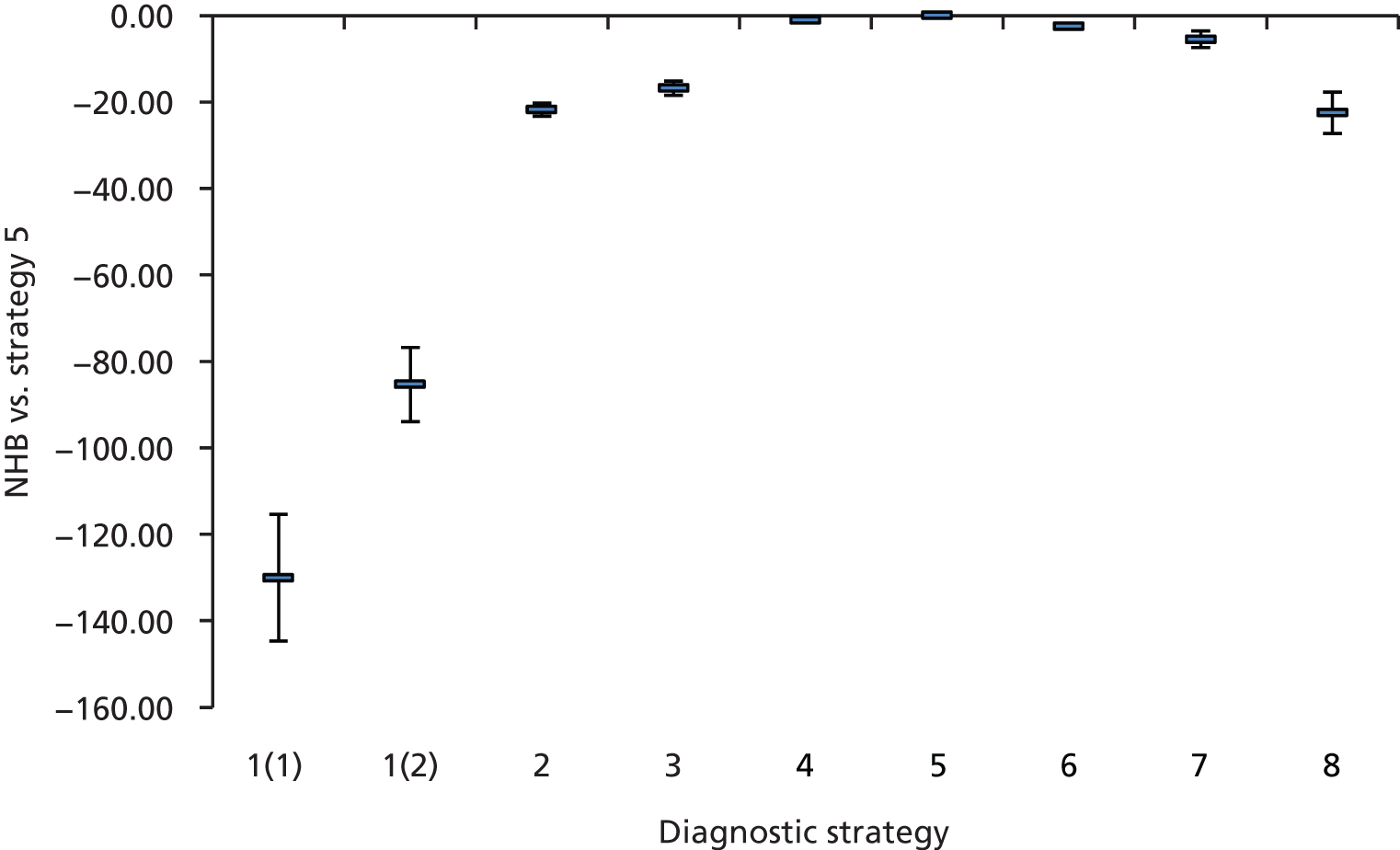
Additional results (base case)
We now present the following important results for each testing strategy from our cost-effectiveness analysis: expected number of colonoscopies per annum in England, expected number of CRCs per annum in England and expected number of ECs per annum in England.
Expected number of colonoscopies
Here we investigate the total number of colonoscopies, because the total cost of colonoscopies strongly influences the cost-effectiveness of testing strategies. As expected, the number of colonoscopies increases with the proportion of patients who are diagnosed with LS, because these patients are offered enhanced surveillance. Individuals diagnosed with LS, who adhere to surveillance, receive one colonoscopy every 2 years. Patients with CRC who are diagnosed LS negative or who decline LS surveillance receive one colonoscopy every 5 years. This explains why all patients receive some colonoscopies, even in strategy 1(1), where no testing occurs. This is the minimum number of colonoscopies per patient across the strategies.
The average number of colonoscopies per proband without LS increases from approximately 2.33 under no testing to 2.39 for strategy 8 (Figure 74). The 2.33 colonoscopies represent standard CRC follow-up, with the increase occurring in other strategies due to probands identified incorrectly as LS positive. It therefore follows that strategies with higher false positivity rates (lower specificity) have a higher number of colonoscopies. The number of colonoscopies per LS-negative relative increases from 0.02 to 0.23 across testing strategies. These are similarly all due to FPs. For patients with LS, the number of colonoscopies per proband increases from 2.11 in the no testing strategy to 5.58 in strategy 8 as a greater proportion of probands with LS are diagnosed as such under genetic testing, and hence a greater proportion receive enhanced surveillance. The number of colonoscopies per relative with LS increases from 0.30 given no testing, to 7.0 in strategy 8, for the same reason. The number of colonoscopies per proband with LS is lower than for those without LS in the no testing strategy, as probands with LS have a shorter life expectancy. Furthermore, the risk of metachronous cancer in probands with LS is much higher than that in probands without; the surgery for this is likely to be more aggressive, resulting in the inability to use follow-up colonoscopies. The increase in colonoscopies for relatives with LS between strategies 1(2) and 2 is most likely due to a higher uptake in LS-positive relatives owing to a correct diagnosis by genetic testing, plus the longer life expectancy of these relatives increasing the number of colonoscopies.
FIGURE 74.
Average number of colonoscopies for probands and relatives according to LS status.
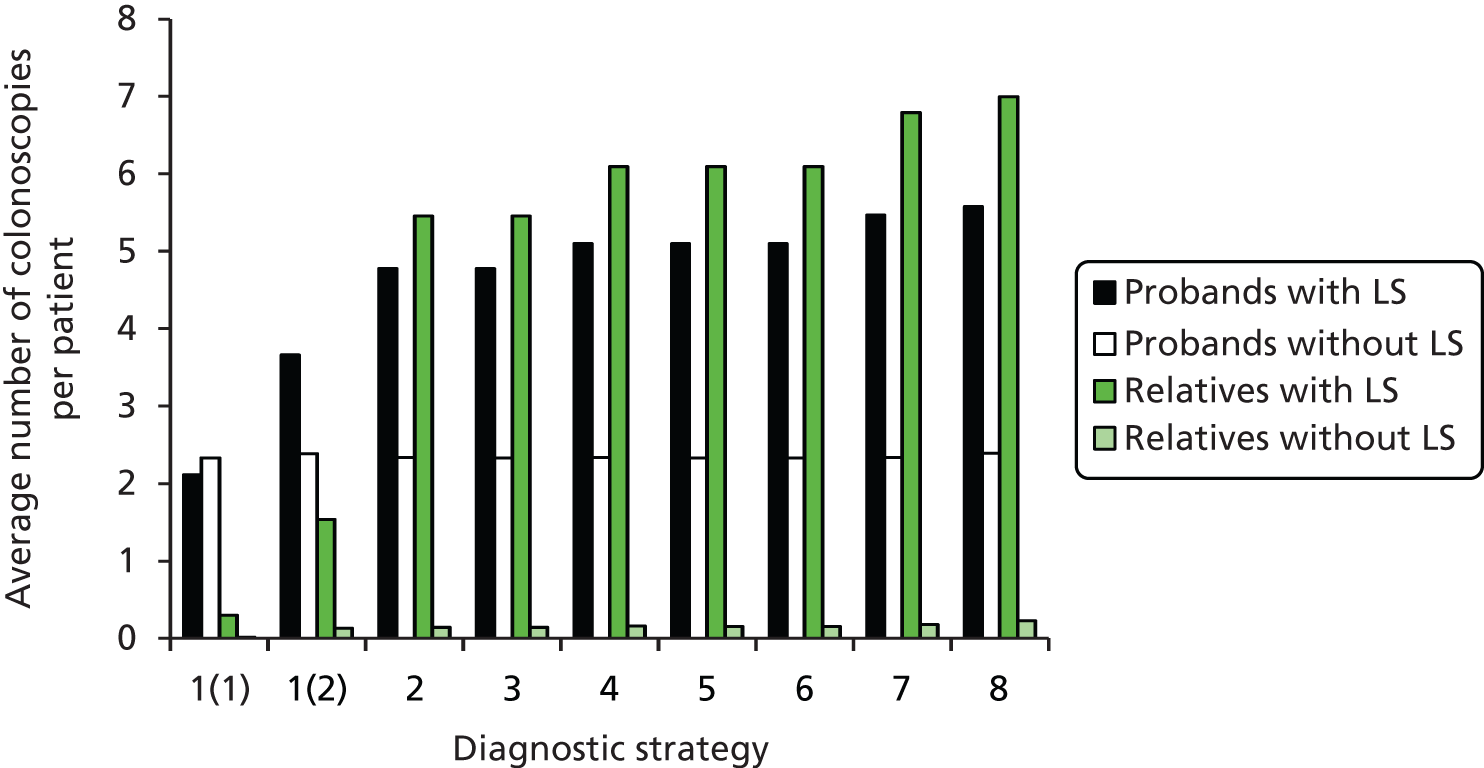
The expected total number of colonoscopies performed in England per annual cohort increases from approximately 4200 given no testing to 8600 in strategy 8 (Figure 75), suggesting that diagnostic strategies for LS may increase the number of colonoscopies twofold.
FIGURE 75.
Total number of colonoscopies conducted in England per cohort in each testing strategy.
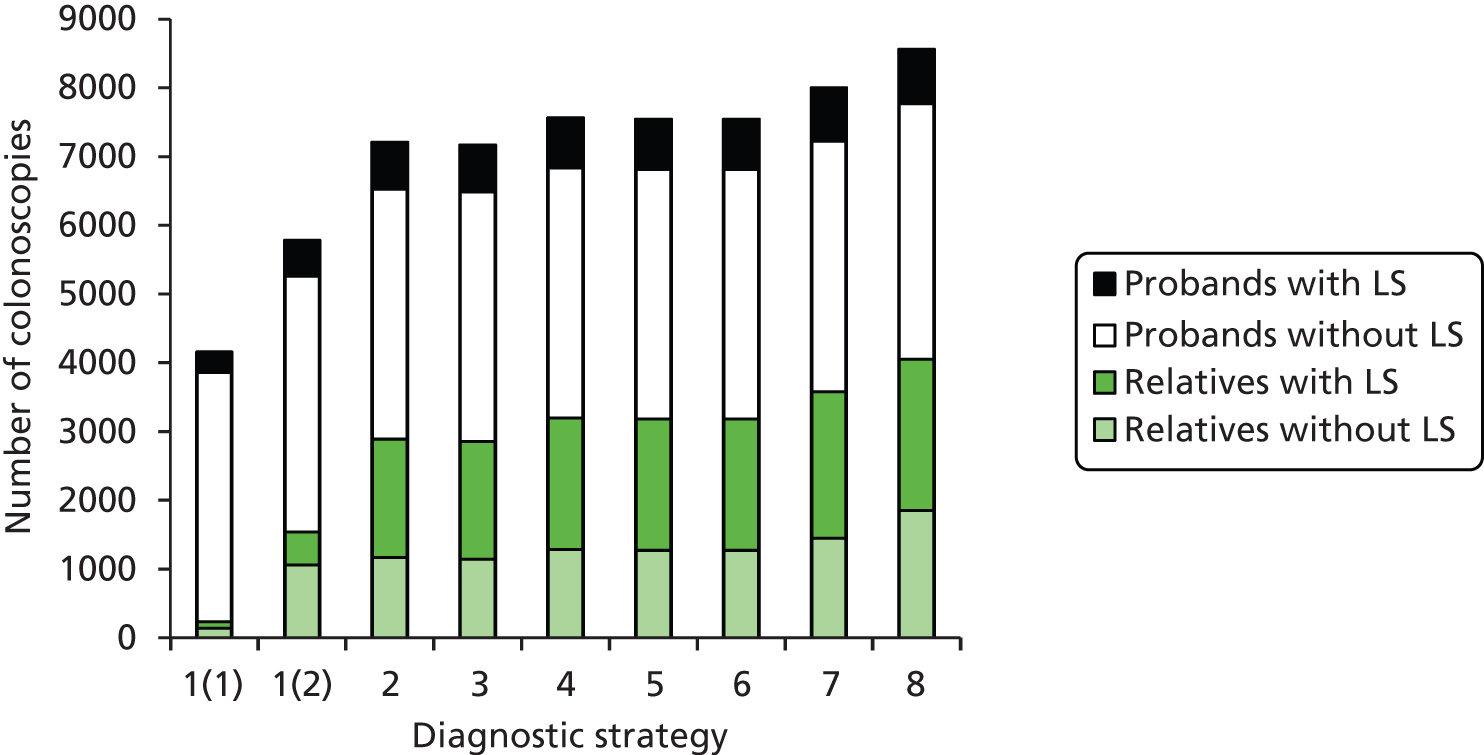
Expected number of colorectal cancers
The number of CRCs in probands clearly does not include the index cancer, as all probands present with this, and its incidence is unaffected by the diagnostic process. Furthermore, relatives are assumed not to have had CRC previously.
The risk of CRC is higher for males than females among probands and relatives (Figure 76), in accordance with our knowledge of CRC risk. The proportion of relatives who develop primary CRC is greater than the proportion of probands who develop metachronous CRC. The probabilities of developing CRC are slightly lower given genetic testing because there is a greater chance that LS is identified.
FIGURE 76.
Proportion of relatives who develop primary CRC and proportion of probands who develop metachronous CRC.
The expected number of new CRCs for the entire cohort of 1699 probands (metachronous CRCs) and 8495 relatives (primary and metachronous CRCs), in England per year, is approximately 665 in the no testing strategy, reducing to a minimum of 633 in strategy 8 (a difference of 32). Given no testing, 79 are metachronous cancers in the proband population and 586 are index and metachronous cancers in the relative population. In strategy 8, 72 metachronous CRCs are expected in the probands and 561 index and metachronous CRCs in the relatives.
The lifetime probabilities of developing metachronous CRC for probands without LS (approximately 1.4%) and primary and metachronous CRC for relatives (approximately 5.5%) remain roughly constant across strategies (Figures 77 and 78). A very small proportion of this group test as FP and consequently undergo enhanced surveillance, accounting for very small differences in the probabilities of having CRC across strategies. As expected, the lifetime probability of developing CRC for patients with LS (24–30% for relatives, 35–40% for probands, depending upon the strategy) is far higher than for those without LS. The probabilities that probands and relatives with LS develop CRC are lower under genetic testing strategies, as these correctly identify many such people as having LS. These patients then receive enhanced surveillance, which acts to reduce the probability of developing CRC. The greater the sensitivity of the test strategy, the lower the probability that probands and relatives develop CRC. For example, the probability of metachronous CRC among probands with LS falls from 40.0% (no testing) to 35.0% (direct genetic testing), and the probability of primary and metachronous CRC among relatives with LS falls from 30.2% (no testing) to 24.1% (direct genetic testing).
FIGURE 77.
Probability that probands develop metachronous CRC, by testing strategy.
FIGURE 78.
Probability that relatives develop primary CRC, by testing strategy.
Expected number of endometrial cancers
No women who are LS negative are assumed to develop EC. Further, all probands and relatives are assumed not to have previously developed EC.
The lifetime risk of EC decreases as more LS-positive relatives and probands are identified by a strategy because these people receive prophylactic TAHBSOs (Figure 79). For example, the probability of EC among probands with LS falls from 11.2% (no testing) to 7.4% (direct genetic testing), and the probability of EC among relatives with LS falls from 23.7% (no testing) to 15.5% (direct genetic testing).
FIGURE 79.
Lifetime risk of EC in females with LS.
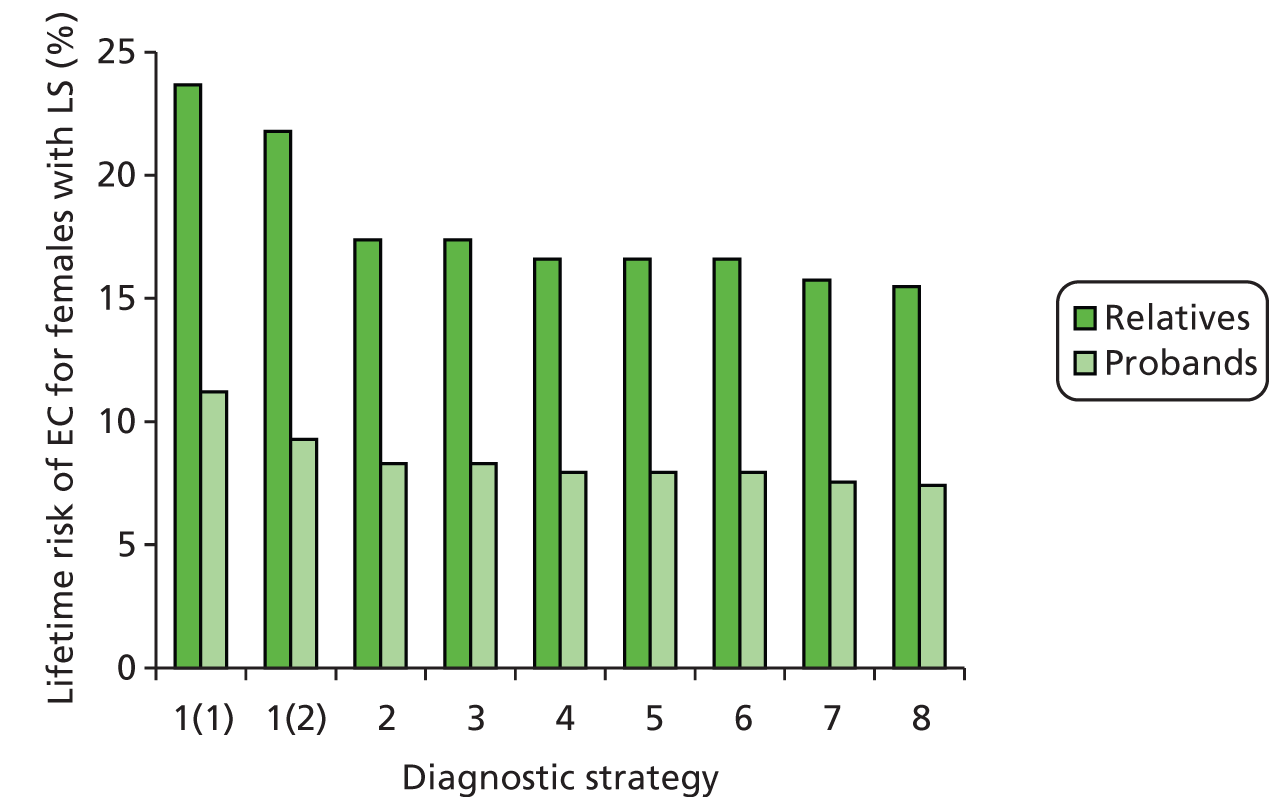
The expected annual number of ECs related to probands aged < 50 years and their relatives in England in the no testing strategy is 54, with 46 occurring in female relatives and 8 in female probands. This reduces to a minimum of 35 in strategy 8 (direct genetic testing), with 30 in female relatives and 5 in the probands. The probability of developing EC is greater for relatives than probands (approximately 17% vs. 8% for genetic strategies) because life expectancy is greater for relatives, which gives a greater time frame in which to develop EC.
Scenario analysis 1 (endometrial cancer excluded) results
In this scenario, we remove EC from the model. In so doing, we exclude the incidence and treatment of EC, as well as the associated mortality rates, costs and QALYs. We also exclude the preventative measures associated with EC, so that there are no longer costs, QALYs or mortality rates for prophylactic TAHBSOs within the model. This allows us to examine the impact of EC on our base-case analysis, as well as providing a scenario that is comparable with previous models of LS, where EC is not accounted for. Additionally, it could be argued that it would be more appropriate to exclude EC from our base-case analysis, given that we do not include OC. This is because the genetic testing strategies incur substantial costs for prophylactic TAHBSO in our base case, but we do not allow for the associated reduction in incidence of OC.
One would imagine that the results for males in this scenario would be as in the base-case analysis, given that males do not get EC. To a very high level of accuracy, this is true. However, there are very slight differences because the model simulates individual patients, and allows for chance in estimating the times of events, such as incidence of CRC and EC.
As expected, the diagnostic results are unaffected by the exclusion of EC.
Life expectancy
The life expectancies of all groups of patients change only very slightly when EC is removed (Figures 80 and 81), which shows that there is little mortality from EC. The life expectancy of male probands remains very similar between the base case and scenario 1. This is expected as the exclusion of EC should not have an impact on the male population. However, the uncertainty inherent in the model due to the individual patient simulations does alter the life expectancy of the male relatives, with a slight increase across all strategies. There are counter-intuitive results for other groups: female probands appear to have a very slightly reduced life expectancy when EC is removed, but female relatives appear to have a very slight increase.
FIGURE 80.
Life expectancy of probands by testing strategy, excluding EC.
FIGURE 81.
Life expectancy of relatives by testing strategy, excluding EC.
This can be partly explained using the results of the cohort’s LS status, as demonstrated in Table 101. Here, as expected, probands and relatives with LS have an increase in life expectancy when EC is removed. Relatives without LS also appear to have an increase in life expectancy, which results from the inherent uncertainty in the model due to the individual patient simulations. Probands without LS appear to have a slightly reduced life expectancy as a result of very small change in the males’ life expectancy and a reduced life expectancy in the female probands. To understand these results more clearly it is important to break down the female subgroups into their relevant diagnoses.
| Output parameter | Scenario | Strategy | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1(1) | 1(2) | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |||
| Life expectancy (years): probands | With LS | Base case | 12.93 | 13.65 | 14.17 | 14.17 | 14.32 | 14.32 | 14.32 | 14.49 | 14.54 |
| Scenario 1 (no EC) | 13.12 | 13.82 | 14.33 | 14.33 | 14.48 | 14.48 | 14.48 | 14.64 | 14.69 | ||
| Without LS | Base case | 13.90 | 13.90 | 13.90 | 13.90 | 13.90 | 13.90 | 13.90 | 13.90 | 13.90 | |
| Scenario 1 (no EC) | 13.86 | 13.86 | 13.86 | 13.86 | 13.86 | 13.86 | 13.86 | 13.86 | 13.86 | ||
| Life expectancy (years): relatives | With LS | Base case | 33.97 | 34.28 | 35.20 | 35.20 | 35.36 | 35.36 | 35.36 | 35.52 | 35.57 |
| Scenario 1 (no EC) | 34.76 | 35.03 | 35.86 | 35.86 | 35.99 | 35.99 | 35.99 | 36.14 | 36.18 | ||
| Without LS | Base case | 37.51 | 37.52 | 37.52 | 37.52 | 37.52 | 37.52 | 37.52 | 37.52 | 37.52 | |
| Scenario 1 (no EC) | 37.55 | 37.55 | 37.55 | 37.55 | 37.55 | 37.55 | 37.55 | 37.55 | 37.55 | ||
When the patients are split according to TP/FP/FN/TN (± surveillance), the causes of differences in life expectancy versus the base case become clearer. For female relatives there is a slight increase in life expectancy (Figure 82; compare with Figure 57), but most noticeably for those diagnosed TP and FN, as mortality due to EC for females with LS is removed. This pattern is repeated for female probands (Figure 83), though the effect is less pronounced.
FIGURE 82.
Life expectancy of female relatives by test result and surveillance, excluding EC.
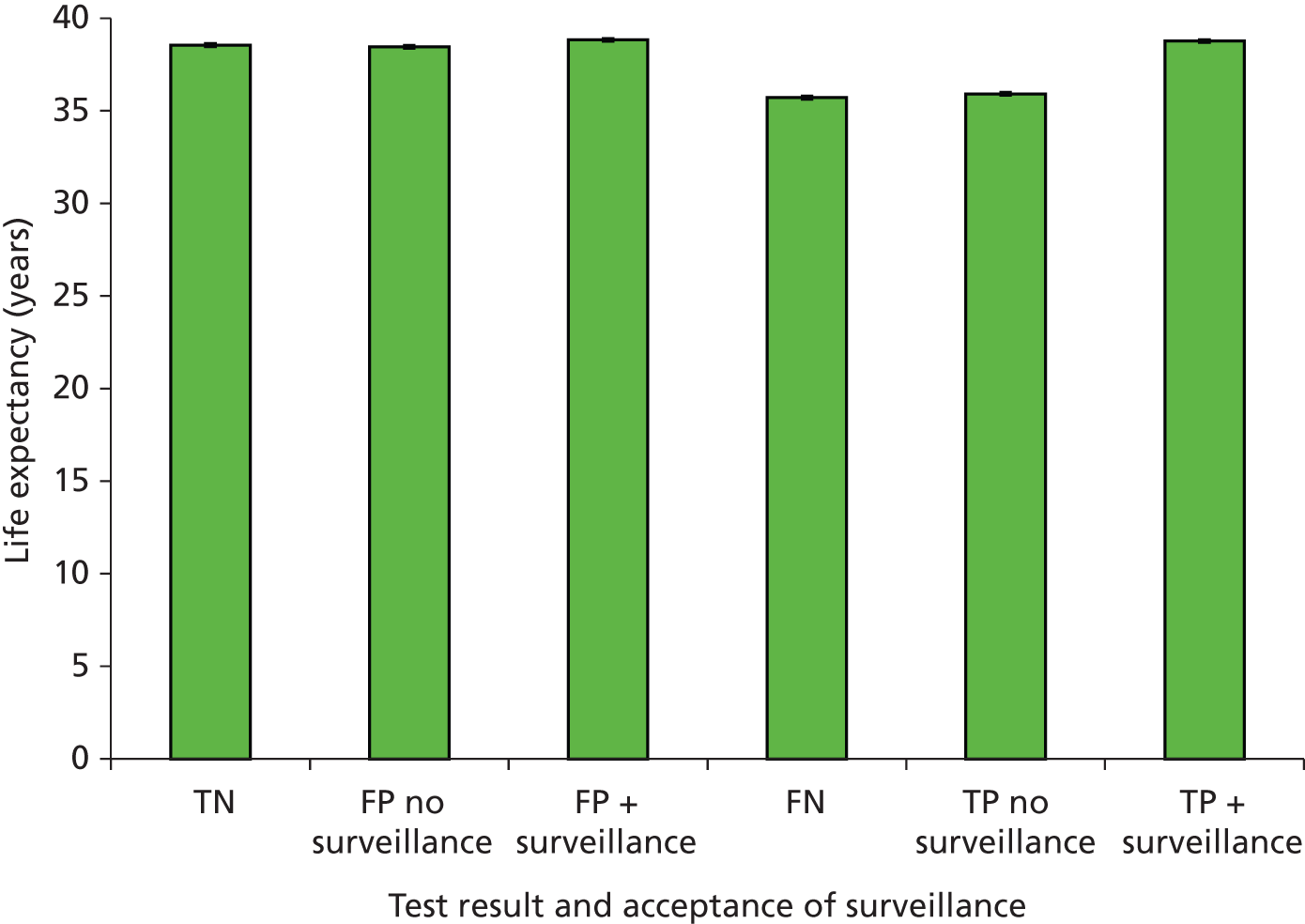
FIGURE 83.
Life expectancy of female probands by test result and surveillance, excluding EC.
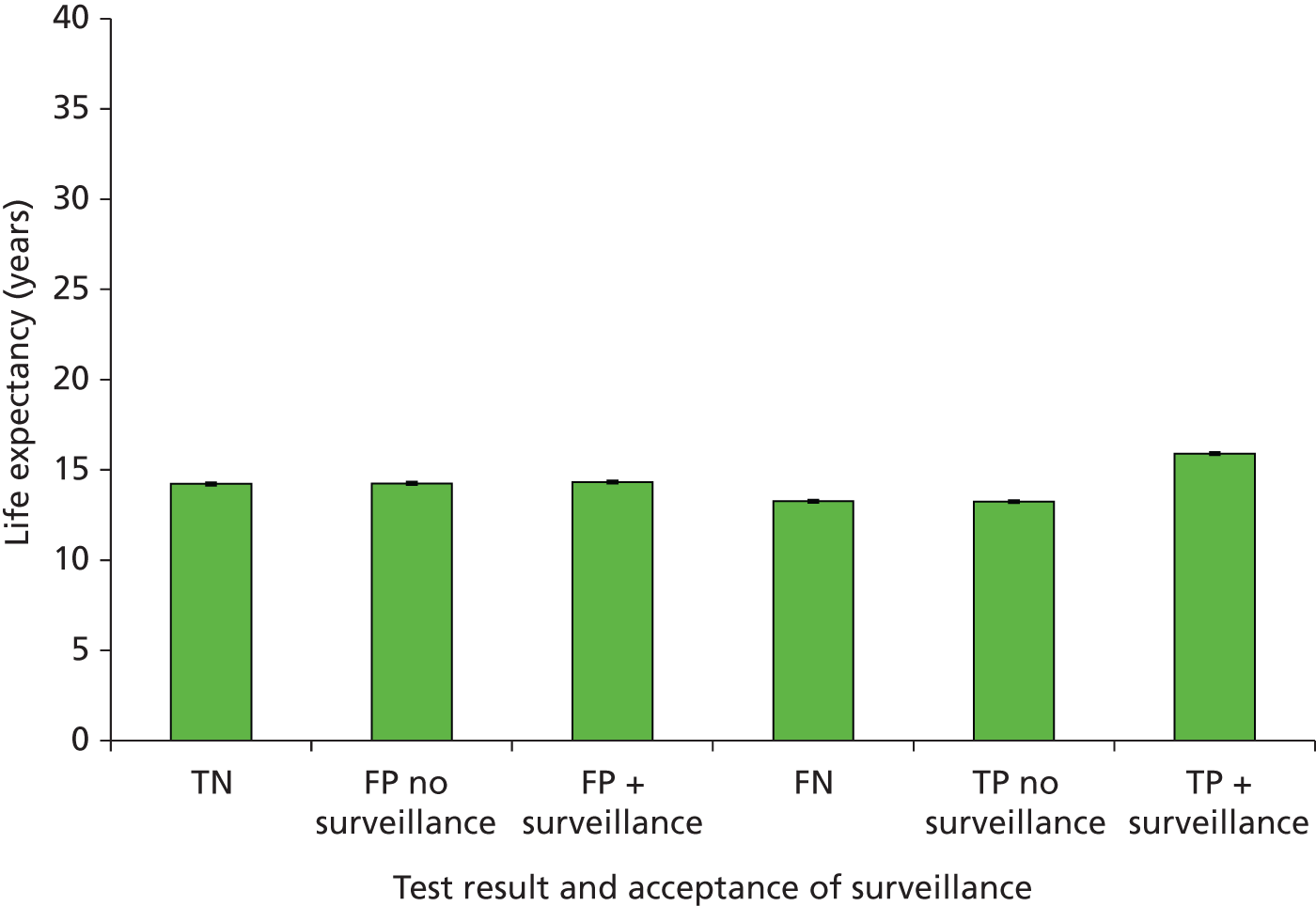
Cost results
As expected, total costs are reduced when EC is excluded, as there are no longer costs for the prevention or treatment of EC. Given the slightly increased life expectancy of female relatives and probands with LS, we expect a slight increase in costs of colonoscopies and CRC treatment, as there are further opportunities for this to occur. Indeed, the undiscounted costs of these increase by between £400,000 and £1,800,000 depending on the strategy (Figure 84; compare with Figure 61).
FIGURE 84.
Undiscounted long-term costs, disaggregated, excluding EC.
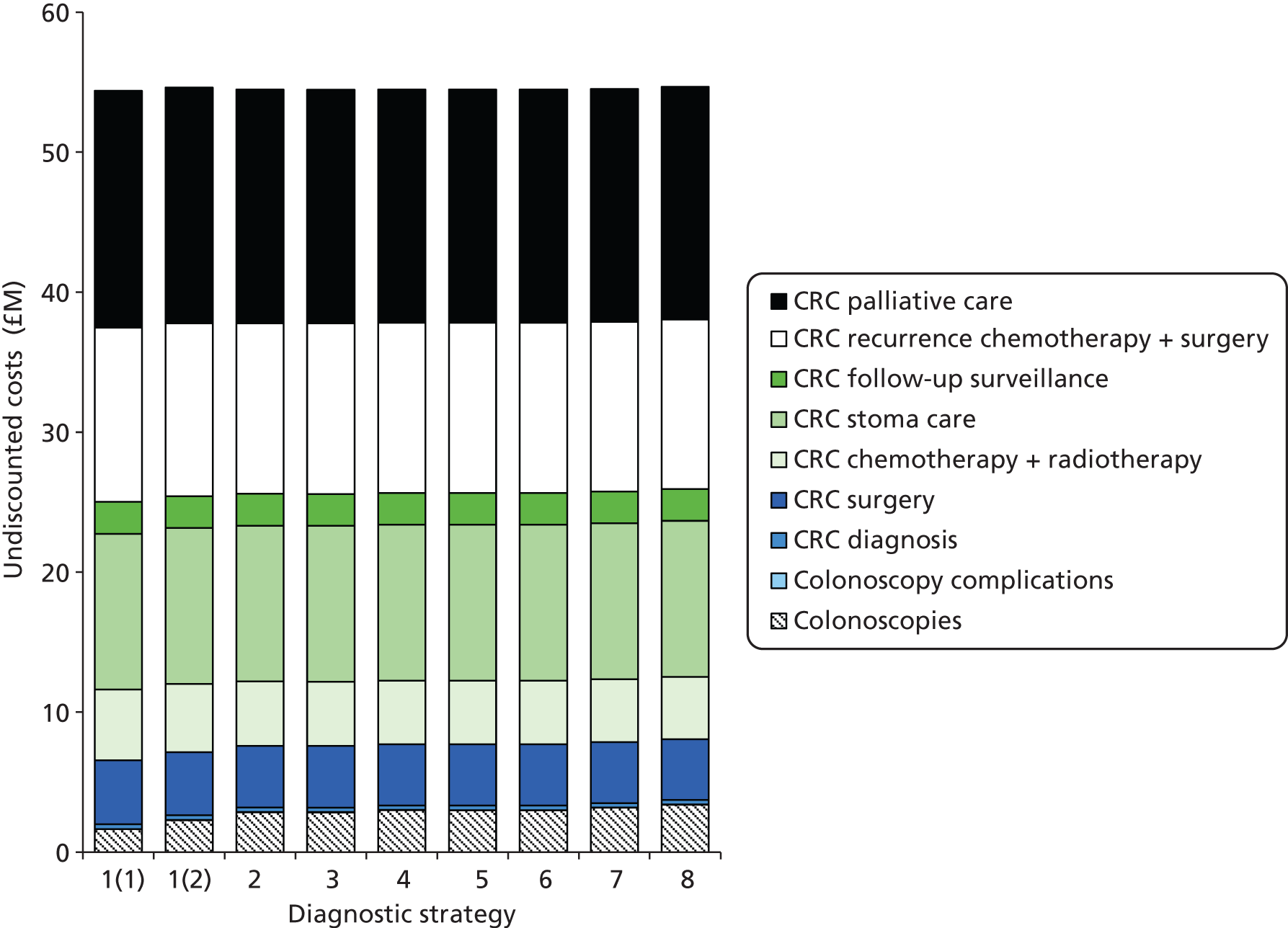
Costs for females diagnosed as having LS (TP, FP) are reduced because the substantial costs of prophylactic TAHBSO are avoided (Figures 85 and 86). Costs for females with LS (FN, TP) are also reduced given that treatment for EC (TAHBSO, chemotherapy and radiotherapy) is avoided.
FIGURE 85.
Undiscounted treatment and prevention costs for female probands, excluding EC.
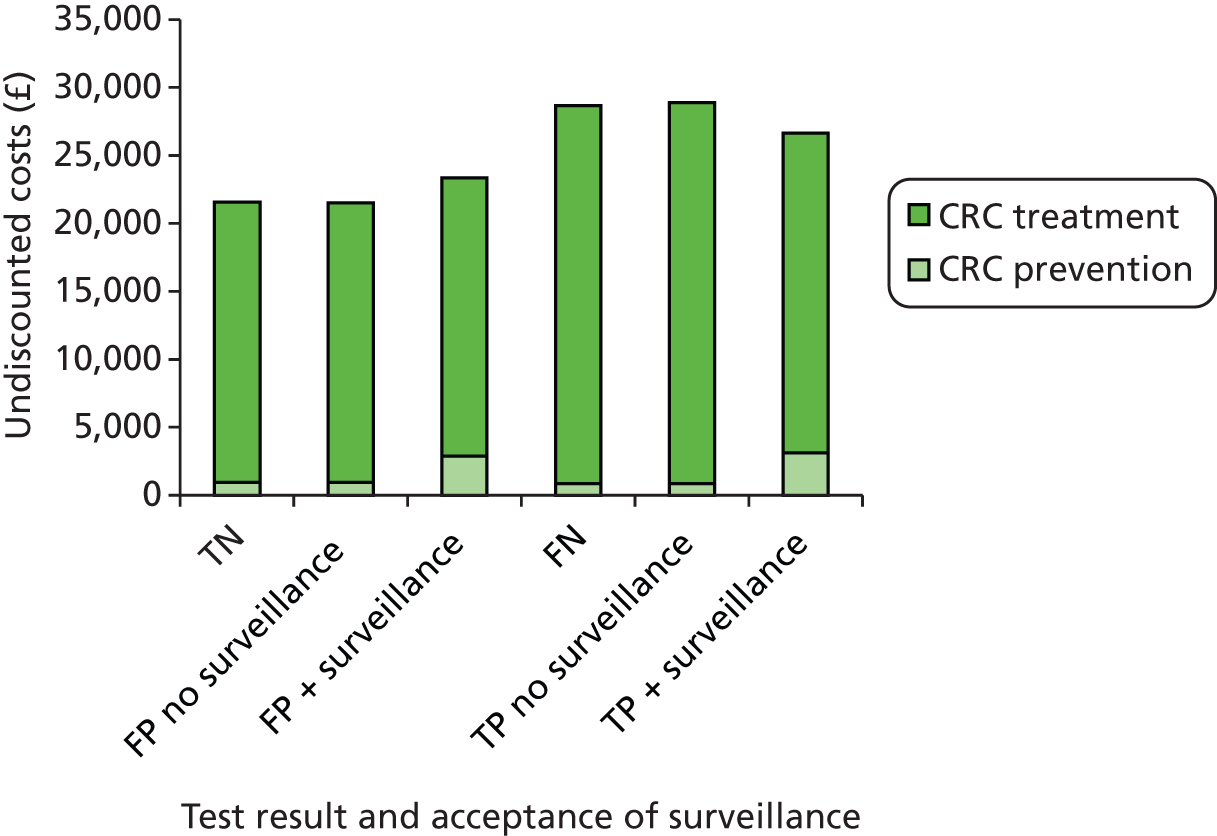
FIGURE 86.
Undiscounted treatment and prevention costs for female relatives, excluding EC
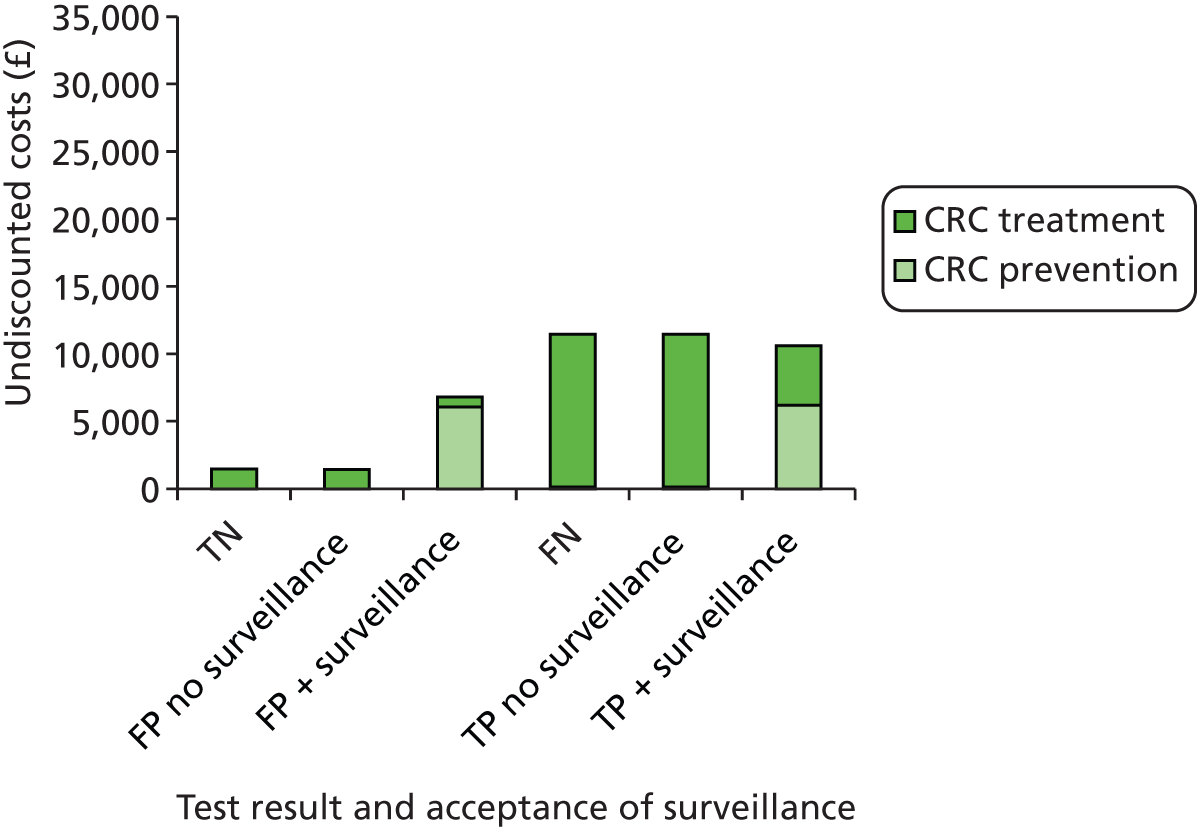
Costs for males and females are now similar, though slightly lower for female relatives, most likely given that they have a reduced risk of CRC compared with male relatives. Similar results occur for female probands.
Additional results
The number of colonoscopies in this scenario is generally very slightly higher than the number of colonoscopies in the base case (Figure 87). This is primarily due to the slightly increased life expectancy for females with LS, who no longer die from EC.
FIGURE 87.
Total number of colonoscopies conducted in England per annum in each testing strategy, excluding EC.
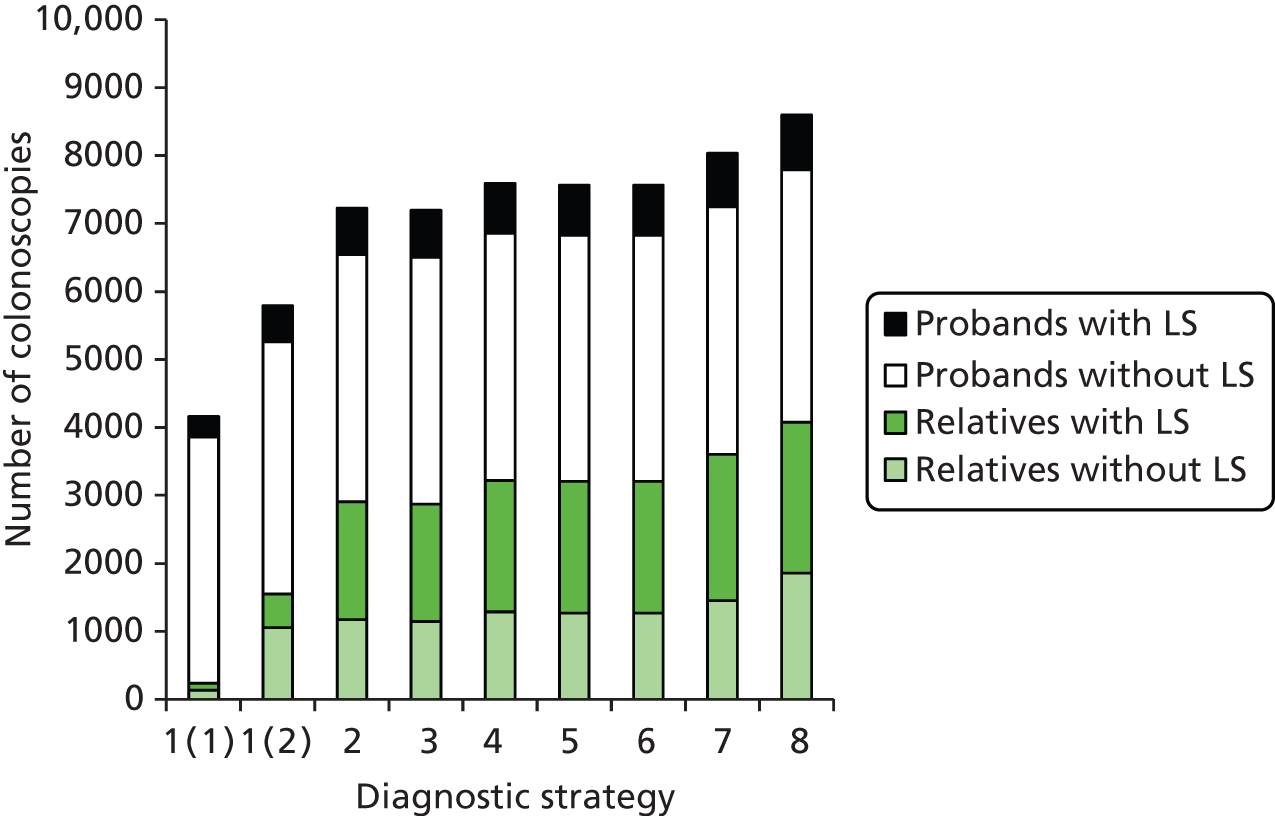
The proportion of female relatives and probands developing CRCs also increases very slightly, for the same reason (Figure 88).
FIGURE 88.
Proportion of relatives who develop primary CRC and proportion of probands who develop metachronous CRC, excluding EC.
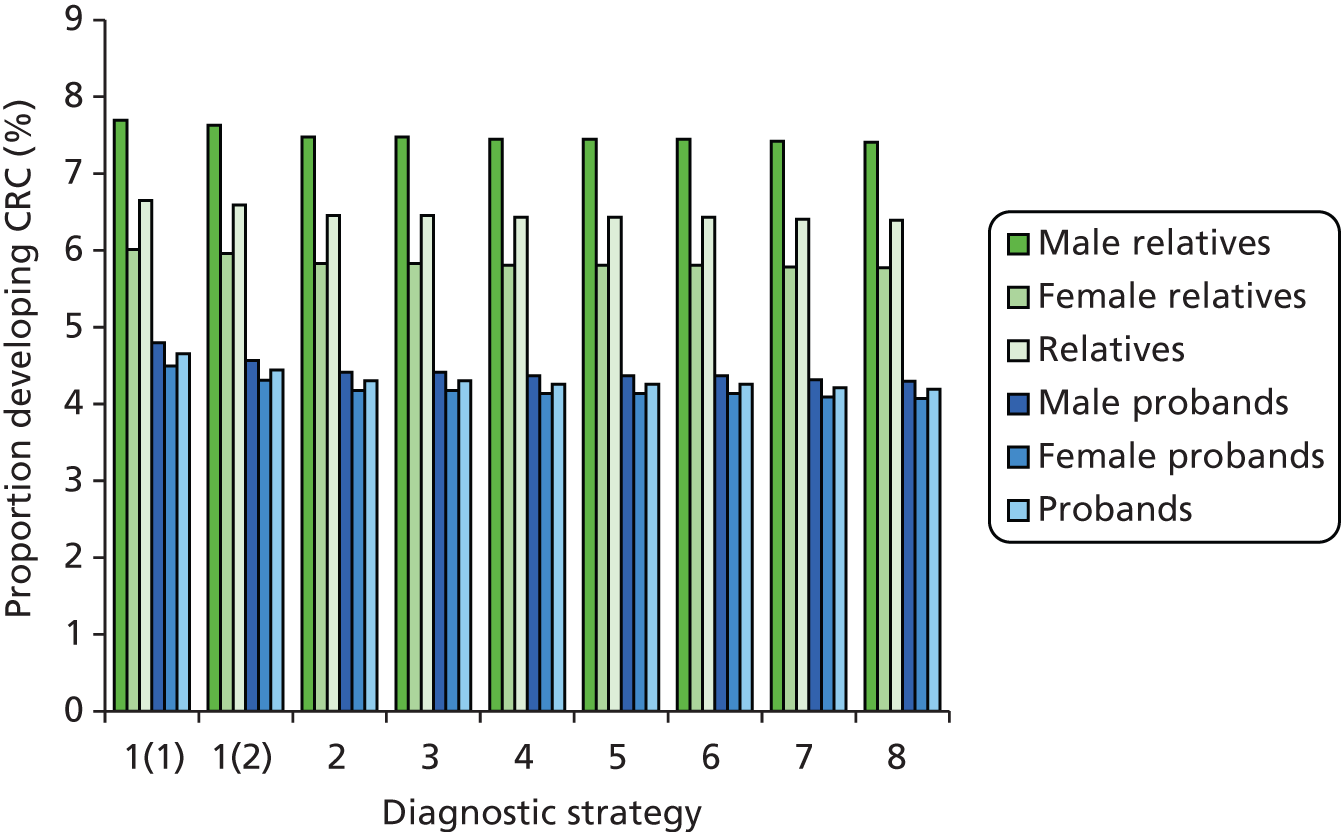
Cost-effectiveness results
Given the reduced costs and slight increase in life expectancy, plus no disutility from EC, the costs for all strategies are reduced from the base case and the QALYs increased. The incremental QALY gains compared with strategy 1(1) are lower for all testing strategies than in the base case (Figure 89), but given the much larger reductions in costs due to the lack of prophylactic TAHBSO (Figures 90 and 91), the ICERs compared with strategy 1(1) are reduced compared with the base case, i.e. all strategies become more cost-effective compared with no testing. This may seem counter-intuitive, as one might expect the cost-effectiveness of testing to improve as more people with LS are correctly identified. In a sense, the result is more a reflection of the cost-effectiveness of treatment for EC. It is important to reiterate that our base-case analysis does not account for the benefit gained from prophylactic TAHBSO in the prevention of OC.
FIGURE 89.
Incremental QALY gains, excluding EC.
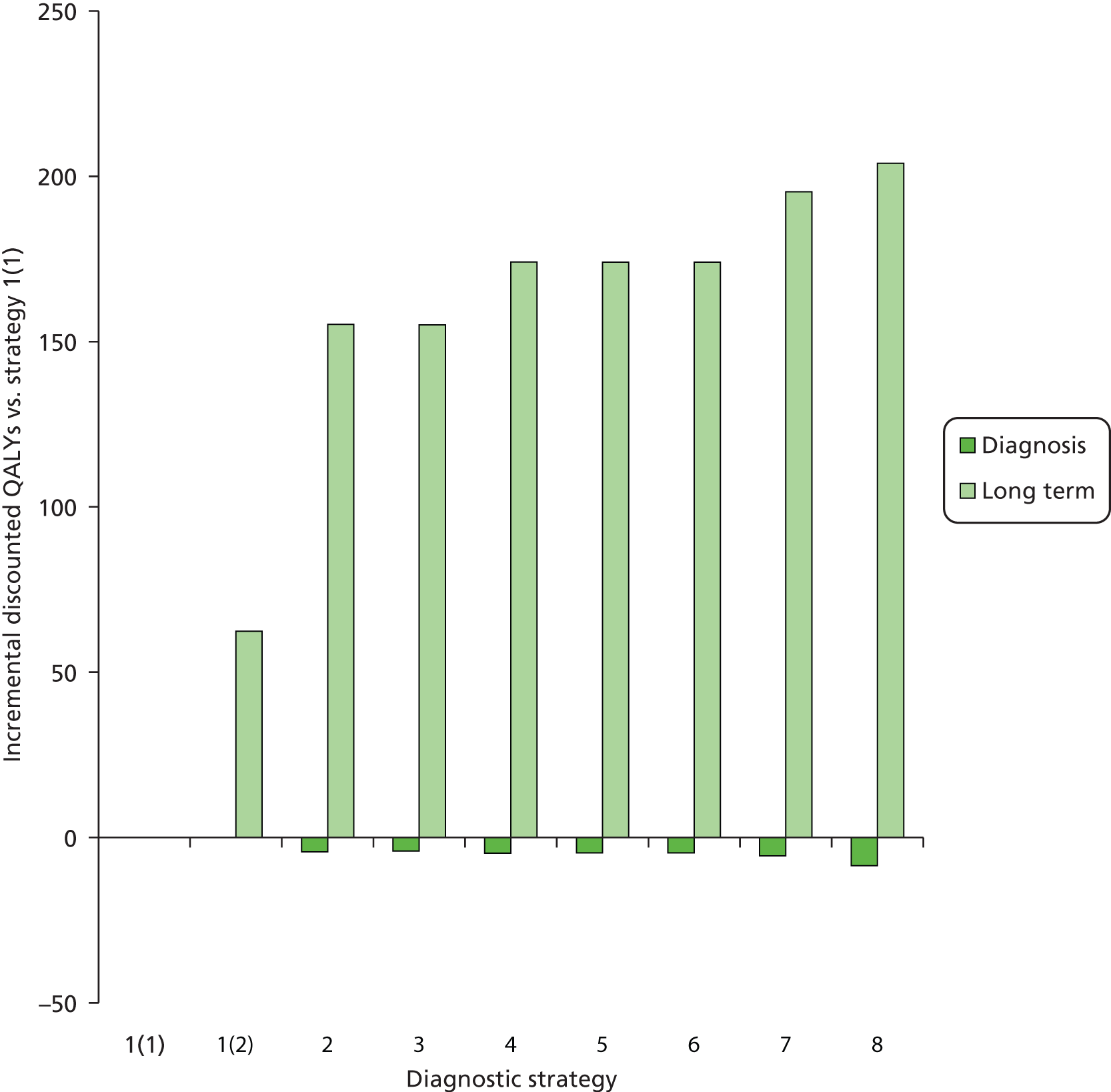
FIGURE 90.
Disaggregated incremental long-term costs, excluding EC.
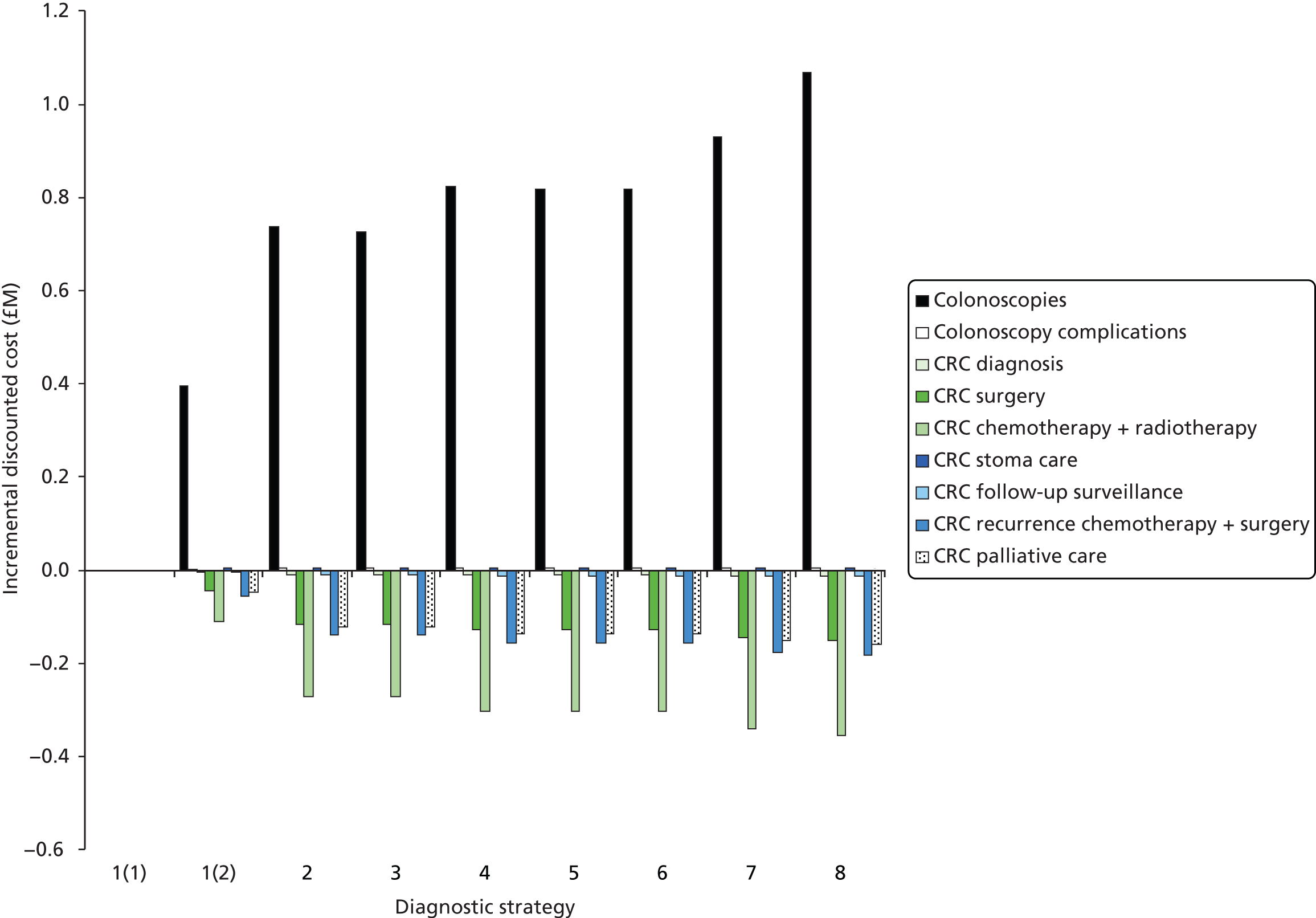
FIGURE 91.
Incremental discounted testing, prevention and treatment costs, excluding EC.
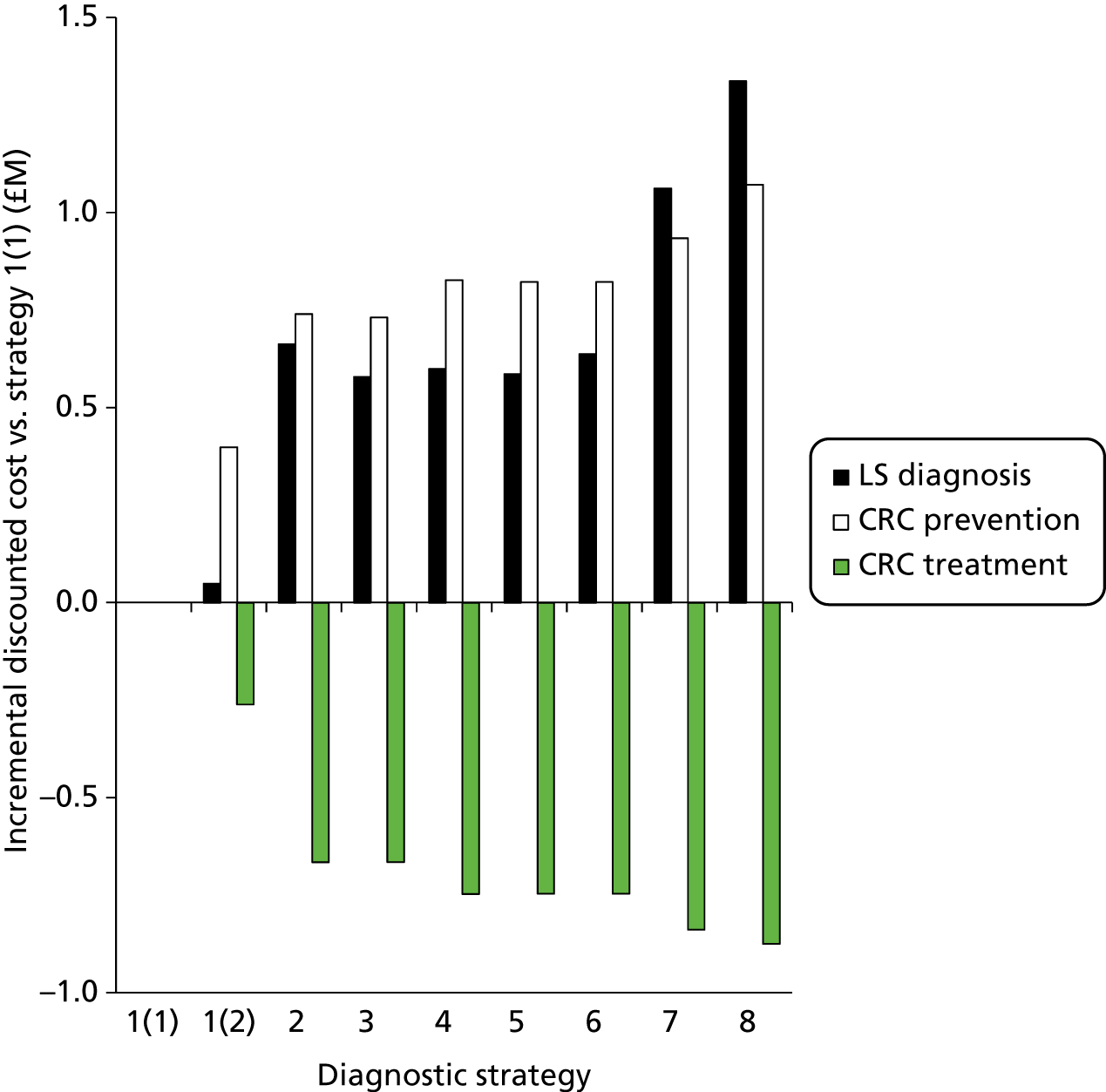
The ranking of cost-effectiveness among strategies remains the same, with strategy 5 still giving the largest net health benefit (Table 102).
| Strategy | Total discounted QALYs | Total discounted cost (£) | Total NHB at £20,000 per QALY | Incremental discounted QALYs vs. strategy 1(1) | Incremental discounted cost vs. strategy 1(1) (£) | INHB at £20,000 per QALY vs. strategy 1(1) | ICER vs. strategy 1(1) (£/QALY) | ICER (£/QALY) |
|---|---|---|---|---|---|---|---|---|
| 1(1) | 151,904 | 36,180,910 | 150,095 | – | – | – | – | – |
| 1(2) | 151,967 | 36,367,105 | 150,148 | 62 | 186,194 | 53.1 | 2985 | 2985 |
| 2 | 152,055 | 36,917,459 | 150,209 | 151 | 736,549 | 114.1 | 4881 | Dominated by strategy 6 |
| 3 | 152,055 | 36,825,081 | 150,214 | 151 | 644,171 | 118.7 | 4268 | Extended dominated by strategies 1(2) and 5 |
| 4 | 152,074 | 36,860,061 | 150,231 | 169 | 679,151 | 135.4 | 4011 | Dominated by strategy 5 |
| 5 | 152,074 | 36,841,931 | 150,232 | 169 | 661,021 | 136.3 | 3903 | 4439 |
| 6 | 152,074 | 36,892,750 | 150,229 | 169 | 711,840 | 133.8 | 4203 | Dominated by strategy 5 |
| 7 | 152,094 | 37,338,139 | 150,227 | 190 | 1,157,229 | 132.0 | 6096 | 24,231 |
| 8 | 152,100 | 37,713,594 | 150,214 | 195 | 1,532,684 | 118.8 | 7844 | 67,327 |
Scenario analysis 2 (BRAF replaced by methylation testing) results
There is currently one further test used in the diagnostic process for LS diagnosis: a methylation test to check for hypermethylation. It is used in a similar manner to BRAF, after MSI or an abnormal MLH1 IHC result to rule out sporadic cancers. As such, we considered it important to conduct a scenario analysis where methylation was used in place of BRAF. Details of this scenario, including in-depth results, can be found in Appendix 15, as we only present the main cost-effectiveness findings here.
Using methylation testing in the place of BRAF only alters the results of the strategies that use BRAF. Table 103 therefore demonstrates little change from the base case, with the ICERs versus strategy 1(1) remaining < £10,000 per QALY. However, the strategies that use methylation now gain fewer QALYs than the similar strategies that do not use methylation (compare strategies 3 and 2, or 5 and 4), thereby altering the order of effectiveness of the strategies. Strategy 5 still has the smallest ICER compared with no testing, but strategy 4 now has the greatest INHB compared with strategy 1(1) at a willingness to pay of £20,000 per QALY.
| Strategy | Total discounted QALYs | Total discounted cost (£) | Total NHB at £20,000 per QALY | Incremental discounted QALYs vs. strategy 1(1) | Incremental discounted cost vs. strategy 1(1) (£) | INHB at £20,000 per QALY | ICER vs. strategy 1(1) (£/QALY) | ICER (£/QALY) |
|---|---|---|---|---|---|---|---|---|
| 1(1) | 151,793 | 36,223,787 | 149,982 | – | – | – | – | – |
| 1(2) | 151,857 | 36,608,672 | 150,027 | 64 | 384,885 | 44.7 | 6021 | Extended dominated by strategies 1(1) and 5 |
| 2 | 151,953 | 37,253,017 | 150,091 | 160 | 1,029,230 | 108.3 | 6444 | Dominated by strategy 4 |
| 3 | 151,949 | 37,144,423 | 150,092 | 156 | 920,635 | 109.9 | 5904 | Dominated by strategy 5 |
| 4 | 151,973 | 37,229,210 | 150,111 | 179 | 1,005,423 | 129.0 | 5610 | 7965 |
| 5 | 151,960 | 37,131,507 | 150,104 | 167 | 907,719 | 121.6 | 5436 | 5436 |
| 6 | 151,960 | 37,165,805 | 150,102 | 167 | 942,018 | 119.9 | 5642 | Dominated by strategy 5 |
| 7 | 151,992 | 37,702,908 | 150,106 | 198 | 1,479,121 | 124.1 | 7466 | 25,108 |
| 8 | 152,000 | 38,198,324 | 150,090 | 206 | 1,974,537 | 107.6 | 9571 | 60,472 |
Comparing strategies simultaneously, strategies 1(2), 2, 3 and 6 are still dominated by other strategies. The ICER for strategy 5 is slightly reduced to £5436 per QALY gained over strategy 1(1), but strategy 4 is no longer extended dominated, instead having an ICER versus strategy 5 of £7965 per QALY gained. The ICERs for strategies 7 and 8 remain above £25,000 per QALY. Observing these results together, it would appear that strategy 4 (MSI followed by genetic testing) is now the most cost-effective strategy at a willingness to pay of £20,000 per QALY.
We conclude that using methylation in place of BRAF can still be cost-effective at a willingness to pay of £20,000 per QALY compared with no testing, but that compared with other strategies (and the results from BRAF), it may not be most cost-effective.
Scenario analysis 3 (changing the age limit for probands)
In an extension of the scope of our work, we examine the consequences of extending the testing for LS to a wider population. We raise the age limit for testing probands from 50 years in our base case to age 60 years, and then to age 70 years.
When the age limit of the probands is raised, the prevalence of LS in probands falls (Table 104) because the incidence of CRC in the general population rises more rapidly than the incidence of CRC in people with LS. Using the study by Hampel and colleagues120 again as our source of LS prevalence in a newly diagnosed population, we find that the prevalence falls from 8.4% at maximum age of 50 years to 5.7% at an age limit of 60 years and 3.8% at an age limit of 70 years. The total annual incidence of cases of CRC in England increases from 1699 at maximum age 50 years, to 5018 at maximum age 60 years, to 13,900 at maximum age 70 years. In addition, the following parameters are affected by the increase in maximum proband age: the proportion of probands who are male, and the proportion of general population patients with colon cancer versus rectal cancer.
| Input parameter | Maximum proband age (years) | Source | ||
|---|---|---|---|---|
| < 50 (base case) | < 60 | < 70 | ||
| Prevalence of LS in probands | 8.4% | 5.7% | 3.8% | Hampel et al. 2008120 |
| Number of probands per annum in England | 1699 | 5018 | 13,900 | ONS Cancer Registration Statistics, England 2010119 |
| Proportion of probands male | 54.8% | 56.0% | 59.5% | ONS Cancer Registration Statistics, England 2006–10119,136–139 |
| Proportion of general population male probands with colon (not rectal) cancer | 58.4% | 54.4% | 56.9% | ONS Cancer Registration Statistics, England 2010119 |
| Proportion of general population female probands with colon (not rectal) cancer | 60.9% | 61.3% | 65.0% | ONS Cancer Registration Statistics, England 2010119 |
The number of relatives increases from 8495 in the base case to 25,090 when the age limit of the probands is raised to 60 years and 69,500 when the age limit is raised to 70 years. As in the base case, the age and other characteristics of relatives are not linked to the age of the probands. Therefore, although the age distribution of the probands alters, the age distribution of the relatives does not.
Maximum age of probands at diagnosis 60 years
Diagnostic results
The change in prevalence of LS in the probands has a surprising effect on the diagnostic results. One might expect that altering the prevalence of a condition in a population should not affect the sensitivity or specificity of a test (or testing strategy), yet our results demonstrate a slight improvement in overall specificity compared with the base case (Figure 92). This is driven by the ratio of the number of false mutation-positive probands to false LS-assumed probands (which significantly alters the diagnoses of the relatives), and occurs as a result of having separate genetic tests for M126 and PMS2.
FIGURE 92.
Receiver operating characteristic plot for all patients (probands and relatives) when age limit of probands is raised to 60 years.
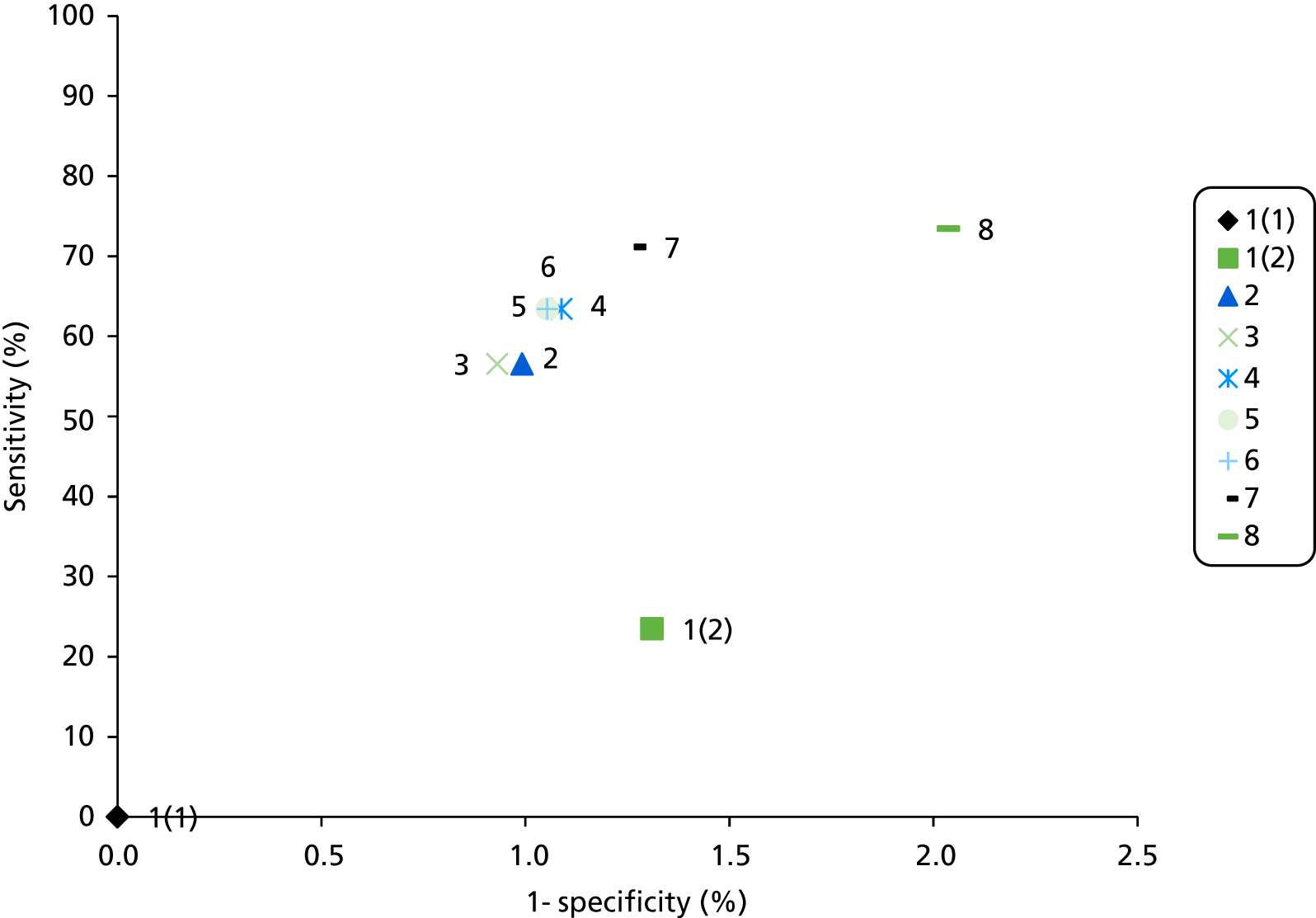
When probands undergo the M126 genetic test, those who are positive for a PMS2 mutation cannot test positive for any other mutation. Therefore, they are part of the test-negative cohort and must make up a section of the cohort who truly test negative on the M126 genetic test. When the prevalence in the probands changes, the ratio is altered between those testing negative because they are LS negative and those testing negative because they are PMS2 positive. As the probands who test negative for M126 go on to further testing (and, if they are not ruled out by FH, become either mutation positive on PMS2 or LS assumed), this alters the ratio between the TN, the FP-assumed and the false mutation-positive probands. The larger number of relatives, whose diagnoses depend on the diagnoses of the probands, exacerbates the results of this difference, resulting in the change in specificity. Sensitivity is unchanged as the FNs and TPs are unaffected.
Life expectancy
The life expectancy of relatives remains fairly similar to the base case, regardless of LS status, owing to the relatives remaining mostly unaffected by the change in the proband age limit (Figure 93). The life expectancy of probands, however, falls regardless of LS status (Figure 94, compared with Figure 53), owing to the higher mean age of probands and consequently shorter life expectancy. The average age of probands in the base case is 41.1 years compared with 49.4 years when the age limit is increased to 60 years.
FIGURE 93.
Life expectancy for relatives, by LS status, when proband age limit increases to 60 years.
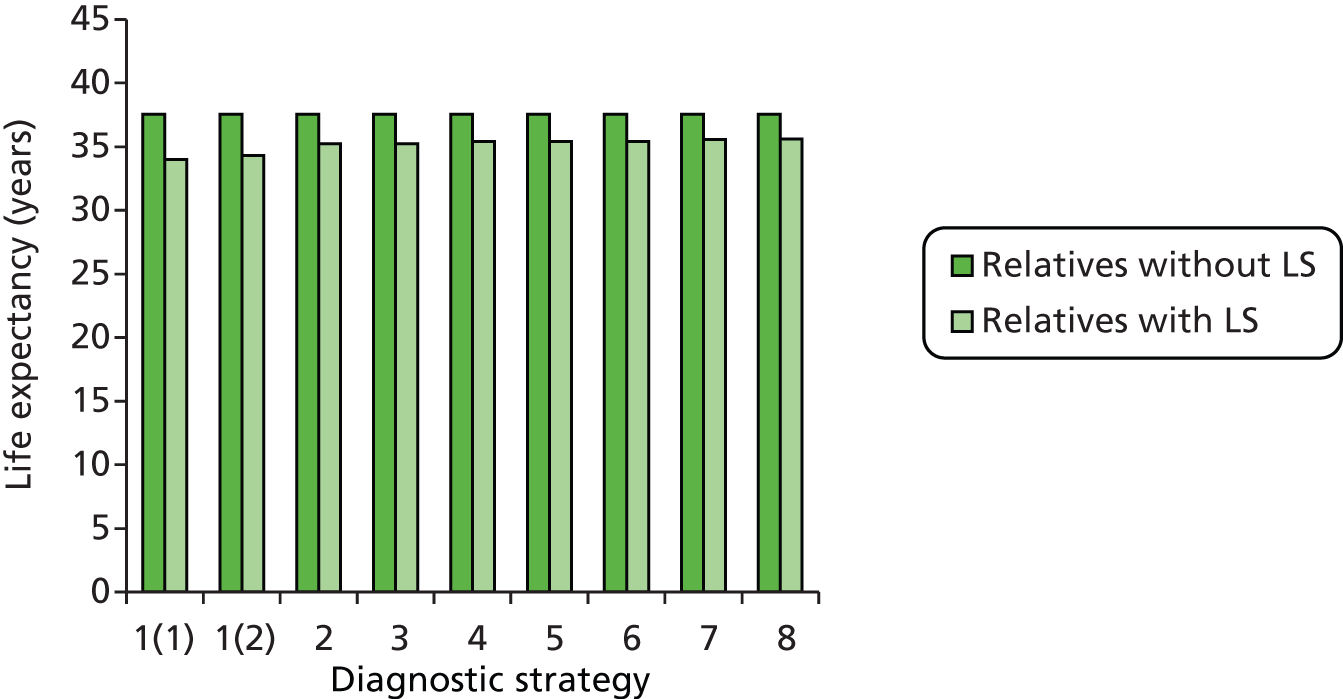
FIGURE 94.
Life expectancy for probands, by LS status, when proband age limit increases to 60 years.
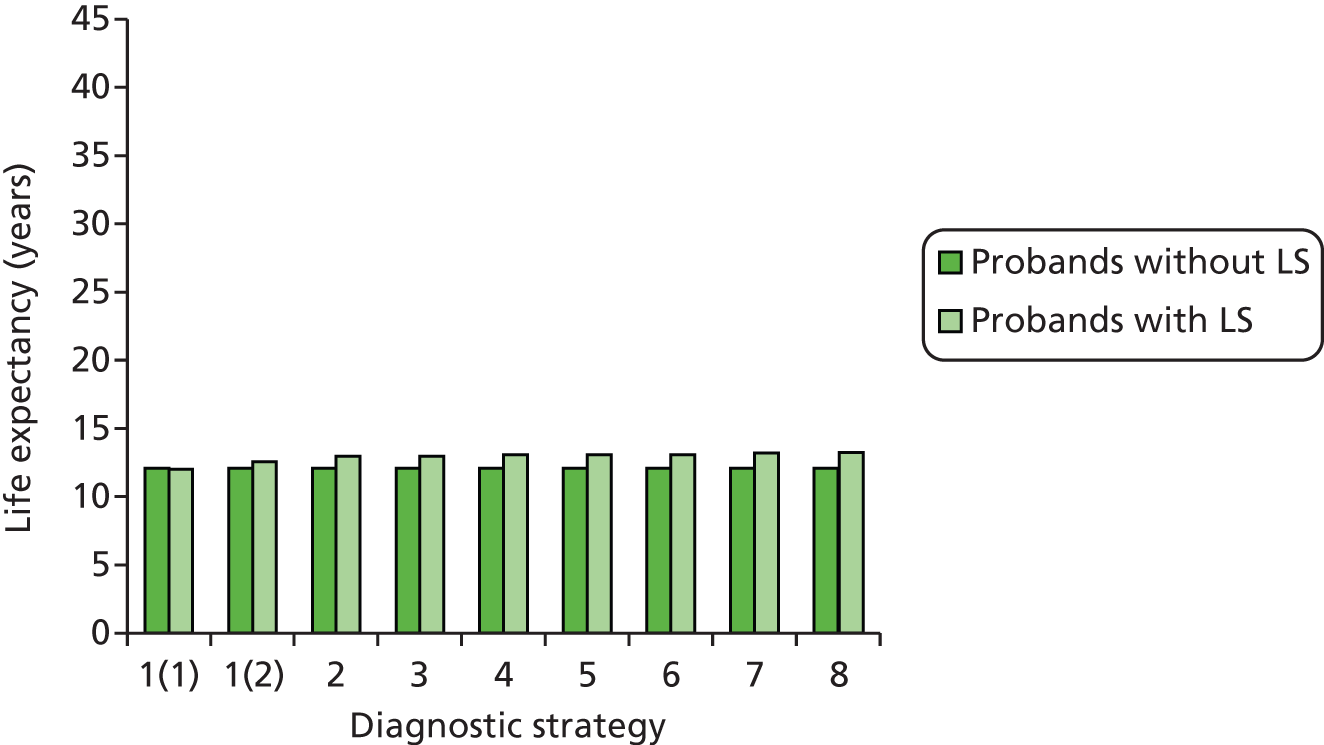
Cost results
As expected, total costs increase dramatically and approximately in proportion to the increase in the number of probands and relatives (Figure 95). The distribution of these costs across strategies is very similar to that in the base case. For example, strategy 5 testing costs are lower than those for strategies 4 or 6 (Figure 96). Differences in long-term costs are also similar across strategies (Figure 97) compared with the base case.
FIGURE 95.
Total undiscounted costs for each strategy.
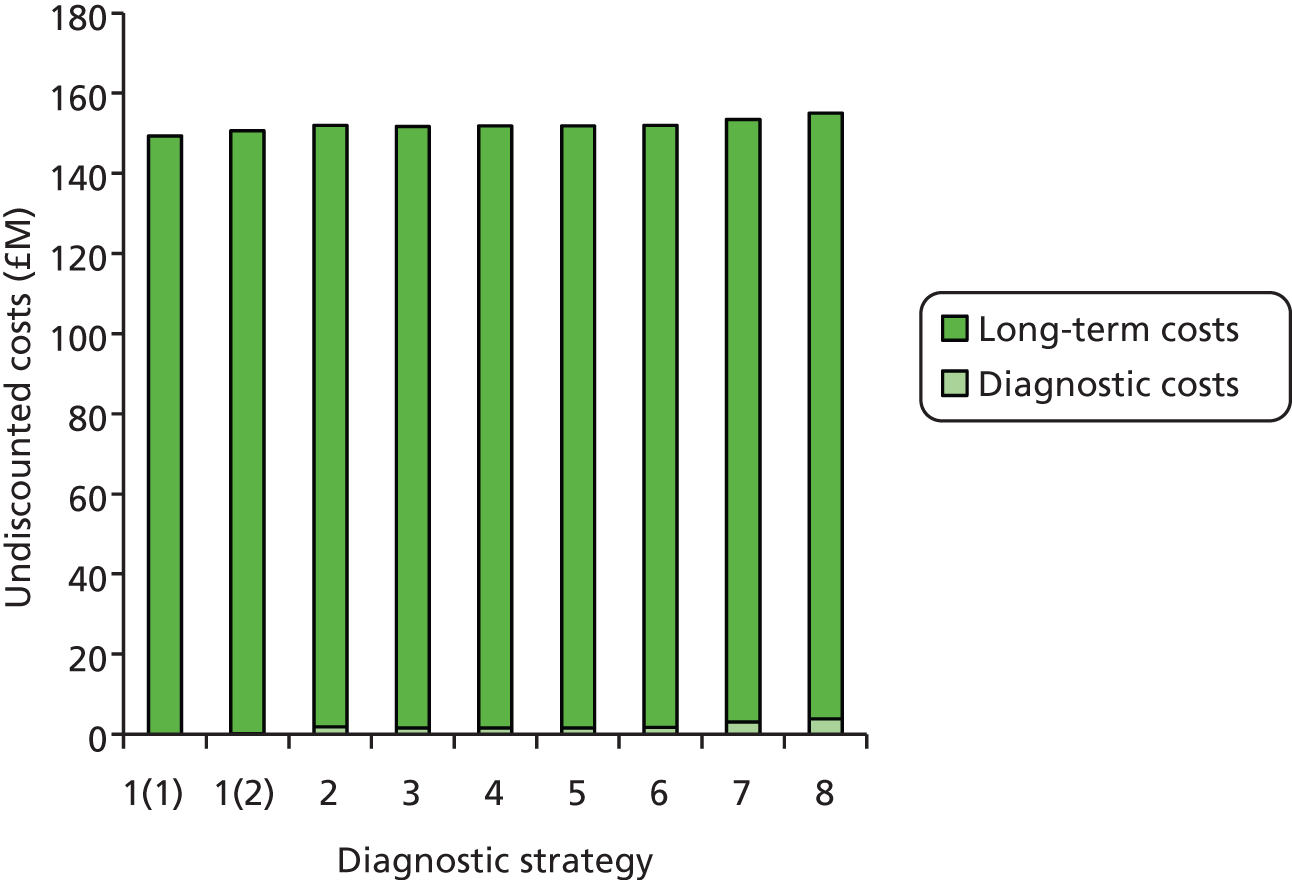
FIGURE 96.
Total costs per annum in England of diagnostic tests for probands and relatives combined.
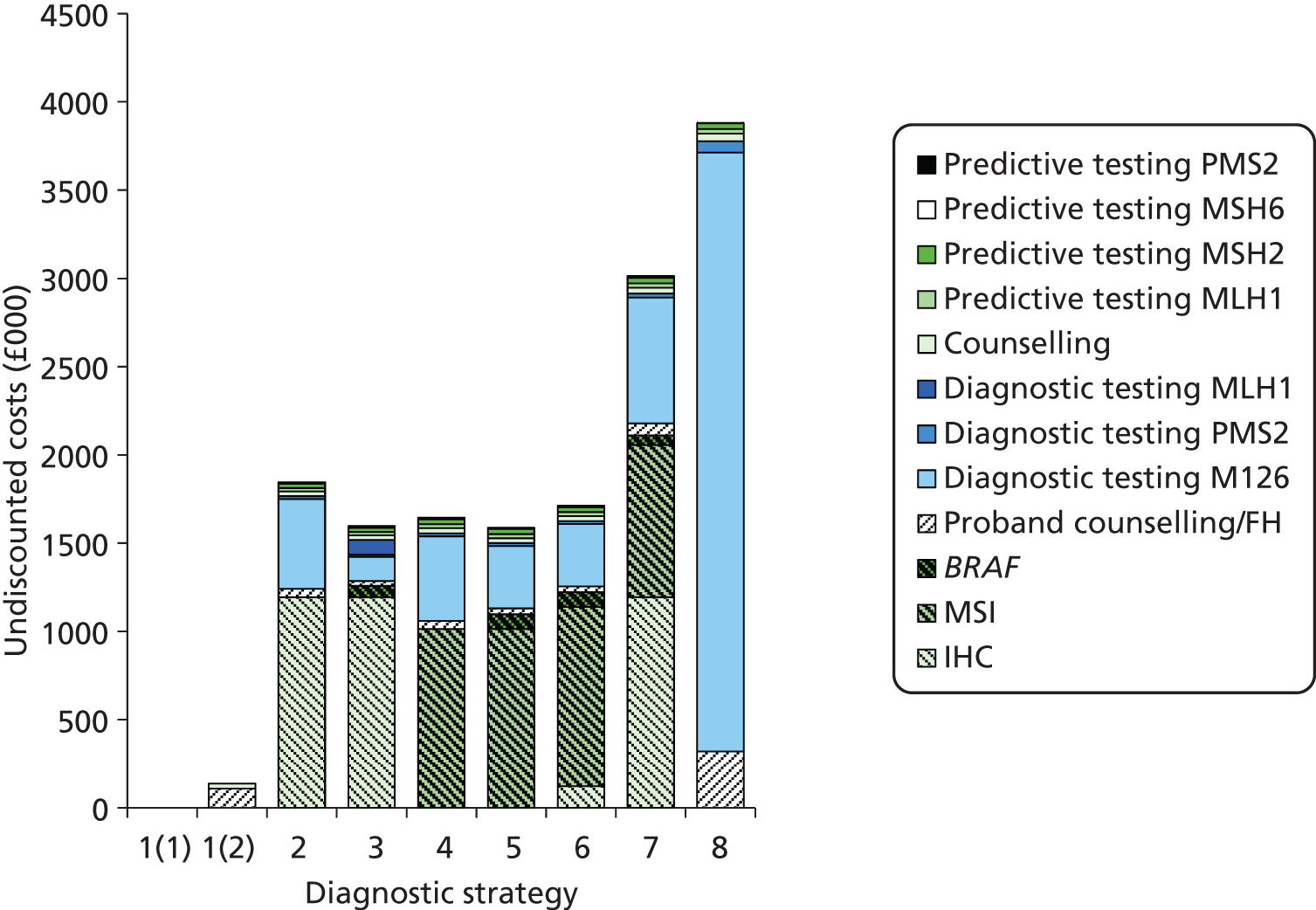
FIGURE 97.
Total undiscounted long-term costs disaggregated by test strategy.
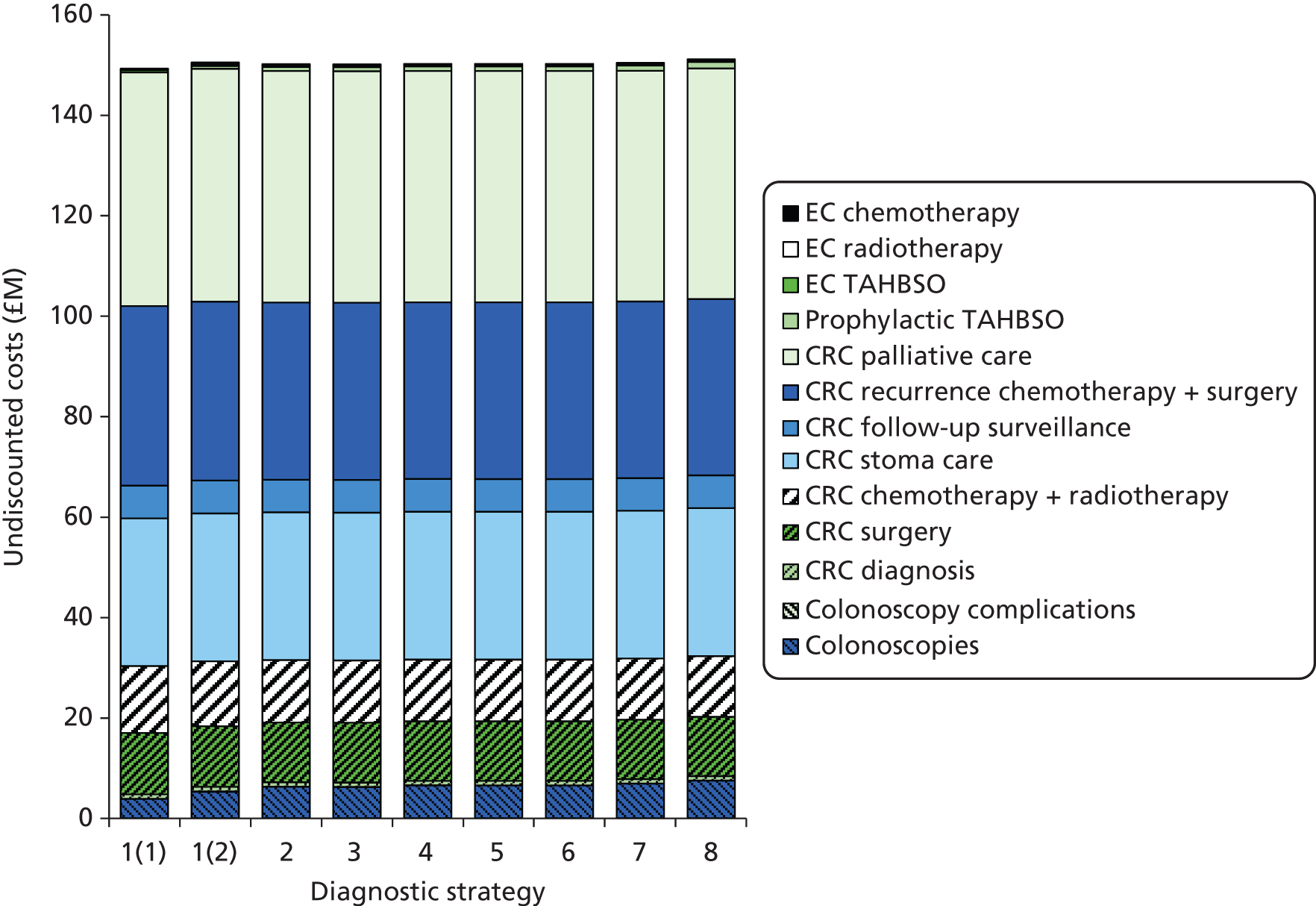
Additional results
Similar to the costs results, the overall number of colonoscopies increases purely on the basis of the number of patients increasing (Figure 98). The proportional increase in the number of colonoscopies is lower than the proportional increase in the number of probands and relatives because a smaller proportion of probands are LS positive. The average number of colonoscopies per patient identified as not having LS decreases for the probands from an average of 2.3–2.7, depending on the strategy, to 1.8–2.0, primarily because of their reduced life expectancy. The number of colonoscopies per relative remains similar to the number in the base case, as their age distribution is assumed unchanged. Figure 99 shows the average number of colonoscopies for individuals with newly diagnosed CRC aged < 60 years and their relatives.
FIGURE 98.
Total number of colonoscopies conducted in England per annum, when proband age limit is increased to 60 years.
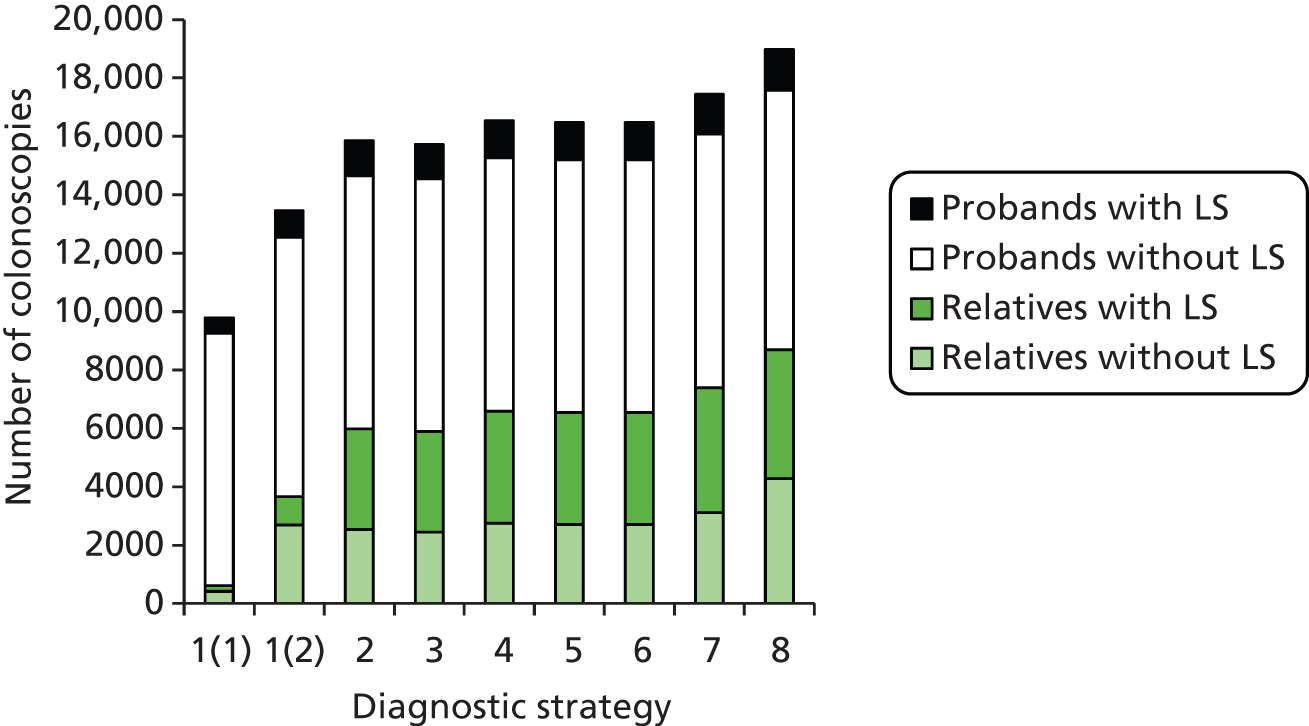
FIGURE 99.
Average number of colonoscopies per patient, when proband age limit is increased to 60 years.
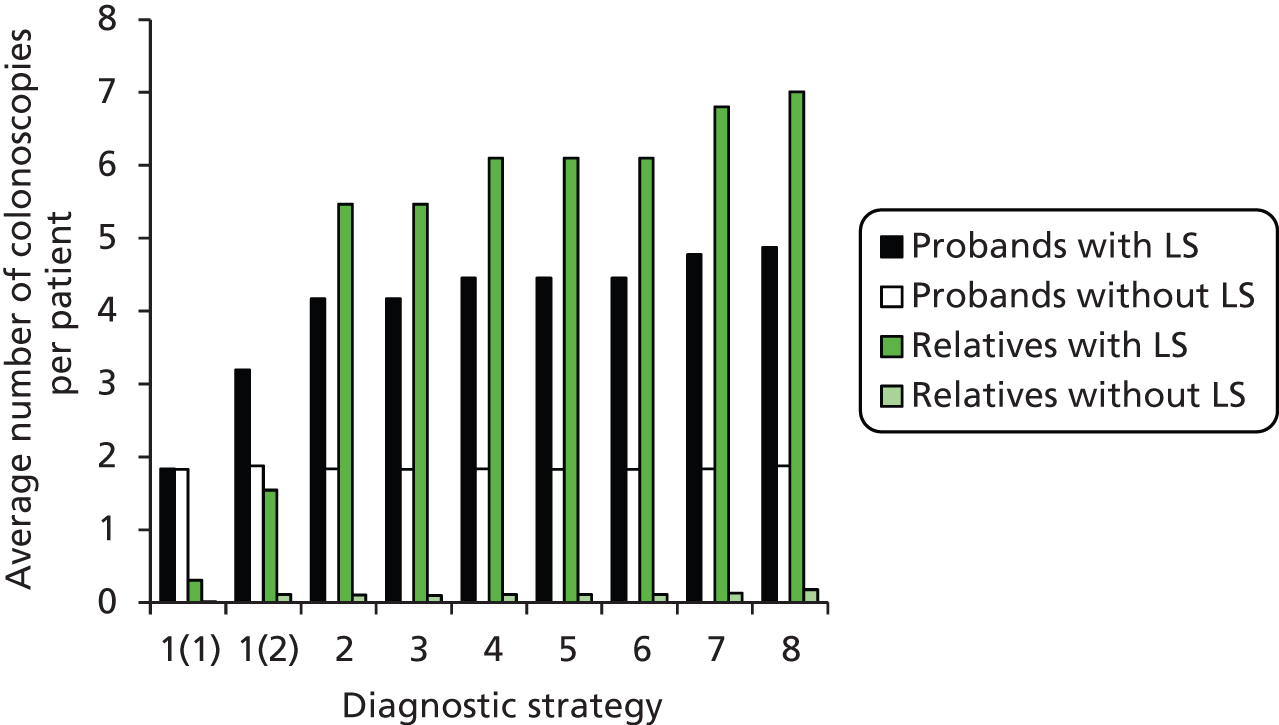
Similarly, the proportion of relatives developing a CRC remains similar to that in the base case, and the proportion of probands developing metachronous CRC reduces (Figure 100). This occurs as the lifetime probability of metachronous CRC reduces for probands, because their life expectancy also reduces (as their mean age at diagnosis increases) (Figure 101; compare with Figure 77).
FIGURE 100.
Proportion of relatives who develop primary CRC and proportion of probands who develop metachronous CRC when age limit of probands is increased to 60 years.
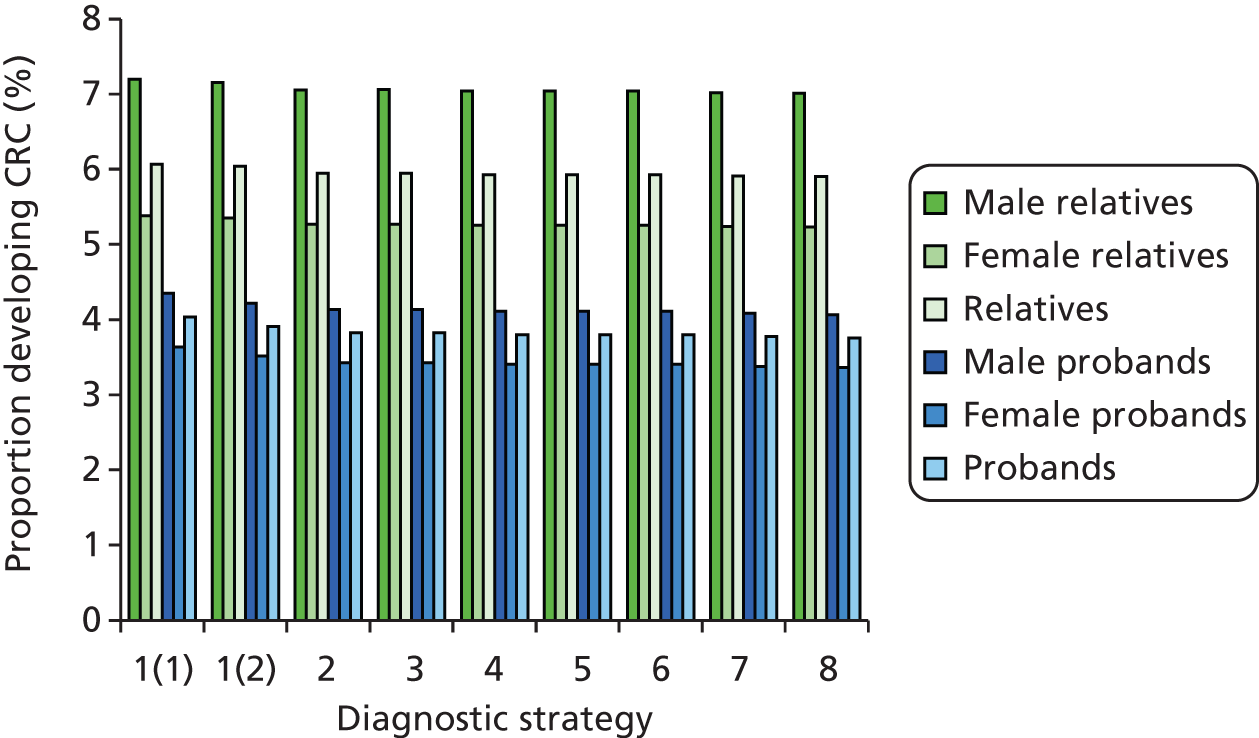
FIGURE 101.
Lifetime risk of metachronous CRC in probands, when the age limit of probands is increased to 60 years.
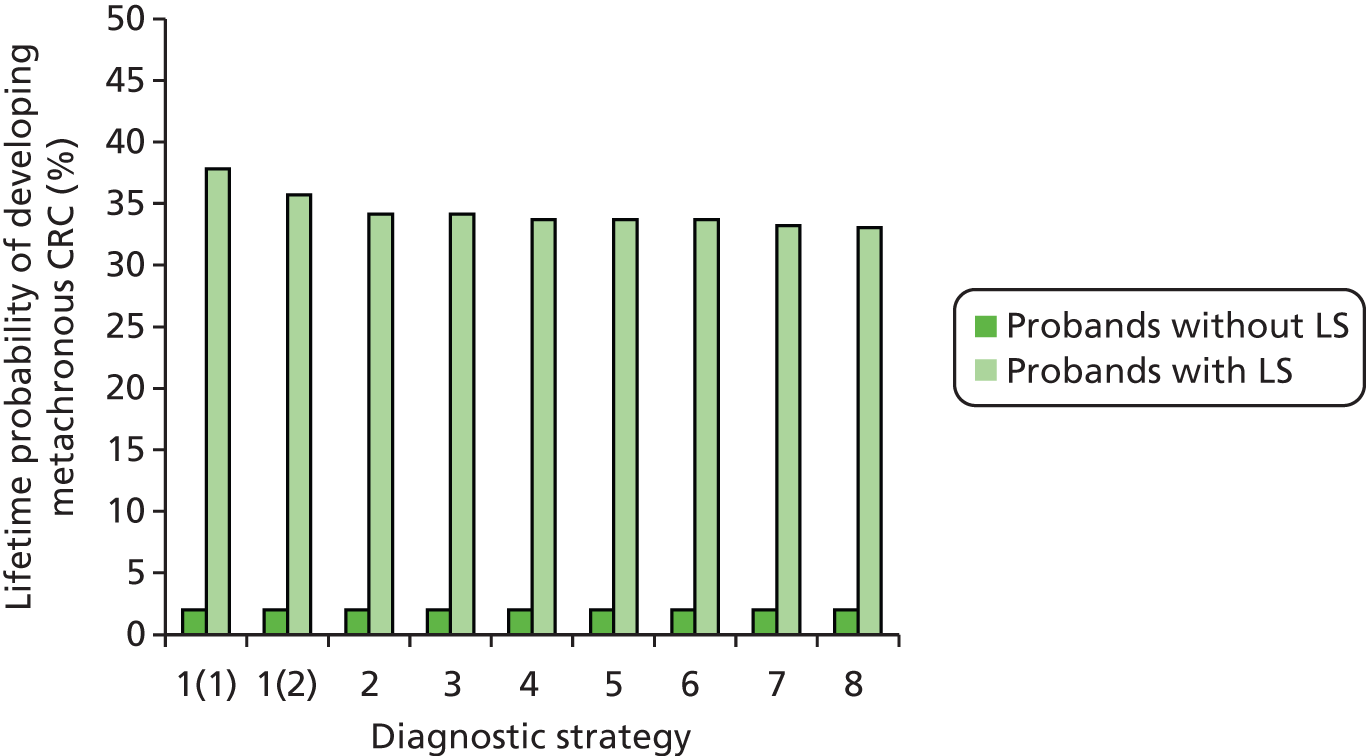
The lifetime risk of EC appears to remain similar to that in the base case for both probands and relatives within the LS population (Figure 102), but there is a reduction in risk for the entire cohort, as there is a smaller proportion of probands and relatives with LS and the probands have a shorter life expectancy.
FIGURE 102.
Lifetime risk of EC when the age limit of probands is increased to 60 years.
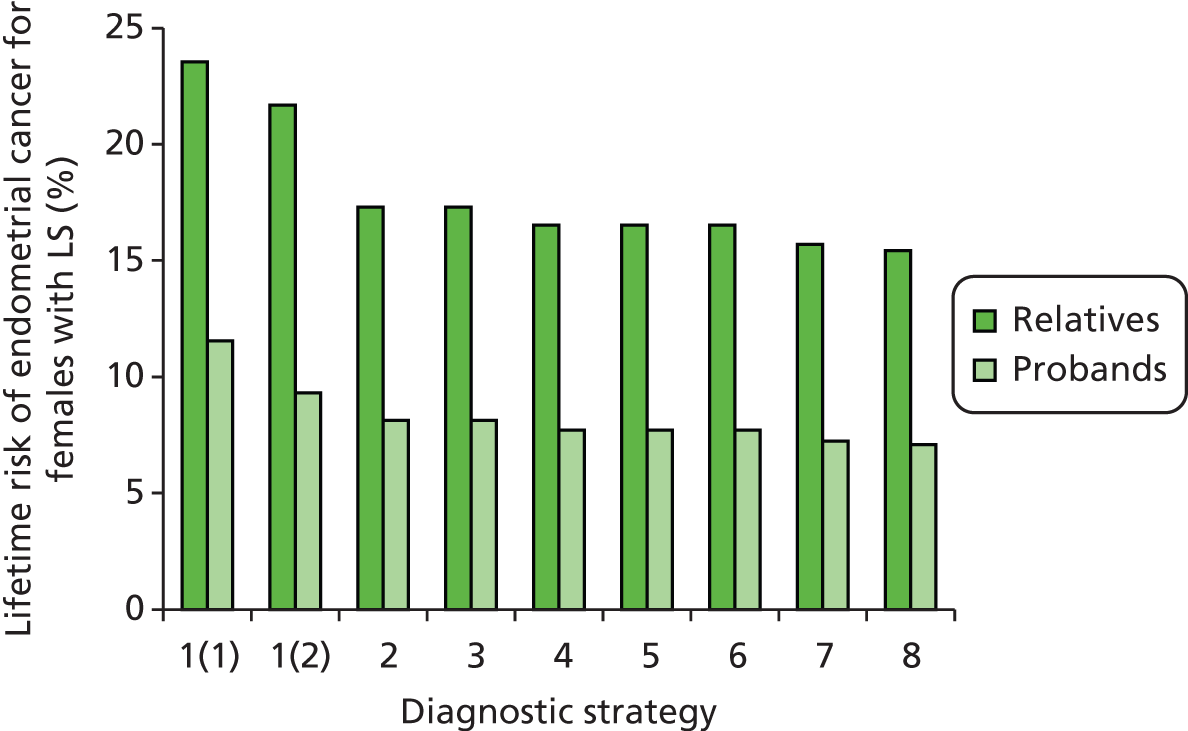
Cost-effectiveness results
The increase in the proband age limit to 60 years increases not only the overall cost per strategy, but also the incremental cost compared with no testing (Figures 103 and 104). The incremental cost of LS diagnosis is now much increased compared with the base case. Indeed, the proportional increase is greater than the proportional increase in the other cost components: CRC prevention and treatment, and EC prevention and treatment. This is due to the lower prevalence of LS within this population compared with the base-case population and, consequently, the smaller proportion offered surveillance colonoscopy for CRC prevention and TAHBSO for EC prevention. In all other respects, despite obvious increases in the costs and cost savings due to the larger population, the pattern of incremental costs across testing strategies remains similar to that seen in the base case.
FIGURE 103.
Disaggregated long-term incremental costs, when the age limit for probands is increased to 60 years.
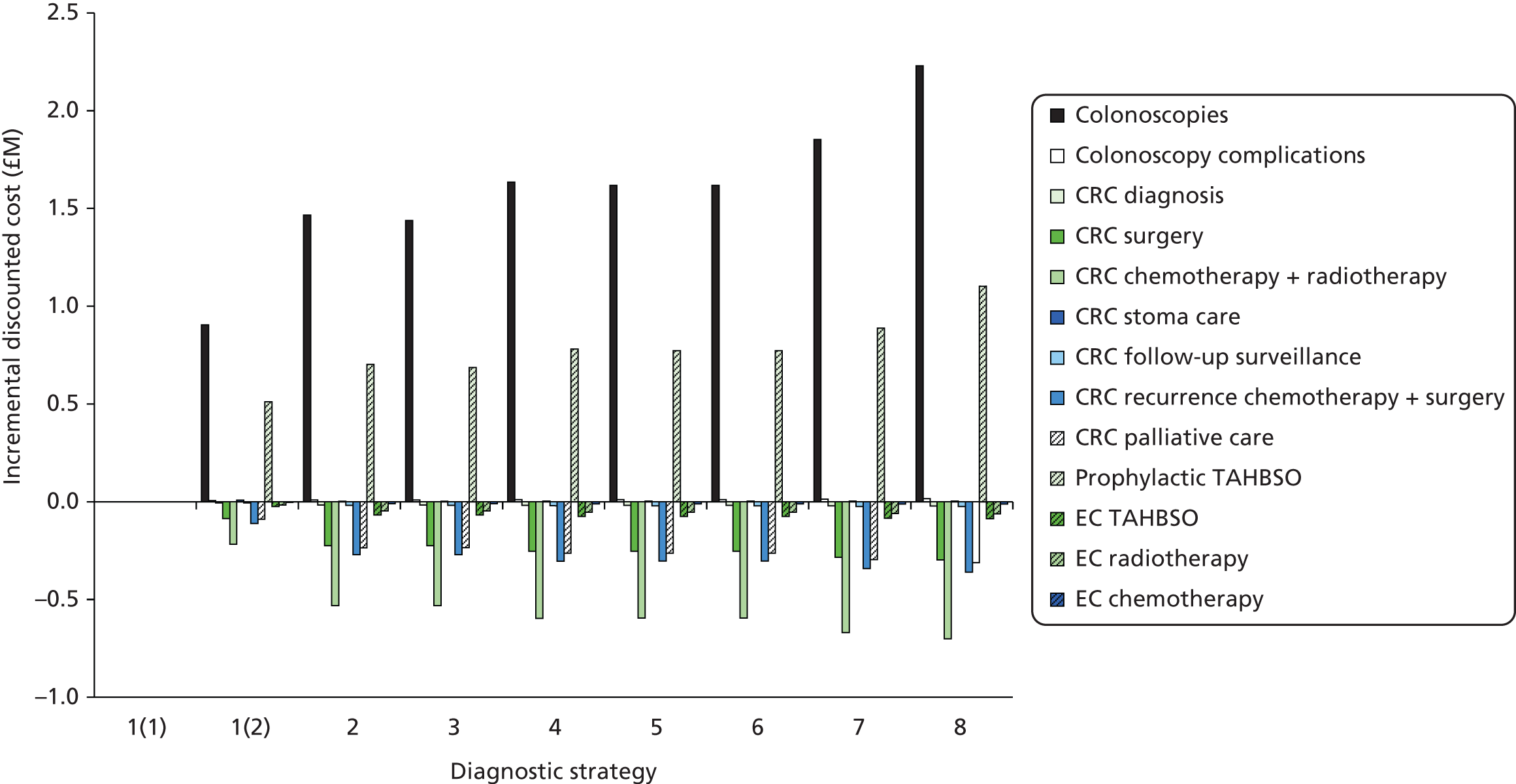
FIGURE 104.
Incremental discounted testing, prevention and treatment costs when the proband age limit is increased to 60 years.
The incremental QALY gains for each strategy compared with strategy 1(1) are also larger than in the base case, though not as pronounced as the increases in costs (Figure 105). The net result is that all test strategies are now slightly worse value for money versus no testing (Table 105). The impact of the QALY loss due to the psychological impact of genetic testing is proportionally larger than in the base case, reflecting the extra numbers undergoing the diagnostic strategies.
FIGURE 105.
Incremental QALY gain compared with strategy 1(1) when the age limit of the probands is increased to 60 years.
| Strategy | Total discounted QALYs | Total discounted cost (£) | Total NHB at £20,000 per QALY | Incremental discounted QALYs vs. strategy 1(1) | Incremental discounted cost vs. strategy 1(1) (£) | INHB at £20,000 per QALY vs. strategy 1(1) | ICER vs. strategy 1(1) (£/QALY) | ICER (£/QALY) |
|---|---|---|---|---|---|---|---|---|
| 1(1) | 445,164 | 102,424,410 | 440,043 | – | – | – | – | – |
| 1(2) | 445,261 | 103,430,520 | 440,089 | 97 | 1,006,110 | 46.3 | 10,414 | Extended dominated by strategies 1(1) and 3 |
| 2 | 445,442 | 105,031,265 | 440,191 | 278 | 2,606,855 | 147.6 | 9380 | Dominated by strategy 6 |
| 3 | 445,443 | 104,742,550 | 440,206 | 279 | 2,318,140 | 163.1 | 8308 | Extended dominated by strategies 1(2) and 5 |
| 4 | 445,477 | 104,906,319 | 440,231 | 312 | 2,481,909 | 188.2 | 7947 | Dominated by strategy 5 |
| 5 | 445,477 | 104,827,821 | 440,236 | 313 | 2,403,411 | 192.8 | 7681 | 7681 |
| 6 | 445,477 | 104,952,238 | 440,230 | 313 | 2,527,829 | 186.5 | 8078 | Dominated by strategy 5 |
| 7 | 445,514 | 106,409,224 | 440,193 | 349 | 3,984,815 | 150.0 | 11,409 | 43,501 |
| 8 | 445,512 | 107,791,681 | 440,122 | 347 | 5,367,271 | 79.0 | 15,453 | Dominated by strategy 7 |
Despite a slight deterioration in cost-effectiveness, the INHB at the population level compared with no testing increases in all strategies except universal genetic testing, compared with the base case. This increase is expected, as the diagnostic testing now benefits a larger group of patients. Universal genetic testing is the exception given that testing costs increase substantially.
All strategies are still cost-effective compared with no testing at a threshold of £20,000 per QALY gained, as demonstrated by Figure 106. Strategy 5 gives the greatest INHB of 193 discounted QALYs, and hence is the most cost-effective, at a willingness to pay of £20,000 per QALY. This compares with 130 discounted QALYs for this strategy in the base-case analysis.
FIGURE 106.
Total discounted costs and QALYs for all probands and relatives per annum in England by testing strategy, when proband age limit is increased to 60 years.
Maximum age of probands at diagnosis 70 years
The changes in the life expectancy and costs described in the previous section, given a maximum proband age at diagnosis of 60 years, follow a pattern similar to that given a maximum proband age of 70 years (Figure 107). Therefore, we present only the main cost-effectiveness results.
FIGURE 107.
Disaggregated incremental long-term costs compared with strategy 1(1) when proband age limit is increased to 70 years.
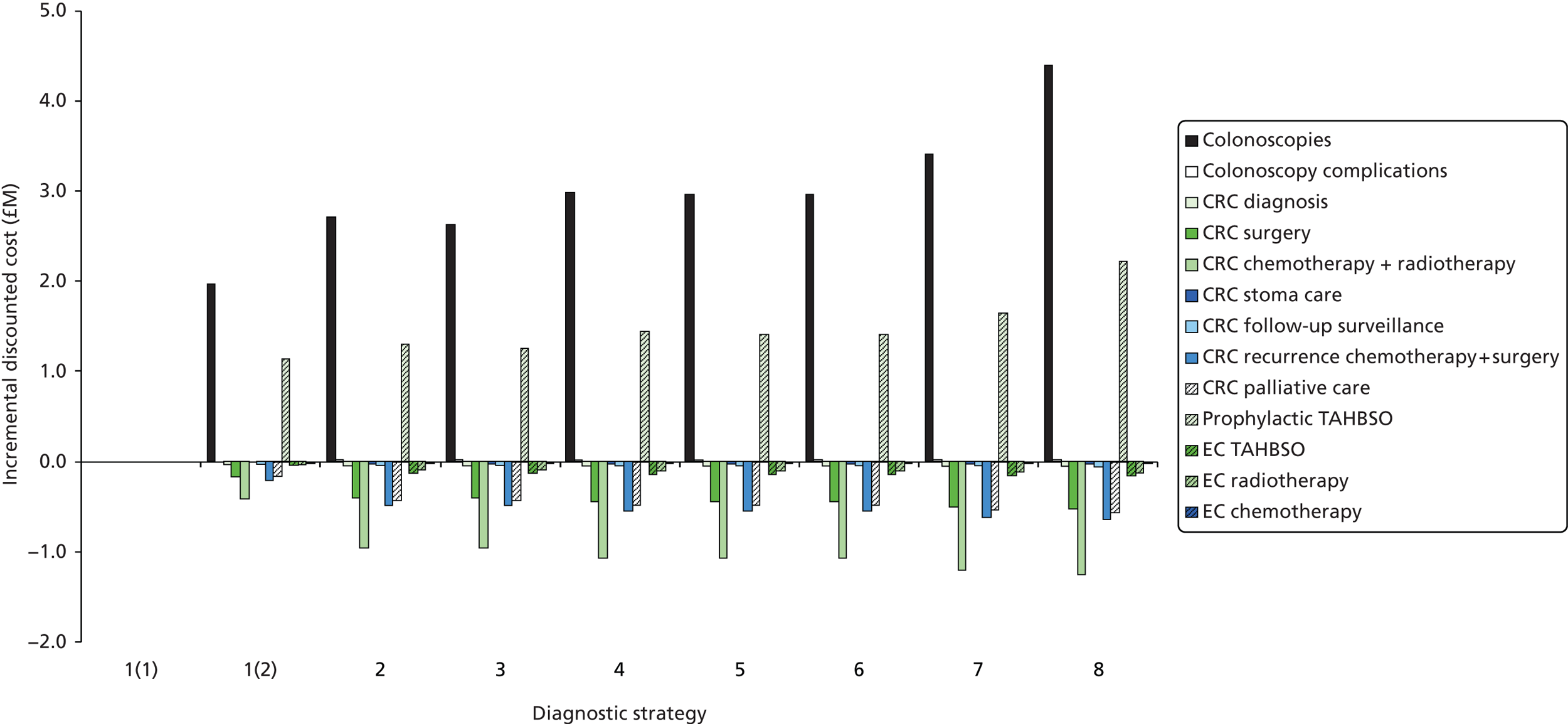
Cost-effectiveness results
As with the 60-year age limit, we find a marked increase in the incremental diagnostic costs for all strategies compared with no testing. Furthermore, the proportional increase in these costs is greater than the proportional changes in the other costs: CRC prevention and treatment, and EC prevention and treatment (Figure 108). Again, this is because of an increase in the proband population and a reduction in the prevalence of LS. In all genetic testing strategies, the incremental cost of LS diagnosis is now higher than the incremental cost of CRC prevention.
FIGURE 108.
Incremental discounted testing, prevention and treatment costs compared with strategy 1(1) when age limit of probands is increased to 70 years.
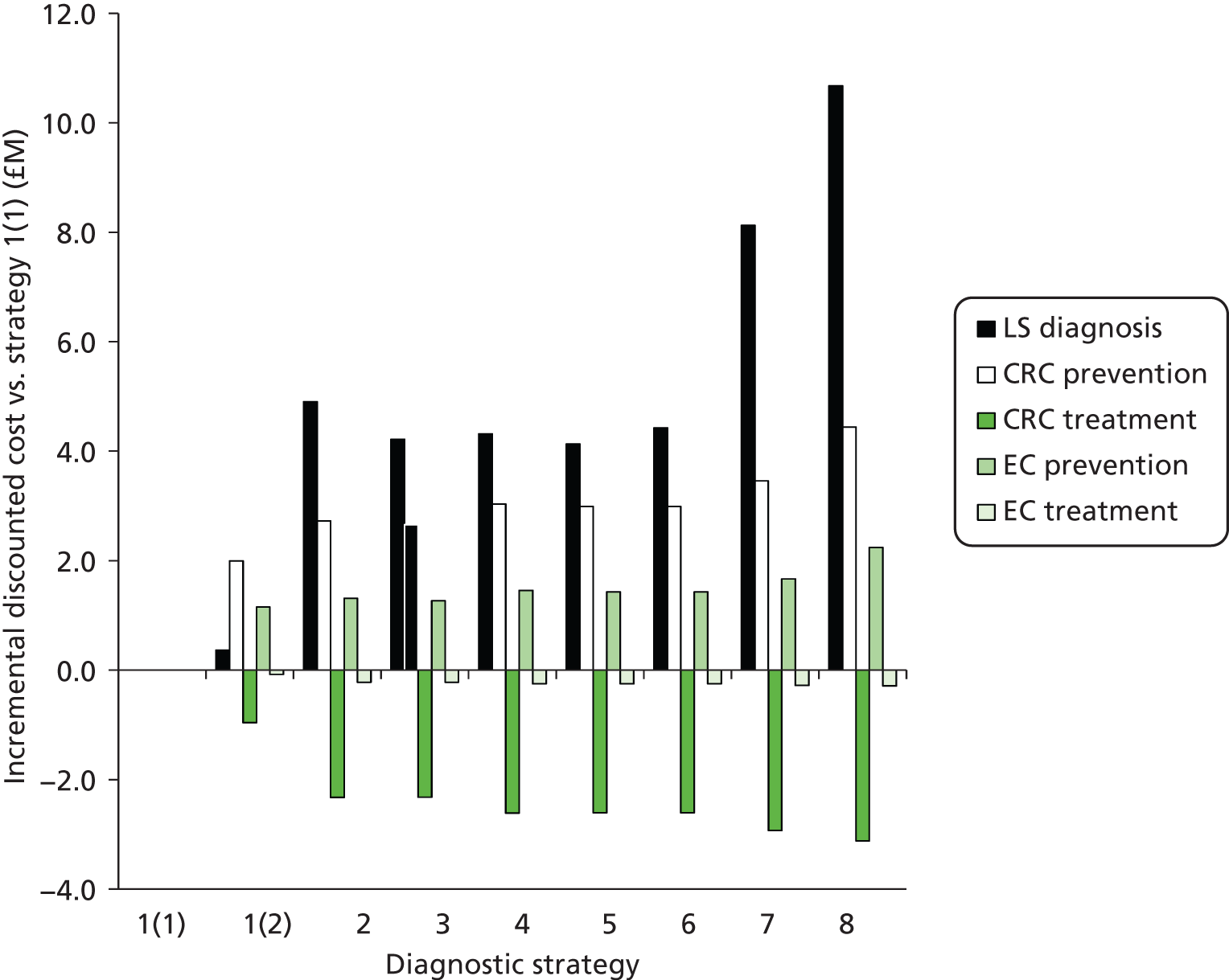
Incremental QALYs versus no testing also increase for all strategies (Figure 109). As with the age limit of 60 years, the QALY loss from LS diagnosis contributes proportionally more compared with the base case, because the ratio of the number of people tested to total health gain increases.
FIGURE 109.
Incremental QALY gain compared with strategy 1(1) when the age limit for probands is increased to 70 years.
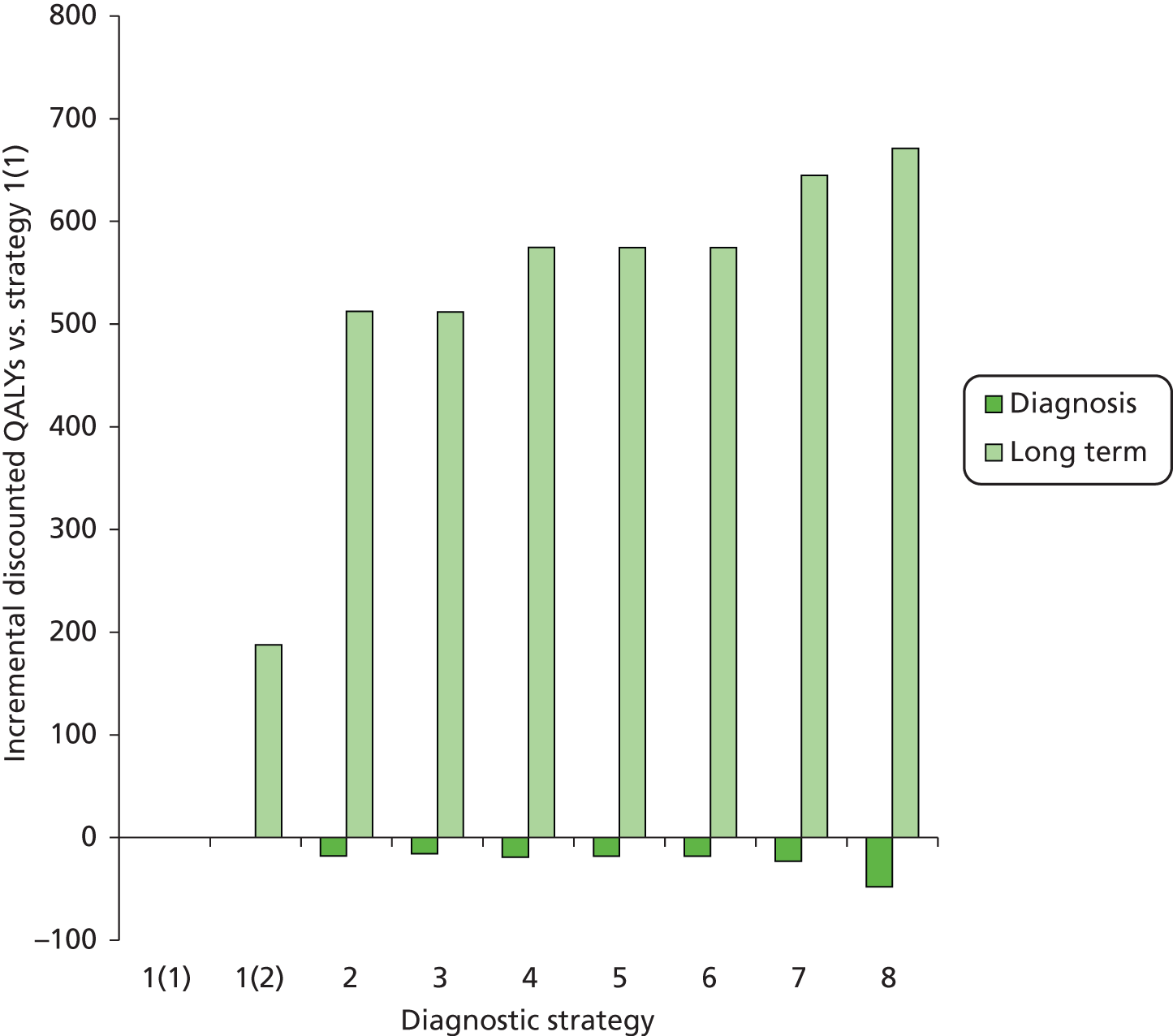
As with the age limit of 60 years, the ICERs of each strategy compared with no testing are larger than in the base case. All ICERs are now > £10,000 per QALY gained. Strategy 5 again has the lowest ICER, at £10,800 per QALY, and the ICER for strategy 8, universal genetic testing, is now above the £20,000-per-QALY threshold (Figure 110 and Table 106), which implies that it is no longer cost-effective versus no testing.
FIGURE 110.
Total discounted costs and QALYs for all probands and relatives per annum in England by testing strategy, when proband age limit is increased to 70 years.
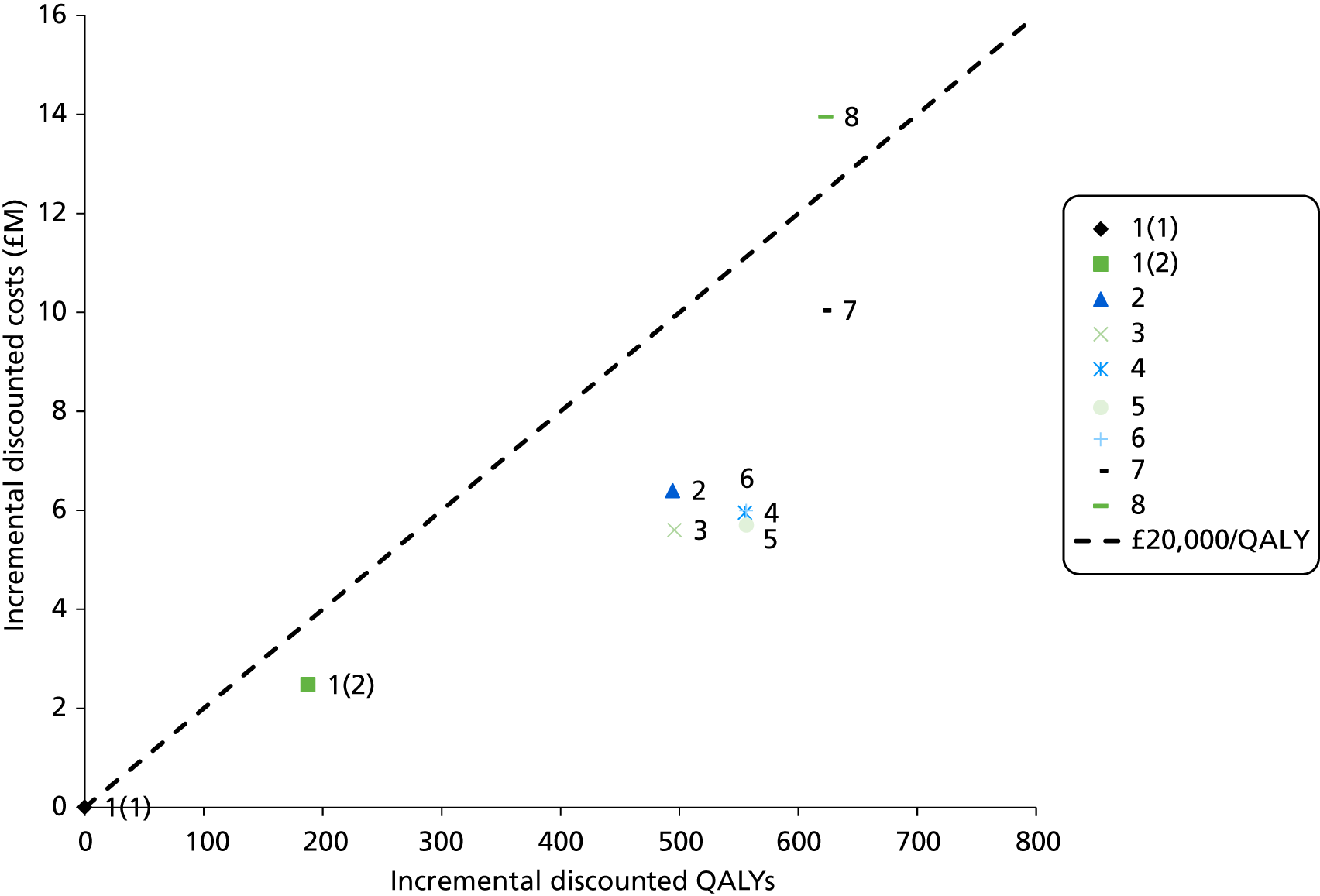
| Strategy | Total discounted QALYs | Total discounted cost (£) | Total NHB at £20,000 per QALY | Incremental discounted QALYs vs. strategy 1(1) | Incremental discounted cost vs. strategy 1(1) (£) | INHB at £20,000 per QALY vs. strategy 1(1) | ICER vs. strategy 1(1) (£/QALY) | ICER (£/QALY) |
|---|---|---|---|---|---|---|---|---|
| 1(1) | 1,218,663 | 269,736,673 | 1,205,177 | – | – | – | – | – |
| 1(2) | 1,218,851 | 272,213,601 | 1,205,240 | 188 | 2,476,928 | 63.9 | 13,194 | Extended dominated by strategies 1(1) and 3 |
| 2 | 1,219,158 | 276,133,925 | 1,205,351 | 494 | 6,397,252 | 174.6 | 12,939 | Dominated by strategy 6 |
| 3 | 1,219,159 | 275,335,255 | 1,205,393 | 496 | 5,598,582 | 216.0 | 11,288 | Extended dominated by strategies 1(2) and 5 |
| 4 | 1,219,219 | 275,685,175 | 1,205,435 | 555 | 5,948,502 | 258.0 | 10,710 | Dominated by strategy 5 |
| 5 | 1,219,220 | 275,437,192 | 1,205,448 | 556 | 5,700,519 | 271.3 | 10,247 | 10,247 |
| 6 | 1,219,220 | 275,731,782 | 1,205,433 | 556 | 5,995,109 | 256.5 | 10,777 | Dominated by strategy 5 |
| 7 | 1,219,285 | 279,774,795 | 1,205,296 | 622 | 10,038,122 | 119.6 | 16,151 | 66,507 |
| 8 | 1,219,287 | 283,685,897 | 1,205,102 | 623 | 13,949,224 | –74.3 | 22,386 | 2,439,898 |
The INHB compared with no testing for all probands and relatives in England per year increases from the base case for all strategies, except strategies 7 and 8. Strategy 5 again gives the maximum INHB, now at approximately 271 discounted QALYs. This compares with 130 discounted QALYs for this strategy in the base-case analysis.
No discounting
In the base case and all scenarios, the costs and benefits of the strategies are discounted over time at a rate of 3.5% per annum, in line with NICE guidance. In this section we present undiscounted results.
The majority of results presented in the base case, for cost and survival, are presented in undiscounted form, with discounts applying only when directly assessing cost-effectiveness. As such, we present only the main cost-effectiveness results (Table 107). The cost-effectiveness of all testing strategies versus no testing improves compared with the base case.
| Strategy | Total undiscounted QALYs | Total undiscounted cost (£) | ICER vs. strategy 1(1) (£/QALY) |
|---|---|---|---|
| 1(1) | 275,654 | 54,336,327 | – |
| 1(2) | 275,825 | 54,852,971 | 3029 |
| 2 | 276,101 | 55,428,502 | 2448 |
| 3 | 276,100 | 55,325,673 | 2219 |
| 4 | 276,155 | 55,410,000 | 2145 |
| 5 | 276,155 | 55,385,965 | 2098 |
| 6 | 276,155 | 55,436,784 | 2199 |
| 7 | 276,216 | 55,944,392 | 2864 |
| 8 | 276,238 | 56,459,582 | 3641 |
As Table 107 shows, not including the discount rate increased both the costs and QALYs of each strategy.
Univariate sensitivity analyses
We conducted several univariate sensitivity analyses to investigate the impact of various parameters on the model results. In the majority of cases, the values used in the sensitivity analyses were not meant to represent the true parameter value, but were chosen to demonstrate the impact of changing the parameter value on the cost-effectiveness of testing for LS.
Cost of tumour testing
For each tumour-based test, we assessed the impact of halving and doubling the cost and present the ICERs compared with strategy 1(1) (no testing), and the INHB at a £20,000 threshold compared with strategy 1(1). As not all tests are used in each strategy, not all strategies change ICER or INHB when the cost of one tumour-based test changes. As expected from the results of the base case, it is the tests which are run first, or on a large proportion of the cohort, that have the greatest impact on the cost-effectiveness of a strategy. As the results in Table 108 (see also Table 109) demonstrate, all strategies are cost-effective at a threshold of £20,000 per QALY compared with no testing, regardless of change in tumour-based test cost. An example of the change in diagnostic costs is given in Figures 111 and 112, which show the change in total diagnostic costs under IHC sensitivity analyses.
| Parameter name | Base parameter | Sensitivity analysis parameter | ICER of each strategy vs. strategy 1(1) (£/QALY) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 (1) | 1 (2) | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |||
| Base case | – | – | – | 6021 | 6444 | 5831 | 5610 | 5491 | 5774 | 7601 | 9571 |
| Cost of IHC (£) | 238 | 119 | – | 6021 | 5178 | 4566 | 5610 | 5491 | 5633 | 6595 | 9571 |
| 476 | – | 6021 | 8975 | 8361 | 5610 | 5491 | 6058 | 9614 | 9571 | ||
| Cost of MSI (£) | 202 | 101 | – | 6021 | 6444 | 5831 | 4652 | 4533 | 4817 | 6890 | 9571 |
| 404 | – | 6021 | 6444 | 5831 | 7525 | 7406 | 7690 | 9024 | 9571 | ||
| Cost of BRAF (£) | 118 | 59 | – | 6021 | 6444 | 5760 | 5610 | 5401 | 5684 | 7553 | 9571 |
| 236 | – | 6021 | 6444 | 5972 | 5610 | 5672 | 5955 | 7697 | 9571 | ||
FIGURE 111.
Diagnostic costs when the cost of IHC is halved.
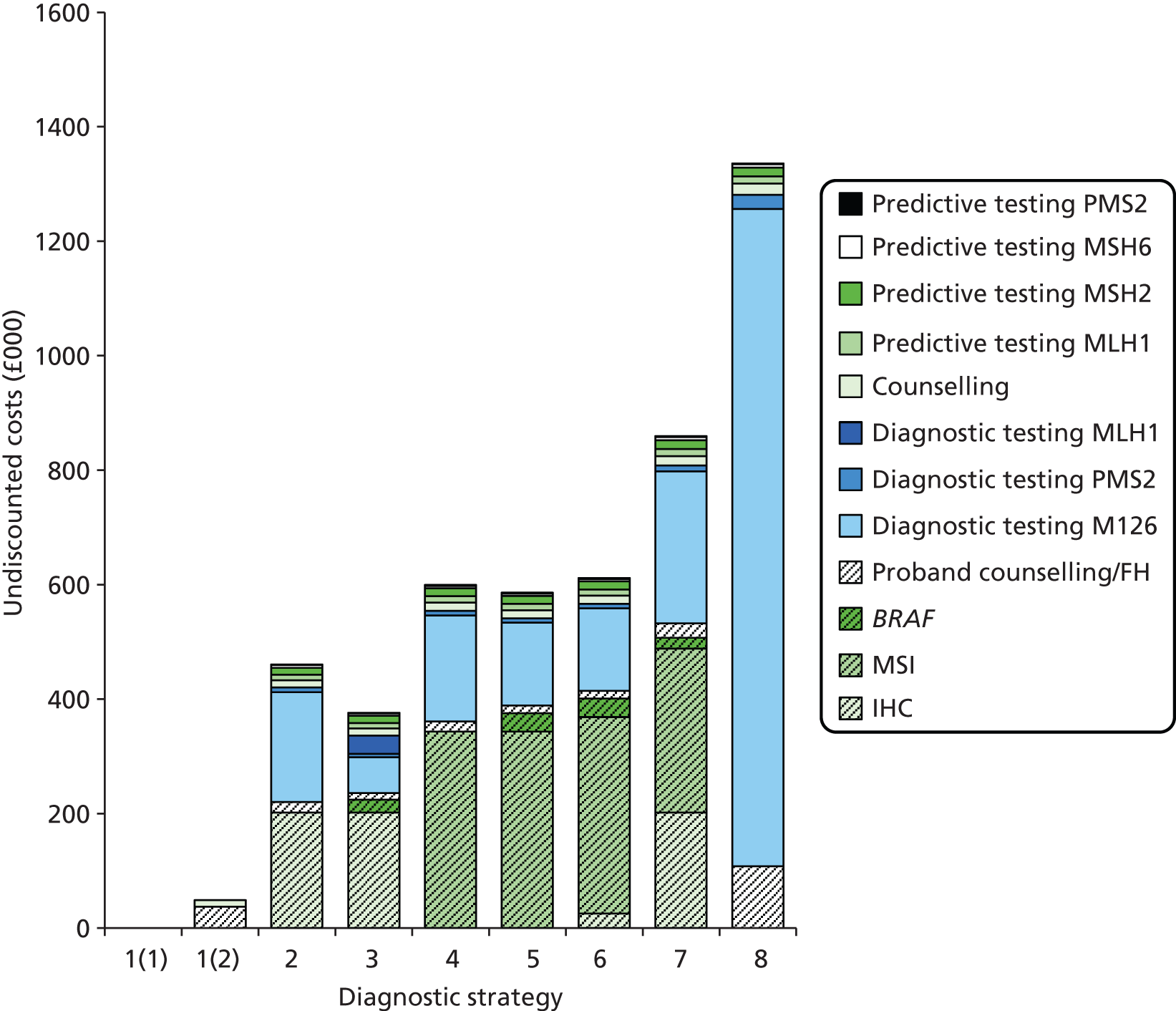
FIGURE 112.
Diagnostic costs when the cost of IHC is doubled.
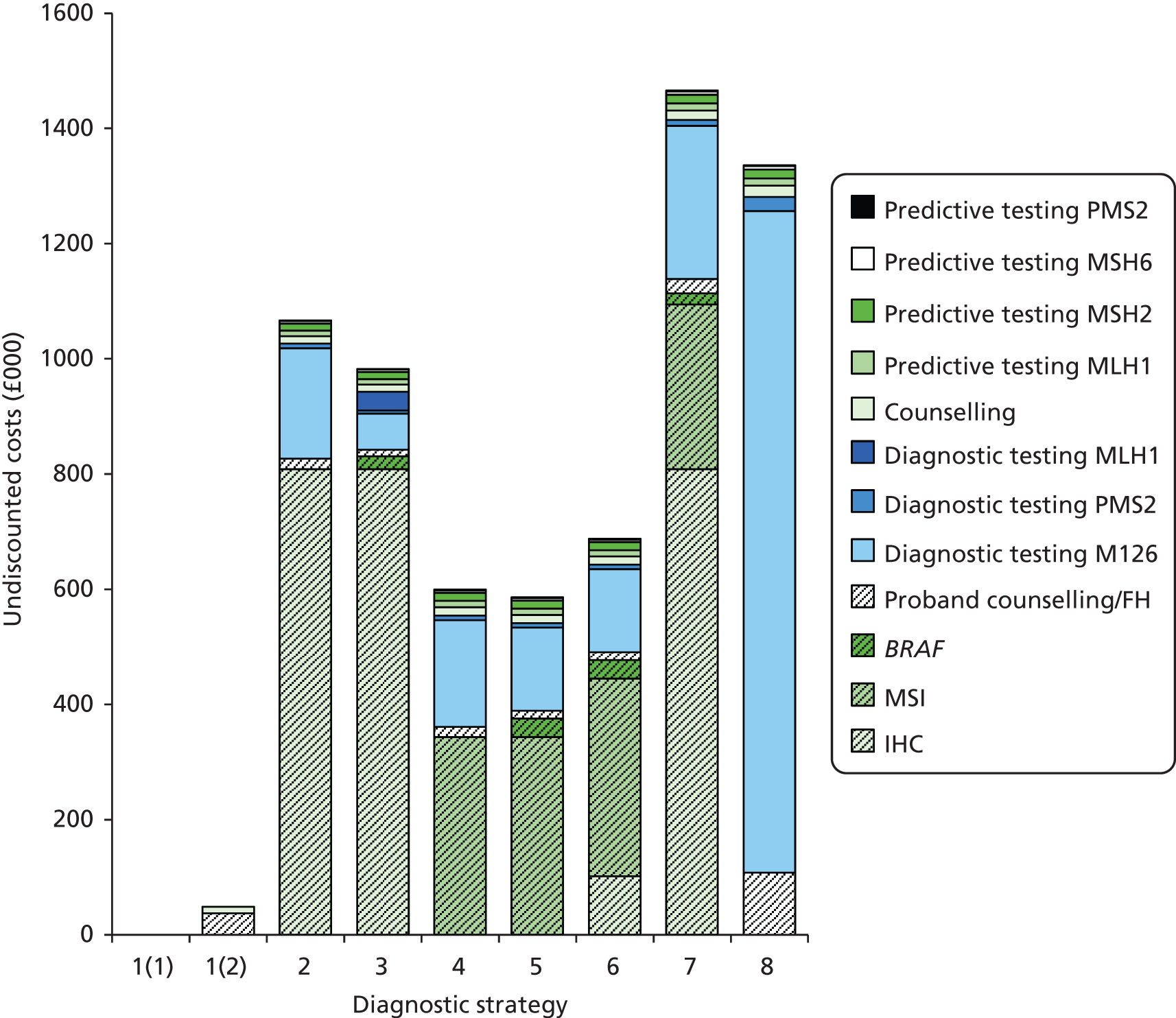
Cost of genetic testing and counselling
In this section, we analyse the impact of costs surrounding genetic testing, counselling and FH assessment by halving and doubling the various costs. The ICERs compared with no testing are given in Table 109.
| Parameter name | Base parameter | Sensitivity analysis parameter | ICER of each strategy vs. strategy 1(1) (£/QALY) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1(1) | 1(2) | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |||
| Base case | – | – | – | 6021 | 6444 | 5831 | 5610 | 5491 | 5774 | 7601 | 9571 |
| Cost of taking FH (£) | 22 | 11 | – | 5729 | 6443 | 5830 | 5608 | 5490 | 5773 | 7600 | 9565 |
| 44 | – | 6604 | 6447 | 5833 | 5612 | 5493 | 5776 | 7604 | 9585 | ||
| Cost of genetic counselling (probands only) (£) | 67 | 33 | – | 6021 | 6389 | 5797 | 5562 | 5454 | 5738 | 7541 | 9317 |
| 134 | – | 6021 | 6554 | 5899 | 5704 | 5565 | 5848 | 7722 | 10,081 | ||
| Cost of genetic counselling (relatives only) (£) | 67 | 33 | – | 5930 | 6404 | 5792 | 5570 | 5452 | 5735 | 7561 | 9523 |
| 134 | – | 6202 | 6524 | 5910 | 5689 | 5570 | 5853 | 7682 | 9668 | ||
| Cost of genetic counselling (probands and relatives) (£) | 67 | 33 | – | 5930 | 6349 | 5758 | 5522 | 5415 | 5698 | 7500 | 9268 |
| 134 | – | 6202 | 6635 | 5978 | 5784 | 5643 | 5927 | 7803 | 10,178 | ||
| Cost of diagnostic genetic testing (probands) (£) | MLH1, 464 | MLH1, 232 | – | 6021 | 5818 | 5517 | 5069 | 5068 | 5351 | 6913 | 6728 |
| PMS2, 735 | PMS2, 367 | ||||||||||
| M126, 812 | M126, 406 | ||||||||||
| MLH1, 929 | – | 6021 | 7697 | 6459 | 6690 | 6338 | 6621 | 8977 | 15,258 | ||
| PMS2, 1469 | |||||||||||
| M126, 1624 | |||||||||||
| Cost of predictive genetic testing (relatives) (£) | MLH1, 169 MSH2, 172 MSH6, 172 PMS2, 176 |
MLH1, 85 | – | 6021 | 6358 | 5745 | 5524 | 5405 | 5689 | 7515 | 9485 |
| MSH2, 86 | |||||||||||
| MSH6, 86 | |||||||||||
| PMS2, 88 | |||||||||||
| MLH1, 339 | – | 6021 | 6616 | 6002 | 5781 | 5663 | 5946 | 7773 | 9745 | ||
| MSH2, 345 | |||||||||||
| MSH6, 345 | |||||||||||
| PMS2, 352 | |||||||||||
Changing the cost of FH assessment affects probands only. This parameter has little effect on the ICERs versus no testing for each strategy, compared with the base case.
Changing the cost of genetic counselling for relatives also changes the cost of counselling for relatives of probands assumed to have LS (and therefore relatives with no possibility of genetic testing), so the change in this cost also affects strategies that do not include genetic testing, i.e. strategy 1(2). Despite this, there is still little change in the ICERs versus strategy 1(1) in the base case. Changing the cost of genetic counselling in probands only affected the strategies that included genetic testing, but again had little impact on the cost-effectiveness of each strategy compared with the base case.
As the cost of genetic counselling for probands and relatives was the same in our base case, we also examined the case when the counselling costs were changed for both at the same time; again there was little change from the base case. The same was true when the cost of genetic testing in relatives was altered.
Changing the cost of diagnostic genetic testing in probands, particularly halving the cost, had a much greater impact on the cost-effectiveness of each strategy, with strategy 8 having lower total costs than strategy 7 (Figure 113). This reduces the ICERs versus no testing for all strategies compared with the base case, except strategy 1(2), which does not include genetic testing. The ICER most altered is that for strategy 8 versus strategy 1(1); strategy 8 in fact becomes the most cost-effective strategy at a cost-effectiveness threshold of £20,000 per QALY. Similar results occur when the cost of genetic testing in probands is doubled (Figure 114), except that the ICERs compared with strategy 1(1) increase compared with the base case.
FIGURE 113.
Total diagnostic costs when the cost of genetic testing in probands is halved.
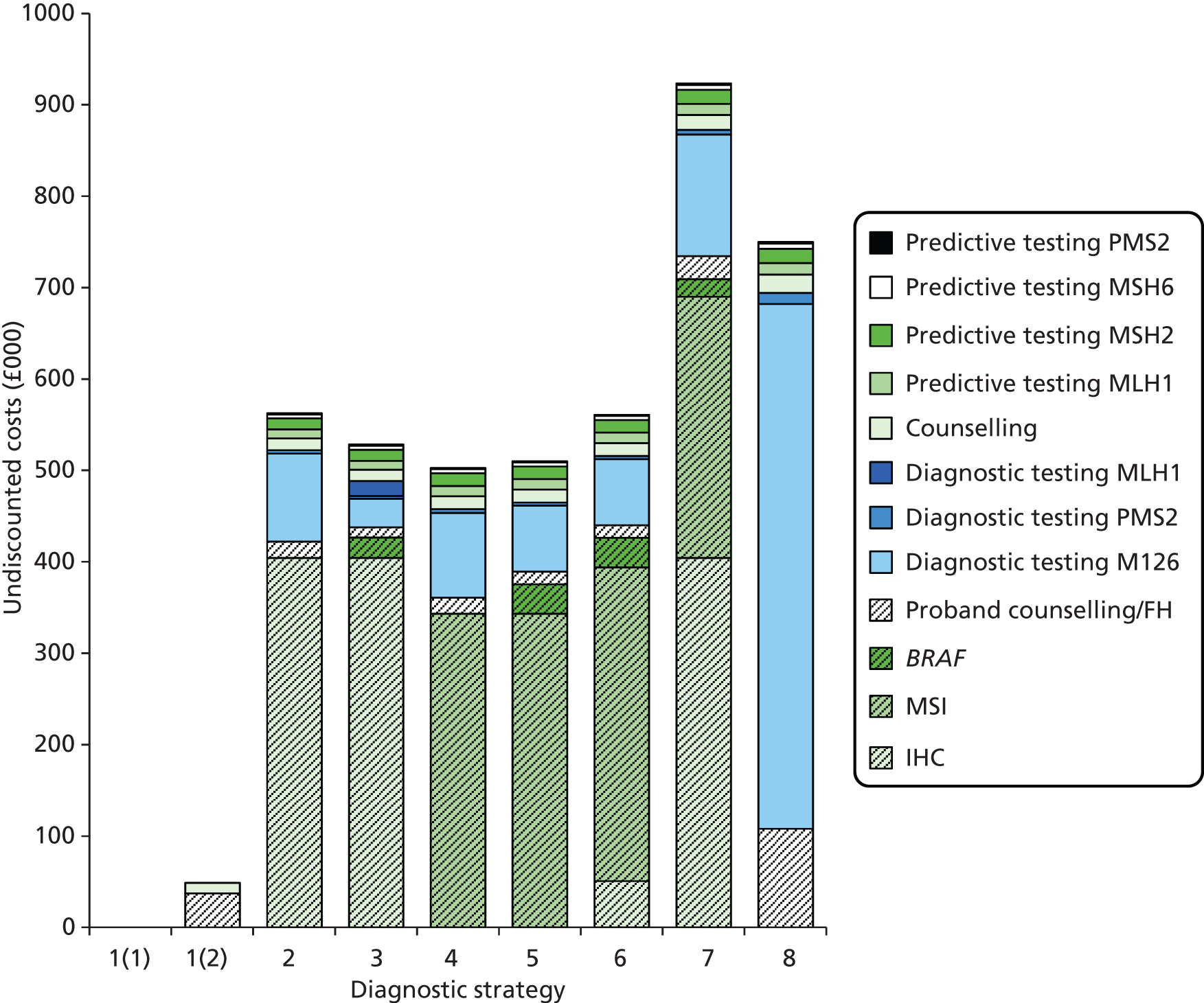
FIGURE 114.
Total diagnostic costs when the cost of genetic testing in probands is doubled.
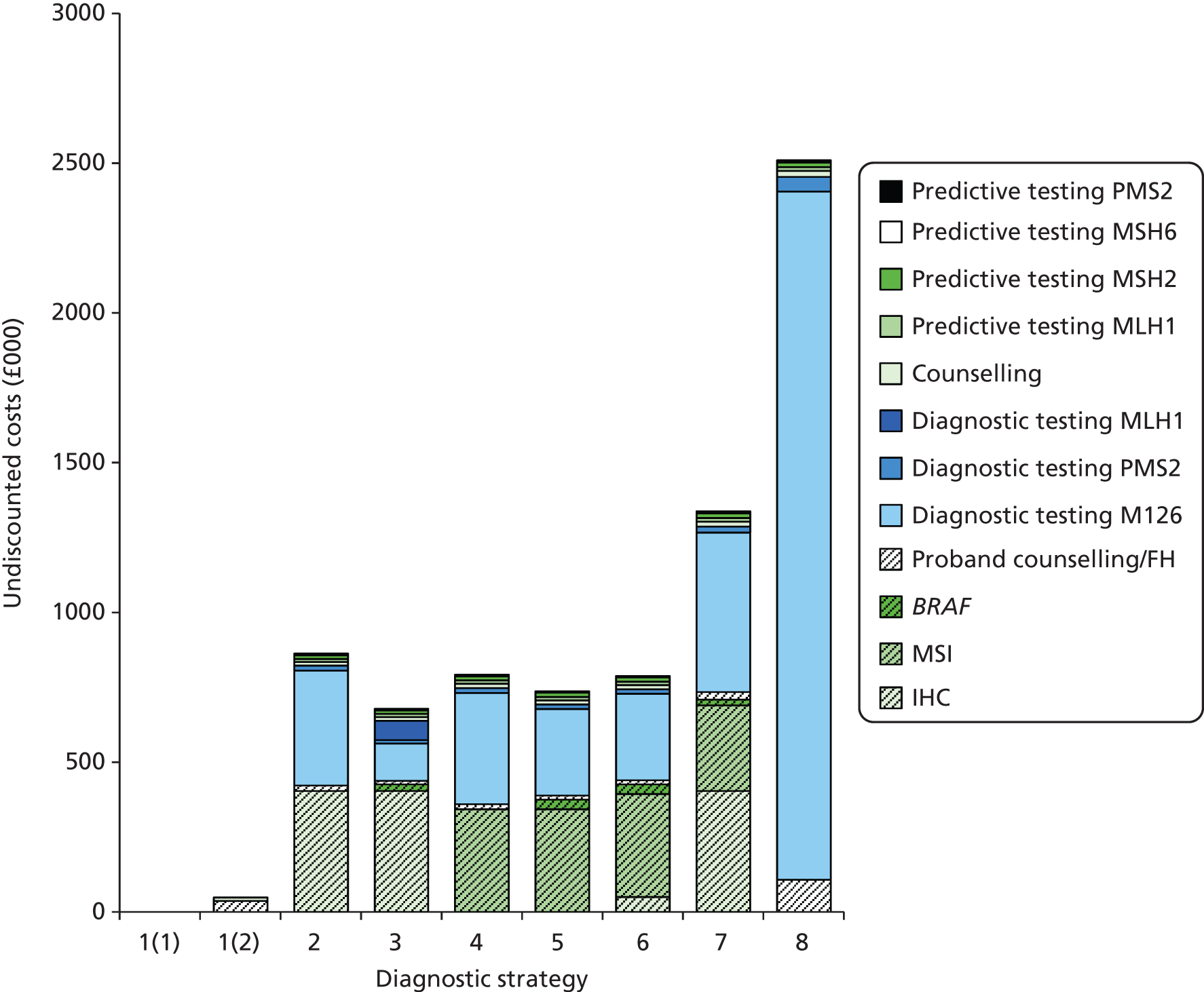
Acceptance of genetic testing
The sensitivity analyses of parameters that influenced the acceptance of genetic testing were conducted by halving the acceptance rate of each parameter and then halving the non-acceptance rate. Results are presented in Table 110. Unlike the costs, relatives’ acceptance of genetic counselling only applies to genetic counselling of relatives with mutation-positive probands, leaving the ICER of strategy 1(2) unaffected.
| Parameter name | Base parameter | Sensitivity analysis parameter | ICER of each strategy vs. strategy 1(1) (£/QALY) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1(1) | 1(2) | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |||
| Base case | – | – | – | 6021 | 6444 | 5831 | 5610 | 5491 | 5774 | 7601 | 9571 |
| Proband adherence to genetic counselling | 92.5% | 46.3% | – | 6021 | 7760 | 7269 | 6592 | 6581 | 7024 | 9488 | 9503 |
| 96.3% | – | 6021 | 6378 | 5758 | 5560 | 5436 | 5711 | 7507 | 9575 | ||
| Relative adherence to genetic counselling | 45% | 23% | – | 6021 | 7395 | 6750 | 6521 | 6396 | 6693 | 8609 | 10686 |
| 73% | – | 6021 | 5416 | 4837 | 4624 | 4513 | 4781 | 6511 | 8366 | ||
| Proband adherence to genetic testing | 90% | 45% | – | 6021 | 7818 | 7305 | 6642 | 6620 | 7063 | 9552 | 9788 |
| 95% | – | 6021 | 6350 | 5730 | 5539 | 5414 | 5686 | 7468 | 9558 | ||
| Relative adherence to genetic testing | 96% | 48% | – | 6021 | 7395 | 6750 | 6521 | 6396 | 6693 | 8609 | 10,686 |
| 98% | – | 6021 | 6405 | 5793 | 5572 | 5453 | 5736 | 7559 | 9525 | ||
| All patients comply with genetic counselling and testing | 100% for all | 6021 | 3994 | 3426 | 3318 | 3192 | 3411 | 4906 | 7025 | ||
Unusually, the change in acceptance of genetic counselling and testing does not appear to have a particularly large impact on the ICERs versus no testing in the base case, with all ICERs compared with no testing remaining < £11,000 per QALY gained in all cases.
Our final sensitivity analysis of the acceptance of genetic testing and counselling is to set the acceptance of both for probands and relatives to 100%. Like the majority of our sensitivity analyses, this was done to demonstrate the importance of acceptance of genetic counselling and testing as parameters in the model, not as a probable real-life scenario. When acceptance was raised to 100% for both counselling and genetic testing in probands and relatives, the ICERs compared with no testing reduced significantly compared with the base case, and strategy 7 had the greatest NHB.
Prevalence of Lynch syndrome in the proband population
As one of the parameters altered in the age-limit-of-probands scenarios, there is already an assumption that the prevalence plays an important role in the cost-effectiveness of strategies to diagnose LS. To assess this this theory we halve, then double, the prevalence.
Halving the prevalence of Lynch syndrome in the proband population (to 4.2%)
As demonstrated in the diagnostic results for the age limit scenarios, reducing the prevalence to 4.2% in the proband population has no effect on the sensitivity, but increases the specificity of the strategies (Figure 115). This appears to be particularly true of the strategies that employ tumour testing, which have higher specificities to begin with.
FIGURE 115.
Receiver operating characteristic plot for all patients, when prevalence of LS in probands is halved to 4.2%.
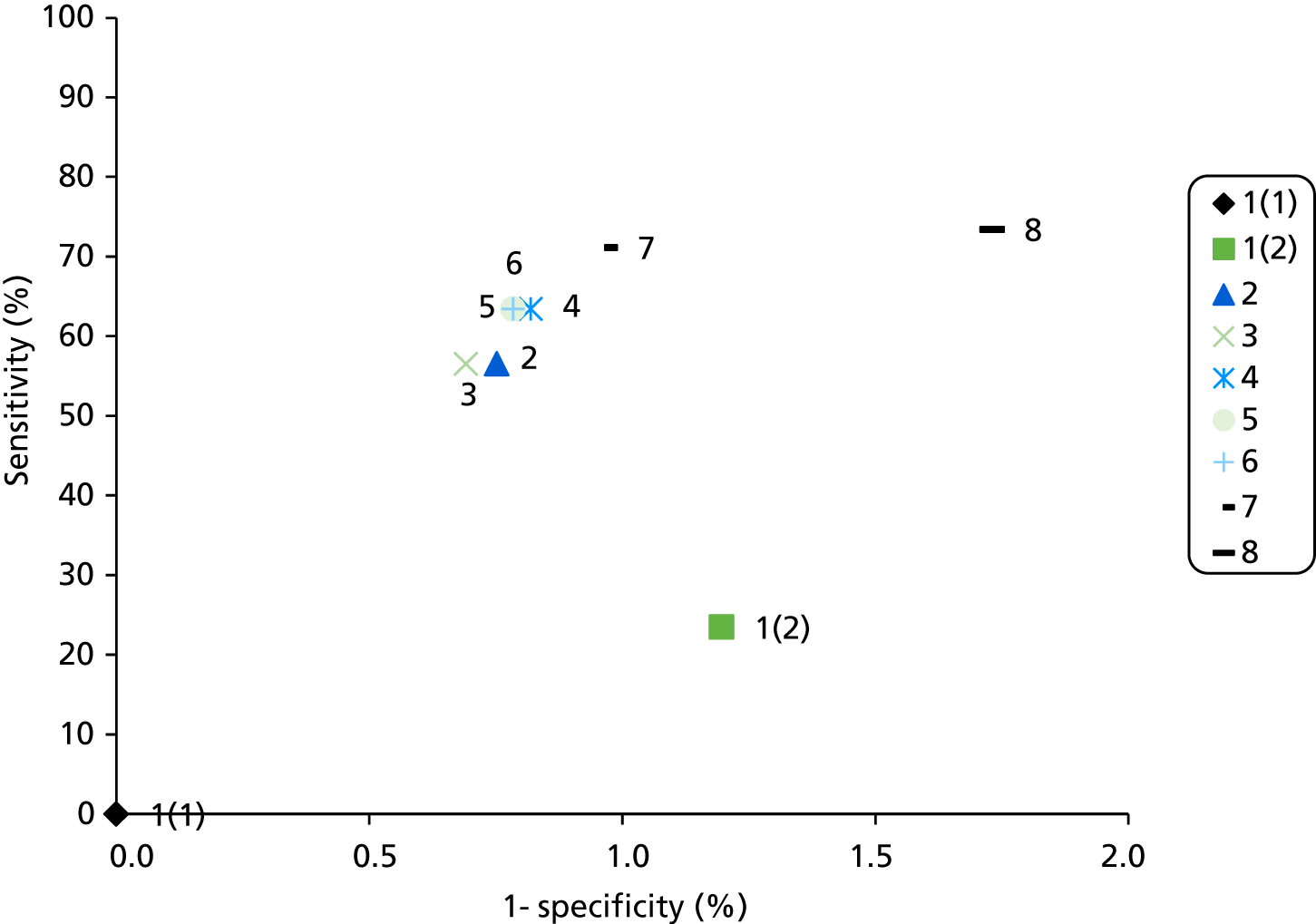
The total costs of LS diagnosis decrease for all strategies except strategy 1(1), because fewer probands test positive in early tests and proceed to diagnostic germline testing (Figure 116). The total costs of CRC prevention decrease substantially for all strategies because surveillance is offered to significantly fewer patients; indeed, in this sensitivity analysis the total cost of CRC prevention is lower than the total cost of LS diagnosis in all strategies that use genetic testing. Given that this sensitivity analysis follows a similar pattern to the results of scenario 3, it is unsurprising that a reduction in prevalence equates to a reduction in the cost-effectiveness of strategies testing for LS.
FIGURE 116.
Incremental discounted costs for prevalence 4.2%.
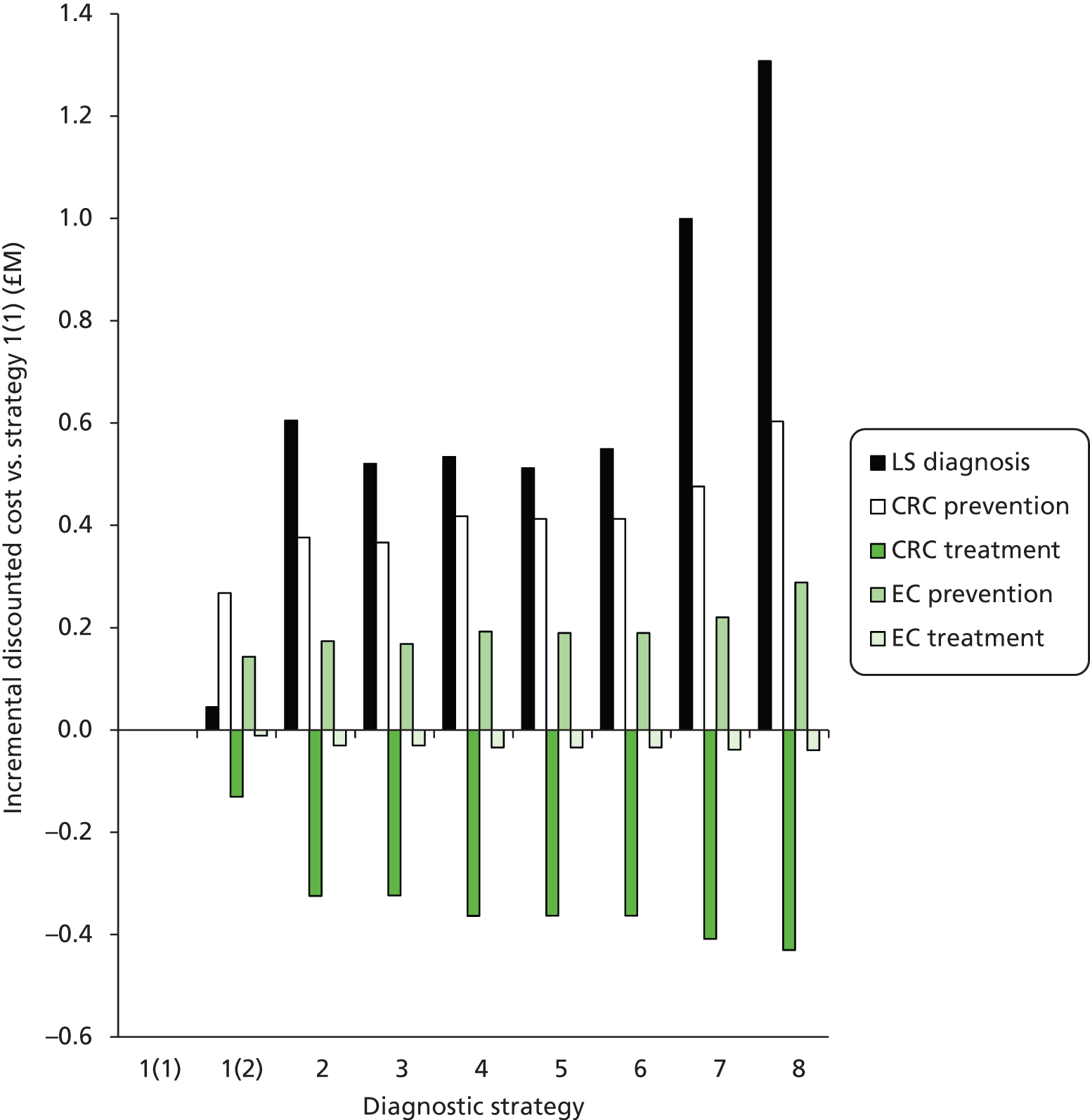
The incremental QALYs for strategies versus strategy 1(1) are approximately halved from the base case (Figure 117).
FIGURE 117.
Incremental discounted QALYs for prevalence 4.2%.
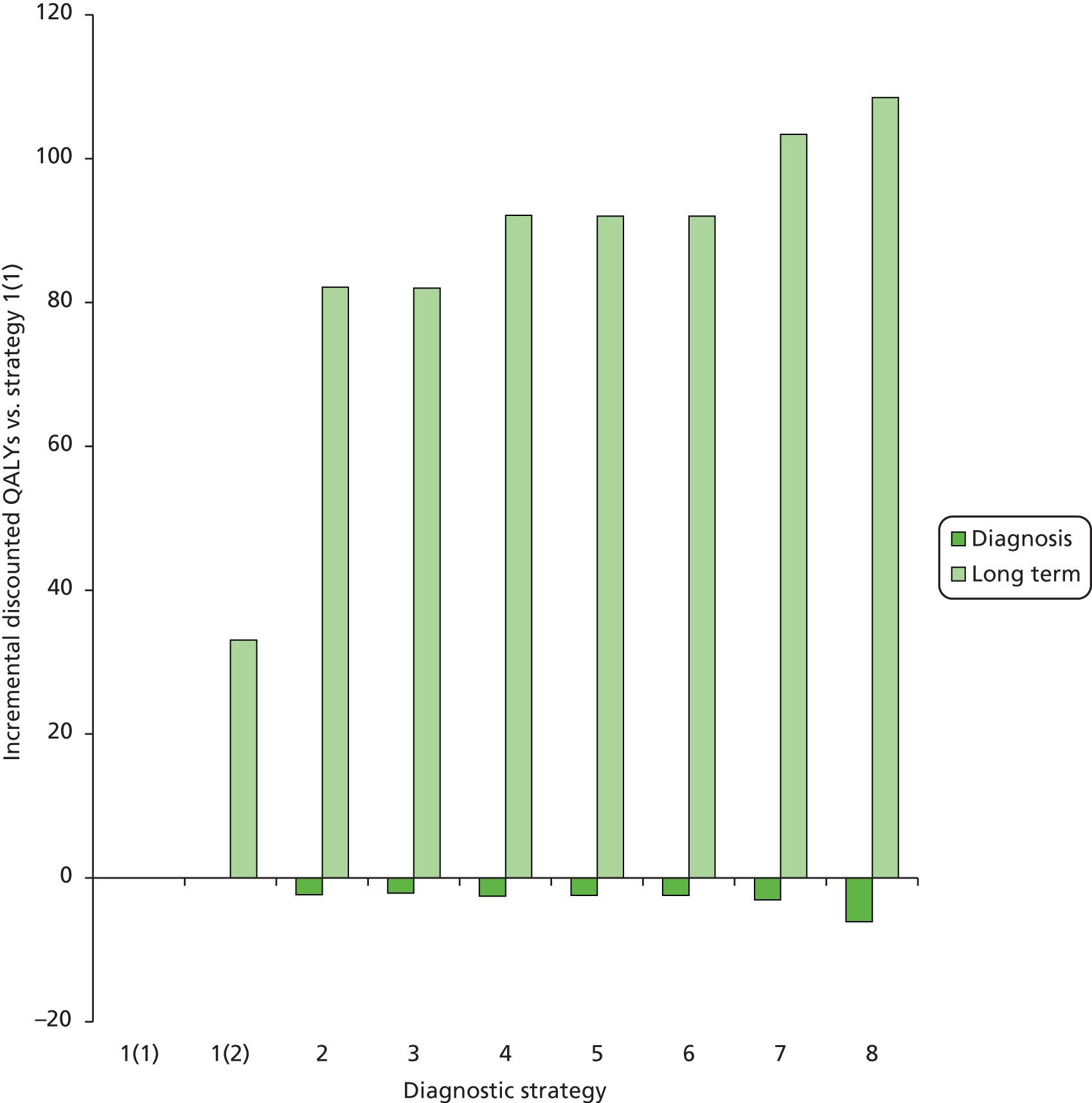
As expected, the results demonstrate a worsening in cost-effectiveness when the prevalence is halved, with ICERs increasing for all strategies compared with strategy 1(1) and the INHB reducing in all cases (Figure 118 and Table 111). However, all strategies are still cost-effective compared with no testing at a threshold of £20,000 per QALY, and at this threshold strategy 5 again has the largest INHB compared with no testing.
FIGURE 118.
Incremental cost-effectiveness ratios compared with strategy 1(1) when the prevalence of LS in the probands is 4.2%.
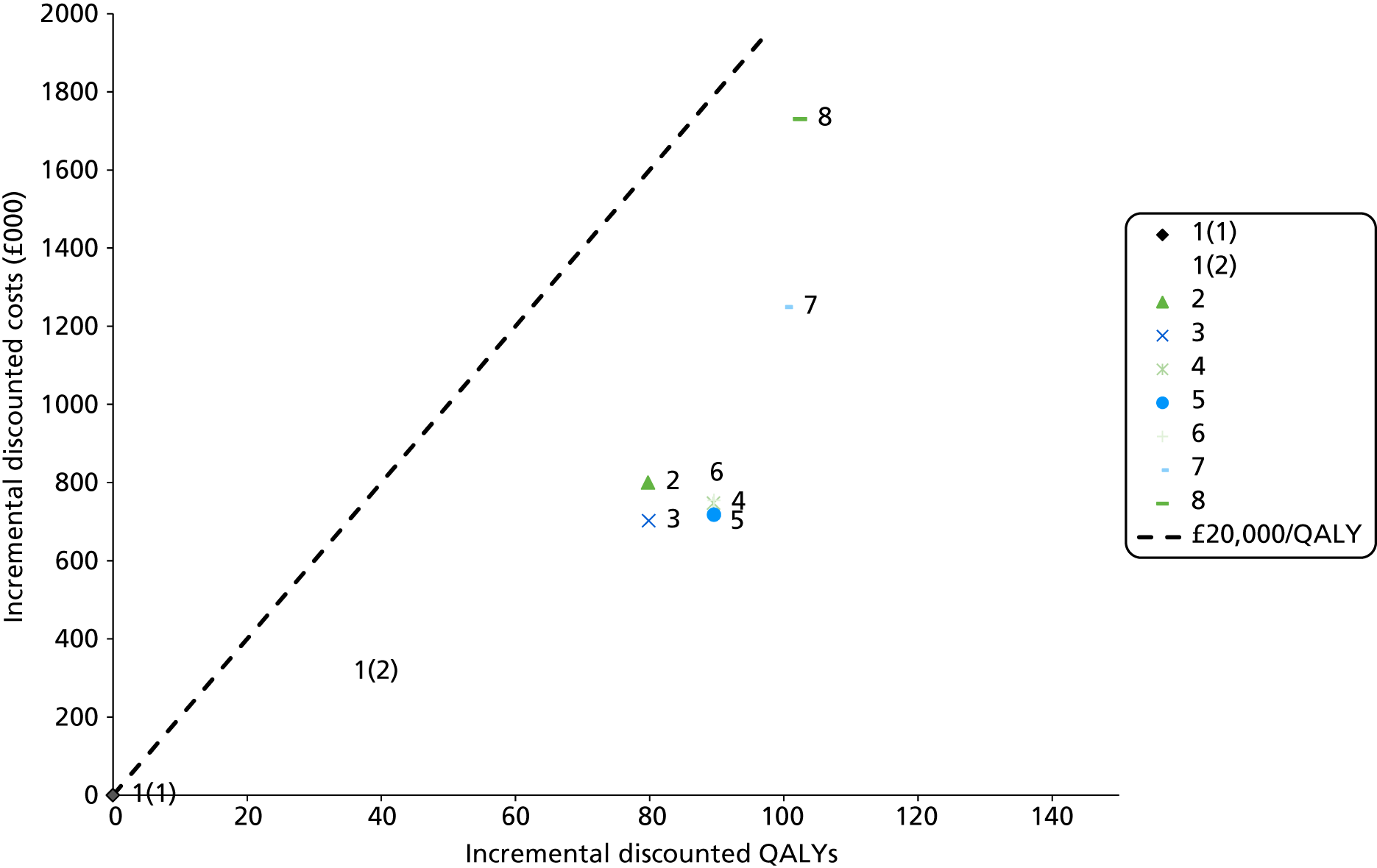
| Strategy | Total discounted QALYs | Total discounted cost (£) | Incremental discounted QALYs vs. strategy 1(1) | Incremental discounted cost vs. strategy 1(1) (£) | ICER vs. strategy 1(1) (£/QALY) | INHB at £20,000 per QALY vs. strategy 1(1) |
|---|---|---|---|---|---|---|
| 1(1) | 151,939 | 35,018,577 | – | – | – | – |
| 1(2) | 151,972 | 35,333,268 | 33 | 314,691 | 9516 | 17.3 |
| 2 | 152,019 | 35,818,538 | 80 | 799,960 | 10,027 | 39.8 |
| 3 | 152,019 | 35,720,435 | 80 | 701,857 | 8787 | 44.8 |
| 4 | 152,029 | 35,765,766 | 90 | 747,188 | 8344 | 52.2 |
| 5 | 152,029 | 35,735,984 | 90 | 717,407 | 8007 | 53.7 |
| 6 | 152,029 | 35,773,280 | 90 | 754,703 | 8423 | 51.9 |
| 7 | 152,040 | 36,267,428 | 100 | 1,248,851 | 12,447 | 37.9 |
| 8 | 152,042 | 36,748,683 | 102 | 1,730,106 | 16,891 | 15.9 |
Doubling the prevalence of Lynch syndrome in probands (to 16.8%)
Doubling the prevalence of LS in probands to 16.8% reduces the specificity of all the strategies and reduces the specificity of those strategies that use tumour-based tests enough that they become less specific than strategy 1(2), which uses only FH to identify LS patients (Figure 119). The incremental costs and QALYs for each strategy are shown in Figures 120 and 121, respectively.
FIGURE 119.
Receiver operating characteristic plot of all patients when the prevalence of LS in patients is 16.8%.
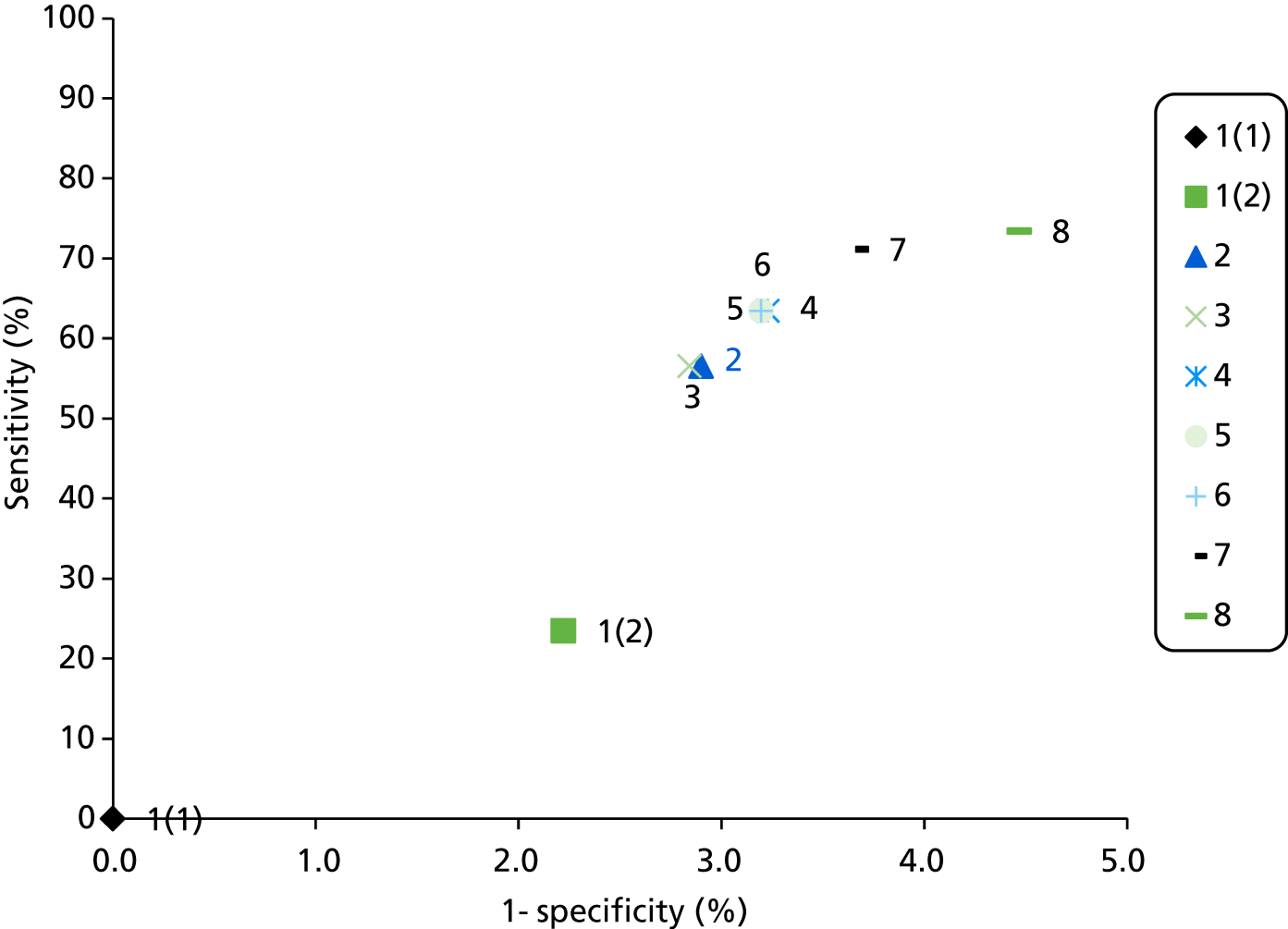
FIGURE 120.
Incremental discounted costs for prevalence 16.8%.
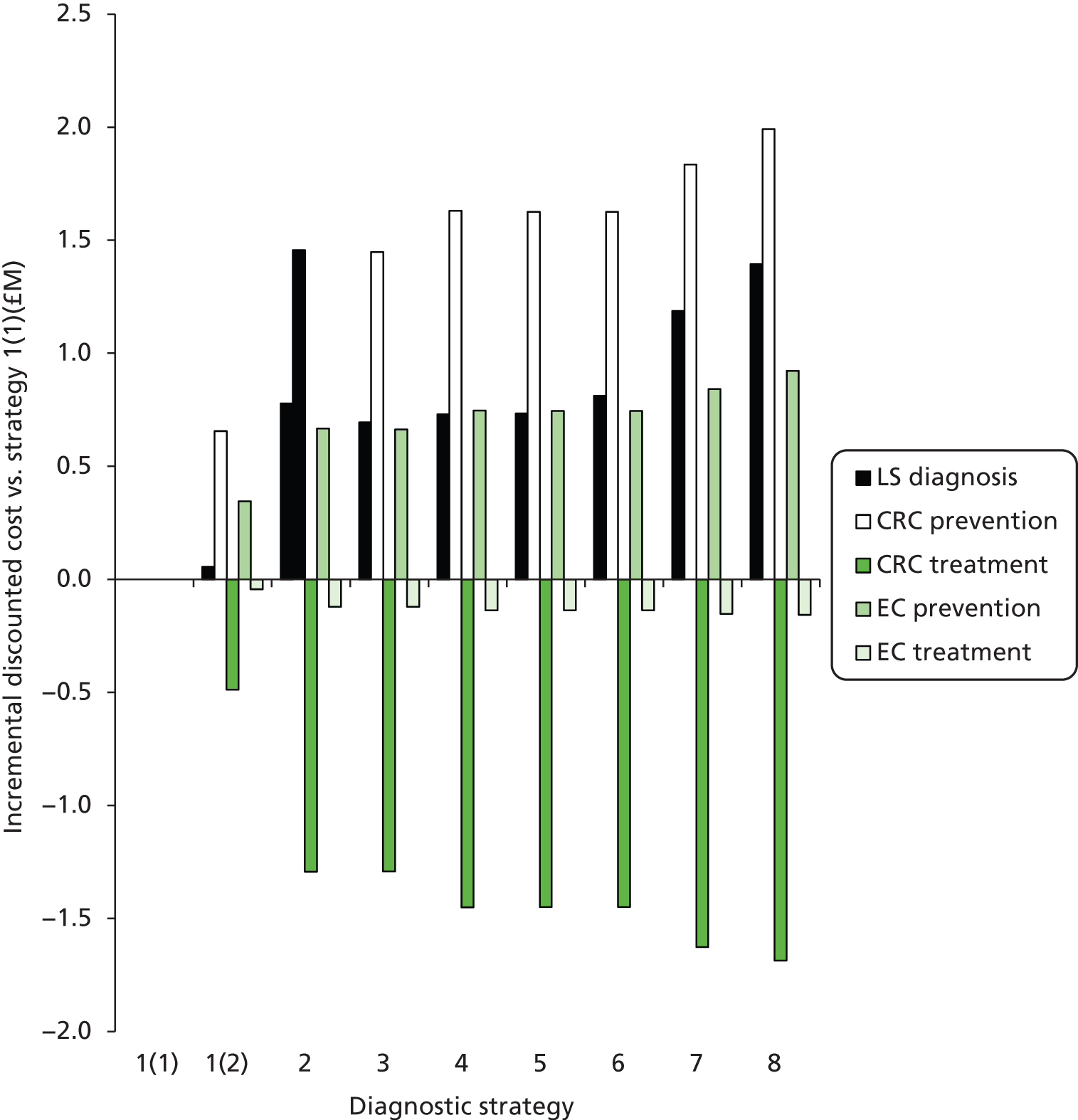
FIGURE 121.
Incremental discounted QALYs for prevalence 16.8%.
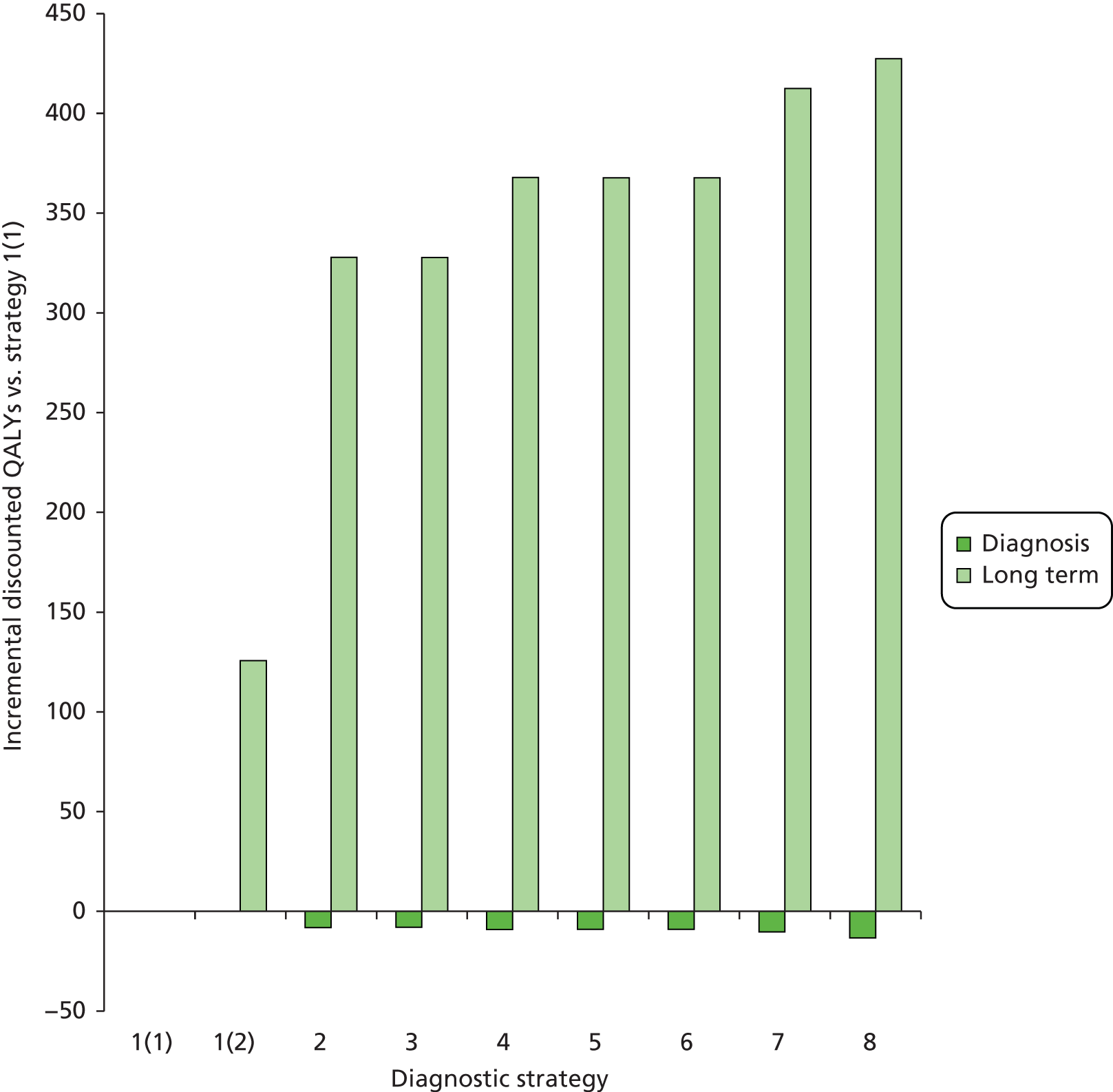
Increasing the prevalence has the expected impact of reducing the ICERs compared with strategy 1(1) and increasing the INHB of each strategy versus strategy 1(1) in the base case; as expected, this is a reversal of the results of halving the prevalence (Figure 122 and Table 112). In this sensitivity analysis, strategy 7 has the greatest NHB.
FIGURE 122.
Incremental cost-effectiveness ratios compared with strategy 1(1) when the prevalence of LS in the probands is 16.8%.
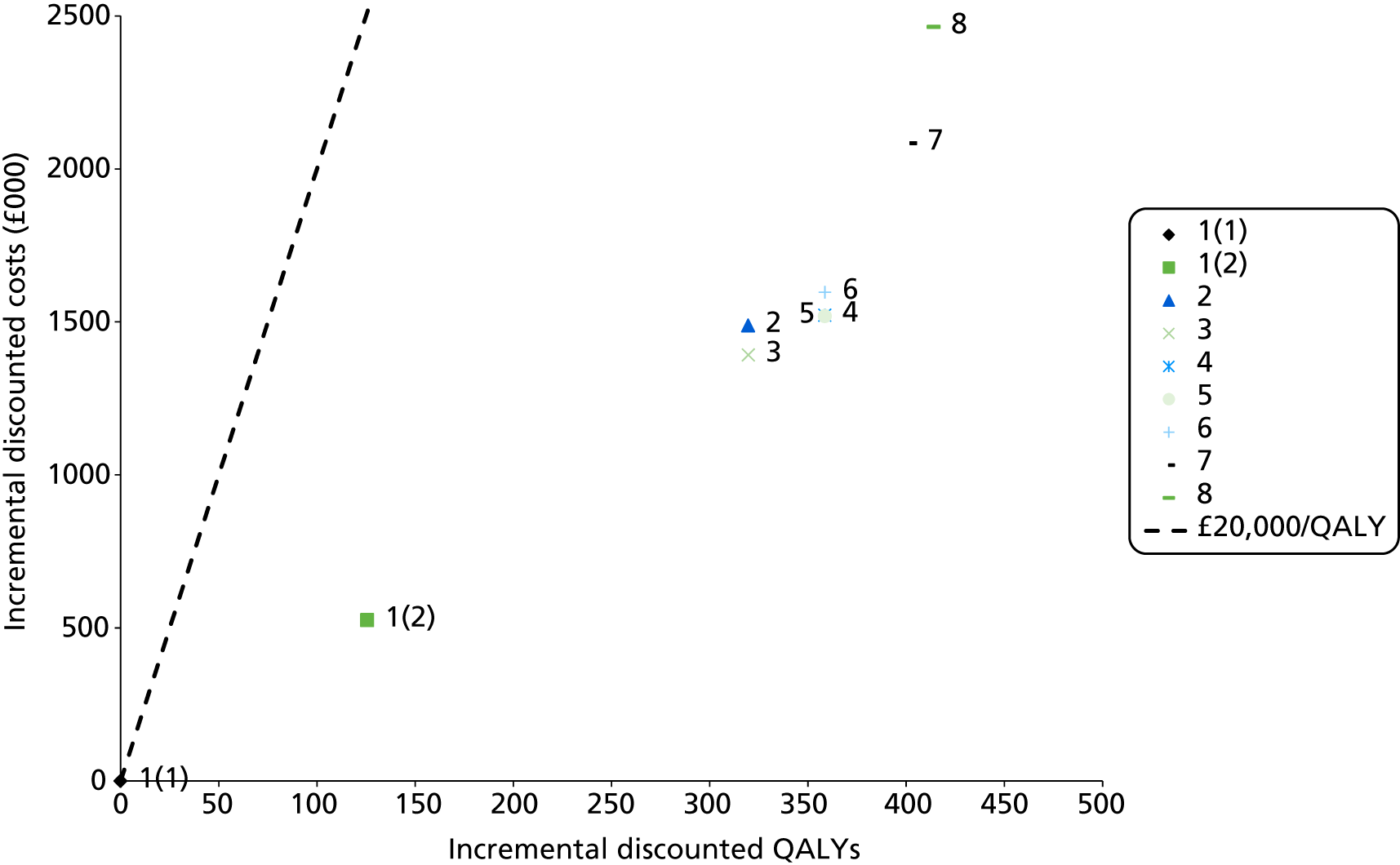
| Strategy | Total discounted QALYs | Total discounted cost (£) | Incremental discounted QALYs vs. strategy 1(1) | Incremental discounted cost vs. strategy 1(1) (£) | ICER vs. strategy 1(1) (£/QALY) | INHB at £20,000 per QALY vs. strategy 1(1) |
|---|---|---|---|---|---|---|
| 1(1) | 151,502 | 38,634,207 | – | – | – | – |
| 1(2) | 151,627 | 39,159,480 | 126 | 525,274 | 4181 | 99.4 |
| 2 | 151,821 | 40,121,977 | 320 | 1,487,770 | 4655 | 245.2 |
| 3 | 151,821 | 40,026,009 | 320 | 1,391,802 | 4354 | 250.1 |
| 4 | 151,860 | 40,156,099 | 359 | 1,521,892 | 4244 | 282.5 |
| 5 | 151,860 | 40,152,774 | 359 | 1,518,568 | 4234 | 282.7 |
| 6 | 151,860 | 40,230,640 | 359 | 1,596,433 | 4451 | 278.8 |
| 7 | 151,904 | 40,717,796 | 402 | 2,083,589 | 5183 | 297.9 |
| 8 | 151,916 | 41,097,605 | 414 | 2,463,398 | 5950 | 290.9 |
Distribution of genes
As genetic testing for LS becomes more commonplace, the distribution of gene mutations identified will alter, given that the mutation distribution is based on the mutations currently identified and the additional ones that may be identified by different diagnostic strategies. In this sensitivity analysis, the gene mutation distribution is taken from Norway, where testing for LS is now routine (Table 113). 184
| Gene mutation | Base case (%) | Sensitivity analysis (%) |
|---|---|---|
| MLH1 | 32 | 18 |
| MSH2 | 39 | 50 |
| MSH6 | 14 | 26 |
| PMS2 | 15 | 6 |
The change in distribution, particularly the reduction in the prevalence of PMS2 mutations, alters the sensitivity and specificity of those strategies that use genetic testing (Figure 123). For these strategies, the sensitivity increases and specificity decreases. This is because the sensitivity is higher for the M126 genes than for PMS2, but when more probands are identified correctly, there will also be a larger proportion of relatives identified incorrectly, thereby increasing the overall FP rate.
FIGURE 123.
Receiver operating characteristic plot for alternative gene mutation distribution.
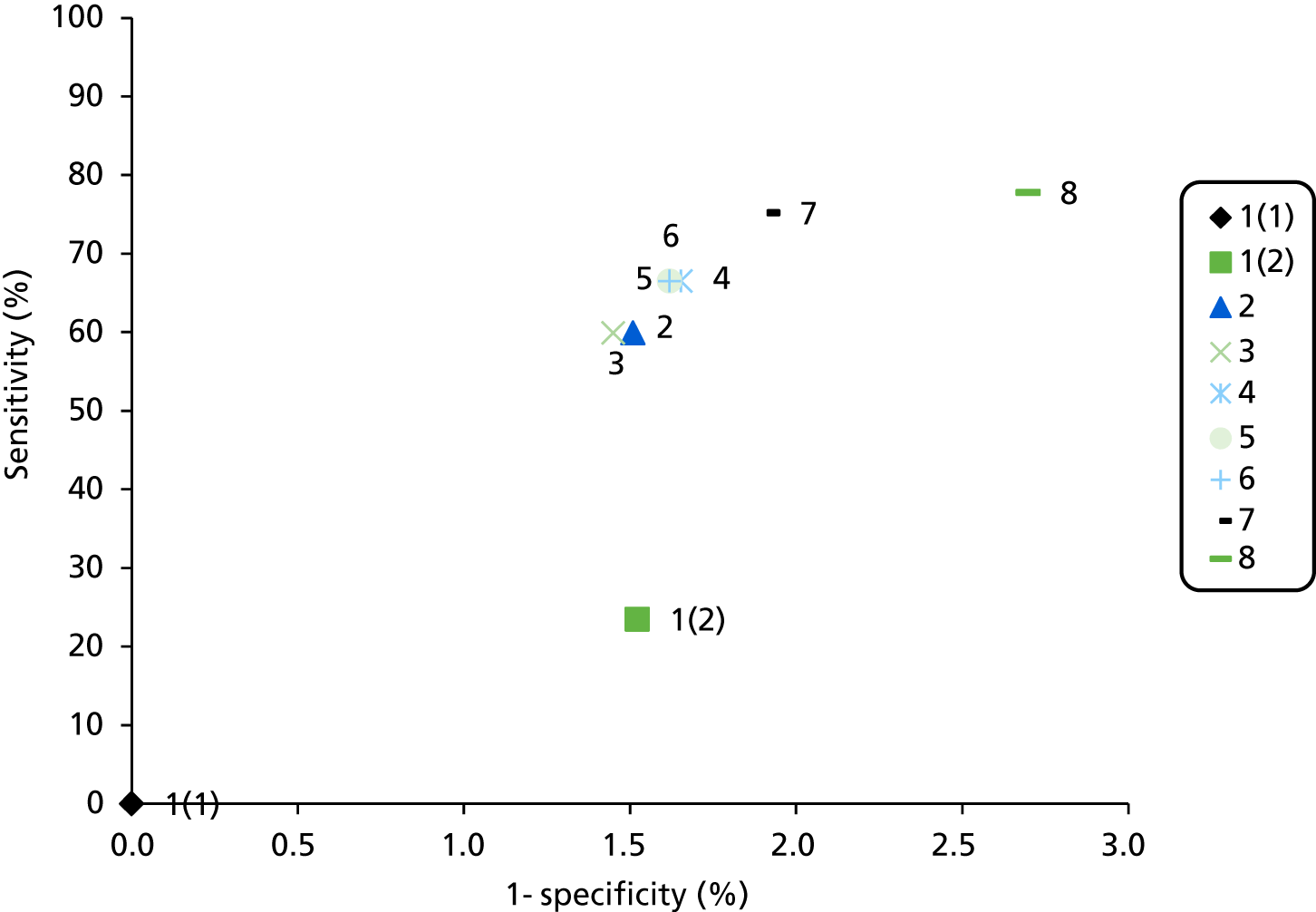
As the type of gene mutation does not affect the long-term outcomes of a patient in our model – only whether or not they have LS and whether or not LS is identified – the only impact on the long-term outcomes occurs as a result of the change in proportions of FNs and FPs. The overall impact on cost-effectiveness is not large, as demonstrated in Table 114; there is an increase in both costs and QALYs gained, but the ICERs remain < £20,000 per QALY for all strategies compared with no testing and decrease slightly in the strategies that use genetic testing. There is also an increase in INHB in these strategies compared with no testing at a threshold of £20,000 per QALY. These results occur primarily because the gain (QALYs gained) resulting from fewer FNs is greater than the loss incurred (costs) by the greater number of FPs.
| Strategy | Total discounted QALYs | Total discounted cost (£) | Incremental discounted QALYs vs. strategy 1(1) | Incremental discounted cost vs. strategy 1(1) (£) | ICER vs. strategy 1(1) (£/QALY) | INHB at £20,000 per QALY vs. strategy 1(1) |
|---|---|---|---|---|---|---|
| 1(1) | 151,793 | 36,223,787 | – | – | – | – |
| 1(2) | 151,857 | 36,608,672 | 64 | 384,885 | 6021 | 44.7 |
| 2 | 151,963 | 37,273,808 | 169 | 1,050,021 | 6201 | 116.8 |
| 3 | 151,963 | 37,179,463 | 169 | 955,676 | 5641 | 121.6 |
| 4 | 151,982 | 37,247,811 | 188 | 1,024,023 | 5445 | 136.9 |
| 5 | 151,982 | 37,226,792 | 188 | 1,003,005 | 5332 | 138.0 |
| 6 | 151,982 | 37,277,498 | 188 | 1,053,710 | 5601 | 135.4 |
| 7 | 152,006 | 37,775,940 | 213 | 1,552,153 | 7303 | 134.9 |
| 8 | 152,012 | 38,225,325 | 219 | 2,001,538 | 9149 | 118.7 |
Probability a relative of a Lynch syndrome-positive proband is affected by Lynch syndrome
In our base case, the proportion of relatives of LS-positive probands who are also affected by LS is 44.0%. Using data direct from Ian Frayling, we examine the case when the proportion reduces to 42.6%. The summary results are given in Table 115; these demonstrate that as the probability decreases, the ICERs compared with no testing increase and the INHB at a £20,000-per-QALY threshold compared with no testing decrease, suggesting a reduction in cost-effectiveness. However, all strategies have a positive INHB and an ICER compared with no testing < £20,000, suggesting they are still cost-effective at a threshold of £20,000 per QALY gained, for this proportion of relatives.
| Strategy | Total discounted QALYs | Total discounted cost (£) | Incremental discounted QALYs vs. strategy 1(1) | Incremental discounted cost vs. strategy 1(1) (£) | ICER vs. strategy 1(1) (£/QALY) | INHB at £20,000 per QALY vs. strategy 1(1) |
|---|---|---|---|---|---|---|
| 1(1) | 151,802 | 36,169,556 | – | – | – | – |
| 1(2) | 151,865 | 36,557,650 | 63 | 388,095 | 6142 | 43.8 |
| 2 | 151,958 | 37,203,902 | 157 | 1,034,346 | 6602 | 105.0 |
| 3 | 151,958 | 37,106,511 | 157 | 936,955 | 5977 | 109.9 |
| 4 | 151,978 | 37,180,712 | 176 | 1,011,156 | 5751 | 125.3 |
| 5 | 151,978 | 37,159,749 | 176 | 990,193 | 5630 | 126.4 |
| 6 | 151,978 | 37,210,568 | 176 | 1,041,012 | 5919 | 123.8 |
| 7 | 151,999 | 37,703,087 | 197 | 1,533,532 | 7781 | 120.4 |
| 8 | 152,004 | 38,150,737 | 202 | 1,981,181 | 9791 | 103.3 |
Number of relatives, proportion first-degree relatives
Another important parameter considered was the number of relatives per proband. As the evidence base for this parameter varied quite widely, we used the extreme values of 0 and 12 relatives to assess the impact of this parameter.
All strategies remain cost-effective compared with no testing at a threshold of £20,000 per QALY when the number of relatives is reduced to 0, suggesting that testing for LS is cost-effective even when it is only conducted on probands. However, ICERs reduce in almost all strategies when the number of relatives increases and therefore the cost-effectiveness improves (Table 116). Figure 124 gives an example of the effect of the number of relatives tested per proband on cost-effectiveness. The only exception to this is strategy 1(2), whose ICER compared with strategy 1(1) increases when the number of relatives increases and reduces when there are no relatives included in the analysis. Presumably, this is due to the larger proportion of incorrectly diagnosed relatives which results when the probands are identified purely on the basis of the FH. There are also fewer probands identified in strategy 1(2) compared with all other testing strategies, resulting in much lower costs.
| Parameter name | Base parameter | Sensitivity analysis parameter | ICER of each strategy vs. strategy 1(1) (£/QALY) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1(1) | 1(2) | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |||
| Base case | – | – | – | 6021 | 6444 | 5831 | 5610 | 5491 | 5774 | 7601 | 9571 |
| Number of relatives | 5 | 0 | – | 907 | 9166 | 7659 | 6966 | 6725 | 7494 | 12,274 | 16,413 |
| 12 | – | 8432 | 5700 | 5329 | 5239 | 5153 | 5303 | 6325 | 7770 | ||
| Proportion of relatives who are FDRs (%) | 42 | 35 | – | 5514 | 6421 | 5814 | 5585 | 5470 | 5756 | 7583 | 9478 |
| 55 | – | 6784 | 6487 | 5863 | 5655 | 5528 | 5809 | 7635 | 9741 | ||
FIGURE 124.
Impact of number of relatives tested per proband on the cost-effectiveness of strategy 5 vs. strategy 1(1).
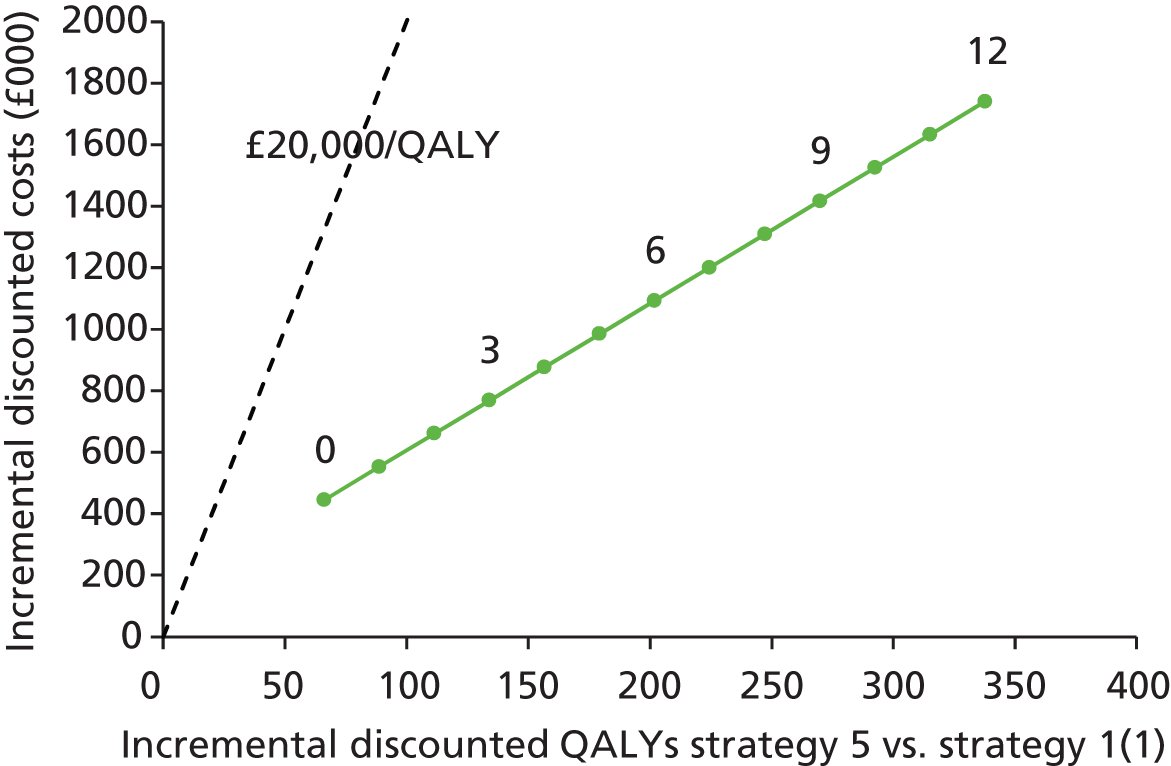
Though an increase in the number of relatives generally appears to improve the cost-effectiveness, an increase in the proportion of FDRs increases the ICERs (i.e. cost-effectiveness worsens). This occurs because the proportion of relatives incorrectly assumed to have LS increases, leading to an increase in unnecessary colonoscopies and prophylactic TAHBSO.
Univariate sensitivity analysis on colorectal cancer
To assess the parameters related to CRC we conduct univariate sensitivity analyses on parameters related to CRC management and CRC prevention. The ICERs compared with no testing for each strategy are presented in Table 117, except the results for changes in the effectiveness and adherence to LS surveillance colonoscopies, which are presented below (see Tables 119 and 120). Where certain analyses represented more than one parameter change, the disaggregation of those parameter groups are given in Table 118, as referenced in Table 117.
| Parameter name | Base parameter | Sensitivity analysis parameter | ICER of each strategy vs. strategy 1(1) (£/QALY) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1(1) | 1(2) | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |||
| Base case | – | – | – | 6021 | 6444 | 5831 | 5610 | 5491 | 5774 | 7601 | 9571 |
| Cost of colonoscopy (£) | £395 | £197 | – | 2939 | 4156 | 3572 | 3331 | 3227 | 3511 | 5305 | 7006 |
| £790 | – | 12,184 | 11,021 | 10,350 | 10,166 | 10,018 | 10,301 | 12,194 | 14,703 | ||
| Cost of colonoscopy complications | Base costs | Halved | – | 6000 | 6429 | 5816 | 5594 | 5476 | 5759 | 7586 | 9554 |
| Doubled | – | 6062 | 6475 | 5861 | 5640 | 5521 | 5805 | 7632 | 9606 | ||
| Cost of CRC diagnosis (£) | 499 | 249 | – | 6045 | 6470 | 5857 | 5635 | 5517 | 5800 | 7627 | 9598 |
| 998 | – | 5972 | 6392 | 5779 | 5558 | 5439 | 5723 | 7549 | 9519 | ||
| Cost of CRC surgery | Base costs | Halved | – | 6355 | 6795 | 6182 | 5961 | 5842 | 6125 | 7953 | 9928 |
| Doubled | – | 5351 | 5742 | 5130 | 4907 | 4790 | 5073 | 6899 | 8858 | ||
| Cost of CRC chemotherapy and radiation | Base costs | Halved | – | 6869 | 7281 | 6666 | 6445 | 6326 | 6610 | 8438 | 10,421 |
| Doubled | – | 4324 | 4771 | 4161 | 3938 | 3821 | 4104 | 5928 | 7872 | ||
| Cost of CRC stoma care (£) | GP, 405; LS, 218 | GP, 202 | – | 5974 | 6428 | 5815 | 5594 | 5475 | 5759 | 7585 | 9556 |
| LS, 109 | |||||||||||
| GP, 810 | – | 6115 | 6476 | 5863 | 5641 | 5523 | 5806 | 7633 | 9603 | ||
| LS, 436 | |||||||||||
| Cost of CRC follow-up surveillance (£) | GP, 264; LS, 268 | GP, 132 | – | 6044 | 6473 | 5860 | 5639 | 5520 | 5803 | 7630 | 9601 |
| LS, 134 | |||||||||||
| GP, 529 | – | 5974 | 6386 | 5773 | 5551 | 5433 | 5716 | 7543 | 9512 | ||
| LS, 536 | |||||||||||
| Cost of CRC recurrence (£) | GP, 12,452; LS, 12,556 | GP, 6226 | – | 6445 | 6874 | 6260 | 6039 | 5920 | 6203 | 8031 | 10,007 |
| LS, 6278 | |||||||||||
| GP, 24,904 | – | 5172 | 5585 | 4973 | 4751 | 4633 | 4917 | 6742 | 8700 | ||
| LS, 25,112 | |||||||||||
| Cost of CRC palliative care (£) | GP, 9826; LS, 10,087 | GP, 4913 | – | 6363 | 6813 | 6199 | 5978 | 5859 | 6143 | 7970 | 9947 |
| LS, 5043 | |||||||||||
| GP, 19,653 | – | 5335 | 5706 | 5095 | 4872 | 4754 | 5038 | 6863 | 8821 | ||
| LS, 20,173 | |||||||||||
| Probability of colonoscopy mortality | 8.3 per 100,000 colonoscopies | 39 per 100,000 colonoscopies | – | 5999 | 6585 | 5967 | 5740 | 5622 | 5909 | 7756 | 9690 |
| CRC incidence | Beta0 = 0.464 males, 0.435 females | Beta0 = 0.303 males, 0.265 females | – | 10,493 | 11,641 | 10,634 | 10,298 | 10,097 | 10,549 | 13,502 | 17,068 |
| 0.715 males, 0.697 females | – | 4665 | 3511 | 3068 | 2915 | 2827 | 3028 | 4334 | 5868 | ||
| HR of LS CRC survival | Dukes’ A/B, 0.57; Dukes’ C/D, 1 | All Dukes’ set to 0.57 | – | 5570 | 6166 | 5571 | 5348 | 5235 | 5514 | 7300 | 9123 |
| All Dukes’ set to 1 | – | 6348 | 7423 | 6729 | 6460 | 6331 | 6660 | 8760 | 10,787 | ||
| CRC utility decrements | Dukes’ A/B/C, 0 (index and metachronous) Dukes’ D, 0.13 (index and metachronous) |
Ness utilities, index and metachronous CRC: | – | 5242 | 5531 | 5022 | 4825 | 4730 | 4971 | 6511 | 8015 |
| Dukes’ A, 0.11 | |||||||||||
| Dukes’ B, 0.23 | |||||||||||
| Dukes’ C, 0.26 | |||||||||||
| Dukes’ D, 0.60 | |||||||||||
| CRC surgery disutility | All surgery disutility 0 | All surgery disutility 0.1 (except segmental resection) | – | 3803 | 5041 | 4587 | 4394 | 4314 | 4539 | 5943 | 7091 |
| Adherence to LS surveillance colonoscopy | See Table 120 | ||||||||||
| Effectiveness of LS surveillance colonoscopy | See Table 119 | ||||||||||
| Probability of aggressive surgery for LS | 0 | 1 | – | 6471 | 5905 | 5310 | 5102 | 4985 | 5256 | 7016 | 9046 |
| Cost parameter group altered | Individual parameter altered | Base cost (£) | Half (£) | Double (£) |
|---|---|---|---|---|
| Cost of colonoscopy complication | Cost of bleeding (mild) | 318 | 159 | 637 |
| Cost of bleeding (moderate) | 490 | 245 | 980 | |
| Cost of bleeding (severe) | 1984 | 992 | 3967 | |
| Cost of perforation | 5134 | 2567 | 10,269 | |
| Cost of mortality | 5134 | 2567 | 10,269 | |
| Cost of CRC surgery | Segmental resection | GP, 6104 | GP, 3052 | GP, 12,209 |
| LS, 6154 | LS, 3077 | LS, 12,308 | ||
| Subtotal colectomy IRA | GP and LS, 7331 | GP and LS, 3666 | GP and LS, 14,662 | |
| Rectal excision | GP and LS, 7399 | GP and LS, 3699 | GP and LS, 14,797 | |
| Proctocolectomy | GP and LS, 7441 | GP and LS, 3720 | GP and LS, 14,882 | |
| Cost of CRC chemotherapy and radiotherapy | Dukes’ A | Colon, 0 | Colon, 0 | Colon, 0 |
| Rectal, 0 | Rectal, 0 | Rectal, 0 | ||
| Dukes’ B | Colon, 5755 | Colon, 2878 | Colon, 11,510 | |
| Rectal, 2848 | Rectal, 1424 | Rectal, 5696 | ||
| Dukes’ C | Colon, 13,133 | Colon, 6567 | Colon, 26,266 | |
| Rectal, 7628 | Rectal, 3814 | Rectal, 15,257 | ||
| Dukes’ D | Colon, 13,133 | Colon, 6567 | Colon, 26,266 | |
| Rectal, 7628 | Rectal, 3814 | Rectal, 15,257 |
Costs were altered using the disaggregated results of the base case, halving and then doubling the costs where appropriate, but all other parameters related to CRC required further runs of the model. Owing to time constraints, these runs were reduced from 32,000 to 4000 simulations, and therefore there is greater uncertainty in them than in the base case. Despite the extra uncertainty, the results are still informative regarding the impact of the parameters upon the model.
Costs associated with colorectal cancer
The general finding of univariate sensitivity analysis on the costs incurred in relation to CRC is that increasing the cost of CRC treatment (diagnosis, surgery, follow-up, etc.) reduces the ICERs for all strategies compared with no testing, thus improving the cost-effectiveness. This is to be expected as the cost savings from preventing CRCs would be greater. In contrast, increasing the cost of CRC prevention (colonoscopies and their complications) increases the ICERs of all strategies compared with no testing. In no circumstance does halving or doubling the cost of prevention or treatment of CRC cause the ICER of any strategy to increase above the £20,000-per-QALY threshold.
The two largest cost drivers appear to be the cost of colonoscopies and the cost of chemotherapy and radiotherapy. This is unsurprising as the cost of chemotherapy and radiotherapy is the single largest treatment cost for CRC, and the cost of colonoscopy, while not incurring a large unit cost, has a large overall cost due to the total number of colonoscopies undertaken in each strategy.
Halving and doubling the cost of colonoscopy complications also demonstrates the effect of halving and doubling the number of adverse events incurred during colonoscopy (except colonoscopy mortality). This is because in the model the only effect of these adverse events is the costs associated with them; there is no additional utility decrement.
Utilities associated with colorectal cancer
In the case of utility decrement associated with CRC stage, we examined a scenario of different plausible values from the study by Ness and colleagues. 200 Under these values, the utility of patients was worse in all of the Dukes’ stages compared with the base case. As such, the cost-effectiveness of all strategies testing for LS is improved, with all ICERs compared with no testing reducing compared with the base case, as identifying patients with LS and preventing CRC, or diagnosing it at an earlier stage through colonoscopy, has a greater benefit than in the base case.
In our base case, the disutility associated with CRC is at its most optimistic, being set at 0 for all types of CRC surgery. In our sensitivity analysis we increase the disutility associated with the more aggressive forms of surgery (subtotal colectomy IRA, rectal excision, proctocolectomy) to 0.1. As with the change in utility associated with CRC stages, this improves the cost-effectiveness of all strategies compared with no testing, compared with the base case. This disutility appears to have a greater impact upon cost-effectiveness than an increase in the disutility of the stages of CRC, with the ICERs reducing by a greater amount compared with the base case under CRC surgery disutility compared with stages disutility.
As both changes to utility increase the cost-effectiveness of the strategies, all strategies have an ICER compared with no testing below the £20,000-per-QALY threshold.
Effectiveness of Lynch syndrome surveillance colonoscopies
The effectiveness of LS surveillance colonoscopies is potentially a very important parameter. We would expect that improving the effectiveness of surveillance colonoscopies would improve the cost-effectiveness of strategies in which individuals are offered surveillance.
To get the best estimate of the impact of the effectiveness of surveillance at preventing index CRC on cost-effectiveness, we held the costs and QALYs for individuals not receiving surveillance and for all probands (as they face no index CRC risk) constant across the sensitivity analyses using the results from the base case (i.e. only the costs and QALYs for relatives receiving surveillance are simulated). We also used 32,000 simulated individuals for each patient group (the same number as in the base case).
We estimated the impact of the effectiveness of surveillance at preventing metachronous CRC by running the whole model with 4000 patients simulated per group.
As expected, when the effectiveness of LS surveillance colonoscopies is increased, the cost-effectiveness of testing increases (Table 119).
| Parameter name | Base parameter | Sensitivity analysis parameter | ICER of each strategy vs. strategy 1(1) (£/QALY) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1(1) | 1(2) | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |||
| Base case | – | – | – | 6021 | 6444 | 5831 | 5610 | 5491 | 5774 | 7601 | 9571 |
| HR for colonoscopy for preventing index CRC | 0.387 | 0.254 | – | 5644 | 5856 | 5272 | 5060 | 4948 | 5218 | 6959 | 8827 |
| 0.590 | – | 6514 | 7247 | 6602 | 6371 | 6246 | 6543 | 8462 | 10,540 | ||
| HR for colonoscopy for preventing metachronous CRC | 0.533 | 1 | – | 11,402 | 9830 | 8974 | 8687 | 8515 | 8898 | 11,407 | 14,443 |
Adherence to Lynch syndrome surveillance to prevent colorectal cancer
Adherence is an important factor for both patients and clinicians. With regards to preventing CRC, we examined both adherence to colonoscopies for patients and the possibility of clinicians conducting a more aggressive colorectal surgery on CRC patients with LS (Table 120).
| Parameter name | Proband/relative | LS diagnosis | Base parameter | Sensitivity analysis parameter | ICER of each strategy vs. strategy 1(1) (£/QALY) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1(1) | 1(2) | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |||||
| Base case | – | – | – | 6021 | 6444 | 5831 | 5610 | 5491 | 5774 | 7601 | 9571 | ||
| Adherence to LS colonoscopy | Proband | LS assumed | 0.7 | 0.35 | – | 8703 | 6641 | 6019 | 5785 | 5666 | 5956 | 7823 | 9789 |
| 0.85 | – | 5258 | 6362 | 5753 | 5537 | 5418 | 5699 | 7509 | 9481 | ||||
| LS mutation positive | 0.8 | 0.40 | – | 6021 | 8083 | 7353 | 7094 | 6953 | 7290 | 9458 | 11,811 | ||
| 0.90 | – | 6021 | 6112 | 5523 | 5309 | 5195 | 5468 | 7225 | 9119 | ||||
| Relative | LS assumed | 0.5 | 0.25 | – | 5437 | 6633 | 5949 | 5680 | 5555 | 5881 | 7959 | 10,042 | |
| 0.75 | – | 6432 | 6299 | 5740 | 5555 | 5442 | 5693 | 7326 | 9212 | ||||
| LS mutation positive | 0.8 | 0.40 | – | 6021 | 7465 | 6756 | 6502 | 6365 | 6692 | 8804 | 11,090 | ||
| 0.90 | – | 6021 | 6231 | 5638 | 5423 | 5308 | 5582 | 7350 | 9254 | ||||
| Probands and relatives | LS assumed or mutation positive | See above | 1.0 | – | 5555 | 5306 | 4848 | 4713 | 4615 | 4813 | 6124 | 7763 | |
Changes in adherence to LS surveillance colonoscopy demonstrate that, for most strategies, a reduction in adherence results in deterioration in cost-effectiveness compared with the base case, with increased ICERs compared with no testing relative to the base case, and that an increase in adherence improves the cost-effectiveness. This is a result of the benefit gained from the additional colonoscopies being greater than the extra cost. Once again it is strategy 1(2) which appears to have a few anomalous results. In the case of strategy 1(2), there is no difference in ICER compared with no testing for patients with confirmed LS mutations (as it does not use genetic testing, there are none in this strategy). For relatives with assumed LS, the cost-effectiveness of strategy 1(2) increases as the adherence to colonoscopies decreases. Though this seems counter-intuitive, especially given both the results of the other strategies and the result of the decreasing adherence for probands in strategy 1(2), this actually reflects reduction in benefit and higher costs from the lower number of TP relatives identified by strategy 1(2) compared with the other strategies. In short, the benefit of identifying some TP patients in strategy 1(2) is outweighed by the costs of not identifying a larger number.
We also looked at the scenario where all patients identified as LS assumed or LS mutation positive are offered and accept LS surveillance colonoscopies. In this instance the cost-effectiveness of all strategies improves.
Colonoscopy mortality
Increasing the probability of mortality due to colonoscopy from 8.3 to 39.0 per 100,000 colonoscopies had a very small impact on cost-effectiveness. The ICERs appear to increase slightly for all strategies versus no testing, except for strategy 1(2). All ICERs compared with no testing remained < £10,000 per QALY.
Aggressive surgery for colorectal cancer patients diagnosed with Lynch syndrome
With regards to the use of aggressive CRC surgery for patients diagnosed with LS (mutation positive or assumed), the values from different sources made it clear that this value could lie anywhere between 0% and 100%. As such, we conduct our sensitivity analysis at 100%, as 0% was used as our base case. As previously stated, a change in surgery is not accounted for in the case of the proband’s index cancer, as we assume that a LS diagnosis will not be achieved until after a proband has been treated. One important aspect of this to consider is that under our other base-case parameters, the disutility for all surgeries is 0, regardless of the extent or functional consequences of surgery. As such, this is a best-case scenario for the use of aggressive surgery.
Again the results for strategy 1(2) differed from the other strategies, owing to the weighting of FPs versus TPs for that strategy. For all the strategies that used genetic testing, an improvement in cost-effectiveness is demonstrated in lower ICERs compared with no testing than in the base case. This is a result of the reduction in CRCs for true LS patients as a result of the more aggressive surgery (as well as a slight reduction in CRCs in the incorrectly diagnosed members of the general population).
Colorectal cancer incidence and survival
The final area that we examined in our sensitivity analysis for CRC was surrounding parameters associated with CRC incidence and survival.
Our results in Table 117 suggest that CRC incidence in LS patients is an important driver of the model. When the incidence was increased, the ICERs of every strategy compared with no testing decreased compared with the base case, implying that when the incidence of CRC is higher it is more beneficial to identify patients with LS. This makes sense as more benefit will be gained from the colonoscopic surveillance of patients with LS. When CRC incidence was reduced, the ICERs greatly increased compared with the base case, with ICERs of all strategies > £10,000 per QALY gained and the ICER of strategy 8 > £17,000.
In our base case, LS patients have an improved survival from CRC in Dukes’ A or B compared with general population patients (with a HR of 0.57) and the same survival for Dukes’ C and D (HR of 1). In our sensitivity analysis of CRC survival based on Dukes’ stage, we examine the case when LS patients have the same survival for all Dukes’ stages as the general population (HR of 1 for all Dukes’ stages) and when LS patients have improved survival for all Dukes’ stages compared with the general population (HR of 0.57 for all Dukes’ stages). As expected, an improvement in survival increases the QALY gain for all LS patients, regardless of strategy, with additional QALYs gained for each strategy compared with their base case [even strategy 1(1)]. There was also a reduction in costs for each strategy compared with the base case. Overall, the strategies that tested for LS had greater benefits compared with the base case than strategy 1(1), resulting in lower ICERs compared with strategy 1(1) than in the base case. Conversely, setting the survival of LS patients to be the same as that of the general population resulted in higher ICERs compared with no testing than in the base case, as the additional benefit of testing for LS (that LS-related CRCs caught earlier would have an increased survival over general population patients in the earlier Dukes’ stages of CRC in the base case) was no longer apparent. However, in both sensitivity analyses all the strategies’ ICERs compared with no testing remained < £11,000 per QALY gained.
Univariate sensitivity analysis on endometrial cancer
As with the parameters influencing CRC, we also conducted univariate sensitivity analyses on the parameters related to both EC treatment and EC prevention. The ICERs compared with no testing for each strategy are presented in Table 121. Changes to EC parameters once again only influence long-term outcomes, with diagnostic results the same as those of the base case.
| Parameter name | Base parameter | Sensitivity analysis parameter | ICER of each strategy vs. strategy 1(1) (£/QALY) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1(1) | 1(2) | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |||
| Base case | – | – | – | 6021 | 6444 | 5831 | 5610 | 5491 | 5774 | 7601 | 9571 |
| Cost of prophylactic TAHBSO (£) | 3322 | 1661 | – | 4375 | 5386 | 4789 | 4557 | 4447 | 4730 | 6539 | 8360 |
| 6644 | – | 9312 | 8561 | 7916 | 7714 | 7580 | 7863 | 9726 | 11,993 | ||
| Cost of EC TAHBSO (£) | 3877 | 1939 | – | 6112 | 6547 | 5933 | 5712 | 5593 | 5877 | 7704 | 9675 |
| 7754 | – | 5837 | 6239 | 5626 | 5404 | 5286 | 5569 | 7396 | 9365 | ||
| Cost of EC radiotherapy (£) | 2753 | 1377 | – | 6086 | 6517 | 5904 | 5682 | 5564 | 5847 | 7674 | 9645 |
| 5506 | – | 5890 | 6299 | 5685 | 5464 | 5345 | 5629 | 7456 | 9425 | ||
| Cost of EC chemotherapy (£) | 541 | 270 | – | 6033 | 6458 | 5845 | 5624 | 5505 | 5789 | 7616 | 9586 |
| 1082 | – | 5995 | 6416 | 5802 | 5581 | 5462 | 5746 | 7573 | 9543 | ||
| EC incidence given LS positive | Age dependent, e.g. cumulative incidence at age 80 years 0.35 | Half | – | 5886 | 7267 | 6608 | 6358 | 6234 | 6545 | 8531 | 10,511 |
| Double | – | 5623 | 5671 | 5099 | 4897 | 4785 | 5047 | 6744 | 8633 | ||
| Adherence to prophylactic TAHBSO (proband and relative) (%) | 55 | 20 | – | 4355 | 5939 | 5280 | 5006 | 4890 | 5211 | 7237 | 9061 |
| 90 | – | 10,377 | 7743 | 7048 | 6857 | 6705 | 6997 | 8959 | 11,782 | ||
| EC and TAHBSO disutility | EC, 0 Prophylactic TAHBSO, 0 |
EC, 0.1 Prophylactic TAHBSO, 0 |
– | 5404 | 6446 | 5875 | 5651 | 5545 | 5818 | 7551 | 9182 |
| EC, 0.1 Prophylactic TAHBSO, 0.1 |
– | –10,014 Dominated by no testing |
–162,330 Dominated by no testing |
–240,283 Dominated by no testing |
–163,915 Dominated by no testing |
–208,191 Dominated by no testing |
–219,492 Dominated by no testing |
–174,638 Dominated by no testing |
–51,515 Dominated by no testing |
||
| EC, 0.2 Prophylactic TAHBSO, 0.1 |
– | –10,057 Dominated by no testing |
–107,066 Dominated by no testing |
–130,097 Dominated by no testing |
–103,329 Dominated by no testing |
–117,727 Dominated by no testing |
–123,529 Dominated by no testing |
–117,683 Dominated by no testing |
–46,072 Dominated by no testing |
||
As with the CRC sensitivity analyses, costs related to the prevention and treatment of EC were again altered using the disaggregated results of the base case, halving and then doubling the costs where appropriate. All other parameters related to CRC required further runs of the model of 4000 simulations, once again resulting in greater uncertainty in them than in the base case, but demonstrating with enough certainty the general pattern of the results.
Costs associated with endometrial cancer
As with the CRC sensitivity analyses, decreasing the cost of EC prevention (prophylactic TAHBSO) resulted in lower ICERs compared with no testing than in the base case, and increasing the cost of prophylactic TAHBSO resulted in higher ICERs compared with no testing than in the base case. However, despite the implications of scenario analysis 1, where EC was not included in the analysis, increasing the cost of prophylactic surgery does not greatly reduce the cost-effectiveness of the strategies, with all of the ICERs remaining < £12,000 per QALY gained. It does, however, have a larger impact on the ICERs than any other single cost parameter related to EC.
Increasing the cost of EC treatment (TAHBSO, radiotherapy or chemotherapy) improves the cost-effectiveness of all the testing strategies compared with the base case; the ICERs compared with no testing are lower than in the base case. Conversely, decreasing the cost of EC treatment reduces the cost-effectiveness of all strategies.
Strategy 5 has the lowest ICER compared with no testing for nearly all EC cost univariate sensitivity analyses, the exception being when the cost of prophylactic TAHBSO is halved.
Utilities associated with endometrial cancer
In the base case there are few disutilities associated with EC. A few relate to psychological disutility associated with being offered a prophylactic TAHBSO, but these are discussed separately in Psychological disutility. The two utility decrements which we evaluate in this section are the impact of a utility decrement associated with undergoing a TAHBSO (either prophylactically or for EC) and a utility decrement associated with having EC. In the sensitivity analyses we consider adjusting these to 0.1.
As Table 121 demonstrates, the addition of a utility decrement for EC improves the cost-effectiveness of strategies that test for LS, with their ICERs compared with no testing reducing from the base case. In this instance, strategy 1(2) had the lowest ICER compared with no testing of £5404 per QALY gained, though several other strategies had very similar ICERs, particularly strategy 5, which had an ICER compared with no testing of £5545 per QALY gained. Strategy 5 had the highest INHB at a threshold of £20,000 compared with no testing.
The single parameter change with the greatest impact upon the cost-effectiveness of testing for LS is the disutility associated with undergoing a TAHBSO, which applies to individuals opting for prophylactic TAHBSO and to EC patients. In our base case this is assumed to be 0, for reasons discussed in Chapter 5, Impact of prophylactic total abdominal hysterectomy with bilateral salpingo-oophorectomy on quality of life, and when it was increased to 0.1 in our sensitivity analysis (chosen arbitrarily), this resulted in all strategies of testing for LS being dominated by no testing, such that they were both more expensive (as in the base case) and had fewer QALYs than strategy 1(1). As Figure 125 shows, some strategies did maintain an incremental QALY gain in the long term compared with strategy 1(1), but when this occurred it was outweighed by the psychological disutility accrued from diagnosing LS. As such, there was no overall incremental QALY gain in any of the strategies. This is a significant change to the outcome of the model, which is not achieved by any other change in parameter value. Given the significant impact of this change, when both EC and TAHBSO disutility are included in the model, the strategies remain dominated by no testing.
FIGURE 125.
Incremental discounted QALYs vs. strategy 1(1) when disutility of prophylactic TAHBSO is set to 0.1.
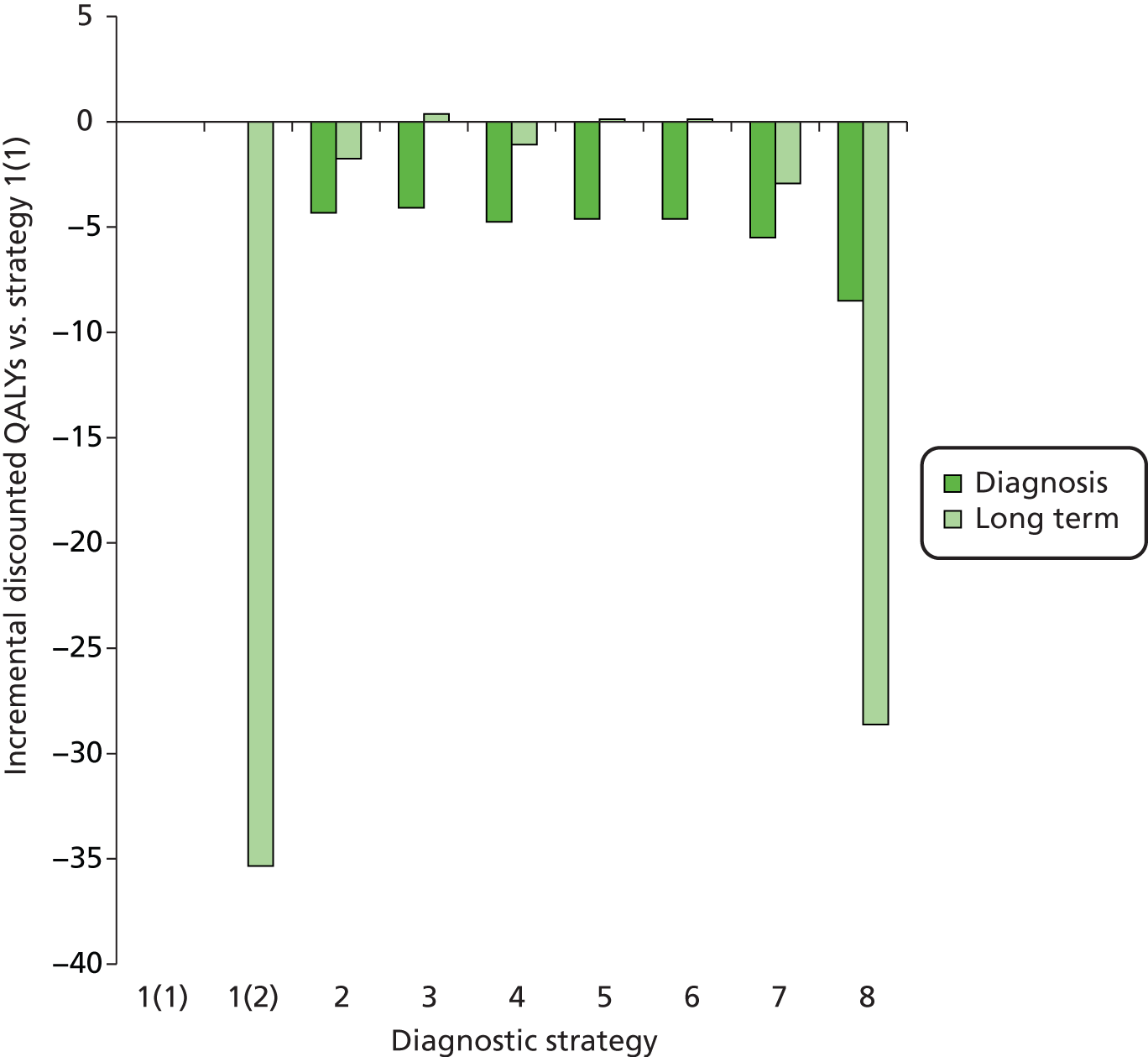
Adherence to Lynch syndrome management to prevent endometrial cancer
Unlike the utility values for prophylactic TAHBSO, the adherence to this prophylactic surgery does not have such a significant impact on the results. It does, however, have an impact on those strategies which identify fewer patients as LS positive, with strategy 1(2) having a greater change in ICER compared with no testing from the base case than any other strategy, particularly when the adherence rate increases to 90%. In general, the ICERs increase with an increase in adherence to prophylactic TAHBSOs, reflecting the additional cost, which is not offset by a particularly significant increase in QALYs.
Psychological disutility
Changing the parameters for the psychological disutility of being tested for LS does not have an impact on the cost of each strategy, though there is some variation from the base case due to having a different set of fewer simulations. The impact of changes to psychological disutility is seen in the change in QALYs gained by each of the LS testing strategies (Table 122). By removing any psychological disutility, we see that the ICERs compared with no testing reduce for all strategies compared with the base case, owing to the reduction of any QALY loss gained from testing for LS, both in the short term with regards to diagnosis and in the long term with regards to being offered a TAHBSO. By doubling the disutility from the base case, we find that the ICERs compared with no testing increase, in accordance with the additional QALY loss accrued in the testing strategies.
| Parameter name | Base parameter | Sensitivity analysis parameter | ICER of each strategy vs. strategy 1(1) (£/QALY) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1(1) | 1(2) | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |||
| Base case | – | – | – | 6021 | 6444 | 5831 | 5610 | 5491 | 5774 | 7601 | 9571 |
| Psychological disutility | GT declined, 0.04 Testing LS positive, 0.02 Being offered TAHBSO: proband, 0.01; relative, 0.04 Declining TAHBSO: proband, 0.06; relative, 0.03 |
0 | – | 4562 | 4899 | 4432 | 4256 | 4168 | 4387 | 5792 | 7214 |
| Double | – | 6045 | 6749 | 6113 | 5889 | 5765 | 6056 | 7941 | 10,068 | ||
| Length of psychological disutility | 4 months | 1 year | – | 13,109 | 9676 | 8660 | 8397 | 8172 | 8590 | 11,433 | 16,181 |
In our base case the time for which the psychological disutilities apply is 4 months. When this is increased to 1 year, the ICERs compared with no testing increase from the base case, particularly in the case of strategy 8, owing to the larger proportion of probands offered genetic testing.
Chapter 7 Discussion
Aim
The question addressed by this project is ‘what is the diagnostic utility and cost-effectiveness of genetic testing for HNPCC in all newly diagnosed persons with CRC under 50 years of age, and of strategies to test their close relatives?’.
This question was addressed by a systematic review of clinical effectiveness and cost-effectiveness studies, and by a de novo economic analysis.
Main findings
Clinical effectiveness
Ten published papers were included in the review, one on MSI only, three on MSI and IHC, four on IHC and one TA commissioned by the US Department of Health and Human Services. The TA, which this review continues from, found minimal published information on the analytical validity of laboratory testing for LS, along with some concern regarding variability between testing facilities. Genomic rearrangements and large deletions were missed when only sequencing and gene screening was performed. Very few studies focused on unselected patients with CRC and many had small sample sizes. Results ranged from 18% to 100% for sensitivity and 25% to 100% for specificity. CIs were very wide.
The majority of studies included in this review were single gate, where only one sample of individuals was assessed by the index test and reference standard. Two studies (which appear to be using the same population) were two gate, recruiting a group of patients with known mutation status and a control group with no known mutation status. All studies were assessed for quality against the QUADAS-2 tool and many were found to be at risk of bias as, owing to cost, the reference standard was only performed on patients with a likely mutation or MSI. Other concerns were small sample size and minimal details on robustness of testing. Some studies recruited from preselected patients (e.g. from registries), whereas others recruited from the general population. This means that a population including patients with sporadic CRC may recruit a higher proportion of patients with methylation of MLH1 (meMLH1). This has consequences for both IHC and MSI testing which will identify meMLH1 as MSI, whereas constitutional genetic testing would identify the sample as negative, thereby leading to FP results.
Owing to the range of study designs – for example a wide range and number of markers of MSI, and targeting different proteins for IHC – pooling of data was not possible. As such, a summary of individual results is displayed in Table 123. The sensitivities and specificities for IHC are wide ranging (from 73.3% to 100% and 12.5% to 100%, respectively), although not all studies searched for all proteins and some of the results were combined. Clearly, specificity is the greatest concern, as a high number of FPs means that individuals may be told they have LS when they do not. Furthermore, those studies recruiting from a population that had no prior testing may include an increased number of FPs due to MLH1 methylation found in sporadic CRC. The sensitivity for MSI ranged from 88% to 100%, and specificity ranged from 68% to 84%. However, no two studies included in this review have used the same panel of markers, and therefore a comparison is difficult and results vary. As with IHC, not all patients received the reference standard.
| IHC | |||||
|---|---|---|---|---|---|
| First author | Patients (n) | Reference standard | Protein | Sensitivity, % (95% CI, %) | Specificity, % (95% CI, %) |
| Barrow 201042 | 68 | Germline testing performed before recruitment | MLH1 | 100 (84 to 100) | 91.5 (79.6 to 97.6) |
| PMS2 | 95.2 (76.2 to 99.9) | 91.5 (79.6 to 97.6) | |||
| MSH2 | 87.5 (61.7 to 98.4) | 88.5 (76.5 to 95.6) | |||
| MSH6 | 81.3 (54.4 to 96.0) | 80.8 (67.5 to 90.4) | |||
| Barrow 201120 | 42 | Germline testing performed before recruitment | MLH1 | 89.5 (66.9 to 98.7) | 78.3 (56.3 to 92.5) |
| MSH2 | 86.7 (59.5 to 98.3) | 77.8 (57.7 to 91.4) | |||
| Becouarn 200543 | 197 | Direct sequencing with search for large mutations if point mutation not identified | MLH1, MSH2 | 73.3 (44.9 to 92.2) | 28.6 (3.7 to 71.0) |
| Limburg 201144 | 195 | Direct sequencing with Southern blot and MLPA | MLH1, MSH2, MSH6 | 85.7 (42.1 to 99.6) | 91.9 (86.3 to 95.7) |
| Niessen 200621 | 281 | Direct sequencing and MLPA | MLH1 | 80 (38 to 96) | 89 (84 to 93) |
| MSH2 | 100 (57 to 100) | 96 (92 to 98) | |||
| MSH6 | 86 (46 to 97) | 93 (88 to 96) | |||
| Shia 200511 | 110 | Direct sequencing and large deletion analysis | MLH1, MSH2, MSH6 | 80.8 (60.6 to 93.4) | 89.5 (75.2 to 97.1) |
| Southey 200539 | 131 | Direct sequencing and MLPA | MLH1, MSH2, MSH6 | 100 (82 to 100) | 91 (83 to 96) |
| Stomorken 200545 | 250 | Direct sequencing and large rearrangement analysis | MLH1, MSH2, MSH6 | 100 (75.3 to 100) | 12.5 (0.3 to 52.7) |
| MSI testing | |||||
| First author | Patients (n) | Reference standard | Panel of markers | Sensitivity, % (95% CI, %) | Specificity, % (95% CI, %) |
| Niessen 200621 | 281 | Direct sequencing and MLPA | Five markers | 88.0 (68.8 to 97.5) | 68.1 (61.7 to 74.0) |
| Shia 200511 | 110 | Direct sequencing and large deletion analysis | Seven markers | 100 (85.8 to 100) | 84.0 (68.0 to 93.8) |
| Southey 200539 | 131 | Direct sequencing and MLPA | 10 markers | 94 (73 to 100) | 80 (71 to 88) |
| Wolf 200646 | 55 | Sequence analysis | Five markers | 100 (71.7 to 100) | 78.6 (62.8 to 89.2) |
Cost-effectiveness model
The results of the cost-effectiveness model are summarised in the scientific summary and in Chapter 6, Summary of Peninsula Technology Assessment Group cost-effectiveness results. Here, we present a section of the results in this summary.
Testing strategies
When testing for LS in probands, specificity is consistently higher than sensitivity across the strategies; all strategies that identify some patients as LS positive have a specificity of ≥ 98%, whereas sensitivity ranges from 39% to 78%. The specificities of all genetic testing strategies are similar and all > 99.8%. Strategy 8 (universal genetic testing) has the highest sensitivity (77.9%). In general, additional tests for a cohort whose previous test result was indicative of LS increases specificity, as each additional test can only reduce the number of FPs. One side effect of this is that it is also likely to reduce the sensitivity; if the tests have < 100% sensitivity then the number of FNs also increases (and the number of TPs decreases) with each additional test. This explains why, in most strategies, the sensitivity of the strategy is lower than the sensitivity of the first test in the sequence, even in strategy 8, where all probands are offered genetic testing, as it includes at least two tests for gene testing plus guidance from the probands’ FH.
For probands, the strategies always identify less than the true proportion of LS-positive probands, but most strategies identify more relatives as LS positive than there actually are. The main driver appears to be the proportion of relatives assumed to have LS, with the majority of these incorrectly assumed to have LS.
The data on test accuracy are limited. It is accepted that gene testing does not have 100% sensitivity or 100% specificity, but often these are used as the ‘gold standard’ for assessing other tests, though some studies use a much broader view including mutation positive and FH indicative of LS as their ‘gold standard’. There is also little evidence regarding the distribution of identified mutations in the FPs from tumour tests such as IHC. By rarely using IHC to identify the testing sequence of genes, this problem is mostly eliminated, though a similar problem then occurs for the distribution of FPs post gene testing.
Little is currently known about the effect of running tests sequentially on the sensitivity and specificity of each component test, particularly tumour-based testing. Only BRAF is currently modelled with values given explicitly after MSI or IHC with MLH1 abnormal. It is unclear whether or not the values for sensitivity and specificity of genetic testing in the literature relate to an enriched group of patients. However, given that it is unlikely that genetic testing in probands would be run on an unselected cohort, the assumed sensitivity and specificity of genetic testing in strategy 8 may not be entirely accurate.
The cost of tests varies substantially across the various genetics centres. Therefore, we varied these parameters widely in our sensitivity analyses.
Evidence for the rates of acceptance of diagnostic tests and genetic counselling is quite varied, though the results from our sensitivity analyses indicate that, even at low rates, LS testing is cost-effective compared with no testing.
Similarly, despite varying the number of relatives per proband widely, all strategies remain cost-effective compared with no testing at a willingness to pay of £20,000 per QALY. This holds even when assuming no relatives, although the net health benefit is greatly reduced.
Clinical outcomes
Life expectancy for probands is similar across the strategies, with females having a higher life expectancy than males. The same is true for relatives, though they have a much higher life expectancy.
Life expectancy is heavily influenced by LS diagnosis and true LS status. Patients who are diagnosed FN or TP with no surveillance have the lowest life expectancy. TP probands who undergo surveillance have the highest life expectancy, as surveillance reduces CRC risk (and CRC is caught at an earlier stage). They also have a higher life expectancy than FP probands with surveillance because, in general, mortality rates for the type of CRC specific to people with LS is lower. Relatives who are TN, FP, FP with surveillance and TP with surveillance have very similar high life expectancies. Relatives who undergo surveillance but are FP have a slightly higher life expectancy than TN and FP relatives, as there is some additional benefit from the colonoscopic surveillance. For male relatives, those who are TP with surveillance have the highest life expectancy. For female relatives, for whom CRC risk is lower in general and the benefit from diagnosis is split between CRC and EC prevention, those who are TP with surveillance have a lower life expectancy than the TN, FP and FP-with-surveillance female relatives. These results suggest that strategies with lower numbers of FN patients will have higher life expectancies overall, which does appear to occur.
When no testing for LS occurs, there are more colonoscopies per relative with LS than per relative without LS, but the number of colonoscopies per proband with LS is lower than for those without LS. The total number of colonoscopies depends on the correct diagnosis of a patient and compliance with surveillance. Strategies that reduce the number of FNs and increase the number of FPs result in more colonoscopies for patients both with and without LS. This is more noticeable in the relatives, as the probands receive follow-up surveillance colonoscopy even if they are diagnosed LS negative.
For male relatives and probands, the proportion developing CRC is significantly higher than their female counterparts, and a higher proportion of relatives develop CRC compared with probands. The expected number of CRCs for the entire cohort of 1699 probands and 8495 relatives reduces as more patients are identified as LS positive by the strategies, with the strategy that offers universal genetic testing (strategy 8) having the lowest number of CRCs, in both the relative and proband populations. This is explained by the impact of colonoscopy surveillance as a preventative measure for CRC.
The lifetime probability of developing CRC for probands (< 2%, where the CRC must be metachronous) and relatives (< 6% develop at least one CRC, depending upon the strategy) without LS remains roughly constant across all strategies. The lifetime probability of developing CRC for patients with LS is unfailingly higher than for those without LS (24–30% of relatives develop index CRC; 35–40% of probands develop metachronous CRC).
The lifetime risk of EC decreases as more LS-positive relatives and probands are identified by a strategy. The expected number of ECs per year in England in the no testing strategy is approximately 54, with 46 for female relatives and 8 for female probands. This reduces to a minimum of 35 in strategy 8, with 30 for female relatives and 5 for probands. This demonstrates that strategies that identify higher numbers of patients as LS positive, and hence result in more prophylactic TAHBSOs, reduce the numbers of ECs.
Aside from correct diagnosis, compliance with preventative measures appears to have a strong influence on clinical outcomes. Even in the base case, where compliance remains the same, the strategies with higher numbers of surveillance colonoscopies appear to have benefits such as few CRCs and a higher life expectancy, because more patients are identified by genetic testing rather than by assumption.
Health-related quality of life following TAHBSO strongly affects total QALYs by testing strategy. In a sensitivity analysis where a disutility of 0.1 is assumed, all strategies result in lower total QALYs than under no testing.
The incidence of CRC for people with LS and the effectiveness of surveillance colonoscopies in preventing metachronous CRCs also strongly affect total QALYs.
Cost-effectiveness
The total diagnostic cost for the cohort demonstrates that the majority of the costs come from the cost of diagnosing the probands, rather than the follow-up diagnostic costs for relatives. This is because the majority of probands will not be diagnosed with LS and therefore the majority of relatives will not undergo any form of testing. The costs are generally driven in each strategy by the tests undertaken by the highest numbers of probands.
Diagnostic costs are small compared with long-term costs, but do influence the difference in costs between strategies. In the long term, strategies 1(2) and 8 have the highest costs, presumably because strategy 8 has a higher cost of colonoscopies and strategy 1(2) has higher CRC treatment costs.
Long-term costs are lowest for TN and FP-with-no-surveillance patients, as these subgroups do not incur the cost of surveillance and have general population risk of CRC. For female patients there is a slight increase in costs for FP with no surveillance over TN, as FP-with-no-surveillance patients are still offered prophylactic TAHBSO. FN and TP-with-no-surveillance patients have the highest costs due to the increased risk of CRC without any measures to improve survival.
In general, across testing strategies, as total QALYs increase, so do total costs. This is expected as the preventative measures used in the strategies increase both costs and QALYs. Discounted QALYs per year in England accrued by both probands and relatives increase from 151,793 when no testing is performed to 152,000 in strategy 8. The total lifetime cost of no testing for the entire cohort is £36,223,787, increasing to £38,198,324 in strategy 8.
The most influential costs in the long-term results are the preventative measures: colonoscopies and prophylactic TAHBSO. Some of these costs are offset by savings in CRC and EC treatment compared with the no testing strategy. In general, those strategies which identify a higher number of patients (probands and relatives) as LS mutation positive or LS assumed have higher CRC and EC preventative costs, as well as increased cost savings from CRC and EC treatment, although the preventative costs always outweigh the savings in treatment. When univariate sensitivity analysis is conducted, the cost of colonoscopies is particularly influential in terms of ICERs, reducing some by nearly half when the cost is halved and increasing them to nearly double when the costs are doubled.
In all strategies that test for LS, there is an increase in QALYs, and those strategies that have a higher sensitivity have the most QALYs. One impact this demonstrates is that the number of FPs is less important to the QALYs than FNs. Long-term outcomes resulting from diagnosis have a much larger impact on the overall QALY gain than the immediate QALY decrement associated with diagnosis.
All strategies compared with no testing have an ICER < £10,000 per QALY gained. The strategy with the lowest ICER compared with no testing [strategy 1(1)] is strategy 5, with an ICER of £5491 per QALY gained. When comparing the strategies with each other in order of most effective, strategy 5 again is the most cost-effective and the ICER remains at £5491 per QALY. Strategy 5 has the largest NHB at the £20,000 threshold, though all strategies that include some form of diagnostic testing have an increase in NHB. The cost-effectiveness results directly reflect the importance of diagnosis in the short term and the long-term cost and benefits that are applied as a result of that diagnosis.
One of the surprising results from the sensitivity analyses is the effect of adding a disutility following TAHBSO. When this disutility was increased to 0.1, strategy 1(1) (no testing) was found to be the cheapest and most effective strategy. As the number of prophylactic TAHBSOs includes those performed in patients who did not need them (patients who tested FP for LS and who accepted prophylactic TAHBSO), and does not eliminate the need for TAHBSOs as treatment for EC (patients who tested FN for LS and those who tested positive but declined prophylactic TAHBSO, who go on to develop EC), it is not unreasonable that there may be an overall QALY loss when comparing these specific groups (without accounting for the effect of LS surveillance colonoscopies). However, the disutility is large enough to negate both the benefit of EC reduction and the benefit from LS surveillance colonoscopies across the entire cohort. The disutility for TAHBSO was not included in the base case for reasons discussed, partly because the quality of the supporting data is poor. Furthermore, we do not account for all benefits of prophylactic TAHBSO, such as preventing OC. It is interesting to note that the cost-effectiveness of the testing strategies is heavily dependent on the cost-effectiveness of the subsequent medical management of CRC and EC. Indeed, this effect has been noted in a previous model of CRC treatment by Pilgrim and colleagues (2009). 251
Strengths and limitations of the systematic review of test accuracy
The strengths of this systematic review are that it was conducted by an independent research team using the latest evidence, to a pre-specified protocol. The search strategy did not restrict by study design and also included forward chasing. The studies were independently screened by two reviewers, with data extraction and quality appraisal performed by one reviewer and checked by a second. Any disagreements were resolved by consensus.
One limitation was the inability to compare or pool data due to variations in study design, for example different selections of biomarkers or genes under observation. A second significant concern was the lack of reference standard testing on individuals considered to be MSS. The reason for this given by many authors was cost. The recruited population was often small, with unclear patient flow. Overall, all studies were deemed to be at risk of bias, according to QUADAS-2.
Strengths and limitations of the systematic review of cost-effectiveness
Again, the strengths of this systematic review are that it was conducted by an independent research team using the latest evidence, to a pre-specified protocol. Abstracts were independently screened by four reviewers and full papers were independently screened by two reviewers, with data extraction and quality appraisal performed by two reviewers. Any disagreements were resolved by consensus.
The review was highly inclusive. This allowed for a wide selection of literature and a comprehensive evidence base, and made us confident about the approach taken in our modelling. By including previous cost-effectiveness reviews conducted in this area, we were confident that the right papers were being identified by our review. The volume of cost-effectiveness evidence currently available did suggest that much of the work in this area had been done before, but the evidence was not always consistent or directly relevant to our study, and so our review indicated that a de novo model would be beneficial to our specific set of circumstances.
Strengths and limitations of the Peninsula Technology Assessment Group economic model
Strengths
-
Our work was not sponsored by any manufacturers of tests for LS.
-
Our economic analysis is the first specifically concerning the UK NHS.
-
Our analysis considers both strategies to identify LS and the long-term consequences of diagnoses.
-
Our model adheres to methodology recommended in the NICE reference case and has been checked extensively. In addition to our four basic scenario analyses, we also present numerous one-way deterministic sensitivity analyses, which we have chosen for plausibility, to reflect key areas of uncertainty and for those parameters that substantially affect cost-effectiveness.
-
The parameter values and structure of our model have been chosen in light of a detailed review of the literature of the cost-effectiveness of testing for LS.
-
Our model uses individual patient simulation, which enables more sophisticated analysis, such as the modelling of competing risks of mortality from CRC and EC.
-
Our analysis includes health state utilities. Only 4 of the 32 published cost-effectiveness analyses for LS have explicitly done this. Furthermore, two of these only looked at long-term management options for patients already diagnosed with LS who were asymptomatic. We also model disutilities due to the psychological impacts of genetic testing.
-
Our analysis includes EC as a long-term outcome. Only 5 of the 32 published cost-effectiveness analyses for LS have done this. Again, two considered long-term management options for patients already diagnosed with LS who were asymptomatic. Furthermore, one of the studies was an update of an earlier study.
-
Several experts in LS have informed the development of our model.
Limitations
It is important to understand that the cost-effectiveness model relies on a considerable number of structural and parametric assumptions, which have been sourced from expert clinical advice and from evidence in the literature. As with any mathematical model which attempts to synthesise a large yet incomplete evidence base, the results of the analysis are subject to considerable uncertainty.
-
We did not model OC and other cancers associated with LS. However, as stated in Chapter 5, Gynaecological cancers, if these cancers were included it is highly likely that the cost-effectiveness of testing for LS would improve.
-
The following parameters are uncertain and strongly influence cost-effectiveness:
-
the effectiveness of LS colonoscopies in preventing metachronous CRC
-
the impact of TAHBSO on HRQoL
-
the psychological impact of LS testing on HRQoL.
-
-
The model does not include chemoprevention (aspirin for CRC and other LS-associated cancers, oral contraceptive pill for gynaecological cancers).
-
We have not produced a probabilistic sensitivity analysis. Although we believe this approach to be appropriate and have chosen to explore the many structural assumptions as thoroughly as possible using deterministic sensitivity analysis, the absence of probabilistic sensitivity analyses might be viewed as a limitation.
-
We have not modelled the specific genes which are mutated. It is suggested in some guidelines (but is not yet recommended by InSiGHT) that colonoscopic surveillance should begin later for carriers of MSH6 and PMS2 mutations than for carriers of MLH1/MSH2 mutations.
-
Compliance with colonoscopy is more complex than the initial acceptance or non-compliance modelled. Some patients may leave excess time between colonoscopies or before starting them. Currently, however, the evidence for how common this behaviour is, and for the impact of larger delays between colonoscopies on the effectiveness of surveillance, is not available. In a Finnish study, a large proportion of people initially declining surveillance subsequently entered the surveillance programme. 94 In this programme, long-term compliance was also good. Data were not available for the UK relating to these behaviours.
-
The psychological impact of genetic testing is poorly understood and utilities in the model are drawn from a single study which has methodological flaws.
-
We do not account for increasing morbidity and mortality from adverse events in colonoscopy with age. The age limit of 75 years in the model reflects a consensus as to when the benefits of colonoscopy are outweighed by the associated complications. Increasing morbidity and mortality could be important, as our model assumes that all relatives and probands receive the same benefits from colonoscopy, regardless of age. If we account for the increasing morbidity and mortality, the cost-effectiveness of colonoscopies (and therefore the cost-effectiveness of diagnosing LS) may decrease. However, given that in a sensitivity analysis where the rate of mortality following colonoscopy was increased from 8.3 to 39 per 100,000 the cost-effectiveness of testing strategies was only marginally worsened, it is likely that cost-effectiveness would not be affected greatly by such a structural change. It might be reasonable to assume that clinicians would accurately balance risks and benefits for patients such that 75 years is an average age of stopping colonoscopies rather than a fixed age, and that morbidity and mortality are not allowed to rise significantly.
-
The added benefits of certain tests are not accounted for. The probable use of IHC to understand results from genetic testing, though discussed, is not modelled owing to a lack of evidence surrounding the impact this has on the sensitivity of gene tests. Additionally, the benefit of MSI testing to determine the chemotherapy regime of a patient is not accounted for.
-
The proportion of de novo mutations is not well documented. Attempts to represent these in our model may not reflect the true proportion.
-
Individuals who decline genetic counselling or genetic testing may still choose to receive surveillance colonoscopies; it is not clear to what extent these patients can give informed consent to this surveillance, as without genetic counselling and genetic testing it is difficult to assess the potential benefits of surveillance. In the case where individuals decline genetic counselling it is not clear who should obtain informed consent for surveillance, and even if individuals receive genetic counselling, there is no set practice for obtaining informed consent when genetic testing is declined.
-
It is assumed that informed consent is taken for tumour testing for LS and that this does not incur any cost or have any psychological impact on HRQoL. The validity of these assumptions has not been explored for tumour testing as it has been for constituent genetic testing.
-
Relatives are assumed not to be screened for LS until age 18 years, but prenatal predictive screening is already offered for LS. DNA collection methods could risk miscarriage, and the presence of mutations may lead to abortions with an accompanying psychological impact which has not been explored in our model.
-
Limited UK data were identified regarding the feasibility of identifying relatives and cascading testing, which led to uncertainty in the number of relatives that would be identified.
-
Risks of CRC and other cancers in the absence of surveillance are not estimated with much precision because it would be unethical to deny surveillance to individuals once they are diagnosed with LS. Statistical methods to adjust for this have low power, although it may be possible to conduct a meta-analysis of different studies using appropriate techniques to obtain more precise estimates. The risks of CRC and other cancers in the presence of surveillance have not been evaluated in a UK setting.
-
We understand that some clinicians would like to investigate the possibility of delaying initial surgical management of CRC until LS status is known. This is not modelled. Although it may be practical to delay surgical management for counselling and predictive genetic testing, i.e. for relatives of known carriers (which can be conducted quickly), our clinical experts have advised that identifying LS in probands can be a lengthy process.
-
There is no high-quality evidence that including a risk-reducing component in surgery for known carriers results in a clinical benefit when any HRQoL disutility is included.
-
In our model, positive diagnostic genetic test results are assumed to be mutations with known or clearly predicted pathogenicity, whereas negative results include results where no mutations are identified and where variants of unknown significance are identified. Although there is a project under way at present attempting to classify variants of unknown significance as pathogenic or non-pathogenic,272 it is possible that variants of unknown significance would result in increased costs as further tests are required which are not necessarily included in the cost of diagnostic testing in the model.
-
No psychological disutility is included for variants of unknown significance, as these are neither explicitly modelled nor investigated in the trial from which psychological disutilities are drawn.
-
The estimates of CRC incidence for individuals with LS are drawn from a study [Bonadona and colleagues (2011)2] with a different distribution of gene mutations to the proportion assumed in the diagnostic model [based on Palomaki and colleagues (2009)65], with the main difference being that 15% of mutations are in the PMS2 gene in the diagnostic model compared with 0% in Bonadona and colleagues (2011).
-
Mutations in other genes lead to familial early-onset CRC [POLE, POLD1; mutations in APC lead to FAP; in MUTYH, MUTYH-associated polyposis; in STK11-LKB1, Peutz-Jeghers syndrome; in SMAD4 and BMPR1A (and, rarely, GREM1 and SGNE1), juvenile polyposis]. 9 As a result, an indicative FH would lead to further investigations in the event of no MMR mutation being found in a proband; such investigations are not included in the model.
-
Constitutional epimutations may not be detected by current methods,273,274 and though at present they would be included in the model as negative diagnostic test results (with LS possibly being assumed on the basis of FH), it is possible that in the future further testing may be recommended for such mutations which could incur extra costs and affect cost-effectiveness. However, a doubling of the cost of diagnostic genetic testing did not have a significant impact on cost-effectiveness, except for strategy 8 (universal genetic testing).
Adaptation of the Peninsula Technology Assessment Group economic model to other countries
Our economic model is written specifically for the UK NHS. However, we believe that it can be adapted reasonably easily for use in other countries because we believe that the great majority of the structure of the model applies to other countries. Examples of parameters that would change include:
-
all unit costs, for example costs of genetic tests and colonoscopies
-
discount rates for costs and benefits
-
numbers of relatives per proband
-
LS surveillance regimes
-
incidences of CRC and EC
-
mortality due to CRC and EC
-
policy of offering prophylactic TAHBSO
-
treatments for CRC and EC.
Examples of parameters that would probably either remain unchanged or change only slightly include:
-
all utilities
-
all test accuracies
-
proband and relative age distributions.
Chapter 8 Conclusions
Implications for service provision
In addition to cost impacts there would be other implications for service provision as a result of implementing reflex testing for LS in CRC patients aged < 50 years.
Impact on colonoscopy services
Based on a cohort of 1599 CRC patients aged < 50 years per year (the 2010 figure for England), were strategy 5 to be adopted (the strategy with the highest NHB) there would be approximately 3400 extra colonoscopies performed over the lifetime of each annual cohort.
Further analysis of the model would be needed to estimate the growth in numbers of colonoscopies as successive cohorts are identified.
Suggested research priorities
One of the attractions of modelling is that it serves to highlight those areas where further research would be of value for future clinical and policy decision-making.
In theory, a formal value-of-information analysis could be performed to quantify the value of research into various components of our cost-effectiveness model. However, this would require quantification of the uncertainty in most of the parameters in this model, which we believe would be time poorly spent. Instead, sensitivity analyses suggest that the following quantities would be worthy of further research. The items are ranked in approximate order of priority.
Incorporate ovarian cancer into the cost-effectiveness model
Ovarian cancer is estimated to affect approximately 9% of women with LS by age 70 years in the absence of preventative action. 2 Prophylactic hysterectomy and bilateral salpingo-oophorectomy effectively removes the risk of OC. 146 Prophylactic hysterectomy and bilateral salpingo-oophorectomy is already incorporated into the cost-effectiveness model for prevention of EC, and hence the costs and surgical mortality risks of this procedure are already included; however, the clinical benefits with respect to OC are not included as OC is not modelled. It is expected that the inclusion of OC would result in an improvement in cost-effectiveness, but the scale of the impact is unknown. A rough estimate for strategy 5, based on a lifetime cost of £25,000,254,275 5-year survival of 44%147 and an assumed 15-year life expectancy from average age of OC, would suggest that 134 women would receive prophylactic surgery, of whom approximately 12 (9%) would have developed OC. This results in undiscounted cost savings of £300,000. Furthermore, we assume that 5.3 (44%) go on to live for 10 years (15 years minus 5 years of survival) at utility 0.75, adding approximately 40 QALYs. The resulting total undiscounted costs are now £55.1M (down from £55.4M, vs. £54.3M for no testing) and the total undiscounted QALYs are now 276,195 (up from 276,155, vs. 275,654 for no testing). The estimated ICER versus no testing (based on undiscounted costs and QALYs) is then only £1500 per QALY, down from £2100 per QALY.
Incorporate aspirin for colorectal cancer chemoprevention into the cost-effectiveness model
There is evidence to suggest that aspirin reduces the risk of CRC for individuals with LS. 17 Further research is being conducted in this area to determine clinically optimum dosage. Incorporation of chemoprevention into the cost-effectiveness model would also allow estimation of the most health-economically efficient dosage and allow estimation of the impact on cost-effectiveness of reflex testing for LS when chemoprevention forms part of the potential management strategy.
It may be the case that acceptance of and adherence to aspirin medication is better than acceptance of and adherence to surveillance colonoscopy.
Model the cost-effectiveness of reflex testing for Lynch syndrome in alternative populations
It has been suggested by our clinical experts and in the literature [e.g. Kwon and colleagues (2011)56] that it may be cost-effective to perform reflex testing for LS in alternative populations, particularly in the incident EC population. An argument is made that identifying LS in women with newly diagnosed EC will benefit them more than such diagnosis in individuals with newly diagnosed CRC as there is greater scope for prevention of CRC (with 5-year survival of 54%), and that more probands will benefit as 5-year survival from EC is high at 77%.
Testing in alternative populations was outside the scope of this project but could result in cost-effective identification of additional LS families. Research is needed to identify the prevalence of LS in such populations, the diagnostic test accuracy of strategies to identify LS in such populations and the cost-effectiveness of such strategies.
Research into the natural history and impact on health-related quality of life of colorectal cancer
We agree with Pilgrim and colleagues (2009)251 that research into the natural history of CRC and disutilities for patients with CRC would be useful.
Impact of hysterectomy and bilateral salpingo-oophorectomy on health-related quality of life
Univariate sensitivity analysis showed that the cost-effectiveness of all strategies to identify LS was very sensitive to the disutility applied to women receiving hysterectomy and bilateral salpingo-oophorectomy, which, when set to 0.1, resulted in all testing strategies being dominated by (i.e. more expensive and less effective than) no testing.
Psychological impact of genetic testing for Lynch syndrome on health-related quality of life
The current evidence for the psychological impact of genetic testing for LS on HRQoL is extremely weak. It is important that the direction, magnitude and duration of this impact is ascertained through good-quality UK studies, or at least by some form of modelling of the psychological impact of genetic testing according to the characteristics of the condition and management options available.
Diagnostic accuracy of tests in combination
Estimates of test accuracy (sensitivity and specificity) were taken from the available literature. Accuracies of tests (e.g. IHC, MSI) are almost always available only for tests in isolation. One exception is the BRAF test, for which we have accuracy information after an IHC test with abnormal MLH1 staining [Palomaki and colleagues (2009)65] and after a MSI test [Domingo and colleagues (2005)183].
Given that information on the accuracy of tests in sequence is generally lacking, the accuracy of such tests was assumed to be the same as if they were applied in isolation. This likely leads to an overestimate of the diagnostic accuracy of strategies in which tests are performed in sequence.
There may be accuracy studies in which tests are performed in sequence within the literature, from which appropriate estimates may be derived. Additionally, it may be appropriate, given the absence of high-quality diagnostic accuracy studies, for new primary research to be conducted in which multiple tests are evaluated for all patients.
We note that the strategy offering the greatest NHB at a willingness to pay of £20,000 per QALY (strategy 5) does not combine IHC and MSI.
Adaptation of the cost-effectiveness model for use in other countries
This evaluation suggests that reflex testing for LS in early-onset CRC patients and their relatives may be cost-effective in the NHS. This naturally suggests that reflex testing may also be cost-effective in other settings. We note that a number of evaluations have already been performed in the USA,22,23,48,49,54,55,57,88 the Netherlands,50 Denmark,52,53 Australia51 and Singapore. 58 We have noted shortcomings of many of these evaluations and suggest that new evaluations could still be worthwhile even in countries where evaluations have already taken place.
Acknowledgements
We would like to thank:
-
Associate Professor Rob Anderson, Deputy Director of PenTAG, for his direct involvement in the specification of the decision problem, development and agreement of the study protocol and development of the modelling approach and structures, and for providing internal peer review of the draft report
-
Dr Ruben Mujica-Mota, Senior Lecturer in Health Economics, for assisting with the screening for the cost-effectiveness review
-
Dr Carole Brewer, Consultant in Clinical Genetics at Royal Devon and Exeter NHS Trust, for her guidance with genetic counselling
-
Mr Ian Daniels, Consultant Surgeon at Royal Devon and Exeter NHS Trust, for his assistance with CRC
-
Mr John Renninson, Clinical Director of Cancer Services at Royal Devon and Exeter NHS Trust, for information regarding EC
-
Dr Mark Arends of the Department of Pathology, University of Cambridge, for providing a cost for IHC
-
Professor Mary Porteous of the University of Edinburgh and South East Scotland Genetic Service for her assistance in costing genetic counselling
-
Dr Mercy Mvundura of PATH (for previous work at the National Center for Chronic Disease Prevention and Health Promotion, Centers for Disease Control and Prevention) and Dr Scott Grosse of the National Center on Birth Defects and Developmental Disabilities, Centers for Disease Control and Prevention, for providing us with a copy of their model of cost-effectiveness of genetic testing for LS
-
Dr Lorraine Cowley of the Institute of Genetic Medicine, Newcastle University, for providing interim results of the Northern Genetic Service audit of LS carriers, including results not shown elsewhere and detailed responses to questions about the results
-
Dr Munaza Ahmed of Wessex Clinical Genetics Service, Princess Anne Hospital, University Hospital Southampton NHS Trust, for providing anonymised details of germline testing for LS and responding to queries
-
Mr Michael Gandy of UCL-Advanced Diagnostics, University College London, for providing a cost for BRAF testing.
We would also like to acknowledge the help of Sue Whiffin and Jenny Lowe for their administrative support throughout the project.
Contributions of authors
Tristan Snowsill assisted with the development of the protocol; contributed to the design of the PenTAG cost-effectiveness model; implemented the long-term management section of the PenTAG cost-effectiveness model; checked some of the diagnostics section of the model; searched the literature for some model parameters; wrote some of the cost-effectiveness methods chapter; contributed to the cost-effectiveness systematic review; and edited all economics chapters.
Nicola Huxley assisted with the development of the protocol; contributed to the design of the PenTAG cost-effectiveness model; implemented the diagnostics section of the PenTAG cost-effectiveness model; searched the literature for the diagnostics parameters; wrote some of the cost-effectiveness methods chapter; led the cost-effectiveness systematic review; and edited all economics chapters.
Martin Hoyle led the economic evaluation, including the design of the PenTAG cost-effectiveness model; searched the literature for some model parameters; checked all parts of the model; wrote some of the cost-effectiveness methods chapter; and edited all economics chapters.
Tracey Jones-Hughes developed the protocol; assessed abstracts and titles for inclusion; led the systematic review of test accuracy; and contributed to the writing and editing of the report.
Helen Coelho assessed abstracts and titles for inclusion, and contributed to the writing and editing of the report.
Chris Cooper designed and carried out literature searches for the systematic reviews and identification of model parameters, and contributed to the writing and editing of the report.
Ian Frayling contributed to the original HTA brief; provided expert advice and liaison with NHS regional genetics centres; provided data on uptake of predictive testing within families; and contributed to the writing and editing of the report.
Chris Hyde contributed to the systematic review, the design of the model, and the writing and editing of the report. He is director of the Technology Assessment Report group at PenTAG and guarantor of the report.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health.
References
- Vasen HFA, Moslein G, Alonso A, Bernstein I, Bertario L, Blanco I, et al. Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis cancer). J Med Genet 2007;44:353-62. http://dx.doi.org/10.1136/jmg.2007.048991.
- Bonadona V, Bonaiti B, Olschwang S, Grandjouan S, Huiart L, Longy M, et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 2011;305:2304-10. http://dx.doi.org/10.1001/jama.2011.743.
- Chung DC, Rustgi AK. The hereditary nonpolyposis colorectal cancer syndrome: genetics and clinical implications. Ann Intern Med 2003;138:560-70. http://dx.doi.org/10.7326/0003-4819-138-7-200304010-00012.
- Bonis PA, Trikalinos TA, Chung M, Chew P, Ip S, DeVine DA, et al. Hereditary nonpolyposis colorectal cancer: diagnostic strategies and their implications. Evid Rep Technol Assess (Full Rep) n.d.:1-180.
- Vasen H, Blanco I, Aktan-Collan K, Gopie J, Alonso A, Aretz S, et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut 2013;62:812-23. http://dx.doi.org/10.1136/gutjnl-2012-304356.
- SEER Cancer Statistics Factsheets: Kidney and Renal Pelvis Cancer. Bethesda, MD: National Cancer Institute; 2014; n.d.
- SEER Cancer Statistics Factsheets: Bladder Cancer. Bethesda, MD: National Cancer Institute; 2014; n.d.
- Cancer Research UK. Lifetime Risk of Cancer 2013. www.cancerresearchuk.org/cancer-info/cancerstats/incidence/risk (accessed July 2014).
- Cairns SR, Scholefield JH, Steele RJ, Dunlop MG, Thomas HJ, Evans GD, et al. Guidelines for colorectal cancer screening and surveillance in moderate and high risk groups (update from 2002). Gut 2010;59:666-89. http://dx.doi.org/10.1136/gut.2009.179804.
- Vasen HFA, Moslein G, Alonso A, Aretz S, Bernstein I, Bertario L, et al. Recommendations to improve identification of hereditary and familial colorectal cancer in Europe. Fam Cancer 2010;9:109-15. http://dx.doi.org/10.1007/s10689-009-9291-3.
- Shia J, Klimstra DS, Nafa K, Offit K, Guillem JG, Markowitz AJ, et al. Value of immunohistochemical detection of DNA mismatch repair proteins in predicting germline mutation in hereditary colorectal neoplasms. Am J Surg Pathol 2005;29:96-104. http://dx.doi.org/10.1097/01.pas.0000146009.85309.3b.
- Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Nat Cancer Inst 2004;96:261-8. http://dx.doi.org/10.1093/jnci/djh034.
- Evaluation of Genomic Applications in Practice and Prevention Working Group . Recommendations from the EGAPP Working Group: genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med 2009;11:35-41. http://dx.doi.org/10.1097/GIM.0b013e31818fa2ff.
- Aarnio M, Mustonen H, Mecklin JP, Jarvinen HJ. Prognosis of colorectal cancer varies in different high-risk conditions. Ann Med 1998;30:75-80. http://dx.doi.org/10.3109/07853899808999387.
- Aarnio M, Mecklin JP, Aaltonen LA, Nystrom-Lahti M, Jarvinen HJ. Life-time risk of different cancers in hereditary non-polyposis colorectal cancer (HNPCC) syndrome. Int J Cancer 1995;64:430-3. http://dx.doi.org/10.1002/ijc.2910640613.
- Jarvinen HJ, Mecklin JP, Sistonen P. Screening reduces colorectal cancer rate in families with hereditary nonpolyposis colorectal cancer. Gastroenterology 1995;108:1405-11. http://dx.doi.org/10.1016/0016-5085(95)90688-6.
- Burn J, Gerdes AM, Macrae F, Mecklin JP, Moeslein G, Olschwang S, et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. The Lancet 2011;378:2081-7. http://dx.doi.org/10.1016/S0140-6736(11)61049-0.
- Muller A, Giuffre G, Edmonston TB, Mathiak M, Roggendorf B, Heinmoller E, et al. Challenges and pitfalls in HNPCC screening by microsatellite analysis and immunohistochemistry. J Mol Diagn 2004;6:308-15. http://dx.doi.org/10.1016/S1525-1578(10)60526-0.
- Brice P. Biomarkers in Familial Colorectal Cancer Screening. Expert Workshop, 14th February 2006. Cambridge: Public Health Genetics Unit; 2006.
- Barrow E, Evans DG, McMahon R, Hill J, Byers R. A comparative study of quantitative immunohistochemistry and quantum dot immunohistochemistry for mutation carrier identification in Lynch syndrome. J Clin Pathol 2011;64:208-14. http://dx.doi.org/10.1136/jcp.2010.084418.
- Niessen RC, Berends MJ, Wu Y, Sijmons RH, Hollema H, Ligtenberg MJ, et al. Identification of mismatch repair gene mutations in young patients with colorectal cancer and in patients with multiple tumours associated with hereditary non-polyposis colorectal cancer. Gut 2006;55:1781-8. http://dx.doi.org/10.1136/gut.2005.090159.
- Brown ML, Kessler LG. Use of gene tests to detect hereditary predisposition to cancer: What do we know about cost effectiveness?. Int J Cancer 1996;69:55-7. http://dx.doi.org/10.1002/(SICI)1097-0215(19960220)69:1<55::AID-IJC14>3.0.CO;2-J.
- Ramsey SD, Clarke L, Etzioni R, Higashi M, Berry K, Urban N. Cost-effectiveness of microsatellite instability screening as a method for detecting hereditary nonpolyposis colorectal cancer. Ann Intern Med 2001;135:577-88. http://dx.doi.org/10.7326/0003-4819-135-8_Part_1-200110160-00008.
- Hitchens MP, Ward RL. Constitutional (germline) MLH1 epimutation as an aetiological mechanism for hereditary non-polyposis colorectal cancer. Review. J Med Genet 2009;46:793-802. http://dx.doi.org/10.1136/jmg.2009.068122.
- Oliveira C, Westra J, Arango D, Ollikainen M, Domingo E, Ferreira A, et al. Distinct patterns of KRAS mutations in colorectal carcinomas according to germline mismatch repair defects and hMLH1 methylation status. Hum Mol Genet 2004;13:2303-11. http://dx.doi.org/10.1093/hmg/ddh238.
- Zhang L. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part II. The utility of microsatellite instability testing. J Mol Diag 2008;10:301-7. http://dx.doi.org/10.2353/jmoldx.2008.080062.
- Help Me Understand Genetics. Bethesda, MD: US National Library of Medicine; 2013.
- Undertaking Systematic Reviews of Research on Effectiveness. York: NHS Centre for Reviews and Dissemination, University of York; 2001.
- Whiting PF, Westwood ME, Rutjes AW, Reitsma JB, Bossuyt PN, Kleijnen J. Evaluation of QUADAS, a tool for the quality assessment of diagnostic accuracy studies. BMC Med Res Methodol 2006;6. http://dx.doi.org/10.1186/1471-2288-6-9.
- Shea BJ, Grimshaw JM, Wells GA, Boers M, Andersson N, Hamel C, et al. Development of AMSTAR: A measurement tool to assess the methodological quality of systematic reviews. BMC Med Res Methodol 2007;7. http://dx.doi.org/10.1186/1471-2288-7-10.
- Calistri D, Presciuttini S, Buonsanti G, Radice P, Gazzoli I, Pensotti V, et al. Microsatellite instability in colorectal-cancer patients with suspected genetic predisposition. Int J Cancer 2000;89:87-91. http://dx.doi.org/10.1002/(SICI)1097-0215(20000120)89:1<87::AID-IJC14>3.0.CO;2-9.
- Christensen M, Katballe N, Wikman F, Primdahl H, Sorensen FB, Laurberg S, et al. Antibody-based screening for hereditary nonpolyposis colorectal carcinoma compared with microsatellite analysis and sequencing. Cancer 2002;95:2422-30. http://dx.doi.org/10.1002/cncr.10979.
- Katballe N, Christensen M, Wikman FP, Orntoft TF, Laurberg S. Frequency of hereditary non-polyposis colorectal cancer in Danish colorectal cancer patients. Gut 2002;50:43-51. http://dx.doi.org/10.1136/gut.50.1.43.
- Debniak T, Kurzawski G, Gorski B, Kladny J, Domagala W, Lubinski J. Value of pedigree/clinical data, immunohistochemistry and microsatellite instability analyses in reducing the cost of determining hMLH1 and hMSH2 gene mutations in patients with colorectal cancer. Eur J Cancer 2000;36:49-54. http://dx.doi.org/10.1016/S0959-8049(99)00208-7.
- Dieumegard B, Grandjouan S, Sabourin JC, Le Bihan ML, Lefrère I, Bellefqih, et al. Extensive molecular screening for hereditary non-polyposis colorectal cancer. Br J Cancer 2000;82:871-80. http://dx.doi.org/10.1054/bjoc.1999.1014.
- Durno C, Aronson M, Bapat B, Cohen Z, Gallinger S. Family history and molecular features of children, adolescents, and young adults with colorectal carcinoma. Gut 2005;54:1146-50. http://dx.doi.org/10.1136/gut.2005.066092.
- Farrington SM, Lin-Goerke J, Ling J, Wang Y, Burczak JD, Robbins DJ, et al. Systematic analysis of hMSH2 and hMLH1 in young colon cancer patients and controls. Am J Hum Genet 1998;63:749-59. http://dx.doi.org/10.1086/301996.
- Peel DJ, Ziogas A, Fox EA, Gildea M, Laham B, Clements E, et al. Characterization of hereditary nonpolyposis colorectal cancer families from a population-based series of cases. J Natl Cancer Inst 2000;92:1517-22. http://dx.doi.org/10.1093/jnci/92.18.1517.
- Southey MC, Jenkins MA, Mead L, Whitty J, Trivett M, Tesoriero AA, et al. Use of molecular tumor characteristics to prioritize mismatch repair gene testing in early-onset colorectal cancer. J Clin Oncol 2005;23:6524-32. http://dx.doi.org/10.1200/JCO.2005.04.671.
- Terdiman JP, Gum JR, Conrad PG, Miller GA, Weinberg V, Crawley SC, et al. Efficient detection of hereditary nonpolyposis colorectal cancer gene carriers by screening for tumor microsatellite instability before germline genetic testing. Gastroenterology 2001;120:21-30. http://dx.doi.org/10.1053/gast.2001.20874.
- Wahlberg SS, Schmeits J, Thomas G, Loda M, Garber J, Syngal S, et al. Evaluation of microsatellite instability and immunohistochemistry for the prediction of germ-line MSH2 and MLH1 mutations in hereditary nonpolyposis colon cancer families. Cancer Res 2002;62:3485-92.
- Barrow E, Jagger E, Brierley J, Wallace A, Evans G, Hill J, et al. Semiquantitative assessment of immunohistochemistry for mismatch repair proteins in Lynch syndrome. Histopathology 2010;56:331-44. http://dx.doi.org/10.1111/j.1365-2559.2010.03485.x.
- Becouarn Y, Rullier A, Gorry P, Smith D, Richard-Molard B, Echinard E, et al. Value of microsatellite instability typing in detecting hereditary non-polyposis colorectal cancer. A prospective multicentric study by the Association Aquitaine Gastro. Gastroenterol Clin Biol 2005;29:667-75. http://dx.doi.org/10.1016/S0399-8320(05)82155-4.
- Limburg PJ, Harmsen WS, Chen HH, Gallinger S, Haile RW, Baron JA, et al. Prevalence of alterations in DNA mismatch repair genes in patients with young-onset colorectal cancer. Clin Gastroenterol Hepatol 2011;9:497-502. http://dx.doi.org/10.1016/j.cgh.2010.10.021.
- Stormorken AT, Bowitz-Lothe IM, Noren T, Kure E, Aase S, Wijnen J, et al. Immunohistochemistry identifies carriers of mismatch repair gene defects causing hereditary nonpolyposis colorectal cancer. J Clin Oncol 2005;23:4705-12. http://dx.doi.org/10.1200/JCO.2005.05.180.
- Wolf B, Gruber S, Hengimueller S, Kappel S, Bergmann M, Wrba F, et al. Efficiency of the revised Bethesda guidelines (2003) for the detection of mutations in mismatch repair genes in Austrian HNPCC patients. Int J Cancer 2006;118:1465-70. http://dx.doi.org/10.1002/ijc.21524.
- Drummond MF, Jefferson TO. Guidelines for authors and peer reviewers of economic submissions to the BMJ. The BMJ Economic Evaluation Working Party. BMJ 1996;313:275-83. http://dx.doi.org/10.1136/bmj.313.7052.275.
- Ladabaum U, Wang G, Terdiman J, Blanco A, Kuppermann M, Boland CR, et al. Strategies to identify the Lynch syndrome among patients with colorectal cancer. Ann Intern Med 2011;155:69-7. http://dx.doi.org/10.7326/0003-4819-155-2-201107190-00002.
- Ramsey SD, Burke W, Clarke L. An economic viewpoint on alternative strategies for identifying persons with hereditary nonpolyposis colorectal cancer. Genet Med 2003;5:353-63. http://dx.doi.org/10.1097/01.GIM.0000086626.03082.B5.
- Kievit W, De Bruin JHFM, Adang EMM, Severens JL, Kleibeuker JH, Sijmons RH, et al. Cost effectiveness of a new strategy to identify HNPCC patients. Gut 2005;54:97-102. http://dx.doi.org/10.1136/gut.2004.039123.
- Breheny N, Geelhoed E, Goldblatt J, Ee H, O’Leary P. Economic evaluation of the familial cancer programme in Western Australia: predictive genetic testing for familial adenomatous polyposis and hereditary non-polyposis colorectal carcinoma. Community Genet 2006;9:98-106. http://dx.doi.org/10.1159/000091487.
- Hereditary Nonpolyposis Colorectal Cancer in Denmark – A Health Technology Assessment (Structured Abstract). Copenhagen: Danish Centre for Evaluation and Health Technology Assessment; 2007.
- Olsen KR, Bojesen SE, Gerdes AM, Lindorff-Larsen K, Bernstein IT. Cost-effectiveness of surveillance programs for families at high and moderate risk of hereditary non-polyposis colorectal cancer. Int J Technol Assess Health Care 2007;23:89-95. http://dx.doi.org/10.1017/S0266462307051616.
- Mvundura M, Grosse SD, Hampel H, Palomaki GE. The cost-effectiveness of genetic testing strategies for Lynch syndrome among newly diagnosed patients with colorectal cancer. Genet Med 2010;12:93-104. http://dx.doi.org/10.1097/GIM.0b013e3181cd666c.
- Dinh TA, Rosner BI, Atwood JC, Boland CR, Syngal S, Vasen HFA, et al. Health benefits and cost-effectiveness of primary genetic screening for Lynch syndrome in the general population. Cancer Prev Res 2011;4:9-22. http://dx.doi.org/10.1158/1940-6207.CAPR-10-0262.
- Kwon JS, Scott JL, Gilks CB, Daniels MS, Sun CC, Lu KH. Testing women with endometrial cancer to detect Lynch syndrome. J Clin Oncol 2011;29:2247-52. http://dx.doi.org/10.1200/JCO.2010.32.9979.
- Wang G, Kuppermann M, Kim B, Phillips KA, Ladabaum U. Influence of patient preferences on the cost-effectiveness of screening for Lynch syndrome. J Oncol Pract 2012;8:e24s-30s. http://dx.doi.org/10.1200/JOP.2011.000535.
- Wang VW, Koh PK, Chow WL, Lim JFY. Predictive genetic testing of first degree relatives of mutation carriers is a cost-effective strategy in preventing hereditary nonpolyposis colorectal cancer in Singapore. Fam Cancer 2012;11:279-89. http://dx.doi.org/10.1007/s10689-012-9513-y.
- Reyes CM, Allen BA, Terdiman JP, Wilson LS. Comparison of selection strategies for genetic testing of patients with hereditary nonpolyposis colorectal carcinoma: effectiveness and cost-effectiveness. Cancer 2002;95:1848-56. http://dx.doi.org/10.1002/cncr.10910.
- Pigatto F, Bateman A, Bunyan D, Strike P, Wilkins E, Curtis C, et al. Economic and practical factors in diagnosing HNPCC using clinical criteria, immunohistochemistry and microsatellite instability analysis. Hered Cancer Clin Pract 2004;2:175-84. http://dx.doi.org/10.1186/1897-4287-2-4-175.
- Pinol V, Castells A, Andreu M, Castellvi-Bel S, Alenda C, Llor X, et al. Accuracy of revised Bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. J Am Med Assoc 2005;293:1986-94. http://dx.doi.org/10.1001/jama.293.16.1986.
- Engel C, Forberg J, Holinski-Feder E, Pagenstecher C, Plaschke J, Kloor M, et al. Novel strategy for optimal sequential application of clinical criteria, immunohistochemistry and microsatellite analysis in the diagnosis of hereditary nonpolyposis colorectal cancer. Int J Cancer 2006;118:115-22. http://dx.doi.org/10.1002/ijc.21313.
- Bessa X, Balleste B, Andreu M, Castells A, Bellosillo B, Balaguer F, et al. A prospective, multicenter, population-based study of BRAF mutational analysis for Lynch syndrome screening. Clin Gastroenterol Hepatol 2008;6:206-14. http://dx.doi.org/10.1016/j.cgh.2007.10.011.
- Yan HL, Hao LQ, Jin HY, Xing QH, Xue G, Mei Q, et al. Clinical features and mismatch repair genes analyses of Chinese suspected hereditary non-polyposis colorectal cancer: a cost-effective screening strategy proposal. Cancer Sci 2008;99:770-80. http://dx.doi.org/10.1111/j.1349-7006.2008.00737.x.
- Palomaki GE, McClain MR, Melillo S, Hampel HL, Thibodeau SN. EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndrome. Genet Med 2009;11:42-65. http://dx.doi.org/10.1097/GIM.0b013e31818fa2db.
- Ramsoekh D, van Leerdam ME, Wagner A, Kuipers EJ, Steyerberg EW. Mutation prediction models in Lynch syndrome: evaluation in a clinical genetic setting. J Med Genet 2009;46:745-51. http://dx.doi.org/10.1136/jmg.2009.066589.
- Resnick K, Straughn JM, Backes F, Hampel H, Matthews KS, Cohn DE. Lynch syndrome screening strategies among newly diagnosed endometrial cancer patients. Obstet Gynecol 2009;114:530-6. http://dx.doi.org/10.1097/AOG.0b013e3181b11ecc.
- Horwitz J, Geurts J, Einstein M, Qureshi J, Elramah M, Leo J, et al. The cost of detecting Lynch syndrome by microsatellite instability testing in community practice. Am J Gastroenterol 2010;105.
- Gudgeon JM, Williams JL, Burt RW, Samowitz WS, Snow GL, Williams MS. Lynch syndrome screening implementation: business analysis by a healthcare system. Am J Manag Care 2011;17:E288-300.
- Perez-Carbonell L, Guarinus C, Soler MR, Sanchez-Fortun C, Sempere-Robles L, Ruiz-Ponte C, et al. Comparison between universal immunohistochemistry for mismatch repair proteins versus revised Bethesda guidelines in the detection of patients with Lynch syndrome. Gastroenterology 2011;140. http://dx.doi.org/10.1016/S0016-5085(11)60394-6.
- Williams MS, Gudgeon JM, Williams JL. Impact of age cutoffs on the efficiency of immunohistochemical tumor screening to identify Lynch syndrome. Familial cancer. Collaborative Group of the Americas on Inherited Colorectal Cancer conference, Montreal, QC, 10–11 October 2011 2011;10:731-2.
- Gausachs M, Mur P, Corral J, Pineda M, Gonzalez S, Benito L, et al. MLH1 promoter hypermethylation in the analytical algorithm of Lynch syndrome: a cost-effectiveness study. Eur J Human Genet 2012;20:762-8. http://dx.doi.org/10.1038/ejhg.2011.277.
- Vasen HFA, Van Ballegooijen M, Buskens E, Kleibeuker JK, Taal BG, Griffioen G, et al. A cost-effectiveness analysis of colorectal screening of hereditary nonpolyposis colorectal carcinoma gene carriers. Cancer 1998;82:1632-7. http://dx.doi.org/10.1002/(SICI)1097-0142(19980501)82:9<1632::AID-CNCR6>3.0.CO;2-C.
- Dunlop MG. Guidance on gastrointestinal surveillance for hereditary non-polyposis colorectal cancer, familial adenomatous polypolis, juvenile polyposis, and Peutz-Jeghers syndrome. Gut 2002;51:21-7. http://dx.doi.org/10.1136/gut.51.suppl_5.v21.
- Kwon JS, Sun CC, Peterson SK, White KG, Daniels MS, Boyd-Rogers SG, et al. Cost-effectiveness analysis of prevention strategies for gynecologic cancers in Lynch syndrome. Cancer 2008;113:326-35. http://dx.doi.org/10.1002/cncr.23554.
- Yang KY, Caughey AB, Little SE, Cheung MK, Chen LM. A cost-effectiveness analysis of prophylactic surgery versus gynecologic surveillance for women from hereditary non-polyposis colorectal cancer (HNPCC) families. Fam Cancer 2011;10:535-43. http://dx.doi.org/10.1007/s10689-011-9444-z.
- Rogowski W. Genetic screening by DNA technology: a systematic review of health economic evidence. Int J Technol Assess Health Care 2006;22:327-37. http://dx.doi.org/10.1017/S0266462306051221.
- Antonanzas F, Rodriguez-Ibeas R, Hutter M, Lorente R, Juarez C, Pinillos M. Genetic testing in the European Union: does economic evaluation matter?. Eur J Health Econ 2012;13:651-61. http://dx.doi.org/10.1007/s10198-011-0319-x.
- Presymptomatic Diagnosis of Hereditary Colorectal Cancer – Early Assessment Briefs (ALERT) (Brief Record). Stockholm: SBU; 2001.
- Phillips KA, Liang SY, Van Bebber S, Afable-Munsuz A, Knight SJ, Kuppermann M, et al. Challenges to the translation of genomic information into clinical practice and health policy: Utilization, preferences and economic value. Curr Opin Mol Ther 2008;10:260-6.
- Barrow E, Alduaij W, Robinson L, Shenton A, Clancy T, Lalloo F, et al. Colorectal cancer in HNPCC: cumulative lifetime incidence, survival and tumour distribution. A report of 121 families with proven mutations. Clin Genet 2008;74:233-42. http://dx.doi.org/10.1111/j.1399-0004.2008.01035.x.
- Hagen A, Hessabi HK, Gorenoi V, Schonermark MP. Cost-effectiveness evaluation of predictive molecular diagnostics using the example of hereditary non-polyposis colorectal cancer (HNPCC). Gesundheitswesen 2008;70:18-27. http://dx.doi.org/10.1055/s-2007-1022526.
- Syngal S, Weeks JC, Schrag D, Garber JE, Kuntz KM. Benefits of colonoscopic surveillance and prophylactic colectomy in patients with hereditary nonpolyposis colorectal cancer mutations. Ann Intern Med 1998;129:787-96. http://dx.doi.org/10.7326/0003-4819-129-10-199811150-00007.
- Domingo E, Laiho P, Ollikainen M, Pinto M, Wang L, French AJ, et al. BRAF screening as a low-cost effective strategy for simplifying HNPCC genetic testing. J Med Genet 2004;41:664-8. http://dx.doi.org/10.1136/jmg.2004.020651.
- Chen LM, Yang KY, Little SE, Cheung MK, Caughey AB. Gynecologic cancer prevention in Lynch syndrome/hereditary nonpolyposis colorectal cancer families. Obstet Gynecol 2007;110:18-25. http://dx.doi.org/10.1097/01.AOG.0000267500.27329.85.
- Maeda T, Cannom RR, Beart RW, Etzioni DA. Decision model of segmental compared with total abdominal colectomy for colon cancer in hereditary nonpolyposis colorectal cancer. J Clin Oncol 2010;28:1175-80. http://dx.doi.org/10.1200/JCO.2009.25.9812.
- De Vos Tot Nederveen Cappel WH, Buskens E, Van Duijvendijk P, Cats A, Menko FH, Griffioen G, et al. Decision analysis in the surgical treatment of colorectal cancer due to a mismatch repair gene defect. Gut 2003;52:1752-5. http://dx.doi.org/10.1136/gut.52.12.1752.
- Brown ML, Kessler LG. The use of gene tests to detect hereditary predisposition to cancer: economic considerations. J Natl Cancer Inst 1995;87:1131-6. http://dx.doi.org/10.1093/jnci/87.15.1131.
- Public-Use CD-ROM (1973–1997). Bethesda, MD: National Cancer Institute; 2000.
- Ponz de Leon M. Descriptive epidemiology of hereditary non-polyposis colorectal cancer. Tumori 1996;82:102-6.
- Lynch HT, Smyrk TC. Hereditary colorectal cancer. Semin Oncol 1999;26:478-84.
- Vasen HF, Mecklin JP, Watson P, Utsunomiya J, Bertario L, Lynch P, et al. Surveillance in hereditary nonpolyposis colorectal cancer: an international cooperative study of 165 families. The International Collaborative Group on HNPCC. Dis Colon Rectum 1993;36:1-4. http://dx.doi.org/10.1007/BF02050292.
- Boland CR, Vogelstein BK, Kinzler KW. The Genetic Basis of Human Cancer. New York, NY: McGraw-Hill; 1998.
- Jarvinen HJ, Aarnio M, Mustonen H, Aktan-Collan K, Aaltonen LA, Peltomaki P, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology 2000;118:829-34. http://dx.doi.org/10.1016/S0016-5085(00)70168-5.
- Cancer in Australia 2000. Canberra: Australian Institute of Health and Welfare and Australasian Association of Cancer Registries; 2003.
- Renkonen-Sinisalo L, Aarnio M, Mecklin JP, Jarvinen HJ. Surveillance improves survival of colorectal cancer in patients with hereditary nonpolyposis colorectal cancer. Cancer Detect Prev 2000;24:137-42.
- Green J, O’Driscoll M, Barnes A, Maher ER, Bridge P, Shields K, et al. Impact of gender and parent of origin on the phenotypic expression of hereditary nonpolyposis colorectal cancer in a large Newfoundland kindred with a common MSH2 mutation. Dis Colon Rectum 2002;45:1223-32. http://dx.doi.org/10.1007/s10350-004-6397-4.
- Diagnosis and Screening. Copenhagen: Danish Institute for Health Technology Assessment; 2001.
- Hendriks YM, Wagner A, Morreau H, Menko F, Stormorken A, Quehenberger F, et al. Cancer risk in hereditary nonpolyposis colorectal cancer due to MSH6 mutations: impact on counseling and surveillance. Gastroenterology 2004;127:17-25. http://dx.doi.org/10.1053/j.gastro.2004.03.068.
- Buttin BM, Powell MA, Mutch DG, Babb SA, Huettner PC, Edmonston TB, et al. Penetrance and expressivity of MSH6 germline mutations in seven kindreds not ascertained by family history. Am J Hum Genet 2004;74:1262-9. http://dx.doi.org/10.1086/421332.
- Dunlop MG, Farrington SM, Carothers AD, Wyllie AH, Sharp L, Burn J, et al. Cancer risk associated with germline DNA mismatch repair gene mutations. Hum Mol Genet 1997;6:105-10. http://dx.doi.org/10.1093/hmg/6.1.105.
- Quehenberger F, Vasen HF, van Houwelingen HC. Risk of colorectal and endometrial cancer for carriers of mutations of the hMLH1 and hMSH2 gene: correction for ascertainment. J Med Genet 2005;42:491-6. http://dx.doi.org/10.1136/jmg.2004.024299.
- Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 2005;352:1851-60. http://dx.doi.org/10.1056/NEJMoa043146.
- Wagner A, Hendriks Y, Meijers-Heijboer EJ, de Leeuw WJ, Morreau H, Hofstra R, et al. Atypical HNPCC owing to MSH6 germline mutations: analysis of a large Dutch pedigree. J Med Genet 2001;38:318-22. http://dx.doi.org/10.1136/jmg.38.5.318.
- Senter L, Clendenning M, Sotamaa K, Hampel H, Green J, Potter JD, et al. The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology 2008;135:419-28. http://dx.doi.org/10.1053/j.gastro.2008.04.026.
- Stoffel E, Mukherjee B, Raymond VM, Tayob N, Kastrinos F, Sparr J, et al. Calculation of risk of colorectal and endometrial cancer among patients with Lynch syndrome. Gastroenterology 2009;137:1621-7. http://dx.doi.org/10.1053/j.gastro.2009.07.039.
- Jarvinen HJ, Renkonen-Sinisalo L, Aktan-Collan K, Peltomaki P, Aaltonen LA, Mecklin J-P. Ten years after mutation testing for Lynch syndrome: cancer incidence and outcome in mutation-positive and mutation-negative family members. J Clin Oncol 2009;27:4793-7. http://dx.doi.org/10.1200/JCO.2009.23.7784.
- Vasen HF, Abdirahman M, Brohet R, Langers AM, Kleibeuker JH, van Kouwen M, et al. One to 2-year surveillance intervals reduce risk of colorectal cancer in families with Lynch syndrome. Gastroenterology 2010;138:2300-6. http://dx.doi.org/10.1053/j.gastro.2010.02.053.
- de Jong AE, Hendriks YM, Kleibeuker JH, de Boer SY, Cats A, Griffioen G, et al. Decrease in mortality in Lynch syndrome families because of surveillance. Gastroenterology 2006;130:665-71. http://dx.doi.org/10.1053/j.gastro.2005.11.032.
- Cotterchio M, Manno M, Klar N, McLaughlin J, Gallinger S. Colorectal screening is associated with reduced colorectal cancer risk: a case-control study within the population-based Ontario Familial Colorectal Cancer Registry. Cancer Causes Control 2005;16:865-75. http://dx.doi.org/10.1007/s10552-005-2370-3.
- Boursi B, Halak A, Umansky M, Galzan L, Guzner-Gur H, Arber N. Colonoscopic screening of an average-risk population for colorectal neoplasia. Endoscopy 2009;41:516-21. http://dx.doi.org/10.1055/s-0029-1214757.
- Rundle AG, Lebwohl B, Vogel R, Levine S, Neugut AI. Colonoscopic screening in average-risk individuals ages 40 to 49 vs 50 to 59 years. Gastroenterology 2008;134:1311-15. http://dx.doi.org/10.1053/j.gastro.2008.02.032.
- Altekruse SF, Kosary CL, Krapcho M, Neyman M, Aminou R, Waldron W, et al. Lifetime Risk, SEER Cancer Statistics Review, 1975–2007. Bethesda, MD: National Cancer Institute; n.d.
- Jenkins MA, Baglietto L, Dowty JG, Van Vliet CM, Smith L, Mead LJ, et al. Cancer risks for mismatch repair gene mutation carriers: a population-based early onset case-family study. Clin Gastroenterol Hepatol 2006;4:489-98. http://dx.doi.org/10.1016/j.cgh.2006.01.002.
- Altekruse SF, Kosary CL, Krapcho M, Neyman M, Waldron W, . SEER Cancer Statistics Review 1975–2007. Bethesda, MD: National Cancer Institute; 2010.
- Vasen HF, Wijnen JT, Menko FH, Kleibeuker JH, Taal BG, Griffioen G, et al. Cancer risk in families with hereditary nonpolyposis colorectal cancer diagnosed by mutation analysis. Gastroenterology 1996;110:1020-7. http://dx.doi.org/10.1053/gast.1996.v110.pm8612988.
- Vasen HF, Taal BG, Nagengast FM, Griffioen G, Menko FH, Kleibeuker JH, et al. Hereditary nonpolyposis colorectal cancer: results of long-term surveillance in 50 families. Eur J Cancer 1995;31A:1145-8. http://dx.doi.org/10.1016/0959-8049(95)00249-I.
- Aarnio M, Sankila R, Pukkala E, Salovaara R, Aaltonen LA, de la Chapelle A, et al. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer 1999;81:214-18. http://dx.doi.org/10.1002/(SICI)1097-0215(19990412)81:2<214::AID-IJC8>3.0.CO;2-L.
- Office for National Statistics . Cancer Statistics Registrations, England (Series MB1) – No. 41, 2010 2012. www.ons.gov.uk/ons/rel/vsob1/cancer-statistics-registrations--england--series-mb1-/no--41--2010/index.html (accessed July 2014).
- Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clinical Oncol 2008;26:5783-8. http://dx.doi.org/10.1200/JCO.2008.17.5950.
- Barrow E, Robinson L, Alduaij W, Shenton A, Clancy T, Lalloo F, et al. Cumulative lifetime incidence of extracolonic cancers in Lynch syndrome: a report of 121 families with proven mutations. Clin Genet 2009;75:141-9. http://dx.doi.org/10.1111/j.1399-0004.2008.01125.x.
- Current Disease Services. Oxford: Oxford University Hospitals NHS Trust; 2011.
- John M. AWMGL Service Price List. Cardiff: All Wales Molecular Genetics Laboratory; 2012.
- Molecular Genetics List of Disorders. Dundee: East of Scotland Regional Genetics Service; 2012.
- Services and Tests Available. Cambridge: Cambridge University Hospitals NHS Foundation Trust; 2012.
- Hereditary Nonpolyposis Colorectal Cancer (HNPCC). Leeds: Leeds Teaching Hospitals; 2012.
- Hall G, Clarkson A, Shi A, Langford E, Leung H, Eckstein RP, et al. Immunohistochemistry for PMS2 and MSH6 alone can replace a four antibody panel for mismatch repair deficiency screening in colorectal adenocarcinoma. Pathology 2010;42:409-13. http://dx.doi.org/10.3109/00313025.2010.493871.
- Klarskov L, Ladelund S, Holck S, Roenlund K, Lindebjerg J, Elebro J, et al. Interobserver variability in the evaluation of mismatch repair protein immunostaining. Human Pathol 2010;41:1387-96. http://dx.doi.org/10.1016/j.humpath.2010.03.003.
- Barton P, Bryan S, Robinson S. Modelling in the economic evaluation of health care: selecting the appropriate approach. J Health Serv Res Pol 2004;9:110-18. http://dx.doi.org/10.1258/135581904322987535.
- Brennan A, Chick SE, Davies R. A taxonomy of model structures for economic evaluation of health technologies. Health Econ 2006;15:1295-310. http://dx.doi.org/10.1002/hec.1148.
- Cancer Staging Manual. New York, NY: Springer; 2010.
- Guide to the Methods of Technology Appraisal. London: National Institute for Health and Care Excellence; 2013.
- Colorectal Cancer: The Diagnosis and Management of Colorectal Cancer. London: National Institute for Health and Care Excellence; 2011.
- Trueman P, Lowson K, Bending M, Ganderton M, Chaplin S, Wright D, et al. Bowel Cancer Services: Costs and Benefits. York: York Health Economics Consortium; 2007.
- Finan P, Smith J, Walker K, van der Meulen J, Greenaway K, Yelland A, et al. National Bowel Cancer Audit Annual Report. Leeds: NHS Information Centre for Health and Social Care; 2011.
- Office for National Statistics . Cancer Statistics Registrations, England (Series MB1) – No. 37, 2006 2008. www.ons.gov.uk/ons/rel/vsob1/cancer-statistics-registrations--england--series-mb1-/no--37--2006/index.html (accessed July 2014).
- Office for National Statistics . Cancer Statistics Registrations, England (Series MB1) – No. 39, 2008 2010. www.ons.gov.uk/ons/rel/vsob1/cancer-statistics-registrations--england--series-mb1-/no--39--2008/index.html (accessed July 2014).
- Office for National Statistics . Cancer Statistics Registrations, England (Series MB1) – No. 38, 2007 2010. www.ons.gov.uk/ons/rel/vsob1/cancer-statistics-registrations--england--series-mb1-/no--38--2007/index.html (accessed July 2014).
- Office for National Statistics . Cancer Statistics Registrations, England (Series MB1) – No. 40, 2009 2011. www.ons.gov.uk/ons/rel/vsob1/cancer-statistics-registrations--england--series-mb1-/no--40--2009/index.html (accessed July 2014).
- Office for National Statistics . Mid-2010 Population Estimates: England and Wales; Estimated Resident Population by Single Year of Age and Sex 2011. www.ons.gov.uk/ons/rel/pop-estimate/population-estimates-for-uk--england-and-wales--scotland-and-northern-ireland/mid-2010-population-estimates/rft---mid-2010-population-estimates.zip (accessed July 2014).
- Office for National Statistics . Mid-2002 to Mid-2010 Population Estimates of the Very Elderly (Including Centenarians) England and Wales; Estimated Resident Population 2011. www.ons.gov.uk/ons/rel/mortality-ageing/population-estimates-of-the-very-elderly/2010/ew-eve-2010.xls (accessed July 2014).
- National Cancer Intelligence Network . One, Five and Ten Year Cancer Prevalence by Cancer Network, UK, 2006 2010. www.ncin.org.uk/view.aspx?rid=76 (accessed July 2014).
- Office for National Statistics . Mortality Statistics: Deaths Registered in 2010 (Series DR) 2011. www.ons.gov.uk/ons/rel/vsob1/mortality-statistics--deaths-registered-in-england-and-wales--series-dr-/2010/dr-tables-2010.xls (accessed July 2014).
- Office for National Statistics . Mid-1971 to Mid-2010 Population Estimates: Quinary Age Groups for Constituent Countries in the United Kingdom; Estimated Resident Population 2011. www.ons.gov.uk/ons/rel/pop-estimate/population-estimates-for-uk--england-and-wales--scotland-and-northern-ireland/population-estimates-timeseries-1971-to-current-year/rft---table-2-quinary-age-groups-constituent-countries.zip (accessed July 2014).
- Welsh Cancer Intelligence and Surveillance Unit . Cancer Incidence in Wales 2006–2010 2012. www.wcisu.wales.nhs.uk/opendoc/226155 (accessed July 2014).
- Schmeler KM, Lynch HT, Chen LM, Munsell MF, Soliman PT, Clark MB, et al. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. New Engl J Med 2006;354:261-9. http://dx.doi.org/10.1056/NEJMoa052627.
- Office for National Statistics . Cancer Survival in England – Patients Diagnosed 2006–2010 and Followed Up to 2011 2012. www.ons.gov.uk/ons/publications/re-reference-tables.html?edition=tcm%3A77-277733 (accessed July 2014).
- Office for National Statistics . Cancer Survival in England – Patients Diagnosed 2005–2009 and Followed Up to 2010 2011. www.ons.gov.uk/ons/publications/re-reference-tables.html?edition=tcm%3A77-239726 (accessed July 2014).
- National Cancer Intelligence Network (NCIN), UK Cancer Information Service (UKCIS) . UK Cancer E-Atlas 2011. www.ncin.org.uk/cancer_information_tools/eatlas/pct/atlas.html?select=Eav&indicator=i0 (accessed July 2014).
- Cancer Research UK. Uterine Cancer Survival Statistics. London: Cancer Research UK; 2012.
- NHS Reference Costs 2011–2012. London: Department of Health; 2012.
- Havrilesky L, Maxwell G, Myers E. Cost-effectiveness analysis of annual screening strategies for endometrial cancer. Am J Obstet Gynecol 2009;200. http://dx.doi.org/10.1016/j.ajog.2009.02.022.
- Commercial Medicines Unit . Electronic Market Information Tool (eMit) 2013. http://cmu.dh.gov.uk/electronic-market-information-tool-emit (accessed July 2014).
- NHS Reference Costs 2008–9. London: Department of Health; 2010.
- National Institute for Health and Care Excellence . Laparoscopic Hysterectomy (Including Laparoscopic Total Hysterectomy and Laparoscopically Assisted Vaginal Hysterectomy) for Endometrial Cancer: Guidance 2010. www.nice.org.uk/guidance/IPG356/Guidance/pdf (accessed July 2014).
- NHS . Treating Uterine (Uterus) Cancer 2013. www.nhs.uk/conditions/Cancer-of-the-uterus/Pages/treatment.aspx (accessed July 2014).
- Baker M, Kessler L, Smucker R, Scheffler RM, Andrews NC, Phillips KA. Cancer Care and Cost: DRGs and Beyond. Ann Arbor, MI: Health Administration Press; 1989.
- National Insitute for Health and Care Excellence . Published Clinical Guidelines 2013. http://guidance.nice.org.uk/CG/Published (accessed July 2014).
- Macmillan Cancer Support . Radiotherapy for Womb Cancer (Endometrial Cancer) 2013. www.macmillan.org.uk/Cancerinformation/Cancertypes/Wombuterus/Treatingwombcancer/Radiotherapy.asp (accessed January 2013).
- Markman M. Chemotherapy in the management of endometrial cancer. J Gynecol Oncol 2007;12:29-32.
- Mulder SA, Kranse R, Damhuis RA, Ouwendijk RJ, Kuipers EJ, van Leerdam ME. The incidence and risk factors of metachronous colorectal cancer: an indication for follow-up. Dis Colon Rectum 2012;55:522-31. http://dx.doi.org/10.1097/DCR.0b013e318249db00.
- Carayol J, Bonaiti-Pellie C. Estimating penetrance from family data using a retrospective likelihood when ascertainment depends on genotype and age of onset. Genet Epidemiol 2004;27:109-17. http://dx.doi.org/10.1002/gepi.20007.
- Rasmussen LJ, Heinen CD, Royer-Pokora B, Drost M, Tavtigian S, Hofstra RM, et al. Pathological assessment of mismatch repair gene variants in Lynch syndrome: past, present, and future. Hum Mutat 2012;33:1617-25. http://dx.doi.org/10.1002/humu.22168.
- van Vliet CM, Dowty JG, van Vliet JL, Smith L, Mead LJ, Macrae FA, et al. Dependence of colorectal cancer risk on the parent-of-origin of mutations in DNA mismatch repair genes. Hum Mutat 2011;32:207-12. http://dx.doi.org/10.1002/humu.21408.
- Alarcon F, Lasset C, Carayol J, Bonadona V, Perdry H, Desseigne F, et al. Estimating cancer risk in HNPCC by the GRL method. Eur J Hum Genet 2007;15:831-6. http://dx.doi.org/10.1038/sj.ejhg.5201843.
- Baglietto L, Lindor NM, Dowty JG, White DM, Wagner A, Gomez Garcia EB, et al. Risks of Lynch syndrome cancers for MSH6 mutation carriers. J Natl Cancer Inst 2010;102:193-201. http://dx.doi.org/10.1093/jnci/djp473.
- Lindor NM, Petersen GM, Hadley DW, Kinney AY, Miesfeldt S, Lu KH, et al. Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome – A systematic review. JAMA 2006;296:1507-17. http://dx.doi.org/10.1001/jama.296.12.1507.
- Parry S, Win AK, Parry B, Macrae FA, Gurrin LC, Church JM, et al. Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut 2011;60:950-7. http://dx.doi.org/10.1136/gut.2010.228056.
- Fajobi O, Yiu CY, Sen-Gupta SB, Boulos PB. Metachronous colorectal cancers. Br J Surg 1998;85:897-901. http://dx.doi.org/10.1046/j.1365-2168.1998.00800.x.
- National Cancer Intelligence Network . Colorectal Cancer Survival by Stage 2009. www.ncin.org.uk/publications/data_briefings/colorectal_cancer_survival_by_stage.aspx (accessed July 2014).
- Cancer Research UK . Bowel Cancer Survival Statistics 2013. www.cancerresearchuk.org/cancer-info/cancerstats/types/bowel/survival (accessed July 2014).
- Coate L, Cuffe S, Horgan A, Hung RJ, Christiani D, Liu G. Germline genetic variation, cancer outcome, and pharmacogenetics. J Clin Oncol 2010;28:4029-37. http://dx.doi.org/10.1200/JCO.2009.27.2336.
- Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol 2005;23:609-18. http://dx.doi.org/10.1200/JCO.2005.01.086.
- Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology 2010;138:2073-87.
- Barnetson RA, Tenesa A, Farrington SM, Nicholl ID, Cetnarskyj R, Porteous ME, et al. Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N Engl J Med 2006;354:2751-63. http://dx.doi.org/10.1056/NEJMoa053493.
- Lin KM, Shashidharan M, Ternent CA, Thorson AG, Blatchford GJ, Christensen MA, et al. Colorectal and extracolonic cancer variations in MLH1/MSH2 hereditary nonpolyposis colorectal cancer kindreds and the general population. Dis Colon Rectum 1998;41:428-33. http://dx.doi.org/10.1007/BF02235755.
- Horner M, Ries L, Krapcho M, Neyman N, Aminou R, Howlader N, et al. SEER Cancer Statistics Review, 1975–2006. Bethesda, MD: National Cancer Institute; 2009.
- Boks DE, Trujillo AP, Voogd AC, Morreau H, Kenter GG, Vasen HF. Survival analysis of endometrial carcinoma associated with hereditary nonpolyposis colorectal cancer. Int J Cancer 2002;102:198-200. http://dx.doi.org/10.1002/ijc.10667.
- Office for National Statistics . England and Wales, Interim Life Tables, 1980–82 to 2008–10 2011. www.ons.gov.uk/ons/rel/lifetables/interim-life-tables/2008-2010/rft-ilt-ew-2008-2010.xls (accessed July 2014).
- Salovaara R, Loukola A, Kristo P, Kaariainen H, Ahtola H, Eskelinen M, et al. Population-based molecular detection of hereditary nonpolyposis colorectal cancer. J Clin Oncol 2000;18:2193-200.
- Green RC, Parfrey PS, Woods MO, Younghusband HB. Prediction of Lynch syndrome in consecutive patients with colorectal cancer. J Natl Cancer Inst 2009;101:331-40. http://dx.doi.org/10.1093/jnci/djn499.
- Balmana J, Balaguer F, Castellvi-Bel S, Steyerberg EW, Andreu M, Llor X, et al. Comparison of predictive models, clinical criteria and molecular tumour screening for the identification of patients with Lynch syndrome in a population-based cohort of colorectal cancer patients. J Med Genet 2008;45:557-63. http://dx.doi.org/10.1136/jmg.2008.059311.
- Domingo E, Niessen RC, Oliveira C, Alhopuro P, Moutinho C, Espin E, et al. BRAF-V600E is not involved in the colorectal tumorigenesis of HNPCC in patients with functional MLH1 and MSH2 genes. Oncogene 2005;24:3995-8. http://dx.doi.org/10.1038/sj.onc.1208569.
- Sjursen W, Haukanes BI, Grindedal EM, Aarset H, Stormorken A, Engebretsen LF, et al. Current clinical criteria for Lynch syndrome are not sensitive enough to identify MSH6 mutation carriers. J Med Genet 2010;47:579-85. http://dx.doi.org/10.1136/jmg.2010.077677.
- Jenkins MA, Baglietto L, Dite GS, Jolley DJ, Southey MC, Whitty J, et al. After hMSH2 and hMLH1 – what next? Analysis of three-generational, population-based, early-onset colorectal cancer families. Int J Cancer 2002;102:166-71. http://dx.doi.org/10.1002/ijc.10670.
- Win AK, Parry S, Parry B, Kalady MF, Macrae FA, Ahnen DJ, et al. Risk of metachronous colon cancer following surgery for rectal cancer in mismatch repair gene mutation carriers. Ann Surg Oncol 2013;20:1829-36. http://dx.doi.org/10.1245/s10434-012-2858-5.
- Cirillo L, Urso ED, Parrinello G, Pucciarelli S, Moneghini D, Agostini M, et al. High risk of rectal cancer and of metachronous colorectal cancer in probands of families fulfilling the Amsterdam Criteria. Ann Surg 2012;257:900-4. http://dx.doi.org/10.1097/SLA.0b013e31826bff79.
- Mecklin JP, Aarnio M, Laara E, Kairaluoma MV, Pylvanainen K, Peltomaki P, et al. Development of colorectal tumors in colonoscopic surveillance in Lynch syndrome. Gastroenterology 2007;133:1093-8. http://dx.doi.org/10.1053/j.gastro.2007.08.019.
- Gavin DR, Valori RM, Anderson JT, Donnelly MT, Williams JG, Swarbrick ET. The national colonoscopy audit: a nationwide assessment of the quality and safety of colonoscopy in the UK. Gut 2013;62:242-9. http://dx.doi.org/10.1136/gutjnl-2011-301848.
- Dove-Edwin I, Boks D, Goff S, Kenter GG, Carpenter R, Vasen HFA, et al. The outcome of endometrial carcinoma surveillance by ultrasound scan in women at risk of hereditary nonpolyposis colorectal carcinoma and familial colorectal carcinoma. Cancer 2002;94:1708-12. http://dx.doi.org/10.1002/cncr.10380.
- Renkonen-Sinisalo L, Butzow R, Leminen A, Lehtovirta P, Mecklin J-P, Jarvinen HJ. Surveillance for endometrial cancer in hereditary nonpolyposis colorectal cancer syndrome. Int J Cancer 2007;120:821-4. http://dx.doi.org/10.1002/ijc.22446.
- Auranen A, Joutsiniemi T. A systematic review of gynecological cancer surveillance in women belonging to hereditary nonpolyposis colorectal cancer (Lynch syndrome) families. Acta Obstet Gynecol Scand 2011;90:437-44. http://dx.doi.org/10.1111/j.1600-0412.2011.01091.x.
- Koornstra JJ, Mourits MJ, Sijmons RH, Leliveld AM, Hollema H, Kleibeuker JH. Management of extracolonic tumours in patients with Lynch syndrome. Lancet Oncol 2009;10:400-8. http://dx.doi.org/10.1016/S1470-2045(09)70041-5.
- Gerritzen LHM, Hoogerbrugge N, Oei ALM, Nagengast FM, van Ham MAPC, Massuger LFAG, et al. Improvement of endometrial biopsy over transvaginal ultrasound alone for endometrial surveillance in women with Lynch syndrome. Fam Cancer 2009;8:391-7. http://dx.doi.org/10.1007/s10689-009-9252-x.
- Lécuru F, Le Frere Belda MA, Bats AS, Tulpin L, Metzger U, Olschwang S, et al. Performance of office hysteroscopy and endometrial biopsy for detecting endometrial disease in women at risk of human non-polyposis colon cancer: a prospective study. Int J Gynecol Cancer 2008;18:1326-31. http://dx.doi.org/10.1111/j.1525-1438.2007.01183.x.
- Rijcken FEM, Mourits MJE, Kleibeuker JH, Hollema H, van der Zee AGJ. Gynecologic screening in hereditary nonpolyposis colorectal cancer. Gynecol Oncol 2003;91:74-80. http://dx.doi.org/10.1016/S0090-8258(03)00371-8.
- Papaioannou D, Brazier J, Paisley S. NICE DSU Technical Support Document 9: The Identification, Review and Synthesis of Health State Utility Values from the Literature. Sheffield: School of Health and Related Research, University of Sheffield; 2010.
- Guide to the Methods of Technology Appraisal. London: National Institute for Health and Care Excellence; 2008.
- Gritz ER, Peterson SK, Vernon SW, Marani SK, Baile WF, Watts BG, et al. Psychological impact of genetic testing for hereditary nonpolyposis colorectal cancer. J Clin Oncol 2005;23:1902-10. http://dx.doi.org/10.1200/JCO.2005.07.102.
- Ness RM, Holmes AM, Klein R, Dittus R. Utility valuations for outcome states of colorectal cancer. Am J Gastroenterol 1999;94:1650-7. http://dx.doi.org/10.1111/j.1572-0241.1999.01157.x.
- van Duijvendijk P, Slors JFM, Taat CW, Oosterveld P, Sprangers MAG, Obertop H, et al. Quality of life after total colectomy with ileorectal anastomosis or proctocolectomy and ileal pouch-anal anastomosis for familial adenomatous polyposis. Br J Surg 2000;87:590-6. http://dx.doi.org/10.1046/j.1365-2168.2000.01442.x.
- Fazio VW, O’Riordain MG, Lavery IC, Church JM, Lau P, Strong SA, et al. Long-term functional outcome and quality of life after stapled restorative proctocolectomy. Ann Surg 1999;230:575-84. http://dx.doi.org/10.1097/00000658-199910000-00013.
- Carter KJ, Ritchey NP, Castro F, Caccamo LP, Kessler E, Erickson BA, et al. Treatment of early-stage breast cancer in the elderly: a health-outcome-based approach. Med Decis Making 1998;18:213-19.
- Grann VR, Sundararajan V, Jacobson JS, Whang W, Heitjan DF, Antman KH, et al. Decision analysis of tamoxifen for the prevention of invasive breast cancer. Cancer J 2000;6:169-78.
- Hillner BE, Hollenberg JP, Pauker SG. Postmenopausal estrogens in prevention of osteoporosis. Benefit virtually without risk if cardiovascular effects are considered. Am J Med 1986;80:1115-27. http://dx.doi.org/10.1016/0002-9343(86)90674-1.
- Weinstein MC, Schiff I. Cost-effectiveness of hormone replacement therapy in the menopause. Obstet Gynecol Surv 1983;38:445-55. http://dx.doi.org/10.1097/00006254-198308000-00001.
- Sun CC, Peterson SK, White KG, Watts BG, Daniels MS, Boyd-Rogers SG, et al. Preferences for cancer prevention strategies (CPS) in women with hereditary nonpolyposis colorectal cancer (HNPCC). ASCO Meeting Abstracts 2006;24.
- Grann VR, Jacobson JS, Thomason D, Hershman D, Heitjan DF, Neugut AI. Effect of prevention strategies on survival and quality-adjusted survival of women with BRCA12 mutations: an updated decision analysis. J Clin Oncol 2002;20:2520-9. http://dx.doi.org/10.1200/JCO.2002.10.101.
- Grann VR, Panageas KS, Whang W, Antman KH, Neugut AI. Decision analysis of prophylactic mastectomy and oophorectomy in BRCA1-positive or BRCA2-positive patients. J Clin Oncol 1998;16:979-85.
- Ortega A, Dranitsaris G, Sturgeon J, Sutherland H, Oza A. Cost-utility analysis of paclitaxel in combination with cisplatin for patients with advanced ovarian cancer. Gynecol Oncol 1997;66:454-63. http://dx.doi.org/10.1006/gyno.1997.4786.
- van Roosmalen MS, Verhoef LCG, Stalmeier PFM, Hoogerbrugge N, van Daal WAJ. Decision analysis of prophylactic surgery or screening for BRCA1 mutation carriers: a more prominent role for oophorectomy. J Clin Oncol 2002;20:2092-100. http://dx.doi.org/10.1200/JCO.2002.08.035.
- Kuppermann M, Wang G, Wong S, Blanco A, Conrad P, Nakagawa S, et al. Preferences for outcomes associated with decisions to undergo or forgo genetic testing for Lynch syndrome. Cancer 2013;119:215-25. http://dx.doi.org/10.1002/cncr.27634.
- Cooper K, Squires H, Carroll C, Papaioannou D, Booth A, Logan R, et al. Chemoprevention of colorectal cancer: systematic review and economic evaluation. Health Technol Assess 2010;14.
- Whyte S, Chilcott J, Halloran S. Reappraisal of the options for colorectal cancer screening in England. Colorectal Dis 2012;14:e547-61. http://dx.doi.org/10.1111/j.1463-1318.2012.03014.x.
- Ara R, Brazier JE. Using health state utility values from the general population to approximate baselines in decision analytic models when condition-specific data are not available. Value Health 2011;14:539-45.
- Ara R, Brazier JE. Populating an economic model with health state utility values: moving towards better practice. Value Health 2010;13:509-18. http://dx.doi.org/10.1111/j.1524-4733.2010.00700.x.
- Ramsey SD, Andersen MR, Etzioni R, Moinpour C, Peacock S, Potosky A, et al. Quality of life in survivors of colorectal carcinoma. Cancer 2000;88:1294-303. http://dx.doi.org/10.1002/(SICI)1097-0142(20000315)88:6<1294::AID-CNCR4>3.0.CO;2-M.
- Ramsey SD, Berry K, Moinpour C, Giedzinska A, Andersen MR. Quality of life in long term survivors of colorectal cancer. Am J Gastroenterol 2002;97:1228-34. http://dx.doi.org/10.1111/j.1572-0241.2002.05694.x.
- Ko CY, Maggard M, Livingston EH. Evaluating health utility in patients with melanoma, breast cancer, colon cancer, and lung cancer: a nationwide, population-based assessment. J Surg Res 2003;114:1-5. http://dx.doi.org/10.1016/S0022-4804(03)00167-7.
- Petrou S, Campbell N. Stabilisation in colorectal cancer. Int J Pall Nurs 1997;3:275-80.
- Seymour MT, Maughan TS, Ledermann JA, Topham C, James R, Gwyther SJ, et al. Different strategies of sequential and combination chemotherapy for patients with poor prognosis advanced colorectal cancer (MRC FOCUS): a randomised controlled trial. Lancet 2007;370:143-52. http://dx.doi.org/10.1016/S0140-6736(07)61087-3.
- Wilke H, Glynne-Jones R, Thaler J, Adenis A, Preusser P, Aguilar EA, et al. Cetuximab plus irinotecan in heavily pretreated metastatic colorectal cancer progressing on irinotecan: MABEL study. J Clin Oncol 2008;26:5335-43. http://dx.doi.org/10.1200/JCO.2008.16.3758.
- Starling N, Tilden D, White J, Cunningham D. Cost-effectiveness analysis of cetuximab/irinotecan vs. active/best supportive care for the treatment of metastatic colorectal cancer patients who have failed previous chemotherapy treatment. Br J Cancer 2007;96:206-12. http://dx.doi.org/10.1038/sj.bjc.6603561.
- Jonker DJ, O’Callaghan CJ, Karapetis CS, Zalcberg JR, Tu DS, Au HJ, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med 2007;357:2040-8. http://dx.doi.org/10.1056/NEJMoa071834.
- Mittmann N, Au HJ, Tu DS, O’Callaghan CJ, Isogai PK, Karapetis CS, et al. Prospective cost-effectiveness analysis of cetuximab in metastatic colorectal cancer: evaluation of National Cancer Institute of Canada Clinical Trials Group CO.17 trial. J Nat Cancer Inst 2009;101:1182-92. http://dx.doi.org/10.1093/jnci/djp232.
- Best JH, Garrison LP, Hollingworth W, Ramsey SD, Veenstra DL. Preference values associated with stage III colon cancer and adjuvant chemotherapy. Qual Life Res 2010;19:391-400. http://dx.doi.org/10.1007/s11136-010-9589-5.
- Kalady M, Dziedzic M, Manilich E, Lynch C, McGannon E, Fay S, et al. Quality of life after surgery for colorectal cancer in HNPCC patients. Fam Cancer 2011;10.
- Ara R, Brazier J. Deriving an algorithm to convert the eight mean SF-36 dimension scores into a mean EQ-5D preference-based score from published studies (where patient level data are not available). Value Health 2008;11:1131-43. http://dx.doi.org/10.1111/j.1524-4733.2008.00352.x.
- Bye A, Trope C, Loge JH, Hjermstad M, Kaasa S. Health-related quality of life and occurrence of intestinal side effects after pelvic radiotherapy – Evaluation of long-term effects of diagnosis and treatment. Acta Oncol 2000;39:173-80. http://dx.doi.org/10.1080/028418600430734.
- De Poll-Franse LVV, Mols F, Essink-Bot ML, Haartsen JE, Vingerhoets JJM, Lybeert MLM, et al. Impact of external beam adjuvant radiotherapy on health-related quality of life for long-term survivors of endometrial adenocarcinoma: a population-based study. Int J Radiat Oncol Biol Physics 2007;69:125-32. http://dx.doi.org/10.1016/j.ijrobp.2007.02.040.
- NHS . Hysterectomy 2013. www.nhs.uk/Conditions/Hysterectomy/Pages/Introduction.aspx (accessed July 2014).
- Collins VR, Meiser B, Ukoumunne OC, Gaff C, John DJS, Halliday JL. The impact of predictive genetic testing for hereditary nonpolyposis colorectal cancer: three years after testing. Genet Med 2007;9:290-7. http://dx.doi.org/10.1097/GIM.0b013e31804b45db.
- Aktan-Collan K, Haukkala A, Mecklin JP, Uutela A, Kaariainen H. Psychological consequences of predictive genetic testing for hereditary non-polyposis colorectal cancer (HNPCC): a prospective follow-up study. Int J Cancer 2001;93:608-11. http://dx.doi.org/10.1002/ijc.1372.
- Shiloh S, Koehly L, Jenkins J, Martin J, Hadley D. Monitoring coping style moderates emotional reactions to genetic testing for hereditary nonpolyposis colorectal cancer: a longitudinal study. Psychooncology 2008;17:746-55. http://dx.doi.org/10.1002/pon.1338.
- Claes E, Evers-Kiebooms G, Decruyenaere M, Denayer L, Boogaerts A, Philippe K, et al. Surveillance behavior and prophylactic surgery after predictive testing for hereditary breast/ovarian cancer. Behav Med 2005;31:93-105. http://dx.doi.org/10.3200/BMED.31.3.93-106.
- Keller M, Jost R, Haunstetter CM, Sattel H, Schroeter C, Bertsch U, et al. Psychosocial outcome following genetic risk counselling for familial colorectal cancer. A comparison of affected patients and family members. Clin Genet 2008;74:414-24. http://dx.doi.org/10.1111/j.1399-0004.2008.01089.x.
- Meiser B, Collins V, Warren R, Gaff C, St John DJ, Young MA, et al. Psychological impact of genetic testing for hereditary non-polyposis colorectal cancer. Clin Genet 2004;66:502-11. http://dx.doi.org/10.1111/j.1399-0004.2004.00339.x.
- Hasenbring MI, Kreddig N, Deges G, Epplen JT, Kunstmann E, Stemmler S, et al. Psychological impact of genetic counseling for hereditary nonpolyposis colorectal cancer: the role of cancer history, gender, age, and psychological distress. Genet Test Mol Biomarkers 2011;15:219-25. http://dx.doi.org/10.1089/gtmb.2010.0165.
- Heshka JT, Palleschi C, Howley H, Wilson B, Wells PS. A systematic review of perceived risks, psychological and behavioral impacts of genetic testing. Genet Med 2008;10:19-32. http://dx.doi.org/10.1097/GIM.0b013e31815f524f.
- van Oostrom I, Meijers-Heijboer H, Duivenvoorden HJ, Brocker-Vriends A, van Asperen CJ, Sijmons RH, et al. Comparison of individuals opting for BRCA1/2 or HNPCC genetic susceptibility testing with regard to coping, illness perceptions, illness experiences, family system characteristics and hereditary cancer distress. Patient Educ Counsel 2007;65:58-6. http://dx.doi.org/10.1016/j.pec.2006.05.006.
- Bleiker EMA, Menko FH, Kluijt I, Taal BG, Gerritsma MA, Wever LD, et al. Colorectal cancer in the family: psychosocial distress and social issues in the years following genetic counselling. Hered Cancer Clin Pract 2007;5:59-66. http://dx.doi.org/10.1186/1897-4287-5-2-59.
- Esplen MJ, Butler K, Aronson M, Rothen-Mund H, Smith K, Way C, et al. Long term adjustment in individuals receiving genetic test results for hereditary non-polyposis colorectal cancer (HNPCC). Psychooncology 2007;16.
- Curtis L. Unit Costs of Health and Social Care 2012. Canterbury: PSSRU, University of Kent; 2012.
- UK Genetic Testing Network . Welcome to the UKGTN 2012. http://ukgtn.nhs.uk/ (accessed July 2014).
- Fireman B, Quesenberry C, Somkin C, Jacobson A, Baer D, West D. The Cost of Care for Cancer in a Health Maintenance Organization. Bethesda, MD: Applied Research Branch, National Cancer Institute; 1994.
- Zauber AG, Lansdorp-Vogelaar I, Wilschut J, Knudsen AB, van Ballegooijen M, Kuntz KM. Cost-Effectiveness of DNA Stool Testing to Screen for Colorectal Cancer: Report to AHRQ and CMS from the Cancer Intervention and Surveillance Modeling Network (CISNET) for MISCAN and SimCRC Models 2007. www.cms.gov/medicare-coverage-database/details/technology-assessments-details.aspx?TAId=52.
- Ferro SA, Myer BS, Wolff DA, Poniewierski MS, Culakova E, Cosler LE, et al. Variation in the cost of medications for the treatment of colorectal cancer. Am J Manag Care 2008;14:717-25.
- Lang K, Lines LM, Lee DW, Korn JR, Earle CC, Menzin J. Lifetime and treatment-phase costs associated with colorectal cancer: evidence from SEER-Medicare data. Clin Gastroenterol Hepatol 2009;7:198-204. http://dx.doi.org/10.1016/j.cgh.2008.08.034.
- Yabroff KR, Warren JL, Schrag D, Mariotto A, Meekins A, Topor M, et al. Comparison of approaches for estimating incidence costs of care for colorectal cancer patients. Med Care 2009;47:S56-63. http://dx.doi.org/10.1097/MLR.0b013e3181a4f482.
- Potosky AL, Riley GF, Lubitz JD, Mentnech RM, Kessler LG. Potential for cancer related health services research using a linked Medicare-tumor registry database. Med Care 1993;31:732-48. http://dx.doi.org/10.1097/00005650-199308000-00006.
- Pilgrim H, Tappenden P, Chilcott J, Bending M, Trueman P, Shorthouse A, et al. The costs and benefits of bowel cancer service developments using discrete event simulation. J Operat Res Soc 2009;60:1305-14. http://dx.doi.org/10.1057/jors.2008.109.
- Health and Social Care Information Centre . HRG4 Code to Group; HRG4 2011 12 Reference Costs Grouper 2012. www.hscic.gov.uk/article/2610/HRG4-201112-Reference-Costs-Grouper-Documentation (accessed July 2014).
- Laparoscopic Techniques for Hysterectomy (IPG239). London: National Institute for Health and Care Excellence; 2007.
- Evidence for Policy and Practice Information and Co-ordinating Centre (EPPI-Centre) . CCEMG – EPPI-Centre Cost Converter (v.1.2) 2013. http://eppi.ioe.ac.uk/costconversion/default.aspx (accessed July 2014).
- Medscape . Endometrial Cancer Treatment Protocols 2011. http://emedicine.medscape.com/article/2001830-overview (accessed July 2014).
- Fleming GF, Brurietto VL, Cella D, Look KY, Reid GCH, Munkarah AR, et al. Phase III trial of doxorubicin plus cisplatin with or without paclitaxel plus filgrastim in advanced endometrial carcinoma: A gynecologic oncology group study. J Clin Oncol 2004;22:2159-66. http://dx.doi.org/10.1200/JCO.2004.07.184.
- Department of Health . NHS Reference Costs 2010–2011 2011. http://data.gov.uk/dataset/nhs-reference-costs-2010-11 (accessed July 2014).
- Podczaski E, Kaminski P, Gurski K, MacNeill C, Stryker JA, Singapuri K, et al. Detection and patterns of treatment failure in 300 consecutive cases of ‘early’ endometrial cancer after primary surgery. Gynecol Oncol 1992;47:323-7. http://dx.doi.org/10.1016/0090-8258(92)90134-5.
- Shumsky AG, Stuart GC, Brasher PM, Nation JG, Robertson DI, Sangkarat S. An evaluation of routine follow-up of patients treated for endometrial carcinoma. Gynecol Oncol 1994;55:229-33. http://dx.doi.org/10.1006/gyno.1994.1282.
- Berchuck A, Anspach C, Evans AC, Soper JT, Rodriguez GC, Dodge R, et al. Postsurgical surveillance of patients with FIGO stage I/II endometrial adenocarcinoma. Gynecol Oncol 1995;59:20-4. http://dx.doi.org/10.1006/gyno.1995.1262.
- Reddoch JM, Burke TW, Morris M, Tornos C, Levenback C, Gershenson DM. Surveillance for recurrent endometrial carcinoma: development of a follow-up scheme. Gynecol Oncol 1995;59:221-5. http://dx.doi.org/10.1006/gyno.1995.0012.
- Owen P, Duncan ID. Is there any value in the long term follow up of women treated for endometrial cancer?. Br J Obstet Gynaecol 1996;103:710-13. http://dx.doi.org/10.1111/j.1471-0528.1996.tb09843.x.
- Allsop JR, Preston J, Crocker S. Is there any value in the long-term follow up of women treated for endometrial cancer?. Br J Obstet Gynaecol 1997;104. http://dx.doi.org/10.1111/j.1471-0528.1997.tb10672.x.
- Salvesen HB, Akslen LA, Iversen T, Iversen OE. Recurrence of endometrial carcinoma and the value of routine follow up. Br J Obstet Gynaecol 1997;104:1302-7. http://dx.doi.org/10.1111/j.1471-0528.1997.tb10979.x.
- Agboola OO, Grunfeld E, Coyle D, Perry GA. Costs and benefits of routine follow-up after curative treatment for endometrial cancer. CMAJ 1997;157:879-86.
- Ng TY, Ngan HY, Cheng DK, Wong LC. Vaginal vault cytology in the routine follow-up of patients treated for endometrial carcinoma: is it useful?. Aust N Z J Obstet Gynaecol 1997;37:104-6. http://dx.doi.org/10.1111/j.1479-828X.1997.tb02229.x.
- Gadducci A, Cosio S, Fanucchi A, Cristofani R, Genazzani AR. An intensive follow-up does not change survival of patients with clinical stage I endometrial cancer. Anticancer Res 2000;20:1977-84.
- Morice P, Levy-Piedbois C, Ajaj S, Pautier P, Haie-Meder C, Lhomme C, et al. Value and cost evaluation of routine follow-up for patients with clinical stage I/II endometrial cancer. Eur J Cancer 2001;37:985-90. http://dx.doi.org/10.1016/S0959-8049(01)00066-1.
- Tjalma WAA, Van Dam PA, Makar AP, Cruickshank DJ. The clinical value and the cost-effectiveness of follow-up in endometrial cancer patients. Int J Gynecol Cancer 2004;14:931-7. http://dx.doi.org/10.1111/j.1048-891X.2004.014532.x.
- Briggs AH, Claxton K, Sculpher MJ. Decision Modelling for Health Economic Evaluation. Oxford: Oxford University Press; 2006.
- Briggs AH, Weinstein MC, Fenwick EA, Karnon J, Sculpher MJ, Paltiel AD, et al. Model parameter estimation and uncertainty analysis: a report of the ISPOR-SMDM Modeling Good Research Practices Task Force Working Group-6. Med Decis Making 2012;32:722-32. http://dx.doi.org/10.1177/0272989X12458348.
- Thompson BA, Spurdle AB, Plazzer JP, Greenblatt MS, Akagi K, Al-Mulla F, et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet 2014;46:107-15. http://dx.doi.org/10.1038/ng.2854.
- Crépin M, Dieu M-C, Lejeune S, Escande F, Boidin D, Porchet N, et al. Evidence of constitutional MLH1 epimutation associated to transgenerational inheritance of cancer susceptability. Hum Mutat 2012;33:180-8. http://dx.doi.org/10.1002/humu.21617.
- Huth C, Kloor M, Voigt AY, Bozukova G, Evers C, Gaspar H, et al. The molecular basis of EPCAM expression loss in Lynch syndrome-associated tumors. Mod Pathol 2012;25:911-16. http://dx.doi.org/10.1038/modpathol.2012.30.
- Kwon JS, Daniels MS, Sun CC, Lu KH. Preventing future cancers by testing women with ovarian cancer for BRCA mutations. J Clin Oncol 2010;28:675-82. http://dx.doi.org/10.1200/JCO.2008.21.4684.
- Doust JA, Pietrzak E, Sanders S, Glasziou PP. Identifying studies for systematic reviews of diagnostic tests was difficult due to the poor sensitivity and precision of methodologic filters and the lack of information in the abstract. J Clin Epidemiol 2005;58:444-9. http://dx.doi.org/10.1016/j.jclinepi.2004.09.011.
- Leeflang MM, Scholten RJ, Rutjes AW, Reitsma JB, Bossuyt PM. Use of methodological search filters to identify diagnostic accuracy studies can lead to the omission of relevant studies. J Clin Epidemiol 2006;59:234-40. http://dx.doi.org/10.1016/j.jclinepi.2005.07.014.
- Bayliss SE, Davenport C. Locating systematic reviews of test accuracy studies: how five specialist review databases measure up. Int J Technol Assess Health Care 2008;22:39-46.
- Glanville J, Paisley S. Identifying economic evaluations for health technology assessment. Int J Technol Assess Health Care 2010;26:436-40. http://dx.doi.org/10.1017/S0266462310000991.
- Sassi F, Archard L, McDaid D. Searching literature databases for health care economic evaluations: how systematic can we afford to be?. Med Care 2002;40:387-94. http://dx.doi.org/10.1097/00005650-200205000-00004.
- Glanville J, Kaunelis D, Mensinkai S. How well do search filters perform in identifying economic evaluations in MEDLINE and EMBASE. Int J Technol Assess Health Care 2009;25:522-9. http://dx.doi.org/10.1017/S0266462309990523.
- Choi YH, Cotterchio M, McKeown-Eyssen G, Neerav M, Bapat B, Boyd K, et al. Penetrance of colorectal cancer among MLH1MSH2 carriers participating in the colorectal cancer familial registry in Ontario. Hered Cancer Clin Pract 2009;7. http://dx.doi.org/10.1186/1897-4287-7-14.
- Bermejo JL, Eng C, Hemminki K. Cancer characteristics in Swedish families fulfilling criteria for hereditary nonpolyposis colorectal cancer. Gastroenterology 2005;129:1889-99. http://dx.doi.org/10.1053/j.gastro.2005.09.012.
- Hampel H, Stephens JA, Pukkala E, Sankila R, Aaltonen LA, Mecklin JP, et al. Cancer risk in hereditary nonpolyposis colorectal cancer syndrome: later age of onset. Gastroenterology 2005;129:415-21. http://dx.doi.org/10.1016/j.gastro.2005.05.011.
- Plaschke J, Engel C, Kruger S, Holinski-Feder E, Pagenstecher C, Mangold E, et al. Lower incidence of colorectal cancer and later age of disease onset in 27 families with pathogenic MSH6 germline mutations compared with families with MLH1 or MSH2 mutations: the German Hereditary Nonpolyposis Colorectal Cancer Consortium. J Clin Oncol 2004;22:4486-94. http://dx.doi.org/10.1200/JCO.2004.02.033.
- Vasen HF, Stormorken A, Menko FH, Nagengast FM, Kleibeuker JH, Griffioen G, et al. MSH2 mutation carriers are at higher risk of cancer than MLH1 mutation carriers: a study of hereditary nonpolyposis colorectal cancer families. J Clin Oncol 2001;19:4074-80.
- Watson P, Lin KM, Rodriguez-Bigas MA, Smyrk T, Lemon S, Shashidharan M, et al. Colorectal carcinoma survival among hereditary nonpolyposis colorectal carcinoma family members. Cancer 1998;83:259-66. http://dx.doi.org/10.1002/(SICI)1097-0142(19980715)83:2<259::AID-CNCR9>3.0.CO;2-L.
- Sankila R, Aaltonen LA, Jarvinen HJ, Mecklin JP. Better survival rates in patients with MLH1-associated hereditary colorectal cancer. Gastroenterology 1996;110:682-7. http://dx.doi.org/10.1053/gast.1996.v110.pm8608876.
- Bertario L, Russo A, Sala P, Eboli M, Radice P, Presciuttini S, et al. Survival of patients with hereditary colorectal cancer: comparison of HNPCC and colorectal cancer in FAP patients with sporadic colorectal cancer. Int J Cancer 1999;80:183-7. http://dx.doi.org/10.1002/(SICI)1097-0215(19990118)80:2<183::AID-IJC4>3.0.CO;2-W.
- Elsakov P, Kurtinaitis J. Survival from colorectal carcinoma in HNPCC families as compared to the general population in Lithuania – initial results. Fam Cancer 2006;5:369-71. http://dx.doi.org/10.1007/s10689-006-0007-7.
- Fujita S, Moriya Y, Sugihara K, Akasu T, Ushio K. Prognosis of hereditary nonpolyposis colorectal cancer (HNPCC) and the role of Japanese criteria for HNPCC. Jpn J Clin Oncol 1996;26:351-5. http://dx.doi.org/10.1093/oxfordjournals.jjco.a023244.
- Haghighi MM, Vahedi M, Mohebbi SR, Pourhoseingholi MA, Fatemi SR, Zali MR. Comparison of survival between patients with hereditary non polyposis colorectal cancer (HNPCC) and sporadic colorectal cancer. Asian Pac J Cancer Prev 2009;10:209-12.
- Myrhøj T, Bisgaard ML, Bernstein I, Svendsen LB, Sondergaard JO, Bulow S. Hereditary non-polyposis colorectal cancer: clinical features and survival. Results from the Danish HNPCC register. Scand J Gastroenterol 1997;32:572-6. http://dx.doi.org/10.3109/00365529709025102.
- Percesepe A, Benatti P, Roncucci L, Sassatelli R, Fante R, Ganazzi D, et al. Survival analysis in families affected by hereditary non-polyposis colorectal cancer. Int J Cancer 1997;71:373-6. http://dx.doi.org/10.1002/(SICI)1097-0215(19970502)71:3<373::AID-IJC12>3.0.CO;2-H.
- Stigliano V, Assisi D, Cosimelli M, Palmirotta R, Giannarelli D, Mottolese M, et al. Survival of hereditary non-polyposis colorectal cancer patients compared with sporadic colorectal cancer patients. J Exp Clin Cancer Res 2008;27. http://dx.doi.org/10.1186/1756-9966-27-39.
- You JF, Hsieh LL, Changchien CR, Chen JS, Chen JR, Chiang JM, et al. Inverse effects of mucin on survival of matched hereditary nonpolyposis colorectal cancer and sporadic colorectal cancer patients. Clin Cancer Res 2006;12:4244-50. http://dx.doi.org/10.1158/1078-0432.CCR-06-0202.
- Russo A, Sala P, Alberici P, Gazzoli I, Radice P, Montefusco C, et al. Prognostic relevance of MLH1 and MSH2 mutations in hereditary non-polyposis colorectal cancer patients. Tumori 2009;95:731-8.
- Guillen-Ponce C, Martinez-Sevila C, Perea R, Arenas M, Molina-Garrido MJ, Goicoechea M, et al. Gynecologic cancer screening in women at high risk of Lynch syndrome. J Clin Oncol 2011;29.
- Helder-Woolderink JM, De Bock GH, Hollema H, Hofstra RMW, Sijmons RH, Mourits MJE. Annual gynaecological surveillance in women with Lynch syndrome: What’s the additional value of microcurettage?. Int J Gynecol Cancer 2011;21.
- Schorge JO, Modesitt SC, Coleman RL, Cohn DE, Kauff ND, Duska LR, et al. SGO white paper on ovarian cancer: Aetiology, screening and surveillance. Gynecol Oncol 2010;119:7-18. http://dx.doi.org/10.1016/j.ygyno.2010.06.003.
- Wood NJ, Duffy SR, Sheridan E, Thomas HJW, Dove-Edwin I, Goff S, et al. The outcome of endometrial carcinoma surveillance by ultrasound scan in women at risk of hereditary nonpolyposis colorectal carcinoma and familial colorectal carcinoma (multiple letters). Cancer 2003;98:1772-4. http://dx.doi.org/10.1002/cncr.11704.
- Boston Medical Center . IPAA Procedure 2013. www.bmc.org/ipaa/procedure.htm (accessed July 2014).
- Bouzourene H, Hutter P, Losi L, Martin P, Benhattar J. Selection of patients with germline MLH1 mutated Lynch syndrome by determination of MLH1 methylation and BRAF mutation. Fam Cancer 2010;9:167-72. http://dx.doi.org/10.1007/s10689-009-9302-4.
- Chang SC, Lin PC, Yang SH, Wang HS, Liang WY, Lin JK. Taiwan hospital-based detection of Lynch syndrome distinguishes 2 types of microsatellite instabilities in colorectal cancers. Surgery 2010;147:720-8. http://dx.doi.org/10.1016/j.surg.2009.10.069.
Appendix 1 Literature search strategy for test accuracy
There remain noted difficulties in searching for, and locating, diagnostic test accuracy studies [Doust et al. (2005);276 Leeflang et al. (2006);277 Bayliss and Davenport (2008)278]. With this advice in mind, we have plotted the search for this review on only the population for this review.
The search strategy was reviewed by the research team and clinical experts prior to running the searches.
| Database | Hits |
|---|---|
| 1. MEDLINE | 4632 |
| 2. MEDLINE In-Process & Other Non-Indexed Citations | 216 |
| 3. EMBASE | 5172 |
| 4. PsycINFO | 49 |
| 5. HMIC | 15 |
| 6. CINAHL | 56 |
| 7. ASSIA | 11 |
| 8. Web of Science | 5274 |
| 9. The Cochrane Library | 75 |
| 10. BNI | 12 |
| Database Results | 15,512 |
| Deduplication | 8361 |
| Unique records to screen | 7151 |
| NRR | 2 |
| Current Controlled Trials | 83 |
| ClinicalTrials.gov | 203 |
| FDA website | 35 |
| EMEA website | 0 |
1. MEDLINE
Host: Ovid.
Data parameters: 1946 to April week 3, 2012.
Date searched: Monday, 30 April 2012.
Searcher: Cooper.
Hits: 4626.
Search strategy
| # | Searches | Results |
|---|---|---|
| 1 | (lynch$ adj3 syndrome).ti,ab. | 895 |
| 2 | (lynch$ adj3 famil$).ti,ab. | 167 |
| 3 | 1 or 2 | 901 |
| 4 | Colorectal Neoplasms, Hereditary Nonpolyposis/ | 2994 |
| 5 | (Hereditary Nonpolyposis Colorectal Cancer or Hereditary Non-polyposis Colorectal Cancer).tw. | 2099 |
| 6 | HNPCC.tw. | 1884 |
| 7 | ((hereditary adj3 nonpolyposis) and (colon$ or colorectal$)).ti,ab. | 1682 |
| 8 | ((hereditary adj3 non-polyposis) and (colon$ or colorectal$)).ti,ab. | 1037 |
| 9 | ((hereditary adj3 (cancer or neoplasm)) and (colon$ or colorectal$)).ti,ab. | 2074 |
| 10 | ((Familial adj3 Nonpolyposis) and (colon$ or colorectal$)).ti,ab. | 14 |
| 11 | ((Familial adj3 Non-polyposis) and (colon$ or colorectal$)).ti,ab. | 17 |
| 12 | or/4-11 | 4196 |
| 13 | ((MLH1 or MSH2 or MSH3 or MSH6 or hMSH2 or hMLH1 or hPMS1 or hPMS2 or hMSH6 or hMLH3 or PMS1 or PMS2) and (colon$ or colorectal or lynch$ or HNPCC or hereditary)).ti,ab. | 2501 |
| 14 | Amsterdam criteria.tw. | 319 |
| 15 | or/13-14 | 2627 |
| 16 | 3 or 12 or 15 | 5271 |
| 17 | limit 16 to english language | 4713 |
| 18 | animals/ not humans/ | 3,611,730 |
| 19 | 17 not 18 | 4632 |
2. MEDLINE In-Process & Other Non-Indexed Citations
Host: Ovid.
Data parameters: added on or before 27 April 2012.
Date searched: Monday, 30 April 2012.
Searcher: Cooper.
Hits: 216.
Search strategy
| # | Searches | Results |
|---|---|---|
| 1 | (lynch$ adj3 syndrome).ti,ab. | 96 |
| 2 | (lynch$ adj3 famil$).ti,ab. | 18 |
| 3 | 1 or 2 | 96 |
| 4 | Colorectal Neoplasms, Hereditary Nonpolyposis/ | 0 |
| 5 | (Hereditary Nonpolyposis Colorectal Cancer or Hereditary Non-polyposis Colorectal Cancer).tw. | 61 |
| 6 | HNPCC.tw. | 63 |
| 7 | ((hereditary adj3 nonpolyposis) and (colon$ or colorectal$)).ti,ab. | 40 |
| 8 | ((hereditary adj3 non-polyposis) and (colon$ or colorectal$)).ti,ab. | 39 |
| 9 | ((hereditary adj3 (cancer or neoplasm)) and (colon or colorectal)).ti,ab. | 70 |
| 10 | ((Familial adj3 Nonpolyposis) and (colon$ or colorectal$)).ti,ab. | 0 |
| 11 | ((Familial adj3 Non-polyposis) and (colon$ or colorectal$)).ti,ab. | 0 |
| 12 | or/4-11 | 115 |
| 13 | ((MLH1 or MSH2 or MSH3 or MSH6 or hMSH2 or hMLH1 or hPMS1 or hPMS2 or hMSH6 or hMLH3 or PMS1 or PMS2) and (colon$ or colorectal or lynch$ or HNPCC or hereditary)).ti,ab. | 103 |
| 14 | Amsterdam criteria.tw. | 7 |
| 15 | or/13-14 | 109 |
| 16 | 3 or 12 or 15 | 228 |
| 17 | limit 16 to english language | 216 |
| 18 | animals/ not humans/ | 1 |
| 19 | 17 not 18 | 216 |
3. EMBASE
Host: Ovid.
Data parameters: 1980 to week 17, 2012.
Date searched: Monday, 30 April 2012.
Searcher: Cooper.
Hits: 1061.
Search strategy
| # | Searches | Results |
|---|---|---|
| 1 | (lynch$ adj3 syndrome).ti,ab. | 1537 |
| 2 | (lynch$ adj3 famil$).ti,ab. | 261 |
| 3 | 1 or 2 | 1545 |
| 4 | hereditary nonpolyposis colorectal cancer/ | 1202 |
| 5 | (Hereditary Nonpolyposis Colorectal Cancer or Hereditary Non-polyposis Colorectal Cancer).tw. | 2629 |
| 6 | HNPCC.tw. | 2469 |
| 7 | ((hereditary adj3 nonpolyposis) and (colon$ or colorectal$)).ti,ab. | 2056 |
| 8 | ((hereditary adj3 non-polyposis) and (colon$ or colorectal$)).ti,ab. | 1344 |
| 9 | ((hereditary adj3 (cancer or neoplasm)) and (colon$ or colorectal$)).ti,ab. | 2687 |
| 10 | ((Familial adj3 Nonpolyposis) and (colon$ or colorectal$)).ti,ab. | 18 |
| 11 | ((Familial adj3 Non-polyposis) and (colon$ or colorectal$)).ti,ab. | 21 |
| 12 | or/4-11 | 4969 |
| 13 | ((MLH1 or MSH2 or MSH3 or MSH6 or hMSH2 or hMLH1 or hPMS1 or hPMS2 or hMSH6 or hMLH3 or PMS1 or PMS2) and (colon$ or colorectal or lynch$ or HNPCC or hereditary)).ti,ab. | 3349 |
| 14 | Amsterdam criteria.tw. | 431 |
| 15 | or/13-14 | 3531 |
| 16 | 3 or 12 or 15 | 6694 |
| 17 | limit 16 to english language | 5979 |
| 18 | limit 17 to human | 5172 |
4. PsycINFO
Host: Ovid.
Data parameters: 1806 to April week 3, 2012.
Date searched: Monday, 30 April 2012.
Searcher: Cooper.
Hits: 49.
Search strategy
| # | Searches | Results |
|---|---|---|
| 1 | (lynch$ adj3 syndrome).ti,ab. | 10 |
| 2 | (lynch$ adj3 famil$).ti,ab. | 3 |
| 3 | 1 or 2 | 11 |
| 4 | Colorectal Neoplasms, Hereditary Nonpolyposis/ | 0 |
| 5 | (Hereditary Nonpolyposis Colorectal Cancer or Hereditary Non-polyposis Colorectal Cancer).tw. | 23 |
| 6 | HNPCC.tw. | 24 |
| 7 | ((hereditary adj3 nonpolyposis) and (colon$ or colorectal$)).ti,ab. | 15 |
| 8 | ((hereditary adj3 non-polyposis) and (colon$ or colorectal$)).ti,ab. | 13 |
| 9 | ((hereditary adj3 (cancer or neoplasm)) and (colon or colorectal)).ti,ab. | 36 |
| 10 | ((Familial adj3 Nonpolyposis) and (colon$ or colorectal$)).ti,ab. | 0 |
| 11 | ((Familial adj3 Non-polyposis) and (colon$ or colorectal$)).ti,ab. | 0 |
| 12 | or/4-11 | 45 |
| 13 | ((MLH1 or MSH2 or MSH3 or MSH6 or hMSH2 or hMLH1 or hPMS1 or hPMS2 or hMSH6 or hMLH3 or PMS1 or PMS2) and (colon$ or colorectal or lynch$ or HNPCC or hereditary)).ti,ab. | 4 |
| 14 | Amsterdam criteria.tw. | 0 |
| 15 | or/13-14 | 4 |
| 16 | 3 or 12 or 15 | 54 |
| 17 | limit 16 to english language | 49 |
| 18 | animals/ not humans/ | 5401 |
| 19 | 17 not 18 | 49 |
5. Health Management Information Consortium
Host: Ovid.
Data parameters: 1979 to March 2012.
Date searched: Monday, 30 April 2012.
Searcher: Cooper.
Hits: 15.
Search strategy
| # | Searches | Results |
|---|---|---|
| 1 | (lynch$ adj3 syndrome).ti,ab. | 4 |
| 2 | (lynch$ adj3 famil$).ti,ab. | 0 |
| 3 | 1 or 2 | 4 |
| 4 | Colorectal Neoplasms, Hereditary Nonpolyposis/ | 0 |
| 5 | (Hereditary Nonpolyposis Colorectal Cancer or Hereditary Non-polyposis Colorectal Cancer).tw. | 5 |
| 6 | HNPCC.tw. | 4 |
| 7 | ((hereditary adj3 nonpolyposis) and (colon$ or colorectal$)).ti,ab. | 4 |
| 8 | ((hereditary adj3 non-polyposis) and (colon$ or colorectal$)).ti,ab. | 2 |
| 9 | ((hereditary adj3 (cancer or neoplasm)) and (colon or colorectal)).ti,ab. | 11 |
| 10 | ((Familial adj3 Nonpolyposis) and (colon$ or colorectal$)).ti,ab. | 0 |
| 11 | ((Familial adj3 Non-polyposis) and (colon$ or colorectal$)).ti,ab. | 0 |
| 12 | or/4-11 | 13 |
| 13 | ((MLH1 or MSH2 or MSH3 or MSH6 or hMSH2 or hMLH1 or hPMS1 or hPMS2 or hMSH6 or hMLH3 or PMS1 or PMS2) and (colon$ or colorectal or lynch$ or HNPCC or hereditary)).ti,ab. | 3 |
| 14 | Amsterdam criteria.tw. | 2 |
| 15 | or/13-14 | 4 |
| 16 | 3 or 12 or 15 | 15 |
| 17 | limit 16 to english language [limit not valid; records were retained] | 15 |
| 18 | animals/ not humans/ | 225 |
| 19 | 17 not 18 | 15 |
6. Cumulative Index to Nursing and Allied Health Literature
Host: EBSCOhost.
Data parameters: 1946 to April week 3, 2012.
Date searched: Monday, 30 April 2012.
Searcher: Cooper.
Hits: 4626.
Search strategy
S1. (lynch* N3 syndrome)
S2. (lynch* N3 famil*)
S3. S1 OR 2S
S4. (MM "Colorectal Neoplasms, Hereditary Nonpolyposis")
S5. ((Hereditary Nonpolyposis Colorectal Cancer) or (Hereditary Non-polyposis Colorectal Cancer))
S6. HNPCC
S7. ((hereditary N3 nonpolyposis) and (colon* or colorectal*))
S8. ((hereditary N3 non-polyposis) and (colon* or colorectal*))
S9. ((hereditary N3 (cancer or neoplasm)) and (colon or colorectal))
S10. ((Familial N3 Nonpolyposis) and (colon* or colorectal*))
S11. ((Familial N3 Non-polyposis) and (colon* or colorectal*))
S12. S4 OR S5 OR S6 OR S7 OR S8 OR S9 OR S10 OR S11
S13. (((MLH1) or (MSH2) or (MSH3) or (MSH6) or (hMSH2) or (hMLH1) or (hPMS1) or (hPMS2) or (hMSH6) or (hMLH3) or (PMS1) or (PMS2)) and (colon* or colorectal or lynch* or HNPCC or hereditary))
S14. (Amsterdam criteria)
S15. S13 OR S14
S16. S3 OR S12 OR S15
Notes: Server-side deduplication carried out to remove MEDLINE hits.
7. Applied Social Sciences Index and Abstracts
Host: CSA.
Data parameters: None specified.
Date searched: Monday, 30 April 2012.
Searcher: Cooper.
Hits: 11.
Search strategy
#1. (lynch* and syndrome)
#2. (lynch* and famil*)
#3. Or/1-2
#4. ((Hereditary Nonpolyposis Colorectal Cancer) or (Hereditary Non-polyposis Colorectal Cancer))
#5. HNPCC
#6. Or/4-5
#7. 3 or 6
8. Web of Science [SCI-EXPANDED, Social Sciences Citation Index (SSCI), Arts & Humanities Citation Index (A&HCI), Conference Proceedings Citation Index – Science (CPCI-S), Conference Proceedings Citation Index – Social Science & Humanities (CPCI-SSH)]
Host: ISI.
Data parameters: None specified.
Date searched: Monday, 30 April 2012.
Searcher: Cooper.
Hits: 5274.
Search strategy
#1. Topic=(("Hereditary Nonpolyposis Colorectal Cancer"))
#2. Topic=(("Hereditary Non-polyposis Colorectal Cancer"))
#3. Topic=(((lynch* near/3 syndrome)))
#4. Topic=((((MLH1) or (MSH2) or (MSH3) or (MSH6) or (hMSH2) or (hMLH1) or (hPMS1) or (hPMS2) or (hMSH6) or (hMLH3) or (PMS1) or (PMS2)) and (colon* or colorectal or lynch* or HNPCC or hereditary)))
#5. Topic=(("Amsterdam criteria"))
#6. #1 or #2 or #3 or #4 or #5
9. The Cochrane Library
Host: www.thecochranelibrary.com/view/0/index.html.
Data parameters: None specified.
Date searched: Monday, 30 April 2012.
Searcher: Cooper.
Hits: Cochrane Reviews (3); other reviews (5); trials (43); methods studies (1); TAs (8); economic evaluations (15); Cochrane Groups (0).
Current search history
#1. (lynch* near/3 syndrome) (19)
#2. (lynch* near/3 famil*) :ti,ab,kw (0)
#3. MeSH descriptor Colorectal Neoplasms, Hereditary Nonpolyposis explode all trees (47)
#4. ((Hereditary Nonpolyposis Colorectal Cancer) or (Hereditary Non-polyposis Colorectal Cancer)):ti,ab,kw (28)
#5. (HNPCC):ti,ab,kw (15)
#6. ((hereditary near/3 nonpolyposis) and (colon* or colorectal*)):ti,ab,kw (51)
#7. ((hereditary near/3 non-polyposis) and (colon* or colorectal*)):ti,ab,kw (9)
#8. ((Familial near/3 Nonpolyposis) and (colon* or colorectal*)):ti,ab,kw (0)
#9. ((Familial near/3 Non-polyposis) and (colon* or colorectal*)):ti,ab,kw (0)
#10. ((hereditary near/3 (cancer or neoplasm)) and (colon* or colorectal*)):ti,ab,kw (56)
#11. (((MLH1) or (MSH2) or (MSH3) or (MSH6) or (hMSH2) or (hMLH1) or (hPMS1) or (hPMS2) or (hMSH6) or (hMLH3) or (PMS1) or (PMS2)) and (colon* or colorectal or lynch* or HNPCC or hereditary)):ti,ab,kw (11)
#12. ("Amsterdam criteria"):ti,ab,kw (3)
#13. (#1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10 OR #11 OR #12) (75)
10. British Nursing Index
Host: ProQuest.
Data parameters: 1994 to current.
Date searched: Monday, 30 April 2012.
Searcher: Cooper.
Hits: 12.
Search strategy
#1. (lynch N/3 syndrome)
#2. (lynch syndrome)
#3. (Hereditary Nonpolyposis Colorectal Cancer or Hereditary Non-polyposis Colorectal Cancer)
#4. HNPCC
#5. ((MLH1 or MSH2 or MSH3 or MSH6 or hMSH2 or hMLH1 or hPMS1 or hPMS2 or hMSH6 or hMLH3 or PMS1 or PMS2) and (colon$ or colorectal or lynch$ or HNPCC or hereditary))
#6. ("Amsterdam criteria")
#7. #1 or #2 or #3 or #4 or #5 or #6
Appendix 2 Clinical effectiveness: blank quality appraisal and data extraction form
| Design | Participants | Tests | Outcomes | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Authors (date): Related references: Objective: Basic design: □ Single gate □ Two gate □ Systematic review □ Other Country: Setting: No. of centres: Funding: |
Sample description/presentation: No. recruited: Selection: □ Consecutive □ Random □ Other □ Unclear Prior testing: Inclusion criteria: Exclusion criteria: |
Index tests included: □ IHC □ MSI □ BRAF □ Methylation Index tests (technical details): Reference standard (technical details): Time intervals between tests: Outcome assessor: Blinding: |
Accuracy outcomes:
Other: Test failures: |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Data | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Sample attrition/dropout:
No. receiving index test (reasons): No. receiving reference standard (reasons): Data excluded (and reasons): |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Notes | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Participant characteristics | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Index test 1Index test 2Index test 3Reference standardNo. of patientsMedian/mean age, yrs (range)No. < 50 yearsNo. meeting AMS IINo. meeting BethesdaGenderMenWomenCancer locationRectumLeft colonRight colonTransverse colon | Index test 1 | Index test 2 | Index test 3 | Reference standard | No. of patients | Median/mean age, yrs (range) | No. < 50 years | No. meeting AMS II | No. meeting Bethesda | Gender |
|
Cancer location |
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Index test 1 | Index test 2 | Index test 3 | Reference standard | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| No. of patients | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Median/mean age, yrs (range) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| No. < 50 years | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| No. meeting AMS II | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| No. meeting Bethesda | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Gender | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cancer location | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Results | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MSI v reference standard: □ Applicable □ Not applicable MSIReference standard+ve–veTotal+ve–veTotal IHC v reference standard: □ Applicable □ Not applicable IHCReference standard+ve–veTotal+ve–veTotal BRAF v reference standard: □ Applicable □ Not applicable BRAFReference standard+ve–veTotal+ve–veTotal Methylation v reference standard: □ Applicable □ Not applicable Not methylatedReference standard+ve–veTotal+ve–veTotal Other results: |
MSI | Reference standard | +ve | –ve | Total | +ve | –ve | Total | IHC | Reference standard | +ve | –ve | Total | +ve | –ve | Total | BRAF | Reference standard | +ve | –ve | Total | +ve | –ve | Total | Not methylated | Reference standard | +ve | –ve | Total | +ve | –ve | Total | ||||||||||||||||||||||||||||||||||||||||||||||||
| MSI | Reference standard | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| +ve | –ve | Total | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| +ve | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| –ve | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Total | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| IHC | Reference standard | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| +ve | –ve | Total | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| +ve | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| –ve | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Total | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| BRAF | Reference standard | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| +ve | –ve | Total | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| +ve | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| –ve | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Total | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Not methylated | Reference standard | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| +ve | –ve | Total | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| +ve | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| –ve | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Total | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Major methodological issues | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Quality appraisal – QUADAS-2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Flow diagram of primary study: | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| DOMAIN 1: PATIENT SELECTION | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Was a consecutive or random sample of patients enrolled? | (Y/N/U) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Was a case–control study design avoided? | (Y/N/U) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Did the study avoid inappropriate exclusions? | (Y/N/U) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Could the selection of patients have introduced bias? | (H/L/U) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Is there concern that the included patients do not match the review question? | (H/L/U) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| DOMAIN 2: INDEX TESTS (complete for each index test) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Were the index test results interpreted without knowledge of the results of the reference standard? | (Y/N/U) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| If a threshold was used, was it pre-specified? | (Y/N/U) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Could the conduct or interpretation of the index test have introduced bias? | (H/L/U) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Is there concern that the index test, its conduct, or interpretation differ from the review question? | (H/L/U) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| DOMAIN 3: REFERENCE STANDARD | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Is the reference standard likely to correctly classify the target condition? | (Y/N/U) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Were the reference standard results interpreted without knowledge of the results of the index test? | (Y/N/U) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | (H/L/U) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Is there concern that the target condition as defined by the reference standard does not match the review question? | (H/L/U) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| DOMAIN 4: FLOW AND TIMING | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Was there an appropriate interval between index test(s) and reference standard? | (Y/N/U) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Did all patients receive a reference standard? | (Y/N/U) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Did all patients receive the same reference standard? | (Y/N/U) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Were all patients included in the analysis? | (Y/N/U) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Could the patient flow have introduced bias? | (H/L/U) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Appendix 3 Clinical effectiveness: excluded studies
Reason for exclusion: population
Abbott DE, Cantor SB, Rodriguez-Bigas MA, Chang GJ, Lynch PM, Feig BW, et al. Detecting hereditary nonpolyposis colorectal cancer syndrome (HNPCC) in colorectal cancer (CRC) patients: optimal strategies at lower costs. Ann Surg Oncol. 65th Annual Cancer Symposium of the Society of Surgical Oncology, Orlando, FL, 21–24 March 2012.
Alemayehu A, Tomkova K, Zavodna K, Ventusova K, Krivulcik T, Bujalkova M, et al. The role of clinical criteria, genetic and epigenetic alterations in Lynch-syndrome diagnosis. Neoplasma 2007;54:391–401.
Benlloch S, Paya A, Alenda C, Bessa X, Andreu M, Jover R, et al. Detection of BRAF V600E mutation in colorectal cancer: comparison of automatic sequencing and real-time chemistry methodology. J Mol Diagn 2006;8:540–3.
Bessa X, Alenda C, Paya A, Alvarez C, Iglesias M, Seoane A, et al. Validation microsatellite path score in a population-based cohort of patients with colorectal cancer. J Clin Oncol 2011;29:3374–80.
Bessa X, Balleste B, Andreu M, Castells A, Bellosillo B, Balaguer F, et al. A prospective, multicenter, population-based study of BRAF mutational analysis for Lynch syndrome screening. Clin Gastroenterol Hepatol 2008;6:206–14.
Bettstetter M, Dechant S, Ruemmele P, Grabowski M, Keller G, Holinski-Feder E, et al. Distinction of hereditary nonpolyposis colorectal cancer and sporadic microsatellite-unstable colorectal cancer through quantification of MLH1 methylation by real-time PCR. Clin Cancer Res 2007;13:3221–8.
Bouzourene H, Taminelli L, Chaubert P, Monnerat C, Seelentag W, Sandmeier D, et al. A cost-effective algorithm for hereditary nonpolyposis colorectal cancer detection. Am J Clin Pathol 2006;125:823–31.
Canard G, Lefevre JH, Colas C, Coulet F, Svrcek M, Lascols O, et al. Screening for Lynch syndrome in colorectal cancer: are we doing enough? Ann Surg Oncol 2012;19:809–16.
Chen S, Watson P, Parmigiani G. Accuracy of MSI testing in predicting germline mutations of MSH2 and MLH1: a case study in Bayesian meta-analysis of diagnostic tests without a gold standard. Biostatistics 2005;6:450–64.
Cicek MS, Lindor NM, Gallinger S, Bapat B, Hopper JL, Jenkins MA, et al. Quality assessment and correlation of microsatellite instability and immunohistochemical markers among population- and clinic-based colorectal tumors results from the Colon Cancer Family Registry. J Mol Diagn 2011;13:271–81.
Esemuede I, Forslund A, Khan SA, Qin LX, Gimbel MI, Nash GM, et al. Improved testing for microsatellite instability in colorectal cancer using a simplified 3-marker assay. Ann Surg Oncol 2010;17:3370–78.
Ewald J, Rodrigue CM, Mourra N, Lefevre JH, Flejou JF, Tiret E, et al. Immunohistochemical staining for mismatch repair proteins, and its relevance in the diagnosis of hereditary non-polyposis colorectal cancer. Br J Surg 2007;94:1020–7.
Frankel WL, Hampel H, LaJeanesse J, Panescu J, Jones S, de la Chapelle A. Immunohistochemical staining for MLH1, MSH2 and MSH6 identifies germline mutations in mismatch repair genes in colorectal and endometrial cancers initially found to be microsatellite stable. Mod Pathol 2005;18:103A.
Glogowski E, Boyar S, Sarrel K, Le Blang C, Utay E, Shia J, et al. Unexplained immunohistochemical DNA mismatch repair deficiency. Collaborative Group of the Americas on Inherited Colorectal Cancer, 15th Annual Meeting, Montreal, QC, 10–11 October 2011.
Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, et al. Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol 2008;26:5783–8.
Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). New Engl J Med 2005;352:1851–60.
Hayes, Inc. BRAF p.Val600Glu (V600E) Testing for Assessment of Treatment Options in Metastatic Colorectal Cancer. Structured abstract. Lansdale, PA: Hayes, Inc.; 2010.
Janavicius R, Matiukaite D, Jakubauskas A, Griskevicius L. Microsatellite instability detection by high-resolution melting analysis. Clin Chem 2010;56:1750–7.
Julie C, Beauchet A, Christophe T, Muti C, Coulet F, Brouquet A, et al. Evaluation of mspath score compared to revised Bethesda guidelines for detecting Lynch syndrome in a consecutive series of 225 colorectal carcinomas. Virchows Arch 2008;452:S30.
Julie C, Beauchet A, Tresallet C, Brouquet A, Muti C, Coulet F, et al. Evaluation of mspath score compared to revised Bethesda guidelines for detecting Lynch syndrome in a consecutive series of 223 colorectal carcinomas. Ann Oncol 2009;20:25.
Kadiyska TK, Konstantinova DV, Atanasov VR, Kremensky IA, Mitev VI. Frequency and application of the hot spot BRAF gene mutation (p.V600E) in the diagnostic strategy for hereditary nonpolyposis colorectal cancer. Cancer Detect Prev 2007;31:254–6.
Kang YN, Kwon SY, Kim SP, Kwon KY, Lee SS. Evaluation of microsatellite instability, hMLH1 expression and hMLH1 promoter hypermethylation in 176 colorectal carcinomas. Lab Invest 2005;85:108A.
Kets CM, Van Krieken JHJM, Hebeda KM, Wezenberg SJ, Goossens M, Brunner HG, et al. Very low prevalence of germline MSH6 mutations in hereditary non-polyposis colorectal cancer suspected patients with colorectal cancer without microsatellite instability. Br J Cancer 2006;95:1678–82.
Lee SC, Gou JY, Lim R, Soo R, Koay E, Salto-Tellez M, et al. Clinical and molecular characteristics of hereditary non-polyposis colorectal cancer families in Southeast Asia. Clin Genet 2005;68:137–45.
Loughrey MB, Waring PM, Tan A, Trivett M, Kovalenko S, Beshay V, et al. Incorporation of somatic BRAF mutation testing into an algorithm for the investigation of hereditary non-polyposis colorectal cancer. Fam Cancer 2007;6:301–10.
Mangold E, Pagenstecher C, Friedl W, Fischer HP, Merkelbach-Bruse S, Ohlendorf M, et al. Tumours from MSH2 mutation carriers show loss of MSH2 expression but many tumours from MLH1 mutation carriers exhibit weak positive MLH1 staining. J Pathol 2005;207:385–95.
Marrupe D, Meizoso T, Lopez E, Mestre MJ, Garcia S, Cortes L, et al. Correlation between clinical and pathologic criteria and immunohistochemical detection of mismatch repair gene proteins in patients with suspected hereditary non-polyposis colorectal cancer. Ann Oncology 2011;22:v112.
Meulemans E, Roemen G, Ruland A, Peeters J, Van Engeland M, De Bruine A, et al. MLH1 promoter hypermethylation, BRAF and K-ras mutation analysis on tumours suspected from Lynch syndrome to prioritize mismatch repair gene testing. EJC Suppl 2008;6:107–8.
Nagasaka T, Koi M, Kloor M, Gebert J, Vilkin A, Nishida N, et al. Mutations in both KRAS and BRAF may contribute to the methylator phenotype in colon cancer. Gastroenterology 2008;134:1950–60.
Nakagawa H, Nagasaka T, Cullings HM, Notohara K, Hoshijima N, Young J, et al. Efficient molecular screening of Lynch syndrome by specific 3’ promoter methylation of the MLH1 or BRAF mutation in colorectal cancer with high-frequency microsatellite instability. Oncol Rep 2009;21:1577–83.
Paya A, Alenda C, Perez-Carbonell L, Rojas E, Soto JL, Guillen C, et al. Utility of p16 immunohistochemistry for the identification of Lynch syndrome. Clin Cancer Res 2009;15:3156–62.
Perez-Carbonell L, Alenda C, Paya A, Castillejo A, Barbera VM, Guillen C, et al. Methylation analysis of MLH1 improves the selection of patients for genetic testing in Lynch syndrome. J Mol Diagn 2010;12:498–504.
Pichler M, Balic M, Stadelmeyer E, Ausch C, Wild M, Guelly C, et al. Evaluation of high-resolution melting analysis as a diagnostic tool to detect the BRAF V600E mutation in colorectal tumors. J Mol Diagn 2009;11:140–7.
Pinol V, Castells A, Andreu M, Castellvi-Bel S, Alenda C, Llor X, et al. Accuracy of revised Bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. JAMA 2005;293:1986–94.
Rahner N, Friedrichs N, Steinke V, Aretz S, Friedl W, Buettner R, et al. Coexisting somatic promoter hypermethylation and pathogenic MLH1 germline mutation in Lynch syndrome. J Pathol 2008;214:10–16.
Theoleyre S, Blayau M, Dugast C, Rioux-Leclercq N, Denis M. Methylation specific multiplex ligation-dependent probe amplification (MS-MLPA): an efficient assay for hMLH1 methylation detection in colorectal cancer. EJC Suppl 2010;8:178.
Valentini AM, Armentano R, Pirrelli M, Gentile M, Caruso ML. Immunohistochemical mismatch repair proteins expression in colorectal cancer. Appl Immunohistochem Mol Morphol 2006;14:42–5.
Xicola RM, Llor X, Pons E, Castells A, Alenda C, Pihol V, et al. Performance of different microsatellite marker panels for detection of mismatch repair-deficient colorectal tumors. J Natl Cancer Inst 2007;99:244–52.
Yearsley M, Hampel H, Lehman A, Nakagawa H, de la Chapelle A, Frankel WL. Histologic features distinguish microsatellite-high from microsatellite-low and microsatellite-stable colorectal carcinomas, but do not differentiate germline mutations from methylation of the MLH1 promoter. Hum Pathol 2006;37:831–8.
Zhang D, Wang Y, Bai Y, Ge Q, Qiao Y, Luo J, et al. A novel method to quantify local CpG methylation density by regional methylation elongation assay on microarray. BMC Genomics 2008;9:59.
Zhou HH, Yan SY, Zhou XY, Du X, Zhang TM, Cai X, et al. MLH1 promoter germline-methylation in selected probands of Chinese hereditary non-polyposis colorectal cancer families. World J Gastroenterol 2008;14:7329–34.
Reason for exclusion: comparator
Bartley AN, Luthra R, Saraiya DS, Urbauer DL, Broaddus RR. Identification of cancer patients with Lynch syndrome: clinically significant discordances and problems in tissue-based mismatch repair testing. Cancer Prev Res 2012;5:320–7.
Bettstetter M, Dechant S, Ruemmele P, Vogel C, Kurz K, Morak M, et al. MethyQESD, a robust and fast method for quantitative methylation analyses in HNPCC diagnostics using formalin-fixed and paraffin-embedded tissue samples. Lab Invest 2008;88:1367–75.
Pavicic W, Perkio E, Kaur S, Peltomaki P. Altered methylation at microRNA-associated CpG islands in hereditary and sporadic carcinomas: a methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA)-based approach. Mol Med 2011;17:726–35.
Perez-Cabornero L, Velasco E, Infante M, Sanz D, Lastra E, Hernandez L, et al. A new strategy to screen MMR genes in Lynch syndrome: HA-CAE, MLPA and RT-PCR. Eur J Cancer 2009;45:1485–93.
Reason for exclusion: outcomes
Alemayehu A, Sebova K, Fridrichova I. Redundant DNA methylation in colorectal cancers of Lynch-syndrome patients. Genes Chromosomes Cancer 2008;47:906–14.
Alenda C, Paya A, Perez L, Alcaraz E, Soto JL, Guillen C, et al. Usefulness of p16 immunohistochemistry in the diagnosis of Lynch’s syndrome. Mod Pathol 2009;22:122A–3A.
Ferreira S, Claro I, Lage P, Filipe B, Fonseca R, Sousa R, et al. Colorectal adenomas in young patients: microsatellite instability is not a useful marker to detect new cases of Lynch syndrome. Dis Colon Rectum 2008;51:909–15.
Guarinos C, Castillejo A, Barbera VM, Perez-Carbonell L, Sanchez-Heras AB, Segura A, et al. EPCAM germ line deletions as causes of Lynch syndrome in Spanish patients. J Mol Diagn 2010;12:765–70.
Hernandez-Losa J, Landolfi S, Cuatrecasas M, Balmana J, Solsona A, Osteso T, et al. Molecular screening in patients with clinical criteria of Lynch syndrome by combination of immunohistochemistry and molecular biology. Virchows Arch 2007;451:276.
Kloor M, Voigt AY, Schackert HK, Schirmacher P, Doeberitz MV, Blaker H. Analysis of EPCAM protein expression in diagnostics of Lynch syndrome. J Clin Oncol 2011;29:223–7.
Reason for exclusion: study design
Chu HT, Chen SN, Louis TA. Random effects models in a meta-analysis of the accuracy of two diagnostic tests without a gold standard. J Am Stat Assoc 2009;104:512–23.
Danish Centre for Evaluation and Health Technology Assessment. Hereditary Nonpolyposis Colorectal Cancer in Denmark – A Health Technology Assessment. Structured abstract. Copenhagen: Danish Centre for Evaluation and Health Technology Assessment; 2007.
Reason for exclusion: within study
Baudhuin LM, Ferber MJ, Winters JL, Steenblock KJ, Swanson RL, French AJ, et al. Characterization of hMLH1 and hMSH2 gene dosage alterations in Lynch syndrome patients. Gastroenterology 2005;129:846–54.
Baudhuin LM, Mai M, French AJ, Kruckeberg KE, Swanson RL, Winters JL, et al. Analysis of hMLH1 and hMSH2 gene dosage alterations in hereditary nonpolyposis colorectal cancer patients by novel methods. J Mol Diagn 2005;7:226–35.
Bujalkova M, Zavodna K, Krivulcik T, Ilencikova D, Wolf B, Kovac M, et al. Multiplex SNaPshot genotyping for detecting loss of heterozygosity in the mismatch-repair genes MLH1 and MSH2 in microsatellite-unstable tumors. Clin Chem 2008;54:1844–54.
Casey G, Lindor NM, Papadopoulos N, Thibodeau SN, Moskow J, Steelman S, et al. Conversion analysis for mutation detection in MLH1 and MSH2 in patients with colorectal cancer. J Am Med Assoc 2005;293:799–809.
Dymerska D, Serrano-Fernandez P, Suchy J, Plawski A, Slomski R, Kaklewski K, et al. Combined iPLEX and TaqMan assays to screen for 45 common mutations in Lynch syndrome and FAP patients. J Mol Diagn 2010;12:82–90.
Hansen TP, Nielsen O, Fenger C. Optimization of antibodies for detection of the mismatch repair proteins MLH1, MSH2, MSH6, and PMS2 using a biotin-free visualization system. Appl Immunohistochem Mol Morphol 2006;14:115–21.
McVety S, Li L, Thiffault I, Gordon P, MacNamara E, Wong N, et al. The value of multi-modal gene screening in HNPCC in Quebec: three mutations in mismatch repair genes that would have not been correctly identified by genomic DNA sequencing alone. Fam Cancer 2006;5:21–8.
Rouleau E, Lefol C, Bourdon V, Coulet F, Noguchi T, Soubrier F, et al. Quantitative PCR high-resolution melting (qPCR-HRM) curve analysis, a new approach to simultaneously screen point mutations and large rearrangements: application to MLH1 germline mutations in Lynch syndrome. Hum Mutat 2009;30:867–75.
Reason for exclusion: other
Frattini M, Molinari F, Perrone F, Lampis A, Sala P, Bassi C, et al. BRAF mutations in hereditary non-polyposis colorectal cancer and Bethesda criteria patients: a pilot study. Ann Oncol 2008;19:I20.
Marginean F, Landolfi S, Hernandez J, de Torres I, Garrido M, Badia D, et al. High feasibility of hMLH1, hMSH2 and hMSH6 protein expression and microsatellite instability analysis (pentaplex system) to screen patients with clinical criteria of Lynch syndrome. Virchows Arch 2008;452:S193–4.
Appendix 4 Literature search strategy for the cost-effectiveness systematic review
This initial search was filtered to capture cost-effectiveness and data on original models, as well as information on guidelines for the review population. With the cost data in mind, the searches were run in the following databases: MEDLINE, MEDLINE In-Process & Other Non-Indexed Citations, EMBASE, CINAHL, The Cochrane Library (all), Web of Science (including conference proceedings) and EconLit. 279,280 The filters used were the NHS EED search filter supplemented by the NHS Quality Improvement filter (brief). 281
The above databases were also searched for guidelines information, as were the British Nursing Index (BNI) and HMIC; however, the latter returned nil results.
Searches were run on Wednesday, 29 February 2012 and returned 2036 title and abstracts.
| Database | Hits |
|---|---|
| 1. MEDLINE | 1061 |
| 2. MEDLINE In-Process & Other Non-Indexed Citations | 51 |
| 3. EMBASE | 1493 |
| 4. CINAHL | 38 |
| 5. The Cochrane Library (all) | 102 |
| 6. Web of Science | 634 |
| 7. EconLit | 1 |
| Total | 3380 |
| Duplicates removed | −1344 |
| Total to screen | 2036 |
1. MEDLINE
Host: Ovid.
Data parameters: 1946 to February week 3, 2012.
Date searched: Wednesday, 29 February 2012.
Searcher: Cooper.
Hits: 1061.
Search strategy
-
(lynch$ adj3 syndrome).ti,ab.
-
((lynch$ adj3 famil$) and (cancer$ or neoplasm$)).ti,ab.
-
Or/1-2
-
Colorectal Neoplasms, Hereditary Nonpolyposis/
-
((Hereditary Nonpolyposis Colorectal Cancer) or (Hereditary Non-polyposis Colorectal Cancer)).tw.
-
HNPCC.tw.
-
((hereditary or inherited) adj3 (colon or colorectal) and (cancer or neoplasm$)).ti,ab.
-
((hereditary adj3 nonpolyposis) and (colon$ or colorectal$)).ti,ab.
-
((hereditary adj3 non-polyposis) and (colon$ or colorectal$)).ti,ab.
-
((hereditary adj3 (cancer or neoplasm)) and (colon or colorectal)).ti,ab.
-
((Familial adj3 Nonpolyposis) and (colon$ or colorectal$)).ti,ab.
-
((Familial adj3 Non-polyposis) and (colon$ or colorectal$)).ti,ab.
-
(familial adj3 (colon$ or colorectal$)).ti,ab.
-
Or/4-13
-
(((MLH1) or (MSH2) or (MSH3) or (MSH6) or (hMSH2) or (hMLH1) or (hPMS1) or (hPMS2) or (hMSH6) or (hMLH3) or (PMS1) or (PMS2)) and (colon$ or colorectal or lynch$ or HNPCC or hereditary)).ti,ab.
-
(Amsterdam criteria).tw.
-
Or/15-16
-
3 OR 14 OR 17
-
exp Economics/
-
ec.fs.
-
economics, medical/
-
economics, nursing/
-
economics, pharmaceutical/
-
exp "economics, hospital"/
-
exp pharmacoeconomics/
-
(economic$ or cost or costs or costly or costing or costed or price or prices or pricing or priced or discount or discounts or discounted or discounting or ration$ or expenditure or expenditures or budget$ or afford$ or pharmacoeconomic or pharmaco-economic$).ti,ab.
-
(cba or cea or cua).ti,ab.
-
exp "fees and charges"/
-
(fee or fees or charge$ or preference$).tw.
-
(fiscal or funding or financial or finance).tw.
-
exp "costs and cost analysis"/
-
exp Health Care Costs/
-
(cost$).ti,ab.
-
(cost$ adj1 (util$ or effective$ or efficac$ or benefit$ or consequence$ or analy$ or minimi$ or saving$ or breakdown or lowering or estimate$ or variable$ or allocation or control or illness or sharing or life or lives or affordabl$ or instrument$ or technolog$ or day$ or fee or fees or charge or charges)).ti,ab.
-
((value or values or valuation) adj3 (money or monetary or life or lives or costs or cost$)).ti,ab.
-
(unit cost or unit-cost or unit-costs or unit costs or drug cost or drug costs or hospital costs or health-care costs or health care cost or medical cost or medical costs).ti,ab.
-
exp decision support techniques/
-
exp models, economic/
-
exp Statistical Model/
-
(markov$).ti,ab. or markov chains/
-
(monte carlo).ti,ab. or monte carlo method/
-
(decision adj2 (tree$ or analy$ or model$)).ti,ab. or decision tree/
-
(survival adj3 analys$).ti,ab.
-
"deductibles and coinsurance"/
-
exp Health expenditures/
-
(uncertain$).ti,ab. or uncertainty/
-
(quality adj3 life).ti,ab. or quality of life/
-
(value adj3 life).ti,ab. or value of life/
-
Quality-adjusted life years/
-
(qol$ or qoly or qolys or hrqol$ or qaly or qalys or qale or qales).ti,ab.
-
((sensitivity analys$) or ("willingness to pay") or (quality-adjusted life year$) or (quality adjusted life year$) or (quality-adjusted life expectanc$) or (quality adjusted life expectanc$)).ti,ab.
-
utilit$.tw.
-
(valu$).tw.
-
exp hospitalization/
-
Or/19-54
-
Guidelines as Topic/
-
Health Planning Guidelines/
-
Practice Guidelines as Topic/
-
Clinical Protocols/
-
Guideline.pt.
-
Practice Guideline/
-
Practice Guideline.pt.
-
Consensus Development Conference.pt.
-
(guideline* or standards).ti.
-
(expert consensus or consensus statement or consensus conference* or practice parameter* or position statement* or policy statement* or cpg or cpgs or best practice*).ti,ab
-
Or/56-65
-
55 or 66
-
18 and 67
-
Animals/ NOT Humans/
-
69 NOT 70
-
limit 70 to english language
2. MEDLINE In-Process & Other Non-Indexed Citations
Host: Ovid.
Data parameters: added on or before 28 February 2012.
Date searched: Wednesday, 29 February 2012.
Searcher: Cooper.
Hits: 51.
Search strategy
-
(lynch$ adj3 syndrome).ti,ab.
-
((lynch$ adj3 famil$) and (cancer$ or neoplasm$)).ti,ab.
-
Or/1-2
-
Colorectal Neoplasms, Hereditary Nonpolyposis/
-
((Hereditary Nonpolyposis Colorectal Cancer) or (Hereditary Non-polyposis Colorectal Cancer)).tw.
-
HNPCC.tw.
-
((hereditary or inherited) adj3 (colon or colorectal) and (cancer or neoplasm$)).ti,ab.
-
((hereditary adj3 nonpolyposis) and (colon$ or colorectal$)).ti,ab.
-
((hereditary adj3 non-polyposis) and (colon$ or colorectal$)).ti,ab.
-
((hereditary adj3 (cancer or neoplasm)) and (colon or colorectal)).ti,ab.
-
((Familial adj3 Nonpolyposis) and (colon$ or colorectal$)).ti,ab.
-
((Familial adj3 Non-polyposis) and (colon$ or colorectal$)).ti,ab.
-
(familial adj3 (colon$ or colorectal$)).ti,ab.
-
Or/4-13
-
(((MLH1) or (MSH2) or (MSH3) or (MSH6) or (hMSH2) or (hMLH1) or (hPMS1) or (hPMS2) or (hMSH6) or (hMLH3) or (PMS1) or (PMS2)) and (colon$ or colorectal or lynch$ or HNPCC or hereditary)).ti,ab.
-
(Amsterdam criteria).tw.
-
Or/15-16
-
3 OR 14 OR 17
-
exp Economics/
-
ec.fs.
-
economics, medical/
-
economics, nursing/
-
economics, pharmaceutical/
-
exp "economics, hospital"/
-
exp pharmacoeconomics/
-
(economic$ or cost or costs or costly or costing or costed or price or prices or pricing or priced or discount or discounts or discounted or discounting or ration$ or expenditure or expenditures or budget$ or afford$ or pharmacoeconomic or pharmaco-economic$).ti,ab.
-
(cba or cea or cua).ti,ab.
-
exp "fees and charges"/
-
(fee or fees or charge$ or preference$).tw.
-
(fiscal or funding or financial or finance).tw.
-
exp "costs and cost analysis"/
-
exp Health Care Costs/
-
(cost$).ti,ab.
-
(cost$ adj1 (util$ or effective$ or efficac$ or benefit$ or consequence$ or analy$ or minimi$ or saving$ or breakdown or lowering or estimate$ or variable$ or allocation or control or illness or sharing or life or lives or affordabl$ or instrument$ or technolog$ or day$ or fee or fees or charge or charges)).ti,ab.
-
((value or values or valuation) adj3 (money or monetary or life or lives or costs or cost$)).ti,ab.
-
(unit cost or unit-cost or unit-costs or unit costs or drug cost or drug costs or hospital costs or health-care costs or health care cost or medical cost or medical costs).ti,ab.
-
exp decision support techniques/
-
exp models, economic/
-
exp Statistical Model/
-
(markov$).ti,ab. or markov chains/
-
(monte carlo).ti,ab. or monte carlo method/
-
(decision adj2 (tree$ or analy$ or model$)).ti,ab. or decision tree/
-
(survival adj3 analys$).ti,ab.
-
"deductibles and coinsurance"/
-
exp Health expenditures/
-
(uncertain$).ti,ab. or uncertainty/
-
(quality adj3 life).ti,ab. or quality of life/
-
(value adj3 life).ti,ab. or value of life/
-
Quality-adjusted life years/
-
(qol$ or qoly or qolys or hrqol$ or qaly or qalys or qale or qales).ti,ab.
-
((sensitivity analys$) or ("willingness to pay") or (quality-adjusted life year$) or (quality adjusted life year$) or (quality-adjusted life expectanc$) or (quality adjusted life expectanc$)).ti,ab.
-
utilit$.tw.
-
(valu$).tw.
-
exp hospitalization/
-
Or/19-54
-
Guidelines as Topic/
-
Health Planning Guidelines/
-
Practice Guidelines as Topic/
-
Clinical Protocols/
-
Guideline.pt.
-
Practice Guideline/
-
Practice Guideline.pt.
-
Consensus Development Conference.pt.
-
(guideline* or standards).ti.
-
(expert consensus or consensus statement or consensus conference* or practice parameter* or position statement* or policy statement* or cpg or cpgs or best practice*).ti,ab
-
Or/56-65
-
55 or 66
-
18 and 67
-
Animals/ NOT Humans/
-
69 NOT 70
-
limit 70 to english language
3. EMBASE
Host: Ovid.
Data parameters: 1980 to week 8, 2012.
Date searched: Wednesday, 29 February 2012.
Searcher: Cooper.
Hits: 1493.
Search strategy
-
limit 70 to engli(lynch$ adj3 syndrome).ti,ab.
-
((lynch$ adj3 famil$) and (cancer$ or neoplasm$)).ti,ab.
-
Or/1-2
-
Colorectal Neoplasms, Hereditary Nonpolyposis/
-
((Hereditary Nonpolyposis Colorectal Cancer) or (Hereditary Non-polyposis Colorectal Cancer)).tw.
-
HNPCC.tw.
-
((hereditary or inherited) adj3 (colon or colorectal) and (cancer or neoplasm$)).ti,ab.
-
((hereditary adj3 nonpolyposis) and (colon$ or colorectal$)).ti,ab.
-
((hereditary adj3 non-polyposis) and (colon$ or colorectal$)).ti,ab.
-
((hereditary adj3 (cancer or neoplasm)) and (colon or colorectal)).ti,ab.
-
((Familial adj3 Nonpolyposis) and (colon$ or colorectal$)).ti,ab.
-
((Familial adj3 Non-polyposis) and (colon$ or colorectal$)).ti,ab.
-
(familial adj3 (colon$ or colorectal$)).ti,ab.
-
Or/4-13
-
(((MLH1) or (MSH2) or (MSH3) or (MSH6) or (hMSH2) or (hMLH1) or (hPMS1) or (hPMS2) or (hMSH6) or (hMLH3) or (PMS1) or (PMS2)) and (colon$ or colorectal or lynch$ or HNPCC or hereditary)).ti,ab.
-
(Amsterdam criteria).tw.
-
Or/15-16
-
3 OR 14 OR 17
-
exp Economics/
-
ec.fs.
-
economics, medical/
-
economics, nursing/
-
economics, pharmaceutical/
-
exp "economics, hospital"/
-
exp pharmacoeconomics/
-
(economic$ or cost or costs or costly or costing or costed or price or prices or pricing or priced or discount or discounts or discounted or discounting or ration$ or expenditure or expenditures or budget$ or afford$ or pharmacoeconomic or pharmaco-economic$).ti,ab.
-
(cba or cea or cua).ti,ab.
-
exp "fees and charges"/
-
(fee or fees or charge$ or preference$).tw.
-
(fiscal or funding or financial or finance).tw.
-
exp "costs and cost analysis"/
-
exp Health Care Costs/
-
(cost$).ti,ab.
-
(cost$ adj1 (util$ or effective$ or efficac$ or benefit$ or consequence$ or analy$ or minimi$ or saving$ or breakdown or lowering or estimate$ or variable$ or allocation or control or illness or sharing or life or lives or affordabl$ or instrument$ or technolog$ or day$ or fee or fees or charge or charges)).ti,ab.
-
((value or values or valuation) adj3 (money or monetary or life or lives or costs or cost$)).ti,ab.
-
(unit cost or unit-cost or unit-costs or unit costs or drug cost or drug costs or hospital costs or health-care costs or health care cost or medical cost or medical costs).ti,ab.
-
exp decision support techniques/
-
exp models, economic/
-
exp Statistical Model/
-
(markov$).ti,ab. or markov chains/
-
(monte carlo).ti,ab. or monte carlo method/
-
(decision adj2 (tree$ or analy$ or model$)).ti,ab. or decision tree/
-
(survival adj3 analys$).ti,ab.
-
"deductibles and coinsurance"/
-
exp Health expenditures/
-
(uncertain$).ti,ab. or uncertainty/
-
(quality adj3 life).ti,ab. or quality of life/
-
(value adj3 life).ti,ab. or value of life/
-
Quality-adjusted life years/
-
(qol$ or qoly or qolys or hrqol$ or qaly or qalys or qale or qales).ti,ab.
-
((sensitivity analys$) or ("willingness to pay") or (quality-adjusted life year$) or (quality adjusted life year$) or (quality-adjusted life expectanc$) or (quality adjusted life expectanc$)).ti,ab.
-
utilit$.tw.
-
(valu$).tw.
-
exp hospitalization/
-
Or/19-54
-
Guidelines as Topic/
-
Health Planning Guidelines/
-
Practice Guidelines as Topic/
-
Clinical Protocols/
-
Guideline.pt.
-
Practice Guideline/
-
Practice Guideline.pt.
-
Consensus Development Conference.pt.
-
(guideline* or standards).ti.
-
(expert consensus or consensus statement or consensus conference* or practice parameter* or position statement* or policy statement* or cpg or cpgs or best practice*).ti,ab
-
Or/56-65
-
55 or 66
-
18 and 67
-
Animals/ NOT Humans/
-
69 NOT 70
-
sh language
4. Cumulative Index to Nursing and Allied Health Literature
Host: EBSCOhost.
Data parameters:
Date searched: 28 February 2012.
Searcher: Cooper.
Hits: 38.
Search strategy
S1. (lynch* N3 syndrome)
S2. ((lynch* N3 famil*) and (cancer* or neoplasm*))
S3. (S1 OR S2)
S4. (MH "Colorectal Neoplasms, Hereditary Nonpolyposis")
S5. ((Hereditary Nonpolyposis Colorectal Cancer) or (Hereditary Non-polyposis Colorectal Cancer))
S6. (HNPCC)
S7. ((hereditary or inherited) N3 (colon or colorectal) and (cancer or neoplasm*))
S8. ((hereditary N3 nonpolyposis) and (colon* or colorectal*))
S9. ((hereditary N3 non-polyposis) and (colon* or colorectal*))
S10. ((hereditary N3 (cancer or neoplasm)) and (colon or colorectal))
S11. ((Familial N3 Nonpolyposis) and (colon* or colorectal*))
S12. ((Familial N3 Non-polyposis) and (colon* or colorectal*))
S13. (familial N3 (colon* or colorectal*))
S14. (S4 OR S5 OR S6 OR S7 OR S8 OR S9 OR S10 OR S11 OR S12 OR S13)
S15. (((MLH1) or (MSH2) or (MSH3) or (MSH6) or (hMSH2) or (hMLH1) or (hPMS1) or (hPMS2) or (hMSH6) or (hMLH3) or (PMS1) or (PMS2)) and (colon* or colorectal or lynch* or HNPCC or hereditary))
S16. ((Amsterdam criteria) and (colon* or colorectal or lynch* or HNPCC or hereditary))
S17. (S15 OR S16)
S18. (S3 OR S14 OR S17)
S19. (MH "Economics+")
S20. (economic* or cost or costs or costly or costing or costed or price or prices or pricing or priced or discount or discounts or discounted or discounting or ration* or expenditure or expenditures or budget* or afford* or pharmacoeconomic or pharmaco-economic*)
S21. (cba or cea or cua)
S22. (MH "Fees and Charges")
S23. (fee or fees or charge* or preference*)
S24. (fiscal or funding or financial or finance)
S25. (MH "Costs and Cost Analysis+")
S26. (cost*)
S27. (cost* N1 (util* or effective* or efficac* or benefit* or consequence* or analy* or minimi* or saving* or breakdown or lowering or estimate* or variable* or allocation or control or illness or sharing or life or lives or affordabl* or instrument* or technolog* or day* or fee or fees or charge or charges))
S28. ((value or values or valuation) N3 (money or monetary or life or lives or costs or cost*))
S29. (unit cost or unit-cost or unit-costs or unit costs or drug cost or drug costs or hospital costs or health-care costs or health care cost or medical cost or medical costs)
S30. (MH "Decision Support Techniques+")
S31. (model*)
S32. (MM "Models, Statistical")
S33. (markov*)
S34. (monte carlo)
S35. (decision N2 (tree* or analy* or model*))
S36. (MH "Decision Trees")
S37. (survival N3 analys*)
S38. (uncertain*)
S39. (quality N3 life)
S40. (MH "Quality of Life")
S41. (value N3 life)
S42. (MM "Quality-Adjusted Life Years")
S43. (qol* or qoly or qolys or hrqol* or qaly or qalys or qale or qales)
S44. ((sensitivity analys*) or ("willingness to pay") or (quality-adjusted life year*) or (quality adjusted life year*) or (quality-adjusted life expectanc*) or (quality adjusted life expectanc*))
S45. utilit*
S46. (valu*)
S47. S20 or S21 or S22 or S23 or S24 or S25 or S26 or S27 or S28 or S29 or S30 or S31 or S32 or S33 or S34 or S35 or S36 or S37 or S38 or S39 or S40 or S41 or S42 or S43 or S44 or S45 or S46 or S47 or S48 or S49
S48. (MM "Practice Guidelines")
S49. (guideline* or standards)
S50. (expert consensus or consensus statement or consensus conference* or practice parameter* or position statement* or policy statement* or cpg or cpgs or best practice*)
S51. S51 or S52 or S53
S52. 55 or 66
5. The Cochrane Library
Host: www.thecochranelibrary.com/view/0/index.html
Data parameters:
Date searched: 28 February 2012.
Searcher: Cooper.
Hits: 102.
Search strategy
#1. (lynch* NEAR/3 syndrome):ti,ab,kw
#2. ((lynch* NEAR/3 famil*) and (cancer* or neoplasm*)):ti,ab,kw
#3. (#1 OR #2)
#4. MeSH descriptor Colorectal Neoplasms, Hereditary Nonpolyposis, this term only
#5. ((Hereditary Nonpolyposis Colorectal Cancer) or (Hereditary Non-polyposis Colorectal Cancer)):ti,ab,kw
#6. (HNPCC):ti,ab,kw
#7. ((hereditary or inherited) NEAR/3 (colon or colorectal) and (cancer or neoplasm*)):ti,ab,kw
#8. ((hereditary NEAR/3 nonpolyposis) and (colon* or colorectal*)):ti,ab,kw
#9. ((hereditary NEAR/3 non-polyposis) and (colon* or colorectal*)):ti,ab,kw
#10. ((hereditary NEAR/3 (cancer or neoplasm)) and (colon or colorectal)):ti,ab,kw
#11. ((Familial NEAR/3 Nonpolyposis) and (colon* or colorectal*)):ti,ab,kw
#12. ((Familial NEAR/3 Non-polyposis) and (colon* or colorectal*)):ti,ab,kw
#13. (familial NEAR/3 (colon* or colorectal*)):ti,ab,kw
#14. (#4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10 OR #11 OR #12 OR #13)
#15. (((MLH1) or (MSH2) or (MSH3) or (MSH6) or (hMSH2) or (hMLH1) or (hPMS1) or (hPMS2) or (hMSH6) or (hMLH3) or (PMS1) or (PMS2)) and (colon* or colorectal or lynch* or HNPCC or hereditary)):ti,ab,kw
#16. ((Amsterdam criteria) and (colon* or colorectal or lynch* or HNPCC or hereditary)):ti,ab,kw
#17. (#15 OR #16)
#18. (#3 OR #14 OR #17)
Notes: line 16 was lost to specificity and so its sensitivity was tightened to the population for this review.
6. Web of Science
Host: ISI.
Data parameters: 1899 to current.
Date searched: Wednesday, 29 February 2012.
Searcher: Cooper.
Hits: 634.
Search strategy
-
Topic=("Hereditary Nonpolyposis Colorectal Cancer")
-
Topic=((lynch* near/3 syndrome))
-
#1 OR #2
-
(economic* or cost or costs or costly or costing or costed or price or prices or pricing or priced or discount or discounts or discounted or discounting or ration* or expenditure or expenditures or budget* or afford* or pharmacoeconomic or pharmaco-economic*)
-
(markov* or monte carlo)
-
(decision near/2 (tree* or analy* or model*))
-
(survival near/3 analys*)
-
(qol* or qoly or qolys or hrqol* or qaly or qalys or qale or qales)
-
((sensitivity analys*) or ("willingness to pay") or (quality-adjusted life year*) or (quality adjusted life year*) or (quality-adjusted life expectanc*) or (quality adjusted life expectanc*))
-
utilit*
-
(valu*)
-
(guideline* or standards)
-
#1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10 OT #11 OR #12
-
#3 AND 13
-
Refined by: Languages=( ENGLISH )
7. EconLit
Host: CSA.
Data parameters:
Date searched: 28 February 2012.
Searcher: Cooper.
Hits: 1.
Search strategy
-
(KW=("Hereditary Nonpolyposis Colorectal Cancer"))
-
KW=(lynch* syndrome)
-
((KW=(hereditary) or KW=(inherited)) and (KW=(colon) or KW=(colorectal)) and (KW=(cancer) or KW=(neoplasm*)))
-
(((KW=(MLH1)) or (KW=(MSH2)) or (KW=(MSH3)) or (KW=(MSH6)) or (KW=(hMSH2)) or (KW=(hMLH1)) or (KW=(hPMS1)) or (KW=(hPMS2)) or (KW=(hMSH6)) or (KW=(hMLH3)) or (KW=(PMS1)) or (KW=(PMS2))) and (KW=(colon*) or KW=(colorectal) or KW=("lynch$") or KW=(HNPCC) or KW=(hereditary)))
-
(KW=(Amsterdam criteria))
Appendix 5 Published estimates of cumulative risk of colorectal cancer for individuals with Lynch syndrome
| Study | Number of families | Selection | Correction for selection/ascertainment bias | Adjustment for interventions | MMR genes included | Cumulative risk of CRC to age 70 years, % (95% CI) |
|---|---|---|---|---|---|---|
| Bonadona et al. 20112 | 537 | 40 cancer genetics clinics | Adjustment for ascertainment within genotype-restricted likelihood mechanism | Risks for CRC censored on first colonoscopy | MLH1 (n = 248), MSH2 (n = 256), MSH6 (n = 33) | MLH1/MSH2/MSH6 M 38 (25 to 59) F 31 (19 to 50) P 35 (25 to 49) MLH1 P 41 (25 to 70) MSH2 P 48 (30 to 77) MSH6 P 12 (8 to 22) |
| van Vliet et al. 2011164 | 17 | Population-based prospective recruitment via early-onset (< 45 years) CRC cases | Adjustment for ascertainment (conditioned on phenotype and genotype of proband) | Censored at first cancer, polypectomy or hysterectomy | MLH1 (n = 9), MSH2 (n = 4), MSH6 (n = 3), PMS2 (n = 1) | Paternal origin M 41 (20 to 71) F 42 (18 to 78) Maternal origin M 81 (49 to 99) F 34 (17 to 63) |
| Baglietto et al. 2010166 | 113 | Family cancer clinics (n = 65) Population-based cancer registries (n = 48) |
Adjustment for ascertainment | Censored at polypectomy or hysterectomy | MSH6 (n = 113) | M 22 (14 to 32) F 10 (5 to 17) |
| Choi et al. 2009282 | 32 | Population-based cancer registry [AC I (n = 27), FH or early-onset CRC (n = 5)] | (Via Bonadona et al. 2011) Incomplete adjustment (conditioned on phenotype of proband and FDRs) |
Not stated | MLH1 (n = 14), MSH2 (n = 17), MSH6 (n = 1) | MLH1/MSH2/MSH6 M 60 (35 to 73) F 47 (27 to 60) P 53 (37 to 64) MLH1 M 67 (27 to 89) F 35 (10 to 59) P 44 (19 to 70) MSH2 M 55 (2 to 75) F 53 (2 to 70) P 54 (3 to 69) |
| Stoffel et al. 2009106 | 147 | Cancer genetics clinic | Base analysis Incomplete adjustment (conditioned on genotype and phenotype of proband and phenotype of FDRs) Most conservative analysisa Adjustment for ascertainment (conditioned on genotype and phenotype of proband and phenotype of all relatives) |
Not censored on polypectomy | MLH1 (n = 55), MSH2 (n = 81), MSH6 (n = 11) | Base analysis MLH1/MSH2/MSH6 M 66 (59 to 76) F 43 (37 to 53) MLH1 M 97 F 53 MSH2 M 52 F 39 Most conservative analysisa MLH1/MSH2/MSH6 M 34 (3 to 54) F 32 (0 to 38) |
| Barrow et al. 200881 | 121 | Cancer genetics clinic | (Via Bonadona et al. 2011) Inappropriate |
Base analysis not censored Sensitivity analysis censored on family ascertainment |
MLH1 (n = 51), MSH2 (n = 59), MSH6 (n = 11) | Base analysis MLH1/MSH2/MSH6 M 54 (51 to 58) F 46 (43 to 50) Sensitivity analysis MLH1/MSH2/MSH6 M 56 (52 to 60) F 44 (40 to 47) |
| Senter et al. 2008105 | 55 | Cancer genetics clinics | Adjustment for ascertainment (conditioned on genotype and phenotype of proband and phenotype of all relatives) | NR | PMS2 | PMS2 M 20 (11 to 34) F 15 (8 to 26) |
| Alarcon et al. 2007165 | 36 | Cancer genetics clinics | Adjustment for ascertainment | Not censored | MLH1 (n = 22), MSH2 (n = 14) | MLH1/MSH2 M 47 (12 to 98) F 33 (24 to 54) |
| Jenkins et al. 2006114 | 17 | Population-based cancer registry recruiting families by probands with CRC < 45 years | Adjusted for ascertainment (conditioned on genotype and phenotype of proband) | Censored on polypectomy if not affected by CRC during lifetime | MLH1 (n = 8), MSH2 (n = 4), MSH6 (n = 4), PMS2 (n = 1) | Any MMR M 45 (29 to 62) F 38 (19 to 51) MLH1/MSH2 M 56 (37 to 75) F 48 (26 to 65) |
| Bermejo et al. 2005283 | 5098 | Cancer registry, families with four generations documented and second generation aged up to 70 years [AC I (n = 21), AC II (n = 42), Bethesda (n = 5095)] | N/A? | Not censored on polypectomy | N/A | (To age 75 years) AC I P 57 (46 to 69) AC II P 41 (33 to 50) Bethesda M 42 (39 to 44) F 23 (22 to 24) |
| Hampel et al. 2005284 | 70 | Cohort 1 (n = 45) CRC between 1980 and 1994 with suggestive clinical characteristics and FH Cohort 2 (n = 25) Population-based recruitment (CRC any age, MSI positive) |
Exclusion of probands (sufficient for cohort 2 but not cohort 1 or combined cohort; analysis published is for combined cohort) | Not censored on polypectomy | MLH1 (n = 65), MSH2 (n = 5) | MLH1/MSH2 M 69 F 52 |
| Quehenberger et al. 2005102 | 84 | HNPCC registry | Adjustment for ascertainment (conditioned on genotype of proband and phenotype of relatives) | Censored on polypectomy | MLH1 (n = 39), MSH2 (n = 45) | MLH1/MSH2 M 27 (13 to 51) F 22 (11 to 44) MLH1 M 22 (7 to 61) F 18 (5 to 42) MSH2 M 30 (13 to 57) F 25 (12 to 50) |
| Hendriks et al. 200499 | (Via Bonadona et al. 2011) 20 |
(Via Bonadona et al. 2011) Multiple-case families |
(Via Bonadona et al. 2011) None |
(Via Bonadona et al. 2011) None |
MSH6 | MSH6 M 69 (42 to 83) F 30 (12 to 44) |
| Plaschke et al. 2004285 | 27 | HNPCC registry (AC I/II, Bethesda), selected MSI-H/L and IHC normal for MLH1 and MSH2 | None | None | MSH6 | (From figure 1) MSH6 P ≈ 80 |
| Green et al. 200297 | 12 | Genetics clinic | Inappropriate exclusion | Censored at entry to surveillance programme | MSH2 founder mutation (943 + 3, A > T) | Founder MSH2 M 81 F 59 |
| Vasen et al. 2001286 | 74 | HNPCC registry (multiple-case families) | None | None | MLH1 (n = 34), MSH2 (n = 40) | (From figures 2 and 3) MLH1 M 65 F 54 P 59 MSH2 M 73 F 54 P 64 |
| Wagner et al. 2001104 | 1 | Referral to clinical genetics on FH | None | None | MSH6 | MSH6 P 32 |
| Aarnio et al. 1999118 | 50 | Known mutation carriers | None | NR | MLH1 (n = 47), MSH2 (n = 3) | MLH1/MSH2 P 82 |
| Dunlop et al. 1997101 | 6 | Relatives of early-onset MSI-H CRC cases (< 35 years) | Partial (exclusion of index cases); not conditioned on phenotype of proband | NR | MLH1 (n = 1), MSH2 (n = 5) | MLH1/MSH2 M 74 F 30 |
| Vasen et al. 1996116 | (Via abstract) 19 |
NR in abstract | NR in abstract | NR in abstract | MLH1, MSH2 | MLH1 P 80 MSH2 P 80 |
| Aarnio et al. 199515 | (Via abstract) 40 |
NR in abstract | NR in abstract | NR in abstract | NR in abstract | P 78 |
Appendix 6 Effect of age at diagnosis on colorectal cancer survival
Table 124 shows the net 1- and 5-year survival of CRC for various age groups.
| Years since diagnosis | All ages (%) | Aged < 70 years (%) | Aged 70–79 years (%) | Aged 80+ years (%) |
|---|---|---|---|---|
| 1 | 73.9 | 83.4 | 74.9 | 58.1 |
| 5 | 54.9 | 62.5 | 56.2 | 41.7 |
We estimated HRs for CRC mortality during the first year since diagnosis by comparing the net survival for the relevant age group with net survival across all ages. The HR for persons aged < 70 years was therefore ln(83.4%)/ln(73.9%) = 0.599. Likewise, for persons aged 70–79 years the HR was ln(74.9%)/ln(74.9%) = 0.956, and for persons aged ≥ 80 years the HR was ln(58.1%)/ln(74.9%) = 1.797.
We estimated HRs for CRC mortality during the subsequent 4 years by first calculating the conditional survival to 5 years given survival to 1 year (Table 125), and then similarly calculating HRs by comparing conditional survival for the relevant age group with conditional survival across all ages. The HR for persons aged < 70 years was therefore ln(74.9%)/ln(74.3%) = 0.972. Likewise, for persons aged 70–79 years the HR was ln(75.1%)/ln(74.3%) = 0.966, and for persons aged ≥ 80 years the HR was ln(71.8%)/ln(74.3%) = 1.116.
| Age | All ages | Aged < 70 years | Aged 70–79 years | Aged 80+ years |
|---|---|---|---|---|
| Conditional survival to 5 years given survival to 1 year | 54.9%/73.9% = 74.3% | 62.5%/83.4% = 74.9% | 56.2%/74.9% = 75.1% | 41.7%/58.1% = 71.8% |
Appendix 7 Selected studies comparing survival of individuals with Lynch syndrome colorectal cancer with that of individuals with sporadic colorectal cancer
| Study | Study populations | Adjusted for age | Adjusted for CRC stage | Estimation method | Censoring | Mortality event | HR |
|---|---|---|---|---|---|---|---|
| Watson et al. 1998287 (primary analysis) | HNPCC cohort (n = 274) Retrospectively identified through HNPCC registries, meeting clinical criteria or being known to carry mutation (20 of 98 families), including synchronous CRCs and metachronous CRCs if index CRC unstaged Tumour registry cohort (n = 820) Consecutive CRC cases at a single hospital, excluding metachronous CRCs |
Partial (non-CRC deaths censored in tumour registry cohort who are generally older) | Yes | Cox (proportional hazards) regression | HNPCC cohort Censored at date of last follow-up of family or at 10 years (whichever is earlier) |
HNPCC cohort Death from any cause before last follow-up of family |
0.67 |
| Tumour registry cohort Censored at date of last follow-up if alive or at death from non-CRC cause |
Tumour registry cohort Death from CRC |
||||||
| Watson et al. 1998287 (additional analysis) | As in primary analysis (row above) | Yes | Yes | Cox (proportional hazards) regression | HNPCC cohort Censored at date of last follow-up of family or at 10 years (whichever is earlier) |
HNPCC cohort Death from any cause before last follow-up of family |
0.63 |
| Tumour registry cohort Censored at date of last follow up |
Tumour registry cohort Death from any cause |
||||||
| Sankila et al. 1996288 (Abstract reviewed only) | HNPCC cohort (n = 175) Sporadic CRC cohort (n = 14,000) |
Partially (sporadic CRC cohort limited to age < 65 years) | ? | Relative survival | ? | ? | Overall 5-year survival rate 65% vs. 44% in favour of HNPCC ‘The relative survival rates of patients with HNPCC were better in every strata analyzed’ |
| Bertario et al. 1999289 | HNPCC cohort (n = 140) Identified through HNPCC registries by AC |
Yes | Yes | Cox proportional hazards regression | Not stated (presumed last follow-up according to definition of ‘observed survival’) | Not stated (presumed death from any cause according to definition of ‘observed survival’) | 1.01 (95% CI 0.72 to 1.32) |
| Sporadic CRC cohort (n = 2035) Consecutive CRC cases at a single hospital, excluding metachronous CRCs and patients with FH of gastrointestinal cancer |
|||||||
| Barnetson et al. 2006175 | Unselected CRC patients (n = 870) aged < 55 years. Germline mutation testing was used to split into two cohorts: HNPCC cohort (n = 38) Sporadic CRC cohort (n = 832) |
No (but all diagnosed < 55 years so unlikely to have effect) | Yes | Log-rank test | End of study (none lost to follow-up) | Death from any cause | Not reported, but no significant difference in survival between carriers and non-carriers when stratified by localised/metastatic (p = 0.6) |
| Aarnio et al. 199814 (abstract reviewed only) | HNPCC cohort (n = 43) Sporadic CRC cohort (n = 122) |
? | ? | ? | ? | ? | 0.41 (p = 0.02) |
| Elsakov and Kurtinaitis 2006290 | HNPCC cohort (n = 8) CRC cases from HNPCC families with proven MLH1/MSH2 mutations Sporadic CRC cohort (n = 263) Retrospectively identified cohort, age matched and with stage II or III CRC |
Yes (age matched) | Partial (limit analysis to stage II/III but proportions different) | Kaplan–Meier Wilcoxon |
End of study period | ? | Improved 10-year survival (87.5% vs. 44.8%) (p = 0.03) |
| Fujita et al. 1996291 | HNPCC cohort (n = 14) CRC cases meeting AC Sporadic CRC cohort (n = 1604) CRC cases without FH of cancer |
No | No | Kaplan–Meier Log-rank test |
? | ? | Improved 5-year survival (92.3% vs. 60.0%) (p < 0.05) |
| Haghighi et al. 2009292 | HNPCC cohort (n = 61) CRC patients from HNPCC families (Bethesda guidelines) Sporadic CRC cohort (n = 60) Consecutive CRC cases without FH |
Partly (stratified by age above or below 50 years) | No | Cox proportional hazards regression | ? | ? | 0.341 (95% CI 0.123 to 0.943) |
| Lin et al. 1998176 | HNPCC cohort (n = 75) CRC cases from HNPCC families with MLH1/MSH2 mutations Sporadic CRC cohort (n = 755) Presumed to be the same group as Watson et al. 1998 |
Partial (non-CRC deaths censored in tumour registry cohort who are generally older) | Yes | Cox proportional hazards regression | Sporadic CRC cohort Non-CRC death |
HNPCC cohort Death from any cause Sporadic CRC cohort CRC death |
0.57 |
| Myrhøj et al. 1997293 (abstract reviewed only) | HNPCC cohort (n = 108) Sporadic CRC cohort (n = 870) |
? | ? | ? | ? | ? | Improved 5-year survival (62% vs. 39%) |
| Percesepe et al. 1997294 | HNPCC cohort (n = 85) CRC cases from families meeting AC Sporadic CRC cohort (n = 377) Consecutive CRC cases not in HNPCC cohort |
Yes | Yes | Cox proportional hazards regression | Non-CRC death | CRC death | 0.741 (95% CI 0.442 to 1.266) |
| Stigliano et al. 2008295 | HNPCC cohort (n = 40) Patients meeting AC I Sporadic CRC cohort (n = 573) Consecutive CRC cases without FH |
No | Yes | Cox proportional hazards regression | Not stated | Any death | HNPCC survival superior (p < 0.0001) |
| You et al. 2006296 | HNPCC cohort (n = 56) CRC cases meeting AC Sporadic CRC cohort (n = 147) CRC cases without FH, age-, sex- and CRC-site matched to HNPCC cohort |
N/A (age matched) | Yes | Cox proportional hazards regression | Loss to follow-up Non-CRC death |
CRC death | No difference (p = 0.774) |
| Russo et al. 2009297 | CRC cases (n = 526) from AC families. Germline mutation testing was used to split into two cohorts: Mutation positive (MLH1/MSH2) (n = 258) Mutation negative (n = 268) |
Yes | Yes | Cox proportional hazards regression | Not reported | Not reported | 0.71 (95% CI 0.51 to 0.98) |
Appendix 8 Analysis of data from Ian Frayling
Dr Ian Frayling MA PhD FRCPath FEBLM, Consultant in Genetic Pathology and Laboratory Director, All Wales Medical Genetics Service
Description of data
Anonymised data were provided by Ian Frayling from the All Wales Medical Genetics Service, including details of predictive tests within 33 families with LS mutations. For each predictive test we were provided with the date when the family mutation was first reported, the date when the predictive test result was reported, the sex of the person receiving the predictive test and whether or not they were diagnosed as a LS carrier. In some cases the data also contained the kinship of the predictive test, i.e. whether the person was a FDR, SDR or more distant relation of the index case.
The data cover reports from October 2000 to November 2012 and include details of 109 predictive tests on members of 33 families. The mean time between diagnostic test and predictive test was 747 days (median 415 days, interquartile range 182–1206 days, range 0–3510 days).
Proportion of relatives who test positive
Of the 109 relatives who received predictive testing, 44 (40%, 95% CI 31% to 50%) tested positive for the family mutation. There is significant evidence to suggest that the true proportion who would test positive is < 0.5 (one-tailed binomial test, p = 0.027), and there are a number of factors which contribute to this, including non-paternity, de novo mutations, mortality bias and offering testing to those at < 50% genetic risk because it is not possible (e.g. owing to death or to testing being declined) or not appropriate to test intervening relatives.
Proportion of relatives who are male
Of the 109 relatives who received predictive testing, 39 (36%, 95% CI 27% to 46%) were male. There is significant evidence to suggest that the true proportion who would be male is not 0.5 (two-tailed binomial test, p = 0.004). Ian Frayling also analysed the sex of the index cases (those receiving diagnostic tests) and found that there were 17 female and 16 male index cases. It is his belief that men are less likely to take up predictive testing than women and we cannot identify any alternative explanations.
Independence of test result and sex
We tested whether or not the test result and sex of the relative were dependent variables by constructing a 2 × 2 table (Table 126) and using a Pearson’s chi-squared test. We found no evidence of dependency (chi-squared = 0.262, degrees of freedom = 1, p = 0.61).
| Sex | Mutation found (n) | Mutation not found (n) |
|---|---|---|
| Male | 17 | 22 |
| Female | 27 | 43 |
Kinship of relatives to proband
For 70 (64%) of the 109 tests, we were provided with the kinship of the relative being tested to the proband. These are summarised in Table 127, which shows that 47% of relatives tested are FDRs while 53% are more distantly related.
| Kinship to proband | Number of predictive tests |
|---|---|
| FDR | 33 |
| SDR | 12 |
| Third- or higher-degree relative | 25 |
Expected number of relatives tested per proband over time
The data provided clearly indicated that relatives are not tested immediately after a positive diagnostic test. It is important to know the expected number of relatives that would be tested for each proband over time as this has an effect on workload and the overall effectiveness of testing (if all relatives waited decades before being tested for the family mutation, most of the potential benefit of testing would have been forgone).
We analysed the number of relatives tested per proband by using a Kaplan–Meier-style estimator. We assumed that all families were censored at the date of the most recent database search (an effective ‘study end’ date of 1 November 2012).
If di is the number of predictive tests at time ti and ni is the number of families still being observed at time ti, our estimator is
Results of applying this method are given in Tables 128 and 129 and Figure 126.
| Time since proband diagnostic test (months) | Expected number of relatives tested |
|---|---|
| 3 | 0.52 |
| 6 | 0.85 |
| 12 | 1.53 |
| 18 | 2.06 |
| 24 | 2.28 |
| 36 | 2.45 |
| 48 | 2.95 |
| 60 | 3.16 |
| 120 | 4.33 |
| Number of relatives tested | Expected time since proband diagnostic test (months) |
|---|---|
| 1 | 6.6 |
| 2 | 16.9 |
| 3 | 52.8 |
| 4 | 97.5 |
FIGURE 126.
Expected number of relatives tested per proband (ticks indicate when families are censored).
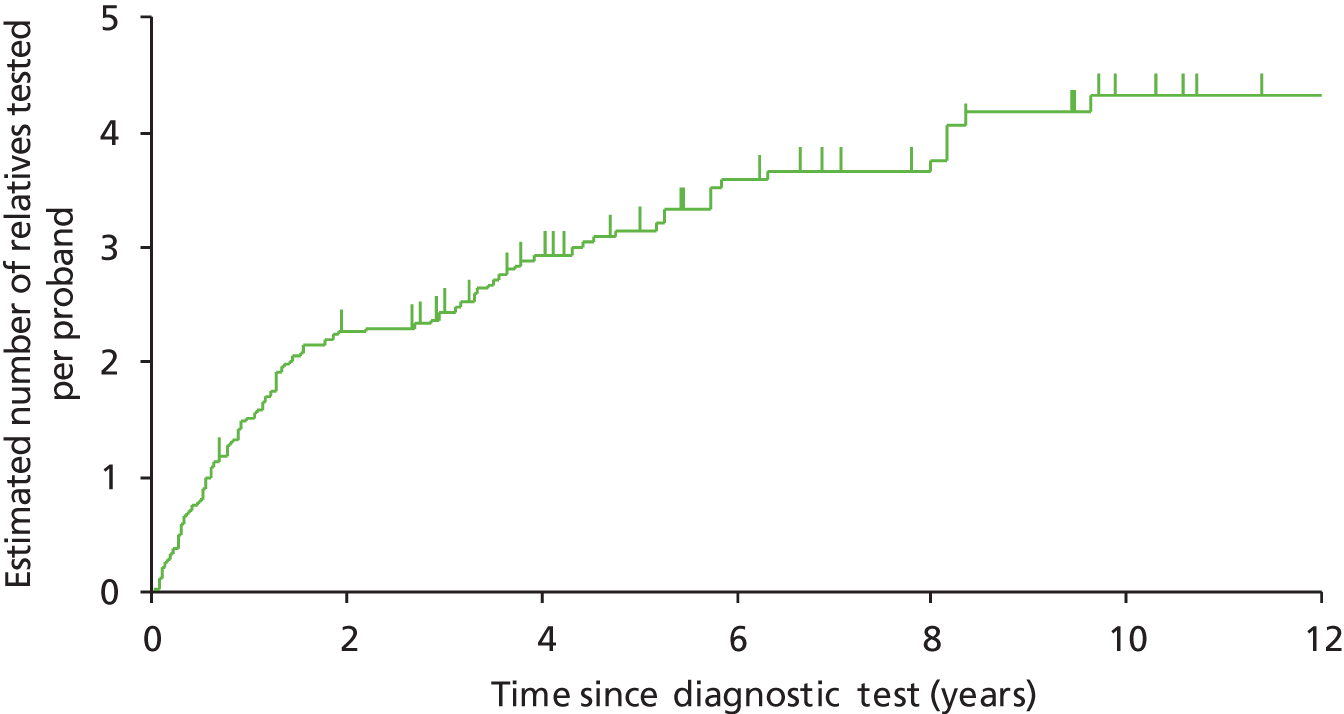
Adjustment for families without predictive tests
The data provided do not include families without any predictive tests. Ian Frayling has advised that the number of such families would be very low as the likelihood of identifying family members for testing is considered during genetic counselling, and, as a result, people without relatives would be less likely to be tested. Ian Frayling gave an estimate that 2% of probands, or fewer, would never identify relatives to be tested. To investigate the effect of this on our analysis, we added a family which would be observed throughout the experiment but would never lead to a predictive test. As there were originally 33 families, this meant that 1 out of 34 = 2.9% of probands would never identify relatives to be tested.
The results with the adjustment are given in Tables 130 and 131 and Figure 127. Figure 128 additionally shows a comparison plot of the estimator with and without the adjustment.
| Time since proband diagnostic test (months) | Expected number of relatives tested |
|---|---|
| 3 | 0.50 |
| 6 | 0.82 |
| 12 | 1.48 |
| 18 | 2.00 |
| 24 | 2.21 |
| 36 | 2.38 |
| 48 | 2.85 |
| 60 | 3.05 |
| 120 | 4.13 |
| Number of relatives tested | Expected time since proband diagnostic test (months) |
|---|---|
| 1 | 7.1 |
| 2 | 18.1 |
| 3 | 54.1 |
| 4 | 99.9 |
FIGURE 127.
Expected number of relatives tested per proband, accounting for the possibility of probands never identifying relatives for testing (ticks indicate when families are censored).
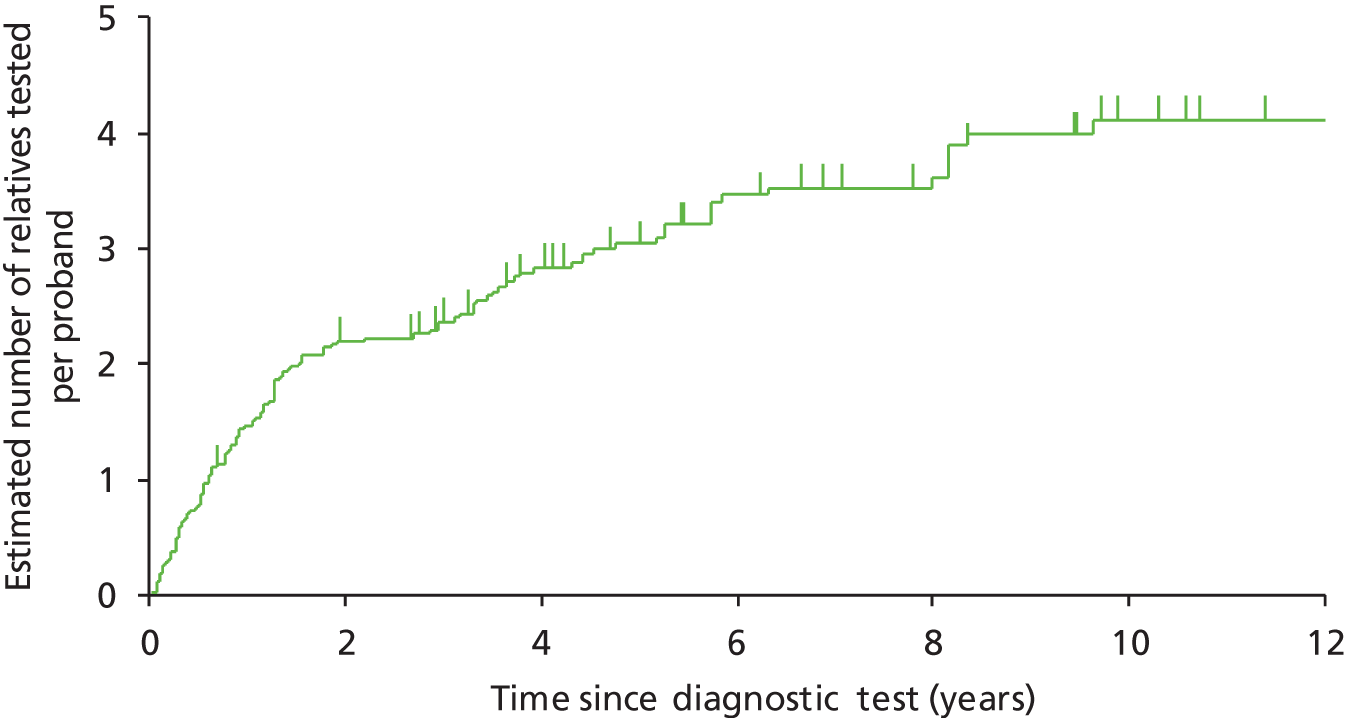
FIGURE 128.
Effect of adjusting for probands never identifying relatives for testing.
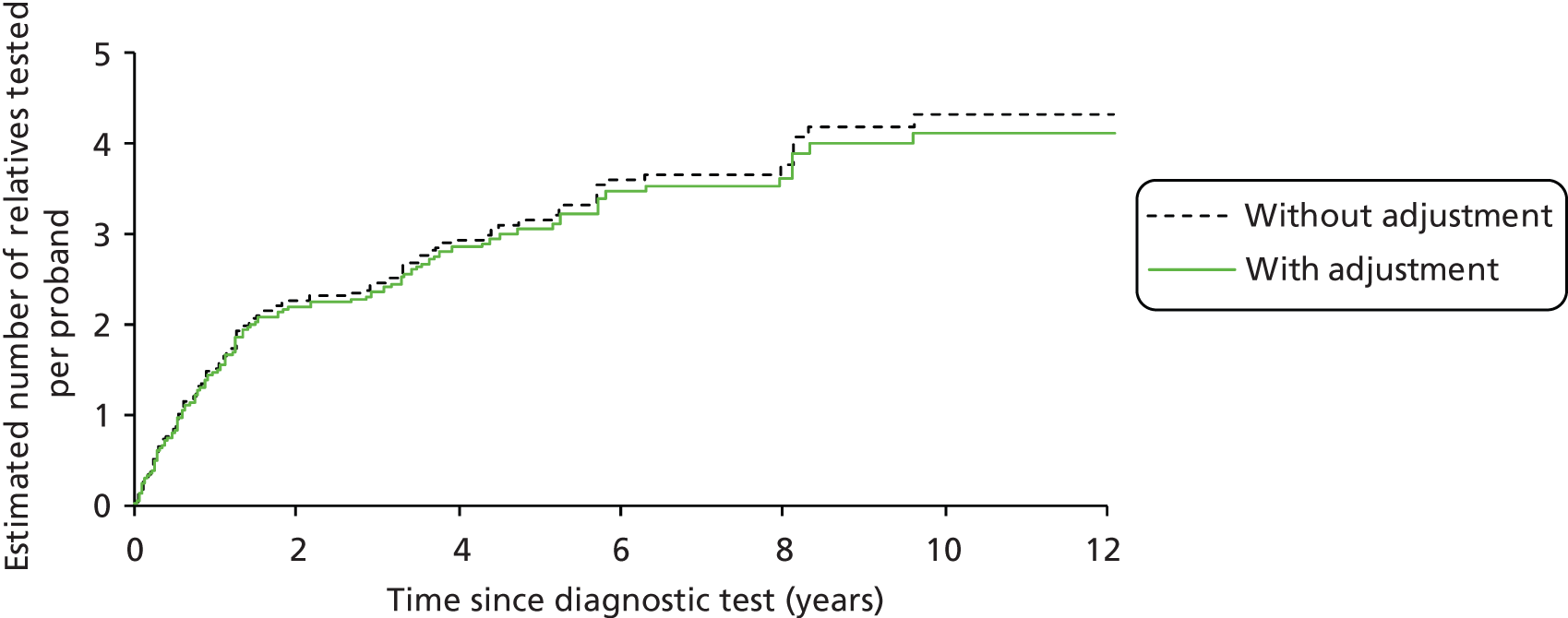
Appendix 9 Data from the Wessex Clinical Genetics Service
Anonymised data were kindly provided by Dr Munaza Ahmed relating to diagnostic and predictive tests for LS performed by the Wessex Clinical Genetics Service. The data related to 208 families, and consisted of 527 test results in total. For each test result, the sex of the subject was recorded, and for 443 results the age of the subject at testing or counselling was also recorded. The data unfortunately did not specify whether the test result was for a diagnostic or a predictive test.
Data processing
We were advised by Dr Ahmed that only the first test record for each family could be a diagnostic test and that it would not have been a diagnostic test if the result was ‘mutation absent’. This allowed us to identify 355 tests as being predictive tests (319 tests were not the first test in the family and 36 were the first test and had the result ‘mutation absent’).
We were further advised that all tests with the result ‘VUS’ (variants of unknown significance) would have been diagnostic tests. This allowed us to identify 11 tests as being diagnostic tests.
Of the 319 predictive tests which were not the first test record for a family, 145 (45%) had the result ‘carrier’ and 174 (55%) had the result ‘mutation absent’ (Table 132). We estimated that 30 of the 161 tests with the result ‘carrier’ which were the first test record for a family but could not be definitely classed as predictive or diagnostic would have been predictive, which gave us the same proportion of carriers in the predictive tests which were the first records as for subsequent records.
| Test result | First test record (n) | Subsequent test record (n) | Total (n) |
|---|---|---|---|
| Carrier | 30 | 145 | 175 |
| Mutation absent | 36 | 174 | 210 |
| Total | 66 | 319 | 385 |
Number of predictive tests for each proband identified
We estimated that in total there were 385 predictive tests and 142 diagnostic tests, which gives a ratio of 2.7 predictive tests for each proband identified.
Sex of relatives receiving predictive testing
We analysed the sex of relatives in predictive tests which were not the first test in a family (as these would have been entirely non-carriers and might affect the sex distribution) (Table 133).
| Sex | Number of relatives (%) |
|---|---|
| Female | 196 (61) |
| Male | 123 (39) |
| Total | 319 (100) |
The data suggest that 38.6% of relatives receiving predictive testing are male (95% CI 33.2% to 44.1%).
Results of predictive testing
We analysed the results of predictive tests which were not the first test in a family (as these would have been carried out entirely among non-carriers) (Table 134).
| Test result | Number of relatives (%) |
|---|---|
| Carrier | 145 (45) |
| Mutation absent | 174 (55) |
| Total | 319 (100) |
Relationship between sex and results of predictive testing
We analysed the relationship between sex and the results of predictive tests which were not the first test in a family (Table 135).
| Sex | Carrier (n) | Mutation absent (n) | Total (n) |
|---|---|---|---|
| Female | 84 | 112 | 196 |
| Male | 61 | 62 | 123 |
| Total | 145 | 174 | 319 |
There is no statistically significant evidence of a relationship between sex and the results of predictive testing (chi-squared p-value = 0.24; risk ratio male vs. female = 1.16, 95% CI 0.91 to 1.47).
Age at predictive testing
We analysed the age at testing or counselling in predictive tests which were not the first test in a family (as these would have been carried out entirely among non-carriers, which might affect the age distribution). The age distribution is shown in Figure 129. It can be seen clearly that there are very few tests on relatives under the age of 20 years, which is to be expected because the test result would be unlikely to have an impact on clinical management at this age. Likewise, testing is less likely to affect clinical management for people aged > 70–75 years, as guidelines suggest that this is the maximum age at which surveillance should be offered.
FIGURE 129.
Age distribution of relatives receiving predictive testing.
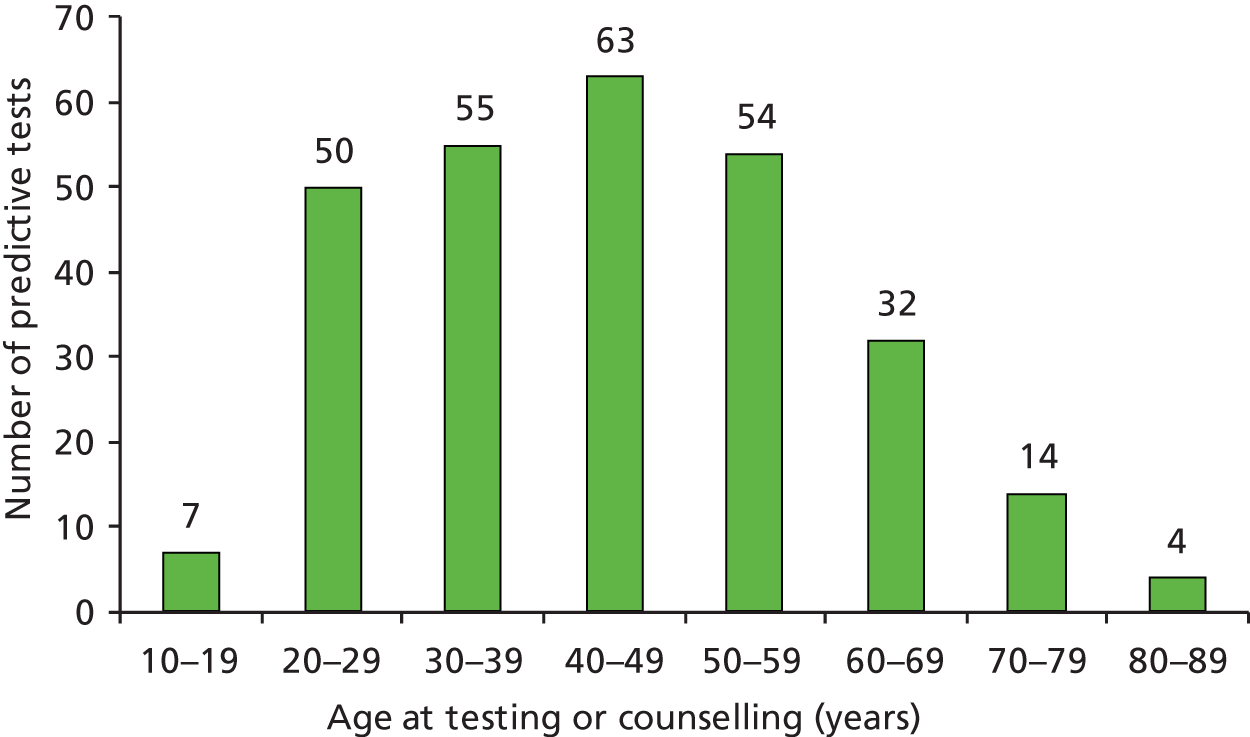
Appendix 10 Deriving the hazard ratio for colorectal cancer incidence due to Lynch syndrome surveillance colonoscopies from Jarvinen and colleagues (2000)
We extracted CRC incidence events from the Kaplan–Meier curves given in figure 1 of Jarvinen and colleagues94 for those with mutations. We estimated time of CRC incidence to 0.1-year accuracy as the figure did not allow more accurate extraction. We confirmed that the number of incident CRCs extracted matched the number described in the text (8/44 in the study group, 19/46 in the control group). We then assumed one censored individual in each group within 15 years to match the reported 15-year Kaplan–Meier survival. The remainder of each cohort was assumed to be censored at 15 years. We believed that very few individuals would have been censored as the steps in the Kaplan–Meier curves were of almost identical height on each curve. We assumed that one individual in the study group was censored between 7.5 years and 11.5 years, and that one individual in the control group was censored at 1.3 years (the minimum follow-up in the control group including non-carriers was 1 year 4 months). Our extracted survival data are presented in Table 136 and Figure 130.
| Group | Incident CRCs (years) | Censoring events (years) | 15-year CRC-free survival |
|---|---|---|---|
| Study group (with surveillance) | 0.0, 0.0, 5.0, 5.1, 5.5, 7.5, 11.5, 11.8 | 10.0 | 0.817 |
| Control group (no surveillance) | 2.7, 3.0, 3.5, 4.1, 4.6, 5.3, 5.5, 5.7, 5.8, 6.1, 6.2, 6.7, 7.7, 8.4, 9.1, 13.3, 14.1, 14.3, 14.5 | 1.3 | 0.578 |
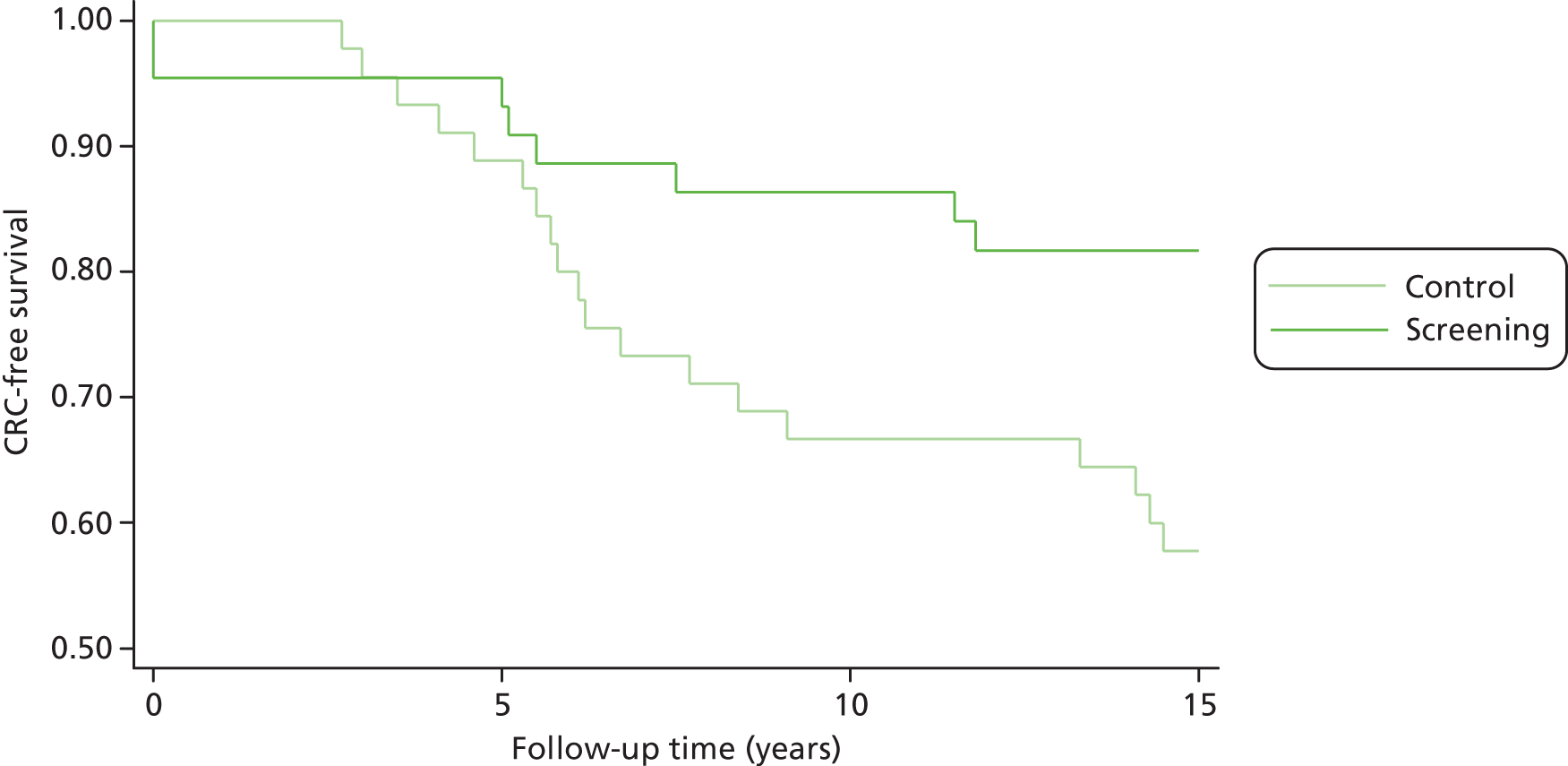
Using the extracted survival data we performed Cox proportional hazards regression and obtained a HR for CRC incidence of 0.387 (95% CI 0.169 to 0.885) for those undergoing surveillance versus those not undergoing surveillance. We tested the proportional hazards assumption using the command ‘estat phtest’ in Stata Version 12 (StataCorp LP, College Station, TX, USA) which gave a p-value of 0.246, indicating that the proportional hazards assumption could not be rejected.
Appendix 11 Literature review of the effectiveness of surveillance for endometrial cancer
We searched the literature for evidence of the effectiveness of gynaecological surveillance for patients with LS. Titles and abstracts were screened by one reviewer (TS) for inclusion (Figure 131).
FIGURE 131.
Included studies.
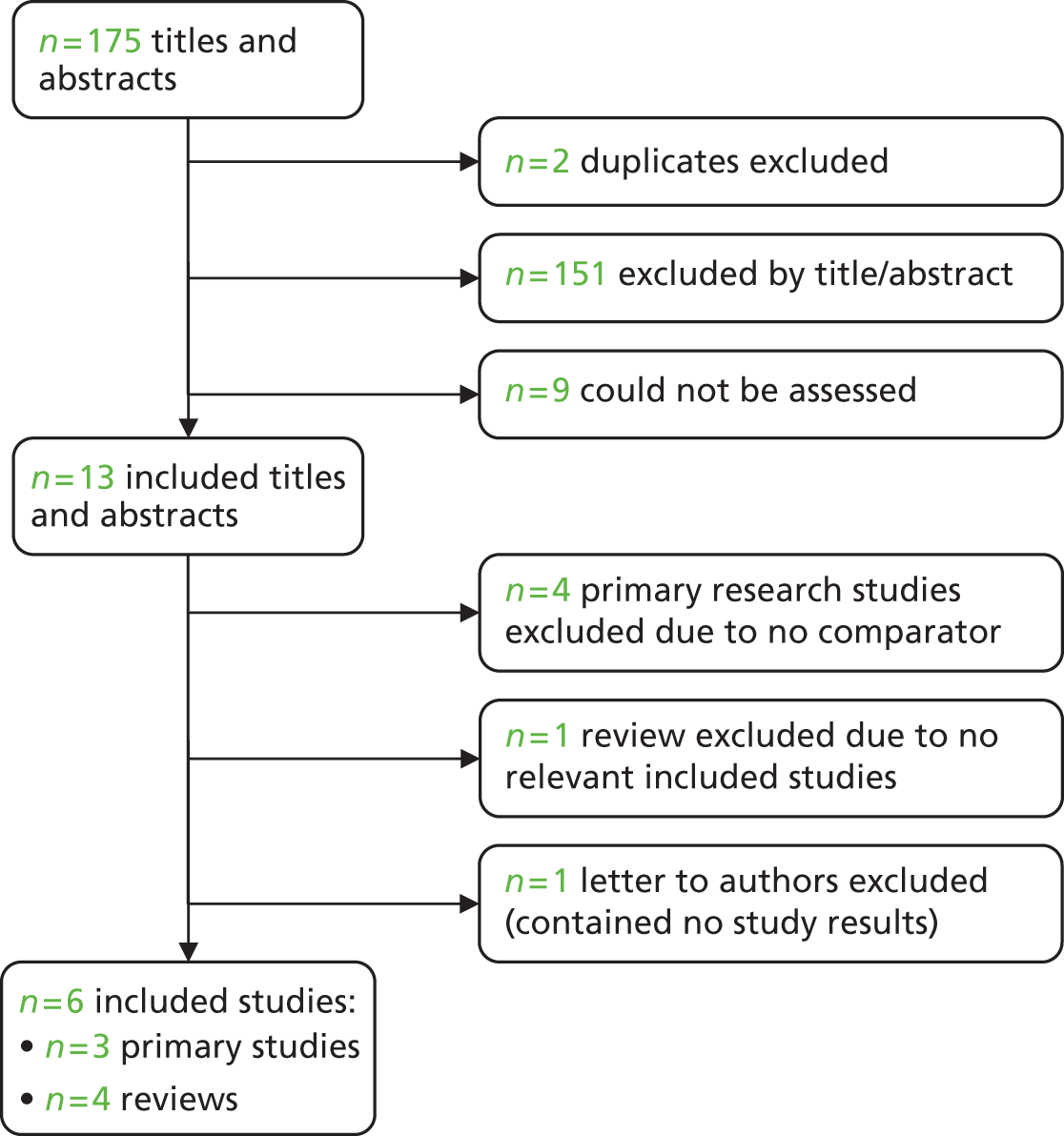
Thirteen references65,107,167,190–194,196,298–301 were included on the basis of title and abstract screening. Of these, seven references107,190,191,194,196,298,299 were primary studies (two298,299 were conference abstracts and the remaining five were peer-reviewed full papers), five references65,167,192,193,300 were papers including a review of the literature and one301 was a letter in response to Dove-Edwin and colleagues. 190
After reviewing full texts, we excluded four primary studies194,196,298,299 as they did not include an appropriate comparator. We excluded the paper by Schorge and colleagues300 as it did not include any relevant studies (all studies related to other hereditary causes of OC). We excluded the letter by Wood and colleagues301 as it did not contain any study results and, as a letter, was not peer-reviewed.
Our final set of included references comprised three primary studies107,190,191 and four reviews. 65,167,192,193
Primary studies
Dove-Edwin and colleagues190 describe the outcomes of surveillance for endometrial carcinoma in 292 patients recruited by the Imperial Cancer Research Fund (ICRF) Family Cancer Clinic at St. Mark’s Hospital, London (n = 184) or the Netherlands Foundation for the Detection of Hereditary Tumours (n = 180). Patient records were examined retrospectively and 269 were informative for the analysis, of which 171 were from families meeting the AC and 98 were from families meeting other clinical criteria suggestive of LS and/or having a LS mutation. Of the 269 women analysed, 222 had at least one scan, meaning that 47 had no TVU scan and could be considered a control group. In the ‘intervention group’ (women receiving at least one TVU scan) there were two endometrial carcinomas, both interval cancers detected following symptomatic presentation and both stage I. In the ‘control group’ (women not receiving a TVU scan) there were no endometrial carcinomas.
Jarvinen and colleagues107 evaluate the effectiveness of surveillance in 242 healthy patients with LS mutations. Of these, 103 were women who were eligible for surveillance when they were recruited. Three women declined surveillance and could be considered a control group. In the ‘intervention group’ (women complying with EC surveillance), 16 ECs were detected during screening (12 at stage I, two at stage II and two at stage III) in addition to one EC at stage I being found following prophylactic hysterectomy. One interval EC was detected at stage I. Three OCs were detected during screening (two at stage I, one at stage II) and two interval OCs were discovered (one at stage I, one at stage III). In the ‘control group’ (women not complying with EC surveillance), one stage I EC and one stage I OC were discovered following symptomatic presentation.
Renkonen-Sinisalo and colleagues191 describe the results of EC screening in 385 women with LS mutations. They also describe the characteristics of 83 women with LS mutations who were affected by EC before surveillance was instituted. The two groups of women cannot be compared in terms of EC incidence as the second group is recruited as 100% affected by EC, but it is possible to compare the stage distributions of ECs in the two groups. The stage distributions for the two groups are shown in Table 137. EC survival was not significantly different for the two groups.
| FIGO stage | Intervention group (n) | Control group (n) |
|---|---|---|
| I | 12 (6× IA, 6× IB) | 67 (27× IA, 32× IB, 8× IC) |
| II | 1 (1× IIB) | 2 (1× IIA, 1× IIB) |
| III | 1 (1× IIIA) | 11 (4× IIIA, 7× IIIC) |
| IV | 0 | 2 (2× IVB) |
| Unknown | 0 | 1 |
Analysis of primary studies
We expect surveillance to reduce the incidence of gynaecological cancers by the following mechanisms:
-
detection and removal by operative hysteroscopy of pre-malignancies195,196
-
detection of pre-malignancies leading to prophylactic hysterectomy (perhaps with bilateral salpingo-oophorectomy)191,194
-
FP screening leading to prophylactic hysterectomy.
We also expect surveillance to improve the stage distribution of gynaecological cancers by the following mechanisms:
-
screen-detected asymptomatic malignancies
-
symptomatic malignancies detected during screening which would not have presented until later and may not have been accurately diagnosed, for example in primary care.
It is also possible that surveillance could improve awareness and response to symptoms, but equally surveillance could give false reassurance.
In our analysis, therefore, we look for reduction in incidence of gynaecological cancers and an improvement in the stage distribution of cancers. We evaluate whether or not a reduction in incidence has been achieved by examining the relative risk of gynaecological cancers individually and combined (Table 138), and evaluate whether or not the stage distribution has been improved using ordered logistic regression (Table 139). Note that in none of the analyses are we able to draw statistically significant conclusions.
| Study | Intervention arm | Control arm | Relative risk of gynaecological cancer (95% CI) | Relative risk of prophylactic hysterectomy (95% CI) |
|---|---|---|---|---|
| Dove-Edwin et al. 2002190 | 222 patients, 2 EC | 47 patients, 0 EC | Not computed | Not computed |
| Jarvinen et al. 2009107 | 100 patients: 17 EC, 5 OC, 47 prophylactic hysterectomy | 3 patients: 1 EC, 1 OC, 1 PH | EC 0.510 (0.097 to 2.677) OC 0.150 (0.024 to 0.920)a Gynaecological cancer 0.330 (0.137 to 0.797)a |
1.410 (0.281 to 7.080) |
| Study | Intervention arm | Control arm | Statistical analysis |
|---|---|---|---|
| Dove-Edwin et al. 2002190 | EC 2× stage I |
EC None |
Not done |
| Jarvinen et al. 2009107 | EC 12× stage I, 2× stage II, 2× stage III |
EC 1× stage I |
Ordered logistic regression cannot be used without more data in control arm |
| OC 3× stage I, 1× stage II, 1× stage III |
OC 1× stage I |
||
| Renkonen-Sinisalo et al. 2007191 | EC 12× stage I, 1× stage II, 1× stage III |
EC 67× stage I, 2× stage II, 11× stage III, 2× stage IV |
Ordered logistic regression with four main stages Coefficient of surveillance not statistically significant (z = –0.45, p = 0.651) Predicted stage distribution: Intervention: 0.864 stage I, 0.025 stage II, 0.095 stage III, 0.015 stage IV Control: 0.815 stage I, 0.032 stage II, 0.130 stage III, 0.022 stage IV |
| Ordered logistic regression with 10 substages Coefficient of surveillance not statistically significant (z = –1.03, p = 0.302) |
Existing reviews
Auranen and Joutsiniemi192 conducted a systematic review of gynaecological cancer screening in women belonging to LS families, which had five included studies. 190,191,194–196 In our review, the study by Lécuru and colleagues195 was excluded because the abstract suggested there was no comparator for the surveillance strategy. Auranen and Joutsiniemi conclude that the studies they included do not allow for evidence-based decision-making.
Koornstra and colleagues193 conducted a review of extracolonic cancers, including gynaecological cancer screening, and included three studies relevant to gynaecological cancer screening. 190,191,196 They conclude that the only evidence of surveillance benefit is that TVU and endometrial sampling detect endometrial tumours in early stages from the study by Renkonen-Sinisalo and colleagues,191 despite this study not reporting a statistical difference by the Pearson chi-squared method.
Lindor and colleagues167 included two studies190,196 and concluded that there was insufficient evidence to argue for or against endometrial sampling or TVU.
Palomaki and colleagues65 included three studies190,191,196 and concluded that TVU is not highly effective at identifying ECs in women with LS but that endometrial biopsy is effective at identifying both pre-malignant and malignant lesions according to the study by Renkonen-Sinisalo and colleagues. 191 Palomaki and colleagues recommend surveillance for women with LS but say that ‘Inadequate data are available to document that transvaginal ultrasound and endometrial biopsy can reduce the incidence of endometrial cancer.’
We conclude that there is insufficient evidence to suggest that gynaecological cancer surveillance reduces the incidence of gynaecological cancers or improves the survival of cancers when individuals are affected.
Appendix 12 Calculating utility scores from van Duijvendijk and colleagues
The study by van Duijvendijk and colleagues201 compares the HRQoL of colectomy and IRA versus proctocolectomy and IPAA in patients receiving major prophylactic surgery for FAP, as well as HRQoL in patients from the general population. Quality of life was measured using the SF-36 questionnaire. The authors report the number of patients in each group, their mean age (and the standard deviation of their age), and the mean values and standard deviations in the eight main categories in the SF-36 questionnaire (Table 140) and the health transition category.
| Abbreviation | Category |
|---|---|
| PF | Physical functioning |
| RP | Role functioning: physical |
| BP | Bodily pain |
| GH | General health perceptions |
| VT | Vitality |
| SF | Social functioning |
| RE | Role functioning: emotional |
| MH | Mental health |
Ara and Brazier228 describe a methodology to calculate utility scores from SF-36 category scores. Their methodology is described by seven models, of which models 1–5 and 7 are special cases of model 6, the most general model supplied, in which all eight categories have corresponding coefficients, as do age and age squared and also the squared values of four categories (PF, SF, MH, BP). Models 4–7 require the squared values of some variables.
The utility score for a single patient, denoted i, is calculated by
where α is the intercept, xi is the vector of questionnaire results and patient characteristics,
and β is the set of coefficients derived and published in the paper.
The mean utility for a set of patients is calculated by
where n is the number of patients. By linearity of expectation, this can be written as
where x¯ is the vector of mean values across all patients. Note that the mean value of Age2 is not generally equal to the square of the mean value of Age (this may explain the discrepancy between our calculated values and those of Dinh and colleagues55), but as standard deviations and patient numbers are provided we can calculate the appropriate mean values of these squared variables. If s is the sample standard deviation of a variable x and we want to calculate E[x2] = ∑(x2)/n we make use of the following formula for s:
to calculate that
and therefore
Using this formula we calculated the additional information (Table 141) necessary to apply all the models in the study by Ara and Brazier228 to calculate utility scores for the three groups.
| Parameter | General population (n = 279) | IRA (n = 161) | IPAA (n = 118) | |||
|---|---|---|---|---|---|---|
| E[x] | E[x2] | E[x] | E[x2] | E[x] | E[x2] | |
| PF | 88.8 | 8141 | 56.3 | 3359 | 55.9 | 3272 |
| SF | 86.4 | 7864 | 67.4 | 4868 | 64.8 | 4675 |
| MH | 77.6 | 6300 | 65.3 | 4522 | 63.7 | 4390 |
| BP | 77.9 | 6573 | 37.2 | 1443 | 36.7 | 1403 |
| Age | 39 | 1716 | 41 | 1905 | 37 | 1512 |
With this information we computed the utility scores for the three groups using the seven different models, and therefore the utility decrement for the two types of surgery using each model (Table 142).
| Calculation model | General population (mean age 39 years) | Group 1: IRA (mean age 41 years) | Group 2: IPAA (mean age 37 years) |
|---|---|---|---|
| Utility score | |||
| Model 1 | 0.873 | 0.583 | 0.575 |
| Model 2 | 0.877 | 0.590 | 0.585 |
| Model 3 | 0.881 | 0.596 | 0.589 |
| Model 4 | 0.909 | 0.650 | 0.642 |
| Model 5 | 0.895 | 0.639 | 0.632 |
| Model 6 | 0.837 | 0.630 | 0.623 |
| Model 7 | 0.852 | 0.641 | 0.633 |
| Disutility from general population | |||
| Model 1 | 0.290 | 0.299 | |
| Model 2 | 0.287 | 0.293 | |
| Model 3 | 0.285 | 0.292 | |
| Model 4 | 0.259 | 0.267 | |
| Model 5 | 0.255 | 0.263 | |
| Model 6 | 0.208 | 0.214 | |
| Model 7 | 0.211 | 0.218 | |
Appendix 13 Estimating costs of colorectal cancer surgery
| Procedure | Relevant OPCS codes | Corresponding HRGs | HRG unit cost (2011–12 prices151) (£) | Unit costs uprated to 2013–14 prices (£) |
|---|---|---|---|---|
| Segmental resection (proximal without exteriorisation) | H061, H062, H063 Extended right hemicolectomy H071, H072, H073 Right hemicolectomy H081, H082, H083 Transverse colectomy |
FZ75 Proximal colon procedures, 19 years and over | 5576 | 5969 |
| Segmental resection (distal without exteriorisation) | H091, H092, H093 Left hemicolectomy H101, H102, H103 Sigmoid colectomy |
FZ76 Distal colon procedures, 19 years and over | 5411 | 5792 |
| Segmental resection (with exteriorisation) | H114 Colectomy and Ileostomy NEC H115 Colectomy and exteriorisation of bowel NEC |
FZ74 Complex large intestine procedures, 19 years and over | 6831 | 7313 |
| Subtotal colectomy with IRA | H051 Total colectomy and anastomosis of ileum to rectum H053 Total colectomy and ileostomy NEC |
FZ74 Complex large intestine procedures, 19 years and over | 6831 | 7313 |
| Anterior resection | H331 Abdominoperineal excision of rectum and end colostomy H332 Proctectomy and anastomosis of colon to anus H333 Anterior resection of rectum and anastomosis of colon to rectum using staples H334 Anterior resection of rectum and anastomosis NEC H336 Anterior resection of rectum and exteriorisation of bowel H338 Other specified excision of rectum |
FZ74 Complex large intestine procedures, 19 years and over | 6831 | 7313 |
| Proctocolectomy with IPAA | H042 Panproctocolectomy and anastomosis of ileum to anus and creation of pouch HFQ | FZ74 Complex large intestine procedures, 19 years and over | 6831 | 7313 |
| Stoma reversal | H154 Closure of colostomy | FZ50 Intermediate large intestine procedures, 19 years and over | 451 | 482 |
The costs of subtotal colectomy with IRA, anterior resection and proctocolectomy with IPAA were therefore £7313 for all patients, plus stoma reversal costing £482 for the proportion of patients who required stomas and whose stomas were subsequently reversed. In the study by Trueman and colleagues,134 67% of rectal cancer patients and 14.5% of colon cancer patients require a stoma, with 26.6% of stomas subsequently reversed. Therefore, the cost of stoma reversal is incurred by 17.8% of rectal cancer patients and 3.9% of colon cancer patients. All subtotal colectomy patients will have colon cancer and hence 3.9% of subtotal colectomies are followed by a stoma reversal, giving a total cost of £7313 + 0.039 × £482 = £7331. All anterior resection patients will have rectal cancer and hence 17.8% of anterior resections are followed by a stoma reversal, giving a total cost of £7313 + 0.178 × £482 = £7399. Proctocolectomy patients may have colon cancer or rectal cancer but the nature of the operation is such that most patients will require a temporary stoma,302 so we assumed that 26.6% of proctocolectomies would be followed by stoma reversal, giving a total cost of £7313 + 0.266 × £482 = £7441.
It is documented by Dinh and colleagues (within an online appendix)55 that LS colon cancers have a greater tendency to be located in the proximal colon (81% of colon cancers in the proximal colon), whereas sporadic colon cancers are more evenly spread (in 2010, 10,499/21,664 = 48% of colon cancers were proximal to the splenic flexure119). The cost for segmental resection in our model is therefore dependent on whether or not the CRC patient has LS.
For LS patients receiving segmental resection, we assumed that 81% of segmental resections would be in the proximal colon with the remainder in the distal colon. We assumed that 14.5% of segmental resections would require a stoma, independent of whether they were proximal or distal. We assumed that 26.6% of segmental resections requiring a stoma would be followed by a stoma reversal. Therefore, the expected cost of a segmental resection in a LS colon cancer patient is 0.145 × (£7313 + 0.266 × £482) + (1 – 0.145) × (0.81 × £5969 + 0.19 × £5792) = £6154.
For sporadic CRC patients receiving segmental resection, we assumed that 48% of segmental resections would be in the proximal colon with the remainder in the distal colon. Similarly to the calculation for LS cost, we calculate 0.145 × (£7313 + 0.266 × £482) + (1 – 0.145) × (0.48 × £5969 + 0.52 × £5792) = £6104.
Appendix 14 Net health benefit
Let Ci denote the total costs of strategy i, and similarly let Bi denote the total health benefits of strategy i (measured in, for example, QALYs). If one is willing to pay λ units of cost for a unit of health benefit then the NHB of strategy i is
The INHB of strategy i versus the base strategy (strategy 1) is
Suppose that Bi and Ci can be expressed as weighted sums across random variables and an intercept term:
We can also then express the INHBs as weighted sums across random variables with intercept terms:
We can therefore calculate the variance of the INHB as a function of the variances of Xj and Yj:
This is applied in our setting where Xj and Yj are, respectively, the discounted total QALYs and the discounted total costs for patient group j and aij is the number of patients in patient group j using strategy i.
Tables 144–146 give the results in the base case.
| j | Proband/relative | LS | LS diagnosed | LS surveillance | Male | X j | Yj (£) | ||
|---|---|---|---|---|---|---|---|---|---|
| Mean | SE | Mean | SE | ||||||
| 1 | Relative | False | False | False | True | 16.316 | 0.0636 | 743 | 39 |
| 2 | Relative | False | False | False | False | 16.394 | 0.0587 | 604 | 36 |
| 3 | Relative | False | True | False | True | 16.284 | 0.0634 | 747 | 39 |
| 4 | Relative | False | True | False | False | 16.402 | 0.0585 | 2061 | 37 |
| 5 | Relative | False | True | True | True | 16.341 | 0.0635 | 5693 | 32 |
| 6 | Relative | False | True | True | False | 16.495 | 0.0586 | 7137 | 32 |
| 7 | Relative | True | False | False | True | 15.590 | 0.0639 | 6032 | 122 |
| 8 | Relative | True | False | False | False | 15.453 | 0.0611 | 6373 | 114 |
| 9 | Relative | True | True | False | True | 15.618 | 0.0642 | 5747 | 118 |
| 10 | Relative | True | True | False | False | 15.532 | 0.0615 | 7552 | 117 |
| 11 | Relative | True | True | True | True | 16.515 | 0.0624 | 7286 | 65 |
| 12 | Relative | True | True | True | False | 16.282 | 0.0598 | 9244 | 67 |
| 13 | Proband | False | False | False | True | 7.611 | 0.0744 | 16,110 | 70 |
| 14 | Proband | False | False | False | False | 7.641 | 0.0758 | 16,403 | 66 |
| 15 | Proband | False | True | False | True | 7.608 | 0.0744 | 16,117 | 69 |
| 16 | Proband | False | True | False | False | 7.497 | 0.0746 | 17,718 | 66 |
| 17 | Proband | False | True | True | True | 7.669 | 0.0747 | 19,046 | 52 |
| 18 | Proband | False | True | True | False | 7.660 | 0.0755 | 20,592 | 50 |
| 19 | Proband | True | False | False | True | 7.458 | 0.0713 | 21,888 | 101 |
| 20 | Proband | True | False | False | False | 7.024 | 0.0687 | 22,850 | 99 |
| 21 | Proband | True | True | False | True | 7.303 | 0.0714 | 22,252 | 99 |
| 22 | Proband | True | True | False | False | 7.109 | 0.0701 | 23,582 | 99 |
| 23 | Proband | True | True | True | True | 8.542 | 0.0803 | 20,504 | 74 |
| 24 | Proband | True | True | True | False | 8.382 | 0.0780 | 22,175 | 73 |
| j | a ij | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1(1) | 1(2) | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
| 1 | 3097 | 3058 | 3050 | 3052 | 3045 | 3046 | 3046 | 3038 | 3015 |
| 2 | 5084 | 5021 | 5008 | 5011 | 5000 | 5002 | 5002 | 4988 | 4950 |
| 3 | 0 | 19 | 23 | 23 | 26 | 25 | 25 | 29 | 41 |
| 4 | 0 | 32 | 38 | 37 | 42 | 41 | 41 | 48 | 67 |
| 5 | 0 | 19 | 23 | 23 | 26 | 25 | 25 | 29 | 41 |
| 6 | 0 | 32 | 38 | 37 | 42 | 41 | 41 | 48 | 67 |
| 7 | 119 | 108 | 57 | 57 | 49 | 49 | 49 | 40 | 38 |
| 8 | 195 | 177 | 93 | 93 | 80 | 80 | 80 | 66 | 62 |
| 9 | 0 | 5 | 23 | 23 | 26 | 26 | 26 | 29 | 30 |
| 10 | 0 | 9 | 37 | 37 | 42 | 42 | 42 | 47 | 48 |
| 11 | 0 | 5 | 40 | 40 | 45 | 45 | 45 | 50 | 51 |
| 12 | 0 | 9 | 65 | 65 | 73 | 73 | 73 | 82 | 84 |
| 13 | 815 | 798 | 813 | 814 | 813 | 814 | 814 | 812 | 798 |
| 14 | 742 | 727 | 740 | 741 | 740 | 741 | 741 | 739 | 727 |
| 15 | 0 | 5 | 1 | 0 | 0 | 0 | 0 | 1 | 5 |
| 16 | 0 | 4 | 1 | 0 | 0 | 0 | 0 | 1 | 4 |
| 17 | 0 | 11 | 1 | 1 | 1 | 1 | 1 | 2 | 12 |
| 18 | 0 | 10 | 1 | 1 | 1 | 1 | 1 | 2 | 11 |
| 19 | 75 | 58 | 33 | 33 | 28 | 28 | 28 | 23 | 21 |
| 20 | 68 | 53 | 30 | 30 | 26 | 26 | 26 | 21 | 19 |
| 21 | 0 | 5 | 9 | 9 | 10 | 10 | 10 | 11 | 11 |
| 22 | 0 | 4 | 8 | 8 | 9 | 9 | 9 | 10 | 10 |
| 23 | 0 | 12 | 33 | 33 | 37 | 37 | 37 | 41 | 42 |
| 24 | 0 | 10 | 30 | 30 | 33 | 33 | 33 | 37 | 39 |
| Calculation | Strategy | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1(1) | 1(2) | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||
| bi (QALY decrement due to genetic testing) | 0 | 0 | −4.122 | −3.939 | −4.545 | −4.412 | −4.412 | −5.267 | −8.241 | |
| ci (diagnostic costs) (£) | 0 | 85,000 | 1,177,000 | 1,106,000 | 1,057,000 | 1,051,000 | 1,149,000 | 1,909,000 | 2,017,000 | |
| INHB vs. strategy 1(1) (ΔNHBi) | Mean | – | 16.2 | 77.7 | 82.2 | 101.3 | 102.3 | 97.3 | 76.1 | 62.3 |
| SE | – | 6.4 | 12.5 | 12.4 | 14.0 | 13.9 | 13.9 | 15.7 | 18.1 | |
| INHB vs. strategy 5 | Mean | −102.3 | −86.1 | −24.5 | −20.1 | −1.0 | – | −4.9 | −26.2 | −39.9 |
| SE | 13.9 | 9.9 | 1.5 | 1.5 | 0.2 | – | 0.0 | 1.9 | 5.2 | |
Appendix 15 Scenario analysis 2 (BRAF replaced by methylation testing)
There is currently one further test used in the diagnostic process for LS: a methylation test to examine for hypermethylation. It is used in a similar manner to BRAF, after MSI or an abnormal MLH1 IHC result, to rule out sporadic cancers. However, instead of testing for a BRAF V600E mutation, the methylation test considers MLH1 promoter hypermethylation as an indicator for sporadic CRCs. As this has only recently been implemented in the UK, we have not included it in the base case, but have included it as a separate scenario analysis. The values of sensitivity and specificity of methylation have been pooled from two 2010 studies: Bouzourene and colleagues303 and Chang and colleagues,304 which were identified from the Gudgeon and colleagues study from 2011. 69
The study by Chang and colleagues (2010)304 from Taiwan identified few MSH6 mutations and no PMS2 mutations among their cohort. The patients were identified for methylation testing using either a MSI result or an abnormal IHC result, but they did not have to fulfil the Revised Bethesda criteria, which probands in our model do automatically (as CRC onset under 50 years of age is one of the criteria that fulfil the Revised Bethesda criteria). Those patients who did not fulfil the Revised Bethesda criteria had to receive a MSI result to then undergo further testing. The value of sensitivity in Chang’s study was 92%, with 72% specificity.
Bouzourene and colleagues’ study303 looked specifically at LS patients in Switzerland with a MLH1 mutation, plus a group of patients identified with sporadic CRC who had MSI and an abnormal MLH1 result on IHC. The sensitivity in Bouzourene’s study was 94% with 100% specificity. Both studies have relatively small sample sizes: 27 patients in Bouzourene’s cohort and 42 in Chang’s. Using these studies, our values for sensitivity and specificity of methylation testing are 93% and 80%, respectively, regardless of which prior tumour test it follows. Methylation therefore has a higher specificity and lower sensitivity than BRAF testing in our evaluation. We received the cost of methylation via personal correspondence with our clinical expert Ian Frayling (£160), and inflated it to 2013–14 costs using methods previously described in Chapter 5, Adjustments to 2013–14 prices, such that the cost of methylation used in the model is £166. This is currently higher than the cost of BRAF used in the model.
As BRAF was only used in strategies 3, 5, 6 and 7, the other strategies remain unchanged by the introduction of methylation in place of BRAF. Furthermore, the long-term simulations based on a patient’s diagnosis and surveillance status remain unchanged; for example, in the model the average long-term cost and survival of a TP patient with surveillance remains the same. However, as methylation has a different sensitivity, specificity and cost compared with BRAF, the short-term costs and distribution of diagnoses (the proportion of patients diagnosed positive or negative and their surveillance status) are altered. As such, the overall costs and QALYs are also different from BRAF’s costs and QALYs. In much the same way, the costs and QALYs of strategies 2 and 4 differ in the base case (same number of tumour tests, but the tests have different costs and accuracies, resulting in different short-term and therefore long-term outcomes).
As the overall ROC plot (Figure 132) for probands and relatives demonstrates, the sensitivity and specificity in strategies 3, 5, 6 and 7 have changed. These differences are highlighted in Table 147. Here, the use of methylation in place of BRAF appears to increase the specificity and reduce the sensitivity in most strategies, which agrees with the difference in the tests themselves.
FIGURE 132.
Overall ROC plot for both probands and their relatives, when BRAF is replaced with methylation.
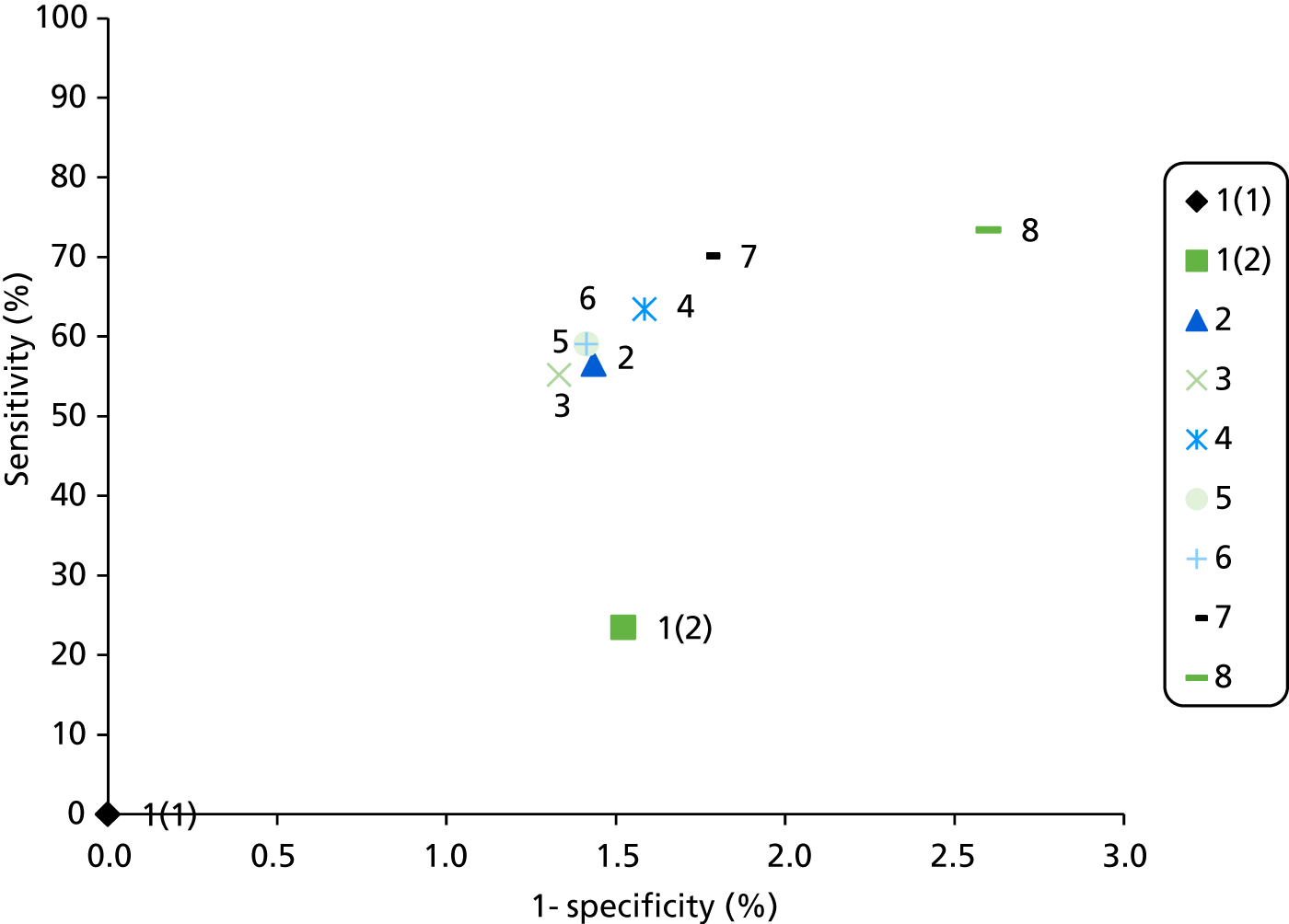
| Strategy | Scenario | Sensitivity (%) | Specificity (%) |
|---|---|---|---|
| 3 | Base case (BRAF) | 56.5 | 98.6 |
| Scenario 2 (methylation) | 55.2 | 98.7 | |
| 5 | Base case (BRAF) | 63.4 | 98.4 |
| Scenario 2 (methylation) | 59.0 | 98.6 | |
| 6 | Base case (BRAF) | 63.4 | 98.4 |
| Scenario 2 (methylation) | 59.0 | 98.6 | |
| 7 | Base case (BRAF) | 71.1 | 98.2 |
| Scenario 2 (methylation) | 70.1 | 98.2 |
Figures 133 and 134 show the proportions of probands and relatives, respectively, testing positive for LS when BRAF is replaced with methylation.
FIGURE 133.
Proportion of probands testing positive for LS when BRAF is replaced with methylation.
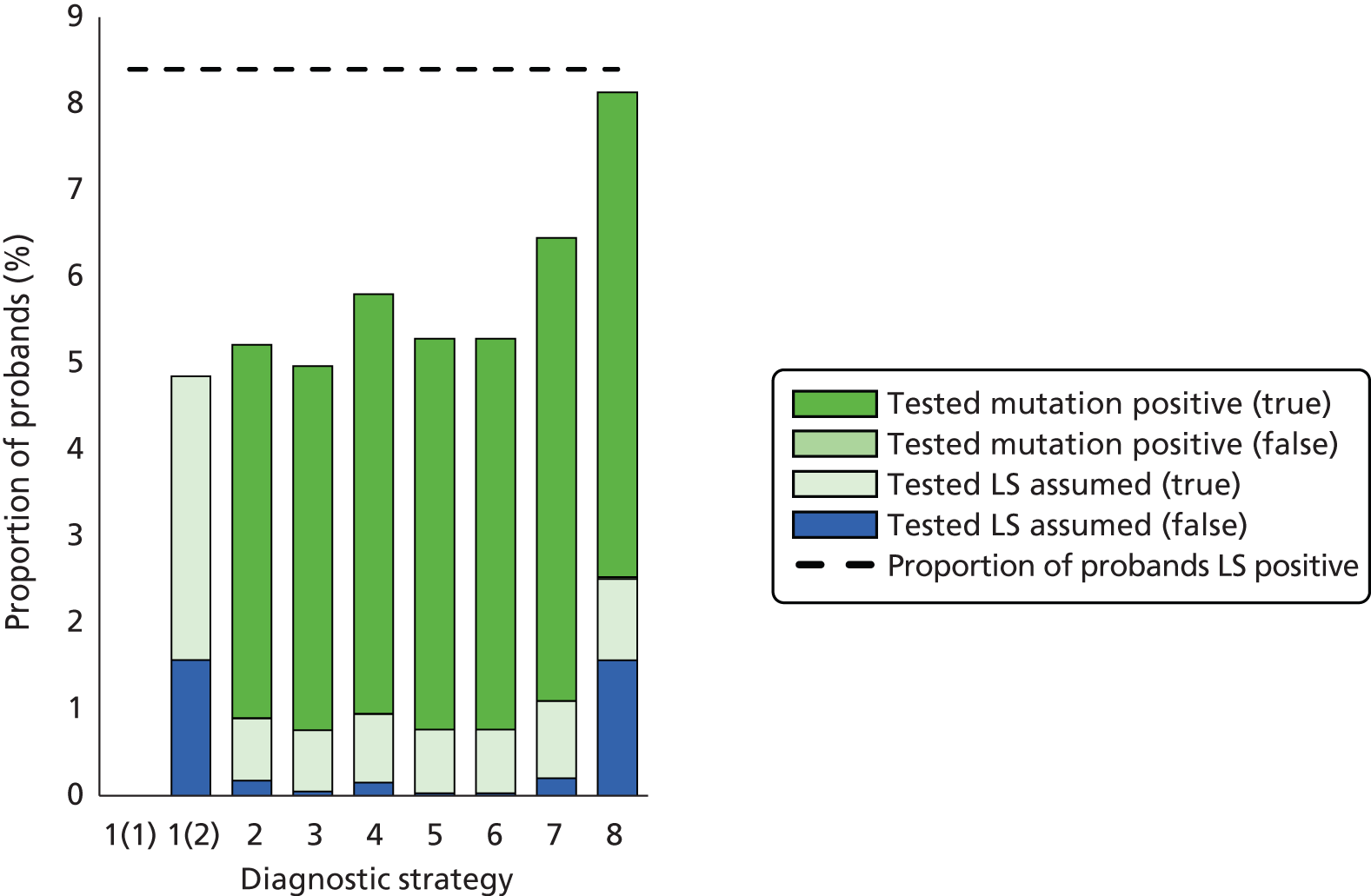
FIGURE 134.
Proportion of relatives testing positive for LS when BRAF is replaced with methylation.
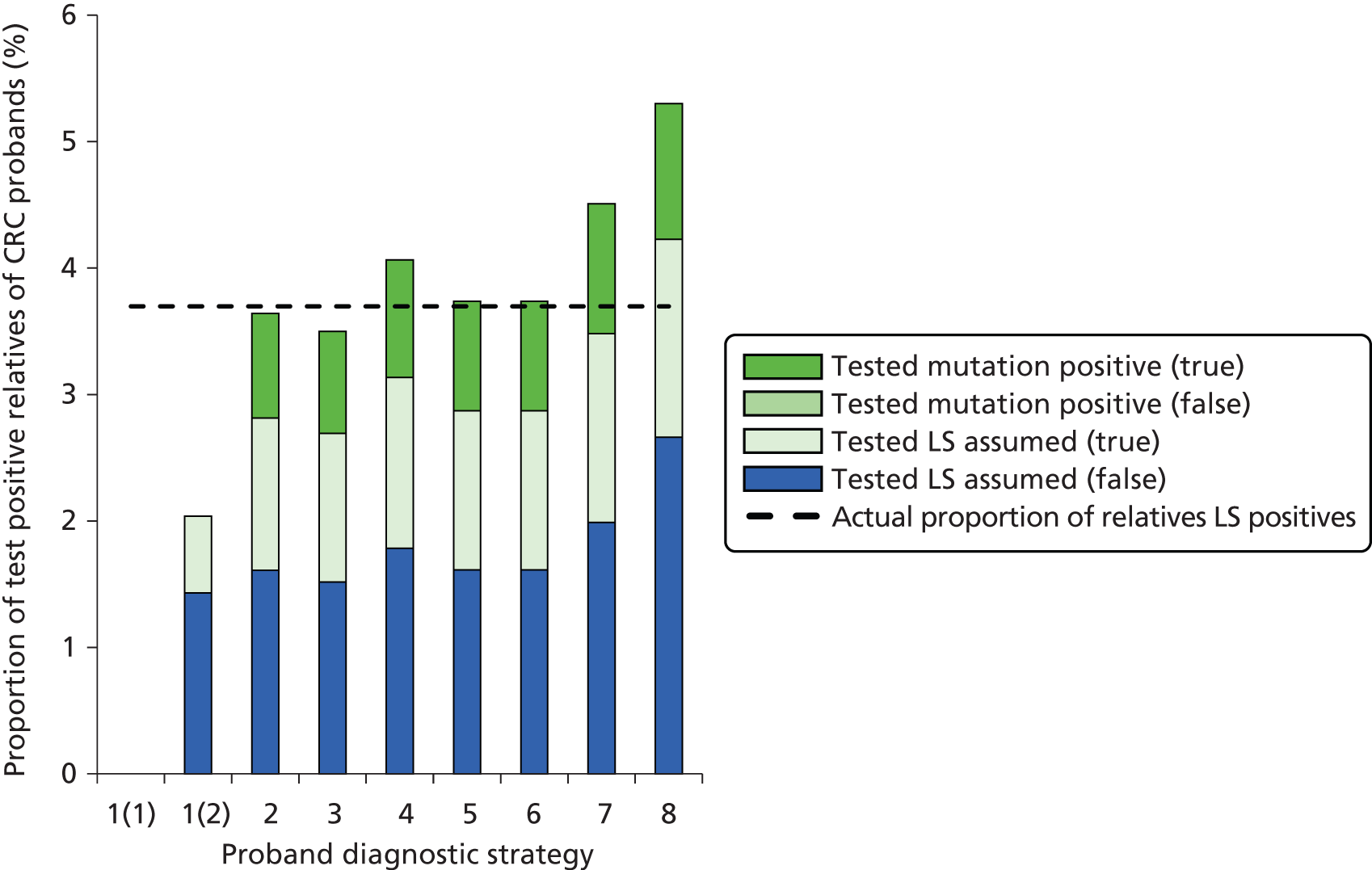
Despite the higher cost of methylation compared with BRAF, in general the diagnostic costs per proband and relative and for the cohort of probands and relatives are reduced in strategies 3, 5, 6 and 7 (Table 148 and Figures 135–137). This happens because a smaller proportion of patients test negative for hypermethylation than for the BRAF V600E mutation, which means that fewer patients go on to further testing, particularly genetic testing which is much more costly. There is a more dramatic cost reduction for strategy 6 than for strategy 5 (despite these having the same diagnostic accuracy) as there is a reduced cost for probands in both the genetic test and the IHC testing.
| Strategy | Scenario | Diagnostic cost per proband (£) | Diagnostic cost per relative (£) | Total diagnostic cost of cohort (£) |
|---|---|---|---|---|
| 3 | Base case (BRAF) | 317 | 4.71 | 578,496 |
| Scenario 2 (methylation) | 317 | 4.59 | 577,767 | |
| 5 | Base case (BRAF) | 318 | 5.29 | 586,043 |
| Scenario 2 (methylation) | 295 | 4.91 | 543,440 | |
| 6 | Base case (BRAF) | 348 | 5.29 | 636,862 |
| Scenario 2 (methylation) | 316 | 4.91 | 577,738 | |
| 7 | Base case (BRAF) | 595 | 5.97 | 1,061,637 |
| Scenario 2 (methylation) | 575 | 5.87 | 1,026,400 |
FIGURE 135.
Cost per proband under methylation scenario.
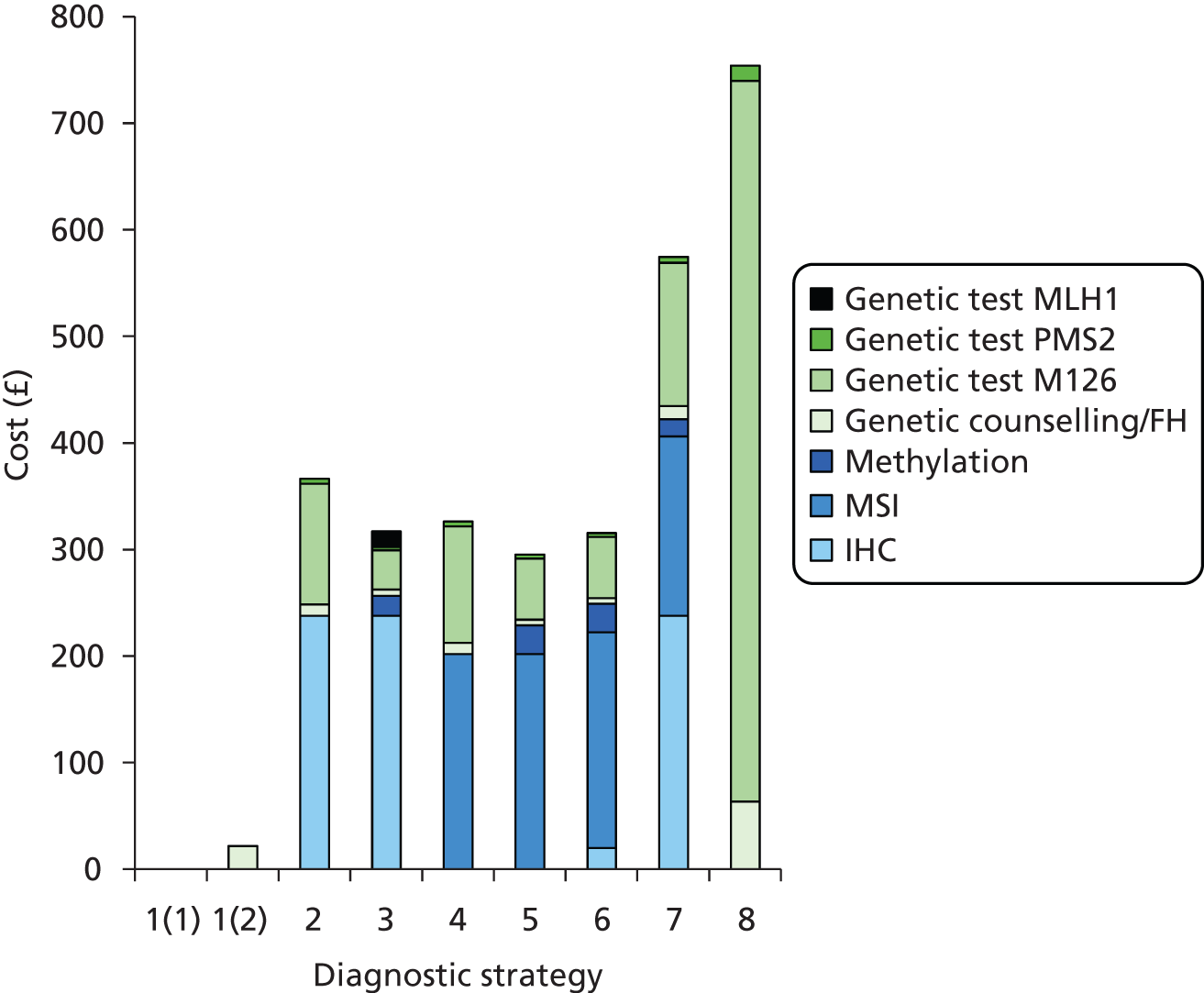
FIGURE 136.
Cost per relative under methylation scenario.
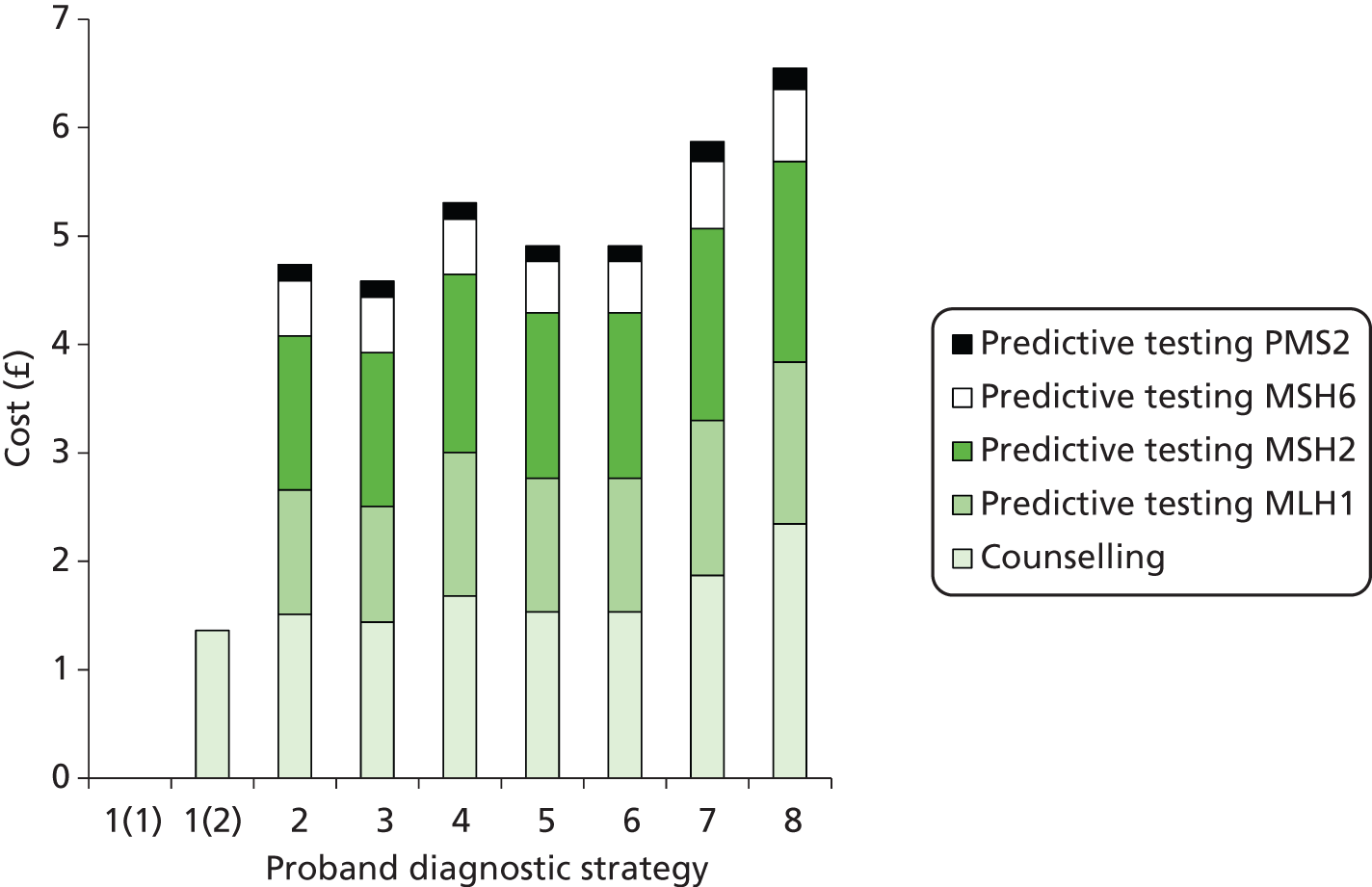
FIGURE 137.
Total diagnostic costs under methylation scenario.
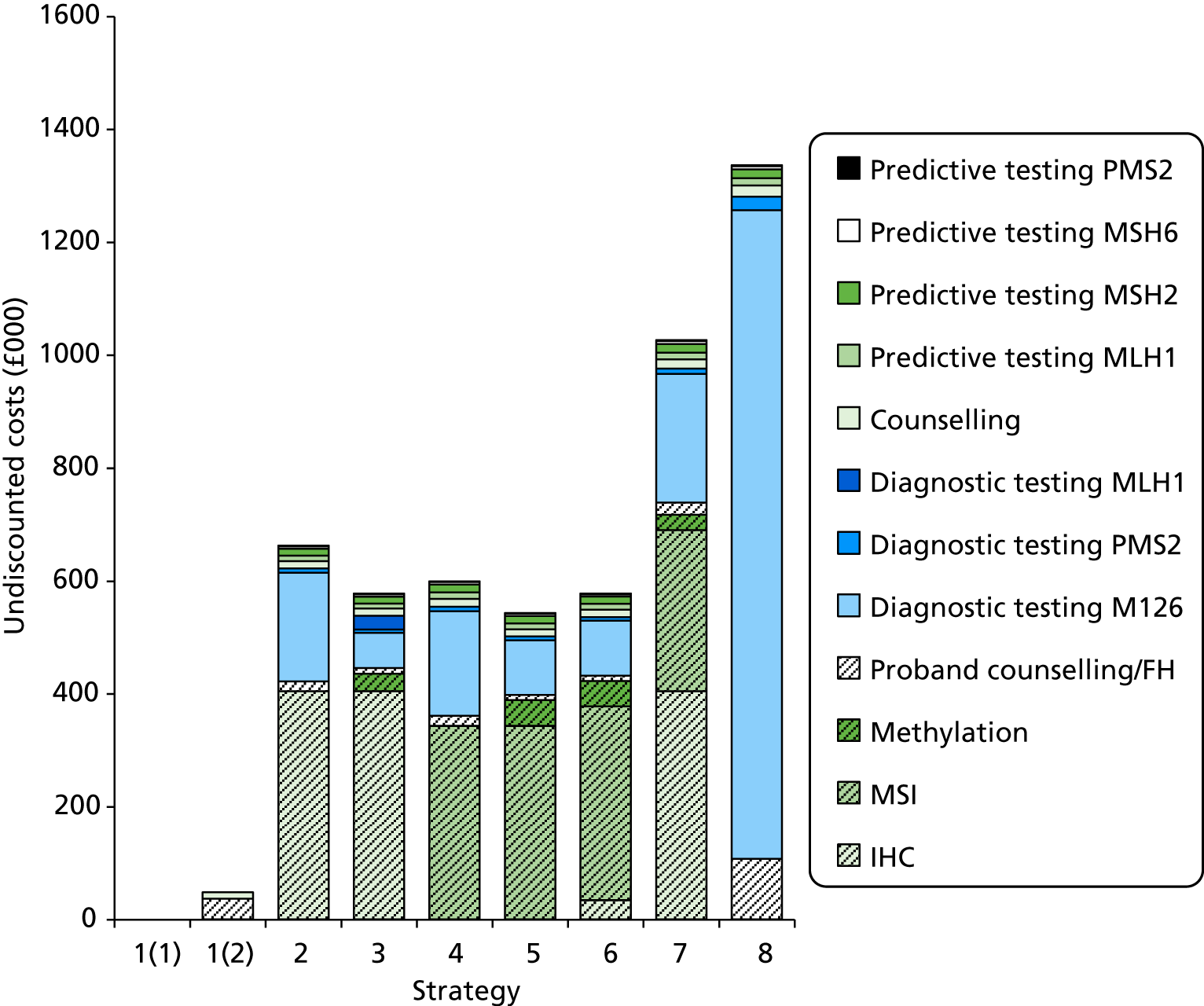
Given the change in number of patients tested positive for LS (and their true LS status), the long-term costs and benefits are also changed. The strategies have reduced long-term QALYs when using methylation compared with BRAF (Figure 138). This would appear to occur because of a decrease in patients testing TP (and an increase in FNs). Similarly, there is a reduction in long-term costs, as fewer patients undergo surveillance colonoscopy and prophylactic TAHBSO (Figure 139).
FIGURE 138.
Incremental discounted QALYs vs. strategy 1(1), when BRAF is replaced with methylation.
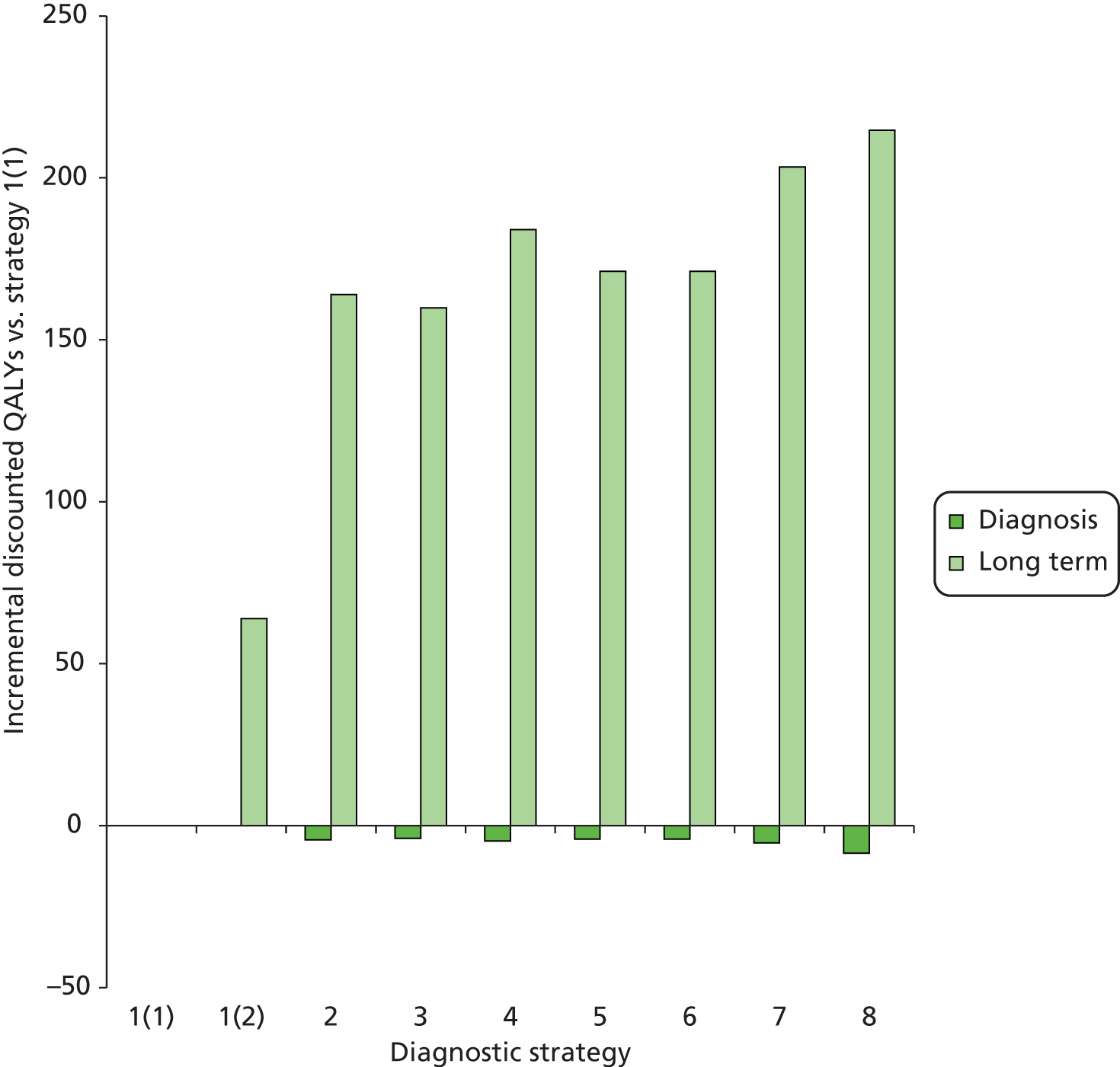
FIGURE 139.
Incremental discounted costs vs. strategy 1(1) when BRAF is replaced with methylation.
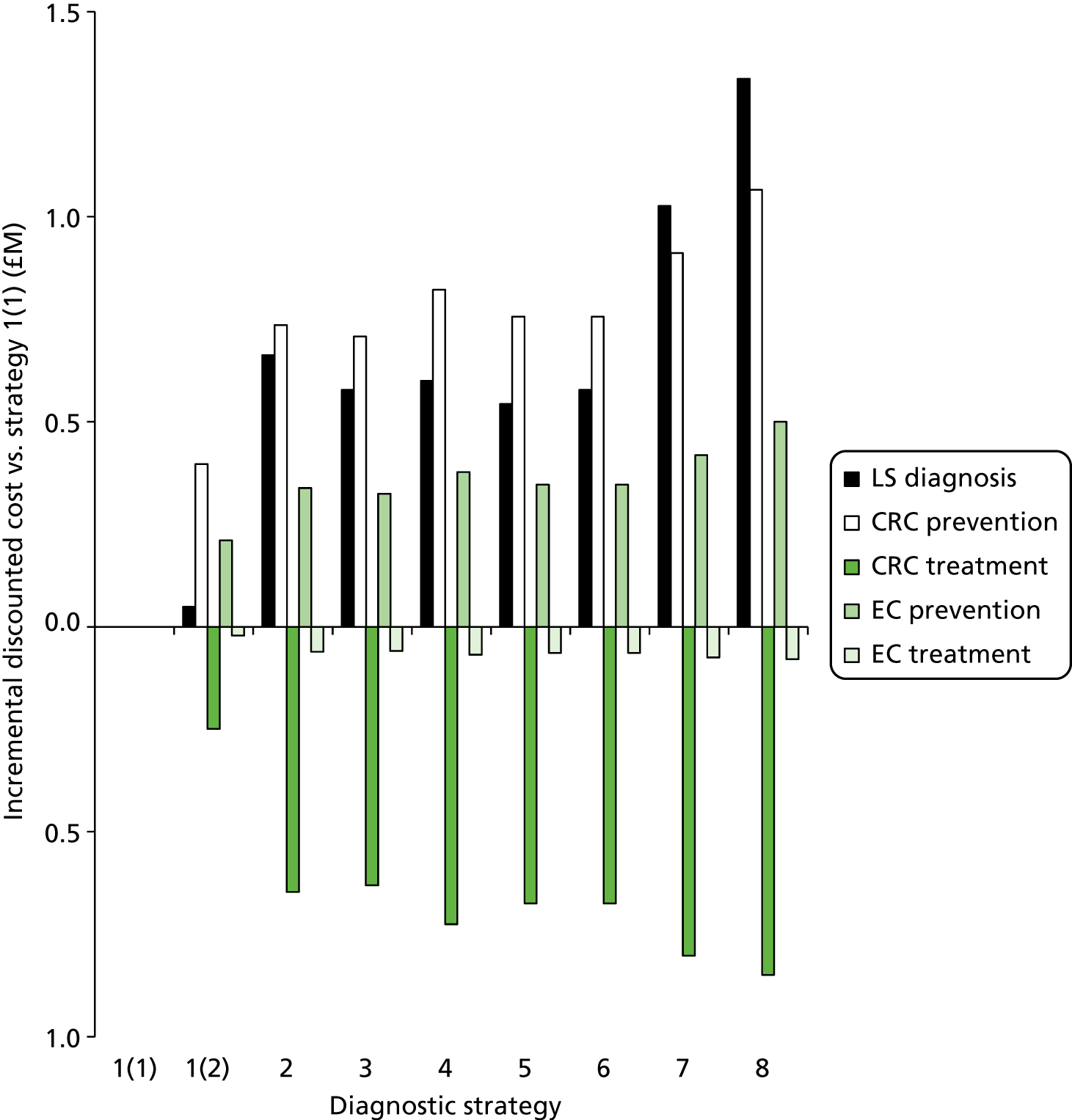
With the reduction in QALYs for those strategies using methylation, there is a change in the order of effectiveness of the strategies. In this scenario, strategy 3 becomes less effective (has a lower QALY gain) than strategy 2, and strategies 5 and 6 become less effective than strategy 4. Strategy 3 has approximately four fewer discounted QALYs than strategy 2, and strategies 5 and 6 have 13 fewer discounted QALYs than strategy 4.
The cost-effectiveness plane showing all strategies when BRAF is replaced with methylation is shown in Figure 140, and the INHB of strategies versus strategy 1(1) in this scenario is shown in Figure 141. These results are also summarised in Table 149.
FIGURE 140.
Incremental discounted costs and QALYs vs. strategy 1(1) when BRAF is replaced with methylation.
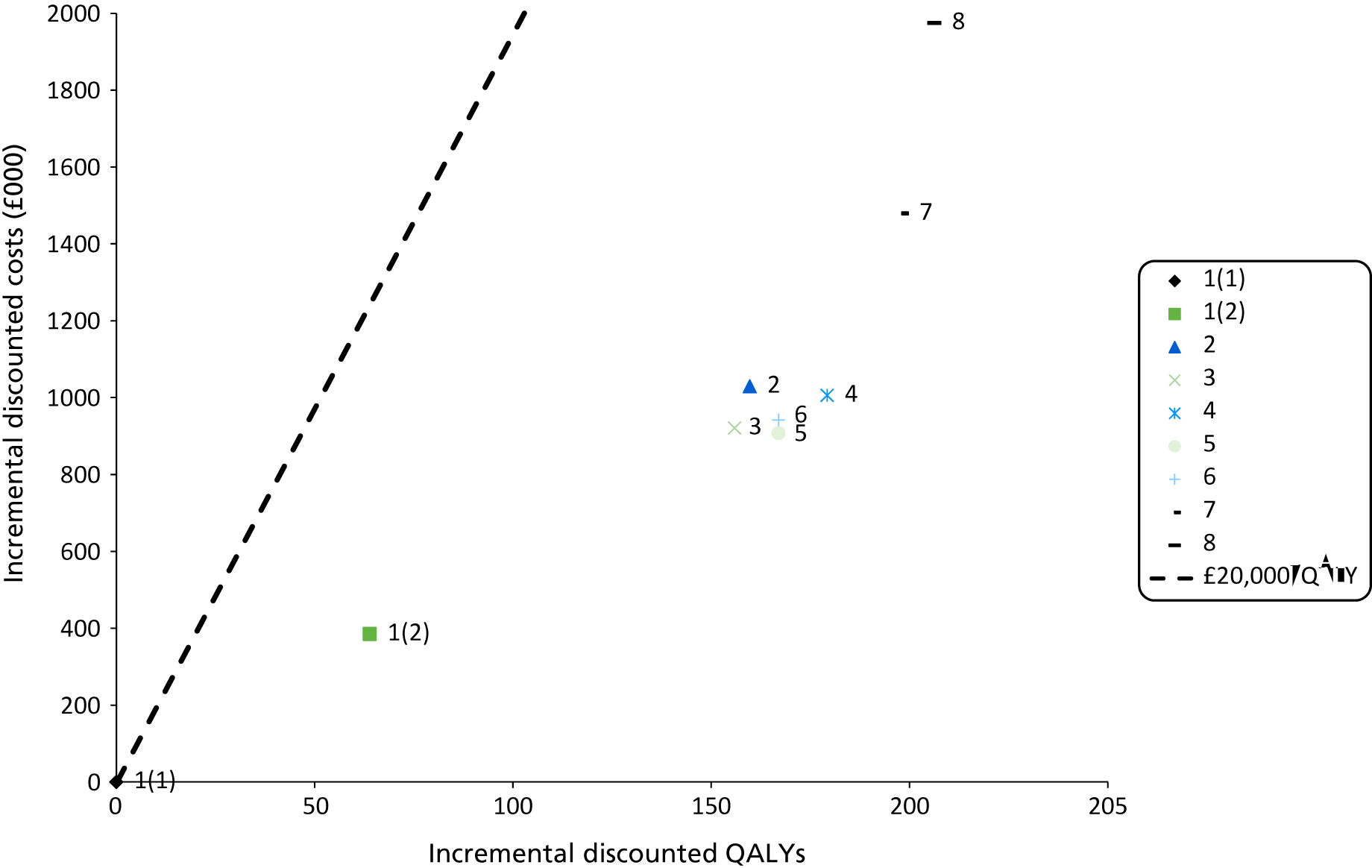
FIGURE 141.
Incremental net health benefit vs. strategy 1(1) when BRAF is replaced with methylation.
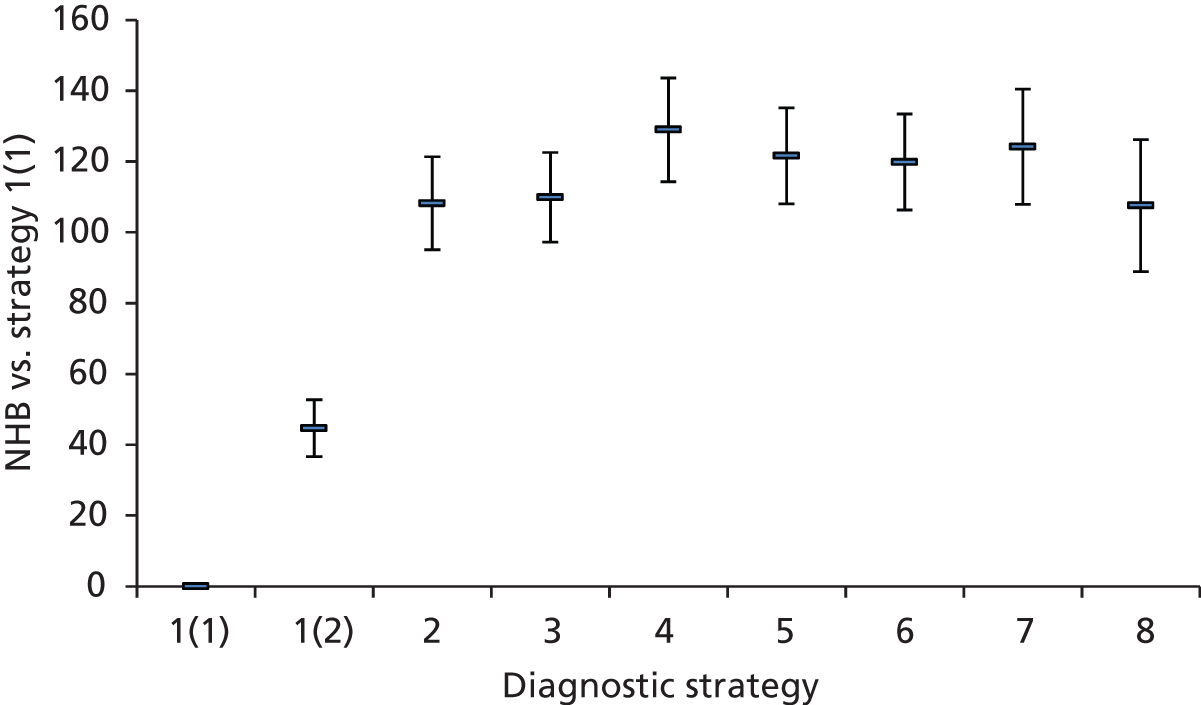
| Strategy | Total discounted QALYs | Total discounted cost (£) | Total NHB at £20,000 per QALY | Incremental discounted QALYs vs. strategy 1(1) | Incremental discounted cost vs. strategy 1(1) (£) | INHB at £20,000 per QALY | ICER vs. strategy 1(1) (£/QALY) | ICER (£/QALY) |
|---|---|---|---|---|---|---|---|---|
| 1(1) | 151,793 | 36,223,787 | 149,982 | – | – | – | – | – |
| 1(2) | 151,857 | 36,608,672 | 150,027 | 64 | 384,885 | 44.7 | 6021 | Extended dominated by strategies 1(1) and 5 |
| 2 | 151,953 | 37,253,017 | 150,091 | 160 | 1,029,230 | 108.3 | 6444 | Dominated by strategy 4 |
| 3 | 151,949 | 37,144,423 | 150,092 | 156 | 920,635 | 109.9 | 5904 | Dominated by strategy 5 |
| 4 | 151,973 | 37,229,210 | 150,111 | 179 | 1,005,423 | 129.0 | 5610 | 7965 |
| 5 | 151,960 | 37,131,507 | 150,104 | 167 | 907,719 | 121.6 | 5436 | 5436 |
| 6 | 151,960 | 37,165,805 | 150,102 | 167 | 942,018 | 119.9 | 5642 | Dominated by strategy 5 |
| 7 | 151,992 | 37,702,908 | 150,106 | 198 | 1,479,121 | 124.1 | 7466 | 25,108 |
| 8 | 152,000 | 38,198,324 | 150,090 | 206 | 1,974,537 | 107.6 | 9571 | 60,472 |
The ICERs versus strategy 1(1) remain < £10,000 per QALY, as they did in the base case. However, the change in order of effectiveness results in a change in the ICERs when the strategies are compared with each other. Strategies 1(2), 2, 3 and 6 are still dominated by other strategies and the ICER for strategy 5 is slightly reduced to £5436 per QALY gained over strategy 1(1), but strategy 4 is no longer extended dominated, instead having an ICER versus strategy 5 of £7965 per QALY gained. The ICERs for strategies 7 and 8 remain > £25,000 per QALY gained, as demonstrated in Table 149. Given these results it would appear that strategy 4, MSI followed by genetic testing, is now the most cost-effective strategy.
Appendix 16 Summary of parameters in the health economic model
| Parameter name | Base-case value | Source | Relevant section in report | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Diagnostic accuracy | |||||||||
| MSI | MLH1 and MSH2 | Sensitivity | 89% | Palomaki et al. 200965 | Chapter 5, Test accuracy | ||||
| Specificity | 90.2% | Palomaki et al. 200965 | |||||||
| MSH6 | Sensitivity | 77% | Palomaki et al. 200965 | ||||||
| Specificity | 90.2% | Palomaki et al. 200965 | |||||||
| PMS2 | Sensitivity | 77% | Assumed | ||||||
| Specificity | 90.2% | Assumed | |||||||
| IHC | Sensitivity | 77.0% | Palomaki et al. 200965 | ||||||
| Specificity | 88.8% | Palomaki et al. 200965 | |||||||
| BRAF after IHC MLH1 abnormal | Sensitivity | 100% | Palomaki et al. 200965 | ||||||
| Specificity | 69% | Palomaki et al. 200965 | |||||||
| BRAF after MSI | Sensitivity | 100% | Domingo et al. 200484 | ||||||
| Specificity | 40% | Domingo et al. 200484 | |||||||
| AC II | Sensitivity | 39% | Hampel et al. 2005,103 Salovaara et al. 2000,180 Green et al. 2009,181 Barnetson et al. 2006,175 Balmana et al. 2008182 | ||||||
| Specificity | 98% | Hampel et al. 2005,103 Salovaara et al. 2000,180 Green et al. 2009,181 Barnetson et al. 2006,175 Balmana et al. 2008182 | |||||||
| Genetic test MLH1, MSH2 and MSH6 (for probands) | Sensitivity | 90% | Dinh et al. 2011,55 Palomaki et al. 200965 | ||||||
| Specificity | 99.97% | Dinh et al. 2011,55 Palomaki et al. 200965 | |||||||
| Genetic test PMS2 (for probands) | Sensitivity | 62% | Dinh et al. 2011,55 Palomaki et al. 200965 | ||||||
| Specificity | 99.97% | Dinh et al. 2011,55 Palomaki et al. 200965 | |||||||
| Targeted genetic test (for relatives) | Sensitivity | 100% | Assumed | ||||||
| Specificity | 100% | Assumed | |||||||
| Numbers of probands and relatives | |||||||||
| Number of probands CRC aged < 50 years | 1699 | ONS cancer registration statistics, England 2010119 | Chapter 5, Numbers of probands | ||||||
| Prevalence of LS in probands | 8.4% | Hampel et al. 2008120 | |||||||
| Distribution of mutations in LS-positive patients | 32% MLH1 | Palomaki et al. 200965 | |||||||
| 39% MSH2 | |||||||||
| 14% MSH6 | |||||||||
| 15% PMS2 | |||||||||
| FP IHC results for probands | 90% MLH1 | Mvundura et al. 201054 | |||||||
| 6% MSH2 | |||||||||
| 2% MSH6 | |||||||||
| 2% PMS2 | |||||||||
| Number of relatives | Five per proband | Barrow et al. 2009,121 unpublished data provided by Ian Frayling | Chapter 5, Numbers of relatives | ||||||
| 8495 total | |||||||||
| Proportion of relatives FDRs | 42% | Jenkins et al. 2006,114 Hampel et al. 2008120 | Chapter 5, Number of first-degree relatives | ||||||
| Proportion of relatives testing positive | 44% | Jenkins et al. 2006,114 Hampel et al. 2008,120 Ian Frayling (unpublished), Munaza Ahmed (unpublished) | Chapter 5, Proportion of relatives testing positive | ||||||
| Adherence to testing and counselling | |||||||||
| MSI | 100% | Ramsey et al. 200349 confirmed by expert Ian Frayling | Chapter 5, Acceptance of testing and surveillance | ||||||
| IHC | 100% | Assumed | |||||||
| BRAF | 100% | Assumed | |||||||
| First genetic test (proband) | 90% | Ladabaum et al. 201148 | |||||||
| Second genetic test (proband) | 100% | Assumed | |||||||
| Genetic counselling (proband) | 92.5% | Clinical experts (Ian Frayling) gave range 90–95% | |||||||
| FH assessment after genetic test (proband) | 100% | Assumed (as included in counselling) | |||||||
| FH assessment when genetic counselling or test declined (proband) | 100% | Assumed | |||||||
| Genetic test (relative) | 96% | Calculated from Palomaki et al. 200965 | |||||||
| Genetic counselling (relative) | 45% | Calculated from Palomaki et al. 200965 | |||||||
| CRC parameters | |||||||||
| Probability of different surgery types for index CRC, for patients not diagnosed with LS | |||||||||
| Colon cancer | Segmental resection | 96% | National Bowel Cancer Audit report 2011135 | Chapter 5, Colorectal cancer surgical management pathways | |||||
| Subtotal colectomy | 4% | ||||||||
| Anterior resection | 0% | ||||||||
| Proctocolectomy | 0% | ||||||||
| Rectal cancer | Segmental resection | 0% | |||||||
| Subtotal colectomy | 0% | ||||||||
| Anterior resection | 98% | ||||||||
| Proctocolectomy | 2% | ||||||||
| Probability index CRC is colon cancer | |||||||||
| With LS | 0.94 males and females | Dinh online appendix55 | Chapter 5, Colorectal cancer incident site | ||||||
| Without LS | 0.58 males | ONS cancer registration statistics, England 2010119 | |||||||
| 0.61 females | |||||||||
| Parameters for CRC incidence model in patients with LS | |||||||||
| β 0 | 0.464 males | Bonadona et al. 20112 | Chapter 5, Incidence rates in patients with Lynch syndrome | ||||||
| 0.435 females | |||||||||
| β 1 | 0.107 males | ||||||||
| 0.108 females | |||||||||
| β 2 | 55.5 males | ||||||||
| 61.3 females | |||||||||
| Proportion with Dukes’ stage at diagnosis | |||||||||
| Dukes’ A | 0.164 | Adapted from table 4.2 of the National Bowel Cancer Audit 2011135 | Chapter 5, Dukes’ stage on diagnosis | ||||||
| Dukes’ B | 0.317 | ||||||||
| Dukes’ C | 0.271 | ||||||||
| Dukes’ D | 0.248 | ||||||||
| Relative survival of patients with CRC by Dukes’ stage across all ages | |||||||||
| 1 year since diagnosis | Dukes’ A | 0.969 | NCIN170 | Chapter 5, Mortality due to colorectal cancer | |||||
| Dukes’ B | 0.917 | ||||||||
| Dukes’ C | 0.815 | ||||||||
| Dukes’ D | 0.380 | ||||||||
| 2 years since diagnosis | Dukes’ A | 0.965 | |||||||
| Dukes’ B | 0.872 | ||||||||
| Dukes’ C | 0.681 | ||||||||
| Dukes’ D | 0.193 | ||||||||
| 3 years since diagnosis | Dukes’ A | 0.957 | |||||||
| Dukes’ B | 0.831 | ||||||||
| Dukes’ C | 0.583 | ||||||||
| Dukes’ D | 0.116 | ||||||||
| 4 years since diagnosis | Dukes’ A | 0.945 | |||||||
| Dukes’ B | 0.799 | ||||||||
| Dukes’ C | 0.522 | ||||||||
| Dukes’ D | 0.083 | ||||||||
| 5 years since diagnosis | Dukes’ A | 0.932 | |||||||
| Dukes’ B | 0.770 | ||||||||
| Dukes’ C | 0.477 | ||||||||
| Dukes’ D | 0.066 | ||||||||
| HRs for CRC mortality by age at diagnosis, compared with CRC mortality across all ages | |||||||||
| < 70 years | First year | 0.599 | ONS cancer survival statistics 2011147 | Chapter 5, Mortality due to colorectal cancer | |||||
| Following 4 years | 0.972 | ||||||||
| Thereafter | 1 | ||||||||
| 70–79 years | First year | 0.956 | |||||||
| Following 4 years | 0.966 | ||||||||
| Thereafter | 1 | ||||||||
| ≥ 80 years | First year | 1.797 | |||||||
| Following 4 years | 1.116 | ||||||||
| Thereafter | 1 | ||||||||
| HR survival for LS carriers | |||||||||
| Dukes’ A | 0.57 | Lin et al. 1998176 | Chapter 5, Mortality due to colorectal cancer for patients with Lynch syndrome | ||||||
| Dukes’ B | 0.57 | ||||||||
| Dukes’ C | 1 | Barnetson et al. 2006175 | |||||||
| Dukes’ D | 1 | ||||||||
| LS surveillance colonoscopy parameters | |||||||||
| Effect of colonoscopy on index CRC incidence rates (HR) | 0.387 | Jarvinen et al. 200094 | Chapter 5, Effect of colonoscopy on index colorectal cancer incidence rates | ||||||
| Stage distribution of CRCs for individuals undergoing colonoscopic surveillance | Dukes’ A | 0.686 | Mecklin et al. 2007188 | Chapter 5, Effect of colonoscopy on Dukes’ stage of incident colorectal cancers | |||||
| Dukes’ B | 0.105 | ||||||||
| Dukes’ C | 0.128 | ||||||||
| Dukes’ D | 0.081 | ||||||||
| Morbidity and mortality of colonoscopy | Mortality | 0.83 per 10,000 | Cairns et al. 20109 | Chapter 5, Morbidity and mortality of Lynch syndrome surveillance colonoscopy | |||||
| Perforation | 4 per 10,000 | Gavin et al. 2013189 | |||||||
| Bleeding | 26 per 10,000 | ||||||||
| of which required admission | 21% | ||||||||
| of which were mild | 73% | ||||||||
| of which were moderate | 18% | ||||||||
| of which were severe | 9% | ||||||||
| Adherence to LS colonoscopic surveillance | |||||||||
| Proband tested LS mutation positive | 80% | Ladabaum et al. 201148 | Chapter 5, Acceptance of testing and surveillance | ||||||
| Proband LS assumed | 70% | Expert opinion from Ladabaum et al. 201148 | |||||||
| Relative tested LS mutation positive | 80% | Ladabaum et al. 201148 | |||||||
| Relative LS assumed | 50% | Expert opinion from Ladabaum et al. 201148 | |||||||
| Effect of colonoscopy on metachronous CRC incidence (HR) | 0.533 | Cirillo et al. 2012187 | Chapter 5, Effect of colonoscopy on metachronous colorectal cancer incidence | ||||||
| CRC surgery | |||||||||
| Effect of CRC surgery on CRC incidence (HRs) | Segmental resection | 1 | Assumed | Chapter 5, Effectiveness of colorectal cancer surgery | |||||
| Anterior resection | 0.94 | Calculated based on CRC incidence | |||||||
| Subtotal colectomy | 0.06 | Calculated based on CRC incidence | |||||||
| Proctocolectomy | 0 | Assumed | |||||||
| Probability patient diagnosed with LS receives more aggressive CRC surgery | 0 | Clinical expert opinion | Chapter 5, More aggressive colorectal cancer surgery for individuals diagnosed with Lynch syndrome | ||||||
| EC | |||||||||
| Cumulative incidence of EC | 0% by age 30 years, 2% by age 40 years, 8% by age 50 years, 23% by age 60 years, 34% by age 70 years, 35% by age 80 years | Bonadona et al. 20112 | Chapter 5, Endometrial cancer incidence | ||||||
| Screening for EC | Not modelled | Assumption | Chapter 5, Screening for endometrial cancer | ||||||
| Probability of dying of EC by year after diagnosis | 9.9% in first year, 5.0% per annum in years 2–3, 2.6% per annum in years 4–5, 0.7% per annum in years 5–10, 0% thereafter | ONS,147 NCIN170 and Cancer Research UK150 | Chapter 5, Mortality due to endometrial cancer | ||||||
| Age when given prophylactic TAHBSO | 45 years for individuals aged ≤ 45 years, otherwise age when tested positive for LS | Expert opinion | Chapter 5, Endometrial cancer in the Peninsula Technology Assessment Group model | ||||||
| Probability of mortality from TAHBSO | 0.0002 | Average over studies reported in Palomaki et al. 200965 | |||||||
| Incidence of EC after TAHBSO | 0% | Schmeler et al. 2006146 | |||||||
| Chemotherapy for EC | Six cycles of TAP (paclitaxel, doxorubicin, cisplatin) | Havrilesky et al. 2009152 | |||||||
| Adherence to EC prevention and treatment | |||||||||
| Adherence to prophylactic TAHBSO for LS-positive probands | 55% | Personal communication with Lorraine Cowley, Northern Genetics Service | Chapter 5, Acceptance of prophylactic total abdominal hysterectomy with bilateral salpingo-oophorectomy | ||||||
| Adherence to prophylactic TAHBSO for LS-positive relatives | 55% | ||||||||
| Adherence to TAHBSO on diagnosis of EC | 100% | Expert opinion | Chapter 5, Endometrial cancer in the Peninsula Technology Assessment Group model | ||||||
| Adherence to radiotherapy | 33% of stage I patients, 100% of stage II and III patients, 50% of stage IV patients | Havrilesky et al. 2009152 | |||||||
| Adherence to chemotherapy | 50% of stage II and III patients, 100% of stage IV patients | ||||||||
| General mortality | |||||||||
| Mortality from other causes | Age specific | ONS life tables 2008–10179 adjusted to remove death from CRC | Chapter 5, Mortality due to other causes | ||||||
| Utilities | |||||||||
| CRC disutilities | Dukes’ A | 0 | Ramsey et al. 2000217 | Chapter 5, Impact of colorectal cancer on quality of life according to Dukes’ stage | |||||
| Dukes’ B | 0 | ||||||||
| Dukes’ C | 0 | ||||||||
| Dukes’ D | 0.13 | Mittman et al. 2009225 | |||||||
| CRC surgery disutility | 0 | Assumed | Chapter 5, Impact of colorectal cancer surgery on quality of life | ||||||
| EC disutility | 0 | Assumed | Chapter 5, Impact of endometrial cancer on quality of life | ||||||
| TAHBSO disutility | 0 | Assumed | Chapter 5, Impact of prophylactictotal abdominal hysterectomy with bilateral salpingo-oophorectomyon quality of life | ||||||
| Colonoscopy disutility | 0 | Assumed based on Whyte et al. 2012214 | Chapter 5, Impact of Lynch syndrome surveillance colonoscopies on quality of life | ||||||
| Psychological disutilities | |||||||||
| Proband | Kuppermann et al. 2013212 | Chapter 5, Psychological impacts of Lynch syndrome testing and management on quality of life | |||||||
| Test declined, surgery not offered | 0.04 | ||||||||
| Test declined, accept TAHBSO (females only) | 0.05 | ||||||||
| Test declined, decline TAHBSO (females only) | 0.11 | ||||||||
| Test accepted, LS negative | 0.00 | ||||||||
| Test accepted, LS positive, surgery not offered | 0.02 | ||||||||
| Test accepted, LS positive, accept TAHBSO (females only) | 0.03 | ||||||||
| Test accepted, LS positive, decline TAHBSO (females only) | 0.09 | ||||||||
| Relative | |||||||||
| Test declined, surgery not offered | 0.04 | ||||||||
| Test declined, accept TAHBSO (females only) | 0.08 | ||||||||
| Test declined, decline TAHBSO (females only) | 0.11 | ||||||||
| Test accepted, LS negative | 0.00 | ||||||||
| Test accepted, LS positive, surgery not offered | 0.02 | ||||||||
| Test accepted, LS positive, accept TAHBSO (females only) | 0.06 | ||||||||
| Test accepted, LS positive, decline TAHBSO (females only) | 0.09 | ||||||||
| Length of time psychological disutility applied for | 4 months | Assumption based on Heshka et al. 2008239 | |||||||
| Costs | |||||||||
| Diagnostic costs | |||||||||
| MSI | £202 | Average of Oxford Medical Genetics Laboratories,122 All Wales Molecular Genetics Laboratory,123 West Midlands Regional Genetics Laboratory (via the UKGTN)244 | Chapter 5, Costs of tumour testing and Cost of genetic testing | ||||||
| IHC | £238 | Dr Mark Arends (Department of Pathology, University of Cambridge) and Dr Ian Frayling (on behalf of All Wales Genetics Service) | |||||||
| BRAF | £118 | Average of personal communication with Mr Michael Gandy (UCL-Advanced Diagnostics), East of Scotland Regional Genetics Service,124 All Wales Molecular Genetics Laboratory123 | |||||||
| Proband genetic test MLH1, MSH2 and MSH6 | £812 | Average of Oxford Medical Genetics Laboratories,122 All Wales Molecular Genetics Laboratory,123 East Anglian Medical Genetics Laboratories,125 Yorkshire Regional Genetics Service126 | |||||||
| Proband genetic test PMS2 | £735 | Yorkshire Regional Genetics Service126 | |||||||
| Proband genetic test MLH1 | £464 | Average of All Wales Molecular Genetics Laboratory,123 East of Scotland Regional Genetics Service,124 East Anglian Medical Genetics Laboratories,125 Yorkshire Regional Genetics Service126 | |||||||
| Proband genetic counselling | £67 | The PSSRU243 and personal communication with Professor Mary Porteous (South East Scotland Genetic Service) | |||||||
| Taking FH | £22 | The PSSRU243 and personal communication with Professor Mary Porteous (South East Scotland Genetic Service) | |||||||
| Targeted genetic test for relatives (MLH1) | £169 | Average of Oxford Medical Genetics Laboratories,122 All Wales Molecular Genetics Laboratory,123 East of Scotland Regional Genetics Service,124 East Anglian Medical Genetics Laboratories,125 Yorkshire Regional Genetics Service126 | |||||||
| Targeted genetic test for relatives (MSH2) | £172 | Average of Oxford Medical Genetics Laboratories,122 All Wales Molecular Genetics Laboratory,123 Yorkshire Regional Genetics Service126 | |||||||
| Targeted genetic test for relatives (MSH6) | £172 | Average of Oxford Medical Genetics Laboratories,122 All Wales Molecular Genetics Laboratory,123 Yorkshire Regional Genetics Service126 | |||||||
| Targeted genetic test for relatives (PMS2) | £176 | Yorkshire Regional Genetics Service126 | |||||||
| Relative genetic counselling | £67 | The PSSRU243 and personal communication with Professor Mary Porteous (South East Scotland Genetic Service) | |||||||
| CRC prevention costs | |||||||||
| Cost of colonoscopy | £395 | Department of Health reference costs 2011–12151 | Chapter 5, Costs of Lynch syndrome surveillance colonoscopies | ||||||
| Cost of morbidity and mortality due to colonoscopies | Mortality | £5134 | Whyte et al. 2012214 | Chapter 5, Costs of morbidity and mortality due to Lynch syndrome surveillance colonoscopies | |||||
| Perforation | £5134 | Department of Health reference costs 2011–12151 | |||||||
| Mild bleeding not requiring admission | £0 | Assumption | |||||||
| Mild bleeding requiring admission | £318 | Whyte et al. 2012214 | |||||||
| Moderate bleeding | £490 | Department of Health reference costs 2011–12151 | |||||||
| Severe bleeding | £1984 | Department of Health reference costs 2011–12151 | |||||||
| CRC treatment costs: colon cancer | |||||||||
| Diagnosis (incurred at diagnosis for all Dukes’ stages) | £499 | Based on Trueman et al. 2007134 | Chapter 5, Costs of colorectal cancer treatment in the Peninsula Technology Assessment Group model | ||||||
| Primary chemotherapy and radiotherapy (incurred at diagnosis) | Dukes’ A | £0 | |||||||
| Dukes’ B | £5755 | ||||||||
| Dukes’ C | £13,133 | ||||||||
| Dukes’ D | £13,133 | ||||||||
| Follow-up surveillance (mean annual cost incurred for maximum 5 years from index CRC for all Dukes’ stages) | £269 | ||||||||
| Recurrence surgery and chemotherapy (incurred in last year of life if patient dies of CRC within 5 years of diagnosis, for all Dukes’ stages) | £12,578 | ||||||||
| Stoma care (annual cost incurred every year after surgery for all Dukes’ stages) | £1684 for 11% of patients | ||||||||
| Palliative care (incurred in last year of life if patient dies of CRC, for all Dukes’ stages) | £10,141 | ||||||||
| CRC treatment costs: rectal cancer | |||||||||
| Diagnosis (incurred at diagnosis for all Dukes’ stages) | £499 | Based on Trueman et al. 2007134 | Chapter 5, Costs of colorectal cancer treatment in the Peninsula Technology Assessment Group model | ||||||
| Primary chemotherapy and radiotherapy (incurred at diagnosis) | Dukes’ A | £0 | |||||||
| Dukes’ B | £2848 | ||||||||
| Dukes’ C | £7628 | ||||||||
| Dukes’ D | £7628 | ||||||||
| Follow-up surveillance (mean annual cost incurred for maximum 5 years from index CRC for all Dukes’ stages) | £256 | ||||||||
| Recurrence surgery and chemotherapy (incurred in last year of life if patient dies of CRC within 5 years of diagnosis, for all Dukes’ stages) | £12,216 | ||||||||
| Stoma care (annual cost incurred every year after surgery for all Dukes’ stages) | £1684 for 49% of patients | ||||||||
| Palliative care (incurred in last year of life if patient dies of CRC, for all Dukes’ stages) | £9236 | ||||||||
| CRC surgery costs | |||||||||
| CRC surgery costs for sporadic CRC | Segmental resection | £6104 | Department of Health reference costs 2011–12151 | Chapter 5, Colorectal cancer surgery costs | |||||
| Subtotal colectomy IRA | £7331 | ||||||||
| Anterior resection | £7399 | ||||||||
| Proctocolectomy IPAA | £7441 | ||||||||
| CRC surgery costs for LS CRC | Segmental resection | £6154 | |||||||
| Subtotal colectomy IRA | £7331 | ||||||||
| Anterior resection | £7399 | ||||||||
| Proctocolectomy IPAA | £7441 | ||||||||
| EC costs | |||||||||
| Cost of prophylactic TAHBSO | £3322 | Department of Health reference costs 2011–12151 | Chapter 5, Cost of endometrial cancer prevention | ||||||
| Cost of EC treatment | TAHBSO | £3877 | Department of Health Reference Costs 2011/12151 | Chapter 5, Cost of surgery for endometrial cancer | |||||
| Radiotherapy | £2753 | Havrilesky et al. 2009152 | Chapter 5, Cost of radiotherapy for endometrial cancer | ||||||
| Chemotherapy | £3005 | Fleming et al. 2004,256 eMit database,153 Department of Health reference costs 2011–12151 | Chapter 5, Cost of chemotherapy for endometrial cancer | ||||||
| Follow-up | £0 | Assumption | Chapter 5, Cost of follow-up management of endometrial cancer | ||||||
| Other parameters | |||||||||
| Discount rate | 3.5% costs and benefits | NICE reference case132 | Chapter 5, Perspective, discounting, time horizon | ||||||
Glossary
- BRAF
- A human gene that makes a protein called B-raf (a member of the Raf kinase family).
- BRAF V600E
- A mutation of the BRAF gene detected in a range of carcinomas, including colorectal cancer.
- Constitutional genetic testing
- Tests for mutations that affect all cells in the body and have been present since conception (also known as germline testing).
- Constitutional mutation
- A genetic mutation present in all cells (also known as germline).
- Cost-effectiveness analysis
- An economic analysis that converts effects into health terms and describes the costs for additional health gain.
- Decision modelling
- A theoretical construct that allows the comparison of the relationship between costs and outcomes of alternative health-care interventions.
- Distant metastases
- Cancer that has spread from the original (primary) tumour to distant organs or distant lymph nodes.
- DNA mismatch repair
- A process that corrects mismatches generated during deoxyribonucleic acid (DNA) replication.
- Dysplastic
- Abnormal development or growth of tissue, organs or cells.
- False negative
- Incorrect negative test result (number of diseased persons with a negative test result).
- False positive
- Incorrect positive test result (number of non-diseased persons with a positive test result).
- Germline
- Inherited material that comes from the eggs or sperm and is passed on to offspring.
- Germline mutation
- A detectable and heritable variation in the lineage of germ cells, which is subsequently transferred to offspring and gives rise to constitutional mutation.
- Immunoreactivity
- A measure of the immune reaction caused by an antigen.
- Incremental cost-effectiveness ratio
- The difference in the mean costs of two interventions in the population of interest divided by the difference in the mean outcomes in the population of interest.
- Index test
- The test whose performance is being evaluated.
- Locoregional metastases
- Metastasis (spread) of a cancer only within the region in which it arose.
- Meta-analysis
- Statistical techniques used to combine the results of two or more studies and obtain a combined estimate of effect.
- Metachronous
- Occurring or starting at different times.
- Metastatic disease
- The spread of cancer from one organ or body part to another.
- Microsatellite instability
- Abnormal patterns of microsatellite repeats observed when DNA is amplified from a tumour with defective mismatch repair compared with DNA amplified from surrounding normal tissue.
- Microsatellite stable
- No evidence of abnormal patterns of microsatellite repeats or defective mismatch repair.
- Net survival
- The survival calculated from the estimated excess hazard of mortality caused by a condition.
- Optimum cut-off
- The cut-off score which demonstrates the best trade-off between sensitivity and specificity.
- Polymerase chain reaction
- A technology used for amplifying DNA sequences.
- Predictive testing
- Testing for known mutations.
- Primary tumour
- A tumour growing at the anatomical site where tumour progression began.
- Proband
- The first affected family member.
- Quality-adjusted life-year
- A measure of health gain, used in economic evaluations, in which survival duration is weighted or adjusted by the patient’s quality of life during the survival period.
- Receiver operating characteristic curve
- A graph which illustrates the trade-offs between sensitivity and specificity which result from varying the diagnostic threshold.
- Reference standard
- The best currently available diagnostic test, against which the index test is compared.
- Regional metastases
- The spread of cancer beyond the initial site to regional lymph nodes.
- Relative survival
- The observed survival within a group (e.g. people with colorectal cancer) as a proportion of the expected survival for a group with the same age and sex distribution.
- Sensitivity
- Proportion of individuals with the target disorder who have a positive test result.
- Single-gate study
- Study in which a single sample of individuals is assessed by both the index test and reference standard.
- Specificity
- Proportion of individuals without the target disorder who have a negative test result.
- Sporadic colorectal cancer
- Colorectal cancer with no apparent hereditary component.
- T1, T2, T3, T4
- Stages of cancer.
- Two-gate study
- Studies which employ separate sampling schemes for diseased and non-diseased participants, with both groups being assessed by the index test.
List of abbreviations
- 5-FU
- fluorouracil
- AC
- Amsterdam criteria
- ACPGBI
- Association of Coloproctology of Great Britain and Ireland
- APER
- abdominoperineal excision of the rectum
- ASSIA
- Applied Social Sciences Index and Abstracts
- BSG
- British Society of Gastroenterology
- CEA
- carcinoembryonic antigen
- CI
- confidence interval
- CINAHL
- Cumulative Index to Nursing and Allied Health Literature
- CRC
- colorectal cancer
- CSA
- Cambridge Scientific Abstracts
- CT
- computerised tomography
- DAB-IHC
- 3,3’-diaminobenzidine immunohistochemistry
- DHPLC
- denaturing high-performance liquid chromatography
- DNA
- deoxyribonucleic acid
- EC
- endometrial cancer
- EGAPP
- Evaluation of Genomic Applications in Practice and Prevention
- EMEA
- European Medicines Agency
- eMit
- electronic Market Information Tool
- EQ-5D
- European Quality of Life-5 Dimensions
- FAP
- familial adenomatous polyposis
- FDA
- Food and Drug Administration
- FDR
- first-degree relative
- FH
- family history
- FN
- false negative
- FP
- false positive
- HCHS
- Hospital and Community Health Services
- HMIC
- Health Management Information Consortium
- HNPCC
- hereditary non-polyposis colorectal cancer
- HR
- hazard ratio
- HRG
- Healthcare Resource Group
- HRQoL
- health-related quality of life
- HTA
- health technology assessment
- HUI3
- Health Utilities Index Mark 3
- ICD-10
- International Classification of Diseases, Tenth Edition
- ICER
- incremental cost-effectiveness ratio
- IHC
- immunohistochemistry
- INHB
- incremental net health benefit
- InSiGHT
- International Society for Gastrointestinal Hereditary Tumours
- IPAA
- ileal pouch–anal anastomosis
- IPS
- individual patient simulation
- IRA
- ileorectal anastomosis
- ISI
- Institute for Scientific Information
- IV
- intravenous
- LAVH
- laparoscopic-assisted vaginal hysterectomy
- LS
- Lynch syndrome
- MLH1
- MutL homologue 1
- MLPA
- multiplex ligation-dependent probe amplification
- MMR
- mismatch repair
- MSH2
- MutS homologue 2
- MSH6
- MutS homologue 6
- MSI
- microsatellite instability
- MSI-H
- microsatellite instability high
- MSI-L
- microsatellite instability low
- MSS
- microsatellite stable
- NCI
- National Cancer Institute
- NCIN
- National Cancer Intelligence Network
- NEQAS
- National External Quality Assessment Scheme
- NHB
- net health benefit
- NHS EED
- NHS Economic Evaluation Database
- NICE
- National Institute for Health and Care Excellence
- NIHR
- National Institute for Health Research
- NPV
- negative predictive value
- NRR
- National Research Register
- OC
- ovarian cancer
- ONS
- Office for National Statistics
- OPCS
- Office of Population, Censuses and Surveys
- PCR
- polymerase chain reaction
- PenTAG
- Peninsula Technology Assessment Group
- PMS2
- postmeiotic segregation increased 2
- PPV
- positive predictive value
- PSS
- Personal Social Services
- PSSRU
- Personal Social Services Research Unit
- QALY
- quality-adjusted life-year
- QD-IHC
- quantum dot immunohistochemistry
- QUADAS-2
- Quality Assessment of Diagnostic Accuracy Studies-2
- RCT
- randomised controlled trial
- ROC
- receiver operating characteristic
- SDR
- second-degree relative
- SEER
- Surveillance, Epidemiology, and End Results
- SF-36
- Short Form questionnaire-36 items
- TA
- technology assessment
- TAHBSO
- total abdominal hysterectomy with bilateral salpingo-oophorectomy
- TLH
- total laparoscopic hysterectomy
- TN
- true negative
- TNM
- tumour node metastasis
- TP
- true positive
- TVU
- transvaginal ultrasound
- UKGTN
- UK Genetic Testing Network
- YHEC
- York Health Economics Consortium