

Notes
Article history
The research reported in this issue of the journal was funded by the HTA programme as project number 13/87/08. The contractual start date was in January 2015. The draft report began editorial review in December 2016 and was accepted for publication in June 2017. The authors have been wholly responsible for all data collection, analysis and interpretation, and for writing up their work. The HTA editors and publisher have tried to ensure the accuracy of the authors’ report and would like to thank the reviewers for their constructive comments on the draft document. However, they do not accept liability for damages or losses arising from material published in this report.
Declared competing interests of authors
Catriona McDaid is a member of the National Institute for Health Research Health Technology Assessment and Efficacy and Mechanism Evaluation Editorial Board. Christine Moffatt has received grant funding from 3M UK PLC and Smith and Nephew, two health science-based technology companies, outside the submitted work.
Permissions
Copyright statement
© Queen’s Printer and Controller of HMSO 2018. This work was produced by Tilbrook et al. under the terms of a commissioning contract issued by the Secretary of State for Health and Social Care. This issue may be freely reproduced for the purposes of private research and study and extracts (or indeed, the full report) may be included in professional journals provided that suitable acknowledgement is made and the reproduction is not associated with any form of advertising. Applications for commercial reproduction should be addressed to: NIHR Journals Library, National Institute for Health Research, Evaluation, Trials and Studies Coordinating Centre, Alpha House, University of Southampton Science Park, Southampton SO16 7NS, UK.
2018 Queen’s Printer and Controller of HMSO
Chapter 1 Background
Chronic venous leg ulcers
Chronic venous leg ulcers (VLUs) are wounds of the lower limb caused by a diseased venous system, which results in swollen legs and damage to the tissues, usually around the ankles. VLUs are most commonly the result of severe varicose veins, a previous deep-vein thrombosis, trauma or failure of the calf muscle pump, all of which result in impaired venous return. Obesity and immobility are additional important factors contributing to venous dysfunction. 1
The VLUs may take many months to heal (with approximately 25% failing to heal completely), during which time they result in significant suffering and reduction in quality of life for patients. 2 VLUs have a tendency to become recurrent, with rates of recurrence estimated at between 18% and 28%. 3 As a result, the management of VLUs represents a substantial cost to the NHS, the majority of which is attributed to nurse time. Estimated lifetime prevalence of VLUs is between 1% and 3% of the elderly population in the USA and Europe. 4 It is estimated that 1% of the adult population will suffer from leg ulcers at some point in their life. 5 Furthermore, incidence and prevalence of ulceration is predicted to increase as a result of the increasing age and obesity of the population in the USA and Europe. A recent cohort study conducted in the UK estimated that 278,000 VLUs per year are managed by the NHS. 6 Furthermore, the annual cost to the NHS of this management was estimated to be £941M, with substantially more cost associated with unhealed wounds. 7
Current treatment strategies
Current treatment strategies for VLUs focus on efforts to reduce venous hypertension. At present, compression is the main treatment for venous ulceration and few additional therapies have robust evidence to suggest they improve healing rates.
Compression therapy
The mainstay of treatment of leg ulcers is graded compression therapy (target pressure of 40 mmHg) and this is the recommended first-line treatment in UK guidelines. 2 The aim of compression therapy is the reduction of venous hypertension, improvement in calf muscle function and the creation of a wound environment conducive to wound healing. Compression therapy in the form of bandages and hosiery has been shown to be effective in many randomised controlled trials (RCTs). 8 However, despite this treatment, patients take many months to heal (with median healing times of approximately 12 weeks in previous trials)3 and for some patients compression therapy does not result in resolution of their leg ulcers. The use of compression (as well as dressings, largely to manage the wound exudate) can be expensive as nurse time is required to change bandages, which can be required weekly or more frequently.
In addition, effective treatment of VLU requires adherence to compression therapy which, for many patients, is uncomfortable and sometimes painful and inconvenient for everyday life (compression is bulky and dressings have to be changed several times weekly). In addition, the use of thicker bandaging systems, such as four-layer bandaging, may restrict movement of the ankle and cause difficulty in wearing shoes. 9
Topical therapies
The most frequently used topical antimicrobials in wound care practice are chlorhexidine, iodine, silver-containing products, mupriocin (Bactroban®, GlaxoSmithKline, Brentford, UK) and fucidic acid. Historically, agents such as acetic acid, honey, hydrogen peroxide, sodium hypochlorite, potassium permanganate and proflavine have all been used. 10 There is currently a lack of reliable evidence to support an association between topical agents and reduction in time to healing in VLUs. 11
Adjunctive drug therapies
A recent Cochrane review has shown pentoxifylline to be an effective adjunct to compression therapy and possibly more effective than placebo or no treatment in the absence of compression. 12 However, pentoxifylline is not commonly prescribed in the NHS13 and has common and intolerable side effects, some of which have the potential to be life-threatening. 14 Other adjunctive drugs, including venoactive drugs, are not recommended owing to insufficient evidence regarding their use and unclear mechanism of action. 15
Surgery
Surgery to treat superficial varicose veins has been shown to prevent recurrence of ulcers once they have healed but does not improve time to healing of existing ulcers. 16 An ongoing RCT is further investigating surgery as a treatment for chronic ulceration, comparing early versus delayed endovenous treatment of superficial venous reflux. 17 This study is due to publish in November 2018. 17
Other therapies
Research into the use of novel cell-based therapies, such as allogenic cells and growth factors, is currently in progress. 18–20 Owing to their cost and associated side effects, it is thought that such therapies are unlikely to be made widely available. 18 If other treatments were able to reduce the time to healing, this would be a significant breakthrough.
Potential role of aspirin as a treatment for venous leg ulcers
Aspirin (also known as acetylsalicylic acid) has been widely used as a medication for > 100 years and is inexpensive, readily available and generally safe to use. Aspirin is a cyclo-oxygenase inhibitor that irreversibly reduces prostaglandin 2 and thromboxane A2. 21 At low doses, it is used very widely to reduce cardiovascular events in those at high risk. 22
The exact mechanism by which aspirin may improve time to healing of VLUs is unclear but it is potentially associated with both the inhibition of platelet activation and the reduction of inflammation. 23
Possible adverse events (AEs) associated with the use of aspirin include gastric ulceration and other gastrointestinal effects. Other effects include liver and renal toxicity, exacerbation of asthma and dermatological reactions. Antiplatelet drugs, when administered in combination with anticoagulants, are associated with a higher risk of gastrointestinal bleeding than that associated with each drug class used alone. 24
Existing evidence on aspirin in the treatment of venous leg ulcers
To date, there have been two small RCTs that have investigated the use of aspirin (300 mg/day) in patients with VLUs of ≥ 2 cm2 in area. The first, a UK-based study in 20 participants, reported healing of 38% of ulcers in the intervention group (aspirin in combination with compression therapy), compared with 0% in the control group (placebo in combination with compression therapy), over a study period of 4 months. The average time to healing was not reported. 25 del Río Solá et al. 26 reported a study of 51 participants to whom aspirin was given in combination with compression therapy (n = 23) compared with compression therapy alone (n = 28). The researchers reported the average time to healing as 12 weeks in the aspirin group compared with 22 weeks in the control group, but that there was no significant difference between groups in the proportion of patients with ulcers healed (74% in the aspirin group and 75% in the control group). These two studies were the only RCTs identified in a recently conducted Cochrane systematic review23 and, owing to variations and limitations in the data, a meta-analysis was not undertaken. Application of GRADE (Grading of Recommendations, Assessment, Development and Evaluations)27 to the data highlighted that the evidence was of low to very low quality.
Explanation of rationale
The Aspirin for Venous leg Ulcers Randomised Trial (AVURT) was undertaken to address the primary question of whether or not the addition of 300 mg of daily aspirin to standard evidence-based therapies demonstrates evidence of a reduction in time to healing of VLUs. This pilot trial was developed to explore this question as well as assessing the feasibility (especially in terms of participant recruitment and treatment compliance) and safety (in terms of aspirin-related AEs) of conducting a larger-scale pragmatic study, powered to investigate the clinical effectiveness and cost-effectiveness of aspirin for VLU healing.
This research is important because leg ulcers are common and costly and result in significant patient suffering. 2 If aspirin, which is commonly used in many patients, was able to reduce the time to healing of VLU with limited risk of treatment-related harm, then this would result in a potentially important reduction in resource use and an improvement in patients’ health-related quality of life. Because aspirin is generally safe, cheap, well tolerated (for most patients) and widely available, the potential impact on this population is large.
Two previously conducted RCTs have been performed on the use of aspirin in the treatment of VLUs. 25,26 The findings of both trials suggested that there may be benefit in patients with VLUs taking aspirin: one reported that a greater proportion of patients healed with 300 mg of aspirin together with standard compression bandaging25 and one reported a shorter time to healing with 300 mg of aspirin in conjunction with gradual compression therapy. 26 However, both trials have been assessed as being at a risk of bias. 23 The authors of the Cochrane review23 concluded that the low-quality and insufficient evidence from the two included trials meant that they were unable to make definitive claims on the benefits and potential harm of oral aspirin, as an adjunct to compression therapy, on the recurrence and healing of VLUs. The Cochrane review23 recommended that further high-quality studies were needed.
A RCT is required to assess the potential effectiveness and safety profile of aspirin in this population. However, it would be premature to conduct a full trial initially, not least as it is not clear how many people with VLUs currently take aspirin or other antiplatelet medications and the potential impact of this on the design and feasibility of any future study.
During the registration of AVURT, we identified two other RCTs investigating aspirin for VLUs. ASPiVLU (ASPirin in Venous Leg Ulcer Healing)28 was a trial being conducted in Australia that was planning to randomise patients with VLU to receive either 300 mg of aspirin or placebo. The primary end point of that study was time to complete healing of reference ulcer at or before 12 weeks post randomisation. Aspirin4VLU (Low Dose Aspirin for Venous Leg Ulcers) was a trial being conducted in New Zealand that was planning to randomise VLU patients to either 150 mg of aspirin or placebo in addition to standard care. The primary end point was time to complete healing of reference/largest ulcer. A secondary outcome of this study is change in estimated reference ulcer area from baseline to 24 weeks. 29
Research objectives
To assess the efficacy of aspirin on time to healing of VLUs, to examine safety issues in this cohort of patients and to assess the feasibility of proceeding from a Phase II trial to a Phase III trial of clinical effectiveness and cost-effectiveness.
Primary objective
To compare the effects of 300 mg of aspirin plus standard care with placebo plus standard care on time to healing of the reference chronic VLU (largest eligible venous ulcer).
Secondary objectives
To assess the safety of aspirin in patients with VLUs and feasibility of leading directly from the pilot Phase II trial into a larger pragmatic study (Phase III) of effectiveness and efficiency, and check, in accordance with the Acceptance Checklist for Clinical Effectiveness Pilot Trials (ACCEPT) criteria,30 whether or not the pilot study fulfilled four criteria:
-
confirming that the effect sizes in the British and Spanish RCTs were too large, but
-
confirming that smaller effect sizes were still plausible, while
-
confirming that the intervention does not lead to unacceptably high rates of serious adverse events (SAEs), and
-
confirming that we can recruit at the planned rate.
Additional objective
To perform an individual patient-level meta-analysis using the data from AVURT and other published25,26,31 and unpublished studies [e.g. A Carolina Weller Barker, I Darby, T Haines, M Underwood, S Ward, P Aldons, E Dapiran, JJ Madan, P Loveland, A Sinha, M Vicaretti, R Wolfe, M Woodward, J McNeiJ (ASPiVLU). School of Public Health and Preventative Medicine, Monash University, Melbourne, VIC, Australia, 2015]. The objective of performing this meta-analysis is to assess the clinical effectiveness on time to healing of VLUs and safety of aspirin use. This will take place following completion of the other trials, which are still recruiting patients28,29 at the time of writing.
Chapter 2 Methods
This chapter reports the methods used to conduct AVURT. It describes the study design and protocol from recruitment of participants to completion in the study, data analysis procedures, quality assurance and governance. The trial protocol has been published. 32
Design
A multicentred, pilot, Phase II randomised double-blind, parallel-group, placebo-controlled efficacy trial.
Setting
Patients presenting at community leg ulcer clinics/hospital outpatients’ clinics, or registered with a leg ulcer clinic but receiving care at home, were recruited. Some sites could use patient identification centres (PICs) to identify patients to take part. Participants were recruited from 10 centres in England, Wales and Scotland (see Appendix 1) from leg ulcer hospital outpatient clinics (n = 5), community leg ulcer clinics or community caseloads (n = 3), a wounds clinic in a university (n = 1) and a primary care leg ulcer clinic (n = 1). At each of the nurse-led community centres (n = 3), a doctor was identified to work with the centre to review and confirm the patient’s eligibility for the trial, to prescribe the investigational medicinal product (IMP) and to review changes to concomitant medication.
Participants
Participant eligibility for the trial was assessed according to the criteria below.
Inclusion criteria
To be eligible for the study, it was necessary for participants to meet all of the following criteria.
-
Having at least one chronic VLU, when chronic venous leg ulceration was defined as any break in the skin that had either (1) been present for > 6 weeks or (2) occurred in a person with a history of venous leg ulceration. Ulcers were considered purely venous if clinically no other aetiology was suspected. The ulcer was required to be venous in appearance (i.e. moist, shallow, of an irregular shape) and lie wholly or partially within the gaiter region of the leg. If the patient had more than one ulcer we chose the largest as the ‘index’ or reference ulcer for purposes of the analysis.
-
Having an ulcer with an area of > 1 cm2.
-
Having had an ankle–brachial pressure index (ABPI) of ≥ 0.8 taken within the previous 3 months or, when the ABPI is incompressible, other accepted forms of assessment included peripheral pulse examination/toe pressure/Duplex ultrasonography in combination with clinical judgement to be used to exclude peripheral arterial disease (PAD).
-
Being aged ≥ 18 years (there was no upper age limit).
-
Being able and willing to give informed consent.
Exclusion criteria
Potential participants were excluded if they fulfilled any of the following criteria.
-
Being unable or unwilling to provide consent.
-
Having a foot (below the ankle) ulcer.
-
Having a leg ulcer of non-venous aetiology (i.e. arterial).
-
Having an ABPI of < 0.8.
-
Using (self-administered or prescribed) regular concomitant aspirin.
-
Having a previous intolerance of aspirin/contraindication to aspirin (decision made according to the prescribers’ clinical judgement).
-
Taking contraindicated medication: probenecid, oral anticoagulants including coumarins (warfarin and acenocoumarol) and phenindione, dabigatran, rivaroxaban, apixiban, heparin, clopidogrel, dipyridamole, sulfinpyrazone and iloprost.
-
Having known lactose intolerance.
-
Being a pregnant or lactating/breastfeeding woman.
-
Being male or a pre-menopausal female of child-bearing potential unwilling to use an effective method of birth control [i.e. either hormonal in the form of the contraceptive pill; barrier method of birth control accompanied by the use of a proprietary spermicidal foam/gel or film; or agreement of true abstinence (withdrawal, calendar, ovulation, symptothermal and post ovulation were not acceptable methods)] from the time consent was signed until 6 weeks after the last dose of IMP. Participants were only considered not of child-bearing potential if they were surgically sterile (i.e. they had undergone a hysterectomy, bilateral tubal ligation, or bilateral oophorectomy) or they were postmenopausal.
-
Currently participating in another study evaluating leg ulcer therapies.
-
Having another reason that excluded them from participating within this trial (decision made according to the nurses’ or prescribers’ clinical judgement).
-
Having previously been recruited to this trial.
Recruitment
Patients were pre-screened on the basis of three criteria (concomitant aspirin, wound size and ulcer duration or history of venous ulceration) by study research nurses to determine those potentially eligible for the study. The reason(s) for ineligibility or not approaching patients were recorded on pre-screening logs (see Appendix 2). Two pre-screening logs were issued. Completion of the first log (version 1.0) was non-mandatory as stipulated by the trial sponsor (based on the sponsor’s belief that the clinics received a heterogeneous referral pattern of mixed aetiology ulcers not thought to be truly representative of the total population of patients with chronic VLUs). Following a recommendation by the Data Monitoring Committee (DMC), the sponsor permitted a new pre-screening log that was made mandatory (version 2.0). Patients attending clinics as part of their routine care and who satisfied the pre-screening criteria were approached by study research nurses or designated health-care professionals and provided with both verbal and written information about the trial in a face-to-face meeting (see Appendix 3). Patients were given a minimum of 24 hours to consider participation in the trial. Study research nurses then obtained voluntary full written consent from those patients who wanted to enter the trial (see Appendix 4). After they gave consent, patients were screened against the study’s full eligibility criteria by the study research nurses or designated health-care professionals using the screening case report form (CRF) (see Appendix 5). The reason(s) for a patient’s ineligibility were recorded. Patients were informed that their eligibility would be subject to confirmation by a medical practitioner and in all cases a medical practitioner determined and confirmed patient eligibility following screening. If a potential participant was not known to the medical practitioner, provision was made for the participant to be contacted by telephone by the medic to check for any possible contraindications. When the medic was satisfied of patient eligibility, they would sign off the prescription for the IMP (see Appendix 6).
Randomisation
Patients were randomly allocated in a 1 : 1 ratio to either aspirin or placebo by the Research Pharmacy (St George’s University Hospitals NHS Foundation Trust, London, UK). Randomisation was stratified according to ulcer size (≤ 5 cm2 or > 5 cm2) as this is the strongest known predictor of outcome. 4
Sequence generation
The aspirin and placebo manufacturer, Sharp Clinical Services (UK) Limited (registered office in Ashby-de-la-zouch, UK), generated the randomisation schedule in advance. They provided one randomisation list to the Research Pharmacy and a copy to the senior trial statistician in the York Trials Unit (YTU; University of York). To facilitate participant allocation according to stratification, the allocation sequence on the randomisation list was mirrored top to bottom bottom to top, and each allocation was referenced 1 to 120 for participant identifier (ID). Where the participant was placed on the randomisation list (top or bottom) depended on the stratification of ulcer size (≤ 5 cm2 or > 5 cm2).
Allocation
After participant consent was taken and baseline data were recorded, the research site faxed the AVURT prescription directly to the Research Pharmacy. The AVURT prescription also indicated ulcer size. On receipt of the original signed prescription by post, the Research Pharmacy allocated the next available randomisation ID. The randomisation ID corresponded to IMP bottle number for allocation (top or bottom), in accordance with the ulcer size stratification as indicated on the prescription. IMP was dispensed by St George’s and sent by courier under temperature-controlled conditions directly to all participants. The date of randomisation, a unique patient ID and a unique screening ID were recorded by the Research Pharmacy on a Microsoft Excel® (Microsoft Corporation, Redmond, WA, USA) spreadsheet, which was sent to the YTU each week or when a participant was randomised.
Blinding
Participants, investigators, research and treating nurses and other attending clinicians were unaware of the trial drug allocation throughout the trial. There was a 24-hour emergency code break facility at the Research Pharmacy for health professionals to contact if they needed to determine whether or not patients were receiving aspirin or placebo for onward clinical management. However, in practical terms, it was expected that most clinicians would treat participants with AEs on the assumption that they had been randomised to receive aspirin.
Interventions
Intervention group
Intervention: 300 mg of daily oral aspirin for 24 weeks (four × 75-mg tablets were encapsulated in size 00 capsules with added lactose and magnesium stearate blend as filler).
Control group
Placebo: daily oral placebo for 24 weeks. Size 00 capsules with lactose and magnesium stearate blend as filler, which were identical in weight, colour and size to the aspirin capsules.
The full course of capsules (190 doses/capsules for 24 weeks’ treatment) were packaged into child-resistant tamper-evident bottles. Participants were advised to take the capsules whole (not crushed or chewed), once a day for 24 weeks or, if the reference ulcer was confirmed as healed before the end of 24 weeks, a member of the medical team would advise them to stop taking the medication. The time of day for taking the trial medication was not specified.
Participants were expected to receive and start their allocated trial treatment from 2 to 7 days after randomisation.
All participants were offered an evidence-based standardised approach to the management of their leg ulcers in accordance with Scottish Intercollegiate Guidelines Network (SIGN) guidance. 33 This consisted of multicomponent compression therapy aiming to deliver 40 mmHg of pressure at the ankle, when possible. The type of dressing used was at the discretion of the health-care professionals managing the participants.
Investigational medicinal product supply
The sponsor had responsibility for the order and purchase of trial medication and for arranging labelling of medication for the trial with St George’s University Hospitals NHS Foundation Trust.
Manufacture, packaging and labelling
Active (aspirin) tablets were manufactured by Intrapharm Laboratories Limited, Maidenhead, UK. Overencapsulation of the 75-mg tablets and production of the matching placebo capsules was performed by Sharp Clinical Services (UK) Limited (MA IMP licence number 10284). Sharp Clinical Services (UK) Limited performed all manufacturing and packaging operations in accordance with good manufacturing practices derived from the rules governing Medicinal Products in the European Community and Good Manufacturing Practice for Medicinal Products. 34–36
The AVURT IMP was assigned an expiry date of 31 May 2016. Following the extension to participant recruitment, the sponsor arranged for stability testing of the IMP with Sharp Clinical Services (UK) Limited. The testing was conducted and the expiry date was extended to 31 January 2017.
The supplies of aspirin and placebo capsules were delivered to the Research Pharmacy, where they were stored and dispatched to all participants.
Outcomes
Primary outcome
Time to healing of the reference ulcer (the largest eligible ulcer).
Secondary outcomes
-
Ulcer size (area) measured in cm2 by specialist software and grid tracings.
-
Following healing of the reference ulcer, recurrence of ulcer on the reference leg (defined as a new ulcer on the reference leg).
-
Adverse events.
-
Ulcer-related pain using a visual analogue scale (VAS).
-
Treatment compliance (capsule count and nurse assessment of compression concordance).
-
Resource use: number of visits to clinic and/or home visits and types of dressings used.
Baseline assessment
Following confirmation of a participant’s eligibility, and before randomisation, a baseline assessment was conducted by the study or research nurse using the baseline CRF (see Appendix 7).
Participant details
Data on ethnicity were collected at baseline. Participants’ date of birth, gender and smoking status were collected at screening. Participants’ contact details (name, address, telephone numbers and e-mail address) and general practitioner (GP) details (name of GP, name of surgery and address) were recorded at the recruiting site only.
Ulcer history and assessment
The last ABPI measurement of the reference leg (leg with the largest eligible ulcer) and date it was taken were recorded, or it was noted that the ABPI was unable to be taken. When ABPI was incompressible, other assessments to exclude PAD were permitted, including peripheral pulse examination/toe pressure/Duplex ultrasonography in combination with clinical judgement, but these forms of assessments were not recorded on the CRF.
Other items recorded were number of ulcers on the reference leg, approximate duration of reference ulcer (years, months and weeks), how long ago since patient developed their first leg ulcer (years, months and weeks), and total number of ulcer episodes on reference leg (leg with largest eligible ulcer) including the reference ulcer (largest eligible ulcer). All ulcers on both legs were drawn onto a leg diagram and the reference ulcer indicated.
Digital photographs and tracings
To measure ulcer area, a photograph and tracing of the reference ulcer were taken at baseline. Photographs were taken with a Nikon Coolpix L3 (Nikon Corporation, Tokyo, Japan), in accordance with trial procedure (see Appendix 8). Anonymised digital photographs were sent to the YTU using a secure electronic method. Sites unable to use this method were able to send anonymised photographs on a memory card via a courier service to the YTU or a collection could be made by one of the trial co-ordinators.
Tracings were taken using a fine-nibbed marker pen on a wound measurement grid composed of 1-cm2 squares (P12v2, ConvaTec, Uxbridge, Middlesex, UK). The wound area was calculated by the treating or research nurse by totalling the number of squares and/or partial squares on the grid contained within the traced ulcer area.
Participant mobility, anthropometry and diabetic status
The level of a participant’s mobility (walking and ankle mobility), their height (feet/inches or centimetres) and weight (stones/pounds or kilograms) were recorded. If both metric and imperial measurements were given, a check was conducted by the YTU to determine if they were equivalent. Any differences were queried with the site. Body mass index [BMI (kg/m2)] was calculated using the formula: weight (kg) divided by height squared (m2). The presence of type of diabetes mellitus (type 1 or 2) was recorded.
Current treatments received
Participants’ medications at baseline were recorded by the study research nurse in a medication diary (see Appendix 9). The medication diaries were then given to participants for recording changes to non-trial medication. The participants were asked to bring their diary along to each clinic assessment for review by the study research nurse and site medical practitioner to check that participants were safe to continue with the IMP. Medication data were not collected for analysis.
Participants’ current treatment(s) for their VLU were recorded (type of compression bandaging), as was the level of ankle pressure compression being aimed for (mmHg) and the primary dressing in contact with the ulcer.
Ulcer-related pain
Participants were asked to rate the intensity of any leg ulcer-related pain over the previous 24 hours using the 21-point Box Scale (BS-21). 37 The BS-21 is a VAS that is divided into units of five and ranges from a value of 0 (no pain) to 100 (worst pain imaginable).
Resource use
The treating or research nurse recorded resource use on the CRFs. They initially recorded the type of dressing administered and level of compression aimed for and subsequently, during follow-up, only recorded a change in the type of dressing administered and/or level of compression.
Outcome assessments
Participants were followed up weekly or fortnightly, depending on their usual pattern of attendance at clinic, for a minimum of 25 weeks post randomisation. Participants were not asked to make any additional visits for the purposes of the trial. The participant weekly data collection file, made up of CRFs and forms, was completed during follow-ups by research nurses or treating nurses (see Appendix 10). In addition, recorded in the file were the weeks in which participants missed or did not have an appointment, the randomisation date, the date that the first IMP dose was taken and the time of day that the IMP was generally taken. A summary flow chart of participant follow-up is shown at Appendix 11.
Planned participant follow-up was for 25 weeks post randomisation, but participants who had a wound initially judged as healed in week 24 or 25 were followed up for 2 further weeks (26 and 27 weeks post randomisation, respectively) to confirm healing. Table 1 summarises the schedule of assessments. All anonymised completed CRFs were faxed or sent via the University of York’s secure electronic system to the YTU.
| Study procedures | Screening | Baseline | During treatment (weekly for 25 weeks post randomisation) | Post treatment (only participants whose reference leg ulcer was judged as healed in weeks 24 and 25) | Post treatment (only participants whose reference leg ulcer was judged as healed in week 25) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Week 1 | Weeks 2–3 | Weeks 4–6 | Weeks 7–24 | Week 25 | Week 26 | Week 27 | |||
| Informed consent | ✓ | ||||||||
| Inclusion/exclusion criteria | ✓ | ✓ | |||||||
| Demographics | ✓ | ||||||||
| Dispensing of IMP | ✓ | ||||||||
| Medical history | ✓ | ✓ | |||||||
| Concomitant medication | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓a | ✓ | ✓ |
| AEs/side effects/change to health status | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| Ulcer photographb | ✓ | ✓ | ✓ | ✓ | ✓ | ✓a | ✓ | ✓ | |
| Tracing of ulcer | ✓ | ✓ | ✓a | ||||||
| Resource use: change to type of usual care/compression bandage administered | ✓ | ✓ | ✓ | ✓ | ✓a | ✓ | ✓ | ||
| Compliance | ✓ | ✓ | ✓ | ✓ | ✓a | ||||
| Pain score | ✓ | ✓ | |||||||
| Ulcer reccurrence (only patients whose leg ulcer was confirmed as healed before week 25) | ✓ | ||||||||
Measurement and verification of primary outcome measure
Time to healing of the reference ulcer
The treating or research nurse identified and monitored the reference ulcer. Healing was defined as complete epithelial cover in the absence of a scab (eschar) with no dressing required. Healing was determined by the treating nurse or research nurse and a digital photograph was taken of the wound area. Healing was reported by the treating site on Form D (see Appendix 10) which was submitted to the YTU. Time to healing was measured in days from the date of randomisation to the date that the ulcer was first assessed as healed. After the treating or research nurse initially judged the ulcer to be healed, participants were followed up for a further 2 weeks, in accordance with the Food and Drug Administration guidelines,38 to confirm healing.
Measurement of secondary outcomes
Ulcer size
The reference ulcer was measured using wound grid tracings at screening, baseline and at final follow-up and at other follow-up visits when a photograph could not be taken. Treating nurses calculated the ulcer size by totalling the number of squares and/or partial squares on the grid contained within the traced ulcer area size and reported the measurement in the CRFs.
Anonymised digital photographs were taken at baseline and at all weekly or fortnightly follow-up visits. All digital images were checked and the ulcer size calculated using SigmaScan® software (Sigma Scan Pro version 5.0, SigmaScan, Systat Software Inc., San Jose, CA, USA) by one researcher, Rachael Forsythe (Specialist Registrar in Vascular Surgery, St George’s Hospital, London, UK) who was blinded to treatment allocation.
Ulcer recurrence
Weekly follow-up CRFs were not completed for participants after their reference ulcer had been confirmed as healed. To collect ulcer recurrence data, participants were given a card with contact details for their recruiting site (see Appendix 12) and were asked to phone the clinic if they developed a new ulcer on their reference leg. In addition, at week 25 post randomisation, the research nurse phoned participants whose reference ulcer had healed, to collect data on leg ulcer recurrence. The date of recurrence of a new venous ulcer on the reference leg was recorded (see Appendix 10, Form E).
Ulcer pain
Participants were asked to rate the intensity of any leg ulcer-related pain over the previous 24 hours using the BS-21. Ulcer-related pain was collected at baseline and at weeks 4, 5 and 6 after randomisation. It was thought that aspirin might have a positive effect on pain. We required one pain score at follow-up but took measurements at three follow-up time points to allow for participants not being seen every week.
Participant compliance with treatment
To monitor treatment concordance with the IMP and compression, the treating nurses recorded in the weekly CRFs (see Appendix 10) how often a participant was taking the capsules and, when applicable, reasons for not taking them every day. Treating nurses also recorded whether or not a participant had fully, had partially, or had not complied with compression therapy, with reason(s) for non-compliance captured when possible.
At the end of the study, the remaining IMP or, in cases when participants had taken all the trial medication, the empty container, was returned to the Research Pharmacy, which undertook a pill count. This information was then forwarded to the YTU for inclusion in the analysis.
Resource use
At follow-up visits (weekly or fortnightly depending on a participant’s usual pattern of care), changes to the level of compression therapy were recorded (see Appendix 10, Form A) and changes to the type of primary dressing or bandaging (see Appendix 10, Form B). The number of times participants had other wound consultations in the previous week was also recorded.
Patient safety
Each participant was regularly reviewed by their treating nurse and/or physician working closely with the AVURT research team and was continually assessed for any increased dyspepsia, other gastrointestinal symptoms, skin rashes and any other possibly linked AEs that could be attributable to the IMP.
Known side effects
Common side effects of aspirin as listed on the summary of product characteristics, which was supplied by the IMP manufacturer (Intrapharm Laboratories Limited), included increased bleeding tendencies and dyspepsia.
Adverse events: definitions
The following definitions were applied in the study.
Adverse event
-
Any untoward medical occurrence in a patient or clinical trial participant who is administered an IMP and which does not necessarily have a causal relationship with this treatment, and which may include an exacerbation of a pre-existing illness.
-
Increase in frequency or severity of pre-existing episodic condition.
-
A condition (regardless of whether or not it was present prior to the start of the trial) that is detected after trial drug administration (this does not include pre-existing conditions recorded as such at baseline, continuous persistent disease or a symptom present at baseline).
Adverse reaction
-
Any untoward and unintended responses to an IMP related to any dose administered.
Serious adverse event or serious adverse reaction
-
Any AE or reaction that, at any dose, results in death.
-
Any AE or reaction that, at any dose, is life-threatening (places the subject, in the view of the investigator, at immediate risk of death).
-
Any AE or reaction that, at any dose, requires hospitalisation or prolongation of existing hospitalisation [hospitalisation is defined as an inpatient admission, regardless of length of stay, even if it is a precautionary measure for observation (including hospitalisation for an elective procedure and for a pre-existing condition)].
-
Any AE or reaction that, at any dose, results in persistent or significant disability or incapacity (substantial disruption of one’s ability to conduct normal life functions).
-
Any AE or reaction that, at any dose, results in a congenital anomaly or birth defect (in offspring of subjects or their parents taking the IMP regardless of time of diagnosis).
-
Any AE or reaction that is related to another important medical condition.
Important medical events that may not be immediately life-threatening or result in death or hospitalisation but may jeopardise the subject or may require intervention to prevent one of the outcomes listed in Serious adverse event or serious adverse reaction was also considered serious.
Suspected unexpected serious adverse reaction
A suspected unexpected serious adverse reaction (SUSAR) is an adverse reaction (AR) that is classed in nature as both serious and unexpected.
An unexpected AR is when both the nature and the severity of the event are not consistent with the reference safety information (RSI) available for the IMP in question.
Assessments
At each follow-up appointment, the treating nurses asked participants if they had experienced any changes in their health and indicated their response in the CRFs. Participants whose reference leg ulcer had healed, and, therefore, were no longer receiving follow-up appointments, were contacted at week 25 post randomisation by a research nurse who collected information on AEs that the participant had experienced since the last data collection point (see Appendix 10, Form E).
Details of the AEs/ARs were recorded in clinic notes and on AE logs held at the recruiting sites. The causality, severity and expectedness assessment was conducted by medically qualified doctors at the sites who were blind to treatment allocation in accordance with the following descriptions.
Causality assessment
-
Definitely: there is clear evidence to suggest a causal relationship, and other possible contributing factors can be ruled out.
-
Probably: there is evidence to suggest a causal relationship, and the influence of other factors is unlikely.
-
Possibly: there is some evidence to suggest a causal relationship (e.g. the event occurred within a reasonable time after administration of the trial medication). However, the influence of other factors may have contributed to the event (i.e. the patient’s clinical condition, other concomitant events).
-
Unlikely: there is little evidence to suggest that there is a causal relationship (e.g. the event did not occur within a reasonable time after administration of the trial medication). There is another reasonable explanation for the event (e.g. the participant’s clinical condition, or other concomitant treatments).
-
Unrelated: there is no evidence of any causal relationship.
-
Not assessable: note – if this description was used, then the sponsor assumed that the event was related to the IMP until follow-up information was received from the investigator to confirm a definitive causality assessment.
Any SUSAR assessed as related to the IMP was required to be reported to the sponsor, irrespective of how long after IMP administration the reaction had occurred.
Expectedness assessment
Assessment was based solely on the available RSI for the IMP and was described using following categories.
-
Expected: an AE that is classed in nature as serious and that is consistent with the information about the IMP listed in the RSI or clearly defined in the study protocol.
-
Unexpected: an AE that is classed in nature as serious and that is not consistent with the information about the IMP listed in the RSI.
All assessments were reviewed by the chief investigator using specific guidance notes from the National Institute for Health Research (NIHR) clinical trials tool kit. 39
Reporting
Non-serious and serious AEs were reported by the sites to the trial manager at the YTU on the sponsor’s AE log. SAEs were recorded on the sponsor’s SAE form and reported directly to the sponsor (St George’s University Hospital) within 24 hours of the local investigators becoming aware. The sponsor followed up SAEs to their resolution and was responsible for reporting the events to Research Ethics Committee (REC), the Medicines and Healthcare products Regulatory Agency (MHRA) and the trial manager at the YTU.
All AEs, ARs, SAEs and serious ARs were reviewed by the sponsor and chief investigator and subsequently the DMC (blinded to allocation), which made the final decision regarding the severity and causality and relationship between the event and treatment.
Withdrawal
Participants were deemed to have exited the trial when they:
-
withdrew consent
-
were lost to follow-up
-
died
-
had completed follow-up (i.e. 25 weeks post randomisation or, for patients whose leg ulcer was first assessed as healed in weeks 24 and 25, weeks 26 and 27, respectively).
If a participant chose to withdraw from the trial then reasonable effort was made to establish the reason for this withdrawal. For participants leaving the trial before final follow-up, nurses completed a change to study status form (see Appendix 10, Form F), giving the main reason for the participant’s exit. No further follow-up data were collected. Participants withdrawing from the study were given the option for their data not to be used.
Participants stopped treatment for any one of the following reasons, but continued with follow-up:
-
Unacceptable treatment toxicity that, in the investigator’s opinion, is attributable to the IMP or a SAE.
-
Intercurrent illness that prevents further protocol treatment.
-
Any change in a participant’s condition that, in the investigator’s opinion, justified the discontinuation of treatment.
-
If a participant became pregnant or suspected that they were pregnant.
-
Reference ulcer confirmed as healed.
-
A participant chose to discontinue treatment.
Participants whose leg ulcer was initially assessed as healed were encouraged to take the IMP during the 2-week observation period. If healing was confirmed after 2 weeks, the participant stopped taking the trial medication and follow-up was suspended until a final follow-up in week 25.
Sample size
The target sample size was 100 participants. This sample size is sufficient to test the feasibility of study procedures, such as recruitment and retention, and is large enough to demonstrate whether or not there is evidence for efficacy in line with two previous trials of aspirin for leg ulcers. 25,26
The primary outcome was time to healing of the largest eligible leg ulcer (reference ulcer). Ulcer area and duration of ulcer are known prognostic factors for healing. In a previous leg ulcer study, Venous leg Ulcer Study IV (VenUS IV), after adjustment for log-area of ulcer and log-duration of ulcer, the standard error for the time to healing estimate was 0.105, with data on 448 participants. 3 Applying this to a smaller sample of 100 participants implies that the standard error of such a sample would be increased to 0.22 [obtained from 0.105 × √(448/100)]. A 95% confidence interval (CI) for the log-hazard ratio (HR) would thus be the estimate of the log (HR) ± 1.96 × 0.222 = log(HR) ± 0.435. The antilog of this is 1.54 and the 95% CI for the HR would be the observed value divided or multiplied by this. Hence, if our HR were the same as that suggested by the existing studies (i.e. about 1.5), then our CI would be 0.97 to 2.31, which just includes 1.00. It would be unlikely that, if the HR is as these two previous smaller studies suggest, we would observe an overall HR of < 1.00. Compliance and follow-up were measured as part of the study and so there is no formal inflation of the recruitment target for drop out.
A secondary outcome was change in wound area. Using data from the Venous leg Ulcer Study I (VenUS I) of compression bandaging,40 ulcer area was measured for 245 participants who were measured within 60 days of recruitment. Ulcer area has a highly skewed distribution, so we calculated a difference in log-area at follow-up, after adjustment for log-ulcer area at baseline and time elapsed until follow-up. The residual standard deviation (SD) was 1.09. Two groups of 50 participants would give us 80% power to detect a difference of 0.62 on the natural-log scale, corresponding to a reduction of 46% in ulcer area at follow-up. In the current study, we had multiple measurements of wound area and so predicted that we should be able to detect smaller differences.
Statistical methods
The statistical methods for the analysis of the trial data were prespecified and detailed in a statistical analysis plan (SAP) before the completion of data collection. The SAP was prepared by the trial statisticians and reviewed by members of the Trial Management Group (TMG) and DMC. However, given that the final number of participants randomised was much lower than the 100 planned (n = 27), many of the pre-planned analyses were infeasible or inappropriate. In this section, we describe the analyses as performed, highlighting any deviations from the SAP.
Pre-screening, screening and eligibility data
The flow of participants through the trial is presented in a Consolidated Standards of Reporting Trials (CONSORT) diagram. 41 The number of patients who were pre-screened, were approached and consented is reported. Reasons for ineligibility at the pre-screening phase and reasons for not consenting are summarised.
Baseline data
Participant characteristics and clinical baseline measurements are summarised descriptively overall and by trial arm. These measures include age, gender, BMI, diagnosis of diabetes mellitus, ethnicity, participant’s level of mobility and ankle mobility, reference ulcer size and corresponding stratification (≤ 5 cm2 or > 5 cm2), time since first ulcer, duration of reference ulcer (actually referring to the duration of the ulcer up to but not beyond randomisation), left/right reference leg, total ulcers on reference leg, ABPI of the reference ulcer, levels of pain from the reference ulcer, current compression and dressing treatments. Continuous measures were summarised using mean, SD, median, minimum, maximum and interquartile range (IQR). Categorical measures were reported as counts and percentages. No formal statistical comparisons of baseline factors by trial arm were undertaken.
Primary outcome
The primary analysis investigated the difference in time to healing by trial arm using Cox’s proportional hazards regression adjusted for ulcer area (cm2) and ulcer duration (days) at baseline, both logarithmically transformed. Ulcer area and ulcer duration tend to have a skewed distribution and, therefore, a logarithmic transformation is used to obtain a distribution that is closer to the normal. It was initially planned to subsequently test for the inclusion of shared centre frailty effects; however, the final distribution of participants across centres (see Table 2) made the frailty model an impractical choice for this analysis. Therefore, only the Kaplan–Meier survival curve, the log-rank test and the Cox’s regression model, both unadjusted and adjusted for the logarithm of the area and of the ulcer duration, were undertaken. HRs, corresponding 95% CIs and p-values for the model covariates are presented.
Secondary outcomes
Adverse events
Adverse events were reported overall and by trial arm in terms of number of participants with at least one event and total number of events. Serious and non-serious events were presented separately and according to whether or not they were thought to be related or unrelated to treatment. For SAEs, reasons for the serious nature of the events were reported. Differences in total number of events by trial arm were compared using negative binomial regression adjusted for size and duration of ulcer (both log transformed).
Ulcer size
The area resulting from the analysis with the SigmaScan was used in statistical analysis whenever available. In the case when a photograph could not be taken, the measure of the ulcer area was obtained by using the tracing of the ulcer, if available. The area at baseline and at each assessment is summarised using descriptive statistics (mean and SD) for each trial arm and overall. A plot containing means and 95% CIs for both trial arms was also produced with lower confidence limits truncated at zero, as wound area can only be positive.
It was planned a priori that the logarithm of the ulcer area would be investigated via a repeated measures mixed model to see if there were any differences by trial arm; however, owing to the low number of participants and the high number of time points, this model was not judged to be appropriate for the final analysis.
Ulcer recurrence
As a recurrence of the reference ulcer was reported for only two participants, the Cox proportional hazards regression initially planned was not performed. For both participants the number of days from healing to recurrence is presented.
Time to first investigational medicinal product dose
The median time in days from randomisation to date the first IMP dose was taken was presented alongside 95% CI by trial arm and overall.
Time of day
The number of participants who reported taking their study drug in the morning, afternoon and evening is summarised using counts and percentages.
Ulcer pain
The VAS scale [from 0 (no pain) to 100 (worst pain imaginable)] to measure pain was used at baseline and at weeks 4, 5 and 6 in order to increase the likelihood of capture, as not all patients were seen weekly. Only one VAS score was used for the analysis: if the week 5 VAS score was present, this was used; if it was not and either only week 4 or only week 6 were provided, then the corresponding VAS score was used; if both week 4 and week 6 were provided but week 5 was not, then the VAS completed on the closest date to week 5 was taken; if both weeks were completed an equidistance from week 5, then week 4 was taken.
Descriptive statistics of VAS score (mean, SD, median, minimum, maximum, IQR) were calculated overall and for each trial arm at baseline and at week 5, obtained as defined above. A plot of the means and 95% CIs for both trial arms at baseline and at week 5 was produced.
The planned linear regression analysis, aimed to compare differences in pain scores between allocated groups, was not performed owing to low numbers.
Participant compliance with treatment
At each assessment visit, compliance with both the compression therapy (for those receiving this treatment) and with the study capsules was recorded. This was via the following two questions: ‘Has the participant complied with their [compression therapy] treatment’ (fully/partially/not at all), and ‘How often has the participant taken their AVURT capsules (300-mg aspirin/placebo per day) this week?’ (every day/most days/some days/not at all). The responses to both of these questions were given numerical values: fully = 1, partially = 2, and not at all = 3; and every day = 1, most days = 2, some days = 3, and not at all = 4. To calculate compliance with compression treatment, the responses across all weeks up to healing/trial exit were summed and divided by the number of visits attended to obtain the mean compliance level for each participant. This compliance level was then categorised as fully compliant if the mean value was 1, partially compliant if the value was between 1 and 3 (not inclusive), and not at all compliant if the value was equal to 3. The number and percentage of participants in each of these categories is presented.
Compliance with study capsules was analysed similarly but only considering responses in the weeks following delivery of the capsules. The compliance level was categorised as fully compliant if the mean value was 1, partially compliant if the value was between 1 and 4 (not inclusive), and not at all compliant if the value was equal to 4. Reasons for lack of full compliance are presented.
The second way that compliance with AVURT capsules was assessed was through the use of the count of the returned capsules at the end of the study. Each participant was given 190 capsules and by subtracting the number of returned pills it was possible to obtain an estimate of the number of capsules actually taken. The number of capsules that should have been taken was calculated starting from the date of first dose until 2 weeks after healing (for those who had healed) or the date of the last visit (for those who did not heal). From this, the percentage of capsules that each participant took (of those they should have taken) was calculated. The level of compliance was split into 11 categories (100%, 90–99%, 80–89%, etc.) and the count and percentage of patients falling in each category is presented (see Tables 13 and 14).
Resource use
Level of compression therapy
The number of changes to compression therapy is presented overall and by trial arm, and also stratified by time to healing (or censoring) using the categories 0–2 months, > 2 to 4 months, and > 4 months. The number and percentage of changes to low/medium/high or no compression therapy are presented overall and by trial arm.
Bandaging and hosiery
The number and percentage of changes to each bandage type are presented overall and by trial arm, as are the number and percentage of patients who received each type of bandaging at least once during the study.
Dressing
The number of changes per participant to dressing type is summarised overall and by trial arm, and stratified by time until healing (or censoring) using the categories 0–2 months, > 2 to 4 months, and > 4 months.
The number and percentage of changes to each dressing type are presented overall and by trial arm, as are the number and percentage of patients who received each type of dressing at least once during the study.
Wound consultations
For each participant, the number of wound consultations per week was calculated by summing the number of consultations the participant had in the previous week, declared on the weekly CRFs, plus the visit in which the CRF was completed and dividing it by the number of visits actually attended. The mean number of wound consultations per week is presented alongside SD, median, minimum, maximum and IQR (see Table 22).
Approvals obtained and governance
Ethics and Medicines and Healthcare products Regulatory Agency approvals
The trial was approved by Nottingham REC on 29 January 2015 (REC reference number 14/EM/1305) and by the University of York Health Science Research Governance Committee on 16 February 2015. The MHRA approved the study on 26 March 2015 (MHRA reference 16745/0221/001-001). The London Local Research Network completed their global checks on 7 May 2015 and thereafter research governance approval was obtained from each trial centre. The trial was registered with ClinicalTrials.gov and assigned the number NCT02333123, and with the European Clinical Trials Database and assigned the European Clinical Trials Database (EudraCT) number 2014-003979-39.
Trial monitoring
The AVURT was monitored by the sponsor, St George’s University of London. The trial was conducted and monitored in compliance with their standard operation procedures:42 International Conference on Harmonisation Harmonised Tripartite Guidelines For Good Clinical Practice E6 (ICH GCP) and the Medicines for Human Use (Clinical Trials) Regulations 2004 (SI 2004/03) (as amended).
The purpose of the monitoring was to ensure:
-
the safety and welfare of trial participants
-
that trial data were accurate and verified from source data when possible
-
that the trial was compliant with good clinical practice and other regulatory requirements.
Trial oversight
The trial was overseen by the TMG, the Trial Steering Committee (TSC) and the DMC.
Trial Management Group
The TMG was responsible for project oversight, directing the management of the trial and reviewing progress. The TMG was chaired by the chief investigator and comprised the trial co-ordinators, trial statisticians and the majority of the coapplicants including a patient representative.
Trial Steering Committee
The TSC provided overall supervision of the progress of the trial towards its interim and overall objectives, to ensure adherence to the protocol and patient safety. The TSC approved the trial protocol prior to participant recruitment, reviewed recruitment, protocol deviations, the trial’s results and recommendations made by the DMC.
The TSC was chaired by an independent representative (Professor Julie Brittenden) and membership consisted of three other independent members, a patient representative and members of the research team, including the chief investigator, the sponsor’s representative, the trial statistician and trial co-ordinators. The TSC met for the first time prior to participant recruitment and then three times during the course of the trial.
Data Monitoring Committee
The main role of the DMC was to ensure the safety of trial participants, to protect the validity of the trial, to advise the investigators and to make recommendations to the TSC about whether or not the trial should continue. The DMC approved the SAP and reviewed recruitment figures, protocol deviations, protocol amendments and AE data.
The DMC was chaired by an independent representative (Professor Peter Franks) and membership consisted of three other independent members and members of the research team, including the chief investigator, the sponsor’s representative, the trial statistician and trial co-ordinators.
Patient and public involvement
At the grant application stage, the views of six patients attending a leg ulcer clinic were elicited. Specifically, they were asked for their views on the likely willingness of patients to take aspirin on a daily basis, given its possible side effects, if it were shown to improve healing of leg ulcers. All responded that they would be willing to take medication if it meant that their ulcer was likely to heal more rapidly. They thought that the risks of aspirin were acceptable given that many patients already take it regularly for cardiovascular disease. In terms of a trial, they thought that they would be happy to receive a dummy tablet (placebo) if it meant that more information could be gleaned about the efficacy of aspirin in terms of healing ulcers – even though a further larger trial might be required to confirm the results. Some patients questioned the benefit about taking a high dose of aspirin. However, the feeling expressed by some was that the perceived increased risks would be worthwhile if it significantly decreased time to healing.
There were two patient coapplicants. Ellie Lindsay, president of the Leg Club Foundation, was a member of the TMG, and our other patient representative, Laurie Williams, was a member of the TSC. Both were involved in the development of the trial during the application stage and throughout the study. They were also involved in the development of the trial’s patient information resources.
Protocol amendments
Amendments to the protocol were required by REC prior to approval. Following approval, no substantial amendments were made to the protocol. Details of all ethics and MHRA amendments are detailed in Appendix 13.
Chapter 3 Results: feasibility of recruitment
The original participant recruitment window was for 6 months and was due to finish on 30 September 2015. Owing to delays in sites opening and to allow the last few sites to open sufficient time to recruit, this was extended to an 8-month recruitment window. Consequently, to allow the full follow-up of all participants, the total duration of the trial was extended by just over 5 months to 14 December 2016 (the project was originally due to close on 30 June 2016).
Site recruitment
Ten sites opened to recruitment. Prior to recruitment, the sites indicated the approximate number of participants they could recruit (see Appendix 1). Recruitment was largely based in leg ulcer community clinics and hospital outpatient clinics. Many of the recruiting sites were chosen as they had been high recruiters to other leg ulcer studies.
Eleven sites were initially interested in participating. Nottingham University Hospitals NHS Trust subsequently declined after undertaking a complete screening review of its patient population, which consisted of 3300 patients referred with chronic oedema of all forms with many suffering from venous leg ulceration. An analysis of their patient profile indicated that they would have little access to non-complex patients. The remaining 10 sites were submitted to REC for approval:
-
St George’s Healthcare NHS Trust London
-
Bradford Teaching Hospitals NHS Foundation Trust
-
Leeds Community Healthcare NHS Trust
-
Newcastle upon Tyne Hospitals NHS Foundation Trust
-
Cardiff & Vale University with Aneurin Bevan University Health Board (Newport)
-
Hull and East Yorkshire Hospitals NHS Trust
-
Harrogate and District NHS Foundation Trust
-
Mid Yorkshire Hospitals NHS Trust (Wakefield)
-
Lancashire Care NHS Foundation Trust
-
Sussex Community NHS Trust (Brighton).
Sites were opened throughout the participant recruitment phase and, of these sites, three (Bradford, Leeds and Wakefield) did not open but were in various stages of contracts, training, site initiation visits and approvals when the trial closed to recruitment. During the recruitment phase, we received interest from three other sites that opened after they received REC and local research and development (R&D) approvals: NHS Tayside (Dundee), NHS Lanarkshire and Kent Community Health NHS Foundation Trust. We were also in discussion with Birmingham Community Healthcare NHS Trust towards the end of the recruitment phase.
Barriers to recruitment
The first site opened to recruitment on 23 June 2015, almost 3 months later than scheduled. Barriers to recruitment included a delayed start due to issues releasing the IMP. Because there was uncertainty about when the IMP would be available, we were unable to confirm a start date for recruitment. Sites were expected to recruit their first patient within 35 days of submission of their site specific information forms to their local R&D. Local checks were likely to include the availability of the IMP. Once the IMP had been released, there was a slow rate of sites opening over the summer owing to staff availability at the sites. At three sites key staff were on long-term leave or had left and were waiting for new staff to be appointed before proceeding.
Participant recruitment
Once open to recruitment there were fewer than expected eligible patients at sites. The pre-screening data from sites indicated various and multiple reasons for patients not being approached (Figure 1). The main reasons for participant ineligibility were:
-
already taking aspirin or other prohibited medication
-
having a small or otherwise ineligible ulcer.
FIGURE 1.
The AVURT pre-screening study flow. a, The actual number of patients pre-screened was higher than 457 as the first version of the pre-screening log was not mandatory; and b, reasons not mutually exclusive.
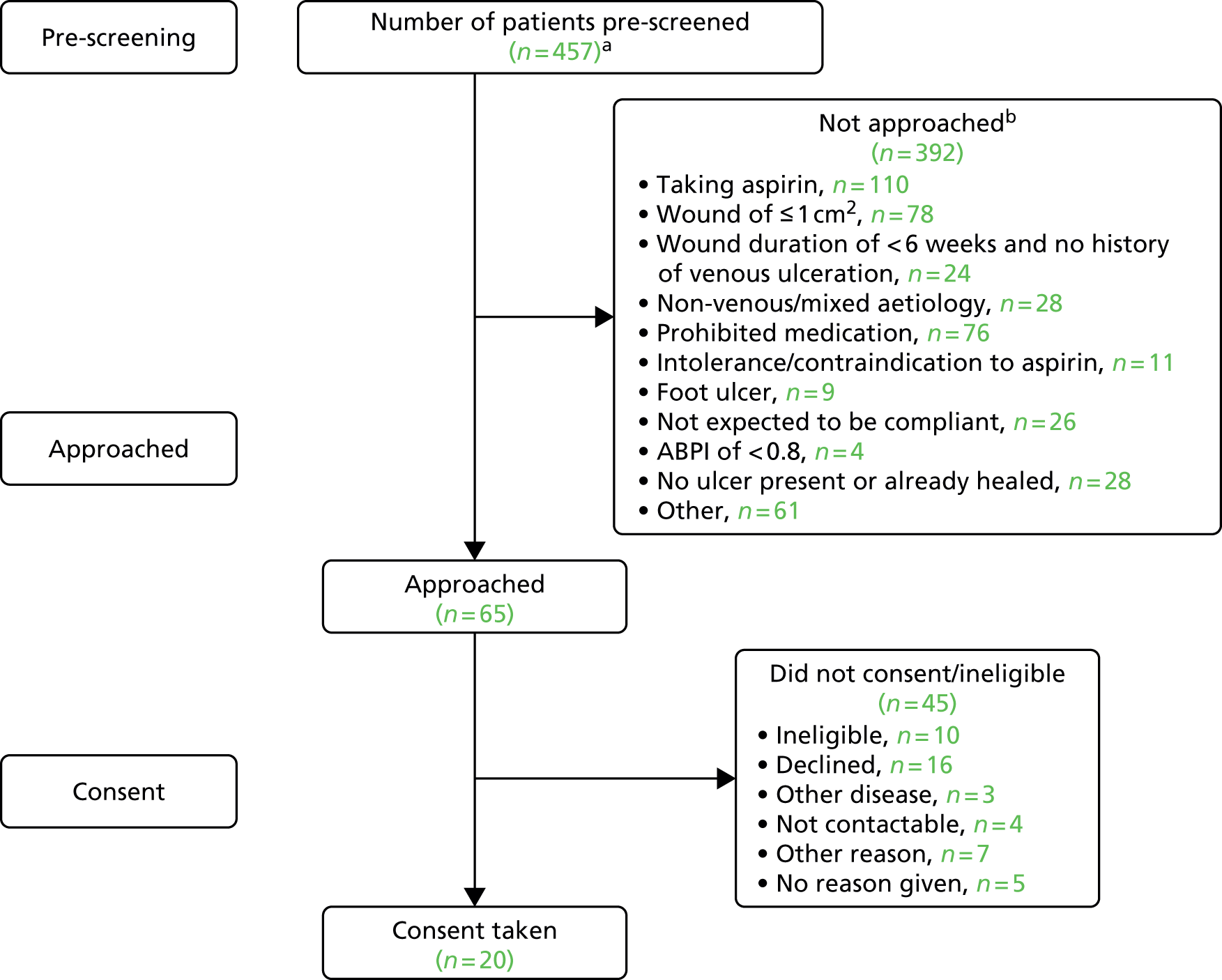
In the last few months of recruitment, when all sites were open, the trial was recruiting three to five participants per month (see Appendix 14). It was generally thought by some sites that a large proportion of leg ulcer patients were receiving treatment in general practices (i.e. in primary care) or outside primary care in specialist clinics and by district nurses. It was therefore likely that the patients being seen by the secondary recruiting sites were older, more likely to have mixed disease and, therefore, more likely to be already taking aspirin.
Strategies to improve recruitment
Strategies to improve recruitment were explored. Modification of the eligibility criteria was assessed with reference to the pre-screening log data. The only acceptable modification to the exclusion and inclusion criteria was to include a smaller wound size. However, those with a wound area of < 1 cm2 were excluded, as these ulcers usually heal very rapidly.
In October and November 2015, the chief investigator and a trial manager contacted the recruiting sites that had been the first to open (Kent, Hull, Brighton, Harrogate and Newcastle) to discuss possible solutions to improve participant recruitment. During meetings with sites, a number of ideas were discussed, including:
-
Advertising via social media.
-
Posters to inform patients about the study.
-
Newsletter for sites.
-
Flyers for sites to remind staff to recruit to the study.
-
Radio advertising (Hull and Harrogate).
-
An amendment to protocol to allow participants to visit sites for follow-up purposes (the protocol stated that patients will not be invited to attend clinics for research purposes), and to introduce per-patient payments for visits that were not part of routine care. This was a particular problem for one site (Newcastle) that did not routinely see patients in clinic after their initial visit.
-
Allowing for more telephone follow-ups so that participants did not need to come in to clinic as regularly as once per week or once per fortnight.
-
Remove minimum ulcer size from the eligibility criteria, in line with the study being conducted in New Zealand. 29
Apart from removing the minimum ulcer size criterion, each of the options considered would have potentially benefited just one or two of the recruiting sites and, therefore, a variety of strategies would need to be implemented across the trial. The funder and REC had required for regular follow-up to monitor patient safety and, therefore, less frequent follow-up was not viewed as a feasible option by the research team.
A flyer was produced and sent to sites to remind clinic staff to recruit to the trial (December 2015) and three electronic newsletters were sent to sites (two during recruitment, in October 2015 and December 2015) to update on participant recruitment in the trial (the third newsletter was sent in June 2016 after recruitment had closed). The flyer and newsletters were relatively cheap to produce and did not require ethics approval and so could be sent out swiftly. During training, sites were reminded to identify potential participants before opening so that the participants could be approached as soon as the sites were given the green light to recruit.
Recruitment from primary care
We explored recruitment from primary care, which was supported by the trial’s DMC. Two options were considered: to use general practices to identify patients and refer them to the secondary care sites already participating in the study (PICs) or to use general practices as recruitment and treatment centres.
The chief investigator approached the NIHR National Speciality Lead for Primary Care in December 2015 to explore how they might be able to support the study and to request some initial pre-screening to see how many patients could be identified in primary care. The NIHR Clinical Research Network, South London, UK, contacted a number of general practices on our behalf with the trial protocol including the inclusion and exclusion criteria and received three responses:
-
One Clinical Commissioning Group provided comments on the difficulty of identifying potential patients using a database search as many of the eligibility criteria, for example size of ulcer and duration of ulcer, are not coded. We were also advised that many patients were taking aspirin over the counter and that this may not currently be recorded in some patients’ records.
-
Two general practices identified a total of three potential participants in total.
We also approached Clinical Research Network Yorkshire and Humber who ran the trial’s inclusion and exclusion criteria through FARSITE (version 0.9.12.2; NorthWest EHealth Limited, Manchester, UK; https://nweh.co.uk/how-we-do-it/our-technology), a web-based anonymous search of patient records. They identified four suitable patients from 12 general practices.
We were unable to obtain details of practice list sizes, Read codes or criteria used in the searches conducted. However, the results indicated that recruitment from primary care was not a viable option using database searches. In addition, two of the recruiting sites (Lanarkshire and Sussex) advised that they would be unable to support referrals from primary care, which at one site was owing to waiting lists already for the service.
Recruitment using general practices as treatment centres was not investigated. Time constraints and budget constraints for implementing this strategy meant that this was option was not explored.
Summary
There were external factors outside the control of the research team that meant that sites were slow to open. Nine out of the 10 recruiting sites were based in secondary care and, once open, there were fewer than anticipated eligible participants. As this was a Clinical Trial of an Investigational Medicinal Product (CTIMP) study, it required more input from a doctor where nurses would otherwise often take the lead. The site make up was very different from other wound trials in which almost all sites were community based with tissue viability nurses acting as principal investigator, which was not possible here. A range of options were considered and explored to improve recruitment, including recruitment from primary care. Preliminary searches of records in primary care also indicated very few potentially eligible participants. In addition, the trial’s short recruitment window and budget constraints meant that many of the options considered to improve recruitment were not viable without a funded extension.
Chapter 4 Results
Analyses were conducted following the principles of intention to treat with all events analysed according to the participant’s original treatment allocation. Analyses were performed using Stata® version 14 (StataCorp LP, College Station, TX, USA). The trial opened to recruitment on 23 June 2015 and closed to recruitment on 29 February 2016. Participant follow-up was completed on 18 August 2016.
Participant flow
The CONSORT diagrams in Figures 1 and 2 show the flow of participants pre-screened and the flow of eligible participants during the trial. Figure 1 illustrates the number of patients pre-screened (n = 457) and the number of those who consented (n = 20). Pre-screening data were unavailable for the nine remaining patients who consented. Pre-screening was under-reported because some sites did not complete the first pre-screening log. The flow of participants in the trial is illustrated in Figure 2, which shows that 29 patients consented. The number of patients randomised by treatment group, receiving the intended treatment, completing the study protocol, and analysed for the primary outcome is presented. Two patients were excluded after they gave consent: one patient developed potential gastric problems and the other patient, affected by multiple comorbidities, was admitted to hospital.
FIGURE 2.
The AVURT CONSORT diagram.
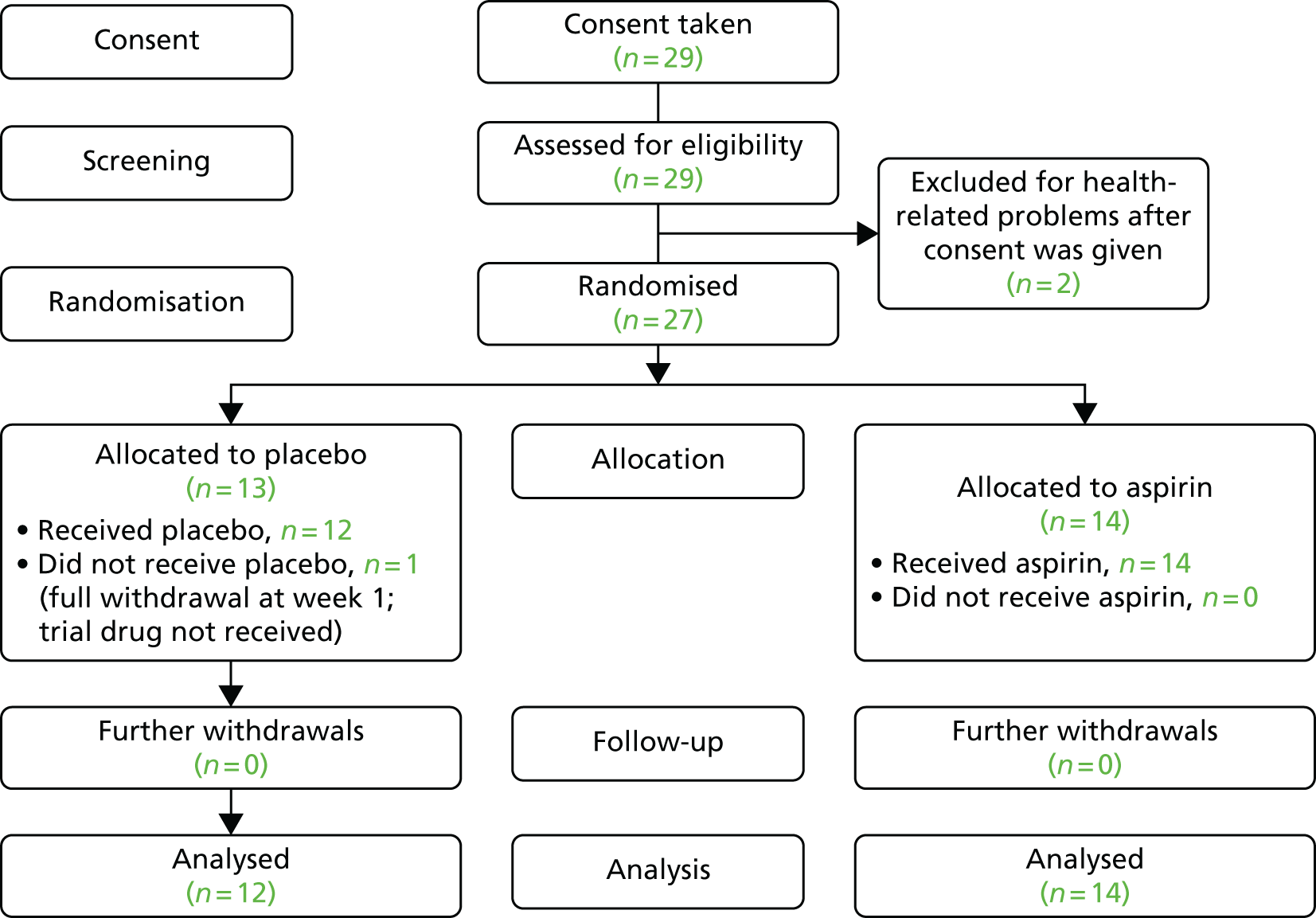
Recruitment
Recruitment took place over 8 months, 2 months longer than originally scheduled. The trial opened on 23 June 2015 and the first participant was recruited on 13 July 2015. Recruitment closed on 29 February 2016. In total, 10 sites were opened and eight randomised a total of 27 patients (Table 2). The sites in Cardiff and Dundee did not recruit any participants. For accumulative recruitment over time see Appendix 14.
| Centre | Participants, n (%) |
|---|---|
| London, St George’s | 7 (25.9) |
| Hull and East Yorkshire | 6 (22.2) |
| Newcastle | 3 (11.1) |
| Lancashire | 3 (11.1) |
| Kent | 3 (11.1) |
| Harrogate | 2 (7.4) |
| Brighton | 2 (7.4) |
| Lanarkshire | 1 (3.7) |
| Cardiff | 0 (0) |
| Dundee/Tayside | 0 (0) |
| Total | 27 (100.0) |
Baseline data
The baseline participant and ulcer-related characteristics, as well as the baseline treatments, are shown in Tables 3–5. The average age of the 27 randomised participants was 62 years (SD 13 years) and two-thirds were male (n = 18). Participants had had their reference ulcer for a median of 15 months and the median size of ulcer was 17.1 cm2. All participants were receiving compression therapy at baseline.
| Participant characteristic | Group | ||
|---|---|---|---|
| Placebo (N = 13) | Aspirin (N = 14) | Overall (N = 27) | |
| Age (years) | |||
| Mean (SD) | 62.1 (15.2) | 62.7 (11.6) | 62.4 (13.2) |
| Median (minimum, maximum) | 66.6 (38.9, 80.8) | 59.2 (47.9, 78.9) | 62.0 (38.9, 80.8) |
| IQR (25%, 75%) | (50.2, 73.4) | (54.0, 74.4) | (50.4, 74.4) |
| Missing, n (%) | 0 (0) | 0 (0) | 0 (0) |
| Gender, n (%) | |||
| Male | 7 (53.9) | 11 (78.6) | 18 (66.7) |
| Female | 6 (46.2) | 3 (21.4) | 9 (33.3) |
| Missing | 0 (0) | 0 (0) | 0 (0) |
| BMI (kg/m2) | |||
| Mean (SD) | 32.1 (8.6) | 36.6 (15.0) | 34.4 (12.3) |
| Median (minimum, maximum) | 28.4 (19.9, 44.1) | 31.6 (20.9, 70.2) | 31.5 (19.9, 70.2) |
| IQR (25%, 75%) | (25.3, 40.6) | (25.9, 40.2) | (25.3, 40.6) |
| Missing, n (%) | 0 (0) | 0 (0) | 0 (0) |
| Mobility, n (%) | |||
| Patient walks freely | 10 (76.9) | 8 (57.1) | 18 (66.7) |
| Patient walks with difficulty | 3 (23.1) | 6 (42.9) | 9 (33.3) |
| Patient is immobile | 0 (0) | 0 (0) | 0 (0) |
| Missing | 0 (0) | 0 (0) | 0 (0) |
| Ankle mobility of reference leg, n (%) | |||
| Patient has full range of motion | 7 (53.9) | 11 (78.6) | 18 (66.7) |
| Reduced range of ankle motion | 6 (46.2) | 3 (21.4) | 9 (33.3) |
| Patient’s ankle is fixed | 0 (0) | 0 (0) | 0 (0) |
| Missing | 0 (0) | 0 (0) | 0 (0) |
| Diabetic, n (%) | |||
| Yesa | 2 (15.4) | 3 (21.4) | 5 (18.5) |
| No | 11 (84.6) | 11 (78.6) | 22 (81.5) |
| Missing | 0 (0) | 0 (0) | 0 (0) |
| Ethnicity, n (%)b | |||
| White British | 11 (84.6) | 12 (85.7) | 23 (85.2) |
| White Irish | 1 (7.7) | 0 (0) | 1 (3.7) |
| Indian | 0 (0) | 1 (7.1) | 1 (3.7) |
| Black African | 1 (7.7) | 1 (7.1) | 2 (7.4) |
| Missing | 0 (0) | 0 (0) | 0 (0) |
| Ulcer-related characteristic | Group | ||
|---|---|---|---|
| Placebo (N = 13) | Aspirin (N = 14) | Overall (N = 27) | |
| Size of ulcer (cm2) | |||
| ≤ 5 cm2, n (%) | 3 (23.1) | 3 (21.4) | 6 (22.2) |
| > 5 cm2, n (%) | 10 (76.9) | 11 (78.6) | 21 (77.8) |
| Mean (SD) | 40.7 (55.1) | 43.1 (47.6) | 42.0 (50.3) |
| Median (minimum, maximum) | 16.0 (2.0, 173.0) | 31.3 (3.8, 155.0) | 17.1 (2.0, 173.0) |
| IQR (25%, 75%) | (6.5, 45.0) | (7.0, 45.0) | (6.5, 45.0) |
| Missing,a n (%) | 0 (0) | 0 (0) | 0 (0) |
| Time since first ulcer (months) | |||
| Mean (SD) | 112.5 (78.5) | 86.4 (86.9) | 99.0 (82.4) |
| Median (minimum, maximum) | 101.0 (11.0, 240.0) | 48 (2.2, 240.0) | 72.0 (2.2, 240.0) |
| IQR (25%, 75%) | (60.0, 168.0) | (18.0, 192.0) | (19.0, 192.0) |
| Missing, n (%) | 0 (0) | 0 (0) | 0 (0) |
| Reference ulcer duration (months) | |||
| Mean (SD) | 58.6 (73.3) | 32.2 (52.0) | 44.9 (63.3) |
| Median (minimum, maximum) | 13.0 (4.0, 234.0) | 16.5 (1.8, 192.0) | 15.0 (1.8, 234.0) |
| IQR (25%, 75%) | (8.0, 72.0) | (3.5, 24.0) | (6.0, 60.0) |
| Missing, n (%) | 0 (0) | 0 (0) | 0 (0) |
| Reference leg, n (%) | |||
| Left | 5 (38.5) | 8 (57.1) | 13 (48.2) |
| Right | 8 (61.5) | 6 (42.9) | 14 (51.9) |
| Missing | 0 (0) | 0 (0) | 0 (0) |
| Ulcers on reference leg | |||
| Mean (SD) | 2.4 (1.7) | 2.4 (1.7) | 2.4 (1.7) |
| Median (minimum, maximum) | 2.0 (1.0, 7.0) | 2.0 (1.0, 6.0) | 2.0 (1.0, 7.0) |
| IQR (25%, 75%) | (1.0, 3.0) | (1.0, 3.0) | (1.0, 3.0) |
| Missing, n (%) | 0 (0) | 0 (0) | 0 (0) |
| Ulcer episodes on reference leg | |||
| Mean (SD) | 1.8 (1.1) | 2.3 (1.9) | 2.1 (1.5) |
| Median (minimum, maximum) | 1.0 (1.0, 4.0) | 1.0 (1.0, 6.0) | 1.0 (1.0, 6.0) |
| IQR (25%, 75%) | (1.0, 2.0) | (1.0, 4.0) | (1.0, 3.0) |
| Missing, n (%) | 0 (0) | 0 (0) | 0 (0) |
| ABPI | |||
| Mean (SD) | 1.1 (0.2) | 1.0 (0.1) | 1.0 (0.2) |
| Median (minimum, maximum) | 1.0 (0.8, 1.5) | 1.0 (0.9, 1.3) | 1.0 (0.8, 1.5) |
| IQR (25%, 75%) | (0.9, 1.2) | (0.9, 1.1) | (0.9, 1.2) |
| Missing, n (%) | 2 (15.4) | 2 (14.3) | 4 (14.8) |
| Ulcer-related pain (0–100 VAS)b | |||
| Mean (SD) | 37.7 (25.9) | 45.4 (36.0) | 41.7 (31.2) |
| Median (minimum, maximum) | 30.0 (5.0, 80.0) | 47.5 (0.0, 100.0) | 35.0 (0.0, 100.0) |
| IQR (25%, 75%) | (15.0, 60.0) | (10.0, 70.0) | (10.0, 70.0) |
| Missing, n (%) | 0 (0) | 0 (0) | 0 (0) |
| Compression treatment characteristic | Group, n (%) | ||
|---|---|---|---|
| Placebo (N = 13) | Aspirin (N = 14) | Overall (N = 27) | |
| Level of ankle pressure compression | |||
| Low (≤ 19 mmHg) | 0 (0) | 0 (0) | 0 (0) |
| Medium (20–39 mmHg) | 0 (0) | 2 (14.3) | 2 (7.4) |
| High (≥ 40 mmHg) | 13 (100) | 12 (85.7) | 25 (92.6) |
| Missing | 0 (0) | 0 (0) | 0 (0) |
| Compression bandaging | |||
| Four layer | 6 (46.2) | 7 (50.0) | 13 (48.2) |
| Three layer | 1 (7.7) | 0 (0) | 1 (3.7) |
| Three-layer reduced compression | 0 (0) | 1 (7.1) | 1 (3.7) |
| Reduced compression | 0 (0) | 1 (7.1) | 1 (3.7) |
| Two-layer hosiery | 2 (15.4) | 0 (0) | 2 (7.4) |
| Reduced compression therapy | 0 (0) | 0 (0) | 0 (0) |
| Othera | 4 (30.8) | 5 (35.7) | 9 (33.3) |
| Missing | 0 (0) | 0 (0) | 0 (0) |
| Primary dressing | |||
| Silver containing | 4 (30.8) | 3 (21.4) | 7 (26.0) |
| Iodine containing | 2 (15.4) | 3 (21.4) | 5 (18.5) |
| Honey containing | 0 (0) | 1 (7.2) | 1 (3.7) |
| Alginate | 1 (7.7) | 0 (0) | 1 (3.7) |
| Soft polymer | 1 (7.7) | 0 (0) | 1 (3.7) |
| Hydrocolloid | 0 (0) | 2 (14.3) | 2 (7.4) |
| Basic wound contact | 3 (23.0) | 3 (21.4) | 6 (22.2) |
| Other antimicrobial dressingb | 1 (7.7) | 0 (0) | 1 (3.7) |
| No dressingc | 1 (7.7) | 2 (14.3) | 3 (11.1) |
| Missing | 0 (0) | 0 (0) | 0 (0) |
Withdrawals and losses to follow-up
Withdrawals and losses to follow-up were recorded on a change of status form (see Appendix 10, Form F) for five patients (placebo, n = 3; aspirin, n = 2). However, there was only one withdrawal during the course of the study (who was in the placebo group) for whom data on primary outcome was not possible to obtain. This was reported at week 2 and so no follow-up data are available for this participant beyond week 1. The other four participants (placebo, n = 2; aspirin, n = 2) either agreed to withdraw from treatment but provided full follow-up until week 25 (i.e. the end of planned follow-up) or healed at a point before withdrawal and, thus, all four provided primary outcome data.
Overall, this means that, in terms of analyses related to primary outcome, only one patient was not included (see Figure 2).
Primary outcomes
Overall, 13 out of the 26 (50.0%) participants followed up were recorded as healing during the course of the study. All the reference ulcers reported to be healed were later confirmed healed approximately 2 weeks later. The first date of reported healing (as reported on the weekly CRF data collection) was used in the statistical analysis (as per the SAP).
Over the course of the trial, 7 out of 12 participants (58.3%) followed in the placebo group were observed to have a healed reference ulcer, and in the aspirin group the corresponding figure was 6 out of 14 (42.9%). It was not possible to estimate median time to healing and/or corresponding 95% CIs as < 50% of participants healed during the 25- to 27-week maximum follow-up period of the study (Table 6). Therefore, the 25th percentile of time to healing was also estimated. Figure 3 shows the unadjusted Kaplan–Meier plot of proportion of reference ulcers healed over time. The unadjusted log-rank test investigating the difference between the survival curves showed no statistically significant difference (test statistic = 1.02; p = 0.30).
| Outcome | Placebo (N = 12)a | Aspirin (N = 14) | Overall (N = 26) |
|---|---|---|---|
| Number healing, n (%) | 7/12 (58.3) | 6/14 (42.9) | 13/26 (50.0) |
| Kaplan–Meier estimate of median time to healing (days) (95% CI) | 98 (21 to NE) | NE (84 to NE) | 147 (97 to NE) |
| Kaplan–Meier estimate of 25th percentile time to healing (days) (95% CI) | 36 (20 to 97) | 111 (69 to NE) | 84 (21 to 111) |
FIGURE 3.
Kaplan–Meier plot of time to ulcer healing by trial arm (unadjusted).
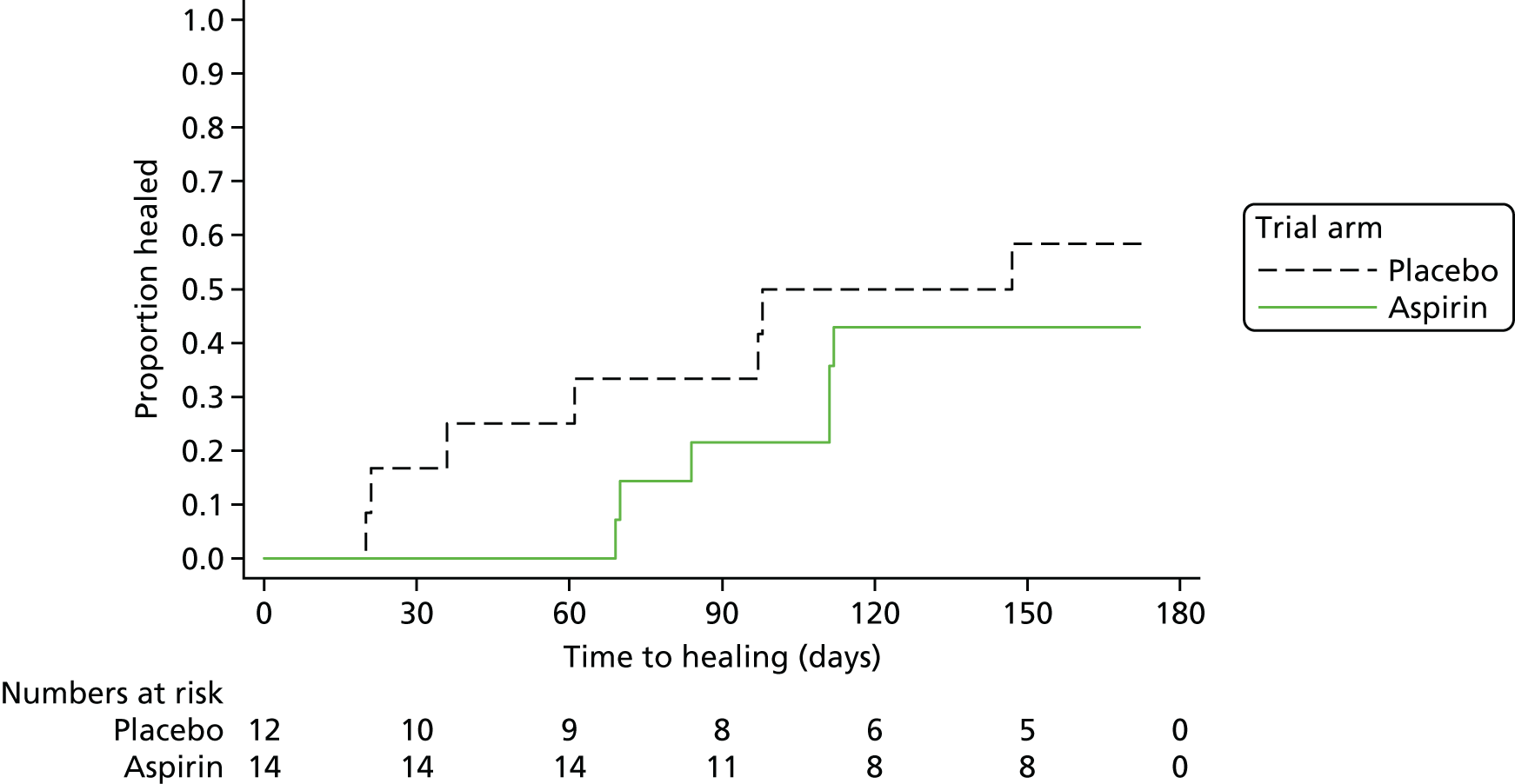
The primary analysis as written in the SAP investigates difference in time to healing using a Cox model adjusting for baseline ulcer area, baseline ulcer duration and centre. The covariates in this model were to be baseline area and duration of the reference ulcer (planned log transformation), randomised allocation and centre as a shared frailty effect. The model was originally chosen with a view to recruiting the target of 100 participants (if successful to follow with a larger definitive study) and was based on analyses performed for the VenUS IV study, which enrolled 457 participants. Given the final distribution of participants across centres (26 participants across eight centres, with six centres contributing three or fewer participants), the shared frailty model is an impractical choice for this analysis. Adjustment with centre as a covariate alongside ulcer area and duration would yield a very low number of events per variable (at approximately four events) and lower than the 10 events that has been previously recommended from simulation studies using Cox regression. 43 Therefore, we present the Kaplan–Meier curves (see Figure 3), log-rank test and unadjusted Cox regression as planned but caution against over-interpretation; we also present the results from the Cox regression adjusted for log-area and log-ulcer duration but without adjustment for centre, giving HRs and corresponding 95% CIs (Table 7).
| Parameter | HR (95% CI) | p-value |
|---|---|---|
| Unadjusted Cox regression | ||
| Aspirin vs. placebo (allocation) | 0.58 (0.19 to 1.72) | 0.322 |
| Adjusted Cox regression | ||
| Aspirin vs. placebo (allocation) | 0.58 (0.18 to 1.85) | 0.357 |
| Area (log-transformed) | 0.42 (0.22 to 0.81) | 0.009 |
| Duration (log-transformed) | 0.61 (0.34 to 1.08) | 0.089 |
Overall, these data do not provide evidence of a difference in time to healing with the addition of aspirin to usual care. The placebo group tended to heal more rapidly but this difference is not statistically significant. The numbers within this feasibility study are small and the results are inconclusive in terms of the primary outcome.
Secondary outcomes
Adverse events
Six out of the 26 (23.1%) participants followed up had no reported AEs (placebo, n = 3; aspirin, n = 3) and the remaining 20 had AEs (placebo, n = 9; aspirin, n = 11) (Table 8). The total number of events experienced by participants was compared by trial arm adjusting for the prognostic factors (log of baseline reference ulcer area and log of baseline reference ulcer duration) using negative binomial regression as per the SAP. There was no evidence that participants receiving aspirin were more likely to suffer an AE than those receiving placebo (incidence rate ratio 1.31, 95% CI 0.51 to 3.41; p = 0.58).
| Group, n (%) | |||
|---|---|---|---|
| Placebo (N = 12) | Aspirin (N = 14) | Overall (N = 26) | |
| SAE, n | 0 | 1 | 1 |
| Number of participants with a SAE | 0 (0.0) | 1 (7.1) | 1 (3.8) |
| Non-serious AEs, n | 36 | 52 | 88 |
| Number of participants with a non-serious AE | 9 (75.0) | 11 (78.6) | 20 (76.9) |
| Relatedness of non-serious AE to treatment (blinded assessment) | |||
| Not related | 21 (58.3) | 30 (57.7) | 51 (58.0) |
| Unlikely | 1 (2.8) | 0 (0) | 1 (1.1) |
| Possibly | 7 (19.4) | 18 (34.6) | 25 (28.4) |
| Probably | 7 (19.4) | 4 (7.7) | 11 (12.5) |
| Definitely | 0 (0) | 0 (0) | 0 (0) |
| Missing | 0 (0) | 0 (0) | 0 (0) |
| Severity of non-serious AE (blinded assessment) | |||
| Mild | 31 (86.1) | 44 (84.6) | 75 (85.3) |
| Moderate | 3 (8.3) | 4 (7.7) | 7 (8.0) |
| Severe | 2 (5.6) | 4 (7.7) | 6 (6.8) |
| Missing | 0 (0) | 0 (0) | 0 (0) |
| Total | 36 (100.0) | 52 (100.0) | 88 (100.0) |
Serious adverse events
One participant suffered one SAE during the course of the study with the description ‘blood transfusion for low Hb’ [haemoglobin]. This SAE was classified as expected and judged as severe in grade and probably related to the blinded trial treatment (aspirin). This participant had 15 other non-serious AEs, of which two subsequent events were thought to be related to the earlier SAE with descriptions ‘colonoscopy: colitis’ and ‘gastroscopy: stomach ulcer’.
Non-serious adverse events
There were 88 non-serious AEs (placebo, n = 36; aspirin, n = 52) recorded in total among 20 participants (placebo, n = 9; aspirin, n = 11).
Ulcer-related pain
The mean baseline VAS score for ulcer-related pain was 37.7 (95% CI 22.0 to 53.4) in the placebo group and was slightly higher at 45.4 (95% CI 24.6 to 66.2) in the aspirin group. At week 5, VAS scores had reduced in both groups with the mean VAS score at week 5 at 13.3 (95% CI 0.3 to 26.3) in the placebo group and 28.5 (95% CI 10.6 to 46.3) in the aspirin group (Table 9 and Figure 4).
| Pain VAS score | Placebo | Aspirin | Overall |
|---|---|---|---|
| Baseline, n | 13 | 14 | 27 |
| Mean (95% CI) | 37.7 (22.0 to 53.4) | 45.4 (24.6 to 66.2) | 41.7 (29.3 to 54.0) |
| Median (minimum, maximum) | 30.0 (5.0, 80.0) | 47.5 (0.0, 100.0) | 35.0 (0.0, 100.0) |
| Week 5, n | 12 | 13 | 25a |
| Mean (95% CI) | 13.3 (0.3 to 26.3) | 28.5 (10.6 to 46.3) | 21.2 (10.4 to 32.0) |
| Median (minimum, maximum) | 2.5 (0.0, 60.0) | 20.0 (0.0, 75.0) | 10.0 (0.0, 75.0) |
FIGURE 4.
Plot of the mean VAS pain score at baseline and at week 5 by trial arm.
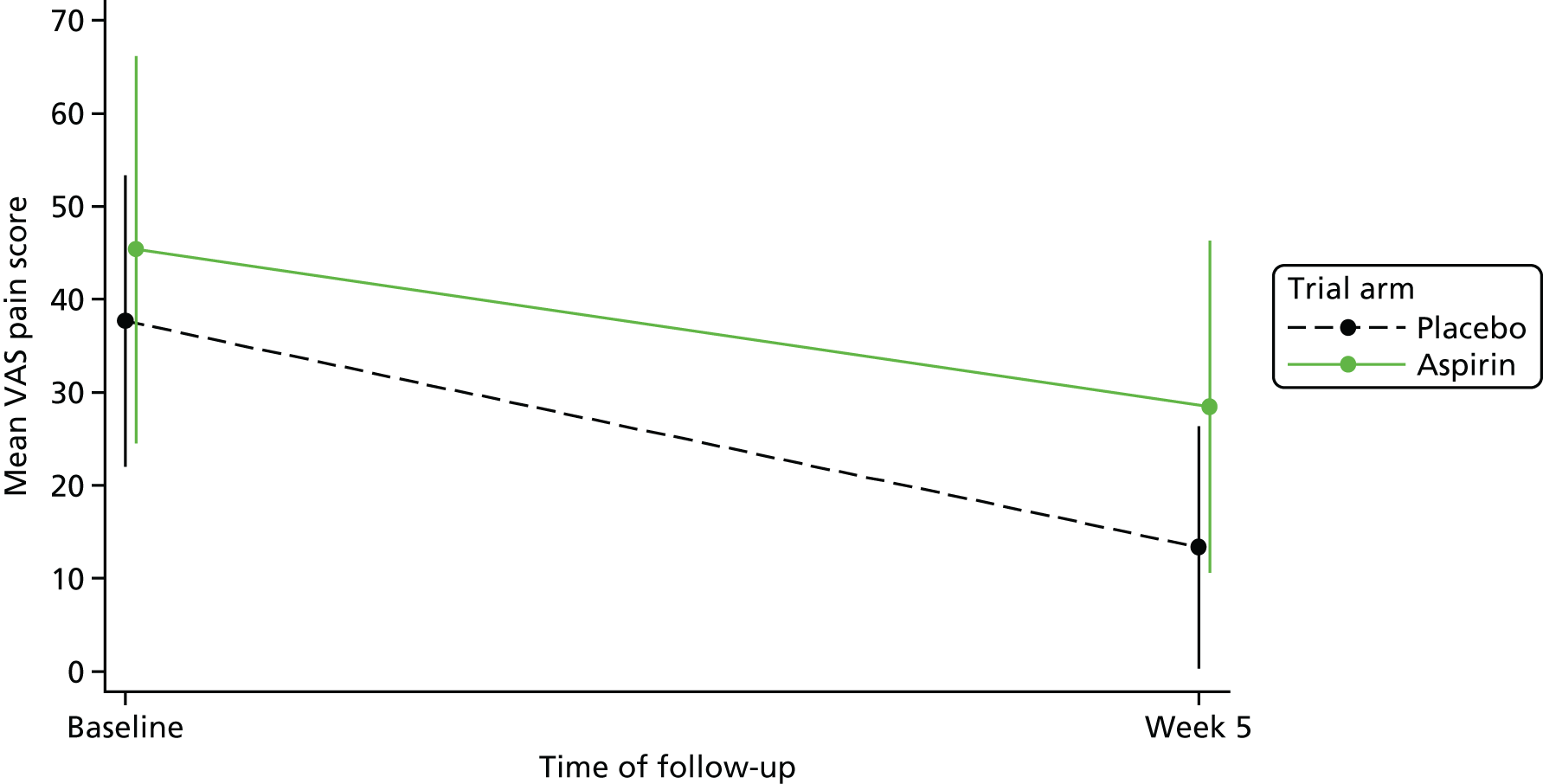
Recurrence
Out of the 13 participants who healed, 12 (92.3%) were assessed for ulcer recurrence using the recurrence assessment form (see Appendix 10, Form E), which was completed in the week 25 follow-up. A recurrence was reported for two participants (placebo, n = 1; aspirin, n = 1). In both cases, the participant was seen in clinic and the ulcer/wound site was clinically assessed. The time in days between ulcer healing and recurrence for these two participants was 126 days (aspirin, healed at week 10) and 158 days (placebo, healed at week 3).
Time to first investigational medicinal product dose
Participants took their first dose of study drug a median of 4 days after randomisation (range 1 to 12 days) (Table 10).
| Time to first dose (days) | Placebo (n = 12) | Aspirin (n = 14) | Overall (n = 26) |
|---|---|---|---|
| Median (minimum, maximum) | 3.0 (1.0, 7.0) | 4.0 (3.0, 12.0) | 4.0 (1.0, 12.0) |
| 95% CI around median | 2.0 to 7.0 | 4.0 to 7.0 | 3.0 to 6.0 |
Time of day
The time of day that participants generally took their IMP was recorded for 24 out of the 27 participants randomised (88.9%; placebo, n = 11; aspirin, n = 13) (Table 11). The majority took the IMP in the morning (70.8%; placebo, n = 9; aspirin, n = 8).
| Time of first dose | Group, n (%) | ||
|---|---|---|---|
| Placebo (N = 11) | Aspirin (N = 13) | Overall (N = 24) | |
| Morning | 9 (81.8) | 8 (61.5) | 17 (70.8) |
| Afternoon | 0 (0) | 3 (23.1) | 3 (12.5) |
| Evening | 2 (18.2) | 2 (15.4) | 4 (16.7) |
Ulcer area
Seven out of the 27 participants (26%; placebo, n = 3; aspirin, n = 4) had missing data for baseline ulcer area, as measured by the analysis of a photograph, and so the measure that was calculated manually by the research nurses was used instead for these participants. Photographs were not available for three measurements during follow-up and so the area calculated from tracings was also used in these cases. Participants who healed during the course of the study contributed to the computation of the ulcer area until healing or 2 weeks after healing, according to the availability of an analysable photograph. Table 12 reports, by trial arm, the number of participants with a valid measure of ulcer area, and the mean and the SD of the ulcer area for each week. The mean ulcer area fluctuates more widely in the placebo group than in the aspirin group (Figure 5) with a distinct fortnightly pattern. These fluctuations are caused by two participants who attended their clinic appointments fortnightly and, coincidentally, in the same calendar weeks, and whose ulcer sizes were particularly large compared with those of the other participants in the placebo group. For example, at baseline, the area of their ulcers was 168 cm2 and 129 cm2, while the mean of the ulcer area of the other placebo participants at baseline was 19.1 cm2 (minimum of 2.14 cm2 and maximum of 78.0 cm2). One of these two participants had an ulcer that was extended around the back of the leg, causing difficulties in the estimation of the area. Figure 6 presents the mean ulcer area over time for the two groups, but with the measurements from these two participants removed.
| Visit week | Ulcer area (cm2) | |||||
|---|---|---|---|---|---|---|
| Placebo (N = 13) | Aspirin (N = 14) | Overall (N = 27) | ||||
| n | Mean (SD) | n | Mean (SD) | n | Mean (SD) | |
| 0 | 13 | 39.0 (53.1) | 14 | 33.8 (37.1) | 27 | 36.3 (44.7) |
| 1 | 9 | 22.9 (31.2) | 11 | 29.7 (25.4) | 20 | 26.6 (27.6) |
| 2 | 10 | 29.8 (43.5) | 12 | 23.8 (19.2) | 22 | 26.5 (31.9) |
| 3 | 8 | 15.7 (12.8) | 11 | 39.2 (42.6) | 19 | 29.3 (34.9) |
| 4 | 10 | 32.7 (48.4) | 10 | 38.7 (38.9) | 20 | 35.7 (42.8) |
| 5 | 6 | 5.9 (7.0) | 12 | 24.9 (24.2) | 18 | 18.6 (21.9) |
| 6 | 8 | 32.8 (52.3) | 7 | 28.1 (25.9) | 15 | 30.6 (40.8) |
| 7 | 5 | 10.0 (6.9) | 10 | 16.9 (18.6) | 15 | 14.6 (15.7) |
| 8 | 8 | 35.1 (53.4) | 10 | 23.3 (14.5) | 18 | 28.5 (36.4) |
| 9 | 5 | 7.2 (7.5) | 12 | 22.3 (16.7) | 17 | 17.9 (16.0) |
| 10 | 9 | 27.1 (43.8) | 12 | 26.0 (24.9) | 21 | 26.5 (33.3) |
| 11 | 5 | 7.3 (7.9) | 12 | 23.6 (24.6) | 17 | 18.8 (22.1) |
| 12 | 6 | 37.9 (51.3) | 11 | 26.3 (22.3) | 17 | 30.4 (34.1) |
| 13 | 3 | 7.7 (9.5) | 11 | 31.4 (31.0) | 14 | 26.3 (29.3) |
| 14 | 7 | 31.7 (42.8) | 9 | 25.2 (22.5) | 16 | 28.1 (31.8) |
| 15 | 5 | 7.0 (8.1) | 8 | 25.0 (18.9) | 13 | 18.1 (17.7) |
| 16 | 5 | 33.0 (37.1) | 9 | 26.4 (18.6) | 14 | 28.7 (25.5) |
| 17 | 1 | 89.2 (.)a | 8 | 37.5 (28.8) | 9 | 43.2 (32.0) |
| 18 | 4 | 24.2 (22.6) | 8 | 25.8 (19.7) | 12 | 25.2 (19.7) |
| 19 | 3 | 14.2 (8.5) | 5 | 27.6 (17.2) | 8 | 22.6 (15.4) |
| 20 | 4 | 41.0 (33.7) | 6 | 26.7 (12.2) | 10 | 32.4 (22.7) |
| 21 | 2 | 11.5 (10.2) | 6 | 44.8 (31.2) | 8 | 36.5 (30.8) |
| 22 | 4 | 39.4 (33.8) | 6 | 22.5 (16.6) | 10 | 29.2 (24.7) |
| 23 | 2 | 10.9 (7.9) | 7 | 29.1 (19.3) | 9 | 25.0 (18.7) |
| 24 | 3 | 34.9 (43.7) | 7 | 27.3 (20.8) | 10 | 29.6 (27.0) |
| 25 | 2 | 12.2 (12.7) | 7 | 33.0 (20.5) | 9 | 28.4 (20.5) |
| 26 | 0 | – | 0 | – | 0 | – |
| 27 | 0 | – | 1 | 4.8 (.)a | 1 | 4.8 (.)a |
FIGURE 5.
Mean of ulcer area and 95% CI for each week of follow-up stratified by allocation group (lower confidence limits truncated at zero).
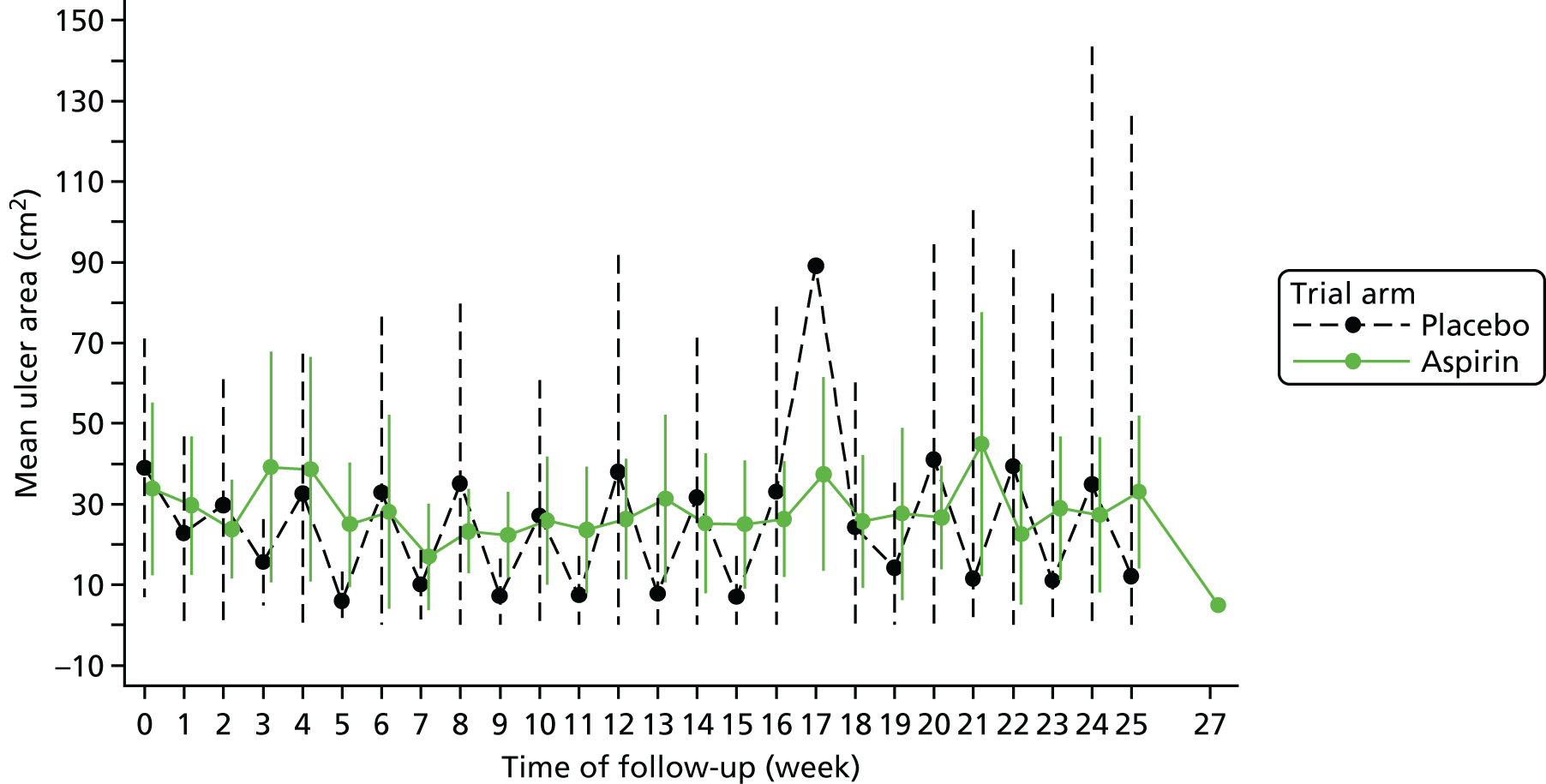
FIGURE 6.
Mean of ulcer area and 95% CI for each week of follow-up stratified by allocation group without the two placebo group participants with rather extended ulcers (lower confidence limits truncated at zero).
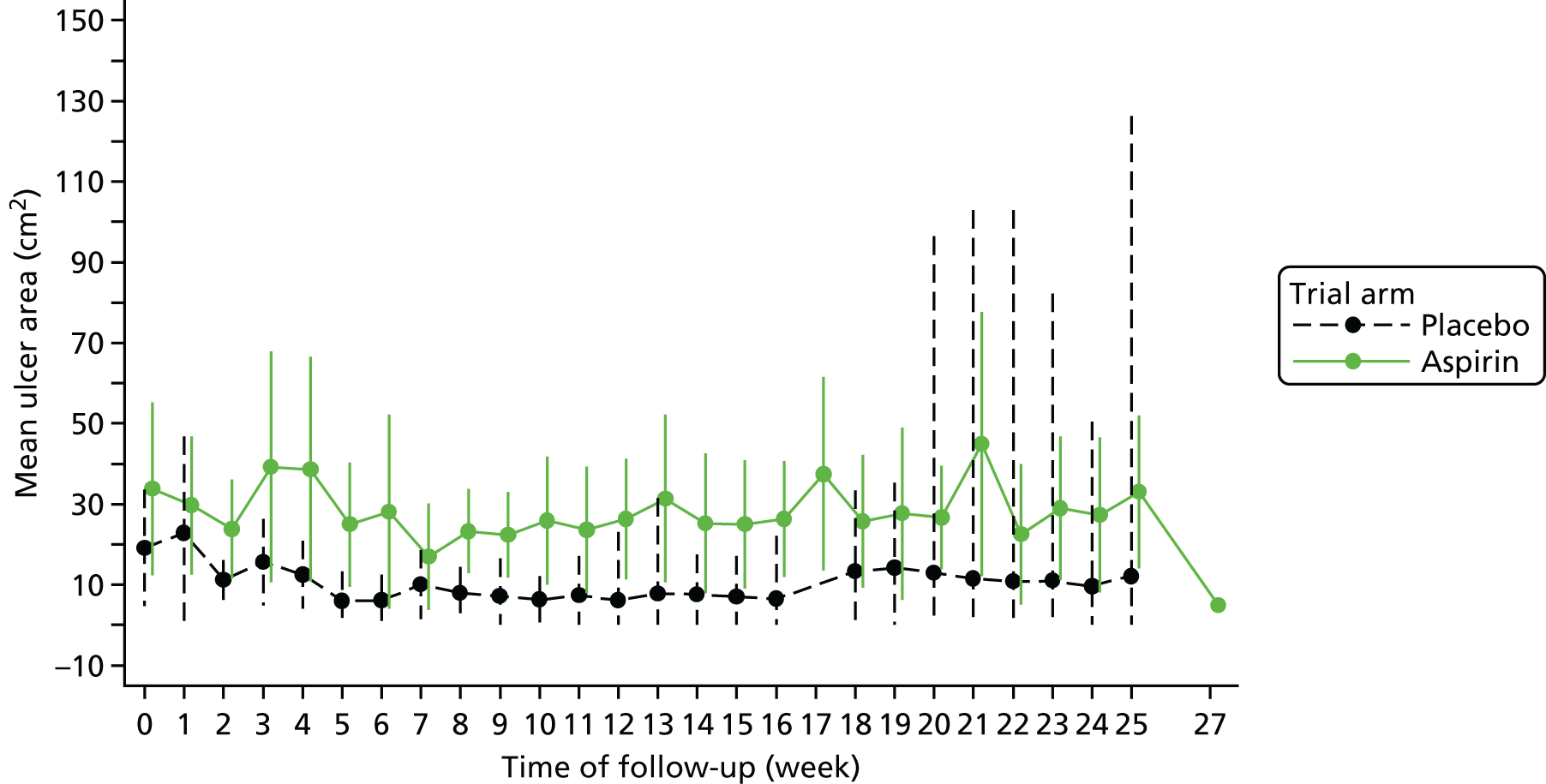
Compliance
Compliance with compression therapy
The mean number of visits attended up until healing or study exit was 13.3 (SD 7.3) and 17.4 (SD 6.8) in the placebo and aspirin groups, respectively. Ten out of 12 participants (83.3%) in the placebo group and 10 out of 14 participants (71.4%) in the aspirin group were fully compliant with their compression therapy (Table 13). Two participants in the placebo group (16.7%) and four participants in the aspirin group (23.1%) were partially compliant. Reasons mentioned by participants for not complying with compression were pain, slipping of the compression bandage, and the participant independently applying the compression garment and, thus, not guaranteeing the correct level of compression. Two of the participants who were classified as partially compliant, one in each group, declared full compliance in half of their visits, while the other four participants did so for at least 89% of their visits.
| Compliance with compression therapy | Group, n (%) | ||
|---|---|---|---|
| Placebo (N = 12) | Aspirin (N = 14) | Overall (N = 26) | |
| Full compliance | 10 (83.3) | 10 (71.4) | 20 (76.9) |
| Partial compliance | 2 (16.7) | 4 (28.6) | 6 (23.1) |
| No compliance at all | 0 (0) | 0 (0) | 0 (0) |
| Total | 12 (100.0) | 14 (100.0) | 26 (100.0) |
Compliance with AVURT capsules
The mean number of visits attended between the administration of the first dose and the date of healing or study exit was 12.8 (SD 7.0) and 17.2 (SD 6.5) in the placebo and aspirin groups, respectively. The imbalance between the groups is probably due to participants in the placebo group healing faster than those in the aspirin group and, therefore, the number of visits in the placebo group is smaller than in the aspirin group.
Eight out of the 12 participants (66.7%) in the placebo group were deemed fully compliant with AVURT capsules and four (33.3%) were partially compliant (Table 14). In the aspirin group, 11 out of the 14 participants (78.6%) were deemed fully compliant and three (21.4%) were partially compliant. Among the partially compliant participants, two in the placebo group and two in the aspirin group were fully compliant for at least 88% of their visits. Reasons for not being fully compliant included illness, forgetting to take the capsule and experiencing an AE. The other three participants (placebo, n = 2; aspirin, n = 1) were deemed to be fully compliant for ≤ 54% of their visits. Two of these participants (placebo, n = 1; aspirin, n = 1) were withdrawn from treatment at week 8 and week 14, respectively, while one participant (in the placebo group) tended to forget to take the capsule.
| Compliance with AVURT capsules | Group, n (%) | ||
|---|---|---|---|
| Placebo (N = 12) | Aspirin (N = 14) | Overall (N = 26) | |
| Full compliance | 8 (66.7) | 11 (78.6) | 19 (73.1) |
| Partial compliance | 4 (33.3) | 3 (21.4) | 7 (26.9) |
| No compliance at all | 0 (0) | 0 (0) | 0 (0) |
| Total | 12 (100.0) | 14 (100.0) | 26 (100.0) |
Table 15 summarises the level of compliance with the study drug, as assessed by the returned pill count. Ten participants in the placebo group (83.4%) and 10 participants in the aspirin group (71.5%) took at least 90% of the AVURT capsules they should have taken.
| Percentage | Group, n (%) | ||
|---|---|---|---|
| Placebo (N = 12) | Aspirin (N = 14) | Total (N = 26) | |
| 100 | 4 (33.4) | 6 (42.9) | 10 (38.5) |
| 90–99 | 6 (50.0) | 4 (28.6) | 10 (38.5) |
| 80–89 | 1 (8.3) | – | 1 (3.8) |
| 70–79 | – | 2 (14.3) | 2 (7.8) |
| 60–69 | – | 1 (7.1) | 1 (3.8) |
| 50–59 | – | – | – |
| 40–49 | 1 (8.3) | – | 1 (3.8) |
| 30–39 | – | – | – |
| 20–29 | – | 1 (7.1) | 1 (3.8) |
| 10–19 | – | – | – |
| 0–9 | – | – | – |
| Total | 12 (100.0) | 14 (100.0) | 26 (100.0) |
Resource use
Resource use: compression therapy
All participants in the placebo group were prescribed high-level compression therapy (≥ 40 mmHg) at baseline. In the aspirin group, 12 were prescribed with high-level compression and two with medium-level compression (20–39 mmHg) (see Table 5).
The total number of changes to the compression therapy prescribed at baseline during the study was 16, with five changes (31.3%) to a medium compression level and nine changes (56.3%) to a high compression level, one change (6.2%) to a low compression level and one change (6.2%) to no compression at all. In the placebo group, four participants had their level of compression changed: two of them changed from a high level of compression to a higher level, one changed from a medium level to a high level and the fourth participant changed from a high level to a higher level, then to a medium level for 2 weeks and then back to a high level. In the aspirin group, seven participants had their level of compression changed: two participants changed from a high level to a higher level, two changed from a high level to a medium level, one changed from a medium level to a high level, one started with a high level, changed to a medium level for 1 week and then went back to a high level, and one went from a medium level to a low level for 2 weeks, then no compression for 10 weeks and after that back to a high level.
The maximum number of changes to level of compression therapy that a participant had during the follow-up period was three (Table 16). Four out of the 12 participants in the placebo group (33.3%) and seven in the aspirin group (50.0%) had at least one change to level of compression therapy; overall, 11 out of the 26 participants (42.3%) had at least one change during their follow-up. The highest number of changes was seen in those participants staying in the study more than 4 months: four out of the six changes (66.7%) in the placebo group and eight out of the 10 changes (80.0%) in the aspirin group were prescribed to participants belonging to this group.
| Period of healing/censoring (months) | Changes to compression therapy, n | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Placebo (N = 12) | Aspirin (N = 14) | Total (N = 26) | ||||||||||
| 0 | 1 | 2 | 3 | 0 | 1 | 2 | 3 | 0 | 1 | 2 | 3 | |
| 0–2 | 2 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 2 | 0 | 0 |
| 2–4 | 2 | 0 | 0 | 0 | 4 | 2 | 0 | 0 | 6 | 2 | 0 | 0 |
| > 4 | 4 | 1 | 0 | 1 | 3 | 3 | 1 | 1 | 7 | 4 | 1 | 2 |
| Total | 8 | 3 | 0 | 1 | 7 | 5 | 1 | 1 | 15 | 8 | 1 | 2 |
Resource use: compression therapy
Table 17 shows that during the follow-up period, a total of 25 changes to type of compression bandaging were made, 11 (44.0%) in the placebo group and 14 (56.0%) in the aspirin group. Overall, the most frequent changes were to other types of bandaging (32.0%), to three-layer bandaging (20.2%) and to two-layer hosiery bandaging (16.0%).
| Compression bandaging | Group, n (%) | ||
|---|---|---|---|
| Placebo (N = 12) | Aspirin (N = 14) | Overall (N = 26) | |
| Four layer | 2 (18.2) | 1 (7.1) | 3 (12.0) |
| Three layer | 2 (18.2) | 3 (21.4) | 5 (20.0) |
| Three-layer reduced compression | 0 (0) | 1 (7.1) | 1 (4.0) |
| Reduced compression | 1 (9.1) | 0 (0) | 1 (4.0) |
| Two-layer hosiery | 2 (18.2) | 2 (14.3) | 4 (16.0) |
| Reduced compression hosiery | 1 (9.1) | 0 (0) | 1 (4.0) |
| Othera | 2 (18.2) | 6 (43.0) | 8 (32.0) |
| No bandaging | 1 (9.1) | 1 (7.1) | 2 (8.0) |
| Total number of changes | 11 (100.0) | 14 (100.0) | 25 (100.0) |
The four-layer bandaging system was the most frequently used type of compression in this study (Table 18), with 16 participants (59.3%) [eight in the placebo group (61.5%) and eight in the aspirin group (57.1%)] receiving it at least once during their follow-up.
| Compression bandaging received at least once, n (%) | Placebo (n = 13) | Aspirin (n = 14) | Overall (n = 27) |
|---|---|---|---|
| Four layer | 8 (61.5) | 8 (57.1) | 16 (59.3) |
| Three layer | 2 (15.4) | 3 (21.4) | 5 (18.5) |
| Three-layer reduced compression | 0 (0) | 2 (14.3) | 2 (7.4) |
| Reduced compression | 1 (7.7) | 1 (7.1) | 2 (7.4) |
| Two-layer hosiery | 4 (30.8) | 2 (14.3) | 6 (22.2) |
| Reduced compression hosiery | 1 (7.7) | 0 (0) | 1 (3.7) |
| Othera | 6 (42.6) | 7 (50.0) | 13 (48.1) |
| No bandaging | 1 (7.7) | 1 (7.1) | 2 (7.4) |
Resource use: dressing
The mean number of changes to the type of primary dressing was higher in those participants with a longer follow-up (> 4 months: mean 1.8, SD 2.0) than in participants with a shorter follow-up (≤ 4 months: mean 1.3, SD 1.5); Table 19). The mean number of changes to type of dressing per participant during the study was 1.6 (SD 1.8) overall, 1.3 (SD 1.5) for the placebo group and 1.9 (SD 2.0) for the aspirin group.
| Number of changes by time period | Group | ||
|---|---|---|---|
| Placebo (n = 12) | Aspirin (n = 14) | Overall (n = 26) | |
| 0–2 months | |||
| Number of participants | 4 | 0 | 4 |
| Mean (SD) | 1.5 (1.0) | – | 1.5 (1.0) |
| Median (minimum, maximum) | 1.0 (1.0, 3.0) | – | 1.0 (1.0, 3.0) |
| IQR (25%, 75%) | (1.0, 2.0) | – | (1.0, 2.0) |
| 2–4 months | |||
| Number of participants | 2 | 6 | 8 |
| Mean (SD) | 1.0 (1.4) | 1.3 (2.0) | 1.3 (1.8) |
| Median (minimum, maximum) | 1.0 (0.0, 2.0) | 0.5 (0.0, 5.0) | 0.5 (0.0, 5.0) |
| IQR (25%, 75%) | (0.0, 2.0) | (0.0, 2.0) | (0.0, 2.0) |
| > 4 months | |||
| Number of participants | 6 | 8 | 14 |
| Mean (SD) | 1.2 (2.0) | 2.3 (2.0) | 1.8 (2.0) |
| Median (minimum, maximum) | 0.0 (0.0, 5.0) | 3.0 (0.0, 5.0) | 1.0 (0.0, 5.0) |
| IQR (25%, 75%) | (0.0, 2.0) | (0.0, 3.5) | (0.0, 3.0) |
| Whole study period | |||
| Number of participants | 12 | 14 | 26 |
| Mean (SD) | 1.3 (1.5) | 1.9 (2.0) | 1.6 (1.8) |
| Median (minimum, maximum) | 1.0 (0.0, 5.0) | 1.5 (0.0, 5.0) | 1.0 (0.0, 5.0) |
| IQR (25%, 75%) | (0.0, 2.0) | (0.0, 3.0) | (0.0, 3.0) |
A total of 41 changes to the primary dressing were recorded during the study (Table 20). A total of 15 out of the 41 changes were in the placebo group (36.6%) while the remaining 26 were in the aspirin group (63.4%). Overall, the most frequent changes were to silver-containing dressing (19.5%) and to basic wound contact dressing (19.5%).
| Type of dressing | Group, n (%) | ||
|---|---|---|---|
| Placebo (N = 12) | Aspirin (N = 14) | Overall (N = 26) | |
| Basic wound contact | 3 (20.0) | 5 (19.2) | 8 (19.5) |
| Silver containing | 1 (6.7) | 7 (26.9) | 8 (19.5) |
| Iodine containing | 2 (13.3) | 3 (11.5) | 5 (12.2) |
| Hydrocolloid | 2 (13.3) | 1 (3.8) | 3 (7.3) |
| Alginate | 1 (6.7) | 1 (3.8) | 2 (4.9) |
| Soft polymer | 0 (0) | 2 (7.7) | 2 (4.9) |
| Honey containing | 0 (0) | 1 (3.8) | 1 (2.4) |
| Hydrogel | 0 (0) | 1 (3.8) | 1 (2.4) |
| Foam | 1 (6.7) | 0 (0) | 1 (2.4) |
| Film | 0 (0) | 0 (0) | 0 (0) |
| Other antimicrobial dressinga | 0 (0) | 1 (3.8) | 1 (2.4) |
| Otherb | 2 (13.3) | 2 (7.7) | 4 (9.8) |
| No dressing | 3 (20.0) | 2 (7.7) | 5 (12.2) |
| Total | 15 (100.0) | 26 (100.0) | 41 (100.0) |
Table 21 shows that, overall, silver-containing dressings and basic wound contact were the most widely used types of dressing in this study: 13 out of the 27 participants (48.1%) had a silver-containing dressing at least once during their follow-up, five participants were in the placebo group (38.5%) and eight in the aspirin group (57.1%); 11 participants (40.7%) had basic wound contact dressing at least once, six of them were in the placebo group (46.2%) and five (35.7%) were in the aspirin group.
| Type of dressing received at least once | Group, n (%) | ||
|---|---|---|---|
| Placebo (N = 13) | Aspirin (N = 14) | Overall (N = 27) | |
| Silver containing | 5 (38.5) | 8 (57.1) | 13 (48.1) |
| Basic wound contact | 6 (46.2) | 5 (35.7) | 11 (40.7) |
| Iodine containing | 3 (23.1) | 5 (35.7) | 8 (29.6) |
| Hydrocolloid | 1 (7.7) | 3 (21.4) | 4 (14.8) |
| Alginate | 2 (15.4) | 1 (7.1) | 3 (11.1) |
| Honey containing | 0 (0) | 2 (14.3) | 2 (7.4) |
| Soft polymer | 1 (7.7) | 1 (7.1) | 2 (7.4) |
| Hydrogel | 0 (0) | 1 (7.1) | 1 (3.7) |
| Foam | 1 (7.7) | 0 (0) | 1 (3.7) |
| Film | 0 (0) | 0 (0) | 0 (0) |
| Other antimicrobial dressinga | 1 (7.7) | 1 (7.1) | 1 (3.7) |
| Otherb | 1 (7.7) | 2 (14.3) | 3 (11.1) |
| No dressing | 4 (30.8) | 4 (28.6) | 8 (29.6) |
Resource use: wound consultations
The mean number of wound consultations per week was 2.1 (SD 1.4) in the placebo group, 1.9 (SD 0.7) in the aspirin group, and 2.0 (SD 1.0) overall (Table 22).
| Number of wound consultations per week | Group | ||
|---|---|---|---|
| Placebo (n = 12) | Aspirin (n = 14) | Overall (n = 26) | |
| Mean (SD) | 2.1 (1.4) | 1.9 (0.7) | 2.0 (1.0) |
| Median (minimum, maximum) | 1.9 (1.0, 5.7) | 1.9 (1.0, 3.5) | 1.9 (1.0, 5.7) |
| IQR (25%, 75%) | (1.1, 2.0) | (1.6, 2.0) | (1.3, 2.0) |
Protocol violations or issues that may have an impact on analysis
One participant was unblinded after the trial had completed treatment and analysis was being undertaken. The participant was unblinded via the emergency unblinding procedure after consultation and agreement with the Independent Steering Committees and DMCs. At the time of writing, the TMG has remained blind to this participant’s allocation.
No protocol violations were reported.
Chapter 5 Discussion
This pilot trial, in which feasibility of recruitment was one of the objectives, was only able to recruit 27% of its target sample size but has important findings for informing the trial design of future CTIMP studies for this patient population.
Summary of findings
Owing to under-recruitment, we were unable to confirm the efficacy of aspirin for VLU healing. Recruitment was more difficult than anticipated owing to the large number of patients already prescribed aspirin medication, predominantly for cardiovascular risk factor management, or who were on concomitant antiplatelet therapies. Others were excluded because they had contraindications to aspirin therapy, had small ulcers of ≤ 1 cm2 (that were anticipated to heal rapidly) or did not want to be enrolled (frequently reported to be associated with the need for regular clinic attendance).
Participants included in the trial had ulcers of a significantly long duration and may have been considered more difficult to heal. The ulcers that most participants had were large, with 80% of them having a surface area of > 5 cm2.
The relatively small number of participants recruited to the trial means that any data should be interpreted with caution. There were a large number of AEs during the trial (n = 89) with most participants (77%) suffering at least one. The majority of these AEs were non-serious (n = 88) and among these 51 (58%) were not related to aspirin. However, there was one SAE of gastrointestinal bleeding requiring blood transfusion. Aspirin was generally well tolerated and there was no evidence of a difference in the number of experienced AEs in the two trial arms.
Compliance with the medication was good, with nearly three-quarters of participants being fully compliant and one-quarter partially compliant. Similarly, the compliance to compression therapy was very good overall and was similar between the treatment groups.
Before study commencement, a decision was made not to collect any data on health-related quality of life and collect only limited resource use health economic data. This was for reasons of brevity and the focus of the trial being feasibility. However, it was hypothesised that participants may receive some benefit in terms of pain relief if they received 300 mg of aspirin daily. The self-reported pain scores at 5 weeks showed no evidence that participants in the aspirin group suffered less pain. The number of clinic visits and dressing/compression changes were also similar among the groups.
The patients taking part in the study tended to have a high BMI, some were experiencing a high degree of ulcer-related pain and the majority receiving high-compression bandaging. A few months into recruitment, and finding that many of the patients were not meeting the trial criteria, we explored recruitment from primary care, which involved a limited database search of records held in primary care. However, very few patients were identified as there were limitations associated with conducting the search on the trial’s eligibility criteria. There may have been potentially eligible patients being treated by nurses in general practices. Further investigation of recruitment from primary care was not undertaken owing to time and budget constraints.
Limitations
There are a number of limitations of the trial. Because the trial under recruited (by 73%), some of the pre-planned analyses were infeasible or inappropriate. Because of the low number of participants, for example, the shared centre frailty effects could not be tested and the repeated measures mixed model for ulcer area could not be estimated. In these cases, the most appropriate analyses were performed. Information concerning compliance with both medication and compression therapy was obtained by participant self-report and pill count.
At the outset of the trial the sponsor did not enforce recruiting sites to record pre-screening log data owing to the disparate nature of the clinics seeing and recruiting patients. It was felt by the sponsor that the heterogeneous nature of the clinics and, therefore, patients would render a pre-screening log meaningless. For example, one site, at St George’s, offered a complex wound service led by a vascular surgeon (high prevalence of PAD and very chronic wounds). In contrast, other sites, such as the one in Brighton, were effectively based in the community with a large number of patients with very small and less complex ulcers that were managed solely by nurses. This omission was rectified later but, consequently, the pre-screening data reported is an under-representation of the number of patients screened. It was not possible to conduct second checks of the pre-screening data and there is the possibility of duplicate and incomplete entries. In addition, the data should be interpreted with caution owing to major differences among demographics between some of the recruiting sites.
It is worth reflecting that the relative rates of healing in this study are very far removed from those seen in the earlier UK and Spanish studies,25,26 which prompted this call for research. One explanation for this may have been the much larger ulcers in this trial or, perhaps, their more chronic nature.
Strengths
Retention and follow-up rates were high and there were few missing data. Both the trial coapplicants and the REC were concerned that participants may suffer a significant number of aspirin-related AEs and SAEs, but this did not appear to be the case. In fact, the number of related events was quite low and the medication appeared to be well tolerated. The frequency of follow-up, once weekly or fortnightly, was to ensure, in part, that information on AEs was identified early (perhaps before progression to more serious AEs). It is possible that some participants suffered other AEs that were related to aspirin, but this seems unlikely given the frequency of assessment.
The aspirin and placebo were manufactured and over-encapsulated effectively in a large capsule. There were some concerns that this may affect participant compliance. However, the sponsor’s previous experience with IMP manufacturer and capsule size was reassuring. Indeed, in this trial participant compliance with medication was generally very good and non-compliance was not associated with the size of the capsule.
Data were captured on ulcer area using two formats: paper tracings and digital image analysis. Both of these techniques are supported by published data3,44 on their reliability and have been used extensively in clinical trials previously. We also decided to use digital image analysis as we thought it may prove easier for the nursing staff. However, many nurses felt more comfortable with wound tracing, especially with the larger ulcers, which may be more difficult to capture in a two-dimensional digital photograph. At the most important time points (baseline and completion of the study), wound tracings were taken to avoid any problems associated with two-dimensional image analysis.
Interpretations
Owing to under-recruitment, we were unable to confirm the effect sizes found in two other published studies, and reliably and confidently establish the safety of aspirin in this population.
It may have been possible to recruit more patients to the trial if other centres could have been rapidly involved. However, the very short nature of the trial meant that it was impossible to involve other centres in a timely fashion.
Generalisability/contribution of this study to the evidence
AVURT was based on plausible results from two other studies investigating aspirin for VLUs. 25,26 This trial has demonstrated that it was possible to randomise those participants who could be recruited, but the study clearly demonstrated that it was not feasible to recruit the necessary participants in the context of a RCT in the UK.
Chapter 6 Conclusions
AVURT was a Phase II randomised pilot trial of aspirin versus placebo for the treatment of patients with chronic venous leg ulceration. It was not possible to recruit the planned number of patients despite an unfunded extension to the trial and, therefore, it can be concluded that a larger Phase III (effectiveness) trial would not be feasible. A future trial would need many centres over a long period of time to get the required numbers if there were no modifications to the inclusion and exclusion criteria.
The Health Technology Assessment (HTA) programme reviewed progress on the study in January 2016 and advised that, in view of the recruitment difficulties that this pilot trial had experienced and was continuing to experience, they did not think it was feasible to recruit the target sample size by the end of July 2016 (the last possible date that participants could be recruited owing to IMP expiry) and, therefore, would not support a funded extension.
There were a number of reasons why patients were excluded or unsuitable for the trial. These included patients with small ulcers at screening and concomitant aspirin therapy (or other concomitant medical therapies that were exclusion criteria). Small ulcers were excluded from the trial because these heal rapidly and the effect of aspirin was unlikely to significantly improve outcome (with potentially increased risks). In retrospect, it may have been possible to randomise patients who were taking 75 mg of aspirin to the larger dose of 300 mg that was used in the trial. However, there are no data (from biological plausibility studies or small trials) to suggest that taking a larger dose of aspirin may have proven effective.
There was a large number of patients already taking aspirin or other antiplatelet medications at screening for cardiovascular indications. It seems likely that this proportion will increase in the future with an ageing population. This suggests that it will likely prove increasingly difficult in the future to recruit to trials of aspirin in patients with chronic venous ulcer of the leg, even if significant changes were made to the present trial design.
The centres recruiting in this trial were identified because they expressed an interest in the trial, estimated that they would be able to recruit sufficient numbers and were the highest recruiters to a previous chronic VLU trial funded by NIHR HTA run through the YTU. The majority of these centres were in a clinic setting (mainly in secondary care locations). It is possible that patients with chronic VLUs are, perhaps increasingly, managed in primary care or in the community. It is also quite possible that younger or fitter patients with easier-to-manage chronic VLUs who may have been eligible for this trial (as they are more likely to have fewer contraindications) were more likely to be managed in the community. Attempts were made to explore recruitment from primary care but this was limited given the relatively short duration of the trial. It is recommended that future studies consider rigorous pre-screening in the design stage of the trial to obtain realistic numbers of potentially eligible patients and to inform recruitment strategy.
Overall, sites found this trial very difficult to recruit to, despite suggesting to the contrary when originally approached. The narrow recruitment window and overall short duration of the trial made it impossible to make changes to the trial recruitment strategies to meet that challenge. The trial’s DMC and TSC both requested that it was reported that the commissioned call for AVURT did not allow sufficient time for the trial to be performed. A 6-month recruitment period was not long enough.
AVURT was designed to very carefully identify AEs. The trial applicants and REC were both concerned that AEs may go unnoticed (e.g. mild gastrointestinal side effects leading to potentially life-threatening complications). There were a large number of AEs in the trial, but most were unrelated to the IMP and only one was serious. The overall safety of 300 mg of aspirin once daily in this group of participants would appear to be reasonable (and when a SAE was noted, it was pre-dated by milder gastrointestinal symptoms). Given these observations, it would appear reasonable to suggest that any further trial of aspirin intervention in chronic VLU might be possible to be performed with fewer clinic visits.
The intervention itself (300 mg of aspirin) appeared feasible and safe in this population but a Phase III RCT would not appear to be feasible in the UK in a hospital clinic-based setting.
Acknowledgements
We would like to thank the participants who took part in this trial. We would also like to thank the principal investigators, research nurses and health-care professionals who screened and recruited participants into the study, collected data and supported the study.
Trial Steering Committee members
We would like to thank external members of the Trial Steering Group: Professor Julie Brittenden, Professor Andrea Nelson, Mrs Angela Oswald (leg ulcer nurse practitioner), Professor Janet Powell and Mr Laurie Williams (coapplicant and patient representative).
Data Monitoring Committee
We would like to thank external members of the DMC: Professor Peter Franks, Dr Sarah Brown, Mr Jonathan Earnshaw and Mr Toby Richards.
Contributions of authors
Helen Tilbrook (Research Fellow) was the lead study trial co-ordinator. She contributed to the development of the trial protocol and the first draft of the report.
Laura Clark (Research Fellow) contributed to the co-ordination of the study.
Liz Cook (Trial Co-ordinator) contributed to the co-ordination of the study and contributed to the first draft of the report.
Martin Bland (Professor of Health Statistics Emeritus) contributed to the development of the grant application and trial protocol and provided statistical expertise.
Hannah Buckley (Medical Statistician) contributed to the design of the study and wrote the SAP.
Ian Chetter (Associate Dean for Research Hull York Medical School/University of Hull) contributed to the development of the grant application and trial protocol and clinical expertise. Professor Chetter was also a principal investigator.
Jo Dumville (Senior Lecturer in Health Sciences) contributed to the development of the grant application and trial protocol and had project oversight.
Chris Fenner (Core Surgical Trainee) reviewed ulcer photographs and calculated ulcer area.
Rachael Forsythe (Clinical Research Fellow) reviewed ulcer photographs and calculated ulcer area.
Rhian Gabe (Reader in Clinical Trials and Senior Trial Statistician) contributed to the development of the grant application, reviewed the SAP and provided statistical oversight.
Keith Harding (Clinical Lead for Wound Healing) contributed to the development of the grant application and trial protocol and provided clinical expertise.
Alison Layton (Consultant Dermatologist) contributed to the development of the grant application and trial protocol, provided clinical expertise and was also a principal investigator.
Ellie Lindsay (President, Leg Club Foundation) contributed to the development of the grant application, trial protocol and participant information, and had project oversight as a member of the TMG.
Catriona McDaid (Senior Research Fellow) contributed to the development of the grant application and trial protocol and had project oversight.
Christine Moffatt (Professor of Clinical Nursing Research) contributed to the development of the grant application and trial protocol.
Debbie Rolfe (Acting Head of Research Governance and Regulatory Assurance Manager) was sponsor representative for the study, contributed to the design of the study protocol and was also responsible for project oversight and monitoring of the study.
Illary Sbizzera (Trainee Statistician) undertook the statistical analysis and contributed to the first draft of the report.
Gerard Stansby (Professor of Vascular Surgery and Honorary Consultant Surgeon) contributed to the development of the grant application and trial protocol and was a principal investigator.
David Torgerson (Director of YTU) provided advice on trial conduct and contributed to the design of the grant application and trial protocol.
Peter Vowden (Honorary Clinical Director NIHR WoundTec HTC) contributed to the development of the grant application and trial protocol and was a principal investigator.
Laurie Williams (patient representative and member of AVURT TSC) contributed to the development of the grant application, trial protocol and participant information, and had project oversight as a member of the TSC.
Robert Hinchliffe (Honorary Consultant in Vascular Surgery) lead applicant and chief investigator for AVURT. He had overall responsibility for the design and implementation of the study and the writing of the first draft of the report with final approval of report submission.
All authors contributed to the final manuscript.
Data-sharing statement
All data requests should be submitted to the corresponding author for consideration. Access to available anonymised data may be granted following review.
Patient data
This work uses data provided by patients and collected by the NHS as part of their care and support. Using patient data is vital to improve health and care for everyone. There is huge potential to make better use of information from people’s patient records, to understand more about disease, develop new treatments, monitor safety, and plan NHS services. Patient data should be kept safe and secure, to protect everyone’s privacy, and it’s important that there are safeguards to make sure that it is stored and used responsibly. Everyone should be able to find out about how patient data are used. #datasaveslives You can find out more about the background to this citation here: https://understandingpatientdata.org.uk/data-citation.
Disclaimers
This report presents independent research funded by the National Institute for Health Research (NIHR). The views and opinions expressed by authors in this publication are those of the authors and do not necessarily reflect those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care. If there are verbatim quotations included in this publication the views and opinions expressed by the interviewees are those of the interviewees and do not necessarily reflect those of the authors, those of the NHS, the NIHR, NETSCC, the HTA programme or the Department of Health and Social Care.
References
- Valencia IC, Falabella A, Kirsner RS, Eaglstein WH. Chronic venous insufficiency and venous leg ulceration. J Am Acad Dermatol 2001;44:401-21. https://doi.org/10.1067/mjd.2001.111633.
- de Araujo T, Valencia I, Federman DG, Kirsner RS. Managing the patient with venous ulcers. Ann Intern Med 2003;138:326-34. https://doi.org/10.7326/0003-4819-138-4-200302180-00012.
- Ashby RL, Gabe R, Ali S, Adderley U, Bland JM, Cullum NA, et al. Clinical and cost-effectiveness of compression hosiery versus compression bandages in treatment of venous leg ulcers (Venous leg Ulcer Study IV, VenUS IV): a randomised controlled trial. Lancet 2014;383:871-9. https://doi.org/10.1016/S0140-6736(13)62368-5.
- Margolis DJ, Allen-Taylor L, Hoffstad O, Berlin JA. The accuracy of venous leg ulcer prognostic models in a wound care system. Wound Repair Regen 2004;12:163-8. https://doi.org/10.1111/j.1067-1927.2004.012207.x.
- Callam M. Prevalence of chronic leg ulceration and severe chronic venous disease in western countries. Phlebology 1992;7:6-12.
- Guest JF, Ayoub N, McIlwraith T, Uchegbu I, Gerrish A, Weidlich D, et al. Health economic burden that wounds impose on the National Health Service in the UK. BMJ Open 2015;5. https://doi.org/10.1136/bmjopen-2015-009283.
- Guest JF, Ayoub N, McIlwraith T, Uchegbu I, Gerrish A, Weidlich D, et al. Health economic burden that different wound types impose on the UK’s National Health Service. Int Wound J 2017;2:322-30.
- O’Meara S, Cullum N, Nelson EA, Dumville JC. Compression for venous leg ulcers. Cochrane Database Syst Rev 2012;11. https://doi.org/10.1002/14651858.CD000265.pub3.
- Madden M. The ghost of Nora Batty: a qualitative exploration of the impact of footwear, bandaging and hosiery interventions in chronic wound care. Chronic Illn 2015;11:218-29. https://doi.org/10.1177/1742395314566824.
- Dogra S, Rai R. Venous leg ulcer: topical treatment, dressings and surgical debridement. Indian Dermatol Online J 2014;5. https://doi.org/10.4103/2229-5178.137820.
- O’Meara S, Richardson R, Lipsky BA. Topical and systemic antimicrobial therapy for venous leg ulcers. JAMA 2014;311:2534-5. https://doi.org/10.1001/jama.2014.4574.
- Jull AB, Arroll B, Parag V, Waters J. Pentoxifylline for treating venous leg ulcers. Cochrane Database Syst Rev 2012;12. https://doi.org/10.1002/14651858.CD001733.pub3.
- Cullum N, Buckley HL, Dumville J, Hall J, Lamb K, Madden MT, et al. Wounds research for patient benefit: a 5 year programme of research. Programme Grants Appl Res 2016;4.
- Nelson EA, Prescott RJ, Harper DR, Gibson B, Brown D, Ruckley CV. A factorial, randomized trial of pentoxifylline or placebo, four-layer or single-layer compression, and knitted viscose or hydrocolloid dressings for venous ulcers. J Vasc Surg 2007;45:134-41. https://doi.org/10.1016/j.jvs.2006.09.043.
- Gohel MS, Davies AH. Pharmacological agents in the treatment of venous disease: an update of the available evidence. Curr Vasc Pharmacol 2009;7:303-8. https://doi.org/10.2174/157016109788340758.
- Barwell JR, Davies CE, Deacon J, Harvey K, Minor J, Sassano A, et al. Comparison of surgery and compression with compression alone in chronic venous ulceration (ESCHAR study): randomised controlled trial. Lancet 2004;363:1854-9. https://doi.org/10.1016/S0140-6736(04)16353-8.
- Davies A, Epstein D, Warwick J, Poskitt K, Gohel M, Bulbulia R, et al. A Randomized Clinical Trial to Compare Early Versus Delayed Treatment of Superficial Venous Reflux in Patients With Chronic Venous Ulceration. Early Venous Reflux Ablation (EVRA) Ulcer Trial [22 November 2016] n.d. www.nets.nihr.ac.uk/projects/hta/11129197.
- Kirsner RS, Baquerizo Nole KL, Fox JD, Liu SN. J Invest Dermatol 2015;135:19-23. https://doi.org/10.1038/jid.2014.444.
- Zuloff-Shani A, Adunsky A, Even-Zahav A, Semo H, Orenstein A, Tamir J, et al. Hard to heal pressure ulcers (stage III-IV): efficacy of injected activated macrophage suspension (AMS) as compared with standard of care (SOC) treatment controlled trial. Arch Gerontol Geriatr 2010;51:268-72. https://doi.org/10.1016/j.archger.2009.11.015.
- Da Costa RM, Ribeiro Jesus FM, Aniceto C, Mendes M. Randomized, double-blind, placebo-controlled, dose-ranging study of granulocyte-macrophage colony stimulating factor in patients with chronic venous leg ulcers. Wound Repair Regen 1999;7:17-25. https://doi.org/10.1046/j.1524-475X.1999.00017.x.
- Vane JR, Botting RM. The mechanism of action of aspirin. Thromb Res 2003;110:255-8. https://doi.org/10.1016/S0049-3848(03)00379-7.
- Campbell CL, Smyth S, Montalescot G, Steinhubl SR. Aspirin dose for the prevention of cardiovascular disease: a systematic review. JAMA 2007;297:2018-24. https://doi.org/10.1001/jama.297.18.2018.
- de Oliveira Carvalho PE, Magolbo NG, De Aquino RF, Weller CD. Oral aspirin for treating venous leg ulcers. Cochrane Database Syst Rev 2016;2. https://doi.org/10.1002/14651858.CD009432.pub2.
- Delaney JA, Opatrny L, Brophy JM, Suissa S. Drug–drug interactions between antithrombotic medications and the risk of gastrointestinal bleeding. CMAJ 2007;177:347-51. https://doi.org/10.1503/cmaj.070186.
- Layton AM, Ibbotson SH, Davies JA, Goodfield MJ. Randomised trial of oral aspirin for chronic venous leg ulcers. Lancet 1994;344:164-5. https://doi.org/10.1016/S0140-6736(94)92759-6.
- del Río Solá ML, Antonio J, Fajardo G, Vaquero Puerta C. Influence of aspirin therapy in the ulcer associated with chronic venous insufficiency. Ann Vasc Surg 2012;26:620-9. https://doi.org/10.1016/j.avsg.2011.02.051.
- Balshem H, Helfand M, Schünemann HJ, Oxman AD, Kunz R, Brozek J, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol 2011;64:401-6. https://doi.org/10.1016/j.jclinepi.2010.07.015.
- Weller CD, Barker A, Darby I, Haines T, Underwood M, Ward S, et al. Aspirin in venous leg ulcer study (ASPiVLU): study protocol for a randomised controlled trial. Trials 2016;17. https://doi.org/10.1186/s13063-016-1314-4.
- Jull A, Wadham A, Bullen C, Parag V, Kerse N, Waters J. Low-dose aspirin as an adjuvant treatment for venous leg ulceration: study protocol for a randomized controlled trial (Aspirin4VLU). J Adv Nurs 2016;72:669-79. https://doi.org/10.1111/jan.12864.
- Charlesworth G, Burnell K, Hoe J, Orrell M, Russell I. Acceptance checklist for clinical effectiveness pilot trials: a systematic approach. BMC 2013;13. https://doi.org/10.1186/1471-2288-13-78.
- Jull A, Wadham A, Bullen C, Parag V, Kerse N, Waters J. Low dose aspirin as adjuvant treatment for venous leg ulceration: pragmatic, randomised, double blind, placebo controlled trial (Aspirin4VLU). BMJ 2017;359. https://doi.org/10.1136/bmj.j5157.
- Tilbrook H, Forsythe RO, Rolfe D, Clark L, Bland M, Buckley H, et al. Aspirin for Venous Ulcers: Randomised Trial (AVURT): study protocol for a randomised controlled trial. Trials 2015;16:1-9. https://doi.org/10.1186/s13063-015-1039-9.
- Scottish Intercollegiate Guidelines Network (SIGN) . Management of Chronic Venous Leg Ulcers. A National Clinical Guideline 2010. www.sign.ac.uk/assets/sign120.pdf (accessed 22 January 2018).
- Council of the European Union . Directive 2001 83 EC of the European Parliament and of the Council of 6 November 2001 on the Community Code Relating to Medicinal Products for Human Use 2001. https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/dir_2001_83_cons2009/2001_83_cons2009_en.pdf (accessed 27 February 2018).
- Council of the European Union . Directive 2001 83 EC of the European Parliament and of the Council of 6 November 2001 on the Community Code Relating to Veterinary Medicinal Products 2001. http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:32001L0082 (accessed 27 February 2018).
- Council of the European Union . Directive 2001 83 EC of the European Parliament and of the Council of 4 April 2001 on the Approximation of Laws, Regulations and Administrative Provisions of the Member States Relating to the Implementation of Good Clinical Practice in the Conduct of Clinical Trials on Medicinal Products for Human Use 2001. http://eur-lex.europa.eu/legal-content/EN/ALL/?uri=celex:32001L0020 (accessed 27 February 2018).
- Jensen MP, Miller L, Fisher LD. Assessment of pain during medical procedures: a comparison of three scales. Clin J Pain 1998;14:343-9. https://doi.org/10.1097/00002508-199812000-00012.
- US Department of Health and Human Services Food and Drug Administration . Guidance for Industry Chronic Cutaneous Ulcer and Burn Wounds – Developing Products for Treatment 2006. www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm064981.htm (accessed 22 January 2018).
- National Institute for Health Research . Clinical Trials Toolkit Safety Reporting n.d. www.ct-toolkit.ac.uk/routemap/safety-reporting (accessed 22 January 2018).
- Iglesias C, Nelson E, Cullum N, Torgerson D. VenUS I: a randomised controlled trial of two types of bandage for treating venous leg ulcers. Health Technol Assess 2004;8. https://doi.org/10.3310/hta8290.
- Schulz KF, Altman DG, Moher D. CONSORT Group . CONSORT 2010 statement: updated guidelines for reporting parallel group randomized trials. Ann Intern Med 2010;152:726-32. https://doi.org/10.7326/0003-4819-152-11-201006010-00232.
- St George’s University Hospitals NHS Foundation Trust . Standard Operating Procedures and Templates 2016. www.stgeorges.nhs.uk/education-and-research/research/standard-operating-procedures-and-templates/ (accessed 22 January 2018).
- Peduzzi P, Concato J, Feinstein AR, Holford TR. Importance of events per independent variable in proportional hazards regression analysis II. Accuracy and precision of regression estimates. J Clin Epidemiol 1995;48:1503-10. https://doi.org/10.1016/0895-4356(95)00048-8.
- Samad A, Hayes S, French L, Dodds S. Digital imaging versus conventional contact tracing for the objective measurement of venous leg ulcers. J Wound Care 2002;11:137-40. https://doi.org/10.12968/jowc.2002.11.4.26385.
Appendix 1 Recruiting sites
| Site ID | Site | Sources of recruitment | Date open to recruitment | Target | Participants recruited |
|---|---|---|---|---|---|
| 14 | Freeman Hospital, Newcastle upon Tyne | Hospital outpatients within vascular attended by vascular nurses and specialist vascular nurses | 23 June 2015 | 5 | 3 |
| 11 | St John’s Therapy Centre, London | Hospital outpatient clinic attended by community tissue viability nurses | 6 July 2015 | 12 | 7 |
| 21 | Sussex Community NHS Trust, Brighton General Hospital, Brighton | Community leg ulcer clinic | 9 July 2015 | 6 | 2 |
| 17 | Hull Royal Infirmary, Hull | Hospital outpatient leg ulcer clinic | 10 July 2015 | 8 | 6 |
| 18 | Harrogate District NHS Trust, Harrogate | Hospital outpatient leg ulcer clinic | 23 July 2015 | 4 | 2 |
| 24 | Kent Community Health NHS Trust | Wound medicine centres (set-up to treat ambulant patients with long term wounds). Centres overseen by tissue viability nurses | 28 September 2015 | 10 | 3 |
| 22 | Monklands Hospital, Lanarkshire | Hospital outpatient leg ulcer clinic | 9 October 2015 | 10 | 1 |
| 15 | Wound Healing Research Unit, Cardiff University, Cardiff | General research clinic for wounds within the university | 14 October 2015 | 15–30 | 0 |
| 23 | Ninewells Hospital & Medical School, Dundee | Primary care leg ulcer clinic | 4 November 2015 | 5–11 | 0 |
| 20 | Lancashire Care NHS Foundation Trust | Community nursing case loads | 11 November 2015 | 4 | 3 |
| Total | 79–100 | 27 |
Appendix 2 Pre-trial screening forms
Appendix 3 Patient information sheet
Appendix 4 Consent form
Appendix 5 Screening form
Appendix 6 Prescription template
Appendix 7 Baseline case report form
Appendix 8 Procedure for taking photographs
Appendix 9 Medication diary (form completed by participants)
Appendix 10 Data collection forms (forms completed by health-care professionals)
Appendix 11 The AVURT flow chart

Appendix 12 Ulcer recurrence card
Appendix 13 Study amendments
Ethics submissions
Amendment 1. Substantial (1). Approval not required.
Amendment 2. Substantial (2) and non-substantial (1). Approved 7 May 2016.
Amendment 3. Non-substantial (2). Approved 4 August 2015.
Amendment 4. Substantial (3). Approval not required.
Amendment 5. Substantial (4). Withdrawn.
Amendment 6. Non-substantial (3). Approved 25 July 2016.
Medicines and Healthcare products Regulatory Agency submissions
Amendment 1. Substantial (1). Approved 8 May 2016.
Amendment 2. Substantial (2) and non-substantial (1). Approval not required.
Amendment 3. Non-substantial (2). Approval not required.
Amendment 4. Substantial (3). Approved 14 December 2015.
Amendment 5. Substantial (4). Approval not required.
Amendment 6. Non-substantial (3). Approval not required.
Non-substantial amendments
Amendment 2: non-substantial (1) was not protocol amendment (protocol version 1.3)
Key changes
Minor changes to correct typographical errors and to make clarifications in protocol – sections: 2. Roles and Responsibilities, 3. Study Synopsis, 6.1 Study disease, 7.1 Overall design, 8.1 IMPs and non-IMPs used in the trial, 10. Subject/Patient Recruitment process, 11.1 Informed Consent, 12.1 Screening assessments, 12.2 Treatment procedure, 12.3 Subsequent assessments, 12.5.1 Obtaining, labelling, storing, 12.6.1 Obtaining, labelling, storing, 12.7.1 Obtaining, labelling, storing, 14.3 Data handling and analysis, 16.4.1 Summary of baseline data and flow of participants and appendix 3 Study Flow Chart and Table of Study Assessments.
Informed consent form amended to facilitate five-digit screening ID (ICF1.2).
Amendment 3: non-substantial (2) was a protocol amendment (protocol 1.4)
Key changes
Clarification of management of patients experiencing AEs/SAEs. Protocol amended to state that patients who develop SAEs (and not AEs) to aspirin (or placebo) will be withdrawn from study treatments.
Changes to sections 2. Roles and Responsibilities, 3. Study Synopsis, 6.2 Investigational Medicinal Product (IMP), 6.5 Assessment & management of potential risk, 8. IMP Dosage regimen and rationale, 11.3 Prescribing & Dispensing IMP, 11.6 Discontinuation/withdrawal of participants and stopping rules and 12.1 Screening assessments.
Amendment 6: non-substantial (3) was a protocol amendment (protocol 1.5)
Section 5. Statement to include that unpublished as well as published studies would be included in meta-analysis.
Substantial amendments
Amendment 1: substantial amendment 1 (Medicines and Healthcare products Regulatory Agency) was not a protocol amendment
Simplified IMP Dossier (v2) required adjustment to IMP capsule target weight range.
Amendment 2: substantial amendment 2 (ethics) dated was not a protocol amendment
Two new recruiting sites (Dundee and Lanarkshire) and a change of principal investigator in Bradford.
Amendment 4: substantial amendment 3 (Medicines and Healthcare products Regulatory Agency) was not a protocol amendment
Change to the expiry date of the IMP following stability information from Sharp Clinical Services (UK) Limited.
Amendment 5: substantial amendment 4 (ethics) was not a protocol amendment
Change to principal investigator at Wakefield site. Application withdrawn as trial recruitment was stopped.
Appendix 14 Accumulative recruitment over time
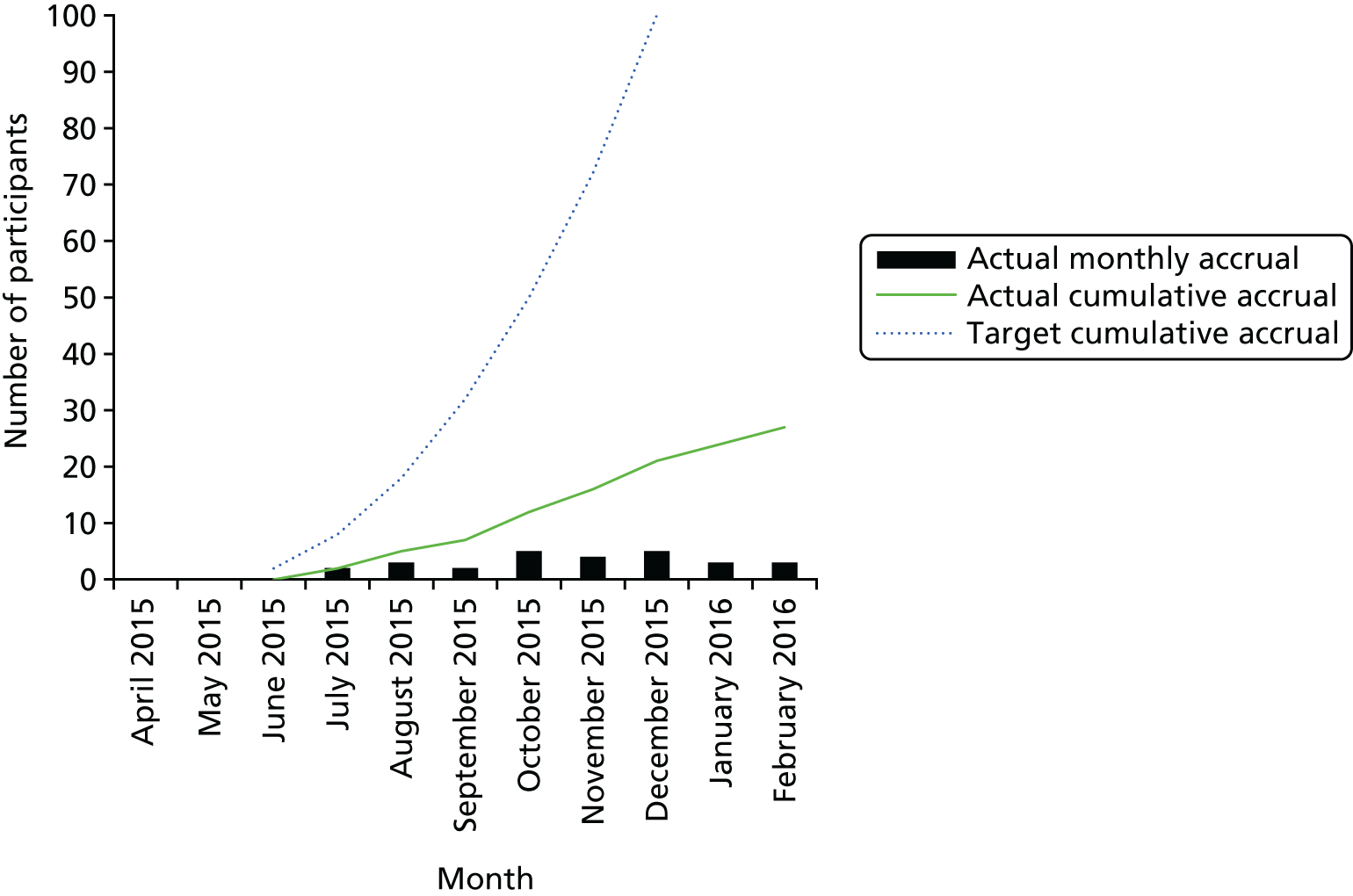
List of abbreviations
- ABPI
- ankle–brachial pressure index
- ACCEPT
- Acceptance Checklist for Clinical Effectiveness Pilot Trials
- AE
- adverse event
- AR
- adverse reaction
- AVURT
- Aspirin for Venous leg Ulcers Randomised Trial
- BMI
- body mass index
- BS-21
- 21-point Box Scale
- CI
- confidence interval
- CONSORT
- Consolidated Standards of Reporting Trials
- CRF
- case report form
- CTIMP
- Clinical Trial of Investigational Medicinal Product
- DMC
- Data Monitoring Committee
- GP
- general practitioner
- HR
- hazard ratio
- HTA
- Health Technology Assessment
- ID
- identifier
- IMP
- investigational medicinal product
- IQR
- interquartile range
- MHRA
- Medicines and Healthcare products Regulatory Agency
- NIHR
- National Institute for Health Research
- PAD
- peripheral arterial disease
- PIC
- patient identification centre
- R&D
- research and development
- RCT
- randomised controlled trial
- REC
- Research Ethics Committee
- RSI
- reference safety information
- SAE
- serious adverse event
- SAP
- statistical analysis plan
- SD
- standard deviation
- SUSAR
- suspected unexpected serious adverse reaction
- TMG
- Trial Management Group
- TSC
- Trial Steering Committee
- VAS
- visual analogue scale
- VenUS IV
- Venous leg Ulcer Study IV
- VLU
- venous leg ulcer
- YTU
- York Trials Unit